
エステルの製造方法
【課題】脱水性並びに耐久性を向上させた分離膜を利用した高効率なエステル化反応プロセスを提供する。
【解決手段】反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜等であり、分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは溶液温度が沸点に達した後に開始すること、からなるエステルの製造方法。
【効果】エステル化反応プロセスを小規模化できるとともに、分離膜の交換等のメンテナンス回数を大幅に低減できる。
【解決手段】反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜等であり、分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは溶液温度が沸点に達した後に開始すること、からなるエステルの製造方法。
【効果】エステル化反応プロセスを小規模化できるとともに、分離膜の交換等のメンテナンス回数を大幅に低減できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、エステルの製造方法に関するものであり、更に詳しくは、触媒共存下で、カルボン酸とアルコールからエステルを製造する際に副生成する水を、水に対して選択透過性をもつ分離膜を利用して、反応器外部に除去することで、高収率でエステルを製造する方法に関するものである。本発明は、分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能とし、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスに関する新技術を提供するものである。
【背景技術】
【0002】
カルボン酸とアルコールからエステルを生成する化学反応は平衡反応であるので、カルボン酸のエステルへの転換効率を向上させるには、大過剰のアルコールを使用する方法と、副生成する水を反応器外部に除去する方法が考えられる。しかし、大過剰のアルコールを使用する方法は、大型の反応容器が必要であり、製造プロセスとして好ましくなく、副生成する水を反応器外部に除去する方法の利用が好ましい。
【0003】
水の除去方法には、蒸留法、吸着法、膜分離法があるが、蒸留法では、大型の蒸留装置が必要であり、消費エネルギーが大きいといった課題に加え、反応溶液が共沸混合物となった場合、消費エネルギーに比して充分に水を除去できないことが指摘されている。
【0004】
一方、吸着法および膜分離法では、脱水に及ぼす共沸の影響は小さく、効率よく水を除去することができる。吸着法では、吸着剤への水の吸着量に限界があるため、吸着剤の再生が不可欠であるのに対して、膜分離法では、分離膜を通過した水が反応系外に排出されるので、連続的に利用することができる等の利点がある。
【0005】
前記のように、連続的に効率よくエステルを製造できることから、近年、膜分離法を利用したエステル化反応に関する研究が盛んに行われている。これらの研究では、分離膜として、ポリビニルアルコール、ポリエーテルイミド等の高分子分離膜と、A型、T型、MER型、PHI型、CHA型ゼオライト等のゼオライト膜に代表される無機多孔質分離膜が利用されているが、高分子膜は、有機溶液に対する耐薬品性および耐熱性が低く、ゼオライト膜等の無機多孔質膜の利用が検討されている。
【0006】
例えば、分離膜にゼオライト膜を利用した研究には、多数の文献(特許文献1〜3、非特許文献1〜4等)があり、多様なカルボン酸とアルコールの組合せで、均一もしくは不均一触媒を利用し、分離膜により溶液もしくは蒸気相から水を選択的に除去することで、効率よくエステルを製造できる。表1にゼオライト膜を利用したエステル化反応の研究例を示す。
【0007】
【表1】

【0008】
表1に示すように、分離膜、特にゼオライト膜を利用することで、効率よくカルボン酸をエスエルに転換できることは知られているが、分離膜の脱水速度および耐久性が不充分であった。
【0009】
非特許文献2および3で説明されるように、分離膜の脱水速度をJ(kg/m2/h)、分離膜の有効面積をS(m2)、反応基質の重量をV(kg)とすれば、分離膜を利用した脱水の効果は JS/Vで説明される。たとえば、乳酸エチル、酢酸エチル等のエステル化反応では、このJS/V値が0.1h−1以上で、効率よくカルボン酸をエステルに転換できる。
【0010】
しかし、利用されるT型ゼオライト膜の脱水速度が、1.0kg/m2/h(特許文献4)と低いため、S/Vを0.1m2/kg以上にする必要があった。これは、基質1トンに対して、有効面積100m2の分離膜が必要であることを意味しており、分離膜の製造メーカーにより多少異なるが、管状分離膜を複数本結束した分離膜モジュールの単位容積当たりの有効膜面積は概ね10m2/m3であるので、反応器容積の約10倍の分離膜が必要になることを示している。表2にゼオライト膜の脱水特性(浸透気化法)を示す。
【0011】
その他、特許文献1では、S/V=0.024m2/kgと比較的小さな膜面積で検討されているが、その他の文献では、S/V=0.1〜40m2/kgであり、反応容器と比べ非常に大きな膜分離器が必要であった。すなわち、分離膜を利用した高効率エステル製造プロセスを小規模化するためには、分離膜の脱水速度向上が必須である。例えば、膜分離器を反応容器と同程度まで小型化するには、分離膜の脱水速度は10kg/m2/h以上が必要である。
【0012】
【表2】
【0013】
充分な脱水速度を有している分離膜であっても、エステル化反応条件下で長期間に渡ってその脱水性能を維持できなければ、頻繁なエステル製造プロセスのメンテナンスが必要となる。すなわち、反応条件下における分離膜の耐久性も重要である。
【0014】
表2に示すように、脱水速度が10kg/m2/h以上となる分離膜は、A型およびCHA型ゼオライト膜であるが、A型ゼオライト膜は酸性条件下における耐久性が著しく低い。非特許文献7によると、A型ゼオライト膜を酸性溶液に接触させた場合には、約10分で分離機能を喪失した。
【0015】
一方、非特許文献8によると、CHA型ゼオライト膜は、類似条件下でも10時間以上水選択透過性を維持しているものの、酸性溶液と接触することで、ゼオライトの構造破壊に起因した脱水速度の低下が顕著であった。
【0016】
耐久性の観点から、エステル化反応へのゼオライト膜の利用に際しては、分離膜と反応溶液の接触を回避できる蒸気透過法が有効であるが、エステル製造プロセスでは、蒸気圧の上限に限界があるため、浸透気化法と比べて、蒸気透過法では脱水速度が低下し、充分な脱水速度が得られなくなるという欠点がある。
【0017】
たとえば、非特許文献3では、T型ゼオライト膜の脱水速度は浸透気化試験では0.8kg/m2/hであったが、蒸気透過試験では0.3〜0.4kg/m2/hに半減している。すなわち、分離膜の脱水速度と耐久性を確保するための反応器の設計および反応操作条件の最適化が必須である。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】特許第4057063号
【特許文献2】特開2002−47213号公報
【特許文献3】特開2006−315991号公報
【特許文献4】特許3686262号
【特許文献5】特開2006−212551号公報
【特許文献6】特開2006−176399号公報
【特許文献7】特開2003−144871号公報
【特許文献8】特開2011−016123号公報
【非特許文献】
【0019】
【非特許文献1】Journal of Membrane Science、2002年、199巻、117−123頁
【非特許文献2】Catalysis Today、2001年、67巻、121−125頁
【非特許文献3】Chemical Engineering Science、2002年、57巻、1577−1584頁
【非特許文献4】Industry and Engineering Chemistry Research、2007年、46巻、3743−3750頁
【非特許文献5】Microporous and Mesoporous Materials,2009年、126巻、107−114頁
【非特許文献6】Journal of Membrane Science、2010年、364巻、318−324頁
【非特許文献7】Journal of Membrane Science、2010年、349巻、189−194頁
【非特許文献8】Journal of Membrane Science、2010年、347巻、193−196頁
【発明の概要】
【発明が解決しようとする課題】
【0020】
以上述べたように、高い脱水速度および耐久性を有する分離膜が開発されていなかったことに加え、エステル製造に不可欠な分離膜の脱水速度および耐久性を向上させうる反応器設計、反応操作条件等に関する検討が不充分であったため、分離膜を利用したエステル化反応プロセスの小規模化が困難であるとともに、分離膜交換等のメンテナンス回数の低減も困難であった。
【0021】
本発明は、分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能にするとともに、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスを提供することを目的とするものである。
【0022】
更に、本発明は、本発明者らの開発した比較的耐酸性が高く、優れた脱水速度を示すCHA型ゼオライト膜(特許文献8)を、エステル化反応に利用するとともに、反応溶液の加熱温度、撹拌速度、分離膜の設置位置、分離膜の温度、分離膜の操作時間等を調整することで、エステル化反応の促進に不可欠な分離膜の脱水速度および耐久性を向上させることにより、エステル化反応プロセスの小規模化、およびメンテナンス回数の低減を実現可能とする新しいエステル化反応プロセスを提供することを目的とするものである。
【課題を解決するための手段】
【0023】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)反応基質を含む反応系から構成される反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始すること、を特徴とするエステルの製造方法。
(2)反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下である、前記(1)記載のエステルの製造方法。
(3)分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去する、前記(1)記載のエステルの製造方法。
(4)(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満である、前記(1)記載のエステルの製造方法。
(5)反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールである、前記(1)記載のエステルの製造方法。
(6)分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜である、前記(1)記載のエステルの製造方法。
(7)多孔質支持体が、0.01〜100μmの大きさの空孔を持ち、その空隙率が10〜70%である、前記(1)記載のエステルの製造方法。
(8)分離器が、反応器内部又は反応器外部に設置される、前記(1)記載のエステルの製造方法。
(9)反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去する、前記(1)記載のエステルの製造方法。
(10)減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させる、前記(1)記載のエステルの製造方法。
(11)反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始する、前記(1)記載のエステルの製造方法。
(12)分離膜の透過側の減圧の度合いが、10kPa未満である、前記(10)記載のエステルの製造方法。
【0024】
次に、本発明について更に詳細に説明する。
本発明は、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を分離膜で除去することによりエステルを製造する方法において、分離膜がたとえばCHA型ゼオライト膜またはNaA型ゼオライト膜であり、分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始することを特徴とするものである。
【0025】
本発明では、上記エステルの製造方法において、分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去すること、反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下であることを好ましい実施の態様としている。
【0026】
また、本発明は、(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満であること、反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールであることを好ましい実施の態様としている。
【0027】
更に、本発明は、分離器が、反応容器外部に設置されること、反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去すること、減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させること、反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始すること、透過側の減圧の度合いが、10kPa未満であること、を好ましい実施の態様としている。
【0028】
本発明は、触媒存在下でのカルボン酸とアルコールを反応させ、副生成した水を分離膜により選択的に分離・除去することで効率的にエステルを製造するものである。本発明で使用されるカルボン酸は、アルコールと反応してエステルを生成するものであればよく、炭素数、カルボシキル基数、その他の置換基の有無等により制限されるものではない。また、カルボン酸は、単独のカルボン酸だけでなく、複数のカルボン酸の混合物であってもよい。
【0029】
例えば、ギ酸、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、ラウリン酸、ミリスチン酸、パルミチン酸等の飽和脂肪酸、オレイン酸、リノール酸、リノレイン酸等の不飽和脂肪酸、安息香酸、フタル酸、イソフタル酸、テレフタル酸、サリチル酸等の芳香族カルボン酸、シュウ酸、コハク酸、グルタル酸、フタル酸、マレイン酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸等のジカルボン酸、乳酸、酒石酸、クエン酸等のヒドロキシ酸、およびこれらの混合物が挙げられる。
【0030】
分離膜が酸により劣化し、その脱水性能が低下することを考慮すると、本発明では、ミリスチン酸、パルミチン酸、シュウ酸、コハク酸、グルタル酸、フタル酸、マレイン酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸等の固体カルボン酸が好適なカルボン酸として用いられる。
【0031】
本発明で使用されるアルコールは、カルボン酸と反応してエステルを生成するものであればよく、炭素数、ヒドロキシル基数、その他の置換基の有無等で制限されるものではない。また、アルコールは、単独でも複数のアルコールの混合物でもよい。
【0032】
例えば、メタノール、エタノール、プロパノール、ブタノール、イソブチルアルコール、ヘプタノール、ヘキサノール等の一級アルコール、2−プロパノール、2−ブタノール、等の2級アルコール、t−ブチルアルコール等の3級アルコール、2−エチルヘキサノール等の高級アルコール、およびこれらの混合物が挙げられる。
【0033】
本発明で使用される触媒は、アルコールとカルボン酸からエステルを生成できるものであればよく、組成、濃度、形状等により制限されるものではない。例えば、エステル化触媒として一般的に利用される硫酸、p−トルエンスルホン酸等の均一触媒、ダウェックス(商品名)、アンバリスト(商品名)、ナフィオン(商品名)等のイオン交換樹脂に代表される有機固体酸、ゼオライト等の無機固体酸といった不均一触媒、およびその混合物が挙げられる。好適には、硫酸、イオン交換樹脂、特にダウェックスシリーズが用いられる。
【0034】
本発明で使用される分離膜は、アルコール、カルボン酸、エステル化合物、および水の混合溶液、もしくは混合溶液の蒸発物から、選択的に水を除去できるものであればよく、分離膜の形状、本数等で制限されるものではない。
【0035】
例えば、多孔質な支持体上に形成されたCHA型、T型、MOR型、A型等のゼオライト膜が使用されるが、これと同等の機能を有するものであれば、有機高分子膜、高分子および炭素の自立膜、およびそれらを複数結束した分離膜モジュールを用いることも適宜可能である。脱水速度、耐酸性、単位容積当たりの有効膜面積等の観点から、本発明では、多孔質管上に形成されたCHA型ゼオライト膜、もしくはそれを複数本結束した分離膜モジュールが好適である。
【0036】
前記多孔質支持体は、0.01〜100μmの大きさの空孔を持ち、その空隙率は10〜70%であればよく、その材質は、アルミナ、ムライト、ステンレス、炭化ケイ素、チッ化ケイ素、有機高分子等を利用でき、管状、平板状等の形状であればよい。また、支持体は、単一、もしくは複数の支持体を一体化したモジュールを利用できる。
【0037】
上記分離膜は、ケイ素源、アルミニウム源、およびアルカリ源を含む水溶液中で、前記多孔質支持体を加熱処理することで得られる。ケイ素源には、コロイダルシリカ、シリカ粉末等を利用でき、アルミニウム源には金属アルミニウムの他、水酸化アルミニウム、硝酸アルミニウム、硫酸アルミニウム等の塩でも、ベーマイト、γ−アルミナ、α−アルミナ等の酸化物でもよい。
【0038】
アルカリ源には、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物、硝酸ナトリウム、硝酸カリウム等の硝酸化物、塩化ナトリウム、塩化カリウム等の塩化物、硫酸ナトリウム、硫酸カリウム等の硫酸塩の他、水酸化ストロンチウム、硝酸ストロンチウム、硫酸ストロンチウム、塩化ストロンチウム等のアルカリ土類金属の塩を利用することができる。
【0039】
これらをアルミニウム1モルに対して、ケイ素を1〜17.5モル、アルカリもしくはアルカリ土類に由来した水酸化物イオンを0.5〜16モル、水を65〜1800モル含むスラリー水溶液が利用される。多孔質支持体には、予め所望のアルミノケイ酸塩の結晶を付着させておくこともできる。加熱温度は、90〜200℃であればよく、加熱時間は加熱温度にもよるが3時間以上であればよい。しかし、本発明の分離膜の調製は、これらの方法に限定されるものではない。
【0040】
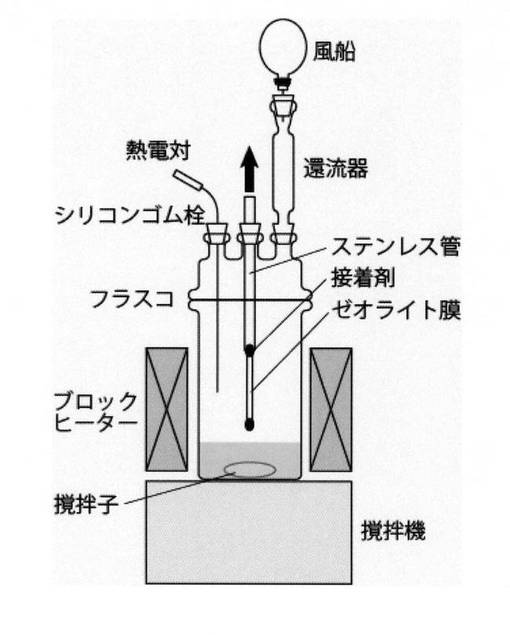
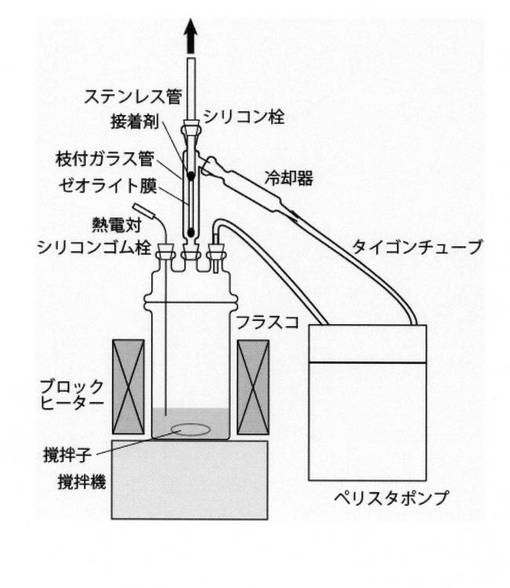
本発明では、反応と分離を並行して進行させることで、カルボン酸からエステルを効率よく製造することが可能となる。ここで、本発明における反応器と分離器の好ましい様態について説明する。まず、分離器は、図1のように反応器内部に設置することもできるし、図2のように反応器の外部に設置してもよい。本発明では、反応器の内容積と分離器容積を考慮し、分離器は、反応器外部に設置することが好ましい。
【0041】
反応器は、カルボン酸、アルコールおよび触媒を均一に撹拌、加熱できる撹拌機構および加熱機構を有する容器であればよく、回分式または連続流通式のいずれでも行うことができるが、本発明では、回分式反応器であることが好ましい。反応器は、反応溶液の沸点Tbより10℃以上高い温度で加熱すればよいが、好ましくは(Tb+15)〜(Tb+30)℃である。
【0042】
分離器は、前記の通り、反応器内部に設置することもできるし、反応容器外部に設置してもよいが、本発明では、反応器外部に設置することが好ましい。分離器では、反応溶液から浸透気化法により水を除去することもできるし、反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去してもよいが、本発明では、分離膜の耐久性維持の観点から、好適には蒸気透過法が利用される。
【0043】
ただし、分離器を反応器内部に設置し、蒸気透過法を利用することも可能であり、その場合には、分離器は、前記の分離膜もしくは分離膜モジュール、分離膜もしくは分離膜モジュールと透過した物質の排気手段、分離膜もしくは分離膜モジュールの保護機構等を備えていればよい。
【0044】
また、飛散した反応溶液が分離膜に付着するのを防止するため、分離膜と反応溶液面の距離が1cm以上であることが好ましく、更に好ましくは3cm以上であればよい。更に、反応溶液の飛散を防ぐために、反応溶液の撹拌速度は400rpm以下であることが好ましい。
【0045】
分離器が反応器外部に設置される場合、分離器は反応器と配管によって接続されている必要がある。すなわち、反応器内の反応溶液もしくは溶液から発生した蒸気が、分離器内の分離膜もしくは分離膜モジュールへと供給される供給経路と、分離膜もしくは分離膜モジュールを透過できなかった物質が、反応器へと還流される還流経路で連結されている必要があり、場合によっては、循環ポンプをいずれかの経路に設置して、反応器と分離器間の流体の循環を促進させてもよい。
【0046】
分離器を反応器外部に設置する場合、分離器は、分離膜もしくは分離膜モジュール、分離膜もしくは分離膜モジュールを収納できるとともに、上記の供給経路および還流経路を備えた容器、分離膜および分離膜モジュールを透過した物質を系外に排出する排出手段、分離器の容器内部で流体の流れもしくは混合を促進するバッフル等を備えていればよい。
【0047】
前記の分離膜もしくは分離膜モジュールを透過した物質を系外に排出する排出方法には、物質が透過した分離膜面(以下、透過側)をポンプで減圧する方法と、窒素、アルゴン、ヘリウム等の不活性ガスを透過側に流通させる方法があるが、本発明では、ポンプ等を利用して透過側を減圧する方法を好適な排出方法としている。
【0048】
また、透過物質の排出は、反応溶液の加熱開始から加熱終了までの期間に渡って実施してよいが、反応溶液が沸点Tbとしたとき、Tb−5(℃)以上に達した時点で、減圧等による透過物質の排出を開始することを更に好ましい排出操作としている。
【0049】
このように、反応溶液の温度がTb−5(℃)以上に到達した後に排出操作を開始することで、分離膜の耐久性を格段に向上させる効果がある。加えて、透過側の減圧の度合いは、反応溶液もしくは溶液から発生した蒸気に含まれる含水率により異なるが、好ましくは10kPa未満、更に好ましくは1kPa未満である。
【発明の効果】
【0050】
本発明によって、以下のような効果が奏される。
(1)分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能とし、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスに関する新技術を提供することができる。
(2)エステル化反応に必要な分離膜面積を従来の1/10〜1/100に低減できる。
(3)分離膜の耐久性を著しく向上させることができる。
(4)触媒共存下で、カルボン酸とアルコールからエステルを製造する際に副生成する水を、水に対して選択透過性をもつ分離膜を利用して、反応器外部に除去することで、高収率でエステルを製造することができる。
【図面の簡単な説明】
【0051】
【図1】蒸気透過試験の概要図を示す。
【図2】蒸気透過試験の他の概要図を示す。
【図3】ヒーターと溶液の温度差が脱水速度に及ぼす影響を示す。
【図4】分離膜と溶液の温度差が脱水速度に及ぼす影響を示す。
【図5】透過側の圧力が脱水速度に及ぼす影響を示す。
【発明を実施するための形態】
【0052】
以下に、実施例を挙げ、本発明を具体的に説明するが、本発明はこれらの例示に限定されるものではない。
【実施例1】
【0053】
硝酸ストロンチウムを溶解させた水溶液にコロイダルシリカを加えて調製したスラリー状水溶液に、水酸化カリウムおよび硝酸アルミニウムを溶解させた水溶液を加えて、膜調製水溶液とした。
【0054】
膜調製水溶液は、モル基準で、アルミニウム1モルに対して、ケイ素を6モル、OH−を3モル、水を390モル含み、溶液のカリウム/ストロンチウムの物質量比は11とした。支持体として多孔質なα−アルミナ管(外径2.0mm、内径1.6mm、長さ23cm、空間率40%、平均孔径0.14μm、長さ22cm)を使用し、その外表面にCHA型ゼオライトの結晶を付着させた。
【0055】
支持体および膜調製水溶液を耐圧容器に入れ、140℃のオーブンで20時間加熱して、アルミナ管の外表面にCHA型ゼオライトの薄膜を形成した。
【0056】
加熱終了後、耐圧容器を水浴に移動して冷却し、ゼオライト・アルミナ複合膜を蒸留水で洗浄し、乾燥させてCHA型ゼオライト分離膜を得た。この分離膜を長さ約5cmに切断し、その片端を接着剤によりステンレス管に接続するとともに、他端を封止し、有効膜面積が2.0cm2のCHA型ゼオライトの分離膜を得た。
【0057】
図1に示す装置を利用し、分離膜Aの蒸気透過特性を評価した。含水率0.2wt%、2wt%、9wt%の2−プロパノール溶液を120mlのセパラブルフラスコに加え、溶液上部に分離膜を設置した。
【0058】
このフラスコを、ブロックヒーター内に設置し、文献(特開2009−125631号公報)に記載の膜性能リアルタイムモニタリング法を利用して脱水性能を測定した。このとき、分離膜下端と溶液面との距離は3cm、透過側圧力は0.5kPa未満とした。
【0059】
その結果、表3に示すCHA型ゼオライト膜Aの蒸気透過法による脱水試験の結果を得た。含水率2wt%の溶液では、ヒーター温度が110℃のとき脱水速度が最大となった。このときのゼオライト膜近傍の蒸気温度を熱電対により測定したところ、91℃であった。
【0060】
一方、ヒーター温度を110℃とし、含水率の異なる溶液で、蒸気透過により脱水特性を評価したところ、溶液の含水率が大きくなるにしたがって、脱水速度は増大した。しかし、分離膜を反応器内部に設置した場合、反応溶液と分離膜の温度を個別に制御することが困難であった。
【0061】
【表3】
【実施例2】
【0062】
次に、実施例1と同様の手順で作製した有効膜面積2.0cm2の分離膜を利用し、図2に示す装置により、脱水性能を評価した。含水率が4wt%のエタノール溶液、28wt%の1−プロパノール溶液、もしくは16wt%の2−プロパノール溶液を加えた120mlのセパラブルフラスコをブロックヒーター内に設置し、分離膜は、フラスコ上部に接続した枝付ガラス管内に設置した。
【0063】
ガラス管外部からリボンヒーターで加熱することで、分離膜の温度を調節した。このとき、ヒーター温度、分離膜温度および透過側圧力が脱水特性に及ぼす影響を調査した。
【0064】
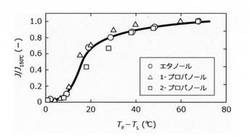
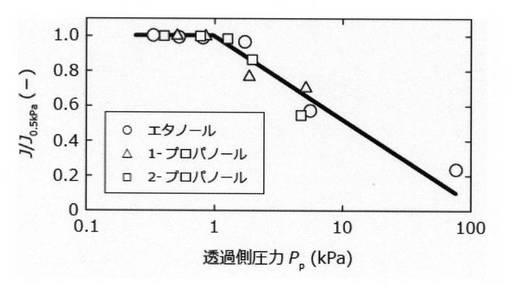
その結果、表4:ヒーター温度が脱水性能に及ぼす影響、表5:分離膜温度が脱水性能に及ぼす影響、表6:透過側圧力が脱水性能に及ぼす影響に示す結果を得た。また、これらの表4〜6の結果を整理することで、それぞれ図3〜5の結果を得た。図3では、横軸にヒーター温度Tfと溶液沸点TLの温度差を、縦軸に脱水速度をヒーター温度150℃での脱水速度で規格化した値J/J150℃をプロットした。
【0065】
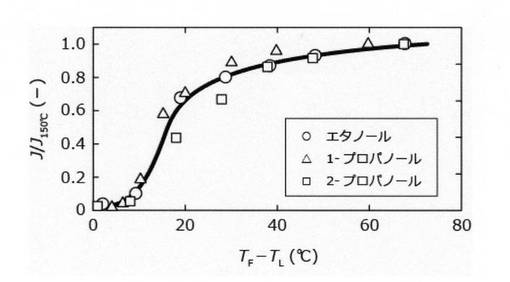
図4では、横軸に分離膜温度TMと溶液沸点TLの差を、縦軸に脱水速度Jをモル濃度に換算した含水率で除した値をプロットした。図5では、横軸に透過側の圧力を、縦軸に透過側圧力が0.5kPa以下としたときの脱水速度で規格化した脱水速度J/J0.5kPaをプロットした。
【0066】
図3に示すように、溶液の種類および含水率に依らず、温度差(Tf−TL)が10〜30℃のとき、J/J150℃が著しく増大したことから、ヒーター温度を、溶液沸点より10℃以上高くすることで、分離膜の脱水速度を著しく向上できることが分かった。
【0067】
また、図4に示すように、分離膜の温度、溶液の種類および含水率に依らず、分離膜の脱水特性が一定であることが分かった。しかし、蒸気の液化を防止するためには、溶液の沸点以上であることが好ましいと考えられた。図5に示すように、透過側圧力が80kPaでも選択的に脱水できたが、溶液の種類や含水率に依らず透過側圧力が1kPa以上になると、脱水速度が著しく低下した。
【0068】
【表4】
【0069】
【表5】
【0070】
【表6】
【0071】
以上、実施例1および実施例2の結果から、分離膜は、反応器の内部もしくは外部のいずれに設置することもできた。しかし、脱水速度制御の観点から、分離膜は、反応器外部に設置することが好ましいことが分かった。また、反応器の加熱温度は、溶液の沸点より10℃以上高ければよいが、10〜30℃高い温度とすることがより好ましかった。
【0072】
分離膜の温度は、特に脱水速度に影響を及ぼさないが、溶液の凝縮を回避するため、溶液の沸点以上で80〜150℃とすることが好ましかった。透過側の圧力は、負圧であればよいが、より高い脱水速度を得るためには、10kPa未満であることが好ましく、更に好ましくは1kPa未満であった。
【実施例3】
【0073】
図1に示す反応器を利用し、濃硫酸を触媒として、アジピン酸と2−プロパノールのエステル化反応を実施した。120mlのセパラブルフラスコにアジピン酸、2−プロパノール、触媒を加え、ヒーター温度110℃で反応させた。ただし、アジピン酸に対する2−プロパノールのモル比は4.3、触媒添加量は基質の1.9wt%、反応時間は4時間とし、分離膜は設置しなかった。
【0074】
溶液の組成を高速イオンクロマトグラフおよびガスクロマトグラフで分析し、アジピン酸の転化率は100−(アジピン酸濃度))/(アジピン酸初期濃度)×100、アジピン酸ジイソプロピルエステルの収率は(アジピン酸ジイソプロピル濃度)/(アジピン酸初期濃度)×100により算出した。その結果、転化率は93.9%、収率は58.6%であった。
【実施例4】
【0075】
触媒添加量を0.3wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は86.2%、収率は39.1%であった。
【実施例5】
【0076】
反応時間を14時間としたことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は92.9%、収率は55.6%であった。
【実施例6】
【0077】
反応時間を40時間としたことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は93.3%、収率は55.9%であった。
【実施例7】
【0078】
触媒としてアンバリスト15H(商品名)を使用し、その添加量を5.8wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は24.9%、収率は2.9%であった。
【実施例8】
【0079】
触媒としてナフィオンNR−50(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は10.7%、収率は1.9%であった。
【実施例9】
【0080】
触媒としてナフィオンSAC−13(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.5%、収率は0.9%であった。
【実施例10】
【0081】
触媒としてダウェックス50W×2(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は42.1%、収率は4.9%であった。
【実施例11】
【0082】
触媒としてダウェックス50W×4(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は48.7%、収率は7.5%であった。
【実施例12】
【0083】
触媒としてダウェックス50W×8(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は27.4%、収率は4.2%であった。
【実施例13】
【0084】
触媒としてH−ZSM−5型ゼオライト(Si/Al=25)を使用し、その添加量を19.2wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は6.8%、収率は0.7%であった。
【実施例14】
【0085】
触媒としてニオブ酸化物を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.1%、収率は1.1%であった。
【実施例15】
【0086】
触媒としてスカンジウムトリフルオロメタンスルホン酸を使用したことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は77.7%、収率は24.8%であった。
【0087】
比較例1
触媒を使用しなかったことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.6%、収率は2.1%であった。
【0088】
実施例3〜15、および比較例1の結果を表7にまとめた。表7に示すように、触媒を加えなかった場合と比べ、硫酸、アンバリスト15H、ダウェックス50W×2、ダウェックス50W×4、ダウェックス50W×8、Sc(OTf)3を加えた場合に、転化率および収率が向上し、ナフィオンNR−50、ナフィオンSAC−13、ゼオライト、およびニオブ酸化物では、転化率および収率の増大は見られなかった。また、硫酸およびSc(OTf)3といった均一触媒が高い触媒活性を有していたのに対して、その他の不均一触媒の転化率および収率は低かった。不均一触媒の中では、ダウェックスシリーズは、比較的高い触媒活性を有していた。
【0089】
【表7】
【実施例16】
【0090】
ダウェックス50Wx2を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は62.4%、収率は11.6%であった。
【実施例17】
【0091】
ダウェックス50Wx4を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果転化率は69.3%、収率は15.2%であった。
【実施例18】
【0092】
ダウェックス50Wx8を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は37.9%、収率は6.8%であった。
【実施例19】
【0093】
触媒添加量を28.5wt%としたことを除いて、実施例17と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は95.3%、収率は61.4%であった。
【0094】
実施例16〜19の結果を表8にまとめた。表8に示すように、イオン交換樹脂であるダウェックスを2−プロパノールで洗浄することで、樹脂に含まれる水分が除去されたため、触媒活性が向上した。洗浄したダウェックス50W×4を更に多く使用すると、硫酸と同等の反応活性が得られた。
【0095】
【表8】
【0096】
以上の結果から、均一触媒である硫酸、Sc(OTf)3の他、イオン交換樹脂等の有機固体酸が触媒として有効であった。特に、均一触媒である硫酸、有機固体酸であるダウェックスがエステル化触媒として有望であった。
【実施例20】
【0097】
実施例1と同様の手順で調製した分離膜を利用し、アジピン酸と2−プロパノールのエステル化反応を実施した。120mlのセパラブルフラスコにアジピン酸、2−プロパノール、硫酸を加え、図1のように反応器内部に分離膜を設置した。
【0098】
アジピン酸に対する2−プロパノールのモル比mは4.3、基質重量に対する膜面積の比S/Vは0.0079m2/kg、基質重量に対する触媒添加量Wは2.0wt%、分離膜下端と溶液面の距離Lは3cm、ヒーター温度Tfは110℃、反応時間tRは4時間、加熱開始から分離膜の透過側の減圧を開始するまでの時間tdは1分、透過側の圧力Ppは0.5kPa未満とした。減圧開始時の溶液温度は31℃であった。その結果、転化率は99.8%、収率は92.0%であった。
【実施例21】
【0099】
以下に示す方法でNaA型ゼオライト膜を調製し、エステル化反応に供した。水ガラスを溶解させた水溶液に、アルミン酸ナトリウムを溶解させた水溶液を加えて膜成長水溶液とし、外表面にNaA型ゼオライトの結晶を擦り込んだ多孔質α−アルミナ管とともに、圧力容器に加えて、100℃で4.5時間加熱し、アルミナ管の外表面にNaA型ゼオライト層を形成した。以下、実施例1と同様に、洗浄、乾燥、接着を行い、有効面積2.0cm2のNaA型ゼオライトの分離膜を得た。
【0100】
分離膜をNaA型ゼオライトの分離膜としたことを除いて、実施例20と同様の条件および操作手順でアジピン酸と2−プロパノールのエステル化反応を実施した。その結果、tR=4時間における転化率は99.5%、収率は97.3%であった。
【実施例22】
【0101】
触媒として2−プロパノールで洗浄後に乾燥させたダウェックス50W×4を28.7wt%使用したことを除けば、実施例20と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は99.9%以上、収率は87.1%であった。
【0102】
表9に、実施例3、および実施例19〜22の結果をまとめた。表9に示すように、分離膜の種類、触媒の種類に依らず、分離膜を利用することで転化率および収率が向上したことから、分離膜による脱水がエステル化反応に有効であることが分かった。
【0103】
【表9】
【実施例23】
【0104】
実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を利用し、アジピン酸と2−プロパノールのエステル化反応を実施した。ただし、w=0.6wt%、tR=8時間、S/V=0.0035m2/kgとした以外は、実施例20と同様の条件および操作手順とした。その結果、転化率は98.5%、収率は79.4%であった。
【実施例24】
【0105】
S/V=0.0050m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は98.2%、収率は87.6%であった。
【実施例25】
【0106】
S/V=0.0060m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は99.9%以上、収率は92.3%であった。
【実施例26】
【0107】
S/V=0.0071m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は98.9%、収率は98.2%であった。
【実施例27】
【0108】
S/V=0.0080m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は99.0%、収率は97.1%であった。
【0109】
表10に、実施例3、実施例6、および実施例23〜27の結果をまとめた。表10に示すように、実施例3および実施例6より、分離膜を使用しない場合、転化率が93〜94%で、収率が55〜59%で平衡であるため、分離膜を利用しなければ、それ以上の転化率および収率を得ることが困難であった。分離膜を利用することで、この平衡を超える転化率および収率を得ることができた。このとき、反応基質重量に対する分離膜面積の比S/Vは0.0035m2/kg以上であればよいことが分かった。
【0110】
【表10】
【実施例28】
【0111】
分離膜と溶液面との距離Lを1cmとしたことを除いて、実施例20と同様の条件でエステル化反応を実施した。分離膜は、実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を使用した。その結果、4時間後の転化率は99.9%以上で、収率は90.8%であった。
【実施例29】
【0112】
分離膜と溶液面との距離Lを30cmとするため、実施例2と同様にフラスコ上部に設置した枝付ガラス管内に分離膜を設置し、ガラス管上部の枝部分から流出した蒸気を冷却、還流させたことを除いて、実施例20と同様の条件および操作手順でエステル化反応を実施した。ただし、分離膜の温度は90℃とした。その結果、転化率は99.4%、収率は94.8%であった。
【0113】
表11に実施例20、28、29の結果をまとめた。表11に示すように、分離膜と溶液面との距離Lは転化率および収率に影響を及ぼさなかった。これは、実施例1および実施例2で検討したように、分離膜の温度が適切で、かつヒーター温度が反応溶液の沸点(90〜95℃)よりも15〜20℃高かったので、分離膜が充分な脱水速度を保持できたためであった。
【0114】
【表11】
【実施例30】
【0115】
分離膜の温度を110℃としたことを除いて、実施例29と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は98.0%、収率は89.6%であった。
【0116】
表12に実施例29、30の結果をまとめた。表12に示すように、分離膜の温度は、転化率および収率に大きな影響を与えなかった。これは、実施例2に示すように、分離膜の脱水速度が、分離膜の温度に影響されないためであった。
【0117】
【表12】
【実施例31】
【0118】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを3分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は51℃であった。その結果、転化率は98.6%、収率は88.3%であった。
【実施例32】
【0119】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを7.5分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は71℃であった。その結果、転化率は98.6%、収率は86.3%であった。
【実施例33】
【0120】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを8分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は80℃であった。その結果、転化率は99.1%、収率は92.5%であった。
【実施例34】
【0121】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを10分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は92℃であった。その結果、転化率は99.0%、収率は91.4%であった。
【実施例35】
【0122】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを30分としたこと、透過側の圧力を1kPaとしたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は91℃であった。その結果、転化率は99.1%、収率は87.2%であった。
【0123】
表13に実施例29、31−35の結果をまとめた。表13に示すように、加熱開始から減圧開始までの時間tdは、転化率および収率にほとんど影響を与えなかった。これは、反応時間(4時間)と比べ、tdが充分に短いためであった。また、透過側圧力を1kPaとしても収率および転化率は高い値を維持していた。
【0124】
【表13】
【実施例36】
【0125】
実施例20、実施例21、実施例28で使用した分離膜について、40℃の2−プロパノール溶液を用いて浸透気化法によりエステル化反応前後の分離膜の脱水速度を測定した。その結果、表14に示す結果を得た。
【0126】
比較例2
実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を使用して、蒸気透過試験を実施した。フラスコに2−プロパノール16.7g、水1.7g、アジピン酸9.4gを加え、ヒーター温度Tf=110℃、分離膜と液面の距離L=1cmとして、蒸気透過試験を実施した。蒸気透過試験の前後に、40℃の2−プロパノール溶液(含水率9%)を用いた浸透気化法により分離膜の脱水速度を測定したところ、蒸気透過前の脱水速度は1.25kg/m2/hであったのに対して、蒸気透過後には1.00kg/m2/hであった。
【0127】
実施例35および比較例2の結果を表14にまとめた。表14に示すように、この結果から、分離膜と液面の距離Lが1cmのとき、反応前後で分離膜の脱水速度は40%以上低下したのに対して、L=3cmとすることでその減少率は20%となった。これは、比較例2の硫酸を含まない溶液に対する減少率と等しかった。
【0128】
この結果から、撹拌速度が400rpm以下であれば、硫酸を含む溶液飛沫が付着による脱水速度の低下を抑制するためには、分離膜と反応溶液面の距離を3cm以上にすればよいことが分かった。
【0129】
一方、NaA型ゼオライトの分離膜は、減少率が約30%とCHA型ゼオライト膜より大きかったことから、NaA型ゼオライトと比べて、CHA型ゼオライトの分離膜の方が耐酸性が高いことが分かった。しかしながら、比較例2から明らかなように、アジピン酸の影響による脱水速度の低下を回避することができなかった。
【0130】
【表14】
【実施例37】
【0131】
実施例31〜34で使用した分離膜についても、40℃の2−プロパノール溶液を用いて浸透気化法によりエステル化反応前後の分離膜の脱水速度を測定した。その結果、表15に示す結果を得た。
【0132】
表15に示すように、加熱開始から1分後に減圧を開始(td=1分)すると、脱水速度の減少率は約20%であったが、td=10分とすると脱水速度は減少しなかった。アジピン酸が溶解した2−プロパノールの沸点は90〜95℃であったことから、溶液温度が沸点に到達した後に分離膜内表面の減圧を開始することで、脱水速度の低下を抑制することができた。すなわち、分離膜内表面の減圧開始は、溶液温度が沸点に到達した後に開始することが好ましいことが分かった。
【0133】
【表15】
【実施例38】
【0134】
有機酸としてミリスチン酸を、アルコールには2−プロパノールを、触媒には硫酸を使用し、ミリスチン酸と2−プロパノールのエステル化反応を実施した。ミリスチン酸に対する2−プロパノールのモル比mは3、硫酸濃度は基質重量の1.0wt%、反応時間は8時間、膜面積と基質重量の比は0.008m2/kg、ヒーター温度は110℃、膜温度は90℃、分離膜内表面の減圧開始時間は15分(溶液温度92.4℃)、減圧度は1kPa未満とした。その結果、ミリスチン酸の転化率(ミリスチン酸イソプロピルの収率)は99.9%であった。
【実施例39】
【0135】
有機酸としてパルミチン酸を使用したことを除いて、実施例38と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は90.6℃)。その結果、パルミチン酸の転化率(=パルミチン酸イソプロピルの収率)は99.7%であった。
【実施例40】
【0136】
有機酸としてセバシン酸を、アルコールとしてエタノールを使用し、セバシン酸に対するエタノールのモル比mを5としたことを除いて、実施例38と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は85.8℃)。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジエチルの収率は99.1%であった。
【実施例41】
【0137】
アルコールとしてブタノールを使用し、ヒーター温度を130℃、膜温度を110℃としたことを除いて、実施例40と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は103.6℃)。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジブチルの収率は98.9%であった。
【0138】
比較例3
分離膜を使用しなかったことを除いて、実施例38と同様の反応条件および操作手順で、ミリスチン酸と2−プロパノールのエステル化反応を実施した。8時間後のミリスチン酸の転化率は(ミリスチン酸イソプロピルの収率)は83.1%であった。
【0139】
比較例4
分離膜を使用しなかったことを除いて、実施例39と同様の反応条件および操作手順で、パルミチン酸と2−プロパノールのエステル化反応を実施した。8時間後のパルミチン酸の転化率は(パルミチン酸イソプロピルの収率)は84.9%であった。
【0140】
比較例5
分離膜を使用しなかったことを除いて、実施例40と同様の反応条件および操作手順で、セバシン酸とエタノールのエステル化反応を実施した。8時間後のセバシン酸の転化率は98.1%、セバシン酸ジエチルの収率は76.2%であった。
【0141】
比較例5
分離膜を使用しなかったことを除いて、実施例40と同様の反応条件および操作手順で、セバシン酸とブタノールのエステル化反応を実施した。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジブチルの収率は84.9%であった。
【0142】
実施例38〜41及び比較例3〜6の結果を表16にまとめたように、分離膜を使用しなかった比較例3〜6では、エステルの収率が76〜85%であったのに対して、分離膜を使用することで収率を99%以上にすることができた。これは、固体有機酸と炭素数4以下のアルコールの組合せであれば、分離膜を使用することで、固体有機酸をエステルに効率よく転換できることを示しており、分離膜が多様なエステルの製造に有効であることが分かった。
【0143】
【表16】
【産業上の利用可能性】
【0144】
以上詳述した通り、本発明は、エステルの製造方法に係るものであり、本発明により、分離膜を利用したエステル化反応プロセスにおいて、反応器設計、反応操作条件設定等の調整により、分離膜の脱水速度および耐久性を向上させることでエステル化反応プロセスの小規模化およびメンテナンス回数の低減を可能にした新しいエステル化反応プロセスを提供できる。本発明は、高い脱水速度および耐久性を有する分離膜を利用したエステル化反応プロセスを構築することでエステル化反応に必要な分離面積を従来の1/10〜1/100に低減することを可能とする分離膜を利用したエステル化反応プロセスに関する新技術を提供するものとして有用である。
【技術分野】
【0001】
本発明は、エステルの製造方法に関するものであり、更に詳しくは、触媒共存下で、カルボン酸とアルコールからエステルを製造する際に副生成する水を、水に対して選択透過性をもつ分離膜を利用して、反応器外部に除去することで、高収率でエステルを製造する方法に関するものである。本発明は、分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能とし、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスに関する新技術を提供するものである。
【背景技術】
【0002】
カルボン酸とアルコールからエステルを生成する化学反応は平衡反応であるので、カルボン酸のエステルへの転換効率を向上させるには、大過剰のアルコールを使用する方法と、副生成する水を反応器外部に除去する方法が考えられる。しかし、大過剰のアルコールを使用する方法は、大型の反応容器が必要であり、製造プロセスとして好ましくなく、副生成する水を反応器外部に除去する方法の利用が好ましい。
【0003】
水の除去方法には、蒸留法、吸着法、膜分離法があるが、蒸留法では、大型の蒸留装置が必要であり、消費エネルギーが大きいといった課題に加え、反応溶液が共沸混合物となった場合、消費エネルギーに比して充分に水を除去できないことが指摘されている。
【0004】
一方、吸着法および膜分離法では、脱水に及ぼす共沸の影響は小さく、効率よく水を除去することができる。吸着法では、吸着剤への水の吸着量に限界があるため、吸着剤の再生が不可欠であるのに対して、膜分離法では、分離膜を通過した水が反応系外に排出されるので、連続的に利用することができる等の利点がある。
【0005】
前記のように、連続的に効率よくエステルを製造できることから、近年、膜分離法を利用したエステル化反応に関する研究が盛んに行われている。これらの研究では、分離膜として、ポリビニルアルコール、ポリエーテルイミド等の高分子分離膜と、A型、T型、MER型、PHI型、CHA型ゼオライト等のゼオライト膜に代表される無機多孔質分離膜が利用されているが、高分子膜は、有機溶液に対する耐薬品性および耐熱性が低く、ゼオライト膜等の無機多孔質膜の利用が検討されている。
【0006】
例えば、分離膜にゼオライト膜を利用した研究には、多数の文献(特許文献1〜3、非特許文献1〜4等)があり、多様なカルボン酸とアルコールの組合せで、均一もしくは不均一触媒を利用し、分離膜により溶液もしくは蒸気相から水を選択的に除去することで、効率よくエステルを製造できる。表1にゼオライト膜を利用したエステル化反応の研究例を示す。
【0007】
【表1】
【0008】
表1に示すように、分離膜、特にゼオライト膜を利用することで、効率よくカルボン酸をエスエルに転換できることは知られているが、分離膜の脱水速度および耐久性が不充分であった。
【0009】
非特許文献2および3で説明されるように、分離膜の脱水速度をJ(kg/m2/h)、分離膜の有効面積をS(m2)、反応基質の重量をV(kg)とすれば、分離膜を利用した脱水の効果は JS/Vで説明される。たとえば、乳酸エチル、酢酸エチル等のエステル化反応では、このJS/V値が0.1h−1以上で、効率よくカルボン酸をエステルに転換できる。
【0010】
しかし、利用されるT型ゼオライト膜の脱水速度が、1.0kg/m2/h(特許文献4)と低いため、S/Vを0.1m2/kg以上にする必要があった。これは、基質1トンに対して、有効面積100m2の分離膜が必要であることを意味しており、分離膜の製造メーカーにより多少異なるが、管状分離膜を複数本結束した分離膜モジュールの単位容積当たりの有効膜面積は概ね10m2/m3であるので、反応器容積の約10倍の分離膜が必要になることを示している。表2にゼオライト膜の脱水特性(浸透気化法)を示す。
【0011】
その他、特許文献1では、S/V=0.024m2/kgと比較的小さな膜面積で検討されているが、その他の文献では、S/V=0.1〜40m2/kgであり、反応容器と比べ非常に大きな膜分離器が必要であった。すなわち、分離膜を利用した高効率エステル製造プロセスを小規模化するためには、分離膜の脱水速度向上が必須である。例えば、膜分離器を反応容器と同程度まで小型化するには、分離膜の脱水速度は10kg/m2/h以上が必要である。
【0012】
【表2】
【0013】
充分な脱水速度を有している分離膜であっても、エステル化反応条件下で長期間に渡ってその脱水性能を維持できなければ、頻繁なエステル製造プロセスのメンテナンスが必要となる。すなわち、反応条件下における分離膜の耐久性も重要である。
【0014】
表2に示すように、脱水速度が10kg/m2/h以上となる分離膜は、A型およびCHA型ゼオライト膜であるが、A型ゼオライト膜は酸性条件下における耐久性が著しく低い。非特許文献7によると、A型ゼオライト膜を酸性溶液に接触させた場合には、約10分で分離機能を喪失した。
【0015】
一方、非特許文献8によると、CHA型ゼオライト膜は、類似条件下でも10時間以上水選択透過性を維持しているものの、酸性溶液と接触することで、ゼオライトの構造破壊に起因した脱水速度の低下が顕著であった。
【0016】
耐久性の観点から、エステル化反応へのゼオライト膜の利用に際しては、分離膜と反応溶液の接触を回避できる蒸気透過法が有効であるが、エステル製造プロセスでは、蒸気圧の上限に限界があるため、浸透気化法と比べて、蒸気透過法では脱水速度が低下し、充分な脱水速度が得られなくなるという欠点がある。
【0017】
たとえば、非特許文献3では、T型ゼオライト膜の脱水速度は浸透気化試験では0.8kg/m2/hであったが、蒸気透過試験では0.3〜0.4kg/m2/hに半減している。すなわち、分離膜の脱水速度と耐久性を確保するための反応器の設計および反応操作条件の最適化が必須である。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】特許第4057063号
【特許文献2】特開2002−47213号公報
【特許文献3】特開2006−315991号公報
【特許文献4】特許3686262号
【特許文献5】特開2006−212551号公報
【特許文献6】特開2006−176399号公報
【特許文献7】特開2003−144871号公報
【特許文献8】特開2011−016123号公報
【非特許文献】
【0019】
【非特許文献1】Journal of Membrane Science、2002年、199巻、117−123頁
【非特許文献2】Catalysis Today、2001年、67巻、121−125頁
【非特許文献3】Chemical Engineering Science、2002年、57巻、1577−1584頁
【非特許文献4】Industry and Engineering Chemistry Research、2007年、46巻、3743−3750頁
【非特許文献5】Microporous and Mesoporous Materials,2009年、126巻、107−114頁
【非特許文献6】Journal of Membrane Science、2010年、364巻、318−324頁
【非特許文献7】Journal of Membrane Science、2010年、349巻、189−194頁
【非特許文献8】Journal of Membrane Science、2010年、347巻、193−196頁
【発明の概要】
【発明が解決しようとする課題】
【0020】
以上述べたように、高い脱水速度および耐久性を有する分離膜が開発されていなかったことに加え、エステル製造に不可欠な分離膜の脱水速度および耐久性を向上させうる反応器設計、反応操作条件等に関する検討が不充分であったため、分離膜を利用したエステル化反応プロセスの小規模化が困難であるとともに、分離膜交換等のメンテナンス回数の低減も困難であった。
【0021】
本発明は、分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能にするとともに、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスを提供することを目的とするものである。
【0022】
更に、本発明は、本発明者らの開発した比較的耐酸性が高く、優れた脱水速度を示すCHA型ゼオライト膜(特許文献8)を、エステル化反応に利用するとともに、反応溶液の加熱温度、撹拌速度、分離膜の設置位置、分離膜の温度、分離膜の操作時間等を調整することで、エステル化反応の促進に不可欠な分離膜の脱水速度および耐久性を向上させることにより、エステル化反応プロセスの小規模化、およびメンテナンス回数の低減を実現可能とする新しいエステル化反応プロセスを提供することを目的とするものである。
【課題を解決するための手段】
【0023】
上記課題を解決するための本発明は、以下の技術的手段から構成される。
(1)反応基質を含む反応系から構成される反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始すること、を特徴とするエステルの製造方法。
(2)反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下である、前記(1)記載のエステルの製造方法。
(3)分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去する、前記(1)記載のエステルの製造方法。
(4)(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満である、前記(1)記載のエステルの製造方法。
(5)反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールである、前記(1)記載のエステルの製造方法。
(6)分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜である、前記(1)記載のエステルの製造方法。
(7)多孔質支持体が、0.01〜100μmの大きさの空孔を持ち、その空隙率が10〜70%である、前記(1)記載のエステルの製造方法。
(8)分離器が、反応器内部又は反応器外部に設置される、前記(1)記載のエステルの製造方法。
(9)反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去する、前記(1)記載のエステルの製造方法。
(10)減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させる、前記(1)記載のエステルの製造方法。
(11)反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始する、前記(1)記載のエステルの製造方法。
(12)分離膜の透過側の減圧の度合いが、10kPa未満である、前記(10)記載のエステルの製造方法。
【0024】
次に、本発明について更に詳細に説明する。
本発明は、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を分離膜で除去することによりエステルを製造する方法において、分離膜がたとえばCHA型ゼオライト膜またはNaA型ゼオライト膜であり、分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始することを特徴とするものである。
【0025】
本発明では、上記エステルの製造方法において、分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去すること、反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下であることを好ましい実施の態様としている。
【0026】
また、本発明は、(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満であること、反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールであることを好ましい実施の態様としている。
【0027】
更に、本発明は、分離器が、反応容器外部に設置されること、反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去すること、減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させること、反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始すること、透過側の減圧の度合いが、10kPa未満であること、を好ましい実施の態様としている。
【0028】
本発明は、触媒存在下でのカルボン酸とアルコールを反応させ、副生成した水を分離膜により選択的に分離・除去することで効率的にエステルを製造するものである。本発明で使用されるカルボン酸は、アルコールと反応してエステルを生成するものであればよく、炭素数、カルボシキル基数、その他の置換基の有無等により制限されるものではない。また、カルボン酸は、単独のカルボン酸だけでなく、複数のカルボン酸の混合物であってもよい。
【0029】
例えば、ギ酸、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、ラウリン酸、ミリスチン酸、パルミチン酸等の飽和脂肪酸、オレイン酸、リノール酸、リノレイン酸等の不飽和脂肪酸、安息香酸、フタル酸、イソフタル酸、テレフタル酸、サリチル酸等の芳香族カルボン酸、シュウ酸、コハク酸、グルタル酸、フタル酸、マレイン酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸等のジカルボン酸、乳酸、酒石酸、クエン酸等のヒドロキシ酸、およびこれらの混合物が挙げられる。
【0030】
分離膜が酸により劣化し、その脱水性能が低下することを考慮すると、本発明では、ミリスチン酸、パルミチン酸、シュウ酸、コハク酸、グルタル酸、フタル酸、マレイン酸、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸等の固体カルボン酸が好適なカルボン酸として用いられる。
【0031】
本発明で使用されるアルコールは、カルボン酸と反応してエステルを生成するものであればよく、炭素数、ヒドロキシル基数、その他の置換基の有無等で制限されるものではない。また、アルコールは、単独でも複数のアルコールの混合物でもよい。
【0032】
例えば、メタノール、エタノール、プロパノール、ブタノール、イソブチルアルコール、ヘプタノール、ヘキサノール等の一級アルコール、2−プロパノール、2−ブタノール、等の2級アルコール、t−ブチルアルコール等の3級アルコール、2−エチルヘキサノール等の高級アルコール、およびこれらの混合物が挙げられる。
【0033】
本発明で使用される触媒は、アルコールとカルボン酸からエステルを生成できるものであればよく、組成、濃度、形状等により制限されるものではない。例えば、エステル化触媒として一般的に利用される硫酸、p−トルエンスルホン酸等の均一触媒、ダウェックス(商品名)、アンバリスト(商品名)、ナフィオン(商品名)等のイオン交換樹脂に代表される有機固体酸、ゼオライト等の無機固体酸といった不均一触媒、およびその混合物が挙げられる。好適には、硫酸、イオン交換樹脂、特にダウェックスシリーズが用いられる。
【0034】
本発明で使用される分離膜は、アルコール、カルボン酸、エステル化合物、および水の混合溶液、もしくは混合溶液の蒸発物から、選択的に水を除去できるものであればよく、分離膜の形状、本数等で制限されるものではない。
【0035】
例えば、多孔質な支持体上に形成されたCHA型、T型、MOR型、A型等のゼオライト膜が使用されるが、これと同等の機能を有するものであれば、有機高分子膜、高分子および炭素の自立膜、およびそれらを複数結束した分離膜モジュールを用いることも適宜可能である。脱水速度、耐酸性、単位容積当たりの有効膜面積等の観点から、本発明では、多孔質管上に形成されたCHA型ゼオライト膜、もしくはそれを複数本結束した分離膜モジュールが好適である。
【0036】
前記多孔質支持体は、0.01〜100μmの大きさの空孔を持ち、その空隙率は10〜70%であればよく、その材質は、アルミナ、ムライト、ステンレス、炭化ケイ素、チッ化ケイ素、有機高分子等を利用でき、管状、平板状等の形状であればよい。また、支持体は、単一、もしくは複数の支持体を一体化したモジュールを利用できる。
【0037】
上記分離膜は、ケイ素源、アルミニウム源、およびアルカリ源を含む水溶液中で、前記多孔質支持体を加熱処理することで得られる。ケイ素源には、コロイダルシリカ、シリカ粉末等を利用でき、アルミニウム源には金属アルミニウムの他、水酸化アルミニウム、硝酸アルミニウム、硫酸アルミニウム等の塩でも、ベーマイト、γ−アルミナ、α−アルミナ等の酸化物でもよい。
【0038】
アルカリ源には、水酸化ナトリウム、水酸化カリウム等のアルカリ金属の水酸化物、硝酸ナトリウム、硝酸カリウム等の硝酸化物、塩化ナトリウム、塩化カリウム等の塩化物、硫酸ナトリウム、硫酸カリウム等の硫酸塩の他、水酸化ストロンチウム、硝酸ストロンチウム、硫酸ストロンチウム、塩化ストロンチウム等のアルカリ土類金属の塩を利用することができる。
【0039】
これらをアルミニウム1モルに対して、ケイ素を1〜17.5モル、アルカリもしくはアルカリ土類に由来した水酸化物イオンを0.5〜16モル、水を65〜1800モル含むスラリー水溶液が利用される。多孔質支持体には、予め所望のアルミノケイ酸塩の結晶を付着させておくこともできる。加熱温度は、90〜200℃であればよく、加熱時間は加熱温度にもよるが3時間以上であればよい。しかし、本発明の分離膜の調製は、これらの方法に限定されるものではない。
【0040】
本発明では、反応と分離を並行して進行させることで、カルボン酸からエステルを効率よく製造することが可能となる。ここで、本発明における反応器と分離器の好ましい様態について説明する。まず、分離器は、図1のように反応器内部に設置することもできるし、図2のように反応器の外部に設置してもよい。本発明では、反応器の内容積と分離器容積を考慮し、分離器は、反応器外部に設置することが好ましい。
【0041】
反応器は、カルボン酸、アルコールおよび触媒を均一に撹拌、加熱できる撹拌機構および加熱機構を有する容器であればよく、回分式または連続流通式のいずれでも行うことができるが、本発明では、回分式反応器であることが好ましい。反応器は、反応溶液の沸点Tbより10℃以上高い温度で加熱すればよいが、好ましくは(Tb+15)〜(Tb+30)℃である。
【0042】
分離器は、前記の通り、反応器内部に設置することもできるし、反応容器外部に設置してもよいが、本発明では、反応器外部に設置することが好ましい。分離器では、反応溶液から浸透気化法により水を除去することもできるし、反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去してもよいが、本発明では、分離膜の耐久性維持の観点から、好適には蒸気透過法が利用される。
【0043】
ただし、分離器を反応器内部に設置し、蒸気透過法を利用することも可能であり、その場合には、分離器は、前記の分離膜もしくは分離膜モジュール、分離膜もしくは分離膜モジュールと透過した物質の排気手段、分離膜もしくは分離膜モジュールの保護機構等を備えていればよい。
【0044】
また、飛散した反応溶液が分離膜に付着するのを防止するため、分離膜と反応溶液面の距離が1cm以上であることが好ましく、更に好ましくは3cm以上であればよい。更に、反応溶液の飛散を防ぐために、反応溶液の撹拌速度は400rpm以下であることが好ましい。
【0045】
分離器が反応器外部に設置される場合、分離器は反応器と配管によって接続されている必要がある。すなわち、反応器内の反応溶液もしくは溶液から発生した蒸気が、分離器内の分離膜もしくは分離膜モジュールへと供給される供給経路と、分離膜もしくは分離膜モジュールを透過できなかった物質が、反応器へと還流される還流経路で連結されている必要があり、場合によっては、循環ポンプをいずれかの経路に設置して、反応器と分離器間の流体の循環を促進させてもよい。
【0046】
分離器を反応器外部に設置する場合、分離器は、分離膜もしくは分離膜モジュール、分離膜もしくは分離膜モジュールを収納できるとともに、上記の供給経路および還流経路を備えた容器、分離膜および分離膜モジュールを透過した物質を系外に排出する排出手段、分離器の容器内部で流体の流れもしくは混合を促進するバッフル等を備えていればよい。
【0047】
前記の分離膜もしくは分離膜モジュールを透過した物質を系外に排出する排出方法には、物質が透過した分離膜面(以下、透過側)をポンプで減圧する方法と、窒素、アルゴン、ヘリウム等の不活性ガスを透過側に流通させる方法があるが、本発明では、ポンプ等を利用して透過側を減圧する方法を好適な排出方法としている。
【0048】
また、透過物質の排出は、反応溶液の加熱開始から加熱終了までの期間に渡って実施してよいが、反応溶液が沸点Tbとしたとき、Tb−5(℃)以上に達した時点で、減圧等による透過物質の排出を開始することを更に好ましい排出操作としている。
【0049】
このように、反応溶液の温度がTb−5(℃)以上に到達した後に排出操作を開始することで、分離膜の耐久性を格段に向上させる効果がある。加えて、透過側の減圧の度合いは、反応溶液もしくは溶液から発生した蒸気に含まれる含水率により異なるが、好ましくは10kPa未満、更に好ましくは1kPa未満である。
【発明の効果】
【0050】
本発明によって、以下のような効果が奏される。
(1)分離膜の脱水速度および耐久性を向上させることで、エステル化反応プロセスの小規模化を可能とし、分離膜交換等のメンテナンス回数を大幅に低減させることを可能とする新しいエステル化反応プロセスに関する新技術を提供することができる。
(2)エステル化反応に必要な分離膜面積を従来の1/10〜1/100に低減できる。
(3)分離膜の耐久性を著しく向上させることができる。
(4)触媒共存下で、カルボン酸とアルコールからエステルを製造する際に副生成する水を、水に対して選択透過性をもつ分離膜を利用して、反応器外部に除去することで、高収率でエステルを製造することができる。
【図面の簡単な説明】
【0051】
【図1】蒸気透過試験の概要図を示す。
【図2】蒸気透過試験の他の概要図を示す。
【図3】ヒーターと溶液の温度差が脱水速度に及ぼす影響を示す。
【図4】分離膜と溶液の温度差が脱水速度に及ぼす影響を示す。
【図5】透過側の圧力が脱水速度に及ぼす影響を示す。
【発明を実施するための形態】
【0052】
以下に、実施例を挙げ、本発明を具体的に説明するが、本発明はこれらの例示に限定されるものではない。
【実施例1】
【0053】
硝酸ストロンチウムを溶解させた水溶液にコロイダルシリカを加えて調製したスラリー状水溶液に、水酸化カリウムおよび硝酸アルミニウムを溶解させた水溶液を加えて、膜調製水溶液とした。
【0054】
膜調製水溶液は、モル基準で、アルミニウム1モルに対して、ケイ素を6モル、OH−を3モル、水を390モル含み、溶液のカリウム/ストロンチウムの物質量比は11とした。支持体として多孔質なα−アルミナ管(外径2.0mm、内径1.6mm、長さ23cm、空間率40%、平均孔径0.14μm、長さ22cm)を使用し、その外表面にCHA型ゼオライトの結晶を付着させた。
【0055】
支持体および膜調製水溶液を耐圧容器に入れ、140℃のオーブンで20時間加熱して、アルミナ管の外表面にCHA型ゼオライトの薄膜を形成した。
【0056】
加熱終了後、耐圧容器を水浴に移動して冷却し、ゼオライト・アルミナ複合膜を蒸留水で洗浄し、乾燥させてCHA型ゼオライト分離膜を得た。この分離膜を長さ約5cmに切断し、その片端を接着剤によりステンレス管に接続するとともに、他端を封止し、有効膜面積が2.0cm2のCHA型ゼオライトの分離膜を得た。
【0057】
図1に示す装置を利用し、分離膜Aの蒸気透過特性を評価した。含水率0.2wt%、2wt%、9wt%の2−プロパノール溶液を120mlのセパラブルフラスコに加え、溶液上部に分離膜を設置した。
【0058】
このフラスコを、ブロックヒーター内に設置し、文献(特開2009−125631号公報)に記載の膜性能リアルタイムモニタリング法を利用して脱水性能を測定した。このとき、分離膜下端と溶液面との距離は3cm、透過側圧力は0.5kPa未満とした。
【0059】
その結果、表3に示すCHA型ゼオライト膜Aの蒸気透過法による脱水試験の結果を得た。含水率2wt%の溶液では、ヒーター温度が110℃のとき脱水速度が最大となった。このときのゼオライト膜近傍の蒸気温度を熱電対により測定したところ、91℃であった。
【0060】
一方、ヒーター温度を110℃とし、含水率の異なる溶液で、蒸気透過により脱水特性を評価したところ、溶液の含水率が大きくなるにしたがって、脱水速度は増大した。しかし、分離膜を反応器内部に設置した場合、反応溶液と分離膜の温度を個別に制御することが困難であった。
【0061】
【表3】
【実施例2】
【0062】
次に、実施例1と同様の手順で作製した有効膜面積2.0cm2の分離膜を利用し、図2に示す装置により、脱水性能を評価した。含水率が4wt%のエタノール溶液、28wt%の1−プロパノール溶液、もしくは16wt%の2−プロパノール溶液を加えた120mlのセパラブルフラスコをブロックヒーター内に設置し、分離膜は、フラスコ上部に接続した枝付ガラス管内に設置した。
【0063】
ガラス管外部からリボンヒーターで加熱することで、分離膜の温度を調節した。このとき、ヒーター温度、分離膜温度および透過側圧力が脱水特性に及ぼす影響を調査した。
【0064】
その結果、表4:ヒーター温度が脱水性能に及ぼす影響、表5:分離膜温度が脱水性能に及ぼす影響、表6:透過側圧力が脱水性能に及ぼす影響に示す結果を得た。また、これらの表4〜6の結果を整理することで、それぞれ図3〜5の結果を得た。図3では、横軸にヒーター温度Tfと溶液沸点TLの温度差を、縦軸に脱水速度をヒーター温度150℃での脱水速度で規格化した値J/J150℃をプロットした。
【0065】
図4では、横軸に分離膜温度TMと溶液沸点TLの差を、縦軸に脱水速度Jをモル濃度に換算した含水率で除した値をプロットした。図5では、横軸に透過側の圧力を、縦軸に透過側圧力が0.5kPa以下としたときの脱水速度で規格化した脱水速度J/J0.5kPaをプロットした。
【0066】
図3に示すように、溶液の種類および含水率に依らず、温度差(Tf−TL)が10〜30℃のとき、J/J150℃が著しく増大したことから、ヒーター温度を、溶液沸点より10℃以上高くすることで、分離膜の脱水速度を著しく向上できることが分かった。
【0067】
また、図4に示すように、分離膜の温度、溶液の種類および含水率に依らず、分離膜の脱水特性が一定であることが分かった。しかし、蒸気の液化を防止するためには、溶液の沸点以上であることが好ましいと考えられた。図5に示すように、透過側圧力が80kPaでも選択的に脱水できたが、溶液の種類や含水率に依らず透過側圧力が1kPa以上になると、脱水速度が著しく低下した。
【0068】
【表4】
【0069】
【表5】
【0070】
【表6】
【0071】
以上、実施例1および実施例2の結果から、分離膜は、反応器の内部もしくは外部のいずれに設置することもできた。しかし、脱水速度制御の観点から、分離膜は、反応器外部に設置することが好ましいことが分かった。また、反応器の加熱温度は、溶液の沸点より10℃以上高ければよいが、10〜30℃高い温度とすることがより好ましかった。
【0072】
分離膜の温度は、特に脱水速度に影響を及ぼさないが、溶液の凝縮を回避するため、溶液の沸点以上で80〜150℃とすることが好ましかった。透過側の圧力は、負圧であればよいが、より高い脱水速度を得るためには、10kPa未満であることが好ましく、更に好ましくは1kPa未満であった。
【実施例3】
【0073】
図1に示す反応器を利用し、濃硫酸を触媒として、アジピン酸と2−プロパノールのエステル化反応を実施した。120mlのセパラブルフラスコにアジピン酸、2−プロパノール、触媒を加え、ヒーター温度110℃で反応させた。ただし、アジピン酸に対する2−プロパノールのモル比は4.3、触媒添加量は基質の1.9wt%、反応時間は4時間とし、分離膜は設置しなかった。
【0074】
溶液の組成を高速イオンクロマトグラフおよびガスクロマトグラフで分析し、アジピン酸の転化率は100−(アジピン酸濃度))/(アジピン酸初期濃度)×100、アジピン酸ジイソプロピルエステルの収率は(アジピン酸ジイソプロピル濃度)/(アジピン酸初期濃度)×100により算出した。その結果、転化率は93.9%、収率は58.6%であった。
【実施例4】
【0075】
触媒添加量を0.3wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は86.2%、収率は39.1%であった。
【実施例5】
【0076】
反応時間を14時間としたことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は92.9%、収率は55.6%であった。
【実施例6】
【0077】
反応時間を40時間としたことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は93.3%、収率は55.9%であった。
【実施例7】
【0078】
触媒としてアンバリスト15H(商品名)を使用し、その添加量を5.8wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は24.9%、収率は2.9%であった。
【実施例8】
【0079】
触媒としてナフィオンNR−50(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は10.7%、収率は1.9%であった。
【実施例9】
【0080】
触媒としてナフィオンSAC−13(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.5%、収率は0.9%であった。
【実施例10】
【0081】
触媒としてダウェックス50W×2(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は42.1%、収率は4.9%であった。
【実施例11】
【0082】
触媒としてダウェックス50W×4(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は48.7%、収率は7.5%であった。
【実施例12】
【0083】
触媒としてダウェックス50W×8(商品名)を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は27.4%、収率は4.2%であった。
【実施例13】
【0084】
触媒としてH−ZSM−5型ゼオライト(Si/Al=25)を使用し、その添加量を19.2wt%としたことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は6.8%、収率は0.7%であった。
【実施例14】
【0085】
触媒としてニオブ酸化物を使用したことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.1%、収率は1.1%であった。
【実施例15】
【0086】
触媒としてスカンジウムトリフルオロメタンスルホン酸を使用したことを除いて、実施例4と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は77.7%、収率は24.8%であった。
【0087】
比較例1
触媒を使用しなかったことを除いて、実施例3と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は13.6%、収率は2.1%であった。
【0088】
実施例3〜15、および比較例1の結果を表7にまとめた。表7に示すように、触媒を加えなかった場合と比べ、硫酸、アンバリスト15H、ダウェックス50W×2、ダウェックス50W×4、ダウェックス50W×8、Sc(OTf)3を加えた場合に、転化率および収率が向上し、ナフィオンNR−50、ナフィオンSAC−13、ゼオライト、およびニオブ酸化物では、転化率および収率の増大は見られなかった。また、硫酸およびSc(OTf)3といった均一触媒が高い触媒活性を有していたのに対して、その他の不均一触媒の転化率および収率は低かった。不均一触媒の中では、ダウェックスシリーズは、比較的高い触媒活性を有していた。
【0089】
【表7】
【実施例16】
【0090】
ダウェックス50Wx2を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は62.4%、収率は11.6%であった。
【実施例17】
【0091】
ダウェックス50Wx4を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果転化率は69.3%、収率は15.2%であった。
【実施例18】
【0092】
ダウェックス50Wx8を2−プロパノールで洗浄、乾燥させ、触媒として使用したことを除いて、実施例3と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は37.9%、収率は6.8%であった。
【実施例19】
【0093】
触媒添加量を28.5wt%としたことを除いて、実施例17と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は95.3%、収率は61.4%であった。
【0094】
実施例16〜19の結果を表8にまとめた。表8に示すように、イオン交換樹脂であるダウェックスを2−プロパノールで洗浄することで、樹脂に含まれる水分が除去されたため、触媒活性が向上した。洗浄したダウェックス50W×4を更に多く使用すると、硫酸と同等の反応活性が得られた。
【0095】
【表8】
【0096】
以上の結果から、均一触媒である硫酸、Sc(OTf)3の他、イオン交換樹脂等の有機固体酸が触媒として有効であった。特に、均一触媒である硫酸、有機固体酸であるダウェックスがエステル化触媒として有望であった。
【実施例20】
【0097】
実施例1と同様の手順で調製した分離膜を利用し、アジピン酸と2−プロパノールのエステル化反応を実施した。120mlのセパラブルフラスコにアジピン酸、2−プロパノール、硫酸を加え、図1のように反応器内部に分離膜を設置した。
【0098】
アジピン酸に対する2−プロパノールのモル比mは4.3、基質重量に対する膜面積の比S/Vは0.0079m2/kg、基質重量に対する触媒添加量Wは2.0wt%、分離膜下端と溶液面の距離Lは3cm、ヒーター温度Tfは110℃、反応時間tRは4時間、加熱開始から分離膜の透過側の減圧を開始するまでの時間tdは1分、透過側の圧力Ppは0.5kPa未満とした。減圧開始時の溶液温度は31℃であった。その結果、転化率は99.8%、収率は92.0%であった。
【実施例21】
【0099】
以下に示す方法でNaA型ゼオライト膜を調製し、エステル化反応に供した。水ガラスを溶解させた水溶液に、アルミン酸ナトリウムを溶解させた水溶液を加えて膜成長水溶液とし、外表面にNaA型ゼオライトの結晶を擦り込んだ多孔質α−アルミナ管とともに、圧力容器に加えて、100℃で4.5時間加熱し、アルミナ管の外表面にNaA型ゼオライト層を形成した。以下、実施例1と同様に、洗浄、乾燥、接着を行い、有効面積2.0cm2のNaA型ゼオライトの分離膜を得た。
【0100】
分離膜をNaA型ゼオライトの分離膜としたことを除いて、実施例20と同様の条件および操作手順でアジピン酸と2−プロパノールのエステル化反応を実施した。その結果、tR=4時間における転化率は99.5%、収率は97.3%であった。
【実施例22】
【0101】
触媒として2−プロパノールで洗浄後に乾燥させたダウェックス50W×4を28.7wt%使用したことを除けば、実施例20と同様の反応条件および操作手順でエステル化反応を実施した。その結果、転化率は99.9%以上、収率は87.1%であった。
【0102】
表9に、実施例3、および実施例19〜22の結果をまとめた。表9に示すように、分離膜の種類、触媒の種類に依らず、分離膜を利用することで転化率および収率が向上したことから、分離膜による脱水がエステル化反応に有効であることが分かった。
【0103】
【表9】
【実施例23】
【0104】
実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を利用し、アジピン酸と2−プロパノールのエステル化反応を実施した。ただし、w=0.6wt%、tR=8時間、S/V=0.0035m2/kgとした以外は、実施例20と同様の条件および操作手順とした。その結果、転化率は98.5%、収率は79.4%であった。
【実施例24】
【0105】
S/V=0.0050m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は98.2%、収率は87.6%であった。
【実施例25】
【0106】
S/V=0.0060m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は99.9%以上、収率は92.3%であった。
【実施例26】
【0107】
S/V=0.0071m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は98.9%、収率は98.2%であった。
【実施例27】
【0108】
S/V=0.0080m2/kgとしたことを除いて、実施例23と同様の反応条件および操作手順とした。その結果、転化率は99.0%、収率は97.1%であった。
【0109】
表10に、実施例3、実施例6、および実施例23〜27の結果をまとめた。表10に示すように、実施例3および実施例6より、分離膜を使用しない場合、転化率が93〜94%で、収率が55〜59%で平衡であるため、分離膜を利用しなければ、それ以上の転化率および収率を得ることが困難であった。分離膜を利用することで、この平衡を超える転化率および収率を得ることができた。このとき、反応基質重量に対する分離膜面積の比S/Vは0.0035m2/kg以上であればよいことが分かった。
【0110】
【表10】
【実施例28】
【0111】
分離膜と溶液面との距離Lを1cmとしたことを除いて、実施例20と同様の条件でエステル化反応を実施した。分離膜は、実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を使用した。その結果、4時間後の転化率は99.9%以上で、収率は90.8%であった。
【実施例29】
【0112】
分離膜と溶液面との距離Lを30cmとするため、実施例2と同様にフラスコ上部に設置した枝付ガラス管内に分離膜を設置し、ガラス管上部の枝部分から流出した蒸気を冷却、還流させたことを除いて、実施例20と同様の条件および操作手順でエステル化反応を実施した。ただし、分離膜の温度は90℃とした。その結果、転化率は99.4%、収率は94.8%であった。
【0113】
表11に実施例20、28、29の結果をまとめた。表11に示すように、分離膜と溶液面との距離Lは転化率および収率に影響を及ぼさなかった。これは、実施例1および実施例2で検討したように、分離膜の温度が適切で、かつヒーター温度が反応溶液の沸点(90〜95℃)よりも15〜20℃高かったので、分離膜が充分な脱水速度を保持できたためであった。
【0114】
【表11】
【実施例30】
【0115】
分離膜の温度を110℃としたことを除いて、実施例29と同様の条件および操作手順でエステル化反応を実施した。その結果、転化率は98.0%、収率は89.6%であった。
【0116】
表12に実施例29、30の結果をまとめた。表12に示すように、分離膜の温度は、転化率および収率に大きな影響を与えなかった。これは、実施例2に示すように、分離膜の脱水速度が、分離膜の温度に影響されないためであった。
【0117】
【表12】
【実施例31】
【0118】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを3分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は51℃であった。その結果、転化率は98.6%、収率は88.3%であった。
【実施例32】
【0119】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを7.5分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は71℃であった。その結果、転化率は98.6%、収率は86.3%であった。
【実施例33】
【0120】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを8分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は80℃であった。その結果、転化率は99.1%、収率は92.5%であった。
【実施例34】
【0121】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを10分としたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は92℃であった。その結果、転化率は99.0%、収率は91.4%であった。
【実施例35】
【0122】
反応溶液の加熱開始から分離膜の内表面の減圧開始までの時間tdを30分としたこと、透過側の圧力を1kPaとしたことを除いて、実施例29と同様の反応条件および操作手順でエステル化反応を実施した。減圧開始時の溶液温度は91℃であった。その結果、転化率は99.1%、収率は87.2%であった。
【0123】
表13に実施例29、31−35の結果をまとめた。表13に示すように、加熱開始から減圧開始までの時間tdは、転化率および収率にほとんど影響を与えなかった。これは、反応時間(4時間)と比べ、tdが充分に短いためであった。また、透過側圧力を1kPaとしても収率および転化率は高い値を維持していた。
【0124】
【表13】
【実施例36】
【0125】
実施例20、実施例21、実施例28で使用した分離膜について、40℃の2−プロパノール溶液を用いて浸透気化法によりエステル化反応前後の分離膜の脱水速度を測定した。その結果、表14に示す結果を得た。
【0126】
比較例2
実施例1と同様の手順で調製したCHA型ゼオライトの分離膜を使用して、蒸気透過試験を実施した。フラスコに2−プロパノール16.7g、水1.7g、アジピン酸9.4gを加え、ヒーター温度Tf=110℃、分離膜と液面の距離L=1cmとして、蒸気透過試験を実施した。蒸気透過試験の前後に、40℃の2−プロパノール溶液(含水率9%)を用いた浸透気化法により分離膜の脱水速度を測定したところ、蒸気透過前の脱水速度は1.25kg/m2/hであったのに対して、蒸気透過後には1.00kg/m2/hであった。
【0127】
実施例35および比較例2の結果を表14にまとめた。表14に示すように、この結果から、分離膜と液面の距離Lが1cmのとき、反応前後で分離膜の脱水速度は40%以上低下したのに対して、L=3cmとすることでその減少率は20%となった。これは、比較例2の硫酸を含まない溶液に対する減少率と等しかった。
【0128】
この結果から、撹拌速度が400rpm以下であれば、硫酸を含む溶液飛沫が付着による脱水速度の低下を抑制するためには、分離膜と反応溶液面の距離を3cm以上にすればよいことが分かった。
【0129】
一方、NaA型ゼオライトの分離膜は、減少率が約30%とCHA型ゼオライト膜より大きかったことから、NaA型ゼオライトと比べて、CHA型ゼオライトの分離膜の方が耐酸性が高いことが分かった。しかしながら、比較例2から明らかなように、アジピン酸の影響による脱水速度の低下を回避することができなかった。
【0130】
【表14】
【実施例37】
【0131】
実施例31〜34で使用した分離膜についても、40℃の2−プロパノール溶液を用いて浸透気化法によりエステル化反応前後の分離膜の脱水速度を測定した。その結果、表15に示す結果を得た。
【0132】
表15に示すように、加熱開始から1分後に減圧を開始(td=1分)すると、脱水速度の減少率は約20%であったが、td=10分とすると脱水速度は減少しなかった。アジピン酸が溶解した2−プロパノールの沸点は90〜95℃であったことから、溶液温度が沸点に到達した後に分離膜内表面の減圧を開始することで、脱水速度の低下を抑制することができた。すなわち、分離膜内表面の減圧開始は、溶液温度が沸点に到達した後に開始することが好ましいことが分かった。
【0133】
【表15】
【実施例38】
【0134】
有機酸としてミリスチン酸を、アルコールには2−プロパノールを、触媒には硫酸を使用し、ミリスチン酸と2−プロパノールのエステル化反応を実施した。ミリスチン酸に対する2−プロパノールのモル比mは3、硫酸濃度は基質重量の1.0wt%、反応時間は8時間、膜面積と基質重量の比は0.008m2/kg、ヒーター温度は110℃、膜温度は90℃、分離膜内表面の減圧開始時間は15分(溶液温度92.4℃)、減圧度は1kPa未満とした。その結果、ミリスチン酸の転化率(ミリスチン酸イソプロピルの収率)は99.9%であった。
【実施例39】
【0135】
有機酸としてパルミチン酸を使用したことを除いて、実施例38と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は90.6℃)。その結果、パルミチン酸の転化率(=パルミチン酸イソプロピルの収率)は99.7%であった。
【実施例40】
【0136】
有機酸としてセバシン酸を、アルコールとしてエタノールを使用し、セバシン酸に対するエタノールのモル比mを5としたことを除いて、実施例38と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は85.8℃)。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジエチルの収率は99.1%であった。
【実施例41】
【0137】
アルコールとしてブタノールを使用し、ヒーター温度を130℃、膜温度を110℃としたことを除いて、実施例40と同様の反応条件および操作手順でエステル化反応を実施した(減圧開始時の溶液温度は103.6℃)。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジブチルの収率は98.9%であった。
【0138】
比較例3
分離膜を使用しなかったことを除いて、実施例38と同様の反応条件および操作手順で、ミリスチン酸と2−プロパノールのエステル化反応を実施した。8時間後のミリスチン酸の転化率は(ミリスチン酸イソプロピルの収率)は83.1%であった。
【0139】
比較例4
分離膜を使用しなかったことを除いて、実施例39と同様の反応条件および操作手順で、パルミチン酸と2−プロパノールのエステル化反応を実施した。8時間後のパルミチン酸の転化率は(パルミチン酸イソプロピルの収率)は84.9%であった。
【0140】
比較例5
分離膜を使用しなかったことを除いて、実施例40と同様の反応条件および操作手順で、セバシン酸とエタノールのエステル化反応を実施した。8時間後のセバシン酸の転化率は98.1%、セバシン酸ジエチルの収率は76.2%であった。
【0141】
比較例5
分離膜を使用しなかったことを除いて、実施例40と同様の反応条件および操作手順で、セバシン酸とブタノールのエステル化反応を実施した。8時間後のセバシン酸の転化率は99.9%以上、セバシン酸ジブチルの収率は84.9%であった。
【0142】
実施例38〜41及び比較例3〜6の結果を表16にまとめたように、分離膜を使用しなかった比較例3〜6では、エステルの収率が76〜85%であったのに対して、分離膜を使用することで収率を99%以上にすることができた。これは、固体有機酸と炭素数4以下のアルコールの組合せであれば、分離膜を使用することで、固体有機酸をエステルに効率よく転換できることを示しており、分離膜が多様なエステルの製造に有効であることが分かった。
【0143】
【表16】
【産業上の利用可能性】
【0144】
以上詳述した通り、本発明は、エステルの製造方法に係るものであり、本発明により、分離膜を利用したエステル化反応プロセスにおいて、反応器設計、反応操作条件設定等の調整により、分離膜の脱水速度および耐久性を向上させることでエステル化反応プロセスの小規模化およびメンテナンス回数の低減を可能にした新しいエステル化反応プロセスを提供できる。本発明は、高い脱水速度および耐久性を有する分離膜を利用したエステル化反応プロセスを構築することでエステル化反応に必要な分離面積を従来の1/10〜1/100に低減することを可能とする分離膜を利用したエステル化反応プロセスに関する新技術を提供するものとして有用である。
【特許請求の範囲】
【請求項1】
反応基質を含む反応系から構成される反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始すること、を特徴とするエステルの製造方法。
【請求項2】
反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下である、請求項1記載のエステルの製造方法。
【請求項3】
分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去する、請求項1記載のエステルの製造方法。
【請求項4】
(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満である、請求項1記載のエステルの製造方法。
【請求項5】
反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールである、請求項1記載のエステルの製造方法。
【請求項6】
分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜である、請求項1記載のエステルの製造方法。
【請求項7】
多孔質支持体が、0.01〜100μmの大きさの空孔を持ち、その空隙率が10〜70%である、請求項1記載のエステルの製造方法。
【請求項8】
分離器が、反応器内部又は反応器外部に設置される、請求項1記載のエステルの製造方法。
【請求項9】
反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去する、請求項1記載のエステルの製造方法。
【請求項10】
減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させる、請求項1記載のエステルの製造方法。
【請求項11】
反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始する、請求項1記載のエステルの製造方法。
【請求項12】
分離膜の透過側の減圧の度合いが、10kPa未満である、請求項10記載のエステルの製造方法。
【請求項1】
反応基質を含む反応系から構成される反応溶液を収容した反応器において、触媒存在下で、カルボン酸とアルコールを反応させると同時に、反応によって生成した水を多孔質支持体上に支持された分離膜を有する分離器で除去することによりエステルを製造する方法において、
分離膜が反応系から水を除去するための減圧操作を、反応溶液の加熱開始から10分以上後もしくは反応溶液温度が沸点に達した後に開始すること、を特徴とするエステルの製造方法。
【請求項2】
反応溶液の加熱温度が、反応溶液の沸点より10〜20℃高く、分離膜の温度が、反応溶液の沸点以上で、且つ80〜150℃の範囲内であり、反応溶液の撹拌速度が400rpm以下である、請求項1記載のエステルの製造方法。
【請求項3】
分離膜を反応溶液面から3cm以上上部に取り付け、反応溶液上部に取り付けた分離膜により、反応溶液から発生した蒸気中から水を選択的に除去する、請求項1記載のエステルの製造方法。
【請求項4】
(分離膜面積)/(反応溶液の重量)が0.01m2/kg未満である、請求項1記載のエステルの製造方法。
【請求項5】
反応基質のカルボン酸は室温で固体カルボン酸、アルコールはC1〜C4アルコールである、請求項1記載のエステルの製造方法。
【請求項6】
分離膜がCHA型ゼオライト膜またはNaA型ゼオライト膜である、請求項1記載のエステルの製造方法。
【請求項7】
多孔質支持体が、0.01〜100μmの大きさの空孔を持ち、その空隙率が10〜70%である、請求項1記載のエステルの製造方法。
【請求項8】
分離器が、反応器内部又は反応器外部に設置される、請求項1記載のエステルの製造方法。
【請求項9】
反応溶液から発生した蒸気から蒸気透過法によって水蒸気を除去する、請求項1記載のエステルの製造方法。
【請求項10】
減圧ポンプを利用して分離膜の透過側を減圧することにより水を排出させる、請求項1記載のエステルの製造方法。
【請求項11】
反応溶液の沸点をTbとしたときにTb−5(℃)以上に達した時点で、水の排出を開始する、請求項1記載のエステルの製造方法。
【請求項12】
分離膜の透過側の減圧の度合いが、10kPa未満である、請求項10記載のエステルの製造方法。
【図1】

【図2】
【図3】
【図4】

【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−214387(P2012−214387A)
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2011−79143(P2011−79143)
【出願日】平成23年3月31日(2011.3.31)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【出願人】(000228729)日本サーファクタント工業株式会社 (44)
【Fターム(参考)】
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願日】平成23年3月31日(2011.3.31)
【出願人】(301021533)独立行政法人産業技術総合研究所 (6,529)
【出願人】(000228729)日本サーファクタント工業株式会社 (44)
【Fターム(参考)】
[ Back to top ]