
オキシントモジュリン誘導体
修飾されたオキシントモジュリン誘導体。かかる誘導体は、糖尿病及び肥満症のような代謝性疾患の治療のために使用可能である。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、オキシントモジュリン誘導体、それらの合成、及び糖尿病及び肥満症のような代謝障害の治療のためのそれらの使用に関する。
【背景技術】
【0002】
ホルモンであるオキシントモジュリン(OXM、グルカゴン−37)は、腸及び中枢神経系(CNS)におけるプレプログルカゴンプロセシングの翻訳後産物であり、摂食に応じて腸管のL−細胞から分泌される。1983年に発見されたOXMは、摂食及びエネルギー消費の調節と関係づけられてきた。ラットにおけるOXMの中枢又は末梢投与は、短期間の摂食の減少を、胃内容排出に対する最小の影響と共に引き起す(デイキン(Daikin)ら著、「エンドクリノロジー」、2001年、第142巻、p4244−4250、デイキンら著、「エンドクリノロジー」、2004年、第145巻、p.2687−2695)。ラットにおけるOXMの反復の脳血管内投与は、結果として、ペアフェッドの動物に比較して、核心温度の上昇及び体重増加の減少を生じ、このことはカロリー摂取及びエネルギー消費の双方に対する影響を示唆している(デイキンら著、「アメリカン・ジャーナル・オブ・フィジオロジー、エンドクリノロジー・アンド・メタボリズム」、2002年、第283巻、E1173−E1177)。
【0003】
OXMは、37アミノ酸のペプチドである。胃酸分泌阻害におけるOXMの効果は、SP−1として知られる8残基のC−末端フラグメントOXM(30−37)により、模倣可能であることが報告されている(カルレス−ボネ(Carles−Bonnet)ら著、「ペプタイズ」、1996年、第17巻、p.557−561)。ヒトでは、健康な正常体重の被験者におけるOXMの単回の90分間の静脈内注入が、ハンガースコア及び立食における摂食を〜19%まで低減した。累積12時間のカロリー摂取は、何らの嘔気又は食物嗜好性の変化の報告なしに、〜11%まで低減された(コーエン(Cohen)ら著、「ザ・ジャーナル・オブ・クリニカル・エンドクリノロジー・アンド・メタボリズム(J.Clin.Endocrinol.Metab.)」、2003年、第88巻、p.4696−4701)。より最近では、肥満の健康なボランティア(BMI〜33)における4週間にわたるOXMの食事前注射は、処置の第1日目に有意なカロリー摂取の低減(〜25%)をもたらし、それは研究の期間にわたって維持された(4週間後、35%減少)(ワイン(Wynne)ら著、「ダイアベーツ(Diabetes)」、2005年、第54巻、p.2390−2395)。処置された被験者では、研究の最後に強い体重減少が観察された(1.9%、プラセボ補正)。OXMの血漿レベルは、注入研究において観察されたものと同様であった(ピーク濃度〜950pM)。インビボでのOXMの不十分な安定性(血漿t1/2<12分間)により必要とされる比較的高い用量にもかかわらず、何らタキフィラキシーがないこと、及び、軽度で一過性の嘔気の発生率が低いこと(〜3%)は、このホルモンを、ヒューマンバリデーション及び魅力的な耐性プロフィールの双方を備えた数少ない肥満症ターゲットの1つにしている。
【0004】
OXMは、非常に短い半減期をもち、細胞表面ジペプチジルペプチダーゼIV(以降DP−IV)により迅速に不活性化される。しかしながら、DP−IV阻害剤は、臨床ではウェイトニュートラル(体重に対し中立)であり、生理的なレベルを超えたOXM(900−1000pM)が、ヒトの体重減少を達成するために必要かもしれないことを示唆している。
【0005】
オキシントモジュリンは、それ故、糖尿病及び肥満症のような代謝障害の治療剤としての可能性を示す。しかしながら、インビボでのOXMの不十分な安定性の故に、糖尿病及び肥満症のような代謝性疾患の治療に対して、安全かつ有効に投与されうるOXM誘導体を開発することが必要である。もし、安定性及び薬物動態を改善する部位への結合により修飾された、又はより具体的にはDP−IV切断に対し耐性を与える修飾により修飾された類似体又は誘導体が開発されるならさらに望ましいであろう。本発明は、OXMポリペプチド誘導体、及び本明細書に記述された誘導体を投与することによる、肥満症及び糖尿病のような代謝障害の治療又は予防の方法を提供する。
【発明の開示】
【0006】
(発明の要旨)
本発明は、式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;
X6は、任意のアミノ酸であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]
を包含するポリペプチドを提供する。医薬的に許容されるそれらの塩は、同様に考えられる。
【0007】
さらに、X3、X4、X6−X8、及びX10−X14のいずれか1又は2個は、Cys(mPEG)、Cys(コレステリル)でよく;Cys(mPEG)teineはまたC1;C2;C3又はC6でもよく、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、かつ各々は、側鎖チオールを介し、示された分子量の直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する。
【0008】
本発明は、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3
[式中、C3=Cys[(mPEG)240kDa]であり、各々は、側鎖チオールを介し、示されたMWの分枝したmPEG[(mPEG)2]によりPEG化されたアミド化システイン残基に相当し;αは、α−アミノイソ酪酸(aib)であり;そして、m=メチオニンスルホキシド(Met(O))である]のOXMポリペプチド誘導体に関する。
【0009】
本発明のもう1つの実施態様においては、
HxSQGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、Hxは、His;HI=イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis;N−メチル−His(Me−H)、N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H)、ベンジルHis(Bzl−H)及びPheからなる群より選択される]を包含するポリペプチドが提供される。
【0010】
本発明のなおもう1つの実施態様においては、
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg、Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含するポリペプチドが提供される。
【0011】
本発明はさらに、
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含するポリペプチドを提供する。
【0012】
本発明のもう1つの実施態様においては、
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)の任意の1つであり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)の任意の1つであり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、
ここで、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)である]を包含するポリペプチドが提供される。
【0013】
本発明のもう1つの実施態様においては、
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Cys(N−エチルマレイミジル)、Q又はCであり;
X17は、m又はMであり;
X18は、アミド化されたk又はKである]を包含するポリペプチドが提供される。
【0014】
本発明のもう1つの実施態様においては、
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、X19は、C又はC8、Cys(N−エチルマレイミジル)である]を包含するポリペプチドが提供される。
【0015】
本発明のなおもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
のポリペプチドが提供される。
【0016】
本発明のもう1つの実施態様においては、式:
HaDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH
[式中、Ttdsは、1−アミノ−4,7,10−トリオキサ−13−トリデカンアミン−スクシンイミン酸(succinimic acid)である]のポリペプチドが提供される。
【0017】
本発明のもう1つの実施態様においては、患者における代謝性疾患の治療のための方法であって、上記記載のポリペプチドを該患者へ投与することを包含する方法が提供される。該代謝性疾患は、糖尿病、メタボリックシンドローム、高血糖症及び肥満症からなる群より選択されてよく、脳までの末梢経路、例えば経口、経粘膜、バッカル、舌下、経鼻 直腸、皮下、経皮 静脈内、筋肉内又は腹腔内経路により投与されてよい。
【0018】
本発明のなおもう1つの実施態様においては、上記記載のポリペプチドと、医薬的に好適な担体とを包含する医薬組成物が提供される。
【0019】
本発明はさらに、必要とする患者に本発明のポリペプチド及び医薬組成物を投与することによる、糖尿病又は肥満症のような代謝障害の治療又は予防に有用な医薬品の調製における、本発明のポリペプチドの使用に関する。
【0020】
(発明の詳細な記載)
本発明は、修飾されたOXM誘導体に関する。OXM誘導体は、安定性及び薬物動態を改善するためのPEG化又は他の部位又は担体タンパク質への結合により、及び/又は、DP−IV切断に対しペプチドを抵抗性にするためのアミノ酸残基の置換基の組み込みにより開発される。さらに、安定化されたOXM誘導体は、グルカゴン受容体アゴニスト活性を示さず、それにより、糖尿病又は前糖尿病患者における高血糖症及び肥満症の治療において一定の利点を提供しうる。これらの患者については、グルカゴン受容体シグナリングの上方制御は、そのことが結果として血中グルコースレベルの上昇を生じ得るので、回避されねばならない。
【0021】
他に特定されない限り、用語「ポリペプチド」、「タンパク質」、及び「ペプチド」は、当業者により、種々の修飾及び/又は安定化された型も包含することが理解される。かかる修飾された型は、PEG化型、パルミトイル化型、コレステロール修飾型、その他を含む化学修飾型であってよく、それに限定されない。修飾はまた、分子内架橋、及び脂質、フラビン、ビオチン、ポリエチレングリコール誘導体、その他のような種々の部位への共有結合も包含する。加えて、修飾はまた、環化、分枝及び架橋を包含してもよい。さらに、遺伝子によってコードされた通常の20アミノ酸以外のアミノ酸もまた、ポリペプチド中に包含されうる。
【0022】
OXM誘導体の構造
本発明は、修飾されたOXM誘導体を提供する。特に、本発明は、式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;
X6は、任意のアミノ酸であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]の、新規な安定化された修飾されOXMポリペプチド誘導体に関する。
【0023】
さらに、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)でよく;Cys(mPEG)teineはまたC1;C2;C3又はC6でもよく、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、そして各々が、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する。
【0024】
本発明はさらに、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3
[式中、C3=Cys[(mPEG)240kDa]であり、各々が、側鎖チオールを介し、示されたMWの分枝したmPEG[(mPEG)2]によりPEG化されたアミド化システイン残基に相当し;αはα−アミノイソ酪酸(aib)であり;そして、m=メチオニンスルホキシド[Met(O)である]の、OXMポリペプチド誘導体を提供する。
【0025】
本発明のもう1つの実施態様においては、
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg;Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含するポリペプチドが提供される。
【0026】
本発明はさらに、
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含するポリペプチドを提供する。
【0027】
本発明のもう1つの実施態様においては、
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、
ここで、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)である]を包含するポリペプチドが提供される。
【0028】
本発明のもう1つの実施態様においては、
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Q又はCであり;
X17は、m又はMであり;
X18はアミド化されたk又はKである]を包含するポリペプチドが提供される。
【0029】
本発明のもう1つの実施態様においては、
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、X19は、C又はC8である]を包含するポリペプチドが提供される。
【0030】
本発明のなおもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
のポリペプチドが提供される。
【0031】
本発明のもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH
のポリペプチドが提供される。
【0032】
本明細書で用いる場合、アミノ酸残基の略語は、以下のように示される:
【0033】
【表1】
【0034】
他に具体的に指定しない限り、全てのアミノ酸残基はL−型である。
【0035】
野生型OXMに比較して、本発明のOXM誘導体は、いくつかのアミノ酸置換基を含有し、及び/又は、PEG化されるか又は別の方法で修飾可能(例えばコレステロール部位)である。類似体は、例えばコレステロール及びPEGの双方により、二重に結合されてもよい。かかるOXM誘導体は、切断に抵抗性であり、ジペプチジルペプチダーゼIV(DP−IV)に不活性である。
【0036】
「受容体アゴニスト」とは、受容体、例えばGLP−1R又はグルカゴン受容体と相互作用し、それにより受容体活性化の薬理学的又は生化学的応答特性を開始し得る、任意の内在又は外来性(薬剤)物質又は化合物が意味される。典型的には、本発明のOXM誘導体は、ヒトGLP−1Rに対する親和性により特徴づけられ、この受容体に対し0.1pM−1μMの範囲のEC50を示す。本発明のOXM誘導体はまた、GcgRに対するその親和性によっても特徴づけられ、EC50>1μMを示す。
【0037】
本発明のOXM誘導体は、摂食及び体重増加の低減において有用であり、また膵島からのグルコース刺激インスリン分泌(GSIS)を仲介し得、それにより例えば肥満症、糖尿病、メタボリックシンドロームX,高血糖症、空腹時グルコース障害、及び他の前糖尿病状態のような代謝障害にかかっている個体のための、治療オプションを提供する。
【0038】
【表2】
【0039】
【表3】
【0040】
【表4】
【0041】
【表5】
【0042】
【表6】
【0043】
【表7】
【0044】
【表8】
【0045】
【表9】
【0046】
α=α−アミノイソ酪酸(Aib);a=D−Ala;S=D−Ser;n=L−ノルロイシン(Nle), X=O−メチル−ホモセリン;C1=Cys(mPEG)5kDa, C2=Cys(mPEG)20kDa, C3=Cys(mPEG)240kDa,各々、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG[(mPEG)2]によりPEG化されたシステイン残基に相当する;C4=Cys(コレステリル),側鎖チオールを介しコレステロールへ結合されたシステイン残基に相当する;C5=Cys(CH2CONH2),側鎖チオールがヨードアセトアミドと反応したシステイン残基に相当する;C6=Cys(mPEG)260kDa、各々、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2mPEG[(mPEG)2]によりPEG化されたシステイン残基に相当する;H1=イミダゾール−乳酸(ImiH);H2=デスアミノ−His(ΔNH2−H) 1Ac=アセチル;Pyr=ピログルタミル;Me−H=N−メチル−His;Me2−H=N,N−ジメチル−His;Bz=ベンゾイル(C7H5O);Bzl=ベンジル(C7H7);m=メチオニンスルホキシド, C7=(Cys)2(mPEG)2−40kDa,各々、側鎖チオールを介し、同じ1つの直鎖メトキシPEG(mPEG)又は1つの分枝鎖mPEG[(mPEG)2]によりPEG化された2つのシステイン残基に相当する, C8=Cys(N−エチルマレイミジル);Ttds,1−アミノ−4,7,10−トリオキサ−13−トリデカンアミン スクシンイミン酸;k,D−リジン。
【0047】
1.1.アミノ酸置換及び修飾
X1(OXMの2位)における置換は、OXM及びGLP−1を含む多くのペプチドの分解において重要な役割を果たすDP−IVによるタンパク質分解に対し、OXM誘導体の抵抗性を改善するためにデザインされている。GLP−1における2位のSerのGlyによる置換が、DP−IV切断に対する抵抗性を改善することが報告されてきた(ロッテ(Lotte,B.K.)著、「ジャーナル・オブ・メディカル・ケミストリー」、2004年、第47巻、p.4128−4134)。OXMとGLP−1との間の、高度の配列相同性にもかかわらず、2位におけるSer→Gly置換が、修飾されたOXMに対し同様の効果を与えることは見出されなかった。しかしながら、2位のSerの、Val、Ile、Asp、Glu、Met、Trp、Asn、D−Ala、D−Ser又はα−アミノイソ酪酸による置換は、実施例において以下に議論されたように、対応するOXM誘導体を、DP−IVに対し野生型OXMよりもさらに抵抗性にした。X1(OXMの2位)における置換をもつペプチド類には、OXM4−7、12、23、28−59、及びOXM33−36及びOXM55−59の前駆体を包含する。
【0048】
X2(OXMの3位)おける置換は、GcgRの活性が最少か又は全くないGLP−1Rの選択的アゴニストであるOXM誘導体を創成するためにデザインされている。かかるOXM誘導体は、肥満性糖尿病患者の治療時に有利であろう。X2(OXMの3位)に置換のあるペプチド類には、OXM8−12、15、23、53、95、96及び97を包含する。
【0049】
同様に、X3及びX5(11位及び16位)におけるAlaに対する置換は、GLP−1Rに対し選択的であって、かつGcgRに対し何ら活性をもたないOXM誘導体を創成するためにデザインされている。X3及びX5(11位及び16位)におけるAlaに対するかかる置換の一例は、OXM24である。
【0050】
X3、X4、X6−X8及びX10−X14の任意の1個以上におけるシステインに対する置換は、OXM誘導体の特定の位置におけるPEG化又はコレステリル化を可能にする。他の置換又は修飾は、当該技術分野において公知であり、活性薬剤の表面には物理的に付着するが、活性薬剤と化学結合もしくは相互作用しないものを包含する。2個以上のかかる修飾が使用可能であって、種々のポリマー、低分子量オリゴマー、天然産物及び界面活性剤を含む公知の有機及び無機の医薬用賦形剤から選択されてよい。
【0051】
1.2.PEG化及び/又はコレステリル化
本発明は、二官能性及びマルチアーム型N−マレイミジルPEG誘導体により例示される、多官能性ポリマー誘導体の使用を意図する。広く多様なポリエチレングリコール(PEG)種が、本発明の新規なOXM誘導体のPEG化のために使用されうる。実質的に任意の好適な反応性PEG試薬が使用可能であり、好適な化学種は、NOFコーポレーション(NOF Corporation)(〒150−6019 東京都渋谷区恵比寿4丁目20−3恵比寿ガーデンタワー)のドラッグデリバリーシステム)カタログにおいて市販されているもの、及び、例として、ネクター・セラピューティクス(アラバマ州35806、ハンツビル、490ディスカバリー・ドライブ)のモレキュラーエンジニアリングカタログのものを包含するが、これに限定されない。例として、そして制限されることなく、以下のPEG試薬が種々の実施態様においてしばしば好ましい:マルチ−アーム型PEG(multi−ArmPEG)、mPEG(MAL)2、mPEG2(MAL)、任意のサンブライト(SUNBRIGHT)活性化PEG(カルボキシル−PEG類、p−NP−PEG類、トレシル−PEG類,アルデヒドPEG類、アセタール−PEG類、アミノ−PEG類、チオール−PEG類、マレイミド−PEG類、ヒドロキシ−PEGアミン、アミノ−PEG−COOH、ヒドロキシ−PEG−アルデヒド、カルボン酸無水物型PEG、官能化PEG−リン脂質を包含するが、これに限定されない)、及び、その特定の適用及び使用のために当業者により選択される他の類似の及び/又は好適な反応性PEG。
【0052】
本発明の新規なOXM誘導体ペプチドはまた、スペーサー部位に対し、カルバメート又はアミド結合を介し共有結合される、2つのPEG部位を含有することも可能であり、ここでスペーサー部位は、該ペプチドの第三級アミドリンカーへ共有結合される。本発明のかかる実施態様において使用される2つのPEG部位の各々は、直鎖であってよく、一の結合点において一緒に結合されてもよい。本発明の1つの実施態様においては、各PEG部位は、約10キロダルトン(10K)−約60Kの分子量を有する(用語「約」は、PEGの標品において、ある分子は標準的な分子量よりもさらに重く、あるものはさらに軽いことを示している)。2つのPEG部位の各々は、約20K−約40Kの分子量を有してもよい。当業者は、所望の用量;循環時間;タンパク質分解に対する抵抗性;もしあれば、生物活性に対する影響;取り扱いの容易さ;抗原性の程度又は欠如;及び治療用ペプチドに対する他の公知のPEGの影響のような考慮に基づき、所望のポリマーサイズを選択することができるであろう。
【0053】
本発明の1つの実施態様においては、N−マレイミジルポリマー誘導体のポリマー主鎖は、ポリ(アルキレングリコール)、そのコポリマー、そのターポリマー、又はそれらの混合物である。例としては、ポリ(エチレングリコール)、ポリ(プロピレングリコール)、及びエチレングリコール及びプロピレングリコールのコポリマーを包含する。以下に、より詳細に説明されるように、本発明のさらに好ましい実施態様は、例えば、二官能性PEG、マルチアーム型PEG、フォーク状PEG、分枝状PEG、ペンダント状PEG、及びその中に分解可能な結合をもつPEGのようなPEGポリマーを利用する。しかしながら、他の関連ポリマーもまた本発明の実施において好適であること、及び、用語PEG又はポリ(エチレングリコール)の使用が、この点について包括的であって排他的でないことを意図したものであることが理解されるべきである。用語PEGは、二官能性PEG、マルチアーム型PEG、フォーク状PEG、分枝状PEG、ペンダント状PEG(すなわち、ポリマー主鎖に対して垂れ下った1個以上の官能基を有するPEG又は関連ポリマー)、又はその中に分解可能な結合をもつPEGを含む、そのどのような形状のポリ(エチレングリコール)をも包含する。
【0054】
ポリマー主鎖は、直鎖又は分枝鎖でよい。PEGは一般に、分枝型で使用され、それは、例えばグリセロール、グリセロールオリゴマー、ペンタエリスリトール及びソルビトールのような種々のポリオールに対するエチレンオキシドの添加により調製可能である。中心の枝分かれ部位はまた、例えばリジンのようないくつかのアミノ酸から誘導可能である。分枝したポリ(エチレングリコール)は、R(−PEG−OH)m
[式中、Rは例えばグリセロール、グリセロールオリゴマー又はペンタエリスリトールのようなコア部位であり、mはアームの数を表わす]のような一般式で表わし得る。マルチアーム型PEG分子、例えば、米国特許第5,932,462号に記載されたものもまた、ポリマー主鎖として使用可能であり、ここに参考することによりその全てが本明細書に含まれる。
【0055】
当業者は、実質的に水溶性かつ非ペプチド性のポリマー主鎖に関する上記のリストが、決して完全なものではなく、例示的なものにすぎないこと、及び上記の特性を有する全てのポリマー素材が意図されていることを認識するであろう。
【0056】
本発明のOXM誘導体上のPEG化位置は、OXMの構造及びそのグルカゴン及びGLP−1受容体との相互作用を考慮して選択される。それ故、PEG化は好ましくは位置特異的である。システインのチオール側鎖におけるPEG化は、広く報告されてきた(例えば、カリケティ及びベロネーゼ(Caliceti&Veronese)、2003年参照)。ペプチド中にCys残基がない場合、置換により導入可能である。本発明のOXM誘導体は、システインの側鎖によりPEG化されてもよい。OXM誘導体は、Cys(mPEG)teineを含有してもよい。Cys(mPEG)teine中のmPEGは、種々の分子量を有し得る。分子量の範囲は、好ましくは5kDa−200kDa、さらには5kDa−100kDa、さらに好ましくは20kDa−60kDaである。mPEGは、直鎖又は分枝鎖でよい。例えば、本発明のCys(mPEG)teineは、C1、C2、C3又はC6でよい。本明細書で例示される場合、C1は、分子量5kDaの直鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)5kDa)(例えば、MPEG−MAL−5000、NEKTAR 2F2MOH01)であり;C2は、分子量20kDaの直鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)20kDa)(例えば、MPEG−MAL−20K、NEKTAR 2F2M0P01)であり;C3は、分子量40kDaの分枝鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)240kDa)(例えば、MPEG2−MAL−40K、NEKTAR 2D3Y0T01、又は、Y型PEGマレイミド、MW40K(ジェンケム・テクノロジー(JenKem Technology)、商品番号Y−MAL−40K、又はサンブライト(SUNBRIGHT)GHT GL2−400MAマレイミド(NOFコーポレーション)であり、そしてC6は分子量60kDaの分枝鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)260kDa)(例えば、MPEG2−MAL−60K、NEKTAR 2D3Y0V01)である。
【0057】
別法として、OXM誘導体のシステイン残基はまた、側鎖チオールを介し、コレステロールにより誘導体化されてもよい。コレステリルOXM誘導体の例は、OXM36、OXM59、OXM65、OXM70、OXM76、OXM82、OXM88及びOXM101を包含する。
【0058】
1.3.他の修飾
N−末端ヒスチジンHxは、His、H1=イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H)、及びPheからなる群より選択される誘導体により変異可能である。N−末端におけるアセチル化及び他の修飾、及び、N−末端キャッピング基は、DP−IV切断に対しOXMを安定化させ、一方C−末端のアミド化は、カルボキシペプチダーゼによるインビボの潜在的な分解を防止しうる。N−末端修飾をもつOXM誘導体には、OXM14及びOXM16−22を包含する。
【0059】
実施例において示されたように、単一用量のOXM2及びOXM3は、自由(ad libitum)に摂食したマウスにおける一晩の体重増加の低減に有効であったのに対し、単一用量のOXM1及び野生型又は天然の(wt)OXMは、このモデルでは有効ではなかった。OXM3は、一晩の体重増加の用量依存性の低減において最も高いインビボ有効性を有しており、受容体結合を妨害しそうなよりかさのあるPEG基を有するOXM2に比較して、GLP−1Rに対するその高い効力を反映しているようであった。OXM2及びOXM3の双方が、インビトロではGLP−1Rに対し、天然のOXMよりも有意に効力が低いことから、PEG化されたOXM誘導体については、wtOXMに比較して増大されたインビボ有効性は、タンパク質分解、及び/又は腎クリアランスに対する何らかの安定化がPEG化のみによって誘導されることを示唆している。
【0060】
さらに、血液成分は、当該ペプチドを安定化するために利用されてよい。好ましい血液成分は、例えば免疫グロブリン、血清アルブミン、フェリチン、ステロイド結合タンパク質、トランスフェリン、チロキシン結合タンパク質、α−2−マクログロブリン等のようなタンパク質を含む。
【0061】
2.OXM誘導体の合成
2.1.ペプチド類の合成
以下の一般的な方法が、いくつかのOXM誘導体を合成するために使用された。固相ペプチド合成は、Fmoc化学を用いて、バッチ又は連続フロー条件下に(例えば、ペニントン及びダン(Pennington&Dunn)著、「ペプチド・シンセシス・プロトコールズ」、1994年、第35巻参照)、PEG−ポリスチレン樹脂を用いて行なった。ペプチド類を樹脂から切断し、トリフルオロ酢酸(TFA)、及びフェノールトリイソプロピルシランのようなカチオンスカベンジャー、及び水を用いて脱保護した。ペプチド類を、冷メチル−t−ブチルエーテルを用いて沈澱させ、沈澱したペプチドを、凍結乾燥に先立ち冷エーテルで2回洗浄した。ペプチドは、0.1%TFAを加えた水/アセトニトリルを典型的な移動相として使用したC4カラム上での逆相HPLC、及びエレクトロスプレー質量分析により確認した。ペプチド類は逆相HPLCにより≧95%まで精製した。
【0062】
2.2.ペプチド類のPEG化
ペプチド類をまず合成し、次にシステインのチオール側鎖をPEG化した。以下の一般的な方法を、ペプチド類のPEG化に使用した。
【0063】
PEG化反応は、チオール化されたペプチド前駆体と、マレイミド−mPEGとの間で行ない、チオエーテル結合を形成した。反応はpH7.3で行い、マレイミド−mPEG量は、チオール化されたペプチドに対し、0.5から10倍モル過剰までの範囲であった。PEG化されたOXMペプチドを、次に、逆相HPLC又はイオン交換クロマトグラフィー、及びそれに続いてサイズ排除クロマトグラフィーを用いて単離した。最後に、PEG化ペプチド類を、分析用RP−HPLC及びMALDAI(マルディ)tof質量分析によりキャラクタライズした。
【0064】
3.OXMをベースとする療法の影響
OXMをベースとする療法は、肥満症及び糖尿病の双方に都合よく影響を及ぼす可能性を有する。OXMの末梢投与時の体重減少及び摂食低減は、ヒトにおいて充分に実証されてきた。本発明者らによる研究は、末梢投与されたブタOXMが、マウスにおいて短期間の摂食及び一晩の体重増加を低減するに充分であることを示してきた。OXMのインクレチン(抗血糖上昇)活性は今日まで十分に研究されてきていないが、マウスの腹腔内グルコース負荷試験(IPGTT)において、OXMのグルコース低下活性がGLP−1のそれに匹敵することが、初めて証明されている。GLP−1と同様、OXMは、静的な単離ネズミ膵島及び灌流ラット膵臓から、大量のグルコース刺激インスリン分泌(GSIS)を誘導し(ジャルーセ(Jarrousse)ら著、「エンドクリノロジー」、1984年、第115巻、p.102−105)、通常のインスリン分泌促進薬に比較して、ローリスクの低血糖を示唆している。ラットでは、OXMの、胃内容排出に対する無視できるほど小さい影響が報告されている(デイキン(Dakin)ら著、「エンドクリノロジー」、2004年、第145巻、p.2687−2695)。マウスでは、OXMは、グルコース低下に最大有効用量において、胃内容排出を〜25%まで低減し、これは、GLP−1受容体アゴニストであるエキセンジン4の最大有効用量によってもたらされるもの(47%低減)よりも少ない。それ故、ヒトにおける胃内容排出に対するOXMの潜在的な良性の作用が、現在のGLP−1模倣薬に比較してこのペプチドホルモンの増強された忍容性に役割を果たしているのかもしれない。
【0065】
本発明のポリペプチドは、肥満症及び/又は糖尿病の治療に有用でありうることが示唆される。第2の適応は、メタボリックシンドローム、高血糖症、空腹時グルコース障害、及び他の前糖尿病状態である。本発明のポリペプチドに関する別の適応は、例えば過敏性腸症候群及び他の腸の吸収疾患、虚血、卒中、及び不安、認知障害及びアルツハイマー病を含む神経障害のような任意の及び全てのGLP−1の適応を包含する。
【0066】
OXMのペプチド性の性質は、天然のホルモンを用いた経口療法を排除する。対照的に、本明細書に提示されたOXM誘導体は、本発明のポリペプチドの1つを、制限されることなく、経口、鼻腔内、舌下、十二指腸内、皮下、バッカル、結腸内、直腸、経膣、経粘膜、経肺、経皮、皮内、非経口、静脈内、筋肉内及び眼内を含む、種々の経路による投与に好適な、医薬的に許容される担体と組合せて含んでなる医薬組成物として、0.001mg/kg−10mg/kg、好ましくは1μg/kg−200mg/kgの用量範囲で、1日2回から1週間以上に1回の投与頻度で投与されてよい。当該ペプチド医薬組成物は、投与方法に依存して、多様な単位用量で投与可能である。好適な単位剤形は、粉末、タブレット、ピル、カプセル、ロゼンジ、坐剤、パッチ、鼻スプレー、注射可能薬物、移植可能な徐放性製剤、脂質複合体、その他を包含し、これに制限されない。ペプチドは、典型的には、医薬的に許容される担体又は賦形剤と組合され、これは、組成物を安定化するため、又は活性薬剤の吸収を増減するために作用する1以上の生理的に許容される化合物を含有し得る。生理的に許容される化合物には、例えばグルコース、スクロース又はデキストランのような炭水化物、例えばアスコルビン酸又はグルタチオンのような酸化防止剤、キレート剤、低分子タンパク質、例えば脂質のような保護及び取込みのエンハンサー、活性薬剤のクリアランス又は加水分解を低減する組成物、又は賦形剤、又は他の安定化剤及び/又は緩衝剤を包含し得る。他の生理的に許容される化合物には、湿潤剤、乳化剤、分散剤又は微生物の増殖又は活動を防止するために特に有用な保存料を包含する。種々の保存料が周知であり、例えば、フェノール及びアスコルビン酸を包含する。当業者は、生理的に許容される化合物を含め、医薬的に許容される担体の選択が、例えば活性薬剤の投与経路及び活性薬剤の特定の物理化学的性質に依存することを理解するであろう。
【0067】
ペプチド類は、天然の形態で、又は所望であれば、該塩が医薬的に許容されるという条件付きで塩の形態で、投与可能である。活性薬剤の塩は、合成有機化学の当業者に周知の標準法を用いて調製されうる。本発明のポリペプチド類は、実施例において詳述されるように、酢酸塩として調製された。
【0068】
本発明のOXMポリペプチドは、GLP1−Rに関与する疾病の治療又は予防において使用される他の薬剤と組合せて使用可能である。本発明のポリペプチドとの組合せにおいて役立つ具体的な化合物には、シンバスタチン、メバスタチン、エゼチミブ、アトロバスタチン、シタグリプチン、メトホルミン、シブトラミン、オルリスタット、Qnexa、トピラマート、ナルトレキソン、ブプリオピオン、フェンテルミン及びロサルタン、ヒドロクロロチアジドとロサルタンの組み合わせを包含する。本発明のポリペプチドとの組合せにおいて役立つ具体的なCB1アンタゴニスト/インバースアゴニストには、N−[3−(4−クロロフェニル)−2(S)−フェニル−1(S)−メチルプロピル]−2−(4−トリフルオロメチル−2−ピリミジルオキシ)−2−メチルプロパンアミド、N−[3−(4−クロロフェニル)−2−(3−シアノフェニル)−1−メチルプロピル]−2−(5−トリフルオロメチル−2−ピリジルオキシ)−2−メチルプロパンアミド、N−[3−(4−クロロフェニル)−2−(5−クロロ−3−ピリジル)−1−メチルプロピル]−2−(5−トリフルオロメチル−2−ピリジルオキシ)−2−メチルプロパンアミド、及び医薬的に許容されるそれらの塩を含む、WO03/077847に記載されたもの;並びに以下:3−{1−[ビス(4−クロロフェニル)メチル]アゼチジン−3−イリデン}−3−(3,5−ジフルオロフェニル)−2,2−ジメチルプロパンニトリル、1−{1−[1−(4−クロロフェニル)ペンチル]アゼチジン−3−イル}−1−(3,5−ジフルオロフェニル)−2−メチルプロパン−2−オール、3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−ヒドロキシ−2−メチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((4−クロロフェニル){3−[1−(3,5−ジフルオロフェニル)−2、2−ジメチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((1S)−1−{1−[(S)−(3−シアノフェニル)(4−シアノフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル)−5−フルオロベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(4H−1,2,4−トリアゾール−4−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、及び5−((4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)チオフェン−3−カルボニトリル、及び医薬的に許容されるそれらの塩を含む、WO05/000809に記載されたもの;並びに3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(3−{(1S)−1−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル}アゼチジン−1−イル)(4−クロロフェニル)メチル]ベンゾニトリル、3−[(S)−(4−シアノフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(3−{(1S)−1−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル}アゼチジン−1−イル)(4−シアノフェニル)メチル]ベンゾニトリル、3−[(S)−(4−シアノフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,2,4−オキサジアゾール−3−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]−メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−1H−テトラゾール、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−1−メチル−1H−テトラゾール、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−2−メチル−2H−テトラゾール、3−[(4−クロロフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(2−メチル−2H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−クロロフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(1−メチル−1H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−シアノフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(1−メチル−1H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−シアノフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(2−メチル−2H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、5−{3−[(S)−{3−[(1S)−1−(3−ブロモ−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}(4−クロロフェニル)メチル]フェニル}−1,3,4−オキサジアゾール−2(3H)−オン、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−{1−[(S)−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)フェニル](4−クロロフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−{1−[(S)−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)フェニル](4−シアノフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、5−[3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)フェニル]−1,3,4−オキサジアゾール−2(3H)−オン、5−[3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)フェニル]−1,3,4−オキサジアゾール−2(3H)−オン、4−{(S)−{3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}−ベンゾニトリル、及び医薬的に許容されるそれらの塩を包含する。
【0069】
本発明のポリペプチドとの組合せにおいて役立つ具体的なNPY5アンタゴニストには、3−オキソ−N−(5−フェニル−2−ピラジニル)−スピロ[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、3−オキソ−N−(7−トリフルオロメチルピリド[3,2−b]ピリジン−2−イル)スピロ−[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、N−[5−(3−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ−[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、トランス−3’−オキソ−N−(5−フェニル−2−ピリミジニル)スピロ[シクロヘキサン−1,1’(3’H)−イソベンゾフラン]−4−カルボキサミド、トランス−3−オキソ−N−[1−(3−キノリル)−4−イミダゾリル]スピロ[シクロヘキサン−1,1’(3’H)−イソベンゾフラン]−4−カルボキサミド、トランス−3−オキソ−N−(5−フェニル−2−ピラジニル)スピロ[4−アザイソ−ベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[5−(3−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ−[5−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[5−(2−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ[5−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[1−(3,5−ジフルオロフェニル)−4−イミダゾリル]−3−オキソスピロ[7−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(1−フェニル−4−ピラゾリル)スピロ[4−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[1−(2−フルオロフェニル)−3−ピラゾリル]−3−オキソスピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(1−フェニル−3−ピラゾリル)スピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(2−フェニル−1,2,3−トリアゾール−4−イル]スピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、及び医薬的に許容されるそれらの塩及びエステルを包含する。
【0070】
本発明のポリペプチドとの組合せにおいて役立つ具体的なACC−1/2阻害剤には、1’−[(4,8−ジメトキシキノリン−2−イル)カルボニル]−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、(5−{1’−[(4,8−ジメトキシキノリン−2−イル)カルボニル]−4−オキソスピロ[クロマン−2,4’−ピペリジン]−6−イル}−2H−テトラゾール−2−イル)メチルピバレート、(5−{1’−[(8−シクロプロピル−4−メトキシキノリン−2−イル)カルボニル]−4−オキソスピロ[クロマン−2,4’−ピペリジン]−6−イル}−ニコチン酸、1’−(8−メトキシ−4−モルホリン−4−イル−2−ナフトイル)−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、及び1’−[(4−エトキシ−8−エチルキノリン−2−イル)カルボニル]−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、及び医薬的に許容されるそれらの塩を包含する。
【0071】
本発明のポリペプチドとの組合せにおいて役立つ具体的なMCH1Rアンタゴニストには、1−{4−[(1−エチルアゼチジン−3−イル)オキシ]フェニル}−4−[(4−フルオロベンジル)オキシ]ピリジン−2(1H)−オン、4−[(4−フルオロベンジル)オキシ]−1−{4−[(1−イソプロピルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、1−[4−(アゼチジン−3−イルオキシ)フェニル]−4−[(5−クロロピリジン−2−イル)メトキシ]ピリジン−2(1H)−オン、4−[(5−クロロピリジン−2−イル)メトキシ]−1−{4−[(1−エチルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、4−[(5−クロロピリジン−2−イル)メトキシ]−1−{4−[(1−プロピルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、及び4−[(5−クロロピリジン−2−イル)メトキシ]−1−(4−{[((2S)−1−エチルアゼチジン−2−イル)メトキシ]フェニル}ピリジン−2(1H)−オン、又は医薬的に許容されるそれらの塩を包含する。
【0072】
本発明のポリペプチドとの組合せにおいて役立つ具体的なDP−IV阻害剤には、7−[(3R)−3−アミノ−4−(2,4,5−トリフルオロフェニル)ブタノイル]−3−(トリフルオロメチル)−5,6,7,8−テトラヒドロ−1,2,4−トリアゾロ[4,3−a]ピラジンから選択される。特に、式Iの化合物は、7−[(3R)−3−アミノ−4−(2,4,5−トリフルオロフェニル)ブタノイル]−3−(トリフルオロメチル)−5,6,7,8−テトラヒドロ−1,2,4−トリアゾロ[4,3−a]ピラジン、及び医薬的に許容されるそれらの塩を包含する。
【0073】
さらに、他のペプチド類似体及びインクレチンホルモングルカゴン様ペプチド1(GLP−1)の模倣薬もまた、本発明のポリペプチドとの組合せにおいて役立つ。
【0074】
本発明の他の特徴及び利点は、異なる実施例を含む本明細書に提供された追加的な記載から明らかである。提供された実施例は、本発明の実施において有用な、種々の成分及び方法論を示している。実施例は、クレームされた本発明を制限するものではない。本開示に基づき、当業者は、本発明の実施において有用な他の成分及び方法論を特定しかつ用いることができる。
【実施例】
【0075】
実施例1:オキシントモジュリン(OXM)類似体の合成
ペプチドOXN類似体(表1参照)は、固相により、Fmoc/tBu化学を用いて、ペプチドマルチシンセサイザーAPEX396(アドバンスド・ケムテック)にて、40ウェル反応ブロックを用いて合成した。各ペプチドを、単一のウェル中で合成した。ペプチドアミド用には、0.1gの、1%架橋された樹脂Fmoc−リンカーAM−チャンピオン(バイオサーチ・テクノロジーズ・インク)、及び、修飾されたリンク(Rink)リンカー p−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化された、PEG−PSベースの樹脂(リンク(Rink、H)著、「テトラヘドロン・レターズ」、1987年、第28巻、p.3787−3789;ベルナトビッツ(Bernatowicz,M.S.)ら著、「テトラヘドロン・レターズ」、1989年、第30巻、p.4645−4667)を使用した。ペプチド酸用には、0.1gの1%架橋チャンピオン樹脂(バイオサーチ・テクノロジーズ・インク)を使用し、それは、あらかじめヒドロキシメチルフェノキシメチル処理により誘導体化した。全てのアミノ酸は、0.5M HOBt(ヒドロキシベンゾトリアゾール)のDMF中の溶液中に、0.5Mの濃度で溶解した。アシル化反応は、樹脂の遊離アミノ基の6倍過剰の活性化アミノ酸を用いて、60分間にわたり行なった。アミノ酸は、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)及び2倍モル過剰のDIEA(N,N−ジイソプロピルエチルアミン)により活性化した。
【0076】
別法として、ペプチドは、Fmoc/tBu化学を用いて、パイオニアペプチドシンセサイザー(アプライド・バイオシステムズ)を用いて合成した。この場合、全てのアシル化反応は、樹脂の遊離アミノ基の4倍過剰の活性化アミノ酸を用いて、60分間にわたり行ない、シンセサイザーでのペプチドアセンブリーの終了に続いた。側鎖保護基は、Asp、Glu、Ser、Thr及びTyrにはtert−ブチル;Asn、Cys、Gln及びHisにはトリチル;Lys、Trpにはtert−ブトキシ−カルボニル;及び、Argには2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニルであった。OXM2及びOXM3ペプチド類については、アセチル化反応は、ペプチドアセンブリーの終了時に、DMF中の10倍過剰の無水酢酸を用いた反応により行なった。
【0077】
OXM14用には、L−ピログルタミン酸を、等モル量のDIPC(ジイソプロピルカルボジイミド)とHOBt(N−ヒドロキシベンゾトリアゾール)との、樹脂遊離アミノ基の4倍過剰の活性化されたアシル作用物(acylant)との反応によりアシル化した。
【0078】
OXM16用には、イミダゾール−乳酸(Imi−H)を、等モル量のPyBOP(ベンゾトリアゾール−1−イル−オキシ−トリス−ピロリジノ−ホスホニウムヘキサフルオロホスフェート)、HOBt及び2倍モル過剰のDIEA(N,N−ジイソプロピルエチルアミン)との、樹脂遊離アミノ基の4倍過剰の活性化されたアシル作用物との反応によりアシル化した。
【0079】
OXM17用には、N−メチル−His(Me−H)を、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)と2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、180分間にわたり行った。
【0080】
OXM18用には、デスアミノ−His(ΔNH2−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、180分間にわたり行った。
【0081】
OXM19用には、N,N−ジメチル−His(Me2−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、一晩にわたり行った。
【0082】
OXM20用には、ベンゾイル−His(Bz−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、240分間にわたり行った。
【0083】
OXM21用には、ベンジル−His(Bzl−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、一晩にわたり行った。
【0084】
合成の終わりに、乾燥ペプチド樹脂は、20mLの切断混合物、88%TFA、5%フェノール、2%トリイソプロピルシラン、及び5%水(ソール及びバラニー(Sole、N.A.&G.Barany,)著、「ザ・ジャーナル・オブ・オーガニック・ケミストリー(J.Org.Chem.)」、1992年、第57巻、p.5399−5403)を用いて、室温で1.5時間にわたり個別に処理した。各樹脂を濾過し、ペプチドを沈澱するため、溶液を冷メチル−t−ブチルエーテルへ添加した。遠心分離後、沈澱ペレットを、新鮮な冷メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリル中に再懸濁し、凍結乾燥した。
【0085】
ペプチドOXM54の合成は、チオール含有OXMペプチド前駆体(配列番号41)を、pH8の0.1MトリスHCl、6M塩化グアニジニウム中に溶解することにより行った。10倍モル過剰のヨードアセトアミドを添加した。1時間のインキュベーション後、ペプチド溶液をHPLCにより精製した。
【0086】
ペプチドOXM55の合成は、チオール含有OXMペプチド前駆体(配列番号64)を、pH8の0.1MトリスHCl、6M塩化グアニジニウム中に溶解することにより行った。この溶液に対し、10倍モル過剰のヨードアセトアミドを添加した。1時間のインキュベーション後、ペプチド溶液をHPLCにより精製した。
【0087】
粗ペプチドを、逆相HPLCにより、セミ分取用ォーターズ(Waters)RCMデルタPakTMC4カートリッジ(40x200mm、15μm)を用い、溶出液として、(A)水中の0.1%TFA、及び(B)アセトニトリル中の0.1%TFAを用いて精製した。以下の溶出液(B)の勾配を使用した:5分間にわたり20%−20%、さらに20分間にわたり20%−35%、流速80mL/分。分析用HPLCは、フェノメネックス、ジュピターC4カラム(150x4.6mm、5μm)にて、以下の溶出液(B)の勾配を使用した:20%−40%B(20分間)−80%(3分間)、流速1mL/分。精製したペプチドを、マイクロマス(Micromass)LCZプラットフォーム上でエレクトロスプレー質量分析によりキャラクタライズした。
【0088】
実施例2:オキシントモジュリン(OXM)類似体のPEG化
PEG化反応は、チオエステル結合形成を可能にする条件下に行なった。次にPEG化されたOXMペプチドを、逆相HPLC又はイオン交換クロマトグラフィー、及びサイズ排除クロマトグラフィー(SEC)を用いて単離した。PEG化されたOXM類似体は、RP−HPLC、HPLC−SEC及びMALDI−Tof質量分析を用いてキャラクタライズした。
【0089】
OXM33、34、35、36及び54ペプチド類は、チオール含有OXMペプチド前駆体(配列番号41)から合成し、チオエーテル結合により共有結合したPEGをもつ誘導体を調製した。
【0090】
OXM33の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.4mLのpH7.3の0.1M HEPES中に溶解した22mgのMPEG−MAL−5000(NEKTAR 2F2MOH01)(4.4μmol)(1:2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0091】
OXM34の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.5mLのpH7.3の0.1M HEPES中に溶解した、80mgのMPEG−MAL−20K(NEKTAR 2F2M0P01)(4.0μmol)(1:1.8 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0092】
OXM35の合成
10mgのペプチド前駆体(0.92μmol)を、0.4mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.8mLのpH7.3の0.1M HEPES中に溶解した、70mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(1.7μmol)を、1:1.8 mol/molのPEG:ペプチド比で添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0093】
コントロールペプチドOXM54は、チオール含有ペプチド前駆体を、pH7.5の0.1M トリスHCl、6M塩化グアニジニウム中、10eqのヨードアセトアミドとインキュベートすることにより調製した。30分間のインキュベーション後、ペプチドをRP−HPLCにより精製し、そしてエレクトロスプレー質量分析によりキャラクタライズした。
【0094】
ペプチドOXM56、57、58は、チオール含有OXMペプチド前駆体(配列番号64)から合成し、チオエーテル結合により共有結合したPEGをもつ誘導体を調製した。
【0095】
OXM56の合成
5mgのペプチド前駆体(1.1μmol)を、0.2mLのpH7.3の0.1M HEPESに溶解した。この溶液に対し、0.4mLのpH7.3の0.1M HEPESに溶解した、57mgのMPEG−MAL−5000(NEKTAR 2F2MOH01)(11.4μmol)(1:10 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を1%酢酸で酸性化し、フラクトゲルTSK CM−650S上での陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、サイズ排出クロマトグラフィー(SEC)によりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0096】
OXM57の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのDMSOに溶解した。この溶液に対し、0.6mLのpH7.3の0.1M HEPES、0.3Mトリス(2−カルボキシ−エチル)ホスフィン中に溶解した、50mgのMPEG−MAL−20K(NEKTAR 2F2M0P01)(2.5μmol)を、1:1.13mol/molのPEG:ペプチド比で添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0097】
OXM58の合成
10mgのペプチド前駆体(0.92μmol)を、0.4mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.8mLのpH7.3の0.1M HEPES中に溶解した70mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(1.7μmol)(1:1.8 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドを、RP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0098】
コントロールペプチドOXM102、OXM112及びOXM116は、チオール含有ペプチド前駆体を、pH7.5の0.1M トリスHCl、6M塩化グアニジニウム中、10eq.のヨードアセトアミドとインキュベートすることにより調製した。30分間のインキュベーション後、ペプチドをRP−HPLCにより精製し、そしてエレクトロスプレー質量分析によりキャラクタライズした。
【0099】
OXM103、OXM105、OXM107、OXM113の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH7.3の0.1M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2Oに溶解した109mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.71μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0100】
OXM109の合成
10mgの対応するペプチド前駆体(2.25μmol)を、2mLの、8M尿素、pH7.3の0.1M HEPES、2mM EDTA中に溶解した。この溶液に対し、2mLのH2O中に溶解した108mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.7μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0101】
OXM117の合成
10mgの対応するペプチド前駆体(2.19μmol)を、2mLの、8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した105mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.63μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0102】
OXM125の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH6.5の0.25M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した105mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.71μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8まで酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0103】
OXM129の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH6.5の0.25M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した109mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.72μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸で酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0104】
実施例3 ペプチド配列のデザイン及び合成
種々のPEGサイズ(mPEG)5kDa、(mPEG)20kDa、(mPEG)240kDa及び(mPEG)260kDaの生物活性を、一連の実験において比較し、最大かつ耐久性のある活性をマウスにおいて与えるために最適なPEGサイズが、一般に40kDaであることを証明した。
【0105】
ペプチドOXM103(2位Aib(α))は、オキシントモジュリン配列内の20位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM102はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(20位のシステイン)は、ヨードアセトアミドと反応した。
【0106】
ペプチドOXM105(2位Aib(α))は、オキシントモジュリン配列内の21位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM104はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(21位のシステイン)は、ヨードアセトアミドと反応した。
【0107】
ペプチドOXM107(2位Aib(α)、27位Met(O))は、オキシントモジュリン配列内の24位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM106はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(24位のシステイン)は、ヨードアセトアミドと反応した。
【0108】
ペプチドOXM109(2位Aib(α))は、オキシントモジュリン配列内の28位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM108はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(28位のシステイン)は、ヨードアセトアミドと反応した。
【0109】
OXM141と呼ばれるペプチドは、3位においてGlnからAspへの変異を有し、このことがGLP−1受容体に対する特異的選択性を与えている。3位にGlnからAspへの変異を、27位にMet(O)を、及び、20位及び38位に2つの結合部位を有するペプチドOXM141は、C38位にコレステロール基及びC20位にPEGの双方に結合されたペプチドを有する可能性を調査するためにデザインした。
【0110】
ペプチドOXM142(Aib(α)、27位にMet(O)、及び20位及び38位に2つの結合部位)は、C38位にコレステロール基及びC20位にPEGの双方に結合されたペプチドを有する可能性を調査するためにデザインした。
【0111】
図14は、GLP1R及びGCG受容体に関するインビトロ活性のデータ(GLP−1R及びGLGRの特異性も示している)を、表形式に要約している。C20位における(mPEG)240kDa結合は、双方の受容体に対し活性を保持しており、それ故、OXM103は、+/+類似体と呼ばれ、それに対し他の全ての(mPEG)240kDa結合は、GCG受容体において能力を失っている。特に、38、24及び28位における(mPEG)240kDa結合については、Gcg受容体に対し2−3オーダーの大きさの選択性が、GLP−1受容体に対し存在する。それ故、類似体OXM99、107及び109は、+/0類似体と呼ばれる。
【0112】
図15は、DIOマウスでの、摂食及び体重減少に対する、(mPEG)240kDa結合のインビボ活性を示す。自由に摂食したDIO(各〜51g)の、雄のC57BL/6マウスに、暗期開始の〜30分前に、ビヒクル(水)又はOxm類似体99、103、105、107及び109を、腹腔内(i.p.)投与した。摂食は、1日目で〜2時間及び18時間(一晩)後に、2日目で26及び42時間(一晩)後に測定した。*P<0.05 対ビヒクル、群当たりn=5−6。
【0113】
ペプチドOXM110(2位D−Ser)は、オキシントモジュリン配列内の38位を、パルミトイル基のような脂質との結合用の部位として調査するためにデザインした。パルミトイル基を、オキシントモジュリン配列のC−末端に付加されたリジンのε−アミノ基へアシル化した。
【0114】
ペプチドOXM113(1位デスアミノ−His(ΔNH2−H)、2位Aib(α)、27位Met(O)、結合部位38位)は、DPPIVタンパク質分解から保護するための、1位における野生型(wt)Hisの、デスアミノ−Hisによる置換の可能性を調査するためにデザインした。OXM113は、(mPEG)240kDa結合であり、OXM114は、コレステリル結合であり、そしてOXM112はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(38位システイン)は、ヨードアセトアミドと反応した。(OXM111は、チオール化されたペプチド前駆体である)。
【0115】
ペプチドOXM117及びOXM118(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位38位)は、3位における野生型Glnの、Aspによる置換の可能性を調査するためにデザインした。上記のように、この変異は、GLP−1Rに対する特異的選択性を与える。OXM117は、(mPEG)240kDa結合であり、OXM118は、コレステリル結合である。(OXM116は、チオール化されたペプチド前駆体である)。
【0116】
ペプチドOXM121(2位Aib(α)、27位Met(O)、結合部位11位)は、オキシントモジュリン配列内の11位を、結合用の位置として調査するためにデザインした。OXM121は、(mPEG)240kDa結合であり、OXM119は、チオール化されたペプチド前駆体であり、そしてOXM120は、コントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(11位のシステイン)は、ヨードアセトアミドと反応した。
【0117】
ペプチドOXM124(2位Aib(α)、27位Met(O)、結合部位12位)は、オキシントモジュリン配列内の12位を、結合用の部位として調査するためにデザインした。OXM121は、(mPEG)240kDa結合であり、OXM122は、チオール化されたペプチド前駆体であり、かつOXM123は、コントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(12位のシステイン)は、ヨードアセトアミドと反応した。
【0118】
ペプチドOXM125(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、20位を(mPEG)240kDaの結合用の部位として、並びに、GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換を調査するためにデザインした。
【0119】
ペプチドOXM127(2位Aib(α)、27位Met(O)、結合部位22位)は、オキシントモジュリン配列内の22位をコレステロール用の結合のための部位として調査するためにデザインした。(OXM126は、チオール化されたペプチド前駆体である)。
【0120】
ペプチドOXM129(1位デスアミノ−His(ΔNH2−H)、2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、以下の組合せの可能性を調査するためにデザインした:DPPIVタンパク質分解からの保護のため1位において野生型Hisをデスアミノ−Hisで置換すること、GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換、及び、20位における結合。
【0121】
ペプチドOXM134(2位Aib(α)、27位Met(O)、結合部位20位)は、強力な+/+OXM103類似体に類似の、(mPEG)240kDa結合であり、メチオニン置換がメチオニンスルホキシドへの点についてのみ異なっている。このペプチドは、20位において結合されており、したがって、GLP1R/GcgR選択性については、+/+パターンを示す。
【0122】
実施例4 C−末端切断類似体又はGcgK類似体
C−末端切断類似体
一連の、ペプチドOXMC−末端切断類似体がデザインされた。GLP1受容体(GLP1R)及びGcg受容体(GcgR)の双方に対するインビトロ活性の分析は、OXM配列がC−末端において、wtグルカゴンに対し1つだけ余分のリジン残基をもつよう切断可能であり、GLP1R及びGcgRの双方に対し極めて効力のある、OXM93のようなペプチドを生じることを証明した。OXM93の効力は、wtOXMのそれのよりも、少なくとも1オーダー(及び恐らくは2オーダー)大きい。Gcgに対し1つだけ余分のリジン残基があることから、この新規なクラスはGcgK類似体と呼ばれる。図16は、GLP1及びGcg受容体において作用する、C−末端切断類似体に関するインビトロの効力を表形式で例示している。
【0123】
選択的GcgK類似体であるC−末端切断類似体
ペプチドOXM130及びOXM131は、インビトロ分析を確認するため、及び安定性及び好適な特性を与えることが知られている他の変異を導入するためデザインした。OXM130及びOXM131は、OXM93と同じ長さのC−末端切断類似体である。OXM130は、2位にAib(α)を、一方OXM131は、2位にAib(α)を、また3位に、GLP1Rに対する選択性を与えるためのGlnからAspへの変異を有する。
【0124】
以下のPEG化された類似体を、これらの切断類似体に基づきデザインした:
【0125】
ペプチドOXM136(2位Aib(α)、27位Met(O)、結合部位20位)は、(mPEG)240kDaの結合である。先の研究(上記参照)から、20位におけるPEG結合の選択は、GLP1R及びGcgRの双方の受容体に対する活性を与えることが知られており、それ故OXM136は、プロトタイプGcg+/+類似体として定義されるであろう。(OXM135は、チオール化されたペプチド前駆体である)。
【0126】
ペプチドOXM138(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、以下の組合せの可能性を調査するためにデザインした:GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換;20位における結合、及び3位におけるwtGlnからAspへの変異。それ故、OXM137は、プロトタイプGcgK+/0類似体として定義されるであろう(OXM137は、チオール化されたペプチド前駆体である)。
【0127】
図17は、PEG化されたOXM類似体選択のための、GLP1及びGCG受容体におけるインビトロの効力のデータを提供する。
【0128】
実施例5:代替のPEG成分をもつペプチド
ペプチドOXM145(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、ジェンケム・セラピューティクス(JenKem Therapeutics)からのY型PEGマレイミドとの結合部位38位)を、代替の分枝した40kDaPEG基を用いる可能性を調査するために調製した。これは、ジェンケム・セラピューティクスからの(mPEG)240kDa(Y型PEGマレイミド、MW40K、商品番号Y−MAL−40K)を用いて得た結合体であり、その構造は、以下に示される:
【0129】
【化1】
【0130】
ペプチドOXM146は、2位におけるAib(α)、3位におけるGlnからAspへの変異、27位におけるMet(O)、NOFコーポレーションからのサンブライトGL2−400MAマレイミドとの38位の結合によりキャラクタライズされる。これは、NOFコーポレーションからの(mPEG)240kDa(サンブライト GL2−400MAマレイミド)を用いて得た結合体であり、その構造は、以下に示される:
【0131】
【化2】
【0132】
ペプチドOXM151(2位Aib(α)、3位GlnからAspへの変異、17位ArgからGluへの変異、18位ArgからAlaへの変異、27位Met(O)、結合部位38位)は、ペプチド配列への、インビボでの増大された安定性を与えるためにデザインした。
【0133】
詳細な研究を行い、ここではOXM139を10%のマウス又はヒト血漿を含有するPBS中、37℃においてインキュベートした。試料調製は、1μLの試験溶液と、1μLのマトリックス(α−シアノ)とを、試料プレート上で直接混合することにより遂行した。結晶化後、Tofスペクトルを収集した:30、60、120及び720分の時点で分析し、同じ時点のコントロール試験溶液と比較した。ペプチド配列内では、Arg17とArg18との間の結合は、第1の加水分解部位であると特定された。Arg18とAla19との間の結合もまた、第2の加水分解部位であると特定されたため、部位17及び18に変異を導入することを決定した。特異的に、Arg17をGluへ変異させ、Arg18をAlaへ変異させた。ペプチドOXM153は、OXM151のN−エチルマレイミド類似体である。ペプチドOXM154は、NOFコーポレーションからの(mPEG)240kDa(サンブライトGL2−400MAマレイミド)を用いて得られた結合体である。
【0134】
ペプチドOXM152は、オキシントモジュリン配列の残基1−16を繋いでおり(2位Aib(α)、3位GlnからAspへの変異、17位スペーサーとしてTtds、18位に結合のためのCys)、1−16配列に対し特異的な抗体を上げるため、担体タンパク質へ結合可能なペプチドである。
【0135】
ペプチドOXM155(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、38位Ttds、グルタミン酸残基39−43、44位に結合のためのCys)は、抗体を上げるための担体タンパク質へ結合可能なもう1つのペプチドである。C末端へのグルタミン酸の付加は、2003年12月18日に出願された、仮出願第60/530867号(高い免疫原性をもつペプチド−担体結合体の作成法)」(これを参照とすることによりその全を本明細書に組み込む)に記載のように、担体タンパク質への結合を可能にするpI修飾に必要である。
【0136】
実施例6 新規な切断類似体又はGcgK類似体
ペプチドOXM143(Aib(α)、27位Met(O)、結合部位20位)は、OXM135のN−エチルマレイミド類似体である。
【0137】
ペプチドOXM144(Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、OXM137のN−エチルマレイミド類似体である。
【0138】
ペプチドOXM147(2位Aib(α)、結合部位20位)は、C20位に結合部位を有するGcgK類似体シリーズ内で、27位に天然のメチオニン残基をもつようデザインした。このデザインの根拠は、天然メチオニンをもつペプチド類似体がグルカゴン受容体に対しより活性があることであった。
【0139】
ペプチドOXM148(2位Aib(α)、結合部位20位)は、OXM147のN−エチルマレイミド類似体である。
【0140】
ペプチドOXM149(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位、及び30位D−リジン置換)は、ペプチドC−末端の、インビボでの酵素分解からの保護を与えるためデザインした。ペプチドOXM150は、OXM149のN−エチルマレイミド類似体である。
【0141】
同様の安定性の研究を、GcgKシリーズの類似体OXM144に対し、10%のマウス又はヒトのいずれかの血漿を含有するPBSとのインキュベーションにより、第1の加水分解部位を決定するために行なった。この例では、Arg17とArg18との間の結合を第1の加水分解部位として特定した。また、Arg18とAla19との間の結合も、第2の加水分解部位として特定した。このことから、GcgK類似体シリーズにおいても、部位17及び18に変異を導入することが決定された。
【0142】
OXM145の合成
54mgの対応するペプチド前駆体(11.8μmol)を、2mLの8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した569mg(14.2μmol)のY型PEGマレイミドMW40K(ジェンケム・テクノロジー、商品番号Y−MAL−40K)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8に酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0143】
OXM146の合成
55mgの対応するペプチド前駆体(12μmol)を、2mLの8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した531mg(13.2μmol)のサンブライトGL2−400MAマレイミド(NOFコーポレーション)(1:1.1 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8に酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした
【0144】
実施例7:サイクリックAMP(cAMP)を用いたGLP−1受容体(GLP−1R)シグナリングの測定
ホモジニアス時間分解蛍光(HTRF)アッセイ及びDP−IVに対する抵抗性の評価
天然の受容体と同様の生物活性をもつヒトGLP−1Rの変異型により安定にトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞系を、ウシ胎児血清(FBS)、ペニシリン−ストレプトマイシン、ヒポキサンチン−チミジン及びG418を含有する、完全なイスコブ改変ダルベッコ培地(IMDM)培地中に維持した。GLP−1受容体活性化に対するホモジニアス時間分解蛍光(HTRF)アッセイを使用して、細胞をリガンド、XL−665及び抗−cAMPクリプテートと37℃でインキュベートしたことを除いて製造業者(シスバイオ(CisBio))の指示に従って、本発明のペプチドとのインキュベーション時のトランスフェクト細胞におけるcAMP蓄積を測定した。アッセイは、96ハーフウェルプレートフォーマット中で、パーキン・エルマーエンビジョンプレートリーダを使用してプレートを読み取った。本発明のポリペプチド及びポリペプチドフラグメント/誘導体については、cAMP HTRFアッセイにおけるGLP−1受容体の「活性化」は、最大活性の誘導であり、それは、天然のヒトOXM配列により誘導される最大活性の少なくとも約60%から約200%までであり、少なくとも0.04%から約1000%までの相対効力である。「相対効力」とは、天然のヒトOXMのEC50を本発明のポリペプチドのEC50で割り、100倍したものである。「EC50」とは、最大活性の50%が達成されるポリペプチド濃度である。
【0145】
DP−IV切断に対する抵抗性を測定するため、5μMのペプチド溶液を、10nMの組換え可溶性ヒトDP−IVと、100μlのアッセイバッファー(10mM HEPES、pH7.5、0.05%BSA)中で、37℃で2時間プレインキュベートした。続いて、hGLP−1Rの活性化を、シスバイオHTRFcAMPアッセイを用いて測定し、DP−IVなしに37℃で2時間プレインキュベートしたコントロールペプチドと比較した。本発明のポリペプチド類及びそれらのフラグメント/誘導体については、本実験における「DP−IVに対する抵抗性」は、0.1から35までの効力比として定義され、ここで「効力比」とは、DP−IVとプレインキュベートしたペプチドのEC50をDP−IVなしでプレインキュベートした本発明の同じポリペプチドのEC50で割ったものである(図2)。
【0146】
実施例8:サイクリックAMPフラッシュプレートアッセイを用いたグルカゴン受容体(GcgR)シグナリングの測定
クローニングされたヒトグルカゴン受容体を発現しているCHO細胞(CHO−hGCGR)(カシエリ(Cascieri)ら著、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー」、1999年、第274巻、p.8694−8697)を、10%FBS、1mM L−グルタミン、ペニシリン−ストレプトマイシン(100U/ml)、及びG418(500μg/ml)を供えたIMDM中に維持した。本発明のペプチドとのインキュベーション時のトランスフェクト細胞におけるcAMPレベルを、フラッシュプレートアッセイ(SMP−004B、パーキン・エルマー・ライフ・サイエンス)を利用して、製造業者の指示に従って測定した。細胞刺激を、細胞溶解剤と125I−標識cAMPトレーサーとを含有する等量の検出バッファーの添加により停止した。プレートに結合した125I−cAMPを、液体シンチレーションカウンタを用いて測定し、各試料中に存在するcAMP量を定量するために使用した。本発明のポリペプチド類及びポリペプチドフラグメント/誘導体については、cAMPフラッシュプレートアッセイにおけるGcg受容体の「活性化」は、最大活性の誘導であり、それは、天然のグルカゴンペプチドにより誘導される最大活性の少なくとも約60%から約200%までであり、少なくとも0.04%から約10000%までの相対効力である。「相対効力」は、天然グルカゴンのEC50(EC50=70pM)を本発明のポリペプチドのEC50で割り、100倍したものである。「EC50」は、最大活性の50%が達成されるポリペプチド濃度である。天然のブタOXMは、このアッセイにおいて、2.4nMのEC50でグルカゴン受容体を活性化した。
【0147】
実施例9:やせたマウスにおける腹腔内グルコース負荷試験(IPGTT)の間の血中グルコース変位に対する効果
雄のC57BL/6Nマウスを、体重別に処理群へ分配し、実験開始の約5時間前に断食させた。ベースライン(t=〜30分)の血中グルコース濃度を、尾部切開の血液からグルコメータにより測定した。次いで動物に、ビヒクル(食塩水)又は本発明のポリペプチド(0.01−10mg/kg)を腹腔内(i.p.)注射した。血中グルコース濃度を、処理(t=0分)の30分後に測定し、次いでマウスを、デキストロース(2g/kg、10mL/kg)のi.p.により誘発した。ビヒクル処理マウスの1群を、ネガティブコントロールとして通常の食塩水で誘発した。血中グルコースレベルを、デキストロース誘発の20、40、60及び120分後に尾部の血から測定した。t=0からt=120分までの血中グルコース変位プロフィールを用いて、各処理について曲線下面積(AUC)を積分した。各処理群についてのグルコース変位のパーセント阻害は、式:
【0148】
【数1】
【0149】
[式中、
AUCdex=ビヒクル処理され、デキストロース誘発された動物の平均AUC、
AUCpeptide=ペプチド処理され、デキストロース誘発された動物の平均AUC、及び
AUCsaline=ビヒクル処理され、食塩水誘発された動物の平均AUC]に従って、水で誘発したコントロールによりノーマライズしたAUCデータから計算した。
【0150】
IPGTTにおける本発明のポリペプチドのインクレチン活性は、グルコース変位のパーセント阻害における用量依存性の増加として明示されており、10mg/kgの投与において少なくとも30%に達する(図2)。
【0151】
実施例10:やせたマウスにおける摂食及び体重に対する急性効果
約3か月齢の、自由摂食させた雄のC57BL/6Nマウスを秤量し、ビヒクル(水)又はビヒクル中に溶解したOXM2又はOXM3のいずれかを、光周期の暗期開始の〜30分前に、i.p.注射により投与した。光周期の暗期開始の〜5分前に、あらかじめ秤量した量のげっ歯類用固形飼料(テクラド7012)を、ワイヤーケージ上のフードホッパーに供給し、光周期の暗期開始の2及び18時間(一晩)後に秤量した。摂食における絶対変化は、各動物につき、特定の時点においてフードホッパー内に残留する餌の量を対応する元の秤量前の量のそれから引くことにより計算した。体重における絶対変化は、各動物につき、投与前の動物の体重を特定の時点における対応する動物の体重から引くことにより計算した。全ての値は、平均±SEMとして報告し、ペプチド処理群は、ビヒクル処理動物を基準としてツーテイルド・アンペアード・スチューデント検定(two tailed unpaired student’s test)により分析した。任意の時点における摂食、及び/又は一晩の体重増加における低減は、p値≦0.05として統計的に有意であって、このモデルにおける対応するOXMポリペプチド(OXM2又はOXM3)の効力を示しているとみなされる(図3)。
【0152】
実施例11:マウスにおけるグルコース刺激インスリン分泌の増大
16mmol/lグルコースでのインスリン分泌の仲介における天然OXMのインビトロの効力を、野生型C57BL/6マウスからの膵島及び、強いGSIS活性をもつマウスインスリノーマ細胞系、MIN6c4(ミナミ(Minami K)ら著、「アメリカン・ジャーナル・オブ・フィジオロジー、エンドクリノロジー・アンド・メタボリズム(Am J Physiol Endocrinol Metab.)」、2000年、第279巻、E773−E781)において、天然OXMペプチドの濃度を増加しながら、16mmol/lグルコースにおいて、グルコース刺激インスリン分泌(GSIS)を測定することにより評価した。膵臓ランゲルハンス島を、正常なC57BL/6Jマウス(ジャクソン・ラボラトリー、メイン州)から、レイシー及びコスチアノフスキーの原法(レイシーら著、「ダイアベーツ」、1967年、第16巻、p.35−39)の変法である、コラゲナーゼ消化及び不連続フィコール勾配分離により単離した。GSISアッセイに先立ち、膵島を、RPMI1640培地(11mMグルコース)中で一晩培養した。GSISを測定するために、膵島をまず、2mMグルコース入りのクレブス・リンガー重炭酸塩(KRB)バッファー中で30分間プレインキュベートした(ペトリ皿中)。KRB培地は、143.5mM Na+、5.8mM K+、2.5mM Ca+、1.2mM Mg2+、124.1mM Cl−、1.2mM PO43−、1.2mM SO42+、25mM CO32−、2mg/mlのウシ血清アルブミン(pH7.4)を含有する。膵島を、次に96穴プレートに移し(1膵島/ウェル)、2又は16mMグルコース入りの200μlのKRBバッファー中で、試験されるべき他の薬剤、例えばGLP−1及びOXMと共に37℃で60分間インキュベートした(ゾウ(Zhou)ら著、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー」、2003年、第278巻、p.51316−51323)。インスリンを、市販のキット(ALPCOダイアグノスティクス、ニューハンプシャー州ウィンダム)によるELISAにより、インキュベーションバッファーの少量中で測定した。MIN6c4細胞におけるインスリン分泌を、同様の方法で96穴プレートに播種した細胞において測定した。
【0153】
図4に示したように、天然のOXMは、マウスの膵島及びMIN6c4細胞において、GSISを有意に増大した。GSISに対するOXMのEC50は、各々、ネズミ膵島で約2.9nM(図4A)、及び155pM(図4B)であった。天然のGLP−1を、この実験におけるポジティブコントロールとして使用した。2つのペプチドの最大GSIS効果は、膵島及びMIN6細胞の双方において類似していた。
【0154】
OXMは、実施例7及び8に記載のように、CHO細胞において異種発現されたGLP−1R及びGCG−R双方を活性化し、そして双方の受容体は、膵臓β細胞において機能性であることが知られている。OXMのインクレチン作用におけるこれら2つのG−タンパク質共役受容体の潜在的役割を識別するため、GSISに対するOXM、GLP−1及びGCGの効果を、GLP−1R−/−マウス(スクロッチ(Scrocchi LA)ら著、「ネイチャー・メディスン」、1996年、第2巻、p.1254−1258)、及び、齢の合致したWT C57BL/6マウスからの膵島において調べた。先の結果と一致して、WTネズミ膵島からの16mmol/lグルコースにおけるGSISの増大においては、3つのペプチドは全て(各10nM)等しく有効であった(図5A)。GSIS及びGCGによるGSISの相乗作用は、GLP−1R−/−膵島では損なわれなかったのに対し、GLP−1及びOXMの双方は、後者においてGSISを全く増大することができなかった。OXMのインクレチン作用におけるGLP−1Rの関与はまた、広く使用されるGLP−1Rのペプチドアンタゴニストであるエキセンジン−9によるこの活性の拮抗作用により示された。OXM及びGLP−1によるGSISの相乗作用は、0.5μMのエキセンジン−9により、WT膵島において完全に阻止された(図5B)。
【0155】
OXMのインクレチン作用におけるGCG−Rの潜在的な関与を、ペプチド仲介GSISを、WT及びGCG−R−/−マウスからの膵島において16mmol/lグルコースにより比較することにより試験した(ゲリング(Gelling XQ)ら著、「プロシーディングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ」、2003年、第100巻、p.1438−1443)。先に記載の通り、3つのペプチドの全て(GLP−1、OXM及びGCG、各10nM)が、等しい効力で、WT膵島におけるGSISを増強した(図6A)。しかしながら、サイズ合致したWT膵島に比較して、2及び16mMグルコースの双方におけるインスリン分泌は、GCG−R−/−膵島では〜2倍まで低減された。膵島のインスリン含有量もまた、GCG−R−/−膵島ではWTに比較して>3倍まで低減された(図6B)。GCG(10nM)は、16mMグルコースにおいてはGCG−R−/−膵島ではGSISを増強しなかったのに対し、GLP−1及びOXM(10nM)は、GSISを、このアッセイにおいて有意に増大させた。データをフラクショナルGSIS(全膵島インスリン含有量に比較した%インスリン放出)として表わすと、OXMにより仲介されるGSISにおける倍数的増加(fold−increase)は、GCG−R−/−膵島ではWTに比較してわずか32%(1.7対2.5倍)に低減された(図6C)のに対し、GLP−1によるGSISの倍数的刺激は、同じに保持された(2.5倍)。対照的に、GCGは、GCG−R−/−膵島では、ベースライン(DMSO)を超えてフラクショナルGSISを増大することはなかった。これらのデータは、GSISに対するOXMの作用において、GCG−Rが限定された役割を果たすかもしれないことを示唆している。
【0156】
OXMのグルコース低下効果(上記記載のように、また図2に示したように)が、増大されたインビボGSISに対し二次的であったかどうかを決定するため、血漿グルコース及びインスリンレベルに対するOXMの効果を、WT及びGLP−1R−/−マウスにおけるIPGTTの間に分析した。一晩絶食したマウスに、グルコース誘発に先立ち、0.3mpk(kg体重当たりのmgペプチド)の天然OXMを前投与した(i.p.)。GLP−1の模倣薬である、エキセンジン−4(0.02mpkでi.p.投与)(トレンス(Thorens,B)ら著、「ダイアベーツ」、1993年、第42巻、p.1678−1682)を、この研究における比較対象として用いた。図7Aに示したように、エキセンジン−4及びOXMの双方は、WTマウスにおいてIPGTTの間に、グルコースレベルを有意に低減し、エキセンジン−4は、グルコース変位の抑制においてより強力であった。0.3mpkのOXM処理群におけるグルコース変位についての曲線下面積(AUC)は、ビヒクル群に比較して約30%低減された[13025±524対19928±811mg/dl/60分間]、p<0.001、n=10(ビヒクル)又は5(OXM)]のに対し、エキセンジン−4処理群におけるグルコースAUCの低減は、>60%(AUC=6601±179mg/dl/60分間)であった。対照的に、同じ用量のOXM及びエキセンジン−4は、GLP−1R−/−マウスにおけるIPGTTにおいて、グルコース変位に影響を及ぼさなかった(図7B)。
【0157】
インビボのGSISでのi.p.のOXM及びエキセンジン−4の効果を、IPGTT研究において、グルコース誘発の前(0分)及び後(10分)の血漿インスリンレベルを測定することにより評価した。OXMは、WTマウスにおいて、ベース(0分)の血漿インスリンレベルを4倍に増大し、i.p.のグルコース誘発に対するインスリン応答を有意に増幅した(図7C)。同様な結果は、WTマウスにおいて、エキセンジン−4を用いて観察された。対照的に、GLP−1R−/−マウスに対するOXM又はエキセンジン−4の投与は、ベースのインスリンレベルに影響を及ぼさず、i.p.のグルコース誘発に対するインスリン応答も改善しなかった(図7D)。
【0158】
医薬組成物の例
本発明の新規なポリペプチドの経口組成物の1つの具体的な実施態様として、5mgの、式
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC10−CONH2、
により記載されるポリペプチドを、サイズ0ハードゲルカプセルを満たすように、全量580−590mgを提供すべく充分な超微粒子状のラクトースと共に製剤する。
【0159】
本発明の新規なポリペプチドの経口組成物の、もう1つの具体的な実施態様として、2.5mgの、式
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC10−CONH2、
により記載されるポリペプチドを、サイズ0ハードゲルカプセルを満たすように、全量580−590mgを提供すべく充分な超微粒子状のラクトースと共に製剤する。
【0160】
他の実施態様は、以下のクレームの範囲内にある。いくつかの実施態様が示され、記載されてきたが、本発明の精神及び範囲から離れることなく、種々の変更が行われてもよい。
【図面の簡単な説明】
【0161】
【図1】変異型のヒトGLP−1受容体の、(a)天然のブタOXM、及び(b)PEG化されたOXM2による活性化、及び、DP−IVとのプレインキュベーションによる、当該ペプチドの効力の減少を描いた説明図である。
【図2】やせたマウスの腹腔内グルコース負荷試験(IPGTT)における、ブタオキシントモジュリンのインクレチン活性を示す図である。グルコース変位の%阻害が、各群について示されている。「ctrl」=ビヒクル処理された食塩水誘発マウス、「veh」=ビヒクル処理されたデキストロース誘発マウス。
【図3】配列OXM2及びOXM3により示されたポリペプチドの、やせたマウスにおける一晩の摂食及び体重増加の低減における効果を例示する図である。*ビヒクル群を基準として、p≦0.05。
【図4】膵島及びMIN6細胞におけるグルコース刺激インスリン分泌(GSIS)に対する、GLP−1及びOXMの効果を描いた図である。
【図5】膵島におけるOXM、GCG及びGLP−1仲介GSISに対する、GLP−1R欠失及び受容体拮抗作用の影響を示す図である。
【図6】グルカゴン受容体−/−膵島でのGSISに対するOXMの効果を示す図である。
【図7】野生型及びGLP−1R−/−マウスにおける、IPGTTの間の血中グルコース及びインスリンレベルに対するOXM及びエキセンジン−4の効果を示す図である。
【図8】やせたマウスのIPGTTにおける、OXM99の急性のグルコース低下効果を示す図である。
【図9】やせたマウスでの、血中グルコースの低減におけるOXM99の効果を描いた図である。
【図10】やせたマウスのIPGTTにおける、OXM117のグルコース低下効果を示す図である。
【図11】エキセンジン−4と比較した、OXM類似体であるOXM117及びOXM99の、活性の持続時間を示す図である。
【図12】ラットにおける皮下投与を用いたOXM117の薬物動態を描いた図である。
【図13】OXM117ペプチドが、インビトロで何らグルカゴン様活性を示さないことを示す研究結果を示す図である。
【図14】GLP1及びGCG受容体におけるインビトロ活性のデータを表形式に要約している図である。
【図15】DIOマウスモデルにおける摂食及び体重減少に対する(mPEG)240kDa結合体のインビボ活性を示す図である。
【図16】GLP1及びGCG受容体において作用するC−末端切断類似体についてのインビトロの効力のデータを示す図である。
【図17】GLP1及びGCG受容体において作用する選択されたPEG化OXM類似体についてのインビトロの効力のデータを表わす図である。
【技術分野】
【0001】
本発明は、オキシントモジュリン誘導体、それらの合成、及び糖尿病及び肥満症のような代謝障害の治療のためのそれらの使用に関する。
【背景技術】
【0002】
ホルモンであるオキシントモジュリン(OXM、グルカゴン−37)は、腸及び中枢神経系(CNS)におけるプレプログルカゴンプロセシングの翻訳後産物であり、摂食に応じて腸管のL−細胞から分泌される。1983年に発見されたOXMは、摂食及びエネルギー消費の調節と関係づけられてきた。ラットにおけるOXMの中枢又は末梢投与は、短期間の摂食の減少を、胃内容排出に対する最小の影響と共に引き起す(デイキン(Daikin)ら著、「エンドクリノロジー」、2001年、第142巻、p4244−4250、デイキンら著、「エンドクリノロジー」、2004年、第145巻、p.2687−2695)。ラットにおけるOXMの反復の脳血管内投与は、結果として、ペアフェッドの動物に比較して、核心温度の上昇及び体重増加の減少を生じ、このことはカロリー摂取及びエネルギー消費の双方に対する影響を示唆している(デイキンら著、「アメリカン・ジャーナル・オブ・フィジオロジー、エンドクリノロジー・アンド・メタボリズム」、2002年、第283巻、E1173−E1177)。
【0003】
OXMは、37アミノ酸のペプチドである。胃酸分泌阻害におけるOXMの効果は、SP−1として知られる8残基のC−末端フラグメントOXM(30−37)により、模倣可能であることが報告されている(カルレス−ボネ(Carles−Bonnet)ら著、「ペプタイズ」、1996年、第17巻、p.557−561)。ヒトでは、健康な正常体重の被験者におけるOXMの単回の90分間の静脈内注入が、ハンガースコア及び立食における摂食を〜19%まで低減した。累積12時間のカロリー摂取は、何らの嘔気又は食物嗜好性の変化の報告なしに、〜11%まで低減された(コーエン(Cohen)ら著、「ザ・ジャーナル・オブ・クリニカル・エンドクリノロジー・アンド・メタボリズム(J.Clin.Endocrinol.Metab.)」、2003年、第88巻、p.4696−4701)。より最近では、肥満の健康なボランティア(BMI〜33)における4週間にわたるOXMの食事前注射は、処置の第1日目に有意なカロリー摂取の低減(〜25%)をもたらし、それは研究の期間にわたって維持された(4週間後、35%減少)(ワイン(Wynne)ら著、「ダイアベーツ(Diabetes)」、2005年、第54巻、p.2390−2395)。処置された被験者では、研究の最後に強い体重減少が観察された(1.9%、プラセボ補正)。OXMの血漿レベルは、注入研究において観察されたものと同様であった(ピーク濃度〜950pM)。インビボでのOXMの不十分な安定性(血漿t1/2<12分間)により必要とされる比較的高い用量にもかかわらず、何らタキフィラキシーがないこと、及び、軽度で一過性の嘔気の発生率が低いこと(〜3%)は、このホルモンを、ヒューマンバリデーション及び魅力的な耐性プロフィールの双方を備えた数少ない肥満症ターゲットの1つにしている。
【0004】
OXMは、非常に短い半減期をもち、細胞表面ジペプチジルペプチダーゼIV(以降DP−IV)により迅速に不活性化される。しかしながら、DP−IV阻害剤は、臨床ではウェイトニュートラル(体重に対し中立)であり、生理的なレベルを超えたOXM(900−1000pM)が、ヒトの体重減少を達成するために必要かもしれないことを示唆している。
【0005】
オキシントモジュリンは、それ故、糖尿病及び肥満症のような代謝障害の治療剤としての可能性を示す。しかしながら、インビボでのOXMの不十分な安定性の故に、糖尿病及び肥満症のような代謝性疾患の治療に対して、安全かつ有効に投与されうるOXM誘導体を開発することが必要である。もし、安定性及び薬物動態を改善する部位への結合により修飾された、又はより具体的にはDP−IV切断に対し耐性を与える修飾により修飾された類似体又は誘導体が開発されるならさらに望ましいであろう。本発明は、OXMポリペプチド誘導体、及び本明細書に記述された誘導体を投与することによる、肥満症及び糖尿病のような代謝障害の治療又は予防の方法を提供する。
【発明の開示】
【0006】
(発明の要旨)
本発明は、式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;
X6は、任意のアミノ酸であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]
を包含するポリペプチドを提供する。医薬的に許容されるそれらの塩は、同様に考えられる。
【0007】
さらに、X3、X4、X6−X8、及びX10−X14のいずれか1又は2個は、Cys(mPEG)、Cys(コレステリル)でよく;Cys(mPEG)teineはまたC1;C2;C3又はC6でもよく、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、かつ各々は、側鎖チオールを介し、示された分子量の直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する。
【0008】
本発明は、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3
[式中、C3=Cys[(mPEG)240kDa]であり、各々は、側鎖チオールを介し、示されたMWの分枝したmPEG[(mPEG)2]によりPEG化されたアミド化システイン残基に相当し;αは、α−アミノイソ酪酸(aib)であり;そして、m=メチオニンスルホキシド(Met(O))である]のOXMポリペプチド誘導体に関する。
【0009】
本発明のもう1つの実施態様においては、
HxSQGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、Hxは、His;HI=イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis;N−メチル−His(Me−H)、N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H)、ベンジルHis(Bzl−H)及びPheからなる群より選択される]を包含するポリペプチドが提供される。
【0010】
本発明のなおもう1つの実施態様においては、
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg、Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含するポリペプチドが提供される。
【0011】
本発明はさらに、
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含するポリペプチドを提供する。
【0012】
本発明のもう1つの実施態様においては、
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)の任意の1つであり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)の任意の1つであり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、
ここで、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)である]を包含するポリペプチドが提供される。
【0013】
本発明のもう1つの実施態様においては、
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Cys(N−エチルマレイミジル)、Q又はCであり;
X17は、m又はMであり;
X18は、アミド化されたk又はKである]を包含するポリペプチドが提供される。
【0014】
本発明のもう1つの実施態様においては、
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、X19は、C又はC8、Cys(N−エチルマレイミジル)である]を包含するポリペプチドが提供される。
【0015】
本発明のなおもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
のポリペプチドが提供される。
【0016】
本発明のもう1つの実施態様においては、式:
HaDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH
[式中、Ttdsは、1−アミノ−4,7,10−トリオキサ−13−トリデカンアミン−スクシンイミン酸(succinimic acid)である]のポリペプチドが提供される。
【0017】
本発明のもう1つの実施態様においては、患者における代謝性疾患の治療のための方法であって、上記記載のポリペプチドを該患者へ投与することを包含する方法が提供される。該代謝性疾患は、糖尿病、メタボリックシンドローム、高血糖症及び肥満症からなる群より選択されてよく、脳までの末梢経路、例えば経口、経粘膜、バッカル、舌下、経鼻 直腸、皮下、経皮 静脈内、筋肉内又は腹腔内経路により投与されてよい。
【0018】
本発明のなおもう1つの実施態様においては、上記記載のポリペプチドと、医薬的に好適な担体とを包含する医薬組成物が提供される。
【0019】
本発明はさらに、必要とする患者に本発明のポリペプチド及び医薬組成物を投与することによる、糖尿病又は肥満症のような代謝障害の治療又は予防に有用な医薬品の調製における、本発明のポリペプチドの使用に関する。
【0020】
(発明の詳細な記載)
本発明は、修飾されたOXM誘導体に関する。OXM誘導体は、安定性及び薬物動態を改善するためのPEG化又は他の部位又は担体タンパク質への結合により、及び/又は、DP−IV切断に対しペプチドを抵抗性にするためのアミノ酸残基の置換基の組み込みにより開発される。さらに、安定化されたOXM誘導体は、グルカゴン受容体アゴニスト活性を示さず、それにより、糖尿病又は前糖尿病患者における高血糖症及び肥満症の治療において一定の利点を提供しうる。これらの患者については、グルカゴン受容体シグナリングの上方制御は、そのことが結果として血中グルコースレベルの上昇を生じ得るので、回避されねばならない。
【0021】
他に特定されない限り、用語「ポリペプチド」、「タンパク質」、及び「ペプチド」は、当業者により、種々の修飾及び/又は安定化された型も包含することが理解される。かかる修飾された型は、PEG化型、パルミトイル化型、コレステロール修飾型、その他を含む化学修飾型であってよく、それに限定されない。修飾はまた、分子内架橋、及び脂質、フラビン、ビオチン、ポリエチレングリコール誘導体、その他のような種々の部位への共有結合も包含する。加えて、修飾はまた、環化、分枝及び架橋を包含してもよい。さらに、遺伝子によってコードされた通常の20アミノ酸以外のアミノ酸もまた、ポリペプチド中に包含されうる。
【0022】
OXM誘導体の構造
本発明は、修飾されたOXM誘導体を提供する。特に、本発明は、式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;
X6は、任意のアミノ酸であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]の、新規な安定化された修飾されOXMポリペプチド誘導体に関する。
【0023】
さらに、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)でよく;Cys(mPEG)teineはまたC1;C2;C3又はC6でもよく、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、そして各々が、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する。
【0024】
本発明はさらに、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3
[式中、C3=Cys[(mPEG)240kDa]であり、各々が、側鎖チオールを介し、示されたMWの分枝したmPEG[(mPEG)2]によりPEG化されたアミド化システイン残基に相当し;αはα−アミノイソ酪酸(aib)であり;そして、m=メチオニンスルホキシド[Met(O)である]の、OXMポリペプチド誘導体を提供する。
【0025】
本発明のもう1つの実施態様においては、
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg;Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含するポリペプチドが提供される。
【0026】
本発明はさらに、
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含するポリペプチドを提供する。
【0027】
本発明のもう1つの実施態様においては、
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、
ここで、X3、X4、X6−X8、及びX10−X14の1又は2個は、Cys(mPEG)又はCys(コレステリル)である]を包含するポリペプチドが提供される。
【0028】
本発明のもう1つの実施態様においては、
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Q又はCであり;
X17は、m又はMであり;
X18はアミド化されたk又はKである]を包含するポリペプチドが提供される。
【0029】
本発明のもう1つの実施態様においては、
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、X19は、C又はC8である]を包含するポリペプチドが提供される。
【0030】
本発明のなおもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
のポリペプチドが提供される。
【0031】
本発明のもう1つの実施態様においては、式:
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH
のポリペプチドが提供される。
【0032】
本明細書で用いる場合、アミノ酸残基の略語は、以下のように示される:
【0033】
【表1】
【0034】
他に具体的に指定しない限り、全てのアミノ酸残基はL−型である。
【0035】
野生型OXMに比較して、本発明のOXM誘導体は、いくつかのアミノ酸置換基を含有し、及び/又は、PEG化されるか又は別の方法で修飾可能(例えばコレステロール部位)である。類似体は、例えばコレステロール及びPEGの双方により、二重に結合されてもよい。かかるOXM誘導体は、切断に抵抗性であり、ジペプチジルペプチダーゼIV(DP−IV)に不活性である。
【0036】
「受容体アゴニスト」とは、受容体、例えばGLP−1R又はグルカゴン受容体と相互作用し、それにより受容体活性化の薬理学的又は生化学的応答特性を開始し得る、任意の内在又は外来性(薬剤)物質又は化合物が意味される。典型的には、本発明のOXM誘導体は、ヒトGLP−1Rに対する親和性により特徴づけられ、この受容体に対し0.1pM−1μMの範囲のEC50を示す。本発明のOXM誘導体はまた、GcgRに対するその親和性によっても特徴づけられ、EC50>1μMを示す。
【0037】
本発明のOXM誘導体は、摂食及び体重増加の低減において有用であり、また膵島からのグルコース刺激インスリン分泌(GSIS)を仲介し得、それにより例えば肥満症、糖尿病、メタボリックシンドロームX,高血糖症、空腹時グルコース障害、及び他の前糖尿病状態のような代謝障害にかかっている個体のための、治療オプションを提供する。
【0038】
【表2】
【0039】
【表3】
【0040】
【表4】
【0041】
【表5】
【0042】
【表6】
【0043】
【表7】
【0044】
【表8】
【0045】
【表9】
【0046】
α=α−アミノイソ酪酸(Aib);a=D−Ala;S=D−Ser;n=L−ノルロイシン(Nle), X=O−メチル−ホモセリン;C1=Cys(mPEG)5kDa, C2=Cys(mPEG)20kDa, C3=Cys(mPEG)240kDa,各々、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG[(mPEG)2]によりPEG化されたシステイン残基に相当する;C4=Cys(コレステリル),側鎖チオールを介しコレステロールへ結合されたシステイン残基に相当する;C5=Cys(CH2CONH2),側鎖チオールがヨードアセトアミドと反応したシステイン残基に相当する;C6=Cys(mPEG)260kDa、各々、側鎖チオールを介し、示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2mPEG[(mPEG)2]によりPEG化されたシステイン残基に相当する;H1=イミダゾール−乳酸(ImiH);H2=デスアミノ−His(ΔNH2−H) 1Ac=アセチル;Pyr=ピログルタミル;Me−H=N−メチル−His;Me2−H=N,N−ジメチル−His;Bz=ベンゾイル(C7H5O);Bzl=ベンジル(C7H7);m=メチオニンスルホキシド, C7=(Cys)2(mPEG)2−40kDa,各々、側鎖チオールを介し、同じ1つの直鎖メトキシPEG(mPEG)又は1つの分枝鎖mPEG[(mPEG)2]によりPEG化された2つのシステイン残基に相当する, C8=Cys(N−エチルマレイミジル);Ttds,1−アミノ−4,7,10−トリオキサ−13−トリデカンアミン スクシンイミン酸;k,D−リジン。
【0047】
1.1.アミノ酸置換及び修飾
X1(OXMの2位)における置換は、OXM及びGLP−1を含む多くのペプチドの分解において重要な役割を果たすDP−IVによるタンパク質分解に対し、OXM誘導体の抵抗性を改善するためにデザインされている。GLP−1における2位のSerのGlyによる置換が、DP−IV切断に対する抵抗性を改善することが報告されてきた(ロッテ(Lotte,B.K.)著、「ジャーナル・オブ・メディカル・ケミストリー」、2004年、第47巻、p.4128−4134)。OXMとGLP−1との間の、高度の配列相同性にもかかわらず、2位におけるSer→Gly置換が、修飾されたOXMに対し同様の効果を与えることは見出されなかった。しかしながら、2位のSerの、Val、Ile、Asp、Glu、Met、Trp、Asn、D−Ala、D−Ser又はα−アミノイソ酪酸による置換は、実施例において以下に議論されたように、対応するOXM誘導体を、DP−IVに対し野生型OXMよりもさらに抵抗性にした。X1(OXMの2位)における置換をもつペプチド類には、OXM4−7、12、23、28−59、及びOXM33−36及びOXM55−59の前駆体を包含する。
【0048】
X2(OXMの3位)おける置換は、GcgRの活性が最少か又は全くないGLP−1Rの選択的アゴニストであるOXM誘導体を創成するためにデザインされている。かかるOXM誘導体は、肥満性糖尿病患者の治療時に有利であろう。X2(OXMの3位)に置換のあるペプチド類には、OXM8−12、15、23、53、95、96及び97を包含する。
【0049】
同様に、X3及びX5(11位及び16位)におけるAlaに対する置換は、GLP−1Rに対し選択的であって、かつGcgRに対し何ら活性をもたないOXM誘導体を創成するためにデザインされている。X3及びX5(11位及び16位)におけるAlaに対するかかる置換の一例は、OXM24である。
【0050】
X3、X4、X6−X8及びX10−X14の任意の1個以上におけるシステインに対する置換は、OXM誘導体の特定の位置におけるPEG化又はコレステリル化を可能にする。他の置換又は修飾は、当該技術分野において公知であり、活性薬剤の表面には物理的に付着するが、活性薬剤と化学結合もしくは相互作用しないものを包含する。2個以上のかかる修飾が使用可能であって、種々のポリマー、低分子量オリゴマー、天然産物及び界面活性剤を含む公知の有機及び無機の医薬用賦形剤から選択されてよい。
【0051】
1.2.PEG化及び/又はコレステリル化
本発明は、二官能性及びマルチアーム型N−マレイミジルPEG誘導体により例示される、多官能性ポリマー誘導体の使用を意図する。広く多様なポリエチレングリコール(PEG)種が、本発明の新規なOXM誘導体のPEG化のために使用されうる。実質的に任意の好適な反応性PEG試薬が使用可能であり、好適な化学種は、NOFコーポレーション(NOF Corporation)(〒150−6019 東京都渋谷区恵比寿4丁目20−3恵比寿ガーデンタワー)のドラッグデリバリーシステム)カタログにおいて市販されているもの、及び、例として、ネクター・セラピューティクス(アラバマ州35806、ハンツビル、490ディスカバリー・ドライブ)のモレキュラーエンジニアリングカタログのものを包含するが、これに限定されない。例として、そして制限されることなく、以下のPEG試薬が種々の実施態様においてしばしば好ましい:マルチ−アーム型PEG(multi−ArmPEG)、mPEG(MAL)2、mPEG2(MAL)、任意のサンブライト(SUNBRIGHT)活性化PEG(カルボキシル−PEG類、p−NP−PEG類、トレシル−PEG類,アルデヒドPEG類、アセタール−PEG類、アミノ−PEG類、チオール−PEG類、マレイミド−PEG類、ヒドロキシ−PEGアミン、アミノ−PEG−COOH、ヒドロキシ−PEG−アルデヒド、カルボン酸無水物型PEG、官能化PEG−リン脂質を包含するが、これに限定されない)、及び、その特定の適用及び使用のために当業者により選択される他の類似の及び/又は好適な反応性PEG。
【0052】
本発明の新規なOXM誘導体ペプチドはまた、スペーサー部位に対し、カルバメート又はアミド結合を介し共有結合される、2つのPEG部位を含有することも可能であり、ここでスペーサー部位は、該ペプチドの第三級アミドリンカーへ共有結合される。本発明のかかる実施態様において使用される2つのPEG部位の各々は、直鎖であってよく、一の結合点において一緒に結合されてもよい。本発明の1つの実施態様においては、各PEG部位は、約10キロダルトン(10K)−約60Kの分子量を有する(用語「約」は、PEGの標品において、ある分子は標準的な分子量よりもさらに重く、あるものはさらに軽いことを示している)。2つのPEG部位の各々は、約20K−約40Kの分子量を有してもよい。当業者は、所望の用量;循環時間;タンパク質分解に対する抵抗性;もしあれば、生物活性に対する影響;取り扱いの容易さ;抗原性の程度又は欠如;及び治療用ペプチドに対する他の公知のPEGの影響のような考慮に基づき、所望のポリマーサイズを選択することができるであろう。
【0053】
本発明の1つの実施態様においては、N−マレイミジルポリマー誘導体のポリマー主鎖は、ポリ(アルキレングリコール)、そのコポリマー、そのターポリマー、又はそれらの混合物である。例としては、ポリ(エチレングリコール)、ポリ(プロピレングリコール)、及びエチレングリコール及びプロピレングリコールのコポリマーを包含する。以下に、より詳細に説明されるように、本発明のさらに好ましい実施態様は、例えば、二官能性PEG、マルチアーム型PEG、フォーク状PEG、分枝状PEG、ペンダント状PEG、及びその中に分解可能な結合をもつPEGのようなPEGポリマーを利用する。しかしながら、他の関連ポリマーもまた本発明の実施において好適であること、及び、用語PEG又はポリ(エチレングリコール)の使用が、この点について包括的であって排他的でないことを意図したものであることが理解されるべきである。用語PEGは、二官能性PEG、マルチアーム型PEG、フォーク状PEG、分枝状PEG、ペンダント状PEG(すなわち、ポリマー主鎖に対して垂れ下った1個以上の官能基を有するPEG又は関連ポリマー)、又はその中に分解可能な結合をもつPEGを含む、そのどのような形状のポリ(エチレングリコール)をも包含する。
【0054】
ポリマー主鎖は、直鎖又は分枝鎖でよい。PEGは一般に、分枝型で使用され、それは、例えばグリセロール、グリセロールオリゴマー、ペンタエリスリトール及びソルビトールのような種々のポリオールに対するエチレンオキシドの添加により調製可能である。中心の枝分かれ部位はまた、例えばリジンのようないくつかのアミノ酸から誘導可能である。分枝したポリ(エチレングリコール)は、R(−PEG−OH)m
[式中、Rは例えばグリセロール、グリセロールオリゴマー又はペンタエリスリトールのようなコア部位であり、mはアームの数を表わす]のような一般式で表わし得る。マルチアーム型PEG分子、例えば、米国特許第5,932,462号に記載されたものもまた、ポリマー主鎖として使用可能であり、ここに参考することによりその全てが本明細書に含まれる。
【0055】
当業者は、実質的に水溶性かつ非ペプチド性のポリマー主鎖に関する上記のリストが、決して完全なものではなく、例示的なものにすぎないこと、及び上記の特性を有する全てのポリマー素材が意図されていることを認識するであろう。
【0056】
本発明のOXM誘導体上のPEG化位置は、OXMの構造及びそのグルカゴン及びGLP−1受容体との相互作用を考慮して選択される。それ故、PEG化は好ましくは位置特異的である。システインのチオール側鎖におけるPEG化は、広く報告されてきた(例えば、カリケティ及びベロネーゼ(Caliceti&Veronese)、2003年参照)。ペプチド中にCys残基がない場合、置換により導入可能である。本発明のOXM誘導体は、システインの側鎖によりPEG化されてもよい。OXM誘導体は、Cys(mPEG)teineを含有してもよい。Cys(mPEG)teine中のmPEGは、種々の分子量を有し得る。分子量の範囲は、好ましくは5kDa−200kDa、さらには5kDa−100kDa、さらに好ましくは20kDa−60kDaである。mPEGは、直鎖又は分枝鎖でよい。例えば、本発明のCys(mPEG)teineは、C1、C2、C3又はC6でよい。本明細書で例示される場合、C1は、分子量5kDaの直鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)5kDa)(例えば、MPEG−MAL−5000、NEKTAR 2F2MOH01)であり;C2は、分子量20kDaの直鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)20kDa)(例えば、MPEG−MAL−20K、NEKTAR 2F2M0P01)であり;C3は、分子量40kDaの分枝鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)240kDa)(例えば、MPEG2−MAL−40K、NEKTAR 2D3Y0T01、又は、Y型PEGマレイミド、MW40K(ジェンケム・テクノロジー(JenKem Technology)、商品番号Y−MAL−40K、又はサンブライト(SUNBRIGHT)GHT GL2−400MAマレイミド(NOFコーポレーション)であり、そしてC6は分子量60kDaの分枝鎖mPEGをもつCys(mPEG)teine(Cys(mPEG)260kDa)(例えば、MPEG2−MAL−60K、NEKTAR 2D3Y0V01)である。
【0057】
別法として、OXM誘導体のシステイン残基はまた、側鎖チオールを介し、コレステロールにより誘導体化されてもよい。コレステリルOXM誘導体の例は、OXM36、OXM59、OXM65、OXM70、OXM76、OXM82、OXM88及びOXM101を包含する。
【0058】
1.3.他の修飾
N−末端ヒスチジンHxは、His、H1=イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis(PyrH);N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H)、及びPheからなる群より選択される誘導体により変異可能である。N−末端におけるアセチル化及び他の修飾、及び、N−末端キャッピング基は、DP−IV切断に対しOXMを安定化させ、一方C−末端のアミド化は、カルボキシペプチダーゼによるインビボの潜在的な分解を防止しうる。N−末端修飾をもつOXM誘導体には、OXM14及びOXM16−22を包含する。
【0059】
実施例において示されたように、単一用量のOXM2及びOXM3は、自由(ad libitum)に摂食したマウスにおける一晩の体重増加の低減に有効であったのに対し、単一用量のOXM1及び野生型又は天然の(wt)OXMは、このモデルでは有効ではなかった。OXM3は、一晩の体重増加の用量依存性の低減において最も高いインビボ有効性を有しており、受容体結合を妨害しそうなよりかさのあるPEG基を有するOXM2に比較して、GLP−1Rに対するその高い効力を反映しているようであった。OXM2及びOXM3の双方が、インビトロではGLP−1Rに対し、天然のOXMよりも有意に効力が低いことから、PEG化されたOXM誘導体については、wtOXMに比較して増大されたインビボ有効性は、タンパク質分解、及び/又は腎クリアランスに対する何らかの安定化がPEG化のみによって誘導されることを示唆している。
【0060】
さらに、血液成分は、当該ペプチドを安定化するために利用されてよい。好ましい血液成分は、例えば免疫グロブリン、血清アルブミン、フェリチン、ステロイド結合タンパク質、トランスフェリン、チロキシン結合タンパク質、α−2−マクログロブリン等のようなタンパク質を含む。
【0061】
2.OXM誘導体の合成
2.1.ペプチド類の合成
以下の一般的な方法が、いくつかのOXM誘導体を合成するために使用された。固相ペプチド合成は、Fmoc化学を用いて、バッチ又は連続フロー条件下に(例えば、ペニントン及びダン(Pennington&Dunn)著、「ペプチド・シンセシス・プロトコールズ」、1994年、第35巻参照)、PEG−ポリスチレン樹脂を用いて行なった。ペプチド類を樹脂から切断し、トリフルオロ酢酸(TFA)、及びフェノールトリイソプロピルシランのようなカチオンスカベンジャー、及び水を用いて脱保護した。ペプチド類を、冷メチル−t−ブチルエーテルを用いて沈澱させ、沈澱したペプチドを、凍結乾燥に先立ち冷エーテルで2回洗浄した。ペプチドは、0.1%TFAを加えた水/アセトニトリルを典型的な移動相として使用したC4カラム上での逆相HPLC、及びエレクトロスプレー質量分析により確認した。ペプチド類は逆相HPLCにより≧95%まで精製した。
【0062】
2.2.ペプチド類のPEG化
ペプチド類をまず合成し、次にシステインのチオール側鎖をPEG化した。以下の一般的な方法を、ペプチド類のPEG化に使用した。
【0063】
PEG化反応は、チオール化されたペプチド前駆体と、マレイミド−mPEGとの間で行ない、チオエーテル結合を形成した。反応はpH7.3で行い、マレイミド−mPEG量は、チオール化されたペプチドに対し、0.5から10倍モル過剰までの範囲であった。PEG化されたOXMペプチドを、次に、逆相HPLC又はイオン交換クロマトグラフィー、及びそれに続いてサイズ排除クロマトグラフィーを用いて単離した。最後に、PEG化ペプチド類を、分析用RP−HPLC及びMALDAI(マルディ)tof質量分析によりキャラクタライズした。
【0064】
3.OXMをベースとする療法の影響
OXMをベースとする療法は、肥満症及び糖尿病の双方に都合よく影響を及ぼす可能性を有する。OXMの末梢投与時の体重減少及び摂食低減は、ヒトにおいて充分に実証されてきた。本発明者らによる研究は、末梢投与されたブタOXMが、マウスにおいて短期間の摂食及び一晩の体重増加を低減するに充分であることを示してきた。OXMのインクレチン(抗血糖上昇)活性は今日まで十分に研究されてきていないが、マウスの腹腔内グルコース負荷試験(IPGTT)において、OXMのグルコース低下活性がGLP−1のそれに匹敵することが、初めて証明されている。GLP−1と同様、OXMは、静的な単離ネズミ膵島及び灌流ラット膵臓から、大量のグルコース刺激インスリン分泌(GSIS)を誘導し(ジャルーセ(Jarrousse)ら著、「エンドクリノロジー」、1984年、第115巻、p.102−105)、通常のインスリン分泌促進薬に比較して、ローリスクの低血糖を示唆している。ラットでは、OXMの、胃内容排出に対する無視できるほど小さい影響が報告されている(デイキン(Dakin)ら著、「エンドクリノロジー」、2004年、第145巻、p.2687−2695)。マウスでは、OXMは、グルコース低下に最大有効用量において、胃内容排出を〜25%まで低減し、これは、GLP−1受容体アゴニストであるエキセンジン4の最大有効用量によってもたらされるもの(47%低減)よりも少ない。それ故、ヒトにおける胃内容排出に対するOXMの潜在的な良性の作用が、現在のGLP−1模倣薬に比較してこのペプチドホルモンの増強された忍容性に役割を果たしているのかもしれない。
【0065】
本発明のポリペプチドは、肥満症及び/又は糖尿病の治療に有用でありうることが示唆される。第2の適応は、メタボリックシンドローム、高血糖症、空腹時グルコース障害、及び他の前糖尿病状態である。本発明のポリペプチドに関する別の適応は、例えば過敏性腸症候群及び他の腸の吸収疾患、虚血、卒中、及び不安、認知障害及びアルツハイマー病を含む神経障害のような任意の及び全てのGLP−1の適応を包含する。
【0066】
OXMのペプチド性の性質は、天然のホルモンを用いた経口療法を排除する。対照的に、本明細書に提示されたOXM誘導体は、本発明のポリペプチドの1つを、制限されることなく、経口、鼻腔内、舌下、十二指腸内、皮下、バッカル、結腸内、直腸、経膣、経粘膜、経肺、経皮、皮内、非経口、静脈内、筋肉内及び眼内を含む、種々の経路による投与に好適な、医薬的に許容される担体と組合せて含んでなる医薬組成物として、0.001mg/kg−10mg/kg、好ましくは1μg/kg−200mg/kgの用量範囲で、1日2回から1週間以上に1回の投与頻度で投与されてよい。当該ペプチド医薬組成物は、投与方法に依存して、多様な単位用量で投与可能である。好適な単位剤形は、粉末、タブレット、ピル、カプセル、ロゼンジ、坐剤、パッチ、鼻スプレー、注射可能薬物、移植可能な徐放性製剤、脂質複合体、その他を包含し、これに制限されない。ペプチドは、典型的には、医薬的に許容される担体又は賦形剤と組合され、これは、組成物を安定化するため、又は活性薬剤の吸収を増減するために作用する1以上の生理的に許容される化合物を含有し得る。生理的に許容される化合物には、例えばグルコース、スクロース又はデキストランのような炭水化物、例えばアスコルビン酸又はグルタチオンのような酸化防止剤、キレート剤、低分子タンパク質、例えば脂質のような保護及び取込みのエンハンサー、活性薬剤のクリアランス又は加水分解を低減する組成物、又は賦形剤、又は他の安定化剤及び/又は緩衝剤を包含し得る。他の生理的に許容される化合物には、湿潤剤、乳化剤、分散剤又は微生物の増殖又は活動を防止するために特に有用な保存料を包含する。種々の保存料が周知であり、例えば、フェノール及びアスコルビン酸を包含する。当業者は、生理的に許容される化合物を含め、医薬的に許容される担体の選択が、例えば活性薬剤の投与経路及び活性薬剤の特定の物理化学的性質に依存することを理解するであろう。
【0067】
ペプチド類は、天然の形態で、又は所望であれば、該塩が医薬的に許容されるという条件付きで塩の形態で、投与可能である。活性薬剤の塩は、合成有機化学の当業者に周知の標準法を用いて調製されうる。本発明のポリペプチド類は、実施例において詳述されるように、酢酸塩として調製された。
【0068】
本発明のOXMポリペプチドは、GLP1−Rに関与する疾病の治療又は予防において使用される他の薬剤と組合せて使用可能である。本発明のポリペプチドとの組合せにおいて役立つ具体的な化合物には、シンバスタチン、メバスタチン、エゼチミブ、アトロバスタチン、シタグリプチン、メトホルミン、シブトラミン、オルリスタット、Qnexa、トピラマート、ナルトレキソン、ブプリオピオン、フェンテルミン及びロサルタン、ヒドロクロロチアジドとロサルタンの組み合わせを包含する。本発明のポリペプチドとの組合せにおいて役立つ具体的なCB1アンタゴニスト/インバースアゴニストには、N−[3−(4−クロロフェニル)−2(S)−フェニル−1(S)−メチルプロピル]−2−(4−トリフルオロメチル−2−ピリミジルオキシ)−2−メチルプロパンアミド、N−[3−(4−クロロフェニル)−2−(3−シアノフェニル)−1−メチルプロピル]−2−(5−トリフルオロメチル−2−ピリジルオキシ)−2−メチルプロパンアミド、N−[3−(4−クロロフェニル)−2−(5−クロロ−3−ピリジル)−1−メチルプロピル]−2−(5−トリフルオロメチル−2−ピリジルオキシ)−2−メチルプロパンアミド、及び医薬的に許容されるそれらの塩を含む、WO03/077847に記載されたもの;並びに以下:3−{1−[ビス(4−クロロフェニル)メチル]アゼチジン−3−イリデン}−3−(3,5−ジフルオロフェニル)−2,2−ジメチルプロパンニトリル、1−{1−[1−(4−クロロフェニル)ペンチル]アゼチジン−3−イル}−1−(3,5−ジフルオロフェニル)−2−メチルプロパン−2−オール、3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−ヒドロキシ−2−メチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((4−クロロフェニル){3−[1−(3,5−ジフルオロフェニル)−2、2−ジメチルプロピル]アゼチジン−1−イル}メチル)ベンゾニトリル、3−((1S)−1−{1−[(S)−(3−シアノフェニル)(4−シアノフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル)−5−フルオロベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(4H−1,2,4−トリアゾール−4−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、及び5−((4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)チオフェン−3−カルボニトリル、及び医薬的に許容されるそれらの塩を含む、WO05/000809に記載されたもの;並びに3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(3−{(1S)−1−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル}アゼチジン−1−イル)(4−クロロフェニル)メチル]ベンゾニトリル、3−[(S)−(4−シアノフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(3−{(1S)−1−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル}アゼチジン−1−イル)(4−シアノフェニル)メチル]ベンゾニトリル、3−[(S)−(4−シアノフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,3,4−オキサジアゾール−2−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(S)−(4−クロロフェニル)(3−{(1S)−2−フルオロ−1−[3−フルオロ−5−(1,2,4−オキサジアゾール−3−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]−メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−1H−テトラゾール、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−1−メチル−1H−テトラゾール、5−(3−{1−[1−(ジフェニルメチル)アゼチジン−3−イル]−2−フルオロ−2−メチルプロピル}−5−フルオロフェニル)−2−メチル−2H−テトラゾール、3−[(4−クロロフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(2−メチル−2H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−クロロフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(1−メチル−1H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−シアノフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(1−メチル−1H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、3−[(4−シアノフェニル)(3−{2−フルオロ−1−[3−フルオロ−5−(2−メチル−2H−テトラゾール−5−イル)フェニル]−2−メチルプロピル}アゼチジン−1−イル)メチル]ベンゾニトリル、5−{3−[(S)−{3−[(1S)−1−(3−ブロモ−5−フルオロフェニル]−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}(4−クロロフェニル)メチル]フェニル}−1,3,4−オキサジアゾール−2(3H)−オン、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(1,3,4−オキサジアゾール−2−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−{1−[(S)−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)フェニル](4−クロロフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−{1−[(S)−[3−(5−アミノ−1,3,4−オキサジアゾール−2−イル)フェニル](4−シアノフェニル)メチル]アゼチジン−3−イル}−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−シアノフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、3−[(1S)−1−(1−{(S)−(4−クロロフェニル)[3−(1,2,4−オキサジアゾール−3−イル)フェニル]メチル}アゼチジン−3−イル)−2−フルオロ−2−メチルプロピル]−5−フルオロベンゾニトリル、5−[3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)フェニル]−1,3,4−オキサジアゾール−2(3H)−オン、5−[3−((S)−(4−クロロフェニル){3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}メチル)フェニル]−1,3,4−オキサジアゾール−2(3H)−オン、4−{(S)−{3−[(1S)−1−(3,5−ジフルオロフェニル)−2−フルオロ−2−メチルプロピル]アゼチジン−1−イル}[3−(5−オキソ−4,5−ジヒドロ−1,3,4−オキサジアゾール−2−イル)フェニル]メチル}−ベンゾニトリル、及び医薬的に許容されるそれらの塩を包含する。
【0069】
本発明のポリペプチドとの組合せにおいて役立つ具体的なNPY5アンタゴニストには、3−オキソ−N−(5−フェニル−2−ピラジニル)−スピロ[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、3−オキソ−N−(7−トリフルオロメチルピリド[3,2−b]ピリジン−2−イル)スピロ−[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、N−[5−(3−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ−[イソベンゾフラン−1(3H),4’−ピペリジン]−1’−カルボキサミド、トランス−3’−オキソ−N−(5−フェニル−2−ピリミジニル)スピロ[シクロヘキサン−1,1’(3’H)−イソベンゾフラン]−4−カルボキサミド、トランス−3−オキソ−N−[1−(3−キノリル)−4−イミダゾリル]スピロ[シクロヘキサン−1,1’(3’H)−イソベンゾフラン]−4−カルボキサミド、トランス−3−オキソ−N−(5−フェニル−2−ピラジニル)スピロ[4−アザイソ−ベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[5−(3−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ−[5−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[5−(2−フルオロフェニル)−2−ピリミジニル]−3−オキソスピロ[5−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[1−(3,5−ジフルオロフェニル)−4−イミダゾリル]−3−オキソスピロ[7−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(1−フェニル−4−ピラゾリル)スピロ[4−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−N−[1−(2−フルオロフェニル)−3−ピラゾリル]−3−オキソスピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(1−フェニル−3−ピラゾリル)スピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、トランス−3−オキソ−N−(2−フェニル−1,2,3−トリアゾール−4−イル]スピロ[6−アザイソベンゾフラン−1(3H),1’−シクロヘキサン]−4’−カルボキサミド、及び医薬的に許容されるそれらの塩及びエステルを包含する。
【0070】
本発明のポリペプチドとの組合せにおいて役立つ具体的なACC−1/2阻害剤には、1’−[(4,8−ジメトキシキノリン−2−イル)カルボニル]−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、(5−{1’−[(4,8−ジメトキシキノリン−2−イル)カルボニル]−4−オキソスピロ[クロマン−2,4’−ピペリジン]−6−イル}−2H−テトラゾール−2−イル)メチルピバレート、(5−{1’−[(8−シクロプロピル−4−メトキシキノリン−2−イル)カルボニル]−4−オキソスピロ[クロマン−2,4’−ピペリジン]−6−イル}−ニコチン酸、1’−(8−メトキシ−4−モルホリン−4−イル−2−ナフトイル)−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、及び1’−[(4−エトキシ−8−エチルキノリン−2−イル)カルボニル]−6−(1H−テトラゾール−5−イル)スピロ[クロマン−2,4’−ピペリジン]−4−オン、及び医薬的に許容されるそれらの塩を包含する。
【0071】
本発明のポリペプチドとの組合せにおいて役立つ具体的なMCH1Rアンタゴニストには、1−{4−[(1−エチルアゼチジン−3−イル)オキシ]フェニル}−4−[(4−フルオロベンジル)オキシ]ピリジン−2(1H)−オン、4−[(4−フルオロベンジル)オキシ]−1−{4−[(1−イソプロピルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、1−[4−(アゼチジン−3−イルオキシ)フェニル]−4−[(5−クロロピリジン−2−イル)メトキシ]ピリジン−2(1H)−オン、4−[(5−クロロピリジン−2−イル)メトキシ]−1−{4−[(1−エチルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、4−[(5−クロロピリジン−2−イル)メトキシ]−1−{4−[(1−プロピルアゼチジン−3−イル)オキシ]フェニル}ピリジン−2(1H)−オン、及び4−[(5−クロロピリジン−2−イル)メトキシ]−1−(4−{[((2S)−1−エチルアゼチジン−2−イル)メトキシ]フェニル}ピリジン−2(1H)−オン、又は医薬的に許容されるそれらの塩を包含する。
【0072】
本発明のポリペプチドとの組合せにおいて役立つ具体的なDP−IV阻害剤には、7−[(3R)−3−アミノ−4−(2,4,5−トリフルオロフェニル)ブタノイル]−3−(トリフルオロメチル)−5,6,7,8−テトラヒドロ−1,2,4−トリアゾロ[4,3−a]ピラジンから選択される。特に、式Iの化合物は、7−[(3R)−3−アミノ−4−(2,4,5−トリフルオロフェニル)ブタノイル]−3−(トリフルオロメチル)−5,6,7,8−テトラヒドロ−1,2,4−トリアゾロ[4,3−a]ピラジン、及び医薬的に許容されるそれらの塩を包含する。
【0073】
さらに、他のペプチド類似体及びインクレチンホルモングルカゴン様ペプチド1(GLP−1)の模倣薬もまた、本発明のポリペプチドとの組合せにおいて役立つ。
【0074】
本発明の他の特徴及び利点は、異なる実施例を含む本明細書に提供された追加的な記載から明らかである。提供された実施例は、本発明の実施において有用な、種々の成分及び方法論を示している。実施例は、クレームされた本発明を制限するものではない。本開示に基づき、当業者は、本発明の実施において有用な他の成分及び方法論を特定しかつ用いることができる。
【実施例】
【0075】
実施例1:オキシントモジュリン(OXM)類似体の合成
ペプチドOXN類似体(表1参照)は、固相により、Fmoc/tBu化学を用いて、ペプチドマルチシンセサイザーAPEX396(アドバンスド・ケムテック)にて、40ウェル反応ブロックを用いて合成した。各ペプチドを、単一のウェル中で合成した。ペプチドアミド用には、0.1gの、1%架橋された樹脂Fmoc−リンカーAM−チャンピオン(バイオサーチ・テクノロジーズ・インク)、及び、修飾されたリンク(Rink)リンカー p−[(R,S)−α−[9H−フルオレン−9−イル−メトキシホルムアミド]−2,4−ジメトキシベンジル]−フェノキシ酢酸で誘導体化された、PEG−PSベースの樹脂(リンク(Rink、H)著、「テトラヘドロン・レターズ」、1987年、第28巻、p.3787−3789;ベルナトビッツ(Bernatowicz,M.S.)ら著、「テトラヘドロン・レターズ」、1989年、第30巻、p.4645−4667)を使用した。ペプチド酸用には、0.1gの1%架橋チャンピオン樹脂(バイオサーチ・テクノロジーズ・インク)を使用し、それは、あらかじめヒドロキシメチルフェノキシメチル処理により誘導体化した。全てのアミノ酸は、0.5M HOBt(ヒドロキシベンゾトリアゾール)のDMF中の溶液中に、0.5Mの濃度で溶解した。アシル化反応は、樹脂の遊離アミノ基の6倍過剰の活性化アミノ酸を用いて、60分間にわたり行なった。アミノ酸は、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)及び2倍モル過剰のDIEA(N,N−ジイソプロピルエチルアミン)により活性化した。
【0076】
別法として、ペプチドは、Fmoc/tBu化学を用いて、パイオニアペプチドシンセサイザー(アプライド・バイオシステムズ)を用いて合成した。この場合、全てのアシル化反応は、樹脂の遊離アミノ基の4倍過剰の活性化アミノ酸を用いて、60分間にわたり行ない、シンセサイザーでのペプチドアセンブリーの終了に続いた。側鎖保護基は、Asp、Glu、Ser、Thr及びTyrにはtert−ブチル;Asn、Cys、Gln及びHisにはトリチル;Lys、Trpにはtert−ブトキシ−カルボニル;及び、Argには2,2,4,6,7−ペンタメチルジヒドロベンゾフラン−5−スルホニルであった。OXM2及びOXM3ペプチド類については、アセチル化反応は、ペプチドアセンブリーの終了時に、DMF中の10倍過剰の無水酢酸を用いた反応により行なった。
【0077】
OXM14用には、L−ピログルタミン酸を、等モル量のDIPC(ジイソプロピルカルボジイミド)とHOBt(N−ヒドロキシベンゾトリアゾール)との、樹脂遊離アミノ基の4倍過剰の活性化されたアシル作用物(acylant)との反応によりアシル化した。
【0078】
OXM16用には、イミダゾール−乳酸(Imi−H)を、等モル量のPyBOP(ベンゾトリアゾール−1−イル−オキシ−トリス−ピロリジノ−ホスホニウムヘキサフルオロホスフェート)、HOBt及び2倍モル過剰のDIEA(N,N−ジイソプロピルエチルアミン)との、樹脂遊離アミノ基の4倍過剰の活性化されたアシル作用物との反応によりアシル化した。
【0079】
OXM17用には、N−メチル−His(Me−H)を、等モル量のHBTU(2−(1H−ベンゾトリアゾール−1−イル)−1,1,3,3−テトラメチルウロニウムヘキサフルオロホスフェート)と2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、180分間にわたり行った。
【0080】
OXM18用には、デスアミノ−His(ΔNH2−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、180分間にわたり行った。
【0081】
OXM19用には、N,N−ジメチル−His(Me2−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、一晩にわたり行った。
【0082】
OXM20用には、ベンゾイル−His(Bz−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、240分間にわたり行った。
【0083】
OXM21用には、ベンジル−His(Bzl−H)を、等モル量のHBTUと2倍モル過剰のDIEAとの反応によりアシル化した。アシル化反応は、樹脂遊離アミノ基の3倍過剰の活性化されたアシル作用物を用いて、一晩にわたり行った。
【0084】
合成の終わりに、乾燥ペプチド樹脂は、20mLの切断混合物、88%TFA、5%フェノール、2%トリイソプロピルシラン、及び5%水(ソール及びバラニー(Sole、N.A.&G.Barany,)著、「ザ・ジャーナル・オブ・オーガニック・ケミストリー(J.Org.Chem.)」、1992年、第57巻、p.5399−5403)を用いて、室温で1.5時間にわたり個別に処理した。各樹脂を濾過し、ペプチドを沈澱するため、溶液を冷メチル−t−ブチルエーテルへ添加した。遠心分離後、沈澱ペレットを、新鮮な冷メチル−t−ブチルエーテルで洗浄して有機スカベンジャーを除去した。このプロセスを2回繰返した。最終ペレットを乾燥し、H2O、20%アセトニトリル中に再懸濁し、凍結乾燥した。
【0085】
ペプチドOXM54の合成は、チオール含有OXMペプチド前駆体(配列番号41)を、pH8の0.1MトリスHCl、6M塩化グアニジニウム中に溶解することにより行った。10倍モル過剰のヨードアセトアミドを添加した。1時間のインキュベーション後、ペプチド溶液をHPLCにより精製した。
【0086】
ペプチドOXM55の合成は、チオール含有OXMペプチド前駆体(配列番号64)を、pH8の0.1MトリスHCl、6M塩化グアニジニウム中に溶解することにより行った。この溶液に対し、10倍モル過剰のヨードアセトアミドを添加した。1時間のインキュベーション後、ペプチド溶液をHPLCにより精製した。
【0087】
粗ペプチドを、逆相HPLCにより、セミ分取用ォーターズ(Waters)RCMデルタPakTMC4カートリッジ(40x200mm、15μm)を用い、溶出液として、(A)水中の0.1%TFA、及び(B)アセトニトリル中の0.1%TFAを用いて精製した。以下の溶出液(B)の勾配を使用した:5分間にわたり20%−20%、さらに20分間にわたり20%−35%、流速80mL/分。分析用HPLCは、フェノメネックス、ジュピターC4カラム(150x4.6mm、5μm)にて、以下の溶出液(B)の勾配を使用した:20%−40%B(20分間)−80%(3分間)、流速1mL/分。精製したペプチドを、マイクロマス(Micromass)LCZプラットフォーム上でエレクトロスプレー質量分析によりキャラクタライズした。
【0088】
実施例2:オキシントモジュリン(OXM)類似体のPEG化
PEG化反応は、チオエステル結合形成を可能にする条件下に行なった。次にPEG化されたOXMペプチドを、逆相HPLC又はイオン交換クロマトグラフィー、及びサイズ排除クロマトグラフィー(SEC)を用いて単離した。PEG化されたOXM類似体は、RP−HPLC、HPLC−SEC及びMALDI−Tof質量分析を用いてキャラクタライズした。
【0089】
OXM33、34、35、36及び54ペプチド類は、チオール含有OXMペプチド前駆体(配列番号41)から合成し、チオエーテル結合により共有結合したPEGをもつ誘導体を調製した。
【0090】
OXM33の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.4mLのpH7.3の0.1M HEPES中に溶解した22mgのMPEG−MAL−5000(NEKTAR 2F2MOH01)(4.4μmol)(1:2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0091】
OXM34の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.5mLのpH7.3の0.1M HEPES中に溶解した、80mgのMPEG−MAL−20K(NEKTAR 2F2M0P01)(4.0μmol)(1:1.8 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0092】
OXM35の合成
10mgのペプチド前駆体(0.92μmol)を、0.4mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.8mLのpH7.3の0.1M HEPES中に溶解した、70mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(1.7μmol)を、1:1.8 mol/molのPEG:ペプチド比で添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0093】
コントロールペプチドOXM54は、チオール含有ペプチド前駆体を、pH7.5の0.1M トリスHCl、6M塩化グアニジニウム中、10eqのヨードアセトアミドとインキュベートすることにより調製した。30分間のインキュベーション後、ペプチドをRP−HPLCにより精製し、そしてエレクトロスプレー質量分析によりキャラクタライズした。
【0094】
ペプチドOXM56、57、58は、チオール含有OXMペプチド前駆体(配列番号64)から合成し、チオエーテル結合により共有結合したPEGをもつ誘導体を調製した。
【0095】
OXM56の合成
5mgのペプチド前駆体(1.1μmol)を、0.2mLのpH7.3の0.1M HEPESに溶解した。この溶液に対し、0.4mLのpH7.3の0.1M HEPESに溶解した、57mgのMPEG−MAL−5000(NEKTAR 2F2MOH01)(11.4μmol)(1:10 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を1%酢酸で酸性化し、フラクトゲルTSK CM−650S上での陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、サイズ排出クロマトグラフィー(SEC)によりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0096】
OXM57の合成
10mgのペプチド前駆体(2.2μmol)を、0.2mLのDMSOに溶解した。この溶液に対し、0.6mLのpH7.3の0.1M HEPES、0.3Mトリス(2−カルボキシ−エチル)ホスフィン中に溶解した、50mgのMPEG−MAL−20K(NEKTAR 2F2M0P01)(2.5μmol)を、1:1.13mol/molのPEG:ペプチド比で添加した。1時間のインキュベーション後、PEG化されたペプチドをRP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0097】
OXM58の合成
10mgのペプチド前駆体(0.92μmol)を、0.4mLのpH7.3の0.1M HEPES、6M塩化グアニジニウム、2mM EDTA中に溶解した。この溶液に対し、0.8mLのpH7.3の0.1M HEPES中に溶解した70mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(1.7μmol)(1:1.8 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチドを、RP−HPLCにより精製し、そしてMALDI−Tofによりキャラクタライズした。
【0098】
コントロールペプチドOXM102、OXM112及びOXM116は、チオール含有ペプチド前駆体を、pH7.5の0.1M トリスHCl、6M塩化グアニジニウム中、10eq.のヨードアセトアミドとインキュベートすることにより調製した。30分間のインキュベーション後、ペプチドをRP−HPLCにより精製し、そしてエレクトロスプレー質量分析によりキャラクタライズした。
【0099】
OXM103、OXM105、OXM107、OXM113の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH7.3の0.1M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2Oに溶解した109mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.71μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0100】
OXM109の合成
10mgの対応するペプチド前駆体(2.25μmol)を、2mLの、8M尿素、pH7.3の0.1M HEPES、2mM EDTA中に溶解した。この溶液に対し、2mLのH2O中に溶解した108mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.7μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0101】
OXM117の合成
10mgの対応するペプチド前駆体(2.19μmol)を、2mLの、8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した105mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.63μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、1%酢酸で酸性化し、TSK CM−650Sでの陽イオン交換クロマトグラフィー(IXC)により、pH4.8の50mM酢酸ナトリウム中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0102】
OXM125の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH6.5の0.25M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した105mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.71μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8まで酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0103】
OXM129の合成
10mgの対応するペプチド前駆体(2.26μmol)を、2mLの、8M尿素、pH6.5の0.25M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した109mgのMPEG2−MAL−40K(NEKTAR 2D3Y0T01)(2.72μmol)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸で酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0104】
実施例3 ペプチド配列のデザイン及び合成
種々のPEGサイズ(mPEG)5kDa、(mPEG)20kDa、(mPEG)240kDa及び(mPEG)260kDaの生物活性を、一連の実験において比較し、最大かつ耐久性のある活性をマウスにおいて与えるために最適なPEGサイズが、一般に40kDaであることを証明した。
【0105】
ペプチドOXM103(2位Aib(α))は、オキシントモジュリン配列内の20位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM102はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(20位のシステイン)は、ヨードアセトアミドと反応した。
【0106】
ペプチドOXM105(2位Aib(α))は、オキシントモジュリン配列内の21位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM104はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(21位のシステイン)は、ヨードアセトアミドと反応した。
【0107】
ペプチドOXM107(2位Aib(α)、27位Met(O))は、オキシントモジュリン配列内の24位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM106はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(24位のシステイン)は、ヨードアセトアミドと反応した。
【0108】
ペプチドOXM109(2位Aib(α))は、オキシントモジュリン配列内の28位を、(mPEG)240kDaとの結合用の部位として調査するためにデザインした。OXM108はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(28位のシステイン)は、ヨードアセトアミドと反応した。
【0109】
OXM141と呼ばれるペプチドは、3位においてGlnからAspへの変異を有し、このことがGLP−1受容体に対する特異的選択性を与えている。3位にGlnからAspへの変異を、27位にMet(O)を、及び、20位及び38位に2つの結合部位を有するペプチドOXM141は、C38位にコレステロール基及びC20位にPEGの双方に結合されたペプチドを有する可能性を調査するためにデザインした。
【0110】
ペプチドOXM142(Aib(α)、27位にMet(O)、及び20位及び38位に2つの結合部位)は、C38位にコレステロール基及びC20位にPEGの双方に結合されたペプチドを有する可能性を調査するためにデザインした。
【0111】
図14は、GLP1R及びGCG受容体に関するインビトロ活性のデータ(GLP−1R及びGLGRの特異性も示している)を、表形式に要約している。C20位における(mPEG)240kDa結合は、双方の受容体に対し活性を保持しており、それ故、OXM103は、+/+類似体と呼ばれ、それに対し他の全ての(mPEG)240kDa結合は、GCG受容体において能力を失っている。特に、38、24及び28位における(mPEG)240kDa結合については、Gcg受容体に対し2−3オーダーの大きさの選択性が、GLP−1受容体に対し存在する。それ故、類似体OXM99、107及び109は、+/0類似体と呼ばれる。
【0112】
図15は、DIOマウスでの、摂食及び体重減少に対する、(mPEG)240kDa結合のインビボ活性を示す。自由に摂食したDIO(各〜51g)の、雄のC57BL/6マウスに、暗期開始の〜30分前に、ビヒクル(水)又はOxm類似体99、103、105、107及び109を、腹腔内(i.p.)投与した。摂食は、1日目で〜2時間及び18時間(一晩)後に、2日目で26及び42時間(一晩)後に測定した。*P<0.05 対ビヒクル、群当たりn=5−6。
【0113】
ペプチドOXM110(2位D−Ser)は、オキシントモジュリン配列内の38位を、パルミトイル基のような脂質との結合用の部位として調査するためにデザインした。パルミトイル基を、オキシントモジュリン配列のC−末端に付加されたリジンのε−アミノ基へアシル化した。
【0114】
ペプチドOXM113(1位デスアミノ−His(ΔNH2−H)、2位Aib(α)、27位Met(O)、結合部位38位)は、DPPIVタンパク質分解から保護するための、1位における野生型(wt)Hisの、デスアミノ−Hisによる置換の可能性を調査するためにデザインした。OXM113は、(mPEG)240kDa結合であり、OXM114は、コレステリル結合であり、そしてOXM112はコントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(38位システイン)は、ヨードアセトアミドと反応した。(OXM111は、チオール化されたペプチド前駆体である)。
【0115】
ペプチドOXM117及びOXM118(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位38位)は、3位における野生型Glnの、Aspによる置換の可能性を調査するためにデザインした。上記のように、この変異は、GLP−1Rに対する特異的選択性を与える。OXM117は、(mPEG)240kDa結合であり、OXM118は、コレステリル結合である。(OXM116は、チオール化されたペプチド前駆体である)。
【0116】
ペプチドOXM121(2位Aib(α)、27位Met(O)、結合部位11位)は、オキシントモジュリン配列内の11位を、結合用の位置として調査するためにデザインした。OXM121は、(mPEG)240kDa結合であり、OXM119は、チオール化されたペプチド前駆体であり、そしてOXM120は、コントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(11位のシステイン)は、ヨードアセトアミドと反応した。
【0117】
ペプチドOXM124(2位Aib(α)、27位Met(O)、結合部位12位)は、オキシントモジュリン配列内の12位を、結合用の部位として調査するためにデザインした。OXM121は、(mPEG)240kDa結合であり、OXM122は、チオール化されたペプチド前駆体であり、かつOXM123は、コントロールペプチド(CH2CONH2)であり、ここで側鎖チオール(12位のシステイン)は、ヨードアセトアミドと反応した。
【0118】
ペプチドOXM125(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、20位を(mPEG)240kDaの結合用の部位として、並びに、GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換を調査するためにデザインした。
【0119】
ペプチドOXM127(2位Aib(α)、27位Met(O)、結合部位22位)は、オキシントモジュリン配列内の22位をコレステロール用の結合のための部位として調査するためにデザインした。(OXM126は、チオール化されたペプチド前駆体である)。
【0120】
ペプチドOXM129(1位デスアミノ−His(ΔNH2−H)、2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、以下の組合せの可能性を調査するためにデザインした:DPPIVタンパク質分解からの保護のため1位において野生型Hisをデスアミノ−Hisで置換すること、GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換、及び、20位における結合。
【0121】
ペプチドOXM134(2位Aib(α)、27位Met(O)、結合部位20位)は、強力な+/+OXM103類似体に類似の、(mPEG)240kDa結合であり、メチオニン置換がメチオニンスルホキシドへの点についてのみ異なっている。このペプチドは、20位において結合されており、したがって、GLP1R/GcgR選択性については、+/+パターンを示す。
【0122】
実施例4 C−末端切断類似体又はGcgK類似体
C−末端切断類似体
一連の、ペプチドOXMC−末端切断類似体がデザインされた。GLP1受容体(GLP1R)及びGcg受容体(GcgR)の双方に対するインビトロ活性の分析は、OXM配列がC−末端において、wtグルカゴンに対し1つだけ余分のリジン残基をもつよう切断可能であり、GLP1R及びGcgRの双方に対し極めて効力のある、OXM93のようなペプチドを生じることを証明した。OXM93の効力は、wtOXMのそれのよりも、少なくとも1オーダー(及び恐らくは2オーダー)大きい。Gcgに対し1つだけ余分のリジン残基があることから、この新規なクラスはGcgK類似体と呼ばれる。図16は、GLP1及びGcg受容体において作用する、C−末端切断類似体に関するインビトロの効力を表形式で例示している。
【0123】
選択的GcgK類似体であるC−末端切断類似体
ペプチドOXM130及びOXM131は、インビトロ分析を確認するため、及び安定性及び好適な特性を与えることが知られている他の変異を導入するためデザインした。OXM130及びOXM131は、OXM93と同じ長さのC−末端切断類似体である。OXM130は、2位にAib(α)を、一方OXM131は、2位にAib(α)を、また3位に、GLP1Rに対する選択性を与えるためのGlnからAspへの変異を有する。
【0124】
以下のPEG化された類似体を、これらの切断類似体に基づきデザインした:
【0125】
ペプチドOXM136(2位Aib(α)、27位Met(O)、結合部位20位)は、(mPEG)240kDaの結合である。先の研究(上記参照)から、20位におけるPEG結合の選択は、GLP1R及びGcgRの双方の受容体に対する活性を与えることが知られており、それ故OXM136は、プロトタイプGcg+/+類似体として定義されるであろう。(OXM135は、チオール化されたペプチド前駆体である)。
【0126】
ペプチドOXM138(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、以下の組合せの可能性を調査するためにデザインした:GLP−1Rに対する特異的選択性を与える3位におけるGlnからAspへの置換;20位における結合、及び3位におけるwtGlnからAspへの変異。それ故、OXM137は、プロトタイプGcgK+/0類似体として定義されるであろう(OXM137は、チオール化されたペプチド前駆体である)。
【0127】
図17は、PEG化されたOXM類似体選択のための、GLP1及びGCG受容体におけるインビトロの効力のデータを提供する。
【0128】
実施例5:代替のPEG成分をもつペプチド
ペプチドOXM145(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、ジェンケム・セラピューティクス(JenKem Therapeutics)からのY型PEGマレイミドとの結合部位38位)を、代替の分枝した40kDaPEG基を用いる可能性を調査するために調製した。これは、ジェンケム・セラピューティクスからの(mPEG)240kDa(Y型PEGマレイミド、MW40K、商品番号Y−MAL−40K)を用いて得た結合体であり、その構造は、以下に示される:
【0129】
【化1】
【0130】
ペプチドOXM146は、2位におけるAib(α)、3位におけるGlnからAspへの変異、27位におけるMet(O)、NOFコーポレーションからのサンブライトGL2−400MAマレイミドとの38位の結合によりキャラクタライズされる。これは、NOFコーポレーションからの(mPEG)240kDa(サンブライト GL2−400MAマレイミド)を用いて得た結合体であり、その構造は、以下に示される:
【0131】
【化2】
【0132】
ペプチドOXM151(2位Aib(α)、3位GlnからAspへの変異、17位ArgからGluへの変異、18位ArgからAlaへの変異、27位Met(O)、結合部位38位)は、ペプチド配列への、インビボでの増大された安定性を与えるためにデザインした。
【0133】
詳細な研究を行い、ここではOXM139を10%のマウス又はヒト血漿を含有するPBS中、37℃においてインキュベートした。試料調製は、1μLの試験溶液と、1μLのマトリックス(α−シアノ)とを、試料プレート上で直接混合することにより遂行した。結晶化後、Tofスペクトルを収集した:30、60、120及び720分の時点で分析し、同じ時点のコントロール試験溶液と比較した。ペプチド配列内では、Arg17とArg18との間の結合は、第1の加水分解部位であると特定された。Arg18とAla19との間の結合もまた、第2の加水分解部位であると特定されたため、部位17及び18に変異を導入することを決定した。特異的に、Arg17をGluへ変異させ、Arg18をAlaへ変異させた。ペプチドOXM153は、OXM151のN−エチルマレイミド類似体である。ペプチドOXM154は、NOFコーポレーションからの(mPEG)240kDa(サンブライトGL2−400MAマレイミド)を用いて得られた結合体である。
【0134】
ペプチドOXM152は、オキシントモジュリン配列の残基1−16を繋いでおり(2位Aib(α)、3位GlnからAspへの変異、17位スペーサーとしてTtds、18位に結合のためのCys)、1−16配列に対し特異的な抗体を上げるため、担体タンパク質へ結合可能なペプチドである。
【0135】
ペプチドOXM155(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、38位Ttds、グルタミン酸残基39−43、44位に結合のためのCys)は、抗体を上げるための担体タンパク質へ結合可能なもう1つのペプチドである。C末端へのグルタミン酸の付加は、2003年12月18日に出願された、仮出願第60/530867号(高い免疫原性をもつペプチド−担体結合体の作成法)」(これを参照とすることによりその全を本明細書に組み込む)に記載のように、担体タンパク質への結合を可能にするpI修飾に必要である。
【0136】
実施例6 新規な切断類似体又はGcgK類似体
ペプチドOXM143(Aib(α)、27位Met(O)、結合部位20位)は、OXM135のN−エチルマレイミド類似体である。
【0137】
ペプチドOXM144(Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位)は、OXM137のN−エチルマレイミド類似体である。
【0138】
ペプチドOXM147(2位Aib(α)、結合部位20位)は、C20位に結合部位を有するGcgK類似体シリーズ内で、27位に天然のメチオニン残基をもつようデザインした。このデザインの根拠は、天然メチオニンをもつペプチド類似体がグルカゴン受容体に対しより活性があることであった。
【0139】
ペプチドOXM148(2位Aib(α)、結合部位20位)は、OXM147のN−エチルマレイミド類似体である。
【0140】
ペプチドOXM149(2位Aib(α)、3位GlnからAspへの変異、27位Met(O)、結合部位20位、及び30位D−リジン置換)は、ペプチドC−末端の、インビボでの酵素分解からの保護を与えるためデザインした。ペプチドOXM150は、OXM149のN−エチルマレイミド類似体である。
【0141】
同様の安定性の研究を、GcgKシリーズの類似体OXM144に対し、10%のマウス又はヒトのいずれかの血漿を含有するPBSとのインキュベーションにより、第1の加水分解部位を決定するために行なった。この例では、Arg17とArg18との間の結合を第1の加水分解部位として特定した。また、Arg18とAla19との間の結合も、第2の加水分解部位として特定した。このことから、GcgK類似体シリーズにおいても、部位17及び18に変異を導入することが決定された。
【0142】
OXM145の合成
54mgの対応するペプチド前駆体(11.8μmol)を、2mLの8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した569mg(14.2μmol)のY型PEGマレイミドMW40K(ジェンケム・テクノロジー、商品番号Y−MAL−40K)(1:1.2 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8に酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした。
【0143】
OXM146の合成
55mgの対応するペプチド前駆体(12μmol)を、2mLの8M尿素、pH6.5の0.2M HEPES、2mM EDTA中に溶解した。この溶液に対し、H2O中に溶解した531mg(13.2μmol)のサンブライトGL2−400MAマレイミド(NOFコーポレーション)(1:1.1 mol/molのPEG:ペプチド比)を添加した。1時間のインキュベーション後、PEG化されたペプチド溶液を、0.2%蟻酸でpH2.8に酸性化し、TSK SP−5PWでの陽イオン交換クロマトグラフィー(IXC)により、0.2%蟻酸中のNaClの直線勾配を用いて精製した。IXC精製したPEG化ペプチドを、SECによりさらに精製し、そしてMALDI−Tofによりキャラクタライズした
【0144】
実施例7:サイクリックAMP(cAMP)を用いたGLP−1受容体(GLP−1R)シグナリングの測定
ホモジニアス時間分解蛍光(HTRF)アッセイ及びDP−IVに対する抵抗性の評価
天然の受容体と同様の生物活性をもつヒトGLP−1Rの変異型により安定にトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞系を、ウシ胎児血清(FBS)、ペニシリン−ストレプトマイシン、ヒポキサンチン−チミジン及びG418を含有する、完全なイスコブ改変ダルベッコ培地(IMDM)培地中に維持した。GLP−1受容体活性化に対するホモジニアス時間分解蛍光(HTRF)アッセイを使用して、細胞をリガンド、XL−665及び抗−cAMPクリプテートと37℃でインキュベートしたことを除いて製造業者(シスバイオ(CisBio))の指示に従って、本発明のペプチドとのインキュベーション時のトランスフェクト細胞におけるcAMP蓄積を測定した。アッセイは、96ハーフウェルプレートフォーマット中で、パーキン・エルマーエンビジョンプレートリーダを使用してプレートを読み取った。本発明のポリペプチド及びポリペプチドフラグメント/誘導体については、cAMP HTRFアッセイにおけるGLP−1受容体の「活性化」は、最大活性の誘導であり、それは、天然のヒトOXM配列により誘導される最大活性の少なくとも約60%から約200%までであり、少なくとも0.04%から約1000%までの相対効力である。「相対効力」とは、天然のヒトOXMのEC50を本発明のポリペプチドのEC50で割り、100倍したものである。「EC50」とは、最大活性の50%が達成されるポリペプチド濃度である。
【0145】
DP−IV切断に対する抵抗性を測定するため、5μMのペプチド溶液を、10nMの組換え可溶性ヒトDP−IVと、100μlのアッセイバッファー(10mM HEPES、pH7.5、0.05%BSA)中で、37℃で2時間プレインキュベートした。続いて、hGLP−1Rの活性化を、シスバイオHTRFcAMPアッセイを用いて測定し、DP−IVなしに37℃で2時間プレインキュベートしたコントロールペプチドと比較した。本発明のポリペプチド類及びそれらのフラグメント/誘導体については、本実験における「DP−IVに対する抵抗性」は、0.1から35までの効力比として定義され、ここで「効力比」とは、DP−IVとプレインキュベートしたペプチドのEC50をDP−IVなしでプレインキュベートした本発明の同じポリペプチドのEC50で割ったものである(図2)。
【0146】
実施例8:サイクリックAMPフラッシュプレートアッセイを用いたグルカゴン受容体(GcgR)シグナリングの測定
クローニングされたヒトグルカゴン受容体を発現しているCHO細胞(CHO−hGCGR)(カシエリ(Cascieri)ら著、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー」、1999年、第274巻、p.8694−8697)を、10%FBS、1mM L−グルタミン、ペニシリン−ストレプトマイシン(100U/ml)、及びG418(500μg/ml)を供えたIMDM中に維持した。本発明のペプチドとのインキュベーション時のトランスフェクト細胞におけるcAMPレベルを、フラッシュプレートアッセイ(SMP−004B、パーキン・エルマー・ライフ・サイエンス)を利用して、製造業者の指示に従って測定した。細胞刺激を、細胞溶解剤と125I−標識cAMPトレーサーとを含有する等量の検出バッファーの添加により停止した。プレートに結合した125I−cAMPを、液体シンチレーションカウンタを用いて測定し、各試料中に存在するcAMP量を定量するために使用した。本発明のポリペプチド類及びポリペプチドフラグメント/誘導体については、cAMPフラッシュプレートアッセイにおけるGcg受容体の「活性化」は、最大活性の誘導であり、それは、天然のグルカゴンペプチドにより誘導される最大活性の少なくとも約60%から約200%までであり、少なくとも0.04%から約10000%までの相対効力である。「相対効力」は、天然グルカゴンのEC50(EC50=70pM)を本発明のポリペプチドのEC50で割り、100倍したものである。「EC50」は、最大活性の50%が達成されるポリペプチド濃度である。天然のブタOXMは、このアッセイにおいて、2.4nMのEC50でグルカゴン受容体を活性化した。
【0147】
実施例9:やせたマウスにおける腹腔内グルコース負荷試験(IPGTT)の間の血中グルコース変位に対する効果
雄のC57BL/6Nマウスを、体重別に処理群へ分配し、実験開始の約5時間前に断食させた。ベースライン(t=〜30分)の血中グルコース濃度を、尾部切開の血液からグルコメータにより測定した。次いで動物に、ビヒクル(食塩水)又は本発明のポリペプチド(0.01−10mg/kg)を腹腔内(i.p.)注射した。血中グルコース濃度を、処理(t=0分)の30分後に測定し、次いでマウスを、デキストロース(2g/kg、10mL/kg)のi.p.により誘発した。ビヒクル処理マウスの1群を、ネガティブコントロールとして通常の食塩水で誘発した。血中グルコースレベルを、デキストロース誘発の20、40、60及び120分後に尾部の血から測定した。t=0からt=120分までの血中グルコース変位プロフィールを用いて、各処理について曲線下面積(AUC)を積分した。各処理群についてのグルコース変位のパーセント阻害は、式:
【0148】
【数1】
【0149】
[式中、
AUCdex=ビヒクル処理され、デキストロース誘発された動物の平均AUC、
AUCpeptide=ペプチド処理され、デキストロース誘発された動物の平均AUC、及び
AUCsaline=ビヒクル処理され、食塩水誘発された動物の平均AUC]に従って、水で誘発したコントロールによりノーマライズしたAUCデータから計算した。
【0150】
IPGTTにおける本発明のポリペプチドのインクレチン活性は、グルコース変位のパーセント阻害における用量依存性の増加として明示されており、10mg/kgの投与において少なくとも30%に達する(図2)。
【0151】
実施例10:やせたマウスにおける摂食及び体重に対する急性効果
約3か月齢の、自由摂食させた雄のC57BL/6Nマウスを秤量し、ビヒクル(水)又はビヒクル中に溶解したOXM2又はOXM3のいずれかを、光周期の暗期開始の〜30分前に、i.p.注射により投与した。光周期の暗期開始の〜5分前に、あらかじめ秤量した量のげっ歯類用固形飼料(テクラド7012)を、ワイヤーケージ上のフードホッパーに供給し、光周期の暗期開始の2及び18時間(一晩)後に秤量した。摂食における絶対変化は、各動物につき、特定の時点においてフードホッパー内に残留する餌の量を対応する元の秤量前の量のそれから引くことにより計算した。体重における絶対変化は、各動物につき、投与前の動物の体重を特定の時点における対応する動物の体重から引くことにより計算した。全ての値は、平均±SEMとして報告し、ペプチド処理群は、ビヒクル処理動物を基準としてツーテイルド・アンペアード・スチューデント検定(two tailed unpaired student’s test)により分析した。任意の時点における摂食、及び/又は一晩の体重増加における低減は、p値≦0.05として統計的に有意であって、このモデルにおける対応するOXMポリペプチド(OXM2又はOXM3)の効力を示しているとみなされる(図3)。
【0152】
実施例11:マウスにおけるグルコース刺激インスリン分泌の増大
16mmol/lグルコースでのインスリン分泌の仲介における天然OXMのインビトロの効力を、野生型C57BL/6マウスからの膵島及び、強いGSIS活性をもつマウスインスリノーマ細胞系、MIN6c4(ミナミ(Minami K)ら著、「アメリカン・ジャーナル・オブ・フィジオロジー、エンドクリノロジー・アンド・メタボリズム(Am J Physiol Endocrinol Metab.)」、2000年、第279巻、E773−E781)において、天然OXMペプチドの濃度を増加しながら、16mmol/lグルコースにおいて、グルコース刺激インスリン分泌(GSIS)を測定することにより評価した。膵臓ランゲルハンス島を、正常なC57BL/6Jマウス(ジャクソン・ラボラトリー、メイン州)から、レイシー及びコスチアノフスキーの原法(レイシーら著、「ダイアベーツ」、1967年、第16巻、p.35−39)の変法である、コラゲナーゼ消化及び不連続フィコール勾配分離により単離した。GSISアッセイに先立ち、膵島を、RPMI1640培地(11mMグルコース)中で一晩培養した。GSISを測定するために、膵島をまず、2mMグルコース入りのクレブス・リンガー重炭酸塩(KRB)バッファー中で30分間プレインキュベートした(ペトリ皿中)。KRB培地は、143.5mM Na+、5.8mM K+、2.5mM Ca+、1.2mM Mg2+、124.1mM Cl−、1.2mM PO43−、1.2mM SO42+、25mM CO32−、2mg/mlのウシ血清アルブミン(pH7.4)を含有する。膵島を、次に96穴プレートに移し(1膵島/ウェル)、2又は16mMグルコース入りの200μlのKRBバッファー中で、試験されるべき他の薬剤、例えばGLP−1及びOXMと共に37℃で60分間インキュベートした(ゾウ(Zhou)ら著、「ザ・ジャーナル・オブ・バイオロジカル・ケミストリー」、2003年、第278巻、p.51316−51323)。インスリンを、市販のキット(ALPCOダイアグノスティクス、ニューハンプシャー州ウィンダム)によるELISAにより、インキュベーションバッファーの少量中で測定した。MIN6c4細胞におけるインスリン分泌を、同様の方法で96穴プレートに播種した細胞において測定した。
【0153】
図4に示したように、天然のOXMは、マウスの膵島及びMIN6c4細胞において、GSISを有意に増大した。GSISに対するOXMのEC50は、各々、ネズミ膵島で約2.9nM(図4A)、及び155pM(図4B)であった。天然のGLP−1を、この実験におけるポジティブコントロールとして使用した。2つのペプチドの最大GSIS効果は、膵島及びMIN6細胞の双方において類似していた。
【0154】
OXMは、実施例7及び8に記載のように、CHO細胞において異種発現されたGLP−1R及びGCG−R双方を活性化し、そして双方の受容体は、膵臓β細胞において機能性であることが知られている。OXMのインクレチン作用におけるこれら2つのG−タンパク質共役受容体の潜在的役割を識別するため、GSISに対するOXM、GLP−1及びGCGの効果を、GLP−1R−/−マウス(スクロッチ(Scrocchi LA)ら著、「ネイチャー・メディスン」、1996年、第2巻、p.1254−1258)、及び、齢の合致したWT C57BL/6マウスからの膵島において調べた。先の結果と一致して、WTネズミ膵島からの16mmol/lグルコースにおけるGSISの増大においては、3つのペプチドは全て(各10nM)等しく有効であった(図5A)。GSIS及びGCGによるGSISの相乗作用は、GLP−1R−/−膵島では損なわれなかったのに対し、GLP−1及びOXMの双方は、後者においてGSISを全く増大することができなかった。OXMのインクレチン作用におけるGLP−1Rの関与はまた、広く使用されるGLP−1Rのペプチドアンタゴニストであるエキセンジン−9によるこの活性の拮抗作用により示された。OXM及びGLP−1によるGSISの相乗作用は、0.5μMのエキセンジン−9により、WT膵島において完全に阻止された(図5B)。
【0155】
OXMのインクレチン作用におけるGCG−Rの潜在的な関与を、ペプチド仲介GSISを、WT及びGCG−R−/−マウスからの膵島において16mmol/lグルコースにより比較することにより試験した(ゲリング(Gelling XQ)ら著、「プロシーディングズ・オブ・ザ・ナショナル・アカデミー・オブ・サイエンシズ」、2003年、第100巻、p.1438−1443)。先に記載の通り、3つのペプチドの全て(GLP−1、OXM及びGCG、各10nM)が、等しい効力で、WT膵島におけるGSISを増強した(図6A)。しかしながら、サイズ合致したWT膵島に比較して、2及び16mMグルコースの双方におけるインスリン分泌は、GCG−R−/−膵島では〜2倍まで低減された。膵島のインスリン含有量もまた、GCG−R−/−膵島ではWTに比較して>3倍まで低減された(図6B)。GCG(10nM)は、16mMグルコースにおいてはGCG−R−/−膵島ではGSISを増強しなかったのに対し、GLP−1及びOXM(10nM)は、GSISを、このアッセイにおいて有意に増大させた。データをフラクショナルGSIS(全膵島インスリン含有量に比較した%インスリン放出)として表わすと、OXMにより仲介されるGSISにおける倍数的増加(fold−increase)は、GCG−R−/−膵島ではWTに比較してわずか32%(1.7対2.5倍)に低減された(図6C)のに対し、GLP−1によるGSISの倍数的刺激は、同じに保持された(2.5倍)。対照的に、GCGは、GCG−R−/−膵島では、ベースライン(DMSO)を超えてフラクショナルGSISを増大することはなかった。これらのデータは、GSISに対するOXMの作用において、GCG−Rが限定された役割を果たすかもしれないことを示唆している。
【0156】
OXMのグルコース低下効果(上記記載のように、また図2に示したように)が、増大されたインビボGSISに対し二次的であったかどうかを決定するため、血漿グルコース及びインスリンレベルに対するOXMの効果を、WT及びGLP−1R−/−マウスにおけるIPGTTの間に分析した。一晩絶食したマウスに、グルコース誘発に先立ち、0.3mpk(kg体重当たりのmgペプチド)の天然OXMを前投与した(i.p.)。GLP−1の模倣薬である、エキセンジン−4(0.02mpkでi.p.投与)(トレンス(Thorens,B)ら著、「ダイアベーツ」、1993年、第42巻、p.1678−1682)を、この研究における比較対象として用いた。図7Aに示したように、エキセンジン−4及びOXMの双方は、WTマウスにおいてIPGTTの間に、グルコースレベルを有意に低減し、エキセンジン−4は、グルコース変位の抑制においてより強力であった。0.3mpkのOXM処理群におけるグルコース変位についての曲線下面積(AUC)は、ビヒクル群に比較して約30%低減された[13025±524対19928±811mg/dl/60分間]、p<0.001、n=10(ビヒクル)又は5(OXM)]のに対し、エキセンジン−4処理群におけるグルコースAUCの低減は、>60%(AUC=6601±179mg/dl/60分間)であった。対照的に、同じ用量のOXM及びエキセンジン−4は、GLP−1R−/−マウスにおけるIPGTTにおいて、グルコース変位に影響を及ぼさなかった(図7B)。
【0157】
インビボのGSISでのi.p.のOXM及びエキセンジン−4の効果を、IPGTT研究において、グルコース誘発の前(0分)及び後(10分)の血漿インスリンレベルを測定することにより評価した。OXMは、WTマウスにおいて、ベース(0分)の血漿インスリンレベルを4倍に増大し、i.p.のグルコース誘発に対するインスリン応答を有意に増幅した(図7C)。同様な結果は、WTマウスにおいて、エキセンジン−4を用いて観察された。対照的に、GLP−1R−/−マウスに対するOXM又はエキセンジン−4の投与は、ベースのインスリンレベルに影響を及ぼさず、i.p.のグルコース誘発に対するインスリン応答も改善しなかった(図7D)。
【0158】
医薬組成物の例
本発明の新規なポリペプチドの経口組成物の1つの具体的な実施態様として、5mgの、式
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC10−CONH2、
により記載されるポリペプチドを、サイズ0ハードゲルカプセルを満たすように、全量580−590mgを提供すべく充分な超微粒子状のラクトースと共に製剤する。
【0159】
本発明の新規なポリペプチドの経口組成物の、もう1つの具体的な実施態様として、2.5mgの、式
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC10−CONH2、
により記載されるポリペプチドを、サイズ0ハードゲルカプセルを満たすように、全量580−590mgを提供すべく充分な超微粒子状のラクトースと共に製剤する。
【0160】
他の実施態様は、以下のクレームの範囲内にある。いくつかの実施態様が示され、記載されてきたが、本発明の精神及び範囲から離れることなく、種々の変更が行われてもよい。
【図面の簡単な説明】
【0161】
【図1】変異型のヒトGLP−1受容体の、(a)天然のブタOXM、及び(b)PEG化されたOXM2による活性化、及び、DP−IVとのプレインキュベーションによる、当該ペプチドの効力の減少を描いた説明図である。
【図2】やせたマウスの腹腔内グルコース負荷試験(IPGTT)における、ブタオキシントモジュリンのインクレチン活性を示す図である。グルコース変位の%阻害が、各群について示されている。「ctrl」=ビヒクル処理された食塩水誘発マウス、「veh」=ビヒクル処理されたデキストロース誘発マウス。
【図3】配列OXM2及びOXM3により示されたポリペプチドの、やせたマウスにおける一晩の摂食及び体重増加の低減における効果を例示する図である。*ビヒクル群を基準として、p≦0.05。
【図4】膵島及びMIN6細胞におけるグルコース刺激インスリン分泌(GSIS)に対する、GLP−1及びOXMの効果を描いた図である。
【図5】膵島におけるOXM、GCG及びGLP−1仲介GSISに対する、GLP−1R欠失及び受容体拮抗作用の影響を示す図である。
【図6】グルカゴン受容体−/−膵島でのGSISに対するOXMの効果を示す図である。
【図7】野生型及びGLP−1R−/−マウスにおける、IPGTTの間の血中グルコース及びインスリンレベルに対するOXM及びエキセンジン−4の効果を示す図である。
【図8】やせたマウスのIPGTTにおける、OXM99の急性のグルコース低下効果を示す図である。
【図9】やせたマウスでの、血中グルコースの低減におけるOXM99の効果を描いた図である。
【図10】やせたマウスのIPGTTにおける、OXM117のグルコース低下効果を示す図である。
【図11】エキセンジン−4と比較した、OXM類似体であるOXM117及びOXM99の、活性の持続時間を示す図である。
【図12】ラットにおける皮下投与を用いたOXM117の薬物動態を描いた図である。
【図13】OXM117ペプチドが、インビトロで何らグルカゴン様活性を示さないことを示す研究結果を示す図である。
【図14】GLP1及びGCG受容体におけるインビトロ活性のデータを表形式に要約している図である。
【図15】DIOマウスモデルにおける摂食及び体重減少に対する(mPEG)240kDa結合体のインビボ活性を示す図である。
【図16】GLP1及びGCG受容体において作用するC−末端切断類似体についてのインビトロの効力のデータを示す図である。
【図17】GLP1及びGCG受容体において作用する選択されたPEG化OXM類似体についてのインビトロの効力のデータを表わす図である。
【特許請求の範囲】
【請求項1】
式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis;N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;X6は、Arg、Cys、Cys(mPEG)、又はCys(コレステリル)であり;
X6は、Arg、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、カルボン酸(COOH)、カルボキサミド、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]
を包含するポリペプチド、又は医薬的に許容されるその塩。
【請求項2】
X3、X4、X6−X8、及びX10−X14の1個以上が、Cys(mPEG)又はCys(コレステリル)である、請求項1記載のポリペプチド。
【請求項3】
Cys(mPEG)がC1、C2、C3又はC6であり、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、各々が、側鎖チオールを介し、およそ示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する、請求項1記載のポリペプチド。
【請求項4】
式:
HxSQGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、Hxは、His、イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H)、アセチルHis、ピログルタミルHis、N−メチル−His(Me−H)、N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H)、ベンジルHis(Bzl−H)及びPheからなる群より選択される]からなる、請求項1記載のポリペプチド。
【請求項5】
式:
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg、Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含する、請求項1記載のポリペプチド。
【請求項6】
式:
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含する、請求項1記載のポリペプチド。
【請求項7】
式:
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、そして
X3、X4、X6−X8、及びX10−X14の1又は2個が、Cys(mPEG)又はCys(コレステリル)である]を包含する、請求項1記載のポリペプチド。
【請求項8】
式:
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Cys(N−エチルマレイミジル)、Q又はCであり;
X17は、m又はMであり;
X18は、アミド化されたk又はKである]を包含するポリペプチド、又は医薬的に許容されるその塩。
【請求項9】
式:
HαQGTFTSDYSKYLDSRRAQDFVQWLANTKRNRNNIAC3−CONH2;
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
HαQGTFTSDYSKYLDSRRAQDFVQWLVNTKRNRNNIAC3−CONH2;
HαQGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3−
CONH2;
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH;
HαQGTFTSDYSKYLDSRRAQDFVQWLXNTKRNRNNIAC3−
CONH2;
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3;及び
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、C3=Cys[(mPEG)240kDa]であり、側鎖チオールを介し、示されたMWの分枝鎖mPEG[(mPEG)2]によりPEG化された、アミド化されたシステイン残基に相当し;αは、α−アミノイソ酪酸(aib)であり;そしてm=メチオニンスルホキシド[Met(O)]であり;X19は、C又はC8(Cys(N−エチルマレイミジル)である]
からなる群より選択されるポリペプチド、及び医薬的に許容されるその塩。
【請求項10】
配列番号1−159のいずれか1つからなる群より選択されるアミノ酸配列を包含するポリペプチド、及び医薬的に許容されるその塩。
【請求項11】
請求項1記載のポリペプチドを患者へ投与するステップを含む、それを必要とする患者における代謝性疾患の治療方法。
【請求項12】
請求項1記載のポリペプチドと、医薬的に好適な担体とを包含する医薬組成物。
【請求項13】
請求項1記載のポリペプチドを包含する、グルコースレベルを低下させるための薬剤。
【請求項14】
請求項1記載のポリペプチドを投与するステップを包含する、グルコースレベルを低下させる方法。
【請求項1】
式:
HxX1X2GTFTSDYX3X4YLDX5X6X6AX7X8FVX7WLX9X10X11KRNRNNX12X13X14
[式中、Hxは、His;イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H);アセチルHis;ピログルタミルHis;N−メチル−His(Me−H);N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H);ベンジルHis(Bzl−H);及びPheからなる群より選択され;
X1は、Ser;Gly;Ala;Arg;Asn;Asp;Glu;Gln;His;Ile;Lys;Met;Phe;Pro;Thr;Trp;Tyr;Val;D−Ala;D−Ser;及びα−アミノイソ酪酸からなる群より選択され;
X2は、Gln、Asp、Glu、Pro、Leu又はL−ノルロイシンであり;
X3は、Ser、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys,Cys(mPEG)又はCys(コレステリル)であり;
X5は、Ser又はAlaであり;X6は、Arg、Cys、Cys(mPEG)、又はCys(コレステリル)であり;
X6は、Arg、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X9は、Met、Met(O)、Val、ノルロイシン、アラニン、α−アミノイソ酪酸又はO−メチル−ホモセリンであり;
X10は、Asn、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、カルボン酸(COOH)、カルボキサミド、第二級アミド、Ala、K(パルミトイル)、Cys、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーである]
を包含するポリペプチド、又は医薬的に許容されるその塩。
【請求項2】
X3、X4、X6−X8、及びX10−X14の1個以上が、Cys(mPEG)又はCys(コレステリル)である、請求項1記載のポリペプチド。
【請求項3】
Cys(mPEG)がC1、C2、C3又はC6であり、ここで、C1=Cys(mPEG)5kDa、C2=Cys(mPEG)20kDa、C3=Cys(mPEG)240kDa、C6=Cys(MPEG)260kDaであり、各々が、側鎖チオールを介し、およそ示されたMWの直鎖メトキシPEG(mPEG)又は分枝鎖mPEG2によりPEG化されたシステイン残基に相当する、請求項1記載のポリペプチド。
【請求項4】
式:
HxSQGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、Hxは、His、イミダゾール−乳酸(ImiH);デスアミノ−His(ΔNH2−H)、アセチルHis、ピログルタミルHis、N−メチル−His(Me−H)、N,N−ジメチル−His(Me2−H);ベンゾイルHis(Bz−H)、ベンジルHis(Bzl−H)及びPheからなる群より選択される]からなる、請求項1記載のポリペプチド。
【請求項5】
式:
HX1QGTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X1は、Ser、Gly、Ala、Arg、Asn、Asp、Glu、Gln、His、Ile、Lys、Met、Phe、Pro、Thr、Trp、Tyr、Val、D−Ala、D−Ser及びα−アミノイソ酪酸からなる群より選択される]を包含する、請求項1記載のポリペプチド。
【請求項6】
式:
HSX2GTFTSDYSKYLDSRRAQDFVQWLMNTKRNRNNIA
[式中、X2は、Gln、Asp、Glu、Pro、Leu及びL−ノルロイシンからなる群より選択される]を包含する、請求項1記載のポリペプチド。
【請求項7】
式:
HSQGTFTSDYX3X4YLDSX6X6AX7X8FVX7WLMX10X11KRNRNNX12X13X14
[式中、X3は、Ser、Ala、Cys(mPEG)又はCys(コレステリル)であり;
X4は、Lys、Cys(mPEG)又はCys(コレステリル)であり;
X6は、Arg、Cys(mPEG)又はCys(コレステリル)であり;
X7は、Gln、Cys(mPEG)又はCys(コレステリル)であり;
X8は、Asp、Cys(mPEG)又はCys(コレステリル)であり;
X10は、Asn、Cys(mPEG)又はCys(コレステリル)であり;
X11は、Thr、Cys(mPEG)又はCys(コレステリル)であり;
X12は、Ile、Cys(mPEG)又はCys(コレステリル)であり;
X13は、Ala、Cys(mPEG)又はCys(コレステリル)であり;そして
X14は、アミド、カルボキシレート、第二級アミド、Ala、K(パルミトイル)、Cys(mPEG)、Cys(コレステリル)、又は、mPEG又はコレステロールが化学結合により結合した任意のリンカーであり、そして
X3、X4、X6−X8、及びX10−X14の1又は2個が、Cys(mPEG)又はCys(コレステリル)である]を包含する、請求項1記載のポリペプチド。
【請求項8】
式:
HαX15GTFTSDYSKYLDSZZAX16DFVQWLX17NTX18
[式中、X15は、D又はQであり;
Zは、任意のアミノ酸であり;
X16は、C8、Cys(N−エチルマレイミジル)、Q又はCであり;
X17は、m又はMであり;
X18は、アミド化されたk又はKである]を包含するポリペプチド、又は医薬的に許容されるその塩。
【請求項9】
式:
HαQGTFTSDYSKYLDSRRAQDFVQWLANTKRNRNNIAC3−CONH2;
HαDGTFTSDYSKYLDS−TtdsEC−CONH2
HαQGTFTSDYSKYLDSRRAQDFVQWLVNTKRNRNNIAC3−CONH2;
HαQGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3−
CONH2;
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIA−Ttds−EEEEEC−COOH;
HαQGTFTSDYSKYLDSRRAQDFVQWLXNTKRNRNNIAC3−
CONH2;
HαDGTFTSDYSKYLDSRRAQDFVQWLmNTKRNRNNIAC3;及び
HαDGTFTSDYSKYDSZZAQDFVQWLmNTKRNRNNIAX19
[式中、C3=Cys[(mPEG)240kDa]であり、側鎖チオールを介し、示されたMWの分枝鎖mPEG[(mPEG)2]によりPEG化された、アミド化されたシステイン残基に相当し;αは、α−アミノイソ酪酸(aib)であり;そしてm=メチオニンスルホキシド[Met(O)]であり;X19は、C又はC8(Cys(N−エチルマレイミジル)である]
からなる群より選択されるポリペプチド、及び医薬的に許容されるその塩。
【請求項10】
配列番号1−159のいずれか1つからなる群より選択されるアミノ酸配列を包含するポリペプチド、及び医薬的に許容されるその塩。
【請求項11】
請求項1記載のポリペプチドを患者へ投与するステップを含む、それを必要とする患者における代謝性疾患の治療方法。
【請求項12】
請求項1記載のポリペプチドと、医薬的に好適な担体とを包含する医薬組成物。
【請求項13】
請求項1記載のポリペプチドを包含する、グルコースレベルを低下させるための薬剤。
【請求項14】
請求項1記載のポリペプチドを投与するステップを包含する、グルコースレベルを低下させる方法。
【図1】

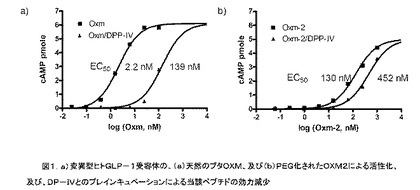
【図2】
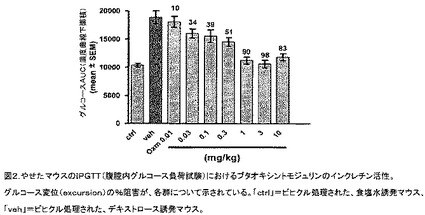
【図3】
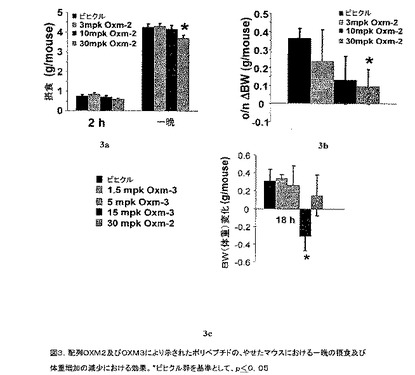
【図4】
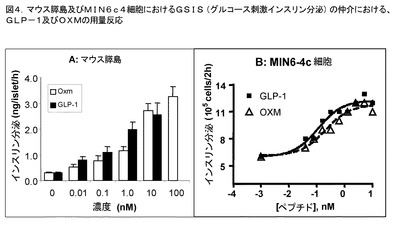
【図5】

【図6】
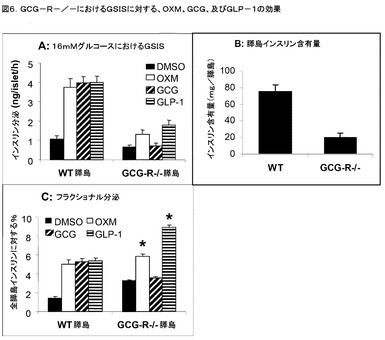
【図7】
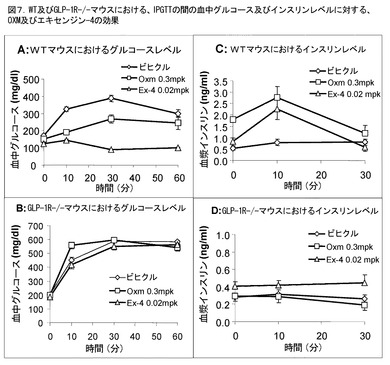
【図8】
【図9】
【図10】
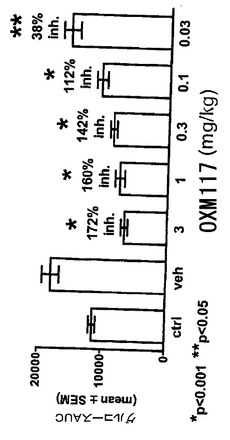
【図11】
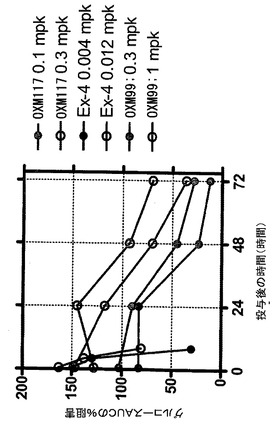
【図12】

【図13】
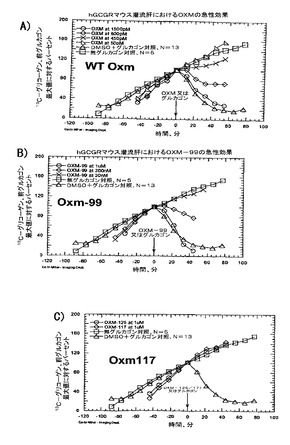
【図14】
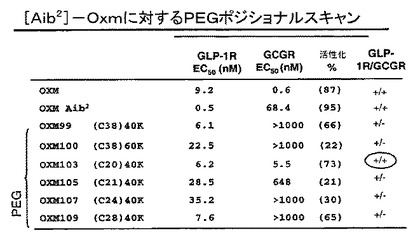
【図15】
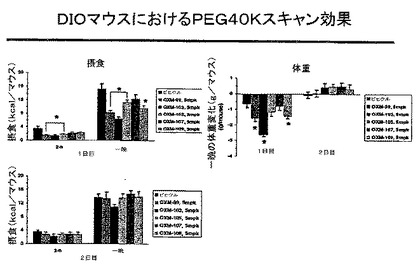
【図16】
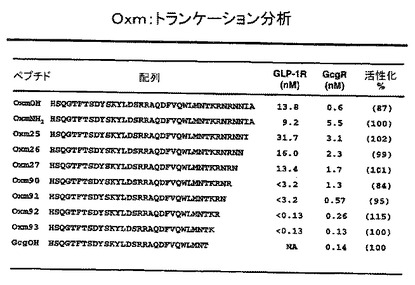
【図17】
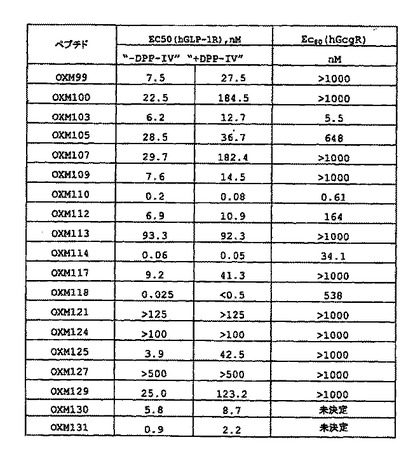
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公表番号】特表2009−527558(P2009−527558A)
【公表日】平成21年7月30日(2009.7.30)
【国際特許分類】
【出願番号】特願2008−556379(P2008−556379)
【出願日】平成19年2月16日(2007.2.16)
【国際出願番号】PCT/US2007/004306
【国際公開番号】WO2007/100535
【国際公開日】平成19年9月7日(2007.9.7)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
【公表日】平成21年7月30日(2009.7.30)
【国際特許分類】
【出願日】平成19年2月16日(2007.2.16)
【国際出願番号】PCT/US2007/004306
【国際公開番号】WO2007/100535
【国際公開日】平成19年9月7日(2007.9.7)
【出願人】(390023526)メルク エンド カムパニー インコーポレーテッド (924)
【氏名又は名称原語表記】MERCK & COMPANY INCOPORATED
【出願人】(501209427)イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー (90)
【Fターム(参考)】
[ Back to top ]