
カルジオトロフィンの幹細胞増殖調節への使用
細胞をカルジオトロフィン−1(CT−1)と接触させることを含む幹細胞増殖促進方法を提供する。本方法は、さらに幹細胞を、幹細胞の分化を促進する一つ以上の幹細胞モジュレーターと接触させることを含んでもよい。また、細胞を一つ以上のCT−1インヒビターと接触させることを含む幹細胞増殖抑制方法も提供する。本方法は、幹細胞増殖をインビトロ及びインビボで促進あるいは抑制するのに使用できる。また、損傷や障害のある組織の交換、あるいは不適当な細胞増殖の抑制における本方法の治療的応用も提供する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、幹細胞治療の分野に関係し、特に幹細胞の増殖調節方法に関係する。
【背景技術】
【0002】
幹細胞とは、様々な特定のタイプの細胞を作り出し、究極的には最終的な分化細胞を作り出す能力を持つ未分化の、あるいは未成熟の細胞である。幹細胞は、他の細胞と異なり、基本的に必要な時に成熟細胞を無限に供給するように自己更新することができる。この自己更新能があることにより、幹細胞は組織の再生及び修復において治療的に有用である。
【0003】
幹細胞は、様々な臨床設定において恩恵を与える可能性を持つ。多くの可能性のある応用への制限は、十分な数の標的細胞を得ること、また、これら幹細胞の成熟した組織特異的細胞への最終的な分化を刺激することであった。
【0004】
成人の骨髄及び多くの他の体細胞組織は、様々な表現型の細胞に分化することができる多能性幹細胞群を含むと考えられている。例えば、成体マウスの心臓内の多能性幹細胞様の細胞群(SP)が最近報告された(Hierlihy, A.M., et al., (2002) FEBS Letters, 530:239-243)。減衰した成長の環境下で、これら細胞は活性化し、心筋細胞に分化する。心臓組織あるいは他の組織に由来する体細胞性幹細胞の投与、及び幹細胞因子(SCF)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、間質細胞由来ファクター−1、スチールファクター、血管内皮成長因子、マクロファージコロニー刺激因子、顆粒球マクロファージ刺激因子あるいはインターロイキン−3などのサイトカインの投与による、損傷した心筋の修復あるいは再生方法が最近開示されている(米国特許出願第20030054973号)。
【0005】
サイトカインは、生物内の細胞間の伝達において重要な役割を果たすこと、ならびに成長及び分化を調節することができる細胞メディエーターとして作用することが知られている。サイトカインを幹細胞の分化促進に使用することが開示されている。例えば、米国特許出願第20030027330号には、幹細胞を成長中あるいは成長した同種あるいは異種の細胞とともに、さらに任意的にはサイトカインや成長因子あるいはケモカインも一緒に共培養することにより、幹細胞から分化した哺乳類細胞あるいは組織を生成する方法が記載されている。米国特許出願第20030103951号には、間葉幹細胞の投与により心筋を再生する方法が記載され、これは、サイトカイン、成長因子、筋原性因子及び転写因子などの分化に重要なタンパクを生成するように遺伝学的に修飾されてもよい。米国特許出願第20020142457号には、心筋細胞に分化する可能性を持つ細胞を増殖させる方法、及び様々なサイトカイン及び転写因子を用いてそれらの心筋細胞への分化を調節する方法が記載されている。
【0006】
カルジオトロフィン−1(CT−1)は、IL−6ファミリーサイトカインの一つであり、比較的心臓に限定された方法で発現する。ヒトとマウスの両方のCT−1をエンコードする遺伝子がクローニングされている(Pennica, D., et al., (1996) Cytokine, 8:183-189; Pennica, D., et al., (1995) Proc. Natl. Acad. Sci. USA, 92:1142-1146)。元々は心臓肥大因子(CHF)として知られていたように、CT−1は心臓肥大の誘導を示し、CT−1及びそのアンタゴニストの心不全、不整脈疾患、変力性疾患あるいは末梢神経障害における使用が記載されており(米国特許第5,534,615号;5,571,675号;5,571,893号;5,624,806号及び5,679,545号参照)、ガンの診断及び治療における使用も記載されている(米国特許出願第20020146707号)。虚血前にCT−1を投与することにより、損傷から成体ラットの心臓を保護することも報告されている(Liao、Z.、et al.、(2002) Cardiovasc. Res.、53:902-910)。
【0007】
この背景情報は、出願人が本発明と関連の可能性があると考える公知情報を作成することを目的として提供するものである。いかなる前記情報も本発明に対して従来技術を構成すると必ずしも自認することを意図するものではなく、また解釈されるべきではない。
【0008】
本発明の目的は、従来技術の欠点を解決することである。
メインクレームの特徴の組み合わせは前記目的に合致し、サブクレームは本発明のさらに有利な実施形態を開示する。
【発明の開示】
【0009】
本発明は、幹細胞治療の分野、特に幹細胞の増殖調節方法に関係する。
【0010】
本発明に従って、幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドと接触させることを含む、幹細胞の増殖促進方法が提供される。
【0011】
別の実施形態においては、どのような形でも限定することを意味するものではないが、幹細胞を一つ以上のカルジオトロフィン−1インヒビターと接触させることを含む、幹細胞の増殖抑制方法が提供される。
【0012】
本発明の別の態様に従って、幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチド、及び一つ以上の幹細胞モジュレーターと接触させることを含み、前記一つ以上の幹細胞モジュレーターが、ポリペプチドあるいはポリペプチドをエンコードするポリヌクレオチドである幹細胞の増殖及び分化の促進方法が提供される。
【0013】
本発明の別の態様に従って、カルジオトロフィン−1の類似体、誘導体、変異体又は活性フラグメントであって、幹細胞の増殖促進能力がある、分離されたポリペプチド、及び該ポリペプチドをエンコードするポリヌクレオチド、及び該ポリヌクレオチドを含むベクターが提供される。
【0014】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を投与することを含む、哺乳類の心臓幹細胞増殖の促進方法が提供される。
【0015】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1インヒビターの有効量を投与することを含む、哺乳類の心臓幹細胞増殖の抑制方法が提供される。
【0016】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を投与することを含み、前記ポリペプチドが心臓幹細胞の増殖を促進する能力があるものである、哺乳類の心臓組織の修復又は再生方法が提供される。
【0017】
本発明のこの要約は、必ずしも発明の全ての必要な特徴を記述しておらず、本発明は、記載された特徴のサブコンビネーションに存在してもよい。
これら及び他の本発明の特徴は、添付の図面を参照した下記の記載からより明らかになるであろう。
【好ましい実施形態の開示】
【0018】
以下の記載は実施例による好適な実施形態の一例に過ぎず、本発明の効果を発揮するために必要な特徴の組み合わせを限定するものではない。
【0019】
本発明は、カルジオトロフィン(CT−1)の活性を調節することにより、幹細胞の増殖を調節する方法を提供する。従って、本発明は、CT−1、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは内因性のCT−1を活性化する化合物に幹細胞を接触させることを含む、幹細胞の増殖、分化あるいはその両方を促進する方法を提供する。また、本発明は、幹細胞を、CT−1インヒビターと接触させることを含む幹細胞の増殖、分化あるいはその両方を抑制する方法も提供する。幹細胞の増殖促進方法は、幹細胞の増殖及び/又は分化を促進するために、幹細胞を一つ以上の幹細胞モジュレーターに接触させることをさらに含んでもよい。本発明のこの実施形態に従って、細胞をCT−1(あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいはCT−1アクティベーター)と一つ以上の幹細胞モジュレーターとに同時に接触させてもよく、あるいは順次接触させてもよい。
【0020】
本方法は、インビトロで幹細胞増殖、幹細胞分化あるいはその両方を促進するために用いてもよい。あるいは、幹細胞増殖、幹細胞分化、あるいはその両方をインビボで促進するために用いてもよい。また、本発明はインビトロ及びインビボで行うことができる方法も意図する。本発明の治療的応用は幹細胞増殖及び/又は分化を促進する必要がある疾病及び疾患、例えば、損傷したあるいは障害のある組織の置換に限定されることを意味しない。ここで提供される方法は、治療方法あるいは予防方法として用いてもよい。また、本発明の方法は幹細胞増殖を抑制するために採用してよく、従って、不適当な細胞増殖により特徴付けられる疾患、例えば、これに限定されるものではないが心臓肥大などの治療における適用がある。
【0021】
定義
他に定義されていない限り、ここで用いる全ての技術的、科学的用語は、本発明が関係する分野の通常の技術者により一般に理解されるのと同じ意味を有する。
【0022】
ここで用いる「幹細胞モジュレーター」との用語は、幹細胞の増殖、分化、あるいは増殖と分化の両方を刺激又は抑制する能力を持つ化合物をいう。
【0023】
ここで用いる「幹細胞」との用語は、一つ以上の分化した細胞型に分化する能力を持つ細胞を言う。幹細胞は、全能性のあるいは多能性の細胞であってよい。全能性幹細胞は、典型的にどのような細胞型にも発展する能力を持つ。全能性幹細胞は、通常胚に由来する。多能性細胞は、典型的には幾つかの異なる、最終的に分化した細胞型に分化する能力を有する幹細胞系の細胞である。多能性幹細胞は様々な組織あるいは器官系を起源とすることができ、これに限定されるものではないが、血液、神経、心筋、骨格筋、皮膚、腸、骨、腎臓、肝臓、膵臓、胸腺などが挙げられる。
【0024】
ここで用いる「前駆細胞」との用語は、特定の細胞系列に決定された細胞であって、一連の細胞分裂により分化細胞型の特に制限された範囲を作り出す細胞を言う。前駆細胞の例として筋芽細胞が挙げられ、これは唯一つの細胞型に分化する能力を持つが、それ自体は、完全に成熟しておらず、あるいは完全に分化していない。
【0025】
ここで細胞に関して同義的に用いられている「増殖」及び「拡張」との用語は、分裂による同型の細胞の数の増加を言う。
【0026】
ここで用いる「分化」との用語は、それによって細胞が特定機能に特殊化する発展過程を言い、例えば、細胞が最初の細胞型とは異なった一つ以上の形態学的特徴及び/又は機能を獲得する過程をいう。「分化」という用語は、系統義務及び最終分化の両方のプロセスを含む。分化は、例えば、免疫組織化学あるいは当該分野の技術者に知られた他の手法を用いて、系列マーカーの有無をモニターすることにより評価することができる。前駆細胞に由来する分化した子孫細胞は、必ずしもそうである必要はないが、幹細胞の起源組織と同じ胚葉あるいは組織に関係していてよい。例えば、神経前駆細胞及び筋前駆細胞は、造血細胞系列に分化することができる。
【0027】
ここで同義的に用いる「系列決定」及び「特殊化」との用語は、幹細胞が経験するプロセスであり、幹細胞が、分化細胞型の特に制限された範囲を形成するよう決められた前駆細胞を生じるプロセスを言う。約束された前駆細胞は、しばしば自己再生あるいは細胞分裂する能力がある。
【0028】
ここで用いる「最終分化」との用語は、成熟した完全に分化した細胞への細胞の最後の分化を言う。例えば、神経前駆細胞及び筋前駆細胞は、造血細胞系列へ分化することができ、その最終分化は、特定の細胞型の成熟した血液細胞を導く。通常、最終分化は、細胞周期及び増殖休止からの離脱に関係する。
【0029】
ここで用いる「自然発生」との用語は、ポリペプチドあるいはポリヌクレオチドに適用されるように、そのポリペプチドあるいはポリヌクレオチドを自然界で見い出すことができる事実を言う。例えば、生物に存在し、自然界の起源から分離することができ、実験室でヒトにより故意に修飾されたことのないポリペプチドあるいはポリヌクレオチドの配列は、自然発生である。
【0030】
カルジオトロフィン−1(CT−1)を用いた幹細胞増殖調節方法
本発明は、CT−1を用いた幹細胞の増殖、分化、あるいは増殖と分化の両方を促進する方法を提供する。CT−1は、単独で、あるいは一つ以上の幹細胞モジュレーターと組み合わせて、幹細胞の増殖及び/又は分化の促進のために用いてよい。CT−1、及び任意成分としての一つ以上の幹細胞モジュレーターは、幹細胞に直接的に与えてもよく、あるいは、例えばこれに限定されないが、CT−1及び/又は一つ以上のモジュレーターを発現可能な細胞と共培養することなどにより間接的に与えることもできる。また、本発明の方法においては、幹細胞あるいは幹細胞と共培養された細胞の内因性CT−1を活性化する化合物の使用も意図する。
【0031】
本発明は、さらに、CT−1の一つ以上のインヒビターを用いて幹細胞増殖を抑制する方法も提供する。CT−1インヒビターは、幹細胞に直接的に与えることができ、あるいは、例えばこれに限定されるものではないが、一つ以上のインヒビターを発現し、好ましくは分泌する他の細胞と共培養することなどにより間接的に与えることができる。
【0032】
胚幹細胞及び成体幹細胞の両方、あるいはそれらを組み合わせて本発明の方法に用いてもよい。幹細胞は、何れの細胞型にも発展する能力を有する全能性幹細胞であってよく、あるいは、特定の組織あるいは器官、例えば、血液、神経、骨格筋、心筋、骨髄、皮膚、腸、骨、腎臓、肝臓、膵臓、胸腺などに由来する多能性幹細胞であってもよい。
【0033】
一つの実施形態において、本発明の方法は、成体幹細胞に適用される。別の実施形態においては、本発明の方法は心筋幹細胞、骨格筋幹細胞、あるいはその両方を用いる。
【0034】
I.CT−1
本発明に従って、CT−1は哺乳類のCT−1(心臓肥大因子、あるいはCHFとしても知られている)を言う。多くのカルジオトロフィンタンパクが当該分野において知られており、例えば、これに限定されるものではないが、マウスCT−1(Pennica, D., et al., Proc. Natl. Acad. Sci. USA, 92:1142-1146)及びヒトCT−1(Pennica, D., et al., (1996) Cytokine, 8:183-189)がある。
【0035】
CT−1は、ポリペプチドとして与えてもよく、あるいはそのポリペプチドをエンコードし、且つ発現することのできるポリヌクレオチドとして与えてもよい。様々なCT−1タンパクの配列が当該分野において知られており、CT−1ポリペプチドの調製の基礎として使用することができる(例えば、前記参照文献ならびにGenBank Accession Nos AAC52173及びNP_ 031821 (マウス); AAD12173及びAAA85229 (ヒト)により提供される)。
【0036】
また、本発明は、自然発生型(野生型)CT−1のポリペプチド類似体、誘導体及び変異体も意図するものであり、また、CT−1の活性ペプチドフラグメント、及びそのペプチドフラグメントの類似体、変異体及びペプチド模倣体も意図するものである。
【0037】
活性フラグメントは、野生型タンパクと実質的に同じ活性を保持する自然発生(野生型)タンパクのフラグメントである。フラグメントは、通常少なくとも約20のアミノ酸長さである。本発明の一つの実施形態において、フラグメントは少なくとも約50のアミノ酸長さである。別の実施形態においては、フラグメントは少なくとも約70のアミノ酸長さである。さらなる実施形態においては、フラグメントは少なくとも約100のアミノ酸長さである。そしてさらに別の実施形態では、フラグメントは少なくとも約150のアミノ酸長さである。「フラグメント」との用語は、野生型の配列のN末端、C末端、あるいはN−、C−両末端から1〜約50アミノ酸が欠損した野生型タンパクに対応するポリペプチドも包含する。候補フラグメントは、自然発生タンパクから生じたランダムフラグメントから選択することができ、あるいは、特に設計することもできる。フラグメントの活性を試験して、野生型タンパクと比較し、自然発生タンパクと実質的に同じ活性を持つフラグメントを選択する。ポリペプチドフラグメントの生成方法は、当該分野においてよく知られており、野生型タンパクあるいはその組み換え体の酵素的、化学的、あるいは機械的切断、そのようなフラグメントをエンコードする核酸の発現などがある。
【0038】
当該分野において知られているように、ポリペプチドの類似体及び誘導体、ならびにペプチド模倣化合物は、自然発生体よりも大きな利点を有していてもよく、例えば、より高い化学的安定性や、増強されたタンパク分解抵抗性、強化された薬理学的特性(例えば、半減期、吸収、作用強度、有効性など)、変化した特異性(例えば、生物学的活性の広域性)、及び/又は減少した抗原性などが挙げられる。
【0039】
「誘導体」は、通常は自然発生の配列の一部ではない化学的あるいは生化学的な追加部分を含むポリペプチドあるいはペプチドである。誘導体は、アミノ末端及び/又はカルボキシ末端及び/又は一つ以上のアミノ酸側鎖が適当な化学的置換基で誘導体化されているポリペプチド及びペプチドを含み、また、環状、2重ならびに多量体のポリペプチド及びペプチド、他のタンパクあるいは担体に融合したポリペプチド及びペプチド、グリコシル化あるいはリン酸化したポリペプチド及びペプチド、親油性部分(例えば、カプロイル、ラウリル、ステアロイル部分)に結合したポリペプチド及びペプチド、ならびに抗体あるいは他の生物学的リガンドに結合したポリペプチド及びペプチドも含む。
【0040】
ポリペプチド及びペプチドの誘導体化に用いることができる化学的置換基の例としては、これらに限定されるものではないが、アルキル基、シクロアルキル基及びアリール基;アルカノイル基やアロイル基を含むアシル基;エステル;アミド;ハロゲン;ヒドロキシル;カルバミルなどがある。また、置換基は、Fmoc(フルオレニルメチル−O−CO−)、カルボベンゾキシ(ベンジル−O−CO−)、モノメトキシスクシニル、ナフチル−NH−CO−、アセチルアミノ−カプロイル及びアダマンチル−NH−CO−などの保護基であってもよい。他の誘導体としては、C−末端ヒドロキシメチル誘導体、O−修飾誘導体(例えば、C−末端ヒドロキシメチルベンジルエーテル)及びアルキルアミドやヒドラジドのような置換アミドなどのN−末端修飾誘導体が挙げられる。
【0041】
「環状」ポリペプチドあるいはペプチドとの用語は、例えば、環化に適当な2以上の追加アミノ酸残基が付加したポリペプチドあるいはペプチドなどの環状誘導体を言う。これらの追加アミノ酸は、カルボキシル末端及びアミノ末端に付加してよく、あるいは、内部の位置にあってもよい。あるいは、環状ポリペプチド/ペプチドは、アミノ酸配列中に自然に生じるシステイン残基を利用してジスルフィド結合を形成し、これにより、ポリペプチド/ペプチドを環化してもよい。環状ポリペプチド/ペプチドは、分子内ジスルフィド結合、すなわち、−S−S−;2つの付加残基の間の分子内アミド結合、すなわち、−CONH−あるいは−NHCO−;あるいは分子内S−アルキル結合、すなわち、−S−(CH2)−CONH−あるいは−NH−CO(CH2)nS−、nは1、2、あるいはそれ以上、の何れかを含むことができる。
【0042】
分子内ジスルフィド結合を含む環状ポリペプチド/ペプチドは、従来の固相合成法により、環化のために選択された位置で適当なS−保護システインあるいはホモシステイン残基を合体させながら調製してもよい(例えば、Sahm et al., 1996, J. Pharm. Pharmacol. 48:197参照)。鎖組み立て完了後、遊離の対応するSH−官能基の支持体上の酸化を結果として生じるS−保護基の選択的な除去によりS−S結合を形成し、次いで支持体から生成物を従来通り離し、適当な方法で精製することにより環化を行うことができ、あるいは完全な側鎖脱保護と共にポリペプチド/ペプチドを支持体から離し、次いで遊離のSH−官能基を高希釈水溶液中で酸化することにより、環化を行うことができる。同様に、分子内アミド結合を有する環状誘導体は、従来の固相合成法により、環化のために選択された位置で適当なアミノ側鎖及びカルボキシル側鎖保護アミノ酸誘導体を合体させながら調製してよく、また、分子内−S−アルキル結合を有する環状ポリペプチド/ペプチドは、従来の固相合成法により、環化のために選択された位置でアミノ酸残基を適当なアミノ保護側鎖及び適当なS−保護システインあるいはホモシステイン残基と合体させながら調製することができる。
【0043】
2重ポリペプチド/ペプチドは、直接的に、あるいはアラニン残基やタンパク分解の推定部位の短い範囲のようなスペーサーを介して、互いに共有結合した2つの同じあるいは2つの異なるポリペプチド/ペプチドからなる(例えば、米国特許第5,126,249号及び欧州特許第495,049号参照)。多量体は、多くの同じあるいは異なるポリペプチド/ペプチドあるいはその誘導体から形成された高分子である。重合は、0.1%グルタルアルデヒドのような適当な重合試薬で行われる(例えば、Audibert et al., 1981, Nature 289:593参照)。
【0044】
「類似体」は、一つ以上の非自然発生のアミノ酸を含むポリペプチド/ペプチドである。例えば、本発明のポリペプチド/ペプチド類似体は、対応するD−アミノ酸残基により置換された、あるいは別の非自然発生アミノ酸で置換された一つ以上のアミノ酸残基を有してよい。非自然発生アミノ酸の例としては、これに限定されるものではないが、N−α−メチルアミノ酸、C−α−メチルアミノ酸、β−メチルアミノ酸、β−アラニン(β−Ala)、ノルバリン(Nva)、ノルロイシン(Nle)、4−アミノ酪酸(γ−Abu)、2−アミノイソ酪酸(Aib)、6−アミノヘキサン酸(ε−Ahx)、オルニチン(orn)、ヒドロキシプロリン(Hyp)、サルコシン、シトルリン、システイン酸、シクロヘキシルアラニン、α−アミノイソ酪酸、t−ブチルグリシン、t−ブチルアラニン、3−アミノプロピオン酸、2,3−ジアミノプロピオン酸(2,3−diaP)、フェニルグリシン、2−ナフチルアラニン(2−Nal)、1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Tic)、β−2−チエニルアラニン(Thi)、メチオニンスルホキシド(MSO)及びホモアルギニン(Har)などが挙げられる。
【0045】
当該分野において知られているように、ペプチド内の全てのL−アミノ酸を全てD−アミノ酸に置換することにより、「インベルソ」(inverso)ペプチドあるいは「レトロインベルソ」(retro-inverso)ペプチドとすることができ(Goodman et al. "Perspectives in Peptide Chemistry" pp. 283-294 (1981); 米国特許第4,522,752号参照)、これらは両方とも本発明との関係において類似体と見なされる。「インベルソ」ペプチドは、配列のL−アミノ酸全てがD−アミノ酸で置換されたものであり、「レトロインベルソ」ペプチドは、アミノ酸の配列が逆転し(「レトロ」)、且つ全てのL−アミノ酸がD−アミノ酸で置換されたものである。例えば、親ペプチドがThr−Ala−Tyrであれば、そのレトロ体はTyr−Ala−Thrであり、インベルソ体はthr−ala−tyrであり、レトロインベルソ体はtyr−ala−thrである(小文字はD−アミノ酸を示す)。親ペプチドに対して、レトロインベルソペプチドは側鎖の実質的にオリジナルな空間的配置を保有しながら逆転した主鎖を持つので、親ペプチドに非常に似たトポロジーのイソマーである。
【0046】
ペプチド模倣体は、ポリペプチド/ペプチドと構造的に同じで、かつ本発明のポリペプチドあるいはペプチドの機能を模倣した化学的部位を含む化合物である。例えば、ポリペプチドが機能的活性を有する2つの帯電した化学的部位を有する場合、模倣体は、帯電した化学的機能が3次元空間内に維持されるように、空間的定位及び強制的構造内に2つの帯電した化学的部位を持つ。従って、ペプチド模倣体との用語は、アイソスターを含むことを意図する。ここで用いる「アイソスター」との用語は、化学構造の立体配置がペプチドあるいはポリペプチドと同じであるために、そのポリペプチドあるいはペプチドと置換可能な化学構造を言い、例えば、その構造はポリペプチドあるいはペプチドに特異的な結合部位に適合する。ペプチド模倣体の例としては、一つ以上の主鎖修飾を含むペプチド(すなわち、アミド結合模倣体)あり、これは当該分野においてよく知られている。アミド結合模倣体の例としては、これに限定されるものではないが、−CH2NH−、−CH2S−、−CH2CH2−、−CH=CH−(シス及びトランス)、−COCH2−、−CH(OH)CH2−、及び−CH2SO−が挙げられる(例えば、Spatola, Vega Data Vol. 1, Issue 3, (1983); Spatola, in Chemistry and Biochemistry of Amino Acids Peptides and Proteins, Weinstein, ed., Marcel Dekker, New York, p. 267 (1983); Morley, J. S., Trends Pharm. Sci. pp. 463-468 (1980); Hudson et al., Int. J. Pept. Prot. Res. 14:177-185 (1979); Spatola et al., Life Sci. 38:1243-1249 (1986); Hann, J. Chem. Soc. Perkin Trans. I 307-314 (1982); Almquist et al., J. Med. Chem. 23:1392-1398 (1980); Jennings-White et al., Tetrahedron Lett. 23:2533 (1982); Szelke et al., EP 45665 (1982); Holladay et al., Tetrahedron Lett. 24:4401-4404 (1983);及びHruby, Life Sci. 31:189-199 (1982)参照)。ペプチド模倣体の他の例としては、一つ以上のベンゾジアゼピン分子で置換されたペプチド(例えば、James, G. L. et al. (1993) Science 260:1937-1942参照)、及び架橋してラクタムあるいは他の環状構造を形成した主鎖を含むペプチドが挙げられる。
【0047】
当該分野の技術者は、ペプチドあるいはポリペプチドの全てのアミノ酸が修飾される必要はないということを理解するであろう。同様に、全てのアミノ酸が同じ方法で修飾される必要はない。このように本発明のポリペプチド/ペプチド誘導体、類似体及びペプチド模倣体は、2つ以上の化学的に別個の領域を有するキメラ分子を含み、それぞれの領域は少なくとも一つのアミノ酸あるいはその修飾体を含む。
【0048】
変異体のポリペプチドあるいはペプチドは、一つ以上のアミノ酸残基が削除され、付加され、あるいは自然発生タンパクのアミノ酸配列にあるアミノ酸残基が置換されたものである。本発明に関連して、変異体はまた、自然発生タンパクと実質的に同じ活性を保持している。通常、変異体が一つ以上のアミノ酸置換を有する場合、それらは「同類」置換である。同類置換は、一つのアミノ酸残基を同様の側鎖特性を有する別の残基で置き換えることを伴う。当該分野において知られているように、20の自然発生アミノ酸はその側鎖の物理化学的性質によって分類することができる。適当な分類としては、アラニン、バリン、ロイシン、イソロイシン、プロリン、メチオニン、フェニルアラニン及びトリプトファン(疎水性側鎖);グリシン、セリン、スレオニン、システイン、チロシン、アスパラギン、及びグルタミン(極性非帯電側鎖);アスパラギン酸及びグルタミン酸(酸性側鎖)及びロイシン、アルギニン及びヒスチジン(塩基性側鎖)がある。アミノ酸の別の分類はフェニルアラニン、トリプトファン、及びチロシン(芳香族側鎖)である。同類置換は、アミノ酸を同じグループの別のアミノ酸で置換することを包含する。
【0049】
本発明に従って、類似体、誘導体、変異体あるいは活性フラグメントは、自然発生CT−1タンパクに比べて実質的に同じかあるいは向上した活性を有する。「実質的に同等の活性」との用語は、自然発生のCT−1タンパクの活性の約50%の活性を示す。一つの実施形態においては、実質的に同等な活性は自然発生CT−1タンパクの活性の約60%の活性を示す。別の実施形態においては、それは自然発生CT−1 タンパクの活性の約75%の活性を示す。また別の実施形態においては、類似体、誘導体、変異体あるいは活性フラグメントは自然発生のCT−1タンパク、好ましくはヒトCT−1タンパクに比べて高められた(増加した)活性を示す。CT−1の活性は、例えば、その幹細胞増殖促進能力を測定することにより決定できる。本発明の一つの実施形態においては、CT−1の活性は、その心臓幹細胞増殖促進能力を言う。幹細胞増殖の増加を測定する方法は、当該分野において知られており、ここで提供されるものを含む。
【0050】
本発明のポリペプチドは、細胞抽出物からの精製あるいは組み換え技術の使用など、当該分野で公知の方法により調製することができる。また、短鎖の配列は、当該分野で公知の方法により化学的に合成することもでき、これに限定されるものではないが、排他的固相合成法、部分的固相合成法、フラグメント縮合あるいは古典的液相合成などが挙げられる(Merrifield (1963) J. Am. Chem. Soc. 85:2149; Merrifield (1986) Science 232:341)。本発明のポリペプチドは、クロマトグラフィー(例えば、イオン交換、アフィニティ、及びサイジングカラムクロマトグラフィーあるいは高速液体クロマトグラフィー)、遠心分離、溶解度差などの標準的技術を用いて、あるいは当該分野の技術者によく知られた他の技術によって、精製することができる。
【0051】
また、ポリペプチドは、組み換え技術によって得ることもできる。通常これは、タンパクあるいはポリペプチドをエンコードするポリヌクレオチドを含む発現ベクターでの、適当な宿主細胞の形質転換(形質移入、形質導入、あるいは感染を含む)を含む。様々なCT−1遺伝子の核酸配列が、当該分野において公知であり(例えば、Pennica et al. Ibid.、米国特許第5,723,585号、5,679,545号、5,627,073号(マウス) 及び GenBank Accession Nos. Q16619及びNM_001330 (ヒト))、本発明のポリヌクレオチドの基礎として使用してよい。
【0052】
ポリヌクレオチドは、標準的技術により適当な起源から誘導あるいは精製することができる。ポリヌクレオチドは、ゲノムDNAあるいはRNAであることができ、また分離したmRNAから調製したcDNAであることができる。あるいは、標準的技術を用いて様々な起源からCT−1ポリペプチドをエンコードする他の核酸配列を得るために、公知の配列を用いてプローブを調製してもよい。核酸を得るための適当な起源は、心筋細胞などの本発明のタンパクを発現することが知られている細胞であり、また、骨格筋組織及び測定可能なCT−1転写産物を持つ他の組織である。
【0053】
自然発生のCT−1タンパクのフラグメントあるいは変異体をエンコードするポリヌクレオチドは、部位特異的突然変異誘発技術などの標準的技術を用い、コード配列内で一つ以上のヌクレオチドの置換により、削除、付加、及び/又は構築することができる。
【0054】
また、本発明のポリペプチド及びペプチドは、融合タンパクとして得ることもできる。このような融合タンパクの用途の一つは、ポリペプチドあるいはペプチドの精製あるいは検出を改善することである。例えば、ポリペプチドあるいはペプチドは、免疫グロブリンのFcドメインに融合することができ、得られた融合タンパクは、タンパクAカラムを用いて容易に精製することができる。融合タンパクの他の例としては、ヒスチジンタグに融合したポリペプチドあるいはペプチド(Nie+樹脂カラムで精製可能)、グルタチオン−S−トランスフェラーゼに融合したポリペプチドあるいはペプチド(グルタチオンカラムで精製可能)あるいはビオチンに融合したポリペプチドあるいはペプチド(ストレプトアビジンカラムあるいはストレプトアビジン標識マグネティックビーズで精製可能)が挙げられる。融合タンパクを一旦精製した後、当該分野で知られている化学的あるいは酵素的方法を用いた部位特異的切断により、タグを除去してもよい。
【0055】
特定の開始シグナルがクローニングされたポリヌクレオチドの効率的な翻訳に要求されるかもしれない。これらのシグナルは、ATG開始コドン及び隣接配列を含む。それ自体の開始コドン及び隣接配列を含む完全な野生型遺伝子あるいはcDNAを適当な発現ベクターに挿入する場合、追加の翻訳コントロールシグナルは必要でないかもしれない、別のケースでは、おそらくATG開始コドンを含む外因性の翻訳コントロールシグナルが与えられなければならない。さらに、その開始コドンは、挿入部分の完全な翻訳を確実にするために、目的とするコード配列の読み取り枠と一致している必要がある。外因性の翻訳コントロールシグナル及び開始コドンは、天然あるいは合成であることができる。発現の効率を、適当な転写促進因子要素及び/又は転写終止区の包有により高めてもよい(Bittner et al. (1987) Methods in Enzymol. 153, 516)。
【0056】
本発明の核酸配列と用いる適当な発現ベクターは、これに限定されるものではないが、プラスミド、ファージミド、ウィルス粒子及びベクター、ファージなどが挙げられる。昆虫細胞としては、バクロウィルス発現ベクターが適当である。植物細胞としては、ウィルス発現ベクター(例えば、カリフラワーモザイクウィルス及びタバコモザイクウィルスなど)及びプラスミド発現ベクター(例えば、Tiプラスミドなど)が適当である。完全な発現ベクター、あるいはその一部は、宿主細胞ゲノムに組み込むことができる。ある状況下では、当該分野において公知の誘発性発現ベクターを用いることが望ましい。
【0057】
分子生物学の分野の技術者は、組み換えポリペプチドあるいはペプチドを得るために、広範な様々の発現システムを用いることができるということを理解するであろう。使用される厳密な宿主細胞は、本発明には重要ではない。ポリペプチドあるいはペプチドは、原核宿主中 (例えば、E. coliあるいはB. subtilis)あるいは真核宿主中(例えば、SaccharomycesあるいはPichia;COS、NIH 3T3、CHO、BHK、293、あるいはHeLa細胞などの哺乳類細胞;昆虫細胞;あるいは植物細胞)で生成できる。形質転換あるいは形質移入の方法及び発現ベクターの選択は、選択した宿主系に依存し、当該分野の技術者により容易に決定することができる。形質転換及び形質移入の方法は、例えば、Ausubel et al. (1994) Current Protocols in Molecular Biology, John Wiley & Sons、New Yorkに記載されている;また様々な発現ベクターは、例えば、Cloning Vectors: A Laboratory Manual (Pouwels et al., 1985, Supp. 1987)及び様々な販売業者により提供されるもののなかから選択することができる。
【0058】
また、宿主細胞は、挿入された配列の発現を調節するもの、あるいは特定の所望のやり方で遺伝子産物を修飾及び処理するものを選択してよい。そのようなタンパク産物の修飾(例えば、グリコシル化)及び処理(例えば、切断)は、タンパクの活性にとって重要であるかもしれない。種々の宿主細胞は、タンパク及び遺伝子産物の翻訳後処理及び修飾のための特徴的な特定のメカニズムを有する。発現した異種タンパクの正確な修飾及び処理を確実にするために、適当な細胞系あるいは宿主系を当該分野の技術者により選択することができる。
【0059】
発現ビヒクルを抱いた宿主細胞は、選択された遺伝子の活性化、選択された遺伝子の抑制、形質転換体の選択、あるいは選択された遺伝子の増幅の必要性に応じて、公知の方法に従って、従来の栄養培地中で培養することができる。
【0060】
II.CT−1を活性化する化合物
本発明は、CT−1を活性化し、内因性CT−1を増加させるあるいは内因性CT−1の活性を増加させる化合物を用いて、幹細胞増殖を促進する方法も意図する。CT−1アクティベーターは、インビボでCT−1の上流に作用してCT−1の発現あるいは活性を上方制御するポリペプチドあるいはポリペプチドをエンコードする遺伝子であってよく、あるいは小さな分子アクティベーターでもよい。CT−1アクティベーターは、遺伝子レベルで作用して、例えばCT−1をエンコードする遺伝子の発現を上昇させてよく、あるいはタンパクレベルで作用してCT−1ポリペプチドの活性を増加させるあるいはCT−1インヒビターの活性を減少させてもよい。CT−1アクティベーターは、例えば、ポリペプチド及びペプチド(あるいは、前記のようなポリペプチドに対応する類似体、誘導体、変異体あるいはペプチド模倣化合物)、ポリヌクレオチド、オリゴヌクレオチド、抗体あるいは抗体フラグメント、あるいは有機又は無機の小分子であることができる。
【0061】
候補アクティベーターを同定するために、当該分野において知られている様々なスクリーニング方法を採用することができる。例えば、標的遺伝子を上方制御あるいは下方制御するアクティベーターは、候補アクティベーターで処理した細胞の標的遺伝子の発現増加あるいは減少をモニターすることにより同定できる。ノーザンブロット分析、定量的RT−PCRあるいはマイクロアレイ解析などの方法を、この目的のために用いることができる。あるいは、例えば、対応するタンパクレベルの増加あるいは減少を、ウエスタンブロット分析によりモニターすることができる。
【0062】
特定のタンパクに結合するポリペプチドあるいはペプチドアクティベーター(あるいはポリペプチドに対応する類似体、誘導体、変異体あるいはペプチド模倣化合物)について、その結合能を当該分野において知られている様々な結合分析の一つを用いて決定することができる(例えば、Coligan et al., (eds.) Current Protocols in Protein Science, J. Wiley & Sons, New York, NY参照)。
【0063】
抗体あるいは抗体フラグメントアクティベーターについて、様々な免疫学的測定法を用いることができる。確立された特異性を持つポリクロナール抗体あるいはモノクロナール抗体の何れかを用いた、競合結合分析あるいは免疫放射定量測定法の多数のプロトコルが、当該分野においてよく知られている。そのような免疫学的検定法は、通常、標的タンパクとその特異的抗体との複合体形成の測定を含む。そのような技術の例としては、ELISAs、放射線免疫検定法(RIAs)、及び蛍光活性化細胞分類(FACS)などが挙げられる。あるいは、2つの非干渉エピトープに反応性を持つモノクローナル抗体を利用したツーサイドモノクローナルベース免疫学的検定法、あるいは競合結合分析を用いることができる (Maddox, D. E. et al. (1983) J. Exp. Med. 158:1211-1216参照)。そのような分析法は当該分野においてよく知られている(例えば、Hampton, R. et al. (1990) Serological Methods: A Laboratory Manual, APS Press, St Paul, Minn., Section IV; Coligan, J. E. et al. (1997, and periodic supplements) Current Protcols in Immunology, Wiley & Sons, New York, N.Y.; Maddox, D. E. et al. (1983) J. Exp. Med. 158:1211-1216参照)。
【0064】
III.CT−1インヒビター
本発明は、CT−1を抑制し、その結果内因性CT−1を減少させる、あるいは内因性CT−1活性を減少させる化合物を用いた幹細胞増殖抑制方法も意図する。CT−1インヒビターは、インビボでCT−1の上流に作用してCT−1の発現あるいは活性を上方制御するポリペプチドあるいはポリペプチドをエンコードする遺伝子であってよく、あるいは小分子のインヒビターであってもよい。CT−1インヒビターは、遺伝子レベルで作用し、例えば、CT−1をエンコードする遺伝子の発現を下方制御してよく、あるいはタンパクレベルで作用してCT−1ポリペプチドの活性を減少させてもよい。CT−1インヒビターは、例えば、野生型タンパクの活性を妨げるCT−1の不活性フラグメントなどを含むポリペプチド及びペプチド(あるいは、前記のようなポリペプチドに対応する類似体、誘導体、変異体又はペプチド模倣化合物)、ポリヌクレオチド、オリゴヌクレオチド、抗体あるいは抗体フラグメント、あるいは有機又は無機の小分子であることができる。
【0065】
CT−1アクティベーターの同定のための前記記載の当該分野で公知の様々なスクリーニング方法は、候補インヒビターの同定に採用することもできる。
【0066】
IV.幹細胞モジュレーター
また、本発明の方法は、CT−1(あるいはCT−1アクティベーター)に加えて、一つ以上の幹細胞モジュレーターの使用も意図する。幹細胞モジュレーターは、幹細胞の増殖及び/又は分化を促進してよい。従って、モジュレーターは、CT−1の活性の増加、及び幹細胞群の増殖増加のために使用することができ、あるいはCT−1での処理により予め拡張した幹細胞群の分化を促進するのに使用してもよい。幹細胞分化を促進するモジュレーターは、細胞の系統決定を刺激してよく、あるいは系列決定された前駆細胞の最終分化を刺激してもよい。
【0067】
本発明の方法において用いることができるモジュレーターの例としては、これに限定されるものではないが、ポリペプチドWntファミリーのメンバー(Wnt1、Wnt2、Wnt3、Wnt4、Wnt5a、Wnt5b、Wnt7a及びWnt7b、及びマウスWnt1、Wnt2、Wnt3a、Wnt3b、Wnt4、Wnt5a、Wnt5b、Wnt6、Wnt7a、Wnt7b、Wnt8a、Wnt8b、Wnt10a、Wnt10b、Wnt11及びWnt12などがある)及びPax7が挙げられる。一つ以上の転写因子、例えば、これに限定されるものではないがPax7、NKX2.5、GATA4、5あるいは6、MEF2C、Handl及びHand2(心筋幹細胞に対して)及びMyoD、ミオゲニン、MRF4及びMyf5(骨格筋幹細胞に対して)を、単独であるいはWnt、Pax7、あるいはその両方と組み合わせて最終分化を促進するために用いてもよい。
【0068】
モジュレーターは、完全長のポリペプチドとして、あるいは活性フラグメントあるいはその変異体(前述)として提供されてよく、あるいは完全長のポリペプチド、活性フラグメントあるいは変異体をエンコードし発現することができるポリヌクレオチドとして提供されてもよい。様々なWntタンパク、Pax7及び多くの転写因子のアミノ酸及び核酸配列が当該分野において知られている(例えば、GenBank Accession Nos. NM_002584及びNP_002575 (Pax7))。
【0069】
本発明の一つの実施形態においては、モジュレーターは幹細胞分化を促進する。別の実施形態においては、幹細胞モジュレーターはWntポリペプチドあるいはWntポリペプチドをエンコードするポリヌクレオチドである。さらなる実施形態においては、幹細胞モジュレーターはWnt11ポリペプチドあるいはWnt11ポリペプチドをエンコードするポリヌクレオチドである。また別の実施形態においては、幹細胞モジュレーターはPax7ポリペプチドあるいはPax7ポリペプチドをエンコードするポリヌクレオチドである。
【0070】
幹細胞増殖試験
CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの、単独でのあるいは幹細胞モジュレーターと組み合わせたときの幹細胞増殖促進能力は、標準的技術を用いてインビトロあるいはインビボで試験することができ、これに限定されるものではないが、ここで記載された技術が挙げられる。一つ以上のCT−1インヒビターによる幹細胞増殖の抑制も、インビトロあるいはインビボで測定することができる。
【0071】
I.インビトロ試験
通常、幹細胞は試験化合物の存在下及び非存在下で培養し、次いで細胞の少なくとも一つの増殖指標をモニターして試験化合物に暴露した細胞での増殖が刺激あるいは抑制されたか否かを決定する。あるいは、幹細胞群を「エジュケーター」細胞と共培養することができ、エジュケーター細胞を、試験化合物に暴露し、次いで幹細胞中少なくとも一つの増殖指標をモニターする。エジュケーター細胞は、共培養の前あるいは共培養中に試験化合物に暴露してよい。様々な組織に由来する成体幹細胞あるいは前駆細胞を用いることができる。例としては、これに限定されるものではないが、心筋、骨格筋、膵臓組織、神経組織、肝臓組織あるいは骨髄、造血細胞、筋芽細胞、肝細胞、胸腺細胞、心筋細胞などからの幹細胞が挙げられる。胚幹細胞を用いてもよい。
【0072】
幹細胞を培地中維持する方法は、当該分野において知られている(例えば、Madlambayan, G.J., et al., (2001) J. Hematother. Stem Cell Res. 10, 481-492; Hierlihy, A.M., et al., (2002) FEBS Lett. 530, 239-243; Asakura, A., et al., (2002) J Cell Biol. 159, 123-134参照)。幹細胞は、単一培養として単独で培養することができ、あるいはエジュケーター細胞と共培養することもできる。培養期間の前、間、あるいは後に、細胞群の同定や分離あるいは分析の成功に寄与するためのステップなど追加のステップをスクリーニング方法中に含んでもよい。例えば、幹細胞群を分離及び拡張するために、成長因子あるいは他の化合物を用いてよい。EGF及びFGFは、Gritti et al(J. Neurosci. (1999) 19:3287-3297)により記載のように神経幹細胞にこの目的のために使用されており、また、Bcl−2は「筋幹細胞」群の分離に用いられている (米国特許第6,337,184号参照)。
【0073】
一般に、化合物は通常約1000倍に及ぶ濃度範囲にわたって試験され、適当な暴露プロトコルは、当該分野の技術者によって容易に確立することができる。共培養を用いる場合、エジュケーター細胞への幹細胞の最初の暴露の前、間あるいは後に、化合物へ幹細胞を暴露することができる。あるいは、試験化合物がポリヌクレオチドあるいはポリヌクレオチドによりエンコードされたポリペプチドあるいはペプチドのような化合物である場合、試験化合物が内因的に生成されるように、ここで記載のあるいは他の標準的な方法を用いて、幹細胞をその核酸あるいはそのポリヌクレオチドを含む発現ベクターで形質移入することができる。さらに、ポリヌクレオチド、あるいはポリヌクレオチドを含む発現ベクターで形質移入された、試験化合物を発現する別の細胞系と幹細胞とを共培養することにより、幹細胞を試験化合物に暴露することができる。
【0074】
上記のように、幹細胞を化合物に直接的に暴露しなくてもよいこともさらに意図する。例えば、エジュケーター細胞群あるいは第3の細胞型をまず化合物と処理し、次いで幹細胞と共培養することができる。あるいは、幹細胞をそのようなただしそれ自身は共培養中に含まれない細胞群により調整された培地の添加によって間接的に暴露することができる。さらに、幹細胞を非液体培養培地中、例えば、寒天、高分子足場、マトリックスあるいは他の構成物のような固体、ゲル状あるいは半固体の成長支持体中に取り込まれた化合物に暴露してよいことも意図される。
【0075】
定性的あるいは定量的にモニターすることができる幹細胞増殖の指標としては、例えば、全体の形態の変化、総細胞数、組織学、組織化学あるいは免疫組織化学、あるいは特異的な細胞マーカー (例えば、増殖マーカーとして、サイクリンDl、ホスホ−ヒストンH1及びH3、E2F及びPCNA)の存在、非存在あるいは相対レベルなどが挙げられる。
【0076】
形態学及び/又は総細胞数の変化は、例えば、適当な染料(ヘキスト染料の一つなど)、BrdU取り込みを用いたFACS分析により、あるいはトリチウムチミジン取り込みにより、モニターできる。細胞マーカーの存在、非存在あるいは相対レベルは、例えば、組織化学的技術、免疫学的技術、電気泳動、ウエスタンブロット分析、FACS分析、フローサイトメトリーなどにより分析できる。あるいは、例えば、PCR技術、ノーザンブロット分析、適当なオリゴヌクレオチドプローブの使用などを用いて、細胞マーカータンパクをエンコードする遺伝子から発現したmRNAの存在を検出することができる。
【0077】
CT−1(あるいはCT−1アクティベーター)及び幹細胞モジュレーターの両方で処理されたこれら細胞について、一つ以上の分化指標をモジュレーターで処理した後の幹細胞群においてモニターしてもよい。通常、分化は前記のような全体の形態変化により、あるいは系統特異的な細胞マーカーの存在によりモニターされ、これは、前記のような多数の標準的技術を用いて分析できる。
【0078】
モニター可能な適当な系統特異的細胞マーカーは、当該分野において知られている。例えば、心筋幹細胞の分化の誘導は、コネキシン−43、MEF2C及びミオシン重鎖のような心筋細胞特異的マーカーの出現をモニターすることにより決定できる;筋由来幹細胞の分化の誘導は、ミオシン重鎖、次リン酸化MyoD、ミオゲニン、Myf5及びトロポニンTのような一つ以上の筋細胞マーカータンパクの発現について細胞を試験することにより測定でき、また、ニューロスフェアあるいはSP細胞フラクションとして誘導された神経幹細胞の分化の誘導は、GFAP、MAP2及びβ−IIIチューブリンの発現をモニターすることにより決定できる(例えば、Hitoshi, S., et al., (2002) Genes & Dev. 16, 846-858参照)。
【0079】
II.インビトロ試験
また、CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの幹細胞の増殖及び/又は分化の促進能力は、例えば、一つ以上の幹細胞モジュレーターと組み合わせて使用した場合に限らず、適当な試験動物中の常在幹細胞群についてインビボで試験してもよい。同様に、CT−1インヒビターの幹細胞増殖抑制能力をインビボで試験してもよい。通常、試験化合物は、試験組織に、例えば注射により直接的に投与される。適当な期間の後、細胞が動物から採取され、幹細胞群が前記のように分析される。必要であれば、インヒビターの増殖予防能あるいは低下能を決定するために、常在幹細胞をCT−1インヒビター及び増殖を刺激する化合物と接触させることによりインヒビターを試験することができる。化合物及びインヒビターは幹細胞に同時に与えてもよく、あるいはインヒビターの前あるいは後に化合物を与えてもよい。
【0080】
本発明の一つの実施形態においては、化合物の幹細胞増殖促進能力はマウスの心臓組織においてインビボで試験される。処理マウスから分離した心臓幹細胞様群(SP)の増加がモニターされ、非処理のあるいは緩衝液や生理食塩水などのプラセボ処理の何れかであるコントロールマウスの場合と比較される。本発明のこの実施形態に従って、SPが少なくとも約2倍まで増加した場合に、化合物が幹細胞増殖を促進すると見なされる。SPの増加は、少なくとも24時間、より典型的には少なくとも96時間にわたって測定される。
【0081】
CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの損傷組織の修復能力は、適当な動物モデルで試験できる。例えば、化合物の損傷心筋組織修復能力は、冠状動脈結紮誘導性心臓障害のマウスに化合物を投与し、損傷を受けた心筋の修復をモニターすることにより、試験することができる (Guo et al., (1999) Proc. Natl. Acad. Sci. USA, 96:11507参照)。同様に、化合物の骨格筋損傷修復能力は、凍結粉砕あるいはカルジオトキシン投与により誘導された筋損傷の動物において決定できる(Megeney et al., (1996) Genes Dev., 10:1173-1183; Asakura et al., (2000) J. Cell Biol., 159:123-134参照)。
【0082】
応用
本発明は、細胞を直接的あるいは間接的にCT−1、その類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーターと接触させることにより、幹細胞の増殖を促進する方法を提供する。本発明は、さらに、細胞を直接的あるいは間接的にCT−1(あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーター)及び一つ以上の幹細胞モジュレーターと接触させることにより、幹細胞の増殖及び分化を促進する方法を提供する。幹細胞増殖抑制方法は、細胞を直接的あるいは間接的に一つ以上のCT−1インヒビターと接触させることを含む。本発明により提供される方法には多くの応用がある。例えば、本方法は、幹細胞の増殖促進のためにインビトロで使用することができ、この場合細胞をさらにインビトロで使用するため、例えば、研究目的のために使用することを予定している。本方法は、インビトロで幹細胞培養を維持するのに使用でき、また、薬物試験のための新しいインビトロモデルの開発における応用の可能性もある。
【0083】
あるいは、増殖及び/又は分化の促進方法は、幹細胞のエクスビボでの拡張及び/又は分化を刺激するのに用いることもでき、これにより、それを必要とする対象への移植あるいは投与に適した細胞群を提供することができる。幹細胞のエクスビボ拡張は、多くの疾病状態の治療のために治療的に必要なものである。
【0084】
また、増殖及び/又は分化の促進方法は、組織の常在幹細胞の増殖及び/又は分化を促進するためにインビボで使用してもよく、これにより、疾病や疾患の結果の、あるいは外科手術や傷害の後の損傷組織の交換あるいは修復を助ける。
【0085】
同様に、一つ以上のCT−1インヒビターを用いた増殖抑制方法は、インビボでの幹細胞増殖を抑制するのに用いることができ、これにより、組織中の不適当な細胞増殖が抑制あるいは最小化される。
【0086】
また、幹細胞の増殖及び続く分化を促進する連続的な方法も意図される。例えば、細胞を直接的あるいは間接的にCT−1あるいはCT−1アクティベーターと接触させることにより、幹細胞群をエクスビボで拡張してよい。拡張した細胞群を、次いで対象に投し、元の位置で幹細胞の分化を促進する一つ以上の幹細胞モジュレーターとインビボで処理する。あるいは、対象への細胞の投与の前に、両方のステップをエクスビボで行ってもよい。
【0087】
インビボ及びエクスビボでの方法について、幹細胞は自家系、同種異型、あるいは異種系であることができる。
【0088】
幹細胞増殖(及び任意には分化)を促進する方法の治療的応用は、通常、失われたあるいは損傷した組織の交換が必要な状況に関連し、例えば、化学療法あるいは放射線治療の後、筋傷害の後、あるいは疾病及び疾患の治療中や管理中に関連している。例えば、本方法は、変性筋疾患、筋ジストロフィー、神経筋変性疾患、HIV感染などの治療、管理、あるいは予防における骨格筋幹細胞;パーキンソン病やアルツハイマー病などの神経変性疾患の治療、管理あるいは予防における神経幹細胞;及び、変性あるいは虚血性心疾患、動脈硬化、高血圧、再狭窄、狭心症、リウマチ性心疾患、先天性心臓血管異常、動脈炎及び動脈、細動脈及び毛細血管の他の疾患などの治療、管理あるいは予防や、弁、通道組織あるいは血管平滑筋の再生や、あるいは冠動脈バイパス移植を受けている対象のさらなる疾病の予防における心筋細胞に用いることができる。本発明の方法を用いて治療あるいは予防してよい他の疾病あるいは疾患としては、これに限定されるものではないが、肝硬変、肝炎、糖尿などの変性肝臓疾患が挙げられる。
【0089】
幹細胞増殖抑制方法の治療的応用は、不適当な細胞増殖を防止あるいは最小化する必要がある状況に常に関係するものではないが、典型的には、例えば心臓肥大の治療あるいは予防がある。
【0090】
本発明の一つの実施形態においては、本方法は、心臓幹細胞に適用される。別の実施形態においては、本方法は、成体心臓幹細胞に適用される。またさらに別の実施形態においては、いかなる方法でも限定することを意味するものではないが、本方法は骨格筋幹細胞に適用される。
【0091】
さらに、本発明は、CT−1、その類似体、誘導体、変異体あるいは活性フラグメント、CT−1アクティベーターあるいはCT−1インヒビター、及び薬学的に許容可能な希釈剤あるいは賦形剤を含む医薬組成物も提供する。本医薬組成物は、さらに一つ以上の幹細胞モジュレーター、一つ以上の幹細胞、あるいはそれらの組み合わせを任意に含んでもよい。医薬組成物及びその調製方法は、当該分野において知られており、例えば、"Remington: The Science and Practice of Pharmacy" (formerly "Remingtons Pharmaceutical Sciences"); Gennaro, A., Lippincott, Williams & Wilkins, Philadelphia, PA (2000)に記載されている。
【0092】
医薬組成物の投与は、局所的あるいは全身的治療のどちらを望むかによって、また治療する領域によって多くのルートにより行ってよい。通常、組成物は治療すべき領域に局所投与される。投与は、局所的 (眼投与ならびに膣送達及び直腸送達などの粘膜投与を含む)、経肺的 (例えば、ネブライザーによるものなど粉末のあるいはエアゾールの吸入あるいは吹送による)、気管内、鼻腔内、表皮的、経皮的、経口的あるいは非経口的であってよい。非経口的投与としては、静脈内、動脈内、皮下、腹腔内あるいは筋肉内注射が挙げられ、例えば、これに限定されるものではないが、心臓内注射あるいは注入、あるいは頭蓋骨内投与、例えば、クモ膜下腔内あるいは脳室内投与が挙げられる。
【0093】
本発明の組成物は、薬学的に許容可能なビヒクルと組み合わせて送達されてもよい。好ましくは、そのようなビヒクルが安定性及び/又は送達性を高める。例としては、リポソーム、微粒子あるいはマイクロカプセルなどが挙げられる。本発明の様々な実施形態において、そのようなビヒクルの使用は、活性化合物の徐放性を達成するのに有利であるかもしれない。
【0094】
非経口的注射剤として製剤化する場合、医薬組成物は、好ましくは、他の溶質、例えば溶液を等張にするための十分な生理食塩水あるいはグルコースなどを含む滅菌溶液の形態で使用される。
【0095】
吸入あるいは吹送による投与のために、医薬組成物は水性あるいは部分的に水性の溶液に製剤化することができ、これはその後エアゾールの形態で利用できる。局所的使用のために、モジュレーターあるいはこれを含む医薬組成物を、薬学的に許容可能なビヒクル中の粉剤、クリームあるいはローションとして製剤化でき、これは皮膚の疾患部分に適用される。
【0096】
医薬組成物の必要用量は、採用した特定の組成物、投与経路、及び特定の治療対象により変化する。必要用量は、当該分野の技術者に知られている標準的な臨床的技術により決定することができる。治療は、一般的に各化合物の適量よりも少ない少量で開始される。その後、その状況下で最適な効果に到達するまで用量が増量される。一般に、医薬組成物は、通常いかなる有害なあるいは悪影響のある副作用も引き起こすことなく有効な結果を与える濃度で投与される。投与は、1回ユニットの投与量とすることができ、あるいは、望むのであれば一日を通して適当な時に投与するのに便利なサブユニットに分けることができる。
【0097】
幹細胞のエクスビボでの処理方法を採用する場合、幹細胞は様々な方法により対象に投与できる。通常、幹細胞の投与は局所に制限される。幹細胞は、薬学的に許容可能な液媒体中の細胞懸濁液として注射により投与することができる。あるいは、幹細胞は、元の位置で半固体あるいは固体マトリックスである、あるいはそのようになる生体適合性媒体中投与することができる。例えば、マトリックスは、コラーゲン及び/又はその誘導体、ポリ乳酸あるいはポリグリコール酸を含むマトリックスのような組織損傷部位で半固体ゲルを形成するような注射可能な液体であってもよく、あるいは含浸繊維マトリックスのようなその最終形態において移植される柔軟な固体マトリックスの一つ以上の層を含んでもよい。そのようなマトリックスは当該分野において知られており(例えば、ゲルフォーム(Gelfoam)、Upjohn, Kalamazoo, Mich.から入手可能)、傷害部位の位置に細胞を保つ機能を有し、投与した細胞が増殖及び分化する機会を増加させる。
【0098】
遺伝子治療
また、本発明は、CT−1(あるいはその変異体又は活性フラグメント、あるいはCT−1アクティベーター)及び任意的に幹細胞モジュレーターをエンコードするポリヌクレオチドの投与も意図し、これはその後当該分野において知られた様々な「遺伝子治療」方法によりインビボでエンコードされた産物を発現する。CT−1の投与方法は、当該分野において知られており、例えば、運動神経変性を治療するために、CT−1はアデノウィルスに用られた(Lesbordes et al., (2003), Hum. Molec. Gen. 12, 1223-1229)。遺伝子治療は、エクスビボ及びインビボの両方の技術を含む。従って、宿主細胞は、ポリヌクレオチドでエクスビボで遺伝的に設計することができ、そして設計された細胞はその後前記のように治療すべき患者に投与される。
【0099】
あるいは、当該分野において知られている技術を用いてポリヌクレオチドを投与することにより、インビボで細胞を設計することができる。例えば、「裸の」ポリヌクレオチド(Feigner及びRhodes、(1991) Nature 349:351-352; 米国特許第5,679,647号)や、細胞のポリヌクレオチドの取り込みを容易にする一つ以上の他の作用物質、例えばサポニン(例えば、米国特許第5,739,118号参照)あるいはカチオン性ポリアミン(例えば、米国特許第5,837,533号参照)などを含む組成物中で処方されたポリヌクレオチドを直接注射することにより;微粒子の爆撃により(例えば、「遺伝子銃」; Biolistic, Dupontの使用により);脂質、細胞表面受容体あるいはトランスフェクト剤でポリヌクレオチドをコーティングすることにより;リポソーム、微粒子、あるいはマイクロカプセル中にポリヌクレオチドを内包させることにより;核に入りこむことが知られているペプチドと結合したポリヌクレオチドを投与することにより;あるいは受容体介在性エンドサイトーシスを受けやすいリガンドと結合したポリヌクレオチドを投与することにより(例えば、Wu and Wu, (1987) J. Biol. Chem. 262:4429-4432参照)、受容体を特異的に発現する標的細胞型に用いることができる。
【0100】
あるいは、リガンドがエンドソームを混乱させる融合ウィルスペプチドを含み、ポリヌクレオチドがリソソーム分解を避けるのを可能にするポリヌクレオチド−リガンド複合体;あるいは、そのポリヌクレオチドが特定の受容体を標的とすることによってインビボで細胞特異的な取り込み及び発現の標的となり得るポリヌクレオチド−リガンド複合体が形成できる(例えば、国際特許出願WO92/06180、WO92/22635、WO92/20316、WO93/14188及びWO 93/20221参照)。また、本発明は、ポリヌクレオチドの細胞内導入、及び続く相同的組み換えによる発現用宿主細胞DNA内への組み込みも意図する (例えば、Koller and Smithies (1989) Proc. Natl. Acad. Sci. USA 86:8932-8935; Zijlstra et al. (1989) Nature 342:435-438参照)。
【0101】
ポリヌクレオチドは、適当な発現ベクターに組み込むこともできる。遺伝子治療の応用に適当な多くのベクターが当該分野において知られている(例えば、Viral Vectors: Basic Science and Gene Therapy, Eaton Publishing Co. (2000)参照)。
【0102】
発現ベクターは、プラスミドベクターでもあってよい。プラスミドDNAを生成及び精製する方法は、迅速かつ簡単である。さらに、プラスミドDNAは、通常宿主細胞のゲノムに統合せず、染色体組み込みを高めるかもしれないという遺伝毒性の問題を除去する別個の存在としてエピソーム部位に維持される。
【0103】
様々なプラスミドが現在は容易に市販品を入手でき、Escherichia coli及びBacillus subtilisに由来し、哺乳類系での使用のために特に設計されたものがある。本発明で使用できるプラスミドの例としては、これに限定されるものではないが、真核発現ベクターpRc/CMV(Invitrogen)、pCR2.1(Invitrogen)、pAd/CMV及びpAd/TR5/GFPq(Massie et al., (1998) Cytotechnology 28:53-64)が挙げられる。代表的な実施形態において、プラスミドはpRc/CMV、pRc/CMV2(Invitrogen)、pAdCMV5(IRB-NRC)、pcDNA (Invitrogen)、pAdMLP5(IRB-NRC)、あるいはpVAX(Invitrogen)である。
【0104】
あるいは、発現ベクターは、ウィルスベースのベクターであることができる。ウィルスベースのベクターとしては、これに限定されるものではないが、複製欠損性レトロウィルス、レンチウィルス、アデノウィルス及びアデノ随伴ウィルス由来のものが挙げられる。レトロウィルスベクター及びアデノ随伴ウィルスベクターは、現在、インビボで外因性遺伝子を特にヒトへトランスファーするための好ましい組み換え遺伝子送達システムである。これらのベクターは、細胞に遺伝子を効果的に送達し、トランスファーされたポリヌクレオチドは、宿主の染色体DNAに安定に組み込まれる。レトロウィルスを使用する主要な必要条件は、特に細胞群中の野生型ウィルスの拡散の可能性に関して、それらの使用の安全性、確保することである。レトロウィルスベクターが由来するレトロウィルスとしては、これに限定されるものではないが、モロニーマウス白血病ウィルス、脾臓壊死ウィルス、ラウス肉腫ウィルスのようなレトロウィルス、ハーベイ肉腫ウィルス、トリ白血病ウィルス、テナガザル白血病ウィルス、ヒト免疫不全ウィルス、アデノウィルス、骨髄増殖性肉腫ウィルス、及び乳癌ウィルスなどが挙げられる。具体的なレトロウィルスとしては、pLJ、pZIP、pWE及びpEMが挙げられ、これらは当該分野の技術者によく知られている。
【0105】
ポリヌクレオチドは通常、インビボでのエンコードされたポリヌクレオチドの発現を可能にする適当なプロモーターのコントロール下で、ベクターに組み込まれる。採用することができる適当なプロモーターとしては、これに限定されるものではないが、アデノウィルスプロモーター、例えばアデノウィルス主要後期プロモーター、E1Aプロモーター、主要後期プロモーター(MLP)及び関連リーダー配列、あるいはE3プロモーターなど;サイトメガロウィルス(CMV)プロモーター;呼吸器合胞体ウィルス(RSV)プロモーター;誘発性プロモーター、例えばMMTプロモーター、メタロチオネインプロモーターなど;ヒートショックプロモーター;アルブミンプロモーター;ApoAIプロモーター;ヒトグロブリンプロモーター;ウィルスチミジンキナーゼプロモーター、例えば単純ヘルペスチミジンキナーゼプロモーターなど;レトロウィルスLTR;ヒストン、pol III、及び(β−アクチンプロモーター;B19パルボウィルスプロモーター;SV40プロモーター;及びヒト成長ホルモンプロモーターが挙げられる。また、プロモーターは、目的遺伝子の天然プロモーターであってもよい。適当なプロモーターの選択は、ベクター、宿主細胞及びエンコードされたタンパクに依存し、当該分野の通常の技術内とみされる。
【0106】
複製欠損レトロウィルスのみを産生する特殊化した細胞系(「パッケージング細胞」という)の開発は、遺伝子治療へのレトロウィルスの利用を増加させ、欠損レトロウィルスは遺伝子治療を目的とする遺伝子トランスファーにおける使用を特徴とする(参考として、Miller, A. D. (1990) Blood 76:271参照)。従って、組み換えレトロウィルスは、レトロウィルスコード配列(gag、pol、env)の一部を被験ポリヌクレオチドにより置換され、レトロウィルスを複製欠損にするように構築することができる。複製欠損レトロウィルスは、その後、標準的技術によってヘルパーウィルスを使用し、標的細胞に感染させるのに使用可能なウィルス粒子中にパッケージされる。組み換えレトロウィルスを得るためのプロトコル及び細胞にインビトロあるいはインビボでそのようなウィルスを感染させるプロトコルは、Current Protocols in Molecular Biology, Ausubel, F. M. et al. (eds.), J. Wiley & Sons, (1989), Sections 9.10-9.14及び他の標準的な実験マニュアル中に認められる。環境栄養性及び両栄養性レトロウィルス系の両方を調製するための適当なパッケージングウィルス系の例としては、Crip、Cre、2及びAmが挙げられる。他のパッケージング細胞の例としては、これに限定されるものではないが、PE501、PA317、Ψ−2、Ψ−AM、PA12、T19−14X、VT−19−17−H2、ΨCRE、ΨCRIP、GP+E−86、GP+envAml2、及びMiller, Human Gene Therapy, Vol. 1, pgs. 5-14 (1990)に記載されたDAN細胞系が挙げられる。
【0107】
さらに、ウィルス粒子の表面でウイルスパッケージングタンパクを修飾することにより、レトロウィルス及び結果としてのレトロウィルスベースベクターの感染スペクトルを、制限することができることが示されている(例えば、PCT公開公報WO93/25234及びWO94/06920参照)。例えば、レトロウィルスベクターの感染スペクトルの修飾方法としては次のようなものが挙げられる:細胞表面抗原に特異的な抗体をウィルス性envタンパクにカップリングする(Roux et al. (1989) PNAS 86:9079-9083; Julan et al. (1992) J. Gen Virol 73:3251-3255;及びGoud et al. (1983) Virology 163:251-254);あるいは細胞表面受容体リガンドをウィルス性envタンパクにカップリングする (Neda et al. (1991) J Biol Chem 266:14143-14146)。カップリングは、タンパクあるいは他の種類(例えば、envタンパクをアシアログリコプロテインに変換するための乳糖)と化学的に架橋した形態とすることができ、また、融合タンパクの生成によることもできる(例えば、単鎖抗体/env融合タンパク)。この技術は、感染をある組織型に制限するあるいは指示するのに有用であり、また、環境栄養性ベクターを両栄養性ベクターに変換するのに用いることもできる。
【0108】
加えて、レトロウィルス遺伝子の送達の使用は、ベクター中に含まれるポリヌクレオチドの発現をコントロールする組織特異的なあるいは細胞特異的な転写調節配列の使用によりさらに高めることができる。
【0109】
遺伝子治療技術に有用な別のウィルスベクターは、アデノウィルス由来のベクターである。アデノウィルスのゲノムは、目的遺伝子産物をエンコード及び発現するが、通常の溶菌ウィルスライフサイクルにおけるその複製能力は不活化されるよう操作することができる。例えば、Berkner et al. (1988) BioTechniques 6:616; Rosenfeld et al. (1991) Science 252:431-434;及びRosenfeld et al. (1992) Cell 68:143-155参照。アデノウィルス株Ad type 5 d1324あるいは他のアデノウィルス株(例えば、Ad2、Ad3、Adzなど)に由来する適当なアデノウィルスベクターは当該分野の技術者によく知られている。組み換えアデノウィルスは、広い多様な細胞型、例えば末梢神経細胞などへの感染に使用できるようなある種の条件において有利であり得る。さらに、ウィルス粒子は比較的安定で、精製及び濃縮でき、前記のように感染性スペクトルに影響を及ぼすように修飾することができる。さらに、導入されたアデノウィルスDNA(及びそこに含まれる外来DNA)は、宿主細胞のゲノムに組み込まれるのではなく、エピソームにとどまり、これにより、導入されたDNAが宿主ゲノム(例えば、レトロウィルスDNA)に組み込まれるようになるような状況において挿入突然変異の結果として生じ得る潜在的な問題を回避する。さらに、外来DNAに対するアデノウィルスゲノムの運搬量(8キロベースまで)は他の遺伝子送達ベクターに比べて大きい(Berkner et al. cited supra; Haj-Ahmand and Graham (1986) J. Virol. 57:267)。現在使用され、また本発明により意図されるほとんどの複製欠損アデノウィルスベクターは、ウィルス性E2及びE3遺伝子の全てあるいは一部が削除されているが、アデノウィルスの遺伝物質の80%と同じ程度を維持している(例えば、Jones et al. (1979) Cell 16:683; 前記Berkner et al.;及びGraham et al. in Methods in Molecular Biology, E. J. Murray, Ed. (Humana, Clifton, N.J., 1991) vol. 7. pp. 109-127参照)。
【0110】
複製欠損ヒトアデノウィルスベクターの作成及び増殖には、特有のヘルパー細胞系が要求される。ヘルパー細胞系は、ヒト胚腎臓細胞、筋細胞、造血細胞あるいは他のヒト胚間葉細胞あるいは上皮細胞などのヒト細胞由来であってよい。あるいは、ヘルパー細胞は、ヒトアデノウィルスに許容される、すなわち、複製欠損性ウィルスの複製を可能にするのに必要な配列を与える他の哺乳動物種の細胞に由来してもよい。そのような細胞としては、例えば、293細胞、ベロ細胞あるいは他のサル胚間葉細胞あるいは上皮細胞が挙げられる。また、ブタあるいはウシアデノウィルスベクターのような非ヒトアデノウィルスベクターの使用も意図される。適当なウィルスベクター及びヘルパー細胞系の選択は、当該分野の一般的な技術内である。
【0111】
本発明の一つの実施形態においては、遺伝子治療ベクターはアデノウィルス由来ベクターである。
【0112】
キット
さらに、本発明は、CT−1、その類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーター、及び任意的に一つ以上の幹細胞モジュレーターを医薬組成物中に含む治療キットを提供する。一つ以上のCT−1インヒビターを医薬組成物中に含有するキットも提供される。キットの個々の成分は別々の容器に包装され、そのような容器とともに、医薬品あるいは生物学的製品の製造、使用又は販売を規制する行政機関指定の様式の通知を有することがあり、通知はヒト投与のために製造、使用あるいは販売する機関による認可を反映する。
【0113】
キットの成分が一つ以上の液体溶液で与えられる場合、液体溶液は水溶液、例えば、滅菌水溶液であることができる。この場合、容器手段はそれ自体が吸入器、注射器、ピペット、点眼ビン、あるいは組成物を患者にそこから投与してもよい他のそのような器具であってもよい。
【0114】
キットの成分は、乾燥状態あるいは凍結乾燥状態で提供されてもよく、また、キットはさらに凍結乾燥された成分の再溶解のために適当な溶媒を含むことができる。容器の数あるいはタイプに関係なく、本発明のキットは、患者への組成物の投与を補助するための器具を含んでいてもよい。そのような器具は、吸入器、注射器、ピペット、ピンセット、計量スプーン、点眼ビンあるいは医療的に認められているそのようないかなる送達ビヒクルでもよい。
【0115】
結果
適当なヌクレオチド構築物で形質転換された細胞から生成及び分泌されたCT−1は、幹細胞の増殖及び/又は分化を促進する。ここで図3及び図4を参照すると、心臓内注射後24、48、72及び96時間でのマウス心臓細胞におけるCT−1の効果を表す経時変化の結果が示されている。このデータは、SP細胞がCT−1治療に反応して増殖することを示唆する。従って、本発明は、CT−1を含む組成物あるいはCT−1を生成することができる組成物を対象に投与することを含む、対象の幹細胞の増殖促進方法を提供する。
【0116】
前記方法の一つの実施形態において、幹細胞は心臓細胞、さらに好ましくは心臓SP細胞を含む。しかしながら、幹細胞が他の流体、組織及び/又は器官に由来する幹細胞、例えば、これに限定されるものではないが、骨格筋幹細胞、骨髄幹細胞、肝臓幹細胞などに由来する幹細胞を含んでもよいことも意図される。本発明の別の実施形態においては、いかなる方法でも限定することを意味しないが、 幹細胞が骨格筋幹細胞を含んでよい。好ましくは幹細胞はヒト幹細胞である。
【0117】
CT−1を含むあるいはエンコードする組成物は、当該分野において公知の何れの経路でも対象に投与してよく、例えば、これに限定されるものではないが注射あるいは注入により投与してよい。いかなる方法でも限定することを望むものではないが、そのような注射経路としては、これに限定されるものではないが、筋肉内、皮下、腹腔内、静脈内、動脈内注射などが挙げられる。本発明の一つの実施形態において、組成物は筋肉内注射により投与される。さらなる本発明の実施形態においては、組成物は心臓内注射あるいは注入により投与される。
【0118】
前述のように、図3及び図4に示される結果は、CT−1が細胞、例えば、これに限定されるものではないがそれで処理された対象の幹細胞の増殖を促進することを示す。CT−1を発現及び分泌する細胞は、対象へのCT−1の連続的投与が可能であるが、ここで得られた結果は、CT−1の連続的投与後にSP細胞群の一時的な増加があることを示す。さらに、この結果は、SP細胞の総数が、CT−1処理後約24時間から約96時間の間にわたって、コントロールに比べて増加することを示す。これらの結果は、幹細胞がCT−1処理に非反応性である期間を回避するために、CT−1が反復投与されることを示唆する。さらに、CT−1の反復投与は、CT−1の単独投与あるいは連続投与に比較して、幹細胞のより大きな増殖及び/又は分化を促進するかしれない。また、ここで記載したような間隔をあけた反復投与は、例えば約24時間間隔の反復投与に比べて幹細胞のより大きな増殖及び/又は分化を促進するかもしれない。
【0119】
本発明は、CT−1を含む2以上の組成物を対象に投与することを含む、対象の幹細胞の増殖促進方法を提供する。好ましくは、対象へ投与する最初及び2回目の投与の間の時間、ならびに任意的にCT−1の何れの2回の投与の間の時間は、約24時間より長く、より好ましくは少なくとも約48時間、さらにより好ましくは少なくとも約72時間、もっとより好ましくは少なくとも約96時間あるいはそれ以上である。また、本発明は、対象にCT−1を2回以上投与することも意図し、この場合、何れの2回の投与の間の時間は、約2日、3日、4日、5日、6日、7日、10日、14日、21日、28日、1ヶ月、2ヶ月、3ヶ月、6ヶ月、1年、あるいはその中間の何れかの時間である。2回以上の投与は、例えば、CT−1の量、剤型、組成物中の成分、あるいはこれらの組み合わせにおいて同じであっても、あるいは異なっていてもよい。また、本発明は、対象にCT−1を含む3、4、5、6、7、あるいはそれ以上の組成物を投与することも意図する。
【0120】
本発明は、次のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法も提供する:
a)化合物を試験群に属する一つ以上の対象に投与する;
b)その一つ以上の対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離する;
c)複数の細胞をFACS分析し、一つ以上の確定した特徴を有する細胞の数を定量する;
d)その一つ以上の確定した特徴を含む細胞の数を一つ以上のコントロール対象から得られた結果と比較する。
【0121】
細胞は、一つの細胞型、あるいは多数の細胞型の組み合わせを含んでもよい。一つの実施形態において、細胞は幹細胞を含む。別の実施形態においては、細胞は心臓細胞、あるいは骨格筋細胞を含む。また別の実施形態においては、細胞は心臓幹細胞を含む。さらに別の実施形態においては、細胞は心臓SP細胞を含む。
【0122】
分析ステップ(ステップc)は、さらに細胞を分離することを含んでよいことも意図される。
【0123】
別の実施形態において、投与ステップ(ステップa)は、一つ以上の化合物を投与することを含んでもよく、その場合、少なくとも一つの化合物はCT−1ポリペプチドあるいはCT−1ポリペプチドと実質的に同じ生物学的活性を発揮する化合物であり、一つ以上の化合物がさらに少なくとも一つの幹細胞モジュレーターを含んでよく、例えば、これに限定されるものではないが、一つ以上のシグナル伝達分子、例えばこれに限定されるものではないが一つ以上のWntポリペプチド、Pax7、あるいはその両方を含んでよい。
【0124】
別の実施形態においては、次のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法が提供される:
a)一つ以上の試験対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離し、さらに任意的に一つ以上の特定の細胞群を濃縮するためにその複数の細胞を精製する;
b)その複数の細胞あるいは一つ以上の濃縮された細胞群を化合物と処理する;
c)細胞あるいは細胞群を増殖及び/又は分化するのに十分な時間培養する、及び;
d)培養ステップの結果得られたコロニー細胞の数を、一つ以上のコントロールから得られた結果と比較する。
【0125】
比較ステップ(ステップd)は、一つ以上の特定の細胞群、例えば、これに限定されるものではないが、SP細胞を同定するために、細胞をFACS分析することをさらに含んでもよい。
【0126】
上記説明は、いかなる方法でもクレームされた発明を限定することを意図せず、さらには、検討された特徴の組み合わせは、本発明の問題解決に絶対的に必要ではないかもしれない。
【0127】
本発明は、さらに以下の実施例において説明される。ただし、これらの実施例は単に説明の目的のものであることが理解されるべきであり、いかなる方法によっても本発明の範囲を制限するのに用いられるべきではない。
【実施例】
【0128】
実施例1:カルジオトロフィン−1が心臓幹細胞様群(SP)を増加させる
偏在するRSVプロモーターにより誘導された完全長CT−1を含むアデノウィルス構築物を構築した(ad−CT−1)(図1)。CT−1は、細胞からのCT−1の分泌を促進するように神経増殖シグナルに融合した。7.5×107PFUのad−CT−1を2ヶ月齢のマウスの心臓に注射した。50μLの滅菌PBSを用いて並列コントロール実験も行った。注射後72時間に細胞を分離しFACS分析した。図2は、ヘキスト33342染料で染色し、ヘキスト33342HSC染色及び幹細胞精製プロトコル(Goodell, M., et al. (1996) J Exp Med 183, 1797-806参照、これは参照することによりここに折り込まれるものである)に従って分析した心臓細胞懸濁液のFACS分析を表す。細胞は、3つの心臓から採集した。生理食塩水を注射した心臓は、約0.93%のSP群を示し(図2A)、カルジオトロフィンを注射した心臓は約3.61%のSP群を示した(図2B)。
【0129】
図2Cは、感染したH9C2細胞におけるCT−1アデノウィルス発現の証拠を表す。細胞はアデノウィルスと処理し、72時間後、これら細胞からの培地を非処理のコントロールH9C2細胞培地とともに抗CT−1抗体を用いたウエスタンブロット分析のために採集した。約24.5kDaのCT−1タンパクがブロットにおいて示されている。
【0130】
図2Dは、コントロール注射後24時間の心臓細胞のFACS分析から得られた結果を表す。図2Eは、コントロールアデノウィルス注射後24時間の心臓細胞のFACS分析の結果を表す。ベラパミルは、SP群を同定するのに使用してよい薬剤(チャンネル遮断薬)である。例えば、ベラパミルで処理した細胞はヘキスト染料を排除し、SP群はFACS分析で検出されなくなる。コントロールアデノウィルスで処理した後ベラパミル感受性SP群には全く変化がない。
【0131】
t=24、48、72及び96時間でのマウス心臓におけるad−CT−1の効果を示す経時変化を図3Aに示す。t=24時間、48時間、72時間及び96時間での心臓細胞におけるCT−1の効果を示す2つめの経時的変化を図3Bに示す。心臓細胞懸濁液をヘキスト33342染料で染色し、FACS分析した。様々な組織源を用いた先の研究により、ヘキスト染料染色細胞懸濁液がヘキスト流出サブ細胞群(サイドポピュレーションあるいはSP細胞)を示すことが決定された。これらの細胞は、分化マーカーの幹細胞様の活性、減少あるいは非存在を有し、また多剤耐性様タンパクのインヒビターであるベラパミルの存在に対する感受性によっても特徴付けられる[Goodell et al. (1997), Nat. Med. 3, 1337-1343; Jackson et al. (1999), Proc.Natl.Acad.Sci.USA 96, 14482-14486.]。従って、常在の心筋幹細胞群の存在を評価するために、成体マウスの心臓(〜2ヶ月)を単細胞懸濁液中に酵素的に解離させ、FACS分析した。FACSは、デュアルレーザーを装備したDakoCytomation MoFloフローサイトメーターを用いて行った。蛍光を2波長で測定した。前方及び側方散乱を488nmで測定した(Spectraphysic Argon laser)。ヘキスト染料を350nmで励起した(I90C UV laser from Coherent)。青色発光を424nmで測定し(424/44バンドパスフィルタ)、赤色発光を675nm以上で測定した(675AGLPロングパスフィルタ)。510DLP二色性ミラーを用いてこれら2波長を分割した。心筋細胞懸濁液のFACS分析は、マウス心臓組織からの強いヘキスト染料排除SP群を示す。全細胞は、直接心臓内注射により7.5×107PFUのad−CT−1あるいはlacZアデノウィルスを受けた2ヶ月齢のマウスの心臓から分離した。
【0132】
ここで図4を参照すると、心臓内注射後4日の回復期間中、AdCT−1処理した心臓において認められるSP細胞の相対的な数を表す結果が示されている。このデータは、各時点でコントロールを注射した心臓で得られたデータに対して標準化した。このデータは、比較的短時間内、例えば、約48時間から約72時間前後で見られる大きな増加とともに、長時間心臓SP細胞における約3倍の増加があることを示す。
【0133】
ここで表1を参照すると、CT−1で処理した対象の体重に対する心臓重量の比率をコントロールと比較して示されている。
【表1】
【0134】
統計解析は、コントロール群とCT−1処理群で得られた比率の間に有意差が全くないことを示している(p>0.05)。これらの結果は、様々な病気あるいは疾病、例えば、これに限定されるものではないが、心臓の病気あるいは疾病を、対象の心臓肥大を促進することなく予防あるいは治療するために、CT−1を対象に投与してもよいことを示唆するものである。
【0135】
24時間、48時間、72時間及び96時間の時点での骨格筋、骨髄及び肝臓におけるad−CT−1の心臓内投与の効果を図5A〜Cに示す。この結果は、ad−CT−1の心臓内注射が、対象の心臓幹細胞の増殖を選択的に促進する適当な方法を提供することを示す。同様に、いかなる方法でも制限することを望むものではないが、CT−1あるいは対象の特定の筋肉内でCT−1を産生する能力のある化合物の筋肉内注射が、その筋肉内で、二次領域、例えばこれに限定されるものではないが、心臓及び他の筋肉あるいは組織などよりも幹細胞の選択的な増殖を結果として生じるかもしれない。一方、制限することを望むものではないが、CT−1が様々な組織あるいは器官系と接触するのを可能にする一つ以上の経路により対象にCT−1、あるいはCT−1を産生する能力がある化合物を投与することは、一つ以上の組織あるいは器官系、例えば、これに限定されるものではないが心臓及び骨格筋組織において幹細胞の非選択的な増殖を結果として生じるかもしれない。これらの結論は、メチルセルロースあるいはコロニー形成促進に必要な因子を含むゲル状培地を用いたMethocult(R)分析の結果により支持される。この分析は、幹細胞群がそのライフサイクルの間の同じ点で活性化されるのであれば、確認に用いることができる。簡単に言えば、実験は製造業者の指示(StemCell Technologies、カタログ#28405、Version 3、2004年2月)に従って、注射後24、48、72あるいは96時間にadCT−1あるいはadLaczを注射したマウスについて、約10000細胞/プレートをプレーティングすることにより行った。心臓、骨格筋、及び 骨髄からのサイドポピュレーション(SP)及びメインポピュレーション(MP)の両方のフラクションを試験した。細胞は、2週間培養し、次いでコロニーを計数し、様々な細胞マーカー、例えば造血性マーカーなどを染色した。Ad−CT−1ベクターのインビボ投与は、一つ以上の骨格筋幹細胞群の増殖を増加させた。例えば、Ad−CT−1暴露骨格筋のSPの増加は小さかったが、骨格筋に由来し、かつ許容状態(すなわち、メソカルト培地)で成長した一次細胞培養は、コントロールで処理した培養に比較して、造血性コロニーの数が約10倍増加した。
【0136】
ここで図6を参照すると、CT−1の投与が、約3週齢以上のマウス対象における細胞増殖を促進することを示唆する結果が示されている。従って、本発明は、ここで全体にわたって記載される方法を意図し、その場合対象が3週齢以上、好ましくは2ヶ月齢以上である。対象がヒトである一つの実施形態において、本発明は、ここで全体にわたって記載の方法を意図し、その場合対象が約1ヶ月以上、好ましくは約2ヶ月以上、さらに好ましくは約6ヶ月以上、よりさらに好ましくは約1歳以上の年齢である。別の実施形態において、対象は成人のヒトである。
【0137】
ここで図7を参照すると、CT−1が成体の心臓幹細胞の分化を促進することを示唆する結果が示されている。構成的に発現されるGFP導入遺伝子を発現するマウスに、CT−1を注射した。心臓は24時間後に採取し、これら心臓から分離したSP細胞を正常な一次心筋細胞と共培養した。結果は、GFP標識SP細胞のコネキシン−43陽性心筋細胞への強い変換を示す。この結果は、対象の心臓幹細胞の増殖及び/又は分化を促進するためいに、CT−1を用いることができるという証拠を与える。さらに、実験から得られた結果に基づいて、本発明は、次のステップを含む、心臓幹細胞の増殖、分化あるいは増殖と分化の両方を調節する化合物のスクリーニング方法も提供する:
a)対象から心臓幹細胞を含む複数の細胞を分離する;
b)その細胞を一つ以上の化合物で処理する;
c)ステップbで処理した細胞を、前記細胞が増殖、分化あるいはその両方を受けるのに十分な期間、一つ以上の正常な一次心筋細胞と培養する、及び;
d)増殖した細胞、分化した細胞、あるいは増殖と分化の両方をした細胞の数を計数する。
処理ステップは、CT−1、あるいはそのフラグメントあるいは誘導体、CT−1活性モジュレーター、wntポリペプチド、あるいはwnt活性を発揮するそのフラグメント又は誘導体、Pax7、あるいはその何れかの組み合わせに細胞を接触させることを含んでもよい。また、他の単形質導入分子も用いてよい。分離ステップに先立って、一つ以上の幹細胞群の増殖を促進するために、対象をCT−1で処理することも意図される。さらなる実施形態においては、心臓幹細胞を含む複数の細胞は、それらが正常な一次心筋細胞から分化されるのを可能にするようなマーカーなどを含んでもよい。例えば、心臓幹細胞を含む複数の細胞は、GFP標識してもよく、一方、正常な一次心筋細胞はGFP標識されない。心臓幹細胞を含む複数の細胞と正常な心筋細胞との間の何れの分化方法もここで記載の方法において用いてよい。
【0138】
ここで図8を参照すると、Ad−CT−1の心臓内注射が損傷を受けた骨格筋の修復に影響することができるか否かを決定するための実験手順を一般的に表した図が示されている。実験は、メソカルト実験で得られた観察結果を調査するために行われた。それらの実験において、CT−1の心臓内注射がメソカルト培地上に形成された骨格筋からの「幹細胞様」コロニーの数を増加させることが認められた。これらの結果は、CT−1が骨格筋における幹細胞様群を活性化させるということ、ならびにこの群が骨格筋修復プロセスに寄与するあるいはこのプロセスを高めるかもしれないということを示唆するものである。これらの観察結果を裏づけるために、マウス後肢への損傷をヘビ毒カルジオトキシンの注射により誘導した。カルジオトキシンは、非常に短期間の内に(約1〜2日)極度の筋肉変性を誘導する。しかしながら、骨格筋は、損傷後数日で十分に自己修復することができることがよく知られている。ここでは、カルジオトキシン損傷誘導後直ちにCT−1を注射した。さらに、カルジオトキシン注射後1〜3日にCT−1を投与する同様の実験を行った。
【0139】
ここで図9を参照すると、コントロール注射、カルジオトキシン(ctx)注射、あるいはカルジオトキシンとAd−CT−1の両方の注射後約6日目の損傷範囲を表す結果が示されている。これらの実験においては、1ヶ月齢マウスからの後肢の右前脛骨筋(TA)に10μモルのカルジオトキシンを注射した。反対側面の肢をコントロールとして用いた。別のマウス群では、CT−1をカルジオトキシン注射後心臓内に投与した。6日後、TA筋を解剖し、切片作成のために凍結した。筋切片をヘマトキシリン/エオシンで染色し、膜成分及び構成成分を可視化した。CT−1をカルジオトキシンとほぼ同時に投与した場合には、筋はより無傷状態であることがわかる。
【0140】
ここで図10を参照すると、前記のように処理し、抗体アルファ−アクチニンで染色した筋肉断面切片が示されている。アルファ−アクチニンは、骨格筋のサルコメアのz−バンドを染色し、筋サルコメアの完全性の一般に認められた指標である。結果は、CT−1で処理した筋肉は、カルジオトキシン(ctx)単独で処理した筋肉よりもよくアルファ−アクチニン染色されることを示唆している。いかなる方法でも理論により結合されることあるいは制限することを望むものではないが、これらの結果は、損傷後の骨格筋の回復を高めるためにCT−1を用いることができること、及び/又は、CT−1は損傷後の変性から筋肉を保護することができることを示唆する。
【0141】
一般的プロトコル
骨髄分離
マウスから肢を切り離す。マウスの上肢の長骨(大腿骨)を離し、骨髄細胞数を増加させるために、下肢も分離してよい。
【0142】
1)周辺の筋肉及び他の組織を除去する。
2)できるだけ高く骨を切る。
3)ひざ周辺の組織を除去し、長骨を切除する。
4)一旦骨をフリーにし、インシュリン注射器を使用して5cmの組織培養皿に骨髄を押し出す。
5)5mLのPBSで骨を押し出し、確実に両末端を押し出す。
6)必要に応じて繰り返す。
7)一旦骨髄を集め、18ゲージ針で皿中で粉砕する。
8)細胞を15mLのファルコンチューブに移す。氷冷維持。
9)FACSのために、細胞を遠沈した点からのプロトコルを続け、その後、DMEM+で洗浄する*消化は全く要求されない*
【0143】
FACSプロトコルのための抗体染色
1)HBSS+中再懸濁する直前、最後の洗浄までヘキストプロトコルに完全に従う。
2)1ml/心臓でPBS+2%FBS中に再懸濁する。
3)血球計数器を用いて細胞を計数する。
式=#細胞/mm3×希釈×10000(#細胞/mL)
4)飽和曲線のために:
>細胞をPBS+2%FBSを用いて終濃度3000000細胞/mLに希釈する
>100μLを8チューブそれぞれに添加する(コントロール2本及び様々な抗体濃度6本)
>次に、抗体溶液を希釈する(例:初期濃度100μg/mLで、5μg/mLの400μLにする)
5(400)=100(x)x=20μL
20μLの抗体を380μLのdH2Oに入れる
【0144】
>100μLを取り、細胞に添加する。これを2°抗体を含まないコントロール#1と呼ぶ。
>別の100μLを取り、細胞に添加し、2°抗体を添加する。
>残った抗体(200μL)とともに、200μLのdH2Oを添加する。これは、2.5μg/mLの400μLとなる。
>適当な回数の希釈を完了するまで続ける。
>コントロール#2は、1°抗体を持たない細胞である。
【0145】
5)細胞を一次抗体と15分間氷上でインキュベートする。
6)細胞を遠心し、上清を除去する。
7)細胞をPBS+2%FBSで洗浄する。
8)200μLのPBS+2%FBS中に再懸濁する。
9)二次抗体を添加し、15分間氷上でインキュベートする。
10)細胞を再度PBS+2%FBSで洗浄する。
11)500μLのHBSS+中に再懸濁する。
12)小さな丸底のファルコンチューブに移す*FACSのため*
13)lccのインシュリン注射器を用いて何れの細胞凝集塊も除去する。
14)細胞を集めるのに用いるために、1mLの培地を入れた15mLチューブを用意することを忘れないこと。.
15)FACSを行う。
【0146】
FACS用処方
ベラパミル(100×)
100×の最終原液を作る
95%エタノールで懸濁する
最終50μM
【0147】
ヘキスト33342(200×)
200×の最終原液を作る
水で懸濁する
最終1mg/mL
【0148】
DMEM+
388mL DMEM
8mL FBS
4mL 1M Hepes
【0149】
HBSS+
100mL HBSS(Hanks Balanced Salt Solution)
2mL FBS
1mL 1M Hepes
【0150】
コラゲナーゼ−ディスパーゼ
フード内で作る
全体500mgのコラゲナーゼビンを5mLのIX PBSで溶解する
5mLづつのコラゲナーゼ−ディスパーゼ溶液各々に対し、下記を添加する:
>4.34mLディスパーゼ 2(t.c.冷凍庫)
>500μLの懸濁コラゲナーゼ(+4℃冷蔵庫)
>125μLの100mM CaCl2
【0151】
PBS+2%FBS*抗体染色時に使用*
500mL PBS
10mL FBS
【0152】
ヘキスト33342幹細胞染色及び精製プロトコル
1)フードを向いてコラゲナーゼ/ディスパーゼを37℃の水槽中で解凍する。
2)マウスを屠殺し、エタノールをスプレーし、心臓を解剖する。
3)心臓を冷PBS中に入れる(4℃に維持した標準PBS)
4)ピンセットを用いて、心臓から過剰な血液を搾り出す。
5)消化の準備ができるまで、冷PBS中で保存する。
6)フード内で、2つの刃物で心臓を細かく切る。(心臓が湿っているようにする〜もしそれらがネバネバしたら100μLのPBSを添加する)
7)その後、細切した心臓を小さな組織培養皿に移す。
8)4〜5mLのコラゲナーゼ/ディスパーゼを1から4心臓当たり添加する。
9)ピペットで吸吐してよく混ぜ、細胞凝集塊をばらばらにする。
10)37℃の培養器内に35〜38分間置く。
11)75ミクロンのメッシュフィルターを通す(ネットウエルプレートから)。
12)フィルターを5mLのPBSで浸漬し、50mLのチューブに集める。
13)50mLチューブの内容物を15mLチューブに移す。
14)1.2の設定で10分間遠心し、上清を除去、8mLの冷DMEM+中に再懸濁し、再度1.2の設定で10分間遠心する。
15)注意深く上清を除去し、沈殿物を心臓当たりそれぞれ2mLのDMEM+中に再懸濁する(細胞の数を見積もる)
16)ヘキスト及びベラパミルで染色する(両方とも冷凍庫保存)
5μL/mLの200×ヘキストを全ての細胞に添加する
1mLのヘキスト染色細胞を取り、新しい15mLチューブに入れる
15μL/mLの100×ベラパミルを添加する
17)37℃の培養器中に90分間入れる。温度一定を確保。
18)氷上に5分間置く。
19)1.2の設定で10分間遠心し、上清を除去し、8mLの冷DMEM+に再懸濁し、1.2の設定で10分間再度遠心する。
20)上清を除去し、HBSS+中再懸濁する(〜200μL/心臓)。
21)FACSのために小さな簡易栓チューブに移し、1ccのインシュリン注射器を用いて何れの細胞凝集塊も除去する。
22)この時点から氷上を維持する(生物学的に不活性)
23)細胞を集めるのに用いるために1mLの培地を入れた15mLチューブを用意するのを忘れないこと。
24)経時的に生じ得るどんな変化も最小限にするように、FACS分析をできるだけ速やかに行う。
【0153】
一次心筋細胞培養培地
培地は、使用前に新しく調製すべきであるが、冷蔵庫内で約1週間保存できる。
【0154】
50 mLのDMEM−F12に下記を添加する:
d−グルコース 0.3g
l−グルタミン 500μLの200mM原液
pen−strep 500μLの5000単位原液
インシュリン 250μLの5mg/mL原液
アポトランスフェリン 100μLの50mg/mL原液
プロゲステロン 1μLの1mM原液
プトレシン 60μLの8mg/mL原液
セレニウム 15μLの100μM原液
フンギソン 50μLの12.5μg/mL原液
ヘパリン 10μLの10mg/mL原液
【0155】
培地を0.2μmフィルターで濾過する。40μLの25μg/mL bFGF原液を濾過後添加する。EGFは任意である。使用するなら、濾過前の培地中、10μLの100μg/mL原液を50mLのDMEM−F12に入れる。
【0156】
心筋細胞分離用の処方
MEM−JM(500mL)
400mL dH2O
5.65g MEM−JM
1.0g 炭酸水素Na
NaOHでpH7.3の溶液、H2Oで終量500mL、濾過滅菌。
【0157】
2×コラゲナーゼ(新鮮なものを調製)
30mg コラゲナーゼ
15mL MEM−JM
濾過滅菌(注射器)
【0158】
DMEM−10%FBS
400mL DMEM
45mL FBS
4.5mL pen/strep
【0159】
DMEM−F12+ITS(500mL)
半パッケージのDMEM−F12(〜7.8g)
2.5mL pen/strep
500μL ITS
0.6g NaHCO3
【0160】
一次心筋細胞分離
調製:
培地を調製し、30mgのコラゲナーゼを15mLのJMEMに添加し、濾過滅菌し、37℃の水槽中に置く(〜6日齢未満の20匹のマウス)
【0161】
心臓の解剖:
1)心房を避けるようにマウスから心臓を切り取り、冷DMEM−JMの50mLチューブ中に入れる。
2)心臓をピペット吸吐によりJMEM培地中で洗浄する。
3)心臓をペトリ皿に移し、必要であればPBSで保湿する。
4)2つの手術用メスを用いて、心臓を細かく切る。
【0162】
コラゲナーゼ消化:
1)静かに心室組織をペトリ皿中で20mLのJMEMにより25mLの広口ピペットを用いて洗浄する。数サイクル後、組織及び培地をピペットで吸う。組織を落ち着かせ、組織のみを清浄な50mLチューブ中に「吐き」出す。赤血球をほとんど含む培地を廃棄する。
【0163】
2)同じ25mLピペットを用いて、50mLチューブ中の組織から残った液体を除去し、適量のJMEM及びコラゲナーゼ2×を添加する(表参照、用いた組織の量を考慮)。
【表2】
【0164】
3)37℃で静かに20−30回転/分で15分間インキュベートする。その間に、10%FBS+DMEMを入れた2−50mLチューブを用意する。
4)最初のインキュベーション後、組織及び培地の両方を25mLピペットで約3回ゆっくり粉砕する。1分待って組織を落ち着かせ、ゆっくりと上清を吸引し、廃棄する(最初ほとんどRBC及び破片)。10mLの新鮮なJMEMで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清を廃棄する。組織の量を測定し、JMEMを添加して適当な量に調製する(表参照)。コラゲナーゼを添加し、インキュベートする。
5)後のインキュベーションの終わりに、10mLピペットでゆっくりと組織及び培地の両方を吸い上げ、2回粉砕する。1分間待って、ゆっくりと上清を吸引し、10mLFBSを入れた50mLチューブに移して、コラゲナーゼ消化を停止させる。宿主組織を新鮮な10mLのJMEMで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清をFBSを入れた同じ50mLチューブに移す。組織の量を測定し、JMEMを添加して適当な量に調整し、その後コラゲナーゼを添加する。
6) FBS及び2回の洗浄から得られた上清を含む50mLチューブに、十分なDMEM+10%FBSを添加してチューブを満たす。エアープレーン中50gで5分間遠心分離する。上清を減圧吸引し、沈殿物を10mLのDMEM−20%FBS中に再懸濁する。室温でフード内に保存する。
7)ステップ4及び5をもう2回(あるいは3回)繰り返す。
8)最後のインキュベーション後、ゆっくりと2回10mLピペットで粉砕する。宿主組織を新鮮な10mLのDMEM+10%FBSで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清をFBSを入れた同じ50mLチューブに移す。
9)FBS及び2回の洗浄からの上清を含む50mLチューブに、十分なDMEM+10%FBSを添加してチューブを満たし、500gで5分間遠心分離する。
【0165】
前プレーティング手順:
1)上清を除去する。
2)20mLのDMEM+10%FBS中に再懸濁する。
3)ナイテックス(nytex)ナイロンフィルターを50mL滅菌チューブの上にかぶせて濾過の準備をする。濾過した懸濁液を一つの150mm滅菌ペトリ皿上に平らに分散し、37℃で15分間インキュベートする(これは第1プレーティングである)。
4)インキュベーション後、ゆっくり粉砕し、上清(〜20mL)を2番目の皿に移す。
5)古い皿を新鮮な10mLDMEM+10%FBSで洗浄し、2番目の皿に移す(これは第2プレーティングである)。37℃で15分間インキュベートする。
6)沈殿物を10mL以下のDMEM+10%FBSで再懸濁する。より少量の再懸濁液量は、細胞数計測手順の間より多い細胞を用いることを意味し、その濃度はプレーティング工程の間用いる終濃度よりも高くあるべきということに留意する。
7)細胞を〜5mLのDMEM+10%FBS中2mm×6mmの皿上にプレーティングする。コートされたプレート上で20時間インキュベートする。前プレーティングした細胞の2番目のプレートはDMEM+10%FBSで維持した。
8)20時間インキュベーション後、プレートの培地をDMEM F12+ITSに換える。
9)増殖第1日目の後、静かにプレートをすすぎ、15mLチューブに浮遊細胞を含む成長培地を移した。細胞を遠沈し、コーティングしたプレート上の新しいDMEM+10%FBS培地上に再プレーティングした。2番目の前プレートはDMEM+10%FBS中で3日間増殖させた。
【0166】
細胞数計測:
1)1.5mLのエッペンドルフチューブ中、200μLの細胞懸濁液を400μLの0.4%トリパンブルー溶液及び400μLのDMEM+10%FBSに添加する。2分間インキュベートし、血球計数器で計数する。生存心筋細胞及び非生存心筋細胞を別々に計数する。生存筋細胞がかなり多く、有核あるいは多核であるべきである。非生存細胞は、青く染色される。RBC及び他の小さな破片は無視する。
2)計数、及びプレーティング後、細胞を予め37℃に加温したDMEM+10%FBS中で終夜あるいは少なくとも16時間インキュベートする。培地を含む血清中でインキュベートするために24時間が最長である。
【0167】
本発明は、記載されたように、同じことが多くの方法において変更されてよいことは明らかである。そのような変更は、本発明の精神及び範囲を逸脱するものと見なされるべきでなく、当該分野の技術者に明白であるような全てのそのような修飾は、請求項の範囲内に含まれることを意図する。
【0168】
全ての引用は、参照することによりここに組み込まれる。
【0169】
本発明は、好ましい実施形態について記載した。しかしながら、多くの変更及び修飾がここに記載の発明の範囲を逸脱することなく為すことが可能であるということは、当該分野の技術者に明らかであろう。
【図面の簡単な説明】
【0170】
【図1】図1は、本発明で採用したアデノウィルス構成ad−CT−1の略図である。
【0171】
【図2A】図2Aは、生理食塩水注射により処理し、次いでヘキスト33342染料で染色した心臓細胞懸濁液のFACS分析を表す。
【図2B】図2Bは、カルジオトロフィン−1注射により処理し、次いでヘキスト33342染料で染色した心臓細胞懸濁液のFACS分析を表す。
【図2C】図2Cは、感染したH9C2細胞におけるCT−1アデノウィルス発現を表すウエスタンブロットを表す。約24.5kDaのCT−1タンパクがブロット中に示されている。
【図2D】図2Dは、コントロール注射後24時間の心臓細胞FACS分析から得られたコントロールの結果を表す。
【図2E】図2Eはアデノウィルス注射後24時間の心臓細胞のFACS分析のコントロール結果を表す。
【0172】
【図3A】図3Aは、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す。
【図3B−1】図3B−1は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す。
【図3B−2】図3B−2は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−3】図3B−3は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−4】図3B−4は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−5】図3B−5は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−6】図3B−6は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−7】図3B−7は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−8】図3B−8は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【0173】
【図4】図4は、心臓内注射後、4日の回復期間の間、AdCT−1処理した心臓に見られたSP細胞の相対数を表す結果を示す。本データは、各点で、コントロール注射の心臓で得られたデータに対して標準化した。コントロール注射は、CT−1をエンコードしていない同等のAdベクターからなっていた。
【0174】
【図5A】図5Aは、24時間、48時間、72時間及び96時間の時点での、骨格筋からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【図5B】図5Bは、24時間、48時間、72時間及び96時間の時点での、骨髄からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【図5C】図5Cは、24時間、48時間、72時間及び96時間の時点での、肝臓からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【0175】
【図6】図6は、3週、4ヶ月及び1年時点でのマウス心臓のコントロール及びCT−1注射後のFACS分析の結果を示し、マウス心臓におけるCT−1の年齢依存性効果の可能性を示唆するものである。
【0176】
【図7】図7は、CT−1が成体心臓幹細胞の分化を促進することを示唆する結果を示す。構成的に発現されるGFP導入遺伝子を発現するマウスにCT−1を注射した。心臓を24時間後に採取し、その心臓から分離したSP細胞を正常な一次心筋細胞と共培養した。結果は、GFP標識したSP細胞のコネキシン−43陽性心筋細胞への強い変換を示す。
【0177】
【図8】図8は、Ad−CT−1の心臓内注射が損傷を受けた骨格筋の修復に影響し得るか否かを確認するための実験手順の概略を表す。
【0178】
【図9】図9は、コントロール注射、カルジオトキシン(ctx)注射、あるいはカルジオトキシンとAd−CT−1の両方の注射後約6日の損傷の範囲を表す結果を示す。これらの実験において、1ヶ月齢マウスからの後肢右前脛骨筋(TA)に10マイクロモルのカルジオトキシンを注射した。反対側の肢をコントロールとして用いた。別のマウスグループに、カルジオトキシン注射後、CT−1を心臓内投与した。6日後、TA筋を解剖し、切片作成のため凍結した。筋肉切片をヘマトキシリン/エオシンで染色し膜及び構成成分を可視化した。
【0179】
【図10】図10は、アルファ−アクチニン抗体で筋肉切片を染色した結果を示す。この抗体は、骨格筋のサルコメアのz−バンドを染色し、筋肉サルコメアの完全性の一般に認められた指標である。
【技術分野】
【0001】
本発明は、幹細胞治療の分野に関係し、特に幹細胞の増殖調節方法に関係する。
【背景技術】
【0002】
幹細胞とは、様々な特定のタイプの細胞を作り出し、究極的には最終的な分化細胞を作り出す能力を持つ未分化の、あるいは未成熟の細胞である。幹細胞は、他の細胞と異なり、基本的に必要な時に成熟細胞を無限に供給するように自己更新することができる。この自己更新能があることにより、幹細胞は組織の再生及び修復において治療的に有用である。
【0003】
幹細胞は、様々な臨床設定において恩恵を与える可能性を持つ。多くの可能性のある応用への制限は、十分な数の標的細胞を得ること、また、これら幹細胞の成熟した組織特異的細胞への最終的な分化を刺激することであった。
【0004】
成人の骨髄及び多くの他の体細胞組織は、様々な表現型の細胞に分化することができる多能性幹細胞群を含むと考えられている。例えば、成体マウスの心臓内の多能性幹細胞様の細胞群(SP)が最近報告された(Hierlihy, A.M., et al., (2002) FEBS Letters, 530:239-243)。減衰した成長の環境下で、これら細胞は活性化し、心筋細胞に分化する。心臓組織あるいは他の組織に由来する体細胞性幹細胞の投与、及び幹細胞因子(SCF)、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、間質細胞由来ファクター−1、スチールファクター、血管内皮成長因子、マクロファージコロニー刺激因子、顆粒球マクロファージ刺激因子あるいはインターロイキン−3などのサイトカインの投与による、損傷した心筋の修復あるいは再生方法が最近開示されている(米国特許出願第20030054973号)。
【0005】
サイトカインは、生物内の細胞間の伝達において重要な役割を果たすこと、ならびに成長及び分化を調節することができる細胞メディエーターとして作用することが知られている。サイトカインを幹細胞の分化促進に使用することが開示されている。例えば、米国特許出願第20030027330号には、幹細胞を成長中あるいは成長した同種あるいは異種の細胞とともに、さらに任意的にはサイトカインや成長因子あるいはケモカインも一緒に共培養することにより、幹細胞から分化した哺乳類細胞あるいは組織を生成する方法が記載されている。米国特許出願第20030103951号には、間葉幹細胞の投与により心筋を再生する方法が記載され、これは、サイトカイン、成長因子、筋原性因子及び転写因子などの分化に重要なタンパクを生成するように遺伝学的に修飾されてもよい。米国特許出願第20020142457号には、心筋細胞に分化する可能性を持つ細胞を増殖させる方法、及び様々なサイトカイン及び転写因子を用いてそれらの心筋細胞への分化を調節する方法が記載されている。
【0006】
カルジオトロフィン−1(CT−1)は、IL−6ファミリーサイトカインの一つであり、比較的心臓に限定された方法で発現する。ヒトとマウスの両方のCT−1をエンコードする遺伝子がクローニングされている(Pennica, D., et al., (1996) Cytokine, 8:183-189; Pennica, D., et al., (1995) Proc. Natl. Acad. Sci. USA, 92:1142-1146)。元々は心臓肥大因子(CHF)として知られていたように、CT−1は心臓肥大の誘導を示し、CT−1及びそのアンタゴニストの心不全、不整脈疾患、変力性疾患あるいは末梢神経障害における使用が記載されており(米国特許第5,534,615号;5,571,675号;5,571,893号;5,624,806号及び5,679,545号参照)、ガンの診断及び治療における使用も記載されている(米国特許出願第20020146707号)。虚血前にCT−1を投与することにより、損傷から成体ラットの心臓を保護することも報告されている(Liao、Z.、et al.、(2002) Cardiovasc. Res.、53:902-910)。
【0007】
この背景情報は、出願人が本発明と関連の可能性があると考える公知情報を作成することを目的として提供するものである。いかなる前記情報も本発明に対して従来技術を構成すると必ずしも自認することを意図するものではなく、また解釈されるべきではない。
【0008】
本発明の目的は、従来技術の欠点を解決することである。
メインクレームの特徴の組み合わせは前記目的に合致し、サブクレームは本発明のさらに有利な実施形態を開示する。
【発明の開示】
【0009】
本発明は、幹細胞治療の分野、特に幹細胞の増殖調節方法に関係する。
【0010】
本発明に従って、幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドと接触させることを含む、幹細胞の増殖促進方法が提供される。
【0011】
別の実施形態においては、どのような形でも限定することを意味するものではないが、幹細胞を一つ以上のカルジオトロフィン−1インヒビターと接触させることを含む、幹細胞の増殖抑制方法が提供される。
【0012】
本発明の別の態様に従って、幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチド、及び一つ以上の幹細胞モジュレーターと接触させることを含み、前記一つ以上の幹細胞モジュレーターが、ポリペプチドあるいはポリペプチドをエンコードするポリヌクレオチドである幹細胞の増殖及び分化の促進方法が提供される。
【0013】
本発明の別の態様に従って、カルジオトロフィン−1の類似体、誘導体、変異体又は活性フラグメントであって、幹細胞の増殖促進能力がある、分離されたポリペプチド、及び該ポリペプチドをエンコードするポリヌクレオチド、及び該ポリヌクレオチドを含むベクターが提供される。
【0014】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を投与することを含む、哺乳類の心臓幹細胞増殖の促進方法が提供される。
【0015】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1インヒビターの有効量を投与することを含む、哺乳類の心臓幹細胞増殖の抑制方法が提供される。
【0016】
本発明の別の態様に従って、哺乳類にカルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を投与することを含み、前記ポリペプチドが心臓幹細胞の増殖を促進する能力があるものである、哺乳類の心臓組織の修復又は再生方法が提供される。
【0017】
本発明のこの要約は、必ずしも発明の全ての必要な特徴を記述しておらず、本発明は、記載された特徴のサブコンビネーションに存在してもよい。
これら及び他の本発明の特徴は、添付の図面を参照した下記の記載からより明らかになるであろう。
【好ましい実施形態の開示】
【0018】
以下の記載は実施例による好適な実施形態の一例に過ぎず、本発明の効果を発揮するために必要な特徴の組み合わせを限定するものではない。
【0019】
本発明は、カルジオトロフィン(CT−1)の活性を調節することにより、幹細胞の増殖を調節する方法を提供する。従って、本発明は、CT−1、あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいは内因性のCT−1を活性化する化合物に幹細胞を接触させることを含む、幹細胞の増殖、分化あるいはその両方を促進する方法を提供する。また、本発明は、幹細胞を、CT−1インヒビターと接触させることを含む幹細胞の増殖、分化あるいはその両方を抑制する方法も提供する。幹細胞の増殖促進方法は、幹細胞の増殖及び/又は分化を促進するために、幹細胞を一つ以上の幹細胞モジュレーターに接触させることをさらに含んでもよい。本発明のこの実施形態に従って、細胞をCT−1(あるいはその類似体、誘導体、変異体又は活性フラグメント、あるいはCT−1アクティベーター)と一つ以上の幹細胞モジュレーターとに同時に接触させてもよく、あるいは順次接触させてもよい。
【0020】
本方法は、インビトロで幹細胞増殖、幹細胞分化あるいはその両方を促進するために用いてもよい。あるいは、幹細胞増殖、幹細胞分化、あるいはその両方をインビボで促進するために用いてもよい。また、本発明はインビトロ及びインビボで行うことができる方法も意図する。本発明の治療的応用は幹細胞増殖及び/又は分化を促進する必要がある疾病及び疾患、例えば、損傷したあるいは障害のある組織の置換に限定されることを意味しない。ここで提供される方法は、治療方法あるいは予防方法として用いてもよい。また、本発明の方法は幹細胞増殖を抑制するために採用してよく、従って、不適当な細胞増殖により特徴付けられる疾患、例えば、これに限定されるものではないが心臓肥大などの治療における適用がある。
【0021】
定義
他に定義されていない限り、ここで用いる全ての技術的、科学的用語は、本発明が関係する分野の通常の技術者により一般に理解されるのと同じ意味を有する。
【0022】
ここで用いる「幹細胞モジュレーター」との用語は、幹細胞の増殖、分化、あるいは増殖と分化の両方を刺激又は抑制する能力を持つ化合物をいう。
【0023】
ここで用いる「幹細胞」との用語は、一つ以上の分化した細胞型に分化する能力を持つ細胞を言う。幹細胞は、全能性のあるいは多能性の細胞であってよい。全能性幹細胞は、典型的にどのような細胞型にも発展する能力を持つ。全能性幹細胞は、通常胚に由来する。多能性細胞は、典型的には幾つかの異なる、最終的に分化した細胞型に分化する能力を有する幹細胞系の細胞である。多能性幹細胞は様々な組織あるいは器官系を起源とすることができ、これに限定されるものではないが、血液、神経、心筋、骨格筋、皮膚、腸、骨、腎臓、肝臓、膵臓、胸腺などが挙げられる。
【0024】
ここで用いる「前駆細胞」との用語は、特定の細胞系列に決定された細胞であって、一連の細胞分裂により分化細胞型の特に制限された範囲を作り出す細胞を言う。前駆細胞の例として筋芽細胞が挙げられ、これは唯一つの細胞型に分化する能力を持つが、それ自体は、完全に成熟しておらず、あるいは完全に分化していない。
【0025】
ここで細胞に関して同義的に用いられている「増殖」及び「拡張」との用語は、分裂による同型の細胞の数の増加を言う。
【0026】
ここで用いる「分化」との用語は、それによって細胞が特定機能に特殊化する発展過程を言い、例えば、細胞が最初の細胞型とは異なった一つ以上の形態学的特徴及び/又は機能を獲得する過程をいう。「分化」という用語は、系統義務及び最終分化の両方のプロセスを含む。分化は、例えば、免疫組織化学あるいは当該分野の技術者に知られた他の手法を用いて、系列マーカーの有無をモニターすることにより評価することができる。前駆細胞に由来する分化した子孫細胞は、必ずしもそうである必要はないが、幹細胞の起源組織と同じ胚葉あるいは組織に関係していてよい。例えば、神経前駆細胞及び筋前駆細胞は、造血細胞系列に分化することができる。
【0027】
ここで同義的に用いる「系列決定」及び「特殊化」との用語は、幹細胞が経験するプロセスであり、幹細胞が、分化細胞型の特に制限された範囲を形成するよう決められた前駆細胞を生じるプロセスを言う。約束された前駆細胞は、しばしば自己再生あるいは細胞分裂する能力がある。
【0028】
ここで用いる「最終分化」との用語は、成熟した完全に分化した細胞への細胞の最後の分化を言う。例えば、神経前駆細胞及び筋前駆細胞は、造血細胞系列へ分化することができ、その最終分化は、特定の細胞型の成熟した血液細胞を導く。通常、最終分化は、細胞周期及び増殖休止からの離脱に関係する。
【0029】
ここで用いる「自然発生」との用語は、ポリペプチドあるいはポリヌクレオチドに適用されるように、そのポリペプチドあるいはポリヌクレオチドを自然界で見い出すことができる事実を言う。例えば、生物に存在し、自然界の起源から分離することができ、実験室でヒトにより故意に修飾されたことのないポリペプチドあるいはポリヌクレオチドの配列は、自然発生である。
【0030】
カルジオトロフィン−1(CT−1)を用いた幹細胞増殖調節方法
本発明は、CT−1を用いた幹細胞の増殖、分化、あるいは増殖と分化の両方を促進する方法を提供する。CT−1は、単独で、あるいは一つ以上の幹細胞モジュレーターと組み合わせて、幹細胞の増殖及び/又は分化の促進のために用いてよい。CT−1、及び任意成分としての一つ以上の幹細胞モジュレーターは、幹細胞に直接的に与えてもよく、あるいは、例えばこれに限定されないが、CT−1及び/又は一つ以上のモジュレーターを発現可能な細胞と共培養することなどにより間接的に与えることもできる。また、本発明の方法においては、幹細胞あるいは幹細胞と共培養された細胞の内因性CT−1を活性化する化合物の使用も意図する。
【0031】
本発明は、さらに、CT−1の一つ以上のインヒビターを用いて幹細胞増殖を抑制する方法も提供する。CT−1インヒビターは、幹細胞に直接的に与えることができ、あるいは、例えばこれに限定されるものではないが、一つ以上のインヒビターを発現し、好ましくは分泌する他の細胞と共培養することなどにより間接的に与えることができる。
【0032】
胚幹細胞及び成体幹細胞の両方、あるいはそれらを組み合わせて本発明の方法に用いてもよい。幹細胞は、何れの細胞型にも発展する能力を有する全能性幹細胞であってよく、あるいは、特定の組織あるいは器官、例えば、血液、神経、骨格筋、心筋、骨髄、皮膚、腸、骨、腎臓、肝臓、膵臓、胸腺などに由来する多能性幹細胞であってもよい。
【0033】
一つの実施形態において、本発明の方法は、成体幹細胞に適用される。別の実施形態においては、本発明の方法は心筋幹細胞、骨格筋幹細胞、あるいはその両方を用いる。
【0034】
I.CT−1
本発明に従って、CT−1は哺乳類のCT−1(心臓肥大因子、あるいはCHFとしても知られている)を言う。多くのカルジオトロフィンタンパクが当該分野において知られており、例えば、これに限定されるものではないが、マウスCT−1(Pennica, D., et al., Proc. Natl. Acad. Sci. USA, 92:1142-1146)及びヒトCT−1(Pennica, D., et al., (1996) Cytokine, 8:183-189)がある。
【0035】
CT−1は、ポリペプチドとして与えてもよく、あるいはそのポリペプチドをエンコードし、且つ発現することのできるポリヌクレオチドとして与えてもよい。様々なCT−1タンパクの配列が当該分野において知られており、CT−1ポリペプチドの調製の基礎として使用することができる(例えば、前記参照文献ならびにGenBank Accession Nos AAC52173及びNP_ 031821 (マウス); AAD12173及びAAA85229 (ヒト)により提供される)。
【0036】
また、本発明は、自然発生型(野生型)CT−1のポリペプチド類似体、誘導体及び変異体も意図するものであり、また、CT−1の活性ペプチドフラグメント、及びそのペプチドフラグメントの類似体、変異体及びペプチド模倣体も意図するものである。
【0037】
活性フラグメントは、野生型タンパクと実質的に同じ活性を保持する自然発生(野生型)タンパクのフラグメントである。フラグメントは、通常少なくとも約20のアミノ酸長さである。本発明の一つの実施形態において、フラグメントは少なくとも約50のアミノ酸長さである。別の実施形態においては、フラグメントは少なくとも約70のアミノ酸長さである。さらなる実施形態においては、フラグメントは少なくとも約100のアミノ酸長さである。そしてさらに別の実施形態では、フラグメントは少なくとも約150のアミノ酸長さである。「フラグメント」との用語は、野生型の配列のN末端、C末端、あるいはN−、C−両末端から1〜約50アミノ酸が欠損した野生型タンパクに対応するポリペプチドも包含する。候補フラグメントは、自然発生タンパクから生じたランダムフラグメントから選択することができ、あるいは、特に設計することもできる。フラグメントの活性を試験して、野生型タンパクと比較し、自然発生タンパクと実質的に同じ活性を持つフラグメントを選択する。ポリペプチドフラグメントの生成方法は、当該分野においてよく知られており、野生型タンパクあるいはその組み換え体の酵素的、化学的、あるいは機械的切断、そのようなフラグメントをエンコードする核酸の発現などがある。
【0038】
当該分野において知られているように、ポリペプチドの類似体及び誘導体、ならびにペプチド模倣化合物は、自然発生体よりも大きな利点を有していてもよく、例えば、より高い化学的安定性や、増強されたタンパク分解抵抗性、強化された薬理学的特性(例えば、半減期、吸収、作用強度、有効性など)、変化した特異性(例えば、生物学的活性の広域性)、及び/又は減少した抗原性などが挙げられる。
【0039】
「誘導体」は、通常は自然発生の配列の一部ではない化学的あるいは生化学的な追加部分を含むポリペプチドあるいはペプチドである。誘導体は、アミノ末端及び/又はカルボキシ末端及び/又は一つ以上のアミノ酸側鎖が適当な化学的置換基で誘導体化されているポリペプチド及びペプチドを含み、また、環状、2重ならびに多量体のポリペプチド及びペプチド、他のタンパクあるいは担体に融合したポリペプチド及びペプチド、グリコシル化あるいはリン酸化したポリペプチド及びペプチド、親油性部分(例えば、カプロイル、ラウリル、ステアロイル部分)に結合したポリペプチド及びペプチド、ならびに抗体あるいは他の生物学的リガンドに結合したポリペプチド及びペプチドも含む。
【0040】
ポリペプチド及びペプチドの誘導体化に用いることができる化学的置換基の例としては、これらに限定されるものではないが、アルキル基、シクロアルキル基及びアリール基;アルカノイル基やアロイル基を含むアシル基;エステル;アミド;ハロゲン;ヒドロキシル;カルバミルなどがある。また、置換基は、Fmoc(フルオレニルメチル−O−CO−)、カルボベンゾキシ(ベンジル−O−CO−)、モノメトキシスクシニル、ナフチル−NH−CO−、アセチルアミノ−カプロイル及びアダマンチル−NH−CO−などの保護基であってもよい。他の誘導体としては、C−末端ヒドロキシメチル誘導体、O−修飾誘導体(例えば、C−末端ヒドロキシメチルベンジルエーテル)及びアルキルアミドやヒドラジドのような置換アミドなどのN−末端修飾誘導体が挙げられる。
【0041】
「環状」ポリペプチドあるいはペプチドとの用語は、例えば、環化に適当な2以上の追加アミノ酸残基が付加したポリペプチドあるいはペプチドなどの環状誘導体を言う。これらの追加アミノ酸は、カルボキシル末端及びアミノ末端に付加してよく、あるいは、内部の位置にあってもよい。あるいは、環状ポリペプチド/ペプチドは、アミノ酸配列中に自然に生じるシステイン残基を利用してジスルフィド結合を形成し、これにより、ポリペプチド/ペプチドを環化してもよい。環状ポリペプチド/ペプチドは、分子内ジスルフィド結合、すなわち、−S−S−;2つの付加残基の間の分子内アミド結合、すなわち、−CONH−あるいは−NHCO−;あるいは分子内S−アルキル結合、すなわち、−S−(CH2)−CONH−あるいは−NH−CO(CH2)nS−、nは1、2、あるいはそれ以上、の何れかを含むことができる。
【0042】
分子内ジスルフィド結合を含む環状ポリペプチド/ペプチドは、従来の固相合成法により、環化のために選択された位置で適当なS−保護システインあるいはホモシステイン残基を合体させながら調製してもよい(例えば、Sahm et al., 1996, J. Pharm. Pharmacol. 48:197参照)。鎖組み立て完了後、遊離の対応するSH−官能基の支持体上の酸化を結果として生じるS−保護基の選択的な除去によりS−S結合を形成し、次いで支持体から生成物を従来通り離し、適当な方法で精製することにより環化を行うことができ、あるいは完全な側鎖脱保護と共にポリペプチド/ペプチドを支持体から離し、次いで遊離のSH−官能基を高希釈水溶液中で酸化することにより、環化を行うことができる。同様に、分子内アミド結合を有する環状誘導体は、従来の固相合成法により、環化のために選択された位置で適当なアミノ側鎖及びカルボキシル側鎖保護アミノ酸誘導体を合体させながら調製してよく、また、分子内−S−アルキル結合を有する環状ポリペプチド/ペプチドは、従来の固相合成法により、環化のために選択された位置でアミノ酸残基を適当なアミノ保護側鎖及び適当なS−保護システインあるいはホモシステイン残基と合体させながら調製することができる。
【0043】
2重ポリペプチド/ペプチドは、直接的に、あるいはアラニン残基やタンパク分解の推定部位の短い範囲のようなスペーサーを介して、互いに共有結合した2つの同じあるいは2つの異なるポリペプチド/ペプチドからなる(例えば、米国特許第5,126,249号及び欧州特許第495,049号参照)。多量体は、多くの同じあるいは異なるポリペプチド/ペプチドあるいはその誘導体から形成された高分子である。重合は、0.1%グルタルアルデヒドのような適当な重合試薬で行われる(例えば、Audibert et al., 1981, Nature 289:593参照)。
【0044】
「類似体」は、一つ以上の非自然発生のアミノ酸を含むポリペプチド/ペプチドである。例えば、本発明のポリペプチド/ペプチド類似体は、対応するD−アミノ酸残基により置換された、あるいは別の非自然発生アミノ酸で置換された一つ以上のアミノ酸残基を有してよい。非自然発生アミノ酸の例としては、これに限定されるものではないが、N−α−メチルアミノ酸、C−α−メチルアミノ酸、β−メチルアミノ酸、β−アラニン(β−Ala)、ノルバリン(Nva)、ノルロイシン(Nle)、4−アミノ酪酸(γ−Abu)、2−アミノイソ酪酸(Aib)、6−アミノヘキサン酸(ε−Ahx)、オルニチン(orn)、ヒドロキシプロリン(Hyp)、サルコシン、シトルリン、システイン酸、シクロヘキシルアラニン、α−アミノイソ酪酸、t−ブチルグリシン、t−ブチルアラニン、3−アミノプロピオン酸、2,3−ジアミノプロピオン酸(2,3−diaP)、フェニルグリシン、2−ナフチルアラニン(2−Nal)、1,2,3,4−テトラヒドロイソキノリン−3−カルボン酸(Tic)、β−2−チエニルアラニン(Thi)、メチオニンスルホキシド(MSO)及びホモアルギニン(Har)などが挙げられる。
【0045】
当該分野において知られているように、ペプチド内の全てのL−アミノ酸を全てD−アミノ酸に置換することにより、「インベルソ」(inverso)ペプチドあるいは「レトロインベルソ」(retro-inverso)ペプチドとすることができ(Goodman et al. "Perspectives in Peptide Chemistry" pp. 283-294 (1981); 米国特許第4,522,752号参照)、これらは両方とも本発明との関係において類似体と見なされる。「インベルソ」ペプチドは、配列のL−アミノ酸全てがD−アミノ酸で置換されたものであり、「レトロインベルソ」ペプチドは、アミノ酸の配列が逆転し(「レトロ」)、且つ全てのL−アミノ酸がD−アミノ酸で置換されたものである。例えば、親ペプチドがThr−Ala−Tyrであれば、そのレトロ体はTyr−Ala−Thrであり、インベルソ体はthr−ala−tyrであり、レトロインベルソ体はtyr−ala−thrである(小文字はD−アミノ酸を示す)。親ペプチドに対して、レトロインベルソペプチドは側鎖の実質的にオリジナルな空間的配置を保有しながら逆転した主鎖を持つので、親ペプチドに非常に似たトポロジーのイソマーである。
【0046】
ペプチド模倣体は、ポリペプチド/ペプチドと構造的に同じで、かつ本発明のポリペプチドあるいはペプチドの機能を模倣した化学的部位を含む化合物である。例えば、ポリペプチドが機能的活性を有する2つの帯電した化学的部位を有する場合、模倣体は、帯電した化学的機能が3次元空間内に維持されるように、空間的定位及び強制的構造内に2つの帯電した化学的部位を持つ。従って、ペプチド模倣体との用語は、アイソスターを含むことを意図する。ここで用いる「アイソスター」との用語は、化学構造の立体配置がペプチドあるいはポリペプチドと同じであるために、そのポリペプチドあるいはペプチドと置換可能な化学構造を言い、例えば、その構造はポリペプチドあるいはペプチドに特異的な結合部位に適合する。ペプチド模倣体の例としては、一つ以上の主鎖修飾を含むペプチド(すなわち、アミド結合模倣体)あり、これは当該分野においてよく知られている。アミド結合模倣体の例としては、これに限定されるものではないが、−CH2NH−、−CH2S−、−CH2CH2−、−CH=CH−(シス及びトランス)、−COCH2−、−CH(OH)CH2−、及び−CH2SO−が挙げられる(例えば、Spatola, Vega Data Vol. 1, Issue 3, (1983); Spatola, in Chemistry and Biochemistry of Amino Acids Peptides and Proteins, Weinstein, ed., Marcel Dekker, New York, p. 267 (1983); Morley, J. S., Trends Pharm. Sci. pp. 463-468 (1980); Hudson et al., Int. J. Pept. Prot. Res. 14:177-185 (1979); Spatola et al., Life Sci. 38:1243-1249 (1986); Hann, J. Chem. Soc. Perkin Trans. I 307-314 (1982); Almquist et al., J. Med. Chem. 23:1392-1398 (1980); Jennings-White et al., Tetrahedron Lett. 23:2533 (1982); Szelke et al., EP 45665 (1982); Holladay et al., Tetrahedron Lett. 24:4401-4404 (1983);及びHruby, Life Sci. 31:189-199 (1982)参照)。ペプチド模倣体の他の例としては、一つ以上のベンゾジアゼピン分子で置換されたペプチド(例えば、James, G. L. et al. (1993) Science 260:1937-1942参照)、及び架橋してラクタムあるいは他の環状構造を形成した主鎖を含むペプチドが挙げられる。
【0047】
当該分野の技術者は、ペプチドあるいはポリペプチドの全てのアミノ酸が修飾される必要はないということを理解するであろう。同様に、全てのアミノ酸が同じ方法で修飾される必要はない。このように本発明のポリペプチド/ペプチド誘導体、類似体及びペプチド模倣体は、2つ以上の化学的に別個の領域を有するキメラ分子を含み、それぞれの領域は少なくとも一つのアミノ酸あるいはその修飾体を含む。
【0048】
変異体のポリペプチドあるいはペプチドは、一つ以上のアミノ酸残基が削除され、付加され、あるいは自然発生タンパクのアミノ酸配列にあるアミノ酸残基が置換されたものである。本発明に関連して、変異体はまた、自然発生タンパクと実質的に同じ活性を保持している。通常、変異体が一つ以上のアミノ酸置換を有する場合、それらは「同類」置換である。同類置換は、一つのアミノ酸残基を同様の側鎖特性を有する別の残基で置き換えることを伴う。当該分野において知られているように、20の自然発生アミノ酸はその側鎖の物理化学的性質によって分類することができる。適当な分類としては、アラニン、バリン、ロイシン、イソロイシン、プロリン、メチオニン、フェニルアラニン及びトリプトファン(疎水性側鎖);グリシン、セリン、スレオニン、システイン、チロシン、アスパラギン、及びグルタミン(極性非帯電側鎖);アスパラギン酸及びグルタミン酸(酸性側鎖)及びロイシン、アルギニン及びヒスチジン(塩基性側鎖)がある。アミノ酸の別の分類はフェニルアラニン、トリプトファン、及びチロシン(芳香族側鎖)である。同類置換は、アミノ酸を同じグループの別のアミノ酸で置換することを包含する。
【0049】
本発明に従って、類似体、誘導体、変異体あるいは活性フラグメントは、自然発生CT−1タンパクに比べて実質的に同じかあるいは向上した活性を有する。「実質的に同等の活性」との用語は、自然発生のCT−1タンパクの活性の約50%の活性を示す。一つの実施形態においては、実質的に同等な活性は自然発生CT−1タンパクの活性の約60%の活性を示す。別の実施形態においては、それは自然発生CT−1 タンパクの活性の約75%の活性を示す。また別の実施形態においては、類似体、誘導体、変異体あるいは活性フラグメントは自然発生のCT−1タンパク、好ましくはヒトCT−1タンパクに比べて高められた(増加した)活性を示す。CT−1の活性は、例えば、その幹細胞増殖促進能力を測定することにより決定できる。本発明の一つの実施形態においては、CT−1の活性は、その心臓幹細胞増殖促進能力を言う。幹細胞増殖の増加を測定する方法は、当該分野において知られており、ここで提供されるものを含む。
【0050】
本発明のポリペプチドは、細胞抽出物からの精製あるいは組み換え技術の使用など、当該分野で公知の方法により調製することができる。また、短鎖の配列は、当該分野で公知の方法により化学的に合成することもでき、これに限定されるものではないが、排他的固相合成法、部分的固相合成法、フラグメント縮合あるいは古典的液相合成などが挙げられる(Merrifield (1963) J. Am. Chem. Soc. 85:2149; Merrifield (1986) Science 232:341)。本発明のポリペプチドは、クロマトグラフィー(例えば、イオン交換、アフィニティ、及びサイジングカラムクロマトグラフィーあるいは高速液体クロマトグラフィー)、遠心分離、溶解度差などの標準的技術を用いて、あるいは当該分野の技術者によく知られた他の技術によって、精製することができる。
【0051】
また、ポリペプチドは、組み換え技術によって得ることもできる。通常これは、タンパクあるいはポリペプチドをエンコードするポリヌクレオチドを含む発現ベクターでの、適当な宿主細胞の形質転換(形質移入、形質導入、あるいは感染を含む)を含む。様々なCT−1遺伝子の核酸配列が、当該分野において公知であり(例えば、Pennica et al. Ibid.、米国特許第5,723,585号、5,679,545号、5,627,073号(マウス) 及び GenBank Accession Nos. Q16619及びNM_001330 (ヒト))、本発明のポリヌクレオチドの基礎として使用してよい。
【0052】
ポリヌクレオチドは、標準的技術により適当な起源から誘導あるいは精製することができる。ポリヌクレオチドは、ゲノムDNAあるいはRNAであることができ、また分離したmRNAから調製したcDNAであることができる。あるいは、標準的技術を用いて様々な起源からCT−1ポリペプチドをエンコードする他の核酸配列を得るために、公知の配列を用いてプローブを調製してもよい。核酸を得るための適当な起源は、心筋細胞などの本発明のタンパクを発現することが知られている細胞であり、また、骨格筋組織及び測定可能なCT−1転写産物を持つ他の組織である。
【0053】
自然発生のCT−1タンパクのフラグメントあるいは変異体をエンコードするポリヌクレオチドは、部位特異的突然変異誘発技術などの標準的技術を用い、コード配列内で一つ以上のヌクレオチドの置換により、削除、付加、及び/又は構築することができる。
【0054】
また、本発明のポリペプチド及びペプチドは、融合タンパクとして得ることもできる。このような融合タンパクの用途の一つは、ポリペプチドあるいはペプチドの精製あるいは検出を改善することである。例えば、ポリペプチドあるいはペプチドは、免疫グロブリンのFcドメインに融合することができ、得られた融合タンパクは、タンパクAカラムを用いて容易に精製することができる。融合タンパクの他の例としては、ヒスチジンタグに融合したポリペプチドあるいはペプチド(Nie+樹脂カラムで精製可能)、グルタチオン−S−トランスフェラーゼに融合したポリペプチドあるいはペプチド(グルタチオンカラムで精製可能)あるいはビオチンに融合したポリペプチドあるいはペプチド(ストレプトアビジンカラムあるいはストレプトアビジン標識マグネティックビーズで精製可能)が挙げられる。融合タンパクを一旦精製した後、当該分野で知られている化学的あるいは酵素的方法を用いた部位特異的切断により、タグを除去してもよい。
【0055】
特定の開始シグナルがクローニングされたポリヌクレオチドの効率的な翻訳に要求されるかもしれない。これらのシグナルは、ATG開始コドン及び隣接配列を含む。それ自体の開始コドン及び隣接配列を含む完全な野生型遺伝子あるいはcDNAを適当な発現ベクターに挿入する場合、追加の翻訳コントロールシグナルは必要でないかもしれない、別のケースでは、おそらくATG開始コドンを含む外因性の翻訳コントロールシグナルが与えられなければならない。さらに、その開始コドンは、挿入部分の完全な翻訳を確実にするために、目的とするコード配列の読み取り枠と一致している必要がある。外因性の翻訳コントロールシグナル及び開始コドンは、天然あるいは合成であることができる。発現の効率を、適当な転写促進因子要素及び/又は転写終止区の包有により高めてもよい(Bittner et al. (1987) Methods in Enzymol. 153, 516)。
【0056】
本発明の核酸配列と用いる適当な発現ベクターは、これに限定されるものではないが、プラスミド、ファージミド、ウィルス粒子及びベクター、ファージなどが挙げられる。昆虫細胞としては、バクロウィルス発現ベクターが適当である。植物細胞としては、ウィルス発現ベクター(例えば、カリフラワーモザイクウィルス及びタバコモザイクウィルスなど)及びプラスミド発現ベクター(例えば、Tiプラスミドなど)が適当である。完全な発現ベクター、あるいはその一部は、宿主細胞ゲノムに組み込むことができる。ある状況下では、当該分野において公知の誘発性発現ベクターを用いることが望ましい。
【0057】
分子生物学の分野の技術者は、組み換えポリペプチドあるいはペプチドを得るために、広範な様々の発現システムを用いることができるということを理解するであろう。使用される厳密な宿主細胞は、本発明には重要ではない。ポリペプチドあるいはペプチドは、原核宿主中 (例えば、E. coliあるいはB. subtilis)あるいは真核宿主中(例えば、SaccharomycesあるいはPichia;COS、NIH 3T3、CHO、BHK、293、あるいはHeLa細胞などの哺乳類細胞;昆虫細胞;あるいは植物細胞)で生成できる。形質転換あるいは形質移入の方法及び発現ベクターの選択は、選択した宿主系に依存し、当該分野の技術者により容易に決定することができる。形質転換及び形質移入の方法は、例えば、Ausubel et al. (1994) Current Protocols in Molecular Biology, John Wiley & Sons、New Yorkに記載されている;また様々な発現ベクターは、例えば、Cloning Vectors: A Laboratory Manual (Pouwels et al., 1985, Supp. 1987)及び様々な販売業者により提供されるもののなかから選択することができる。
【0058】
また、宿主細胞は、挿入された配列の発現を調節するもの、あるいは特定の所望のやり方で遺伝子産物を修飾及び処理するものを選択してよい。そのようなタンパク産物の修飾(例えば、グリコシル化)及び処理(例えば、切断)は、タンパクの活性にとって重要であるかもしれない。種々の宿主細胞は、タンパク及び遺伝子産物の翻訳後処理及び修飾のための特徴的な特定のメカニズムを有する。発現した異種タンパクの正確な修飾及び処理を確実にするために、適当な細胞系あるいは宿主系を当該分野の技術者により選択することができる。
【0059】
発現ビヒクルを抱いた宿主細胞は、選択された遺伝子の活性化、選択された遺伝子の抑制、形質転換体の選択、あるいは選択された遺伝子の増幅の必要性に応じて、公知の方法に従って、従来の栄養培地中で培養することができる。
【0060】
II.CT−1を活性化する化合物
本発明は、CT−1を活性化し、内因性CT−1を増加させるあるいは内因性CT−1の活性を増加させる化合物を用いて、幹細胞増殖を促進する方法も意図する。CT−1アクティベーターは、インビボでCT−1の上流に作用してCT−1の発現あるいは活性を上方制御するポリペプチドあるいはポリペプチドをエンコードする遺伝子であってよく、あるいは小さな分子アクティベーターでもよい。CT−1アクティベーターは、遺伝子レベルで作用して、例えばCT−1をエンコードする遺伝子の発現を上昇させてよく、あるいはタンパクレベルで作用してCT−1ポリペプチドの活性を増加させるあるいはCT−1インヒビターの活性を減少させてもよい。CT−1アクティベーターは、例えば、ポリペプチド及びペプチド(あるいは、前記のようなポリペプチドに対応する類似体、誘導体、変異体あるいはペプチド模倣化合物)、ポリヌクレオチド、オリゴヌクレオチド、抗体あるいは抗体フラグメント、あるいは有機又は無機の小分子であることができる。
【0061】
候補アクティベーターを同定するために、当該分野において知られている様々なスクリーニング方法を採用することができる。例えば、標的遺伝子を上方制御あるいは下方制御するアクティベーターは、候補アクティベーターで処理した細胞の標的遺伝子の発現増加あるいは減少をモニターすることにより同定できる。ノーザンブロット分析、定量的RT−PCRあるいはマイクロアレイ解析などの方法を、この目的のために用いることができる。あるいは、例えば、対応するタンパクレベルの増加あるいは減少を、ウエスタンブロット分析によりモニターすることができる。
【0062】
特定のタンパクに結合するポリペプチドあるいはペプチドアクティベーター(あるいはポリペプチドに対応する類似体、誘導体、変異体あるいはペプチド模倣化合物)について、その結合能を当該分野において知られている様々な結合分析の一つを用いて決定することができる(例えば、Coligan et al., (eds.) Current Protocols in Protein Science, J. Wiley & Sons, New York, NY参照)。
【0063】
抗体あるいは抗体フラグメントアクティベーターについて、様々な免疫学的測定法を用いることができる。確立された特異性を持つポリクロナール抗体あるいはモノクロナール抗体の何れかを用いた、競合結合分析あるいは免疫放射定量測定法の多数のプロトコルが、当該分野においてよく知られている。そのような免疫学的検定法は、通常、標的タンパクとその特異的抗体との複合体形成の測定を含む。そのような技術の例としては、ELISAs、放射線免疫検定法(RIAs)、及び蛍光活性化細胞分類(FACS)などが挙げられる。あるいは、2つの非干渉エピトープに反応性を持つモノクローナル抗体を利用したツーサイドモノクローナルベース免疫学的検定法、あるいは競合結合分析を用いることができる (Maddox, D. E. et al. (1983) J. Exp. Med. 158:1211-1216参照)。そのような分析法は当該分野においてよく知られている(例えば、Hampton, R. et al. (1990) Serological Methods: A Laboratory Manual, APS Press, St Paul, Minn., Section IV; Coligan, J. E. et al. (1997, and periodic supplements) Current Protcols in Immunology, Wiley & Sons, New York, N.Y.; Maddox, D. E. et al. (1983) J. Exp. Med. 158:1211-1216参照)。
【0064】
III.CT−1インヒビター
本発明は、CT−1を抑制し、その結果内因性CT−1を減少させる、あるいは内因性CT−1活性を減少させる化合物を用いた幹細胞増殖抑制方法も意図する。CT−1インヒビターは、インビボでCT−1の上流に作用してCT−1の発現あるいは活性を上方制御するポリペプチドあるいはポリペプチドをエンコードする遺伝子であってよく、あるいは小分子のインヒビターであってもよい。CT−1インヒビターは、遺伝子レベルで作用し、例えば、CT−1をエンコードする遺伝子の発現を下方制御してよく、あるいはタンパクレベルで作用してCT−1ポリペプチドの活性を減少させてもよい。CT−1インヒビターは、例えば、野生型タンパクの活性を妨げるCT−1の不活性フラグメントなどを含むポリペプチド及びペプチド(あるいは、前記のようなポリペプチドに対応する類似体、誘導体、変異体又はペプチド模倣化合物)、ポリヌクレオチド、オリゴヌクレオチド、抗体あるいは抗体フラグメント、あるいは有機又は無機の小分子であることができる。
【0065】
CT−1アクティベーターの同定のための前記記載の当該分野で公知の様々なスクリーニング方法は、候補インヒビターの同定に採用することもできる。
【0066】
IV.幹細胞モジュレーター
また、本発明の方法は、CT−1(あるいはCT−1アクティベーター)に加えて、一つ以上の幹細胞モジュレーターの使用も意図する。幹細胞モジュレーターは、幹細胞の増殖及び/又は分化を促進してよい。従って、モジュレーターは、CT−1の活性の増加、及び幹細胞群の増殖増加のために使用することができ、あるいはCT−1での処理により予め拡張した幹細胞群の分化を促進するのに使用してもよい。幹細胞分化を促進するモジュレーターは、細胞の系統決定を刺激してよく、あるいは系列決定された前駆細胞の最終分化を刺激してもよい。
【0067】
本発明の方法において用いることができるモジュレーターの例としては、これに限定されるものではないが、ポリペプチドWntファミリーのメンバー(Wnt1、Wnt2、Wnt3、Wnt4、Wnt5a、Wnt5b、Wnt7a及びWnt7b、及びマウスWnt1、Wnt2、Wnt3a、Wnt3b、Wnt4、Wnt5a、Wnt5b、Wnt6、Wnt7a、Wnt7b、Wnt8a、Wnt8b、Wnt10a、Wnt10b、Wnt11及びWnt12などがある)及びPax7が挙げられる。一つ以上の転写因子、例えば、これに限定されるものではないがPax7、NKX2.5、GATA4、5あるいは6、MEF2C、Handl及びHand2(心筋幹細胞に対して)及びMyoD、ミオゲニン、MRF4及びMyf5(骨格筋幹細胞に対して)を、単独であるいはWnt、Pax7、あるいはその両方と組み合わせて最終分化を促進するために用いてもよい。
【0068】
モジュレーターは、完全長のポリペプチドとして、あるいは活性フラグメントあるいはその変異体(前述)として提供されてよく、あるいは完全長のポリペプチド、活性フラグメントあるいは変異体をエンコードし発現することができるポリヌクレオチドとして提供されてもよい。様々なWntタンパク、Pax7及び多くの転写因子のアミノ酸及び核酸配列が当該分野において知られている(例えば、GenBank Accession Nos. NM_002584及びNP_002575 (Pax7))。
【0069】
本発明の一つの実施形態においては、モジュレーターは幹細胞分化を促進する。別の実施形態においては、幹細胞モジュレーターはWntポリペプチドあるいはWntポリペプチドをエンコードするポリヌクレオチドである。さらなる実施形態においては、幹細胞モジュレーターはWnt11ポリペプチドあるいはWnt11ポリペプチドをエンコードするポリヌクレオチドである。また別の実施形態においては、幹細胞モジュレーターはPax7ポリペプチドあるいはPax7ポリペプチドをエンコードするポリヌクレオチドである。
【0070】
幹細胞増殖試験
CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの、単独でのあるいは幹細胞モジュレーターと組み合わせたときの幹細胞増殖促進能力は、標準的技術を用いてインビトロあるいはインビボで試験することができ、これに限定されるものではないが、ここで記載された技術が挙げられる。一つ以上のCT−1インヒビターによる幹細胞増殖の抑制も、インビトロあるいはインビボで測定することができる。
【0071】
I.インビトロ試験
通常、幹細胞は試験化合物の存在下及び非存在下で培養し、次いで細胞の少なくとも一つの増殖指標をモニターして試験化合物に暴露した細胞での増殖が刺激あるいは抑制されたか否かを決定する。あるいは、幹細胞群を「エジュケーター」細胞と共培養することができ、エジュケーター細胞を、試験化合物に暴露し、次いで幹細胞中少なくとも一つの増殖指標をモニターする。エジュケーター細胞は、共培養の前あるいは共培養中に試験化合物に暴露してよい。様々な組織に由来する成体幹細胞あるいは前駆細胞を用いることができる。例としては、これに限定されるものではないが、心筋、骨格筋、膵臓組織、神経組織、肝臓組織あるいは骨髄、造血細胞、筋芽細胞、肝細胞、胸腺細胞、心筋細胞などからの幹細胞が挙げられる。胚幹細胞を用いてもよい。
【0072】
幹細胞を培地中維持する方法は、当該分野において知られている(例えば、Madlambayan, G.J., et al., (2001) J. Hematother. Stem Cell Res. 10, 481-492; Hierlihy, A.M., et al., (2002) FEBS Lett. 530, 239-243; Asakura, A., et al., (2002) J Cell Biol. 159, 123-134参照)。幹細胞は、単一培養として単独で培養することができ、あるいはエジュケーター細胞と共培養することもできる。培養期間の前、間、あるいは後に、細胞群の同定や分離あるいは分析の成功に寄与するためのステップなど追加のステップをスクリーニング方法中に含んでもよい。例えば、幹細胞群を分離及び拡張するために、成長因子あるいは他の化合物を用いてよい。EGF及びFGFは、Gritti et al(J. Neurosci. (1999) 19:3287-3297)により記載のように神経幹細胞にこの目的のために使用されており、また、Bcl−2は「筋幹細胞」群の分離に用いられている (米国特許第6,337,184号参照)。
【0073】
一般に、化合物は通常約1000倍に及ぶ濃度範囲にわたって試験され、適当な暴露プロトコルは、当該分野の技術者によって容易に確立することができる。共培養を用いる場合、エジュケーター細胞への幹細胞の最初の暴露の前、間あるいは後に、化合物へ幹細胞を暴露することができる。あるいは、試験化合物がポリヌクレオチドあるいはポリヌクレオチドによりエンコードされたポリペプチドあるいはペプチドのような化合物である場合、試験化合物が内因的に生成されるように、ここで記載のあるいは他の標準的な方法を用いて、幹細胞をその核酸あるいはそのポリヌクレオチドを含む発現ベクターで形質移入することができる。さらに、ポリヌクレオチド、あるいはポリヌクレオチドを含む発現ベクターで形質移入された、試験化合物を発現する別の細胞系と幹細胞とを共培養することにより、幹細胞を試験化合物に暴露することができる。
【0074】
上記のように、幹細胞を化合物に直接的に暴露しなくてもよいこともさらに意図する。例えば、エジュケーター細胞群あるいは第3の細胞型をまず化合物と処理し、次いで幹細胞と共培養することができる。あるいは、幹細胞をそのようなただしそれ自身は共培養中に含まれない細胞群により調整された培地の添加によって間接的に暴露することができる。さらに、幹細胞を非液体培養培地中、例えば、寒天、高分子足場、マトリックスあるいは他の構成物のような固体、ゲル状あるいは半固体の成長支持体中に取り込まれた化合物に暴露してよいことも意図される。
【0075】
定性的あるいは定量的にモニターすることができる幹細胞増殖の指標としては、例えば、全体の形態の変化、総細胞数、組織学、組織化学あるいは免疫組織化学、あるいは特異的な細胞マーカー (例えば、増殖マーカーとして、サイクリンDl、ホスホ−ヒストンH1及びH3、E2F及びPCNA)の存在、非存在あるいは相対レベルなどが挙げられる。
【0076】
形態学及び/又は総細胞数の変化は、例えば、適当な染料(ヘキスト染料の一つなど)、BrdU取り込みを用いたFACS分析により、あるいはトリチウムチミジン取り込みにより、モニターできる。細胞マーカーの存在、非存在あるいは相対レベルは、例えば、組織化学的技術、免疫学的技術、電気泳動、ウエスタンブロット分析、FACS分析、フローサイトメトリーなどにより分析できる。あるいは、例えば、PCR技術、ノーザンブロット分析、適当なオリゴヌクレオチドプローブの使用などを用いて、細胞マーカータンパクをエンコードする遺伝子から発現したmRNAの存在を検出することができる。
【0077】
CT−1(あるいはCT−1アクティベーター)及び幹細胞モジュレーターの両方で処理されたこれら細胞について、一つ以上の分化指標をモジュレーターで処理した後の幹細胞群においてモニターしてもよい。通常、分化は前記のような全体の形態変化により、あるいは系統特異的な細胞マーカーの存在によりモニターされ、これは、前記のような多数の標準的技術を用いて分析できる。
【0078】
モニター可能な適当な系統特異的細胞マーカーは、当該分野において知られている。例えば、心筋幹細胞の分化の誘導は、コネキシン−43、MEF2C及びミオシン重鎖のような心筋細胞特異的マーカーの出現をモニターすることにより決定できる;筋由来幹細胞の分化の誘導は、ミオシン重鎖、次リン酸化MyoD、ミオゲニン、Myf5及びトロポニンTのような一つ以上の筋細胞マーカータンパクの発現について細胞を試験することにより測定でき、また、ニューロスフェアあるいはSP細胞フラクションとして誘導された神経幹細胞の分化の誘導は、GFAP、MAP2及びβ−IIIチューブリンの発現をモニターすることにより決定できる(例えば、Hitoshi, S., et al., (2002) Genes & Dev. 16, 846-858参照)。
【0079】
II.インビトロ試験
また、CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの幹細胞の増殖及び/又は分化の促進能力は、例えば、一つ以上の幹細胞モジュレーターと組み合わせて使用した場合に限らず、適当な試験動物中の常在幹細胞群についてインビボで試験してもよい。同様に、CT−1インヒビターの幹細胞増殖抑制能力をインビボで試験してもよい。通常、試験化合物は、試験組織に、例えば注射により直接的に投与される。適当な期間の後、細胞が動物から採取され、幹細胞群が前記のように分析される。必要であれば、インヒビターの増殖予防能あるいは低下能を決定するために、常在幹細胞をCT−1インヒビター及び増殖を刺激する化合物と接触させることによりインヒビターを試験することができる。化合物及びインヒビターは幹細胞に同時に与えてもよく、あるいはインヒビターの前あるいは後に化合物を与えてもよい。
【0080】
本発明の一つの実施形態においては、化合物の幹細胞増殖促進能力はマウスの心臓組織においてインビボで試験される。処理マウスから分離した心臓幹細胞様群(SP)の増加がモニターされ、非処理のあるいは緩衝液や生理食塩水などのプラセボ処理の何れかであるコントロールマウスの場合と比較される。本発明のこの実施形態に従って、SPが少なくとも約2倍まで増加した場合に、化合物が幹細胞増殖を促進すると見なされる。SPの増加は、少なくとも24時間、より典型的には少なくとも96時間にわたって測定される。
【0081】
CT−1、その類似体、誘導体、変異体及び活性フラグメント、あるいはCT−1アクティベーターの損傷組織の修復能力は、適当な動物モデルで試験できる。例えば、化合物の損傷心筋組織修復能力は、冠状動脈結紮誘導性心臓障害のマウスに化合物を投与し、損傷を受けた心筋の修復をモニターすることにより、試験することができる (Guo et al., (1999) Proc. Natl. Acad. Sci. USA, 96:11507参照)。同様に、化合物の骨格筋損傷修復能力は、凍結粉砕あるいはカルジオトキシン投与により誘導された筋損傷の動物において決定できる(Megeney et al., (1996) Genes Dev., 10:1173-1183; Asakura et al., (2000) J. Cell Biol., 159:123-134参照)。
【0082】
応用
本発明は、細胞を直接的あるいは間接的にCT−1、その類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーターと接触させることにより、幹細胞の増殖を促進する方法を提供する。本発明は、さらに、細胞を直接的あるいは間接的にCT−1(あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーター)及び一つ以上の幹細胞モジュレーターと接触させることにより、幹細胞の増殖及び分化を促進する方法を提供する。幹細胞増殖抑制方法は、細胞を直接的あるいは間接的に一つ以上のCT−1インヒビターと接触させることを含む。本発明により提供される方法には多くの応用がある。例えば、本方法は、幹細胞の増殖促進のためにインビトロで使用することができ、この場合細胞をさらにインビトロで使用するため、例えば、研究目的のために使用することを予定している。本方法は、インビトロで幹細胞培養を維持するのに使用でき、また、薬物試験のための新しいインビトロモデルの開発における応用の可能性もある。
【0083】
あるいは、増殖及び/又は分化の促進方法は、幹細胞のエクスビボでの拡張及び/又は分化を刺激するのに用いることもでき、これにより、それを必要とする対象への移植あるいは投与に適した細胞群を提供することができる。幹細胞のエクスビボ拡張は、多くの疾病状態の治療のために治療的に必要なものである。
【0084】
また、増殖及び/又は分化の促進方法は、組織の常在幹細胞の増殖及び/又は分化を促進するためにインビボで使用してもよく、これにより、疾病や疾患の結果の、あるいは外科手術や傷害の後の損傷組織の交換あるいは修復を助ける。
【0085】
同様に、一つ以上のCT−1インヒビターを用いた増殖抑制方法は、インビボでの幹細胞増殖を抑制するのに用いることができ、これにより、組織中の不適当な細胞増殖が抑制あるいは最小化される。
【0086】
また、幹細胞の増殖及び続く分化を促進する連続的な方法も意図される。例えば、細胞を直接的あるいは間接的にCT−1あるいはCT−1アクティベーターと接触させることにより、幹細胞群をエクスビボで拡張してよい。拡張した細胞群を、次いで対象に投し、元の位置で幹細胞の分化を促進する一つ以上の幹細胞モジュレーターとインビボで処理する。あるいは、対象への細胞の投与の前に、両方のステップをエクスビボで行ってもよい。
【0087】
インビボ及びエクスビボでの方法について、幹細胞は自家系、同種異型、あるいは異種系であることができる。
【0088】
幹細胞増殖(及び任意には分化)を促進する方法の治療的応用は、通常、失われたあるいは損傷した組織の交換が必要な状況に関連し、例えば、化学療法あるいは放射線治療の後、筋傷害の後、あるいは疾病及び疾患の治療中や管理中に関連している。例えば、本方法は、変性筋疾患、筋ジストロフィー、神経筋変性疾患、HIV感染などの治療、管理、あるいは予防における骨格筋幹細胞;パーキンソン病やアルツハイマー病などの神経変性疾患の治療、管理あるいは予防における神経幹細胞;及び、変性あるいは虚血性心疾患、動脈硬化、高血圧、再狭窄、狭心症、リウマチ性心疾患、先天性心臓血管異常、動脈炎及び動脈、細動脈及び毛細血管の他の疾患などの治療、管理あるいは予防や、弁、通道組織あるいは血管平滑筋の再生や、あるいは冠動脈バイパス移植を受けている対象のさらなる疾病の予防における心筋細胞に用いることができる。本発明の方法を用いて治療あるいは予防してよい他の疾病あるいは疾患としては、これに限定されるものではないが、肝硬変、肝炎、糖尿などの変性肝臓疾患が挙げられる。
【0089】
幹細胞増殖抑制方法の治療的応用は、不適当な細胞増殖を防止あるいは最小化する必要がある状況に常に関係するものではないが、典型的には、例えば心臓肥大の治療あるいは予防がある。
【0090】
本発明の一つの実施形態においては、本方法は、心臓幹細胞に適用される。別の実施形態においては、本方法は、成体心臓幹細胞に適用される。またさらに別の実施形態においては、いかなる方法でも限定することを意味するものではないが、本方法は骨格筋幹細胞に適用される。
【0091】
さらに、本発明は、CT−1、その類似体、誘導体、変異体あるいは活性フラグメント、CT−1アクティベーターあるいはCT−1インヒビター、及び薬学的に許容可能な希釈剤あるいは賦形剤を含む医薬組成物も提供する。本医薬組成物は、さらに一つ以上の幹細胞モジュレーター、一つ以上の幹細胞、あるいはそれらの組み合わせを任意に含んでもよい。医薬組成物及びその調製方法は、当該分野において知られており、例えば、"Remington: The Science and Practice of Pharmacy" (formerly "Remingtons Pharmaceutical Sciences"); Gennaro, A., Lippincott, Williams & Wilkins, Philadelphia, PA (2000)に記載されている。
【0092】
医薬組成物の投与は、局所的あるいは全身的治療のどちらを望むかによって、また治療する領域によって多くのルートにより行ってよい。通常、組成物は治療すべき領域に局所投与される。投与は、局所的 (眼投与ならびに膣送達及び直腸送達などの粘膜投与を含む)、経肺的 (例えば、ネブライザーによるものなど粉末のあるいはエアゾールの吸入あるいは吹送による)、気管内、鼻腔内、表皮的、経皮的、経口的あるいは非経口的であってよい。非経口的投与としては、静脈内、動脈内、皮下、腹腔内あるいは筋肉内注射が挙げられ、例えば、これに限定されるものではないが、心臓内注射あるいは注入、あるいは頭蓋骨内投与、例えば、クモ膜下腔内あるいは脳室内投与が挙げられる。
【0093】
本発明の組成物は、薬学的に許容可能なビヒクルと組み合わせて送達されてもよい。好ましくは、そのようなビヒクルが安定性及び/又は送達性を高める。例としては、リポソーム、微粒子あるいはマイクロカプセルなどが挙げられる。本発明の様々な実施形態において、そのようなビヒクルの使用は、活性化合物の徐放性を達成するのに有利であるかもしれない。
【0094】
非経口的注射剤として製剤化する場合、医薬組成物は、好ましくは、他の溶質、例えば溶液を等張にするための十分な生理食塩水あるいはグルコースなどを含む滅菌溶液の形態で使用される。
【0095】
吸入あるいは吹送による投与のために、医薬組成物は水性あるいは部分的に水性の溶液に製剤化することができ、これはその後エアゾールの形態で利用できる。局所的使用のために、モジュレーターあるいはこれを含む医薬組成物を、薬学的に許容可能なビヒクル中の粉剤、クリームあるいはローションとして製剤化でき、これは皮膚の疾患部分に適用される。
【0096】
医薬組成物の必要用量は、採用した特定の組成物、投与経路、及び特定の治療対象により変化する。必要用量は、当該分野の技術者に知られている標準的な臨床的技術により決定することができる。治療は、一般的に各化合物の適量よりも少ない少量で開始される。その後、その状況下で最適な効果に到達するまで用量が増量される。一般に、医薬組成物は、通常いかなる有害なあるいは悪影響のある副作用も引き起こすことなく有効な結果を与える濃度で投与される。投与は、1回ユニットの投与量とすることができ、あるいは、望むのであれば一日を通して適当な時に投与するのに便利なサブユニットに分けることができる。
【0097】
幹細胞のエクスビボでの処理方法を採用する場合、幹細胞は様々な方法により対象に投与できる。通常、幹細胞の投与は局所に制限される。幹細胞は、薬学的に許容可能な液媒体中の細胞懸濁液として注射により投与することができる。あるいは、幹細胞は、元の位置で半固体あるいは固体マトリックスである、あるいはそのようになる生体適合性媒体中投与することができる。例えば、マトリックスは、コラーゲン及び/又はその誘導体、ポリ乳酸あるいはポリグリコール酸を含むマトリックスのような組織損傷部位で半固体ゲルを形成するような注射可能な液体であってもよく、あるいは含浸繊維マトリックスのようなその最終形態において移植される柔軟な固体マトリックスの一つ以上の層を含んでもよい。そのようなマトリックスは当該分野において知られており(例えば、ゲルフォーム(Gelfoam)、Upjohn, Kalamazoo, Mich.から入手可能)、傷害部位の位置に細胞を保つ機能を有し、投与した細胞が増殖及び分化する機会を増加させる。
【0098】
遺伝子治療
また、本発明は、CT−1(あるいはその変異体又は活性フラグメント、あるいはCT−1アクティベーター)及び任意的に幹細胞モジュレーターをエンコードするポリヌクレオチドの投与も意図し、これはその後当該分野において知られた様々な「遺伝子治療」方法によりインビボでエンコードされた産物を発現する。CT−1の投与方法は、当該分野において知られており、例えば、運動神経変性を治療するために、CT−1はアデノウィルスに用られた(Lesbordes et al., (2003), Hum. Molec. Gen. 12, 1223-1229)。遺伝子治療は、エクスビボ及びインビボの両方の技術を含む。従って、宿主細胞は、ポリヌクレオチドでエクスビボで遺伝的に設計することができ、そして設計された細胞はその後前記のように治療すべき患者に投与される。
【0099】
あるいは、当該分野において知られている技術を用いてポリヌクレオチドを投与することにより、インビボで細胞を設計することができる。例えば、「裸の」ポリヌクレオチド(Feigner及びRhodes、(1991) Nature 349:351-352; 米国特許第5,679,647号)や、細胞のポリヌクレオチドの取り込みを容易にする一つ以上の他の作用物質、例えばサポニン(例えば、米国特許第5,739,118号参照)あるいはカチオン性ポリアミン(例えば、米国特許第5,837,533号参照)などを含む組成物中で処方されたポリヌクレオチドを直接注射することにより;微粒子の爆撃により(例えば、「遺伝子銃」; Biolistic, Dupontの使用により);脂質、細胞表面受容体あるいはトランスフェクト剤でポリヌクレオチドをコーティングすることにより;リポソーム、微粒子、あるいはマイクロカプセル中にポリヌクレオチドを内包させることにより;核に入りこむことが知られているペプチドと結合したポリヌクレオチドを投与することにより;あるいは受容体介在性エンドサイトーシスを受けやすいリガンドと結合したポリヌクレオチドを投与することにより(例えば、Wu and Wu, (1987) J. Biol. Chem. 262:4429-4432参照)、受容体を特異的に発現する標的細胞型に用いることができる。
【0100】
あるいは、リガンドがエンドソームを混乱させる融合ウィルスペプチドを含み、ポリヌクレオチドがリソソーム分解を避けるのを可能にするポリヌクレオチド−リガンド複合体;あるいは、そのポリヌクレオチドが特定の受容体を標的とすることによってインビボで細胞特異的な取り込み及び発現の標的となり得るポリヌクレオチド−リガンド複合体が形成できる(例えば、国際特許出願WO92/06180、WO92/22635、WO92/20316、WO93/14188及びWO 93/20221参照)。また、本発明は、ポリヌクレオチドの細胞内導入、及び続く相同的組み換えによる発現用宿主細胞DNA内への組み込みも意図する (例えば、Koller and Smithies (1989) Proc. Natl. Acad. Sci. USA 86:8932-8935; Zijlstra et al. (1989) Nature 342:435-438参照)。
【0101】
ポリヌクレオチドは、適当な発現ベクターに組み込むこともできる。遺伝子治療の応用に適当な多くのベクターが当該分野において知られている(例えば、Viral Vectors: Basic Science and Gene Therapy, Eaton Publishing Co. (2000)参照)。
【0102】
発現ベクターは、プラスミドベクターでもあってよい。プラスミドDNAを生成及び精製する方法は、迅速かつ簡単である。さらに、プラスミドDNAは、通常宿主細胞のゲノムに統合せず、染色体組み込みを高めるかもしれないという遺伝毒性の問題を除去する別個の存在としてエピソーム部位に維持される。
【0103】
様々なプラスミドが現在は容易に市販品を入手でき、Escherichia coli及びBacillus subtilisに由来し、哺乳類系での使用のために特に設計されたものがある。本発明で使用できるプラスミドの例としては、これに限定されるものではないが、真核発現ベクターpRc/CMV(Invitrogen)、pCR2.1(Invitrogen)、pAd/CMV及びpAd/TR5/GFPq(Massie et al., (1998) Cytotechnology 28:53-64)が挙げられる。代表的な実施形態において、プラスミドはpRc/CMV、pRc/CMV2(Invitrogen)、pAdCMV5(IRB-NRC)、pcDNA (Invitrogen)、pAdMLP5(IRB-NRC)、あるいはpVAX(Invitrogen)である。
【0104】
あるいは、発現ベクターは、ウィルスベースのベクターであることができる。ウィルスベースのベクターとしては、これに限定されるものではないが、複製欠損性レトロウィルス、レンチウィルス、アデノウィルス及びアデノ随伴ウィルス由来のものが挙げられる。レトロウィルスベクター及びアデノ随伴ウィルスベクターは、現在、インビボで外因性遺伝子を特にヒトへトランスファーするための好ましい組み換え遺伝子送達システムである。これらのベクターは、細胞に遺伝子を効果的に送達し、トランスファーされたポリヌクレオチドは、宿主の染色体DNAに安定に組み込まれる。レトロウィルスを使用する主要な必要条件は、特に細胞群中の野生型ウィルスの拡散の可能性に関して、それらの使用の安全性、確保することである。レトロウィルスベクターが由来するレトロウィルスとしては、これに限定されるものではないが、モロニーマウス白血病ウィルス、脾臓壊死ウィルス、ラウス肉腫ウィルスのようなレトロウィルス、ハーベイ肉腫ウィルス、トリ白血病ウィルス、テナガザル白血病ウィルス、ヒト免疫不全ウィルス、アデノウィルス、骨髄増殖性肉腫ウィルス、及び乳癌ウィルスなどが挙げられる。具体的なレトロウィルスとしては、pLJ、pZIP、pWE及びpEMが挙げられ、これらは当該分野の技術者によく知られている。
【0105】
ポリヌクレオチドは通常、インビボでのエンコードされたポリヌクレオチドの発現を可能にする適当なプロモーターのコントロール下で、ベクターに組み込まれる。採用することができる適当なプロモーターとしては、これに限定されるものではないが、アデノウィルスプロモーター、例えばアデノウィルス主要後期プロモーター、E1Aプロモーター、主要後期プロモーター(MLP)及び関連リーダー配列、あるいはE3プロモーターなど;サイトメガロウィルス(CMV)プロモーター;呼吸器合胞体ウィルス(RSV)プロモーター;誘発性プロモーター、例えばMMTプロモーター、メタロチオネインプロモーターなど;ヒートショックプロモーター;アルブミンプロモーター;ApoAIプロモーター;ヒトグロブリンプロモーター;ウィルスチミジンキナーゼプロモーター、例えば単純ヘルペスチミジンキナーゼプロモーターなど;レトロウィルスLTR;ヒストン、pol III、及び(β−アクチンプロモーター;B19パルボウィルスプロモーター;SV40プロモーター;及びヒト成長ホルモンプロモーターが挙げられる。また、プロモーターは、目的遺伝子の天然プロモーターであってもよい。適当なプロモーターの選択は、ベクター、宿主細胞及びエンコードされたタンパクに依存し、当該分野の通常の技術内とみされる。
【0106】
複製欠損レトロウィルスのみを産生する特殊化した細胞系(「パッケージング細胞」という)の開発は、遺伝子治療へのレトロウィルスの利用を増加させ、欠損レトロウィルスは遺伝子治療を目的とする遺伝子トランスファーにおける使用を特徴とする(参考として、Miller, A. D. (1990) Blood 76:271参照)。従って、組み換えレトロウィルスは、レトロウィルスコード配列(gag、pol、env)の一部を被験ポリヌクレオチドにより置換され、レトロウィルスを複製欠損にするように構築することができる。複製欠損レトロウィルスは、その後、標準的技術によってヘルパーウィルスを使用し、標的細胞に感染させるのに使用可能なウィルス粒子中にパッケージされる。組み換えレトロウィルスを得るためのプロトコル及び細胞にインビトロあるいはインビボでそのようなウィルスを感染させるプロトコルは、Current Protocols in Molecular Biology, Ausubel, F. M. et al. (eds.), J. Wiley & Sons, (1989), Sections 9.10-9.14及び他の標準的な実験マニュアル中に認められる。環境栄養性及び両栄養性レトロウィルス系の両方を調製するための適当なパッケージングウィルス系の例としては、Crip、Cre、2及びAmが挙げられる。他のパッケージング細胞の例としては、これに限定されるものではないが、PE501、PA317、Ψ−2、Ψ−AM、PA12、T19−14X、VT−19−17−H2、ΨCRE、ΨCRIP、GP+E−86、GP+envAml2、及びMiller, Human Gene Therapy, Vol. 1, pgs. 5-14 (1990)に記載されたDAN細胞系が挙げられる。
【0107】
さらに、ウィルス粒子の表面でウイルスパッケージングタンパクを修飾することにより、レトロウィルス及び結果としてのレトロウィルスベースベクターの感染スペクトルを、制限することができることが示されている(例えば、PCT公開公報WO93/25234及びWO94/06920参照)。例えば、レトロウィルスベクターの感染スペクトルの修飾方法としては次のようなものが挙げられる:細胞表面抗原に特異的な抗体をウィルス性envタンパクにカップリングする(Roux et al. (1989) PNAS 86:9079-9083; Julan et al. (1992) J. Gen Virol 73:3251-3255;及びGoud et al. (1983) Virology 163:251-254);あるいは細胞表面受容体リガンドをウィルス性envタンパクにカップリングする (Neda et al. (1991) J Biol Chem 266:14143-14146)。カップリングは、タンパクあるいは他の種類(例えば、envタンパクをアシアログリコプロテインに変換するための乳糖)と化学的に架橋した形態とすることができ、また、融合タンパクの生成によることもできる(例えば、単鎖抗体/env融合タンパク)。この技術は、感染をある組織型に制限するあるいは指示するのに有用であり、また、環境栄養性ベクターを両栄養性ベクターに変換するのに用いることもできる。
【0108】
加えて、レトロウィルス遺伝子の送達の使用は、ベクター中に含まれるポリヌクレオチドの発現をコントロールする組織特異的なあるいは細胞特異的な転写調節配列の使用によりさらに高めることができる。
【0109】
遺伝子治療技術に有用な別のウィルスベクターは、アデノウィルス由来のベクターである。アデノウィルスのゲノムは、目的遺伝子産物をエンコード及び発現するが、通常の溶菌ウィルスライフサイクルにおけるその複製能力は不活化されるよう操作することができる。例えば、Berkner et al. (1988) BioTechniques 6:616; Rosenfeld et al. (1991) Science 252:431-434;及びRosenfeld et al. (1992) Cell 68:143-155参照。アデノウィルス株Ad type 5 d1324あるいは他のアデノウィルス株(例えば、Ad2、Ad3、Adzなど)に由来する適当なアデノウィルスベクターは当該分野の技術者によく知られている。組み換えアデノウィルスは、広い多様な細胞型、例えば末梢神経細胞などへの感染に使用できるようなある種の条件において有利であり得る。さらに、ウィルス粒子は比較的安定で、精製及び濃縮でき、前記のように感染性スペクトルに影響を及ぼすように修飾することができる。さらに、導入されたアデノウィルスDNA(及びそこに含まれる外来DNA)は、宿主細胞のゲノムに組み込まれるのではなく、エピソームにとどまり、これにより、導入されたDNAが宿主ゲノム(例えば、レトロウィルスDNA)に組み込まれるようになるような状況において挿入突然変異の結果として生じ得る潜在的な問題を回避する。さらに、外来DNAに対するアデノウィルスゲノムの運搬量(8キロベースまで)は他の遺伝子送達ベクターに比べて大きい(Berkner et al. cited supra; Haj-Ahmand and Graham (1986) J. Virol. 57:267)。現在使用され、また本発明により意図されるほとんどの複製欠損アデノウィルスベクターは、ウィルス性E2及びE3遺伝子の全てあるいは一部が削除されているが、アデノウィルスの遺伝物質の80%と同じ程度を維持している(例えば、Jones et al. (1979) Cell 16:683; 前記Berkner et al.;及びGraham et al. in Methods in Molecular Biology, E. J. Murray, Ed. (Humana, Clifton, N.J., 1991) vol. 7. pp. 109-127参照)。
【0110】
複製欠損ヒトアデノウィルスベクターの作成及び増殖には、特有のヘルパー細胞系が要求される。ヘルパー細胞系は、ヒト胚腎臓細胞、筋細胞、造血細胞あるいは他のヒト胚間葉細胞あるいは上皮細胞などのヒト細胞由来であってよい。あるいは、ヘルパー細胞は、ヒトアデノウィルスに許容される、すなわち、複製欠損性ウィルスの複製を可能にするのに必要な配列を与える他の哺乳動物種の細胞に由来してもよい。そのような細胞としては、例えば、293細胞、ベロ細胞あるいは他のサル胚間葉細胞あるいは上皮細胞が挙げられる。また、ブタあるいはウシアデノウィルスベクターのような非ヒトアデノウィルスベクターの使用も意図される。適当なウィルスベクター及びヘルパー細胞系の選択は、当該分野の一般的な技術内である。
【0111】
本発明の一つの実施形態においては、遺伝子治療ベクターはアデノウィルス由来ベクターである。
【0112】
キット
さらに、本発明は、CT−1、その類似体、誘導体、変異体あるいは活性フラグメント、あるいはCT−1アクティベーター、及び任意的に一つ以上の幹細胞モジュレーターを医薬組成物中に含む治療キットを提供する。一つ以上のCT−1インヒビターを医薬組成物中に含有するキットも提供される。キットの個々の成分は別々の容器に包装され、そのような容器とともに、医薬品あるいは生物学的製品の製造、使用又は販売を規制する行政機関指定の様式の通知を有することがあり、通知はヒト投与のために製造、使用あるいは販売する機関による認可を反映する。
【0113】
キットの成分が一つ以上の液体溶液で与えられる場合、液体溶液は水溶液、例えば、滅菌水溶液であることができる。この場合、容器手段はそれ自体が吸入器、注射器、ピペット、点眼ビン、あるいは組成物を患者にそこから投与してもよい他のそのような器具であってもよい。
【0114】
キットの成分は、乾燥状態あるいは凍結乾燥状態で提供されてもよく、また、キットはさらに凍結乾燥された成分の再溶解のために適当な溶媒を含むことができる。容器の数あるいはタイプに関係なく、本発明のキットは、患者への組成物の投与を補助するための器具を含んでいてもよい。そのような器具は、吸入器、注射器、ピペット、ピンセット、計量スプーン、点眼ビンあるいは医療的に認められているそのようないかなる送達ビヒクルでもよい。
【0115】
結果
適当なヌクレオチド構築物で形質転換された細胞から生成及び分泌されたCT−1は、幹細胞の増殖及び/又は分化を促進する。ここで図3及び図4を参照すると、心臓内注射後24、48、72及び96時間でのマウス心臓細胞におけるCT−1の効果を表す経時変化の結果が示されている。このデータは、SP細胞がCT−1治療に反応して増殖することを示唆する。従って、本発明は、CT−1を含む組成物あるいはCT−1を生成することができる組成物を対象に投与することを含む、対象の幹細胞の増殖促進方法を提供する。
【0116】
前記方法の一つの実施形態において、幹細胞は心臓細胞、さらに好ましくは心臓SP細胞を含む。しかしながら、幹細胞が他の流体、組織及び/又は器官に由来する幹細胞、例えば、これに限定されるものではないが、骨格筋幹細胞、骨髄幹細胞、肝臓幹細胞などに由来する幹細胞を含んでもよいことも意図される。本発明の別の実施形態においては、いかなる方法でも限定することを意味しないが、 幹細胞が骨格筋幹細胞を含んでよい。好ましくは幹細胞はヒト幹細胞である。
【0117】
CT−1を含むあるいはエンコードする組成物は、当該分野において公知の何れの経路でも対象に投与してよく、例えば、これに限定されるものではないが注射あるいは注入により投与してよい。いかなる方法でも限定することを望むものではないが、そのような注射経路としては、これに限定されるものではないが、筋肉内、皮下、腹腔内、静脈内、動脈内注射などが挙げられる。本発明の一つの実施形態において、組成物は筋肉内注射により投与される。さらなる本発明の実施形態においては、組成物は心臓内注射あるいは注入により投与される。
【0118】
前述のように、図3及び図4に示される結果は、CT−1が細胞、例えば、これに限定されるものではないがそれで処理された対象の幹細胞の増殖を促進することを示す。CT−1を発現及び分泌する細胞は、対象へのCT−1の連続的投与が可能であるが、ここで得られた結果は、CT−1の連続的投与後にSP細胞群の一時的な増加があることを示す。さらに、この結果は、SP細胞の総数が、CT−1処理後約24時間から約96時間の間にわたって、コントロールに比べて増加することを示す。これらの結果は、幹細胞がCT−1処理に非反応性である期間を回避するために、CT−1が反復投与されることを示唆する。さらに、CT−1の反復投与は、CT−1の単独投与あるいは連続投与に比較して、幹細胞のより大きな増殖及び/又は分化を促進するかしれない。また、ここで記載したような間隔をあけた反復投与は、例えば約24時間間隔の反復投与に比べて幹細胞のより大きな増殖及び/又は分化を促進するかもしれない。
【0119】
本発明は、CT−1を含む2以上の組成物を対象に投与することを含む、対象の幹細胞の増殖促進方法を提供する。好ましくは、対象へ投与する最初及び2回目の投与の間の時間、ならびに任意的にCT−1の何れの2回の投与の間の時間は、約24時間より長く、より好ましくは少なくとも約48時間、さらにより好ましくは少なくとも約72時間、もっとより好ましくは少なくとも約96時間あるいはそれ以上である。また、本発明は、対象にCT−1を2回以上投与することも意図し、この場合、何れの2回の投与の間の時間は、約2日、3日、4日、5日、6日、7日、10日、14日、21日、28日、1ヶ月、2ヶ月、3ヶ月、6ヶ月、1年、あるいはその中間の何れかの時間である。2回以上の投与は、例えば、CT−1の量、剤型、組成物中の成分、あるいはこれらの組み合わせにおいて同じであっても、あるいは異なっていてもよい。また、本発明は、対象にCT−1を含む3、4、5、6、7、あるいはそれ以上の組成物を投与することも意図する。
【0120】
本発明は、次のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法も提供する:
a)化合物を試験群に属する一つ以上の対象に投与する;
b)その一つ以上の対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離する;
c)複数の細胞をFACS分析し、一つ以上の確定した特徴を有する細胞の数を定量する;
d)その一つ以上の確定した特徴を含む細胞の数を一つ以上のコントロール対象から得られた結果と比較する。
【0121】
細胞は、一つの細胞型、あるいは多数の細胞型の組み合わせを含んでもよい。一つの実施形態において、細胞は幹細胞を含む。別の実施形態においては、細胞は心臓細胞、あるいは骨格筋細胞を含む。また別の実施形態においては、細胞は心臓幹細胞を含む。さらに別の実施形態においては、細胞は心臓SP細胞を含む。
【0122】
分析ステップ(ステップc)は、さらに細胞を分離することを含んでよいことも意図される。
【0123】
別の実施形態において、投与ステップ(ステップa)は、一つ以上の化合物を投与することを含んでもよく、その場合、少なくとも一つの化合物はCT−1ポリペプチドあるいはCT−1ポリペプチドと実質的に同じ生物学的活性を発揮する化合物であり、一つ以上の化合物がさらに少なくとも一つの幹細胞モジュレーターを含んでよく、例えば、これに限定されるものではないが、一つ以上のシグナル伝達分子、例えばこれに限定されるものではないが一つ以上のWntポリペプチド、Pax7、あるいはその両方を含んでよい。
【0124】
別の実施形態においては、次のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法が提供される:
a)一つ以上の試験対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離し、さらに任意的に一つ以上の特定の細胞群を濃縮するためにその複数の細胞を精製する;
b)その複数の細胞あるいは一つ以上の濃縮された細胞群を化合物と処理する;
c)細胞あるいは細胞群を増殖及び/又は分化するのに十分な時間培養する、及び;
d)培養ステップの結果得られたコロニー細胞の数を、一つ以上のコントロールから得られた結果と比較する。
【0125】
比較ステップ(ステップd)は、一つ以上の特定の細胞群、例えば、これに限定されるものではないが、SP細胞を同定するために、細胞をFACS分析することをさらに含んでもよい。
【0126】
上記説明は、いかなる方法でもクレームされた発明を限定することを意図せず、さらには、検討された特徴の組み合わせは、本発明の問題解決に絶対的に必要ではないかもしれない。
【0127】
本発明は、さらに以下の実施例において説明される。ただし、これらの実施例は単に説明の目的のものであることが理解されるべきであり、いかなる方法によっても本発明の範囲を制限するのに用いられるべきではない。
【実施例】
【0128】
実施例1:カルジオトロフィン−1が心臓幹細胞様群(SP)を増加させる
偏在するRSVプロモーターにより誘導された完全長CT−1を含むアデノウィルス構築物を構築した(ad−CT−1)(図1)。CT−1は、細胞からのCT−1の分泌を促進するように神経増殖シグナルに融合した。7.5×107PFUのad−CT−1を2ヶ月齢のマウスの心臓に注射した。50μLの滅菌PBSを用いて並列コントロール実験も行った。注射後72時間に細胞を分離しFACS分析した。図2は、ヘキスト33342染料で染色し、ヘキスト33342HSC染色及び幹細胞精製プロトコル(Goodell, M., et al. (1996) J Exp Med 183, 1797-806参照、これは参照することによりここに折り込まれるものである)に従って分析した心臓細胞懸濁液のFACS分析を表す。細胞は、3つの心臓から採集した。生理食塩水を注射した心臓は、約0.93%のSP群を示し(図2A)、カルジオトロフィンを注射した心臓は約3.61%のSP群を示した(図2B)。
【0129】
図2Cは、感染したH9C2細胞におけるCT−1アデノウィルス発現の証拠を表す。細胞はアデノウィルスと処理し、72時間後、これら細胞からの培地を非処理のコントロールH9C2細胞培地とともに抗CT−1抗体を用いたウエスタンブロット分析のために採集した。約24.5kDaのCT−1タンパクがブロットにおいて示されている。
【0130】
図2Dは、コントロール注射後24時間の心臓細胞のFACS分析から得られた結果を表す。図2Eは、コントロールアデノウィルス注射後24時間の心臓細胞のFACS分析の結果を表す。ベラパミルは、SP群を同定するのに使用してよい薬剤(チャンネル遮断薬)である。例えば、ベラパミルで処理した細胞はヘキスト染料を排除し、SP群はFACS分析で検出されなくなる。コントロールアデノウィルスで処理した後ベラパミル感受性SP群には全く変化がない。
【0131】
t=24、48、72及び96時間でのマウス心臓におけるad−CT−1の効果を示す経時変化を図3Aに示す。t=24時間、48時間、72時間及び96時間での心臓細胞におけるCT−1の効果を示す2つめの経時的変化を図3Bに示す。心臓細胞懸濁液をヘキスト33342染料で染色し、FACS分析した。様々な組織源を用いた先の研究により、ヘキスト染料染色細胞懸濁液がヘキスト流出サブ細胞群(サイドポピュレーションあるいはSP細胞)を示すことが決定された。これらの細胞は、分化マーカーの幹細胞様の活性、減少あるいは非存在を有し、また多剤耐性様タンパクのインヒビターであるベラパミルの存在に対する感受性によっても特徴付けられる[Goodell et al. (1997), Nat. Med. 3, 1337-1343; Jackson et al. (1999), Proc.Natl.Acad.Sci.USA 96, 14482-14486.]。従って、常在の心筋幹細胞群の存在を評価するために、成体マウスの心臓(〜2ヶ月)を単細胞懸濁液中に酵素的に解離させ、FACS分析した。FACSは、デュアルレーザーを装備したDakoCytomation MoFloフローサイトメーターを用いて行った。蛍光を2波長で測定した。前方及び側方散乱を488nmで測定した(Spectraphysic Argon laser)。ヘキスト染料を350nmで励起した(I90C UV laser from Coherent)。青色発光を424nmで測定し(424/44バンドパスフィルタ)、赤色発光を675nm以上で測定した(675AGLPロングパスフィルタ)。510DLP二色性ミラーを用いてこれら2波長を分割した。心筋細胞懸濁液のFACS分析は、マウス心臓組織からの強いヘキスト染料排除SP群を示す。全細胞は、直接心臓内注射により7.5×107PFUのad−CT−1あるいはlacZアデノウィルスを受けた2ヶ月齢のマウスの心臓から分離した。
【0132】
ここで図4を参照すると、心臓内注射後4日の回復期間中、AdCT−1処理した心臓において認められるSP細胞の相対的な数を表す結果が示されている。このデータは、各時点でコントロールを注射した心臓で得られたデータに対して標準化した。このデータは、比較的短時間内、例えば、約48時間から約72時間前後で見られる大きな増加とともに、長時間心臓SP細胞における約3倍の増加があることを示す。
【0133】
ここで表1を参照すると、CT−1で処理した対象の体重に対する心臓重量の比率をコントロールと比較して示されている。
【表1】
【0134】
統計解析は、コントロール群とCT−1処理群で得られた比率の間に有意差が全くないことを示している(p>0.05)。これらの結果は、様々な病気あるいは疾病、例えば、これに限定されるものではないが、心臓の病気あるいは疾病を、対象の心臓肥大を促進することなく予防あるいは治療するために、CT−1を対象に投与してもよいことを示唆するものである。
【0135】
24時間、48時間、72時間及び96時間の時点での骨格筋、骨髄及び肝臓におけるad−CT−1の心臓内投与の効果を図5A〜Cに示す。この結果は、ad−CT−1の心臓内注射が、対象の心臓幹細胞の増殖を選択的に促進する適当な方法を提供することを示す。同様に、いかなる方法でも制限することを望むものではないが、CT−1あるいは対象の特定の筋肉内でCT−1を産生する能力のある化合物の筋肉内注射が、その筋肉内で、二次領域、例えばこれに限定されるものではないが、心臓及び他の筋肉あるいは組織などよりも幹細胞の選択的な増殖を結果として生じるかもしれない。一方、制限することを望むものではないが、CT−1が様々な組織あるいは器官系と接触するのを可能にする一つ以上の経路により対象にCT−1、あるいはCT−1を産生する能力がある化合物を投与することは、一つ以上の組織あるいは器官系、例えば、これに限定されるものではないが心臓及び骨格筋組織において幹細胞の非選択的な増殖を結果として生じるかもしれない。これらの結論は、メチルセルロースあるいはコロニー形成促進に必要な因子を含むゲル状培地を用いたMethocult(R)分析の結果により支持される。この分析は、幹細胞群がそのライフサイクルの間の同じ点で活性化されるのであれば、確認に用いることができる。簡単に言えば、実験は製造業者の指示(StemCell Technologies、カタログ#28405、Version 3、2004年2月)に従って、注射後24、48、72あるいは96時間にadCT−1あるいはadLaczを注射したマウスについて、約10000細胞/プレートをプレーティングすることにより行った。心臓、骨格筋、及び 骨髄からのサイドポピュレーション(SP)及びメインポピュレーション(MP)の両方のフラクションを試験した。細胞は、2週間培養し、次いでコロニーを計数し、様々な細胞マーカー、例えば造血性マーカーなどを染色した。Ad−CT−1ベクターのインビボ投与は、一つ以上の骨格筋幹細胞群の増殖を増加させた。例えば、Ad−CT−1暴露骨格筋のSPの増加は小さかったが、骨格筋に由来し、かつ許容状態(すなわち、メソカルト培地)で成長した一次細胞培養は、コントロールで処理した培養に比較して、造血性コロニーの数が約10倍増加した。
【0136】
ここで図6を参照すると、CT−1の投与が、約3週齢以上のマウス対象における細胞増殖を促進することを示唆する結果が示されている。従って、本発明は、ここで全体にわたって記載される方法を意図し、その場合対象が3週齢以上、好ましくは2ヶ月齢以上である。対象がヒトである一つの実施形態において、本発明は、ここで全体にわたって記載の方法を意図し、その場合対象が約1ヶ月以上、好ましくは約2ヶ月以上、さらに好ましくは約6ヶ月以上、よりさらに好ましくは約1歳以上の年齢である。別の実施形態において、対象は成人のヒトである。
【0137】
ここで図7を参照すると、CT−1が成体の心臓幹細胞の分化を促進することを示唆する結果が示されている。構成的に発現されるGFP導入遺伝子を発現するマウスに、CT−1を注射した。心臓は24時間後に採取し、これら心臓から分離したSP細胞を正常な一次心筋細胞と共培養した。結果は、GFP標識SP細胞のコネキシン−43陽性心筋細胞への強い変換を示す。この結果は、対象の心臓幹細胞の増殖及び/又は分化を促進するためいに、CT−1を用いることができるという証拠を与える。さらに、実験から得られた結果に基づいて、本発明は、次のステップを含む、心臓幹細胞の増殖、分化あるいは増殖と分化の両方を調節する化合物のスクリーニング方法も提供する:
a)対象から心臓幹細胞を含む複数の細胞を分離する;
b)その細胞を一つ以上の化合物で処理する;
c)ステップbで処理した細胞を、前記細胞が増殖、分化あるいはその両方を受けるのに十分な期間、一つ以上の正常な一次心筋細胞と培養する、及び;
d)増殖した細胞、分化した細胞、あるいは増殖と分化の両方をした細胞の数を計数する。
処理ステップは、CT−1、あるいはそのフラグメントあるいは誘導体、CT−1活性モジュレーター、wntポリペプチド、あるいはwnt活性を発揮するそのフラグメント又は誘導体、Pax7、あるいはその何れかの組み合わせに細胞を接触させることを含んでもよい。また、他の単形質導入分子も用いてよい。分離ステップに先立って、一つ以上の幹細胞群の増殖を促進するために、対象をCT−1で処理することも意図される。さらなる実施形態においては、心臓幹細胞を含む複数の細胞は、それらが正常な一次心筋細胞から分化されるのを可能にするようなマーカーなどを含んでもよい。例えば、心臓幹細胞を含む複数の細胞は、GFP標識してもよく、一方、正常な一次心筋細胞はGFP標識されない。心臓幹細胞を含む複数の細胞と正常な心筋細胞との間の何れの分化方法もここで記載の方法において用いてよい。
【0138】
ここで図8を参照すると、Ad−CT−1の心臓内注射が損傷を受けた骨格筋の修復に影響することができるか否かを決定するための実験手順を一般的に表した図が示されている。実験は、メソカルト実験で得られた観察結果を調査するために行われた。それらの実験において、CT−1の心臓内注射がメソカルト培地上に形成された骨格筋からの「幹細胞様」コロニーの数を増加させることが認められた。これらの結果は、CT−1が骨格筋における幹細胞様群を活性化させるということ、ならびにこの群が骨格筋修復プロセスに寄与するあるいはこのプロセスを高めるかもしれないということを示唆するものである。これらの観察結果を裏づけるために、マウス後肢への損傷をヘビ毒カルジオトキシンの注射により誘導した。カルジオトキシンは、非常に短期間の内に(約1〜2日)極度の筋肉変性を誘導する。しかしながら、骨格筋は、損傷後数日で十分に自己修復することができることがよく知られている。ここでは、カルジオトキシン損傷誘導後直ちにCT−1を注射した。さらに、カルジオトキシン注射後1〜3日にCT−1を投与する同様の実験を行った。
【0139】
ここで図9を参照すると、コントロール注射、カルジオトキシン(ctx)注射、あるいはカルジオトキシンとAd−CT−1の両方の注射後約6日目の損傷範囲を表す結果が示されている。これらの実験においては、1ヶ月齢マウスからの後肢の右前脛骨筋(TA)に10μモルのカルジオトキシンを注射した。反対側面の肢をコントロールとして用いた。別のマウス群では、CT−1をカルジオトキシン注射後心臓内に投与した。6日後、TA筋を解剖し、切片作成のために凍結した。筋切片をヘマトキシリン/エオシンで染色し、膜成分及び構成成分を可視化した。CT−1をカルジオトキシンとほぼ同時に投与した場合には、筋はより無傷状態であることがわかる。
【0140】
ここで図10を参照すると、前記のように処理し、抗体アルファ−アクチニンで染色した筋肉断面切片が示されている。アルファ−アクチニンは、骨格筋のサルコメアのz−バンドを染色し、筋サルコメアの完全性の一般に認められた指標である。結果は、CT−1で処理した筋肉は、カルジオトキシン(ctx)単独で処理した筋肉よりもよくアルファ−アクチニン染色されることを示唆している。いかなる方法でも理論により結合されることあるいは制限することを望むものではないが、これらの結果は、損傷後の骨格筋の回復を高めるためにCT−1を用いることができること、及び/又は、CT−1は損傷後の変性から筋肉を保護することができることを示唆する。
【0141】
一般的プロトコル
骨髄分離
マウスから肢を切り離す。マウスの上肢の長骨(大腿骨)を離し、骨髄細胞数を増加させるために、下肢も分離してよい。
【0142】
1)周辺の筋肉及び他の組織を除去する。
2)できるだけ高く骨を切る。
3)ひざ周辺の組織を除去し、長骨を切除する。
4)一旦骨をフリーにし、インシュリン注射器を使用して5cmの組織培養皿に骨髄を押し出す。
5)5mLのPBSで骨を押し出し、確実に両末端を押し出す。
6)必要に応じて繰り返す。
7)一旦骨髄を集め、18ゲージ針で皿中で粉砕する。
8)細胞を15mLのファルコンチューブに移す。氷冷維持。
9)FACSのために、細胞を遠沈した点からのプロトコルを続け、その後、DMEM+で洗浄する*消化は全く要求されない*
【0143】
FACSプロトコルのための抗体染色
1)HBSS+中再懸濁する直前、最後の洗浄までヘキストプロトコルに完全に従う。
2)1ml/心臓でPBS+2%FBS中に再懸濁する。
3)血球計数器を用いて細胞を計数する。
式=#細胞/mm3×希釈×10000(#細胞/mL)
4)飽和曲線のために:
>細胞をPBS+2%FBSを用いて終濃度3000000細胞/mLに希釈する
>100μLを8チューブそれぞれに添加する(コントロール2本及び様々な抗体濃度6本)
>次に、抗体溶液を希釈する(例:初期濃度100μg/mLで、5μg/mLの400μLにする)
5(400)=100(x)x=20μL
20μLの抗体を380μLのdH2Oに入れる
【0144】
>100μLを取り、細胞に添加する。これを2°抗体を含まないコントロール#1と呼ぶ。
>別の100μLを取り、細胞に添加し、2°抗体を添加する。
>残った抗体(200μL)とともに、200μLのdH2Oを添加する。これは、2.5μg/mLの400μLとなる。
>適当な回数の希釈を完了するまで続ける。
>コントロール#2は、1°抗体を持たない細胞である。
【0145】
5)細胞を一次抗体と15分間氷上でインキュベートする。
6)細胞を遠心し、上清を除去する。
7)細胞をPBS+2%FBSで洗浄する。
8)200μLのPBS+2%FBS中に再懸濁する。
9)二次抗体を添加し、15分間氷上でインキュベートする。
10)細胞を再度PBS+2%FBSで洗浄する。
11)500μLのHBSS+中に再懸濁する。
12)小さな丸底のファルコンチューブに移す*FACSのため*
13)lccのインシュリン注射器を用いて何れの細胞凝集塊も除去する。
14)細胞を集めるのに用いるために、1mLの培地を入れた15mLチューブを用意することを忘れないこと。.
15)FACSを行う。
【0146】
FACS用処方
ベラパミル(100×)
100×の最終原液を作る
95%エタノールで懸濁する
最終50μM
【0147】
ヘキスト33342(200×)
200×の最終原液を作る
水で懸濁する
最終1mg/mL
【0148】
DMEM+
388mL DMEM
8mL FBS
4mL 1M Hepes
【0149】
HBSS+
100mL HBSS(Hanks Balanced Salt Solution)
2mL FBS
1mL 1M Hepes
【0150】
コラゲナーゼ−ディスパーゼ
フード内で作る
全体500mgのコラゲナーゼビンを5mLのIX PBSで溶解する
5mLづつのコラゲナーゼ−ディスパーゼ溶液各々に対し、下記を添加する:
>4.34mLディスパーゼ 2(t.c.冷凍庫)
>500μLの懸濁コラゲナーゼ(+4℃冷蔵庫)
>125μLの100mM CaCl2
【0151】
PBS+2%FBS*抗体染色時に使用*
500mL PBS
10mL FBS
【0152】
ヘキスト33342幹細胞染色及び精製プロトコル
1)フードを向いてコラゲナーゼ/ディスパーゼを37℃の水槽中で解凍する。
2)マウスを屠殺し、エタノールをスプレーし、心臓を解剖する。
3)心臓を冷PBS中に入れる(4℃に維持した標準PBS)
4)ピンセットを用いて、心臓から過剰な血液を搾り出す。
5)消化の準備ができるまで、冷PBS中で保存する。
6)フード内で、2つの刃物で心臓を細かく切る。(心臓が湿っているようにする〜もしそれらがネバネバしたら100μLのPBSを添加する)
7)その後、細切した心臓を小さな組織培養皿に移す。
8)4〜5mLのコラゲナーゼ/ディスパーゼを1から4心臓当たり添加する。
9)ピペットで吸吐してよく混ぜ、細胞凝集塊をばらばらにする。
10)37℃の培養器内に35〜38分間置く。
11)75ミクロンのメッシュフィルターを通す(ネットウエルプレートから)。
12)フィルターを5mLのPBSで浸漬し、50mLのチューブに集める。
13)50mLチューブの内容物を15mLチューブに移す。
14)1.2の設定で10分間遠心し、上清を除去、8mLの冷DMEM+中に再懸濁し、再度1.2の設定で10分間遠心する。
15)注意深く上清を除去し、沈殿物を心臓当たりそれぞれ2mLのDMEM+中に再懸濁する(細胞の数を見積もる)
16)ヘキスト及びベラパミルで染色する(両方とも冷凍庫保存)
5μL/mLの200×ヘキストを全ての細胞に添加する
1mLのヘキスト染色細胞を取り、新しい15mLチューブに入れる
15μL/mLの100×ベラパミルを添加する
17)37℃の培養器中に90分間入れる。温度一定を確保。
18)氷上に5分間置く。
19)1.2の設定で10分間遠心し、上清を除去し、8mLの冷DMEM+に再懸濁し、1.2の設定で10分間再度遠心する。
20)上清を除去し、HBSS+中再懸濁する(〜200μL/心臓)。
21)FACSのために小さな簡易栓チューブに移し、1ccのインシュリン注射器を用いて何れの細胞凝集塊も除去する。
22)この時点から氷上を維持する(生物学的に不活性)
23)細胞を集めるのに用いるために1mLの培地を入れた15mLチューブを用意するのを忘れないこと。
24)経時的に生じ得るどんな変化も最小限にするように、FACS分析をできるだけ速やかに行う。
【0153】
一次心筋細胞培養培地
培地は、使用前に新しく調製すべきであるが、冷蔵庫内で約1週間保存できる。
【0154】
50 mLのDMEM−F12に下記を添加する:
d−グルコース 0.3g
l−グルタミン 500μLの200mM原液
pen−strep 500μLの5000単位原液
インシュリン 250μLの5mg/mL原液
アポトランスフェリン 100μLの50mg/mL原液
プロゲステロン 1μLの1mM原液
プトレシン 60μLの8mg/mL原液
セレニウム 15μLの100μM原液
フンギソン 50μLの12.5μg/mL原液
ヘパリン 10μLの10mg/mL原液
【0155】
培地を0.2μmフィルターで濾過する。40μLの25μg/mL bFGF原液を濾過後添加する。EGFは任意である。使用するなら、濾過前の培地中、10μLの100μg/mL原液を50mLのDMEM−F12に入れる。
【0156】
心筋細胞分離用の処方
MEM−JM(500mL)
400mL dH2O
5.65g MEM−JM
1.0g 炭酸水素Na
NaOHでpH7.3の溶液、H2Oで終量500mL、濾過滅菌。
【0157】
2×コラゲナーゼ(新鮮なものを調製)
30mg コラゲナーゼ
15mL MEM−JM
濾過滅菌(注射器)
【0158】
DMEM−10%FBS
400mL DMEM
45mL FBS
4.5mL pen/strep
【0159】
DMEM−F12+ITS(500mL)
半パッケージのDMEM−F12(〜7.8g)
2.5mL pen/strep
500μL ITS
0.6g NaHCO3
【0160】
一次心筋細胞分離
調製:
培地を調製し、30mgのコラゲナーゼを15mLのJMEMに添加し、濾過滅菌し、37℃の水槽中に置く(〜6日齢未満の20匹のマウス)
【0161】
心臓の解剖:
1)心房を避けるようにマウスから心臓を切り取り、冷DMEM−JMの50mLチューブ中に入れる。
2)心臓をピペット吸吐によりJMEM培地中で洗浄する。
3)心臓をペトリ皿に移し、必要であればPBSで保湿する。
4)2つの手術用メスを用いて、心臓を細かく切る。
【0162】
コラゲナーゼ消化:
1)静かに心室組織をペトリ皿中で20mLのJMEMにより25mLの広口ピペットを用いて洗浄する。数サイクル後、組織及び培地をピペットで吸う。組織を落ち着かせ、組織のみを清浄な50mLチューブ中に「吐き」出す。赤血球をほとんど含む培地を廃棄する。
【0163】
2)同じ25mLピペットを用いて、50mLチューブ中の組織から残った液体を除去し、適量のJMEM及びコラゲナーゼ2×を添加する(表参照、用いた組織の量を考慮)。
【表2】
【0164】
3)37℃で静かに20−30回転/分で15分間インキュベートする。その間に、10%FBS+DMEMを入れた2−50mLチューブを用意する。
4)最初のインキュベーション後、組織及び培地の両方を25mLピペットで約3回ゆっくり粉砕する。1分待って組織を落ち着かせ、ゆっくりと上清を吸引し、廃棄する(最初ほとんどRBC及び破片)。10mLの新鮮なJMEMで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清を廃棄する。組織の量を測定し、JMEMを添加して適当な量に調製する(表参照)。コラゲナーゼを添加し、インキュベートする。
5)後のインキュベーションの終わりに、10mLピペットでゆっくりと組織及び培地の両方を吸い上げ、2回粉砕する。1分間待って、ゆっくりと上清を吸引し、10mLFBSを入れた50mLチューブに移して、コラゲナーゼ消化を停止させる。宿主組織を新鮮な10mLのJMEMで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清をFBSを入れた同じ50mLチューブに移す。組織の量を測定し、JMEMを添加して適当な量に調整し、その後コラゲナーゼを添加する。
6) FBS及び2回の洗浄から得られた上清を含む50mLチューブに、十分なDMEM+10%FBSを添加してチューブを満たす。エアープレーン中50gで5分間遠心分離する。上清を減圧吸引し、沈殿物を10mLのDMEM−20%FBS中に再懸濁する。室温でフード内に保存する。
7)ステップ4及び5をもう2回(あるいは3回)繰り返す。
8)最後のインキュベーション後、ゆっくりと2回10mLピペットで粉砕する。宿主組織を新鮮な10mLのDMEM+10%FBSで置換する。洗浄ステップを一回繰り返す。1分間組織を落ち着かせ、上清をFBSを入れた同じ50mLチューブに移す。
9)FBS及び2回の洗浄からの上清を含む50mLチューブに、十分なDMEM+10%FBSを添加してチューブを満たし、500gで5分間遠心分離する。
【0165】
前プレーティング手順:
1)上清を除去する。
2)20mLのDMEM+10%FBS中に再懸濁する。
3)ナイテックス(nytex)ナイロンフィルターを50mL滅菌チューブの上にかぶせて濾過の準備をする。濾過した懸濁液を一つの150mm滅菌ペトリ皿上に平らに分散し、37℃で15分間インキュベートする(これは第1プレーティングである)。
4)インキュベーション後、ゆっくり粉砕し、上清(〜20mL)を2番目の皿に移す。
5)古い皿を新鮮な10mLDMEM+10%FBSで洗浄し、2番目の皿に移す(これは第2プレーティングである)。37℃で15分間インキュベートする。
6)沈殿物を10mL以下のDMEM+10%FBSで再懸濁する。より少量の再懸濁液量は、細胞数計測手順の間より多い細胞を用いることを意味し、その濃度はプレーティング工程の間用いる終濃度よりも高くあるべきということに留意する。
7)細胞を〜5mLのDMEM+10%FBS中2mm×6mmの皿上にプレーティングする。コートされたプレート上で20時間インキュベートする。前プレーティングした細胞の2番目のプレートはDMEM+10%FBSで維持した。
8)20時間インキュベーション後、プレートの培地をDMEM F12+ITSに換える。
9)増殖第1日目の後、静かにプレートをすすぎ、15mLチューブに浮遊細胞を含む成長培地を移した。細胞を遠沈し、コーティングしたプレート上の新しいDMEM+10%FBS培地上に再プレーティングした。2番目の前プレートはDMEM+10%FBS中で3日間増殖させた。
【0166】
細胞数計測:
1)1.5mLのエッペンドルフチューブ中、200μLの細胞懸濁液を400μLの0.4%トリパンブルー溶液及び400μLのDMEM+10%FBSに添加する。2分間インキュベートし、血球計数器で計数する。生存心筋細胞及び非生存心筋細胞を別々に計数する。生存筋細胞がかなり多く、有核あるいは多核であるべきである。非生存細胞は、青く染色される。RBC及び他の小さな破片は無視する。
2)計数、及びプレーティング後、細胞を予め37℃に加温したDMEM+10%FBS中で終夜あるいは少なくとも16時間インキュベートする。培地を含む血清中でインキュベートするために24時間が最長である。
【0167】
本発明は、記載されたように、同じことが多くの方法において変更されてよいことは明らかである。そのような変更は、本発明の精神及び範囲を逸脱するものと見なされるべきでなく、当該分野の技術者に明白であるような全てのそのような修飾は、請求項の範囲内に含まれることを意図する。
【0168】
全ての引用は、参照することによりここに組み込まれる。
【0169】
本発明は、好ましい実施形態について記載した。しかしながら、多くの変更及び修飾がここに記載の発明の範囲を逸脱することなく為すことが可能であるということは、当該分野の技術者に明らかであろう。
【図面の簡単な説明】
【0170】
【図1】図1は、本発明で採用したアデノウィルス構成ad−CT−1の略図である。
【0171】
【図2A】図2Aは、生理食塩水注射により処理し、次いでヘキスト33342染料で染色した心臓細胞懸濁液のFACS分析を表す。
【図2B】図2Bは、カルジオトロフィン−1注射により処理し、次いでヘキスト33342染料で染色した心臓細胞懸濁液のFACS分析を表す。
【図2C】図2Cは、感染したH9C2細胞におけるCT−1アデノウィルス発現を表すウエスタンブロットを表す。約24.5kDaのCT−1タンパクがブロット中に示されている。
【図2D】図2Dは、コントロール注射後24時間の心臓細胞FACS分析から得られたコントロールの結果を表す。
【図2E】図2Eはアデノウィルス注射後24時間の心臓細胞のFACS分析のコントロール結果を表す。
【0172】
【図3A】図3Aは、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す。
【図3B−1】図3B−1は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す。
【図3B−2】図3B−2は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−3】図3B−3は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−4】図3B−4は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−5】図3B−5は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−6】図3B−6は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−7】図3B−7は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【図3B−8】図3B−8は、ad−CT−1注射後t=24、48、72及び96時間のマウス心臓におけるad−CT−1の効果を表す経時変化の結果を示す(続き)。
【0173】
【図4】図4は、心臓内注射後、4日の回復期間の間、AdCT−1処理した心臓に見られたSP細胞の相対数を表す結果を示す。本データは、各点で、コントロール注射の心臓で得られたデータに対して標準化した。コントロール注射は、CT−1をエンコードしていない同等のAdベクターからなっていた。
【0174】
【図5A】図5Aは、24時間、48時間、72時間及び96時間の時点での、骨格筋からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【図5B】図5Bは、24時間、48時間、72時間及び96時間の時点での、骨髄からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【図5C】図5Cは、24時間、48時間、72時間及び96時間の時点での、肝臓からのSP細胞におけるad−CT−1の心臓内投与の結果を示す。
【0175】
【図6】図6は、3週、4ヶ月及び1年時点でのマウス心臓のコントロール及びCT−1注射後のFACS分析の結果を示し、マウス心臓におけるCT−1の年齢依存性効果の可能性を示唆するものである。
【0176】
【図7】図7は、CT−1が成体心臓幹細胞の分化を促進することを示唆する結果を示す。構成的に発現されるGFP導入遺伝子を発現するマウスにCT−1を注射した。心臓を24時間後に採取し、その心臓から分離したSP細胞を正常な一次心筋細胞と共培養した。結果は、GFP標識したSP細胞のコネキシン−43陽性心筋細胞への強い変換を示す。
【0177】
【図8】図8は、Ad−CT−1の心臓内注射が損傷を受けた骨格筋の修復に影響し得るか否かを確認するための実験手順の概略を表す。
【0178】
【図9】図9は、コントロール注射、カルジオトキシン(ctx)注射、あるいはカルジオトキシンとAd−CT−1の両方の注射後約6日の損傷の範囲を表す結果を示す。これらの実験において、1ヶ月齢マウスからの後肢右前脛骨筋(TA)に10マイクロモルのカルジオトキシンを注射した。反対側の肢をコントロールとして用いた。別のマウスグループに、カルジオトキシン注射後、CT−1を心臓内投与した。6日後、TA筋を解剖し、切片作成のため凍結した。筋肉切片をヘマトキシリン/エオシンで染色し膜及び構成成分を可視化した。
【0179】
【図10】図10は、アルファ−アクチニン抗体で筋肉切片を染色した結果を示す。この抗体は、骨格筋のサルコメアのz−バンドを染色し、筋肉サルコメアの完全性の一般に認められた指標である。
【特許請求の範囲】
【請求項1】
幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドと接触させることを含む、幹細胞群の増殖促進方法。
【請求項2】
幹細胞を、一つ以上のカルジオトロフィン−1インヒビターと接触させることを含む、幹細胞増殖抑制方法。
【請求項3】
幹細胞を、カルジオトロフィン−1アミノ酸配列を含む第一のポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記第一のポリペプチドをエンコードするポリヌクレオチド、及び一つ以上の幹細胞モジュレーターと接触させることを含み、前記一つ以上の幹細胞モジュレーターがポリペプチドあるいはポリペプチドをエンコードするポリヌクレオチドである、幹細胞の増殖及び分化の促進方法。
【請求項4】
前記幹細胞モジュレーターがWntポリペプチドである、前記請求項3記載の方法。
【請求項5】
前記幹細胞モジュレーターがPax7である、前記請求項3記載の方法。
【請求項6】
前記一つ以上の幹細胞モジュレーターが少なくとも一つのWntポリペプチド及びPax7を含むことを特徴とする、前記請求項3記載の方法。
【請求項7】
前記方法がインビトロで行われる、前記請求項1記載の方法。
【請求項8】
前記方法がインビボで行われる、前記請求項1記載の方法。
【請求項9】
前記幹細胞が心臓幹細胞である、前記請求項1記載の方法。
【請求項10】
カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を哺乳類に投与することを含む、哺乳類の心臓幹細胞の増殖促進方法。
【請求項11】
さらに、前記心臓幹細胞の増殖及び/又は分化を誘導する一つ以上の幹細胞モジュレーターを投与することを含む、前記請求項10記載の方法。
【請求項12】
カルジオトロフィン−1インヒビターの有効量を哺乳類に投与することを含む、哺乳類の心臓幹細胞の増殖抑制方法。
【請求項13】
カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を哺乳類に投与することを含み、前記ポリペプチドが心臓幹細胞の増殖を促進する能力を有する、哺乳類の心臓組織の修復あるいは再生方法。
【請求項14】
カルジオトロフィン−1活性を有する少なくとも一つのペプチド、あるいはカルジオトロフィン−1活性を有するペプチドを発現可能なヌクレオチド配列を含む少なくとも一つの組成物を対象に投与することを含む、対象の幹細胞群の増殖、分化あるいは増殖及び分化の両方を促進する方法。
【請求項15】
前記幹細胞が心臓幹細胞あるいは骨格筋幹細胞を含む、前記請求項14記載の方法。
【請求項16】
前記幹細胞が心臓幹細胞を含む、前記請求項15記載の方法。
【請求項17】
前記幹細胞が骨格筋幹細胞を含む、前記請求項16記載の方法。
【請求項18】
前記投与ステップが、皮下注射、筋肉内注射、腹腔内注射、あるいは静脈内注射を含む、前記請求項14記載の方法。
【請求項19】
前記筋肉内注射が心臓内注射を含む、前記請求項18記載の方法。
【請求項20】
前記ヌクレオチド配列がウィルスのヌクレオチド配列を含む、前記請求項14記載の方法。
【請求項21】
前記ウィルスのヌクレオチド配列が、アデノウィルスあるいはレトロウィルスのヌクレオチド配列である、前記請求項20記載の方法。
【請求項22】
前記ヌクレオチド配列が、前記対象の細胞からカルジオトロフィン−1活性を有するペプチドの分泌を促進する分泌ヌクレオチド配列を含む、前記請求項20記載の方法。
【請求項23】
前記分泌ヌクレオチド配列が、神経増殖シグナル配列を含む、前記請求項22記載の方法。
【請求項24】
前記投与ステップが、それぞれがカルジオトロフィン活性を有する2以上の組成物を注射することを含み、前記2以上の注射が少なくとも約96時間の時間的間隔により分けられている、前記請求項14記載の方法。
【請求項25】
下記a及びbを含む組成物:
a)カルジオトロフィン−1、あるいはカルジオトロフィン−1をエンコードするするヌクレオチド配列;
b)次の群から選ばれる、少なくとも一つの幹細胞増殖、分化あるいはその両方のモジュレーター:
i)一つ以上のWntポリペプチド;
ii)一つ以上のWntポリペプチドをエンコードする一つ以上のヌクレオチド配列;
iii)Pax7ポリペプチド;
iv)Pax7ポリペプチドをエンコードするヌクレオチド配列;あるいは
それらの組み合わせ。
【請求項26】
下記ステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法:
a)化合物を試験グループに属する一つ以上の対象に投与する;
b)前記一つ以上の対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離する;
c)複数の細胞をFACS分析し、一つ以上の確定した特徴を含む細胞の数を定量する、及び;
d)その一つ以上の確定した特徴を持つ細胞の数を一つ以上のコントロール対象から得られた結果と比較する。
【請求項27】
下記のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法:
a)一つ以上の試験対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離し、任意的に一つ以上の特定の細胞群を濃縮するために前記複数の細胞を精製する;
b)前記複数の細胞あるいは前記一つ以上の濃縮された細胞群を化合物と処理する;
c)前記細胞あるいは細胞群を、増殖及び/又は分化するのに十分な時間培養する、及び;
d)培養ステップの結果得られたコロニー細胞の数を一つ以上のコントロールから得られた結果と比較する。
【請求項1】
幹細胞を、カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドと接触させることを含む、幹細胞群の増殖促進方法。
【請求項2】
幹細胞を、一つ以上のカルジオトロフィン−1インヒビターと接触させることを含む、幹細胞増殖抑制方法。
【請求項3】
幹細胞を、カルジオトロフィン−1アミノ酸配列を含む第一のポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記第一のポリペプチドをエンコードするポリヌクレオチド、及び一つ以上の幹細胞モジュレーターと接触させることを含み、前記一つ以上の幹細胞モジュレーターがポリペプチドあるいはポリペプチドをエンコードするポリヌクレオチドである、幹細胞の増殖及び分化の促進方法。
【請求項4】
前記幹細胞モジュレーターがWntポリペプチドである、前記請求項3記載の方法。
【請求項5】
前記幹細胞モジュレーターがPax7である、前記請求項3記載の方法。
【請求項6】
前記一つ以上の幹細胞モジュレーターが少なくとも一つのWntポリペプチド及びPax7を含むことを特徴とする、前記請求項3記載の方法。
【請求項7】
前記方法がインビトロで行われる、前記請求項1記載の方法。
【請求項8】
前記方法がインビボで行われる、前記請求項1記載の方法。
【請求項9】
前記幹細胞が心臓幹細胞である、前記請求項1記載の方法。
【請求項10】
カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を哺乳類に投与することを含む、哺乳類の心臓幹細胞の増殖促進方法。
【請求項11】
さらに、前記心臓幹細胞の増殖及び/又は分化を誘導する一つ以上の幹細胞モジュレーターを投与することを含む、前記請求項10記載の方法。
【請求項12】
カルジオトロフィン−1インヒビターの有効量を哺乳類に投与することを含む、哺乳類の心臓幹細胞の増殖抑制方法。
【請求項13】
カルジオトロフィン−1アミノ酸配列を含むポリペプチド、あるいはその類似体、誘導体、変異体あるいは活性フラグメント、あるいは前記ポリペプチドをエンコードするポリヌクレオチドの有効量を哺乳類に投与することを含み、前記ポリペプチドが心臓幹細胞の増殖を促進する能力を有する、哺乳類の心臓組織の修復あるいは再生方法。
【請求項14】
カルジオトロフィン−1活性を有する少なくとも一つのペプチド、あるいはカルジオトロフィン−1活性を有するペプチドを発現可能なヌクレオチド配列を含む少なくとも一つの組成物を対象に投与することを含む、対象の幹細胞群の増殖、分化あるいは増殖及び分化の両方を促進する方法。
【請求項15】
前記幹細胞が心臓幹細胞あるいは骨格筋幹細胞を含む、前記請求項14記載の方法。
【請求項16】
前記幹細胞が心臓幹細胞を含む、前記請求項15記載の方法。
【請求項17】
前記幹細胞が骨格筋幹細胞を含む、前記請求項16記載の方法。
【請求項18】
前記投与ステップが、皮下注射、筋肉内注射、腹腔内注射、あるいは静脈内注射を含む、前記請求項14記載の方法。
【請求項19】
前記筋肉内注射が心臓内注射を含む、前記請求項18記載の方法。
【請求項20】
前記ヌクレオチド配列がウィルスのヌクレオチド配列を含む、前記請求項14記載の方法。
【請求項21】
前記ウィルスのヌクレオチド配列が、アデノウィルスあるいはレトロウィルスのヌクレオチド配列である、前記請求項20記載の方法。
【請求項22】
前記ヌクレオチド配列が、前記対象の細胞からカルジオトロフィン−1活性を有するペプチドの分泌を促進する分泌ヌクレオチド配列を含む、前記請求項20記載の方法。
【請求項23】
前記分泌ヌクレオチド配列が、神経増殖シグナル配列を含む、前記請求項22記載の方法。
【請求項24】
前記投与ステップが、それぞれがカルジオトロフィン活性を有する2以上の組成物を注射することを含み、前記2以上の注射が少なくとも約96時間の時間的間隔により分けられている、前記請求項14記載の方法。
【請求項25】
下記a及びbを含む組成物:
a)カルジオトロフィン−1、あるいはカルジオトロフィン−1をエンコードするするヌクレオチド配列;
b)次の群から選ばれる、少なくとも一つの幹細胞増殖、分化あるいはその両方のモジュレーター:
i)一つ以上のWntポリペプチド;
ii)一つ以上のWntポリペプチドをエンコードする一つ以上のヌクレオチド配列;
iii)Pax7ポリペプチド;
iv)Pax7ポリペプチドをエンコードするヌクレオチド配列;あるいは
それらの組み合わせ。
【請求項26】
下記ステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法:
a)化合物を試験グループに属する一つ以上の対象に投与する;
b)前記一つ以上の対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離する;
c)複数の細胞をFACS分析し、一つ以上の確定した特徴を含む細胞の数を定量する、及び;
d)その一つ以上の確定した特徴を持つ細胞の数を一つ以上のコントロール対象から得られた結果と比較する。
【請求項27】
下記のステップを含む、対象の細胞増殖を高める化合物のスクリーニング方法:
a)一つ以上の試験対象の少なくとも一つの流体、組織あるいは器官から複数の細胞を分離し、任意的に一つ以上の特定の細胞群を濃縮するために前記複数の細胞を精製する;
b)前記複数の細胞あるいは前記一つ以上の濃縮された細胞群を化合物と処理する;
c)前記細胞あるいは細胞群を、増殖及び/又は分化するのに十分な時間培養する、及び;
d)培養ステップの結果得られたコロニー細胞の数を一つ以上のコントロールから得られた結果と比較する。
【図1】







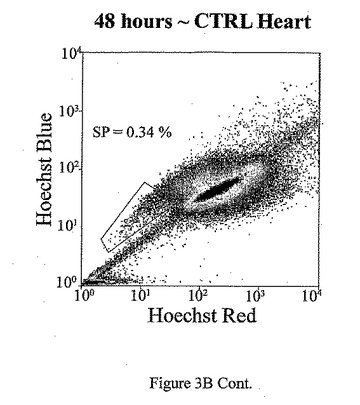
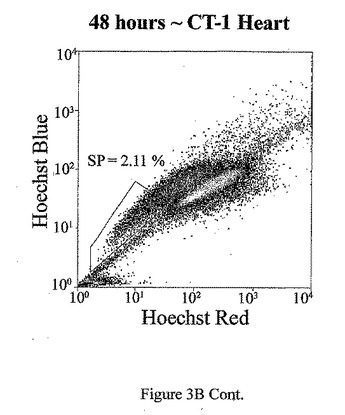
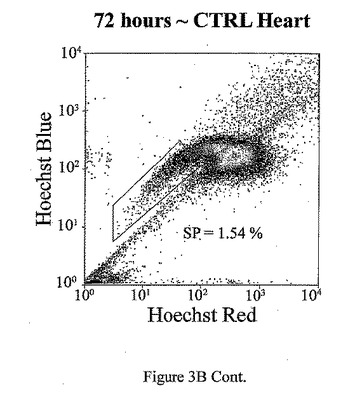
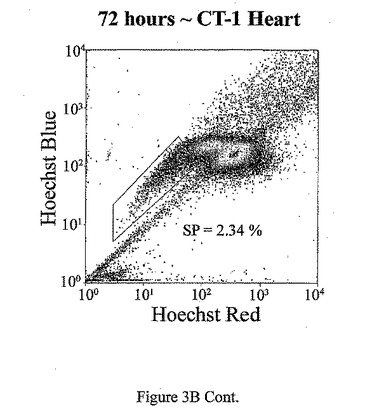
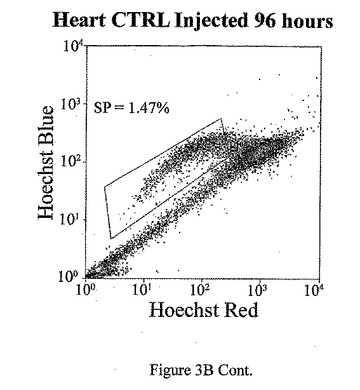
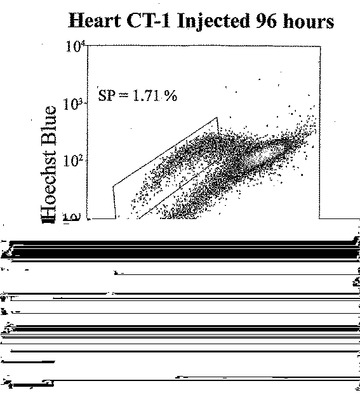
【図4】



【図6】

【図7】

【図8】
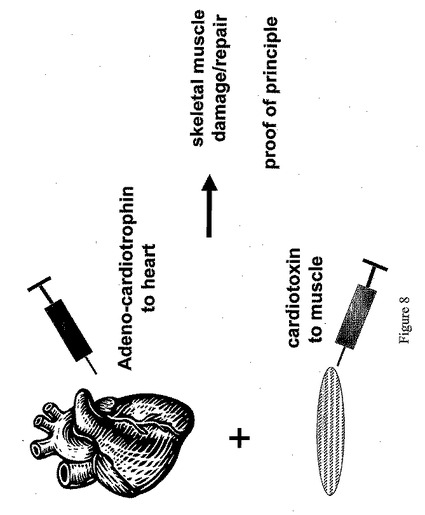
【図9】
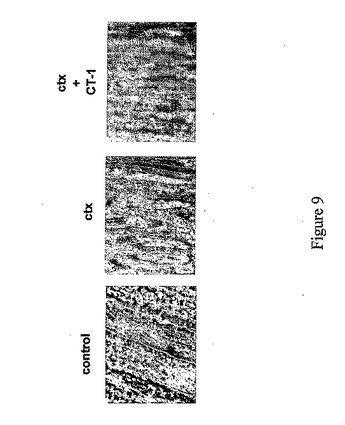
【図10】
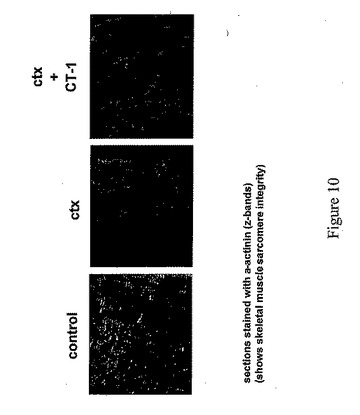
【図4】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2007−537692(P2007−537692A)
【公表日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願番号】特願2006−515608(P2006−515608)
【出願日】平成16年6月25日(2004.6.25)
【国際出願番号】PCT/CA2004/000940
【国際公開番号】WO2004/113380
【国際公開日】平成16年12月29日(2004.12.29)
【出願人】(505471037)オタワ ヘルス リサーチ インスティテュート (2)
【氏名又は名称原語表記】OTTAWA HEALTH RESEARCH INSTITUTE
【住所又は居所原語表記】Technology Transfer Office,725 Parkdale Avenue, Ottawa, Ontario K1Y 4E9, Canada
【Fターム(参考)】
【公表日】平成19年12月27日(2007.12.27)
【国際特許分類】
【出願日】平成16年6月25日(2004.6.25)
【国際出願番号】PCT/CA2004/000940
【国際公開番号】WO2004/113380
【国際公開日】平成16年12月29日(2004.12.29)
【出願人】(505471037)オタワ ヘルス リサーチ インスティテュート (2)
【氏名又は名称原語表記】OTTAWA HEALTH RESEARCH INSTITUTE
【住所又は居所原語表記】Technology Transfer Office,725 Parkdale Avenue, Ottawa, Ontario K1Y 4E9, Canada
【Fターム(参考)】
[ Back to top ]