
ガバペンチンプロドラッグ持続放出経口薬剤
【課題】ガバペンチンプロドラッグ、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤、及びかかる薬剤を投与することによる、ガバペンチンが治療に有効である疾患、及び障害を治療する又は予防する方法を提供すること。
【解決手段】本発明は、ガバペンチンプロドラッグ、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤を開示する。当該薬剤は、ガバペンチンが治療に有効である疾患、及び障害を治療する又は予防するために有用である。
【解決手段】本発明は、ガバペンチンプロドラッグ、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤を開示する。当該薬剤は、ガバペンチンが治療に有効である疾患、及び障害を治療する又は予防するために有用である。
【発明の詳細な説明】
【技術分野】
【0001】
分野
本願の開示は、ガバペンチンプロドラッグ、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤、及びかかる薬剤を投与することによる、ガバペンチンが治療に有効である疾患、及び障害を治療する又は予防する方法に関する。
【背景技術】
【0002】
本出願は、2004年11月4日に提出された米国仮出願番号第60/625,737号の利益を主張し、その全内容を本願明細書中に援用する。
【0003】
背景
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸(1)、ガンマアミノ酪酸(GABA)類似体であるガバペンチンのプロドラッグ(2)は、経口的、あるいは哺乳類の結腸に直接的のいずれかで投薬されるとき、ガバペンチンとして高い生体内利用性を有する(Gallop et al., 国際特許公開WO02/100347号;Cundy et al.,J pharmacol Exp Ther.2004,311:315−323;Cundy et al.,J pharmacol Exp Ther.2004,311:324−333)。当該高い生体内利用性は、化合物(1)を、てんかん、痛み(特に神経障害性の痛み、並びに筋肉痛、及び骨格痛)、うつ、不安、精神病、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、炎症性障害(すなわち関節症)、不眠症、胃腸障害、ほてり、下肢静止不能症候群、尿失禁、及びエタノール離脱症候群を治療する又は予防するために有用な経口薬剤(持続放出薬剤を含む)の重要な成分とさせる。
【0004】
【化1】

【0005】
Gallop et al.,国際特許公開WO02/100347号に記載のとおり製造された化合物(1)は、含水アセトニトリルから、凍結乾燥後、ガラス状固体として単離された。この過程により入手される材料は、部分的に又は全体的に非晶質であり、あるアルカリ金属塩型は、吸湿性である。さらに最近、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の結晶性形状、及びその合成方法が記載されている(Estrada et al.,国際特許公開第WO2005/037784号)。当該化合物(1)の結晶性形状は、製剤過程、及び医薬組成物において有用な物理化学的性質を改良した。
【発明の概要】
【0006】
要約
本願は、化合物(1)の持続放出経口薬剤を開示する。ある態様において、化合物(1)の持続放出経口薬剤は、約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する。
【0007】
薬剤は、例えば、(a)約10重量%〜約80重量%の化合物(1)、及び
(b)約1重量%〜約50重量%の放出速度改良ポリマーを含むタブレットとなり得、ここで重量%は当該製剤の総乾燥重量に基づく。好適な放出速度改良ポリマーは、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、及びステアロイルマクロゴールグリセリドの如きグリセリルエステルを含む。他の好適な放出速度改良ポリマーはメタクリレート共重合体、アンモニオアルキルメタクリレート共重合体、並びにそれらの共重合体、及び組み合わせを含む。
【0008】
患者に経口(すなわち、当該患者がタブレットを嚥下することによる)で投与されるとき、当該製剤は、時間とともに血しょう中のガバペンチンの濃度曲線を提供し得、当該曲線は、図に示される最大血しょう濃度(Tmax)の形と時間を有する。
【図面の簡単な説明】
【0009】
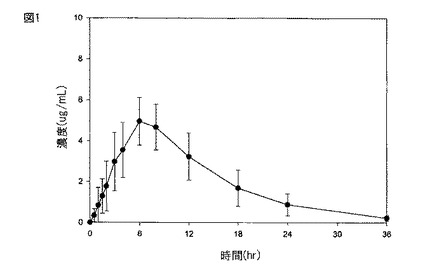
【図1】図1は、実施例3に記載されるように、絶食した患者に化合物(1)を含む持続放出タブレット(2×600mg)を経口投与した(処置A)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
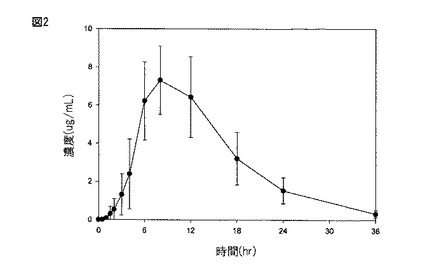
【図2】図2は、実施例3に記載されるように、食物を与えられた患者に化合物(1)を含む持続放出タブレット(2×600mg)を経口投与した(処置B)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
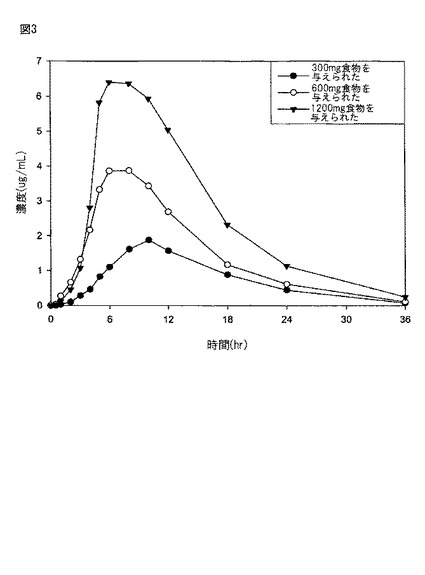
【図3】図3は、実施例4に記載されるように、食物を与えられた患者に化合物(1)を含む持続放出タブレット(1×300mg;1×600mg;及び2×600mg)を経口投与した(処置B)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
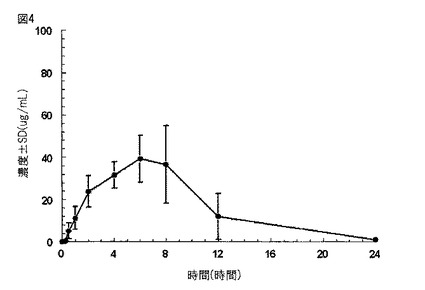
【図4】図4は、実施例5に記載されるように、絶食させた成人雄カニクイザルに化合物(1)を含む持続放出タブレット(1×600mg)を経口投与した後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図5】図5は、実施例7に記載されるように、絶食させた成人雄カニクイザルに化合物(1)を含む持続放出タブレット(1×600mg)を経口投与した後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
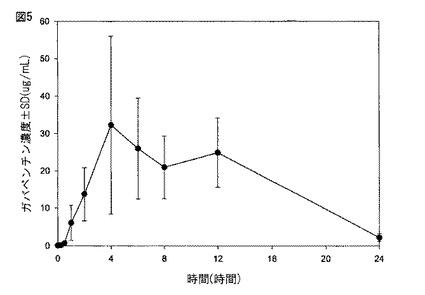
【図6】図6は、実施例1に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
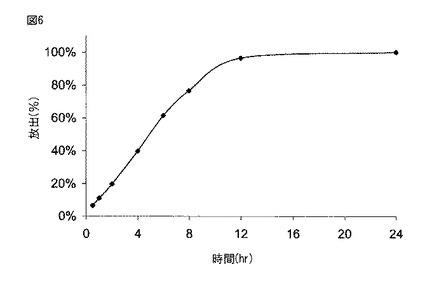
【図7A】図7Aは、実施例2に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
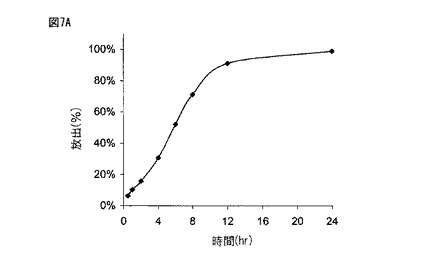
【図7B】図7Bは、実施例2に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
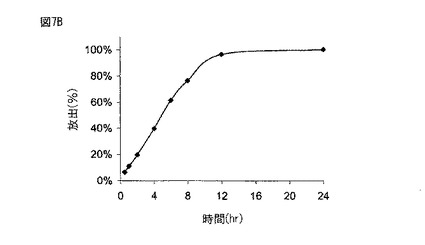
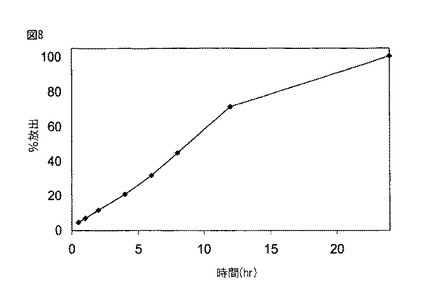
【図8】図8は、実施例6に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
【発明を実施するための形態】
【0010】
定義
「AUC」は、患者への当該化合物の投与後の時間の関数として、患者における体液中の当該化合物又はそれらの代謝体の濃度を示す曲線下の面積である。ある態様において、当該化合物はプロドラッグとなり得、及び代謝体は薬物となり得る。体液の例は、血液、及び血しょうを含む。AUCは、様々な時間間隔で、液体クロマトグラフィータンデム質量分析(LC/MS/MS)の如き方法を使用して、血しょう又は血液の如き体液中の化合物又はそれらの代謝体の濃度を測定し、及び血しょうの濃度対時間曲線下の面積を計算することにより、決定され得る。薬物の濃度対時間曲線からAUCを計算するための好適な方法は、本分野においてよく知られているだろう。本願明細書の開示に関して、ガバペンチンについてのAUCは、化合物(1)、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む薬剤の経口投与後の患者の血しょう、及び/又は血液中のガバペンチンの濃度を測定することにより決定され得る。
【0011】
「生体内利用性」は、患者への当該薬物又はプロドラッグの投与後の当該患者の全身循環に達する薬物の比率、及び量をいい、そして、例えば化合物についての血しょう及び/又は血液の濃度対時間のプロフィールを評価することにより決定され得る。血しょう及び/又は血液濃度対時間曲線を特徴付けるのに有用なパラメータは、当該曲線下の面積(AUC)、ピーク濃度までの時間(Tmax)、及び最大薬物濃度(Cmax)を含む。
【0012】
「生物学的に同等」は、患者への薬物又はプロドラッグの等量を投与した後の薬物吸収の速度及び程度が等しいことをいう。本明細書に記載される通り、もし2つのプロフィールの平均応答の比率についての90%信頼区間が0.8〜1.25の範囲内であれば、当該2つの血しょう又は血液濃度プロフィールは生物学的に同等である。当該平均応答は、AUC、Tmax、及びCmaxの如き、プロフィールの特徴的パラメータの少なくとも1つを含む。
【0013】
「Cmax」は、患者への薬物又はプロドラッグの投与後の当該患者の血しょう又は血液中の当該薬物の最大濃度である。
【0014】
「Tmax」は、患者への薬物又はプロドラッグの投与後の当該患者の血しょう又は血液中の当該薬物の最大濃度(Cmax)までの時間である。
【0015】
「絶食した患者」は、薬剤が投与される時点で、及び投与後少なくとも4時間の間に患者の胃が実質的に食物の無い状態である患者をいう。患者の胃について食事後に食物が実質的に無くなる時間は、例えば、カロリー量の如き食事の大きさ、脂肪含量の如き食事の内容、当該患者の健康状態、及び患者の胃腸管の状態を含む多くの因子に依存し得る。健康な被験者の胃は、食物の摂取後約4〜約8時間後に、通常、実質的に食物がなくなる。
【0016】
ある態様において、絶食した患者は、投薬前約10時間から、投薬後約4時間まで全く食物を食べず(しかし、適当量の水又は澄んだ液体を摂取し得る)、投薬前約2時間、及び投薬前約1時間に約250mlの水を飲み、及び投薬後約2時間に約250mlの水を飲み、投薬後約4時間に昼食を食べ、そして投薬後約10時間に夕飯を食べる。
【0017】
「食物を与えられた患者」は、その胃が食物を含む患者である。ある態様において、食物を与えられた患者は、投薬前約30分に試験食を食べ始め、投与前約5分に当該試験食を食べることを完全に終わらせ、投薬後4時間に昼食を食べ、そして投薬後約10時間に夕飯を食べる。当該試験食は、例えば、バターで揚げた2つの卵、ベーコン2切れ、バター付の小麦のトースト2枚、ハッシュトポテトの4オンス、及び全乳8オンスの如き、高脂肪(当該試験食中におけるカロリーの総量の約50%)、及び高カロリー(約1000総カロリー)の朝食を含む。当該試験食は、約150タンパク質カロリー、250炭水化物カロリー、及び約500〜600脂肪カロリーを含む。
【0018】
「患者」は、人間の如き、哺乳類をいう。
【0019】
「医薬として許容される」とは、連邦政府又は州政府の監督庁により承認される又は承認見込みであること、あるいは、動物に、より特には人間における使用として米薬局方又は他の一般的に認識される薬局方に挙げられることをいう。
【0020】
「医薬として許容される塩」は、医薬として許容され、及び親化合物の所望の医薬活性を有する本願に開示の化合物の塩をいう。かかる塩は、以下の:(1)塩酸、臭化水素酸、硫酸、硝酸、リン酸などの如き無機酸と形成される;又は酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、3−(4−ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、1,2−エタン−ジスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、4−クロロベンゼンスルホン酸、2−ナフタレンスルホン酸、4−トルエンスルホン酸、カンファースルホン酸、4−メチルビシクロ[2,2,2]−オクト−2−エン−1−カルボン酸、グルコヘプトン酸、3−フェニルプロピオン酸、トリメチル酢酸、第3ブチル酢酸、ラウリルスルホン酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸などの如き有機酸と形成される酸付加塩;又は(2)当該親化合物中に存在する酸性プロトンが、アルカリ金属イオン、アルカリ土類金属イオン、又はアルミニウムイオンの如き金属イオンにより置換される、あるいはエタノールアミン、ジエタノールアミン、トリエタノールアミン、N−メチルグルカミンなどの如き有機塩基と配位結合する、いずれかのときに形成される塩を含む。
【0021】
「予防する」又は「予防」とは、疾患又は障害に陥るリスクにおける低減をいう(すなわち、疾患にさらされる又は疾患にかかりやすいかもしれないが、当該疾患を未だ経験せず又は当該疾患の症状を未だ示さない患者において、臨床的症状の少なくとも1つを発現しないようにする)。
【0022】
「プロドラッグ」は、体内で活性薬物を放出するような転換を必要とする薬物分子をいう。化合物(1)は、患者の体内で当該親薬物であるガバペンチンを形成するよう代謝されるプロドラッグであり、それ故、化合物(1)はガバペンチンのプロドラッグである。化合物(1)、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸は、医薬として許容される塩、又は化合物(1)の遊離酸型の医薬として許容される溶媒和物、並びに上述の結晶性形状を含む。
【0023】
「溶媒和物」は、化合物と化学量論的又は非化学量論的な量の1若しくは複数の溶媒分子との分子複合体をいう。かかる溶媒分子は、医薬分野で一般的に使用されるものであり、そしてそれは、水、エタノールの如き受容者にとって無害であるとして知られるものである。化合物又は化合物の一部と溶媒との分子複合体は、例えば静電気力、ファンデルワールス力又は水素結合の如き非共有分子内力により、安定され得る。「水和物」は、当該1又は複数の溶媒分子が水であるときの複合体をいう。
【0024】
「持続放出」とは、化合物(1)の即時放出製剤の経口投与により達成される場合と比較してより長時間に渡る、全身性血液循環における化合物(1)又はそれらの活性代謝体の治療又は予防濃度を達成するのに効果的な速度での薬剤からの化合物(1)の放出をいう。ある態様において、化合物(1)の放出は、少なくとも6時間超、ある態様においては少なくとも12時間超、ある態様においては少なくとも18時間超、ある態様においては24時間超の時間に渡り生ずる。
【0025】
「治療有効量」とは、疾患を治療する又は予防するために患者に投与されたとき、当該疾患のかかる治療又は予防に十分に有効である化合物(1)の量を意味する。当該「治療有効量」は、当該疾患、及びその重篤度、並びに当該治療又は予防される疾患を有する患者の年齢、体重などに依存して変化するだろう。
【0026】
ある疾患又は障害の「処置」又は「治療」は、ある態様において、当該疾患又は障害を改善することをいう(すなわち、当該疾患又はそれらの臨床的症状の少なくとも1つの進行を止めること、又は低減すること)。他の態様において、「処置」又は「治療」は、少なくとも1つの物理的パラメータを改善することをいい、そしてそれは当該患者により認識され得又は認識され得ない。ある態様において、「処置」又は「治療」は、物理的(例えば、認識される症状の安定化)、生理学的(例えば、物理的パラメータの安定化)、又はその両方のいずれかで、当該疾患又は障害を阻害することをいう。
【0027】
「Wt%」は、重量パーセントの如き、組成物又は薬剤の総乾燥重量と比べた成分又は原料の重量をいう。例えば、40wt%の化合物(1)を含み、かつ1000mgの重量を有する薬剤は、400mgの化合物(1)を含む。化合物(1)の塩、及び/又は溶媒和物についていうとき、当該wt%は、化合物(1)の医薬として許容される塩、及び/又は医薬として許容される溶媒和物を形成する1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の質量当量をいう。
【0028】
ここに参照されるように、薬剤及び方法のある態様に対して詳細に理解されるだろう。この開示された態様は本願の請求項を制限することを意図しない。反対に、当該請求項が、当該開示された態様の全ての代替物、改良物、そして等価物を保護することを意図する。
【0029】
持続放出経口薬剤
本明細書に開示されるある態様は、タブレット形状における持続放出経口薬剤を提供するけれども、持続放出経口薬剤の分野における当業者は、粉末、粒子、サシェ、液体懸濁液、及び/又は再構成のためのカプセルの如き他の持続放出経口薬剤も使用され得ることを理解するだろう。ある態様において、薬剤はタブレットとなり得、それは、回転楕円形、角ばった楕円形又は長円体の如き薬剤の経口投与に適したいずれかの形状の内の1つとなり得る。1つのタブレットは、化合物(1)の1単位用量を含み得、又は1単位用量未満を含むミニタブレットの場合、ミニタブレットは、1単位用量を提供するためにカプセルに入れ込まれ得る。
【0030】
ある態様において、タブレットは、複数層のタブレットとなり得、当該異なる層は、各タブレット層からの化合物(1)の放出特性に影響を与える異なる粒子群、及び/又は異なる賦形剤を含む。タブレットの例は、崩壊タブレット、速溶性タブレット、発泡性タブレット、速融解タブレット、チュアブルタブレット、破砕可能タブレット、及びミニタブレットである。
【0031】
一般的に、タブレット薬剤は、少なくとも約15キロポンド(kp)(147.1ニュートンと等量)の硬さに圧宿され得る。薬剤は、本分野において知られた方法により製造され得、そして医薬として許容される賦形剤を、必要に応じてさらに含み得る。
【0032】
ある態様において、持続放出経口薬剤は、(a)約10重量%〜約80重量%の化合物(1)、及び(b)約1重量%〜約30重量%の放出速度改良ポリマー、を含む。
【0033】
ある態様において、持続放出経口薬剤は、(a)約10重量%〜約80重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマー、を含む。
【0034】
ある態様において、持続放出経口薬剤は、(a)約30重量%〜約75重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマーを含む。
【0035】
ある態様において、持続放出経口薬剤は、(a)約40重量%〜約65重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマーを含む。
【0036】
ある態様において、持続放出経口薬剤は、(a)約50重量%〜約60重量%の化合物(1)、及び(b)約20重量%〜約50重量%の放出速度改良ポリマーを含む。
【0037】
本発明に開示される薬剤は、放出速度改良ポリマー、場合により賦形剤において化合物(1)が均一に分散されるマトリクス系である。
【0038】
マトリクス系は、例えば、「Handbook of Pharmaceutical Controlled Release Technology,」ed. D.L.Wise,Marcel Dekker,Inc.(2000)、及び「Treatise on Controlled Drug Delivery,Fundamentals,Optimization, and Applications,」ed.A.Kydonieus,Marcel Dekker,Inc.(1992)に記載されるように本分野においてよく知られる。
【0039】
放出速度改良ポリマーは、薬剤からの薬物の放出を遅らせ得る。好適な放出速度改良ポリマーは、pH感受性ポリマー、pH非感受性ポリマー、水又は水性媒体と接触したとき高い膨張度を有する親水性ポリマー、水又は水性媒体と接触することでゲルを形成するポリマー、水又は水性媒体と接触したときに膨張特性、及びゲル化特性の両方を示すポリマー、ワックスの如き脂肪族化合物、及び生物分解性ポリマーを非制限的に含む。
【0040】
ある態様において、放出速度改良ポリマーは、アクリル酸、及びメタクリル酸ポリマーや共重合体、メチルメタクリレート共重合体、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミド共重合体、ポリ(メチルメタクリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)共重合体、ポリアクリルアミド、アミノアルキルメタクリレート共重合体、ポリ(メタクリル酸無水物)、グリシジルメタクリレート共重合体、アンモニオアルキルメタクリレート共重合体、及び上述のいずれかの組み合わせの如きpH感受性ポリマーとなり得る。
【0041】
ある態様において、pH依存性ポリマーは、ジエチルアミノエチルメタクリレート、及び他の天然メタクリル酸エステルから合成される共重合体となり得、Eudragit(登録商標)(Rohm Pharma)として商業的に入手可能な、メタクリル酸共重合体又はポリマーメタクリレートとしても知られる。
【0042】
ある態様において、pH非感受性ポリマーは、Eudragit(登録商標)RS、及びEudragit(登録商標)RLの如きアンモニオアルキルメタクリレート共重合体であり、それは、第4アンモニウム基の低い含有量のアクリル及びメタクリル酸エステルの共重合体を含むアクリル樹脂である。
【0043】
高い膨張性を示す親水性放出速度改良ポリマーの例は、架橋されたソジウムカルボキシメチルセルロース、架橋されたヒドロキシプロピルセルロース、高分子量ヒドロキシプロピルメチルセルロース、カルボキシメチルアミド、ポタシュームメタクリレートジビニルベンゼン共重合体、ポリメチルメタクリレート、ポリビニルピロリドン、高分子量ポリビニルアルコール、メチルセルロース、ビニルアセテート共重合体などを含む。
【0044】
水と接触してゲル化する放出速度改良ポリマーの例は、メチルセルロース、カルボキシメチルセルロース、低分子量ヒドロキシプロピルメチルセルロース、低分子量ポリビニルアルコール、ポリオキシエチレングリコール、非架橋ポリビニルピロリドン、キサンタンガムなどを含む。
【0045】
膨張、及びゲル化特性の両方を示す放出速度改良ポリマーの例は、中間粘性ヒドロキシプロピルメチルセルロース、及び中間粘性ポリビニルアルコールを含む。
【0046】
ある態様において、当該放出速度改良ポリマーは、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、ステアロイルマクロゴールグリセリド、又は上述のいずれかの組み合わせの如きグリセリルエステルである。ある態様において、当該放出速度改良ポリマーは、グリセリルベヘネートである。他の脂肪、及び/又はワックス放出速度改良ポリマーは、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール、セトステアリルアルコール、パルミトイルアルコール、ouricuryワックス、硬化植物油、カンデリラワックス、エスパルトワックス、ステアリン酸、パラフィンワックス、蜜ろう、グリコワックス、キャスターワックス、及びカルナバワックスを含む。
【0047】
生体内分解性ポリマーの例は、コラーゲン、ゼラチン、ポリビニルアルコール、ポリオルトエステル、ポリアセチル、ポリオルトカルボネート、ポリアミド、ポリアミノ酸、ポリエステル、ポリ乳酸、ポリグリコール酸、ポリカルボハイドレート、ポリオルトエステル、ポリオルトカルボネート、ポリアセチル、ポリ無水物、ポリデヒドロピラン、ポリジオキシノンなどを含む。
【0048】
本発明に開示の薬剤に組み込まれ得る他の好適な放出改良ポリマーは、天然又は合成ゴムの如き親水コロイド、アカシアの如き炭水化物を基材とした物質、トラガカントゴム、ローカストビーンガム、グアガム、アガー、ペクチン、カラギーナン、可溶性及び不溶性アルギナート、カルボキシポリメチレン、カゼイン、ゼイン、ポリエチレンオキシド、無水マレイン酸/メチルビニルエーテル共重合体、及びゼラチンの如きタンパク性物質を含む。
【0049】
放出速度改良ポリマーは、単独で又は1若しくは複数の他の放出速度改良ポリマーとの組み合わせで使用され得、そして/あるいは、複数の放出速度改良ポリマーの共重合体となり得る。
【0050】
化合物(1)、並びに1又は複数の放出速度改良ポリマーの製剤は、湿式造粒法、流動造粒法、乾燥造粒法、及び直接圧縮(「Remington’s Pharmaceutical Sciences,」 Lippincott Williams & Wilkins,889−928,(2005)を参照のこと)の如き本分野においてよく知られる標準技術を使用して製造され得る。例えば、マトリクス製剤は、放出改良ポリマー、充填剤、化合物(1)、及び賦形剤を乾式混合し、その後、適した造粒が得られるまで、アルコールを使用して当該混合物を粒状にすることにより製造され得る。当該造粒は、本分野において知られた方法により実施され得る。湿った顆粒を、流動乾燥機内で乾燥させ、ふるいにかけ、そして適当な大きさの粉にすることができる。平滑剤を乾燥された顆粒と混合し、最終製剤を得ることができる。ある態様において、かかる製剤を、本分野においてよく知られた方法によりタブレット型薬剤に圧縮し得る。
【0051】
ある態様において、薬剤中の化合物(1)の量は、約50mgから約800mg、ある態様において、約100mgから約800mg、及びある態様において、約300mgから約700mgの範囲となる。医薬として許容される塩、及び/又は化合物(1)の溶媒和物を含む薬剤について、薬剤中の化合物(1)の量は、化合物(1)の質量当量をいう。薬剤中に含まれる化合物(1)の量又は荷重は、治療される特定の症状、及びその後吸収される荷重プロドラッグから産生されるガバペンチンの量に依存し得る。
【0052】
本明細書中に記載される化合物(1)、及び当該放出速度改良ポリマーに加えて、薬剤は、界面活性剤、滑剤、希釈剤、抗粘着剤、流動促進剤、緩衝液、染料、湿潤剤、乳化剤、pH緩衝剤、安定剤、増粘剤、崩壊剤、及び着色剤の如き1又は複数の医薬として許容される賦形剤を含む。かかる賦形剤は、でんぷん、糖、ゼラチン、モルト、米、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、グリセロールモノステアレート、滑石、塩化ナトリウム、グリセロール、プロピレングリコール、水、エタノールなどを含む。
【0053】
希釈剤は、薬剤を圧縮のため実用的な大きさとするために添加され得、容積を増大し得る。有用な希釈剤の例は、第2リン酸カルシウム、第2リン酸カルシウムジハイドライド、硫酸カルシウム、第2リン酸カルシウム、第3リン酸カルシウム、乳糖、微晶性セルロ−スを含むセルロース、カオリン、マンニトール、塩化ナトリウム、乾燥でんぷん、アルファでんぷん、圧縮性の糖、マンニトール、及び上述のいずれかの組み合わせを含む。ある態様において、単一の希釈剤は、第2リン酸カルシウム、及び微晶性セルロ−スから選択される。当該希釈剤が第2リン酸カルシウムである態様において、薬剤は、約30重量%〜約50重量%、及びある態様においては約35重量%〜約45重量%の範囲である希釈剤の量を含み得る。当該希釈剤が微晶性セルロースである態様において、薬剤は、約5重量%〜約20重量%、及びある態様においては約10重量%〜約16重量%の範囲である希釈剤の量を含み得る。
【0054】
流動促進剤は、本発明の薬剤中に含まれ得、製造、膜形成、及び/又は乾燥の間、付着効果を低減する。有用な流動促進剤の例は、滑石、ステアリン酸マグネシウム、グリセリルモノステアレート、コロイド状シリコンジオキシド、沈降シリコンジオキシド、上述のいずれかの組み合わせである。ある態様において、流動促進剤は、コロイド状シリコンジオキシドである。薬剤は、約2重量%未満の流動促進剤、ある態様において、約1重量%未満の流動促進剤を含み得る。
【0055】
滑剤、及び抗粘着剤は、加工に役立つために本発明の薬剤中に含まれ得る。有用な滑剤、及び/又は抗粘着剤の例は、ステアリン酸カルシウム、グリセリルベヘネート、グリセリルモノステアレート、ステアリン酸マグネシウム、鉱油、ポリエチレングリコール、ソジウムステアリルフマレート、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ステアリン酸、滑石、硬化植物油、ステアリン酸亜鉛、及び上述のいずれかの組み合わせである。ある態様において、当該滑剤は、グリセリルモノステアレートである。ある態様において、滑剤は、ステアリン酸マグネシウムである。薬剤は、約1重量%〜約13重量%の、ある態様において、約4重量%〜約10重量%の範囲の滑剤、及び/又は抗粘着剤を含み得る。
【0056】
本発明の薬剤において有用な界面活性剤の例は、医薬として許容される、アニオン性界面活性剤、カチオン性界面活性剤、両性{両親媒性(amphiphatic)/両親媒性(amphiphilic)}界面活性剤、非イオン性界面活性剤、ポリエチレングリコールエステル又はエーテル、及び上述のいずれかの組み合わせを含む。好適な医薬として許容されるアニオン性界面活性剤は、1価アルキルカルボキシレート、アシルラクチレート、アルキルエーテルカルボキシレート、N−アシルサルコシネート、多価アルキルカルボネート、N−アシルグルタメート、脂肪酸−ポリペプチド凝縮物、硫酸エステル、ラウリル硫酸ナトリウム及びドデシル硫酸ナトリウムの如きアルキルスルフェイト、エトキシル化アルキルスルフェイト、ドキュセートナトリウム及びジオクチルソジウムスクシネートの如きエステル結合スルホネート、アルファオレフィンスルホネート、又はリン酸化エトキシル化アルコールを含む。
【0057】
好適な医薬として許容されるカチオン性界面活性剤の例は、モノアルキル第4アンモニウム塩、ジアルキル第4アンモニウム化合物、アミドアミン、及びアミンイミドを含む。
【0058】
好適な医薬として許容される両性界面活性剤の例は、N−置換アルキルアミド、N−アルキルベタイン、スルホベタイン、及びN−アルキル−6−アミノプロピオネートを含む。
【0059】
好適な医薬として許容されるポリエチレングリコールエステル又はエーテルの例は、ポリエトキシル化キャスター油、ポリエトキシル化水素化キャスター油を含む。ある態様において、界面活性剤は、ラウリル硫酸ナトリウム、及びドデシル硫酸ナトリウムから選択される。ある態様において、薬剤は、約3重量%未満、ある態様においては約2重量%未満の界面活性剤を含み得る。
【0060】
タブレット薬剤の如き本発明の薬剤は、1又は複数のコーティングをさらに含む。1又は複数の追加のコーティングの目的は、物理的保護、審美、嚥下の際の容易さ、識別、及び/又は当該粒子のさらなる処理を促進するためのものである。一定のコーティングが適用され胃腸管の中における当該薬剤からの化合物(1)の放出を改良又はそれらに影響を与え得るが、一方、他のものはかかる効果を有し得ない。コーティングは、水分に対して不浸透性又は水分浸透性となり得る。
【0061】
水分浸透性の外側タブレットコーティングは、乾燥剤の存在下で包装された薬剤中の低い水分含量を維持するために有用となり得、及びそれ故、例えば当該薬剤の保存性を促進し得る。これらの追加のコーティングは、本分野においてよく知られた方法により本発明の薬剤に適用され得る。物理的保護のためのコーティングにおいて有用な材料の例は、ヒドロキシプロピルメチルセルロ−ス、ヒドロキシプロピルセルロース、ヒドロキシプロピルエチルセルロース、及びキサンタンガムの如き浸透性又は溶解性材料を含む。さらなる加工を促進するためのコーティングにおいて有用な材料の例は、滑石、コロイド状シリカ、ポリビニルアルコール、二酸化チタン、微粒シリカ、グリセロールモノステアレート、マグネシウムトリシリケート、及びステアリン酸マグネシウムを含む。コーティングは、単一材料又は本明細書中に記載されたもののいずれかを含む複数の材料を含み得る。
【0062】
本発明の薬剤は、化合物(1)、及び/又はガバペンチンの分子内環化反応により形成されるラクタム副生物を実質的に含まないものとなり得る。薬剤は、実質的にラクタム形成なし(好ましくはラクタム0.5重量%未満、より好ましくはラクタム0.2重量%未満、最も好ましくはラクタム0.1重量%未満)で、拡張保存(より好ましくは、1年超)について好ましくは安定的である。
【0063】
ある態様において、当該薬剤中の化合物(1)は、2005年7月14日に刊行されたEstrada et al,米国特許出願公開US2005/0154057号に記載された結晶性形状である。
【0064】
患者に経口で投与されたとき(すなわち、患者が当該タブレットを嚥下することにより)、本発明の薬剤は、時間とともに血しょう又は血液中のガバペンチン濃度のプロフィールを提供し得る。実施例1に記載される組成物及び荷重を有する薬剤について、ガバペンチン血しょう濃度プロフィールは、患者に化合物(1)を投与した後の、絶食した、及び食物を与えられた人間の当該患者それぞれについて、図1及び2に示される形状、大きさ、及びAUCを有する。これらのプロフィールは、ガバペンチンのみの投与後に得られたプロフィールと異なっている。1つの重要な相違は、最大血液濃度(Cmax)に達するまでの時間(Tmax)である。
【0065】
絶食した患者において、本発明の持続放出経口薬剤についてのTmaxは、約4時間超である。食物を与えられた患者において、持続放出経口薬剤についてのTmaxは、約6時間超である。対照的に、絶食した、及び食物を与えられた患者にガバペンチンのみを投与した後のTmaxは、約2〜4時間である。本発明の持続放出経口薬剤のもう1つの重要な有利点は、当該ガバペンチン代謝体の生物学的利用能である。化合物(1)の1200mgの荷重用量で、本発明の薬剤は、ガバペンチンの当モル用量の投与と比較して、絶食した患者において、少なくとも20%より高い、そしてある態様においては、少なくとも25%より高いガバペンチン生物学的利用能を提供し得る。化合物(1)の1200mgの荷重用量で、本発明の薬剤は、ガバペンチンの当モル用量の投与と比較して、食物を与えられた患者において、少なくとも50%より高い、そしてある態様においては、少なくとも100%より高いガバペンチン生物学的利用能を提供し得る。
【0066】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約3μg/mL〜約6μg/mLの範囲のCmax、約4時間〜約7時間の範囲のTmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0067】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の食物を与えられた人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約5μg/mL〜約8μg/mLの範囲のCmax、約6時間〜約11時間の範囲のTmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0068】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で絶食した人間の患者群に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約3μg/mL〜約6μg/mLの範囲の平均Cmax、約4時間〜約7時間の範囲の平均Tmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し、そして、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で食物を与えられた人間の患者群に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約5μg/mL〜約8μg/mLの範囲の平均Cmax、約6時間〜約11時間の範囲の平均Tmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0069】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約3μg/mL〜約6μg/mLの範囲のCmax、約4時間〜約7時間の範囲のTmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0070】
ある態様において、1又は複数の食物を与えられた人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約5μg/mL〜約8μg/mLの範囲のCmax、約6時間〜約11時間の範囲のTmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0071】
ある態様において、絶食した人間の患者群への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約3μg/mL〜約6μg/mLの範囲の平均Cmax、約4時間〜約7時間の範囲の平均Tmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得、そして、食物を与えられた人間の患者群への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約5μg/mL〜約8μg/mLの範囲の平均Cmax、約6時間〜約11時間の範囲の平均Tmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0072】
本発明の薬剤は、吸収の比率及び程度に関して、本明細書中に記載される薬剤と生物学的に同等の薬剤を含み、例えばそれはU.S.Food and Drug Administrationにより定義され、及び「Guidance for Industry−Bioavailability and Bioequivalence Studies for Orally Administered Drug Products」(2003)において議論されるようなものである。
【0073】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、あるいは、1又は複数の食物を与えられた人間の患者に投与したとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0074】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、かつ、1又は複数の食物を与えられた人間の患者に投与したとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0075】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、あるいは、1又は複数の食物を与えられた人間の患者への同様の薬剤の経口投与は、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0076】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、かつ、1又は複数の食物を与えられた人間の患者への同様の薬剤の経口投与は、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0077】
経口投与後、本発明の薬剤は、少なくとも約6時間の間、ある態様においては、少なくとも約12時間の間、ある態様においては、少なくとも約18時間の間、及び他の態様においては、少なくとも24時間の間、患者の血液及び/又は血しょう中のガバペンチンの治療濃度又は予防的濃度を提供し得る。患者の血液及び/又は血しょう中のガバペンチンの治療有効濃度又は予防的有効濃度は、例えば、治療される疾患、当該疾患の重篤度、当該患者の体重、当該患者の健康状態などを含む多数の要因に依存し得る。
【0078】
ある態様において、本発明の薬剤は、1日2度投与され得、ある態様においては、1日に1度投与され得る。
【0079】
治療的使用
本発明の持続放出経口薬剤は、親薬物であるガバペンチンが治療に有効だと知られている又は今後そのことが発見されるいずれかの疾患又は障害に苦しむ患者に投与され得る。ガバペンチンが処方される症状、及びそれ故本発明の薬剤もまた有効である症状は、てんかん、うつ、不安、精神病、認知、統合失調症、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、痛み{特に、神経障害性の痛み(例えばヘルペス後神経痛)、筋肉痛、及び骨格痛}、下肢静止不能症候群、ほてり、尿失禁、炎症性障害(すなわち関節症)、不眠症、胃腸障害、アルコール/コカイン中毒、エタノール離脱症候群、外陰病変、早漏、及びグルタミン酸作動性のようなものを含む。
【0080】
当該薬剤は、上記疾患又は障害に対する予防措置としても、患者に投与され得る。それ故、当該薬剤は、てんかん、うつ、不安、精神病、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、痛み(特に、神経障害性の痛み、及び筋肉痛、及び骨格痛)、炎症性障害(すなわち、関節症)、不眠症、胃腸障害、エタノール離脱症候群、早漏、及び外陰病変の傾向を有する患者へ、予防措置として投与され得る。
【0081】
従って、当該薬剤は、ある疾患又は障害の予防のために、同時に他の疾患又は障害を治療するために使用され得る(例えば、精神病の予防、一方、胃腸障害の治療;神経障害性の痛みの予防、一方、エタノール離脱症候群の治療)。当該薬剤は、初期ウイルス性感染の間、抗ウイルス薬の如き他の薬物との組み合わせにおいて使用され、神経病の障害の結果生じる事態を阻害又は低減し得る。
【0082】
さらに、当該薬剤は、副作用として神経病の障害を引き起こすと知られるもの自体である他の薬物との組み合わせで使用され得、それ故、前記副作用の発生を阻害し、又は低減する。
【0083】
上記疾患、及び症状を治療する又は予防する際における当該薬剤の適合性は、本分野において記載される方法により決定される得る(例えば、Satzinger et al,米国特許第4,024,175号;Satzinger et al,米国特許第4,087,544号;Woodruff,米国特許第5,084,169号;Silverman et al,米国特許第5,563,175;Singh,米国特許第6,001,876号;Horwell et al,米国特許第6,020,370号;Silverman et al,米国特許第6,028,214号;Horwell et al,米国特許第6,103,932号;Silverman et al,米国特許第6,117,906号;Silverman,国際特許公開第WO92/09560号;Silverman et al,国際特許公開第WO93/23383号;Horwell et al,国際特許公開第WO97/29101号,Horwell et al,国際特許公開第WO97/33858号;Horwell et al,国際特許公開第WO97/33859号;Bryans et al,国際特許公開第WO98/17627号;Guglietta et al,国際特許公開第WO99/08671号;Bryans et al,国際特許公開第WO99/21824;Bryans et al,国際特許公開第WO99/31057号;Magnus−Miller et al,国際特許公開第WO99/37296号;Bryans et al,国際特許公開第WO99/31075号;Bryans et al,国際特許公開第WO99/61424号;Pande,国際特許公開第WO00/23067;Bryans,国際特許公開第WO00/31020;Bryans et al,国際特許公開第WO00/50027;Bryans et al,国際特許公開第WO02/00209;2005年8月5日に出願されたTran,米国特許出願公開第60/711,477号;及び2005年8月5日に出願されたTran,米国特許出願公開第60/710,963号を参照のこと。)。
【0084】
投薬
本明細書中に記載される特定の疾患、障害又症状の治療において有効であろう化合物(1)の量は、当該疾患又は症状の性質に、少なくとも部分的に、依存するだろうし、前術のような本分野において知られる標準的臨床技術により決定され得る。さらに、生体外又は生体内アッセイが場合により使用され、最適な用量範囲の特定を補助する。投与されるプロドラッグの量は、もちろん、他の要因間で、治療される疾患を患う対象、当該対象の重さ、当該苦痛の重篤度、投与様式、及び処方する医師の判断に依存するだろう。
【0085】
ある態様において、持続放出経口薬剤は、1日あたり1〜3回、患者に投与されるように適合される。他の態様において、持続放出経口薬剤は、1日あたり1〜2回、患者に投与されるように適合される。投薬は、それだけで、又は他の薬物との併用で提供され得、そして当該疾患又は障害の効果的な治療又は予防に必要とされる限り続けられ得る。
【0086】
ガバペンチンの経口投与のための好適な用量範囲は、通常、約100mg/日〜約3600mg/日であり、そして化合物(1)あるいはそれらの医薬として許容される塩又は医薬として許容される溶媒和物は、ガバペンチンの等モル量を提供するように調節され得る。用量範囲は、当業者に知られる方法により容易に決定され得る。
【0087】
以下の実施例は、持続放出経口薬剤、持続放出経口薬剤を製造する際に使用される方法、及び材料、並びに、患者に対する当該薬剤の投与により達成された結果をさらに例証する。
【実施例】
【0088】
実施例1
化合物(1)を含む持続放出経口薬剤タブレットを表1に示される成分を有するように製造した。
【0089】
【表1】
【0090】
当該タブレットを、以下のステップに従い製造した。化合物(1)、第2リン酸カルシウム、グリセリルベヘネート、滑石、及びコロイド状シリコンジオキシドの重さを量り、#20メッシュスクリーンを通してスクリーニングし、そしてV−ブレンダー中で15分間混合した。ラウリル硫酸ナトリウムのスラッギング部分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのスラッギング部分の重さを量り、そして#40メッシュスクリーンを通した。スクリーニングされたラウリル硫酸ナトリウム、及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして5分間混合した。当該混合物を排出し、そして、タブレット圧縮機上で約400mg重のスラッグに圧縮した。次いで、当該スラッグを、Comil 194 Ultraミル(Quadro Engineering,Inc.,Millburn,NJ)に通し、さらなる圧縮のための製粉された材料を得た。ラウリル酸硫酸ナトリウムのタブレット分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのタブレット分の重さを量り、そして#40メッシュスクリーンを通した。当該製粉された材料、及び当該ラウリル硫酸ナトリウム、及びステアリン酸マグネシウムのタブレット分をV−ブレンダーに添加し、そして3分間混合した。当該混合された材料を排出し、そして、圧縮し、1310mgの総重量、及び600mg(45.8重量%)の化合物(1)積載量を有するタブレットを形成させた。当該タブレットは、16.1〜22.2kp(158〜218ニュートン)の平均最終硬度を有した。
【0091】
実施例2
表1に示される成分を有する持続放出経口製剤タブレットを、以下のステップに従う異なる方法を使用して製造した。化合物(1)、第2リン酸カルシウム、グリセリルベヘネート、滑石、及びコロイド状シリコンジオキシドの重さを量り、#20メッシュスクリーンを通してスクリーニングし、そしてV−ブレンダー中で15分間混合した。ラウリル硫酸ナトリウムの圧縮分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムの圧縮分の重さを量り、そして#40メッシュスクリーンを通した。スクリーニングされたラウリル硫酸ナトリウム、及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして5分間混合した。当該混合物を排出し、そして、Chilsonator機(FitzPatrick、Elmhurst,ILによるローラー圧縮機)上でコンパクトに圧縮した。次いで、得られたコンパクトを、ハンマーミル(FitzPatrick,Elmhurst,NJ)に通し、さらなる圧縮のための製粉された材料を得た。ラウリル酸硫酸ナトリウムのタブレット分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのタブレット分の重さを量り、そして#40メッシュスクリーンを通した。当該製粉された材料、及び当該ラウリル硫酸ナトリウム、及びステアリン酸マグネシウムのタブレット分をV−ブレンダーに添加し、そして3分間混合した。当該混合された材料を排出し、そして、圧縮し、1310mgの総重量、及び600mg(45.8重量%)の化合物(1)積載量を有するタブレットを形成した。当該タブレットを、300mgの化合物(1)積載量を有する655mgのタブレットにも圧縮した。当該タブレットは、各々、15.7〜18.9kp、そして11.1〜13.7kpの平均最終硬度を有した。
【0092】
実施例3
成人健常者における化合物(1)の持続放出経口薬剤の、安全性、耐容性、及び薬物動態についての、無作為化交差、絶食した/食物を与えられた単回投与試験を実施した。実施例1の持続放出経口薬剤を使用した。当該試験を、商業的なガバペンチンカプセル製剤(Neurontin)(登録商標)と比較して人間におけるこの薬剤の能力を評価するように計画した。12人の成人健常者(7人の男性、及び5人の女性)がこの試験に参加した。平均体重は、75.6kgだった。全患者は、治療と治療との間に1週間のウォッシュアウトを有し、無秩序な順番に異なる治療を受けた。当該2つの治療とは、(A)一晩食物を与えられた後の実施例1のタブレットの単回投与{2×600mg化合物(1)};及び(B)高脂肪朝食の後に実施例1の単回投与{2×600mg化合物(1)}であった。
【0093】
投与に先立って、及び投与後0.5時間、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、12時間、18時間、36時間で、全患者から血液、及び血しょうサンプルを回収した。尿サンプルを全患者から投与に先立ち回収し、そして、完全尿排出量を、投与後、0〜4時間、4〜8時間、8〜12時間、12〜18時間、18〜24時間、及び24〜36時間の間隔で得た。血液サンプルをメタノールですぐに急冷し、≦−70℃で冷凍保存した。サンプルアリコートを、高感度、及び特異的LC/MS/MS方法を使用するガバペンチン、及び化合物(1)の分析のために準備した。
【0094】
絶食した、及び食物を与えられた成人健常患者に対する、実施例1に従って製造された持続放出経口薬剤の経口投与後のガバペンチンの血しょう濃度±1SDを、それぞれ図1及び2に示す。
【0095】
当該タブレットの経口投与後(絶食)の血しょう中のガバペンチンについての平均±SD Cmaxは、4.21±1.15μg/mLだった。高脂肪の朝食後の当該タブレットの投与後、血しょう中のガバペンチンについてのCmaxは、6.24±1.55μg/mLにさらに増大した。
【0096】
当該タブレットの経口投与後(絶食)の血しょう中のガバペンチンについての平均±SD AUCは、54.5±12.2μg・hr/mLだった。高脂肪の朝食後の当該タブレットの投与後、血しょう中のガバペンチンのAUCは、83.0±21.8μg・hr/mLにさらに増大した。食物の存在下、当該タブレットの投与後のガバペンチンへの暴露は、絶食した患者におけるものと比較してさらに52%増大した。
【0097】
ガバペンチンのピーク血しょう濃度までの時間(Tmax)は、当該タブレットの経口投与後、著しく遅延した。絶食した患者において、当該タブレットの経口投与は、5.08±1.62hrのガバペンチンTmaxを与えた。これは、約2〜4hrの即時放出ガバペンチンの典型的Tmaxと比較する。当該タブレットの投与後のガバペンチンTmaxは、食物の存在下、8.40±2.07hrにさらに遅延した。
【0098】
血しょう中のガバペンチンについての明らかな最終的な排出半減期は、全ての処置:絶食した患者における当該タブレットについて6.47±0.77hr、及び食物を与えられた患者における当該タブレットについて5.38±0.80hrを超えて同じであった。
【0099】
当該タブレットの経口投与後、尿中に回収された当該ガバペンチン用量のパーセントは、絶食した患者について46.5±15.8%であり、食物を与えられた患者について73.7±7.2%だった。
【0100】
当該タブレットの経口投与後の血しょう中の完全なプロドラッグへの暴露は、低かった。絶食した患者における当該タブレットの経口投与後、血しょう中の完全な化合物(1)の濃度は、対応するピークガバペンチン濃度の約1.0%である、最大0.040μg/mLに達した。同様に、これらの患者の血しょう中の化合物(1)のAUCは、対応する血しょう中のガバペンチンのAUCの0.3%だった。食物を与えられた患者における当該タブレットの経口投与後、血しょう中の完全な化合物(1)の濃度は、対応するピークガバペンチン濃度の約0.3%である、最大0.018μg/mLに達した。同様に、これらの患者の血しょう中の化合物(1)のAUCは、血しょう中のガバペンチンのAUCの<0.1%だった。
【0101】
実施例4
食物を与えられた人間の患者に対する、実施例2に従って製造された持続放出経口薬剤の経口投与後、ガバペンチンの平均血しょう濃度を、図3に示す。12人の人間の患者におけるガバペンチンの平均血しょう濃度を、(a)300mgの化合物(1)を含む1つのタブレット、(b)600mgの化合物(1)を含む1つのタブレット、及び(c)各タブレットが600mgの化合物(1)を含む2つのタブレット中の1200mgの化合物(1)の投与後、実施例3に記載される方法に従って測定した。
【0102】
実施例5
カニクイザルに対する、実施例1に従って製造された持続放出経口薬剤の経口投与後のガバペンチンの血中濃度±1SDを、図4に示す。カニクイザルにおけるガバペンチンの血中濃度を、以下の手順に従い測定した。
【0103】
投与プロトコール
化合物(1)を含むタブレット{タブレットあたり1×600mg化合物(1)}を、4匹の成人で雄のカニクイザル(Macaca fascicularis)(約3kgの体重)の一群に経口投薬により投与した。各サルに1つのタブレットを投与した。動物に、試験前の一晩、及び投薬後約4時間、絶食させた。血液サンプル(1.0mL)を、経口投薬後、24時間に渡り、大腿静脈経由で得た。
【0104】
血液を、メタノールを使用してすぐに急冷し、分析するまで−20℃で凍結した。試験化合物を、投薬セッションの間、最小72時間のウォッシュアウト期間を有するサルに投与した。
【0105】
吸収された薬物についてのサンプル製造
300μLのメタノールを、サンプル及びスタンダードの製造のために、1.5mLのエッペンドルフチューブに添加した。
【0106】
サンプル製造:血液を異なる時点で回収し、即座に、100μLの血液を、300μLのメタノールを含むエッペンドルフチューブに添加し、そしてボルテックスにより混合した。
【0107】
スタンダード製造:90μLの血液を、エッペンドルフチューブ中の300μLのメタノールに添加した。10μLのガバペンチンのスタンダード溶液(0.04、0.2、1、5、25、及び100μg/mL)を各チューブに添加し、最終的較正スタンダード(0.004、0.02、0.1、0.5、0.25、及び10μg/mL)を作成した。
【0108】
20μLのp−クロロフェニルアラニンを、全サンプル、及びスタンダードに添加した。当該サンプルをボルテックスし、14,000rpmで20分間遠心した。当該上清を、LC/MS/MS分析のために使用した。
【0109】
LC/MS/MS分析
当該サルの血液中のガバペンチンの濃度を、Shimadzu SLC−10AVP、及びLEAPオートサンプラーを装備したAPI2000LC/MS/MS器具を使用して測定した。カラムは、室温で動作するZorbax C8XDB4.6×150mmカラムだった。移動相は、(A)水中に0.1%ギ酸、及び(B)アセトニトリル中に0.1%ギ酸だった。勾配条件は、3.5分間の2%B、3.5分間で95%Bに増大し2分間維持、次いで5.6分間で2%Bまで低減、そして2.3分間維持だった。30μLのサンプルを当該カラム中に注入した。Turbo−IonSpray源を使用し、そしてガバペンチンを、172/137のMRMトランジションについて陽イオンモードにおいて検出した。ピークを、Analyst1.2定量ソフトウェアを使用して調整した。
【0110】
実施例6
化合物(1)を含む持続放出経口薬剤を、下記の表2に示される成分を有するように製造した。
【表2】
【0111】
当該タブレットを以下のステップに従って作成した。化合物(1)、微晶性セルロース(MCC PH113)、グリセリルベヘネート、滑石、コロイド状シリコンジオキシド、及びドデシル硫酸ナトリウム(SDS)(第1混合分)及びステアリン酸マグネシウム(第1混合分)の重さを量り、#20メッシュスクリーンを通じてスクリーニングし、そしてV−ブレンダー中で7分間混合した{MaxiBlend Lab Blender MB−1(Globepharma)}。当該混合された成分を、ローラー圧縮機(BO50PH圧縮機、クロップドロール/クローズドエンド、3.9−in ロール直径、1.5−in ロール幅、11.6kN ロール力、12rpm ロールスピード、及び7rpm 水平スクリューフィードスピード)のフィードホッパー中に注入した。
【0112】
次いで、当該圧縮された材料を、Quadro Underdriven Comilモデル U5ミル(Quadro Engineering,Inc.,Millburn,NJ,0.079−in グレイター孔サイズ、1607 インペラスタイル、2500 インペラrpm)を通して、さらなる圧縮のために製粉された材料を得た。当該製粉された材料を、ブレンダー{MaxiBlend Lab ブレンダーモデル MB−1(Globepharma)、25rpm シェルスピード}に移し、そして5分間混合した。追加のSDS(第2混合分)、及び/又はステアリン酸マグネシウム(第2混合分)を、必要に応じて添加し、特定の量を得た。当該混合された材料を、排出し、そして圧縮し、総計1100mg、及び600mgの化合物(1)積載量(54.55%)を有するタブレットを製造した。当該タブレットは、14〜17kp(137〜214ニュートン)の平均最終硬度を有した。
【0113】
実施例7
実施例6に従って製造された持続放出経口薬剤の、カニクイザルに対する経口投薬後(1×600mg)のガバペンチンの血中濃度±1SDを、図5に示す。カニクイザルにおけるガバペンチンの血中濃度を、実施例5に記載された方法に従い測定した。
【0114】
実施例8
以下のステップを使用し、実施例1、2、及び6に従って製造された薬剤の生体外溶解プロフィールを測定した。薬剤を、37℃で、10mMリン酸2水素カリウム緩衝液(KH2PO4、pH7.4)及び1%(wt/体積)ラウリル硫酸ナトリウムの900mLを含む溶解容器中に配置した。当該溶解媒体を50rpm(USP,タイプII、パドル)で撹拌した。サンプルを0.5、1、2、4、6、8、12、及び24時間で回収し、溶液中の化合物(1)の含有量を、210nmでのフォトダイオード検出を有する、C18カラム、及びリン酸緩衝液/アセトニトリル/水の等張移動相を使用する逆相HPLCにより測定した。
【0115】
図6に示されるように、実施例1に従って製造された薬剤は、約2時間後に約20%、約5時間後に約50%、そして約8時間後に約80%の化合物(1)を放出した。図7Aに示されるように、実施例2に従って製造された300mgの化合物(1)を含む薬剤は、約2時間後に約20%、約6時間後に約50%、そして約10時間後に約80%の化合物(1)を放出した。図7Bに示されるように、実施例2に従って製造された600mgの化合物(1)を含む薬剤は、約2時間後に約20%、約5時間後に約50%、そして約8時間後に約80%の化合物(1)を放出した。図8に示されるように、実施例6に従って製造された薬剤は、約5時間後に約30%、約10時間後に約60%、そして約15時間後に約80%の化合物(1)を放出した。
【0116】
最後に、本発明を実施する他の方法があることに留意すべきである。従って、本明細書中に記載の態様は、例証、及び非制限的として考慮され、そして、本発明は、本明細書中に記載の詳細に制限されるべきではない、しかし、本明細書から生じる本発明の請求の範囲、及びそれと同等の範囲内において改良し得る。
【0117】
本明細書中に記載された刊行物、及び特許の全内容を、本願明細書中に援用する。
【技術分野】
【0001】
分野
本願の開示は、ガバペンチンプロドラッグ、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤、及びかかる薬剤を投与することによる、ガバペンチンが治療に有効である疾患、及び障害を治療する又は予防する方法に関する。
【背景技術】
【0002】
本出願は、2004年11月4日に提出された米国仮出願番号第60/625,737号の利益を主張し、その全内容を本願明細書中に援用する。
【0003】
背景
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸(1)、ガンマアミノ酪酸(GABA)類似体であるガバペンチンのプロドラッグ(2)は、経口的、あるいは哺乳類の結腸に直接的のいずれかで投薬されるとき、ガバペンチンとして高い生体内利用性を有する(Gallop et al., 国際特許公開WO02/100347号;Cundy et al.,J pharmacol Exp Ther.2004,311:315−323;Cundy et al.,J pharmacol Exp Ther.2004,311:324−333)。当該高い生体内利用性は、化合物(1)を、てんかん、痛み(特に神経障害性の痛み、並びに筋肉痛、及び骨格痛)、うつ、不安、精神病、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、炎症性障害(すなわち関節症)、不眠症、胃腸障害、ほてり、下肢静止不能症候群、尿失禁、及びエタノール離脱症候群を治療する又は予防するために有用な経口薬剤(持続放出薬剤を含む)の重要な成分とさせる。
【0004】
【化1】
【0005】
Gallop et al.,国際特許公開WO02/100347号に記載のとおり製造された化合物(1)は、含水アセトニトリルから、凍結乾燥後、ガラス状固体として単離された。この過程により入手される材料は、部分的に又は全体的に非晶質であり、あるアルカリ金属塩型は、吸湿性である。さらに最近、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の結晶性形状、及びその合成方法が記載されている(Estrada et al.,国際特許公開第WO2005/037784号)。当該化合物(1)の結晶性形状は、製剤過程、及び医薬組成物において有用な物理化学的性質を改良した。
【発明の概要】
【0006】
要約
本願は、化合物(1)の持続放出経口薬剤を開示する。ある態様において、化合物(1)の持続放出経口薬剤は、約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する。
【0007】
薬剤は、例えば、(a)約10重量%〜約80重量%の化合物(1)、及び
(b)約1重量%〜約50重量%の放出速度改良ポリマーを含むタブレットとなり得、ここで重量%は当該製剤の総乾燥重量に基づく。好適な放出速度改良ポリマーは、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、及びステアロイルマクロゴールグリセリドの如きグリセリルエステルを含む。他の好適な放出速度改良ポリマーはメタクリレート共重合体、アンモニオアルキルメタクリレート共重合体、並びにそれらの共重合体、及び組み合わせを含む。
【0008】
患者に経口(すなわち、当該患者がタブレットを嚥下することによる)で投与されるとき、当該製剤は、時間とともに血しょう中のガバペンチンの濃度曲線を提供し得、当該曲線は、図に示される最大血しょう濃度(Tmax)の形と時間を有する。
【図面の簡単な説明】
【0009】
【図1】図1は、実施例3に記載されるように、絶食した患者に化合物(1)を含む持続放出タブレット(2×600mg)を経口投与した(処置A)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図2】図2は、実施例3に記載されるように、食物を与えられた患者に化合物(1)を含む持続放出タブレット(2×600mg)を経口投与した(処置B)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図3】図3は、実施例4に記載されるように、食物を与えられた患者に化合物(1)を含む持続放出タブレット(1×300mg;1×600mg;及び2×600mg)を経口投与した(処置B)後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図4】図4は、実施例5に記載されるように、絶食させた成人雄カニクイザルに化合物(1)を含む持続放出タブレット(1×600mg)を経口投与した後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図5】図5は、実施例7に記載されるように、絶食させた成人雄カニクイザルに化合物(1)を含む持続放出タブレット(1×600mg)を経口投与した後の血しょう中のガバペンチンの平均濃度±SDのグラフである。
【図6】図6は、実施例1に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
【図7A】図7Aは、実施例2に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
【図7B】図7Bは、実施例2に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
【図8】図8は、実施例6に記載されるように製造された本開示の薬剤の生体外溶解プロフィールを示す。
【発明を実施するための形態】
【0010】
定義
「AUC」は、患者への当該化合物の投与後の時間の関数として、患者における体液中の当該化合物又はそれらの代謝体の濃度を示す曲線下の面積である。ある態様において、当該化合物はプロドラッグとなり得、及び代謝体は薬物となり得る。体液の例は、血液、及び血しょうを含む。AUCは、様々な時間間隔で、液体クロマトグラフィータンデム質量分析(LC/MS/MS)の如き方法を使用して、血しょう又は血液の如き体液中の化合物又はそれらの代謝体の濃度を測定し、及び血しょうの濃度対時間曲線下の面積を計算することにより、決定され得る。薬物の濃度対時間曲線からAUCを計算するための好適な方法は、本分野においてよく知られているだろう。本願明細書の開示に関して、ガバペンチンについてのAUCは、化合物(1)、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む薬剤の経口投与後の患者の血しょう、及び/又は血液中のガバペンチンの濃度を測定することにより決定され得る。
【0011】
「生体内利用性」は、患者への当該薬物又はプロドラッグの投与後の当該患者の全身循環に達する薬物の比率、及び量をいい、そして、例えば化合物についての血しょう及び/又は血液の濃度対時間のプロフィールを評価することにより決定され得る。血しょう及び/又は血液濃度対時間曲線を特徴付けるのに有用なパラメータは、当該曲線下の面積(AUC)、ピーク濃度までの時間(Tmax)、及び最大薬物濃度(Cmax)を含む。
【0012】
「生物学的に同等」は、患者への薬物又はプロドラッグの等量を投与した後の薬物吸収の速度及び程度が等しいことをいう。本明細書に記載される通り、もし2つのプロフィールの平均応答の比率についての90%信頼区間が0.8〜1.25の範囲内であれば、当該2つの血しょう又は血液濃度プロフィールは生物学的に同等である。当該平均応答は、AUC、Tmax、及びCmaxの如き、プロフィールの特徴的パラメータの少なくとも1つを含む。
【0013】
「Cmax」は、患者への薬物又はプロドラッグの投与後の当該患者の血しょう又は血液中の当該薬物の最大濃度である。
【0014】
「Tmax」は、患者への薬物又はプロドラッグの投与後の当該患者の血しょう又は血液中の当該薬物の最大濃度(Cmax)までの時間である。
【0015】
「絶食した患者」は、薬剤が投与される時点で、及び投与後少なくとも4時間の間に患者の胃が実質的に食物の無い状態である患者をいう。患者の胃について食事後に食物が実質的に無くなる時間は、例えば、カロリー量の如き食事の大きさ、脂肪含量の如き食事の内容、当該患者の健康状態、及び患者の胃腸管の状態を含む多くの因子に依存し得る。健康な被験者の胃は、食物の摂取後約4〜約8時間後に、通常、実質的に食物がなくなる。
【0016】
ある態様において、絶食した患者は、投薬前約10時間から、投薬後約4時間まで全く食物を食べず(しかし、適当量の水又は澄んだ液体を摂取し得る)、投薬前約2時間、及び投薬前約1時間に約250mlの水を飲み、及び投薬後約2時間に約250mlの水を飲み、投薬後約4時間に昼食を食べ、そして投薬後約10時間に夕飯を食べる。
【0017】
「食物を与えられた患者」は、その胃が食物を含む患者である。ある態様において、食物を与えられた患者は、投薬前約30分に試験食を食べ始め、投与前約5分に当該試験食を食べることを完全に終わらせ、投薬後4時間に昼食を食べ、そして投薬後約10時間に夕飯を食べる。当該試験食は、例えば、バターで揚げた2つの卵、ベーコン2切れ、バター付の小麦のトースト2枚、ハッシュトポテトの4オンス、及び全乳8オンスの如き、高脂肪(当該試験食中におけるカロリーの総量の約50%)、及び高カロリー(約1000総カロリー)の朝食を含む。当該試験食は、約150タンパク質カロリー、250炭水化物カロリー、及び約500〜600脂肪カロリーを含む。
【0018】
「患者」は、人間の如き、哺乳類をいう。
【0019】
「医薬として許容される」とは、連邦政府又は州政府の監督庁により承認される又は承認見込みであること、あるいは、動物に、より特には人間における使用として米薬局方又は他の一般的に認識される薬局方に挙げられることをいう。
【0020】
「医薬として許容される塩」は、医薬として許容され、及び親化合物の所望の医薬活性を有する本願に開示の化合物の塩をいう。かかる塩は、以下の:(1)塩酸、臭化水素酸、硫酸、硝酸、リン酸などの如き無機酸と形成される;又は酢酸、プロピオン酸、ヘキサン酸、シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、コハク酸、リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、3−(4−ヒドロキシベンゾイル)安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、1,2−エタン−ジスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、4−クロロベンゼンスルホン酸、2−ナフタレンスルホン酸、4−トルエンスルホン酸、カンファースルホン酸、4−メチルビシクロ[2,2,2]−オクト−2−エン−1−カルボン酸、グルコヘプトン酸、3−フェニルプロピオン酸、トリメチル酢酸、第3ブチル酢酸、ラウリルスルホン酸、グルコン酸、グルタミン酸、ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸などの如き有機酸と形成される酸付加塩;又は(2)当該親化合物中に存在する酸性プロトンが、アルカリ金属イオン、アルカリ土類金属イオン、又はアルミニウムイオンの如き金属イオンにより置換される、あるいはエタノールアミン、ジエタノールアミン、トリエタノールアミン、N−メチルグルカミンなどの如き有機塩基と配位結合する、いずれかのときに形成される塩を含む。
【0021】
「予防する」又は「予防」とは、疾患又は障害に陥るリスクにおける低減をいう(すなわち、疾患にさらされる又は疾患にかかりやすいかもしれないが、当該疾患を未だ経験せず又は当該疾患の症状を未だ示さない患者において、臨床的症状の少なくとも1つを発現しないようにする)。
【0022】
「プロドラッグ」は、体内で活性薬物を放出するような転換を必要とする薬物分子をいう。化合物(1)は、患者の体内で当該親薬物であるガバペンチンを形成するよう代謝されるプロドラッグであり、それ故、化合物(1)はガバペンチンのプロドラッグである。化合物(1)、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸は、医薬として許容される塩、又は化合物(1)の遊離酸型の医薬として許容される溶媒和物、並びに上述の結晶性形状を含む。
【0023】
「溶媒和物」は、化合物と化学量論的又は非化学量論的な量の1若しくは複数の溶媒分子との分子複合体をいう。かかる溶媒分子は、医薬分野で一般的に使用されるものであり、そしてそれは、水、エタノールの如き受容者にとって無害であるとして知られるものである。化合物又は化合物の一部と溶媒との分子複合体は、例えば静電気力、ファンデルワールス力又は水素結合の如き非共有分子内力により、安定され得る。「水和物」は、当該1又は複数の溶媒分子が水であるときの複合体をいう。
【0024】
「持続放出」とは、化合物(1)の即時放出製剤の経口投与により達成される場合と比較してより長時間に渡る、全身性血液循環における化合物(1)又はそれらの活性代謝体の治療又は予防濃度を達成するのに効果的な速度での薬剤からの化合物(1)の放出をいう。ある態様において、化合物(1)の放出は、少なくとも6時間超、ある態様においては少なくとも12時間超、ある態様においては少なくとも18時間超、ある態様においては24時間超の時間に渡り生ずる。
【0025】
「治療有効量」とは、疾患を治療する又は予防するために患者に投与されたとき、当該疾患のかかる治療又は予防に十分に有効である化合物(1)の量を意味する。当該「治療有効量」は、当該疾患、及びその重篤度、並びに当該治療又は予防される疾患を有する患者の年齢、体重などに依存して変化するだろう。
【0026】
ある疾患又は障害の「処置」又は「治療」は、ある態様において、当該疾患又は障害を改善することをいう(すなわち、当該疾患又はそれらの臨床的症状の少なくとも1つの進行を止めること、又は低減すること)。他の態様において、「処置」又は「治療」は、少なくとも1つの物理的パラメータを改善することをいい、そしてそれは当該患者により認識され得又は認識され得ない。ある態様において、「処置」又は「治療」は、物理的(例えば、認識される症状の安定化)、生理学的(例えば、物理的パラメータの安定化)、又はその両方のいずれかで、当該疾患又は障害を阻害することをいう。
【0027】
「Wt%」は、重量パーセントの如き、組成物又は薬剤の総乾燥重量と比べた成分又は原料の重量をいう。例えば、40wt%の化合物(1)を含み、かつ1000mgの重量を有する薬剤は、400mgの化合物(1)を含む。化合物(1)の塩、及び/又は溶媒和物についていうとき、当該wt%は、化合物(1)の医薬として許容される塩、及び/又は医薬として許容される溶媒和物を形成する1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の質量当量をいう。
【0028】
ここに参照されるように、薬剤及び方法のある態様に対して詳細に理解されるだろう。この開示された態様は本願の請求項を制限することを意図しない。反対に、当該請求項が、当該開示された態様の全ての代替物、改良物、そして等価物を保護することを意図する。
【0029】
持続放出経口薬剤
本明細書に開示されるある態様は、タブレット形状における持続放出経口薬剤を提供するけれども、持続放出経口薬剤の分野における当業者は、粉末、粒子、サシェ、液体懸濁液、及び/又は再構成のためのカプセルの如き他の持続放出経口薬剤も使用され得ることを理解するだろう。ある態様において、薬剤はタブレットとなり得、それは、回転楕円形、角ばった楕円形又は長円体の如き薬剤の経口投与に適したいずれかの形状の内の1つとなり得る。1つのタブレットは、化合物(1)の1単位用量を含み得、又は1単位用量未満を含むミニタブレットの場合、ミニタブレットは、1単位用量を提供するためにカプセルに入れ込まれ得る。
【0030】
ある態様において、タブレットは、複数層のタブレットとなり得、当該異なる層は、各タブレット層からの化合物(1)の放出特性に影響を与える異なる粒子群、及び/又は異なる賦形剤を含む。タブレットの例は、崩壊タブレット、速溶性タブレット、発泡性タブレット、速融解タブレット、チュアブルタブレット、破砕可能タブレット、及びミニタブレットである。
【0031】
一般的に、タブレット薬剤は、少なくとも約15キロポンド(kp)(147.1ニュートンと等量)の硬さに圧宿され得る。薬剤は、本分野において知られた方法により製造され得、そして医薬として許容される賦形剤を、必要に応じてさらに含み得る。
【0032】
ある態様において、持続放出経口薬剤は、(a)約10重量%〜約80重量%の化合物(1)、及び(b)約1重量%〜約30重量%の放出速度改良ポリマー、を含む。
【0033】
ある態様において、持続放出経口薬剤は、(a)約10重量%〜約80重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマー、を含む。
【0034】
ある態様において、持続放出経口薬剤は、(a)約30重量%〜約75重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマーを含む。
【0035】
ある態様において、持続放出経口薬剤は、(a)約40重量%〜約65重量%の化合物(1)、及び(b)約1重量%〜約50重量%の放出速度改良ポリマーを含む。
【0036】
ある態様において、持続放出経口薬剤は、(a)約50重量%〜約60重量%の化合物(1)、及び(b)約20重量%〜約50重量%の放出速度改良ポリマーを含む。
【0037】
本発明に開示される薬剤は、放出速度改良ポリマー、場合により賦形剤において化合物(1)が均一に分散されるマトリクス系である。
【0038】
マトリクス系は、例えば、「Handbook of Pharmaceutical Controlled Release Technology,」ed. D.L.Wise,Marcel Dekker,Inc.(2000)、及び「Treatise on Controlled Drug Delivery,Fundamentals,Optimization, and Applications,」ed.A.Kydonieus,Marcel Dekker,Inc.(1992)に記載されるように本分野においてよく知られる。
【0039】
放出速度改良ポリマーは、薬剤からの薬物の放出を遅らせ得る。好適な放出速度改良ポリマーは、pH感受性ポリマー、pH非感受性ポリマー、水又は水性媒体と接触したとき高い膨張度を有する親水性ポリマー、水又は水性媒体と接触することでゲルを形成するポリマー、水又は水性媒体と接触したときに膨張特性、及びゲル化特性の両方を示すポリマー、ワックスの如き脂肪族化合物、及び生物分解性ポリマーを非制限的に含む。
【0040】
ある態様において、放出速度改良ポリマーは、アクリル酸、及びメタクリル酸ポリマーや共重合体、メチルメタクリレート共重合体、エトキシエチルメタクリレート、シアノエチルメタクリレート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミド共重合体、ポリ(メチルメタクリレート)、ポリメタクリレート、ポリ(メチルメタクリレート)共重合体、ポリアクリルアミド、アミノアルキルメタクリレート共重合体、ポリ(メタクリル酸無水物)、グリシジルメタクリレート共重合体、アンモニオアルキルメタクリレート共重合体、及び上述のいずれかの組み合わせの如きpH感受性ポリマーとなり得る。
【0041】
ある態様において、pH依存性ポリマーは、ジエチルアミノエチルメタクリレート、及び他の天然メタクリル酸エステルから合成される共重合体となり得、Eudragit(登録商標)(Rohm Pharma)として商業的に入手可能な、メタクリル酸共重合体又はポリマーメタクリレートとしても知られる。
【0042】
ある態様において、pH非感受性ポリマーは、Eudragit(登録商標)RS、及びEudragit(登録商標)RLの如きアンモニオアルキルメタクリレート共重合体であり、それは、第4アンモニウム基の低い含有量のアクリル及びメタクリル酸エステルの共重合体を含むアクリル樹脂である。
【0043】
高い膨張性を示す親水性放出速度改良ポリマーの例は、架橋されたソジウムカルボキシメチルセルロース、架橋されたヒドロキシプロピルセルロース、高分子量ヒドロキシプロピルメチルセルロース、カルボキシメチルアミド、ポタシュームメタクリレートジビニルベンゼン共重合体、ポリメチルメタクリレート、ポリビニルピロリドン、高分子量ポリビニルアルコール、メチルセルロース、ビニルアセテート共重合体などを含む。
【0044】
水と接触してゲル化する放出速度改良ポリマーの例は、メチルセルロース、カルボキシメチルセルロース、低分子量ヒドロキシプロピルメチルセルロース、低分子量ポリビニルアルコール、ポリオキシエチレングリコール、非架橋ポリビニルピロリドン、キサンタンガムなどを含む。
【0045】
膨張、及びゲル化特性の両方を示す放出速度改良ポリマーの例は、中間粘性ヒドロキシプロピルメチルセルロース、及び中間粘性ポリビニルアルコールを含む。
【0046】
ある態様において、当該放出速度改良ポリマーは、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、ステアロイルマクロゴールグリセリド、又は上述のいずれかの組み合わせの如きグリセリルエステルである。ある態様において、当該放出速度改良ポリマーは、グリセリルベヘネートである。他の脂肪、及び/又はワックス放出速度改良ポリマーは、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール、セトステアリルアルコール、パルミトイルアルコール、ouricuryワックス、硬化植物油、カンデリラワックス、エスパルトワックス、ステアリン酸、パラフィンワックス、蜜ろう、グリコワックス、キャスターワックス、及びカルナバワックスを含む。
【0047】
生体内分解性ポリマーの例は、コラーゲン、ゼラチン、ポリビニルアルコール、ポリオルトエステル、ポリアセチル、ポリオルトカルボネート、ポリアミド、ポリアミノ酸、ポリエステル、ポリ乳酸、ポリグリコール酸、ポリカルボハイドレート、ポリオルトエステル、ポリオルトカルボネート、ポリアセチル、ポリ無水物、ポリデヒドロピラン、ポリジオキシノンなどを含む。
【0048】
本発明に開示の薬剤に組み込まれ得る他の好適な放出改良ポリマーは、天然又は合成ゴムの如き親水コロイド、アカシアの如き炭水化物を基材とした物質、トラガカントゴム、ローカストビーンガム、グアガム、アガー、ペクチン、カラギーナン、可溶性及び不溶性アルギナート、カルボキシポリメチレン、カゼイン、ゼイン、ポリエチレンオキシド、無水マレイン酸/メチルビニルエーテル共重合体、及びゼラチンの如きタンパク性物質を含む。
【0049】
放出速度改良ポリマーは、単独で又は1若しくは複数の他の放出速度改良ポリマーとの組み合わせで使用され得、そして/あるいは、複数の放出速度改良ポリマーの共重合体となり得る。
【0050】
化合物(1)、並びに1又は複数の放出速度改良ポリマーの製剤は、湿式造粒法、流動造粒法、乾燥造粒法、及び直接圧縮(「Remington’s Pharmaceutical Sciences,」 Lippincott Williams & Wilkins,889−928,(2005)を参照のこと)の如き本分野においてよく知られる標準技術を使用して製造され得る。例えば、マトリクス製剤は、放出改良ポリマー、充填剤、化合物(1)、及び賦形剤を乾式混合し、その後、適した造粒が得られるまで、アルコールを使用して当該混合物を粒状にすることにより製造され得る。当該造粒は、本分野において知られた方法により実施され得る。湿った顆粒を、流動乾燥機内で乾燥させ、ふるいにかけ、そして適当な大きさの粉にすることができる。平滑剤を乾燥された顆粒と混合し、最終製剤を得ることができる。ある態様において、かかる製剤を、本分野においてよく知られた方法によりタブレット型薬剤に圧縮し得る。
【0051】
ある態様において、薬剤中の化合物(1)の量は、約50mgから約800mg、ある態様において、約100mgから約800mg、及びある態様において、約300mgから約700mgの範囲となる。医薬として許容される塩、及び/又は化合物(1)の溶媒和物を含む薬剤について、薬剤中の化合物(1)の量は、化合物(1)の質量当量をいう。薬剤中に含まれる化合物(1)の量又は荷重は、治療される特定の症状、及びその後吸収される荷重プロドラッグから産生されるガバペンチンの量に依存し得る。
【0052】
本明細書中に記載される化合物(1)、及び当該放出速度改良ポリマーに加えて、薬剤は、界面活性剤、滑剤、希釈剤、抗粘着剤、流動促進剤、緩衝液、染料、湿潤剤、乳化剤、pH緩衝剤、安定剤、増粘剤、崩壊剤、及び着色剤の如き1又は複数の医薬として許容される賦形剤を含む。かかる賦形剤は、でんぷん、糖、ゼラチン、モルト、米、小麦粉、チョーク、シリカゲル、ステアリン酸ナトリウム、グリセロールモノステアレート、滑石、塩化ナトリウム、グリセロール、プロピレングリコール、水、エタノールなどを含む。
【0053】
希釈剤は、薬剤を圧縮のため実用的な大きさとするために添加され得、容積を増大し得る。有用な希釈剤の例は、第2リン酸カルシウム、第2リン酸カルシウムジハイドライド、硫酸カルシウム、第2リン酸カルシウム、第3リン酸カルシウム、乳糖、微晶性セルロ−スを含むセルロース、カオリン、マンニトール、塩化ナトリウム、乾燥でんぷん、アルファでんぷん、圧縮性の糖、マンニトール、及び上述のいずれかの組み合わせを含む。ある態様において、単一の希釈剤は、第2リン酸カルシウム、及び微晶性セルロ−スから選択される。当該希釈剤が第2リン酸カルシウムである態様において、薬剤は、約30重量%〜約50重量%、及びある態様においては約35重量%〜約45重量%の範囲である希釈剤の量を含み得る。当該希釈剤が微晶性セルロースである態様において、薬剤は、約5重量%〜約20重量%、及びある態様においては約10重量%〜約16重量%の範囲である希釈剤の量を含み得る。
【0054】
流動促進剤は、本発明の薬剤中に含まれ得、製造、膜形成、及び/又は乾燥の間、付着効果を低減する。有用な流動促進剤の例は、滑石、ステアリン酸マグネシウム、グリセリルモノステアレート、コロイド状シリコンジオキシド、沈降シリコンジオキシド、上述のいずれかの組み合わせである。ある態様において、流動促進剤は、コロイド状シリコンジオキシドである。薬剤は、約2重量%未満の流動促進剤、ある態様において、約1重量%未満の流動促進剤を含み得る。
【0055】
滑剤、及び抗粘着剤は、加工に役立つために本発明の薬剤中に含まれ得る。有用な滑剤、及び/又は抗粘着剤の例は、ステアリン酸カルシウム、グリセリルベヘネート、グリセリルモノステアレート、ステアリン酸マグネシウム、鉱油、ポリエチレングリコール、ソジウムステアリルフマレート、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ステアリン酸、滑石、硬化植物油、ステアリン酸亜鉛、及び上述のいずれかの組み合わせである。ある態様において、当該滑剤は、グリセリルモノステアレートである。ある態様において、滑剤は、ステアリン酸マグネシウムである。薬剤は、約1重量%〜約13重量%の、ある態様において、約4重量%〜約10重量%の範囲の滑剤、及び/又は抗粘着剤を含み得る。
【0056】
本発明の薬剤において有用な界面活性剤の例は、医薬として許容される、アニオン性界面活性剤、カチオン性界面活性剤、両性{両親媒性(amphiphatic)/両親媒性(amphiphilic)}界面活性剤、非イオン性界面活性剤、ポリエチレングリコールエステル又はエーテル、及び上述のいずれかの組み合わせを含む。好適な医薬として許容されるアニオン性界面活性剤は、1価アルキルカルボキシレート、アシルラクチレート、アルキルエーテルカルボキシレート、N−アシルサルコシネート、多価アルキルカルボネート、N−アシルグルタメート、脂肪酸−ポリペプチド凝縮物、硫酸エステル、ラウリル硫酸ナトリウム及びドデシル硫酸ナトリウムの如きアルキルスルフェイト、エトキシル化アルキルスルフェイト、ドキュセートナトリウム及びジオクチルソジウムスクシネートの如きエステル結合スルホネート、アルファオレフィンスルホネート、又はリン酸化エトキシル化アルコールを含む。
【0057】
好適な医薬として許容されるカチオン性界面活性剤の例は、モノアルキル第4アンモニウム塩、ジアルキル第4アンモニウム化合物、アミドアミン、及びアミンイミドを含む。
【0058】
好適な医薬として許容される両性界面活性剤の例は、N−置換アルキルアミド、N−アルキルベタイン、スルホベタイン、及びN−アルキル−6−アミノプロピオネートを含む。
【0059】
好適な医薬として許容されるポリエチレングリコールエステル又はエーテルの例は、ポリエトキシル化キャスター油、ポリエトキシル化水素化キャスター油を含む。ある態様において、界面活性剤は、ラウリル硫酸ナトリウム、及びドデシル硫酸ナトリウムから選択される。ある態様において、薬剤は、約3重量%未満、ある態様においては約2重量%未満の界面活性剤を含み得る。
【0060】
タブレット薬剤の如き本発明の薬剤は、1又は複数のコーティングをさらに含む。1又は複数の追加のコーティングの目的は、物理的保護、審美、嚥下の際の容易さ、識別、及び/又は当該粒子のさらなる処理を促進するためのものである。一定のコーティングが適用され胃腸管の中における当該薬剤からの化合物(1)の放出を改良又はそれらに影響を与え得るが、一方、他のものはかかる効果を有し得ない。コーティングは、水分に対して不浸透性又は水分浸透性となり得る。
【0061】
水分浸透性の外側タブレットコーティングは、乾燥剤の存在下で包装された薬剤中の低い水分含量を維持するために有用となり得、及びそれ故、例えば当該薬剤の保存性を促進し得る。これらの追加のコーティングは、本分野においてよく知られた方法により本発明の薬剤に適用され得る。物理的保護のためのコーティングにおいて有用な材料の例は、ヒドロキシプロピルメチルセルロ−ス、ヒドロキシプロピルセルロース、ヒドロキシプロピルエチルセルロース、及びキサンタンガムの如き浸透性又は溶解性材料を含む。さらなる加工を促進するためのコーティングにおいて有用な材料の例は、滑石、コロイド状シリカ、ポリビニルアルコール、二酸化チタン、微粒シリカ、グリセロールモノステアレート、マグネシウムトリシリケート、及びステアリン酸マグネシウムを含む。コーティングは、単一材料又は本明細書中に記載されたもののいずれかを含む複数の材料を含み得る。
【0062】
本発明の薬剤は、化合物(1)、及び/又はガバペンチンの分子内環化反応により形成されるラクタム副生物を実質的に含まないものとなり得る。薬剤は、実質的にラクタム形成なし(好ましくはラクタム0.5重量%未満、より好ましくはラクタム0.2重量%未満、最も好ましくはラクタム0.1重量%未満)で、拡張保存(より好ましくは、1年超)について好ましくは安定的である。
【0063】
ある態様において、当該薬剤中の化合物(1)は、2005年7月14日に刊行されたEstrada et al,米国特許出願公開US2005/0154057号に記載された結晶性形状である。
【0064】
患者に経口で投与されたとき(すなわち、患者が当該タブレットを嚥下することにより)、本発明の薬剤は、時間とともに血しょう又は血液中のガバペンチン濃度のプロフィールを提供し得る。実施例1に記載される組成物及び荷重を有する薬剤について、ガバペンチン血しょう濃度プロフィールは、患者に化合物(1)を投与した後の、絶食した、及び食物を与えられた人間の当該患者それぞれについて、図1及び2に示される形状、大きさ、及びAUCを有する。これらのプロフィールは、ガバペンチンのみの投与後に得られたプロフィールと異なっている。1つの重要な相違は、最大血液濃度(Cmax)に達するまでの時間(Tmax)である。
【0065】
絶食した患者において、本発明の持続放出経口薬剤についてのTmaxは、約4時間超である。食物を与えられた患者において、持続放出経口薬剤についてのTmaxは、約6時間超である。対照的に、絶食した、及び食物を与えられた患者にガバペンチンのみを投与した後のTmaxは、約2〜4時間である。本発明の持続放出経口薬剤のもう1つの重要な有利点は、当該ガバペンチン代謝体の生物学的利用能である。化合物(1)の1200mgの荷重用量で、本発明の薬剤は、ガバペンチンの当モル用量の投与と比較して、絶食した患者において、少なくとも20%より高い、そしてある態様においては、少なくとも25%より高いガバペンチン生物学的利用能を提供し得る。化合物(1)の1200mgの荷重用量で、本発明の薬剤は、ガバペンチンの当モル用量の投与と比較して、食物を与えられた患者において、少なくとも50%より高い、そしてある態様においては、少なくとも100%より高いガバペンチン生物学的利用能を提供し得る。
【0066】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約3μg/mL〜約6μg/mLの範囲のCmax、約4時間〜約7時間の範囲のTmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0067】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の食物を与えられた人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約5μg/mL〜約8μg/mLの範囲のCmax、約6時間〜約11時間の範囲のTmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0068】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で絶食した人間の患者群に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約3μg/mL〜約6μg/mLの範囲の平均Cmax、約4時間〜約7時間の範囲の平均Tmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し、そして、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で食物を与えられた人間の患者群に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、約5μg/mL〜約8μg/mLの範囲の平均Cmax、約6時間〜約11時間の範囲の平均Tmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0069】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約3μg/mL〜約6μg/mLの範囲のCmax、約4時間〜約7時間の範囲のTmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0070】
ある態様において、1又は複数の食物を与えられた人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約5μg/mL〜約8μg/mLの範囲のCmax、約6時間〜約11時間の範囲のTmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0071】
ある態様において、絶食した人間の患者群への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約3μg/mL〜約6μg/mLの範囲の平均Cmax、約4時間〜約7時間の範囲の平均Tmax、及び約30μg・hr/mL〜約70μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得、そして、食物を与えられた人間の患者群への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、約5μg/mL〜約8μg/mLの範囲の平均Cmax、約6時間〜約11時間の範囲の平均Tmax、及び約60μg・hr/mL〜約110μg・hr/mLの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供し得る。
【0072】
本発明の薬剤は、吸収の比率及び程度に関して、本明細書中に記載される薬剤と生物学的に同等の薬剤を含み、例えばそれはU.S.Food and Drug Administrationにより定義され、及び「Guidance for Industry−Bioavailability and Bioequivalence Studies for Orally Administered Drug Products」(2003)において議論されるようなものである。
【0073】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、あるいは、1又は複数の食物を与えられた人間の患者に投与したとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0074】
ある態様において、約1100mg〜約1300mgの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で1又は複数の絶食した人間の患者に投与されたとき、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、かつ、1又は複数の食物を与えられた人間の患者に投与したとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0075】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、あるいは、1又は複数の食物を与えられた人間の患者への同様の薬剤の経口投与は、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0076】
ある態様において、1又は複数の絶食した人間の患者への、各薬剤が600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む2つの持続放出経口薬剤の経口投与は、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得、かつ、1又は複数の食物を与えられた人間の患者への同様の薬剤の経口投与は、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供し得る。
【0077】
経口投与後、本発明の薬剤は、少なくとも約6時間の間、ある態様においては、少なくとも約12時間の間、ある態様においては、少なくとも約18時間の間、及び他の態様においては、少なくとも24時間の間、患者の血液及び/又は血しょう中のガバペンチンの治療濃度又は予防的濃度を提供し得る。患者の血液及び/又は血しょう中のガバペンチンの治療有効濃度又は予防的有効濃度は、例えば、治療される疾患、当該疾患の重篤度、当該患者の体重、当該患者の健康状態などを含む多数の要因に依存し得る。
【0078】
ある態様において、本発明の薬剤は、1日2度投与され得、ある態様においては、1日に1度投与され得る。
【0079】
治療的使用
本発明の持続放出経口薬剤は、親薬物であるガバペンチンが治療に有効だと知られている又は今後そのことが発見されるいずれかの疾患又は障害に苦しむ患者に投与され得る。ガバペンチンが処方される症状、及びそれ故本発明の薬剤もまた有効である症状は、てんかん、うつ、不安、精神病、認知、統合失調症、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、痛み{特に、神経障害性の痛み(例えばヘルペス後神経痛)、筋肉痛、及び骨格痛}、下肢静止不能症候群、ほてり、尿失禁、炎症性障害(すなわち関節症)、不眠症、胃腸障害、アルコール/コカイン中毒、エタノール離脱症候群、外陰病変、早漏、及びグルタミン酸作動性のようなものを含む。
【0080】
当該薬剤は、上記疾患又は障害に対する予防措置としても、患者に投与され得る。それ故、当該薬剤は、てんかん、うつ、不安、精神病、失神発作、運動機能減少、頭蓋障害、神経変性障害、パニック、痛み(特に、神経障害性の痛み、及び筋肉痛、及び骨格痛)、炎症性障害(すなわち、関節症)、不眠症、胃腸障害、エタノール離脱症候群、早漏、及び外陰病変の傾向を有する患者へ、予防措置として投与され得る。
【0081】
従って、当該薬剤は、ある疾患又は障害の予防のために、同時に他の疾患又は障害を治療するために使用され得る(例えば、精神病の予防、一方、胃腸障害の治療;神経障害性の痛みの予防、一方、エタノール離脱症候群の治療)。当該薬剤は、初期ウイルス性感染の間、抗ウイルス薬の如き他の薬物との組み合わせにおいて使用され、神経病の障害の結果生じる事態を阻害又は低減し得る。
【0082】
さらに、当該薬剤は、副作用として神経病の障害を引き起こすと知られるもの自体である他の薬物との組み合わせで使用され得、それ故、前記副作用の発生を阻害し、又は低減する。
【0083】
上記疾患、及び症状を治療する又は予防する際における当該薬剤の適合性は、本分野において記載される方法により決定される得る(例えば、Satzinger et al,米国特許第4,024,175号;Satzinger et al,米国特許第4,087,544号;Woodruff,米国特許第5,084,169号;Silverman et al,米国特許第5,563,175;Singh,米国特許第6,001,876号;Horwell et al,米国特許第6,020,370号;Silverman et al,米国特許第6,028,214号;Horwell et al,米国特許第6,103,932号;Silverman et al,米国特許第6,117,906号;Silverman,国際特許公開第WO92/09560号;Silverman et al,国際特許公開第WO93/23383号;Horwell et al,国際特許公開第WO97/29101号,Horwell et al,国際特許公開第WO97/33858号;Horwell et al,国際特許公開第WO97/33859号;Bryans et al,国際特許公開第WO98/17627号;Guglietta et al,国際特許公開第WO99/08671号;Bryans et al,国際特許公開第WO99/21824;Bryans et al,国際特許公開第WO99/31057号;Magnus−Miller et al,国際特許公開第WO99/37296号;Bryans et al,国際特許公開第WO99/31075号;Bryans et al,国際特許公開第WO99/61424号;Pande,国際特許公開第WO00/23067;Bryans,国際特許公開第WO00/31020;Bryans et al,国際特許公開第WO00/50027;Bryans et al,国際特許公開第WO02/00209;2005年8月5日に出願されたTran,米国特許出願公開第60/711,477号;及び2005年8月5日に出願されたTran,米国特許出願公開第60/710,963号を参照のこと。)。
【0084】
投薬
本明細書中に記載される特定の疾患、障害又症状の治療において有効であろう化合物(1)の量は、当該疾患又は症状の性質に、少なくとも部分的に、依存するだろうし、前術のような本分野において知られる標準的臨床技術により決定され得る。さらに、生体外又は生体内アッセイが場合により使用され、最適な用量範囲の特定を補助する。投与されるプロドラッグの量は、もちろん、他の要因間で、治療される疾患を患う対象、当該対象の重さ、当該苦痛の重篤度、投与様式、及び処方する医師の判断に依存するだろう。
【0085】
ある態様において、持続放出経口薬剤は、1日あたり1〜3回、患者に投与されるように適合される。他の態様において、持続放出経口薬剤は、1日あたり1〜2回、患者に投与されるように適合される。投薬は、それだけで、又は他の薬物との併用で提供され得、そして当該疾患又は障害の効果的な治療又は予防に必要とされる限り続けられ得る。
【0086】
ガバペンチンの経口投与のための好適な用量範囲は、通常、約100mg/日〜約3600mg/日であり、そして化合物(1)あるいはそれらの医薬として許容される塩又は医薬として許容される溶媒和物は、ガバペンチンの等モル量を提供するように調節され得る。用量範囲は、当業者に知られる方法により容易に決定され得る。
【0087】
以下の実施例は、持続放出経口薬剤、持続放出経口薬剤を製造する際に使用される方法、及び材料、並びに、患者に対する当該薬剤の投与により達成された結果をさらに例証する。
【実施例】
【0088】
実施例1
化合物(1)を含む持続放出経口薬剤タブレットを表1に示される成分を有するように製造した。
【0089】
【表1】
【0090】
当該タブレットを、以下のステップに従い製造した。化合物(1)、第2リン酸カルシウム、グリセリルベヘネート、滑石、及びコロイド状シリコンジオキシドの重さを量り、#20メッシュスクリーンを通してスクリーニングし、そしてV−ブレンダー中で15分間混合した。ラウリル硫酸ナトリウムのスラッギング部分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのスラッギング部分の重さを量り、そして#40メッシュスクリーンを通した。スクリーニングされたラウリル硫酸ナトリウム、及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして5分間混合した。当該混合物を排出し、そして、タブレット圧縮機上で約400mg重のスラッグに圧縮した。次いで、当該スラッグを、Comil 194 Ultraミル(Quadro Engineering,Inc.,Millburn,NJ)に通し、さらなる圧縮のための製粉された材料を得た。ラウリル酸硫酸ナトリウムのタブレット分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのタブレット分の重さを量り、そして#40メッシュスクリーンを通した。当該製粉された材料、及び当該ラウリル硫酸ナトリウム、及びステアリン酸マグネシウムのタブレット分をV−ブレンダーに添加し、そして3分間混合した。当該混合された材料を排出し、そして、圧縮し、1310mgの総重量、及び600mg(45.8重量%)の化合物(1)積載量を有するタブレットを形成させた。当該タブレットは、16.1〜22.2kp(158〜218ニュートン)の平均最終硬度を有した。
【0091】
実施例2
表1に示される成分を有する持続放出経口製剤タブレットを、以下のステップに従う異なる方法を使用して製造した。化合物(1)、第2リン酸カルシウム、グリセリルベヘネート、滑石、及びコロイド状シリコンジオキシドの重さを量り、#20メッシュスクリーンを通してスクリーニングし、そしてV−ブレンダー中で15分間混合した。ラウリル硫酸ナトリウムの圧縮分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムの圧縮分の重さを量り、そして#40メッシュスクリーンを通した。スクリーニングされたラウリル硫酸ナトリウム、及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして5分間混合した。当該混合物を排出し、そして、Chilsonator機(FitzPatrick、Elmhurst,ILによるローラー圧縮機)上でコンパクトに圧縮した。次いで、得られたコンパクトを、ハンマーミル(FitzPatrick,Elmhurst,NJ)に通し、さらなる圧縮のための製粉された材料を得た。ラウリル酸硫酸ナトリウムのタブレット分の重さを量り、そして#30メッシュスクリーンを通した。ステアリン酸マグネシウムのタブレット分の重さを量り、そして#40メッシュスクリーンを通した。当該製粉された材料、及び当該ラウリル硫酸ナトリウム、及びステアリン酸マグネシウムのタブレット分をV−ブレンダーに添加し、そして3分間混合した。当該混合された材料を排出し、そして、圧縮し、1310mgの総重量、及び600mg(45.8重量%)の化合物(1)積載量を有するタブレットを形成した。当該タブレットを、300mgの化合物(1)積載量を有する655mgのタブレットにも圧縮した。当該タブレットは、各々、15.7〜18.9kp、そして11.1〜13.7kpの平均最終硬度を有した。
【0092】
実施例3
成人健常者における化合物(1)の持続放出経口薬剤の、安全性、耐容性、及び薬物動態についての、無作為化交差、絶食した/食物を与えられた単回投与試験を実施した。実施例1の持続放出経口薬剤を使用した。当該試験を、商業的なガバペンチンカプセル製剤(Neurontin)(登録商標)と比較して人間におけるこの薬剤の能力を評価するように計画した。12人の成人健常者(7人の男性、及び5人の女性)がこの試験に参加した。平均体重は、75.6kgだった。全患者は、治療と治療との間に1週間のウォッシュアウトを有し、無秩序な順番に異なる治療を受けた。当該2つの治療とは、(A)一晩食物を与えられた後の実施例1のタブレットの単回投与{2×600mg化合物(1)};及び(B)高脂肪朝食の後に実施例1の単回投与{2×600mg化合物(1)}であった。
【0093】
投与に先立って、及び投与後0.5時間、1時間、1.5時間、2時間、3時間、4時間、6時間、8時間、12時間、18時間、36時間で、全患者から血液、及び血しょうサンプルを回収した。尿サンプルを全患者から投与に先立ち回収し、そして、完全尿排出量を、投与後、0〜4時間、4〜8時間、8〜12時間、12〜18時間、18〜24時間、及び24〜36時間の間隔で得た。血液サンプルをメタノールですぐに急冷し、≦−70℃で冷凍保存した。サンプルアリコートを、高感度、及び特異的LC/MS/MS方法を使用するガバペンチン、及び化合物(1)の分析のために準備した。
【0094】
絶食した、及び食物を与えられた成人健常患者に対する、実施例1に従って製造された持続放出経口薬剤の経口投与後のガバペンチンの血しょう濃度±1SDを、それぞれ図1及び2に示す。
【0095】
当該タブレットの経口投与後(絶食)の血しょう中のガバペンチンについての平均±SD Cmaxは、4.21±1.15μg/mLだった。高脂肪の朝食後の当該タブレットの投与後、血しょう中のガバペンチンについてのCmaxは、6.24±1.55μg/mLにさらに増大した。
【0096】
当該タブレットの経口投与後(絶食)の血しょう中のガバペンチンについての平均±SD AUCは、54.5±12.2μg・hr/mLだった。高脂肪の朝食後の当該タブレットの投与後、血しょう中のガバペンチンのAUCは、83.0±21.8μg・hr/mLにさらに増大した。食物の存在下、当該タブレットの投与後のガバペンチンへの暴露は、絶食した患者におけるものと比較してさらに52%増大した。
【0097】
ガバペンチンのピーク血しょう濃度までの時間(Tmax)は、当該タブレットの経口投与後、著しく遅延した。絶食した患者において、当該タブレットの経口投与は、5.08±1.62hrのガバペンチンTmaxを与えた。これは、約2〜4hrの即時放出ガバペンチンの典型的Tmaxと比較する。当該タブレットの投与後のガバペンチンTmaxは、食物の存在下、8.40±2.07hrにさらに遅延した。
【0098】
血しょう中のガバペンチンについての明らかな最終的な排出半減期は、全ての処置:絶食した患者における当該タブレットについて6.47±0.77hr、及び食物を与えられた患者における当該タブレットについて5.38±0.80hrを超えて同じであった。
【0099】
当該タブレットの経口投与後、尿中に回収された当該ガバペンチン用量のパーセントは、絶食した患者について46.5±15.8%であり、食物を与えられた患者について73.7±7.2%だった。
【0100】
当該タブレットの経口投与後の血しょう中の完全なプロドラッグへの暴露は、低かった。絶食した患者における当該タブレットの経口投与後、血しょう中の完全な化合物(1)の濃度は、対応するピークガバペンチン濃度の約1.0%である、最大0.040μg/mLに達した。同様に、これらの患者の血しょう中の化合物(1)のAUCは、対応する血しょう中のガバペンチンのAUCの0.3%だった。食物を与えられた患者における当該タブレットの経口投与後、血しょう中の完全な化合物(1)の濃度は、対応するピークガバペンチン濃度の約0.3%である、最大0.018μg/mLに達した。同様に、これらの患者の血しょう中の化合物(1)のAUCは、血しょう中のガバペンチンのAUCの<0.1%だった。
【0101】
実施例4
食物を与えられた人間の患者に対する、実施例2に従って製造された持続放出経口薬剤の経口投与後、ガバペンチンの平均血しょう濃度を、図3に示す。12人の人間の患者におけるガバペンチンの平均血しょう濃度を、(a)300mgの化合物(1)を含む1つのタブレット、(b)600mgの化合物(1)を含む1つのタブレット、及び(c)各タブレットが600mgの化合物(1)を含む2つのタブレット中の1200mgの化合物(1)の投与後、実施例3に記載される方法に従って測定した。
【0102】
実施例5
カニクイザルに対する、実施例1に従って製造された持続放出経口薬剤の経口投与後のガバペンチンの血中濃度±1SDを、図4に示す。カニクイザルにおけるガバペンチンの血中濃度を、以下の手順に従い測定した。
【0103】
投与プロトコール
化合物(1)を含むタブレット{タブレットあたり1×600mg化合物(1)}を、4匹の成人で雄のカニクイザル(Macaca fascicularis)(約3kgの体重)の一群に経口投薬により投与した。各サルに1つのタブレットを投与した。動物に、試験前の一晩、及び投薬後約4時間、絶食させた。血液サンプル(1.0mL)を、経口投薬後、24時間に渡り、大腿静脈経由で得た。
【0104】
血液を、メタノールを使用してすぐに急冷し、分析するまで−20℃で凍結した。試験化合物を、投薬セッションの間、最小72時間のウォッシュアウト期間を有するサルに投与した。
【0105】
吸収された薬物についてのサンプル製造
300μLのメタノールを、サンプル及びスタンダードの製造のために、1.5mLのエッペンドルフチューブに添加した。
【0106】
サンプル製造:血液を異なる時点で回収し、即座に、100μLの血液を、300μLのメタノールを含むエッペンドルフチューブに添加し、そしてボルテックスにより混合した。
【0107】
スタンダード製造:90μLの血液を、エッペンドルフチューブ中の300μLのメタノールに添加した。10μLのガバペンチンのスタンダード溶液(0.04、0.2、1、5、25、及び100μg/mL)を各チューブに添加し、最終的較正スタンダード(0.004、0.02、0.1、0.5、0.25、及び10μg/mL)を作成した。
【0108】
20μLのp−クロロフェニルアラニンを、全サンプル、及びスタンダードに添加した。当該サンプルをボルテックスし、14,000rpmで20分間遠心した。当該上清を、LC/MS/MS分析のために使用した。
【0109】
LC/MS/MS分析
当該サルの血液中のガバペンチンの濃度を、Shimadzu SLC−10AVP、及びLEAPオートサンプラーを装備したAPI2000LC/MS/MS器具を使用して測定した。カラムは、室温で動作するZorbax C8XDB4.6×150mmカラムだった。移動相は、(A)水中に0.1%ギ酸、及び(B)アセトニトリル中に0.1%ギ酸だった。勾配条件は、3.5分間の2%B、3.5分間で95%Bに増大し2分間維持、次いで5.6分間で2%Bまで低減、そして2.3分間維持だった。30μLのサンプルを当該カラム中に注入した。Turbo−IonSpray源を使用し、そしてガバペンチンを、172/137のMRMトランジションについて陽イオンモードにおいて検出した。ピークを、Analyst1.2定量ソフトウェアを使用して調整した。
【0110】
実施例6
化合物(1)を含む持続放出経口薬剤を、下記の表2に示される成分を有するように製造した。
【表2】
【0111】
当該タブレットを以下のステップに従って作成した。化合物(1)、微晶性セルロース(MCC PH113)、グリセリルベヘネート、滑石、コロイド状シリコンジオキシド、及びドデシル硫酸ナトリウム(SDS)(第1混合分)及びステアリン酸マグネシウム(第1混合分)の重さを量り、#20メッシュスクリーンを通じてスクリーニングし、そしてV−ブレンダー中で7分間混合した{MaxiBlend Lab Blender MB−1(Globepharma)}。当該混合された成分を、ローラー圧縮機(BO50PH圧縮機、クロップドロール/クローズドエンド、3.9−in ロール直径、1.5−in ロール幅、11.6kN ロール力、12rpm ロールスピード、及び7rpm 水平スクリューフィードスピード)のフィードホッパー中に注入した。
【0112】
次いで、当該圧縮された材料を、Quadro Underdriven Comilモデル U5ミル(Quadro Engineering,Inc.,Millburn,NJ,0.079−in グレイター孔サイズ、1607 インペラスタイル、2500 インペラrpm)を通して、さらなる圧縮のために製粉された材料を得た。当該製粉された材料を、ブレンダー{MaxiBlend Lab ブレンダーモデル MB−1(Globepharma)、25rpm シェルスピード}に移し、そして5分間混合した。追加のSDS(第2混合分)、及び/又はステアリン酸マグネシウム(第2混合分)を、必要に応じて添加し、特定の量を得た。当該混合された材料を、排出し、そして圧縮し、総計1100mg、及び600mgの化合物(1)積載量(54.55%)を有するタブレットを製造した。当該タブレットは、14〜17kp(137〜214ニュートン)の平均最終硬度を有した。
【0113】
実施例7
実施例6に従って製造された持続放出経口薬剤の、カニクイザルに対する経口投薬後(1×600mg)のガバペンチンの血中濃度±1SDを、図5に示す。カニクイザルにおけるガバペンチンの血中濃度を、実施例5に記載された方法に従い測定した。
【0114】
実施例8
以下のステップを使用し、実施例1、2、及び6に従って製造された薬剤の生体外溶解プロフィールを測定した。薬剤を、37℃で、10mMリン酸2水素カリウム緩衝液(KH2PO4、pH7.4)及び1%(wt/体積)ラウリル硫酸ナトリウムの900mLを含む溶解容器中に配置した。当該溶解媒体を50rpm(USP,タイプII、パドル)で撹拌した。サンプルを0.5、1、2、4、6、8、12、及び24時間で回収し、溶液中の化合物(1)の含有量を、210nmでのフォトダイオード検出を有する、C18カラム、及びリン酸緩衝液/アセトニトリル/水の等張移動相を使用する逆相HPLCにより測定した。
【0115】
図6に示されるように、実施例1に従って製造された薬剤は、約2時間後に約20%、約5時間後に約50%、そして約8時間後に約80%の化合物(1)を放出した。図7Aに示されるように、実施例2に従って製造された300mgの化合物(1)を含む薬剤は、約2時間後に約20%、約6時間後に約50%、そして約10時間後に約80%の化合物(1)を放出した。図7Bに示されるように、実施例2に従って製造された600mgの化合物(1)を含む薬剤は、約2時間後に約20%、約5時間後に約50%、そして約8時間後に約80%の化合物(1)を放出した。図8に示されるように、実施例6に従って製造された薬剤は、約5時間後に約30%、約10時間後に約60%、そして約15時間後に約80%の化合物(1)を放出した。
【0116】
最後に、本発明を実施する他の方法があることに留意すべきである。従って、本明細書中に記載の態様は、例証、及び非制限的として考慮され、そして、本発明は、本明細書中に記載の詳細に制限されるべきではない、しかし、本明細書から生じる本発明の請求の範囲、及びそれと同等の範囲内において改良し得る。
【0117】
本明細書中に記載された刊行物、及び特許の全内容を、本願明細書中に援用する。
【特許請求の範囲】
【請求項1】
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤であって、以下:
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
前記持続放出経口薬剤。
【請求項2】
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤であって、以下:
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する、
前記持続放出経口薬剤。
【請求項3】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項1、又は2のいずれか1項に記載の薬剤。
【請求項4】
前記薬剤がタブレットを含む、請求項3に記載の薬剤。
【請求項5】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約30重量%〜約75重量%であり、前記放出速度改良ポリマーが約1重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項6】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約40重量%〜約65重量%であり、前記放出速度改良ポリマーが約1重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項7】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約50重量%〜約60重量%であり、前記放出速度改良ポリマーが約20重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項8】
前記放出速度改良ポリマーが、脂肪族化合物、及びメタクリル酸共重合体から選択される、請求項3に記載の薬剤。
【請求項9】
前記脂肪族化合物が、グリセリルエステルである、請求項8に記載の薬剤。
【請求項10】
前記グリセリルエステルが、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、ステアロイルマクロゴールグリセリド、及び前述のいずれかの組み合わせから選択される、請求項9に記載の薬剤。
【請求項11】
前記グリセリルエステルが、グリセリルベヘネートである、請求項10に記載の薬剤。
【請求項12】
前記脂肪族化合物が、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール、セトステアリルアルコール、ステアリン酸、パラフィンワックス、蜜ろう、グリコワックス、キャスターワックス、カルナバワックス、及び前述のいずれかの組み合わせから選択される請求項8に記載の薬剤。
【請求項13】
前記メタクリル酸共重合体が、アクリル酸エステル共重合体、メタクリル酸エステル共重合体、及び前述のいずれかの組み合わせから選択される、請求項8に記載の薬剤。
【請求項14】
約50mg〜約800mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項15】
約100mg〜約800mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項16】
約300mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項17】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が、結晶性形状である、請求項3に記載の薬剤。
【請求項18】
希釈剤、滑剤、抗粘着剤、流動促進剤、界面活性剤、崩壊剤、及び前述のいずれかの組み合わせから選択される、1又は複数の医薬として許容される賦形剤をさらに含む、請求項3に記載の薬剤。
【請求項19】
前記希釈剤が、第2リン酸カルシウム、及び微晶性セルロ−スから選択される、請求項18に記載の薬剤。
【請求項20】
前記薬剤が、約600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含むタブレットであり、かつ、その投与量が、2つの前記タブレット及び約1200mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項1又は2のいずれか1項に記載の薬剤。
【請求項21】
コーティングを含む、請求項3に記載の薬剤。
【請求項22】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;及び
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
請求項1に記載の薬剤。
【請求項23】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記絶食した人間の患者群に投与するとき、約3μg/mLから約6μg/mLまでの範囲の平均Cmax、約4時間から約7時間までの範囲の平均Tmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;及び、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記食物を与えられた人間の患者群に投与するとき、約5μg/mLから約8μg/mLまでの範囲の平均Cmax、約6時間から約11時間までの範囲の平均Tmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
請求項1に記載の薬剤。
【請求項24】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の絶食した人間の患者に投与するとき、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する;及び
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の食物を与えられた人間の患者に投与するとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する、
請求項2に記載の薬剤。
【請求項25】
前記1又は複数の絶食した人間の患者は、前記薬剤を投与する前の約10時間から投薬後約4時間までいかなる食物をも食べず、投薬前約2時間と約1時間に約250mLの水を飲み、及び投薬後約2時間に約250mLの水を飲み、投薬後約4時間に昼食を食べ、そして投薬後約10時間に夕食を食べる;並びに、
前記1又は複数の食物を与えられた人間の患者は、前記薬剤を投与する前の約30分前に試験食を食べ始め、当該薬剤を投与する前の約5分前に当該試験食を食べ終え、投薬後約4時間に昼食を食べ、そして投薬後約10時間後に夕食を食べる、ここで当該試験食は、その中で約500カロリーが脂肪のカロリーを構成する約1000総カロリーを含む、
請求項1又は2のいずれか1項に記載の薬剤。
【請求項26】
かかる治療を必要とする患者に、請求項1又は2のいずれか1項に記載の薬剤を投与することを含む、神経障害性の痛み、てんかん、下肢静止不能症候群、ほてり、尿失禁、早漏、及び外陰病変から選択される疾患又は症状を治療する方法。
【請求項27】
前記痛みが、ヘルペス後神経痛を含む、請求項26に記載の方法。
【請求項28】
10mMのリン酸2水素カリウム緩衝液、及びpH7.4で1%(重量/体積)のラウリル硫酸ナトリウムの中に置かれ、そして37℃、50rpm(USP、タイプII)で撹拌されたとき、約2時間後に前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の約20%、約5時間後に約50%、及び約8時間後に約80%を放出する、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤。
【請求項29】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項28に記載の薬剤。
【請求項30】
約500mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項29に記載の薬剤。
【請求項31】
第2リン酸カルシウムをさらに含む、請求項29に記載の薬剤。
【請求項32】
10mMのリン酸2水素カリウム緩衝液、及びpH7.4で1%(重量/体積)のラウリル硫酸ナトリウムの中に置かれ、そして37℃、50rpm(USP、タイプII)で撹拌されたとき、約5時間後に前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の約30%、約10時間後に約60%、及び約15時間後に約80%を放出する、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤。
【請求項33】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項32に記載の薬剤。
【請求項34】
約500mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項33に記載の薬剤。
【請求項35】
微晶性セルロースをさらに含む、請求項33に記載の薬剤。
【請求項1】
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤であって、以下:
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
前記持続放出経口薬剤。
【請求項2】
1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤であって、以下:
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の絶食した人間の患者に投与するとき、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する;又は、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、1又は複数の食物を与えられた人間の患者に投与するとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する、
前記持続放出経口薬剤。
【請求項3】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項1、又は2のいずれか1項に記載の薬剤。
【請求項4】
前記薬剤がタブレットを含む、請求項3に記載の薬剤。
【請求項5】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約30重量%〜約75重量%であり、前記放出速度改良ポリマーが約1重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項6】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約40重量%〜約65重量%であり、前記放出速度改良ポリマーが約1重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項7】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が約50重量%〜約60重量%であり、前記放出速度改良ポリマーが約20重量%〜約50重量%である、請求項3に記載の薬剤。
【請求項8】
前記放出速度改良ポリマーが、脂肪族化合物、及びメタクリル酸共重合体から選択される、請求項3に記載の薬剤。
【請求項9】
前記脂肪族化合物が、グリセリルエステルである、請求項8に記載の薬剤。
【請求項10】
前記グリセリルエステルが、グリセリルモノステアレート、グリセリルベヘネート、グリセリルパルミトステアレート、ラウロイルマクロゴールグリセリド、ステアロイルマクロゴールグリセリド、及び前述のいずれかの組み合わせから選択される、請求項9に記載の薬剤。
【請求項11】
前記グリセリルエステルが、グリセリルベヘネートである、請求項10に記載の薬剤。
【請求項12】
前記脂肪族化合物が、ラウリルアルコール、ミリスチルアルコール、ステアリルアルコール、セチルアルコール、セトステアリルアルコール、ステアリン酸、パラフィンワックス、蜜ろう、グリコワックス、キャスターワックス、カルナバワックス、及び前述のいずれかの組み合わせから選択される請求項8に記載の薬剤。
【請求項13】
前記メタクリル酸共重合体が、アクリル酸エステル共重合体、メタクリル酸エステル共重合体、及び前述のいずれかの組み合わせから選択される、請求項8に記載の薬剤。
【請求項14】
約50mg〜約800mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項15】
約100mg〜約800mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項16】
約300mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項3に記載の薬剤。
【請求項17】
前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸が、結晶性形状である、請求項3に記載の薬剤。
【請求項18】
希釈剤、滑剤、抗粘着剤、流動促進剤、界面活性剤、崩壊剤、及び前述のいずれかの組み合わせから選択される、1又は複数の医薬として許容される賦形剤をさらに含む、請求項3に記載の薬剤。
【請求項19】
前記希釈剤が、第2リン酸カルシウム、及び微晶性セルロ−スから選択される、請求項18に記載の薬剤。
【請求項20】
前記薬剤が、約600mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含むタブレットであり、かつ、その投与量が、2つの前記タブレット及び約1200mgの1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項1又は2のいずれか1項に記載の薬剤。
【請求項21】
コーティングを含む、請求項3に記載の薬剤。
【請求項22】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の絶食した人間の患者に投与するとき、約3μg/mLから約6μg/mLまでの範囲のCmax、約4時間から約7時間までの範囲のTmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;及び
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の食物を与えられた人間の患者に投与するとき、約5μg/mLから約8μg/mLまでの範囲のCmax、約6時間から約11時間までの範囲のTmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲のAUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
請求項1に記載の薬剤。
【請求項23】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記絶食した人間の患者群に投与するとき、約3μg/mLから約6μg/mLまでの範囲の平均Cmax、約4時間から約7時間までの範囲の平均Tmax、及び約30μg・hr/mLから約70μg・hr/mLまでの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する;及び、
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記食物を与えられた人間の患者群に投与するとき、約5μg/mLから約8μg/mLまでの範囲の平均Cmax、約6時間から約11時間までの範囲の平均Tmax、及び約60μg・hr/mLから約110μg・hr/mLまでの範囲の平均AUCにより特徴付けられるガバペンチン血しょう濃度プロフィールを提供する、
請求項1に記載の薬剤。
【請求項24】
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の絶食した人間の患者に投与するとき、図1に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する;及び
約1100mgから約1300mgまでの範囲の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の用量で、前記1又は複数の食物を与えられた人間の患者に投与するとき、図2に示されるプロフィールと生物学的に同等なガバペンチン血しょう濃度プロフィールを提供する、
請求項2に記載の薬剤。
【請求項25】
前記1又は複数の絶食した人間の患者は、前記薬剤を投与する前の約10時間から投薬後約4時間までいかなる食物をも食べず、投薬前約2時間と約1時間に約250mLの水を飲み、及び投薬後約2時間に約250mLの水を飲み、投薬後約4時間に昼食を食べ、そして投薬後約10時間に夕食を食べる;並びに、
前記1又は複数の食物を与えられた人間の患者は、前記薬剤を投与する前の約30分前に試験食を食べ始め、当該薬剤を投与する前の約5分前に当該試験食を食べ終え、投薬後約4時間に昼食を食べ、そして投薬後約10時間後に夕食を食べる、ここで当該試験食は、その中で約500カロリーが脂肪のカロリーを構成する約1000総カロリーを含む、
請求項1又は2のいずれか1項に記載の薬剤。
【請求項26】
かかる治療を必要とする患者に、請求項1又は2のいずれか1項に記載の薬剤を投与することを含む、神経障害性の痛み、てんかん、下肢静止不能症候群、ほてり、尿失禁、早漏、及び外陰病変から選択される疾患又は症状を治療する方法。
【請求項27】
前記痛みが、ヘルペス後神経痛を含む、請求項26に記載の方法。
【請求項28】
10mMのリン酸2水素カリウム緩衝液、及びpH7.4で1%(重量/体積)のラウリル硫酸ナトリウムの中に置かれ、そして37℃、50rpm(USP、タイプII)で撹拌されたとき、約2時間後に前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の約20%、約5時間後に約50%、及び約8時間後に約80%を放出する、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤。
【請求項29】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項28に記載の薬剤。
【請求項30】
約500mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項29に記載の薬剤。
【請求項31】
第2リン酸カルシウムをさらに含む、請求項29に記載の薬剤。
【請求項32】
10mMのリン酸2水素カリウム緩衝液、及びpH7.4で1%(重量/体積)のラウリル硫酸ナトリウムの中に置かれ、そして37℃、50rpm(USP、タイプII)で撹拌されたとき、約5時間後に前記1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の約30%、約10時間後に約60%、及び約15時間後に約80%を放出する、1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸の持続放出経口薬剤。
【請求項33】
以下の:
(a)約10重量%〜約80重量%の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸;及び
(b)約1重量%〜約50重量%の放出速度改良ポリマー、
を含み、ここで重量%は当該製剤の総乾燥重量に基づく、請求項32に記載の薬剤。
【請求項34】
約500mg〜約700mgの範囲の量の1−{[(α−イソブタノイルオキシエトキシ)カルボニル]アミノメチル}−1−シクロヘキサン酢酸を含む、請求項33に記載の薬剤。
【請求項35】
微晶性セルロースをさらに含む、請求項33に記載の薬剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7A】
【図7B】
【図8】
【公開番号】特開2013−107900(P2013−107900A)
【公開日】平成25年6月6日(2013.6.6)
【国際特許分類】
【外国語出願】
【出願番号】特願2013−34943(P2013−34943)
【出願日】平成25年2月25日(2013.2.25)
【分割の表示】特願2007−539363(P2007−539363)の分割
【原出願日】平成17年11月3日(2005.11.3)
【出願人】(503455503)ゼノポート,インコーポレイティド (22)
【Fターム(参考)】
【公開日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2013−34943(P2013−34943)
【出願日】平成25年2月25日(2013.2.25)
【分割の表示】特願2007−539363(P2007−539363)の分割
【原出願日】平成17年11月3日(2005.11.3)
【出願人】(503455503)ゼノポート,インコーポレイティド (22)
【Fターム(参考)】
[ Back to top ]