
シトシンの化学修飾により微生物核酸を簡素化するための方法
シトシンを修飾する作用物質で微生物ゲノムまたは核酸を処理し、微生物核酸誘導体を形成する工程と、微生物核酸誘導体を増幅して微生物ゲノムまたは核酸の簡素化形態を生成する工程とを含む、微生物ゲノムまたは微生物核酸を簡素化するための方法。
【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本発明は、微生物の検出のための核酸検出アッセイに関する。本発明は、微生物検出のために特異的リガンドの使用と組み合わせて微生物ゲノムの複雑さを低減することを目的とした核酸の化学処理のための方法にも関する。
【背景技術】
【0002】
背景技術
現在、特異的核酸分子の検出のために多数の手順が利用可能である。これらの手順は、典型的には標的核酸と、長さで短いオリゴヌクレオチド(20塩基またはそれ未満)から何キロベース(kb)もの配列の範囲でありうる核酸プローブとの間の配列依存的なハイブリダイゼーションに依存する。
【0003】
核酸配列の集団から特異的配列を増幅するための最も広く用いられる方法は、ポリメラーゼ連鎖反応(PCR)の方法である(Dieffenbach, C and Dveksler, G. eds. PCR Primer: A Laboratory Manual. Cold Spring Harbor Press, Plainview NY)。この増幅方法では、相補的DNA鎖上および増幅されるべき領域のいずれかの末端にて一般に長さが20〜30ヌクレオチドであるオリゴヌクレオチドを使用することで変性された一本鎖DNA上でDNA合成がプライミングされる。熱安定性DNAポリメラーゼを用いた変性、プライマーハイブリダイゼーションおよびDNA鎖合成の連続サイクルにより、プライマー間の配列の指数関数的増幅が可能である。RNA配列をまず逆転写酵素を用いた複製によって増幅することで相補的DNA(cDNA)複製物が生成されうる。ゲル電気泳動、標識プローブを伴うハイブリダイゼーション、引き続く(例えば酵素結合アッセイによる)同定を可能にするタグ付プライマーの使用、および標的DNAとのハイブリダイゼーション時にシグナルを生成する蛍光タグ付プライマーの使用(例えばBeaconおよびTaqMan系)を含む種々の手段により、増幅DNA断片の検出が可能である。
【0004】
PCRと同様、特異的ヌクレオチド配列の検出および増幅を目的とした種々の他の技術が開発されている。一例としてリガーゼ連鎖反応が挙げられる(1991 , Barany, F. et al., Proc. Natl. Acad. Sci. USA 88, 189-193)。
【0005】
別の例として1992年に最初に記載された等温増幅が挙げられ(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992))、鎖置換増幅(Strand Displacement Amplification;SDA)と称される。それ以来、RNAポリメラーゼを用いてRNA配列を複製するが対応するゲノムDNAを複製しない転写媒介増幅(Transcription Mediated Amplification;TMA)および核酸配列ベース増幅(Nucleic Acid Sequence Based Amplification;NASBA)を含む多数の他の等温増幅技術についての記載がなされている(Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richmann DD and Gingeras TR. 「レトロウイルス複製後にモデル化された多酵素反応によるin vitroでの核酸の等温増幅」 PNAS 87: 1874-1878 (1990): Kievits T, van Gemen B, van Strijp D, Schukkink R, Dircks M, Adriaanse H, Malek L, Sooknanan R, Lens P. NASBA 「HIV-1感染の診断用に最適化されたin vitroでの酵素による核酸の等温増幅」 J Virol Methods. 1991 Dec; 35(3):273-86)。
【0006】
他のDNAに基づく等温技術には、DNAポリメラーゼが環状鋳型に方向付けられたプライマーを伸長させるローリングサークル増幅(RCA)(Fire A and Xu SQ. 「短いサークルのローリング複製」 PNAS 92: 4641-4645 (1995))、標的検出用に環状プローブを用いる分岐増幅(RAM)(Zhang W, Cohenford M, Lentrichia B, lsenberg HD, Simson E, Li H, Yi J, Zhang DY. 「等温分岐増幅法によるトラコーマクラミジアの検出:フィージビリティ・スタディ」 J Clin Microbiol. 2002 Jan; 40(1): 128-32.)およびより最近では、熱の代わりにヘリカーゼを用いてDNA鎖を巻き戻すヘリカーゼ依存性等温DNA増幅(HDA)(Vincent M, Xu Y, Kong H. 「ヘリカーゼ依存性等温DNA増幅」 EMBO Rep. 2004 Aug; 5(8):795-800.) が含まれる。
【0007】
最近、DNA増幅の等温法についての記載がなされてきている(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992))。従来の増幅技術は、標的分子の変性および再生のサイクルを増幅反応の各サイクルで継続させることに依存している。DNAの熱処理によってある程度のDNA分子の剪断が生じることから、DNAが例えば発達中の胚細胞由来の少数の細胞からのDNAの単離など限定的である場合、または特にDNAが既に組織切片、パラフィンブロックおよび古いDNA試料などの断片化形態である場合、この加熱−冷却サイクルによってDNAがさらに損傷を受け、かつ増幅シグナルの低下がもたらされる可能性がある。等温法は、さらなる増幅から得られる鋳型として働く一本鎖分子を生成するための鋳型DNAの継続する変性に依存するのではなく、一定温度での特異的制限エンドヌクレアーゼによるDNA分子の酵素によるニッキングに依存する。
【0008】
鎖置換増幅(SDA)と称される技術は、特定の制限酵素における半修飾DNAの未修飾鎖をニッキングする能力および5’−3’エキソヌクレアーゼ欠損ポリメラーゼにおける下流の鎖に対する延長能および置換能に依存する。次いで、センス反応からの鎖置換がアンチセンス反応のための鋳型としての働きをする、センスおよびアンチセンス反応を連結することにより、指数関数的増幅が行われる(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅」 PNAS 89: 392-396 (1992))。かかる技術は、結核菌(Mycobacterium tuberculosis)(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992), 「HIV−1、C型肝炎およびHPV−16」 Nuovo G. J., 2000)、トラコーマクラミジア(Chlamydia trachomatis)(Spears PA, Linn P, Woodard DL and Walker GT. 「トラコーマクラミジアの同時鎖置換増幅および蛍光偏光検出」 Anal. Biochem. 247: 130-137 (1997))の増幅の成功のために用いられている。
【0009】
これまでSDAの使用は、修飾鎖上であれば酵素切断に抵抗性を示すヘミホスホロチオエートDNA二本鎖を生成するため、ホスホロチオエートヌクレオチドの修飾に依存しており、消化ではなく酵素によるニッキングが生じ置換反応が促進される。しかし最近になり、数種類の「ニッカーゼ(nickase)」酵素の設計が行われている。これらの酵素は、従来の様式でDNAを切断することなく、一方のDNA鎖上にニックを生じさせる。「ニッカーゼ」酵素は、N.Alw1(Xu Y, Lunnen KD and Kong H. 「ドメインスワッピングによるニッキングエンドヌクレアーゼN.AlwIの設計」 PNAS 98: 12990-12995 (2001))、N.BstNBI(Morgan RD, Calvet C, Demeter M, Agra R, Kong H. 「制限エンドヌクレアーゼの特異的DNAニッキング活性の特徴づけ」 N.BstNBI. Biol Chem. 2000 Nov;381 (11 ):1123-5.)およびMly1(Besnier CE, Kong H. 「Mly1エンドヌクレアーゼのオリゴマー形成状態を変化させることによるニッキング酵素への変換」 EMBO Rep. 2001 Sep;2(9):782-6. Epub 2001 Aug 23)を含む。したがって、かかる酵素を使用すれば、SDA法が簡素化されることになる。
【0010】
さらに、熱安定性を示す制限酵素(Ava1)と熱安定性を示すExo−ポリメラーゼ(Bstポリメラーゼ)の併用によってSDAは改良されている。この併用により、この技術を用いて特有の単一の複製分子が増幅されうるように、108倍の増幅から1010倍の増幅へ反応の増幅効率が高まることが判明している。熱安定性を示すポリメラーゼ/酵素の併用の結果得られる増幅因子は約109である(Milla M. A., Spears P. A., Pearson R. E. and Walker G. T. 「鎖置換増幅における制限酵素Ava1およびExo−Bstポリメラーゼの使用」 Biotechniques 1997 24:392- 396)。
【0011】
これまであらゆる等温DNA増幅技術は、初期の二本鎖鋳型DNA分子が増幅の開始に先立ち変性されることを必要とする。さらに、増幅は各プライミング事象から1回開始されるのみである。
【0012】
直接検出のため、標的核酸は、最も一般的には、標的配列に相補的なプローブとのハイブリダイゼーション(サザンおよびノーザンブロッティング)に先立ち、ゲル電気泳動によるサイズを基準に分離され、固体支持体に移動される。プローブは、天然核酸あるいはペプチド核酸(PNA)または固定化核酸(LNA)またはインターカレーティング核酸(INA)などの類似体でありうる。プローブは、(例えば32Pで)間接標識されるかまたは直接検出法が用いられる場合がある。間接法は通常、ビオチンもしくはジゴキシゲニンなどの「タグ」のプローブへの取り込みに依存し、次いでプローブは酵素結合性の基質変換または化学発光などの手段によって検出される。
【0013】
広く用いられている核酸の直接検出のための別の方法は「サンドイッチ」ハイブリダイゼーションである。この方法では、捕捉プローブが固体支持体に連結され、溶液中の標的核酸が結合プローブとハイブリダイズする。結合していない標的核酸が洗い流され、結合した核酸が標的配列にハイブリダイズする第2のプローブを使用して検出される。検出には上で概説したように直接法または間接法を用いることができる。かかる方法の例として、サンドイッチハイブリダイゼーションの原理を用いる例である「分岐DNA」シグナル検出系が挙げられる(1991, Urdea, M. S., et al., Nucleic Acids Symp. Ser. 24,197-200)。核酸配列の直接検出を目的として核酸ハイブリダイゼーションを用いる急成長分野は、DNAマイクロアレイ分野である(2002, Nature Genetics, 32, [Supplement]; 2004, Cope, L.M., et al., Bioinformatics, 20, 323-331; 2004, Kendall, S.L., et al., Trends in Microbiology, 12, 537-544)。このプロセスでは、短いオリゴヌクレオチド(典型的にはアフィメトリクス(Affymetrix)システムにおける25−mers)からより長いオリゴヌクレオチド(典型的にはアプライドバイオシステムズ(Applied Biosystems)およびアジレント(Agilent)のプラットフォームにおける60−mers)や、cDNAクローンなどのさらにより長い配列に及ぶ範囲でありうる個々の核酸種が、固体支持体に格子パターンで固定されるかあるいは固体支持体上にフォトリソグラフィー的に合成される。次いで、タグが付加されるかまたは標識された核酸集団がアレイとハイブリダイズし、アレイ内の各スポットとのハイブリダイゼーションのレベルが定量化される。化学発光などの他の検出系の使用が可能であるが、最も一般的には、ハイブリダイゼーションにおいて放射性標識または蛍光標識された核酸(例えばcRNAまたはcDNA)が使用される。
【0014】
核酸配列の直接検出を目的として核酸ハイブリダイゼーションを用いる急成長分野はDNAマイクロアレイである(Young RA 「DNAアレイによる生物医学的発見」 Cell 102: 9-15 (2000); Watson 「新ツール:新型ハイテク捜査」 Science 289:850-854 (2000))。このプロセスでは、オリゴヌクレオチドから相補的DNA(cDNA)クローンなどのより長い配列に及ぶ範囲でありうる個々の核酸種が固体支持体に格子パターンで固定される。次いで、タグが付加されるかまたは標識された核酸集団がアレイとハイブリダイズし、アレイ内の各スポットのハイブリダイゼーションのレベルが定量化される。他の検出系が用いられたが、最も一般的には、ハイブリダイゼーションにおいて放射性標識または蛍光標識された核酸(例えばcDNA)が使用された。
【0015】
細菌、酵母および真菌などの微生物を検出するための従来の方法は、選択的な栄養培地上での微生物の培養と、それに続く、従来の光学顕微鏡下で見られる、大きさ、形状、胞子生成、生化学または酵素反応などの特徴および(グラム染色などの)特異的染色特性に基づく微生物の分類を含む。ウイルス種は、分化した組織または細胞内で成長し、次いで電子顕微鏡によって判定されたそれらの構造と大きさに基づいて分類する必要がある。かかる技術の主な欠点は、かかるアプローチの有用性を制限する従来の培養または細胞条件下で成長するのは微生物のすべてではないという点である。例えば髄膜炎菌(Neisseria meningitidis)、ストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)およびインフルエンザ桿菌(Haemophilus influenzae)(全部が髄膜炎を誘発し、それらの中で髄膜炎菌は髄膜炎と劇症髄膜炎菌血症のいずれも誘発する)などの細菌の場合、3つの種全部は培養することが困難である。最大7日間にわたり毎日定期的に血液培養ボトルが検査され、継代培養が必要とされる。インフルエンザ桿菌は、ニコチンアミドアデニンジヌクレオチドとヘミンの双方を含有する特定の培地およびチョコレート寒天プレート上での成長を必要とする。血液培養物は、トリプチカーゼソイブロス(trypticase soy broth)または脳心臓浸出物(brain heart infusion)およびポリアネトールスルホン酸ナトリウムなどの様々な添加物の添加を必要とする。重度の食中毒およびフロッピーベビー症候群を誘発するボツリヌス菌(Clostridium botulinum)などの微生物における毒素の同定には、食物抽出物または培養上清のマウスへの注射および2日後での結果の可視化が含まれる。さらに、特定の培地上での潜在的な微生物の培養には1週間かかる。黄色ブドウ球菌(Staphylococcus aureus)エンテロトキシン(食中毒ならびに皮膚感染、血液感染、肺炎、骨髄炎、関節炎および脳膿瘍の原因)は、イオン交換樹脂を介する毒素の選択的吸収またはモノクローナル抗体を用いた逆受身ラテックス凝集(Reverse Passive Latex Agglutination)によって微量に検出される。それに対し、表皮ブドウ球菌(S.epidermis)は血液感染をもたらし、病院内の機器や床・壁・天井(surfaces)および医療設備や医療器具を汚染する。
【0016】
非ウイルス微生物についても、グルコース、マルトースまたはスクロースなどの基質上での発酵反応の間での特異的アミノ酸または代謝産物の産生などのそれらの代謝特性に基づく分類が可能である。あるいは、微生物では抗生物質に対するそれらの感受性に基づく分類が可能である。細胞表面抗原に対する特異的抗体または毒素などの排泄タンパク質を使用しても微生物が同定または分類される。しかし、上記方法はいずれも、後の試験に先立つ微生物の培養に左右される。微生物の培養は高価で多大な時間を要し、選好性が低めの微生物による汚染または過成長を被る可能性もある。これらの技術はまた、確定診断に達するために同じ試料に対して多数の試験を行う必要がある点であまり洗練されているとはいえない。大部分の微生物は既知の培地内では容易に成長できず、それ故に微生物の異種の典型的な混合集団が野生でまたはより高等な生物に随伴して存在する場合には検出レベルを下回る。
【0017】
病原微生物を検出および同定するための他の方法は、微生物による感染に応答して抗体が産生されるという血清学的アプローチに基づく。例えば髄膜炎菌(Meningococci)は、それらの莢膜多糖類における構造的な違いに基づいて分類可能である。これらは異なる抗原性を有し、5つの主な血清型(A、B、C、YおよびW−135)の判定を可能にする。酵素結合免疫吸着アッセイ(ELISA)またはラジオイムノアッセイ(RIA)は、かかる抗体の産生を評価可能である。これらの方法はいずれも、感染の過程において宿主動物によって産生される特異的抗体の存在を検出する。これらの方法は、宿主動物によって抗体が産生されるまである程度時間がかかることから非常に早期の感染が見逃されることが多いという欠点を有する。さらに、かかるアッセイを使用しても過去の感染と現在の(active)感染を確実に区別することはできない。
【0018】
より最近では、感染疾患の診断における分子法の使用に多大な関心が寄せられている。これらの方法は、病原微生物の高感度かつ特異的な検出を提供する。かかる方法の例として、「分岐DNA」シグナル検出系が挙げられる。この方法は、サンドイッチハイブリダイゼーションの原理を使用する例である(Urdea MS et al. 「ヒトHIVおよび肝炎ウイルスの高感度な直接検出のための分岐DNA増幅多量体」 Nucleic Acids Symp Ser. 1991 ;(24):197-200)。
【0019】
細菌を検出および分類するための別の方法は、16SリボソームRNA配列の増幅である。16SrRNAは、種々の臨床または環境試料中での細菌種の検出を目的としたPCR増幅アッセイにおける使用に適する標的であることが報じられており、かつ16SrRNA遺伝子が種特異的な多型性を示すことから様々な特異的微生物を同定するのに使用されていることが多い(Cloud, J. L., H. Neal, R. Rosenberry, C. Y. Turenne, M. Jama, D. R. Hillyard, and K. C. Carroll. 2002. J. Clin. Microbiol. 40:400-406)。しかし、細菌の純粋培養が必要とされ、試料はPCR増幅後にさらに配列決定されるかまたは種の判定のためにマイクロアレイ型デバイスにハイブリダイズされる必要がある(Fukushima M, Kakinuma K, Hayashi H, Nagai H, lto K, Kawaguchi R. J Clin Microbiol. 2003 Jun; 41(6):2605-15)。かかる方法は高価であり、多大な時間や労力を要する。
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明者らは、任意の微生物種を対象とした一般的検出または初期スクリーニングアッセイに適合しうる、微生物を検出するための新たな方法を開発している。
【課題を解決するための手段】
【0021】
発明の開示
一般的な態様では、本発明は、シトシンを修飾する作用物質で微生物核酸を処理し、かつ処理された核酸を増幅することでゲノムまたは核酸の簡素化(simplified)形態を生成することにより、微生物ゲノムまたは核酸の塩基構成の複雑さを低減することに関する。
【0022】
第1の態様では、本発明は、
シトシンを修飾する作用物質で微生物ゲノムまたは核酸を処理し、微生物核酸誘導体を形成する工程と、
微生物核酸誘導体を増幅することで微生物ゲノムまたは核酸の簡素化形態を生成する工程と、
を含む、微生物ゲノムまたは微生物核酸を簡素化するための方法を提供する。
【0023】
第2の態様では、本発明は、
シトシンを修飾する作用物質で微生物由来のDNAを含有する試料を処理し、微生物核酸誘導体を形成する工程と、
微生物核酸誘導体の少なくとも一部を増幅することで、対応する未処理の微生物核酸と比べてシトシンの総数が低下している、微生物または微生物型に特異的な核酸配列を含む簡素化された核酸分子を形成する工程と、
を含む、微生物に特異的な核酸分子を生成するための方法を提供する。
【0024】
第3の態様では、本発明は、
微生物からDNA配列を得る工程と、
実質的にアデニン、グアニンおよびチミンの塩基を有するように、各シトシンをチミンに変化させることによって微生物DNA配列の変換を行うことにより、微生物DNA配列の簡素化形態を形成する工程と、
微生物DNAの簡素化形態から微生物に特異的な核酸分子を選択する工程と、
を含む、微生物に特異的な核酸分子を生成するための方法を提供する。
【0025】
第4の態様では、本発明は、本発明の第3の態様に記載の方法によって得られる、微生物に特異的な核酸分子を提供する。
【0026】
第5の態様では、本発明は、試験もしくはアッセイにおいて微生物に特異的な核酸分子に結合または該核酸分子を増幅するためのプローブあるいはプライマーを得ることを目的とした本発明の第3の態様に記載の方法の使用を提供する。
【0027】
第6の態様では、本発明は本発明の第5の態様によって得られるプローブまたはプライマーを提供する。
【0028】
第7の態様では、本発明は、試料中での微生物の存在を検出するための方法であって、
微生物を含有すると見られる試料から微生物DNAを得る工程と、
シトシンを修飾する作用物質によって微生物核酸を処理し、微生物核酸誘導体を形成する工程と、
所望の微生物に特異的な核酸分子の微生物核酸誘導体への増幅を可能にする能力を有するプライマーを提供する工程と、
微生物核酸誘導体に対して増幅反応を行うことで簡素化された核酸を形成する工程と、
所望の微生物に特異的な核酸分子を有する増幅核酸産物の存在についてアッセイし、所望の微生物に特異的な核酸分子の検出が試料中での微生物の存在を示す工程と、
を含む、方法を提供する。
【0029】
ゲノムまたは微生物核酸がDNAである場合、それを処理することでDNA誘導体を形成可能であり、次いで増幅することでDNAの簡素化形態が形成される。
【0030】
ゲノムまたは微生物核酸がRNAである場合、微生物ゲノムまたは核酸の処理に先立ちそれをDNAに変換してもよい。あるいは、微生物RNAを処理してRNA誘導体分子を生成してもよく、次いでそれは増幅に先立ちDNA誘導体分子に変換される。RNAからDNAへの変換方法は周知であり、cDNAを形成するための逆転写酵素の使用を含む。
【0031】
微生物ゲノムまたは核酸を、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から入手してもよい。
【0032】
微生物ゲノムまたは核酸を、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択してもよい。好ましくは、リボソーム遺伝子領域は、原核生物における16Sまたは23Sおよび単細胞真核微生物の場合の18Sおよび28Sである。作用物質を、重亜硫酸塩、酢酸塩またはクエン酸塩から選択してもよい。好ましくは、作用物質は重亜硫酸ナトリウムである。
【0033】
好ましくは、作用物質は、非相補的微生物核酸分子ではなく、2つの誘導体を形成する相補的二本鎖の微生物ゲノムDNAの各鎖内でシトシンをウラシルに修飾する。好ましい形態では、シトシンは典型的には微生物核酸内で見られるようにメチル化されていない。
【0034】
好ましくは、微生物核酸誘導体におけるシトシンの総数が対応する未処理の微生物ゲノムまたは核酸と比べて低下している。
【0035】
好ましくは、微生物ゲノムまたは核酸の簡素化形態におけるシトシンの総数が対応する未処理の微生物ゲノムまたは核酸と比べて低下している。
【0036】
好ましい一形態では、微生物核酸誘導体は、アデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未処理の微生物ゲノムまたは核酸と実質的に総数が同じ塩基を有する。
【0037】
別の好ましい形態では、微生物ゲノムまたは核酸の簡素化形態は、実質的にアデニン(A)、グアニン(G)およびチミン(T)の塩基からなる。
【0038】
好ましくは、増幅はポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって行われる。
【0039】
本発明の第2の態様に記載の方法は、微生物に特異的な核酸分子を検出する工程をさらに含みうる。
【0040】
好ましい形態では、微生物に特異的な核酸分子は、
微生物に特異的な核酸分子の標的領域に対する結合能を有する検出リガンドを提供し、かつ検出リガンドが標的領域に結合するのに十分な時間を与える工程と、
検出リガンドの標的領域に対する結合を測定し、微生物に特異的な核酸分子の存在を検出する工程と、
によって検出される。
【0041】
別の好ましい形態では、微生物に特異的な核酸分子は、増幅産物を分離しかつ分離産物を可視化することによって検出される。好ましくは、増幅産物は電気泳動によって分離され、ゲル上の1つもしくは複数のバンドを可視化することによって検出される。
【0042】
好ましくは、微生物に特異的な核酸分子は微生物内で自然発生しない。
【0043】
好ましい形態では、微生物に特異的な核酸分子は、微生物の分類レベルを示す核酸配列を有する。微生物の分類レベルは、科、属、種、株、タイプ、あるいは同じもしくは異なる地理的集団または底生性集団由来の異なる集団を含むがこれらに限定されない。
【0044】
本発明の第3の態様に記載の方法の好ましい形態では、2種以上の微生物DNA配列の簡素化形態が得られ、かつ少なくとも1種の微生物に特異的な核酸分子を得るために2種以上の配列が比較される。
【0045】
本発明の第7の態様の好ましい形態では、核酸分子は、
微生物に特異的な核酸分子の領域に対する結合能を有する検出リガンドを提供し、かつ検出リガンドが領域に結合するのに十分な時間を与える工程と、
検出リガンドの核酸分子に対する結合を測定し、核酸分子の存在を検出する工程と、
によって検出される。
【0046】
別の好ましい形態では、核酸分子は、増幅産物を分離しかつ分離産物を可視化することによって検出される。
【0047】
微生物がDNAゲノムまたは微生物ゲノムを有しないかまたは核酸がRNA、例えばRNAウイルスである場合には、DNAを作用物質で処理することを目的に、まずRNAウイルスゲノムをcDNAに変換してもよい。RNAを処理することも可能であり、増幅に先立ちRNA誘導体がDNAに変換される。
【0048】
好ましくは、核酸誘導体はアデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有する。重要なことには、核酸誘導体分子は、微生物DNAが任意のシトシン(C)にてメチル化されなかったことを前提として実質的にシトシンを有しない。
【0049】
好ましくは、増幅された核酸誘導体はA、TおよびGの塩基を実質的に有し、かつ対応する核酸誘導体(および未修飾の微生物核酸)と実質的に総数が同じ塩基を有する。増幅された核酸誘導体は簡素化された核酸と称される。
【0050】
好ましい形態では、微生物に特異的な核酸分子は微生物の分類レベルを示す核酸配列を有する。微生物の分類レベルは、科、属、種、株、タイプ、あるいは同じもしくは異なる地理的集団または底生性集団由来の異なる集団を含みうる。細菌の場合、発明者らは、細菌、プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目(Neisseriales);ナイセリア科(Neisseriaceae);ナイセリア属(Neisseria)などの一般に認められたスキーマに従うことができる。異なる集団は、微生物内部の細胞内形態(プラスミドもしくはファージミド)または病原性島などの微生物ゲノムの多形性の染色体領域内に存在するDNA分子内に存在する単一のヌクレオチド変化または変異に対して多形性を示しうる。
【0051】
本発明を用いて微生物およびウイルスゲノムの流動性を認識することも可能であり、かつ独立要素でありうるウイルスゲノムのキメラ性を認識することが可能であることから、新規に生じる株は、異なる動物由来のゲノム領域を再び類別することから生じる。例えば、他の哺乳類または鳥ウイルスゲノムから採取されるセグメントのキメラとしての新たなヒトインフルエンザ株が挙げられる。
【0052】
方法を微生物の既知の核酸配列からin silicoで実施することが可能であり、元の配列内の1個もしくは複数個のシトシンがチミンに変換されて簡素化された核酸が得られることは理解されるであろう。変換された配列から配列同一性の判定が可能である。かかるin silicoにおける方法は処理および増幅工程を再現する。
【0053】
微生物に特異的な核酸分子がこの方法によって任意の所定の微生物において得られている場合、プローブまたはプライマーを増幅反応における対象領域の増幅を保証するように設計してもよい。したがって、プローブまたはプライマーが設計されている場合、試料に対して臨床的または科学的アッセイを実施することで、所定の分類レベルで所定の微生物を検出することが可能になろう。
【0054】
微生物に特異的な核酸分子は特有であるかまたは分類レベル内で高い類似性を有する可能性がある。本発明の1つの利点は、例えば、配列類似性が高い1個もしくは複数個の分子に対し、微生物の分類レベル間または分類レベル内での潜在的な塩基の相違を著しく簡素化できる点である。特異的プライマーまたは数が減少した変性プライマーを使用して所定の試料中の微生物に特異的な核酸分子を増幅してもよい。
【0055】
シトシンを有する二本鎖DNAにおいて、処理工程により2個の核酸誘導体(各相補鎖に対して1個)が生成され、それぞれはアデニン、グアニン、チミンおよびウラシルの塩基を有する。2個の核酸誘導体は、二本鎖DNAの2本の一本鎖から生成される。2個の核酸誘導体は、好ましくはシトシンを全く有しないが、依然として元の未処理のDNA分子と同じ塩基の総数および配列長を有する。重要なことには、2個の核酸誘導体は互いに相補的でなく、増幅における上鎖および下鎖の鋳型を形成する。1本もしくは複数本の鎖を増幅における標的として用いることで簡素化された核酸分子の生成が可能である。核酸誘導体の増幅の間、核酸の対応する増幅された簡素化形態において上部鎖(または下鎖)内のウラシルがチミンによって置換される。増幅が継続するにつれて、新たな各相補鎖がアデニン、グアニン、チミンの塩基のみを有するようになることから上鎖(および/またはもし増幅される場合に下鎖)が希釈されることになる。
【0056】
本発明のこの態様が、微生物に特異的な核酸分子に対して相補的な配列を有する核酸分子、および好ましくは厳しい条件下で微生物に特異的な核酸分子にハイブリダイズ可能な核酸分子も含むことが理解されるであろう。
【0057】
本発明では、任意の微生物が所定の試料中に存在するか否かを判定するのに使用可能な、代表的な微生物型を示すプローブまたはプライマーを使用してもよい。さらに、微生物型に特異的なプローブを使用し、微生物の所定のタイプ、サブタイプ、変異体および遺伝子型の例を実際に検出するかまたは同定することが可能である。
【0058】
任意の所定の微生物において微生物に特異的な核酸分子が得られているかまたは同定されている場合、プローブまたはプライマーを増幅反応における対象領域の増幅を保証するように設計してもよい。処理または変換によって配列の非対称性がもたらされることから、プライマー設計を目的とする、処理によって変換されたゲノム(以降、「核酸誘導体」と称される)の両鎖を分析可能であることに注目することは重要であり、それ故に同一遺伝子座の「上」鎖および「下」鎖(「ワトソン」鎖および「クリック」鎖としても知られる)の検出にとって異なるプライマー配列が必要とされる。したがって、変換直後に存在するものとしての変換されたゲノムである2つの分子集団および核酸誘導体が従来の酵素学的手段(PCR)または等温増幅などの方法によって複製された後に生じる分子集団が存在する。プライマーは典型的には便宜上、変換された上鎖に対して設計されるが、下鎖に対してもプライマーを作製してもよい。したがって、試料に対して臨床的または科学的アッセイを実施することで所定の微生物を検出することが可能になろう。
【0059】
核酸誘導体の特異的領域の増幅を可能にするようにプライマーまたはプローブを設計してもよい。好ましい形態では、プライマーは微生物に特異的な核酸分子の増幅をもたらす。
【0060】
第7の態様では、本発明は、増幅反応のための1種もしくは複数種の試薬または成分とともに本発明の第5の態様に記載のプライマーまたはプローブを含む、微生物に特異的な核酸分子を検出するためのキットを提供する。
【0061】
好ましくは、微生物は、Kingdom Protoctista by Margulis, L., et al 1990, Handbook of Protoctista, Jones and Bartlett, Publishers, Boston USAなど、いかに様々に分類されようと、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、単細胞生物、または任意の他の微生物、あるいはHarrisons Principles of Internal Medicine, 12th Edition, edited by J D Wilson et al., McGraw Hill Incおよび後版で定義されたヒトに随伴する微生物から選択される。それは、OMIM、Online Mendelian Inheritance in Man, www.ncbi.govにおいて定義されたヒトの状態と関連して記載されたすべての微生物も含む。
【0062】
微生物は、細胞内外で成熟もしくは胞子形態である、またはキメラ生物形態に関連する、または苔癬に関連した微生物または細菌膜に関連した微生物などの2種以上の生物形態の間で外部片利共生的に(ectocommensally)存在する、病原体、自然発生的な環境試料、水生生物または空中浮遊生物(または液体または気体媒体中で存在するかもしくは運搬される生物)でありうる。
【0063】
RNAウイルスまたはウイロイドの存在については、まずそれらのRNAゲノムを逆転写によってcDNA形態に変換し、次いで試薬によってcDNAを修飾することにより、アッセイすることが可能である。これにより、RNAウイルス中のシトシンに生じる任意のメチル化の問題が、逆転写酵素がそれらを正常なシトシンであるかの如く複製する際に克服される。
【0064】
好ましくは、作用物質は非メチル化シトシンをウラシルに修飾し、次いでウラシルは核酸誘導体の増幅の間にチミンと置換される。好ましくは、シトシンを修飾するために使用される作用物質は重亜硫酸ナトリウムである。本発明の方法では、メチル化シトシンではなく、非メチル化シトシンを同様に修飾する他の作用物質もまた使用してもよい。例として、重亜硫酸塩、酢酸塩またはクエン酸塩が挙げられるがこれらに限定されない。好ましくは、作用物質は、水の存在下でシトシンをウラシルに修飾する試薬である重亜硫酸ナトリウムである。
【0065】
重亜硫酸ナトリウム(NaHSO3)は、シトシンの5,6−二重結合と容易に反応して脱アミノ化を受けやすいスルホン化されたシトシン反応中間体を形成し、水の存在下でウラシルサルファイト(uracil sulfite)を生成する。必要であれば、亜硫酸基を弱アルカリ性の条件下で除去する結果としてウラシルを形成してもよい。したがって、可能性としてはすべてのシトシンがウラシルに変換されることになる。しかし、メチル化による保護に起因する試薬の修飾により、どのメチル化シトシンをも変換することはできない。
【0066】
本発明を、同一細菌種の分離株間の膨大な想定外のゲノム変異によって明らかになる発生する問題の一部の回避に役立つように適合させてもよい(2005, Tettelin , H., et al., Proc. Natl. Acad. Sci. USA. 102, 13950-13955;「ストレプトコッカス・アガラクチアエ(Streptococcus agalacticiae)の複数の病原性分離株のゲノム分析:微生物“全ゲノム(pan-genome)”の意味するもの」)。この細菌種のすべての分離株は、約80%の遺伝子プールに加え、部分的に共有されかつ株特異的タンパク質をコードする遺伝子からなる可欠ゲノムを示す、タンパク質をコードする遺伝子の「コア」ゲノムを有する。本発明に記載の方法によって細菌集団内に存在する23S遺伝子を処理することにより、発明者らは、すべての細菌分離株内に存在するタンパク質をコードしないコア成分を扱うことができる。
【0067】
本発明は、まず生物が属する一般集団を決定することを目的とし、種レベルを超えるまたは種レベルでの初期の同一性が有用である場合での微生物についての臨床、環境、法医学、生物戦、または科学的アッセイに適する。例として、任意の生物における疾患(脊椎動物、無脊椎動物、原核または真核細胞、例えば植物および家畜の疾患、養魚場およびカキ養殖場などのヒトの食料源での疾患)の診断、スクリーニングあるいは体外受精診療施設内でのヒト胚細胞の産生もしくは動物飼育における細胞培養物または体外受精卵の汚染について判定する、自然または汚染状態である環境源のサンプリングが挙げられる。法医学的状況または生物戦に関連した微生物の検出は特に重要である。
【0068】
本明細書全体を通し、もし文脈上、別の意味を必要としない場合、「含む(comprise)」あるいは「含む(comprises)」または「含む(comprising)」などの変形は、述べられる要素、整数または工程、あるいは要素、整数または工程の群を含むだけでなく、任意の要素、整数または工程、あるいは要素、整数または工程の群を除外しないことを意味することが理解されるであろう。
【0069】
本明細書中に含められている文書、行為、材料、デバイス、記事などに関するいずれの説明も、専ら本発明に対する文脈をもたらすことを目的とするものである。これらの事項のいずれかまたはすべてが、先行技術基盤の一部を形成しているとか、あるいは本発明の構築に先立ちオーストラリア内に存在したかのように本発明に関する分野における共通の一般的知識であったという承認として解釈されるべきものではない。
【0070】
本発明がより明確に理解されうるように、好ましい実施形態が以下の図面および実施例と関連して記載されることになる。
【発明を実施するための最良の形態】
【0071】
本発明を実施するための形態
定義
本明細書において用いられる「ゲノムの簡素化」という用語は、ゲノム(または他の)核酸が4つの塩基のアデニン(A)、グアニン(G)、チミン(T)およびシトシン(C)からなる状態から修飾されることで、実質的に塩基のアデニン(A)、グアニン(G)、チミン(T)を有するがやはり実質的に総数が同じ塩基を有することを意味する。
【0072】
本明細書において用いられる「核酸誘導体」という用語は、塩基のA、G、TおよびU(または何らかの他の非A、非Gもしくは非T塩基または塩基様実体(base−like entity))を実質的に有し、かつ対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有する核酸を意味する。微生物DNA内の実質的にすべてのシトシンが、作用物質による処理の間、ウラシルに変換されていることになる。例えばメチル化などにより改変されたシトシンが必ずしもウラシル(または何らかの他の非A、非Gもしくは非T塩基または塩基様実体)に変換されない場合があることは理解されるであろう。微生物核酸が典型的にはメチル化シトシン(または他のシトシン改変物)を有しないことから、処理工程では好ましくはすべてのシトシンが変換される。好ましくは、シトシンはウラシルに修飾される。
【0073】
本明細書において用いられる「簡素化された核酸」という用語は、核酸誘導体の増幅後に結果的に得られる核酸産物を意味する。次いで、核酸誘導体内のウラシルは核酸誘導体の増幅の間にチミン(T)と置換されて簡素化された核酸分子が形成される。結果産物は、対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有するが、実質的に3つの塩基(A、GおよびT)の組み合わせから構成される。
【0074】
本明細書において用いられる「簡素化配列」という用語は、核酸誘導体を増幅して簡素化された核酸が形成された後に結果的に得られる核酸配列を意味する。得られる簡素化配列は、対応する未修飾の微生物核酸配列と実質的に総数が同じ塩基を有するが、実質的に3つの塩基(A、GおよびT)の組み合わせから構成される。
【0075】
本明細書において用いられる「変換されていない配列」という用語は、処理および増幅に先立つ微生物核酸の核酸配列を意味する。変換されていない配列は、典型的には自然発生的な微生物核酸配列である。
【0076】
本明細書において用いられる「修飾する」という用語は、シトシンの別のヌクレオチドへの変換を意味する。好ましくは、作用物質は非メチル化シトシンをウラシルに修飾し、核酸誘導体を形成する。
【0077】
本明細書において用いられる「シトシンを修飾する作用物質」という用語は、シトシンを別の化学物質に変換する能力がある作用物質を意味する。好ましくは、作用物質は、シトシンをウラシルに修飾し、次いでウラシルは核酸誘導体の増幅の間にチミンと置換される。好ましくは、シトシンを修飾するために使用される作用物質は重亜硫酸ナトリウムである。メチル化シトシンではなく、シトシンを同様に修飾する他の作用物質もまた本発明の方法において使用してもよい。例として、重亜硫酸塩、酢酸塩またはクエン酸塩が挙げられるがこれらに限定されない。好ましくは、作用物質は、酸性の水性条件の存在下でシトシンをウラシルに修飾する試薬の重亜硫酸ナトリウムである。重亜硫酸ナトリウム(NaHSO3)は、シトシンの5,6−二重結合と容易に反応して脱アミノ化の作用を受けやすいスルホン化されたシトシン反応中間体を形成し、水の存在下でウラシルサルファイトを生成する。必要であれば、亜硫酸基を弱アルカリ性の条件下で除去する結果としてウラシルを形成してもよい。したがって、可能性としてはすべてのシトシンがウラシルに変換されることになる。しかし、メチル化による保護に起因する試薬の修飾により、あらゆるメチル化シトシンを変換することはできない。シトシン(または任意の他の塩基)を、本発明によって教示される核酸誘導体を得るための酵素的手段によるならば修飾可能であることが理解されるであろう。
【0078】
核酸内塩基に対する化学的および酵素的修飾を可能にするのに、2つの幅広い一般的方法がある。したがって、天然酵素あるいはいまだ報じられていない人工的に構築されるかまたは選択される酵素により、本発明の修飾を行ってもよい。重亜硫酸塩の方法論などの化学的処理により、適切な化学的工程を介してシトシンをウラシルに変換してもよい。同様に、例えばシトシンデアミナーゼが変換を行うことで核酸誘導体を形成しうる。発明者らの知見によると、シトシンデアミナーゼに関する最初の報告は1932, Schmidt, G., Z. physiol. Chem., 208, 185である(1950, Wang, T.P., Sable, H.Z., Lampen, J.O., J. Biol. Chem, 184, 17-28,「シトシンヌクレオチドの酵素による脱アミノ化」も参照)。この初期の研究では、他のヌクレオ−デアミナーゼなしにシトシンデアミナーゼは得られなかったが、ワン(Wang)らは酵母および大腸菌からかかる活性を精製することができた。したがって、シトシンの任意の酵素的変換によって核酸誘導体が形成され、最終的にシトシンとは異なる塩基の挿入がその位置での次の複製の間にもたらされ、簡素化されたゲノムが生成されることになる。誘導体とそれに続く簡素化されたゲノムを生成するための化学的および酵素的変換は、微生物の自然発生的な核酸内のプリンであれピリミジンであれ任意のヌクレオ塩基に適用可能である。
【0079】
本明細書において用いられる「ゲノムまたは核酸の簡素化形態」という用語は、自然発生であれ合成であれ、通常4つの共通の塩基G、A、TおよびCを有するゲノムまたは核酸が、ゲノム内のCの大部分もしくはすべてが適切な化学修飾およびそれに続く増幅手順によってTに変換されていることからこの段階では主に3つの塩基G、AおよびTのみからなることを意味する。ゲノムの簡素化形態は、相対的なゲノムの複雑さが4つの塩基の基盤から3つの塩基組成物に向けて低減されることを意味する。
【0080】
本明細書において用いられる「塩基様実体(base−like entity)」という用語は、シトシンの修飾によって形成される実体を意味する。塩基様実体は、核酸誘導体の増幅の間にDNAポリメラーゼによって認識可能であり、ポリメラーゼにより、A、GまたはTが核酸誘導体内の塩基様実体の対向位置にある新たに形成された相補的DNA鎖上に配置される。典型的には、塩基様実体は、対応する未処理の微生物核酸内のシトシンから修飾を受けているウラシルである。塩基様実体の例として、プリンであれピリミジンであれ、任意のヌクレオ塩基が挙げられる。
【0081】
本明細書において用いられる「相対的な複雑さの低減」という用語は、プローブ長、すなわち同じ大きさの2つのゲノムにおいて、第1のゲノムが「変化しない状態」であって4つの塩基G、A、TおよびCからなる一方、第2のゲノムが正確に同じ長さであっても一部のシトシン(望ましくはすべてのシトシン)がチミンに変換されている場合の所定の一まとまりの分子条件下における、特異的遺伝子座に対するプローブのハイブリダイゼーションの同じ特異性およびレベルを得るのに必要な平均プローブ長の増加に関する。試験される遺伝子座は、元の変換されていないゲノムおよび変換されたゲノムにおける同じ位置に存在する。平均で11−merプローブが、4つの塩基G、A、TおよびCからなる4,194,304(411が4,194,304に相当)個の塩基の正規のゲノム内で完全にハイブリダイズする特有の位置を有することになる。しかし、一旦4,194,304個の塩基のかかる正規のゲノムが重亜硫酸塩または他の適切な手段によって変換されていると、この段階ではこの変換されたゲノムは3つの塩基のみからなり、明らかに複雑さが低減される。しかし、ゲノムの複雑さにおけるこの低減の結果として、かつて発明者ら独自のものであった11−merのプローブが簡素化されたゲノム内でハイブリダイズ可能である特有の部位をもはや有しないということである。この段階では、重亜硫酸塩変換の結果として新規に生じている11個の塩基配列からなる多数の他の相当位置が存在する可能性がある。ここでは、元の遺伝子座を見出しかつハイブリダイズするのに14−merのプローブが必要になる。当初、それが直感に反するように見られるかもしれないが、より多くのゲノムが同様と見られる(より類似した配列を有する)ことから、この段階で簡素化された3つの塩基からなるゲノムにおける元の位置を検出するのにプローブ長の増加が必要である。したがって、相対的なゲノムの複雑さの低減(または3つの塩基からなるゲノムの簡素さ)とは、元の特有部位を見出すためにより長いプローブを設計する必要があるという意味である。
【0082】
本明細書において用いられる用語「相対的なゲノムの複雑さの低減」は、未修飾DNAと比較して微生物に特異性を示すことが可能なプローブ長の増加によって測定されうる。この用語は、微生物の存在を判定するのに用いられるプローブ配列のタイプについても包含する。これらのプローブは、例えばPNAまたはLNAにおける従来とは異なる骨格、あるいは例えばINAにおいて示された骨格に対して修飾された付加物を有する場合がある。したがって、ゲノムでは、プローブが例えばINAにおけるインターカレーティングシュードヌクレオチド(intercalating pseudonucleotide)などの付加成分を有するか否かに無関係に、相対的な複雑さが低減していると考えられる。例として、DNA、RNA、固定化核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、インターカレーティング核酸(INA)、シクロヘキサニル核酸(CNA)およびこれらの混合物やこれらのハブリッド、ならびにこれらのリン原子修飾物、例えばホスホロチオエート、メチルホスホレート、ホスホアミダイト(phosphoramidites)、ホスホロジチエート(phosphorodithiates)、ホスホロセレノエート(phosphoroselenoates)、ホスホトリエステルおよびホスホボラノエート(phosphoboranoates)など(これらに限定されない)が挙げられるがこれらに限定されない。非天然ヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−ザイロ−LNA、β−D−ザイロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、ビシクロ−DNA、6−アミノ−ビシクロ−DNA、5−エピ−ビシクロ−DNA、α−ビシクロ−DNA、トリシクロ−DNA、ビシクロ[4.3.0]−DNA、ビシクロ[3.2.1]−DNA、ビシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキソピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内部に含まれるヌクレオチドが含まれるがこれらに限定されない。さらに、非リンを含有する化合物を、メチルイミノメチル、ホルムアセテート(formacetate)、チオホルムアセテート(thioformacetate)およびアミドを有する連結基などの(これらに限定されない)ヌクレオチドへの連結のために使用することが可能である。特に核酸および核酸類似体は、1つもしくは複数のインターカレーターシュードヌクレオチド(intercalator pseudonucleotide)(IPN)を含みうる。IPNの存在は、核酸分子における複雑さの一部を示すものではなく、例えばPNAにおけるその複雑さの骨格部分でもない。
【0083】
「INA」とは、本明細書中に参照により援用される国際公開第03/051901号パンフレット、国際公開第03/052132号パンフレット、国際公開第03/052133号パンフレットおよび国際公開第03/052134号パンフレット(ユネスト(Unest A/S))の教示に記載のインターカレーティング核酸を意味する。INAは、1種もしくは複数種のインターカレーターシュードヌクレオチド(IPN)分子を有するオリゴヌクレオチドまたはオリゴヌクレオチド類似体である。
【0084】
「HNA」とは、例えばVan Aetschot et al., 1995に記載の核酸を意味する。
【0085】
「MNA」とは、Hossain et al, 1998に記載の核酸を意味する。
【0086】
「ANA」はAllert et al, 1999に記載の核酸を示す。
【0087】
「LNA」は国際公開第99/14226号パンフレット(エキシコン(Exiqon))に記載の任意のLNA分子でありうる。好ましくは、LNAは国際公開第99/14226号パンフレットの要約書中に示された分子から選択される。より好ましくは、LNAはSingh et al, 1998, Koshkin et al, 1998またはObika et al., 1997に記載の核酸である。
【0088】
「PNA」は、例えばNielsen et al, 1991に記載のペプチド核酸を示す。
【0089】
本明細書において用いられる「相対的複雑さの低減」は、ATATATATATATAT(配列番号1)の配列と同じ長さのAAAAAAATTTTTTT(配列番号2)の配列の間の任意の数学的な複雑さの違いなど、塩基が生じる順序を示すのではなく、相対的ゲノムサイズ(および推定的にはゲノムの複雑さ)を有する元の再連結データを示すものでもない。このことはWaring, M. & Britten R. J.1966, Science, 154, 791-794; and Britten, RJ and Kohne D E., 1968, Science, 161 , 529-540およびそれらの中のCarnegie Institution of Washington Yearbook reportsを出所とする初期の参考文献によって科学論文に紹介されている。
【0090】
本明細書において用いられる「相対的なゲノムの複雑さ」とは、分子プローブによって接近される、2つのゲノム内の塩基の変化しない位置を示す(元のゲノムと変換されていないゲノムの双方が不変位置の1〜nで塩基を有する)。特定のヒト女性の30億塩基対のハプロイドヒトゲノムの場合、不変位置が1〜n(nは3,000,000,000)であると定義される。もし配列1〜nにおいてith塩基が元のゲノム内でCである場合、ith塩基は変換されたゲノム内でTである。
【0091】
本明細書において用いられる「ゲノム核酸」という用語は、微生物(原核生物および単細胞真核生物)RNA、DNA、タンパク質をコードする核酸、タンパク質をコードしない核酸、ならびに原核生物および単細胞真核微生物のリボソーム遺伝子領域を含む。
【0092】
本明細書において用いられる「微生物ゲノム」という用語は、染色体および染色体外核酸、ならびにプラスミド、バクテリオファージおよび最も幅広い意味での可動要素などのそのゲノムの一時的寄生物をカバーする。「ゲノム」は、例えばS.galactiaeのようなコア成分を有するとともに、異なる分離株間で変化するコーディングおよび非コーディング要素を有する可能性がある。
【0093】
本明細書において用いられる「微生物由来のDNA」という用語は、微生物から直接的に得られるか、あるいは逆転写酵素など既知であるかまたは適切な方法によって微生物RNAをDNAに変換することにより間接的に得られるDNAを含む。
【0094】
本明細書において用いられる「微生物」という用語は、Kingdom Protoctista by Margulis, L., et al 1990, Handbook of Protoctista, Jones and Bartlett, Publishers, Boston USAなど、いかに様々に分類されようと、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、単細胞生物、または任意の他の微生物を含むか、あるいはHarrisons Principles of Internal Medicine, 12th Edition, edited by J D Wilson et al., McGraw Hill Incおよび後版で定義されたヒトに関連した微生物を含む。それは、OMIM、Online Mendelian Inheritance in Man, www.ncbi.govにおいて定義されたヒト疾患と関連して記載されたすべての微生物も含む。
【0095】
本明細書において用いられる「微生物に特異的な核酸分子」という用語は、微生物に特異的な1つもしくは複数の配列を有する、本発明に記載の方法を用いて判定または取得されている分子を意味する。
【0096】
本明細書において用いられる「微生物の分類レベル」という用語は、同じもしくは異なる地理的集団または底生性集団由来の科、属、種、株、タイプ、または異なる集団を含む。細菌の場合、細菌、プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目;ナイセリア科;ナイセリア属などの一般に認められたスキーマが用いられる。異なる集団は、微生物内部の細胞内形態(プラスミドもしくはファージミド)内、または病原性島などの微生物ゲノムの多形性染色体領域内に存在するDNA分子内に存在する単一のヌクレオチド変化あるいは変異に対して多形性を示しうる。微生物およびウイルスゲノムの流動性は認識されており、ウイルスゲノムのキメラ性を含み、それは独立した核酸片の状態でありうる。それ故、異なる動物由来のゲノム領域の混ぜ合わせから新たに生じる株として、例えば他の哺乳類または鳥ウイルスゲノムから採取された断片のキメラとしての新たなヒトインフルエンザ株が挙げられる。
【0097】
本明細書において用いられる「高い配列類似性」という用語は、相対的な配列の複雑さおよび尺度としてのプローブ長に関する上記定義を含む。
【実施例】
【0098】
材料および方法
DNAの抽出
一般に、微生物DNA(またはウイルスRNA)は任意の適切な供給源から入手可能である。例として、細胞培養物、培養液、環境試料、臨床試料、体液、液体試料、組織などの固体試料が挙げられるがこれらに限定されない。試料由来の微生物DNAを標準的手順によって入手可能である。適切な抽出の一例は以下の通りである。目的の試料を、7M塩酸グアニジン400μl、5mM EDTA、100mMトリス/HCI pH6.4、1%トリトン−X−100、50mMプロテイナーゼK(シグマ(Sigma))、100μg/mlの酵母tRNAの中に置く。試料を1.5mlの使い捨て乳棒で十分に均一化し、60℃で48時間放置する。インキュベーション後、試料に対して5回のドライアイスでの凍結/解凍サイクルを5分/95℃で5分間施す。次いで試料をマイクロチューブ内で2分間回転およびボルテックスし、細胞残屑をペレット化する。上清を無菌チューブ内に移し、希釈して塩濃度を低下させ、フェノール:クロロホルム抽出し、エタノール沈殿させ、50μlの10mMトリス/0.1mM EDTAに再懸濁する。
【0099】
特に、(各々の種に特異的な栄養所要量を有する)標準的な寒天プレート上で成長させたグラム陽性およびグラム陰性細菌からのDNA抽出を以下のように行った。
【0100】
グラム陰性細菌からのDNA抽出におけるプロトコルは以下のようであった。
a)無菌のトゥースピックを使用し、細菌コロニーを培養プレートから無菌にした1.5mlの遠心管内にこすり落とした。
b)チオシアン酸グアニジン抽出緩衝液(7Mチオシアン酸グアニジン、5mM EDTA(pH8.0)、40mMトリス/HCl pH7.6、1%トリトン−X−100)180μlを添加し、試料を混合して細菌コロニーを再懸濁した。
c)20μl(20mg/ml)のプロテイナーゼKを添加し、試料を十分に混合した。
d)試料を55℃で3時間インキュベートし、細胞を溶解させた。
e)水200μlを各試料に添加し、ゆっくりとしたピペット操作により試料を混合した。
f)フェノール/クロロホルム/イソアミルアルコール(25:24:1)400μlを添加し、試料を15秒間で2回ボルテックスした。
g)次いで、試料をマイクロチューブ内、14,000rpmで4分間回転させた。
h)水相を無菌にした1.5mlの遠心管に移した。
i)フェノール/クロロホルム/イソアミルアルコール(25:24:1)400μlを添加し、試料を15秒間で2回ボルテックスした。
j)次いで、試料をマイクロチューブ内、14,000rpmで4分間回転させた。
k)水相を無菌にした1.5mlの遠心管に移した。
l)100%エタノール800μlを各試料に添加し、試料を短時間ボルテックスし、次いで−20℃で1時間放置した。
m)試料をマイクロチューブ内、4℃、14,000rpmで4分間回転させた。
n)DNAペレットを70%エタノール500μlで洗浄した。
o)試料をマイクロチューブ内、4℃、14,000rpmで5分間回転させ、エタノールを廃棄し、ペレットを5分間風乾した。
p)最後にDNAを100μlの10mMトリス/HCI pH8.0、1mM EDTA pH8.0に再懸濁した。
q)溶液の吸光度を230、260、280nmで測定することにより、DNAの濃度および純度を計算した。
【0101】
グラム陽性細菌からのDNA抽出におけるプロトコルは以下のようであった。
a)無菌のトゥースピックを使用し、細菌コロニーを培養プレートから無菌にした1.5mlの遠心管内にこすり落とした。
b)20mg/mlのLysozyme(シグマ(Sigma))180μlおよびLysostaphin(シグマ(Sigma))200μgを各試料に添加し、試料を徐々に混合して細菌コロニーを再懸濁した。
c)試料を37℃で30分間インキュベートし、細胞壁を分解した。
d)次いで、グラム陽性細菌に対するQIAamp DNAミニキットプロトコルに従って試料を処理し、DNAを抽出した。
【0102】
患者由来の細胞学的試料からのDNA抽出
a)試料を手動で激しく振とうさせて沈降した細胞を再懸濁しかつ溶液の均一性を確保した。
b)再懸濁した細胞4mlを15mlのコスター(Costar)遠心管に移した。
c)管をスイングアウト(swing−out)式バケットローター内、3000×gで15分間遠心した。
d)ペレット化した細胞物質を乱さないように、上清を注意深くデカントして廃棄した。
e)ペレット化した細胞を溶解緩衝液(100mMトリス/HCI pH8.0、2mM EDTA pH8.0、0.5%SDS、0.5%トリトン−X−100)200μlに再懸濁し、溶液が均一になるまで十分に混合した。
f)試料80μlを96ウェルの試料調製プレートに移した。
g)プロテイナーゼKを20μl添加し、溶液を55℃で1時間インキュベートした(この手順により細胞溶解物が得られる)。
【0103】
尿試料からのDNA抽出
QIAamp UltraSens(商標) Virus Handbookに従い、尿の開始容量1mlからDNAを抽出した。
【0104】
DNA試料の重亜硫酸塩処理
MethylEasy(商標) High Throughput DNA重亜硫酸塩修飾キット(ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures)、オーストラリア)に従い、重亜硫酸塩処理を行った。下記も参照のこと。
【0105】
意外なことに、例えばヒト細胞の試料中に微生物DNAが存在する場合、微生物DNAを核酸の他のソースから分離する必要性が全くないことが本発明者らによって見出されている。異なるDNAタイプの膨大な混合物に対して処理工程を利用してもよく、さらに微生物に特異的な核酸についても本発明によって同定してもよい。複雑なDNA混合物中での検出の限界が標的核酸分子の単一の複製物に至るまで対応可能である標準的なPCR検出の限界であると推定される。
【0106】
試料
本発明において任意の適切な試料を使用してもよい。例として、微生物培養物、臨床試料、獣医学的試料(veterinary samples)、体液、組織培養試料、環境試料、水試料、流出物が挙げられるがこれらに限定されない。本発明が任意の微生物の検出に適応可能であることから、このリストは網羅的と考えるべきではない。
【0107】
キット
本発明を様々なキットの形態でまたはキットの併用によって実施し、手動、半自動または完全にロボット化されたプラットフォームの点から具体化してもよい。好ましい形態では、MethyEasy(商標)またはHighThroughput MethylEasy(商標)キット(ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures Pty Ltd),オーストラリア)は、EpMotionなどのロボット化されたプラットフォームを使用して96または384プレート内での核酸の変換を可能にする。
【0108】
重亜硫酸塩処理
核酸の有効な重亜硫酸塩処理のための典型的なプロトコルを下記に示す。プロトコルにより、実質的にすべての処理されたDNAが保持されることになる。本明細書では、この方法は、ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures)(HGS)法とも称される。試料または試薬の容量または量が変化しうることは理解されるであろう。
【0109】
重亜硫酸塩処理の好ましい方法を、本明細書中に参照により援用される米国特許出願第10/428310号明細書またはPCT/AU2004/000549号明細書において見出すことができる。
【0110】
望まれる場合に適切な制限酵素で予備消化可能なDNA2μgに対し、2μl(1/10容量)の3M NaOH(水50ml中6g、作製後間もない)を最終容量が20μlになるように添加した。この工程により、重亜硫酸塩試薬が好ましくも一本鎖分子と反応することから二本鎖DNA分子が一本鎖形態に変性される。混合物を37℃で15分間インキュベートした。室温を超える温度でインキュベーションを用いることで変性の効率を改善してもよい。
【0111】
インキュベーション後、2Mメタ重亜硫酸ナトリウム(10N NaOH 416mlを有する水20ml中7.6g;BDH AnalaR #10356.4D;作製後間もない)208μlと10mMキノール(水50ml中0.055g、BDH AnalR #103122E;作製後間もない)12μlを連続的に添加した。キノールは還元剤であり、試薬の酸化の低減を促進する。ジチオトレイトール(DTT)、メルカプトエタノール、キノン(ヒドロキノン)、または他の適切な還元剤を例とする他の還元剤もまた使用してもよい。試料を鉱油200μlとオーバーレイした。鉱油のオーバーレイは試薬の蒸発および酸化を防止するが必須ではない。次いで、試料を55℃で一晩インキュベートした。あるいは、試料を、以下のように約4時間または一晩インキュベートしてサーマルサイクラー内でサイクル動作してもよい。PCR装置内で、工程1として55℃/2時間、工程2として95℃/2分サイクル動作させる。工程1を約37℃〜約9O℃の任意の温度で行い、5分〜8時間の長さで変化させてもよい。工程2を約70℃〜約99℃の任意の温度で行い、約1秒〜60分の長さまたはそれより長い時間で変化させてもよい。
【0112】
メタ重亜硫酸ナトリウムによる処理後、オイルを除去し、もしDNA濃度が低い場合、tRNA(20mg/ml)1μlまたはグリコーゲン2μlを添加した。これらの添加物は任意であり、特にDNAが低濃度で存在する場合、これらを使用することで標的DNAとの共沈によって得られるDNAの収量を改善することが可能である。核酸量が0.5μg未満である場合、より効率的な核酸の沈降を目的とした担体としての添加物の使用は一般に望ましい。
【0113】
イソプロパノールによるクリーンアップ処理を以下のように行った。水800μlを試料に添加し、混合し、次いでイソプロパノール1mlを添加した。水または緩衝液は、反応槽内の重亜硫酸塩の濃度を塩が目的の標的核酸と共沈しないレベルまで低下させる。本明細書に開示されるように、塩濃度が所望の範囲未満に希釈される限り、希釈率は一般に約1/4〜1/1000である。
【0114】
試料を再び混合し、4℃で最低5分間放置した。試料をマイクロチューブ内で10〜15分間回転させ、ペレットを70%ETOHで2回洗浄し、各回ボルテックスした。この洗浄処理により、核酸と共沈した任意の残留塩が除去される。
【0115】
ペレットを乾燥させておき、次いで50μlなどの適切な容量のT/E(10mMトリス/0.1mM EDTA) pH7.0〜12.5で再懸濁した。pH10.5の緩衝液が特に有効であることが判明している。試料を、核酸の懸濁に対する必要性に応じ、37℃〜95℃で1分〜96時間インキュベートした。
【0116】
重亜硫酸塩処理に関する別の例を、シトシンからウラシルへの変換に対する方法および材料を提供する国際公開第2005021778号パンフレット(本明細書中に参照により援用される)に見出すことができる。一部の実施形態では、gDNAなどの核酸は重亜硫酸塩およびトリアミンまたはテトラアミンなどのポリアミン触媒と反応する。場合により、重亜硫酸塩は重亜硫酸マグネシウムを含有する。他の実施形態では、核酸は、場合によってポリアミン触媒および/または第四アミン触媒の存在下で重亜硫酸マグネシウムと反応する。本発明の方法を実施するのに使用可能なキットも提供される。これらの方法を処理工程で用いれば本発明にも適することが理解されるであろう。
【0117】
増幅
PCR増幅を、重亜硫酸塩処理したゲノムDNAを2μl含有する反応混合物25μlにおいてPromega PCR master mix、6ng/μlの各プライマーを使用して行った。増幅のために鎖特異的な入れ子(nested)プライマーを使用した。PCRプライマー1および4を使用して第1ラウンドのPCR増幅を行った(下記参照)。第1ラウンドの増幅後、増幅された物質1μlをPCRプライマー2および3を有する第2ラウンドのPCRプレミックスに移し、上記のように増幅した。PCR産物の試料を、95℃で4分間の1サイクルの後、95℃で1分間、50℃で2分間および72℃で2分間の30サイクル;72℃で10分間の1サイクルといった条件下で、ThermoHybaid PX2サーマルサイクラー内で増幅した。
【0118】
【表1】

【0119】
多重増幅
もし検出にとって多重増幅が必要である場合、以下の方法論を実施してもよい。
【0120】
重亜硫酸塩処理したDNA1μlを、反応容量25μl中の以下の成分、すなわち×1 Qiagen multiplex master mix、5〜100ngの各々の第1ラウンド用のINAまたはオリゴヌクレオチドプライマー、1.5〜4.0mM MgSO4、400μMの各dNTPおよび0.5〜2単位のポリメラーゼの混合物に添加する。次いで成分をホットリッドのサーマルサイクラー内で以下のようにサイクル動作させる。各増幅反応では、典型的には最大で200の独立したプライマー配列がありうる。
【0121】
【表2】
【0122】
次いで、第1ラウンドの増幅物から分注した1μlを酵素反応混合物および適切な第2ラウンド用プライマーを有する第2ラウンドの反応管に移して第2ラウンドの増幅を行う。次いで上記のようにサイクル動作を行う。
【0123】
プライマー
本発明において任意の適切なPCRプライマーを使用してもよい。プライマーは、典型的には増幅対象の配列に対する相補配列を有する。プライマーは、典型的にはオリゴヌクレオチドであるが、オリゴヌクレオチド類似体であってもよい。
【0124】
プローブ
プローブは任意の適切な核酸分子または核酸類似体でありうる。例として、DNA、RNA、固定化核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、インターカレーティング核酸(INA)、シクロヘキサニル核酸(CNA)およびこれらの混合物やこれらのハブリッド、ならびにこれらのリン原子修飾物、例えばホスホロチオエート、メチルホスホレート、ホスホアミダイト、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなど(これらに限定されない)が挙げられるがこれらに限定されない。非天然ヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−ザイロ−LNA、β−D−ザイロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、ビシクロ−DNA、6−アミノ−ビシクロ−DNA、5−エピ−ビシクロ−DNA、α−ビシクロ−DNA、トリシクロ−DNA、ビシクロ[4.3.0]−DNA、ビシクロ[3.2.1]−DNA、ビシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキソピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内部に含まれるヌクレオチドが含まれるがこれらに限定されない。さらに、非リンを含有する化合物を、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミドを有する連結基などの(これらに限定されない)ヌクレオチドへの連結のために使用することが可能である。特に核酸および核酸類似体は、1つもしくは複数のインターカレーターシュードヌクレオチドを含みうる。
【0125】
好ましくは、プローブは、INAを形成する1種もしくは複数種の内部IPNを有するDNAまたはDNAオリゴヌクレオチドである。
【0126】
電気泳動
E−Gelシステムのユーザーガイド(www.invitrogen.doc)に従って試料の電気泳動を行った。
【0127】
検出方法
所望の試料の状態を測定するのに可能な極めて多数の検出系が存在する。本発明では核酸分子を検出するための既知の系または方法であればいずれも使用可能であることが理解されるであろう。検出系には、以下のものが含まれるがそれらに限定されない。
I.10から200,000に及ぶ個々の成分に対する選択が可能と思われるマイクロアレイ型デバイスに対する適切に標識したDNAのハイブリダイゼーション。ガラス、プラスチック、雲母、ナイロン、ビード、磁気ビード、蛍光ビードもしくは膜などの任意の適切な固体表面上に、これらのアレイをINA、PNAまたはヌクレオチドもしくは修飾ヌクレオチドアレイから構成できると思われる。
II.サザンブロット型検出系
III.例えばアガロースゲル、GeneScan分析などの蛍光読み出しといった標準的なPCR検出系。サンドイッチハイブリダイゼーションアッセイ、エチジウムブロミドなどのDNA染色試薬、サイバーグリーン(Syber green)、抗体検出、ELISAプレートリーダー型デバイス、蛍光分光デバイス
IV.特異的増幅もしくは多重増幅されたゲノム断片またはそれに対する任意の変異におけるリアルタイムPCR定量
V.国際公開第2004/065625号パンフレットの中で概説された蛍光ビーズ、酵素抱合体、放射性ビーズなどの検出系のいずれか
Vl.リガーゼ連鎖反応などの増幅工程または鎖置換増幅(SDA)などの等温DNA増幅技術を利用する任意の他の検出系
VII.マルチフォトン検出系
VIII.ゲルにおける電気泳動および可視化
IX.核酸を検出するのに使用されるまたは使用可能と思われる任意の検出プラットフォーム
【0128】
インターカレーティング核酸
インターカレーティング核酸(INA)は、配列特異性を有する核酸(DNAおよびRNA)にハイブリダイズ可能な非天然型ポリヌクレオチドである。INAは、いくつかの望ましい特性を示すことから、プローブに基づくハイブリダイゼーションアッセイにおける核酸プローブに対する代替物/置換物としての候補である。INAは、核酸にハイブリダイズすることで対応する天然核酸/核酸複合体よりも熱力学的に安定なハイブリッドを形成するポリマーである。それらはペプチドまたは核酸を分解することで知られる酵素に対する基質ではない。したがって、INAであれば、生物学的試料中でより安定であることに加え、天然核酸断片よりも長い貯蔵寿命(shelf−life)を有するはずである。INAの核酸とのハイブリダイゼーションは、イオン強度に強く依存する核酸ハイブリダイゼーションと異なり、イオン強度にほとんど依存せず、天然核酸の核酸に対するハイブリダイゼーションにとって極めて不適当な条件下の低いイオン強度で有利である。INAの結合強度は、分子内に設計された挿入基の数、ならびに二本鎖構造の特異的な様式で積み重なった塩基間の水素結合からの通常の相互作用に依存する。配列の識別は、DNAを認識するDNAの場合よりもINAを認識するDNAの場合の方がより効率的である。
【0129】
好ましくは、INAは(S)−1−O−(4,4’−ジメトキシトリフェニルメチル)−3−O−(1−ピレニルメチル)−グリセロールのホスホアミダイトである。
【0130】
INAは、商業的に利用可能な形式で標準的なオリゴヌクレオチドの合成手順を適合させることによって合成される。INAおよびそれらの合成の完全な定義について、本明細書中に参照により援用される国際公開第03/051901号パンフレット、国際公開第03/052132号パンフレット、国際公開第03/052133号パンフレットおよび国際公開第03/052134号パンフレット(ユネスト(Unest A/S))の中に見出すことができる。
【0131】
INAプローブと標準的核酸プローブの間には確かに多数の違いがある。これらの違いは、便宜上、生物学的、構造的、および物理−化学的な違いに分かれる。上記および下記で考察されるように、これらの生物学的、構造的、および物理−化学的な違いは、核酸が通常利用されている応用においてINAプローブの使用を試みる場合に予測不能な結果をもたらす場合がある。異なる組成物における相違点は、化学技術において観察されることが多い。
【0132】
生物学的違いに関しては、核酸は、遺伝的伝達および発現の作用物質として生物種の生命における中心的役割を担う生物学的物質である。それらのin vivoでの特性は、かなりよく理解されている。しかし、INAは最近開発された完全に人工的な分子であり、化学者の頭の中で考え出され、合成有機化学を用いて作製されたものである。それは既知の生物学的機能を全く有しない。
【0133】
構造的には、INAはまた核酸とは格段に異なる。双方ともに共通のヌクレオ塩基(A、C、G、TおよびU)の利用が可能であるが、これらの分子の組成物は構造的に多様である。RNA、DNAおよびINAの骨格は、リン酸ジエステルリボースおよび2−デオキシリボース単位の反復からなる。INAは、リンカー分子を介してポリマーに付着された1つもしくは複数の大きな均一分子を有する点でDNAまたはRNAと異なる。均一分子は、二本鎖構造をなすINAに対する相補的DNA鎖内の塩基間に挿入する。
【0134】
INAとDNAまたはRNAとの物理/化学的な違いは重要でもある。INAは同じ標的配列に結合する核酸プローブよりも迅速に相補的DNAに結合する。DNAまたはRNAの断片と異なり、もし挿入基が末端位置に位置しない場合、INAのRNAに対する結合は不十分である。相補的DNA鎖上の挿入基と塩基の間の強い相互作用のため、INA/DNA複合体の安定性は、類似体のDNA/DNAまたはRNA/DNA複合体のそれよりも高い。
【0135】
DNAもしくはRNAの断片またはPNAなどの他の核酸と異なり、INAは自己凝集または結合特性を示さない。
【0136】
INAは配列特異性を有する核酸にハイブリダイズすることから、INAはプローブに基づくアッセイの開発にとっての有用な候補であり、特にキットおよびスクリーニングアッセイに適合する。しかし、INAプローブは核酸プローブと同等ではない。それ故、プローブに基づくアッセイの特異性、感受性および信頼性を改善可能と思われる任意の方法、キットまたは組成物は、DNAを含有する試料の検出、分析および定量において有用となろう。INAはこのために必要な特性を有する。
【0137】
結果
微生物(細菌、ウイルスまたは真菌株など)の検出は、その種内の多数の微生物の各株によって阻害されることが多い。
【0138】
本発明のin silicoでの一般的原理について、髄膜炎菌、淋菌(Neisseria gonorrhoeae)、インフルエンザ桿菌、連鎖球菌(Streptococcus sp)およびブドウ球菌(Staphylococcus)の細菌を用いて教示する(図1〜5)。本発明の一般的原理について、インフルエンザウイルスおよびロタウイルスを用いて教示している(図6および7)。
【0139】
本発明を教示および支持するための一般的な生化学的データを、臨床的意義のあるグラム陽性およびグラム陰性細菌を用いて図8〜18に記載する。
【0140】
細菌
図1は、髄膜炎菌内のigaプロテアーゼ遺伝子の34個のヌクレオチド領域および淋菌内の対応する遺伝子座を示す(これらの領域はそれらの天然細菌ゲノム内に存在するものとする)(完全な分類;細菌;プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目;ナイセリア科;髄膜炎菌、Z2491血清型Aおよび完全な遺伝子座の特徴;iga、IgAIプロテアーゼ;GeneID906889、ローカスタグ(Locus Tag)NMA0905;RefSeq登録番号NC_003116.1;PMID10761919;Parkhill J et al., 2000, Nature, 404, 502- 506)。これら2つのナイセリア属の34個のヌクレオチド配列の間には74%の配列類似性がある。両方の細菌種内のこれらの領域を増幅するためにPCRに基づくプライマーを作製するとしたら、512通りの組み合わせが可能な変性プライマーが必要となろう。PCR増幅の一部として共通配列を使用するとしたら、それは34個のヌクレオチド配列GYAATYW AGGYCGYCTY GAAGAYTAYA AYATGGC(配列番号3)であろう。この場合、異なる位置を指定するための標準的コードを以下に示す。N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはCおよびW=AまたはT。
【0141】
しかし、これらの2つの種由来の細菌DNAを重亜硫酸塩試薬(シトシンのチミンへの変換をもたらす)で処理する場合、自然発生配列が、コーディングの可能性が全くなく天然に存在しない誘導配列に変換される。この段階で誘導配列は97%の配列類似性を有する。PCRに基づくプライマーは、これらの細菌の遺伝子座双方におけるPCR増幅を1回の試験で可能なように設計されると、この段階では2つのプライマーのみを組み合わせる必要があるにすぎない。位置7の塩基のみがアデニンまたはチミン(Wで示される)である場合、組み合わせは配列GTAATTW AGGTTGTTTT GAAGATTATA ATATGGT(配列番号4)に基づくであろう。したがって、重亜硫酸塩変換により、相対的なゲノムの複雑さが512通りから2通りのプライマータイプへと低減される。この大幅な低減により、関連する細菌種からの同じ遺伝子座の増幅が簡素化される。
【0142】
さらに、これら2種の細菌種由来の領域を増幅するのに場合によってINAプローブを使用し、再び同じ遺伝子座を使用することにより、利点がもたらされる。図2は、図1に示した髄膜炎菌および淋菌のiga遺伝子の同じ34個のヌクレオチド領域を図示し、これはINAプローブをさらに用いてもプローブ長や複雑さが低減可能であるという対象範囲を追加的に明らかにしている。短いINAの16merの配列AGGYCGYCTY GAAGAY(配列番号5)であれば、この領域を検出するのに16通りの可能なプライマーの組み合わせが必要となろうが、重亜硫酸塩による変換後には、特有のプライマー配列AGGTTGTTTT GAAGAT(配列番号6)で十分であろう。INA分子の利点は、骨格内に組み込まれるインターカレーティングシュードヌクレオチドのおかげで、標準オリゴヌクレオチドに対するINAのTmの増大により、正確な遺伝子座へのハイブリダイゼーションを非特異的結合と識別することが著しく容易になることである。そうであっても、標準オリゴヌクレオチドが依然として適切に機能することが理解されるであろう。
【0143】
関連性が高い細菌種が類似の臨床症状をもたらす場合、重亜硫酸塩で変換したDNAを再び使用して、特異的細菌タイプの存在についてアッセイするためのより簡素化したプローブを設計することができる。図3は、3種の細菌種内のiga遺伝子におけるDNAアラインメントを示し、それらの1種であるインフルエンザ桿菌は異なる分類群由来である。細菌DNAの重亜硫酸塩処理により、プローブの組み合わせ数が極めて少なくなった。この比較は、1回の試験で関連性のない種についてのアッセイが可能であることの重要性を示す。髄膜炎菌とインフルエンザ桿菌の双方が髄膜炎を誘発することから、1回の試験で同じ臨床症状を誘発するすべての微生物についてアッセイ可能であることは有利である。
【0144】
同じ分類群からの多数の異なる細菌種の分析は、本発明によってやはり容易になる。図4は、連鎖球菌群、すなわちストレプトコッカス・オラリス(S.oralis)、ストレプトコッカス・ミチス(S.mitis)、ストレプトコッカス・ディスガラクティエ(S.dysgalactiae)、ストレプトコッカス・クリスタツス(S.cristatus)、ストレプトコッカス・ゴルドニー(S.gordonii)、ストレプトコッカス・パラウベリス(S.parauberis)、ストレプトコッカス・ニューモニエ、ストレプトコッカス・ボビス(S.bovis)、ストレプトコッカス・ベスチブラリス(S.vestivularis)およびストレプトコッカス・ウベリス(S.uberis)の10種の細菌種内のtuf遺伝子の40個のヌクレオチドセグメントを示す。この領域は、10種の間で約68%の配列類似性を有し、1回の試験で10種について同時にアッセイするのに12,288通りのプライマーの組み合わせを必要とする。重亜硫酸塩変換された配列は、これらの種の間で85%の配列類似性を有し、ここで必要とする可能なプライマーの組み合わせは単に64通りである。
【0145】
同じ細菌種に属する異なる株の分析もまた本発明によって簡素化される。図5は、黄色ブドウ球菌のエンテロトキシン遺伝子の23個のヌクレオチドセグメントを図示する。この遺伝子領域の天然配列は、全部で7種の株の間に56%しか配列類似性を有さず、1536通りのプライマーの組み合わせを必要とする一方、重亜硫酸塩で変換した配列は74%の配列類似性を有し、必要とするプライマーの組み合わせは単に64通りである。
【0146】
ウイルス核酸分析および相対的なゲノムの複雑さの低減
相対的なゲノムの複雑さを低減する原理を、DNAゲノムを有するインフルエンザウイルスなどのウイルス群と同様に、(RNAは逆転写酵素によってDNAに変換され、次いでそれによって重亜硫酸塩処理されうることから)RNAゲノムを有するウイルス群にも適用してもよい。ウイルス検出への適用を例示するため、分節RNAゲノムを有する、インフルエンザウイルス株(オルトミクソウイルス科(Family Orthomyxoviridae))のノイラミニダーゼ遺伝子およびロタウイルス株(レオウイルス科(Family Reoviridae))の表面タンパク質をコードするVP4遺伝子の両ウイルスを使用している。インフルエンザウイルスの分類は複雑であり、例えばタイプA、BおよびCが抗原特性に基づき、かつさらなるサブタイプが複製起点部位、単離年、単離数およびサブタイプに基づいている。これにより、第一にインフルエンザウイルスを集団として同定し、次いで結果的にかなり下位の分類レベル(sub−sub−classification level)の分析にまで掘り下げられるという需要が高まる。
【0147】
ロタウイルスの分類もまた複雑である。ロタウイルス血清型の数は多く、PおよびG血清型という認められた2つの主な血清型を伴う。最小でも14種の異なるG血清型が存在し、小児医学ではそれらを明確に検出することが重要である。世界中のほぼすべての小児が、3歳になるまでに既に少なくとも1回ロタウイルスによる感染を、たとえこれらの感染が不顕性であり、胃腸管に対して軽度の作用を有するにすぎない場合であっても被っていると推定される。
【0148】
臨床レベルでのインフルエンザによる感染の結果はよく知られており、ほぼ毎冬で罹患率および死亡率が有意な状況である。しかし、特にストレプトコッカス・ニューモニエ、インフルエンザ桿菌および黄色ブドウ球菌による感染後に重度の二次合併症が発症しうる。肺炎合併症が細菌およびウイルス感染症の特徴が混ざり合うことから発症しうることや、速やかな抗生治療が有効な治療でありうることから、ウイルス感染症と細菌感染症の双方について同時分析できることが有利であることは極めて明らかである。
【0149】
9種の異なるインフルエンザ株における相対的なゲノムの複雑さの低減について図6に示す。インフルエンザウイルスのノイラミニダーゼ遺伝子の20個のヌクレオチド領域をそのDNA形態で示す。これらの9種の単離物の間には50%の配列類似性がある。重亜硫酸塩変換後、配列類似性は75%に増加している。その元の形態では、これらの9種の株を分析するには2048通りの可能なプライマーの組み合わせが必要であると思われる一方、重亜硫酸塩変換後に必要なプライマーの組み合わせは48通りにすぎない。
【0150】
3種の異なるロタウイルス株のVP4遺伝子における相対的なゲノムの複雑さの低減について図7に示す。VP4遺伝子の20個のヌクレオチド領域は、変換前に52%および変換後に74%の配列類似性を有する。プライマーの組み合わせ数は512から32に減少する。
【0151】
病院や病理学実験室で一般に遭遇する臨床的意義のある微生物種を用いた、微生物を検出する一手段として微生物ゲノムを簡素化するin silicoでのアプローチを支持する分子データを図8〜15に図示する。
【0152】
もし試料中に存在するのがグラム陽性細菌かまたはグラム陰性細菌かを初期に決定できるとしたら、それは汚染性微生物の迅速な検出にとって明らかな利点であり、臨床的意義が大きい。本明細書に記載の方法では、異なる細菌種の23Sリボソーム遺伝子を用いたかかる試験が提供され、増幅反応を介し、簡素化したゲノム上でプライマーセットを利用することによりグラム陽性細菌またはグラム陰性細菌の検出を可能にするかかるプライマーが生成される。23S配列は、例えば上記S.galactiaeの例において見られる一部の細菌ゲノムに任意に付加される一部のタンパク質コーディング配列とは異なり、すべての細菌種内で生じることから、かかる高レベルの識別にとって望ましい。多数のタンパク質をコードする微生物配列は、ゲノムの「がらくた(flotsam and jetsam)」に類するものであり、それらの有用性は、異なる微生物株、タイプまたは単離物などのより低いレベルの分類カテゴリの間、あるいはウイルスの場合、異なるタイプまたは新しく生じた変異体の間で識別することにある。これら成分すべての正規および簡素化されたゲノム配列、タンパク質をコードしないリボソームRNA遺伝子、および細菌のタンパク質をコードするrecA遺伝子をそれぞれ図15および16に示す。23S細菌アンプリコンに対する増幅反応の実施に使用されるプライマー配列を表1に示す。recAアンプリコンに対する増幅反応の実施に使用されるプライマー配列を表2に示す。すべてのプライマーを重亜硫酸塩で処理したDNAに対して作製し、5’から3’の向きで示す。
【0153】
表1は、グラム陽性(Pos)、グラム陰性(Neg)の検出用プライマーを生成するためのアラインメントを用いた、23SリボソームRNA遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる適切な細菌用プライマー配列を示す。さらに、マイコプラズマ(Mycoplasma spp)(Myc)、ブドウ球菌(Staphylococcus spp)(Staph)、連鎖球菌(Streptococcus spp)(Strep)、ナイセリア(Neisseria spp)(NG)、クラミジア(Chlamydia)(CT)、ならびに大腸菌(Escherichia coli)(EC)およびストレプトコッカス・ニューモニエの特異的検出用にプライマーを設計した。
【0154】
以下の記号は以下の塩基付加を指定する。N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはCおよびW=AまたはT。
【0155】
使用されるすべてのプライマーは重亜硫酸塩で簡素化したDNA配列に基づいた。
【0156】
【表3】
【0157】
【表4】
【0158】
【表5】
【0159】
【表6】
【0160】
【表7】
【0161】
【表8】
【0162】
表2は、特有の細菌の分類を目的とした、黄色ブドウ球菌(SA)、表皮ブドウ球菌(Staphylococcus epidermidis)(SE)、レイ菌(Serratia marscesens) (SM)、大腸菌(Escherichia coli)(EC)およびエンテロコリティカ菌(Yersinia enterocolitica)(YE)由来の、アラインメントを用いた、recAタンパク質をコードする遺伝子由来の簡素化DNAの増幅に用いられる細菌用プライマー配列を示す。
【0163】
【表9】
【0164】
【表10】
【0165】
表1は、グラム陽性(Posで示す)細菌およびグラム陰性(Negで示す)細菌の検出用の最適なプライマーを生成するための多重アラインメントを用いた、23SリボソームRNA遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる細菌用プライマー配列を示す。さらに、種の群および個々の種の特異的検出用にもプライマーを設計した。これらの細菌用プライマー群に対し、大腸菌および肺炎桿菌(EC)、Neisseria spp(NG)、クラミジア(CT)、マイコプラズマ(Myc)、連鎖球菌(Strep)およびブドウ球菌(Staph)のように指定する。下位指定のFおよびRのは、それぞれフォワードプライマーおよびリバースプライマーである。さらに、2種以上の可能な塩基が所定のヌクレオチド位置で必要である場合、塩基の縮重が、N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはC;およびW=AまたはTのコードによって与えられる。繰り返して言えば、本発明において用いられるすべてのプライマーは重亜硫酸塩で簡素化したDNA配列に基づいている。
【0166】
表2は、特有の細菌の分類を目的とした、黄色ブドウ球菌(SA)、表皮ブドウ球菌(SE)、レイ菌(SM)、大腸菌(Escherichia coli)(EC)およびエンテロコリティカ菌(YE)由来の、アラインメントを用いた、recAタンパク質をコードする遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる細菌用プライマー配列を示す。
【0167】
図8は、グラム陽性およびグラム陰性細菌のゲノム的に簡素化した23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示し、ここで適切にサイジングしたアンプリコンはアガロースゲル電気泳動による特異的な長さのバンドとして検出される。矢印は、マーカーのレーン(M)内を動く、標準的なサイジングしたマーカーに対するアンプリコンの想定サイズを示す。グラム陰性細菌に特異的なプライマーを使用すると、大腸菌、淋菌、肺炎桿菌、カタル球菌(Moraxella catarrhalis)、緑膿菌(Pseudomonas aeruginosa)およびプロテウス・ブルガリス(Proteus vulgaris)における6種のグラム陰性のレーン1〜6(上パネル)に限ってバンドが出現する。グラム陽性細菌に特異的なプライマーを使用すると、腸球菌(Enterococcus faecalis)、表皮ブドウ球菌、黄色ブドウ球菌、スタフィロコッカッス・キシロサス(Staphylococcus xylosis)、ストレプトコッカス・ニューモニエおよび溶血性連鎖球菌(Streptococcus haemolyticus)における6種のグラム陽性のレーン7〜12(下パネル)に限ってバンドが出現する。
【0168】
図9は、(この例では)大腸菌および肺炎桿菌(K.pneumoniae)という2種のみのグラム陰性細菌種を由来とするアンプリコンを検出するように設計した、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。増幅方法論の特異性について、大腸菌および肺炎桿菌を示すレーン1および3におけるアンプリコンの存在、ならびにレーン2およびレーン4〜12において増幅産物が存在しないこと(これら10個の空のレーンは試験で用いられる細菌の残りの10種を示す)によって図示する。
【0169】
図10は、1種のみのナイセリア細菌群に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。ゲノムを簡素化する方法論の特異性について、淋菌を示すレーン2のみにおけるアンプリコンの存在およびレーン1およびレーン3〜12において増幅産物が存在しないこと(これら11個の空のレーンは試験で用いられる細菌の残りの11種を示す)によって図示する。
【0170】
個々の微生物種の分析においては、タンパク質をコードする遺伝子についても、微生物の異なる株が問題の遺伝子配列におけるそれらの存在/非存在に対して多形性を示さないという条件で必要に応じて用いてもよい。
【0171】
図11は、大腸菌の細菌recA遺伝子に対するプライマーの使用について図示する。アンプリコンの特異性を、レーン1における正確にサイジングしたアンプリコンの存在および細菌の他の11種を示す残りのレーン2〜12においてそれが存在しないことによって図示する。
【0172】
図12のデータは、ブドウ球菌(Staphylococci)などのより大きい細菌群のメンバーを明らかにするプライマーの特異性をさらに図示する。ブドウ球菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物は、表皮ブドウ球菌、黄色ブドウ球菌およびスタフィロコッカッス・キシロサスを示すレーン8、9および10でのみアンプリコンを示す。レーン1〜7およびレーン11〜12において増幅産物が存在しないことは反応の特異性を証明している。9つの空のレーンは、試験で用いられる9種の非ブドウ球菌細菌を示す。
【0173】
図13は、連鎖球菌(Streptococcal)細菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。連鎖球菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物は、ストレプトコッカス・ニューモニエおよび溶血性連鎖球菌を示すレーン11および12でのみアンプリコンを示す。レーン1〜10において増幅産物が存在しないことは反応の特異性を示している。これらの10個の空のレーンは、試験で用いられる10種の非連鎖球菌細菌を示す。
【0174】
図14は、表皮ブドウ球菌(レーン8)のrecA遺伝子のゲノム的に簡素化した領域由来のタンパク質をコードする遺伝子からPCRによって得られた増幅産物を示す。2つのバンド(矢印)は、第1ラウンド(上部バンド)および第2ラウンド(下部バンド)のPCR増幅から得られた繰り越し(carry over)アンプリコンを示す。レーン1〜7および9〜12においてアンプリコンが存在しないことは方法の特異性を示し、タンパク質をコードする遺伝子を特殊環境下でゲノムの非コーディング成分の代わりに使用することで1種の細菌種に限って検出がなされるという点を強調している。
【0175】
図15は、クラミジアのゲノム的に簡素化した23Sリボソーム遺伝子を標的にする特異的プライマーを用いたアンプリコンの検出を示す。DNAの開始量が少ないことからPCR反応を2通りに行った。レーン番号5は、既知の陰性の個人の尿から抽出したDNAであった。2通りのうちのいずれかでバンドが出現したことは、クラミジアDNAの存在に対する陽性反応であると考えられた。
【0176】
図16は、大腸菌由来の23SリボソームRNA遺伝子の正規のヌクレオチド配列および図示目的として全シトシンがチミンと置換されている場合のゲノムの簡素化後の同配列を示す。
【0177】
図17は、大腸菌由来のrecA遺伝子の正規のヌクレオチド配列および図示目的として全シトシンがチミンと置換されている場合のゲノムの簡素化後の同配列を示す。
【0178】
要するに、微生物源由来の重亜硫酸塩で処理したDNAは、ゲノム的に簡素化したプライマーを用いて増幅される場合、オリゴヌクレオチドまたはINAなどの修飾核酸であると、試料中で任意のタイプの微生物を見出すための最高レベルの検出系を提供し、それはヒト臨床物質由来の試料であるかまたは汚染水などの環境源由来の別の極端な状況にある。本発明については、広範な異なる細菌種や臨床的意義のあるウイルスに対して実証されている。酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)またはその近縁種などの単細胞真核微生物の検出は、本方法を単純に拡張したものである。それは、18または28Sリボソーム配列などの類似のゲノム配列ソースあるいは上記のように所定の種、タイプ、株または変異体もしくは多型に対して特異的なタンパク質をコードするコーディング配列を必要とする。
【0179】
本発明に記載の検出系の実際の意味もまた重要である。本明細書において詳細に記載された原理が増幅用PCRを用いて示されている一方、当該技術で既知の任意の方法論を介して読み出しを連携させてもよい。重亜硫酸塩処理がゲノムの複雑さを低減し、それ故に微生物のさらなるクラスを対象とした、検出器の数がより少ない(という特徴を備えた)マイクロアレイ上での試験が可能になることから、現在重視されているマイクロアレイ検出系とともに、ゲノム的に簡素化されたDNAを用いることで微生物のはるかに広大な範囲を検出することができるであろう。
【0180】
もし、例えばマイクロアレイが250,000種程度の異なる微生物を1回の試験で検出するように構築されなければならないとしたら、現行の方法論は検出プラットフォームの物理的制限によって圧倒されることから、十分な実用的検出プラットフォームを提供できないであろう。しかし、ゲノムの簡素化を併せるならば、小型マイクロアレイは1000種程度の異なる高レベルの細菌カテゴリを検出可能であろう。次いで、あくまで初期試験において陽性であったそれらの群を代表するものを有する別のアレイを用いれば、かかる試験からの陽性について評価することができるであろう。
【0181】
当業者であれば、特定の実施形態において示されるように、極めて多くの変異および/または修飾が、幅広く記載された本発明の趣旨または範囲から逸脱することなく本発明に対してなされうることを理解するであろう。したがって、本実施形態は、例示としてかつ限定されることなく、あらゆる点から考えられるべきである。
【図面の簡単な説明】
【0182】
【図1】ゲノムの簡素化の前後における髄膜炎菌および淋菌のiga遺伝子の一部のアラインメントを示す。図に示されるように、ゲノムの簡素化に先立ち、ナイセリア種のユニバーサルな検出にとって全部で512通りのプローブの組み合わせ(74%の配列類似性)が必要であるのに対し、核酸誘導体を形成する簡素化後では、単に2通りの組み合わせ(97%の配列類似性)で十分であると思われる(配列番号は各配列の後に列挙される)。
【図2】INAプローブが標準オリゴヌクレオチドプローブより短い長さを有しうることから簡素化配列の配列類似性をさらに高めるためのINAプローブの使用を示す。ゲノムの簡素化方法をINAプローブと組み合わせると、標的配列に対して100%の配列類似性を有するプローブの選択および使用が可能になる(配列番号は各配列の後に列挙される)。
【図3】ナイセリアおよびヘモフィルス由来のiga遺伝子のアラインメントを用いた、近縁種である種間の区別をするためのゲノムの簡素化について示す。図に示されるように、本発明の方法は、種特異的プローブを生成するためのゲノム物質の簡素化を可能にする。さらに、それはゲノムDNAを簡素化するが、ナイセリア種と、近縁種であるヘモフィルス種の間の区別も可能にする(配列番号は各配列の後に列挙される)。
【図4】連鎖球菌の異なる10種におけるゲノムの簡素化の前後での連鎖球菌のtuf遺伝子のアラインメントについて示す。処理前には、tuf遺伝子のユニバーサルプライマーにとって全部で12,288通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは64通りにすぎないと思われる。さらに、配列類似性は簡素化前には67.5%にすぎず、簡素化後には85%に高まる(配列番号は各配列の後に列挙される)。
【図5】ゲノムの簡素化の前後でのブドウ球菌のエンテロトキシン遺伝子のアラインメントについて示す。重亜硫酸塩処理前には、ブドウ球菌のエンテロトキシン遺伝子のユニバーサルプライマーにとって全部で1,536通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは64通りにすぎないと思われる(配列番号は各配列の後に列挙される)。
【図6】ゲノムの簡素化の前後での様々なインフルエンザ株のインフルエンザ群AおよびBのノイラミニダーゼ遺伝子のアラインメントについて示す。処理前には、群AおよびBのノイラミニダーゼ遺伝子のユニバーサルプライマーにとって全部で2,048通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは48通りにすぎないと思われる。さらに、配列類似性は簡素化前には50%にすぎず、簡素化後には75%に高まる(配列番号は各配列の後に列挙される)。
【図7】ゲノムの簡素化の前後でのロタウイルスVP4遺伝子のアラインメントについて示す。処理前には、ロタウイルスVP4遺伝子のユニバーサルプライマーにとって全部で512通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは32通りにすぎないと思われる(配列番号は各配列の後に列挙される)。
【図8】グラム陽性およびグラム陰性細菌のゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示し、ここでは適切なアンプリコンがアガロースゲル電気泳動による特異的長さのバンドとして検出される。矢印は、マーカーレーン(M)内を動く、標準的なサイジングされたマーカーに対するアンプリコンの想定される大きさを示す。グラム陰性細菌に特異的なプライマーを用いると、6種のグラム陰性のレーンのみにバンドが出現する(上パネル)。グラム陽性細菌に特異的なプライマーを用いると、6種のグラム陽性のレーンのみにバンドが出現する(下パネル)。
【図9】大腸菌(レーン1)および肺炎桿菌(レーン3)のゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。増幅の特異性は、残りの10種の細菌由来の増幅産物が存在しないことによって図示される。
【図10】ナイセリアに特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図11】大腸菌のrecA遺伝子のゲノム的に簡素化された領域由来のタンパク質をコードする遺伝子からのPCRによって得られた増幅産物を示す。アンプリコンの特異性は、大腸菌のrecAアンプリコンの存在および他の11種の細菌からのその出現がないことによって図示される。
【図12】ブドウ球菌に特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図13】連鎖球菌に特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図14】表皮ブドウ球菌のrecA遺伝子のゲノム的に簡素化された領域由来のタンパク質をコードする遺伝子からのPCRによって得られた増幅産物を示す。2つのバンド(矢印)は、第1ラウンド(上部バンド)および第2ラウンド(下部バンド)のPCR増幅から得られた繰り越しアンプリコンを示す。
【図15】トラコーマクラミジアのゲノム的に簡素化された23Sリボソーム遺伝子を標的にする特異的プライマーを用いたアンプリコンの検出を示す。
【図16】表皮ブドウ球菌由来の正規のゲノム配列およびゲノム的に簡素化された23S rDNA配列を示す(配列番号は各配列の後に列挙される)。
【図17】大腸菌recA遺伝子のゲノム配列およびゲノム的に簡素化された配列を示す(配列番号は各配列の後に列挙される)。
【技術分野】
【0001】
技術分野
本発明は、微生物の検出のための核酸検出アッセイに関する。本発明は、微生物検出のために特異的リガンドの使用と組み合わせて微生物ゲノムの複雑さを低減することを目的とした核酸の化学処理のための方法にも関する。
【背景技術】
【0002】
背景技術
現在、特異的核酸分子の検出のために多数の手順が利用可能である。これらの手順は、典型的には標的核酸と、長さで短いオリゴヌクレオチド(20塩基またはそれ未満)から何キロベース(kb)もの配列の範囲でありうる核酸プローブとの間の配列依存的なハイブリダイゼーションに依存する。
【0003】
核酸配列の集団から特異的配列を増幅するための最も広く用いられる方法は、ポリメラーゼ連鎖反応(PCR)の方法である(Dieffenbach, C and Dveksler, G. eds. PCR Primer: A Laboratory Manual. Cold Spring Harbor Press, Plainview NY)。この増幅方法では、相補的DNA鎖上および増幅されるべき領域のいずれかの末端にて一般に長さが20〜30ヌクレオチドであるオリゴヌクレオチドを使用することで変性された一本鎖DNA上でDNA合成がプライミングされる。熱安定性DNAポリメラーゼを用いた変性、プライマーハイブリダイゼーションおよびDNA鎖合成の連続サイクルにより、プライマー間の配列の指数関数的増幅が可能である。RNA配列をまず逆転写酵素を用いた複製によって増幅することで相補的DNA(cDNA)複製物が生成されうる。ゲル電気泳動、標識プローブを伴うハイブリダイゼーション、引き続く(例えば酵素結合アッセイによる)同定を可能にするタグ付プライマーの使用、および標的DNAとのハイブリダイゼーション時にシグナルを生成する蛍光タグ付プライマーの使用(例えばBeaconおよびTaqMan系)を含む種々の手段により、増幅DNA断片の検出が可能である。
【0004】
PCRと同様、特異的ヌクレオチド配列の検出および増幅を目的とした種々の他の技術が開発されている。一例としてリガーゼ連鎖反応が挙げられる(1991 , Barany, F. et al., Proc. Natl. Acad. Sci. USA 88, 189-193)。
【0005】
別の例として1992年に最初に記載された等温増幅が挙げられ(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992))、鎖置換増幅(Strand Displacement Amplification;SDA)と称される。それ以来、RNAポリメラーゼを用いてRNA配列を複製するが対応するゲノムDNAを複製しない転写媒介増幅(Transcription Mediated Amplification;TMA)および核酸配列ベース増幅(Nucleic Acid Sequence Based Amplification;NASBA)を含む多数の他の等温増幅技術についての記載がなされている(Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richmann DD and Gingeras TR. 「レトロウイルス複製後にモデル化された多酵素反応によるin vitroでの核酸の等温増幅」 PNAS 87: 1874-1878 (1990): Kievits T, van Gemen B, van Strijp D, Schukkink R, Dircks M, Adriaanse H, Malek L, Sooknanan R, Lens P. NASBA 「HIV-1感染の診断用に最適化されたin vitroでの酵素による核酸の等温増幅」 J Virol Methods. 1991 Dec; 35(3):273-86)。
【0006】
他のDNAに基づく等温技術には、DNAポリメラーゼが環状鋳型に方向付けられたプライマーを伸長させるローリングサークル増幅(RCA)(Fire A and Xu SQ. 「短いサークルのローリング複製」 PNAS 92: 4641-4645 (1995))、標的検出用に環状プローブを用いる分岐増幅(RAM)(Zhang W, Cohenford M, Lentrichia B, lsenberg HD, Simson E, Li H, Yi J, Zhang DY. 「等温分岐増幅法によるトラコーマクラミジアの検出:フィージビリティ・スタディ」 J Clin Microbiol. 2002 Jan; 40(1): 128-32.)およびより最近では、熱の代わりにヘリカーゼを用いてDNA鎖を巻き戻すヘリカーゼ依存性等温DNA増幅(HDA)(Vincent M, Xu Y, Kong H. 「ヘリカーゼ依存性等温DNA増幅」 EMBO Rep. 2004 Aug; 5(8):795-800.) が含まれる。
【0007】
最近、DNA増幅の等温法についての記載がなされてきている(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992))。従来の増幅技術は、標的分子の変性および再生のサイクルを増幅反応の各サイクルで継続させることに依存している。DNAの熱処理によってある程度のDNA分子の剪断が生じることから、DNAが例えば発達中の胚細胞由来の少数の細胞からのDNAの単離など限定的である場合、または特にDNAが既に組織切片、パラフィンブロックおよび古いDNA試料などの断片化形態である場合、この加熱−冷却サイクルによってDNAがさらに損傷を受け、かつ増幅シグナルの低下がもたらされる可能性がある。等温法は、さらなる増幅から得られる鋳型として働く一本鎖分子を生成するための鋳型DNAの継続する変性に依存するのではなく、一定温度での特異的制限エンドヌクレアーゼによるDNA分子の酵素によるニッキングに依存する。
【0008】
鎖置換増幅(SDA)と称される技術は、特定の制限酵素における半修飾DNAの未修飾鎖をニッキングする能力および5’−3’エキソヌクレアーゼ欠損ポリメラーゼにおける下流の鎖に対する延長能および置換能に依存する。次いで、センス反応からの鎖置換がアンチセンス反応のための鋳型としての働きをする、センスおよびアンチセンス反応を連結することにより、指数関数的増幅が行われる(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅」 PNAS 89: 392-396 (1992))。かかる技術は、結核菌(Mycobacterium tuberculosis)(Walker GT, Little MC, Nadeau JG and Shank D.「制限酵素/DNAポリメラーゼ系によるin vitroでのDNAの等温増幅 」 PNAS 89: 392-396 (1992), 「HIV−1、C型肝炎およびHPV−16」 Nuovo G. J., 2000)、トラコーマクラミジア(Chlamydia trachomatis)(Spears PA, Linn P, Woodard DL and Walker GT. 「トラコーマクラミジアの同時鎖置換増幅および蛍光偏光検出」 Anal. Biochem. 247: 130-137 (1997))の増幅の成功のために用いられている。
【0009】
これまでSDAの使用は、修飾鎖上であれば酵素切断に抵抗性を示すヘミホスホロチオエートDNA二本鎖を生成するため、ホスホロチオエートヌクレオチドの修飾に依存しており、消化ではなく酵素によるニッキングが生じ置換反応が促進される。しかし最近になり、数種類の「ニッカーゼ(nickase)」酵素の設計が行われている。これらの酵素は、従来の様式でDNAを切断することなく、一方のDNA鎖上にニックを生じさせる。「ニッカーゼ」酵素は、N.Alw1(Xu Y, Lunnen KD and Kong H. 「ドメインスワッピングによるニッキングエンドヌクレアーゼN.AlwIの設計」 PNAS 98: 12990-12995 (2001))、N.BstNBI(Morgan RD, Calvet C, Demeter M, Agra R, Kong H. 「制限エンドヌクレアーゼの特異的DNAニッキング活性の特徴づけ」 N.BstNBI. Biol Chem. 2000 Nov;381 (11 ):1123-5.)およびMly1(Besnier CE, Kong H. 「Mly1エンドヌクレアーゼのオリゴマー形成状態を変化させることによるニッキング酵素への変換」 EMBO Rep. 2001 Sep;2(9):782-6. Epub 2001 Aug 23)を含む。したがって、かかる酵素を使用すれば、SDA法が簡素化されることになる。
【0010】
さらに、熱安定性を示す制限酵素(Ava1)と熱安定性を示すExo−ポリメラーゼ(Bstポリメラーゼ)の併用によってSDAは改良されている。この併用により、この技術を用いて特有の単一の複製分子が増幅されうるように、108倍の増幅から1010倍の増幅へ反応の増幅効率が高まることが判明している。熱安定性を示すポリメラーゼ/酵素の併用の結果得られる増幅因子は約109である(Milla M. A., Spears P. A., Pearson R. E. and Walker G. T. 「鎖置換増幅における制限酵素Ava1およびExo−Bstポリメラーゼの使用」 Biotechniques 1997 24:392- 396)。
【0011】
これまであらゆる等温DNA増幅技術は、初期の二本鎖鋳型DNA分子が増幅の開始に先立ち変性されることを必要とする。さらに、増幅は各プライミング事象から1回開始されるのみである。
【0012】
直接検出のため、標的核酸は、最も一般的には、標的配列に相補的なプローブとのハイブリダイゼーション(サザンおよびノーザンブロッティング)に先立ち、ゲル電気泳動によるサイズを基準に分離され、固体支持体に移動される。プローブは、天然核酸あるいはペプチド核酸(PNA)または固定化核酸(LNA)またはインターカレーティング核酸(INA)などの類似体でありうる。プローブは、(例えば32Pで)間接標識されるかまたは直接検出法が用いられる場合がある。間接法は通常、ビオチンもしくはジゴキシゲニンなどの「タグ」のプローブへの取り込みに依存し、次いでプローブは酵素結合性の基質変換または化学発光などの手段によって検出される。
【0013】
広く用いられている核酸の直接検出のための別の方法は「サンドイッチ」ハイブリダイゼーションである。この方法では、捕捉プローブが固体支持体に連結され、溶液中の標的核酸が結合プローブとハイブリダイズする。結合していない標的核酸が洗い流され、結合した核酸が標的配列にハイブリダイズする第2のプローブを使用して検出される。検出には上で概説したように直接法または間接法を用いることができる。かかる方法の例として、サンドイッチハイブリダイゼーションの原理を用いる例である「分岐DNA」シグナル検出系が挙げられる(1991, Urdea, M. S., et al., Nucleic Acids Symp. Ser. 24,197-200)。核酸配列の直接検出を目的として核酸ハイブリダイゼーションを用いる急成長分野は、DNAマイクロアレイ分野である(2002, Nature Genetics, 32, [Supplement]; 2004, Cope, L.M., et al., Bioinformatics, 20, 323-331; 2004, Kendall, S.L., et al., Trends in Microbiology, 12, 537-544)。このプロセスでは、短いオリゴヌクレオチド(典型的にはアフィメトリクス(Affymetrix)システムにおける25−mers)からより長いオリゴヌクレオチド(典型的にはアプライドバイオシステムズ(Applied Biosystems)およびアジレント(Agilent)のプラットフォームにおける60−mers)や、cDNAクローンなどのさらにより長い配列に及ぶ範囲でありうる個々の核酸種が、固体支持体に格子パターンで固定されるかあるいは固体支持体上にフォトリソグラフィー的に合成される。次いで、タグが付加されるかまたは標識された核酸集団がアレイとハイブリダイズし、アレイ内の各スポットとのハイブリダイゼーションのレベルが定量化される。化学発光などの他の検出系の使用が可能であるが、最も一般的には、ハイブリダイゼーションにおいて放射性標識または蛍光標識された核酸(例えばcRNAまたはcDNA)が使用される。
【0014】
核酸配列の直接検出を目的として核酸ハイブリダイゼーションを用いる急成長分野はDNAマイクロアレイである(Young RA 「DNAアレイによる生物医学的発見」 Cell 102: 9-15 (2000); Watson 「新ツール:新型ハイテク捜査」 Science 289:850-854 (2000))。このプロセスでは、オリゴヌクレオチドから相補的DNA(cDNA)クローンなどのより長い配列に及ぶ範囲でありうる個々の核酸種が固体支持体に格子パターンで固定される。次いで、タグが付加されるかまたは標識された核酸集団がアレイとハイブリダイズし、アレイ内の各スポットのハイブリダイゼーションのレベルが定量化される。他の検出系が用いられたが、最も一般的には、ハイブリダイゼーションにおいて放射性標識または蛍光標識された核酸(例えばcDNA)が使用された。
【0015】
細菌、酵母および真菌などの微生物を検出するための従来の方法は、選択的な栄養培地上での微生物の培養と、それに続く、従来の光学顕微鏡下で見られる、大きさ、形状、胞子生成、生化学または酵素反応などの特徴および(グラム染色などの)特異的染色特性に基づく微生物の分類を含む。ウイルス種は、分化した組織または細胞内で成長し、次いで電子顕微鏡によって判定されたそれらの構造と大きさに基づいて分類する必要がある。かかる技術の主な欠点は、かかるアプローチの有用性を制限する従来の培養または細胞条件下で成長するのは微生物のすべてではないという点である。例えば髄膜炎菌(Neisseria meningitidis)、ストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)およびインフルエンザ桿菌(Haemophilus influenzae)(全部が髄膜炎を誘発し、それらの中で髄膜炎菌は髄膜炎と劇症髄膜炎菌血症のいずれも誘発する)などの細菌の場合、3つの種全部は培養することが困難である。最大7日間にわたり毎日定期的に血液培養ボトルが検査され、継代培養が必要とされる。インフルエンザ桿菌は、ニコチンアミドアデニンジヌクレオチドとヘミンの双方を含有する特定の培地およびチョコレート寒天プレート上での成長を必要とする。血液培養物は、トリプチカーゼソイブロス(trypticase soy broth)または脳心臓浸出物(brain heart infusion)およびポリアネトールスルホン酸ナトリウムなどの様々な添加物の添加を必要とする。重度の食中毒およびフロッピーベビー症候群を誘発するボツリヌス菌(Clostridium botulinum)などの微生物における毒素の同定には、食物抽出物または培養上清のマウスへの注射および2日後での結果の可視化が含まれる。さらに、特定の培地上での潜在的な微生物の培養には1週間かかる。黄色ブドウ球菌(Staphylococcus aureus)エンテロトキシン(食中毒ならびに皮膚感染、血液感染、肺炎、骨髄炎、関節炎および脳膿瘍の原因)は、イオン交換樹脂を介する毒素の選択的吸収またはモノクローナル抗体を用いた逆受身ラテックス凝集(Reverse Passive Latex Agglutination)によって微量に検出される。それに対し、表皮ブドウ球菌(S.epidermis)は血液感染をもたらし、病院内の機器や床・壁・天井(surfaces)および医療設備や医療器具を汚染する。
【0016】
非ウイルス微生物についても、グルコース、マルトースまたはスクロースなどの基質上での発酵反応の間での特異的アミノ酸または代謝産物の産生などのそれらの代謝特性に基づく分類が可能である。あるいは、微生物では抗生物質に対するそれらの感受性に基づく分類が可能である。細胞表面抗原に対する特異的抗体または毒素などの排泄タンパク質を使用しても微生物が同定または分類される。しかし、上記方法はいずれも、後の試験に先立つ微生物の培養に左右される。微生物の培養は高価で多大な時間を要し、選好性が低めの微生物による汚染または過成長を被る可能性もある。これらの技術はまた、確定診断に達するために同じ試料に対して多数の試験を行う必要がある点であまり洗練されているとはいえない。大部分の微生物は既知の培地内では容易に成長できず、それ故に微生物の異種の典型的な混合集団が野生でまたはより高等な生物に随伴して存在する場合には検出レベルを下回る。
【0017】
病原微生物を検出および同定するための他の方法は、微生物による感染に応答して抗体が産生されるという血清学的アプローチに基づく。例えば髄膜炎菌(Meningococci)は、それらの莢膜多糖類における構造的な違いに基づいて分類可能である。これらは異なる抗原性を有し、5つの主な血清型(A、B、C、YおよびW−135)の判定を可能にする。酵素結合免疫吸着アッセイ(ELISA)またはラジオイムノアッセイ(RIA)は、かかる抗体の産生を評価可能である。これらの方法はいずれも、感染の過程において宿主動物によって産生される特異的抗体の存在を検出する。これらの方法は、宿主動物によって抗体が産生されるまである程度時間がかかることから非常に早期の感染が見逃されることが多いという欠点を有する。さらに、かかるアッセイを使用しても過去の感染と現在の(active)感染を確実に区別することはできない。
【0018】
より最近では、感染疾患の診断における分子法の使用に多大な関心が寄せられている。これらの方法は、病原微生物の高感度かつ特異的な検出を提供する。かかる方法の例として、「分岐DNA」シグナル検出系が挙げられる。この方法は、サンドイッチハイブリダイゼーションの原理を使用する例である(Urdea MS et al. 「ヒトHIVおよび肝炎ウイルスの高感度な直接検出のための分岐DNA増幅多量体」 Nucleic Acids Symp Ser. 1991 ;(24):197-200)。
【0019】
細菌を検出および分類するための別の方法は、16SリボソームRNA配列の増幅である。16SrRNAは、種々の臨床または環境試料中での細菌種の検出を目的としたPCR増幅アッセイにおける使用に適する標的であることが報じられており、かつ16SrRNA遺伝子が種特異的な多型性を示すことから様々な特異的微生物を同定するのに使用されていることが多い(Cloud, J. L., H. Neal, R. Rosenberry, C. Y. Turenne, M. Jama, D. R. Hillyard, and K. C. Carroll. 2002. J. Clin. Microbiol. 40:400-406)。しかし、細菌の純粋培養が必要とされ、試料はPCR増幅後にさらに配列決定されるかまたは種の判定のためにマイクロアレイ型デバイスにハイブリダイズされる必要がある(Fukushima M, Kakinuma K, Hayashi H, Nagai H, lto K, Kawaguchi R. J Clin Microbiol. 2003 Jun; 41(6):2605-15)。かかる方法は高価であり、多大な時間や労力を要する。
【発明の開示】
【発明が解決しようとする課題】
【0020】
本発明者らは、任意の微生物種を対象とした一般的検出または初期スクリーニングアッセイに適合しうる、微生物を検出するための新たな方法を開発している。
【課題を解決するための手段】
【0021】
発明の開示
一般的な態様では、本発明は、シトシンを修飾する作用物質で微生物核酸を処理し、かつ処理された核酸を増幅することでゲノムまたは核酸の簡素化(simplified)形態を生成することにより、微生物ゲノムまたは核酸の塩基構成の複雑さを低減することに関する。
【0022】
第1の態様では、本発明は、
シトシンを修飾する作用物質で微生物ゲノムまたは核酸を処理し、微生物核酸誘導体を形成する工程と、
微生物核酸誘導体を増幅することで微生物ゲノムまたは核酸の簡素化形態を生成する工程と、
を含む、微生物ゲノムまたは微生物核酸を簡素化するための方法を提供する。
【0023】
第2の態様では、本発明は、
シトシンを修飾する作用物質で微生物由来のDNAを含有する試料を処理し、微生物核酸誘導体を形成する工程と、
微生物核酸誘導体の少なくとも一部を増幅することで、対応する未処理の微生物核酸と比べてシトシンの総数が低下している、微生物または微生物型に特異的な核酸配列を含む簡素化された核酸分子を形成する工程と、
を含む、微生物に特異的な核酸分子を生成するための方法を提供する。
【0024】
第3の態様では、本発明は、
微生物からDNA配列を得る工程と、
実質的にアデニン、グアニンおよびチミンの塩基を有するように、各シトシンをチミンに変化させることによって微生物DNA配列の変換を行うことにより、微生物DNA配列の簡素化形態を形成する工程と、
微生物DNAの簡素化形態から微生物に特異的な核酸分子を選択する工程と、
を含む、微生物に特異的な核酸分子を生成するための方法を提供する。
【0025】
第4の態様では、本発明は、本発明の第3の態様に記載の方法によって得られる、微生物に特異的な核酸分子を提供する。
【0026】
第5の態様では、本発明は、試験もしくはアッセイにおいて微生物に特異的な核酸分子に結合または該核酸分子を増幅するためのプローブあるいはプライマーを得ることを目的とした本発明の第3の態様に記載の方法の使用を提供する。
【0027】
第6の態様では、本発明は本発明の第5の態様によって得られるプローブまたはプライマーを提供する。
【0028】
第7の態様では、本発明は、試料中での微生物の存在を検出するための方法であって、
微生物を含有すると見られる試料から微生物DNAを得る工程と、
シトシンを修飾する作用物質によって微生物核酸を処理し、微生物核酸誘導体を形成する工程と、
所望の微生物に特異的な核酸分子の微生物核酸誘導体への増幅を可能にする能力を有するプライマーを提供する工程と、
微生物核酸誘導体に対して増幅反応を行うことで簡素化された核酸を形成する工程と、
所望の微生物に特異的な核酸分子を有する増幅核酸産物の存在についてアッセイし、所望の微生物に特異的な核酸分子の検出が試料中での微生物の存在を示す工程と、
を含む、方法を提供する。
【0029】
ゲノムまたは微生物核酸がDNAである場合、それを処理することでDNA誘導体を形成可能であり、次いで増幅することでDNAの簡素化形態が形成される。
【0030】
ゲノムまたは微生物核酸がRNAである場合、微生物ゲノムまたは核酸の処理に先立ちそれをDNAに変換してもよい。あるいは、微生物RNAを処理してRNA誘導体分子を生成してもよく、次いでそれは増幅に先立ちDNA誘導体分子に変換される。RNAからDNAへの変換方法は周知であり、cDNAを形成するための逆転写酵素の使用を含む。
【0031】
微生物ゲノムまたは核酸を、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から入手してもよい。
【0032】
微生物ゲノムまたは核酸を、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択してもよい。好ましくは、リボソーム遺伝子領域は、原核生物における16Sまたは23Sおよび単細胞真核微生物の場合の18Sおよび28Sである。作用物質を、重亜硫酸塩、酢酸塩またはクエン酸塩から選択してもよい。好ましくは、作用物質は重亜硫酸ナトリウムである。
【0033】
好ましくは、作用物質は、非相補的微生物核酸分子ではなく、2つの誘導体を形成する相補的二本鎖の微生物ゲノムDNAの各鎖内でシトシンをウラシルに修飾する。好ましい形態では、シトシンは典型的には微生物核酸内で見られるようにメチル化されていない。
【0034】
好ましくは、微生物核酸誘導体におけるシトシンの総数が対応する未処理の微生物ゲノムまたは核酸と比べて低下している。
【0035】
好ましくは、微生物ゲノムまたは核酸の簡素化形態におけるシトシンの総数が対応する未処理の微生物ゲノムまたは核酸と比べて低下している。
【0036】
好ましい一形態では、微生物核酸誘導体は、アデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未処理の微生物ゲノムまたは核酸と実質的に総数が同じ塩基を有する。
【0037】
別の好ましい形態では、微生物ゲノムまたは核酸の簡素化形態は、実質的にアデニン(A)、グアニン(G)およびチミン(T)の塩基からなる。
【0038】
好ましくは、増幅はポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって行われる。
【0039】
本発明の第2の態様に記載の方法は、微生物に特異的な核酸分子を検出する工程をさらに含みうる。
【0040】
好ましい形態では、微生物に特異的な核酸分子は、
微生物に特異的な核酸分子の標的領域に対する結合能を有する検出リガンドを提供し、かつ検出リガンドが標的領域に結合するのに十分な時間を与える工程と、
検出リガンドの標的領域に対する結合を測定し、微生物に特異的な核酸分子の存在を検出する工程と、
によって検出される。
【0041】
別の好ましい形態では、微生物に特異的な核酸分子は、増幅産物を分離しかつ分離産物を可視化することによって検出される。好ましくは、増幅産物は電気泳動によって分離され、ゲル上の1つもしくは複数のバンドを可視化することによって検出される。
【0042】
好ましくは、微生物に特異的な核酸分子は微生物内で自然発生しない。
【0043】
好ましい形態では、微生物に特異的な核酸分子は、微生物の分類レベルを示す核酸配列を有する。微生物の分類レベルは、科、属、種、株、タイプ、あるいは同じもしくは異なる地理的集団または底生性集団由来の異なる集団を含むがこれらに限定されない。
【0044】
本発明の第3の態様に記載の方法の好ましい形態では、2種以上の微生物DNA配列の簡素化形態が得られ、かつ少なくとも1種の微生物に特異的な核酸分子を得るために2種以上の配列が比較される。
【0045】
本発明の第7の態様の好ましい形態では、核酸分子は、
微生物に特異的な核酸分子の領域に対する結合能を有する検出リガンドを提供し、かつ検出リガンドが領域に結合するのに十分な時間を与える工程と、
検出リガンドの核酸分子に対する結合を測定し、核酸分子の存在を検出する工程と、
によって検出される。
【0046】
別の好ましい形態では、核酸分子は、増幅産物を分離しかつ分離産物を可視化することによって検出される。
【0047】
微生物がDNAゲノムまたは微生物ゲノムを有しないかまたは核酸がRNA、例えばRNAウイルスである場合には、DNAを作用物質で処理することを目的に、まずRNAウイルスゲノムをcDNAに変換してもよい。RNAを処理することも可能であり、増幅に先立ちRNA誘導体がDNAに変換される。
【0048】
好ましくは、核酸誘導体はアデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有する。重要なことには、核酸誘導体分子は、微生物DNAが任意のシトシン(C)にてメチル化されなかったことを前提として実質的にシトシンを有しない。
【0049】
好ましくは、増幅された核酸誘導体はA、TおよびGの塩基を実質的に有し、かつ対応する核酸誘導体(および未修飾の微生物核酸)と実質的に総数が同じ塩基を有する。増幅された核酸誘導体は簡素化された核酸と称される。
【0050】
好ましい形態では、微生物に特異的な核酸分子は微生物の分類レベルを示す核酸配列を有する。微生物の分類レベルは、科、属、種、株、タイプ、あるいは同じもしくは異なる地理的集団または底生性集団由来の異なる集団を含みうる。細菌の場合、発明者らは、細菌、プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目(Neisseriales);ナイセリア科(Neisseriaceae);ナイセリア属(Neisseria)などの一般に認められたスキーマに従うことができる。異なる集団は、微生物内部の細胞内形態(プラスミドもしくはファージミド)または病原性島などの微生物ゲノムの多形性の染色体領域内に存在するDNA分子内に存在する単一のヌクレオチド変化または変異に対して多形性を示しうる。
【0051】
本発明を用いて微生物およびウイルスゲノムの流動性を認識することも可能であり、かつ独立要素でありうるウイルスゲノムのキメラ性を認識することが可能であることから、新規に生じる株は、異なる動物由来のゲノム領域を再び類別することから生じる。例えば、他の哺乳類または鳥ウイルスゲノムから採取されるセグメントのキメラとしての新たなヒトインフルエンザ株が挙げられる。
【0052】
方法を微生物の既知の核酸配列からin silicoで実施することが可能であり、元の配列内の1個もしくは複数個のシトシンがチミンに変換されて簡素化された核酸が得られることは理解されるであろう。変換された配列から配列同一性の判定が可能である。かかるin silicoにおける方法は処理および増幅工程を再現する。
【0053】
微生物に特異的な核酸分子がこの方法によって任意の所定の微生物において得られている場合、プローブまたはプライマーを増幅反応における対象領域の増幅を保証するように設計してもよい。したがって、プローブまたはプライマーが設計されている場合、試料に対して臨床的または科学的アッセイを実施することで、所定の分類レベルで所定の微生物を検出することが可能になろう。
【0054】
微生物に特異的な核酸分子は特有であるかまたは分類レベル内で高い類似性を有する可能性がある。本発明の1つの利点は、例えば、配列類似性が高い1個もしくは複数個の分子に対し、微生物の分類レベル間または分類レベル内での潜在的な塩基の相違を著しく簡素化できる点である。特異的プライマーまたは数が減少した変性プライマーを使用して所定の試料中の微生物に特異的な核酸分子を増幅してもよい。
【0055】
シトシンを有する二本鎖DNAにおいて、処理工程により2個の核酸誘導体(各相補鎖に対して1個)が生成され、それぞれはアデニン、グアニン、チミンおよびウラシルの塩基を有する。2個の核酸誘導体は、二本鎖DNAの2本の一本鎖から生成される。2個の核酸誘導体は、好ましくはシトシンを全く有しないが、依然として元の未処理のDNA分子と同じ塩基の総数および配列長を有する。重要なことには、2個の核酸誘導体は互いに相補的でなく、増幅における上鎖および下鎖の鋳型を形成する。1本もしくは複数本の鎖を増幅における標的として用いることで簡素化された核酸分子の生成が可能である。核酸誘導体の増幅の間、核酸の対応する増幅された簡素化形態において上部鎖(または下鎖)内のウラシルがチミンによって置換される。増幅が継続するにつれて、新たな各相補鎖がアデニン、グアニン、チミンの塩基のみを有するようになることから上鎖(および/またはもし増幅される場合に下鎖)が希釈されることになる。
【0056】
本発明のこの態様が、微生物に特異的な核酸分子に対して相補的な配列を有する核酸分子、および好ましくは厳しい条件下で微生物に特異的な核酸分子にハイブリダイズ可能な核酸分子も含むことが理解されるであろう。
【0057】
本発明では、任意の微生物が所定の試料中に存在するか否かを判定するのに使用可能な、代表的な微生物型を示すプローブまたはプライマーを使用してもよい。さらに、微生物型に特異的なプローブを使用し、微生物の所定のタイプ、サブタイプ、変異体および遺伝子型の例を実際に検出するかまたは同定することが可能である。
【0058】
任意の所定の微生物において微生物に特異的な核酸分子が得られているかまたは同定されている場合、プローブまたはプライマーを増幅反応における対象領域の増幅を保証するように設計してもよい。処理または変換によって配列の非対称性がもたらされることから、プライマー設計を目的とする、処理によって変換されたゲノム(以降、「核酸誘導体」と称される)の両鎖を分析可能であることに注目することは重要であり、それ故に同一遺伝子座の「上」鎖および「下」鎖(「ワトソン」鎖および「クリック」鎖としても知られる)の検出にとって異なるプライマー配列が必要とされる。したがって、変換直後に存在するものとしての変換されたゲノムである2つの分子集団および核酸誘導体が従来の酵素学的手段(PCR)または等温増幅などの方法によって複製された後に生じる分子集団が存在する。プライマーは典型的には便宜上、変換された上鎖に対して設計されるが、下鎖に対してもプライマーを作製してもよい。したがって、試料に対して臨床的または科学的アッセイを実施することで所定の微生物を検出することが可能になろう。
【0059】
核酸誘導体の特異的領域の増幅を可能にするようにプライマーまたはプローブを設計してもよい。好ましい形態では、プライマーは微生物に特異的な核酸分子の増幅をもたらす。
【0060】
第7の態様では、本発明は、増幅反応のための1種もしくは複数種の試薬または成分とともに本発明の第5の態様に記載のプライマーまたはプローブを含む、微生物に特異的な核酸分子を検出するためのキットを提供する。
【0061】
好ましくは、微生物は、Kingdom Protoctista by Margulis, L., et al 1990, Handbook of Protoctista, Jones and Bartlett, Publishers, Boston USAなど、いかに様々に分類されようと、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、単細胞生物、または任意の他の微生物、あるいはHarrisons Principles of Internal Medicine, 12th Edition, edited by J D Wilson et al., McGraw Hill Incおよび後版で定義されたヒトに随伴する微生物から選択される。それは、OMIM、Online Mendelian Inheritance in Man, www.ncbi.govにおいて定義されたヒトの状態と関連して記載されたすべての微生物も含む。
【0062】
微生物は、細胞内外で成熟もしくは胞子形態である、またはキメラ生物形態に関連する、または苔癬に関連した微生物または細菌膜に関連した微生物などの2種以上の生物形態の間で外部片利共生的に(ectocommensally)存在する、病原体、自然発生的な環境試料、水生生物または空中浮遊生物(または液体または気体媒体中で存在するかもしくは運搬される生物)でありうる。
【0063】
RNAウイルスまたはウイロイドの存在については、まずそれらのRNAゲノムを逆転写によってcDNA形態に変換し、次いで試薬によってcDNAを修飾することにより、アッセイすることが可能である。これにより、RNAウイルス中のシトシンに生じる任意のメチル化の問題が、逆転写酵素がそれらを正常なシトシンであるかの如く複製する際に克服される。
【0064】
好ましくは、作用物質は非メチル化シトシンをウラシルに修飾し、次いでウラシルは核酸誘導体の増幅の間にチミンと置換される。好ましくは、シトシンを修飾するために使用される作用物質は重亜硫酸ナトリウムである。本発明の方法では、メチル化シトシンではなく、非メチル化シトシンを同様に修飾する他の作用物質もまた使用してもよい。例として、重亜硫酸塩、酢酸塩またはクエン酸塩が挙げられるがこれらに限定されない。好ましくは、作用物質は、水の存在下でシトシンをウラシルに修飾する試薬である重亜硫酸ナトリウムである。
【0065】
重亜硫酸ナトリウム(NaHSO3)は、シトシンの5,6−二重結合と容易に反応して脱アミノ化を受けやすいスルホン化されたシトシン反応中間体を形成し、水の存在下でウラシルサルファイト(uracil sulfite)を生成する。必要であれば、亜硫酸基を弱アルカリ性の条件下で除去する結果としてウラシルを形成してもよい。したがって、可能性としてはすべてのシトシンがウラシルに変換されることになる。しかし、メチル化による保護に起因する試薬の修飾により、どのメチル化シトシンをも変換することはできない。
【0066】
本発明を、同一細菌種の分離株間の膨大な想定外のゲノム変異によって明らかになる発生する問題の一部の回避に役立つように適合させてもよい(2005, Tettelin , H., et al., Proc. Natl. Acad. Sci. USA. 102, 13950-13955;「ストレプトコッカス・アガラクチアエ(Streptococcus agalacticiae)の複数の病原性分離株のゲノム分析:微生物“全ゲノム(pan-genome)”の意味するもの」)。この細菌種のすべての分離株は、約80%の遺伝子プールに加え、部分的に共有されかつ株特異的タンパク質をコードする遺伝子からなる可欠ゲノムを示す、タンパク質をコードする遺伝子の「コア」ゲノムを有する。本発明に記載の方法によって細菌集団内に存在する23S遺伝子を処理することにより、発明者らは、すべての細菌分離株内に存在するタンパク質をコードしないコア成分を扱うことができる。
【0067】
本発明は、まず生物が属する一般集団を決定することを目的とし、種レベルを超えるまたは種レベルでの初期の同一性が有用である場合での微生物についての臨床、環境、法医学、生物戦、または科学的アッセイに適する。例として、任意の生物における疾患(脊椎動物、無脊椎動物、原核または真核細胞、例えば植物および家畜の疾患、養魚場およびカキ養殖場などのヒトの食料源での疾患)の診断、スクリーニングあるいは体外受精診療施設内でのヒト胚細胞の産生もしくは動物飼育における細胞培養物または体外受精卵の汚染について判定する、自然または汚染状態である環境源のサンプリングが挙げられる。法医学的状況または生物戦に関連した微生物の検出は特に重要である。
【0068】
本明細書全体を通し、もし文脈上、別の意味を必要としない場合、「含む(comprise)」あるいは「含む(comprises)」または「含む(comprising)」などの変形は、述べられる要素、整数または工程、あるいは要素、整数または工程の群を含むだけでなく、任意の要素、整数または工程、あるいは要素、整数または工程の群を除外しないことを意味することが理解されるであろう。
【0069】
本明細書中に含められている文書、行為、材料、デバイス、記事などに関するいずれの説明も、専ら本発明に対する文脈をもたらすことを目的とするものである。これらの事項のいずれかまたはすべてが、先行技術基盤の一部を形成しているとか、あるいは本発明の構築に先立ちオーストラリア内に存在したかのように本発明に関する分野における共通の一般的知識であったという承認として解釈されるべきものではない。
【0070】
本発明がより明確に理解されうるように、好ましい実施形態が以下の図面および実施例と関連して記載されることになる。
【発明を実施するための最良の形態】
【0071】
本発明を実施するための形態
定義
本明細書において用いられる「ゲノムの簡素化」という用語は、ゲノム(または他の)核酸が4つの塩基のアデニン(A)、グアニン(G)、チミン(T)およびシトシン(C)からなる状態から修飾されることで、実質的に塩基のアデニン(A)、グアニン(G)、チミン(T)を有するがやはり実質的に総数が同じ塩基を有することを意味する。
【0072】
本明細書において用いられる「核酸誘導体」という用語は、塩基のA、G、TおよびU(または何らかの他の非A、非Gもしくは非T塩基または塩基様実体(base−like entity))を実質的に有し、かつ対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有する核酸を意味する。微生物DNA内の実質的にすべてのシトシンが、作用物質による処理の間、ウラシルに変換されていることになる。例えばメチル化などにより改変されたシトシンが必ずしもウラシル(または何らかの他の非A、非Gもしくは非T塩基または塩基様実体)に変換されない場合があることは理解されるであろう。微生物核酸が典型的にはメチル化シトシン(または他のシトシン改変物)を有しないことから、処理工程では好ましくはすべてのシトシンが変換される。好ましくは、シトシンはウラシルに修飾される。
【0073】
本明細書において用いられる「簡素化された核酸」という用語は、核酸誘導体の増幅後に結果的に得られる核酸産物を意味する。次いで、核酸誘導体内のウラシルは核酸誘導体の増幅の間にチミン(T)と置換されて簡素化された核酸分子が形成される。結果産物は、対応する未修飾の微生物核酸と実質的に総数が同じ塩基を有するが、実質的に3つの塩基(A、GおよびT)の組み合わせから構成される。
【0074】
本明細書において用いられる「簡素化配列」という用語は、核酸誘導体を増幅して簡素化された核酸が形成された後に結果的に得られる核酸配列を意味する。得られる簡素化配列は、対応する未修飾の微生物核酸配列と実質的に総数が同じ塩基を有するが、実質的に3つの塩基(A、GおよびT)の組み合わせから構成される。
【0075】
本明細書において用いられる「変換されていない配列」という用語は、処理および増幅に先立つ微生物核酸の核酸配列を意味する。変換されていない配列は、典型的には自然発生的な微生物核酸配列である。
【0076】
本明細書において用いられる「修飾する」という用語は、シトシンの別のヌクレオチドへの変換を意味する。好ましくは、作用物質は非メチル化シトシンをウラシルに修飾し、核酸誘導体を形成する。
【0077】
本明細書において用いられる「シトシンを修飾する作用物質」という用語は、シトシンを別の化学物質に変換する能力がある作用物質を意味する。好ましくは、作用物質は、シトシンをウラシルに修飾し、次いでウラシルは核酸誘導体の増幅の間にチミンと置換される。好ましくは、シトシンを修飾するために使用される作用物質は重亜硫酸ナトリウムである。メチル化シトシンではなく、シトシンを同様に修飾する他の作用物質もまた本発明の方法において使用してもよい。例として、重亜硫酸塩、酢酸塩またはクエン酸塩が挙げられるがこれらに限定されない。好ましくは、作用物質は、酸性の水性条件の存在下でシトシンをウラシルに修飾する試薬の重亜硫酸ナトリウムである。重亜硫酸ナトリウム(NaHSO3)は、シトシンの5,6−二重結合と容易に反応して脱アミノ化の作用を受けやすいスルホン化されたシトシン反応中間体を形成し、水の存在下でウラシルサルファイトを生成する。必要であれば、亜硫酸基を弱アルカリ性の条件下で除去する結果としてウラシルを形成してもよい。したがって、可能性としてはすべてのシトシンがウラシルに変換されることになる。しかし、メチル化による保護に起因する試薬の修飾により、あらゆるメチル化シトシンを変換することはできない。シトシン(または任意の他の塩基)を、本発明によって教示される核酸誘導体を得るための酵素的手段によるならば修飾可能であることが理解されるであろう。
【0078】
核酸内塩基に対する化学的および酵素的修飾を可能にするのに、2つの幅広い一般的方法がある。したがって、天然酵素あるいはいまだ報じられていない人工的に構築されるかまたは選択される酵素により、本発明の修飾を行ってもよい。重亜硫酸塩の方法論などの化学的処理により、適切な化学的工程を介してシトシンをウラシルに変換してもよい。同様に、例えばシトシンデアミナーゼが変換を行うことで核酸誘導体を形成しうる。発明者らの知見によると、シトシンデアミナーゼに関する最初の報告は1932, Schmidt, G., Z. physiol. Chem., 208, 185である(1950, Wang, T.P., Sable, H.Z., Lampen, J.O., J. Biol. Chem, 184, 17-28,「シトシンヌクレオチドの酵素による脱アミノ化」も参照)。この初期の研究では、他のヌクレオ−デアミナーゼなしにシトシンデアミナーゼは得られなかったが、ワン(Wang)らは酵母および大腸菌からかかる活性を精製することができた。したがって、シトシンの任意の酵素的変換によって核酸誘導体が形成され、最終的にシトシンとは異なる塩基の挿入がその位置での次の複製の間にもたらされ、簡素化されたゲノムが生成されることになる。誘導体とそれに続く簡素化されたゲノムを生成するための化学的および酵素的変換は、微生物の自然発生的な核酸内のプリンであれピリミジンであれ任意のヌクレオ塩基に適用可能である。
【0079】
本明細書において用いられる「ゲノムまたは核酸の簡素化形態」という用語は、自然発生であれ合成であれ、通常4つの共通の塩基G、A、TおよびCを有するゲノムまたは核酸が、ゲノム内のCの大部分もしくはすべてが適切な化学修飾およびそれに続く増幅手順によってTに変換されていることからこの段階では主に3つの塩基G、AおよびTのみからなることを意味する。ゲノムの簡素化形態は、相対的なゲノムの複雑さが4つの塩基の基盤から3つの塩基組成物に向けて低減されることを意味する。
【0080】
本明細書において用いられる「塩基様実体(base−like entity)」という用語は、シトシンの修飾によって形成される実体を意味する。塩基様実体は、核酸誘導体の増幅の間にDNAポリメラーゼによって認識可能であり、ポリメラーゼにより、A、GまたはTが核酸誘導体内の塩基様実体の対向位置にある新たに形成された相補的DNA鎖上に配置される。典型的には、塩基様実体は、対応する未処理の微生物核酸内のシトシンから修飾を受けているウラシルである。塩基様実体の例として、プリンであれピリミジンであれ、任意のヌクレオ塩基が挙げられる。
【0081】
本明細書において用いられる「相対的な複雑さの低減」という用語は、プローブ長、すなわち同じ大きさの2つのゲノムにおいて、第1のゲノムが「変化しない状態」であって4つの塩基G、A、TおよびCからなる一方、第2のゲノムが正確に同じ長さであっても一部のシトシン(望ましくはすべてのシトシン)がチミンに変換されている場合の所定の一まとまりの分子条件下における、特異的遺伝子座に対するプローブのハイブリダイゼーションの同じ特異性およびレベルを得るのに必要な平均プローブ長の増加に関する。試験される遺伝子座は、元の変換されていないゲノムおよび変換されたゲノムにおける同じ位置に存在する。平均で11−merプローブが、4つの塩基G、A、TおよびCからなる4,194,304(411が4,194,304に相当)個の塩基の正規のゲノム内で完全にハイブリダイズする特有の位置を有することになる。しかし、一旦4,194,304個の塩基のかかる正規のゲノムが重亜硫酸塩または他の適切な手段によって変換されていると、この段階ではこの変換されたゲノムは3つの塩基のみからなり、明らかに複雑さが低減される。しかし、ゲノムの複雑さにおけるこの低減の結果として、かつて発明者ら独自のものであった11−merのプローブが簡素化されたゲノム内でハイブリダイズ可能である特有の部位をもはや有しないということである。この段階では、重亜硫酸塩変換の結果として新規に生じている11個の塩基配列からなる多数の他の相当位置が存在する可能性がある。ここでは、元の遺伝子座を見出しかつハイブリダイズするのに14−merのプローブが必要になる。当初、それが直感に反するように見られるかもしれないが、より多くのゲノムが同様と見られる(より類似した配列を有する)ことから、この段階で簡素化された3つの塩基からなるゲノムにおける元の位置を検出するのにプローブ長の増加が必要である。したがって、相対的なゲノムの複雑さの低減(または3つの塩基からなるゲノムの簡素さ)とは、元の特有部位を見出すためにより長いプローブを設計する必要があるという意味である。
【0082】
本明細書において用いられる用語「相対的なゲノムの複雑さの低減」は、未修飾DNAと比較して微生物に特異性を示すことが可能なプローブ長の増加によって測定されうる。この用語は、微生物の存在を判定するのに用いられるプローブ配列のタイプについても包含する。これらのプローブは、例えばPNAまたはLNAにおける従来とは異なる骨格、あるいは例えばINAにおいて示された骨格に対して修飾された付加物を有する場合がある。したがって、ゲノムでは、プローブが例えばINAにおけるインターカレーティングシュードヌクレオチド(intercalating pseudonucleotide)などの付加成分を有するか否かに無関係に、相対的な複雑さが低減していると考えられる。例として、DNA、RNA、固定化核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、インターカレーティング核酸(INA)、シクロヘキサニル核酸(CNA)およびこれらの混合物やこれらのハブリッド、ならびにこれらのリン原子修飾物、例えばホスホロチオエート、メチルホスホレート、ホスホアミダイト(phosphoramidites)、ホスホロジチエート(phosphorodithiates)、ホスホロセレノエート(phosphoroselenoates)、ホスホトリエステルおよびホスホボラノエート(phosphoboranoates)など(これらに限定されない)が挙げられるがこれらに限定されない。非天然ヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−ザイロ−LNA、β−D−ザイロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、ビシクロ−DNA、6−アミノ−ビシクロ−DNA、5−エピ−ビシクロ−DNA、α−ビシクロ−DNA、トリシクロ−DNA、ビシクロ[4.3.0]−DNA、ビシクロ[3.2.1]−DNA、ビシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキソピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内部に含まれるヌクレオチドが含まれるがこれらに限定されない。さらに、非リンを含有する化合物を、メチルイミノメチル、ホルムアセテート(formacetate)、チオホルムアセテート(thioformacetate)およびアミドを有する連結基などの(これらに限定されない)ヌクレオチドへの連結のために使用することが可能である。特に核酸および核酸類似体は、1つもしくは複数のインターカレーターシュードヌクレオチド(intercalator pseudonucleotide)(IPN)を含みうる。IPNの存在は、核酸分子における複雑さの一部を示すものではなく、例えばPNAにおけるその複雑さの骨格部分でもない。
【0083】
「INA」とは、本明細書中に参照により援用される国際公開第03/051901号パンフレット、国際公開第03/052132号パンフレット、国際公開第03/052133号パンフレットおよび国際公開第03/052134号パンフレット(ユネスト(Unest A/S))の教示に記載のインターカレーティング核酸を意味する。INAは、1種もしくは複数種のインターカレーターシュードヌクレオチド(IPN)分子を有するオリゴヌクレオチドまたはオリゴヌクレオチド類似体である。
【0084】
「HNA」とは、例えばVan Aetschot et al., 1995に記載の核酸を意味する。
【0085】
「MNA」とは、Hossain et al, 1998に記載の核酸を意味する。
【0086】
「ANA」はAllert et al, 1999に記載の核酸を示す。
【0087】
「LNA」は国際公開第99/14226号パンフレット(エキシコン(Exiqon))に記載の任意のLNA分子でありうる。好ましくは、LNAは国際公開第99/14226号パンフレットの要約書中に示された分子から選択される。より好ましくは、LNAはSingh et al, 1998, Koshkin et al, 1998またはObika et al., 1997に記載の核酸である。
【0088】
「PNA」は、例えばNielsen et al, 1991に記載のペプチド核酸を示す。
【0089】
本明細書において用いられる「相対的複雑さの低減」は、ATATATATATATAT(配列番号1)の配列と同じ長さのAAAAAAATTTTTTT(配列番号2)の配列の間の任意の数学的な複雑さの違いなど、塩基が生じる順序を示すのではなく、相対的ゲノムサイズ(および推定的にはゲノムの複雑さ)を有する元の再連結データを示すものでもない。このことはWaring, M. & Britten R. J.1966, Science, 154, 791-794; and Britten, RJ and Kohne D E., 1968, Science, 161 , 529-540およびそれらの中のCarnegie Institution of Washington Yearbook reportsを出所とする初期の参考文献によって科学論文に紹介されている。
【0090】
本明細書において用いられる「相対的なゲノムの複雑さ」とは、分子プローブによって接近される、2つのゲノム内の塩基の変化しない位置を示す(元のゲノムと変換されていないゲノムの双方が不変位置の1〜nで塩基を有する)。特定のヒト女性の30億塩基対のハプロイドヒトゲノムの場合、不変位置が1〜n(nは3,000,000,000)であると定義される。もし配列1〜nにおいてith塩基が元のゲノム内でCである場合、ith塩基は変換されたゲノム内でTである。
【0091】
本明細書において用いられる「ゲノム核酸」という用語は、微生物(原核生物および単細胞真核生物)RNA、DNA、タンパク質をコードする核酸、タンパク質をコードしない核酸、ならびに原核生物および単細胞真核微生物のリボソーム遺伝子領域を含む。
【0092】
本明細書において用いられる「微生物ゲノム」という用語は、染色体および染色体外核酸、ならびにプラスミド、バクテリオファージおよび最も幅広い意味での可動要素などのそのゲノムの一時的寄生物をカバーする。「ゲノム」は、例えばS.galactiaeのようなコア成分を有するとともに、異なる分離株間で変化するコーディングおよび非コーディング要素を有する可能性がある。
【0093】
本明細書において用いられる「微生物由来のDNA」という用語は、微生物から直接的に得られるか、あるいは逆転写酵素など既知であるかまたは適切な方法によって微生物RNAをDNAに変換することにより間接的に得られるDNAを含む。
【0094】
本明細書において用いられる「微生物」という用語は、Kingdom Protoctista by Margulis, L., et al 1990, Handbook of Protoctista, Jones and Bartlett, Publishers, Boston USAなど、いかに様々に分類されようと、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、単細胞生物、または任意の他の微生物を含むか、あるいはHarrisons Principles of Internal Medicine, 12th Edition, edited by J D Wilson et al., McGraw Hill Incおよび後版で定義されたヒトに関連した微生物を含む。それは、OMIM、Online Mendelian Inheritance in Man, www.ncbi.govにおいて定義されたヒト疾患と関連して記載されたすべての微生物も含む。
【0095】
本明細書において用いられる「微生物に特異的な核酸分子」という用語は、微生物に特異的な1つもしくは複数の配列を有する、本発明に記載の方法を用いて判定または取得されている分子を意味する。
【0096】
本明細書において用いられる「微生物の分類レベル」という用語は、同じもしくは異なる地理的集団または底生性集団由来の科、属、種、株、タイプ、または異なる集団を含む。細菌の場合、細菌、プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目;ナイセリア科;ナイセリア属などの一般に認められたスキーマが用いられる。異なる集団は、微生物内部の細胞内形態(プラスミドもしくはファージミド)内、または病原性島などの微生物ゲノムの多形性染色体領域内に存在するDNA分子内に存在する単一のヌクレオチド変化あるいは変異に対して多形性を示しうる。微生物およびウイルスゲノムの流動性は認識されており、ウイルスゲノムのキメラ性を含み、それは独立した核酸片の状態でありうる。それ故、異なる動物由来のゲノム領域の混ぜ合わせから新たに生じる株として、例えば他の哺乳類または鳥ウイルスゲノムから採取された断片のキメラとしての新たなヒトインフルエンザ株が挙げられる。
【0097】
本明細書において用いられる「高い配列類似性」という用語は、相対的な配列の複雑さおよび尺度としてのプローブ長に関する上記定義を含む。
【実施例】
【0098】
材料および方法
DNAの抽出
一般に、微生物DNA(またはウイルスRNA)は任意の適切な供給源から入手可能である。例として、細胞培養物、培養液、環境試料、臨床試料、体液、液体試料、組織などの固体試料が挙げられるがこれらに限定されない。試料由来の微生物DNAを標準的手順によって入手可能である。適切な抽出の一例は以下の通りである。目的の試料を、7M塩酸グアニジン400μl、5mM EDTA、100mMトリス/HCI pH6.4、1%トリトン−X−100、50mMプロテイナーゼK(シグマ(Sigma))、100μg/mlの酵母tRNAの中に置く。試料を1.5mlの使い捨て乳棒で十分に均一化し、60℃で48時間放置する。インキュベーション後、試料に対して5回のドライアイスでの凍結/解凍サイクルを5分/95℃で5分間施す。次いで試料をマイクロチューブ内で2分間回転およびボルテックスし、細胞残屑をペレット化する。上清を無菌チューブ内に移し、希釈して塩濃度を低下させ、フェノール:クロロホルム抽出し、エタノール沈殿させ、50μlの10mMトリス/0.1mM EDTAに再懸濁する。
【0099】
特に、(各々の種に特異的な栄養所要量を有する)標準的な寒天プレート上で成長させたグラム陽性およびグラム陰性細菌からのDNA抽出を以下のように行った。
【0100】
グラム陰性細菌からのDNA抽出におけるプロトコルは以下のようであった。
a)無菌のトゥースピックを使用し、細菌コロニーを培養プレートから無菌にした1.5mlの遠心管内にこすり落とした。
b)チオシアン酸グアニジン抽出緩衝液(7Mチオシアン酸グアニジン、5mM EDTA(pH8.0)、40mMトリス/HCl pH7.6、1%トリトン−X−100)180μlを添加し、試料を混合して細菌コロニーを再懸濁した。
c)20μl(20mg/ml)のプロテイナーゼKを添加し、試料を十分に混合した。
d)試料を55℃で3時間インキュベートし、細胞を溶解させた。
e)水200μlを各試料に添加し、ゆっくりとしたピペット操作により試料を混合した。
f)フェノール/クロロホルム/イソアミルアルコール(25:24:1)400μlを添加し、試料を15秒間で2回ボルテックスした。
g)次いで、試料をマイクロチューブ内、14,000rpmで4分間回転させた。
h)水相を無菌にした1.5mlの遠心管に移した。
i)フェノール/クロロホルム/イソアミルアルコール(25:24:1)400μlを添加し、試料を15秒間で2回ボルテックスした。
j)次いで、試料をマイクロチューブ内、14,000rpmで4分間回転させた。
k)水相を無菌にした1.5mlの遠心管に移した。
l)100%エタノール800μlを各試料に添加し、試料を短時間ボルテックスし、次いで−20℃で1時間放置した。
m)試料をマイクロチューブ内、4℃、14,000rpmで4分間回転させた。
n)DNAペレットを70%エタノール500μlで洗浄した。
o)試料をマイクロチューブ内、4℃、14,000rpmで5分間回転させ、エタノールを廃棄し、ペレットを5分間風乾した。
p)最後にDNAを100μlの10mMトリス/HCI pH8.0、1mM EDTA pH8.0に再懸濁した。
q)溶液の吸光度を230、260、280nmで測定することにより、DNAの濃度および純度を計算した。
【0101】
グラム陽性細菌からのDNA抽出におけるプロトコルは以下のようであった。
a)無菌のトゥースピックを使用し、細菌コロニーを培養プレートから無菌にした1.5mlの遠心管内にこすり落とした。
b)20mg/mlのLysozyme(シグマ(Sigma))180μlおよびLysostaphin(シグマ(Sigma))200μgを各試料に添加し、試料を徐々に混合して細菌コロニーを再懸濁した。
c)試料を37℃で30分間インキュベートし、細胞壁を分解した。
d)次いで、グラム陽性細菌に対するQIAamp DNAミニキットプロトコルに従って試料を処理し、DNAを抽出した。
【0102】
患者由来の細胞学的試料からのDNA抽出
a)試料を手動で激しく振とうさせて沈降した細胞を再懸濁しかつ溶液の均一性を確保した。
b)再懸濁した細胞4mlを15mlのコスター(Costar)遠心管に移した。
c)管をスイングアウト(swing−out)式バケットローター内、3000×gで15分間遠心した。
d)ペレット化した細胞物質を乱さないように、上清を注意深くデカントして廃棄した。
e)ペレット化した細胞を溶解緩衝液(100mMトリス/HCI pH8.0、2mM EDTA pH8.0、0.5%SDS、0.5%トリトン−X−100)200μlに再懸濁し、溶液が均一になるまで十分に混合した。
f)試料80μlを96ウェルの試料調製プレートに移した。
g)プロテイナーゼKを20μl添加し、溶液を55℃で1時間インキュベートした(この手順により細胞溶解物が得られる)。
【0103】
尿試料からのDNA抽出
QIAamp UltraSens(商標) Virus Handbookに従い、尿の開始容量1mlからDNAを抽出した。
【0104】
DNA試料の重亜硫酸塩処理
MethylEasy(商標) High Throughput DNA重亜硫酸塩修飾キット(ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures)、オーストラリア)に従い、重亜硫酸塩処理を行った。下記も参照のこと。
【0105】
意外なことに、例えばヒト細胞の試料中に微生物DNAが存在する場合、微生物DNAを核酸の他のソースから分離する必要性が全くないことが本発明者らによって見出されている。異なるDNAタイプの膨大な混合物に対して処理工程を利用してもよく、さらに微生物に特異的な核酸についても本発明によって同定してもよい。複雑なDNA混合物中での検出の限界が標的核酸分子の単一の複製物に至るまで対応可能である標準的なPCR検出の限界であると推定される。
【0106】
試料
本発明において任意の適切な試料を使用してもよい。例として、微生物培養物、臨床試料、獣医学的試料(veterinary samples)、体液、組織培養試料、環境試料、水試料、流出物が挙げられるがこれらに限定されない。本発明が任意の微生物の検出に適応可能であることから、このリストは網羅的と考えるべきではない。
【0107】
キット
本発明を様々なキットの形態でまたはキットの併用によって実施し、手動、半自動または完全にロボット化されたプラットフォームの点から具体化してもよい。好ましい形態では、MethyEasy(商標)またはHighThroughput MethylEasy(商標)キット(ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures Pty Ltd),オーストラリア)は、EpMotionなどのロボット化されたプラットフォームを使用して96または384プレート内での核酸の変換を可能にする。
【0108】
重亜硫酸塩処理
核酸の有効な重亜硫酸塩処理のための典型的なプロトコルを下記に示す。プロトコルにより、実質的にすべての処理されたDNAが保持されることになる。本明細書では、この方法は、ヒューマン・ジェネティック・シグネチャーズ(Human Genetic Signatures)(HGS)法とも称される。試料または試薬の容量または量が変化しうることは理解されるであろう。
【0109】
重亜硫酸塩処理の好ましい方法を、本明細書中に参照により援用される米国特許出願第10/428310号明細書またはPCT/AU2004/000549号明細書において見出すことができる。
【0110】
望まれる場合に適切な制限酵素で予備消化可能なDNA2μgに対し、2μl(1/10容量)の3M NaOH(水50ml中6g、作製後間もない)を最終容量が20μlになるように添加した。この工程により、重亜硫酸塩試薬が好ましくも一本鎖分子と反応することから二本鎖DNA分子が一本鎖形態に変性される。混合物を37℃で15分間インキュベートした。室温を超える温度でインキュベーションを用いることで変性の効率を改善してもよい。
【0111】
インキュベーション後、2Mメタ重亜硫酸ナトリウム(10N NaOH 416mlを有する水20ml中7.6g;BDH AnalaR #10356.4D;作製後間もない)208μlと10mMキノール(水50ml中0.055g、BDH AnalR #103122E;作製後間もない)12μlを連続的に添加した。キノールは還元剤であり、試薬の酸化の低減を促進する。ジチオトレイトール(DTT)、メルカプトエタノール、キノン(ヒドロキノン)、または他の適切な還元剤を例とする他の還元剤もまた使用してもよい。試料を鉱油200μlとオーバーレイした。鉱油のオーバーレイは試薬の蒸発および酸化を防止するが必須ではない。次いで、試料を55℃で一晩インキュベートした。あるいは、試料を、以下のように約4時間または一晩インキュベートしてサーマルサイクラー内でサイクル動作してもよい。PCR装置内で、工程1として55℃/2時間、工程2として95℃/2分サイクル動作させる。工程1を約37℃〜約9O℃の任意の温度で行い、5分〜8時間の長さで変化させてもよい。工程2を約70℃〜約99℃の任意の温度で行い、約1秒〜60分の長さまたはそれより長い時間で変化させてもよい。
【0112】
メタ重亜硫酸ナトリウムによる処理後、オイルを除去し、もしDNA濃度が低い場合、tRNA(20mg/ml)1μlまたはグリコーゲン2μlを添加した。これらの添加物は任意であり、特にDNAが低濃度で存在する場合、これらを使用することで標的DNAとの共沈によって得られるDNAの収量を改善することが可能である。核酸量が0.5μg未満である場合、より効率的な核酸の沈降を目的とした担体としての添加物の使用は一般に望ましい。
【0113】
イソプロパノールによるクリーンアップ処理を以下のように行った。水800μlを試料に添加し、混合し、次いでイソプロパノール1mlを添加した。水または緩衝液は、反応槽内の重亜硫酸塩の濃度を塩が目的の標的核酸と共沈しないレベルまで低下させる。本明細書に開示されるように、塩濃度が所望の範囲未満に希釈される限り、希釈率は一般に約1/4〜1/1000である。
【0114】
試料を再び混合し、4℃で最低5分間放置した。試料をマイクロチューブ内で10〜15分間回転させ、ペレットを70%ETOHで2回洗浄し、各回ボルテックスした。この洗浄処理により、核酸と共沈した任意の残留塩が除去される。
【0115】
ペレットを乾燥させておき、次いで50μlなどの適切な容量のT/E(10mMトリス/0.1mM EDTA) pH7.0〜12.5で再懸濁した。pH10.5の緩衝液が特に有効であることが判明している。試料を、核酸の懸濁に対する必要性に応じ、37℃〜95℃で1分〜96時間インキュベートした。
【0116】
重亜硫酸塩処理に関する別の例を、シトシンからウラシルへの変換に対する方法および材料を提供する国際公開第2005021778号パンフレット(本明細書中に参照により援用される)に見出すことができる。一部の実施形態では、gDNAなどの核酸は重亜硫酸塩およびトリアミンまたはテトラアミンなどのポリアミン触媒と反応する。場合により、重亜硫酸塩は重亜硫酸マグネシウムを含有する。他の実施形態では、核酸は、場合によってポリアミン触媒および/または第四アミン触媒の存在下で重亜硫酸マグネシウムと反応する。本発明の方法を実施するのに使用可能なキットも提供される。これらの方法を処理工程で用いれば本発明にも適することが理解されるであろう。
【0117】
増幅
PCR増幅を、重亜硫酸塩処理したゲノムDNAを2μl含有する反応混合物25μlにおいてPromega PCR master mix、6ng/μlの各プライマーを使用して行った。増幅のために鎖特異的な入れ子(nested)プライマーを使用した。PCRプライマー1および4を使用して第1ラウンドのPCR増幅を行った(下記参照)。第1ラウンドの増幅後、増幅された物質1μlをPCRプライマー2および3を有する第2ラウンドのPCRプレミックスに移し、上記のように増幅した。PCR産物の試料を、95℃で4分間の1サイクルの後、95℃で1分間、50℃で2分間および72℃で2分間の30サイクル;72℃で10分間の1サイクルといった条件下で、ThermoHybaid PX2サーマルサイクラー内で増幅した。
【0118】
【表1】
【0119】
多重増幅
もし検出にとって多重増幅が必要である場合、以下の方法論を実施してもよい。
【0120】
重亜硫酸塩処理したDNA1μlを、反応容量25μl中の以下の成分、すなわち×1 Qiagen multiplex master mix、5〜100ngの各々の第1ラウンド用のINAまたはオリゴヌクレオチドプライマー、1.5〜4.0mM MgSO4、400μMの各dNTPおよび0.5〜2単位のポリメラーゼの混合物に添加する。次いで成分をホットリッドのサーマルサイクラー内で以下のようにサイクル動作させる。各増幅反応では、典型的には最大で200の独立したプライマー配列がありうる。
【0121】
【表2】
【0122】
次いで、第1ラウンドの増幅物から分注した1μlを酵素反応混合物および適切な第2ラウンド用プライマーを有する第2ラウンドの反応管に移して第2ラウンドの増幅を行う。次いで上記のようにサイクル動作を行う。
【0123】
プライマー
本発明において任意の適切なPCRプライマーを使用してもよい。プライマーは、典型的には増幅対象の配列に対する相補配列を有する。プライマーは、典型的にはオリゴヌクレオチドであるが、オリゴヌクレオチド類似体であってもよい。
【0124】
プローブ
プローブは任意の適切な核酸分子または核酸類似体でありうる。例として、DNA、RNA、固定化核酸(LNA)、ペプチド核酸(PNA)、MNA、アルトリトール核酸(ANA)、ヘキシトール核酸(HNA)、インターカレーティング核酸(INA)、シクロヘキサニル核酸(CNA)およびこれらの混合物やこれらのハブリッド、ならびにこれらのリン原子修飾物、例えばホスホロチオエート、メチルホスホレート、ホスホアミダイト、ホスホロジチエート、ホスホロセレノエート、ホスホトリエステルおよびホスホボラノエートなど(これらに限定されない)が挙げられるがこれらに限定されない。非天然ヌクレオチドには、DNA、RNA、PNA、INA、HNA、MNA、ANA、LNA、CNA、CeNA、TNA、(2’−NH)−TNA、(3’−NH)−TNA、α−L−リボ−LNA、α−L−ザイロ−LNA、β−D−ザイロ−LNA、α−D−リボ−LNA、[3.2.1]−LNA、ビシクロ−DNA、6−アミノ−ビシクロ−DNA、5−エピ−ビシクロ−DNA、α−ビシクロ−DNA、トリシクロ−DNA、ビシクロ[4.3.0]−DNA、ビシクロ[3.2.1]−DNA、ビシクロ[4.3.0]アミド−DNA、β−D−リボピラノシル−NA、α−L−リキソピラノシル−NA、2’−R−RNA、α−L−RNAまたはα−D−RNA、β−D−RNA内部に含まれるヌクレオチドが含まれるがこれらに限定されない。さらに、非リンを含有する化合物を、メチルイミノメチル、ホルムアセテート、チオホルムアセテートおよびアミドを有する連結基などの(これらに限定されない)ヌクレオチドへの連結のために使用することが可能である。特に核酸および核酸類似体は、1つもしくは複数のインターカレーターシュードヌクレオチドを含みうる。
【0125】
好ましくは、プローブは、INAを形成する1種もしくは複数種の内部IPNを有するDNAまたはDNAオリゴヌクレオチドである。
【0126】
電気泳動
E−Gelシステムのユーザーガイド(www.invitrogen.doc)に従って試料の電気泳動を行った。
【0127】
検出方法
所望の試料の状態を測定するのに可能な極めて多数の検出系が存在する。本発明では核酸分子を検出するための既知の系または方法であればいずれも使用可能であることが理解されるであろう。検出系には、以下のものが含まれるがそれらに限定されない。
I.10から200,000に及ぶ個々の成分に対する選択が可能と思われるマイクロアレイ型デバイスに対する適切に標識したDNAのハイブリダイゼーション。ガラス、プラスチック、雲母、ナイロン、ビード、磁気ビード、蛍光ビードもしくは膜などの任意の適切な固体表面上に、これらのアレイをINA、PNAまたはヌクレオチドもしくは修飾ヌクレオチドアレイから構成できると思われる。
II.サザンブロット型検出系
III.例えばアガロースゲル、GeneScan分析などの蛍光読み出しといった標準的なPCR検出系。サンドイッチハイブリダイゼーションアッセイ、エチジウムブロミドなどのDNA染色試薬、サイバーグリーン(Syber green)、抗体検出、ELISAプレートリーダー型デバイス、蛍光分光デバイス
IV.特異的増幅もしくは多重増幅されたゲノム断片またはそれに対する任意の変異におけるリアルタイムPCR定量
V.国際公開第2004/065625号パンフレットの中で概説された蛍光ビーズ、酵素抱合体、放射性ビーズなどの検出系のいずれか
Vl.リガーゼ連鎖反応などの増幅工程または鎖置換増幅(SDA)などの等温DNA増幅技術を利用する任意の他の検出系
VII.マルチフォトン検出系
VIII.ゲルにおける電気泳動および可視化
IX.核酸を検出するのに使用されるまたは使用可能と思われる任意の検出プラットフォーム
【0128】
インターカレーティング核酸
インターカレーティング核酸(INA)は、配列特異性を有する核酸(DNAおよびRNA)にハイブリダイズ可能な非天然型ポリヌクレオチドである。INAは、いくつかの望ましい特性を示すことから、プローブに基づくハイブリダイゼーションアッセイにおける核酸プローブに対する代替物/置換物としての候補である。INAは、核酸にハイブリダイズすることで対応する天然核酸/核酸複合体よりも熱力学的に安定なハイブリッドを形成するポリマーである。それらはペプチドまたは核酸を分解することで知られる酵素に対する基質ではない。したがって、INAであれば、生物学的試料中でより安定であることに加え、天然核酸断片よりも長い貯蔵寿命(shelf−life)を有するはずである。INAの核酸とのハイブリダイゼーションは、イオン強度に強く依存する核酸ハイブリダイゼーションと異なり、イオン強度にほとんど依存せず、天然核酸の核酸に対するハイブリダイゼーションにとって極めて不適当な条件下の低いイオン強度で有利である。INAの結合強度は、分子内に設計された挿入基の数、ならびに二本鎖構造の特異的な様式で積み重なった塩基間の水素結合からの通常の相互作用に依存する。配列の識別は、DNAを認識するDNAの場合よりもINAを認識するDNAの場合の方がより効率的である。
【0129】
好ましくは、INAは(S)−1−O−(4,4’−ジメトキシトリフェニルメチル)−3−O−(1−ピレニルメチル)−グリセロールのホスホアミダイトである。
【0130】
INAは、商業的に利用可能な形式で標準的なオリゴヌクレオチドの合成手順を適合させることによって合成される。INAおよびそれらの合成の完全な定義について、本明細書中に参照により援用される国際公開第03/051901号パンフレット、国際公開第03/052132号パンフレット、国際公開第03/052133号パンフレットおよび国際公開第03/052134号パンフレット(ユネスト(Unest A/S))の中に見出すことができる。
【0131】
INAプローブと標準的核酸プローブの間には確かに多数の違いがある。これらの違いは、便宜上、生物学的、構造的、および物理−化学的な違いに分かれる。上記および下記で考察されるように、これらの生物学的、構造的、および物理−化学的な違いは、核酸が通常利用されている応用においてINAプローブの使用を試みる場合に予測不能な結果をもたらす場合がある。異なる組成物における相違点は、化学技術において観察されることが多い。
【0132】
生物学的違いに関しては、核酸は、遺伝的伝達および発現の作用物質として生物種の生命における中心的役割を担う生物学的物質である。それらのin vivoでの特性は、かなりよく理解されている。しかし、INAは最近開発された完全に人工的な分子であり、化学者の頭の中で考え出され、合成有機化学を用いて作製されたものである。それは既知の生物学的機能を全く有しない。
【0133】
構造的には、INAはまた核酸とは格段に異なる。双方ともに共通のヌクレオ塩基(A、C、G、TおよびU)の利用が可能であるが、これらの分子の組成物は構造的に多様である。RNA、DNAおよびINAの骨格は、リン酸ジエステルリボースおよび2−デオキシリボース単位の反復からなる。INAは、リンカー分子を介してポリマーに付着された1つもしくは複数の大きな均一分子を有する点でDNAまたはRNAと異なる。均一分子は、二本鎖構造をなすINAに対する相補的DNA鎖内の塩基間に挿入する。
【0134】
INAとDNAまたはRNAとの物理/化学的な違いは重要でもある。INAは同じ標的配列に結合する核酸プローブよりも迅速に相補的DNAに結合する。DNAまたはRNAの断片と異なり、もし挿入基が末端位置に位置しない場合、INAのRNAに対する結合は不十分である。相補的DNA鎖上の挿入基と塩基の間の強い相互作用のため、INA/DNA複合体の安定性は、類似体のDNA/DNAまたはRNA/DNA複合体のそれよりも高い。
【0135】
DNAもしくはRNAの断片またはPNAなどの他の核酸と異なり、INAは自己凝集または結合特性を示さない。
【0136】
INAは配列特異性を有する核酸にハイブリダイズすることから、INAはプローブに基づくアッセイの開発にとっての有用な候補であり、特にキットおよびスクリーニングアッセイに適合する。しかし、INAプローブは核酸プローブと同等ではない。それ故、プローブに基づくアッセイの特異性、感受性および信頼性を改善可能と思われる任意の方法、キットまたは組成物は、DNAを含有する試料の検出、分析および定量において有用となろう。INAはこのために必要な特性を有する。
【0137】
結果
微生物(細菌、ウイルスまたは真菌株など)の検出は、その種内の多数の微生物の各株によって阻害されることが多い。
【0138】
本発明のin silicoでの一般的原理について、髄膜炎菌、淋菌(Neisseria gonorrhoeae)、インフルエンザ桿菌、連鎖球菌(Streptococcus sp)およびブドウ球菌(Staphylococcus)の細菌を用いて教示する(図1〜5)。本発明の一般的原理について、インフルエンザウイルスおよびロタウイルスを用いて教示している(図6および7)。
【0139】
本発明を教示および支持するための一般的な生化学的データを、臨床的意義のあるグラム陽性およびグラム陰性細菌を用いて図8〜18に記載する。
【0140】
細菌
図1は、髄膜炎菌内のigaプロテアーゼ遺伝子の34個のヌクレオチド領域および淋菌内の対応する遺伝子座を示す(これらの領域はそれらの天然細菌ゲノム内に存在するものとする)(完全な分類;細菌;プロテオバクテリア;ベータプロテオバクテリア;ナイセリア目;ナイセリア科;髄膜炎菌、Z2491血清型Aおよび完全な遺伝子座の特徴;iga、IgAIプロテアーゼ;GeneID906889、ローカスタグ(Locus Tag)NMA0905;RefSeq登録番号NC_003116.1;PMID10761919;Parkhill J et al., 2000, Nature, 404, 502- 506)。これら2つのナイセリア属の34個のヌクレオチド配列の間には74%の配列類似性がある。両方の細菌種内のこれらの領域を増幅するためにPCRに基づくプライマーを作製するとしたら、512通りの組み合わせが可能な変性プライマーが必要となろう。PCR増幅の一部として共通配列を使用するとしたら、それは34個のヌクレオチド配列GYAATYW AGGYCGYCTY GAAGAYTAYA AYATGGC(配列番号3)であろう。この場合、異なる位置を指定するための標準的コードを以下に示す。N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはCおよびW=AまたはT。
【0141】
しかし、これらの2つの種由来の細菌DNAを重亜硫酸塩試薬(シトシンのチミンへの変換をもたらす)で処理する場合、自然発生配列が、コーディングの可能性が全くなく天然に存在しない誘導配列に変換される。この段階で誘導配列は97%の配列類似性を有する。PCRに基づくプライマーは、これらの細菌の遺伝子座双方におけるPCR増幅を1回の試験で可能なように設計されると、この段階では2つのプライマーのみを組み合わせる必要があるにすぎない。位置7の塩基のみがアデニンまたはチミン(Wで示される)である場合、組み合わせは配列GTAATTW AGGTTGTTTT GAAGATTATA ATATGGT(配列番号4)に基づくであろう。したがって、重亜硫酸塩変換により、相対的なゲノムの複雑さが512通りから2通りのプライマータイプへと低減される。この大幅な低減により、関連する細菌種からの同じ遺伝子座の増幅が簡素化される。
【0142】
さらに、これら2種の細菌種由来の領域を増幅するのに場合によってINAプローブを使用し、再び同じ遺伝子座を使用することにより、利点がもたらされる。図2は、図1に示した髄膜炎菌および淋菌のiga遺伝子の同じ34個のヌクレオチド領域を図示し、これはINAプローブをさらに用いてもプローブ長や複雑さが低減可能であるという対象範囲を追加的に明らかにしている。短いINAの16merの配列AGGYCGYCTY GAAGAY(配列番号5)であれば、この領域を検出するのに16通りの可能なプライマーの組み合わせが必要となろうが、重亜硫酸塩による変換後には、特有のプライマー配列AGGTTGTTTT GAAGAT(配列番号6)で十分であろう。INA分子の利点は、骨格内に組み込まれるインターカレーティングシュードヌクレオチドのおかげで、標準オリゴヌクレオチドに対するINAのTmの増大により、正確な遺伝子座へのハイブリダイゼーションを非特異的結合と識別することが著しく容易になることである。そうであっても、標準オリゴヌクレオチドが依然として適切に機能することが理解されるであろう。
【0143】
関連性が高い細菌種が類似の臨床症状をもたらす場合、重亜硫酸塩で変換したDNAを再び使用して、特異的細菌タイプの存在についてアッセイするためのより簡素化したプローブを設計することができる。図3は、3種の細菌種内のiga遺伝子におけるDNAアラインメントを示し、それらの1種であるインフルエンザ桿菌は異なる分類群由来である。細菌DNAの重亜硫酸塩処理により、プローブの組み合わせ数が極めて少なくなった。この比較は、1回の試験で関連性のない種についてのアッセイが可能であることの重要性を示す。髄膜炎菌とインフルエンザ桿菌の双方が髄膜炎を誘発することから、1回の試験で同じ臨床症状を誘発するすべての微生物についてアッセイ可能であることは有利である。
【0144】
同じ分類群からの多数の異なる細菌種の分析は、本発明によってやはり容易になる。図4は、連鎖球菌群、すなわちストレプトコッカス・オラリス(S.oralis)、ストレプトコッカス・ミチス(S.mitis)、ストレプトコッカス・ディスガラクティエ(S.dysgalactiae)、ストレプトコッカス・クリスタツス(S.cristatus)、ストレプトコッカス・ゴルドニー(S.gordonii)、ストレプトコッカス・パラウベリス(S.parauberis)、ストレプトコッカス・ニューモニエ、ストレプトコッカス・ボビス(S.bovis)、ストレプトコッカス・ベスチブラリス(S.vestivularis)およびストレプトコッカス・ウベリス(S.uberis)の10種の細菌種内のtuf遺伝子の40個のヌクレオチドセグメントを示す。この領域は、10種の間で約68%の配列類似性を有し、1回の試験で10種について同時にアッセイするのに12,288通りのプライマーの組み合わせを必要とする。重亜硫酸塩変換された配列は、これらの種の間で85%の配列類似性を有し、ここで必要とする可能なプライマーの組み合わせは単に64通りである。
【0145】
同じ細菌種に属する異なる株の分析もまた本発明によって簡素化される。図5は、黄色ブドウ球菌のエンテロトキシン遺伝子の23個のヌクレオチドセグメントを図示する。この遺伝子領域の天然配列は、全部で7種の株の間に56%しか配列類似性を有さず、1536通りのプライマーの組み合わせを必要とする一方、重亜硫酸塩で変換した配列は74%の配列類似性を有し、必要とするプライマーの組み合わせは単に64通りである。
【0146】
ウイルス核酸分析および相対的なゲノムの複雑さの低減
相対的なゲノムの複雑さを低減する原理を、DNAゲノムを有するインフルエンザウイルスなどのウイルス群と同様に、(RNAは逆転写酵素によってDNAに変換され、次いでそれによって重亜硫酸塩処理されうることから)RNAゲノムを有するウイルス群にも適用してもよい。ウイルス検出への適用を例示するため、分節RNAゲノムを有する、インフルエンザウイルス株(オルトミクソウイルス科(Family Orthomyxoviridae))のノイラミニダーゼ遺伝子およびロタウイルス株(レオウイルス科(Family Reoviridae))の表面タンパク質をコードするVP4遺伝子の両ウイルスを使用している。インフルエンザウイルスの分類は複雑であり、例えばタイプA、BおよびCが抗原特性に基づき、かつさらなるサブタイプが複製起点部位、単離年、単離数およびサブタイプに基づいている。これにより、第一にインフルエンザウイルスを集団として同定し、次いで結果的にかなり下位の分類レベル(sub−sub−classification level)の分析にまで掘り下げられるという需要が高まる。
【0147】
ロタウイルスの分類もまた複雑である。ロタウイルス血清型の数は多く、PおよびG血清型という認められた2つの主な血清型を伴う。最小でも14種の異なるG血清型が存在し、小児医学ではそれらを明確に検出することが重要である。世界中のほぼすべての小児が、3歳になるまでに既に少なくとも1回ロタウイルスによる感染を、たとえこれらの感染が不顕性であり、胃腸管に対して軽度の作用を有するにすぎない場合であっても被っていると推定される。
【0148】
臨床レベルでのインフルエンザによる感染の結果はよく知られており、ほぼ毎冬で罹患率および死亡率が有意な状況である。しかし、特にストレプトコッカス・ニューモニエ、インフルエンザ桿菌および黄色ブドウ球菌による感染後に重度の二次合併症が発症しうる。肺炎合併症が細菌およびウイルス感染症の特徴が混ざり合うことから発症しうることや、速やかな抗生治療が有効な治療でありうることから、ウイルス感染症と細菌感染症の双方について同時分析できることが有利であることは極めて明らかである。
【0149】
9種の異なるインフルエンザ株における相対的なゲノムの複雑さの低減について図6に示す。インフルエンザウイルスのノイラミニダーゼ遺伝子の20個のヌクレオチド領域をそのDNA形態で示す。これらの9種の単離物の間には50%の配列類似性がある。重亜硫酸塩変換後、配列類似性は75%に増加している。その元の形態では、これらの9種の株を分析するには2048通りの可能なプライマーの組み合わせが必要であると思われる一方、重亜硫酸塩変換後に必要なプライマーの組み合わせは48通りにすぎない。
【0150】
3種の異なるロタウイルス株のVP4遺伝子における相対的なゲノムの複雑さの低減について図7に示す。VP4遺伝子の20個のヌクレオチド領域は、変換前に52%および変換後に74%の配列類似性を有する。プライマーの組み合わせ数は512から32に減少する。
【0151】
病院や病理学実験室で一般に遭遇する臨床的意義のある微生物種を用いた、微生物を検出する一手段として微生物ゲノムを簡素化するin silicoでのアプローチを支持する分子データを図8〜15に図示する。
【0152】
もし試料中に存在するのがグラム陽性細菌かまたはグラム陰性細菌かを初期に決定できるとしたら、それは汚染性微生物の迅速な検出にとって明らかな利点であり、臨床的意義が大きい。本明細書に記載の方法では、異なる細菌種の23Sリボソーム遺伝子を用いたかかる試験が提供され、増幅反応を介し、簡素化したゲノム上でプライマーセットを利用することによりグラム陽性細菌またはグラム陰性細菌の検出を可能にするかかるプライマーが生成される。23S配列は、例えば上記S.galactiaeの例において見られる一部の細菌ゲノムに任意に付加される一部のタンパク質コーディング配列とは異なり、すべての細菌種内で生じることから、かかる高レベルの識別にとって望ましい。多数のタンパク質をコードする微生物配列は、ゲノムの「がらくた(flotsam and jetsam)」に類するものであり、それらの有用性は、異なる微生物株、タイプまたは単離物などのより低いレベルの分類カテゴリの間、あるいはウイルスの場合、異なるタイプまたは新しく生じた変異体の間で識別することにある。これら成分すべての正規および簡素化されたゲノム配列、タンパク質をコードしないリボソームRNA遺伝子、および細菌のタンパク質をコードするrecA遺伝子をそれぞれ図15および16に示す。23S細菌アンプリコンに対する増幅反応の実施に使用されるプライマー配列を表1に示す。recAアンプリコンに対する増幅反応の実施に使用されるプライマー配列を表2に示す。すべてのプライマーを重亜硫酸塩で処理したDNAに対して作製し、5’から3’の向きで示す。
【0153】
表1は、グラム陽性(Pos)、グラム陰性(Neg)の検出用プライマーを生成するためのアラインメントを用いた、23SリボソームRNA遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる適切な細菌用プライマー配列を示す。さらに、マイコプラズマ(Mycoplasma spp)(Myc)、ブドウ球菌(Staphylococcus spp)(Staph)、連鎖球菌(Streptococcus spp)(Strep)、ナイセリア(Neisseria spp)(NG)、クラミジア(Chlamydia)(CT)、ならびに大腸菌(Escherichia coli)(EC)およびストレプトコッカス・ニューモニエの特異的検出用にプライマーを設計した。
【0154】
以下の記号は以下の塩基付加を指定する。N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはCおよびW=AまたはT。
【0155】
使用されるすべてのプライマーは重亜硫酸塩で簡素化したDNA配列に基づいた。
【0156】
【表3】
【0157】
【表4】
【0158】
【表5】
【0159】
【表6】
【0160】
【表7】
【0161】
【表8】
【0162】
表2は、特有の細菌の分類を目的とした、黄色ブドウ球菌(SA)、表皮ブドウ球菌(Staphylococcus epidermidis)(SE)、レイ菌(Serratia marscesens) (SM)、大腸菌(Escherichia coli)(EC)およびエンテロコリティカ菌(Yersinia enterocolitica)(YE)由来の、アラインメントを用いた、recAタンパク質をコードする遺伝子由来の簡素化DNAの増幅に用いられる細菌用プライマー配列を示す。
【0163】
【表9】
【0164】
【表10】
【0165】
表1は、グラム陽性(Posで示す)細菌およびグラム陰性(Negで示す)細菌の検出用の最適なプライマーを生成するための多重アラインメントを用いた、23SリボソームRNA遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる細菌用プライマー配列を示す。さらに、種の群および個々の種の特異的検出用にもプライマーを設計した。これらの細菌用プライマー群に対し、大腸菌および肺炎桿菌(EC)、Neisseria spp(NG)、クラミジア(CT)、マイコプラズマ(Myc)、連鎖球菌(Strep)およびブドウ球菌(Staph)のように指定する。下位指定のFおよびRのは、それぞれフォワードプライマーおよびリバースプライマーである。さらに、2種以上の可能な塩基が所定のヌクレオチド位置で必要である場合、塩基の縮重が、N=A、G、TまたはC;D=A、GまたはT;H=A、TまたはC;B=G、TまたはC;V=G、AまたはC;K=GまたはT;S=CまたはG;Y=TまたはC;R=AまたはG;M=AまたはC;およびW=AまたはTのコードによって与えられる。繰り返して言えば、本発明において用いられるすべてのプライマーは重亜硫酸塩で簡素化したDNA配列に基づいている。
【0166】
表2は、特有の細菌の分類を目的とした、黄色ブドウ球菌(SA)、表皮ブドウ球菌(SE)、レイ菌(SM)、大腸菌(Escherichia coli)(EC)およびエンテロコリティカ菌(YE)由来の、アラインメントを用いた、recAタンパク質をコードする遺伝子由来の重亜硫酸塩で簡素化したDNAの増幅に用いられる細菌用プライマー配列を示す。
【0167】
図8は、グラム陽性およびグラム陰性細菌のゲノム的に簡素化した23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示し、ここで適切にサイジングしたアンプリコンはアガロースゲル電気泳動による特異的な長さのバンドとして検出される。矢印は、マーカーのレーン(M)内を動く、標準的なサイジングしたマーカーに対するアンプリコンの想定サイズを示す。グラム陰性細菌に特異的なプライマーを使用すると、大腸菌、淋菌、肺炎桿菌、カタル球菌(Moraxella catarrhalis)、緑膿菌(Pseudomonas aeruginosa)およびプロテウス・ブルガリス(Proteus vulgaris)における6種のグラム陰性のレーン1〜6(上パネル)に限ってバンドが出現する。グラム陽性細菌に特異的なプライマーを使用すると、腸球菌(Enterococcus faecalis)、表皮ブドウ球菌、黄色ブドウ球菌、スタフィロコッカッス・キシロサス(Staphylococcus xylosis)、ストレプトコッカス・ニューモニエおよび溶血性連鎖球菌(Streptococcus haemolyticus)における6種のグラム陽性のレーン7〜12(下パネル)に限ってバンドが出現する。
【0168】
図9は、(この例では)大腸菌および肺炎桿菌(K.pneumoniae)という2種のみのグラム陰性細菌種を由来とするアンプリコンを検出するように設計した、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。増幅方法論の特異性について、大腸菌および肺炎桿菌を示すレーン1および3におけるアンプリコンの存在、ならびにレーン2およびレーン4〜12において増幅産物が存在しないこと(これら10個の空のレーンは試験で用いられる細菌の残りの10種を示す)によって図示する。
【0169】
図10は、1種のみのナイセリア細菌群に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。ゲノムを簡素化する方法論の特異性について、淋菌を示すレーン2のみにおけるアンプリコンの存在およびレーン1およびレーン3〜12において増幅産物が存在しないこと(これら11個の空のレーンは試験で用いられる細菌の残りの11種を示す)によって図示する。
【0170】
個々の微生物種の分析においては、タンパク質をコードする遺伝子についても、微生物の異なる株が問題の遺伝子配列におけるそれらの存在/非存在に対して多形性を示さないという条件で必要に応じて用いてもよい。
【0171】
図11は、大腸菌の細菌recA遺伝子に対するプライマーの使用について図示する。アンプリコンの特異性を、レーン1における正確にサイジングしたアンプリコンの存在および細菌の他の11種を示す残りのレーン2〜12においてそれが存在しないことによって図示する。
【0172】
図12のデータは、ブドウ球菌(Staphylococci)などのより大きい細菌群のメンバーを明らかにするプライマーの特異性をさらに図示する。ブドウ球菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物は、表皮ブドウ球菌、黄色ブドウ球菌およびスタフィロコッカッス・キシロサスを示すレーン8、9および10でのみアンプリコンを示す。レーン1〜7およびレーン11〜12において増幅産物が存在しないことは反応の特異性を証明している。9つの空のレーンは、試験で用いられる9種の非ブドウ球菌細菌を示す。
【0173】
図13は、連鎖球菌(Streptococcal)細菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物を示す。連鎖球菌に特異的なプライマーを用いた、ゲノム的に簡素化した23Sリボソーム遺伝子領域からPCRによって得られた増幅産物は、ストレプトコッカス・ニューモニエおよび溶血性連鎖球菌を示すレーン11および12でのみアンプリコンを示す。レーン1〜10において増幅産物が存在しないことは反応の特異性を示している。これらの10個の空のレーンは、試験で用いられる10種の非連鎖球菌細菌を示す。
【0174】
図14は、表皮ブドウ球菌(レーン8)のrecA遺伝子のゲノム的に簡素化した領域由来のタンパク質をコードする遺伝子からPCRによって得られた増幅産物を示す。2つのバンド(矢印)は、第1ラウンド(上部バンド)および第2ラウンド(下部バンド)のPCR増幅から得られた繰り越し(carry over)アンプリコンを示す。レーン1〜7および9〜12においてアンプリコンが存在しないことは方法の特異性を示し、タンパク質をコードする遺伝子を特殊環境下でゲノムの非コーディング成分の代わりに使用することで1種の細菌種に限って検出がなされるという点を強調している。
【0175】
図15は、クラミジアのゲノム的に簡素化した23Sリボソーム遺伝子を標的にする特異的プライマーを用いたアンプリコンの検出を示す。DNAの開始量が少ないことからPCR反応を2通りに行った。レーン番号5は、既知の陰性の個人の尿から抽出したDNAであった。2通りのうちのいずれかでバンドが出現したことは、クラミジアDNAの存在に対する陽性反応であると考えられた。
【0176】
図16は、大腸菌由来の23SリボソームRNA遺伝子の正規のヌクレオチド配列および図示目的として全シトシンがチミンと置換されている場合のゲノムの簡素化後の同配列を示す。
【0177】
図17は、大腸菌由来のrecA遺伝子の正規のヌクレオチド配列および図示目的として全シトシンがチミンと置換されている場合のゲノムの簡素化後の同配列を示す。
【0178】
要するに、微生物源由来の重亜硫酸塩で処理したDNAは、ゲノム的に簡素化したプライマーを用いて増幅される場合、オリゴヌクレオチドまたはINAなどの修飾核酸であると、試料中で任意のタイプの微生物を見出すための最高レベルの検出系を提供し、それはヒト臨床物質由来の試料であるかまたは汚染水などの環境源由来の別の極端な状況にある。本発明については、広範な異なる細菌種や臨床的意義のあるウイルスに対して実証されている。酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)またはその近縁種などの単細胞真核微生物の検出は、本方法を単純に拡張したものである。それは、18または28Sリボソーム配列などの類似のゲノム配列ソースあるいは上記のように所定の種、タイプ、株または変異体もしくは多型に対して特異的なタンパク質をコードするコーディング配列を必要とする。
【0179】
本発明に記載の検出系の実際の意味もまた重要である。本明細書において詳細に記載された原理が増幅用PCRを用いて示されている一方、当該技術で既知の任意の方法論を介して読み出しを連携させてもよい。重亜硫酸塩処理がゲノムの複雑さを低減し、それ故に微生物のさらなるクラスを対象とした、検出器の数がより少ない(という特徴を備えた)マイクロアレイ上での試験が可能になることから、現在重視されているマイクロアレイ検出系とともに、ゲノム的に簡素化されたDNAを用いることで微生物のはるかに広大な範囲を検出することができるであろう。
【0180】
もし、例えばマイクロアレイが250,000種程度の異なる微生物を1回の試験で検出するように構築されなければならないとしたら、現行の方法論は検出プラットフォームの物理的制限によって圧倒されることから、十分な実用的検出プラットフォームを提供できないであろう。しかし、ゲノムの簡素化を併せるならば、小型マイクロアレイは1000種程度の異なる高レベルの細菌カテゴリを検出可能であろう。次いで、あくまで初期試験において陽性であったそれらの群を代表するものを有する別のアレイを用いれば、かかる試験からの陽性について評価することができるであろう。
【0181】
当業者であれば、特定の実施形態において示されるように、極めて多くの変異および/または修飾が、幅広く記載された本発明の趣旨または範囲から逸脱することなく本発明に対してなされうることを理解するであろう。したがって、本実施形態は、例示としてかつ限定されることなく、あらゆる点から考えられるべきである。
【図面の簡単な説明】
【0182】
【図1】ゲノムの簡素化の前後における髄膜炎菌および淋菌のiga遺伝子の一部のアラインメントを示す。図に示されるように、ゲノムの簡素化に先立ち、ナイセリア種のユニバーサルな検出にとって全部で512通りのプローブの組み合わせ(74%の配列類似性)が必要であるのに対し、核酸誘導体を形成する簡素化後では、単に2通りの組み合わせ(97%の配列類似性)で十分であると思われる(配列番号は各配列の後に列挙される)。
【図2】INAプローブが標準オリゴヌクレオチドプローブより短い長さを有しうることから簡素化配列の配列類似性をさらに高めるためのINAプローブの使用を示す。ゲノムの簡素化方法をINAプローブと組み合わせると、標的配列に対して100%の配列類似性を有するプローブの選択および使用が可能になる(配列番号は各配列の後に列挙される)。
【図3】ナイセリアおよびヘモフィルス由来のiga遺伝子のアラインメントを用いた、近縁種である種間の区別をするためのゲノムの簡素化について示す。図に示されるように、本発明の方法は、種特異的プローブを生成するためのゲノム物質の簡素化を可能にする。さらに、それはゲノムDNAを簡素化するが、ナイセリア種と、近縁種であるヘモフィルス種の間の区別も可能にする(配列番号は各配列の後に列挙される)。
【図4】連鎖球菌の異なる10種におけるゲノムの簡素化の前後での連鎖球菌のtuf遺伝子のアラインメントについて示す。処理前には、tuf遺伝子のユニバーサルプライマーにとって全部で12,288通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは64通りにすぎないと思われる。さらに、配列類似性は簡素化前には67.5%にすぎず、簡素化後には85%に高まる(配列番号は各配列の後に列挙される)。
【図5】ゲノムの簡素化の前後でのブドウ球菌のエンテロトキシン遺伝子のアラインメントについて示す。重亜硫酸塩処理前には、ブドウ球菌のエンテロトキシン遺伝子のユニバーサルプライマーにとって全部で1,536通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは64通りにすぎないと思われる(配列番号は各配列の後に列挙される)。
【図6】ゲノムの簡素化の前後での様々なインフルエンザ株のインフルエンザ群AおよびBのノイラミニダーゼ遺伝子のアラインメントについて示す。処理前には、群AおよびBのノイラミニダーゼ遺伝子のユニバーサルプライマーにとって全部で2,048通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは48通りにすぎないと思われる。さらに、配列類似性は簡素化前には50%にすぎず、簡素化後には75%に高まる(配列番号は各配列の後に列挙される)。
【図7】ゲノムの簡素化の前後でのロタウイルスVP4遺伝子のアラインメントについて示す。処理前には、ロタウイルスVP4遺伝子のユニバーサルプライマーにとって全部で512通りのプローブの組み合わせが必要であると思われる。ゲノムの簡素化後、ユニバーサルな検出にとって必要とされるプローブの組み合わせは32通りにすぎないと思われる(配列番号は各配列の後に列挙される)。
【図8】グラム陽性およびグラム陰性細菌のゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示し、ここでは適切なアンプリコンがアガロースゲル電気泳動による特異的長さのバンドとして検出される。矢印は、マーカーレーン(M)内を動く、標準的なサイジングされたマーカーに対するアンプリコンの想定される大きさを示す。グラム陰性細菌に特異的なプライマーを用いると、6種のグラム陰性のレーンのみにバンドが出現する(上パネル)。グラム陽性細菌に特異的なプライマーを用いると、6種のグラム陽性のレーンのみにバンドが出現する(下パネル)。
【図9】大腸菌(レーン1)および肺炎桿菌(レーン3)のゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。増幅の特異性は、残りの10種の細菌由来の増幅産物が存在しないことによって図示される。
【図10】ナイセリアに特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図11】大腸菌のrecA遺伝子のゲノム的に簡素化された領域由来のタンパク質をコードする遺伝子からのPCRによって得られた増幅産物を示す。アンプリコンの特異性は、大腸菌のrecAアンプリコンの存在および他の11種の細菌からのその出現がないことによって図示される。
【図12】ブドウ球菌に特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図13】連鎖球菌に特異的なプライマーを用いた、ゲノム的に簡素化された23Sリボソーム遺伝子領域からのPCRによって得られた増幅産物を示す。
【図14】表皮ブドウ球菌のrecA遺伝子のゲノム的に簡素化された領域由来のタンパク質をコードする遺伝子からのPCRによって得られた増幅産物を示す。2つのバンド(矢印)は、第1ラウンド(上部バンド)および第2ラウンド(下部バンド)のPCR増幅から得られた繰り越しアンプリコンを示す。
【図15】トラコーマクラミジアのゲノム的に簡素化された23Sリボソーム遺伝子を標的にする特異的プライマーを用いたアンプリコンの検出を示す。
【図16】表皮ブドウ球菌由来の正規のゲノム配列およびゲノム的に簡素化された23S rDNA配列を示す(配列番号は各配列の後に列挙される)。
【図17】大腸菌recA遺伝子のゲノム配列およびゲノム的に簡素化された配列を示す(配列番号は各配列の後に列挙される)。
【特許請求の範囲】
【請求項1】
微生物ゲノムまたは微生物核酸を簡素化するための方法であって、
シトシンを修飾する作用物質で微生物ゲノムまたは核酸を処理し、微生物核酸誘導体を形成する工程と、
前記微生物核酸誘導体を増幅して前記微生物ゲノムまたは核酸の簡素化形態を生成する工程と、
を含む、方法。
【請求項2】
前記微生物ゲノムまたは核酸の処理に先立ち、微生物RNAをDNAに変換する工程を含む請求項1に記載の方法。
【請求項3】
微生物RNAを処理してRNA誘導体分子を生成し、次いで前記RNA誘導体を変換してDNA誘導体分子を形成する工程を含む請求項1に記載の方法。
【請求項4】
前記微生物ゲノムまたは核酸が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から得られるものである請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記微生物ゲノムまたは核酸が、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択される請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記リボソーム遺伝子領域が原核生物内の16Sまたは23Sおよび単細胞真核微生物内の18Sまたは28Sである請求項5に記載の方法。
【請求項7】
前記作用物質が非メチル化シトシンを修飾する請求項1〜6のいずれか一項に記載の方法。
【請求項8】
前記作用物質が重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記作用物質が重亜硫酸ナトリウムである請求項8に記載の方法。
【請求項10】
前記作用物質が、相補的二本鎖の微生物ゲノムDNAの各鎖においてシトシンをウラシルに修飾することで、2つの非相補的な微生物核酸誘導体分子を形成する請求項1〜9のいずれか一項に記載の方法。
【請求項11】
前記微生物核酸誘導体におけるシトシンの総数が、対応する未処理の微生物ゲノムまたは核酸に比べて低下している請求項1〜10のいずれか一項に記載の方法。
【請求項12】
前記微生物ゲノムまたは核酸の簡素化形態におけるシトシンの総数が、対応する未処理の微生物ゲノムまたは核酸に比べて低下している請求項1〜11のいずれか一項に記載の方法。
【請求項13】
前記微生物核酸誘導体がアデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未処理の微生物ゲノムまたは核酸と塩基の総数が実質的に等しい請求項1〜12のいずれか一項に記載の方法。
【請求項14】
前記微生物ゲノムまたは核酸の簡素化形態が、実質的にアデニン(A)、グアニン(G)およびチミン(T)の塩基からなる請求項1〜13のいずれか一項に記載の方法。
【請求項15】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項1〜14のいずれか一項に記載の方法。
【請求項16】
微生物に特異的な核酸分子を生成するための方法であって、
シトシンを修飾する作用物質で微生物由来のDNAを含有する試料を処理し、微生物核酸誘導体を形成する工程と、
前記微生物核酸誘導体の少なくとも一部を増幅することで、対応する未処理の微生物核酸に比べてシトシンの総数が低下しており、微生物または微生物型に特異的な核酸配列を含む簡素化された核酸分子を形成する工程と、
を含む、方法。
【請求項17】
前記微生物が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から選択される請求項16に記載の方法。
【請求項18】
前記微生物ゲノムまたは核酸が、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択される請求項16または17に記載の方法。
【請求項19】
前記リボソーム遺伝子領域が原核生物内の16Sまたは23Sおよび単細胞真核微生物内の18Sまたは28Sである請求項18に記載の方法。
【請求項20】
前記作用物質が非メチル化シトシンを修飾する請求項16〜19のいずれか一項に記載の方法。
【請求項21】
前記作用物質が、重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項16〜20のいずれか一項に記載の方法。
【請求項22】
前記作用物質が重亜硫酸ナトリウムである請求項21に記載の方法。
【請求項23】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項16〜22のいずれか一項に記載の方法。
【請求項24】
前記微生物に特異的な核酸分子を検出する工程をさらに含む請求項16〜23のいずれか一項に記載の方法。
【請求項25】
前記微生物に特異的な核酸分子が、
前記微生物に特異的な核酸分子の標的領域に結合可能な検出リガンドを提供し、かつ前記検出リガンドが前記標的領域に結合するのに十分な時間を与える工程と、
前記検出リガンドの前記標的領域に対する結合を測定し、前記微生物に特異的な核酸分子の存在を検出する工程と、
によって検出される請求項24に記載の方法。
【請求項26】
前記微生物に特異的な核酸分子が、増幅産物を分離しかつ前記分離された産物を可視化することによって検出される請求項24に記載の方法。
【請求項27】
前記増幅産物が電気泳動によって分離され、かつゲル上の1つもしくは複数のバンドを可視化することによって検出される請求項26に記載の方法。
【請求項28】
前記簡素化された核酸分子が実質的にシトシンを全く有しない請求項16〜27のいずれか一項に記載の方法。
【請求項29】
前記微生物に特異的な核酸分子が前記微生物において自然発生しないものである請求項28に記載の方法。
【請求項30】
前記微生物に特異的な核酸分子が前記微生物の分類レベルを示す核酸配列を有する請求項16〜29のいずれか一項に記載の方法。
【請求項31】
前記微生物の前記分類レベルが同一もしくは異なる地理的集団または底生性集団由来の科、属、種、株、タイプ、あるいは異なる集団を含む請求項30に記載の方法。
【請求項32】
微生物からDNA配列を得る工程と、
アデニン、グアニンおよびチミンの塩基を実質的に含むように、各シトシンをチミンに変化させることによって前記微生物DNA配列の変換を行うことにより、前記微生物DNA配列の簡素化形態を形成する工程と、
前記微生物DNAの簡素化形態から微生物に特異的な核酸分子を選択する工程と、
を含む微生物に特異的な核酸分子を生成するための方法。
【請求項33】
前記変換がin silicoで行われる請求項32に記載の方法。
【請求項34】
2種以上の微生物DNA配列の簡素化形態が得られ、かつ前記2種以上の配列を比較することで少なくとも1種の微生物に特異的な核酸分子が得られる請求項32または33に記載の方法。
【請求項35】
請求項32〜34のいずれか一項に記載の方法によって得られる微生物に特異的な核酸分子。
【請求項36】
試験もしくはアッセイにおいて前記微生物に特異的な核酸分子を結合または増幅するためのプローブあるいはプライマーを得ることを目的とした請求項32〜34のいずれか一項に記載の方法の使用。
【請求項37】
試料中の微生物の存在を検出するための方法であって、
前記微生物を含有すると見られる試料から微生物DNAを得る工程と、
シトシンを修飾する作用物質で前記微生物核酸を処理し、微生物核酸誘導体を形成する工程と、
所望の微生物に特異的な核酸分子の増幅を可能にする能力を有するプライマーを、前記微生物核酸誘導体に提供する工程と、
前記微生物核酸誘導体に対して増幅反応を行うことで簡素化された核酸を形成する工程と、
前記所望の微生物に特異的な核酸分子を有する増幅核酸産物の存在についてアッセイし、前記所望の微生物に特異的な核酸分子の検出が前記試料中での前記微生物の存在を示す工程と、
を含む、方法。
【請求項38】
前記微生物が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から選択される請求項37に記載の方法。
【請求項39】
前記作用物質が非メチル化シトシンを修飾する請求項37または38に記載の方法。
【請求項40】
前記作用物質が、重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項37〜39のいずれか一項に記載の方法。
【請求項41】
前記作用物質が重亜硫酸ナトリウムである請求項40に記載の方法。
【請求項42】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項37〜41のいずれか一項に記載の方法。
【請求項43】
前記核酸分子が、
前記核酸分子の領域に結合可能な検出リガンドを提供し、かつ前記検出リガンドが前記領域に結合するのに十分な時間を与える工程と、
前記検出リガンドの前記核酸分子に対する結合を測定し、前記核酸分子の存在を検出する工程と、
によって検出される請求項37〜42のいずれか一項に記載の方法。
【請求項44】
前記核酸分子が、増幅産物を分離しかつ前記分離された産物を可視化することによって検出される請求項37〜43のいずれか一項に記載の方法。
【請求項1】
微生物ゲノムまたは微生物核酸を簡素化するための方法であって、
シトシンを修飾する作用物質で微生物ゲノムまたは核酸を処理し、微生物核酸誘導体を形成する工程と、
前記微生物核酸誘導体を増幅して前記微生物ゲノムまたは核酸の簡素化形態を生成する工程と、
を含む、方法。
【請求項2】
前記微生物ゲノムまたは核酸の処理に先立ち、微生物RNAをDNAに変換する工程を含む請求項1に記載の方法。
【請求項3】
微生物RNAを処理してRNA誘導体分子を生成し、次いで前記RNA誘導体を変換してDNA誘導体分子を形成する工程を含む請求項1に記載の方法。
【請求項4】
前記微生物ゲノムまたは核酸が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から得られるものである請求項1〜3のいずれか一項に記載の方法。
【請求項5】
前記微生物ゲノムまたは核酸が、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択される請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記リボソーム遺伝子領域が原核生物内の16Sまたは23Sおよび単細胞真核微生物内の18Sまたは28Sである請求項5に記載の方法。
【請求項7】
前記作用物質が非メチル化シトシンを修飾する請求項1〜6のいずれか一項に記載の方法。
【請求項8】
前記作用物質が重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記作用物質が重亜硫酸ナトリウムである請求項8に記載の方法。
【請求項10】
前記作用物質が、相補的二本鎖の微生物ゲノムDNAの各鎖においてシトシンをウラシルに修飾することで、2つの非相補的な微生物核酸誘導体分子を形成する請求項1〜9のいずれか一項に記載の方法。
【請求項11】
前記微生物核酸誘導体におけるシトシンの総数が、対応する未処理の微生物ゲノムまたは核酸に比べて低下している請求項1〜10のいずれか一項に記載の方法。
【請求項12】
前記微生物ゲノムまたは核酸の簡素化形態におけるシトシンの総数が、対応する未処理の微生物ゲノムまたは核酸に比べて低下している請求項1〜11のいずれか一項に記載の方法。
【請求項13】
前記微生物核酸誘導体がアデニン(A)、グアニン(G)、チミン(T)およびウラシル(U)の塩基を実質的に有し、かつ対応する未処理の微生物ゲノムまたは核酸と塩基の総数が実質的に等しい請求項1〜12のいずれか一項に記載の方法。
【請求項14】
前記微生物ゲノムまたは核酸の簡素化形態が、実質的にアデニン(A)、グアニン(G)およびチミン(T)の塩基からなる請求項1〜13のいずれか一項に記載の方法。
【請求項15】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項1〜14のいずれか一項に記載の方法。
【請求項16】
微生物に特異的な核酸分子を生成するための方法であって、
シトシンを修飾する作用物質で微生物由来のDNAを含有する試料を処理し、微生物核酸誘導体を形成する工程と、
前記微生物核酸誘導体の少なくとも一部を増幅することで、対応する未処理の微生物核酸に比べてシトシンの総数が低下しており、微生物または微生物型に特異的な核酸配列を含む簡素化された核酸分子を形成する工程と、
を含む、方法。
【請求項17】
前記微生物が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から選択される請求項16に記載の方法。
【請求項18】
前記微生物ゲノムまたは核酸が、タンパク質をコードする核酸、タンパク質をコードしない核酸、原核生物または単細胞真核微生物のリボソーム遺伝子領域から選択される請求項16または17に記載の方法。
【請求項19】
前記リボソーム遺伝子領域が原核生物内の16Sまたは23Sおよび単細胞真核微生物内の18Sまたは28Sである請求項18に記載の方法。
【請求項20】
前記作用物質が非メチル化シトシンを修飾する請求項16〜19のいずれか一項に記載の方法。
【請求項21】
前記作用物質が、重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項16〜20のいずれか一項に記載の方法。
【請求項22】
前記作用物質が重亜硫酸ナトリウムである請求項21に記載の方法。
【請求項23】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項16〜22のいずれか一項に記載の方法。
【請求項24】
前記微生物に特異的な核酸分子を検出する工程をさらに含む請求項16〜23のいずれか一項に記載の方法。
【請求項25】
前記微生物に特異的な核酸分子が、
前記微生物に特異的な核酸分子の標的領域に結合可能な検出リガンドを提供し、かつ前記検出リガンドが前記標的領域に結合するのに十分な時間を与える工程と、
前記検出リガンドの前記標的領域に対する結合を測定し、前記微生物に特異的な核酸分子の存在を検出する工程と、
によって検出される請求項24に記載の方法。
【請求項26】
前記微生物に特異的な核酸分子が、増幅産物を分離しかつ前記分離された産物を可視化することによって検出される請求項24に記載の方法。
【請求項27】
前記増幅産物が電気泳動によって分離され、かつゲル上の1つもしくは複数のバンドを可視化することによって検出される請求項26に記載の方法。
【請求項28】
前記簡素化された核酸分子が実質的にシトシンを全く有しない請求項16〜27のいずれか一項に記載の方法。
【請求項29】
前記微生物に特異的な核酸分子が前記微生物において自然発生しないものである請求項28に記載の方法。
【請求項30】
前記微生物に特異的な核酸分子が前記微生物の分類レベルを示す核酸配列を有する請求項16〜29のいずれか一項に記載の方法。
【請求項31】
前記微生物の前記分類レベルが同一もしくは異なる地理的集団または底生性集団由来の科、属、種、株、タイプ、あるいは異なる集団を含む請求項30に記載の方法。
【請求項32】
微生物からDNA配列を得る工程と、
アデニン、グアニンおよびチミンの塩基を実質的に含むように、各シトシンをチミンに変化させることによって前記微生物DNA配列の変換を行うことにより、前記微生物DNA配列の簡素化形態を形成する工程と、
前記微生物DNAの簡素化形態から微生物に特異的な核酸分子を選択する工程と、
を含む微生物に特異的な核酸分子を生成するための方法。
【請求項33】
前記変換がin silicoで行われる請求項32に記載の方法。
【請求項34】
2種以上の微生物DNA配列の簡素化形態が得られ、かつ前記2種以上の配列を比較することで少なくとも1種の微生物に特異的な核酸分子が得られる請求項32または33に記載の方法。
【請求項35】
請求項32〜34のいずれか一項に記載の方法によって得られる微生物に特異的な核酸分子。
【請求項36】
試験もしくはアッセイにおいて前記微生物に特異的な核酸分子を結合または増幅するためのプローブあるいはプライマーを得ることを目的とした請求項32〜34のいずれか一項に記載の方法の使用。
【請求項37】
試料中の微生物の存在を検出するための方法であって、
前記微生物を含有すると見られる試料から微生物DNAを得る工程と、
シトシンを修飾する作用物質で前記微生物核酸を処理し、微生物核酸誘導体を形成する工程と、
所望の微生物に特異的な核酸分子の増幅を可能にする能力を有するプライマーを、前記微生物核酸誘導体に提供する工程と、
前記微生物核酸誘導体に対して増幅反応を行うことで簡素化された核酸を形成する工程と、
前記所望の微生物に特異的な核酸分子を有する増幅核酸産物の存在についてアッセイし、前記所望の微生物に特異的な核酸分子の検出が前記試料中での前記微生物の存在を示す工程と、
を含む、方法。
【請求項38】
前記微生物が、ファージ、ウイルス、ウイロイド、細菌、真菌、藻、原生動物、スピロヘータ、または単細胞生物から選択される請求項37に記載の方法。
【請求項39】
前記作用物質が非メチル化シトシンを修飾する請求項37または38に記載の方法。
【請求項40】
前記作用物質が、重亜硫酸塩、酢酸塩またはクエン酸塩から選択される請求項37〜39のいずれか一項に記載の方法。
【請求項41】
前記作用物質が重亜硫酸ナトリウムである請求項40に記載の方法。
【請求項42】
ポリメラーゼ連鎖反応(PCR)、等温増幅、またはシグナル増幅などの任意の適切な手段によって増幅が行われる請求項37〜41のいずれか一項に記載の方法。
【請求項43】
前記核酸分子が、
前記核酸分子の領域に結合可能な検出リガンドを提供し、かつ前記検出リガンドが前記領域に結合するのに十分な時間を与える工程と、
前記検出リガンドの前記核酸分子に対する結合を測定し、前記核酸分子の存在を検出する工程と、
によって検出される請求項37〜42のいずれか一項に記載の方法。
【請求項44】
前記核酸分子が、増幅産物を分離しかつ前記分離された産物を可視化することによって検出される請求項37〜43のいずれか一項に記載の方法。
【図1】


【図2】

【図3】

【図4】

【図5】

【図6】

【図7】

【図8】
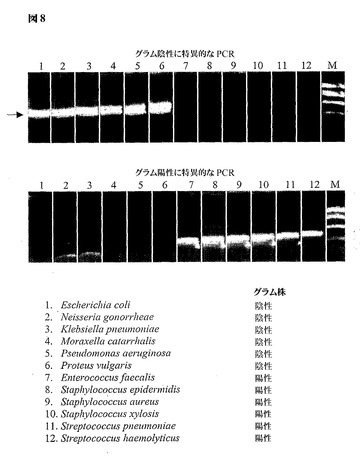
【図9】
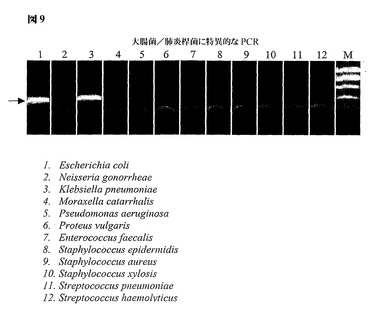
【図10】
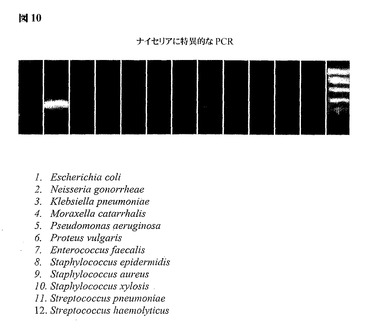
【図11】
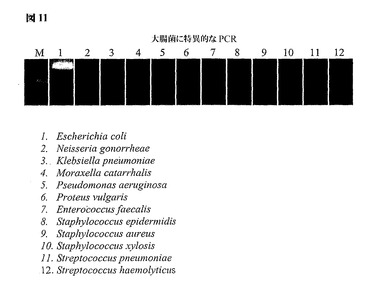
【図12】
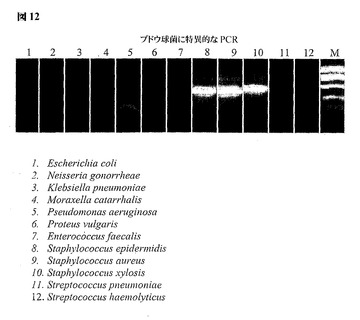
【図13】
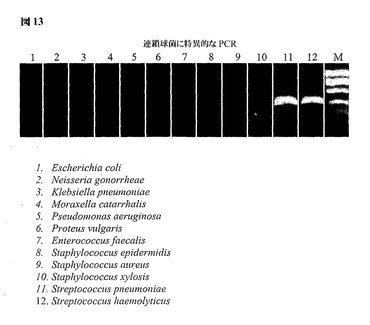
【図14】
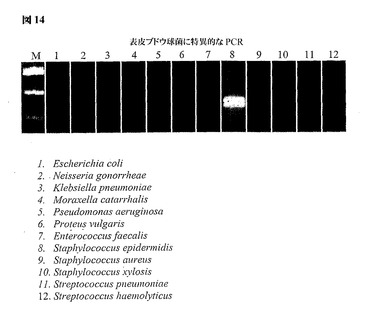
【図15】

【図16A】

【図16B】

【図17A】

【図17B】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A】
【図16B】
【図17A】
【図17B】
【公表番号】特表2008−521413(P2008−521413A)
【公表日】平成20年6月26日(2008.6.26)
【国際特許分類】
【出願番号】特願2007−543659(P2007−543659)
【出願日】平成17年12月5日(2005.12.5)
【国際出願番号】PCT/AU2005/001840
【国際公開番号】WO2006/058393
【国際公開日】平成18年6月8日(2006.6.8)
【出願人】(505188906)ヒューマン ジェネティック シグネチャーズ ピーティーワイ リミテッド (15)
【Fターム(参考)】
【公表日】平成20年6月26日(2008.6.26)
【国際特許分類】
【出願日】平成17年12月5日(2005.12.5)
【国際出願番号】PCT/AU2005/001840
【国際公開番号】WO2006/058393
【国際公開日】平成18年6月8日(2006.6.8)
【出願人】(505188906)ヒューマン ジェネティック シグネチャーズ ピーティーワイ リミテッド (15)
【Fターム(参考)】
[ Back to top ]