
シブトラミンのスルホン酸塩
物理化学的性質に優れたシブトラミンの新規スルホン酸塩を開示する。また、前記化合物の製造方法、および前記化合物を含む薬学的組成物を開示する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、シブトラミンの新規スルホン酸塩、前記化合物の製造方法、および前記化合物を有効成分として含む薬学的組成物に関する。
【背景技術】
【0002】
シブトラミン(N−[1−[1−(4−クロロフェニル)シクロブチル]−3−メチルブチル]−N,N−ジメチルアミン)は、うつ病、パーキンソン病、肥満症、インスリン非依存性糖尿病、癲癇などの治療に有用な治療剤であって、生体内5−ヒドロキシトリプタミン及びノルアドレナリン再吸収抑制剤である(Neuropharmacology, 28, p129-134)。また、シブトラミンは、満腹感を増加させて食物の摂取を減少させ、熱発生を促進させてエネルギー消費を増加させる、この二重作用により体重を軽減させる(Int. J. Obesity, 19, p145; Brit. J. Pharmacol. 114, p388)。シブトラミンのうつ病治療の使用は、英国特許第2098602号に記載されており、シブトラミンのパーキンソン病治療の使用は、PCT国際特許公開第88/06444号公報に記載されており、シブトラミンの脳機能疾患治療の使用は、米国特許第4939175号に記載されており、肥満の治療におけるシブトラミン塩酸塩の使用は、ヨーロッパ特許第397831号に記載されており、損傷した耐糖力、またはインスリン非依存性糖尿病に苦しんでいる患者の耐糖力を改善させるためのシブトラミンの使用は、PCT国際特許公開第95/20949号公報に記載されている。
【0003】
一般に、製剤学的に優れた物性の塩を製造するためには、(1)優れた溶解度、(2)優れた安定性、(3)優れた非吸湿性、および(4)錠剤剤形への加工性といった物理化学的基準を満足しなければならない。
【0004】
シブトラミンは、低い融点のために精製することが難しいので、シブトラミンを含む薬学的組成物の製造のためには、再結晶化によって精製できる結晶型物質を使用することが好ましい。韓国特許公報第1990−0000274号には、シブトラミンは、薬学的に許容される陰イオンを含有する非毒性酸付加塩を形成する酸から形成される塩として、例えば、塩酸塩、リンゴ酸塩、酢酸塩、クエン酸塩、フマル酸塩、酒石酸塩、琥珀酸塩、アスパラギン酸塩、グルタミン酸塩の形態で、使用できると開示されている。
【0005】
ところが、シブトラミン塩酸塩は、吸湿性があって製剤学的に取り扱うことが難しいため、薬物の製造に使用することが好ましくない。薬物の製造の際に、それぞれの剤形に一定重量の活性化合物が含有されるようにしなければならないが、周囲から水を吸収する活性成分は、そのような一貫性を実現することが難しい。韓国特許公報第94−8913号には、シブトラミン塩酸塩を一水和物として製造する場合、カプセル剤、錠剤および他の薬学的剤形の製造に適切な非吸湿性生成物を得ることができると開示されている。また、この韓国特許公報には、シブトラミン塩酸塩一水和物は、シブトラミン塩酸塩を、水からなる又は水を含有する媒質(水との不混和性溶媒又は水との混和性溶媒)と接触させることにより製造することができると記載されている。
【0006】
このように現在商用化されているシブトラミン塩酸塩一水和物は、これを製造するために、反応に一定量の水を投入して製造するか、あるいは先ずシブトラミン塩酸塩無水物を製造した後、水含有溶媒で長時間懸濁攪拌して一水和物を製造する一連の複雑な工程を経なければならないうえ、全く吸湿性のない正確な一水和物を製造するための工程上の難点がある。
【0007】
そこで、本発明者らは、かかる問題点を解決することが可能な新規シブトラミン塩を開発するために鋭意研究を重ねた結果、シブトラミンスルホン酸塩のうち、水和物の製造のために一定量の水を含ませなければならない煩わしい製造過程が不要な無水物形態のシブトラミンの、ベンゼンスルホン酸塩(ベシラート、benzenesulfonic acid salt)、カンファースルホン酸塩(カンシラート、(+)-(1S)-cmphor-10-sulfonic acid salt)、p−トルエンスルホン酸塩(トシラート、p-toluenesulfonic acid salt)およびエタンジスルホン酸塩(エジシラート、1,2-ethane disulfonic acid salt)が、優れた物理化学的性質(溶解度、非吸湿性、安定性)を持つことを見出した。また、シブトラミンスルホン酸のうち、水和物形態のシブトラミンエタンスルホン酸塩(エシラート、ethansulfonic acid salt)半水和物が、非吸湿性であり、安定性を持ち、水に対する溶解度がシブトラミン塩酸塩一水和物に比べて著しく高いことを見出した。
【発明の開示】
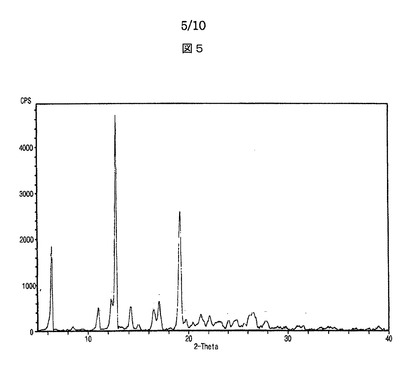
【0008】
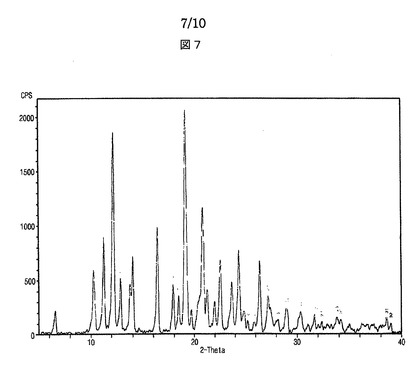
本発明の目的は、新規なシブトラミンスルホン酸塩を提供することにある。
【0009】
本発明の他の目的は、前記シブトラミンスルホン酸塩の製造方法を提供することにある。
【0010】
本発明の別の目的は、前記シブトラミンスルホン酸塩を活性成分として含む、肥満および肥満関連疾患の治療および予防のための薬学的組成物を提供することにある。本発明の別の目的は、前記シブトラミンスルホン酸塩を有効成分とする、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療するための薬学的組成物を提供することである。
【0011】
本発明の別の目的は、前記シブトラミンスルホン酸塩を活性成分として含む薬学的組成物を投与し、肥満および肥満関連疾患の病的状態を治療および予防する方法、並びにうつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療する方法を提供することである。
【0012】
(発明を実施するための最良の態様)
一つの態様として、本発明は、シブトラミンベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、およびエタンスルホン酸塩半水和物からなるシブトラミンスルホン酸塩の中から選択されるシブトラミンスルホン酸塩に関する。
【0013】
シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、およびエタンスルホン酸塩半水和物は、下記化学式1で表される。
【化002】

【0014】
本発明によるシブトラミンのベンゼンスルホン酸、p−トルエンスルホン酸塩、エタンジスルホン酸塩は、市販されているシブトラミン塩酸塩一水和物に比べて同等以上の非吸湿性、化学的および熱力学的安定性、製剤加工性(formulability)を示し、溶解度の面ではシブトラミン塩酸塩一水和物に比べて多少低下するが、許容可能な希釈剤または担体を含む薬学的組成物を製造した場合、製剤学的溶出または生体利用率の面では問題がない程度の十分な溶解度を示した。本発明によるシブトラミンのカンファースルホン酸塩、エタンスルホン酸塩半水和物は、シブトラミン塩酸塩一水和物に比べて同等以上の溶解度、非吸湿性、化学的および熱力学的安定性、製剤加工性を示し、特に、エタンスルホン酸塩半水和物は、蒸留水およびpH1.2、pH4.0、pH5.3、pH6.8、pH7.4の各水溶液でシブトラミン塩酸塩に比べて2倍以上の非常に優れた溶解度を示した。前記スルホン酸塩は、非吸湿性の面では10%、75%、90%の相対湿度で7日以上放置しても、水分増加または水分減少を全く示しておらず、安定性の面では60℃の温度で一ヶ月以上放置しても、不純物の生成および含量の変化がなく、光安定性の面でもやはり優れた結果を示した。
【0015】
本発明のシブトラミンスルホン酸塩の製造に使用されたベンゼンスルホン酸(besylate)、カンファースルホン酸(camsylate)、p−トルエンスルホン酸(tosylate)、エタンジスルホン酸(edisylate)、エタンスルホン酸(esylate)は、一般に医薬品に多用されており、米国FDAで認証された薬学的に使用可能な有機酸である。前記酸は、長期的な使用例と安全性が立証された毒性の少ない酸であって、シブトラミンの新規塩として長期間服用するには有用である。
【0016】
本発明のシブトラミンスルホン酸塩は、シブトラミン塩酸塩と同様に、ラセミ化合物であって、シブトラミンスルホン酸塩の光学的に純粋な(+)と(−)の光学異性体の約1:1混合物である。
【0017】
本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、結晶型または非結晶型である。結晶型のシブトラミンスルホン酸塩が、非吸湿性、熱力学的な安定性、流動性などの物性の面でさらに好ましい。
【0018】
具体的に、シブトラミンベンゼンスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.6、10.9、13.4、14.5、16.2、17.1、17.4、19.7、20.1、21.4、22.0、22.7、23.5、24.6、24.9、25.7、26.6、27.3及び32.9を示すことを特徴とする。
【0019】
シブトラミンカンファースルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.7、8.1、12.5、12.8、13.3、15.0、16.0、16.6、18.4、19.0、20.0、21.3、22.5、22.8、24.2、24.7、25.0、26.6、28.3及び33.8を示すことを特徴とする。
【0020】
シブトラミンp−トルエンスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.4、11.0、11.1、12.3、12.8、14.2、16.5、17.1、19.2、19.8、21.3、22.1、24.1、24.9、26.3、26.5及び27.8を示すことを特徴とする。
【0021】
シブトラミンエタンジスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.6、10.2、11.2、12.2、12.8、13.7、14.0、16.4、18.0、18.5、19.2、19.8、20.9、21.3、22.0、22.6、23.7、24.4、25.0、26.5、27.3、29.0及び30.3を示すことを特徴とする。
【0022】
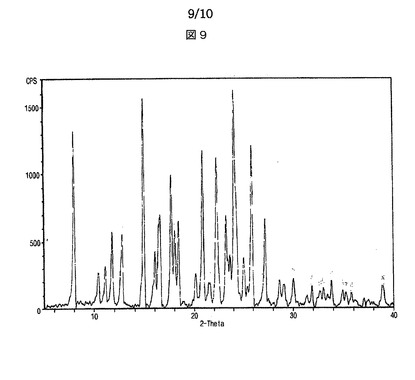
シブトラミンエタンスルホン酸塩半水和物は、X線回折分析においてピーク強度200以上の場合における2θの値が、8.0、10.4、11.1、11.7、12.7、14.9、16.0、16.5、17.6、18.0、18.4、20.1、20.8、22.3、23.2、23.4、23.6、24.1、25.0、25.9、27.2、28.6、30.0及び33.8を示すことを特徴とする。
【0023】
別の態様として、本発明は、前記シブトラミンスルホン酸塩の製造方法に関する。
【0024】
具体的に、本発明は、不活性溶媒中でシブトラミンと、ベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸およびエタンスルホン酸の中から選択されるスルホン酸とを反応させる段階を含む、シブトラミンスルホン酸塩の製造方法を含む。
【0025】
具体的に前記反応を反応式で表わすと、下記反応式1の通りである。
【化003】
【0026】
反応物として使用されるシブトラミンは、(−)−シブトラミンと(+)−シブトラミンの1:1混合物である。スルホン酸の中でも、カンファースルホン酸は、ラセミ化合物または光学的純粋物質であり、光学的に純粋な(+)−(1S)−カンファースルホン酸が好ましい。
【0027】
本発明の製造方法に使用されるスルホン酸は、以下のようなLD50およびLDL0値を持つことが公知である:ベンゼンスルホン酸は、1,157mg/kgのLD50(ラット、経口投与)、カンファースルホン酸は、2,502mg/kgのLD50(マウス、皮下注射)、p−トルエンスルホン酸は、2,480mg/kgのLD50(ラット、経口投与)、エタンジスルホン酸は、68mg/kgのLD50(マウス、静脈投与)、エタンスルホン酸は、48mg/kgのLD50(マウス、静脈投与)をそれぞれ持つ。これらの有機酸は、米国FDAで承認を受けて薬学的に使用可能な塩として様々な医薬品に用いられており、特に高血圧治療剤としてのアムロジピンとスルタミシリン、トスフロキサシン、クロールメチアゾール、エルゴトキシンなど、多様な適応症を持つ医薬品で長期間安全に使用されてきた。
【0028】
本発明の製造方法に使用可能な不活性溶媒には、酢酸エチル、メタノール、エタノール、イソプロパノール、アセトニトリル、ヘキサン、イソプロピルエーテル、t−ブチルメチルエーテルなどがあり、好ましくは酢酸エチルまたはエタノールである。これらの不活性溶媒は単独でまたは配合して使用することができる。
【0029】
前記不活性溶媒の中で、シブトラミン1当量に対して、スルホン酸1〜2当量、好ましくは1.02〜1.2当量を、−5〜40℃、好ましくは20〜30℃の反応温度で、0.5〜5時間、好ましくは1〜2時間、反応させることができる。
【0030】
前述したような本発明の製造方法によって、シブトラミンのスルホン酸塩を90%以上の収率と99%以上の高純度で製造することができる。
【0031】
本発明は、治療学的に有効な量のシブトラミンスルホン酸塩と薬学的に許容可能な希釈剤または担体を含む薬学的組成物、およびこれを投与して疾病を治療または予防する方法に関する。一態様として、本発明は、治療学的に有効な量のシブトラミンスルホン酸塩と薬学的に許容可能な希釈剤または担体を含む、肥満および肥満関連疾患を治療または予防するための薬学的組成物、およびこれを用いて肥満および肥満関連疾患の病的状態を治療または予防する方法を含む。
【0032】
本発明は、また、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含むうつ病治療用薬学的組成物、およびこれを投与してうつ病を治療する方法を含む。
【0033】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む、パーキンソン病を治療または予防するための薬学的組成物、およびこれを投与してパーキンソン病を治療する方法を含む。
【0034】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含むインスリン非依存性糖尿病治療用薬学的組成物、およびこれを投与してインスリン非依存性糖尿病を治療する方法を含む。
【0035】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む癲癇治療用薬学的組成物、およびこれを投与して癲癇を治療する方法を含む。
【0036】
本発明によるシブトラミンスルホン酸塩を活性成分として含む薬学的組成物において、好適な投与形態は経口投与であり、このような投与手段としては例えば、錠剤またはカプセルを挙げることができる。
【0037】
錠剤は、活性成分が、担体、希釈剤または賦形剤などと混合した後、錠剤化して製造することができる。この際、使用される適切な担体、希釈剤または賦形剤の実例としては、例えば、澱粉、糖およびマンニトールなどの崩解剤、燐酸カルシウムおよび珪酸誘導体などの充填剤および増量剤、例えばカルボキシメチルセルロースおよび他のセルロース誘導体、ゼラチンおよびポリビニルピロリドンなどの結合剤、並びに例えば滑石、カルシウムおよびステアリン酸マグネシウム、および固相ポリエチレングリコールなどの潤滑剤などを挙げることができる。また、前記の担体、希釈剤または賦形剤などの添加剤なしでまたは添加剤と共に活性成分を含有する硬質または軟質ゼラチンカプセルを、通常の方法によって製造することができる。
【0038】
薬学的組成物の活性成分として、化学式1における結晶型シブトラミンスルホン酸塩は、組成物250重量部に対して1〜50重量部を含むことが好ましい。例えば、総重量250mgの本発明による薬学的組成物の製造の際に活性成分として化学式1の結晶型シブトラミンスルホン酸塩10mg(シブトラミン含量基準)、微細結晶セルロース115mg、ラクトース115mg、二酸化珪素5mg、およびステアリン酸マグネシウム5mgを含むように製造することができる。しかしながら、前記薬学的組成物の組成比は、一つの例示に過ぎないので、これにより本発明の範囲が制限されるものではない。
【0039】
以下、本発明を実施例によってより詳細に説明する。しかし、これらの実施例は本発明を例示的に説明するためのもので、本発明の範囲を限定するものではない。
【0040】
(実施例)
本発明の製造方法によって、シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物を製造した後、吸湿性、溶解度、安定性、光安定性および結晶性などの物性をシブトラミン塩酸塩水和物と比較した。また、製剤加工性と溶出性パターンを調べるために、シブトラミンスルホン酸塩のカプセル剤を製造した。
【0041】
参考例1:シブトラミン塩酸塩一水和物の製造
韓国特許公告第90−00274号に記載された方法によって、シブトラミン塩酸塩無水和物を製造した。その後、韓国特許公告第94−08913号に記載の方法によって、前記で製造されたシブトラミン塩酸塩無水和物10gを、アセトン110mLおよび水1.2mLの沸騰混合物に溶解させた後、溶液を熱濾過(hot-filtration)し、溶媒80mLを蒸留除去して、濾液の容量を減少させた。濃縮液を濾過し、生成された固体を濾過して回収した後、真空中で乾燥させて、融点195℃の化学式2の化合物9.2g(収率:87%)を得た。
【実施例1】
【0042】
(シブトラミンベンゼンスルホン酸塩の製造)
シブトラミン(22.4g、0.08モル)を、酢酸エチル224mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、ベンゼンスルホン酸(12.64g、0.08モル)の酢酸エチル溶液124mLを徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物33.99g(収率:97.0%)を得た。
【0043】
得られたシブトラミンベンゼンスルホン酸塩の元素分析結果および融点は、次の通りである。
【表1】
【0044】
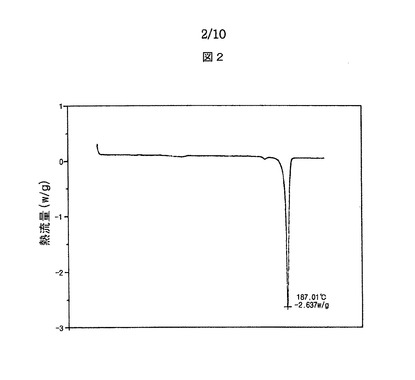
融点:187℃
NMR(δ、DMSO−d6):8.42(1H、s)、7.63〜7.32(9H、m)、3.76(1H、t)、2.83(3H、d)、2.55〜2.50(1H、d)、2.32〜2.30(2H、m)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.41(2H、m)、0.99(6H、t)
【実施例2】
【0045】
(シブトラミンカンファースルホン酸塩の製造)
シブトラミン(30.0g、0.107モル)を、酢酸エチル300mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、(1S)−(+)−カンファー−10−スルホン酸(24.9g、0.107モル)の酢酸エチル溶液250mLを、徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物52.31g(収率:95.4%)を得た。
【0046】
得られたシブトラミンカンファースルホン酸塩の元素分析結果および融点は、次の通りである。
【表2】
【0047】
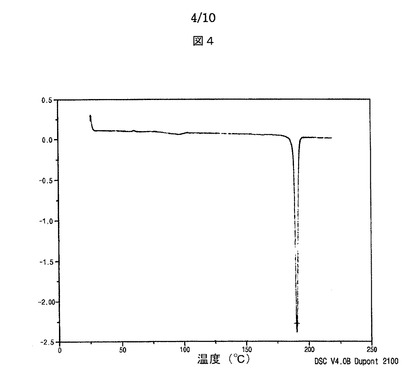
融点:190℃
NMR(δ、DMSO−d6):8.49(1H、s)、7.56〜7.49(4H、m)、3.76(1H、t)、2.87(1H、d)、2.83(3H、d)、2.75(1H、t)、2.55〜2.50(1H、d)、2.39(1H、d)、2.32〜2.20(3H、m)、2.13(3H、d)、1.95〜1.70(6H、m)、1.50〜1.28(4H、m)、1.06(3H、s)、0.99(6H、t)、0.76(3H、s)
【実施例3】
【0048】
(シブトラミンp−トルエンスルホン酸塩の製造)
シブトラミン(22.4g、0.08モル)を、酢酸エチル224mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、p−トルエンスルホン酸一水和物(15.2g、0.08モル)の酢酸エチル溶液160mLを、徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物35.14g(収率:97.1%)を得た。
【0049】
得られたシブトラミンp−トルエンスルホン酸塩の元素分析結果および融点は、次の通りである。
【表3】
【0050】
融点:185℃
NMR(δ、DMSO−d6):8.42(1H、s)、7.54〜7.48(6H、m)、7.13(2H、d)、3.76(1H、t)、2.83(3H、d)、2.50(1H、d)2.32〜2.30(2H、m)、2.30(3H、d)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例4】
【0051】
(シブトラミンエタンジスルホン酸塩の製造)
シブトラミン(24.0g、0.086モル)を、酢酸エチル240mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、1,2−エタンジスルホン酸(8.16g、0.043モル)の酢酸エチル溶液72mLを徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物27.40g(収率:85.0%)を得た。得られたシブトラミンエタンジスルホン酸塩の元素分析結果および融点は次の通りである。
【表4】
【0052】
融点:270℃
NMR(δ、DMSO−d6):8.51(1H、s)、7.55〜7.49(4H、m)、3.76(1H、t)、2.83(3H、d)、2.68(2H、s)、2.50(2H、d)、2.32(2H、t)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例5】
【0053】
(シブトラミンエタンスルホン酸塩半水和物の製造)
シブトラミン(20.0g、0.071モル)を、酢酸エチル80mL、t−ブチルメチルエーテル80mL及び蒸留水2mLの混合液に攪拌して溶解させた。溶液の温度を25℃に調節した後、エタンスルホン酸(8.68g、0.078モル)を徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的化合物26.0g(収率:93.9%)を得た。
【0054】
得られたシブトラミンエタンジスルホン酸塩半水和物の元素分析結果および融点は、次の通りである。
【表5】
【0055】
融点:174℃
NMR(δ、DMSO−d6):8.53(1H、s)、7.55〜7.48(4H、m)、3.76(1H、t)、2.83(3H、d)、2.51(2H、d)、2.42(2H、q)、2.33(2H、m)、2.13(3H、t)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例6】
【0056】
(シブトラミンベンゼンスルホン酸塩を含むカプセル剤の製造)
以下の表6に記載の配合の成分を混合し、シブトラミンベンゼンスルホン酸塩を含むカプセル剤を製造した。
【表6】
【0057】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例7】
【0058】
(シブトラミンカンファースルホン酸塩を含むカプセル剤の製造)
以下の表7に記載の配合の成分を混合し、シブトラミンカンファースルホン酸塩を含むカプセル剤を製造した。
【表7】
【0059】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例8】
【0060】
(シブトラミンp−トルエンスルホン酸塩を含むカプセル剤の製造)
以下の表8に記載の配合の成分を混合し、シブトラミンp−トルエンスルホン酸塩を含むカプセル剤を製造した。
【表8】
【0061】
前記各分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例9】
【0062】
(シブトラミンエタンジスルホン酸塩を含むカプセル剤の製造)
以下の表9に記載の配合の成分を混合し、シブトラミンエタンジスルホン酸塩を含むカプセル剤を製造した。
【表9】
【0063】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例10】
【0064】
(シブトラミンエタンスルホン酸塩半水和物を含むカプセル剤の製造)
以下の表10に記載の配合の成分を混合し、シブトラミンエタンスルホン酸塩半水和物を含むカプセル剤を製造した。
【表10】
【0065】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例11】
【0066】
(シブトラミンスルホン酸塩の吸湿性評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物の含水量(K.F.水分%)を、25℃にて様々な湿度条件(10%、75%及び90%RH)で3日間又は1週間曝露した後に測定し、その測定結果を表11に示した。
【表11】
【0067】
表11に示すように、シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、様々な湿度条件でシブトラミン塩酸塩一水和物と同様に水分の変化が殆どないことを示した。
【実施例12】
【0068】
(シブトラミンスルホン酸塩の溶解度評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物の溶解度を、様々なpH値の水溶液条件の下で測定し、その測定結果を表12に示した。
【表12】
【0069】
表12に示すように、蒸留水(DW)と様々なpHの緩衝液でシブトラミンのベンゼンスルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩の溶解度は、シブトラミン塩酸塩一水和物と比較して多少劣るが、カプセルのような薬学的剤形を製造するのに十分な溶解度(3mg/mL)以上であった。シブトラミンカンファースルホン酸塩の溶解度はシブトラミン塩酸塩一水和物と同様であった。シブトラミンエタンスルホン酸塩半水和物は、シブトラミン塩酸塩一水和物に比べて非常に優れた溶解度を示した。更に、5種のシブトラミンのスルホン酸塩のカプセル剤形はいずれも、比較溶出評価の結果、シブトラミン塩酸塩一水和物と同一の結果を得ることができた。
【実施例13】
【0070】
(シブトラミンスルホン酸塩の安定性評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物を、厳しい60℃での熱処理試験を行い、その結果を表13にまとめた。
【表13】
【0071】
HPLC分析条件は、次の通りである。
【0072】
UV検出の波長:225nm
カラム:オクタデシル・シリカゲルC18(4.6×150mm、5μm)
移動相:一水素化リン酸アンモニウム水溶液(0.05M、リン酸でpH6に調節) :アセトニトリル=35:65
流速 :1.0mL/分
表13に示すように、シブトラミンスルホン酸塩は、シブトラミン塩酸塩一水和物と同様に、厳しい60℃での熱処理試験で、含量の変化が殆どなかった。したがって、シブトラミンスルホン酸塩は、塩酸塩一水和物と同様に、高温での化学的安定性に優れることが分かった。
【実施例14】
【0073】
(シブトラミンスルホン酸塩の光安定性評価)
本実施例では、光安定性試験のために、実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物を、25℃でICHガイドラインに適した光安定性試験機器を用いて、蛍光に露出させたままで4週間保管した。その結果を表14に示す。
【表14】
【0074】
表14に示すように、シブトラミンスルホン酸塩の光安定性の評価のために、含量の変化をHPLCにより分析したところ、シブトラミンスルホン酸塩は、シブトラミン塩酸塩一水和物と同様に、光安定性に優れることが分かった。
【産業上の利用可能性】
【0075】
本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、優れた物理化学的性質、すなわち非吸湿性、溶解度、安定性、製剤加工性、結晶性を持つ。これらのシブトラミンスルホン酸塩は、水和物を製造するための追加工程が不要であり、エタンスルホン酸塩半水和物は、塩酸塩一水和物に比べて相当優れた溶解度を示す。したがって、本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、製造工程が単純であり、長期間保管が可能であり、薬学的剤形の製造に適切な一貫性を保つことができ、生態利用率が増進するなどの利点を持つ。
【0076】
また、新規なシブトラミンスルホン酸塩の製造の際に使用されるベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸、エタンスルホン酸は、製薬学的に長期間使用された例と安定性が立証された毒性の少ない酸であるので、前記製造された新規なシブトラミンスルホン酸塩を、毒性のおそれなしに長期間服用することができる。
【図面の簡単な説明】
【0077】
本発明の前記および他の目的、特徴および利点は、添付図面を参照して、詳細な説明より明らかに理解されるであろう。
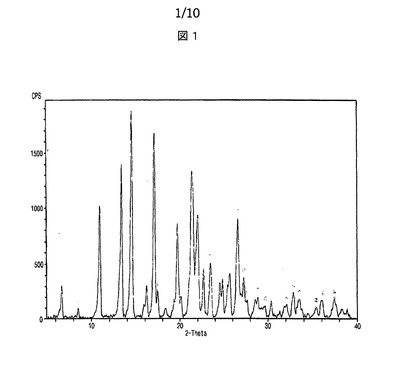
【図1】図1は、本発明によるシブトラミンベンゼンスルホン酸塩のX線回折スペクトルを示す。
【図2】図2は、本発明によるシブトラミンベンゼンスルホン酸塩の示差走査熱量計サーモグラムを示す。
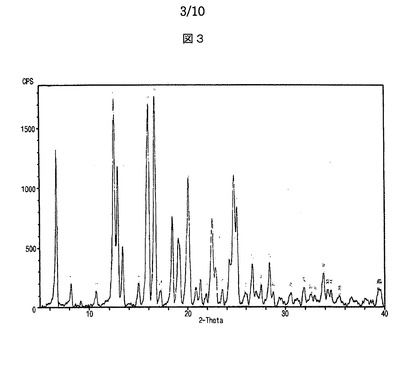
【図3】図3は、本発明によるシブトラミンカンファースルホン酸塩のX線回折スペクトルを示す。
【図4】図4は、本発明によるシブトラミンカンファースルホン酸塩の示差走査熱量計サーモグラムを示す。
【図5】図5は、本発明によるシブトラミンp−トルエンスルホン酸塩のX線回折スペクトルを示す。
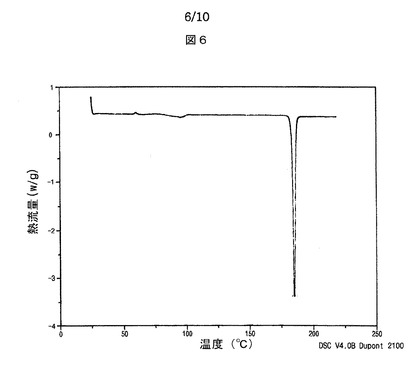
【図6】図6は、本発明によるシブトラミンp−トルエンスルホン酸塩の示差走査熱量計サーモグラムを示す。
【図7】図7は、本発明によるシブトラミンエタンジスルホン酸塩のX線回折スペクトルを示す。
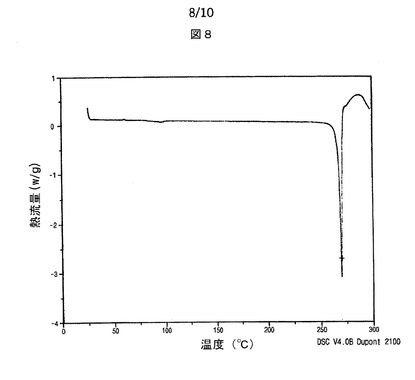
【図8】図8は、本発明によるシブトラミンエタンジスルホン酸塩の示差走査熱量計サーモグラムを示す。
【図9】図9は、本発明による本発明に係るシブトラミンエタンスルホン酸塩半水和物のX線回折スペクトルを示す。
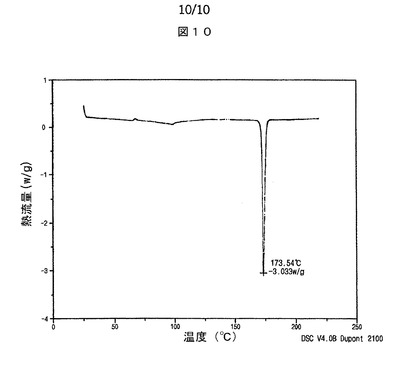
【図10】図10は、本発明によるシブトラミンエタンスルホン酸塩半水和物の示差走査熱量計サーモグラムを示す。
【技術分野】
【0001】
本発明は、シブトラミンの新規スルホン酸塩、前記化合物の製造方法、および前記化合物を有効成分として含む薬学的組成物に関する。
【背景技術】
【0002】
シブトラミン(N−[1−[1−(4−クロロフェニル)シクロブチル]−3−メチルブチル]−N,N−ジメチルアミン)は、うつ病、パーキンソン病、肥満症、インスリン非依存性糖尿病、癲癇などの治療に有用な治療剤であって、生体内5−ヒドロキシトリプタミン及びノルアドレナリン再吸収抑制剤である(Neuropharmacology, 28, p129-134)。また、シブトラミンは、満腹感を増加させて食物の摂取を減少させ、熱発生を促進させてエネルギー消費を増加させる、この二重作用により体重を軽減させる(Int. J. Obesity, 19, p145; Brit. J. Pharmacol. 114, p388)。シブトラミンのうつ病治療の使用は、英国特許第2098602号に記載されており、シブトラミンのパーキンソン病治療の使用は、PCT国際特許公開第88/06444号公報に記載されており、シブトラミンの脳機能疾患治療の使用は、米国特許第4939175号に記載されており、肥満の治療におけるシブトラミン塩酸塩の使用は、ヨーロッパ特許第397831号に記載されており、損傷した耐糖力、またはインスリン非依存性糖尿病に苦しんでいる患者の耐糖力を改善させるためのシブトラミンの使用は、PCT国際特許公開第95/20949号公報に記載されている。
【0003】
一般に、製剤学的に優れた物性の塩を製造するためには、(1)優れた溶解度、(2)優れた安定性、(3)優れた非吸湿性、および(4)錠剤剤形への加工性といった物理化学的基準を満足しなければならない。
【0004】
シブトラミンは、低い融点のために精製することが難しいので、シブトラミンを含む薬学的組成物の製造のためには、再結晶化によって精製できる結晶型物質を使用することが好ましい。韓国特許公報第1990−0000274号には、シブトラミンは、薬学的に許容される陰イオンを含有する非毒性酸付加塩を形成する酸から形成される塩として、例えば、塩酸塩、リンゴ酸塩、酢酸塩、クエン酸塩、フマル酸塩、酒石酸塩、琥珀酸塩、アスパラギン酸塩、グルタミン酸塩の形態で、使用できると開示されている。
【0005】
ところが、シブトラミン塩酸塩は、吸湿性があって製剤学的に取り扱うことが難しいため、薬物の製造に使用することが好ましくない。薬物の製造の際に、それぞれの剤形に一定重量の活性化合物が含有されるようにしなければならないが、周囲から水を吸収する活性成分は、そのような一貫性を実現することが難しい。韓国特許公報第94−8913号には、シブトラミン塩酸塩を一水和物として製造する場合、カプセル剤、錠剤および他の薬学的剤形の製造に適切な非吸湿性生成物を得ることができると開示されている。また、この韓国特許公報には、シブトラミン塩酸塩一水和物は、シブトラミン塩酸塩を、水からなる又は水を含有する媒質(水との不混和性溶媒又は水との混和性溶媒)と接触させることにより製造することができると記載されている。
【0006】
このように現在商用化されているシブトラミン塩酸塩一水和物は、これを製造するために、反応に一定量の水を投入して製造するか、あるいは先ずシブトラミン塩酸塩無水物を製造した後、水含有溶媒で長時間懸濁攪拌して一水和物を製造する一連の複雑な工程を経なければならないうえ、全く吸湿性のない正確な一水和物を製造するための工程上の難点がある。
【0007】
そこで、本発明者らは、かかる問題点を解決することが可能な新規シブトラミン塩を開発するために鋭意研究を重ねた結果、シブトラミンスルホン酸塩のうち、水和物の製造のために一定量の水を含ませなければならない煩わしい製造過程が不要な無水物形態のシブトラミンの、ベンゼンスルホン酸塩(ベシラート、benzenesulfonic acid salt)、カンファースルホン酸塩(カンシラート、(+)-(1S)-cmphor-10-sulfonic acid salt)、p−トルエンスルホン酸塩(トシラート、p-toluenesulfonic acid salt)およびエタンジスルホン酸塩(エジシラート、1,2-ethane disulfonic acid salt)が、優れた物理化学的性質(溶解度、非吸湿性、安定性)を持つことを見出した。また、シブトラミンスルホン酸のうち、水和物形態のシブトラミンエタンスルホン酸塩(エシラート、ethansulfonic acid salt)半水和物が、非吸湿性であり、安定性を持ち、水に対する溶解度がシブトラミン塩酸塩一水和物に比べて著しく高いことを見出した。
【発明の開示】
【0008】
本発明の目的は、新規なシブトラミンスルホン酸塩を提供することにある。
【0009】
本発明の他の目的は、前記シブトラミンスルホン酸塩の製造方法を提供することにある。
【0010】
本発明の別の目的は、前記シブトラミンスルホン酸塩を活性成分として含む、肥満および肥満関連疾患の治療および予防のための薬学的組成物を提供することにある。本発明の別の目的は、前記シブトラミンスルホン酸塩を有効成分とする、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療するための薬学的組成物を提供することである。
【0011】
本発明の別の目的は、前記シブトラミンスルホン酸塩を活性成分として含む薬学的組成物を投与し、肥満および肥満関連疾患の病的状態を治療および予防する方法、並びにうつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療する方法を提供することである。
【0012】
(発明を実施するための最良の態様)
一つの態様として、本発明は、シブトラミンベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、およびエタンスルホン酸塩半水和物からなるシブトラミンスルホン酸塩の中から選択されるシブトラミンスルホン酸塩に関する。
【0013】
シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、およびエタンスルホン酸塩半水和物は、下記化学式1で表される。
【化002】
【0014】
本発明によるシブトラミンのベンゼンスルホン酸、p−トルエンスルホン酸塩、エタンジスルホン酸塩は、市販されているシブトラミン塩酸塩一水和物に比べて同等以上の非吸湿性、化学的および熱力学的安定性、製剤加工性(formulability)を示し、溶解度の面ではシブトラミン塩酸塩一水和物に比べて多少低下するが、許容可能な希釈剤または担体を含む薬学的組成物を製造した場合、製剤学的溶出または生体利用率の面では問題がない程度の十分な溶解度を示した。本発明によるシブトラミンのカンファースルホン酸塩、エタンスルホン酸塩半水和物は、シブトラミン塩酸塩一水和物に比べて同等以上の溶解度、非吸湿性、化学的および熱力学的安定性、製剤加工性を示し、特に、エタンスルホン酸塩半水和物は、蒸留水およびpH1.2、pH4.0、pH5.3、pH6.8、pH7.4の各水溶液でシブトラミン塩酸塩に比べて2倍以上の非常に優れた溶解度を示した。前記スルホン酸塩は、非吸湿性の面では10%、75%、90%の相対湿度で7日以上放置しても、水分増加または水分減少を全く示しておらず、安定性の面では60℃の温度で一ヶ月以上放置しても、不純物の生成および含量の変化がなく、光安定性の面でもやはり優れた結果を示した。
【0015】
本発明のシブトラミンスルホン酸塩の製造に使用されたベンゼンスルホン酸(besylate)、カンファースルホン酸(camsylate)、p−トルエンスルホン酸(tosylate)、エタンジスルホン酸(edisylate)、エタンスルホン酸(esylate)は、一般に医薬品に多用されており、米国FDAで認証された薬学的に使用可能な有機酸である。前記酸は、長期的な使用例と安全性が立証された毒性の少ない酸であって、シブトラミンの新規塩として長期間服用するには有用である。
【0016】
本発明のシブトラミンスルホン酸塩は、シブトラミン塩酸塩と同様に、ラセミ化合物であって、シブトラミンスルホン酸塩の光学的に純粋な(+)と(−)の光学異性体の約1:1混合物である。
【0017】
本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、結晶型または非結晶型である。結晶型のシブトラミンスルホン酸塩が、非吸湿性、熱力学的な安定性、流動性などの物性の面でさらに好ましい。
【0018】
具体的に、シブトラミンベンゼンスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.6、10.9、13.4、14.5、16.2、17.1、17.4、19.7、20.1、21.4、22.0、22.7、23.5、24.6、24.9、25.7、26.6、27.3及び32.9を示すことを特徴とする。
【0019】
シブトラミンカンファースルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.7、8.1、12.5、12.8、13.3、15.0、16.0、16.6、18.4、19.0、20.0、21.3、22.5、22.8、24.2、24.7、25.0、26.6、28.3及び33.8を示すことを特徴とする。
【0020】
シブトラミンp−トルエンスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.4、11.0、11.1、12.3、12.8、14.2、16.5、17.1、19.2、19.8、21.3、22.1、24.1、24.9、26.3、26.5及び27.8を示すことを特徴とする。
【0021】
シブトラミンエタンジスルホン酸塩は、X線回折分析においてピーク強度200以上の場合における2θの値が、6.6、10.2、11.2、12.2、12.8、13.7、14.0、16.4、18.0、18.5、19.2、19.8、20.9、21.3、22.0、22.6、23.7、24.4、25.0、26.5、27.3、29.0及び30.3を示すことを特徴とする。
【0022】
シブトラミンエタンスルホン酸塩半水和物は、X線回折分析においてピーク強度200以上の場合における2θの値が、8.0、10.4、11.1、11.7、12.7、14.9、16.0、16.5、17.6、18.0、18.4、20.1、20.8、22.3、23.2、23.4、23.6、24.1、25.0、25.9、27.2、28.6、30.0及び33.8を示すことを特徴とする。
【0023】
別の態様として、本発明は、前記シブトラミンスルホン酸塩の製造方法に関する。
【0024】
具体的に、本発明は、不活性溶媒中でシブトラミンと、ベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸およびエタンスルホン酸の中から選択されるスルホン酸とを反応させる段階を含む、シブトラミンスルホン酸塩の製造方法を含む。
【0025】
具体的に前記反応を反応式で表わすと、下記反応式1の通りである。
【化003】
【0026】
反応物として使用されるシブトラミンは、(−)−シブトラミンと(+)−シブトラミンの1:1混合物である。スルホン酸の中でも、カンファースルホン酸は、ラセミ化合物または光学的純粋物質であり、光学的に純粋な(+)−(1S)−カンファースルホン酸が好ましい。
【0027】
本発明の製造方法に使用されるスルホン酸は、以下のようなLD50およびLDL0値を持つことが公知である:ベンゼンスルホン酸は、1,157mg/kgのLD50(ラット、経口投与)、カンファースルホン酸は、2,502mg/kgのLD50(マウス、皮下注射)、p−トルエンスルホン酸は、2,480mg/kgのLD50(ラット、経口投与)、エタンジスルホン酸は、68mg/kgのLD50(マウス、静脈投与)、エタンスルホン酸は、48mg/kgのLD50(マウス、静脈投与)をそれぞれ持つ。これらの有機酸は、米国FDAで承認を受けて薬学的に使用可能な塩として様々な医薬品に用いられており、特に高血圧治療剤としてのアムロジピンとスルタミシリン、トスフロキサシン、クロールメチアゾール、エルゴトキシンなど、多様な適応症を持つ医薬品で長期間安全に使用されてきた。
【0028】
本発明の製造方法に使用可能な不活性溶媒には、酢酸エチル、メタノール、エタノール、イソプロパノール、アセトニトリル、ヘキサン、イソプロピルエーテル、t−ブチルメチルエーテルなどがあり、好ましくは酢酸エチルまたはエタノールである。これらの不活性溶媒は単独でまたは配合して使用することができる。
【0029】
前記不活性溶媒の中で、シブトラミン1当量に対して、スルホン酸1〜2当量、好ましくは1.02〜1.2当量を、−5〜40℃、好ましくは20〜30℃の反応温度で、0.5〜5時間、好ましくは1〜2時間、反応させることができる。
【0030】
前述したような本発明の製造方法によって、シブトラミンのスルホン酸塩を90%以上の収率と99%以上の高純度で製造することができる。
【0031】
本発明は、治療学的に有効な量のシブトラミンスルホン酸塩と薬学的に許容可能な希釈剤または担体を含む薬学的組成物、およびこれを投与して疾病を治療または予防する方法に関する。一態様として、本発明は、治療学的に有効な量のシブトラミンスルホン酸塩と薬学的に許容可能な希釈剤または担体を含む、肥満および肥満関連疾患を治療または予防するための薬学的組成物、およびこれを用いて肥満および肥満関連疾患の病的状態を治療または予防する方法を含む。
【0032】
本発明は、また、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含むうつ病治療用薬学的組成物、およびこれを投与してうつ病を治療する方法を含む。
【0033】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む、パーキンソン病を治療または予防するための薬学的組成物、およびこれを投与してパーキンソン病を治療する方法を含む。
【0034】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含むインスリン非依存性糖尿病治療用薬学的組成物、およびこれを投与してインスリン非依存性糖尿病を治療する方法を含む。
【0035】
本発明は、治療学的に有効な量のシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む癲癇治療用薬学的組成物、およびこれを投与して癲癇を治療する方法を含む。
【0036】
本発明によるシブトラミンスルホン酸塩を活性成分として含む薬学的組成物において、好適な投与形態は経口投与であり、このような投与手段としては例えば、錠剤またはカプセルを挙げることができる。
【0037】
錠剤は、活性成分が、担体、希釈剤または賦形剤などと混合した後、錠剤化して製造することができる。この際、使用される適切な担体、希釈剤または賦形剤の実例としては、例えば、澱粉、糖およびマンニトールなどの崩解剤、燐酸カルシウムおよび珪酸誘導体などの充填剤および増量剤、例えばカルボキシメチルセルロースおよび他のセルロース誘導体、ゼラチンおよびポリビニルピロリドンなどの結合剤、並びに例えば滑石、カルシウムおよびステアリン酸マグネシウム、および固相ポリエチレングリコールなどの潤滑剤などを挙げることができる。また、前記の担体、希釈剤または賦形剤などの添加剤なしでまたは添加剤と共に活性成分を含有する硬質または軟質ゼラチンカプセルを、通常の方法によって製造することができる。
【0038】
薬学的組成物の活性成分として、化学式1における結晶型シブトラミンスルホン酸塩は、組成物250重量部に対して1〜50重量部を含むことが好ましい。例えば、総重量250mgの本発明による薬学的組成物の製造の際に活性成分として化学式1の結晶型シブトラミンスルホン酸塩10mg(シブトラミン含量基準)、微細結晶セルロース115mg、ラクトース115mg、二酸化珪素5mg、およびステアリン酸マグネシウム5mgを含むように製造することができる。しかしながら、前記薬学的組成物の組成比は、一つの例示に過ぎないので、これにより本発明の範囲が制限されるものではない。
【0039】
以下、本発明を実施例によってより詳細に説明する。しかし、これらの実施例は本発明を例示的に説明するためのもので、本発明の範囲を限定するものではない。
【0040】
(実施例)
本発明の製造方法によって、シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物を製造した後、吸湿性、溶解度、安定性、光安定性および結晶性などの物性をシブトラミン塩酸塩水和物と比較した。また、製剤加工性と溶出性パターンを調べるために、シブトラミンスルホン酸塩のカプセル剤を製造した。
【0041】
参考例1:シブトラミン塩酸塩一水和物の製造
韓国特許公告第90−00274号に記載された方法によって、シブトラミン塩酸塩無水和物を製造した。その後、韓国特許公告第94−08913号に記載の方法によって、前記で製造されたシブトラミン塩酸塩無水和物10gを、アセトン110mLおよび水1.2mLの沸騰混合物に溶解させた後、溶液を熱濾過(hot-filtration)し、溶媒80mLを蒸留除去して、濾液の容量を減少させた。濃縮液を濾過し、生成された固体を濾過して回収した後、真空中で乾燥させて、融点195℃の化学式2の化合物9.2g(収率:87%)を得た。
【実施例1】
【0042】
(シブトラミンベンゼンスルホン酸塩の製造)
シブトラミン(22.4g、0.08モル)を、酢酸エチル224mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、ベンゼンスルホン酸(12.64g、0.08モル)の酢酸エチル溶液124mLを徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物33.99g(収率:97.0%)を得た。
【0043】
得られたシブトラミンベンゼンスルホン酸塩の元素分析結果および融点は、次の通りである。
【表1】
【0044】
融点:187℃
NMR(δ、DMSO−d6):8.42(1H、s)、7.63〜7.32(9H、m)、3.76(1H、t)、2.83(3H、d)、2.55〜2.50(1H、d)、2.32〜2.30(2H、m)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.41(2H、m)、0.99(6H、t)
【実施例2】
【0045】
(シブトラミンカンファースルホン酸塩の製造)
シブトラミン(30.0g、0.107モル)を、酢酸エチル300mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、(1S)−(+)−カンファー−10−スルホン酸(24.9g、0.107モル)の酢酸エチル溶液250mLを、徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物52.31g(収率:95.4%)を得た。
【0046】
得られたシブトラミンカンファースルホン酸塩の元素分析結果および融点は、次の通りである。
【表2】
【0047】
融点:190℃
NMR(δ、DMSO−d6):8.49(1H、s)、7.56〜7.49(4H、m)、3.76(1H、t)、2.87(1H、d)、2.83(3H、d)、2.75(1H、t)、2.55〜2.50(1H、d)、2.39(1H、d)、2.32〜2.20(3H、m)、2.13(3H、d)、1.95〜1.70(6H、m)、1.50〜1.28(4H、m)、1.06(3H、s)、0.99(6H、t)、0.76(3H、s)
【実施例3】
【0048】
(シブトラミンp−トルエンスルホン酸塩の製造)
シブトラミン(22.4g、0.08モル)を、酢酸エチル224mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、p−トルエンスルホン酸一水和物(15.2g、0.08モル)の酢酸エチル溶液160mLを、徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物35.14g(収率:97.1%)を得た。
【0049】
得られたシブトラミンp−トルエンスルホン酸塩の元素分析結果および融点は、次の通りである。
【表3】
【0050】
融点:185℃
NMR(δ、DMSO−d6):8.42(1H、s)、7.54〜7.48(6H、m)、7.13(2H、d)、3.76(1H、t)、2.83(3H、d)、2.50(1H、d)2.32〜2.30(2H、m)、2.30(3H、d)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例4】
【0051】
(シブトラミンエタンジスルホン酸塩の製造)
シブトラミン(24.0g、0.086モル)を、酢酸エチル240mLに攪拌して溶解させた。溶液の温度を25℃に調節した後、1,2−エタンジスルホン酸(8.16g、0.043モル)の酢酸エチル溶液72mLを徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的物27.40g(収率:85.0%)を得た。得られたシブトラミンエタンジスルホン酸塩の元素分析結果および融点は次の通りである。
【表4】
【0052】
融点:270℃
NMR(δ、DMSO−d6):8.51(1H、s)、7.55〜7.49(4H、m)、3.76(1H、t)、2.83(3H、d)、2.68(2H、s)、2.50(2H、d)、2.32(2H、t)、2.13(3H、d)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例5】
【0053】
(シブトラミンエタンスルホン酸塩半水和物の製造)
シブトラミン(20.0g、0.071モル)を、酢酸エチル80mL、t−ブチルメチルエーテル80mL及び蒸留水2mLの混合液に攪拌して溶解させた。溶液の温度を25℃に調節した後、エタンスルホン酸(8.68g、0.078モル)を徐々に滴下した。反応溶液を25℃で2時間攪拌して、沈澱を生成させ、4℃で1時間攪拌した。生成された固体を減圧濾過し、酢酸エチル100mLで洗浄した。40℃で真空乾燥させて、目的化合物26.0g(収率:93.9%)を得た。
【0054】
得られたシブトラミンエタンジスルホン酸塩半水和物の元素分析結果および融点は、次の通りである。
【表5】
【0055】
融点:174℃
NMR(δ、DMSO−d6):8.53(1H、s)、7.55〜7.48(4H、m)、3.76(1H、t)、2.83(3H、d)、2.51(2H、d)、2.42(2H、q)、2.33(2H、m)、2.13(3H、t)、1.90(1H、m)、1.69(2H、m)、1.39(2H、m)、0.99(6H、t)
【実施例6】
【0056】
(シブトラミンベンゼンスルホン酸塩を含むカプセル剤の製造)
以下の表6に記載の配合の成分を混合し、シブトラミンベンゼンスルホン酸塩を含むカプセル剤を製造した。
【表6】
【0057】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例7】
【0058】
(シブトラミンカンファースルホン酸塩を含むカプセル剤の製造)
以下の表7に記載の配合の成分を混合し、シブトラミンカンファースルホン酸塩を含むカプセル剤を製造した。
【表7】
【0059】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例8】
【0060】
(シブトラミンp−トルエンスルホン酸塩を含むカプセル剤の製造)
以下の表8に記載の配合の成分を混合し、シブトラミンp−トルエンスルホン酸塩を含むカプセル剤を製造した。
【表8】
【0061】
前記各分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例9】
【0062】
(シブトラミンエタンジスルホン酸塩を含むカプセル剤の製造)
以下の表9に記載の配合の成分を混合し、シブトラミンエタンジスルホン酸塩を含むカプセル剤を製造した。
【表9】
【0063】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例10】
【0064】
(シブトラミンエタンスルホン酸塩半水和物を含むカプセル剤の製造)
以下の表10に記載の配合の成分を混合し、シブトラミンエタンスルホン酸塩半水和物を含むカプセル剤を製造した。
【表10】
【0065】
前記各成分を混合し、カプセル充填器(Bosche社製)を用いて硬質カプセルに充填した。
【実施例11】
【0066】
(シブトラミンスルホン酸塩の吸湿性評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物の含水量(K.F.水分%)を、25℃にて様々な湿度条件(10%、75%及び90%RH)で3日間又は1週間曝露した後に測定し、その測定結果を表11に示した。
【表11】
【0067】
表11に示すように、シブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、様々な湿度条件でシブトラミン塩酸塩一水和物と同様に水分の変化が殆どないことを示した。
【実施例12】
【0068】
(シブトラミンスルホン酸塩の溶解度評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物の溶解度を、様々なpH値の水溶液条件の下で測定し、その測定結果を表12に示した。
【表12】
【0069】
表12に示すように、蒸留水(DW)と様々なpHの緩衝液でシブトラミンのベンゼンスルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩の溶解度は、シブトラミン塩酸塩一水和物と比較して多少劣るが、カプセルのような薬学的剤形を製造するのに十分な溶解度(3mg/mL)以上であった。シブトラミンカンファースルホン酸塩の溶解度はシブトラミン塩酸塩一水和物と同様であった。シブトラミンエタンスルホン酸塩半水和物は、シブトラミン塩酸塩一水和物に比べて非常に優れた溶解度を示した。更に、5種のシブトラミンのスルホン酸塩のカプセル剤形はいずれも、比較溶出評価の結果、シブトラミン塩酸塩一水和物と同一の結果を得ることができた。
【実施例13】
【0070】
(シブトラミンスルホン酸塩の安定性評価)
実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物を、厳しい60℃での熱処理試験を行い、その結果を表13にまとめた。
【表13】
【0071】
HPLC分析条件は、次の通りである。
【0072】
UV検出の波長:225nm
カラム:オクタデシル・シリカゲルC18(4.6×150mm、5μm)
移動相:一水素化リン酸アンモニウム水溶液(0.05M、リン酸でpH6に調節) :アセトニトリル=35:65
流速 :1.0mL/分
表13に示すように、シブトラミンスルホン酸塩は、シブトラミン塩酸塩一水和物と同様に、厳しい60℃での熱処理試験で、含量の変化が殆どなかった。したがって、シブトラミンスルホン酸塩は、塩酸塩一水和物と同様に、高温での化学的安定性に優れることが分かった。
【実施例14】
【0073】
(シブトラミンスルホン酸塩の光安定性評価)
本実施例では、光安定性試験のために、実施例1〜5で製造したシブトラミンスルホン酸塩およびシブトラミン塩酸塩一水和物を、25℃でICHガイドラインに適した光安定性試験機器を用いて、蛍光に露出させたままで4週間保管した。その結果を表14に示す。
【表14】
【0074】
表14に示すように、シブトラミンスルホン酸塩の光安定性の評価のために、含量の変化をHPLCにより分析したところ、シブトラミンスルホン酸塩は、シブトラミン塩酸塩一水和物と同様に、光安定性に優れることが分かった。
【産業上の利用可能性】
【0075】
本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、優れた物理化学的性質、すなわち非吸湿性、溶解度、安定性、製剤加工性、結晶性を持つ。これらのシブトラミンスルホン酸塩は、水和物を製造するための追加工程が不要であり、エタンスルホン酸塩半水和物は、塩酸塩一水和物に比べて相当優れた溶解度を示す。したがって、本発明のシブトラミンのベンゼンスルホン酸塩、カンファースルホン酸塩、p−トルエンスルホン酸塩、エタンジスルホン酸塩、エタンスルホン酸塩半水和物は、製造工程が単純であり、長期間保管が可能であり、薬学的剤形の製造に適切な一貫性を保つことができ、生態利用率が増進するなどの利点を持つ。
【0076】
また、新規なシブトラミンスルホン酸塩の製造の際に使用されるベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸、エタンスルホン酸は、製薬学的に長期間使用された例と安定性が立証された毒性の少ない酸であるので、前記製造された新規なシブトラミンスルホン酸塩を、毒性のおそれなしに長期間服用することができる。
【図面の簡単な説明】
【0077】
本発明の前記および他の目的、特徴および利点は、添付図面を参照して、詳細な説明より明らかに理解されるであろう。
【図1】図1は、本発明によるシブトラミンベンゼンスルホン酸塩のX線回折スペクトルを示す。
【図2】図2は、本発明によるシブトラミンベンゼンスルホン酸塩の示差走査熱量計サーモグラムを示す。
【図3】図3は、本発明によるシブトラミンカンファースルホン酸塩のX線回折スペクトルを示す。
【図4】図4は、本発明によるシブトラミンカンファースルホン酸塩の示差走査熱量計サーモグラムを示す。
【図5】図5は、本発明によるシブトラミンp−トルエンスルホン酸塩のX線回折スペクトルを示す。
【図6】図6は、本発明によるシブトラミンp−トルエンスルホン酸塩の示差走査熱量計サーモグラムを示す。
【図7】図7は、本発明によるシブトラミンエタンジスルホン酸塩のX線回折スペクトルを示す。
【図8】図8は、本発明によるシブトラミンエタンジスルホン酸塩の示差走査熱量計サーモグラムを示す。
【図9】図9は、本発明による本発明に係るシブトラミンエタンスルホン酸塩半水和物のX線回折スペクトルを示す。
【図10】図10は、本発明によるシブトラミンエタンスルホン酸塩半水和物の示差走査熱量計サーモグラムを示す。
【特許請求の範囲】
【請求項1】
下記化学式1の構造を持つシブトラミンのスルホン酸塩。
【化001】
【請求項2】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.6、10.9、13.4、14.5、16.2、17.1、17.4、19.7、20.1、21.4、22.0、22.7、23.5、24.6、24.9、25.7、26.6、27.3、32.9を示すシブトラミンベンゼンスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項3】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.7、8.1、12.5、12.8、13.3、15.0、16.0、16.6、18.4、19.0、20.0、21.3、22.5、22.8、24.2、24.7、25.0、26.6、28.3、33.8を示すシブトラミンカンファースルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項4】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.4、11.0、11.1、12.3、12.8、14.2、16.5、17.1、19.2、19.8、21.3、22.1、24.1、24.9、26.3、26.5、27.8を示すシブトラミンp−トルエンスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項5】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.6、10.2、11.2、12.2、12.8、13.7、14.0、16.4、18.0、18.5、19.2、19.8、20.9、21.3、22.0、22.6、23.7、24.4、25.0、26.5、27.3、29.0、30.3を示すシブトラミンエタンジスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項6】
X線回折分析において、ピーク強度200以上の場合における2θの値が、8.0、10.4、11.1、11.7、12.7、14.9、16.0、16.5、17.6、18.0、18.4、20.1、20.8、22.3、23.2、23.4、23.6、24.1、25.0、25.9、27.2、28.6、30.0、33.8を示すシブトラミンエタンスルホン酸塩半水和物であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項7】
不活性溶媒中でシブトラミンに、ベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸およびエタンスルホン酸の中から選択されるスルホン酸を反応させる段階を含み、請求項1に記載の化学式1で表されるシブトラミンのスルホン酸塩を製造する方法。
【請求項8】
請求項1に記載の化学式1で表されるシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む、肥満および肥満関連疾患、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇の治療または予防のための薬学的組成物。
【請求項9】
錠剤またはカプセル剤の剤形であることを特徴とする、請求項8に記載の薬学的組成物。
【請求項10】
請求項8に記載の薬学的組成物を投与して、肥満および肥満関連疾患、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療または予防する方法。
【請求項1】
下記化学式1の構造を持つシブトラミンのスルホン酸塩。
【化001】
【請求項2】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.6、10.9、13.4、14.5、16.2、17.1、17.4、19.7、20.1、21.4、22.0、22.7、23.5、24.6、24.9、25.7、26.6、27.3、32.9を示すシブトラミンベンゼンスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項3】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.7、8.1、12.5、12.8、13.3、15.0、16.0、16.6、18.4、19.0、20.0、21.3、22.5、22.8、24.2、24.7、25.0、26.6、28.3、33.8を示すシブトラミンカンファースルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項4】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.4、11.0、11.1、12.3、12.8、14.2、16.5、17.1、19.2、19.8、21.3、22.1、24.1、24.9、26.3、26.5、27.8を示すシブトラミンp−トルエンスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項5】
X線回折分析において、ピーク強度200以上の場合における2θの値が、6.6、10.2、11.2、12.2、12.8、13.7、14.0、16.4、18.0、18.5、19.2、19.8、20.9、21.3、22.0、22.6、23.7、24.4、25.0、26.5、27.3、29.0、30.3を示すシブトラミンエタンジスルホン酸塩であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項6】
X線回折分析において、ピーク強度200以上の場合における2θの値が、8.0、10.4、11.1、11.7、12.7、14.9、16.0、16.5、17.6、18.0、18.4、20.1、20.8、22.3、23.2、23.4、23.6、24.1、25.0、25.9、27.2、28.6、30.0、33.8を示すシブトラミンエタンスルホン酸塩半水和物であることを特徴とする、請求項1に記載のシブトラミンのスルホン酸塩。
【請求項7】
不活性溶媒中でシブトラミンに、ベンゼンスルホン酸、カンファースルホン酸、p−トルエンスルホン酸、エタンジスルホン酸およびエタンスルホン酸の中から選択されるスルホン酸を反応させる段階を含み、請求項1に記載の化学式1で表されるシブトラミンのスルホン酸塩を製造する方法。
【請求項8】
請求項1に記載の化学式1で表されるシブトラミンのスルホン酸塩および薬学的に許容可能な希釈剤または担体を含む、肥満および肥満関連疾患、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇の治療または予防のための薬学的組成物。
【請求項9】
錠剤またはカプセル剤の剤形であることを特徴とする、請求項8に記載の薬学的組成物。
【請求項10】
請求項8に記載の薬学的組成物を投与して、肥満および肥満関連疾患、うつ病、パーキンソン病、インスリン非依存性糖尿病または癲癇を治療または予防する方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2008−526835(P2008−526835A)
【公表日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願番号】特願2007−550300(P2007−550300)
【出願日】平成18年1月6日(2006.1.6)
【国際出願番号】PCT/KR2006/000072
【国際公開番号】WO2006/073291
【国際公開日】平成18年7月13日(2006.7.13)
【出願人】(508075890)シージェー チェイルジェダン コーポレーション (4)
【Fターム(参考)】
【公表日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願日】平成18年1月6日(2006.1.6)
【国際出願番号】PCT/KR2006/000072
【国際公開番号】WO2006/073291
【国際公開日】平成18年7月13日(2006.7.13)
【出願人】(508075890)シージェー チェイルジェダン コーポレーション (4)
【Fターム(参考)】
[ Back to top ]