
ポリヒドロシランの製造方法
【課題】SiH4を高活性且つ高効率に重合して高分子量のポリヒドロシランを製造する方法を提供すること。
【解決手段】上記方法は、4族メタロセンと、アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と、を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする。上記方法によって製造されたポリヒドロシランは、シリコンの前駆体として好適に使用することができる。
【解決手段】上記方法は、4族メタロセンと、アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と、を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする。上記方法によって製造されたポリヒドロシランは、シリコンの前駆体として好適に使用することができる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ポリヒドロシランの製造方法に関する。より詳しくは、メタロセン触媒を用いて、シラン(SiH4)を高活性且つ高効率に重合してポリヒドロシランを製造する方法に関する。
【背景技術】
【0002】
集積回路、薄膜トランジスタなどの半導体デバイスに応用されるシリコン膜の形成は、CVD(Chemical Vapor Deposition)法などの気相プロセスにより行われている。しかしこの方法によると、大掛かりな真空装置が必要であること、原料の使用効率が低いことなどのコスト上の問題がある。
近年、シリコンの前駆体としてポリヒドロシランを用いる液相プロセスが提案された(非特許文献1および2)。この技術は、ポリヒドロシランの溶液を基板上に塗布して得た塗膜を非酸化性雰囲気下で加熱することにより、シリコン膜を形成する技術である。この技術によると、真空装置が不要であり、原料化合物の使用効率が高いことに加え、液体特有の性質を利用した加工法(例えば印刷法を利用したパターン状シリコン膜の形成)が可能であることなど、種々の利点があり、実用化が期待されている。
シリコンの前駆体である上記ポリヒドロシランは、ケイ素原子数5または6のシクロシラン(SinH2n、n=5または6)の光重合によって合成される(特許文献1)。このシクロシランは、例えばジフェニルジクロロシランを適当なアルカリ金属(例えばナトリウム、リチウムなど)の存在下で環化した後、適当な水素化剤(例えば水素化リチウムアルミニウム)によって水素化することによって合成することができる(特許文献2)。しかしながら、原料のジフェニルジクロロシラン自体が高価であり、その水素化工程において多量の還元剤を必要とするほか、環化工程・水素化工程の双方において多量の廃棄物を生じるため、コストおよび環境の両面からの課題を有する。
【0003】
ポリヒドロシランを用いる場合に生ずるこれらの問題を解決するには、ポリヒドロシランを安価なシラン(モノシラン、SiH4)から直接に合成することが最も望ましい。
SiH4からポリヒドロシランを合成した例が今までに皆無であったわけではない。例えば非特許文献3には、SiH4ガス中で無声放電を行うことによりポリヒドロシランが生成することが記載されている。また特許文献3には、SiH4を350〜460℃に加熱することによるポリヒドロシランの合成方法が記載されている。しかし、これらの文献に記載された方法の主生成物はいずれもジシランであり、トリシラン以上のポリヒドロシランの収率は極めて低い。これらの方法においては、無声放電または高温加熱によって高分子量のポリヒドロシランが生成したとしても、このような高エネルギー条件下ではポリヒドロシランが容易に分解すると考えられる。従って、本願が所期する用途に必要な量の高分子量のポリヒドロシランを、これらの方法によって製造しようとすることは現実的ではない。
そこで、遷移金属触媒を使用する脱水素縮合重合により、SiH4から高分子量のポリヒドロシランを直接合成することが望まれる。触媒反応は、一般に穏和な条件で進行するため、一旦生成した高分子量ポリヒドロシランの分解を回避することができる。また、脱水素縮合重合の副生成物は水素のみであるから、クリーンな反応が期待できる。
この点、有機シランの触媒的脱水素縮合重合は多数報告されており、触媒として遷移金属錯体を用いた場合に比較的良好な結果が得られることが知られている。例えば二塩化ジルコノセンをn−ブチルリチウムで処理して得られる「根岸試薬」は、一級または二級の有機シランの脱水素縮合重合に高い活性を示す(非特許文献4)。
しかしながら、SiH4の触媒的脱水素縮合重合を効率的に行って、高分子量のポリヒドロシランを製造することのできる技術は、未だ提案されていない。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−313299号公報
【特許文献2】特開2001−262058号公報
【特許文献3】特表2008−536784号公報
【非特許文献】
【0005】
【非特許文献1】Nature,440,pp783〜786(2006)
【非特許文献2】J.Non−Cryst.Solids,354,pp2623〜2626(2008)
【非特許文献3】Inorg.Chem.,1,pp432〜433(1962)
【非特許文献4】Organometallics,10,pp924〜930(1991)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明者らの検討により、根岸試薬などの公知のメタロセン触媒を用いてSiH4の重合を行っても、触媒の初期活性が十分ではなく、さらに重合開始後すぐに触媒の失活が起こるため、ポリヒドロシランを高収率で得ることはできないことが分かった。
本発明は、このような事実が明らかになったことに基づいてなされたものであり、その目的は、SiH4を高活性且つ高効率に重合してポリヒドロシランを製造する方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明によれば、本発明の上記目的および利点は、
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする、前記方法によって達成される。
【発明の効果】
【0008】
本発明の方法で用いられる触媒は、初期活性が高く、且つ触媒寿命が長いものである。従って、このような触媒を用いて行う本発明の方法によってSiH4を重合することにより、高活性、高収率でポリヒドロシランを製造することができる。
本発明の方法によって製造されたポリヒドロシランは、シリコンの前駆体として、例えば半導体装置の製造などに好適に使用することができる。
【図面の簡単な説明】
【0009】
【図1】実施例2で得られた固体を試料として測定したATR−IRチャート。
【図2】実施例2で得られた固体を試料として測定した29Si CP−MAS NMRチャート。
【図3】実施例4の実験例a〜dにおける、重合反応転化率の経時的変化を示すグラフ。
【図4】実施例4の実験例e〜iにおける、重合反応転化率の経時的変化を示すグラフ。
【図5】実施例4の実験例jにおける、重合反応転化率の経時的変化を示すグラフ。
【図6】実施例5の実験例a〜dおよび比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図7】実施例5の実験例e〜gおよび比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図8】実施例6の実験例a〜dおよび実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図9】実施例6の実験例eおよびfならびに実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図10】実施例6の実験例gおよびhならびに実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図11】実施例7で得られた0価シリコンのXPSチャート。
【図12】実施例9で得られた膜状物のラマン分光チャート。
【発明を実施するための形態】
【0010】
本発明は上記のとおり、
4族メタロセンと
アルキルリチウムおよびアルキルアルミニウムから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法に関する。
上記4族メタロセンとアルキル化剤との接触は、SiH4が存在する条件下で行われる。4族メタロセンとアルキル化剤との接触は、SiH4のほかにさらにルイス塩基が存在する条件下で行ってもよい。
<4族メタロセン>
本発明で使用される4族メタロセンとしては、中心金属がチタニウム、ジルコニウムまたはハフニウムであるメタロセンを好ましく使用することができ、例えば下記式(1−1)および(1−2)
【0011】
【化1】

【0012】
(上記式中、Mはチタニウム、ジルコニウムまたはハフニウムであり;
L1およびL2は、それぞれ独立に、シクロペンタジエニル基、インデニル基またはフルオレニル基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
L3およびL4は、それぞれ独立に、シクロペンタジエニレン基、インデニレン基またはフルオレニレン基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
Rはメチレン基、炭素数2〜4のアルキレン基またはジメチルシリレン基であり;そして
Xはハロゲン原子である。)
のそれぞれで表される錯体などを挙げることができ、これらのうちから選択される少なくとも1種を使用することができる。
上記式(1−1)中のL1およびL2としては、それぞれ、シクロペンタジエニル基、t−ブチルシクロペンタジエニル基、トリメチルシリルシクロペンタジエニル基、ペンタメチルシクロペンタジエニル基またはインデニル基であることが;
上記式(1−2)中のL3およびL4としては、それぞれ、インデニレン基であることが好ましい。
上記式(1−1)および(1−2)中のXとしては、塩素原子、臭素原子またはヨウ素原子であることが好ましく、塩素原子であることがより好ましい。
【0013】
本発明で使用される4族メタロセンの具体例としては、例えば
ジシクロペンタジエニルチタニウムジクロリド(Cp2TiCl2)、
ビス(t−ブチルシクロペンタジエニル)チタニウムジクロリド(tBuCp2TiCl2)、
ビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)、
ビス(インデニル)チタニウムジクロリド(Ind2TiCl2)、
ジシクロペンタジエニルジルコニウムジクロリド(Cp2ZrCl2)、
ビス(t−ブチルシクロペンタジエニル)ジルコニウムジクロリド(tBuCp2ZrCl2)、
ビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)、
ビス(インデニル)ジルコニウムジクロリド(Ind2ZrCl2)、
ansa−エチレンビス(インデニル)ジルコニウムジクロリド(ansa−Ind2ZrCl2)、
ビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBuCp2HfCl2)などを挙げることができ、これらのうちから選択される1種以上を好ましく使用することができる。
4族メタロセンの使用割合は、SiH41モルに対して、1×10−4〜1×10−1モルとすることが好ましく、1×10−3〜5×10−2モルとすることがより好ましい。
【0014】
<アルキル化剤>
本発明で使用されるアルキル化剤は、アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選択される。
上記アルキルリチウムとしては、炭素数2〜6のアルキル基を有するアルキルリチウムが好ましい。このようなアルキルリチウムとしては、例えばエチルリチウム、n−プロピルリチウム、i−プロピルリチウム、n−ブチルリチウム、sec−ブチルリチウム、iso−ブチルリチウム、n−ペンチルリチウム、n−ヘキシルリチウムなどを挙げることができる。
上記アルキルアルミニウムとしては、炭素数2〜6のアルキル基を有するジアルキルアルミニウムヒドリド、炭素数2〜6のアルキル基を有するトリアルキルアルミニウムなどを挙げることができる。ここで、炭素数2〜6のアルキル基としては、例えばエチル基、n−プロピル基、i−プロピル基、n−ブチル基、sec−ブチル基、iso−ブチル基、n−ペンチル基、n−ヘキシル基などを挙げることができる。アルキルアルミニウムとしては、炭素数3〜5のアルキル基を有するトリアルキルアルミニウムが好ましい。
上記アルキルマグネシウムとしては、例えば炭素数2〜6のアルキル基を有するアルキルマグネシウムを挙げることができる。
上記アルキルマグネシウムハライドとしては、例えば炭素数2〜6のアルキル基を有するアルキルマグネシウムクロリド、アルキルマグネシウムブロミド、アルキルマグネシウムヨージドを挙げることができる。
本発明におけるアルキル化剤としては、アルキルリチウムおよびアルキルアルミニウムから選択されるアルキル化剤を使用することが好ましく、炭素数3〜5の直鎖のアルキル基を有するアルキルリチウムを使用することがより好ましく、特に好ましくはn−ブチルリチウムである。
アルキル化剤の使用割合は、4族メタロセン1モルに対して、1〜20モルとすることが好ましく、1.5〜10モルとすることがより好ましく、2〜3モルとすることがさらに好ましい。
【0015】
<ルイス塩基>
本発明において任意的に使用されるルイス塩基としては、窒素原子、酸素原子、リン原子または硫黄原子を1〜6個有する有機化合物を挙げることができ、これらのうちから選択される少なくとも1種を使用することが好ましい。
上記窒素原子を有する有機化合物としては、芳香族環を構成するメンバーとして窒素原子を有する複素芳香族化合物、トリアルキルアミン、テトラアルキルアルキレンジアミン、ペンタアルキルジアルキレントリアミンなどを挙げることができる。このトリアルキルアミン、テトラアルキルアルキレンジアミン、ペンタアルキルジアルキレントリアミンの有するアルキル基としては、炭素数1〜6のアルキル基であることが好ましく、炭素数1〜3のアルキル基であることがより好ましく;
アルキレン基としては、炭素数2〜6のアルキル基であることが好ましく、1,2−エチレン基または1,2−プロピレン基であることがより好ましい。
【0016】
本発明におけるルイス塩基としての窒素原子を有する有機化合物の具体例としては、上記窒素原子を有する複素芳香族化合物として例えばピリジン、ピリミジン、ピラジン、トリアジン、ビピリジン、ターピリジン、ビキノリン、フェナントロリンなどを;
トリアルキルアミンとして例えばトリメチルアミン、トリエチルアミン、トリイソペンチルメチルアミン、ピペラジン、1,8−ビス(ジメチルアミノ)ナフタレンなどを;
テトラアルキルアルキレンジアミンとして例えばN,N,N’,N’−テトラメチルエチレンジアミンなどを;
ペンタアルキルジアルキレントリアミンとして例えばN,N,N’,N”,N”−ペンタメチルジエチレントリアミンなどを、それぞれ挙げることができる。
酸素原子を有する有機化合物としては、例えば芳香族環を構成するメンバーとして酸素原子を有する複素芳香族化合物、エーテルなどを挙げることができる。上記エーテルとしては、ジアルキルエーテル、ジアルコキシアルキレン、ポリアルキレンオキシドジアルキルエーテル、環状エーテルなどを挙げることができる。上記ジアルキルエーテル、ジアルコキシアルキレンおよびポリアルキレンオキシドジアルキルエーテルの有するアルキル基またはアルコキシ基の炭素数は、1〜6であることが好ましく、1〜3であることがより好ましい。
【0017】
本発明におけるルイス塩基としての酸素原子を有する有機化合物の具体例としては、
上記酸素原子を有する複素芳香族化合物として例えばフランなどを;
ジアルキルエーテル鎖状のモノエーテルとして例えばジメチルエーテル、ジエチルエーテル、メチルエチルエーテル、ジイソプロピルエーテルなどを;
ジアルコキシアルキレンとして例えばジメトキシエタン、ジエトキシエタンなどを;
ポリアルキレンオキシドジアルキルエーテルとして例えばジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテルなどを;
環状エーテルとして例えばテトラヒドロフラン、テトラヒドロピランなどを、それぞれ挙げることができる。
【0018】
リン原子を有する有機化合物としては、例えば下記式(P−1)または(P−2)
R1R2R3P (P−1)
R4R5P−R8−PR6R7 (P−2)
(上記式中、R1〜R7は、それぞれ独立に、炭素数1〜6のアルキル基または炭素数とヘテロ原子数との合計が6〜12の芳香族基であり;
R8はメチレン基または炭素数2〜6のアルキレン基である。)
で表される化合物を挙げることができる。
上記式(P−1)で表される化合物としては、例えばジフェニルメチルホスフィン、トリフェニルホスフィン、ジフェニルピリジルホスフィン、トリス(ジメチルアミノ)ホスフィンなどを;
上記式(P−2)で表される化合物としては、例えば1,2−ビス(ジフェニルホスフィノ)エタン、1,2−ビス(ジメチルホスフィノ)エタンなどを、それぞれ挙げることができる。
硫黄原子を有する有機化合物としては、例えばジメチルスルフィド、ジエチルスルフィド、チオフェン、チオベンゾフェノン、スルフランなどを挙げることができる。
【0019】
本発明におけるルイス塩基としては、窒素原子、酸素原子またはリン原子を1〜6個有する有機化合物よりなる群から選択される少なくとも1種を使用することが好ましく、特にトリエチルアミン、N,N,N’,N’−テトラメチルエチレンジアミン、N,N,N’,N”,N”−ペンタメチルジエチレントリアミン、ピリジン、ビピリジン、ビキノリン、ジメトキシエタン、ジフェニルメチルホスフィン、1,2−ビス(ジフェニルホスフィノ)エタンなどを挙げることができ、これらのうちから選択される少なくとも1種を使用することがより好ましい。
ルイス塩基の使用割合は、4族メタロセン1モルに対して、100モル以下とすることが好ましく、0.1〜50モルとすることがより好ましい。ルイス塩基のさらに好ましい使用割合は、使用するルイス塩基の種類に応じて異なり、4族メタロセン1モルに対する使用量としてそれぞれ以下のとおりである。
窒素原子を有する有機化合物:さらに好ましくは0.5〜10モル、特に好ましくは1〜5モル
酸素原子または硫黄原子を有する有機化合物:さらに好ましくは1〜50モル、特に好ましくは5〜20モル
リン原子を有する有機化合物:さらに好ましくは0.5〜10モル、特に好ましくは1〜5モル
【0020】
<4族メタロセンとアルキル化剤との接触>
本発明のポリヒドロシランの製造方法において使用される触媒は、上記のごとき4族メタロセンと、アルキル化剤とを、SiH4が存在する条件下で接触させることにより調製される。4族メタロセンとアルキル化剤との接触は、SiH4のほかにさらにルイス塩基が存在する条件下で行ってもよい。
このSiH4および任意的にルイス塩基の存在下における4族メタロセンとアルキル化剤との接触は、SiH4の重合を行う重合反応容器中で行ってもよく、あるいは重合反応容器以外の別容器中で接触を行った後に、接触後の混合物を重合反応容器中に添加してもよい。この4族メタロセンとアルキル化剤との接触は、溶媒の存在下または不存在下に行うことができるが、より高活性の種を均一に生成するとの観点から、溶媒の存在下に行うことが好ましい。
4族メタロセンとアルキル化剤との接触を重合反応容器以外の別容器中で行う場合、該容器中に存在すべきSiH4の量は、4族メタロセンの1モルに対して、2モル以上とすることが好ましく、5〜10モルとすることがより好ましい。
【0021】
4族メタロセンとアルキル化剤との接触において使用することのできる溶媒としては、脂肪族炭化水素溶媒、芳香族炭化水素溶媒などを挙げることができる。上記脂肪族炭化水素溶媒の具体例としては例えばn−ヘキサン、シクロヘキサン、シクロヘキセン、n−ヘプタン、n−オクタン、シクロオクタン、シクロオクテン、n−デカン、デカリンなどを;
上記芳香族炭化水素溶媒の具体例としては例えばベンゼン、トルエン、キシレン、インダン、テトラリンなどを、それぞれ挙げることができ、これらのうちから選択される1種以上を使用することができる。
溶媒の使用割合としては、4族メタロセン1モルに対して、10L以上とすることが好ましく、80〜1,000Lとすることがより好ましく、さらに100〜1,000Lとすることが好ましい。なお、上記式(1−1)および(1−2)のそれぞれで表される、ジハロゲン化物である4族メタロセンは上記の溶媒に対する溶解度が低く、溶媒の使用割合を4族メタロセン1モルに対して80L未満とすると4族メタロセンは溶解しきらずに固体として残存することがある。しかしながらアルキル化剤と接触してアルキル化された後の4族メタロセンの溶解度は一般に高いから、溶媒の使用割合が少ない場合であっても接触後には均一な溶液を得ることができる。
接触温度は−78〜50℃とすることが好ましく、−78〜30℃とすることがより好ましい。
【0022】
<SiH4の重合>
4族メタロセンとアルキル化剤との接触を重合反応容器中で行った場合には、場合により一定の誘導期間を経た後、SiH4の重合がそのまま開始される。
一方、4族メタロセンとアルキル化剤との接触を重合反応容器以外の別容器中で行う場合、その接触時間を有意に長くする必要はなく、接触後直ちに(通常は数秒以内に)触媒活性種が生成するから、該接触混合物を重合反応容器に注入することによって、SiH4の重合が開始される。
SiH4の重合に際しては、重合反応容器に、4族メタロセンとアルキル化剤との接触の際に使用することのできる溶媒として上記したものと同様の溶媒を添加して希釈下で重合することも許容される。
SiH4の重合の際に使用することのできる溶媒の割合は、溶液中のSiH4仕込み量が20重量%以下となる割合とすることが好ましく、10重量%以下となる割合とすることがより好ましく、特に5重量%以下となる割合とすることが好ましい。
重合温度は、好ましくは−78〜100℃であり、より好ましくは−5〜50℃である。
重合時間は、好ましくは0.5〜72時間であり、より好ましくは4〜20時間である。
【0023】
<ポリヒドロシラン>
上記のようにして、好ましくは固体状のポリヒドロシランを得ることができる。このポリヒドロシランは、必要に応じて適当な精製方法によって触媒残滓などの不純物を除去した後、シリコンの前駆体として好適に使用することができる。
上記精製方法としては、例えば溶媒洗浄を挙げることができる。この溶媒洗浄に使用される溶媒としては、例えばエーテル、芳香族炭化水素、アミンなどを挙げることができる。これらの具体例としては、エーテルとして例えばテトラヒドロフラン、テトラヒドロピラン、ジオキサン、1,2−ジメトキシエタン、アニソールなどを;
芳香族炭化水素として例えばトルエン、キシレン、テトラリンなどを;
アミンとして例えばトリエチルアミン、N,N,N’,N’−テトラメチルエチレンジアミン、N,N,N’,N”,N”−ペンタメチルジエチレントリアミンなどを、それぞれ挙げることができる。溶媒の使用割合は、ポリヒドロシラン100重量部に対して、100〜10,000重量部とすることが好ましく、2,000〜5,000重量部とすることがより好ましい。
溶媒洗浄の際には、例えば震とう、回転翼の使用、超音波の印加などの適当な撹拌手段により、洗浄液を撹拌することが好ましい。
溶媒洗浄は、使用する溶媒が液体状態を維持することのできる温度および圧力において、10分以上行うことが好ましく、30〜90分行うことがより好ましい。
溶媒洗浄は、1回または複数回行うことができ、ポリヒドロシラン中の不純物量を可及的に低減する観点から、2回以上行うことが好ましく、3〜5回行うことがより好ましい。
上記の如き本発明の方法で得られ、好ましくは精製されたポリヒドロシランは、これを非酸化性雰囲気中で加熱することにより、容易に0価シリコンに変換することができる。非酸化性雰囲気とは、例えば不活性気体中または不活性気体と還元性気体との混合気体中を挙げることができ、前記不活性気体としては、例えば窒素、ヘリウム、アルゴンなどを;
前記還元性気体としては、例えば水素などを、それぞれ挙げることができる。加熱温度は例えば340℃以上、好ましくは400〜600℃とすることができ、加熱時間は例えば15分以上、好ましくは30〜90分とすることができる。
【実施例】
【0024】
シラン(SiH4)は高圧ガス保安法にて指定された特殊高圧ガスに該当するため、以下の重合反応はすべて、国立大学法人北陸先端科学技術大学院大学(石川県能美市)内に設置され、石川県知事の許可を受けたオートクレーブ(内容量約27mLまたは約70mL、耐圧約0.3MPaG)内で、室温(23〜27℃)にて行った。このオートクレーブは、チャージ/サンプリング用セプタム、給気管および排気管を有し、排気管から排出されたガスは乾式除害設備によって無害化されたうえで外気に排出される。
【0025】
実施例1および比較例1では、触媒成分を混合する順番の影響を調べた。実施例1は本発明の方法であり、比較例1はメタロセン触媒による重合において通常適用される方法である。
実施例1
圧力0.01MPaGのSiH4ガスを充填した内容積27mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)のトルエン溶液(濃度7.5mmol/L)の4mLをシリンジを用いて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジを用いて注入し、重合を開始した。ここで使用したCp*2TiCl2およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、3モル%および6モル%に相当する。
重合開始から4時間後に、セプタムから気体をサンプリングし、これをガスクロマトグラフィー(GC)で分析してSiH4の脱水素縮合重合により発生した水素の量を定量し、この値を用いた計算によりSiH4の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して74.3モル%であった。反応系から少量の固体を回収した。
【0026】
比較例1
内容量6mLのガラス容器中で、Cp*2TiCl2のトルエン溶液(濃度7.5mmol/L)の4mLおよびn−BuLiのトルエン溶液(濃度60mmol/L)の1mLを混合して触媒溶液を得た。
圧力0.01MPaGのSiH4ガスを充填したオートクレーブ内に、セプタムから上記触媒溶液の全量をシリンジを用いて注入して重合を開始し、実施例1と同様にして反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率はSiH4の仕込み量に対して8.5モル%であった。反応系から少量の固体を回収した。
【0027】
実施例2および3では、使用するメタロセンの種類を変えて検討した。
実施例2
Cp*2TiCl2の代わりにビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)を使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を行い、反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して76.2モル%であった。反応系から少量の固体を回収した。
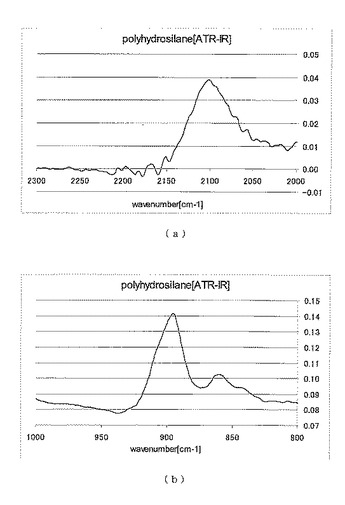
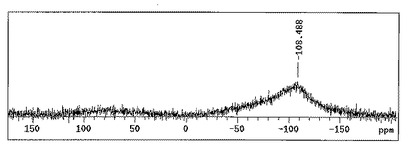
この反応系から回収した固体を試料として測定したATR−IR(減衰全反射赤外分光)チャートを図1(a)および(b)に、
固体NMR(29Si CP−MAS(交差分極−マジック角回転) NMR、80MHz、装置はVarian Inc.製を使用)チャートを図2に、それぞれ示した。
これらの結果から、回収された固体はポリヒドロシランであると考えられる。
【0028】
実施例3
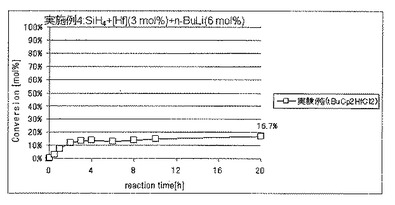
Cp*2TiCl2の代わりにビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBu−Cp2HfCl2)を使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を行い、反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して16.7モル%であった。反応系から少量の固体を回収した。
【0029】
実施例4では、種々のメタロセンを用いて、SiH4の重合反応転化率の経時的変化を追跡した。
実施例4
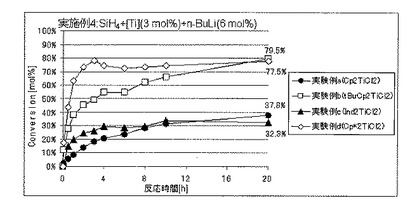
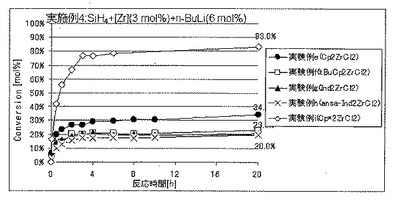
Cp*2TiCl2の代わりに各種のメタロセンを使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を開始した。実施例1における、セプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実験例の、反応開始20時間後におけるSiH4の仕込み量に対する重合反応転化率を表1に、SiH4の重合反応転化率の経時的変化を図3〜5に、それぞれ示した。
【0030】
各実験例において使用したメタロセンの種類は、以下のとおりである。カッコ内は、表1および図3〜5における表記である。
実験例a:ジシクロペンタジエニルチタニウムジクロリド(Cp2TiCl2)
実験例b:ビス(t−ブチルシクロペンタジエニル)チタニウムジクロリド(tBuCp2TiCl2)
実験例c:ビス(インデニル)チタニウムジクロリド(Ind2TiCl2)
実験例d:ビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)
実験例e:ジシクロペンタジエニルジルコニウムジクロリド(Cp2ZrCl2)
実験例f:ビス(t−ブチルシクロペンタジエニル)ジルコニウムジクロリド(tBuCp2ZrCl2)
実験例g:ビス(インデニル)ジルコニウムジクロリド(Ind2ZrCl2)
実験例h:ansa−エチレンビス(インデニル)ジルコニウムジクロリド(ansa−Ind2ZrCl2)
実験例i:ビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)
実験例j:ビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBuCp2HfCl2)
【0031】
【表1】
【0032】
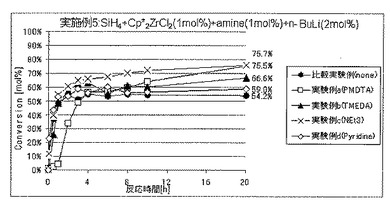
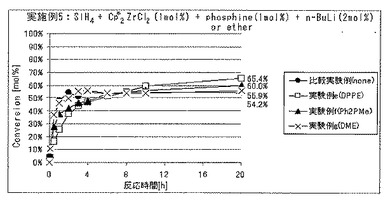
実施例5では、ルイス塩基を添加した場合の効果について調べた。実験例a〜dがルイス塩基としてアミンを添加した場合であり、
実施例e〜fがルイス塩基としてホスフィンを添加した場合であり、
実施例gがルイス塩基としてエーテルを添加した場合である。比較実験例はルイス塩基を添加しなかった場合である。
実施例5(実験例a〜g)
圧力0.01MPaGのSiH4ガスを充填した内容積25mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)のトルエン溶液(濃度10mmol/L)の3mLおよび各種ルイス塩基のトルエン溶液(溶液濃度および溶液使用量は後述)をシリンジにて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジにて注入し、重合を開始した。ここで使用したCp*2ZrCl2、ルイス塩基およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、1モル%、1モル%および2モル%に相当する。
反応開始後、実施例4と同様にしてセプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実験例における、ルイス塩基の種類、反応開始20時間後のSiH4の仕込み量に対する重合反応転化率を表2に、SiH4の重合反応転化率の経時的変化を図6および7に、それぞれ示した。
【0033】
各実験例において使用したルイス塩基の種類、溶液濃度および溶液使用量は以下のとおりである。カッコ内は、表2ならびに図6および7における表記である。
実験例a:N,N,N’,N”,N”−ペンタメチルジエチレントリアミン(PMDTA)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例b:N,N,N’,N’−テトラメチルエチレンジアミン(TMEDA)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例c:トリエチルアミン(NEt3)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例d:ピリジン(Py)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例e:1,2−ビス(ジフェニルホスフィノ)エタン(DPPE)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例f:ジフェニルメチルホスフィン(Ph2PMe)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例g:1,2−ジメトキシエタン(DME)、溶液濃度=30mmol/L、溶液使用量=1mL
実施例5(比較実験例)
ルイス塩基を使用しなかったほかは、上記実験例a〜gと同様にして実施し、SiH4の重合反応転化率を経時的に求めた。このときの、反応開始20時間後におけるSiH4の仕込み量に対する重合反応転化率を表2に、重合反応転化率の経時的変化を図6および7に示した。
【0034】
【表2】
【0035】
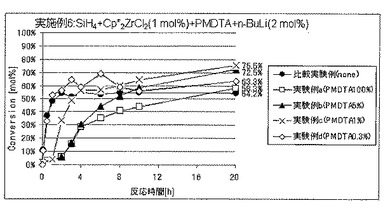
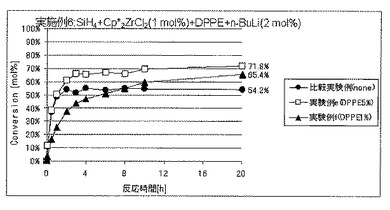
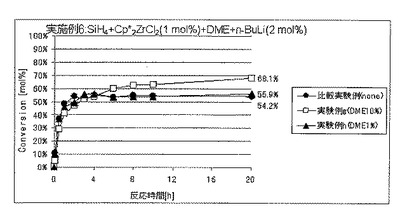
実施例6では、ルイス塩基の添加量の効果について調べた。
実験例a〜dはルイス塩基としてN,N,N’,N”,N”−ペンタメチルジエチレントリアミン(PMDTA)を用いた場合であり、
実験例eおよびfはルイス塩基として1,2−ビス(ジフェニルホスフィノ)エタン(DPPE)を用いた場合であり、そして
実験例gおよびhはルイス塩基として1,2−ジメトキシエタン(DME)を用いた場合である。
実施例6(実験例a〜h)
圧力0.01MPaGのSiH4ガスを充填した内容積25mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)のトルエン溶液(濃度10mol/L)の3mLおよび各種ルイス塩基のトルエン溶液(溶液濃度はそれぞれ上記実施例5におけるのと同じであり、溶液使用量は変量としてルイス塩基量がそれぞれ表3に記載の値となるようにした。)をシリンジにて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジにて注入し、重合を開始した。ここで使用したCp*2ZrCl2およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、1モル%および2モル%に相当する。
反応開始後、実施例4と同様にしてセプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実施例における、ルイス塩基の種類およびSiH4の仕込み量に対する添加量、反応開始20時間後のSiH4の仕込み量に対する重合反応転化率を表3に、SiH4の重合反応転化率の経時的変化を図8〜10に、それぞれ示した。表3ならびに図8〜10には、上記実施例5における比較実験例の結果も合わせて示した。
【0036】
【表3】
【0037】
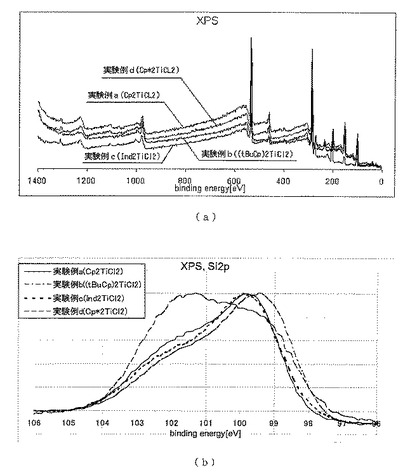
実施例7
本実施例では、本発明の方法によって得られたポリヒドロシランがシリコンの前駆体として好適であることを検証した。
上記実施例4の実験例a〜dで回収した各固体を、窒素雰囲気下、450℃において1.5時間加熱することにより、暗褐色の粉末をそれぞれ得た。
この粉末を試料として、以下の条件でX線光電子分光(XPS)分析を行った。
測定装置:Phi5600 ESCA System(ULVAC−PHI Inc.製)
得られたXPSチャートを図11(a)および(b)に示した。結合エネルギー99〜100eV付近にSi(0価)のピークが、102〜104eV付近にSi(4価)の微弱なピークが、それぞれ観察されたことから、上記加熱後の粉末は0価のシリコンであることが確認され、本発明の方法によって得られたポリヒドロシランがシリコンの前駆体として好適であることが分かった。
【0038】
実施例8
本実施例では、本発明の方法によって得られたポリヒドロシランを前駆体として製造されたシリコンが高純度のものであることを検証した。
上記実施例2で回収した固体約30mgにテトラヒドロフラン2mLを加え、懸濁液とした。得られた懸濁液につき、超音波液体処理機Sonifier150(Branson Ultrasonics社製、23kHz)を用いて出力2〜4Wにて30分間超音波洗浄した。洗浄後の懸濁液につき、小型微量遠心機PMC−060((株)トミー精工製、遠心加速度約2,000G)を用いて固体と洗浄液とに遠心分離した。この固体を回収し、同様の手法でさらに2回超音波洗浄/遠心分離操作を行った。
上記のようにして合計3回洗浄した固体を窒素雰囲気のグローブボックス中に搬入し、450℃において1時間加熱したところ、25.1mgの暗褐色の粉末が得られた。
得られた粉末を試料として、誘導結合プラズマ質量分析(ICP−MS)によって金属不純物の含有量を分析したところ、Zr=16mg/g、Li=2.4mg/gであった。
【0039】
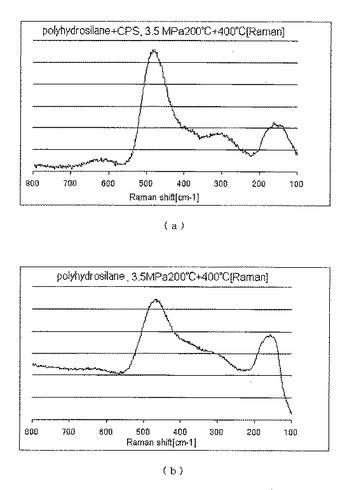
実施例9
本実施例では、本発明の方法によって得られたポリヒドロシランを用いてシリコン膜を形成した。
上記実施例2で回収した固体を、実施例8におけるのと同様にしてテトラヒドロフランで洗浄したうえ、その約20mgをシクロペンタシラン約20mgと混合して、膜形成用組成物を得た。
窒素雰囲気のグローブボックス中で、シリコン基板上に、上記の膜形成用組成物を配置し、その上に、約1cm四方の平滑なSiO2製鋳型を載せて、室温において0.5MPaGにて5分、次いで200℃において3.5MPaGにて15分加圧して型押しした。その後鋳型を除去し、基板上の成形物を、ホットプレートにて窒素下、400℃において1時間加熱したところ、膜状物が得られた。
この膜状物について測定したラマン分光チャートを図12(a)に示した。
また、シクロペンタシランを用いなかったほかは上記と同様にして得られたものについて測定したラマン分光チャートを図12(b)に示した。
【技術分野】
【0001】
本発明は、ポリヒドロシランの製造方法に関する。より詳しくは、メタロセン触媒を用いて、シラン(SiH4)を高活性且つ高効率に重合してポリヒドロシランを製造する方法に関する。
【背景技術】
【0002】
集積回路、薄膜トランジスタなどの半導体デバイスに応用されるシリコン膜の形成は、CVD(Chemical Vapor Deposition)法などの気相プロセスにより行われている。しかしこの方法によると、大掛かりな真空装置が必要であること、原料の使用効率が低いことなどのコスト上の問題がある。
近年、シリコンの前駆体としてポリヒドロシランを用いる液相プロセスが提案された(非特許文献1および2)。この技術は、ポリヒドロシランの溶液を基板上に塗布して得た塗膜を非酸化性雰囲気下で加熱することにより、シリコン膜を形成する技術である。この技術によると、真空装置が不要であり、原料化合物の使用効率が高いことに加え、液体特有の性質を利用した加工法(例えば印刷法を利用したパターン状シリコン膜の形成)が可能であることなど、種々の利点があり、実用化が期待されている。
シリコンの前駆体である上記ポリヒドロシランは、ケイ素原子数5または6のシクロシラン(SinH2n、n=5または6)の光重合によって合成される(特許文献1)。このシクロシランは、例えばジフェニルジクロロシランを適当なアルカリ金属(例えばナトリウム、リチウムなど)の存在下で環化した後、適当な水素化剤(例えば水素化リチウムアルミニウム)によって水素化することによって合成することができる(特許文献2)。しかしながら、原料のジフェニルジクロロシラン自体が高価であり、その水素化工程において多量の還元剤を必要とするほか、環化工程・水素化工程の双方において多量の廃棄物を生じるため、コストおよび環境の両面からの課題を有する。
【0003】
ポリヒドロシランを用いる場合に生ずるこれらの問題を解決するには、ポリヒドロシランを安価なシラン(モノシラン、SiH4)から直接に合成することが最も望ましい。
SiH4からポリヒドロシランを合成した例が今までに皆無であったわけではない。例えば非特許文献3には、SiH4ガス中で無声放電を行うことによりポリヒドロシランが生成することが記載されている。また特許文献3には、SiH4を350〜460℃に加熱することによるポリヒドロシランの合成方法が記載されている。しかし、これらの文献に記載された方法の主生成物はいずれもジシランであり、トリシラン以上のポリヒドロシランの収率は極めて低い。これらの方法においては、無声放電または高温加熱によって高分子量のポリヒドロシランが生成したとしても、このような高エネルギー条件下ではポリヒドロシランが容易に分解すると考えられる。従って、本願が所期する用途に必要な量の高分子量のポリヒドロシランを、これらの方法によって製造しようとすることは現実的ではない。
そこで、遷移金属触媒を使用する脱水素縮合重合により、SiH4から高分子量のポリヒドロシランを直接合成することが望まれる。触媒反応は、一般に穏和な条件で進行するため、一旦生成した高分子量ポリヒドロシランの分解を回避することができる。また、脱水素縮合重合の副生成物は水素のみであるから、クリーンな反応が期待できる。
この点、有機シランの触媒的脱水素縮合重合は多数報告されており、触媒として遷移金属錯体を用いた場合に比較的良好な結果が得られることが知られている。例えば二塩化ジルコノセンをn−ブチルリチウムで処理して得られる「根岸試薬」は、一級または二級の有機シランの脱水素縮合重合に高い活性を示す(非特許文献4)。
しかしながら、SiH4の触媒的脱水素縮合重合を効率的に行って、高分子量のポリヒドロシランを製造することのできる技術は、未だ提案されていない。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開2003−313299号公報
【特許文献2】特開2001−262058号公報
【特許文献3】特表2008−536784号公報
【非特許文献】
【0005】
【非特許文献1】Nature,440,pp783〜786(2006)
【非特許文献2】J.Non−Cryst.Solids,354,pp2623〜2626(2008)
【非特許文献3】Inorg.Chem.,1,pp432〜433(1962)
【非特許文献4】Organometallics,10,pp924〜930(1991)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明者らの検討により、根岸試薬などの公知のメタロセン触媒を用いてSiH4の重合を行っても、触媒の初期活性が十分ではなく、さらに重合開始後すぐに触媒の失活が起こるため、ポリヒドロシランを高収率で得ることはできないことが分かった。
本発明は、このような事実が明らかになったことに基づいてなされたものであり、その目的は、SiH4を高活性且つ高効率に重合してポリヒドロシランを製造する方法を提供することにある。
【課題を解決するための手段】
【0007】
本発明によれば、本発明の上記目的および利点は、
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする、前記方法によって達成される。
【発明の効果】
【0008】
本発明の方法で用いられる触媒は、初期活性が高く、且つ触媒寿命が長いものである。従って、このような触媒を用いて行う本発明の方法によってSiH4を重合することにより、高活性、高収率でポリヒドロシランを製造することができる。
本発明の方法によって製造されたポリヒドロシランは、シリコンの前駆体として、例えば半導体装置の製造などに好適に使用することができる。
【図面の簡単な説明】
【0009】
【図1】実施例2で得られた固体を試料として測定したATR−IRチャート。
【図2】実施例2で得られた固体を試料として測定した29Si CP−MAS NMRチャート。
【図3】実施例4の実験例a〜dにおける、重合反応転化率の経時的変化を示すグラフ。
【図4】実施例4の実験例e〜iにおける、重合反応転化率の経時的変化を示すグラフ。
【図5】実施例4の実験例jにおける、重合反応転化率の経時的変化を示すグラフ。
【図6】実施例5の実験例a〜dおよび比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図7】実施例5の実験例e〜gおよび比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図8】実施例6の実験例a〜dおよび実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図9】実施例6の実験例eおよびfならびに実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図10】実施例6の実験例gおよびhならびに実施例5の比較実験例における、重合反応転化率の経時的変化を示すグラフ。
【図11】実施例7で得られた0価シリコンのXPSチャート。
【図12】実施例9で得られた膜状物のラマン分光チャート。
【発明を実施するための形態】
【0010】
本発明は上記のとおり、
4族メタロセンと
アルキルリチウムおよびアルキルアルミニウムから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法に関する。
上記4族メタロセンとアルキル化剤との接触は、SiH4が存在する条件下で行われる。4族メタロセンとアルキル化剤との接触は、SiH4のほかにさらにルイス塩基が存在する条件下で行ってもよい。
<4族メタロセン>
本発明で使用される4族メタロセンとしては、中心金属がチタニウム、ジルコニウムまたはハフニウムであるメタロセンを好ましく使用することができ、例えば下記式(1−1)および(1−2)
【0011】
【化1】
【0012】
(上記式中、Mはチタニウム、ジルコニウムまたはハフニウムであり;
L1およびL2は、それぞれ独立に、シクロペンタジエニル基、インデニル基またはフルオレニル基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
L3およびL4は、それぞれ独立に、シクロペンタジエニレン基、インデニレン基またはフルオレニレン基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
Rはメチレン基、炭素数2〜4のアルキレン基またはジメチルシリレン基であり;そして
Xはハロゲン原子である。)
のそれぞれで表される錯体などを挙げることができ、これらのうちから選択される少なくとも1種を使用することができる。
上記式(1−1)中のL1およびL2としては、それぞれ、シクロペンタジエニル基、t−ブチルシクロペンタジエニル基、トリメチルシリルシクロペンタジエニル基、ペンタメチルシクロペンタジエニル基またはインデニル基であることが;
上記式(1−2)中のL3およびL4としては、それぞれ、インデニレン基であることが好ましい。
上記式(1−1)および(1−2)中のXとしては、塩素原子、臭素原子またはヨウ素原子であることが好ましく、塩素原子であることがより好ましい。
【0013】
本発明で使用される4族メタロセンの具体例としては、例えば
ジシクロペンタジエニルチタニウムジクロリド(Cp2TiCl2)、
ビス(t−ブチルシクロペンタジエニル)チタニウムジクロリド(tBuCp2TiCl2)、
ビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)、
ビス(インデニル)チタニウムジクロリド(Ind2TiCl2)、
ジシクロペンタジエニルジルコニウムジクロリド(Cp2ZrCl2)、
ビス(t−ブチルシクロペンタジエニル)ジルコニウムジクロリド(tBuCp2ZrCl2)、
ビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)、
ビス(インデニル)ジルコニウムジクロリド(Ind2ZrCl2)、
ansa−エチレンビス(インデニル)ジルコニウムジクロリド(ansa−Ind2ZrCl2)、
ビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBuCp2HfCl2)などを挙げることができ、これらのうちから選択される1種以上を好ましく使用することができる。
4族メタロセンの使用割合は、SiH41モルに対して、1×10−4〜1×10−1モルとすることが好ましく、1×10−3〜5×10−2モルとすることがより好ましい。
【0014】
<アルキル化剤>
本発明で使用されるアルキル化剤は、アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選択される。
上記アルキルリチウムとしては、炭素数2〜6のアルキル基を有するアルキルリチウムが好ましい。このようなアルキルリチウムとしては、例えばエチルリチウム、n−プロピルリチウム、i−プロピルリチウム、n−ブチルリチウム、sec−ブチルリチウム、iso−ブチルリチウム、n−ペンチルリチウム、n−ヘキシルリチウムなどを挙げることができる。
上記アルキルアルミニウムとしては、炭素数2〜6のアルキル基を有するジアルキルアルミニウムヒドリド、炭素数2〜6のアルキル基を有するトリアルキルアルミニウムなどを挙げることができる。ここで、炭素数2〜6のアルキル基としては、例えばエチル基、n−プロピル基、i−プロピル基、n−ブチル基、sec−ブチル基、iso−ブチル基、n−ペンチル基、n−ヘキシル基などを挙げることができる。アルキルアルミニウムとしては、炭素数3〜5のアルキル基を有するトリアルキルアルミニウムが好ましい。
上記アルキルマグネシウムとしては、例えば炭素数2〜6のアルキル基を有するアルキルマグネシウムを挙げることができる。
上記アルキルマグネシウムハライドとしては、例えば炭素数2〜6のアルキル基を有するアルキルマグネシウムクロリド、アルキルマグネシウムブロミド、アルキルマグネシウムヨージドを挙げることができる。
本発明におけるアルキル化剤としては、アルキルリチウムおよびアルキルアルミニウムから選択されるアルキル化剤を使用することが好ましく、炭素数3〜5の直鎖のアルキル基を有するアルキルリチウムを使用することがより好ましく、特に好ましくはn−ブチルリチウムである。
アルキル化剤の使用割合は、4族メタロセン1モルに対して、1〜20モルとすることが好ましく、1.5〜10モルとすることがより好ましく、2〜3モルとすることがさらに好ましい。
【0015】
<ルイス塩基>
本発明において任意的に使用されるルイス塩基としては、窒素原子、酸素原子、リン原子または硫黄原子を1〜6個有する有機化合物を挙げることができ、これらのうちから選択される少なくとも1種を使用することが好ましい。
上記窒素原子を有する有機化合物としては、芳香族環を構成するメンバーとして窒素原子を有する複素芳香族化合物、トリアルキルアミン、テトラアルキルアルキレンジアミン、ペンタアルキルジアルキレントリアミンなどを挙げることができる。このトリアルキルアミン、テトラアルキルアルキレンジアミン、ペンタアルキルジアルキレントリアミンの有するアルキル基としては、炭素数1〜6のアルキル基であることが好ましく、炭素数1〜3のアルキル基であることがより好ましく;
アルキレン基としては、炭素数2〜6のアルキル基であることが好ましく、1,2−エチレン基または1,2−プロピレン基であることがより好ましい。
【0016】
本発明におけるルイス塩基としての窒素原子を有する有機化合物の具体例としては、上記窒素原子を有する複素芳香族化合物として例えばピリジン、ピリミジン、ピラジン、トリアジン、ビピリジン、ターピリジン、ビキノリン、フェナントロリンなどを;
トリアルキルアミンとして例えばトリメチルアミン、トリエチルアミン、トリイソペンチルメチルアミン、ピペラジン、1,8−ビス(ジメチルアミノ)ナフタレンなどを;
テトラアルキルアルキレンジアミンとして例えばN,N,N’,N’−テトラメチルエチレンジアミンなどを;
ペンタアルキルジアルキレントリアミンとして例えばN,N,N’,N”,N”−ペンタメチルジエチレントリアミンなどを、それぞれ挙げることができる。
酸素原子を有する有機化合物としては、例えば芳香族環を構成するメンバーとして酸素原子を有する複素芳香族化合物、エーテルなどを挙げることができる。上記エーテルとしては、ジアルキルエーテル、ジアルコキシアルキレン、ポリアルキレンオキシドジアルキルエーテル、環状エーテルなどを挙げることができる。上記ジアルキルエーテル、ジアルコキシアルキレンおよびポリアルキレンオキシドジアルキルエーテルの有するアルキル基またはアルコキシ基の炭素数は、1〜6であることが好ましく、1〜3であることがより好ましい。
【0017】
本発明におけるルイス塩基としての酸素原子を有する有機化合物の具体例としては、
上記酸素原子を有する複素芳香族化合物として例えばフランなどを;
ジアルキルエーテル鎖状のモノエーテルとして例えばジメチルエーテル、ジエチルエーテル、メチルエチルエーテル、ジイソプロピルエーテルなどを;
ジアルコキシアルキレンとして例えばジメトキシエタン、ジエトキシエタンなどを;
ポリアルキレンオキシドジアルキルエーテルとして例えばジエチレングリコールジメチルエーテル、ジエチレングリコールジエチルエーテルなどを;
環状エーテルとして例えばテトラヒドロフラン、テトラヒドロピランなどを、それぞれ挙げることができる。
【0018】
リン原子を有する有機化合物としては、例えば下記式(P−1)または(P−2)
R1R2R3P (P−1)
R4R5P−R8−PR6R7 (P−2)
(上記式中、R1〜R7は、それぞれ独立に、炭素数1〜6のアルキル基または炭素数とヘテロ原子数との合計が6〜12の芳香族基であり;
R8はメチレン基または炭素数2〜6のアルキレン基である。)
で表される化合物を挙げることができる。
上記式(P−1)で表される化合物としては、例えばジフェニルメチルホスフィン、トリフェニルホスフィン、ジフェニルピリジルホスフィン、トリス(ジメチルアミノ)ホスフィンなどを;
上記式(P−2)で表される化合物としては、例えば1,2−ビス(ジフェニルホスフィノ)エタン、1,2−ビス(ジメチルホスフィノ)エタンなどを、それぞれ挙げることができる。
硫黄原子を有する有機化合物としては、例えばジメチルスルフィド、ジエチルスルフィド、チオフェン、チオベンゾフェノン、スルフランなどを挙げることができる。
【0019】
本発明におけるルイス塩基としては、窒素原子、酸素原子またはリン原子を1〜6個有する有機化合物よりなる群から選択される少なくとも1種を使用することが好ましく、特にトリエチルアミン、N,N,N’,N’−テトラメチルエチレンジアミン、N,N,N’,N”,N”−ペンタメチルジエチレントリアミン、ピリジン、ビピリジン、ビキノリン、ジメトキシエタン、ジフェニルメチルホスフィン、1,2−ビス(ジフェニルホスフィノ)エタンなどを挙げることができ、これらのうちから選択される少なくとも1種を使用することがより好ましい。
ルイス塩基の使用割合は、4族メタロセン1モルに対して、100モル以下とすることが好ましく、0.1〜50モルとすることがより好ましい。ルイス塩基のさらに好ましい使用割合は、使用するルイス塩基の種類に応じて異なり、4族メタロセン1モルに対する使用量としてそれぞれ以下のとおりである。
窒素原子を有する有機化合物:さらに好ましくは0.5〜10モル、特に好ましくは1〜5モル
酸素原子または硫黄原子を有する有機化合物:さらに好ましくは1〜50モル、特に好ましくは5〜20モル
リン原子を有する有機化合物:さらに好ましくは0.5〜10モル、特に好ましくは1〜5モル
【0020】
<4族メタロセンとアルキル化剤との接触>
本発明のポリヒドロシランの製造方法において使用される触媒は、上記のごとき4族メタロセンと、アルキル化剤とを、SiH4が存在する条件下で接触させることにより調製される。4族メタロセンとアルキル化剤との接触は、SiH4のほかにさらにルイス塩基が存在する条件下で行ってもよい。
このSiH4および任意的にルイス塩基の存在下における4族メタロセンとアルキル化剤との接触は、SiH4の重合を行う重合反応容器中で行ってもよく、あるいは重合反応容器以外の別容器中で接触を行った後に、接触後の混合物を重合反応容器中に添加してもよい。この4族メタロセンとアルキル化剤との接触は、溶媒の存在下または不存在下に行うことができるが、より高活性の種を均一に生成するとの観点から、溶媒の存在下に行うことが好ましい。
4族メタロセンとアルキル化剤との接触を重合反応容器以外の別容器中で行う場合、該容器中に存在すべきSiH4の量は、4族メタロセンの1モルに対して、2モル以上とすることが好ましく、5〜10モルとすることがより好ましい。
【0021】
4族メタロセンとアルキル化剤との接触において使用することのできる溶媒としては、脂肪族炭化水素溶媒、芳香族炭化水素溶媒などを挙げることができる。上記脂肪族炭化水素溶媒の具体例としては例えばn−ヘキサン、シクロヘキサン、シクロヘキセン、n−ヘプタン、n−オクタン、シクロオクタン、シクロオクテン、n−デカン、デカリンなどを;
上記芳香族炭化水素溶媒の具体例としては例えばベンゼン、トルエン、キシレン、インダン、テトラリンなどを、それぞれ挙げることができ、これらのうちから選択される1種以上を使用することができる。
溶媒の使用割合としては、4族メタロセン1モルに対して、10L以上とすることが好ましく、80〜1,000Lとすることがより好ましく、さらに100〜1,000Lとすることが好ましい。なお、上記式(1−1)および(1−2)のそれぞれで表される、ジハロゲン化物である4族メタロセンは上記の溶媒に対する溶解度が低く、溶媒の使用割合を4族メタロセン1モルに対して80L未満とすると4族メタロセンは溶解しきらずに固体として残存することがある。しかしながらアルキル化剤と接触してアルキル化された後の4族メタロセンの溶解度は一般に高いから、溶媒の使用割合が少ない場合であっても接触後には均一な溶液を得ることができる。
接触温度は−78〜50℃とすることが好ましく、−78〜30℃とすることがより好ましい。
【0022】
<SiH4の重合>
4族メタロセンとアルキル化剤との接触を重合反応容器中で行った場合には、場合により一定の誘導期間を経た後、SiH4の重合がそのまま開始される。
一方、4族メタロセンとアルキル化剤との接触を重合反応容器以外の別容器中で行う場合、その接触時間を有意に長くする必要はなく、接触後直ちに(通常は数秒以内に)触媒活性種が生成するから、該接触混合物を重合反応容器に注入することによって、SiH4の重合が開始される。
SiH4の重合に際しては、重合反応容器に、4族メタロセンとアルキル化剤との接触の際に使用することのできる溶媒として上記したものと同様の溶媒を添加して希釈下で重合することも許容される。
SiH4の重合の際に使用することのできる溶媒の割合は、溶液中のSiH4仕込み量が20重量%以下となる割合とすることが好ましく、10重量%以下となる割合とすることがより好ましく、特に5重量%以下となる割合とすることが好ましい。
重合温度は、好ましくは−78〜100℃であり、より好ましくは−5〜50℃である。
重合時間は、好ましくは0.5〜72時間であり、より好ましくは4〜20時間である。
【0023】
<ポリヒドロシラン>
上記のようにして、好ましくは固体状のポリヒドロシランを得ることができる。このポリヒドロシランは、必要に応じて適当な精製方法によって触媒残滓などの不純物を除去した後、シリコンの前駆体として好適に使用することができる。
上記精製方法としては、例えば溶媒洗浄を挙げることができる。この溶媒洗浄に使用される溶媒としては、例えばエーテル、芳香族炭化水素、アミンなどを挙げることができる。これらの具体例としては、エーテルとして例えばテトラヒドロフラン、テトラヒドロピラン、ジオキサン、1,2−ジメトキシエタン、アニソールなどを;
芳香族炭化水素として例えばトルエン、キシレン、テトラリンなどを;
アミンとして例えばトリエチルアミン、N,N,N’,N’−テトラメチルエチレンジアミン、N,N,N’,N”,N”−ペンタメチルジエチレントリアミンなどを、それぞれ挙げることができる。溶媒の使用割合は、ポリヒドロシラン100重量部に対して、100〜10,000重量部とすることが好ましく、2,000〜5,000重量部とすることがより好ましい。
溶媒洗浄の際には、例えば震とう、回転翼の使用、超音波の印加などの適当な撹拌手段により、洗浄液を撹拌することが好ましい。
溶媒洗浄は、使用する溶媒が液体状態を維持することのできる温度および圧力において、10分以上行うことが好ましく、30〜90分行うことがより好ましい。
溶媒洗浄は、1回または複数回行うことができ、ポリヒドロシラン中の不純物量を可及的に低減する観点から、2回以上行うことが好ましく、3〜5回行うことがより好ましい。
上記の如き本発明の方法で得られ、好ましくは精製されたポリヒドロシランは、これを非酸化性雰囲気中で加熱することにより、容易に0価シリコンに変換することができる。非酸化性雰囲気とは、例えば不活性気体中または不活性気体と還元性気体との混合気体中を挙げることができ、前記不活性気体としては、例えば窒素、ヘリウム、アルゴンなどを;
前記還元性気体としては、例えば水素などを、それぞれ挙げることができる。加熱温度は例えば340℃以上、好ましくは400〜600℃とすることができ、加熱時間は例えば15分以上、好ましくは30〜90分とすることができる。
【実施例】
【0024】
シラン(SiH4)は高圧ガス保安法にて指定された特殊高圧ガスに該当するため、以下の重合反応はすべて、国立大学法人北陸先端科学技術大学院大学(石川県能美市)内に設置され、石川県知事の許可を受けたオートクレーブ(内容量約27mLまたは約70mL、耐圧約0.3MPaG)内で、室温(23〜27℃)にて行った。このオートクレーブは、チャージ/サンプリング用セプタム、給気管および排気管を有し、排気管から排出されたガスは乾式除害設備によって無害化されたうえで外気に排出される。
【0025】
実施例1および比較例1では、触媒成分を混合する順番の影響を調べた。実施例1は本発明の方法であり、比較例1はメタロセン触媒による重合において通常適用される方法である。
実施例1
圧力0.01MPaGのSiH4ガスを充填した内容積27mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)のトルエン溶液(濃度7.5mmol/L)の4mLをシリンジを用いて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジを用いて注入し、重合を開始した。ここで使用したCp*2TiCl2およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、3モル%および6モル%に相当する。
重合開始から4時間後に、セプタムから気体をサンプリングし、これをガスクロマトグラフィー(GC)で分析してSiH4の脱水素縮合重合により発生した水素の量を定量し、この値を用いた計算によりSiH4の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して74.3モル%であった。反応系から少量の固体を回収した。
【0026】
比較例1
内容量6mLのガラス容器中で、Cp*2TiCl2のトルエン溶液(濃度7.5mmol/L)の4mLおよびn−BuLiのトルエン溶液(濃度60mmol/L)の1mLを混合して触媒溶液を得た。
圧力0.01MPaGのSiH4ガスを充填したオートクレーブ内に、セプタムから上記触媒溶液の全量をシリンジを用いて注入して重合を開始し、実施例1と同様にして反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率はSiH4の仕込み量に対して8.5モル%であった。反応系から少量の固体を回収した。
【0027】
実施例2および3では、使用するメタロセンの種類を変えて検討した。
実施例2
Cp*2TiCl2の代わりにビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)を使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を行い、反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して76.2モル%であった。反応系から少量の固体を回収した。
この反応系から回収した固体を試料として測定したATR−IR(減衰全反射赤外分光)チャートを図1(a)および(b)に、
固体NMR(29Si CP−MAS(交差分極−マジック角回転) NMR、80MHz、装置はVarian Inc.製を使用)チャートを図2に、それぞれ示した。
これらの結果から、回収された固体はポリヒドロシランであると考えられる。
【0028】
実施例3
Cp*2TiCl2の代わりにビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBu−Cp2HfCl2)を使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を行い、反応開始4時間後の重合反応転化率を求めた。その結果、反応開始4時間後の重合反応転化率は、SiH4の仕込み量に対して16.7モル%であった。反応系から少量の固体を回収した。
【0029】
実施例4では、種々のメタロセンを用いて、SiH4の重合反応転化率の経時的変化を追跡した。
実施例4
Cp*2TiCl2の代わりに各種のメタロセンを使用したほかは、実施例1と同様にしてSiH4の脱水素縮合重合を開始した。実施例1における、セプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実験例の、反応開始20時間後におけるSiH4の仕込み量に対する重合反応転化率を表1に、SiH4の重合反応転化率の経時的変化を図3〜5に、それぞれ示した。
【0030】
各実験例において使用したメタロセンの種類は、以下のとおりである。カッコ内は、表1および図3〜5における表記である。
実験例a:ジシクロペンタジエニルチタニウムジクロリド(Cp2TiCl2)
実験例b:ビス(t−ブチルシクロペンタジエニル)チタニウムジクロリド(tBuCp2TiCl2)
実験例c:ビス(インデニル)チタニウムジクロリド(Ind2TiCl2)
実験例d:ビス(ペンタメチルシクロペンタジエニル)チタニウムジクロリド(Cp*2TiCl2)
実験例e:ジシクロペンタジエニルジルコニウムジクロリド(Cp2ZrCl2)
実験例f:ビス(t−ブチルシクロペンタジエニル)ジルコニウムジクロリド(tBuCp2ZrCl2)
実験例g:ビス(インデニル)ジルコニウムジクロリド(Ind2ZrCl2)
実験例h:ansa−エチレンビス(インデニル)ジルコニウムジクロリド(ansa−Ind2ZrCl2)
実験例i:ビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)
実験例j:ビス(t−ブチルシクロペンタジエニル)ハフニウムジクロリド(tBuCp2HfCl2)
【0031】
【表1】
【0032】
実施例5では、ルイス塩基を添加した場合の効果について調べた。実験例a〜dがルイス塩基としてアミンを添加した場合であり、
実施例e〜fがルイス塩基としてホスフィンを添加した場合であり、
実施例gがルイス塩基としてエーテルを添加した場合である。比較実験例はルイス塩基を添加しなかった場合である。
実施例5(実験例a〜g)
圧力0.01MPaGのSiH4ガスを充填した内容積25mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)のトルエン溶液(濃度10mmol/L)の3mLおよび各種ルイス塩基のトルエン溶液(溶液濃度および溶液使用量は後述)をシリンジにて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジにて注入し、重合を開始した。ここで使用したCp*2ZrCl2、ルイス塩基およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、1モル%、1モル%および2モル%に相当する。
反応開始後、実施例4と同様にしてセプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実験例における、ルイス塩基の種類、反応開始20時間後のSiH4の仕込み量に対する重合反応転化率を表2に、SiH4の重合反応転化率の経時的変化を図6および7に、それぞれ示した。
【0033】
各実験例において使用したルイス塩基の種類、溶液濃度および溶液使用量は以下のとおりである。カッコ内は、表2ならびに図6および7における表記である。
実験例a:N,N,N’,N”,N”−ペンタメチルジエチレントリアミン(PMDTA)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例b:N,N,N’,N’−テトラメチルエチレンジアミン(TMEDA)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例c:トリエチルアミン(NEt3)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例d:ピリジン(Py)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例e:1,2−ビス(ジフェニルホスフィノ)エタン(DPPE)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例f:ジフェニルメチルホスフィン(Ph2PMe)、溶液濃度=30mmol/L、溶液使用量=1mL
実験例g:1,2−ジメトキシエタン(DME)、溶液濃度=30mmol/L、溶液使用量=1mL
実施例5(比較実験例)
ルイス塩基を使用しなかったほかは、上記実験例a〜gと同様にして実施し、SiH4の重合反応転化率を経時的に求めた。このときの、反応開始20時間後におけるSiH4の仕込み量に対する重合反応転化率を表2に、重合反応転化率の経時的変化を図6および7に示した。
【0034】
【表2】
【0035】
実施例6では、ルイス塩基の添加量の効果について調べた。
実験例a〜dはルイス塩基としてN,N,N’,N”,N”−ペンタメチルジエチレントリアミン(PMDTA)を用いた場合であり、
実験例eおよびfはルイス塩基として1,2−ビス(ジフェニルホスフィノ)エタン(DPPE)を用いた場合であり、そして
実験例gおよびhはルイス塩基として1,2−ジメトキシエタン(DME)を用いた場合である。
実施例6(実験例a〜h)
圧力0.01MPaGのSiH4ガスを充填した内容積25mLのオートクレーブ内に、セプタムからビス(ペンタメチルシクロペンタジエニル)ジルコニウムジクロリド(Cp*2ZrCl2)のトルエン溶液(濃度10mol/L)の3mLおよび各種ルイス塩基のトルエン溶液(溶液濃度はそれぞれ上記実施例5におけるのと同じであり、溶液使用量は変量としてルイス塩基量がそれぞれ表3に記載の値となるようにした。)をシリンジにて注入し、十分に撹拌した。次いでここに、セプタムからn−ブチルリチウム(n−BuLi)のトルエン溶液(濃度60mmol/L)の1mLをシリンジにて注入し、重合を開始した。ここで使用したCp*2ZrCl2およびn−BuLiの量は、SiH4の仕込み量に対して、それぞれ、1モル%および2モル%に相当する。
反応開始後、実施例4と同様にしてセプタムからの気体のサンプリングおよびGC分析を経時的に行うことにより、SiH4の重合反応転化率を経時的に求めた。
各実施例における、ルイス塩基の種類およびSiH4の仕込み量に対する添加量、反応開始20時間後のSiH4の仕込み量に対する重合反応転化率を表3に、SiH4の重合反応転化率の経時的変化を図8〜10に、それぞれ示した。表3ならびに図8〜10には、上記実施例5における比較実験例の結果も合わせて示した。
【0036】
【表3】
【0037】
実施例7
本実施例では、本発明の方法によって得られたポリヒドロシランがシリコンの前駆体として好適であることを検証した。
上記実施例4の実験例a〜dで回収した各固体を、窒素雰囲気下、450℃において1.5時間加熱することにより、暗褐色の粉末をそれぞれ得た。
この粉末を試料として、以下の条件でX線光電子分光(XPS)分析を行った。
測定装置:Phi5600 ESCA System(ULVAC−PHI Inc.製)
得られたXPSチャートを図11(a)および(b)に示した。結合エネルギー99〜100eV付近にSi(0価)のピークが、102〜104eV付近にSi(4価)の微弱なピークが、それぞれ観察されたことから、上記加熱後の粉末は0価のシリコンであることが確認され、本発明の方法によって得られたポリヒドロシランがシリコンの前駆体として好適であることが分かった。
【0038】
実施例8
本実施例では、本発明の方法によって得られたポリヒドロシランを前駆体として製造されたシリコンが高純度のものであることを検証した。
上記実施例2で回収した固体約30mgにテトラヒドロフラン2mLを加え、懸濁液とした。得られた懸濁液につき、超音波液体処理機Sonifier150(Branson Ultrasonics社製、23kHz)を用いて出力2〜4Wにて30分間超音波洗浄した。洗浄後の懸濁液につき、小型微量遠心機PMC−060((株)トミー精工製、遠心加速度約2,000G)を用いて固体と洗浄液とに遠心分離した。この固体を回収し、同様の手法でさらに2回超音波洗浄/遠心分離操作を行った。
上記のようにして合計3回洗浄した固体を窒素雰囲気のグローブボックス中に搬入し、450℃において1時間加熱したところ、25.1mgの暗褐色の粉末が得られた。
得られた粉末を試料として、誘導結合プラズマ質量分析(ICP−MS)によって金属不純物の含有量を分析したところ、Zr=16mg/g、Li=2.4mg/gであった。
【0039】
実施例9
本実施例では、本発明の方法によって得られたポリヒドロシランを用いてシリコン膜を形成した。
上記実施例2で回収した固体を、実施例8におけるのと同様にしてテトラヒドロフランで洗浄したうえ、その約20mgをシクロペンタシラン約20mgと混合して、膜形成用組成物を得た。
窒素雰囲気のグローブボックス中で、シリコン基板上に、上記の膜形成用組成物を配置し、その上に、約1cm四方の平滑なSiO2製鋳型を載せて、室温において0.5MPaGにて5分、次いで200℃において3.5MPaGにて15分加圧して型押しした。その後鋳型を除去し、基板上の成形物を、ホットプレートにて窒素下、400℃において1時間加熱したところ、膜状物が得られた。
この膜状物について測定したラマン分光チャートを図12(a)に示した。
また、シクロペンタシランを用いなかったほかは上記と同様にして得られたものについて測定したラマン分光チャートを図12(b)に示した。
【特許請求の範囲】
【請求項1】
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする、前記方法。
【請求項2】
前記4族メタロセンとアルキル化剤との接触が、SiH4のほかにさらにルイス塩基が存在する条件下で行われる、請求項1に記載の方法。
【請求項3】
前記ルイス塩基が、窒素原子、酸素原子、リン原子または硫黄原子を1〜4個有する有機化合物よりなる群から選択される少なくとも1種である、請求項2に記載の方法。
【請求項4】
前記4族メタロセンが、下記式(1−1)および(1−2)
【化1】
(上記式中、Mはチタニウム、ジルコニウムまたはハフニウムであり;
L1およびL2は、それぞれ独立に、シクロペンタジエニル基、インデニル基またはフルオレニル基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
L3およびL4は、それぞれ独立に、シクロペンタジエニレン基、インデニレン基またはフルオレニレン基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
Rはメチレン基、炭素数2〜4のアルキレン基またはジメチルシリレン基であり;そして
Xはハロゲン原子である。)
のそれぞれで表される錯体よりなる群から選択される少なくとも1種である、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われ、そして
SiH4を重合してポリヒドロシランを製造するために用いられることを特徴とする、前記触媒。
【請求項1】
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒によってSiH4を重合する、ポリヒドロシランの製造方法であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われることを特徴とする、前記方法。
【請求項2】
前記4族メタロセンとアルキル化剤との接触が、SiH4のほかにさらにルイス塩基が存在する条件下で行われる、請求項1に記載の方法。
【請求項3】
前記ルイス塩基が、窒素原子、酸素原子、リン原子または硫黄原子を1〜4個有する有機化合物よりなる群から選択される少なくとも1種である、請求項2に記載の方法。
【請求項4】
前記4族メタロセンが、下記式(1−1)および(1−2)
【化1】
(上記式中、Mはチタニウム、ジルコニウムまたはハフニウムであり;
L1およびL2は、それぞれ独立に、シクロペンタジエニル基、インデニル基またはフルオレニル基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
L3およびL4は、それぞれ独立に、シクロペンタジエニレン基、インデニレン基またはフルオレニレン基であり、ただしこれらの基は炭素数1〜4のアルキル基またはトリメチルシリル基で置換されていてもよく;
Rはメチレン基、炭素数2〜4のアルキレン基またはジメチルシリレン基であり;そして
Xはハロゲン原子である。)
のそれぞれで表される錯体よりなる群から選択される少なくとも1種である、請求項1〜3のいずれか一項に記載の方法。
【請求項5】
4族メタロセンと
アルキルリチウム、アルキルアルミニウム、アルキルマグネシウムおよびアルキルマグネシウムハライドから選ばれるアルキル化剤と
を接触させて得られる触媒であって、
前記4族メタロセンとアルキル化剤との接触が、SiH4が存在する条件下で行われ、そして
SiH4を重合してポリヒドロシランを製造するために用いられることを特徴とする、前記触媒。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2012−207152(P2012−207152A)
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願番号】特願2011−74587(P2011−74587)
【出願日】平成23年3月30日(2011.3.30)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(000004178)JSR株式会社 (3,320)
【Fターム(参考)】
【公開日】平成24年10月25日(2012.10.25)
【国際特許分類】
【出願日】平成23年3月30日(2011.3.30)
【出願人】(503360115)独立行政法人科学技術振興機構 (1,734)
【出願人】(000004178)JSR株式会社 (3,320)
【Fターム(参考)】
[ Back to top ]