
ルシフェラーゼバイオセンサー
【課題】環境に感受性のあるレポータータンパク質である修飾された甲虫ルシフェラーゼタンパク質が提供される。
【解決手段】関心のある分子と相互作用するアミノ酸配列を含む挿入を含み、修飾に寛容性のある親甲虫ルシフェラーゼ配列の残基又は領域において環状置換された甲虫ルシフェラーゼをコードするポリヌクレオチド。前記ポリヌクレオチドによってコードされる修飾された甲虫ルシフェラーゼ。前記ポリヌクレオチドを細胞に形質導入し、細胞内の関心のある分子を検出する方法。
【解決手段】関心のある分子と相互作用するアミノ酸配列を含む挿入を含み、修飾に寛容性のある親甲虫ルシフェラーゼ配列の残基又は領域において環状置換された甲虫ルシフェラーゼをコードするポリヌクレオチド。前記ポリヌクレオチドによってコードされる修飾された甲虫ルシフェラーゼ。前記ポリヌクレオチドを細胞に形質導入し、細胞内の関心のある分子を検出する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本出願は、米国特許法第119条(e)の下、参照により本明細書に組み込まれた2003年10月10日に出願の米国特許出願第60/510187号の出願日の権益を請求する。
【0002】
本発明は、生化学アッセイ及び試薬の分野に関するものである。より具体的には、本発明は発光性レポータータンパク質などの修飾されたレポータータンパク質、及びそれらの使用方法に関するものである。
【背景技術】
【0003】
ルシフェラーゼは、光量子の放出を伴う基質(例えばルシフェリン)の酸化を触媒する酵素である。ルシフェラーゼは、鞘翅類節足動物及び多くの海洋生物を含む多数の種から単離されている。検出が容易でありその活性は高精度で定量化することが可能なので、ルシフェラーゼ/基質対は遺伝子発現及びタンパク質の所在を研究するために広く使用されてきた。発色団の形成に最高30分を要する他のレポータータンパク質である緑色蛍光タンパク質(GFP)と異なり、ルシフェラーゼ生成物はポリペプチド鎖の合成(基質及び酸素も存在する場合)終了後直ちに検出することが可能である。更に、酵素活性のために翻訳後修飾は必要ではなく、また、この酵素は補欠分子団、結合コファクター及びジスルフィド結合を含まない。ルシフェラーゼは、多数の種及び多種多様な細胞における有用なレポーターである。
【0004】
ルシフェラーゼは、バイオセンシングのためのレポーター分子、すなわち、生物学的の性状を明らかにする分子としてそれらを特に有用なものにする追加機能を有する。バイオセンサー(生物学的構成要素を含むセンサー)におけるシグナル伝達は、通常二段階過程を含む。即ち、生物学的構成要素を通してのシグナル生成並びに電気的構成要素を通してのシグナル伝達及び増幅である。シグナル生成は、一般的に結合又は触媒作用を通して達成される。電気信号へのこれらの生化学事象の変換は一般的に電気化学的又は熱検出法に基づいているが、これらは生化学反応の自由エネルギーの変化によって制限される。大部分の反応については、これはATP2分子の加水分解のエネルギー、すなわち約70kJ/モルより少ない。しかし、ルシフェラーゼによって導き出される発光は、非常に高いエネルギー含量を有する。例えば、ホタルルシフェラーゼによって触媒される反応(560nm)は、214kJ/モルのエネルギーを発する。更に、ルシフェラーゼによって触媒される反応は知られている最も効率的な生物発光反応の1つであり、約0.9の量子収率を有する。ルシフェラーゼは、したがって極めて効率的な化学エネルギー変換器である。
【0005】
ルシフェラーゼバイオセンサーが記載されている。例えば、Sala−Newbyら(1991年)は、サイクリックAMP依存性タンパク質キナーゼリン酸化部位を生成するためにPhotinus pyralisルシフェラーゼcDNAをin vitroで増幅したことを開示している。特に、部位RRFSを生成するために217位置のバリンがアルギニンに変異され、ブタピルビン酸キナーゼのリン酸化部位であるヘプタペプチドケンプチドがルシフェラーゼのN又はC末端に加えられた。Sala−Newbyらは、リン酸化部位を運ぶタンパク質の比活性、pI、発光色に及ぼすpHの影響、及びATP存在下でのタンパク質キナーゼAの触媒サブユニットの影響について特徴を明らかにしたと述べている。組換えタンパク質(RRFS)の1つだけが野生型ルシフェラーゼとかなり異なり、そのRRFS変異体は低い比活性、低い最適pHを有し、低いpHでより濃い緑の光を発し、且つリン酸化されるとその活性が最高80%減少することが発見された。後者の影響はホスファターゼによって逆転されたことが開示されている。
【0006】
Waudら(1996年)は、Photinus pyralisルシフェラーゼcDNAに、タンパク質キナーゼ認識配列及びプロテイナーゼ部位を導入した。ルシフェラーゼの2つの領域が、Waudらによって修飾された。1つはアミノ酸209と227との間で、他はC末端のアミノ酸537と550との間である。Waudらは、残基209及び227の間のアミノ酸の変異は生物発光活性を野生型組換え体の1%未満に低下させたが、C末端におけるペプチド配列の導入は野生型組換えルシフェラーゼの0.06%〜120%の範囲の比活性をもたらしたことを開示している。Waudらは、サイクリックAMP依存性タンパク質キナーゼ触媒サブユニットの、キナーゼ認識配列LRRASLG(配列番号107)が組み込まれ、セリンがアミノ酸位置543にある変異型ルシフェラーゼへの添加は、30%の活性低下をもたらしたことも開示している。アルカリホスファターゼ処理は活性を回復させた。Waudらは、トロンビン認識配列LVPRES(配列番号108)を含み、切断部位がアミノ酸542及び543の間に位置する変異型ルシフェラーゼの生物発光活性は、トロンビン存在下でインキュベートされると50%減少したことも開示している。
【0007】
Ozawaら(2001年)は、ホタルルシフェラーゼの合理的に設計された断片のタンパク質スプライシング誘発性の相補に基づくバイオセンサーを記載している。タンパク質スプライシングは、それを通してインテイン(intein)(内部タンパク質)が前駆体融合タンパク質から切除されて連なるエクステイン(extein)(外部タンパク質)が連結されて連続したポリペプチドになる、翻訳後タンパク質修飾である。Synechocystis属種PCC6803からのN及びC末端のインテインDnaEは、それぞれルシフェラーゼのN及びC末端の断片に融合されたことが開示されている。タンパク質対タンパク相互作用はDnaEインテインの折畳みを引き起こしてタンパク質スプライシングをもたらし、それにより、連結されたルシフェラーゼのエクステインはその酵素活性を取り戻す。Ozawaらは、公知の結合パートナーであるリン酸化インスリン受容体基質1(IRS−1)及びその標的であるPI3−キナーゼのN末端SH2ドメインの間の相互作用が、インスリン存在下で分割されたルシフェラーゼを使用して観察されたことを開示している。
【0008】
Paulmuruganら(2002年)は分割されたホタルルシフェラーゼに基づくアッセイを使用し、相補性方策及びインテイン媒介再構成手法を使用して細胞培養及びマウスで2つのタンパク質、即ちMyoD及びIdの相互作用を観察した。レポーター活性を保持するために、相補性手法では融合タンパク質はタンパク相互作用、即ち、タンパク質パートナーMyoD及びIdの相互作用を必要とするが、再構成手法においてはインテイン媒介スプライシングを通して形成された新しい完全なレポータータンパク質がタンパク質パートナーの間の継続的な相互作用のない場合でさえその活性を維持する。
【0009】
タンパク質断片相補性アッセイは、Michnickら(米国特許第6270964号、第6294330号及び第6428951号)で開示されている。具体的には、Michnickは、DHFRのN末端断片及びDHFRのC末端断片がそれぞれGCN4ロイシンジッパー配列に融合している、分割されたマウスのジヒドロ葉酸レダクターゼ(DHFR)遺伝子に基づくアッセイを記載している。DHFR活性は、両方の融合タンパク質を発現していた細胞で検出された。Michnickらは、アミノグリコシドキナーゼ(AK)遺伝子内のS1ヌクレアーゼ生成欠失の入れ子セットがロイシンジッパー構築物に導入され、得られた構築物セットが細胞に導入されてAK活性がスクリーニングされた他の相補性手法も記載している。
【発明の概要】
【発明が解決しようとする課題】
【0010】
必要なものは、例えばタンパク質対タンパク質相互作用などの細胞現象を高い特異性及び高量子収率で検出する際にバイオセンサーとして使用するための、改良された組換えレポータータンパク質である。
【課題を解決するための手段】
【0011】
本発明は改良された遺伝子産物、例えば他の分子(1つ又は複数の関心のある分子)の存在下で、又はある条件下で1つ又は複数の変更された活性を有する修飾されたレポータータンパク質、例えば修飾された甲虫ルシフェラーゼを提供する。一実施形態では、修飾されたレポータータンパク質のアミノ酸配列は、修飾に寛容性のある、例えば挿入、欠失、環状置換又はその任意の組み合わせに寛容性のある部位(残基)又はある領域における1つ又は複数の修飾の結果として、対応する修飾されていない(天然、野生型又は親の)レポータータンパク質のアミノ酸配列とは異なる。1つ又は複数の修飾は修飾されていないレポータータンパク質のN又はC末端に対して内部でもよく、且つ/又は修飾されていないレポータータンパク質のN及び/又はC末端におけるものでもよく、例えば1つ又は複数のアミノ酸残基の欠失及び/又は挿入であり、それにより修飾されたレポータータンパク質が与えられる。修飾としては、直接又は間接的に関心のある分子と相互作用し、且つ/又はさもなければ条件の変化に感受性のある1つ又は複数の別々の(分離された)アミノ酸配列の導入があり、任意選択に、例えば修飾に寛容性のある部位又は領域、例えば修飾されていないレポータータンパク質のN及び/又はC末端における1つ又は複数のアミノ酸の欠失があるが、この場合、得られた修飾されたレポータータンパク質は外因性の剤などの関心のある分子との相互作用又は条件変化の前及び/又は後にレポーター活性を有するものとする。例えば、修飾されたレポータータンパク質は、対応する修飾されていないレポータータンパク質に対する、1から約10又は15の残基、或いはその中間の任意の整数のN又はC末端位置の欠失を含んでもよい。修飾は、修飾されたレポータータンパク質における、対応する修飾されていないレポータータンパク質においてペプチド結合を通して結合された2つのアミノ酸の間のペプチド結合の欠如であってもよく、並びに修飾されたレポータータンパク質における、対応する修飾されていないレポータータンパク質のN末端及びC末端の残基において又はその近くで見られる残基の間のペプチド結合の欠如でもよく、これらからは環状置換されたレポータータンパク質が与えられ、このレポータータンパク質は任意選択に直接又は間接的に関心のある分子と相互作用するかさもなければ条件変化に感受性であるアミノ酸配列を含む。修飾されたレポータータンパク質は、したがって可逆的相互作用、例えば2つ以上の分子の結合、ジスルフィド結合の形成又は他の高次構造変化又はpH、温度若しくは溶媒疎水性などの条件の変化、或いは修飾されたレポータータンパク質の活性の変化、例えば光強度、色又は動態学的プロフィールの変化によるペプチド結合の切断などの不可逆的相互作用を検出するために使用することができる。
【0012】
本明細書で以下に記載されているように、コメツキムシルシフェラーゼ核酸配列への19のアミノ酸をコードするDNA挿入のライブラリーを調製するために、Tn5を使用した。挿入を有する416のクローンの分析により、クローンの約10%(52)は部分活性、例えば野生型の最高で2%の活性を有することが明らかになった。52クローンの内27クローンはルシフェラーゼ読取り枠内に挿入を有し、それらの挿入の内の16は残基398から409の間(「ヒンジ」領域)にあった。特に、検出可能な活性を有する修飾されたコメツキムシルシフェラーゼをもたらすインフレーム挿入は、コメツキムシルシフェラーゼの残基21、25、117、358、376、379、398、399、400、401、402、403、405、406、407、409又は490の位置であり、即ち、それらの残基及び/又はそれらの残基の近くの領域は挿入を含む修飾に寛容性である。したがって、本発明は、コメツキムシルシフェラーゼの例えば残基21、25、117、358、376、379、398、399、400、401、402、403、405、406、407、409又は490などの残基、或いは、残基15から30、例えば残基21若しくは25、残基112から122、例えば残基117、残基352から362、例えば残基358、残基371から384、例えば残基379、残基393から414又は残基485から495に対応する領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。対応する位置は、ルシフェラーゼ配列を一列に並べることによって特定することができる。詳細には、本発明は甲虫ルシフェラーゼのヒンジ領域、例えばコメツキムシルシフェラーゼの残基390から409に対応する残基並びに修飾に寛容性のある他の領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。
【0013】
また本明細書で記載されているように、ホタルルシフェラーゼ核酸配列への挿入のライブラリーを調製するために、Tn7を使用した。検出可能な活性を有する修飾されたホタルルシフェラーゼをもたらすインフレーム挿入は、ホタルルシフェラーゼの残基7、121、233、267、294、303、361、540又は541の位置であり、即ち、それらの残基及び/又はそれらの残基の近くの領域は挿入を含む修飾に寛容性である。したがって、本発明はホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546と対応する残基又は領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。対応する位置は、ルシフェラーゼ配列を一列に並べることによって特定することができる。
【0014】
したがって、一実施形態ではレポータータンパク質は甲虫ルシフェラーゼであり、修飾された甲虫ルシフェラーゼのアミノ酸配列は、修飾に寛容性のある部位又はある領域における1つ又は複数の修飾の結果として、対応する修飾されていない甲虫ルシフェラーゼのアミノ酸配列とは異なる。例えば、一実施形態では、修飾された甲虫ルシフェラーゼは検出可能な活性を有し、また対応する修飾されていない甲虫ルシフェラーゼに対する修飾に寛容性のある部位又は領域における1つ又は複数のアミノ酸の挿入を含み、この挿入は修飾された甲虫ルシフェラーゼのN及びC末端の内部にある。一実施形態では、修飾された甲虫ルシフェラーゼは、2つ以上、例えば3、4、5、10、20、50、100、200、300以上で約500未満の、又はその中間の任意の整数のアミノ酸残基の挿入を含む。一実施形態では、本発明の修飾された甲虫ルシフェラーゼは修飾に寛容性のある残基又は領域に少なくとも4つのアミノ酸の内部挿入を含み、この挿入は関心のある分子と直接的に相互作用するアミノ酸配列を含み、例としては関心のある分子の認識配列を含む挿入又は関心のある分子に間接的に、例えば他の分子を通して作用する挿入がある。一実施形態では、内部挿入を有する修飾された甲虫ルシフェラーゼは、甲虫ルシフェラーゼ配列の内部欠失、例えば1つ又は複数で約100未満、例えば50、40、30、20、10又は5未満又はその中間の任意の整数の残基の欠失を更に含む。
【0015】
一実施形態では、修飾された甲虫ルシフェラーゼは対応する修飾されていない甲虫ルシフェラーゼに対する、修飾に寛容性のある部位又は領域における欠失を含む。一実施形態では、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼに対する少なくとも50、例えば少なくとも100の連続したアミノ酸残基の欠失を含み、即ち、前記修飾された甲虫ルシフェラーゼは完全長の修飾されていない甲虫ルシフェラーゼ配列の断片、例えば少なくとも50、例えば少なくとも100の連続したアミノ酸残基の断片であり、例えば対応する完全長の修飾されていない甲虫ルシフェラーゼよりも少なくとも5%、例えば10%少ない残基、及び関心のある分子と直接又は間接的に相互作用するかさもなければ条件に感受性のあるアミノ酸配列の挿入を有する断片である。そのような修飾された甲虫ルシフェラーゼは、タンパク質相補性アッセイで、例えば関心のある分子に連結されたルシフェラーゼの他の断片の存在下でルシフェラーゼの検出可能な活性が増加する場合に、又はインテイン媒介組換えなどのタンパク質組換えアッセイで使用することができる。一実施形態では、甲虫ルシフェラーゼ断片(1つ又は複数の異種配列を有さず)は対応する完全長の修飾されていない甲虫ルシフェラーゼの活性の例えば約0.001%、0.01%、0.1%又は1%未満の検出可能な活性を有し、また相補断片(1つ又は複数の異種配列を有さず)と結合すると、いずれの断片と比較しても3倍を超える、例えば10又は50から100倍又はそれ以上の活性増加を有する。例えば、一実施形態では、N末端甲虫ルシフェラーゼ断片は対応する完全長の修飾されていない甲虫ルシフェラーゼの少なくとも0.001%で1%未満の、また、C末端甲虫ルシフェラーゼ断片は少なくとも0.01%で5%未満の活性を有する。他の実施形態において、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼに対する少なくとも50、例えば少なくとも100の連続したアミノ酸残基の欠失、関心のある分子と直接又は間接的に相互作用するかさもなければ条件に感受性のあるアミノ酸配列の挿入、及び異種、例えば非甲虫ルシフェラーゼ配列の挿入を有する断片であり、これらの挿入は好ましくは甲虫ルシフェラーゼ断片の活性を増加しないが個々に、又は一緒にそれを減少させてもよいが、一旦取り除かれると修飾された甲虫ルシフェラーゼに対して活性の増加したトランケーションされた甲虫ルシフェラーゼをもたらす。
【0016】
本明細書で更に記載されているように、対応する非環状で置換されていない甲虫ルシフェラーゼ内の修飾に寛容性のある残基又は領域にN末端を有し、任意選択に、関心のある分子、例えばプロテアーゼ認識部位若しくはキナーゼ部位と直接又は間接的に相互作用するアミノ酸配列を含む、環状置換された蛍及びコメツキムシのルシフェラーゼが調製され、それらは検出可能な活性を有することが示されたが、この活性は関心のある分子、例えば環状置換ルシフェラーゼ内でプロテアーゼ認識部位又はキナーゼ部位をそれぞれコードしていた構築物内の適当なプロテアーゼ又はキナーゼの存在下で変化した。それ故に、一実施形態では、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼのアミノ酸配列に対して環状置換されたアミノ酸配列を含み、その結果修飾された甲虫ルシフェラーゼ内に新しいN及びC末端を含み、少なくともその1つは修飾に寛容性のある部位又は領域にある。他の実施形態において、環状置換された甲虫ルシフェラーゼは他の修飾、例えばそれには限定されないが、環状置換された甲虫ルシフェラーゼのN又はC末端の内部の挿入及び/又は欠失、例えば対応する修飾されていない甲虫ルシフェラーゼのN及びC末端又はその近くの挿入及び/又は欠失、例えば修飾されていない甲虫ルシフェラーゼのN末端の残基1から約10若しくは15又はその中間の任意の整数と対応する残基、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応する残基における挿入及び/又は欠失を含む。したがって、レポータータンパク質のN及びC末端は環状置換を通して変更することが可能であり、得られた置換された分子は置換されないレポータータンパク質の1つ又は複数の活性を有することができる。したがって、環状置換されたレポータータンパク質は、タンパク質相補性アッセイ又はタンパク質組換えアッセイで使用することができる。さらに、環状置換されたレポータータンパク質は、直接又は間接的に関心のある分子と相互作用するかさもなければ条件変化に感受性のあるアミノ酸配列を導入することによって、官能性を有するようにすることができる。一実施形態では、本発明の環状置換されたレポータータンパク質は、チモーゲンである。
【0017】
一実施形態では、関心のある分子が存在しない場合、修飾された甲虫ルシフェラーゼのような修飾されたレポータータンパク質の活性は対応する修飾されていないレポータータンパク質の活性よりも小さく、例えば修飾された甲虫ルシフェラーゼのレポーター活性は、対応する修飾されていない甲虫ルシフェラーゼのそれの約0.001%、0.01%、0.1%、1%、10%、20%、50%、70%又はそれ以上で100%未満であり、その修飾されたレポータータンパク質の活性は任意選択に検出可能である。他の実施形態では、関心のある分子が存在しない場合、修飾された甲虫ルシフェラーゼのような修飾されたレポータータンパク質の活性は対応する修飾されていないレポータータンパク質の活性よりも大きく、例えば修飾された甲虫ルシフェラーゼのレポーター活性は、対応する修飾されていない甲虫ルシフェラーゼのそれの約1.5倍、例えば少なくとも2倍、3倍又は5倍又はそれ以上である。関心のある分子の存在下では、修飾されたレポータータンパク質の活性は検出可能に変更される。例えば、関心のある分子の存在下での修飾された甲虫ルシフェラーゼの活性における検出可能な変化は、関心のある分子がない場合の修飾された甲虫ルシフェラーゼの活性と比較して、少なくとも0.001%、0.01%、0.1%、1%、10%又は100%、及び最高で2倍、4倍、10倍、100倍、1,000倍、10,000倍、又はそれ以上の変化である。したがって、修飾されたレポータータンパク質に存在するが対応する修飾されていないレポータータンパク質には存在しない修飾と相互作用する関心のある分子の物理的近接性は、修飾されたレポータータンパク質の活性を変化、例えば減少、除去又は増加させる。例えば、修飾された甲虫ルシフェラーゼは対応する修飾されていない甲虫ルシフェラーゼに対する内部挿入を含んでもよく、この挿入はプロテアーゼ認識部位、即ちプロテアーゼによって切断される部位を含む。プロテアーゼ存在下でのそのような修飾された甲虫ルシフェラーゼの発光シグナルは、プロテアーゼが存在しない場合の修飾された甲虫ルシフェラーゼの発光シグナルと比較して、又は関心のある分子の存在下又は非存在下における対応する修飾されていない甲虫ルシフェラーゼの発光シグナルと比較して、減少させ、除去し又は増加させることができる。或いは、対応する修飾されていない甲虫ルシフェラーゼに対する欠失を含む修飾された甲虫ルシフェラーゼは、関心のある分子と相互作用するリガンドと融合することができる。甲虫ルシフェラーゼの相補性の第2の断片が関心のある分子と融合され、2つの融合の相互作用が可能になり、この相互作用は、例えばいずれかの融合単独の活性と比較して得られた複合体の活性を変化、例えば増加させる。一実施形態では、甲虫ルシフェラーゼの1つの断片は、ホタルルシフェラーゼの残基約1から約126、約1から約238、約1から約272、約1から約308、約1から約366、約116から約550、約228から約550、約262から約550、約289から約550、又は約356から約550、又はその中間の任意の整数の残基、或いはコメツキムシルシフェラーゼの約1から約122、約1から約362、約1から約384、約1から約414、約352から約542、約371から約542、又は約393から約542、又はその中間の任意の整数の残基と対応する残基を有する。
【0018】
本発明は、レポータータンパク質のN末端及びC末端に異種配列を含む修飾されたレポータータンパク質も提供し、即ち、前記修飾されたタンパク質は融合タンパク質であり、また前記異種配列は非共有結合で相互作用し、つまり2つの異種配列は結合パートナーである。一実施形態では、修飾されたレポータータンパク質はN末端及びC末端に異種配列を含む、環状置換された甲虫ルシフェラーゼである。一実施形態では、1つ又は複数の外因性因子(少なくともその1つは関心のある分子でよく、例えば試料中で検出又は特定されるもの)がない場合、1つはN末端に他はC末端にある両方の異種配列を有する修飾されたレポータータンパク質は、対応する修飾されていないレポータータンパク質より小さいか、同じか又は大きな活性を有する。一実施形態では、修飾されたレポータータンパク質は、修飾されていないレポータータンパク質のN及び/又はC末端に存在する1つ又は複数のアミノ酸を欠いてもよく、その欠落は修飾されたレポータータンパク質のレポーター活性を実質的に変更せず、例えば修飾されたレポータータンパク質のレポーター部分の活性は、欠失のない対応するレポータータンパク質の活性の少なくとも0.001%、0.01%、0.1%、1%、10%、50%、100%、又はそれ以上である。一実施形態では、1つ又は複数の外因性因子の存在下で、又は特定条件下で、異種配列のない対応するレポータータンパク質(即ち対応する修飾されていないレポータータンパク質)ではなく両方の異種配列を有する修飾されたレポータータンパク質の活性は、例えば少なくとも2倍、5倍又は10倍又はそれ以上検出可能に変更される。例えば、ラパマイシン存在下で、ラパマイシン結合タンパク質(FRB)及びFK506結合タンパク質(FKBP)に融合されたルシフェラーゼは、FRB及びFKBPを欠くルシフェラーゼと比較して活性は低下している。一実施形態では、外因性因子がない場合又は異なる条件下で、修飾されたレポータータンパク質は検出可能な活性を有さないが他の実施形態においては検出可能な活性を有し、その活性は少なくとも1つの外因性因子の存在下で又は特定条件下で強化することができる。例えば、外因性因子がない場合、修飾されたレポータータンパク質の活性はほとんど又は全くないが、2つの異種配列の非共有結合的相互作用を強化する選択された外因性因子を添加すると、修飾されたレポータータンパク質の活性は強化される。或いは、両方の異種配列を有する修飾されたレポータータンパク質の活性は、少なくとも1つの外因性因子の存在下又は特定条件下では抑制されてもよい。一実施形態では、1つの異種配列は、他の異種配列の領域と非共有結合的に相互作用する、任意選択にリン酸化などにより共有結合的に修飾されてもよい例えば3以上のアミノ酸残基などの領域を含む。甲虫ルシフェラーゼに融合する場合の結合パートナーとして有用な異種配列としては、それには限定されないが、in vitro及び/又はin vivoで相互作用し、また任意選択に例えばタンパク質モデリングに基づき、結合に加わらないが結合パートナーの相互作用を変化させる外因性因子の存在下又は非存在下で選択された距離離れており、甲虫ルシフェラーゼの末端へのそれらの融合は調整可能な甲虫ルシフェラーゼをもたらす結合配列を有するものがある。例示的な異種配列としては、それには限定されないが、FRB及びFKBP内のそれらのもの、タンパク質キナーゼの調節サブユニット(PKa−R)及びタンパク質キナーゼの触媒サブユニット(PKa−C)、src相同領域(SH2)及びリン酸化が可能な配列、例えばチロシン含有配列、14−3−3のアイソフォーム、例えば14−3−3t(Milsら、2000年を参照)、及びリン酸化が可能な配列、WW領域(プロリンが豊富な分子と結合するタンパク質内の配列(Ilsleyら、2002年;Einbondら、1996年を参照))を有するタンパク質、並びにリン酸化が可能な異種配列、例えばセリン及び/又はトレオニン含有配列、並びにジヒドロ葉酸レダクターゼ(DHFR)及びギラーゼB(GyrB)内の配列などがある。
【0019】
他の実施形態において、1つの(第1の)外因性因子の存在下では、結合パートナーであるN末端及びC末端の異種配列を含む修飾されたレポータータンパク質は、前記外因性因子がない場合の活性に対して変化した活性を有し、また異なる(第2の)外因性因子の存在下では、前記修飾されたレポータータンパク質の活性は、前記第1の外因性因子の存在下で、例えば前記第2の外因性因子が前記第1の外因性因子と競合する条件下での活性に対して変化する。一実施形態では、第1の外因性因子がない場合、修飾されたレポータータンパク質は検出可能な活性を全く示さないか低い活性を示し、第1の外因性因子の添加は修飾されたレポータータンパク質の活性の増加をもたらし、この増加は第2の外因性因子の添加により戻すことが可能である。他の実施形態では、第1の外因性因子がない場合、修飾されたレポータータンパク質は検出可能な活性を有し、第1の外因性因子の添加は検出可能な活性の低下又は喪失をもたらすか、或いは検出可能な活性の増加をもたらし、これは第2の外因性因子の添加により戻すことが可能である。修飾されたレポータータンパク質は、任意選択に修飾されていないレポータータンパク質に対してN及び/又はC末端の1つ又は複数のアミノ酸を欠いてもよく、例としては対応する修飾されていないレポータータンパク質のN末端の残基1又は残基1から約10若しくは15、又はその中間の任意の整数、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応する残基の欠失がある。
【0020】
他の実施形態では、修飾されたレポータータンパク質は、修飾されたレポータータンパク質の活性を抑制するなど変更する異種配列をN末端又はC末端に含み、前記活性は第1の外因性因子の添加によって例えば少なくとも部分的に取り戻されるなど変更される。任意選択に、第1の外因性因子の影響は、第2の外因性因子によって可逆的に変更される。一実施形態では、異種配列は基質の進入を抑制することができ、また、異種配列の高次構造は第1の外因性因子の存在下で実質的に変更されて、修飾されたレポータータンパク質はその基質と相互作用することが可能になる。修飾されたレポータータンパク質は、任意選択に修飾されていないレポータータンパク質のN及び/又はC末端の1つ又は複数のアミノ酸を欠いてもよく、例としては対応する修飾されていないレポータータンパク質のN末端の残基1から約10若しくは15、又はその中間の任意の整数の残基、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応するものがある。この実施形態で有用な異種配列は、カルモジュリン(CaM)である。
【0021】
したがって、修飾されたレポータータンパク質は結合パートナーの可逆的相互作用又は異種配列の可逆的高次構造変化を検出するために使用することができ、これは1つ又は複数の因子又はイオン強度若しくは温度などの条件の変化によって強化又は抑制することができる。
【0022】
したがって、本発明の修飾された甲虫ルシフェラーゼは、バイオセンサーとして使用することができる。
【0023】
本発明は、本発明の修飾されたレポータータンパク質をコードしている核酸配列を含んでいる単離された核酸分子(ポリヌクレオチド)も提供する。修飾されたレポータータンパク質及び修飾されたレポータータンパク質のN末端(N末端融合パートナー)及び/又はC末端(C末端融合パートナー)の1つ又は複数のアミノ酸残基を含んでいる融合タンパク質をコードする核酸配列を含んでいる、単離された核酸分子も更に提供されている。したがって、本明細書で使用されるように、「融合タンパク質」は本発明の修飾されたレポータータンパク質のN末端及び/又はC末端に1つ又は複数のアミノ酸を含む、ポリペプチドである。好ましくは、融合タンパク質内の1つ又は複数の融合パートナーの存在は、対応する修飾されたレポータータンパク質に対して、融合タンパク質の検出可能な活性を実質的に変更しない。一実施形態では、融合タンパク質は少なくとも2つの異なる融合パートナーを、1つは修飾されたレポータータンパク質のN末端に、他はC末端に含む。N又はC末端の融合パートナーは、精製のために使用される配列、例えばグルタチオンSトランスフェラーゼ(GST)又はpolyHis配列、修飾されたレポータータンパク質の特性を変えることを意図した配列、例えば修飾されたレポータータンパク質内のキナーゼ部位のための修飾に寛容性のある残基又は領域のタンパク質不安定化配列又はキナーゼ結合ドメイン、或いは融合タンパク質内のレポータータンパク質の1つ又は複数の特性と区別できる特性を有する配列でよい。一実施形態では、融合タンパク質は修飾された甲虫ルシフェラーゼ及び甲虫ルシフェラーゼとは異なるレポータータンパク質である融合パートナーを含み、このレポータータンパク質は分子内対照、例えば蛍光タンパク質として有用である。他の実施形態において、本発明は本発明の修飾された甲虫ルシフェラーゼを含んでいる融合タンパク質をコードする核酸配列及び前記修飾された甲虫ルシフェラーゼの甲虫ルシフェラーゼとは異なるレポータータンパク質をコードする核酸断片を含んでいるベクターを含む。任意選択に、少なくとも甲虫ルシフェラーゼをコードするヒトコドンの最適化された配列などの最適化された核酸配列、及び好ましくは修飾された甲虫ルシフェラーゼ又は修飾された甲虫ルシフェラーゼを含む融合タンパク質は、それらの最適化された配列は甲虫ルシフェラーゼのシグナル強度を高めることができるので、本発明の核酸分子で使用される。核酸配列の最適化は当技術分野で知られており、例えば国際公開02/16944を参照。
【0024】
本発明は、本発明の甲虫ルシフェラーゼなどの修飾されたレポータータンパク質又は融合タンパク質を発現する安定した細胞系、並びに本発明の修飾されたレポータータンパク質又は融合タンパク質をコードする核酸分子を含んでいる発現カセット、及び本発明の核酸分子を宿主細胞で発現することができるベクターも含む。好ましくは、前記発現カセットは、作動可能的に核酸配列に結合されたプロモーター、例えば構成的又は調節可能なプロモーターを含む。一実施形態では、発現カセットは、誘導的プロモーターを含む。本発明の発現カセット又はベクターを含む宿主細胞、例えば原核細胞又は植物若しくは脊椎動物の細胞、例えばそれには限定されないがヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ウマ、ヒツジ、又は齧歯類(例えばウサギ、ラット、白イタチ若しくはマウス)の細胞を含む哺乳類細胞などの真核細胞、及び核酸分子、発現カセット、ベクター、宿主細胞又は本発明の修飾された甲虫ルシフェラーゼ若しくは融合タンパク質を含むキットも提供される。
【0025】
本発明の修飾されたレポータータンパク質は、修飾されていないレポータータンパク質が例えば機能的レポーターとして関心のある分子の様々な状態及び/又は分子を測定及び検出することが不可能な用途で使用することができる。例えば、プロテアーゼ切断認識部位の挿入を含んでいる修飾された甲虫ルシフェラーゼをコードするベクター、又はこの修飾された甲虫ルシフェラーゼは、細胞、細胞溶解物、in vitro転写/翻訳混合物、又は上清に導入され、修飾された甲虫ルシフェラーゼの活性は、例えば1つ又は複数の時点で、対応する修飾されていない甲虫ルシフェラーゼと比較されて検出又は測定される。細胞、細胞溶解物、in vitro転写/翻訳混合物又は上清における発光活性の経時的及び/又は対照、例えば対応する修飾されていない甲虫ルシフェラーゼを有する細胞に対する変化は、プロテアーゼの存在を示す。例えば、本発明は重症急性呼吸器症候群と関連するウイルスを検出するための方法を含む。この方法は、生物学的試料、例えば生理学的組織又は流体試料を、対応する修飾されていないレポータータンパク質に対する内部挿入を含む修飾されたレポータータンパク質、例えば修飾された甲虫ルシフェラーゼと接触させることを含み、前記修飾されたレポータータンパク質は検出可能な活性を有する。前記挿入は、レポータータンパク質配列内の、修飾に寛容性がありウイルスのプロテアーゼに対するアミノ酸認識配列を含む残基又は領域で起こる。修飾されたレポータータンパク質の活性が試料存在下で変更されるか否かが検出又は決定され、それによって試料がウイルスを含むかどうかが示される。
【0026】
本発明は関心のある分子の存在を検出する方法も提供する。例えば、細胞を、調節可能なプロモーターなどのプロモーター、及び関心のある分子と相互作用する挿入を含んでいる本発明の修飾されたレポータータンパク質をコードする核酸配列を含んでいるベクターと接触させる。一実施形態では、形質移入細胞はプロモーターが修飾されたレポータータンパク質の一時的発現を誘発する条件下で培養され、修飾されたレポータータンパク質の検出可能な活性が測定される。
【0027】
また、修飾されたレポータータンパク質をコードする選択された変異ポリヌクレオチドを調製する方法も提供される。この方法は、検出可能な活性を有する修飾されたレポータータンパク質をコードする親ポリヌクレオチドに変異を起こさせて、変異を起こした修飾されたレポータータンパク質をコードする1つ又は複数の変異ポリヌクレオチドを生成することを含む。親ポリヌクレオチドは、対応する修飾されていないレポータータンパク質に対して修飾に寛容性のある残基又は領域が修飾されている、修飾されたレポータータンパク質の読取り枠を含む。修飾されたレポータータンパク質は、直接又は間接的に関心のある分子と相互作用するか、さもなければ対応する修飾されていないレポータータンパク質と比較して条件に感受性を示すアミノ酸配列を含む。関心のある分子との相互作用又はある条件下での活性が、修飾されたレポータータンパク質の相互作用又は活性に対して変更された、変異修飾レポータータンパク質をコードする、1つ又は複数の変異ポリヌクレオチドが選択される。他の実施形態において、本発明は、本発明の修飾されたレポータータンパク質を分子のライブラリーと接触させて、1つ又は複数の分子が、修飾と又は前記修飾されたレポータータンパク質内の非レポータータンパク質配列と相互作用するかどうかを検出又は決定することを含む方法を提供する。
【図面の簡単な説明】
【0028】
【図1】EZ:TNインフレームリンカー挿入プロトコルの概要を示す図である。
【図2】cbg69遺伝子へのTn5挿入突然変異誘発の結果を示す図である。cbg69によってコードされるタンパク質は、野生型コメツキムシルシフェラーゼと比較して位置409(I409V)に1つのアミノ酸置換を有する(図3を参照)。
【図3】コメツキムシルシフェラーゼ(配列番号89)におけるTn5挿入位置(太字)を示す図である。
【図4】Tn5挿入で修飾されたコメツキムシルシフェラーゼの活性を示す図である。
【図5A】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。cbg69ss又はcbg69DEVDによるカスパーゼアッセイにおける相対光単位(RLU)。
【図5B】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。コメツキムシルシフェラーゼ及びカスパーゼ阻害剤(Ac−DEVD−CHO)によるカスパーゼアッセイにおけるRLU。
【図5C】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。様々な量のカスパーゼ−3及びcbg69DEVDによるアッセイにおける経時的RLU。
【図6A】プロテアーゼ認識部位、キナーゼ認識部位、抗体結合部位及び金属結合部位を含むヒンジ領域における修飾を有するコメツキムシルシフェラーゼの配列及び活性を示す図である。6His=6×His−標識;FLAG=DYKDDDDK(配列番号4);DEVD(配列番号106)=カスパーゼ3/7認識部位、Pka=Pkaキナーゼ部位(配列番号90〜96)。挿入は、SnaBI及びSalIを使用してCbgLuc(I409V)のヒンジ領域に導入された。
【図6B】SARSウイルスプロテアーゼ認識部位を有する修飾されたコメツキムシルシフェラーゼの存在下におけるSARSウイルス3CLプロテアーゼ活性を示す図である。
【図6C】ヒンジ領域(配列番号90〜91及び97〜102)にSARSウイルスプロテアーゼ認識部位を有するコメツキムシルシフェラーゼの配列及び活性を示す図である。挿入は、SnaBI及びSalIを使用してCbgLuc(I409V)のヒンジ領域に導入された。
【図7A】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。親(修飾されていない)ホタルルシフェラーゼ(luc+)(配列番号103)のアミノ酸配列。
【図7B】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基233の後にGly(3)Asp(4)LysGly(3)挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図7C】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基233の後にProGlyProGIy(3)Asp(4)LysGly(3)ProGlyPro挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図7D】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基541の後にAsp(4)Lys挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図8】エンテロキナーゼ部位を有する環状置換されたホタルルシフェラーゼのエンテロキナーゼ活性化を示す図である。
【図9】カスパーゼ−3部位を有する環状置換されたホタルルシフェラーゼによる経時的カスパーゼ−3活性化を示す図である。
【図10A】様々な量のカスパーゼ−3及びカスパーゼ−3認識部位を有する環状置換されたホタルルシフェラーゼによるカスパーゼアッセイにおけるRLUを示す図である。
【図10B】様々な量のカスパーゼ−3及びカスパーゼ−3認識部位を有する環状置換されたホタルルシフェラーゼによるカスパーゼアッセイにおけるRLUを示す図である。
【図11】エンテロキナーゼ部位又はカスパーゼ−3部位を有する環状置換されたホタルルシフェラーゼのデータの比較を示す図である。
【図12】SARSウイルスプロテアーゼ認識部位を有する環状置換されたコメツキムシ(CP1:R=Asn401及びCP2:R=Arg223)及びホタル(CP:R=Asp234)ルシフェラーゼによるSARSウイルス3CLプロテアーゼ活性を示すグラフである。
【図13】カスパーゼ−3部位を有しTRAILで処理された環状置換されたルシフェラーゼのRLUを示す図である。
【図14】二重ルシフェラーゼカスパーゼアッセイのためのベクターの概略図である。
【図15】pBINDベクター及び対照ルシフェラーゼ構築物並びに自己集合のためのN又はC末端のルシフェラーゼ構築物の概略図である。
【図16A】完全長ホタルルシフェラーゼ、ホタルルシフェラーゼのN末端部分、ホタルルシフェラーゼのC末端の部分又はN末端及びC末端部分の混合物のSDS−PAGE分析を示す図である。
【図16B】完全長ホタルルシフェラーゼ、ホタルルシフェラーゼのN末端部分、ホタルルシフェラーゼのC末端の部分又はN末端及びC末端部分の混合物のin vitro活性を示す図である。
【図17】CHO又は293の哺乳類細胞抽出物内のルシフェラーゼタンパク質のin vivo活性を示す図である。
【図18】ルシフェラーゼの結合パートナーX又はYとの融合を発現する構築物を調製するためのクローニング方策を示す図である。
【図19】修飾されていないルシフェラーゼタンパク質及びin vitro転写/翻訳反応を使用して生成した1つ又は複数の異種配列とのルシフェラーゼの融合のSDS−PAGEゲル分析を示す図である。
【図20A】ラパマイシンの存在下又は非存在下における、修飾されていないルシフェラーゼ(Luc2)、FRB(ラパマイシン結合タンパク質)と融合されたルシフェラーゼ、FKBP(FK506結合タンパク質)と融合されたルシフェラーゼ並びにFRB及びFKBPと融合されたルシフェラーゼのルシフェラーゼ活性を示す図である。
【図20B】上昇する濃度のFK506の存在下におけるルシフェラーゼとFRBとFKBPとの融合のルシフェラーゼ活性を示す図である。
【図21A】ホタルルシフェラーゼ(Luc2)、コメツキムシルシフェラーゼ(Cbg及びCbr)及びRenilla(RLuc)ルシフェラーゼのFRB及びFKBPとの融合のSDS−PAGE分析を示す図である。
【図21B】ラパマイシンの存在下又は非存在下におけるFRB及びFKBPのホタルルシフェラーゼ、コメツキムシルシフェラーゼ及びRenillaルシフェラーゼとの融合のルシフェラーゼ活性を示す図である。
【図22】Tkプロモーターからルシフェラーゼを発現する構築物を示す図である。
【図23】FK506によるラパマイシン媒介調整の抑制を示している、FRB及びFKBP(FRB1−luc2−FKBP)と融合されたルシフェラーゼで形質移入されたD293細胞におけるラパマイシン存在下でのFK506の滴定を示す図である。
【図24】ラパマイシンの存在下又は非存在下における、TKプロモーター及びFRB−ルシフェラーゼ−FKBP融合のコード領域を有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25A】ラパマイシンの存在下又は非存在下における、FRB−ルシフェラーゼ−FKBP融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25B】ラパマイシンの存在下又は非存在下における、FRB−ルシフェラーゼ融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25C】ラパマイシンの存在下又は非存在下における、ルシフェラーゼのコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25D】ラパマイシンの存在下又は非存在下における、ルシフェラーゼ−FKBP融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図26】EGTA又はCa2+存在下におけるカルモジュリン−ルシフェラーゼ融合の相対発光を示す図である。
【発明を実施するための形態】
【0029】
定義
本明細書で使用される用語「核酸分子」、「ポリヌクレオチド」又は「核酸配列」は、ポリペプチド又はタンパク質前駆体の生産のために必要なコード配列を含む核酸、DNA又はRNAを指す。コードされたポリペプチドは完全長ポリペプチド、その断片(完全長より短い)、又は完全長ポリペプチド若しくはその断片と他のポリペプチドとの融合から生じる融合ポリペプチドであってもよい。
【0030】
本明細書で使用されるように、「核酸」は、1つのヌクレオチドのペントースの3’位置がリン酸ジエステル基によって次のペントースの5’位置と連結され、またヌクレオチド残基(塩基)が特定の配列順序、即ち線状ヌクレオチドで連結されている、共有結合で結ばれたヌクレオチド配列である。本明細書で使用されるように、「ポリヌクレオチド」は長さが約100ヌクレオチドを超える配列を含んでいる核酸である。本明細書で使用されるように、「オリゴヌクレオチド」又は「プライマー」は短いポリヌクレオチド、又はポリヌクレオチドの一部である。オリゴヌクレオチドは、一般的に約2から約100の塩基の配列を含む。語「オリゴ」は語「オリゴヌクレオチド」の代わりに時々使用される。
【0031】
核酸ホスホジエステル結合が置換基モノヌクレオチドのペントース環の5’炭素及び3’炭素で起こるので、核酸分子は「5’末端」(5’端)及び「3’末端」(3’端)を有すると言われる。新しい結合が5’炭素に対して起こるポリヌクレオチドの末端は、その5’末端ヌクレオチドである。新しい結合が3’炭素に対して起こるポリヌクレオチドの末端は、その3’末端ヌクレオチドである。本明細書で使用されるように、末端ヌクレオチドは3’又は5’末端の末端位置にあるヌクレオチドである。
【0032】
モノヌクレオチドが反応して、1つのモノヌクレオチドペントース環の5’リン酸がホスホジエステル結合を通して1方向にその隣の3’酸素と結合するようにオリゴヌクレオチドを形成するので、DNA分子は「5’端」及び「3’端」を有すると言われる。したがって、オリゴヌクレオチドの末端は、その5’リン酸がモノヌクレオチドペントース環の3’酸素と結合しない場合は「5’端」と呼ばれ、その3’酸素が以降のモノヌクレオチドペントース環の5’リン酸と結合しない場合は「3’端」と呼ばれる。
【0033】
本明細書で使用されるように、たとえより大きなオリゴヌクレオチド又はポリヌクレオチドの内部にある場合でも、核酸配列は5’端及び3’端を有すると言うことができる。線状又は環状DNA分子のいずれの場合も、独立した要素は「上流」又は「下流」の5’側又は3’要素と称される。この専門用語は、転写がDNA鎖に沿って5’から3’にかけて進行するという事実を反映する。一般的に、連結された遺伝子(例えば読取り枠又はコード領域)の転写を指示するプロモーター及びエンハンサー要素は、通常、コード領域の5’側又は上流に位置する。しかし、エンハンサー要素は、プロモーター要素及びコード領域の3’側に位置する場合でも、それらの影響を発揮することができる。転写終結及びポリアデニル化シグナルは、コード領域の3’側又は下流に位置する。
【0034】
本明細書で使用する用語「コドン」は、ポリペプチド鎖に取り込まれる特定のアミノ酸、又は開始若しくは終止シグナルを指定する3つのヌクレオチドの配列からなる基本的な遺伝暗号単位である。構造遺伝子に関連して使用される場合の用語「コード領域」は、mRNA分子の翻訳の結果として発達中のポリペプチドで見られるアミノ酸をコードするヌクレオチド配列を指す。一般的に、コード領域は5’側は開始因子メチオニンをコードするヌクレオチドトリプレット「ATG」が結合し、3’側は終止コドン(例えばTAA、TAG、TGA)が結合する。場合によっては、コード領域はヌクレオチドトリプレット「TTC」から始まることも知られている。
【0035】
用語「遺伝子」は、コード配列及び任意選択にDNA配列からのポリペプチドの生産に必要な制御配列を含む、DNA配列を指す。
【0036】
本明細書で使用されるように、用語「異種」核酸配列又はタンパク質は、基準配列に対して由来の異なる配列、例えば外来種に起源を有するものを指し、又は同じ種からのものであっても原形からかなり変化したものでもよい。
【0037】
核酸は、異なる種類の変異を含むことが知られている。「点」突然変異は、野生型配列に対するヌクレオチド配列内の1塩基位置の変化を指す。変異は、1つ又は複数の塩基の挿入又は欠失により核酸配列が野生型などの基準配列と異なるものも指す。
【0038】
本明細書で使用されるように、用語「ハイブリダイズする」及び「ハイブリダイゼーション」は、標的核酸への相補配列のアニーリング、即ち相補配列を含んでいる2つの核酸ポリマー(ポリヌクレオチド)の塩基対合を通してアニーリングする能力を指す。用語「アニールされた」及び「ハイブリダイズされた」は本明細書全体を通して互換的に使用され、部分相補性だけを有する領域の結合を含む、相補配列及び標的核酸の間の任意の特異的且つ再生可能な相互作用を包含するものとする。普通は天然核酸では見られないある種の塩基が本発明の核酸に含まれてもよく、例としてはイノシン及び7−デアザグアニンがある。核酸技術の当業者ならば、二重鎖の安定性は多くの変数、例えば相補配列の長さ、オリゴヌクレオチドの塩基組成及び配列順序、イオン強度並びにミスマッチ塩基対の発生率などを考慮して経験的に決めることができる。核酸二重鎖の安定性は、融点又は「Tm」で測定される。特定の条件下での特定の核酸二重鎖のTmは、平均して塩基の半分が分離する温度である。
【0039】
用語「組換えDNA分子」は、自然界では通常一緒に見られない少なくとも2つのヌクレオチド配列を含んでいるハイブリッドDNA配列を意味する。用語「ベクター」は、DNAの断片を挿入又はクローニングすることができる核酸分子に関して使用され、細胞にDNAセグメントを移すために使用でき、また細胞内での複製が可能である。ベクターは、プラスミド、バクテリオファージ、ウイルス、コスミドなどから得ることができる。
【0040】
本明細書で使用されるように、用語「組換えベクター」及び「発現ベクター」は、所望のコード配列及び特定の宿主生物における作動可能的に結合されたコード配列の発現のために必要な、適当なDNA又はRNA配列を含むDNA又はRNA配列を指す。原核生物の発現ベクターは、プロモーター、リボソーム結合部位、宿主細胞における自律複製のための複製起点、及び恐らく他の配列、例えば任意選択のオペレーター配列、任意選択の制限酵素部位を含む。プロモーターは、RNAポリメラーゼにDNAとの結合及びRNA合成の開始を指示するDNA配列と定義される。真核生物の発現ベクターは、プロモーター、任意選択にポリアデニル化シグナル及び任意選択にエンハンサー配列を含む。
【0041】
タンパク質又はポリペプチドをコードするヌクレオチド配列を有するポリヌクレオチドは、遺伝子のコード領域を含んでいる核酸配列を意味し、又は、核酸配列は遺伝子産物をコードすると換言することができる。コード領域は、cDNA、ゲノムDNA又はRNAのいずれの形態でも存在することができる。DNA形態で存在するとき、オリゴヌクレオチドは一本鎖(即ちセンス鎖)又は二本鎖でよい。エンハンサー/プロモーター、スプライス部位、ポリアデニル化シグナルその他などの適当な調節要素は、適切な転写開始を可能にし且つ/又は一次RNA転写産物のプロセシングを修正するために、必要ならば遺伝子のコード領域に近接して置くことができる。或いは、本発明の発現ベクターで利用されるコード領域は、内因性のエンハンサー/プロモーター、スプライス部位、介在配列、ポリアデニル化シグナルその他を含んでもよい。更なる実施形態において、コード領域は内因性及び外因性の調節要素の組み合わせを含んでもよい。
【0042】
用語「転写調節要素」又は「転写調節配列」は、核酸配列の発現の一部の態様を調節する遺伝的要素又は配列を指す。例えば、プロモーターは作動可能的に結合されたコード領域の転写の開始を容易にする調節要素である。他の調節要素としては、それには限定されないが、転写因子結合部位、スプライシングシグナル、ポリアデニル化シグナル、終止シグナル及びエンハンサー要素がある。
【0043】
真核生物における転写調節シグナルは、「プロモーター」及び「エンハンサー」要素を含む。プロモーター及びエンハンサーは、転写に関係する細胞タンパク質と特異的に相互作用するDNA配列の短い配列からなる。プロモーター及びエンハンサー要素は、酵母、昆虫及び哺乳類細胞の遺伝子を含む様々な真核生物の供与源から単離された。プロモーター及びエンハンサー要素はウイルスからも単離され、類似の調節要素、例えばプロモーターも原核生物で見られる。特定のプロモーター及びエンハンサーの選択は、関係するタンパク質を発現するために使用される細胞の種類によって決まる。真核生物のプロモーター及びエンハンサーは広い宿主域を有するものもあれば、限られたサブセットの細胞種で機能的なものもある。例えば、SV40初期遺伝子エンハンサーは、多くの哺乳類の種からの多種多様な細胞種で非常に活性であり、哺乳類細胞内でのタンパク質の発現のために広く使われてきた。広範な哺乳類細胞種で活性を示すプロモーター/エンハンサー要素の他の2つの例は、ヒト伸長因子I遺伝子及びRous肉腫ウイルスの末端反復配列、並びにヒトサイトメガロウイルスからのものである。
【0044】
用語「プロモーター/エンハンサー」は、プロモーター及びエンハンサー機能(即ち先に述べたようにプロモーター要素及びエンハンサー要素によって提供される機能)を提供することができる配列を含んでいる、DNAセグメントを意味する。例えば、レトロウイルスの末端反復配列は、プロモーター及びエンハンサーの両方の機能を含む。エンハンサー/プロモーターは「内因性」又は「外因性」又は「異種性」でよい。「内因性」エンハンサー/プロモーターは、ゲノム内の与えられた遺伝子と自然に連結しているものである。「外因性」又は「異種性」エンハンサー/プロモーターは、遺伝子の転写が結合したエンハンサー/プロモーターによって指示されるように、遺伝子操作(即ち分子生物学的技術)によって遺伝子の近くに置かれたものである。
【0045】
発現ベクター上の「スプライシングシグナル」の存在は、真核宿主細胞における組換え転写産物のより高い発現レベルをしばしばもたらす。スプライシングシグナルは一次RNA転写産物からのイントロンの除去を媒介し、スプライス供与部及び受容部位からなる。通常使用されるスプライス供与部及び受容体部位は、SV40の16S RNAからのスプライス部位である。
【0046】
真核細胞における組換えDNA配列の効率的な発現のためには、得られた転写産物の効率的な終結及びポリアデニル化を指示するシグナルの発現を必要とする。転写終止シグナルは、通常、ポリアデニル化シグナルの下流に見られ、長さは2、3百ヌクレオチドである。本明細書で使用する用語「ポリ(A)部位」又は「ポリ(A)配列」は、発達中のRNA転写物の終結及びポリアデニル化を指示するDNA配列を意味する。ポリ(A)テールがない転写産物は不安定であり速やかに分解するので、組換え転写産物の効率的なポリアデニル化が望ましい。発現ベクターで利用されるポリ(A)シグナルは「異種性」又は「内因性」でよい。内因性のポリ(A)シグナルは、ゲノム内の与えられた遺伝子のコード領域の3’末端で自然に見られるものである。異種ポリ(A)シグナルは、1つの遺伝子から単離され、他の遺伝子に対して3’に位置するものである。通常使用される異種ポリ(A)シグナルは、SV40 ポリ(A)シグナルである。SV40 ポリ(A)シグナルは、237bpのBamH I/Bcl I制限断片に含まれ、終結及びポリアデニル化を指示する。
【0047】
真核生物の発現ベクターは、また「ウイルスのレプリコン」又は「ウイルスの複製開始点」を含んでもよい。ウイルスのレプリコンは、適当な複製因子を発現している宿主細胞内でのベクターの染色体外複製を可能にする、ウイルスのDNA配列である。SV40又はポリオーマウイルスの複製起点を含んでいるベクターは、適当なウイルスのT抗原を発現する細胞内で高いコピー数(最高104の複製/細胞)で複製する。対照的に、ウシの乳頭腫ウイルス又はEBウイルスからのレプリコンを含んでいるベクターは、染色体外で低コピー数(約100の複製/細胞)を複製する。
【0048】
用語「in vitro」は、人工環境及び人工環境内で起こる過程又は反応を指す。in vitro環境としては、それには限定されないが、試験管及び細胞溶解物がある。用語「in vivo」は自然環境(例えば動物又は細胞)、及び自然環境内で起こる過程又は反応を指す。
【0049】
用語「発現系」は、関心のある遺伝子の発現を決定する(例えば、検出する)ための任意のアッセイ又は系を指す。分子生物学の分野に熟練している者は、多種多様な発現系のいずれも使用されることを理解しよう。広範囲にわたる適当な哺乳類細胞が広範囲の供給源(例えばアメリカンタイプカルチャーコレクション、Rockland、メリーランド州)から入手可能である。形質転換又は形質移入の方法及び発現媒体の選択は、選択された宿主系によって決まる。形質転換及び形質移入の方法は、当技術分野で公知である。発現系は、関心のある遺伝子(例えばレポーター遺伝子)が調節配列に結合され、遺伝子の発現は遺伝子の発現を抑制又は誘発する剤による処理の後に観察される、in vitro遺伝子発現アッセイを含む。遺伝子発現の検出は任意の適当な手段、例えばそれには限定されないが、発現されたmRNA又はタンパク質(例えばレポーター遺伝子の検出可能な生成物)の検出を通して、又は関心のある遺伝子を発現している細胞の表現型の検出可能な変化を通して可能である。発現系は、切断事象又は他の核酸若しくは細胞の変化が検出されるアッセイを含んでもよい。
【0050】
本明細書で使用されるように用語「野生型」は、天然の供与源から単離されたその遺伝子又は遺伝子産物の特性を有する遺伝子又は遺伝子産物を指す。野生型遺伝子は、集団で最もしばしば観察され、したがって任意にその遺伝子の「野生型」形態と称されるものである。対照的に、用語「突然変異体」は、野生型の遺伝子又は遺伝子産物に対して、配列及び/又は機能的性状(即ち、特性変化)の変化を示す遺伝子又は遺伝子産物を指す。天然の突然変異体を単離することが可能であると指摘されている。これらは野生型の遺伝子又は遺伝子産物に対して変化した特性を有することで確認される。
【0051】
「単離されたオリゴヌクレオチド」又は「単離されたポリヌクレオチド」のように核酸に関して使用される場合の用語「単離された」は、その供与源内で通常関連している少なくとも1つの混在物から識別及び分離される核酸配列を指す。したがって、単離された核酸は、自然界でそれが見られるものとは異なる形態又はセッティングで存在する。対照的に、単離されていない核酸(例えばDNA及びRNA)は、それらが自然界で存在する状態で見られる。例えば、与えられたDNA配列(例えば遺伝子)は、宿主細胞染色体上で近隣の遺伝子に近接して見られる。RNA配列(例えば、特定のタンパク質をコードする特定のmRNA配列)は、多くのタンパク質をコードする他の多数のmRNAとの混合物として細胞内で見られる。しかし、単離された核酸としては、例えばその核酸が自然の細胞のそれと異なる染色体上の位置にあるか、さもなければ自然で見られるものとは異なる核酸配列に連結している、通常その核酸を発現している細胞内の核酸がある。単離された核酸又はオリゴヌクレオチドは、一本鎖又は二本鎖の形態で存在することができる。単離された核酸又はオリゴヌクレオチドがタンパク質を発現するために利用される場合は、オリゴヌクレオチドは最低限センス又はコード鎖(即ち、オリゴヌクレオチドは一本鎖でよい)を含むが、センス及びアンチセンスの両方の鎖(即ち、オリゴヌクレオチドは二本鎖でよい)を含んでもよい。
【0052】
「ペプチド」、「タンパク質」及び「ポリペプチド」は、長さ又は翻訳後修飾(例えばグリコシル化又はリン酸化)に関係なく任意のアミノ酸鎖を意味する。本発明の核酸分子は、天然のタンパク質の変異体又はそのポリペプチド断片をコードしてもよく、それは、それが由来する天然の(生来若しくは野生型)タンパク質のアミノ酸配列と少なくとも85%、90%、95%又は99%同一であるアミノ酸配列を有する。用語「融合ポリペプチド」又は「融合タンパク質」は、N及び/又はC末端が1つ又は複数の異種配列(例えば非ルシフェラーゼポリペプチド)に結合されている参照タンパク質(例えばルシフェラーゼ)を含んでいるキメラタンパク質を指す。一部の実施形態において、本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長ポリペプチドの一部は、対応する完全長機能的(非キメラ)ポリペプチドの活性の少なくとも一部を保持してもよい。他の実施形態において、外因性因子又は関心のある分子がない場合、本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長機能的ポリペプチドの一部は、対応する完全長機能的ポリペプチドに対して活性を失ってもよい。他の実施形態において、外因性因子の存在下で本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長機能的ポリペプチドの一部は、対応する完全長機能的ポリペプチドに対して活性の少なくとも一部若しくは実質的に同じ活性を保持してもよく、或いは、活性を失ってもよい。
【0053】
ポリペプチド分子は、ペプチド結合が第1のアミノ酸残基の骨格アミノ基及び第2のアミノ酸残基の骨格カルボキシル基の間で起こるので、「アミノ末端」(N末端)及び「カルボキシ末端」(C末端)を有すると言われる。ポリペプチド配列に関した用語「N末端」及び「C末端」は、それぞれポリペプチドのN末端及びC末端領域の一部を含むポリペプチド領域を指す。ポリペプチドのN末端領域の一部を含む配列は、主にポリペプチド鎖のN末端半分からのアミノ酸を含むが、そのような配列に限定はされない。例えば、N末端配列は、ポリペプチドのN末端及びC末端の両半分からの塩基を含むポリペプチド配列の内側の部分を含んでもよい。同じことは、C末端領域にも当てはまる。N末端及びC末端の領域は、その必要はないが、ポリペプチドのそれぞれ最終N末端及びC末端を規定しているアミノ酸を含んでもよい。
【0054】
本明細書で使用されるように用語「組換えタンパク質」又は「組換えポリペプチド」は、組換えDNA分子から発現されたタンパク質分子を指す。対照的に、用語「未変性タンパク質」は、本明細書では天然の(即ち遺伝的組換えではない)供与源から単離されたタンパク質の意味で使用される。タンパク質の未変性形態に対して同一の特性を有するタンパク質の組換え体を生成するために、分子生物学的技術を使用することができる。
【0055】
本明細書で使用されるように用語「細胞」、「細胞系」及び「宿主細胞」は互換的に使用され、そのような呼称は全てこれらの呼称の後代又は潜在的後代を含む。「形質転換細胞」は、それに(又はその先祖に)本発明の核酸分子が導入された細胞を意味する。任意選択に、本発明の核酸分子は、遺伝子によってコードされたタンパク質又はポリペプチドを産生することができる安定的に形質移入された細胞系を作製するために、適当な細胞系に導入してもよい。ベクター、細胞及びそのような細胞系を構築する方法は、当技術分野で公知である。用語「形質転換体」又は「形質転換細胞」は、経代数に関係なく元の形質転換細胞に由来する一次形質転換細胞を含む。故意又は偶発的な突然変異のために、必ずしも全ての後代においてDNAの内容が正確に同一ではないかもしれない。それにもかかわらず、形質転換体の定義には、当初の形質転換細胞でスクリーニングされたものと同じ機能を有する変異後代が含まれる。
【0056】
用語「相同性」は、2つ以上の配列の間の相補性の程度を指す。部分相同性又は完全な相同性(即ち同一性)があってもよい。相同性は、しばしば配列解析ソフトウェア(例えばGenetics Computer Group、ウィスコンシン大学バイオテクノロジーセンター、1710 University Avenue、マジソン、ウィスコンシン州、53705、のSequence Analysis Softwareパッケージ)を使用して測定される。そのようなソフトウェアは、相同性の程度を様々な置換、欠失、挿入、その他の修飾に割り当てることによって類似の配列を比較する。保存的な置換は、一般に下記群内の置換を含む:グリシン、アラニン;バリン、イソロイシン、ロイシン;アスパラギン酸、グルタミン酸、アスパラギン、グルタミン;セリン、スレオニン;リジン、アルギニン;及び、フェニルアラニン、チロシン。
【0057】
「単離されたタンパク質」又は「単離されたポリペプチド」のようにポリペプチドに関して使用される場合の用語「単離された」は、その供与源内で通常関連している少なくとも1つの混在物から識別及び分離されるポリペプチドを指す。したがって、単離されたポリペプチドは、自然界でそれが見られるものとは異なる形態又はセッティングで存在する。対照的に、単離されていないポリペプチド(例えばタンパク質及び酵素)は、それらが自然界で存在する状態で見られる。
【0058】
用語「精製された」又は「精製する」は、関係する構成要素、例えばタンパク質又は核酸から混在物の一部を除去する任意の過程の結果を意味する。精製された構成要素の試料中のパーセントは、それによって増加される。
【0059】
本明細書で使用されるように、「純粋」は対象種が存在する支配的な種(即ち、モルベースでは組成物中の他のいかなる個々の種よりも豊富)であることを意味し、また好ましくは実質的に精製された分画は、存在する全ての高分子種の少なくとも約50パーセント(モルベースで)を対象種が占める組成物である。通常、「実質的に純粋な」組成物は、組成物中に存在する全ての高分子種の約80パーセント、より好ましくは約85%、約90%、約95%及び約99%を超える割合を含む。最も好ましくは、対象種は、組成物が本質的に単一の高分子種からなる本質的に均質状態(従来の検出法によって混在種は組成物内に検出されない)になるまで精製される。
【0060】
本明細書で使用されるように用語「作動可能的に結合された」は、与えられた遺伝子の転写及び/又は所望のタンパク質分子の合成を指示することができる核酸分子が生成されるような核酸配列の結合を指す。この用語は、機能的(例えば、酵素的に活性、結合パートナーとの結合が可能、抑制することが可能、その他)タンパク質又はポリペプチドが生成されるような、アミノ酸をコードする配列の結合を指す。
【0061】
本明細書で使用されるように、用語「ポリヒスチジントラクト」又は(Hisタグ)は、2から10のヒスチジン残基を含んでいる分子、例えば5から10の残基のポリヒスチジントラクトを指す。ポリヒスチジントラクトは、固定化金属、例えばニッケル、亜鉛、コバルト又は銅、キレートカラムの上の共有結合した分子のアフィニティ精製、又は他の分子(例えばHisタグと反応する抗体)との相互作用を通した精製を可能にする。
【0062】
「タンパク質不安定化配列」としては、それには限定されないが、PEST配列、例えば有糸分裂サイクリンなどのサイクリン、ウラシル透過酵素又はODCからのPEST配列、ODCなど短寿命タンパク質のC末端領域、初期応答タンパク質、例えばサイトカイン、リンフォカイン、プロトオンコジーン、例えばc−myc若しくはc−fos、MyoD、HMG CoAレダクターゼ、又はS−アデノシルメチオニンデカルボキシラーゼ、CL配列、サイクリン破壊ボックス又はN−デグロン(degron)からの配列などがある。
【0063】
本明細書で使用されるように、「マーカー遺伝子」又は「レポーター遺伝子」はその遺伝子を発現している細胞に異なった表現型を与える遺伝子であり、したがって、その遺伝子を有しない細胞からのその遺伝子を有する細胞の区別を可能にする。化学的手段、例えば選択剤(例えば除草剤、抗生物質、その他)の使用を通してそれについて「選択」することのできる形質をマーカーが付与するのか、或いはそれは単に観察又は検査を通して、即ち「スクリーニング」により特定することができる「レポーター」形質に過ぎないのかによって、そのような遺伝子は、選択可能な又はスクリーニングが可能なマーカーのいずれかをコードすることができる。本開示の要素は、特定のマーカー遺伝子の使用を通して詳細に例証される。当然ながら、適当なマーカー遺伝子又はレポーター遺伝子の多くの例は当分野で公知であり、本発明の実施において使用することが可能である。したがって、以下の説明は網羅的というよりは例示的であることは理解されよう。本明細書で開示されている技術及び当技術分野で公知の一般的組換え技術に照らして、本発明は任意の遺伝子の変更を可能にする。例示的な修飾されたレポータータンパク質は、修飾されたレポーター遺伝子、例えばそれには限定されないが、ネオ遺伝子、β−gal遺伝子、gus遺伝子、cat遺伝子、gpt遺伝子、hyg遺伝子、hisD遺伝子、ble遺伝子、mprt遺伝子、bar遺伝子、ニトリラーゼ遺伝子、ガラクトピラノシド遺伝子、キシロシダーゼ遺伝子、チミジンキナーゼ遺伝子、アラビノシダーゼ遺伝子、突然変異アセトラクテートシンターゼ遺伝子(ALS)若しくはアセトアシッドシンターゼ遺伝子(AAS)、メトトレキセート抵抗性dhfr遺伝子、ダラポンデハロゲナーゼ遺伝子、5−メチルトリプトファンに抵抗性を付与する突然変異アントラニル酸シンターゼ遺伝子(国際公開97/26366)、R座位遺伝子、βラクタマーゼ遺伝子、xylE遺伝子、α−アミラーゼ遺伝子、チロシナーゼ遺伝子、ルシフェラーゼ(luc)遺伝子(例えばRenilla reniformisルシフェラーゼ遺伝子、ホタルルシフェラーゼ遺伝子若しくはコメツキムシルシフェラーゼ(Pyrophorus plagiophthalamus)遺伝子)、エクオリン遺伝子、赤色蛍光タンパク質遺伝子、又は緑色蛍光タンパク質遺伝子などの修飾を含んでいる核酸分子によってコードされる。
【0064】
本明細書で特定される全てのアミノ酸残基は、天然のL−コンフィギュレーションである。標準ポリペプチド命名法に従い、アミノ酸残基の略記号は以下の対応表に示す通りである。
【表1】

【0065】
I.修飾に寛容性のあるレポータータンパク質の残基又は領域を特定する方法
破壊されるなど修飾されても転写及び翻訳されたときに所望の、例えば容易に検出可能な遺伝子産物を生成することができるレポータータンパク質遺伝子内の部位及び/又は領域を特定するために、多数の方法が利用できる。例えば、1つ又は複数のアミノ酸残基のヌクレオチドをレポータータンパク質遺伝子に対して削除及び/又は挿入するために、増幅反応を使用することができる。或いは、挿入変異のライブラリーを調製するためにトランスポゾンを使用することができる。トランスポゾンは、原核生物及び真核生物のゲノムで見られる移動性のDNA配列である。トランスポゾン標識は、プライマー結合部位をランダムに分散し、遺伝子「ノックアウト」を作製し、物理的標識又は遺伝標識を大きな標的DNAに導入するための強力な研究手段として、長く認められてきた。本発明の修飾されたレポータータンパク質を調製するために有用なレポーター遺伝子内の挿入は、レポータータンパク質のコード領域内部のインフレーム挿入である。以下の実施例は例示のために示したものにすぎないが、レポーター遺伝子内の挿入に寛容性のある領域を特定するための、Tn5ベースの系(Epicentre、マジソン、ウィスコンシン州からのEZ:TN(商標))及びTn7ベースの系(GPS−M Mutagenesis System、New England Biolabs、Inc.)の使用を記載する。
【0066】
A.Tn−5挿入突然変異
頻繁に使われる転位系の1つは、グラム陰性菌から単離されたTn5系である。Tn5トランスポザーゼは、クローニングされ、高比活性に精製された小さな、単一のサブユニット酵素であり、宿主細胞因子を必要とせずに転位を実行する。さらに、標的DNAへのTn5トランスポゾンの挿入は非常にランダムであり、また単純な過程で進行する。Tn5トランスポザーゼは、その短い19の塩基対Mosaic End(ME)Tn5トランスポザーゼ認識配列の間に含まれるいかなるDNA配列も転位させる。EZ:TNインフレームリンカー挿入プロトコルの概要を図1に示す。
【0067】
i.トランスポゾン挿入反応
標的DNAの調製。標的レポーターDNAは、トランスポゾン遺伝子、例えばカナマイシン抵抗性遺伝子によってコードされていないものを選択する。トランスポゾン挿入反応は標的DNA調製物内の高レベルのRNA汚染によってかなり抑制されることはないが、標的DNAが標的転位の直接の競合者である染色体DNAによって高度に汚染されるならば、クローン数は減少する。プラスミド及びコスミドクローンは、標準の微小溶解手法によって精製することが可能であり、挿入反応で標的DNAとして使用することができる。BAC又はコスミドクローンなどの低いコピー数のベクターは、より高いモル比率の大腸菌染色体DNAによってしばしば汚染され、したがってトランスポゾン挿入頻度が低下する。したがって、挿入反応の前に染色体DNAを除去するためにBAC及びコスミドDNAを精製することが好ましい。
【0068】
in vitroトランスポゾン挿入反応。複数の挿入事象を最小にしてトランスポゾン挿入の効率を最大にするために、反応条件が最適化される。例えば、等モル量のトランスポゾンがそのモル数の標的DNAに加えられる。
1.以下の順序で加えることによってトランスポゾン挿入反応混合物を調製する:
1μl 10×反応緩衝液
0.2μg 標的DNA*
xμl モル当量トランスポゾン
9μlの反応体積にxμlの滅菌水
1μl トランスポザーゼ
10μl 総反応体積
2.反応混合物を37℃で2時間インキュベートする。
3.μl停止液を加えて反応を止める。
混合して70℃で10分間加熱する。
反応混合物は−20℃で保存することができる。
【0069】
ii.トランスポゾン挿入クローンの選択
形質転換及び回収。1回の反応で得られるトランスポゾン挿入クローンの数は、とりわけ使用されるコンピテント細胞の形質転換効率によって決まる。コンピテント細胞の形質転換効率がより高いほど、より多くの挿入クローンが得られる。ベクターの多量体形態を生成する可能性をなくすために、大腸菌のrecA−系統が好まれる。また、宿主系統はトランスポゾンで存在するいかなる抗生物質耐性マーカー、例えばカナマイシン抵抗性マーカーを発現してはならない。
1.挿入反応混合物の1μlを使用して、recA−大腸菌、例えばエレクトロコンピテント細胞を形質転換する。
2.エレクトロポレーションの直後にSOC培地を1mlの最終体積までエレクトロポレーションセルに加えることによって、エレクトロポレーションされた細胞を回収する。培地/細胞をピペットで穏やかに混ぜる。管に移して37℃の撹拌機で30〜60分間インキュベートして細胞成長を促進する。
【0070】
形質転換体の平板培養及び選択。トランスポゾン挿入クローンは、抗生物質を含んでいるプレート上で選択される。Tn5については、カナマイシンを含んでいるプレートを使用してもよいが、トランスポゾンはネオマイシン及びG418抵抗性を大腸菌に付与することもできる。
1.細胞の一部を50μg/mlのカナマイシンを含んでいるLBプレート上で平板培養する。
2.トランスポゾン挿入効率を測定するために、形質転換反応の同一の希釈液及び希釈アリコートを、標的DNAを特異的に選択する抗生物質(例えば対照DNAでは100μg/mlのアンピシリン)を含んでいる第2のプレート上で平板培養する。転位頻度は、対照DNAのKanR/AmpRのクローン比率によって与えられる。
3.37℃で一晩プレートを増殖させる。1%のトランスポゾン挿入効率及び高純度の標的DNA(即ち染色体DNAの汚染がほとんど又は全くない)の使用を仮定すると、1プレートにつき約100〜500KanRクローンが存在する。
【0071】
iii.インフレーム19コドン挿入の生成
トランスポゾン挿入のマッピング。Tn5は、ランダムに標的DNAに挿入される。したがって、各クローンにおけるトランスポゾン挿入部位は、3つの方法の1つによって制限酵素消化、例えばNotI消化の前に決定しなければならない:
1.挿入クローンは、順方向及び逆方向のトランスポゾン特異プライマーを使用して二方向に配列決定をすることが可能である。各クローンの挿入部位も、配列決定の前にマッピングすることが可能である。
2.挿入部位は、コロニー微小溶解DNAをテンプレートとして使用して、PCR産物のサイズ分析によってマッピングすることが可能である。挿入部位をマッピングするために、順方向又は逆方向のトランスポゾン特異プライマー及びベクター特異フランキングプライマーを使用してもよい。
3.或いは、挿入部位は制限酵素消化物によってマッピングすることが可能である。
【0072】
所望のクローンのトランスポゾン挿入部位が一旦決定されると、それらのクローンは制限酵素、例えばNotIで個々に消化してDNAを線状にする。線状にされたDNAは次に精製される(例えば、アガロースゲル電気泳動、カラム精製、などによって)。
【0073】
再連結及び形質転換。線状にされたクローンは、T4 DNAリガーゼを使用して再連結される。再連結が成功すると、単一の制限部位、例えばNotIが再生され、3つ全ての読取り枠への57ヌクレオチド(19コドン)の挿入が作製される。再連結されたDNAは選択された細胞に形質転換され、元のクローニングベクター上に存在する抗生物質マーカー(例えば対照DNAではアンピシリン)を使用して組換え体が選択される。
【0074】
19コドン挿入クローンの分析。57のヌクレオチドのうちの9は、トランスポゾン挿入部位に直接連なっている9bp配列の複製の結果である。標的DNAによってコードされるタンパク質のアミノ酸配列は、19コドン挿入の両側で保存される。
【0075】
iv.トランスポゾン挿入クローンのDNA配列決定
プライマーの考慮。プライマーは、通常使用されるクローニングベクターとの相同性を最小にするように構築されなければならず、また、各プライマーの配列は、ベクターとの配列相同性が極小になるように使用者の特定のクローニングベクターのそれと比較しなければならない。
【0076】
標的部位複製。Tn5の触媒によるトランスポゾン挿入は、1コピーが挿入されたトランスポゾンの各側に直接連なる、9bp標的部位配列の複製をもたらす。
【0077】
挿入配列のためのトランスポゾン配列の識別。プライマーがトランスポゾン末端近くの領域にアニールするならば、各シークエンシング反応から得られる第1の配列データはトランスポゾンDNAのそれである。
【0078】
B.Tn7ベースの挿入突然変異
GPS−M Mutagenesis Systemは、Tn7ベースのトランスポゾンをDNA標的にランダムに挿入するために、TnsABC*トランスポザーゼを使用する。標的DNAは、プラスミド、コスミド、BAC又は精製された染色体DNAであってもよい。挿入部位が翻訳される遺伝子セグメント内にあるならば、これは通常無効な(機能の損失)変異をもたらす。挿入に対しては最小限の部位による好みがあるので、任意の読取り枠の破壊は可能である。標的の免疫性のために、in vivoでは約190kbの範囲でDNA分子1つにつき挿入は1つだけ起こる。したがって、in vitro反応は、それぞれ異なる位置に転位因子を含む標的DNA分子の集団を生じる。
【0079】
トランスポゾン供与体は、カナマイシンなどの抗生物質抵抗性マーカーを増やすか又は取り替えることによって修飾することが可能である。供与体プラスミドは標準の大腸菌実験系統で増殖させることができ、ベクター骨格はAmprなどのトランスポゾンと異なる抗生物質マーカー及び複製起点を有す。反応しない供与体分子を破壊して望ましくない反応生成物を避けるために、供与体はレアカッティング酵素、例えばPI−SceI(VDE)による消化で破壊することができる。突然変異を起こすDNAが元来コンピテントな生物(一本鎖DNAを取り込む)へ形質転換される適用については、ギャップが埋められて連結される。
【0080】
i.反応プロトコル
1.以下の試薬を混合する(20μl反応液あたり):
2μl 10×緩衝液
1μl スーパーコイルカスタム供与体(0.02μg)
0.08μg 標的DNA
dH2O
18μl 総体積
ピペット操作を2、3回行ってよく混ぜる。
2.1μlトランスポザーゼを各管に加える。再び混合する。
3.37℃で10分間インキュベートする。これは、集合反応である。
4.1μl開始溶液を各管に加える。ピペット操作を2、3回行ってよく混ぜる。
5.37℃で1時間インキュベートする。これは、鎖転移反応である。
6.75℃で10分間加熱不活化する。注:65℃は十分でない。
7.任意選択のギャップ修復。
8.5μl 10×Pl−SceI緩衝液を加える
0.5μl BSA
18.5μl dH2O
6μl Pl−SceI(VDE)(6単位)
9.37℃で1〜2時間インキュベートする。
10.75℃で10分間インキュベートする。
11.形質転換する。サブクローニング効率細胞(pUC1マイクログラムにつき107)による化学的形質転換のためには、非希釈反応液の1μl及び10μlを形質転換する。エレクトロポレーション(pUC1マイクログラムにつき>109)のためには、dH2Oで10倍に希釈して1μl及び10μlを形質転換する。増殖のためには、1mlのLBに又は製造業者の指示通りに形質転換混合物を希釈し、37℃で1時間通気下でインキュベートする。選択のないこの期間は、薬剤特にカナマイシンに対する耐性の発現のために必要である。
【0081】
ii.一般的考慮
標的の量。標的DNAの推奨質量(1反応につき0.08μg)は、プラスミド標的に対して有効である。コスミド及びBACについては、2:1(供与体対標的)のモル比が有効である。比率を4:1に増やすと、僅かに効率が低下する。
【0082】
供与体対標的比率。推奨されている供与体:標的質量比(1:4、20μl反応液につき0.08μg標的)が最適である。逸脱が小さければ、回収された生成物の数の小さな変化がもたらされるだけである。しかし、供与体の飽和量は反応を抑制し、二重挿入の蓄積をもたらす可能性がある。
【0083】
添加順序。水、標的DNA、緩衝液及び供与体プラスミドを先ず加え、次にトランスポザーゼを加える。開始溶液は、集合反応の後に加えなければならない。
【0084】
集合反応。この工程を省略すると、複合生成物の比率が増加する。
【0085】
インキュベーション時間。反応物は37℃で少なくとも1時間で線状になる。極めて長いインキュベーション時間は、二重挿入の蓄積をもたらす可能性がある。
【0086】
インキュベーション温度。室温及び30℃では、反応はより遅い速度であるが進行する。BACとの反応では、30℃が推奨される。
【0087】
熱殺傷。75℃で10分間の加熱処理は反応複合体を有効に破壊する。65℃で20分間の熱処理は十分でない。フェノール/クロロホルム抽出後のアルコール沈殿も有効である。
【0088】
手順の拡大縮小。最終体積及び全ての構成要素の体積を同じパーセンテージによって増加又は減少させる;2つのDNA種及びタンパク質の相対濃度は、緩衝液条件と同様に非常に重要である。
【0089】
酵素名。Pl−SceI(VDE)はl−SceIと同じでない。供与体の消化のためにはPl−SceI(VDE)を使用し、得られた挿入のマッピングのためにはScelを使用する。
【0090】
ギャップ修復。この工程は大腸菌への形質転換には必要でなく、所望の適用が元来コンピテントな細菌への形質転換を含むときだけ必要である。元来コンピテントな細菌としては、ナイセリア(Neisseria)、ヘモフィルス(Haemophilus)、バシラス(Bacillus)、ニューモコッカス(Pneumococcus)、スタフィロコッカス(Staphylococcus)及びストレプトコッカス(Streptococcus)属の種がある。これらの生物へのDNA取込みは、他の鎖の内部移行に伴う1本の鎖の分解を含む。ギャップ修復なしの場合、トランスポゾン挿入部位の5塩基ギャップは、トランスポゾン挿入を一方又は他の側のフランキングDNAから解き放す。コンピテンスが化学的に又はエレクトロポレーションによって誘発される生物(例えば大腸菌及び他の腸内細菌の組織培養細胞、その他)は、両方のDNA鎖を取り込む。挿入部位のギャップは、細胞内の機構により有効に修復される。
【0091】
iii.ギャップ修復プロトコル
7.フェノール/クロロホルム抽出(50μl)。
8.エタノール沈殿:
6μl 3M NaAcetate
100μl EtOH
−20℃で20分間インキュベートする
微量遠心管で10分間遠心分離する
9.15μl TEで再懸濁する。
10.1μl DNAポリメラーゼI(大腸菌)(10単位)
3μl 10×EcoPol緩衝液
9μl dNTP(各ヌクレオチド100μM;最終濃度それぞれ33μM)
11.室温で15分間インキュベートする。
12.1μl T4 DNAリガーゼ(400単位)及びATPを1mMの最終濃度まで加える。
13.16℃で4時間インキュベートする。
14.フェノール/クロロホルム抽出。
15.アルコール沈殿。
16.20μl TEで再懸濁する。
17.5μl OX Pl−SceI緩衝液を加える
0.5μl BSA
18.5μl dH2O
6μl Pl−SceI(VDE)(6単位)
18.37℃で1〜2時間インキュベートする。
19.75℃で10分間インキュベートする。
20.適当な方法によって形質転換する。
【0092】
iv.供与体操作
1.トランスポゾン供与体はスーパーコイルでなければならない。弛緩した又は線状の供与体を使用した反応の効率は、約100倍低下する。供与体の調製物は良質でなければならないが、CsCl精製は必要でない。
2.トランスポザーゼに必須の認識要素は必須である。終止コドンはトランスポゾンに読み込まれる全ての枠に存在してもよい。転写は外側から非必須領域へ容易に進行することが可能である。
3.転位効率は、トランスポゾンが長くなるに従い若干減少する可能性がある。
4.最良の結果のために、トランスポゾン供与体プラスミドは単量体であることを確認する。
5.供与体調製物が単量体且つスーパーコイルであり、また、供与体分子が宿主生物内で複製しないならば、Pl−SceI消化工程は省略してもよい。
【0093】
v.標的DNAの要件
配列決定をするためのプラスミド標的は、回収を容易にするために環状でなければならない。線状の(例えば、染色体の)DNAは、効率的な基質である。本来形質転換が可能な生物を使用するときは、修復及びライゲーション工程が形質転換の前に必要である。大きなプラスミド、例えばコスミド及びBACは使用可能な標的である。標的DNAは、1×TEのような無塩緩衝液では少なくとも5μg/mlでなければならない。濃度は、公知の濃度のDNAとのアガロースゲル帯強度の比較によって、又は、260における吸光度によって推定することが可能である。
【0094】
II.修飾の例
一旦レポータータンパク質における修飾に寛容性のある部位又は領域が特定されるならば、その部位又は領域は1つ又は複数の残基の欠失、1つ又は複数の残基の挿入及び/又は環状置換又はその任意の組み合わせによって修飾することができる。一実施形態では、修飾は加水分解酵素、例えばそれには限定されないがプロテアーゼ、ペプチダーゼ、エステラーゼ(例えばコレステロールエステラーゼ)、ホスファターゼ(例えばアルカリホスファターゼ)などの認識部位の導入であってよい。例えば、加水分解酵素としてはそれには限定されないが、ペプチド結合に作用する酵素(ペプチドヒドロラーゼ)、例えばアミノペプチダーゼ、ジペプチダーゼ、ジペプチジルペプチダーゼ及びトリペプチジルペプチダーゼ、ペプチジルジペプチダーゼ、セリン型カルボキシペプチダーゼ、メタロカルボキシペプチダーゼ、システイン型カルボキシペプチダーゼ、ωペプチダーゼ、セリンエンドペプチダーゼ、システインエンドペプチダーゼ、アスパラギン酸エンドペプチダーゼ、メタロエンドペプチダーゼ、トレオニンエンドペプチダーゼ及び未知の触媒メカニズムのエンドペプチダーゼがある。例えば、本発明の修飾された甲虫ルシフェラーゼは、エンテロキナーゼ切断部位、カスパーゼ切断部位、コロナウイルスプロテアーゼ部位(STLQ−SGLRKMA;配列番号10)、キナーゼ部位、HIV−1プロテアーゼ部位(SQNY−PIVQ又はKAVRL−AEAMS;それぞれ配列番号11及び配列番号12)、HCVプロテアーゼ部位(AEDVVCC−SMSYS;配列番号13)(例えばLeeら、2003年を参照)、SARSウイルスプロテアーゼ部位(例えばQTSITSAVLQSGFRKMAFPS;配列番号16又はVRQCSGVTFQGKFKKIVKGT;配列番号17)、ライノウイルスプロテアーゼ部位、例えば、ライノウイルス3Cプロテアーゼ部位、プロホルモンコンベルターゼ部位、インターロイキン16変換酵素部位、CMV集合部位、リーシュマンジシン(leishmandysin)部位、B.アントラシス(B.anthracis)致死因子、ボツリヌス神経毒素軽鎖プロテアーゼ部位、アミロイド前駆体タンパク質のためのβ−セクレターゼ部位(VKM−DAEF;配列番号14)、前立腺特異性抗原配列、トロンビン部位、レニン及びアンジオテンシン変換酵素部位、カテプシンD部位、マトリックスメタロプロテイナーゼ部位、uPA部位、プラスミン部位、カチオンのための結合部位、例えばカルシウム結合ドメイン、カルモジュリン結合ドメイン、セルロース結合ドメイン、キチン質結合ドメイン、マルトース結合タンパク質ドメイン又はビオチン結合ドメインを含むことができる。他の実施形態では、本発明の修飾されたレポータータンパク質は、リガンドによって認識される配列、例えば抗体、又はカルシウムのような金属を含んでもよい。
【0095】
III.ポリヌクレオチド及びタンパク質の例
本発明は検出可能な活性、例えば発光活性を有するポリペプチドを提供する任意のアミノ酸配列を含んでいる修飾されたレポータータンパク質、並びに組換えにより又は合成的に合成されたそのタンパク質断片を含む。修飾されたレポータータンパク質のレポータータンパク質配列は、対応する修飾されていないレポータータンパク質のアミノ酸配列と同じか又は実質的に同じである。実質的に同じ配列を有するポリペプチド又はペプチドは、アミノ酸配列が完全にではないがほとんど同じであり、それと関連する配列の機能的活性を保持することを意味する。一般に、2つのアミノ酸配列は、それらが少なくとも70%同一であるならば、例えば少なくとも80%、90%、95%又はそれ以上の同一性を有する場合は実質的に同じ又は実質的に相同である。
【0096】
相同性又は同一性は、しばしば配列解析ソフトウェア(例えばGenetics Computer Group、ウィスコンシン大学バイオテクノロジーセンター、1710 University Avenue、マジソン、ウィスコンシン州 53705、のSequence Analysis Softwareパッケージ)を使用して測定される。そのようなソフトウェアは、相同性の程度を様々な欠失、置換及びその他の修飾に割り当てることによって類似の配列を比較する。2つ以上の核酸又はポリペプチド配列との関連で用語「相同性」及び「同一性」は、任意の数の配列比較アルゴリズムを用いることにより又は手動アラインメント及び目視検査により測定されるような、比較ウィンドウ又は指定領域上で最大の対応関係について比較及び整列したときに、同じであるか又は特定割合の同じアミノ酸残基若しくはヌクレオチドを有する2つ以上の配列又は部分配列を指す。
【0097】
配列比較のために、一般的に1つの配列は試験配列が比較される基準配列としての役割を果たす。配列比較アルゴリズムを使用するときは、試験配列及び基準配列がコンピュータに入力され、必要に応じて部分配列座標が指定され、配列アルゴリズムプログラムパラメータが指定される。デフォルトのプログラムパラメータが使用でき、又は代替パラメータを指定することができる。次に、配列比較アルゴリズムはプログラムパラメータに基づいて、基準配列と比較した試験配列の配列同一割合を計算する。
【0098】
比較のための配列アラインメントの方法は、当技術分野で公知である。比較のために最適な配列アラインメントは、Smithら(1981年)のローカルホモロジーアルゴリズム、Needlemanら(1970年)のホモロジーアラインメントアルゴリズム、Personら(1988年)の類似性検索法、これらのアルゴリズムのコンピュータ上での実行(Wisconsin Genetics Softwareパッケージ、Genetics Computer Group、575 Science Dr.、マジソン、ウィスコンシン州、内のGAP、BESTFIT、FASTA及びTFASTA)、又は手動アラインメント及び目視検査によって行うことができる。
【0099】
これらの数値計算用アルゴリズムのコンピュータによる実行は、配列同一性の決定のために配列の比較のために利用することが可能である。そのような実行例としては、それには限定されないが、PC/Geneプログラム(Intelligenetics、マウンテンビュー、カリフォルニアから入手可能)のCLUSTAL、並びにWisconsin Genetics Softwareパッケージ、バージョン8(Genetics Computer Group(GCG)、575 Science Drive、Madison、Wisconsin、USAから入手可能)のALIGNプログラム(バージョン2.0)及びGAP、BESTFIT、BLAST、FASTA及びTFASTAがある。これらのプログラムを使用したアラインメントは、デフォルトパラメータを使用して実施することが可能である。CLUSTALプログラムは、Higginsら(1988年)、Higginsら(1989年)、Corpetら(1988年)、Huangら(1992年)及びPearsonら(1994年)が適切に記載している。ALIGNプログラムは、Myers及びMiller(1988年)のアルゴリズムに基づく。Altschulら(1990年)のBLASTプログラムは、Karlin及びAltschul(1990年)のアルゴリズムに基づく。
【0100】
BLAST分析を実行するためのソフトウェアは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)から公的に入手することができる。このアルゴリズムは、第1に、データベース配列内の同じ長さのワードと並べたときに正の値の閾値スコアTにマッチするかそれを満たす、クエリー配列内の長さWのショートワードを特定することによって、高いスコアの配列の対(HSP)を特定することを含む。Tは、近傍ワードスコア閾値と称される(Altschulら、1990年)。これらの初期近傍ワードヒットは、それらを含むより長いHSPの検索を開始するためのシードとして機能する。ワードヒットは、次に累積アラインメントスコアが増加するまで、各配列に沿って両方向に拡張される。累積スコアは、ヌクレオチド配列のためにはパラメータM(一対のマッチング残基のリワードスコア;常に>0)及びパラメータN(ミスマッチ残基のペナルティスコア;常に<0)を使用して計算される。アミノ酸配列については、累積スコアの計算のためにはスコアリングマトリックスが用いられる。各方向へのワードヒットの拡張は、累積アラインメントスコアがその最大到達値から量X分低下する場合、累積スコアが1つ又は複数の負のスコアの残基アラインメントの蓄積によってゼロ以下になる場合、或いはいずれかの配列の末端に到達した場合に停止される。
【0101】
配列同一割合を計算することに加えて、BLASTアルゴリズムは2つの配列間の類似性の統計解析も実行する(例えばKarlin及びAltschul(1993年)を参照)。BLASTアルゴリズムによって提供される類似性測定手段の1つは、最小合計確率(P(N))であり、それは2つのヌクレオチド又はアミノ酸配列間の一致が偶然に起こる確率の指標となる。例えば、試験核酸と基準核酸との比較において最小合計確率が約0.1未満、より好ましくは約0.01未満、最も好ましくは約0.001未満のとき、試験核酸配列は基準配列と類似しているとみなされる。
【0102】
比較目的のためのギャップアラインメントを得るために、Altschulら(1997年)が記載しているようにGapped BLAST(BLAST 2.0に含まれる)を利用することが可能である。或いは、分子間の距離関係を検出する反復検索を実施するために、PSI−BLAST(BLAST 2.0に含まれる)を使用することが可能である。Altschulら、上記、を参照。BLAST、Gapped BLAST、PSI−BLASTを利用するときは、それぞれのプログラムのデフォルトパラメータを使用することが可能である(例えばヌクレオチド配列のためのBLASTN、タンパク質のためのBLASTX)。BLASTNプログラム(ヌクレオチド配列のための)は、デフォルトとして11のワード長(W)、10の期待値(E)、100のカットオフ値、M=5、N=−4、及び両方の鎖の比較を用いる。アミノ酸配列については、BLASTPプログラムはデフォルトとして3のワード長(W)、10の期待値(E)及びBLOSUM62スコアリングマトリックスを用いる(Henikoff及びHenikoff、1989年、を参照)。http://www.ncbi.nlm.nih.govを参照。
【0103】
詳細には、ポリペプチドは保存的な変異を除けばかなり相関している。保存的な変異は、他の生物学上類似した残基によるアミノ酸残基の置換を意味する。保存的な変異の例としては、1つの疎水性残基、例えばイソロイシン、バリン、ロイシン又はメチオニンによる他の残基の置換、1つの極性残基による他の残基の置換、例えばアルギニンによるリジンの置換、グルタミン酸によるアスパラギン酸の置換、又はグルタミンによるアスパラギンの置換、その他がある。保存的な置換の他の実例としては、下記変更が含まれる:アラニンからセリン;アルギニンからリジン;アスパラギンからグルタミン又はヒスチジン;アスパラギン酸からグルタミン酸;システインからセリン;グルタミンからアスパラギン;グルタミン酸からアスパラギン酸;グリシンからプロリン;ヒスチジンからアスパラギン又はグルタミン;イソロイシンからロイシン又はバリン;ロイシンからバリン又はイソロイシン;リジンからアルギニン、グルタミン又はグルタミン酸;メチオニンからロイシン又はイソロイシン;フェニルアラニンからチロシン、ロイシン又はメチオニン;セリンからトレオニン;トレオニンからセリン;トリプトファンからチロシン;チロシンからトリプトファン又はフェニルアラニン;バリンからイソロイシン、ロイシン。
【0104】
一実施形態では、本発明のポリヌクレオチドは、特定の宿主での発現のために最適化される。本明細書で使用されるように、最適化はコドン最適化、並びに真核細胞においてはコザック配列及び/又は1つ又は複数のイントロンの導入を含む。したがって、核酸分子は、修飾されていない甲虫ルシフェラーゼをコードする野生型核酸配列とは、コドンの30%、35%、40%又は45%以上、例えば50%、55%、60%若しくはそれ以上を超える割合で異なるコドン組成物を有してもよい。本発明に用いられる好ましいコドンは、特定の生物において同じアミノ酸の少なくとも1つの他のコドンよりも高頻度で使用され、またより好ましくはその生物において低利用コドンではなく、また核酸分子の発現のためにクローニング又はスクリーニングするために使用する生物においても低利用コドンではないものである。更に、あるアミノ酸(即ち、3つ以上のコドンを有するアミノ酸)の好ましいコドンは、他の(好ましくない)コドンよりも高頻度で使用される2つ以上のコドンを含んでもよい。1つの生物において他の生物におけるよりも高頻度で使用される核酸分子内のコドンの存在は、それらのコドンをより高頻度で使用する生物の細胞に導入されたとき、それらの細胞における野生型又は親の核酸配列の発現よりも高いレベルでそれらの細胞において発現される核酸分子をもたらす。
【0105】
本発明の一実施形態では、異なるコドンとは、哺乳類においてより高頻度で使用されるものであり、一方、他の実施形態においては、異なるコドンとは植物においてより高頻度で使用されるものである。特定の種類の哺乳類、例えばヒトは、他の種類の哺乳類とは異なる好ましいコドンセットを有している可能性がある。同様に、特定の種類の植物は、他の種類の植物とは異なる好ましいコドンセットを有しているかもしれない。本発明の一実施形態では、異なるコドンのほとんどは、所望の宿主細胞における好ましいコドンである。哺乳類(例えばヒト)及び植物の好ましいコドンは、当技術分野で公知である(例えばWadaら、1990年)。例えば、ヒトの好ましいコドンとして、それには限定されないが以下がある。
【化1】
(Wadaら、1990年)。
したがって、本発明の好ましい「ヒト化」合成の核酸分子は、増加した数の好ましいヒトコドンを有することによって野生型核酸配列と異なるコドン組成を有する。例えば、
【化2】
又はそのいかなる組み合わせがある。例えば、本発明の核酸分子は、野生型核酸配列と比較して増加した数のCTG又はTTGのロイシンエンコーディングコドン、GTG又はGTCのバリンエンコーディングコドン、GGC又はGGTのグリシンエンコーディングコドン、ATC又はATTのイソロイシンエンコーディングコドン、CCA又はCCTのプロリンエンコーディングコドン、CGC又はCGTのアルギニンエンコーディングコドン、AGC又はTCTのセリンエンコーディングコドン、ACC又はACTのトレオニンエンコーディングコドン、GCC又はGCTのアラニンエンコーディングコドン、或いはその任意の組み合わせを有することができる。同様に、植物においてより高頻度に使用されるコドンをより多く有す核酸分子は、増加した数の植物コドンを有することによって野生型核酸配列と異なるコドン組成を有し、このようなコドンとしては、それには限定されないが、
【化3】
又はその任意の組み合わせがある(Murrayら、1989年)。好ましいコドンは、植物の種類が異なれば異なってもよい(Wadaら、1990年)。
【0106】
本発明の修飾された甲虫ルシフェラーゼタンパク質又は融合タンパク質は、組換え方法又は固相化学ペプチド合成法によって調製することができる。そのような方法は1960年代初期から当技術分野で公知であり(Merrifield、1963年)(Stewartら、固相ペプチド合成(Solid Phase Peptide Synthesis)、第2版、Pierce Chemical Co.、ロックフォード、Ill.、11〜12頁も参照)、また、市販のラボラトリペプチドデザイン及び合成キット(Cambridge Research Biochemicals)でも最近採用された。そのような市販のラボラトリキットは、通常、Geysenら(1984年)の教示を利用しており、全て単一のプレートに接続されている多数の「ロッド」又は「ピン」の先端でペプチドを合成することを可能にする。そのような系が利用されるときは、ロッド又はピンのプレートは逆にされて、適当なアミノ酸をピン又はロッドの先端に結合又は固定するための溶液を含んでいる、対応するウェル又は貯蔵器の第2のプレート内に挿入される。そのような工程を繰り返すことにより、例えばロッド及びピンの先端を逆にして適当な溶液に挿入することにより、アミノ酸は所望のペプチドに構築される。更に、いくつかのFMOCペプチド合成系が利用できる。例えば、ポリペプチド又は断片の集合は、Applied Biosystems、Inc.Model 431A自動ペプチド合成機を使用して固体支持体上で実行することが可能である。そのような設備は、直接合成又は他の公知の技術を使用して結合することができる一連の断片の合成による、本発明のペプチドへの到達を容易にする。
【0107】
IV.本発明の修飾されたレポータータンパク質との有用な融合パートナー
修飾されたレポータータンパク質をコードする本発明のポリヌクレオチドは、他の核酸配列、例えばcDNAなどの未変性の配列、又はN末端、C末端又はN及びC末端の融合タンパク質、例えば選択可能なマーカーを含む異なるレポーター遺伝子によってコードされるタンパク質との融合を調製するためにin vitroで操作されたものと一緒に使用することができる。適当な融合パートナーの多くの例は当技術分野で公知であり、本発明の実施で使用することが可能である。
【0108】
融合パートナーとしては、それには限定されないが、親和性ドメイン又は他の機能性タンパク質配列、例えば酵素活性を有するものがある。例えば、機能性タンパク質配列はキナーゼ触媒ドメインをコードして(Hanks及びHunter、1995年)、リン酸部分を酵素的に特定のアミノ酸に加えることができる融合タンパク質を生成することができ、又は、Src Homology 2(SH2)ドメインをコードして(Sadowskiら、1986年;Mayer及びBaltimore、1993年)、リン酸化されたチロシンと特異的に結合する融合タンパク質を生成することができる。
【0109】
親和性ドメインは、通常、固体支持体上で固定されるものなどの結合パートナーと相互作用することが可能なペプチド配列である。複数の連続的な単一アミノ酸、例えばヒスチジンをコードするDNA配列は、発現タンパク質に融合される場合、ニッケルセファロースなどの樹脂カラムとの高親和性結合による組換えタンパク質の一段精製のために使用してもよい。キチン結合ドメイン(キチンと結合する)、グルタチオン−S転移酵素(グルタチオンと結合する)、ビオチン(アビジン及びストレプトアビジンと結合する)、その他などのペプチドをコードする配列も、関係するタンパク質の精製を容易にするために使用することができる。親和性ドメインは、インテイン(タンパク質セルフスプライシング要素(Chongら、1997年)の使用など、当業者に公知の方法によって関係するタンパク質から分離することが可能である。例示的な親和性ドメインとしては、HisV5(HHHHH)(配列番号1)、HisX6(HHHHHH)(配列番号2)、C−myc(EQKLISEEDL)(配列番号3)、Flag(DYKDDDDK)(配列番号4)、SteptTag(WSHPQFEK)(配列番号5)、血液凝集素、例えばHA Tag(YPYDVPDYA)(配列番号6)、GST、チオレドキシン、セルロース結合ドメイン、RYIRS(配列番号104)、Phe−His−His−Thr(配列番号105)、キチン結合ドメイン、S−ペプチド、T7ペプチド、SH2ドメイン、C末端RNA標識、WEAAAREACCRECCARA(配列番号8)、金属結合ドメイン、例えば、亜鉛結合ドメイン又はカルシウム結合ドメイン、例えばカルモジュリン、トロポニンC、カルシニューリンB、ミオシン軽鎖、リカバリン、S−モジュリン、ビシニン(visinin)、VILIP、ニューロカルシン(neurocalcin)、ヒポカルシン(hippocalcin)、フリケニン(frequenin)、カルトラシン(caltractin)、カルパイン大サブユニット、S100タンパク質、パルブアルブミン、カルビンジンD9K、カルビンジンD28K及びカルレチニンなどのカルシウム結合タンパク質からのもの、インテイン、ビオチン、ストレプトアビジン、MyoD、Id、ロイシンジッパー配列並びにマルトース結合タンパク質がある。一実施形態では、融合パートナーはHis又はGST標識など融合タンパク質を精製するために有用な配列であり、一実施形態では、精製標識は環状置換されたレポータータンパク質のN又はC末端に融合される。
【0110】
他のクラスの融合パートナーとしては、レポーター遺伝子、例えばそれには限定されないがネオ遺伝子、β−gal遺伝子、gus遺伝子、cat遺伝子、gpt遺伝子、hyg遺伝子、hisD遺伝子、ble遺伝子、mprt遺伝子、bar遺伝子、ニトリラーゼ遺伝子、ガラクトピラノシド遺伝子、キシロシダーゼ遺伝子、チミジンキナーゼ遺伝子、アラビノシダーゼ遺伝子、突然変異アセトラクテートシンターゼ遺伝子(ALS)若しくはアセトアシッドシンターゼ遺伝子(AAS)、メトトレキセート抵抗性dhfr遺伝子、ダラポンデハロゲナーゼ遺伝子、5−メチルトリプトファン抵抗性を付与する突然変異アントラニル酸シンターゼ遺伝子(国際公開97/26366)、R座位遺伝子、βラクタマーゼ遺伝子、xylE遺伝子、α−アミラーゼ遺伝子、チロシナーゼ遺伝子、花虫類ルシフェラーゼ(luc)遺伝子(例えばRenilla reniformisルシフェラーゼ遺伝子、エクオリン遺伝子、赤色蛍光タンパク質遺伝子、又は緑色蛍光タンパク質遺伝子などによってコードされるたんぱく質がある。選択可能な又はスクリーニング可能なマーカー遺伝子という用語には、その分泌が形質転換細胞の特定又は選択の手段として検出することが可能な「分泌可能なマーカー」をコードする遺伝子も含まれる。例には、抗体相互作用によって特定することが可能な分泌可能な抗原をコードするマーカー、又はそれらの触媒活性によって検出することができる分泌可能な酵素をコードするマーカーさえ含まれる。分泌可能なタンパク質はいくつかのクラスに分けられ、例としては例えばELISAによって検出可能な小さな、分散性タンパク質、及び細胞膜に挿入されるか捕捉されるタンパク質がある。
【0111】
V.修飾されたレポータータンパク質又はその融合物をコードするベクター及び宿主細胞
修飾されたレポータータンパク質又はその融合物をコードする望ましい核酸分子が一旦調製されると、その修飾されたレポータータンパク質又はその修飾されたレポータータンパク質を含んでいる融合タンパク質をコードする発現カセットが調製される。例えば、修飾された甲虫ルシフェラーゼをコードする核酸配列を含んでいる核酸分子は、発現カセットを形成するために任意選択に転写調節配列、例えば1つ又は複数のエンハンサー、プロモーター、転写終結配列又はその組み合わせと作動可能的に結合される。核酸分子又は発現カセットは、プラスミド又はウイルスベクターなどの任意選択に選択可能なマーカー遺伝子を含んでいるベクターに導入し、このベクターを関係する細胞、例えば大腸菌、ストレプトミセス属、バシラス属、スタフィロコッカス属などの原核細胞、並びに植物(双子葉植物又は単子葉植物)、真菌、酵母、例えばピチア、サッカロミセス又はシゾサッカロミセス属、或いは哺乳類細胞などの真核細胞に導入することができる。好ましい哺乳類細胞としては、ウシ、ヤギ、羊、イヌ、ネコ、サルなどヒト以外の霊長類、及びヒトの細胞がある。好ましい哺乳動物細胞系には、それには限定されないが、CHO、COS、293、Hela、CV−1、SH−SY5Y、HEK293及びNIH3T3細胞が含まれる。
【0112】
コードされる修飾されたレポータータンパク質の発現は、原核細胞又は真核細胞で発現可能な任意のプロモーターによって調節することができる。好ましい原核プロモーターとしては、それには限定されないが、SP6、T7、T5、tac、bla、trp、gal、lac又はマルトースプロモーターが含まれる。好ましい真核生物のプロモーターとしては、それには限定されないが、構成プロモーター、例えばCMV、SV40及びRSVプロモーターなどのウイルスのプロモーター、並びに調節可能なプロモーター、例えばtetプロモーター、hsp70プロモーター及びCREによって調節される合成プロモーターなどの誘導又は抑制が可能なプロモーターが含まれる。本発明の核酸分子、発現カセット及び/又はベクターは、任意の方法、例えばそれには限定されないが、カルシウム媒介性形質転換、エレクトロポレーション、顕微注射、リポフェクションなどによって細胞に導入することができる。
【0113】
VI.使用例
修飾されたレポータータンパク質又はその融合物は任意の目的、例えばそれには限定されないが、特定分子の量及び存在の検出(バイオセンサー)、特定分子の単離、特定分子の高次構造の例えば結合、リン酸化又はイオン化による変化の検出、pH又は温度などの条件の検出、高スループット又は低スループットのスクリーニングの促進、タンパク質−タンパク質、タンパク質−DNA又は他のタンパク質ベースの相互作用の検出、或いはバイオセンサーの選択又は開発などのために有用である。例えば、修飾されたレポータータンパク質又はその融合物は以下のために有用である。例えばin vitro又は細胞ベースのアッセイにおける特定のキナーゼ(例えば、キナーゼ部位をレポータータンパク質に挿入することによる)、RNAi(例えば、RNAiによって認識されることが疑われる配列をレポータータンパク質のコード配列に挿入し、次にRNAiを添加した後にレポーター活性を監視することによる)、又はプロテアーゼの量、存在又は活性の検出であり、例えば特定のウイルスプロテアーゼの存在を検出するためのものがあり、これは今度はウイルス又は抗体の存在の指標となる;阻害剤、例えばプロテアーゼインヒビターのスクリーニング;認識部位の特定、又は例えば選択された認識配列を有する修飾されたルシフェラーゼ又は単一の関心のある分子を有する異なる複数の配列若しくは複数の(例えばライブラリー)分子を有する修飾されたルシフェラーゼのライブラリーを使用した基質特異性の検出;バイオセンサー又は関心のある分子、例えばプロテアーゼの選択又は開発;或いは、in vitro又は細胞ベースの方法による相補性又は結合を通したタンパク質−タンパク質相互作用の検出。一実施形態では、挿入を含む修飾された甲虫ルシフェラーゼが分子のランダムなライブラリー又は変異ライブラリーと接触させられ、その挿入と相互作用する分子が特定される。他の実施形態において、複数の挿入を有する修飾されたルシフェラーゼのライブラリーが分子と接触させられ、その分子と相互作用する修飾されたルシフェラーゼが特定される。
【0114】
本発明は、細胞内の分子の発現、場所及び/又はトラフィッキングを監視するための、並びに細胞内の微小環境の変化を監視するための、修飾された甲虫ルシフェラーゼ又はその融合タンパク質を使用した方法も提供する。例えば、一実施形態では、修飾された甲虫ルシフェラーゼは、ある条件下で互いと相互作用する2つのドメインを含んでいる内部挿入を含む。一実施形態では、挿入内の1ドメインはリン酸化が可能なアミノ酸を含み、他のドメインはホスホアミノ酸の結合ドメインである。適当なキナーゼ又はホスファターゼの存在下で、挿入内の2つのドメインは相互作用して修飾された甲虫ルシフェラーゼの高次構造を変え、その結果修飾された甲虫ルシフェラーゼの検出可能な活性の変化が起こる。他の実施形態において、修飾された甲虫ルシフェラーゼは分子の認識部位を含み、その分子がその認識部位と相互作用すると活性が増加するので、他の分子の存在又は量を検出又は決定するために使用することができる。
【0115】
2−ハイブリッド系は、in vivoでタンパク質:タンパク質相互作用を検出するための、並びにタンパク質:タンパク質相互作用に関係する残基/ドメインを特定するための極めて強力な方法である。2−ハイブリッド系の基礎は、一部の転写因子で見られるモジュラードメインである。即ち、特定のDNA配列と結合するDNA結合ドメイン、及び基礎の転写機構と相互作用する転写活性化ドメインである(Sadowski、1988年)。DNA結合ドメインと共同した転写活性化ドメインは、TATAボックスでのRNAポリメラーゼII複合体の集合を進めて転写を増やすことができる。CheckMate(商標)Mammalian Two−Hybridシステム(Promega Corp.、マジソン、ウィスコンシン州)において、別々のプラスミドによって生じたDNA結合ドメイン及び転写活性化ドメインは、DNA結合ドメインに融合された1つのタンパク質(「X」)が転写活性化ドメインに融合された第2のタンパク質(「Y」)と相互作用するときは、緊密に関連する。この系において、タンパク質X及びYの間の相互作用は、レポーター遺伝子又は選択可能なマーカー遺伝子の転写をもたらす。特に、pBINDベクターは複数のクローニング領域の上流に酵母GAL4のDNA結合ドメインを含み、pACTベクターは複数のクローニング領域の上流に単純疱疹ウイルスVP16の活性化ドメインを含む。更に、pBINDベクターは、Renilla reniformisルシフェラーゼを発現する。関係する相互作用する可能性のある2つのタンパク質をコードする2つの遺伝子は、pBIND及びpACTベクターにクローニングされ、それぞれGAL4のDNA結合ドメイン及びVP16の活性化ドメインと融合タンパク質を生成する。pG5lucベクターは、ホタルルシフェラーゼ遺伝子(luc+)の上流にある最小TATAボックスの更に上流に、5つのGAL4結合部位を含む。pGAL4及びpVP16融合構築物は、pG5lucベクターと共に哺乳類細胞に形質移入される。形質移入の2〜3日後に細胞は溶解され、Renillaルシフェラーゼ及びホタルルシフェラーゼの量はDual−Luciferase(登録商標)Reporter Assay System(Promega Cat.#E1910)を使用して定量することが可能である。GAL4及びVP16融合構築物のような2つの試験タンパク質の間の相互作用は、陰性対照を上回るホタルルシフェラーゼ発現の増加をもたらす。本発明の修飾された甲虫ルシフェラーゼ、例えば、修飾に寛容性のある1部位又は領域(N末端断片)で欠失が起きているものはDNA結合ドメインと融合され、甲虫ルシフェラーゼの残り(C末端の断片)は転写活性ドメインと融合される。
【0116】
本発明は、関心のある分子の活性を調整することができる剤(「試験」剤)についてスクリーニングする方法も提供する。「調整」は、生物的活性又は過程(例えば酵素活性)の機能的特性を強めるか抑制する能力を指し、そのような強化又は抑制は特定の事象、例えばシグナル伝達経路の活性化の発生に付随するものでもよく、且つ/又は特定の細胞種だけで現れるものでもよい。「モジュレーター」は作用剤(天然又は非天然の)を指し、例えば生体高分子(例えば核酸、タンパク質、非ペプチド又は有機分子)、小分子、或いは細菌、植物、真菌類又は動物(特に哺乳動物)の細胞又は組織などの生体物質からの抽出物を指す。モジュレーターは、本明細書で記載されているスクリーニングアッセイに含めることにより、生物学的過程の(直接的又は間接的)阻害剤又は活性剤(例えばアゴニスト、部分アンタゴニスト、部分アゴニスト、アンタゴニスト、抗腫瘍薬、細胞傷害剤、腫瘍性転化又は細胞増殖の阻害剤、細胞増殖促進剤、など)としての潜在的な活性について評価される。モジュレーターの活性は、知られていても、未知であっても、又は部分的に知られていてもよい。そのようなモジュレーターは、本発明の方法を使用してスクリーニングすることができる。用語「試験剤」は、推定されるモジュレーターとして本発明の1つ又は複数のスクリーニング法によって試験される剤を指す。通常、0.01μM、0.1μM、1.0μM及び10.0μMなどの様々な所定濃度がスクリーニングで使用される。対照は、試験剤がない場合のシグナル測定、標的を調整することが公知の剤との比較、又は試験剤との接触の前、その間、及び/又は後の試料(例えば細胞、組織又は生物体)との比較を含むことができる。
【0117】
一実施形態では、この方法はプロテアーゼ活性を調整する剤についてスクリーニングすることを含む。例えば、一実施形態では、アポトーシスを調整することができる剤を特定する方法が提供される。カスパーゼファミリープロテアーゼは、アポトーシスと関連付けられている。したがって、この方法はカスパーゼファミリープロテアーゼを含むことが疑われる試料を、カスパーゼ活性を調整することが疑われる剤、及びカスパーゼによる切断が可能な切断部位を有する修飾されたレポータータンパク質と接触させることを含む。試料中の修飾されたレポータータンパク質の活性は、試験剤との接触の前後に検出される。剤との接触後の活性増加は、アポトーシスを抑制する剤の指標であり、活性減少はアポトーシスを活性化する剤の指標である。
【0118】
したがって、本発明は本発明の修飾されたレポータータンパク質内に存在する認識配列の切断を調整する剤を特定してその活性を検出するための、有用なスクリーニング系を提供する。この系は、プロテアーゼ活性モジュレーターの迅速なスクリーニングを可能にする。本明細書で記載されているスクリーニング系を利用すると、プロテアーゼ、例えばカスパーゼファミリープロテアーゼを調整(例えば抑制又は活性化)する剤を特定するための感度が高く迅速な手段が提供される。
【0119】
したがって本発明の修飾されたレポータータンパク質は、修飾されたレポータータンパク質内の挿入及び関心のある分子との間の相互作用を調整する剤又は条件を研究するための基質として有用である。詳細には、本発明は、挿入が関係する酵素の切断部位であるアミノ酸配列を含んでいる、修飾されたルシフェラーゼタンパク質を企図する。したがって関心のある分子がプロテアーゼであるとき、挿入はそのプロテアーゼの切断認識配列を含んでいるペプチドを含む。プロテアーゼの切断認識配列は、タンパク分解性切断の間にそのプロテアーゼによって認識される特異的アミノ酸配列である。したがって、本発明は、試料を本発明の修飾されたルシフェラーゼポリペプチドと接触させてルシフェラーゼ活性の変化を測定することにより、試料中のプロテアーゼの量を測定する方法を提供する。本発明の修飾されたルシフェラーゼタンパク質は、とりわけ修飾されたルシフェラーゼを発現する細胞内のプロテアーゼ活性を監視するために使用することができる。
【0120】
本発明のアッセイは、修飾されたレポータータンパク質を切断するプロテアーゼ活性を変える化合物を特定するための薬剤をスクリーニングするために使用することができる。一実施形態では、アッセイはプロテアーゼを含んで試料に対してin vitroで実施される。既知量のプロテアーゼを含んでいる試料は、本発明の修飾されたレポータータンパク質、及び試験剤と混合される。次に、試料中のプロテアーゼ活性量が先に述べたように測定される。その後、試験剤の存在下におけるプロテアーゼ1モルあたりの活性量が、試験剤がない場合のプロテアーゼ1モルあたりの活性と比較される。相違は、試験剤がプロテアーゼ活性を変化させることを示す。したがって、変化は修飾されたレポータータンパク質活性の減少をもたらすプロテアーゼ活性の増加或いは修飾されたレポータータンパク質活性の増加又は維持と対応するプロテアーゼ活性の低下である。
【0121】
一実施形態では、プロテアーゼ活性を変化させる剤の能力が決定される。このアッセイにおいて、細胞はプロテアーゼ活性を調整することが疑われる剤で条件づけられるか又は接触させられる。培養内の細胞は溶解されて、プロテアーゼ活性が測定される。例えば、既知又は未知の量のプロテアーゼを含んでいる溶解細胞試料は、本発明の修飾されたレポータータンパク質と混合される。次に、上記したように対照又は未処理の試料及び処理された溶解細胞試料の中の修飾されたレポータータンパク質活性の程度を測定することにより、試料中のプロテアーゼ活性量が測定される。活性又は抑制は、試料中のタンパク質1マイクログラム又は1ミリグラムをベースにして計算することができる。したがって、プロテアーゼ活性の調整は、修飾されたレポータータンパク質活性の減少をもたらすプロテアーゼ活性の増加或いは修飾されたレポータータンパク質活性の増加又は維持と対応するプロテアーゼ活性の低下を含む。一般的に、プロテアーゼ活性の絶対量を与えるために、相違は標準の測定値に対して較正される。プロテアーゼの活性又は発現を抑制又はブロックする試験剤は、非処理の対照と比較した処理された細胞内の修飾されたレポータータンパク質活性の増加によって検出することができる。
【0122】
他の実施形態では、in vivoでプロテアーゼ活性を変化させる剤の能力が決定される。in vivoアッセイにおいて、本発明の修飾されたレポータータンパク質をコードする発現ベクターで形質移入された細胞は異なる量の試験剤に曝され、細胞内のレポータータンパク質活性への影響を決定することができる。一般的に、プロテアーゼ活性の絶対量を与えるために、相違は標準の測定値に対して較正される。プロテアーゼの活性又は発現を抑制又はブロックする試験剤は、非処理の対照と比較した処理された細胞内の修飾されたレポータータンパク質活性の増加によって検出することができる。
【0123】
本発明のアッセイに用いられる物質及び組成物は、キットの調製に適していることが理想である。そのようなキットは、1つ又は複数の容器手段、例えばバイアル、管、その他を含んでいる担体手段を含んでもよく、各容器手段はその方法で使用される別々の要素の1つを含んでいる。容器の1つは、本発明の修飾されたレポータータンパク質又はポリヌクレオチドを(例えば、ベクターの形で)含む。第2の容器は、修飾されたレポータータンパク質の基質を含んでもよい。
【0124】
本発明は、以下のそれには限定されない実施例で更に説明される。
【実施例1】
【0125】
コメツキムシルシフェラーゼ遺伝子のTn5挿入突然変異
A.トランスポゾン挿入反応
標的DNAの調製。コメツキムシルシフェラーゼ遺伝子(cbg69)を大腸菌T7発現ベクターにクローニングし、得られたプラスミド(pJLC1)をトランスポゾン突然変異反応のために標的DNAとして使用した。
In Vitroトランスポゾン挿入反応。複数の挿入事象を最小にしてトランスポゾン挿入の効率を最大にするために、反応条件を最適化した。例えば、等モル量のトランスポゾンをそのモル数の標的DNAに加えた。
【0126】
1.以下の順序で加えることによってトランスポゾン挿入反応混合物を調製する:
1μl 10×反応緩衝液
0.35μg 標的DNA(pJLC1)(7μl)
1μl モル当量トランスポゾン
1μl トランスポザーゼ
10μl 総反応体積
2.反応混合物を37℃で2時間インキュベートする。
3.1μl停止液を加えて反応を止める。
混合して65℃で15分間加熱する。
反応混合物は−20℃で保存した。
【0127】
B.トランスポゾン挿入クローンの選択
形質転換及び回収。1回の反応で得られるトランスポゾン挿入クローンの数は、とりわけ使用されるコンピテント細胞の形質転換効率によって決まる。コンピテント細胞の形質転換効率がより高いほど、より多くの挿入クローンが得られる。大腸菌のrecA−系統(EpicentreからのEC100コンピテント細胞)を形質転換のために使用した。
3.挿入反応混合物の1μlを使用して、EC100エレクトロコンピテント細胞へ形質転換する。
4.エレクトロポレーションの直後にSOC培地を1mlの最終体積までエレクトロポレーションセルに加えることによって、エレクトロポレーションされた細胞を回収する。培地/細胞をピペットで穏やかに混ぜる。管に移して37℃の撹拌機で30〜60分間インキュベートして細胞成長を促進する。
5.細胞の一部を50μg/mlのカナマイシンを含んでいるLBプレート上で平板培養する。
6.37℃で一晩プレートを増殖させる。
【0128】
C.トランスポゾン挿入マッピング。数千の挿入コロニーが得られた。27の挿入クローンを選択し、cbg69遺伝子の2つの末端のプライマーセットを使用してTn5トランスポゾンを含んでいるコメツキムシluc遺伝子をPCR増幅した。PCR産物を、同じプライマーセットを使用して配列決定した。Tn5挿入の位置はランダムであることが明らかになった(図2〜3)。
【0129】
D.トランスポゾン挿入を有するluc遺伝子のプラスミドライブラリーの作製。
luc遺伝子内に挿入を有するクローンは、プラスミド骨格に挿入を有するものから分離する必要がある。このため、全ての形質転換体をプールして、プラスミドDNAを精製した。トランスポゾン挿入を有するcbg69遺伝子を含んでいるDNAフラグメントを放出するために、得られたプラスミドDNAを一対の制限酵素(例えばNdeI及びEcoRI)で消化した。このDNAフラグメントはトランスポゾン挿入を含まない大腸菌T7発現ベクターのそれぞれの制限酵素部位に再クローニングし、Tn5挿入を有するluc遺伝子を含んでいるプラスミドライブラリーを得た。
【0130】
E.インフレーム19コドン挿入ライブラリーの作製
Tn5トランスポゾンの除去。一旦トランスポゾン挿入を有するluc遺伝子のプラスミドライブラリーが作製されたならば、制限酵素、例えばNotIによる消化によってTn5トランスポゾンを除去した。線状にされたDNAは、アガロースゲル電気泳動によってTn5を含んでいるDNA断片から分離し、次に精製した。
【0131】
再連結及び形質転換。線状にされたDNAは、T4 DNAリガーゼを使用して再連結した。再連結が成功すると、単一の制限部位、例えばNotIが再生され、3つの読取り枠の1つへの57ヌクレオチド(19コドン)の挿入が作製された。再連結されたDNAはEC100細胞に形質転換し、元のクローニングベクター上に存在する抗生物質マーカー(例えば対照DNAではアンピシリン)を使用して組換え体を選択した。
【0132】
F.活性リンカー挿入クローンの選択。個々のリンカー挿入クローンを使用して100μg/mlのアンピシリンを含んでいる1mlのLB培地に接種し、37℃で一晩増殖させた。ルシフェラーゼ活性は、一晩培養したもの100μlとPromega Corp.(マジソン、ウィスコンシン州)からのBright−Glo試薬100μlとを混ぜることによって測定した。発光は、5分後に照度計で記録した。
【0133】
G.活性リンカー挿入クローンのDNA配列決定。400以上のクローンがスクリーニングされた。バックグラウンドより20倍よりも強いルシフェラーゼ活性を有していたリンカー挿入クローンを選択した。リンカー挿入位置は、リンカー挿入を含んでいるluc遺伝子のPCR産物の配列決定により決定した。各活性リンカー挿入クローンの位置及び相対活性は、図3〜4に示す。
【実施例2】
【0134】
ホタルルシフェラーゼ遺伝子のTn−7挿入突然変異
市販のキット(New England Biolabs(NEB)からのGPS(商標)−M GPS Mutagenesis System)を使用して、Tn7ベースのトランスポゾンをホタルルシフェラーゼDNAにランダムに挿入した。この挿入断片の大部分を次に制限酵素消化により切り取り、再連結によって5アミノ酸の挿入を得た。最初に、コロニーを増殖させて切り取りの前にルシフェラーゼ活性の消失についてスクリーニングした。ルシフェラーゼ活性を有していた培養物内のプラスミドを次に切り取り、細胞へ形質転換で戻し、ルシフェラーゼ活性の復活がないかコロニーを調べた。その後、ランダム位置に大きな挿入を含んでいるゲル精製ルシフェラーゼ断片がベクターにクローニングされ、ベクター集団の大量切り取りが実施されたより効率的な方法が使用された。ここで、切り取られたベクターによる形質転換の後にルシフェラーゼ活性を発現したコロニーを選択した。トランスポゾンはカナマイシン耐性を有していたので、挿入を含まないベクター分子を除去することが可能であった。
【0135】
第1の方法では、反応は次のように組み立てられた:
【化4】
カナマイシン耐性遺伝子を有するpGPS5(NEB)は供与体プラスミドであり、アンピシリン耐性遺伝子を有するpSP−Luc+(Promega Corp.)は受容体であった。転位が成功すると、受容体プラスミドにカナマイシン耐性カセットが挿入された。反応体をピペット操作で混合し、次にTnsABC Transposaseの1μlを加えて反応体を更に混合した。反応体は、37℃で1時間インキュベートし、75℃で10分間加熱してから氷上に置いた。次に5μlを100μlの高効率コンピテント大腸菌JM109(Promega Corp.)に形質転換した。氷上での10分間のインキュベーションの後、細胞を42℃で45秒間の温熱ショックで処理し、次に氷上で2分間インキュベーションした。1mlのLuria Broth(LB)を次に加え、細胞を37℃で1時間振盪した。次に、40μlずつを100μg/mlのアンピシリン及び25μg/mlのカナマイシンを含んでいるLB寒天プレート上で平板培養した。
【0136】
その翌日、コロニーをそれらのプレートから拾い、3mlのLB/amp/kan+0.5mM IPTG内で個々に生育させた。一晩増殖の後、培養物の10μlをpH5.5の100mMクエン酸ナトリウム中の1mMルシフェリンの100μlに加えることによってこれらの培養物をルシフェラーゼ活性について分析し、Turner20/20照度計で数値を読み取った。
【0137】
プラスミドは低活性培養物(Promega Wizard Plus Miniprepsキット)から調製し、制限酵素PmeI(NEB)で消化して大多数の挿入断片を切り取り、次に再連結した。一般的に、これらの反応は次の通りであった。
【化5】
インキュベーションは37℃で1時間であった。次に反応体を65℃で20分間加熱して制限酵素を不活性化し、ライゲーション反応を下記のように組み立てた。
1μl 上の反応液
3μl 10×リガーゼ緩衝液(Promega)
1μl 3 U/μl T4 DNAリガーゼ(Promega)
25μl H2O
30μl
ライゲーションは16℃で少なくとも2時間インキュベートし、次に先に述べたように3μlをJM109に形質転換した。各形質転換の50μlをLB/ampプレート又はこれらのプレートの上に重層されたニトロセルロースフィルター上に平板培養した。37℃で一晩増殖させた後に、フィルターを取り出して40℃にセットしたスライドウォーマー(Fisher Scientific)上のpH5.5の100mMクエン酸ナトリウム中の1mMルシフェリン溶液の1mlの上に置いた。部屋を暗くし、フィルターの発光を観察した。輝くのが観察されたピックからのコロニーはLB/ampプレートから成長させ、プラスミドを単離し、次に制限酵素切断及びシークエンシングによって分析した。大きなカナマイシン挿入断片の切り出しの後、単一のPmeI部位が挿入部位に残る。したがって、PmeI及び他の制限酵素による切断は、挿入部位のマッピングを可能にする。
【0138】
第2の方法において、挿入ライブラリーをゲル精製ルシフェラーゼ断片内で単離し、タンパク質の切り出し及び発現のためにベクターにクローニングした。具体的には、pSPLuc+への転位を先に述べたように達成し、次に、先に述べたように3×5μlを3×100μl高効率JM109に形質転換した。各管からの40μlをLB/amp/kan上に平板培養し、この管の残り並びに他の管からの細胞を50mlのLB/amp/kanに加えて37℃で一晩生育させた。プレートは約7,000の異なるプラスミドのライブラリーに相当する93のコロニーを生成したが、その約1,400の挿入はルシフェラーゼコード配列内にあると予想された。プラスミドは、液体培養の8mlから単離された。ルシフェラーゼ遺伝子と連結するKpnI及びEcoRIによるプラスミドの消化は、ベクター骨格及びルシフェラーゼコード配列と対応する4つの断片をもたらし、それぞれはカナマイシン挿入断片を有すか有さなかった。関係するバンドは長さ3,438bpであり、転位されたルシフェラーゼ遺伝子断片と一致した。ライブラリーからの約2μgのプラスミドをKpnI及びEcoRIで消化し、1μg/mlのエチジウムブロミドを含んでいる1%アガロースゲル上で電気泳動にかけた。3,438bpバンドはUV照明による可視化の後にゲルから切り取り、Wizard PCR Preps(Promega Corp.)を使用してアガロース切片から精製した。次に標準手順に従ってこのDNAをKpnI及びEcoRIで消化したpGEM−3Z(Promega Corp.)にクローニングした。これによりルシフェラーゼ遺伝子はベクター内のLacプロモーターの支配下に置かれる。大多数のカナマイシン挿入断片は、PmeIで切断してライブラリーから切り取った。
2μl 0.25μg/μl pGEM−3Z−luc−kanライブラリー
2μl 10×緩衝液C(Promega)
1.5μl 10 U/μl PmeI(NEB)
14.5μl H2O
20μl
この反応体を37℃で1時間インキュベートし、次に65℃で20分間加熱して下記のように連結した。
2μl 上記消化液
3μl 10×リガーゼ緩衝液(Promega)
1μl 3 U/μl T4 DNAリガーゼ(Promega)
24μl H2O
30μl
ライゲーション反応体を16℃で一晩インキュベートし、次に個々のコロニーを得るためにコンピテントなJM 109に形質転換した。アンピシリンのみ又はアンピシリンとカナマイシンの両方を含んでいるプレート上に平板培養することによって、アンピシリンプレート上の形質転換体の約90%はカナマイシンに感受性であり、したがって挿入断片の切り取りに成功したと推測することができた。個々のコロニーを3mlのLB+100μg/mlアンピシリンで培養し、培養物のルシフェラーゼ活性を分析した。
【0139】
結果
第1の方法については、培養物の約20%はルシフェラーゼ活性が大きく低下しており、このことはトランスポゾンがpSP−Luc+プラスミド内のルシフェラーゼコード領域に挿入されていることと一致している。第2の方法については、個々のコロニーからの培養物の約15%でかなりの活性が観察された。プラスミドは活性のある培養物から調製され、PmeI部位挿入断片のおおよその位置を特定するために制限マッピングを実施した。次にこれらの試料に対して、アイオワ大学DNAシーケンシング施設で標準のジデオキシ配列決定法を実施した。活性クローンの約半分は、ルシフェラーゼコード領域の直ぐ外側に挿入断片を含んでいた。残りは、コード領域内の様々な場所に挿入断片を有していた。上で議論した2つの異なる方法からの結果を合わせたものを下で提示するが、挿入の位置及び残存している活性のおおよその割合が示されている。
【表2】
【実施例3】
【0140】
ヒンジ領域に修飾を有する修飾されたコメツキムシルシフェラーゼ
トランスポゾン突然変異研究によって特定された位置に関係する様々な部位を都合よく挿入するために、コメツキムシルシフェラーゼ遺伝子(cbg69)を修飾して、ヒンジ領域をコードする配列に連続している2つの独特の制限酵素部位、SnaBI(TACGTA)及びSalI(GTCGAC)を生成した。具体的には、2つのオリゴヌクレオチド:
【化6】
を使用してGeneEditor(Promega)によるcbg69遺伝子の修飾を実施した。得られたコメツキムシルシフェラーゼCbg69ssはIle409のValへの1アミノ酸置換を有し、野生型Cbg69の2倍の活性を示すことが明らかになった。Cbg69ssを有すプラスミド(pJLC1ss)は、ヒンジ領域に修飾を有する他のルシフェラーゼを生成するためのテンプレートとして使用した。
そのために、以下のオリゴヌクレオチド対を合成した:
【化7】
各オリゴヌクレオチドは、以下の反応条件を使用してリン酸化した。
オリゴヌクレオチド 30pmol
10× T4ポリヌクレオチドキナーゼ緩衝液 2.5μl
10mM ATP 2.5μl
T4オリゴヌクレオチドキナーゼ(1μ/μl) 0.5μl
水 25μlまで
37℃で30分間インキュベートし、70℃で10分間不活性化する。
【0141】
各リンカーにつき、一対のリン酸化オリゴヌクレオチド(上記反応体からの10μl)を95℃で5分間加熱して1時間で37℃に冷却してアニールした。次に各リンカーをpJLC1ssのSnaBI及びSalI部位にクローニングした。
【0142】
結果
A.コメツキムシルシフェラーゼを残基400の後で修飾してカスパーゼ−3認識部位(DEVD)を含むようにし、Cbg69DEVDが得られた。Cbg69ss及びCbg69DEVDは細菌宿主で発現された。細菌溶解物をカスパーゼ−3の様々な量(0、6.25、12.5、25、50、100又は200ng)或いは200ngのカスパーゼ−3及び0.1mMカスパーゼ阻害剤Ac−DEVD−CHOと混合し、ルシフェラーゼ活性を監視した。図5Aは、カスパーゼ−3濃度の上昇に従って、Cbg69ssのそれではなくCbg69DEVDの活性が減少したことを示す。さらに、カスパーゼ阻害剤の存在下では活性低下は観察されなかった(図5B)。さらに、ルシフェラーゼ活性は時間と共に低下した(図5C)。
【0143】
B.SARSウイルス3CLプロテアーゼはSARSコロナウイルスのシステインプロテアーゼであり、抗SARSウイルス薬剤の潜在的な標的である。2つのコメツキムシルシフェラーゼを残基400の後で修飾して、2つのSARSプロテアーゼ認識部位(Cbg69SARS3及びCbg69SARS6)の1つを含むようにした。Cbg69ss、Cbg69SARS3及びCbg69SARS6は、ウサギ網状赤血球溶解物及び/又はコムギ胚抽出物(Promega)などのin vitro翻訳系を使用して生成した。SARSプロテアーゼは、New England BiolabsからのpMAL精製系を使用して部分的に精製した。コメツキムシルシフェラーゼを含んでいる溶解物をSARSプロテアーゼと混合して、ルシフェラーゼ活性を監視した。図6Cは、室温で1時間のインキュベーションの後、Cbg69ssではなくCbg69SARSは、SARSプロテアーゼ(約0.3μg)で処理したとき非処理の試料と比較して活性が低下したことを示す。
【0144】
C.図6A〜Cで示すように、Asn400の後に様々な挿入部位を有する修飾されたコメツキムシルシフェラーゼは全て活性であった。これらの修飾されたルシフェラーゼは、Cbg69ssと比較して12〜64%の活性を有していた。したがって、コメツキムシルシフェラーゼのヒンジ領域における修飾は、活性を保持している修飾されたルシフェラーゼを与えることができる。
【実施例4】
【0145】
内部エンテロキナーゼ部位を有する修飾されたホタルルシフェラーゼ
ホタルルシフェラーゼのアミノ酸233及び541の後の5アミノ酸挿入は酵素活性の最大分画を保持していた(実施例2)ので、それらの部位を詳細解析のために選択した。プロテアーゼによる切断の後のルシフェラーゼ活性に対する影響を検討するために、GeneEditor(商標)in vitro Site−Directed Mutagenesis System(Promega Corp.)を使用してin vitro突然変異誘発を行い、これらの部位にプロテアーゼ切断部位を挿入した。先ず、標準の手法を使用してルシフェラーゼ遺伝子を発現ベクターpRSET−B(Invitrogen)のNcoI及びHindIII部位の間にクローニングした。luc+遺伝子(図7Aで示すタンパク質配列をコードする)をNcoI−EcoRV断片上でpSPLuc+から切り取り、HindIII部位を埋めて平滑末端にした後にpRSET−B内のNcoI及びHindIIIの間にクローニングした。この構築物は、ルシフェラーゼアミノ酸配列をアミノ末端6Xヒスチジンタグと融合させる。
【0146】
luc+のPro233の後のpRSET−B−luc+にエンテロキナーゼプロテアーゼ切断部位(Asp(4)Lys)を挿入するために、配列
【化8】
のオリゴヌクレオチドを使用した。プラスミドテンプレートを先ず下記のように変性した。
【化9】
この混合物を室温で5分間インキュベートし、次に2μlの2M酢酸アンモニウム及び75μlの95%エタノールを加え、得られた混合物を−20℃で30分間インキュベートした。次に混合物を最高速度の微量遠心機により4℃で5分間遠心分離した。ペレットを次に−20℃の70%エタノール150μlを用いて洗浄し、2分間遠心分離にかけ、減圧下乾燥し、100μlのTEに溶かした。突然変異誘発性オリゴヌクレオチドを、変性させたテンプレートに以下の反応でアニールさせた:
10μl 変性テンプレート(上記)
1μl 2.9ng/μl上部鎖選択オリゴヌクレオチド(0.25pmole)
1μl 28ng/μl上記突然変異誘発性オリゴヌクレオチド(1.25pmole)
2μl アニーリング 10×緩衝液
6μl H2O
20μl
このアニーリング反応体を75℃の水200mlを含んでいるビーカーに置き、次にその水の中で37℃に冷却させた。次に以下の成分を添加した。
5μl H2O
3μl 10×合成緩衝液
1μl 7.7 U/μl T4 DNAポリメラーゼ
1μl 3 U/μl T4 DNAリガーゼ
30μl
この反応体を37℃で90分間インキュベートし、その後GeneEditor(商標)技術マニュアルで記載されているように反応体の5μlをコンピテントなBMH 71−18 mutSに形質転換した。その翌日、得られた培養物からプラスミドを調製してJM109に再度形質転換した。得られた個々のコロニーを拾い、増殖させ、プラスミドを調製した。突然変異体のスクリーニングは、BanII及びSphIでプラスミドを消化し、エチジウムブロミドで染色した6%ポリアクリルアミドゲル(Novex、Invitrogen)上で生成物を電気泳動にかけることによって実施した。消化物は野生型遺伝子(WT)の場合は361bpの断片を生じ、エンテロキナーゼ部位を含んでいる挿入突然変異体では376bpの断片を生じる。このようにして特定された突然変異体は、次に配列決定をすることによって確認された。この実験では、7/8のクローンが所望の挿入を含んでいた。
【0147】
WT luc+遺伝子又はエンテロキナーゼ部位挿入をコードするプラスミドは、BL21(DE3)pLysS(Novagen)に形質転換した。形質転換された培養物を37℃で約0.5のA600まで増殖させ、次に、1mMのIPTGで誘導し、37℃で更に3〜4時間増殖を継続させた。細胞を次にペレットにし、MagneHis樹脂(Promega Corp.)を使用して酵素精製した。一般的には、2mlの細胞を微量遠心機で2分間遠心分離にかけてペレットにした。ペレットは100μlのMagneHis Wash/Binding緩衝液中で再懸濁し、次に、10μlの10×MLR(製品番号V583A)を加えて細胞を溶解した。5μlの1U/μl RQI DNase(Promega Corp.)及び3μlの7U/μl RNase One(Promega Corp.)を溶解細胞に加え、時々混合しながら氷上で10分間インキュベーションした後、溶解物を4℃で5分間微量遠心機で回転させた。40μlのMagneHis樹脂を上清に加え、次に、得られた混合物を時折混合しながら5分間室温でインキュベートした。樹脂は、次に磁場を加えることによって管壁の上で濃縮し、MagneHis Wash/Binding緩衝液中で再懸濁及び磁化のサイクルを3回繰り返して洗浄した。タンパク質は、最後に、pH7.5の100mM HEPES中の500mMイミダゾールの100μlで溶出した。この手順により、WT又は修飾されたタンパク質の約5μgが得られた。
【0148】
修飾されたタンパク質はエンテロキナーゼ部位を取り込んだけれども、対応するプロテアーゼは酵素活性に影響を及ぼさず、またPro233の後で変異タンパク質を切断することもなかった。WT及び変異タンパク質は、タンパク質からの6Xヒスチジンタグの除去を認めるアミノ末端の他のエンテロキナーゼ部位も含んでいた。ゲル分析は、両方のタンパク質でこの部位がエンテロキナーゼによって利用されることを示した。
【0149】
潜在的にエンテロキナーゼ部位の利用をより容易にするGly(3)Asp(4)LysGly(3)部位がPro233の後に挿入されている、他の修飾されたタンパク質を調製した。突然変異誘発は、配列
【化10】
を有する突然変異誘発性オリゴヌクレオチドを利用して上のように実施した。
【0150】
消化反応は次の通りに組み立てた。
【表3】
エンテロキナーゼ(EKMax)及びその10×緩衝液は、Invitrogenから入手した。反応体は室温でインキュベートし、15分時及び30分時に反応体の1μlを100μlのLuciferase Assay Reagent(Promega Corp.)に加えた。各試料は、次にTurner20/20照度計で読み取った。
これからは、以下のデータが得られた:
【表4】
Gly(3)Asp(4)LysGly(3)部位を有する修飾されたタンパク質をエンテロキナーゼで処理すると、ルシフェラーゼ活性は50〜100%増加することがわかった(図7B)。対照的に、エンテロキナーゼはWT酵素の活性に対して影響を及ぼさなかった。したがって、修飾されたルシフェラーゼ骨格のニッキングは、酵素活性を破壊しなかった。さらに、挿入断片のアミノ酸配列は修飾されたタンパク質に対してプロテアーゼによるニッキングにより軽減されるストレスを起こすこともあり、その結果酵素活性が増加する。
【0151】
エンテロキナーゼ部位、即ちProGlyProGly(3)Asp(4)LysGly(3)ProGlyProを含んでいるより大きな挿入断片を、Luc+のPro233の後に挿入した。タンパク質に対する捻り応力を更に高めるためにProGlyProが含まれた。この挿入を作製するために使用したオリゴヌクレオチドは、
【化11】
であった。pRSET−B−Luc+を開始プラスミドとして使用することより、上のように突然変異誘発を実施した。この場合、得られた突然変異体プラスミドは、ウサギ網状赤血球(Promega TnT(登録商標)Coupled Reticulocyte Lysate System)を使用してin vitroで、以下のような反応で翻訳した;
【表5】
反応体は、30℃で1時間インキュベートしてから、下の通りエンテロキナーゼ(EKMax、Invitrogen)で処理した:
【表6】
エンテロキナーゼを加える前に、次にプロテアーゼ添加後の様々な時間に室温で、100μlのLuciferase Assay Reagent(LAR)で1μlを分析した。得られたデータは、図7Cで示す。WT酵素の活性はプロテアーゼの影響を受けなかったが、修飾された酵素はプロテアーゼ処理で不活性化された。
【0152】
LUC+のLys541の後へのエンテロキナーゼ部位の挿入の影響も、測定した。この場合、前記したようにオリゴヌクレオチド
【化12】
をpRSET−B−luc+プラスミド及びGeneEditorキットと使用して、カルボキシル末端から9番目のアミノ酸であるLys541の後にエンテロキナーゼ部位を導入した。突然変異体プラスミドは、上述のそれらと類似の反応で対照のWTと共に転写及び翻訳し、その後エンテロキナーゼで消化した(図7D)。エンテロキナーゼによる修飾された酵素の処理はその活性を約75%減らしたが、WT酵素の活性は変化しなかった。
【実施例5】
【0153】
欠失及び異種挿入を有する修飾されたホタルルシフェラーゼ
in vitro又はin vivoプロテアーゼアッセイで有用なルシフェラーゼチモーゲンを調製するために、また、特定のタンパク質分解、例えばアポトーシスによって引き起こされるかそれに依存する細胞事象を監視する際に、Lys541(550のアミノ酸のうち)の後に挿入された9のアミノ酸を有するホタルルシフェラーゼ突然変異体を構築した。この9つのアミノ酸は5残基のエンテロキナーゼプロテアーゼ部位をコードし、その後に2つのグリシン、次に(DDDDKGGDI;配列番号58)をクローニングするためのEcoRV部位をコードする2つのアミノ酸が続いた。ベクターは、遺伝子の3’末端の外側に、クローニング部位として使用されたEcoRI部位も有していた。この塩基構築物によって特定されたタンパク質がエンテロキナーゼで切断されると、カルボキシ末端の9アミノ酸が除去され、約10%のWT酵素活性を有する酵素が生成された。大腸菌DNAのEcoRV及びEcoRI断片のライブラリーを、基礎ベクター内のこれらの部位の間にクローニングした。100コロニーを選んでルシフェラーゼ活性を分析した。7コロニーは、WTと比較して活性が100〜1000倍低下していることがわかった。これらの7コロニーを培養してプラスミドを調製した。プラスミドは、それぞれ大きさが約0.2から3kbの大腸菌DNAの挿入断片を含むことがわかった。これらのプラスミドをTNTウサギ網状赤血球溶解物内で翻訳すると、分子量のより高いルシフェラーゼをコードすることがわかった。タンパク質の1つのエンテロキナーゼ切断により、ルシフェラーゼ活性が最大で40倍増加することがわかった。最大の活性化を示す修飾されたタンパク質は分子量が約68kDであり、約60の残基がルシフェラーゼに追加されてチモーゲンを形成していることを示している。
【実施例6】
【0154】
環状置換されている修飾されたホタルルシフェラーゼ
Plainkumら(2003年)は、新しいN及びC末端及び元のN及びC末端を連結するプロテアーゼ認識部位を含んでいるペプチドリンカーを有するリボヌクレアーゼAの環状置換形態は、活性部位の位置的閉塞のためにリボヌクレアーゼ活性が低下したことを報告した。Plainkumらは、プロテアーゼによる環状置換リボヌクレアーゼAの切断は、恐らく活性部位の障害を除去することによってタンパク質の活性を増加させることを明らかにした。
【0155】
ルシフェラーゼの場合、N及びC末端は約40オングストローム離れており、その距離は5〜6のアミノ酸に相当する。ペプチド鎖でルシフェラーゼのN及びC末端を連結することは、タンパク質のカルボキシル末端ドメインによって形成された「リッド」ドメインの閉鎖を阻止することによって、その活性を崩壊させる可能性がある。したがって、ホタルルシフェラーゼluc+遺伝子の頭−尾結合二量体を構築した。PCRプライマーは、増幅された上流側プライマーがAsp(234)から始まり、増幅された下流側プライマーがPro(233)から始まるように設計された。上流側プライマーはAsp(234)の直前にメチオニンのATGコドンを含み、下流側プライマーは終止コドンを含んでいた。元のC及びN末端の間の終止コドンを除去してこれらの末端をプロテアーゼ認識部位をコードする配列で連結するために、in vitro突然変異誘発を使用した。得られたPCR産物をクローニングするために、上流側及び下流側の両方のプライマーは制限酵素部位もコードしていた。
【0156】
方法
頭−尾結合(head to tail)luc+二量体は、次のように構築した。ベクターpSPLuc+(Promega Corp.)をNcoIで消化し、T4 DNAポリメラーゼを使用して末端を埋め、平滑末端線状化ベクターはEcoRIで消化した。受容媒体の役目を果たすために、pSPLuc+はXbaIで消化し、T4 DNAポリメラーゼを使用して末端を埋め、次にEcoRIで消化した。第1の消化物からのルシフェラーゼ断片をこのベクターにクローニングし、同じベクター内の2つのluc+遺伝子の頭−尾結合配列を得た。具体的には、pSPLuc+は次のような反応で消化した。
【化13】
反応体は、37℃で1時間インキュベートし、65℃で15分間加熱してから氷上で短時間冷却した。次に、5μlの10mM dNTP及び1μlの9U/μl T4 DNAポリメラーゼ(Promega Corp.)を加えて、反応体を37℃で20分間インキュベートした。Wizard Clean−Upキット(Promega Corp.)を使用して反応体を精製した。Clean−Up樹脂からの50μlにおける65℃での溶出の後、混合物を冷却し、DNAは5μlの10×緩衝液H(Promega Corp.)及び1μlの12U/μl EcoRI(Promega Corp.)を加えることによって消化した。反応体は、37℃で1時間インキュベートし、次に65℃で15分間加熱した。受容ベクターは、次に以下のように調製した。
【化14】
上の反応体を37℃で1時間インキュベートし、次にPromega Wizard Clean−Upキットを使用して精製し、溶出は65℃の50μl中で行った。以下を精製したDNAに加えた。
【化15】
反応体は、37℃で20分間インキュベートし、次に上で述べたように精製した。5μlの10×緩衝液H及び1μlの12U/μl EcoRIを、Clean−Up樹脂からの溶出液に加えた。反応体は、37℃で1時間インキュベートし、次に65℃で15分間加熱して制限酵素を不活性化した。このDNAを次に下の通り上記の消化したDNAと混合した:
15μl XbaI切断、埋める、EcoRI切断、pSPLuc+加熱
25μl NcoI切断、埋める、EcoRI切断、pSPLuc+加熱
5μl 10×リガーゼ緩衝液(Promega Corp.)
2μl 3U/μl T4 DNAリガーゼ
16℃で一晩のライゲーションの後、1μlを高効率コンピテント大腸菌JM109(Promega Corp.)に形質転換し、細胞をLB/ampプレートに平板培養した。正しい大きさのプラスミドを含んでいる形質転換体を特定した。それらの形質転換体を展開し、そこからプラスミドを単離し、制限酵素消化によってプラスミドの同一性を確認した。
【0157】
新しいN末端がAsp(234)に、新しいC末端がPro(233)にある置換されたルシフェラーゼのPCR増幅のために、上で構築された頭−尾結合二量体Luc+DNAをテンプレートとして使用した。この増幅で使用されたプライマーは、下記配列を有した。
上流側プライマー
【化16】
下流側プライマー
【化17】
増幅反応は、次の通りであった。
5μl 10×PfuUltra緩衝液(Stratagene)
1μl 10mM dNTP
1μl 5ng/μl 上のLuc+二量体構築物DNA
1μl 100ng/μl 上流側プライマー
1μl 100ng/μl 下流側プライマー
40μl H2O
49μl
反応体を混合し、鉱油を重層し、95℃のPE480サーマルサイクラーに入れた。この温度で2分後に、1μlの2.5U/μl PfuUltra DNAポリメラーゼ(Stratagene)を加え、95℃で30秒、50℃で30秒、72℃で1分のサイクルを20回繰り返し、その後ブロックを4℃にした。完了した反応体は次にPromegaのWizard PCR Prepsキットを使用して精製し、Wizard樹脂から50μlのH2Oで溶出した。生成物に取り込まれたPCRプライマーは、NdeI(上流側プライマー)又はBamHI(下流側プライマー)のための部位を有する。PCR産物をこれらの酵素で消化し、下の通りT7発現ベクターpET−3a(Novagen)にクローニングした。
【表7】
【0158】
上の反応体を37℃で1時間インキュベートし、次にそれぞれPromega Wizard Clean−Upキットを使用して精製し、DNAの溶出は50μlのTE中で65℃で行った。2つの精製されたDNAを混合し、下の通り連結した。
5μl 10×リガーゼ緩衝液
20μl 溶出1
10μl 溶出2
2μl 3U/μl T4 DNAリガーゼ
13μl H2O
50μl
ライゲーション反応体を16℃で2時間インキュベートし、次に5μlをコンピテントなJM109に形質転換し、細胞をLB/ampに平板培養した。適当な大きさのプラスミドを含んでいるコロニーを展開し、プラスミドを調製し、正しく挿入されているかについて各調製物を制限消化により確認した。プラスミドはPCR産物の挿入を含んでいるのがわかったので、これは、終止コドンが除かれ2つのルシフェラーゼ遺伝子部分を分けている接合部でC及びN末端を連結している、in vitro突然変異誘発のための基礎ベクターとして使用した。
【0159】
最初の突然変異誘発は、Asp(4)Lysのカルボキシル末端側を切断するプロテアーゼエンテロキナーゼの認識部位を含んでいる突然変異誘発性オリゴヌクレオチドを利用し、Promega Corp.からのGene Editorキットを使用して実施した。このオリゴヌクレオチドは、下記配列を有していた。
【化18】
突然変異誘発手順の第2回目の形質転換からの6コロニーを個々に増殖させて、それらからプラスミドを調製した。これらのプラスミドは、TnTウサギ網状赤血球溶解物(Promega Corp.)内での共役転写/翻訳による突然変異誘発性オリゴヌクレオチドの取り込みについてスクリーニングした。正しい突然変異体は、ルシフェラーゼドメインのC及びN末端が融合し、完全長ルシフェラーゼタンパク質を生成した。翻訳反応は、次のように実施した。
25μl TnTウサギ網状赤血球溶解物
2μl TnT反応緩衝液
1μl T7 RNAポリメラーゼ
1μl 完全アミノ酸配合
1μl Fluorotect Lys tRNA
1μl 40U/μl rRNasin
5μl ミニプレップDNA
14μl
50μl
翻訳反応体は、30℃で60分間インキュベートしてから、下の通りエンテロキナーゼ(EK)(EKMax、Invitrogen)で処理した(又は処理しなかった)。
2μl 10×EKMax緩衝液
5μl 上の翻訳反応体
+/−1μl 1U/μl EKMax
12μl H2O
20μl
これらの消化は室温で30分間行われ、その後1μlを100μlのルシフェラーゼ検定用試薬(Promega Corp.)に加えて検定した。データ収集は、Turner20/20照度計で行った。5μlの4×SDS試料緩衝液を各反応体の残りに加え、試料を65℃で2分間加熱した。次に試料を4〜20%Novexトリス−グリシンゲルで電気泳動にかけ、ゲルは高感度のMolecular Dynamics FluorImagerでスキャンした。結果は、融合した完全長タンパク質は6クローンの内の2つで形成されたことを示し、突然変異誘発が成功したことを示した。さらに、融合した変異タンパク質の活性は、エンテロキナーゼによる処理で約150倍増加した。さらに、ゲルは、プロテアーゼが完全長タンパク質を消化して断片化したことを示した。
【0160】
蛍光リジン誘導体の取り込みによって標識されていない突然変異体ルシフェラーゼの活性に及ぼすEK処理の影響を検討するために、翻訳反応を上記のように実施したが、Fluorotect Lys tRNAは反応から省略された。この場合、EKで酵素を処理するとルシフェラーゼ活性の約90倍の活性化が観察された(図7)。活性化の後、変異酵素はWT活性の約0.5%を回復した。
【0161】
2つのルシフェラーゼドメインの間にカスパーゼ−3 DEVD切断部位を挿入するために、他の突然変異誘発を実施した。Promega Gene Editorキットは、以下の突然変異誘発性オリゴヌクレオチドと使用された。
【化19】
【0162】
この場合、所望の突然変異体は5/8クローンで見られ、これらはin vitro転写/翻訳によってスクリーニングした。カスパーゼ−3による活性化倍率は、エンテロキナーゼで以前に観察された活性化倍率より高いことが明らかになった。また、切断によって回復された活性の割合も高くなっていた。
【0163】
in vitro翻訳は、Promega TnTウサギ網状赤血球溶解物を使用して、カスパーゼ−3 DEVD切断部位を含んでいる置換されたルシフェラーゼをコードするプラスミド又はWTルシフェラーゼを含んでいる反応で実施した。これらの反応体の一部を次にカスパーゼ−3(100単位、BioMol)で消化し、図9〜11で示すデータが生成された。WT酵素の活性はプロテアーゼの影響を受けなかった。対照的に、変異酵素の活性はカスパーゼ−3による処理で大きく増加した。活性化倍率はこの場合約500倍であり、活性化された酵素の活性はWTの活性の約17%であった。
【0164】
置換された酵素のカスパーゼ−3活性を検出する能力も、発光プロテアーゼアッセイで検討した。カスパーゼ反応は以下のように実施した。
10μl 2×カスパーゼ緩衝液
5μl in vitro翻訳タンパク質
1μl 希釈カスパーゼ−3
4μl H2O
20μl
反応体は9.6から2333pgのカスパーゼ−3を含み、これらは室温で90分間インキュベートし、その後1μlを取り出して100μlのルシフェラーゼ検定用試薬に加え、Turner 20/20照度計で読み取った。図9Aは、得られたデータを示す。プロテアーゼの低量点を再プロットすると(図9B)、このアッセイで低ピコグラムのカスパーゼ−3を検出することができることが示される。さらに、インキュベーション時間を90分から一晩に長くすると、アッセイの感度は更に4倍高まった(データは示さず)。
【0165】
置換されたルシフェラーゼの合成及び活性化も、TnT Wheat Germ抽出物(Promega Corp.)で検討した。反応には以下が含まれた。
25μl TnT T7 WG抽出物
2μl TnT反応緩衝液
1μl T7 RNAポリメラーゼ
1μl アミノ酸配合
1μl 40U/μl rRNasin
5μl 50ng/μlルシフェラーゼプラスミド
15μl H2O
50μl
反応体は30℃で90分間インキュベートした後、以下の通りプロテアーゼで消化した。
10μl 2×緩衝液(100mM HEPES pH7.5、200mM NaCl、0.2%CHAPS、2mM EDTA、20%グリセリン、20mM DTT)
10μl in vitro翻訳反応体
1μl 1U/μl EKMax又は1μl 100U/μl カスパーゼ−3
【0166】
プロテアーゼ消化物は室温でインキュベートし、その後様々な時間で1μlを100μlのルシフェラーゼ検定用試薬に加え、Turner 20/20照度計で読み取った。この実験で、カスパーゼ−3は置換カスパーゼ−ルシフェラーゼの活性をWTのそれの約4分の1まで約3000倍高め、EKはEK−ルシフェラーゼの活性をWTの約1.1%まで約300倍高めた(図8及び11)。一例ではDEVDG、他ではDDDDGで、両方の挿入断片は同じ大きさである点に注意する。したがって、ルシフェラーゼの3つのN末端アミノ酸及び6つのC末端アミノ酸を元のタンパク質の末端からそれぞれ置換することによって、より長い配列が取り込まれる可能性がある。このことは、少なくとも14〜15のアミノ酸部位が2つのルシフェラーゼドメインの間に取り込まれるのを可能にするはずである。上で言及した9つの残基は対応する結晶構造に現れず、したがって非常に柔軟であり、酵素活性に悪影響を及ぼさずに置換することが可能な点に注意する。
【実施例7】
【0167】
追加の環状置換構築物
A.PSAは、配列Ala−Asn−Lys−Ile−Ser−Tyr−Gln−Ser−Ser−Ser−Thr−Glu(配列番号21)内のGln及びSerの間でSemenogelin Iを切断するプロテアーゼである。PSAの切断基質を有する修飾されたルシフェラーゼを生成するために、関連する12量体ペプチドAla−Asn−Lys−Ala−Ser−Tyr−Gln−Ser−Ala−Ser−Thr−Glu(配列番号22)のオリゴヌクレオチドを、実施例6で記載したプラスミド構築物内のXhoI及びNcoI部位の間にクローニングした。以下の配列
【化20】
を有するオリゴヌクレオチドを、以下の配列を有するオリゴヌクレオチドにハイブリダイズした。
【化21】
ハイブリダイズされたオリゴヌクレオチドはXhoI及びNcoI適合末端を有する二本鎖断片を生成するが、NcoI部位は改造されているのに対してXhoI部位は破壊されている。ベクターをXhoI及びNcoIで消化してアニールしたオリゴヌクレオチドに連結し、次に大腸菌に形質転換した。ミニプレップDNAを個々のコロニーから調製し、プロテアーゼ部位を含んでいるオリゴヌクレオチドの取り込みを示す、XhoIではなくNcoIによる消化についてプラスミドをスクリーニングした。所望の構築物はコムギ胚(WG)翻訳抽出物又はウサギ網状赤血球溶解物内でin vitroで翻訳し、得られたタンパク質は精製PSA(シグマ)で処理した。翻訳を実施した。開裂反応は以下の通り実施した。
【表8】
反応体は室温で20分又は40分間インキュベートした。各反応の1μlを100μlのルシフェラーゼ検定用試薬(LAR)に加え、光出力をTurner20/0照度計で記録した。下記データを得た。
【表9】
PSAの添加は、光出力の実質的な増加をもたらした。20分時には、修飾されたルシフェラーゼの活性化倍率はウサギ網状赤血球溶解物内で合成された修飾されたルシフェラーゼの場合は658倍であり、コムギ胚抽出物で合成された修飾されたルシフェラーゼの場合は1,110倍であった。
【0168】
B.PreScissionプロテアーゼは、GST(グルタチオンSトランスフェラーゼ)及びライノウイルス3Cプロテアーゼ(Amersham)で構成される融合タンパク質である。このプロテアーゼは、配列Leu−Glu−Val−Leu−Phe−Gln−Gly−Pro(配列番号25)内のGln及びGly残基の間で切断することができる。この配列を規定しているオリゴヌクレオチドが設計され、下記配列(トップストランド)
【化22】
を有していた。これらのオリゴヌクレオチドのアニーリングはXhoI及びNcoI適合末端を有する二本鎖断片を生成し、XhoI部位は保持されているのに対してNcoI部位は破壊されている。上記の例の場合のように、アニールされたオリゴヌクレオチドは、XhoI及びNcoIで切断されたベクターにクローニングされた。所望のクローンの濃縮のために、形質転換の前にライゲーション混合体をNcoIで再切断した。所望のプラスミドを選択して、前記したようにウサギ網状赤血球溶解物内でin vitro翻訳を行った。消化反応体は、以下の通り調製した。
【表10】
反応体は室温でインキュベートし、その後様々な時間で1μlを100μlのLARに加え、試料をTurner 20/20照度計で読み取った。以下のデータが生成された。
【表11】
PreScissionプロテアーゼによるルシフェラーゼの活性化は速やかに起こり、プロテアーゼの存在下で4,000倍を超える発光の増加をもたらした。
【0169】
C.真核細胞が含まれない溶解物内で合成された置換されたルシフェラーゼのタンパク分解性処理によって高度な活性化が観察されたが、非融合タンパク質が大腸菌で合成されたときの活性化の程度は非常に小さかった。面白いことに、大腸菌調製物の部分精製は、プロテアーゼによって活性化される能力の高くなったタンパク質をもたらした。細菌細胞から環状置換されたルシフェラーゼを有効に精製するために、ベクターpGEX−6P3(Amersham)内でカスパーゼ−3部位を有する環状置換されたルシフェラーゼがGSTに融合されているベクターを調製した。PCR反応体は以下を含んでいた。
5μl 10×PfuUltra緩衝液
1μl 10mM dNTP
1μl 5ng/μlカスパーゼ−3部位プラスミド
1μl 100ng/μl 上流側オリゴヌクレオチド
1μl 100ng/μl 下流側オリゴヌクレオチド
40μl H2O
50μl
1μlの2.5μ/μl PfuUltra DNAポリメラーゼ(Stratagene)を加えてPCRを開始し、95℃で30秒、50℃で30秒、72℃で1分のサイクルを20回繰り返し、その後4℃にした。
【0170】
上流側オリゴヌクレオチドはBamH1を含み、下記配列を有し
【化23】
下流側オリゴヌクレオチドはEcoRI部位を含み、下記配列を有す
【化24】
得られたPCR産物はEcoRI及びBamH1で消化し、ベクター内のこれらの部位の間にクローニングすると、GSTとルシフェラーゼとのインフレーム融合がもたらされる。所望のプラスミドを特定し、大腸菌系統Rosetta(Novagen)に形質転換した。細胞をLB培地で増殖させ、IPTGを1mMまで添加することによって誘導した。最良の増殖条件は25〜26℃で一晩の誘導であることが明らかになった。細胞を収集して超音波処理で溶解した。遠心分離による清浄の後、上清を固定化グルタチオンを含んでいるカラムに加え、遊離のグルタチオンを含んでいる緩衝液で溶出した。融合タンパク質の収率は、初期の培養物1リットルにつき約1ミリグラムであった。カスパーゼ−3による活性化は、活性化反応の条件に従い約1,200倍以上で、最高50,000倍(氷上で一晩の活性化)であった。
【0171】
D.SARSウイルスプロテアーゼ部位TSAVLQSGFR(配列番号19)を含んでいる3つの環状置換されたルシフェラーゼを生成した。2つはコメツキムシのルシフェラーゼ(CP1:R=Asn401及びCP2:R=Arg223)で1つはホタルルシフェラーゼ(CP:R=Asp234)のものである。CP2は、ホタルルシフェラーゼの位置234と対応するコメツキムシルシフェラーゼの位置に挿入を有する。
【0172】
SARSウイルスプロテアーゼ部位を有する環状置換されたコメツキムシルシフェラーゼは、次のように構築された。先に述べたようにNdeI及びBamHI部位の間に挿入突然変異体cbg69SARS3遺伝子を、及びSnaBI及びSalIの間にSARSウイルスプロテアーゼ部位をコードする配列を含んでいるプラスミドpJLC33を開始ベクターとして使用した。以下のプライマーセットを使用して、野生型cbg69を含んでいるpJLC1からPCR断片を増幅した。
【化25】
【0173】
CP1−a(又はCP2−a)のPCR産物をSalI及びBamHIで消化し、pJLC33のそれぞれの部位にクローニングし、pJLC−cp1a(又はpJLC−cp2a)を得た。CP1−b(又はCP2−b)のPCR産物をNdeI及びSnaBIで消化し、pJLC−cp1a(又はpJLC−cp2a)のそれぞれの部位にクローニングした。得られたプラスミドpJLC47(又はpJLC48)は、SARSウイルスプロテアーゼ部位を有するコメツキムシルシフェラーゼの環状置換された突然変異体1(又は2)を含む。
【0174】
置換されたホタルルシフェラーゼについては、置換されたベクターは、元のN及びC末端のためにDNAを分離しているXhoI及びNcoI部位を有するリンカーを取り込むように修飾した。このリンカーは、
【化26】
であり、これは第1のドメインのC末端から6アミノ酸を除去し、第2のドメインのN末端から3アミノ酸を除去する。SARSウイルスN末端の自己切断部位は、SITSAVLQSGFRKMA(配列番号53)である。この配列を規定しているオリゴヌクレオチドは、次のように設計された。
【化27】
アニールされたオリゴヌクレオチドはNcoI部位を保持し、XhoI部位を欠く。アニールされ消化されたオリゴヌクレオチドは、上記したように基礎ベクターにクローニングした。
【0175】
SARSウイルスプロテアーゼ部位を有する3つの環状置換されたルシフェラーゼCbg69CP1、Cbg69CP2及びFfCPの全ては、ウサギ網状赤血球溶解物及び/又はコムギ胚抽出物(Promega)などのin vitro翻訳系を使用して生成した。SARSウイルスプロテアーゼは、New England BiolabsからのpMAL精製系を使用して部分的に精製した。変異ルシフェラーゼを含んでいる溶解物をSARSウイルスプロテアーゼと混合して、ルシフェラーゼ活性を監視した。約0.3μgのSARSウイルスプロテアーゼとの室温で1時間のインキュベーションの後、Cbg69CP2及びFfCPはそれぞれ20〜30倍及び60〜200倍に活性化された(図12)が、Cbg69CP1は活性化されなかった(データは示さず)。
【0176】
E.活性カスパーゼ−3を生成するためのプロカスパーゼ−3の活性化は、生細胞におけるアポトーシス誘導の代理である。修飾されたルシフェラーゼが細胞内のアポトーシスを監視するために使用することができるかどうか確認するために、カスパーゼ−3切断部位を含んでいる環状置換されたルシフェラーゼをCMVプロモーターの制御下の哺乳類発現ベクターにクローニングし、一過性形質移入を通してHela細胞に導入した。細胞を次にタンパク質TRAILで処理し、死受容体の活性化を通してアポトーシスを誘発して活性カスパーゼ−8を形成し、これは次にプロカスパーゼ−3を活性化してカスパーゼ−3にする。したがって、活性カスパーゼ−3の出現は、ルシフェラーゼ基質が切断されて酵素によって活性化されるに従い、発光の増加を伴うはずである。
【0177】
カスパーゼ−3切断部位を含んでいる置換されたルシフェラーゼのPCR断片は、NheI及びEcoRIのための部位を含んでいるプライマーを使用して作製され、ベクターpCI−neo(Promega)のこれらの部位の間にクローニングした。増幅は、上記したように下記上流側プライマーを用いて実施した。
【化28】
得られた構築物は、一般式ANNATGGの最適コザック配列を有していた。DNAは、TransFast形質移入試薬(Promega)を使用してHela細胞に形質移入し、DMEM+10%Cosmic Calf Serum中の1μg/μlのTRAILタンパク質(Biomol)を加えることによりアポトーシスを開始した。一部のウェルを、初期SV40プロモーター/エンハンサーの制御下の天然のホタルルシフェラーゼ遺伝子(非置換)を運ぶプラスミドpGL3−controlで形質移入した。示された時間に100μlのBright−Glo試薬をウェルに加え、Orion照度計(0.5秒の読取り)で発光を記録した。
【表12】
【0178】
図13のデータは、置換されたルシフェラーゼ−カスパーゼ−3プラスミドを受け取る細胞では、発光がなくなってから1〜2時間の間にTRAILタンパク質が発光活性を誘導することを示す。この影響は、培地だけを含んでいるウェルでは見られない。pGL3−controlプラスミドを含んでいるウェルは、培地単独と培地+TRAILとの間で相違を示さなかった。培地を変えたときに見られるLUの全般的増加の原因はタンパク合成の誘導である可能性があり、pGL3−controlではルシフェラーゼは活性型であるが、pCI−neo−カスパーゼプラスミドではルシフェラーゼは不活性型である。形質移入のストレスによる死細胞バックグラウンドが観察されているので、この培地単独の場合に見られる小さな増加の原因は、アッセイの全過程にわたる死細胞の蓄積にあるかもしれない。
【実施例8】
【0179】
修飾されたルシフェラーゼによる細胞ベースのアッセイ
分子内制御をコードし、カスパーゼ−3活性を検出するベクターを提供するために、本発明の融合タンパク質をコードするベクターを調製した。Renillaルシフェラーゼ(対照)を、残基400の後にDEVDを含んでいる修飾されたコメツキムシルシフェラーゼ(Cbg69DEVD)のN末端又はC末端に融合した。リンカー配列(Gly(2)SerGly(4)SerGly(4)SerGly(2))を2つのタンパク質の間に置いた。
【0180】
rLuc−リンカー−Cbg69DEVD融合を作製するために、一対のオリゴヌクレオチド
【化29】
を使用してプラスミドpJLC6から完全長Renillaルシフェラーゼ遺伝子(rLuc)を増幅した。得られたPCR断片をNdeI及びAseIで消化して、Cbg69DEVDをコードするpJLC23のNdeI部位にクローニングした。
【0181】
Cbg69DEVD−リンカー−rLuc融合を作製するために、一対のオリゴヌクレオチド
【化30】
を使用してプラスミドpJLC23から完全長Cbg69DEVD遺伝子を増幅した。得られたPCR断片をNdeI及びAseIで消化して、rLucを含んでいるpJLC6のNdeI部位にクローニングした。
【0182】
図14は、各融合タンパク質がRenillaルシフェラーゼ並びにコメツキムシルシフェラーゼの活性を有していたことを示す。
【実施例9】
【0183】
結合して機能性ルシフェラーゼを形成するルシフェラーゼ断片の能力の評価
一実施形態では、本発明は、ルシフェラーゼの2つの独立した断片が互いに相補して機能性タンパク質を生成することのできる系を提供する。
【0184】
材料及び方法
ルシフェラーゼのN及びC末端の断片の、in vitro及びin vivoで結合して機能性ルシフェラーゼタンパク質を形成する能力を評価するために、3つの構築物が設計された(図15)。ホタルルシフェラーゼ遺伝子のN末端の699ヌクレオチド(アミノ酸1〜233)を、順方向プライマー
【化31】
及び逆方向プライマー
【化32】
を使用して以下の条件でpSP−luc+(Promega Corp.)から増幅した。パーキンエルマー2400サーマルサイクラーを使用して、95℃で2分間、1サイクルが95℃で30秒間、50℃で30秒間及び72℃で2分間を25サイクル、その後72℃で10分間。NheI制限部位は順方向プライマーの5’末端へ導入し、BamHI制限部位は逆方向プライマーの5’末端へ導入した。得られたN末端ルシフェラーゼ断片は次に確立された技術(Sambrookら、1989年)を使用してpBINDベクターのNheI及びBamHI制限部位にクローニングし、発現ベクターpJLC62(n luc)を得た。
【0185】
同様に、ホタルルシフェラーゼ遺伝子のC末端の951ヌクレオチド(アミノ酸234〜550)を、順方向プライマー
【化33】
及び逆方向プライマー5’TTGGCGCGCCGGATCCTTACACGGCGATCTTTCCGCCCTTCTTG3’(配列番号63)を使用して、N末端のクローニングについて前記したのと同じPCR条件下でpSP−luc+から増幅した。NheI及びBamHI制限部位はN末端プライマーについて前記したようにプライマーに導入し、C末端のルシフェラーゼ断片はpBINDベクターのNheI及びBamHIにクローニングして発現ベクターpJLC63(c luc)を得た。
【0186】
ルシフェラーゼ遺伝子全体(1650ヌクレオチド、550アミノ酸)を、順方向プライマー
【化34】
及び、逆方向プライマー
【化35】
を使用し、また上記同じPCR条件を使用してN及びC末端クローンで使用されたのと同じ方法でpBINDベクターにクローニングした。得られた発現ベクターpJLC64(完全長FF)をタンパク質相補性実験のための対照として使用した。
【0187】
全ての構築物は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して、正しいタンパク質の大きさについて検証した。
【0188】
in vitroタンパク質相補性試験は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して実施した。翻訳の後、各試料の2μlを100μlのルシフェラーゼ検定用試薬に加え、Veritas照度計を使用して発光を測定した。
【0189】
in vivo相補性試験は、チャイニーズハムスター卵巣(CHO)及び293のヒト胎生期腎臓組織培養細胞で実施した。CHO又は293の細胞のいずれかの組織培養細胞を6穴の組織培養プレートに播種し、37℃で一晩、5%CO2で増殖させ、翌日80%のコンフルエンシーで形質移入した。形質移入は、TransFast(商標)形質移入試薬(Promega Corporation)を使用してメーカーの推奨に従って実施した。つまり、対照反応では、各形質移入の最終濃度が2μgの全DNAとなるように、pJLC62、pJLC63又はpJLC64のいずれかの1μgを1μgのpBIND対照プラスミド(ホタルルシフェラーゼ遺伝子を含まない元のベクター)で形質移入した(3μl TransFast(商標)試薬/μg DNA)。タンパク質相補性試験については、1μgのpJLC62及び1μgのpJLC63を同じプロトコルに従って形質移入した。形質移入後24時間時に、細胞をトリプシン処理して各形質移入条件のために2群に分けた。250μlの1×Passive Lysis緩衝液(Promega Corporation、PLB)を1つの群に加え、250μlの1リン酸緩衝生理食塩水(PBS)を他の群に加えた。PLB群は組織培養細胞の溶解を確実にするために−80℃の凍結融解で1回処理し、PBS群は凍結融解をしなかったので非溶解細胞が維持された。全ての群からの発光は、Dual−Luciferase (登録商標)レポーターアッセイシステムを使用し、メーカーの推奨に従って測定した。基本的には、各群からの20μlを3反復の白の96穴プレートに加え、Veritas照度計でアッセイを実施した。全てのホタルルシフェラーゼデータは、Renillaルシフェラーゼシグナルに正規化した。
【0190】
結果
図15で示す3つの構築物の全ては、正しい大きさのタンパク質を与えた(図16A)。本明細書で先に記載したプロテアーゼ切断後の環状置換ホタルルシフェラーゼの活性化は、ルシフェラーゼの断片が酵素活性を相補し且つ再構成することができることを示唆した。図16B(同じTnT反応におけるルシフェラーゼのN及びC断片)で見られるように、N及びC末端のルシフェラーゼ断片のin vitroタンパク質相補性は、完全長ルシフェラーゼタンパク質と比較して機能性タンパク質を与えた。さらに、in vivoタンパク質相補性は、CHO及び293組織培養細胞の両方で起こった(図17)。たとえ組織培養細胞が溶解しなかった場合でも類似の傾向が見られた(PBS;データは示さず)。
【実施例10】
【0191】
モジュレーター系におけるルシフェラーゼ融合タンパク質の非共有結合的結合の検出
一実施形態では、本発明は、2つの部分の結合を誘発又は強化、或いは抑制する外因性因子(エフェクターA)、及び任意選択に前記2つの部分の結合をそれぞれ解離又は強化する他の外因性因子(エフェクターB)を有するモジュレーター系を提供する。例えば、そのような系は、ルシフェラーゼに融合されたFKBP及びFRBの結合の誘導物質としてラパマイシンを、また結合の解離物質としてFK506を使用することができる。
【0192】
A.ルシフェラーゼモジュレーター系を示すin vitro実験
材料及び方法
ヒトコドン最適化ホタルルシフェラーゼ遺伝子(luc2.0)を、順方向プライマー
【化36】
及び逆方向プライマー
【化37】
を使用してポリメラーゼ連鎖反応(PCR)により以下の条件でpGL4.10(luc2)(Promega Corporation)(配列番号66)から増幅した:95℃で2分間、次に1サイクルが95℃で30秒間、50℃で30秒間及び72℃で2分間のものを25サイクル、72℃で10分間の最終展開。ルシフェラーゼタンパク質とのN末端融合の生成を容易にするために、NcoI及びEcoRV制限酵素部位を順方向プライマーの5’末端に導入した。ルシフェラーゼタンパク質とのC末端融合の生成を容易にするために、SnaBI、NotI及びSacI制限酵素部位を逆方向プライマーの5’に導入した。5’及び3’末端上の更なるクローニング部位を有する増幅されたルシフェラーゼ遺伝子をルシフェラーゼT7対照ベクター(Promega Corp.、Cat No#L4821)のHindIII/SacI部位にクローニングし、対照ベクターに通常存在するルシフェラーゼ遺伝子を置換した。得られたベクターは、pJLC65と呼ばれた。in vitro発現ルシフェラーゼT7対照ベクターへのクローニングの一般的スキームは、図18で見ることができる。
【0193】
いくつかの発現構築物がpJLC65ベクターを使用して作製された;ホタルルシフェラーゼへのFRBのN末端融合(pJLC66)、ホタルルシフェラーゼへのFKBPのC末端融合(pJLC67)及びホタルルシフェラーゼへのFRB(N末端)及びFKBP(C末端)の二重融合(pJLC68)。FRBは、
【化38】
の合成遺伝子を含んでいるBlue Heronからのプラスミドから得られた。FRBは、5’末端のNcoI制限酵素部位及び3’末端のEcoRV制限部位を使用してBlue Heronベクターから切断され、公知の分子生物学的技術(Sambrookら、1989年)を使用してルシフェラーゼ遺伝子のN末端にクローニングした。
【0194】
FKBPは、
【化39】
の合成遺伝子を含んでいるBlue Heronからのプラスミドから得られた。FKBP断片がルシフェラーゼ遺伝子のC末端にクローニングされるように、FKBPを遺伝子の5’末端のSnaBI制限酵素部位及び3’末端のNotI制限酵素部位を使用してBlue Heronベクターから切断した。二重融合は、FRB及びFKBPをそれぞれN末端及びC末端に含んでいた。
【0195】
4つのルシフェラーゼ構築物は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して、発現されたタンパク質の正しい大きさについて評価した。簡潔には、4つの各反応において、1μgの適当なDNAをFluoroTect(商標)GreenLys tRNAを含む50μlの反応体に加えた。各反応体(5μl)からの試料を、NuPAGE(登録商標)技術ガイド(バージョンE、IM−1001)で記載されているように、1×NuPAGE(登録商標)MES SDSランニング緩衝液を使用して10%のNuPAGE(登録商標)Novex Pre−Cast Bis−トリスゲル(Invitrogen Corporation)に流した。FluorImager SI(Molecular Dynamics)を使用してゲルを画像化した。
【0196】
in vitroアッセイについては、上述の各TnT(登録商標)反応からの5μlを、0.2μMラパマイシン(BioMol)を含んでいるか含まない95μlの1×Passive Lysis緩衝液(Promega Corporation)に別々に加えた。ラパマイシンの添加の後、各試料からの10μlを100μlのルシフェラーゼ検定用試薬(TnT(登録商標)システムを設置)に加え、Turner20/20照度計(Turner BioSystems)を使用して発光を測定した。
【0197】
FRB及びFKBPの間の相互作用が調整することができるかどうか研究するために、ラパマイシンと競合して融合パートナー間の相互作用を抑制することが公知であるFK506をin vitro実験で使用した。二重融合FRB−luc2−FKBPを先に述べたように転写及び翻訳した。翻訳の後、試料の4μlを5μlの2×FLICE緩衝液(100mM HEPES、pH7.5、200mM NaCl、0.2%CHAPS、2mM EDTA、20%グリセリン、20mM DTT)及び1μlのラパマイシン(10nM)、及び0、1、2、5、10、20及び40nM(0、0.82、1.64、4.1、8.2、16.4及び32.8ng/mlのタクロリムスに相当)の様々な濃度のFK506(タクロリムス、Antibioticplus.com)と混合した。試料は室温で15分間インキュベートし、その後試料の5μlを100μlのルシフェラーゼ検定用試薬で希釈してTurner20/20照度計で発光を測定した。
【0198】
結果
下記4つの構築物を調製した:luc2(ホタルルシフェラーゼをコードする;550アミノ酸)、FRB−luc2(FRB及びホタルルシフェラーゼの融合をコードする;644アミノ酸)、luc2−FKBP(ホタルルシフェラーゼ及びFKBPの融合をコードする;657アミノ酸)及びFRB−luc2−FKBP(二重融合をコードする;771アミノ酸)。4つの構築物(3つは対照、1つは二重融合)は、TnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling Systemと一緒に使用して、発現されたタンパク質の正しい大きさについて評価した。4つの構築物の全ては、正しい大きさのタンパク質を与えた(図19)。
【0199】
次に2つの融合パートナーFRB及びFKBPの間の相互作用をin vitro系で検出するために、これらの構築物を実験で使用した。誘導物質ラパマイシンの存在下で、2つの融合パートナーが結合して発光は減少するはずである。対照反応と比較して、ラパマイシンの添加により二重融合FRB−luc2−FKBPでは相対発光は20倍減少した(図20A)。したがって、ラパマイシンの存在下では、ルシフェラーゼとの二重融合における2つの結合パートナーは結合し、それによってルシフェラーゼ酵素を制限し、その結果酵素はルシフェリン基質と有効に相互作用することができなくなった。逆に、FK506の添加量が増加すると、二重融合反応の発光はFK506の増加の量に応じて増加した(図20B)。
【0200】
B.他のルシフェラーゼモジュレーター系
材料及び方法
クローニングは、以下の例外を除いて先に述べたように実施した。赤色コメツキムシ遺伝子(cbr)は、順方向プライマー
【化40】
及び、逆方向プライマー
【化41】
を使用してpCBR−Basic(Promega Corporation)から増幅した。EcoRV部位を順方向プライマーの5’末端に、BglIIを逆方向プライマーの5’末端に導入し、その後、対応する増幅された断片をpJLC68の対応する部位にクローニングした。緑色コメツキムシ遺伝子(cbg)は、順方向プライマー
【化42】
及び、逆方向プライマー
【化43】
を使用してpCBG68−Basic(Promega Corporation)から増幅した。赤色コメツキムシでpJLC68ベクターにクローニングしたのと同じ制限部位をこれらのプライマーに導入した。Renillaルシフェラーゼ遺伝子(Rluc)は、順方向プライマー
【化44】
及び、逆方向プライマー
【化45】
を使用してphRL−null(Promega Corporation)から増幅した。pJLC68ベクターのEcoRV(SnaBIとの平滑末端ライゲーション)及びBglII上でクローニングするために、SnaBI部位を順方向プライマーの5’末端に、BglII部位を逆方向プライマーの5’末端に導入した。pJLC68へのcbg、cbr及びRlucのクローニングは、FRB−ルシフェラーゼ−FKBP型の二重融合をもたらした。正しいタンパク質の大きさについてクローンを検証した(図21A)。先に述べたように二重融合を転写及び翻訳し、発光を測定したが、例外として、ラパマイシンの実験では0.2μMのラパマイシンだけが使用された。
【0201】
結果
ホタルルシフェラーゼタンパク質で見られたFRB及びFKBP系の類似した調整が他の種類のルシフェラーゼでも見られるかどうかを決定するために、ホタルルシフェラーゼ遺伝子をPyrophorus plagiophalamからの赤及び緑の2つの修飾されたコメツキムシ遺伝子、並びにRenilla reniformisからのルシフェラーゼ遺伝子で置換した。pJLC68へのcbg、cbr及びRlucのクローニングは、FRB−ルシフェラーゼ−FKBPの二重融合をもたらした。正しいタンパク質の大きさについてクローンを検証した(図21A)。先に述べたように二重融合を転写及び翻訳し、発光を測定した。
【0202】
図21Bで見られるように、緑及び赤のコメツキムシ二重融合は、ラパマイシンが存在したときは対照(Luc2)と比較して同じ相対効果を示した。発光はラパマイシンに応じて減少したが、Renillaルシフェラーゼはラパマイシンに反応しなかった。
【0203】
C.ルシフェラーゼモジュレーター系のin vivo証明
実施例X.AからのpJLC68をテンプレートとして使用して、FRBのルシフェラーゼとのN末端融合(FRB−Luc2)の断片、ルシフェラーゼのFKBPとのC末端融合(Luc2−FKBP)、又は二重融合(FRB−Luc2−FKBP)を、95℃で2分間、続いて1サイクルが95℃で30秒、50℃で30秒間及び72℃で2分間のものを25サイクル、また72℃で10分間の最終展開からなるPCRプログラムに従って増幅した。増幅のための全ての順方向プライマーはプライマーの5’末端にNheI制限酵素を含むように設計され、全ての逆方向プライマーはプライマーの5’末端にBamHI制限酵素部位を含むように設計され、それによりpBINDベクターへのクローニングのための5’末端がNheI部位及び3’末端のBamHI部位と連なる増幅断片が形成された。増幅のためのプライマーは、次の通りである:
Luc2:
順方向プライマー:
【化46】
逆方向プライマー:
【化47】
FRB−Luc2:
順方向プライマー:
【化48】
逆方向プライマー:
【化49】
Luc2−FKBP:
順方向プライマー;
【化50】
逆方向プライマー;
【化51】
FRP−Luc2−FKBP:
順方向プライマー:
【化52】
逆方向プライマー:
【化53】
【0204】
phRL−TKベクター(Promega Corporation)をTKプロモーター及びベクター骨格の供与源として使用して(図22A)、ベクターで見られるRenillaルシフェラーゼを除去してpBIND−FRB−Luc2−FKBPから増幅したFRB−Luc2−FKBP配列で置換し、断片の5’末端にNheI制限酵素を、断片の3’末端にXbaI部位を生成した。確立された分子生物学的手法(Sambrookら、1989年)を使用して、以下のプライマーで以降のphRL−TKベクターへの挿入のための生成物を生成した。TK FRP−Luc2−FKBP;
順方向プライマー:
【化54】
逆方向プライマー
【化55】
【0205】
ラパマイシンへのFRB−FKBP相互作用のin vivo反応を、D293細胞(cAMP刺激に対する反応の増加のために以前に選択された親ATCC CRL−1573 HEK293細胞の下位集団)を使用して研究した。全てのin vivo実験について、形質移入の前にD293細胞を96穴組織培養プレート上に5,000細胞/ウェルの密度で播種し、37℃及び10%のCO2で少なくとも8時間インキュベートした。プロトコルで記載されているように、TransIT(登録商標)LT1形質移入試薬(Mirus Corporation)を使用し、96穴プレートの1ウェルにつき0.1μgDNA/0.3μl形質移入試薬の濃度を使用して、pBIND構築物及びTK二重融合構築物をD293細胞に形質移入した。形質移入の約24時間後に(図24〜25)、10μlの50mMルシフェリンEF(Promega Corporation、無エンドトキシン)を各ウェルに加え、少なくとも15分間細胞を平衡化させた。初期の発光読取値を各試料(時間は0分)から測定し、次に、OptiMEM組織培養培地(Invitrogen)で希釈した0.2μMラパマイシン原液の10μlをラパマイシン試験ウェルに加え、対照ウェルにはラパマイシンを加えなかった。ラパマイシンの添加後、またラパマイシンの添加の後最高で1時間までの約15分ごとにプレートを読み取った。図24で示すデータについては、反応体に0〜50μMの範囲でFK506を滴定すると、ラパマイシン濃度は1μMであった。Veritas照度計を使用して発光を直接測定した。
【0206】
結果
FRB−ルシフェラーゼ−FKBPのラパマイシンを介した調整は、in vivoで観察された(図23〜25)。TK又はCMVプロモーター系を使用して、発光シグナルのそれぞれ最高5倍及び2倍の減少が観察された。対照構築物はラパマイシンに反応を示さなかった(図25B〜D)。さらに、FKBPとの結合においてラパマイシンと競合するFK506は、滴定可能な方法でラパマイシンの影響を打ち消す(図24)。
【実施例11】
【0207】
C末端調整ルシフェラーゼ融合系によるin vitro実験
一実施形態では、ルシフェラーゼ活性は、ルシフェラーゼのN又はC末端の融合によって調整することができる。例えば、カルモジュリンとのルシフェラーゼC末端の融合は、カルモジュリンを調整する剤によって調整される。
【0208】
材料及び方法
ヒトカルモジュリン遺伝子(CaM)は、順方向プライマー
【化56】
及び、逆方向プライマー
【化57】
を使用して、下記プログラムに従ってベクターpOTB7(ATCC(登録商標)Global Resource Center、MGC−1447)から増幅した:95℃で5分間、次に1サイクルが95℃で30秒間、60℃で30秒間及び72℃で1分間10秒を20サイクル。SnaBI部位は順方向プライマーの5’末端へ導入し、XhoI部位は逆方向プライマーの5’末端へ導入した。CaM遺伝子はLuc2遺伝子(先に述べたように)をSnaBI/XhoI部位に有するルシフェラーゼT7対照ベクターのC末端へクローニングして、Luc2−CaM融合構築物を作製した。融合タンパク質は、TnT(登録商標)Coupled Reticulocyte Lysate System(Promega Corp.)を使用してメーカーのプロトコルに従ってin vitroで発現させた。発光は、Turner20/20照度計で測定した。
【0209】
結合したCaMタンパク質によるルシフェラーゼタンパク質の調整を検定するために、EGTA及びCaCl2を順番にin vitro Luc2−CaM融合タンパク質溶解物に加えた。最初に、TnT(登録商標)反応からのLuc2−CaM溶解物の1μlを100μlのルシフェラーゼ検定用試薬(LAR、Promega Corp.)に加え、混合物の25μlを使用してEGTA及びCaCl2の添加前のベースライン発光を確定した。最初の発光を測定した後、1μlの75mM EGTA溶液(最終濃度3mM)を溶解物/LARに加えて発光を測定した。一旦EGTAの添加に応じた発光を測定した後、1μlの100mM CaCl2溶液を溶解物/LARに加えて発光を測定した。したがって、Luc2−CaM融合構築物の発光測定が3つある;1)EGTA又はCa+2を添加しないベースライン、2)EGTAの添加の後、及び3)Ca+2の添加の後。
【0210】
結果
カルモジュリンタンパク質はカルシウムに応じて構造が大きく変化し、それによりC末端の融合を通してルシフェラーゼ活性を調整する他の可能性を提供する。EGTA又はCa+2が存在しない場合、CaMはルシフェラーゼ及びその基質の間の相互作用を制限する(図27、「試料」)。しかし、EGTAの添加後、この制限は解放され(図27、「EGTA」)、発光は約9倍増加する。この発光増加は、Ca+2の添加によって逆転される(図27、「CaCl2」)。したがって、CaMの高次構造は、Luc2−CaM融合におけるルシフェラーゼ活性に影響を及ぼすようであった。
【0211】
(参考文献)
【0212】
全ての刊行物、特許及び特許出願は、参照により本明細書で組み込まれている。以上の明細書において、本発明は特定の好ましい実施形態に関して記載され、多くの詳細が例示目的のために述べられているが、本発明に更なる実施形態を加えることが可能なこと、また本明細書の詳細の一部は本発明の基本原理から逸脱することなくかなりの程度変更することが可能なことは、当業者にとって明らかであろう。
【技術分野】
【0001】
本出願は、米国特許法第119条(e)の下、参照により本明細書に組み込まれた2003年10月10日に出願の米国特許出願第60/510187号の出願日の権益を請求する。
【0002】
本発明は、生化学アッセイ及び試薬の分野に関するものである。より具体的には、本発明は発光性レポータータンパク質などの修飾されたレポータータンパク質、及びそれらの使用方法に関するものである。
【背景技術】
【0003】
ルシフェラーゼは、光量子の放出を伴う基質(例えばルシフェリン)の酸化を触媒する酵素である。ルシフェラーゼは、鞘翅類節足動物及び多くの海洋生物を含む多数の種から単離されている。検出が容易でありその活性は高精度で定量化することが可能なので、ルシフェラーゼ/基質対は遺伝子発現及びタンパク質の所在を研究するために広く使用されてきた。発色団の形成に最高30分を要する他のレポータータンパク質である緑色蛍光タンパク質(GFP)と異なり、ルシフェラーゼ生成物はポリペプチド鎖の合成(基質及び酸素も存在する場合)終了後直ちに検出することが可能である。更に、酵素活性のために翻訳後修飾は必要ではなく、また、この酵素は補欠分子団、結合コファクター及びジスルフィド結合を含まない。ルシフェラーゼは、多数の種及び多種多様な細胞における有用なレポーターである。
【0004】
ルシフェラーゼは、バイオセンシングのためのレポーター分子、すなわち、生物学的の性状を明らかにする分子としてそれらを特に有用なものにする追加機能を有する。バイオセンサー(生物学的構成要素を含むセンサー)におけるシグナル伝達は、通常二段階過程を含む。即ち、生物学的構成要素を通してのシグナル生成並びに電気的構成要素を通してのシグナル伝達及び増幅である。シグナル生成は、一般的に結合又は触媒作用を通して達成される。電気信号へのこれらの生化学事象の変換は一般的に電気化学的又は熱検出法に基づいているが、これらは生化学反応の自由エネルギーの変化によって制限される。大部分の反応については、これはATP2分子の加水分解のエネルギー、すなわち約70kJ/モルより少ない。しかし、ルシフェラーゼによって導き出される発光は、非常に高いエネルギー含量を有する。例えば、ホタルルシフェラーゼによって触媒される反応(560nm)は、214kJ/モルのエネルギーを発する。更に、ルシフェラーゼによって触媒される反応は知られている最も効率的な生物発光反応の1つであり、約0.9の量子収率を有する。ルシフェラーゼは、したがって極めて効率的な化学エネルギー変換器である。
【0005】
ルシフェラーゼバイオセンサーが記載されている。例えば、Sala−Newbyら(1991年)は、サイクリックAMP依存性タンパク質キナーゼリン酸化部位を生成するためにPhotinus pyralisルシフェラーゼcDNAをin vitroで増幅したことを開示している。特に、部位RRFSを生成するために217位置のバリンがアルギニンに変異され、ブタピルビン酸キナーゼのリン酸化部位であるヘプタペプチドケンプチドがルシフェラーゼのN又はC末端に加えられた。Sala−Newbyらは、リン酸化部位を運ぶタンパク質の比活性、pI、発光色に及ぼすpHの影響、及びATP存在下でのタンパク質キナーゼAの触媒サブユニットの影響について特徴を明らかにしたと述べている。組換えタンパク質(RRFS)の1つだけが野生型ルシフェラーゼとかなり異なり、そのRRFS変異体は低い比活性、低い最適pHを有し、低いpHでより濃い緑の光を発し、且つリン酸化されるとその活性が最高80%減少することが発見された。後者の影響はホスファターゼによって逆転されたことが開示されている。
【0006】
Waudら(1996年)は、Photinus pyralisルシフェラーゼcDNAに、タンパク質キナーゼ認識配列及びプロテイナーゼ部位を導入した。ルシフェラーゼの2つの領域が、Waudらによって修飾された。1つはアミノ酸209と227との間で、他はC末端のアミノ酸537と550との間である。Waudらは、残基209及び227の間のアミノ酸の変異は生物発光活性を野生型組換え体の1%未満に低下させたが、C末端におけるペプチド配列の導入は野生型組換えルシフェラーゼの0.06%〜120%の範囲の比活性をもたらしたことを開示している。Waudらは、サイクリックAMP依存性タンパク質キナーゼ触媒サブユニットの、キナーゼ認識配列LRRASLG(配列番号107)が組み込まれ、セリンがアミノ酸位置543にある変異型ルシフェラーゼへの添加は、30%の活性低下をもたらしたことも開示している。アルカリホスファターゼ処理は活性を回復させた。Waudらは、トロンビン認識配列LVPRES(配列番号108)を含み、切断部位がアミノ酸542及び543の間に位置する変異型ルシフェラーゼの生物発光活性は、トロンビン存在下でインキュベートされると50%減少したことも開示している。
【0007】
Ozawaら(2001年)は、ホタルルシフェラーゼの合理的に設計された断片のタンパク質スプライシング誘発性の相補に基づくバイオセンサーを記載している。タンパク質スプライシングは、それを通してインテイン(intein)(内部タンパク質)が前駆体融合タンパク質から切除されて連なるエクステイン(extein)(外部タンパク質)が連結されて連続したポリペプチドになる、翻訳後タンパク質修飾である。Synechocystis属種PCC6803からのN及びC末端のインテインDnaEは、それぞれルシフェラーゼのN及びC末端の断片に融合されたことが開示されている。タンパク質対タンパク相互作用はDnaEインテインの折畳みを引き起こしてタンパク質スプライシングをもたらし、それにより、連結されたルシフェラーゼのエクステインはその酵素活性を取り戻す。Ozawaらは、公知の結合パートナーであるリン酸化インスリン受容体基質1(IRS−1)及びその標的であるPI3−キナーゼのN末端SH2ドメインの間の相互作用が、インスリン存在下で分割されたルシフェラーゼを使用して観察されたことを開示している。
【0008】
Paulmuruganら(2002年)は分割されたホタルルシフェラーゼに基づくアッセイを使用し、相補性方策及びインテイン媒介再構成手法を使用して細胞培養及びマウスで2つのタンパク質、即ちMyoD及びIdの相互作用を観察した。レポーター活性を保持するために、相補性手法では融合タンパク質はタンパク相互作用、即ち、タンパク質パートナーMyoD及びIdの相互作用を必要とするが、再構成手法においてはインテイン媒介スプライシングを通して形成された新しい完全なレポータータンパク質がタンパク質パートナーの間の継続的な相互作用のない場合でさえその活性を維持する。
【0009】
タンパク質断片相補性アッセイは、Michnickら(米国特許第6270964号、第6294330号及び第6428951号)で開示されている。具体的には、Michnickは、DHFRのN末端断片及びDHFRのC末端断片がそれぞれGCN4ロイシンジッパー配列に融合している、分割されたマウスのジヒドロ葉酸レダクターゼ(DHFR)遺伝子に基づくアッセイを記載している。DHFR活性は、両方の融合タンパク質を発現していた細胞で検出された。Michnickらは、アミノグリコシドキナーゼ(AK)遺伝子内のS1ヌクレアーゼ生成欠失の入れ子セットがロイシンジッパー構築物に導入され、得られた構築物セットが細胞に導入されてAK活性がスクリーニングされた他の相補性手法も記載している。
【発明の概要】
【発明が解決しようとする課題】
【0010】
必要なものは、例えばタンパク質対タンパク質相互作用などの細胞現象を高い特異性及び高量子収率で検出する際にバイオセンサーとして使用するための、改良された組換えレポータータンパク質である。
【課題を解決するための手段】
【0011】
本発明は改良された遺伝子産物、例えば他の分子(1つ又は複数の関心のある分子)の存在下で、又はある条件下で1つ又は複数の変更された活性を有する修飾されたレポータータンパク質、例えば修飾された甲虫ルシフェラーゼを提供する。一実施形態では、修飾されたレポータータンパク質のアミノ酸配列は、修飾に寛容性のある、例えば挿入、欠失、環状置換又はその任意の組み合わせに寛容性のある部位(残基)又はある領域における1つ又は複数の修飾の結果として、対応する修飾されていない(天然、野生型又は親の)レポータータンパク質のアミノ酸配列とは異なる。1つ又は複数の修飾は修飾されていないレポータータンパク質のN又はC末端に対して内部でもよく、且つ/又は修飾されていないレポータータンパク質のN及び/又はC末端におけるものでもよく、例えば1つ又は複数のアミノ酸残基の欠失及び/又は挿入であり、それにより修飾されたレポータータンパク質が与えられる。修飾としては、直接又は間接的に関心のある分子と相互作用し、且つ/又はさもなければ条件の変化に感受性のある1つ又は複数の別々の(分離された)アミノ酸配列の導入があり、任意選択に、例えば修飾に寛容性のある部位又は領域、例えば修飾されていないレポータータンパク質のN及び/又はC末端における1つ又は複数のアミノ酸の欠失があるが、この場合、得られた修飾されたレポータータンパク質は外因性の剤などの関心のある分子との相互作用又は条件変化の前及び/又は後にレポーター活性を有するものとする。例えば、修飾されたレポータータンパク質は、対応する修飾されていないレポータータンパク質に対する、1から約10又は15の残基、或いはその中間の任意の整数のN又はC末端位置の欠失を含んでもよい。修飾は、修飾されたレポータータンパク質における、対応する修飾されていないレポータータンパク質においてペプチド結合を通して結合された2つのアミノ酸の間のペプチド結合の欠如であってもよく、並びに修飾されたレポータータンパク質における、対応する修飾されていないレポータータンパク質のN末端及びC末端の残基において又はその近くで見られる残基の間のペプチド結合の欠如でもよく、これらからは環状置換されたレポータータンパク質が与えられ、このレポータータンパク質は任意選択に直接又は間接的に関心のある分子と相互作用するかさもなければ条件変化に感受性であるアミノ酸配列を含む。修飾されたレポータータンパク質は、したがって可逆的相互作用、例えば2つ以上の分子の結合、ジスルフィド結合の形成又は他の高次構造変化又はpH、温度若しくは溶媒疎水性などの条件の変化、或いは修飾されたレポータータンパク質の活性の変化、例えば光強度、色又は動態学的プロフィールの変化によるペプチド結合の切断などの不可逆的相互作用を検出するために使用することができる。
【0012】
本明細書で以下に記載されているように、コメツキムシルシフェラーゼ核酸配列への19のアミノ酸をコードするDNA挿入のライブラリーを調製するために、Tn5を使用した。挿入を有する416のクローンの分析により、クローンの約10%(52)は部分活性、例えば野生型の最高で2%の活性を有することが明らかになった。52クローンの内27クローンはルシフェラーゼ読取り枠内に挿入を有し、それらの挿入の内の16は残基398から409の間(「ヒンジ」領域)にあった。特に、検出可能な活性を有する修飾されたコメツキムシルシフェラーゼをもたらすインフレーム挿入は、コメツキムシルシフェラーゼの残基21、25、117、358、376、379、398、399、400、401、402、403、405、406、407、409又は490の位置であり、即ち、それらの残基及び/又はそれらの残基の近くの領域は挿入を含む修飾に寛容性である。したがって、本発明は、コメツキムシルシフェラーゼの例えば残基21、25、117、358、376、379、398、399、400、401、402、403、405、406、407、409又は490などの残基、或いは、残基15から30、例えば残基21若しくは25、残基112から122、例えば残基117、残基352から362、例えば残基358、残基371から384、例えば残基379、残基393から414又は残基485から495に対応する領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。対応する位置は、ルシフェラーゼ配列を一列に並べることによって特定することができる。詳細には、本発明は甲虫ルシフェラーゼのヒンジ領域、例えばコメツキムシルシフェラーゼの残基390から409に対応する残基並びに修飾に寛容性のある他の領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。
【0013】
また本明細書で記載されているように、ホタルルシフェラーゼ核酸配列への挿入のライブラリーを調製するために、Tn7を使用した。検出可能な活性を有する修飾されたホタルルシフェラーゼをもたらすインフレーム挿入は、ホタルルシフェラーゼの残基7、121、233、267、294、303、361、540又は541の位置であり、即ち、それらの残基及び/又はそれらの残基の近くの領域は挿入を含む修飾に寛容性である。したがって、本発明はホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546と対応する残基又は領域における修飾を有する、修飾された甲虫ルシフェラーゼを含む。対応する位置は、ルシフェラーゼ配列を一列に並べることによって特定することができる。
【0014】
したがって、一実施形態ではレポータータンパク質は甲虫ルシフェラーゼであり、修飾された甲虫ルシフェラーゼのアミノ酸配列は、修飾に寛容性のある部位又はある領域における1つ又は複数の修飾の結果として、対応する修飾されていない甲虫ルシフェラーゼのアミノ酸配列とは異なる。例えば、一実施形態では、修飾された甲虫ルシフェラーゼは検出可能な活性を有し、また対応する修飾されていない甲虫ルシフェラーゼに対する修飾に寛容性のある部位又は領域における1つ又は複数のアミノ酸の挿入を含み、この挿入は修飾された甲虫ルシフェラーゼのN及びC末端の内部にある。一実施形態では、修飾された甲虫ルシフェラーゼは、2つ以上、例えば3、4、5、10、20、50、100、200、300以上で約500未満の、又はその中間の任意の整数のアミノ酸残基の挿入を含む。一実施形態では、本発明の修飾された甲虫ルシフェラーゼは修飾に寛容性のある残基又は領域に少なくとも4つのアミノ酸の内部挿入を含み、この挿入は関心のある分子と直接的に相互作用するアミノ酸配列を含み、例としては関心のある分子の認識配列を含む挿入又は関心のある分子に間接的に、例えば他の分子を通して作用する挿入がある。一実施形態では、内部挿入を有する修飾された甲虫ルシフェラーゼは、甲虫ルシフェラーゼ配列の内部欠失、例えば1つ又は複数で約100未満、例えば50、40、30、20、10又は5未満又はその中間の任意の整数の残基の欠失を更に含む。
【0015】
一実施形態では、修飾された甲虫ルシフェラーゼは対応する修飾されていない甲虫ルシフェラーゼに対する、修飾に寛容性のある部位又は領域における欠失を含む。一実施形態では、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼに対する少なくとも50、例えば少なくとも100の連続したアミノ酸残基の欠失を含み、即ち、前記修飾された甲虫ルシフェラーゼは完全長の修飾されていない甲虫ルシフェラーゼ配列の断片、例えば少なくとも50、例えば少なくとも100の連続したアミノ酸残基の断片であり、例えば対応する完全長の修飾されていない甲虫ルシフェラーゼよりも少なくとも5%、例えば10%少ない残基、及び関心のある分子と直接又は間接的に相互作用するかさもなければ条件に感受性のあるアミノ酸配列の挿入を有する断片である。そのような修飾された甲虫ルシフェラーゼは、タンパク質相補性アッセイで、例えば関心のある分子に連結されたルシフェラーゼの他の断片の存在下でルシフェラーゼの検出可能な活性が増加する場合に、又はインテイン媒介組換えなどのタンパク質組換えアッセイで使用することができる。一実施形態では、甲虫ルシフェラーゼ断片(1つ又は複数の異種配列を有さず)は対応する完全長の修飾されていない甲虫ルシフェラーゼの活性の例えば約0.001%、0.01%、0.1%又は1%未満の検出可能な活性を有し、また相補断片(1つ又は複数の異種配列を有さず)と結合すると、いずれの断片と比較しても3倍を超える、例えば10又は50から100倍又はそれ以上の活性増加を有する。例えば、一実施形態では、N末端甲虫ルシフェラーゼ断片は対応する完全長の修飾されていない甲虫ルシフェラーゼの少なくとも0.001%で1%未満の、また、C末端甲虫ルシフェラーゼ断片は少なくとも0.01%で5%未満の活性を有する。他の実施形態において、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼに対する少なくとも50、例えば少なくとも100の連続したアミノ酸残基の欠失、関心のある分子と直接又は間接的に相互作用するかさもなければ条件に感受性のあるアミノ酸配列の挿入、及び異種、例えば非甲虫ルシフェラーゼ配列の挿入を有する断片であり、これらの挿入は好ましくは甲虫ルシフェラーゼ断片の活性を増加しないが個々に、又は一緒にそれを減少させてもよいが、一旦取り除かれると修飾された甲虫ルシフェラーゼに対して活性の増加したトランケーションされた甲虫ルシフェラーゼをもたらす。
【0016】
本明細書で更に記載されているように、対応する非環状で置換されていない甲虫ルシフェラーゼ内の修飾に寛容性のある残基又は領域にN末端を有し、任意選択に、関心のある分子、例えばプロテアーゼ認識部位若しくはキナーゼ部位と直接又は間接的に相互作用するアミノ酸配列を含む、環状置換された蛍及びコメツキムシのルシフェラーゼが調製され、それらは検出可能な活性を有することが示されたが、この活性は関心のある分子、例えば環状置換ルシフェラーゼ内でプロテアーゼ認識部位又はキナーゼ部位をそれぞれコードしていた構築物内の適当なプロテアーゼ又はキナーゼの存在下で変化した。それ故に、一実施形態では、本発明の修飾された甲虫ルシフェラーゼは、対応する修飾されていない甲虫ルシフェラーゼのアミノ酸配列に対して環状置換されたアミノ酸配列を含み、その結果修飾された甲虫ルシフェラーゼ内に新しいN及びC末端を含み、少なくともその1つは修飾に寛容性のある部位又は領域にある。他の実施形態において、環状置換された甲虫ルシフェラーゼは他の修飾、例えばそれには限定されないが、環状置換された甲虫ルシフェラーゼのN又はC末端の内部の挿入及び/又は欠失、例えば対応する修飾されていない甲虫ルシフェラーゼのN及びC末端又はその近くの挿入及び/又は欠失、例えば修飾されていない甲虫ルシフェラーゼのN末端の残基1から約10若しくは15又はその中間の任意の整数と対応する残基、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応する残基における挿入及び/又は欠失を含む。したがって、レポータータンパク質のN及びC末端は環状置換を通して変更することが可能であり、得られた置換された分子は置換されないレポータータンパク質の1つ又は複数の活性を有することができる。したがって、環状置換されたレポータータンパク質は、タンパク質相補性アッセイ又はタンパク質組換えアッセイで使用することができる。さらに、環状置換されたレポータータンパク質は、直接又は間接的に関心のある分子と相互作用するかさもなければ条件変化に感受性のあるアミノ酸配列を導入することによって、官能性を有するようにすることができる。一実施形態では、本発明の環状置換されたレポータータンパク質は、チモーゲンである。
【0017】
一実施形態では、関心のある分子が存在しない場合、修飾された甲虫ルシフェラーゼのような修飾されたレポータータンパク質の活性は対応する修飾されていないレポータータンパク質の活性よりも小さく、例えば修飾された甲虫ルシフェラーゼのレポーター活性は、対応する修飾されていない甲虫ルシフェラーゼのそれの約0.001%、0.01%、0.1%、1%、10%、20%、50%、70%又はそれ以上で100%未満であり、その修飾されたレポータータンパク質の活性は任意選択に検出可能である。他の実施形態では、関心のある分子が存在しない場合、修飾された甲虫ルシフェラーゼのような修飾されたレポータータンパク質の活性は対応する修飾されていないレポータータンパク質の活性よりも大きく、例えば修飾された甲虫ルシフェラーゼのレポーター活性は、対応する修飾されていない甲虫ルシフェラーゼのそれの約1.5倍、例えば少なくとも2倍、3倍又は5倍又はそれ以上である。関心のある分子の存在下では、修飾されたレポータータンパク質の活性は検出可能に変更される。例えば、関心のある分子の存在下での修飾された甲虫ルシフェラーゼの活性における検出可能な変化は、関心のある分子がない場合の修飾された甲虫ルシフェラーゼの活性と比較して、少なくとも0.001%、0.01%、0.1%、1%、10%又は100%、及び最高で2倍、4倍、10倍、100倍、1,000倍、10,000倍、又はそれ以上の変化である。したがって、修飾されたレポータータンパク質に存在するが対応する修飾されていないレポータータンパク質には存在しない修飾と相互作用する関心のある分子の物理的近接性は、修飾されたレポータータンパク質の活性を変化、例えば減少、除去又は増加させる。例えば、修飾された甲虫ルシフェラーゼは対応する修飾されていない甲虫ルシフェラーゼに対する内部挿入を含んでもよく、この挿入はプロテアーゼ認識部位、即ちプロテアーゼによって切断される部位を含む。プロテアーゼ存在下でのそのような修飾された甲虫ルシフェラーゼの発光シグナルは、プロテアーゼが存在しない場合の修飾された甲虫ルシフェラーゼの発光シグナルと比較して、又は関心のある分子の存在下又は非存在下における対応する修飾されていない甲虫ルシフェラーゼの発光シグナルと比較して、減少させ、除去し又は増加させることができる。或いは、対応する修飾されていない甲虫ルシフェラーゼに対する欠失を含む修飾された甲虫ルシフェラーゼは、関心のある分子と相互作用するリガンドと融合することができる。甲虫ルシフェラーゼの相補性の第2の断片が関心のある分子と融合され、2つの融合の相互作用が可能になり、この相互作用は、例えばいずれかの融合単独の活性と比較して得られた複合体の活性を変化、例えば増加させる。一実施形態では、甲虫ルシフェラーゼの1つの断片は、ホタルルシフェラーゼの残基約1から約126、約1から約238、約1から約272、約1から約308、約1から約366、約116から約550、約228から約550、約262から約550、約289から約550、又は約356から約550、又はその中間の任意の整数の残基、或いはコメツキムシルシフェラーゼの約1から約122、約1から約362、約1から約384、約1から約414、約352から約542、約371から約542、又は約393から約542、又はその中間の任意の整数の残基と対応する残基を有する。
【0018】
本発明は、レポータータンパク質のN末端及びC末端に異種配列を含む修飾されたレポータータンパク質も提供し、即ち、前記修飾されたタンパク質は融合タンパク質であり、また前記異種配列は非共有結合で相互作用し、つまり2つの異種配列は結合パートナーである。一実施形態では、修飾されたレポータータンパク質はN末端及びC末端に異種配列を含む、環状置換された甲虫ルシフェラーゼである。一実施形態では、1つ又は複数の外因性因子(少なくともその1つは関心のある分子でよく、例えば試料中で検出又は特定されるもの)がない場合、1つはN末端に他はC末端にある両方の異種配列を有する修飾されたレポータータンパク質は、対応する修飾されていないレポータータンパク質より小さいか、同じか又は大きな活性を有する。一実施形態では、修飾されたレポータータンパク質は、修飾されていないレポータータンパク質のN及び/又はC末端に存在する1つ又は複数のアミノ酸を欠いてもよく、その欠落は修飾されたレポータータンパク質のレポーター活性を実質的に変更せず、例えば修飾されたレポータータンパク質のレポーター部分の活性は、欠失のない対応するレポータータンパク質の活性の少なくとも0.001%、0.01%、0.1%、1%、10%、50%、100%、又はそれ以上である。一実施形態では、1つ又は複数の外因性因子の存在下で、又は特定条件下で、異種配列のない対応するレポータータンパク質(即ち対応する修飾されていないレポータータンパク質)ではなく両方の異種配列を有する修飾されたレポータータンパク質の活性は、例えば少なくとも2倍、5倍又は10倍又はそれ以上検出可能に変更される。例えば、ラパマイシン存在下で、ラパマイシン結合タンパク質(FRB)及びFK506結合タンパク質(FKBP)に融合されたルシフェラーゼは、FRB及びFKBPを欠くルシフェラーゼと比較して活性は低下している。一実施形態では、外因性因子がない場合又は異なる条件下で、修飾されたレポータータンパク質は検出可能な活性を有さないが他の実施形態においては検出可能な活性を有し、その活性は少なくとも1つの外因性因子の存在下で又は特定条件下で強化することができる。例えば、外因性因子がない場合、修飾されたレポータータンパク質の活性はほとんど又は全くないが、2つの異種配列の非共有結合的相互作用を強化する選択された外因性因子を添加すると、修飾されたレポータータンパク質の活性は強化される。或いは、両方の異種配列を有する修飾されたレポータータンパク質の活性は、少なくとも1つの外因性因子の存在下又は特定条件下では抑制されてもよい。一実施形態では、1つの異種配列は、他の異種配列の領域と非共有結合的に相互作用する、任意選択にリン酸化などにより共有結合的に修飾されてもよい例えば3以上のアミノ酸残基などの領域を含む。甲虫ルシフェラーゼに融合する場合の結合パートナーとして有用な異種配列としては、それには限定されないが、in vitro及び/又はin vivoで相互作用し、また任意選択に例えばタンパク質モデリングに基づき、結合に加わらないが結合パートナーの相互作用を変化させる外因性因子の存在下又は非存在下で選択された距離離れており、甲虫ルシフェラーゼの末端へのそれらの融合は調整可能な甲虫ルシフェラーゼをもたらす結合配列を有するものがある。例示的な異種配列としては、それには限定されないが、FRB及びFKBP内のそれらのもの、タンパク質キナーゼの調節サブユニット(PKa−R)及びタンパク質キナーゼの触媒サブユニット(PKa−C)、src相同領域(SH2)及びリン酸化が可能な配列、例えばチロシン含有配列、14−3−3のアイソフォーム、例えば14−3−3t(Milsら、2000年を参照)、及びリン酸化が可能な配列、WW領域(プロリンが豊富な分子と結合するタンパク質内の配列(Ilsleyら、2002年;Einbondら、1996年を参照))を有するタンパク質、並びにリン酸化が可能な異種配列、例えばセリン及び/又はトレオニン含有配列、並びにジヒドロ葉酸レダクターゼ(DHFR)及びギラーゼB(GyrB)内の配列などがある。
【0019】
他の実施形態において、1つの(第1の)外因性因子の存在下では、結合パートナーであるN末端及びC末端の異種配列を含む修飾されたレポータータンパク質は、前記外因性因子がない場合の活性に対して変化した活性を有し、また異なる(第2の)外因性因子の存在下では、前記修飾されたレポータータンパク質の活性は、前記第1の外因性因子の存在下で、例えば前記第2の外因性因子が前記第1の外因性因子と競合する条件下での活性に対して変化する。一実施形態では、第1の外因性因子がない場合、修飾されたレポータータンパク質は検出可能な活性を全く示さないか低い活性を示し、第1の外因性因子の添加は修飾されたレポータータンパク質の活性の増加をもたらし、この増加は第2の外因性因子の添加により戻すことが可能である。他の実施形態では、第1の外因性因子がない場合、修飾されたレポータータンパク質は検出可能な活性を有し、第1の外因性因子の添加は検出可能な活性の低下又は喪失をもたらすか、或いは検出可能な活性の増加をもたらし、これは第2の外因性因子の添加により戻すことが可能である。修飾されたレポータータンパク質は、任意選択に修飾されていないレポータータンパク質に対してN及び/又はC末端の1つ又は複数のアミノ酸を欠いてもよく、例としては対応する修飾されていないレポータータンパク質のN末端の残基1又は残基1から約10若しくは15、又はその中間の任意の整数、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応する残基の欠失がある。
【0020】
他の実施形態では、修飾されたレポータータンパク質は、修飾されたレポータータンパク質の活性を抑制するなど変更する異種配列をN末端又はC末端に含み、前記活性は第1の外因性因子の添加によって例えば少なくとも部分的に取り戻されるなど変更される。任意選択に、第1の外因性因子の影響は、第2の外因性因子によって可逆的に変更される。一実施形態では、異種配列は基質の進入を抑制することができ、また、異種配列の高次構造は第1の外因性因子の存在下で実質的に変更されて、修飾されたレポータータンパク質はその基質と相互作用することが可能になる。修飾されたレポータータンパク質は、任意選択に修飾されていないレポータータンパク質のN及び/又はC末端の1つ又は複数のアミノ酸を欠いてもよく、例としては対応する修飾されていないレポータータンパク質のN末端の残基1から約10若しくは15、又はその中間の任意の整数の残基、及び/或いはC末端の最後の残基又は最後の約15、又は1から15の間の任意の整数の残基と対応するものがある。この実施形態で有用な異種配列は、カルモジュリン(CaM)である。
【0021】
したがって、修飾されたレポータータンパク質は結合パートナーの可逆的相互作用又は異種配列の可逆的高次構造変化を検出するために使用することができ、これは1つ又は複数の因子又はイオン強度若しくは温度などの条件の変化によって強化又は抑制することができる。
【0022】
したがって、本発明の修飾された甲虫ルシフェラーゼは、バイオセンサーとして使用することができる。
【0023】
本発明は、本発明の修飾されたレポータータンパク質をコードしている核酸配列を含んでいる単離された核酸分子(ポリヌクレオチド)も提供する。修飾されたレポータータンパク質及び修飾されたレポータータンパク質のN末端(N末端融合パートナー)及び/又はC末端(C末端融合パートナー)の1つ又は複数のアミノ酸残基を含んでいる融合タンパク質をコードする核酸配列を含んでいる、単離された核酸分子も更に提供されている。したがって、本明細書で使用されるように、「融合タンパク質」は本発明の修飾されたレポータータンパク質のN末端及び/又はC末端に1つ又は複数のアミノ酸を含む、ポリペプチドである。好ましくは、融合タンパク質内の1つ又は複数の融合パートナーの存在は、対応する修飾されたレポータータンパク質に対して、融合タンパク質の検出可能な活性を実質的に変更しない。一実施形態では、融合タンパク質は少なくとも2つの異なる融合パートナーを、1つは修飾されたレポータータンパク質のN末端に、他はC末端に含む。N又はC末端の融合パートナーは、精製のために使用される配列、例えばグルタチオンSトランスフェラーゼ(GST)又はpolyHis配列、修飾されたレポータータンパク質の特性を変えることを意図した配列、例えば修飾されたレポータータンパク質内のキナーゼ部位のための修飾に寛容性のある残基又は領域のタンパク質不安定化配列又はキナーゼ結合ドメイン、或いは融合タンパク質内のレポータータンパク質の1つ又は複数の特性と区別できる特性を有する配列でよい。一実施形態では、融合タンパク質は修飾された甲虫ルシフェラーゼ及び甲虫ルシフェラーゼとは異なるレポータータンパク質である融合パートナーを含み、このレポータータンパク質は分子内対照、例えば蛍光タンパク質として有用である。他の実施形態において、本発明は本発明の修飾された甲虫ルシフェラーゼを含んでいる融合タンパク質をコードする核酸配列及び前記修飾された甲虫ルシフェラーゼの甲虫ルシフェラーゼとは異なるレポータータンパク質をコードする核酸断片を含んでいるベクターを含む。任意選択に、少なくとも甲虫ルシフェラーゼをコードするヒトコドンの最適化された配列などの最適化された核酸配列、及び好ましくは修飾された甲虫ルシフェラーゼ又は修飾された甲虫ルシフェラーゼを含む融合タンパク質は、それらの最適化された配列は甲虫ルシフェラーゼのシグナル強度を高めることができるので、本発明の核酸分子で使用される。核酸配列の最適化は当技術分野で知られており、例えば国際公開02/16944を参照。
【0024】
本発明は、本発明の甲虫ルシフェラーゼなどの修飾されたレポータータンパク質又は融合タンパク質を発現する安定した細胞系、並びに本発明の修飾されたレポータータンパク質又は融合タンパク質をコードする核酸分子を含んでいる発現カセット、及び本発明の核酸分子を宿主細胞で発現することができるベクターも含む。好ましくは、前記発現カセットは、作動可能的に核酸配列に結合されたプロモーター、例えば構成的又は調節可能なプロモーターを含む。一実施形態では、発現カセットは、誘導的プロモーターを含む。本発明の発現カセット又はベクターを含む宿主細胞、例えば原核細胞又は植物若しくは脊椎動物の細胞、例えばそれには限定されないがヒト、非ヒト霊長類、イヌ、ネコ、ウシ、ウマ、ヒツジ、又は齧歯類(例えばウサギ、ラット、白イタチ若しくはマウス)の細胞を含む哺乳類細胞などの真核細胞、及び核酸分子、発現カセット、ベクター、宿主細胞又は本発明の修飾された甲虫ルシフェラーゼ若しくは融合タンパク質を含むキットも提供される。
【0025】
本発明の修飾されたレポータータンパク質は、修飾されていないレポータータンパク質が例えば機能的レポーターとして関心のある分子の様々な状態及び/又は分子を測定及び検出することが不可能な用途で使用することができる。例えば、プロテアーゼ切断認識部位の挿入を含んでいる修飾された甲虫ルシフェラーゼをコードするベクター、又はこの修飾された甲虫ルシフェラーゼは、細胞、細胞溶解物、in vitro転写/翻訳混合物、又は上清に導入され、修飾された甲虫ルシフェラーゼの活性は、例えば1つ又は複数の時点で、対応する修飾されていない甲虫ルシフェラーゼと比較されて検出又は測定される。細胞、細胞溶解物、in vitro転写/翻訳混合物又は上清における発光活性の経時的及び/又は対照、例えば対応する修飾されていない甲虫ルシフェラーゼを有する細胞に対する変化は、プロテアーゼの存在を示す。例えば、本発明は重症急性呼吸器症候群と関連するウイルスを検出するための方法を含む。この方法は、生物学的試料、例えば生理学的組織又は流体試料を、対応する修飾されていないレポータータンパク質に対する内部挿入を含む修飾されたレポータータンパク質、例えば修飾された甲虫ルシフェラーゼと接触させることを含み、前記修飾されたレポータータンパク質は検出可能な活性を有する。前記挿入は、レポータータンパク質配列内の、修飾に寛容性がありウイルスのプロテアーゼに対するアミノ酸認識配列を含む残基又は領域で起こる。修飾されたレポータータンパク質の活性が試料存在下で変更されるか否かが検出又は決定され、それによって試料がウイルスを含むかどうかが示される。
【0026】
本発明は関心のある分子の存在を検出する方法も提供する。例えば、細胞を、調節可能なプロモーターなどのプロモーター、及び関心のある分子と相互作用する挿入を含んでいる本発明の修飾されたレポータータンパク質をコードする核酸配列を含んでいるベクターと接触させる。一実施形態では、形質移入細胞はプロモーターが修飾されたレポータータンパク質の一時的発現を誘発する条件下で培養され、修飾されたレポータータンパク質の検出可能な活性が測定される。
【0027】
また、修飾されたレポータータンパク質をコードする選択された変異ポリヌクレオチドを調製する方法も提供される。この方法は、検出可能な活性を有する修飾されたレポータータンパク質をコードする親ポリヌクレオチドに変異を起こさせて、変異を起こした修飾されたレポータータンパク質をコードする1つ又は複数の変異ポリヌクレオチドを生成することを含む。親ポリヌクレオチドは、対応する修飾されていないレポータータンパク質に対して修飾に寛容性のある残基又は領域が修飾されている、修飾されたレポータータンパク質の読取り枠を含む。修飾されたレポータータンパク質は、直接又は間接的に関心のある分子と相互作用するか、さもなければ対応する修飾されていないレポータータンパク質と比較して条件に感受性を示すアミノ酸配列を含む。関心のある分子との相互作用又はある条件下での活性が、修飾されたレポータータンパク質の相互作用又は活性に対して変更された、変異修飾レポータータンパク質をコードする、1つ又は複数の変異ポリヌクレオチドが選択される。他の実施形態において、本発明は、本発明の修飾されたレポータータンパク質を分子のライブラリーと接触させて、1つ又は複数の分子が、修飾と又は前記修飾されたレポータータンパク質内の非レポータータンパク質配列と相互作用するかどうかを検出又は決定することを含む方法を提供する。
【図面の簡単な説明】
【0028】
【図1】EZ:TNインフレームリンカー挿入プロトコルの概要を示す図である。
【図2】cbg69遺伝子へのTn5挿入突然変異誘発の結果を示す図である。cbg69によってコードされるタンパク質は、野生型コメツキムシルシフェラーゼと比較して位置409(I409V)に1つのアミノ酸置換を有する(図3を参照)。
【図3】コメツキムシルシフェラーゼ(配列番号89)におけるTn5挿入位置(太字)を示す図である。
【図4】Tn5挿入で修飾されたコメツキムシルシフェラーゼの活性を示す図である。
【図5A】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。cbg69ss又はcbg69DEVDによるカスパーゼアッセイにおける相対光単位(RLU)。
【図5B】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。コメツキムシルシフェラーゼ及びカスパーゼ阻害剤(Ac−DEVD−CHO)によるカスパーゼアッセイにおけるRLU。
【図5C】カスパーゼ−3認識部位挿入(cbg69DEVD)で修飾されたコメツキムシルシフェラーゼの活性を示す図である。様々な量のカスパーゼ−3及びcbg69DEVDによるアッセイにおける経時的RLU。
【図6A】プロテアーゼ認識部位、キナーゼ認識部位、抗体結合部位及び金属結合部位を含むヒンジ領域における修飾を有するコメツキムシルシフェラーゼの配列及び活性を示す図である。6His=6×His−標識;FLAG=DYKDDDDK(配列番号4);DEVD(配列番号106)=カスパーゼ3/7認識部位、Pka=Pkaキナーゼ部位(配列番号90〜96)。挿入は、SnaBI及びSalIを使用してCbgLuc(I409V)のヒンジ領域に導入された。
【図6B】SARSウイルスプロテアーゼ認識部位を有する修飾されたコメツキムシルシフェラーゼの存在下におけるSARSウイルス3CLプロテアーゼ活性を示す図である。
【図6C】ヒンジ領域(配列番号90〜91及び97〜102)にSARSウイルスプロテアーゼ認識部位を有するコメツキムシルシフェラーゼの配列及び活性を示す図である。挿入は、SnaBI及びSalIを使用してCbgLuc(I409V)のヒンジ領域に導入された。
【図7A】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。親(修飾されていない)ホタルルシフェラーゼ(luc+)(配列番号103)のアミノ酸配列。
【図7B】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基233の後にGly(3)Asp(4)LysGly(3)挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図7C】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基233の後にProGlyProGIy(3)Asp(4)LysGly(3)ProGlyPro挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図7D】エンテロキナーゼ認識部位で修飾されたホタルルシフェラーゼの活性を示す図である。残基541の後にAsp(4)Lys挿入を有する修飾されたホタルルシフェラーゼ又は親のホタルルシフェラーゼ(WT)によるエンテロキナーゼアッセイにおけるRLU。
【図8】エンテロキナーゼ部位を有する環状置換されたホタルルシフェラーゼのエンテロキナーゼ活性化を示す図である。
【図9】カスパーゼ−3部位を有する環状置換されたホタルルシフェラーゼによる経時的カスパーゼ−3活性化を示す図である。
【図10A】様々な量のカスパーゼ−3及びカスパーゼ−3認識部位を有する環状置換されたホタルルシフェラーゼによるカスパーゼアッセイにおけるRLUを示す図である。
【図10B】様々な量のカスパーゼ−3及びカスパーゼ−3認識部位を有する環状置換されたホタルルシフェラーゼによるカスパーゼアッセイにおけるRLUを示す図である。
【図11】エンテロキナーゼ部位又はカスパーゼ−3部位を有する環状置換されたホタルルシフェラーゼのデータの比較を示す図である。
【図12】SARSウイルスプロテアーゼ認識部位を有する環状置換されたコメツキムシ(CP1:R=Asn401及びCP2:R=Arg223)及びホタル(CP:R=Asp234)ルシフェラーゼによるSARSウイルス3CLプロテアーゼ活性を示すグラフである。
【図13】カスパーゼ−3部位を有しTRAILで処理された環状置換されたルシフェラーゼのRLUを示す図である。
【図14】二重ルシフェラーゼカスパーゼアッセイのためのベクターの概略図である。
【図15】pBINDベクター及び対照ルシフェラーゼ構築物並びに自己集合のためのN又はC末端のルシフェラーゼ構築物の概略図である。
【図16A】完全長ホタルルシフェラーゼ、ホタルルシフェラーゼのN末端部分、ホタルルシフェラーゼのC末端の部分又はN末端及びC末端部分の混合物のSDS−PAGE分析を示す図である。
【図16B】完全長ホタルルシフェラーゼ、ホタルルシフェラーゼのN末端部分、ホタルルシフェラーゼのC末端の部分又はN末端及びC末端部分の混合物のin vitro活性を示す図である。
【図17】CHO又は293の哺乳類細胞抽出物内のルシフェラーゼタンパク質のin vivo活性を示す図である。
【図18】ルシフェラーゼの結合パートナーX又はYとの融合を発現する構築物を調製するためのクローニング方策を示す図である。
【図19】修飾されていないルシフェラーゼタンパク質及びin vitro転写/翻訳反応を使用して生成した1つ又は複数の異種配列とのルシフェラーゼの融合のSDS−PAGEゲル分析を示す図である。
【図20A】ラパマイシンの存在下又は非存在下における、修飾されていないルシフェラーゼ(Luc2)、FRB(ラパマイシン結合タンパク質)と融合されたルシフェラーゼ、FKBP(FK506結合タンパク質)と融合されたルシフェラーゼ並びにFRB及びFKBPと融合されたルシフェラーゼのルシフェラーゼ活性を示す図である。
【図20B】上昇する濃度のFK506の存在下におけるルシフェラーゼとFRBとFKBPとの融合のルシフェラーゼ活性を示す図である。
【図21A】ホタルルシフェラーゼ(Luc2)、コメツキムシルシフェラーゼ(Cbg及びCbr)及びRenilla(RLuc)ルシフェラーゼのFRB及びFKBPとの融合のSDS−PAGE分析を示す図である。
【図21B】ラパマイシンの存在下又は非存在下におけるFRB及びFKBPのホタルルシフェラーゼ、コメツキムシルシフェラーゼ及びRenillaルシフェラーゼとの融合のルシフェラーゼ活性を示す図である。
【図22】Tkプロモーターからルシフェラーゼを発現する構築物を示す図である。
【図23】FK506によるラパマイシン媒介調整の抑制を示している、FRB及びFKBP(FRB1−luc2−FKBP)と融合されたルシフェラーゼで形質移入されたD293細胞におけるラパマイシン存在下でのFK506の滴定を示す図である。
【図24】ラパマイシンの存在下又は非存在下における、TKプロモーター及びFRB−ルシフェラーゼ−FKBP融合のコード領域を有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25A】ラパマイシンの存在下又は非存在下における、FRB−ルシフェラーゼ−FKBP融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25B】ラパマイシンの存在下又は非存在下における、FRB−ルシフェラーゼ融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25C】ラパマイシンの存在下又は非存在下における、ルシフェラーゼのコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図25D】ラパマイシンの存在下又は非存在下における、ルシフェラーゼ−FKBP融合のコード領域と結合されたCMVプロモーターを有する構築物で形質移入されたD293細胞における経時的な相対発光を示す図である。
【図26】EGTA又はCa2+存在下におけるカルモジュリン−ルシフェラーゼ融合の相対発光を示す図である。
【発明を実施するための形態】
【0029】
定義
本明細書で使用される用語「核酸分子」、「ポリヌクレオチド」又は「核酸配列」は、ポリペプチド又はタンパク質前駆体の生産のために必要なコード配列を含む核酸、DNA又はRNAを指す。コードされたポリペプチドは完全長ポリペプチド、その断片(完全長より短い)、又は完全長ポリペプチド若しくはその断片と他のポリペプチドとの融合から生じる融合ポリペプチドであってもよい。
【0030】
本明細書で使用されるように、「核酸」は、1つのヌクレオチドのペントースの3’位置がリン酸ジエステル基によって次のペントースの5’位置と連結され、またヌクレオチド残基(塩基)が特定の配列順序、即ち線状ヌクレオチドで連結されている、共有結合で結ばれたヌクレオチド配列である。本明細書で使用されるように、「ポリヌクレオチド」は長さが約100ヌクレオチドを超える配列を含んでいる核酸である。本明細書で使用されるように、「オリゴヌクレオチド」又は「プライマー」は短いポリヌクレオチド、又はポリヌクレオチドの一部である。オリゴヌクレオチドは、一般的に約2から約100の塩基の配列を含む。語「オリゴ」は語「オリゴヌクレオチド」の代わりに時々使用される。
【0031】
核酸ホスホジエステル結合が置換基モノヌクレオチドのペントース環の5’炭素及び3’炭素で起こるので、核酸分子は「5’末端」(5’端)及び「3’末端」(3’端)を有すると言われる。新しい結合が5’炭素に対して起こるポリヌクレオチドの末端は、その5’末端ヌクレオチドである。新しい結合が3’炭素に対して起こるポリヌクレオチドの末端は、その3’末端ヌクレオチドである。本明細書で使用されるように、末端ヌクレオチドは3’又は5’末端の末端位置にあるヌクレオチドである。
【0032】
モノヌクレオチドが反応して、1つのモノヌクレオチドペントース環の5’リン酸がホスホジエステル結合を通して1方向にその隣の3’酸素と結合するようにオリゴヌクレオチドを形成するので、DNA分子は「5’端」及び「3’端」を有すると言われる。したがって、オリゴヌクレオチドの末端は、その5’リン酸がモノヌクレオチドペントース環の3’酸素と結合しない場合は「5’端」と呼ばれ、その3’酸素が以降のモノヌクレオチドペントース環の5’リン酸と結合しない場合は「3’端」と呼ばれる。
【0033】
本明細書で使用されるように、たとえより大きなオリゴヌクレオチド又はポリヌクレオチドの内部にある場合でも、核酸配列は5’端及び3’端を有すると言うことができる。線状又は環状DNA分子のいずれの場合も、独立した要素は「上流」又は「下流」の5’側又は3’要素と称される。この専門用語は、転写がDNA鎖に沿って5’から3’にかけて進行するという事実を反映する。一般的に、連結された遺伝子(例えば読取り枠又はコード領域)の転写を指示するプロモーター及びエンハンサー要素は、通常、コード領域の5’側又は上流に位置する。しかし、エンハンサー要素は、プロモーター要素及びコード領域の3’側に位置する場合でも、それらの影響を発揮することができる。転写終結及びポリアデニル化シグナルは、コード領域の3’側又は下流に位置する。
【0034】
本明細書で使用する用語「コドン」は、ポリペプチド鎖に取り込まれる特定のアミノ酸、又は開始若しくは終止シグナルを指定する3つのヌクレオチドの配列からなる基本的な遺伝暗号単位である。構造遺伝子に関連して使用される場合の用語「コード領域」は、mRNA分子の翻訳の結果として発達中のポリペプチドで見られるアミノ酸をコードするヌクレオチド配列を指す。一般的に、コード領域は5’側は開始因子メチオニンをコードするヌクレオチドトリプレット「ATG」が結合し、3’側は終止コドン(例えばTAA、TAG、TGA)が結合する。場合によっては、コード領域はヌクレオチドトリプレット「TTC」から始まることも知られている。
【0035】
用語「遺伝子」は、コード配列及び任意選択にDNA配列からのポリペプチドの生産に必要な制御配列を含む、DNA配列を指す。
【0036】
本明細書で使用されるように、用語「異種」核酸配列又はタンパク質は、基準配列に対して由来の異なる配列、例えば外来種に起源を有するものを指し、又は同じ種からのものであっても原形からかなり変化したものでもよい。
【0037】
核酸は、異なる種類の変異を含むことが知られている。「点」突然変異は、野生型配列に対するヌクレオチド配列内の1塩基位置の変化を指す。変異は、1つ又は複数の塩基の挿入又は欠失により核酸配列が野生型などの基準配列と異なるものも指す。
【0038】
本明細書で使用されるように、用語「ハイブリダイズする」及び「ハイブリダイゼーション」は、標的核酸への相補配列のアニーリング、即ち相補配列を含んでいる2つの核酸ポリマー(ポリヌクレオチド)の塩基対合を通してアニーリングする能力を指す。用語「アニールされた」及び「ハイブリダイズされた」は本明細書全体を通して互換的に使用され、部分相補性だけを有する領域の結合を含む、相補配列及び標的核酸の間の任意の特異的且つ再生可能な相互作用を包含するものとする。普通は天然核酸では見られないある種の塩基が本発明の核酸に含まれてもよく、例としてはイノシン及び7−デアザグアニンがある。核酸技術の当業者ならば、二重鎖の安定性は多くの変数、例えば相補配列の長さ、オリゴヌクレオチドの塩基組成及び配列順序、イオン強度並びにミスマッチ塩基対の発生率などを考慮して経験的に決めることができる。核酸二重鎖の安定性は、融点又は「Tm」で測定される。特定の条件下での特定の核酸二重鎖のTmは、平均して塩基の半分が分離する温度である。
【0039】
用語「組換えDNA分子」は、自然界では通常一緒に見られない少なくとも2つのヌクレオチド配列を含んでいるハイブリッドDNA配列を意味する。用語「ベクター」は、DNAの断片を挿入又はクローニングすることができる核酸分子に関して使用され、細胞にDNAセグメントを移すために使用でき、また細胞内での複製が可能である。ベクターは、プラスミド、バクテリオファージ、ウイルス、コスミドなどから得ることができる。
【0040】
本明細書で使用されるように、用語「組換えベクター」及び「発現ベクター」は、所望のコード配列及び特定の宿主生物における作動可能的に結合されたコード配列の発現のために必要な、適当なDNA又はRNA配列を含むDNA又はRNA配列を指す。原核生物の発現ベクターは、プロモーター、リボソーム結合部位、宿主細胞における自律複製のための複製起点、及び恐らく他の配列、例えば任意選択のオペレーター配列、任意選択の制限酵素部位を含む。プロモーターは、RNAポリメラーゼにDNAとの結合及びRNA合成の開始を指示するDNA配列と定義される。真核生物の発現ベクターは、プロモーター、任意選択にポリアデニル化シグナル及び任意選択にエンハンサー配列を含む。
【0041】
タンパク質又はポリペプチドをコードするヌクレオチド配列を有するポリヌクレオチドは、遺伝子のコード領域を含んでいる核酸配列を意味し、又は、核酸配列は遺伝子産物をコードすると換言することができる。コード領域は、cDNA、ゲノムDNA又はRNAのいずれの形態でも存在することができる。DNA形態で存在するとき、オリゴヌクレオチドは一本鎖(即ちセンス鎖)又は二本鎖でよい。エンハンサー/プロモーター、スプライス部位、ポリアデニル化シグナルその他などの適当な調節要素は、適切な転写開始を可能にし且つ/又は一次RNA転写産物のプロセシングを修正するために、必要ならば遺伝子のコード領域に近接して置くことができる。或いは、本発明の発現ベクターで利用されるコード領域は、内因性のエンハンサー/プロモーター、スプライス部位、介在配列、ポリアデニル化シグナルその他を含んでもよい。更なる実施形態において、コード領域は内因性及び外因性の調節要素の組み合わせを含んでもよい。
【0042】
用語「転写調節要素」又は「転写調節配列」は、核酸配列の発現の一部の態様を調節する遺伝的要素又は配列を指す。例えば、プロモーターは作動可能的に結合されたコード領域の転写の開始を容易にする調節要素である。他の調節要素としては、それには限定されないが、転写因子結合部位、スプライシングシグナル、ポリアデニル化シグナル、終止シグナル及びエンハンサー要素がある。
【0043】
真核生物における転写調節シグナルは、「プロモーター」及び「エンハンサー」要素を含む。プロモーター及びエンハンサーは、転写に関係する細胞タンパク質と特異的に相互作用するDNA配列の短い配列からなる。プロモーター及びエンハンサー要素は、酵母、昆虫及び哺乳類細胞の遺伝子を含む様々な真核生物の供与源から単離された。プロモーター及びエンハンサー要素はウイルスからも単離され、類似の調節要素、例えばプロモーターも原核生物で見られる。特定のプロモーター及びエンハンサーの選択は、関係するタンパク質を発現するために使用される細胞の種類によって決まる。真核生物のプロモーター及びエンハンサーは広い宿主域を有するものもあれば、限られたサブセットの細胞種で機能的なものもある。例えば、SV40初期遺伝子エンハンサーは、多くの哺乳類の種からの多種多様な細胞種で非常に活性であり、哺乳類細胞内でのタンパク質の発現のために広く使われてきた。広範な哺乳類細胞種で活性を示すプロモーター/エンハンサー要素の他の2つの例は、ヒト伸長因子I遺伝子及びRous肉腫ウイルスの末端反復配列、並びにヒトサイトメガロウイルスからのものである。
【0044】
用語「プロモーター/エンハンサー」は、プロモーター及びエンハンサー機能(即ち先に述べたようにプロモーター要素及びエンハンサー要素によって提供される機能)を提供することができる配列を含んでいる、DNAセグメントを意味する。例えば、レトロウイルスの末端反復配列は、プロモーター及びエンハンサーの両方の機能を含む。エンハンサー/プロモーターは「内因性」又は「外因性」又は「異種性」でよい。「内因性」エンハンサー/プロモーターは、ゲノム内の与えられた遺伝子と自然に連結しているものである。「外因性」又は「異種性」エンハンサー/プロモーターは、遺伝子の転写が結合したエンハンサー/プロモーターによって指示されるように、遺伝子操作(即ち分子生物学的技術)によって遺伝子の近くに置かれたものである。
【0045】
発現ベクター上の「スプライシングシグナル」の存在は、真核宿主細胞における組換え転写産物のより高い発現レベルをしばしばもたらす。スプライシングシグナルは一次RNA転写産物からのイントロンの除去を媒介し、スプライス供与部及び受容部位からなる。通常使用されるスプライス供与部及び受容体部位は、SV40の16S RNAからのスプライス部位である。
【0046】
真核細胞における組換えDNA配列の効率的な発現のためには、得られた転写産物の効率的な終結及びポリアデニル化を指示するシグナルの発現を必要とする。転写終止シグナルは、通常、ポリアデニル化シグナルの下流に見られ、長さは2、3百ヌクレオチドである。本明細書で使用する用語「ポリ(A)部位」又は「ポリ(A)配列」は、発達中のRNA転写物の終結及びポリアデニル化を指示するDNA配列を意味する。ポリ(A)テールがない転写産物は不安定であり速やかに分解するので、組換え転写産物の効率的なポリアデニル化が望ましい。発現ベクターで利用されるポリ(A)シグナルは「異種性」又は「内因性」でよい。内因性のポリ(A)シグナルは、ゲノム内の与えられた遺伝子のコード領域の3’末端で自然に見られるものである。異種ポリ(A)シグナルは、1つの遺伝子から単離され、他の遺伝子に対して3’に位置するものである。通常使用される異種ポリ(A)シグナルは、SV40 ポリ(A)シグナルである。SV40 ポリ(A)シグナルは、237bpのBamH I/Bcl I制限断片に含まれ、終結及びポリアデニル化を指示する。
【0047】
真核生物の発現ベクターは、また「ウイルスのレプリコン」又は「ウイルスの複製開始点」を含んでもよい。ウイルスのレプリコンは、適当な複製因子を発現している宿主細胞内でのベクターの染色体外複製を可能にする、ウイルスのDNA配列である。SV40又はポリオーマウイルスの複製起点を含んでいるベクターは、適当なウイルスのT抗原を発現する細胞内で高いコピー数(最高104の複製/細胞)で複製する。対照的に、ウシの乳頭腫ウイルス又はEBウイルスからのレプリコンを含んでいるベクターは、染色体外で低コピー数(約100の複製/細胞)を複製する。
【0048】
用語「in vitro」は、人工環境及び人工環境内で起こる過程又は反応を指す。in vitro環境としては、それには限定されないが、試験管及び細胞溶解物がある。用語「in vivo」は自然環境(例えば動物又は細胞)、及び自然環境内で起こる過程又は反応を指す。
【0049】
用語「発現系」は、関心のある遺伝子の発現を決定する(例えば、検出する)ための任意のアッセイ又は系を指す。分子生物学の分野に熟練している者は、多種多様な発現系のいずれも使用されることを理解しよう。広範囲にわたる適当な哺乳類細胞が広範囲の供給源(例えばアメリカンタイプカルチャーコレクション、Rockland、メリーランド州)から入手可能である。形質転換又は形質移入の方法及び発現媒体の選択は、選択された宿主系によって決まる。形質転換及び形質移入の方法は、当技術分野で公知である。発現系は、関心のある遺伝子(例えばレポーター遺伝子)が調節配列に結合され、遺伝子の発現は遺伝子の発現を抑制又は誘発する剤による処理の後に観察される、in vitro遺伝子発現アッセイを含む。遺伝子発現の検出は任意の適当な手段、例えばそれには限定されないが、発現されたmRNA又はタンパク質(例えばレポーター遺伝子の検出可能な生成物)の検出を通して、又は関心のある遺伝子を発現している細胞の表現型の検出可能な変化を通して可能である。発現系は、切断事象又は他の核酸若しくは細胞の変化が検出されるアッセイを含んでもよい。
【0050】
本明細書で使用されるように用語「野生型」は、天然の供与源から単離されたその遺伝子又は遺伝子産物の特性を有する遺伝子又は遺伝子産物を指す。野生型遺伝子は、集団で最もしばしば観察され、したがって任意にその遺伝子の「野生型」形態と称されるものである。対照的に、用語「突然変異体」は、野生型の遺伝子又は遺伝子産物に対して、配列及び/又は機能的性状(即ち、特性変化)の変化を示す遺伝子又は遺伝子産物を指す。天然の突然変異体を単離することが可能であると指摘されている。これらは野生型の遺伝子又は遺伝子産物に対して変化した特性を有することで確認される。
【0051】
「単離されたオリゴヌクレオチド」又は「単離されたポリヌクレオチド」のように核酸に関して使用される場合の用語「単離された」は、その供与源内で通常関連している少なくとも1つの混在物から識別及び分離される核酸配列を指す。したがって、単離された核酸は、自然界でそれが見られるものとは異なる形態又はセッティングで存在する。対照的に、単離されていない核酸(例えばDNA及びRNA)は、それらが自然界で存在する状態で見られる。例えば、与えられたDNA配列(例えば遺伝子)は、宿主細胞染色体上で近隣の遺伝子に近接して見られる。RNA配列(例えば、特定のタンパク質をコードする特定のmRNA配列)は、多くのタンパク質をコードする他の多数のmRNAとの混合物として細胞内で見られる。しかし、単離された核酸としては、例えばその核酸が自然の細胞のそれと異なる染色体上の位置にあるか、さもなければ自然で見られるものとは異なる核酸配列に連結している、通常その核酸を発現している細胞内の核酸がある。単離された核酸又はオリゴヌクレオチドは、一本鎖又は二本鎖の形態で存在することができる。単離された核酸又はオリゴヌクレオチドがタンパク質を発現するために利用される場合は、オリゴヌクレオチドは最低限センス又はコード鎖(即ち、オリゴヌクレオチドは一本鎖でよい)を含むが、センス及びアンチセンスの両方の鎖(即ち、オリゴヌクレオチドは二本鎖でよい)を含んでもよい。
【0052】
「ペプチド」、「タンパク質」及び「ポリペプチド」は、長さ又は翻訳後修飾(例えばグリコシル化又はリン酸化)に関係なく任意のアミノ酸鎖を意味する。本発明の核酸分子は、天然のタンパク質の変異体又はそのポリペプチド断片をコードしてもよく、それは、それが由来する天然の(生来若しくは野生型)タンパク質のアミノ酸配列と少なくとも85%、90%、95%又は99%同一であるアミノ酸配列を有する。用語「融合ポリペプチド」又は「融合タンパク質」は、N及び/又はC末端が1つ又は複数の異種配列(例えば非ルシフェラーゼポリペプチド)に結合されている参照タンパク質(例えばルシフェラーゼ)を含んでいるキメラタンパク質を指す。一部の実施形態において、本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長ポリペプチドの一部は、対応する完全長機能的(非キメラ)ポリペプチドの活性の少なくとも一部を保持してもよい。他の実施形態において、外因性因子又は関心のある分子がない場合、本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長機能的ポリペプチドの一部は、対応する完全長機能的ポリペプチドに対して活性を失ってもよい。他の実施形態において、外因性因子の存在下で本発明の修飾されたポリペプチド、融合ポリペプチド又は完全長機能的ポリペプチドの一部は、対応する完全長機能的ポリペプチドに対して活性の少なくとも一部若しくは実質的に同じ活性を保持してもよく、或いは、活性を失ってもよい。
【0053】
ポリペプチド分子は、ペプチド結合が第1のアミノ酸残基の骨格アミノ基及び第2のアミノ酸残基の骨格カルボキシル基の間で起こるので、「アミノ末端」(N末端)及び「カルボキシ末端」(C末端)を有すると言われる。ポリペプチド配列に関した用語「N末端」及び「C末端」は、それぞれポリペプチドのN末端及びC末端領域の一部を含むポリペプチド領域を指す。ポリペプチドのN末端領域の一部を含む配列は、主にポリペプチド鎖のN末端半分からのアミノ酸を含むが、そのような配列に限定はされない。例えば、N末端配列は、ポリペプチドのN末端及びC末端の両半分からの塩基を含むポリペプチド配列の内側の部分を含んでもよい。同じことは、C末端領域にも当てはまる。N末端及びC末端の領域は、その必要はないが、ポリペプチドのそれぞれ最終N末端及びC末端を規定しているアミノ酸を含んでもよい。
【0054】
本明細書で使用されるように用語「組換えタンパク質」又は「組換えポリペプチド」は、組換えDNA分子から発現されたタンパク質分子を指す。対照的に、用語「未変性タンパク質」は、本明細書では天然の(即ち遺伝的組換えではない)供与源から単離されたタンパク質の意味で使用される。タンパク質の未変性形態に対して同一の特性を有するタンパク質の組換え体を生成するために、分子生物学的技術を使用することができる。
【0055】
本明細書で使用されるように用語「細胞」、「細胞系」及び「宿主細胞」は互換的に使用され、そのような呼称は全てこれらの呼称の後代又は潜在的後代を含む。「形質転換細胞」は、それに(又はその先祖に)本発明の核酸分子が導入された細胞を意味する。任意選択に、本発明の核酸分子は、遺伝子によってコードされたタンパク質又はポリペプチドを産生することができる安定的に形質移入された細胞系を作製するために、適当な細胞系に導入してもよい。ベクター、細胞及びそのような細胞系を構築する方法は、当技術分野で公知である。用語「形質転換体」又は「形質転換細胞」は、経代数に関係なく元の形質転換細胞に由来する一次形質転換細胞を含む。故意又は偶発的な突然変異のために、必ずしも全ての後代においてDNAの内容が正確に同一ではないかもしれない。それにもかかわらず、形質転換体の定義には、当初の形質転換細胞でスクリーニングされたものと同じ機能を有する変異後代が含まれる。
【0056】
用語「相同性」は、2つ以上の配列の間の相補性の程度を指す。部分相同性又は完全な相同性(即ち同一性)があってもよい。相同性は、しばしば配列解析ソフトウェア(例えばGenetics Computer Group、ウィスコンシン大学バイオテクノロジーセンター、1710 University Avenue、マジソン、ウィスコンシン州、53705、のSequence Analysis Softwareパッケージ)を使用して測定される。そのようなソフトウェアは、相同性の程度を様々な置換、欠失、挿入、その他の修飾に割り当てることによって類似の配列を比較する。保存的な置換は、一般に下記群内の置換を含む:グリシン、アラニン;バリン、イソロイシン、ロイシン;アスパラギン酸、グルタミン酸、アスパラギン、グルタミン;セリン、スレオニン;リジン、アルギニン;及び、フェニルアラニン、チロシン。
【0057】
「単離されたタンパク質」又は「単離されたポリペプチド」のようにポリペプチドに関して使用される場合の用語「単離された」は、その供与源内で通常関連している少なくとも1つの混在物から識別及び分離されるポリペプチドを指す。したがって、単離されたポリペプチドは、自然界でそれが見られるものとは異なる形態又はセッティングで存在する。対照的に、単離されていないポリペプチド(例えばタンパク質及び酵素)は、それらが自然界で存在する状態で見られる。
【0058】
用語「精製された」又は「精製する」は、関係する構成要素、例えばタンパク質又は核酸から混在物の一部を除去する任意の過程の結果を意味する。精製された構成要素の試料中のパーセントは、それによって増加される。
【0059】
本明細書で使用されるように、「純粋」は対象種が存在する支配的な種(即ち、モルベースでは組成物中の他のいかなる個々の種よりも豊富)であることを意味し、また好ましくは実質的に精製された分画は、存在する全ての高分子種の少なくとも約50パーセント(モルベースで)を対象種が占める組成物である。通常、「実質的に純粋な」組成物は、組成物中に存在する全ての高分子種の約80パーセント、より好ましくは約85%、約90%、約95%及び約99%を超える割合を含む。最も好ましくは、対象種は、組成物が本質的に単一の高分子種からなる本質的に均質状態(従来の検出法によって混在種は組成物内に検出されない)になるまで精製される。
【0060】
本明細書で使用されるように用語「作動可能的に結合された」は、与えられた遺伝子の転写及び/又は所望のタンパク質分子の合成を指示することができる核酸分子が生成されるような核酸配列の結合を指す。この用語は、機能的(例えば、酵素的に活性、結合パートナーとの結合が可能、抑制することが可能、その他)タンパク質又はポリペプチドが生成されるような、アミノ酸をコードする配列の結合を指す。
【0061】
本明細書で使用されるように、用語「ポリヒスチジントラクト」又は(Hisタグ)は、2から10のヒスチジン残基を含んでいる分子、例えば5から10の残基のポリヒスチジントラクトを指す。ポリヒスチジントラクトは、固定化金属、例えばニッケル、亜鉛、コバルト又は銅、キレートカラムの上の共有結合した分子のアフィニティ精製、又は他の分子(例えばHisタグと反応する抗体)との相互作用を通した精製を可能にする。
【0062】
「タンパク質不安定化配列」としては、それには限定されないが、PEST配列、例えば有糸分裂サイクリンなどのサイクリン、ウラシル透過酵素又はODCからのPEST配列、ODCなど短寿命タンパク質のC末端領域、初期応答タンパク質、例えばサイトカイン、リンフォカイン、プロトオンコジーン、例えばc−myc若しくはc−fos、MyoD、HMG CoAレダクターゼ、又はS−アデノシルメチオニンデカルボキシラーゼ、CL配列、サイクリン破壊ボックス又はN−デグロン(degron)からの配列などがある。
【0063】
本明細書で使用されるように、「マーカー遺伝子」又は「レポーター遺伝子」はその遺伝子を発現している細胞に異なった表現型を与える遺伝子であり、したがって、その遺伝子を有しない細胞からのその遺伝子を有する細胞の区別を可能にする。化学的手段、例えば選択剤(例えば除草剤、抗生物質、その他)の使用を通してそれについて「選択」することのできる形質をマーカーが付与するのか、或いはそれは単に観察又は検査を通して、即ち「スクリーニング」により特定することができる「レポーター」形質に過ぎないのかによって、そのような遺伝子は、選択可能な又はスクリーニングが可能なマーカーのいずれかをコードすることができる。本開示の要素は、特定のマーカー遺伝子の使用を通して詳細に例証される。当然ながら、適当なマーカー遺伝子又はレポーター遺伝子の多くの例は当分野で公知であり、本発明の実施において使用することが可能である。したがって、以下の説明は網羅的というよりは例示的であることは理解されよう。本明細書で開示されている技術及び当技術分野で公知の一般的組換え技術に照らして、本発明は任意の遺伝子の変更を可能にする。例示的な修飾されたレポータータンパク質は、修飾されたレポーター遺伝子、例えばそれには限定されないが、ネオ遺伝子、β−gal遺伝子、gus遺伝子、cat遺伝子、gpt遺伝子、hyg遺伝子、hisD遺伝子、ble遺伝子、mprt遺伝子、bar遺伝子、ニトリラーゼ遺伝子、ガラクトピラノシド遺伝子、キシロシダーゼ遺伝子、チミジンキナーゼ遺伝子、アラビノシダーゼ遺伝子、突然変異アセトラクテートシンターゼ遺伝子(ALS)若しくはアセトアシッドシンターゼ遺伝子(AAS)、メトトレキセート抵抗性dhfr遺伝子、ダラポンデハロゲナーゼ遺伝子、5−メチルトリプトファンに抵抗性を付与する突然変異アントラニル酸シンターゼ遺伝子(国際公開97/26366)、R座位遺伝子、βラクタマーゼ遺伝子、xylE遺伝子、α−アミラーゼ遺伝子、チロシナーゼ遺伝子、ルシフェラーゼ(luc)遺伝子(例えばRenilla reniformisルシフェラーゼ遺伝子、ホタルルシフェラーゼ遺伝子若しくはコメツキムシルシフェラーゼ(Pyrophorus plagiophthalamus)遺伝子)、エクオリン遺伝子、赤色蛍光タンパク質遺伝子、又は緑色蛍光タンパク質遺伝子などの修飾を含んでいる核酸分子によってコードされる。
【0064】
本明細書で特定される全てのアミノ酸残基は、天然のL−コンフィギュレーションである。標準ポリペプチド命名法に従い、アミノ酸残基の略記号は以下の対応表に示す通りである。
【表1】
【0065】
I.修飾に寛容性のあるレポータータンパク質の残基又は領域を特定する方法
破壊されるなど修飾されても転写及び翻訳されたときに所望の、例えば容易に検出可能な遺伝子産物を生成することができるレポータータンパク質遺伝子内の部位及び/又は領域を特定するために、多数の方法が利用できる。例えば、1つ又は複数のアミノ酸残基のヌクレオチドをレポータータンパク質遺伝子に対して削除及び/又は挿入するために、増幅反応を使用することができる。或いは、挿入変異のライブラリーを調製するためにトランスポゾンを使用することができる。トランスポゾンは、原核生物及び真核生物のゲノムで見られる移動性のDNA配列である。トランスポゾン標識は、プライマー結合部位をランダムに分散し、遺伝子「ノックアウト」を作製し、物理的標識又は遺伝標識を大きな標的DNAに導入するための強力な研究手段として、長く認められてきた。本発明の修飾されたレポータータンパク質を調製するために有用なレポーター遺伝子内の挿入は、レポータータンパク質のコード領域内部のインフレーム挿入である。以下の実施例は例示のために示したものにすぎないが、レポーター遺伝子内の挿入に寛容性のある領域を特定するための、Tn5ベースの系(Epicentre、マジソン、ウィスコンシン州からのEZ:TN(商標))及びTn7ベースの系(GPS−M Mutagenesis System、New England Biolabs、Inc.)の使用を記載する。
【0066】
A.Tn−5挿入突然変異
頻繁に使われる転位系の1つは、グラム陰性菌から単離されたTn5系である。Tn5トランスポザーゼは、クローニングされ、高比活性に精製された小さな、単一のサブユニット酵素であり、宿主細胞因子を必要とせずに転位を実行する。さらに、標的DNAへのTn5トランスポゾンの挿入は非常にランダムであり、また単純な過程で進行する。Tn5トランスポザーゼは、その短い19の塩基対Mosaic End(ME)Tn5トランスポザーゼ認識配列の間に含まれるいかなるDNA配列も転位させる。EZ:TNインフレームリンカー挿入プロトコルの概要を図1に示す。
【0067】
i.トランスポゾン挿入反応
標的DNAの調製。標的レポーターDNAは、トランスポゾン遺伝子、例えばカナマイシン抵抗性遺伝子によってコードされていないものを選択する。トランスポゾン挿入反応は標的DNA調製物内の高レベルのRNA汚染によってかなり抑制されることはないが、標的DNAが標的転位の直接の競合者である染色体DNAによって高度に汚染されるならば、クローン数は減少する。プラスミド及びコスミドクローンは、標準の微小溶解手法によって精製することが可能であり、挿入反応で標的DNAとして使用することができる。BAC又はコスミドクローンなどの低いコピー数のベクターは、より高いモル比率の大腸菌染色体DNAによってしばしば汚染され、したがってトランスポゾン挿入頻度が低下する。したがって、挿入反応の前に染色体DNAを除去するためにBAC及びコスミドDNAを精製することが好ましい。
【0068】
in vitroトランスポゾン挿入反応。複数の挿入事象を最小にしてトランスポゾン挿入の効率を最大にするために、反応条件が最適化される。例えば、等モル量のトランスポゾンがそのモル数の標的DNAに加えられる。
1.以下の順序で加えることによってトランスポゾン挿入反応混合物を調製する:
1μl 10×反応緩衝液
0.2μg 標的DNA*
xμl モル当量トランスポゾン
9μlの反応体積にxμlの滅菌水
1μl トランスポザーゼ
10μl 総反応体積
2.反応混合物を37℃で2時間インキュベートする。
3.μl停止液を加えて反応を止める。
混合して70℃で10分間加熱する。
反応混合物は−20℃で保存することができる。
【0069】
ii.トランスポゾン挿入クローンの選択
形質転換及び回収。1回の反応で得られるトランスポゾン挿入クローンの数は、とりわけ使用されるコンピテント細胞の形質転換効率によって決まる。コンピテント細胞の形質転換効率がより高いほど、より多くの挿入クローンが得られる。ベクターの多量体形態を生成する可能性をなくすために、大腸菌のrecA−系統が好まれる。また、宿主系統はトランスポゾンで存在するいかなる抗生物質耐性マーカー、例えばカナマイシン抵抗性マーカーを発現してはならない。
1.挿入反応混合物の1μlを使用して、recA−大腸菌、例えばエレクトロコンピテント細胞を形質転換する。
2.エレクトロポレーションの直後にSOC培地を1mlの最終体積までエレクトロポレーションセルに加えることによって、エレクトロポレーションされた細胞を回収する。培地/細胞をピペットで穏やかに混ぜる。管に移して37℃の撹拌機で30〜60分間インキュベートして細胞成長を促進する。
【0070】
形質転換体の平板培養及び選択。トランスポゾン挿入クローンは、抗生物質を含んでいるプレート上で選択される。Tn5については、カナマイシンを含んでいるプレートを使用してもよいが、トランスポゾンはネオマイシン及びG418抵抗性を大腸菌に付与することもできる。
1.細胞の一部を50μg/mlのカナマイシンを含んでいるLBプレート上で平板培養する。
2.トランスポゾン挿入効率を測定するために、形質転換反応の同一の希釈液及び希釈アリコートを、標的DNAを特異的に選択する抗生物質(例えば対照DNAでは100μg/mlのアンピシリン)を含んでいる第2のプレート上で平板培養する。転位頻度は、対照DNAのKanR/AmpRのクローン比率によって与えられる。
3.37℃で一晩プレートを増殖させる。1%のトランスポゾン挿入効率及び高純度の標的DNA(即ち染色体DNAの汚染がほとんど又は全くない)の使用を仮定すると、1プレートにつき約100〜500KanRクローンが存在する。
【0071】
iii.インフレーム19コドン挿入の生成
トランスポゾン挿入のマッピング。Tn5は、ランダムに標的DNAに挿入される。したがって、各クローンにおけるトランスポゾン挿入部位は、3つの方法の1つによって制限酵素消化、例えばNotI消化の前に決定しなければならない:
1.挿入クローンは、順方向及び逆方向のトランスポゾン特異プライマーを使用して二方向に配列決定をすることが可能である。各クローンの挿入部位も、配列決定の前にマッピングすることが可能である。
2.挿入部位は、コロニー微小溶解DNAをテンプレートとして使用して、PCR産物のサイズ分析によってマッピングすることが可能である。挿入部位をマッピングするために、順方向又は逆方向のトランスポゾン特異プライマー及びベクター特異フランキングプライマーを使用してもよい。
3.或いは、挿入部位は制限酵素消化物によってマッピングすることが可能である。
【0072】
所望のクローンのトランスポゾン挿入部位が一旦決定されると、それらのクローンは制限酵素、例えばNotIで個々に消化してDNAを線状にする。線状にされたDNAは次に精製される(例えば、アガロースゲル電気泳動、カラム精製、などによって)。
【0073】
再連結及び形質転換。線状にされたクローンは、T4 DNAリガーゼを使用して再連結される。再連結が成功すると、単一の制限部位、例えばNotIが再生され、3つ全ての読取り枠への57ヌクレオチド(19コドン)の挿入が作製される。再連結されたDNAは選択された細胞に形質転換され、元のクローニングベクター上に存在する抗生物質マーカー(例えば対照DNAではアンピシリン)を使用して組換え体が選択される。
【0074】
19コドン挿入クローンの分析。57のヌクレオチドのうちの9は、トランスポゾン挿入部位に直接連なっている9bp配列の複製の結果である。標的DNAによってコードされるタンパク質のアミノ酸配列は、19コドン挿入の両側で保存される。
【0075】
iv.トランスポゾン挿入クローンのDNA配列決定
プライマーの考慮。プライマーは、通常使用されるクローニングベクターとの相同性を最小にするように構築されなければならず、また、各プライマーの配列は、ベクターとの配列相同性が極小になるように使用者の特定のクローニングベクターのそれと比較しなければならない。
【0076】
標的部位複製。Tn5の触媒によるトランスポゾン挿入は、1コピーが挿入されたトランスポゾンの各側に直接連なる、9bp標的部位配列の複製をもたらす。
【0077】
挿入配列のためのトランスポゾン配列の識別。プライマーがトランスポゾン末端近くの領域にアニールするならば、各シークエンシング反応から得られる第1の配列データはトランスポゾンDNAのそれである。
【0078】
B.Tn7ベースの挿入突然変異
GPS−M Mutagenesis Systemは、Tn7ベースのトランスポゾンをDNA標的にランダムに挿入するために、TnsABC*トランスポザーゼを使用する。標的DNAは、プラスミド、コスミド、BAC又は精製された染色体DNAであってもよい。挿入部位が翻訳される遺伝子セグメント内にあるならば、これは通常無効な(機能の損失)変異をもたらす。挿入に対しては最小限の部位による好みがあるので、任意の読取り枠の破壊は可能である。標的の免疫性のために、in vivoでは約190kbの範囲でDNA分子1つにつき挿入は1つだけ起こる。したがって、in vitro反応は、それぞれ異なる位置に転位因子を含む標的DNA分子の集団を生じる。
【0079】
トランスポゾン供与体は、カナマイシンなどの抗生物質抵抗性マーカーを増やすか又は取り替えることによって修飾することが可能である。供与体プラスミドは標準の大腸菌実験系統で増殖させることができ、ベクター骨格はAmprなどのトランスポゾンと異なる抗生物質マーカー及び複製起点を有す。反応しない供与体分子を破壊して望ましくない反応生成物を避けるために、供与体はレアカッティング酵素、例えばPI−SceI(VDE)による消化で破壊することができる。突然変異を起こすDNAが元来コンピテントな生物(一本鎖DNAを取り込む)へ形質転換される適用については、ギャップが埋められて連結される。
【0080】
i.反応プロトコル
1.以下の試薬を混合する(20μl反応液あたり):
2μl 10×緩衝液
1μl スーパーコイルカスタム供与体(0.02μg)
0.08μg 標的DNA
dH2O
18μl 総体積
ピペット操作を2、3回行ってよく混ぜる。
2.1μlトランスポザーゼを各管に加える。再び混合する。
3.37℃で10分間インキュベートする。これは、集合反応である。
4.1μl開始溶液を各管に加える。ピペット操作を2、3回行ってよく混ぜる。
5.37℃で1時間インキュベートする。これは、鎖転移反応である。
6.75℃で10分間加熱不活化する。注:65℃は十分でない。
7.任意選択のギャップ修復。
8.5μl 10×Pl−SceI緩衝液を加える
0.5μl BSA
18.5μl dH2O
6μl Pl−SceI(VDE)(6単位)
9.37℃で1〜2時間インキュベートする。
10.75℃で10分間インキュベートする。
11.形質転換する。サブクローニング効率細胞(pUC1マイクログラムにつき107)による化学的形質転換のためには、非希釈反応液の1μl及び10μlを形質転換する。エレクトロポレーション(pUC1マイクログラムにつき>109)のためには、dH2Oで10倍に希釈して1μl及び10μlを形質転換する。増殖のためには、1mlのLBに又は製造業者の指示通りに形質転換混合物を希釈し、37℃で1時間通気下でインキュベートする。選択のないこの期間は、薬剤特にカナマイシンに対する耐性の発現のために必要である。
【0081】
ii.一般的考慮
標的の量。標的DNAの推奨質量(1反応につき0.08μg)は、プラスミド標的に対して有効である。コスミド及びBACについては、2:1(供与体対標的)のモル比が有効である。比率を4:1に増やすと、僅かに効率が低下する。
【0082】
供与体対標的比率。推奨されている供与体:標的質量比(1:4、20μl反応液につき0.08μg標的)が最適である。逸脱が小さければ、回収された生成物の数の小さな変化がもたらされるだけである。しかし、供与体の飽和量は反応を抑制し、二重挿入の蓄積をもたらす可能性がある。
【0083】
添加順序。水、標的DNA、緩衝液及び供与体プラスミドを先ず加え、次にトランスポザーゼを加える。開始溶液は、集合反応の後に加えなければならない。
【0084】
集合反応。この工程を省略すると、複合生成物の比率が増加する。
【0085】
インキュベーション時間。反応物は37℃で少なくとも1時間で線状になる。極めて長いインキュベーション時間は、二重挿入の蓄積をもたらす可能性がある。
【0086】
インキュベーション温度。室温及び30℃では、反応はより遅い速度であるが進行する。BACとの反応では、30℃が推奨される。
【0087】
熱殺傷。75℃で10分間の加熱処理は反応複合体を有効に破壊する。65℃で20分間の熱処理は十分でない。フェノール/クロロホルム抽出後のアルコール沈殿も有効である。
【0088】
手順の拡大縮小。最終体積及び全ての構成要素の体積を同じパーセンテージによって増加又は減少させる;2つのDNA種及びタンパク質の相対濃度は、緩衝液条件と同様に非常に重要である。
【0089】
酵素名。Pl−SceI(VDE)はl−SceIと同じでない。供与体の消化のためにはPl−SceI(VDE)を使用し、得られた挿入のマッピングのためにはScelを使用する。
【0090】
ギャップ修復。この工程は大腸菌への形質転換には必要でなく、所望の適用が元来コンピテントな細菌への形質転換を含むときだけ必要である。元来コンピテントな細菌としては、ナイセリア(Neisseria)、ヘモフィルス(Haemophilus)、バシラス(Bacillus)、ニューモコッカス(Pneumococcus)、スタフィロコッカス(Staphylococcus)及びストレプトコッカス(Streptococcus)属の種がある。これらの生物へのDNA取込みは、他の鎖の内部移行に伴う1本の鎖の分解を含む。ギャップ修復なしの場合、トランスポゾン挿入部位の5塩基ギャップは、トランスポゾン挿入を一方又は他の側のフランキングDNAから解き放す。コンピテンスが化学的に又はエレクトロポレーションによって誘発される生物(例えば大腸菌及び他の腸内細菌の組織培養細胞、その他)は、両方のDNA鎖を取り込む。挿入部位のギャップは、細胞内の機構により有効に修復される。
【0091】
iii.ギャップ修復プロトコル
7.フェノール/クロロホルム抽出(50μl)。
8.エタノール沈殿:
6μl 3M NaAcetate
100μl EtOH
−20℃で20分間インキュベートする
微量遠心管で10分間遠心分離する
9.15μl TEで再懸濁する。
10.1μl DNAポリメラーゼI(大腸菌)(10単位)
3μl 10×EcoPol緩衝液
9μl dNTP(各ヌクレオチド100μM;最終濃度それぞれ33μM)
11.室温で15分間インキュベートする。
12.1μl T4 DNAリガーゼ(400単位)及びATPを1mMの最終濃度まで加える。
13.16℃で4時間インキュベートする。
14.フェノール/クロロホルム抽出。
15.アルコール沈殿。
16.20μl TEで再懸濁する。
17.5μl OX Pl−SceI緩衝液を加える
0.5μl BSA
18.5μl dH2O
6μl Pl−SceI(VDE)(6単位)
18.37℃で1〜2時間インキュベートする。
19.75℃で10分間インキュベートする。
20.適当な方法によって形質転換する。
【0092】
iv.供与体操作
1.トランスポゾン供与体はスーパーコイルでなければならない。弛緩した又は線状の供与体を使用した反応の効率は、約100倍低下する。供与体の調製物は良質でなければならないが、CsCl精製は必要でない。
2.トランスポザーゼに必須の認識要素は必須である。終止コドンはトランスポゾンに読み込まれる全ての枠に存在してもよい。転写は外側から非必須領域へ容易に進行することが可能である。
3.転位効率は、トランスポゾンが長くなるに従い若干減少する可能性がある。
4.最良の結果のために、トランスポゾン供与体プラスミドは単量体であることを確認する。
5.供与体調製物が単量体且つスーパーコイルであり、また、供与体分子が宿主生物内で複製しないならば、Pl−SceI消化工程は省略してもよい。
【0093】
v.標的DNAの要件
配列決定をするためのプラスミド標的は、回収を容易にするために環状でなければならない。線状の(例えば、染色体の)DNAは、効率的な基質である。本来形質転換が可能な生物を使用するときは、修復及びライゲーション工程が形質転換の前に必要である。大きなプラスミド、例えばコスミド及びBACは使用可能な標的である。標的DNAは、1×TEのような無塩緩衝液では少なくとも5μg/mlでなければならない。濃度は、公知の濃度のDNAとのアガロースゲル帯強度の比較によって、又は、260における吸光度によって推定することが可能である。
【0094】
II.修飾の例
一旦レポータータンパク質における修飾に寛容性のある部位又は領域が特定されるならば、その部位又は領域は1つ又は複数の残基の欠失、1つ又は複数の残基の挿入及び/又は環状置換又はその任意の組み合わせによって修飾することができる。一実施形態では、修飾は加水分解酵素、例えばそれには限定されないがプロテアーゼ、ペプチダーゼ、エステラーゼ(例えばコレステロールエステラーゼ)、ホスファターゼ(例えばアルカリホスファターゼ)などの認識部位の導入であってよい。例えば、加水分解酵素としてはそれには限定されないが、ペプチド結合に作用する酵素(ペプチドヒドロラーゼ)、例えばアミノペプチダーゼ、ジペプチダーゼ、ジペプチジルペプチダーゼ及びトリペプチジルペプチダーゼ、ペプチジルジペプチダーゼ、セリン型カルボキシペプチダーゼ、メタロカルボキシペプチダーゼ、システイン型カルボキシペプチダーゼ、ωペプチダーゼ、セリンエンドペプチダーゼ、システインエンドペプチダーゼ、アスパラギン酸エンドペプチダーゼ、メタロエンドペプチダーゼ、トレオニンエンドペプチダーゼ及び未知の触媒メカニズムのエンドペプチダーゼがある。例えば、本発明の修飾された甲虫ルシフェラーゼは、エンテロキナーゼ切断部位、カスパーゼ切断部位、コロナウイルスプロテアーゼ部位(STLQ−SGLRKMA;配列番号10)、キナーゼ部位、HIV−1プロテアーゼ部位(SQNY−PIVQ又はKAVRL−AEAMS;それぞれ配列番号11及び配列番号12)、HCVプロテアーゼ部位(AEDVVCC−SMSYS;配列番号13)(例えばLeeら、2003年を参照)、SARSウイルスプロテアーゼ部位(例えばQTSITSAVLQSGFRKMAFPS;配列番号16又はVRQCSGVTFQGKFKKIVKGT;配列番号17)、ライノウイルスプロテアーゼ部位、例えば、ライノウイルス3Cプロテアーゼ部位、プロホルモンコンベルターゼ部位、インターロイキン16変換酵素部位、CMV集合部位、リーシュマンジシン(leishmandysin)部位、B.アントラシス(B.anthracis)致死因子、ボツリヌス神経毒素軽鎖プロテアーゼ部位、アミロイド前駆体タンパク質のためのβ−セクレターゼ部位(VKM−DAEF;配列番号14)、前立腺特異性抗原配列、トロンビン部位、レニン及びアンジオテンシン変換酵素部位、カテプシンD部位、マトリックスメタロプロテイナーゼ部位、uPA部位、プラスミン部位、カチオンのための結合部位、例えばカルシウム結合ドメイン、カルモジュリン結合ドメイン、セルロース結合ドメイン、キチン質結合ドメイン、マルトース結合タンパク質ドメイン又はビオチン結合ドメインを含むことができる。他の実施形態では、本発明の修飾されたレポータータンパク質は、リガンドによって認識される配列、例えば抗体、又はカルシウムのような金属を含んでもよい。
【0095】
III.ポリヌクレオチド及びタンパク質の例
本発明は検出可能な活性、例えば発光活性を有するポリペプチドを提供する任意のアミノ酸配列を含んでいる修飾されたレポータータンパク質、並びに組換えにより又は合成的に合成されたそのタンパク質断片を含む。修飾されたレポータータンパク質のレポータータンパク質配列は、対応する修飾されていないレポータータンパク質のアミノ酸配列と同じか又は実質的に同じである。実質的に同じ配列を有するポリペプチド又はペプチドは、アミノ酸配列が完全にではないがほとんど同じであり、それと関連する配列の機能的活性を保持することを意味する。一般に、2つのアミノ酸配列は、それらが少なくとも70%同一であるならば、例えば少なくとも80%、90%、95%又はそれ以上の同一性を有する場合は実質的に同じ又は実質的に相同である。
【0096】
相同性又は同一性は、しばしば配列解析ソフトウェア(例えばGenetics Computer Group、ウィスコンシン大学バイオテクノロジーセンター、1710 University Avenue、マジソン、ウィスコンシン州 53705、のSequence Analysis Softwareパッケージ)を使用して測定される。そのようなソフトウェアは、相同性の程度を様々な欠失、置換及びその他の修飾に割り当てることによって類似の配列を比較する。2つ以上の核酸又はポリペプチド配列との関連で用語「相同性」及び「同一性」は、任意の数の配列比較アルゴリズムを用いることにより又は手動アラインメント及び目視検査により測定されるような、比較ウィンドウ又は指定領域上で最大の対応関係について比較及び整列したときに、同じであるか又は特定割合の同じアミノ酸残基若しくはヌクレオチドを有する2つ以上の配列又は部分配列を指す。
【0097】
配列比較のために、一般的に1つの配列は試験配列が比較される基準配列としての役割を果たす。配列比較アルゴリズムを使用するときは、試験配列及び基準配列がコンピュータに入力され、必要に応じて部分配列座標が指定され、配列アルゴリズムプログラムパラメータが指定される。デフォルトのプログラムパラメータが使用でき、又は代替パラメータを指定することができる。次に、配列比較アルゴリズムはプログラムパラメータに基づいて、基準配列と比較した試験配列の配列同一割合を計算する。
【0098】
比較のための配列アラインメントの方法は、当技術分野で公知である。比較のために最適な配列アラインメントは、Smithら(1981年)のローカルホモロジーアルゴリズム、Needlemanら(1970年)のホモロジーアラインメントアルゴリズム、Personら(1988年)の類似性検索法、これらのアルゴリズムのコンピュータ上での実行(Wisconsin Genetics Softwareパッケージ、Genetics Computer Group、575 Science Dr.、マジソン、ウィスコンシン州、内のGAP、BESTFIT、FASTA及びTFASTA)、又は手動アラインメント及び目視検査によって行うことができる。
【0099】
これらの数値計算用アルゴリズムのコンピュータによる実行は、配列同一性の決定のために配列の比較のために利用することが可能である。そのような実行例としては、それには限定されないが、PC/Geneプログラム(Intelligenetics、マウンテンビュー、カリフォルニアから入手可能)のCLUSTAL、並びにWisconsin Genetics Softwareパッケージ、バージョン8(Genetics Computer Group(GCG)、575 Science Drive、Madison、Wisconsin、USAから入手可能)のALIGNプログラム(バージョン2.0)及びGAP、BESTFIT、BLAST、FASTA及びTFASTAがある。これらのプログラムを使用したアラインメントは、デフォルトパラメータを使用して実施することが可能である。CLUSTALプログラムは、Higginsら(1988年)、Higginsら(1989年)、Corpetら(1988年)、Huangら(1992年)及びPearsonら(1994年)が適切に記載している。ALIGNプログラムは、Myers及びMiller(1988年)のアルゴリズムに基づく。Altschulら(1990年)のBLASTプログラムは、Karlin及びAltschul(1990年)のアルゴリズムに基づく。
【0100】
BLAST分析を実行するためのソフトウェアは、National Center for Biotechnology Information(http://www.ncbi.nlm.nih.gov/)から公的に入手することができる。このアルゴリズムは、第1に、データベース配列内の同じ長さのワードと並べたときに正の値の閾値スコアTにマッチするかそれを満たす、クエリー配列内の長さWのショートワードを特定することによって、高いスコアの配列の対(HSP)を特定することを含む。Tは、近傍ワードスコア閾値と称される(Altschulら、1990年)。これらの初期近傍ワードヒットは、それらを含むより長いHSPの検索を開始するためのシードとして機能する。ワードヒットは、次に累積アラインメントスコアが増加するまで、各配列に沿って両方向に拡張される。累積スコアは、ヌクレオチド配列のためにはパラメータM(一対のマッチング残基のリワードスコア;常に>0)及びパラメータN(ミスマッチ残基のペナルティスコア;常に<0)を使用して計算される。アミノ酸配列については、累積スコアの計算のためにはスコアリングマトリックスが用いられる。各方向へのワードヒットの拡張は、累積アラインメントスコアがその最大到達値から量X分低下する場合、累積スコアが1つ又は複数の負のスコアの残基アラインメントの蓄積によってゼロ以下になる場合、或いはいずれかの配列の末端に到達した場合に停止される。
【0101】
配列同一割合を計算することに加えて、BLASTアルゴリズムは2つの配列間の類似性の統計解析も実行する(例えばKarlin及びAltschul(1993年)を参照)。BLASTアルゴリズムによって提供される類似性測定手段の1つは、最小合計確率(P(N))であり、それは2つのヌクレオチド又はアミノ酸配列間の一致が偶然に起こる確率の指標となる。例えば、試験核酸と基準核酸との比較において最小合計確率が約0.1未満、より好ましくは約0.01未満、最も好ましくは約0.001未満のとき、試験核酸配列は基準配列と類似しているとみなされる。
【0102】
比較目的のためのギャップアラインメントを得るために、Altschulら(1997年)が記載しているようにGapped BLAST(BLAST 2.0に含まれる)を利用することが可能である。或いは、分子間の距離関係を検出する反復検索を実施するために、PSI−BLAST(BLAST 2.0に含まれる)を使用することが可能である。Altschulら、上記、を参照。BLAST、Gapped BLAST、PSI−BLASTを利用するときは、それぞれのプログラムのデフォルトパラメータを使用することが可能である(例えばヌクレオチド配列のためのBLASTN、タンパク質のためのBLASTX)。BLASTNプログラム(ヌクレオチド配列のための)は、デフォルトとして11のワード長(W)、10の期待値(E)、100のカットオフ値、M=5、N=−4、及び両方の鎖の比較を用いる。アミノ酸配列については、BLASTPプログラムはデフォルトとして3のワード長(W)、10の期待値(E)及びBLOSUM62スコアリングマトリックスを用いる(Henikoff及びHenikoff、1989年、を参照)。http://www.ncbi.nlm.nih.govを参照。
【0103】
詳細には、ポリペプチドは保存的な変異を除けばかなり相関している。保存的な変異は、他の生物学上類似した残基によるアミノ酸残基の置換を意味する。保存的な変異の例としては、1つの疎水性残基、例えばイソロイシン、バリン、ロイシン又はメチオニンによる他の残基の置換、1つの極性残基による他の残基の置換、例えばアルギニンによるリジンの置換、グルタミン酸によるアスパラギン酸の置換、又はグルタミンによるアスパラギンの置換、その他がある。保存的な置換の他の実例としては、下記変更が含まれる:アラニンからセリン;アルギニンからリジン;アスパラギンからグルタミン又はヒスチジン;アスパラギン酸からグルタミン酸;システインからセリン;グルタミンからアスパラギン;グルタミン酸からアスパラギン酸;グリシンからプロリン;ヒスチジンからアスパラギン又はグルタミン;イソロイシンからロイシン又はバリン;ロイシンからバリン又はイソロイシン;リジンからアルギニン、グルタミン又はグルタミン酸;メチオニンからロイシン又はイソロイシン;フェニルアラニンからチロシン、ロイシン又はメチオニン;セリンからトレオニン;トレオニンからセリン;トリプトファンからチロシン;チロシンからトリプトファン又はフェニルアラニン;バリンからイソロイシン、ロイシン。
【0104】
一実施形態では、本発明のポリヌクレオチドは、特定の宿主での発現のために最適化される。本明細書で使用されるように、最適化はコドン最適化、並びに真核細胞においてはコザック配列及び/又は1つ又は複数のイントロンの導入を含む。したがって、核酸分子は、修飾されていない甲虫ルシフェラーゼをコードする野生型核酸配列とは、コドンの30%、35%、40%又は45%以上、例えば50%、55%、60%若しくはそれ以上を超える割合で異なるコドン組成物を有してもよい。本発明に用いられる好ましいコドンは、特定の生物において同じアミノ酸の少なくとも1つの他のコドンよりも高頻度で使用され、またより好ましくはその生物において低利用コドンではなく、また核酸分子の発現のためにクローニング又はスクリーニングするために使用する生物においても低利用コドンではないものである。更に、あるアミノ酸(即ち、3つ以上のコドンを有するアミノ酸)の好ましいコドンは、他の(好ましくない)コドンよりも高頻度で使用される2つ以上のコドンを含んでもよい。1つの生物において他の生物におけるよりも高頻度で使用される核酸分子内のコドンの存在は、それらのコドンをより高頻度で使用する生物の細胞に導入されたとき、それらの細胞における野生型又は親の核酸配列の発現よりも高いレベルでそれらの細胞において発現される核酸分子をもたらす。
【0105】
本発明の一実施形態では、異なるコドンとは、哺乳類においてより高頻度で使用されるものであり、一方、他の実施形態においては、異なるコドンとは植物においてより高頻度で使用されるものである。特定の種類の哺乳類、例えばヒトは、他の種類の哺乳類とは異なる好ましいコドンセットを有している可能性がある。同様に、特定の種類の植物は、他の種類の植物とは異なる好ましいコドンセットを有しているかもしれない。本発明の一実施形態では、異なるコドンのほとんどは、所望の宿主細胞における好ましいコドンである。哺乳類(例えばヒト)及び植物の好ましいコドンは、当技術分野で公知である(例えばWadaら、1990年)。例えば、ヒトの好ましいコドンとして、それには限定されないが以下がある。
【化1】
(Wadaら、1990年)。
したがって、本発明の好ましい「ヒト化」合成の核酸分子は、増加した数の好ましいヒトコドンを有することによって野生型核酸配列と異なるコドン組成を有する。例えば、
【化2】
又はそのいかなる組み合わせがある。例えば、本発明の核酸分子は、野生型核酸配列と比較して増加した数のCTG又はTTGのロイシンエンコーディングコドン、GTG又はGTCのバリンエンコーディングコドン、GGC又はGGTのグリシンエンコーディングコドン、ATC又はATTのイソロイシンエンコーディングコドン、CCA又はCCTのプロリンエンコーディングコドン、CGC又はCGTのアルギニンエンコーディングコドン、AGC又はTCTのセリンエンコーディングコドン、ACC又はACTのトレオニンエンコーディングコドン、GCC又はGCTのアラニンエンコーディングコドン、或いはその任意の組み合わせを有することができる。同様に、植物においてより高頻度に使用されるコドンをより多く有す核酸分子は、増加した数の植物コドンを有することによって野生型核酸配列と異なるコドン組成を有し、このようなコドンとしては、それには限定されないが、
【化3】
又はその任意の組み合わせがある(Murrayら、1989年)。好ましいコドンは、植物の種類が異なれば異なってもよい(Wadaら、1990年)。
【0106】
本発明の修飾された甲虫ルシフェラーゼタンパク質又は融合タンパク質は、組換え方法又は固相化学ペプチド合成法によって調製することができる。そのような方法は1960年代初期から当技術分野で公知であり(Merrifield、1963年)(Stewartら、固相ペプチド合成(Solid Phase Peptide Synthesis)、第2版、Pierce Chemical Co.、ロックフォード、Ill.、11〜12頁も参照)、また、市販のラボラトリペプチドデザイン及び合成キット(Cambridge Research Biochemicals)でも最近採用された。そのような市販のラボラトリキットは、通常、Geysenら(1984年)の教示を利用しており、全て単一のプレートに接続されている多数の「ロッド」又は「ピン」の先端でペプチドを合成することを可能にする。そのような系が利用されるときは、ロッド又はピンのプレートは逆にされて、適当なアミノ酸をピン又はロッドの先端に結合又は固定するための溶液を含んでいる、対応するウェル又は貯蔵器の第2のプレート内に挿入される。そのような工程を繰り返すことにより、例えばロッド及びピンの先端を逆にして適当な溶液に挿入することにより、アミノ酸は所望のペプチドに構築される。更に、いくつかのFMOCペプチド合成系が利用できる。例えば、ポリペプチド又は断片の集合は、Applied Biosystems、Inc.Model 431A自動ペプチド合成機を使用して固体支持体上で実行することが可能である。そのような設備は、直接合成又は他の公知の技術を使用して結合することができる一連の断片の合成による、本発明のペプチドへの到達を容易にする。
【0107】
IV.本発明の修飾されたレポータータンパク質との有用な融合パートナー
修飾されたレポータータンパク質をコードする本発明のポリヌクレオチドは、他の核酸配列、例えばcDNAなどの未変性の配列、又はN末端、C末端又はN及びC末端の融合タンパク質、例えば選択可能なマーカーを含む異なるレポーター遺伝子によってコードされるタンパク質との融合を調製するためにin vitroで操作されたものと一緒に使用することができる。適当な融合パートナーの多くの例は当技術分野で公知であり、本発明の実施で使用することが可能である。
【0108】
融合パートナーとしては、それには限定されないが、親和性ドメイン又は他の機能性タンパク質配列、例えば酵素活性を有するものがある。例えば、機能性タンパク質配列はキナーゼ触媒ドメインをコードして(Hanks及びHunter、1995年)、リン酸部分を酵素的に特定のアミノ酸に加えることができる融合タンパク質を生成することができ、又は、Src Homology 2(SH2)ドメインをコードして(Sadowskiら、1986年;Mayer及びBaltimore、1993年)、リン酸化されたチロシンと特異的に結合する融合タンパク質を生成することができる。
【0109】
親和性ドメインは、通常、固体支持体上で固定されるものなどの結合パートナーと相互作用することが可能なペプチド配列である。複数の連続的な単一アミノ酸、例えばヒスチジンをコードするDNA配列は、発現タンパク質に融合される場合、ニッケルセファロースなどの樹脂カラムとの高親和性結合による組換えタンパク質の一段精製のために使用してもよい。キチン結合ドメイン(キチンと結合する)、グルタチオン−S転移酵素(グルタチオンと結合する)、ビオチン(アビジン及びストレプトアビジンと結合する)、その他などのペプチドをコードする配列も、関係するタンパク質の精製を容易にするために使用することができる。親和性ドメインは、インテイン(タンパク質セルフスプライシング要素(Chongら、1997年)の使用など、当業者に公知の方法によって関係するタンパク質から分離することが可能である。例示的な親和性ドメインとしては、HisV5(HHHHH)(配列番号1)、HisX6(HHHHHH)(配列番号2)、C−myc(EQKLISEEDL)(配列番号3)、Flag(DYKDDDDK)(配列番号4)、SteptTag(WSHPQFEK)(配列番号5)、血液凝集素、例えばHA Tag(YPYDVPDYA)(配列番号6)、GST、チオレドキシン、セルロース結合ドメイン、RYIRS(配列番号104)、Phe−His−His−Thr(配列番号105)、キチン結合ドメイン、S−ペプチド、T7ペプチド、SH2ドメイン、C末端RNA標識、WEAAAREACCRECCARA(配列番号8)、金属結合ドメイン、例えば、亜鉛結合ドメイン又はカルシウム結合ドメイン、例えばカルモジュリン、トロポニンC、カルシニューリンB、ミオシン軽鎖、リカバリン、S−モジュリン、ビシニン(visinin)、VILIP、ニューロカルシン(neurocalcin)、ヒポカルシン(hippocalcin)、フリケニン(frequenin)、カルトラシン(caltractin)、カルパイン大サブユニット、S100タンパク質、パルブアルブミン、カルビンジンD9K、カルビンジンD28K及びカルレチニンなどのカルシウム結合タンパク質からのもの、インテイン、ビオチン、ストレプトアビジン、MyoD、Id、ロイシンジッパー配列並びにマルトース結合タンパク質がある。一実施形態では、融合パートナーはHis又はGST標識など融合タンパク質を精製するために有用な配列であり、一実施形態では、精製標識は環状置換されたレポータータンパク質のN又はC末端に融合される。
【0110】
他のクラスの融合パートナーとしては、レポーター遺伝子、例えばそれには限定されないがネオ遺伝子、β−gal遺伝子、gus遺伝子、cat遺伝子、gpt遺伝子、hyg遺伝子、hisD遺伝子、ble遺伝子、mprt遺伝子、bar遺伝子、ニトリラーゼ遺伝子、ガラクトピラノシド遺伝子、キシロシダーゼ遺伝子、チミジンキナーゼ遺伝子、アラビノシダーゼ遺伝子、突然変異アセトラクテートシンターゼ遺伝子(ALS)若しくはアセトアシッドシンターゼ遺伝子(AAS)、メトトレキセート抵抗性dhfr遺伝子、ダラポンデハロゲナーゼ遺伝子、5−メチルトリプトファン抵抗性を付与する突然変異アントラニル酸シンターゼ遺伝子(国際公開97/26366)、R座位遺伝子、βラクタマーゼ遺伝子、xylE遺伝子、α−アミラーゼ遺伝子、チロシナーゼ遺伝子、花虫類ルシフェラーゼ(luc)遺伝子(例えばRenilla reniformisルシフェラーゼ遺伝子、エクオリン遺伝子、赤色蛍光タンパク質遺伝子、又は緑色蛍光タンパク質遺伝子などによってコードされるたんぱく質がある。選択可能な又はスクリーニング可能なマーカー遺伝子という用語には、その分泌が形質転換細胞の特定又は選択の手段として検出することが可能な「分泌可能なマーカー」をコードする遺伝子も含まれる。例には、抗体相互作用によって特定することが可能な分泌可能な抗原をコードするマーカー、又はそれらの触媒活性によって検出することができる分泌可能な酵素をコードするマーカーさえ含まれる。分泌可能なタンパク質はいくつかのクラスに分けられ、例としては例えばELISAによって検出可能な小さな、分散性タンパク質、及び細胞膜に挿入されるか捕捉されるタンパク質がある。
【0111】
V.修飾されたレポータータンパク質又はその融合物をコードするベクター及び宿主細胞
修飾されたレポータータンパク質又はその融合物をコードする望ましい核酸分子が一旦調製されると、その修飾されたレポータータンパク質又はその修飾されたレポータータンパク質を含んでいる融合タンパク質をコードする発現カセットが調製される。例えば、修飾された甲虫ルシフェラーゼをコードする核酸配列を含んでいる核酸分子は、発現カセットを形成するために任意選択に転写調節配列、例えば1つ又は複数のエンハンサー、プロモーター、転写終結配列又はその組み合わせと作動可能的に結合される。核酸分子又は発現カセットは、プラスミド又はウイルスベクターなどの任意選択に選択可能なマーカー遺伝子を含んでいるベクターに導入し、このベクターを関係する細胞、例えば大腸菌、ストレプトミセス属、バシラス属、スタフィロコッカス属などの原核細胞、並びに植物(双子葉植物又は単子葉植物)、真菌、酵母、例えばピチア、サッカロミセス又はシゾサッカロミセス属、或いは哺乳類細胞などの真核細胞に導入することができる。好ましい哺乳類細胞としては、ウシ、ヤギ、羊、イヌ、ネコ、サルなどヒト以外の霊長類、及びヒトの細胞がある。好ましい哺乳動物細胞系には、それには限定されないが、CHO、COS、293、Hela、CV−1、SH−SY5Y、HEK293及びNIH3T3細胞が含まれる。
【0112】
コードされる修飾されたレポータータンパク質の発現は、原核細胞又は真核細胞で発現可能な任意のプロモーターによって調節することができる。好ましい原核プロモーターとしては、それには限定されないが、SP6、T7、T5、tac、bla、trp、gal、lac又はマルトースプロモーターが含まれる。好ましい真核生物のプロモーターとしては、それには限定されないが、構成プロモーター、例えばCMV、SV40及びRSVプロモーターなどのウイルスのプロモーター、並びに調節可能なプロモーター、例えばtetプロモーター、hsp70プロモーター及びCREによって調節される合成プロモーターなどの誘導又は抑制が可能なプロモーターが含まれる。本発明の核酸分子、発現カセット及び/又はベクターは、任意の方法、例えばそれには限定されないが、カルシウム媒介性形質転換、エレクトロポレーション、顕微注射、リポフェクションなどによって細胞に導入することができる。
【0113】
VI.使用例
修飾されたレポータータンパク質又はその融合物は任意の目的、例えばそれには限定されないが、特定分子の量及び存在の検出(バイオセンサー)、特定分子の単離、特定分子の高次構造の例えば結合、リン酸化又はイオン化による変化の検出、pH又は温度などの条件の検出、高スループット又は低スループットのスクリーニングの促進、タンパク質−タンパク質、タンパク質−DNA又は他のタンパク質ベースの相互作用の検出、或いはバイオセンサーの選択又は開発などのために有用である。例えば、修飾されたレポータータンパク質又はその融合物は以下のために有用である。例えばin vitro又は細胞ベースのアッセイにおける特定のキナーゼ(例えば、キナーゼ部位をレポータータンパク質に挿入することによる)、RNAi(例えば、RNAiによって認識されることが疑われる配列をレポータータンパク質のコード配列に挿入し、次にRNAiを添加した後にレポーター活性を監視することによる)、又はプロテアーゼの量、存在又は活性の検出であり、例えば特定のウイルスプロテアーゼの存在を検出するためのものがあり、これは今度はウイルス又は抗体の存在の指標となる;阻害剤、例えばプロテアーゼインヒビターのスクリーニング;認識部位の特定、又は例えば選択された認識配列を有する修飾されたルシフェラーゼ又は単一の関心のある分子を有する異なる複数の配列若しくは複数の(例えばライブラリー)分子を有する修飾されたルシフェラーゼのライブラリーを使用した基質特異性の検出;バイオセンサー又は関心のある分子、例えばプロテアーゼの選択又は開発;或いは、in vitro又は細胞ベースの方法による相補性又は結合を通したタンパク質−タンパク質相互作用の検出。一実施形態では、挿入を含む修飾された甲虫ルシフェラーゼが分子のランダムなライブラリー又は変異ライブラリーと接触させられ、その挿入と相互作用する分子が特定される。他の実施形態において、複数の挿入を有する修飾されたルシフェラーゼのライブラリーが分子と接触させられ、その分子と相互作用する修飾されたルシフェラーゼが特定される。
【0114】
本発明は、細胞内の分子の発現、場所及び/又はトラフィッキングを監視するための、並びに細胞内の微小環境の変化を監視するための、修飾された甲虫ルシフェラーゼ又はその融合タンパク質を使用した方法も提供する。例えば、一実施形態では、修飾された甲虫ルシフェラーゼは、ある条件下で互いと相互作用する2つのドメインを含んでいる内部挿入を含む。一実施形態では、挿入内の1ドメインはリン酸化が可能なアミノ酸を含み、他のドメインはホスホアミノ酸の結合ドメインである。適当なキナーゼ又はホスファターゼの存在下で、挿入内の2つのドメインは相互作用して修飾された甲虫ルシフェラーゼの高次構造を変え、その結果修飾された甲虫ルシフェラーゼの検出可能な活性の変化が起こる。他の実施形態において、修飾された甲虫ルシフェラーゼは分子の認識部位を含み、その分子がその認識部位と相互作用すると活性が増加するので、他の分子の存在又は量を検出又は決定するために使用することができる。
【0115】
2−ハイブリッド系は、in vivoでタンパク質:タンパク質相互作用を検出するための、並びにタンパク質:タンパク質相互作用に関係する残基/ドメインを特定するための極めて強力な方法である。2−ハイブリッド系の基礎は、一部の転写因子で見られるモジュラードメインである。即ち、特定のDNA配列と結合するDNA結合ドメイン、及び基礎の転写機構と相互作用する転写活性化ドメインである(Sadowski、1988年)。DNA結合ドメインと共同した転写活性化ドメインは、TATAボックスでのRNAポリメラーゼII複合体の集合を進めて転写を増やすことができる。CheckMate(商標)Mammalian Two−Hybridシステム(Promega Corp.、マジソン、ウィスコンシン州)において、別々のプラスミドによって生じたDNA結合ドメイン及び転写活性化ドメインは、DNA結合ドメインに融合された1つのタンパク質(「X」)が転写活性化ドメインに融合された第2のタンパク質(「Y」)と相互作用するときは、緊密に関連する。この系において、タンパク質X及びYの間の相互作用は、レポーター遺伝子又は選択可能なマーカー遺伝子の転写をもたらす。特に、pBINDベクターは複数のクローニング領域の上流に酵母GAL4のDNA結合ドメインを含み、pACTベクターは複数のクローニング領域の上流に単純疱疹ウイルスVP16の活性化ドメインを含む。更に、pBINDベクターは、Renilla reniformisルシフェラーゼを発現する。関係する相互作用する可能性のある2つのタンパク質をコードする2つの遺伝子は、pBIND及びpACTベクターにクローニングされ、それぞれGAL4のDNA結合ドメイン及びVP16の活性化ドメインと融合タンパク質を生成する。pG5lucベクターは、ホタルルシフェラーゼ遺伝子(luc+)の上流にある最小TATAボックスの更に上流に、5つのGAL4結合部位を含む。pGAL4及びpVP16融合構築物は、pG5lucベクターと共に哺乳類細胞に形質移入される。形質移入の2〜3日後に細胞は溶解され、Renillaルシフェラーゼ及びホタルルシフェラーゼの量はDual−Luciferase(登録商標)Reporter Assay System(Promega Cat.#E1910)を使用して定量することが可能である。GAL4及びVP16融合構築物のような2つの試験タンパク質の間の相互作用は、陰性対照を上回るホタルルシフェラーゼ発現の増加をもたらす。本発明の修飾された甲虫ルシフェラーゼ、例えば、修飾に寛容性のある1部位又は領域(N末端断片)で欠失が起きているものはDNA結合ドメインと融合され、甲虫ルシフェラーゼの残り(C末端の断片)は転写活性ドメインと融合される。
【0116】
本発明は、関心のある分子の活性を調整することができる剤(「試験」剤)についてスクリーニングする方法も提供する。「調整」は、生物的活性又は過程(例えば酵素活性)の機能的特性を強めるか抑制する能力を指し、そのような強化又は抑制は特定の事象、例えばシグナル伝達経路の活性化の発生に付随するものでもよく、且つ/又は特定の細胞種だけで現れるものでもよい。「モジュレーター」は作用剤(天然又は非天然の)を指し、例えば生体高分子(例えば核酸、タンパク質、非ペプチド又は有機分子)、小分子、或いは細菌、植物、真菌類又は動物(特に哺乳動物)の細胞又は組織などの生体物質からの抽出物を指す。モジュレーターは、本明細書で記載されているスクリーニングアッセイに含めることにより、生物学的過程の(直接的又は間接的)阻害剤又は活性剤(例えばアゴニスト、部分アンタゴニスト、部分アゴニスト、アンタゴニスト、抗腫瘍薬、細胞傷害剤、腫瘍性転化又は細胞増殖の阻害剤、細胞増殖促進剤、など)としての潜在的な活性について評価される。モジュレーターの活性は、知られていても、未知であっても、又は部分的に知られていてもよい。そのようなモジュレーターは、本発明の方法を使用してスクリーニングすることができる。用語「試験剤」は、推定されるモジュレーターとして本発明の1つ又は複数のスクリーニング法によって試験される剤を指す。通常、0.01μM、0.1μM、1.0μM及び10.0μMなどの様々な所定濃度がスクリーニングで使用される。対照は、試験剤がない場合のシグナル測定、標的を調整することが公知の剤との比較、又は試験剤との接触の前、その間、及び/又は後の試料(例えば細胞、組織又は生物体)との比較を含むことができる。
【0117】
一実施形態では、この方法はプロテアーゼ活性を調整する剤についてスクリーニングすることを含む。例えば、一実施形態では、アポトーシスを調整することができる剤を特定する方法が提供される。カスパーゼファミリープロテアーゼは、アポトーシスと関連付けられている。したがって、この方法はカスパーゼファミリープロテアーゼを含むことが疑われる試料を、カスパーゼ活性を調整することが疑われる剤、及びカスパーゼによる切断が可能な切断部位を有する修飾されたレポータータンパク質と接触させることを含む。試料中の修飾されたレポータータンパク質の活性は、試験剤との接触の前後に検出される。剤との接触後の活性増加は、アポトーシスを抑制する剤の指標であり、活性減少はアポトーシスを活性化する剤の指標である。
【0118】
したがって、本発明は本発明の修飾されたレポータータンパク質内に存在する認識配列の切断を調整する剤を特定してその活性を検出するための、有用なスクリーニング系を提供する。この系は、プロテアーゼ活性モジュレーターの迅速なスクリーニングを可能にする。本明細書で記載されているスクリーニング系を利用すると、プロテアーゼ、例えばカスパーゼファミリープロテアーゼを調整(例えば抑制又は活性化)する剤を特定するための感度が高く迅速な手段が提供される。
【0119】
したがって本発明の修飾されたレポータータンパク質は、修飾されたレポータータンパク質内の挿入及び関心のある分子との間の相互作用を調整する剤又は条件を研究するための基質として有用である。詳細には、本発明は、挿入が関係する酵素の切断部位であるアミノ酸配列を含んでいる、修飾されたルシフェラーゼタンパク質を企図する。したがって関心のある分子がプロテアーゼであるとき、挿入はそのプロテアーゼの切断認識配列を含んでいるペプチドを含む。プロテアーゼの切断認識配列は、タンパク分解性切断の間にそのプロテアーゼによって認識される特異的アミノ酸配列である。したがって、本発明は、試料を本発明の修飾されたルシフェラーゼポリペプチドと接触させてルシフェラーゼ活性の変化を測定することにより、試料中のプロテアーゼの量を測定する方法を提供する。本発明の修飾されたルシフェラーゼタンパク質は、とりわけ修飾されたルシフェラーゼを発現する細胞内のプロテアーゼ活性を監視するために使用することができる。
【0120】
本発明のアッセイは、修飾されたレポータータンパク質を切断するプロテアーゼ活性を変える化合物を特定するための薬剤をスクリーニングするために使用することができる。一実施形態では、アッセイはプロテアーゼを含んで試料に対してin vitroで実施される。既知量のプロテアーゼを含んでいる試料は、本発明の修飾されたレポータータンパク質、及び試験剤と混合される。次に、試料中のプロテアーゼ活性量が先に述べたように測定される。その後、試験剤の存在下におけるプロテアーゼ1モルあたりの活性量が、試験剤がない場合のプロテアーゼ1モルあたりの活性と比較される。相違は、試験剤がプロテアーゼ活性を変化させることを示す。したがって、変化は修飾されたレポータータンパク質活性の減少をもたらすプロテアーゼ活性の増加或いは修飾されたレポータータンパク質活性の増加又は維持と対応するプロテアーゼ活性の低下である。
【0121】
一実施形態では、プロテアーゼ活性を変化させる剤の能力が決定される。このアッセイにおいて、細胞はプロテアーゼ活性を調整することが疑われる剤で条件づけられるか又は接触させられる。培養内の細胞は溶解されて、プロテアーゼ活性が測定される。例えば、既知又は未知の量のプロテアーゼを含んでいる溶解細胞試料は、本発明の修飾されたレポータータンパク質と混合される。次に、上記したように対照又は未処理の試料及び処理された溶解細胞試料の中の修飾されたレポータータンパク質活性の程度を測定することにより、試料中のプロテアーゼ活性量が測定される。活性又は抑制は、試料中のタンパク質1マイクログラム又は1ミリグラムをベースにして計算することができる。したがって、プロテアーゼ活性の調整は、修飾されたレポータータンパク質活性の減少をもたらすプロテアーゼ活性の増加或いは修飾されたレポータータンパク質活性の増加又は維持と対応するプロテアーゼ活性の低下を含む。一般的に、プロテアーゼ活性の絶対量を与えるために、相違は標準の測定値に対して較正される。プロテアーゼの活性又は発現を抑制又はブロックする試験剤は、非処理の対照と比較した処理された細胞内の修飾されたレポータータンパク質活性の増加によって検出することができる。
【0122】
他の実施形態では、in vivoでプロテアーゼ活性を変化させる剤の能力が決定される。in vivoアッセイにおいて、本発明の修飾されたレポータータンパク質をコードする発現ベクターで形質移入された細胞は異なる量の試験剤に曝され、細胞内のレポータータンパク質活性への影響を決定することができる。一般的に、プロテアーゼ活性の絶対量を与えるために、相違は標準の測定値に対して較正される。プロテアーゼの活性又は発現を抑制又はブロックする試験剤は、非処理の対照と比較した処理された細胞内の修飾されたレポータータンパク質活性の増加によって検出することができる。
【0123】
本発明のアッセイに用いられる物質及び組成物は、キットの調製に適していることが理想である。そのようなキットは、1つ又は複数の容器手段、例えばバイアル、管、その他を含んでいる担体手段を含んでもよく、各容器手段はその方法で使用される別々の要素の1つを含んでいる。容器の1つは、本発明の修飾されたレポータータンパク質又はポリヌクレオチドを(例えば、ベクターの形で)含む。第2の容器は、修飾されたレポータータンパク質の基質を含んでもよい。
【0124】
本発明は、以下のそれには限定されない実施例で更に説明される。
【実施例1】
【0125】
コメツキムシルシフェラーゼ遺伝子のTn5挿入突然変異
A.トランスポゾン挿入反応
標的DNAの調製。コメツキムシルシフェラーゼ遺伝子(cbg69)を大腸菌T7発現ベクターにクローニングし、得られたプラスミド(pJLC1)をトランスポゾン突然変異反応のために標的DNAとして使用した。
In Vitroトランスポゾン挿入反応。複数の挿入事象を最小にしてトランスポゾン挿入の効率を最大にするために、反応条件を最適化した。例えば、等モル量のトランスポゾンをそのモル数の標的DNAに加えた。
【0126】
1.以下の順序で加えることによってトランスポゾン挿入反応混合物を調製する:
1μl 10×反応緩衝液
0.35μg 標的DNA(pJLC1)(7μl)
1μl モル当量トランスポゾン
1μl トランスポザーゼ
10μl 総反応体積
2.反応混合物を37℃で2時間インキュベートする。
3.1μl停止液を加えて反応を止める。
混合して65℃で15分間加熱する。
反応混合物は−20℃で保存した。
【0127】
B.トランスポゾン挿入クローンの選択
形質転換及び回収。1回の反応で得られるトランスポゾン挿入クローンの数は、とりわけ使用されるコンピテント細胞の形質転換効率によって決まる。コンピテント細胞の形質転換効率がより高いほど、より多くの挿入クローンが得られる。大腸菌のrecA−系統(EpicentreからのEC100コンピテント細胞)を形質転換のために使用した。
3.挿入反応混合物の1μlを使用して、EC100エレクトロコンピテント細胞へ形質転換する。
4.エレクトロポレーションの直後にSOC培地を1mlの最終体積までエレクトロポレーションセルに加えることによって、エレクトロポレーションされた細胞を回収する。培地/細胞をピペットで穏やかに混ぜる。管に移して37℃の撹拌機で30〜60分間インキュベートして細胞成長を促進する。
5.細胞の一部を50μg/mlのカナマイシンを含んでいるLBプレート上で平板培養する。
6.37℃で一晩プレートを増殖させる。
【0128】
C.トランスポゾン挿入マッピング。数千の挿入コロニーが得られた。27の挿入クローンを選択し、cbg69遺伝子の2つの末端のプライマーセットを使用してTn5トランスポゾンを含んでいるコメツキムシluc遺伝子をPCR増幅した。PCR産物を、同じプライマーセットを使用して配列決定した。Tn5挿入の位置はランダムであることが明らかになった(図2〜3)。
【0129】
D.トランスポゾン挿入を有するluc遺伝子のプラスミドライブラリーの作製。
luc遺伝子内に挿入を有するクローンは、プラスミド骨格に挿入を有するものから分離する必要がある。このため、全ての形質転換体をプールして、プラスミドDNAを精製した。トランスポゾン挿入を有するcbg69遺伝子を含んでいるDNAフラグメントを放出するために、得られたプラスミドDNAを一対の制限酵素(例えばNdeI及びEcoRI)で消化した。このDNAフラグメントはトランスポゾン挿入を含まない大腸菌T7発現ベクターのそれぞれの制限酵素部位に再クローニングし、Tn5挿入を有するluc遺伝子を含んでいるプラスミドライブラリーを得た。
【0130】
E.インフレーム19コドン挿入ライブラリーの作製
Tn5トランスポゾンの除去。一旦トランスポゾン挿入を有するluc遺伝子のプラスミドライブラリーが作製されたならば、制限酵素、例えばNotIによる消化によってTn5トランスポゾンを除去した。線状にされたDNAは、アガロースゲル電気泳動によってTn5を含んでいるDNA断片から分離し、次に精製した。
【0131】
再連結及び形質転換。線状にされたDNAは、T4 DNAリガーゼを使用して再連結した。再連結が成功すると、単一の制限部位、例えばNotIが再生され、3つの読取り枠の1つへの57ヌクレオチド(19コドン)の挿入が作製された。再連結されたDNAはEC100細胞に形質転換し、元のクローニングベクター上に存在する抗生物質マーカー(例えば対照DNAではアンピシリン)を使用して組換え体を選択した。
【0132】
F.活性リンカー挿入クローンの選択。個々のリンカー挿入クローンを使用して100μg/mlのアンピシリンを含んでいる1mlのLB培地に接種し、37℃で一晩増殖させた。ルシフェラーゼ活性は、一晩培養したもの100μlとPromega Corp.(マジソン、ウィスコンシン州)からのBright−Glo試薬100μlとを混ぜることによって測定した。発光は、5分後に照度計で記録した。
【0133】
G.活性リンカー挿入クローンのDNA配列決定。400以上のクローンがスクリーニングされた。バックグラウンドより20倍よりも強いルシフェラーゼ活性を有していたリンカー挿入クローンを選択した。リンカー挿入位置は、リンカー挿入を含んでいるluc遺伝子のPCR産物の配列決定により決定した。各活性リンカー挿入クローンの位置及び相対活性は、図3〜4に示す。
【実施例2】
【0134】
ホタルルシフェラーゼ遺伝子のTn−7挿入突然変異
市販のキット(New England Biolabs(NEB)からのGPS(商標)−M GPS Mutagenesis System)を使用して、Tn7ベースのトランスポゾンをホタルルシフェラーゼDNAにランダムに挿入した。この挿入断片の大部分を次に制限酵素消化により切り取り、再連結によって5アミノ酸の挿入を得た。最初に、コロニーを増殖させて切り取りの前にルシフェラーゼ活性の消失についてスクリーニングした。ルシフェラーゼ活性を有していた培養物内のプラスミドを次に切り取り、細胞へ形質転換で戻し、ルシフェラーゼ活性の復活がないかコロニーを調べた。その後、ランダム位置に大きな挿入を含んでいるゲル精製ルシフェラーゼ断片がベクターにクローニングされ、ベクター集団の大量切り取りが実施されたより効率的な方法が使用された。ここで、切り取られたベクターによる形質転換の後にルシフェラーゼ活性を発現したコロニーを選択した。トランスポゾンはカナマイシン耐性を有していたので、挿入を含まないベクター分子を除去することが可能であった。
【0135】
第1の方法では、反応は次のように組み立てられた:
【化4】
カナマイシン耐性遺伝子を有するpGPS5(NEB)は供与体プラスミドであり、アンピシリン耐性遺伝子を有するpSP−Luc+(Promega Corp.)は受容体であった。転位が成功すると、受容体プラスミドにカナマイシン耐性カセットが挿入された。反応体をピペット操作で混合し、次にTnsABC Transposaseの1μlを加えて反応体を更に混合した。反応体は、37℃で1時間インキュベートし、75℃で10分間加熱してから氷上に置いた。次に5μlを100μlの高効率コンピテント大腸菌JM109(Promega Corp.)に形質転換した。氷上での10分間のインキュベーションの後、細胞を42℃で45秒間の温熱ショックで処理し、次に氷上で2分間インキュベーションした。1mlのLuria Broth(LB)を次に加え、細胞を37℃で1時間振盪した。次に、40μlずつを100μg/mlのアンピシリン及び25μg/mlのカナマイシンを含んでいるLB寒天プレート上で平板培養した。
【0136】
その翌日、コロニーをそれらのプレートから拾い、3mlのLB/amp/kan+0.5mM IPTG内で個々に生育させた。一晩増殖の後、培養物の10μlをpH5.5の100mMクエン酸ナトリウム中の1mMルシフェリンの100μlに加えることによってこれらの培養物をルシフェラーゼ活性について分析し、Turner20/20照度計で数値を読み取った。
【0137】
プラスミドは低活性培養物(Promega Wizard Plus Miniprepsキット)から調製し、制限酵素PmeI(NEB)で消化して大多数の挿入断片を切り取り、次に再連結した。一般的に、これらの反応は次の通りであった。
【化5】
インキュベーションは37℃で1時間であった。次に反応体を65℃で20分間加熱して制限酵素を不活性化し、ライゲーション反応を下記のように組み立てた。
1μl 上の反応液
3μl 10×リガーゼ緩衝液(Promega)
1μl 3 U/μl T4 DNAリガーゼ(Promega)
25μl H2O
30μl
ライゲーションは16℃で少なくとも2時間インキュベートし、次に先に述べたように3μlをJM109に形質転換した。各形質転換の50μlをLB/ampプレート又はこれらのプレートの上に重層されたニトロセルロースフィルター上に平板培養した。37℃で一晩増殖させた後に、フィルターを取り出して40℃にセットしたスライドウォーマー(Fisher Scientific)上のpH5.5の100mMクエン酸ナトリウム中の1mMルシフェリン溶液の1mlの上に置いた。部屋を暗くし、フィルターの発光を観察した。輝くのが観察されたピックからのコロニーはLB/ampプレートから成長させ、プラスミドを単離し、次に制限酵素切断及びシークエンシングによって分析した。大きなカナマイシン挿入断片の切り出しの後、単一のPmeI部位が挿入部位に残る。したがって、PmeI及び他の制限酵素による切断は、挿入部位のマッピングを可能にする。
【0138】
第2の方法において、挿入ライブラリーをゲル精製ルシフェラーゼ断片内で単離し、タンパク質の切り出し及び発現のためにベクターにクローニングした。具体的には、pSPLuc+への転位を先に述べたように達成し、次に、先に述べたように3×5μlを3×100μl高効率JM109に形質転換した。各管からの40μlをLB/amp/kan上に平板培養し、この管の残り並びに他の管からの細胞を50mlのLB/amp/kanに加えて37℃で一晩生育させた。プレートは約7,000の異なるプラスミドのライブラリーに相当する93のコロニーを生成したが、その約1,400の挿入はルシフェラーゼコード配列内にあると予想された。プラスミドは、液体培養の8mlから単離された。ルシフェラーゼ遺伝子と連結するKpnI及びEcoRIによるプラスミドの消化は、ベクター骨格及びルシフェラーゼコード配列と対応する4つの断片をもたらし、それぞれはカナマイシン挿入断片を有すか有さなかった。関係するバンドは長さ3,438bpであり、転位されたルシフェラーゼ遺伝子断片と一致した。ライブラリーからの約2μgのプラスミドをKpnI及びEcoRIで消化し、1μg/mlのエチジウムブロミドを含んでいる1%アガロースゲル上で電気泳動にかけた。3,438bpバンドはUV照明による可視化の後にゲルから切り取り、Wizard PCR Preps(Promega Corp.)を使用してアガロース切片から精製した。次に標準手順に従ってこのDNAをKpnI及びEcoRIで消化したpGEM−3Z(Promega Corp.)にクローニングした。これによりルシフェラーゼ遺伝子はベクター内のLacプロモーターの支配下に置かれる。大多数のカナマイシン挿入断片は、PmeIで切断してライブラリーから切り取った。
2μl 0.25μg/μl pGEM−3Z−luc−kanライブラリー
2μl 10×緩衝液C(Promega)
1.5μl 10 U/μl PmeI(NEB)
14.5μl H2O
20μl
この反応体を37℃で1時間インキュベートし、次に65℃で20分間加熱して下記のように連結した。
2μl 上記消化液
3μl 10×リガーゼ緩衝液(Promega)
1μl 3 U/μl T4 DNAリガーゼ(Promega)
24μl H2O
30μl
ライゲーション反応体を16℃で一晩インキュベートし、次に個々のコロニーを得るためにコンピテントなJM 109に形質転換した。アンピシリンのみ又はアンピシリンとカナマイシンの両方を含んでいるプレート上に平板培養することによって、アンピシリンプレート上の形質転換体の約90%はカナマイシンに感受性であり、したがって挿入断片の切り取りに成功したと推測することができた。個々のコロニーを3mlのLB+100μg/mlアンピシリンで培養し、培養物のルシフェラーゼ活性を分析した。
【0139】
結果
第1の方法については、培養物の約20%はルシフェラーゼ活性が大きく低下しており、このことはトランスポゾンがpSP−Luc+プラスミド内のルシフェラーゼコード領域に挿入されていることと一致している。第2の方法については、個々のコロニーからの培養物の約15%でかなりの活性が観察された。プラスミドは活性のある培養物から調製され、PmeI部位挿入断片のおおよその位置を特定するために制限マッピングを実施した。次にこれらの試料に対して、アイオワ大学DNAシーケンシング施設で標準のジデオキシ配列決定法を実施した。活性クローンの約半分は、ルシフェラーゼコード領域の直ぐ外側に挿入断片を含んでいた。残りは、コード領域内の様々な場所に挿入断片を有していた。上で議論した2つの異なる方法からの結果を合わせたものを下で提示するが、挿入の位置及び残存している活性のおおよその割合が示されている。
【表2】
【実施例3】
【0140】
ヒンジ領域に修飾を有する修飾されたコメツキムシルシフェラーゼ
トランスポゾン突然変異研究によって特定された位置に関係する様々な部位を都合よく挿入するために、コメツキムシルシフェラーゼ遺伝子(cbg69)を修飾して、ヒンジ領域をコードする配列に連続している2つの独特の制限酵素部位、SnaBI(TACGTA)及びSalI(GTCGAC)を生成した。具体的には、2つのオリゴヌクレオチド:
【化6】
を使用してGeneEditor(Promega)によるcbg69遺伝子の修飾を実施した。得られたコメツキムシルシフェラーゼCbg69ssはIle409のValへの1アミノ酸置換を有し、野生型Cbg69の2倍の活性を示すことが明らかになった。Cbg69ssを有すプラスミド(pJLC1ss)は、ヒンジ領域に修飾を有する他のルシフェラーゼを生成するためのテンプレートとして使用した。
そのために、以下のオリゴヌクレオチド対を合成した:
【化7】
各オリゴヌクレオチドは、以下の反応条件を使用してリン酸化した。
オリゴヌクレオチド 30pmol
10× T4ポリヌクレオチドキナーゼ緩衝液 2.5μl
10mM ATP 2.5μl
T4オリゴヌクレオチドキナーゼ(1μ/μl) 0.5μl
水 25μlまで
37℃で30分間インキュベートし、70℃で10分間不活性化する。
【0141】
各リンカーにつき、一対のリン酸化オリゴヌクレオチド(上記反応体からの10μl)を95℃で5分間加熱して1時間で37℃に冷却してアニールした。次に各リンカーをpJLC1ssのSnaBI及びSalI部位にクローニングした。
【0142】
結果
A.コメツキムシルシフェラーゼを残基400の後で修飾してカスパーゼ−3認識部位(DEVD)を含むようにし、Cbg69DEVDが得られた。Cbg69ss及びCbg69DEVDは細菌宿主で発現された。細菌溶解物をカスパーゼ−3の様々な量(0、6.25、12.5、25、50、100又は200ng)或いは200ngのカスパーゼ−3及び0.1mMカスパーゼ阻害剤Ac−DEVD−CHOと混合し、ルシフェラーゼ活性を監視した。図5Aは、カスパーゼ−3濃度の上昇に従って、Cbg69ssのそれではなくCbg69DEVDの活性が減少したことを示す。さらに、カスパーゼ阻害剤の存在下では活性低下は観察されなかった(図5B)。さらに、ルシフェラーゼ活性は時間と共に低下した(図5C)。
【0143】
B.SARSウイルス3CLプロテアーゼはSARSコロナウイルスのシステインプロテアーゼであり、抗SARSウイルス薬剤の潜在的な標的である。2つのコメツキムシルシフェラーゼを残基400の後で修飾して、2つのSARSプロテアーゼ認識部位(Cbg69SARS3及びCbg69SARS6)の1つを含むようにした。Cbg69ss、Cbg69SARS3及びCbg69SARS6は、ウサギ網状赤血球溶解物及び/又はコムギ胚抽出物(Promega)などのin vitro翻訳系を使用して生成した。SARSプロテアーゼは、New England BiolabsからのpMAL精製系を使用して部分的に精製した。コメツキムシルシフェラーゼを含んでいる溶解物をSARSプロテアーゼと混合して、ルシフェラーゼ活性を監視した。図6Cは、室温で1時間のインキュベーションの後、Cbg69ssではなくCbg69SARSは、SARSプロテアーゼ(約0.3μg)で処理したとき非処理の試料と比較して活性が低下したことを示す。
【0144】
C.図6A〜Cで示すように、Asn400の後に様々な挿入部位を有する修飾されたコメツキムシルシフェラーゼは全て活性であった。これらの修飾されたルシフェラーゼは、Cbg69ssと比較して12〜64%の活性を有していた。したがって、コメツキムシルシフェラーゼのヒンジ領域における修飾は、活性を保持している修飾されたルシフェラーゼを与えることができる。
【実施例4】
【0145】
内部エンテロキナーゼ部位を有する修飾されたホタルルシフェラーゼ
ホタルルシフェラーゼのアミノ酸233及び541の後の5アミノ酸挿入は酵素活性の最大分画を保持していた(実施例2)ので、それらの部位を詳細解析のために選択した。プロテアーゼによる切断の後のルシフェラーゼ活性に対する影響を検討するために、GeneEditor(商標)in vitro Site−Directed Mutagenesis System(Promega Corp.)を使用してin vitro突然変異誘発を行い、これらの部位にプロテアーゼ切断部位を挿入した。先ず、標準の手法を使用してルシフェラーゼ遺伝子を発現ベクターpRSET−B(Invitrogen)のNcoI及びHindIII部位の間にクローニングした。luc+遺伝子(図7Aで示すタンパク質配列をコードする)をNcoI−EcoRV断片上でpSPLuc+から切り取り、HindIII部位を埋めて平滑末端にした後にpRSET−B内のNcoI及びHindIIIの間にクローニングした。この構築物は、ルシフェラーゼアミノ酸配列をアミノ末端6Xヒスチジンタグと融合させる。
【0146】
luc+のPro233の後のpRSET−B−luc+にエンテロキナーゼプロテアーゼ切断部位(Asp(4)Lys)を挿入するために、配列
【化8】
のオリゴヌクレオチドを使用した。プラスミドテンプレートを先ず下記のように変性した。
【化9】
この混合物を室温で5分間インキュベートし、次に2μlの2M酢酸アンモニウム及び75μlの95%エタノールを加え、得られた混合物を−20℃で30分間インキュベートした。次に混合物を最高速度の微量遠心機により4℃で5分間遠心分離した。ペレットを次に−20℃の70%エタノール150μlを用いて洗浄し、2分間遠心分離にかけ、減圧下乾燥し、100μlのTEに溶かした。突然変異誘発性オリゴヌクレオチドを、変性させたテンプレートに以下の反応でアニールさせた:
10μl 変性テンプレート(上記)
1μl 2.9ng/μl上部鎖選択オリゴヌクレオチド(0.25pmole)
1μl 28ng/μl上記突然変異誘発性オリゴヌクレオチド(1.25pmole)
2μl アニーリング 10×緩衝液
6μl H2O
20μl
このアニーリング反応体を75℃の水200mlを含んでいるビーカーに置き、次にその水の中で37℃に冷却させた。次に以下の成分を添加した。
5μl H2O
3μl 10×合成緩衝液
1μl 7.7 U/μl T4 DNAポリメラーゼ
1μl 3 U/μl T4 DNAリガーゼ
30μl
この反応体を37℃で90分間インキュベートし、その後GeneEditor(商標)技術マニュアルで記載されているように反応体の5μlをコンピテントなBMH 71−18 mutSに形質転換した。その翌日、得られた培養物からプラスミドを調製してJM109に再度形質転換した。得られた個々のコロニーを拾い、増殖させ、プラスミドを調製した。突然変異体のスクリーニングは、BanII及びSphIでプラスミドを消化し、エチジウムブロミドで染色した6%ポリアクリルアミドゲル(Novex、Invitrogen)上で生成物を電気泳動にかけることによって実施した。消化物は野生型遺伝子(WT)の場合は361bpの断片を生じ、エンテロキナーゼ部位を含んでいる挿入突然変異体では376bpの断片を生じる。このようにして特定された突然変異体は、次に配列決定をすることによって確認された。この実験では、7/8のクローンが所望の挿入を含んでいた。
【0147】
WT luc+遺伝子又はエンテロキナーゼ部位挿入をコードするプラスミドは、BL21(DE3)pLysS(Novagen)に形質転換した。形質転換された培養物を37℃で約0.5のA600まで増殖させ、次に、1mMのIPTGで誘導し、37℃で更に3〜4時間増殖を継続させた。細胞を次にペレットにし、MagneHis樹脂(Promega Corp.)を使用して酵素精製した。一般的には、2mlの細胞を微量遠心機で2分間遠心分離にかけてペレットにした。ペレットは100μlのMagneHis Wash/Binding緩衝液中で再懸濁し、次に、10μlの10×MLR(製品番号V583A)を加えて細胞を溶解した。5μlの1U/μl RQI DNase(Promega Corp.)及び3μlの7U/μl RNase One(Promega Corp.)を溶解細胞に加え、時々混合しながら氷上で10分間インキュベーションした後、溶解物を4℃で5分間微量遠心機で回転させた。40μlのMagneHis樹脂を上清に加え、次に、得られた混合物を時折混合しながら5分間室温でインキュベートした。樹脂は、次に磁場を加えることによって管壁の上で濃縮し、MagneHis Wash/Binding緩衝液中で再懸濁及び磁化のサイクルを3回繰り返して洗浄した。タンパク質は、最後に、pH7.5の100mM HEPES中の500mMイミダゾールの100μlで溶出した。この手順により、WT又は修飾されたタンパク質の約5μgが得られた。
【0148】
修飾されたタンパク質はエンテロキナーゼ部位を取り込んだけれども、対応するプロテアーゼは酵素活性に影響を及ぼさず、またPro233の後で変異タンパク質を切断することもなかった。WT及び変異タンパク質は、タンパク質からの6Xヒスチジンタグの除去を認めるアミノ末端の他のエンテロキナーゼ部位も含んでいた。ゲル分析は、両方のタンパク質でこの部位がエンテロキナーゼによって利用されることを示した。
【0149】
潜在的にエンテロキナーゼ部位の利用をより容易にするGly(3)Asp(4)LysGly(3)部位がPro233の後に挿入されている、他の修飾されたタンパク質を調製した。突然変異誘発は、配列
【化10】
を有する突然変異誘発性オリゴヌクレオチドを利用して上のように実施した。
【0150】
消化反応は次の通りに組み立てた。
【表3】
エンテロキナーゼ(EKMax)及びその10×緩衝液は、Invitrogenから入手した。反応体は室温でインキュベートし、15分時及び30分時に反応体の1μlを100μlのLuciferase Assay Reagent(Promega Corp.)に加えた。各試料は、次にTurner20/20照度計で読み取った。
これからは、以下のデータが得られた:
【表4】
Gly(3)Asp(4)LysGly(3)部位を有する修飾されたタンパク質をエンテロキナーゼで処理すると、ルシフェラーゼ活性は50〜100%増加することがわかった(図7B)。対照的に、エンテロキナーゼはWT酵素の活性に対して影響を及ぼさなかった。したがって、修飾されたルシフェラーゼ骨格のニッキングは、酵素活性を破壊しなかった。さらに、挿入断片のアミノ酸配列は修飾されたタンパク質に対してプロテアーゼによるニッキングにより軽減されるストレスを起こすこともあり、その結果酵素活性が増加する。
【0151】
エンテロキナーゼ部位、即ちProGlyProGly(3)Asp(4)LysGly(3)ProGlyProを含んでいるより大きな挿入断片を、Luc+のPro233の後に挿入した。タンパク質に対する捻り応力を更に高めるためにProGlyProが含まれた。この挿入を作製するために使用したオリゴヌクレオチドは、
【化11】
であった。pRSET−B−Luc+を開始プラスミドとして使用することより、上のように突然変異誘発を実施した。この場合、得られた突然変異体プラスミドは、ウサギ網状赤血球(Promega TnT(登録商標)Coupled Reticulocyte Lysate System)を使用してin vitroで、以下のような反応で翻訳した;
【表5】
反応体は、30℃で1時間インキュベートしてから、下の通りエンテロキナーゼ(EKMax、Invitrogen)で処理した:
【表6】
エンテロキナーゼを加える前に、次にプロテアーゼ添加後の様々な時間に室温で、100μlのLuciferase Assay Reagent(LAR)で1μlを分析した。得られたデータは、図7Cで示す。WT酵素の活性はプロテアーゼの影響を受けなかったが、修飾された酵素はプロテアーゼ処理で不活性化された。
【0152】
LUC+のLys541の後へのエンテロキナーゼ部位の挿入の影響も、測定した。この場合、前記したようにオリゴヌクレオチド
【化12】
をpRSET−B−luc+プラスミド及びGeneEditorキットと使用して、カルボキシル末端から9番目のアミノ酸であるLys541の後にエンテロキナーゼ部位を導入した。突然変異体プラスミドは、上述のそれらと類似の反応で対照のWTと共に転写及び翻訳し、その後エンテロキナーゼで消化した(図7D)。エンテロキナーゼによる修飾された酵素の処理はその活性を約75%減らしたが、WT酵素の活性は変化しなかった。
【実施例5】
【0153】
欠失及び異種挿入を有する修飾されたホタルルシフェラーゼ
in vitro又はin vivoプロテアーゼアッセイで有用なルシフェラーゼチモーゲンを調製するために、また、特定のタンパク質分解、例えばアポトーシスによって引き起こされるかそれに依存する細胞事象を監視する際に、Lys541(550のアミノ酸のうち)の後に挿入された9のアミノ酸を有するホタルルシフェラーゼ突然変異体を構築した。この9つのアミノ酸は5残基のエンテロキナーゼプロテアーゼ部位をコードし、その後に2つのグリシン、次に(DDDDKGGDI;配列番号58)をクローニングするためのEcoRV部位をコードする2つのアミノ酸が続いた。ベクターは、遺伝子の3’末端の外側に、クローニング部位として使用されたEcoRI部位も有していた。この塩基構築物によって特定されたタンパク質がエンテロキナーゼで切断されると、カルボキシ末端の9アミノ酸が除去され、約10%のWT酵素活性を有する酵素が生成された。大腸菌DNAのEcoRV及びEcoRI断片のライブラリーを、基礎ベクター内のこれらの部位の間にクローニングした。100コロニーを選んでルシフェラーゼ活性を分析した。7コロニーは、WTと比較して活性が100〜1000倍低下していることがわかった。これらの7コロニーを培養してプラスミドを調製した。プラスミドは、それぞれ大きさが約0.2から3kbの大腸菌DNAの挿入断片を含むことがわかった。これらのプラスミドをTNTウサギ網状赤血球溶解物内で翻訳すると、分子量のより高いルシフェラーゼをコードすることがわかった。タンパク質の1つのエンテロキナーゼ切断により、ルシフェラーゼ活性が最大で40倍増加することがわかった。最大の活性化を示す修飾されたタンパク質は分子量が約68kDであり、約60の残基がルシフェラーゼに追加されてチモーゲンを形成していることを示している。
【実施例6】
【0154】
環状置換されている修飾されたホタルルシフェラーゼ
Plainkumら(2003年)は、新しいN及びC末端及び元のN及びC末端を連結するプロテアーゼ認識部位を含んでいるペプチドリンカーを有するリボヌクレアーゼAの環状置換形態は、活性部位の位置的閉塞のためにリボヌクレアーゼ活性が低下したことを報告した。Plainkumらは、プロテアーゼによる環状置換リボヌクレアーゼAの切断は、恐らく活性部位の障害を除去することによってタンパク質の活性を増加させることを明らかにした。
【0155】
ルシフェラーゼの場合、N及びC末端は約40オングストローム離れており、その距離は5〜6のアミノ酸に相当する。ペプチド鎖でルシフェラーゼのN及びC末端を連結することは、タンパク質のカルボキシル末端ドメインによって形成された「リッド」ドメインの閉鎖を阻止することによって、その活性を崩壊させる可能性がある。したがって、ホタルルシフェラーゼluc+遺伝子の頭−尾結合二量体を構築した。PCRプライマーは、増幅された上流側プライマーがAsp(234)から始まり、増幅された下流側プライマーがPro(233)から始まるように設計された。上流側プライマーはAsp(234)の直前にメチオニンのATGコドンを含み、下流側プライマーは終止コドンを含んでいた。元のC及びN末端の間の終止コドンを除去してこれらの末端をプロテアーゼ認識部位をコードする配列で連結するために、in vitro突然変異誘発を使用した。得られたPCR産物をクローニングするために、上流側及び下流側の両方のプライマーは制限酵素部位もコードしていた。
【0156】
方法
頭−尾結合(head to tail)luc+二量体は、次のように構築した。ベクターpSPLuc+(Promega Corp.)をNcoIで消化し、T4 DNAポリメラーゼを使用して末端を埋め、平滑末端線状化ベクターはEcoRIで消化した。受容媒体の役目を果たすために、pSPLuc+はXbaIで消化し、T4 DNAポリメラーゼを使用して末端を埋め、次にEcoRIで消化した。第1の消化物からのルシフェラーゼ断片をこのベクターにクローニングし、同じベクター内の2つのluc+遺伝子の頭−尾結合配列を得た。具体的には、pSPLuc+は次のような反応で消化した。
【化13】
反応体は、37℃で1時間インキュベートし、65℃で15分間加熱してから氷上で短時間冷却した。次に、5μlの10mM dNTP及び1μlの9U/μl T4 DNAポリメラーゼ(Promega Corp.)を加えて、反応体を37℃で20分間インキュベートした。Wizard Clean−Upキット(Promega Corp.)を使用して反応体を精製した。Clean−Up樹脂からの50μlにおける65℃での溶出の後、混合物を冷却し、DNAは5μlの10×緩衝液H(Promega Corp.)及び1μlの12U/μl EcoRI(Promega Corp.)を加えることによって消化した。反応体は、37℃で1時間インキュベートし、次に65℃で15分間加熱した。受容ベクターは、次に以下のように調製した。
【化14】
上の反応体を37℃で1時間インキュベートし、次にPromega Wizard Clean−Upキットを使用して精製し、溶出は65℃の50μl中で行った。以下を精製したDNAに加えた。
【化15】
反応体は、37℃で20分間インキュベートし、次に上で述べたように精製した。5μlの10×緩衝液H及び1μlの12U/μl EcoRIを、Clean−Up樹脂からの溶出液に加えた。反応体は、37℃で1時間インキュベートし、次に65℃で15分間加熱して制限酵素を不活性化した。このDNAを次に下の通り上記の消化したDNAと混合した:
15μl XbaI切断、埋める、EcoRI切断、pSPLuc+加熱
25μl NcoI切断、埋める、EcoRI切断、pSPLuc+加熱
5μl 10×リガーゼ緩衝液(Promega Corp.)
2μl 3U/μl T4 DNAリガーゼ
16℃で一晩のライゲーションの後、1μlを高効率コンピテント大腸菌JM109(Promega Corp.)に形質転換し、細胞をLB/ampプレートに平板培養した。正しい大きさのプラスミドを含んでいる形質転換体を特定した。それらの形質転換体を展開し、そこからプラスミドを単離し、制限酵素消化によってプラスミドの同一性を確認した。
【0157】
新しいN末端がAsp(234)に、新しいC末端がPro(233)にある置換されたルシフェラーゼのPCR増幅のために、上で構築された頭−尾結合二量体Luc+DNAをテンプレートとして使用した。この増幅で使用されたプライマーは、下記配列を有した。
上流側プライマー
【化16】
下流側プライマー
【化17】
増幅反応は、次の通りであった。
5μl 10×PfuUltra緩衝液(Stratagene)
1μl 10mM dNTP
1μl 5ng/μl 上のLuc+二量体構築物DNA
1μl 100ng/μl 上流側プライマー
1μl 100ng/μl 下流側プライマー
40μl H2O
49μl
反応体を混合し、鉱油を重層し、95℃のPE480サーマルサイクラーに入れた。この温度で2分後に、1μlの2.5U/μl PfuUltra DNAポリメラーゼ(Stratagene)を加え、95℃で30秒、50℃で30秒、72℃で1分のサイクルを20回繰り返し、その後ブロックを4℃にした。完了した反応体は次にPromegaのWizard PCR Prepsキットを使用して精製し、Wizard樹脂から50μlのH2Oで溶出した。生成物に取り込まれたPCRプライマーは、NdeI(上流側プライマー)又はBamHI(下流側プライマー)のための部位を有する。PCR産物をこれらの酵素で消化し、下の通りT7発現ベクターpET−3a(Novagen)にクローニングした。
【表7】
【0158】
上の反応体を37℃で1時間インキュベートし、次にそれぞれPromega Wizard Clean−Upキットを使用して精製し、DNAの溶出は50μlのTE中で65℃で行った。2つの精製されたDNAを混合し、下の通り連結した。
5μl 10×リガーゼ緩衝液
20μl 溶出1
10μl 溶出2
2μl 3U/μl T4 DNAリガーゼ
13μl H2O
50μl
ライゲーション反応体を16℃で2時間インキュベートし、次に5μlをコンピテントなJM109に形質転換し、細胞をLB/ampに平板培養した。適当な大きさのプラスミドを含んでいるコロニーを展開し、プラスミドを調製し、正しく挿入されているかについて各調製物を制限消化により確認した。プラスミドはPCR産物の挿入を含んでいるのがわかったので、これは、終止コドンが除かれ2つのルシフェラーゼ遺伝子部分を分けている接合部でC及びN末端を連結している、in vitro突然変異誘発のための基礎ベクターとして使用した。
【0159】
最初の突然変異誘発は、Asp(4)Lysのカルボキシル末端側を切断するプロテアーゼエンテロキナーゼの認識部位を含んでいる突然変異誘発性オリゴヌクレオチドを利用し、Promega Corp.からのGene Editorキットを使用して実施した。このオリゴヌクレオチドは、下記配列を有していた。
【化18】
突然変異誘発手順の第2回目の形質転換からの6コロニーを個々に増殖させて、それらからプラスミドを調製した。これらのプラスミドは、TnTウサギ網状赤血球溶解物(Promega Corp.)内での共役転写/翻訳による突然変異誘発性オリゴヌクレオチドの取り込みについてスクリーニングした。正しい突然変異体は、ルシフェラーゼドメインのC及びN末端が融合し、完全長ルシフェラーゼタンパク質を生成した。翻訳反応は、次のように実施した。
25μl TnTウサギ網状赤血球溶解物
2μl TnT反応緩衝液
1μl T7 RNAポリメラーゼ
1μl 完全アミノ酸配合
1μl Fluorotect Lys tRNA
1μl 40U/μl rRNasin
5μl ミニプレップDNA
14μl
50μl
翻訳反応体は、30℃で60分間インキュベートしてから、下の通りエンテロキナーゼ(EK)(EKMax、Invitrogen)で処理した(又は処理しなかった)。
2μl 10×EKMax緩衝液
5μl 上の翻訳反応体
+/−1μl 1U/μl EKMax
12μl H2O
20μl
これらの消化は室温で30分間行われ、その後1μlを100μlのルシフェラーゼ検定用試薬(Promega Corp.)に加えて検定した。データ収集は、Turner20/20照度計で行った。5μlの4×SDS試料緩衝液を各反応体の残りに加え、試料を65℃で2分間加熱した。次に試料を4〜20%Novexトリス−グリシンゲルで電気泳動にかけ、ゲルは高感度のMolecular Dynamics FluorImagerでスキャンした。結果は、融合した完全長タンパク質は6クローンの内の2つで形成されたことを示し、突然変異誘発が成功したことを示した。さらに、融合した変異タンパク質の活性は、エンテロキナーゼによる処理で約150倍増加した。さらに、ゲルは、プロテアーゼが完全長タンパク質を消化して断片化したことを示した。
【0160】
蛍光リジン誘導体の取り込みによって標識されていない突然変異体ルシフェラーゼの活性に及ぼすEK処理の影響を検討するために、翻訳反応を上記のように実施したが、Fluorotect Lys tRNAは反応から省略された。この場合、EKで酵素を処理するとルシフェラーゼ活性の約90倍の活性化が観察された(図7)。活性化の後、変異酵素はWT活性の約0.5%を回復した。
【0161】
2つのルシフェラーゼドメインの間にカスパーゼ−3 DEVD切断部位を挿入するために、他の突然変異誘発を実施した。Promega Gene Editorキットは、以下の突然変異誘発性オリゴヌクレオチドと使用された。
【化19】
【0162】
この場合、所望の突然変異体は5/8クローンで見られ、これらはin vitro転写/翻訳によってスクリーニングした。カスパーゼ−3による活性化倍率は、エンテロキナーゼで以前に観察された活性化倍率より高いことが明らかになった。また、切断によって回復された活性の割合も高くなっていた。
【0163】
in vitro翻訳は、Promega TnTウサギ網状赤血球溶解物を使用して、カスパーゼ−3 DEVD切断部位を含んでいる置換されたルシフェラーゼをコードするプラスミド又はWTルシフェラーゼを含んでいる反応で実施した。これらの反応体の一部を次にカスパーゼ−3(100単位、BioMol)で消化し、図9〜11で示すデータが生成された。WT酵素の活性はプロテアーゼの影響を受けなかった。対照的に、変異酵素の活性はカスパーゼ−3による処理で大きく増加した。活性化倍率はこの場合約500倍であり、活性化された酵素の活性はWTの活性の約17%であった。
【0164】
置換された酵素のカスパーゼ−3活性を検出する能力も、発光プロテアーゼアッセイで検討した。カスパーゼ反応は以下のように実施した。
10μl 2×カスパーゼ緩衝液
5μl in vitro翻訳タンパク質
1μl 希釈カスパーゼ−3
4μl H2O
20μl
反応体は9.6から2333pgのカスパーゼ−3を含み、これらは室温で90分間インキュベートし、その後1μlを取り出して100μlのルシフェラーゼ検定用試薬に加え、Turner 20/20照度計で読み取った。図9Aは、得られたデータを示す。プロテアーゼの低量点を再プロットすると(図9B)、このアッセイで低ピコグラムのカスパーゼ−3を検出することができることが示される。さらに、インキュベーション時間を90分から一晩に長くすると、アッセイの感度は更に4倍高まった(データは示さず)。
【0165】
置換されたルシフェラーゼの合成及び活性化も、TnT Wheat Germ抽出物(Promega Corp.)で検討した。反応には以下が含まれた。
25μl TnT T7 WG抽出物
2μl TnT反応緩衝液
1μl T7 RNAポリメラーゼ
1μl アミノ酸配合
1μl 40U/μl rRNasin
5μl 50ng/μlルシフェラーゼプラスミド
15μl H2O
50μl
反応体は30℃で90分間インキュベートした後、以下の通りプロテアーゼで消化した。
10μl 2×緩衝液(100mM HEPES pH7.5、200mM NaCl、0.2%CHAPS、2mM EDTA、20%グリセリン、20mM DTT)
10μl in vitro翻訳反応体
1μl 1U/μl EKMax又は1μl 100U/μl カスパーゼ−3
【0166】
プロテアーゼ消化物は室温でインキュベートし、その後様々な時間で1μlを100μlのルシフェラーゼ検定用試薬に加え、Turner 20/20照度計で読み取った。この実験で、カスパーゼ−3は置換カスパーゼ−ルシフェラーゼの活性をWTのそれの約4分の1まで約3000倍高め、EKはEK−ルシフェラーゼの活性をWTの約1.1%まで約300倍高めた(図8及び11)。一例ではDEVDG、他ではDDDDGで、両方の挿入断片は同じ大きさである点に注意する。したがって、ルシフェラーゼの3つのN末端アミノ酸及び6つのC末端アミノ酸を元のタンパク質の末端からそれぞれ置換することによって、より長い配列が取り込まれる可能性がある。このことは、少なくとも14〜15のアミノ酸部位が2つのルシフェラーゼドメインの間に取り込まれるのを可能にするはずである。上で言及した9つの残基は対応する結晶構造に現れず、したがって非常に柔軟であり、酵素活性に悪影響を及ぼさずに置換することが可能な点に注意する。
【実施例7】
【0167】
追加の環状置換構築物
A.PSAは、配列Ala−Asn−Lys−Ile−Ser−Tyr−Gln−Ser−Ser−Ser−Thr−Glu(配列番号21)内のGln及びSerの間でSemenogelin Iを切断するプロテアーゼである。PSAの切断基質を有する修飾されたルシフェラーゼを生成するために、関連する12量体ペプチドAla−Asn−Lys−Ala−Ser−Tyr−Gln−Ser−Ala−Ser−Thr−Glu(配列番号22)のオリゴヌクレオチドを、実施例6で記載したプラスミド構築物内のXhoI及びNcoI部位の間にクローニングした。以下の配列
【化20】
を有するオリゴヌクレオチドを、以下の配列を有するオリゴヌクレオチドにハイブリダイズした。
【化21】
ハイブリダイズされたオリゴヌクレオチドはXhoI及びNcoI適合末端を有する二本鎖断片を生成するが、NcoI部位は改造されているのに対してXhoI部位は破壊されている。ベクターをXhoI及びNcoIで消化してアニールしたオリゴヌクレオチドに連結し、次に大腸菌に形質転換した。ミニプレップDNAを個々のコロニーから調製し、プロテアーゼ部位を含んでいるオリゴヌクレオチドの取り込みを示す、XhoIではなくNcoIによる消化についてプラスミドをスクリーニングした。所望の構築物はコムギ胚(WG)翻訳抽出物又はウサギ網状赤血球溶解物内でin vitroで翻訳し、得られたタンパク質は精製PSA(シグマ)で処理した。翻訳を実施した。開裂反応は以下の通り実施した。
【表8】
反応体は室温で20分又は40分間インキュベートした。各反応の1μlを100μlのルシフェラーゼ検定用試薬(LAR)に加え、光出力をTurner20/0照度計で記録した。下記データを得た。
【表9】
PSAの添加は、光出力の実質的な増加をもたらした。20分時には、修飾されたルシフェラーゼの活性化倍率はウサギ網状赤血球溶解物内で合成された修飾されたルシフェラーゼの場合は658倍であり、コムギ胚抽出物で合成された修飾されたルシフェラーゼの場合は1,110倍であった。
【0168】
B.PreScissionプロテアーゼは、GST(グルタチオンSトランスフェラーゼ)及びライノウイルス3Cプロテアーゼ(Amersham)で構成される融合タンパク質である。このプロテアーゼは、配列Leu−Glu−Val−Leu−Phe−Gln−Gly−Pro(配列番号25)内のGln及びGly残基の間で切断することができる。この配列を規定しているオリゴヌクレオチドが設計され、下記配列(トップストランド)
【化22】
を有していた。これらのオリゴヌクレオチドのアニーリングはXhoI及びNcoI適合末端を有する二本鎖断片を生成し、XhoI部位は保持されているのに対してNcoI部位は破壊されている。上記の例の場合のように、アニールされたオリゴヌクレオチドは、XhoI及びNcoIで切断されたベクターにクローニングされた。所望のクローンの濃縮のために、形質転換の前にライゲーション混合体をNcoIで再切断した。所望のプラスミドを選択して、前記したようにウサギ網状赤血球溶解物内でin vitro翻訳を行った。消化反応体は、以下の通り調製した。
【表10】
反応体は室温でインキュベートし、その後様々な時間で1μlを100μlのLARに加え、試料をTurner 20/20照度計で読み取った。以下のデータが生成された。
【表11】
PreScissionプロテアーゼによるルシフェラーゼの活性化は速やかに起こり、プロテアーゼの存在下で4,000倍を超える発光の増加をもたらした。
【0169】
C.真核細胞が含まれない溶解物内で合成された置換されたルシフェラーゼのタンパク分解性処理によって高度な活性化が観察されたが、非融合タンパク質が大腸菌で合成されたときの活性化の程度は非常に小さかった。面白いことに、大腸菌調製物の部分精製は、プロテアーゼによって活性化される能力の高くなったタンパク質をもたらした。細菌細胞から環状置換されたルシフェラーゼを有効に精製するために、ベクターpGEX−6P3(Amersham)内でカスパーゼ−3部位を有する環状置換されたルシフェラーゼがGSTに融合されているベクターを調製した。PCR反応体は以下を含んでいた。
5μl 10×PfuUltra緩衝液
1μl 10mM dNTP
1μl 5ng/μlカスパーゼ−3部位プラスミド
1μl 100ng/μl 上流側オリゴヌクレオチド
1μl 100ng/μl 下流側オリゴヌクレオチド
40μl H2O
50μl
1μlの2.5μ/μl PfuUltra DNAポリメラーゼ(Stratagene)を加えてPCRを開始し、95℃で30秒、50℃で30秒、72℃で1分のサイクルを20回繰り返し、その後4℃にした。
【0170】
上流側オリゴヌクレオチドはBamH1を含み、下記配列を有し
【化23】
下流側オリゴヌクレオチドはEcoRI部位を含み、下記配列を有す
【化24】
得られたPCR産物はEcoRI及びBamH1で消化し、ベクター内のこれらの部位の間にクローニングすると、GSTとルシフェラーゼとのインフレーム融合がもたらされる。所望のプラスミドを特定し、大腸菌系統Rosetta(Novagen)に形質転換した。細胞をLB培地で増殖させ、IPTGを1mMまで添加することによって誘導した。最良の増殖条件は25〜26℃で一晩の誘導であることが明らかになった。細胞を収集して超音波処理で溶解した。遠心分離による清浄の後、上清を固定化グルタチオンを含んでいるカラムに加え、遊離のグルタチオンを含んでいる緩衝液で溶出した。融合タンパク質の収率は、初期の培養物1リットルにつき約1ミリグラムであった。カスパーゼ−3による活性化は、活性化反応の条件に従い約1,200倍以上で、最高50,000倍(氷上で一晩の活性化)であった。
【0171】
D.SARSウイルスプロテアーゼ部位TSAVLQSGFR(配列番号19)を含んでいる3つの環状置換されたルシフェラーゼを生成した。2つはコメツキムシのルシフェラーゼ(CP1:R=Asn401及びCP2:R=Arg223)で1つはホタルルシフェラーゼ(CP:R=Asp234)のものである。CP2は、ホタルルシフェラーゼの位置234と対応するコメツキムシルシフェラーゼの位置に挿入を有する。
【0172】
SARSウイルスプロテアーゼ部位を有する環状置換されたコメツキムシルシフェラーゼは、次のように構築された。先に述べたようにNdeI及びBamHI部位の間に挿入突然変異体cbg69SARS3遺伝子を、及びSnaBI及びSalIの間にSARSウイルスプロテアーゼ部位をコードする配列を含んでいるプラスミドpJLC33を開始ベクターとして使用した。以下のプライマーセットを使用して、野生型cbg69を含んでいるpJLC1からPCR断片を増幅した。
【化25】
【0173】
CP1−a(又はCP2−a)のPCR産物をSalI及びBamHIで消化し、pJLC33のそれぞれの部位にクローニングし、pJLC−cp1a(又はpJLC−cp2a)を得た。CP1−b(又はCP2−b)のPCR産物をNdeI及びSnaBIで消化し、pJLC−cp1a(又はpJLC−cp2a)のそれぞれの部位にクローニングした。得られたプラスミドpJLC47(又はpJLC48)は、SARSウイルスプロテアーゼ部位を有するコメツキムシルシフェラーゼの環状置換された突然変異体1(又は2)を含む。
【0174】
置換されたホタルルシフェラーゼについては、置換されたベクターは、元のN及びC末端のためにDNAを分離しているXhoI及びNcoI部位を有するリンカーを取り込むように修飾した。このリンカーは、
【化26】
であり、これは第1のドメインのC末端から6アミノ酸を除去し、第2のドメインのN末端から3アミノ酸を除去する。SARSウイルスN末端の自己切断部位は、SITSAVLQSGFRKMA(配列番号53)である。この配列を規定しているオリゴヌクレオチドは、次のように設計された。
【化27】
アニールされたオリゴヌクレオチドはNcoI部位を保持し、XhoI部位を欠く。アニールされ消化されたオリゴヌクレオチドは、上記したように基礎ベクターにクローニングした。
【0175】
SARSウイルスプロテアーゼ部位を有する3つの環状置換されたルシフェラーゼCbg69CP1、Cbg69CP2及びFfCPの全ては、ウサギ網状赤血球溶解物及び/又はコムギ胚抽出物(Promega)などのin vitro翻訳系を使用して生成した。SARSウイルスプロテアーゼは、New England BiolabsからのpMAL精製系を使用して部分的に精製した。変異ルシフェラーゼを含んでいる溶解物をSARSウイルスプロテアーゼと混合して、ルシフェラーゼ活性を監視した。約0.3μgのSARSウイルスプロテアーゼとの室温で1時間のインキュベーションの後、Cbg69CP2及びFfCPはそれぞれ20〜30倍及び60〜200倍に活性化された(図12)が、Cbg69CP1は活性化されなかった(データは示さず)。
【0176】
E.活性カスパーゼ−3を生成するためのプロカスパーゼ−3の活性化は、生細胞におけるアポトーシス誘導の代理である。修飾されたルシフェラーゼが細胞内のアポトーシスを監視するために使用することができるかどうか確認するために、カスパーゼ−3切断部位を含んでいる環状置換されたルシフェラーゼをCMVプロモーターの制御下の哺乳類発現ベクターにクローニングし、一過性形質移入を通してHela細胞に導入した。細胞を次にタンパク質TRAILで処理し、死受容体の活性化を通してアポトーシスを誘発して活性カスパーゼ−8を形成し、これは次にプロカスパーゼ−3を活性化してカスパーゼ−3にする。したがって、活性カスパーゼ−3の出現は、ルシフェラーゼ基質が切断されて酵素によって活性化されるに従い、発光の増加を伴うはずである。
【0177】
カスパーゼ−3切断部位を含んでいる置換されたルシフェラーゼのPCR断片は、NheI及びEcoRIのための部位を含んでいるプライマーを使用して作製され、ベクターpCI−neo(Promega)のこれらの部位の間にクローニングした。増幅は、上記したように下記上流側プライマーを用いて実施した。
【化28】
得られた構築物は、一般式ANNATGGの最適コザック配列を有していた。DNAは、TransFast形質移入試薬(Promega)を使用してHela細胞に形質移入し、DMEM+10%Cosmic Calf Serum中の1μg/μlのTRAILタンパク質(Biomol)を加えることによりアポトーシスを開始した。一部のウェルを、初期SV40プロモーター/エンハンサーの制御下の天然のホタルルシフェラーゼ遺伝子(非置換)を運ぶプラスミドpGL3−controlで形質移入した。示された時間に100μlのBright−Glo試薬をウェルに加え、Orion照度計(0.5秒の読取り)で発光を記録した。
【表12】
【0178】
図13のデータは、置換されたルシフェラーゼ−カスパーゼ−3プラスミドを受け取る細胞では、発光がなくなってから1〜2時間の間にTRAILタンパク質が発光活性を誘導することを示す。この影響は、培地だけを含んでいるウェルでは見られない。pGL3−controlプラスミドを含んでいるウェルは、培地単独と培地+TRAILとの間で相違を示さなかった。培地を変えたときに見られるLUの全般的増加の原因はタンパク合成の誘導である可能性があり、pGL3−controlではルシフェラーゼは活性型であるが、pCI−neo−カスパーゼプラスミドではルシフェラーゼは不活性型である。形質移入のストレスによる死細胞バックグラウンドが観察されているので、この培地単独の場合に見られる小さな増加の原因は、アッセイの全過程にわたる死細胞の蓄積にあるかもしれない。
【実施例8】
【0179】
修飾されたルシフェラーゼによる細胞ベースのアッセイ
分子内制御をコードし、カスパーゼ−3活性を検出するベクターを提供するために、本発明の融合タンパク質をコードするベクターを調製した。Renillaルシフェラーゼ(対照)を、残基400の後にDEVDを含んでいる修飾されたコメツキムシルシフェラーゼ(Cbg69DEVD)のN末端又はC末端に融合した。リンカー配列(Gly(2)SerGly(4)SerGly(4)SerGly(2))を2つのタンパク質の間に置いた。
【0180】
rLuc−リンカー−Cbg69DEVD融合を作製するために、一対のオリゴヌクレオチド
【化29】
を使用してプラスミドpJLC6から完全長Renillaルシフェラーゼ遺伝子(rLuc)を増幅した。得られたPCR断片をNdeI及びAseIで消化して、Cbg69DEVDをコードするpJLC23のNdeI部位にクローニングした。
【0181】
Cbg69DEVD−リンカー−rLuc融合を作製するために、一対のオリゴヌクレオチド
【化30】
を使用してプラスミドpJLC23から完全長Cbg69DEVD遺伝子を増幅した。得られたPCR断片をNdeI及びAseIで消化して、rLucを含んでいるpJLC6のNdeI部位にクローニングした。
【0182】
図14は、各融合タンパク質がRenillaルシフェラーゼ並びにコメツキムシルシフェラーゼの活性を有していたことを示す。
【実施例9】
【0183】
結合して機能性ルシフェラーゼを形成するルシフェラーゼ断片の能力の評価
一実施形態では、本発明は、ルシフェラーゼの2つの独立した断片が互いに相補して機能性タンパク質を生成することのできる系を提供する。
【0184】
材料及び方法
ルシフェラーゼのN及びC末端の断片の、in vitro及びin vivoで結合して機能性ルシフェラーゼタンパク質を形成する能力を評価するために、3つの構築物が設計された(図15)。ホタルルシフェラーゼ遺伝子のN末端の699ヌクレオチド(アミノ酸1〜233)を、順方向プライマー
【化31】
及び逆方向プライマー
【化32】
を使用して以下の条件でpSP−luc+(Promega Corp.)から増幅した。パーキンエルマー2400サーマルサイクラーを使用して、95℃で2分間、1サイクルが95℃で30秒間、50℃で30秒間及び72℃で2分間を25サイクル、その後72℃で10分間。NheI制限部位は順方向プライマーの5’末端へ導入し、BamHI制限部位は逆方向プライマーの5’末端へ導入した。得られたN末端ルシフェラーゼ断片は次に確立された技術(Sambrookら、1989年)を使用してpBINDベクターのNheI及びBamHI制限部位にクローニングし、発現ベクターpJLC62(n luc)を得た。
【0185】
同様に、ホタルルシフェラーゼ遺伝子のC末端の951ヌクレオチド(アミノ酸234〜550)を、順方向プライマー
【化33】
及び逆方向プライマー5’TTGGCGCGCCGGATCCTTACACGGCGATCTTTCCGCCCTTCTTG3’(配列番号63)を使用して、N末端のクローニングについて前記したのと同じPCR条件下でpSP−luc+から増幅した。NheI及びBamHI制限部位はN末端プライマーについて前記したようにプライマーに導入し、C末端のルシフェラーゼ断片はpBINDベクターのNheI及びBamHIにクローニングして発現ベクターpJLC63(c luc)を得た。
【0186】
ルシフェラーゼ遺伝子全体(1650ヌクレオチド、550アミノ酸)を、順方向プライマー
【化34】
及び、逆方向プライマー
【化35】
を使用し、また上記同じPCR条件を使用してN及びC末端クローンで使用されたのと同じ方法でpBINDベクターにクローニングした。得られた発現ベクターpJLC64(完全長FF)をタンパク質相補性実験のための対照として使用した。
【0187】
全ての構築物は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して、正しいタンパク質の大きさについて検証した。
【0188】
in vitroタンパク質相補性試験は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して実施した。翻訳の後、各試料の2μlを100μlのルシフェラーゼ検定用試薬に加え、Veritas照度計を使用して発光を測定した。
【0189】
in vivo相補性試験は、チャイニーズハムスター卵巣(CHO)及び293のヒト胎生期腎臓組織培養細胞で実施した。CHO又は293の細胞のいずれかの組織培養細胞を6穴の組織培養プレートに播種し、37℃で一晩、5%CO2で増殖させ、翌日80%のコンフルエンシーで形質移入した。形質移入は、TransFast(商標)形質移入試薬(Promega Corporation)を使用してメーカーの推奨に従って実施した。つまり、対照反応では、各形質移入の最終濃度が2μgの全DNAとなるように、pJLC62、pJLC63又はpJLC64のいずれかの1μgを1μgのpBIND対照プラスミド(ホタルルシフェラーゼ遺伝子を含まない元のベクター)で形質移入した(3μl TransFast(商標)試薬/μg DNA)。タンパク質相補性試験については、1μgのpJLC62及び1μgのpJLC63を同じプロトコルに従って形質移入した。形質移入後24時間時に、細胞をトリプシン処理して各形質移入条件のために2群に分けた。250μlの1×Passive Lysis緩衝液(Promega Corporation、PLB)を1つの群に加え、250μlの1リン酸緩衝生理食塩水(PBS)を他の群に加えた。PLB群は組織培養細胞の溶解を確実にするために−80℃の凍結融解で1回処理し、PBS群は凍結融解をしなかったので非溶解細胞が維持された。全ての群からの発光は、Dual−Luciferase (登録商標)レポーターアッセイシステムを使用し、メーカーの推奨に従って測定した。基本的には、各群からの20μlを3反復の白の96穴プレートに加え、Veritas照度計でアッセイを実施した。全てのホタルルシフェラーゼデータは、Renillaルシフェラーゼシグナルに正規化した。
【0190】
結果
図15で示す3つの構築物の全ては、正しい大きさのタンパク質を与えた(図16A)。本明細書で先に記載したプロテアーゼ切断後の環状置換ホタルルシフェラーゼの活性化は、ルシフェラーゼの断片が酵素活性を相補し且つ再構成することができることを示唆した。図16B(同じTnT反応におけるルシフェラーゼのN及びC断片)で見られるように、N及びC末端のルシフェラーゼ断片のin vitroタンパク質相補性は、完全長ルシフェラーゼタンパク質と比較して機能性タンパク質を与えた。さらに、in vivoタンパク質相補性は、CHO及び293組織培養細胞の両方で起こった(図17)。たとえ組織培養細胞が溶解しなかった場合でも類似の傾向が見られた(PBS;データは示さず)。
【実施例10】
【0191】
モジュレーター系におけるルシフェラーゼ融合タンパク質の非共有結合的結合の検出
一実施形態では、本発明は、2つの部分の結合を誘発又は強化、或いは抑制する外因性因子(エフェクターA)、及び任意選択に前記2つの部分の結合をそれぞれ解離又は強化する他の外因性因子(エフェクターB)を有するモジュレーター系を提供する。例えば、そのような系は、ルシフェラーゼに融合されたFKBP及びFRBの結合の誘導物質としてラパマイシンを、また結合の解離物質としてFK506を使用することができる。
【0192】
A.ルシフェラーゼモジュレーター系を示すin vitro実験
材料及び方法
ヒトコドン最適化ホタルルシフェラーゼ遺伝子(luc2.0)を、順方向プライマー
【化36】
及び逆方向プライマー
【化37】
を使用してポリメラーゼ連鎖反応(PCR)により以下の条件でpGL4.10(luc2)(Promega Corporation)(配列番号66)から増幅した:95℃で2分間、次に1サイクルが95℃で30秒間、50℃で30秒間及び72℃で2分間のものを25サイクル、72℃で10分間の最終展開。ルシフェラーゼタンパク質とのN末端融合の生成を容易にするために、NcoI及びEcoRV制限酵素部位を順方向プライマーの5’末端に導入した。ルシフェラーゼタンパク質とのC末端融合の生成を容易にするために、SnaBI、NotI及びSacI制限酵素部位を逆方向プライマーの5’に導入した。5’及び3’末端上の更なるクローニング部位を有する増幅されたルシフェラーゼ遺伝子をルシフェラーゼT7対照ベクター(Promega Corp.、Cat No#L4821)のHindIII/SacI部位にクローニングし、対照ベクターに通常存在するルシフェラーゼ遺伝子を置換した。得られたベクターは、pJLC65と呼ばれた。in vitro発現ルシフェラーゼT7対照ベクターへのクローニングの一般的スキームは、図18で見ることができる。
【0193】
いくつかの発現構築物がpJLC65ベクターを使用して作製された;ホタルルシフェラーゼへのFRBのN末端融合(pJLC66)、ホタルルシフェラーゼへのFKBPのC末端融合(pJLC67)及びホタルルシフェラーゼへのFRB(N末端)及びFKBP(C末端)の二重融合(pJLC68)。FRBは、
【化38】
の合成遺伝子を含んでいるBlue Heronからのプラスミドから得られた。FRBは、5’末端のNcoI制限酵素部位及び3’末端のEcoRV制限部位を使用してBlue Heronベクターから切断され、公知の分子生物学的技術(Sambrookら、1989年)を使用してルシフェラーゼ遺伝子のN末端にクローニングした。
【0194】
FKBPは、
【化39】
の合成遺伝子を含んでいるBlue Heronからのプラスミドから得られた。FKBP断片がルシフェラーゼ遺伝子のC末端にクローニングされるように、FKBPを遺伝子の5’末端のSnaBI制限酵素部位及び3’末端のNotI制限酵素部位を使用してBlue Heronベクターから切断した。二重融合は、FRB及びFKBPをそれぞれN末端及びC末端に含んでいた。
【0195】
4つのルシフェラーゼ構築物は、メーカーのプロトコルに従ってTnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling System(Promega Corporation)と一緒に使用して、発現されたタンパク質の正しい大きさについて評価した。簡潔には、4つの各反応において、1μgの適当なDNAをFluoroTect(商標)GreenLys tRNAを含む50μlの反応体に加えた。各反応体(5μl)からの試料を、NuPAGE(登録商標)技術ガイド(バージョンE、IM−1001)で記載されているように、1×NuPAGE(登録商標)MES SDSランニング緩衝液を使用して10%のNuPAGE(登録商標)Novex Pre−Cast Bis−トリスゲル(Invitrogen Corporation)に流した。FluorImager SI(Molecular Dynamics)を使用してゲルを画像化した。
【0196】
in vitroアッセイについては、上述の各TnT(登録商標)反応からの5μlを、0.2μMラパマイシン(BioMol)を含んでいるか含まない95μlの1×Passive Lysis緩衝液(Promega Corporation)に別々に加えた。ラパマイシンの添加の後、各試料からの10μlを100μlのルシフェラーゼ検定用試薬(TnT(登録商標)システムを設置)に加え、Turner20/20照度計(Turner BioSystems)を使用して発光を測定した。
【0197】
FRB及びFKBPの間の相互作用が調整することができるかどうか研究するために、ラパマイシンと競合して融合パートナー間の相互作用を抑制することが公知であるFK506をin vitro実験で使用した。二重融合FRB−luc2−FKBPを先に述べたように転写及び翻訳した。翻訳の後、試料の4μlを5μlの2×FLICE緩衝液(100mM HEPES、pH7.5、200mM NaCl、0.2%CHAPS、2mM EDTA、20%グリセリン、20mM DTT)及び1μlのラパマイシン(10nM)、及び0、1、2、5、10、20及び40nM(0、0.82、1.64、4.1、8.2、16.4及び32.8ng/mlのタクロリムスに相当)の様々な濃度のFK506(タクロリムス、Antibioticplus.com)と混合した。試料は室温で15分間インキュベートし、その後試料の5μlを100μlのルシフェラーゼ検定用試薬で希釈してTurner20/20照度計で発光を測定した。
【0198】
結果
下記4つの構築物を調製した:luc2(ホタルルシフェラーゼをコードする;550アミノ酸)、FRB−luc2(FRB及びホタルルシフェラーゼの融合をコードする;644アミノ酸)、luc2−FKBP(ホタルルシフェラーゼ及びFKBPの融合をコードする;657アミノ酸)及びFRB−luc2−FKBP(二重融合をコードする;771アミノ酸)。4つの構築物(3つは対照、1つは二重融合)は、TnT(登録商標)Coupled Wheat Germ Extract SystemをFluoroTect(商標)GreenLys in vitro Translation Labeling Systemと一緒に使用して、発現されたタンパク質の正しい大きさについて評価した。4つの構築物の全ては、正しい大きさのタンパク質を与えた(図19)。
【0199】
次に2つの融合パートナーFRB及びFKBPの間の相互作用をin vitro系で検出するために、これらの構築物を実験で使用した。誘導物質ラパマイシンの存在下で、2つの融合パートナーが結合して発光は減少するはずである。対照反応と比較して、ラパマイシンの添加により二重融合FRB−luc2−FKBPでは相対発光は20倍減少した(図20A)。したがって、ラパマイシンの存在下では、ルシフェラーゼとの二重融合における2つの結合パートナーは結合し、それによってルシフェラーゼ酵素を制限し、その結果酵素はルシフェリン基質と有効に相互作用することができなくなった。逆に、FK506の添加量が増加すると、二重融合反応の発光はFK506の増加の量に応じて増加した(図20B)。
【0200】
B.他のルシフェラーゼモジュレーター系
材料及び方法
クローニングは、以下の例外を除いて先に述べたように実施した。赤色コメツキムシ遺伝子(cbr)は、順方向プライマー
【化40】
及び、逆方向プライマー
【化41】
を使用してpCBR−Basic(Promega Corporation)から増幅した。EcoRV部位を順方向プライマーの5’末端に、BglIIを逆方向プライマーの5’末端に導入し、その後、対応する増幅された断片をpJLC68の対応する部位にクローニングした。緑色コメツキムシ遺伝子(cbg)は、順方向プライマー
【化42】
及び、逆方向プライマー
【化43】
を使用してpCBG68−Basic(Promega Corporation)から増幅した。赤色コメツキムシでpJLC68ベクターにクローニングしたのと同じ制限部位をこれらのプライマーに導入した。Renillaルシフェラーゼ遺伝子(Rluc)は、順方向プライマー
【化44】
及び、逆方向プライマー
【化45】
を使用してphRL−null(Promega Corporation)から増幅した。pJLC68ベクターのEcoRV(SnaBIとの平滑末端ライゲーション)及びBglII上でクローニングするために、SnaBI部位を順方向プライマーの5’末端に、BglII部位を逆方向プライマーの5’末端に導入した。pJLC68へのcbg、cbr及びRlucのクローニングは、FRB−ルシフェラーゼ−FKBP型の二重融合をもたらした。正しいタンパク質の大きさについてクローンを検証した(図21A)。先に述べたように二重融合を転写及び翻訳し、発光を測定したが、例外として、ラパマイシンの実験では0.2μMのラパマイシンだけが使用された。
【0201】
結果
ホタルルシフェラーゼタンパク質で見られたFRB及びFKBP系の類似した調整が他の種類のルシフェラーゼでも見られるかどうかを決定するために、ホタルルシフェラーゼ遺伝子をPyrophorus plagiophalamからの赤及び緑の2つの修飾されたコメツキムシ遺伝子、並びにRenilla reniformisからのルシフェラーゼ遺伝子で置換した。pJLC68へのcbg、cbr及びRlucのクローニングは、FRB−ルシフェラーゼ−FKBPの二重融合をもたらした。正しいタンパク質の大きさについてクローンを検証した(図21A)。先に述べたように二重融合を転写及び翻訳し、発光を測定した。
【0202】
図21Bで見られるように、緑及び赤のコメツキムシ二重融合は、ラパマイシンが存在したときは対照(Luc2)と比較して同じ相対効果を示した。発光はラパマイシンに応じて減少したが、Renillaルシフェラーゼはラパマイシンに反応しなかった。
【0203】
C.ルシフェラーゼモジュレーター系のin vivo証明
実施例X.AからのpJLC68をテンプレートとして使用して、FRBのルシフェラーゼとのN末端融合(FRB−Luc2)の断片、ルシフェラーゼのFKBPとのC末端融合(Luc2−FKBP)、又は二重融合(FRB−Luc2−FKBP)を、95℃で2分間、続いて1サイクルが95℃で30秒、50℃で30秒間及び72℃で2分間のものを25サイクル、また72℃で10分間の最終展開からなるPCRプログラムに従って増幅した。増幅のための全ての順方向プライマーはプライマーの5’末端にNheI制限酵素を含むように設計され、全ての逆方向プライマーはプライマーの5’末端にBamHI制限酵素部位を含むように設計され、それによりpBINDベクターへのクローニングのための5’末端がNheI部位及び3’末端のBamHI部位と連なる増幅断片が形成された。増幅のためのプライマーは、次の通りである:
Luc2:
順方向プライマー:
【化46】
逆方向プライマー:
【化47】
FRB−Luc2:
順方向プライマー:
【化48】
逆方向プライマー:
【化49】
Luc2−FKBP:
順方向プライマー;
【化50】
逆方向プライマー;
【化51】
FRP−Luc2−FKBP:
順方向プライマー:
【化52】
逆方向プライマー:
【化53】
【0204】
phRL−TKベクター(Promega Corporation)をTKプロモーター及びベクター骨格の供与源として使用して(図22A)、ベクターで見られるRenillaルシフェラーゼを除去してpBIND−FRB−Luc2−FKBPから増幅したFRB−Luc2−FKBP配列で置換し、断片の5’末端にNheI制限酵素を、断片の3’末端にXbaI部位を生成した。確立された分子生物学的手法(Sambrookら、1989年)を使用して、以下のプライマーで以降のphRL−TKベクターへの挿入のための生成物を生成した。TK FRP−Luc2−FKBP;
順方向プライマー:
【化54】
逆方向プライマー
【化55】
【0205】
ラパマイシンへのFRB−FKBP相互作用のin vivo反応を、D293細胞(cAMP刺激に対する反応の増加のために以前に選択された親ATCC CRL−1573 HEK293細胞の下位集団)を使用して研究した。全てのin vivo実験について、形質移入の前にD293細胞を96穴組織培養プレート上に5,000細胞/ウェルの密度で播種し、37℃及び10%のCO2で少なくとも8時間インキュベートした。プロトコルで記載されているように、TransIT(登録商標)LT1形質移入試薬(Mirus Corporation)を使用し、96穴プレートの1ウェルにつき0.1μgDNA/0.3μl形質移入試薬の濃度を使用して、pBIND構築物及びTK二重融合構築物をD293細胞に形質移入した。形質移入の約24時間後に(図24〜25)、10μlの50mMルシフェリンEF(Promega Corporation、無エンドトキシン)を各ウェルに加え、少なくとも15分間細胞を平衡化させた。初期の発光読取値を各試料(時間は0分)から測定し、次に、OptiMEM組織培養培地(Invitrogen)で希釈した0.2μMラパマイシン原液の10μlをラパマイシン試験ウェルに加え、対照ウェルにはラパマイシンを加えなかった。ラパマイシンの添加後、またラパマイシンの添加の後最高で1時間までの約15分ごとにプレートを読み取った。図24で示すデータについては、反応体に0〜50μMの範囲でFK506を滴定すると、ラパマイシン濃度は1μMであった。Veritas照度計を使用して発光を直接測定した。
【0206】
結果
FRB−ルシフェラーゼ−FKBPのラパマイシンを介した調整は、in vivoで観察された(図23〜25)。TK又はCMVプロモーター系を使用して、発光シグナルのそれぞれ最高5倍及び2倍の減少が観察された。対照構築物はラパマイシンに反応を示さなかった(図25B〜D)。さらに、FKBPとの結合においてラパマイシンと競合するFK506は、滴定可能な方法でラパマイシンの影響を打ち消す(図24)。
【実施例11】
【0207】
C末端調整ルシフェラーゼ融合系によるin vitro実験
一実施形態では、ルシフェラーゼ活性は、ルシフェラーゼのN又はC末端の融合によって調整することができる。例えば、カルモジュリンとのルシフェラーゼC末端の融合は、カルモジュリンを調整する剤によって調整される。
【0208】
材料及び方法
ヒトカルモジュリン遺伝子(CaM)は、順方向プライマー
【化56】
及び、逆方向プライマー
【化57】
を使用して、下記プログラムに従ってベクターpOTB7(ATCC(登録商標)Global Resource Center、MGC−1447)から増幅した:95℃で5分間、次に1サイクルが95℃で30秒間、60℃で30秒間及び72℃で1分間10秒を20サイクル。SnaBI部位は順方向プライマーの5’末端へ導入し、XhoI部位は逆方向プライマーの5’末端へ導入した。CaM遺伝子はLuc2遺伝子(先に述べたように)をSnaBI/XhoI部位に有するルシフェラーゼT7対照ベクターのC末端へクローニングして、Luc2−CaM融合構築物を作製した。融合タンパク質は、TnT(登録商標)Coupled Reticulocyte Lysate System(Promega Corp.)を使用してメーカーのプロトコルに従ってin vitroで発現させた。発光は、Turner20/20照度計で測定した。
【0209】
結合したCaMタンパク質によるルシフェラーゼタンパク質の調整を検定するために、EGTA及びCaCl2を順番にin vitro Luc2−CaM融合タンパク質溶解物に加えた。最初に、TnT(登録商標)反応からのLuc2−CaM溶解物の1μlを100μlのルシフェラーゼ検定用試薬(LAR、Promega Corp.)に加え、混合物の25μlを使用してEGTA及びCaCl2の添加前のベースライン発光を確定した。最初の発光を測定した後、1μlの75mM EGTA溶液(最終濃度3mM)を溶解物/LARに加えて発光を測定した。一旦EGTAの添加に応じた発光を測定した後、1μlの100mM CaCl2溶液を溶解物/LARに加えて発光を測定した。したがって、Luc2−CaM融合構築物の発光測定が3つある;1)EGTA又はCa+2を添加しないベースライン、2)EGTAの添加の後、及び3)Ca+2の添加の後。
【0210】
結果
カルモジュリンタンパク質はカルシウムに応じて構造が大きく変化し、それによりC末端の融合を通してルシフェラーゼ活性を調整する他の可能性を提供する。EGTA又はCa+2が存在しない場合、CaMはルシフェラーゼ及びその基質の間の相互作用を制限する(図27、「試料」)。しかし、EGTAの添加後、この制限は解放され(図27、「EGTA」)、発光は約9倍増加する。この発光増加は、Ca+2の添加によって逆転される(図27、「CaCl2」)。したがって、CaMの高次構造は、Luc2−CaM融合におけるルシフェラーゼ活性に影響を及ぼすようであった。
【0211】
(参考文献)
【0212】
全ての刊行物、特許及び特許出願は、参照により本明細書で組み込まれている。以上の明細書において、本発明は特定の好ましい実施形態に関して記載され、多くの詳細が例示目的のために述べられているが、本発明に更なる実施形態を加えることが可能なこと、また本明細書の詳細の一部は本発明の基本原理から逸脱することなくかなりの程度変更することが可能なことは、当業者にとって明らかであろう。
【特許請求の範囲】
【請求項1】
修飾された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記修飾された甲虫ルシフェラーゼは、関心のある分子と相互作用するアミノ酸配列を含む挿入を含み、前記修飾された甲虫ルシフェラーゼは、修飾に寛容性のある親甲虫ルシフェラーゼ配列の残基又は領域において環状置換され、前記修飾された甲虫ルシフェラーゼの活性は、前記関心のある分子と挿入配列が相互作用した後に活性に関して相互作用の前と比べて変化し、前記修飾された甲虫ルシフェラーゼはホタル又はコメツキムシルシフェラーゼであり、前記修飾された甲虫ルシフェラーゼがホタルルシフェラーゼの場合、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546と対応する領域において環状置換されており、前記修飾された甲虫ルシフェラーゼがコメツキムシルシフェラーゼの場合、コメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応する領域において環状置換されている、前記ポリヌクレオチド。
【請求項2】
前記修飾された甲虫ルシフェラーゼのN末端、C末端又はその両方における少なくとも1つのアミノ酸の標識をコードするポリヌクレオチドを更に含んでいる、請求項1に記載のポリヌクレオチド。
【請求項3】
前記修飾された甲虫ルシフェラーゼは挿入に加えて甲虫ルシフェラーゼ残基N末端及び/又はC末端の欠失を更に含む、請求項1又は2に記載のポリヌクレオチド。
【請求項4】
前記欠失は甲虫ルシフェラーゼ配列の15残基以下である、請求項3に記載のポリヌクレオチド。
【請求項5】
前記修飾された甲虫ルシフェラーゼは親甲虫ルシフェラーゼのN末端及び/又はC末端と対応する甲虫ルシフェラーゼ残基の欠失を更に含む、請求項1又は2に記載のポリヌクレオチド。
【請求項6】
前記欠失は甲虫ルシフェラーゼ配列の15残基以下である、請求項5に記載のポリヌクレオチド。
【請求項7】
前記挿入は親甲虫ルシフェラーゼのN末端及び/又はC末端におけるものである、請求項1又は2に記載のポリヌクレオチド。
【請求項8】
前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼの活性の少なくとも約50%を有する、請求項1又は2に記載のポリヌクレオチド。
【請求項9】
前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼの活性の少なくとも約1%を有する、請求項1又は2に記載のポリヌクレオチド。
【請求項10】
前記標識はPEST配列、GST配列、ポリヒスチジン配列又はレポータータンパク質である、請求項2に記載のポリヌクレオチド。
【請求項11】
前記挿入は約4から約50のアミノ酸残基である、請求項1又は2に記載のポリヌクレオチド。
【請求項12】
前記甲虫ルシフェラーゼはコメツキムシルシフェラーゼである、請求項1又は2に記載のポリヌクレオチド。
【請求項13】
前記甲虫ルシフェラーゼはホタルルシフェラーゼである、請求項1又は2に記載のポリヌクレオチド。
【請求項14】
請求項1又は2に記載のポリヌクレオチドを含むベクター。
【請求項15】
請求項14に記載のベクターを含む宿主細胞。
【請求項16】
請求項1に記載のポリヌクレオチドによってコードされる修飾された甲虫ルシフェラーゼ。
【請求項17】
請求項2に記載のポリヌクレオチドによってコードされる融合タンパク質。
【請求項18】
細胞内の関心のある分子を検出する方法であって、a)細胞を請求項14に記載のベクターと接触させ、前記ベクターを前記細胞に形質移入し、前記挿入は前記分子によって認識されるステップと、b)前記ベクターによってコードされる前記修飾された甲虫ルシフェラーゼの活性を検出又は決定し、それによって前記細胞内の前記分子の存在又は量を検出又は決定するステップとを含む方法。
【請求項19】
前記甲虫ルシフェラーゼはホタルルシフェラーゼである、請求項18に記載の方法。
【請求項20】
前記甲虫ルシフェラーゼはコメツキムシルシフェラーゼである、請求項18に記載の方法。
【請求項21】
修飾された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼに対する少なくとも2つの修飾を含み、前記少なくとも2つの修飾のそれぞれは修飾に寛容性のある甲虫ルシフェラーゼ配列の残基又は領域にあり、前記修飾された甲虫ルシフェラーゼは環状置換されており、前記修飾に寛容性のある前記残基又は領域は、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546に対応し、又はコメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応するものである、前記ポリヌクレオチド。
【請求項22】
プロテアーゼ切断部位を含んでいる環状置換された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記ルシフェラーゼは対応する非置換の甲虫ルシフェラーゼにおいては修飾に寛容性のある部位で置換されており、前記修飾に寛容性のある部位は、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546に対応し、又はコメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応するものである、前記ポリヌクレオチド。
【請求項23】
前記プロテアーゼ切断部位は対応する非置換ルシフェラーゼのN及びC末端と対応している残基の位置又はその近くで挿入される、請求項22に記載のポリヌクレオチド。
【請求項24】
前記プロテアーゼ切断部位での切断は発光の増加をもたらす、請求項22に記載のポリヌクレオチド。
【請求項25】
前記プロテアーゼ切断部位はカスパーゼ3切断部位、カスパーゼ8切断部位、エンテロキナーゼ切断部位、前立腺血清抗原切断部位、SARSウイルスプロテアーゼ切断部位、TEVプロテアーゼ切断部位(NLYFQG)、又はライノウイルスプロテアーゼ切断部位である、請求項22に記載のポリヌクレオチド。
【請求項26】
請求項22又は23に記載のポリヌクレオチドによってコードされるルシフェラーゼ。
【請求項27】
請求項22又は23に記載のポリヌクレオチドを含むベクター。
【請求項28】
請求項27に記載のベクターを含む宿主細胞。
【請求項1】
修飾された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記修飾された甲虫ルシフェラーゼは、関心のある分子と相互作用するアミノ酸配列を含む挿入を含み、前記修飾された甲虫ルシフェラーゼは、修飾に寛容性のある親甲虫ルシフェラーゼ配列の残基又は領域において環状置換され、前記修飾された甲虫ルシフェラーゼの活性は、前記関心のある分子と挿入配列が相互作用した後に活性に関して相互作用の前と比べて変化し、前記修飾された甲虫ルシフェラーゼはホタル又はコメツキムシルシフェラーゼであり、前記修飾された甲虫ルシフェラーゼがホタルルシフェラーゼの場合、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546と対応する領域において環状置換されており、前記修飾された甲虫ルシフェラーゼがコメツキムシルシフェラーゼの場合、コメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応する領域において環状置換されている、前記ポリヌクレオチド。
【請求項2】
前記修飾された甲虫ルシフェラーゼのN末端、C末端又はその両方における少なくとも1つのアミノ酸の標識をコードするポリヌクレオチドを更に含んでいる、請求項1に記載のポリヌクレオチド。
【請求項3】
前記修飾された甲虫ルシフェラーゼは挿入に加えて甲虫ルシフェラーゼ残基N末端及び/又はC末端の欠失を更に含む、請求項1又は2に記載のポリヌクレオチド。
【請求項4】
前記欠失は甲虫ルシフェラーゼ配列の15残基以下である、請求項3に記載のポリヌクレオチド。
【請求項5】
前記修飾された甲虫ルシフェラーゼは親甲虫ルシフェラーゼのN末端及び/又はC末端と対応する甲虫ルシフェラーゼ残基の欠失を更に含む、請求項1又は2に記載のポリヌクレオチド。
【請求項6】
前記欠失は甲虫ルシフェラーゼ配列の15残基以下である、請求項5に記載のポリヌクレオチド。
【請求項7】
前記挿入は親甲虫ルシフェラーゼのN末端及び/又はC末端におけるものである、請求項1又は2に記載のポリヌクレオチド。
【請求項8】
前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼの活性の少なくとも約50%を有する、請求項1又は2に記載のポリヌクレオチド。
【請求項9】
前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼの活性の少なくとも約1%を有する、請求項1又は2に記載のポリヌクレオチド。
【請求項10】
前記標識はPEST配列、GST配列、ポリヒスチジン配列又はレポータータンパク質である、請求項2に記載のポリヌクレオチド。
【請求項11】
前記挿入は約4から約50のアミノ酸残基である、請求項1又は2に記載のポリヌクレオチド。
【請求項12】
前記甲虫ルシフェラーゼはコメツキムシルシフェラーゼである、請求項1又は2に記載のポリヌクレオチド。
【請求項13】
前記甲虫ルシフェラーゼはホタルルシフェラーゼである、請求項1又は2に記載のポリヌクレオチド。
【請求項14】
請求項1又は2に記載のポリヌクレオチドを含むベクター。
【請求項15】
請求項14に記載のベクターを含む宿主細胞。
【請求項16】
請求項1に記載のポリヌクレオチドによってコードされる修飾された甲虫ルシフェラーゼ。
【請求項17】
請求項2に記載のポリヌクレオチドによってコードされる融合タンパク質。
【請求項18】
細胞内の関心のある分子を検出する方法であって、a)細胞を請求項14に記載のベクターと接触させ、前記ベクターを前記細胞に形質移入し、前記挿入は前記分子によって認識されるステップと、b)前記ベクターによってコードされる前記修飾された甲虫ルシフェラーゼの活性を検出又は決定し、それによって前記細胞内の前記分子の存在又は量を検出又は決定するステップとを含む方法。
【請求項19】
前記甲虫ルシフェラーゼはホタルルシフェラーゼである、請求項18に記載の方法。
【請求項20】
前記甲虫ルシフェラーゼはコメツキムシルシフェラーゼである、請求項18に記載の方法。
【請求項21】
修飾された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記修飾された甲虫ルシフェラーゼは対応する親甲虫ルシフェラーゼに対する少なくとも2つの修飾を含み、前記少なくとも2つの修飾のそれぞれは修飾に寛容性のある甲虫ルシフェラーゼ配列の残基又は領域にあり、前記修飾された甲虫ルシフェラーゼは環状置換されており、前記修飾に寛容性のある前記残基又は領域は、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546に対応し、又はコメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応するものである、前記ポリヌクレオチド。
【請求項22】
プロテアーゼ切断部位を含んでいる環状置換された甲虫ルシフェラーゼをコードするポリヌクレオチドであって、前記ルシフェラーゼは対応する非置換の甲虫ルシフェラーゼにおいては修飾に寛容性のある部位で置換されており、前記修飾に寛容性のある部位は、ホタルルシフェラーゼの残基2から12、残基116から126、残基228から238、残基262から272、残基289から308、残基356から366又は残基535から546に対応し、又はコメツキムシルシフェラーゼの残基15から30、残基112から122、残基352から362、残基371から384、残基393から414又は残基485から495に対応するものである、前記ポリヌクレオチド。
【請求項23】
前記プロテアーゼ切断部位は対応する非置換ルシフェラーゼのN及びC末端と対応している残基の位置又はその近くで挿入される、請求項22に記載のポリヌクレオチド。
【請求項24】
前記プロテアーゼ切断部位での切断は発光の増加をもたらす、請求項22に記載のポリヌクレオチド。
【請求項25】
前記プロテアーゼ切断部位はカスパーゼ3切断部位、カスパーゼ8切断部位、エンテロキナーゼ切断部位、前立腺血清抗原切断部位、SARSウイルスプロテアーゼ切断部位、TEVプロテアーゼ切断部位(NLYFQG)、又はライノウイルスプロテアーゼ切断部位である、請求項22に記載のポリヌクレオチド。
【請求項26】
請求項22又は23に記載のポリヌクレオチドによってコードされるルシフェラーゼ。
【請求項27】
請求項22又は23に記載のポリヌクレオチドを含むベクター。
【請求項28】
請求項27に記載のベクターを含む宿主細胞。
【図1】

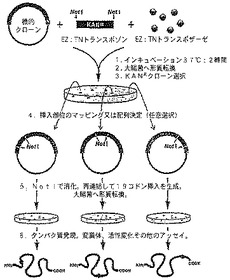
【図2】

【図3】

【図4】
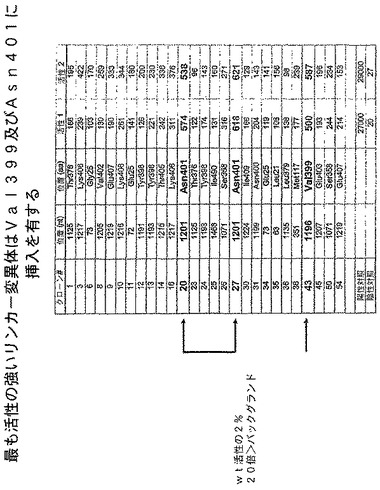
【図5A】
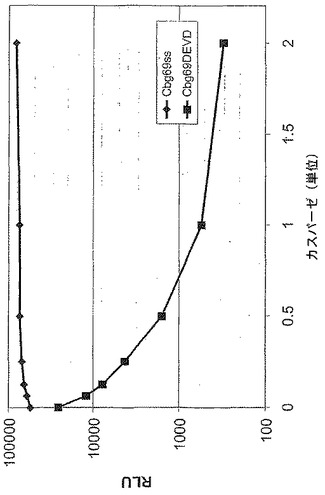
【図5B】
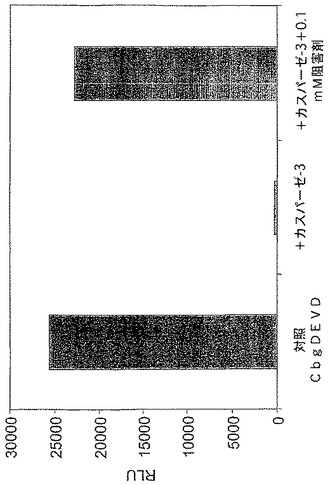
【図5C】
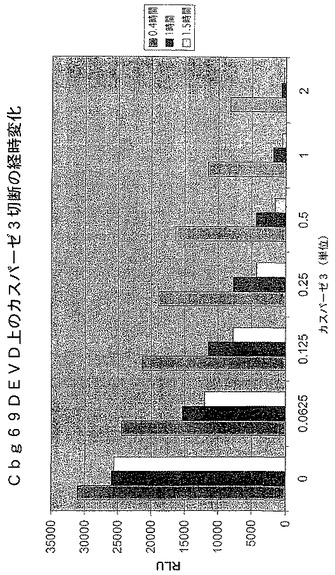
【図6A】

【図6B】
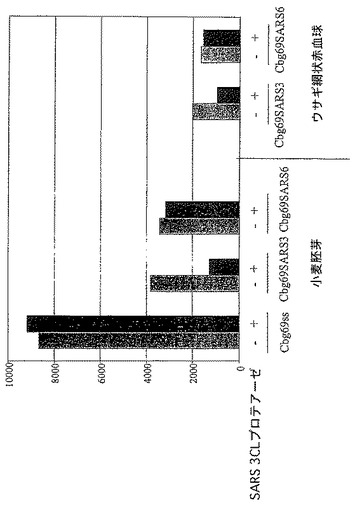
【図6C】

【図7A】

【図7B】
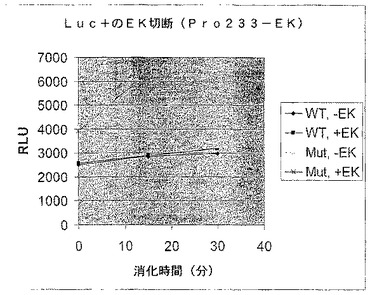
【図7C】
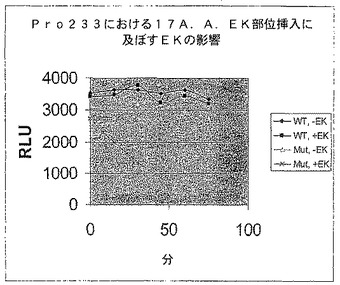
【図7D】
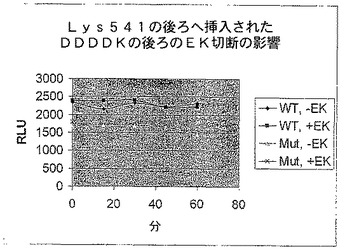
【図8】
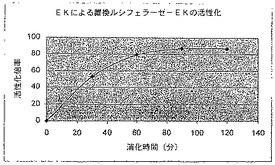
【図9】
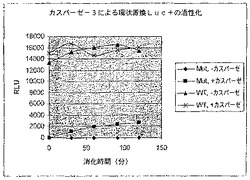
【図10A】
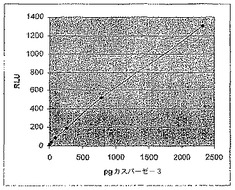
【図10B】
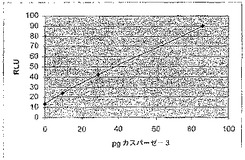
【図11】
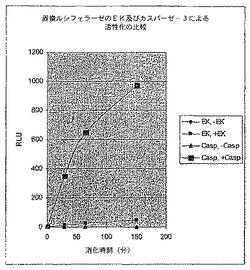
【図12】
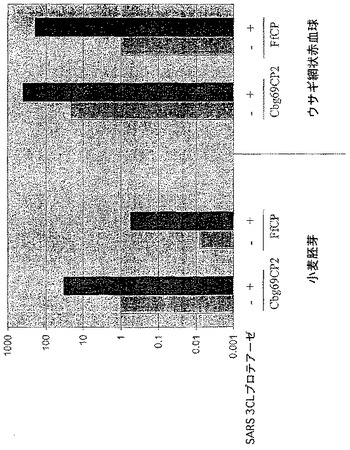
【図13】
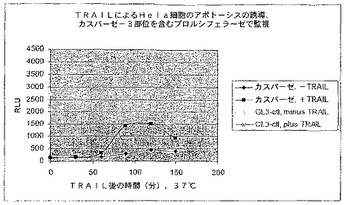
【図14】
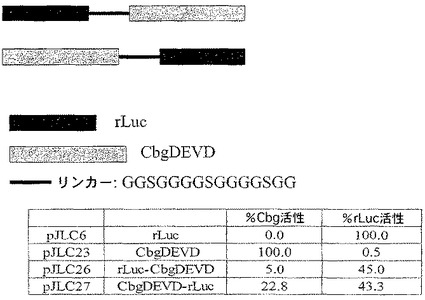
【図15】
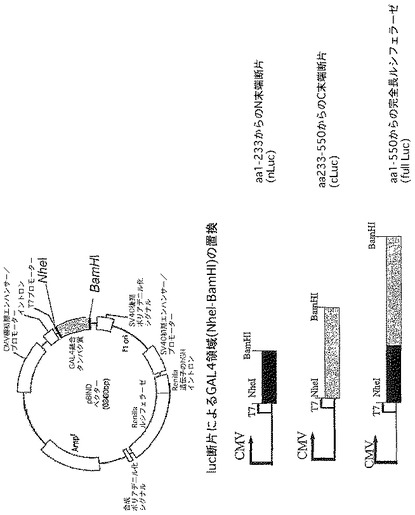
【図16A】
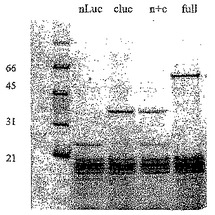
【図16B】

【図17】
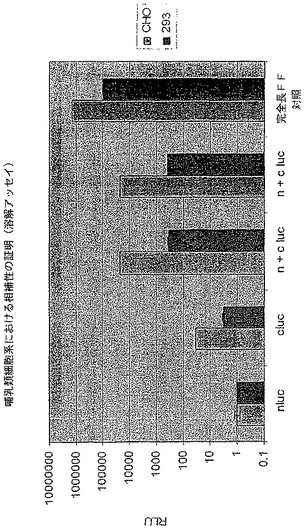
【図18】
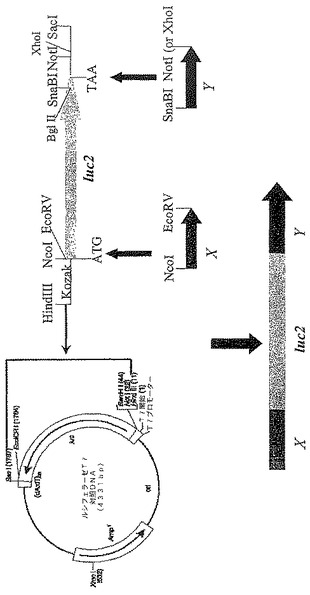
【図19】
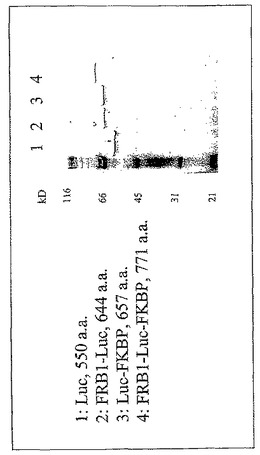
【図20A】

【図20B】
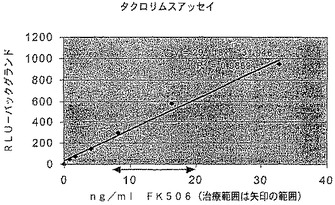
【図21A】
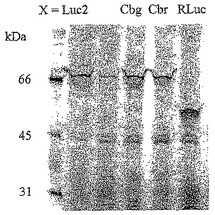
【図21B】
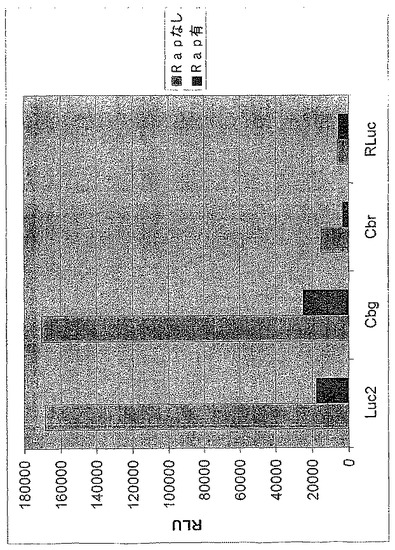
【図22】
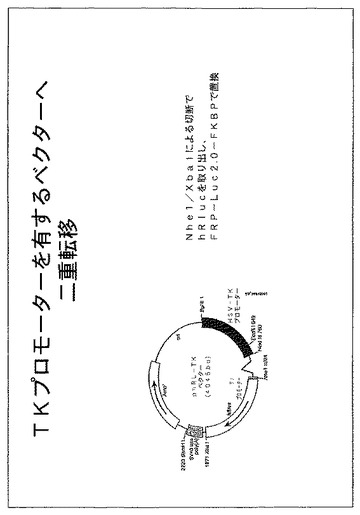
【図23】
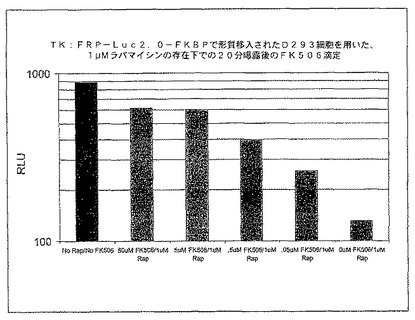
【図24】
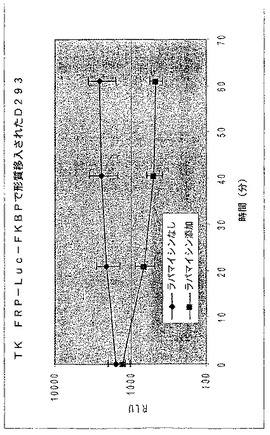
【図25A】
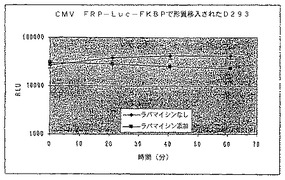
【図25B】
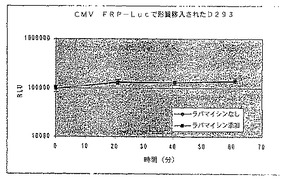
【図25C】
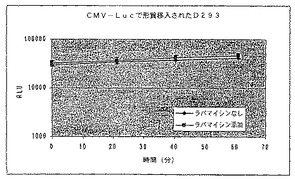
【図25D】
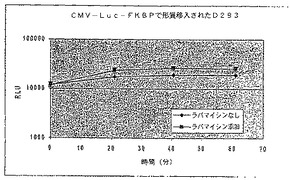
【図26】
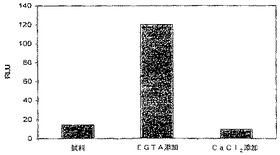
【図2】
【図3】
【図4】
【図5A】
【図5B】
【図5C】
【図6A】
【図6B】
【図6C】
【図7A】
【図7B】
【図7C】
【図7D】
【図8】
【図9】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A】
【図16B】
【図17】
【図18】
【図19】
【図20A】
【図20B】
【図21A】
【図21B】
【図22】
【図23】
【図24】
【図25A】
【図25B】
【図25C】
【図25D】
【図26】
【公開番号】特開2011−135892(P2011−135892A)
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願番号】特願2011−43966(P2011−43966)
【出願日】平成23年3月1日(2011.3.1)
【分割の表示】特願2006−534242(P2006−534242)の分割
【原出願日】平成16年10月1日(2004.10.1)
【出願人】(593089149)プロメガ コーポレイション (57)
【氏名又は名称原語表記】Promega Corporation
【Fターム(参考)】
【公開日】平成23年7月14日(2011.7.14)
【国際特許分類】
【出願日】平成23年3月1日(2011.3.1)
【分割の表示】特願2006−534242(P2006−534242)の分割
【原出願日】平成16年10月1日(2004.10.1)
【出願人】(593089149)プロメガ コーポレイション (57)
【氏名又は名称原語表記】Promega Corporation
【Fターム(参考)】
[ Back to top ]