
人工多能性幹細胞(iPS細胞)の作製
本発明は人工多能性幹細胞(iPS細胞)の作製法に関し、方法は1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち該標的細胞の人工多能性幹細胞への再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞はOct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。更に、本発明は本発明の方法によって作製された人工多能性幹細胞、および標的細胞の人工多能性幹細胞への再プログラミングに関与する化合物の同定法に関する。また、トランスジェニック非ヒト動物の作製法、および本発明の方法によって作製したiPS細胞を含む遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための組成物の作製法も意図される。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、人工多能性幹細胞(iPS細胞)の作製法に関するものであり、該方法は1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち人工多能性幹細胞への該標的細胞の再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSoxに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。更に、本発明は本発明の方法によって作製した人工多能性幹細胞、および人工多能性幹細胞への標的細胞の再プログラミングに関与する化合物の同定法に関する。また、トランスジェニック非ヒト動物の作製法、および本発明の方法によって作製したiPS細胞を含む遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための組成物の作製法も意図される。
【0002】
本明細書中でいくつかの文書を引用する。本明細書に引用する文書(製造者の仕様書、説明書などを含む)の開示内容は、参照により本明細書に組み込まれる。
【背景技術】
【0003】
胚性幹細胞(ES細胞)のような多能性細胞は、その自己再生能力および種々の細胞型への分化能力を特徴とする。ES細胞はin vitroにおいて、物理的および生物学的誘導因子の存在下で3つの胚葉層(内胚葉、中胚葉、および外胚葉)全ての特殊化(specialized)細胞系統に分化させることができる。これまでのところ、多くの有望な研究により、動物モデルにおいて種々の疾患の寛解におけるESCの分化誘導体の治療可能性が示されてきた。その結果、多能性幹細胞は組織工学および移植医療への使用に関して、非常に大きな可能性を有している。これらの細胞を特定の細胞型に分化させることができれば、多くの深刻な変性疾患(例えば糖尿病、パーキンソン病、およびアルツハイマー病)の治療を目的とする移植のための、ほぼ無制限の細胞供与源となりうる(Biswasら, 2007;Kimら, 2007;Zimmermannら, 2007)。
【0004】
体細胞を表現型、遺伝子型、および多能性の点でES細胞に類似した状態に再分化するように遺伝子的に修飾しうることが明らかにされたのはごく最近のことである(TakahashiおよびYamanaka, 2006;Okitaら, 2007;Wernigら, 2007)。いわゆる体細胞の「再プログラミング」は多能性の再獲得の機構を理解する有益な手段であると同時に、患者に特異的な多能性幹細胞を作製するという可能性を開くものでもある。マウスおよびヒト体細胞の多能性幹細胞(人工多能性幹細胞(iPS細胞)と表記する)への再プログラミングは、4つの転写因子、Oct4、Sox2、c-Myc、およびKlf4の発現によって可能とされてきた。
【0005】
現在、iPS細胞が医学的応用(例えば患者特異的再生細胞療法)において多大な可能性を有していることは広く知られているが、現行のiPS細胞の作製法は医療分野での使用を妨げる。特に、いくつかの再プログラミング因子を組み合わせて導入および発現するのに使用されるレトロウィルスベクターは、複数のコピーでランダムにゲノムに(傾向的には近傍または活性な内因性遺伝子に(into the vicinity or into active endogenous genes))組み込まれ、そのため癌遺伝子または腫瘍抑制遺伝子のそれぞれ活性化または不活性化変異体を生じうる。従って、標的細胞のゲノムを改変する度合いを最小化させた方法を用いたiPS細胞の作製により、この手段の臨床的に安全な適用が助長されうる。
【発明の概要】
【課題を解決するための手段】
【0006】
従って、本発明の第1の態様では人工多能性幹細胞(iPS細胞)の作製法に関し、該方法は1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち該標的細胞の人工多能性幹細胞への再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。
【0007】
「人工多能性幹細胞(iPS細胞)」は胚性幹細胞(ESC)に類似した特徴を示す細胞である。該特徴には例えばin vitroにおける無制限の自己再生、正常な核型、Oct3/4、Sox2、Nanog、アルカリホスファターゼ(ALP)、および幹細胞特異的抗原3および4(SSEA3/4)のような幹細胞マーカー遺伝子を含む特徴的な遺伝子発現パターン、ならびに特定の細胞型への分化能がある(Hanna, J.ら (2007), Science 318(5858): 1920-3;Meissner, A.ら (2007), Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら (2007), Nat Biotechnol.; Okita, K.ら (2007), Nature 448(7151): 313-7;Takahashi, K.,ら (2007) Cell 131 (5): 861-72;Wernig, M.ら (2007), Nature 448(7151): 318-24;Yu, J.ら (2007), Science 318(5858): 1917-20;Park, I. H.ら (2008), Nature 451(7175): 141-6))。最先端の線維芽細胞培養物からのiPS細胞作製についてはTakahashi, Okita, Nakagawa, Yamanaka (2007) Nature Protocols 2(12)に報告されている。マウスiPS細胞の多能性は、例えばin vitroにおける神経細胞、グリア細胞、および心臓細胞への分化、ならびに胚盤胞注入による生殖系列キメラマウスの作製によって試験することができる。ヒトiPS細胞株は、in vitroにおける神経細胞、グリア細胞、および心臓細胞への分化によって分析することができ、そのin vivoにおける分化能は免疫不全SCIDマウスへの注射およびテラトーマとして生じる腫瘍のキャラクタリゼーションによって試験できる。
【0008】
iPS細胞は一般に、以下の細胞生物学的特徴に従って評価および分類できる:
形態:iPS細胞は形態学的に胚性幹細胞(ESC)と類似している。各細胞は円形であり、大型核を有し,細胞質が少ない。iPS細胞のコロニーもESCのものと類似している。ヒトiPS細胞はhESCと同様の、境界が明瞭で扁平かつ密集したコロニーを形成し、一方、マウスiPS細胞はmESCと同様に、hESCほど扁平ではなく、より凝集したコロニーを形成する。
【0009】
増殖性:幹細胞はその定義の一部として自己再生しなければならないため、倍加時間および有糸分裂活性はESCに不可欠なものである。iPS細胞は有糸分裂的に活性であり、ESCと同等の速度で活発に自己再生、増殖、および分裂する。
【0010】
幹細胞マーカー:iPS細胞はESCで発現される細胞表面抗原マーカーを発現する。ヒトiPSCはhESCに特異的なマーカー(例えばSSEA-3、SSEA-4、TRA-1-60、TRA-1-81、TRA-2-49/6E、およびNanog)を発現する。マウスiPS細胞はmESCと同様、SSEA-1は発現するが、SSEA-3およびSSEA-4はいずれも発現しない。
【0011】
幹細胞遺伝子:iPS細胞は未分化ESC中で発現される遺伝子(例えばOct3/4、Sox2、Nanog、GDF3、REX1、FGF4、ESG1、DPPA2、DPPA4、およびhTERT)を発現する。
【0012】
テロメラーゼ活性:テロメラーゼは、ヘイフリック(Hayflick)限界(〜50回の細胞分裂)に制限されずに細胞分裂を持続するのに必要とされる。hESCは自己再生および増殖を持続する高いテロメラーゼ活性を発現し、またiPS細胞も高テロメラーゼ活性を示し、テロメラーゼタンパク質複合体の必要構成要素であるhTERT(ヒトテロメラーゼ逆転写酵素)を発現する。
【0013】
多能性:iPS細胞は、ESCと同様の方法で(in a fashion)、完全に分化した組織に分化する能力を有する。例えば免疫不全マウスに注射したiPS細胞は、9週間後に自然発生的にテラトーマを形成する。テラトーマは3つの胚葉(内胚葉、中胚葉、および外胚葉)から誘導される組織を含む複数系統の腫瘍である;これは、他の腫瘍が通常1つの細胞型のみのものであるのとは異なっている。テラトーマ形成は重要な多能性の試験である。更に培養下のhESCは「胚様体」と呼ばれる球状の胚様構造を自発的に形成し、これは有糸分裂的に活性な分化hESCの中核、およびそれを囲む3つの胚葉全てに由来する完全に分化した細胞から成る。iPS細胞も胚様体を形成し、外縁部に分化細胞を有する。
【0014】
胚盤胞注入:hESCは、天然では胚盤胞の内部細胞塊(胚結節)内に存在して胚結節内で胚に分化し、一方、胚盤胞の外殻(栄養膜)は胚外組織に分化する。内部が空洞となった栄養膜は生きた胚を形成することができず、従ってこれは胚性幹細胞が胚結節内で分化して胚を形成するのに必要とされるものである。iPS細胞をマイクロピペットで栄養膜内に注入することができ、胚盤胞はメスのレシピエントに移植される。それによって生存キメラマウスの子、すなわちiPS細胞誘導体が種々の程度のキメラ化で全身にわたって導入されたマウスを作製することができる。
【0015】
プロモーターの脱メチル化:メチル化とは、DNA塩基へのメチル基の転移、一般的にはCpG部位(シトシン/グアニンが隣接した配列)のシトシン分子へのメチル基の転移である。遺伝子の広範なメチル化は、発現タンパク質活性の阻害または発現に干渉する酵素のリクルートによって、発現に干渉する。従って遺伝子のメチル化により、転写が阻害されることによって効果的にその発現が停止される。多能性関連遺伝子(例えばOct3/4、Rex1、およびNanog)のプロモーターはiPS細胞中で脱メチル化され、iPS細胞においてそのプロモーター活性、並びに活性な多能性関連遺伝子の促進(promotion)および発現を示す。
【0016】
ヒストン脱メチル化:ヒストンは圧縮タンパク質であり、構造的にはDNA配列に局在し、種々のクロマチン関連修飾を介してその活性がもたらされる。例えばOct3/4、Sox2、およびNanogに関連するH3ヒストンは脱メチル化されてOct3/4、Sox2、およびNanogを発現する。
【0017】
本発明に関して使用する「導入する」という用語は、コード配列を標的細胞に取り入れさせ、その後、該コード配列を標的細胞のゲノムDNAに組み込ませる工程に関する。この工程は一般に安定トランスフェクションとして知られ、安定トランスフェクションのための方法は当業者に公知であり、例えばBonetta, L., (2005), Nature Methods 2, 875-883に報告されている。トランスフェクトされた細胞で起こる再プログラミング事象は低率であるため、効率の高い安定トランスフェクション法を用いるのが有利である。そのため、トランスフェクション/感染効率の高い方法によってコード配列を標的細胞に導入するのが好ましい。例えばトランスフェクション/感染効率が少なくとも30%、少なくとも50%、または少なくとも80%であるのが好ましい。好適な方法には、例えばリポフェクション、エレクトロポレーション、ヌクレオフェクション(nucleofection)、マグネトフェクション(magnetofection)、またはウィルスベクター感染がある。レトロウィルスを使用して標的細胞の安定トランスフェクションを行うのが好ましいが、これは該ベクターが、標的細胞へのコード配列の効率的な侵入だけでなく、標的細胞のゲノムDNAへの融合も仲介するためである。明らかにされているところによれば、レトロウィルスベクターによって種々の動物種由来の広範な型の細胞の形質導入を行い、ベクターが運ぶ遺伝物質を標的細胞に組み込み、形質導入されたコード配列を高レベルで発現させることができ、そして好都合なことに、レトロウィルスベクターは感染後に伝播またはウィルスタンパク質を生成することがない。好適なレトロウィルスベクター系は当業者に公知であり、それらは例えばMoMuLV LTR、MESV LTRを有するレトロウィルスベクター、CMVプロモーターのような種々の内部プロモーターを含有する(好ましくはエンハンサー/プロモーターを組み合わせて含有する)レンチウィルスベクターで、胚/多能性細胞中で導入遺伝子発現の抑制を示すものである。アデノウィルスベクターのようなエピソームベクター系、他の非組込み型ベクター、エピソーム複製プラスミド(episomally replicating plasmids)を使用することもできる。好ましくは、レトロウィルスMXベクター系を本発明の方法に使用する(Kitamuraら, (2003), Exp Hematol., 31(11):1007-1014)。
【0018】
本発明の方法で使用する標的細胞は既存の細胞株由来のものであるか、または種々の方法(例えば、初代細胞株を樹立するために組織サンプルを得る)によって得てもよい。種々の組織からサンプルを得る方法および初代細胞株を樹立する方法は、当業界で公知である(例えばJonesおよびWise, Methods Mol Biol. 1997参照)。好適な体細胞株を様々な供給者(例えば American tissue culture collection (ATCC)、the German Collection of Microorganisms and Cell Cultures (DSMZ)、またはPromoCell社(Sickingenstr. 63/65, D-69126、ハイデルベルグ))から購入してもよい。本発明の方法によれば、好適な標的細胞はOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、該因子は、これを補完する因子群(すなわち Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子)から選択される外因的に導入される因子と組み合わされて、非多能性標的細胞をiPS細胞に再プログラミングする能力を有する。1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。少なくとも2つの上記因子が標的細胞中で内因的に発現されるか否かを確認する方法は、当業者に公知である。それらの方法には、例えばウェスタンブロッティング、リアルタイムPCR、または細胞間染色がある。更に当業者は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する細胞を作製して標的細胞のiPS細胞への再プログラミングを開始させるため、内因的に発現される因子群を補完するのにいずれの外因性因子(単数または複数)が必要であるかを容易に認識できる。従って発現可能な形態でコード配列(単数または複数)を導入した細胞は、Oct3/4並びにMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つずつの因子から成る因子群を発現する。
【0019】
本発明は、既に標的細胞中に内因的に存在するコード配列を導入する態様も包含する。これは、例えば相当する因子が標的細胞の再プログラミングに関与しない、または十分関与しない影響で内因性コード配列が低レベルでしか発現されないような場合、有効でありうる。
【0020】
「コード配列」という用語は、転写の際にコードされた産物を生じさせるヌクレオチド配列を示す。本発明にかかるコード配列の転写は、好適なプロモーターと関連して容易に達成される。好ましくはコード配列は、転写の際に標的細胞の人工多能性幹細胞への再プログラミングに関与する因子を生じさせる遺伝子のcDNA配列に相当し、本発明の方法にかかる再プログラミング因子は、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される。
【0021】
「標的細胞の人工多能性幹細胞への再プログラミングに関与する因子」とは、標的細胞の人工多能性幹細胞への再プログラミングの誘導に関与する能力を有する因子を示し、該因子はOct3/4並びにMyc、Klf、およびSox因子ファミリーに属する因子から選択される。それらの再プログラミング因子には、例えばOct3/4、Sox2、Sox1、Sox3、c-Myc、n-Myc、I-Myc、Klf1、Klf2、Klf4、Klf5など、または再プログラミング能を保持するそれらの変異体がある。該再プログラミングへの関与は、例えば細胞のメチル化パターンを胚性幹細胞と同様のものに変化させること、細胞の発現プロファイルを胚性幹細胞の発現プロファイルのように変更すること、またはヒストン結合の調節によって凝集した核DNAのコンフォメーションを胚性幹細胞で観察されるのと同様にすることという形態であってもよく、該変化はそれぞれ、好適な再プログラミング因子によって単独で、または組み合わされて起こってもよい。上記の因子以外に、当業者に認識されるように、更なる好適な再プログラミング因子を同定する方法、例えばバイサルファイトゲノムシーケンシング、RT-PCR、リアルタイムPCR、マイクロアレイ分析、核型分析、テラトーマ形成、アルカリホスファターゼ染色があり、それらは全て当業者に公知であり、例えば以下に記載されている:Okita, K.ら(2007), Nature 448(7151): 313-7;Park, I. H.ら(2008), Nature 451(7175): 141-6;Takahashi, K.ら(2007), Cell 131(5): 861-72;Wernig, M.ら(2007), Nature 448(7151): 318-24;Takahashi, K.ら(2007), Nat Protoc 2(12): 3081-9;またはHogan, B.ら(1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press。
【0022】
Oct3/4はオクタマー(「Oct」)転写因子ファミリーに属し、多能性の維持に関与する。通常はOct3/4を発現する細胞(例えば割球および胚性幹細胞)においてOct3/4が存在しなければ、自発的な栄養膜への分化が誘導される。従って、Oct3/4の存在は胚性幹細胞の多能性および分化能に関与する。「Oct」ファミリーの種々の他の遺伝子(Oct1およびOct6など)は誘導を起こさず、これは誘導過程に対するOct3/4の排他性を示している。本明細書では、「Oct4」という用語は「Oct3/4」と互換的に使用する。
【0023】
Sox遺伝子ファミリーはOct3/4同様、多能性の維持に関係するが、多能性幹細胞で排他的に発現されるOct3/4とは異なり、複能性(multipotency)および単能性幹細胞と関連する。
【0024】
Klf遺伝子ファミリーのKlf4は初めにマウスiPS細胞作製のための因子として同定され、ヒトiPS細胞の作製因子であることが明らかにされた。
【0025】
Mycファミリーに属する遺伝子は、癌に関係する癌原遺伝子である。c-MycがマウスiPS細胞の作製に関係する因子であり、ヒトiPS細胞の作製に関係する因子でもあることが証明された。iPS細胞作製のための標的細胞への「Myc」遺伝子ファミリーの導入は、臨床治療としてのiPS細胞の成り行きに困難を与えており、これは、c-Myc誘導iPS細胞を移植したマウスの25%で致命的なテラトーマが発生したためである。N-MycおよびI-Mycは同様の効率でc-mycに代わるものとして同定されている。
【0026】
本発明に関して使用する「再プログラミング」という用語は、遺伝子型的および/または表現型的に胚性幹細胞と同様の細胞となるような細胞の遺伝子型的および表現型的プロファイルの変化の過程に関する。該変化は、例えば上記のようなメチル化パターンの変化、発現プロファイルの変更、または凝集した核DNAのコンフォメーション変化を含む。上記は本明細書で以下に記載する他の態様にも準用される。
【0027】
本発明の方法は、2つの再プログラミング因子のみの導入によってiPS細胞を得ることができるという、驚くべき発見に基づく。この発見がなされるまで、従前技術の定説は、in vivo実験で機能的である、すなわち3つの胚葉層に関与する能力を有する生存iPS細胞は少なくとも3つ、より効率的には4つの再プログラミング因子を組み合わせて導入しなければ作製できないというものであった。
【0028】
例として、レトロウィルスMXベクター系を用いて、4つ(4F)、3つ(3F)、および2つのみ(2F)の再プログラミング因子の組み合わせ、並びに1つの再プログラミング因子のみの導入によって、マウス神経幹細胞(NSC)の再プログラミングを行うことができることが明らかになった。NSCを成体OG2/Rosa26ヘテロ接合トランスジェニックマウスの脳から樹立し(Ryan, A. K. & Rosenfeld, M. G., Genes Dev 11 , 1207-25 (1997);Do, J. T. & Scholer, H. R., Stem Cells 22, 941-9 (2004);Pollard, S. M., Conti, L., Sun, Y., Goffredo, D. & Smith, A., Cereb Cortex 16 Suppl 1 , i112-20 (2006))、Oct4プロモーターの制御下でGFP(Oct4-GFP)を、構成的(constitutive)Rosa26遺伝子座からLacZ導入遺伝子を発現させた。
【0029】
GFP+コロニーの観察を、まずOct4およびKlf4に感染させたNSC培地で行い(2F OK)、1-2週間後にOct4およびc-mycに感染させた培地で行った(2F OM)(表1)。
【0030】
【表1】
【0031】
2F OM iPS細胞を更に分析したところ、ESC様の発現パターンおよびテラトーマの3つの胚葉への関与が明らかになった。
【0032】
2F OK iPS細胞を4F(4つの再プログラミング因子を導入してiPS細胞を作製する標準的な方法を用いて作製)iPS細胞およびESCと比較した。感染14日後に5個のGFP+コロニーを分離し、ESC培養条件下で増殖させ(図1c、f)、3個(すなわち60%)のESCと形態学的に識別不能な2F OK iPS細胞クローン(B-2、D-7、およびF-4)を得た(図1d、g)。コントロールウィルス(MX)に感染させたNSCからはコロニーは形成されなかった(図1e、h)。2F OK iPSおよび4F iPSの再プログラミング効率の経時変化を、Oct4-GFP+コロニー数およびNSCのMX-GFPコントロールウィルスでの形質転換速度から見積もった(図1i、j)。その結果、再プログラミング効率はNSCの4F再プログラミングでは3.6%、2因子法では0.11%と算出され、これは選択を行った場合の線維芽細胞の再プログラミング効率(0.08%未満、Takahashi, K.およびYamanaka, S., Cell 126, 663-76 (2006);Okita, K., lchisaka, T.およびYamanaka, S., Nature 448, 313-7 (2007);Wernig, M.ら, Nature 448, 318-24 (2007))および選択を行わない場合(0.5%; Meissner, A., Wernig, M.およびJaenisch, R., Nat Biotechnol 25, 1177-81 (2007))に匹敵するものであった(図1j)。4因子全てでの形質転換はタイミング(timing)およびGFP+コロニー数にプラスの影響を与えた。ウィルス導入遺伝子の組込みはジェノタイピングPCR(genotyping PCR)で確認した。4因子全てのウィルス導入遺伝子が4F iPS細胞で検出され、一方、2F OK iPS細胞はOct4およびKlf4導入遺伝子のみを含有した。
【0033】
2F OK iPS細胞は、SSEA-1およびアルカリホスファターゼに関して陽性に染色され、4F iPS細胞およびESCと同様のES細胞マーカー遺伝子発現パターンを示した(図2a)。qRT-PCRの結果は、2F OK iPS細胞における内因性Oct4、Sox2、c-Myc、およびKlf4発現がESCに匹敵し、2F OK iPS細胞中のウィルス転写物のサイレンシング(30日後で1000倍低下)が起こることを示した。2F iPSの総体的な遺伝子発現もESCおよび4F iPSに類似している(clusters close to)(図2b)。DNAマイクロアレイ分析の散布図は、2F iPS細胞およびESC間の類似性が2F iPS細胞およびNSC間の類似性よりも高いことを示した(図2c、d)。従って、2F iPS細胞(クローンF-4)は総体的な転写レベルでマウスESCと非常に類似していると考えられた。
【0034】
2F OK iPS細胞の分化能をin vitroにおける胚様体(EB)への分化によって確認した。これらの細胞は外胚葉(Tuj1)、内胚葉(α-フェトプロテイン)、および中胚葉マーカーFlk1(心筋細胞を擬態する拍動細胞(beating cell)によって発現される)を発現した(図3a)。テラトーマは3つの胚葉全ての誘導体を含有し(図3b)、3つの胚葉のマーカーを発現した。ドナー細胞(NSC)からはテラトーマは形成されなかった。これらのデータは、2F OK iPS細胞がin vitroおよびin vivoにおいて多能性表現型を示すことを示している。
【0035】
分化能を検討するために、2F OK iPS細胞を8細胞期胚と凝集させた。iPS細胞は分化中の胚盤胞において内部細胞塊の形成に関与した(図4a)。凝集した胚盤胞を偽妊娠メスに移植した後、E13.5で16個の生存胚を得、そのうち2個はOct4-GFP発現による判定で胎仔生殖腺における生殖細胞の関与を示した(図4b)。全胚からの胚組織のX-gal染色(Rosaβ-geo26(lacZ導入遺伝子)を保有するNSCドナー細胞を可視化)により、得られたキメラにおいて、2F OK iPS細胞は3つの胚葉全ての分化に関与していることが明らかになった(図4c、e)。最も厳密な試験で4倍体(4N)胚凝集体の分化能を試験したところ(n=122)、E13.5で2個の死亡(停止)胚が確認された(図4d)。これは4N胚凝集体の正常率以内であり、導入した細胞の多能性欠損には関係しなかった。これらのデータは、iPS細胞が後期胚の組織の全てを生ずることができることを示している。2倍体(2N)凝集体では、PCR遺伝子型同定により、13個のキメラのうち2個がドナー細胞のOct4-GFP対立遺伝子陽性であることが明らかになった(図4fおよびg(上段))。2F OK iPS細胞が生殖細胞系列に関与できるか否かを評価するために、キメラをCD-1メスと交配した。12匹の子のうち2匹がOct4-GFP対立遺伝子を有し、12匹中1匹がlacZ対立遺伝子を有した。ドナー細胞はヘテロ接合性マウス(Oct4+/- Rosa26+/-)に由来するので、それらはOct4およびKlf4導入遺伝子も有する(図4g(下段))。成体キメラおよびF1マウス(それぞれ17週齢および3週齢まで)で腫瘍形成は観察されなかった。この知見は、2F OK iPS細胞がキメラの発生全期に関与して次世代(F1)の生存可能な子を生じることができることを示しており、従ってiPS細胞がESCと同様の分化特性を有することを示唆している。
【0036】
以下の実施例9に詳細を記載するが、出願人は、2つまたは1つだけの再プログラミング因子の導入によってヒト細胞を多能性幹細胞に転換できることも明らかにした。該再プログラミング因子はOct4またはOct4およびKlf4である。
【0037】
結論として、上記の知見は、2つの再プログラミング因子または1つだけの再プログラミング因子を使用してiPS細胞を作製することに成功したことを示している。本発明の方法の利点は、1つまたは2つの再プログラミング因子の安定トランスフェクションのために2つまたは1つしかレトロウィルスを使用しない点にある。従前技術による方法に比較して少ない数のレトロウィルスベクターでiPS細胞を誘導できることは、再プログラミングすべき最初の細胞集団の遺伝子調節を最小限にすることへの大きな一歩である。従って、異常細胞および腫瘍原性細胞形成のリスクが大幅に低減され、それにより、特に治療目的に適したiPS細胞の作製が可能となる。
【0038】
本発明の方法の好ましい態様では、Myc、Klf、およびSox因子ファミリーに属し、標的細胞に導入されるべきコード配列によって内因的に発現される、またはコードされる因子は、I-Myc、n-Myc、c-Myc、Klf1、Klf2、Klf4、Klf15、Sox1、Sox2、Sox3、Sox15、およびSox18から成る群から選択される。
【0039】
例えばマウスOct3/4、Sox2、c-Myc、およびKlf4のコード配列はそれぞれSEQ ID NO: 1、5、9、および13に見られる。マウスOct3/4、Sox2、c-Myc、およびKlf4のタンパク質配列はそれぞれSEQ ID NO: 2、6、10、および14に見られる。ヒトOct3/4、Sox2、c-Myc、およびKlf4のコード配列はそれぞれSEQ ID NO: 3、7、11、および15に見られる。ヒトOct3/4、Sox2、c-Myc、およびKlf4のタンパク質配列はそれぞれSEQ ID NO: 4、8、12、および16に見られる。当業者は、当該分野で公知の方法を用いて任意の標的種の再プログラミング因子のコード配列を決定する立場にある。例えば、データベース(例えばNational Center for Biotechnology Information (NCBI)が維持しており、ワールドワイドウェブ(http://www.ncbi.nlm.nih.gov/)からアクセスできるデータベース)から、配列および機能に関するデータを検索することができる。更に、比較ゲノミクスのためのデータベースには、限定されるわけではないが以下がある:NCBIが維持するデータベース(http://www.dcode.org/)、全ての完全にシーケンシングが行われた生物のタンパク質アノテーションのデータベース(http://supfam.org/SUPERFAMILY/)、様々な種のゲノム情報を含むデータベース(http://www.cbs.dtu.dk/services/GenomeAtlas/)、または遺伝子クラスターを含むデータベース(http://phigs.jgi-psf.org/)。該データベースにより、当業者はマウスおよびヒトで知られている配列から開始して、例えば異種間の配列アラインメントを実施して相同遺伝子を同定することによって、他の種における再プログラミング因子のコード配列を同定することができる。
【0040】
ごく最近になって発表されたいくつかの科学論文(Hanna, J.ら(2007) Science 318(5858): 1920-3;Meissner, A.ら(2007) Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら(2007) Nat Biotechnol.;Okita, K.ら(2007), Nature 448(7151): 313-7;Takahashi, K.ら(2007), Cell 131 (5): 861-72;Wernig, M.ら(2007) Nature 448(7151): 318-24;Yu, J.ら(2007) Science 318(5858): 1917-20;Park, I. H.ら(2008) Nature 451(7175): 141-6)により、Oct、Sox、Klf、およびMycファミリーに属する転写因子がマウス並びにヒト体細胞における再プログラミングの誘導に寄与する能力を有することが明らかになった。
【0041】
別の好ましい態様では、標的細胞は、該標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しない。
【0042】
因子の内因的発現を評価する方法は当業者に公知であり、本明細書の別の箇所にも記載されている。本発明の方法に従ってiPS細胞を作製するために、標的細胞は標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しなくてもよい。例えば標的細胞であるマウス神経幹細胞においてOct3/4は発現されず、Sox2、Klf4、およびc-Mycは内因的に発現されるということが示される可能性もある。Oct3/4の外来性の導入およびそれに続く発現は、4つ組の再プログラミング因子を補完し、iPS細胞の生成を誘導するのに十分であった。
【0043】
別の好ましい態様では、標的細胞は複能性幹細胞である。
【0044】
複能性幹細胞はいくつかの他の細胞型を生じるが、それらの型の数には限りがある。これは多能性幹細胞が任意の細胞型に分化する能力を有するのと全く対照的である。複能性細胞の一例として、例えば骨髄、臍帯血、または循環中に見られる造血細胞があり、これはいくつかの型の血液細胞となれるが、他の型の細胞にはなれない。複能性細胞の別の例は神経幹細胞である。複能性細胞は亢進的に調節された再プログラミング因子を既に保有するため、再プログラミング標的細胞として特に好適である。
【0045】
より好ましい態様では、複能性幹細胞は外胚葉細胞である。
【0046】
外胚葉は3つの主要な胚細胞層の最も外側のものであり(他の2つは中胚葉および内胚葉である)、ごく初期の胚を構成する。これは分化して多くの重要な組織および構造、例えば皮膚の外層およびその付属器官(汗腺、毛髪、および爪)、歯、眼のレンズ、内耳の各部、神経組織、脳、および脊髄を生じる。外胚葉細胞は神経幹細胞のように既に再プログラミング因子を内因的に発現するため、複能性幹細胞としての外胚葉細胞は標的細胞として特に好適である。
【0047】
別の好ましい態様では、標的細胞は神経幹細胞(NSC)である。
【0048】
神経幹細胞は発生途中の哺乳動物神経系のみならず、全ての哺乳動物(ヒトを含む)の成体の神経系にも存在する。神経幹細胞はより原始的な胚性幹細胞からも誘導されうる。成体において生存可能な位置の数は限定されているものの、分化するためにそれらの子孫が移動する成体幹細胞の位置および脳領域はまだ解明されていない(総説はGage(2000)参照)。神経幹細胞は既に再プログラミング因子を内因的に発現するため、標的細胞として特に好適である。
【0049】
より好ましい態様では、導入すべきコード配列はOct3/4因子をコードする。
【0050】
上記に概説し、後述の実施例に記載するように、Oct3/4のみを神経幹細胞に導入すれば、iPS細胞を作製するには十分であった。C-Mycはキメラの子における発癌性を増加するため(Okita, K., Ichisaka, T.およびYamanaka, S., Nature 448, 313-7 (2007))、最近のc-Mycレトロウィルスの組み込みを用いないiPS細胞の作製を示す研究(Nakagawa, M.ら, Nat Biotechnol 26, 101-106 (2008);Wernig, M., Meissner, A., Cassady, J. P.およびJaenisch, R., Cell Stem Cells 2, 11-12 (2008))は著しい改善を見せている。しかしながら、この態様で示すようにc-Mycを用いずに少ない数のレトロウィルスベクターと組み合わせてiPS細胞を誘導する可能性は、再プログラミングすべき最初の細胞集団の遺伝子調節を最小限にすることへの大きな一歩である。
【0051】
同じ標的細胞を、2つの因子だけの導入によって再プログラミングすることもできた。従って、別のより好ましい態様では、導入すべき2つのコード配列はOct3/4およびc-Myc、またはOct3/4およびKlf4をコードする。
【0052】
更により好ましい態様では、標的細胞はc-Myc、Klf4、およびSox2因子を内因的に発現する。
【0053】
標的細胞が上記の再プログラミング因子の組み合わせを内因的に発現する場合、1つまたは2つの外来再プログラミング因子(例えばOct3/4のみ、またはOct3/4およびc-Myc、またはOct3/4およびKlf4)を導入した時に再プログラミングが起きやすいことも明らかにされた。
【0054】
更に好ましい態様では、標的細胞はc-Myc、Klf4、およびSox2因子を、標的細胞と同じ属の胚性幹細胞における相当する発現レベルに比較して、最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する。
【0055】
本発明の方法によれば、内因性再プログラミング因子の発現レベルが標的細胞と同じ属のESCにおける発現レベルに比較して、一定の範囲内にある場合が有利である。好ましくは、標的細胞は再プログラミング因子であるc-Myc、Klf4、およびSox2を、ESCにおける該因子の相当する発現レベルに比較して、最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する。より好ましくは、標的細胞と同じ属に属するESCの場合よりSox2の発現が約2倍高く、c-Mycが約10倍高く、そして/またはKlf4が約8倍低い。本発明に関連して使用する「約」という用語は、平均偏差が最大+/-20%、好ましくは+/-10%であることをいう。また、発現レベルが該ESCに比較して少なくても8、6、5、4、3、もしくは2倍低い、または多くても8、6、5、4、3、もしくは2倍高い、あるいはその間の任意の数である場合も意図される。
【0056】
より好ましい態様では、標的細胞はマウスまたはヒト神経幹細胞である。
【0057】
更に本発明は、本発明の方法によって作製した人工多能性幹細胞に関する。
【0058】
本発明の方法によって作製した多能性幹細胞は、種々の実験設定および治療設定において有用であり得る。例えば、iPS細胞、分化によってそれから誘導された細胞、または該iPS細胞もしくはそれから誘導された細胞から作製した組織の、治療(therapeuticum)または診断(diagnosticum)としての使用、遺伝子または細胞移植治療中での使用、ゲノム標的の同定および検証のための使用、並びに薬剤スクリーニング法への使用も意図される。
【0059】
iPS細胞の培養条件は相当する種の胚性幹細胞で確立されたものと同じであり、当業者に公知である。一般に、細胞培養法(例えば培地の成分、マーカーの選択肢および選択、細胞の定量および単離)は当該分野で公知であり、例えば、"Practical Cell Culture Techniques", Boulton et Baker編, Humana Press (1992), ISBN 0896032140;"Human Cell Culture Protocols", Gareth E. Jones, Humana Press (1996), ISBN 089603335X、および実施例の項に例証されている。培地中で細胞を培養および維持する方法は当該分野で公知である;増殖培養液および他の細胞培養に関係する物質、並びに細胞の培養を成功させるための説明書および方法は、例えばSigma-AldrichまたはInvitrogen社から入手できる。
【0060】
更に、本発明は標的細胞の人工多能性幹細胞への再プログラミングに関与する化合物の同定法に関するものであり、該方法は以下の工程:(a)本発明の方法に従って標的細胞を再プログラミングすること(1つの導入されるべきコード配列を、試験する化合物で置き換える);および(b)試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価することを含み、ここで、試験する化合物を導入した標的細胞からiPS細胞が形成されれば、その化合物が標的細胞の人工多能性幹細胞への再プログラミングに関与していることを示す。
【0061】
本発明によれば、試験する化合物は1つまたはそれ以上の核酸、例えばDNA、cDNA、RNA、dsRNA、siRNA、shRNA、miRNA、タンパク質、ペプチド、小分子(有機または無機)、化学物質、または任意のそれらの組み合わせであってもよい。
【0062】
本発明の方法にかかる標的細胞の再プログラミングについては既に上述した。標的細胞へ化合物を導入する工程に関しては、試験する化合物の性質によっては本発明の方法を改変する必要があり得る。例えば他の転写因子を評価すべき場合、改変せずに上記のように相当するコード配列を導入してもよい。これに対して化学物質または小分子は、各化合物を細胞培養液に外来的に添加し、細胞の受動的または能動的な取り込み機構を利用することにより、導入してもよい。化合物が実際にそれが代替する因子の代わりとなって標的細胞の再プログラミングを誘導するか否かを試験するために、試験する化合物を細胞(好ましくは核)に導入する方法は当業者に公知である。核酸、例えばDNA、cDNA、RNA、dsRNA、siRNA、shRNA、miRNAはトランスフェクションまたは感染によって導入することができ、小分子(有機または無機)、化学物質は単に膜を通じた浸透によって導入できる。
【0063】
試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価する方法は当業者に公知である。iPS細胞の分類の基準は当業者に公知であり、既に上述した。評価すべき基準によって方法は種々であり、例えば顕微鏡による観察(visual control)、マーカーの発現分析、テラトーマ形成を、単独で、または組み合わせて含みうる。
【0064】
ある細胞の再プログラミングに関与する一組の因子を内因的に発現する該細胞を、更なる因子を外来的に添加することによって補完し、細胞に4つ組の再プログラミング因子(すなわちOct3/4、並びにMyc、Klf、およびSox因子ファミリーそれぞれからの因子)を発現させ、それによって標的細胞の再プログラミングを誘導してもよいという本発明の発見により、再プログラミング過程においてある因子を代替できる化合物の同定が有意に簡易化される。スクリーニングに好適な細胞を作製するために、当該分野で公知の4つの因子のうちの3つまたは全部ではなく、1つまたは2つだけを導入すればよいため、時間、コスト、および実験の困難さが大幅に削減される。新規の再プログラミング因子のためのハイスループットスクリーニング法は、導入する必要のある因子のセットが少ないことによって、時間および効率の点で明らかに改善される。
【0065】
また、本発明はトランスジェニック非ヒト動物の作製法に関し、該方法は以下:(a)本発明の、または本発明の方法によって作製された人工多能性幹細胞を非ヒト着床前胚に導入すること;(b)工程(a)の胚をメス非ヒト動物の子宮に移植すること;および(c)胚を分化および出生させることを含む。
【0066】
本発明に関して使用する「トランスジェニック非ヒト動物」という用語は、本明細書に記載する方法によってそのゲノムの意図的な改変を実施した動物をいう。
【0067】
本発明のトランスジェニック非ヒト動物の作製法は、好ましくは既に構築されている胚性幹細胞を使用したトランスジェニック非ヒト動物の作製法に従い、胚性幹細胞の代わりに本発明のiPS細胞を使用して実施する。該方法は当該分野で公知である(Hogan, B., R. Beddingtonら(1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press;Hanna, J.ら(2007), Science 318(5858): 1920-3;Meissner, A.ら(2007), Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら(2007), Nat Biotechnol.;Okita, K.ら(2007), Nature 448(7151): 313-7;Takahashi, K.ら(2007), Cell 131 (5): 861-72;Wernig, M.ら(2007), Nature 448(7151): 318-24;Yu, J.ら(2007), Science 318(5858): 1917-20;Park, I. H.ら(2008), Nature 451(7175): 141-6)。簡潔に記載すると、iPS細胞の非ヒト着床前胚(例えば桑実胚または胚盤胞)への導入は、好ましくは桑実胚もしくは胚盤胞へのマイクロインジェクション、または8細胞期胚もしくは桑実胚とiPS細胞との凝集によって行う。その後、該キメラ胚を偽妊娠非ヒト・メスの子宮に移植し、胚に分化させ最終的に出生させる(実施例8参照)。
【0068】
iPS細胞からのトランスジェニック非ヒト動物系の作製は、該iPS細胞の多能性(すなわちホストへの注入に際して胚(例えば胚盤胞または桑実胚)に分化し、胚形成に関与し、得られる動物の生殖細胞に寄与する能力)に基づく。上記のように、注入されたiPS細胞を含有する胚盤胞は偽妊娠非ヒト・メスの子宮内で分化し、キメラとして出生する。得られるトランスジェニック非ヒト動物はiPS細胞に由来する細胞のキメラであり、DNAセグメントの組み合わせがホモ接合性であるトランスジェニック動物を同定するために、これを野生型非ヒト動物と戻し交配し(backcrossed)、iPS細胞の遺伝的内容のみを保有する動物をスクリーニングする。
【0069】
トランスジェニック非ヒト動物は、例えばトランスジェニックマウス、ラット、ハムスター、イヌ、サル、ウサギ、ブタ、またはウシであってもよい。好ましくは該トランスジェニック非ヒト動物はマウスである。
【0070】
従って本発明は、本発明の方法に従って作製したトランスジェニック非ヒト動物にも関する。
【0071】
最後に、本発明は、遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための、本発明の方法によって作製したiPS細胞を含む組成物に関する。
【0072】
本明細書で使用する組成物は、iPS細胞、および好ましくは該細胞の生存能力を維持する更なる成分を含有する組成物に関する。それらの成分は当業者に公知であり、例えば細胞培養液成分を含む。更に、意図する適用によって、組成物は更なる成分、例えば患者への投与を容易にする成分を含んでもよい。
【0073】
本発明のiPS細胞を含む組成物(本発明のiPS細胞自体と同様)を種々の実験のシナリオおよび治療のシナリオに使用することができる。相対的に少数のトランスジェニック発現要素を有し、癌性細胞に分化するリスクが全体的に低い本発明のiPS細胞は、遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングにおいて有益であると考えられる。
【0074】
遺伝子治療は、遺伝的欠損を修正するためにex vivoまたはin vivoの手法によって治療のためのDNAコンストラクトを生殖系細胞に導入することに基づくが、これは遺伝子移入の最も重要な適用の1つである。in vitroまたはin vivo遺伝子治療に好適なベクターおよび方法は文献に報告されており、当業者に公知である(Davis PB, Cooper MJ., AAPS J. (2007), 19;9(1):E11-7;Li S, Ma Z., Curr Gene Ther. (2001),1 (2):201-26)。本発明によれば、患者から得た細胞を、例えば当該分野で既知の方法によって遺伝子的に修正した後、本発明の方法によってES細胞の表現型および遺伝子型を有するiPS細胞に再プログラミングすることができうる。これは遺伝子治療および/または細胞療法におけるiPS細胞の適用性を証明するものである。再生医療を用いて、損傷した組織をin vivoで再生させるか、または組織および臓器をin vitroで成長させた後、それを患者に移植することによって、機能不全の、損傷した、または欠陥を有する組織に起因する任意の疾病を治療できる可能性がある。実質的に任意の組織(外胚葉、中胚葉、内胚葉細胞)に分化できる本発明のiPS細胞は、再生医療のあらゆる観点で使用することができ、それによってES細胞の必要性は大幅に低減される。
【0075】
本発明のiPS細胞を用いて薬剤ターゲットを同定し、可能性のある薬物療法学を試験することができ、それによってES細胞およびin vivoにおける研究の必要性は低減される。可能性のある薬剤の効果(例えば標的部位および標的特異性、毒性、生物学的利用能)の同定および/または評価のための実験手順および方法は、当業者に公知である。
【0076】
更に、iPS細胞を使用して出生異常の予防および治療の研究、または細胞分化の研究を行ってもよい。
【0077】
また、本発明のiPS細胞は(治療的適用ではなく)実験設定において、標的細胞の再プログラミングの誘導の際の脱分化に関する種々の観点(例えば遺伝子の発現パターンまたはメチル化パターンの時空間的変化、または凝集挙動に変化を与える形態学的変化)を研究するのに有用でありうる。さらに、iPS細胞で、例えば以下に関係する研究を行うこともできる:遺伝子治療、遺伝子ターゲティング、分化の研究、薬剤の安全性および有効性に関する試験、自己または同種異系(allogenic)再生させた組織の移植、組織修復(例えば神経系、心筋)、疾病(例えばパーキンソン病、心臓発作、糖尿病、癌、白血病、または脊髄損傷)、胚性遺伝子発現、胚性遺伝子の遺伝子操作、初期発生学および胎児発達、胚細胞マーカーの同定、細胞移動またはアポトーシス。
【0078】
図1:OG2/Rosa26トランスジェニックマウスの成体NSCからの2F Oct4/Klf4(OK)iPS細胞の作製。
a.ESCおよびNSCにおけるOct4、Nanog、Klf4、Sox2、およびc-MycのRT-PCRおよびqRT-PCR分析。ローディング・コントロールとしてβ-アクチンを使用した。b.ESCおよびNSCにおける4因子のウェスタンブロット分析。ローディング・コントロールとして抗アクチン抗体を使用した。c.感染14日後の2F OK iPS細胞コロニーの形態。ESC様コロニーがOct4-GFPを発現(f)。d.樹立した2F OK iPS細胞(クローンF-4)の感染30日後の形態(放射線照射したMEFで増殖)。位相定数およびOct4-GFP(g)を示す。e.感染30日後のNSCおよび擬似感染の形態(h)。i.2F OKおよび4F感染の7日後、14日後、および21日後のGFP+コロニーの生成(n=3;エラーバーはs.d.を表す)。j.2Fおよび4F iPS細胞作製の再プログラミング効率(n=3)。感染の7日後、14日後、および21日後の50,000播種NSCあたりのGFP+コロニー総数を示す。
【0079】
図2:iPS細胞の遺伝子発現プロファイル。
a.ESC、4F iPS細胞(クローンA-2c)、2F OK iPS細胞(クローンB-2、D-7、およびF-4)、およびNSCにおけるES細胞マーカー遺伝子発現のRT-PCR分析。プライマーは各内因性遺伝子座からの転写物に特異的である。β-アクチンをローディング・コントロールとして使用した。b.NSC、2F (OK) iPS、4F iPS、およびESCでの種々の発現遺伝子のヒートマップ。市街地距離(cityblock distance)および平均連結法を用いて遺伝子の階層的クラスタリングを行った。c.DNAマイクロアレイを用いて2F iPS細胞(クローンF-4)およびESC間、並びに2F iPS細胞(クローンF-4)およびNSC間で総体的な遺伝子発現パターンを比較した。d.黒線は2つの細胞型間の遺伝子発現レベルの2倍の変化を示している。NSCまたはESCと比較して2F iPS細胞(クローンF-4)で過剰発現している遺伝子を青で示す;低発現のものを赤で示す。散布図に多能性遺伝子Oct4、Nanog、Sox2、c-Myc、Klf4、およびLin28の位置を示す。遺伝子発現レベルはlog2で測定する(scaled)。
【0080】
図3:2F Oct4/Klf4(OK)iPS細胞(クローンF-4)は多能性であり、in vitroおよびin vivoにおいて分化する。
a.in vitroにおける3つの胚葉全てへの分化。胚様体形成後、凝集体をゼラチンコーティングしたプレート上に移し、更に10日間分化させた。細胞を抗Tuj1、抗α-フェトプロテイン(AFP)、または抗Flk1で染色した。核はDAPIで染色した。b.3つの胚葉全てを含有するF-4 iPS細胞のテラトーマ。F-4 iPS細胞(1.5 x 106細胞)をヌードマウスの皮下に接種した。4週間後、テラトーマをヘマトキシリンおよびエオシン色素で染色した。神経ロゼット(外胚葉)、筋肉(中胚葉)、および円柱上皮(内胚葉)を含有するテラトーマを示す。
【0081】
図4:2F Oct4/Klf4(OK)iPS細胞(クローンF-4)のin vivoにおける分化能。
a.F-4 iPS細胞のキメラ胚は凝集の24時間後に胚盤胞に分化した。蛍光光学は、Oct4-GFP細胞が胚盤胞の内部細胞塊中に存在することを示している。b.Oct4-GFPの発現によって示される、F-4 iPS細胞のマウス胚発生への生殖系列の関与。胚をE13.5で蛍光顕微鏡を用いて分析した。c、d.13.5 dpcのキメラ胚(コントロール、2N、および4N)をX-gal溶液で染色した。e.lacZ染色した13.5 dpcのキメラ胚(2N)の組織学的分析。f.F-4 iPS細胞によって作製したキメラマウス(8週齢)。F-4 iPS細胞に由来するアグーチ毛色(agouti coat colour)。g.F-4 iPS細胞に由来するキメラのPCRジェノタイピング。Oct4-GFPについてPCR分析を行った(上段パネル)。F-4 iPS細胞の生殖細胞系伝達。CD-1メスと交配させたキメラ・オスの子孫の遺伝子型同定(genotyping)により、Oct4-GFPおよびlacZ対立遺伝子の存在、ならびにOct4およびKlf4のウィルス組込みが明らかになった(下段パネル)。略語:Gastroint. tract.:胃腸管。
【0082】
図5:1因子hNSC由来iPS(1F hNiPS)細胞のコロニー形成および細胞株のキャラクタリゼーション。
(A)NSC培地中で増殖させたhNSCの形態。(B)hOCT4感染細胞の感染10週間後のコロニー形成。(C)コロニーはhESC様形態を成すが、コロニーの中央は再プログラミングされていない神経ロゼットのままである。(D)継代1で機械的に単離した後のフィーダー上での典型的なhESC様iPSコロニー形成(1F hNiPSクローンC)。(E)継代10でのiPSコロニーの高倍率画像。(F)1F hNiPSコロニーをAPで染色した。スケールバーは250μm。(G)2F hNiPS(クローンA)および1F hNiPS(クローンC)細胞における多能性マーカー(OCT4、SSEA4、TRA-1-60、およびTRA-1-81)の免疫細胞化学分析。核をDAPIで染色する(青)。スケールバーは250μm。
【0083】
図6:hNSC由来iPS(hNiPS)細胞における多能性マーカーの発現レベルおよびDNAメチル化分析。
(A)H1 hESC、hNSC、2F hNiPSクローン(A、B、およびC)、および1F hNiPSクローン(AおよびC)における多能性マーカーの定量PCR分析。データは、内因性転写物に特異的なプライマーを使用し、H9 hESCに対する相対発現で示す。RNA発現レベルは対数尺度で示す。転写レベルはβ-アクチンレベルで正規化した。エラーバーは3回行った試験(triplicates)からのs.d.を示す。(B)H9 hESC、hNSC、2F hNiPSクローン(A、B、およびC)、および1F hNiPSクローン(AおよびC)におけるOCT4およびNANOGプロモーター領域のバイサルファイトシーケンシング分析。所与の増副産物に関するそれぞれの丸印の列は、その領域での1つの細菌クローンの各CpGのメチル化状態を表す。白丸はメチル化されていないCpGを表し、黒丸はメチル化されたCpGを表す。各列の下部の数字は、転写開始位置(TSS;+1)に対するCpGジヌクレオチドの位置を示す。
【0084】
図7:in vitroにおけるhNSC誘導iPS(hNiPS)細胞の3つの胚葉への分化。
(A)免疫蛍光分析は、2Fおよび1F hNiPS細胞が3つの胚葉:内胚葉(α-フェトプロテイン;AFP)、中胚葉(α-平滑筋アクチン;α-SMA)、および外胚葉(β-チューブリン IIIb;Tuj1)の全てに分化することを示している。DAPIで核を染色する(青)。スケールバーは100μm。(B)2F hNiPS(クローンA)および1F hNiPS(クローンC)細胞に由来する1ヶ月胚様体(EB)分化の定量PCR分析。内胚葉(AFP、GATA6、およびSox17)、中胚葉(FOXF1およびHAND1)、および外胚葉(NCAM1、PAX6、およびSox1)。データはそれぞれの未分化hNiPS親細胞に対する相対発現で示す。RNA発現レベルを対数尺度で表す。転写レベルはβ-アクチンレベルに対して正規化した。
【0085】
図8:in vivoにおけるhNSC由来iPS(hNiPS)細胞の多能性および総体的遺伝子発現プロファイル。
(A)2F hNipS(クローンA)および1F hNiPS(クローンC)細胞をSCIDマウスに移植した後のテラトーマ形成。6-8週間でテラトーマを切片化し、ヘマトキシリンおよびエオシンで染色した。3つの胚葉:内胚葉(呼吸上皮;r)、中胚葉(骨格筋;m、軟骨;c)、および外胚葉(神経上皮;n)全てを示すことが同定された細胞の組織切片。拡大部は呼吸上皮、筋肉および神経上皮を示す(矢印で表示)。スケールバーは100μm。(B)hNSC、1F hNiPS(クローンC)、2F hNiPS(クローンA)、H9 hESC、およびH1 hESC(左)からの総体的な遺伝子発現のヒートマップ(左パネル)および階層的クラスター分析(右パネル)。(C)1F hNiPS(クローンC)およびH9 hESC間(左パネル)、2F hNiPS(クローンA)およびH9 hESC間(中央パネル)、そしてhNSCおよび1F hNiPS(クローンC)間(右パネル)間の総体的な遺伝子発現プロファイルを比較する散布図。黒線は2つの細胞集団間の遺伝子発現レベルが2倍の相違であることを示す。転写発現レベルはlog2スケールである。
【図面の簡単な説明】
【0086】
【図1】図1は、OG2/Rosa26トランスジェニックマウスの成体NSCからの2F Oct4/Klf4(OK)iPS細胞の作製を示す。
【図2】図2は、iPS細胞の遺伝子発現プロファイルを示す。
【図3】図3は、2F Oct4/Klf4(OK)iPS細胞(クローンF-4)は多能性であり、in vitroおよびin vivoにおいて分化することを示す。
【図4】図4は、2F Oct4/Klf4(OK)iPS細胞(クローンF-4)のin vivoにおける分化能を示す。
【図5】図5は、1因子hNSC由来iPS(1F hNiPS)細胞のコロニー形成および細胞株のキャラクタリゼーションを示す。
【図6】図6は、hNSC由来iPS(hNiPS)細胞における多能性マーカーの発現レベルおよびDNAメチル化分析を示す。
【図7】図7は、in vitroにおけるhNSC誘導iPS(hNiPS)細胞の3つの胚葉への分化を示す。
【図8】図8は、in vivoにおけるhNSC由来iPS(hNiPS)細胞の多能性および総体的遺伝子発現プロファイルを示す。
【発明を実施するための形態】
【0087】
実施例にて本発明を例証する。
【0088】
実施例1:OG2マウスの作製
OG2系統をROSA26トランスジェニック系統と数代にわたって交配させ(Do, J. T.およびScholer, H. R., Stem Cells 22, 941-9 (2004);Szabo, P. E., Hubner, K., Scholer, H.およびMann, J. R., Mech Dev 115, 157-60 (2002))、neo/lacZおよびOct4-GFP導入遺伝子の複合ホモ接合性マウスを作製した。NSCを誘導するために、ホモ接合性OG2 x ROSA26オス・マウスをICRメスと交配させてヘテロ接合性の子を出生させる。5日齢のOG2 x ROSA26ヘテロ接合性マウスから脳組織を回収した。
【0089】
実施例2:人工多能性幹細胞の作製
iPS細胞およびESCをESC培養液(15% FBS、非必須アミノ酸、L-グルタミン、ペニシリン/ストレプトマイシン、β-メルカプトエタノール、および1,000 U/ml 白血病抑制因子(LIF)を補充したDMEM)中、放射線照射したMEF上で増殖させた。Oct4、Sox2、Klf4、およびc-MycのマウスcDNAをコードするpMXに基づくレトロウィルスベクターを、Fugene 6トランスフェクション試薬(Roche社)を用いて、パッケージング欠損ヘルパープラスミドと共に293細胞に個別にコトランスフェクトした。48時間後、従前の報告(Zaehres, H.およびDaley, G. Q., (2006), Methods Enzymol 420, 49-64)のようにウィルス上清を回収した。OG2/Rosa26トランスジェニックマウスから誘導したNSCを6-ウェルプレートあたり5 x 104細胞の密度で播種し、4因子(1:1:1:1)またはOct4およびKlf4(1:1)のウィルス含有上清に6μg/mlの硫酸プロタミン(Sigma社)を補充したものと共に24時間インキュベートした。pMX-GFPコントロールウィルスを用いて形質導入効率を算出した。細胞を新たな神経増殖培養液に再播種した。感染の2日後、更なる選択を行わずに、細胞をLIF含有ESC培養液中、放射線照射MEF上で更に継代培養した。Oct4-GFP陽性コロニーを機械的に単離し、個々の細胞を分離した後、MEF上に再播種した。コロニーを選択して増殖させた。
【0090】
実施例3:qRT-PCR分析
MiniRNeasy Kit(Qiagen社、ドイツ、ヒルデン市;http://www.qiagen.com)を用い、製造者の説明書に従って細胞から総RNAを抽出した。High Capacity cDNA Archive Kit(Applied Biosystems社、ドイツ、ダルムシュタット市;http://www.appliedbiosystems.com)を用い、製造者の説明書に従い、反応液容量を20μlにスケールダウンして相補的DNA合成を行った。ABI PRISM Sequence Detection System 7900(Applied BioSystems社)およびready-to-use 5'-nuclease Assays-on-Demandを用いて転写レベルを測定した。各リアルタイム増幅で、テンプレートは5ngの総RNA相当であった。測定は3回重複して(triplicate)行った;各サンプルのRTブランクおよびテンプレートなしのブランクをネガティブコントロールとした。増幅曲線および遺伝子発現をハウスキーピング遺伝子Hprt(内部標準として使用)に対して正規化した。
【0091】
以下の遺伝子の検出のために、Taqman Assay-on-Demandによってオリゴヌクレオチドを設計した:Pou5f1 (Oct3/4) (Mm00658129_gH)、Sox2 (Mm00488369_s1)、c-Myc (Mm00487803_m1)、Klf4 (Mm00516104_m1)、B-Act (Mm00607939_s1)、およびHprt1 (Mm00446968_m1)。Nanogおよびウィルス配列を検出するためのオリゴは注文設計した。ΔΔCt法(ABI PRISM 7700 Sequence Detection System, ユーザー会報#2)を用いて各プローブ/プライマーセットに関して得られた増幅曲線のlog直線相内で内因性Hprt遺伝子について定量を正規化した。
【0092】
ウィルス特異的qRT-PCRのプライマー配列
【0093】
【化1】
【0094】
実施例4:マイクロアレイ分析
Affymetrix Mouse Genome 430 2.0 GeneChip arrays(Affymetrix社、カリフォルニア州サンタクララ市)を用い、基本的に従前の報告(Ruau, D.ら,(2008), Stem Cells)に基づいてマイクロアレイ研究を行った。簡潔に記載すると、DNAアーゼ消化を含むRNAeasy kit(Qiagen社、ドイツ、ヒルデン市)を用いて細胞から総RNAを抽出した。GeneChip One-Cycle標識キット(Affymetrix)を用いてビオチン標識したcRNAを3μgの総RNAから得た。15μgのcRNAを断片化し、45℃で16時間、Affymetrix 430 2.0 GeneChipアレイにハイブリダイズさせた。DNAチップを洗浄し、染色してAffymetrix Fluidics装置およびGCS3000スキャナーを用いてスキャンし、得られた画像をGCOSソフトウェアを用いて分析した。ESCおよびiPS細胞では3回重複して、NSCに関しては2回重複して実験を行った。BioConductorで実行されたRMAアルゴリズム(Irizarry, R. A.ら, (2003), Nucleic Acids Res 31 , e15)を用いて、正規化を行った。
【0095】
実施例5:in vitroにおけるiPS細胞の分化
Oct4-GFP細胞をFACS分析によって回収し、胚様体(EB)のin vitro分化に用いた(LIFを含有しないESC培養液中、懸滴で実施)。3日後、EBをゼラチンコーとした4-ウェルプレートに播種し、さらに10日間培養した。細胞を抗Tuj1抗体(1:100。Chemicon)、抗αフェトプロテイン(AFP)抗体(1:100。R&D Systems)、または抗FlK1抗体(1:100。R&D Systems)で染色した。
【0096】
実施例6:ウェスタンブロット分析、SSEA-1、およびAP染色
ESCおよびNSCから調製した総細胞ライセート(2 x 106)を、Oct4 (Santa Cruz)、Sox2 (Santa Cruz)、Klf4 (Abcam)、およびc-Myc (Abcam)の発現に関するウェスタンブロット分析に供した。全てのサンプルで、β-アクチン発現レベルをローディングコントロールとして使用した(Abcam)。
【0097】
ES Cell Characterization Kit (Chemicon)を用い、製造者のプロトコルに従って、SSEA-1およびアルカリホスファターゼ (AP)染色を行った。
【0098】
実施例7:テラトーマの形成
iPS細胞およびNSC細胞(1.5 x 106細胞/マウス)をヌードマウスの背側側腹部に皮下注射した。注射4週間後、形成されたテラトーマを4% PFAで一晩固定し、パラフィンに包埋した。切片をヘマトキシリンおよびエオシン色素で染色した。
【0099】
実施例8:キメラの形成
iPS細胞を凝集させ、裸出されたコンパクション後の(denuded post-compacted)8細胞期マウス胚と共に培養した。簡潔に記載すると、2細胞期胚を1.5dpcのマウス((C57BL/6 x C3H)F1メス x CD1オス)から洗い出し(flushed)、M2培地中に配して、0.1% BSA含有KSOM培地中で一晩、8細胞期まで培養した。短時間のトリプシン処理をした2日目の培地から緩く結合したiPS細胞塊(10-20細胞)を選択し、鉱油下の10% FCS含有KSOM培地の懸滴に移した;各塊を懸滴中のくぼみに(in a depression)配した。それと同時に、30-40個の胚のバッチを酸性化タイロード液と共に、透明帯が分解するまで短時間インキュベートした。1個の胚を細胞塊上に配した。全ての凝集体をこの方法で集め、5% CO2/大気雰囲気下、37℃で培養した。培養24時間後、凝集体の多くが胚盤胞を形成していた。総数64個の凝集した胚盤胞(2.5 dpc)を5検体の偽妊娠マウス(CD-1バックグラウンド)の子宮角に移植した。
【0100】
実施例9:Oct4によるヒト神経幹細胞の再プログラミング
ヒト胎児脳組織から誘導したhNSCを従前の報告(Kimら, Exp Neurol 199, 222 (2006);Parkら, Nat Biotechnol 20, 1111 (2002))に従って無血清NSC培地で増殖させた(図5A参照)。まずhNSCをヒトOCT4およびKLF4(2F)またはOCT4(1F)をコードするpMXに感染させた。次いで、感染させたhNSCを7日間までNSC培地中に維持した(Kimら, Exp Neurol 199, 222 (2006))。感染8日後、10ng/ml bFGF含有hESC培地およびMEF条件培地(CM)(1:1)中で細胞を支持細胞(feeder cell)層上に再播種し、hESC様コロニーが出現するまで培養を続けた。感染後10-11週間以内に、hESC様iPSコロニーを同定したが、コロニーの中央は神経ロゼッタの様相のままであった(図5B参照)。更に5-6日以内に、コロニーは増殖して大きくなり、典型的なhESC様形態を示したが、コロニーの中央には神経ロゼッタが残存していた(図5C参照)。神経ロゼッタをコロニーから分離する。その後、コロニーの一片を機械的単離によって支持細胞層上に移植した(図5D参照)。OCT4に感染させたhNSCからのピッキングによって、3つのhESC様コロニーから2つのクローンを樹立することに成功した(1F hNiPSクローンAおよびC。再プログラミング効率0.02%)。他方、我々は2F感染hNSCで、感染後7-8週間以内に、5つのhESC様コロニーから3つのクローンを樹立した(2F hNiPS A、B、およびC。再プログラミング効率0.15%)。これらは全て、hESC培養条件で増殖することができた。1F hNiPS細胞はhESCと形態学的に類似しており、アルカリホスファターゼ染色陽性であった(図5EおよびF)。免疫蛍光染色により、2Fおよび1F hNiPS細胞が多能性マーカー(OCT4、SSEA4、TRA-1-60、およびTRA-1-81を含む)を一様に発現していることが確認された(図5G参照)。これらの結果は、OCT4およびKLF4、並びにOCT4のみによって、hNSCからヒトiPS細胞を作製できることを示している。
【0101】
次に我々は、定量RT-PCR分析により、これらのiPS細胞における多能性マーカー遺伝子のmRNA発現レベルを分子レベルで試験した。hESC特異的マーカーを内因的に発現する2Fおよび1F hNiPS細胞は、H9およびH1 hESCに類似しており、親hNSCと比較して有意に亢進的に調節されていた(図6A参照)。遺伝子型同定PCRは、1F hNiPSクローンがゲノム中にOCT4導入遺伝子のみを保有し、2F hNiPSクローンがOCT4およびKLF4導入遺伝子を保有することを示した。我々はまた、2F hNiPSクローンBからのOCT4発現を除いて、導入遺伝子OCT4またはKLF4の発現レベルが2Fおよび1F hNiPSクローンにおいて有意に抑制されていることも確認した。サザンブロット分析により、2Fおよび1F hNiPSクローンにおけるOCT4導入遺伝子の組込みが確認された。実験室でのhESCからの混入によってiPSクローンが生じた可能性を排除するために、DNAフィンガープリント分析を行って、hNiPS細胞がドナーhNSCと厳密に相関することを確認した(表2参照)。
【0102】
再プログラミングした細胞からのOCT4およびNANOGプロモーターのエピジェネティックな改変を確認するために、バイサルファイトシークエンシングを行って両プロモーターの脱メチル化を測定した。OCT4およびNANOGプロモーター領域はドナーhNSCに対して2Fおよび1F hNiPS細胞では脱メチル化されており、hESCと類似していた。これらをまとめると、hNSCはOCT4単独の形質導入によって分子レベルでhESCに類似したiPS細胞に再プログラミングできる。
【0103】
次に我々は、胚様体(EB)分化および直接分化によってin vitroにおける2Fおよび1F hNiPS細胞の多能性を試験した。hNiPS細胞はEB分化によって容易に内胚葉(AFP)、中胚葉(a-SMA)、および外胚葉(Tuj1)に分化し(図5A参照)、我々は定量RT-PCR分析によって直接分化から3つの胚葉マーカー全ての発現を確認した(図7B参照)。in vivoにおけるこれらのヒトiPS細胞の多能性を確認するために、細胞を重度の複合型免疫不全(SCID)マウスに皮下移植した。移植の6-8週後、2Fおよび1F hNiPS細胞は3つの胚葉全てを(呼吸器、骨格筋、軟骨組織、および神経上皮を含む)含有するテラトーマを生じた(図8A参照)。これらの結果は2Fおよび1F hNiPS細胞がhESCのようにin vitroおよびin vivoにおいて多能性を有することを示している。
【0104】
最後に我々は、cDNAマイクロアレイによって、hNSC、hNSCに由来する2Fおよび1F hNiPS細胞、H9およびH1 hESCの総体的な遺伝子発現分析を行った。ヒートマップは、2Fおよび1F hNiPS細胞がhESCと同様であり、他方、親hNSCは多能性集団から分離されることを示し(図8B、左パネル参照)、階層的クラスター分析は、hNiPS細胞がhESCとクラスターを形成し、親hNSCとは区別されることを示している(図8B、右パネル参照)。散布図分析は、hNiPS細胞が異なるhESC間のように、親hNSCよりhESCとより高い類似性を有することを示している(図8C参照)。1Fおよび2F hNiPS細胞もH1 hESCとの類似性を示す。これらのデータはhNiPS細胞が総体的な遺伝子発現プロファイルにおいてhESCと類似していることを示している。結果は1Fおよび2F hNiPS細胞が分子レベルおよび多能性に関してhESCに酷似していることを示している。
【0105】
【表2】
【0106】
試料および方法
細胞培養
ヒトNSCは終脳(HFT13)に由来し、従前の報告(Kimら, Exp Neurol 199, 222 (2006))に従って樹立した。簡潔に記載すると、終脳組織を新たに切り出し、0.1%トリプシン中で30分間解離させ、NSC培地中、200,000細胞/mlの密度で10cmプレートに播種した。これらの細胞を従前の報告(Kimら, Exp Neurol 199, 222 (2006);Parkら, Nat Biotechnol 20, 1111 (2002))に従ってNSC培地中で培養した。ヒトESおよびiPS細胞を、マイトマイシンC処理したCF1マウス支持細胞層(Millipore)上で、ヒトESC培地(20% ノックアウト血清代替添加物(Invitrogen)、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、ペニシリン/ストレプトマイシン、および10ng/ml ヒト塩基性線維芽細胞成長因子(bFGF) (Invitrogen)を補充したノックアウトDMEM (Invitrogen)を含有)中、従前の報告(Takahashiら, Cell 131 , 861 (2007))に従って維持した。
【0107】
1F hNiPS細胞および2F hNiPS細胞の導入
OCT4およびKLF4のヒトcDNAをコードするpMXに基づくレトロウィルスベクター(TakahashiおよびYamanaka, Cell 126, 663 (2006))をFugeneトランスフェクション試薬(Roche社)を用いて、パッケージング欠損ヘルパープラスミドと共に293細胞にコトランスフェクトし、従前の報告(ZaehresおよびDaley, Methods Enzymol 420, 49 (2006))に従って水疱性口内炎ウィルス(VSV)Gタンパク質偽型ウィルスを生成させた。トランスフェクションの48時間後にウィルス上清を回収し、超遠心によって濃縮し、ヒトNSCに感染させた。iPSの作製のために、ヒトNSCを6-ウェルプレートあたり5 x 104細胞の密度で播種し、Oct4またはOct4およびKLF4のウィルス含有上清に6μg/mlの硫酸プロタミン(Sigma社)を補充したものと共に24時間インキュベートした。翌日、感染後1日目に培地をフレッシュなNSC培地で交換し、感染後7日目まで維持した。感染後8日目から、細胞をヒトESC培地中で更に培養した。感染後2ヶ月または2.5ヶ月でiPSコロニーを機械的に単離した後、ヒトESC培地中、CF1マウス支持細胞層 (Millipore)上に播種し、維持した。
【0108】
定量RT-PCR
バルクの細胞培養液サンプルまたは手でピックアップした未分化コロニーから、オンカラムDNA消化を含むRNeasyカラム(Qiagen)を用いて総RNAを単離した。オリゴ-dT15プライミングおよびM-MLV逆転写酵素(UBS社)を用い、製造者の説明書に従って42℃で1時間、cDNAを生成させた。通常、20μlのSYBR Green PCR反応液に約50ngの総RNA相当物をテンプレートとして使用し、反応液は更に0.375μMの各プライマーおよび10μlのSYBR Green PCR mix (ABI)を含有する(95℃で15秒/60℃で60秒を40サイクル。Applied Biosystems 7300装置)。使用したプライマーは全て、副産物を生成せずに最適に近い効率で予期する産物を増幅することを確認した。プライマー配列を表3に示す。比較Ct法を用い、生体コントロールサンプルおよび正規化のための2つのハウスキーピング遺伝子に基づいて相対発現レベルを算出した。エラーバーは生体複製物(マーカー遺伝子発現データ)または正規化のための個別のハウスキーピング遺伝子(導入遺伝子サイレンシングデータ)を用いて得られた標準誤差を表す。
【0109】
総体的な遺伝子発現分析
転写分析を、400ngの総DNA未含有RNAを標識cRNA合成(Illumina TotalPrep RNA Amplification Kit - Ambion)のインプットとして使用し、製造者の説明書に従って行った(IVT:10h)。クォリティーを確認したcRNAサンプルを生物学的または技術的な複製(duplicate)としてHumanRef-8 v3 expression BeadChips(Illumina)上に18時間ハイブリダイズさせ、洗浄、染色、およびスキャンした(ガイドラインに従い、製造者が提供/推奨する物質/装置を用いた)。マイクロアレイ・データはGEO(Gene Expression Omnibus)ウェブサイト(受入番号:GSE GSE15355)から入手できる。
【0110】
バイサルファイトシーケンシング
DNeasyカラム(Qiagen)を用いてバルクの細胞培養液サンプルまたは手でピックアップした未分化コロニーからゲノムDNAを単離した。300ngを亜硫酸水素変換のインプットとして使用した(EpiTect Bisulfite Kit - Qiagen)。50ngの変換DNAを従来型のネステッドPCRのテンプレートとして使用し、Oct4およびNANOGプロモーターのそれぞれ467および336bpの領域を増幅した。プライマーはセンスDNA鎖の変換に特異的であり、表3に記載する。精製したPCRをpCR2.1-TOPO (Invitrogen)にTA-クローニングした。ランダムにピックアップしたクローンのインサート配列をBiQ Analyzerプログラムを用いて分析し、必要によりクォリティーチェックに基づく示唆に従って、個々のクローンを除外(drop)した。OCT4転写開始コドンから+20位のCpG部位からのデータは、情報を与えないため記載していない。
【0111】
ショートタンデムリピート(STR)分析
DNeasyカラム(Qiagen)を用いて培養細胞サンプルからゲノムDNAを単離した。これをSTR分析(PowerPlex 16 System (Promega)およびABI PRISM装置を使用)のテンプレートとして使用した。数字は15の常染色体フラグメントの長さ(bp)を示す。Eurofins Medigenomix (ドイツ マルティンスリード)で分析を実施した。
【0112】
テラトーマ形成
hNiPS細胞およびhNSC(3-5 x 106 細胞/マウス)をSCIDマウスの背側側腹部に皮下注射した。注射の6-8週間後にテラトーマを4% PFAで一晩固定し、パラフィンに包埋した。切片をヘマトキシリンおよびエオシン色素で染色した。
【0113】
アルカリホスファターゼ(AP)および免疫蛍光染色
アルカリホスファターゼ(AP)染色をES Cell Characterization Kit(Chemicon)で製造者のプロトコルに従って実施した。以下の一次抗体を使用して免疫蛍光染色を行った:AFP(Sigma、1:100)、a-SMA(Sigma、1:50)、TuJ1(Chemicon、1:500)、OCT4(Santa Cruz、1:200)、SSEA4(Chemicon、1:200)、TRA-1-60(Chemicon、1:200)、TRA-1-81(Chemicon、1:200)。
【0114】
【表3】
【0115】
【表4】
【0116】
サザンブロット分析
BamHIで消化した1F hNiPS、hNSC、および2F hNiPS細胞からのゲノムDNAを0.8%アガロースゲル上で分離し、Biodyne Bナイロン膜(PALL Life Sciences)に転写した。DecaLabelTM DNA Labeling Kit (Fermentas)を用いてDNAをOCT4の32P標識したフラグメント(Eco81I (Saul)ヒトOCT4 cDNAフラグメント)とハイブリダイズさせた。標識したラムダHindIII消化DNAをマーカーとした。
【0117】
in vitroにおけるヒトiPS細胞の分化
免疫細胞化学で、懸滴法を用い、MEF条件培地中でiPS細胞から胚様体(EB)を作製した。5日後、EBをゼラチンコーティングしたプレートに移し、20% FBS、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、およびペニシリン/ストレプトマイシンを補充したノックアウトDMEM (Invitrogen)中で更に14日間培養した。qRT-PCRで、iPSコロニーを機械的に単離し、MEF条件培地中、マトリゲルをコーティングしたプレート上に再播種した。2日後、培地を3つの胚葉分化培地で置換した。内胚葉分化では、細胞を、2% FBS、100ng/ml アクチビンA (R&D Systems)、L-グルタミン、およびペニシリン/ストレプトマイシンを補充したRPMI1640培地中で3週間維持した(Huangfuら, Nat Biotechnol 26, 1269 (2008))。中胚葉分化では、100uM アスコルビン酸(Sigma)、20%-FBS、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、およびペニシリン/ストレプトマイシンを補充したノックアウトDMEM中で3週間維持した(Aasenら, Nat Biotechnol 26, 1276 (2008))。外胚葉分化では、細胞をN2B27培地中で7日間維持し、2週間の間、10ng/ml bFGF2(peprotech)、100ng/ml ソニックヘッジホッグ(R&D Systems)、10ng/ml PDFG(R&D Systems)、L-グルタミン、およびペニシリン/ストレプトマイシンを補充したN2培地で置換した。培地は1日おきに交換した。プライマー配列を表3に示す。
【0118】
参考文献
Biswas, A. and Hutchins, R. (2007). Embryonic stem cells. Stem Cells Dev 16(2):213-22.
Davis, P. B., Cooper, M.J. (2007). Vectors for airway gene delivery. AAPS J 19;9(1):E11-7.
Do, J. T. & Scholer, H. R. (2004). Nuclei of embryonic stem cells reprogram somatic cells. Stem Cells 22:941-9.
Gage, F. H. (2000). Mammalian neural stem cells. Science 287(5457): 1433-8.
Hanna, J., Wernig, M., Markoulaki, S., Sun, C.W., Meissner, A., Cassady, J. P., Beard, C, Brambrink, T., Wu, L.C., Townes, T.M. and Jaenisch, R. (2007).
Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318(5858): 1920-3.
Hogan, B., R. Beddington, et al. (1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press.
Jones, G. E., Wise, CJ. (1997). Establishment, maintenance, and cloning of human dermal fibroblasts. Methods MoI Biol. 75:13-21.
Kim, D. S., Kim, J.Y., Kang, M., Cho, M.S., Kim, D.W. (2007). Derivation of functional dopamine neurons from embryonic stem cells. Cell Transplant 16(2): 117-23.
Li, S. and Ma, Z. (2001). Nonviral gene therapy. Curr Gene Ther.,1(2):201-26. Meissner, A., Wernig, M. and Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol 25(10): 1177-81.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., lchisaka, T., Aoi, T., Okita, K., Mochiduki, Y., Takizawa, N. and Yamanaka, S. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 26(1): 101 -6.
Okita, K., lchisaka, T. and Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature 448(7151):313-7.
Park, I. H., Zhao, R., West, J.A., Yabuuchi, A., Huo, H., Ince, T.A., Lerou, P.H., Lensch, M.W., Daley, G. Q. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451 (7175): 141 -6.
Pollard, S. M., Conti, L., Sun, Y., Goffredo, D. & Smith, A. (2006). Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 16 Suppl 1 , i112-20.
Ryan, A. K. and Rosenfeld, M. G. (1997). POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev 11 :1207-25.
Takahashi, K. and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663-76.
Takahashi, K., Okita, K., Nakagawa, M., and Yamanaka, S. (2007). Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2(12):3081-9. Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., lchisaka, T., Tomoda, K. and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861-72.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., Bernstein, B. E. and Jaenisch, R. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448:318-24.
Wernig, M., Meissner, A., Cassady, J. P. and Jaenisch, R. (2008). c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cells 2:10-12.
Yu, J., Vodyanik, M.A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir, G.A., Ruotti, V., Stewart, R., Slukvin, I.I. and Thomson, J.A. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858): 1917-20.
Zimmermann, W. H. and Eschenhagen, T. (2007). Embryonic stem cells for cardiac muscle engineering. Trends Cardiovasc Med 17(4): 134-40.
H. T. Kim et al., Exp Neurol 199, 222 (May, 2006)
K. I. Park, Y. D. Teng, E. Y. Snyder, Nat Biotechnol 20, 1111 (Nov, 2002)
H. Zaehres, G. Q. Daley, Methods Enzymol 420, 49 (2006)
D. Huangfu et al., Nat Biotechnol 26, 1269 (Nov, 2008)
T. Aasen et al., Nat Biotechnol 26, 1276 (Nov, 2008)
【技術分野】
【0001】
本発明は、人工多能性幹細胞(iPS細胞)の作製法に関するものであり、該方法は1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち人工多能性幹細胞への該標的細胞の再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSoxに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。更に、本発明は本発明の方法によって作製した人工多能性幹細胞、および人工多能性幹細胞への標的細胞の再プログラミングに関与する化合物の同定法に関する。また、トランスジェニック非ヒト動物の作製法、および本発明の方法によって作製したiPS細胞を含む遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための組成物の作製法も意図される。
【0002】
本明細書中でいくつかの文書を引用する。本明細書に引用する文書(製造者の仕様書、説明書などを含む)の開示内容は、参照により本明細書に組み込まれる。
【背景技術】
【0003】
胚性幹細胞(ES細胞)のような多能性細胞は、その自己再生能力および種々の細胞型への分化能力を特徴とする。ES細胞はin vitroにおいて、物理的および生物学的誘導因子の存在下で3つの胚葉層(内胚葉、中胚葉、および外胚葉)全ての特殊化(specialized)細胞系統に分化させることができる。これまでのところ、多くの有望な研究により、動物モデルにおいて種々の疾患の寛解におけるESCの分化誘導体の治療可能性が示されてきた。その結果、多能性幹細胞は組織工学および移植医療への使用に関して、非常に大きな可能性を有している。これらの細胞を特定の細胞型に分化させることができれば、多くの深刻な変性疾患(例えば糖尿病、パーキンソン病、およびアルツハイマー病)の治療を目的とする移植のための、ほぼ無制限の細胞供与源となりうる(Biswasら, 2007;Kimら, 2007;Zimmermannら, 2007)。
【0004】
体細胞を表現型、遺伝子型、および多能性の点でES細胞に類似した状態に再分化するように遺伝子的に修飾しうることが明らかにされたのはごく最近のことである(TakahashiおよびYamanaka, 2006;Okitaら, 2007;Wernigら, 2007)。いわゆる体細胞の「再プログラミング」は多能性の再獲得の機構を理解する有益な手段であると同時に、患者に特異的な多能性幹細胞を作製するという可能性を開くものでもある。マウスおよびヒト体細胞の多能性幹細胞(人工多能性幹細胞(iPS細胞)と表記する)への再プログラミングは、4つの転写因子、Oct4、Sox2、c-Myc、およびKlf4の発現によって可能とされてきた。
【0005】
現在、iPS細胞が医学的応用(例えば患者特異的再生細胞療法)において多大な可能性を有していることは広く知られているが、現行のiPS細胞の作製法は医療分野での使用を妨げる。特に、いくつかの再プログラミング因子を組み合わせて導入および発現するのに使用されるレトロウィルスベクターは、複数のコピーでランダムにゲノムに(傾向的には近傍または活性な内因性遺伝子に(into the vicinity or into active endogenous genes))組み込まれ、そのため癌遺伝子または腫瘍抑制遺伝子のそれぞれ活性化または不活性化変異体を生じうる。従って、標的細胞のゲノムを改変する度合いを最小化させた方法を用いたiPS細胞の作製により、この手段の臨床的に安全な適用が助長されうる。
【発明の概要】
【課題を解決するための手段】
【0006】
従って、本発明の第1の態様では人工多能性幹細胞(iPS細胞)の作製法に関し、該方法は1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち該標的細胞の人工多能性幹細胞への再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。
【0007】
「人工多能性幹細胞(iPS細胞)」は胚性幹細胞(ESC)に類似した特徴を示す細胞である。該特徴には例えばin vitroにおける無制限の自己再生、正常な核型、Oct3/4、Sox2、Nanog、アルカリホスファターゼ(ALP)、および幹細胞特異的抗原3および4(SSEA3/4)のような幹細胞マーカー遺伝子を含む特徴的な遺伝子発現パターン、ならびに特定の細胞型への分化能がある(Hanna, J.ら (2007), Science 318(5858): 1920-3;Meissner, A.ら (2007), Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら (2007), Nat Biotechnol.; Okita, K.ら (2007), Nature 448(7151): 313-7;Takahashi, K.,ら (2007) Cell 131 (5): 861-72;Wernig, M.ら (2007), Nature 448(7151): 318-24;Yu, J.ら (2007), Science 318(5858): 1917-20;Park, I. H.ら (2008), Nature 451(7175): 141-6))。最先端の線維芽細胞培養物からのiPS細胞作製についてはTakahashi, Okita, Nakagawa, Yamanaka (2007) Nature Protocols 2(12)に報告されている。マウスiPS細胞の多能性は、例えばin vitroにおける神経細胞、グリア細胞、および心臓細胞への分化、ならびに胚盤胞注入による生殖系列キメラマウスの作製によって試験することができる。ヒトiPS細胞株は、in vitroにおける神経細胞、グリア細胞、および心臓細胞への分化によって分析することができ、そのin vivoにおける分化能は免疫不全SCIDマウスへの注射およびテラトーマとして生じる腫瘍のキャラクタリゼーションによって試験できる。
【0008】
iPS細胞は一般に、以下の細胞生物学的特徴に従って評価および分類できる:
形態:iPS細胞は形態学的に胚性幹細胞(ESC)と類似している。各細胞は円形であり、大型核を有し,細胞質が少ない。iPS細胞のコロニーもESCのものと類似している。ヒトiPS細胞はhESCと同様の、境界が明瞭で扁平かつ密集したコロニーを形成し、一方、マウスiPS細胞はmESCと同様に、hESCほど扁平ではなく、より凝集したコロニーを形成する。
【0009】
増殖性:幹細胞はその定義の一部として自己再生しなければならないため、倍加時間および有糸分裂活性はESCに不可欠なものである。iPS細胞は有糸分裂的に活性であり、ESCと同等の速度で活発に自己再生、増殖、および分裂する。
【0010】
幹細胞マーカー:iPS細胞はESCで発現される細胞表面抗原マーカーを発現する。ヒトiPSCはhESCに特異的なマーカー(例えばSSEA-3、SSEA-4、TRA-1-60、TRA-1-81、TRA-2-49/6E、およびNanog)を発現する。マウスiPS細胞はmESCと同様、SSEA-1は発現するが、SSEA-3およびSSEA-4はいずれも発現しない。
【0011】
幹細胞遺伝子:iPS細胞は未分化ESC中で発現される遺伝子(例えばOct3/4、Sox2、Nanog、GDF3、REX1、FGF4、ESG1、DPPA2、DPPA4、およびhTERT)を発現する。
【0012】
テロメラーゼ活性:テロメラーゼは、ヘイフリック(Hayflick)限界(〜50回の細胞分裂)に制限されずに細胞分裂を持続するのに必要とされる。hESCは自己再生および増殖を持続する高いテロメラーゼ活性を発現し、またiPS細胞も高テロメラーゼ活性を示し、テロメラーゼタンパク質複合体の必要構成要素であるhTERT(ヒトテロメラーゼ逆転写酵素)を発現する。
【0013】
多能性:iPS細胞は、ESCと同様の方法で(in a fashion)、完全に分化した組織に分化する能力を有する。例えば免疫不全マウスに注射したiPS細胞は、9週間後に自然発生的にテラトーマを形成する。テラトーマは3つの胚葉(内胚葉、中胚葉、および外胚葉)から誘導される組織を含む複数系統の腫瘍である;これは、他の腫瘍が通常1つの細胞型のみのものであるのとは異なっている。テラトーマ形成は重要な多能性の試験である。更に培養下のhESCは「胚様体」と呼ばれる球状の胚様構造を自発的に形成し、これは有糸分裂的に活性な分化hESCの中核、およびそれを囲む3つの胚葉全てに由来する完全に分化した細胞から成る。iPS細胞も胚様体を形成し、外縁部に分化細胞を有する。
【0014】
胚盤胞注入:hESCは、天然では胚盤胞の内部細胞塊(胚結節)内に存在して胚結節内で胚に分化し、一方、胚盤胞の外殻(栄養膜)は胚外組織に分化する。内部が空洞となった栄養膜は生きた胚を形成することができず、従ってこれは胚性幹細胞が胚結節内で分化して胚を形成するのに必要とされるものである。iPS細胞をマイクロピペットで栄養膜内に注入することができ、胚盤胞はメスのレシピエントに移植される。それによって生存キメラマウスの子、すなわちiPS細胞誘導体が種々の程度のキメラ化で全身にわたって導入されたマウスを作製することができる。
【0015】
プロモーターの脱メチル化:メチル化とは、DNA塩基へのメチル基の転移、一般的にはCpG部位(シトシン/グアニンが隣接した配列)のシトシン分子へのメチル基の転移である。遺伝子の広範なメチル化は、発現タンパク質活性の阻害または発現に干渉する酵素のリクルートによって、発現に干渉する。従って遺伝子のメチル化により、転写が阻害されることによって効果的にその発現が停止される。多能性関連遺伝子(例えばOct3/4、Rex1、およびNanog)のプロモーターはiPS細胞中で脱メチル化され、iPS細胞においてそのプロモーター活性、並びに活性な多能性関連遺伝子の促進(promotion)および発現を示す。
【0016】
ヒストン脱メチル化:ヒストンは圧縮タンパク質であり、構造的にはDNA配列に局在し、種々のクロマチン関連修飾を介してその活性がもたらされる。例えばOct3/4、Sox2、およびNanogに関連するH3ヒストンは脱メチル化されてOct3/4、Sox2、およびNanogを発現する。
【0017】
本発明に関して使用する「導入する」という用語は、コード配列を標的細胞に取り入れさせ、その後、該コード配列を標的細胞のゲノムDNAに組み込ませる工程に関する。この工程は一般に安定トランスフェクションとして知られ、安定トランスフェクションのための方法は当業者に公知であり、例えばBonetta, L., (2005), Nature Methods 2, 875-883に報告されている。トランスフェクトされた細胞で起こる再プログラミング事象は低率であるため、効率の高い安定トランスフェクション法を用いるのが有利である。そのため、トランスフェクション/感染効率の高い方法によってコード配列を標的細胞に導入するのが好ましい。例えばトランスフェクション/感染効率が少なくとも30%、少なくとも50%、または少なくとも80%であるのが好ましい。好適な方法には、例えばリポフェクション、エレクトロポレーション、ヌクレオフェクション(nucleofection)、マグネトフェクション(magnetofection)、またはウィルスベクター感染がある。レトロウィルスを使用して標的細胞の安定トランスフェクションを行うのが好ましいが、これは該ベクターが、標的細胞へのコード配列の効率的な侵入だけでなく、標的細胞のゲノムDNAへの融合も仲介するためである。明らかにされているところによれば、レトロウィルスベクターによって種々の動物種由来の広範な型の細胞の形質導入を行い、ベクターが運ぶ遺伝物質を標的細胞に組み込み、形質導入されたコード配列を高レベルで発現させることができ、そして好都合なことに、レトロウィルスベクターは感染後に伝播またはウィルスタンパク質を生成することがない。好適なレトロウィルスベクター系は当業者に公知であり、それらは例えばMoMuLV LTR、MESV LTRを有するレトロウィルスベクター、CMVプロモーターのような種々の内部プロモーターを含有する(好ましくはエンハンサー/プロモーターを組み合わせて含有する)レンチウィルスベクターで、胚/多能性細胞中で導入遺伝子発現の抑制を示すものである。アデノウィルスベクターのようなエピソームベクター系、他の非組込み型ベクター、エピソーム複製プラスミド(episomally replicating plasmids)を使用することもできる。好ましくは、レトロウィルスMXベクター系を本発明の方法に使用する(Kitamuraら, (2003), Exp Hematol., 31(11):1007-1014)。
【0018】
本発明の方法で使用する標的細胞は既存の細胞株由来のものであるか、または種々の方法(例えば、初代細胞株を樹立するために組織サンプルを得る)によって得てもよい。種々の組織からサンプルを得る方法および初代細胞株を樹立する方法は、当業界で公知である(例えばJonesおよびWise, Methods Mol Biol. 1997参照)。好適な体細胞株を様々な供給者(例えば American tissue culture collection (ATCC)、the German Collection of Microorganisms and Cell Cultures (DSMZ)、またはPromoCell社(Sickingenstr. 63/65, D-69126、ハイデルベルグ))から購入してもよい。本発明の方法によれば、好適な標的細胞はOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、該因子は、これを補完する因子群(すなわち Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子)から選択される外因的に導入される因子と組み合わされて、非多能性標的細胞をiPS細胞に再プログラミングする能力を有する。1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する。少なくとも2つの上記因子が標的細胞中で内因的に発現されるか否かを確認する方法は、当業者に公知である。それらの方法には、例えばウェスタンブロッティング、リアルタイムPCR、または細胞間染色がある。更に当業者は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する細胞を作製して標的細胞のiPS細胞への再プログラミングを開始させるため、内因的に発現される因子群を補完するのにいずれの外因性因子(単数または複数)が必要であるかを容易に認識できる。従って発現可能な形態でコード配列(単数または複数)を導入した細胞は、Oct3/4並びにMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つずつの因子から成る因子群を発現する。
【0019】
本発明は、既に標的細胞中に内因的に存在するコード配列を導入する態様も包含する。これは、例えば相当する因子が標的細胞の再プログラミングに関与しない、または十分関与しない影響で内因性コード配列が低レベルでしか発現されないような場合、有効でありうる。
【0020】
「コード配列」という用語は、転写の際にコードされた産物を生じさせるヌクレオチド配列を示す。本発明にかかるコード配列の転写は、好適なプロモーターと関連して容易に達成される。好ましくはコード配列は、転写の際に標的細胞の人工多能性幹細胞への再プログラミングに関与する因子を生じさせる遺伝子のcDNA配列に相当し、本発明の方法にかかる再プログラミング因子は、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される。
【0021】
「標的細胞の人工多能性幹細胞への再プログラミングに関与する因子」とは、標的細胞の人工多能性幹細胞への再プログラミングの誘導に関与する能力を有する因子を示し、該因子はOct3/4並びにMyc、Klf、およびSox因子ファミリーに属する因子から選択される。それらの再プログラミング因子には、例えばOct3/4、Sox2、Sox1、Sox3、c-Myc、n-Myc、I-Myc、Klf1、Klf2、Klf4、Klf5など、または再プログラミング能を保持するそれらの変異体がある。該再プログラミングへの関与は、例えば細胞のメチル化パターンを胚性幹細胞と同様のものに変化させること、細胞の発現プロファイルを胚性幹細胞の発現プロファイルのように変更すること、またはヒストン結合の調節によって凝集した核DNAのコンフォメーションを胚性幹細胞で観察されるのと同様にすることという形態であってもよく、該変化はそれぞれ、好適な再プログラミング因子によって単独で、または組み合わされて起こってもよい。上記の因子以外に、当業者に認識されるように、更なる好適な再プログラミング因子を同定する方法、例えばバイサルファイトゲノムシーケンシング、RT-PCR、リアルタイムPCR、マイクロアレイ分析、核型分析、テラトーマ形成、アルカリホスファターゼ染色があり、それらは全て当業者に公知であり、例えば以下に記載されている:Okita, K.ら(2007), Nature 448(7151): 313-7;Park, I. H.ら(2008), Nature 451(7175): 141-6;Takahashi, K.ら(2007), Cell 131(5): 861-72;Wernig, M.ら(2007), Nature 448(7151): 318-24;Takahashi, K.ら(2007), Nat Protoc 2(12): 3081-9;またはHogan, B.ら(1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press。
【0022】
Oct3/4はオクタマー(「Oct」)転写因子ファミリーに属し、多能性の維持に関与する。通常はOct3/4を発現する細胞(例えば割球および胚性幹細胞)においてOct3/4が存在しなければ、自発的な栄養膜への分化が誘導される。従って、Oct3/4の存在は胚性幹細胞の多能性および分化能に関与する。「Oct」ファミリーの種々の他の遺伝子(Oct1およびOct6など)は誘導を起こさず、これは誘導過程に対するOct3/4の排他性を示している。本明細書では、「Oct4」という用語は「Oct3/4」と互換的に使用する。
【0023】
Sox遺伝子ファミリーはOct3/4同様、多能性の維持に関係するが、多能性幹細胞で排他的に発現されるOct3/4とは異なり、複能性(multipotency)および単能性幹細胞と関連する。
【0024】
Klf遺伝子ファミリーのKlf4は初めにマウスiPS細胞作製のための因子として同定され、ヒトiPS細胞の作製因子であることが明らかにされた。
【0025】
Mycファミリーに属する遺伝子は、癌に関係する癌原遺伝子である。c-MycがマウスiPS細胞の作製に関係する因子であり、ヒトiPS細胞の作製に関係する因子でもあることが証明された。iPS細胞作製のための標的細胞への「Myc」遺伝子ファミリーの導入は、臨床治療としてのiPS細胞の成り行きに困難を与えており、これは、c-Myc誘導iPS細胞を移植したマウスの25%で致命的なテラトーマが発生したためである。N-MycおよびI-Mycは同様の効率でc-mycに代わるものとして同定されている。
【0026】
本発明に関して使用する「再プログラミング」という用語は、遺伝子型的および/または表現型的に胚性幹細胞と同様の細胞となるような細胞の遺伝子型的および表現型的プロファイルの変化の過程に関する。該変化は、例えば上記のようなメチル化パターンの変化、発現プロファイルの変更、または凝集した核DNAのコンフォメーション変化を含む。上記は本明細書で以下に記載する他の態様にも準用される。
【0027】
本発明の方法は、2つの再プログラミング因子のみの導入によってiPS細胞を得ることができるという、驚くべき発見に基づく。この発見がなされるまで、従前技術の定説は、in vivo実験で機能的である、すなわち3つの胚葉層に関与する能力を有する生存iPS細胞は少なくとも3つ、より効率的には4つの再プログラミング因子を組み合わせて導入しなければ作製できないというものであった。
【0028】
例として、レトロウィルスMXベクター系を用いて、4つ(4F)、3つ(3F)、および2つのみ(2F)の再プログラミング因子の組み合わせ、並びに1つの再プログラミング因子のみの導入によって、マウス神経幹細胞(NSC)の再プログラミングを行うことができることが明らかになった。NSCを成体OG2/Rosa26ヘテロ接合トランスジェニックマウスの脳から樹立し(Ryan, A. K. & Rosenfeld, M. G., Genes Dev 11 , 1207-25 (1997);Do, J. T. & Scholer, H. R., Stem Cells 22, 941-9 (2004);Pollard, S. M., Conti, L., Sun, Y., Goffredo, D. & Smith, A., Cereb Cortex 16 Suppl 1 , i112-20 (2006))、Oct4プロモーターの制御下でGFP(Oct4-GFP)を、構成的(constitutive)Rosa26遺伝子座からLacZ導入遺伝子を発現させた。
【0029】
GFP+コロニーの観察を、まずOct4およびKlf4に感染させたNSC培地で行い(2F OK)、1-2週間後にOct4およびc-mycに感染させた培地で行った(2F OM)(表1)。
【0030】
【表1】
【0031】
2F OM iPS細胞を更に分析したところ、ESC様の発現パターンおよびテラトーマの3つの胚葉への関与が明らかになった。
【0032】
2F OK iPS細胞を4F(4つの再プログラミング因子を導入してiPS細胞を作製する標準的な方法を用いて作製)iPS細胞およびESCと比較した。感染14日後に5個のGFP+コロニーを分離し、ESC培養条件下で増殖させ(図1c、f)、3個(すなわち60%)のESCと形態学的に識別不能な2F OK iPS細胞クローン(B-2、D-7、およびF-4)を得た(図1d、g)。コントロールウィルス(MX)に感染させたNSCからはコロニーは形成されなかった(図1e、h)。2F OK iPSおよび4F iPSの再プログラミング効率の経時変化を、Oct4-GFP+コロニー数およびNSCのMX-GFPコントロールウィルスでの形質転換速度から見積もった(図1i、j)。その結果、再プログラミング効率はNSCの4F再プログラミングでは3.6%、2因子法では0.11%と算出され、これは選択を行った場合の線維芽細胞の再プログラミング効率(0.08%未満、Takahashi, K.およびYamanaka, S., Cell 126, 663-76 (2006);Okita, K., lchisaka, T.およびYamanaka, S., Nature 448, 313-7 (2007);Wernig, M.ら, Nature 448, 318-24 (2007))および選択を行わない場合(0.5%; Meissner, A., Wernig, M.およびJaenisch, R., Nat Biotechnol 25, 1177-81 (2007))に匹敵するものであった(図1j)。4因子全てでの形質転換はタイミング(timing)およびGFP+コロニー数にプラスの影響を与えた。ウィルス導入遺伝子の組込みはジェノタイピングPCR(genotyping PCR)で確認した。4因子全てのウィルス導入遺伝子が4F iPS細胞で検出され、一方、2F OK iPS細胞はOct4およびKlf4導入遺伝子のみを含有した。
【0033】
2F OK iPS細胞は、SSEA-1およびアルカリホスファターゼに関して陽性に染色され、4F iPS細胞およびESCと同様のES細胞マーカー遺伝子発現パターンを示した(図2a)。qRT-PCRの結果は、2F OK iPS細胞における内因性Oct4、Sox2、c-Myc、およびKlf4発現がESCに匹敵し、2F OK iPS細胞中のウィルス転写物のサイレンシング(30日後で1000倍低下)が起こることを示した。2F iPSの総体的な遺伝子発現もESCおよび4F iPSに類似している(clusters close to)(図2b)。DNAマイクロアレイ分析の散布図は、2F iPS細胞およびESC間の類似性が2F iPS細胞およびNSC間の類似性よりも高いことを示した(図2c、d)。従って、2F iPS細胞(クローンF-4)は総体的な転写レベルでマウスESCと非常に類似していると考えられた。
【0034】
2F OK iPS細胞の分化能をin vitroにおける胚様体(EB)への分化によって確認した。これらの細胞は外胚葉(Tuj1)、内胚葉(α-フェトプロテイン)、および中胚葉マーカーFlk1(心筋細胞を擬態する拍動細胞(beating cell)によって発現される)を発現した(図3a)。テラトーマは3つの胚葉全ての誘導体を含有し(図3b)、3つの胚葉のマーカーを発現した。ドナー細胞(NSC)からはテラトーマは形成されなかった。これらのデータは、2F OK iPS細胞がin vitroおよびin vivoにおいて多能性表現型を示すことを示している。
【0035】
分化能を検討するために、2F OK iPS細胞を8細胞期胚と凝集させた。iPS細胞は分化中の胚盤胞において内部細胞塊の形成に関与した(図4a)。凝集した胚盤胞を偽妊娠メスに移植した後、E13.5で16個の生存胚を得、そのうち2個はOct4-GFP発現による判定で胎仔生殖腺における生殖細胞の関与を示した(図4b)。全胚からの胚組織のX-gal染色(Rosaβ-geo26(lacZ導入遺伝子)を保有するNSCドナー細胞を可視化)により、得られたキメラにおいて、2F OK iPS細胞は3つの胚葉全ての分化に関与していることが明らかになった(図4c、e)。最も厳密な試験で4倍体(4N)胚凝集体の分化能を試験したところ(n=122)、E13.5で2個の死亡(停止)胚が確認された(図4d)。これは4N胚凝集体の正常率以内であり、導入した細胞の多能性欠損には関係しなかった。これらのデータは、iPS細胞が後期胚の組織の全てを生ずることができることを示している。2倍体(2N)凝集体では、PCR遺伝子型同定により、13個のキメラのうち2個がドナー細胞のOct4-GFP対立遺伝子陽性であることが明らかになった(図4fおよびg(上段))。2F OK iPS細胞が生殖細胞系列に関与できるか否かを評価するために、キメラをCD-1メスと交配した。12匹の子のうち2匹がOct4-GFP対立遺伝子を有し、12匹中1匹がlacZ対立遺伝子を有した。ドナー細胞はヘテロ接合性マウス(Oct4+/- Rosa26+/-)に由来するので、それらはOct4およびKlf4導入遺伝子も有する(図4g(下段))。成体キメラおよびF1マウス(それぞれ17週齢および3週齢まで)で腫瘍形成は観察されなかった。この知見は、2F OK iPS細胞がキメラの発生全期に関与して次世代(F1)の生存可能な子を生じることができることを示しており、従ってiPS細胞がESCと同様の分化特性を有することを示唆している。
【0036】
以下の実施例9に詳細を記載するが、出願人は、2つまたは1つだけの再プログラミング因子の導入によってヒト細胞を多能性幹細胞に転換できることも明らかにした。該再プログラミング因子はOct4またはOct4およびKlf4である。
【0037】
結論として、上記の知見は、2つの再プログラミング因子または1つだけの再プログラミング因子を使用してiPS細胞を作製することに成功したことを示している。本発明の方法の利点は、1つまたは2つの再プログラミング因子の安定トランスフェクションのために2つまたは1つしかレトロウィルスを使用しない点にある。従前技術による方法に比較して少ない数のレトロウィルスベクターでiPS細胞を誘導できることは、再プログラミングすべき最初の細胞集団の遺伝子調節を最小限にすることへの大きな一歩である。従って、異常細胞および腫瘍原性細胞形成のリスクが大幅に低減され、それにより、特に治療目的に適したiPS細胞の作製が可能となる。
【0038】
本発明の方法の好ましい態様では、Myc、Klf、およびSox因子ファミリーに属し、標的細胞に導入されるべきコード配列によって内因的に発現される、またはコードされる因子は、I-Myc、n-Myc、c-Myc、Klf1、Klf2、Klf4、Klf15、Sox1、Sox2、Sox3、Sox15、およびSox18から成る群から選択される。
【0039】
例えばマウスOct3/4、Sox2、c-Myc、およびKlf4のコード配列はそれぞれSEQ ID NO: 1、5、9、および13に見られる。マウスOct3/4、Sox2、c-Myc、およびKlf4のタンパク質配列はそれぞれSEQ ID NO: 2、6、10、および14に見られる。ヒトOct3/4、Sox2、c-Myc、およびKlf4のコード配列はそれぞれSEQ ID NO: 3、7、11、および15に見られる。ヒトOct3/4、Sox2、c-Myc、およびKlf4のタンパク質配列はそれぞれSEQ ID NO: 4、8、12、および16に見られる。当業者は、当該分野で公知の方法を用いて任意の標的種の再プログラミング因子のコード配列を決定する立場にある。例えば、データベース(例えばNational Center for Biotechnology Information (NCBI)が維持しており、ワールドワイドウェブ(http://www.ncbi.nlm.nih.gov/)からアクセスできるデータベース)から、配列および機能に関するデータを検索することができる。更に、比較ゲノミクスのためのデータベースには、限定されるわけではないが以下がある:NCBIが維持するデータベース(http://www.dcode.org/)、全ての完全にシーケンシングが行われた生物のタンパク質アノテーションのデータベース(http://supfam.org/SUPERFAMILY/)、様々な種のゲノム情報を含むデータベース(http://www.cbs.dtu.dk/services/GenomeAtlas/)、または遺伝子クラスターを含むデータベース(http://phigs.jgi-psf.org/)。該データベースにより、当業者はマウスおよびヒトで知られている配列から開始して、例えば異種間の配列アラインメントを実施して相同遺伝子を同定することによって、他の種における再プログラミング因子のコード配列を同定することができる。
【0040】
ごく最近になって発表されたいくつかの科学論文(Hanna, J.ら(2007) Science 318(5858): 1920-3;Meissner, A.ら(2007) Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら(2007) Nat Biotechnol.;Okita, K.ら(2007), Nature 448(7151): 313-7;Takahashi, K.ら(2007), Cell 131 (5): 861-72;Wernig, M.ら(2007) Nature 448(7151): 318-24;Yu, J.ら(2007) Science 318(5858): 1917-20;Park, I. H.ら(2008) Nature 451(7175): 141-6)により、Oct、Sox、Klf、およびMycファミリーに属する転写因子がマウス並びにヒト体細胞における再プログラミングの誘導に寄与する能力を有することが明らかになった。
【0041】
別の好ましい態様では、標的細胞は、該標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しない。
【0042】
因子の内因的発現を評価する方法は当業者に公知であり、本明細書の別の箇所にも記載されている。本発明の方法に従ってiPS細胞を作製するために、標的細胞は標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しなくてもよい。例えば標的細胞であるマウス神経幹細胞においてOct3/4は発現されず、Sox2、Klf4、およびc-Mycは内因的に発現されるということが示される可能性もある。Oct3/4の外来性の導入およびそれに続く発現は、4つ組の再プログラミング因子を補完し、iPS細胞の生成を誘導するのに十分であった。
【0043】
別の好ましい態様では、標的細胞は複能性幹細胞である。
【0044】
複能性幹細胞はいくつかの他の細胞型を生じるが、それらの型の数には限りがある。これは多能性幹細胞が任意の細胞型に分化する能力を有するのと全く対照的である。複能性細胞の一例として、例えば骨髄、臍帯血、または循環中に見られる造血細胞があり、これはいくつかの型の血液細胞となれるが、他の型の細胞にはなれない。複能性細胞の別の例は神経幹細胞である。複能性細胞は亢進的に調節された再プログラミング因子を既に保有するため、再プログラミング標的細胞として特に好適である。
【0045】
より好ましい態様では、複能性幹細胞は外胚葉細胞である。
【0046】
外胚葉は3つの主要な胚細胞層の最も外側のものであり(他の2つは中胚葉および内胚葉である)、ごく初期の胚を構成する。これは分化して多くの重要な組織および構造、例えば皮膚の外層およびその付属器官(汗腺、毛髪、および爪)、歯、眼のレンズ、内耳の各部、神経組織、脳、および脊髄を生じる。外胚葉細胞は神経幹細胞のように既に再プログラミング因子を内因的に発現するため、複能性幹細胞としての外胚葉細胞は標的細胞として特に好適である。
【0047】
別の好ましい態様では、標的細胞は神経幹細胞(NSC)である。
【0048】
神経幹細胞は発生途中の哺乳動物神経系のみならず、全ての哺乳動物(ヒトを含む)の成体の神経系にも存在する。神経幹細胞はより原始的な胚性幹細胞からも誘導されうる。成体において生存可能な位置の数は限定されているものの、分化するためにそれらの子孫が移動する成体幹細胞の位置および脳領域はまだ解明されていない(総説はGage(2000)参照)。神経幹細胞は既に再プログラミング因子を内因的に発現するため、標的細胞として特に好適である。
【0049】
より好ましい態様では、導入すべきコード配列はOct3/4因子をコードする。
【0050】
上記に概説し、後述の実施例に記載するように、Oct3/4のみを神経幹細胞に導入すれば、iPS細胞を作製するには十分であった。C-Mycはキメラの子における発癌性を増加するため(Okita, K., Ichisaka, T.およびYamanaka, S., Nature 448, 313-7 (2007))、最近のc-Mycレトロウィルスの組み込みを用いないiPS細胞の作製を示す研究(Nakagawa, M.ら, Nat Biotechnol 26, 101-106 (2008);Wernig, M., Meissner, A., Cassady, J. P.およびJaenisch, R., Cell Stem Cells 2, 11-12 (2008))は著しい改善を見せている。しかしながら、この態様で示すようにc-Mycを用いずに少ない数のレトロウィルスベクターと組み合わせてiPS細胞を誘導する可能性は、再プログラミングすべき最初の細胞集団の遺伝子調節を最小限にすることへの大きな一歩である。
【0051】
同じ標的細胞を、2つの因子だけの導入によって再プログラミングすることもできた。従って、別のより好ましい態様では、導入すべき2つのコード配列はOct3/4およびc-Myc、またはOct3/4およびKlf4をコードする。
【0052】
更により好ましい態様では、標的細胞はc-Myc、Klf4、およびSox2因子を内因的に発現する。
【0053】
標的細胞が上記の再プログラミング因子の組み合わせを内因的に発現する場合、1つまたは2つの外来再プログラミング因子(例えばOct3/4のみ、またはOct3/4およびc-Myc、またはOct3/4およびKlf4)を導入した時に再プログラミングが起きやすいことも明らかにされた。
【0054】
更に好ましい態様では、標的細胞はc-Myc、Klf4、およびSox2因子を、標的細胞と同じ属の胚性幹細胞における相当する発現レベルに比較して、最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する。
【0055】
本発明の方法によれば、内因性再プログラミング因子の発現レベルが標的細胞と同じ属のESCにおける発現レベルに比較して、一定の範囲内にある場合が有利である。好ましくは、標的細胞は再プログラミング因子であるc-Myc、Klf4、およびSox2を、ESCにおける該因子の相当する発現レベルに比較して、最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する。より好ましくは、標的細胞と同じ属に属するESCの場合よりSox2の発現が約2倍高く、c-Mycが約10倍高く、そして/またはKlf4が約8倍低い。本発明に関連して使用する「約」という用語は、平均偏差が最大+/-20%、好ましくは+/-10%であることをいう。また、発現レベルが該ESCに比較して少なくても8、6、5、4、3、もしくは2倍低い、または多くても8、6、5、4、3、もしくは2倍高い、あるいはその間の任意の数である場合も意図される。
【0056】
より好ましい態様では、標的細胞はマウスまたはヒト神経幹細胞である。
【0057】
更に本発明は、本発明の方法によって作製した人工多能性幹細胞に関する。
【0058】
本発明の方法によって作製した多能性幹細胞は、種々の実験設定および治療設定において有用であり得る。例えば、iPS細胞、分化によってそれから誘導された細胞、または該iPS細胞もしくはそれから誘導された細胞から作製した組織の、治療(therapeuticum)または診断(diagnosticum)としての使用、遺伝子または細胞移植治療中での使用、ゲノム標的の同定および検証のための使用、並びに薬剤スクリーニング法への使用も意図される。
【0059】
iPS細胞の培養条件は相当する種の胚性幹細胞で確立されたものと同じであり、当業者に公知である。一般に、細胞培養法(例えば培地の成分、マーカーの選択肢および選択、細胞の定量および単離)は当該分野で公知であり、例えば、"Practical Cell Culture Techniques", Boulton et Baker編, Humana Press (1992), ISBN 0896032140;"Human Cell Culture Protocols", Gareth E. Jones, Humana Press (1996), ISBN 089603335X、および実施例の項に例証されている。培地中で細胞を培養および維持する方法は当該分野で公知である;増殖培養液および他の細胞培養に関係する物質、並びに細胞の培養を成功させるための説明書および方法は、例えばSigma-AldrichまたはInvitrogen社から入手できる。
【0060】
更に、本発明は標的細胞の人工多能性幹細胞への再プログラミングに関与する化合物の同定法に関するものであり、該方法は以下の工程:(a)本発明の方法に従って標的細胞を再プログラミングすること(1つの導入されるべきコード配列を、試験する化合物で置き換える);および(b)試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価することを含み、ここで、試験する化合物を導入した標的細胞からiPS細胞が形成されれば、その化合物が標的細胞の人工多能性幹細胞への再プログラミングに関与していることを示す。
【0061】
本発明によれば、試験する化合物は1つまたはそれ以上の核酸、例えばDNA、cDNA、RNA、dsRNA、siRNA、shRNA、miRNA、タンパク質、ペプチド、小分子(有機または無機)、化学物質、または任意のそれらの組み合わせであってもよい。
【0062】
本発明の方法にかかる標的細胞の再プログラミングについては既に上述した。標的細胞へ化合物を導入する工程に関しては、試験する化合物の性質によっては本発明の方法を改変する必要があり得る。例えば他の転写因子を評価すべき場合、改変せずに上記のように相当するコード配列を導入してもよい。これに対して化学物質または小分子は、各化合物を細胞培養液に外来的に添加し、細胞の受動的または能動的な取り込み機構を利用することにより、導入してもよい。化合物が実際にそれが代替する因子の代わりとなって標的細胞の再プログラミングを誘導するか否かを試験するために、試験する化合物を細胞(好ましくは核)に導入する方法は当業者に公知である。核酸、例えばDNA、cDNA、RNA、dsRNA、siRNA、shRNA、miRNAはトランスフェクションまたは感染によって導入することができ、小分子(有機または無機)、化学物質は単に膜を通じた浸透によって導入できる。
【0063】
試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価する方法は当業者に公知である。iPS細胞の分類の基準は当業者に公知であり、既に上述した。評価すべき基準によって方法は種々であり、例えば顕微鏡による観察(visual control)、マーカーの発現分析、テラトーマ形成を、単独で、または組み合わせて含みうる。
【0064】
ある細胞の再プログラミングに関与する一組の因子を内因的に発現する該細胞を、更なる因子を外来的に添加することによって補完し、細胞に4つ組の再プログラミング因子(すなわちOct3/4、並びにMyc、Klf、およびSox因子ファミリーそれぞれからの因子)を発現させ、それによって標的細胞の再プログラミングを誘導してもよいという本発明の発見により、再プログラミング過程においてある因子を代替できる化合物の同定が有意に簡易化される。スクリーニングに好適な細胞を作製するために、当該分野で公知の4つの因子のうちの3つまたは全部ではなく、1つまたは2つだけを導入すればよいため、時間、コスト、および実験の困難さが大幅に削減される。新規の再プログラミング因子のためのハイスループットスクリーニング法は、導入する必要のある因子のセットが少ないことによって、時間および効率の点で明らかに改善される。
【0065】
また、本発明はトランスジェニック非ヒト動物の作製法に関し、該方法は以下:(a)本発明の、または本発明の方法によって作製された人工多能性幹細胞を非ヒト着床前胚に導入すること;(b)工程(a)の胚をメス非ヒト動物の子宮に移植すること;および(c)胚を分化および出生させることを含む。
【0066】
本発明に関して使用する「トランスジェニック非ヒト動物」という用語は、本明細書に記載する方法によってそのゲノムの意図的な改変を実施した動物をいう。
【0067】
本発明のトランスジェニック非ヒト動物の作製法は、好ましくは既に構築されている胚性幹細胞を使用したトランスジェニック非ヒト動物の作製法に従い、胚性幹細胞の代わりに本発明のiPS細胞を使用して実施する。該方法は当該分野で公知である(Hogan, B., R. Beddingtonら(1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press;Hanna, J.ら(2007), Science 318(5858): 1920-3;Meissner, A.ら(2007), Nat Biotechnol 25(10): 1177-81;Nakagawa, M.ら(2007), Nat Biotechnol.;Okita, K.ら(2007), Nature 448(7151): 313-7;Takahashi, K.ら(2007), Cell 131 (5): 861-72;Wernig, M.ら(2007), Nature 448(7151): 318-24;Yu, J.ら(2007), Science 318(5858): 1917-20;Park, I. H.ら(2008), Nature 451(7175): 141-6)。簡潔に記載すると、iPS細胞の非ヒト着床前胚(例えば桑実胚または胚盤胞)への導入は、好ましくは桑実胚もしくは胚盤胞へのマイクロインジェクション、または8細胞期胚もしくは桑実胚とiPS細胞との凝集によって行う。その後、該キメラ胚を偽妊娠非ヒト・メスの子宮に移植し、胚に分化させ最終的に出生させる(実施例8参照)。
【0068】
iPS細胞からのトランスジェニック非ヒト動物系の作製は、該iPS細胞の多能性(すなわちホストへの注入に際して胚(例えば胚盤胞または桑実胚)に分化し、胚形成に関与し、得られる動物の生殖細胞に寄与する能力)に基づく。上記のように、注入されたiPS細胞を含有する胚盤胞は偽妊娠非ヒト・メスの子宮内で分化し、キメラとして出生する。得られるトランスジェニック非ヒト動物はiPS細胞に由来する細胞のキメラであり、DNAセグメントの組み合わせがホモ接合性であるトランスジェニック動物を同定するために、これを野生型非ヒト動物と戻し交配し(backcrossed)、iPS細胞の遺伝的内容のみを保有する動物をスクリーニングする。
【0069】
トランスジェニック非ヒト動物は、例えばトランスジェニックマウス、ラット、ハムスター、イヌ、サル、ウサギ、ブタ、またはウシであってもよい。好ましくは該トランスジェニック非ヒト動物はマウスである。
【0070】
従って本発明は、本発明の方法に従って作製したトランスジェニック非ヒト動物にも関する。
【0071】
最後に、本発明は、遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための、本発明の方法によって作製したiPS細胞を含む組成物に関する。
【0072】
本明細書で使用する組成物は、iPS細胞、および好ましくは該細胞の生存能力を維持する更なる成分を含有する組成物に関する。それらの成分は当業者に公知であり、例えば細胞培養液成分を含む。更に、意図する適用によって、組成物は更なる成分、例えば患者への投与を容易にする成分を含んでもよい。
【0073】
本発明のiPS細胞を含む組成物(本発明のiPS細胞自体と同様)を種々の実験のシナリオおよび治療のシナリオに使用することができる。相対的に少数のトランスジェニック発現要素を有し、癌性細胞に分化するリスクが全体的に低い本発明のiPS細胞は、遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングにおいて有益であると考えられる。
【0074】
遺伝子治療は、遺伝的欠損を修正するためにex vivoまたはin vivoの手法によって治療のためのDNAコンストラクトを生殖系細胞に導入することに基づくが、これは遺伝子移入の最も重要な適用の1つである。in vitroまたはin vivo遺伝子治療に好適なベクターおよび方法は文献に報告されており、当業者に公知である(Davis PB, Cooper MJ., AAPS J. (2007), 19;9(1):E11-7;Li S, Ma Z., Curr Gene Ther. (2001),1 (2):201-26)。本発明によれば、患者から得た細胞を、例えば当該分野で既知の方法によって遺伝子的に修正した後、本発明の方法によってES細胞の表現型および遺伝子型を有するiPS細胞に再プログラミングすることができうる。これは遺伝子治療および/または細胞療法におけるiPS細胞の適用性を証明するものである。再生医療を用いて、損傷した組織をin vivoで再生させるか、または組織および臓器をin vitroで成長させた後、それを患者に移植することによって、機能不全の、損傷した、または欠陥を有する組織に起因する任意の疾病を治療できる可能性がある。実質的に任意の組織(外胚葉、中胚葉、内胚葉細胞)に分化できる本発明のiPS細胞は、再生医療のあらゆる観点で使用することができ、それによってES細胞の必要性は大幅に低減される。
【0075】
本発明のiPS細胞を用いて薬剤ターゲットを同定し、可能性のある薬物療法学を試験することができ、それによってES細胞およびin vivoにおける研究の必要性は低減される。可能性のある薬剤の効果(例えば標的部位および標的特異性、毒性、生物学的利用能)の同定および/または評価のための実験手順および方法は、当業者に公知である。
【0076】
更に、iPS細胞を使用して出生異常の予防および治療の研究、または細胞分化の研究を行ってもよい。
【0077】
また、本発明のiPS細胞は(治療的適用ではなく)実験設定において、標的細胞の再プログラミングの誘導の際の脱分化に関する種々の観点(例えば遺伝子の発現パターンまたはメチル化パターンの時空間的変化、または凝集挙動に変化を与える形態学的変化)を研究するのに有用でありうる。さらに、iPS細胞で、例えば以下に関係する研究を行うこともできる:遺伝子治療、遺伝子ターゲティング、分化の研究、薬剤の安全性および有効性に関する試験、自己または同種異系(allogenic)再生させた組織の移植、組織修復(例えば神経系、心筋)、疾病(例えばパーキンソン病、心臓発作、糖尿病、癌、白血病、または脊髄損傷)、胚性遺伝子発現、胚性遺伝子の遺伝子操作、初期発生学および胎児発達、胚細胞マーカーの同定、細胞移動またはアポトーシス。
【0078】
図1:OG2/Rosa26トランスジェニックマウスの成体NSCからの2F Oct4/Klf4(OK)iPS細胞の作製。
a.ESCおよびNSCにおけるOct4、Nanog、Klf4、Sox2、およびc-MycのRT-PCRおよびqRT-PCR分析。ローディング・コントロールとしてβ-アクチンを使用した。b.ESCおよびNSCにおける4因子のウェスタンブロット分析。ローディング・コントロールとして抗アクチン抗体を使用した。c.感染14日後の2F OK iPS細胞コロニーの形態。ESC様コロニーがOct4-GFPを発現(f)。d.樹立した2F OK iPS細胞(クローンF-4)の感染30日後の形態(放射線照射したMEFで増殖)。位相定数およびOct4-GFP(g)を示す。e.感染30日後のNSCおよび擬似感染の形態(h)。i.2F OKおよび4F感染の7日後、14日後、および21日後のGFP+コロニーの生成(n=3;エラーバーはs.d.を表す)。j.2Fおよび4F iPS細胞作製の再プログラミング効率(n=3)。感染の7日後、14日後、および21日後の50,000播種NSCあたりのGFP+コロニー総数を示す。
【0079】
図2:iPS細胞の遺伝子発現プロファイル。
a.ESC、4F iPS細胞(クローンA-2c)、2F OK iPS細胞(クローンB-2、D-7、およびF-4)、およびNSCにおけるES細胞マーカー遺伝子発現のRT-PCR分析。プライマーは各内因性遺伝子座からの転写物に特異的である。β-アクチンをローディング・コントロールとして使用した。b.NSC、2F (OK) iPS、4F iPS、およびESCでの種々の発現遺伝子のヒートマップ。市街地距離(cityblock distance)および平均連結法を用いて遺伝子の階層的クラスタリングを行った。c.DNAマイクロアレイを用いて2F iPS細胞(クローンF-4)およびESC間、並びに2F iPS細胞(クローンF-4)およびNSC間で総体的な遺伝子発現パターンを比較した。d.黒線は2つの細胞型間の遺伝子発現レベルの2倍の変化を示している。NSCまたはESCと比較して2F iPS細胞(クローンF-4)で過剰発現している遺伝子を青で示す;低発現のものを赤で示す。散布図に多能性遺伝子Oct4、Nanog、Sox2、c-Myc、Klf4、およびLin28の位置を示す。遺伝子発現レベルはlog2で測定する(scaled)。
【0080】
図3:2F Oct4/Klf4(OK)iPS細胞(クローンF-4)は多能性であり、in vitroおよびin vivoにおいて分化する。
a.in vitroにおける3つの胚葉全てへの分化。胚様体形成後、凝集体をゼラチンコーティングしたプレート上に移し、更に10日間分化させた。細胞を抗Tuj1、抗α-フェトプロテイン(AFP)、または抗Flk1で染色した。核はDAPIで染色した。b.3つの胚葉全てを含有するF-4 iPS細胞のテラトーマ。F-4 iPS細胞(1.5 x 106細胞)をヌードマウスの皮下に接種した。4週間後、テラトーマをヘマトキシリンおよびエオシン色素で染色した。神経ロゼット(外胚葉)、筋肉(中胚葉)、および円柱上皮(内胚葉)を含有するテラトーマを示す。
【0081】
図4:2F Oct4/Klf4(OK)iPS細胞(クローンF-4)のin vivoにおける分化能。
a.F-4 iPS細胞のキメラ胚は凝集の24時間後に胚盤胞に分化した。蛍光光学は、Oct4-GFP細胞が胚盤胞の内部細胞塊中に存在することを示している。b.Oct4-GFPの発現によって示される、F-4 iPS細胞のマウス胚発生への生殖系列の関与。胚をE13.5で蛍光顕微鏡を用いて分析した。c、d.13.5 dpcのキメラ胚(コントロール、2N、および4N)をX-gal溶液で染色した。e.lacZ染色した13.5 dpcのキメラ胚(2N)の組織学的分析。f.F-4 iPS細胞によって作製したキメラマウス(8週齢)。F-4 iPS細胞に由来するアグーチ毛色(agouti coat colour)。g.F-4 iPS細胞に由来するキメラのPCRジェノタイピング。Oct4-GFPについてPCR分析を行った(上段パネル)。F-4 iPS細胞の生殖細胞系伝達。CD-1メスと交配させたキメラ・オスの子孫の遺伝子型同定(genotyping)により、Oct4-GFPおよびlacZ対立遺伝子の存在、ならびにOct4およびKlf4のウィルス組込みが明らかになった(下段パネル)。略語:Gastroint. tract.:胃腸管。
【0082】
図5:1因子hNSC由来iPS(1F hNiPS)細胞のコロニー形成および細胞株のキャラクタリゼーション。
(A)NSC培地中で増殖させたhNSCの形態。(B)hOCT4感染細胞の感染10週間後のコロニー形成。(C)コロニーはhESC様形態を成すが、コロニーの中央は再プログラミングされていない神経ロゼットのままである。(D)継代1で機械的に単離した後のフィーダー上での典型的なhESC様iPSコロニー形成(1F hNiPSクローンC)。(E)継代10でのiPSコロニーの高倍率画像。(F)1F hNiPSコロニーをAPで染色した。スケールバーは250μm。(G)2F hNiPS(クローンA)および1F hNiPS(クローンC)細胞における多能性マーカー(OCT4、SSEA4、TRA-1-60、およびTRA-1-81)の免疫細胞化学分析。核をDAPIで染色する(青)。スケールバーは250μm。
【0083】
図6:hNSC由来iPS(hNiPS)細胞における多能性マーカーの発現レベルおよびDNAメチル化分析。
(A)H1 hESC、hNSC、2F hNiPSクローン(A、B、およびC)、および1F hNiPSクローン(AおよびC)における多能性マーカーの定量PCR分析。データは、内因性転写物に特異的なプライマーを使用し、H9 hESCに対する相対発現で示す。RNA発現レベルは対数尺度で示す。転写レベルはβ-アクチンレベルで正規化した。エラーバーは3回行った試験(triplicates)からのs.d.を示す。(B)H9 hESC、hNSC、2F hNiPSクローン(A、B、およびC)、および1F hNiPSクローン(AおよびC)におけるOCT4およびNANOGプロモーター領域のバイサルファイトシーケンシング分析。所与の増副産物に関するそれぞれの丸印の列は、その領域での1つの細菌クローンの各CpGのメチル化状態を表す。白丸はメチル化されていないCpGを表し、黒丸はメチル化されたCpGを表す。各列の下部の数字は、転写開始位置(TSS;+1)に対するCpGジヌクレオチドの位置を示す。
【0084】
図7:in vitroにおけるhNSC誘導iPS(hNiPS)細胞の3つの胚葉への分化。
(A)免疫蛍光分析は、2Fおよび1F hNiPS細胞が3つの胚葉:内胚葉(α-フェトプロテイン;AFP)、中胚葉(α-平滑筋アクチン;α-SMA)、および外胚葉(β-チューブリン IIIb;Tuj1)の全てに分化することを示している。DAPIで核を染色する(青)。スケールバーは100μm。(B)2F hNiPS(クローンA)および1F hNiPS(クローンC)細胞に由来する1ヶ月胚様体(EB)分化の定量PCR分析。内胚葉(AFP、GATA6、およびSox17)、中胚葉(FOXF1およびHAND1)、および外胚葉(NCAM1、PAX6、およびSox1)。データはそれぞれの未分化hNiPS親細胞に対する相対発現で示す。RNA発現レベルを対数尺度で表す。転写レベルはβ-アクチンレベルに対して正規化した。
【0085】
図8:in vivoにおけるhNSC由来iPS(hNiPS)細胞の多能性および総体的遺伝子発現プロファイル。
(A)2F hNipS(クローンA)および1F hNiPS(クローンC)細胞をSCIDマウスに移植した後のテラトーマ形成。6-8週間でテラトーマを切片化し、ヘマトキシリンおよびエオシンで染色した。3つの胚葉:内胚葉(呼吸上皮;r)、中胚葉(骨格筋;m、軟骨;c)、および外胚葉(神経上皮;n)全てを示すことが同定された細胞の組織切片。拡大部は呼吸上皮、筋肉および神経上皮を示す(矢印で表示)。スケールバーは100μm。(B)hNSC、1F hNiPS(クローンC)、2F hNiPS(クローンA)、H9 hESC、およびH1 hESC(左)からの総体的な遺伝子発現のヒートマップ(左パネル)および階層的クラスター分析(右パネル)。(C)1F hNiPS(クローンC)およびH9 hESC間(左パネル)、2F hNiPS(クローンA)およびH9 hESC間(中央パネル)、そしてhNSCおよび1F hNiPS(クローンC)間(右パネル)間の総体的な遺伝子発現プロファイルを比較する散布図。黒線は2つの細胞集団間の遺伝子発現レベルが2倍の相違であることを示す。転写発現レベルはlog2スケールである。
【図面の簡単な説明】
【0086】
【図1】図1は、OG2/Rosa26トランスジェニックマウスの成体NSCからの2F Oct4/Klf4(OK)iPS細胞の作製を示す。
【図2】図2は、iPS細胞の遺伝子発現プロファイルを示す。
【図3】図3は、2F Oct4/Klf4(OK)iPS細胞(クローンF-4)は多能性であり、in vitroおよびin vivoにおいて分化することを示す。
【図4】図4は、2F Oct4/Klf4(OK)iPS細胞(クローンF-4)のin vivoにおける分化能を示す。
【図5】図5は、1因子hNSC由来iPS(1F hNiPS)細胞のコロニー形成および細胞株のキャラクタリゼーションを示す。
【図6】図6は、hNSC由来iPS(hNiPS)細胞における多能性マーカーの発現レベルおよびDNAメチル化分析を示す。
【図7】図7は、in vitroにおけるhNSC誘導iPS(hNiPS)細胞の3つの胚葉への分化を示す。
【図8】図8は、in vivoにおけるhNSC由来iPS(hNiPS)細胞の多能性および総体的遺伝子発現プロファイルを示す。
【発明を実施するための形態】
【0087】
実施例にて本発明を例証する。
【0088】
実施例1:OG2マウスの作製
OG2系統をROSA26トランスジェニック系統と数代にわたって交配させ(Do, J. T.およびScholer, H. R., Stem Cells 22, 941-9 (2004);Szabo, P. E., Hubner, K., Scholer, H.およびMann, J. R., Mech Dev 115, 157-60 (2002))、neo/lacZおよびOct4-GFP導入遺伝子の複合ホモ接合性マウスを作製した。NSCを誘導するために、ホモ接合性OG2 x ROSA26オス・マウスをICRメスと交配させてヘテロ接合性の子を出生させる。5日齢のOG2 x ROSA26ヘテロ接合性マウスから脳組織を回収した。
【0089】
実施例2:人工多能性幹細胞の作製
iPS細胞およびESCをESC培養液(15% FBS、非必須アミノ酸、L-グルタミン、ペニシリン/ストレプトマイシン、β-メルカプトエタノール、および1,000 U/ml 白血病抑制因子(LIF)を補充したDMEM)中、放射線照射したMEF上で増殖させた。Oct4、Sox2、Klf4、およびc-MycのマウスcDNAをコードするpMXに基づくレトロウィルスベクターを、Fugene 6トランスフェクション試薬(Roche社)を用いて、パッケージング欠損ヘルパープラスミドと共に293細胞に個別にコトランスフェクトした。48時間後、従前の報告(Zaehres, H.およびDaley, G. Q., (2006), Methods Enzymol 420, 49-64)のようにウィルス上清を回収した。OG2/Rosa26トランスジェニックマウスから誘導したNSCを6-ウェルプレートあたり5 x 104細胞の密度で播種し、4因子(1:1:1:1)またはOct4およびKlf4(1:1)のウィルス含有上清に6μg/mlの硫酸プロタミン(Sigma社)を補充したものと共に24時間インキュベートした。pMX-GFPコントロールウィルスを用いて形質導入効率を算出した。細胞を新たな神経増殖培養液に再播種した。感染の2日後、更なる選択を行わずに、細胞をLIF含有ESC培養液中、放射線照射MEF上で更に継代培養した。Oct4-GFP陽性コロニーを機械的に単離し、個々の細胞を分離した後、MEF上に再播種した。コロニーを選択して増殖させた。
【0090】
実施例3:qRT-PCR分析
MiniRNeasy Kit(Qiagen社、ドイツ、ヒルデン市;http://www.qiagen.com)を用い、製造者の説明書に従って細胞から総RNAを抽出した。High Capacity cDNA Archive Kit(Applied Biosystems社、ドイツ、ダルムシュタット市;http://www.appliedbiosystems.com)を用い、製造者の説明書に従い、反応液容量を20μlにスケールダウンして相補的DNA合成を行った。ABI PRISM Sequence Detection System 7900(Applied BioSystems社)およびready-to-use 5'-nuclease Assays-on-Demandを用いて転写レベルを測定した。各リアルタイム増幅で、テンプレートは5ngの総RNA相当であった。測定は3回重複して(triplicate)行った;各サンプルのRTブランクおよびテンプレートなしのブランクをネガティブコントロールとした。増幅曲線および遺伝子発現をハウスキーピング遺伝子Hprt(内部標準として使用)に対して正規化した。
【0091】
以下の遺伝子の検出のために、Taqman Assay-on-Demandによってオリゴヌクレオチドを設計した:Pou5f1 (Oct3/4) (Mm00658129_gH)、Sox2 (Mm00488369_s1)、c-Myc (Mm00487803_m1)、Klf4 (Mm00516104_m1)、B-Act (Mm00607939_s1)、およびHprt1 (Mm00446968_m1)。Nanogおよびウィルス配列を検出するためのオリゴは注文設計した。ΔΔCt法(ABI PRISM 7700 Sequence Detection System, ユーザー会報#2)を用いて各プローブ/プライマーセットに関して得られた増幅曲線のlog直線相内で内因性Hprt遺伝子について定量を正規化した。
【0092】
ウィルス特異的qRT-PCRのプライマー配列
【0093】
【化1】
【0094】
実施例4:マイクロアレイ分析
Affymetrix Mouse Genome 430 2.0 GeneChip arrays(Affymetrix社、カリフォルニア州サンタクララ市)を用い、基本的に従前の報告(Ruau, D.ら,(2008), Stem Cells)に基づいてマイクロアレイ研究を行った。簡潔に記載すると、DNAアーゼ消化を含むRNAeasy kit(Qiagen社、ドイツ、ヒルデン市)を用いて細胞から総RNAを抽出した。GeneChip One-Cycle標識キット(Affymetrix)を用いてビオチン標識したcRNAを3μgの総RNAから得た。15μgのcRNAを断片化し、45℃で16時間、Affymetrix 430 2.0 GeneChipアレイにハイブリダイズさせた。DNAチップを洗浄し、染色してAffymetrix Fluidics装置およびGCS3000スキャナーを用いてスキャンし、得られた画像をGCOSソフトウェアを用いて分析した。ESCおよびiPS細胞では3回重複して、NSCに関しては2回重複して実験を行った。BioConductorで実行されたRMAアルゴリズム(Irizarry, R. A.ら, (2003), Nucleic Acids Res 31 , e15)を用いて、正規化を行った。
【0095】
実施例5:in vitroにおけるiPS細胞の分化
Oct4-GFP細胞をFACS分析によって回収し、胚様体(EB)のin vitro分化に用いた(LIFを含有しないESC培養液中、懸滴で実施)。3日後、EBをゼラチンコーとした4-ウェルプレートに播種し、さらに10日間培養した。細胞を抗Tuj1抗体(1:100。Chemicon)、抗αフェトプロテイン(AFP)抗体(1:100。R&D Systems)、または抗FlK1抗体(1:100。R&D Systems)で染色した。
【0096】
実施例6:ウェスタンブロット分析、SSEA-1、およびAP染色
ESCおよびNSCから調製した総細胞ライセート(2 x 106)を、Oct4 (Santa Cruz)、Sox2 (Santa Cruz)、Klf4 (Abcam)、およびc-Myc (Abcam)の発現に関するウェスタンブロット分析に供した。全てのサンプルで、β-アクチン発現レベルをローディングコントロールとして使用した(Abcam)。
【0097】
ES Cell Characterization Kit (Chemicon)を用い、製造者のプロトコルに従って、SSEA-1およびアルカリホスファターゼ (AP)染色を行った。
【0098】
実施例7:テラトーマの形成
iPS細胞およびNSC細胞(1.5 x 106細胞/マウス)をヌードマウスの背側側腹部に皮下注射した。注射4週間後、形成されたテラトーマを4% PFAで一晩固定し、パラフィンに包埋した。切片をヘマトキシリンおよびエオシン色素で染色した。
【0099】
実施例8:キメラの形成
iPS細胞を凝集させ、裸出されたコンパクション後の(denuded post-compacted)8細胞期マウス胚と共に培養した。簡潔に記載すると、2細胞期胚を1.5dpcのマウス((C57BL/6 x C3H)F1メス x CD1オス)から洗い出し(flushed)、M2培地中に配して、0.1% BSA含有KSOM培地中で一晩、8細胞期まで培養した。短時間のトリプシン処理をした2日目の培地から緩く結合したiPS細胞塊(10-20細胞)を選択し、鉱油下の10% FCS含有KSOM培地の懸滴に移した;各塊を懸滴中のくぼみに(in a depression)配した。それと同時に、30-40個の胚のバッチを酸性化タイロード液と共に、透明帯が分解するまで短時間インキュベートした。1個の胚を細胞塊上に配した。全ての凝集体をこの方法で集め、5% CO2/大気雰囲気下、37℃で培養した。培養24時間後、凝集体の多くが胚盤胞を形成していた。総数64個の凝集した胚盤胞(2.5 dpc)を5検体の偽妊娠マウス(CD-1バックグラウンド)の子宮角に移植した。
【0100】
実施例9:Oct4によるヒト神経幹細胞の再プログラミング
ヒト胎児脳組織から誘導したhNSCを従前の報告(Kimら, Exp Neurol 199, 222 (2006);Parkら, Nat Biotechnol 20, 1111 (2002))に従って無血清NSC培地で増殖させた(図5A参照)。まずhNSCをヒトOCT4およびKLF4(2F)またはOCT4(1F)をコードするpMXに感染させた。次いで、感染させたhNSCを7日間までNSC培地中に維持した(Kimら, Exp Neurol 199, 222 (2006))。感染8日後、10ng/ml bFGF含有hESC培地およびMEF条件培地(CM)(1:1)中で細胞を支持細胞(feeder cell)層上に再播種し、hESC様コロニーが出現するまで培養を続けた。感染後10-11週間以内に、hESC様iPSコロニーを同定したが、コロニーの中央は神経ロゼッタの様相のままであった(図5B参照)。更に5-6日以内に、コロニーは増殖して大きくなり、典型的なhESC様形態を示したが、コロニーの中央には神経ロゼッタが残存していた(図5C参照)。神経ロゼッタをコロニーから分離する。その後、コロニーの一片を機械的単離によって支持細胞層上に移植した(図5D参照)。OCT4に感染させたhNSCからのピッキングによって、3つのhESC様コロニーから2つのクローンを樹立することに成功した(1F hNiPSクローンAおよびC。再プログラミング効率0.02%)。他方、我々は2F感染hNSCで、感染後7-8週間以内に、5つのhESC様コロニーから3つのクローンを樹立した(2F hNiPS A、B、およびC。再プログラミング効率0.15%)。これらは全て、hESC培養条件で増殖することができた。1F hNiPS細胞はhESCと形態学的に類似しており、アルカリホスファターゼ染色陽性であった(図5EおよびF)。免疫蛍光染色により、2Fおよび1F hNiPS細胞が多能性マーカー(OCT4、SSEA4、TRA-1-60、およびTRA-1-81を含む)を一様に発現していることが確認された(図5G参照)。これらの結果は、OCT4およびKLF4、並びにOCT4のみによって、hNSCからヒトiPS細胞を作製できることを示している。
【0101】
次に我々は、定量RT-PCR分析により、これらのiPS細胞における多能性マーカー遺伝子のmRNA発現レベルを分子レベルで試験した。hESC特異的マーカーを内因的に発現する2Fおよび1F hNiPS細胞は、H9およびH1 hESCに類似しており、親hNSCと比較して有意に亢進的に調節されていた(図6A参照)。遺伝子型同定PCRは、1F hNiPSクローンがゲノム中にOCT4導入遺伝子のみを保有し、2F hNiPSクローンがOCT4およびKLF4導入遺伝子を保有することを示した。我々はまた、2F hNiPSクローンBからのOCT4発現を除いて、導入遺伝子OCT4またはKLF4の発現レベルが2Fおよび1F hNiPSクローンにおいて有意に抑制されていることも確認した。サザンブロット分析により、2Fおよび1F hNiPSクローンにおけるOCT4導入遺伝子の組込みが確認された。実験室でのhESCからの混入によってiPSクローンが生じた可能性を排除するために、DNAフィンガープリント分析を行って、hNiPS細胞がドナーhNSCと厳密に相関することを確認した(表2参照)。
【0102】
再プログラミングした細胞からのOCT4およびNANOGプロモーターのエピジェネティックな改変を確認するために、バイサルファイトシークエンシングを行って両プロモーターの脱メチル化を測定した。OCT4およびNANOGプロモーター領域はドナーhNSCに対して2Fおよび1F hNiPS細胞では脱メチル化されており、hESCと類似していた。これらをまとめると、hNSCはOCT4単独の形質導入によって分子レベルでhESCに類似したiPS細胞に再プログラミングできる。
【0103】
次に我々は、胚様体(EB)分化および直接分化によってin vitroにおける2Fおよび1F hNiPS細胞の多能性を試験した。hNiPS細胞はEB分化によって容易に内胚葉(AFP)、中胚葉(a-SMA)、および外胚葉(Tuj1)に分化し(図5A参照)、我々は定量RT-PCR分析によって直接分化から3つの胚葉マーカー全ての発現を確認した(図7B参照)。in vivoにおけるこれらのヒトiPS細胞の多能性を確認するために、細胞を重度の複合型免疫不全(SCID)マウスに皮下移植した。移植の6-8週後、2Fおよび1F hNiPS細胞は3つの胚葉全てを(呼吸器、骨格筋、軟骨組織、および神経上皮を含む)含有するテラトーマを生じた(図8A参照)。これらの結果は2Fおよび1F hNiPS細胞がhESCのようにin vitroおよびin vivoにおいて多能性を有することを示している。
【0104】
最後に我々は、cDNAマイクロアレイによって、hNSC、hNSCに由来する2Fおよび1F hNiPS細胞、H9およびH1 hESCの総体的な遺伝子発現分析を行った。ヒートマップは、2Fおよび1F hNiPS細胞がhESCと同様であり、他方、親hNSCは多能性集団から分離されることを示し(図8B、左パネル参照)、階層的クラスター分析は、hNiPS細胞がhESCとクラスターを形成し、親hNSCとは区別されることを示している(図8B、右パネル参照)。散布図分析は、hNiPS細胞が異なるhESC間のように、親hNSCよりhESCとより高い類似性を有することを示している(図8C参照)。1Fおよび2F hNiPS細胞もH1 hESCとの類似性を示す。これらのデータはhNiPS細胞が総体的な遺伝子発現プロファイルにおいてhESCと類似していることを示している。結果は1Fおよび2F hNiPS細胞が分子レベルおよび多能性に関してhESCに酷似していることを示している。
【0105】
【表2】
【0106】
試料および方法
細胞培養
ヒトNSCは終脳(HFT13)に由来し、従前の報告(Kimら, Exp Neurol 199, 222 (2006))に従って樹立した。簡潔に記載すると、終脳組織を新たに切り出し、0.1%トリプシン中で30分間解離させ、NSC培地中、200,000細胞/mlの密度で10cmプレートに播種した。これらの細胞を従前の報告(Kimら, Exp Neurol 199, 222 (2006);Parkら, Nat Biotechnol 20, 1111 (2002))に従ってNSC培地中で培養した。ヒトESおよびiPS細胞を、マイトマイシンC処理したCF1マウス支持細胞層(Millipore)上で、ヒトESC培地(20% ノックアウト血清代替添加物(Invitrogen)、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、ペニシリン/ストレプトマイシン、および10ng/ml ヒト塩基性線維芽細胞成長因子(bFGF) (Invitrogen)を補充したノックアウトDMEM (Invitrogen)を含有)中、従前の報告(Takahashiら, Cell 131 , 861 (2007))に従って維持した。
【0107】
1F hNiPS細胞および2F hNiPS細胞の導入
OCT4およびKLF4のヒトcDNAをコードするpMXに基づくレトロウィルスベクター(TakahashiおよびYamanaka, Cell 126, 663 (2006))をFugeneトランスフェクション試薬(Roche社)を用いて、パッケージング欠損ヘルパープラスミドと共に293細胞にコトランスフェクトし、従前の報告(ZaehresおよびDaley, Methods Enzymol 420, 49 (2006))に従って水疱性口内炎ウィルス(VSV)Gタンパク質偽型ウィルスを生成させた。トランスフェクションの48時間後にウィルス上清を回収し、超遠心によって濃縮し、ヒトNSCに感染させた。iPSの作製のために、ヒトNSCを6-ウェルプレートあたり5 x 104細胞の密度で播種し、Oct4またはOct4およびKLF4のウィルス含有上清に6μg/mlの硫酸プロタミン(Sigma社)を補充したものと共に24時間インキュベートした。翌日、感染後1日目に培地をフレッシュなNSC培地で交換し、感染後7日目まで維持した。感染後8日目から、細胞をヒトESC培地中で更に培養した。感染後2ヶ月または2.5ヶ月でiPSコロニーを機械的に単離した後、ヒトESC培地中、CF1マウス支持細胞層 (Millipore)上に播種し、維持した。
【0108】
定量RT-PCR
バルクの細胞培養液サンプルまたは手でピックアップした未分化コロニーから、オンカラムDNA消化を含むRNeasyカラム(Qiagen)を用いて総RNAを単離した。オリゴ-dT15プライミングおよびM-MLV逆転写酵素(UBS社)を用い、製造者の説明書に従って42℃で1時間、cDNAを生成させた。通常、20μlのSYBR Green PCR反応液に約50ngの総RNA相当物をテンプレートとして使用し、反応液は更に0.375μMの各プライマーおよび10μlのSYBR Green PCR mix (ABI)を含有する(95℃で15秒/60℃で60秒を40サイクル。Applied Biosystems 7300装置)。使用したプライマーは全て、副産物を生成せずに最適に近い効率で予期する産物を増幅することを確認した。プライマー配列を表3に示す。比較Ct法を用い、生体コントロールサンプルおよび正規化のための2つのハウスキーピング遺伝子に基づいて相対発現レベルを算出した。エラーバーは生体複製物(マーカー遺伝子発現データ)または正規化のための個別のハウスキーピング遺伝子(導入遺伝子サイレンシングデータ)を用いて得られた標準誤差を表す。
【0109】
総体的な遺伝子発現分析
転写分析を、400ngの総DNA未含有RNAを標識cRNA合成(Illumina TotalPrep RNA Amplification Kit - Ambion)のインプットとして使用し、製造者の説明書に従って行った(IVT:10h)。クォリティーを確認したcRNAサンプルを生物学的または技術的な複製(duplicate)としてHumanRef-8 v3 expression BeadChips(Illumina)上に18時間ハイブリダイズさせ、洗浄、染色、およびスキャンした(ガイドラインに従い、製造者が提供/推奨する物質/装置を用いた)。マイクロアレイ・データはGEO(Gene Expression Omnibus)ウェブサイト(受入番号:GSE GSE15355)から入手できる。
【0110】
バイサルファイトシーケンシング
DNeasyカラム(Qiagen)を用いてバルクの細胞培養液サンプルまたは手でピックアップした未分化コロニーからゲノムDNAを単離した。300ngを亜硫酸水素変換のインプットとして使用した(EpiTect Bisulfite Kit - Qiagen)。50ngの変換DNAを従来型のネステッドPCRのテンプレートとして使用し、Oct4およびNANOGプロモーターのそれぞれ467および336bpの領域を増幅した。プライマーはセンスDNA鎖の変換に特異的であり、表3に記載する。精製したPCRをpCR2.1-TOPO (Invitrogen)にTA-クローニングした。ランダムにピックアップしたクローンのインサート配列をBiQ Analyzerプログラムを用いて分析し、必要によりクォリティーチェックに基づく示唆に従って、個々のクローンを除外(drop)した。OCT4転写開始コドンから+20位のCpG部位からのデータは、情報を与えないため記載していない。
【0111】
ショートタンデムリピート(STR)分析
DNeasyカラム(Qiagen)を用いて培養細胞サンプルからゲノムDNAを単離した。これをSTR分析(PowerPlex 16 System (Promega)およびABI PRISM装置を使用)のテンプレートとして使用した。数字は15の常染色体フラグメントの長さ(bp)を示す。Eurofins Medigenomix (ドイツ マルティンスリード)で分析を実施した。
【0112】
テラトーマ形成
hNiPS細胞およびhNSC(3-5 x 106 細胞/マウス)をSCIDマウスの背側側腹部に皮下注射した。注射の6-8週間後にテラトーマを4% PFAで一晩固定し、パラフィンに包埋した。切片をヘマトキシリンおよびエオシン色素で染色した。
【0113】
アルカリホスファターゼ(AP)および免疫蛍光染色
アルカリホスファターゼ(AP)染色をES Cell Characterization Kit(Chemicon)で製造者のプロトコルに従って実施した。以下の一次抗体を使用して免疫蛍光染色を行った:AFP(Sigma、1:100)、a-SMA(Sigma、1:50)、TuJ1(Chemicon、1:500)、OCT4(Santa Cruz、1:200)、SSEA4(Chemicon、1:200)、TRA-1-60(Chemicon、1:200)、TRA-1-81(Chemicon、1:200)。
【0114】
【表3】
【0115】
【表4】
【0116】
サザンブロット分析
BamHIで消化した1F hNiPS、hNSC、および2F hNiPS細胞からのゲノムDNAを0.8%アガロースゲル上で分離し、Biodyne Bナイロン膜(PALL Life Sciences)に転写した。DecaLabelTM DNA Labeling Kit (Fermentas)を用いてDNAをOCT4の32P標識したフラグメント(Eco81I (Saul)ヒトOCT4 cDNAフラグメント)とハイブリダイズさせた。標識したラムダHindIII消化DNAをマーカーとした。
【0117】
in vitroにおけるヒトiPS細胞の分化
免疫細胞化学で、懸滴法を用い、MEF条件培地中でiPS細胞から胚様体(EB)を作製した。5日後、EBをゼラチンコーティングしたプレートに移し、20% FBS、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、およびペニシリン/ストレプトマイシンを補充したノックアウトDMEM (Invitrogen)中で更に14日間培養した。qRT-PCRで、iPSコロニーを機械的に単離し、MEF条件培地中、マトリゲルをコーティングしたプレート上に再播種した。2日後、培地を3つの胚葉分化培地で置換した。内胚葉分化では、細胞を、2% FBS、100ng/ml アクチビンA (R&D Systems)、L-グルタミン、およびペニシリン/ストレプトマイシンを補充したRPMI1640培地中で3週間維持した(Huangfuら, Nat Biotechnol 26, 1269 (2008))。中胚葉分化では、100uM アスコルビン酸(Sigma)、20%-FBS、1mM L-グルタミン、1% 非必須アミノ酸、0.1mM β-メルカプトエタノール、およびペニシリン/ストレプトマイシンを補充したノックアウトDMEM中で3週間維持した(Aasenら, Nat Biotechnol 26, 1276 (2008))。外胚葉分化では、細胞をN2B27培地中で7日間維持し、2週間の間、10ng/ml bFGF2(peprotech)、100ng/ml ソニックヘッジホッグ(R&D Systems)、10ng/ml PDFG(R&D Systems)、L-グルタミン、およびペニシリン/ストレプトマイシンを補充したN2培地で置換した。培地は1日おきに交換した。プライマー配列を表3に示す。
【0118】
参考文献
Biswas, A. and Hutchins, R. (2007). Embryonic stem cells. Stem Cells Dev 16(2):213-22.
Davis, P. B., Cooper, M.J. (2007). Vectors for airway gene delivery. AAPS J 19;9(1):E11-7.
Do, J. T. & Scholer, H. R. (2004). Nuclei of embryonic stem cells reprogram somatic cells. Stem Cells 22:941-9.
Gage, F. H. (2000). Mammalian neural stem cells. Science 287(5457): 1433-8.
Hanna, J., Wernig, M., Markoulaki, S., Sun, C.W., Meissner, A., Cassady, J. P., Beard, C, Brambrink, T., Wu, L.C., Townes, T.M. and Jaenisch, R. (2007).
Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin. Science 318(5858): 1920-3.
Hogan, B., R. Beddington, et al. (1994), "Manipulating the Mouse Embryo: A Laboratory Manual", Cold Spring Harbour Press.
Jones, G. E., Wise, CJ. (1997). Establishment, maintenance, and cloning of human dermal fibroblasts. Methods MoI Biol. 75:13-21.
Kim, D. S., Kim, J.Y., Kang, M., Cho, M.S., Kim, D.W. (2007). Derivation of functional dopamine neurons from embryonic stem cells. Cell Transplant 16(2): 117-23.
Li, S. and Ma, Z. (2001). Nonviral gene therapy. Curr Gene Ther.,1(2):201-26. Meissner, A., Wernig, M. and Jaenisch, R. (2007). Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol 25(10): 1177-81.
Nakagawa, M., Koyanagi, M., Tanabe, K., Takahashi, K., lchisaka, T., Aoi, T., Okita, K., Mochiduki, Y., Takizawa, N. and Yamanaka, S. (2008). Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 26(1): 101 -6.
Okita, K., lchisaka, T. and Yamanaka, S. (2007). Generation of germline-competent induced pluripotent stem cells. Nature 448(7151):313-7.
Park, I. H., Zhao, R., West, J.A., Yabuuchi, A., Huo, H., Ince, T.A., Lerou, P.H., Lensch, M.W., Daley, G. Q. (2008). Reprogramming of human somatic cells to pluripotency with defined factors. Nature 451 (7175): 141 -6.
Pollard, S. M., Conti, L., Sun, Y., Goffredo, D. & Smith, A. (2006). Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 16 Suppl 1 , i112-20.
Ryan, A. K. and Rosenfeld, M. G. (1997). POU domain family values: flexibility, partnerships, and developmental codes. Genes Dev 11 :1207-25.
Takahashi, K. and Yamanaka, S. (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663-76.
Takahashi, K., Okita, K., Nakagawa, M., and Yamanaka, S. (2007). Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2(12):3081-9. Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., lchisaka, T., Tomoda, K. and Yamanaka, S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861-72.
Wernig, M., Meissner, A., Foreman, R., Brambrink, T., Ku, M., Hochedlinger, K., Bernstein, B. E. and Jaenisch, R. (2007). In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature 448:318-24.
Wernig, M., Meissner, A., Cassady, J. P. and Jaenisch, R. (2008). c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cells 2:10-12.
Yu, J., Vodyanik, M.A., Smuga-Otto, K., Antosiewicz-Bourget, J., Frane, J. L., Tian, S., Nie, J., Jonsdottir, G.A., Ruotti, V., Stewart, R., Slukvin, I.I. and Thomson, J.A. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318(5858): 1917-20.
Zimmermann, W. H. and Eschenhagen, T. (2007). Embryonic stem cells for cardiac muscle engineering. Trends Cardiovasc Med 17(4): 134-40.
H. T. Kim et al., Exp Neurol 199, 222 (May, 2006)
K. I. Park, Y. D. Teng, E. Y. Snyder, Nat Biotechnol 20, 1111 (Nov, 2002)
H. Zaehres, G. Q. Daley, Methods Enzymol 420, 49 (2006)
D. Huangfu et al., Nat Biotechnol 26, 1269 (Nov, 2008)
T. Aasen et al., Nat Biotechnol 26, 1276 (Nov, 2008)
【特許請求の範囲】
【請求項1】
人工多能性幹細胞(iPS細胞)を作製する方法であって、1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち該標的細胞の人工多能性幹細胞への再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する、上記方法。
【請求項2】
Myc、Klf、およびSox因子ファミリーに属し、標的細胞に導入されるべきコード配列によって内因的に発現またはコードされる因子が、I-Myc、n-Myc、c-Myc、Klf1、Klf2、Klf4、Klf15、Sox1、Sox2、Sox3、Sox15、およびSox18から成る群から選択される、請求項1に記載の方法。
【請求項3】
標的細胞が、該標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しない、請求項1または2に記載の方法。
【請求項4】
標的細胞が複能性(multipotent)幹細胞である、請求項1から3のいずれか一項に記載される方法。
【請求項5】
複能性幹細胞が外胚葉細胞である、請求項4に記載の方法。
【請求項6】
標的細胞が神経幹細胞(NSC)である、請求項1から5のいずれか一項に記載される方法。
【請求項7】
導入されるべきコード配列がOct3/4因子をコードする、請求項6に記載の方法。
【請求項8】
導入されるべき2つのコード配列がOct3/4およびc-MycまたはOct3/4およびKlf4因子をコードする、請求項6に記載の方法。
【請求項9】
標的細胞がc-Myc、Klf4、およびSox2因子を内因的に発現する、請求項7または8に記載される方法。
【請求項10】
標的細胞がc-Myc、Klf4、およびSox2因子を、標的細胞と同じ属(genus)の胚性幹細胞での相当する発現レベルに比較して最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する、請求項9に記載の方法。
【請求項11】
標的細胞がマウス神経幹細胞である、請求項7から10のいずれか一項に記載される方法。
【請求項12】
請求項1から11のいずれか一項に記載される方法によって作製した人工多能性幹細胞。
【請求項13】
標的細胞の人工多能性幹細胞への再プログラミングに関与する化合物の同定法であって、以下の工程:
(a)請求項1から11のいずれかに記載される方法に従って標的細胞を再プログラミングすること(1つの導入されるべきコード配列を、試験する化合物で置き換える);および
(b)試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価すること
を含み、ここで、試験する化合物を導入した標的細胞からiPS細胞が形成されれば、その化合物が標的細胞の人工多能性幹細胞への再プログラミングに関与していることを示す、上記方法。
【請求項14】
トランスジェニック非ヒト動物を作製する方法であって、以下の工程:
(a)請求項1から11のいずれか、または請求項12に記載される方法によって作製した人工多能性幹細胞を非ヒト着床前胚に導入すること;
(b)工程(a)の胚をメス非ヒト動物の子宮に移植すること;および
(c)胚を分化および出生させること
を含む、上記方法。
【請求項15】
請求項14に記載の方法で作製したトランスジェニック非ヒト動物。
【請求項16】
遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための、請求項1から11のいずれか一項に記載される方法で作製したiPS細胞を含む組成物。
【請求項1】
人工多能性幹細胞(iPS細胞)を作製する方法であって、1つまたは2つのコード配列を標的細胞に導入する工程を含み、コード配列はそれぞれ、ある因子、すなわち該標的細胞の人工多能性幹細胞への再プログラミングに関与し、Oct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を転写の際に生じさせ、ここで、標的細胞は少なくとも、導入されるコード配列にコードされておらずOct3/4またはMyc、Klf、およびSox因子ファミリーに属する因子から選択される因子を内因的に発現し、1つまたは2つのコード配列の導入によって得られる細胞は、Oct3/4因子をMyc、Klf、およびSoxの群から選択される各因子ファミリーの少なくとも1つの因子と組み合わせて発現する、上記方法。
【請求項2】
Myc、Klf、およびSox因子ファミリーに属し、標的細胞に導入されるべきコード配列によって内因的に発現またはコードされる因子が、I-Myc、n-Myc、c-Myc、Klf1、Klf2、Klf4、Klf15、Sox1、Sox2、Sox3、Sox15、およびSox18から成る群から選択される、請求項1に記載の方法。
【請求項3】
標的細胞が、該標的細胞に導入されるべき1つまたは2つのコード配列にコードされる因子の1つを内因的に発現しない、請求項1または2に記載の方法。
【請求項4】
標的細胞が複能性(multipotent)幹細胞である、請求項1から3のいずれか一項に記載される方法。
【請求項5】
複能性幹細胞が外胚葉細胞である、請求項4に記載の方法。
【請求項6】
標的細胞が神経幹細胞(NSC)である、請求項1から5のいずれか一項に記載される方法。
【請求項7】
導入されるべきコード配列がOct3/4因子をコードする、請求項6に記載の方法。
【請求項8】
導入されるべき2つのコード配列がOct3/4およびc-MycまたはOct3/4およびKlf4因子をコードする、請求項6に記載の方法。
【請求項9】
標的細胞がc-Myc、Klf4、およびSox2因子を内因的に発現する、請求項7または8に記載される方法。
【請求項10】
標的細胞がc-Myc、Klf4、およびSox2因子を、標的細胞と同じ属(genus)の胚性幹細胞での相当する発現レベルに比較して最低でも10倍低いレベルで、または最高でも10倍高いレベルで内因的に発現する、請求項9に記載の方法。
【請求項11】
標的細胞がマウス神経幹細胞である、請求項7から10のいずれか一項に記載される方法。
【請求項12】
請求項1から11のいずれか一項に記載される方法によって作製した人工多能性幹細胞。
【請求項13】
標的細胞の人工多能性幹細胞への再プログラミングに関与する化合物の同定法であって、以下の工程:
(a)請求項1から11のいずれかに記載される方法に従って標的細胞を再プログラミングすること(1つの導入されるべきコード配列を、試験する化合物で置き換える);および
(b)試験する化合物の存在下および非存在下でiPS細胞が形成されるか否かを評価すること
を含み、ここで、試験する化合物を導入した標的細胞からiPS細胞が形成されれば、その化合物が標的細胞の人工多能性幹細胞への再プログラミングに関与していることを示す、上記方法。
【請求項14】
トランスジェニック非ヒト動物を作製する方法であって、以下の工程:
(a)請求項1から11のいずれか、または請求項12に記載される方法によって作製した人工多能性幹細胞を非ヒト着床前胚に導入すること;
(b)工程(a)の胚をメス非ヒト動物の子宮に移植すること;および
(c)胚を分化および出生させること
を含む、上記方法。
【請求項15】
請求項14に記載の方法で作製したトランスジェニック非ヒト動物。
【請求項16】
遺伝子治療、再生医療、細胞療法、または薬剤スクリーニングのための、請求項1から11のいずれか一項に記載される方法で作製したiPS細胞を含む組成物。
【図1】

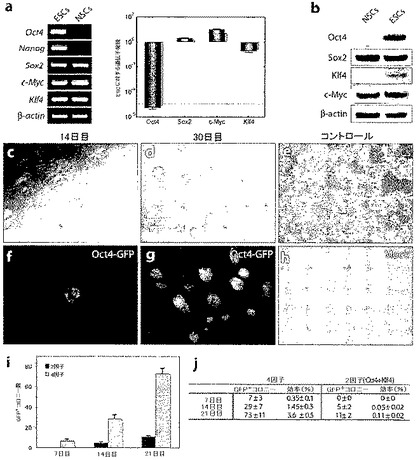
【図2】
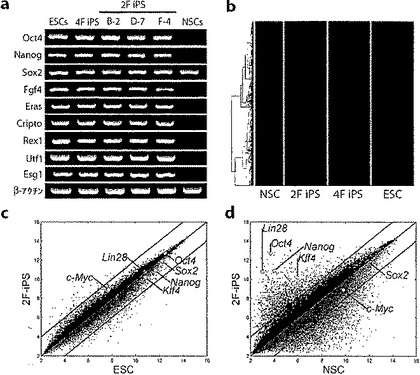
【図3】
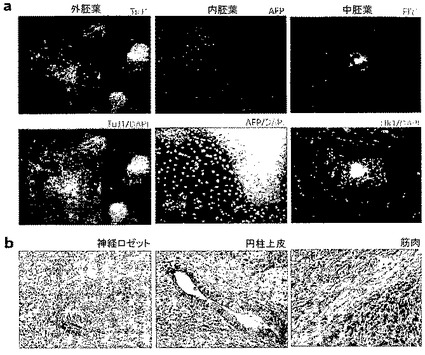
【図4】
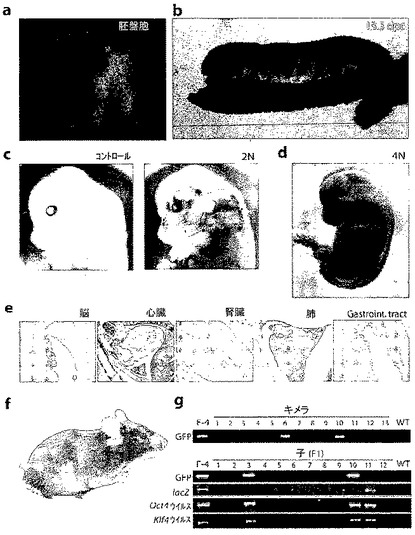
【図5】
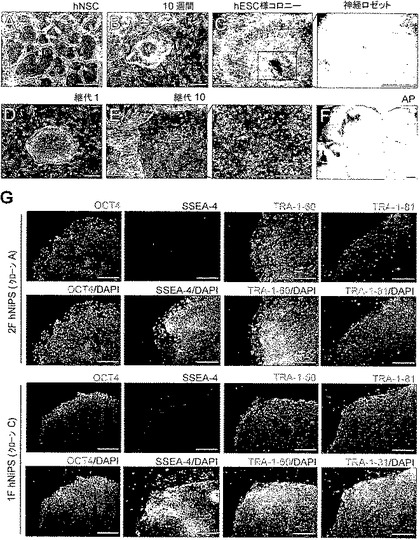
【図6】
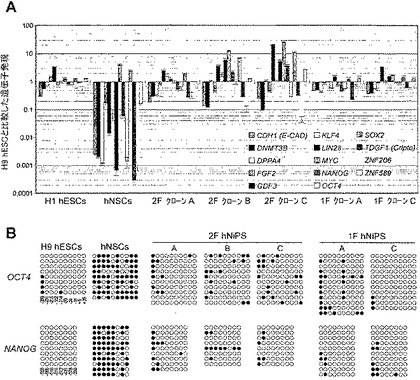
【図7】
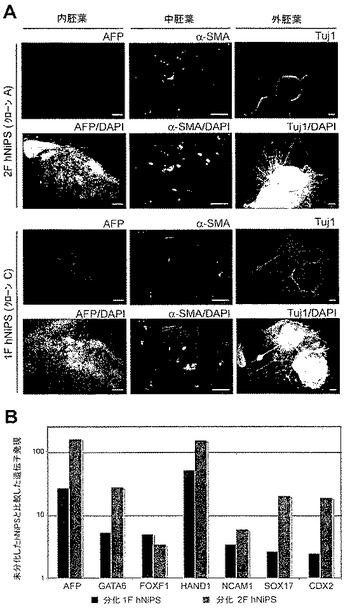
【図8】
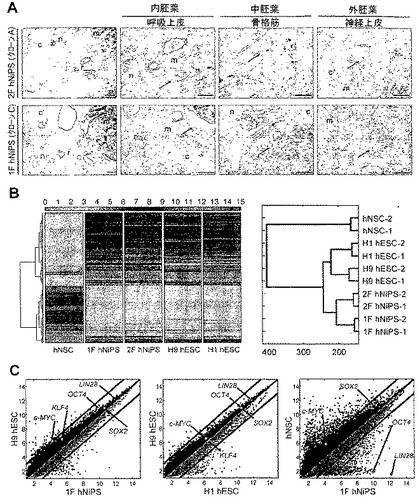
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2011−524160(P2011−524160A)
【公表日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願番号】特願2011−510886(P2011−510886)
【出願日】平成21年5月26日(2009.5.26)
【国際出願番号】PCT/EP2009/003735
【国際公開番号】WO2009/144008
【国際公開日】平成21年12月3日(2009.12.3)
【出願人】(596070445)マックス−プランク−ゲゼルシャフト ツール フォーデルング デル ヴィッセンシャフテン エー.ヴェー. (13)
【Fターム(参考)】
【公表日】平成23年9月1日(2011.9.1)
【国際特許分類】
【出願日】平成21年5月26日(2009.5.26)
【国際出願番号】PCT/EP2009/003735
【国際公開番号】WO2009/144008
【国際公開日】平成21年12月3日(2009.12.3)
【出願人】(596070445)マックス−プランク−ゲゼルシャフト ツール フォーデルング デル ヴィッセンシャフテン エー.ヴェー. (13)
【Fターム(参考)】
[ Back to top ]