
代謝関連障害を処置するためのGPR43およびその調節因子
本発明は、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が調節されるかどうかを決定することによって代謝安定化化合物を同定する方法であって、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法に関する。また、本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が増加するかどうかを決定することを含み、GPR43機能の増加が、その候補化合物が代謝安定化化合物であることを示す方法に関する。さらに本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が減少するかどうかを決定することを含み、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、ある化合物がGPR43機能を調節するかどうかを決定することによって、代謝安定化化合物を同定する方法に関する。したがって本発明の化合物は、低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患などの代謝関連障害の予防または処置に有用である。
【背景技術】
【0002】
(発明の背景)
細胞は主要エネルギー源としてグルコースを使用する。したがって食物は利用される前にまず身体によってグルコースに分解される。次にグルコースは腸から血中に放出されて、血中グルコースレベルの上昇をもたらす。このグルコースレベルの上昇に応答して、膵ベータ島細胞は、そのインスリン産生およびインスリン分泌を増加させる。インスリンは血液を介して循環し、脂肪組織、筋肉および肝臓などのインスリン応答器官にシグナルを送る伝達物質として作用して、それらのグルコース摂取を増加させる。このようにして、血中グルコースの上昇は、それに続くベータ細胞からのインスリン分泌の増加を伴う。血中グルコースレベルを正常に戻すように作用するのは、インスリンの上昇である。健常な個体では、血中グルコースレベルがかなり一定に保たれる。正常血糖(正常グルコースレベル)と呼ばれるこの平衡状態は、インスリンによって厳しく制御される。
【0003】
糖尿病などの疾患では、血中グルコースレベルのこの厳しい調節が失われて、糖尿病患者に観察される増加した血中グルコースレベルをもたらす。高血糖(高グルコースレベル)状態は、膵ベータ細胞によるインスリン産生の不足、および/または筋肉、肝臓および脂肪などの標的器官によるグルコースの不十分な取り込みによって起こりうる。最終結果は、血中グルコースレベルの増加である。したがって糖尿病は、2タイプの欠陥、すなわちベータ細胞からのインスリン分泌異常、および主要インスリン応答器官によるインスリン感受性異常の結果であると考えることができる。(器官がインスリンの作用に抵抗するので)インスリン抵抗性とも呼ばれているこのインスリン感受性異常は、標的器官がそのグルコース取り込みを増加させるのに、より多くのインスリンが要求されることを意味する。インスリン抵抗性はベータ細胞に対する圧力の増加をもたらす。なぜならベータ細胞は、インスリン抵抗性を補償するために、そのインスリン分泌を増加させる必要があるからである。これは段階的に拡大する問題であって、まず最初は耐糖能異常をもたらし、そして最終的には、増え続けるインスリンの需要に膵臓が応じきれないために起こるインスリン分泌の完全な喪失をもたらす。
【0004】
糖尿病は、血中グルコースの上昇をもたらす異常グルコース恒常性を特徴とする一群の障害を表す診断用語である。糖尿病には多くのタイプがあるが、最も一般的な2タイプが、インスリン依存性糖尿病またはIDDMとも呼ばれるI型と、インスリン非依存性糖尿病またはNIDDMとも呼ばれるII型である。I型糖尿病は、主として、発症年齢が低い疾患であり、膵臓中のインスリン分泌ベータ細胞が免疫系によって破壊されることによるものである。この場合、身体は膵ベータ細胞を自己と認識することができず、自分自身の細胞を破壊してしまう。ベータ細胞が破壊されるとインスリン分泌が完全に失われ、そのような患者は、生存するために、インスリンへの依存を強いられる。II型糖尿病は、主として、発症年齢が高い疾患であり、通常は40歳以降であるが、近年は、II型糖尿病と診断される若い人々も増えている。これはインスリン抵抗性およびベータ細胞疲弊を主たる特徴とし、多くの場合、肥満に関連する。II型糖尿病はI型糖尿病より一般的であり、世界中で診断される全糖尿病例の90〜95%を占める。
【0005】
高血糖への組織の慢性的曝露は、神経障害、網膜症および腎症の微小血管問題ならびに脳卒中、冠動脈心疾患、および末梢血管疾患の大血管性合併症を含む多様な合併症をもたらしうる。血中グルコースレベルの不適切な制御は、糖尿病以外の疾患、例えば肥満およびX症候群などの特徴でもある。例えば、X症候群の特徴の1つは、インスリン抵抗性またはグルコース不耐性である。また、肥満は、高インスリン血症およびインスリン抵抗性を特徴とし、これはNIDDM、高血圧およびアテローム性動脈硬化にも共通する特徴である。さらに肥満はNIDDMの主要リスク因子である。30%以上の過体重である被験者では、NIDDMを発症するリスクが3倍になり、NIDDM患者の4分の3は過体重である。
【0006】
カロリー摂取とエネルギー支出との間の不均衡の結果である肥満は、実験動物およびヒトにおいて、インスリン抵抗性および糖尿病と高い相関関係にある。しかし、肥満−糖尿病症候群に関与する分子機序は、まだ調査中である。肥満の初期発症中は、インスリン分泌の増加がインスリン抵抗性を相殺し、患者を高血糖から保護する(非特許文献1)。しかし、時が経つにつれてベータ細胞機能が劣化し、肥満個体の約20%でインスリン非依存性糖尿病が発症する(非特許文献2および非特許文献3)。したがって、現在社会におけるその高い罹患率を考えると、肥満はNIDDMの最も重要なリスク因子になっている(非特許文献4)。しかし、脂肪蓄積に呼応したインスリン分泌の変化を一部の患者で起こしやすくしている因子は、まだわかっていない。残念ながら、肥満の処置に利用することができる有効な長期治療はまだない。
【0007】
糖尿病は世界中で数百万人を苦しめている。米国だけで糖尿病患者は1800万人を超え、毎年600,000例が新たに診断される。糖尿病を持つ人々は、心臓疾患、失明、腎不全、感染、四肢切断、および他の状態のリスクが高くなっている。米国では糖尿病に起因すると考えられる直接医療支出および間接支出が、2002年の場合、1320億ドルだった。総合的にみて、糖尿病合併症は米国の主要死因の1つである。
【0008】
例えばα−グルコシダーゼ阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類および外因性インスリンなど、糖尿病を処置するための治療法は存在する。しかしこれらの治療法は有効性が限られており、低血糖エピソード、体重増加、胃腸障害および貧血など、重大な安全性および忍容性の問題を伴う。また、処置選択肢の多くは、注射または毎日複数回の投薬を必要とし、それが服薬遵守の問題を生む。
【非特許文献1】Le Stunffら,Diabetes(1989)43:696−702
【非特許文献2】Pederson,P.,Diab.Metab.Rev.(1989)5:505−509
【非特許文献3】Brancati,F.L.ら,Arch.Intern.Med.(1999)159:957−963
【非特許文献4】Hill,J.O.ら,Science(1998)280:1371−1374
【発明の開示】
【発明が解決しようとする課題】
【0009】
したがって、例えば糖尿病、アテローム性動脈硬化および肥満などの代謝関連障害を安全かつ効果的に処置する薬剤の同定が必要とされている。本発明は、この必要を満たすと共に、関連する利点を提供するものである。
【課題を解決するための手段】
【0010】
(要旨)
本出願人は、GPR43が膵島で発現され、db/db糖尿病マウスおよびob/ob肥満マウスでは、GPR43がアップレギュレートされることを、予想外に見いだした。また本出願人は、分化した脂肪細胞ではGPR43が強く誘導されることを開示する。さらに本出願人は、インスリン抵抗性、耐糖能異常および糖尿病などの代謝関連障害を処置するために、GPR43のインバースアゴニストまたはアンタゴニストを使用できることを開示する。
【0011】
第1態様として、本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が調節されるかどうかを決定することを含み、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法を特徴とする。一部の実施形態では、前記GPR43がヒト由来である。一部の実施形態では、前記決定が二次メッセンジャーアッセイを含む。
【0012】
第2態様として、本発明は、第1態様の方法に従って同定される代謝安定化化合物を特徴とする。一部の実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。
【0013】
第3態様として、本発明は、組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、前記化合物が第1態様の方法によって同定される方法を特徴とする。
【0014】
第4態様として、本発明は、第2態様の化合物を含むか、または本質的に第2態様の化合物からなるか、または第2態様の化合物からなる、医薬組成物を特徴とする。
【0015】
第5態様として、本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、第4態様の化合物の有効量を前記個体に投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、第5態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、第4態様の化合物の有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、個体が哺乳動物であり、一部の実施形態では、個体がヒトである。
【0016】
第6態様として、本発明は、代謝安定化化合物として使用するための第4態様の化合物を含む医薬を製造する方法、および代謝関連障害の処置に使用するための第4態様の化合物を含む医薬を製造する方法を特徴とする。
【0017】
第7態様として、本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が減少するかどうかを決定することを含み、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法を特徴とする。一部の実施形態では、前記GPR43がヒト由来である。一部の実施形態では、前記決定が二次メッセンジャーアッセイを含む。
【0018】
第8態様として、本発明は、第7態様の方法に従って同定される代謝安定化化合物を特徴とする。一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。
【0019】
第9態様として、本発明は、組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、前記化合物が第7態様の方法によって同定される方法を特徴とする。
【0020】
第10態様として、本発明は、第8態様の化合物を含むか、または本質的に第8態様の化合物からなるか、または第8態様の化合物からなる、医薬組成物を特徴とする。
【0021】
第11態様として、本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、第10態様の化合物の有効量を前記個体に投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、第11態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、第10態様の化合物の有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、個体が哺乳動物であり、一部の実施形態では、個体がヒトである。
【0022】
第12態様として、本発明は、代謝安定化化合物として使用するための第10態様の化合物を含む医薬を製造する方法、および代謝関連障害の処置に使用するための第10態様の化合物を含む医薬を製造する方法を特徴とする。
【0023】
第13態様として、本発明は、代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が低血糖または加齢であり、前記調節因子がアゴニストである。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患であり、前記調節因子がインバースアゴニストまたはアンタゴニストである。一部の実施形態では、第13態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。
【0024】
第14態様として、本発明は、GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法を特徴とする。一部の実施形態では、前記障害が代謝関連障害、例えばインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害がII型糖尿病である。一部の実施形態では、第14態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。
【発明を実施するための最良の形態】
【0025】
(詳細な説明)
本出願人は、マウスGPR43が脂肪、大腸、および膵島細胞において強く発現されること(図1参照)、ならびにdb/db糖尿病マウスおよびob/ob肥満マウスから単離された膵島では、野生型マウスの膵島と比較して、マウスGPR43がアップレギュレートされていること(図1参照)を、ここに開示する。また本出願人は、分化した3T3−L1脂肪細胞においてマウスGPRが強く誘導されることも、ここに開示する(図1参照)。さらに本出願人は、ヒトGPR43が膵臓を含むいくつかの組織で発現されること(図2上図参照)、ならびにマウスGPR43がいくつかの組織および膵島細胞株で発現されること(図2下図参照)を開示する。
【0026】
ヒトにはいくつかの受容体クラスが存在するが、群を抜いて豊富に存在し、治療的に重要なクラスは、Gタンパク質共役受容体(GPCR)クラスに代表される。ヒトゲノムにはおよそ30,000〜40,000の遺伝子があると見積られており、そのうちの約2%はGPCRをコードすると見積られている。GPCRは医薬品の開発にとって重要な分野であり、100の既知GPCRのうち約20から、全処方薬の約60%が開発されている。
【0027】
GPCRは、22〜24個の疎水性アミノ酸からなる7つの配列を持ち、それらは7つのαヘリックスを形成して、そのそれぞれが膜をまたいでいる(各区間は、膜貫通領域1(TM−1)、膜貫通領域2(TM−2)などというように、数字で識別される)という、共通の構造モチーフを有する。細胞膜の外側、すなわち「細胞外」側では、膜貫通ヘリックスが、膜貫通領域2と膜貫通領域3の間、膜貫通領域4と膜貫通領域5の間、および膜貫通領域6と膜貫通領域7の間にあるアミノ酸の鎖によってつながれる(これらは、それぞれ「細胞外」領域1、2および3(EC−1、EC−2およびEC−3)と呼ばれる)。細胞膜の内側、すなわち「細胞内」側でも、膜貫通ヘリックスは、膜貫通領域1と膜貫通領域2の間、膜貫通領域3と膜貫通領域4の間、および膜貫通領域5と膜貫通領域6の間にあるアミノ酸の鎖によってつながれる(これらは、それぞれ「細胞内」領域1、2および3(IC−1、IC−2およびIC−3)と呼ばれる)。受容体の「カルボキシ」(「C」)末端は細胞内の細胞内空間にあり、受容体の「アミノ」(「N」)末端は細胞外の細胞外空間にある。
【0028】
一般に、リガンドが受容体と結合すると(これはしばしば受容体の「活性化」と呼ばれる)、受容体のコンフォメーションに、細胞内領域と細胞内「Gタンパク質」との共役を促進するような変化が起こる。GPCRはGタンパク質に関して「プロミスキャス(promiscuous)」である、すなわちGPCRは2以上のGタンパク質と相互作用することができると報告されている。Kenakin,T.,43 Life Sciences 1095(1988)参照。Gタンパク質は他にも存在するが、現在のところ、Gq、Gs、Gi、GzおよびGoが、同定されているGタンパク質である。リガンド活性化GPCRのGタンパク質との共役は、シグナル伝達カスケード過程(「シグナル伝達」という)を開始させる。常態では、シグナル伝達が最終的に細胞活性化または細胞阻害をもたらす。理論に束縛されることは望まないが、IC−3ループおよび受容体のカルボキシ末端がGタンパク質と相互作用すると考えられる。
【0029】
数クラスのGPCRをホスホリパーゼC経路に共役させるプロミスキャスGタンパク質、例えばGα15またはGα16(OffermannsおよびSimon,J Biol Chem 270:15175−80(1995))や、同じ経路、例えばホスホリパーゼCに、多数の異なるGPCRを共役させるように設計されたキメラGタンパク質(MilliganおよびRees,Trends in Pharmaceutical Sciences20:118−24(1999))もある。
【0030】
Gi共役GPCRは細胞内cAMPレベルを低下させる。黒色素胞技術(下記参照)は、Gi共役GPCRを同定するのに役立ち、前記Gi共役GPCRの調節因子を同定するのにも役立つ。
【0031】
生理的条件下で、GPCRは、2つの異なるコンフォメーション、すなわち「不活性」状態と「活性」状態の間の平衡状態で、細胞膜中に存在する。不活性状態の受容体は、細胞内シグナル伝達経路に連結して生物学的応答につながるシグナル伝達を開始させることができない。受容体コンフォメーションを活性状態に変化させると(Gタンパク質による)伝達経路への連結が可能になり、生物学的応答が生成する。
【0032】
リガンドまたは薬物などの化合物により、受容体を活性状態で安定化することができる。受容体のアミノ酸配列の改変を含む(ただしこれだけに限定されるわけではない)最近の発見により、活性状態コンフォメーションの受容体を助長し、安定化するための、リガンドまたは薬物以外の手段が与えられる。これらの手段は、受容体へのリガンド結合の作用をまねることにより、活性状態の受容体を効果的に安定化する。そのようなリガンド非依存的手段による安定化は「構成的受容体活性化」と呼ばれる。
【0033】
GPR43の配列は、Sawzdargoらによる文献で初めて公表された(Sawzdargoら,Biochem.Biophys.Res.Commun.,239:543−547(1997))。Sawzdargoらは、ヒトおよびラットガラニン受容体1(GALR1)ならびにラットGALR2内の保存された配列に基づく縮重プライマーによるPCRを使って、ヒトゲノムDNAを増幅した。1つの産物は、ヒトCD22遺伝子の3プライム領域の一部に対して100%のホモロジーを示すセグメントを含有した。著者らは、この領域とオーバーラップし、新規GPCR遺伝子GPR43を含有するPACクローンを、配列データベース中に同定した。このイントロンレスGPR43遺伝子は、GPR42遺伝子の約77kb下流に位置する。GPR43は7つの膜貫通ドメインを持つ推定330アミノ酸のタンパク質をコードする。GPR43タンパク質はGPR40との間に28%のアミノ酸一致度を持つが、GALR類との類似性はほとんどない。また、GPR43はGPR41との間に43%のアミノ酸一致度を持つ。第3のファミリーメンバーGPR42は、おそらくはGPR41の最近の遺伝子重複であり、偽遺伝子であるかもしれない。
【0034】
GPR43はオーファン受容体に分類された。これは、この受容体のリガンドが同定されていなかったことを意味する。最近になって、Brownらが、GPR43は酢酸および他の単鎖カルボン酸アニオンによって活性されると報告した(Brownら,J.Biol.Chem.,278:11312−11319(2003))。またBrownらは、GPR43が、Gタンパク質のGi、GqおよびG12ファミリーを活性化することも示している。さらにBrownらは、GPR43が好中球および単球などの免疫細胞中に、最も高いレベルで見いだされると報告している。
【0035】
(定義)
受容体周辺で発展してきた科学文献は、受容体に対して種々の影響を有するリガンドを言及するために多数の用語を採用した。明確さおよび一貫性のために、以下の定義が、本特許文書全体を通して用いられる。
【0036】
「アゴニスト」は、受容体に結合した際に細胞内応答を活性化する物質(例えば、リガンド、候補化合物)を意味するものとする。特定の実施形態では、アゴニストは、受容体に結合する際に細胞内応答を活性化する(例えば、膜へのGTPγS結合を増強するかまたは細胞内cAMPレベルを上昇させる)ことが以前には公知でなかった物質である。特定の実施形態では、アゴニストは、受容体に結合する際に、血中グルコースレベルを下げることが以前には公知でなかった物質である。用語「アゴニスト」はまた、受容体に結合する際に、完全なアゴニストよりも低い程度または小さい範囲で細胞内応答を活性化する、部分アゴニストを包含する。
【0037】
「アンタゴニスト」は、アゴニストと同じ部位で受容体に競合的に結合するが、細胞内応答を活性化せず、それによってアゴニストにより誘発される細胞内応答を阻害し得る、物質(例えば、リガンド、候補化合物)を意味するものとする。アンタゴニストは、アゴニストの非存在下ではベースラインの細胞内応答を減少させない。特定の実施形態では、アンタゴニストは、受容体に結合した際にアゴニストと競合して細胞応答を阻害することが以前には公知でなかった物質である(例えば、細胞応答は、膜へのGTPγS結合または細胞内cAMPレベルの上昇である)。
【0038】
「抗体」は、本明細書中では、モノクローナル抗体およびポリクローナル抗体を包含することが意図される。抗体はさらに、IgG、IgA、IgD、IgE、およびIgMを包含することが意図される。抗体は、抗体全体(単鎖抗体の全体が挙げられる)およびそれらの抗原結合フラグメント(Fab、Fab’、F(ab)2およびF(ab’)2が挙げられる)を包含する。抗体は、任意の天然起源または合成起源(例えば、ヒト、マウス、ウサギ、ヤギ、モルモット、ハムスター、ラクダ、ロバ、ヒツジ、ウマまたはニワトリ)に由来し得る。抗体は、例えば、5×10−6M未満、10−6M未満、5×10−7M未満、10−7M未満、5×10−8M未満、10−8M未満、5×10−9M未満、10−9M未満、5×10−10M未満、10−10M未満、5×10−11M未満、10−11M未満、5×10−12M未満、10−12M未満、5×10−13M未満、10−13M未満、5×10−14M未満、10−14M未満、5×10−15M未満および10−15M未満の解離定数すなわちKd値を有する結合親和性を有する。本発明の抗体は、当該分野で公知の任意の適切な方法により調製され得る。
【0039】
「候補化合物」とは、スクリーニング技術に従う分子(例えば、化合物)を意味するものとする。用語「候補化合物」は、特に、GPR43を調節することが既に公知のいかなる化合物(例えば、GPR43の公知のアゴニスト)も除外する。
【0040】
「組成物」とは、少なくとも2つの化合物または2つの成分を含む物質を意味する。例えば、医薬組成物は、組成物である。
【0041】
「化合物の効力」とは、受容体結合活性とは反対に、受容体の機能を化合物が阻害または刺激する能力の尺度を意味するものとする。
【0042】
「構成的に活性化された受容体」とは、構成的な活性化に供された受容体を意味する。
【0043】
「構成的な受容体活性化」とは、その内因性リガンドまたはその化学的等価物による結合以外による、活性状態の受容体の安定化を意味する。
【0044】
「接触」、または「接触する」とは、インビトロ系においてであろうとインビボ系においてであろうと、少なくとも2つの部分を合わせることを意味する。
【0045】
本明細書で使用される「糖尿病」とは、例えば、以下のリストを含む、任意の方法からなされる糖尿病の通常の診断を包含することが意図される:糖尿病の症状(例えば、多尿症、多渇症、多食症)と200mg/dl以上である平常時の血中グルコースレベル(ここで、平常時の血中グルコースレベル、食事または飲料の消費の時期にかかわらず、一日の任意の時間に定義される);または、126mg/dl以上である8時間絶食時の血中グルコースレベル;または、水に溶解させた75gの無水グルコースの経口投与から2時間後、200mg/dl以上である血中グルコースレベル。さらに、本明細書で使用される用語「糖尿病」はまた、American Diabates Associationにより定義され、100mg/dl〜125mg/dlの空腹時血中グルコースレベルであるか、またはグルコースの経口投与から2時間後の140mg/dl〜199mg/dlの血中グルコースレベルである「前糖尿病」状態も包含する。糖尿病は、いくつかの状態(例えば、ベータ島細胞の自己免疫破壊、ベータ細胞のアポトーシス、または妊娠糖尿病が挙げられる)により促進され得る。
【0046】
「内因性」とは、哺乳動物が天然に産生する物質を意味する。内因性に関して、例としてであり限定ではなく、用語「受容体」は、哺乳動物(例としてであり限定ではなく、ヒト)またはウイルスによって天然に産生されたものを意味する。対照的に、この状況における用語「非内因性」は、哺乳動物(例としてであり限定ではなく、ヒト)によってもウイルスによっても天然に産生されないものを意味する。例としてであり限定ではなく、その内因性形態において構成的に活性でないが、操作される場合に構成的に活性になる受容体は、最も好ましくは、本明細書において「非内因性の、構成的に活性化された受容体」と称される。両方の用語は、「インビボ」系および「インビトロ」系の両方を記述するために使用され得る。例としてであり限定ではなく、スクリーニングアプローチにおいて、内因性受容体または非内因性受容体がインビトロスクリーニング系に参照され得る。
【0047】
「有効量」とは、本明細書において使用される場合、研究者、医師または他の医療従事者によって探索されるべき、組織、系、もしくは個体において望ましい生物学的または医学的な応答を誘発する、活性化合物または医薬組成物の量を意味する。例えば、有効用量は、代謝関連障害を処置することのできる量であり得る。また、例えば、有効用量は、代謝関連障害を予防することのできる量であり得る。
【0048】
「グルコース寛容減損(IGT)」は、本明細書において使用される場合、明白な2型糖尿病と正常なグルコース寛容(NGT)との中間であるインスリン抵抗性に関連する条件を示すことを意図される。IGTは、罹患した人物の食後グルコース反応が、食後2時間での血漿グルコースレベルによって評価される場合に異常であると決定される手順によって診断される。この試験において、測定された量のグルコースが患者に与えられ、血中グルコースレベルが、定期的な間隔(通常、最初の2時間は30分毎、その後1時間毎)で測定される。「正常」な、または非IGTの個体において、最初の2時間のグルコースレベルは、140mg/dl未満まで上昇し、次いで急速に低下する。IGT個体においては、血中グルコースレベルは、より高く上昇し、そしてよりゆっくりとした速度で落ちる。
【0049】
「予防または処置の必要がある」とは、本明細書において使用される場合、ヒトにおいては介護者(例えば、医師、看護士、ナースプラクティショナーなど)によって、動物(非ヒト哺乳動物が挙げられる)においては獣医によってなされる、個体もしくは動物が処置を必要とするかまたはその処置から利益を受けるという判断を指す。この判断は、介護者の専門的意見の領域であるが、個体または動物が病気であるか病気になり、結果として本発明の化合物によって処置可能な状態であるという知見を含む、種々の要因に基づいてなされる。
【0050】
本明細書において使用される場合、「個体」とは、哺乳動物(好ましくは、マウス、ラット、他のげっ歯類、ウサギ、イヌ、ネコ、ブタ、ウシ、ヒツジ、ウマ、または霊長類、最も好ましくはヒト)を含む、任意の動物を指す。
【0051】
「阻害」または「阻害する」とは、用語「反応(応答)」に関連して、化合物の非存在下に対して、化合物の存在下で、反応が減少または防止されることを意味する。
【0052】
「インスリン抵抗性」は、本明細書において使用される場合、多くの方法のいずれかによって行われるインスリン抵抗性の通常の診断を包含することを意図される。上記方法としては、静脈内グルコース寛容試験または空腹時インスリンレベルの測定が挙げられるが、これらに制限されない。空腹時インスリンレベルとインスリン抵抗性の程度との間に強い相関が存在することは、周知である。したがって、高い空腹時インスリンレベルを、正常なグルコース寛容(NGT)個体がインスリン抵抗性を有することを同定する目的のため、インスリン抵抗性の代理のマーカーとして使用することができる。インスリン抵抗性の診断はまた、オイグリセミックグルコースクランプ試験を使用して行われ得る。
【0053】
「インバースアゴニスト」は、受容体の内因性形態または構成的に活性化された形態のいずれかに結合して、アゴニストの非存在下で観察されるその受容体のベースラインの細胞内応答を低下させる物質(例えば、リガンド、候補化合物)を意味する。細胞内応答は、例えば、膜へのGTPの結合の調節、または二次メッセンジャー(例えば、cAMPまたはIP3)のレベルの調節であり得る。特定の実施形態において、インバースアゴニストは、アゴニストの非存在下で観察される受容体のベースラインの細胞内応答を低下させることが既知ではない物質である。
【0054】
「リガンド」とは、内因性の天然に存在する受容体に特異的な、内因性の天然に存在するリガンドを意味する。
【0055】
「代謝関連障害」とは、代謝の障害を意味する。本明細書で使用されるように、本明細書において、代謝関連障害は、例えば、以下を包含することが意図される:低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患。
【0056】
「代謝安定化化合物」とは、代謝パラメータを安定化する化合物を意味することが意図される。代謝パラメータとしては、代謝プロセスに応答する脂質、糖、酵素または他のタンパク質のレベルのようないかなる尺度も挙げられる。例えば、代謝安定化化合物は、個体中の血中グルコースレベルを安定化し得る。
【0057】
本明細書において使用される場合、用語「調節する」または「改変する」は、特に活性、機能または分子の、量、質、応答または効果の増加または減少を指すことを意味する。「GPR43調節因子」は、GPR43受容体を調節する因子である。
【0058】
「医薬組成物」とは、少なくとも1種の化合物と薬学的に受容可能な担体とを含有する組成物を意味する。例えば、医薬組成物は、少なくとも1種の活性成分を含み、これによって、この組成物は、動物(例えば、限定ではなく、ヒト)における特定の効果のある結果についての調査を受けることができる。当業者は、活性成分が当業者の必要性に基づく所望の効果のある結果を有するか否かを決定するのに適切な技術を、理解および認識する。
【0059】
「受容体機能」は、刺激を受容して細胞の効果(遺伝子転写の調節、イオン流の調節、触媒作用の実行、および/またはGタンパク質を介した調節性活性が挙げられるが、これらに限定されない)を調節する、受容体の正常な働きを指す。GPR43の機能は、例えば、Gタンパク質(例えば、Gi、GqまたはG12)に結合すること、二次メッセンジャー(例えば、cAMP、IP3またはカルシウム)を介してのシグナル伝達、GPR43特異的抗体への結合、またはGPR43アゴニストのような化合物への結合であり得る。
【0060】
「二次メッセンジャー」とは、受容体活性化の結果として生じる細胞内応答を意味する。二次メッセンジャーとしては、例えば、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、およびCa2+が挙げられ得る。二次メッセンジャー応答は、受容体活性化の決定に関して測定され得る。さらに、二次メッセンジャー応答は、候補化合物(例えば、インバースアゴニスト、部分アゴニスト、アゴニスト、およびアンタゴニストが挙げられる)を同定するために測定され得る。
【0061】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が調節されるかどうかを決定することを含み、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法を提供する。
【0062】
本明細書にいう「GPR43」は、配列番号2に示すアミノ酸配列を持つポリペプチド、またはこの配列の変種もしくはオルソログであって、配列番号2に記載のアミノ酸配列を持つポリペプチドの機能を実質的に保っているものを指す。
【0063】
GPR43には、その機能を破壊することなく、限定的な変異または改変を加えることができると理解される。例えばGPR43は、他のGPR43ポリペプチド、例えばヒトGPR43ポリペプチドの哺乳動物種オルソログを包含するものとする。ヒトGPR43の種オルソログのヌクレオチド配列およびアミノ酸配列はデータベース中に存在する。例えばGPR43のマウスオルソログはGenBankのアクセッション番号NM_146187に見いだすことができ、GPR43のラットオルソログは、GenBankのアクセッション番号AB106675に見いだすことができる。また、GPR43は、GPR43の対立遺伝子変種、スプライス変種および保存的アミノ酸置換変種も包含する。例えばGPR43は、野生型GPR43ポリペプチドの機能、例えばGαi、GαqまたはGα12を介してシグナルする能力、GPR43特異的抗体に結合する能力、または既知のリガンドもしくはアゴニストなどといった化合物に結合する能力を、実質的に保っている変種を包含する。GPR43変種は野生型GPR43と同レベルに機能する必要はなく、野生型GPR43の全ての機能を含有する必要もない。
【0064】
アミノ酸配列への保存的および非保存的なアミノ酸変化、ギャップ、および挿入は、Basic Local Alignment Search Tool(「BLAST」)などの入手可能なアルゴリズムおよびプログラムを使って、デフォルト設定で、基準配列と比較することができる[例えばKarlinおよびAltschul,Proc Natl Acad Sci USA(1990)87:2264−8;Altschulら,J MoI Biol(1990)215:403−410;Altschulら,Nature Genetics(1993)3:266−72;およびAltschulら,Nucleic Acids Res(1997)25:3389−3402参照]。
【0065】
ポリペプチド全体の機能を実質的に保っているGPR43の断片は、この定義に包含されると理解される。例えば、GPR43のシグナル生成ドメインまたはGPR43の化合物結合ドメインを、ポリペプチド全体の代わりに使用することができる。またGPR43は、エピトープタグまたは他の融合ポリペプチドなどといった異種配列を含有することもできる。さらにGPR43は、ラベル、例えば放射性ラベル、蛍光ラベルまたは酵素ラベルなどを含有することができる。
【0066】
ある実施形態では、配列番号2に対して99%、98%、95%、92%、90%、85%、80%、または75%の配列一致度を含むポリペプチドを使って、本発明の方法を応用することができる。
【0067】
一部の実施形態では、GPR43の前記変種が、GPR43の非内在性かつ構成的に活性化された突然変異体である。ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0068】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と、またはそのような宿主細胞の膜と、接触させることを含み、宿主細胞は、当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知アゴニストの存在下で行なわれる。
【0069】
特定の実施形態では、前記方法が、候補化合物によって引き起こされる受容体の調節を、受容体を受容体の既知調節因子と接触させることによって引き起こされる受容体の第2の調節と比較することを、さらに含む。特定の実施形態では、前記既知調節因子がアゴニストである。
【0070】
一部の実施形態では、前記代謝安定化化合物が血中グルコース安定化化合物である。一部の実施形態では、前記代謝安定化化合物がインスリン分泌調節因子である。
【0071】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定は、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびカルシウム(Ca2+)からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定がCRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、例えばFLIPRアッセイを使った、細胞内カルシウム(Ca2+)の測定によって行なわれる。
【0072】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0073】
特定の実施形態では、前記決定が黒色素胞アッセイを使用することによって行なわれる。
【0074】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0075】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0076】
上述のように、受容体機能とは、刺激を受取り、細胞における作用を加減する(例えば、Gタンパク質を介して遺伝子転写を調節する、イオンの内向きフラックスまたは外向きフラックスを調節する、触媒反応を達成する、および/または活性を調節するなどであるが、これらに限定されるわけではない)ための、受容体の正常な働きを指す。GPR43機能として、例えば、Gi、GqまたはG12などのGタンパク質を結合すること、cAMP、IP3、もしくはカルシウムなどの二次メッセンジャーを介してシグナルすること、GPR43特異的抗体に結合すること、またはGPR43アゴニストなどの化合物に結合することを挙げることができる。
【0077】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0078】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0079】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0080】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法であって、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物を提供する。
【0081】
ある実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0082】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。
【0083】
特定の実施形態では、前記EC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のEC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0084】
一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つ、GPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。
【0085】
特定の実施形態では、前記IC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0086】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0087】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法も提供する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法も提供する。
【0088】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物も提供する。
【0089】
化合物は、当業者に周知の技法を使って、医薬組成物に製剤化することができる。本明細書で言及する担体の他にも、適切な医薬的に許容できる担体を利用することができる。例えば「Remington’s Pharmaceutical Sciences」第16版,1980,Mack Publishing Co.,(Osloら編)を参照されたい。
【0090】
予防または処置に使用する場合、本発明の方法によって同定される化合物は、代替的に、原薬または純薬として投与することができると考えられるが、その化合物または活性成分を、医薬的に許容できる担体をさらに含む医薬製剤または医薬組成物として提供することが有用な場合もある。
【0091】
したがって本発明は、本発明の方法によって同定される化合物または医薬的に許容できるその塩もしくは誘導体を、1以上の医薬的に許容できるその担体および/または予防成分と共に含む医薬製剤を、さらに提供する。担体は、製剤の他の成分と適合し、その受容者にとって過度に有害でないという意味で、「許容できる」。
【0092】
医薬製剤には、経口、直腸、鼻腔、局所(口腔粘膜および舌下を含む)、膣または非経口(筋肉内、皮下および静脈内を含む)投与に適したもの、または吸入もしくは吹送による投与に適した形態にあるものが包含される。
【0093】
したがって、本発明の化合物は、従来の佐剤、担体、または希釈剤と共に、医薬製剤およびその単位剤の形態にすることができ、そのような形態で、錠剤もしくは充填カプセル剤などの固形物として、または溶液剤、懸濁剤、乳剤、エリキシル剤、ゲル剤もしくはそれらを充填したカプセル剤などの流動物として、いずれも経口用途に使用するか、または直腸投与用の坐剤の形態で使用するか、または非経口(皮下を含む)用の滅菌注射可能溶液の形態で使用することができる。そのような医薬組成物およびその単位剤形は、追加活性化合物または追加有効成分と共に、または追加活性化合物または追加有効成分を伴わずに、従来の成分を従来の比率で含むことができ、そのような単位剤形は、使用しようとしている1日量範囲に見合った任意の適切な有効量の活性成分を含有することができる。
【0094】
経口投与の場合、医薬組成物は、例えば錠剤、カプセル剤、懸濁剤または液剤の形態をとることができる。医薬組成物は、特定量の活性成分を含有する投薬単位の形態で製造することができる。そのような投薬単位の例は、乳糖、マンニトール、トウモロコシデンプンまたはバレイショデンプンなどの従来の添加剤;結晶セルロース、セルロース誘導体、アラビアゴム、トウモロコシデンプンまたはゼラチンなどの結合剤;トウモロコシデンプン、バレイショデンプンまたはカルボキシメチル−セルロースナトリウムなどの崩壊剤;およびタルクまたはステアリン酸マグネシウムなどの潤滑剤を含む、カプセル剤、錠剤、粉末剤、顆粒剤または懸濁剤である。活性成分は、組成物として注射によって投与することもでき、その場合は、例えば食塩水、デキストロースまたは水を、適切な医薬的に許容できる担体として使用することができる。
【0095】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、投与される化合物が、GPR43インバースアゴニストまたはアンタゴニストを含む。ある実施形態では、本方法が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。例えば、ある実施形態では、本方法が、GPR43インバースアゴニストを含有する医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。ある実施形態では、前記代謝関連障害が低血糖であり、投与される前記化合物がGPR43アゴニストを含む。
【0096】
障害に関して本明細書で使用する「処置する」という用語は、ある特定の障害に付随する1以上の症状の重症度の軽減を意味する。したがって、ある障害を処置するとは、必ずしも、障害に付随する全ての症状の重症度の軽減を意味するわけではなく、また必ずしも、障害に付随する1以上の症状の重症度の完全な軽減を意味するわけでもない。同様に、「予防する」という用語は、ある特定の障害に付随する1以上の症状の出現または発生の予防を意味し、必ずしも、ある障害の完全な予防を意味するわけではない。本発明の方法は、例えば糖尿病を含む代謝関連障害を処置するために使用することができる。
【0097】
本発明の方法によって同定される化合物を使用する場合、用量は広範囲に変動することができ、慣行のとおり、また医師には知られているとおり、個々の症例で個々の状態に合わせて調節されるべきである。これは、例えば、処置すべき病気の性質および重症度、患者の状態、使用する化合物、または急性疾患もしくは慢性疾患を処置するのか、それとも予防を行なうのか、または本発明の方法によって同定される化合物に加えて、さらなる活性化合物を投与するかどうかなどに依存する。本発明の代表的な用量には、約0.01mg〜約1000mg、約0.01〜約750mg、約0.01〜約500mg、0.01〜約250mg、0.01mg〜約200mg、約0.01mg〜150mg、約0.01mg〜約100mg、および約0.01mg〜約75mgが含まれる。投与は一日に複数回行なうことができ、特に比較的多量に必要であると考えられる場合は、例えば2、3または4回投与することができる。適当と認められる場合は、個々の挙動に応じて、また適宜、患者の医師もしくは介護者に従って、上記1日量を加減することが必要な場合がある。
【0098】
処置に使用する必要がある活性成分またはその活性塩もしくは誘導体の量は、選択したその塩によって変動するだけでなく、投与経路、処置される状態の性質、患者の年齢および状態によっても変動し、最終的には担当の医師または臨床家の判断によるだろう。一般に当業者は、モデル系(典型的には動物モデル)で得られたインビボデータを他の系、例えばヒトなどに外挿する方法を理解している。通例、動物モデルには、齧歯類糖尿病モデルが含まれるが、これらに限定されるわけではない(他の動物モデルが、ReedおよびScribnerにより、Diabetes,Obestiy and Metabolism,1:75−86(1999)に報告されている)。状況によっては、これらの外挿が、単に、哺乳動物などの他の系、例えばヒトと比較した動物モデルの体重に基づく場合もあるが、多くの場合、これらの外挿は、体重だけに基づくのではなく、さまざまな因子を加味して行なわれる。代表的な因子には、患者のタイプ、年齢、体重、性別、食餌および医学的状態、疾患の重症度、投与経路、薬理学的事項、例えば使用する特定化合物の活性、抗力、薬物動態および毒性プロファイル、薬物送達系を利用するかどうか、急性もしくは慢性疾患状態を処置しているのか、それとも予防を行なうのか、または本発明の方法によって同定される化合物に加えて、さらなる活性化合物を、複合薬の一部として投与するかどうかが含まれる。本発明の化合物および/または組成物を使って疾患状態を処置するための投薬レジメンは、上述したさまざまな因子に応じて選択される。したがって、使用される実際の投薬レジメンは広い範囲にわたって多様であることができ、それゆえに好ましい投薬レジメンから逸脱する場合もあり、これらの典型的範囲から外れた投薬量および投薬レジメンも試験することができ、それが適当と認められる場合には、本発明の方法において使用できることは、当業者には理解されるだろう。
【0099】
望ましい用量は、1回量として、または適当な間隔で投与される分割量として、例えば1日に2回、3回、4回もしくはそれ以上の部分量として、便利に提示することができる。部分量自体をさらに、例えばいくつかの不連続で大まかな間隔の投与に、分割することもできる。1日量は、比較的大量に投与することが適当であるとみなされる場合は特に、数回、例えば2回、3回または4回の部分投与に分割することができる。適当と認められる場合は、個々の挙動に応じて、上記1日量を加減することが必要な場合がある。
【0100】
本発明の方法によって同定される化合物は、広範囲にわたるさまざまな経口剤形および非経口剤形で投与することができる。以下の剤形が、活性成分として、本明細書に開示する化合物、または本発明の方法によって同定される化合物もしくは本発明の方法によって同定される化合物の医薬的に許容できる塩を含みうることは、当業者には明白だろう。
【0101】
本発明の方法によって同定される化合物から医薬組成物を調製する場合、適切な医薬的に許容できる担体の選択は、固体、液体、または両者の混合物であることができる。固形調製物には、粉末剤、錠剤、丸剤、カプセル剤、カシェ剤、坐剤、および分散性顆粒剤が含まれる。固形担体は、希釈剤、着香剤、溶解剤、潤滑剤、懸濁化剤、結合剤、保存剤、錠剤崩壊剤、または封入材料としても作用しうる1以上の物質であることができる。
【0102】
粉末剤の場合、担体は、微粉活性成分と混合された微粉固体である。錠剤の場合、活性成分は必要な結合能を持つ担体と適切な比率で混合され、所望の外形およびサイズに圧縮される。
【0103】
粉末剤および錠剤は、さまざまなパーセンテージ量の活性化合物を含有することができる。粉末剤および錠剤中の代表的な量は0.5〜約90%の活性化合物を含有しうるが、この範囲外の量がいつ必要になるかは、当業者にはわかるだろう。粉末剤用および錠剤用の適切な担体は、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、ペクチン、デキストリン、デンプン、ゼラチン、トラガカント、メチルセルロース、カルボキシメチルセルロースナトリウム、低融点ワックス、カカオ脂などである。「調製物」という用語は、カプセル剤を与える封入材料を担体とする活性化合物の製剤(このカプセル剤では、活性成分が、担体と共に、または担体を伴わずに、担体によって取り囲まれ、担体はそのようにして活性成分と関係している)を包含するものとする。同様に、カシェ剤および口中錠も包含される。錠剤、粉末剤、カプセル剤、丸剤、カシェ剤、および口中錠は、経口投与に適した固体剤形として使用することができる。
【0104】
坐剤を調製するには、低融点ワックス、例えば脂肪酸グリセリドまたはカカオ脂の混合物などを、まず融解し、そこに活性成分を撹拌するなどして均一に分散させる。次にその融解均一混合物を都合のよいサイズの鋳型に注ぎ込み、冷ますことによって固化させる。
【0105】
膣投与に適した製剤は、活性成分の他に当技術分野で適当であることが知られているような担体を含有する膣坐薬、タンポン、クリーム剤、ゲル剤、ペースト剤、フォーム剤またはスプレー剤として提示することができる。
【0106】
液体調製物には、溶液剤、懸濁剤、および乳剤、例えば水溶液または水−プロピレングリコール溶液が含まれる。例えば非経口注射液体調製物は、水性ポリエチレングリコール溶液中の溶液剤として製剤化することができる。注射可能調製物、例えば滅菌注射可能水性または油性懸濁剤は、適切な分散剤または湿潤剤および懸濁化剤を使用し、既知の技術に従って製剤化することができる。滅菌注射可能調製物は、無毒性の非経口的に許容できる希釈剤または溶媒中の滅菌注射可能溶液剤または懸濁剤、例えば1,3−ブタンジオール中の溶液剤であることもできる。使用することができる許容できるビヒクルおよび溶媒には、水、リンゲル液、および等張食塩溶液などがある。また、滅菌した固定油も、溶媒または分散媒として、便利に使用される。この目的には、合成モノ−またはジ−グリセリドを含む任意の無刺激性固定油を使用することができる。また、オレイン酸などの脂肪酸も、注射剤の調製に使用することができる。
【0107】
本発明の化合物はこのように非経口投与(例えばボーラス注射または持続注入などの注射による投与)用に製剤化することができ、アンプル、充填済注射器、少量注入容器に入れて1回量型として提示するか、または保存剤を添加した多用量型容器に入れて提示することができる。医薬組成物は、油性または水性ビヒクル中の懸濁剤、溶液剤、または乳剤などといった形態をとることができ、懸濁化剤、安定剤および/または分散剤などの調剤用薬剤を含有することができる。あるいは、活性成分は、滅菌固体の無菌的単離または溶液からの凍結乾燥によって得られる粉末であって、使用前に適切なビヒクル、例えば滅菌パイロジェンフリー水などで復元される形態であることもできる。
【0108】
経口使用に適した水性溶液剤は、活性成分を水に溶解し、希望に応じて、適切な着色剤、着香剤、安定剤および増粘剤を加えることによって調製することができる。
【0109】
経口使用に適した水性懸濁剤は、微粉活性成分を粘性材料、例えば天然ゴムもしくは合成ゴム、樹脂、メチルセルロース、カルボキシメチルセルロースナトリウム、または他の周知の懸濁化剤などと共に、水に分散させることによって作ることができる。
【0110】
使用直前に経口投与用の液状調製物に変換されることを意図した固形調製物も包含される。そのような液体剤形には、溶液剤、懸濁剤、および乳剤が含まれる。これらの調製物は、活性成分の他に、着色剤、着香剤、安定剤、緩衝剤、人工および天然甘味剤、分散剤、増粘剤、溶解剤などを含有することができる。
【0111】
表皮への局所投与用に、本発明の化合物を、軟膏、クリーム剤もしくはローション剤として、または経皮貼付剤として製剤化することができる。
【0112】
軟膏およびクリーム剤は、例えば、水性または油性基剤を使って、適切な増粘および/またはゲル化剤を添加して、製剤化することができる。ローション剤は水性または油性基剤を使って製剤化することができ、一般的には1以上の乳化剤、安定剤、分散剤、懸濁化剤、増粘剤、または着色剤も含有するだろう。
【0113】
口内の局所投与に適した製剤には、着香した基剤(通常はスクロースおよびアラビアゴムまたはトラガカント)中に活性剤を含む口中錠;ゼラチンおよびグリセリンまたはスクロースおよびアラビアゴムなどの不活性基剤中に活性成分を含むトローチ剤;ならびに適切な液状担体中に活性成分を含む洗口剤が含まれる。
【0114】
溶液剤または懸濁剤は、従来の手段により、例えば滴ビン、ピペットまたはスプレーを使って、鼻腔に直接適用される。製剤は単回量剤形または複数回量剤形で提供することができる。滴ビンまたはピペットで後者の場合、これは、適当な所定の体積の溶液剤または懸濁剤を患者が投与することによって達成することができる。スプレーの場合、これは、例えば計量式噴霧スプレーポンプを使って達成することができる。
【0115】
気道への投与は、エアロゾル製剤を使って達成することもでき、この製剤では、活性成分が適切な噴射剤と共に加圧容器に入れて提供される。本発明の方法によって同定される化合物またはそれらを含む医薬組成物をエアロゾル剤として、例えば鼻エアロゾル剤として、または吸入によって投与する場合、これは、例えばスプレー、ネブライザー、ポンプネブライザー、吸入装置、計量式吸入器または乾燥粉末吸入器を使って行なうことができる。本発明の方法によって同定される化合物をエアロゾルとして投与するための医薬剤形は、当業者に周知の方法によって調製することができる。それらの調製には、例えば、本発明の方法によって同定される化合物の、水、水/アルコール混合物または適切な食塩溶液中の溶液または分散液を、慣例の添加剤、例えばベンジルアルコールまたは他の適切な保存剤、生物学的利用能を増加させるための吸収促進剤、溶解剤、分散剤などを使って、そしてまた、それが適当な場合には、通例の噴射剤、例えば二酸化炭素、CFC類(例えばジクロロジフルオロメタン、トリクロロフルオロメタン、またはジクロロテトラフルオロエタンなど)を使って、使用することができる。エアロゾル剤はレシチンなどの界面活性剤も都合よく含有することができる。薬物の用量は計量式バルブを備えることによって制御することができる。
【0116】
気道への投与を意図する製剤では、鼻腔内製剤を含めて、化合物は一般に小さな粒径、例えば10ミクロン未満程度の粒径を持つだろう。そのような粒径は当技術分野では既知の手段によって、例えば微粒子化によって、得ることができる。所望であれば、活性成分の徐放が得られるように適合させた製剤を使用することができる。
【0117】
あるいは、活性成分は、乾燥粉末の形態で、例えば、ラクトース、デンプン、デンプン誘導体(ヒドロキシプロピルメチルセルロースなど)およびポリビニルピロリドン(PVP)などの適切な粉末基剤中の化合物の粉末混合物という形態で、提供することもできる。好都合なことに、粉末担体は、鼻腔内でゲルを形成するだろう。粉末組成物は、例えば、ゼラチン製のカプセルもしくはカートリッジまたはブリスター包装に入った、そこから吸入器を使って粉末を投与することができる、1回量型として提示することができる。
【0118】
医薬調製物は単位剤形をとることができる。そのような剤形では、調製物は、適当な量の活性成分を含有する1回量に細分される。単位剤形は、包装済みの錠剤、カプセル剤、およびバイアルまたはアンプル中の粉末剤など、包装が分離した調製物量を含有している包装済みの調製物であることができる。また、単位剤形はカプセル剤、錠剤、カシェ剤、または口中錠そのものであることができ、あるいは包装された形態にある、適当な数の、これらのいずれかであることができる。
【0119】
経口投与用の錠剤またはカプセル剤および静脈内投与用の液剤は、特に有用な組成物である。
【0120】
代謝関連障害には、例えば低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患が含まれる。
【0121】
低血糖は、異常に低い血中グルコースと定義される。低血糖は、例えば過剰なインスリンまたは貧しい食餌などに起因しうる。例えば低血糖は、糖尿病を持つ人がインスリンを多量に注射しすぎた場合、食物摂取量が少なすぎた場合、または余分な食物を摂らずに運動した場合などに起こりうる。低血糖の症状には、例えば、緊張感または脱力感、頭痛、かすみ目、空腹感、および過剰発汗などが含まれる。
【0122】
加齢は生物が年をとるにつれて起こる生理学的過程である。カロリー制限はインスリン分泌をダウンレギュレートし、これらの作用は、カロリー制限が長寿に及ぼすよい影響の重要な媒介因子ではないかと疑うに足りる理由がある。また、インスリン中またはインスリンシグナル伝達経路中の突然変異は、C.elegansにおける加齢に影響を及ぼす。したがって、インスリンをダウンレギュレートするための戦略は、老化の過程を遅らせ、寿命を延ばすのに役立ちうる。
【0123】
糖尿病、肥満、ならびにインスリン抵抗性、耐糖能異常および高血糖などの関連状態については上述した。一部の実施形態では、前記代謝関連障害に、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、X症候群および末梢血管疾患が含まれる。
【0124】
膵臓中のベータ細胞はインスリンを産生する。インスリンは体内で血液から細胞へのグルコースの取り込みを刺激する。上述のように、身体の細胞がインスリンの作用に対して抵抗性である場合、それをインスリン抵抗性と呼ぶ。インスリン抵抗性の結果として、膵臓は正常よりはるかに多くのインスリンを産生する。これを高インスリン血症という。
【0125】
異常脂質血症は、体内の脂質レベルの調節不全を意味する一般用語である。高脂質血症は、血流中の脂質(脂肪)の上昇である。これらの脂質には、コレステロール、コレステロールエステル(化合物)、リン脂質およびトリグリセリドが含まれる。それらは血中ではリポタンパク質とよばれる大分子の一部として輸送される。血中(血漿)リポタンパク質には5つの主要ファミリー、すなわちカイロミクロン、超低密度リポタンパク質(VLDL)、中間密度リポタンパク質(IDL)、低密度リポタンパク質(LDL)、高密度リポタンパク質(HDL)がある。高脂血症を、血中で上昇するリポタンパク質の1以上のクラスという観点から定義する場合は、高リポタンパク質血症という用語が用いられる。高コレステロール血症は、高い血中コレステロールレベルを表す用語である。高トリグリセリド血症は、高い血中トリグリセリドレベルを指す。
【0126】
アテローム性動脈硬化は、脂肪物質、コレステロールおよび他の物質の沈着が動脈の内層に蓄積する過程である。この蓄積をプラークをいう。破裂するプラークは血塊の形成を引き起こし、それが心臓(心臓発作)または脳(脳卒中)への血流を遮断しうる。心臓発作は、米国では男女とも第1位の死因であり、脳卒中は第3位の死因である[例えばNature Medicine,Special Focus on Atherosclerosis,(2002)8:1209−1262を参照されたい]。異常に高い循環脂質レベルは、アテローム性動脈硬化の発症における主要素因である。上昇した低密度リポタンパク質(LDL)コレステロールレベル、上昇したトリグリセリドレベル、または低い高密度リポタンパク質(HDL)コレステロールレベルは、独立して、アテローム性動脈硬化および関連病変のリスク因子である。
【0127】
心臓疾患には、心不全、冠不全、冠状動脈疾患、および高い血圧(高血圧)が含まれるが、これらに限定されるわけではない。末梢血管疾患とは、心臓外および脳外にある血管の疾患を指す。器質的末梢血管疾患は、炎症および組織損傷などといった血管の構造変化によって引き起こされる。末梢動脈疾患はその一例である。末梢動脈疾患(PAD)は冠状動脈疾患および頚動脈疾患に似た状態である。PADでは、主に脚および足に通じる動脈において、脂肪沈着が動脈壁に沿って蓄積し、血液循環に影響を及ぼす。その初期段階では、一般的な症状は、活動中に起こる脚部および臀部の痙攣または疲労である。そのような痙攣は、その人が動かずにじっとしている時にはおさまる。これを「間欠性跛行」という。PADを持つ人々は、血塊のリスクがあるため、脳卒中および心臓発作で死亡するリスクが高い。
【0128】
X症候群は、代謝症候群とも呼ばれ、一人における一群の代謝リスク因子を特徴とする。それらには、中心性肥満(腹部および腹部周辺の過剰な脂肪組織)、アテローム生成的異常脂質血症(血中脂肪障害−主に高トリグリセリドおよび低HDLコレステロール)、上昇した血圧(130/85mmHg以上)、インスリン抵抗性またはグルコース不耐性、易血栓性状態(例えば血中の高いフィブリノゲンまたはプラスミノゲン活性化因子阻害因子[−1])、および易炎症状態(例えば、血中の上昇した高感度C反応性タンパク質)が含まれる。
【0129】
高血圧は、血圧が異常に高い状態(140/90mmHg以上の読み)を保つ一般的な障害である。高血圧を処置することができる薬物はいくつか販売されている。高血圧は脳卒中を含むいくつかの深刻な状態のリスク因子である。
【0130】
末梢血管疾患は、心臓外の動脈における動脈硬化プラークの蓄積である。末梢血管疾患の症状は、どの動脈が冒されるか、および血流の低下がどの程度深刻であるかに依存する。例えば、鈍い痙攣痛、しびれもしくは刺痛、または皮膚の色の変化を起しうる。臨床試験により、糖尿病または喫煙など、末梢血管疾患のリスクを増加させる因子が同定されている。末梢血管疾患は、例えば、足関節上腕血圧比(ABI)検査、超音波ドップラー検査、血管造影図などを使って診断することができる。末梢血管疾患は、薬物療法、外科、低侵襲インターベンション術、またはこれらの治療法の組み合わせによって処置することができる。
【0131】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、1以上の薬剤と、例えば糖尿病、血中脂質障害、または肥満の処置に使用される薬剤などと、組み合わせて使用することもできる。例えば、GPR43インバースアゴニストまたはアンタゴニストなどの化合物は、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類、インスリン、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤、アンギオテンシン変換酵素(ACE)阻害剤、リパーゼ阻害剤、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤として知られている薬物クラスに属する1以上の薬剤と組み合わせて使用することができる。
【0132】
α−グルコシダーゼ阻害剤は、膵臓または小腸において、例えばα−アミラーゼ、マルターゼ、α−デキストリナーゼ、スクラーゼなどの消化酵素を競合的に阻害する薬物クラスに属する。α−グルコシダーゼ阻害剤による可逆的阻害は、デンプンおよび糖の消化を遅らせることにより、血中グルコースレベルを遅延、減少、または他の形で低下させる。α−グルコシダーゼ阻害剤の代表例をいくつか挙げると、アカルボース、N−(1,3−ジヒドロキシ−2−プロピル)バリオールアミン(一般名:ボグリボーズ)、ミグリトール、および当技術分野で知られるα−グルコシダーゼ阻害剤がある。
【0133】
アルドース還元酵素阻害剤クラスは、ポリオール経路の第1段階律速酵素を阻害し、それによって糖尿病合併症を予防または抑止する薬物である。糖尿病の高血糖状態では、ポリオール経路におけるグルコースの利用が増加し、その結果、細胞内に蓄積した過剰なソルビトールが組織毒として作用することにより、糖尿病性神経障害、網膜症、および腎症などの合併症の発症を惹起する。アルドース還元酵素阻害剤の例には、トルレスタット、エパルレスタット、3,4−ジヒドロ−2,8−ジイソプロピル−3−チオキソ−2H−1,4−ベンゾオキサジン−4−酢酸、2,7−ジフルオロスピロ(9H−フルオレン−9,4’−イミダゾリジン)−2’,5’−ジオン(一般名:イミレスタット)、3−[(4−ブロモ−2−フルオロフェニル)メチル]−7−クロロ−3,4−ジヒドロ−2,4−ジオキソ−1(2H)−キナゾリン酢酸(一般名:ゼナレスタット)、6−フルオロ−2,3−ジヒドロ−2’,5’−ジオキソ−スピロ[4H−1−ベンゾピラン−4,4’−イミダゾリジン]−2−カルボキサミド(SNK−860)、ゾポルレスタット、ソルビニル、および1−[(3−ブロモ−2−ベンゾフラニル)スルホニル]−2,4−イミダゾリジンジオン(M−16209)、ならびに当技術分野で知られるアルドース還元酵素阻害剤がある。
【0134】
ビグアニド類は、嫌気的解糖を刺激し、末梢組織におけるインスリンに対する感度を増加させ、腸からのグルコース吸収を阻害し、肝糖新生を抑制し、脂肪酸酸化を阻害する薬物クラスである。ビグアニド類の例には、フェンホルミン、メトホルミン、ブホルミン、および当技術分野で知られるビグアニド類がある。
【0135】
インスリン分泌促進剤は、膵ベータ細胞からのインスリンの分泌を促進する性質を持つ薬物クラスに属する。インスリン分泌促進剤の例にはスルホニル尿素類(SU)がある。スルホニル尿素類(SU)は、細胞膜中のSU受容体を介してインスリン分泌のシグナルを伝達することにより、膵ベータ細胞からのインスリンの分泌を促進する薬物である。スルホニル尿素類の例には、トルブタミド、クロルプロパミド、トラザミド、アセトヘキサミド、4−クロロ−N−[(1−ピロリジニルアミノ)カルボニル]−ベンゼンスルホンアミド(一般名:グリコピラミド)またはそのアンモニウム塩、グリベンクラミド(グリブリド)、グリクラジド、1−ブチル−3−メタニリル尿素、カルブタミド、グリボヌリド(glibonuride)、グリピジド、グリキドン、グリソキセピド、グリブチアゾール、グリブゾール、グリへキサミド、グリミジン、グリピナミド、フェンブタミド、トルシクラミド、グリメピリド、および当技術分野で知られる他のインスリン分泌促進剤がある。他のインスリン分泌促進剤には、N−[[4−(1−メチルエチル)シクロヘキシル)カルボニル]−D−フェニルアラニン(ナテグリニド)、(2S)−2−ベンジル−3−(cis−ヘキサヒドロ−2−イソインドリニルカルボニル)プロピオン酸カルシウム二水和物(ミチグリニド,KAD−1229)、および当技術分野で知られる他のインスリン分泌促進剤がある。
【0136】
チアゾリジンジオン類は、より一般的にはTZD類として知られている薬物クラスに属する。チアゾリジンジオン類は、インスリンに対する細胞の感受性を増加させることによって血糖を低下させる2型糖尿病のための薬物クラスである。その結果、インスリンは、エネルギーを得るためにグルコースを血液から細胞内へと移動させることができる。これらの薬物はHDLを増加させることもできる。
【0137】
チアゾリジンジオン類の例には、ロジグリタゾン、ピオグリタゾン、および当技術分野で知られるチアゾリジンジオン類がある。レズリン(トログリタゾン)は、米国ではこのクラスの最初の薬物だったが、肝毒性のために市場から撤収された。より良い安全性プロファイルを持つ現在利用可能な姉妹化合物には、アクトス(ピオグリタゾン)およびアバンディア(ロジグリタゾン)がある。これらの薬物療法の使用に対する主な禁忌には、肝疾患および心不全がある。これらの薬物は体液貯留の有意な増加を引き起こすことによって、心不全のリスクを増加させることもできる。
【0138】
メグリチニド類は、2型糖尿病を持つ人が食事をとった直後に起こりうる血糖の急速な上昇を止めるために用いられる。これらの化合物は、例えばレパグリニド(プランジン)およびナテグリニド(スターリックス)などを含めて、スルホニル尿素薬物療法が機能する方法と同様に、膵臓によって産生されるインスリンの量を増加させることによって機能する。メグリチニド類は食事をとる前に投与される。この薬物クラスに付随する副作用には、低血糖、鼻炎症状を含む上気道炎、頭痛、関節痛および背痛、悪心、下痢および便秘がある。
【0139】
異なるタイプのインスリンは、それらがどのくらい早く作用し始めるか(発現)、およびそれらがどのくらい長く作用し続けるか(持続時間)に応じて分類される。現在利用できるタイプには、速効型、短時間作用型、中時間作用型、および長時間作用型インスリンがある。70/30インスリンと呼ばれる70%中時間作用型(NPH)および30%短時間作用型レギュラーインスリン;50/50インスリンと呼ばれる50%中時間作用型(NPH)および50%短時間作用型レギュラーインスリン;75/25インスリンと呼ばれる75%中時間作用型(NPH)および25%速効型ヒューマログ(リスプロ);ノボログミックス(NovoLog Mix)70/30と呼ばれる70%中時間作用型(NPH)および30%速効型ノボログ(インスリンアスパルト)など、混合済みの速効型および中時間作用型インスリンを利用することができる。インスリンは通常、皮下組織への注射として与えられる(皮下)。インスリンポンプまたはジェットインジェクター(皮膚内に医薬品を噴霧する装置)によって与えることもできる。
【0140】
インスリンは糖(グルコース)を細胞に入らせ、そこで糖はエネルギーを得るために使用される。インスリンがないと、血糖レベルは身体にとって安全であるレベルを上回ってしまう。通常は、身体が必要とする一定レベルおよび可変レベルのインスリンを提供するために、速効型または短時間作用型インスリンおよび中時間作用型または長時間作用型インスリンが投与される。短時間作用型インスリンは血糖レベルを急速に低下させてから消失する。一部の長時間作用型インスリンは速効型または短時間作用型インスリンが消失し始めた時に効き始める。新しい長時間作用型インスリン・ランタスは、投与されてから数分以内に作用し始め、約24時間は同じ速度で作用し続ける。
【0141】
速効型または短時間作用型インスリンと中時間作用型または長時間作用型インスリンとの併用は、血糖レベルを、身体にとって安全な範囲内に、終日維持するのに役立つ。したがって、インスリンは、1型糖尿病を持つ人々、2型糖尿病を持つ人々であってその膵臓がインスリンをほとんどまたは全く産生しないか、経口薬物療法ではその血糖が管理されない人々を処置するために使用することができる。これらの人々にはインスリンを単独で、または経口薬物療法と一緒に投与することができる。2型糖尿病を持つ人々であって、重病または大手術のために血糖レベルが高い人々、2型糖尿病を持つ婦人であって、妊娠中であるか授乳中であり、その血糖レベルを食餌と運動で安全域内に保つことができない人々。妊娠中の使用について調べられた経口糖尿病薬物療法は1つだけである(グリブリド)。
【0142】
インスリンの主要副作用としては、危険なほど低い血糖レベル(重症低血糖)を挙げることができる。極めて低い血糖レベルが10〜15分以内に発生しうる。インスリンは、既に過体重である2型糖尿病を持つ人々では特に、体重増加の一因となりうる。長期インスリン使用の他の考えうる副作用には、インスリンが注射される部分の脂肪組織の喪失(リポジストロフィ)、そして稀に、腫脹(浮腫)を含むアレルギー反応がある。
【0143】
スタチン化合物は、ヒドロキシメチルグルタリルCoA(HMG−CoA)還元酵素を阻害することによって血中コレステロールレベルを低下させる薬物クラスに属する。HMG−CoA還元酵素はコレステロール生合成における律速酵素である。この還元酵素を阻害するスタチンは、血液からのLDLの浄化を担うLDL受容体の活性をアップレギュレートすることにより、血清LDL濃度を低下させる。スタチン化合物の例には、ロスバスタチン、プラバスタチンおよびそのナトリウム塩、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチン、セリバスタチン、および当技術分野で知られるHMG−CoA還元酵素阻害剤がある。
【0144】
スクアレン合成阻害剤は、スクアレンの合成を阻害することによって血中コレステロールレベルを低下させる薬物クラスに属する。スクアレン合成阻害剤の例には、(S)−α−[ビス[2,2−ジメチル−1−オキソプロポキシ)メトキシ]ホスフィニル]−3−フェノキシベンゼンブタンスルホン酸・一カリウム塩(BMS−188494)および当技術分野で知られるスクアレン合成阻害剤がある。
【0145】
フィブラート化合物は、肝臓におけるトリグリセリドの合成および分泌を阻害し、リポタンパク質リパーゼを活性化することによって、血中コレステロールレベルを低下させる薬物クラスに属する。フィブラート類は、ペルオキシソーム増殖因子活性化受容体を活性化し、リポタンパク質リパーゼ発現を誘導することが知られている。フィブラート化合物の例には、ベザフィブラート、ベクロブラート、ビニフィブラート、シプロフィブラート、クリノフィブラート、クロフィブラート、クロフィブリン酸、エトフィブラート、フェノフィブラート、ゲムフィブロジル、ニコフィブラート、ピリフィブラート、ロニフィブラート、シンフィブラート、テオフィブラート(theofibrate)、および当技術分野で知られるフィブラート類がある。
【0146】
LDL(低密度リポタンパク質)異化促進剤は、LDL(低密度リポタンパク質)受容体の数を増加させることによって血中コレステロールレベルを低下させる薬物クラスに属し、当技術分野で知られるLDL異化促進剤がその例に含まれる。
【0147】
アンギオテンシン変換酵素(ACE)阻害剤は、アンギオテンシン変換酵素を阻害することによって、血圧を低下させると共に、血中グルコースレベルもある程度は低下させる薬物クラスに属する。アンギオテンシン変換酵素阻害剤の例には、カプトプリル、エナラプリル、アラセプリル、デラプリル;ラミプリル、リシノプリル、イミダプリル、ベナゼプリル、セロナプリル、シラザプリル、エナラプリラート、フォシノプリル、モベルトプリル(moveltopril)、ペリンドプリル、キナプリル、スピラプリル、テモカプリル、トランドラプリル、および当技術分野で知られるアンギオテンシン変換酵素阻害剤がある。
【0148】
リパーゼ阻害剤には、例えば、オルリスタット(XENICAL(商標))などの抗肥満化合物が含まれる。オルリスタットは、脂肪吸収を直接阻害するが、下痢および鼓腸などの不快な胃副作用も高い発生率で生じる傾向がある。
【0149】
抗肥満薬のもう1つのクラスには、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤がある。例えばシブトラミン(Meridia(商標))は混合性5−HT/ノルアドレナリン再取り込み阻害剤である。シブトラミンの主な副作用として、一部の患者における血圧の増加および心拍数の増加を挙げることができる。セロトニン放出薬/再取り込み阻害剤フェンフルラミン(Pondimin(商標))およびデキスフェンフルラミン(Redux(商標))は、長期間(6ヶ月超)にわたって食物摂取量および体重を減少させると報告されている。しかしどちらも製品も、その使用に関連する心臓弁異常を示す予備的証拠が報告された後は、使用が中止されている。
【0150】
本発明の一部の実施形態は、本明細書に開示する化合物もしくは本発明の方法によって同定される化合物または医薬的に許容できるその塩を、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤およびアンギオテンシン変換酵素阻害剤からなる群より選択される少なくとも1つのメンバーと組み合わせて含む医薬組成物を包含する。もう1つの実施形態では、HMG−CoA還元酵素阻害剤が、プレバスタチン(prevastatin)、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンおよびリピトールからなる群より選択される。
【0151】
本発明によれば、各活性成分を全て一緒にして、または個別に、上述の生理学的に許容できる担体、賦形剤、結合剤、希釈剤などと混合し、その混合物または混合物群を医薬組成物として経口投与または非経口投与することにより、これらの組み合わせを使用することができる。化合物または化合物の混合物を、別の活性化合物と共に、併用治療または併用予防として投与する場合、それらの治療剤は、同じ時点または異なる時点で与えられる別個の医薬組成物として製剤化するか、それらの治療剤を単一の組成物として与えることができる。
【0152】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法も提供する。
【0153】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法を提供する。
【0154】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が増加するかどうかを決定することを含み、GPR43機能の増加が、その候補化合物が代謝安定化化合物であることを示す方法に関する。
【0155】
ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0156】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知のアゴニストまたは本明細書に開示するアゴニストの存在下で行なわれる。
【0157】
特定の実施形態では、前記方法が、候補化合物によって引き起こされる受容体機能の増加を、受容体を受容体の既知リガンドまたは既知アゴニストと接触させることによって引き起こされる受容体機能の第2の増加と比較することを、さらに含む。
【0158】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびCa2+からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が、全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が、細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定が、CRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、細胞内Ca2+の測定によって行なわれる。
【0159】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0160】
特定の実施形態では、前記決定が、黒色素胞アッセイの使用によって行なわれる。
【0161】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRのアゴニストの存在下で行なわれる。
【0162】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0163】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0164】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0165】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0166】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0167】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法であって、GPR43機能の増加が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物に関する。
【0168】
ある実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0169】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。
【0170】
特定の実施形態では、前記EC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のEC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、上記GPCRに対して選択的である。
【0171】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0172】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法に関する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法に関する。
【0173】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物にも関係する。
【0174】
本発明の一部の実施形態は、本明細書に開示する化合物実施形態のいずれかに従う少なくとも1つの化合物と医薬的に許容できる担体とを混合することを含む医薬組成物の生産方法を包含する。
【0175】
化合物は、当業者に周知の技法および本明細書に記載する技法を使って、医薬組成物に製剤化することができる。
【0176】
予防または処置に使用する場合、本明細書に開示する化合物または本発明の方法によって同定される化合物は、代替的に、原薬または純薬として投与することができると考えられるが、その化合物または活性成分を、医薬的に許容できる担体をさらに含む医薬製剤として提供することが有用な場合もある。
【0177】
したがって本発明は、本明細書に開示する化合物もしくは本発明の方法によって同定される化合物または医薬的に許容できるその塩もしくは誘導体を、1以上の医薬的に許容できるその担体および/または予防成分と共に含む医薬製剤を、さらに提供する。担体は、製剤の他の成分と適合し、その受容者にとって過度に有害でないという意味で、「許容できる」。
【0178】
医薬製剤、投与経路、および投与量については上述した。
【0179】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害が低血糖または加齢である。ある実施形態では、投与される化合物が、GRP43アゴニストを含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0180】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、それらを1以上の薬剤と組み合わせて使用することもできる。
【0181】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法にも関係する。
【0182】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法にも関係する。
【0183】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が減少するかどうかを決定することを含み、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法を提供する。
【0184】
一部の実施形態では、前記代謝安定化化合物が血中グルコース安定化化合物である。一部の実施形態では、前記代謝安定化化合物がインスリン分泌調節因子である。
【0185】
ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0186】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知アゴニストの存在下で行なわれる。
【0187】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびCa2+からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が、全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が、細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定が、CRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、細胞内Ca2+の測定によって、例えばFLIPRアッセイを使って行なわれる。
【0188】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0189】
特定の実施形態では、前記決定が、黒色素胞アッセイの使用によって行なわれる。
【0190】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0191】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0192】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0193】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0194】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0195】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法であって、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物を提供する。
【0196】
ある実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストである。ある実施形態では、前記代謝安定化化合物がGPR43アンタゴニストである。
【0197】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つGPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。
【0198】
特定の実施形態では、前記IC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0199】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0200】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法に関する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法を提供する。
【0201】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物も提供する。
【0202】
本発明の一部の実施形態は、本明細書に開示する化合物実施形態のいずれかに従う少なくとも1つの化合物と医薬的に許容できる担体とを混合することを含む医薬組成物の生産方法を包含する。
【0203】
医薬製剤、投与経路、および投与量については上述した。
【0204】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害が高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、投与される化合物が、GRP43インバースアゴニストまたはアンタゴニストを含む。
【0205】
ある実施形態では、本方法が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。例えば、ある実施形態では、本方法が、GPR43インバースアゴニストを含有する医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。
【0206】
ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0207】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、それらを1以上の薬剤と、例えば糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤などと、組み合わせて使用することもできる。例えば、GPR43インバースアゴニストまたはアンタゴニストなどの化合物は、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類、インスリン、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤、アンギオテンシン変換酵素(ACE)阻害剤、リパーゼ阻害剤、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤として知られている薬物クラスに属する1以上の薬剤と組み合わせて使用することができる。
【0208】
本発明の一部の実施形態は、本発明の方法によって同定される化合物または医薬的に許容できるその塩を、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤およびアンギオテンシン変換酵素阻害剤からなる群より選択される少なくとも1つのメンバーと組み合わせて含む医薬組成物を包含する。もう1つの実施形態では、HMG−CoA還元酵素阻害剤が、プレバスタチン、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンおよびリピトールからなる群より選択される。
【0209】
本発明によれば、各活性成分を全て一緒にして、または個別に、上述の生理学的に許容できる担体、賦形剤、結合剤、希釈剤などと混合し、その混合物または混合物群を医薬組成物として経口投与または非経口投与することにより、これらの組み合わせを使用することができる。化合物または化合物の混合物を、別の活性化合物と共に、併用治療または併用予防として投与する場合、それらの治療剤は、同じ時点または異なる時点で与えられる別個の医薬組成物として製剤化するか、それらの治療剤を単一の組成物として与えることができる。
【0210】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法も提供する。
【0211】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法を提供する。
【0212】
本発明は、GPR43機能を増加させる方法であって、GPR43をGPR43アゴニストの有効量と接触させることを含む方法にも関係する。本発明は、細胞中のGPR43機能を増加させる方法であって、GPR43を発現させる細胞をGPR43アゴニストの有効量と接触させることを含む方法にも関係する。細胞は例えばある個体の中にあってもよいし、細胞は単離された細胞であってもよい。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0213】
本発明は、GPR43機能を減少させる方法であって、GPR43をGPR43インバースアゴニストまたはアンタゴニストの有効量と接触させることを含む方法にも関係する。本発明は、細胞中のGPR43機能を減少させる方法であって、GPR43を発現させる細胞をGPR43インバースアゴニストまたはアンタゴニストの有効量と接触させることを含む方法にも関係する。細胞は例えばある個体の中にあってもよいし、細胞は単離された細胞であってもよい。
【0214】
本発明は、代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法を提供する。
【0215】
ある実施形態では、前記代謝関連障害が低血糖または加齢である。ある実施形態では、前記調節因子がアゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0216】
ある実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害に、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患が含まれる。ある実施形態では、前記調節因子が、インバースアゴニストまたはアンタゴニストである。ある実施形態では、前記方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。ある実施形態では、前記代謝関連障害が、インスリン抵抗性、耐糖能異常または糖尿病である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0217】
本発明は、GPR43機能を増加させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43アゴニストの有効量を投与することを含む方法に関する。ある実施形態では、前記障害が代謝関連障害である。一部の実施形態では、前記代謝関連障害が低血糖または加齢である。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0218】
本発明は、GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法を提供する。ある実施形態では、前記障害が代謝関連障害である。ある実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、前記方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0219】
本発明は、その必要がある個体の血中グルコースレベルを増加させる方法であって、その個体にGPR43アゴニストの有効量を投与することを含む方法にも関係する。本発明は、その必要がある個体の血中グルコースレベルを減少させる方法であって、その個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法にも関係する。
【0220】
また本発明は、その必要がある個体におけるインスリン分泌を減少させる方法であって、その個体にGPR43アゴニストの有効量を投与することを含む方法に関する。
【0221】
本発明の目的の1つは、a)代謝安定化化合物を同定するために本発明の方法を実行し、(b)要すれば、その化合物の構造を決定し、(c)その化合物またはその化合物の名称もしくは構造を提供する方法に関する。また本発明は、a)代謝安定化化合物を同定するために本発明の方法を実行し、(b)要すれば、その化合物の構造を決定し、(c)要すれば、その化合物の名称または構造を提供し、(d)その化合物を生産または合成する方法に関する。さらに本発明は、GPCRの機能を調節する方法であって、代謝安定化化合物を同定するために本発明の方法を実行し、次に当該GPCRをその代謝安定化化合物と接触させるか、その代謝安定化化合物を、当該GPCRの機能を調節するのに十分な条件で個体に投与することを含む方法に関する。
【0222】
本出願人は、本発明の実施形態のいずれかから、任意の1以上の候補化合物を除外する権利を留保する。本出願人は、本発明の実施形態のいずれかから、任意の1以上の調節因子を除外する権利も留保する。さらに本出願人は、本発明の実施形態のいずれかから任意のポリヌクレオチドまたはポリペプチドを除外する権利を留保する。本出願人は、本発明の実施形態のいずれかから任意の代謝関連障害を除外する権利も留保する。
【0223】
ここに開示する受容体および方法の他の用途は、とりわけ、この特許文書を再検討することにより、当業者には明らかになるだろう。
【0224】
以下の実施例は本発明を例示するために記載するものであり、決して包括的なものではない。
【実施例】
【0225】
本発明をさらに詳しく記述するために実施例を挙げるが、これらの実施例の詳細に本発明が限定されるわけではない。
【0226】
(実施例1)
マウス成体およびマウス胎児の組織および細胞におけるマウスGPR43発現のAffymetrixチップ解析
この実施例では、Affymetrix遺伝子チップを使って、いくつかのマウス成体およびマウス胎児の組織および細胞で、マウスGPR43の発現レベルを決定した(左から右に:胎児脳、橋脊髄、脊髄下部、SN橋視床、嗅球、視床、海馬、スイス3T3、3T3−LI脂肪細胞、BV2+LPS 24時間、NIH−3T3、3T3−LI前脂肪細胞、NIT−1、NIE−115分化型、E14TG2A、BV2、NIE−115、BV2+LPS 4時間、NIT CTL、C57BL6 ES、D3 ES、リンパ節、骨髄、T細胞、CD4+ 卵白アルブミン、脾臓、T細胞、CD4+ ナイーブ、胸腺、十二指腸、皮膚脂肪、褐色脂肪、精巣上体脂肪、心室、心房、大動脈、線維芽細胞、新生仔心臓、心室筋細胞、低酸素−再酸素化、新生仔、心室筋細胞、正常酸素圧、新生仔、心室筋細胞、心室筋細胞、低酸素、新生仔、心室、左TAC、心室、左シャム、大腸近位部、胃フォンデュ(fondues)、小腸、大腸遠位部、胃洞、白血球、肝臓、肺、気管、唾液腺、骨、線維芽細胞、真皮、副腎、骨格筋、皮膚、膀胱、皮膚、口、口腔粘膜、口、表皮、食道、腎臓、胆嚢、β島、ob/ob 16週、β島、ob/ob 6週、MIN6、βTC6、β島、C57Bl/6、αTC9、β島、db/db、子宮、臍帯、卵巣)。
【0227】
1.AFFYMETRIX GENECHIP(登録商標)技術
いくつかのGタンパク質共役受容体(GPCR)に対応するヌクレオチド配列をAffymetrixに提出した。Affymetrixでは、そのGeneChip(登録商標)技術によって、さまざまな組織におけるこれらの受容体のmRNA発現レベルを測定するために、オリゴヌクレオチドマイクロアレイが設計され、製造された。製造者の指示に従って、多数の組織および細胞タイプから得たRNA試料を増幅し、標識し、そのマイクロアレイにハイブリダイズさせ、データ解析した。
【0228】
(製造者の指示に基づき従って)発現指数(expression index)が50より大きければ、GPCRは発現していると決定した。データは解析され、50より大きい発現指数によるGPCRの分類が合理的であることを示していた。なぜなら、いくつかの既知GPCRは、ニューロン組織において、50より大きい発現指数で発現すると、先に報告されていたからである。
【0229】
GeneChip(登録商標)を使って、本出願人は、GPR43が脂肪、大腸、膵島および島細胞株ならびに3T3脂肪細胞に高い発現レベルを持つことを見いだした(図1参照)。
【0230】
(実施例2)
ヒトおよびマウスの組織および細胞におけるGPR43発現のRT−PCR解析
この実施例では、RT−PCRアッセイを使って、いくつかのヒトおよびマウスの組織および細胞タイプにおけるヒトおよびマウスGPR43の発現レベルを決定した。図2上図に示すように、例えば胎盤、肺、肝臓、腎臓、膵臓、脾臓、前立腺および白血球を含むいくつかのヒト組織および細胞で、ヒトGPR43遺伝子発現が観察された。また、図2下図に示すように、例えば肺、膵臓、骨格筋、小腸、脾臓、胃、島、3T3−L1分化脂肪細胞、Nit−1細胞、Min6細胞およびβTC−6細胞を含むいくつかのマウス組織、細胞および細胞株で、マウスGPR43遺伝子発現が観察された。
【0231】
この実験のために、ヒトcDNAをHuman MTC Panel IおよびHuman MTC Panel II(Clontech)から得た。マウスポリA+RNAをClontechから得た。次に、iScript cDNA Synthesis Kit(Bio−Rad)を使って、製造者のプロトコールに従って、cDNAを合成した。反応のために、1μlの1:10希釈マウスポリA+、8μlの5×iScript Reaction Mixおよび2μlのiScript Reverse Transcriptaseを水と共に合わせて、総液量を40μlにした。反応をPCR機中、25℃、5分;42℃、30分:85℃、5分で行なった。
【0232】
細胞株RNAおよびマウス島RNAはトリゾールを使って単離し、さらにDNAフリーキット(Ambion)を使って、製造者のプロトコールに従って、DNアーゼで処理した。iScript cDNA Synthesis Kit(Bio−Rad)を使って、2μgのDNアーゼ処理RNAを、40μlの総反応液量で使用した。
【0233】
ヒトおよびマウスGPR43のためのPCR反応を、InvitorgenのPlatinum PCR Supermixを使って行なった。各反応につき、2μlのcDNA、48μlのSupermix、0.2μlの各プライマー(100μMストック)を合わせて、液量を合計50μlにした。ヒトGPR43のための反応は、PCR機中、95℃で4分間の変性後、95℃、1分;60℃、30秒;72℃、1分を30サイクル行い、最後に72℃で7分間の伸長で終えた。マウスGPR35のために使用したアニーリング温度は63℃である。
【0234】
Clontechにより、Human MTC Panel IおよびIIに、G3PDH 5’および3’PCRプライマーが、対照として用意された。G3PDH PCR反応のために、2μlのcDNA、45μlのPlatinum PCR Supermix(Invitrogen)、1μlの各プライマー(10μMストック)を合わせて、液量を合計50μlにした。反応はPCR機中、95℃で1分間の変性後、95℃、30秒;68℃、3分を23サイクル行い、最後に68℃で3分間の伸長で終えた。
ヒトGPR43 RT−PCRプライマー対:
【0235】
【化1】
マウスGPR43 RT−PCRプライマー対:
【0236】
【化2】
(実施例3)
GPR43調節因子の同定
この実施例では、黒色素胞でのスクリーニングプロトコールを使って、GPR43調節因子を同定する。
【0237】
1.黒色素胞技術
黒色素胞は下等脊椎動物に見出される皮膚細胞である。それらはメラノソームと呼ばれる色素性細胞小器官を含有する。黒色素胞は、Gタンパク質共役受容体(GPCR)活性化時に、これらのメラノソームを微小管ネットワークに沿って再分配することができる。この色素移動の結果は、細胞の明白な明化または暗化である。黒色素胞では、Gi共役受容体の活性化に起因する細胞内cAMPレベルの減少が、メラノソームを細胞の中心に移動させ、その結果、色の劇的な明化が起こる。次に、Gs共役受容体の活性化に続いてcAMPレベルが上昇すると、メラノソームは再び分散し、細胞は再び暗く見えるようになる。Gq共役受容体の活性化に起因するジアシルグリセロールレベルの増加も、この再分散を誘発することができる。また、この技術は、一定の受容体チロシンキナーゼの研究にも適している。黒色素胞の応答は受容体活性化から数分以内に起こり、単純で強い色変化をもたらす。この応答は、従来の吸光度マイクロプレートリーダーまたは簡単な撮像システムを使って、容易に検出することができる。他の皮膚細胞とは異なり、黒色素胞は神経堤に由来し、シグナル伝達タンパク質の全てを発現させるようである。特に、これらの細胞は、極めて広い範囲のGタンパク質を発現させるので、ほとんど全てのGPCRを機能的に発現させることができる。
【0238】
黒色素胞は、GPCRに結合し、そして/またはGPCRを活性化する化合物(天然リガンドを含む)を同定するために利用することができる。この方法は、特異的刺激に応答してその色素を分散または凝集させることおよびGPCRをコードする外来クローンを発現させることができる色素細胞株の試験細胞を導入することによって行なうことができる。色素沈着の初期状態は、例えばメラトニン、MSHまたは光などを使って設定することができる。次に試験細胞を化学化合物と接触させ、細胞における色素沈着が色素沈着の初期状態から変化したかどうかを決定する。候補化合物(リガンドを含むが、これに限定されるわけではない)がGPCRに結合したことによる色素細胞の分散はペトリ皿上で暗く見え、一方、色素細胞の凝集は明るく見えるだろう。
【0239】
材料および方法は米国特許第5,462,856号および米国特許第6,051,386号の開示に従った。これらの特許明細書は参照によりその全体が本明細書に組み入れられる。
【0240】
マウスまたはヒトGPR43のコード配列を含有するプラスミドを、エレクトロポレーションによって、黒色素胞にトランスフェクトする。細胞を96ウェルプレートにプレーティングする。トランスフェクションの48時間後に、各プレート上の細胞の半分を10nMメラトニンで処理する。メラトニンは黒色素胞中の内在性Gi共役受容体を活性化し、黒色素胞がその色素を凝集させるようにする。細胞の残り半分を無血清培地0.7×L−15(Gibco)に移す。1時間後に、無血清培地中の細胞が色素分散状態にあるのに対して、メラトニン処理細胞は色素凝集状態にある。この時点で、細胞を、140,000〜150,000の有機小分子化合物を含有する専有化合物ライブラリーからのさまざまな化合物で処理する。GPR43がその化合物に結合すれば、黒色素胞は、その化合物に応答して、例えば色素凝集による色変化を起こすと予想される。
【0241】
(実施例4)
経口ブドウ糖負荷試験
アゴニスト、アンタゴニストまたはインバースアゴニストなどのGPR43調節因子を、経口グルコース投与後の血漿グルコースに対するその作用について試験することができる。
【0242】
例えば、67日齢の雄C57bl/6マウスを18時間絶食させ、無作為にグループ分けして、選択した用量でGPR43調節因子を与えるか、ビヒクル(80%PEG、10%Tween80、および10%エタノールを含有するPET)を与える。GPR43調節因子は強制栄養針(gavaga needle)を通して経口的に送達する(経口,液量100μl)。GPR43調節因子投与またはビヒクル投与の30分後に、3g/kgの用量でデキストロースをマウスに経口投与する。血中グルコースのレベルを、Glucometer Elite XL(Bayer)を使って、数時点で決定する。
【0243】
グルコース耐性は、グルコースの腹腔内送達を使って試験することもできる。例えば、68日齢の雄C57Bl/6マウスを、18時間の絶食後に、100mg/kgのGPR43調節因子またはPETビヒクルで処置する。GPR43調節因子投与またはビヒクル投与の30分後に、2g/kgの用量でマウスにデキストロースを腹腔内投与する。血中グルコースのレベルを、Glucometer Elite XL(Bayer)を使って、選択した時点で決定する。
【0244】
(実施例5)
3T3−L1脂肪細胞におけるインスリン刺激によるグルコース取り込み
この実施例では、脂肪細胞におけるインスリン刺激によるグルコース取り込みにGPR43調節因子が及ぼす作用を、3H−2−デオキシグルコース取り込みアッセイを使って試験する。
【0245】
簡単に述べると、標準的プロトコールを使って、まず、3T3−L1細胞を脂肪細胞に分化させる(PatelおよびLane,Proc.Natl.Acad.Sci.U.S.A.96:1279−1284(1999))。次に、ビヒクルまたは5μMのGPR43調節因子を含有する無血清培地で、細胞を3時間刺激する。次に、細胞をクレブスリンゲルリン酸緩衝液中で2回洗浄し、クレブスリンゲルリン酸緩衝液中、10nMインスリンの存在下または不在下で、20分間インキュベートする。0.05mM(0.5μCi/mol)3H−2−デオキシグルコースおよび0.05mM非放射性2−デオキシグルコースを細胞に37℃で5分間添加することにより、2−デオキシグルコース輸送を測定する。輸送反応を停止させるために、細胞を氷冷PBSで3回洗浄し、1%トリトンXに可溶化する。溶解物中の放射能レベルをシンチレーション計数によって決定する。
【0246】
(実施例6)
GPCR活性化を決定するためのアッセイ
ヒトGPCRの活性化の評価には、さまざまなアプローチを利用することができる。以下に実例を挙げる。当業者であれば、その当業者のニーズにとって優先的に有益である技法を決定する能力を持つと考えられる。
【0247】
1.膜結合アッセイ:[35S]GTPγSアッセイ
Gタンパク質共役受容体が、リガンド結合の結果として、または構成的活性化の結果として、その活性状態にある場合、受容体はGタンパク質に共役し、GDPの放出と、それに続くGTPのGタンパク質への結合を刺激する。Gタンパク質−受容体複合体のαサブユニットはGTPアーゼとして作用して、GTPをGDPにゆっくりと加水分解し、通常その時点で、受容体は不活化される。活性化受容体はGDPをGTPに交換し続ける。非加水分解性GTP類似体[35S]GTPγSを利用して、活性化受容体を発現させる膜への[35S]GTPγSの結合の増進を実証することができる。[35S]GTPγS結合を使って活性化を測定することの利点は、(a)全てのGタンパク質共役受容体に広く応用することができること;(b)膜表面に近接しているので、細胞内カスケードに影響を及ぼす分子を拾い上げる可能性が少なくなることである。
【0248】
このアッセイでは、関連受容体を発現させる膜への[35S]GTPγS結合を刺激するという、Gタンパク質共役受容体の能力を利用する。したがってこのアッセイは、内在性GPCRおよび非内在性構成的活性化GPCRに対して候補化合物をスクリーニングするための直接的同定方法に使用することができる。このアッセイは汎用であり、全てのGタンパク質共役受容体における創薬に応用することができる。
【0249】
[35S]GTPγSアッセイを、20mM HEPESおよび1〜約20mM MgCl2(この量は結果を最適化するために調節することができるが、20mMが好ましい)pH7.4、約0.3〜約1.2nM[35S]GTPγS(この量は結果を最適化するために調節することできるが、1.2が好ましい)および12.5〜75μgの膜タンパク質(例えばGPR43を発現させる293細胞;この量は結果を最適化するために調節することができる)および10μM GDP(この量は結果を最適化するために調節することができる)を含む結合緩衝液中で、1時間インキュベートする。次に、コムギ胚芽凝集素ビーズ(25μl;Amersham)を加えた後、その混合物を室温でさらに30分間ンインキュベートする。次に、チューブを1500×g、室温で5分間遠心分離した後、シンチレーション計数器で計数する。
【0250】
2.アデニリルシクラーゼ
細胞ベースのアッセイ用に設計されたFlash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)を、粗製形質膜で使用するために改変することができる。Flash Plateウェルは、cAMPを認識する特異的抗体も含有するシンチラントコーティングを含有することができる。ウェル内で生成したcAMPは、放射性cAMPトレーサーのcAMP抗体への結合に関する直接的競合によって、定量することができる。以下の記述は、受容体を発現させる全細胞中のcAMPレベルの変化を測定するための簡潔なプロトコールとして使用することができる。
【0251】
トランスフェクト細胞を一過性トランスフェクションの約24時間後に収集する。培地を注意深く吸引して捨てる。10mlのPBSを細胞の各ディッシュに穏やかに加えた後、注意深く吸引する。1mlのSigma細胞解離緩衝液および3mlのPBSを各プレートに加える。細胞をピペットでプレートから剥がし、細胞懸濁液を50ml円錐形遠心管に集める。次に、細胞を1,100rpm、室温で、5分間遠心分離する。細胞ペレットを適当な液量のPBS(約3ml/プレート)に注意深く再懸濁する。次に、血球計を使って細胞を計数し、適当な細胞数になるようにPBSを追加する(最終液量を約50μl/ウェルにする)。
【0252】
cAMP標準および検出緩衝液(検出緩衝液11mlに対して1μCiのトレーサー[125I]cAMP(50μl)を含む)を調製し、製造者の指示にしたがって維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、50μlの刺激緩衝液、3μlの候補化合物(最終アッセイ濃度12μM)および50μlの細胞を含有する。アッセイ緩衝液は使用するまで氷上に保存する。50μlのcAMP標準を適当なウェルに加えた後、50μlのPBSAをウェルH11およびH12に添加することにより、(好ましくは例えば96ウェルプレートで行なわれる)アッセイを開始する。50μlの刺激緩衝液を全てのウェルに加える。3μlの化合物溶液を分注することができるピンツールを使って、DMSO(または選択した候補化合物)を適当なウェルに加え、候補化合物の最終アッセイ濃度を12μMに、そして総アッセイ液量を100μlにする。次に、細胞をウェルに加え、室温で60分間インキュベートする。次に、トレーサーcAMPを含有する100μlの検出混合物をウェルに加える。次にプレートをさらに2時間インキュベートした後、Wallac MicroBetaシンチレーション計数器で計数する。次に、各アッセイプレート内に含まれる標準cAMP曲線から、cAMP/ウェルの値を外挿する。
【0253】
3.Gi共役標的GPCRのための細胞ベースのcAMP
TSHRは、活性化時にcAMPの蓄積を引き起こすGs共役GPCRである。TSHRは、アミノ酸残基623を突然変異させることにより(すなわちアラニン残基をイソロイシン残基に変えることにより)、構成的に活性化することができる。Gi共役受容体はアデニリルシクラーゼを阻害し、したがってcAMP産生レベルを減少させると予想され、これがcAMPレベルの評価を困難にする場合がある。Gi共役受容体の活性の指標としてcAMP産生量の減少を測定するための効果的な技法は、cAMPのベースラインレベルを確定するために、「シグナル増強物質」としての非内在性構成的活性化TSHR(TSHR−A623I)(または内在性構成的活性化Gs共役受容体)を、Gi連結標的GPCRと同時トランスフェクトすることによって達成することができる。内在型または非内在型のGi共役受容体を作製したら、その標的GPCRをシグナル増強物質と同時トランスフェクトする。スクリーニングに使用することができるのは、この物質である。一部の実施形態では、このアプローチが、Gi共役受容体に抗する候補化合物の直接的同定に、好ましく使用される。Gi共役GPCRの場合、このアプローチを使用すると、標的GPCRのインバースアゴニストがcAMPシグナルを増加させ、アゴニストがcAMPシグナルを減少させることを注記しておく。
【0254】
1日目に、2×104個の293細胞/ウェルをプレーティングする。2日目に、2本の反応チューブを調製する(各チューブについて以下に示す比率はプレート1枚あたりである):チューブAは、哺乳動物細胞にトランスフェクトする各受容体のDNA2μgで、合計4μgのDNA(例えばpCMVベクター;pCMVベクターと突然変異型THSR(TSHR−A623I);TSHR−A623IおよびGPCRなど)を、1.2mlの無血清DMEM(Irvine Scientific,カリフォルニア州アービン)に混合することによって調製される;チューブBは、120μlのリポフェクトアミン(Gibco BRL)を、1.2mlの無血清DMEMに混合することによって調製される。次に、チューブAおよびBを反転(数回)によって混合した後、室温で30〜45分間インキュベートする。この混合物を「トランスフェクション混合物」という。プレーティングした293細胞を1×PBSで洗浄した後、10mlの無血清DMEMを加える。次に、2.4mlのトランスフェクション混合物を細胞に加えた後、37℃/5%CO2で4時間インキュベートする。次にトランスフェクション混合物を吸引によって除去した後、25mlのDMEM/10%ウシ胎仔血清を加える。次に、細胞を37℃/5%CO2でインキュベートする。24時間のインキュベーション後に、細胞を収集し、解析に利用する。
【0255】
Flash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)は細胞ベースのアッセイ用に設計されているが、当業者の必要に応じて、粗製形質膜で使用するために改変することができる。Flash Plateウェルは、cAMPを認識する特異的抗体も含有するシンチラントコーティングを含有する。ウェル内で生成したcAMPは、放射性cAMPトレーサーのcAMP抗体への結合に関する直接的競合によって、定量することができる。以下の記述は、目的の受容体を発現させる全細胞中のcAMPレベルの変化を測定するための簡潔なプロトコールとして使用することができる。
【0256】
トランスフェクト細胞を一過性トランスフェクションの約24時間後に収集する。培地を注意深く吸引して捨てる。10mlのPBSを細胞の各ディッシュに穏やかに加えた後、注意深く吸引する。1mlのSigma細胞解離緩衝液および3mlのPBSを各プレートに加える。細胞をピペットでプレートから剥がし、細胞懸濁液を50ml円錐形遠心管に集める。次に、細胞を1,100rpm、室温で、5分間遠心分離する。細胞ペレットを適当な液量のPBS(約3ml/プレート)に注意深く再懸濁する。次に、血球計を使って細胞を計数し、適当な細胞数になるようにPBSを追加する(最終液量を約50μl/ウェルにする)。
【0257】
製造者の指示に従って、cAMP標準および検出緩衝液(検出緩衝液11mlに対して1μCiのトレーサー[125I]cAMP(50μl)を含む)を調製し、維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、50μlの刺激緩衝液、3μlの候補化合物(最終アッセイ濃度12μM)および50μlの細胞を含有する。アッセイ緩衝液は使用するまで氷上に保存することができる。50μlのcAMP標準を適当なウェルに加えた後、50μlのPBSAをウェルH11およびH12に添加することにより、アッセイを開始する。50μlの刺激緩衝液を全てのウェルに加える。3μlの化合物溶液を分注することができるピンツールを使って、選択した化合物(例えばTSH)を適当なウェルに加え、候補化合物の最終アッセイ濃度を12μMに、そして総アッセイ液量を100μlにする。次に、細胞をウェルに加え、室温で60分間インキュベートする。次に、トレーサーcAMPを含有する100μlの検出混合物をウェルに加える。次にプレートをさらに2時間インキュベートした後、Wallac MicroBetaシンチレーション計数器で計数する。各アッセイプレート内に含まれる標準cAMP曲線から、cAMP/ウェルの値を外挿する。
【0258】
4.レポーターベースのアッセイ
a.CRE−LUCレポーターアッセイ(Gs関連受容体)
293細胞または293T細胞を96ウェルプレートに1ウェルあたり2×104細胞の密度でプレーティングし、その翌日に、製造者の指示に従い、リポフェクトアミン試薬(BRL)を使ってトランスフェクトする。DNA/脂質混合物を各6ウェルトランスフェクションについて以下のように調製する:DMEM100μl中のプラスミドDNA260ngをDMEM100μl中の脂質2μlと穏やかに混合する(このプラスミドDNA260ngは、8×CRE−Lucレポータープラスミド200ng、内在性受容体もしくは非内在性受容体を含むpCMVまたはpCMVのみ50ng、およびGPRS発現プラスミド(pcDNA3(Invitrogen)中のGPRS)10ngからなる)。8×CRE−Lucレポータープラスミドは以下のように調製される:pβgal−Basicベクター(Clontech)中のBglV−HindIII部位にラットソマトスタチンプロモーター(−71/+51)をクローニングすることにより、ベクターSRIF−β−galを得る。8コピーのcAMP応答配列を、アデノウイルステンプレートAdpCF126CCRE8(Suzukiら,Hum Gene Ther 7:1883−1893(1996)を参照されたい;この文献の開示は参照によりその全体が本明細書に組み入れられる)からPCRによって取得し、それをSRIF−β−galベクター中に、Kpn−BglV部位でクローニングすることにより、8×CRE−β−galレポーターベクターを得る。8×CRE−β−galレポーターベクター中のベータ−ガラクトシダーゼ遺伝子を、HindIII−BamHI部位で、pGL3−basicベクター(Promega)から得たルシフェラーゼ遺伝子で置き換えることにより、8×CRE−Lucレポータープラスミドを生成させる。室温で30分間インキュベートした後、そのDNA/脂質混合物をDMEM400μlで希釈し、その希釈混合物100μlを各ウェルに加える。細胞培養インキュベータで4時間インキュベートした後、10%FCSを含むDMEM100μlを各ウェルに加える。翌日、トランスフェクト細胞を、10%FCSを含むDMEM200μl/ウェルに換える。8時間後に、ウェルをPBSで1回洗浄した後、フェノールレッド不含DMEM100μl/ウェルに換える。翌日、LucLite(商標)レポーター遺伝子アッセイキット(Packard)を使用し、製造者の指示に従って、ルシフェラーゼ活性を測定し、1450MicroBeta(商標)シンチレーションおよびルミネセンスカウンター(Wallac)で読み取る。
【0259】
b.AP1レポーターアッセイ(Gq関連受容体)
Gq刺激を検出するための方法は、そのプロモーター中にAP1配列を含有する遺伝子の活性化を引き起こすというGq依存性ホスホリパーゼCの既知の性質に依存する。リン酸カルシウム沈殿物の成分が410ngのpAP1−Luc、80ngのpCMV−受容体発現プラスミド、および20ngのCMV−SEAPである点以外は、CREBレポーターアッセイに関する上述のプロトコールに従って、Pathdetect(商標)AP−1cis−Reporting System(Stratagene,カタログ番号219073)を利用することができる。
【0260】
c.SRF−LUCレポーターアッセイ(Gq関連受容体)
Gq刺激を検出するための一方法は、そのプロモーター中に血清応答因子を含有する遺伝子の活性化を引き起こすというGq依存性ホスホリパーゼCの既知の性質に依存する。Pathdetect(商標)SRF−Luc−Reporting System(Stratagene)は、例えばCOS7細胞におけるGq共役活性のアッセイに利用することができる。このシステムのプラスミド成分および内在性または非内在性GPCRをコードする表示の発現プラスミドを、Mammalian Transfection(商標)Kit(Stratagene,カタログ番号200285)を使用し、製造者の指示に従って、細胞にトランスフェクトする。簡単に述べると、SRF−Luc410ng、pCMV−受容体発現プラスミド80ng、およびCMV−SEAP(分泌型アルカリホスファターゼ発現プラスミド;試料間のトランスフェクション効率のばらつきについて管理するためにトランスフェクト細胞中のアルカリホスファターゼ活性を測定する)20ngを、製造者の指示に従って、リン酸カルシウム沈殿物中で組み合わせる。沈殿物の半分を96ウェルプレート中の3ウェルに均等に分配し、無血清培地中の細胞上に24時間保つ。最後の5時間は、細胞を例えば1μMの候補混合物と共にインキュベートする。次に細胞を溶解し、Luclite(商標)Kit(Packard,カタログ番号6016911)および「Trilux 1450 Microbeta」液体シンチレーションおよびルミネセンスカウンター(Wallac)を使って、製造者の指示に従ってルシフェラーゼ活性をアッセイする。データはGraphPad Prism(商標)2.0a(GraphPad Software Inc.)を使って解析することができる。
【0261】
d.細胞内IP3蓄積アッセイ(Gq関連受容体)
1日目に、目的の受容体(内在性または非内在性)を含む細胞を24ウェルプレートに、通常は1×105細胞/ウェルの密度(ただしこの数字は最適化することができる)でプレーティングすることができる。2日目に、まず、1ウェルにつき無血清DMEM50μl中のDNA0.25μgと1ウェルにつき無血清DMEM50μl中のリポフェクトアミン2μlとを混合することによって、細胞をトランスフェクトすることができる。その溶液を穏やかに混合し、室温で15〜30分間インキュベートする。細胞を0.5mlのPBSで洗浄し、400μlの無血清培地をトランスフェクション培地と混合して、細胞に加える。次に、細胞を37℃/5%CO2で3〜4時間インキュベートしてから、トランスフェクション培地を除去し、1ml/ウェルの通常成長培地で置き換える。3日目に、細胞を3H−ミオイノシトールで標識する。簡単に述べると、培地を除去し、細胞を0.5mlのPBSで洗浄する。次に、1ウェルにつき0.5mlのイノシトール不含無血清培地(GIBCO BRL)を、1ウェルにつき0.25μCiの3H−ミオイノシトールと共に、細胞を37℃/5%CO2で16〜18時間、終夜インキュベートする。4日目、細胞を0.5mlのPBSで洗浄し、イノシトール不含無血清培地、10μMパージリン、10mM塩化リチウムを含有する0.45mlのアッセイ培地を加えるか、またはセロトニン受容体を含有する対照コンストラクトを使用する場合は、0.4mlのアッセイ培地および50μlの10×ケタンセリン(ket)を、最終濃度が10μMになるように加える。次に、細胞を37℃で30分間インキュベートする。次に、細胞を0.5mlのPBSで洗浄し、1ウェルにつき200μlの新鮮な氷冷停止溶液(1M KOH;18mMホウ酸Na;3.8mM EDTA)を加える。その溶液を氷上に5〜10分間または細胞が溶解するまで保ち、次に200μlの新鮮な氷冷中和溶液(7.5%HCL)で中和する。次に、溶解物を1.5mlエッペンドルフチューブに移し、チューブ1本につき1mlのクロロホルム/メタノール(1:2)を加える。その溶液を15秒間ボルテックスし、上相をBiorad AG1−X8(商標)アニオン交換樹脂(100〜200メッシュ)に適用する。まず、樹脂を1:1.25W/Vの水で洗浄し、0.9mlの上相をカラムにのせる。カラムを10mlの5mMミオイノシトールおよび10mlの5mMホウ酸Na/60mMギ酸Naで洗浄する。10mlのシンチレーションカクテルが2mlの0.1Mギ酸/1Mギ酸アンモニウムと共に入っているシンチレーションバイアルに、イノシトール三リン酸を溶出させる。10mlの0.1Mギ酸/3Mギ酸アンモニウムで洗浄することによってカラムを再生し、ddH2Oで2回すすぎ、水中、4℃で保存する。
【0262】
(実施例7)
融合タンパク質調製
a.GPCR:Gs融合コンストラクト
GPCR−Gタンパク質融合コンストラクトの設計は、以下のように行なうことができる:ラットGタンパク質Gsα(長型;Itoh,H.ら,Proc.Natl.Acad.Sci.83:3776(1986))の5’末端および3’末端をどちらも、そこにHindIII配列が含まれるように操作する。正しい配列を(隣接HindIII配列を含めて)確認した後、全配列を、pcDNA3.1(−)(Invitrogen,カタログ番号V795−20)中に、そのベクターのHindIII制限部位を使ってサブクローニングすることによってシャトル(shuttle)する。pcDNA3.1(−)へのサブクローニング後に、Gsα配列について、正しい向きを決定する。次に、HindIII配列にラットGsα遺伝子を含有する改変pcDNA3.1(−)を確認する。このベクターは、この時点で、「ユニバーサル」Gsαタンパク質ベクターとして利用することができる。pcDNA3.1(−)ベクターはHindIII部位の上流にさまざまな周知の制限部位を含有するので、Gsタンパク質の上流に目的の受容体のコード配列を挿入することができるという利益が得られる。これと同じアプローチを利用して、他の「ユニバーサル」Gタンパク質ベクターを作製することができ、もちろん、当業者に知られている他の市販ベクターまたは専有ベクターを利用することもできる。重要な基準は、GPCRの配列がGタンパク質の配列に対して上流にあり、かつインフレームであることである。
【0263】
b.Gq(6アミノ酸欠失)/Gi融合コンストラクト
Gq(欠失)/Gi融合コンストラクトの設計は、以下のように行なうことができる:GαqサブユニットのN末端の6アミノ酸(TLESIM(配列番号7)という配列を持つアミノ酸2〜7)を欠失させ、EYNLV(配列番号8)という配列を持つC末端の5アミノ酸を、DCGLF(配列番号9)という配列を持つGαiタンパク質の対応するアミノ酸で置き換える。この融合コンストラクトは、以下のプライマー:
【0264】
【化3】
と、マウスGαq−野生型をヘマグルチニンと共に含有するテンプレートとしてのプラスミド63313とを用いるPCRによって、取得することができる。
【0265】
TaqPlus Precision DNAポリメラーゼ(Stratagene)を、以下のサイクルによる増幅に利用することができる(第2〜第4ステップを35回繰り返す):95℃で2分;95℃で20秒;56℃で20秒;72℃で2分;および72℃で7分。PCR産物をpCRII−TOPOベクター(Invitrogen)にクローニングし、ABI Big Dye Terminatorキット(P.E.Biosystems)を使って配列決定することができる。融合コンストラクトの配列を含有するTOPOクローンからのインサートは、2段階クローニング法により、発現ベクターpcDNA3.1(+)中のHindIII/BamHI部位にシャトルすることができる。WO02068600として2002年9月6日に公開されたPCT出願番号PCT/US02/05625も参照されたい。この文献の開示は参照によりその全体が本明細書に組み入れられる。
【0266】
(実施例8)
[35S]GTPγSアッセイ
A.膜調製
一部の実施形態では、例えばアゴニスト、インバースアゴニストまたはアンタゴニストなどの候補化合物の同定で使用するために、目的の標的GPCRを含む膜を、以下のように調製する。
【0267】
a.材料
「膜掻爬緩衝液」は20mM HEPESおよび10mM EDTA、pH7.4から構成され;「膜洗浄緩衝液」は20mM HEPESおよび0.1mM EDTA、pH7.4から構成され;「結合緩衝液」は20mM HEPES、100mM NaCl、および10mM MgCl2、pH7.4から構成される。
【0268】
b.手順
この手順中は全ての材料を常に氷上に保つ。まず、細胞のコンフルエント単層から培地を吸引し、次に10mlの冷PBSですすいだ後、吸引する。次いで、細胞を掻爬するために5mlの膜掻爬緩衝液を加える。次に、細胞抽出物を50ml遠心管に移す(4℃、20,000rpmで17分間遠心分離する)。次いで、上清を吸引し、ペレットを30mlの膜洗浄緩衝液に再懸濁し、次に4℃、20,000rpmで17分間遠心分離する。次に上清を吸引し、ペレットを結合緩衝液に再懸濁する。次にこれをBrinkman Polytron(商標)ホモジナイザーを使ってホモジナイズする(全ての物質が懸濁状態になるまで15〜20秒間のバースト)。これを、ここでは「膜タンパク質」という。
【0269】
ブラッドフォードタンパク質アッセイ
ホモジナイゼーション後に、ブラッドフォードタンパク質アッセイを使って、膜のタンパク質濃度を決定する(タンパク質を約1.5mg/mlに希釈し、小分けし、後で使用するために凍結(−80℃)することができる。凍結した場合、使用のためのプロトコールは以下のとおりになる:アッセイ当日に、凍結膜タンパク質を室温で融解し、次にボルテックスしてから、Polytronを使って、約12×1,000rpm(約5〜10秒)で、ホモジナイズする。調製物が複数の場合、異なる調製物のホモジナイゼーション間で、ホモジナイザーを十分に洗浄すべきであることを注記しておく)。
【0270】
a.材料
結合緩衝液(上記のとおり);ブラッドフォード色素試薬;ブラッドフォードタンパク質標準を、製造者の指示に従って利用する(Biorad,カタログ番号500−0006)。
【0271】
b.手順
一対のチューブを調製する。1つは膜を含み、もう1つは対照「ブランク」とする。各チューブは800μlの結合緩衝液を含有する。次いで、10μlのブラッドフォードタンパク質標準(1mg/ml)を各チューブに加えた後、10μlの膜タンパク質を一方のチューブだけに加える(ブランクには加えない)。次いで、200μlのブラッドフォード色素試薬を各チューブに加えた後、各チューブをボルテックスする。5分後に、チューブを再びボルテックスし、その中の材料をキュベットに移す。そのキュベットを、CECIL3041分光光度計を使って、波長595で読み取る。
【0272】
同定アッセイ
a.材料
GDP緩衝液は37.5mlの結合緩衝液および2mgのGDP(Sigma,カタログ番号G−7127)からなり、次に結合緩衝液での連続希釈により、0.2μM GDPにする(各ウェルにおけるGDPの最終濃度は0.1μM GDPである);候補化合物を含む各ウェルは、GDP緩衝液100μl(最終濃度0.1μM GDP)、結合緩衝液中の膜タンパク質50μl、および結合緩衝液中の[35S]GTPγS(0.6nM)(結合緩衝液10mlにつき2.5μlの[35S]GTPγS)50μlからなる200μlの最終液量を持つ。
【0273】
b.手順
候補化合物は96ウェルプレート形式を使ってスクリーニングすることができる(これらは−80℃で凍結させることができる)。膜タンパク質(または対照として、標的GPCRを除外した発現ベクターによる膜)を懸濁状態になるまで手短にホモジナイズする。上述のブラッドフォードタンパク質アッセイを使ってタンパク質濃度を決定する。膜タンパク質(および対照)を結合緩衝液で0.25mg/mlに希釈する(最終アッセイ濃度,12.5μg/ウェル)。次いで、100μlのGDP緩衝液をWallac Scintistrip(商標)(Wallac)の各ウェルに加える。5μlピンツールを使って、5μlの候補化合物を上記のウェルに移す(すなわち、総アッセイ液量200μl中の5μlは、候補化合物の最終濃度が10μMになるように、1:40の比である)。ここでも、混入を避けるために、各移送ステップ後に、水(1回)、エタノール(1回)および水(2回)を含む3つのレザバーで、ピンツールをすすぐべきであり、各すすぎ後に過剰な液体をツールから振り払い、紙およびキムワイプで乾燥させるべきである。次いで、50μlの膜タンパク質を各ウェルに加え(標的GPCRを伴わない膜を含む対照ウェルも利用する)、室温で5〜10分間、プレインキュベートする。次いで、結合緩衝液中の[35S]GTPγS(0.6nM)50μlを各ウェルに加えた後、振とう機上、室温で60分間インキュベートする(プレートを箔で覆う)。次に、4000RPM、22℃で15分間、プレートを遠心することによって、アッセイを停止する。プレートを8チャンネル多岐管で吸引し、プレートカバーで密封する。プレートを、Wallac1450で、設定「Prot.#37」を使って、(製造者の指示どおりに)読み取る。
【0274】
(実施例9)
サイクリックAMPアッセイ
例えばアゴニスト、インバースアゴニスト、またはアンタゴニストなどの候補化合物を同定するためのもう1つのアッセイアプローチは、シクラーゼベースのアッセイを利用することによって達成することができる。このアッセイアプローチは、直接的同定だけでなく、上記の実施例で説明した[35S]GTPγSアプローチで得られる結果を確認するための間接的アプローチとしても利用することができる。
【0275】
候補化合物を目的の受容体に対するインバースアゴニストおよびアゴニストとして直接同定するには、改変Flash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)を、以下のプロトコールに従って利用することができる。
【0276】
トランスフェクト細胞をトランスフェクションの約3日後に収集する。20mM HEPES(pH7.4)および10mM MgCl2を含有する緩衝液中の懸濁細胞をホモジナイズすることにより、膜を調製する。ホモジナイゼーションは、Brinkman Polytron(商標)を使って氷上で約10秒間行なう。得られたホモジネートを49,000×g、4℃で15分間遠心分離する。次に、得られたペレットを、20mM HEPES(pH7.4)および0.1mM EDTAを含有する緩衝液に再懸濁した後、49,000×g、4℃で15分間遠心分離する。次に、得られたペレットを使用時まで−80℃で保存する。直接的同定スクリーニングの当日に、膜ペレットを室温でゆっくり融解し、20mM HEPES(pH7.4)および10mM MgCl2を含有する緩衝液に再懸濁して、最終タンパク質濃度を0.60mg/mlにする(再懸濁した膜は使用時まで氷上に置く)。
【0277】
製造者の指示に従って、cAMP標準および検出緩衝液(検出緩衝液11mlに対して2μCiのトレーサー[125I]cAMP(100μl)を含む)を調製し、維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、20mM HEPES(pH7.4)、10mM MgCl2、20mMホスホクレアチン(Sigma)、0.1単位/mlクレアチンホスホキナーゼ(Sigma)、50μM GTP(Sigma)、および0.2mM ATP(Sigma)を含有する。次に、アッセイ緩衝液を使用時まで氷上に保存する。
【0278】
例えば96ウェルプレートのウェルに、候補化合物(3μl/ウェル;最終アッセイ濃度12μM)を、40μlの膜タンパク質(30μg/ウェル)および50μlのアッセイ緩衝液と共に加える。次に、この混合物を、穏やかに振とうしながら、室温で30分間インキュベートする。
【0279】
インキュベーション後に、100μlの検出緩衝液を各ウェルに加え、次に2〜24時間インキュベートする。次にプレートを、Wallac MicroBeta(商標)プレートリーダーで、「Prot.#31」を使って、(製造者の指示どおりに)計数する。
【0280】
(実施例10)
細胞内カルシウム濃度を測定するための蛍光イメージングプレートリーダー(FLIPR)アッセイ
標的受容体(実験)およびpCMV(陰性対照)を安定にトランスフェクトした、各クローン株由来の細胞を、ポリ−D−リジン前処理96ウェルプレート(Becton−Dickinson,#356640)に、5.5×104細胞/ウェルの密度で完全培養培地(10%FBS、2mM L−グルタミン、1mMピルビン酸ナトリウムを含むDMEM)と共に播種し、翌日アッセイする。GPR43はGαqに共役するので、検出可能なカルシウムフラックスを引き起こすのに、Gα15、Gα16などのプロミスキャスGタンパク質、またはキメラGq/Giαサブユニットを必要としない。Fluo4−AM(Molecular Probe,#F14202)インキュベーション緩衝液ストックを調製するために、1mgのFluo4−AMを467μlのDMSOおよび467μlのプルオロニックアシッド(Pluoronic acid)(Molecular Probe,#P3000)に溶解して、1mMストック溶液を得る。このストック溶液は−20℃で1ヶ月保存することができる。Fluo4−AMは蛍光カルシウム指示薬色素である。
【0281】
候補化合物を洗浄緩衝液(1×HBSS/2.5mMプロベニシド(Probenicid)/20mM HEPES、pH7.4)中に調製する。
【0282】
アッセイ時に、培養培地をウェルから除去し、細胞に100μlの4μM Fluo4−AM/2.5mMプロベニシド(Sigma,#P8761)/20mM HEPES/完全培地、pH7.4を負荷する。37℃/5%CO2でのインキュベーションを60分間進行させる。
【0283】
1時間のインキュベーション後に、Fluo4−AMインキュベーション緩衝液を除去し、細胞を100μlの洗浄緩衝液で2回洗浄する。各ウェルに100μlの洗浄緩衝液を残す。プレートを37℃/5%CO2のインキュベータに60分間戻す。
【0284】
50μlの候補化合物を30秒目に添加し、その候補化合物が惹起する細胞内カルシウム濃度([Ca2+])の一過性変化をさらに150秒にわたって記録するように、FLIPR(蛍光イメージングプレートリーダー;Molecular Device)をプログラムする。FLIPRソフトウェアを使用し、総蛍光変化カウントを使ってアゴニスト活性を決定する。この計器ソフトウェアは蛍光値を標準化して、初期値を等しくゼロにする。
【0285】
上記は、安定トランスフェクト細胞を使ったアゴニスト活性に関するFLIPRアッセイであるが、当業者であれば、アンタゴニスト活性を特徴づけるために、このアッセイを容易に改変することができるだろう。あるいは、一過性トランスフェクト細胞も使用できることは、前記当業者には容易に理解されるだろう。
【0286】
(実施例11)
MAPキナーゼアッセイ
MPAキナーゼ(マイトジェン活性化キナーゼ)を監視することで、受容体活性化を評価することができる。MAPキナーゼはいくつかのアプローチによって検出することができる。1つのアプローチは、非リン酸化(不活性)状態か、リン酸化(活性)状態かという、リン酸化状態の評価に基づく。リン酸化されたタンパク質はSDS−PAGEでの移動度が遅くなるので、ウェスタンブロット法を使って、刺激されていないタンパク質と比較することができる。あるいは、リン酸化されたタンパク質に特異的な抗体を入手することができ(New England Biolabs)、それを使ってリン酸化されたキナーゼの増加を検出することができる。どちらの方法でも、細胞を候補化合物で刺激した後、Laemmli緩衝液で抽出する。可溶性画分をSDS−PAGEゲルに適用し、タンパク質を電気泳動的にニトロセルロースまたはイモビリン(Immobilin)に転写する。免疫反応性バンドを標準的ウェスタンブロット技術によって検出する。可視シグナルまたは化学発光シグナルをフィルムに記録する。これはデンシトメトリーによって定量することができる。
【0287】
もう1つのアプローチは、リン酸化アッセイによるMAPキナーゼ活性の評価に基づく。細胞を候補化合物で刺激し、可溶性抽出物を調製する。その抽出物を、ガンマ−32P−ATP、ATP再生系、およびMAPキナーゼの特異的基質、例えばPHAS(phoshporylated heat and acid stable protein regulated by insulin:インスリンによって調節されるリン酸化耐熱耐酸性タンパク質)−Iなどと共に、30℃で10分間インキュベートする。H3PO4の添加によって反応を停止させ、試料を氷に移す。リン酸化されたタンパク質を保持するワットマンP81クロマトグラフィーペーパーに一部をスポットする。そのクロマトグラフィーペーパーを洗浄し、液体シンチレーション計数器で32Pについて計数する。あるいは、細胞抽出物を、ガンマ−32P−ATP、ATP再生系、およびストレプトアビジンによって濾紙支持体に結合されたビオチン化ミエリン塩基性タンパク質と共に、インキュベートする。ミエリン塩基性タンパク質は活性化されたMAPキナーゼの基質である。リン酸化反応を30℃で10分間行なう。次に、リン酸化されたミエリン塩基性タンパク質を保持する濾紙を通して、抽出物を吸引する。濾紙を洗浄し、液体シンチレーション計数により、32Pについて計数する。
【0288】
(実施例12)
受容体結合アッセイ
本明細書に記載する方法の他に、候補化合物を評価するためのもう1つの手段は、GPR43受容体への結合親和性を決定することによる。このタイプのアッセイは一般に、GPR43受容体に対する放射標識リガンドを必要とする。GPR43受容体の既知リガンドおよびその放射性ラベルを使用することの他に、GPR43アゴニスト化合物を放射性同位体で標識して、GPR43受容体に対する候補化合物の親和性を評価するためのアッセイに使用することもできる。
【0289】
化合物を同定/評価するために、GPR43アゴニストなどの放射標識GPR43化合物をスクリーニングアッセイで使用することができる。一般論として、新たに合成または同定された化合物(すなわち候補化合物)を、GPR43受容体に対する放射標識GPR43アゴニストの結合を減少させるその能力について、評価することができる。したがって、GPR43受容体への結合に関して放射標識GPR43アゴニストと競合する能力は、GPR43受容体に対する候補化合物の結合親和性に、直接的に相関する。
【0290】
GPR43に関する受容体結合を決定するためのアッセイプロトコール
A.GPR43受容体調製
例えば、ここで述べるように、HEK293細胞(ヒト腎臓,ATCC)にGPR43を一過性にまたは安定にトランスフェクトすることができる。例えば293細胞に10μgのヒトGPR43受容体および60μlのリポフェクトアミン(15cmディッシュ1枚あたり)をトランスフェクトし、培地を交換してそのディッシュ中で24時間(75%コンフルエント)成長させることができる。10ml/ディッシュのHepes−EDTA緩衝液(20mM Hepes+10mM EDTA,pH7.4)を使って細胞を取り出す。次に、細胞をBeckman Coulter遠心機中、17,000rpmで20分間、遠心分離する(JA−25.50ローター)。次に、ペレットを20mM Hepes+1mM EDTA,pH7.4に再懸濁し、50mlダウンスホモジナイザーでホモジナイズし、再び遠心分離する。上清を除去した後、結合アッセイに使用するまでペレットを−80℃で保存する。アッセイに使用する時は、膜を氷上で20分間融解したのち、10mLのインキュベーション緩衝液(20mM Hepes,1mM MgCl2、100mM NaCl、pH7.4)を加える。次に、粗製膜ペレットを再懸濁するために膜をボルテックスし、Brinkmann PT−3100 Polytronホモジナイザーを使って、設定6で15秒間、ホモジナイズする。膜タンパク質の濃度をBRLブラッドフォードタンパク質アッセイを使って決定する。
【0291】
B.結合アッセイ
総結合量については、総液量50μlの適当に希釈した膜(50mMトリスHCl(pH7.4)、10mM MgCl2および1mM EDTAを含有するアッセイ緩衝液に希釈したもの;タンパク質5〜50μg)を、96ウェルポリプロピレンマイクロタイタープレートに加え、次に100μlのアッセイ緩衝液および50μlの放射標識GPR43アゴニストを加える。非特異的結合量については、100μlではなく50μlのアッセイ緩衝液を加え、さらに50μlの10μM非放射性GPR43を加えてから、50μlの放射標識GPR43アゴニストを加える。次にプレートを室温で60〜120分間インキュベートする。Brandell 96ウェルプレートハーベスターで、Microplate Devices GF/C Unifilter濾過プレートを通して、アッセイプレートを濾過した後、0.9%NaClを含有する冷50mMトリスHCl(pH7.4)で洗浄することにより、結合反応を停止させる。次に、濾過プレートの底を密封し、50μlのOptiphase Supermixを各ウェルに加え、プレートの上部を密封し、プレートをTrilux MicroBetaシンチレーション計数器で計数する。化合物競合試験については、100μlのアッセイ緩衝液を加える代わりに、100μlの適当に希釈した候補化合物を、適当なウェルに加えた後、50μlの放射標識GPR43アゴニストを加える。
【0292】
C.計算
候補化合物を最初は1および0.1μMでアッセイし、次に、中央用量が放射標識GPR43アゴニスト結合のおよそ50%阻害(すなわちIC50)を引き起こすように選択した濃度範囲でアッセイする。候補化合物の不在下での特異的結合量(BO)は、総結合量(BT)−非特異的結合量(NSB)の差であり、同様に特異的結合量(候補化合物の存在下)(B)は、置換結合量(BD)−非特異的結合量(NSB)の差である。IC50は阻害応答曲線、%B/BO対候補化合物濃度のロジット−ログプロットから決定される。
【0293】
KiはChengおよびPrustoff変換によって計算される:
Ki=IC50/(1+[L]/KD)
式中、[L]は、アッセイに使用する放射標識GPR43アゴニストの濃度であり、KDは、同じ結合条件下で独立して決定された放射標識GPR43アゴニストの解離定数である。
【0294】
(実施例13)
齧歯類糖尿病モデル
肥満およびインスリン抵抗性に関連するII型糖尿病の齧歯類モデルが開発されている。疾患の病態生理を理解し、候補治療化合物を試験するために、マウスにおけるdb/dbおよびob/ob[Diabetes(1982)31:1−6参照]ならびにズッカーラットにおけるfa/faなどの遺伝的モデルが開発されている[Diabetes(1983)32:830−838;Annu Rep Sankyo Res Lab(1994)46:1−57]。Jackson Laboratoryによって開発されたホモ接合体動物C57BL/KsJ−db/dbマウスは、肥満、高血糖、高インスリン血症およびインスリン抵抗性であるのに対して[J Clin Invest(1990)85:962−967]、ヘテロ接合体は痩せていて正常血糖である。db/dbモデルの場合、マウスは、加齢に伴って徐々に、インスリン欠乏(糖レベルの管理が不十分な場合、ヒトII型糖尿病の後期に共通して観察される特徴)を発症する。このモデルはヒトII型糖尿病に似ているので、例えば血漿グルコースおよびトリグリセリドを低下させることなどの活性(ただしこれに限定されるわけではない)について、本発明の代謝安定化化合物を、試験する。ズッカー(fa/fa)ラットは重度の肥満、高インスリン血症、およびインスリン抵抗性であり{Coleman,Diabetes(1982)31:1;E Shafrir「Diabetes Mellitus」H RifkinおよびD Porte,Jr編[Elsevier Science Publishing Co,New York,ed.4(1990),pp.299−340]}、fa/fa突然変異は、マウスdb突然変異のラット等価物であるかもしれない[Friedmanら,Cell(1992)69:217−220;Truettら,Proc Natl Acad Sci USA(1991)88:7806]。タビー(tubby)(tub/tub)マウスは、高血糖を伴わない肥満、中等度のインスリン抵抗性および高インスリン血症を特徴とする[Colemanら,Heredity(1990)81:424]。
【0295】
本発明は、上記齧歯類糖尿病モデルのいずれかもしくは全て、II型糖尿病もしくは他の好ましい代謝関連障害もしくは上述した脂質代謝の障害を持つヒト、または他の哺乳動物に基づくモデルにおけるインスリン抵抗性および高血糖を低下させるための、本発明化合物の使用を包含する。血漿グルコースおよびインスリンレベル、ならびに血漿遊離脂肪酸およびトリグリセリドを含む(ただしこれに限定されるわけではない)他の因子を、試験することができる。
【0296】
本発明の化合物の抗高血糖活性に関するインビボアッセイ
遺伝子改変肥満糖尿病マウス(db/db)(雄、7〜9週齢)を、標準的実験室条件下に、22℃および相対湿度50%で飼育し(7〜9匹/ケージ)、Purina齧歯類飼料の食餌および水を自由に摂取させて維持する。処置に先だって、血液を各動物の尾静脈から収集し、One Touch Basic Glucose Monitor System(Lifescan)を使って血中グルコース濃度を決定する。250〜500mg/dlの血漿グルコースレベルを持つマウスを使用する。各処置群は、平均グルコースレベルが研究の開始時に各群で等しくなるように分配した7匹のマウスからなる。イソフルラン麻酔を使って挿入した微小浸透圧ポンプでdb/dbマウスに投薬することにより、本発明の化合物、食塩水、または無関係な化合物をマウスの皮下(s.c.)に与える。その後、定期的に、血液試料を尾静脈から採取し、血中グルコース濃度について解析する。群間の有意差(本発明の化合物を食塩水処置と比較する)をスチューデントt検定を使って評価する。
【0297】
上記は限定ではなく例示を目的としている。II型糖尿病の典型的齧歯類モデルは他にも記述されている[Moller DE,Nature(2001)414:821−7およびその中の引用文献;ならびにReed MJら,Diabetes,ObesityおよびMetabolism(1999)1:75−86およびその中の引用文献;これら各文献の開示は参照によりその全体が本明細書に組み入れられる]。
【0298】
本明細書に記載した典型的実施例には、本発明の要旨から逸脱することなく、さまざまな変更、付加、置換および変異を加えることができ、したがってそれらも本発明の範囲に包含されるとみなされることは、当業者には理解されるだろう。上で言及した文献は、印刷刊行物および暫定的、通常の特許出願(ただしこれに限るわけではない)を含めていずれも、参照として、その全体が本明細書に援用される。
【図面の簡単な説明】
【0299】
【図1】マウス成体およびマウス胎児の組織および細胞におけるマウスGPR43発現のAffymetrix遺伝子チップ解析を表す。
【図2】選ばれたヒト組織およびヒト細胞におけるヒトGPR43発現(上図)ならびに選ばれたマウス組織、マウス細胞およびマウス細胞株におけるマウスGPR43発現(下図)のRT−PCR解析を表す。
【技術分野】
【0001】
(発明の分野)
本発明は、ある化合物がGPR43機能を調節するかどうかを決定することによって、代謝安定化化合物を同定する方法に関する。したがって本発明の化合物は、低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患などの代謝関連障害の予防または処置に有用である。
【背景技術】
【0002】
(発明の背景)
細胞は主要エネルギー源としてグルコースを使用する。したがって食物は利用される前にまず身体によってグルコースに分解される。次にグルコースは腸から血中に放出されて、血中グルコースレベルの上昇をもたらす。このグルコースレベルの上昇に応答して、膵ベータ島細胞は、そのインスリン産生およびインスリン分泌を増加させる。インスリンは血液を介して循環し、脂肪組織、筋肉および肝臓などのインスリン応答器官にシグナルを送る伝達物質として作用して、それらのグルコース摂取を増加させる。このようにして、血中グルコースの上昇は、それに続くベータ細胞からのインスリン分泌の増加を伴う。血中グルコースレベルを正常に戻すように作用するのは、インスリンの上昇である。健常な個体では、血中グルコースレベルがかなり一定に保たれる。正常血糖(正常グルコースレベル)と呼ばれるこの平衡状態は、インスリンによって厳しく制御される。
【0003】
糖尿病などの疾患では、血中グルコースレベルのこの厳しい調節が失われて、糖尿病患者に観察される増加した血中グルコースレベルをもたらす。高血糖(高グルコースレベル)状態は、膵ベータ細胞によるインスリン産生の不足、および/または筋肉、肝臓および脂肪などの標的器官によるグルコースの不十分な取り込みによって起こりうる。最終結果は、血中グルコースレベルの増加である。したがって糖尿病は、2タイプの欠陥、すなわちベータ細胞からのインスリン分泌異常、および主要インスリン応答器官によるインスリン感受性異常の結果であると考えることができる。(器官がインスリンの作用に抵抗するので)インスリン抵抗性とも呼ばれているこのインスリン感受性異常は、標的器官がそのグルコース取り込みを増加させるのに、より多くのインスリンが要求されることを意味する。インスリン抵抗性はベータ細胞に対する圧力の増加をもたらす。なぜならベータ細胞は、インスリン抵抗性を補償するために、そのインスリン分泌を増加させる必要があるからである。これは段階的に拡大する問題であって、まず最初は耐糖能異常をもたらし、そして最終的には、増え続けるインスリンの需要に膵臓が応じきれないために起こるインスリン分泌の完全な喪失をもたらす。
【0004】
糖尿病は、血中グルコースの上昇をもたらす異常グルコース恒常性を特徴とする一群の障害を表す診断用語である。糖尿病には多くのタイプがあるが、最も一般的な2タイプが、インスリン依存性糖尿病またはIDDMとも呼ばれるI型と、インスリン非依存性糖尿病またはNIDDMとも呼ばれるII型である。I型糖尿病は、主として、発症年齢が低い疾患であり、膵臓中のインスリン分泌ベータ細胞が免疫系によって破壊されることによるものである。この場合、身体は膵ベータ細胞を自己と認識することができず、自分自身の細胞を破壊してしまう。ベータ細胞が破壊されるとインスリン分泌が完全に失われ、そのような患者は、生存するために、インスリンへの依存を強いられる。II型糖尿病は、主として、発症年齢が高い疾患であり、通常は40歳以降であるが、近年は、II型糖尿病と診断される若い人々も増えている。これはインスリン抵抗性およびベータ細胞疲弊を主たる特徴とし、多くの場合、肥満に関連する。II型糖尿病はI型糖尿病より一般的であり、世界中で診断される全糖尿病例の90〜95%を占める。
【0005】
高血糖への組織の慢性的曝露は、神経障害、網膜症および腎症の微小血管問題ならびに脳卒中、冠動脈心疾患、および末梢血管疾患の大血管性合併症を含む多様な合併症をもたらしうる。血中グルコースレベルの不適切な制御は、糖尿病以外の疾患、例えば肥満およびX症候群などの特徴でもある。例えば、X症候群の特徴の1つは、インスリン抵抗性またはグルコース不耐性である。また、肥満は、高インスリン血症およびインスリン抵抗性を特徴とし、これはNIDDM、高血圧およびアテローム性動脈硬化にも共通する特徴である。さらに肥満はNIDDMの主要リスク因子である。30%以上の過体重である被験者では、NIDDMを発症するリスクが3倍になり、NIDDM患者の4分の3は過体重である。
【0006】
カロリー摂取とエネルギー支出との間の不均衡の結果である肥満は、実験動物およびヒトにおいて、インスリン抵抗性および糖尿病と高い相関関係にある。しかし、肥満−糖尿病症候群に関与する分子機序は、まだ調査中である。肥満の初期発症中は、インスリン分泌の増加がインスリン抵抗性を相殺し、患者を高血糖から保護する(非特許文献1)。しかし、時が経つにつれてベータ細胞機能が劣化し、肥満個体の約20%でインスリン非依存性糖尿病が発症する(非特許文献2および非特許文献3)。したがって、現在社会におけるその高い罹患率を考えると、肥満はNIDDMの最も重要なリスク因子になっている(非特許文献4)。しかし、脂肪蓄積に呼応したインスリン分泌の変化を一部の患者で起こしやすくしている因子は、まだわかっていない。残念ながら、肥満の処置に利用することができる有効な長期治療はまだない。
【0007】
糖尿病は世界中で数百万人を苦しめている。米国だけで糖尿病患者は1800万人を超え、毎年600,000例が新たに診断される。糖尿病を持つ人々は、心臓疾患、失明、腎不全、感染、四肢切断、および他の状態のリスクが高くなっている。米国では糖尿病に起因すると考えられる直接医療支出および間接支出が、2002年の場合、1320億ドルだった。総合的にみて、糖尿病合併症は米国の主要死因の1つである。
【0008】
例えばα−グルコシダーゼ阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類および外因性インスリンなど、糖尿病を処置するための治療法は存在する。しかしこれらの治療法は有効性が限られており、低血糖エピソード、体重増加、胃腸障害および貧血など、重大な安全性および忍容性の問題を伴う。また、処置選択肢の多くは、注射または毎日複数回の投薬を必要とし、それが服薬遵守の問題を生む。
【非特許文献1】Le Stunffら,Diabetes(1989)43:696−702
【非特許文献2】Pederson,P.,Diab.Metab.Rev.(1989)5:505−509
【非特許文献3】Brancati,F.L.ら,Arch.Intern.Med.(1999)159:957−963
【非特許文献4】Hill,J.O.ら,Science(1998)280:1371−1374
【発明の開示】
【発明が解決しようとする課題】
【0009】
したがって、例えば糖尿病、アテローム性動脈硬化および肥満などの代謝関連障害を安全かつ効果的に処置する薬剤の同定が必要とされている。本発明は、この必要を満たすと共に、関連する利点を提供するものである。
【課題を解決するための手段】
【0010】
(要旨)
本出願人は、GPR43が膵島で発現され、db/db糖尿病マウスおよびob/ob肥満マウスでは、GPR43がアップレギュレートされることを、予想外に見いだした。また本出願人は、分化した脂肪細胞ではGPR43が強く誘導されることを開示する。さらに本出願人は、インスリン抵抗性、耐糖能異常および糖尿病などの代謝関連障害を処置するために、GPR43のインバースアゴニストまたはアンタゴニストを使用できることを開示する。
【0011】
第1態様として、本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が調節されるかどうかを決定することを含み、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法を特徴とする。一部の実施形態では、前記GPR43がヒト由来である。一部の実施形態では、前記決定が二次メッセンジャーアッセイを含む。
【0012】
第2態様として、本発明は、第1態様の方法に従って同定される代謝安定化化合物を特徴とする。一部の実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。
【0013】
第3態様として、本発明は、組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、前記化合物が第1態様の方法によって同定される方法を特徴とする。
【0014】
第4態様として、本発明は、第2態様の化合物を含むか、または本質的に第2態様の化合物からなるか、または第2態様の化合物からなる、医薬組成物を特徴とする。
【0015】
第5態様として、本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、第4態様の化合物の有効量を前記個体に投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、第5態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、第4態様の化合物の有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、個体が哺乳動物であり、一部の実施形態では、個体がヒトである。
【0016】
第6態様として、本発明は、代謝安定化化合物として使用するための第4態様の化合物を含む医薬を製造する方法、および代謝関連障害の処置に使用するための第4態様の化合物を含む医薬を製造する方法を特徴とする。
【0017】
第7態様として、本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が減少するかどうかを決定することを含み、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法を特徴とする。一部の実施形態では、前記GPR43がヒト由来である。一部の実施形態では、前記決定が二次メッセンジャーアッセイを含む。
【0018】
第8態様として、本発明は、第7態様の方法に従って同定される代謝安定化化合物を特徴とする。一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。
【0019】
第9態様として、本発明は、組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、前記化合物が第7態様の方法によって同定される方法を特徴とする。
【0020】
第10態様として、本発明は、第8態様の化合物を含むか、または本質的に第8態様の化合物からなるか、または第8態様の化合物からなる、医薬組成物を特徴とする。
【0021】
第11態様として、本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、第10態様の化合物の有効量を前記個体に投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、第11態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、第10態様の化合物の有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、個体が哺乳動物であり、一部の実施形態では、個体がヒトである。
【0022】
第12態様として、本発明は、代謝安定化化合物として使用するための第10態様の化合物を含む医薬を製造する方法、および代謝関連障害の処置に使用するための第10態様の化合物を含む医薬を製造する方法を特徴とする。
【0023】
第13態様として、本発明は、代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法を特徴とする。一部の実施形態では、前記代謝関連障害が低血糖または加齢であり、前記調節因子がアゴニストである。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患であり、前記調節因子がインバースアゴニストまたはアンタゴニストである。一部の実施形態では、第13態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。
【0024】
第14態様として、本発明は、GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法を特徴とする。一部の実施形態では、前記障害が代謝関連障害、例えばインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害がII型糖尿病である。一部の実施形態では、第14態様の方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。
【発明を実施するための最良の形態】
【0025】
(詳細な説明)
本出願人は、マウスGPR43が脂肪、大腸、および膵島細胞において強く発現されること(図1参照)、ならびにdb/db糖尿病マウスおよびob/ob肥満マウスから単離された膵島では、野生型マウスの膵島と比較して、マウスGPR43がアップレギュレートされていること(図1参照)を、ここに開示する。また本出願人は、分化した3T3−L1脂肪細胞においてマウスGPRが強く誘導されることも、ここに開示する(図1参照)。さらに本出願人は、ヒトGPR43が膵臓を含むいくつかの組織で発現されること(図2上図参照)、ならびにマウスGPR43がいくつかの組織および膵島細胞株で発現されること(図2下図参照)を開示する。
【0026】
ヒトにはいくつかの受容体クラスが存在するが、群を抜いて豊富に存在し、治療的に重要なクラスは、Gタンパク質共役受容体(GPCR)クラスに代表される。ヒトゲノムにはおよそ30,000〜40,000の遺伝子があると見積られており、そのうちの約2%はGPCRをコードすると見積られている。GPCRは医薬品の開発にとって重要な分野であり、100の既知GPCRのうち約20から、全処方薬の約60%が開発されている。
【0027】
GPCRは、22〜24個の疎水性アミノ酸からなる7つの配列を持ち、それらは7つのαヘリックスを形成して、そのそれぞれが膜をまたいでいる(各区間は、膜貫通領域1(TM−1)、膜貫通領域2(TM−2)などというように、数字で識別される)という、共通の構造モチーフを有する。細胞膜の外側、すなわち「細胞外」側では、膜貫通ヘリックスが、膜貫通領域2と膜貫通領域3の間、膜貫通領域4と膜貫通領域5の間、および膜貫通領域6と膜貫通領域7の間にあるアミノ酸の鎖によってつながれる(これらは、それぞれ「細胞外」領域1、2および3(EC−1、EC−2およびEC−3)と呼ばれる)。細胞膜の内側、すなわち「細胞内」側でも、膜貫通ヘリックスは、膜貫通領域1と膜貫通領域2の間、膜貫通領域3と膜貫通領域4の間、および膜貫通領域5と膜貫通領域6の間にあるアミノ酸の鎖によってつながれる(これらは、それぞれ「細胞内」領域1、2および3(IC−1、IC−2およびIC−3)と呼ばれる)。受容体の「カルボキシ」(「C」)末端は細胞内の細胞内空間にあり、受容体の「アミノ」(「N」)末端は細胞外の細胞外空間にある。
【0028】
一般に、リガンドが受容体と結合すると(これはしばしば受容体の「活性化」と呼ばれる)、受容体のコンフォメーションに、細胞内領域と細胞内「Gタンパク質」との共役を促進するような変化が起こる。GPCRはGタンパク質に関して「プロミスキャス(promiscuous)」である、すなわちGPCRは2以上のGタンパク質と相互作用することができると報告されている。Kenakin,T.,43 Life Sciences 1095(1988)参照。Gタンパク質は他にも存在するが、現在のところ、Gq、Gs、Gi、GzおよびGoが、同定されているGタンパク質である。リガンド活性化GPCRのGタンパク質との共役は、シグナル伝達カスケード過程(「シグナル伝達」という)を開始させる。常態では、シグナル伝達が最終的に細胞活性化または細胞阻害をもたらす。理論に束縛されることは望まないが、IC−3ループおよび受容体のカルボキシ末端がGタンパク質と相互作用すると考えられる。
【0029】
数クラスのGPCRをホスホリパーゼC経路に共役させるプロミスキャスGタンパク質、例えばGα15またはGα16(OffermannsおよびSimon,J Biol Chem 270:15175−80(1995))や、同じ経路、例えばホスホリパーゼCに、多数の異なるGPCRを共役させるように設計されたキメラGタンパク質(MilliganおよびRees,Trends in Pharmaceutical Sciences20:118−24(1999))もある。
【0030】
Gi共役GPCRは細胞内cAMPレベルを低下させる。黒色素胞技術(下記参照)は、Gi共役GPCRを同定するのに役立ち、前記Gi共役GPCRの調節因子を同定するのにも役立つ。
【0031】
生理的条件下で、GPCRは、2つの異なるコンフォメーション、すなわち「不活性」状態と「活性」状態の間の平衡状態で、細胞膜中に存在する。不活性状態の受容体は、細胞内シグナル伝達経路に連結して生物学的応答につながるシグナル伝達を開始させることができない。受容体コンフォメーションを活性状態に変化させると(Gタンパク質による)伝達経路への連結が可能になり、生物学的応答が生成する。
【0032】
リガンドまたは薬物などの化合物により、受容体を活性状態で安定化することができる。受容体のアミノ酸配列の改変を含む(ただしこれだけに限定されるわけではない)最近の発見により、活性状態コンフォメーションの受容体を助長し、安定化するための、リガンドまたは薬物以外の手段が与えられる。これらの手段は、受容体へのリガンド結合の作用をまねることにより、活性状態の受容体を効果的に安定化する。そのようなリガンド非依存的手段による安定化は「構成的受容体活性化」と呼ばれる。
【0033】
GPR43の配列は、Sawzdargoらによる文献で初めて公表された(Sawzdargoら,Biochem.Biophys.Res.Commun.,239:543−547(1997))。Sawzdargoらは、ヒトおよびラットガラニン受容体1(GALR1)ならびにラットGALR2内の保存された配列に基づく縮重プライマーによるPCRを使って、ヒトゲノムDNAを増幅した。1つの産物は、ヒトCD22遺伝子の3プライム領域の一部に対して100%のホモロジーを示すセグメントを含有した。著者らは、この領域とオーバーラップし、新規GPCR遺伝子GPR43を含有するPACクローンを、配列データベース中に同定した。このイントロンレスGPR43遺伝子は、GPR42遺伝子の約77kb下流に位置する。GPR43は7つの膜貫通ドメインを持つ推定330アミノ酸のタンパク質をコードする。GPR43タンパク質はGPR40との間に28%のアミノ酸一致度を持つが、GALR類との類似性はほとんどない。また、GPR43はGPR41との間に43%のアミノ酸一致度を持つ。第3のファミリーメンバーGPR42は、おそらくはGPR41の最近の遺伝子重複であり、偽遺伝子であるかもしれない。
【0034】
GPR43はオーファン受容体に分類された。これは、この受容体のリガンドが同定されていなかったことを意味する。最近になって、Brownらが、GPR43は酢酸および他の単鎖カルボン酸アニオンによって活性されると報告した(Brownら,J.Biol.Chem.,278:11312−11319(2003))。またBrownらは、GPR43が、Gタンパク質のGi、GqおよびG12ファミリーを活性化することも示している。さらにBrownらは、GPR43が好中球および単球などの免疫細胞中に、最も高いレベルで見いだされると報告している。
【0035】
(定義)
受容体周辺で発展してきた科学文献は、受容体に対して種々の影響を有するリガンドを言及するために多数の用語を採用した。明確さおよび一貫性のために、以下の定義が、本特許文書全体を通して用いられる。
【0036】
「アゴニスト」は、受容体に結合した際に細胞内応答を活性化する物質(例えば、リガンド、候補化合物)を意味するものとする。特定の実施形態では、アゴニストは、受容体に結合する際に細胞内応答を活性化する(例えば、膜へのGTPγS結合を増強するかまたは細胞内cAMPレベルを上昇させる)ことが以前には公知でなかった物質である。特定の実施形態では、アゴニストは、受容体に結合する際に、血中グルコースレベルを下げることが以前には公知でなかった物質である。用語「アゴニスト」はまた、受容体に結合する際に、完全なアゴニストよりも低い程度または小さい範囲で細胞内応答を活性化する、部分アゴニストを包含する。
【0037】
「アンタゴニスト」は、アゴニストと同じ部位で受容体に競合的に結合するが、細胞内応答を活性化せず、それによってアゴニストにより誘発される細胞内応答を阻害し得る、物質(例えば、リガンド、候補化合物)を意味するものとする。アンタゴニストは、アゴニストの非存在下ではベースラインの細胞内応答を減少させない。特定の実施形態では、アンタゴニストは、受容体に結合した際にアゴニストと競合して細胞応答を阻害することが以前には公知でなかった物質である(例えば、細胞応答は、膜へのGTPγS結合または細胞内cAMPレベルの上昇である)。
【0038】
「抗体」は、本明細書中では、モノクローナル抗体およびポリクローナル抗体を包含することが意図される。抗体はさらに、IgG、IgA、IgD、IgE、およびIgMを包含することが意図される。抗体は、抗体全体(単鎖抗体の全体が挙げられる)およびそれらの抗原結合フラグメント(Fab、Fab’、F(ab)2およびF(ab’)2が挙げられる)を包含する。抗体は、任意の天然起源または合成起源(例えば、ヒト、マウス、ウサギ、ヤギ、モルモット、ハムスター、ラクダ、ロバ、ヒツジ、ウマまたはニワトリ)に由来し得る。抗体は、例えば、5×10−6M未満、10−6M未満、5×10−7M未満、10−7M未満、5×10−8M未満、10−8M未満、5×10−9M未満、10−9M未満、5×10−10M未満、10−10M未満、5×10−11M未満、10−11M未満、5×10−12M未満、10−12M未満、5×10−13M未満、10−13M未満、5×10−14M未満、10−14M未満、5×10−15M未満および10−15M未満の解離定数すなわちKd値を有する結合親和性を有する。本発明の抗体は、当該分野で公知の任意の適切な方法により調製され得る。
【0039】
「候補化合物」とは、スクリーニング技術に従う分子(例えば、化合物)を意味するものとする。用語「候補化合物」は、特に、GPR43を調節することが既に公知のいかなる化合物(例えば、GPR43の公知のアゴニスト)も除外する。
【0040】
「組成物」とは、少なくとも2つの化合物または2つの成分を含む物質を意味する。例えば、医薬組成物は、組成物である。
【0041】
「化合物の効力」とは、受容体結合活性とは反対に、受容体の機能を化合物が阻害または刺激する能力の尺度を意味するものとする。
【0042】
「構成的に活性化された受容体」とは、構成的な活性化に供された受容体を意味する。
【0043】
「構成的な受容体活性化」とは、その内因性リガンドまたはその化学的等価物による結合以外による、活性状態の受容体の安定化を意味する。
【0044】
「接触」、または「接触する」とは、インビトロ系においてであろうとインビボ系においてであろうと、少なくとも2つの部分を合わせることを意味する。
【0045】
本明細書で使用される「糖尿病」とは、例えば、以下のリストを含む、任意の方法からなされる糖尿病の通常の診断を包含することが意図される:糖尿病の症状(例えば、多尿症、多渇症、多食症)と200mg/dl以上である平常時の血中グルコースレベル(ここで、平常時の血中グルコースレベル、食事または飲料の消費の時期にかかわらず、一日の任意の時間に定義される);または、126mg/dl以上である8時間絶食時の血中グルコースレベル;または、水に溶解させた75gの無水グルコースの経口投与から2時間後、200mg/dl以上である血中グルコースレベル。さらに、本明細書で使用される用語「糖尿病」はまた、American Diabates Associationにより定義され、100mg/dl〜125mg/dlの空腹時血中グルコースレベルであるか、またはグルコースの経口投与から2時間後の140mg/dl〜199mg/dlの血中グルコースレベルである「前糖尿病」状態も包含する。糖尿病は、いくつかの状態(例えば、ベータ島細胞の自己免疫破壊、ベータ細胞のアポトーシス、または妊娠糖尿病が挙げられる)により促進され得る。
【0046】
「内因性」とは、哺乳動物が天然に産生する物質を意味する。内因性に関して、例としてであり限定ではなく、用語「受容体」は、哺乳動物(例としてであり限定ではなく、ヒト)またはウイルスによって天然に産生されたものを意味する。対照的に、この状況における用語「非内因性」は、哺乳動物(例としてであり限定ではなく、ヒト)によってもウイルスによっても天然に産生されないものを意味する。例としてであり限定ではなく、その内因性形態において構成的に活性でないが、操作される場合に構成的に活性になる受容体は、最も好ましくは、本明細書において「非内因性の、構成的に活性化された受容体」と称される。両方の用語は、「インビボ」系および「インビトロ」系の両方を記述するために使用され得る。例としてであり限定ではなく、スクリーニングアプローチにおいて、内因性受容体または非内因性受容体がインビトロスクリーニング系に参照され得る。
【0047】
「有効量」とは、本明細書において使用される場合、研究者、医師または他の医療従事者によって探索されるべき、組織、系、もしくは個体において望ましい生物学的または医学的な応答を誘発する、活性化合物または医薬組成物の量を意味する。例えば、有効用量は、代謝関連障害を処置することのできる量であり得る。また、例えば、有効用量は、代謝関連障害を予防することのできる量であり得る。
【0048】
「グルコース寛容減損(IGT)」は、本明細書において使用される場合、明白な2型糖尿病と正常なグルコース寛容(NGT)との中間であるインスリン抵抗性に関連する条件を示すことを意図される。IGTは、罹患した人物の食後グルコース反応が、食後2時間での血漿グルコースレベルによって評価される場合に異常であると決定される手順によって診断される。この試験において、測定された量のグルコースが患者に与えられ、血中グルコースレベルが、定期的な間隔(通常、最初の2時間は30分毎、その後1時間毎)で測定される。「正常」な、または非IGTの個体において、最初の2時間のグルコースレベルは、140mg/dl未満まで上昇し、次いで急速に低下する。IGT個体においては、血中グルコースレベルは、より高く上昇し、そしてよりゆっくりとした速度で落ちる。
【0049】
「予防または処置の必要がある」とは、本明細書において使用される場合、ヒトにおいては介護者(例えば、医師、看護士、ナースプラクティショナーなど)によって、動物(非ヒト哺乳動物が挙げられる)においては獣医によってなされる、個体もしくは動物が処置を必要とするかまたはその処置から利益を受けるという判断を指す。この判断は、介護者の専門的意見の領域であるが、個体または動物が病気であるか病気になり、結果として本発明の化合物によって処置可能な状態であるという知見を含む、種々の要因に基づいてなされる。
【0050】
本明細書において使用される場合、「個体」とは、哺乳動物(好ましくは、マウス、ラット、他のげっ歯類、ウサギ、イヌ、ネコ、ブタ、ウシ、ヒツジ、ウマ、または霊長類、最も好ましくはヒト)を含む、任意の動物を指す。
【0051】
「阻害」または「阻害する」とは、用語「反応(応答)」に関連して、化合物の非存在下に対して、化合物の存在下で、反応が減少または防止されることを意味する。
【0052】
「インスリン抵抗性」は、本明細書において使用される場合、多くの方法のいずれかによって行われるインスリン抵抗性の通常の診断を包含することを意図される。上記方法としては、静脈内グルコース寛容試験または空腹時インスリンレベルの測定が挙げられるが、これらに制限されない。空腹時インスリンレベルとインスリン抵抗性の程度との間に強い相関が存在することは、周知である。したがって、高い空腹時インスリンレベルを、正常なグルコース寛容(NGT)個体がインスリン抵抗性を有することを同定する目的のため、インスリン抵抗性の代理のマーカーとして使用することができる。インスリン抵抗性の診断はまた、オイグリセミックグルコースクランプ試験を使用して行われ得る。
【0053】
「インバースアゴニスト」は、受容体の内因性形態または構成的に活性化された形態のいずれかに結合して、アゴニストの非存在下で観察されるその受容体のベースラインの細胞内応答を低下させる物質(例えば、リガンド、候補化合物)を意味する。細胞内応答は、例えば、膜へのGTPの結合の調節、または二次メッセンジャー(例えば、cAMPまたはIP3)のレベルの調節であり得る。特定の実施形態において、インバースアゴニストは、アゴニストの非存在下で観察される受容体のベースラインの細胞内応答を低下させることが既知ではない物質である。
【0054】
「リガンド」とは、内因性の天然に存在する受容体に特異的な、内因性の天然に存在するリガンドを意味する。
【0055】
「代謝関連障害」とは、代謝の障害を意味する。本明細書で使用されるように、本明細書において、代謝関連障害は、例えば、以下を包含することが意図される:低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患。
【0056】
「代謝安定化化合物」とは、代謝パラメータを安定化する化合物を意味することが意図される。代謝パラメータとしては、代謝プロセスに応答する脂質、糖、酵素または他のタンパク質のレベルのようないかなる尺度も挙げられる。例えば、代謝安定化化合物は、個体中の血中グルコースレベルを安定化し得る。
【0057】
本明細書において使用される場合、用語「調節する」または「改変する」は、特に活性、機能または分子の、量、質、応答または効果の増加または減少を指すことを意味する。「GPR43調節因子」は、GPR43受容体を調節する因子である。
【0058】
「医薬組成物」とは、少なくとも1種の化合物と薬学的に受容可能な担体とを含有する組成物を意味する。例えば、医薬組成物は、少なくとも1種の活性成分を含み、これによって、この組成物は、動物(例えば、限定ではなく、ヒト)における特定の効果のある結果についての調査を受けることができる。当業者は、活性成分が当業者の必要性に基づく所望の効果のある結果を有するか否かを決定するのに適切な技術を、理解および認識する。
【0059】
「受容体機能」は、刺激を受容して細胞の効果(遺伝子転写の調節、イオン流の調節、触媒作用の実行、および/またはGタンパク質を介した調節性活性が挙げられるが、これらに限定されない)を調節する、受容体の正常な働きを指す。GPR43の機能は、例えば、Gタンパク質(例えば、Gi、GqまたはG12)に結合すること、二次メッセンジャー(例えば、cAMP、IP3またはカルシウム)を介してのシグナル伝達、GPR43特異的抗体への結合、またはGPR43アゴニストのような化合物への結合であり得る。
【0060】
「二次メッセンジャー」とは、受容体活性化の結果として生じる細胞内応答を意味する。二次メッセンジャーとしては、例えば、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、およびCa2+が挙げられ得る。二次メッセンジャー応答は、受容体活性化の決定に関して測定され得る。さらに、二次メッセンジャー応答は、候補化合物(例えば、インバースアゴニスト、部分アゴニスト、アゴニスト、およびアンタゴニストが挙げられる)を同定するために測定され得る。
【0061】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が調節されるかどうかを決定することを含み、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法を提供する。
【0062】
本明細書にいう「GPR43」は、配列番号2に示すアミノ酸配列を持つポリペプチド、またはこの配列の変種もしくはオルソログであって、配列番号2に記載のアミノ酸配列を持つポリペプチドの機能を実質的に保っているものを指す。
【0063】
GPR43には、その機能を破壊することなく、限定的な変異または改変を加えることができると理解される。例えばGPR43は、他のGPR43ポリペプチド、例えばヒトGPR43ポリペプチドの哺乳動物種オルソログを包含するものとする。ヒトGPR43の種オルソログのヌクレオチド配列およびアミノ酸配列はデータベース中に存在する。例えばGPR43のマウスオルソログはGenBankのアクセッション番号NM_146187に見いだすことができ、GPR43のラットオルソログは、GenBankのアクセッション番号AB106675に見いだすことができる。また、GPR43は、GPR43の対立遺伝子変種、スプライス変種および保存的アミノ酸置換変種も包含する。例えばGPR43は、野生型GPR43ポリペプチドの機能、例えばGαi、GαqまたはGα12を介してシグナルする能力、GPR43特異的抗体に結合する能力、または既知のリガンドもしくはアゴニストなどといった化合物に結合する能力を、実質的に保っている変種を包含する。GPR43変種は野生型GPR43と同レベルに機能する必要はなく、野生型GPR43の全ての機能を含有する必要もない。
【0064】
アミノ酸配列への保存的および非保存的なアミノ酸変化、ギャップ、および挿入は、Basic Local Alignment Search Tool(「BLAST」)などの入手可能なアルゴリズムおよびプログラムを使って、デフォルト設定で、基準配列と比較することができる[例えばKarlinおよびAltschul,Proc Natl Acad Sci USA(1990)87:2264−8;Altschulら,J MoI Biol(1990)215:403−410;Altschulら,Nature Genetics(1993)3:266−72;およびAltschulら,Nucleic Acids Res(1997)25:3389−3402参照]。
【0065】
ポリペプチド全体の機能を実質的に保っているGPR43の断片は、この定義に包含されると理解される。例えば、GPR43のシグナル生成ドメインまたはGPR43の化合物結合ドメインを、ポリペプチド全体の代わりに使用することができる。またGPR43は、エピトープタグまたは他の融合ポリペプチドなどといった異種配列を含有することもできる。さらにGPR43は、ラベル、例えば放射性ラベル、蛍光ラベルまたは酵素ラベルなどを含有することができる。
【0066】
ある実施形態では、配列番号2に対して99%、98%、95%、92%、90%、85%、80%、または75%の配列一致度を含むポリペプチドを使って、本発明の方法を応用することができる。
【0067】
一部の実施形態では、GPR43の前記変種が、GPR43の非内在性かつ構成的に活性化された突然変異体である。ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0068】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と、またはそのような宿主細胞の膜と、接触させることを含み、宿主細胞は、当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知アゴニストの存在下で行なわれる。
【0069】
特定の実施形態では、前記方法が、候補化合物によって引き起こされる受容体の調節を、受容体を受容体の既知調節因子と接触させることによって引き起こされる受容体の第2の調節と比較することを、さらに含む。特定の実施形態では、前記既知調節因子がアゴニストである。
【0070】
一部の実施形態では、前記代謝安定化化合物が血中グルコース安定化化合物である。一部の実施形態では、前記代謝安定化化合物がインスリン分泌調節因子である。
【0071】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定は、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびカルシウム(Ca2+)からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定がCRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、例えばFLIPRアッセイを使った、細胞内カルシウム(Ca2+)の測定によって行なわれる。
【0072】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0073】
特定の実施形態では、前記決定が黒色素胞アッセイを使用することによって行なわれる。
【0074】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0075】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0076】
上述のように、受容体機能とは、刺激を受取り、細胞における作用を加減する(例えば、Gタンパク質を介して遺伝子転写を調節する、イオンの内向きフラックスまたは外向きフラックスを調節する、触媒反応を達成する、および/または活性を調節するなどであるが、これらに限定されるわけではない)ための、受容体の正常な働きを指す。GPR43機能として、例えば、Gi、GqまたはG12などのGタンパク質を結合すること、cAMP、IP3、もしくはカルシウムなどの二次メッセンジャーを介してシグナルすること、GPR43特異的抗体に結合すること、またはGPR43アゴニストなどの化合物に結合することを挙げることができる。
【0077】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0078】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0079】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0080】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法であって、GPR43機能の調節が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物を提供する。
【0081】
ある実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0082】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。
【0083】
特定の実施形態では、前記EC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のEC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0084】
一部の実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つ、GPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。
【0085】
特定の実施形態では、前記IC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0086】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0087】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法も提供する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法も提供する。
【0088】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物も提供する。
【0089】
化合物は、当業者に周知の技法を使って、医薬組成物に製剤化することができる。本明細書で言及する担体の他にも、適切な医薬的に許容できる担体を利用することができる。例えば「Remington’s Pharmaceutical Sciences」第16版,1980,Mack Publishing Co.,(Osloら編)を参照されたい。
【0090】
予防または処置に使用する場合、本発明の方法によって同定される化合物は、代替的に、原薬または純薬として投与することができると考えられるが、その化合物または活性成分を、医薬的に許容できる担体をさらに含む医薬製剤または医薬組成物として提供することが有用な場合もある。
【0091】
したがって本発明は、本発明の方法によって同定される化合物または医薬的に許容できるその塩もしくは誘導体を、1以上の医薬的に許容できるその担体および/または予防成分と共に含む医薬製剤を、さらに提供する。担体は、製剤の他の成分と適合し、その受容者にとって過度に有害でないという意味で、「許容できる」。
【0092】
医薬製剤には、経口、直腸、鼻腔、局所(口腔粘膜および舌下を含む)、膣または非経口(筋肉内、皮下および静脈内を含む)投与に適したもの、または吸入もしくは吹送による投与に適した形態にあるものが包含される。
【0093】
したがって、本発明の化合物は、従来の佐剤、担体、または希釈剤と共に、医薬製剤およびその単位剤の形態にすることができ、そのような形態で、錠剤もしくは充填カプセル剤などの固形物として、または溶液剤、懸濁剤、乳剤、エリキシル剤、ゲル剤もしくはそれらを充填したカプセル剤などの流動物として、いずれも経口用途に使用するか、または直腸投与用の坐剤の形態で使用するか、または非経口(皮下を含む)用の滅菌注射可能溶液の形態で使用することができる。そのような医薬組成物およびその単位剤形は、追加活性化合物または追加有効成分と共に、または追加活性化合物または追加有効成分を伴わずに、従来の成分を従来の比率で含むことができ、そのような単位剤形は、使用しようとしている1日量範囲に見合った任意の適切な有効量の活性成分を含有することができる。
【0094】
経口投与の場合、医薬組成物は、例えば錠剤、カプセル剤、懸濁剤または液剤の形態をとることができる。医薬組成物は、特定量の活性成分を含有する投薬単位の形態で製造することができる。そのような投薬単位の例は、乳糖、マンニトール、トウモロコシデンプンまたはバレイショデンプンなどの従来の添加剤;結晶セルロース、セルロース誘導体、アラビアゴム、トウモロコシデンプンまたはゼラチンなどの結合剤;トウモロコシデンプン、バレイショデンプンまたはカルボキシメチル−セルロースナトリウムなどの崩壊剤;およびタルクまたはステアリン酸マグネシウムなどの潤滑剤を含む、カプセル剤、錠剤、粉末剤、顆粒剤または懸濁剤である。活性成分は、組成物として注射によって投与することもでき、その場合は、例えば食塩水、デキストロースまたは水を、適切な医薬的に許容できる担体として使用することができる。
【0095】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、投与される化合物が、GPR43インバースアゴニストまたはアンタゴニストを含む。ある実施形態では、本方法が、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。例えば、ある実施形態では、本方法が、GPR43インバースアゴニストを含有する医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。ある実施形態では、前記代謝関連障害が低血糖であり、投与される前記化合物がGPR43アゴニストを含む。
【0096】
障害に関して本明細書で使用する「処置する」という用語は、ある特定の障害に付随する1以上の症状の重症度の軽減を意味する。したがって、ある障害を処置するとは、必ずしも、障害に付随する全ての症状の重症度の軽減を意味するわけではなく、また必ずしも、障害に付随する1以上の症状の重症度の完全な軽減を意味するわけでもない。同様に、「予防する」という用語は、ある特定の障害に付随する1以上の症状の出現または発生の予防を意味し、必ずしも、ある障害の完全な予防を意味するわけではない。本発明の方法は、例えば糖尿病を含む代謝関連障害を処置するために使用することができる。
【0097】
本発明の方法によって同定される化合物を使用する場合、用量は広範囲に変動することができ、慣行のとおり、また医師には知られているとおり、個々の症例で個々の状態に合わせて調節されるべきである。これは、例えば、処置すべき病気の性質および重症度、患者の状態、使用する化合物、または急性疾患もしくは慢性疾患を処置するのか、それとも予防を行なうのか、または本発明の方法によって同定される化合物に加えて、さらなる活性化合物を投与するかどうかなどに依存する。本発明の代表的な用量には、約0.01mg〜約1000mg、約0.01〜約750mg、約0.01〜約500mg、0.01〜約250mg、0.01mg〜約200mg、約0.01mg〜150mg、約0.01mg〜約100mg、および約0.01mg〜約75mgが含まれる。投与は一日に複数回行なうことができ、特に比較的多量に必要であると考えられる場合は、例えば2、3または4回投与することができる。適当と認められる場合は、個々の挙動に応じて、また適宜、患者の医師もしくは介護者に従って、上記1日量を加減することが必要な場合がある。
【0098】
処置に使用する必要がある活性成分またはその活性塩もしくは誘導体の量は、選択したその塩によって変動するだけでなく、投与経路、処置される状態の性質、患者の年齢および状態によっても変動し、最終的には担当の医師または臨床家の判断によるだろう。一般に当業者は、モデル系(典型的には動物モデル)で得られたインビボデータを他の系、例えばヒトなどに外挿する方法を理解している。通例、動物モデルには、齧歯類糖尿病モデルが含まれるが、これらに限定されるわけではない(他の動物モデルが、ReedおよびScribnerにより、Diabetes,Obestiy and Metabolism,1:75−86(1999)に報告されている)。状況によっては、これらの外挿が、単に、哺乳動物などの他の系、例えばヒトと比較した動物モデルの体重に基づく場合もあるが、多くの場合、これらの外挿は、体重だけに基づくのではなく、さまざまな因子を加味して行なわれる。代表的な因子には、患者のタイプ、年齢、体重、性別、食餌および医学的状態、疾患の重症度、投与経路、薬理学的事項、例えば使用する特定化合物の活性、抗力、薬物動態および毒性プロファイル、薬物送達系を利用するかどうか、急性もしくは慢性疾患状態を処置しているのか、それとも予防を行なうのか、または本発明の方法によって同定される化合物に加えて、さらなる活性化合物を、複合薬の一部として投与するかどうかが含まれる。本発明の化合物および/または組成物を使って疾患状態を処置するための投薬レジメンは、上述したさまざまな因子に応じて選択される。したがって、使用される実際の投薬レジメンは広い範囲にわたって多様であることができ、それゆえに好ましい投薬レジメンから逸脱する場合もあり、これらの典型的範囲から外れた投薬量および投薬レジメンも試験することができ、それが適当と認められる場合には、本発明の方法において使用できることは、当業者には理解されるだろう。
【0099】
望ましい用量は、1回量として、または適当な間隔で投与される分割量として、例えば1日に2回、3回、4回もしくはそれ以上の部分量として、便利に提示することができる。部分量自体をさらに、例えばいくつかの不連続で大まかな間隔の投与に、分割することもできる。1日量は、比較的大量に投与することが適当であるとみなされる場合は特に、数回、例えば2回、3回または4回の部分投与に分割することができる。適当と認められる場合は、個々の挙動に応じて、上記1日量を加減することが必要な場合がある。
【0100】
本発明の方法によって同定される化合物は、広範囲にわたるさまざまな経口剤形および非経口剤形で投与することができる。以下の剤形が、活性成分として、本明細書に開示する化合物、または本発明の方法によって同定される化合物もしくは本発明の方法によって同定される化合物の医薬的に許容できる塩を含みうることは、当業者には明白だろう。
【0101】
本発明の方法によって同定される化合物から医薬組成物を調製する場合、適切な医薬的に許容できる担体の選択は、固体、液体、または両者の混合物であることができる。固形調製物には、粉末剤、錠剤、丸剤、カプセル剤、カシェ剤、坐剤、および分散性顆粒剤が含まれる。固形担体は、希釈剤、着香剤、溶解剤、潤滑剤、懸濁化剤、結合剤、保存剤、錠剤崩壊剤、または封入材料としても作用しうる1以上の物質であることができる。
【0102】
粉末剤の場合、担体は、微粉活性成分と混合された微粉固体である。錠剤の場合、活性成分は必要な結合能を持つ担体と適切な比率で混合され、所望の外形およびサイズに圧縮される。
【0103】
粉末剤および錠剤は、さまざまなパーセンテージ量の活性化合物を含有することができる。粉末剤および錠剤中の代表的な量は0.5〜約90%の活性化合物を含有しうるが、この範囲外の量がいつ必要になるかは、当業者にはわかるだろう。粉末剤用および錠剤用の適切な担体は、炭酸マグネシウム、ステアリン酸マグネシウム、タルク、糖、ラクトース、ペクチン、デキストリン、デンプン、ゼラチン、トラガカント、メチルセルロース、カルボキシメチルセルロースナトリウム、低融点ワックス、カカオ脂などである。「調製物」という用語は、カプセル剤を与える封入材料を担体とする活性化合物の製剤(このカプセル剤では、活性成分が、担体と共に、または担体を伴わずに、担体によって取り囲まれ、担体はそのようにして活性成分と関係している)を包含するものとする。同様に、カシェ剤および口中錠も包含される。錠剤、粉末剤、カプセル剤、丸剤、カシェ剤、および口中錠は、経口投与に適した固体剤形として使用することができる。
【0104】
坐剤を調製するには、低融点ワックス、例えば脂肪酸グリセリドまたはカカオ脂の混合物などを、まず融解し、そこに活性成分を撹拌するなどして均一に分散させる。次にその融解均一混合物を都合のよいサイズの鋳型に注ぎ込み、冷ますことによって固化させる。
【0105】
膣投与に適した製剤は、活性成分の他に当技術分野で適当であることが知られているような担体を含有する膣坐薬、タンポン、クリーム剤、ゲル剤、ペースト剤、フォーム剤またはスプレー剤として提示することができる。
【0106】
液体調製物には、溶液剤、懸濁剤、および乳剤、例えば水溶液または水−プロピレングリコール溶液が含まれる。例えば非経口注射液体調製物は、水性ポリエチレングリコール溶液中の溶液剤として製剤化することができる。注射可能調製物、例えば滅菌注射可能水性または油性懸濁剤は、適切な分散剤または湿潤剤および懸濁化剤を使用し、既知の技術に従って製剤化することができる。滅菌注射可能調製物は、無毒性の非経口的に許容できる希釈剤または溶媒中の滅菌注射可能溶液剤または懸濁剤、例えば1,3−ブタンジオール中の溶液剤であることもできる。使用することができる許容できるビヒクルおよび溶媒には、水、リンゲル液、および等張食塩溶液などがある。また、滅菌した固定油も、溶媒または分散媒として、便利に使用される。この目的には、合成モノ−またはジ−グリセリドを含む任意の無刺激性固定油を使用することができる。また、オレイン酸などの脂肪酸も、注射剤の調製に使用することができる。
【0107】
本発明の化合物はこのように非経口投与(例えばボーラス注射または持続注入などの注射による投与)用に製剤化することができ、アンプル、充填済注射器、少量注入容器に入れて1回量型として提示するか、または保存剤を添加した多用量型容器に入れて提示することができる。医薬組成物は、油性または水性ビヒクル中の懸濁剤、溶液剤、または乳剤などといった形態をとることができ、懸濁化剤、安定剤および/または分散剤などの調剤用薬剤を含有することができる。あるいは、活性成分は、滅菌固体の無菌的単離または溶液からの凍結乾燥によって得られる粉末であって、使用前に適切なビヒクル、例えば滅菌パイロジェンフリー水などで復元される形態であることもできる。
【0108】
経口使用に適した水性溶液剤は、活性成分を水に溶解し、希望に応じて、適切な着色剤、着香剤、安定剤および増粘剤を加えることによって調製することができる。
【0109】
経口使用に適した水性懸濁剤は、微粉活性成分を粘性材料、例えば天然ゴムもしくは合成ゴム、樹脂、メチルセルロース、カルボキシメチルセルロースナトリウム、または他の周知の懸濁化剤などと共に、水に分散させることによって作ることができる。
【0110】
使用直前に経口投与用の液状調製物に変換されることを意図した固形調製物も包含される。そのような液体剤形には、溶液剤、懸濁剤、および乳剤が含まれる。これらの調製物は、活性成分の他に、着色剤、着香剤、安定剤、緩衝剤、人工および天然甘味剤、分散剤、増粘剤、溶解剤などを含有することができる。
【0111】
表皮への局所投与用に、本発明の化合物を、軟膏、クリーム剤もしくはローション剤として、または経皮貼付剤として製剤化することができる。
【0112】
軟膏およびクリーム剤は、例えば、水性または油性基剤を使って、適切な増粘および/またはゲル化剤を添加して、製剤化することができる。ローション剤は水性または油性基剤を使って製剤化することができ、一般的には1以上の乳化剤、安定剤、分散剤、懸濁化剤、増粘剤、または着色剤も含有するだろう。
【0113】
口内の局所投与に適した製剤には、着香した基剤(通常はスクロースおよびアラビアゴムまたはトラガカント)中に活性剤を含む口中錠;ゼラチンおよびグリセリンまたはスクロースおよびアラビアゴムなどの不活性基剤中に活性成分を含むトローチ剤;ならびに適切な液状担体中に活性成分を含む洗口剤が含まれる。
【0114】
溶液剤または懸濁剤は、従来の手段により、例えば滴ビン、ピペットまたはスプレーを使って、鼻腔に直接適用される。製剤は単回量剤形または複数回量剤形で提供することができる。滴ビンまたはピペットで後者の場合、これは、適当な所定の体積の溶液剤または懸濁剤を患者が投与することによって達成することができる。スプレーの場合、これは、例えば計量式噴霧スプレーポンプを使って達成することができる。
【0115】
気道への投与は、エアロゾル製剤を使って達成することもでき、この製剤では、活性成分が適切な噴射剤と共に加圧容器に入れて提供される。本発明の方法によって同定される化合物またはそれらを含む医薬組成物をエアロゾル剤として、例えば鼻エアロゾル剤として、または吸入によって投与する場合、これは、例えばスプレー、ネブライザー、ポンプネブライザー、吸入装置、計量式吸入器または乾燥粉末吸入器を使って行なうことができる。本発明の方法によって同定される化合物をエアロゾルとして投与するための医薬剤形は、当業者に周知の方法によって調製することができる。それらの調製には、例えば、本発明の方法によって同定される化合物の、水、水/アルコール混合物または適切な食塩溶液中の溶液または分散液を、慣例の添加剤、例えばベンジルアルコールまたは他の適切な保存剤、生物学的利用能を増加させるための吸収促進剤、溶解剤、分散剤などを使って、そしてまた、それが適当な場合には、通例の噴射剤、例えば二酸化炭素、CFC類(例えばジクロロジフルオロメタン、トリクロロフルオロメタン、またはジクロロテトラフルオロエタンなど)を使って、使用することができる。エアロゾル剤はレシチンなどの界面活性剤も都合よく含有することができる。薬物の用量は計量式バルブを備えることによって制御することができる。
【0116】
気道への投与を意図する製剤では、鼻腔内製剤を含めて、化合物は一般に小さな粒径、例えば10ミクロン未満程度の粒径を持つだろう。そのような粒径は当技術分野では既知の手段によって、例えば微粒子化によって、得ることができる。所望であれば、活性成分の徐放が得られるように適合させた製剤を使用することができる。
【0117】
あるいは、活性成分は、乾燥粉末の形態で、例えば、ラクトース、デンプン、デンプン誘導体(ヒドロキシプロピルメチルセルロースなど)およびポリビニルピロリドン(PVP)などの適切な粉末基剤中の化合物の粉末混合物という形態で、提供することもできる。好都合なことに、粉末担体は、鼻腔内でゲルを形成するだろう。粉末組成物は、例えば、ゼラチン製のカプセルもしくはカートリッジまたはブリスター包装に入った、そこから吸入器を使って粉末を投与することができる、1回量型として提示することができる。
【0118】
医薬調製物は単位剤形をとることができる。そのような剤形では、調製物は、適当な量の活性成分を含有する1回量に細分される。単位剤形は、包装済みの錠剤、カプセル剤、およびバイアルまたはアンプル中の粉末剤など、包装が分離した調製物量を含有している包装済みの調製物であることができる。また、単位剤形はカプセル剤、錠剤、カシェ剤、または口中錠そのものであることができ、あるいは包装された形態にある、適当な数の、これらのいずれかであることができる。
【0119】
経口投与用の錠剤またはカプセル剤および静脈内投与用の液剤は、特に有用な組成物である。
【0120】
代謝関連障害には、例えば低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患が含まれる。
【0121】
低血糖は、異常に低い血中グルコースと定義される。低血糖は、例えば過剰なインスリンまたは貧しい食餌などに起因しうる。例えば低血糖は、糖尿病を持つ人がインスリンを多量に注射しすぎた場合、食物摂取量が少なすぎた場合、または余分な食物を摂らずに運動した場合などに起こりうる。低血糖の症状には、例えば、緊張感または脱力感、頭痛、かすみ目、空腹感、および過剰発汗などが含まれる。
【0122】
加齢は生物が年をとるにつれて起こる生理学的過程である。カロリー制限はインスリン分泌をダウンレギュレートし、これらの作用は、カロリー制限が長寿に及ぼすよい影響の重要な媒介因子ではないかと疑うに足りる理由がある。また、インスリン中またはインスリンシグナル伝達経路中の突然変異は、C.elegansにおける加齢に影響を及ぼす。したがって、インスリンをダウンレギュレートするための戦略は、老化の過程を遅らせ、寿命を延ばすのに役立ちうる。
【0123】
糖尿病、肥満、ならびにインスリン抵抗性、耐糖能異常および高血糖などの関連状態については上述した。一部の実施形態では、前記代謝関連障害に、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、X症候群および末梢血管疾患が含まれる。
【0124】
膵臓中のベータ細胞はインスリンを産生する。インスリンは体内で血液から細胞へのグルコースの取り込みを刺激する。上述のように、身体の細胞がインスリンの作用に対して抵抗性である場合、それをインスリン抵抗性と呼ぶ。インスリン抵抗性の結果として、膵臓は正常よりはるかに多くのインスリンを産生する。これを高インスリン血症という。
【0125】
異常脂質血症は、体内の脂質レベルの調節不全を意味する一般用語である。高脂質血症は、血流中の脂質(脂肪)の上昇である。これらの脂質には、コレステロール、コレステロールエステル(化合物)、リン脂質およびトリグリセリドが含まれる。それらは血中ではリポタンパク質とよばれる大分子の一部として輸送される。血中(血漿)リポタンパク質には5つの主要ファミリー、すなわちカイロミクロン、超低密度リポタンパク質(VLDL)、中間密度リポタンパク質(IDL)、低密度リポタンパク質(LDL)、高密度リポタンパク質(HDL)がある。高脂血症を、血中で上昇するリポタンパク質の1以上のクラスという観点から定義する場合は、高リポタンパク質血症という用語が用いられる。高コレステロール血症は、高い血中コレステロールレベルを表す用語である。高トリグリセリド血症は、高い血中トリグリセリドレベルを指す。
【0126】
アテローム性動脈硬化は、脂肪物質、コレステロールおよび他の物質の沈着が動脈の内層に蓄積する過程である。この蓄積をプラークをいう。破裂するプラークは血塊の形成を引き起こし、それが心臓(心臓発作)または脳(脳卒中)への血流を遮断しうる。心臓発作は、米国では男女とも第1位の死因であり、脳卒中は第3位の死因である[例えばNature Medicine,Special Focus on Atherosclerosis,(2002)8:1209−1262を参照されたい]。異常に高い循環脂質レベルは、アテローム性動脈硬化の発症における主要素因である。上昇した低密度リポタンパク質(LDL)コレステロールレベル、上昇したトリグリセリドレベル、または低い高密度リポタンパク質(HDL)コレステロールレベルは、独立して、アテローム性動脈硬化および関連病変のリスク因子である。
【0127】
心臓疾患には、心不全、冠不全、冠状動脈疾患、および高い血圧(高血圧)が含まれるが、これらに限定されるわけではない。末梢血管疾患とは、心臓外および脳外にある血管の疾患を指す。器質的末梢血管疾患は、炎症および組織損傷などといった血管の構造変化によって引き起こされる。末梢動脈疾患はその一例である。末梢動脈疾患(PAD)は冠状動脈疾患および頚動脈疾患に似た状態である。PADでは、主に脚および足に通じる動脈において、脂肪沈着が動脈壁に沿って蓄積し、血液循環に影響を及ぼす。その初期段階では、一般的な症状は、活動中に起こる脚部および臀部の痙攣または疲労である。そのような痙攣は、その人が動かずにじっとしている時にはおさまる。これを「間欠性跛行」という。PADを持つ人々は、血塊のリスクがあるため、脳卒中および心臓発作で死亡するリスクが高い。
【0128】
X症候群は、代謝症候群とも呼ばれ、一人における一群の代謝リスク因子を特徴とする。それらには、中心性肥満(腹部および腹部周辺の過剰な脂肪組織)、アテローム生成的異常脂質血症(血中脂肪障害−主に高トリグリセリドおよび低HDLコレステロール)、上昇した血圧(130/85mmHg以上)、インスリン抵抗性またはグルコース不耐性、易血栓性状態(例えば血中の高いフィブリノゲンまたはプラスミノゲン活性化因子阻害因子[−1])、および易炎症状態(例えば、血中の上昇した高感度C反応性タンパク質)が含まれる。
【0129】
高血圧は、血圧が異常に高い状態(140/90mmHg以上の読み)を保つ一般的な障害である。高血圧を処置することができる薬物はいくつか販売されている。高血圧は脳卒中を含むいくつかの深刻な状態のリスク因子である。
【0130】
末梢血管疾患は、心臓外の動脈における動脈硬化プラークの蓄積である。末梢血管疾患の症状は、どの動脈が冒されるか、および血流の低下がどの程度深刻であるかに依存する。例えば、鈍い痙攣痛、しびれもしくは刺痛、または皮膚の色の変化を起しうる。臨床試験により、糖尿病または喫煙など、末梢血管疾患のリスクを増加させる因子が同定されている。末梢血管疾患は、例えば、足関節上腕血圧比(ABI)検査、超音波ドップラー検査、血管造影図などを使って診断することができる。末梢血管疾患は、薬物療法、外科、低侵襲インターベンション術、またはこれらの治療法の組み合わせによって処置することができる。
【0131】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、1以上の薬剤と、例えば糖尿病、血中脂質障害、または肥満の処置に使用される薬剤などと、組み合わせて使用することもできる。例えば、GPR43インバースアゴニストまたはアンタゴニストなどの化合物は、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類、インスリン、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤、アンギオテンシン変換酵素(ACE)阻害剤、リパーゼ阻害剤、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤として知られている薬物クラスに属する1以上の薬剤と組み合わせて使用することができる。
【0132】
α−グルコシダーゼ阻害剤は、膵臓または小腸において、例えばα−アミラーゼ、マルターゼ、α−デキストリナーゼ、スクラーゼなどの消化酵素を競合的に阻害する薬物クラスに属する。α−グルコシダーゼ阻害剤による可逆的阻害は、デンプンおよび糖の消化を遅らせることにより、血中グルコースレベルを遅延、減少、または他の形で低下させる。α−グルコシダーゼ阻害剤の代表例をいくつか挙げると、アカルボース、N−(1,3−ジヒドロキシ−2−プロピル)バリオールアミン(一般名:ボグリボーズ)、ミグリトール、および当技術分野で知られるα−グルコシダーゼ阻害剤がある。
【0133】
アルドース還元酵素阻害剤クラスは、ポリオール経路の第1段階律速酵素を阻害し、それによって糖尿病合併症を予防または抑止する薬物である。糖尿病の高血糖状態では、ポリオール経路におけるグルコースの利用が増加し、その結果、細胞内に蓄積した過剰なソルビトールが組織毒として作用することにより、糖尿病性神経障害、網膜症、および腎症などの合併症の発症を惹起する。アルドース還元酵素阻害剤の例には、トルレスタット、エパルレスタット、3,4−ジヒドロ−2,8−ジイソプロピル−3−チオキソ−2H−1,4−ベンゾオキサジン−4−酢酸、2,7−ジフルオロスピロ(9H−フルオレン−9,4’−イミダゾリジン)−2’,5’−ジオン(一般名:イミレスタット)、3−[(4−ブロモ−2−フルオロフェニル)メチル]−7−クロロ−3,4−ジヒドロ−2,4−ジオキソ−1(2H)−キナゾリン酢酸(一般名:ゼナレスタット)、6−フルオロ−2,3−ジヒドロ−2’,5’−ジオキソ−スピロ[4H−1−ベンゾピラン−4,4’−イミダゾリジン]−2−カルボキサミド(SNK−860)、ゾポルレスタット、ソルビニル、および1−[(3−ブロモ−2−ベンゾフラニル)スルホニル]−2,4−イミダゾリジンジオン(M−16209)、ならびに当技術分野で知られるアルドース還元酵素阻害剤がある。
【0134】
ビグアニド類は、嫌気的解糖を刺激し、末梢組織におけるインスリンに対する感度を増加させ、腸からのグルコース吸収を阻害し、肝糖新生を抑制し、脂肪酸酸化を阻害する薬物クラスである。ビグアニド類の例には、フェンホルミン、メトホルミン、ブホルミン、および当技術分野で知られるビグアニド類がある。
【0135】
インスリン分泌促進剤は、膵ベータ細胞からのインスリンの分泌を促進する性質を持つ薬物クラスに属する。インスリン分泌促進剤の例にはスルホニル尿素類(SU)がある。スルホニル尿素類(SU)は、細胞膜中のSU受容体を介してインスリン分泌のシグナルを伝達することにより、膵ベータ細胞からのインスリンの分泌を促進する薬物である。スルホニル尿素類の例には、トルブタミド、クロルプロパミド、トラザミド、アセトヘキサミド、4−クロロ−N−[(1−ピロリジニルアミノ)カルボニル]−ベンゼンスルホンアミド(一般名:グリコピラミド)またはそのアンモニウム塩、グリベンクラミド(グリブリド)、グリクラジド、1−ブチル−3−メタニリル尿素、カルブタミド、グリボヌリド(glibonuride)、グリピジド、グリキドン、グリソキセピド、グリブチアゾール、グリブゾール、グリへキサミド、グリミジン、グリピナミド、フェンブタミド、トルシクラミド、グリメピリド、および当技術分野で知られる他のインスリン分泌促進剤がある。他のインスリン分泌促進剤には、N−[[4−(1−メチルエチル)シクロヘキシル)カルボニル]−D−フェニルアラニン(ナテグリニド)、(2S)−2−ベンジル−3−(cis−ヘキサヒドロ−2−イソインドリニルカルボニル)プロピオン酸カルシウム二水和物(ミチグリニド,KAD−1229)、および当技術分野で知られる他のインスリン分泌促進剤がある。
【0136】
チアゾリジンジオン類は、より一般的にはTZD類として知られている薬物クラスに属する。チアゾリジンジオン類は、インスリンに対する細胞の感受性を増加させることによって血糖を低下させる2型糖尿病のための薬物クラスである。その結果、インスリンは、エネルギーを得るためにグルコースを血液から細胞内へと移動させることができる。これらの薬物はHDLを増加させることもできる。
【0137】
チアゾリジンジオン類の例には、ロジグリタゾン、ピオグリタゾン、および当技術分野で知られるチアゾリジンジオン類がある。レズリン(トログリタゾン)は、米国ではこのクラスの最初の薬物だったが、肝毒性のために市場から撤収された。より良い安全性プロファイルを持つ現在利用可能な姉妹化合物には、アクトス(ピオグリタゾン)およびアバンディア(ロジグリタゾン)がある。これらの薬物療法の使用に対する主な禁忌には、肝疾患および心不全がある。これらの薬物は体液貯留の有意な増加を引き起こすことによって、心不全のリスクを増加させることもできる。
【0138】
メグリチニド類は、2型糖尿病を持つ人が食事をとった直後に起こりうる血糖の急速な上昇を止めるために用いられる。これらの化合物は、例えばレパグリニド(プランジン)およびナテグリニド(スターリックス)などを含めて、スルホニル尿素薬物療法が機能する方法と同様に、膵臓によって産生されるインスリンの量を増加させることによって機能する。メグリチニド類は食事をとる前に投与される。この薬物クラスに付随する副作用には、低血糖、鼻炎症状を含む上気道炎、頭痛、関節痛および背痛、悪心、下痢および便秘がある。
【0139】
異なるタイプのインスリンは、それらがどのくらい早く作用し始めるか(発現)、およびそれらがどのくらい長く作用し続けるか(持続時間)に応じて分類される。現在利用できるタイプには、速効型、短時間作用型、中時間作用型、および長時間作用型インスリンがある。70/30インスリンと呼ばれる70%中時間作用型(NPH)および30%短時間作用型レギュラーインスリン;50/50インスリンと呼ばれる50%中時間作用型(NPH)および50%短時間作用型レギュラーインスリン;75/25インスリンと呼ばれる75%中時間作用型(NPH)および25%速効型ヒューマログ(リスプロ);ノボログミックス(NovoLog Mix)70/30と呼ばれる70%中時間作用型(NPH)および30%速効型ノボログ(インスリンアスパルト)など、混合済みの速効型および中時間作用型インスリンを利用することができる。インスリンは通常、皮下組織への注射として与えられる(皮下)。インスリンポンプまたはジェットインジェクター(皮膚内に医薬品を噴霧する装置)によって与えることもできる。
【0140】
インスリンは糖(グルコース)を細胞に入らせ、そこで糖はエネルギーを得るために使用される。インスリンがないと、血糖レベルは身体にとって安全であるレベルを上回ってしまう。通常は、身体が必要とする一定レベルおよび可変レベルのインスリンを提供するために、速効型または短時間作用型インスリンおよび中時間作用型または長時間作用型インスリンが投与される。短時間作用型インスリンは血糖レベルを急速に低下させてから消失する。一部の長時間作用型インスリンは速効型または短時間作用型インスリンが消失し始めた時に効き始める。新しい長時間作用型インスリン・ランタスは、投与されてから数分以内に作用し始め、約24時間は同じ速度で作用し続ける。
【0141】
速効型または短時間作用型インスリンと中時間作用型または長時間作用型インスリンとの併用は、血糖レベルを、身体にとって安全な範囲内に、終日維持するのに役立つ。したがって、インスリンは、1型糖尿病を持つ人々、2型糖尿病を持つ人々であってその膵臓がインスリンをほとんどまたは全く産生しないか、経口薬物療法ではその血糖が管理されない人々を処置するために使用することができる。これらの人々にはインスリンを単独で、または経口薬物療法と一緒に投与することができる。2型糖尿病を持つ人々であって、重病または大手術のために血糖レベルが高い人々、2型糖尿病を持つ婦人であって、妊娠中であるか授乳中であり、その血糖レベルを食餌と運動で安全域内に保つことができない人々。妊娠中の使用について調べられた経口糖尿病薬物療法は1つだけである(グリブリド)。
【0142】
インスリンの主要副作用としては、危険なほど低い血糖レベル(重症低血糖)を挙げることができる。極めて低い血糖レベルが10〜15分以内に発生しうる。インスリンは、既に過体重である2型糖尿病を持つ人々では特に、体重増加の一因となりうる。長期インスリン使用の他の考えうる副作用には、インスリンが注射される部分の脂肪組織の喪失(リポジストロフィ)、そして稀に、腫脹(浮腫)を含むアレルギー反応がある。
【0143】
スタチン化合物は、ヒドロキシメチルグルタリルCoA(HMG−CoA)還元酵素を阻害することによって血中コレステロールレベルを低下させる薬物クラスに属する。HMG−CoA還元酵素はコレステロール生合成における律速酵素である。この還元酵素を阻害するスタチンは、血液からのLDLの浄化を担うLDL受容体の活性をアップレギュレートすることにより、血清LDL濃度を低下させる。スタチン化合物の例には、ロスバスタチン、プラバスタチンおよびそのナトリウム塩、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチン、セリバスタチン、および当技術分野で知られるHMG−CoA還元酵素阻害剤がある。
【0144】
スクアレン合成阻害剤は、スクアレンの合成を阻害することによって血中コレステロールレベルを低下させる薬物クラスに属する。スクアレン合成阻害剤の例には、(S)−α−[ビス[2,2−ジメチル−1−オキソプロポキシ)メトキシ]ホスフィニル]−3−フェノキシベンゼンブタンスルホン酸・一カリウム塩(BMS−188494)および当技術分野で知られるスクアレン合成阻害剤がある。
【0145】
フィブラート化合物は、肝臓におけるトリグリセリドの合成および分泌を阻害し、リポタンパク質リパーゼを活性化することによって、血中コレステロールレベルを低下させる薬物クラスに属する。フィブラート類は、ペルオキシソーム増殖因子活性化受容体を活性化し、リポタンパク質リパーゼ発現を誘導することが知られている。フィブラート化合物の例には、ベザフィブラート、ベクロブラート、ビニフィブラート、シプロフィブラート、クリノフィブラート、クロフィブラート、クロフィブリン酸、エトフィブラート、フェノフィブラート、ゲムフィブロジル、ニコフィブラート、ピリフィブラート、ロニフィブラート、シンフィブラート、テオフィブラート(theofibrate)、および当技術分野で知られるフィブラート類がある。
【0146】
LDL(低密度リポタンパク質)異化促進剤は、LDL(低密度リポタンパク質)受容体の数を増加させることによって血中コレステロールレベルを低下させる薬物クラスに属し、当技術分野で知られるLDL異化促進剤がその例に含まれる。
【0147】
アンギオテンシン変換酵素(ACE)阻害剤は、アンギオテンシン変換酵素を阻害することによって、血圧を低下させると共に、血中グルコースレベルもある程度は低下させる薬物クラスに属する。アンギオテンシン変換酵素阻害剤の例には、カプトプリル、エナラプリル、アラセプリル、デラプリル;ラミプリル、リシノプリル、イミダプリル、ベナゼプリル、セロナプリル、シラザプリル、エナラプリラート、フォシノプリル、モベルトプリル(moveltopril)、ペリンドプリル、キナプリル、スピラプリル、テモカプリル、トランドラプリル、および当技術分野で知られるアンギオテンシン変換酵素阻害剤がある。
【0148】
リパーゼ阻害剤には、例えば、オルリスタット(XENICAL(商標))などの抗肥満化合物が含まれる。オルリスタットは、脂肪吸収を直接阻害するが、下痢および鼓腸などの不快な胃副作用も高い発生率で生じる傾向がある。
【0149】
抗肥満薬のもう1つのクラスには、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤がある。例えばシブトラミン(Meridia(商標))は混合性5−HT/ノルアドレナリン再取り込み阻害剤である。シブトラミンの主な副作用として、一部の患者における血圧の増加および心拍数の増加を挙げることができる。セロトニン放出薬/再取り込み阻害剤フェンフルラミン(Pondimin(商標))およびデキスフェンフルラミン(Redux(商標))は、長期間(6ヶ月超)にわたって食物摂取量および体重を減少させると報告されている。しかしどちらも製品も、その使用に関連する心臓弁異常を示す予備的証拠が報告された後は、使用が中止されている。
【0150】
本発明の一部の実施形態は、本明細書に開示する化合物もしくは本発明の方法によって同定される化合物または医薬的に許容できるその塩を、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤およびアンギオテンシン変換酵素阻害剤からなる群より選択される少なくとも1つのメンバーと組み合わせて含む医薬組成物を包含する。もう1つの実施形態では、HMG−CoA還元酵素阻害剤が、プレバスタチン(prevastatin)、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンおよびリピトールからなる群より選択される。
【0151】
本発明によれば、各活性成分を全て一緒にして、または個別に、上述の生理学的に許容できる担体、賦形剤、結合剤、希釈剤などと混合し、その混合物または混合物群を医薬組成物として経口投与または非経口投与することにより、これらの組み合わせを使用することができる。化合物または化合物の混合物を、別の活性化合物と共に、併用治療または併用予防として投与する場合、それらの治療剤は、同じ時点または異なる時点で与えられる別個の医薬組成物として製剤化するか、それらの治療剤を単一の組成物として与えることができる。
【0152】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法も提供する。
【0153】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が調節されるかどうかを決定する方法(この場合、GPR43機能の調節は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法を提供する。
【0154】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が増加するかどうかを決定することを含み、GPR43機能の増加が、その候補化合物が代謝安定化化合物であることを示す方法に関する。
【0155】
ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0156】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知のアゴニストまたは本明細書に開示するアゴニストの存在下で行なわれる。
【0157】
特定の実施形態では、前記方法が、候補化合物によって引き起こされる受容体機能の増加を、受容体を受容体の既知リガンドまたは既知アゴニストと接触させることによって引き起こされる受容体機能の第2の増加と比較することを、さらに含む。
【0158】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびCa2+からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が、全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が、細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定が、CRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、細胞内Ca2+の測定によって行なわれる。
【0159】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0160】
特定の実施形態では、前記決定が、黒色素胞アッセイの使用によって行なわれる。
【0161】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRのアゴニストの存在下で行なわれる。
【0162】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0163】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0164】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0165】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0166】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0167】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法であって、GPR43機能の増加が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物に関する。
【0168】
ある実施形態では、前記代謝安定化化合物がGPR43アゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0169】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つGPR43アゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。
【0170】
特定の実施形態では、前記EC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のEC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のEC50を持つアゴニストである。一部の実施形態では、前記代謝安定化化合物が、上記GPCRに対して選択的である。
【0171】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0172】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法に関する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法に関する。
【0173】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物にも関係する。
【0174】
本発明の一部の実施形態は、本明細書に開示する化合物実施形態のいずれかに従う少なくとも1つの化合物と医薬的に許容できる担体とを混合することを含む医薬組成物の生産方法を包含する。
【0175】
化合物は、当業者に周知の技法および本明細書に記載する技法を使って、医薬組成物に製剤化することができる。
【0176】
予防または処置に使用する場合、本明細書に開示する化合物または本発明の方法によって同定される化合物は、代替的に、原薬または純薬として投与することができると考えられるが、その化合物または活性成分を、医薬的に許容できる担体をさらに含む医薬製剤として提供することが有用な場合もある。
【0177】
したがって本発明は、本明細書に開示する化合物もしくは本発明の方法によって同定される化合物または医薬的に許容できるその塩もしくは誘導体を、1以上の医薬的に許容できるその担体および/または予防成分と共に含む医薬製剤を、さらに提供する。担体は、製剤の他の成分と適合し、その受容者にとって過度に有害でないという意味で、「許容できる」。
【0178】
医薬製剤、投与経路、および投与量については上述した。
【0179】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害が低血糖または加齢である。ある実施形態では、投与される化合物が、GRP43アゴニストを含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0180】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、それらを1以上の薬剤と組み合わせて使用することもできる。
【0181】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法にも関係する。
【0182】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が増加するかどうかを決定する方法(この場合、GPR43機能の増加は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法にも関係する。
【0183】
本発明は、代謝安定化化合物を同定する方法であって、a)候補化合物をGPR43と接触させること、およびb)GPR43機能が減少するかどうかを決定することを含み、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法を提供する。
【0184】
一部の実施形態では、前記代謝安定化化合物が血中グルコース安定化化合物である。一部の実施形態では、前記代謝安定化化合物がインスリン分泌調節因子である。
【0185】
ある実施形態では、前記GPR43が哺乳動物に由来する。もう1つの実施形態では、前記GPR43がヒト由来である。
【0186】
特定の実施形態では、前記GPR43が組換え体である。特定の実施形態では、前記接触が、当該GPCRを発現させる宿主細胞と接触させること、または当該GPCRを発現させる宿主細胞の膜と接触させることを含み、宿主細胞が当該受容体をコードするポリヌクレオチドを含む発現ベクターを含む。一部の実施形態では、前記接触が、当該GPCRの既知アゴニストの存在下で行なわれる。
【0187】
一部の実施形態では、前記決定が二次メッセンジャーアッセイを含み、例えば決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。特定の実施形態では、前記GTPγSが[35S]で標識される。特定の実施形態では、前記決定が、サイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ活性、およびCa2+からなる群より選択される二次メッセンジャーのレベルの測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがcAMPである。特定の実施形態では、前記cAMPの測定が、全細胞アデニリルシクラーゼアッセイを使って行なわれる。特定の実施形態では、前記cAMPの測定が、前記GPCRを含む膜を使って行なわれる。特定の実施形態では、前記決定が、細胞内IP3の測定によって行なわれる。特定の実施形態では、前記二次メッセンジャーがMAPキナーゼ活性である。一部の実施形態では、前記決定が、CRE−レポーターアッセイによって行なわれる。特定の実施形態では、前記レポーターがルシフェラーゼである。一部の実施形態では、前記レポーターがβ−ガラクトシダーゼである。特定の実施形態では、前記決定または前記比較が、細胞内Ca2+の測定によって、例えばFLIPRアッセイを使って行なわれる。
【0188】
一部の実施形態では、前記決定が、哺乳動物から得られる脂肪細胞によるグルコース取り込みの測定によって行なわれる。
【0189】
特定の実施形態では、前記決定が、黒色素胞アッセイの使用によって行なわれる。
【0190】
本発明の方法では、応答の特異性を示すために、対照反応を行なうことができる。例えば、モックトランスフェクト細胞を、GPR43トランスフェクト細胞と比較して、GPR43受容体に対する応答の特異性を示すことができる。
【0191】
本発明の方法では、特定の実施形態において、前記候補化合物が抗体ではなく、その抗原結合性誘導体でもない。特定の実施形態では、前記候補化合物がペプチドではない。特定の実施形態では、前記候補化合物がポリペプチドではない。
【0192】
本発明の方法において、決定は、二次メッセンジャーアッセイを含むことができる。細胞内シグナルの開始は、例えばサイクリックAMP(cAMP)、サイクリックGMP(cGMP)、イノシトール三リン酸(IP3)、ジアシルグリセロール(DAG)、MAPキナーゼ、またはカルシウムなどの二次メッセンジャーのレベルの測定によって決定することができる。当技術分野では、これらの二次メッセンジャーを測定するために、例えばcAMPアッセイ、IP3アッセイ、FLIPRアッセイ、黒色素胞アッセイ、またはCRE−レポーターアッセイなど、いくつかのアッセイがよく知られている。また、二次メッセンジャーアッセイの例を、本明細書の実施例6〜11にも開示する。特定の実施形態では、前記二次メッセンジャーがcAMPである。別の実施形態では、前記二次メッセンジャーがIP3である。さらなる実施形態では、前記二次メッセンジャーがカルシウムである。
【0193】
ある実施形態では、前記決定が、前記GPCRを含む膜へのGTPγS結合の測定によって行なわれる。そのようなアッセイは当技術分野ではよく知られており、本明細書では実施例6および8に例示する。特定の実施形態では、前記GTPγSが[35S]で標識される。
【0194】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができる代謝安定化化合物にも関係する。
【0195】
例えば本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法であって、GPR43機能の減少が、その候補化合物が代謝安定化化合物であることを示す方法によって同定される代謝安定化化合物を提供する。
【0196】
ある実施形態では、前記代謝安定化化合物がGPR43インバースアゴニストである。ある実施形態では、前記代謝安定化化合物がGPR43アンタゴニストである。
【0197】
一部の実施形態では、前記代謝安定化化合物が、10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つGPR43インバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。
【0198】
特定の実施形態では、前記IC50が、以下のアッセイからなる群より選択されるアッセイを使って決定される:組換えGPR43ポリペプチドを発現させるトランスフェクトHEK293細胞を使って行なわれるIP3アッセイ;および組換えGPR43ポリペプチドを発現させるトランスフェクト黒色素胞を使って行なわれる黒色素胞アッセイ。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満、1μM未満、100nM未満、または10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて9μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて8μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて7μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて6μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて5μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて4μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて3μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて2μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて1μM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて900nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて800nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて700nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて600nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて500nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて400nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて300nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて200nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて100nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて90nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて80nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて70nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて60nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて50nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて40nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて30nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて20nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて10nM未満のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜1μMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜100nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、前記アッセイにおいて、1nM〜10nMの区間から選択される値のIC50を持つインバースアゴニストまたはアンタゴニストである。一部の実施形態では、前記代謝安定化化合物が、当該GPCRに対して選択的である。
【0199】
一部の実施形態では、前記代謝安定化化合物が経口生物学的利用能を持つ。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも1%、少なくとも5%、少なくとも10%、少なくとも15%、少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能が、腹腔内投与と比較して少なくとも20%、少なくとも25%、少なくとも30%、少なくとも35%、少なくとも40%、または少なくとも45%である。一部の実施形態では、前記経口生物学的利用能を持つ代謝安定化化合物がさらに、血液脳関門を横切ることもできる。
【0200】
また本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定することができるものである方法に関する。例えば本発明は、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含む、組成物の調製方法であって、前記化合物が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される方法を提供する。
【0201】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる、医薬組成物も提供する。
【0202】
本発明の一部の実施形態は、本明細書に開示する化合物実施形態のいずれかに従う少なくとも1つの化合物と医薬的に許容できる担体とを混合することを含む医薬組成物の生産方法を包含する。
【0203】
医薬製剤、投与経路、および投与量については上述した。
【0204】
本発明は、処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される化合物の有効量を、前記個体に投与することを含む方法を提供する。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害が高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、投与される化合物が、GRP43インバースアゴニストまたはアンタゴニストを含む。
【0205】
ある実施形態では、本方法が、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。例えば、ある実施形態では、本方法が、GPR43インバースアゴニストを含有する医薬組成物の有効量と組み合わせて、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、前記個体に投与することをさらに含む。
【0206】
ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0207】
本発明の方法によって同定される化合物は、上述のように、唯一の活性医薬剤として投与することができるが、それらを1以上の薬剤と、例えば糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤などと、組み合わせて使用することもできる。例えば、GPR43インバースアゴニストまたはアンタゴニストなどの化合物は、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド類、チアゾリジンジオン類、メグリチニド類、スルホニル尿素類、インスリン、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤、アンギオテンシン変換酵素(ACE)阻害剤、リパーゼ阻害剤、セロトニンおよび/またはノルアドレナリン放出薬または再取り込み阻害剤として知られている薬物クラスに属する1以上の薬剤と組み合わせて使用することができる。
【0208】
本発明の一部の実施形態は、本発明の方法によって同定される化合物または医薬的に許容できるその塩を、α−グルコシダーゼ阻害剤、アルドース還元酵素阻害剤、ビグアニド、HMG−CoA還元酵素阻害剤、スクアレン合成阻害剤、フィブラート化合物、LDL異化促進剤およびアンギオテンシン変換酵素阻害剤からなる群より選択される少なくとも1つのメンバーと組み合わせて含む医薬組成物を包含する。もう1つの実施形態では、HMG−CoA還元酵素阻害剤が、プレバスタチン、シンバスタチン、ロバスタチン、アトルバスタチン、フルバスタチンおよびリピトールからなる群より選択される。
【0209】
本発明によれば、各活性成分を全て一緒にして、または個別に、上述の生理学的に許容できる担体、賦形剤、結合剤、希釈剤などと混合し、その混合物または混合物群を医薬組成物として経口投与または非経口投与することにより、これらの組み合わせを使用することができる。化合物または化合物の混合物を、別の活性化合物と共に、併用治療または併用予防として投与する場合、それらの治療剤は、同じ時点または異なる時点で与えられる別個の医薬組成物として製剤化するか、それらの治療剤を単一の組成物として与えることができる。
【0210】
本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝安定化化合物として使用するための医薬を製造する方法も提供する。
【0211】
さらに本発明は、a)候補化合物をGPR43と接触させ、b)GPR43機能が減少するかどうかを決定する方法(この場合、GPR43機能の減少は、その候補化合物が代謝安定化化合物であることを示す)によって同定される代謝安定化化合物を含むか、または本質的に前記代謝安定化化合物からなるか、または前記代謝安定化化合物からなる医薬組成物を含む、代謝関連疾患の処置に使用するための医薬を製造する方法を提供する。
【0212】
本発明は、GPR43機能を増加させる方法であって、GPR43をGPR43アゴニストの有効量と接触させることを含む方法にも関係する。本発明は、細胞中のGPR43機能を増加させる方法であって、GPR43を発現させる細胞をGPR43アゴニストの有効量と接触させることを含む方法にも関係する。細胞は例えばある個体の中にあってもよいし、細胞は単離された細胞であってもよい。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0213】
本発明は、GPR43機能を減少させる方法であって、GPR43をGPR43インバースアゴニストまたはアンタゴニストの有効量と接触させることを含む方法にも関係する。本発明は、細胞中のGPR43機能を減少させる方法であって、GPR43を発現させる細胞をGPR43インバースアゴニストまたはアンタゴニストの有効量と接触させることを含む方法にも関係する。細胞は例えばある個体の中にあってもよいし、細胞は単離された細胞であってもよい。
【0214】
本発明は、代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法を提供する。
【0215】
ある実施形態では、前記代謝関連障害が低血糖または加齢である。ある実施形態では、前記調節因子がアゴニストである。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0216】
ある実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。一部の実施形態では、前記代謝関連障害に、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患が含まれる。ある実施形態では、前記調節因子が、インバースアゴニストまたはアンタゴニストである。ある実施形態では、前記方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。ある実施形態では、前記代謝関連障害が、インスリン抵抗性、耐糖能異常または糖尿病である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0217】
本発明は、GPR43機能を増加させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43アゴニストの有効量を投与することを含む方法に関する。ある実施形態では、前記障害が代謝関連障害である。一部の実施形態では、前記代謝関連障害が低血糖または加齢である。一部の実施形態において、前記アゴニストは、それがGPR43受容体に結合した時に細胞内応答を活性化することが今まで知られていなかった物質である。
【0218】
本発明は、GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法を提供する。ある実施形態では、前記障害が代謝関連障害である。ある実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である。一部の実施形態では、前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である。ある実施形態では、前記代謝関連障害がII型糖尿病である。ある実施形態では、前記方法が、糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することを、さらに含む。ある実施形態では、個体が哺乳動物であり、もう1つの実施形態では、個体がヒトである。
【0219】
本発明は、その必要がある個体の血中グルコースレベルを増加させる方法であって、その個体にGPR43アゴニストの有効量を投与することを含む方法にも関係する。本発明は、その必要がある個体の血中グルコースレベルを減少させる方法であって、その個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法にも関係する。
【0220】
また本発明は、その必要がある個体におけるインスリン分泌を減少させる方法であって、その個体にGPR43アゴニストの有効量を投与することを含む方法に関する。
【0221】
本発明の目的の1つは、a)代謝安定化化合物を同定するために本発明の方法を実行し、(b)要すれば、その化合物の構造を決定し、(c)その化合物またはその化合物の名称もしくは構造を提供する方法に関する。また本発明は、a)代謝安定化化合物を同定するために本発明の方法を実行し、(b)要すれば、その化合物の構造を決定し、(c)要すれば、その化合物の名称または構造を提供し、(d)その化合物を生産または合成する方法に関する。さらに本発明は、GPCRの機能を調節する方法であって、代謝安定化化合物を同定するために本発明の方法を実行し、次に当該GPCRをその代謝安定化化合物と接触させるか、その代謝安定化化合物を、当該GPCRの機能を調節するのに十分な条件で個体に投与することを含む方法に関する。
【0222】
本出願人は、本発明の実施形態のいずれかから、任意の1以上の候補化合物を除外する権利を留保する。本出願人は、本発明の実施形態のいずれかから、任意の1以上の調節因子を除外する権利も留保する。さらに本出願人は、本発明の実施形態のいずれかから任意のポリヌクレオチドまたはポリペプチドを除外する権利を留保する。本出願人は、本発明の実施形態のいずれかから任意の代謝関連障害を除外する権利も留保する。
【0223】
ここに開示する受容体および方法の他の用途は、とりわけ、この特許文書を再検討することにより、当業者には明らかになるだろう。
【0224】
以下の実施例は本発明を例示するために記載するものであり、決して包括的なものではない。
【実施例】
【0225】
本発明をさらに詳しく記述するために実施例を挙げるが、これらの実施例の詳細に本発明が限定されるわけではない。
【0226】
(実施例1)
マウス成体およびマウス胎児の組織および細胞におけるマウスGPR43発現のAffymetrixチップ解析
この実施例では、Affymetrix遺伝子チップを使って、いくつかのマウス成体およびマウス胎児の組織および細胞で、マウスGPR43の発現レベルを決定した(左から右に:胎児脳、橋脊髄、脊髄下部、SN橋視床、嗅球、視床、海馬、スイス3T3、3T3−LI脂肪細胞、BV2+LPS 24時間、NIH−3T3、3T3−LI前脂肪細胞、NIT−1、NIE−115分化型、E14TG2A、BV2、NIE−115、BV2+LPS 4時間、NIT CTL、C57BL6 ES、D3 ES、リンパ節、骨髄、T細胞、CD4+ 卵白アルブミン、脾臓、T細胞、CD4+ ナイーブ、胸腺、十二指腸、皮膚脂肪、褐色脂肪、精巣上体脂肪、心室、心房、大動脈、線維芽細胞、新生仔心臓、心室筋細胞、低酸素−再酸素化、新生仔、心室筋細胞、正常酸素圧、新生仔、心室筋細胞、心室筋細胞、低酸素、新生仔、心室、左TAC、心室、左シャム、大腸近位部、胃フォンデュ(fondues)、小腸、大腸遠位部、胃洞、白血球、肝臓、肺、気管、唾液腺、骨、線維芽細胞、真皮、副腎、骨格筋、皮膚、膀胱、皮膚、口、口腔粘膜、口、表皮、食道、腎臓、胆嚢、β島、ob/ob 16週、β島、ob/ob 6週、MIN6、βTC6、β島、C57Bl/6、αTC9、β島、db/db、子宮、臍帯、卵巣)。
【0227】
1.AFFYMETRIX GENECHIP(登録商標)技術
いくつかのGタンパク質共役受容体(GPCR)に対応するヌクレオチド配列をAffymetrixに提出した。Affymetrixでは、そのGeneChip(登録商標)技術によって、さまざまな組織におけるこれらの受容体のmRNA発現レベルを測定するために、オリゴヌクレオチドマイクロアレイが設計され、製造された。製造者の指示に従って、多数の組織および細胞タイプから得たRNA試料を増幅し、標識し、そのマイクロアレイにハイブリダイズさせ、データ解析した。
【0228】
(製造者の指示に基づき従って)発現指数(expression index)が50より大きければ、GPCRは発現していると決定した。データは解析され、50より大きい発現指数によるGPCRの分類が合理的であることを示していた。なぜなら、いくつかの既知GPCRは、ニューロン組織において、50より大きい発現指数で発現すると、先に報告されていたからである。
【0229】
GeneChip(登録商標)を使って、本出願人は、GPR43が脂肪、大腸、膵島および島細胞株ならびに3T3脂肪細胞に高い発現レベルを持つことを見いだした(図1参照)。
【0230】
(実施例2)
ヒトおよびマウスの組織および細胞におけるGPR43発現のRT−PCR解析
この実施例では、RT−PCRアッセイを使って、いくつかのヒトおよびマウスの組織および細胞タイプにおけるヒトおよびマウスGPR43の発現レベルを決定した。図2上図に示すように、例えば胎盤、肺、肝臓、腎臓、膵臓、脾臓、前立腺および白血球を含むいくつかのヒト組織および細胞で、ヒトGPR43遺伝子発現が観察された。また、図2下図に示すように、例えば肺、膵臓、骨格筋、小腸、脾臓、胃、島、3T3−L1分化脂肪細胞、Nit−1細胞、Min6細胞およびβTC−6細胞を含むいくつかのマウス組織、細胞および細胞株で、マウスGPR43遺伝子発現が観察された。
【0231】
この実験のために、ヒトcDNAをHuman MTC Panel IおよびHuman MTC Panel II(Clontech)から得た。マウスポリA+RNAをClontechから得た。次に、iScript cDNA Synthesis Kit(Bio−Rad)を使って、製造者のプロトコールに従って、cDNAを合成した。反応のために、1μlの1:10希釈マウスポリA+、8μlの5×iScript Reaction Mixおよび2μlのiScript Reverse Transcriptaseを水と共に合わせて、総液量を40μlにした。反応をPCR機中、25℃、5分;42℃、30分:85℃、5分で行なった。
【0232】
細胞株RNAおよびマウス島RNAはトリゾールを使って単離し、さらにDNAフリーキット(Ambion)を使って、製造者のプロトコールに従って、DNアーゼで処理した。iScript cDNA Synthesis Kit(Bio−Rad)を使って、2μgのDNアーゼ処理RNAを、40μlの総反応液量で使用した。
【0233】
ヒトおよびマウスGPR43のためのPCR反応を、InvitorgenのPlatinum PCR Supermixを使って行なった。各反応につき、2μlのcDNA、48μlのSupermix、0.2μlの各プライマー(100μMストック)を合わせて、液量を合計50μlにした。ヒトGPR43のための反応は、PCR機中、95℃で4分間の変性後、95℃、1分;60℃、30秒;72℃、1分を30サイクル行い、最後に72℃で7分間の伸長で終えた。マウスGPR35のために使用したアニーリング温度は63℃である。
【0234】
Clontechにより、Human MTC Panel IおよびIIに、G3PDH 5’および3’PCRプライマーが、対照として用意された。G3PDH PCR反応のために、2μlのcDNA、45μlのPlatinum PCR Supermix(Invitrogen)、1μlの各プライマー(10μMストック)を合わせて、液量を合計50μlにした。反応はPCR機中、95℃で1分間の変性後、95℃、30秒;68℃、3分を23サイクル行い、最後に68℃で3分間の伸長で終えた。
ヒトGPR43 RT−PCRプライマー対:
【0235】
【化1】
マウスGPR43 RT−PCRプライマー対:
【0236】
【化2】
(実施例3)
GPR43調節因子の同定
この実施例では、黒色素胞でのスクリーニングプロトコールを使って、GPR43調節因子を同定する。
【0237】
1.黒色素胞技術
黒色素胞は下等脊椎動物に見出される皮膚細胞である。それらはメラノソームと呼ばれる色素性細胞小器官を含有する。黒色素胞は、Gタンパク質共役受容体(GPCR)活性化時に、これらのメラノソームを微小管ネットワークに沿って再分配することができる。この色素移動の結果は、細胞の明白な明化または暗化である。黒色素胞では、Gi共役受容体の活性化に起因する細胞内cAMPレベルの減少が、メラノソームを細胞の中心に移動させ、その結果、色の劇的な明化が起こる。次に、Gs共役受容体の活性化に続いてcAMPレベルが上昇すると、メラノソームは再び分散し、細胞は再び暗く見えるようになる。Gq共役受容体の活性化に起因するジアシルグリセロールレベルの増加も、この再分散を誘発することができる。また、この技術は、一定の受容体チロシンキナーゼの研究にも適している。黒色素胞の応答は受容体活性化から数分以内に起こり、単純で強い色変化をもたらす。この応答は、従来の吸光度マイクロプレートリーダーまたは簡単な撮像システムを使って、容易に検出することができる。他の皮膚細胞とは異なり、黒色素胞は神経堤に由来し、シグナル伝達タンパク質の全てを発現させるようである。特に、これらの細胞は、極めて広い範囲のGタンパク質を発現させるので、ほとんど全てのGPCRを機能的に発現させることができる。
【0238】
黒色素胞は、GPCRに結合し、そして/またはGPCRを活性化する化合物(天然リガンドを含む)を同定するために利用することができる。この方法は、特異的刺激に応答してその色素を分散または凝集させることおよびGPCRをコードする外来クローンを発現させることができる色素細胞株の試験細胞を導入することによって行なうことができる。色素沈着の初期状態は、例えばメラトニン、MSHまたは光などを使って設定することができる。次に試験細胞を化学化合物と接触させ、細胞における色素沈着が色素沈着の初期状態から変化したかどうかを決定する。候補化合物(リガンドを含むが、これに限定されるわけではない)がGPCRに結合したことによる色素細胞の分散はペトリ皿上で暗く見え、一方、色素細胞の凝集は明るく見えるだろう。
【0239】
材料および方法は米国特許第5,462,856号および米国特許第6,051,386号の開示に従った。これらの特許明細書は参照によりその全体が本明細書に組み入れられる。
【0240】
マウスまたはヒトGPR43のコード配列を含有するプラスミドを、エレクトロポレーションによって、黒色素胞にトランスフェクトする。細胞を96ウェルプレートにプレーティングする。トランスフェクションの48時間後に、各プレート上の細胞の半分を10nMメラトニンで処理する。メラトニンは黒色素胞中の内在性Gi共役受容体を活性化し、黒色素胞がその色素を凝集させるようにする。細胞の残り半分を無血清培地0.7×L−15(Gibco)に移す。1時間後に、無血清培地中の細胞が色素分散状態にあるのに対して、メラトニン処理細胞は色素凝集状態にある。この時点で、細胞を、140,000〜150,000の有機小分子化合物を含有する専有化合物ライブラリーからのさまざまな化合物で処理する。GPR43がその化合物に結合すれば、黒色素胞は、その化合物に応答して、例えば色素凝集による色変化を起こすと予想される。
【0241】
(実施例4)
経口ブドウ糖負荷試験
アゴニスト、アンタゴニストまたはインバースアゴニストなどのGPR43調節因子を、経口グルコース投与後の血漿グルコースに対するその作用について試験することができる。
【0242】
例えば、67日齢の雄C57bl/6マウスを18時間絶食させ、無作為にグループ分けして、選択した用量でGPR43調節因子を与えるか、ビヒクル(80%PEG、10%Tween80、および10%エタノールを含有するPET)を与える。GPR43調節因子は強制栄養針(gavaga needle)を通して経口的に送達する(経口,液量100μl)。GPR43調節因子投与またはビヒクル投与の30分後に、3g/kgの用量でデキストロースをマウスに経口投与する。血中グルコースのレベルを、Glucometer Elite XL(Bayer)を使って、数時点で決定する。
【0243】
グルコース耐性は、グルコースの腹腔内送達を使って試験することもできる。例えば、68日齢の雄C57Bl/6マウスを、18時間の絶食後に、100mg/kgのGPR43調節因子またはPETビヒクルで処置する。GPR43調節因子投与またはビヒクル投与の30分後に、2g/kgの用量でマウスにデキストロースを腹腔内投与する。血中グルコースのレベルを、Glucometer Elite XL(Bayer)を使って、選択した時点で決定する。
【0244】
(実施例5)
3T3−L1脂肪細胞におけるインスリン刺激によるグルコース取り込み
この実施例では、脂肪細胞におけるインスリン刺激によるグルコース取り込みにGPR43調節因子が及ぼす作用を、3H−2−デオキシグルコース取り込みアッセイを使って試験する。
【0245】
簡単に述べると、標準的プロトコールを使って、まず、3T3−L1細胞を脂肪細胞に分化させる(PatelおよびLane,Proc.Natl.Acad.Sci.U.S.A.96:1279−1284(1999))。次に、ビヒクルまたは5μMのGPR43調節因子を含有する無血清培地で、細胞を3時間刺激する。次に、細胞をクレブスリンゲルリン酸緩衝液中で2回洗浄し、クレブスリンゲルリン酸緩衝液中、10nMインスリンの存在下または不在下で、20分間インキュベートする。0.05mM(0.5μCi/mol)3H−2−デオキシグルコースおよび0.05mM非放射性2−デオキシグルコースを細胞に37℃で5分間添加することにより、2−デオキシグルコース輸送を測定する。輸送反応を停止させるために、細胞を氷冷PBSで3回洗浄し、1%トリトンXに可溶化する。溶解物中の放射能レベルをシンチレーション計数によって決定する。
【0246】
(実施例6)
GPCR活性化を決定するためのアッセイ
ヒトGPCRの活性化の評価には、さまざまなアプローチを利用することができる。以下に実例を挙げる。当業者であれば、その当業者のニーズにとって優先的に有益である技法を決定する能力を持つと考えられる。
【0247】
1.膜結合アッセイ:[35S]GTPγSアッセイ
Gタンパク質共役受容体が、リガンド結合の結果として、または構成的活性化の結果として、その活性状態にある場合、受容体はGタンパク質に共役し、GDPの放出と、それに続くGTPのGタンパク質への結合を刺激する。Gタンパク質−受容体複合体のαサブユニットはGTPアーゼとして作用して、GTPをGDPにゆっくりと加水分解し、通常その時点で、受容体は不活化される。活性化受容体はGDPをGTPに交換し続ける。非加水分解性GTP類似体[35S]GTPγSを利用して、活性化受容体を発現させる膜への[35S]GTPγSの結合の増進を実証することができる。[35S]GTPγS結合を使って活性化を測定することの利点は、(a)全てのGタンパク質共役受容体に広く応用することができること;(b)膜表面に近接しているので、細胞内カスケードに影響を及ぼす分子を拾い上げる可能性が少なくなることである。
【0248】
このアッセイでは、関連受容体を発現させる膜への[35S]GTPγS結合を刺激するという、Gタンパク質共役受容体の能力を利用する。したがってこのアッセイは、内在性GPCRおよび非内在性構成的活性化GPCRに対して候補化合物をスクリーニングするための直接的同定方法に使用することができる。このアッセイは汎用であり、全てのGタンパク質共役受容体における創薬に応用することができる。
【0249】
[35S]GTPγSアッセイを、20mM HEPESおよび1〜約20mM MgCl2(この量は結果を最適化するために調節することができるが、20mMが好ましい)pH7.4、約0.3〜約1.2nM[35S]GTPγS(この量は結果を最適化するために調節することできるが、1.2が好ましい)および12.5〜75μgの膜タンパク質(例えばGPR43を発現させる293細胞;この量は結果を最適化するために調節することができる)および10μM GDP(この量は結果を最適化するために調節することができる)を含む結合緩衝液中で、1時間インキュベートする。次に、コムギ胚芽凝集素ビーズ(25μl;Amersham)を加えた後、その混合物を室温でさらに30分間ンインキュベートする。次に、チューブを1500×g、室温で5分間遠心分離した後、シンチレーション計数器で計数する。
【0250】
2.アデニリルシクラーゼ
細胞ベースのアッセイ用に設計されたFlash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)を、粗製形質膜で使用するために改変することができる。Flash Plateウェルは、cAMPを認識する特異的抗体も含有するシンチラントコーティングを含有することができる。ウェル内で生成したcAMPは、放射性cAMPトレーサーのcAMP抗体への結合に関する直接的競合によって、定量することができる。以下の記述は、受容体を発現させる全細胞中のcAMPレベルの変化を測定するための簡潔なプロトコールとして使用することができる。
【0251】
トランスフェクト細胞を一過性トランスフェクションの約24時間後に収集する。培地を注意深く吸引して捨てる。10mlのPBSを細胞の各ディッシュに穏やかに加えた後、注意深く吸引する。1mlのSigma細胞解離緩衝液および3mlのPBSを各プレートに加える。細胞をピペットでプレートから剥がし、細胞懸濁液を50ml円錐形遠心管に集める。次に、細胞を1,100rpm、室温で、5分間遠心分離する。細胞ペレットを適当な液量のPBS(約3ml/プレート)に注意深く再懸濁する。次に、血球計を使って細胞を計数し、適当な細胞数になるようにPBSを追加する(最終液量を約50μl/ウェルにする)。
【0252】
cAMP標準および検出緩衝液(検出緩衝液11mlに対して1μCiのトレーサー[125I]cAMP(50μl)を含む)を調製し、製造者の指示にしたがって維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、50μlの刺激緩衝液、3μlの候補化合物(最終アッセイ濃度12μM)および50μlの細胞を含有する。アッセイ緩衝液は使用するまで氷上に保存する。50μlのcAMP標準を適当なウェルに加えた後、50μlのPBSAをウェルH11およびH12に添加することにより、(好ましくは例えば96ウェルプレートで行なわれる)アッセイを開始する。50μlの刺激緩衝液を全てのウェルに加える。3μlの化合物溶液を分注することができるピンツールを使って、DMSO(または選択した候補化合物)を適当なウェルに加え、候補化合物の最終アッセイ濃度を12μMに、そして総アッセイ液量を100μlにする。次に、細胞をウェルに加え、室温で60分間インキュベートする。次に、トレーサーcAMPを含有する100μlの検出混合物をウェルに加える。次にプレートをさらに2時間インキュベートした後、Wallac MicroBetaシンチレーション計数器で計数する。次に、各アッセイプレート内に含まれる標準cAMP曲線から、cAMP/ウェルの値を外挿する。
【0253】
3.Gi共役標的GPCRのための細胞ベースのcAMP
TSHRは、活性化時にcAMPの蓄積を引き起こすGs共役GPCRである。TSHRは、アミノ酸残基623を突然変異させることにより(すなわちアラニン残基をイソロイシン残基に変えることにより)、構成的に活性化することができる。Gi共役受容体はアデニリルシクラーゼを阻害し、したがってcAMP産生レベルを減少させると予想され、これがcAMPレベルの評価を困難にする場合がある。Gi共役受容体の活性の指標としてcAMP産生量の減少を測定するための効果的な技法は、cAMPのベースラインレベルを確定するために、「シグナル増強物質」としての非内在性構成的活性化TSHR(TSHR−A623I)(または内在性構成的活性化Gs共役受容体)を、Gi連結標的GPCRと同時トランスフェクトすることによって達成することができる。内在型または非内在型のGi共役受容体を作製したら、その標的GPCRをシグナル増強物質と同時トランスフェクトする。スクリーニングに使用することができるのは、この物質である。一部の実施形態では、このアプローチが、Gi共役受容体に抗する候補化合物の直接的同定に、好ましく使用される。Gi共役GPCRの場合、このアプローチを使用すると、標的GPCRのインバースアゴニストがcAMPシグナルを増加させ、アゴニストがcAMPシグナルを減少させることを注記しておく。
【0254】
1日目に、2×104個の293細胞/ウェルをプレーティングする。2日目に、2本の反応チューブを調製する(各チューブについて以下に示す比率はプレート1枚あたりである):チューブAは、哺乳動物細胞にトランスフェクトする各受容体のDNA2μgで、合計4μgのDNA(例えばpCMVベクター;pCMVベクターと突然変異型THSR(TSHR−A623I);TSHR−A623IおよびGPCRなど)を、1.2mlの無血清DMEM(Irvine Scientific,カリフォルニア州アービン)に混合することによって調製される;チューブBは、120μlのリポフェクトアミン(Gibco BRL)を、1.2mlの無血清DMEMに混合することによって調製される。次に、チューブAおよびBを反転(数回)によって混合した後、室温で30〜45分間インキュベートする。この混合物を「トランスフェクション混合物」という。プレーティングした293細胞を1×PBSで洗浄した後、10mlの無血清DMEMを加える。次に、2.4mlのトランスフェクション混合物を細胞に加えた後、37℃/5%CO2で4時間インキュベートする。次にトランスフェクション混合物を吸引によって除去した後、25mlのDMEM/10%ウシ胎仔血清を加える。次に、細胞を37℃/5%CO2でインキュベートする。24時間のインキュベーション後に、細胞を収集し、解析に利用する。
【0255】
Flash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)は細胞ベースのアッセイ用に設計されているが、当業者の必要に応じて、粗製形質膜で使用するために改変することができる。Flash Plateウェルは、cAMPを認識する特異的抗体も含有するシンチラントコーティングを含有する。ウェル内で生成したcAMPは、放射性cAMPトレーサーのcAMP抗体への結合に関する直接的競合によって、定量することができる。以下の記述は、目的の受容体を発現させる全細胞中のcAMPレベルの変化を測定するための簡潔なプロトコールとして使用することができる。
【0256】
トランスフェクト細胞を一過性トランスフェクションの約24時間後に収集する。培地を注意深く吸引して捨てる。10mlのPBSを細胞の各ディッシュに穏やかに加えた後、注意深く吸引する。1mlのSigma細胞解離緩衝液および3mlのPBSを各プレートに加える。細胞をピペットでプレートから剥がし、細胞懸濁液を50ml円錐形遠心管に集める。次に、細胞を1,100rpm、室温で、5分間遠心分離する。細胞ペレットを適当な液量のPBS(約3ml/プレート)に注意深く再懸濁する。次に、血球計を使って細胞を計数し、適当な細胞数になるようにPBSを追加する(最終液量を約50μl/ウェルにする)。
【0257】
製造者の指示に従って、cAMP標準および検出緩衝液(検出緩衝液11mlに対して1μCiのトレーサー[125I]cAMP(50μl)を含む)を調製し、維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、50μlの刺激緩衝液、3μlの候補化合物(最終アッセイ濃度12μM)および50μlの細胞を含有する。アッセイ緩衝液は使用するまで氷上に保存することができる。50μlのcAMP標準を適当なウェルに加えた後、50μlのPBSAをウェルH11およびH12に添加することにより、アッセイを開始する。50μlの刺激緩衝液を全てのウェルに加える。3μlの化合物溶液を分注することができるピンツールを使って、選択した化合物(例えばTSH)を適当なウェルに加え、候補化合物の最終アッセイ濃度を12μMに、そして総アッセイ液量を100μlにする。次に、細胞をウェルに加え、室温で60分間インキュベートする。次に、トレーサーcAMPを含有する100μlの検出混合物をウェルに加える。次にプレートをさらに2時間インキュベートした後、Wallac MicroBetaシンチレーション計数器で計数する。各アッセイプレート内に含まれる標準cAMP曲線から、cAMP/ウェルの値を外挿する。
【0258】
4.レポーターベースのアッセイ
a.CRE−LUCレポーターアッセイ(Gs関連受容体)
293細胞または293T細胞を96ウェルプレートに1ウェルあたり2×104細胞の密度でプレーティングし、その翌日に、製造者の指示に従い、リポフェクトアミン試薬(BRL)を使ってトランスフェクトする。DNA/脂質混合物を各6ウェルトランスフェクションについて以下のように調製する:DMEM100μl中のプラスミドDNA260ngをDMEM100μl中の脂質2μlと穏やかに混合する(このプラスミドDNA260ngは、8×CRE−Lucレポータープラスミド200ng、内在性受容体もしくは非内在性受容体を含むpCMVまたはpCMVのみ50ng、およびGPRS発現プラスミド(pcDNA3(Invitrogen)中のGPRS)10ngからなる)。8×CRE−Lucレポータープラスミドは以下のように調製される:pβgal−Basicベクター(Clontech)中のBglV−HindIII部位にラットソマトスタチンプロモーター(−71/+51)をクローニングすることにより、ベクターSRIF−β−galを得る。8コピーのcAMP応答配列を、アデノウイルステンプレートAdpCF126CCRE8(Suzukiら,Hum Gene Ther 7:1883−1893(1996)を参照されたい;この文献の開示は参照によりその全体が本明細書に組み入れられる)からPCRによって取得し、それをSRIF−β−galベクター中に、Kpn−BglV部位でクローニングすることにより、8×CRE−β−galレポーターベクターを得る。8×CRE−β−galレポーターベクター中のベータ−ガラクトシダーゼ遺伝子を、HindIII−BamHI部位で、pGL3−basicベクター(Promega)から得たルシフェラーゼ遺伝子で置き換えることにより、8×CRE−Lucレポータープラスミドを生成させる。室温で30分間インキュベートした後、そのDNA/脂質混合物をDMEM400μlで希釈し、その希釈混合物100μlを各ウェルに加える。細胞培養インキュベータで4時間インキュベートした後、10%FCSを含むDMEM100μlを各ウェルに加える。翌日、トランスフェクト細胞を、10%FCSを含むDMEM200μl/ウェルに換える。8時間後に、ウェルをPBSで1回洗浄した後、フェノールレッド不含DMEM100μl/ウェルに換える。翌日、LucLite(商標)レポーター遺伝子アッセイキット(Packard)を使用し、製造者の指示に従って、ルシフェラーゼ活性を測定し、1450MicroBeta(商標)シンチレーションおよびルミネセンスカウンター(Wallac)で読み取る。
【0259】
b.AP1レポーターアッセイ(Gq関連受容体)
Gq刺激を検出するための方法は、そのプロモーター中にAP1配列を含有する遺伝子の活性化を引き起こすというGq依存性ホスホリパーゼCの既知の性質に依存する。リン酸カルシウム沈殿物の成分が410ngのpAP1−Luc、80ngのpCMV−受容体発現プラスミド、および20ngのCMV−SEAPである点以外は、CREBレポーターアッセイに関する上述のプロトコールに従って、Pathdetect(商標)AP−1cis−Reporting System(Stratagene,カタログ番号219073)を利用することができる。
【0260】
c.SRF−LUCレポーターアッセイ(Gq関連受容体)
Gq刺激を検出するための一方法は、そのプロモーター中に血清応答因子を含有する遺伝子の活性化を引き起こすというGq依存性ホスホリパーゼCの既知の性質に依存する。Pathdetect(商標)SRF−Luc−Reporting System(Stratagene)は、例えばCOS7細胞におけるGq共役活性のアッセイに利用することができる。このシステムのプラスミド成分および内在性または非内在性GPCRをコードする表示の発現プラスミドを、Mammalian Transfection(商標)Kit(Stratagene,カタログ番号200285)を使用し、製造者の指示に従って、細胞にトランスフェクトする。簡単に述べると、SRF−Luc410ng、pCMV−受容体発現プラスミド80ng、およびCMV−SEAP(分泌型アルカリホスファターゼ発現プラスミド;試料間のトランスフェクション効率のばらつきについて管理するためにトランスフェクト細胞中のアルカリホスファターゼ活性を測定する)20ngを、製造者の指示に従って、リン酸カルシウム沈殿物中で組み合わせる。沈殿物の半分を96ウェルプレート中の3ウェルに均等に分配し、無血清培地中の細胞上に24時間保つ。最後の5時間は、細胞を例えば1μMの候補混合物と共にインキュベートする。次に細胞を溶解し、Luclite(商標)Kit(Packard,カタログ番号6016911)および「Trilux 1450 Microbeta」液体シンチレーションおよびルミネセンスカウンター(Wallac)を使って、製造者の指示に従ってルシフェラーゼ活性をアッセイする。データはGraphPad Prism(商標)2.0a(GraphPad Software Inc.)を使って解析することができる。
【0261】
d.細胞内IP3蓄積アッセイ(Gq関連受容体)
1日目に、目的の受容体(内在性または非内在性)を含む細胞を24ウェルプレートに、通常は1×105細胞/ウェルの密度(ただしこの数字は最適化することができる)でプレーティングすることができる。2日目に、まず、1ウェルにつき無血清DMEM50μl中のDNA0.25μgと1ウェルにつき無血清DMEM50μl中のリポフェクトアミン2μlとを混合することによって、細胞をトランスフェクトすることができる。その溶液を穏やかに混合し、室温で15〜30分間インキュベートする。細胞を0.5mlのPBSで洗浄し、400μlの無血清培地をトランスフェクション培地と混合して、細胞に加える。次に、細胞を37℃/5%CO2で3〜4時間インキュベートしてから、トランスフェクション培地を除去し、1ml/ウェルの通常成長培地で置き換える。3日目に、細胞を3H−ミオイノシトールで標識する。簡単に述べると、培地を除去し、細胞を0.5mlのPBSで洗浄する。次に、1ウェルにつき0.5mlのイノシトール不含無血清培地(GIBCO BRL)を、1ウェルにつき0.25μCiの3H−ミオイノシトールと共に、細胞を37℃/5%CO2で16〜18時間、終夜インキュベートする。4日目、細胞を0.5mlのPBSで洗浄し、イノシトール不含無血清培地、10μMパージリン、10mM塩化リチウムを含有する0.45mlのアッセイ培地を加えるか、またはセロトニン受容体を含有する対照コンストラクトを使用する場合は、0.4mlのアッセイ培地および50μlの10×ケタンセリン(ket)を、最終濃度が10μMになるように加える。次に、細胞を37℃で30分間インキュベートする。次に、細胞を0.5mlのPBSで洗浄し、1ウェルにつき200μlの新鮮な氷冷停止溶液(1M KOH;18mMホウ酸Na;3.8mM EDTA)を加える。その溶液を氷上に5〜10分間または細胞が溶解するまで保ち、次に200μlの新鮮な氷冷中和溶液(7.5%HCL)で中和する。次に、溶解物を1.5mlエッペンドルフチューブに移し、チューブ1本につき1mlのクロロホルム/メタノール(1:2)を加える。その溶液を15秒間ボルテックスし、上相をBiorad AG1−X8(商標)アニオン交換樹脂(100〜200メッシュ)に適用する。まず、樹脂を1:1.25W/Vの水で洗浄し、0.9mlの上相をカラムにのせる。カラムを10mlの5mMミオイノシトールおよび10mlの5mMホウ酸Na/60mMギ酸Naで洗浄する。10mlのシンチレーションカクテルが2mlの0.1Mギ酸/1Mギ酸アンモニウムと共に入っているシンチレーションバイアルに、イノシトール三リン酸を溶出させる。10mlの0.1Mギ酸/3Mギ酸アンモニウムで洗浄することによってカラムを再生し、ddH2Oで2回すすぎ、水中、4℃で保存する。
【0262】
(実施例7)
融合タンパク質調製
a.GPCR:Gs融合コンストラクト
GPCR−Gタンパク質融合コンストラクトの設計は、以下のように行なうことができる:ラットGタンパク質Gsα(長型;Itoh,H.ら,Proc.Natl.Acad.Sci.83:3776(1986))の5’末端および3’末端をどちらも、そこにHindIII配列が含まれるように操作する。正しい配列を(隣接HindIII配列を含めて)確認した後、全配列を、pcDNA3.1(−)(Invitrogen,カタログ番号V795−20)中に、そのベクターのHindIII制限部位を使ってサブクローニングすることによってシャトル(shuttle)する。pcDNA3.1(−)へのサブクローニング後に、Gsα配列について、正しい向きを決定する。次に、HindIII配列にラットGsα遺伝子を含有する改変pcDNA3.1(−)を確認する。このベクターは、この時点で、「ユニバーサル」Gsαタンパク質ベクターとして利用することができる。pcDNA3.1(−)ベクターはHindIII部位の上流にさまざまな周知の制限部位を含有するので、Gsタンパク質の上流に目的の受容体のコード配列を挿入することができるという利益が得られる。これと同じアプローチを利用して、他の「ユニバーサル」Gタンパク質ベクターを作製することができ、もちろん、当業者に知られている他の市販ベクターまたは専有ベクターを利用することもできる。重要な基準は、GPCRの配列がGタンパク質の配列に対して上流にあり、かつインフレームであることである。
【0263】
b.Gq(6アミノ酸欠失)/Gi融合コンストラクト
Gq(欠失)/Gi融合コンストラクトの設計は、以下のように行なうことができる:GαqサブユニットのN末端の6アミノ酸(TLESIM(配列番号7)という配列を持つアミノ酸2〜7)を欠失させ、EYNLV(配列番号8)という配列を持つC末端の5アミノ酸を、DCGLF(配列番号9)という配列を持つGαiタンパク質の対応するアミノ酸で置き換える。この融合コンストラクトは、以下のプライマー:
【0264】
【化3】
と、マウスGαq−野生型をヘマグルチニンと共に含有するテンプレートとしてのプラスミド63313とを用いるPCRによって、取得することができる。
【0265】
TaqPlus Precision DNAポリメラーゼ(Stratagene)を、以下のサイクルによる増幅に利用することができる(第2〜第4ステップを35回繰り返す):95℃で2分;95℃で20秒;56℃で20秒;72℃で2分;および72℃で7分。PCR産物をpCRII−TOPOベクター(Invitrogen)にクローニングし、ABI Big Dye Terminatorキット(P.E.Biosystems)を使って配列決定することができる。融合コンストラクトの配列を含有するTOPOクローンからのインサートは、2段階クローニング法により、発現ベクターpcDNA3.1(+)中のHindIII/BamHI部位にシャトルすることができる。WO02068600として2002年9月6日に公開されたPCT出願番号PCT/US02/05625も参照されたい。この文献の開示は参照によりその全体が本明細書に組み入れられる。
【0266】
(実施例8)
[35S]GTPγSアッセイ
A.膜調製
一部の実施形態では、例えばアゴニスト、インバースアゴニストまたはアンタゴニストなどの候補化合物の同定で使用するために、目的の標的GPCRを含む膜を、以下のように調製する。
【0267】
a.材料
「膜掻爬緩衝液」は20mM HEPESおよび10mM EDTA、pH7.4から構成され;「膜洗浄緩衝液」は20mM HEPESおよび0.1mM EDTA、pH7.4から構成され;「結合緩衝液」は20mM HEPES、100mM NaCl、および10mM MgCl2、pH7.4から構成される。
【0268】
b.手順
この手順中は全ての材料を常に氷上に保つ。まず、細胞のコンフルエント単層から培地を吸引し、次に10mlの冷PBSですすいだ後、吸引する。次いで、細胞を掻爬するために5mlの膜掻爬緩衝液を加える。次に、細胞抽出物を50ml遠心管に移す(4℃、20,000rpmで17分間遠心分離する)。次いで、上清を吸引し、ペレットを30mlの膜洗浄緩衝液に再懸濁し、次に4℃、20,000rpmで17分間遠心分離する。次に上清を吸引し、ペレットを結合緩衝液に再懸濁する。次にこれをBrinkman Polytron(商標)ホモジナイザーを使ってホモジナイズする(全ての物質が懸濁状態になるまで15〜20秒間のバースト)。これを、ここでは「膜タンパク質」という。
【0269】
ブラッドフォードタンパク質アッセイ
ホモジナイゼーション後に、ブラッドフォードタンパク質アッセイを使って、膜のタンパク質濃度を決定する(タンパク質を約1.5mg/mlに希釈し、小分けし、後で使用するために凍結(−80℃)することができる。凍結した場合、使用のためのプロトコールは以下のとおりになる:アッセイ当日に、凍結膜タンパク質を室温で融解し、次にボルテックスしてから、Polytronを使って、約12×1,000rpm(約5〜10秒)で、ホモジナイズする。調製物が複数の場合、異なる調製物のホモジナイゼーション間で、ホモジナイザーを十分に洗浄すべきであることを注記しておく)。
【0270】
a.材料
結合緩衝液(上記のとおり);ブラッドフォード色素試薬;ブラッドフォードタンパク質標準を、製造者の指示に従って利用する(Biorad,カタログ番号500−0006)。
【0271】
b.手順
一対のチューブを調製する。1つは膜を含み、もう1つは対照「ブランク」とする。各チューブは800μlの結合緩衝液を含有する。次いで、10μlのブラッドフォードタンパク質標準(1mg/ml)を各チューブに加えた後、10μlの膜タンパク質を一方のチューブだけに加える(ブランクには加えない)。次いで、200μlのブラッドフォード色素試薬を各チューブに加えた後、各チューブをボルテックスする。5分後に、チューブを再びボルテックスし、その中の材料をキュベットに移す。そのキュベットを、CECIL3041分光光度計を使って、波長595で読み取る。
【0272】
同定アッセイ
a.材料
GDP緩衝液は37.5mlの結合緩衝液および2mgのGDP(Sigma,カタログ番号G−7127)からなり、次に結合緩衝液での連続希釈により、0.2μM GDPにする(各ウェルにおけるGDPの最終濃度は0.1μM GDPである);候補化合物を含む各ウェルは、GDP緩衝液100μl(最終濃度0.1μM GDP)、結合緩衝液中の膜タンパク質50μl、および結合緩衝液中の[35S]GTPγS(0.6nM)(結合緩衝液10mlにつき2.5μlの[35S]GTPγS)50μlからなる200μlの最終液量を持つ。
【0273】
b.手順
候補化合物は96ウェルプレート形式を使ってスクリーニングすることができる(これらは−80℃で凍結させることができる)。膜タンパク質(または対照として、標的GPCRを除外した発現ベクターによる膜)を懸濁状態になるまで手短にホモジナイズする。上述のブラッドフォードタンパク質アッセイを使ってタンパク質濃度を決定する。膜タンパク質(および対照)を結合緩衝液で0.25mg/mlに希釈する(最終アッセイ濃度,12.5μg/ウェル)。次いで、100μlのGDP緩衝液をWallac Scintistrip(商標)(Wallac)の各ウェルに加える。5μlピンツールを使って、5μlの候補化合物を上記のウェルに移す(すなわち、総アッセイ液量200μl中の5μlは、候補化合物の最終濃度が10μMになるように、1:40の比である)。ここでも、混入を避けるために、各移送ステップ後に、水(1回)、エタノール(1回)および水(2回)を含む3つのレザバーで、ピンツールをすすぐべきであり、各すすぎ後に過剰な液体をツールから振り払い、紙およびキムワイプで乾燥させるべきである。次いで、50μlの膜タンパク質を各ウェルに加え(標的GPCRを伴わない膜を含む対照ウェルも利用する)、室温で5〜10分間、プレインキュベートする。次いで、結合緩衝液中の[35S]GTPγS(0.6nM)50μlを各ウェルに加えた後、振とう機上、室温で60分間インキュベートする(プレートを箔で覆う)。次に、4000RPM、22℃で15分間、プレートを遠心することによって、アッセイを停止する。プレートを8チャンネル多岐管で吸引し、プレートカバーで密封する。プレートを、Wallac1450で、設定「Prot.#37」を使って、(製造者の指示どおりに)読み取る。
【0274】
(実施例9)
サイクリックAMPアッセイ
例えばアゴニスト、インバースアゴニスト、またはアンタゴニストなどの候補化合物を同定するためのもう1つのアッセイアプローチは、シクラーゼベースのアッセイを利用することによって達成することができる。このアッセイアプローチは、直接的同定だけでなく、上記の実施例で説明した[35S]GTPγSアプローチで得られる結果を確認するための間接的アプローチとしても利用することができる。
【0275】
候補化合物を目的の受容体に対するインバースアゴニストおよびアゴニストとして直接同定するには、改変Flash Plate(商標)アデニリルシクラーゼキット(New England Nuclear;カタログ番号SMP004A)を、以下のプロトコールに従って利用することができる。
【0276】
トランスフェクト細胞をトランスフェクションの約3日後に収集する。20mM HEPES(pH7.4)および10mM MgCl2を含有する緩衝液中の懸濁細胞をホモジナイズすることにより、膜を調製する。ホモジナイゼーションは、Brinkman Polytron(商標)を使って氷上で約10秒間行なう。得られたホモジネートを49,000×g、4℃で15分間遠心分離する。次に、得られたペレットを、20mM HEPES(pH7.4)および0.1mM EDTAを含有する緩衝液に再懸濁した後、49,000×g、4℃で15分間遠心分離する。次に、得られたペレットを使用時まで−80℃で保存する。直接的同定スクリーニングの当日に、膜ペレットを室温でゆっくり融解し、20mM HEPES(pH7.4)および10mM MgCl2を含有する緩衝液に再懸濁して、最終タンパク質濃度を0.60mg/mlにする(再懸濁した膜は使用時まで氷上に置く)。
【0277】
製造者の指示に従って、cAMP標準および検出緩衝液(検出緩衝液11mlに対して2μCiのトレーサー[125I]cAMP(100μl)を含む)を調製し、維持する。スクリーニング用にアッセイ緩衝液を新しく調製する。これは、20mM HEPES(pH7.4)、10mM MgCl2、20mMホスホクレアチン(Sigma)、0.1単位/mlクレアチンホスホキナーゼ(Sigma)、50μM GTP(Sigma)、および0.2mM ATP(Sigma)を含有する。次に、アッセイ緩衝液を使用時まで氷上に保存する。
【0278】
例えば96ウェルプレートのウェルに、候補化合物(3μl/ウェル;最終アッセイ濃度12μM)を、40μlの膜タンパク質(30μg/ウェル)および50μlのアッセイ緩衝液と共に加える。次に、この混合物を、穏やかに振とうしながら、室温で30分間インキュベートする。
【0279】
インキュベーション後に、100μlの検出緩衝液を各ウェルに加え、次に2〜24時間インキュベートする。次にプレートを、Wallac MicroBeta(商標)プレートリーダーで、「Prot.#31」を使って、(製造者の指示どおりに)計数する。
【0280】
(実施例10)
細胞内カルシウム濃度を測定するための蛍光イメージングプレートリーダー(FLIPR)アッセイ
標的受容体(実験)およびpCMV(陰性対照)を安定にトランスフェクトした、各クローン株由来の細胞を、ポリ−D−リジン前処理96ウェルプレート(Becton−Dickinson,#356640)に、5.5×104細胞/ウェルの密度で完全培養培地(10%FBS、2mM L−グルタミン、1mMピルビン酸ナトリウムを含むDMEM)と共に播種し、翌日アッセイする。GPR43はGαqに共役するので、検出可能なカルシウムフラックスを引き起こすのに、Gα15、Gα16などのプロミスキャスGタンパク質、またはキメラGq/Giαサブユニットを必要としない。Fluo4−AM(Molecular Probe,#F14202)インキュベーション緩衝液ストックを調製するために、1mgのFluo4−AMを467μlのDMSOおよび467μlのプルオロニックアシッド(Pluoronic acid)(Molecular Probe,#P3000)に溶解して、1mMストック溶液を得る。このストック溶液は−20℃で1ヶ月保存することができる。Fluo4−AMは蛍光カルシウム指示薬色素である。
【0281】
候補化合物を洗浄緩衝液(1×HBSS/2.5mMプロベニシド(Probenicid)/20mM HEPES、pH7.4)中に調製する。
【0282】
アッセイ時に、培養培地をウェルから除去し、細胞に100μlの4μM Fluo4−AM/2.5mMプロベニシド(Sigma,#P8761)/20mM HEPES/完全培地、pH7.4を負荷する。37℃/5%CO2でのインキュベーションを60分間進行させる。
【0283】
1時間のインキュベーション後に、Fluo4−AMインキュベーション緩衝液を除去し、細胞を100μlの洗浄緩衝液で2回洗浄する。各ウェルに100μlの洗浄緩衝液を残す。プレートを37℃/5%CO2のインキュベータに60分間戻す。
【0284】
50μlの候補化合物を30秒目に添加し、その候補化合物が惹起する細胞内カルシウム濃度([Ca2+])の一過性変化をさらに150秒にわたって記録するように、FLIPR(蛍光イメージングプレートリーダー;Molecular Device)をプログラムする。FLIPRソフトウェアを使用し、総蛍光変化カウントを使ってアゴニスト活性を決定する。この計器ソフトウェアは蛍光値を標準化して、初期値を等しくゼロにする。
【0285】
上記は、安定トランスフェクト細胞を使ったアゴニスト活性に関するFLIPRアッセイであるが、当業者であれば、アンタゴニスト活性を特徴づけるために、このアッセイを容易に改変することができるだろう。あるいは、一過性トランスフェクト細胞も使用できることは、前記当業者には容易に理解されるだろう。
【0286】
(実施例11)
MAPキナーゼアッセイ
MPAキナーゼ(マイトジェン活性化キナーゼ)を監視することで、受容体活性化を評価することができる。MAPキナーゼはいくつかのアプローチによって検出することができる。1つのアプローチは、非リン酸化(不活性)状態か、リン酸化(活性)状態かという、リン酸化状態の評価に基づく。リン酸化されたタンパク質はSDS−PAGEでの移動度が遅くなるので、ウェスタンブロット法を使って、刺激されていないタンパク質と比較することができる。あるいは、リン酸化されたタンパク質に特異的な抗体を入手することができ(New England Biolabs)、それを使ってリン酸化されたキナーゼの増加を検出することができる。どちらの方法でも、細胞を候補化合物で刺激した後、Laemmli緩衝液で抽出する。可溶性画分をSDS−PAGEゲルに適用し、タンパク質を電気泳動的にニトロセルロースまたはイモビリン(Immobilin)に転写する。免疫反応性バンドを標準的ウェスタンブロット技術によって検出する。可視シグナルまたは化学発光シグナルをフィルムに記録する。これはデンシトメトリーによって定量することができる。
【0287】
もう1つのアプローチは、リン酸化アッセイによるMAPキナーゼ活性の評価に基づく。細胞を候補化合物で刺激し、可溶性抽出物を調製する。その抽出物を、ガンマ−32P−ATP、ATP再生系、およびMAPキナーゼの特異的基質、例えばPHAS(phoshporylated heat and acid stable protein regulated by insulin:インスリンによって調節されるリン酸化耐熱耐酸性タンパク質)−Iなどと共に、30℃で10分間インキュベートする。H3PO4の添加によって反応を停止させ、試料を氷に移す。リン酸化されたタンパク質を保持するワットマンP81クロマトグラフィーペーパーに一部をスポットする。そのクロマトグラフィーペーパーを洗浄し、液体シンチレーション計数器で32Pについて計数する。あるいは、細胞抽出物を、ガンマ−32P−ATP、ATP再生系、およびストレプトアビジンによって濾紙支持体に結合されたビオチン化ミエリン塩基性タンパク質と共に、インキュベートする。ミエリン塩基性タンパク質は活性化されたMAPキナーゼの基質である。リン酸化反応を30℃で10分間行なう。次に、リン酸化されたミエリン塩基性タンパク質を保持する濾紙を通して、抽出物を吸引する。濾紙を洗浄し、液体シンチレーション計数により、32Pについて計数する。
【0288】
(実施例12)
受容体結合アッセイ
本明細書に記載する方法の他に、候補化合物を評価するためのもう1つの手段は、GPR43受容体への結合親和性を決定することによる。このタイプのアッセイは一般に、GPR43受容体に対する放射標識リガンドを必要とする。GPR43受容体の既知リガンドおよびその放射性ラベルを使用することの他に、GPR43アゴニスト化合物を放射性同位体で標識して、GPR43受容体に対する候補化合物の親和性を評価するためのアッセイに使用することもできる。
【0289】
化合物を同定/評価するために、GPR43アゴニストなどの放射標識GPR43化合物をスクリーニングアッセイで使用することができる。一般論として、新たに合成または同定された化合物(すなわち候補化合物)を、GPR43受容体に対する放射標識GPR43アゴニストの結合を減少させるその能力について、評価することができる。したがって、GPR43受容体への結合に関して放射標識GPR43アゴニストと競合する能力は、GPR43受容体に対する候補化合物の結合親和性に、直接的に相関する。
【0290】
GPR43に関する受容体結合を決定するためのアッセイプロトコール
A.GPR43受容体調製
例えば、ここで述べるように、HEK293細胞(ヒト腎臓,ATCC)にGPR43を一過性にまたは安定にトランスフェクトすることができる。例えば293細胞に10μgのヒトGPR43受容体および60μlのリポフェクトアミン(15cmディッシュ1枚あたり)をトランスフェクトし、培地を交換してそのディッシュ中で24時間(75%コンフルエント)成長させることができる。10ml/ディッシュのHepes−EDTA緩衝液(20mM Hepes+10mM EDTA,pH7.4)を使って細胞を取り出す。次に、細胞をBeckman Coulter遠心機中、17,000rpmで20分間、遠心分離する(JA−25.50ローター)。次に、ペレットを20mM Hepes+1mM EDTA,pH7.4に再懸濁し、50mlダウンスホモジナイザーでホモジナイズし、再び遠心分離する。上清を除去した後、結合アッセイに使用するまでペレットを−80℃で保存する。アッセイに使用する時は、膜を氷上で20分間融解したのち、10mLのインキュベーション緩衝液(20mM Hepes,1mM MgCl2、100mM NaCl、pH7.4)を加える。次に、粗製膜ペレットを再懸濁するために膜をボルテックスし、Brinkmann PT−3100 Polytronホモジナイザーを使って、設定6で15秒間、ホモジナイズする。膜タンパク質の濃度をBRLブラッドフォードタンパク質アッセイを使って決定する。
【0291】
B.結合アッセイ
総結合量については、総液量50μlの適当に希釈した膜(50mMトリスHCl(pH7.4)、10mM MgCl2および1mM EDTAを含有するアッセイ緩衝液に希釈したもの;タンパク質5〜50μg)を、96ウェルポリプロピレンマイクロタイタープレートに加え、次に100μlのアッセイ緩衝液および50μlの放射標識GPR43アゴニストを加える。非特異的結合量については、100μlではなく50μlのアッセイ緩衝液を加え、さらに50μlの10μM非放射性GPR43を加えてから、50μlの放射標識GPR43アゴニストを加える。次にプレートを室温で60〜120分間インキュベートする。Brandell 96ウェルプレートハーベスターで、Microplate Devices GF/C Unifilter濾過プレートを通して、アッセイプレートを濾過した後、0.9%NaClを含有する冷50mMトリスHCl(pH7.4)で洗浄することにより、結合反応を停止させる。次に、濾過プレートの底を密封し、50μlのOptiphase Supermixを各ウェルに加え、プレートの上部を密封し、プレートをTrilux MicroBetaシンチレーション計数器で計数する。化合物競合試験については、100μlのアッセイ緩衝液を加える代わりに、100μlの適当に希釈した候補化合物を、適当なウェルに加えた後、50μlの放射標識GPR43アゴニストを加える。
【0292】
C.計算
候補化合物を最初は1および0.1μMでアッセイし、次に、中央用量が放射標識GPR43アゴニスト結合のおよそ50%阻害(すなわちIC50)を引き起こすように選択した濃度範囲でアッセイする。候補化合物の不在下での特異的結合量(BO)は、総結合量(BT)−非特異的結合量(NSB)の差であり、同様に特異的結合量(候補化合物の存在下)(B)は、置換結合量(BD)−非特異的結合量(NSB)の差である。IC50は阻害応答曲線、%B/BO対候補化合物濃度のロジット−ログプロットから決定される。
【0293】
KiはChengおよびPrustoff変換によって計算される:
Ki=IC50/(1+[L]/KD)
式中、[L]は、アッセイに使用する放射標識GPR43アゴニストの濃度であり、KDは、同じ結合条件下で独立して決定された放射標識GPR43アゴニストの解離定数である。
【0294】
(実施例13)
齧歯類糖尿病モデル
肥満およびインスリン抵抗性に関連するII型糖尿病の齧歯類モデルが開発されている。疾患の病態生理を理解し、候補治療化合物を試験するために、マウスにおけるdb/dbおよびob/ob[Diabetes(1982)31:1−6参照]ならびにズッカーラットにおけるfa/faなどの遺伝的モデルが開発されている[Diabetes(1983)32:830−838;Annu Rep Sankyo Res Lab(1994)46:1−57]。Jackson Laboratoryによって開発されたホモ接合体動物C57BL/KsJ−db/dbマウスは、肥満、高血糖、高インスリン血症およびインスリン抵抗性であるのに対して[J Clin Invest(1990)85:962−967]、ヘテロ接合体は痩せていて正常血糖である。db/dbモデルの場合、マウスは、加齢に伴って徐々に、インスリン欠乏(糖レベルの管理が不十分な場合、ヒトII型糖尿病の後期に共通して観察される特徴)を発症する。このモデルはヒトII型糖尿病に似ているので、例えば血漿グルコースおよびトリグリセリドを低下させることなどの活性(ただしこれに限定されるわけではない)について、本発明の代謝安定化化合物を、試験する。ズッカー(fa/fa)ラットは重度の肥満、高インスリン血症、およびインスリン抵抗性であり{Coleman,Diabetes(1982)31:1;E Shafrir「Diabetes Mellitus」H RifkinおよびD Porte,Jr編[Elsevier Science Publishing Co,New York,ed.4(1990),pp.299−340]}、fa/fa突然変異は、マウスdb突然変異のラット等価物であるかもしれない[Friedmanら,Cell(1992)69:217−220;Truettら,Proc Natl Acad Sci USA(1991)88:7806]。タビー(tubby)(tub/tub)マウスは、高血糖を伴わない肥満、中等度のインスリン抵抗性および高インスリン血症を特徴とする[Colemanら,Heredity(1990)81:424]。
【0295】
本発明は、上記齧歯類糖尿病モデルのいずれかもしくは全て、II型糖尿病もしくは他の好ましい代謝関連障害もしくは上述した脂質代謝の障害を持つヒト、または他の哺乳動物に基づくモデルにおけるインスリン抵抗性および高血糖を低下させるための、本発明化合物の使用を包含する。血漿グルコースおよびインスリンレベル、ならびに血漿遊離脂肪酸およびトリグリセリドを含む(ただしこれに限定されるわけではない)他の因子を、試験することができる。
【0296】
本発明の化合物の抗高血糖活性に関するインビボアッセイ
遺伝子改変肥満糖尿病マウス(db/db)(雄、7〜9週齢)を、標準的実験室条件下に、22℃および相対湿度50%で飼育し(7〜9匹/ケージ)、Purina齧歯類飼料の食餌および水を自由に摂取させて維持する。処置に先だって、血液を各動物の尾静脈から収集し、One Touch Basic Glucose Monitor System(Lifescan)を使って血中グルコース濃度を決定する。250〜500mg/dlの血漿グルコースレベルを持つマウスを使用する。各処置群は、平均グルコースレベルが研究の開始時に各群で等しくなるように分配した7匹のマウスからなる。イソフルラン麻酔を使って挿入した微小浸透圧ポンプでdb/dbマウスに投薬することにより、本発明の化合物、食塩水、または無関係な化合物をマウスの皮下(s.c.)に与える。その後、定期的に、血液試料を尾静脈から採取し、血中グルコース濃度について解析する。群間の有意差(本発明の化合物を食塩水処置と比較する)をスチューデントt検定を使って評価する。
【0297】
上記は限定ではなく例示を目的としている。II型糖尿病の典型的齧歯類モデルは他にも記述されている[Moller DE,Nature(2001)414:821−7およびその中の引用文献;ならびにReed MJら,Diabetes,ObesityおよびMetabolism(1999)1:75−86およびその中の引用文献;これら各文献の開示は参照によりその全体が本明細書に組み入れられる]。
【0298】
本明細書に記載した典型的実施例には、本発明の要旨から逸脱することなく、さまざまな変更、付加、置換および変異を加えることができ、したがってそれらも本発明の範囲に包含されるとみなされることは、当業者には理解されるだろう。上で言及した文献は、印刷刊行物および暫定的、通常の特許出願(ただしこれに限るわけではない)を含めていずれも、参照として、その全体が本明細書に援用される。
【図面の簡単な説明】
【0299】
【図1】マウス成体およびマウス胎児の組織および細胞におけるマウスGPR43発現のAffymetrix遺伝子チップ解析を表す。
【図2】選ばれたヒト組織およびヒト細胞におけるヒトGPR43発現(上図)ならびに選ばれたマウス組織、マウス細胞およびマウス細胞株におけるマウスGPR43発現(下図)のRT−PCR解析を表す。
【特許請求の範囲】
【請求項1】
代謝安定化化合物を同定する方法であって、
a)候補化合物をGPR43と接触させること、および
b)GPR43機能が調節されるかどうかを決定すること
を含み、GPR43機能の調節が、該候補化合物が代謝安定化化合物であることを示す方法。
【請求項2】
前記GPR43がヒト由来である、請求項1に記載の方法。
【請求項3】
前記決定が二次メッセンジャーアッセイを含む、請求項1に記載の方法。
【請求項4】
請求項1に記載の方法に従って同定される代謝安定化化合物。
【請求項5】
前記代謝安定化化合物がGPR43アゴニストである、請求項4に記載の化合物。
【請求項6】
前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである、請求項4に記載の化合物。
【請求項7】
組成物を調製する方法であって、代謝安定化化合物を同定し、次に該化合物を担体と混合することを含み、該化合物が請求項1に記載の方法によって同定される方法。
【請求項8】
請求項4に記載の化合物を含むか、または本質的に請求項4に記載の化合物からなるか、または請求項4に記載の化合物からなる、医薬組成物。
【請求項9】
処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、請求項8に記載の化合物の有効量を該個体に投与することを含む方法。
【請求項10】
前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項9に記載の方法。
【請求項11】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、請求項8に記載の化合物の有効量と組み合わせて前記個体に投与することをさらに含む、請求項9に記載の方法。
【請求項12】
前記個体が哺乳動物である、請求項9に記載の方法。
【請求項13】
前記個体がヒトである、請求項9に記載の方法。
【請求項14】
代謝安定化化合物として使用するための請求項8に記載の化合物を含む医薬を製造する方法。
【請求項15】
代謝関連障害の処置に使用するための請求項8に記載の化合物を含む医薬を製造する方法。
【請求項16】
代謝安定化化合物を同定する方法であって、
a)候補化合物をGPR43と接触させること、および
b)GPR43機能が減少するかどうかを決定すること
を含み、GPR43機能の減少が、該候補化合物が代謝安定化化合物であることを示す方法。
【請求項17】
前記GPR43がヒト由来である、請求項16に記載の方法。
【請求項18】
前記決定が二次メッセンジャーアッセイを含む、請求項16に記載の方法。
【請求項19】
請求項16に記載の方法に従って同定される代謝安定化化合物。
【請求項20】
前記代謝安定化化合物がGPR43インバースアゴニストである、請求項19に記載の化合物。
【請求項21】
前記代謝安定化化合物がGPR43アンタゴニストである、請求項19に記載の化合物。
【請求項22】
組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、該化合物が請求項16に記載の方法によって同定される方法。
【請求項23】
請求項19に記載の化合物を含むか、または本質的に請求項19に記載の化合物からなるか、または請求項19に記載の化合物からなる、医薬組成物。
【請求項24】
処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、請求項23に記載の化合物の有効量を該個体に投与することを含む方法。
【請求項25】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項24に記載の方法。
【請求項26】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項24に記載の方法。
【請求項27】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、請求項23に記載の化合物の有効量と組み合わせて該個体に投与することをさらに含む、請求項24に記載の方法。
【請求項28】
前記個体が哺乳動物である、請求項24に記載の方法。
【請求項29】
前記個体がヒトである、請求項24に記載の方法。
【請求項30】
代謝安定化化合物として使用するための請求項23に記載の化合物を含む医薬を製造する方法。
【請求項31】
代謝関連障害の処置に使用するための請求項23に記載の化合物を含む医薬を製造する方法。
【請求項32】
代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法。
【請求項33】
前記代謝関連障害が低血糖または加齢である、請求項32に記載の方法。
【請求項34】
前記調節因子がアゴニストである、請求項33に記載の方法。
【請求項35】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項32に記載の方法。
【請求項36】
前記調節因子がインバースアゴニストまたはアンタゴニストである、請求項35に記載の方法。
【請求項37】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することをさらに含む、請求項35に記載の方法。
【請求項38】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項32に記載の方法。
【請求項39】
GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法。
【請求項40】
前記障害が代謝関連障害である、請求項39に記載の方法。
【請求項41】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項39に記載の方法。
【請求項42】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項39に記載の方法。
【請求項43】
前記代謝関連障害がII型糖尿病である、請求項39に記載の方法。
【請求項44】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することをさらに含む、請求項39に記載の方法。
【請求項1】
代謝安定化化合物を同定する方法であって、
a)候補化合物をGPR43と接触させること、および
b)GPR43機能が調節されるかどうかを決定すること
を含み、GPR43機能の調節が、該候補化合物が代謝安定化化合物であることを示す方法。
【請求項2】
前記GPR43がヒト由来である、請求項1に記載の方法。
【請求項3】
前記決定が二次メッセンジャーアッセイを含む、請求項1に記載の方法。
【請求項4】
請求項1に記載の方法に従って同定される代謝安定化化合物。
【請求項5】
前記代謝安定化化合物がGPR43アゴニストである、請求項4に記載の化合物。
【請求項6】
前記代謝安定化化合物がGPR43インバースアゴニストまたはアンタゴニストである、請求項4に記載の化合物。
【請求項7】
組成物を調製する方法であって、代謝安定化化合物を同定し、次に該化合物を担体と混合することを含み、該化合物が請求項1に記載の方法によって同定される方法。
【請求項8】
請求項4に記載の化合物を含むか、または本質的に請求項4に記載の化合物からなるか、または請求項4に記載の化合物からなる、医薬組成物。
【請求項9】
処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、請求項8に記載の化合物の有効量を該個体に投与することを含む方法。
【請求項10】
前記代謝関連障害が低血糖、加齢、インスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項9に記載の方法。
【請求項11】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、請求項8に記載の化合物の有効量と組み合わせて前記個体に投与することをさらに含む、請求項9に記載の方法。
【請求項12】
前記個体が哺乳動物である、請求項9に記載の方法。
【請求項13】
前記個体がヒトである、請求項9に記載の方法。
【請求項14】
代謝安定化化合物として使用するための請求項8に記載の化合物を含む医薬を製造する方法。
【請求項15】
代謝関連障害の処置に使用するための請求項8に記載の化合物を含む医薬を製造する方法。
【請求項16】
代謝安定化化合物を同定する方法であって、
a)候補化合物をGPR43と接触させること、および
b)GPR43機能が減少するかどうかを決定すること
を含み、GPR43機能の減少が、該候補化合物が代謝安定化化合物であることを示す方法。
【請求項17】
前記GPR43がヒト由来である、請求項16に記載の方法。
【請求項18】
前記決定が二次メッセンジャーアッセイを含む、請求項16に記載の方法。
【請求項19】
請求項16に記載の方法に従って同定される代謝安定化化合物。
【請求項20】
前記代謝安定化化合物がGPR43インバースアゴニストである、請求項19に記載の化合物。
【請求項21】
前記代謝安定化化合物がGPR43アンタゴニストである、請求項19に記載の化合物。
【請求項22】
組成物を調製する方法であって、代謝安定化化合物を同定し、次に前記化合物を担体と混合することを含み、該化合物が請求項16に記載の方法によって同定される方法。
【請求項23】
請求項19に記載の化合物を含むか、または本質的に請求項19に記載の化合物からなるか、または請求項19に記載の化合物からなる、医薬組成物。
【請求項24】
処置または予防を必要とする個体の代謝関連障害を処置または予防する方法であって、請求項23に記載の化合物の有効量を該個体に投与することを含む方法。
【請求項25】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項24に記載の方法。
【請求項26】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項24に記載の方法。
【請求項27】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、請求項23に記載の化合物の有効量と組み合わせて該個体に投与することをさらに含む、請求項24に記載の方法。
【請求項28】
前記個体が哺乳動物である、請求項24に記載の方法。
【請求項29】
前記個体がヒトである、請求項24に記載の方法。
【請求項30】
代謝安定化化合物として使用するための請求項23に記載の化合物を含む医薬を製造する方法。
【請求項31】
代謝関連障害の処置に使用するための請求項23に記載の化合物を含む医薬を製造する方法。
【請求項32】
代謝関連障害を処置または予防する方法であって、その必要がある個体にGPR43調節因子の有効量を投与することを含む方法。
【請求項33】
前記代謝関連障害が低血糖または加齢である、請求項32に記載の方法。
【請求項34】
前記調節因子がアゴニストである、請求項33に記載の方法。
【請求項35】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項32に記載の方法。
【請求項36】
前記調節因子がインバースアゴニストまたはアンタゴニストである、請求項35に記載の方法。
【請求項37】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することをさらに含む、請求項35に記載の方法。
【請求項38】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項32に記載の方法。
【請求項39】
GPR43機能を減少させることによって処置できるまたは予防できる障害を処置または予防する方法であって、その必要がある個体にGPR43インバースアゴニストまたはアンタゴニストの有効量を投与することを含む方法。
【請求項40】
前記障害が代謝関連障害である、請求項39に記載の方法。
【請求項41】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、糖尿病、高血糖、高インスリン血症、高脂質血症、高トリグリセリド血症、高コレステロール血症、異常脂質血症、肥満、X症候群、アテローム性動脈硬化、心臓疾患、脳卒中、高血圧、または末梢血管疾患である、請求項39に記載の方法。
【請求項42】
前記代謝関連障害がインスリン抵抗性、耐糖能異常、または糖尿病である、請求項39に記載の方法。
【請求項43】
前記代謝関連障害がII型糖尿病である、請求項39に記載の方法。
【請求項44】
糖尿病、血中脂質障害、または肥満の処置に用いられる薬剤の有効量を、GPR43インバースアゴニストまたはアンタゴニストの有効量と組み合わせて前記個体に投与することをさらに含む、請求項39に記載の方法。
【図1】


【図2】
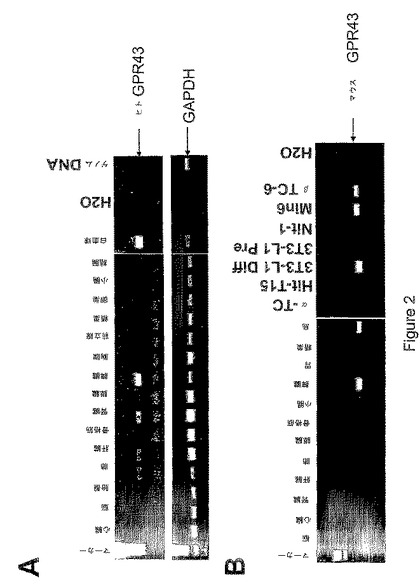
【図2】
【公表番号】特表2008−513792(P2008−513792A)
【公表日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願番号】特願2007−532638(P2007−532638)
【出願日】平成17年9月21日(2005.9.21)
【国際出願番号】PCT/US2005/033795
【国際公開番号】WO2006/036688
【国際公開日】平成18年4月6日(2006.4.6)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
【公表日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願日】平成17年9月21日(2005.9.21)
【国際出願番号】PCT/US2005/033795
【国際公開番号】WO2006/036688
【国際公開日】平成18年4月6日(2006.4.6)
【出願人】(500478097)アリーナ ファーマシューティカルズ, インコーポレイテッド (97)
【Fターム(参考)】
[ Back to top ]