
光学活性な環式アミノ酸の調製
【課題】(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸等の環式アミノ酸の光学活性な化合物もしくはその薬学的に許容できる塩またはその化合物もしくはその薬学的に許容できる塩の反対の鏡像異性体を調製するための方法および物質の提供。
【解決手段】対応するシアノ化合物、例えば2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸もしくはその反対の鏡像異性体またはその化合物の塩またはその反対の鏡像異性体のシアノ部分を還元してアミノ部分にするステップを包含する。
【解決手段】対応するシアノ化合物、例えば2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸もしくはその反対の鏡像異性体またはその化合物の塩またはその反対の鏡像異性体のシアノ部分を還元してアミノ部分にするステップを包含する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、疼痛ならびに様々な精神障害および睡眠障害を治療するために有用なキラル環式アミノ酸を調製するための物質および方法に関する。
【背景技術】
【0002】
Bryansらに付与された米国特許第6635673B1号明細書(’673号特許)は、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸を包含するいくつかの光学活性な環式アミノ酸およびその薬学的に許容できる塩を記載している。これらの化合物は、カルシウムチャネルのアルファ−2−デルタ(α2δ)サブユニットに結合する。これらは特に、不眠症、てんかん、失神発作、運動機能減少症、うつ病、不安、パニック、疼痛、過敏性腸症候群および関節炎を包含するいくつかの疾患を治療するために有用である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
’673号特許は、光学活性な環式アミノ酸を調製するためのいくつかの方法を記載している。これらの方法の多くは、中間体または出発原料として、(S,S)−3,4−ジメチル−シクロペンタノンを包含するキラル3,4−二置換シクロペンタノンを使用する。シクロペンタノンを調製する方法は知られているが、効率および経費の関係により、またはプロセスが市販されていない出発原料を使用することにより、これらのプロセスの多くは、市場規模での製造には問題がある。例えば、Blakemoreらに付与された米国特許第6872856号明細書参照。さらに、キラルシクロペンタノンを所望の光学活性な環式アミノ酸に変換するためには、数多くのステップが必要となりうる。したがって、キラル環式アミノ酸を調製するための改善された方法が望ましい。
【課題を解決するための手段】
【0004】
本発明は、市販の出発原料から光学活性な環式アミノ酸(下記の式1)を調製するための比較的効率的で費用対効果の大きい方法を提供する。例えば、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸を、(2R,3R)−1,4−ジブロモ−2,3−ジメチル−ブタンまたは(2R,3R)−2,3−ジメチル−ブタン−1,4−ジイル−ジトシレートから3または4ステップで調製することができる。
【0005】
本発明の一態様は、式1の化合物もしくは薬学的に許容できるその塩または式1の化合物の反対の鏡像異性体もしくは薬学的に許容できるその塩を製造する方法を提供し:
【0006】
【化1】

[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]、
ここで、前記方法は、
(a)式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩のシアノ部分をアミノ部分に還元するステップと
【0007】
【化2】
[式5中のR1およびR2は、式1に関する前記と同様に定義され、R4は水素原子およびC1〜6アルキルから選択される]、
(b)任意選択で、式1の化合物またはその反対の鏡像異性体を式1の化合物またはその反対の鏡像異性体の薬学的に許容できる塩に変換するステップを含む。
【0008】
本発明の他の態様は、式4の化合物またはその反対の鏡像異性体を提供する:
【0009】
【化3】
[式中、式1に関する前記と同様に定義され、
R3は、
【0010】
【化4】
により表される構造を有するカルボン酸またはエステル保護基である
(式7および式8中の「A」は、式4の化合物の残りの部分に結合している結合点を表し、
R6、R7、R8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成し、
Z1およびZ2はそれぞれ独立に、OおよびSから選択され、
R9は、C1〜6アルキルであり、
R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、
R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する)]。
【0011】
本発明の他の態様は、式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩を提供する:
【0012】
【化5】
[式中、
R1およびR2は、式1に関する前記と同様に定義され、
R4は、H、C1〜6アルキル、アリール−C1〜3アルキル、第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオンから選択され、
ここで、前記各アリール−C1〜3アルキル基中のアリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6−アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]。
【0013】
本発明は、可能であれば、薬学的に許容できるかできないかにかかわらず、前記化合物のすべての塩、複合体、溶媒和物、水和物および多形体を包含する。
【発明を実施するための最良の形態】
【0014】
定義および略語
他に示されていない限り、本開示は、下記に提示される定義を使用する。いくつかの定義および式は、原子間の結合または指名されているか指名されていない原子もしくは原子の基への結合点を示すためにダッシュ(「−」)を包含することもある。他の定義および式は、それぞれ二重結合または三重結合を示すために等号(「=」)または識別記号(「≡」)を包含することもある。他の式は、1つまたは複数の波形結合
【0015】
【化6】
を包含することもある。立体中心に結合している場合、波形結合は、個々の、または混合物としての両方の立体異性体に関している。同様に、二重結合に結合している場合、波形結合は、Z異性体、E異性体またはZおよびE異性体の混合物を指している。いくつかの式は、単結合または二重結合を示すために破線結合
【0016】
【化7】
を包含することもある。
【0017】
「置換されている」基は、1個または複数の水素原子が1個または複数の非水素原子または基で置換されているものであるが、ただし、価要求は満たしていて、置換により、化学的に安定な化合物が生じている。
【0018】
測定可能な数値変数に関連して使用される場合、「約」または「ほぼ」は、示されている変数の値および示されている値の実験誤差の範囲内(例えば平均値に関して95%の信頼区間内)または示されている値の10%の範囲内で、いずれにしろより高い変数の値すべてを指している。
【0019】
「アルキル」は、規定の数の炭素原子を通常は有する直鎖および分枝鎖飽和炭化水素基を指している(即ち、C1〜6アルキルは、1、2、3、4、5または6個の炭素原子を有するアルキル基を指している)。アルキル基の例には、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、s−ブチル、i−ブチル、t−ブチル、ペンチ−1−イル、ペンチ−2−イル、ペンチ−3−イル、3−メチルブチ−1−イル、3−メチルブチ−2−イル、2−メチルブチ−2−イル、2,2,2−トリメチルエチ−1−イルおよびn−ヘキシルが包含される。
【0020】
「アルケニル」は、1個または複数の不飽和炭素−炭素結合を有し、規定の数の炭素原子を通常は有する直鎖および分枝鎖炭化水素基を指している。アルケニル基の例には、エテニル、1−プロペン−1−イル、1−プロペン−2−イル、2−プロペン−1−イル、1−ブテン−1−イル、1−ブテン−2−イル、3−ブテン−1−イル、3−ブテン−2−イル、2−ブテン−1−イル、2−ブテン−2−イル、2−メチル−1−プロペン−1−イル、2−メチル−2−プロペン−1−イル、1,3−ブタジエン−1−イルおよび1,3−ブタジエン−2−イルが包含される。
【0021】
「アルキニル」は、1個または複数の三重炭素−炭素結合を有し、規定の数の炭素原子を通常は有する直鎖または分枝鎖炭化水素基を指している。アルキニル基の例には、エチニル、1−プロピン−1−イル、2−プロピン−1−イル、1−ブチン−1−イル、3−ブチン−1−イル、3−ブチン−2−イルおよび2−ブチン−1−イルが包含される。
【0022】
「アルカノイル」は、アルキルが前記のように定義され、規定の数の炭素原子を通常は包含するアルキル−C(O)−を指しており、カルボニル炭素を包含する。アルカノイル基の例には、ホルミル、アセチル、プロピオニル、ブチリル、ペンタノイルおよびヘキサノイルが包含される。
【0023】
「アルコキシ」および「アルコキシカルボニル」はそれぞれ、アルキルが前記のように定義されるアルキル−O−およびアルキル−O−C(O)−を指している。アルコキシ基の例には、メトキシ、エトキシ、n−プロポキシ、i−プロポキシ、n−ブトキシ、s−ブトキシ、t−ブトキシ、n−ペントキシおよびs−ペントキシが包含される。アルコキシカルボニル基の例には、メトキシカルボニル、エトキシカルボニル、n−プロポキシカルボニル、i−プロポキシカルボニル、n−ブトキシカルボニル、s−ブトキシカルボニル、t−ブトキシカルボニル、n−ペントキシカルボニルおよびs−ペントキシカルボニルが包含される。
【0024】
「ハロ」、「ハロゲン」および「ハロゲノ」は、互換的に使用することができ、フルオロ、クロロ、ブロモおよびヨードを指している。
【0025】
「ハロアルキル」は、1個または複数のハロゲン原子で置換されているアルキル基を指しており、ここで、アルキルは前記のように定義される。ハロアルキル基の例には、トリフルオロメチル、トリクロロメチル、ペンタフルオロエチルおよびペンタクロロエチルが包含される。
【0026】
「シクロアルキル」は、環を含み、規定の数の炭素原子を一般に有する飽和単環式および二環式炭化水素環を指している(即ち、C3〜7シクロアルキルは環員として3、4、5、6または7個の炭素原子を有するシクロアルキル基を指している)。シクロアルキルは、そのような結合が原子価要求に反しない限り、親基に結合していてもよいし、任意の環原子の所で基質に結合していてもよい。同様に、シクロアルキル基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたようなアルキル、アルコキシ、アルコキシカルボニル、アルカノイルおよびハロならびにヒドロキシ、メルカプト、ニトロおよびアミノが包含される。
【0027】
単環式シクロアルキル基の例には、シクロプロピル、シクロブチル、シクロペンチルおよびシクロヘキシルが包含される。二環式シクロアルキル基の例には、ビシクロ[1.1.0]ブチル、ビシクロ[1.1.1]ペンチル、ビシクロ[2.1.0]ペンチル、ビシクロ[2.1.1]ヘキシル、ビシクロ[3.1.0]ヘキシル、ビシクロ[2.2.1]ヘプチル、ビシクロ[3.2.0]ヘプチル、ビシクロ[3.1.1]ヘプチル、ビシクロ[4.1.0]ヘプチル、ビシクロ[2.2.2]オクチル、ビシクロ[3.2.1]オクチル、ビシクロ[4.1.1]オクチル、ビシクロ[3.3.0]オクチル、ビシクロ[4.2.0]オクチル、ビシクロ[3.3.1]ノニル、ビシクロ[4.2.1]ノニル、ビシクロ[4.3.0]ノニル、ビシクロ[3.3.2]デシル、ビシクロ[4.2.2]デシル、ビシクロ[4.3.1]デシル、ビシクロ[4.4.0]デシル、ビシクロ[3.3.3]ウンデシル、ビシクロ[4.3.2]ウンデシルおよびビシクロ[4.3.3]ドデシルが包含される。
【0028】
「シクロアルケニル」は、環を含み、1個または複数の不飽和炭素−炭素結合を有し、規定の数の炭素原子を通常は有する単環式および二環式炭化水素環を指している(即ち、C3〜7シクロアルケニルは、環員として3、4、5、6または7個の炭素原子を有するシクロアルケニル基を指している)。シクロアルケニルは、そのような結合が原子価要求に反しない限り、親基に結合していてもよいし、任意の環原子の所で基質に結合していてもよい。同様に、シクロアルケニル基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたようなアルキル、アルケニル、アルキニル、アルコキシ、アルコキシカルボニル、アルカノイルおよびハロならびにヒドロキシ、メルカプト、ニトロおよびアミノが包含される。
【0029】
「シクロアルカノイル」および「シクロアルケノイル」は、それぞれシクロアルキル−C(O)−およびシクロアルケニル−C(O)−を指し、ここで、シクロアルキルおよびシクロアルケニルは前記のように定義される。シクロアルカノイルおよびシクロアルケノイルに関する言及は通常、カルボニル炭素を除く規定の数の炭素原子を包含する。シクロアルカノイル基の例には、シクロプロパノイル、シクロブタノイル、シクロペンタノイル、シクロヘキサノイル、シクロヘプタノイル、1−シクロブテノイル、2−シクロブテノイル、1−シクロペンテノイル、2−シクロペンテノイル、3−シクロペンテノイル、1−シクロヘキセノイル、2−シクロヘキセノイルおよび3−シクロヘキセノイルが包含される。
【0030】
「シクロアルコキシ」および「シクロアルコキシカルボニル」は、それぞれシクロアルキル−O−およびシクロアルケニル−Oならびにシクロアルキル−O−C(O)−およびシクロアルケニル−O−C(O)−を指し、ここで、シクロアルキルおよびシクロアルケニルは前記のように定義される。シクロアルコキシおよびシクロアルコキシカルボニルに関する言及は通常、カルボニル炭素を除く規定の数の炭素原子を包含する。シクロアルコキシ基の例には、シクロプロポキシ、シクロブトキシ、シクロペントキシ、シクロヘキソキシ、1−シクロブテノキシ、2−シクロブテノキシ、1−シクロペンテノキシ、2−シクロペンテノキシ、3−シクロペンテノキシ、1−シクロヘキセノキシ、2−シクロヘキセノキシおよび3−シクロヘキセノキシが包含される。シクロアルコキシカルボニル基の例には、シクロプロポキシカルボニル、シクロブトキシカルボニル、シクロペントキシカルボニル、シクロヘキソキシカルボニル、1−シクロブテノキシカルボニル、2−シクロブテノキシカルボニル、1−シクロペンテノキシカルボニル、2−シクロペンテノキシカルボニル、3−シクロペンテノキシカルボニル、1−シクロヘキセノキシカルボニル、2−シクロヘキセノキシカルボニルおよび3−シクロヘキセノキシカルボニルが包含される。
【0031】
「アリール」および「アリーレン」は、窒素、酸素およびイオウから独立に選択される0から4個のヘテロ原子を含有する5員および6員の単環式芳香族基を包含するそれぞれ一価および二価の芳香族基を指している。単環式アリール基の例には、フェニル、ピロリル、フラニル、チオフェネイル、チアゾリル、イソチアゾリル、イミダゾリル、トリアゾリル、テトラゾリル、ピラゾリル、オキサゾリル、イソオキサゾリル、ピリジニル、ピラジニル、ピリダジニルおよびピリミジニルが包含される。アリールおよびアリーレン基にはさらに、前記の縮合5員および6員の環を包含する二環式および三環式基が包含される。多環式アリール基の例には、ナフチル、ビフェニル、アントラセニル、ピレニル、カルバゾリル、ベンゾオキサゾリル、ベンゾジオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、ベンゾチオフェネイル、キノリニル、イソキノリニル、インドリル、ベンゾフラニル、プリニルおよびインドリジニルが包含される。これらのアリールおよびアリーレン基は、そのような結合が原子価要求に反しない限り、任意の環原子の所で他の基に結合していてもよい。アリールおよびアリーレン基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アルコキシ、シクロアルコキシ、アルカノイル、シクロアルカノイル、シクロアルケノイル、アルコキシカルボニル、シクロアルコキシカルボニルおよびハロならびにヒドロキシ、メルカプト、ニトロ、アミノおよびアルキルアミノが包含される。
【0032】
「ヘテロアリール」および「ヘテロアリーレン」は、少なくとも1個のヘテロ原子を含有する前記で定義されたそれぞれ一価および二価のアリールおよびアリーレン基を指している。
【0033】
「複素環」および「ヘテロシクリル」は、それぞれ、5員から7員または7員から11員の環員を有する飽和、部分不飽和または不飽和単環式または二環式環を指している。単環式および二環式基はそれぞれ、炭素原子および独立に窒素、酸素またはイオウである1から4個または1から6個のヘテロ原子からなる環員を有し、前記で定義された任意の単環式複素環がベンゼン環に縮合している任意の二環式基を含有してもよい。窒素およびイオウヘテロ原子は、酸化されていてもよい。複素環は、そのような結合が原子価要求に反しない限り、任意のヘテロ原子または炭素原子の所で他の基に結合していてもよい。任意の炭素または窒素環員は、そのような置換が原子価要求に反しない限り、非水素置換基を包含してよい。有用な置換基には、前記で定義されたアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アルコキシ、シクロアルコキシ、アルカノイル、シクロアルカノイル、シクロアルケノイル、アルコキシカルボニル、シクロアルコキシカルボニルおよびハロならびにヒドロキシ、メルカプト、ニトロ、アミノおよびアルキルアミノが包含される。
【0034】
複素環の例には、アクリジニル、アゾシニル、ベンズイミダゾリル、ベンゾフラニル、ベンゾチオフラニル、ベンゾチオフェニル、ベンゾオキサゾリル、ベンズチアゾリル、ベンズトリアゾリル、ベンズテトラゾリル、ベンズイソオキサゾリル、ベンズイソチアゾリル、ベンズイミダゾリニル、カルバゾリル、4aH−カルバゾリル、カルボリニル、クロマニル、クロメニル、シノリニル、デカヒドロキノリニル、2H,6H−1,5,2−ジチアジニル、ジヒドロフロ[2,3−b]テトラヒドロフラン、フラニル、フラザニル、イミダゾリジニル、イミダゾリニル、イミダゾリル、1H−インダゾリル、インドレニル、インドリニル、インドリジニル、インドリル、3H−インドリル、イソベンゾフラニル、イソクロマニル、イソインダゾリル、イソインドリニル、イソインドリル、イソキノリニル、イソチアゾリル、イソオキサゾリル、モルホリニル、ナフチリジニル、オクタヒドロイソキノリニル、オキサジアゾリル、1,2,3−オキサジアゾリル、1,2,4−オキサジアゾリル、1,2,5−オキサジアゾリル、1,3,4−オキサジアゾリル、オキサゾリジニル、オキサゾリル、オキサゾリジニル、ピリミジニル、フェナントリジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、フタラジニル、ピペラジニル、ピペリジニル、プテリジニル、プリニル、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリドオキサゾール、ピリドイミダゾール、ピリドチアゾール、ピリジニル、ピリジル、ピリミジニル、ピロリジニル、ピロリニル、2H−ピロリル、ピロリル、キナゾリニル、キノリニル、4H−キノリジニル、キノキサリニル、キヌクリジニル、テトラヒドロフラニル、テトラヒドロイソキノリニル、テトラヒドロキノリニル、6H−1,2,5−チアジアジニル、1,2,3−チアジアゾリル、1,2,4−チアジアゾリル、1,2,5−チアジアゾリル、1,3,4−チアジアゾリル、チアントレニル、チアゾリル、チエニル、チエノチアゾリル、チエノオキサゾリル、チエノイミダゾリル、チオフェニル、トリアジニル、1,2,3−トリアゾリル、1,2,4−トリアゾリル、1,2,5−トリアゾリル、1,3,4−トリアゾリルおよびキサンテニルが包含される。
【0035】
「アリールアルキル」および「ヘテロアリールアルキル」はそれぞれ、アリール−アルキルおよびヘテロアリール−アルキルを指し、ここで、アリール、ヘテロアリールおよびアルキルは前記のように定義される。例には、ベンジル、フルオレニルメチルおよびイミダゾール−2−イル−メチルが包含される。
【0036】
「脱離基」は、置換反応、脱離反応、付加−脱離反応を包含する分裂プロセスの間に分子から脱離する任意の基を指している。脱離基は、脱離基と分子の間の結合として前は役立っていた電子対と共に基が離れる離核性であってもよいし、基が電子対なしに離れる離電性であってもよい。離核性脱離基の脱離する能力は、その塩基強度に左右され、その際、最も強い塩基が、最も弱い脱離基である。一般的な離核性脱離基には、窒素(例えばジアゾニウム塩から)、アルキルスルホネート(例えばメシレート)、フルオロアルキルスルホネート(例えばトリフレート、ヘキサフレート、ノナフレートおよびトレシレート)およびアリールスルホネート(例えばトシレート、ブロシレート、クロシレートおよびノシレート)を包含するスルホネートが包含される。他には、カルボネート、ハライドイオン、カルボキシレートアニオン、フェノレートイオンおよびアルコキシドが包含される。NH2−およびOH−などのいくつかの比較的強い塩基を、酸で処理することにより、より良好な脱離基にすることができる。一般的な離電性脱離基には、プロトン、CO2および金属が包含される。
【0037】
「鏡像異性体過剰率」または「ee」は、所定の試料での、キラル化合物のラセミ試料に対する一方の鏡像異性体の過剰率の尺度であり、パーセンテージで表される。鏡像異性体過剰率は、100×(er−1)/(er+1)として定義され、ここで、「er」は、多い方の鏡像異性体と少ない方の鏡像異性体との比である。
【0038】
「ジアステレオ異性体過剰率」または「de」は、所定の試料での、等量のジアステレオ異性体を有する試料に対する一方のジアステレオ異性体の過剰率の尺度であり、パーセンテージで表される。ジアステレオ異性体過剰率は、100×(dr−1)/(dr+1)として定義され、ここで、「dr」は、多い方のジアステレオ異性体と少ない方のジアステレオ異性体との比である。
【0039】
「立体選択的」、「エナンチオ選択的」、「ジアステレオ選択的」およびそれらの変形は、それぞれ、他方に対して一方の立体異性体、鏡像異性体またはジアステレオ異性体を多くもたらす所定のプロセス(例えば水素化)を指している。
【0040】
「高度立体選択性」、「高度エナンチオ選択性」、「高度ジアステレオ選択性」およびそれらの変形は、それぞれ、その生成物の少なくとも約90%を構成する一方の立体異性体、鏡像異性体またはジアステレオ異性体の過剰率を有する生成物をもたらす所定のプロセスを指している。一対の鏡像異性体またはジアステレオ異性体では、高度エナンチオ選択性またはジアステレオ選択性は、少なくとも約80%のeeまたはdeに対応するであろう。
【0041】
「立体異性的に富化された」、「鏡像異性的に富化された」、「ジアステレオ異性的に富化された」およびそれらの変形はそれぞれ、他方よりも一方の立体異性体、鏡像異性体またはジアステレオ異性体を多く有する化合物の試料を指している。富化の程度は、全生成物に対して、または一対の鏡像異性体またはジアステレオ異性体に関する%により、またはeeもしくはdeにより測定することができる。
【0042】
「実質的に純粋な立体異性体」、「実質的に純粋な鏡像異性体」、「実質的に純粋なジアステレオ異性体」およびそれらの変形はそれぞれ、試料の少なくとも約95%を構成する立体異性体、鏡像異性体またはジアステレオ異性体を含有する試料を指している。鏡像異性体またはジアステレオ異性体の対では、実質的に純粋な鏡像異性体およびジアステレオ異性体は、約90%以上のeeまたはdeを有する試料に対応するであろう。
【0043】
「純粋な立体異性体」、「純粋な鏡像異性体」、「純粋なジアステレオ異性体」およびその変形はそれぞれ、試料の少なくとも約99.5%を構成する立体異性体、鏡像異性体またはジアステレオ異性体を含有する試料を指している。鏡像異性体およびジアステレオ異性体の対では、純粋な鏡像異性体または純粋なジアステレオ異性体は、約99%以上のeeまたはdeを有する試料に対応するであろう。
【0044】
「反対の鏡像異性体」は、基準分子に対して重なり合わない鏡像である分子を指しており、これは、対照分子の立体由来中心のすべてを逆にすると得ることができる。例えば、基準分子がS絶対立体化学的配置を有する場合、その反対の鏡像異性体は、R絶対立体化学的配置を有する。同様に、基準分子がS,S絶対立体化学的配置を有する場合、その反対の鏡像異性体は、R,R立体化学的配置を有するなどである。
【0045】
規定の化合物の「立体異性体」は、化合物の反対の鏡像異性体を、さらに化合物の任意のジアステレオ異性体または幾何異性体(Z/E)を指している。例えば、規定の化合物がS,R,Z立体化学的配置を有する場合、その立体異性体は、R,S,Z立体配置を有するその反対の鏡像異性体、S,S,Z立体配置およびR,R,Z立体配置を有するそのジアステレオ異性体ならびにS,R,E立体配置、R,S,E,立体配置、S,S,E立体配置およびR,R,E立体配置を有するその幾何異性体を包含するであろう。
【0046】
「溶媒和物」は、開示または請求されている化合物と化学量論的量または非化学量論的量の1種または複数の溶媒分子(例えばEtOH、アセトン、水)を含む分子複合体を指している。
【0047】
「水和物」は、開示または請求されている化合物と化学量論的量または非化学量論的量の水を含む溶媒和物を指している。
【0048】
「薬学的に許容できる複合体、塩、溶媒和物または水和物」は、正当な医学的判断の範囲内であり、過度の毒性、刺激およびアレルギー応答を伴うことなく、患者の組織と接触させて使用するために適していて、妥当な損益比と釣り合い、その意図されている使用に有効である、請求および開示されている化合物の複合体、酸もしくは塩基付加塩、溶媒和物または水和物を指している。
【0049】
「治療する」は、このような用語が適用される障害または状態の進行を逆転、緩和、阻害することもしくはそれらを予防することを、またはこのような障害または状態の1つまたは複数の症状を予防することを指している。
【0050】
「治療」は、直前で定義された「治療する」の行為を指している。
【0051】
表1は、本明細書を通して使用される略語を列記している。
【0052】
【表1−1】
【0053】
【表1−2】
【0054】
下記のいくつかのスキームおよび実施例は、有機化学の当業者には知られている酸化および還元を包含する一般的な反応の詳細を省略していることもある。このような反応の詳細は、Richard Larock、Comprehensive Organic Transformations(1999年)およびMichael B.Smithらによって編集されている複数巻シリーズ、Compendium of Organic Synthetic Methods(1974年以降)を包含する複数の論文で見ることができる。出発原料および試薬は、市場の供給源から得るか、文献方法を使用して調製することができる。例えば、下記に記載の出発原料の1種、(R)−2−メチルコハク酸4−メチルエステルは、対応するジエステルのエステラーゼ仲介加水分解を介して、または適切な不飽和モノエステルの不斉水素化を介して得ることができる。S.G.Cohen&A.J.Milovanovic、J.Am.Chem.Soc.90:3495(1968年)およびM.Ostermeierら、Eur.J.Org.Chem.17:3453(2003年)参照。
【0055】
下記のいくつかの反応スキームおよび実施例では、ある種の化合物は、他の反応部位での望ましくない化学反応を防ぐ保護基を使用して調製することができる。保護基を使用して、溶解性を増強するか、他の場合には化合物の物理的特性を変更することもできる。保護基計画の検討、保護基を導入および除去するための物質および方法の記載ならびにアミン、カルボン酸、アルコール、ケトンおよびアルデヒドを包含する一般的な官能基のために有用な保護基の編集に関しては、T.W.GreeneおよびP.G.Wuts、Protective Groups in Organic Chemistry(1999年)およびP.Kocienski、Protective Groups(2000年)参照。
【0056】
通常、本明細書を通して記載される化学的変換は、実質的に化学量論的量の反応成分を使用して実施することができるが、ある種の反応は、1種または複数の反応成分を過剰に使用することから利益を得ることもある。加えて、多くの反応は、ほぼ室温で実施することができるが、特定の反応は、反応動態、収率および他の事情に応じて、より高い温度(例えば還流まで)またはより低い温度(例えば0℃以下)の使用を必要とすることもある。多くの化学的変換は、反応速度および収率に影響を及ぼしうる1種または複数の相容性の溶媒を使用することもでき、反応成分の性質に応じて、極性プロトン性溶媒(例えば水、MeOH、EtOH、PrOH、i−PrOH、ギ酸、HOAc、ホルムアミド)、極性非プロトン性溶媒(例えばアセトン、THF、MEK、EtOAc、ACN、DMF、DMSO)、非極性溶媒(例えばヘキサン、ベンゼン、トルエン、ジエチルエーテル、CH2Cl2、CHCl3、CCl4)またはこれらのいくつかの組合せであってもよい。濃度範囲、温度範囲またはpH範囲を包含する範囲に関する本開示における任意の言及は、示されている終点を包含する。
【0057】
この開示は、式1の光学活性な環式アミノ酸、その反対の鏡像異性体ならびに式1の化合物およびその反対の鏡像異性体の薬学的に許容できる複合体、塩、溶媒和物および水和物を調製するための物質および方法に関する。前記のように、式1中のR1およびR2は、Me、Et、Pr、i−Pr、n−Bu、s−Bu、t−BuなどのC1〜6アルキル、さらにBn、フェニル−エチルおよびフェニル−プロピルなどのアリール−C1〜3アルキルを包含しうる。したがって代表的な式1の化合物には、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸、(3S,4S)−(1−アミノメチル−3,4−ジエチル−シクロペンチル)−酢酸、(3S,4S)−(1−アミノメチル−3,4−ジプロピル−シクロペンチル)−酢酸および(3S,4S)−(1−アミノメチル−3,4−ジベンジル−シクロペンチル)−酢酸、これらの反対の鏡像異性体ならびに前記化合物の薬学的に許容できる複合体、塩、溶媒和物および水和物が包含される。
【0058】
一般的な反応スキーム
【0059】
【化8】
スキーム1は、式1の化合物およびその反対の鏡像異性体を調製するための方法を示している。方法は、キラル1,4−ビス−求電子体(式2)を保護(マスキング)されているカルボン酸(式3)と反応させて、光学活性なシクロペンタンカルボニトリル(式4)を得ることを包含する。カルボキシ部分の脱保護(非マスキング)により、1−カルボキシル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。ニトリルエステル(式5、R4はHではないか、R4はカチオン、M1ではない)を還元すると、アミンが得られ、これを環化してラクタム(式6)を得、これを加水分解すると、所望の環式アミノ酸(式1)が得られる。別法では、ニトリルエステルの加水分解(必要な場合)、それに続く酸またはその塩(式5、R4=HまたはM1)を還元すると、環式アミノ酸(式1)が得られる。
【0060】
式2および式4〜6中の置換基R1およびR2は、式1に関する前記と同様に定義され、式5中のR4は、H、C1〜6アルキル、アリール−C1〜3アルキルまたはカチオン、M1(例えば第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオン)であり、式2中のX1は、ハロゲノ(例えばCl、Br、I)などの脱離基またはR5O−(ここで、R5はC1〜6アルキルスルホニル(例えばメシル)、フルオロ−C1〜6アルキルスルホニル(例えばトリフリル)またはアリールスルホニル(例えばトシル、ブロシル、クロシルまたはノシル)である)である。式2および式3中の置換基R3は、カルボン酸またはエステルのための保護基である。有用なR3には、
【0061】
【化9】
により表される構造を有するカルボン酸またはエステル保護基が包含され、ここで、「A」は、式3または4の化合物の残りの部分への結合点を表している。式7では、R6、R7およびR8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成する。式8では、Z1およびZ2はそれぞれ独立に、OおよびSから選択され、R9はC1〜6アルキルであり、R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する。有用なR3には、C1〜3アルキルオルトエステル、オルトビシクロオクチルエステルおよび1,1−ビス−C1〜3アルキルスルファニル−メタン−1−イリデンが包含される。
【0062】
式2および式4〜6中の代表的なR1およびR2には、C1〜3アルキルおよびアリール−C1〜3アルキルが包含され、式2中の代表的なX1にはBrおよびトシレートが包含され、式3および式4中の代表的なR3には、4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イルなどのオルトエステルおよびトリメトキシ−メタン−1−イル、トリエトキシ−メタン−1−イルなどのトリC1〜3アルコキシエステルが包含され、式5中の代表的なR4には、H、Me、Et、Pr、i−Pr、Bn、Li+、Na+およびK+が包含される。したがって有用なキラル1,4−ビス−求電子体(式2)には、(R,R)−1,4−ジブロモ−2,3−ジメチル−ブタン、(R,R)−3,4−ビス−ブロモメチル−ヘキサン、(R,R)−4,5−ビス−ブロモメチル−オクタン、(R,R)−1,4−ジブロモ−2,3−ジベンジル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジエチル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジプロピル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジベンジル−ブタンおよびこれらの反対の鏡像異性体が包含される。
【0063】
代表的なマスキングされているカルボン酸(式3)には、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イル)−プロピオニトリル、4,4,4−トリメトキシ−ブチロニトリルおよび4,4,4−トリエトキシ−ブチロニトリルが包含される。代表的な光学活性なシクロペンタンカルボニトリル(式4)には、(S,S)−3,4−ジメチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジメチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジメチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリルおよびこれらの反対の鏡像異性体が包含される。
【0064】
式5の代表的な化合物には、(3S,4S)−(3,4−ジメチル−1−シアノ−シクロペンチル)−酢酸、(3S,4S)−(3,4−ジエチル−1−シアノ−シクロペンチル)−酢酸、(3S,4S)−(3,4−ジプロピル−1−シアノ−シクロペンチル)−酢酸および(3S,4S)−(3,4−ジベンジル−1−シアノ−シクロペンチル)−酢酸、前記の酸のリチウム、ナトリウムおよびカリウム塩、前記の酸のメチル、エチル、プロピル、イソプロピルおよびベンジルエステルならびに前記の酸、塩およびエステルの反対の鏡像異性体が包含される。
【0065】
代表的なラクタム(式6)には、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン、(7S,8S)−7,8−ジエチル−2−アザ−スピロ[4.4]ノナン−3−オン、(7S,8S)−7,8−ジプロピル−2−アザ−スピロ[4.4]ノナン−3−オンおよび(7S,8S)−7,8−ジベンジル−2−アザ−スピロ[4.4]ノナン−3−オンが包含され、これらの反対の鏡像異性体が包含される。
【0066】
前記のように、スキームIに記載の方法は、キラル1,4−ビス−求電子体(式2)と保護(マスキング)されているカルボン酸(式3)とを反応させて、光学的に活性なシクロペンタンカルボニトリル(式4)を得ることを包含し、カルボキシ部分の脱保護(非マスキング)により、1−カルボキシメチル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。シクロペンタンカルボニトリル(式4)を典型的には、極性非プロトン性溶媒(例えばTHF)中、塩基条件下、約−30℃からほぼ室温または約−20℃から約5℃の範囲の温度で調製する。有用な塩基には、LiHMDS、LDAおよびリチウムジエチルアミドなどのヒンダード非求核性塩基が包含される。
【0067】
マスキングされているカルボン酸(式3)は、文献から適合させたプロセスを使用して調製することができる。例えば、CH2CH2/MTBE中、約7〜18℃で約1.5時間、次いで約9℃で約20時間、スクシノニトリルをMeOHおよびHClと反応させて、メチル3−シアノプロパンイミデートクロリドを得ることにより、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イル)−プロピオニトリルを調製することができる。このイミデートを、THF中、約40℃で約15時間、2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオールと反応させて、オルトビシクロオクチルエステルを得る。E.J.Corey&K.Shimoji、J.Am.Chem.Soc.105:1662(1983年)参照。同様に、文献に記載されている条件を使用してメチル3−シアノプロパンイミデートクロリドをメタノリシスすることにより、4,4,4−トリメトキシ−ブチロニトリルを調製することができる。S.M.McElvain&J.P.Schroeder、J.Am.Chem.Soc.71:40(1949年)参照。
【0068】
スキームIに示されているように、式4の化合物のカルボキシ部分の脱保護(非マスキング)により、1−カルボキシメチル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。反応条件は、保護基(R3)の選択に左右され、脱保護を通常、酸加水分解を介して実施する。例えば、室温で約5時間、酸水溶液(例えば6NのHCl、5%クエン酸、42%H3PO4または50%HOAc)と接触させることを介しての中程度の酸性条件(例えば約3以下のpH)が、オルトエステル(例えば4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル、トリメトキシ−メタン−1−イル、トリエトキシ−メタン−1−イル)および1,1−ビス−C1〜3アルキルスルファニル−メタン−1−イリデン(例えば1,3−ジチオラン−2−イリデン)を脱保護するためには十分である。
【0069】
スキームIに従い、シアノエステル(式5、R4はHではないかMではない)を還元し、還元剤で処理することによりその場で環化して、ラクタム(式6)を得る。反応を典型的には、MeOH、EtOHまたはi−PrOHなどのアルコール溶媒中、金属触媒を用いて、水素ガスの存在下、大気圧から250psigの範囲の圧力、ほぼ室温から還流までの範囲の温度で行う。有用な金属触媒には、ニッケル触媒が包含される。反応を慣用の「バッチ法」で行うことができるが、ここで、触媒および実質的にすべての基質(式5)を初めに反応容器に充填し、その後に水素ガスを加えて、変換を行う。別法では、副生成物を低減し、収率を上げるために、反応を「半バッチ」法で実施することができる。半バッチ法では、触媒および水素が、反応の開始時に容器中に存在し、その後に、シアノエステル(式5)を、還元の速度に相応する速度で反応器に供給する。バッチ法においてのように、水素ガスも、ニトリル基の還元の間に反応容器に加える。
【0070】
スキームIに示されているラクタム(式6)を、ほぼ室温からほぼ還流まで、または約80℃から約95℃の範囲の温度で、酸で処理することを介して加水分解して、所望のアミノ酸(式1)またはその塩を得ることができる。酸濃度は、約1%から約50%で変動してよく、モル比は、約1:1から約10:1で変動してよい。有用な酸には、HCl、H2SO4、HBr、HlおよびHNO3などの無機酸ならびにTFAおよびTCAなどの有機酸が包含される。
【0071】
前記のように、シアノエステル(式5)を加水分解して、生じた酸または酸塩を還元すると、式1の化合物を得ることができる。シアノエステル(式5)を、酸または塩基での処理により、または塩基(または酸)での処理により、それに続く酸(または塩基)での処理により加水分解することができる。例えば、シアノエステル(式5)をHCl、H2SO4などで過剰のH2Oと共に処理することにより、酸が生じ、これを塩基(例えばKOH)で処理すると、塩基付加塩を得ることができる。シアノエステル(式5)をLiOH、KOH、NaOH、CsOH、Na2CO3、K2CO3、Cs2CO3などの無機塩基水溶液で、任意選択の極性溶媒(例えばTHF、MeOH、EtOH、アセトン、ACNなど)中で処理することにより、塩基付加塩が得られ、これを酸で処理すると、遊離酸を得ることができる。エステル加水分解は、室温または還流温度までの温度で実施することができる。次いで、生じた遊離酸または塩基付加塩を、シアノエステル(式6)からラクタム(式6)の調製に関して前記された条件下に、還元剤で、例えばH2と接触させて処理することを介して還元する。
【0072】
前記のように、有用な式2のキラル1,4−ビス−求電子体の1種は、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンである。この化合物の2種の結晶形態(形態Aおよび形態B)は、単離されていて、FT−IR、FT−ラマン、PXRDおよびDSCにより特徴づけられている。
【0073】
Smart Golden Gate(商標)単反射ATRアクセサリ(セレン化亜鉛光学部品を備えたダイヤモンドATR結晶)およびd−TGS KBr検出器を備えたThermoNicolet Avatar 360FT−IR分光計を使用して、形態Aおよび形態BのFT−IRスペクトルを得た。スペクトルを2cm−1解像度および128走査の同時付加で集めた。Happ−Genzelアポダイゼーションを使用した。単反射ATRを使用してFT−IRスペクトルを記録したので、試料調製は必要なかった。ATR FT−IRを使用することにより、KBrディスクまたはヌジョール試料調製を使用する吸光FT−IRスペクトルで見られるものとは異なる赤外バンドの相対強度が生じる。ATR FT−IRの性質により、比較的低い波数でのバンドは、比較的高い波数でのバンドよりも強い。他に記載のない限り、実験誤差は±2cm−1であった。ThermoNicolet Omnic 6.1aソフトウェアを使用して、ピークを選んだ。強度の割り当ては、スペクトルでの主なバンドに対してである(即ち、これらは、基線から測定された絶対値をベースとするのではない)。スプリットピークを評価する場合には、その強度値を基線から取ったが、この場合にも、強度は、スペクトルにおける最も強いバンドに対して評価した。
【0074】
1064nmNdYAGレーザーおよびLN−Germanium検出器を備えたRamll FT−Ramanモジュールを伴うBruker Vertex70 FT−IR分光計を使用して、形態Aおよび形態BのFT−ラマンスペクトルを集めた。スペクトルすべてを、2cm−1解像度およびBlackman−Harris 4項アポダイゼーション、350mWレーザー出力および2048走査を使用して記録した。各試料をガラスバイアルから直接計測し、レーザー放射に曝露した。データは、強度およびラマンシフトの関数として表されている。他に記載のない限り、実験誤差は2cm−1であった。ThermoNicolet Omnic 6.1aソフトウェアを使用して、ピークを選んだ。強度の割り当ては、スペクトルでの主なバンドに対してである(即ち、これらは、基線から測定された絶対値をベースとするのではない)。スプリットピークを評価する場合には、その強度値を基線から取ったが、この場合にも、強度は、スペクトルにおける最も強いバンドに対して割り当てた。
【0075】
自動試料チェンジャー、シータ−シータゴニオメーター、自動ビーム広がりスリットおよびPSDVantec−1検出器を備えたBruker−AXS Ltd.D4粉末X線回折計を使用して、粉末X線回折パターンを決定した。低バックグラウンドシリコンウェハ(キャビティを伴う)に試料量をマウントすることにより、試料を分析のために調製した。試料を回転させたが、その間、40kV/35mAで運転されるX線管を用いて、銅K−アルファ1X線(波長=1.5406Å)を照射した。2°から50°の2シータ範囲にわたって0.018°ステップ当たり0.2秒カウントに設定された連続モードで運転するゴニオメーターを用いて、分析を行った。得られたピークを、シリコン参照標準に対して整列させた。1の閾値および0.3°2シータのピーク幅を伴うBruker−AXS Ltd.Evaluationソフトウェアを使用して、ピークを選択した。データを21℃で集めた。他に記載のない限り、実験誤差は、±0.1度2θであった。
【0076】
当業者であれば理解されるように、下記に示されている表2および3の範囲内の様々なピークの相対強度は、例えばX線ビームにおける結晶の配向効果または分析される物質の純度または試料の結晶度などのいくつかのファクターによって変動しうる。ピーク位置も、試料高さの変化でシフトすることがあるが、ピーク位置は、示されている表に定義されているままに実質的にはとどまる。
【0077】
当業者であれば、異なる波長を使用しての測定は、Bragg式−nλ=2d sinθに従い、異なるシフトをもたらすであろうことも理解されるであろう。
【0078】
別の波長を使用することにより生じるこのような他のPXRDパターンは、本発明の結晶物質のPXRDパターンの別の表現であると考えられ、それ自体、本発明の範囲内である。
【0079】
DSCサーモグラムを得るために、TA Instruments Q 1000DSCを使用して、ふたを備えたアルミニウムパン中、窒素パージガスを用いて、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの試料を、1分当たり10℃で20から150℃に加熱した。
【0080】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、932、844、756および1160cm−1に吸収バンド振動数を伴うFT−IRスペクトルを有する。
【0081】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、956、1170、832および1256cm−1に吸収バンド振動数を伴うFT−IRスペクトルを有する。
【0082】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、1163、233、800および165cm−1にラマンバンド振動数を伴うFT−ラマンスペクトルを有する。
【0083】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、1174、1007、740、108および794cm−1にラマンバンド振動数を伴うFT−ラマンスペクトルを有する。
【0084】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、6.5、13.1および21.9度2θに粉末X線回折ピークを有する。
【0085】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、14.5、25.1および26.0度2θに粉末x線回折ピークを有する。
【0086】
図1および図2に示されている形態AのFT−IRスペクトルは、次の波数に吸収バンドを有する(cm−1、w:弱、m:中、s:強):3070w、2979w、2911w、1597w、1492w、1468w、1394w、1374w、1369w、1345s、1303w、1293w、1282w、1242w、1212w、1188s、1160s、1135w、1111w、1097m、1040w、1018w、971w、945s、932s、881w、857m、852m、844s、817s、756m、710wおよび669s。
【0087】
図3および図4に示されている形態AのFT−ラマンスペクトルは、次のラマンシフトにラマンバンドを有する(cm−1、w:弱、m:中、s:強、vs:極強):3191w、3145w、3070vs、3061s、3041w、3011w、2996m、2982m、2948m、2926s、2909s、2886vs、2849w、2789w、2762w、2746w、2731w、2585w、1598s、1575w、1499w、1473m、1417w、1384w、1371w、1352m、1341w、1304w、1293w、1284w、1250w、1212w、1190s、1163vs、1137w、1117w、1101m、1042w、1019w、965w、950w、942w、934w、882w、859w、848w、822w、800s、757m、712w、672w、638s、621w、610w、556w、504w、495w、471w、418w、396m、372w、351w、334w、292s、233m、221m、191w、165s、123vs、91vs、80s、72vsおよび54s。
【0088】
図5および図6に示されている形態BのFT−IRスペクトルは、次の波数に吸収バンドを有する(cm−1、w:弱、m:中、s:強):3055w、2976w、2961w、2919w、2888w、1598w、1495w、1473w、1449w、1404w、1388w、1355s、1344s、1309m、1294m、1256w、1244w、1212w、1189m、1170s、1123w、1096m、1042w、1018m、1007m、965s、956s、922s、857w、832s、812s、791s、705m、668sおよび663s。
【0089】
図7および図8に示されている形態BのFT−ラマンスペクトルは、次のラマンシフトにラマンバンドを有する(cm−1、w:弱、m:中、s:強、vs:極強):3191w、3148w、3073vs、3056m、3040m、3033m、2996s、2986s、2967s、2958s、2929s、2918s、2887m、2848w、2760w、2718w、2591w、1598s、1577w、1496w、1473m、1468m、1456w、1407w、1381w、1356w、1310w、1295w、1263w、1255w、1212w、1190m、1174vs、1149w、1098s、1007m、976w、948w、924w、899w、856w、846w、832w、816m、794vs、740m、707w、670w、665m、635s、585m、574m、562w、556m、530w、509m、480m、420m、407m、375m、365m、342m、317s、293vs、266m、244m、213m、176m、108vs、89vsおよび77vs。
【0090】
好ましい配向効果の結果として、大抵のPXRDピークは強度10%未満である。図9(フルスケール)および図10(30000cpsまで)に示されているパターンから、形態Aの最も強いPXRDピーク20を下記で表2に列挙する。
【0091】
【表2】
【0092】
図11(フルスケール)および図12(30000cpsまで)に示されているパターンから、形態Bの最も強いPXRDピーク20を下記で表3に列挙する。
【0093】
【表3】
【0094】
形態Aおよび形態BのDSCサーモグラムをそれぞれ、図13および図14に示す。(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、87℃±2℃の開始で、89℃±2℃に鋭い吸熱ピークを示す。(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、90℃±2℃の開始で、92℃±2℃に鋭い吸熱ピークを示す。
【0095】
【化10】
スキームIIは、その活性化キラルジオール(式2a)、キラル二ハロゲン化物(式2b)および反対の鏡像異性体を包含するキラル1,4−ビス−求電子体(式2)を調製する方法を示している。方法は、光学活性な2−置換コハク酸モノエステルまたはスクシンアミド酸(式9)をアルキル化剤(式10)と反応させて、2,3−二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を得ることを包含する。二置換モノエステルまたはスクシンアミド酸の還元により、ジオール(式12)が得られ、続いてこれを、例えばスルホニル化剤(式13)と反応させることを介して活性化させる。生じた活性化キラルジオール(式2a)をハライド源(例えば式16)と反応させて、キラル二ハロゲン化物(式2b)を得ることもできる。式9〜12中の置換基R1およびR2は、式1に関する前記と同様に定義され、式9および11中のR11はR1O−またはアミノであり、式2aおよび13中のR5は、式2中のX1に関する前記と同様に定義され、式10中のX2および式13中のX3は脱離基(例えば、ハロゲノ、R5O−)であり、X4はハロゲノであり、M2はカチオン(例えばLi+、Na+、K+などの第1族金属イオンまたはアンモニウム塩)である。
【0096】
式9〜12中の代表的なR1およびR2には、C1〜6アルキルおよびアリール−C1〜3アルキルが包含され、式9中の代表的なR11には、アミノ、メトキシ、エトキシ、n−プロポキシ、i−プロポキシおよびt−ブトキシなどのC1〜6アルコキシならびにベンズオキシなどのアリール−C1〜3アルコキシが包含される。したがって有用な出発原料(式9)には、(R)−2−メチル−コハク酸4−メチルエステル、(R)−2−メチル−コハク酸4−エチルエステル、(R)−2−メチル−コハク酸4−プロピルエステル、(R)−2−メチル−コハク酸4−イソプロピルエステル、(R)−2−メチル−コハク酸4−tert−ブチルエステル、(R)−2−メチル−スクシンアミド酸、(R)−2−メチル−コハク酸4−メチルエステル、(R)−2−エチル−コハク酸4−エチルエステル、(R)−2−エチル−コハク酸4−プロピルエステル、(R)−2−エチル−コハク酸4−イソプロピルエステル、(R)−2−エチル−コハク酸4−tert−ブチルエステルおよび(R)−2−エチル−スクシンアミド酸が包含され、これらの反対の鏡像異性体が包含される。
【0097】
したがって同様に、代表的な2,3−二置換コハク酸モノエステルまたはスクシンアミド酸(式11)には、(R,R)−2,3−ジメチルコハク酸4−メチルエステル、(R,R)−2,3−ジエチル−コハク酸4−メチルエステル、(R,R)−2,3−ジプロピル−コハク酸4−メチルエステル、(R,R)−2,3−ジイソプロピル−コハク酸4−メチルエステル、(R,R)−2,3−ジベンジル−コハク酸4−メチルエステル、(R,R)−2,3−ジメチル−コハク酸4−エチルエステル、(R,R)−2,3−ジエチル−コハク酸4−エチルエステル、(R,R)−2,3−ジプロピル−コハク酸4−エチルエステル、(R,R)−2,3−ジイソプロピル−コハク酸4−エチルエステル、(R,R)−2,3−ジベンジル−コハク酸4−エチルエステル、(R,R)−2,3−ジメチル−スクシンアミド酸、(R,R)−2,3−ジエチルスクシンアミド酸、(R,R)−2,3−ジプロピル−スクシンアミド酸、(R,R)−2,3−ジイソプロピル−スクシンアミド酸および(R,R)−2,3−ジベンジル−スクシンアミド酸が包含され、これらの反対の鏡像異性体が包含される。
【0098】
一置換コハク酸モノエステルまたはスクシンアミド酸(式9)のアルキル化は、相容性の溶媒中で適切な塩基を使用して実施する。適切な塩基には、エステルまたはアミド部分(式9)に隣接する(α)メチレン基を脱保護しうるものが包含される。これらには、非求核性またはヒンダード塩基が包含され、LDA、LHMDS、KHMDS,LICA、LTMP、LiNET2、リチウムジシクロヘキシルアミドなどのリチウムアミド塩基ならびに(i−Pr)2NMgClおよびEt2NMgClなどの対応するマグネシウムアミド塩基が包含される。リチウムおよびマグネシウムアミド塩基はそれぞれ、LiNR1R2およびR1R2NMgX4により表すことができ、ここで、R1およびR2は、式1に関する前記と同様に定義され、X4はハロゲノである。相容性の溶媒には、その共役酸がpKa≦9、典型的には≦4および往々には≦1を有するものが包含される。このような溶媒には例えば、THF、Et2O、DMSO、ACN、DMFおよびアセトンが包含されるが、アンモニアは包含されない。
【0099】
これらの群の塩基および溶媒を使用することにより、所望のanti−ジアステレオ異性体(式11に図示)が過剰に得られる。典型的には、anti−ジアステレオ異性体とsyn−ジアステレオ異性体との比は、約85:15、90:10または92:8に等しいか、それ以上である。したがって、下記の実施例に示されているように、THF中でLHMDSを使用する(R)−2−メチル−コハク酸4−メチルエステルのアルキル化により、anti−ジアステレオ異性体である(R,R)−2,3−ジメチル−コハク酸モノメチルエステルが約80%以上のdeで得られた。反対に、NH3およびEt2O中でLiNH2を使用する(R)−2−メチル−コハク酸4−メチルエステルのアルキル化により、望ましくないsyn−ジアステレオ異性体が過剰に得られる。W.G.Kofron& L.G.Wideman、J.Org.Chem.37:555(1972年)参照。
【0100】
前記のように、アルキル化剤(式10)は、脱離基(X2)を包含し、これには、Cl、BrおよびIなどのハロ置換基ならびにトルエン−p−スルホネート、メチルスルホネート、p−ブロモ−ベンゼン−スルホネートおよびトリフレートなどのスルホネート置換基が包含されうる。したがって、代表的なアルキル化剤(式10)には、MeCl、MeBr、MeI、EtCl、EtBr、EtI、n−PrCl、n−PrBr、n−PrI、i−PrCl、i−PrBrおよびi−PrIなどのC1〜6アルキルハロゲン化物ならびにMeOTs、MeOMs、MeOBs、MeOTf、EtOTs、EtOMs、EtOBs、EtOTf、n−PrOTs、n−PrOMs、n−PrOBs、n−PrOTf、i−PrTs、i−PrMs、i−PrBsおよびi−PrTfなどのC1〜6アルキルスルホネートエステルが包含される。アルキル化剤は、市場供給源から得られるか、知られている方法を使用して調製することができる。
【0101】
アルキル化反応は、化学量論的量の反応成分(即ち、2−置換コハク酸モノエステルまたはスクシンアミド酸とアルキル化剤とのモル比は1:1)を使用すればよいが、変換率を改善し、副生成物を最小化するなどのために、アルキル化ステップは一方の反応成分を過剰に使用することもできる(例えば、1:1.1から1.1:1、1:1.5から1.5:1、2:1から1:2、3:1から1:3のモル比)。同様に、アルキル化反応は、化学量論的量の塩基(即ち2:1の塩基と基質とのモル比)を使用すればよいが、過剰の塩基を使用することもできる(例えば2.1:1、2.5:1、3:1のモル比)。
【0102】
アルキル化を約−30℃から還流の温度で実施することができる。反応を典型的には、室温で実施するが、より高いか低い温度から利益を得ることもある。例えば、実施例に記載されているように、出発原料(式9)を塩基に加え、続いて、アルキル化剤(式10)を加える間、反応混合物を約−30℃から約−25℃に冷却することができる。次いで、生じた混合物を、完了まで室温で反応させることができる。
【0103】
接触スキームは収率に影響を及ぼしうる。実施例に記載されているように、出発原料(式9)およびアルキル化剤(式10)の表面下添加は、表面上反応成分添加に比較すると、anti−ジアステレオ異性体(式10)のdeを上昇させうる。
【0104】
スキームIIに示されているように、THF、MTBEおよびEt2Oなどの1種または複数のエーテル(無水)溶媒中でのLAHとの反応を介して、二置換コハク酸モノエステル(式11)をジオール(式12)に還元する。他の有用な還元剤および溶媒には、ジグリム中のNaBH4およびAlCl3、THF中のB2H6、THF中の9−BBN、THF中のLiAlH(OMe)3、THF中のAlH3、THF中のDIBAL−HならびにトルエンまたはTHF中のRed−Alが包含される。反応は通常、モル過剰の還元剤(例えば>4当量のLAH)を使用し、ほぼ室温から還流までの範囲の温度で行われる。
【0105】
アルキル化においてのように、還元処理の接触スキームは収率に影響を及ぼしうる。LAHを使用する還元後の従来の(Fieser)処理、後続のH2O、15%NaOH水溶液およびH2Oの反応混合物への連続添加は、大きな(kg)規模で行う場合には処理の困難をもたらしうる。例えば、最初の水クエンチは、大量の水素ガスの迅速な放出をもたらし、さらに、かなりの生成物(式12)のフラクションを固体副生成物に捕捉する。多少の捕捉された生成物は、固体の洗浄および濾過により回収することができるが、プロセスは非効率的で、時間を浪費する。それというのも、洗浄液の多くは、フィルターケークを通過するよりもむしろ、その周りを流れるためである。さらに、液体を除去すると、フィルターケークは往々にして、可逆的にひび割れる。これらのひび割れは、洗浄液をフィルターケークの内部から流し、このことがさらに、回収プロセスの有効性を低下させる。
【0106】
実施例に示されているように、反応混合物をかなり過剰の塩基水溶液に供給するように従来の接触スキームを変更することにより、水素の発生速度が低下し、ジオール(式12)の収率および回収が上昇するようである。反応混合物を塩基水溶液に加えるので、アルミニウムアルコキシド中間体は、塩基触媒される加水分解を受けて、所望のジオール(式12)、さらに、溶液から沈殿する水酸化アルミニウムをもたらす。ジオールは溶液中に残るので、これを、液相をデカンテーションすることにより、沈殿物から分離することができる。さらに、反応混合物を塩基水溶液に加える速度を慎重に制御することにより、水素ガス発生の速度を緊密に調節することができる。
【0107】
スキームIIに示されているように、方法は、エステルまたはアミド部分の酸または塩基加水分解を介して、二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を二酸(式14)またはその塩に変換するために準備されていてもよい。例えば、エステルまたはアミドをHClまたはH2SO4で、および過剰のH2Oで処理すると、二酸が生じる。同様に、コハク酸モノエステルまたはスクシンアミド酸をLiOH、KOH、NaOH、CsOH、Na2CO3、K2CO3またはCs2CO3などの無機塩基水溶液で、任意選択の極性溶媒(例えばTHF、MeOH、EtOH、アセトンまたはACN)中で処理すると、二酸の塩基付加塩が得られ、これを酸で処理すると、遊離二酸が生じる。通常、過剰の酸または塩基を使用し、エステルおよびアミド加水分解を室温で、または還流までの温度で実施する。
【0108】
二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を加水分解した後に、生じた二酸(式14)またはその塩を無水酢酸で処理すると、環式無水物(式15)が得られる。反応は通常、THFなどの非プロトン性極性溶媒中、ほぼ室温から還流までの範囲の温度で行うが、約50℃から約75℃の範囲の反応温度を使用することもできる。過剰の無水酢酸(例えば1.5当量以上)を使用して、ジエステルの完全な変換を保証することもできる。環式無水物(式15)を還元するか、コハク酸モノエステル、スクシンアミド酸(式11)または二酸(式14)を還元することにより、ジオールを調製することができる。反応を典型的には、過剰の還元剤(例えばLAH)を極性非プロトン性溶媒(例えばTHF)中、約40℃から還流までの範囲の温度で用いて行う。
【0109】
代表的な反応基質(式14)には、(R,R)−2,3−ジメチル−コハク酸、(R,R)−2,3−ジエチル−コハク酸、(R,R)−2,3−ジプロピル−コハク酸、(R,R)−2,3−ジイソプロピル−コハク酸および(R,R)−2,3−ジベンジル−コハク酸が包含され、それらの塩を包含する。代表的な環式無水物(式15)には、(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン、ジエチル−ジヒドロ−フラン−2,5−ジオン、(R,R)−3,4−ジプロピル−ジヒドロ−フラン−2,5−ジオン、(R,R)−3,4−ジイソプロピル−ジヒドロ−フラン−2,5−ジオンおよび(R,R)−3,4−ジベンジル−ジヒドロ−フラン−2,5−ジオンが包含され、これらの反対の鏡像異性体を包含する。
【0110】
環式無水物(式15)を介してのジオール(式12)の調製は、モノエステルまたはアミド(式11)からジオール(式12)への直接的な還元を上回る利点をもたらしうる。例えば、モノエステルとは逆に、環式無水物は容易に再結晶化するので、還元の前に単離することができる。環式無水物の再結晶化は、モノアルキル化副生成物および不所望のジアステレオ異性体の形成を抑制することにより、活性化ジオール(式9a)の下流単離の効率を改善するようである。加えて、還元剤(例えばLAH)が、不純物によって、またはカルボン酸部分によって消費されないので、結晶環式無水物のより高い純度は、還元ステップの改善された処理量をもたらすはずである。ジオール(式12)の比較的高い純度によっても、再結晶化を介しての単離が可能になる。
【0111】
スキームIIに示されているように、ジオール(式12)を、式13の化合物との反応を介して活性化させる。有用なジオールには、(R,R)−2,3−ジメチル−ブタン−1,4−ジオール、(R,R)−2,3−ジエチル−ブタン−1,4−ジオール、(R,R)−2,3−ジプロピル−ブタン−1,4−ジオール、(R,R)−2,3−ジイソプロピル−ブタン−1,4−ジオールおよび(R,R)−2,3−ジベンジル−ブタン−1,4−ジオールが包含され、これらの反対の鏡像異性体を包含する。式13の有用な化合物には、TsCl、MsCl、BsCl、NsClおよびTfClなどのスルホニル化剤ならびにこれらの対応する無水物(例えば無水p−トルエンスルホン酸)が包含される。
【0112】
式12の化合物を、ピリジンまたはEt3Nおよび酢酸エチル、CH2Cl2、ACNまたはTHFなどの非プロトン性溶媒の存在下にTsClまたはMsClと反応させると、前記のようなジトシレートまたはジメシレート、例えば、(R,R)−1,4−ビス−(メタンスルホニルオキシ)−2,3−ジメチル−ブタン、(R,R)−2,3−ジエチル−1,4−ビス−(メタンスルホニルオキシ)ブタン、(R,R)−1,4−ビス−(メタンスルホニルオキシ)−2,3−ジムプロピル−ブタン、(R,R)−2,3−ジイソプロピル−1,4−ビス−(メタンスルホニルオキシ)−ブタンまたは(R,R)−2,3−ジベンジル−1,4−ビス−(メタンスルホニルオキシ)−ブタン(これらの反対の鏡像異性体を包含する)を得ることができる。典型的には、反応を過剰(例えば2.5当量以上)のスルホニル化剤(式13)を用いて、過剰の塩基(例えば3当量以上)を用いて、ほぼ室温以下(例えば約0℃)の温度で実施する。
【0113】
前記のように、活性化ジオール(式2a)をハライド源(式16)と反応させると、キラル二ハロゲン化物(式2b)を得ることができる。ハライド塩(例えばLiBr、NaBr、KBr、LiCl、NaCl、KCl、LiI、NaI、KIなど)を極性非プロトン性溶媒(例えばアセトン)中、または非極性溶媒(例えばトルエン)中、少量の水および相転位触媒(例えばnBu4NBr、nBu4NCl、nBu4NIなど)と共に使用して、反応を実施することができる。反応を典型的には、化学量論的に過剰のハライド塩(例えば3当量以上)を用いて、還流までの温度で実施する。
【0114】
本開示に記載の化合物の多くは、薬学的に許容できる塩を形成しうる。これらの塩には、酸付加塩(二酸を包含する)および塩基塩が包含される。薬学的に許容できる酸付加塩には、塩酸、硝酸、リン酸、硫酸、臭化水素酸、ヨウ化水素酸、フッ化水素酸および亜リン酸などの無機酸に由来する非毒性の塩、さらに、脂肪族モノカルボン酸およびジカルボン酸、フェニル置換アルカン酸、ヒドロキシアルカン酸、アルカン二酸、芳香族酸、脂肪族および芳香族スルホン酸などの有機酸に由来する非毒性の塩が包含される。したがってこのような塩には、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、硝酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、トリフルオロ酢酸塩、プロピオン酸塩、カプリル酸塩、イソ酪酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、マンデル酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、フタル酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸、フェニル酢酸塩、クエン酸塩、乳酸塩、リンゴ酸塩、酒石酸塩およびメタンスルホン酸塩が包含される。
【0115】
薬学的に許容できる塩基塩には、アルカリ金属またはアルカリ土類金属カチオンなどの金属カチオン、さらにアミンを包含する塩基に由来する非毒性塩が包含される。適切な金属カチオンの例には、ナトリウムカチオン(Na+)、カリウムカチオン(K+)、マグネシウムカチオン(Mg2+)およびカルシウムカチオン(Ca2+)が包含される。適切なアミンの例には、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミン、N−メチルグルカミンおよびプロカインが包含される。有用な酸付加塩および塩基塩の検討に関しては、S.M.Bergeら、「Pharmaceutical Salts」、J.Pharm.Sci.、66:1〜19(1977年)参照、さらにStahlおよびWermuth、Handbook of Pharmaceutical Salts:Properties,Selection,and Use(2002年)参照。
【0116】
化合物の遊離塩基(または遊離酸)を十分な量の所望の酸(または塩基)と接触させて、非毒性の塩を製造することにより、酸付加塩(または塩基塩)を調製することができる。次いで、溶液から沈殿する場合には濾過により、または蒸発により、塩を回収して、塩を単離することができる。さらに、酸付加塩を塩基と(または塩基塩を酸と)接触させることにより、遊離塩基(または遊離酸)を再生することもできる。化合物の遊離塩基、遊離酸または両性イオンのいくつかの物理的特性(例えば可溶性、結晶構造、吸湿性)は、その酸または塩基付加塩とは異なることがある。しかしながら通常は、化合物の遊離酸、遊離塩基または両性イオンに対する言及は、その酸および塩基付加塩を包含する。
【0117】
開示および請求されている化合物は、非溶媒和形態および溶媒和形態の両方で、さらに塩以外の他のタイプの複合体としても存在しうる。有用な複合体には、包接化合物または化合物−ホスト包接複合体が包含され、ここで、化合物およびホストは、化学量論的または非化学量論的量で存在する。有用な複合体はさらに、2種以上の有機、無機または有機および無機成分を化学量論的または非化学量論的量で含有してもよい。生じる複合体は、電離しているか、部分的に電離しているか、非電離であってよい。このような複合体の総説に関しては、J.K.Haleblian、J Pharm Sci.64(8):1269〜1288(1975年)参照。薬学的に許容できる溶媒和物は、結晶溶媒が同位体置換されている、例えばD2O、d6−アセトン、d6−DMSOである水和物および溶媒和物も包含する。通常、この開示では、化合物の非溶媒和形態に関する言及は、化合物の対応する溶媒和形態または水和形態も包含する。
【0118】
本出願に開示されている化合物のうちの数種は、不斉炭素、イオウまたはリン原子(立体由来中心)を含有することがあるので、光学的に活性な立体異性体(即ち鏡像異性体対の一方の鏡像異性体)として存在しうる。化合物のうちの数種はさらに、アルケニルまたは環式基を含有することがあるので、シス/トランスまたは(Z/E)立体異性体(ジアステレオ異性体)が可能になる。さらに他の化合物は、2個以上の立体由来中心を含有することがあるので、それぞれが光学活性でありうる(即ち、鏡像異性体対の一方の鏡像異性体を含む)ジアステレオ異性体が可能になる。最後に、化合物のうちの数種は、ケトまたはオキシム基を含有することもあるので、互変異性が生じることもある。このような場合、本開示の範囲は、すべての互変異性体およびすべての立体異性体を包含し、これには、純粋であるか、実質的に純粋であるか、混合物であるかにかかわらず、鏡像異性体、ジアステレオ異性体およびZ/E異性体を包含する。
【0119】
本明細書に開示されている任意の化合物の所望の鏡像異性体を、従来の分割、キラルクロマトグラフィーまたは再結晶化を介してさらに富化することもできる。例えば、鏡像異性体の混合物を鏡像異性的に純粋な化合物(例えば酸または塩基)と反応させると、それぞれが単一の鏡像異性体から構成されるジアステレオ異性体対を得ることができ、これらを、分別再結晶またはクロマトグラフィーを介して分離する。続いて、所望の鏡像異性体を、適切なジアステレオ異性体から再生することができる。加えて、鏡像異性体を十分な量で利用することができる場合には、所望の鏡像異性体を適切な溶媒中で再結晶化することにより、さらに富化することができる(例えば典型的には、ee約85%以上、場合によってはee約90%以上)。
【0120】
開示されている化合物はさらに、少なくとも1個の原子が、同じ原子数を有するが、自然に通常存在する原子質量とは異なる原子質量を有する原子に置換されているすべての薬学的に許容できる同位体バリエーションを包含する。開示されている化合物中に含まれるために適している同位体の例には、2Hおよび3Hなどの水素の同位体、13Cおよび14Cなどの炭素の同位体、15Nなどの窒素の同位体、17Oおよび18Oなどの酸素の同位体、31Pおよび32Pなどのリンの同位体、35Sなどのイオウの同位体、18Fなどのフッ素の同位体、36Clなどの塩素の同位体が含まれる。同位体バリエーション(例えばジュウテリウム、2H)の使用は、より大きな代謝安定性、例えば高いインビボ半減期または低い用量要求から生じるある種の治療的利点をもたらしうる。加えて、開示されている化合物のある種の同位体バリエーションは、放射性同位体(例えばトリチウム、3Hまたは14C)を含んでもよく、これは、薬物および/または基質組織分布研究で有用でありうる。
【実施例】
【0121】
次の実施例は、例示的かつ非限定的であることが意図されており、本発明の具体的な実施例を表している。
【0122】
(実施例1)
(R,R)−2,3−ジメチル−コハク酸モノメチルエステルの調製
(R)−2−メチルコハク酸4−メチルエステル(24.5105g、0.1677mol)のTHF(25mL)溶液を濾過して、白色の固体(26.3mg、水に可溶性、CH2Cl2に不溶性)を除去し、LHMDS/THF(1.0M、360mL、0.360mol、2.15当量)の−30℃溶液に、温度が−25℃未満に維持される速度で加えた(1.5時間)。混合物を−10℃に加温し、1時間攪拌し、−30℃に再び冷却し、ヨウ化メチル(25.12g、0.1770mol、1.06当量)のTHF(25mL)溶液で、温度が−25℃を超えないような速度で処理した(1.5時間)。混合物を−25℃で2時間攪拌し、次いで室温に加温し、15.5時間攪拌し、0℃に冷却し、NH4Cl(25g、0.467mol、2.79当量)のH2O(75mL)溶液で、温度が約0℃を超えないような速度で、慎重にクエンチした(14℃までの短いエクスカーションは除く。最初の2mLを30分にわたって加え、残りを1時間にわたって加える)。混合物をH2O(100mL)で希釈して、固体を溶かし、層を分離した。有機層をEt3N(2.4mL)で処理して、残りのヨウ化メチルをクエンチし、廃棄した。水性層を6NのHClでpH1.92まで酸性化し、MTBE(4×150mL)で抽出した。水性層(約pH3)を廃棄した。有機抽出物を合わせ、真空濃縮して、13C−NMRおよび1H−NMRにより前記表題化合物と識別される暗コハク色のオイルにした。anti/syn/モノメチルの比は、GCにより88.5:7.8:3.7であると決定された。重量:28.01g;13C−NMR(100MHz,CDCl3):δ180.97(s);175.67(s);51.80(q);41.36(d);41.23(d);13.50(q);13.40(q);1H−NMR(400MHz,CDCl3):δ8.53(1H,br s);3.69(3H,s);2.84(2H,重複した多重線);1.20(3H,d,J=4.3Hz);1.18(3H,d,J=4.4Hz)。
【0123】
(実施例2)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
実施例1からの暗コハク色のオイル(27.82g)をTHF(137mL、不溶物を除去するために濾過、57.1mg)で希釈し、0℃のLAH(16.48g、0.4343mol、2.61当量)のTHF(434mL)懸濁液に40分にわたって加えた。混合物を5℃で1時間、次いで30℃で17.5時間攪拌し、次いで、0℃に冷却した。H2O(16.5g、0.916mol、5.50当量)を70分にわたって、続いて、15%NaOH水溶液(16.5mL)を10分にわたって、続いてH2O(50mL)を10分にわたって加えることにより、混合物を慎重にクエンチした。3種の溶液をすべて、内部温度が5℃から15℃に維持される速度で加えた。オフホワイト色のスラリーを濾過し(粗いフリット、ゆっくりとした濾過)、THF(165mL)ですすいだ。濾液をオイルに濃縮した。水を共沸により除去するために、オイルをトルエン(135mL)で希釈し、蒸留して、13C−NMRおよび1H−NMRにより前記表題化合物と識別される淡褐色のオイルにした。dl:メソ:モノメチルの比は、GCにより92.65:5.91:1.43であると決定された。重量:17.16g(0.1452mol、(R)−2−メチル−コハク酸4−メチルエステルから全体で87.2%;13C−NMR(100MHz,CDCl3):δ65.56(t);37.18(d);13.13(q);1H−NMR(400MHz,CDCl3):δ0.85(6H,d,J=6.6Hz);1.72(2H,多重線);3.45(2H,dd,J=10.9,6.5Hz);3.55(2H,dd,J=10.9,4.4Hz);4.10(2H,d,J=7.4Hz)。
【0124】
(実施例3)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(15.75g、0.1333mol)および塩化p−トルエンスルホニル(63.53g、0.3332mol、2.50当量)のACN(169mL)溶液を0℃に冷却し、そのままのEt3N(56mL、40.66g、0.4018mol、3.01当量)で、温度が5℃を上回らない速度で処理した(35分)。混合物を0℃で2時間、次いで室温で19時間攪拌した。混合物をEtOAc(107mL)およびH2O(103mL)で希釈した。相を分離し、水性フラクションをEtOAc(2×92mL)で抽出した。有機層を合わせ、H2O(115mL)で、次いで10%NaHCO3水溶液(103mL)および25%NaCl水溶液80mLで洗浄し、オフホワイト色/黄色の固体に濃縮した。固体をMTBE(345mL)およびEtOH(27g)に60℃で溶かし、7時間にわたって0℃に冷却し、0℃で16時間攪拌し、濾過した。ケークを0℃のMTBE(2×25mL)で洗浄し、N2流により乾燥させると、13C−NMRおよび1H−NMRにより前記表題化合物として識別される白色の固体が得られた。dl:メソ:モノメチルの比は、HPLCにより96.72:1.79:1.49であると決定された。重量:38.20g(0.8956mol、67.2%);13C−NMR(100MHz,CDCl3):δ144.91(s);132.71(s);129.90(d);127.82(d);72.59(t);32.97(d);21.62(q);11.20(q);1H−NMR(400MHz,CDCl3):δ0.75(6H,d,J=6.7Hz);1.97(2H,多重線);2.47(6H,s);3.84(4H,d,J=5.5Hz);7.36(4H,d,J=8.1Hz);7.77(4H,d,J=8.2Hz)。
【0125】
(実施例4)
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンの調製
−22℃の(R)−2−メチル−コハク酸4−メチルエステル(133.5kg、912mol)のTHF(125kg)溶液を、−34℃のTHF(1190L)中のLHMDS(322.6kg、1928mol、2.11当量)に1.5時間にわたって加えたが、この間、反応混合物を−34℃から−26℃に維持した。基質をTHF(10kg)ですすいだ。溶液を−26℃から−30℃で1時間攪拌し、次いで、1.5時間にわたって12℃に加温した。混合物を−12℃から−10℃で5分間攪拌し、次いで、7.8時間にわたって−34℃に冷却した。−21℃のMeI(140kg、986mol、1.08当量)のTHF(153L)溶液を、ジアニオン溶液に5時間にわたって加えたが、この間、−34℃から−27℃を維持し、THF(45L)ですすいだ。混合物を−27℃から−29℃に4時間攪拌し、1.5時間にわたって20℃に加温し、20℃から21℃で12時間攪拌し、5℃に冷却した。NH4Cl(136kg、2543mol、2.79当量)の水(400L)溶液を7.3時間にわたって加えたが、その間、混合物の温度を5℃から25℃に維持した。水(540L)を加え、上部相を廃棄した。下部水性相に、水(240L)を加え、続いて、pHを、HCl(300kg、35重量%、2880mol、3.16当量)で1に調節したが、この間、反応混合物の温度を4℃から11℃に維持した。水(10L)ですすいだ後に、生成物をMTBE(4×304kg)で抽出し、濃縮すると、(R,R)−2,3−ジメチル−コハク酸モノメチルエステルがオイル(167kg、内部標準GCにより78.6重量%、収率89.7%、シス6.4面積%、デス−メチル0.4%、トリメチル3.3%)として得られた。
【0126】
粗製(R,R)−2,3−ジメチル−コハク酸モノメチルエステル(350.81g、78.6重量%、1.72mol)の試料を、水(500mL)と混合して、二相混合物を得た。この混合物に、NaOH(50重量%、351.45g、4.39mol、2.55当量)を加えたが、この間、混合物の温度を45℃以下に維持した。混合物を45℃で10分間攪拌し、次いで、pHを、HCl(438g、37.5重量%、4.50mol、2.62当量)で10.4から0.5に調節したが、その間、混合物の温度を30℃以下に維持した。溶液をEtOAc(3×1L)で抽出し、MgSO4上で乾燥させ、粘稠なスラリー(正味重量412g)に濃縮した。無水酢酸(250mL、2.645mol、1.54当量)を加え、混合物を109℃に加温した。NMRは、環式無水物への完全な変換を示し、後続の実験は、環化反応が75℃で迅速であることを示した。溶液を50℃に冷却し、シーディングして、スラリーを得た。tert−アミルアルコール(1l)を加え、スラリーを−8℃に冷却した。沈殿物を、真空濾過により集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させて、(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンが白色の固体(196.91g、89.4%、(R)−2−メチル−コハク酸4−メチルエステルから80.2%)として得られた。融点103.5〜105.4℃、[α]25D=103.07°(ジオキサン、c=1.00)。1H NMR(400MHz,DMSO−d6)δ1.23(d,J=7Hz,6H)、2.98(八重線,J=4Hz,2H);13C NMR(100MHz,DMSO−d6)δ12.73、42.12、174.57;MS(EICI)m/z(相対強度)127[(M−H)−,100];元素分析 C6H8O3の計算値:C,56.25;H,6.29;N,0.00;実測値:C,56.24;H,6.25;N,<0.05;(R,R)−2,3−ジメチル−コハク酸モノメチルエステルへのメタノリシスは、GCにより1.3%のシス異性体および<0.1%の何らかの他の関連不純物を示した。
【0127】
(実施例5)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
45℃の(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン(40.04g、312.52mmol)のMTBE(440mL)およびTHF(58mL)溶液に、LAHのTHF溶液(175mL、2.4M、420mmol)を、滴下漏斗を介して0.5時間にわたって滴加したが、この間、反応混合物を45℃から54℃(還流)の温度に維持し、続いて、THFですすいだ(10mL)。LAH添加の初めの150mLでは、混合物は、攪拌可能なスラリーであるが、添加の終了時には、溶液になった。強く、やや遅い発熱が、添加の初めの150mLで存在し、続いて、最後の25mLでは非常に弱い吸熱が存在した。生じた溶液を、NaOH(50%、1.38g、17.25mmol、0.055当量)、水(55mL)およびTHF(275mL)の−7℃二相混合物に40分にわたってカニューレ添加したが、この間、反応混合物を13℃以下の温度に維持した。残りの溶液をTHF(40mL)ですすぎ、生じたスラリーを1時間にわたって55℃に加温し、55℃で2時間攪拌した。沈殿物を真空濾過により55℃で除去し(濾過時間4分)、MTBE(330mL)およびMeOH(28mL)の55℃混合物で2回洗浄した(各洗浄で濾過15分)。合わせた濾液をMgSO4上で乾燥させ、透明にし、真空濃縮して軽いオイル(47.43g)にした。分枝鎖オクタン(100g)を加え、二相混合物を20℃でシーディングすると、5分間攪拌した後にスラリーが得られた。分枝鎖オクタン(200g)を加え、混合物を3℃に冷却した。真空濾過により、沈殿物を集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(34.24g、92.7%)として得られた。mp42.5〜44.5℃、[α]25D=103.07°(ジエチルエーテル、c=1.00);1H NMR(400MHz,CDCl3)δ0.84(d,J=7Hz,6H)、1.71(m,2H)、3.44(dd,J=6.5Hz,J=11Hz,2H)、3.54(dd,J=6.5Hz,J=11Hz,2H);13C NMR(100MHz,CDCl3)δ13.21、37.25、65.53;MS(EICI)m/z(相対強度)117[(M−H)−,100];元素分析 C6H14O2の計算値:C,60.98;H,11.94;N,0.00;実測値:C,60.91;H,12.27;N,<0.05。
【0128】
(実施例6)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
−10℃の塩化p−トルエンスルホニル(20.255g、106.24mmol、2.52当量)のACN(75mL)スラリーに、(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(4.982g、42.16mmol)を加えた。Et3N(17.7mL、127.0mmol、3.01当量)を1分にわたって加えたが、その間、反応混合物を0℃以下の温度に維持した。スラリーを0℃から5℃で3.5時間攪拌し、20℃に加温した。水(40mL)およびEtOAc(60mL)を順次加え、相を29℃で分離した。水性層をEtOAc(2×50mL)で洗浄し、合わせた有機層を水(40mL)、10%NaHCO3水溶液(40mL)および飽和NaCl水溶液(40mL)で洗浄した。有機フラクションをMgSO4上で乾燥させ、真空濃縮して、オイル22.56gにした。トルエン(100mL)を加えて、溶液を得た。分枝鎖オクタン(50mL)を加えた後に、生成物を10分にわたって結晶化させた。分枝鎖オクタン(150mL)を加え、真空濾過により、沈殿物を集めた。固体を分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(15.563g、86.5%)として得られた;融点83.1〜83.6℃、[α]25D=−5.55(酢酸エチル、c=100);1H NMR(400MHz,CDCl3)δ0.75(d,J=6Hz,6H)、1.97(m,2H)、2.47(s,6H)、3.85(d,J=6Hz,4H)、7.37(d,J=8Hz,4H)、7.78(d,J=8Hz,4H);13C NMR(100MHz,CDCl3)δ11.20、21.64、32.96、72.51、127.84、129.90、132.70、144.90;MS(TSP)m/z(相対強度)427[(M+H)+,20]、444[(M+H2O)+,100];元素分析 C20H26O6S2の計算値:C,56.32;H,6.14;N,0.00;実測値:C,56.25;H,6.00;N,0.05;HPLC:(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、99.6面積%;(R,S)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、0.08面積%;(R)−2−メチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、0.08面積%。
【0129】
(実施例7)
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンの調製
−30℃のLHMDSのTHF溶液(24.4重量%、518.5g、756.1mmol、2.12当量)に、THF(52mL)中の(R)−2−メチルコハク酸4−メチルエステル(52.127g、356.69mmol)を0.5時間にわたって加えたが、この間、反応混合物温度を−29℃に維持した。基質をTHF(6mL)ですすいだ。混合物を−29℃で25分間攪拌し、次いで、MeI(53.32g、375.65mmol、1.05当量)のTHF(105mL)溶液を0.5時間にわたって加えたが、この間、反応混合物を−30℃に維持した。MeIをTHF(15mL)ですすいだ。混合物を−30℃で2時間攪拌し、0℃に加温し、1時間攪拌した。GCは、(R,R)−2,3−ジメチル−コハク酸モノメチルエステル78.8面積%、(R)−2−メチルコハク酸4−メチルエステル15.0面積%およびメソ異性体(R,S)−2,3−ジメチル−コハク酸6.2面積%の混合物を示した。
【0130】
NaOH(50.0%、57.0g、713mmol、2.00当量)および水(200mL)の混合物を10分にわたって加えたが、この間、反応混合物の温度を0℃に維持した。混合物を20℃で16時間攪拌し、亜硫酸水素ナトリウム(SO266.6%、1.68g、17.5mmol、0.049当量)を加えた。pHをHCl(37.5重量%、208.6g、2.145mol、3.01当量)で10.67から0.11に調節したが、この間、反応混合物の温度を25℃以下に維持した。相を分離し、トルエン(200mL)を、続いて飽和NaCl水溶液(50mL)を有機層に加えた。相を分離し、水性層をEtOAc(750mL)で抽出した。合わせた有機フラクションをMgSO4上で乾燥させ、真空濃縮して、物質90.91gにした。トルエン(150mL)を加え、スラリーを75℃に加温すると、溶液が得られた。無水酢酸(44.67g、437.6mmol、1.23当量)を5分にわたって加え、混合物を75℃で0.5時間攪拌すると、この時点で、NMRは完全な変換を示した。溶液を30℃に冷却し、分枝鎖オクタン(200mL)およびt−アミルアルコール(200mL)を加えた。シーディングの後に、生成物を結晶化させ、分枝鎖オクタン(200mL)を加えた。スラリーを−8℃に冷却し、真空濾過により、沈殿物を集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、(R,R)−2,3−ジメチル−コハク酸がベージュ色の固体(29.38g、64.3%)として得られた;1H NMR(400MHz,DMSO−d6)δ1.23(d,J=7Hz,6H)、2.98(八重線,J=4Hz,2H);13C NMR(100MHz,DMSO−d6)δ12.73、42.12、174.58;13C NMRは、93.7%の(R,R)−2,3−ジメチル−コハク酸、2.9%のメソ−異性体(10.75、38.14ppm)および3.5%のデス−メチル(14.74、35.33、35.74ppm)を示した。
【0131】
(実施例8)
(R,R)−2,3−ジメチルコハク酸モノメチルエステルの調製
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン(40.06g、312.6mmol)のMeOH(400mL)溶液を65℃で6時間還流させた。生じた溶液を真空濃縮すると、前記表題化合物が淡ベージュ色のオイル(49.80g、99.4%)として得られた;1H NMR(400MHz,CDCl3)δ1.19(t,J=7Hz,6H)、2.82(q,J=7Hz,1H)、2.88(q,J=7Hz,1H)、3.70(s,3H)、10.61(bs,1H);13C NMR(100MHz,CDCl3)δ13.41、13.52、41.20、41.38、51.92、175.59、181.43;MS(EICI)m/z(相対強度)159[(M−H)−,100];元素分析 C7H12O4の計算値:C,52.49;H,7.55;N,0.00;実測値:C,52.20;H,7.76;N,<0.05。
【0132】
(実施例9)
(R,R)−2,3−ジメチルコハク酸の調製
粗製(R,R)−2,3−ジメチルコハク酸モノメチルエステル(3.2177g、20.09mmol GC:85.9面積%、6.1面積%のシス異性体、1.8%面積%のデスメチル不純物、3.3面積%のトリメチル不純物)および水(11mL)の二相混合物に、50重量%NaOH水溶液(4.08g、50.94mmol、2.54当量)を加えたが、その間、反応混合物の温度を25℃以下に維持した。生じた溶液を20℃で39分間攪拌し、次いで、HCl(37.5重量%、5.36g、55.1mmol)を加えた。生じた溶液をCH2Cl2(5mL、次いで2×10mL)およびEtOAc(3×35mL)で抽出した。有機層をMgSO4上で乾燥させ、乾燥まで濃縮した。粗製の固体をi−PrOH(10mL)に溶かし、分枝鎖オクタン(30mL)を加えた。溶液を全体体積20mLまで真空濃縮し、冷却すると、スラリーが得られた。分枝鎖オクタン(10mL)を加え、混合物を0℃に冷却した。沈殿物を真空濾過により集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(1.4592g、49.7%)として得られた;1H NMR(400MHz,DMSO−d6)δ1.02(d,J=7Hz,6H)、2.58(m,2H);13C NMR(100MHz,DMSO−d6)δ13.40、40.96、176.38;13C−NMRは、1.6%のメソ異性体(14.77ppm、41.87ppm)を示した。
【0133】
(実施例10〜21)
表面下添加を介しての(R,R)−2,3−ジメチル−コハク酸モノメチルエステルの調製
LHMDS(1300kg、24.9重量%。1.93kg−mol、2.1当量)のTHF溶液をタンクに充填し、−30℃に冷却した。(R)−2−メチル−コハク酸4−メチルエステル(133kg、0.91kg−mol、1当量)を、等体積のTHFと混合し、その出口が、容器攪拌機の先端から約0.3mであるように反応器に取り付けられている浸漬管を使用して、リチウム試薬に対して表面下で加えた。基質を加えている間、反応混合物の温度を−25℃以下に維持した。生じた混合物を攪拌し、−10℃に加温し、次いで−30℃に冷却した。MeI(136kg、0.96kg−mol、1.05当量)を2体積のTHFと混合し、反応混合物に対して表面下で供給したが、その間、反応混合物の温度を−25℃以下に維持した。温度を8時間にわたって室温に調節した。NH4Cl(136kg)を水(400L)に溶かし、反応器容器に徐々に供給して、反応をクエンチした。さらなる水(550L)を攪拌しながら加え、次いで、攪拌機を止めて、相を分離した。有機相を廃棄した。水性相を37%HCl(300kg)および水(250L)の混合物で酸性化して、MTBE(4×400L)で抽出した。MTBE相を合わせ、蒸留すると、前記表題化合物がオイルとして得られた。
【0134】
表4は、表面下反応成分添加を使用しての所望のanti−ジアステレオ異性体(R,R)−2,3−ジメチル−コハク酸モノメチルエステルおよび不所望のsyn−ジアステレオ異性体(R,S)−2,3−ジメチルコハク酸モノメチルエステルの収率を示している。比較のために、表4は、先行する段落に記載されているプロセスと類似しているプロセスを使用するが、表面上添加を介して反応成分を加える2種のジアステレオ異性体の収率も示している。
【0135】
【表4】
【0136】
(実施例22〜28)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
(R,R)−2,3−ジメチル−コハク酸モノメチルエステル(150kg、936mol)をTHF(260L)およびMTBE(1500L)で希釈し、60℃に加熱した。10%LAH溶液(THF中530kg、1.33kg−mol)をこの溶液に供給すると、軽いアルミニウムアルコキシド中間体を含有するスラリーが生じた。反応により生じる熱を、溶媒沸騰および凝縮により除去した。第2のタンクに、THF(約970L)、水(220L)および50%NaOH水溶液(5kg)を充填し、約50℃に加熱した。アルミニウムアルコキシドスラリーを、THF、水およびNaOHを含有するタンクに制御して供給した。次いで、攪拌機を止め、水酸化アルミニウムを10分から15分間沈降させ、生成物をデカンテーションにより除去した。固体をMTBE(3×600L)で洗浄して、追加の生成物を抽出した。有機液体を集め、蒸留すると、前記表題化合物が得られた。
【0137】
表5は、先行する段落に記載された処理(即ち、過剰の塩基の添加、実施例22〜26)を使用する(R,R)−2,3−ジメチル−コハク酸モノメチルエステルのLAH還元を介しての(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの収率を示している。比較のために、表5は、Fieser処理(H2O、15%NaOH水溶液およびH2Oの連続添加、続いてLAH還元)を使用する(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの収率も示している。
【0138】
【表5】
【0139】
(実施例29)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
反応器容器に塩化p−トルエンスルホニル(400kg、2.14kg−mol、2.5当量)を無水充填した。アセトニトリル(1000L)を続いて加え、生じたスラリーを0℃に冷却した。(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(100kg、0.85kg−mol、1当量)を反応器に加えた。続いて、Et3N(260kg、2.5kg−mol、3当量)を、反応器温度を5℃以下に維持する速度で反応器に供給した。EtOAc(660L)および水(640L)を攪拌しながら加えて、反応をクエンチした。攪拌を止めて、有機相および水性相を分離した。水性相をEtOAc(560L)で洗浄し、生じた有機相を、反応クエンチからの有機相と合わせた。合わせた有機相を10%NaHCO3水溶液(720kg)および25%NaCl水溶液(670kg)で順次洗浄した。EtOAcを大気圧で留去すると、約400Lの液体体積が得られ、これに、MTBE(1100L)およびEtOH(170kg)を加えた。混合物を還流に加熱し、続いて、約20℃に冷却して、粗製生成物を結晶化させ、これを、濾過により集めた。粗製生成物をMTBE(2200L)に分散させ、混合物を還流まで加熱して、固体を溶解させた。溶解の後に、水(200L)を攪拌しながら加え、相を分離した。水性相を廃棄した。EtOH(150kg)を有機相に加え、混合物を20℃に冷却して、前記表題化合物を結晶化させ、これを濾過により集めた。
【0140】
(実施例30)
LHMDSによる(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの分解
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(269mg、0.631mmol)のTHF(1.5mL)溶液をLHMDSのTHF溶液(1.35Mの溶液1.0mL、1.35mmol、2.14当量)で処理し、17℃で6.5時間攪拌したが、この時点で、定量HPLC分析によると、元々のジトシレートの55.5%が残っていた。
【0141】
(実施例31)
メチル3−シアノプロパンイミデートクロリドの調製
スクシノニトリル(12.124g、0.1514mol)を、最小体積の温ジオキサン(10mL)に溶かした。その間に、MeOH(6.2mL、4.90g、0.153mol、1.01当量)を、HClのEt2O溶液(2Mの溶液76mL、0.152mol、1.00当量)に10℃で加えた。スクシノニトリル溶液を、HCl/MeOH溶液に3分にわたって加えた。次いで、さらなるHCl溶液を加えた(15mL、0.030mol、0.20当量)。約10分の後に、固体が沈殿し始めた。さらなる15分の後に、スラリーが粘稠性になった。混合物を10℃で26時間攪拌し、次いで、Et2O(蒸発を補償するために30mL)で希釈し、濾過した。ケークをEt2O(3×20mL)で洗浄し、N2流により乾燥させて、白色の結晶固体としての前記表題化合物(17.196g、76.4化学%)にした。13C−NMR(100MHz,DMSO−d6):δ171.28(s);120.47(s);51.83(q);29.05(t);12.46(t);1H−NMR(400MHz,DMSO−d6):δ1.86(2H,t,J=7.4Hz);2.46(2H,t,J=7.4Hz);3.35(3H,s)。
【0142】
(実施例32)
3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリルの調製
メチル3−シアノプロパンイミデートクロリド(15.29g、0.1029mol)および2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオール(12.363g、0.1029mol、1.00当量)の無水THF(300mL)懸濁液を40℃で13.5時間攪拌し、次いで、不溶性物質を濾別した。濾液を、より小さい規模の実験(イミデート0.0035molから)からの濾液と合わせた。合わせた濾液を濃縮して白色の半固体残渣にし、これを、CH2Cl2/EtOAc(約15mL)に溶かし、シリカゲルパッド(40g)に施与し、EtOAc/シクロヘキサン(10%を500mL、続いて20%を500mL)で溶離した。濾液を濃縮して粘稠性のスラリーにし、ヘプタン(100mL)で希釈し、再濃縮し(2回)、次いで、濾過した。ケークをヘプタン(3×8mL)で洗浄し、N2流により乾燥させると、前記表題化合物が白色の結晶固体(9.417g、48.3化学%)として得られた。13C−NMR(100MHz,CDCl3):δ119.62(s);107.32(s);72.74(t);32.35(t);30.32(s);14.31(q);11.78(t);1H−NMR(400MHz,CDCl3):δ0.81(3H,s);2.01(2H,t,J=7.8Hz);2.48(2H,t,J=7.8Hz);3.89(6H,s)。
【0143】
(実施例33)
3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル
刊行された手順に従った。S.M McElvain&J.P.Schoeder、J.Am.Chem.Soc.71:40(1949年)参照。スクシノニトリル(107.53g、1.3426mol)のジオキサン(120mL)溶液をEt2O(930mL)で処理した。二相混合物をMeOH(43.4g、1.3546mol、1.01当量)で処理し、8℃に冷却した。生じた1相溶液を7℃に冷却し、温度が8℃未満に維持されるような速度で、ガス分散管を介して、無水HCl(58g、1.59mol、1.18当量)を溶液に散布する(添加は発熱性である)。混合物を8℃で一晩攪拌し、次いで濾過した。ケークをEt2O(500mL)で洗浄し、窒素流により乾燥させた。ケークは、13C−NMR(CD3CO2D)によりメチル3−シアノプロパンイミデートクロリド(103.3g、0.6952mol、51.8M%、純度>95%)と識別された。イミデート(1.0g、6.73mmol)、2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオール(2.4g、19.98mmol、2.97当量)およびBHT(24mg、0.109mmol、0.016当量)の無水THF(10.5mL)懸濁液を40℃で17時間攪拌し、次いで、不溶性物質を濾別した。前記表題化合物の収率は、GCによる濾液の定量分析により80.5M%であると決定された。
【0144】
(実施例34)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(5.375g、0.01260mol)およびLiBr(3.2839g、0.03782mol、3.00当量)のトルエン(50mL)中の混合物を還流に加熱した。18時間後に、TLC(溶離液:20%EtOAc/シクロヘキサン、チャー:PMA、Rf[ジトシレート]=0.31、Rf[ジブロミド]=0.76)によると、前記表題化合物の変換が完了した。不溶性固体(トシル酸リチウム)を、セライトの小さいパッドで濾別し、トルエン(4×5mL)ですすいだ。13C−NMR(100MHz,CDCl3):δ39.19(t);37.02(d);14.41(q);1H−NMR(400MHz,CDCl3):δ0.99(6H,d,J=6.4Hz);2.05(2H,多重線);3.38(2H,dd,J=10.2,5.0Hz);3.46(2H,dd,J=10.4,5.2Hz)。
【0145】
(実施例35)
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリルの調製
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンを含有する濾液に、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル(3.0017g、0.01638mol、1.30当量)を加えた。次いで、混合物を室温で2時間にわたって、LiN(SiMe3)2のTHF溶液(1.28Mの溶液34mL、0.0435mol、3.45当量)で滴加処理した。30分後に、GCおよびLC分析により、未反応の二臭化物(GCにより25.4化学%、LCにより32.9化学%)および未反応のシアノオルトエステル(0.090当量)が、生成物(46.5化学%)と共に示された。さらなるシアノオルトエステル(1.151g、0.00628mol、0.50当量)を加え、混合物をさらに16時間攪拌したが、この時点で、GCによると反応は完了したので(二臭化物3.4化学%およびシアノオルトエステル10.2化学%)、反応を水(50mL)でクエンチした。上部有機相を分離し、水性相をCH2Cl2(2×50mL)で抽出した。有機相を合わせ、濃縮すると、前記表題化合物がオレンジ−茶色の結晶固体(5.7532g、収率94.7化学%、ESTD GC、ジトシレートから全体で)が得られた。13C−NMR(100MHz,CDCl3):δ126.24(s);107.68(s);72.60(t);48.46(t);47.25(t);45.30(t);41.52(d);40.55(d);36.72(s);30.40(s);18.49(q);17.42(q);14.50(q);1H−NMR(400MHz,CDCl3):δ0.70(3H,s);0.87(3H,d,J=6.4Hz);0.92(3H,d,J=6.8Hz);1.2〜2.3(8H,多重線);3.80(6H,s)。
【0146】
(実施例36)
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリルの調製
21℃の(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(214.4mg、0.5026mmol)および3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル(239.9mg、1.3094mmol、2.61当量)のTHF(4.0mL)溶液をLiN(SiMe3)2のTHF溶液(1.28Mの溶液2.50mL、3.20mmol、6.37当量)で2時間にわたって滴加処理し、室温で62時間攪拌した。混合物をH2O(1mL)でクエンチし、室温で30分間攪拌し、次いで、H2O(40mL)に注ぎ、CH2Cl2(3×10mL)で抽出した。抽出物を、アセトニトリルを用いてメスフラスコ中で100.0mLまでにした。GCおよびLCによる定量分析により、前記表題化合物の不在が示された。
【0147】
(実施例37)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸2,2−ジメトキシプロピルの調製
粗製(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリル(2.5131g、ジトシレート5.5044mmolに由来)をCH2Cl2(25mL)に溶かし、6NのHCl水溶液(25mL)で処理した。二相混合物を室温で30分間攪拌したが、この時点で、TLCによると、反応成分(Rf=88)の前記表題化合物(Rf=0.72)への加水分解は完了した。混合物を分離漏斗に移した。他の分離漏斗に、水(25mL)を加えた。第1分離漏斗中の有機相を、第2分離漏斗に移し、混合物を振盪し、CH2Cl2相を除去した。次いで、2個の分離漏斗中の水性相をCH2Cl2で順次抽出した(4×25mL)。抽出物を合わせ、淡褐色のオイルまで濃縮し、これを、放置して結晶化させると、前記表題化合物がオフホワイト色の固体として純粋な形態(1.4177g、5.003mmol、ジトシレートから全体で90.9化学%)で得られた。13C−NMR(100MHz,CDCl3):δ170.02(s);125.35(s);67.28(t);66.97(t);47.47(t);46.23(t);43.91(t);41.83(d);40.68(d);40.39(s);36.96(s);17.89(q);17.19(q);16.80(q);1H−NMR(400MHz,CDCl3):δ0.88(3H,s);1.02(3H,d,J=6.4Hz);1.06(3H,d,J=6.5Hz);1.3〜2.7(8H,多重線);3.61(4H,s);3.91(2H,t,J=7.1Hz);4.24(2H,s)。
【0148】
(実施例38)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の調製
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸2,2−ジメトキシプロピル(1.0034g、3.541mmol)を、KOH(純度約87.3%の物質247.7mg、216mg、3.85mmol、1.09当量)の水(1.5mL)溶液に加えた。懸濁液を21℃で1時間攪拌し、この時点で、すべての固体は溶解しており、TLC分析により、反応成分(Rf=0.75)から前記表題化合物(Rf=0.36)へ、および1,1,1−トリス(ヒドロキシメチル)エタン(Rf=0.56)への加水分解が完了したことが示された。溶液をMTBE(3×0.5mL)で洗浄して、非極性不純物を除去した。13C−NMR(100MHz,CDCl3):δ175.41(s);125.00(s);47.44(t);46.16(t);43.35(t);41.79(d);40.75(d);36.80(s);17.96(q);17.55(q);1H−NMR(400MHz,CDCl3):δ1.02(3H,d,J=6.4Hz);1.05(3H,d,J=6.4Hz);1.3〜2.7(8H,多重線);9.9(1H,br s)。
【0149】
(実施例39)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の調製
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の水溶液(シアノエステル201.4mg[0.7107mmol]および水酸化カリウム[純度約87.3%の物質49.6mg、43.3mg、0.772mmol、1.09当量]に由来、水0.3mL中)をMeOH(3mL)で希釈し、スポンジニッケルA−7000(128mg)で処理し、H2(100psig)下に35℃で攪拌した。14時間後に、シアノ酸(Rf=0.41)から前記表題化合物(Rf=0.29)への還元は、TLCによると約75%完了した。スラリーを濾過し、水(0.3mL)で洗浄した。濾液を黄色がかったオイルまで濃縮し、これを、水(0.5mL)に溶かし、氷酢酸(3滴)で処理すると、粘稠性のスラリーが生じ、これを、水(0.5mL)で薄めた。スラリーを70℃に加熱し、十分なイソプロパノールで希釈して、すべての固体(0.25mL)を溶かし、次いで1.5時間にわたって徐々に0℃に冷却した。生じたスラリーを濾過すると、前記表題化合物が、基準標準とのTLC比較によると、反応成分および1,1,1−トリス(ヒドロキシメチル)エタンで汚染された固体として得られた。
【0150】
(実施例40)
((3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンチルメタンアミンの調製
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリル(302.8mg、1.1411mmol)の無水EtOH(4mL)溶液を、NaOH水溶液(50%溶液135mg、67.5mg、1.687mmol、1.48当量)で、続いてスポンジニッケルA−7000(183mg)で処理した。混合物をH2(50psig)下に室温で21時間攪拌し、次いで濾過し、水(3×1.5mL)ですすいだ。濾液を水(5mL)で希釈し、MTBE(5mL)で抽出し、無水K2CO3上で乾燥させ、濃縮すると、前記表題化合物が淡黄色の固体残渣(333.9mg、1.2395mmol、収率108.6化学%)として得られた。TLC溶離液:70:20:5:5のEtOAc:MeOH:濃NH4OH:H2O、チャー:50%H2SO4水溶液、Rf[アミン]=0.66、Rf[ニトリル]=0.79);13C−NMR(100MHz,CDCl3):δ107.46(s);70.37(t);49.29(t);45.23(t);45.01(t);42.59(t);42.28(s);39.81(d);39.49(d);28.23(s);16.34(q);15.98(q);12.10(q)。
【0151】
(実施例41)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの調製
((3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンチル)メタンアミン(200.5mg、0.7443mmol)のHCl水溶液(1.0Mの溶液2.3mL、2.3mmol、3.1当量)溶液を室温で75分間攪拌したが、この時点で、TLC(Rf[アミノオルトエステル]=0.74、Rf[アミノエステル]=0.64)によると、アミノエステル、2−((3S,4S)−1−(アミノメチル)−3,4−ジメチルシクロペンチル)酢酸3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロピルへの加水分解が完了した。反応を固体炭酸水素ナトリウム(315.4mg、3.754mmol、5.0当量)でクエンチし、硫酸ナトリウム(103mg)で処理し、THF(3×2mL)で抽出した。抽出物を合わせ、50℃で4.5時間加熱したが、この時点で、TLCによると、アミノエステル(Rf=0.66)から前記表題ラクタム(Rf=0.80)および1,1,1−トリス(ヒドロキシメチル)エタン(Rf=0.56)への変換が完了した。混合物はさらに、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(Rf=0.21)およびBHT(Rf=0.91)の痕跡も示す。混合物を濃縮して黄色のオイルにし、これを、塩化メチレン(3mL)に溶かし、水(3×3mL)で洗浄してトリオールを除去し、次いで、濃縮して油性残渣にし、これを、ヘプタン(0.5mL)に70℃で溶かし、−20℃に冷却して、結晶を分離した。母液をデカンテーションし、これを、真空乾燥すると、前記表題化合物が結晶固体(78.9mg、0.472mmol、アミノオルトエステルから63.4化学%、シアノオルトエステルから全体で68.9化学%)として得られた。LC純度:基準試料と比較して97.5面積%。13C−NMR(100MHz,CDCl3):δ178.14(s);56.13(t);48.35(t);47.91(t);45.74(t);45.04(s);41.63(d);41.57(d);18.60(q);18.49(q);1H−NMR(400MHz,CDCl3):δ0.977(3H,d,J=6.0Hz);0.983(3H,d,J=6.0Hz);1.3〜2.3(8H,多重線);3.21(1H,d,J=9.6Hz);3.24(1H,d,J=10.4Hz);6.02(1H,br s)。
【0152】
(実施例42)
塩化メチル3−シアノプロパンイミデートの調製
スクラバー(水)に対して出口を有する口を1つ備えた1Lジャケット付き三つ口フラスコに、スクシノニトリル(Aldrich、mw80.09、103.06g、1.2868mol)、塩化メチレン(E&M)200mL、メチル−t−ブチルエーテル(E&M)200mLおよびMeOH(E&M、mw32.04、41.5g、1.2953mol、1.01当量)を充填した。溶液を6.5℃に冷却し、温度が12℃未満に維持されるような速度で、ガス分散管を介して、無水塩化水素(mw36.46、55.5g、1.5222mol、1.18当量)を溶液に散布する(添加は発熱性である)。塩化水素の重量を、レクチャーボトルを秤量することにより測定した。レクチャーボトルを天秤上のスタンドに保持した。これを、ガス分散管に、レクチャーボトルに至る管セグメントにおける張力を最小にするようにクランプされているPVC管を介して接続した。流れを、AGA Model LB 165−40−2F−BV腐食性ガス調節器を介して調節した。レクチャーボトルの圧力が高すぎるので、単純なバルブを使用しても、流れを十分に制御することができなかった。添加は4時間かかった。添加のほぼ中程で、生成物イミデートが結晶化し始めた。添加が完了したら、ジャケット温度を12℃に調節した。25時間攪拌した後に、スラリーが溶媒蒸発により非常に粘稠性になったので、塩化メチレン175mLを加えた。粘稠性ではあるが、スラリーは容易に攪拌可能および濾過可能であった。混合物を濾過した。ケークを塩化メチレン325mLで洗浄し、室温窒素により乾燥させた。物質は、GCアッセイにより前記表題化合物と識別された(カラム:60メートルDB−1、内径0.25mm w 1ミクロンフィルム、温度勾配:150℃で3分間、次いで、10℃/分で225℃まで勾配させ、次いで、10分間保持;試料調製:イミデート約100mg(正確に秤量)に1NのHCl0.5mLを添加し、溶液を5分間放置し、溶液を10mLメスフラスコに2mL水と共に移し、MeOHを用いて10mLまでにした;保持時間:スクシノニトリル、9.29分;メチル−3−シアノプロピオネート、9.78分;コハク酸ジメチル、10.68分)。冷蔵庫での貯蔵が推奨される。褐色ビンで室温貯蔵されたイミデートの多くは、1〜7日後に、著しいガス発生を伴って茶色の固体に分解した。重量:178.97g(mw148.59;1.2045mol、収率93.6化学%[未修正])。
【0153】
(実施例43)
4,4,4−トリメトキシブタンニトリルの調製
3L三つ口フラスコに、塩化メチル3−シアノプロパンイミデート(mw148.59、176.77g、1.1896mol)およびMeOH(E&M)1.7Lを充填した。攪拌すると、イミデートが溶けて、均一な溶液が生じた。溶液を室温で攪拌した。約2時間後に、塩化アンモニウムが沈殿し始めた。明朝(13時間後)、混合物は、顆粒固体の白色のスラリーになっていた。この反応を、NMR(アリコットを取り、約20%のCD3ODで希釈)により監視することができる。(塩化アンモニウムがオルトエステルとゆっくりと反応して、3−シアノプロピオン酸メチルを形成してしまうので、スラリーの加熱は回避すべきである。例えば、反応混合物を真空濃縮して(ポット温度33℃)、塩化アンモニウムを沈殿させることにより反応を完了させる別の実験では、4%の3−シアノプロピオン酸メチルが形成された)。スラリーを、トリエチルアミン(mw101.19;d0.726、180mL、130.68g、1.2914mol、1.09当量)を含有する受器へと濾過し、少量のMeOHですすいだ。ケーク(塩化アンモニウム)は19.76gの重さであった(mw53.49、0.3694mol、理論の31.1%)。濾液をNMRによりチェックする(約20体積%のCD3ODで希釈されたそのままのアリコットで行う)。メチルエステルが存在する場合、濾液を一晩攪拌すると、遊離したアンモニアがメチルエステルをアンモニア分解する。濾液を水酸化カリウムペレット(mw56.11、55.2g、87.3重量%、48.19g、0.8588mol、0.72当量)の水160mL溶液で処理すると、このことにより、固体が沈殿した(おそらくKCl)。スラリーを濾過した。ケークの重量は50.32gであった(KClならばmw74.56、0.6749mol、0.57当量)。濾液に、炭酸ナトリウム(mw105.99、32g、0.3019mol、0.25当量)の水200mL溶液を加えた。混合物を回転蒸発(浴温度47℃)させて、MeOHを除去すると、均一な溶液が残った(体積約575mL)。溶液をH2O95mLで希釈し、MTBE(700mL、次いで360mL)で抽出した。相分割は優れていた。MTBE抽出物を合わせ、無水炭酸カリウム(2.7g)で処理し、二相混合物に濃縮し、さらなる炭酸カリウム(53g)で処理すると、多少の固体を伴う二相混合物が得られた。MTBE相を分離し、水性相をMTBE(50mL)で抽出した。MTBE抽出物を合わせ、無水粉砕炭酸カリウム(14g)上で乾燥させ、無水炭酸カリウム(5g)から蒸留した。中間留分(116.52g、沸点92℃/26mm)は、NMRおよびGCにより表題化合物と識別される無色の液体(KF0.025)であった。重量:116.52g(mw159.19、94.60重量%、110.23g、0.6924mol)。初期留分(forecut)(重量41.46g、沸点92℃/27mm)は数滴の水(KF0.479)を含有し、トルエン25mLおよびか焼無水炭酸カリウム0.3gからの再蒸留により再処理すると、さらなる無色の液体(沸点82〜83.6℃/25mm)が得られた。トルエン共沸蒸留後および生成物蒸留前に得られたKFは、0.011%であった。重量:37.2g(mw159.19、95.93重量%、35.69g、0.2242mol)。(抽出における水残存の問題は、抽出溶媒としてMTBEの代わりにトルエンを使用することにより回避することができる)。GCアッセイ(カラム:30メートルDB−5、内径0.25mm、w 1ミクロンフィルム、温度勾配:70℃で5分間、次いで、10℃/分で320℃まで勾配させ、次いで2分間保持、保持時間:15.88分)によると、いずれの留分も、1.7面積%より多い何らかの不純物を含有しなかった。3−シアノプロピオン酸メチルのレベルは、中間留分では0.05面積%であり、再蒸留された初期留分では検出されなかった。収率(合わせて):0.9166mol、77.1化学%(純度修正)。13C−NMR(100MHz,CD3OD):δ121.49(s);115.98(s);50.64(q);28.00(t);12.59(t);1H−NMR(400MHz,CD3OD):δ2.11(2H,t,J=7.4Hz);2.42(2H,t,J=7.4Hz);3.25(9H,s)。
【0154】
(実施例44)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55、200.0g、0.4689mol)および臭化ナトリウム(mw102.89、145.0g、1.4093mol、3.01当量)のトルエン1L中の混合物を70℃に加熱すると、均一なスラリーが得られた。臭化テトラブチルアンモニウム(mw322.38、15.0g、0.0465mol、0.099当量)のH2O15.0mL溶液を加えた。8分後に、固体が沈殿し始めた(トシル酸ナトリウム)。スラリーを70℃で3時間、次いで80℃で17時間攪拌した。混合物を50℃に冷却し、H2O(350mL、次いで2×150mL)で洗浄し、回転蒸発により濃縮して、199.1gの重量のオイルにした。
【0155】
(実施例45)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
リチウムヘキサメチルジシラジドのTHF溶液(Chemetall、mw167.33、0.226g/mL、1058.4mL、239.2g、1.4295mol、3.07当量)を真空蒸留(ジャケット温度25℃)して、非常に粘稠性なスラリー(体積:400mL)にし、これを、THF150mLで希釈した。このスラリーを0℃に冷却し、先行する実施例からの(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの一部(197.4g、ジトシレート0.4649molから)で20分にわたって処理し、THF50mLですすいだ。添加は、やや発熱性であった(0℃に低下する前に+15℃までの短時間のエクスカーション)。添加の終了時には、混合物は澄明な溶液になった。この溶液に、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、105.84g、0.6649mol、1.43当量)を徐々に6時間にわたって加えた。添加の間、ポット温度は0℃であった。添加の終了時に、温度を−5℃に低下させた。混合物を14時間攪拌し、次いで、H2O(252mL)でクエンチした。最初の13mLを50分にわたって加え(+4℃までの発熱)、次いで残りを、5分にわたって加えた。混合物を23℃に加温すると、界面に暗色のオイル(リチウムヘキサメチルジシラジド/THF試薬における鉱油、不純物と識別された)を伴う二相混合物が得られた。水性層を分離した。有機層を350〜400mLの体積まで濃縮し、ヘプタン350mLで処理し、500〜550mLの体積まで濃縮し、ヘプタン400mLで処理し、次いで、450mLの体積まで濃縮した。ヘプタン溶液を0.05NのNaOH(325mL、次いで175mL)で洗浄した。他の実験からの溶液の一部を濃縮して、13C−NMRおよび1H−NMRにより前記表題化合物と識別されるオイルにした。13C−NMR(100MHz,CD3CD2OD):δ125.44(s);113.16(s);48.66(3C,q);47.50(t);46.44(t);41.12(d);39.76(d);38.76(t);37.06(s);18.08(q);16.82(q);1H−NMR(400MHz,CD3CD2OD):δ1.05(3H,d,J=6.4Hz);1.09(3H,d,J=6.7Hz);1.46(1H,t,J=12.6Hz);1.5〜2.3(7H,多重線);3.29(9H,s)。
【0156】
(実施例46)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの調製
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルを含有する先行する実施例からのヘプタン溶液全体を、十分な3NのHClで処理して、pHを<3に調節した(d1.0495、207g、197.2mL、0.592mol、1.27当量)。混合物を室温で40分間攪拌し、次いで、水性層を分離して、ヘプタン100mLで抽出した。合わせた有機層をH2O(50mL)で洗浄した。重量:425.19g。同様の実験からの溶液を濃縮し、蒸留すると(沸点145〜147℃/35.1mm)、13C−NMRおよび1H−NMRおよびGCにより本質的に純粋な形態の前記表題化合物と識別される液体が得られた(99.0A%)。13C−NMR(100MHz,CDCl3):δ169.63(s);124.91(s);51.62(q);47.15(t);45.92(t);43.07(t);41.56(d);40.39(d);36.77(s);17.77(q);16.97(q);1H−NMR(400MHz,CDCl3):δ0.93(3H,d,J=6.4Hz);0.97(3H,d,J=6.6Hz);1.29(1H,t,J=12.4Hz);1.46(1H,多重線);1.67(1H,多重線);1.84(1H,dd,J=13.8,9.4Hz);2.05(1H,dd,J=13.8,8.5Hz);2.38(1H,dd,J=13.2,6.5Hz);2.58(2H,s);3.65(3H,s)。
【0157】
(実施例47)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの調製
モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、20.0mLで33.33g、計算乾燥重量:15.55g[0.35g/g])を、MeOH13mLで処理し、攪拌し、沈降させ、上澄み13mLをデカンテーションした。触媒洗浄手順を2回繰り返した。メタノール触媒スラリーを、450mLガラス攪拌反応器にMeOH60mLと共に入れた。容器を密封し、H2でパージし、60psigまで加圧し、攪拌しながら85℃に加熱した。反応器温度が85℃に達したら、圧力を調節して、60psigに維持した。先行する実施例からのヘプタン溶液の一部(208.60g、ジトシレート0.2281molから、mw195.26、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル44.535gを理論的に含有)を濃縮してオイル(重量:62.43g)にし、これを、MeOH119mLで希釈すると、黄色−白色のエマルションが得られ、これを、水素化反応器に4.9時間にわたって計量供給した。次いで、計量供給系をMeOH(3×5mL)で反応器へとすすぎ、完成した混合物を85℃/60psigにさらに5時間維持した。室温まで冷却した後に、反応混合物を真空濾過した。反応器および触媒ケークをMeOHですすいだ。合わせた濾液および洗浄液の重量は、399.11gであった。このほぼ無色の溶液の一部(396.29g、ジトシレート0.2265molから)を真空蒸留(ポット温度37℃)して、150mLの体積にした。次いで、溶液を大気圧で蒸留したが、この間、留出物の沸点が101℃で一様になるまでヘプタンを少量ずつ加えた(3×100mL)。生じた濁った溶液を−5℃に冷却した(結晶化が42℃で生じた)。スラリーを−5℃に予め冷却されたフリット上で濾過した。ケークを室温のヘプタン80mLで洗浄し、N2圧力により乾燥させた。生成物は、基準試料とのESTD LC比較により前記表題化合物と識別された(濃度:96.49W%)。収率:33.80g(mw167.25、32.61g(濃度修正)、0.1950mol、ジトシレートから全体で86.1M%)。濾液のESTD LC分析により、ラクタムが判明した(ジトシレートからの全体収率3.2M%)。
【0158】
(実施例48)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からの(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(mw167.25、純度96.49W%の物質32.80g、31.65g、0.1892mol)を、37%HCl(mw36.46、37W%溶液57.4g、21.24g、0.5825mol、3.08当量)およびRO水32.8mLで処理した。混合物を>88℃で24時間攪拌した。LCによる分析により、残渣ラクタム2.06M%が判明した。混合物を60℃に冷却し、50%水酸化ナトリウム(30.85g、水酸化ナトリウム15.425g[mw40.00、0.3856mol、2.04当量]含有)を加えることにより、pHを約2(紙)に調節した。混合物をH2O43mLで希釈し、溶液が濁るまで、真空蒸留した(ポット温度48℃)(最終体積約115mL)。50%水酸化ナトリウム(14.81g、水酸化ナトリウム7.405g[mw40.00、0.1851mol、0.98当量]含有)を加えることにより、pHを6.5(紙)に調節した。混合物を<5℃に冷却し、30分間攪拌し、濾過した。フラスコを濾液で2回すすいだ。ケークを<10℃のRO水33mLで洗浄し、N2流により乾燥させた。生成物は、基準試料とのESTD LC比較により、前記表題化合物と識別された(濃度:88.6W%)。金属に関するスクリーニングによりMo(0.1ppm)およびAl(1ppm)が検出された。重量:35.43g(mw185.27、31.39g[濃度修正]、0.1694mol、89.6M%)。濾液を濃縮して残渣にした(重量:32.94g)。表題化合物に関するLC分析により、濃度2.1W%が判明した(0.692g、0.00373mol、ラクタムから2.0M%)。
【0159】
粗製生成物の一部(mw185.27、純度88.6W%の物質34.0g、30.124g、0.1626mol)をRO水120mLおよびイソプロパノール40gに懸濁させた。混合物を還流(85℃)に加熱したが、この時点で、溶液が形成された。溶液を<5℃に4時間にわたって冷却し、1時間攪拌し、濾過した。ケークを<5℃のイソプロパノール70mLで洗浄し、N2流により17時間乾燥させた。生成物は、基準試料とのESTD LC比較により、前記表題化合物と識別された(濃度:99.6W%)。重量:29.15g(mw185.27、29.03g[濃度修正]、0.1567mol、粗製生成物から96.4M%)。濾液を濃縮して残渣(重量:5.19g)にした。表題化合物に関するLC分析により、濃度51.6W%が判明した(2.678g、0.01445mol、粗製生成物から8.9M%)。13C−NMR(100MHz,4/1 CD3OD/D2O):δ182.49(s);53.04(t);52.33(t);48.80(t);48.33(t);44.13(d);43.81(d);43.74(s);19.99(q);19.86(q);1H−NMR(400MHz,4/1 CD3OD/D2O):δ0.98(3H,d,J=6.3Hz);0.99(3H,d,J=6.2Hz);1.14(1H,dd,J=12.6,10.2Hz);1.19(1H,dd,J=12.9,10.2Hz);1.52(2H,重複した多重線);1.88(1H,t,J=13.6Hz);1.90(1H,t,J=13.9Hz);2.47(1H,d,J=15.2Hz);2.52(1H,d,J=15.2Hz);2.92(1H,d,J=12.9Hz);2.99(1H,d,J=12.9Hz)。
【0160】
(実施例49)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からのヘプタン溶液の一部(208.53g、ジトシレート0.2280molから、mw195.26、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル44.520gを理論的に含有)を水酸化カリウム(mw56.11、純度88W%の物質29.46g、25.925g、0.4620mol、2.03当量)のH2O125mL溶液で処理した。二相混合物を2時間激しく攪拌したが、この時点で、TLCによると加水分解は完了した。2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩を含有する水性相を分離した。重量:189.02g。
【0161】
450mLガラス製攪拌反応器に、モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、20.0mL、重量33.03g、乾燥重量:15.20g[0.31g/g])およびMeOH60mLを充填した。容器を密封し、H2でパージし、H2で25psigまで加圧した。反応器を攪拌し、約50℃に加温したがが、この時点で、既知の体積の高圧レザバーから供給することにより、圧力を50psigに調節および維持した。前記で調製された水性相の一部(185.72g、ジトシレート0.2240molから、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸[mw219.33、49.130g]含有)を、水素化容器に2.7時間にわたって計量供給した。供給系をMeOH(3×5mL)で反応器へとすすいだ。最後のすすぎの完了後約15分後で、水素摂取は完了したようである。反応器を50℃にさらに5時間維持し、その後、室温まで冷却した。反応混合物を濾過し、触媒ケークをMeOHおよび水ですすいだ。(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸のカリウム塩を含有する濾液の重量は610.92gであった。
【0162】
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸のカリウム塩(608.55g、ジトシレート0.2231molから)を含有する前記濾液の一部を回転蒸発により濃縮して、188.04gにし、ジャケット付きの三つ口フラスコに移し、H2O(2×10mL)ですすいだ。氷酢酸(mw60.05、27.14g、0.4520mol、2.03当量)を加えて、pHを>14から6.72に調節した。スラリーを0℃に冷却し、0℃で3時間攪拌し、濾過し、ケークをN2流により乾燥させた。結晶は、TLCおよびLCにより前記表題化合物として識別された。重量:40.51g。
【0163】
粗製(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の一部(38.86g、ジトシレート0.2140molから)をイソプロパノール307mLと混合した。スラリーを80℃に2時間加熱し、次いで、1.5時間にわたって0℃に冷却し、濾過した。濾液を使用して固体をフラスコからフィルター上にすすいだ。ケークをN2流により乾燥させた。結晶はオフホワイト色だった。重量:33.05g。
【0164】
前記(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸結晶を25W%i−PrOH/H2O165mLと混合した。スラリーを80℃に加熱すると、澄明な溶液が、あるとしても非常に僅かな濁りのみを伴って生じた。溶液を0℃に4時間にわたって冷却し、0℃で1時間攪拌し、次いで濾過した。ケークを濾液で洗浄し、N2流により乾燥させた。LCによると、結晶は0.1A%を上回る不純物は含有しなかった(濃度:99.6W%)。重量:30.55g(mw185.27;30.43g、0.1642mol、ジトシレートから全体で76.7M%)。
【0165】
(実施例50)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55;200.0g、0.4689mol)および臭化カリウム(mw119.01、169.0g、1.4200mol、3.03当量)のトルエン400mL中の混合物をN2で45分間脱ガスし、次いで、50℃に加熱し、臭化テトラブチルアンモニウム(mw322.38、30.0g、0.09306mol、0.198当量)のH2O30mL溶液で加えて処理した。混合物を21時間還流し、次いで50℃に冷却し、H2O(400mL、次いで2×100mL)で洗浄した。有機層をトルエン250mLで希釈し、回転蒸発により濃縮すると(浴温度51℃)、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンが重量208.7gのオイルとして得られた。
【0166】
リチウムヘキサメチルジシラジドのTHF溶液(Chemetall、mw167.33、0.226g/mL、1082.4mL、244.6g、1.4619mol、3.17当量)を真空蒸留して、500mLの体積にし、−2℃に冷却し、1時間にわたって一部の(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(205g、ジトシレート0.4606molから)で処理した。混合物を−2℃で10分間攪拌し、次いで、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、105.84g、0.6649mol、1.44当量)で徐々に6時間にわたって処理した。添加の間、ポット温度を0℃に維持した。混合物を14時間攪拌し、次いで、H2O(250mL)でクエンチした。初めの20mLは1時間にわたって加え(+5℃まで発熱)、次いで残りは5分間にわたって加えた。混合物を18℃に加温し、層を分離した。水性層をTHF50mLで逆抽出し、抽出物を初めの有機層と合わせた。前記表題化合物を含有する合わせた有機層の重量は、786.7gであった。
【0167】
(実施例51)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
先行する実施例からの粗製(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルのTHF溶液の一部(188.18g、ジトシレート0.1102molから)を37%HCl水溶液(mw36.46、d1.2g/mL、29mL、34.8g、塩化水素12.88g[0.3532mol、3.20当量]含有)で処理して、pHを−1に調節した。添加の間、温度は52℃に上昇し、固体が沈殿した。水(65mL)を加えて、固体を溶かし、混合物を室温に低下させた。混合物を(攪拌することなく)室温に20時間維持し、次いで水性相(pH0.55)を分離した。2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルを含有する有機相に、水酸化カリウム(mw56.11、87.3W%の物質14.0g、12.22g、0.2178mol、1.98当量)のH2O50mL溶液を加えた。添加の間に、温度は36℃に上昇した。混合物を室温で2時間攪拌したが、この時点で、TLC分析により、けん化が完了したことが示された。相を分離し、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩を含有する水性相を濃縮して、60mLの体積にした(MeOHおよび残留THFを除去するため)。
【0168】
HCl水溶液(37%、mw36.46、d1.2g/mL、19mL、22.8g、塩化水素8.436g[0.2314mol、2.10当量]含有)をH2O57mLで希釈し、0.3℃に冷却し、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩の結晶でシーディングした。この混合物に、75分にわたってシアノ酸カリウム塩の濃水溶液を滴加し、その際、ポット温度を−3℃から−1℃に維持した。添加を通して、結晶(非粘着性)を分離した。スラリーを−1℃で1時間攪拌し、次いで濾過した。ケークを0℃のH2O35mLで洗浄し、N2流によりKF0.135%まで乾燥させた。結晶は、13C−NMRおよび1H−NMRにより前記表題化合物と識別された。重量18.78g(mw181.24、0.1036mol、ジトシレートからの全体収率94.0M%)。
【0169】
(実施例52)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55、200.76g、0.4707mol)、臭化ナトリウム(mw102.89、145.3g、1.4122mol、3.00当量)および臭化テトラブチルアンモニウム(mw322.38、15.0g、0.0465mol、0.099当量)のトルエン1.5LおよびH2O84mL中の混合物を93℃に加熱した。フラスコのヘッドスペースはフォームでほぼ充満した。温度を92℃に低下させると、このことにより、フォームの量が低減した。混合物を89℃で攪拌した。反応の進行をTLCにより監視した(溶離液:30%EtOAc/シクロヘキサン;Rf[ジトシレート]=0.38、Rf[モノトシレート−一臭化物]=0.54)。t=2時間で、多くのモノトシレート−一臭化物および痕跡量のジトシレートが検出可能であった。t=6.5時間で、痕跡量のモノトシレート−一臭化物が検出可能であった。t=24時間で、ジトシレートまたはモノトシレート−一臭化物が検出されなかったので、混合物を室温に冷却し、濾過し、ケーク(トシル酸ナトリウム)をトルエン250mLで洗浄した。トルエン抽出物を合わせ、H2O(2×250mL)で洗浄し、真空濃縮(53℃/150mm)して約200mLの体積にした。生成物は、ISTD GCにより(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンとして識別された(濃度:55.9W%)。重量:204.8g(mw243.97、二臭化物114.48g[0.4693mol、収率99.7M%]含有)。
【0170】
そのままのヘキサメチルジシラザン(mw161.40、79.2g、0.4907mol、3.32当量)を−11℃に冷却し、n−ブチルリチウムのヘキサン溶液(2.5Mの溶液177mL、0.4425mol、3.00当量)で、温度が4℃未満に維持される速度で処理した(35分)。生じたスラリーを25℃に加温して、澄明な溶液を得、これを真空蒸留して粘稠なスラリー(体積約100mL)にし、−3℃に冷却し、THF130mLで処理すると、44℃までの発熱が生じた。混合物を−2℃に冷却し、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの前記で調製されたトルエン溶液の一部(mw243.97、純度55.9W%の物質64.4g、36.00g、0.1476mol)ですべて一度に処理した。混合物を−2℃で10分間攪拌し、次いで、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、37.4g、0.2349mol、1.59当量)で18分にわたって処理した。(12℃までの短時間のエクスカーション)。混合物を−2℃で攪拌した。t=1.5時間で、ISTD GC分析により、2,3−ジメチルブタジエン(0.62W%)、3−シアノプロピオン酸メチル(18.8基準M%)、二臭化物(10.0基準M%)、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(69.7基準M%)および2:1付加生成物(0.85W%)の存在が示された。t=18時間で、ISTD GC分析により、ジメチルブタジエン(1.8W%)、3−シアノプロピオン酸メチル(1.4基準M%)、二臭化物(2.3基準M%)、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(93.5基準M%)、2:1付加生成物(1.0W%)およびMeO2CCH2CH2COCH(CN)CH2CO2Me(0.57A%)の存在が示された。混合物をH2O100mLでクエンチし、25℃に加温すると、二相混合物が得られた。水性相を分離し、ヘプタン50mLで抽出した。2つの有機相を合わせると、前記表題化合物(重量328.12g)を含有する溶液が得られた。
【0171】
(実施例53)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
先行する実施例からの(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの溶液の一部(183.84g、二臭化物0.08270molから)を1NのHCl(41.4mL、0.0414mol、0.50当量)で、続いて6NのHCl(37.7mL、0.2262mol、2.74当量)で処理して、pHを約11から約2に調節した(紙)。二相混合物を室温で1時間攪拌したが、この時点で、GCによると、オルトエステル(保持時間22.0分)から2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(保持時間19.7分)への加水分解が完了した。水性相を分離し、廃棄した。有機相を、50%水酸化ナトリウム水溶液(mw40.00、14.0g、水酸化ナトリウム7.0g[0.175mol、2.12当量]含有)およびH2O40mLで処理した。塩基と接触すると、有機相はオレンジ色から暗茶色に変化した。混合物を室温で3時間攪拌したが、この時点で、TLCによると、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のナトリウム塩への加水分解が完了した。有機相を分離し、H2O20mLで抽出した。水性相を合わせ、6NのHCl(28.25mL、0.1695mol、2.05当量)を加えることにより、pHを13.2から2.1に調節し、トルエン(3×40mL)で抽出した。トルエン抽出物を合わせ、H2O40mLで洗浄して、極性の未知物質(Rf=0.02および0.06)を除去し、回転蒸発により濃縮して、粘稠な赤コハク色のオイルにしたが、これは、迅速かつ発熱性に結晶化した。結晶は、TLCによると、本質的に均一な形態の前記表題化合物と識別された(Rf=0.44、溶離液:70/20/5/5のEtOAc/MeOH/濃NH4OH/H2O)。重量:14.8317g(mw181.24、0.08183mol、二臭化物から収率99.0M%)。
【0172】
(実施例54)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からの結晶2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸(mw181.24、14.8317g、0.08183mol)を、冷蔵庫で45日間貯蔵した後に、0℃のMeOH60mLと共に攪拌して、溶液を形成した。この溶液に、水酸化カリウム(mw56.11、純度87.3W%の物質11.0873g、9.6792g、0.1725mol、2.11当量)のMeOH40mL溶液を加えた。モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、乾燥重量5.73g[シアノ酸カリウム塩0.32g/g])を、300mLステンレス鋼オートクレーブにMeOH60mLと共に入れた。容器を密封し、H2でパージし、H2で50psigに加圧し、30℃に加熱した。前記シアノ酸カリウム塩のMeOH溶液(mw219.33、0.08183mol、17.9478g)を水素化容器に2.7時間にわたって計量供給した。混合物を30℃/H250psigに保持した。20時間後に、摂取は本質的に止んだ。
【0173】
全部で44時間後に、混合物を真空濾過し、触媒をMeOHですすいだ。合わせた濾液および洗浄液(重量:249.10g)をH2O33mLで処理し、回転蒸発により濃縮し、H2O28mLで処理し、さらに濃縮して、51gの重量にした。この溶液(pH14.0)を氷酢酸(mw60.05、10.88g、0.1812mol、2.21当量)で処理して、スラリーを得て、これを0〜5℃に冷却して、1時間攪拌し、濾過し、N2流により乾燥させた。TLCにより、結晶は、本質的に純粋な形態の前記表題化合物と識別された(溶離液:70/20/5/5のEtOAc/MeOH/濃NH4OH/H2O;Rf[ヒドロキシ酸]=0.34、Rf=[表題化合物]=0.20)。重量:15.601g。
【0174】
粗製の前記表題化合物の一部(15.48g、シアノ酸0.08120molから)を、イソプロパノール155mLに懸濁させ、80℃(浴温度)で30分間攪拌し、次いで、0〜5℃に冷却し、1時間攪拌し、次いで、濾過した。濾液を使用して、固体をフラスコから洗浄した。ケークをN2流により乾燥させた。重量:10.791g。濾液を回転蒸発により濃縮して、固体(重量:4.299g)にした。
【0175】
粉砕された前記表題化合物の一部(10.67g、シアノ酸0.08029molから)を25%(v/v)のiPrOH/H2O53mLに懸濁させ、80℃に加熱した。固体が完全には溶けなかったので、イソプロパノール3.6mLを加え、温度を87℃に上げた。固体が完全に溶けて、金コハク色の溶液を形成した。溶液を0〜5℃に冷却し(温度が78℃であったときには、固体が結晶化した)、2時間攪拌し、濾過した。ケークを濾液で洗浄し、N2流により乾燥させた。LCにより、生成物は前記表題化合物と識別された(濃度99.8W%)。結晶は、後記溶離ピークの0.18A%(RRT1.84)および初期溶離ピークの0.12W%(RRT0.48)を除いて、0.1W%を超える不純物は含有しなかった。金属スクリーニングにより、Ni(4.2ppm)およびMo(0.6ppm)が検出された。重量:9.634g(mw185.27、9.615g、0.05190mol、シアノ酸からの収率64.6M%)。
【0176】
(実施例55)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの単離
機械式攪拌機を備えていて、87mLレベルにマーキングされている1L四つ口丸底フラスコに、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(理論=17.2g、103mmol)のメタノール溶液を充填した。混合物を真空蒸留により濃縮して、87mLにした。残渣を2回、酢酸エチル(170mL)で希釈し、再蒸留して87mLにした。生じた有機溶液を初めに1NのHCl(87mL)で、次いで、1NのNaOH(87mL)で抽出した。水性相を連続して酢酸エチル(45mL)で洗浄した。合わせた有機相を、1L四つ口丸底フラスコに分枝鎖オクタン(170mL)と共に充填した。混合物を真空蒸留により濃縮して、87mLにした。留出物を分枝鎖オクタン(170mL)で希釈し、再び真空蒸留により濃縮して87mLにした。この希釈/蒸留手順をさらに2回繰り返した。生じた溶液87mLを周囲温度に1時間にわたって冷却したが、この間に、結晶が形成した。生じたスラリーを約0℃に冷却し、30分間攪拌し、その後、真空濾過した。回収された結晶を冷分枝鎖オクタン(3×25mL)で洗浄し、真空下に65℃で1時間乾燥させると、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(11.6g、67%)が得られた。
【0177】
(実施例56)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
頭上攪拌機、還流凝縮器およびPTFEコーティングされたサーモカップルを備えている500mL四つ口丸底フラスコ(RBF)に、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(20.0g、0.12mol、1.0当量)、HCl(37%w/w、35.0g、0.35mol、3.0当量)および水(20.0g)を窒素ブランケット下に充填した。混合物を90℃に加熱し、反応が完了するまで攪拌した(HPLCによると変換率97%、典型的には24時間以内)。生じた溶液を50から60℃の温度に冷却し、トルエン(2×30mL)で抽出した。トルエン洗浄液を廃棄した。水性相を50%NaOH(約19g、0.23mol、2.0当量)で、pH2.0に調節した。活性化炭素(4.0g)、濾過剤(4.0g)および水(10g)をフラスコに加え、内容物を50℃で30分間攪拌した。スラリーを60℃に加熱し、粗いフリットおよび0.5μmPTFE膜で濾過したが、その間、スラリーを>55℃の温度に維持した。フィルターケークを、>50℃の温度に加熱されていた水(25mL)ですすいだ。合わせた水性層を温度<40℃に冷却し、真空蒸留して、約70mLの最終体積にした。生成物を含有する濃縮溶液を、50%NaOH(約8.2g、0.1mol、0.9当量)でpH6.5から7.5に調節すると、沈殿物が形成した。混合物を氷浴で温度<5℃に冷却した。生じたスラリーを温度<5℃で60分間攪拌し、次いで、濾過して、粗製(固体)生成物を単離した。ケークをまだ湿っている間に水(20g)ですすぎ、次いで、フィルター上で24から48時間乾燥させた。
【0178】
粗製(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(20g)およびイソプロパノール/水(40重量%i−PrOH水溶液125mL)を、頭上攪拌、還流凝縮器およびサーモカップルを備えた500mL四つ口RBFに窒素ブランケット下に充填した。スラリーを還流(約85℃)に加熱した。溶液を15分間攪拌し、窒素圧力を使用して、0.5μmPTFE膜および焼結ガラスフリット(60℃に加熱されていた)で濾過することにより澄明にした。イソプロパノール/水(40wt%i−PrOH水溶液20mL)をフラスコに充填し、還流まで加熱した。すすぎ液を膜およびフリットを介して窒素圧力で移し、生成物溶液濾液と合わせた。合わせた濾液を、頭上攪拌、還流凝縮器およびサーモカップルを備えていて、予めマーキングされている(102mLの所)500mL四つ口RBFに窒素ブランケット下に移した。溶液を<40℃に冷却した。スラリーを40から50℃で真空蒸留して、102mLの全体積にした。イソプロパノール内容物を24から27重量%i−PrOHに調節した。スラリーを還流(約87℃)に再加熱し、固体がすべて溶解するまで保持した。溶液を20℃/hの速度で5℃まで徐々に冷却し、60分間保持して、生成物を沈殿させた。最終(固体)生成物を真空濾過により単離し、イソプロパノール(30mL、<5℃に冷却)で洗浄した。フィルターケークを40℃で真空下に24時間乾燥させて、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(21.0g、95%)を得た。
【0179】
(実施例57)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
乾燥不活性容器に、(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.56、100kg、234mol、1.00当量)、臭化カリウム(mw119.00、60Kg、504mol、2.15当量)およびトルエン180Lを充填した。スラリーを攪拌し、水6L中の臭化テトラブチルアンモニウム(mw322.37、6kg、18.60mol、0.08当量)を加えた。スラリーを89〜93℃に少なくとも8時間加熱し、次いで、60〜65℃に冷却し、トルエン50Lおよび水130Lを加えた。水性相を除去し、有機相を50℃で、水50Lポーションで2回洗浄した。有機相を含有する生じた生成物を25℃未満に冷却した。真空を適用し、有機溶液を蒸留して、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(57.1kg、234mol)を含有する約50wt%溶液にした。
【0180】
(実施例58)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの調製
乾燥不活性容器に、リチウムヘキサメチルジシラジドのTHF溶液(mw167.33、24重量%、500Kg、3.3当量)を充填した。溶液を真空蒸留して、その元の体積の約50%にした。先行する実施例からの(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(mw243.97、57.1Kg、234mol、1当量)のトルエン溶液を加え、その際、温度を5℃未満に維持した。4,4,4−トリメトキシブチロニトリル(mw159.19、51.0Kg、320mol、1.4当量)を6時間にわたって加え、その際、温度を0から5℃に維持した。GCにより、反応が完了したと思われたら、水15Lを制御添加することにより、反応をクエンチし、次いで、水135Lを加え、混合物を20℃に加温した。二相混合物を分離し、下部水性相を廃棄した。THF溶媒を、全部で300Lのn−ヘプタンの3回の真空蒸留追跡により置換した。次いで、(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルを含有する有機混合物を水100Lポーションで2回洗浄し、水60Lおよびトルエン5Lを加え、混合物を激しく攪拌した。濃塩酸(mw36.46;37%溶液35kg、355mol)の制御添加を行って、pHをpH3未満に調節し、混合物を30分間攪拌した。下部水性相を除去し、水洗浄を行い、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルを含有する有機溶液を次のステップに入れた。
【0181】
(実施例59)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の調製
先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの溶液を45wt%水酸化カリウム水溶液(mw56.11、溶液29Kg、233mol)で約1時間にわたって処理し、次いで、けん化が完了するまで、さらに1時間攪拌した。下部水性相を除去し、有機相を水で洗浄し、水性相を合わせた。前記表題化合物を含有する水溶液の重量は、約100Kgであった。
【0182】
(実施例60)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
水素化オートクレーブに、モリブデン促進スポンジニッケル(Johnson Matthey A−7000、4Kg)、45wt%水酸化カリウム水溶液(mw56.11、11Kg、88.21mol)および水20Lを充填した。これに、先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の溶液約50Kgを加えた。溶液を水素で50psigで処理したが、この間、温度を30℃に保持した。反応は12時間後に完了し、触媒を濾過により除去した。先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の溶液の残りの50Kgを同じ方法で水素化した。2つの水素化溶液を合わせ、L−酒石酸(mw150.09、2.5Kg、16.66mol)を含有するタンクに充填し、酸が溶けるまで攪拌した。溶液のpHを、濃塩酸(mw36.46、37%溶液35Kg、355mol)で約pH11.0に調節したが、この際、温度を20〜25℃に維持した。この時点で、粗製生成物が沈殿し始めた。次いで、混合物を、氷酢酸(mw60.05、2.5Kg、41.67mol)でpH6〜8に調節した。粗製生成物を濾過により単離し、プロパン−2−オール92Kgで洗浄し、40℃窒素で乾燥させると、粗製の(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、37Kg、199mol)が得られた。
【0183】
粗製の(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、37Kg、199mol)を約80℃で、2−プロパノール130Lおよび水130Lの混合物に溶かし、溶液を研磨フィルターで濾過した。溶液を50〜55℃に冷却したが、この際、生成物が結晶化し始めた。さらに20℃に冷却した後に、スラリーを真空蒸留したが、この間に、2−プロパノール35Kgポーションを2回加えて、水と置換した。約4L/Kgの最終蒸留体積に達したら、スラリーを0〜5℃に冷却し、濾過した。フィルターケークを2−プロパノール40Kgで洗浄し、40℃窒素で乾燥させると、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、30Kg、161.91mol)が得られた。
【0184】
(実施例61)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
清潔な不活性容器に、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩(mw219.3、48%溶液106Kg、232mol)および水35Lの溶液を充填した。溶液を攪拌し、真空蒸留して、35Lまで体積を低減し、次いで、温度を0〜5℃に維持しながら、5.7Mの塩酸水溶液92.8Kgを含有する容器に徐々に移し、2−((3S、4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の結晶をシーディングした。さらなる水10Lを加え、移動が完了したら、生じたスラリーを0℃で1時間攪拌し、濾過し、冷水75Lで洗浄した。ケークを窒素掃引で約1時間、部分的に乾燥させると、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の水湿潤ケーク86.82Kgが得られた。
【0185】
(実施例62)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の調製
先行する実施例からの水で湿潤させた2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸86Kgを、45wt%水酸化カリウム水溶液(mw56.11、溶液63.2Kg、506mol)で約1時間にわたって処理すると、2((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩を含有する均一な溶液が得られた。
【0186】
(実施例63)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸tert−ブチルアミン塩の調製
窒素入口および機械式頭上攪拌機を備えた250mL三つ口丸底フラスコ(RBF)に、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩(mw219.3、13.2g、0.06mol)を含有する水溶液25gおよび水酸化カリウム(mw56.11、0.68g、0.012mol)、水25mLおよびトルエン55mLを充填した。塩酸水溶液(mw36.43、37%溶液7.3g、0.075mol)を約3分にわたって加えて、pHをpH2未満に調節した。5分間攪拌した後に、相を分離し、下部水性相を除去した。生成物を含有する有機相の20mL水洗浄を行い、生じたトルエン溶液に、2−プロパノール10mLを加えた。溶液を20〜25℃に維持して、tert−ブチルアミン(mw73.14、5.2g、0.07mol、1.2当量)を約5分にわたって加えた。生じたスラリーを75℃に加温すると、濁った混合物が得られた。約20℃/時間の速度で5℃に冷却すると、塩が沈殿した。0〜5℃で1時間攪拌した後に、固体を濾過し、トルエン50mLで洗浄し、真空炉中40℃で乾燥させると、固体の2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸tert−ブチルアミン塩(mw254.1、12.8g、0.05mol、83%)が得られた。
【0187】
本明細書および添付の請求項において使用される場合、「a」、「an」および「the」などの単数冠詞は、文脈で他に明確に記載されていない限り、1個の対象または複数の対象を指しうることを特記する。したがって例えば、「(a)化合物」を含む組成物に関する言及は、単一化合物または2種以上の化合物を含みうる。
【0188】
前記記載は、詳述を意図しており、制限的ではないことを意図していることを理解されたい。前記記載を読めば、当業者には、多くの実施形態が明らかであろう。したがって本発明の範囲は、添付の請求項に関する言及と共に、このような請求項が権利を有する同等の全範囲により決定されるべきである。
【図面の簡単な説明】
【0189】
【図1】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(全スペクトル)。
【図2】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(フィンガープリント領域)。
【図3】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(全スペクトル)。
【図4】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(フィンガープリント領域)。
【図5】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(全スペクトル)。
【図6】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(フィンガープリント領域)。
【図7】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(全スペクトル)。
【図8】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(フィンガープリント領域)。
【図9】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの粉末X線回折(PXRD)パターン(フルスケール)。
【図10】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(30000cpsまで)。
【図11】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(フルスケール)。
【図12】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(30000cpsまで)。
【図13】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの示差走査熱分析(DSC)サーモグラム。
【図14】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのDSCサーモグラム。
【技術分野】
【0001】
本発明は、疼痛ならびに様々な精神障害および睡眠障害を治療するために有用なキラル環式アミノ酸を調製するための物質および方法に関する。
【背景技術】
【0002】
Bryansらに付与された米国特許第6635673B1号明細書(’673号特許)は、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸を包含するいくつかの光学活性な環式アミノ酸およびその薬学的に許容できる塩を記載している。これらの化合物は、カルシウムチャネルのアルファ−2−デルタ(α2δ)サブユニットに結合する。これらは特に、不眠症、てんかん、失神発作、運動機能減少症、うつ病、不安、パニック、疼痛、過敏性腸症候群および関節炎を包含するいくつかの疾患を治療するために有用である。
【発明の開示】
【発明が解決しようとする課題】
【0003】
’673号特許は、光学活性な環式アミノ酸を調製するためのいくつかの方法を記載している。これらの方法の多くは、中間体または出発原料として、(S,S)−3,4−ジメチル−シクロペンタノンを包含するキラル3,4−二置換シクロペンタノンを使用する。シクロペンタノンを調製する方法は知られているが、効率および経費の関係により、またはプロセスが市販されていない出発原料を使用することにより、これらのプロセスの多くは、市場規模での製造には問題がある。例えば、Blakemoreらに付与された米国特許第6872856号明細書参照。さらに、キラルシクロペンタノンを所望の光学活性な環式アミノ酸に変換するためには、数多くのステップが必要となりうる。したがって、キラル環式アミノ酸を調製するための改善された方法が望ましい。
【課題を解決するための手段】
【0004】
本発明は、市販の出発原料から光学活性な環式アミノ酸(下記の式1)を調製するための比較的効率的で費用対効果の大きい方法を提供する。例えば、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸を、(2R,3R)−1,4−ジブロモ−2,3−ジメチル−ブタンまたは(2R,3R)−2,3−ジメチル−ブタン−1,4−ジイル−ジトシレートから3または4ステップで調製することができる。
【0005】
本発明の一態様は、式1の化合物もしくは薬学的に許容できるその塩または式1の化合物の反対の鏡像異性体もしくは薬学的に許容できるその塩を製造する方法を提供し:
【0006】
【化1】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]、
ここで、前記方法は、
(a)式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩のシアノ部分をアミノ部分に還元するステップと
【0007】
【化2】
[式5中のR1およびR2は、式1に関する前記と同様に定義され、R4は水素原子およびC1〜6アルキルから選択される]、
(b)任意選択で、式1の化合物またはその反対の鏡像異性体を式1の化合物またはその反対の鏡像異性体の薬学的に許容できる塩に変換するステップを含む。
【0008】
本発明の他の態様は、式4の化合物またはその反対の鏡像異性体を提供する:
【0009】
【化3】
[式中、式1に関する前記と同様に定義され、
R3は、
【0010】
【化4】
により表される構造を有するカルボン酸またはエステル保護基である
(式7および式8中の「A」は、式4の化合物の残りの部分に結合している結合点を表し、
R6、R7、R8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成し、
Z1およびZ2はそれぞれ独立に、OおよびSから選択され、
R9は、C1〜6アルキルであり、
R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、
R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する)]。
【0011】
本発明の他の態様は、式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩を提供する:
【0012】
【化5】
[式中、
R1およびR2は、式1に関する前記と同様に定義され、
R4は、H、C1〜6アルキル、アリール−C1〜3アルキル、第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオンから選択され、
ここで、前記各アリール−C1〜3アルキル基中のアリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6−アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]。
【0013】
本発明は、可能であれば、薬学的に許容できるかできないかにかかわらず、前記化合物のすべての塩、複合体、溶媒和物、水和物および多形体を包含する。
【発明を実施するための最良の形態】
【0014】
定義および略語
他に示されていない限り、本開示は、下記に提示される定義を使用する。いくつかの定義および式は、原子間の結合または指名されているか指名されていない原子もしくは原子の基への結合点を示すためにダッシュ(「−」)を包含することもある。他の定義および式は、それぞれ二重結合または三重結合を示すために等号(「=」)または識別記号(「≡」)を包含することもある。他の式は、1つまたは複数の波形結合
【0015】
【化6】
を包含することもある。立体中心に結合している場合、波形結合は、個々の、または混合物としての両方の立体異性体に関している。同様に、二重結合に結合している場合、波形結合は、Z異性体、E異性体またはZおよびE異性体の混合物を指している。いくつかの式は、単結合または二重結合を示すために破線結合
【0016】
【化7】
を包含することもある。
【0017】
「置換されている」基は、1個または複数の水素原子が1個または複数の非水素原子または基で置換されているものであるが、ただし、価要求は満たしていて、置換により、化学的に安定な化合物が生じている。
【0018】
測定可能な数値変数に関連して使用される場合、「約」または「ほぼ」は、示されている変数の値および示されている値の実験誤差の範囲内(例えば平均値に関して95%の信頼区間内)または示されている値の10%の範囲内で、いずれにしろより高い変数の値すべてを指している。
【0019】
「アルキル」は、規定の数の炭素原子を通常は有する直鎖および分枝鎖飽和炭化水素基を指している(即ち、C1〜6アルキルは、1、2、3、4、5または6個の炭素原子を有するアルキル基を指している)。アルキル基の例には、メチル、エチル、n−プロピル、i−プロピル、n−ブチル、s−ブチル、i−ブチル、t−ブチル、ペンチ−1−イル、ペンチ−2−イル、ペンチ−3−イル、3−メチルブチ−1−イル、3−メチルブチ−2−イル、2−メチルブチ−2−イル、2,2,2−トリメチルエチ−1−イルおよびn−ヘキシルが包含される。
【0020】
「アルケニル」は、1個または複数の不飽和炭素−炭素結合を有し、規定の数の炭素原子を通常は有する直鎖および分枝鎖炭化水素基を指している。アルケニル基の例には、エテニル、1−プロペン−1−イル、1−プロペン−2−イル、2−プロペン−1−イル、1−ブテン−1−イル、1−ブテン−2−イル、3−ブテン−1−イル、3−ブテン−2−イル、2−ブテン−1−イル、2−ブテン−2−イル、2−メチル−1−プロペン−1−イル、2−メチル−2−プロペン−1−イル、1,3−ブタジエン−1−イルおよび1,3−ブタジエン−2−イルが包含される。
【0021】
「アルキニル」は、1個または複数の三重炭素−炭素結合を有し、規定の数の炭素原子を通常は有する直鎖または分枝鎖炭化水素基を指している。アルキニル基の例には、エチニル、1−プロピン−1−イル、2−プロピン−1−イル、1−ブチン−1−イル、3−ブチン−1−イル、3−ブチン−2−イルおよび2−ブチン−1−イルが包含される。
【0022】
「アルカノイル」は、アルキルが前記のように定義され、規定の数の炭素原子を通常は包含するアルキル−C(O)−を指しており、カルボニル炭素を包含する。アルカノイル基の例には、ホルミル、アセチル、プロピオニル、ブチリル、ペンタノイルおよびヘキサノイルが包含される。
【0023】
「アルコキシ」および「アルコキシカルボニル」はそれぞれ、アルキルが前記のように定義されるアルキル−O−およびアルキル−O−C(O)−を指している。アルコキシ基の例には、メトキシ、エトキシ、n−プロポキシ、i−プロポキシ、n−ブトキシ、s−ブトキシ、t−ブトキシ、n−ペントキシおよびs−ペントキシが包含される。アルコキシカルボニル基の例には、メトキシカルボニル、エトキシカルボニル、n−プロポキシカルボニル、i−プロポキシカルボニル、n−ブトキシカルボニル、s−ブトキシカルボニル、t−ブトキシカルボニル、n−ペントキシカルボニルおよびs−ペントキシカルボニルが包含される。
【0024】
「ハロ」、「ハロゲン」および「ハロゲノ」は、互換的に使用することができ、フルオロ、クロロ、ブロモおよびヨードを指している。
【0025】
「ハロアルキル」は、1個または複数のハロゲン原子で置換されているアルキル基を指しており、ここで、アルキルは前記のように定義される。ハロアルキル基の例には、トリフルオロメチル、トリクロロメチル、ペンタフルオロエチルおよびペンタクロロエチルが包含される。
【0026】
「シクロアルキル」は、環を含み、規定の数の炭素原子を一般に有する飽和単環式および二環式炭化水素環を指している(即ち、C3〜7シクロアルキルは環員として3、4、5、6または7個の炭素原子を有するシクロアルキル基を指している)。シクロアルキルは、そのような結合が原子価要求に反しない限り、親基に結合していてもよいし、任意の環原子の所で基質に結合していてもよい。同様に、シクロアルキル基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたようなアルキル、アルコキシ、アルコキシカルボニル、アルカノイルおよびハロならびにヒドロキシ、メルカプト、ニトロおよびアミノが包含される。
【0027】
単環式シクロアルキル基の例には、シクロプロピル、シクロブチル、シクロペンチルおよびシクロヘキシルが包含される。二環式シクロアルキル基の例には、ビシクロ[1.1.0]ブチル、ビシクロ[1.1.1]ペンチル、ビシクロ[2.1.0]ペンチル、ビシクロ[2.1.1]ヘキシル、ビシクロ[3.1.0]ヘキシル、ビシクロ[2.2.1]ヘプチル、ビシクロ[3.2.0]ヘプチル、ビシクロ[3.1.1]ヘプチル、ビシクロ[4.1.0]ヘプチル、ビシクロ[2.2.2]オクチル、ビシクロ[3.2.1]オクチル、ビシクロ[4.1.1]オクチル、ビシクロ[3.3.0]オクチル、ビシクロ[4.2.0]オクチル、ビシクロ[3.3.1]ノニル、ビシクロ[4.2.1]ノニル、ビシクロ[4.3.0]ノニル、ビシクロ[3.3.2]デシル、ビシクロ[4.2.2]デシル、ビシクロ[4.3.1]デシル、ビシクロ[4.4.0]デシル、ビシクロ[3.3.3]ウンデシル、ビシクロ[4.3.2]ウンデシルおよびビシクロ[4.3.3]ドデシルが包含される。
【0028】
「シクロアルケニル」は、環を含み、1個または複数の不飽和炭素−炭素結合を有し、規定の数の炭素原子を通常は有する単環式および二環式炭化水素環を指している(即ち、C3〜7シクロアルケニルは、環員として3、4、5、6または7個の炭素原子を有するシクロアルケニル基を指している)。シクロアルケニルは、そのような結合が原子価要求に反しない限り、親基に結合していてもよいし、任意の環原子の所で基質に結合していてもよい。同様に、シクロアルケニル基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたようなアルキル、アルケニル、アルキニル、アルコキシ、アルコキシカルボニル、アルカノイルおよびハロならびにヒドロキシ、メルカプト、ニトロおよびアミノが包含される。
【0029】
「シクロアルカノイル」および「シクロアルケノイル」は、それぞれシクロアルキル−C(O)−およびシクロアルケニル−C(O)−を指し、ここで、シクロアルキルおよびシクロアルケニルは前記のように定義される。シクロアルカノイルおよびシクロアルケノイルに関する言及は通常、カルボニル炭素を除く規定の数の炭素原子を包含する。シクロアルカノイル基の例には、シクロプロパノイル、シクロブタノイル、シクロペンタノイル、シクロヘキサノイル、シクロヘプタノイル、1−シクロブテノイル、2−シクロブテノイル、1−シクロペンテノイル、2−シクロペンテノイル、3−シクロペンテノイル、1−シクロヘキセノイル、2−シクロヘキセノイルおよび3−シクロヘキセノイルが包含される。
【0030】
「シクロアルコキシ」および「シクロアルコキシカルボニル」は、それぞれシクロアルキル−O−およびシクロアルケニル−Oならびにシクロアルキル−O−C(O)−およびシクロアルケニル−O−C(O)−を指し、ここで、シクロアルキルおよびシクロアルケニルは前記のように定義される。シクロアルコキシおよびシクロアルコキシカルボニルに関する言及は通常、カルボニル炭素を除く規定の数の炭素原子を包含する。シクロアルコキシ基の例には、シクロプロポキシ、シクロブトキシ、シクロペントキシ、シクロヘキソキシ、1−シクロブテノキシ、2−シクロブテノキシ、1−シクロペンテノキシ、2−シクロペンテノキシ、3−シクロペンテノキシ、1−シクロヘキセノキシ、2−シクロヘキセノキシおよび3−シクロヘキセノキシが包含される。シクロアルコキシカルボニル基の例には、シクロプロポキシカルボニル、シクロブトキシカルボニル、シクロペントキシカルボニル、シクロヘキソキシカルボニル、1−シクロブテノキシカルボニル、2−シクロブテノキシカルボニル、1−シクロペンテノキシカルボニル、2−シクロペンテノキシカルボニル、3−シクロペンテノキシカルボニル、1−シクロヘキセノキシカルボニル、2−シクロヘキセノキシカルボニルおよび3−シクロヘキセノキシカルボニルが包含される。
【0031】
「アリール」および「アリーレン」は、窒素、酸素およびイオウから独立に選択される0から4個のヘテロ原子を含有する5員および6員の単環式芳香族基を包含するそれぞれ一価および二価の芳香族基を指している。単環式アリール基の例には、フェニル、ピロリル、フラニル、チオフェネイル、チアゾリル、イソチアゾリル、イミダゾリル、トリアゾリル、テトラゾリル、ピラゾリル、オキサゾリル、イソオキサゾリル、ピリジニル、ピラジニル、ピリダジニルおよびピリミジニルが包含される。アリールおよびアリーレン基にはさらに、前記の縮合5員および6員の環を包含する二環式および三環式基が包含される。多環式アリール基の例には、ナフチル、ビフェニル、アントラセニル、ピレニル、カルバゾリル、ベンゾオキサゾリル、ベンゾジオキサゾリル、ベンゾチアゾリル、ベンゾイミダゾリル、ベンゾチオフェネイル、キノリニル、イソキノリニル、インドリル、ベンゾフラニル、プリニルおよびインドリジニルが包含される。これらのアリールおよびアリーレン基は、そのような結合が原子価要求に反しない限り、任意の環原子の所で他の基に結合していてもよい。アリールおよびアリーレン基は、そのような置換が原子価要求に反しない限り、1個または複数の非水素置換基を包含してよい。有用な置換基には、前記で定義されたアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アルコキシ、シクロアルコキシ、アルカノイル、シクロアルカノイル、シクロアルケノイル、アルコキシカルボニル、シクロアルコキシカルボニルおよびハロならびにヒドロキシ、メルカプト、ニトロ、アミノおよびアルキルアミノが包含される。
【0032】
「ヘテロアリール」および「ヘテロアリーレン」は、少なくとも1個のヘテロ原子を含有する前記で定義されたそれぞれ一価および二価のアリールおよびアリーレン基を指している。
【0033】
「複素環」および「ヘテロシクリル」は、それぞれ、5員から7員または7員から11員の環員を有する飽和、部分不飽和または不飽和単環式または二環式環を指している。単環式および二環式基はそれぞれ、炭素原子および独立に窒素、酸素またはイオウである1から4個または1から6個のヘテロ原子からなる環員を有し、前記で定義された任意の単環式複素環がベンゼン環に縮合している任意の二環式基を含有してもよい。窒素およびイオウヘテロ原子は、酸化されていてもよい。複素環は、そのような結合が原子価要求に反しない限り、任意のヘテロ原子または炭素原子の所で他の基に結合していてもよい。任意の炭素または窒素環員は、そのような置換が原子価要求に反しない限り、非水素置換基を包含してよい。有用な置換基には、前記で定義されたアルキル、アルケニル、アルキニル、シクロアルキル、シクロアルケニル、アルコキシ、シクロアルコキシ、アルカノイル、シクロアルカノイル、シクロアルケノイル、アルコキシカルボニル、シクロアルコキシカルボニルおよびハロならびにヒドロキシ、メルカプト、ニトロ、アミノおよびアルキルアミノが包含される。
【0034】
複素環の例には、アクリジニル、アゾシニル、ベンズイミダゾリル、ベンゾフラニル、ベンゾチオフラニル、ベンゾチオフェニル、ベンゾオキサゾリル、ベンズチアゾリル、ベンズトリアゾリル、ベンズテトラゾリル、ベンズイソオキサゾリル、ベンズイソチアゾリル、ベンズイミダゾリニル、カルバゾリル、4aH−カルバゾリル、カルボリニル、クロマニル、クロメニル、シノリニル、デカヒドロキノリニル、2H,6H−1,5,2−ジチアジニル、ジヒドロフロ[2,3−b]テトラヒドロフラン、フラニル、フラザニル、イミダゾリジニル、イミダゾリニル、イミダゾリル、1H−インダゾリル、インドレニル、インドリニル、インドリジニル、インドリル、3H−インドリル、イソベンゾフラニル、イソクロマニル、イソインダゾリル、イソインドリニル、イソインドリル、イソキノリニル、イソチアゾリル、イソオキサゾリル、モルホリニル、ナフチリジニル、オクタヒドロイソキノリニル、オキサジアゾリル、1,2,3−オキサジアゾリル、1,2,4−オキサジアゾリル、1,2,5−オキサジアゾリル、1,3,4−オキサジアゾリル、オキサゾリジニル、オキサゾリル、オキサゾリジニル、ピリミジニル、フェナントリジニル、フェナントロリニル、フェナジニル、フェノチアジニル、フェノキサチイニル、フェノキサジニル、フタラジニル、ピペラジニル、ピペリジニル、プテリジニル、プリニル、ピラニル、ピラジニル、ピラゾリジニル、ピラゾリニル、ピラゾリル、ピリダジニル、ピリドオキサゾール、ピリドイミダゾール、ピリドチアゾール、ピリジニル、ピリジル、ピリミジニル、ピロリジニル、ピロリニル、2H−ピロリル、ピロリル、キナゾリニル、キノリニル、4H−キノリジニル、キノキサリニル、キヌクリジニル、テトラヒドロフラニル、テトラヒドロイソキノリニル、テトラヒドロキノリニル、6H−1,2,5−チアジアジニル、1,2,3−チアジアゾリル、1,2,4−チアジアゾリル、1,2,5−チアジアゾリル、1,3,4−チアジアゾリル、チアントレニル、チアゾリル、チエニル、チエノチアゾリル、チエノオキサゾリル、チエノイミダゾリル、チオフェニル、トリアジニル、1,2,3−トリアゾリル、1,2,4−トリアゾリル、1,2,5−トリアゾリル、1,3,4−トリアゾリルおよびキサンテニルが包含される。
【0035】
「アリールアルキル」および「ヘテロアリールアルキル」はそれぞれ、アリール−アルキルおよびヘテロアリール−アルキルを指し、ここで、アリール、ヘテロアリールおよびアルキルは前記のように定義される。例には、ベンジル、フルオレニルメチルおよびイミダゾール−2−イル−メチルが包含される。
【0036】
「脱離基」は、置換反応、脱離反応、付加−脱離反応を包含する分裂プロセスの間に分子から脱離する任意の基を指している。脱離基は、脱離基と分子の間の結合として前は役立っていた電子対と共に基が離れる離核性であってもよいし、基が電子対なしに離れる離電性であってもよい。離核性脱離基の脱離する能力は、その塩基強度に左右され、その際、最も強い塩基が、最も弱い脱離基である。一般的な離核性脱離基には、窒素(例えばジアゾニウム塩から)、アルキルスルホネート(例えばメシレート)、フルオロアルキルスルホネート(例えばトリフレート、ヘキサフレート、ノナフレートおよびトレシレート)およびアリールスルホネート(例えばトシレート、ブロシレート、クロシレートおよびノシレート)を包含するスルホネートが包含される。他には、カルボネート、ハライドイオン、カルボキシレートアニオン、フェノレートイオンおよびアルコキシドが包含される。NH2−およびOH−などのいくつかの比較的強い塩基を、酸で処理することにより、より良好な脱離基にすることができる。一般的な離電性脱離基には、プロトン、CO2および金属が包含される。
【0037】
「鏡像異性体過剰率」または「ee」は、所定の試料での、キラル化合物のラセミ試料に対する一方の鏡像異性体の過剰率の尺度であり、パーセンテージで表される。鏡像異性体過剰率は、100×(er−1)/(er+1)として定義され、ここで、「er」は、多い方の鏡像異性体と少ない方の鏡像異性体との比である。
【0038】
「ジアステレオ異性体過剰率」または「de」は、所定の試料での、等量のジアステレオ異性体を有する試料に対する一方のジアステレオ異性体の過剰率の尺度であり、パーセンテージで表される。ジアステレオ異性体過剰率は、100×(dr−1)/(dr+1)として定義され、ここで、「dr」は、多い方のジアステレオ異性体と少ない方のジアステレオ異性体との比である。
【0039】
「立体選択的」、「エナンチオ選択的」、「ジアステレオ選択的」およびそれらの変形は、それぞれ、他方に対して一方の立体異性体、鏡像異性体またはジアステレオ異性体を多くもたらす所定のプロセス(例えば水素化)を指している。
【0040】
「高度立体選択性」、「高度エナンチオ選択性」、「高度ジアステレオ選択性」およびそれらの変形は、それぞれ、その生成物の少なくとも約90%を構成する一方の立体異性体、鏡像異性体またはジアステレオ異性体の過剰率を有する生成物をもたらす所定のプロセスを指している。一対の鏡像異性体またはジアステレオ異性体では、高度エナンチオ選択性またはジアステレオ選択性は、少なくとも約80%のeeまたはdeに対応するであろう。
【0041】
「立体異性的に富化された」、「鏡像異性的に富化された」、「ジアステレオ異性的に富化された」およびそれらの変形はそれぞれ、他方よりも一方の立体異性体、鏡像異性体またはジアステレオ異性体を多く有する化合物の試料を指している。富化の程度は、全生成物に対して、または一対の鏡像異性体またはジアステレオ異性体に関する%により、またはeeもしくはdeにより測定することができる。
【0042】
「実質的に純粋な立体異性体」、「実質的に純粋な鏡像異性体」、「実質的に純粋なジアステレオ異性体」およびそれらの変形はそれぞれ、試料の少なくとも約95%を構成する立体異性体、鏡像異性体またはジアステレオ異性体を含有する試料を指している。鏡像異性体またはジアステレオ異性体の対では、実質的に純粋な鏡像異性体およびジアステレオ異性体は、約90%以上のeeまたはdeを有する試料に対応するであろう。
【0043】
「純粋な立体異性体」、「純粋な鏡像異性体」、「純粋なジアステレオ異性体」およびその変形はそれぞれ、試料の少なくとも約99.5%を構成する立体異性体、鏡像異性体またはジアステレオ異性体を含有する試料を指している。鏡像異性体およびジアステレオ異性体の対では、純粋な鏡像異性体または純粋なジアステレオ異性体は、約99%以上のeeまたはdeを有する試料に対応するであろう。
【0044】
「反対の鏡像異性体」は、基準分子に対して重なり合わない鏡像である分子を指しており、これは、対照分子の立体由来中心のすべてを逆にすると得ることができる。例えば、基準分子がS絶対立体化学的配置を有する場合、その反対の鏡像異性体は、R絶対立体化学的配置を有する。同様に、基準分子がS,S絶対立体化学的配置を有する場合、その反対の鏡像異性体は、R,R立体化学的配置を有するなどである。
【0045】
規定の化合物の「立体異性体」は、化合物の反対の鏡像異性体を、さらに化合物の任意のジアステレオ異性体または幾何異性体(Z/E)を指している。例えば、規定の化合物がS,R,Z立体化学的配置を有する場合、その立体異性体は、R,S,Z立体配置を有するその反対の鏡像異性体、S,S,Z立体配置およびR,R,Z立体配置を有するそのジアステレオ異性体ならびにS,R,E立体配置、R,S,E,立体配置、S,S,E立体配置およびR,R,E立体配置を有するその幾何異性体を包含するであろう。
【0046】
「溶媒和物」は、開示または請求されている化合物と化学量論的量または非化学量論的量の1種または複数の溶媒分子(例えばEtOH、アセトン、水)を含む分子複合体を指している。
【0047】
「水和物」は、開示または請求されている化合物と化学量論的量または非化学量論的量の水を含む溶媒和物を指している。
【0048】
「薬学的に許容できる複合体、塩、溶媒和物または水和物」は、正当な医学的判断の範囲内であり、過度の毒性、刺激およびアレルギー応答を伴うことなく、患者の組織と接触させて使用するために適していて、妥当な損益比と釣り合い、その意図されている使用に有効である、請求および開示されている化合物の複合体、酸もしくは塩基付加塩、溶媒和物または水和物を指している。
【0049】
「治療する」は、このような用語が適用される障害または状態の進行を逆転、緩和、阻害することもしくはそれらを予防することを、またはこのような障害または状態の1つまたは複数の症状を予防することを指している。
【0050】
「治療」は、直前で定義された「治療する」の行為を指している。
【0051】
表1は、本明細書を通して使用される略語を列記している。
【0052】
【表1−1】
【0053】
【表1−2】
【0054】
下記のいくつかのスキームおよび実施例は、有機化学の当業者には知られている酸化および還元を包含する一般的な反応の詳細を省略していることもある。このような反応の詳細は、Richard Larock、Comprehensive Organic Transformations(1999年)およびMichael B.Smithらによって編集されている複数巻シリーズ、Compendium of Organic Synthetic Methods(1974年以降)を包含する複数の論文で見ることができる。出発原料および試薬は、市場の供給源から得るか、文献方法を使用して調製することができる。例えば、下記に記載の出発原料の1種、(R)−2−メチルコハク酸4−メチルエステルは、対応するジエステルのエステラーゼ仲介加水分解を介して、または適切な不飽和モノエステルの不斉水素化を介して得ることができる。S.G.Cohen&A.J.Milovanovic、J.Am.Chem.Soc.90:3495(1968年)およびM.Ostermeierら、Eur.J.Org.Chem.17:3453(2003年)参照。
【0055】
下記のいくつかの反応スキームおよび実施例では、ある種の化合物は、他の反応部位での望ましくない化学反応を防ぐ保護基を使用して調製することができる。保護基を使用して、溶解性を増強するか、他の場合には化合物の物理的特性を変更することもできる。保護基計画の検討、保護基を導入および除去するための物質および方法の記載ならびにアミン、カルボン酸、アルコール、ケトンおよびアルデヒドを包含する一般的な官能基のために有用な保護基の編集に関しては、T.W.GreeneおよびP.G.Wuts、Protective Groups in Organic Chemistry(1999年)およびP.Kocienski、Protective Groups(2000年)参照。
【0056】
通常、本明細書を通して記載される化学的変換は、実質的に化学量論的量の反応成分を使用して実施することができるが、ある種の反応は、1種または複数の反応成分を過剰に使用することから利益を得ることもある。加えて、多くの反応は、ほぼ室温で実施することができるが、特定の反応は、反応動態、収率および他の事情に応じて、より高い温度(例えば還流まで)またはより低い温度(例えば0℃以下)の使用を必要とすることもある。多くの化学的変換は、反応速度および収率に影響を及ぼしうる1種または複数の相容性の溶媒を使用することもでき、反応成分の性質に応じて、極性プロトン性溶媒(例えば水、MeOH、EtOH、PrOH、i−PrOH、ギ酸、HOAc、ホルムアミド)、極性非プロトン性溶媒(例えばアセトン、THF、MEK、EtOAc、ACN、DMF、DMSO)、非極性溶媒(例えばヘキサン、ベンゼン、トルエン、ジエチルエーテル、CH2Cl2、CHCl3、CCl4)またはこれらのいくつかの組合せであってもよい。濃度範囲、温度範囲またはpH範囲を包含する範囲に関する本開示における任意の言及は、示されている終点を包含する。
【0057】
この開示は、式1の光学活性な環式アミノ酸、その反対の鏡像異性体ならびに式1の化合物およびその反対の鏡像異性体の薬学的に許容できる複合体、塩、溶媒和物および水和物を調製するための物質および方法に関する。前記のように、式1中のR1およびR2は、Me、Et、Pr、i−Pr、n−Bu、s−Bu、t−BuなどのC1〜6アルキル、さらにBn、フェニル−エチルおよびフェニル−プロピルなどのアリール−C1〜3アルキルを包含しうる。したがって代表的な式1の化合物には、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸、(3S,4S)−(1−アミノメチル−3,4−ジエチル−シクロペンチル)−酢酸、(3S,4S)−(1−アミノメチル−3,4−ジプロピル−シクロペンチル)−酢酸および(3S,4S)−(1−アミノメチル−3,4−ジベンジル−シクロペンチル)−酢酸、これらの反対の鏡像異性体ならびに前記化合物の薬学的に許容できる複合体、塩、溶媒和物および水和物が包含される。
【0058】
一般的な反応スキーム
【0059】
【化8】
スキーム1は、式1の化合物およびその反対の鏡像異性体を調製するための方法を示している。方法は、キラル1,4−ビス−求電子体(式2)を保護(マスキング)されているカルボン酸(式3)と反応させて、光学活性なシクロペンタンカルボニトリル(式4)を得ることを包含する。カルボキシ部分の脱保護(非マスキング)により、1−カルボキシル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。ニトリルエステル(式5、R4はHではないか、R4はカチオン、M1ではない)を還元すると、アミンが得られ、これを環化してラクタム(式6)を得、これを加水分解すると、所望の環式アミノ酸(式1)が得られる。別法では、ニトリルエステルの加水分解(必要な場合)、それに続く酸またはその塩(式5、R4=HまたはM1)を還元すると、環式アミノ酸(式1)が得られる。
【0060】
式2および式4〜6中の置換基R1およびR2は、式1に関する前記と同様に定義され、式5中のR4は、H、C1〜6アルキル、アリール−C1〜3アルキルまたはカチオン、M1(例えば第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオン)であり、式2中のX1は、ハロゲノ(例えばCl、Br、I)などの脱離基またはR5O−(ここで、R5はC1〜6アルキルスルホニル(例えばメシル)、フルオロ−C1〜6アルキルスルホニル(例えばトリフリル)またはアリールスルホニル(例えばトシル、ブロシル、クロシルまたはノシル)である)である。式2および式3中の置換基R3は、カルボン酸またはエステルのための保護基である。有用なR3には、
【0061】
【化9】
により表される構造を有するカルボン酸またはエステル保護基が包含され、ここで、「A」は、式3または4の化合物の残りの部分への結合点を表している。式7では、R6、R7およびR8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成する。式8では、Z1およびZ2はそれぞれ独立に、OおよびSから選択され、R9はC1〜6アルキルであり、R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する。有用なR3には、C1〜3アルキルオルトエステル、オルトビシクロオクチルエステルおよび1,1−ビス−C1〜3アルキルスルファニル−メタン−1−イリデンが包含される。
【0062】
式2および式4〜6中の代表的なR1およびR2には、C1〜3アルキルおよびアリール−C1〜3アルキルが包含され、式2中の代表的なX1にはBrおよびトシレートが包含され、式3および式4中の代表的なR3には、4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イルなどのオルトエステルおよびトリメトキシ−メタン−1−イル、トリエトキシ−メタン−1−イルなどのトリC1〜3アルコキシエステルが包含され、式5中の代表的なR4には、H、Me、Et、Pr、i−Pr、Bn、Li+、Na+およびK+が包含される。したがって有用なキラル1,4−ビス−求電子体(式2)には、(R,R)−1,4−ジブロモ−2,3−ジメチル−ブタン、(R,R)−3,4−ビス−ブロモメチル−ヘキサン、(R,R)−4,5−ビス−ブロモメチル−オクタン、(R,R)−1,4−ジブロモ−2,3−ジベンジル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジエチル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジプロピル−ブタン、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジベンジル−ブタンおよびこれらの反対の鏡像異性体が包含される。
【0063】
代表的なマスキングされているカルボン酸(式3)には、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イル)−プロピオニトリル、4,4,4−トリメトキシ−ブチロニトリルおよび4,4,4−トリエトキシ−ブチロニトリルが包含される。代表的な光学活性なシクロペンタンカルボニトリル(式4)には、(S,S)−3,4−ジメチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジメチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジメチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジエチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イルメチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル、(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリルおよびこれらの反対の鏡像異性体が包含される。
【0064】
式5の代表的な化合物には、(3S,4S)−(3,4−ジメチル−1−シアノ−シクロペンチル)−酢酸、(3S,4S)−(3,4−ジエチル−1−シアノ−シクロペンチル)−酢酸、(3S,4S)−(3,4−ジプロピル−1−シアノ−シクロペンチル)−酢酸および(3S,4S)−(3,4−ジベンジル−1−シアノ−シクロペンチル)−酢酸、前記の酸のリチウム、ナトリウムおよびカリウム塩、前記の酸のメチル、エチル、プロピル、イソプロピルおよびベンジルエステルならびに前記の酸、塩およびエステルの反対の鏡像異性体が包含される。
【0065】
代表的なラクタム(式6)には、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン、(7S,8S)−7,8−ジエチル−2−アザ−スピロ[4.4]ノナン−3−オン、(7S,8S)−7,8−ジプロピル−2−アザ−スピロ[4.4]ノナン−3−オンおよび(7S,8S)−7,8−ジベンジル−2−アザ−スピロ[4.4]ノナン−3−オンが包含され、これらの反対の鏡像異性体が包含される。
【0066】
前記のように、スキームIに記載の方法は、キラル1,4−ビス−求電子体(式2)と保護(マスキング)されているカルボン酸(式3)とを反応させて、光学的に活性なシクロペンタンカルボニトリル(式4)を得ることを包含し、カルボキシ部分の脱保護(非マスキング)により、1−カルボキシメチル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。シクロペンタンカルボニトリル(式4)を典型的には、極性非プロトン性溶媒(例えばTHF)中、塩基条件下、約−30℃からほぼ室温または約−20℃から約5℃の範囲の温度で調製する。有用な塩基には、LiHMDS、LDAおよびリチウムジエチルアミドなどのヒンダード非求核性塩基が包含される。
【0067】
マスキングされているカルボン酸(式3)は、文献から適合させたプロセスを使用して調製することができる。例えば、CH2CH2/MTBE中、約7〜18℃で約1.5時間、次いで約9℃で約20時間、スクシノニトリルをMeOHおよびHClと反応させて、メチル3−シアノプロパンイミデートクロリドを得ることにより、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクチ−1−イル)−プロピオニトリルを調製することができる。このイミデートを、THF中、約40℃で約15時間、2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオールと反応させて、オルトビシクロオクチルエステルを得る。E.J.Corey&K.Shimoji、J.Am.Chem.Soc.105:1662(1983年)参照。同様に、文献に記載されている条件を使用してメチル3−シアノプロパンイミデートクロリドをメタノリシスすることにより、4,4,4−トリメトキシ−ブチロニトリルを調製することができる。S.M.McElvain&J.P.Schroeder、J.Am.Chem.Soc.71:40(1949年)参照。
【0068】
スキームIに示されているように、式4の化合物のカルボキシ部分の脱保護(非マスキング)により、1−カルボキシメチル−シクロペンタンカルボニトリルまたはその塩もしくはエステル(式5)が得られる。反応条件は、保護基(R3)の選択に左右され、脱保護を通常、酸加水分解を介して実施する。例えば、室温で約5時間、酸水溶液(例えば6NのHCl、5%クエン酸、42%H3PO4または50%HOAc)と接触させることを介しての中程度の酸性条件(例えば約3以下のpH)が、オルトエステル(例えば4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル、トリメトキシ−メタン−1−イル、トリエトキシ−メタン−1−イル)および1,1−ビス−C1〜3アルキルスルファニル−メタン−1−イリデン(例えば1,3−ジチオラン−2−イリデン)を脱保護するためには十分である。
【0069】
スキームIに従い、シアノエステル(式5、R4はHではないかMではない)を還元し、還元剤で処理することによりその場で環化して、ラクタム(式6)を得る。反応を典型的には、MeOH、EtOHまたはi−PrOHなどのアルコール溶媒中、金属触媒を用いて、水素ガスの存在下、大気圧から250psigの範囲の圧力、ほぼ室温から還流までの範囲の温度で行う。有用な金属触媒には、ニッケル触媒が包含される。反応を慣用の「バッチ法」で行うことができるが、ここで、触媒および実質的にすべての基質(式5)を初めに反応容器に充填し、その後に水素ガスを加えて、変換を行う。別法では、副生成物を低減し、収率を上げるために、反応を「半バッチ」法で実施することができる。半バッチ法では、触媒および水素が、反応の開始時に容器中に存在し、その後に、シアノエステル(式5)を、還元の速度に相応する速度で反応器に供給する。バッチ法においてのように、水素ガスも、ニトリル基の還元の間に反応容器に加える。
【0070】
スキームIに示されているラクタム(式6)を、ほぼ室温からほぼ還流まで、または約80℃から約95℃の範囲の温度で、酸で処理することを介して加水分解して、所望のアミノ酸(式1)またはその塩を得ることができる。酸濃度は、約1%から約50%で変動してよく、モル比は、約1:1から約10:1で変動してよい。有用な酸には、HCl、H2SO4、HBr、HlおよびHNO3などの無機酸ならびにTFAおよびTCAなどの有機酸が包含される。
【0071】
前記のように、シアノエステル(式5)を加水分解して、生じた酸または酸塩を還元すると、式1の化合物を得ることができる。シアノエステル(式5)を、酸または塩基での処理により、または塩基(または酸)での処理により、それに続く酸(または塩基)での処理により加水分解することができる。例えば、シアノエステル(式5)をHCl、H2SO4などで過剰のH2Oと共に処理することにより、酸が生じ、これを塩基(例えばKOH)で処理すると、塩基付加塩を得ることができる。シアノエステル(式5)をLiOH、KOH、NaOH、CsOH、Na2CO3、K2CO3、Cs2CO3などの無機塩基水溶液で、任意選択の極性溶媒(例えばTHF、MeOH、EtOH、アセトン、ACNなど)中で処理することにより、塩基付加塩が得られ、これを酸で処理すると、遊離酸を得ることができる。エステル加水分解は、室温または還流温度までの温度で実施することができる。次いで、生じた遊離酸または塩基付加塩を、シアノエステル(式6)からラクタム(式6)の調製に関して前記された条件下に、還元剤で、例えばH2と接触させて処理することを介して還元する。
【0072】
前記のように、有用な式2のキラル1,4−ビス−求電子体の1種は、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンである。この化合物の2種の結晶形態(形態Aおよび形態B)は、単離されていて、FT−IR、FT−ラマン、PXRDおよびDSCにより特徴づけられている。
【0073】
Smart Golden Gate(商標)単反射ATRアクセサリ(セレン化亜鉛光学部品を備えたダイヤモンドATR結晶)およびd−TGS KBr検出器を備えたThermoNicolet Avatar 360FT−IR分光計を使用して、形態Aおよび形態BのFT−IRスペクトルを得た。スペクトルを2cm−1解像度および128走査の同時付加で集めた。Happ−Genzelアポダイゼーションを使用した。単反射ATRを使用してFT−IRスペクトルを記録したので、試料調製は必要なかった。ATR FT−IRを使用することにより、KBrディスクまたはヌジョール試料調製を使用する吸光FT−IRスペクトルで見られるものとは異なる赤外バンドの相対強度が生じる。ATR FT−IRの性質により、比較的低い波数でのバンドは、比較的高い波数でのバンドよりも強い。他に記載のない限り、実験誤差は±2cm−1であった。ThermoNicolet Omnic 6.1aソフトウェアを使用して、ピークを選んだ。強度の割り当ては、スペクトルでの主なバンドに対してである(即ち、これらは、基線から測定された絶対値をベースとするのではない)。スプリットピークを評価する場合には、その強度値を基線から取ったが、この場合にも、強度は、スペクトルにおける最も強いバンドに対して評価した。
【0074】
1064nmNdYAGレーザーおよびLN−Germanium検出器を備えたRamll FT−Ramanモジュールを伴うBruker Vertex70 FT−IR分光計を使用して、形態Aおよび形態BのFT−ラマンスペクトルを集めた。スペクトルすべてを、2cm−1解像度およびBlackman−Harris 4項アポダイゼーション、350mWレーザー出力および2048走査を使用して記録した。各試料をガラスバイアルから直接計測し、レーザー放射に曝露した。データは、強度およびラマンシフトの関数として表されている。他に記載のない限り、実験誤差は2cm−1であった。ThermoNicolet Omnic 6.1aソフトウェアを使用して、ピークを選んだ。強度の割り当ては、スペクトルでの主なバンドに対してである(即ち、これらは、基線から測定された絶対値をベースとするのではない)。スプリットピークを評価する場合には、その強度値を基線から取ったが、この場合にも、強度は、スペクトルにおける最も強いバンドに対して割り当てた。
【0075】
自動試料チェンジャー、シータ−シータゴニオメーター、自動ビーム広がりスリットおよびPSDVantec−1検出器を備えたBruker−AXS Ltd.D4粉末X線回折計を使用して、粉末X線回折パターンを決定した。低バックグラウンドシリコンウェハ(キャビティを伴う)に試料量をマウントすることにより、試料を分析のために調製した。試料を回転させたが、その間、40kV/35mAで運転されるX線管を用いて、銅K−アルファ1X線(波長=1.5406Å)を照射した。2°から50°の2シータ範囲にわたって0.018°ステップ当たり0.2秒カウントに設定された連続モードで運転するゴニオメーターを用いて、分析を行った。得られたピークを、シリコン参照標準に対して整列させた。1の閾値および0.3°2シータのピーク幅を伴うBruker−AXS Ltd.Evaluationソフトウェアを使用して、ピークを選択した。データを21℃で集めた。他に記載のない限り、実験誤差は、±0.1度2θであった。
【0076】
当業者であれば理解されるように、下記に示されている表2および3の範囲内の様々なピークの相対強度は、例えばX線ビームにおける結晶の配向効果または分析される物質の純度または試料の結晶度などのいくつかのファクターによって変動しうる。ピーク位置も、試料高さの変化でシフトすることがあるが、ピーク位置は、示されている表に定義されているままに実質的にはとどまる。
【0077】
当業者であれば、異なる波長を使用しての測定は、Bragg式−nλ=2d sinθに従い、異なるシフトをもたらすであろうことも理解されるであろう。
【0078】
別の波長を使用することにより生じるこのような他のPXRDパターンは、本発明の結晶物質のPXRDパターンの別の表現であると考えられ、それ自体、本発明の範囲内である。
【0079】
DSCサーモグラムを得るために、TA Instruments Q 1000DSCを使用して、ふたを備えたアルミニウムパン中、窒素パージガスを用いて、(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの試料を、1分当たり10℃で20から150℃に加熱した。
【0080】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、932、844、756および1160cm−1に吸収バンド振動数を伴うFT−IRスペクトルを有する。
【0081】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、956、1170、832および1256cm−1に吸収バンド振動数を伴うFT−IRスペクトルを有する。
【0082】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、1163、233、800および165cm−1にラマンバンド振動数を伴うFT−ラマンスペクトルを有する。
【0083】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、1174、1007、740、108および794cm−1にラマンバンド振動数を伴うFT−ラマンスペクトルを有する。
【0084】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、6.5、13.1および21.9度2θに粉末X線回折ピークを有する。
【0085】
(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、14.5、25.1および26.0度2θに粉末x線回折ピークを有する。
【0086】
図1および図2に示されている形態AのFT−IRスペクトルは、次の波数に吸収バンドを有する(cm−1、w:弱、m:中、s:強):3070w、2979w、2911w、1597w、1492w、1468w、1394w、1374w、1369w、1345s、1303w、1293w、1282w、1242w、1212w、1188s、1160s、1135w、1111w、1097m、1040w、1018w、971w、945s、932s、881w、857m、852m、844s、817s、756m、710wおよび669s。
【0087】
図3および図4に示されている形態AのFT−ラマンスペクトルは、次のラマンシフトにラマンバンドを有する(cm−1、w:弱、m:中、s:強、vs:極強):3191w、3145w、3070vs、3061s、3041w、3011w、2996m、2982m、2948m、2926s、2909s、2886vs、2849w、2789w、2762w、2746w、2731w、2585w、1598s、1575w、1499w、1473m、1417w、1384w、1371w、1352m、1341w、1304w、1293w、1284w、1250w、1212w、1190s、1163vs、1137w、1117w、1101m、1042w、1019w、965w、950w、942w、934w、882w、859w、848w、822w、800s、757m、712w、672w、638s、621w、610w、556w、504w、495w、471w、418w、396m、372w、351w、334w、292s、233m、221m、191w、165s、123vs、91vs、80s、72vsおよび54s。
【0088】
図5および図6に示されている形態BのFT−IRスペクトルは、次の波数に吸収バンドを有する(cm−1、w:弱、m:中、s:強):3055w、2976w、2961w、2919w、2888w、1598w、1495w、1473w、1449w、1404w、1388w、1355s、1344s、1309m、1294m、1256w、1244w、1212w、1189m、1170s、1123w、1096m、1042w、1018m、1007m、965s、956s、922s、857w、832s、812s、791s、705m、668sおよび663s。
【0089】
図7および図8に示されている形態BのFT−ラマンスペクトルは、次のラマンシフトにラマンバンドを有する(cm−1、w:弱、m:中、s:強、vs:極強):3191w、3148w、3073vs、3056m、3040m、3033m、2996s、2986s、2967s、2958s、2929s、2918s、2887m、2848w、2760w、2718w、2591w、1598s、1577w、1496w、1473m、1468m、1456w、1407w、1381w、1356w、1310w、1295w、1263w、1255w、1212w、1190m、1174vs、1149w、1098s、1007m、976w、948w、924w、899w、856w、846w、832w、816m、794vs、740m、707w、670w、665m、635s、585m、574m、562w、556m、530w、509m、480m、420m、407m、375m、365m、342m、317s、293vs、266m、244m、213m、176m、108vs、89vsおよび77vs。
【0090】
好ましい配向効果の結果として、大抵のPXRDピークは強度10%未満である。図9(フルスケール)および図10(30000cpsまで)に示されているパターンから、形態Aの最も強いPXRDピーク20を下記で表2に列挙する。
【0091】
【表2】
【0092】
図11(フルスケール)および図12(30000cpsまで)に示されているパターンから、形態Bの最も強いPXRDピーク20を下記で表3に列挙する。
【0093】
【表3】
【0094】
形態Aおよび形態BのDSCサーモグラムをそれぞれ、図13および図14に示す。(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Aは、87℃±2℃の開始で、89℃±2℃に鋭い吸熱ピークを示す。(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの形態Bは、90℃±2℃の開始で、92℃±2℃に鋭い吸熱ピークを示す。
【0095】
【化10】
スキームIIは、その活性化キラルジオール(式2a)、キラル二ハロゲン化物(式2b)および反対の鏡像異性体を包含するキラル1,4−ビス−求電子体(式2)を調製する方法を示している。方法は、光学活性な2−置換コハク酸モノエステルまたはスクシンアミド酸(式9)をアルキル化剤(式10)と反応させて、2,3−二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を得ることを包含する。二置換モノエステルまたはスクシンアミド酸の還元により、ジオール(式12)が得られ、続いてこれを、例えばスルホニル化剤(式13)と反応させることを介して活性化させる。生じた活性化キラルジオール(式2a)をハライド源(例えば式16)と反応させて、キラル二ハロゲン化物(式2b)を得ることもできる。式9〜12中の置換基R1およびR2は、式1に関する前記と同様に定義され、式9および11中のR11はR1O−またはアミノであり、式2aおよび13中のR5は、式2中のX1に関する前記と同様に定義され、式10中のX2および式13中のX3は脱離基(例えば、ハロゲノ、R5O−)であり、X4はハロゲノであり、M2はカチオン(例えばLi+、Na+、K+などの第1族金属イオンまたはアンモニウム塩)である。
【0096】
式9〜12中の代表的なR1およびR2には、C1〜6アルキルおよびアリール−C1〜3アルキルが包含され、式9中の代表的なR11には、アミノ、メトキシ、エトキシ、n−プロポキシ、i−プロポキシおよびt−ブトキシなどのC1〜6アルコキシならびにベンズオキシなどのアリール−C1〜3アルコキシが包含される。したがって有用な出発原料(式9)には、(R)−2−メチル−コハク酸4−メチルエステル、(R)−2−メチル−コハク酸4−エチルエステル、(R)−2−メチル−コハク酸4−プロピルエステル、(R)−2−メチル−コハク酸4−イソプロピルエステル、(R)−2−メチル−コハク酸4−tert−ブチルエステル、(R)−2−メチル−スクシンアミド酸、(R)−2−メチル−コハク酸4−メチルエステル、(R)−2−エチル−コハク酸4−エチルエステル、(R)−2−エチル−コハク酸4−プロピルエステル、(R)−2−エチル−コハク酸4−イソプロピルエステル、(R)−2−エチル−コハク酸4−tert−ブチルエステルおよび(R)−2−エチル−スクシンアミド酸が包含され、これらの反対の鏡像異性体が包含される。
【0097】
したがって同様に、代表的な2,3−二置換コハク酸モノエステルまたはスクシンアミド酸(式11)には、(R,R)−2,3−ジメチルコハク酸4−メチルエステル、(R,R)−2,3−ジエチル−コハク酸4−メチルエステル、(R,R)−2,3−ジプロピル−コハク酸4−メチルエステル、(R,R)−2,3−ジイソプロピル−コハク酸4−メチルエステル、(R,R)−2,3−ジベンジル−コハク酸4−メチルエステル、(R,R)−2,3−ジメチル−コハク酸4−エチルエステル、(R,R)−2,3−ジエチル−コハク酸4−エチルエステル、(R,R)−2,3−ジプロピル−コハク酸4−エチルエステル、(R,R)−2,3−ジイソプロピル−コハク酸4−エチルエステル、(R,R)−2,3−ジベンジル−コハク酸4−エチルエステル、(R,R)−2,3−ジメチル−スクシンアミド酸、(R,R)−2,3−ジエチルスクシンアミド酸、(R,R)−2,3−ジプロピル−スクシンアミド酸、(R,R)−2,3−ジイソプロピル−スクシンアミド酸および(R,R)−2,3−ジベンジル−スクシンアミド酸が包含され、これらの反対の鏡像異性体が包含される。
【0098】
一置換コハク酸モノエステルまたはスクシンアミド酸(式9)のアルキル化は、相容性の溶媒中で適切な塩基を使用して実施する。適切な塩基には、エステルまたはアミド部分(式9)に隣接する(α)メチレン基を脱保護しうるものが包含される。これらには、非求核性またはヒンダード塩基が包含され、LDA、LHMDS、KHMDS,LICA、LTMP、LiNET2、リチウムジシクロヘキシルアミドなどのリチウムアミド塩基ならびに(i−Pr)2NMgClおよびEt2NMgClなどの対応するマグネシウムアミド塩基が包含される。リチウムおよびマグネシウムアミド塩基はそれぞれ、LiNR1R2およびR1R2NMgX4により表すことができ、ここで、R1およびR2は、式1に関する前記と同様に定義され、X4はハロゲノである。相容性の溶媒には、その共役酸がpKa≦9、典型的には≦4および往々には≦1を有するものが包含される。このような溶媒には例えば、THF、Et2O、DMSO、ACN、DMFおよびアセトンが包含されるが、アンモニアは包含されない。
【0099】
これらの群の塩基および溶媒を使用することにより、所望のanti−ジアステレオ異性体(式11に図示)が過剰に得られる。典型的には、anti−ジアステレオ異性体とsyn−ジアステレオ異性体との比は、約85:15、90:10または92:8に等しいか、それ以上である。したがって、下記の実施例に示されているように、THF中でLHMDSを使用する(R)−2−メチル−コハク酸4−メチルエステルのアルキル化により、anti−ジアステレオ異性体である(R,R)−2,3−ジメチル−コハク酸モノメチルエステルが約80%以上のdeで得られた。反対に、NH3およびEt2O中でLiNH2を使用する(R)−2−メチル−コハク酸4−メチルエステルのアルキル化により、望ましくないsyn−ジアステレオ異性体が過剰に得られる。W.G.Kofron& L.G.Wideman、J.Org.Chem.37:555(1972年)参照。
【0100】
前記のように、アルキル化剤(式10)は、脱離基(X2)を包含し、これには、Cl、BrおよびIなどのハロ置換基ならびにトルエン−p−スルホネート、メチルスルホネート、p−ブロモ−ベンゼン−スルホネートおよびトリフレートなどのスルホネート置換基が包含されうる。したがって、代表的なアルキル化剤(式10)には、MeCl、MeBr、MeI、EtCl、EtBr、EtI、n−PrCl、n−PrBr、n−PrI、i−PrCl、i−PrBrおよびi−PrIなどのC1〜6アルキルハロゲン化物ならびにMeOTs、MeOMs、MeOBs、MeOTf、EtOTs、EtOMs、EtOBs、EtOTf、n−PrOTs、n−PrOMs、n−PrOBs、n−PrOTf、i−PrTs、i−PrMs、i−PrBsおよびi−PrTfなどのC1〜6アルキルスルホネートエステルが包含される。アルキル化剤は、市場供給源から得られるか、知られている方法を使用して調製することができる。
【0101】
アルキル化反応は、化学量論的量の反応成分(即ち、2−置換コハク酸モノエステルまたはスクシンアミド酸とアルキル化剤とのモル比は1:1)を使用すればよいが、変換率を改善し、副生成物を最小化するなどのために、アルキル化ステップは一方の反応成分を過剰に使用することもできる(例えば、1:1.1から1.1:1、1:1.5から1.5:1、2:1から1:2、3:1から1:3のモル比)。同様に、アルキル化反応は、化学量論的量の塩基(即ち2:1の塩基と基質とのモル比)を使用すればよいが、過剰の塩基を使用することもできる(例えば2.1:1、2.5:1、3:1のモル比)。
【0102】
アルキル化を約−30℃から還流の温度で実施することができる。反応を典型的には、室温で実施するが、より高いか低い温度から利益を得ることもある。例えば、実施例に記載されているように、出発原料(式9)を塩基に加え、続いて、アルキル化剤(式10)を加える間、反応混合物を約−30℃から約−25℃に冷却することができる。次いで、生じた混合物を、完了まで室温で反応させることができる。
【0103】
接触スキームは収率に影響を及ぼしうる。実施例に記載されているように、出発原料(式9)およびアルキル化剤(式10)の表面下添加は、表面上反応成分添加に比較すると、anti−ジアステレオ異性体(式10)のdeを上昇させうる。
【0104】
スキームIIに示されているように、THF、MTBEおよびEt2Oなどの1種または複数のエーテル(無水)溶媒中でのLAHとの反応を介して、二置換コハク酸モノエステル(式11)をジオール(式12)に還元する。他の有用な還元剤および溶媒には、ジグリム中のNaBH4およびAlCl3、THF中のB2H6、THF中の9−BBN、THF中のLiAlH(OMe)3、THF中のAlH3、THF中のDIBAL−HならびにトルエンまたはTHF中のRed−Alが包含される。反応は通常、モル過剰の還元剤(例えば>4当量のLAH)を使用し、ほぼ室温から還流までの範囲の温度で行われる。
【0105】
アルキル化においてのように、還元処理の接触スキームは収率に影響を及ぼしうる。LAHを使用する還元後の従来の(Fieser)処理、後続のH2O、15%NaOH水溶液およびH2Oの反応混合物への連続添加は、大きな(kg)規模で行う場合には処理の困難をもたらしうる。例えば、最初の水クエンチは、大量の水素ガスの迅速な放出をもたらし、さらに、かなりの生成物(式12)のフラクションを固体副生成物に捕捉する。多少の捕捉された生成物は、固体の洗浄および濾過により回収することができるが、プロセスは非効率的で、時間を浪費する。それというのも、洗浄液の多くは、フィルターケークを通過するよりもむしろ、その周りを流れるためである。さらに、液体を除去すると、フィルターケークは往々にして、可逆的にひび割れる。これらのひび割れは、洗浄液をフィルターケークの内部から流し、このことがさらに、回収プロセスの有効性を低下させる。
【0106】
実施例に示されているように、反応混合物をかなり過剰の塩基水溶液に供給するように従来の接触スキームを変更することにより、水素の発生速度が低下し、ジオール(式12)の収率および回収が上昇するようである。反応混合物を塩基水溶液に加えるので、アルミニウムアルコキシド中間体は、塩基触媒される加水分解を受けて、所望のジオール(式12)、さらに、溶液から沈殿する水酸化アルミニウムをもたらす。ジオールは溶液中に残るので、これを、液相をデカンテーションすることにより、沈殿物から分離することができる。さらに、反応混合物を塩基水溶液に加える速度を慎重に制御することにより、水素ガス発生の速度を緊密に調節することができる。
【0107】
スキームIIに示されているように、方法は、エステルまたはアミド部分の酸または塩基加水分解を介して、二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を二酸(式14)またはその塩に変換するために準備されていてもよい。例えば、エステルまたはアミドをHClまたはH2SO4で、および過剰のH2Oで処理すると、二酸が生じる。同様に、コハク酸モノエステルまたはスクシンアミド酸をLiOH、KOH、NaOH、CsOH、Na2CO3、K2CO3またはCs2CO3などの無機塩基水溶液で、任意選択の極性溶媒(例えばTHF、MeOH、EtOH、アセトンまたはACN)中で処理すると、二酸の塩基付加塩が得られ、これを酸で処理すると、遊離二酸が生じる。通常、過剰の酸または塩基を使用し、エステルおよびアミド加水分解を室温で、または還流までの温度で実施する。
【0108】
二置換コハク酸モノエステルまたはスクシンアミド酸(式11)を加水分解した後に、生じた二酸(式14)またはその塩を無水酢酸で処理すると、環式無水物(式15)が得られる。反応は通常、THFなどの非プロトン性極性溶媒中、ほぼ室温から還流までの範囲の温度で行うが、約50℃から約75℃の範囲の反応温度を使用することもできる。過剰の無水酢酸(例えば1.5当量以上)を使用して、ジエステルの完全な変換を保証することもできる。環式無水物(式15)を還元するか、コハク酸モノエステル、スクシンアミド酸(式11)または二酸(式14)を還元することにより、ジオールを調製することができる。反応を典型的には、過剰の還元剤(例えばLAH)を極性非プロトン性溶媒(例えばTHF)中、約40℃から還流までの範囲の温度で用いて行う。
【0109】
代表的な反応基質(式14)には、(R,R)−2,3−ジメチル−コハク酸、(R,R)−2,3−ジエチル−コハク酸、(R,R)−2,3−ジプロピル−コハク酸、(R,R)−2,3−ジイソプロピル−コハク酸および(R,R)−2,3−ジベンジル−コハク酸が包含され、それらの塩を包含する。代表的な環式無水物(式15)には、(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン、ジエチル−ジヒドロ−フラン−2,5−ジオン、(R,R)−3,4−ジプロピル−ジヒドロ−フラン−2,5−ジオン、(R,R)−3,4−ジイソプロピル−ジヒドロ−フラン−2,5−ジオンおよび(R,R)−3,4−ジベンジル−ジヒドロ−フラン−2,5−ジオンが包含され、これらの反対の鏡像異性体を包含する。
【0110】
環式無水物(式15)を介してのジオール(式12)の調製は、モノエステルまたはアミド(式11)からジオール(式12)への直接的な還元を上回る利点をもたらしうる。例えば、モノエステルとは逆に、環式無水物は容易に再結晶化するので、還元の前に単離することができる。環式無水物の再結晶化は、モノアルキル化副生成物および不所望のジアステレオ異性体の形成を抑制することにより、活性化ジオール(式9a)の下流単離の効率を改善するようである。加えて、還元剤(例えばLAH)が、不純物によって、またはカルボン酸部分によって消費されないので、結晶環式無水物のより高い純度は、還元ステップの改善された処理量をもたらすはずである。ジオール(式12)の比較的高い純度によっても、再結晶化を介しての単離が可能になる。
【0111】
スキームIIに示されているように、ジオール(式12)を、式13の化合物との反応を介して活性化させる。有用なジオールには、(R,R)−2,3−ジメチル−ブタン−1,4−ジオール、(R,R)−2,3−ジエチル−ブタン−1,4−ジオール、(R,R)−2,3−ジプロピル−ブタン−1,4−ジオール、(R,R)−2,3−ジイソプロピル−ブタン−1,4−ジオールおよび(R,R)−2,3−ジベンジル−ブタン−1,4−ジオールが包含され、これらの反対の鏡像異性体を包含する。式13の有用な化合物には、TsCl、MsCl、BsCl、NsClおよびTfClなどのスルホニル化剤ならびにこれらの対応する無水物(例えば無水p−トルエンスルホン酸)が包含される。
【0112】
式12の化合物を、ピリジンまたはEt3Nおよび酢酸エチル、CH2Cl2、ACNまたはTHFなどの非プロトン性溶媒の存在下にTsClまたはMsClと反応させると、前記のようなジトシレートまたはジメシレート、例えば、(R,R)−1,4−ビス−(メタンスルホニルオキシ)−2,3−ジメチル−ブタン、(R,R)−2,3−ジエチル−1,4−ビス−(メタンスルホニルオキシ)ブタン、(R,R)−1,4−ビス−(メタンスルホニルオキシ)−2,3−ジムプロピル−ブタン、(R,R)−2,3−ジイソプロピル−1,4−ビス−(メタンスルホニルオキシ)−ブタンまたは(R,R)−2,3−ジベンジル−1,4−ビス−(メタンスルホニルオキシ)−ブタン(これらの反対の鏡像異性体を包含する)を得ることができる。典型的には、反応を過剰(例えば2.5当量以上)のスルホニル化剤(式13)を用いて、過剰の塩基(例えば3当量以上)を用いて、ほぼ室温以下(例えば約0℃)の温度で実施する。
【0113】
前記のように、活性化ジオール(式2a)をハライド源(式16)と反応させると、キラル二ハロゲン化物(式2b)を得ることができる。ハライド塩(例えばLiBr、NaBr、KBr、LiCl、NaCl、KCl、LiI、NaI、KIなど)を極性非プロトン性溶媒(例えばアセトン)中、または非極性溶媒(例えばトルエン)中、少量の水および相転位触媒(例えばnBu4NBr、nBu4NCl、nBu4NIなど)と共に使用して、反応を実施することができる。反応を典型的には、化学量論的に過剰のハライド塩(例えば3当量以上)を用いて、還流までの温度で実施する。
【0114】
本開示に記載の化合物の多くは、薬学的に許容できる塩を形成しうる。これらの塩には、酸付加塩(二酸を包含する)および塩基塩が包含される。薬学的に許容できる酸付加塩には、塩酸、硝酸、リン酸、硫酸、臭化水素酸、ヨウ化水素酸、フッ化水素酸および亜リン酸などの無機酸に由来する非毒性の塩、さらに、脂肪族モノカルボン酸およびジカルボン酸、フェニル置換アルカン酸、ヒドロキシアルカン酸、アルカン二酸、芳香族酸、脂肪族および芳香族スルホン酸などの有機酸に由来する非毒性の塩が包含される。したがってこのような塩には、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、硝酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、トリフルオロ酢酸塩、プロピオン酸塩、カプリル酸塩、イソ酪酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、マンデル酸塩、安息香酸塩、クロロ安息香酸塩、メチル安息香酸塩、ジニトロ安息香酸塩、フタル酸塩、ベンゼンスルホン酸塩、トルエンスルホン酸、フェニル酢酸塩、クエン酸塩、乳酸塩、リンゴ酸塩、酒石酸塩およびメタンスルホン酸塩が包含される。
【0115】
薬学的に許容できる塩基塩には、アルカリ金属またはアルカリ土類金属カチオンなどの金属カチオン、さらにアミンを包含する塩基に由来する非毒性塩が包含される。適切な金属カチオンの例には、ナトリウムカチオン(Na+)、カリウムカチオン(K+)、マグネシウムカチオン(Mg2+)およびカルシウムカチオン(Ca2+)が包含される。適切なアミンの例には、N,N’−ジベンジルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、ジシクロヘキシルアミン、エチレンジアミン、N−メチルグルカミンおよびプロカインが包含される。有用な酸付加塩および塩基塩の検討に関しては、S.M.Bergeら、「Pharmaceutical Salts」、J.Pharm.Sci.、66:1〜19(1977年)参照、さらにStahlおよびWermuth、Handbook of Pharmaceutical Salts:Properties,Selection,and Use(2002年)参照。
【0116】
化合物の遊離塩基(または遊離酸)を十分な量の所望の酸(または塩基)と接触させて、非毒性の塩を製造することにより、酸付加塩(または塩基塩)を調製することができる。次いで、溶液から沈殿する場合には濾過により、または蒸発により、塩を回収して、塩を単離することができる。さらに、酸付加塩を塩基と(または塩基塩を酸と)接触させることにより、遊離塩基(または遊離酸)を再生することもできる。化合物の遊離塩基、遊離酸または両性イオンのいくつかの物理的特性(例えば可溶性、結晶構造、吸湿性)は、その酸または塩基付加塩とは異なることがある。しかしながら通常は、化合物の遊離酸、遊離塩基または両性イオンに対する言及は、その酸および塩基付加塩を包含する。
【0117】
開示および請求されている化合物は、非溶媒和形態および溶媒和形態の両方で、さらに塩以外の他のタイプの複合体としても存在しうる。有用な複合体には、包接化合物または化合物−ホスト包接複合体が包含され、ここで、化合物およびホストは、化学量論的または非化学量論的量で存在する。有用な複合体はさらに、2種以上の有機、無機または有機および無機成分を化学量論的または非化学量論的量で含有してもよい。生じる複合体は、電離しているか、部分的に電離しているか、非電離であってよい。このような複合体の総説に関しては、J.K.Haleblian、J Pharm Sci.64(8):1269〜1288(1975年)参照。薬学的に許容できる溶媒和物は、結晶溶媒が同位体置換されている、例えばD2O、d6−アセトン、d6−DMSOである水和物および溶媒和物も包含する。通常、この開示では、化合物の非溶媒和形態に関する言及は、化合物の対応する溶媒和形態または水和形態も包含する。
【0118】
本出願に開示されている化合物のうちの数種は、不斉炭素、イオウまたはリン原子(立体由来中心)を含有することがあるので、光学的に活性な立体異性体(即ち鏡像異性体対の一方の鏡像異性体)として存在しうる。化合物のうちの数種はさらに、アルケニルまたは環式基を含有することがあるので、シス/トランスまたは(Z/E)立体異性体(ジアステレオ異性体)が可能になる。さらに他の化合物は、2個以上の立体由来中心を含有することがあるので、それぞれが光学活性でありうる(即ち、鏡像異性体対の一方の鏡像異性体を含む)ジアステレオ異性体が可能になる。最後に、化合物のうちの数種は、ケトまたはオキシム基を含有することもあるので、互変異性が生じることもある。このような場合、本開示の範囲は、すべての互変異性体およびすべての立体異性体を包含し、これには、純粋であるか、実質的に純粋であるか、混合物であるかにかかわらず、鏡像異性体、ジアステレオ異性体およびZ/E異性体を包含する。
【0119】
本明細書に開示されている任意の化合物の所望の鏡像異性体を、従来の分割、キラルクロマトグラフィーまたは再結晶化を介してさらに富化することもできる。例えば、鏡像異性体の混合物を鏡像異性的に純粋な化合物(例えば酸または塩基)と反応させると、それぞれが単一の鏡像異性体から構成されるジアステレオ異性体対を得ることができ、これらを、分別再結晶またはクロマトグラフィーを介して分離する。続いて、所望の鏡像異性体を、適切なジアステレオ異性体から再生することができる。加えて、鏡像異性体を十分な量で利用することができる場合には、所望の鏡像異性体を適切な溶媒中で再結晶化することにより、さらに富化することができる(例えば典型的には、ee約85%以上、場合によってはee約90%以上)。
【0120】
開示されている化合物はさらに、少なくとも1個の原子が、同じ原子数を有するが、自然に通常存在する原子質量とは異なる原子質量を有する原子に置換されているすべての薬学的に許容できる同位体バリエーションを包含する。開示されている化合物中に含まれるために適している同位体の例には、2Hおよび3Hなどの水素の同位体、13Cおよび14Cなどの炭素の同位体、15Nなどの窒素の同位体、17Oおよび18Oなどの酸素の同位体、31Pおよび32Pなどのリンの同位体、35Sなどのイオウの同位体、18Fなどのフッ素の同位体、36Clなどの塩素の同位体が含まれる。同位体バリエーション(例えばジュウテリウム、2H)の使用は、より大きな代謝安定性、例えば高いインビボ半減期または低い用量要求から生じるある種の治療的利点をもたらしうる。加えて、開示されている化合物のある種の同位体バリエーションは、放射性同位体(例えばトリチウム、3Hまたは14C)を含んでもよく、これは、薬物および/または基質組織分布研究で有用でありうる。
【実施例】
【0121】
次の実施例は、例示的かつ非限定的であることが意図されており、本発明の具体的な実施例を表している。
【0122】
(実施例1)
(R,R)−2,3−ジメチル−コハク酸モノメチルエステルの調製
(R)−2−メチルコハク酸4−メチルエステル(24.5105g、0.1677mol)のTHF(25mL)溶液を濾過して、白色の固体(26.3mg、水に可溶性、CH2Cl2に不溶性)を除去し、LHMDS/THF(1.0M、360mL、0.360mol、2.15当量)の−30℃溶液に、温度が−25℃未満に維持される速度で加えた(1.5時間)。混合物を−10℃に加温し、1時間攪拌し、−30℃に再び冷却し、ヨウ化メチル(25.12g、0.1770mol、1.06当量)のTHF(25mL)溶液で、温度が−25℃を超えないような速度で処理した(1.5時間)。混合物を−25℃で2時間攪拌し、次いで室温に加温し、15.5時間攪拌し、0℃に冷却し、NH4Cl(25g、0.467mol、2.79当量)のH2O(75mL)溶液で、温度が約0℃を超えないような速度で、慎重にクエンチした(14℃までの短いエクスカーションは除く。最初の2mLを30分にわたって加え、残りを1時間にわたって加える)。混合物をH2O(100mL)で希釈して、固体を溶かし、層を分離した。有機層をEt3N(2.4mL)で処理して、残りのヨウ化メチルをクエンチし、廃棄した。水性層を6NのHClでpH1.92まで酸性化し、MTBE(4×150mL)で抽出した。水性層(約pH3)を廃棄した。有機抽出物を合わせ、真空濃縮して、13C−NMRおよび1H−NMRにより前記表題化合物と識別される暗コハク色のオイルにした。anti/syn/モノメチルの比は、GCにより88.5:7.8:3.7であると決定された。重量:28.01g;13C−NMR(100MHz,CDCl3):δ180.97(s);175.67(s);51.80(q);41.36(d);41.23(d);13.50(q);13.40(q);1H−NMR(400MHz,CDCl3):δ8.53(1H,br s);3.69(3H,s);2.84(2H,重複した多重線);1.20(3H,d,J=4.3Hz);1.18(3H,d,J=4.4Hz)。
【0123】
(実施例2)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
実施例1からの暗コハク色のオイル(27.82g)をTHF(137mL、不溶物を除去するために濾過、57.1mg)で希釈し、0℃のLAH(16.48g、0.4343mol、2.61当量)のTHF(434mL)懸濁液に40分にわたって加えた。混合物を5℃で1時間、次いで30℃で17.5時間攪拌し、次いで、0℃に冷却した。H2O(16.5g、0.916mol、5.50当量)を70分にわたって、続いて、15%NaOH水溶液(16.5mL)を10分にわたって、続いてH2O(50mL)を10分にわたって加えることにより、混合物を慎重にクエンチした。3種の溶液をすべて、内部温度が5℃から15℃に維持される速度で加えた。オフホワイト色のスラリーを濾過し(粗いフリット、ゆっくりとした濾過)、THF(165mL)ですすいだ。濾液をオイルに濃縮した。水を共沸により除去するために、オイルをトルエン(135mL)で希釈し、蒸留して、13C−NMRおよび1H−NMRにより前記表題化合物と識別される淡褐色のオイルにした。dl:メソ:モノメチルの比は、GCにより92.65:5.91:1.43であると決定された。重量:17.16g(0.1452mol、(R)−2−メチル−コハク酸4−メチルエステルから全体で87.2%;13C−NMR(100MHz,CDCl3):δ65.56(t);37.18(d);13.13(q);1H−NMR(400MHz,CDCl3):δ0.85(6H,d,J=6.6Hz);1.72(2H,多重線);3.45(2H,dd,J=10.9,6.5Hz);3.55(2H,dd,J=10.9,4.4Hz);4.10(2H,d,J=7.4Hz)。
【0124】
(実施例3)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(15.75g、0.1333mol)および塩化p−トルエンスルホニル(63.53g、0.3332mol、2.50当量)のACN(169mL)溶液を0℃に冷却し、そのままのEt3N(56mL、40.66g、0.4018mol、3.01当量)で、温度が5℃を上回らない速度で処理した(35分)。混合物を0℃で2時間、次いで室温で19時間攪拌した。混合物をEtOAc(107mL)およびH2O(103mL)で希釈した。相を分離し、水性フラクションをEtOAc(2×92mL)で抽出した。有機層を合わせ、H2O(115mL)で、次いで10%NaHCO3水溶液(103mL)および25%NaCl水溶液80mLで洗浄し、オフホワイト色/黄色の固体に濃縮した。固体をMTBE(345mL)およびEtOH(27g)に60℃で溶かし、7時間にわたって0℃に冷却し、0℃で16時間攪拌し、濾過した。ケークを0℃のMTBE(2×25mL)で洗浄し、N2流により乾燥させると、13C−NMRおよび1H−NMRにより前記表題化合物として識別される白色の固体が得られた。dl:メソ:モノメチルの比は、HPLCにより96.72:1.79:1.49であると決定された。重量:38.20g(0.8956mol、67.2%);13C−NMR(100MHz,CDCl3):δ144.91(s);132.71(s);129.90(d);127.82(d);72.59(t);32.97(d);21.62(q);11.20(q);1H−NMR(400MHz,CDCl3):δ0.75(6H,d,J=6.7Hz);1.97(2H,多重線);2.47(6H,s);3.84(4H,d,J=5.5Hz);7.36(4H,d,J=8.1Hz);7.77(4H,d,J=8.2Hz)。
【0125】
(実施例4)
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンの調製
−22℃の(R)−2−メチル−コハク酸4−メチルエステル(133.5kg、912mol)のTHF(125kg)溶液を、−34℃のTHF(1190L)中のLHMDS(322.6kg、1928mol、2.11当量)に1.5時間にわたって加えたが、この間、反応混合物を−34℃から−26℃に維持した。基質をTHF(10kg)ですすいだ。溶液を−26℃から−30℃で1時間攪拌し、次いで、1.5時間にわたって12℃に加温した。混合物を−12℃から−10℃で5分間攪拌し、次いで、7.8時間にわたって−34℃に冷却した。−21℃のMeI(140kg、986mol、1.08当量)のTHF(153L)溶液を、ジアニオン溶液に5時間にわたって加えたが、この間、−34℃から−27℃を維持し、THF(45L)ですすいだ。混合物を−27℃から−29℃に4時間攪拌し、1.5時間にわたって20℃に加温し、20℃から21℃で12時間攪拌し、5℃に冷却した。NH4Cl(136kg、2543mol、2.79当量)の水(400L)溶液を7.3時間にわたって加えたが、その間、混合物の温度を5℃から25℃に維持した。水(540L)を加え、上部相を廃棄した。下部水性相に、水(240L)を加え、続いて、pHを、HCl(300kg、35重量%、2880mol、3.16当量)で1に調節したが、この間、反応混合物の温度を4℃から11℃に維持した。水(10L)ですすいだ後に、生成物をMTBE(4×304kg)で抽出し、濃縮すると、(R,R)−2,3−ジメチル−コハク酸モノメチルエステルがオイル(167kg、内部標準GCにより78.6重量%、収率89.7%、シス6.4面積%、デス−メチル0.4%、トリメチル3.3%)として得られた。
【0126】
粗製(R,R)−2,3−ジメチル−コハク酸モノメチルエステル(350.81g、78.6重量%、1.72mol)の試料を、水(500mL)と混合して、二相混合物を得た。この混合物に、NaOH(50重量%、351.45g、4.39mol、2.55当量)を加えたが、この間、混合物の温度を45℃以下に維持した。混合物を45℃で10分間攪拌し、次いで、pHを、HCl(438g、37.5重量%、4.50mol、2.62当量)で10.4から0.5に調節したが、その間、混合物の温度を30℃以下に維持した。溶液をEtOAc(3×1L)で抽出し、MgSO4上で乾燥させ、粘稠なスラリー(正味重量412g)に濃縮した。無水酢酸(250mL、2.645mol、1.54当量)を加え、混合物を109℃に加温した。NMRは、環式無水物への完全な変換を示し、後続の実験は、環化反応が75℃で迅速であることを示した。溶液を50℃に冷却し、シーディングして、スラリーを得た。tert−アミルアルコール(1l)を加え、スラリーを−8℃に冷却した。沈殿物を、真空濾過により集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させて、(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンが白色の固体(196.91g、89.4%、(R)−2−メチル−コハク酸4−メチルエステルから80.2%)として得られた。融点103.5〜105.4℃、[α]25D=103.07°(ジオキサン、c=1.00)。1H NMR(400MHz,DMSO−d6)δ1.23(d,J=7Hz,6H)、2.98(八重線,J=4Hz,2H);13C NMR(100MHz,DMSO−d6)δ12.73、42.12、174.57;MS(EICI)m/z(相対強度)127[(M−H)−,100];元素分析 C6H8O3の計算値:C,56.25;H,6.29;N,0.00;実測値:C,56.24;H,6.25;N,<0.05;(R,R)−2,3−ジメチル−コハク酸モノメチルエステルへのメタノリシスは、GCにより1.3%のシス異性体および<0.1%の何らかの他の関連不純物を示した。
【0127】
(実施例5)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
45℃の(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン(40.04g、312.52mmol)のMTBE(440mL)およびTHF(58mL)溶液に、LAHのTHF溶液(175mL、2.4M、420mmol)を、滴下漏斗を介して0.5時間にわたって滴加したが、この間、反応混合物を45℃から54℃(還流)の温度に維持し、続いて、THFですすいだ(10mL)。LAH添加の初めの150mLでは、混合物は、攪拌可能なスラリーであるが、添加の終了時には、溶液になった。強く、やや遅い発熱が、添加の初めの150mLで存在し、続いて、最後の25mLでは非常に弱い吸熱が存在した。生じた溶液を、NaOH(50%、1.38g、17.25mmol、0.055当量)、水(55mL)およびTHF(275mL)の−7℃二相混合物に40分にわたってカニューレ添加したが、この間、反応混合物を13℃以下の温度に維持した。残りの溶液をTHF(40mL)ですすぎ、生じたスラリーを1時間にわたって55℃に加温し、55℃で2時間攪拌した。沈殿物を真空濾過により55℃で除去し(濾過時間4分)、MTBE(330mL)およびMeOH(28mL)の55℃混合物で2回洗浄した(各洗浄で濾過15分)。合わせた濾液をMgSO4上で乾燥させ、透明にし、真空濃縮して軽いオイル(47.43g)にした。分枝鎖オクタン(100g)を加え、二相混合物を20℃でシーディングすると、5分間攪拌した後にスラリーが得られた。分枝鎖オクタン(200g)を加え、混合物を3℃に冷却した。真空濾過により、沈殿物を集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(34.24g、92.7%)として得られた。mp42.5〜44.5℃、[α]25D=103.07°(ジエチルエーテル、c=1.00);1H NMR(400MHz,CDCl3)δ0.84(d,J=7Hz,6H)、1.71(m,2H)、3.44(dd,J=6.5Hz,J=11Hz,2H)、3.54(dd,J=6.5Hz,J=11Hz,2H);13C NMR(100MHz,CDCl3)δ13.21、37.25、65.53;MS(EICI)m/z(相対強度)117[(M−H)−,100];元素分析 C6H14O2の計算値:C,60.98;H,11.94;N,0.00;実測値:C,60.91;H,12.27;N,<0.05。
【0128】
(実施例6)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
−10℃の塩化p−トルエンスルホニル(20.255g、106.24mmol、2.52当量)のACN(75mL)スラリーに、(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(4.982g、42.16mmol)を加えた。Et3N(17.7mL、127.0mmol、3.01当量)を1分にわたって加えたが、その間、反応混合物を0℃以下の温度に維持した。スラリーを0℃から5℃で3.5時間攪拌し、20℃に加温した。水(40mL)およびEtOAc(60mL)を順次加え、相を29℃で分離した。水性層をEtOAc(2×50mL)で洗浄し、合わせた有機層を水(40mL)、10%NaHCO3水溶液(40mL)および飽和NaCl水溶液(40mL)で洗浄した。有機フラクションをMgSO4上で乾燥させ、真空濃縮して、オイル22.56gにした。トルエン(100mL)を加えて、溶液を得た。分枝鎖オクタン(50mL)を加えた後に、生成物を10分にわたって結晶化させた。分枝鎖オクタン(150mL)を加え、真空濾過により、沈殿物を集めた。固体を分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(15.563g、86.5%)として得られた;融点83.1〜83.6℃、[α]25D=−5.55(酢酸エチル、c=100);1H NMR(400MHz,CDCl3)δ0.75(d,J=6Hz,6H)、1.97(m,2H)、2.47(s,6H)、3.85(d,J=6Hz,4H)、7.37(d,J=8Hz,4H)、7.78(d,J=8Hz,4H);13C NMR(100MHz,CDCl3)δ11.20、21.64、32.96、72.51、127.84、129.90、132.70、144.90;MS(TSP)m/z(相対強度)427[(M+H)+,20]、444[(M+H2O)+,100];元素分析 C20H26O6S2の計算値:C,56.32;H,6.14;N,0.00;実測値:C,56.25;H,6.00;N,0.05;HPLC:(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、99.6面積%;(R,S)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、0.08面積%;(R)−2−メチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン、0.08面積%。
【0129】
(実施例7)
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオンの調製
−30℃のLHMDSのTHF溶液(24.4重量%、518.5g、756.1mmol、2.12当量)に、THF(52mL)中の(R)−2−メチルコハク酸4−メチルエステル(52.127g、356.69mmol)を0.5時間にわたって加えたが、この間、反応混合物温度を−29℃に維持した。基質をTHF(6mL)ですすいだ。混合物を−29℃で25分間攪拌し、次いで、MeI(53.32g、375.65mmol、1.05当量)のTHF(105mL)溶液を0.5時間にわたって加えたが、この間、反応混合物を−30℃に維持した。MeIをTHF(15mL)ですすいだ。混合物を−30℃で2時間攪拌し、0℃に加温し、1時間攪拌した。GCは、(R,R)−2,3−ジメチル−コハク酸モノメチルエステル78.8面積%、(R)−2−メチルコハク酸4−メチルエステル15.0面積%およびメソ異性体(R,S)−2,3−ジメチル−コハク酸6.2面積%の混合物を示した。
【0130】
NaOH(50.0%、57.0g、713mmol、2.00当量)および水(200mL)の混合物を10分にわたって加えたが、この間、反応混合物の温度を0℃に維持した。混合物を20℃で16時間攪拌し、亜硫酸水素ナトリウム(SO266.6%、1.68g、17.5mmol、0.049当量)を加えた。pHをHCl(37.5重量%、208.6g、2.145mol、3.01当量)で10.67から0.11に調節したが、この間、反応混合物の温度を25℃以下に維持した。相を分離し、トルエン(200mL)を、続いて飽和NaCl水溶液(50mL)を有機層に加えた。相を分離し、水性層をEtOAc(750mL)で抽出した。合わせた有機フラクションをMgSO4上で乾燥させ、真空濃縮して、物質90.91gにした。トルエン(150mL)を加え、スラリーを75℃に加温すると、溶液が得られた。無水酢酸(44.67g、437.6mmol、1.23当量)を5分にわたって加え、混合物を75℃で0.5時間攪拌すると、この時点で、NMRは完全な変換を示した。溶液を30℃に冷却し、分枝鎖オクタン(200mL)およびt−アミルアルコール(200mL)を加えた。シーディングの後に、生成物を結晶化させ、分枝鎖オクタン(200mL)を加えた。スラリーを−8℃に冷却し、真空濾過により、沈殿物を集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、(R,R)−2,3−ジメチル−コハク酸がベージュ色の固体(29.38g、64.3%)として得られた;1H NMR(400MHz,DMSO−d6)δ1.23(d,J=7Hz,6H)、2.98(八重線,J=4Hz,2H);13C NMR(100MHz,DMSO−d6)δ12.73、42.12、174.58;13C NMRは、93.7%の(R,R)−2,3−ジメチル−コハク酸、2.9%のメソ−異性体(10.75、38.14ppm)および3.5%のデス−メチル(14.74、35.33、35.74ppm)を示した。
【0131】
(実施例8)
(R,R)−2,3−ジメチルコハク酸モノメチルエステルの調製
(R,R)−3,4−ジメチル−ジヒドロ−フラン−2,5−ジオン(40.06g、312.6mmol)のMeOH(400mL)溶液を65℃で6時間還流させた。生じた溶液を真空濃縮すると、前記表題化合物が淡ベージュ色のオイル(49.80g、99.4%)として得られた;1H NMR(400MHz,CDCl3)δ1.19(t,J=7Hz,6H)、2.82(q,J=7Hz,1H)、2.88(q,J=7Hz,1H)、3.70(s,3H)、10.61(bs,1H);13C NMR(100MHz,CDCl3)δ13.41、13.52、41.20、41.38、51.92、175.59、181.43;MS(EICI)m/z(相対強度)159[(M−H)−,100];元素分析 C7H12O4の計算値:C,52.49;H,7.55;N,0.00;実測値:C,52.20;H,7.76;N,<0.05。
【0132】
(実施例9)
(R,R)−2,3−ジメチルコハク酸の調製
粗製(R,R)−2,3−ジメチルコハク酸モノメチルエステル(3.2177g、20.09mmol GC:85.9面積%、6.1面積%のシス異性体、1.8%面積%のデスメチル不純物、3.3面積%のトリメチル不純物)および水(11mL)の二相混合物に、50重量%NaOH水溶液(4.08g、50.94mmol、2.54当量)を加えたが、その間、反応混合物の温度を25℃以下に維持した。生じた溶液を20℃で39分間攪拌し、次いで、HCl(37.5重量%、5.36g、55.1mmol)を加えた。生じた溶液をCH2Cl2(5mL、次いで2×10mL)およびEtOAc(3×35mL)で抽出した。有機層をMgSO4上で乾燥させ、乾燥まで濃縮した。粗製の固体をi−PrOH(10mL)に溶かし、分枝鎖オクタン(30mL)を加えた。溶液を全体体積20mLまで真空濃縮し、冷却すると、スラリーが得られた。分枝鎖オクタン(10mL)を加え、混合物を0℃に冷却した。沈殿物を真空濾過により集め、分枝鎖オクタンで洗浄し、窒素流中で乾燥させると、前記表題化合物が白色の固体(1.4592g、49.7%)として得られた;1H NMR(400MHz,DMSO−d6)δ1.02(d,J=7Hz,6H)、2.58(m,2H);13C NMR(100MHz,DMSO−d6)δ13.40、40.96、176.38;13C−NMRは、1.6%のメソ異性体(14.77ppm、41.87ppm)を示した。
【0133】
(実施例10〜21)
表面下添加を介しての(R,R)−2,3−ジメチル−コハク酸モノメチルエステルの調製
LHMDS(1300kg、24.9重量%。1.93kg−mol、2.1当量)のTHF溶液をタンクに充填し、−30℃に冷却した。(R)−2−メチル−コハク酸4−メチルエステル(133kg、0.91kg−mol、1当量)を、等体積のTHFと混合し、その出口が、容器攪拌機の先端から約0.3mであるように反応器に取り付けられている浸漬管を使用して、リチウム試薬に対して表面下で加えた。基質を加えている間、反応混合物の温度を−25℃以下に維持した。生じた混合物を攪拌し、−10℃に加温し、次いで−30℃に冷却した。MeI(136kg、0.96kg−mol、1.05当量)を2体積のTHFと混合し、反応混合物に対して表面下で供給したが、その間、反応混合物の温度を−25℃以下に維持した。温度を8時間にわたって室温に調節した。NH4Cl(136kg)を水(400L)に溶かし、反応器容器に徐々に供給して、反応をクエンチした。さらなる水(550L)を攪拌しながら加え、次いで、攪拌機を止めて、相を分離した。有機相を廃棄した。水性相を37%HCl(300kg)および水(250L)の混合物で酸性化して、MTBE(4×400L)で抽出した。MTBE相を合わせ、蒸留すると、前記表題化合物がオイルとして得られた。
【0134】
表4は、表面下反応成分添加を使用しての所望のanti−ジアステレオ異性体(R,R)−2,3−ジメチル−コハク酸モノメチルエステルおよび不所望のsyn−ジアステレオ異性体(R,S)−2,3−ジメチルコハク酸モノメチルエステルの収率を示している。比較のために、表4は、先行する段落に記載されているプロセスと類似しているプロセスを使用するが、表面上添加を介して反応成分を加える2種のジアステレオ異性体の収率も示している。
【0135】
【表4】
【0136】
(実施例22〜28)
(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの調製
(R,R)−2,3−ジメチル−コハク酸モノメチルエステル(150kg、936mol)をTHF(260L)およびMTBE(1500L)で希釈し、60℃に加熱した。10%LAH溶液(THF中530kg、1.33kg−mol)をこの溶液に供給すると、軽いアルミニウムアルコキシド中間体を含有するスラリーが生じた。反応により生じる熱を、溶媒沸騰および凝縮により除去した。第2のタンクに、THF(約970L)、水(220L)および50%NaOH水溶液(5kg)を充填し、約50℃に加熱した。アルミニウムアルコキシドスラリーを、THF、水およびNaOHを含有するタンクに制御して供給した。次いで、攪拌機を止め、水酸化アルミニウムを10分から15分間沈降させ、生成物をデカンテーションにより除去した。固体をMTBE(3×600L)で洗浄して、追加の生成物を抽出した。有機液体を集め、蒸留すると、前記表題化合物が得られた。
【0137】
表5は、先行する段落に記載された処理(即ち、過剰の塩基の添加、実施例22〜26)を使用する(R,R)−2,3−ジメチル−コハク酸モノメチルエステルのLAH還元を介しての(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの収率を示している。比較のために、表5は、Fieser処理(H2O、15%NaOH水溶液およびH2Oの連続添加、続いてLAH還元)を使用する(R,R)−2,3−ジメチル−ブタン−1,4−ジオールの収率も示している。
【0138】
【表5】
【0139】
(実施例29)
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの調製
反応器容器に塩化p−トルエンスルホニル(400kg、2.14kg−mol、2.5当量)を無水充填した。アセトニトリル(1000L)を続いて加え、生じたスラリーを0℃に冷却した。(R,R)−2,3−ジメチル−ブタン−1,4−ジオール(100kg、0.85kg−mol、1当量)を反応器に加えた。続いて、Et3N(260kg、2.5kg−mol、3当量)を、反応器温度を5℃以下に維持する速度で反応器に供給した。EtOAc(660L)および水(640L)を攪拌しながら加えて、反応をクエンチした。攪拌を止めて、有機相および水性相を分離した。水性相をEtOAc(560L)で洗浄し、生じた有機相を、反応クエンチからの有機相と合わせた。合わせた有機相を10%NaHCO3水溶液(720kg)および25%NaCl水溶液(670kg)で順次洗浄した。EtOAcを大気圧で留去すると、約400Lの液体体積が得られ、これに、MTBE(1100L)およびEtOH(170kg)を加えた。混合物を還流に加熱し、続いて、約20℃に冷却して、粗製生成物を結晶化させ、これを、濾過により集めた。粗製生成物をMTBE(2200L)に分散させ、混合物を還流まで加熱して、固体を溶解させた。溶解の後に、水(200L)を攪拌しながら加え、相を分離した。水性相を廃棄した。EtOH(150kg)を有機相に加え、混合物を20℃に冷却して、前記表題化合物を結晶化させ、これを濾過により集めた。
【0140】
(実施例30)
LHMDSによる(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタンの分解
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(269mg、0.631mmol)のTHF(1.5mL)溶液をLHMDSのTHF溶液(1.35Mの溶液1.0mL、1.35mmol、2.14当量)で処理し、17℃で6.5時間攪拌したが、この時点で、定量HPLC分析によると、元々のジトシレートの55.5%が残っていた。
【0141】
(実施例31)
メチル3−シアノプロパンイミデートクロリドの調製
スクシノニトリル(12.124g、0.1514mol)を、最小体積の温ジオキサン(10mL)に溶かした。その間に、MeOH(6.2mL、4.90g、0.153mol、1.01当量)を、HClのEt2O溶液(2Mの溶液76mL、0.152mol、1.00当量)に10℃で加えた。スクシノニトリル溶液を、HCl/MeOH溶液に3分にわたって加えた。次いで、さらなるHCl溶液を加えた(15mL、0.030mol、0.20当量)。約10分の後に、固体が沈殿し始めた。さらなる15分の後に、スラリーが粘稠性になった。混合物を10℃で26時間攪拌し、次いで、Et2O(蒸発を補償するために30mL)で希釈し、濾過した。ケークをEt2O(3×20mL)で洗浄し、N2流により乾燥させて、白色の結晶固体としての前記表題化合物(17.196g、76.4化学%)にした。13C−NMR(100MHz,DMSO−d6):δ171.28(s);120.47(s);51.83(q);29.05(t);12.46(t);1H−NMR(400MHz,DMSO−d6):δ1.86(2H,t,J=7.4Hz);2.46(2H,t,J=7.4Hz);3.35(3H,s)。
【0142】
(実施例32)
3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリルの調製
メチル3−シアノプロパンイミデートクロリド(15.29g、0.1029mol)および2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオール(12.363g、0.1029mol、1.00当量)の無水THF(300mL)懸濁液を40℃で13.5時間攪拌し、次いで、不溶性物質を濾別した。濾液を、より小さい規模の実験(イミデート0.0035molから)からの濾液と合わせた。合わせた濾液を濃縮して白色の半固体残渣にし、これを、CH2Cl2/EtOAc(約15mL)に溶かし、シリカゲルパッド(40g)に施与し、EtOAc/シクロヘキサン(10%を500mL、続いて20%を500mL)で溶離した。濾液を濃縮して粘稠性のスラリーにし、ヘプタン(100mL)で希釈し、再濃縮し(2回)、次いで、濾過した。ケークをヘプタン(3×8mL)で洗浄し、N2流により乾燥させると、前記表題化合物が白色の結晶固体(9.417g、48.3化学%)として得られた。13C−NMR(100MHz,CDCl3):δ119.62(s);107.32(s);72.74(t);32.35(t);30.32(s);14.31(q);11.78(t);1H−NMR(400MHz,CDCl3):δ0.81(3H,s);2.01(2H,t,J=7.8Hz);2.48(2H,t,J=7.8Hz);3.89(6H,s)。
【0143】
(実施例33)
3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル
刊行された手順に従った。S.M McElvain&J.P.Schoeder、J.Am.Chem.Soc.71:40(1949年)参照。スクシノニトリル(107.53g、1.3426mol)のジオキサン(120mL)溶液をEt2O(930mL)で処理した。二相混合物をMeOH(43.4g、1.3546mol、1.01当量)で処理し、8℃に冷却した。生じた1相溶液を7℃に冷却し、温度が8℃未満に維持されるような速度で、ガス分散管を介して、無水HCl(58g、1.59mol、1.18当量)を溶液に散布する(添加は発熱性である)。混合物を8℃で一晩攪拌し、次いで濾過した。ケークをEt2O(500mL)で洗浄し、窒素流により乾燥させた。ケークは、13C−NMR(CD3CO2D)によりメチル3−シアノプロパンイミデートクロリド(103.3g、0.6952mol、51.8M%、純度>95%)と識別された。イミデート(1.0g、6.73mmol)、2−(ヒドロキシメチル)−2−メチルプロパン−1,3−ジオール(2.4g、19.98mmol、2.97当量)およびBHT(24mg、0.109mmol、0.016当量)の無水THF(10.5mL)懸濁液を40℃で17時間攪拌し、次いで、不溶性物質を濾別した。前記表題化合物の収率は、GCによる濾液の定量分析により80.5M%であると決定された。
【0144】
(実施例34)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(5.375g、0.01260mol)およびLiBr(3.2839g、0.03782mol、3.00当量)のトルエン(50mL)中の混合物を還流に加熱した。18時間後に、TLC(溶離液:20%EtOAc/シクロヘキサン、チャー:PMA、Rf[ジトシレート]=0.31、Rf[ジブロミド]=0.76)によると、前記表題化合物の変換が完了した。不溶性固体(トシル酸リチウム)を、セライトの小さいパッドで濾別し、トルエン(4×5mL)ですすいだ。13C−NMR(100MHz,CDCl3):δ39.19(t);37.02(d);14.41(q);1H−NMR(400MHz,CDCl3):δ0.99(6H,d,J=6.4Hz);2.05(2H,多重線);3.38(2H,dd,J=10.2,5.0Hz);3.46(2H,dd,J=10.4,5.2Hz)。
【0145】
(実施例35)
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリルの調製
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンを含有する濾液に、3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル(3.0017g、0.01638mol、1.30当量)を加えた。次いで、混合物を室温で2時間にわたって、LiN(SiMe3)2のTHF溶液(1.28Mの溶液34mL、0.0435mol、3.45当量)で滴加処理した。30分後に、GCおよびLC分析により、未反応の二臭化物(GCにより25.4化学%、LCにより32.9化学%)および未反応のシアノオルトエステル(0.090当量)が、生成物(46.5化学%)と共に示された。さらなるシアノオルトエステル(1.151g、0.00628mol、0.50当量)を加え、混合物をさらに16時間攪拌したが、この時点で、GCによると反応は完了したので(二臭化物3.4化学%およびシアノオルトエステル10.2化学%)、反応を水(50mL)でクエンチした。上部有機相を分離し、水性相をCH2Cl2(2×50mL)で抽出した。有機相を合わせ、濃縮すると、前記表題化合物がオレンジ−茶色の結晶固体(5.7532g、収率94.7化学%、ESTD GC、ジトシレートから全体で)が得られた。13C−NMR(100MHz,CDCl3):δ126.24(s);107.68(s);72.60(t);48.46(t);47.25(t);45.30(t);41.52(d);40.55(d);36.72(s);30.40(s);18.49(q);17.42(q);14.50(q);1H−NMR(400MHz,CDCl3):δ0.70(3H,s);0.87(3H,d,J=6.4Hz);0.92(3H,d,J=6.8Hz);1.2〜2.3(8H,多重線);3.80(6H,s)。
【0146】
(実施例36)
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリルの調製
21℃の(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(214.4mg、0.5026mmol)および3−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)プロパンニトリル(239.9mg、1.3094mmol、2.61当量)のTHF(4.0mL)溶液をLiN(SiMe3)2のTHF溶液(1.28Mの溶液2.50mL、3.20mmol、6.37当量)で2時間にわたって滴加処理し、室温で62時間攪拌した。混合物をH2O(1mL)でクエンチし、室温で30分間攪拌し、次いで、H2O(40mL)に注ぎ、CH2Cl2(3×10mL)で抽出した。抽出物を、アセトニトリルを用いてメスフラスコ中で100.0mLまでにした。GCおよびLCによる定量分析により、前記表題化合物の不在が示された。
【0147】
(実施例37)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸2,2−ジメトキシプロピルの調製
粗製(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリル(2.5131g、ジトシレート5.5044mmolに由来)をCH2Cl2(25mL)に溶かし、6NのHCl水溶液(25mL)で処理した。二相混合物を室温で30分間攪拌したが、この時点で、TLCによると、反応成分(Rf=88)の前記表題化合物(Rf=0.72)への加水分解は完了した。混合物を分離漏斗に移した。他の分離漏斗に、水(25mL)を加えた。第1分離漏斗中の有機相を、第2分離漏斗に移し、混合物を振盪し、CH2Cl2相を除去した。次いで、2個の分離漏斗中の水性相をCH2Cl2で順次抽出した(4×25mL)。抽出物を合わせ、淡褐色のオイルまで濃縮し、これを、放置して結晶化させると、前記表題化合物がオフホワイト色の固体として純粋な形態(1.4177g、5.003mmol、ジトシレートから全体で90.9化学%)で得られた。13C−NMR(100MHz,CDCl3):δ170.02(s);125.35(s);67.28(t);66.97(t);47.47(t);46.23(t);43.91(t);41.83(d);40.68(d);40.39(s);36.96(s);17.89(q);17.19(q);16.80(q);1H−NMR(400MHz,CDCl3):δ0.88(3H,s);1.02(3H,d,J=6.4Hz);1.06(3H,d,J=6.5Hz);1.3〜2.7(8H,多重線);3.61(4H,s);3.91(2H,t,J=7.1Hz);4.24(2H,s)。
【0148】
(実施例38)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の調製
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸2,2−ジメトキシプロピル(1.0034g、3.541mmol)を、KOH(純度約87.3%の物質247.7mg、216mg、3.85mmol、1.09当量)の水(1.5mL)溶液に加えた。懸濁液を21℃で1時間攪拌し、この時点で、すべての固体は溶解しており、TLC分析により、反応成分(Rf=0.75)から前記表題化合物(Rf=0.36)へ、および1,1,1−トリス(ヒドロキシメチル)エタン(Rf=0.56)への加水分解が完了したことが示された。溶液をMTBE(3×0.5mL)で洗浄して、非極性不純物を除去した。13C−NMR(100MHz,CDCl3):δ175.41(s);125.00(s);47.44(t);46.16(t);43.35(t);41.79(d);40.75(d);36.80(s);17.96(q);17.55(q);1H−NMR(400MHz,CDCl3):δ1.02(3H,d,J=6.4Hz);1.05(3H,d,J=6.4Hz);1.3〜2.7(8H,多重線);9.9(1H,br s)。
【0149】
(実施例39)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の調製
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸カリウム塩の水溶液(シアノエステル201.4mg[0.7107mmol]および水酸化カリウム[純度約87.3%の物質49.6mg、43.3mg、0.772mmol、1.09当量]に由来、水0.3mL中)をMeOH(3mL)で希釈し、スポンジニッケルA−7000(128mg)で処理し、H2(100psig)下に35℃で攪拌した。14時間後に、シアノ酸(Rf=0.41)から前記表題化合物(Rf=0.29)への還元は、TLCによると約75%完了した。スラリーを濾過し、水(0.3mL)で洗浄した。濾液を黄色がかったオイルまで濃縮し、これを、水(0.5mL)に溶かし、氷酢酸(3滴)で処理すると、粘稠性のスラリーが生じ、これを、水(0.5mL)で薄めた。スラリーを70℃に加熱し、十分なイソプロパノールで希釈して、すべての固体(0.25mL)を溶かし、次いで1.5時間にわたって徐々に0℃に冷却した。生じたスラリーを濾過すると、前記表題化合物が、基準標準とのTLC比較によると、反応成分および1,1,1−トリス(ヒドロキシメチル)エタンで汚染された固体として得られた。
【0150】
(実施例40)
((3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンチルメタンアミンの調製
(3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンタンカルボニトリル(302.8mg、1.1411mmol)の無水EtOH(4mL)溶液を、NaOH水溶液(50%溶液135mg、67.5mg、1.687mmol、1.48当量)で、続いてスポンジニッケルA−7000(183mg)で処理した。混合物をH2(50psig)下に室温で21時間攪拌し、次いで濾過し、水(3×1.5mL)ですすいだ。濾液を水(5mL)で希釈し、MTBE(5mL)で抽出し、無水K2CO3上で乾燥させ、濃縮すると、前記表題化合物が淡黄色の固体残渣(333.9mg、1.2395mmol、収率108.6化学%)として得られた。TLC溶離液:70:20:5:5のEtOAc:MeOH:濃NH4OH:H2O、チャー:50%H2SO4水溶液、Rf[アミン]=0.66、Rf[ニトリル]=0.79);13C−NMR(100MHz,CDCl3):δ107.46(s);70.37(t);49.29(t);45.23(t);45.01(t);42.59(t);42.28(s);39.81(d);39.49(d);28.23(s);16.34(q);15.98(q);12.10(q)。
【0151】
(実施例41)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの調製
((3S,4S)−3,4−ジメチル−1−((4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクタン−1−イル)メチル)シクロペンチル)メタンアミン(200.5mg、0.7443mmol)のHCl水溶液(1.0Mの溶液2.3mL、2.3mmol、3.1当量)溶液を室温で75分間攪拌したが、この時点で、TLC(Rf[アミノオルトエステル]=0.74、Rf[アミノエステル]=0.64)によると、アミノエステル、2−((3S,4S)−1−(アミノメチル)−3,4−ジメチルシクロペンチル)酢酸3−ヒドロキシ−2−(ヒドロキシメチル)−2−メチルプロピルへの加水分解が完了した。反応を固体炭酸水素ナトリウム(315.4mg、3.754mmol、5.0当量)でクエンチし、硫酸ナトリウム(103mg)で処理し、THF(3×2mL)で抽出した。抽出物を合わせ、50℃で4.5時間加熱したが、この時点で、TLCによると、アミノエステル(Rf=0.66)から前記表題ラクタム(Rf=0.80)および1,1,1−トリス(ヒドロキシメチル)エタン(Rf=0.56)への変換が完了した。混合物はさらに、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(Rf=0.21)およびBHT(Rf=0.91)の痕跡も示す。混合物を濃縮して黄色のオイルにし、これを、塩化メチレン(3mL)に溶かし、水(3×3mL)で洗浄してトリオールを除去し、次いで、濃縮して油性残渣にし、これを、ヘプタン(0.5mL)に70℃で溶かし、−20℃に冷却して、結晶を分離した。母液をデカンテーションし、これを、真空乾燥すると、前記表題化合物が結晶固体(78.9mg、0.472mmol、アミノオルトエステルから63.4化学%、シアノオルトエステルから全体で68.9化学%)として得られた。LC純度:基準試料と比較して97.5面積%。13C−NMR(100MHz,CDCl3):δ178.14(s);56.13(t);48.35(t);47.91(t);45.74(t);45.04(s);41.63(d);41.57(d);18.60(q);18.49(q);1H−NMR(400MHz,CDCl3):δ0.977(3H,d,J=6.0Hz);0.983(3H,d,J=6.0Hz);1.3〜2.3(8H,多重線);3.21(1H,d,J=9.6Hz);3.24(1H,d,J=10.4Hz);6.02(1H,br s)。
【0152】
(実施例42)
塩化メチル3−シアノプロパンイミデートの調製
スクラバー(水)に対して出口を有する口を1つ備えた1Lジャケット付き三つ口フラスコに、スクシノニトリル(Aldrich、mw80.09、103.06g、1.2868mol)、塩化メチレン(E&M)200mL、メチル−t−ブチルエーテル(E&M)200mLおよびMeOH(E&M、mw32.04、41.5g、1.2953mol、1.01当量)を充填した。溶液を6.5℃に冷却し、温度が12℃未満に維持されるような速度で、ガス分散管を介して、無水塩化水素(mw36.46、55.5g、1.5222mol、1.18当量)を溶液に散布する(添加は発熱性である)。塩化水素の重量を、レクチャーボトルを秤量することにより測定した。レクチャーボトルを天秤上のスタンドに保持した。これを、ガス分散管に、レクチャーボトルに至る管セグメントにおける張力を最小にするようにクランプされているPVC管を介して接続した。流れを、AGA Model LB 165−40−2F−BV腐食性ガス調節器を介して調節した。レクチャーボトルの圧力が高すぎるので、単純なバルブを使用しても、流れを十分に制御することができなかった。添加は4時間かかった。添加のほぼ中程で、生成物イミデートが結晶化し始めた。添加が完了したら、ジャケット温度を12℃に調節した。25時間攪拌した後に、スラリーが溶媒蒸発により非常に粘稠性になったので、塩化メチレン175mLを加えた。粘稠性ではあるが、スラリーは容易に攪拌可能および濾過可能であった。混合物を濾過した。ケークを塩化メチレン325mLで洗浄し、室温窒素により乾燥させた。物質は、GCアッセイにより前記表題化合物と識別された(カラム:60メートルDB−1、内径0.25mm w 1ミクロンフィルム、温度勾配:150℃で3分間、次いで、10℃/分で225℃まで勾配させ、次いで、10分間保持;試料調製:イミデート約100mg(正確に秤量)に1NのHCl0.5mLを添加し、溶液を5分間放置し、溶液を10mLメスフラスコに2mL水と共に移し、MeOHを用いて10mLまでにした;保持時間:スクシノニトリル、9.29分;メチル−3−シアノプロピオネート、9.78分;コハク酸ジメチル、10.68分)。冷蔵庫での貯蔵が推奨される。褐色ビンで室温貯蔵されたイミデートの多くは、1〜7日後に、著しいガス発生を伴って茶色の固体に分解した。重量:178.97g(mw148.59;1.2045mol、収率93.6化学%[未修正])。
【0153】
(実施例43)
4,4,4−トリメトキシブタンニトリルの調製
3L三つ口フラスコに、塩化メチル3−シアノプロパンイミデート(mw148.59、176.77g、1.1896mol)およびMeOH(E&M)1.7Lを充填した。攪拌すると、イミデートが溶けて、均一な溶液が生じた。溶液を室温で攪拌した。約2時間後に、塩化アンモニウムが沈殿し始めた。明朝(13時間後)、混合物は、顆粒固体の白色のスラリーになっていた。この反応を、NMR(アリコットを取り、約20%のCD3ODで希釈)により監視することができる。(塩化アンモニウムがオルトエステルとゆっくりと反応して、3−シアノプロピオン酸メチルを形成してしまうので、スラリーの加熱は回避すべきである。例えば、反応混合物を真空濃縮して(ポット温度33℃)、塩化アンモニウムを沈殿させることにより反応を完了させる別の実験では、4%の3−シアノプロピオン酸メチルが形成された)。スラリーを、トリエチルアミン(mw101.19;d0.726、180mL、130.68g、1.2914mol、1.09当量)を含有する受器へと濾過し、少量のMeOHですすいだ。ケーク(塩化アンモニウム)は19.76gの重さであった(mw53.49、0.3694mol、理論の31.1%)。濾液をNMRによりチェックする(約20体積%のCD3ODで希釈されたそのままのアリコットで行う)。メチルエステルが存在する場合、濾液を一晩攪拌すると、遊離したアンモニアがメチルエステルをアンモニア分解する。濾液を水酸化カリウムペレット(mw56.11、55.2g、87.3重量%、48.19g、0.8588mol、0.72当量)の水160mL溶液で処理すると、このことにより、固体が沈殿した(おそらくKCl)。スラリーを濾過した。ケークの重量は50.32gであった(KClならばmw74.56、0.6749mol、0.57当量)。濾液に、炭酸ナトリウム(mw105.99、32g、0.3019mol、0.25当量)の水200mL溶液を加えた。混合物を回転蒸発(浴温度47℃)させて、MeOHを除去すると、均一な溶液が残った(体積約575mL)。溶液をH2O95mLで希釈し、MTBE(700mL、次いで360mL)で抽出した。相分割は優れていた。MTBE抽出物を合わせ、無水炭酸カリウム(2.7g)で処理し、二相混合物に濃縮し、さらなる炭酸カリウム(53g)で処理すると、多少の固体を伴う二相混合物が得られた。MTBE相を分離し、水性相をMTBE(50mL)で抽出した。MTBE抽出物を合わせ、無水粉砕炭酸カリウム(14g)上で乾燥させ、無水炭酸カリウム(5g)から蒸留した。中間留分(116.52g、沸点92℃/26mm)は、NMRおよびGCにより表題化合物と識別される無色の液体(KF0.025)であった。重量:116.52g(mw159.19、94.60重量%、110.23g、0.6924mol)。初期留分(forecut)(重量41.46g、沸点92℃/27mm)は数滴の水(KF0.479)を含有し、トルエン25mLおよびか焼無水炭酸カリウム0.3gからの再蒸留により再処理すると、さらなる無色の液体(沸点82〜83.6℃/25mm)が得られた。トルエン共沸蒸留後および生成物蒸留前に得られたKFは、0.011%であった。重量:37.2g(mw159.19、95.93重量%、35.69g、0.2242mol)。(抽出における水残存の問題は、抽出溶媒としてMTBEの代わりにトルエンを使用することにより回避することができる)。GCアッセイ(カラム:30メートルDB−5、内径0.25mm、w 1ミクロンフィルム、温度勾配:70℃で5分間、次いで、10℃/分で320℃まで勾配させ、次いで2分間保持、保持時間:15.88分)によると、いずれの留分も、1.7面積%より多い何らかの不純物を含有しなかった。3−シアノプロピオン酸メチルのレベルは、中間留分では0.05面積%であり、再蒸留された初期留分では検出されなかった。収率(合わせて):0.9166mol、77.1化学%(純度修正)。13C−NMR(100MHz,CD3OD):δ121.49(s);115.98(s);50.64(q);28.00(t);12.59(t);1H−NMR(400MHz,CD3OD):δ2.11(2H,t,J=7.4Hz);2.42(2H,t,J=7.4Hz);3.25(9H,s)。
【0154】
(実施例44)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55、200.0g、0.4689mol)および臭化ナトリウム(mw102.89、145.0g、1.4093mol、3.01当量)のトルエン1L中の混合物を70℃に加熱すると、均一なスラリーが得られた。臭化テトラブチルアンモニウム(mw322.38、15.0g、0.0465mol、0.099当量)のH2O15.0mL溶液を加えた。8分後に、固体が沈殿し始めた(トシル酸ナトリウム)。スラリーを70℃で3時間、次いで80℃で17時間攪拌した。混合物を50℃に冷却し、H2O(350mL、次いで2×150mL)で洗浄し、回転蒸発により濃縮して、199.1gの重量のオイルにした。
【0155】
(実施例45)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
リチウムヘキサメチルジシラジドのTHF溶液(Chemetall、mw167.33、0.226g/mL、1058.4mL、239.2g、1.4295mol、3.07当量)を真空蒸留(ジャケット温度25℃)して、非常に粘稠性なスラリー(体積:400mL)にし、これを、THF150mLで希釈した。このスラリーを0℃に冷却し、先行する実施例からの(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの一部(197.4g、ジトシレート0.4649molから)で20分にわたって処理し、THF50mLですすいだ。添加は、やや発熱性であった(0℃に低下する前に+15℃までの短時間のエクスカーション)。添加の終了時には、混合物は澄明な溶液になった。この溶液に、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、105.84g、0.6649mol、1.43当量)を徐々に6時間にわたって加えた。添加の間、ポット温度は0℃であった。添加の終了時に、温度を−5℃に低下させた。混合物を14時間攪拌し、次いで、H2O(252mL)でクエンチした。最初の13mLを50分にわたって加え(+4℃までの発熱)、次いで残りを、5分にわたって加えた。混合物を23℃に加温すると、界面に暗色のオイル(リチウムヘキサメチルジシラジド/THF試薬における鉱油、不純物と識別された)を伴う二相混合物が得られた。水性層を分離した。有機層を350〜400mLの体積まで濃縮し、ヘプタン350mLで処理し、500〜550mLの体積まで濃縮し、ヘプタン400mLで処理し、次いで、450mLの体積まで濃縮した。ヘプタン溶液を0.05NのNaOH(325mL、次いで175mL)で洗浄した。他の実験からの溶液の一部を濃縮して、13C−NMRおよび1H−NMRにより前記表題化合物と識別されるオイルにした。13C−NMR(100MHz,CD3CD2OD):δ125.44(s);113.16(s);48.66(3C,q);47.50(t);46.44(t);41.12(d);39.76(d);38.76(t);37.06(s);18.08(q);16.82(q);1H−NMR(400MHz,CD3CD2OD):δ1.05(3H,d,J=6.4Hz);1.09(3H,d,J=6.7Hz);1.46(1H,t,J=12.6Hz);1.5〜2.3(7H,多重線);3.29(9H,s)。
【0156】
(実施例46)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの調製
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルを含有する先行する実施例からのヘプタン溶液全体を、十分な3NのHClで処理して、pHを<3に調節した(d1.0495、207g、197.2mL、0.592mol、1.27当量)。混合物を室温で40分間攪拌し、次いで、水性層を分離して、ヘプタン100mLで抽出した。合わせた有機層をH2O(50mL)で洗浄した。重量:425.19g。同様の実験からの溶液を濃縮し、蒸留すると(沸点145〜147℃/35.1mm)、13C−NMRおよび1H−NMRおよびGCにより本質的に純粋な形態の前記表題化合物と識別される液体が得られた(99.0A%)。13C−NMR(100MHz,CDCl3):δ169.63(s);124.91(s);51.62(q);47.15(t);45.92(t);43.07(t);41.56(d);40.39(d);36.77(s);17.77(q);16.97(q);1H−NMR(400MHz,CDCl3):δ0.93(3H,d,J=6.4Hz);0.97(3H,d,J=6.6Hz);1.29(1H,t,J=12.4Hz);1.46(1H,多重線);1.67(1H,多重線);1.84(1H,dd,J=13.8,9.4Hz);2.05(1H,dd,J=13.8,8.5Hz);2.38(1H,dd,J=13.2,6.5Hz);2.58(2H,s);3.65(3H,s)。
【0157】
(実施例47)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの調製
モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、20.0mLで33.33g、計算乾燥重量:15.55g[0.35g/g])を、MeOH13mLで処理し、攪拌し、沈降させ、上澄み13mLをデカンテーションした。触媒洗浄手順を2回繰り返した。メタノール触媒スラリーを、450mLガラス攪拌反応器にMeOH60mLと共に入れた。容器を密封し、H2でパージし、60psigまで加圧し、攪拌しながら85℃に加熱した。反応器温度が85℃に達したら、圧力を調節して、60psigに維持した。先行する実施例からのヘプタン溶液の一部(208.60g、ジトシレート0.2281molから、mw195.26、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル44.535gを理論的に含有)を濃縮してオイル(重量:62.43g)にし、これを、MeOH119mLで希釈すると、黄色−白色のエマルションが得られ、これを、水素化反応器に4.9時間にわたって計量供給した。次いで、計量供給系をMeOH(3×5mL)で反応器へとすすぎ、完成した混合物を85℃/60psigにさらに5時間維持した。室温まで冷却した後に、反応混合物を真空濾過した。反応器および触媒ケークをMeOHですすいだ。合わせた濾液および洗浄液の重量は、399.11gであった。このほぼ無色の溶液の一部(396.29g、ジトシレート0.2265molから)を真空蒸留(ポット温度37℃)して、150mLの体積にした。次いで、溶液を大気圧で蒸留したが、この間、留出物の沸点が101℃で一様になるまでヘプタンを少量ずつ加えた(3×100mL)。生じた濁った溶液を−5℃に冷却した(結晶化が42℃で生じた)。スラリーを−5℃に予め冷却されたフリット上で濾過した。ケークを室温のヘプタン80mLで洗浄し、N2圧力により乾燥させた。生成物は、基準試料とのESTD LC比較により前記表題化合物と識別された(濃度:96.49W%)。収率:33.80g(mw167.25、32.61g(濃度修正)、0.1950mol、ジトシレートから全体で86.1M%)。濾液のESTD LC分析により、ラクタムが判明した(ジトシレートからの全体収率3.2M%)。
【0158】
(実施例48)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からの(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(mw167.25、純度96.49W%の物質32.80g、31.65g、0.1892mol)を、37%HCl(mw36.46、37W%溶液57.4g、21.24g、0.5825mol、3.08当量)およびRO水32.8mLで処理した。混合物を>88℃で24時間攪拌した。LCによる分析により、残渣ラクタム2.06M%が判明した。混合物を60℃に冷却し、50%水酸化ナトリウム(30.85g、水酸化ナトリウム15.425g[mw40.00、0.3856mol、2.04当量]含有)を加えることにより、pHを約2(紙)に調節した。混合物をH2O43mLで希釈し、溶液が濁るまで、真空蒸留した(ポット温度48℃)(最終体積約115mL)。50%水酸化ナトリウム(14.81g、水酸化ナトリウム7.405g[mw40.00、0.1851mol、0.98当量]含有)を加えることにより、pHを6.5(紙)に調節した。混合物を<5℃に冷却し、30分間攪拌し、濾過した。フラスコを濾液で2回すすいだ。ケークを<10℃のRO水33mLで洗浄し、N2流により乾燥させた。生成物は、基準試料とのESTD LC比較により、前記表題化合物と識別された(濃度:88.6W%)。金属に関するスクリーニングによりMo(0.1ppm)およびAl(1ppm)が検出された。重量:35.43g(mw185.27、31.39g[濃度修正]、0.1694mol、89.6M%)。濾液を濃縮して残渣にした(重量:32.94g)。表題化合物に関するLC分析により、濃度2.1W%が判明した(0.692g、0.00373mol、ラクタムから2.0M%)。
【0159】
粗製生成物の一部(mw185.27、純度88.6W%の物質34.0g、30.124g、0.1626mol)をRO水120mLおよびイソプロパノール40gに懸濁させた。混合物を還流(85℃)に加熱したが、この時点で、溶液が形成された。溶液を<5℃に4時間にわたって冷却し、1時間攪拌し、濾過した。ケークを<5℃のイソプロパノール70mLで洗浄し、N2流により17時間乾燥させた。生成物は、基準試料とのESTD LC比較により、前記表題化合物と識別された(濃度:99.6W%)。重量:29.15g(mw185.27、29.03g[濃度修正]、0.1567mol、粗製生成物から96.4M%)。濾液を濃縮して残渣(重量:5.19g)にした。表題化合物に関するLC分析により、濃度51.6W%が判明した(2.678g、0.01445mol、粗製生成物から8.9M%)。13C−NMR(100MHz,4/1 CD3OD/D2O):δ182.49(s);53.04(t);52.33(t);48.80(t);48.33(t);44.13(d);43.81(d);43.74(s);19.99(q);19.86(q);1H−NMR(400MHz,4/1 CD3OD/D2O):δ0.98(3H,d,J=6.3Hz);0.99(3H,d,J=6.2Hz);1.14(1H,dd,J=12.6,10.2Hz);1.19(1H,dd,J=12.9,10.2Hz);1.52(2H,重複した多重線);1.88(1H,t,J=13.6Hz);1.90(1H,t,J=13.9Hz);2.47(1H,d,J=15.2Hz);2.52(1H,d,J=15.2Hz);2.92(1H,d,J=12.9Hz);2.99(1H,d,J=12.9Hz)。
【0160】
(実施例49)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からのヘプタン溶液の一部(208.53g、ジトシレート0.2280molから、mw195.26、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル44.520gを理論的に含有)を水酸化カリウム(mw56.11、純度88W%の物質29.46g、25.925g、0.4620mol、2.03当量)のH2O125mL溶液で処理した。二相混合物を2時間激しく攪拌したが、この時点で、TLCによると加水分解は完了した。2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩を含有する水性相を分離した。重量:189.02g。
【0161】
450mLガラス製攪拌反応器に、モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、20.0mL、重量33.03g、乾燥重量:15.20g[0.31g/g])およびMeOH60mLを充填した。容器を密封し、H2でパージし、H2で25psigまで加圧した。反応器を攪拌し、約50℃に加温したがが、この時点で、既知の体積の高圧レザバーから供給することにより、圧力を50psigに調節および維持した。前記で調製された水性相の一部(185.72g、ジトシレート0.2240molから、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸[mw219.33、49.130g]含有)を、水素化容器に2.7時間にわたって計量供給した。供給系をMeOH(3×5mL)で反応器へとすすいだ。最後のすすぎの完了後約15分後で、水素摂取は完了したようである。反応器を50℃にさらに5時間維持し、その後、室温まで冷却した。反応混合物を濾過し、触媒ケークをMeOHおよび水ですすいだ。(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸のカリウム塩を含有する濾液の重量は610.92gであった。
【0162】
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸のカリウム塩(608.55g、ジトシレート0.2231molから)を含有する前記濾液の一部を回転蒸発により濃縮して、188.04gにし、ジャケット付きの三つ口フラスコに移し、H2O(2×10mL)ですすいだ。氷酢酸(mw60.05、27.14g、0.4520mol、2.03当量)を加えて、pHを>14から6.72に調節した。スラリーを0℃に冷却し、0℃で3時間攪拌し、濾過し、ケークをN2流により乾燥させた。結晶は、TLCおよびLCにより前記表題化合物として識別された。重量:40.51g。
【0163】
粗製(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の一部(38.86g、ジトシレート0.2140molから)をイソプロパノール307mLと混合した。スラリーを80℃に2時間加熱し、次いで、1.5時間にわたって0℃に冷却し、濾過した。濾液を使用して固体をフラスコからフィルター上にすすいだ。ケークをN2流により乾燥させた。結晶はオフホワイト色だった。重量:33.05g。
【0164】
前記(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸結晶を25W%i−PrOH/H2O165mLと混合した。スラリーを80℃に加熱すると、澄明な溶液が、あるとしても非常に僅かな濁りのみを伴って生じた。溶液を0℃に4時間にわたって冷却し、0℃で1時間攪拌し、次いで濾過した。ケークを濾液で洗浄し、N2流により乾燥させた。LCによると、結晶は0.1A%を上回る不純物は含有しなかった(濃度:99.6W%)。重量:30.55g(mw185.27;30.43g、0.1642mol、ジトシレートから全体で76.7M%)。
【0165】
(実施例50)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55;200.0g、0.4689mol)および臭化カリウム(mw119.01、169.0g、1.4200mol、3.03当量)のトルエン400mL中の混合物をN2で45分間脱ガスし、次いで、50℃に加熱し、臭化テトラブチルアンモニウム(mw322.38、30.0g、0.09306mol、0.198当量)のH2O30mL溶液で加えて処理した。混合物を21時間還流し、次いで50℃に冷却し、H2O(400mL、次いで2×100mL)で洗浄した。有機層をトルエン250mLで希釈し、回転蒸発により濃縮すると(浴温度51℃)、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンが重量208.7gのオイルとして得られた。
【0166】
リチウムヘキサメチルジシラジドのTHF溶液(Chemetall、mw167.33、0.226g/mL、1082.4mL、244.6g、1.4619mol、3.17当量)を真空蒸留して、500mLの体積にし、−2℃に冷却し、1時間にわたって一部の(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(205g、ジトシレート0.4606molから)で処理した。混合物を−2℃で10分間攪拌し、次いで、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、105.84g、0.6649mol、1.44当量)で徐々に6時間にわたって処理した。添加の間、ポット温度を0℃に維持した。混合物を14時間攪拌し、次いで、H2O(250mL)でクエンチした。初めの20mLは1時間にわたって加え(+5℃まで発熱)、次いで残りは5分間にわたって加えた。混合物を18℃に加温し、層を分離した。水性層をTHF50mLで逆抽出し、抽出物を初めの有機層と合わせた。前記表題化合物を含有する合わせた有機層の重量は、786.7gであった。
【0167】
(実施例51)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
先行する実施例からの粗製(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルのTHF溶液の一部(188.18g、ジトシレート0.1102molから)を37%HCl水溶液(mw36.46、d1.2g/mL、29mL、34.8g、塩化水素12.88g[0.3532mol、3.20当量]含有)で処理して、pHを−1に調節した。添加の間、温度は52℃に上昇し、固体が沈殿した。水(65mL)を加えて、固体を溶かし、混合物を室温に低下させた。混合物を(攪拌することなく)室温に20時間維持し、次いで水性相(pH0.55)を分離した。2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルを含有する有機相に、水酸化カリウム(mw56.11、87.3W%の物質14.0g、12.22g、0.2178mol、1.98当量)のH2O50mL溶液を加えた。添加の間に、温度は36℃に上昇した。混合物を室温で2時間攪拌したが、この時点で、TLC分析により、けん化が完了したことが示された。相を分離し、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩を含有する水性相を濃縮して、60mLの体積にした(MeOHおよび残留THFを除去するため)。
【0168】
HCl水溶液(37%、mw36.46、d1.2g/mL、19mL、22.8g、塩化水素8.436g[0.2314mol、2.10当量]含有)をH2O57mLで希釈し、0.3℃に冷却し、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のカリウム塩の結晶でシーディングした。この混合物に、75分にわたってシアノ酸カリウム塩の濃水溶液を滴加し、その際、ポット温度を−3℃から−1℃に維持した。添加を通して、結晶(非粘着性)を分離した。スラリーを−1℃で1時間攪拌し、次いで濾過した。ケークを0℃のH2O35mLで洗浄し、N2流によりKF0.135%まで乾燥させた。結晶は、13C−NMRおよび1H−NMRにより前記表題化合物と識別された。重量18.78g(mw181.24、0.1036mol、ジトシレートからの全体収率94.0M%)。
【0169】
(実施例52)
(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの調製
(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.55、200.76g、0.4707mol)、臭化ナトリウム(mw102.89、145.3g、1.4122mol、3.00当量)および臭化テトラブチルアンモニウム(mw322.38、15.0g、0.0465mol、0.099当量)のトルエン1.5LおよびH2O84mL中の混合物を93℃に加熱した。フラスコのヘッドスペースはフォームでほぼ充満した。温度を92℃に低下させると、このことにより、フォームの量が低減した。混合物を89℃で攪拌した。反応の進行をTLCにより監視した(溶離液:30%EtOAc/シクロヘキサン;Rf[ジトシレート]=0.38、Rf[モノトシレート−一臭化物]=0.54)。t=2時間で、多くのモノトシレート−一臭化物および痕跡量のジトシレートが検出可能であった。t=6.5時間で、痕跡量のモノトシレート−一臭化物が検出可能であった。t=24時間で、ジトシレートまたはモノトシレート−一臭化物が検出されなかったので、混合物を室温に冷却し、濾過し、ケーク(トシル酸ナトリウム)をトルエン250mLで洗浄した。トルエン抽出物を合わせ、H2O(2×250mL)で洗浄し、真空濃縮(53℃/150mm)して約200mLの体積にした。生成物は、ISTD GCにより(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンとして識別された(濃度:55.9W%)。重量:204.8g(mw243.97、二臭化物114.48g[0.4693mol、収率99.7M%]含有)。
【0170】
そのままのヘキサメチルジシラザン(mw161.40、79.2g、0.4907mol、3.32当量)を−11℃に冷却し、n−ブチルリチウムのヘキサン溶液(2.5Mの溶液177mL、0.4425mol、3.00当量)で、温度が4℃未満に維持される速度で処理した(35分)。生じたスラリーを25℃に加温して、澄明な溶液を得、これを真空蒸留して粘稠なスラリー(体積約100mL)にし、−3℃に冷却し、THF130mLで処理すると、44℃までの発熱が生じた。混合物を−2℃に冷却し、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの前記で調製されたトルエン溶液の一部(mw243.97、純度55.9W%の物質64.4g、36.00g、0.1476mol)ですべて一度に処理した。混合物を−2℃で10分間攪拌し、次いで、そのままの4,4,4−トリメトキシブチロニトリル(mw159.19、37.4g、0.2349mol、1.59当量)で18分にわたって処理した。(12℃までの短時間のエクスカーション)。混合物を−2℃で攪拌した。t=1.5時間で、ISTD GC分析により、2,3−ジメチルブタジエン(0.62W%)、3−シアノプロピオン酸メチル(18.8基準M%)、二臭化物(10.0基準M%)、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(69.7基準M%)および2:1付加生成物(0.85W%)の存在が示された。t=18時間で、ISTD GC分析により、ジメチルブタジエン(1.8W%)、3−シアノプロピオン酸メチル(1.4基準M%)、二臭化物(2.3基準M%)、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(93.5基準M%)、2:1付加生成物(1.0W%)およびMeO2CCH2CH2COCH(CN)CH2CO2Me(0.57A%)の存在が示された。混合物をH2O100mLでクエンチし、25℃に加温すると、二相混合物が得られた。水性相を分離し、ヘプタン50mLで抽出した。2つの有機相を合わせると、前記表題化合物(重量328.12g)を含有する溶液が得られた。
【0171】
(実施例53)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
先行する実施例からの(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルの溶液の一部(183.84g、二臭化物0.08270molから)を1NのHCl(41.4mL、0.0414mol、0.50当量)で、続いて6NのHCl(37.7mL、0.2262mol、2.74当量)で処理して、pHを約11から約2に調節した(紙)。二相混合物を室温で1時間攪拌したが、この時点で、GCによると、オルトエステル(保持時間22.0分)から2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチル(保持時間19.7分)への加水分解が完了した。水性相を分離し、廃棄した。有機相を、50%水酸化ナトリウム水溶液(mw40.00、14.0g、水酸化ナトリウム7.0g[0.175mol、2.12当量]含有)およびH2O40mLで処理した。塩基と接触すると、有機相はオレンジ色から暗茶色に変化した。混合物を室温で3時間攪拌したが、この時点で、TLCによると、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸のナトリウム塩への加水分解が完了した。有機相を分離し、H2O20mLで抽出した。水性相を合わせ、6NのHCl(28.25mL、0.1695mol、2.05当量)を加えることにより、pHを13.2から2.1に調節し、トルエン(3×40mL)で抽出した。トルエン抽出物を合わせ、H2O40mLで洗浄して、極性の未知物質(Rf=0.02および0.06)を除去し、回転蒸発により濃縮して、粘稠な赤コハク色のオイルにしたが、これは、迅速かつ発熱性に結晶化した。結晶は、TLCによると、本質的に均一な形態の前記表題化合物と識別された(Rf=0.44、溶離液:70/20/5/5のEtOAc/MeOH/濃NH4OH/H2O)。重量:14.8317g(mw181.24、0.08183mol、二臭化物から収率99.0M%)。
【0172】
(実施例54)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
先行する実施例からの結晶2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸(mw181.24、14.8317g、0.08183mol)を、冷蔵庫で45日間貯蔵した後に、0℃のMeOH60mLと共に攪拌して、溶液を形成した。この溶液に、水酸化カリウム(mw56.11、純度87.3W%の物質11.0873g、9.6792g、0.1725mol、2.11当量)のMeOH40mL溶液を加えた。モリブデン促進スポンジニッケルの水性スラリー(Johnson Matthey A−7000、乾燥重量5.73g[シアノ酸カリウム塩0.32g/g])を、300mLステンレス鋼オートクレーブにMeOH60mLと共に入れた。容器を密封し、H2でパージし、H2で50psigに加圧し、30℃に加熱した。前記シアノ酸カリウム塩のMeOH溶液(mw219.33、0.08183mol、17.9478g)を水素化容器に2.7時間にわたって計量供給した。混合物を30℃/H250psigに保持した。20時間後に、摂取は本質的に止んだ。
【0173】
全部で44時間後に、混合物を真空濾過し、触媒をMeOHですすいだ。合わせた濾液および洗浄液(重量:249.10g)をH2O33mLで処理し、回転蒸発により濃縮し、H2O28mLで処理し、さらに濃縮して、51gの重量にした。この溶液(pH14.0)を氷酢酸(mw60.05、10.88g、0.1812mol、2.21当量)で処理して、スラリーを得て、これを0〜5℃に冷却して、1時間攪拌し、濾過し、N2流により乾燥させた。TLCにより、結晶は、本質的に純粋な形態の前記表題化合物と識別された(溶離液:70/20/5/5のEtOAc/MeOH/濃NH4OH/H2O;Rf[ヒドロキシ酸]=0.34、Rf=[表題化合物]=0.20)。重量:15.601g。
【0174】
粗製の前記表題化合物の一部(15.48g、シアノ酸0.08120molから)を、イソプロパノール155mLに懸濁させ、80℃(浴温度)で30分間攪拌し、次いで、0〜5℃に冷却し、1時間攪拌し、次いで、濾過した。濾液を使用して、固体をフラスコから洗浄した。ケークをN2流により乾燥させた。重量:10.791g。濾液を回転蒸発により濃縮して、固体(重量:4.299g)にした。
【0175】
粉砕された前記表題化合物の一部(10.67g、シアノ酸0.08029molから)を25%(v/v)のiPrOH/H2O53mLに懸濁させ、80℃に加熱した。固体が完全には溶けなかったので、イソプロパノール3.6mLを加え、温度を87℃に上げた。固体が完全に溶けて、金コハク色の溶液を形成した。溶液を0〜5℃に冷却し(温度が78℃であったときには、固体が結晶化した)、2時間攪拌し、濾過した。ケークを濾液で洗浄し、N2流により乾燥させた。LCにより、生成物は前記表題化合物と識別された(濃度99.8W%)。結晶は、後記溶離ピークの0.18A%(RRT1.84)および初期溶離ピークの0.12W%(RRT0.48)を除いて、0.1W%を超える不純物は含有しなかった。金属スクリーニングにより、Ni(4.2ppm)およびMo(0.6ppm)が検出された。重量:9.634g(mw185.27、9.615g、0.05190mol、シアノ酸からの収率64.6M%)。
【0176】
(実施例55)
(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オンの単離
機械式攪拌機を備えていて、87mLレベルにマーキングされている1L四つ口丸底フラスコに、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(理論=17.2g、103mmol)のメタノール溶液を充填した。混合物を真空蒸留により濃縮して、87mLにした。残渣を2回、酢酸エチル(170mL)で希釈し、再蒸留して87mLにした。生じた有機溶液を初めに1NのHCl(87mL)で、次いで、1NのNaOH(87mL)で抽出した。水性相を連続して酢酸エチル(45mL)で洗浄した。合わせた有機相を、1L四つ口丸底フラスコに分枝鎖オクタン(170mL)と共に充填した。混合物を真空蒸留により濃縮して、87mLにした。留出物を分枝鎖オクタン(170mL)で希釈し、再び真空蒸留により濃縮して87mLにした。この希釈/蒸留手順をさらに2回繰り返した。生じた溶液87mLを周囲温度に1時間にわたって冷却したが、この間に、結晶が形成した。生じたスラリーを約0℃に冷却し、30分間攪拌し、その後、真空濾過した。回収された結晶を冷分枝鎖オクタン(3×25mL)で洗浄し、真空下に65℃で1時間乾燥させると、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(11.6g、67%)が得られた。
【0177】
(実施例56)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
頭上攪拌機、還流凝縮器およびPTFEコーティングされたサーモカップルを備えている500mL四つ口丸底フラスコ(RBF)に、(7S,8S)−7,8−ジメチル−2−アザ−スピロ[4.4]ノナン−3−オン(20.0g、0.12mol、1.0当量)、HCl(37%w/w、35.0g、0.35mol、3.0当量)および水(20.0g)を窒素ブランケット下に充填した。混合物を90℃に加熱し、反応が完了するまで攪拌した(HPLCによると変換率97%、典型的には24時間以内)。生じた溶液を50から60℃の温度に冷却し、トルエン(2×30mL)で抽出した。トルエン洗浄液を廃棄した。水性相を50%NaOH(約19g、0.23mol、2.0当量)で、pH2.0に調節した。活性化炭素(4.0g)、濾過剤(4.0g)および水(10g)をフラスコに加え、内容物を50℃で30分間攪拌した。スラリーを60℃に加熱し、粗いフリットおよび0.5μmPTFE膜で濾過したが、その間、スラリーを>55℃の温度に維持した。フィルターケークを、>50℃の温度に加熱されていた水(25mL)ですすいだ。合わせた水性層を温度<40℃に冷却し、真空蒸留して、約70mLの最終体積にした。生成物を含有する濃縮溶液を、50%NaOH(約8.2g、0.1mol、0.9当量)でpH6.5から7.5に調節すると、沈殿物が形成した。混合物を氷浴で温度<5℃に冷却した。生じたスラリーを温度<5℃で60分間攪拌し、次いで、濾過して、粗製(固体)生成物を単離した。ケークをまだ湿っている間に水(20g)ですすぎ、次いで、フィルター上で24から48時間乾燥させた。
【0178】
粗製(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(20g)およびイソプロパノール/水(40重量%i−PrOH水溶液125mL)を、頭上攪拌、還流凝縮器およびサーモカップルを備えた500mL四つ口RBFに窒素ブランケット下に充填した。スラリーを還流(約85℃)に加熱した。溶液を15分間攪拌し、窒素圧力を使用して、0.5μmPTFE膜および焼結ガラスフリット(60℃に加熱されていた)で濾過することにより澄明にした。イソプロパノール/水(40wt%i−PrOH水溶液20mL)をフラスコに充填し、還流まで加熱した。すすぎ液を膜およびフリットを介して窒素圧力で移し、生成物溶液濾液と合わせた。合わせた濾液を、頭上攪拌、還流凝縮器およびサーモカップルを備えていて、予めマーキングされている(102mLの所)500mL四つ口RBFに窒素ブランケット下に移した。溶液を<40℃に冷却した。スラリーを40から50℃で真空蒸留して、102mLの全体積にした。イソプロパノール内容物を24から27重量%i−PrOHに調節した。スラリーを還流(約87℃)に再加熱し、固体がすべて溶解するまで保持した。溶液を20℃/hの速度で5℃まで徐々に冷却し、60分間保持して、生成物を沈殿させた。最終(固体)生成物を真空濾過により単離し、イソプロパノール(30mL、<5℃に冷却)で洗浄した。フィルターケークを40℃で真空下に24時間乾燥させて、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(21.0g、95%)を得た。
【0179】
(実施例57)
(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタンの調製
乾燥不活性容器に、(R,R)−2,3−ジメチル−1,4−ビス−(トルエン−4−スルホニルオキシ)−ブタン(mw426.56、100kg、234mol、1.00当量)、臭化カリウム(mw119.00、60Kg、504mol、2.15当量)およびトルエン180Lを充填した。スラリーを攪拌し、水6L中の臭化テトラブチルアンモニウム(mw322.37、6kg、18.60mol、0.08当量)を加えた。スラリーを89〜93℃に少なくとも8時間加熱し、次いで、60〜65℃に冷却し、トルエン50Lおよび水130Lを加えた。水性相を除去し、有機相を50℃で、水50Lポーションで2回洗浄した。有機相を含有する生じた生成物を25℃未満に冷却した。真空を適用し、有機溶液を蒸留して、(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(57.1kg、234mol)を含有する約50wt%溶液にした。
【0180】
(実施例58)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの調製
乾燥不活性容器に、リチウムヘキサメチルジシラジドのTHF溶液(mw167.33、24重量%、500Kg、3.3当量)を充填した。溶液を真空蒸留して、その元の体積の約50%にした。先行する実施例からの(2R,3R)−1,4−ジブロモ−2,3−ジメチルブタン(mw243.97、57.1Kg、234mol、1当量)のトルエン溶液を加え、その際、温度を5℃未満に維持した。4,4,4−トリメトキシブチロニトリル(mw159.19、51.0Kg、320mol、1.4当量)を6時間にわたって加え、その際、温度を0から5℃に維持した。GCにより、反応が完了したと思われたら、水15Lを制御添加することにより、反応をクエンチし、次いで、水135Lを加え、混合物を20℃に加温した。二相混合物を分離し、下部水性相を廃棄した。THF溶媒を、全部で300Lのn−ヘプタンの3回の真空蒸留追跡により置換した。次いで、(3S,4S)−3,4−ジメチル−1−(2,2,2−トリメトキシエチル)シクロペンタンカルボニトリルを含有する有機混合物を水100Lポーションで2回洗浄し、水60Lおよびトルエン5Lを加え、混合物を激しく攪拌した。濃塩酸(mw36.46;37%溶液35kg、355mol)の制御添加を行って、pHをpH3未満に調節し、混合物を30分間攪拌した。下部水性相を除去し、水洗浄を行い、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルを含有する有機溶液を次のステップに入れた。
【0181】
(実施例59)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の調製
先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸メチルの溶液を45wt%水酸化カリウム水溶液(mw56.11、溶液29Kg、233mol)で約1時間にわたって処理し、次いで、けん化が完了するまで、さらに1時間攪拌した。下部水性相を除去し、有機相を水で洗浄し、水性相を合わせた。前記表題化合物を含有する水溶液の重量は、約100Kgであった。
【0182】
(実施例60)
(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸の調製
水素化オートクレーブに、モリブデン促進スポンジニッケル(Johnson Matthey A−7000、4Kg)、45wt%水酸化カリウム水溶液(mw56.11、11Kg、88.21mol)および水20Lを充填した。これに、先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の溶液約50Kgを加えた。溶液を水素で50psigで処理したが、この間、温度を30℃に保持した。反応は12時間後に完了し、触媒を濾過により除去した。先行する実施例からの2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の溶液の残りの50Kgを同じ方法で水素化した。2つの水素化溶液を合わせ、L−酒石酸(mw150.09、2.5Kg、16.66mol)を含有するタンクに充填し、酸が溶けるまで攪拌した。溶液のpHを、濃塩酸(mw36.46、37%溶液35Kg、355mol)で約pH11.0に調節したが、この際、温度を20〜25℃に維持した。この時点で、粗製生成物が沈殿し始めた。次いで、混合物を、氷酢酸(mw60.05、2.5Kg、41.67mol)でpH6〜8に調節した。粗製生成物を濾過により単離し、プロパン−2−オール92Kgで洗浄し、40℃窒素で乾燥させると、粗製の(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、37Kg、199mol)が得られた。
【0183】
粗製の(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、37Kg、199mol)を約80℃で、2−プロパノール130Lおよび水130Lの混合物に溶かし、溶液を研磨フィルターで濾過した。溶液を50〜55℃に冷却したが、この際、生成物が結晶化し始めた。さらに20℃に冷却した後に、スラリーを真空蒸留したが、この間に、2−プロパノール35Kgポーションを2回加えて、水と置換した。約4L/Kgの最終蒸留体積に達したら、スラリーを0〜5℃に冷却し、濾過した。フィルターケークを2−プロパノール40Kgで洗浄し、40℃窒素で乾燥させると、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸(mw185.27、30Kg、161.91mol)が得られた。
【0184】
(実施例61)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の調製
清潔な不活性容器に、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩(mw219.3、48%溶液106Kg、232mol)および水35Lの溶液を充填した。溶液を攪拌し、真空蒸留して、35Lまで体積を低減し、次いで、温度を0〜5℃に維持しながら、5.7Mの塩酸水溶液92.8Kgを含有する容器に徐々に移し、2−((3S、4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の結晶をシーディングした。さらなる水10Lを加え、移動が完了したら、生じたスラリーを0℃で1時間攪拌し、濾過し、冷水75Lで洗浄した。ケークを窒素掃引で約1時間、部分的に乾燥させると、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸の水湿潤ケーク86.82Kgが得られた。
【0185】
(実施例62)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩の調製
先行する実施例からの水で湿潤させた2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸86Kgを、45wt%水酸化カリウム水溶液(mw56.11、溶液63.2Kg、506mol)で約1時間にわたって処理すると、2((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩を含有する均一な溶液が得られた。
【0186】
(実施例63)
2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸tert−ブチルアミン塩の調製
窒素入口および機械式頭上攪拌機を備えた250mL三つ口丸底フラスコ(RBF)に、2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸カリウム塩(mw219.3、13.2g、0.06mol)を含有する水溶液25gおよび水酸化カリウム(mw56.11、0.68g、0.012mol)、水25mLおよびトルエン55mLを充填した。塩酸水溶液(mw36.43、37%溶液7.3g、0.075mol)を約3分にわたって加えて、pHをpH2未満に調節した。5分間攪拌した後に、相を分離し、下部水性相を除去した。生成物を含有する有機相の20mL水洗浄を行い、生じたトルエン溶液に、2−プロパノール10mLを加えた。溶液を20〜25℃に維持して、tert−ブチルアミン(mw73.14、5.2g、0.07mol、1.2当量)を約5分にわたって加えた。生じたスラリーを75℃に加温すると、濁った混合物が得られた。約20℃/時間の速度で5℃に冷却すると、塩が沈殿した。0〜5℃で1時間攪拌した後に、固体を濾過し、トルエン50mLで洗浄し、真空炉中40℃で乾燥させると、固体の2−((3S,4S)−1−シアノ−3,4−ジメチルシクロペンチル)酢酸tert−ブチルアミン塩(mw254.1、12.8g、0.05mol、83%)が得られた。
【0187】
本明細書および添付の請求項において使用される場合、「a」、「an」および「the」などの単数冠詞は、文脈で他に明確に記載されていない限り、1個の対象または複数の対象を指しうることを特記する。したがって例えば、「(a)化合物」を含む組成物に関する言及は、単一化合物または2種以上の化合物を含みうる。
【0188】
前記記載は、詳述を意図しており、制限的ではないことを意図していることを理解されたい。前記記載を読めば、当業者には、多くの実施形態が明らかであろう。したがって本発明の範囲は、添付の請求項に関する言及と共に、このような請求項が権利を有する同等の全範囲により決定されるべきである。
【図面の簡単な説明】
【0189】
【図1】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(全スペクトル)。
【図2】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(フィンガープリント領域)。
【図3】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(全スペクトル)。
【図4】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(フィンガープリント領域)。
【図5】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(全スペクトル)。
【図6】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−IRスペクトル(フィンガープリント領域)。
【図7】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(全スペクトル)。
【図8】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのFT−ラマンスペクトル(フィンガープリント領域)。
【図9】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの粉末X線回折(PXRD)パターン(フルスケール)。
【図10】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(30000cpsまで)。
【図11】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(フルスケール)。
【図12】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのPXRDパターン(30000cpsまで)。
【図13】形態Aの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンの示差走査熱分析(DSC)サーモグラム。
【図14】形態Bの(R,R)−1,4−ジ−p−トルエンスルホニルオキシ−2,3−ジメチル−ブタンのDSCサーモグラム。
【特許請求の範囲】
【請求項1】
式1の化合物もしくは薬学的に許容できるその塩または式1の化合物の反対の鏡像異性体もしくは薬学的に許容できるその塩を製造する方法であって:
【化1】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]、
前記方法が、
(a)式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩のシアノ部分をアミノ部分に還元するステップと
【化2】
[式5中のR1およびR2は、式1に関する前記と同様に定義され、R4は水素原子およびC1〜6アルキルから選択される]、
(b)任意選択で、式1の化合物またはその反対の鏡像異性体を式1の化合物またはその反対の鏡像異性体の薬学的に許容できる塩に変換するステップを含む方法。
【請求項2】
式6の化合物またはその反対の鏡像異性体:
【化3】
[式6中のR1およびR2は、式1に関する前記と同様に定義される]
を加水分解するステップをさらに含む請求項1に記載の方法。
【請求項3】
式5の化合物を加水分解するステップをさらに含む請求項1に記載の方法。
【請求項4】
式4の化合物またはその反対の鏡像異性体:
【化4】
[式4中のR1およびR2は、式1に関する前記と同様に定義され、式4中のR3はカルボン酸またはエステルのための保護基である]
を脱保護して、式5の化合物またはその反対の鏡像異性体を得るステップをさらに含む請求項1から3のいずれか一項に記載の方法。
【請求項5】
式2の化合物またはその反対の鏡像異性体:
【化5】
を式3の化合物:
【化6】
と塩基の存在下に反応させて、式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩を得るステップをさらに含む請求項4に記載の方法
[式2中のR1およびR2は、式1に関する前記と同様に定義され、式2中のX1は脱離基であり、式3中のR3は式4に関する前記と同様に定義される]。
【請求項6】
式4の化合物またはその反対の鏡像異性体:
【化7】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよく、
R3は、
【化8】
により表される構造を有するカルボン酸またはエステル保護基である
(式7および式8中の「A」は、式4の化合物の残りの部分に結合している結合点を表し、
R6、R7、R8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成し、
Z1およびZ2はそれぞれ独立に、OおよびSから選択され、
R9は、C1〜6アルキルであり、
R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、
R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する)]。
【請求項7】
(S,S)−3,4−ジメチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジメチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジメチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
から選択される請求項6に記載の化合物、前記化合物の反対の鏡像異性体ならびに前記化合物およびその反対の鏡像異性体の塩。
【請求項8】
式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩:
【化9】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、
R4は、H、C1〜6アルキル、アリール−C1〜3アルキル、第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオンから選択され、
ここで、前記各アリール−C1〜3アルキル基中のアリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6−アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]。
【請求項9】
(3S,4S)−(3,4−ジメチル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジエチル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジプロピル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジベンジル−1−シアノ−シクロペンチル)−酢酸;
から選択される請求項8に記載の化合物、
前記酸のメチル、エチル、プロピル、イソプロピルおよびベンジルエステル、
前記酸の塩ならびに
前記酸、塩およびエステルの反対の鏡像異性体。
【請求項1】
式1の化合物もしくは薬学的に許容できるその塩または式1の化合物の反対の鏡像異性体もしくは薬学的に許容できるその塩を製造する方法であって:
【化1】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]、
前記方法が、
(a)式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩のシアノ部分をアミノ部分に還元するステップと
【化2】
[式5中のR1およびR2は、式1に関する前記と同様に定義され、R4は水素原子およびC1〜6アルキルから選択される]、
(b)任意選択で、式1の化合物またはその反対の鏡像異性体を式1の化合物またはその反対の鏡像異性体の薬学的に許容できる塩に変換するステップを含む方法。
【請求項2】
式6の化合物またはその反対の鏡像異性体:
【化3】
[式6中のR1およびR2は、式1に関する前記と同様に定義される]
を加水分解するステップをさらに含む請求項1に記載の方法。
【請求項3】
式5の化合物を加水分解するステップをさらに含む請求項1に記載の方法。
【請求項4】
式4の化合物またはその反対の鏡像異性体:
【化4】
[式4中のR1およびR2は、式1に関する前記と同様に定義され、式4中のR3はカルボン酸またはエステルのための保護基である]
を脱保護して、式5の化合物またはその反対の鏡像異性体を得るステップをさらに含む請求項1から3のいずれか一項に記載の方法。
【請求項5】
式2の化合物またはその反対の鏡像異性体:
【化5】
を式3の化合物:
【化6】
と塩基の存在下に反応させて、式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩を得るステップをさらに含む請求項4に記載の方法
[式2中のR1およびR2は、式1に関する前記と同様に定義され、式2中のX1は脱離基であり、式3中のR3は式4に関する前記と同様に定義される]。
【請求項6】
式4の化合物またはその反対の鏡像異性体:
【化7】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、ここで、アリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよく、
R3は、
【化8】
により表される構造を有するカルボン酸またはエステル保護基である
(式7および式8中の「A」は、式4の化合物の残りの部分に結合している結合点を表し、
R6、R7、R8はそれぞれ独立に、C1〜6アルキルであるか、それらが結合している原子と一緒になって、酸素および炭素環員のみを有する二環式複素環を形成し、
Z1およびZ2はそれぞれ独立に、OおよびSから選択され、
R9は、C1〜6アルキルであり、
R10は、C1〜6アルキル、シリルおよびC1〜6アルキルシリルから選択されるか、
R9およびR10は、それらが結合している原子と一緒になって、炭素および酸素環員のみ、炭素およびイオウ環員のみ、または炭素、酸素およびイオウ環員のみを有する単環式複素環を形成する)]。
【請求項7】
(S,S)−3,4−ジメチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジメチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジメチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジエチル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジプロピル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(4−メチル−2,6,7−トリオキサ−ビシクロ[2.2.2]オクト−1−イルメチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(2,2,2−トリメトキシ−エチル)−シクロペンタンカルボニトリル;
(S,S)−3,4−ジベンジル−1−(2,2,2−トリエトキシ−エチル)−シクロペンタンカルボニトリル;
から選択される請求項6に記載の化合物、前記化合物の反対の鏡像異性体ならびに前記化合物およびその反対の鏡像異性体の塩。
【請求項8】
式5の化合物もしくはその反対の鏡像異性体または式5の化合物もしくはその反対の鏡像異性体の塩:
【化9】
[式中、
R1およびR2はそれぞれ独立に、C1〜6アルキル、C2〜6アルケニル、C2〜6アルキニル、C3〜6シクロアルキル、C3〜6シクロアルケニル、C3〜6シクロアルキル−C1〜3アルキル、C3〜6シクロアルケニル−C1〜3アルキルまたはアリール−C1〜3アルキルであり、
R4は、H、C1〜6アルキル、アリール−C1〜3アルキル、第1族金属イオン、第2族金属イオン、第1級アンモニウムイオンまたは第2級アンモニウムイオンから選択され、
ここで、前記各アリール−C1〜3アルキル基中のアリールは、C1〜6アルキル、C1〜6アルコキシ、C1〜6−アルコキシカルボニル、カルボキシ、ヒドロキシ、ハロゲノ、フルオロ−C1〜6アルキルおよびニトロから選択される1から3個の置換基で置換されていてもよい]。
【請求項9】
(3S,4S)−(3,4−ジメチル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジエチル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジプロピル−1−シアノ−シクロペンチル)−酢酸;
(3S,4S)−(3,4−ジベンジル−1−シアノ−シクロペンチル)−酢酸;
から選択される請求項8に記載の化合物、
前記酸のメチル、エチル、プロピル、イソプロピルおよびベンジルエステル、
前記酸の塩ならびに
前記酸、塩およびエステルの反対の鏡像異性体。
【図1】

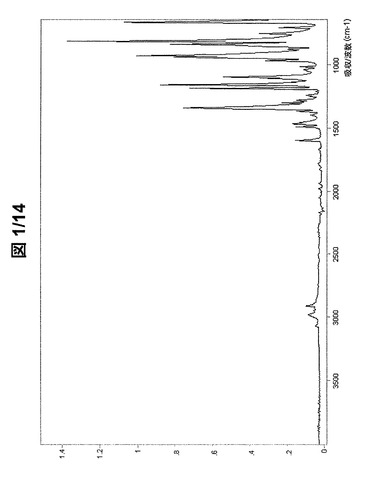
【図2】
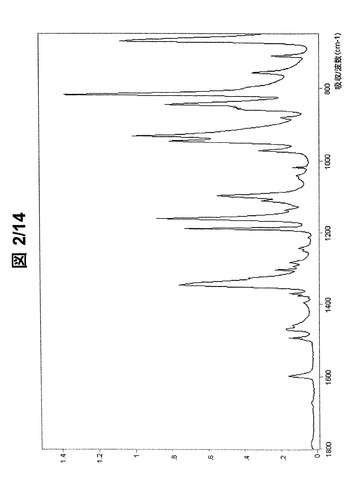
【図3】
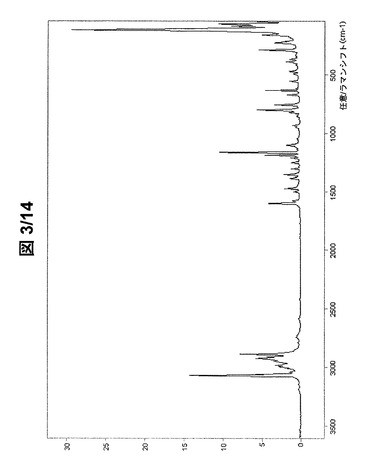
【図4】
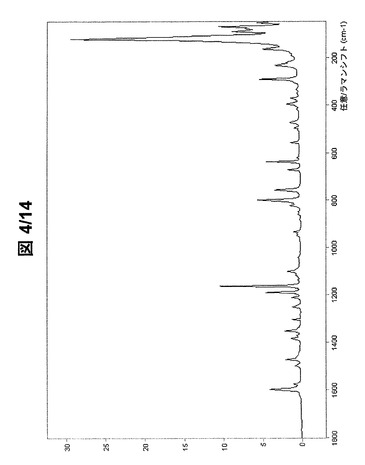
【図5】
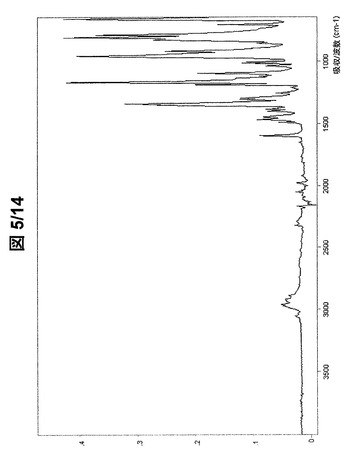
【図6】
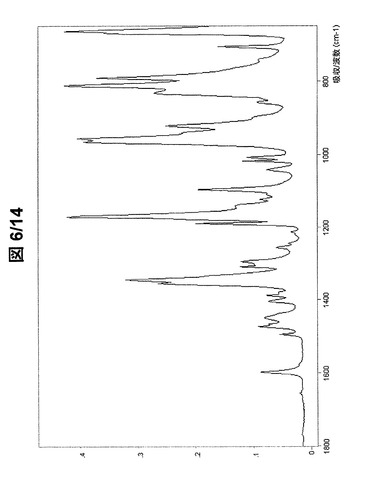
【図7】
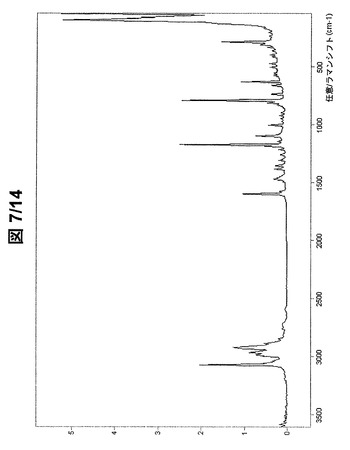
【図8】
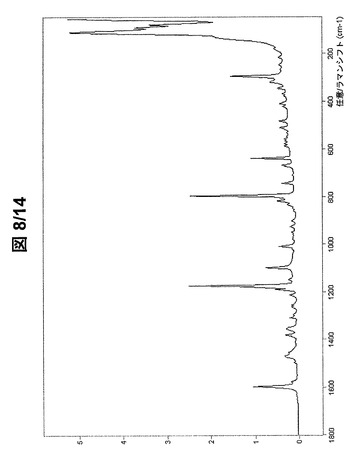
【図9】
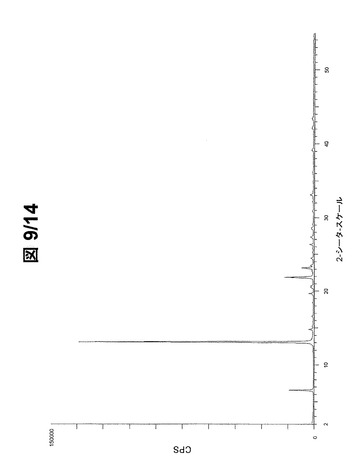
【図10】
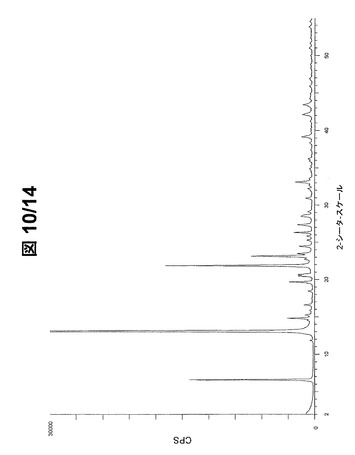
【図11】
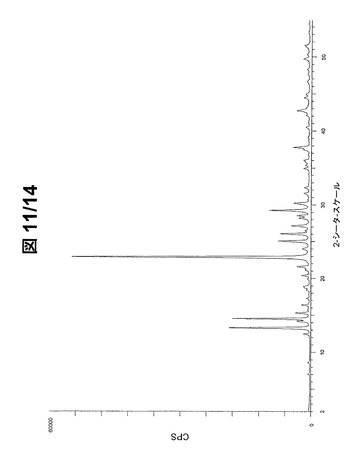
【図12】
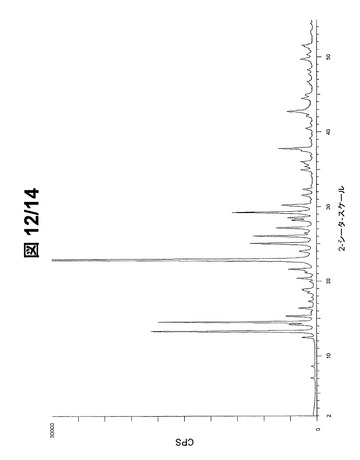
【図13】
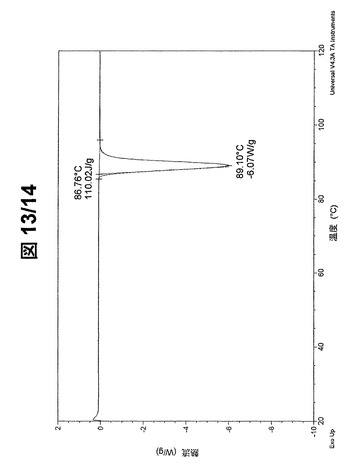
【図14】
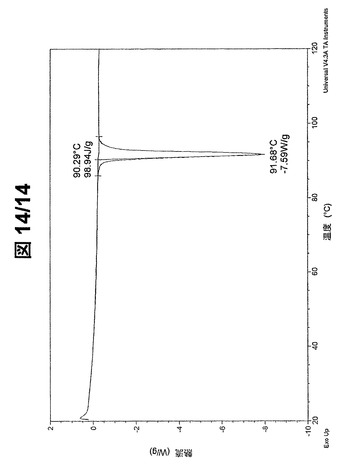
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2009−108024(P2009−108024A)
【公開日】平成21年5月21日(2009.5.21)
【国際特許分類】
【外国語出願】
【出願番号】特願2008−158803(P2008−158803)
【出願日】平成20年6月18日(2008.6.18)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
【公開日】平成21年5月21日(2009.5.21)
【国際特許分類】
【出願番号】特願2008−158803(P2008−158803)
【出願日】平成20年6月18日(2008.6.18)
【出願人】(593141953)ファイザー・インク (302)
【Fターム(参考)】
[ Back to top ]