
分散物及び脳神経変性疾患治療薬
【課題】本発明は、複合粒子を有する分散物を提供することを課題とする。
【解決手段】疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
【解決手段】疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、分散物及び脳神経変性疾患治療薬に関する。
【背景技術】
【0002】
脳は、精神活動と運動制御に関わっており、特定の領域に各機能が局在している。例えば、大脳皮質には思考、記憶、知覚及び言語の機能を担う領域があり、線条体や小脳には主に体性運動や体運動の機能調整を担う領域がある。近年、生活習慣の乱れなどによって、これらの脳領域の恒常性の破綻に伴う、アルツハイマー型認知症などの脳神経変性疾患やうつ病などの精神疾患が増加の一途を辿っている。そのため、疾患治療に向けた薬物輸送システムの技術開発が望まれている。
【0003】
脳は、その恒常性を維持するため、血液脳関門(Blood-Brain-Barrier(以下「BBB」ともいう。))を介して、血液中から脳内に取り込む物質を厳密に制御している。そのため、アルツハイマー型認知症などの脳神経変性疾患の診断や治療のための薬剤の開発においては、BBB透過性を向上させることが課題であり、脳神経変性疾患領域に薬剤を確実に蓄積できるようにデザインする必要がある。その手段の一つとして、人工低密度リポタンパク質キャリアを利用した薬物輸送システムとして、リポソーム表面にアポリポタンパク質E(ApoE:ApolipoproteinE)が結合した薬剤内包リポソームが開発されている(例えば、特許文献1参照)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特表2006−516539号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、前述の薬剤内包リポソームは、その表面にApoEが結合しているものの、該ApoEを認識する低密度リポタンパク質受容体(LDLR:Low Density Lipoprotein Receptor)は、主に肝臓で発現しているため、薬剤内包リポソームは肝臓に多く蓄積すると考えられる。また、ApoEの分子種の一つであるApoE2はLDLRでは認識されない。さらに、BBBを形成している脳血管内皮細胞は、LDLRではなく、LDLRファミリーの一つである低密度リポタンパク質受容体関連タンパク質-8(LRP-8:Lipoprotein Receptor related Protein-8)が主に発現している。また、粒径1nm〜100nmの物質のBBBにおける輸送は、クラスリン被覆ピットやカベオラ被覆ピットが関与していると言われている。そのため、前記薬剤内包リポソーム表面に該ApoEが存在する人工低密度リポタンパク質というだけでは、該キャリアのBBB透過性の効率が一概に上昇するとは限らない。このため、現在においてもBBB透過性を示す薬剤キャリアの更なる開発が待ち望まれている。また、現時点において、脳神経変性疾患領域に薬物を蓄積させることが可能なキャリアは開発されていない。
【0006】
本発明は、上記の状況に鑑みてなされたものであり、以下の目的を達成することを課題とする。
すなわち、本発明は、BBB透過性が良好であるとともに、脳組織の局所に蓄積することが可能な分散物を提供することを課題とする。また、本発明は、該分散物を利用した、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬を提供することを課題とする。さらに、本発明は、該分散物の製造方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、前記の課題を抜本的に解決するため、詳細な実験・検討を行った結果、特定の成分で調製された複合粒子がBBB透過性を持ち、脳組織内の特定領域への局在化を可能とする知見を得た。本発明は、かかる知見に基づいて達成されたものである。
前記の課題を解決するための具体的な手段は以下の通りである。
<1> 疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
<2> 前記複合粒子がリン脂質及びアポリポタンパク質を疎水性粒子表面に有する<1>に記載の複合粒子を有する分散物。
<3> 前記リン脂質の複合粒子の全質量に対する含有量が、0.2質量%〜20質量%である<1>又は<2>に記載の複合粒子を有する分散物。
<4> 前記アポリポタンパク質が、0.1μg/ml〜10μg/mlの量で含まれる<1>〜<3>のいずれかに記載の複合粒子を有する分散物。
<5> 前記アポリポタンパク質が、アポリポタンパク質A1、アポリポタンパク質B100及びアポリポタンパク質E4からなる群より選ばれる少なくとも1種のアポリポタンパク質である<1>〜<4>のいずれかに記載の複合粒子を有する分散物。
<6> 前記疎水性粒子が、金属ナノ粒子又は生分解性ポリマーである<1>〜<5>のいずれかに記載の複合粒子を有する分散物。
<7> 前記疎水性粒子の粒径が1nm〜100nmである<1>〜<6>のいずれかに記載の複合粒子を有する分散物。
<8> 前記複合粒子の粒径が1nm〜100nmである<1>〜<7>のいずれかに記載の複合粒子を有する分散物。
<9> 前記疎水性粒子が、生分解性ポリマーであり、かつ該生分解性ポリマーが薬物を内包している<1>〜<8>のいずれかに記載の複合粒子を有する分散物。
<10> <9>に記載の複合粒子を有する分散物を有効成分として含有する脳神経変性疾患治療薬。
<11> 脳神経変性疾患が、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症からなる群より選ばれる少なくとも1種の疾患である<10>に記載の脳神経変性疾患治療薬。
<12> <1>〜<9>のいずれかに記載の複合粒子を有する分散物を含有する医薬組成物。
<13> <1>〜<9>のいずれかに記載の複合粒子を有する分散物を含有する造影補助試薬。
<14> 疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む複合粒子の分散物の製造方法。
<15> 疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む製造方法により得られる複合粒子の分散物。
【発明の効果】
【0008】
本発明によれば、BBB透過性が良好であるとともに、脳組織内の特定領域への局在化を可能とする分散物を提供することができる。また、本発明によれば、該分散物を利用した、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬を提供することができる。さらに、本発明によれば、該分散物の製造方法を提供することができる。
【図面の簡単な説明】
【0009】
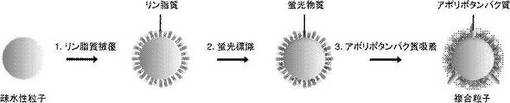
【図1】本実施例にかかる複合粒子の作製手順を示した模式図である。
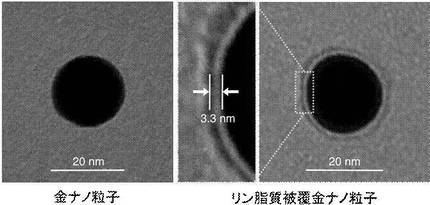
【図2】本実施例にかかる金ナノ粒子及びリン脂質被覆金ナノ粒子を観察した透過型電子顕微鏡(TEM:Transmission Electron Microscopy)像である。
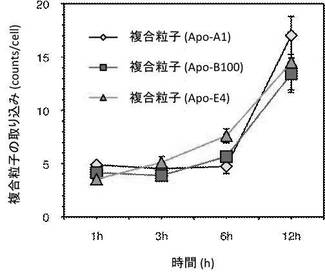
【図3】本実施例にかかる各複合粒子(Apo-A1、Apo-B100、Apo-E4)の脳血管内皮細胞における時間経過に伴う取り込み量を示したグラフである。
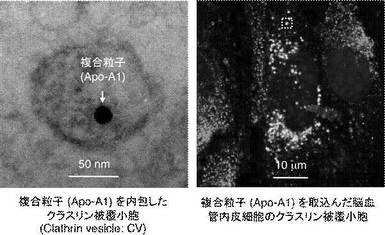
【図4】本実施例にかかる複合粒子(Apo-A1)が脳血管内皮細胞のクラスリン被覆小胞内に存在しているところを観察したTEM像並びにレーザー共焦点顕微鏡像である。
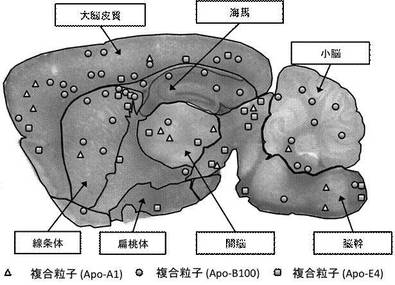
【図5】各複合粒子(Apo-A1、Apo-B100、Apo-E4)の各脳領域における分布図である。
【図6】脳連続切片(100枚)における複合粒子(Apo-B100)を取込んだ脳神経細胞数を示したグラフである。
【図7】複合粒子(Apo-B100)を取込んだ脳領域別における脳神経細胞数を示したグラフである。
【図8】複合粒子(Apo-B100)を取込んだ終脳の領域別における脳神経細胞数を示したグラフである。
【発明を実施するための形態】
【0010】
以下、本発明の分散物、分散物の製造方法、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬について詳述する。
以下に記載する構成要件の説明は、本発明の代表的な実施態様に基づいてなされることがあるが、本発明はそのような実施態様に限定されるものではない。
なお、本発明において「〜」を用いて表される数値範囲は、「〜」の前後に記載される数値を下限値及び上限値として含む範囲を意味する。また、本明細書において「工程」との語は、独立した工程だけではなく、他の工程と明確に区別できない場合であってもその工程の所期の作用が達成されれば、本用語に含まれる。
【0011】
本発明は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物に関する。
本発明にかかる複合粒子は、粒径1nm〜100nmの疎水性粒子と、特定量のリン脂質及びアポリポタンパク質を含む構成としたことで、ナノサイズの疎水性粒子が、生体内で安定に分散でき、かつBBBにおける物質の輸送機構に関わる脳血管内皮細胞のクラスリン被覆ピットやカベオラ被覆ピットに認識されるようになった。このメカニズムにより、良好なBBB透過性を実現すると同時に、脳組織内の特定領域への局在化を可能とする複合粒子を提供するものである。
【0012】
≪分散物≫
本発明の分散物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有しており、必要に応じて他の成分を含んでいてもよい。
本発明の分散物は、水性相に複合粒子が分散しているものである。
以下、複合粒子を構成する成分である疎水性粒子、リン脂質、アポリポタンパク質及びその他の成分、並びに水性相を構成する成分について説明する。
<疎水性粒子>
本発明にかかる複合粒子に含まれる疎水性粒子としては、粒子と水などの媒質との間の親和性の強弱により、疎水性の傾向を示すコロイドであり、粒子表面の帯電に起因する静電気的な反発及びブラウン運動の能力を有する粒子である限り、特に制限はなく、公知のものを利用することができる。具体的には、金属ナノ粒子、生分解性ポリマー、デンドリマー、量子ドット、ラテックス粒子、シリカ粒子、疎水性アミノ酸を含有するタンパク質粒子(例えば、アルブミン粒子)等が挙げられ、脳神経変性疾患治療薬や医薬組成物及び造影補助試薬を提供することの観点より、金属ナノ粒子又は生分解性ポリマーが好ましい。
【0013】
また、金属ナノ粒子に使用しうる金属元素としては、アルカリ金属(周期律表第1族元素)、アルカリ土類金属(周期律表第2族元素)、遷移金属等が挙げられ、中でも金又は磁性体(酸化鉄、酸化クロム、コバルト、フェライト)が好ましく、生体への影響などの観点より、金がより好ましい。
【0014】
中でも、疎水性粒子としては、所望する粒径及び均一サイズの粒子を容易に作製が可能であり、リン脂質との親和性に優れ、大きな電気陰性度を持つことや水溶液中で安定に分散できることなどの観点より、金ナノ粒子がより好ましい。
【0015】
生分解性ポリマーとしては、Poly-ε-caprolactone(PCL)、Poly(lactic acid)(PLA)、Poly (glycolic acid)(PGA)、Ester-terminated poly(DL-lactic-co-glycolic acid) (PLGA)等が挙げられ、生分解性ポリマーの分解速度による薬物放出量の制御や薬物の運搬性及び生体による利用効率(バイオアベイラビィリティ)の観点より、PLGA又はPLAが好ましく、PLGAがより好ましい。
【0016】
前記疎水性粒子の粒径は、1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
1nm〜100nmの範囲とすることで、脳血管内皮細胞表面に存在しているクラスリン被覆ピット (サイズ:100nm〜150nm)やカベオラ被覆ピット(サイズ:50nm〜80nm)を介したエンドサイトーシスが容易となるため好ましい。
【0017】
前記疎水性粒子が、金属ナノ粒子である場合には、その粒径は1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、肝臓における非特異的な蓄積の低減の観点から、1nm〜20nmであることがさらに好ましい。
【0018】
前記疎水性粒子が、生分解性ポリマーである場合には、その粒径は1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
前記疎水性粒子が、生分解性ポリマーである場合には、その粒径は、生体に存在する粒径5nm〜15nmの高密度リポタンパク質(HDL:High Density Lipoprotein)や粒径18nm〜28nmの低密度リポタンパク質(LDL:Low Density Lipoprotein )と同等サイズであるとの観点から、1nm〜100nmであることが好ましい。
【0019】
疎水性粒子の粒径は、疎水性粒子の種類に応じた公知の方法により、適宜所望の大きさに調整することができる。例えば、金ナノ粒子の場合には、塩化金酸塩溶液の還元によるファラデー法などにより、粒径を調整することが可能である。
本明細書において、粒径とは、特に断らない限り、平均粒子径を表す。平均粒子径は、TEM(日本電子社製)等により、任意に500個の粒子を選択して、個別に粒子サイズを測定して、その測定値の算術平均値をとったものである。
【0020】
なお、前記疎水性粒子の製造方法としては、疎水性粒子の種類に応じた公知の方法を使用し、製造することができる。
例えば、金ナノ粒子は、Surface Science. 1982, 120, 435−455などに記載の方法により製造することができる。
また、生分解性ポリマーであるPLGAは、Langmuir 2010, 26(22), 16958−16962などに記載の方法により製造することができる。
また、本発明において疎水性粒子は、市販されているものを使用することもできる。
【0021】
<リン脂質>
本発明にかかる複合粒子には、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質が含まれる。
リン脂質としては、グリセロールを骨格とするグリセロリン脂質やスフィンゴシンを骨格とするスフィンゴリン脂質である限り、特に制限はなく、公知のものを利用することができる。具体的には、ホスファチジルコリン、ホスファチジル酸、ホスファチジルグリコシド、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ホスファチジルセリン、ホスファチジルグリセロール、ジホスファチジルグリセロール、スフィンゴミエリン等が挙げられ、リポタンパク質を構成する代表的なリン脂質であることの観点より、ホスファチジルコリンが好ましい。
【0022】
本発明にかかる複合粒子には、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質が含まれる。また、臨界ミセル濃度としては0.5μg/ml〜8.0μg/mlが好ましく、1.0μg/ml〜6.0μg/mlがより好ましく、2.0μg/ml〜4.0μg/mlがさらに好ましい。
リン脂質の臨界ミセル濃度が0.1μg/ml未満である場合には、疎水性粒子表面を十分に被覆することができず、10μg/mlより高い臨界ミセル濃度を用いた場合には、リン脂質の水溶性が高すぎるため、疎水性粒子表面にリン脂質が吸着しない。
また、リン脂質の臨界ミセル濃度を2.0μg/ml以上とすることで、リン脂質の疎水性粒子表面被覆率が100%となるため好ましい。
前記リン脂質は、複合粒子の全質量に対して0.2質量%〜20質量%含まれていることが好ましく、リン脂質による疎水性粒子の表面被覆率という観点より、2.0質量%〜20質量%含まれていることがより好ましい。
【0023】
<アポリポタンパク質>
本発明にかかる複合粒子には、アポリポタンパク質が含まれる。
本発明にかかる複合粒子に含まれるアポリポタンパク質としては、血漿アポリポタンパク質である限り、特に制限はなく、公知のものを利用することができる。具体的には、アポリポタンパク質-A1〜4、アポリポタンパク質-B48、アポリポタンパク質-B100、アポリポタンパク質-C1〜3、アポリポタンパク質-D、アポリポタンパク質-E2〜4、アポリポタンパク質-J等が挙げられ、脳神経変性疾患治療薬や医薬組成物及び造影補助試薬を提供することの観点より、アポリポタンパク質-A1、アポリポタンパク質-B100又はアポリポタンパク質-E4が好ましく、中でも、アポリポタンパク質-B100がより好ましい。
【0024】
アポリポタンパク質は、上記アポリポタンパク質のうち、1種単独のアポリポタンパク質を含んでいてもよく、2種以上のアポリポタンパク質を含んでいてもよい。
【0025】
本発明にかかる複合粒子には、脳組織の特異的な領域への蓄積という観点から、好ましくは0.1μg/ml〜10μg/mlのアポリポタンパク質が含まれ、より好ましくは1.0μg/ml〜10μg/mlのアポリポタンパク質が含まれ、1.0μg/mlのアポリポタンパク質が含まれることがさらに好ましい。
前記アポリポタンパク質は、複合粒子の分散物の全質量に対して0.2質量%〜20質量%含まれていることが好ましく、2.0質量%〜20質量%含まれていることがより好ましく、2.0質量%含まれていることがさらに好ましい。
2.0質量%〜20質量%の範囲とすることで、脳組織内の特定領域への局在化が可能であり、好ましい。
【0026】
本発明にかかる複合粒子は、疎水性粒子を核として、該疎水性粒子の表面をリン脂質が被覆している。該リン脂質は、該疎水性粒子上に層(以下、「リン脂質層」ともいう)を形成する。該リン脂質層は、単層であっても複数層であってもよいが、複合粒子の調製が容易であることの観点より、単層である場合が好ましい。
【0027】
アポリポタンパク質は、機能を損なわない限り複合粒子中の如何なる領域に存在していてもよい。例えば、リン脂質層中に、アポリポタンパク質の一部が入り込んでもよいし、リン脂質層の外側、すなわち複合粒子の表面に存在してもよい。複合粒子は、脳組織内の特定領域への局在化を可能とすることの観点より、その表面にアポリポタンパク質を有している場合が好ましい。
【0028】
本発明にかかる複合粒子の粒径は、1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜27nmであることがさらに好ましい。
1nm〜100nmの範囲とすることで、脳血管内皮細胞表面に存在しているクラスリン被覆ピット(サイズ:100nm〜150nm)やカベオラ被覆ピット(サイズ:50nm〜80nm) を介したエンドサイトーシスが容易であり好ましい。
【0029】
<その他の成分>
本発明にかかる複合粒子に含まれるその他の成分としては、前記疎水性粒子、前記リン脂質及び前記アポリポタンパク質の他に、必要に応じて他の成分を含んでいてもよい。
他の成分としては、脂肪酸、ラベル用試薬、タンパク質、脂溶性ビタミン、コレステロール、中性脂肪 (トリグリセリド)、ホルモン、糖鎖、遺伝子、薬剤等が挙げられる。
脂肪酸としては、リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等が挙げられ、脳神経機能の維持及び促進の観点から、アラキドン酸やドコサエキサエン酸等が好ましい。
【0030】
ラベル用試薬としては、蛍光物質、放射線標識化合物、核磁気共鳴画像用造影剤などが挙げられ、蛍光顕微鏡、レーザー共焦点顕微鏡、二光子顕微鏡、質量顕微鏡、電子顕微鏡、陽電子放射断層撮影法(PET)、単一光子放射コンピュータ断層撮影法(SPECT)、機能的核磁気共鳴画像法(fMRI)等を用いた顕微観察の用途に用いることができる。
タンパク質としては、BBB透過性の能力を持つアルブミン、トランスフェリン、成長因子等が挙げられる。
ビタミンとしては、ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等が挙げられ、脳神経機能の維持・促進の用途に用いることができる。
前記その他の成分は、複合粒子の核の部分、リン脂質層又は複合粒子の表面のいずれに存在してもよい。例えば、他の成分がラベル用試薬である蛍光物質などの場合には、リン脂質及びアポリポタンパク質に結合して存在しても、複合粒子の表面に存在してもよい。
【0031】
(薬剤)
本発明にかかる複合粒子としては、前記疎水性粒子が、前記生分解性ポリマーであり、かつ該生分解性ポリマーが薬物を内包している場合が挙げられる。
生分解性ポリマーに内包される薬剤としては、人体に対して何らかの治療効果を奏する物質であれば特に制限はなく、神経変性疾患治療剤、抗がん剤、精神疾患治療剤(抗うつ剤等)などが挙げられ、神経変性疾患治療剤又は精神疾患治療剤が好ましく、本発明にかかる複合粒子の脳領域への局在性の観点より、神経変性疾患治療剤がより好ましい。
神経変性疾患治療剤としては、アルツハイマー型認知症治療剤、パーキンソン症候群治療剤及び脊髄小脳変性症治療剤などが挙げられ、具体的には、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等、ドネペジル塩酸塩(アリセプト(登録商標))などが挙げられる。
【0032】
抗がん剤としては、アルキル化剤、代謝拮抗剤などが挙げられ、具体的には、テモゾロミド (テモダール(登録商標)) 、ニムスチン (ニドラン(登録商標))、エノシタビン(サンラビン(登録商標))などが挙げられる。
抗うつ剤としては、三環系抗うつ剤、選択的セロトニン再取り込み阻害剤、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-,γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等が挙げられる。
【0033】
本発明における薬剤としては、上記薬剤は、1種単独でもよく、2種以上であってもよい。
本発明における薬剤の含有量は、特に制限はなく、各薬剤の有効投与量により適宜設定できる。例えば、薬剤としてビタミンA1を用いる場合には、疎水性粒子である生分解性ポリマーに対して、5.0×10−5質量%〜5.0×10−4質量%であることが好ましい。
【0034】
本発明において、薬剤はどのような形で複合粒子に含まれていてもよいが、好ましくは生分解性ポリマーに内包されている。
本発明において、上記薬剤を、生分解性ポリマーに内包させる方法としては、公知の方法を利用することができる。例えば、ACS NANO. 2008, 2(8), 1696−1702等に記載の方法に従い、生分解性ポリマーに薬剤を内包させることができる。
【0035】
<水性相を構成する成分>
水性相を構成する成分としては、水、生理食塩水、エタノール等が挙げられる。これらは、1種でもよく、2種以上を混合して使用してもよい。溶媒としては、生体組織を傷つけないなどの観点より、生理食塩水がより好ましい。
【0036】
本発明の分散物を得る方法のうち、好ましい方法としては以下に述べる製造方法が挙げられる。
≪分散物の製造方法≫
本発明にかかる複合粒子の分散物は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製すること(以下、「リン脂質被覆粒子調製工程」ともいう。)と、該リン脂質被覆粒子及びアポリポタンパク質を混合すること(以下、「混合工程」ともいう。)とを含む製造方法により製造することができる。
本発明にかかる複合粒子の分散物の製造方法は、リン脂質被覆粒子調製工程と、混合工程とを含むことにより、簡便で効率のよい複合粒子の製造方法を提供しうるものである。
【0037】
<リン脂質被覆粒子調製工程>
本発明にかかる複合粒子の分散物の製造方法は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製するリン脂質被覆粒子調製工程を含む。
リン脂質被覆粒子調製工程においては、疎水性粒子表面におけるリン脂質の自己集合の原理を利用することで、リン脂質被覆粒子を調製することが可能である。
【0038】
疎水性粒子の粒径は、好ましくは1nm〜100nmであり、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
疎水性粒子の粒径が1nm〜100nmの範囲内であれば、疎水性粒子表面のリン脂質の被覆率の制御が容易であり好ましい。
疎水性粒子の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載した疎水性粒子と同様のものが挙げられ、好ましい範囲も同様である。
【0039】
リン脂質の臨界ミセル濃度は0.1μg/ml〜10μg/mlである。
リン脂質の臨界ミセル濃度が0.1μg/ml未満である場合には、疎水性粒子表面を十分に被覆することができず、10μg/mlより高い臨界ミセル濃度を用いた場合には、リン脂質の水溶性が高すぎるため、疎水性粒子表面にリン脂質が吸着しない。
【0040】
なお、上記臨界ミセル濃度は、リン脂質のアシル基の長さにより適宜、上記範囲内において、最適の値に変更することができる。例えば、リン脂質としてホスファチジルコリン(PC)を用いた場合、アシル基の長さと臨界ミセル濃度との関係としては、PC(10:0/10:0)の場合には、2.0μg/ml〜6.0μg/mlであり、PC(12:0/12:0)の場合には、0.1μg/ml〜1.0μg/mlである。
ここで、PC(10:0/10:0)とは、ホスファチジルコリン中における2本のアシル基が飽和状態であり、該アシル基の炭素数が10であることを意味する。
【0041】
本明細書において、臨界ミセル濃度は、エレクトロスプレーイオン化質量分析法に従い測定する。溶媒はメタノール、クロロホルム及び純水(ppbレベル)の混合液である。
リン脂質の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載したリン脂質と同様のものが挙げられ、好ましい範囲も同様である。
【0042】
前記疎水性粒子及び前記リン脂質を混合する際の温度としては、特に制限はされないが、35℃〜37℃が好ましい。また、リン脂質被覆粒子調製工程の際には、約1分間のボルテックを必要とする。調整する時間としては、特に制限はされないが、3時間〜12時間が好ましい。また、疎水性粒子分散水溶液に既にクエン酸ナトリウムなどの分散安定剤が付加されている場合には、疎水性粒子を一度遠心し沈殿させ、その上澄みを純水に交換することが好ましい。
【0043】
<混合工程>
本発明にかかる複合粒子の分散物の製造方法は、前記リン脂質被覆粒子及びアポリポタンパク質を混合する混合工程を含む。
【0044】
アポリポタンパク質の添加量は、好ましくは0.1μg/ml〜10μg/mlであり、脳組織内の特定領域への蓄積の観点より1.0μg/ml〜10μg/mlであることがより好ましく、さらに好ましくは1.0μg/mlである。
アポリポタンパク質の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載したアポリポタンパク質と同様のものが挙げられ、好ましい範囲も同様である。
【0045】
本発明における混合工程において、温度は特に制限はされないが、35℃〜37℃が好ましい。また、混合工程の際にはボルテックを必要としない。混合時間としては、特に制限はされないが、3時間〜5時間が挙げられる。
【0046】
なお、疎水性粒子である生分解性ポリマーに薬剤が内包されている場合には、例えば、前記リン脂質被覆粒子調製工程の前に、生分解性ポリマーに薬剤を内包する薬剤内包工程を含んでいてもよい。
薬剤内包工程としては、公知の方法を利用することができ、例えば、ACS NANO. 2008, 2(8), 1696−1702等に記載の方法に従い、生分解性ポリマーに薬剤を内包させることができる。
【0047】
さらに、別の態様として、本発明は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製すること(以下、「リン脂質被覆粒子調製工程」ともいう。)と、該リン脂質被覆粒子及びアポリポタンパク質を混合すること(以下、「混合工程」ともいう。)とを含む製造方法により得られる複合粒子を提供する。
【0048】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製するリン脂質被覆粒子調製工程と、該リン脂質被覆粒子及びアポリポタンパク質を混合する混合工程とは、前記複合粒子の製造方法に記載のリポタンパク質作製工程及び混合工程と同義であり、好ましい範囲等も同様である。
【0049】
≪脳神経変性疾患治療薬≫
本発明の脳神経変性疾患治療薬は、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を有効成分として含有する。
また、本発明における別の態様としては、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を患者に投与することを含む脳神経変性疾患の診断及び治療の方法を提供しうる。
本発明の脳神経変性疾患治療薬は、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を有効成分として含有することにより、脳内への移行性が良好で、優れた治療効果を発揮しうる、脳神経変性疾患治療薬を提供しうるものである。
【0050】
脳神経変性疾患としては、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症が挙げられる。
【0051】
薬剤としては、神経変性疾患治療剤が挙げられる。
神経変性疾患治療剤としては、アルツハイマー型認知症治療剤、パーキンソン症候群治療剤及び脊髄小脳変性症治療剤などが挙げられ、具体的には、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等、ドネペジル塩酸塩 (アリセプト(登録商標))などが挙げられ、脳神経機能の恒常性の維持及び促進の観点より、脂溶性ビタミン又は脂肪酸が好ましい。
【0052】
本発明の神経変性疾患治療薬の有効成分である分散物における前記複合粒子の含有量は、用途により適宜設定しうるが、治療効果の観点より、例えば、前記複合粒子を、分散物全量に対して、好ましくは1質量%〜10質量%含有する。
本発明の神経変性疾患治療薬の有効成分である分散物における薬剤の含有量は、特に制限はなく、各薬剤の有効投与量により適宜設定できる。例えば、薬剤としてビタミンA1を用いる場合には、疎水性粒子である生分解性ポリマーに対して、5.0×10−5質量%〜5.0×10−4質量%であることが好ましい。
【0053】
生分解性ポリマー、リン脂質及びアポリポタンパク質については、本発明の神経変性疾患治療薬の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【0054】
≪医薬組成物≫
本発明の医薬組成物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有する。
本発明の医薬組成物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有することにより、脳内への移行性が良好で、副作用の発現頻度を抑制しうる医薬組成物を提供しうるものである。
【0055】
本発明の医薬組成物における前記複合粒子の含有量は、用途により適宜設定しうるが、治療効果の観点より、例えば、前記複合粒子を、医薬組成物の全固形分に対して、好ましくは1質量%〜10質量%含有する。
本明細書において、全固形分とは、医薬組成物中、溶液や生理食塩水を除いた、全成分の合計含量のことである。
【0056】
本発明の医薬組成物は、神経変性疾患治療、抗がん治療、精神疾患治療(抗うつ治療等)等の各種疾患の治療のために使用することができる。
具体的には、薬学上許容しうる担体(希釈剤、溶解補助剤など)と混合して得られる医薬組成物あるいは製剤の形態で、血管内投与、膀胱投与、腹腔内投与、局所投与等の方法で患者に投与することができる。
本発明の医薬組成物の投与量は、疎水性粒子や該疎水性粒子である生分解性ポリマーに内包された薬剤の種類や活性により、最適な投与量を決定することができる。例えば薬剤として、ビタミンA1を生分解性ポリマーに内包する場合を例に取れば、投与量はビタミンA1量として5mg/kg〜170mg/kgが好ましい。
【0057】
疎水性粒子、リン脂質及びアポリポタンパク質については、本発明の医薬組成物の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【0058】
≪造影補助試薬≫
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有する。
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有することにより、BBB透過性が良好で、脳の恒常性の変化(脳の自己組織化)が生じていると考えられる領域の特定などに有用な造影補助試薬を提供しうるものである。
【0059】
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子を含む。前記ラベル用疎水性粒子としては、蛍光顕微鏡、レーザー共焦点顕微鏡、二光子顕微鏡、質量顕微鏡、電子顕微鏡、陽電子放射断層撮影法(PET)、単一光子放射コンピュータ断層撮影法(SPECT)、機能的核磁気共鳴画像法(fMRI)等により画像解析が可能なコントラスト像を生じさせる、公知のもの全てが挙げられる。中でも、金属ナノ粒子、ラベル用試薬を内包する生分解性ポリマー、量子ドット等が良好な画像化能を有する観点より好ましい。
【0060】
前記ラベル用試薬を内包する生分解性ポリマーにおける、ラベル用試薬としては、各種顕微観察技術でコントラスト像を生じさせるものであれば、特に限定されない。例えば、蛍光色素(例えば、Cell Tracker CM-DiI、Alexa Fluor-488、-594(登録商標)等)、放射線標識化合物(例えば、In-111、Tc-99m、I-123、I-125 F-18、Ga-67、Ga-68等)、不対スピン原子又はフリーラジカル(例えば、Fe、ランタイド類(lanthides)、Gd等)及び核磁気共鳴画像用造影剤(例えば、キレート化した(DTPA)マンガン等)等が挙げられる。
また、前記ラベル用試薬は、ラベル用疎水性粒子としての生分解性ポリマーに内包されるだけではなく、該生分解性ポリマーとは独立して、造影補助試薬に含有されていてもよい。造影補助試薬にかかる複合粒子の分散物が、ラベル用疎水性粒子として金属ナノ粒子や、量子ドットの他に、さらに、前記ラベル用試薬を含んでいる態様も本発明の造影補助試薬に含まれる。
【0061】
本発明にかかる造影補助試薬が、疎水性粒子に加えてさらにラベル用試薬を含む場合の製造方法について、ラベル用試薬が蛍光色素である場合を例に挙げて簡単に説明する。すなわち、本発明におけるリン脂質被覆粒子調製工程後に、該リン脂質被覆粒子が含まれる溶液に、適当量の蛍光色素を添加し、該蛍光色素で表面がラベルされたリン脂質被覆粒子と、アポリポタンパク質とを混合して、造影補助試薬を得ることもできる。
なお、本発明の造影補助試薬の製造方法はこれに制限されるものではなく、公知の方法を利用することができる。
【0062】
疎水性粒子、リン脂質及びアポリポタンパク質については、本発明の造影補助試薬の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【実施例】
【0063】
以下、本発明を実施例により更に具体的に説明するが、本発明はその主旨を越えない限り、以下の実施例に限定されるものではない。
【0064】
<実施例1> 複合粒子(分散物)の調製
平均粒径20nmのコロイド状金ナノ粒子(G1652、Sigma−Aldrich社製)を最終濃度が50μg/mlとなるように、クエン酸ナトリウムなどの分散安定剤を遠心し除去した後、純水(ppbレベル)に分散させた。また、35℃の純水(ppb レベル)にPC(10:0/10:0)(850325P、Avanti Polar Lipids社製)を、最終濃度が1.0mg/mlとなるように溶解した後、0.1μmのカットオフ特性を持つ親水性メンブレンフィルターで、リポソーム及び凝集物を除去した。この調整したPC溶液(臨界ミセル濃度2.8μg/ml)及びコロイド状金ナノ粒子溶液を混合し、37℃で一晩攪拌 (約10時間撹拌) することにより、コロイド状金ナノ粒子表面に1層のPCが吸着した、リン脂質被覆金ナノ粒子を得た。
【0065】
次に、リン脂質被覆金ナノ粒子と、1.0μg/mlに調整した蛍光物質(商品名Cell Tracker CM-DiI、Molecular probes社製)とを30分間反応させた後、1.0μg/mlのアポリポタンパク質(A1、B100又はE4)をリン脂質被覆金ナノ粒子に吸着させた。
最後に、37℃、20,000×gで5分間の遠心を2回行い、上清を除いた沈殿を生理食塩水によって再分散させた後に、0.1μmのフィルターによって濾過して、複合粒子を得た。なお、リン脂質の複合粒子の全質量に対する含有量は4.5質量%である。
【0066】
図1に実施例1にかかる複合粒子の作製手順を模式的に示す。
図1は、コロイド状の金ナノ粒子をリン脂質で被覆し、リン脂質被覆金ナノ粒子を調製し、その後、蛍光物質で該リン脂質被覆金ナノ粒子をラベルし、最後にアポリポタンパク質をリン脂質被覆金ナノ粒子に吸着させ、複合粒子を得る工程を模式化した図である。
【0067】
作製したリン脂質被覆金ナノ粒子をTEMにより観察した。また、各複合粒子の平均粒径を動的光散乱法 (DLS:Dynamic Light Scattering)により評価した。
【0068】
図2は本実施例にかかるリン脂質被覆金ナノ粒子を観察したTEM像を示す。TEMで観察した結果、各複合粒子は、いずれも20nmの金ナノ粒子の表面に、3.3nmの1層のPCが吸着した形状を示すことが明らかとなった。
また、DLS測定の結果、各複合粒子の平均粒径は、それぞれ、51+/−0.0nm(複合粒子(Apo-A1))、30+/−1.9nm(複合粒子(Apo-B100))、38+/−3.8nm(複合粒子(Apo-E4))であった。
【0069】
<実施例2> 複合粒子の脳血管内皮細胞への取り込み機構の解明
細胞培養培地(商品名ダルベッコイーグル培地、和光社製)に、10%ウシ胎児血清(商品名Fetal Bovine Serum、Bio West社製)及び0.1%ゲンタマイシン(Sigma−Aldrich社製)を添加して、マウスの脳組織由来の血管内皮細胞(ATCC No.bEnd.3)を培養した。培養は37℃、5%CO2環境下で行なった。
細胞免疫化学的解析に供するために、ラット由来のコラーゲンでコーティングした1.1cm2のガラスプレートに、血管内皮細胞を播種し、24ウェルプレート(面積1.55×1.55cm2、高さ1.75cm)において、約2.5×104細胞/cm2のサブコンフルエンス状態まで培養させた。その後、前記細胞培養培地に、複合粒子の最終濃度が2.5μg/mlとなるように、実施例1で調整した複合粒子を添加し、1時間、3時間、6時間又は12時間培養して、各サンプルを得た。
その後、各サンプルをPBSで2度洗浄し、4%パラホルムアルデヒドで固定化し、10ng/mlのジギトニンで処理し、3% normal goat serum(Millipore社製)でブロッキングした。1時間後、各サンプルを、Clathirn HC H−300(sc−9069、Santa Cruz Biotechnology社製)又はLDL receptor (C-term)(1956-1、Epitomics社製)のいずれかとともに4℃で一晩反応させた。続いてAlexa Fluor-488 Goat Anti‐rabbit IgG (A11034、Molecular probes社製)を添加して反応させ、ビスベンズイミド H33342 三塩酸塩(商品名B2261、Sigma−Aldrich社製)を用いて核染色を行った。
【0070】
図3は本実施例にかかる各複合粒子(Apo-A1、Apo-B100及びApo-E4)の脳血管内皮細胞における時間経過に伴う取り込み量を示す。図3中、縦軸は複合粒子の血管内皮細胞への取り込み量(カウント/細胞)を示し、横軸は用いたサンプルの培養時間を示す。また、レーザー共焦点顕微鏡(ライカSP−II、ライカ社製)を用いて、1時間後、3時間後、6時間後及び12時間後それぞれの処理時間における、各複合粒子(Apo-A1、Apo-B100及びApo-E4)の血管内皮細胞への取り込み量の増加が確認された。
【0071】
図4は、本実施例にかかる複合粒子(Apo-A1)が脳血管内皮細胞のクラスリン被覆小胞(CV:Clathrin Vesicle)内に存在しているところを観察したTEM像並びにレーザー共焦点顕微鏡(ライカSP−II、ライカ社製)像を示す。12時間培養後の脳血管内皮細胞サンプルを観察した結果、各複合粒子(Apo-A1、Apo-B100及びApo-E4)が、クラスリンにより被覆され、血管内皮細胞に取り込まれたことを見出した。また同様に、各複合粒子(Apo-A1、Apo-B100及びApo-E4)は、LDLRを介して血管内皮細胞内へ輸送されるものではないことも見出した。
【0072】
これらの結果より、各複合粒子は、開口分泌されたクラスリン経路を介して、血液中から脳実質内に取り込まれる可能性があることが明らかとなった。
【0073】
<実施例3> 複合粒子の脳における自己組織化
5週齢のC57BL/6J雄性マウスに、生理食塩水で希釈した25μg/ml〜125μg/mlの各複合粒子 (Apo-A1、Apo-B100、Apo-E4) 溶液100μlを、尾静脈より投与した。なお、コントロールには生理食塩水を使用した。
【0074】
24時間後、50μg/mlのペントバルビタールを腹腔内注射し、麻酔下で切開し、0.1Mのリン酸緩衝生理食塩水で毎分1mlの流速で血液を灌流し、4%パラホルムアルデヒド溶液で脳組織を固定した。固定後、5%スクロースで脱水し、矢状方向に半割した脳試料を厚さ約10μmでミクロトームを用い連続切片を作製した。メタノール洗浄により自家蛍光を持つ付着物を除去したスライドガラスに固定化し、蛍光顕微鏡による観察に供した。
【0075】
図5は各複合粒子 (Apo-A1、Apo-B100、Apo-E4)の脳組織内における分布図を示す。また、脳組織領域別における各複合粒子の蓄積度合を比較した結果、複合粒子(Apo-B100)は、複合粒子(Apo-A1或いはApo-E4)に比べ、大脳皮質、線条体、小脳及び海馬に局在化することが可能であった。
【0076】
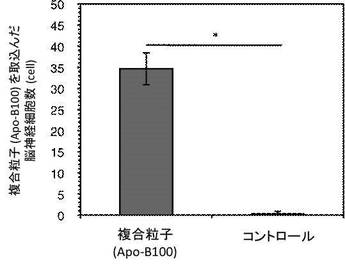
図6は脳連続切片(100枚)における複合粒子(Apo-B100)を取込んだ脳神経細胞数を示す。コントロールは生理食塩水を投与した。エラーバーは各群マウス3匹あたりの標準偏差であり、 図6中、*P<0.0005はStudent's t-testを用い示した。この結果、複合粒子(Apo-B100)は、統計学的解析より確実にBBB透過性能を持っていることが証明された。
【0077】
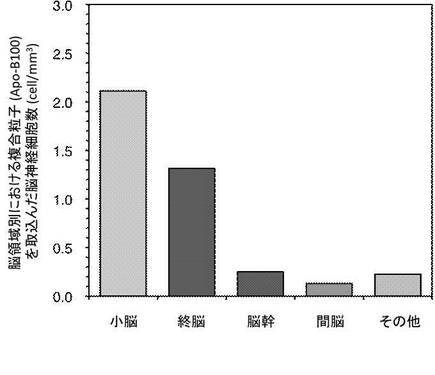
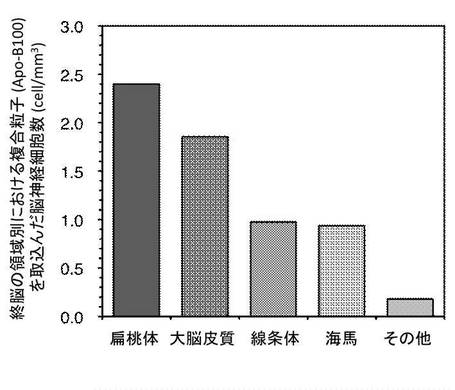
図7及び図8は複合粒子(Apo-B100)を取込んだ脳組織領域別における脳神経細胞数を示す。
この結果、複合粒子(Apo-B100)は、小脳及び終脳の特定領域に局在化することが可能であった。
【0078】
以上の結果より、本発明にかかる複合粒子は、BBB透過性が良好であるとともに、脳組織内の特定領域への局在化が可能であることが明らかとなった。
【技術分野】
【0001】
本発明は、分散物及び脳神経変性疾患治療薬に関する。
【背景技術】
【0002】
脳は、精神活動と運動制御に関わっており、特定の領域に各機能が局在している。例えば、大脳皮質には思考、記憶、知覚及び言語の機能を担う領域があり、線条体や小脳には主に体性運動や体運動の機能調整を担う領域がある。近年、生活習慣の乱れなどによって、これらの脳領域の恒常性の破綻に伴う、アルツハイマー型認知症などの脳神経変性疾患やうつ病などの精神疾患が増加の一途を辿っている。そのため、疾患治療に向けた薬物輸送システムの技術開発が望まれている。
【0003】
脳は、その恒常性を維持するため、血液脳関門(Blood-Brain-Barrier(以下「BBB」ともいう。))を介して、血液中から脳内に取り込む物質を厳密に制御している。そのため、アルツハイマー型認知症などの脳神経変性疾患の診断や治療のための薬剤の開発においては、BBB透過性を向上させることが課題であり、脳神経変性疾患領域に薬剤を確実に蓄積できるようにデザインする必要がある。その手段の一つとして、人工低密度リポタンパク質キャリアを利用した薬物輸送システムとして、リポソーム表面にアポリポタンパク質E(ApoE:ApolipoproteinE)が結合した薬剤内包リポソームが開発されている(例えば、特許文献1参照)。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特表2006−516539号公報
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかしながら、前述の薬剤内包リポソームは、その表面にApoEが結合しているものの、該ApoEを認識する低密度リポタンパク質受容体(LDLR:Low Density Lipoprotein Receptor)は、主に肝臓で発現しているため、薬剤内包リポソームは肝臓に多く蓄積すると考えられる。また、ApoEの分子種の一つであるApoE2はLDLRでは認識されない。さらに、BBBを形成している脳血管内皮細胞は、LDLRではなく、LDLRファミリーの一つである低密度リポタンパク質受容体関連タンパク質-8(LRP-8:Lipoprotein Receptor related Protein-8)が主に発現している。また、粒径1nm〜100nmの物質のBBBにおける輸送は、クラスリン被覆ピットやカベオラ被覆ピットが関与していると言われている。そのため、前記薬剤内包リポソーム表面に該ApoEが存在する人工低密度リポタンパク質というだけでは、該キャリアのBBB透過性の効率が一概に上昇するとは限らない。このため、現在においてもBBB透過性を示す薬剤キャリアの更なる開発が待ち望まれている。また、現時点において、脳神経変性疾患領域に薬物を蓄積させることが可能なキャリアは開発されていない。
【0006】
本発明は、上記の状況に鑑みてなされたものであり、以下の目的を達成することを課題とする。
すなわち、本発明は、BBB透過性が良好であるとともに、脳組織の局所に蓄積することが可能な分散物を提供することを課題とする。また、本発明は、該分散物を利用した、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬を提供することを課題とする。さらに、本発明は、該分散物の製造方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者らは、前記の課題を抜本的に解決するため、詳細な実験・検討を行った結果、特定の成分で調製された複合粒子がBBB透過性を持ち、脳組織内の特定領域への局在化を可能とする知見を得た。本発明は、かかる知見に基づいて達成されたものである。
前記の課題を解決するための具体的な手段は以下の通りである。
<1> 疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
<2> 前記複合粒子がリン脂質及びアポリポタンパク質を疎水性粒子表面に有する<1>に記載の複合粒子を有する分散物。
<3> 前記リン脂質の複合粒子の全質量に対する含有量が、0.2質量%〜20質量%である<1>又は<2>に記載の複合粒子を有する分散物。
<4> 前記アポリポタンパク質が、0.1μg/ml〜10μg/mlの量で含まれる<1>〜<3>のいずれかに記載の複合粒子を有する分散物。
<5> 前記アポリポタンパク質が、アポリポタンパク質A1、アポリポタンパク質B100及びアポリポタンパク質E4からなる群より選ばれる少なくとも1種のアポリポタンパク質である<1>〜<4>のいずれかに記載の複合粒子を有する分散物。
<6> 前記疎水性粒子が、金属ナノ粒子又は生分解性ポリマーである<1>〜<5>のいずれかに記載の複合粒子を有する分散物。
<7> 前記疎水性粒子の粒径が1nm〜100nmである<1>〜<6>のいずれかに記載の複合粒子を有する分散物。
<8> 前記複合粒子の粒径が1nm〜100nmである<1>〜<7>のいずれかに記載の複合粒子を有する分散物。
<9> 前記疎水性粒子が、生分解性ポリマーであり、かつ該生分解性ポリマーが薬物を内包している<1>〜<8>のいずれかに記載の複合粒子を有する分散物。
<10> <9>に記載の複合粒子を有する分散物を有効成分として含有する脳神経変性疾患治療薬。
<11> 脳神経変性疾患が、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症からなる群より選ばれる少なくとも1種の疾患である<10>に記載の脳神経変性疾患治療薬。
<12> <1>〜<9>のいずれかに記載の複合粒子を有する分散物を含有する医薬組成物。
<13> <1>〜<9>のいずれかに記載の複合粒子を有する分散物を含有する造影補助試薬。
<14> 疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む複合粒子の分散物の製造方法。
<15> 疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む製造方法により得られる複合粒子の分散物。
【発明の効果】
【0008】
本発明によれば、BBB透過性が良好であるとともに、脳組織内の特定領域への局在化を可能とする分散物を提供することができる。また、本発明によれば、該分散物を利用した、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬を提供することができる。さらに、本発明によれば、該分散物の製造方法を提供することができる。
【図面の簡単な説明】
【0009】
【図1】本実施例にかかる複合粒子の作製手順を示した模式図である。
【図2】本実施例にかかる金ナノ粒子及びリン脂質被覆金ナノ粒子を観察した透過型電子顕微鏡(TEM:Transmission Electron Microscopy)像である。
【図3】本実施例にかかる各複合粒子(Apo-A1、Apo-B100、Apo-E4)の脳血管内皮細胞における時間経過に伴う取り込み量を示したグラフである。
【図4】本実施例にかかる複合粒子(Apo-A1)が脳血管内皮細胞のクラスリン被覆小胞内に存在しているところを観察したTEM像並びにレーザー共焦点顕微鏡像である。
【図5】各複合粒子(Apo-A1、Apo-B100、Apo-E4)の各脳領域における分布図である。
【図6】脳連続切片(100枚)における複合粒子(Apo-B100)を取込んだ脳神経細胞数を示したグラフである。
【図7】複合粒子(Apo-B100)を取込んだ脳領域別における脳神経細胞数を示したグラフである。
【図8】複合粒子(Apo-B100)を取込んだ終脳の領域別における脳神経細胞数を示したグラフである。
【発明を実施するための形態】
【0010】
以下、本発明の分散物、分散物の製造方法、脳神経変性疾患治療薬、医薬組成物及び造影補助試薬について詳述する。
以下に記載する構成要件の説明は、本発明の代表的な実施態様に基づいてなされることがあるが、本発明はそのような実施態様に限定されるものではない。
なお、本発明において「〜」を用いて表される数値範囲は、「〜」の前後に記載される数値を下限値及び上限値として含む範囲を意味する。また、本明細書において「工程」との語は、独立した工程だけではなく、他の工程と明確に区別できない場合であってもその工程の所期の作用が達成されれば、本用語に含まれる。
【0011】
本発明は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物に関する。
本発明にかかる複合粒子は、粒径1nm〜100nmの疎水性粒子と、特定量のリン脂質及びアポリポタンパク質を含む構成としたことで、ナノサイズの疎水性粒子が、生体内で安定に分散でき、かつBBBにおける物質の輸送機構に関わる脳血管内皮細胞のクラスリン被覆ピットやカベオラ被覆ピットに認識されるようになった。このメカニズムにより、良好なBBB透過性を実現すると同時に、脳組織内の特定領域への局在化を可能とする複合粒子を提供するものである。
【0012】
≪分散物≫
本発明の分散物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有しており、必要に応じて他の成分を含んでいてもよい。
本発明の分散物は、水性相に複合粒子が分散しているものである。
以下、複合粒子を構成する成分である疎水性粒子、リン脂質、アポリポタンパク質及びその他の成分、並びに水性相を構成する成分について説明する。
<疎水性粒子>
本発明にかかる複合粒子に含まれる疎水性粒子としては、粒子と水などの媒質との間の親和性の強弱により、疎水性の傾向を示すコロイドであり、粒子表面の帯電に起因する静電気的な反発及びブラウン運動の能力を有する粒子である限り、特に制限はなく、公知のものを利用することができる。具体的には、金属ナノ粒子、生分解性ポリマー、デンドリマー、量子ドット、ラテックス粒子、シリカ粒子、疎水性アミノ酸を含有するタンパク質粒子(例えば、アルブミン粒子)等が挙げられ、脳神経変性疾患治療薬や医薬組成物及び造影補助試薬を提供することの観点より、金属ナノ粒子又は生分解性ポリマーが好ましい。
【0013】
また、金属ナノ粒子に使用しうる金属元素としては、アルカリ金属(周期律表第1族元素)、アルカリ土類金属(周期律表第2族元素)、遷移金属等が挙げられ、中でも金又は磁性体(酸化鉄、酸化クロム、コバルト、フェライト)が好ましく、生体への影響などの観点より、金がより好ましい。
【0014】
中でも、疎水性粒子としては、所望する粒径及び均一サイズの粒子を容易に作製が可能であり、リン脂質との親和性に優れ、大きな電気陰性度を持つことや水溶液中で安定に分散できることなどの観点より、金ナノ粒子がより好ましい。
【0015】
生分解性ポリマーとしては、Poly-ε-caprolactone(PCL)、Poly(lactic acid)(PLA)、Poly (glycolic acid)(PGA)、Ester-terminated poly(DL-lactic-co-glycolic acid) (PLGA)等が挙げられ、生分解性ポリマーの分解速度による薬物放出量の制御や薬物の運搬性及び生体による利用効率(バイオアベイラビィリティ)の観点より、PLGA又はPLAが好ましく、PLGAがより好ましい。
【0016】
前記疎水性粒子の粒径は、1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
1nm〜100nmの範囲とすることで、脳血管内皮細胞表面に存在しているクラスリン被覆ピット (サイズ:100nm〜150nm)やカベオラ被覆ピット(サイズ:50nm〜80nm)を介したエンドサイトーシスが容易となるため好ましい。
【0017】
前記疎水性粒子が、金属ナノ粒子である場合には、その粒径は1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、肝臓における非特異的な蓄積の低減の観点から、1nm〜20nmであることがさらに好ましい。
【0018】
前記疎水性粒子が、生分解性ポリマーである場合には、その粒径は1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
前記疎水性粒子が、生分解性ポリマーである場合には、その粒径は、生体に存在する粒径5nm〜15nmの高密度リポタンパク質(HDL:High Density Lipoprotein)や粒径18nm〜28nmの低密度リポタンパク質(LDL:Low Density Lipoprotein )と同等サイズであるとの観点から、1nm〜100nmであることが好ましい。
【0019】
疎水性粒子の粒径は、疎水性粒子の種類に応じた公知の方法により、適宜所望の大きさに調整することができる。例えば、金ナノ粒子の場合には、塩化金酸塩溶液の還元によるファラデー法などにより、粒径を調整することが可能である。
本明細書において、粒径とは、特に断らない限り、平均粒子径を表す。平均粒子径は、TEM(日本電子社製)等により、任意に500個の粒子を選択して、個別に粒子サイズを測定して、その測定値の算術平均値をとったものである。
【0020】
なお、前記疎水性粒子の製造方法としては、疎水性粒子の種類に応じた公知の方法を使用し、製造することができる。
例えば、金ナノ粒子は、Surface Science. 1982, 120, 435−455などに記載の方法により製造することができる。
また、生分解性ポリマーであるPLGAは、Langmuir 2010, 26(22), 16958−16962などに記載の方法により製造することができる。
また、本発明において疎水性粒子は、市販されているものを使用することもできる。
【0021】
<リン脂質>
本発明にかかる複合粒子には、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質が含まれる。
リン脂質としては、グリセロールを骨格とするグリセロリン脂質やスフィンゴシンを骨格とするスフィンゴリン脂質である限り、特に制限はなく、公知のものを利用することができる。具体的には、ホスファチジルコリン、ホスファチジル酸、ホスファチジルグリコシド、ホスファチジルエタノールアミン、ホスファチジルイノシトール、ホスファチジルセリン、ホスファチジルグリセロール、ジホスファチジルグリセロール、スフィンゴミエリン等が挙げられ、リポタンパク質を構成する代表的なリン脂質であることの観点より、ホスファチジルコリンが好ましい。
【0022】
本発明にかかる複合粒子には、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質が含まれる。また、臨界ミセル濃度としては0.5μg/ml〜8.0μg/mlが好ましく、1.0μg/ml〜6.0μg/mlがより好ましく、2.0μg/ml〜4.0μg/mlがさらに好ましい。
リン脂質の臨界ミセル濃度が0.1μg/ml未満である場合には、疎水性粒子表面を十分に被覆することができず、10μg/mlより高い臨界ミセル濃度を用いた場合には、リン脂質の水溶性が高すぎるため、疎水性粒子表面にリン脂質が吸着しない。
また、リン脂質の臨界ミセル濃度を2.0μg/ml以上とすることで、リン脂質の疎水性粒子表面被覆率が100%となるため好ましい。
前記リン脂質は、複合粒子の全質量に対して0.2質量%〜20質量%含まれていることが好ましく、リン脂質による疎水性粒子の表面被覆率という観点より、2.0質量%〜20質量%含まれていることがより好ましい。
【0023】
<アポリポタンパク質>
本発明にかかる複合粒子には、アポリポタンパク質が含まれる。
本発明にかかる複合粒子に含まれるアポリポタンパク質としては、血漿アポリポタンパク質である限り、特に制限はなく、公知のものを利用することができる。具体的には、アポリポタンパク質-A1〜4、アポリポタンパク質-B48、アポリポタンパク質-B100、アポリポタンパク質-C1〜3、アポリポタンパク質-D、アポリポタンパク質-E2〜4、アポリポタンパク質-J等が挙げられ、脳神経変性疾患治療薬や医薬組成物及び造影補助試薬を提供することの観点より、アポリポタンパク質-A1、アポリポタンパク質-B100又はアポリポタンパク質-E4が好ましく、中でも、アポリポタンパク質-B100がより好ましい。
【0024】
アポリポタンパク質は、上記アポリポタンパク質のうち、1種単独のアポリポタンパク質を含んでいてもよく、2種以上のアポリポタンパク質を含んでいてもよい。
【0025】
本発明にかかる複合粒子には、脳組織の特異的な領域への蓄積という観点から、好ましくは0.1μg/ml〜10μg/mlのアポリポタンパク質が含まれ、より好ましくは1.0μg/ml〜10μg/mlのアポリポタンパク質が含まれ、1.0μg/mlのアポリポタンパク質が含まれることがさらに好ましい。
前記アポリポタンパク質は、複合粒子の分散物の全質量に対して0.2質量%〜20質量%含まれていることが好ましく、2.0質量%〜20質量%含まれていることがより好ましく、2.0質量%含まれていることがさらに好ましい。
2.0質量%〜20質量%の範囲とすることで、脳組織内の特定領域への局在化が可能であり、好ましい。
【0026】
本発明にかかる複合粒子は、疎水性粒子を核として、該疎水性粒子の表面をリン脂質が被覆している。該リン脂質は、該疎水性粒子上に層(以下、「リン脂質層」ともいう)を形成する。該リン脂質層は、単層であっても複数層であってもよいが、複合粒子の調製が容易であることの観点より、単層である場合が好ましい。
【0027】
アポリポタンパク質は、機能を損なわない限り複合粒子中の如何なる領域に存在していてもよい。例えば、リン脂質層中に、アポリポタンパク質の一部が入り込んでもよいし、リン脂質層の外側、すなわち複合粒子の表面に存在してもよい。複合粒子は、脳組織内の特定領域への局在化を可能とすることの観点より、その表面にアポリポタンパク質を有している場合が好ましい。
【0028】
本発明にかかる複合粒子の粒径は、1nm〜100nmであることが好ましく、1nm〜50nmであることがより好ましく、1nm〜27nmであることがさらに好ましい。
1nm〜100nmの範囲とすることで、脳血管内皮細胞表面に存在しているクラスリン被覆ピット(サイズ:100nm〜150nm)やカベオラ被覆ピット(サイズ:50nm〜80nm) を介したエンドサイトーシスが容易であり好ましい。
【0029】
<その他の成分>
本発明にかかる複合粒子に含まれるその他の成分としては、前記疎水性粒子、前記リン脂質及び前記アポリポタンパク質の他に、必要に応じて他の成分を含んでいてもよい。
他の成分としては、脂肪酸、ラベル用試薬、タンパク質、脂溶性ビタミン、コレステロール、中性脂肪 (トリグリセリド)、ホルモン、糖鎖、遺伝子、薬剤等が挙げられる。
脂肪酸としては、リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等が挙げられ、脳神経機能の維持及び促進の観点から、アラキドン酸やドコサエキサエン酸等が好ましい。
【0030】
ラベル用試薬としては、蛍光物質、放射線標識化合物、核磁気共鳴画像用造影剤などが挙げられ、蛍光顕微鏡、レーザー共焦点顕微鏡、二光子顕微鏡、質量顕微鏡、電子顕微鏡、陽電子放射断層撮影法(PET)、単一光子放射コンピュータ断層撮影法(SPECT)、機能的核磁気共鳴画像法(fMRI)等を用いた顕微観察の用途に用いることができる。
タンパク質としては、BBB透過性の能力を持つアルブミン、トランスフェリン、成長因子等が挙げられる。
ビタミンとしては、ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等が挙げられ、脳神経機能の維持・促進の用途に用いることができる。
前記その他の成分は、複合粒子の核の部分、リン脂質層又は複合粒子の表面のいずれに存在してもよい。例えば、他の成分がラベル用試薬である蛍光物質などの場合には、リン脂質及びアポリポタンパク質に結合して存在しても、複合粒子の表面に存在してもよい。
【0031】
(薬剤)
本発明にかかる複合粒子としては、前記疎水性粒子が、前記生分解性ポリマーであり、かつ該生分解性ポリマーが薬物を内包している場合が挙げられる。
生分解性ポリマーに内包される薬剤としては、人体に対して何らかの治療効果を奏する物質であれば特に制限はなく、神経変性疾患治療剤、抗がん剤、精神疾患治療剤(抗うつ剤等)などが挙げられ、神経変性疾患治療剤又は精神疾患治療剤が好ましく、本発明にかかる複合粒子の脳領域への局在性の観点より、神経変性疾患治療剤がより好ましい。
神経変性疾患治療剤としては、アルツハイマー型認知症治療剤、パーキンソン症候群治療剤及び脊髄小脳変性症治療剤などが挙げられ、具体的には、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等、ドネペジル塩酸塩(アリセプト(登録商標))などが挙げられる。
【0032】
抗がん剤としては、アルキル化剤、代謝拮抗剤などが挙げられ、具体的には、テモゾロミド (テモダール(登録商標)) 、ニムスチン (ニドラン(登録商標))、エノシタビン(サンラビン(登録商標))などが挙げられる。
抗うつ剤としては、三環系抗うつ剤、選択的セロトニン再取り込み阻害剤、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-,γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等が挙げられる。
【0033】
本発明における薬剤としては、上記薬剤は、1種単独でもよく、2種以上であってもよい。
本発明における薬剤の含有量は、特に制限はなく、各薬剤の有効投与量により適宜設定できる。例えば、薬剤としてビタミンA1を用いる場合には、疎水性粒子である生分解性ポリマーに対して、5.0×10−5質量%〜5.0×10−4質量%であることが好ましい。
【0034】
本発明において、薬剤はどのような形で複合粒子に含まれていてもよいが、好ましくは生分解性ポリマーに内包されている。
本発明において、上記薬剤を、生分解性ポリマーに内包させる方法としては、公知の方法を利用することができる。例えば、ACS NANO. 2008, 2(8), 1696−1702等に記載の方法に従い、生分解性ポリマーに薬剤を内包させることができる。
【0035】
<水性相を構成する成分>
水性相を構成する成分としては、水、生理食塩水、エタノール等が挙げられる。これらは、1種でもよく、2種以上を混合して使用してもよい。溶媒としては、生体組織を傷つけないなどの観点より、生理食塩水がより好ましい。
【0036】
本発明の分散物を得る方法のうち、好ましい方法としては以下に述べる製造方法が挙げられる。
≪分散物の製造方法≫
本発明にかかる複合粒子の分散物は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製すること(以下、「リン脂質被覆粒子調製工程」ともいう。)と、該リン脂質被覆粒子及びアポリポタンパク質を混合すること(以下、「混合工程」ともいう。)とを含む製造方法により製造することができる。
本発明にかかる複合粒子の分散物の製造方法は、リン脂質被覆粒子調製工程と、混合工程とを含むことにより、簡便で効率のよい複合粒子の製造方法を提供しうるものである。
【0037】
<リン脂質被覆粒子調製工程>
本発明にかかる複合粒子の分散物の製造方法は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製するリン脂質被覆粒子調製工程を含む。
リン脂質被覆粒子調製工程においては、疎水性粒子表面におけるリン脂質の自己集合の原理を利用することで、リン脂質被覆粒子を調製することが可能である。
【0038】
疎水性粒子の粒径は、好ましくは1nm〜100nmであり、1nm〜50nmであることがより好ましく、1nm〜20nmであることがさらに好ましい。
疎水性粒子の粒径が1nm〜100nmの範囲内であれば、疎水性粒子表面のリン脂質の被覆率の制御が容易であり好ましい。
疎水性粒子の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載した疎水性粒子と同様のものが挙げられ、好ましい範囲も同様である。
【0039】
リン脂質の臨界ミセル濃度は0.1μg/ml〜10μg/mlである。
リン脂質の臨界ミセル濃度が0.1μg/ml未満である場合には、疎水性粒子表面を十分に被覆することができず、10μg/mlより高い臨界ミセル濃度を用いた場合には、リン脂質の水溶性が高すぎるため、疎水性粒子表面にリン脂質が吸着しない。
【0040】
なお、上記臨界ミセル濃度は、リン脂質のアシル基の長さにより適宜、上記範囲内において、最適の値に変更することができる。例えば、リン脂質としてホスファチジルコリン(PC)を用いた場合、アシル基の長さと臨界ミセル濃度との関係としては、PC(10:0/10:0)の場合には、2.0μg/ml〜6.0μg/mlであり、PC(12:0/12:0)の場合には、0.1μg/ml〜1.0μg/mlである。
ここで、PC(10:0/10:0)とは、ホスファチジルコリン中における2本のアシル基が飽和状態であり、該アシル基の炭素数が10であることを意味する。
【0041】
本明細書において、臨界ミセル濃度は、エレクトロスプレーイオン化質量分析法に従い測定する。溶媒はメタノール、クロロホルム及び純水(ppbレベル)の混合液である。
リン脂質の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載したリン脂質と同様のものが挙げられ、好ましい範囲も同様である。
【0042】
前記疎水性粒子及び前記リン脂質を混合する際の温度としては、特に制限はされないが、35℃〜37℃が好ましい。また、リン脂質被覆粒子調製工程の際には、約1分間のボルテックを必要とする。調整する時間としては、特に制限はされないが、3時間〜12時間が好ましい。また、疎水性粒子分散水溶液に既にクエン酸ナトリウムなどの分散安定剤が付加されている場合には、疎水性粒子を一度遠心し沈殿させ、その上澄みを純水に交換することが好ましい。
【0043】
<混合工程>
本発明にかかる複合粒子の分散物の製造方法は、前記リン脂質被覆粒子及びアポリポタンパク質を混合する混合工程を含む。
【0044】
アポリポタンパク質の添加量は、好ましくは0.1μg/ml〜10μg/mlであり、脳組織内の特定領域への蓄積の観点より1.0μg/ml〜10μg/mlであることがより好ましく、さらに好ましくは1.0μg/mlである。
アポリポタンパク質の種類や好ましい範囲等は、前記複合粒子を構成する成分について記載したアポリポタンパク質と同様のものが挙げられ、好ましい範囲も同様である。
【0045】
本発明における混合工程において、温度は特に制限はされないが、35℃〜37℃が好ましい。また、混合工程の際にはボルテックを必要としない。混合時間としては、特に制限はされないが、3時間〜5時間が挙げられる。
【0046】
なお、疎水性粒子である生分解性ポリマーに薬剤が内包されている場合には、例えば、前記リン脂質被覆粒子調製工程の前に、生分解性ポリマーに薬剤を内包する薬剤内包工程を含んでいてもよい。
薬剤内包工程としては、公知の方法を利用することができ、例えば、ACS NANO. 2008, 2(8), 1696−1702等に記載の方法に従い、生分解性ポリマーに薬剤を内包させることができる。
【0047】
さらに、別の態様として、本発明は、疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製すること(以下、「リン脂質被覆粒子調製工程」ともいう。)と、該リン脂質被覆粒子及びアポリポタンパク質を混合すること(以下、「混合工程」ともいう。)とを含む製造方法により得られる複合粒子を提供する。
【0048】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製するリン脂質被覆粒子調製工程と、該リン脂質被覆粒子及びアポリポタンパク質を混合する混合工程とは、前記複合粒子の製造方法に記載のリポタンパク質作製工程及び混合工程と同義であり、好ましい範囲等も同様である。
【0049】
≪脳神経変性疾患治療薬≫
本発明の脳神経変性疾患治療薬は、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を有効成分として含有する。
また、本発明における別の態様としては、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を患者に投与することを含む脳神経変性疾患の診断及び治療の方法を提供しうる。
本発明の脳神経変性疾患治療薬は、疎水性粒子である生分解性ポリマーと、該生分解性ポリマーに内包された薬剤と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を有効成分として含有することにより、脳内への移行性が良好で、優れた治療効果を発揮しうる、脳神経変性疾患治療薬を提供しうるものである。
【0050】
脳神経変性疾患としては、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症が挙げられる。
【0051】
薬剤としては、神経変性疾患治療剤が挙げられる。
神経変性疾患治療剤としては、アルツハイマー型認知症治療剤、パーキンソン症候群治療剤及び脊髄小脳変性症治療剤などが挙げられ、具体的には、脂溶性ビタミン;ビタミンA1、ビタミンD2、ビタミンα-、β-、γ-、δ-E等、脂肪酸;リノール酸、α-、γ-リノレン酸、アラキドン酸、エイコサペンタエン酸、ドコサエキサエン酸等、ドネペジル塩酸塩 (アリセプト(登録商標))などが挙げられ、脳神経機能の恒常性の維持及び促進の観点より、脂溶性ビタミン又は脂肪酸が好ましい。
【0052】
本発明の神経変性疾患治療薬の有効成分である分散物における前記複合粒子の含有量は、用途により適宜設定しうるが、治療効果の観点より、例えば、前記複合粒子を、分散物全量に対して、好ましくは1質量%〜10質量%含有する。
本発明の神経変性疾患治療薬の有効成分である分散物における薬剤の含有量は、特に制限はなく、各薬剤の有効投与量により適宜設定できる。例えば、薬剤としてビタミンA1を用いる場合には、疎水性粒子である生分解性ポリマーに対して、5.0×10−5質量%〜5.0×10−4質量%であることが好ましい。
【0053】
生分解性ポリマー、リン脂質及びアポリポタンパク質については、本発明の神経変性疾患治療薬の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【0054】
≪医薬組成物≫
本発明の医薬組成物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有する。
本発明の医薬組成物は、疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有することにより、脳内への移行性が良好で、副作用の発現頻度を抑制しうる医薬組成物を提供しうるものである。
【0055】
本発明の医薬組成物における前記複合粒子の含有量は、用途により適宜設定しうるが、治療効果の観点より、例えば、前記複合粒子を、医薬組成物の全固形分に対して、好ましくは1質量%〜10質量%含有する。
本明細書において、全固形分とは、医薬組成物中、溶液や生理食塩水を除いた、全成分の合計含量のことである。
【0056】
本発明の医薬組成物は、神経変性疾患治療、抗がん治療、精神疾患治療(抗うつ治療等)等の各種疾患の治療のために使用することができる。
具体的には、薬学上許容しうる担体(希釈剤、溶解補助剤など)と混合して得られる医薬組成物あるいは製剤の形態で、血管内投与、膀胱投与、腹腔内投与、局所投与等の方法で患者に投与することができる。
本発明の医薬組成物の投与量は、疎水性粒子や該疎水性粒子である生分解性ポリマーに内包された薬剤の種類や活性により、最適な投与量を決定することができる。例えば薬剤として、ビタミンA1を生分解性ポリマーに内包する場合を例に取れば、投与量はビタミンA1量として5mg/kg〜170mg/kgが好ましい。
【0057】
疎水性粒子、リン脂質及びアポリポタンパク質については、本発明の医薬組成物の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【0058】
≪造影補助試薬≫
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有する。
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物を含有することにより、BBB透過性が良好で、脳の恒常性の変化(脳の自己組織化)が生じていると考えられる領域の特定などに有用な造影補助試薬を提供しうるものである。
【0059】
本発明の造影補助試薬は、疎水性粒子としてのラベル用疎水性粒子を含む。前記ラベル用疎水性粒子としては、蛍光顕微鏡、レーザー共焦点顕微鏡、二光子顕微鏡、質量顕微鏡、電子顕微鏡、陽電子放射断層撮影法(PET)、単一光子放射コンピュータ断層撮影法(SPECT)、機能的核磁気共鳴画像法(fMRI)等により画像解析が可能なコントラスト像を生じさせる、公知のもの全てが挙げられる。中でも、金属ナノ粒子、ラベル用試薬を内包する生分解性ポリマー、量子ドット等が良好な画像化能を有する観点より好ましい。
【0060】
前記ラベル用試薬を内包する生分解性ポリマーにおける、ラベル用試薬としては、各種顕微観察技術でコントラスト像を生じさせるものであれば、特に限定されない。例えば、蛍光色素(例えば、Cell Tracker CM-DiI、Alexa Fluor-488、-594(登録商標)等)、放射線標識化合物(例えば、In-111、Tc-99m、I-123、I-125 F-18、Ga-67、Ga-68等)、不対スピン原子又はフリーラジカル(例えば、Fe、ランタイド類(lanthides)、Gd等)及び核磁気共鳴画像用造影剤(例えば、キレート化した(DTPA)マンガン等)等が挙げられる。
また、前記ラベル用試薬は、ラベル用疎水性粒子としての生分解性ポリマーに内包されるだけではなく、該生分解性ポリマーとは独立して、造影補助試薬に含有されていてもよい。造影補助試薬にかかる複合粒子の分散物が、ラベル用疎水性粒子として金属ナノ粒子や、量子ドットの他に、さらに、前記ラベル用試薬を含んでいる態様も本発明の造影補助試薬に含まれる。
【0061】
本発明にかかる造影補助試薬が、疎水性粒子に加えてさらにラベル用試薬を含む場合の製造方法について、ラベル用試薬が蛍光色素である場合を例に挙げて簡単に説明する。すなわち、本発明におけるリン脂質被覆粒子調製工程後に、該リン脂質被覆粒子が含まれる溶液に、適当量の蛍光色素を添加し、該蛍光色素で表面がラベルされたリン脂質被覆粒子と、アポリポタンパク質とを混合して、造影補助試薬を得ることもできる。
なお、本発明の造影補助試薬の製造方法はこれに制限されるものではなく、公知の方法を利用することができる。
【0062】
疎水性粒子、リン脂質及びアポリポタンパク質については、本発明の造影補助試薬の効果を損なわない範囲において、前記複合粒子を構成する成分について記載した全ての事項が適用される。
【実施例】
【0063】
以下、本発明を実施例により更に具体的に説明するが、本発明はその主旨を越えない限り、以下の実施例に限定されるものではない。
【0064】
<実施例1> 複合粒子(分散物)の調製
平均粒径20nmのコロイド状金ナノ粒子(G1652、Sigma−Aldrich社製)を最終濃度が50μg/mlとなるように、クエン酸ナトリウムなどの分散安定剤を遠心し除去した後、純水(ppbレベル)に分散させた。また、35℃の純水(ppb レベル)にPC(10:0/10:0)(850325P、Avanti Polar Lipids社製)を、最終濃度が1.0mg/mlとなるように溶解した後、0.1μmのカットオフ特性を持つ親水性メンブレンフィルターで、リポソーム及び凝集物を除去した。この調整したPC溶液(臨界ミセル濃度2.8μg/ml)及びコロイド状金ナノ粒子溶液を混合し、37℃で一晩攪拌 (約10時間撹拌) することにより、コロイド状金ナノ粒子表面に1層のPCが吸着した、リン脂質被覆金ナノ粒子を得た。
【0065】
次に、リン脂質被覆金ナノ粒子と、1.0μg/mlに調整した蛍光物質(商品名Cell Tracker CM-DiI、Molecular probes社製)とを30分間反応させた後、1.0μg/mlのアポリポタンパク質(A1、B100又はE4)をリン脂質被覆金ナノ粒子に吸着させた。
最後に、37℃、20,000×gで5分間の遠心を2回行い、上清を除いた沈殿を生理食塩水によって再分散させた後に、0.1μmのフィルターによって濾過して、複合粒子を得た。なお、リン脂質の複合粒子の全質量に対する含有量は4.5質量%である。
【0066】
図1に実施例1にかかる複合粒子の作製手順を模式的に示す。
図1は、コロイド状の金ナノ粒子をリン脂質で被覆し、リン脂質被覆金ナノ粒子を調製し、その後、蛍光物質で該リン脂質被覆金ナノ粒子をラベルし、最後にアポリポタンパク質をリン脂質被覆金ナノ粒子に吸着させ、複合粒子を得る工程を模式化した図である。
【0067】
作製したリン脂質被覆金ナノ粒子をTEMにより観察した。また、各複合粒子の平均粒径を動的光散乱法 (DLS:Dynamic Light Scattering)により評価した。
【0068】
図2は本実施例にかかるリン脂質被覆金ナノ粒子を観察したTEM像を示す。TEMで観察した結果、各複合粒子は、いずれも20nmの金ナノ粒子の表面に、3.3nmの1層のPCが吸着した形状を示すことが明らかとなった。
また、DLS測定の結果、各複合粒子の平均粒径は、それぞれ、51+/−0.0nm(複合粒子(Apo-A1))、30+/−1.9nm(複合粒子(Apo-B100))、38+/−3.8nm(複合粒子(Apo-E4))であった。
【0069】
<実施例2> 複合粒子の脳血管内皮細胞への取り込み機構の解明
細胞培養培地(商品名ダルベッコイーグル培地、和光社製)に、10%ウシ胎児血清(商品名Fetal Bovine Serum、Bio West社製)及び0.1%ゲンタマイシン(Sigma−Aldrich社製)を添加して、マウスの脳組織由来の血管内皮細胞(ATCC No.bEnd.3)を培養した。培養は37℃、5%CO2環境下で行なった。
細胞免疫化学的解析に供するために、ラット由来のコラーゲンでコーティングした1.1cm2のガラスプレートに、血管内皮細胞を播種し、24ウェルプレート(面積1.55×1.55cm2、高さ1.75cm)において、約2.5×104細胞/cm2のサブコンフルエンス状態まで培養させた。その後、前記細胞培養培地に、複合粒子の最終濃度が2.5μg/mlとなるように、実施例1で調整した複合粒子を添加し、1時間、3時間、6時間又は12時間培養して、各サンプルを得た。
その後、各サンプルをPBSで2度洗浄し、4%パラホルムアルデヒドで固定化し、10ng/mlのジギトニンで処理し、3% normal goat serum(Millipore社製)でブロッキングした。1時間後、各サンプルを、Clathirn HC H−300(sc−9069、Santa Cruz Biotechnology社製)又はLDL receptor (C-term)(1956-1、Epitomics社製)のいずれかとともに4℃で一晩反応させた。続いてAlexa Fluor-488 Goat Anti‐rabbit IgG (A11034、Molecular probes社製)を添加して反応させ、ビスベンズイミド H33342 三塩酸塩(商品名B2261、Sigma−Aldrich社製)を用いて核染色を行った。
【0070】
図3は本実施例にかかる各複合粒子(Apo-A1、Apo-B100及びApo-E4)の脳血管内皮細胞における時間経過に伴う取り込み量を示す。図3中、縦軸は複合粒子の血管内皮細胞への取り込み量(カウント/細胞)を示し、横軸は用いたサンプルの培養時間を示す。また、レーザー共焦点顕微鏡(ライカSP−II、ライカ社製)を用いて、1時間後、3時間後、6時間後及び12時間後それぞれの処理時間における、各複合粒子(Apo-A1、Apo-B100及びApo-E4)の血管内皮細胞への取り込み量の増加が確認された。
【0071】
図4は、本実施例にかかる複合粒子(Apo-A1)が脳血管内皮細胞のクラスリン被覆小胞(CV:Clathrin Vesicle)内に存在しているところを観察したTEM像並びにレーザー共焦点顕微鏡(ライカSP−II、ライカ社製)像を示す。12時間培養後の脳血管内皮細胞サンプルを観察した結果、各複合粒子(Apo-A1、Apo-B100及びApo-E4)が、クラスリンにより被覆され、血管内皮細胞に取り込まれたことを見出した。また同様に、各複合粒子(Apo-A1、Apo-B100及びApo-E4)は、LDLRを介して血管内皮細胞内へ輸送されるものではないことも見出した。
【0072】
これらの結果より、各複合粒子は、開口分泌されたクラスリン経路を介して、血液中から脳実質内に取り込まれる可能性があることが明らかとなった。
【0073】
<実施例3> 複合粒子の脳における自己組織化
5週齢のC57BL/6J雄性マウスに、生理食塩水で希釈した25μg/ml〜125μg/mlの各複合粒子 (Apo-A1、Apo-B100、Apo-E4) 溶液100μlを、尾静脈より投与した。なお、コントロールには生理食塩水を使用した。
【0074】
24時間後、50μg/mlのペントバルビタールを腹腔内注射し、麻酔下で切開し、0.1Mのリン酸緩衝生理食塩水で毎分1mlの流速で血液を灌流し、4%パラホルムアルデヒド溶液で脳組織を固定した。固定後、5%スクロースで脱水し、矢状方向に半割した脳試料を厚さ約10μmでミクロトームを用い連続切片を作製した。メタノール洗浄により自家蛍光を持つ付着物を除去したスライドガラスに固定化し、蛍光顕微鏡による観察に供した。
【0075】
図5は各複合粒子 (Apo-A1、Apo-B100、Apo-E4)の脳組織内における分布図を示す。また、脳組織領域別における各複合粒子の蓄積度合を比較した結果、複合粒子(Apo-B100)は、複合粒子(Apo-A1或いはApo-E4)に比べ、大脳皮質、線条体、小脳及び海馬に局在化することが可能であった。
【0076】
図6は脳連続切片(100枚)における複合粒子(Apo-B100)を取込んだ脳神経細胞数を示す。コントロールは生理食塩水を投与した。エラーバーは各群マウス3匹あたりの標準偏差であり、 図6中、*P<0.0005はStudent's t-testを用い示した。この結果、複合粒子(Apo-B100)は、統計学的解析より確実にBBB透過性能を持っていることが証明された。
【0077】
図7及び図8は複合粒子(Apo-B100)を取込んだ脳組織領域別における脳神経細胞数を示す。
この結果、複合粒子(Apo-B100)は、小脳及び終脳の特定領域に局在化することが可能であった。
【0078】
以上の結果より、本発明にかかる複合粒子は、BBB透過性が良好であるとともに、脳組織内の特定領域への局在化が可能であることが明らかとなった。
【特許請求の範囲】
【請求項1】
疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
【請求項2】
前記複合粒子がアポリポタンパク質を表面に有する請求項1に記載の複合粒子を有する分散物。
【請求項3】
前記リン脂質の複合粒子の全質量に対する含有量が、0.2質量%〜20質量%である請求項1又は請求項2に記載の複合粒子を有する分散物。
【請求項4】
前記アポリポタンパク質が、0.1μg/ml〜10μg/mlの量で含まれる請求項1〜請求項3のいずれか1項に記載の複合粒子を有する分散物。
【請求項5】
前記アポリポタンパク質が、アポリポタンパク質A1、アポリポタンパク質B100及びアポリポタンパク質E4からなる群より選ばれる少なくとも1種のアポリポタンパク質である請求項1〜請求項4のいずれか1項に記載の複合粒子を有する分散物。
【請求項6】
前記疎水性粒子が、金属ナノ粒子又は生分解性ポリマーである請求項1〜請求項5のいずれか1項に記載の複合粒子を有する分散物。
【請求項7】
前記疎水性粒子の粒径が1nm〜100nmである請求項1〜請求項6のいずれか1項に記載の複合粒子を有する分散物。
【請求項8】
前記複合粒子の粒径が1nm〜100nmである請求項1〜請求項7のいずれか1項に記載の複合粒子を有する分散物。
【請求項9】
前記疎水性粒子が、生分解性ポリマーであり、かつ該生分解性ポリマーが薬剤を内包している請求項1〜請求項8のいずれか1項に記載の複合粒子を有する分散物。
【請求項10】
請求項9に記載の複合粒子を有する分散物を有効成分として含有する脳神経変性疾患治療薬。
【請求項11】
脳神経変性疾患が、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症からなる群より選ばれる少なくとも1種の疾患である請求項10に記載の脳神経変性疾患治療薬。
【請求項12】
請求項1〜請求項9のいずれか1項に記載の複合粒子を有する分散物を含有する医薬組成物。
【請求項13】
請求項1〜請求項8のいずれか1項に記載の複合粒子を有する分散物を含有する造影補助試薬。
【請求項14】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む複合粒子を有する分散物の製造方法。
【請求項15】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む製造方法により得られる複合粒子を有する分散物。
【請求項1】
疎水性粒子と、臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質と、アポリポタンパク質と、を含む複合粒子を有する分散物。
【請求項2】
前記複合粒子がアポリポタンパク質を表面に有する請求項1に記載の複合粒子を有する分散物。
【請求項3】
前記リン脂質の複合粒子の全質量に対する含有量が、0.2質量%〜20質量%である請求項1又は請求項2に記載の複合粒子を有する分散物。
【請求項4】
前記アポリポタンパク質が、0.1μg/ml〜10μg/mlの量で含まれる請求項1〜請求項3のいずれか1項に記載の複合粒子を有する分散物。
【請求項5】
前記アポリポタンパク質が、アポリポタンパク質A1、アポリポタンパク質B100及びアポリポタンパク質E4からなる群より選ばれる少なくとも1種のアポリポタンパク質である請求項1〜請求項4のいずれか1項に記載の複合粒子を有する分散物。
【請求項6】
前記疎水性粒子が、金属ナノ粒子又は生分解性ポリマーである請求項1〜請求項5のいずれか1項に記載の複合粒子を有する分散物。
【請求項7】
前記疎水性粒子の粒径が1nm〜100nmである請求項1〜請求項6のいずれか1項に記載の複合粒子を有する分散物。
【請求項8】
前記複合粒子の粒径が1nm〜100nmである請求項1〜請求項7のいずれか1項に記載の複合粒子を有する分散物。
【請求項9】
前記疎水性粒子が、生分解性ポリマーであり、かつ該生分解性ポリマーが薬剤を内包している請求項1〜請求項8のいずれか1項に記載の複合粒子を有する分散物。
【請求項10】
請求項9に記載の複合粒子を有する分散物を有効成分として含有する脳神経変性疾患治療薬。
【請求項11】
脳神経変性疾患が、アルツハイマー型認知症、パーキンソン症候群及び脊髄小脳変性症からなる群より選ばれる少なくとも1種の疾患である請求項10に記載の脳神経変性疾患治療薬。
【請求項12】
請求項1〜請求項9のいずれか1項に記載の複合粒子を有する分散物を含有する医薬組成物。
【請求項13】
請求項1〜請求項8のいずれか1項に記載の複合粒子を有する分散物を含有する造影補助試薬。
【請求項14】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む複合粒子を有する分散物の製造方法。
【請求項15】
疎水性粒子及び臨界ミセル濃度が0.1μg/ml〜10μg/mlのリン脂質を混合してリン脂質被覆粒子を調製することと、該リン脂質被覆粒子及びアポリポタンパク質を混合することとを含む製造方法により得られる複合粒子を有する分散物。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2013−35793(P2013−35793A)
【公開日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願番号】特願2011−174323(P2011−174323)
【出願日】平成23年8月9日(2011.8.9)
【出願人】(803000115)学校法人東京理科大学 (545)
【Fターム(参考)】
【公開日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願日】平成23年8月9日(2011.8.9)
【出願人】(803000115)学校法人東京理科大学 (545)
【Fターム(参考)】
[ Back to top ]