
医薬固形製剤
【課題】本発明は、消化管上部での薬剤の放出を抑え、かつ消化管下部において薬剤の放出を速やかにする高度な放出制御性を備えていながら、可塑剤配合による上記欠点を悉く解消したマトリックス型固形製剤を提供することを課題とする。
【解決手段】本発明の製剤は、(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、前記糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃)の量が4g以下である、医薬固形製剤である。
【解決手段】本発明の製剤は、(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、前記糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃)の量が4g以下である、医薬固形製剤である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、医薬固形製剤に関する。
【背景技術】
【0002】
医薬の分野において、薬剤放出を制御し、薬剤血中濃度を適正レベルに長時間維持させる試みが広く行われている。薬剤血中濃度を適正レベルに長時間維持するためには、薬剤の吸収が長時間持続するような製剤学的工夫(徐放化技術)が必要である。経口投与された固形製剤は、時間の経過に伴って消化管の上部(胃及び小腸上部)から下部(小腸下部及び大腸)へ移動するが,薬剤の吸収性は消化管の上部に比べて下部では低い場合が非常に多い。そのため,固形製剤の滞留時間が最も長い消化管下部において,薬剤が持続的に吸収できるような方策を立てることがなにより重要と考えられる。
【0003】
徐放化技術としては、例えば、拡散制御に基づいた徐放化技術、より具体的には、薬剤を含有する核組成物又は核錠子に水不溶性高分子化合物の被膜を施したメンブランコーティング製剤;水不溶性高分子化合物、ワックス等とともに製造されるマトリックス製剤等が知られている。しかしながら、これら技術では、薬剤の放出が進行するのに従って放出速度が減少してゆくため、服用後数時間経過して到達する消化管下部では薬剤の放出速度が不足し、薬剤血中濃度の維持が困難となる。
【0004】
また、消化管下部における薬剤放出を意図した製剤的手法が施された徐放化技術(例えば、速放性の薬剤含有核組成物に腸溶性被膜を被覆した腸溶性製剤等)が知られている。しかしながら、この技術では、薬剤の放出を被膜によって制御するため、フィルムコーティング操作を必要とする。その結果、医薬製剤の製造工程が煩雑となる。
【0005】
一方、メタクリル酸系の腸溶性高分子を用いて徐放化されたマトリックス製剤が知られている。腸溶性高分子化合物は溶解できるpHより低いpH領域では不溶性物質となり、溶解できるpH以上のpH領域では可溶性物質となる。従って、腸溶性高分子化合物が配合されたマトリックス製剤は消化管上部では薬剤放出を抑制し、消化管下部で速やかに薬剤を放出させることができる。つまり、腸溶性高分子化合物の鋭いpH応答性により、精緻な薬剤放出制御を備える徐放性製剤を提供することが可能になる。
【0006】
例えば、腸溶性高分子化合物及び薬剤を混合した後、圧縮成形法(打錠)によって腸溶性高分子化合物が配合されたマトリックス製剤が知られている(例えば、特許文献1、特許文献2、特許文献3、特許文献4等)。しかしながら、マトリックス製剤からの薬剤放出は,一般に製剤の表面積に依存すると考えられているが、このような打錠により得られるマトリックス製剤は、溶媒と接触する表面積が小さいので、溶解度が低く、溶解速度が遅い難溶性薬剤の場合には、製剤の小さな表面積が薬剤放出の障害となる。
【0007】
これに対し、メタクリル酸コポリマーSを含む混合粉末をエタノールで湿式練合し、押出造粒法によって得られる製剤が知られている(特許文献5)。この製剤は、湿式練合し、押出造粒法を経ることで徐放性の粒状剤となる。このような粒状剤(粒状剤、顆粒剤、散剤は、マルチプルユニット型と呼ばれている)は、製剤の表面積を大きくでき、難溶性薬剤にも適用することができる。さらに、打錠して得られる錠剤のようなシングルユニット型に比べて、粒状剤のようなマルチプルユニット型は、消化管内で適度に分散されるので、錠剤よりも薬剤吸収の個体間変動を小さく抑えることができる。
【0008】
さらに、マルチプルユニット型の製剤として、メタクリル酸コポリマーS、メタクリル酸コポリマーLD、薬剤、ポリビニルピロリドン及び可塑剤としてクエン酸トリエチルを含む混合粉末を水で湿式練合し、得られた練合物を押出造粒し、次いで球形整粒して顆粒が得られることが知られている(非特許文献1)。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特公平4−43049号公報
【特許文献2】特開平6−199657号公報
【特許文献3】米国特許4,968,508号明細書
【特許文献4】米国特許公開2006/0159753号公報
【特許文献5】特開平6−24991号公報
【非特許文献】
【0010】
【非特許文献1】International Journal of Pharmaceutics,2001年,第213巻,7-12頁
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、上記の腸溶性高分子化合物を含有する混合物を、押出造粒法に続いて球形整粒法により、粒状剤や顆粒剤を得る方法では、下記のような問題点がある。
【0012】
まず、メタクリル酸系の腸溶性高分子化合物は、そのガラス転移温度が160℃以上と高温であることから推察されるように堅い腸溶性高分子である。そのため、押出造粒法、球形整粒法によってメタクリル酸系腸溶性高分子化合物を含むマトリックス製剤を製造する場合、押出造粒を容易に行うために、湿式練合物の塑性を補うことが必要となる。
【0013】
上記特許文献5の技術では、メタクリル酸コポリマーSの配合割合を5重量%としているが、得られる医薬製剤のpH応答性を一段と向上させるためにはメタクリル酸コポリマーSの配合量をより多くする必要がある。しかし、配合量を多くすると塑性が不十分となり、押出造粒法、球形整粒法によってメタクリル酸系の腸溶性高分子を含むマトリックス製剤を製造しにくくなる。つまり、塑性が不十分であると、押出造粒する時、練合物を押出す際の抵抗が大きくなり、押出造粒できなくなる。
【0014】
一方、上記非特許文献1に記載の技術では、77重量%のメタクリル酸コポリマーを含有しており、練合物を押出造粒する際に必要な塑性を補うために、可塑剤としてクエン酸トリエチルを約11重量%添加している。しかしながら、この技術では、可塑剤の添加によりメタクリル酸系腸溶性高分子化合物のガラス転移温度が低下し、メタクリル酸系腸溶性高分子化合物の変質・変形が促進されるために、スクリーンの内側に膜を形成する可能性があり、閉塞や装置の故障の原因となりえる。さらに、得られた製剤中に可塑剤が残存するため、メタクリル酸系腸溶性高分子化合物の経時的な変質・変形を促進し、その結果、経時的な溶出変化が生じる可能性が高い。また、非特許文献1に記載の技術では、薬剤との不適合も懸念される。
【0015】
このようにメタクリル酸系腸溶性高分子化合物の配合比率を高くするためには可塑剤が必要となる一方において、可塑剤を添加すると上記のような問題点が生じるのを回避できない。換言すれば、可塑剤を添加することによる問題点を解決しながら腸溶性高分子化合物の配合比率を高くすることは極めて困難である。
【0016】
本発明は、徐放化するためにpH応答性が良好なメタクリル酸系の腸溶性高分子化合物の配合比率を高くしても、上記従来技術のような問題点が生じない医薬固形製剤を提供することである。
【0017】
すなわち、本発明は、消化管上部での薬剤の放出を抑え、かつ消化管下部において薬剤の放出を速やかにする高度な放出制御性を備えていながら、可塑剤配合による上記欠点を悉く解消したマトリックス型固形製剤を提供することを課題とする。
【課題を解決するための手段】
【0018】
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、メタクリル酸系腸溶性高分子化合物と特定の性質を備えた糖及び/又は糖アルコールとを併用することにより所望のマトリックス型固形製剤が得られることを見い出した。本発明は、かかる知見に基づき完成されたものである。
【0019】
本発明は、項1〜項13に係るマトリックス型の医薬固形製剤を提供する。
項1.(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、前記(b)糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である、医薬固形製剤。
項2.糖及び/又は糖アルコールの配合量が、メタクリル酸系腸溶性高分子化合物1重量部に対して、0.1〜10重量部である、項1に記載の医薬固形製剤。
項3.糖及び/又は糖アルコールは、融点が140℃以下である、項1又は項2に記載の医薬固形製剤。
項4.可塑剤を含有していない項1又は項2に記載の医薬固形製剤。
項5.可塑剤を含有していない項3に記載の医薬固形製剤。
項6.押出造粒工程を有する製造方法により製造される、項1又は項2に記載の医薬固形製剤。
項7.押出造粒工程を有する製造方法により製造される、項3に記載の医薬固形製剤。
項8.糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種である、項1〜項7のいずれかに記載の医薬固形製剤。
項9.メタクリル酸系腸溶性高分子化合物の含有量が、6〜50重量%である、項1に記載の医薬固形製剤。
項10.メタクリル酸系腸溶性高分子化合物は、pH5.5以上で溶解する性質を備えている、項1に記載の医薬固形製剤。
項11.メタクリル酸系腸溶性高分子化合物は、ガラス転移温度が100℃以上である、項1に記載の医薬固形製剤。
項12.メタクリル酸系腸溶性高分子化合物は、メタクリル酸コポリマーL、メタクリル酸コポリマーLD及びメタクリル酸コポリマーSからなる群から選ばれた少なくとも1種である、項1〜項11のいずれかに記載の医薬固形製剤。
項13.シロスタゾール、トルバプタン、フェニトイン、アスピリン及びナプロキセンからなる群から選ばれた少なくとも1種の薬剤を含む、項1に記載の医薬固形製剤。
【0020】
医薬固形製剤
本発明のマトリックス型の医薬固形製剤は、(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有する。
【0021】
(b)の糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である性質を備えている。
【0022】
本発明の医薬固形製剤には、上記(a)及び(b)成分の他、他の成分を含んでいてもよく、好ましくは(c)薬剤及び(d)形状維持物質が含まれている。本発明の医薬固形製剤は、可塑剤を含有していない。
【0023】
本発明の医薬固形製剤は、マトリックス型の経口徐放性医薬固形製剤であることが好ましい。
【0024】
(a)メタクリル酸系腸溶性高分子化合物
本発明において、(a)メタクリル酸系腸溶性高分子化合物は、小腸下部及び大腸でのpH環境域で溶解するものである限り、公知のものを広く使用できる。pHが5.5以上、より好ましくはpHが6.0で、かつpHが7.5以下で溶解するメタクリル酸系腸溶性高分子化合物が好ましい。溶解するpHが上記範囲内にあると、当該腸溶性高分子化合物が小腸内及び/又は大腸内で溶解するので、消化管下部において製剤から薬剤を速やかに放出させることができる。
【0025】
更に、本発明において、メタクリル酸系腸溶性高分子化合物のガラス転移温度は、通常100℃以上、好ましくは105℃以上、より好ましくは130℃以上である。該ガラス転移温度は200℃以下であるのが望ましい。ガラス転移温度がこの範囲にあると、常温で変形及び変質することがないので、製剤の経時的な溶出変化の懸念が少なくなり、また押出造粒時に装置へ過度の負荷を発生させずに処理できる等の利点がある。
【0026】
上記メタクリル酸系腸溶性高分子化合物の好ましい具体例としては、メタクリル酸コポリマーLD、メタクリル酸コポリマーL、メタクリル酸コポリマーS等が挙げられる。メタクリル酸コポリマーLDは粉末状であるのが好ましい。粉末状のメタクリル酸コポリマーLDとは、液状や、固形分30%の懸濁液でないものを意味する。また、本発明で用いられるメタクリル酸コポリマーLDは、乾燥している状態、又は多少水分を含んでいる場合でも粉末状であればよい。以下、このような粉末状のメタクリル酸コポリマーLDを、乾燥メタクリル酸コポリマーLDとする場合がある。
【0027】
メタクリル酸系腸溶性高分子化合物は、入手が容易な市販品をいずれも使用でき、例えば、乾燥メタクリル酸コポリマーLDはデグサ社製の「Eudragit L100D55」を、メタクリル酸コポリマーLはデグサ社製の「Eudragit L100」を、メタクリル酸コポリマーSはデグサ社製の「Eudragit S100」を、それぞれ使用できる。これらの腸溶性高分子化合物は、1種単独で又は2種以上混合して使用される。
【0028】
上記腸溶性高分子化合物を2種以上混合することにより、製剤が小腸下部及び大腸で溶解するpH(腸溶性高分子化合物の溶解pH)を5.5〜7の間で任意に設定することができる。例えば、腸溶性高分子化合物の溶解pHを5.5〜6の間で任意に設定する場合は、メタクリル酸コポリマーLD:メタクリル酸コポリマーLの混合比率を1:99〜99:1の範囲で混合すればよい。また、腸溶性高分子化合物の溶解pHを5.5〜7の間で任意に設定する場合は、メタクリル酸コポリマーLD:メタクリル酸コポリマーSの混合比率を1:99〜99:1の範囲で混合すればよい。更に、腸溶性高分子化合物の溶解pHを6〜7の間で任意に設定する場合は、メタクリル酸コポリマーL:メタクリル酸コポリマーSの混合比率を1:99〜99:1の範囲で混合すればよい。
【0029】
本発明において、製剤に含有されるべき腸溶性高分子化合物の量は、通常1〜50重量%であり、好ましくは3〜45重量%であり、より好ましくは6〜40重量%であり、さらに好ましくは10〜35重量%である。腸溶性高分子化合物の含有量が上記範囲であると、薬物放出パターンのバラエティ、生産適性(押出造粒が良好であること)の点で好ましい。ここで、薬物放出パターンのバラエティとは、製剤の設計者が意図する薬物放出パターンを容易に達成しやすくなることを意味する。
【0030】
本発明の医薬固形製剤に使用されるメタクリル酸系腸溶性高分子化合物はpH5.5以上で溶解するのが好ましい。本発明の医薬固形製剤に付与すべきpH応答性は、以下に列記した薬剤の種類、望まれる薬理効果等により適宜調整される。また、そのpH応答性は使用されるメタクリル酸系腸溶性高分子化合物の種類、製剤中の腸溶性高分子化合物の含有量等によって調整可能である。
【0031】
例えば、消化管下部(小腸下部及び大腸)で持続的に吸収できる機能に加え、消化管上部(胃及び小腸上部)から薬剤が緩やかに放出されるように製剤設計する方が好ましい場合がある。その場合には医薬固形製剤のpH応答性を緩和するのが望ましく、具体的には医薬固形製剤中におけるメタクリル酸系腸溶性高分子化合物の含有量を少なめに設定する。
【0032】
また、消化管下部で持続的に吸収できる機能をより充実させたい場合には、医薬固形製剤のpH応答性を鋭くするのが望ましく、具体的には医薬固形製剤中におけるメタクリル酸系腸溶性高分子化合物の含有量を多めに設定する。
【0033】
(b)糖及び/又は糖アルコール
本発明で使用される糖及び/又は糖アルコールは、水に対する特定の溶解性を有している。糖及び/又は糖アルコール1gを溶解するのに要する、20〜25℃の間の水温での水の量は、通常4g以下、好ましくは3.5g以下である。また、糖及び/又は糖アルコール1gを溶解するのに要する、20〜25℃の間の水温での水の量は、1g以上であるのが望ましい。溶解するのに要する水の量がこの範囲であると、製剤製造前の練合混合物に適度な塑性を付与することができる。
【0034】
より好ましい糖及び/又は糖アルコールは、融点が140℃以下、好ましくは130℃以下、より好ましくは125℃以下であり、90℃以上である。融点がこの範囲であると、糖及び/又は糖アルコールが常温で固体形状を採り得るので取り扱い易くなる。他に、当該糖及び/又は糖アルコール自体の堅さが押出造粒時に影響を及ぼさない利点がある。
【0035】
本発明において用いることができる糖及び/又は糖アルコールは、上記のような特性を有するものであればよく、水和物の形態であってもよい。そのような糖及び/又は糖アルコールとしては、例えば、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種が挙げられる。好ましい糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種が挙げられる。より好ましい糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース及びマルトースからなる群から選ばれた少なくとも1種である。さらにより好ましい糖及び/又は糖アルコールは、エリスリトール、ラクチトール一水和物、トレハロース二水和物及びマルトース一水和物からなる群から選ばれた少なくとも1種である。これらの更により好ましい糖及び/又は糖アルコールは、適度な溶解性及び適度な融点を有しており、かつ吸湿性の懸念が少なく、長期保管での安定性が良好である。
【0036】
糖及び/又は糖アルコールは、市販品を広く用いることができる。より具体的には、以下のものが挙げられる。
【0037】
(B-i) エリスリトール及びその水和物
糖アルコールであるエリスリトールは、酵素反応によってブドウ糖から製造される。エリスリトールの融点は119〜122℃、エリスリトール1g溶解するのに要する水(25℃)の量は3.3gである(日研化学社製、エリスリトール技術資料)。エリスリトールとしては、市販品、例えば、日研化学社の「エリスリトール100M」等を使用できる。
【0038】
(B-ii) キシリトール及びその水和物
糖アルコールであるキシリトールは、種々のセルロース原料を加水分解によってキシロースに変換後、水素添加して製造される。キシリトールは、若干の吸湿性を有している。1gのキシリトールを溶解するのに要する水の量(20℃)は1.6gである(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。更に、キシリトールの融点は93〜95℃である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。キシリトールとしては、市販品、例えば、日研化成社の「キシリトールP」、ロケット社の「XYLISORB」、東和化成工業の「キシリットP」等を使用できる。
【0039】
(B-iii) ラクチトール及びその水和物
糖アルコールであるラクチトールは、乳糖を触媒水素添加して製造される。ラクチトールには、無水物、一水和物、二水和物及び三水和物が含まれる。そのうち好ましいのは非吸湿性の一水和物である。ラクチトール一水和物の融点は97℃(Merck Index 第12版)、1gのラクチトールを溶解するのに要する水の量(20℃)は1.8gである(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。ラクチトール一水和物としては、市販品、例えば、日研化成社の「ラクチトールLC−1」等を使用できる。
【0040】
(B-iv) ソルビトール及びその水和物
糖アルコールであるソルビトールは、ブドウ糖又はトウモロコシシロップを高圧水素添加するか、電解還元することによって製造される。ソルビトールは吸湿性が強い。1gのソルビトールを溶解するのに要する水の量(25℃)は0.5ml(g)である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。更に、ソルビトールの融点は97〜112℃である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編、Journal of Thermal Analysis and Calorimetry、第73巻、615-621頁)。ソルビトールとしては、市販品、例えば、日研化成社の「ソルビトールSP」、ロケット社の「NEOSORB Powder」、東和化成工業の「ソルビットDP−10M」等を使用できる。
【0041】
(B-v) トレハロース及びその水和物
糖成分であるトレハロース二水和物は、マルトースと同じくグルコースが2個結合した二糖類である。医薬品添加剤としてのトレハロースは、澱粉部分分解物からトレハロース産生細菌を用いた酵素法によって製造される。トレハロース二水和物は、非吸湿性ではないが、吸湿性が少ない。トレハロース二水和物の融点は、97℃であり、1gのトレハロース二水和物を溶解するのに要する水の量(20℃)は1.2gである(いずれもトレハロース技術資料,林原生物化学研究所)。トレハロース二水和物としては、市販品、例えば、旭化成ケミカルズ社の「トレハロースP」、林原社の「トレハ」等を使用できる。
【0042】
(B-vi) マルトース及びその水和物
糖成分であるマルトース一水和物は、二糖類の炭水化物で、デンプンの酵素分解により製造される。マルトース含量が90%以上であれば医薬品添加物のアメ粉として医薬品にも使用することができる。マルトースには無水物と一水和物があり、一水和物は吸湿性が低い。New Food Industry、第31巻4号、17-22頁によると、1gの含水結晶マルトースを溶解させるのに要する水の量(20℃)は1.2gである。更に、医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編によると、マルトース一水和物の融点(分解)は102〜103℃である。マルトース一水和物としては、市販品、例えば、三和澱粉工業社の「サンマルト−S」,日本食品化工社の「日食結晶マルトース」等を使用できる。
【0043】
本発明においては、(b)の糖及び/又は糖アルコールを配合することにより、可塑剤を使用することなく、メタクリル酸系腸溶性高分子化合物を含有する練合混合物の塑性を高めることができる。本発明では、可塑剤を使用しないので、可塑剤使用に起因する種々の弊害を防止できる。このため、製剤の安定性が向上し、薬剤の溶出性を容易にコントロールすることができる。
【0044】
本発明において、メタクリル酸系腸溶性高分子化合物と糖及び/又は糖アルコールの配合割合は、メタクリル酸系腸溶性高分子化合物1重量部に対して、糖及び/又は糖アルコールが通常0.01〜20重量部、好ましくは0.1〜10重量部、より好ましくは0.2〜5重量部である。糖及び/又は糖アルコールの含有量が上記の範囲にあると、製剤が取り扱い易くなる、服用し易くなる、練合混合物の塑性が向上する、製造適性(製造しやすさ)の点で好ましい。
【0045】
前記具体的に記載したその他の糖及び/又は糖アルコールは、日本医薬添加物協会訳編、医薬品添加物ハンドブック(2001年)によれば、融点並びに糖及び/又は糖アルコールの1gを溶解させるのに要する水量は、ショ糖では160〜186℃、0.5gであり、デキストロースでは83℃、1gであり、フルクトースでは102〜105℃、0.3gであり、マルチトールでは148〜151℃、溶けやすい、である。
【0046】
(c)薬剤
薬剤は、医薬活性成分として疾病の治療あるいは予防に供されるものであれば、特に限定されず、それらの薬剤はフリー体、その塩、溶媒和物(水和物、エタノール和物など)、又は結晶多形を用いることができる。本発明で配合される薬剤は、特に、本発明の徐放化によって副作用の発現が抑えられ、治療効果が高まるような薬剤が適している。更に、クローン病、潰瘍性大腸炎、過敏性大腸炎、結腸癌など消化管下部に疾患部位があるような疾患に対して、薬剤を速やかに放出することが治療効果の増大に繋がるような薬剤が適している。
【0047】
薬剤は、結晶性であっても非結晶性であってもよい。薬剤は、水溶性及び脂溶性のいずれであってもよく、水に対して難溶性であってもよい。薬剤は、弱塩基性、中性又は酸性であるのが好ましい。
【0048】
薬剤が難溶性の場合、難溶性の薬剤の溶解性を改善するために、ナノ粉砕、微粉砕、非晶質化などといった製剤技術を用いることがあるが、前記背景技術に記載の特許文献5に記載のエタノールで湿式練合している技術では、エタノールが薬剤の結晶化、結晶成長化などの問題を引き起こす恐れがある。
【0049】
本発明に用いられる薬剤としては、例えば、5−アミノサリチル酸、アシクロビル、アスピリン、アセチルサリチル酸、アセトアミノフェン、アリピプラゾール、アンピシリン、イソジアニド、イブプロフェン、インドメタシン、エテンザミド、エナラプリル、エリスロマイシン、オメプラゾール、ケトコナゾール、サルブタモール、サラゾスルファピリジン、サラゾピリン、ジアゼパム、ジクロフェナク、ジクロフェナクナトリウム、ジピリダモール、シメチジン、シロスタゾール、シンバスタチン、スクラルファート、スルピリド、スルファサラジン、セレコキシブ、タクロリムス、テオフィリン、テガフール、デキサメタゾン、デキストロメトルファン、テトミラスト、テルフェナジン、ドキソルビシン、トリアムシロノン、トルバプタン、ナジフロキサシン、ナプロキセン、ニフェジピン、尿素、バルプロ酸ナトリウム、ハロペリドール、バロシクロビル、バリペリドン、ヒドロコルチゾン、ファモチジン、フェナセチン、フェニトイン、フェニルプロパノールアミン、プデソニド、プラバスタチン、プラバスタチンナトリウム、フルオロウラシル、プレドニゾロン、プレドニゾン、フロセミド、プロブコール、ベスナリノン、ペニシリン、ペルフェナジン、マレイン酸クロルフェニラミン、ミダゾラム、メシル酸ドキサゾシン、メトトレキセート、モルヒネ、ラニチジン、ランソプラゾール、リシノプリル、リスペリドン、リドカイン、レバミピド、レポドパ、ロチゴチン、ロバスタチン、ロラゼパム、ワーファリン、塩酸アンブロキソール、塩酸カルテオロール、塩酸ジフェンヒドロミン、塩酸タムスロシン、塩酸ニカルジピン、塩酸ヒドララジン、塩酸ブプレノルフィン、塩酸プロカテロール、塩酸モザバプタン、塩酸ラニチジン、塩酸レボカルニチン、酢酸コルチゾン、硫酸サルブタモール等が挙げられる。
【0050】
好ましい薬剤としては、5−アミノサリチル酸、アシクロビル、アスピリン、アセチルサリチル酸、アセトアミノフェン、アリピプラゾール、イブプロフェン、インドメタシン、エテンザミド、オメプラゾール、サラゾスルファピリジン、サラゾピリン、ジアゼパム、ジクロフェナク、ジクロフェナクナトリウム、ジピリダモール、シロスタゾール、シンバスタチン、タクロリムス、テオフィリン、テガフール、テトミラスト、ドキソルビシン、トルバプタン、ハロペリドール、バリペリドン、ヒドロコルチゾン、フェニトイン、プデソニド、プラバスタチン、フルオロウラシル、プレドニゾロン、プレドニゾン、フロセミド、プロブコール、ベスナリノン、ランソプラゾール、リスペリドン、レバミピド、レポドパ、ロチゴチン、ロバスタチン、塩酸カルテオロール、塩酸ニカルジピン、塩酸プロカテロール、塩酸モザバプタン、酢酸コルチゾン、硫酸サルブタモール等が挙げられる。より好ましい薬剤は、シロスタゾール、トルバプタン、フェニトイン、アスピリン及びナプロキセンである。
【0051】
上記薬剤の含有量は、医薬製剤中、通常1〜90重量%、好ましくは5〜80重量%、より好ましくは10〜70重量%である。これらの薬剤は、本発明の医薬製剤に、1種単独で又は2種以上混合して使用される。
【0052】
本発明において、上記のような薬剤を用いて徐放性医薬固形製剤を製造すると、1日1〜2回程度の投与が可能となる。例えば、トルバプタンは、バソプレシン拮抗剤として、例えば、血管拡張作用、血圧降下作用、肝糖放出抑制作用、メサンギウム細胞増殖抑制作用、水利尿作用、血小板凝集抑制作用、嘔吐抑制作用、尿素排泄促進作用、第VIII因子分泌抑制作用、心機能亢進作用、メサンギウム細胞収縮抑制作用、肝糖新生抑制作用、アルドステロン分泌抑制作用、エンドセリン産生抑制作用、レニン分泌調節作用、記憶調節作用、体温調節作用、プロスタグランジン産生調節作用等を有し、血管拡張剤、降圧剤、水利尿剤、血小板凝集抑制剤、尿素排泄促進剤、抗心不全剤、抗腎不全剤等として有利であり、高血圧、浮腫、腹水、心不全、腎機能障害、バソプレシン分泌異常症候群(SIADH)、肝硬変、低ナトリウム血症、低カリウム血症、糖尿病、循環不全、動揺病、水代謝障害、腎不全、各種虚血性疾患等の予防及び/又は治療に有効である。また、トルバプタンは、オキシトシン拮抗剤としては、例えば、子宮平滑筋収縮抑制作用、乳汁放出抑制作用、プロスタグランジン合成及び放出抑制作用、血管拡張作用を有し、オキシトシン関連疾患、特に早期分別、月経困難、帝王切開前の出産防止等の予防及び/又は治療に有効である。また、トルバプタンは、多発性のう胞腎の予防及び/又は治療に有効である。これら薬剤は、本発明の徐放化技術を用いると、1日1回投与の製剤を提供することができる。
【0053】
(d)形状維持物質
本発明において(d)形状維持物質とは、種々の製造工程において製造中の製剤に所望の形状を維持できるようなものであればよく、好ましくは押出造粒法及び球形整粒法を経て製造される医薬製剤の形状を維持できるような物質を意味する。
【0054】
本発明において、(d)形状維持物質としては、保水性があり、膨潤性、塑性を備えている。使用される形状維持物質としては、例えば、ヒドロキシプロピルセルロース、ヒプロメロース、メチルセルロース、ヒドロキシエチルセルロース等の水溶性セルロース;結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、クロスカルメロースナトリウム、カルボキシメチルエチルセルロース、エチルセルロース、ヒプロメロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、酢酸セルロース、酢酸フタル酸セルロース等の水不溶性セルロース;ポリビニルピロリドン、ポリエチレンオキサイド、カルボキシビニルポリマー、ポリビニルアルコール(部分又は完全けん化物)等の水溶性合成高分子;クロスポビドン、ポリカルボフィル、ポリカルボフィルカルシウム、アミノアルキルメタクリレートコポリマーE、アミノアルキルメタクリレートコポリマーRS、メタクリル酸コポリマーL、メタクリル酸コポリマーS、メタクリル酸コポリマーLD、乾燥メタクリル酸コポリマーLD、ポレビニルアセタールジエチルアミノアセート等の水不溶性合成高分子;コムギデンプン、コメデンプン、トウモロコシデンプン、バレイショデンプン、部分アルファ化デンプン、α化デンプン、デキストリン、α−シクロデキストリン、β−シクロデキストリン、マルトデキストリン、イソマルト、ヒドロキシプロピルスターチ、カルボキシメチルスターチナトリウム、プルラン等のデンプン;アラビアゴム、アラビアゴム末、カンテン、カンテン末、ゼラチン、精製ゼラチン、キトサン、キサンタンガム、ペクチン、アルギン酸ナトリウム、ローカストビーンカム、グァーガム等の天然高分子化合物;ステアリン酸、ステアリン酸モノグリセリン、カルナウバロウ、ステアリルアルコール、セタノール、マクロゴール1500、マクロゴール4000、マクロゴール6000等の低融点物質等が挙げられる。これらの形状維持物質は、1種単独で又は2種以上混合して使用される。
【0055】
形状維持物質は、水不溶性で、かつ崩壊作用の少ない物質であるのが好ましい。このような形状維持物質としては、例えば、結晶セルロース、キトサン、アルギン酸ナトリウム、ポリカルボフィル、ポリカルボフィルカルシウム等が挙げられる。最も好ましい形状維持物質は、結晶セルロースである。
【0056】
結晶セルロースとしては、市販品、例えば、旭化成ケミカルズ社の「セオラスPH−101」、「セオラスPH−102」、「セオラスPH−301」、「セオラスPH−302」及び「セオラスKG−802」、FMC社の「アビセルPH−200」、JRS社の「VIVAPUR 12」等を使用できる。
【0057】
上記形状維持物質の含有量は、医薬製剤中、通常1〜90重量%、好ましくは3〜80重量%、より好ましくは5〜50重量%である。
【0058】
その他の成分
本発明の医薬製剤は、例えば、賦形剤、結合剤、pH調整剤、吸収促進剤、滑沢剤、着色剤、矯味剤、香料,剤皮等の固形医薬製剤に配合可能な各種添加剤を含有することができる。これらの成分は、本発明の効果を妨げない範囲内で本発明医薬製剤に配合できる。
【0059】
本発明の医薬固形製剤は、好ましくは押出造粒法及び球形整粒法を経て製造される固形製剤である。その剤形は、散剤、顆粒剤及びカプセル剤であることが好ましい。また、本発明の医薬固形製剤は、「進化する薬物治療 DDS最前線,2002年,金尾義治著,22頁,廣川書店」に記載されているような、スペイスタブ型錠剤(顆粒含有錠剤)のように、上記散剤又は顆粒剤を錠剤に含ませるように成形してもよい。本発明の医薬固形製剤のより好ましい剤形は、顆粒剤、カプセル剤及び顆粒含有錠剤である。剤形がカプセル剤又は顆粒含有錠剤であると、医薬固形製剤の取り扱い易さ及び服用のし易さの点で有利である。また、錠剤強度の向上及び湿度対策の観点から、顆粒含有錠剤に被膜を本発明の効果を妨げない程度に施してもよい。
【0060】
本発明の医薬固形製剤が、散剤又は顆粒剤である場合には、散剤及び顆粒剤の粒度が0.3〜3mmであることが、押出造粒時の製造適正の見地から好ましい。
【0061】
本発明の医薬固形製剤が、カプセル剤である場合には、カプセル剤のサイズが5〜00号であることが、取り扱い易さ及び服用のし易さの観点から好ましい。
【0062】
本発明の医薬固形製剤が、顆粒含有錠剤である場合には、生産性、取り扱い易さ及び服用のし易さの観点から、錠剤の形状は丸形又はカプレット形であるのが好ましく、顆粒含有錠剤の直径又は長径が6〜30mmの範囲にあることが好ましい。
【0063】
本発明の医薬固形製剤の製造方法
以下に本発明の医薬固形製剤の製造方法、特に散剤、顆粒剤及びカプセル剤の製造方法について説明するが、これに限定されるものではない。
【0064】
本発明の医薬固形製剤は、種々の製造方法により製造することができる。本発明において、好ましい剤形である散剤、顆粒剤及びカプセル剤を製造する場合には、少なくとも押出造粒工程を有する製造方法が好ましい。この押出造粒工程に加えて、湿式練合工程及び球形整粒工程を有する製造方法が好ましい。
【0065】
本発明の医薬固形製剤の好ましい製造方法は、湿式練合工程、押出造粒工程、球形整粒工程、乾燥工程、及び必要に応じて整粒工程を経て製造される。本発明の好ましい製造工程は、湿式練合工程、押出造粒工程、球形整粒工程、及び乾燥工程の順である。
【0066】
本発明の医薬固形製剤がカプセル剤である場合、整粒工程の後、更に、流動化剤混合工程及びカプセル充填工程を経て製造される。
【0067】
本発明の医薬固形製剤が顆粒含有錠剤である場合、整粒工程の後、得られる顆粒を適当な添加剤と混合する工程、滑沢剤を混合する工程及び打錠工程を経て製造される。また必要に応じて打錠後にフィルムコーティングを行ってもよい。
【0068】
湿式練合工程
湿式練合工程は、上記成分を結合剤等の他の成分と共に湿式で練合し、前記各成分に水を馴染ませる工程である。この際、(b)成分の糖及び/又は糖アルコールは粉末形態のまま使用してもよいし、予め水に溶解させた溶液の形態で使用してもよい。
【0069】
湿式練合は、主に湿式高剪断造粒法に基づいて操作される。湿式練合工程で用いられる装置としては、例えば、岡田精工社製の「ニュースピードニーダー」、パウレック社製の「バーチカルグラニュレーター」、深江パウテック社製の「ハイスピードミキサー」、奈良機械製作所社製の「高速攪拌型混造粒機NMG」、ミューチュアル社製の「ディオスナ攪拌混合造粒機」、スペクトラム(Spectrium)社製の「エアロマティックフィールダー(Aeromatic-filelder)」等が挙げられる。
【0070】
押出造粒工程
押出造粒工程は、湿式練合工程で得られた湿式練合混合物をスクリーンに向かって押出し、円柱状繊維を得る工程である。押出造粒装置としては、特に限定がなく、例えば、スクリュー供給方式、重力供給方式、ピストン供給方式等のいずれでもよい。スクリュー方式の押出造粒装置としては、不二パウダル社製の「ドームグランDG−L1」、「ツインドームグランTDG−80」、「ツインドームグランTDG−110」等が挙げられる。重力供給方式の押出造粒装置として、ギアロールタイプのホソカワミクロン社製の「ギャペレタイザGCS」、放射状タイプの深江パウテック社製の「FG型円筒造粒機」等が挙げられる。
【0071】
散剤を製造する場合は、スクリーンの孔径を0.3〜0.5mmとすればよい。また顆粒剤及びカプセル剤を製造する場合はスクリーンの孔径を0.3〜3mmとすればよい。
【0072】
球形整粒工程
球形整粒工程は、押出造粒工程で得られた円柱状繊維を適度な大きさに切って球形化し、形を整える工程である。球形整粒工程に用いられる装置としては、例えば、岡田精工社製の「ニュースピードニーダー」、不二パウダル社製の「マルメライザーQJ」、フロイント産業社製の「CF造粒機」、「グラニュレックスGX」等が挙げられる。
【0073】
乾燥工程
乾燥工程は、球形整粒工程で処理された粒子を乾燥し、水分を除去する工程である。乾燥は、直接加熱方式及び間接加熱方式のいずれでもよい。直接加熱方式としては、例えば、棚式送風乾燥機、流動層乾燥機等が挙げられる。間接加熱方式としては、例えば、真空乾燥機、マイクロ波乾燥機、遠赤外線乾燥機等が挙げられる。乾燥工程に用いられる装置としては、具体的には、パウレック社製の「グラット流動層造粒機WST」「マルチプレックス」、不二パウダル社製の「箱型通気平行流式ドライヤー」、「ミゼツトドライヤー」、大川原製作所社製の「スリットフローFBS」、フロイント産業社製の「フロードライヤーNFOD」、中央化工機社製の「振動乾燥機」、タバイエスペック社製の「SPHH−200」等が挙げられる。
【0074】
整粒工程
整粒工程は、乾燥粒子から一定の粒度範囲の粒子を取り出す工程である。この工程には、例えば、篩分け法等が適用できる。
【0075】
流動化剤混合工程
流動化剤混合工程は、整粒工程後の粒子に流動化剤を添加し、粒子と流動化剤とを均質に混合する工程である。流動化剤混合工程には、例えば、拡散式ミキサー方式(容器回転式)が適している。
【0076】
カプセル充填工程
カプセル充填工程は、流動化剤が混合された粒子をカプセルに充填する工程である。カプセル充填工程で使用される装置としては、例えば、クオリカプス社製の「LIQFIL super」シリーズ、ボッシュ包装機社製の「GKF」シリーズ、IMA社の「ZANASI」、「MATIC」シリーズ等が挙げられる。
【発明の効果】
【0077】
本発明においては、特定の性質を有する糖及び/又は糖アルコールを使用することによりメタクリル酸系腸溶性高分子化合物の含有比率が高い練合混合物に適度な塑性を付与することができる。そのため、本発明の医薬固形製剤には、可塑剤を配合する必要がなく、本発明の医薬固形製剤は、可塑剤配合に基づく欠点(即ち、可塑剤の配合によりメタクリル酸系腸溶性高分子化合物の変質及び変形が促進され、スクリーンの内側に該腸溶性高分子化合物からなる膜が形成されやすく、スクリーンの閉塞及び造粒装置の故障の原因となり得る。更に、この固形製剤中には可塑剤が残存するため,メタクリル酸系腸溶性高分子化合物の経時的な変質及び変形を促し、その結果、薬剤の経時的な溶出変化が生じるのが避けられなくなる。)を有していない。
【0078】
本発明の医薬固形製剤は、可塑剤を使用しなくてもメタクリル酸系腸溶性高分子化合物を多量に含有させることができるので、消化管上部での薬剤の放出を抑え、かつ消化管下部において薬剤の放出を速やかにするような高度な放出制御性を備えている。
【0079】
本発明の医薬固形製剤は、pH応答性の良いメタクリル酸系腸溶性高分子化合物を有することで高度な放出制御性を備えている。そのため、腸溶性製剤、時限放出型製剤、結腸特異的放出型製剤等を製造する際に施されるコーティング工程を必要としない。それ故、本発明は所望の徐放性能を備えた医薬固形製剤を、安価でかつ高い生産性をもって製造することができる。
【図面の簡単な説明】
【0080】
【図1】図1は、実施例24、25及び26の溶出試験結果を示すグラフである。
【図2】図2は、絶食下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【図3】図3は、食後下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【図4】図4は、実施例24〜26において、平均滞留時間(MRT)と50%溶出時間(T50)との関係 (In Vitro-In Vivo Correlation)(レベルB)を示すグラフである。
【発明を実施するための形態】
【実施例】
【0081】
以下に実施例及び比較例を掲げて本発明をより一層明らかにする。
【0082】
実施例1
シロスタゾール100g、サンマルト−S(マルトース一水和物、三和澱粉工業社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、180秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0083】
上記押出造粒工程において得られる素麺状造粒物は、本発明の医薬固形製剤の品質及び生産性に大きく影響を及ぼす。少量スケールではスクリーンを通過する練合物の量が少ないため、1回通過では練合物に加わる圧力が小さく、生産性を予測するのは困難である。このため、少量の練合物で生産性を予測する場合、同じ練合物を複数回繰り返しスクリーンを通過させ、スクリーンとスクリューの隙間に滞留する固形組成物の状態を安定化させた状態で製造性を把握するのがよい。以下の実施例では、同じ練合物を3回繰り返し押出造粒を行った後の練合物の温度を各回毎に測定し、これらの温度から練合物が適度の塑性を備えているかどうかを確認した。
【0084】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0085】
【表1】

【0086】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0087】
実施例2
シロスタゾール100g、エリスリトール100M(エリスリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、160秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を15秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0088】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0089】
【表2】
【0090】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0091】
実施例3
シロスタゾール100g、ソルビトールSP(ソルビトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラス PH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0092】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0093】
【表3】
【0094】
上記の結果から、ソルビトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0095】
実施例4
シロスタゾール100g、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、170秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0096】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0097】
【表4】
【0098】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0099】
実施例5
シロスタゾール100g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0100】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0101】
【表5】
【0102】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0103】
実施例6
シロスタゾール100g、キシリトールP(キシリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、115秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0104】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0105】
【表6】
【0106】
上記の結果から、キシリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0107】
比較例1
シロスタゾール100g、WYNDALE 200M乳糖(乳糖一水和物、ラクトースカンパニーニュージーランドリミテッド社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、240秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回繰り返して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0108】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0109】
【表7】
【0110】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、乳糖一水和物を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0111】
比較例2
シロスタゾール100g、ペアリトール50C(D−マンニトール、ロケット社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、300秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0112】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0113】
【表8】
【0114】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、D−マンニトールを配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0115】
実施例7
シロスタゾール100g、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水18gを結合液として投入し、320秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0116】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0117】
【表9】
【0118】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0119】
実施例8
シロスタゾール100g、エリスリトール100M(エリスリトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、100秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0120】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0121】
【表10】
【0122】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0123】
実施例9
シロスタゾール100g、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水16.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0124】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0125】
【表11】
【0126】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0127】
実施例10
シロスタゾール100g、ソルビトールSP(ソルビトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、50秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0128】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0129】
【表12】
【0130】
上記の結果から、ソルビトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0131】
実施例11
シロスタゾール100g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水16.5gを結合液として投入し、110秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0132】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0133】
【表13】
【0134】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0135】
実施例12
シロスタゾール100g、キシリトールP(キシリトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラス PH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、50秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0136】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0137】
【表14】
【0138】
上記の結果から、キシリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0139】
比較例3
シロスタゾール100g、ハンマーミルで粉砕した白糖(グラニュー糖CH、塩水港精糖社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水17gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0140】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0141】
【表15】
【0142】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、白糖を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0143】
参考例1
非晶質化したトルバプタンを次のようにして調製した。即ち、7−クロロ−5−ヒドロキシ−1−[2−メチル−4−(2−メチルベンゾイルアミノ)ベンゾイル]−2,3,4,5−テトラヒドロ−1H−ベンゾアゼピン100g及びヒドロキシプロピルセルロース(HPC−SL、日本曹達社製、ヒドロキシプロポキシル基含量:53〜78重量%)50gをジクロロメタン1390g及びエタノール350gの混合溶液に溶解し、噴霧乾燥機(大川原加工機社製、ODT−8型)で処理した後、直ちに真空乾燥機(タバイエスペック社製、LCV−232)で乾燥し、非晶質粉末(非晶質化トルバプタン)を調製した。
【0144】
実施例13
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水15gを結合液として投入し、110秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0145】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0146】
【表16】
【0147】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0148】
実施例14
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、エリスリトール100M(エリスリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水13gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0149】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0150】
【表17】
【0151】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0152】
実施例15
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水13gを結合液として投入し、120秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0153】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0154】
【表18】
【0155】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0156】
実施例16
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、トレハロースP(トレハロース二水和物、林原生物化学研究所製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水14.5gを結合液として投入し、130秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0157】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0158】
【表19】
【0159】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0160】
比較例4
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、WYNDALE 200M乳糖(乳糖一水和物、ラクトースカンパニーニュージーランドリミテッド社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水17.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0161】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0162】
【表20】
【0163】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、乳糖一水和物を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0164】
実施例17
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水11gを結合液として投入し、180秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入し、40rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約2〜4cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、マトリックス顆粒を得た。篩過により600μm〜1000μmのマトリックス顆粒を製品とした。
【0165】
1.粒子径分布の計測
約5gのマトリックス顆粒を取り、日本薬局方の一般試験法に規定される粒度測定法(乾式ふるい分け法)に準じてロボットシフターRPS−95(セイシン企業社製)で粒度測定を行った。マトリックス顆粒の平均粒子径は770μmであった。
【0166】
2.分解物の測定
実施例17で得られたマトリックス顆粒140.4gに軽質無水ケイ酸(アドソリダー−101、ワイ・ケイ・エフ社製)0.6gを加え、よく混合後、ヒプロメロース3号カプセル(QUALI−Vカプセル、クオリカプス社製)にトルバプタン60mg相当になるよう充填した。マトリックス顆粒含有カプセルをプラスチック容器に入れ、60℃に2週間保存した。
【0167】
トルバプタン30mg相当のマトリックス顆粒を秤取して乳鉢内で粉砕後、メタノールを加え、超音波を照射して完全に顆粒を破壊した。これにメタノールを加え、約0.5μmのメンブランフィルターでろ過し、得られたろ液をLC−2010CTシステム(高速液体クロマトグラフィー、島津製作所製)で測定波長254nm、移動相(アセトニトリル/水/リン酸=500/500/1、容積比)の送液速度約1mlで分解物Aの探索を行った。分解物Aの濃度を面積百分率法で算出した(トルバプタンのピーク面積に対する分解物Aのピーク面積の比率)。
【0168】
保存の前後における分解物Aの濃度を調べたところ、製造直後における分解物Aの濃度が0.01%、60℃で2週間保存した後の分解物Aの濃度は0.02%であり、分解物の生成が抑制されていることが明らかになった。
【0169】
3.溶出試験
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。pH7.4の薄めたMcIlvaine緩衝液にポリソルベート80を1w/v%の濃度となるよう添加した溶液900mlを溶出試験液とした。パドルの回転数は50rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0170】
60℃で2週間保存した前後におけるそれぞれの溶出率及び溶出率の差(Δ溶出率、製造直後−60℃/2週間保存後)を以下に示す。
【0171】
【表21】
【0172】
上記表から、実施例17で得られたマトリックス顆粒含有カプセルからのトルバプタンの溶出率については製造直後及び60℃/2週間保存後の双方において殆ど変化がなく、溶出の経時的安定性が良好であることが明らかである。
【0173】
比較例5
参考例1で調製した非晶質化トルバプタン45g(トルバプタンとして30g)、ペアリトール50C(D−マンニトール、ロケット社製)45g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gを奈良式高速攪拌型混合造粒機 NMG−1L(奈良機械製作所社製)に投入した。次に、4w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)と4w/v%ポリソルベート80水溶液(ポリソルベート80(HM)、日本油脂社製)の混液(1:1重量比)40gを結合液として投入し、30秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜4cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、マトリックス顆粒を得た。篩過により355μm〜850μmのマトリックス顆粒を製品とした。
【0174】
実施例17と同様にして粒子径分布の計測を行ったところ、マトリックス顆粒の平均粒子径は690μmであった。
【0175】
製造直後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した後、ガラス製気密容器に充填し、室温で1年間保管した。室温一年保管後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した。さらに、室温一年保管後のマトリックス顆粒をガラス製気密容器に充填して60℃で2週間保存し、60℃2週間保存後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した。なお、分解物Aの測定は、実施例17と同様にして行った。溶出試験は、以下の方法で行った。
【0176】
溶出試験:
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。pH7.0のMcIlvaine緩衝液にポリソルベート80を1w/v%の濃度となるよう添加した溶液900mlを溶出試験液とした。パドルの回転数は100rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0177】
分解物Aの測定結果を表22に示す。
【0178】
【表22】
【0179】
溶出試験の結果を表23に示す。なお、表中のΔ溶出率は、室温で一年保存後の溶出率から60℃で2週間保存後の溶出率を差し引いたものである。
【0180】
【表23】
【0181】
マトリックス顆粒に可塑剤(ポリソルベート80)が含まれていると、顕著な分解物Aの生成増加が認められ,かつ溶出率の低下も大きかった。また、室温で1年保存している間に分解物Aの生成量が増加し、溶出も遅延する傾向を示していた。そのため、比較例5で得られたマトリックス顆粒は、溶出の経時的安定性に著しく劣っていることが判明した。
【0182】
実施例18
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)18g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水15gを結合液として投入し、250秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を5回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を50秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが7.4重量%の割合で含有されている。
【0183】
実施例19
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水8gを結合液として投入し、160秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約10cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが11.7重量%の割合で含有されている。
【0184】
実施例20
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水11gを結合液として投入し、180秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約3〜5cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが19.2重量%の割合で含有されている。
【0185】
比較例6
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)12.9g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水6gを結合液として投入し、210秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を5回繰り返して行い、長さ約1〜2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSは含有されていない。
【0186】
溶出試験:
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。使用した試験液1及び試験液2は以下の通りである。
試験液1:日本薬局方15 溶出試験液第一液(pH1.2)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
試験液2:薄めたMcIlvaine緩衝液(pH7.4)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
パドルの回転数はいずれも50rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0187】
試験液1を用いて溶出試験を行った結果を表24に示す。表における数値は、試験を3回行った平均値である。
【0188】
【表24】
【0189】
pH1.2の酸性条件では、顆粒からの薬剤の溶出性に全く差は認められず、メタクリル酸高分子化合物の含有割合の違いを確認できなかった。
【0190】
試験液2を用いて溶出試験を行った結果を表25に示す。表における数値は、試験を3回行った平均値である。
【0191】
【表25】
【0192】
メタクリル酸高分子化合物が溶解するpH域では、その含有量に応じて溶出性が速やかになったが、比較例6は酸性条件のときと全く同じ溶出性を示していた。
【0193】
実施例21
フェニトイン(シグマアルドリッチ社製)100g、エリスリトール100M(エリスリトール、日研化成社製)60g、オイドラギットL100(メタクリル酸コポリマーL、デグサ社製)15g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機,岡田精工社製)に投入した。5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース,日本曹達社製)40g及び精製水9.5gを結合液として投入し、150秒湿式練合した。孔径0.6mm,開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を10秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0194】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0195】
【表26】
【0196】
実施例22
アスピリン(アセチルサリチル酸、和光純薬工業社製)100g、ラクチトールLC−1(ラクチトール、日研化成社製)10g、オイドラギットL100D55(乾燥メタクリル酸コポリマーLD、デグサ社製)40g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)40g及び精製水9gを結合液として投入し、130秒湿式練合した。孔径0.6mm,開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0197】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0198】
【表27】
【0199】
実施例23
ナプロキセン(シグマアルドリッチ社製)15g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)30g、オイドラギットL100D55(乾燥メタクリル酸コポリマーLD、デグサ社製)40g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)30gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水4.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm,開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機,不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約4〜5cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機,不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機,タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0200】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0201】
【表28】
【0202】
実施例24
実施例20で得られた乾燥顆粒196gに、アドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.8gを添加混合し、得られた顆粒141mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0203】
実施例25
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをそれぞれスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60gと精製水3gを結合液として投入し、180秒湿式練合した。孔径0.8mm,開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、4回繰り返して押出造粒を行い、長さ約2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)で約1000rpmの回転速度で、この素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で4時間乾燥した。その後篩過処理によって平均粒子径780μmの顆粒を得た。この顆粒179gにアドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.8gを添加混合し、得られた顆粒129mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0204】
実施例26
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)30gをそれぞれスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60gと精製水16gを結合液として投入し、180秒湿式練合した。孔径0.8mm,開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、4回繰り返して押出造粒を行い、長さ約2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)で約1000rpmの回転速度で、この素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で4時間乾燥した。その後篩過処理によって平均粒子径780μmの顆粒を得た。この顆粒217gにアドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.9gを添加混合し、得られた顆粒147mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0205】
溶出試験方法
溶出試験システムNTR−6200A(富山産業社製)を用い、日本薬局方溶出試験方法第二法(パドル法)で実施例24、25及び26で得られるカプセル剤からのトルバプタンの溶出試験を行った。下記に示す試験液を使用した。
【0206】
試験液:日本薬局方15 溶出試験液第二液(pH6.8)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
測定波長:λ1,268nm; λ2,350nm
パドル回転数:100rpm
サンプル数:n=6
サンプリング時点:0.5、1、2、3、4、6、8、10及び12時間
50%溶出時間(D50)の算出
図1よりトルバプタンが50%溶出するのに要した時間を求めた。
【0207】
【表29】
【0208】
経口投与試験
本発明の医薬固形製剤の徐放化効果を確認するため,健康人への経口投与試験を行った。18名の健康な男女(年齢18〜45歳)を無作為に6名ずつA群、B群及びC群に分け、以下の計画に従って3群4期の非完全クロスオーバー試験で行った。
【0209】
【表30】
【0210】
いずれの群においても、第1期は速放錠を一日二回空腹下に投与した。早朝にトルバプタンをトータルで45mg含む2個の錠剤(30mg錠+15mg錠)を空腹下経口投与し、8時間経過後に15mg錠1個を経口投与した。一日投与量はトルバプタン60mgである。第2期では,群ごとに実施例24、25及び26で得られる異なるカプセルを空腹下に経口投与した。第3期では,いずれの群においても第2期とは異なる、実施例24、25及び26で得られるカプセルを空腹下経口投与した。第4期では,第3期に投与したカプセルと同じカプセルを食後経口投与した。食事の内容は米国FDAのガイダンス(Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. U.S. Department of Healthy and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2002)に基づいて設定した高脂肪食であり、食事を摂取後30分以内に実施例24、25及び26で得られるカプセルを経口投与した。速放錠並びに実施例24、25及び26で得られるカプセルを空腹下投与した例数は、それぞれ18例及び12例であり、実施例24、25及び26のカプセルを食後投与した例数は6例である。なお、速放製剤の組成は以下の通りである。
【0211】
比較例7:30mg錠(速放錠)
参考例1で調製した非晶質化したトルバプタン112.5g(トルバプタン量75g)、乳糖一水和物185g、トウモロコシデンプン50g及び結晶セルロース50gを混合し、攪拌流動造粒乾燥機(パウレック社製,マルチプレックスMP−01)に投入した。ヒドロキシプロポキシル基含量が53〜78重量%のヒドロキシプロピルセルロース5w/v%水溶液200gを用いて流動層造粒及び乾燥を行い、造粒物を得た。これにLH−11(低置換ヒドロキシプロピルセルロース)22.5g及びステアリン酸マグネシウム5gを混合し,打錠用顆粒とした。これをロータリー式連続打錠機(菊水製作所製,12HUK−AWC)を用いて打錠圧900kg,回転数40rpmの条件でトルバプタン30mgを含有する重量約174mgの直径8mmの平面錠を製造した。
【0212】
比較例8:15mg錠(速放錠)
参考例1で調製した非晶質化したトルバプタン56.3g(トルバプタン量37.6g)、乳糖一水和物256.3g、トウモロコシデンプン50g及び結晶セルロース50gを混合し、攪拌流動造粒乾燥機(パウレック社製,マルチプレックスMP−01)に投入した。ヒドロキシプロポキシル基含量が53〜78重量%のヒドロキシプロピルセルロース5w/v%水溶液200gを用いて流動層造粒及び乾燥を行い、造粒物を得た。これにLH−11(低置換ヒドロキシプロピルセルロース)22.5g及びステアリン酸マグネシウム5gを混合し、打錠用顆粒とした。これをロータリー式連続打錠機(菊水製作所製,12HUK−AWC)を用いて打錠圧1000kg,回転数50rpmの条件でトルバプタン15mgを含有する重量約180mgの直径8mmの平面錠を製造した。
【0213】
評価
経時的に採血し、血漿中トルバプタン濃度を測定した。薬物動態パラメータは、WinNolin (ver4.0, Pharsight社製ソフトウェア)及びPSAG-CP ((株)アスメディカ社製ソフトウェア)を用いて算出した。
【0214】
図2は、絶食下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。図3は、食後下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【0215】
【表31】
【0216】
図4は、実施例24〜26において、平均滞留時間(MRT)と50%溶出時間(T50)との関係 (In Vitro-In Vivo Correlation)を示すグラフである。米国FDAのガイダンス(Guidance for Industry Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations, U.S. Department of Health and Human Services, Food and Drug Administration, and Center for Drug Evaluation and Research (CDER), September 1997)によると、レベルBは、MRTとT50とのよい関係を示している。
【0217】
長期保存サンプルの色調変化
試験方法
実施例13、14、15及び16の顆粒剤をポリエチレン袋に入れて密閉し、室内に1年以上保管した。このときの温度及び湿度は自然体である。保管の前後で色調の変化を目視で確認した。また顆粒の堅さを手で確認した。
【0218】
【表32】
【0219】
実施例13〜16の顆粒剤は、いずれも色調変化はなく、顆粒の硬さも変化がなかった。
【産業上の利用可能性】
【0220】
本発明の医薬固形製剤は、医薬分野で利用価値が高い。特に、可塑剤を配合しなくても、製剤を製造する際の練合物に良好な塑性を与えることができる。更に、本発明の製剤は、可塑剤を配合する場合に生ずる問題点を解決することができ、極めて有用である。また、本発明の医薬固形製剤は、経口徐放性医薬固形製剤として極めて利用価値が高い。
【技術分野】
【0001】
本発明は、医薬固形製剤に関する。
【背景技術】
【0002】
医薬の分野において、薬剤放出を制御し、薬剤血中濃度を適正レベルに長時間維持させる試みが広く行われている。薬剤血中濃度を適正レベルに長時間維持するためには、薬剤の吸収が長時間持続するような製剤学的工夫(徐放化技術)が必要である。経口投与された固形製剤は、時間の経過に伴って消化管の上部(胃及び小腸上部)から下部(小腸下部及び大腸)へ移動するが,薬剤の吸収性は消化管の上部に比べて下部では低い場合が非常に多い。そのため,固形製剤の滞留時間が最も長い消化管下部において,薬剤が持続的に吸収できるような方策を立てることがなにより重要と考えられる。
【0003】
徐放化技術としては、例えば、拡散制御に基づいた徐放化技術、より具体的には、薬剤を含有する核組成物又は核錠子に水不溶性高分子化合物の被膜を施したメンブランコーティング製剤;水不溶性高分子化合物、ワックス等とともに製造されるマトリックス製剤等が知られている。しかしながら、これら技術では、薬剤の放出が進行するのに従って放出速度が減少してゆくため、服用後数時間経過して到達する消化管下部では薬剤の放出速度が不足し、薬剤血中濃度の維持が困難となる。
【0004】
また、消化管下部における薬剤放出を意図した製剤的手法が施された徐放化技術(例えば、速放性の薬剤含有核組成物に腸溶性被膜を被覆した腸溶性製剤等)が知られている。しかしながら、この技術では、薬剤の放出を被膜によって制御するため、フィルムコーティング操作を必要とする。その結果、医薬製剤の製造工程が煩雑となる。
【0005】
一方、メタクリル酸系の腸溶性高分子を用いて徐放化されたマトリックス製剤が知られている。腸溶性高分子化合物は溶解できるpHより低いpH領域では不溶性物質となり、溶解できるpH以上のpH領域では可溶性物質となる。従って、腸溶性高分子化合物が配合されたマトリックス製剤は消化管上部では薬剤放出を抑制し、消化管下部で速やかに薬剤を放出させることができる。つまり、腸溶性高分子化合物の鋭いpH応答性により、精緻な薬剤放出制御を備える徐放性製剤を提供することが可能になる。
【0006】
例えば、腸溶性高分子化合物及び薬剤を混合した後、圧縮成形法(打錠)によって腸溶性高分子化合物が配合されたマトリックス製剤が知られている(例えば、特許文献1、特許文献2、特許文献3、特許文献4等)。しかしながら、マトリックス製剤からの薬剤放出は,一般に製剤の表面積に依存すると考えられているが、このような打錠により得られるマトリックス製剤は、溶媒と接触する表面積が小さいので、溶解度が低く、溶解速度が遅い難溶性薬剤の場合には、製剤の小さな表面積が薬剤放出の障害となる。
【0007】
これに対し、メタクリル酸コポリマーSを含む混合粉末をエタノールで湿式練合し、押出造粒法によって得られる製剤が知られている(特許文献5)。この製剤は、湿式練合し、押出造粒法を経ることで徐放性の粒状剤となる。このような粒状剤(粒状剤、顆粒剤、散剤は、マルチプルユニット型と呼ばれている)は、製剤の表面積を大きくでき、難溶性薬剤にも適用することができる。さらに、打錠して得られる錠剤のようなシングルユニット型に比べて、粒状剤のようなマルチプルユニット型は、消化管内で適度に分散されるので、錠剤よりも薬剤吸収の個体間変動を小さく抑えることができる。
【0008】
さらに、マルチプルユニット型の製剤として、メタクリル酸コポリマーS、メタクリル酸コポリマーLD、薬剤、ポリビニルピロリドン及び可塑剤としてクエン酸トリエチルを含む混合粉末を水で湿式練合し、得られた練合物を押出造粒し、次いで球形整粒して顆粒が得られることが知られている(非特許文献1)。
【先行技術文献】
【特許文献】
【0009】
【特許文献1】特公平4−43049号公報
【特許文献2】特開平6−199657号公報
【特許文献3】米国特許4,968,508号明細書
【特許文献4】米国特許公開2006/0159753号公報
【特許文献5】特開平6−24991号公報
【非特許文献】
【0010】
【非特許文献1】International Journal of Pharmaceutics,2001年,第213巻,7-12頁
【発明の概要】
【発明が解決しようとする課題】
【0011】
しかしながら、上記の腸溶性高分子化合物を含有する混合物を、押出造粒法に続いて球形整粒法により、粒状剤や顆粒剤を得る方法では、下記のような問題点がある。
【0012】
まず、メタクリル酸系の腸溶性高分子化合物は、そのガラス転移温度が160℃以上と高温であることから推察されるように堅い腸溶性高分子である。そのため、押出造粒法、球形整粒法によってメタクリル酸系腸溶性高分子化合物を含むマトリックス製剤を製造する場合、押出造粒を容易に行うために、湿式練合物の塑性を補うことが必要となる。
【0013】
上記特許文献5の技術では、メタクリル酸コポリマーSの配合割合を5重量%としているが、得られる医薬製剤のpH応答性を一段と向上させるためにはメタクリル酸コポリマーSの配合量をより多くする必要がある。しかし、配合量を多くすると塑性が不十分となり、押出造粒法、球形整粒法によってメタクリル酸系の腸溶性高分子を含むマトリックス製剤を製造しにくくなる。つまり、塑性が不十分であると、押出造粒する時、練合物を押出す際の抵抗が大きくなり、押出造粒できなくなる。
【0014】
一方、上記非特許文献1に記載の技術では、77重量%のメタクリル酸コポリマーを含有しており、練合物を押出造粒する際に必要な塑性を補うために、可塑剤としてクエン酸トリエチルを約11重量%添加している。しかしながら、この技術では、可塑剤の添加によりメタクリル酸系腸溶性高分子化合物のガラス転移温度が低下し、メタクリル酸系腸溶性高分子化合物の変質・変形が促進されるために、スクリーンの内側に膜を形成する可能性があり、閉塞や装置の故障の原因となりえる。さらに、得られた製剤中に可塑剤が残存するため、メタクリル酸系腸溶性高分子化合物の経時的な変質・変形を促進し、その結果、経時的な溶出変化が生じる可能性が高い。また、非特許文献1に記載の技術では、薬剤との不適合も懸念される。
【0015】
このようにメタクリル酸系腸溶性高分子化合物の配合比率を高くするためには可塑剤が必要となる一方において、可塑剤を添加すると上記のような問題点が生じるのを回避できない。換言すれば、可塑剤を添加することによる問題点を解決しながら腸溶性高分子化合物の配合比率を高くすることは極めて困難である。
【0016】
本発明は、徐放化するためにpH応答性が良好なメタクリル酸系の腸溶性高分子化合物の配合比率を高くしても、上記従来技術のような問題点が生じない医薬固形製剤を提供することである。
【0017】
すなわち、本発明は、消化管上部での薬剤の放出を抑え、かつ消化管下部において薬剤の放出を速やかにする高度な放出制御性を備えていながら、可塑剤配合による上記欠点を悉く解消したマトリックス型固形製剤を提供することを課題とする。
【課題を解決するための手段】
【0018】
本発明者らは、上記課題を解決するために鋭意研究を重ねた結果、メタクリル酸系腸溶性高分子化合物と特定の性質を備えた糖及び/又は糖アルコールとを併用することにより所望のマトリックス型固形製剤が得られることを見い出した。本発明は、かかる知見に基づき完成されたものである。
【0019】
本発明は、項1〜項13に係るマトリックス型の医薬固形製剤を提供する。
項1.(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、前記(b)糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である、医薬固形製剤。
項2.糖及び/又は糖アルコールの配合量が、メタクリル酸系腸溶性高分子化合物1重量部に対して、0.1〜10重量部である、項1に記載の医薬固形製剤。
項3.糖及び/又は糖アルコールは、融点が140℃以下である、項1又は項2に記載の医薬固形製剤。
項4.可塑剤を含有していない項1又は項2に記載の医薬固形製剤。
項5.可塑剤を含有していない項3に記載の医薬固形製剤。
項6.押出造粒工程を有する製造方法により製造される、項1又は項2に記載の医薬固形製剤。
項7.押出造粒工程を有する製造方法により製造される、項3に記載の医薬固形製剤。
項8.糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種である、項1〜項7のいずれかに記載の医薬固形製剤。
項9.メタクリル酸系腸溶性高分子化合物の含有量が、6〜50重量%である、項1に記載の医薬固形製剤。
項10.メタクリル酸系腸溶性高分子化合物は、pH5.5以上で溶解する性質を備えている、項1に記載の医薬固形製剤。
項11.メタクリル酸系腸溶性高分子化合物は、ガラス転移温度が100℃以上である、項1に記載の医薬固形製剤。
項12.メタクリル酸系腸溶性高分子化合物は、メタクリル酸コポリマーL、メタクリル酸コポリマーLD及びメタクリル酸コポリマーSからなる群から選ばれた少なくとも1種である、項1〜項11のいずれかに記載の医薬固形製剤。
項13.シロスタゾール、トルバプタン、フェニトイン、アスピリン及びナプロキセンからなる群から選ばれた少なくとも1種の薬剤を含む、項1に記載の医薬固形製剤。
【0020】
医薬固形製剤
本発明のマトリックス型の医薬固形製剤は、(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有する。
【0021】
(b)の糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である性質を備えている。
【0022】
本発明の医薬固形製剤には、上記(a)及び(b)成分の他、他の成分を含んでいてもよく、好ましくは(c)薬剤及び(d)形状維持物質が含まれている。本発明の医薬固形製剤は、可塑剤を含有していない。
【0023】
本発明の医薬固形製剤は、マトリックス型の経口徐放性医薬固形製剤であることが好ましい。
【0024】
(a)メタクリル酸系腸溶性高分子化合物
本発明において、(a)メタクリル酸系腸溶性高分子化合物は、小腸下部及び大腸でのpH環境域で溶解するものである限り、公知のものを広く使用できる。pHが5.5以上、より好ましくはpHが6.0で、かつpHが7.5以下で溶解するメタクリル酸系腸溶性高分子化合物が好ましい。溶解するpHが上記範囲内にあると、当該腸溶性高分子化合物が小腸内及び/又は大腸内で溶解するので、消化管下部において製剤から薬剤を速やかに放出させることができる。
【0025】
更に、本発明において、メタクリル酸系腸溶性高分子化合物のガラス転移温度は、通常100℃以上、好ましくは105℃以上、より好ましくは130℃以上である。該ガラス転移温度は200℃以下であるのが望ましい。ガラス転移温度がこの範囲にあると、常温で変形及び変質することがないので、製剤の経時的な溶出変化の懸念が少なくなり、また押出造粒時に装置へ過度の負荷を発生させずに処理できる等の利点がある。
【0026】
上記メタクリル酸系腸溶性高分子化合物の好ましい具体例としては、メタクリル酸コポリマーLD、メタクリル酸コポリマーL、メタクリル酸コポリマーS等が挙げられる。メタクリル酸コポリマーLDは粉末状であるのが好ましい。粉末状のメタクリル酸コポリマーLDとは、液状や、固形分30%の懸濁液でないものを意味する。また、本発明で用いられるメタクリル酸コポリマーLDは、乾燥している状態、又は多少水分を含んでいる場合でも粉末状であればよい。以下、このような粉末状のメタクリル酸コポリマーLDを、乾燥メタクリル酸コポリマーLDとする場合がある。
【0027】
メタクリル酸系腸溶性高分子化合物は、入手が容易な市販品をいずれも使用でき、例えば、乾燥メタクリル酸コポリマーLDはデグサ社製の「Eudragit L100D55」を、メタクリル酸コポリマーLはデグサ社製の「Eudragit L100」を、メタクリル酸コポリマーSはデグサ社製の「Eudragit S100」を、それぞれ使用できる。これらの腸溶性高分子化合物は、1種単独で又は2種以上混合して使用される。
【0028】
上記腸溶性高分子化合物を2種以上混合することにより、製剤が小腸下部及び大腸で溶解するpH(腸溶性高分子化合物の溶解pH)を5.5〜7の間で任意に設定することができる。例えば、腸溶性高分子化合物の溶解pHを5.5〜6の間で任意に設定する場合は、メタクリル酸コポリマーLD:メタクリル酸コポリマーLの混合比率を1:99〜99:1の範囲で混合すればよい。また、腸溶性高分子化合物の溶解pHを5.5〜7の間で任意に設定する場合は、メタクリル酸コポリマーLD:メタクリル酸コポリマーSの混合比率を1:99〜99:1の範囲で混合すればよい。更に、腸溶性高分子化合物の溶解pHを6〜7の間で任意に設定する場合は、メタクリル酸コポリマーL:メタクリル酸コポリマーSの混合比率を1:99〜99:1の範囲で混合すればよい。
【0029】
本発明において、製剤に含有されるべき腸溶性高分子化合物の量は、通常1〜50重量%であり、好ましくは3〜45重量%であり、より好ましくは6〜40重量%であり、さらに好ましくは10〜35重量%である。腸溶性高分子化合物の含有量が上記範囲であると、薬物放出パターンのバラエティ、生産適性(押出造粒が良好であること)の点で好ましい。ここで、薬物放出パターンのバラエティとは、製剤の設計者が意図する薬物放出パターンを容易に達成しやすくなることを意味する。
【0030】
本発明の医薬固形製剤に使用されるメタクリル酸系腸溶性高分子化合物はpH5.5以上で溶解するのが好ましい。本発明の医薬固形製剤に付与すべきpH応答性は、以下に列記した薬剤の種類、望まれる薬理効果等により適宜調整される。また、そのpH応答性は使用されるメタクリル酸系腸溶性高分子化合物の種類、製剤中の腸溶性高分子化合物の含有量等によって調整可能である。
【0031】
例えば、消化管下部(小腸下部及び大腸)で持続的に吸収できる機能に加え、消化管上部(胃及び小腸上部)から薬剤が緩やかに放出されるように製剤設計する方が好ましい場合がある。その場合には医薬固形製剤のpH応答性を緩和するのが望ましく、具体的には医薬固形製剤中におけるメタクリル酸系腸溶性高分子化合物の含有量を少なめに設定する。
【0032】
また、消化管下部で持続的に吸収できる機能をより充実させたい場合には、医薬固形製剤のpH応答性を鋭くするのが望ましく、具体的には医薬固形製剤中におけるメタクリル酸系腸溶性高分子化合物の含有量を多めに設定する。
【0033】
(b)糖及び/又は糖アルコール
本発明で使用される糖及び/又は糖アルコールは、水に対する特定の溶解性を有している。糖及び/又は糖アルコール1gを溶解するのに要する、20〜25℃の間の水温での水の量は、通常4g以下、好ましくは3.5g以下である。また、糖及び/又は糖アルコール1gを溶解するのに要する、20〜25℃の間の水温での水の量は、1g以上であるのが望ましい。溶解するのに要する水の量がこの範囲であると、製剤製造前の練合混合物に適度な塑性を付与することができる。
【0034】
より好ましい糖及び/又は糖アルコールは、融点が140℃以下、好ましくは130℃以下、より好ましくは125℃以下であり、90℃以上である。融点がこの範囲であると、糖及び/又は糖アルコールが常温で固体形状を採り得るので取り扱い易くなる。他に、当該糖及び/又は糖アルコール自体の堅さが押出造粒時に影響を及ぼさない利点がある。
【0035】
本発明において用いることができる糖及び/又は糖アルコールは、上記のような特性を有するものであればよく、水和物の形態であってもよい。そのような糖及び/又は糖アルコールとしては、例えば、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種が挙げられる。好ましい糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース、マルトース、デキストロース、フルクトース及びマルチトールからなる群から選ばれた少なくとも1種が挙げられる。より好ましい糖及び/又は糖アルコールは、エリスリトール、キシリトール、ラクチトール、ソルビトール、トレハロース及びマルトースからなる群から選ばれた少なくとも1種である。さらにより好ましい糖及び/又は糖アルコールは、エリスリトール、ラクチトール一水和物、トレハロース二水和物及びマルトース一水和物からなる群から選ばれた少なくとも1種である。これらの更により好ましい糖及び/又は糖アルコールは、適度な溶解性及び適度な融点を有しており、かつ吸湿性の懸念が少なく、長期保管での安定性が良好である。
【0036】
糖及び/又は糖アルコールは、市販品を広く用いることができる。より具体的には、以下のものが挙げられる。
【0037】
(B-i) エリスリトール及びその水和物
糖アルコールであるエリスリトールは、酵素反応によってブドウ糖から製造される。エリスリトールの融点は119〜122℃、エリスリトール1g溶解するのに要する水(25℃)の量は3.3gである(日研化学社製、エリスリトール技術資料)。エリスリトールとしては、市販品、例えば、日研化学社の「エリスリトール100M」等を使用できる。
【0038】
(B-ii) キシリトール及びその水和物
糖アルコールであるキシリトールは、種々のセルロース原料を加水分解によってキシロースに変換後、水素添加して製造される。キシリトールは、若干の吸湿性を有している。1gのキシリトールを溶解するのに要する水の量(20℃)は1.6gである(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。更に、キシリトールの融点は93〜95℃である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。キシリトールとしては、市販品、例えば、日研化成社の「キシリトールP」、ロケット社の「XYLISORB」、東和化成工業の「キシリットP」等を使用できる。
【0039】
(B-iii) ラクチトール及びその水和物
糖アルコールであるラクチトールは、乳糖を触媒水素添加して製造される。ラクチトールには、無水物、一水和物、二水和物及び三水和物が含まれる。そのうち好ましいのは非吸湿性の一水和物である。ラクチトール一水和物の融点は97℃(Merck Index 第12版)、1gのラクチトールを溶解するのに要する水の量(20℃)は1.8gである(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。ラクチトール一水和物としては、市販品、例えば、日研化成社の「ラクチトールLC−1」等を使用できる。
【0040】
(B-iv) ソルビトール及びその水和物
糖アルコールであるソルビトールは、ブドウ糖又はトウモロコシシロップを高圧水素添加するか、電解還元することによって製造される。ソルビトールは吸湿性が強い。1gのソルビトールを溶解するのに要する水の量(25℃)は0.5ml(g)である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編)。更に、ソルビトールの融点は97〜112℃である(医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編、Journal of Thermal Analysis and Calorimetry、第73巻、615-621頁)。ソルビトールとしては、市販品、例えば、日研化成社の「ソルビトールSP」、ロケット社の「NEOSORB Powder」、東和化成工業の「ソルビットDP−10M」等を使用できる。
【0041】
(B-v) トレハロース及びその水和物
糖成分であるトレハロース二水和物は、マルトースと同じくグルコースが2個結合した二糖類である。医薬品添加剤としてのトレハロースは、澱粉部分分解物からトレハロース産生細菌を用いた酵素法によって製造される。トレハロース二水和物は、非吸湿性ではないが、吸湿性が少ない。トレハロース二水和物の融点は、97℃であり、1gのトレハロース二水和物を溶解するのに要する水の量(20℃)は1.2gである(いずれもトレハロース技術資料,林原生物化学研究所)。トレハロース二水和物としては、市販品、例えば、旭化成ケミカルズ社の「トレハロースP」、林原社の「トレハ」等を使用できる。
【0042】
(B-vi) マルトース及びその水和物
糖成分であるマルトース一水和物は、二糖類の炭水化物で、デンプンの酵素分解により製造される。マルトース含量が90%以上であれば医薬品添加物のアメ粉として医薬品にも使用することができる。マルトースには無水物と一水和物があり、一水和物は吸湿性が低い。New Food Industry、第31巻4号、17-22頁によると、1gの含水結晶マルトースを溶解させるのに要する水の量(20℃)は1.2gである。更に、医薬品添加物ハンドブック、2001年、日本医薬品添加物協会訳編によると、マルトース一水和物の融点(分解)は102〜103℃である。マルトース一水和物としては、市販品、例えば、三和澱粉工業社の「サンマルト−S」,日本食品化工社の「日食結晶マルトース」等を使用できる。
【0043】
本発明においては、(b)の糖及び/又は糖アルコールを配合することにより、可塑剤を使用することなく、メタクリル酸系腸溶性高分子化合物を含有する練合混合物の塑性を高めることができる。本発明では、可塑剤を使用しないので、可塑剤使用に起因する種々の弊害を防止できる。このため、製剤の安定性が向上し、薬剤の溶出性を容易にコントロールすることができる。
【0044】
本発明において、メタクリル酸系腸溶性高分子化合物と糖及び/又は糖アルコールの配合割合は、メタクリル酸系腸溶性高分子化合物1重量部に対して、糖及び/又は糖アルコールが通常0.01〜20重量部、好ましくは0.1〜10重量部、より好ましくは0.2〜5重量部である。糖及び/又は糖アルコールの含有量が上記の範囲にあると、製剤が取り扱い易くなる、服用し易くなる、練合混合物の塑性が向上する、製造適性(製造しやすさ)の点で好ましい。
【0045】
前記具体的に記載したその他の糖及び/又は糖アルコールは、日本医薬添加物協会訳編、医薬品添加物ハンドブック(2001年)によれば、融点並びに糖及び/又は糖アルコールの1gを溶解させるのに要する水量は、ショ糖では160〜186℃、0.5gであり、デキストロースでは83℃、1gであり、フルクトースでは102〜105℃、0.3gであり、マルチトールでは148〜151℃、溶けやすい、である。
【0046】
(c)薬剤
薬剤は、医薬活性成分として疾病の治療あるいは予防に供されるものであれば、特に限定されず、それらの薬剤はフリー体、その塩、溶媒和物(水和物、エタノール和物など)、又は結晶多形を用いることができる。本発明で配合される薬剤は、特に、本発明の徐放化によって副作用の発現が抑えられ、治療効果が高まるような薬剤が適している。更に、クローン病、潰瘍性大腸炎、過敏性大腸炎、結腸癌など消化管下部に疾患部位があるような疾患に対して、薬剤を速やかに放出することが治療効果の増大に繋がるような薬剤が適している。
【0047】
薬剤は、結晶性であっても非結晶性であってもよい。薬剤は、水溶性及び脂溶性のいずれであってもよく、水に対して難溶性であってもよい。薬剤は、弱塩基性、中性又は酸性であるのが好ましい。
【0048】
薬剤が難溶性の場合、難溶性の薬剤の溶解性を改善するために、ナノ粉砕、微粉砕、非晶質化などといった製剤技術を用いることがあるが、前記背景技術に記載の特許文献5に記載のエタノールで湿式練合している技術では、エタノールが薬剤の結晶化、結晶成長化などの問題を引き起こす恐れがある。
【0049】
本発明に用いられる薬剤としては、例えば、5−アミノサリチル酸、アシクロビル、アスピリン、アセチルサリチル酸、アセトアミノフェン、アリピプラゾール、アンピシリン、イソジアニド、イブプロフェン、インドメタシン、エテンザミド、エナラプリル、エリスロマイシン、オメプラゾール、ケトコナゾール、サルブタモール、サラゾスルファピリジン、サラゾピリン、ジアゼパム、ジクロフェナク、ジクロフェナクナトリウム、ジピリダモール、シメチジン、シロスタゾール、シンバスタチン、スクラルファート、スルピリド、スルファサラジン、セレコキシブ、タクロリムス、テオフィリン、テガフール、デキサメタゾン、デキストロメトルファン、テトミラスト、テルフェナジン、ドキソルビシン、トリアムシロノン、トルバプタン、ナジフロキサシン、ナプロキセン、ニフェジピン、尿素、バルプロ酸ナトリウム、ハロペリドール、バロシクロビル、バリペリドン、ヒドロコルチゾン、ファモチジン、フェナセチン、フェニトイン、フェニルプロパノールアミン、プデソニド、プラバスタチン、プラバスタチンナトリウム、フルオロウラシル、プレドニゾロン、プレドニゾン、フロセミド、プロブコール、ベスナリノン、ペニシリン、ペルフェナジン、マレイン酸クロルフェニラミン、ミダゾラム、メシル酸ドキサゾシン、メトトレキセート、モルヒネ、ラニチジン、ランソプラゾール、リシノプリル、リスペリドン、リドカイン、レバミピド、レポドパ、ロチゴチン、ロバスタチン、ロラゼパム、ワーファリン、塩酸アンブロキソール、塩酸カルテオロール、塩酸ジフェンヒドロミン、塩酸タムスロシン、塩酸ニカルジピン、塩酸ヒドララジン、塩酸ブプレノルフィン、塩酸プロカテロール、塩酸モザバプタン、塩酸ラニチジン、塩酸レボカルニチン、酢酸コルチゾン、硫酸サルブタモール等が挙げられる。
【0050】
好ましい薬剤としては、5−アミノサリチル酸、アシクロビル、アスピリン、アセチルサリチル酸、アセトアミノフェン、アリピプラゾール、イブプロフェン、インドメタシン、エテンザミド、オメプラゾール、サラゾスルファピリジン、サラゾピリン、ジアゼパム、ジクロフェナク、ジクロフェナクナトリウム、ジピリダモール、シロスタゾール、シンバスタチン、タクロリムス、テオフィリン、テガフール、テトミラスト、ドキソルビシン、トルバプタン、ハロペリドール、バリペリドン、ヒドロコルチゾン、フェニトイン、プデソニド、プラバスタチン、フルオロウラシル、プレドニゾロン、プレドニゾン、フロセミド、プロブコール、ベスナリノン、ランソプラゾール、リスペリドン、レバミピド、レポドパ、ロチゴチン、ロバスタチン、塩酸カルテオロール、塩酸ニカルジピン、塩酸プロカテロール、塩酸モザバプタン、酢酸コルチゾン、硫酸サルブタモール等が挙げられる。より好ましい薬剤は、シロスタゾール、トルバプタン、フェニトイン、アスピリン及びナプロキセンである。
【0051】
上記薬剤の含有量は、医薬製剤中、通常1〜90重量%、好ましくは5〜80重量%、より好ましくは10〜70重量%である。これらの薬剤は、本発明の医薬製剤に、1種単独で又は2種以上混合して使用される。
【0052】
本発明において、上記のような薬剤を用いて徐放性医薬固形製剤を製造すると、1日1〜2回程度の投与が可能となる。例えば、トルバプタンは、バソプレシン拮抗剤として、例えば、血管拡張作用、血圧降下作用、肝糖放出抑制作用、メサンギウム細胞増殖抑制作用、水利尿作用、血小板凝集抑制作用、嘔吐抑制作用、尿素排泄促進作用、第VIII因子分泌抑制作用、心機能亢進作用、メサンギウム細胞収縮抑制作用、肝糖新生抑制作用、アルドステロン分泌抑制作用、エンドセリン産生抑制作用、レニン分泌調節作用、記憶調節作用、体温調節作用、プロスタグランジン産生調節作用等を有し、血管拡張剤、降圧剤、水利尿剤、血小板凝集抑制剤、尿素排泄促進剤、抗心不全剤、抗腎不全剤等として有利であり、高血圧、浮腫、腹水、心不全、腎機能障害、バソプレシン分泌異常症候群(SIADH)、肝硬変、低ナトリウム血症、低カリウム血症、糖尿病、循環不全、動揺病、水代謝障害、腎不全、各種虚血性疾患等の予防及び/又は治療に有効である。また、トルバプタンは、オキシトシン拮抗剤としては、例えば、子宮平滑筋収縮抑制作用、乳汁放出抑制作用、プロスタグランジン合成及び放出抑制作用、血管拡張作用を有し、オキシトシン関連疾患、特に早期分別、月経困難、帝王切開前の出産防止等の予防及び/又は治療に有効である。また、トルバプタンは、多発性のう胞腎の予防及び/又は治療に有効である。これら薬剤は、本発明の徐放化技術を用いると、1日1回投与の製剤を提供することができる。
【0053】
(d)形状維持物質
本発明において(d)形状維持物質とは、種々の製造工程において製造中の製剤に所望の形状を維持できるようなものであればよく、好ましくは押出造粒法及び球形整粒法を経て製造される医薬製剤の形状を維持できるような物質を意味する。
【0054】
本発明において、(d)形状維持物質としては、保水性があり、膨潤性、塑性を備えている。使用される形状維持物質としては、例えば、ヒドロキシプロピルセルロース、ヒプロメロース、メチルセルロース、ヒドロキシエチルセルロース等の水溶性セルロース;結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルメロース、カルメロースカルシウム、カルメロースナトリウム、クロスカルメロースナトリウム、カルボキシメチルエチルセルロース、エチルセルロース、ヒプロメロースフタレート、ヒドロキシプロピルメチルセルロースアセテートフタレート、酢酸セルロース、酢酸フタル酸セルロース等の水不溶性セルロース;ポリビニルピロリドン、ポリエチレンオキサイド、カルボキシビニルポリマー、ポリビニルアルコール(部分又は完全けん化物)等の水溶性合成高分子;クロスポビドン、ポリカルボフィル、ポリカルボフィルカルシウム、アミノアルキルメタクリレートコポリマーE、アミノアルキルメタクリレートコポリマーRS、メタクリル酸コポリマーL、メタクリル酸コポリマーS、メタクリル酸コポリマーLD、乾燥メタクリル酸コポリマーLD、ポレビニルアセタールジエチルアミノアセート等の水不溶性合成高分子;コムギデンプン、コメデンプン、トウモロコシデンプン、バレイショデンプン、部分アルファ化デンプン、α化デンプン、デキストリン、α−シクロデキストリン、β−シクロデキストリン、マルトデキストリン、イソマルト、ヒドロキシプロピルスターチ、カルボキシメチルスターチナトリウム、プルラン等のデンプン;アラビアゴム、アラビアゴム末、カンテン、カンテン末、ゼラチン、精製ゼラチン、キトサン、キサンタンガム、ペクチン、アルギン酸ナトリウム、ローカストビーンカム、グァーガム等の天然高分子化合物;ステアリン酸、ステアリン酸モノグリセリン、カルナウバロウ、ステアリルアルコール、セタノール、マクロゴール1500、マクロゴール4000、マクロゴール6000等の低融点物質等が挙げられる。これらの形状維持物質は、1種単独で又は2種以上混合して使用される。
【0055】
形状維持物質は、水不溶性で、かつ崩壊作用の少ない物質であるのが好ましい。このような形状維持物質としては、例えば、結晶セルロース、キトサン、アルギン酸ナトリウム、ポリカルボフィル、ポリカルボフィルカルシウム等が挙げられる。最も好ましい形状維持物質は、結晶セルロースである。
【0056】
結晶セルロースとしては、市販品、例えば、旭化成ケミカルズ社の「セオラスPH−101」、「セオラスPH−102」、「セオラスPH−301」、「セオラスPH−302」及び「セオラスKG−802」、FMC社の「アビセルPH−200」、JRS社の「VIVAPUR 12」等を使用できる。
【0057】
上記形状維持物質の含有量は、医薬製剤中、通常1〜90重量%、好ましくは3〜80重量%、より好ましくは5〜50重量%である。
【0058】
その他の成分
本発明の医薬製剤は、例えば、賦形剤、結合剤、pH調整剤、吸収促進剤、滑沢剤、着色剤、矯味剤、香料,剤皮等の固形医薬製剤に配合可能な各種添加剤を含有することができる。これらの成分は、本発明の効果を妨げない範囲内で本発明医薬製剤に配合できる。
【0059】
本発明の医薬固形製剤は、好ましくは押出造粒法及び球形整粒法を経て製造される固形製剤である。その剤形は、散剤、顆粒剤及びカプセル剤であることが好ましい。また、本発明の医薬固形製剤は、「進化する薬物治療 DDS最前線,2002年,金尾義治著,22頁,廣川書店」に記載されているような、スペイスタブ型錠剤(顆粒含有錠剤)のように、上記散剤又は顆粒剤を錠剤に含ませるように成形してもよい。本発明の医薬固形製剤のより好ましい剤形は、顆粒剤、カプセル剤及び顆粒含有錠剤である。剤形がカプセル剤又は顆粒含有錠剤であると、医薬固形製剤の取り扱い易さ及び服用のし易さの点で有利である。また、錠剤強度の向上及び湿度対策の観点から、顆粒含有錠剤に被膜を本発明の効果を妨げない程度に施してもよい。
【0060】
本発明の医薬固形製剤が、散剤又は顆粒剤である場合には、散剤及び顆粒剤の粒度が0.3〜3mmであることが、押出造粒時の製造適正の見地から好ましい。
【0061】
本発明の医薬固形製剤が、カプセル剤である場合には、カプセル剤のサイズが5〜00号であることが、取り扱い易さ及び服用のし易さの観点から好ましい。
【0062】
本発明の医薬固形製剤が、顆粒含有錠剤である場合には、生産性、取り扱い易さ及び服用のし易さの観点から、錠剤の形状は丸形又はカプレット形であるのが好ましく、顆粒含有錠剤の直径又は長径が6〜30mmの範囲にあることが好ましい。
【0063】
本発明の医薬固形製剤の製造方法
以下に本発明の医薬固形製剤の製造方法、特に散剤、顆粒剤及びカプセル剤の製造方法について説明するが、これに限定されるものではない。
【0064】
本発明の医薬固形製剤は、種々の製造方法により製造することができる。本発明において、好ましい剤形である散剤、顆粒剤及びカプセル剤を製造する場合には、少なくとも押出造粒工程を有する製造方法が好ましい。この押出造粒工程に加えて、湿式練合工程及び球形整粒工程を有する製造方法が好ましい。
【0065】
本発明の医薬固形製剤の好ましい製造方法は、湿式練合工程、押出造粒工程、球形整粒工程、乾燥工程、及び必要に応じて整粒工程を経て製造される。本発明の好ましい製造工程は、湿式練合工程、押出造粒工程、球形整粒工程、及び乾燥工程の順である。
【0066】
本発明の医薬固形製剤がカプセル剤である場合、整粒工程の後、更に、流動化剤混合工程及びカプセル充填工程を経て製造される。
【0067】
本発明の医薬固形製剤が顆粒含有錠剤である場合、整粒工程の後、得られる顆粒を適当な添加剤と混合する工程、滑沢剤を混合する工程及び打錠工程を経て製造される。また必要に応じて打錠後にフィルムコーティングを行ってもよい。
【0068】
湿式練合工程
湿式練合工程は、上記成分を結合剤等の他の成分と共に湿式で練合し、前記各成分に水を馴染ませる工程である。この際、(b)成分の糖及び/又は糖アルコールは粉末形態のまま使用してもよいし、予め水に溶解させた溶液の形態で使用してもよい。
【0069】
湿式練合は、主に湿式高剪断造粒法に基づいて操作される。湿式練合工程で用いられる装置としては、例えば、岡田精工社製の「ニュースピードニーダー」、パウレック社製の「バーチカルグラニュレーター」、深江パウテック社製の「ハイスピードミキサー」、奈良機械製作所社製の「高速攪拌型混造粒機NMG」、ミューチュアル社製の「ディオスナ攪拌混合造粒機」、スペクトラム(Spectrium)社製の「エアロマティックフィールダー(Aeromatic-filelder)」等が挙げられる。
【0070】
押出造粒工程
押出造粒工程は、湿式練合工程で得られた湿式練合混合物をスクリーンに向かって押出し、円柱状繊維を得る工程である。押出造粒装置としては、特に限定がなく、例えば、スクリュー供給方式、重力供給方式、ピストン供給方式等のいずれでもよい。スクリュー方式の押出造粒装置としては、不二パウダル社製の「ドームグランDG−L1」、「ツインドームグランTDG−80」、「ツインドームグランTDG−110」等が挙げられる。重力供給方式の押出造粒装置として、ギアロールタイプのホソカワミクロン社製の「ギャペレタイザGCS」、放射状タイプの深江パウテック社製の「FG型円筒造粒機」等が挙げられる。
【0071】
散剤を製造する場合は、スクリーンの孔径を0.3〜0.5mmとすればよい。また顆粒剤及びカプセル剤を製造する場合はスクリーンの孔径を0.3〜3mmとすればよい。
【0072】
球形整粒工程
球形整粒工程は、押出造粒工程で得られた円柱状繊維を適度な大きさに切って球形化し、形を整える工程である。球形整粒工程に用いられる装置としては、例えば、岡田精工社製の「ニュースピードニーダー」、不二パウダル社製の「マルメライザーQJ」、フロイント産業社製の「CF造粒機」、「グラニュレックスGX」等が挙げられる。
【0073】
乾燥工程
乾燥工程は、球形整粒工程で処理された粒子を乾燥し、水分を除去する工程である。乾燥は、直接加熱方式及び間接加熱方式のいずれでもよい。直接加熱方式としては、例えば、棚式送風乾燥機、流動層乾燥機等が挙げられる。間接加熱方式としては、例えば、真空乾燥機、マイクロ波乾燥機、遠赤外線乾燥機等が挙げられる。乾燥工程に用いられる装置としては、具体的には、パウレック社製の「グラット流動層造粒機WST」「マルチプレックス」、不二パウダル社製の「箱型通気平行流式ドライヤー」、「ミゼツトドライヤー」、大川原製作所社製の「スリットフローFBS」、フロイント産業社製の「フロードライヤーNFOD」、中央化工機社製の「振動乾燥機」、タバイエスペック社製の「SPHH−200」等が挙げられる。
【0074】
整粒工程
整粒工程は、乾燥粒子から一定の粒度範囲の粒子を取り出す工程である。この工程には、例えば、篩分け法等が適用できる。
【0075】
流動化剤混合工程
流動化剤混合工程は、整粒工程後の粒子に流動化剤を添加し、粒子と流動化剤とを均質に混合する工程である。流動化剤混合工程には、例えば、拡散式ミキサー方式(容器回転式)が適している。
【0076】
カプセル充填工程
カプセル充填工程は、流動化剤が混合された粒子をカプセルに充填する工程である。カプセル充填工程で使用される装置としては、例えば、クオリカプス社製の「LIQFIL super」シリーズ、ボッシュ包装機社製の「GKF」シリーズ、IMA社の「ZANASI」、「MATIC」シリーズ等が挙げられる。
【発明の効果】
【0077】
本発明においては、特定の性質を有する糖及び/又は糖アルコールを使用することによりメタクリル酸系腸溶性高分子化合物の含有比率が高い練合混合物に適度な塑性を付与することができる。そのため、本発明の医薬固形製剤には、可塑剤を配合する必要がなく、本発明の医薬固形製剤は、可塑剤配合に基づく欠点(即ち、可塑剤の配合によりメタクリル酸系腸溶性高分子化合物の変質及び変形が促進され、スクリーンの内側に該腸溶性高分子化合物からなる膜が形成されやすく、スクリーンの閉塞及び造粒装置の故障の原因となり得る。更に、この固形製剤中には可塑剤が残存するため,メタクリル酸系腸溶性高分子化合物の経時的な変質及び変形を促し、その結果、薬剤の経時的な溶出変化が生じるのが避けられなくなる。)を有していない。
【0078】
本発明の医薬固形製剤は、可塑剤を使用しなくてもメタクリル酸系腸溶性高分子化合物を多量に含有させることができるので、消化管上部での薬剤の放出を抑え、かつ消化管下部において薬剤の放出を速やかにするような高度な放出制御性を備えている。
【0079】
本発明の医薬固形製剤は、pH応答性の良いメタクリル酸系腸溶性高分子化合物を有することで高度な放出制御性を備えている。そのため、腸溶性製剤、時限放出型製剤、結腸特異的放出型製剤等を製造する際に施されるコーティング工程を必要としない。それ故、本発明は所望の徐放性能を備えた医薬固形製剤を、安価でかつ高い生産性をもって製造することができる。
【図面の簡単な説明】
【0080】
【図1】図1は、実施例24、25及び26の溶出試験結果を示すグラフである。
【図2】図2は、絶食下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【図3】図3は、食後下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【図4】図4は、実施例24〜26において、平均滞留時間(MRT)と50%溶出時間(T50)との関係 (In Vitro-In Vivo Correlation)(レベルB)を示すグラフである。
【発明を実施するための形態】
【実施例】
【0081】
以下に実施例及び比較例を掲げて本発明をより一層明らかにする。
【0082】
実施例1
シロスタゾール100g、サンマルト−S(マルトース一水和物、三和澱粉工業社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、180秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0083】
上記押出造粒工程において得られる素麺状造粒物は、本発明の医薬固形製剤の品質及び生産性に大きく影響を及ぼす。少量スケールではスクリーンを通過する練合物の量が少ないため、1回通過では練合物に加わる圧力が小さく、生産性を予測するのは困難である。このため、少量の練合物で生産性を予測する場合、同じ練合物を複数回繰り返しスクリーンを通過させ、スクリーンとスクリューの隙間に滞留する固形組成物の状態を安定化させた状態で製造性を把握するのがよい。以下の実施例では、同じ練合物を3回繰り返し押出造粒を行った後の練合物の温度を各回毎に測定し、これらの温度から練合物が適度の塑性を備えているかどうかを確認した。
【0084】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0085】
【表1】
【0086】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0087】
実施例2
シロスタゾール100g、エリスリトール100M(エリスリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、160秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を15秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0088】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0089】
【表2】
【0090】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0091】
実施例3
シロスタゾール100g、ソルビトールSP(ソルビトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラス PH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0092】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0093】
【表3】
【0094】
上記の結果から、ソルビトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0095】
実施例4
シロスタゾール100g、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、170秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0096】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0097】
【表4】
【0098】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0099】
実施例5
シロスタゾール100g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0100】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0101】
【表5】
【0102】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0103】
実施例6
シロスタゾール100g、キシリトールP(キシリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水8.5gを結合液として投入し、115秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0104】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0105】
【表6】
【0106】
上記の結果から、キシリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0107】
比較例1
シロスタゾール100g、WYNDALE 200M乳糖(乳糖一水和物、ラクトースカンパニーニュージーランドリミテッド社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、240秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回繰り返して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0108】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0109】
【表7】
【0110】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、乳糖一水和物を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0111】
比較例2
シロスタゾール100g、ペアリトール50C(D−マンニトール、ロケット社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水10gを結合液として投入し、300秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0112】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0113】
【表8】
【0114】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、D−マンニトールを配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0115】
実施例7
シロスタゾール100g、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水18gを結合液として投入し、320秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0116】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0117】
【表9】
【0118】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0119】
実施例8
シロスタゾール100g、エリスリトール100M(エリスリトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、100秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0120】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0121】
【表10】
【0122】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0123】
実施例9
シロスタゾール100g、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水16.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0124】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0125】
【表11】
【0126】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0127】
実施例10
シロスタゾール100g、ソルビトールSP(ソルビトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、50秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0128】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0129】
【表12】
【0130】
上記の結果から、ソルビトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0131】
実施例11
シロスタゾール100g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水16.5gを結合液として投入し、110秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0132】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0133】
【表13】
【0134】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0135】
実施例12
シロスタゾール100g、キシリトールP(キシリトール、日研化成社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラス PH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水15gを結合液として投入し、50秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0136】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0137】
【表14】
【0138】
上記の結果から、キシリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0139】
比較例3
シロスタゾール100g、ハンマーミルで粉砕した白糖(グラニュー糖CH、塩水港精糖社製)25g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)60g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水17gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0140】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0141】
【表15】
【0142】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、白糖を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0143】
参考例1
非晶質化したトルバプタンを次のようにして調製した。即ち、7−クロロ−5−ヒドロキシ−1−[2−メチル−4−(2−メチルベンゾイルアミノ)ベンゾイル]−2,3,4,5−テトラヒドロ−1H−ベンゾアゼピン100g及びヒドロキシプロピルセルロース(HPC−SL、日本曹達社製、ヒドロキシプロポキシル基含量:53〜78重量%)50gをジクロロメタン1390g及びエタノール350gの混合溶液に溶解し、噴霧乾燥機(大川原加工機社製、ODT−8型)で処理した後、直ちに真空乾燥機(タバイエスペック社製、LCV−232)で乾燥し、非晶質粉末(非晶質化トルバプタン)を調製した。
【0144】
実施例13
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水15gを結合液として投入し、110秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0145】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0146】
【表16】
【0147】
上記の結果から、マルトース一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0148】
実施例14
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、エリスリトール100M(エリスリトール、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水13gを結合液として投入し、90秒湿式練合した。孔径0.6mm、開口比19.3%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0149】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0150】
【表17】
【0151】
上記の結果から、エリスリトールを配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0152】
実施例15
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、ラクチトールLC−1(ラクチトール一水和物、日研化成社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水13gを結合液として投入し、120秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0153】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0154】
【表18】
【0155】
上記の結果から、ラクチトール一水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0156】
実施例16
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、トレハロースP(トレハロース二水和物、林原生物化学研究所製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水14.5gを結合液として投入し、130秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を20秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2〜3時間乾燥し、乾燥顆粒を得た。
【0157】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0158】
【表19】
【0159】
上記の結果から、トレハロース二水和物を配合すれば、メタクリル酸コポリマーSの配合量が多量であっても、湿式練合物に適度な塑性を付与できることがわかる。
【0160】
比較例4
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、WYNDALE 200M乳糖(乳糖一水和物、ラクトースカンパニーニュージーランドリミテッド社製)15g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水17.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグラン DG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、2回連続して押出造粒したが、3回目の押出造粒は、DG−L1に対する負荷が大きく、モーターの電流値が大きく変動したため、中止した。
【0161】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0162】
【表20】
【0163】
上記の結果から、メタクリル酸コポリマーSの配合量が多量の場合には、乳糖一水和物を配合しても、湿式練合物に適度な塑性を付与できないことがわかる。
【0164】
実施例17
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水11gを結合液として投入し、180秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入し、40rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約2〜4cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、マトリックス顆粒を得た。篩過により600μm〜1000μmのマトリックス顆粒を製品とした。
【0165】
1.粒子径分布の計測
約5gのマトリックス顆粒を取り、日本薬局方の一般試験法に規定される粒度測定法(乾式ふるい分け法)に準じてロボットシフターRPS−95(セイシン企業社製)で粒度測定を行った。マトリックス顆粒の平均粒子径は770μmであった。
【0166】
2.分解物の測定
実施例17で得られたマトリックス顆粒140.4gに軽質無水ケイ酸(アドソリダー−101、ワイ・ケイ・エフ社製)0.6gを加え、よく混合後、ヒプロメロース3号カプセル(QUALI−Vカプセル、クオリカプス社製)にトルバプタン60mg相当になるよう充填した。マトリックス顆粒含有カプセルをプラスチック容器に入れ、60℃に2週間保存した。
【0167】
トルバプタン30mg相当のマトリックス顆粒を秤取して乳鉢内で粉砕後、メタノールを加え、超音波を照射して完全に顆粒を破壊した。これにメタノールを加え、約0.5μmのメンブランフィルターでろ過し、得られたろ液をLC−2010CTシステム(高速液体クロマトグラフィー、島津製作所製)で測定波長254nm、移動相(アセトニトリル/水/リン酸=500/500/1、容積比)の送液速度約1mlで分解物Aの探索を行った。分解物Aの濃度を面積百分率法で算出した(トルバプタンのピーク面積に対する分解物Aのピーク面積の比率)。
【0168】
保存の前後における分解物Aの濃度を調べたところ、製造直後における分解物Aの濃度が0.01%、60℃で2週間保存した後の分解物Aの濃度は0.02%であり、分解物の生成が抑制されていることが明らかになった。
【0169】
3.溶出試験
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。pH7.4の薄めたMcIlvaine緩衝液にポリソルベート80を1w/v%の濃度となるよう添加した溶液900mlを溶出試験液とした。パドルの回転数は50rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0170】
60℃で2週間保存した前後におけるそれぞれの溶出率及び溶出率の差(Δ溶出率、製造直後−60℃/2週間保存後)を以下に示す。
【0171】
【表21】
【0172】
上記表から、実施例17で得られたマトリックス顆粒含有カプセルからのトルバプタンの溶出率については製造直後及び60℃/2週間保存後の双方において殆ど変化がなく、溶出の経時的安定性が良好であることが明らかである。
【0173】
比較例5
参考例1で調製した非晶質化トルバプタン45g(トルバプタンとして30g)、ペアリトール50C(D−マンニトール、ロケット社製)45g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)10gを奈良式高速攪拌型混合造粒機 NMG−1L(奈良機械製作所社製)に投入した。次に、4w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)と4w/v%ポリソルベート80水溶液(ポリソルベート80(HM)、日本油脂社製)の混液(1:1重量比)40gを結合液として投入し、30秒湿式練合した。孔径0.6mm、開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜4cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、マトリックス顆粒を得た。篩過により355μm〜850μmのマトリックス顆粒を製品とした。
【0174】
実施例17と同様にして粒子径分布の計測を行ったところ、マトリックス顆粒の平均粒子径は690μmであった。
【0175】
製造直後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した後、ガラス製気密容器に充填し、室温で1年間保管した。室温一年保管後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した。さらに、室温一年保管後のマトリックス顆粒をガラス製気密容器に充填して60℃で2週間保存し、60℃2週間保存後のマトリックス顆粒について分解物Aの測定及び溶出試験を実施した。なお、分解物Aの測定は、実施例17と同様にして行った。溶出試験は、以下の方法で行った。
【0176】
溶出試験:
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。pH7.0のMcIlvaine緩衝液にポリソルベート80を1w/v%の濃度となるよう添加した溶液900mlを溶出試験液とした。パドルの回転数は100rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0177】
分解物Aの測定結果を表22に示す。
【0178】
【表22】
【0179】
溶出試験の結果を表23に示す。なお、表中のΔ溶出率は、室温で一年保存後の溶出率から60℃で2週間保存後の溶出率を差し引いたものである。
【0180】
【表23】
【0181】
マトリックス顆粒に可塑剤(ポリソルベート80)が含まれていると、顕著な分解物Aの生成増加が認められ,かつ溶出率の低下も大きかった。また、室温で1年保存している間に分解物Aの生成量が増加し、溶出も遅延する傾向を示していた。そのため、比較例5で得られたマトリックス顆粒は、溶出の経時的安定性に著しく劣っていることが判明した。
【0182】
実施例18
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)18g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水15gを結合液として投入し、250秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を5回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を50秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが7.4重量%の割合で含有されている。
【0183】
実施例19
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水8gを結合液として投入し、160秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約10cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を25秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが11.7重量%の割合で含有されている。
【0184】
実施例20
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60g及び精製水11gを結合液として投入し、180秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を6回繰り返して行い、長さ約3〜5cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSが19.2重量%の割合で含有されている。
【0185】
比較例6
参考例1で調製した非晶質化トルバプタン90g(トルバプタンとして60g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)12.9g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)30g及び精製水6gを結合液として投入し、210秒湿式練合した。孔径0.8mm、開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、押出造粒を5回繰り返して行い、長さ約1〜2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。乾燥顆粒を篩過処理し、600〜1000μmを製品とした。この乾燥顆粒には、メタクリル酸コポリマーSは含有されていない。
【0186】
溶出試験:
溶出試験システムDT−610(日本分光社製)を用いて日本薬局方溶出試験方法第二法(パドル法)によってマトリックス顆粒含有カプセルからのトルバプタンの溶出試験を行った。使用した試験液1及び試験液2は以下の通りである。
試験液1:日本薬局方15 溶出試験液第一液(pH1.2)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
試験液2:薄めたMcIlvaine緩衝液(pH7.4)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
パドルの回転数はいずれも50rpmとし、測定波長に268nm及び350nmの二波長を用いた。
【0187】
試験液1を用いて溶出試験を行った結果を表24に示す。表における数値は、試験を3回行った平均値である。
【0188】
【表24】
【0189】
pH1.2の酸性条件では、顆粒からの薬剤の溶出性に全く差は認められず、メタクリル酸高分子化合物の含有割合の違いを確認できなかった。
【0190】
試験液2を用いて溶出試験を行った結果を表25に示す。表における数値は、試験を3回行った平均値である。
【0191】
【表25】
【0192】
メタクリル酸高分子化合物が溶解するpH域では、その含有量に応じて溶出性が速やかになったが、比較例6は酸性条件のときと全く同じ溶出性を示していた。
【0193】
実施例21
フェニトイン(シグマアルドリッチ社製)100g、エリスリトール100M(エリスリトール、日研化成社製)60g、オイドラギットL100(メタクリル酸コポリマーL、デグサ社製)15g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機,岡田精工社製)に投入した。5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース,日本曹達社製)40g及び精製水9.5gを結合液として投入し、150秒湿式練合した。孔径0.6mm,開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を10秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0194】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0195】
【表26】
【0196】
実施例22
アスピリン(アセチルサリチル酸、和光純薬工業社製)100g、ラクチトールLC−1(ラクチトール、日研化成社製)10g、オイドラギットL100D55(乾燥メタクリル酸コポリマーLD、デグサ社製)40g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)20gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)40g及び精製水9gを結合液として投入し、130秒湿式練合した。孔径0.6mm,開口比19.3%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約2〜3cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0197】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0198】
【表27】
【0199】
実施例23
ナプロキセン(シグマアルドリッチ社製)15g、トレハロースP(トレハロース二水和物、林原生物化学研究所製)30g、オイドラギットL100D55(乾燥メタクリル酸コポリマーLD、デグサ社製)40g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)30gをスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)20g及び精製水4.5gを結合液として投入し、140秒湿式練合した。孔径0.6mm,開口比22.6%のドームダイを装着したドームグランDG−L1(押出造粒機,不二パウダル社製)にこの練合物を徐々に投入した。40rpmのスクリュー速度で、押出造粒を3回繰り返して行い、長さ約4〜5cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機,不二パウダル社製)を使用し、約1000rpmの回転速度でこの素麺状造粒物を30秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機,タバイエスペック社製)で2時間乾燥し、乾燥顆粒を得た。
【0200】
非接触型近赤外温度計IR−101(テクノライン社製)を用い、湿式練合後の練合物の温度並びに各回押出造粒後の湿式練合物の温度を測定した。結果を次表に示す。
【0201】
【表28】
【0202】
実施例24
実施例20で得られた乾燥顆粒196gに、アドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.8gを添加混合し、得られた顆粒141mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0203】
実施例25
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)30g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)18gをそれぞれスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60gと精製水3gを結合液として投入し、180秒湿式練合した。孔径0.8mm,開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、4回繰り返して押出造粒を行い、長さ約2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)で約1000rpmの回転速度で、この素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で4時間乾燥した。その後篩過処理によって平均粒子径780μmの顆粒を得た。この顆粒179gにアドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.8gを添加混合し、得られた顆粒129mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0204】
実施例26
参考例1で調製した非晶質化トルバプタン180g(トルバプタンとして120g)、サンマルト−S(マルトース一水和物、三和澱粉工業社製)25.8g、オイドラギットS100(メタクリル酸コポリマーS、デグサ社製)54g及びセオラスPH−301(結晶セルロース、旭化成ケミカルズ社製)30gをそれぞれスピードニーダーNSK−150(攪拌造粒機、岡田精工社製)に投入した。次に、5w/v%HPC−L水溶液(ヒドロキシプロピルセルロース、日本曹達社製)60gと精製水16gを結合液として投入し、180秒湿式練合した。孔径0.8mm,開口比22.5%のドームダイを装着したドームグランDG−L1(押出造粒機、不二パウダル社製)にこの練合物を徐々に投入した。60rpmのスクリュー速度で、4回繰り返して押出造粒を行い、長さ約2cmの素麺状造粒物を得た。3mm間隔にピッチが刻まれているクロスハッチプレートを装着したマルメライザーQJ−400(球形整粒機、不二パウダル社製)で約1000rpmの回転速度で、この素麺状造粒物を60秒間処理し、湿顆粒を得た。これを70℃に設定したSPHH−200(棚式送風乾燥機、タバイエスペック社製)で4時間乾燥した。その後篩過処理によって平均粒子径780μmの顆粒を得た。この顆粒217gにアドソリダー−101(二酸化ケイ素、ワイ・ケイ・エフ社製)0.9gを添加混合し、得られた顆粒147mgをヒプロメロースカプセル3号に充填した。このカプセル剤は、トルバプタンを60mg含有する。
【0205】
溶出試験方法
溶出試験システムNTR−6200A(富山産業社製)を用い、日本薬局方溶出試験方法第二法(パドル法)で実施例24、25及び26で得られるカプセル剤からのトルバプタンの溶出試験を行った。下記に示す試験液を使用した。
【0206】
試験液:日本薬局方15 溶出試験液第二液(pH6.8)にポリソルベート80を1w/v%の濃度となるよう添加した溶液900ml
測定波長:λ1,268nm; λ2,350nm
パドル回転数:100rpm
サンプル数:n=6
サンプリング時点:0.5、1、2、3、4、6、8、10及び12時間
50%溶出時間(D50)の算出
図1よりトルバプタンが50%溶出するのに要した時間を求めた。
【0207】
【表29】
【0208】
経口投与試験
本発明の医薬固形製剤の徐放化効果を確認するため,健康人への経口投与試験を行った。18名の健康な男女(年齢18〜45歳)を無作為に6名ずつA群、B群及びC群に分け、以下の計画に従って3群4期の非完全クロスオーバー試験で行った。
【0209】
【表30】
【0210】
いずれの群においても、第1期は速放錠を一日二回空腹下に投与した。早朝にトルバプタンをトータルで45mg含む2個の錠剤(30mg錠+15mg錠)を空腹下経口投与し、8時間経過後に15mg錠1個を経口投与した。一日投与量はトルバプタン60mgである。第2期では,群ごとに実施例24、25及び26で得られる異なるカプセルを空腹下に経口投与した。第3期では,いずれの群においても第2期とは異なる、実施例24、25及び26で得られるカプセルを空腹下経口投与した。第4期では,第3期に投与したカプセルと同じカプセルを食後経口投与した。食事の内容は米国FDAのガイダンス(Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies. U.S. Department of Healthy and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), December 2002)に基づいて設定した高脂肪食であり、食事を摂取後30分以内に実施例24、25及び26で得られるカプセルを経口投与した。速放錠並びに実施例24、25及び26で得られるカプセルを空腹下投与した例数は、それぞれ18例及び12例であり、実施例24、25及び26のカプセルを食後投与した例数は6例である。なお、速放製剤の組成は以下の通りである。
【0211】
比較例7:30mg錠(速放錠)
参考例1で調製した非晶質化したトルバプタン112.5g(トルバプタン量75g)、乳糖一水和物185g、トウモロコシデンプン50g及び結晶セルロース50gを混合し、攪拌流動造粒乾燥機(パウレック社製,マルチプレックスMP−01)に投入した。ヒドロキシプロポキシル基含量が53〜78重量%のヒドロキシプロピルセルロース5w/v%水溶液200gを用いて流動層造粒及び乾燥を行い、造粒物を得た。これにLH−11(低置換ヒドロキシプロピルセルロース)22.5g及びステアリン酸マグネシウム5gを混合し,打錠用顆粒とした。これをロータリー式連続打錠機(菊水製作所製,12HUK−AWC)を用いて打錠圧900kg,回転数40rpmの条件でトルバプタン30mgを含有する重量約174mgの直径8mmの平面錠を製造した。
【0212】
比較例8:15mg錠(速放錠)
参考例1で調製した非晶質化したトルバプタン56.3g(トルバプタン量37.6g)、乳糖一水和物256.3g、トウモロコシデンプン50g及び結晶セルロース50gを混合し、攪拌流動造粒乾燥機(パウレック社製,マルチプレックスMP−01)に投入した。ヒドロキシプロポキシル基含量が53〜78重量%のヒドロキシプロピルセルロース5w/v%水溶液200gを用いて流動層造粒及び乾燥を行い、造粒物を得た。これにLH−11(低置換ヒドロキシプロピルセルロース)22.5g及びステアリン酸マグネシウム5gを混合し、打錠用顆粒とした。これをロータリー式連続打錠機(菊水製作所製,12HUK−AWC)を用いて打錠圧1000kg,回転数50rpmの条件でトルバプタン15mgを含有する重量約180mgの直径8mmの平面錠を製造した。
【0213】
評価
経時的に採血し、血漿中トルバプタン濃度を測定した。薬物動態パラメータは、WinNolin (ver4.0, Pharsight社製ソフトウェア)及びPSAG-CP ((株)アスメディカ社製ソフトウェア)を用いて算出した。
【0214】
図2は、絶食下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。図3は、食後下での投与後における血漿中トルバプタン濃度の推移を示すグラフである。
【0215】
【表31】
【0216】
図4は、実施例24〜26において、平均滞留時間(MRT)と50%溶出時間(T50)との関係 (In Vitro-In Vivo Correlation)を示すグラフである。米国FDAのガイダンス(Guidance for Industry Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations, U.S. Department of Health and Human Services, Food and Drug Administration, and Center for Drug Evaluation and Research (CDER), September 1997)によると、レベルBは、MRTとT50とのよい関係を示している。
【0217】
長期保存サンプルの色調変化
試験方法
実施例13、14、15及び16の顆粒剤をポリエチレン袋に入れて密閉し、室内に1年以上保管した。このときの温度及び湿度は自然体である。保管の前後で色調の変化を目視で確認した。また顆粒の堅さを手で確認した。
【0218】
【表32】
【0219】
実施例13〜16の顆粒剤は、いずれも色調変化はなく、顆粒の硬さも変化がなかった。
【産業上の利用可能性】
【0220】
本発明の医薬固形製剤は、医薬分野で利用価値が高い。特に、可塑剤を配合しなくても、製剤を製造する際の練合物に良好な塑性を与えることができる。更に、本発明の製剤は、可塑剤を配合する場合に生ずる問題点を解決することができ、極めて有用である。また、本発明の医薬固形製剤は、経口徐放性医薬固形製剤として極めて利用価値が高い。
【特許請求の範囲】
【請求項1】
(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、
前記(b)糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である、
医薬固形製剤。
【請求項2】
糖及び/又は糖アルコールの配合量が、メタクリル酸系腸溶性高分子化合物1重量部に対して、0.1〜10重量部である、請求項1に記載の医薬固形製剤。
【請求項3】
糖及び/又は糖アルコールは、融点が140℃以下である、請求項1又は請求項2に記載の医薬固形製剤。
【請求項4】
可塑剤を含有していない請求項1又は請求項2に記載の医薬固形製剤。
【請求項5】
可塑剤を含有していない請求項3に記載の医薬固形製剤。
【請求項6】
押出造粒工程を有する製造方法により製造される、請求項1又は請求項2に記載の医薬固形製剤。
【請求項7】
押出造粒工程を有する製造方法により製造される、請求項3に記載の医薬固形製剤。
【請求項1】
(a)メタクリル酸系腸溶性高分子化合物並びに(b)糖及び/又は糖アルコールを含有するマトリックス型の医薬固形製剤であって、
前記(b)糖及び/又は糖アルコールは、その1gを溶解するのに要する水(20〜25℃のいずれかの水温)の量が4g以下である、
医薬固形製剤。
【請求項2】
糖及び/又は糖アルコールの配合量が、メタクリル酸系腸溶性高分子化合物1重量部に対して、0.1〜10重量部である、請求項1に記載の医薬固形製剤。
【請求項3】
糖及び/又は糖アルコールは、融点が140℃以下である、請求項1又は請求項2に記載の医薬固形製剤。
【請求項4】
可塑剤を含有していない請求項1又は請求項2に記載の医薬固形製剤。
【請求項5】
可塑剤を含有していない請求項3に記載の医薬固形製剤。
【請求項6】
押出造粒工程を有する製造方法により製造される、請求項1又は請求項2に記載の医薬固形製剤。
【請求項7】
押出造粒工程を有する製造方法により製造される、請求項3に記載の医薬固形製剤。
【図1】

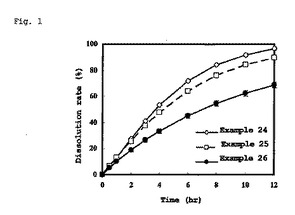
【図2】
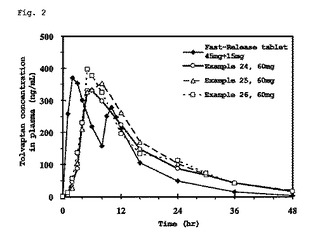
【図3】

【図4】
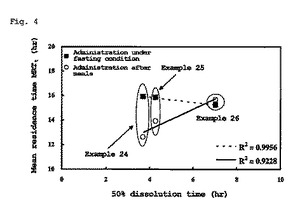
【図2】
【図3】
【図4】
【公表番号】特表2011−500511(P2011−500511A)
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願番号】特願2010−515141(P2010−515141)
【出願日】平成20年9月26日(2008.9.26)
【国際出願番号】PCT/JP2008/067996
【国際公開番号】WO2009/051022
【国際公開日】平成21年4月23日(2009.4.23)
【出願人】(000206956)大塚製薬株式会社 (230)
【Fターム(参考)】
【公表日】平成23年1月6日(2011.1.6)
【国際特許分類】
【出願日】平成20年9月26日(2008.9.26)
【国際出願番号】PCT/JP2008/067996
【国際公開番号】WO2009/051022
【国際公開日】平成21年4月23日(2009.4.23)
【出願人】(000206956)大塚製薬株式会社 (230)
【Fターム(参考)】
[ Back to top ]