
医薬製品
【課題】オピオイド拮抗薬の放出を実質的に防止するオピオイド拮抗薬を含有する経口剤型の提供。
【解決手段】下記を含有する医薬製品:a)第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子、及び押出成形した粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が、たとえば、押出成形した粒子重量の約5%〜約30%を含有する複数の粒子;b)第3疎水性物質内に分散したオピオイド作動薬を含有する複数の粒子;及びc)複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の押出成形した粒子を含むカプセル。
【解決手段】下記を含有する医薬製品:a)第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子、及び押出成形した粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が、たとえば、押出成形した粒子重量の約5%〜約30%を含有する複数の粒子;b)第3疎水性物質内に分散したオピオイド作動薬を含有する複数の粒子;及びc)複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の押出成形した粒子を含むカプセル。
【発明の詳細な説明】
【背景技術】
【0001】
医薬製品は時折乱用の対象となる。たとえば、オピオイド作動薬の特定用量は、非経口
的に投与すると、同じ用量を経口で投与した場合と比較してさらに強力になる場合がある
。一部の製剤は、改変する(tampered)ことによって該製剤に含まれるオピオイド作動薬
を違法な使用に供することができる。放出制御型オピオイド作動薬は時折薬物乱用者によ
って細砕され、それに含まれるオピオイドが経口または非経口投与直後に即時放出に供さ
れる。
【0002】
オピオイド拮抗薬がオピオイド作動薬の非経口的な乱用を抑止するために、特定のオピ
オイド作動薬と組み合わされている。 従来の技術では、即放性ペンタゾシンとナロキソ
ンの併用が、米国で Sanofi-Winthrop 社から Talwin(登録商標)Nx (タルウィン) とし
て市販されている錠剤で利用されている。Talwin(登録商標)Nx (タルウィン) には、50
mg 塩基(base)に相当する即放性塩酸ペンタゾシン及び 0.5 mg 塩基(base)に相当す
る塩酸ナロキソンが含まれる。チリジン (50 mg) 及びナロキソン (4 mg) を含有する固
定併用療法(fixed combination therapy)が 1978 年以来、疼痛管理のためにドイツで
提供されている (Valoron(登録商標)N、Goedecke)。ブプレノルフィンとナロキソンの
固定併用は疼痛治療のためにニュージーランド (Temgesic(登録商標)Nx、Reckitt & Co
lman) で 1991 年に導入された。
【0003】
米 Purdue Pharma 社は現在徐放性オキシコドンを 10、20、40 及び 80 mg の塩酸オキ
シコドンを含む剤型でオキシコンチンの商品名で市販している。
【0004】
米国特許番号第 5,266,331 号; 第 5,508,042 号; 第 5,549,912 及び第 5,656,295 号
により徐放性オキシコドン製剤が開示されている。
【0005】
米国特許番号第 5,472,943 号(Crain らによる)では、作動薬をオピオイド拮抗薬と共
に投与することによって二峰性に作用するオピオイド作動薬の鎮痛効力を高める方法が記
述されている。
【0006】
米国特許特許番号第 6,277,384 号; 第 6,475,494 号; 第 及び第 6,375,957 号(Kaiko
らによる); 及び第 6,228,863 号 (Colucci らによる)では、オピオイド鎮痛剤型に関連
する乱用の可能性を少なくすることを狙いとしている。
【0007】
PCT 公開番号 WO 01/58451 の発明の名称「不正使用防止経口オピオイド作動薬製剤 (T
amper Resistant Oral Opioid Agonist Formulations)」では、オピオイド作動薬剤型内
の隔離したオピオイド拮抗薬を抱合することによってオピオイド鎮痛剤型に関連する乱用
の可能性を少なくすることをその狙いとする。
【0008】
乱用の可能性が低いオピオイド作動薬を含有する経口剤型技術の必要性が継続して存在
する。
【0009】
前述を含む本明細書中で引用したすべての参照を引用することを以って本明細書の一部
となす。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的と要旨
本発明の目的は、オピオイド拮抗薬の放出を実質的に防止するオピオイド拮抗薬を含有
する経口剤型の提供することである。
【課題を解決するための手段】
【0011】
本発明の特定の実施形態の目的は、オピオイド作動薬の乱用の可能性を少なくすること
に有用なオピオイド拮抗薬の処方を含有する経口剤型提供することである。
【0012】
本発明の特定の実施形態の目的は、オピオイド作動薬の鎮痛効果に影響を及ぼすこと、
または退薬を誘発する可能性を受けることなしに、オピオイド作動薬の乱用の可能性を少
なくするために有用なオピオイド拮抗薬の処方を含有する経口剤型を提供することである
。
【0013】
本発明の特定の実施形態の目的は、剤型が無傷で経口投与されるときにオピオイド作動
薬の鎮痛効力を変化させないか、または実質的に変化させないオピオイド作動薬の有効量
及びオピオイド拮抗薬の一回分量を含む経口剤型を提供することである。但し、剤型が改
変されていない場合、オピオイド拮抗薬は大幅に放出され、オピオイド作動薬の効果を妨
害することによって乱用を防止することができる。
【0014】
本発明の特定の実施形態の目的は、剤型が改変された際に作動薬のすべてが即時放出の
ために遊離されないような放出制御製剤内にオピオイド作動薬の有効量を含む経口剤型を
提供することである。
【0015】
本発明の特定の実施形態の目的は、作動薬粒子と拮抗薬粒子が、たとえば、外観、質感
、臭い、味、硬度、形状、サイズおよび/またはその組み合わせにおいて類似するか、ま
たはこれら特質の1つ以上によって互いを実質的に識別できない、オピオイド作動薬粒子
及び隔離した拮抗薬の粒子を含む経口剤型を提供することである。
【0016】
本発明の特定の実施形態の目的は、隔離された、たとえば、形剤の用量が無傷で投与さ
れると生物によって利用可能でないが、剤型が改変される (たとえば、オピオイド作動薬
の用量の誤用が試みられる) と生物によって利用可能な、オピオイド拮抗薬を剤型内に含
むことによって、オピオイド作動薬を含有する経口剤型の乱用を防止するための方法を提
供することである。
【0017】
本発明の特定の実施形態の目的は、経口、非経口、鼻腔内あるいは舌下経路による誤用
の可能性を少なくしたオピオイド作動薬の経口剤型を投与することを含有するヒト患者に
おける疼痛治療の方法を提供することである。
【0018】
上記目的は、数ある中で、複数の粒子、たとえば押出成形した粒子、マトリックス内に
分散したオピオイド拮抗薬を含有する粒子のそれぞれ; 及び剤型内のオピオイド拮抗薬を
環境流体に晒すと、マトリックス及び層が剤型内のオピオイド拮抗薬を隔離する働きをす
る (すなわち、放出または実質的な放出を防止する) 粒子それぞれの周囲に配置した層を
含有する剤型を部分的にその狙いとする本発明によって達成する。
【0019】
特定の実施形態では、マトリックスは疎水性物質を含有する。特定の他の実施形態では
、層は疎水性物質を含有する。
【0020】
特定の実施形態では、本発明は下記を含有する経口剤型医薬品をその狙いとする。a)
粒子、たとえば、第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した
粒子; 及び b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水
性物質が、たとえば、粒子の重量の約 2% 〜約 30%を含有する層。あるいは、第2疎水性
物質が粒子の重量の約 5% 〜約 25%、約 10% 〜約 20%、約 10% 〜約 25%、約 15% 〜約
25%、約 22% 〜約 28%、または約 5% 〜約 15%を含有する。
【0021】
特定の実施形態では、本発明は下記を含有する経口剤型医薬品をその狙いとする。 a)
複数の粒子、たとえば、第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成
形した粒子、及び粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、
該第2疎水性物質が、たとえば、約 2% 〜約 30% 粒子重量を含有する層; b) 第3疎水性
物質内に分散したオピオイド作動薬を含有する複数の粒子; 及び c) 複数個のオピオイド
作動薬粒子及び複数個のオピオイド受容体拮抗薬の粒子を含むカプセル。あるいは、第2
疎水性物質が粒子の重量の約 5% 〜約 25%、約 10% 〜約 20%、約 10% 〜約 25%、約 15%
〜約 25%、約 22% 〜約 28%、または約 5% 〜約 15%を含有する。
【0022】
特定の実施形態では、本発明では、第1マトリックス及びオピオイド作動薬を含有する
複数の粒子を含有する剤型; 及び第2マトリックス及びオピオイド拮抗薬を含有する複数
の粒子 (たとえば、押出成形した粒子)、及びオピオイド拮抗薬を環境流体に晒すと、第
2マトリックス及び層がオピオイド拮抗薬を隔離する働きをするオピオイド拮抗薬を含有
する粒子それぞれの周囲に配置した層をさらにその狙いとする。
【0023】
特定の実施形態では、オピオイド拮抗薬の粒子のマトリックスは疎水性物質を含有する
。特定の他の実施形態では、オピオイド拮抗薬の粒子上の層は疎水性物質を含有する。特
定の実施形態では、マトリックス及び層の両方が疎水性物質を含有する。
【0024】
特定の実施形態では、層を、オピオイド作動薬粒子の外観をオピオイド拮抗薬に類似さ
せるか、またはオピオイド拮抗薬と実質的に識別できないようにするための粒子を含み、
それによって乱用者が粒子を含む作動薬から粒子を含む拮抗薬を物理的に分離する能力を
低下させる粒子を含むオピオイド作動薬の周囲に配置する。作動薬層は、放出制御を提供
するか、または放出制御を増強するために機能層であり得る。あるいは、作動薬層は、た
とえば、放出制御能力を提供しないフィルムコートのような非機能層でもあり得る。
【0025】
特定の実施形態では、本発明は下記を含有する経口剤型をその狙いとする。
(i) 放出可能型の複数の粒子を含有するオピオイド作動薬及び (ii) 複数の粒子、たとえ
ば、疎水性物質を含有するマトリックスを含有する押出成形した粒子、マトリックス内に
分散したオピオイド拮抗薬、及び剤型が無傷で患者に投与されるときにマトリックス及び
層が拮抗薬の放出を防止または実質的に防止するような粒子の周囲に配置した疎水性物質
を含有する層。
【0026】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の 人工胃液 (SGF) 中で 1 時間の剤型の溶出に基づいた、改変後に剤型から放出さ
れる拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率は約 20:1 以上; 約 50:
1 以上; 約 100:1 以上; 約 150:1 以上; または約 1000:1 以上である。
【0027】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の 人工腸液 (SIF ) に切り替えた条件で、
剤型の 2 時間、4 時間、12 時間、24 時間あるいは 36 時間での溶出に基づいた、改変
後に剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率は約
20:1 以上; 約 50:1 以上; 約 100:1 以上; 約 150:1 以上; または約 1000:1 以上であ
る。
【0028】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で剤型の 1 時間での溶出に基づいた無傷剤型から放出される拮抗薬の重
量パーセントは 1.0% 重量未満; 0.5% 重量未満; 0.2% 重量未満; または0.1 重量% 未満
である。
【0029】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 2 時間
での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 2.0% 未満; 1.0%
未満; 0.5% 未満; または0.25% 未満である。
【0030】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 4 時間
での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 2.2% 未満; 1.5%
未満; 1.0% 未満; または 0.75% 未満である。
【0031】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 12 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 3 .0% 未満; 1
.8% 未満; 1.25%; または0.3% 未満である。
【0032】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 24 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 4.8% 未満; 2.
5% 未満; 1.8% 未満; または0.4% 未満である。
【0033】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 36 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 未満7.0% 未満
; 6.5% 未満; 3.0%; または未満1.5%.
【0034】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、1.0% 以下の拮抗薬を 1 時間で、2.0% 以下の拮抗薬を 2 時間で、
2.2% 以下の拮抗薬を 4 時間で、3.0% 以下の拮抗薬を 12 時間で、4.8% 以下の拮抗薬を
24 時間で、及び 7.0% 以下の拮抗薬を 36 時間でそれぞれ放出する。
【0035】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、0.5% 以下の拮抗薬を 1 時間で、1.0% 以下の拮抗薬を 2 時間で、
1.5% 以下の拮抗薬を 4 時間で、1.8% 以下の拮抗薬を 12 時間で、2.5% 以下の拮抗薬 2
4 時間で及び 6.5% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0036】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は 0.2% 以下の拮抗薬を 1 時間で、0.5% 以下の拮抗薬を 2 時間で、1
.0% 以下の拮抗薬を 4 時間で、1.25% 以下の拮抗薬を 12 時間で、1.8% 以下の拮抗薬を
24 時間で、及び 3.0% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0037】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、0.1% 以下の拮抗薬を 1 時間で、0.25% 以下の拮抗薬を 2 時間で
、0.75% 以下の拮抗薬を 4 時間で、0.3% 以下の拮抗薬を 12 時間で、0.4% 以下の拮抗
薬を 24 時間で、及び 1.5% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0038】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で剤型の 1 時間での溶出に基づいた、改変後剤型から放出される作動薬
の重量百分率は、50% 未満、40% 未満、または 35% 未満である。
【0039】
本発明の特定の実施形態では、改変剤型(tampered dosage form)を患者集団に単回投
与した後に得られる拮抗薬の平均 Cmax の無傷剤型を患者集団に単回投与した後に得られ
る拮抗薬の平均 Cmax に対する比率は、約 20:1 以上; 約 50:1 以上; 約 75:1 以上; 約
100:1 以上; 約 125:1 以上; 約 150:1 以上; または約 1000:1 以上である。これらの
値は好ましくは絶食状態で得た値である。
【0040】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 Cmax の無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対
する比率は約 20:1 〜約 1000:1; 約 20:1 〜約 150:1; 約 20:1 〜約 125:1; 約 20:1
〜約 100:1; 約 20:1 〜約 75:1; または約 20:1 〜約 50:1 である。他の実施形態では
、その範囲は約 50:1 〜約 1000:1; 約 75:1 〜約 1000:1; 約 100:1 〜約 1000:1; 約 1
25:1 〜約 1000:1; または約 150:1 〜約 1000:1 である。これらの値は好ましくは絶食
状態で得た値である。
【0041】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC に対
する比率は、約 5:1 以上; 約 25:1 以上; 約 75:1 以上; 約 100:1 以上; 約 150:1 以
上; 約 200:1 以上または約 250:1 以上である。これらの値は好ましくは絶食状態で得た
値である。
【0042】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC に対
する比率は約 5:1 〜約 250:1; 約 5:1 〜約 200:1; 約 5:1 〜約 150:1; 約 5:1to 約 1
00:1;約 5:1 〜約 75:1;または約 5:1to 約 25:1 である。他の実施形態では、その範囲
は約 25:1 〜約 250:1; 約 75:1 〜約 250:1; 約 100:1 〜約 250:1; 約 150: I 〜約 10
00:1; または約 200:1 〜約 250:1 である。これらの値は好ましくは絶食状態で得た値で
ある。
【0043】
特定の実施形態では、本発明は、剤型が改変の対象となり、経口、鼻腔内、非経口ある
いは舌下に投与される場合、オピオイド作動薬の効果がオピオイド受容体拮抗薬の放出に
よって大幅または完全に遮断される本明細書で開示した剤型を利用して、オピオイド作動
薬の乱用を防止する方法をさらにその狙いとする。
【0044】
特定の実施形態では、本発明は、必要とする患者に対する鎮痛薬を含有する、本明細書
で開示した本発明の実施形態の任意の医療製品を、たとえば経口で投与することによる疼
痛治療の方法をさらにその狙いとする。
【0045】
オピオイド作動薬を含有する複数粒子と、オピオイド拮抗薬を含有する複数粒子が類似
しているか、または実質的に互いを識別できない実施形態では、粒子の類似性または実質
的な識別不可能性は、(i) 機能層または非機能層、(ii) 層化する必要のない類似の調製
方法、(iii) 類似、または実質的に識別できない最終製品を結果としてもたら異なる調製
方法、(iv) 異なる最終製品を結果としてもたらし、次いで追加処理工程 (たとえば層化)
の対象となり、類似性または実質的な識別不可能性を提供する異なる調製方法、(v) ま
たは所望の特質 (たとえば、外観、質感、臭い、味、硬度、形状、サイズなど) を結果と
してもたらす任意の他方法のためであり得る。
【0046】
特定の好ましい実施形態では、粒子の平均直径は約 0.1 〜約 12 mm; 約 0.1 〜約 2.5
mm; 約 0.2 〜約 6 mm; 約 0.5 〜約 3 mm; 約 0.5 mm 〜約 2 mm; または約 1 mm 〜約
2 mm である。
【0047】
無傷剤型の投与直後に放出されるオピオイド拮抗薬の量は、存在する場合、剤型が引き
続き鎮痛効果を持ち続けられるような量である。
【0048】
本発明の特定の実施形態では、オピオイド作動薬の隔離したオピオイド拮抗薬に対する
比率は重量で約 1:1 〜約 50:1; 好ましくは重量で約 1: I 〜約 20:1; または重量で約
15:1 〜約 30:1 である。オピオイド作動薬の、活性成分の重量を指すオピオイド拮抗薬
に対する重量比。たとえば、オピオイド拮抗薬の重量には、オピオイド拮抗薬を一緒にな
って隔離する働きをする層及びマトリックスの重量が除かれている。特定の好ましい実施
形態では、作動薬の隔離した拮抗薬に対する重量比は、重量で約 1:1 〜約 10:1 である
。
【0049】
オピオイド拮抗薬の実質的に放出不可能な形態と組み合わせたオピオイド作動薬を含む
本発明の経口剤型には、限定するものではないが、錠剤及びカプセルが含まれる。本発明
の経口剤型には、当業者であれば公知の任意の所望の医薬品賦形剤を含むことができる。
経口剤型は、オピオイド作動薬の即放性および/またはオピオイド作動薬の放出制御を提
供することができる。
【0050】
本発明の乱用防止剤型は、オピオイド作動薬の一回分量を含有し、長時間にわたる放出
を意図した放出制御剤型と関連して有用である。薬物乱用者は、そのような放出制御型の
製品を利用し、粉砕、細砕、抽出あるいは別の方法で、剤型の全含有量を放出させ即時吸
収する目的で製品を損傷する。本発明に係る剤型を改変すると、結果としてオピオイド拮
抗薬も同時に吸収に提供されるようになるので、本発明はそのような乱用を妨げる手段を
提供する。
【0051】
また、本発明は、本明細書で開示した剤型を用いた疼痛治療の方法もその狙いとする。
本方法には、開放可能な形態でオピオイド作動薬及び本明細書で開示したとおりの隔離し
た拮抗薬を含む経口剤型を提供すること、及び無傷経口剤型をそのような治療が必要な哺
乳動物 (たとえば、ヒト) に経口で投与することを含有することができる。
【0052】
特定の実施形態では、本発明は、本明細書で開示した経口剤型の調製方法をさらにその
狙いとする。特定の実施形態では、本発明は、押出成形により疎水性物質を含有するマト
リックス内に分散したオピオイド拮抗薬を含有する複数の粒子を調製すること; 及び剤型
が無傷で投与されるときにマトリックス及び層が拮抗薬を隔離する働きをする疎水性物質
を含有する層を押出成形した粒子の周辺に配置することを含有する経口剤型の調製方法を
含有する。本方法には、隔離した拮抗薬を隔離した拮抗薬の完全性を維持するような方法
でオピオイド作動薬の放出可能形態 (たとえば放出制御) と組み合わせることをさらに含
有することができる。本発明のすべての実施形態では、マトリックスの疎水性物質は層の
疎水性物質と同じであっても同じでなくともよい。
【0053】
本発明の好ましい実施形態では完全にオピオイド拮抗薬の放出を防止する形態でオピオ
イド拮抗薬を含有するが、本発明は拮抗薬も実質的に放出不可能な形態で含まれる。用語
「実質的に放出されない」及び「実質的に放出不可能」は、剤型が意図されたように経口
でヒト投与されるとき、放出量が鎮痛効力に影響を及ぼさない、または実質的に影響を及
ぼさない限りにおいて、少量で放出され得る拮抗薬を参照する。
【0054】
隔離された拮抗薬の粒子では、本発明に従っていくつかの可能性が存在する。第1の可
能性は、マトリックスには層なしで拮抗薬を隔離する能力がいくらかあり、層がその隔離
機能を高めること。第2の可能性は、層にはマトリックスなしで拮抗薬を隔離する能力が
いくらかあり、マトリックスがその隔離機能を高めること。第3の可能性は、マトリック
スには層なしで拮抗薬を隔離する能力がなく、層にはマトリックスなしで拮抗薬を隔離す
る能力がなく、マトリックスと層共には拮抗薬を隔離する能力を持つ(たとえば、マトリ
ックス及び層のそれぞれには、拮抗薬の放出制御を個々に提供する能力があるが、同一剤
型内のマトリックスと層共は拮抗薬を隔離する) ことである。第1及び第2の可能性では
、マトリックスおよび/または層は、拮抗薬の放出制御を個々に提供する能力を持つこと
によって隔離機能を高める。
【0055】
本発明の特定の好ましい実施形態では、拮抗薬の実質的に放出不可能な形態は、遅延性
の結腸通過を管理するために用いられる緩下薬 (たとえば鉱物油) に対して耐性を示し、
そして胃酸欠乏状態に対しても耐性を示す。
【0056】
本発明の好ましい実施形態では、オピオイド拮抗薬の実質的に放出不可能な形態は、経
口剤型の機械的、熱的あるいは化学的改変、たとえば、粉砕、せん断、細砕、咀嚼あるい
は加熱 (たとえば約 45℃ を超える) と組み合わせた溶剤溶出による改変に対して無防備
である。改変がなされると、オピオイド拮抗薬の実質的に放出不可能な形態の完全性が損
なわれ、オピオイド拮抗薬が即時放出に供され、それによって少なくとも部分的に及び好
ましくは実質的にオピオイド作動薬の効果が遮断される。このように、オピオイド作動薬
及びオピオイド拮抗薬を含有する経口剤型が、咀嚼、粉砕、細砕または溶剤溶解及び加熱
され、及び経口、鼻腔内、非経口あるいは舌下に投与されると、オピオイドの鎮痛あるい
は陶酔効果は低下または除去される。
【0057】
本発明は、経口剤型内のオピオイド作動薬の乱用の可能性を少なくする方法をさらにそ
の狙いとする。 本方法は、本明細書に記載したように経口剤型内のオピオイド作動薬を
提供することを含有する。
【0058】
用語「鎮痛有効性」は、本発明では、ヒト患者による決定を基にした副作用の許容レベ
ルと共に、疼痛の満足できる軽減または除去として定義する。
【0059】
句「オピオイド作動薬の鎮痛効果を実質的に遮断していない」は、本発明の目的上、鎮
痛を提供する上で治療上の効果が弱い剤型を与えること応じて、オピオイド拮抗薬が十分
な度合いでオピオイド作動薬の効果を遮断しないことを意味する。
【0060】
用語「改変」は、無傷剤型の特質を変える機械的、急速な放出または即時放出のために
少なくともオピオイド作動薬の部分を遊離するため、またはオピオイド作動薬を不正投与
に利用できるようにするための (たとえば非経口的な投与)、熱的あるいは化学的手段に
よる任意の操作を意味する。無傷剤型を改変する方法は、たとえば、粉砕、せん断、細砕
、咀嚼、溶剤溶出、加熱 (たとえば約 45℃ を超える温度で)、またはこの目的を達成す
るそれらの任意の組み合わせであり得る。
【0061】
特定の実施形態では、剤型の改変を乳鉢と乳棒を用いて粉末に粉砕して行う。他の実施
形態では、改変はをクリューキャップ付き錠剤粉砕器によるか、または2つのステンレス
製大さじを用いることによって行う。
【0062】
特定の実施形態では、乳鉢と乳棒を利用して粉砕を実施し、咀嚼をシミュレートする。
たとえば、乳棒の 3 ストロークは軽度咀嚼をシミュレートし、乳棒のストロークは中度
咀嚼を シミュレートし、及び乳棒のストロークは十分な咀嚼をシミュレートする。特定
の実施形態では、乳鉢と乳棒を用いて、たとえば、乳棒の 24、50、500 または 600 スト
ロークで剤型を粉砕して粉末する。
【0063】
特定の実施形態では、スクリューキャップ付き錠剤粉砕器を利用し、剤型を粉砕器に配
置し、スクリューキャップを回転して剤型を粉砕する。次いで、キャップをゆるめ、粉砕
器を硬面上で軽くたたき、粉砕をさらに 2 回繰り返す。
【0064】
特定の実施形態では、ステンレス製大さじを利用し、剤型を第1スプーンに載置し、第
2スプーンを第1スプーン上に配置し、手圧力を用いて剤型をスプーン間で粉砕する。
【0065】
用語「層は実質的に拮抗薬を欠いている」は、原則的として層にはオピオイド拮抗薬が
含まれていないが、押出成形した成分から移動する少量を含む場合があることを意味する
。
【0066】
用語「少なくとも部分的にオピオイド効果を遮断する」は、本発明の目的上、オピオイ
ド拮抗薬は少なくとも実質的にオピオイド作動薬の陶酔効果を遮断することを意味する。
【0067】
用語「放出制御」は、オピオイド作動薬に適用する場合、本発明の目的上、通常 (すな
わち即放性) 製剤の単回投与に比べてより長い作用期間を提供する放出速度で薬物が製剤
から放出されることを意味する。たとえば、典型的な即放性経口製剤は薬物を、たとえば
、1 時間間隔にわたって放出するが、放出制御型の経口製剤は薬物を、たとえば、4 〜 2
4 時間間隔にわたって放出する。
【0068】
本発明の目的上、用語「オピオイド作動薬」は用語「オピオイド」または「オピオイド
鎮痛薬」と互換可能であるものとし、1つの作動薬または1つ以上のオピオイド作動薬の
組み合わせを含み、さらに、オピオイド塩基、混合した作動薬と拮抗薬、部分的作動薬、
その薬剤的に許容できる塩類、その立体異性体、そのエーテル、そのエステル、及び前述
した物質のいずれかからなる混合物の使用が含まれる
【0069】
本発明の目的上、用語「オピオイド拮抗薬」には、1つの拮抗薬及び1つ以上の拮抗薬
の組み合わせを含めるものとし、さらに、塩基、その薬剤的に許容できる塩類、その立体
異性体、そのエーテル、そのエステル、及び前述した物質のいずれかからなる混合物の使
用も含まれる。
【0070】
本発明の明細書では、開示したオピオイド作動薬及び拮抗薬のすべての薬剤的に許容で
きる塩類の使用も包含することを意図する。薬剤的に許容できる塩類には、限定するもの
ではないが、ナトリウム塩類、カリウム塩類、セシウム (secium) 塩類などのような金属
塩類; カルシウム塩類、マグネシウム塩類などのようなアルカリ土類金属; トリエチルア
ミン塩類、ピリジン塩類、ピコリン塩類、エタノールアミン塩類、トリエタノールアミン
塩類、ジシクロヘキシルアミン塩類、N,N'-ジベンジルエチレンジアミン塩類などのよう
な有機アミン塩類; 塩酸塩、ヒドロブロミド、硫酸塩、リン酸塩などのような無機酸塩類
; ホルマート、アセテート、トリフルオロアセテート、マレアート、酒石酸ラートなどの
ような有機酸塩類; メタンスルホナート、ベンゼンスルホナート、p-トルエンスルホナー
トなどのようなスルホナート; アルギネート、アスパルギネート (asparginate)、グルタ
マートなどのようなアミノ酸塩類が含まれる。
【0071】
本発明に従って用いられる一部のオピオイド作動薬及び拮抗薬には、1つ以上の不斉中
心が含まれ、そして鏡像異性体、ジアステレオマー、または他の立体異性体を作り出す。
また、本発明は、そのようなすべての可能形態の使用、さらには、それらのラセミ形態及
び分解形態及びその混合物を包含することも意図する。本明細書に記載した化合物にオレ
フィン二重結合または他の幾何学的な非対称性の中心が含まれるときには、E 及び Z 幾
何異性体の両方を含むことを意図する。また、すべて互変異性体も本発明によって包含す
ることを意図する。
【0072】
用語「層」は、たとえば、コーティングとしても適用できる、粒子の周囲に配置した材
料を意味する(粒子自体及び、たとえば、シールコートのような1つ以上の任意中間層も
含み得る)。マトリックスの層化は、当分野で周知の工程、たとえば、噴霧コーティング
、浸漬またはエンロービングによって実施することができる。
【0073】
用語「の周囲に配置した」は、中間層または該物質と粒子間の層の有無にかかわらず、
粒子の周囲に配置した材料が少なくとも粒子の一部分を覆うことを意味する。特定の実施
形態では、粒子は材料によって完全に覆われる。
【0074】
本明細書中では、用語「立体異性体」は、空間における原子配向のみが異なる個々分子
のすべて異性体に対する一般用語であり、相互の鏡像ではない1つ以上のキラル中心を有
する化合物の鏡像異性体 及び異性体が含まれる (ジアステレオマー)。
【0075】
用語「キラル中心」は、4 つの異なる群が結合されている炭素原子を指す。
【0076】
用語「鏡像異性体」または「鏡像異性」は、その鏡像で重畳不可能であり、従って光学
的に活性である分子を指すものであり、ここでは、鏡像異性体が 偏光の平面を一方向に
回転し、その鏡像が偏光の平面を逆方向に回転するものとする。
【0077】
用語「ラセミ」は等量の鏡像異性体の混合物を指し、光学的に不活性である。
【0078】
用語「分解能」は、分子の2鏡像異性形態のうちの1つの分離または濃度または消耗を
指す。
【0079】
本発明の粒子の周囲に配置した疎水性物質に関する用語「粒子重量の X% 」または「X%
重量増加」は、疎水性物質が層状粒子合計の重量% ではなく、粒子の重量% として測定
されることを意味する。たとえば、100 mg の非層状粒子は、引き続いて層化して10% 重
量増加すると、層内に 10 mg の疎水性物質を持つ。
【0080】
用語「直径」は、粒子の断面直径を意味し、押出成形工程で用いるオリフィス直径によ
って大きく左右される。
【0081】
用語「長さ」は、押出成形した粒子の長さを意味し、押出成形したストランドの切断間
隔によって大きく左右される。
【0082】
用語「医薬製品」は、投与に好適な剤型または剤型の成分を意味する。
【発明を実施するための最良の形態】
【0083】
本発明は、隔離したオピオイド拮抗薬の粒子が、環境流体に晒されると拮抗薬が無傷形
態から「漏出」することをさらに低下する被覆剤を用いて、押出成形したオピオイド拮抗
薬の粒子にコーティングを施すことによって改善できるという観察に基づくものである。
本発明の効力によって、隔離した拮抗薬がオピオイド作動薬と結合すると、好ましくは無
視できるほどに僅かな量の拮抗薬 (すなわち、作動薬によって提供される鎮痛に影響を及
ぼさない量) が所定の使用条件下で放出される。所定の使用条件下で放出される拮抗薬の
量は、皆無であるか、または測定できないほどに少ないことが最も好ましい。
【0084】
特定の実施形態では、本発明には、剤型が改変されていない場合、オピオイド作動薬の
効果を少なくとも実質的に遮断するための量でオピオイド拮抗薬を含有する、複数の押出
成形し、隔離した粒子と共に、経口で治療学的に効率的な量のオピオイド作動薬を含有す
る複数の粒子を含有する経口剤型が含まれる。好ましくは、オピオイド拮抗薬を含有する
複数個の薬剤的に許容できる粒子、及びオピオイド作動薬を含有する複数個の薬剤的に許
容できる粒子は視覚的に類似し、最も好ましくは、それらが視覚的に識別できないものと
する。
【0085】
特定の実施形態では、オピオイド作動薬のオピオイド拮抗薬に対する比率を、経口剤型
が改変されてオピオイド拮抗薬を含有する粒子の完全性が損なわれた場合、放出される拮
抗薬の量が、被検体に経口、非経口、鼻腔内あるいは舌下に投与されたとき、オピオイド
作動薬の陶酔効果を実質的に低下または除去するように設定する。
【0086】
たとえば、本発明の特定の好ましい実施形態では、剤型が非経口的あるいは舌下に誤用
されると、オピオイド拮抗薬によってオピオイド作動薬の陶酔効果は実質的に低下または
除去される。特定の実施形態では、剤型が咀嚼、粉砕、または溶剤溶解及び加熱され、経
口、鼻腔内、非経口あるいは舌下に投与されるとき、オピオイドの鎮痛または陶酔効果は
オピオイド拮抗薬の放出に起因して実質的に低下または除去される。特定の実施形態では
、オピオイド薬物の効果はオピオイド拮抗薬によって少なくとも部分的に遮断される。特
定の他の実施形態では、オピオイド薬物の効果はオピオイド拮抗薬によって実質的に遮断
される。特定の他の実施形態では、オピオイド薬物の効果はオピオイド拮抗薬によって完
全に遮断される。
【0087】
本発明の無傷経口剤型は、意図されたように正しく投与されるとオピオイド拮抗薬を実
質的に放出しないので、経口投与時にオピオイド拮抗薬が GI システムに放出されるよう
に提供される場合、拮抗薬量の変動の範囲はより大きくなる。
【0088】
マトリックス及び層が拮抗薬を実質的に放出不可能にする隔離形態でのオピオイド拮抗
薬は、層が該粒子のそれぞれの周囲に配置された状態で、マトリックス内に分散したオピ
オイド拮抗薬を含有する複数の押出成形した粒子を含有する。一実施形態では、層は薬剤
的に許容できる疎水性物質を含有する。別の実施形態では、マトリックスは薬剤的に許容
できる疎水性物質を含有する。別の実施形態では、マトリックス及び層の両方が薬剤的に
許容できる疎水性物質を含有する。マトリックスの疎水性物質は層の疎水性物質と同じで
あってもよいし、異なるものであってもよい。疎水性物質は、好ましくは、拮抗薬が被覆
されたマトリックスから放出されないか、または実質的に放出されないような量であって
、したがって、GI システムを介した経口剤型の転移中に吸収されるように使用できない
か、または実質的に使用することができない。
【0089】
本発明の特定の好ましい実施形態では、オピオイド拮抗薬を溶解押出成形によってマト
リックス内に分散する。マトリックスが1つまたはそれ以上の薬剤的に許容できる疎水性
物質を含有する。
【0090】
本発明の特定の実施形態では、オピオイド作動薬を含む粒子は放出制御型の押出成形マ
トリックスの複数微粒子である。特定の実施形態では、放出制御型押出成形マトリックス
の複数微粒子がオピオイド作動薬を即時放出に供するように改変されると、作動薬の一部
分のみが即時放出のために遊離されることが見出されている。特定の実施形態では、50 r
pm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で押出成形された剤型
の 1 時間での溶出に基づいた、改変後剤型から放出される作動薬の重量百分率は 50% 未
満、40% 未満、または 35% 未満である。
【0091】
改変直後にオピオイド拮抗薬の一部分のみが即時放出のためにマトリックスの複数微粒
子から遊離させることができるので、改変直後に本発明の所定の目的に必要な量の拮抗薬
が遊離されることを確実にするため、拮抗薬を高添加することができる。たとえば、本発
明の実施形態で改変直後に拮抗薬の 50% が遊離される場合、もし改変直後に 2 mg の拮
抗薬放出を遊離させる必要があれば、剤型は 4 mg の拮抗薬を添加して製剤することがで
きる。本発明の経口剤型では、無傷剤型の投与直後に拮抗薬が放出または実質的に放出さ
れることがないので、拮抗薬の高添加によっても作動薬の鎮痛効力を妨げる量の拮抗薬が
無傷剤型から放出されるようなことは生じない。
【0092】
本発明の押出成形マトリックスに使用する材料には、たとえば、限定するものではない
が、ゴム、セルロースエーテル、アクリル樹脂、タンパク質由来の材料のような親水性あ
るいは疎水性物質; 脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、無機質及び
野菜油類及びワックス (天然の及び合成)、及びステアリルアルコールのような消化性の
、長鎖 (C8-C50、特にC12-C40)の、置換または非置換炭化水素; 及びポリアルキレングリ
コールが含まれる。マトリックスには、1% 〜 80% (重量で) の少なくとも1つの親水性
または好ましくは少なくとも1つの疎水性物質を含むことができる。
【0093】
押出成形マトリックスが疎水性物質を含有する場合、疎水性物質はこの目的に役立つ任
意の疎水性物質であってもよいが、好ましくはアルキルセルロース、アクリル及びメタク
リル酸重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、またはその
混合物からなる群より選択されるものとする。本発明の特定の好ましい実施形態では、疎
水性物質は薬剤的に許容できるアクリル重合体であって、アクリル酸及びメタクリル酸共
重合体、メチル メタクリラート、メチル メタクリラート共重合体、エトキシエチル (et
hoxyethyl) メタクリラート、シアノエチル メタクリラート、アミノアルキル メタクリ
ラート共重合体、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミン
共重合体、ポリ(メチル メタクリラート)、ポリ(メタクリル酸)(水素化物)、ポリメタク
リラート、ポリアクリルアミド、ポリ(メタクリル酸 水素化物)、及びグリシジル メタク
リラート共重合体のいずれかが含まれるが、これらに限定されるものではない。
【0094】
本発明で有用なアクリル重合体には、限定するものではないが、使用したアクリル及び
メタクリルモノマーのモル毎に約 0.02 〜 0.03 モルのトリ (低級アルキル) アンモニウ
ム基を含むアクリル及びメタクリル酸エステル (たとえば、アクリル酸の低級アルキルエ
ステル及びメタクリル酸の低級アルキルエステルの共重合体) から合成した共重合体を含
有するアクリル樹脂が含まれる。好適なアクリル樹脂の例は、ローム製薬(Rohm Pharma
)有限責任会社によって製造された重合体であり、商標 Eudragit(登録商標)RS で販売
されている。Eudragit(登録商標)RS30D は好ましいものである。Eudragit(登録商標)
RS は、エチル アクリラート(EA)、メチル メタクリラート (MM) 及びトリエチルアンモ
ニウムエチル メタクリラート塩化物 (TAM) の水不溶性共重合体であって、他成分(EA 及
びMM) に対する TAM のモル比は 1:40 である。Eudragit(登録商標)RS のようなアクリ
ル樹脂は水溶性の懸濁液の形で使用することが可能である。
【0095】
他の実施形態では、疎水性物質はハイドロキシプロピルメチルセルロースのような1つ
またはそれ以上のヒドロキシアルキルセルロースのような材料より選択される。
【0096】
特定の実施形態では、本発明で有用な疎水性物質は約 30℃ 〜約 200℃、または約 45
℃ 〜約 90℃の融点を持つ。
【0097】
特定の実施形態では、疎水性物質は、限定はされないが、脂肪酸エステル、脂肪酸グリ
セリド (モノ-、ジ-、及びトリ-グリセリド)、硬化脂肪、炭化水素、正常ワックス、ステ
アリン酸、ステアリルアルコール及び炭化水素主鎖を有する疎水性と親水性の材料を含む
天然または合成ワックス、脂肪アルコール(例: ラウリル、ミリスチル、ステアリル、セ
チルまたはセトステアリルアルコール)、脂肪酸を含有する。好適なワックスには、たと
えば、蜜ロウ、糖ロウ、キャスターロウ及びカルナウバワックスが含まれる。本発明の目
的上、ワックス様物質は、通常は室温で固体であり、約 30 〜約 100 ℃の融点を持つ任
意の材料として定義する。
【0098】
特定の実施形態では、疎水性物質はエチルセルロース、セルロースアセテート、セルロ
ースプロピオナート(低、中または高の分子量)、セルロースアセテートプロピオナート、
セルロースアセテートブチラート、セルロースアセテートフタラート及びセルローストリ
アセテートからなる群より選択されるセルロースポリマーを含有する。エチルセルロース
の例としては、44 〜 55% のエトキシル基含有量を持つものが挙げられる。エチルセルロ
ースはアルコール溶液の形で使用することが可能である。特定の他の実施形態では、疎水
性物質はポリ乳酸、ポリグリコール酸またはポリ乳酸及びポリグリコール酸の共重合体を
含有する。
【0099】
特定の実施形態では、疎水性物質はセルロースエーテル、セルロースエステル、セルロ
ースエステルエーテル、及びセルロースからなる群より選択されるセルロースポリマーを
含有する。特定の実施形態では、セルロースポリマーはアンヒドログルコース ユニット
に対してゼロから最大で 3 までの置換度 (D.S.) を持つ。置換度は、セルロースポリマ
ーを含有するアンヒドログルコース ユニット上に存在している水酸基の平均数が置換群
によって交換されることを意味する。代表的な材料には、セルロースアシレート、セルロ
ースジアシレート、セルローストリアシレート (triacylate)、セルロースアセテート、
セルロースジアセテート、セルローストリアセテート、モノ/ジ/トリセルロース アルカ
ニレート (alkanylate)、モノ/ジ/トリセルロース アロイレート (aroylates)、及びモノ
/ジ/トリセルロースアルケニレート (alkenylate) からなる群より選択される重合体が含
まれる。例示的重合体には、最大で 1 までの D.S. 及び最大で 21% までのアセチル含有
量を有するセルロースアセテート; 最大で 32 〜 39.8% までのアセチル含有量を有する
セルロースアセテート; 1 〜 2 の D.S. 及び 21 〜 35% のアセチル含有量を有するセ
ルロースアセテート; 及び 2 〜 3 の D.S. 及び 35 〜 44.8% のアセチル含有量を有す
るセルロースアセテートが含まれる。
【0100】
特定のセルロースポリマーには、1.8 の D.S.、39.2 〜 45% のプロピル含有量及び 2.
8 〜 5.4% の水酸含有量を有するセルロースプロピオナート; 1.8 の D.S.、13 〜 15%
のアセチル含有量及び 34 〜 39% のブチリル含有量を有するセルロースアセテートブチ
ラート; 2 〜 29% のアセチル含有量、17 〜 53% のブチリル含有量及び 0.5 〜 4.7%
の水酸含有量を有するセルロースアセテートブチラート; 2.9 〜 3 の D.S. を有するセ
ルローストリアセテート、セルロース トリバレラート (tivalerate)、セルローストリラ
ウラート、セルローストリパルミチン酸、セルローストリスクシナート、及びセルロース
トリオクタノアートのようなセルローストリアクレート (triacylate); 2.2 〜 2.6 の
D.S. を有し、セルロースジスクシナート、セルロースジパルミチン酸、セルロースジオ
クタノアート、セルロースジペンタノアートのようなセルロースジアシレート、及びセル
ロースアセテートブチラート、セルロースアセテートオクタノアートブチラート及びセル
ロースアセテートプロピオナートのようなセルロースコエステル (coester)が含まれる。
【0101】
追加セルロースポリマーには、アセトアルデヒドジメチル セルロースアセテート、セ
ルロースアセテートエチルカルバマート、セルロースアセテートメチルカルバマート、及
びセルロースアセテートジメチルアミノセルロースアセテートが含まれる。
【0102】
特定の実施形態では、薬剤的に許容できる疎水性物質には、乳酸及びグリコール酸 (「
PLGA」) の共重合体を含有する生分解性重合体、ポリ乳酸、ポリグリコリド、ポリ無水物
、ポリオルトエステル、ポリカプロラクトン、ポリホスファゼン、多糖類、タンパク質重
合体、ポリエステル、ポリジオキサノン、ポリグルコナート、ポリ乳酸-酸-ポリエチレン
酸化物共重合体、ポリ(ヒドロキシブチラート)、ポリホスホエステル (polyphosphoesthe
r) または前述の任意物質の混合物またはブレンド物が含まれる。
【0103】
特定の実施形態では、PLGA を含有する生分解性重合体は約 2,000 〜約 500,000 ドル
トンの分子量を有する。乳酸のグリコール酸に対する比率は約 100:0 〜約 25:75 である
が、乳酸のグリコール酸に対する比率が約 65:35 であることが好ましい。
【0104】
PLGA は米国特許特許番号第 4,293,539 号 (Ludwig らによる) に示された工程によっ
て調製することが可能であり、その開示を引用することを以って本明細書の一部となす。
手短に言えば、Ludwig は、容易に取り外し可能な重合触媒 (たとえば、Dowex HCR-W2-H
のような強力な酸イオン交換樹脂)の存在下で乳酸酸及びグリコール酸を縮合することに
よって共重合体を調製する。触媒の量は重合にとって重要ではないが、一般には、乳酸と
グリコール酸を組み合わせた総重量に比べて約 0.01 〜約 20 重量部 である。重合反応
は、溶剤を用いずに、約 100℃ 〜約 250℃ の温度で約 48 〜約 96 時間、水分及び副産
物の除去を容易にするために好ましくは低圧下で実行することができる。次いで、PLGA
は、ジクロロメタンまたはアセトンのような有機溶剤中で溶融した反応混合物をろ過する
ことによって回収し、次いでろ過により触媒を除去する。
【0105】
特定の好ましい実施形態では、2つまたはそれ以上の疎水性物質の組み合わせを押出成
形マトリックス内に含む。2つまたはそれ以上の疎水性物質を含む場合、少なくとも1つ
の疎水性物質を、好ましくは天然及び合成ワックス、脂肪酸、脂肪アルコール、及び同物
質の混合物から選択する。その例としては、限定するものではないが、蜜ロウ、カルナウ
バワックス、ステアリン酸及びステアリルアルコールが挙げられる。
【0106】
疎水性物質が炭化水素であるとき、炭化水素は好ましくは 25℃ 〜 90℃ 間の融点を持
つ。長鎖炭化水素材料中、脂肪(脂肪族) アルコールが好ましい。本マトリックスには、
最大で 60% (重量で) までの少なくとも1つの消化性長鎖炭化水素を含むことが可能であ
る。
【0107】
特定の好ましい実施形態では、押出成形マトリックスには最大で 60% (重量で) までの
少なくとも1つのポリアルキレングリコールを含む。
【0108】
1つの好適な押出成形マトリックスは、少なくとも1つの水溶性ヒドロキシアルキルセ
ルロース、少なくとも1つのC12-C36、好ましくは C14-C22、脂肪アルコール及び、任意
選択で、少なくとも1つのポリアルキレングリコールを含有する。ヒドロキシアルキルセ
ルロースは、好ましくはヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセル
ロースまたはヒドロキシエチルセルロースのようなヒドロキシ(C1 〜 C6) アルキルセル
ロースである。経口剤型内のヒドロキシアルキルセルロース量は、特に、所要活性剤の正
確な放出速度によって決定する。脂肪アルコールは、たとえば、ラウリル アルコール、
ミリスチル アルコールまたはステアリル アルコールであってもよい。特に好適な実施形
態では、但し、脂肪アルコールはセチル アルコールまたはセトステアリルアルコールで
ある。経口剤型内の脂肪アルコール量は、特に、所要活性剤の正確な放出速度によって決
定する。これはまた、ポリアルキレングリコールが経口剤型内に存在しているかどうかに
応じても異なる。ポリアルキレングリコールの不在時には、経口剤型は好ましくは脂肪ア
ルコールの約 20% 〜約 50% (重量で) を含む。少なくとも1つのポリアルキレングリコ
ールが経口剤型内に存在するとき、脂肪アルコールとポリアルキレングリコールを組み合
わせた重量は好ましくは全剤型の 20% 〜 50% (重量で) を構成する。
【0109】
一実施形態では、ヒドロキシアルキルセルロースまたはアクリル樹脂の、脂肪アルコー
ル/ ポリアルキレングリコールに対する比率により、活性剤の製剤からの放出速度がかな
りの程度まで決定される。ヒドロキシアルキルセルロースの脂肪アルコール/ポリアルキ
レングリコールに対する比率は 1:2 及び 1:4 が好ましいが、1:3 及び 1:4 の比率は特
に好適である。
【0110】
ポリアルキレングリコールは、たとえば、ポリプロピレングリコールまたはポリエチレ
ングリコールであってよい。ポリアルキレングリコールの平均分子量は、好ましくは約 1
,000 〜約 15,000 であるが、約 1,500 〜約 12,000 が特に好ましい。
【0111】
別の好適な押出成形マトリックスは、アルキルセルロース (特にエチルセルロース)、C
12 〜 C36 脂肪アルコール及び、任意選択で、ポリアルキレングリコールを含有する。
【0112】
別の好適な押出成形マトリックスは、アクリル重合体 (特にEudragit(登録商標)RSPO
)、C12to C36 脂肪アルコール及び、任意選択で、ポリアルキレングリコールを含有する
。
【0113】
特定の好ましい実施形態では、マトリックスには少なくとも2つの薬剤的に許容できる
疎水性材料の組み合わせが含まれる。
【0114】
上記開示したように、オピオイド拮抗薬を含有する薬剤的に許容できるマトリックスを
含有する複数個の押出成形した粒子は、マトリックス材料に加えて、オピオイド拮抗薬を
隔離することができる1つまたはそれ以上の疎水性物質と重層させる。コーティングの疎
水性物質は任意の上述した物質から選択することが可能である。特定の好ましい実施形態
では、疎水性物質はセルロース材料または重合体、アクリル重合体、またはその組み合わ
せである。用語「第1疎水性物質」、「第2疎水性物質」及び「第3疎水性物質」はそれ
ぞれ、少なくとも部分的分散または層状配列 (arraignment) において1つまたはそれ以
上の疎水性物質を包含することを意図する。第1、第2及び第3疎水性物質は同じであっ
ても異なっていてもよい。特定の実施形態では、第1及び第2疎水性物質が同じであり得
; 第1及び第3疎水性物質が同じであり得; 第2及び第3疎水性物質が同じであり得; ま
たは第1、第2及び第3疎水性物質が同じであり得る。
【0115】
実施形態では、層内に1つ以上の疎水性物質がある場合、疎水性物質を相互分散または
部分的に相互分散することができる。あるいは、疎水性物質は層状配列にあってもよい。
たとえば、粒子重量の 25% の量にあたる層は、重量で粒子の15% にあたるエチルセルロ
ース層及び重量で粒子の 10% にあたるエチルセルロース層の周囲に配置したアクリル重
合体層を持つことができる。
【0116】
コーティング組成物は、当分野で周知の任意の好適な噴霧器を用いて噴霧によって複数
個の押出成形した粒子に塗布することができる。たとえば、下部から注入する噴出空気が
被覆材料を流動化し、コーティングを噴霧中に乾燥させる Wurster 流動層システムを使
用してもよい。コーティング厚は、使用する特定のコーティング組成物の特性によって異
なる。
【0117】
本発明の押出成形した粒子を層化するために好適な疎水性物質には、アルキルセルロー
スを始めとするセルロース材料及び重合体が含まれる。好ましいアルキルセルロースポリ
マーの1つとしてエチルセルロースが挙げられるが、他のセルロースあるいはアルキルセ
ルロースポリマーも、本発明に係る疎水性コーティングのすべてまたは一部として単独ま
たは組み合わせて容易に利用することができる。
【0118】
市販されているエチルセルロースの水分散系の1つとして、Aquacoat (登録商標)(FM
C 社、米国ペンシルバニア州フィラデルフィア) が挙げられるが、これは水不混和性有機
溶剤中のエチルセルロースを溶解し、次いで同セルロースを界面活性剤及び安定剤存在下
で水中で乳化することによって作成される。均質化後、サブミクロンの溶滴を作成するに
は、有機溶剤を真空下で蒸発させ、擬似ラテックスを形成する。
【0119】
エチルセルロースの別の水分散系は、Surelease (登録商標)(Colorcon 社、米国ペン
シルバニア州ウェストポイント) として市販されている。この製品は、製造工程中に可塑
剤を分散系内に取り込むことによって作成される。重合体、可塑剤 (ジブチルセバカート
)、及び安定剤 (オレイン酸) の熱溶融を均一な混合物として調製し、次いでアルカリ溶
液で希釈して複数粒子に直接的に塗布できる水分散系を得る。
【0120】
本発明の他の好ましい実施形態では、層の疎水性物質は、アクリル酸及びメタクリル酸
共重合体、メチル メタクリラート共重合体、エトキシエチル (ethoxyethyl) メタクリラ
ート、シアノエチル メタクリラート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタク
リル酸アルキルアミド共重合体、ポリ(メチル メタクリラート)、ポリメタクリラート、
ポリ(メチル メタクリラート) 共重合体、ポリアクリルアミド、アミノアルキルメタクリ
ラート共重合体、ポリ(メタクリル酸酸無水物)、グリシジル メタクリラート共重合体、
及びその組み合わせを含むが、これらに限定されるものではない薬剤的に許容できるアク
リル重合体である。
【0121】
特定の好ましい実施形態では、アクリル重合体は1つまたはそれ以上のアンモニオ メ
タクリラート共重合体からなる。アンモニオ メタクリラート共重合体は当分野で周知の
ものであり、低含有量の四級アンモニウム基を有するアクリル及びメタクリル酸エステル
の完全重合の共重合体として NF XVII に記載されている。
【0122】
特定の好ましい実施形態では、四級アンモニウム基の中性 (メタクリルエステル) アク
リルエステルに対するモル比が異なるような異なる物性を有する2つ以上のアンモニオ
メタクリラート共重合体取り込むことが必要である場合がある。
【0123】
特定の実施形態では、アクリル層は Eudragit(登録商標)RL30D 及び Eudragit(登録
商標)RS30D の商品名で ローム製薬(Rohm Pharma) (ダルムシュタット(Darnastadt)
、ドイツ) から市販されている2つのアクリル樹脂ラッカーの混合物を含有する。Eudrag
it(登録商標)RL30D 及び Eudragit(登録商標)RS30D は低含有量の四級アンモニウム
基を有するアクリル及びメタクリルエステルの共重合体であって、アンモニウム基の残存
中性 (メタクリルエステル) アクリルエステルに対するモル比は Eudragit(登録商標)
RL30D では 1:20 及び Eudragit(登録商標)RS30D では 1:40 である。平均分子量は約
150,000 である。コード記号表示 RL (高透過性) 及び RS (低透過性) はこれら薬剤の透
過性特質を参照する。特定の実施形態では、本発明における Eudragit(登録商標)RS は
、Eudragit(登録商標)RSPM、Eudragit(登録商標)RSPO、Eudragit(登録商標)RS100
、Eudragit(登録商標)RS12.5、及びその混合物からなる群より選択される。用語 「Eud
ragit(登録商標)RSPM」は Eudragit(登録商標)RS の通常の未細砕粉末を表し、用語
「Eudragit(登録商標)RSPO」は Eudragit(登録商標)RS の細砕微粉末を表し、用語
「Eudragit(登録商標)RS100」は Eudragit(登録商標)RS の顆粒を表し、その一方、
用語 「Eudragit(登録商標)RS12.5」は、Eudragit(登録商標)RS が有機溶剤中に溶解
されている Eudragit(登録商標)RS の溶液製品を表す。特定の実施形態では、本発明に
おいて使用した Eudragit(登録商標)RL は、Eudragit(登録商標)RLPM、Eudragit(登
録商標)RLPO、Eudragit(登録商標)RL100、Eudragit(登録商標)RL12.5、及びその混
合物からなる群より選択される。用語 「PM」、「PO」、「100」、及び「12.5」は Eudra
git(登録商標)RS に関して上記のように定義される。また、Eudragit(登録商標)RS
シリーズ及び Eudragit(登録商標)RL シリーズの混合物も、任意の比率で、本発明のア
ンモニオ メタクリラート共重合体として使用する。
【0124】
本発明の Eudragit(登録商標)RL/RS 分散系は望ましい溶解特性を有する隔離製剤を
最終的に得るために互いに所望の比率で混合することができる。たとえば、望ましい製剤
は、現に、100% の Eudragit(登録商標)RL、50% の Eudragit(登録商標)RL 及び 50%
の Eudragit(登録商標)RS、及び 10% の Eudragit(登録商標)RL、Eudragit(登録商
標)90% RS に由来するコーティングから得ることができる。 もとより、当業者であれば
、たとえば、Eudragit(登録商標)L のような他のアクリル重合体を使用することも可能
であることを認識するであろう。
【0125】
層は、有機または水溶液または分散系の形式で適用することができる。所望の隔離を得
るため、層を適用して、オピオイド拮抗薬を含有する複数個の薬剤的に許容できる粒子の
約 2 〜約 25% の重量増加を達成することが可能である。水分散系に由来するコーティン
グは、たとえば、米国特許特許番号第 5,273,760 号及び第 5,286,493 号に詳しく記載さ
れている。本発明に従って使用できるコーティングの他の例としては、米国特許特許番号
第 5,324,351 号; 第5,356,467 号、及び第 5,472,712 号が挙げられる。
【0126】
オピオイド拮抗薬を含有する複数個の押出成形した粒子が疎水性物質の水分散系と重層
する特定の実施形態では、 疎水性物質の水分散系には好ましくは可塑剤有効量が含まれ
る。
【0127】
層を疎水性物質の水分散系から調製する本発明の実施形態では、水分散系中の可塑剤有
効量を包含すると、層の物性をさらに改善することができる。たとえば、エチルセルロー
スは比較的高いガラス転移温度を持ち、コーティング標準条件下で柔軟な膜を形成しない
ので、可塑剤をエチルセルロースのコーティング溶液に取り込むことが好ましい。一般的
に、溶液に含まれる可塑剤量は、フィルム形成物濃度、たとえば、ほとんどの場合、フィ
ルム形成物の重量の約 1 〜約 50 パーセントに基づく。
【0128】
好適なエチルセルロースに対する可塑剤の例には、ジブチルセバカート、ジエチル フ
タラート、クエン酸トリエチル、クエン酸トリブチル、及びトリアセチンのような不水溶
性可塑剤が含まれるが、他の水不溶性可塑剤 (例: アセチル化モノグリセリド、フタラー
トエステル、ヒマシ油など) を使用することも可能である。クエン酸トリエチルは、エチ
ルセルロースの水分散系に対して特に好ましい可塑剤である。
【0129】
本発明のアクリル重合体に潜在的に好適な可塑剤の例としては、限定するものではない
が、クエン酸トリエチル NF XVI、クエン酸トリブチル、ジブチルフタラート、及びプロ
ピレングリコールのようなクエン酸エステルが挙げられる。Eudragit(登録商標)RL/RS
ラッカー液のようなアクリルフィルムから形成し膜弾性を高めるのに好適であると実証さ
れている他の可塑剤には、ポリエチレングリコール、ジエチル フタラート、ヒマシ油、
及びトリアセチンが含まれる。クエン酸トリエチルはアクリル重合体の水分散系に特に好
ましい可塑剤である。
【0130】
可塑化した疎水性物質は、当分野で既知の任意の好適な噴霧器を用いた噴霧によって、
オピオイド拮抗薬含有する複数個の薬剤的に許容できる粒子に適用することができる。好
ましい方法では、下部から注入する噴出空気が被覆材料を流動化し、コーティングを噴霧
中に乾燥させる Wurster 流動層システムを使用する。
【0131】
本発明のコーティング溶液は、疎水性物質、可塑剤、及び溶剤システム (たとえば、水
) に加えて、エレガンスと製品識別を提供するために着色剤をさらに含有することができ
る。好適な着色剤には、アルコールまたはプロピレングリコール系色分散剤、摩砕したア
ルミニウムレーキ及び二酸化チタン及び鉄酸化物色素のような乳白剤が含まれる。着色剤
は、コーティング工程中に疎水性物質の分散系に添加することができる。あるいは、本発
明の製剤に色を提供する他の任意の好適な方法を使用することもできる。たとえば、Opad
ry(登録商標)のような色塗を薬剤的に許容できる被覆粒子に適用することができる。
【0132】
特定の実施形態では、水分散系が処理中に固着する傾向、あるいは研磨剤としての役割
を果たす傾向を低下させるために少量の滑石を使用することが可能である。
【0133】
複数個の薬剤的に許容できる粒子は、たとえば、摂取し胃液に接触し、次いで腸液に接
触するときに一定の期間にわたって制御下でオピオイド作動薬緩慢に放出する放出制御マ
トリックスに分散したオピオイド作動薬を含有することができる。粒子のマトリックスは
、好ましくは約 8 〜約 24 時間、好ましくは約 12 〜約 24 時間にわたって作動薬の放
出制御を提供する。 オピオイド作動薬を含有する粒子に使用する放出制御マトリックス
には、親水性物質あるいは疎水性物質に関して上記に記述した材料 (たとえば、ゴム、セ
ルロースエーテル、アクリル樹脂、タンパク質由来の材料; 消化性の長鎖 (C8-C50 特にC
12-C40)、置換または非置換炭化水素、例: 脂肪酸、脂肪アルコール、脂肪酸のグリセリ
ルエステル、無機質及び野菜油類及びワックス (天然の及び合成)、及びステアリルアル
コール; 及びポリアルキレングリコール) を含むことができる。
【0134】
特定の実施形態では、オピオイド作動薬含有する粒子は、その表面に配置した放出制御
層を有する即放性マトリックスを含有することができる。放出制御層には、上記に記載し
た1つまたはそれ以上の疎水性物質を含むことができる。
【0135】
特定の実施形態では、オピオイド作動薬を含有する複数個の薬剤的に許容できる粒子を
、任意選択で: (i) オピオイド作動薬放出の調節; (ii) 製剤の保護または (iii) 拮抗薬
を含有する被覆粒子のコーティングと実質的に識別できないコーティング提供すること;
または (i)、(ii) または (iii) の組み合わせに好適な1つまたはそれ以上の材料と重層
させる。たとえば、一実施形態では、コーティングを提供して、たとえば、胃腸液と接触
するときに pH-依存型放出または pH-非依存型放出のいずれかを許容する。pH-依存型コ
ーティングは、オピオイドを胃腸管、たとえば、胃腸または小腸の所望領域に放出する役
割を果たし、したがって、少なくとも約 8 時間及び好ましくは約 12 時間、最大で 約 2
4 時間までの鎮痛を患者に提供する能力がある吸収特性を提供することができる。pH-非
依存性層が所望される場合は、層を環境流体、たとえば、胃腸管における pH 変化などに
拘わらずオピオイドの放出を達成することを目的として設計する。また、胃腸管、たとえ
ば、胃腸の 1 所望領域に用量の一部を放出し、胃腸管の別領域、たとえば、小腸に用量
の残存部を放出する組成物を調剤することも可能である。
【0136】
特定の実施形態では、オピオイド作動薬またはオピオイド拮抗薬を含む複数個の薬剤的
に許容できる粒子を硬化させる。好ましくは、粒子は、複数個の薬剤的に許容できる粒子
が安定した溶解 (または無溶解) を提供する終点に至るまで硬化させる。硬化の終点は、
硬化直後の剤型の溶解特性 (曲線) を、たとえば、温度 40℃ 及び相対湿度 75% で少な
くとも 1 ヶ月間の加速した貯蔵条件に晒した後の剤型の溶解特性 (曲線) に対して比較
することによって決定することが可能である。硬化した製剤は、たとえば、米国特許特許
番号第 5,273,760 号; 第 5,286,493 号; 第 5,500,227 号; 第 5,580,578 号; 第 5,639
,476 号; 第 5,681,585 号; 及び第 6,024,982 号に詳しく記載されている。本発明に従
って使用することが可能である放出制御製剤及びコーティングの他の例には、米国特許特
許番号第 5,324,351 号; 第 5,356,467; 及び 5,472,712 に記載された製剤及びコーティ
ングが含まれる。
【0137】
特定の実施形態では、オピオイド作動薬を含有する複数個の薬剤的に許容できる粒子お
よび/またはオピオイド拮抗薬を含有する薬剤的に許容できる粒子には、薬剤的に許容で
きる粒子からのオピオイド作動薬あるいはオピオイド拮抗薬の放出に実質的に影響を及ぼ
さない材料でフィルムコートを施す。特定の実施形態では、Opadry(登録商標)のような
フィルムコートを複数個の薬剤的に許容できる粒子に適用する。フィルムコートは、該当
する場合、好ましくは粒子の凝集を実質的に低下させるため、または作動薬及び拮抗薬を
含む粒子を互いに識別困難にする助けとなるために提供する。好ましくは、本発明のフィ
ルムコーティングには、スムーズでエレガンであって、色素及び他のコーティング添加物
を支持する能力があり、非毒性、不活性、及び不粘着である強力な連続フィルムを作る能
力のあることが望ましい。
【0138】
上記成分に加えて、オピオイド作動薬含有する粒子及び拮抗薬を含有する粒子のいずれ
かまたは両方には、好適な量の他材料、たとえば、医薬技術では慣習的な希釈液、潤滑剤
、結合剤、造粒剤、偏球化剤、着色剤、加味剤 (flavorants) 及び流動促進剤を含むこと
もできる。これらの追加材料の量は所望の効果を所望の製剤に対して提供するために十分
であろう。
【0139】
潤滑剤の例には、限定するものではないが、ステアリン酸マグネシウム、ステアリン酸
ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイ
ン酸、オレイン酸カリウム、カプリル酸、フマル酸ステアリルナトリウム、及びパルミチ
ン酸マグネシウムが含まれる。
【0140】
低粘性の水溶性重合体のような好適な結合剤は、医薬品技術における当業者には周知で
あろう。但し、ヒドロキシプロピルセルロースのような水溶性ヒドロキシ低級アルキルセ
ルロースが好ましい。
【0141】
着色剤には、二酸化チタンおよび/またはF. D. & C として知られている食物に好適な
色素、及び葡萄皮エキス、ビーツの赤粉末、β カロテン、べにの木、カルミン、ウコン
、パプリカ、のような天然着色剤及び前述の任意物質の組み合わせを含むことができる。
【0142】
組成物に組み込む風味は、合成風味油類及び香味料芳香化合物および/または天然の油
類、植物葉、花、および果実の抽出物、及び前述の任意物質の組み合わせから選択するこ
とが可能である。
【0143】
経口剤型調剤するために使用することが可能である薬剤的に許容できる担体、希釈液、
造粒剤、球化剤、流動促進剤及び他の賦形剤の具体例は、医薬品賦形剤ハンドブック (Ha
ndbook of Pharmaceutical Excipients) (米国薬学会) (1986) に記載されている。
【0144】
いくつかのプロセスを用いて、使用する技術が損傷の隔離した拮抗薬の完全性を損なわ
ない限りは本発明の剤型を調製することが可能である (たとえば、拮抗薬の粒子を作動薬
粒子との組み合わせるときに)。隔離した拮抗薬の粒子の完全性を損なうと、作動薬の効
力を損なう量のオピオイド拮抗薬が、無傷剤型の投与直後に放出されることを結果として
もたらすことがある。
【0145】
本発明の粒子を調製するための好ましい工程は、溶融押出成形または溶融造粒技術を介
した工程である。一般的に、溶融造粒技術には、通常は固体の疎水性物質、たとえばワッ
クスを溶融または軟化すること、および散剤をその中に取り込むことが含まれる。特定の
実施形態では、追加の疎水性物質、たとえば、エチルセルロースまたは水不溶性アクリル
重合体を、溶融または軟化した疎水性物質に追加することができる。
【0146】
追加疎水性物質は1つまたはそれ以上のワックス様の熱可塑性物質を含有することがで
きる。特定の実施形態では、製剤内の個々のワックス様の物質は、実質的に分解不可能で
あり、初期放出相中に 胃腸液に不溶性であることが望ましい。有用なワックス様物質は
、約 1:5,000 以下の水溶性を有する物質であってもよい (w/w)。
【0147】
特定の実施形態では、本発明に係る好適な溶解・押出成形マトリックスの調製には、オ
ピオイド作動薬またはオピオイド拮抗薬を、少なくとも1つまたはそれ以上の疎水性物質
と共にブレンドし、均一な混合物を得る工程を含むことができる。次いで、その均一な混
合物を少なくとも押出成形するために十分に軟化するように十分な温度に加熱する。結果
として得られた均一な混合物を次いで押出成型して、たとえば、細長いストランドを形成
する。押出品は、好ましくは冷却し、当分野で周知の任意の手段によって複数微粒子(た
とえば、複数の粒子) に切断する。押出品は、好ましくは約 0.1 〜約 12 mm、約 0.1 〜
約 2.5 mm、約 0.2 〜約 6 mm、0.5 〜約 3 mm; 約 0.5 mm 〜約 2 mm、または約 1 mm
〜約 2 mm の平均直径を持つ。
【0148】
溶解・押出成形マトリックスの調製に有用な好適な疎水性物質には、限定するものでは
ないが、上記のようにアクリル重合体、セルロースポリマー及び脂肪アルコールが含まれ
る。
【0149】
本発明の溶融押出成形を調製するための任意工程では、直接的に疎水性物質、治療活性
剤、及び任意の結合剤を押出機にメータリングし; 成分をブレンド及び加熱して、均一混
合物を形成し; 均一混合物を押出成型して、細長いストランドを形成し; 均一な混合物を
含むストランドを冷却して; ストランドを約 0.1 mm 〜約 12 mm のサイズを有する粒子
に切断する必要がある。本発明のこの実施態様では、比較的に連続的な製造工程が実現す
る。
【0150】
押出機口径または出口の直径は、押出成形したストランドのさまざまな厚みに合わせて
調節することができる。その上、押出機出口は円形である必要はなく、長円形、長方形な
どであってもよい。得られたストランドは、熱線カッター、裁断機などを用いて粒子に細
断することができる。
【0151】
溶融押出成形したマトリックスは、押出機出口の形状により、たとえば、顆粒、偏球ま
たはペレット形状にすることができる。本発明の目的上、用語「溶融押出成形したマトリ
ックス,」及び「溶融押出成形マトリックスシステム」「溶融押出成形した複数微粒子」
及び「溶融押出成形した粒子」はすべて複数単位の粒子を意味し、好ましくは類似サイズ
あるいは類似形状であって、1つまたはそれ以上の活性剤及び1つまたはそれ以上の賦形
剤を含み、好ましくは本明細書に記載したように疎水性物質を含む粒子をいう。この関連
から、溶融押出成形したマトリックスのサイズ範囲は直径あるいは長さにおいて約 0.1
〜約 12 mm; 約 0.1 〜約 2.5 mm; 約 0.2 〜約 6 mm; 約 0.5 〜約 3 mm; 約 0.5 mm 〜
約 2 mm; または約 1 mm 〜約 2 mm である。さらに、溶融押出成形したマトリックスは
、このサイズ範囲内で任意の幾何学的形状であり得ることを理解されるべきである。特定
の実施形態では、押出品は所望の長さに切断し、偏球化工程を必要とせずにオピオイド拮
抗薬またはオピオイド作動薬の単位用量に分割することができる。
【0152】
本発明の他の実施形態では、溶融押出成形した材料は、オピオイド作動薬あるいはオピ
オイド拮抗薬を包含することなく調製し、その後にそれらの薬剤を押出品に対して添加す
る。そのような製剤によると、一般に押出成形マトリックス材料とブレンドした薬物が得
られ、次いで混合物を当分野で周知の方法によって複数微粒子に形成する。そのような製
剤は、たとえば、製剤に含まれているオピオイド作動薬またはオピオイド拮抗薬が、1つ
以上の疎水性物質を軟化するために必要な温度に対して敏感であるとき、有益であり得る
。
【0153】
特定の実施形態では、複数個の薬剤的に許容できる粒子を製造するための工程は押出成
形/偏球化工程である。この工程のため、オピオイド作動薬またはオピオイド拮抗薬は結
合剤で湿潤し、穿孔したプレートまたはダイを介して押出成形し、回転円板に載置する。
押出品は、好ましくは砕けて破片となり、回転円板上で球体、偏球体、または円形棒に丸
める。この方法の好ましい工程及び組成物には、水を用いて、たとえば約 80% 〜 25% の
オピオイド作動薬またはオピオイド拮抗薬とブレンドした、たとえば約 20% 〜 75% のセ
ルロース誘導体を含有するブレンド物を湿潤することが含まれる。
【0154】
特定の実施形態では、複数の薬剤的に許容できる粒子を製造するための工程で有機溶剤
を用いてオピオイド拮抗薬または作動薬をマトリックス材料と混合する補助を行う必要が
ある。この技術は、マトリックス材料を利用しなければ、不適当に高い融点となり、材料
を溶融した状態に利用した場合、薬物またはマトリックス材料の分解を生じるか、または
容認できない溶融粘度が結果としてもたらされ、それによって薬物 (たとえば、オピオイ
ド作動薬またはオピオイド拮抗薬) のマトリックス材料との混合を防止する状態になる場
合に使用することが望ましい。薬物及びマトリックス材料は中程度の量の溶剤と組み合わ
せてペーストを形成することができ、次いで強制的にスクリーンを通して顆粒を形成し、
次いで顆粒から溶剤を除去する。あるいは、薬物及びマトリックス材料を十分な量の溶剤
と組み合わせて、完全にマトリックス材料を溶解し、結果として得られた溶液 (固体薬物
粒子が含まれている可能性がある) を噴霧乾燥して複数個の薬剤的に許容できる粒子を形
成することができる。この技術は、マトリックス材料がセルロースエーテルまたはセルロ
ースエステルのような高分子量の合成重合体のときに好ましい。本工程に典型的に利用さ
れる溶剤にはアセトン、エタノール、アイソプロパノール、エチル アセテート、及びそ
の混合物が含まれる。
【0155】
上述のように、オピオイド拮抗薬を含有する複数個の押出成形した粒子は、粒子のそれ
ぞれの周囲に配置した疎水性物質を好ましくは含有する層を持つ。好ましくは、オピオイ
ド拮抗薬を含有する層状粒子は、オピオイド拮抗薬の放出を実質的に低下またはを防止し
、一方では、オピオイド作動薬を含有する薬剤的に許容できる粒子は、好ましくはオピオ
イド作動薬の放出制御を約 8 〜約 24 時間以上にわたって、最も好ましくは約 12 〜約
24 時間にわたって提供する。
【0156】
好ましい実施形態では、拮抗薬含有マトリックスの周囲に配置した層は、拮抗薬に対し
て不浸透性または実質的に不浸透性であり、GI システム中に不溶性または実質的に不溶
性である。好ましくは、本発明の無傷剤型をヒトに経口投与するとき、オピオイド拮抗薬
は、実質的に放出されず、よって体内吸収に利用できない。このように、オピオイド拮抗
薬は剤型内に存在するが、オピオイド作動薬の鎮痛有効性を実質的に遮断しない。但し、
本発明の経口剤型が改変される場合、それに含まれるオピオイド拮抗薬は放出され、少な
くとも部分的にオピオイド作動薬の効果を遮断する。本発明のこの実施態様は経口剤型内
のオピオイド作動薬の乱用 または転換の可能性を減少することができる。たとえば、個
人が、たとえば、咀嚼、粉砕、細砕または溶剤を加熱溶解 (たとえば約 45℃ 〜約 5O℃
を超える温度で) することによって、本発明の経口剤型に含まれた薬物乱用を試みる場合
、層及びマトリックスは両方とも損傷され、オピオイド拮抗薬を隔離する働きをしなくな
る。改変剤型の投与時、オピオイド拮抗薬は放出され、好ましくはオピオイド作動薬の陶
酔効果を実質的に遮断する。
【0157】
本発明の複数個の薬剤的に許容できる粒子(すなわち、層状オピオイド拮抗薬の押出成
形した粒子及びオピオイド作動薬粒子) は、任意選択で、当分野で周知の従来の賦形剤を
用いて経口剤型にさらに組み込まれる。
【0158】
1つの好ましい実施形態では、経口剤型を調製して、オピオイド作動薬含有粒子及びオ
ピオイド拮抗薬含有粒子の有効量をカプセル内に含ませる。たとえば、複数の薬剤的に許
容できる粒子を、摂取されると有効な持続放出用量を提供するために十分な量でゼラチン
カプセル内に入れることができる。カプセルは密閉型にしてもよいし、粒子を散布できる
ように非密閉型にしてもよい。
【0159】
別の実施形態では、好適な量の層状拮抗薬含有粒子をオピオイド作動薬含有粒子と組み
合わせて、複数個の薬剤的に許容できる粒子の完全性を実質的に崩壊することがないよう
に経口錠剤を作るために圧縮する。
【0160】
別の実施形態では、拮抗薬含有粒子が複数個の薬剤的に許容できる粒子の完全性を崩壊
することなく作動薬マトリックス内に埋め込まれている好適な量の層状拮抗薬含有粒子を
オピオイド製剤 (たとえば、徐放性造粒) と組み合わせて錠剤を作るために圧縮する。
【0161】
錠剤 (圧縮及び成型された) を作成するための技術及び組成物、カプセル (ハードゼラ
チン及びソフトゼラチン) 及び錠剤は、Remington's Pharmaceutical Sciences、(Arthur
Osol 編), 1553-1593 (1980) に記載されている。
【0162】
特定の実施形態では、経口剤型には、迅速な治療効果を提供する量の即放性オピオイド
作動薬が含まれる。特定の実施形態では、即放性オピオイド作動薬を、たとえば、ゼラチ
ンカプセル内に別のペレットとして組み込むか、または剤型の調製後に作動薬含有粒子の
表面を被覆することができる。
【0163】
本発明の放出制御製剤は、たとえば、摂取され、胃液と接触し、次いで腸液と接触する
ときには、好ましくはオピオイド作動薬を緩慢に放出するものである。本発明の製剤の放
出制御特性は、たとえば、遅延剤、すなわち、疎水性物質の量を変えることによって、疎
水性物質に相対して可塑剤の量を変えることによって、追加成分または賦形剤を抱合する
ことによって、製造などの方法を変えることによって改変することができる。
【0164】
好ましい実施形態では、本発明に有用なオピオイド作動薬には、限定するものではない
が、アルフェンタニル、アリルプロジン、アルファプロジン、アニレリジン、ベンジルモ
ルヒネ、ベジトラミド、ブプレノルフィン、ブトルファノール、クロニタゼン、コデイン
、デソモルヒネ、デキストロモルアミド、デゾシン、ジアンプロミド、ジアモルホン、ジ
ヒドロコデイン、ジヒドロモルヒネ、ジメノキサドール、ジメフェプタノール、ジメチル
チアンブテン、ジオキサフェチル ブチラート、ジピパノン、エプタゾシン、エトヘプタ
ジン、エチルメチルチアンブテン、エチルモルヒネ、エトニタゼン、エトルフィン、ジヒ
ドロエトルフィン、フェンタニール及び誘導体、ヘロイン、ヒドロコドン、ヒドロモルホ
ン、ヒドロキシペチジン、イソメタドン、ケトベミドン、レボルファノール、レボフェナ
シルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタゾシン、メサドン
、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノルレボルファノール
、ノルメサドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピパノン、アヘン、
オキシコドン、オキシモルホン、パパベレタム、ペンタゾシン、フェナドキソン、フェノ
モルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミド、プロpヘプタ
ジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニル、チリジン、ト
ラマドル、それらの薬剤的に許容できる塩類、及び前述した物質のいずれかの混合物が含
まれる。特定の実施形態では、剤型内のオピオイド作動薬の量は約 75 ng 〜 750 mg で
あってもよい。
【0165】
好ましい実施形態では、本発明のオピオイド拮抗薬は、ナルトレキソン、ナロキソン、
ナルメフェン、シクラザシン、レバロルファン、それらの薬剤的に許容できる塩類、及び
前述した物質のいずれかの混合物より選択される。特定の好ましい実施形態では、オピオ
イド拮抗薬は、ナルトレキソンまたはその薬剤的に許容できる塩類 (たとえば、塩酸ナル
トレキソン) である。特定の実施形態では、実質的に放出不可能な形態で存在しているオ
ピオイド拮抗薬の量は、約 0.5 mg 〜約 50 mg、約 1 mg 〜約 25 mg、約 2 mg 〜約 20
mg、約 5 mg 〜約 15 mg、約 2 mg 〜約 10 mg、または約 4 mg 〜約 10 mg または約 6
mg 〜約 8 mg であってよい。
【0166】
ナロキソンは、作動薬効果をほとんど無効にするオピオイド拮抗薬である。最大で 12
mg までのナロキソンの皮下投与は識別可能の自覚効果を生ぜず、24 mg のナロキソンは
軽微な傾眠のみを引き起こす。男性の筋肉内または静脈内に与えた少量 (0.4 〜 0.8 mg)
のナロキソンは、モルヒネ様オピオイド作動薬の効果を防止するか、または即座に逆転
する。1 mg のナロキソンの静脈注射により、25 mg のヘロイン効果を完全に遮断するこ
とが報告されている。ナロキソンの効果は静脈内投与のほとんど直後に見られる。経口投
与後薬物は吸収されるが、その肝臓を介した第1通路で代謝されて急速に不活性形態とな
ることが報告されており、非経口的に投与される場合より大幅に低い作用効力を持つこと
が報告されている。1g 以上の経口投薬は、24 時間未満にほとんど完全に代謝されことが
報告されている。また、舌下投与された 25% のナロキソンが吸収されることも報告され
ている。Weinberg らの、Sublingual Absorption of selected Opioid Analgesics, Clin
Pharmacol Ther. (1988); 44:335-340。参照。
【0167】
他のオピオイド拮抗薬、たとえば、窒素のシクロプロピルメチル置換基をそれぞれに持
つシクラゾシン及びナルトレキソンは、経口経路による効力の多くを維持し、その作用時
間ははるかに長く、経口による服用後 24 時間に達する。
【0168】
本発明の好ましい実施形態では、オピオイド作動薬は、オキシコドン、ヒドロコドン、
ヒドロモルホン、モルヒネ、オキシモルホン、コデインまたはそれらの薬剤的に許容でき
る塩類を含有し、オピオイド拮抗薬は、ナルトレキソンまたはその薬剤的に許容できる塩
類を含有し、約 2 mg 〜約 15 mg、約 5 mg 〜約 10 mg、または約 6 mg 〜約 5 mg の量
で存在する。
【0169】
オピオイド作動薬がヒドロコドンまたはその薬剤的に許容できる塩類を含有する実施形
態では、徐放性経口剤型に、鎮痛用量約 5 mg 〜 約 50 mg の ヒドロコドンまたはその
塩類を用量単位毎に含むことができる。ヒドロモルホンまたはその薬剤的に許容できる塩
類が治療学的に活性なオピオイドである徐放性経口剤型では、それは約 2 mg 〜約 64 mg
の量のヒドロモルホンまたはその塩類に含まれる。別の実施形態では、オピオイド作動
薬は、モルヒネまたはその薬剤的に許容できる塩類を含有し、本発明の放出制御経口剤型
には約 2.5 mg 〜約 800 mg のモルヒネまたはその塩類が含まれる。さらに別の実施形態
では、オピオイド作動薬は、オキシコドンまたはその薬剤的に許容できる塩類を含有し、
放出制御経口剤型には約 2.5 mg 〜約 800 mg のオキシコドンまたはその塩類が含まれる
。特定の好ましい実施形態では、徐放性経口剤型には約 5mg、10mg、20mg、40mg、60mg、
80 mg、160 mg または 320 mg の塩酸オキシコドンが含まれる。放出制御オキシコドン製
剤は当分野で周知である。特定の実施形態では、オピオイド作動薬は、トラマドルまたは
その薬剤的に許容できる塩類を含有し、放出制御経口剤型には、用量単位毎に約 25 mg
〜 800 mg のトラマドルを含むことができる。剤型には、1つ以上のオピオイド作動薬を
含むことができ、1つの作動薬製品によって達成される治療効果と比較して等価な治療効
果を提供することが可能である。あるいは、剤型には、本発明で有用なオピオイド作動薬
の他の塩類と等価のモル濃度を含むことが可能である。
【0170】
特定の実施形態では、安定剤を剤型内に含ませ、オピオイド拮抗薬の劣化を防止する。
特定の実施形態では、剤型で使用する安定剤には、たとえば、限定するものではないが、
有機酸、カルボン酸、アミノ酸の酸塩類 (たとえば、システイン、L-システイン、システ
イン塩酸塩、グリシン塩酸塩またはシステイン二塩酸塩)、メタ重亜硫酸ソーダ、アスコ
ルビン酸及びその誘導体、リンゴ酸酸、イソアスコルビン酸、クエン酸、酒石酸酸、パル
ミチン酸、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カルシウム、リン酸水素カルシウ
ム、二酸化硫黄、亜硫酸ナトリウム、重硫酸ナトリウム、トコフェロール、さらには、そ
の水溶性及び脂溶性の誘導体 (たとえばtocofersolan または酢酸トコフェロールのよう
な)、亜硫酸塩剤、重亜硫酸及び亜硫酸水素またはアルカリ金属、アルカリ土類金属及び
他の金属、PHB エステル、ガラート、ブチル化ヒドロキシアニソール (BHA) またはブチ
ル化ヒドロキシトルエン (BHT)、及び2,6-ジ-t-ブチル-アルファ-ジメチルアミノ-p-クレ
ゾール、t-ブチルヒドロキノン、ジ-t-アミルヒドロキノン、ジ-t-ブチルヒドロキノン、
ブチルヒドロキシトルエン、ブチルヒドロキシアニソール、ピロカテコール、ピロガロー
ル、プロピル/ガラート、及びノルジヒドログアヤレト酸、さらには、低級脂肪酸、フル
ーツ酸、リン酸、ソルビン酸及び安息香酸、さらには、それらの塩類、エステル、誘導体
及び異性化合物、アスコルビン酸パルミテート、レシチン、モノ- 及びポリヒドロキシル
化ベンゼン誘導体、エチレンジアミン四酢酸及びその塩類、シトラコン酸、コンアイデン
ドリン(conidendrine)、ジエチル カルボナート、メチレンジオキシフェノール、ケフ
ァリン、β,β'-ジチオプロピオン酸、ビフェニル及び他のフェニル誘導体、それらの薬
剤的に許容できる塩類、及びその混合物が含まれる。
【0171】
本発明の経口剤型には、オピオイド作動薬及び拮抗薬に加えて、それとともに相乗的作
用してもよいし、相乗的作用しなくてもよい1つまたはそれ以上の薬物がさらに含まれる
。このように、特定の実施形態では、オピオイド拮抗薬に加えて、剤型内に2つのオピオ
イド作動薬の組み合わせを含むことができる。たとえば、剤型には、半減期、溶解度、作
用効力のような異なる特質を有する2つのオピオイド作動薬、及び前述の任意物質の組み
合わせを含むことができる。なお、さらなる実施形態では、オピオイド拮抗薬に加えて、
1つまたはそれ以上のオピオイド作動薬を含み、さらに非オピオイド薬物もまた含む。そ
のような非オピオイド薬物は、好ましくは追加鎮痛を提供し、たとえば、アスピリン、ア
セトアミノフェン; 非ステロイド性抗炎症性薬 (「NSAID」)、たとえば、イブプロフェン
、ケトプロフェンなど; N-メチル-D-アスパルタート(NMDA) 受容体拮抗薬、たとえば、モ
ルフィナンのようなデキストロメトルファンまたはデキストロルファン、またはケタミン
; シクロオキシゲナーゼ-II 阻害剤 (「COX-II 阻害剤」); あるいはグリシン受容体拮
抗薬を含む。追加薬剤を、第1作動薬として同じ粒子内または異なる粒子内に含むことが
できる。
【0172】
本発明の特定の好ましい実施形態では、本発明は、NSAID または COX-2 阻害剤のよう
な付加的な非オピオイド作動薬を包含することによって、低用量のオピオイド鎮痛薬の使
用を可能にする。いずれかまたは両方の薬物を低量で用いることによって、ヒトにおける
効果的な疼痛管理に関連する副作用を抑えることが可能である。
【0173】
好適な非ステロイド性抗炎症薬には、イブプロフェン、ジクロフェナク、ナプロキセン
、ベノクサプロフェン (benoxaprofen)、フルルビプロフェン、フェノプロフェン、フ
ルブフェン (flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カル
プロフェン、オキサプロジン、プラモプロフェン (pramoprofen)、ムロプロフェン (m
uroprofen)、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェ
ン酸、フルプロフェン、ブクロクス酸、インドメタシン、スリンダク、トルメチン、ゾメ
ピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オク
スピナク (oxpinac)、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸
、トルフェナム酸、ジフルリサール (diflurisal)、フルフェニサール、ピロキシカム
、スドキシカムまたはイソキシカム、それらの薬剤的に許容できる塩類、その混合物など
が含まれる。これらの薬物の有用な用量は当業者には公知である。
【0174】
N-メチル-D-アスパルタート(NMDA) 受容体拮抗薬は当分野で周知であり、たとえば、デ
キストロメトルファンまたはデキストロルファンのようなモルフィナン、ケタミン、d-メ
サドンまたはそれらの薬剤的に許容できる塩類を包含する。本発明の目的上、用語「NMDA
拮抗薬」もまた、NMDA-受容体活性化、たとえば、GM1 またはGT1b のようなガングリオ
シド、トリフルオペラジンのようなフェノチアジンまたはN(6-アミノテキシル)-5-クロロ
-l-ナフタレンスルホンアミドのようなナフタレンスルホンアミドの主な細胞内の結果物
を遮断する薬物を包含すると見なされる。これらの薬物は、 米国特許番号第 5,321,0
12 号及び第 5,556,838 号 (両方とも Mayer らによる) では添加薬物、たとえば、モル
ヒネ、コデインなどのような麻薬性鎮痛薬に対する耐性あるいは依存性を抑制することを
趣旨としており、米国特許特許番号第 5,502,058 号(Mayer らによる) では慢性疼痛を治
療することを趣旨としている。
【0175】
グリシン受容体拮抗薬の使用を介した慢性疼痛の治療、及びその薬物の同定は、米国特
許特許番号第 5,514,680 号 (Weber らによる) に記載されている。
【0176】
CQX-2 阻害剤は当技術分野で報告されており、多くの化学構造がシクロオキシゲナーゼ
-2 の抑制を実現することが知られている。COX-2 阻害剤は、たとえば米国特許特許番号
第 5,616,601 号; 第 5,604,260 号; 第 5,593,994 号; 第 5,550,142 号; 第 5,536,752
号; 第 5,521,213 号; 第 5,475,995 号; 第 5,639,780 号; 第 5,604,253 号; 第 5,55
2,422 号; 第 5,510,368 号; 第 5,436,265 号; 第 5,409,944 号; 及び第 5,130,311 号
に記載されている。特定の好ましい COX-2 阻害剤には、セレコクシブ (SC-58635)、DUP-
697、フロスリド (CGP-28238)、メロキシカム、6-メトキシ-2 ナフチル酢酸 (6-MNA)、MK
-966 (またバイオックスとしても知られている)、ナブメトン (6-MNA 用のプロドラッグ)
、ニメスリド、NS-398、SC-5766、SC-58215、T-614; それらの薬剤的に許容できる塩類、
及びその組み合わせが含まれる。1日当たり、体重 1 キログラム当たり約 0.005 mg 〜
約 140 mg の COX-2 阻害剤の投与量レベルは、オピオイド鎮痛薬と組み合わせた場合に
治療効果がある。あるいは、 1 日当たり、患者当たり約 0.25 mg 〜約 7 g の COX-2 阻
害剤をオピオイド鎮痛薬と組み合わせて投与する。オピオイド作動薬と COX-2 阻害剤の
組み合わせは、WO 99/13799 に開示されている。
【0177】
なお、さらならる実施形態では、鎮痛薬以外の、たとえば、鎮咳薬、排痰剤、制吐薬、
充血除去剤、抗ヒスタミン薬、局所麻酔などの所望の効果を提供する非オピオイド薬物を
含むことができる。
【0178】
また、本発明は、活性剤の乱用を抑止するために、異なる活性剤/拮抗薬の組み合わせ
(すなわち非オピオイド) を利用する本明細書で開示した剤型をその狙いとするをする。
たとえば、ベンゾジアゼピンを本発明の剤型内で活性剤として用いると、隔離したベンゾ
ジアゼピン拮抗薬を剤型内で製剤することができる。バルビツール酸塩を本発明の剤型内
で活性剤として用いると、隔離したバルビツール酸塩拮抗薬を剤型内で製剤することがで
きる。アンフェタミンを本発明の剤型内で活性剤として用いると、隔離したアンフェタミ
ン拮抗薬を剤型内で製剤することができる。
【0179】
用語「ベンゾジアゼピン」は中枢神経系を抑圧することができるベンゾジアゼピン、及
びベンゾジアゼピンの誘導体である薬物を指す。ベンゾジアゼピンには、限定するもので
はないが、、アルプラゾラム、ブロマゼパム、クロルジアゼポキシド、クロラゼプ酸、ジ
アゼパム、エスタゾラム、フルラゼパム、ハラゼパム、ケタゾラム、ロラゼパム、ニトラ
ゼパム、オキサゼパム、プラゼパム、クァゼパム、テマゼパム、トリアゾラム、メチルフ
ェニデート及びその混合物が含まれる。
【0180】
本発明で使用することができるベンゾジアゼピン拮抗薬には、限定するものではないが
、フルマゼニルが含まれる。
【0181】
バルビツール酸塩はバルビツール酸 (2、4、6、-トリオキソヘキサヒドロピリミジン)
に由来する鎮静薬・催眠薬を意味する。バルビツール酸塩には、限定するものではないが
、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタルビタール、メトヘ
キシタール、メホバルビタール、メタルビタール、ペントバルビタール、フェノバルビタ
ール、セコバルビタール及びその混合物が含まれる。
【0182】
本発明で使用することができるバルビツール酸塩拮抗薬には、限定するものではないが
、本明細書に記載したようにアンフェタミンが含まれる。
【0183】
刺激薬は中枢神経系を刺激する薬物を意味する。刺激薬には、限定するものではないが
、アンフェタミン、デキストロアンフェタミン樹脂複合体、デキストロアンフェタミン、
塩酸メタンフェタミン、メチルフェニデートのようなアンフェタミン類、及びその混合物
が含まれる。
【0184】
本発明で使用することができる刺激拮抗薬には、限定するものではないが本明細書に記
載したようにベンゾジアゼピンが含まれる。
【0185】
本発明はまた、活性剤の乱用を抑止するするために拮抗薬以外の逆作用剤を利用する本
明細書で開示した剤型をその狙いとする。用語「逆作用剤」は非隔離形態で投与して不快
な効果を作り出すことができる任意の薬剤を指す。拮抗薬以外の逆作用剤の例には、催吐
薬、刺激剤及び苦味剤が含まれる。
【0186】
催吐薬には、限定するものではないが、トコン及びアポモルヒネが含まれる。
【0187】
刺激剤には、限定するものではないが、カプサイチン、カプサイチン類似体、及びその
混合物が含まれる。カプサイチン類似体には、レシニフェラトキシン、ティニアトキシン
(tinyatoxin)、ヘプタノイルイソブチルアミド(heptanoylisobutylamide)、ヘプタノ
イルグアイアシルアミド(heptanoyl guaiacylamide)、他のイソブチルアミド類または
グアイアシルアミド(guaiacylamides)類、ジヒドロカプサイチン、ホモバニリル オク
チルエステル、ノナノイル バニリルアミド、及びその混合物が含まれる。
【0188】
苦味剤には、限定するものではないが、風味油類; 香味料芳香化合物; 含油樹脂; 植物
、葉、花に由来するエキス; フルーツ風味; サッカロース誘導体; クロロサッカロース誘
導体; 硫酸キニーネ; 安息香酸デナトニウム; 及びその組み合わせが含まれる。
【0189】
以下に、添付実施例を参照して本発明をより詳細に説明する。ただし、以下の説明は単
なる例示目的にすぎず、いかようにも本発明を限定するものではないことを理解されるべ
きである。
【実施例1】
【0190】
塩酸ナルトレキソン 2 mg カプセル
本実施例は、隔離製品を産生するために溶融押出成形した複数微粒子 (以下「MEM」と
する) として製剤したオピオイド拮抗薬である塩酸ナルトレキソンの比較実施例である。
選択した重合体及び賦形剤に基づいて、ペレットを無傷で分析すると MEM ペレットは極
めて僅かなナルトレキソンを放出するが、改変(粉砕) されると著しい量のナルトレキソ
ンを放出する。この実施例は、実施例 1 に続く実施例においてコーティングがどのよう
に隔離特質を高めることができるかを示すために参照として含むものである。実施例 1
の 塩酸ナルトレキソン製剤を下記の表に記載する。
【表1】

実施例 1 の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機に1.7 kg/時 〜 2.6 kg/時
の速度で連続供給し、押出成形物を回収する(Leistritz ZSE-27)。ブレンド物を 75℃ 〜
100℃ のバレル温度で直径約 1 mm の ストランドに押出成型する。押出成形したストラ
ンドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
7. カプセル化工程: スクリーニングしたペレットを目標重量 121 mg でハードゼラチン
カプセルに充填する。
【0191】
In vitro 溶解工程:
実施例 1 に従って調製した製剤を以下の in vitro 溶解検査方法で検査を行い、以下
の表 1B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 75 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF (人工胃液) で 1 時間、その後、900 ml の SIF (人工腸液) に
切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表2】
【0192】
模擬改変のプロセス及び溶解:
実施例 1 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の in
-vitro 溶解検査方法で検査した。1 時間の溶解結果を表1 C に記載した。改変プロセス
では、この溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (600 ストローク)
を用いて細砕し、粉末にした。
溶解方法: 上記と同じ
結果:
【表3】
【0193】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 1 時間で溶解した % と無傷ペレットが 36 時
間で溶解した % との比率である。
粉砕/無傷比の結果: 33.5% / 6.2% = 5.4:1
【0194】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
上記の工程と製法を用いて製造したカプセル (MEM) を臨床研究に用いて、即放性ナル
トレキソン錠剤と比較した薬物動態学/バイオアベイラビリティを決定した。ヒト被検体
に無傷塩酸ナルトレキソン MEM (全体)、粉砕した塩酸ナルトレキソン MEM (細砕) また
は即放性塩酸ナルトレキソン錠剤 (IR NTX) 剤型のいずれかを投与した。結果は図1 のグ
ラフ表示に示す。即放性ナルトレキソン (IR NTX) 及び粉砕 (細砕) 及び無傷 (全体) の
IR NTX の用量調節 Cmax と比較した無傷 (全体) 及び粉砕 (細砕) の用量調節 (1 mg
の IR NTX 錠剤に) 暴露度 (AUCt) を下記の表 1D に記載する。
【表4】
【0195】
用量調節した血漿濃度は、無傷状態で採取すると MEM 剤型からナルトレキソンの最小
放出があることを示す。ナルトレキソンのレベルは、MEM を粉砕 (細砕) 状態で採取する
と増加する。平均 Cmax に基づくと、粉砕 MEM/無傷 MEM カプセルの比率は、約 8 であ
る。同様に、平均 AUCt、粉砕 MEM/無傷 MEM カプセルの比率は、約 4.4 である。これは
合計及びピーク時の暴露の比率が粉砕後に大幅に増加することを示す。
【実施例2】
【0196】
エチルセルロース被覆の塩酸ナルトレキソン 2 mg ペレット
実施例 2 では、ナルトレキソン MEM を実施例 1 と同様に調製し、次いで MEM をエチ
ルセルロース (Surelease) でさまざまなレベル (5%、10%、15%、及び20% 重量増加) に
被覆した。実施例 2 の 非被覆の塩酸ナルトレキソン製剤を下記の表 2A に記載する。
【表5】
【0197】
実施例 2 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機に 2.9 kg/時 〜 4.8 kg/時
の速度で連続供給する (Leistritz ZSE-27)。ブレンド物を 95℃ 〜 105℃ のバレル温度
で直径約 1 mm の ストランドに押出成型する。押出成形したストランドをコンベア上に
回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収した。
【0198】
In vitro 溶解工程:
実施例 2 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 2B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表6】
【0199】
模擬改変; プロセス及び溶解:
実施例 2 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2C に記載した。改変プロセスでは、この
溶解研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を
用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表7】
【0200】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比の結果: 31% / 5.3% = 5.8:1
【0201】
実施例 2 によって調製し、表 2A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング(Surelease)で被覆して
、5%、10%、15% 及び20% 重量増加し; 及び疎水性コーティング(Surelease) とカラーコ
ーティング(Opadry)で被覆して20% の重量増加を得た。20% の重量増加を得た製剤例のコ
ーティング及びカラーコーティングを以下の表に記載する。
【表8】
実施例 2 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: 水と混合することによって Surelease 懸濁液を15% (w/w)
固体に希釈する。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Surelease 分散液を 700 g スケ
ール以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 7.0 〜 9.0 m/秒
* 吸気温度: 40 〜 50℃
* 分散液噴霧速度: 8 〜 11 g/分
5%、10%、15% 及び20% の重量増加に対する分散の理論量を噴霧したとき、サンプルを採
取した。
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 7.0 m/秒
* 吸気温度: 50℃
* 分散液噴霧速度: 8.5 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。上記の 20% 製法及び工程に従って 5%、10% 及び 15% の重量増加に被
覆したペレットを、単位当たり 6.05、12.1 及び 18.15 mg の Surelease をそれぞれ用
いて調製した。
【0202】
In vitro 溶解工程:
実施例 2 に従って調製した被覆した製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 2E に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表9】
溶解結果から分かるように、ナルトレキソンのペレットの溶解度は一般的に重合体コーテ
ィングレベルの増加に伴って減少した。
【0203】
模擬改変のプロセス及び溶解:
実施例 2 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表10】
【0204】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 2G に記載した。
粉砕/無傷比の結果:
【表11】
溶解結果から分かるように、コーティングレベルが増加するにつれ、粉砕/無傷比も増加
した。
【0205】
実施例 1 と比較した実施例 2 の結果
このように、実施例 1 における非被覆のMEM と同じ製剤であるMEM を実施例 2 で過剰
コーティングすることによって、薬物の放出は 36 時間で 5% 以上から およそ 2% まで
に下落した。結果として、実施例 1 における非被覆の MEM からの拮抗薬の「漏出」も、
機能コーティングを用いることによって大幅に低下した。粉砕/無傷比は、およそ 5:1 か
ら 10:1 に増加し得る。
【実施例3】
【0206】
エチルセルロース被覆の塩酸ナルトレキソン 8 mg ペレット
実施例 3 では、8mg のナルトレキソンを含むペレットを調製し、次いでエチルセルロ
ース (Surelease) でさまざまなレベル (5%、10%、15%、及び20%、25% 及び 30% 重量増
加) に被覆した。実施例 1 における 非被覆の塩酸ナルトレキソン製剤を下記の表に記載
する。
【表12】
実施例 3 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 3.9
kg/時の速度で連続供給する。ブレンド物を 95℃ 〜 100℃ のバレル温度で直径約 1 mm
の ストランドに押出成型する。押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0207】
In vitro 溶解工程:
実施例 3 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 3B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表13】
【0208】
模擬改変のプロセス及び溶解:
実施例 3 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の結果を表 3C に記載した。改変プロセスでは、この溶解
研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用い
て細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表14】
【0209】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比の結果: 57% / 18.7% = 3.0
【0210】
実施例 3 によって調製し、表 3A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング(Surelease)で被覆して
、5%、10%、15%、20% 及び25% 重量増加し; 及び疎水性コーティング(Surelease) とカラ
ーコーティング(Opadry)で被覆して30% の重量増加を得た。30% の重量増加を得た製剤例
の疎水性コーティング及びカラーコーティングを以下の表に記載する。
【表15】
実施例 3 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: 水と混合することによって Surelease 懸濁液を15% (w/w)
固体に希釈する。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Surelease 分散液を 700 g スケ
ール以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 8.6 〜 9.6 m/秒
* 吸気温度: 40 〜 50℃
* 分散液噴霧速度: 9 〜 14.8 g/分
5%、10%、15%、20%、25%、30% の重量増加に対する分散の理論量を噴霧したときに、サ
ンプルを採取した (単位当たり、それぞれおよそ 6.05、12.1、18.15、24.2 及び 30.25
mg の Surelease)
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 8.6 〜 9.0 m/秒
* 吸気温度: 47 ℃
* 分散液噴霧速度: 9.0 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。
【0211】
In vitro 溶解
実施例 3 に従って調製した、疎水性コーティング (Surelease) 及びカラーコーティン
グ (Opadry) で被覆した製剤を以下の in vitro 溶解検査方法で検査を行い、以下の表 3
E に記載した結果を得た。
方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表16】
溶解結果から分かるように、ナルトレキソンのペレットの溶解は一般的に重合体コーティ
ングレベルの増加に伴って減少した。
【0212】
模擬改変のプロセス及び溶解:
実施例 3 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表17】
【0213】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 3G に記載した。
粉砕/無傷比の結果:
【表18】
【0214】
上記の溶解結果から分かるように、コーティングレベルが増加するにつれ、無傷ペレッ
トから放出されるナルトレキソンの量が大幅に減少する (36 時間で 18% 以上から 2% 未
満に減少する) が、粉砕後には、拮抗薬のおよそ 50% が放出され、粉砕/無傷比は大幅に
増加する。
【0215】
コーティング後、8 mg の無傷ペレットでは、ナルトレキソンの放出が非被覆の無傷ペ
レットと比較して著しい減少を示した。但し、粉砕した被覆の 8 mg ペレットからの放出
は、粉砕した非被覆の 2 mg ペレットと比較して高い。
【実施例4】
【0216】
メタクリル共重合体の被覆塩酸ナルトレキソン 8 mg ペレット
実施例 4 では、8mg のナルトレキソンを含むペレットを実施例 3 と同様に調製したが
、メタクリル共重合体 (Eudragit RS 30D) でさまざまなレベル (5%、10%、15%、及び20%
、及び25% 重量増加) に被覆した。実施例 4 の 非被覆の塩酸ナルトレキソン製剤を下記
の表 4A に記載する。
【表19】
実施例 1 の非被覆塩酸ナルトレキソン製剤を
以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 3.9
kg/時の速度で連続供給する。ブレンド物を 95℃ 〜 100℃ のバレル温度で直径約 1 mm
の ストランドに押出成型する。押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0217】
In vitro 溶解工程:
実施例 4 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 4B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表20】
【0218】
模擬改変; プロセス及び溶解:
実施例 4 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 4C に記載した。改変プロセスでは、この
溶解研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を
用いて細砕し、粉末にした。
溶解方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表21】
【0219】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分での溶解分と無傷ペレットが 36 時間で
の溶解分との比率である。
粉砕/無傷比の結果: 57% / 18.7% = 3.0
【0220】
実施例 4 によって調製し、表 4A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング (Eudragit に基づいて)
で被覆して、5%、10%、15% 及び20% 重量増加し; 及び疎水性コーティング (Eudragit
に基づいて) とカラーコーティング (Opadry) で被覆して25% の重量増加を得た。25% の
重量増加を得た製剤例の疎水性コーティング及びカラーコーティングを以下の表に記載す
る。
【表22】
実施例 4 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 700 g スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴霧
する。
* エアスピード: 8.5 〜 9.5 m/秒
* 吸気温度: 35℃
* 分散液噴霧速度: 14 g/分
5%、10%、15%、20%、及び25% の重量増加に対する分散の理論量を噴霧したときに、サン
プルを採取した。
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 8.5 m/秒
* 吸気温度: 35 ℃
* 分散液噴霧速度: 8.5 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。
上記の 20% 製法及び工程に従って 5%、10%、15% 及び20% の重量増加に被覆したペレッ
トを、単位当たり 6.05、12.1、18.15 及び 24.2 mg の Eudragit RS30D (固体) をそれ
ぞれを用いて調製した。
【0221】
In vitro 溶解工程:
実施例 4 に従って調製した、疎水性コーティングで被覆した製剤を以下の in vitro
溶解検査方法で検査を行い、以下の表 4E に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表23】
溶解結果から分かるように、ナルトレキソンのペレットの溶解は一般的に重合体コーテ
ィングレベルの増加に伴って減少した。
【0222】
模擬改変のプロセス及び溶解:
実施例 4 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 4F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
【0223】
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表24】
【0224】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 4G に記載した。
結果:
【表25】
【0225】
上記の溶解結果から分かるように、コーティングレベルが増加するにつれ、無傷ペレッ
トから放出されるナルトレキソンの量が大幅に減少する (36 時間で 18% 以上から約 1%
以下に減少する) が、粉砕後には、拮抗薬のおよそ 50% が依然として放出され、粉砕/無
傷比は増加する。
【0226】
この製品は、コーティングの添加が無傷ペレットからのナルトレキソン放出の著しい減
少を結果としてもたらすが、粉砕されたペレットからはかなりの量の拮抗薬を放出する能
力を保持することを示す。
【実施例5】
【0227】
実施例 5 では、実施例 4 の製剤を GMP 条件下でパイロット規模で繰り返し、In-Vivo
評価に使用した。
【0228】
8 mg の ナルトレキソンを含むペレットを実施例 4 と同様に調製し、疎水性コーティ
ングで 15% 重量増加 (Eudragit RS 30D に基づいて) に被覆した。これらのペレットを
次いでサイズ #2 カプセルに充填した。実施例 5 における 非被覆の塩酸ナルトレキソン
製剤を下記の表に記載する。
【表26】
実施例 5 の非被覆ペレットを以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/分の速度で連続供給する。ブレンド物を 80℃ 〜 100℃ のバレル温度
で直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。押出成形したストランドをコ
ンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0229】
実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティングで15% 重量増加に被覆し
た(Eudragit RS 30D に基づいて)。被覆ペレットは下記の表に記載した。
【表27】
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 700 〜 780 CFM
* 吸気温度: 35 ℃
* 分散液噴霧速度:115 〜 135 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 750 〜 760 CFM
* 吸気温度: 35 ℃
* 分散液噴霧速度: 75 〜 95 g/分
5. スクリーニング: 14 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動分離
器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留した材
料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 149.7 mg でハードゼラチ
ンカプセルに充填する。
【0230】
In vitro 溶解 (無傷ペレット)
実施例 5 において疎水性コーティングで被覆した製剤を、バルクのペレット及びカプ
セル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表 5
C に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表28】
【0231】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 5 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表29】
【0232】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 46.4/0.8 = 58.0
【0233】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
この実施例の上記工程と製法 を用いて製造したカプセルを臨床研究に用いて、異なる
条件下の MEM 製剤の薬物動態学/バイオアベイラビリティを決定し、即放性ナルトレキソ
ン錠剤の薬物動態学/バイオアベイラビリティと比較した。ヒト被検体に無傷塩酸ナルト
レキソン MEM カプセル (1 カプセルまたは 5 カプセル、絶食); 粉砕した塩酸ナルトレ
キソン MEM (1 カプセルの含有量、細砕、絶食); 即放性塩酸ナルトレキソン剤型、錠剤
、絶食; または 1 MEM カプセル、無傷、給餌状態のいずれかを投与した。本研究は、非
盲検、単回投与、5 方向で、クロスオーバー研究を 15 健常者に対して、治療間 14 日間
の洗い出しで行った。治療法は次の通りに設計した:
A. 1x8 mg のナルトレキソン MEM カプセル、無傷、絶食状態
B. 1x8 mg のナルトレキソン MEM カプセル、カプセル内容物粉砕、絶食状態
C. 1x8 mg のナルトレキソン MEM カプセル、無傷、給餌状態
D. 5x8 mg (40 mg) ナルトレキソン MEM カプセル無傷、絶食状態
E. 2x0.5 mg (1 mg) ナルトレキソン即放性錠剤、絶食状態
【0234】
血漿濃度は、無傷状態で採取すると ナルトレキソン MEM ペレットからナルトレキソン
の非常に少量の放出があることを示す。時間曲線データと対比したナルトレキソン濃度 (
pg/ml) は、図2 に示してある。粉砕/細砕したナルトレキソンのペレットを絶食状態で経
口摂取させたとき、ナルトレキソン血漿レベルが大幅に増加した。
平均 Cmax 粉砕した MEM (N=14)/無傷 MEM (N=15) カプセルの比率は112.34 である。
同様に、平均 AUCt 粉砕した MEM(N=14)/無傷 MEM (N=15) カプセルの比率は 31.55 であ
る。
実施例 1 の非被覆 MEM 及び実施例 5 の被覆 MEM の in-vitro 及び in-vivo 比較は
下記の表 5E、及び 5F に示した。
【表30】
【表31】
【実施例6】
【0235】
メタクリル共重合体被覆の 2 mg の塩酸ナルトレキソン MEM
実施例 6 の 非被覆の塩酸ナルトレキソン製剤を下記の表 6A に記載する。
【表32】
実施例 6 の塩酸ナルトレキソン製剤を以下を用いて調製した:
工程:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27に 2.9 k
g/時 〜 4.8 kg/時の速度で連続供給する)。ブレンド物を 95℃ 〜 105℃ のバレル温度
で直径約 1 mm の ストランドに押出成型する。押出成形したストランドをコンベア上に
回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0236】
In vitro 溶解 (無傷ペレット)
実施例 6 に従って調製した製剤を以下の in vitro 溶解検査方法で検査を行い、以下
の表 6B に記載した溶解結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間 / その後 900 ml SIF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表33】
【0237】
模擬改変及び溶解プロセス (粉砕したペレット):
実施例 6 に従って調製した製剤に対して模擬改変プロセスを実施し、以下の in vitro
溶解検査方法で検査した。1 時間の溶解結果を表 6C に記載した。改変プロセスでは、
この溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用い
て細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表34】
【0238】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比: 31% / 5.3% = 5.8:1
【0239】
実施例 6 によって調製し、表 6A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティングで15% 重量増加に被覆し
た(Eudragit RS 30D に基づいて)。15% の重量増加を得た製剤を以下の表に記載する。
【表35】
実施例 6 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 700 g スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴霧
する。
* エアスピード: 9.0 m/秒 .
* 吸気温度: 35 ℃
* 分散液噴霧速度: 8.8 g/分
3. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
【0240】
In vitro 溶解 (無傷ペレット)
実施例 6 に従って調製した、疎水性コーティングで被覆した製剤を以下の in vitro
溶解検査方法で検査を行い、以下の表 6E に記載した結果を得た。
方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー結果:
【表36】
【0241】
模擬改変及び溶解プロセス (粉砕したペレット):
実施例 6 に従って調製した被覆製剤に対して模擬改変プロセスを実施し、次いで以下
の溶解方法で検査した。1 時間の溶解結果を表 6F に記載した。改変プロセスでは、この
溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細
砕し、粉末にした。
溶解方法: 上記結果と同じ
【表37】
【0242】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 1 時間で溶解した % と無傷ペレットが 36 時
間で溶解した % との比率である。
【0243】
粉砕/無傷比: 7% / 0.6% = 12 (注: 検出可能なナルトレキソンが無傷ペレットから観
察されなかったので、粉砕/無傷比 12 をはるかに超える可能性があるが確認していない
。
【実施例7】
【0244】
メタクリル共重合体、次いで Surelease 被覆の 8 mg の塩酸ナルトレキソン MEM
実施例 7 では、MEM の2段階の順次コーティング、最初のコーティングは Eudragit R
S 30D により、重量増加は 15%、次のコーティングは Surelease による 10% の重量増加
(非被覆の押出成形したペレットに基づいて) を調製した。8mg のナルトレキソンを含む
ペレットを実施例 5 と同様に調製し、メタクリル共重合体 (Eudragit RS 30D) で 15%
の重量増加、継いで、エチルセルロース (Surelease) で10% の重量増加に被覆した。こ
の製品は、無傷ペレットからのナルトレキソン放出の著しい減少を結果としてもたらし、
その一方では、粉砕されたペレットからの放出を高めることを示す。実施例 7 の 非被覆
の塩酸ナルトレキソン製剤を下記の表 7A に記載する。
【表38】
実施例 7 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/時の速度で連続供給する。ブレンド物を 85℃ 〜 90℃ のバレル温度で
直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。押出成形したストランドをコン
ベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0245】
実施例 7 によって調製し、表 7A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットをメタクリル共重合体で10% 重量増加に被覆し
、継いでエチルセルロースで 15% 重量増加 (非被覆ペレットに基づいて) に被覆した。
被覆ペレットは下記の表に記載した。
【表39】
被覆塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. メタクリルコーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して
15 分間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の
水に分散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. エチルセルロースコーティングの分散: Surelease を合計 20% (w/w) 固体分散を達成
するのに十分な量の水と混合する。
3. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を達成する。
4. メタクリルコーティングコーティング: 以下のパラメータ指針を用いて Eudragit 分
散液を 700 g スケール以上で調製したナルトレキソンのペレットに流動床プロセッサ (G
PCG-1) を用いて噴霧する。
* エアスピード: 8.8 〜 9.0 m/秒
* 吸気温度: 35 ℃
* 分散液噴霧速度: 9.6 g/分
5. エチルセルロースコーティング: Eudragit コーティングの完了時点で、以下のパラメ
ータ指針を用いて Surelease 分散液を被覆したペレットに噴霧した。
* エアスピード: 9.0 m/秒
* 吸気温度: 40 〜 45℃
* 分散液噴霧速度: 9.2 〜 9.6 g/分
6. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 8.8 〜 9.0 m/秒
* 吸気温度: 50℃
* 分散液噴霧速度: 9.3 g/分
7. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
【0246】
In vitro 溶解 (無傷ペレット)
疎水性コーティング (メタクリル共重合体コーティング及びエチルセルロースコーティ
ング) で被覆し、実施例 7 に従って調製した製剤からは、
以下の in vitro 溶解方法で検査したところ、下記の表 7C に記載の結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表40】
【0247】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 7 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。溶解結果は
下記の表 7D に記載した。
溶解方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表41】
【0248】
粉砕/無傷比:
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 34 時間
で溶解した % との比率である。
粉砕/無傷比: 92.5
【実施例8】
【0249】
Surelease 次いで、メタクリル共重合体被覆の 8 mg の塩酸ナルトレキソン MEM
8mg のナルトレキソンを含むペレットを実施例 5 と同様に調製し、エチルセルロース
(Surelease) で10% 重量増加、継いで、メタクリル共重合体 (Eudragit RS 30D) で15%
重量増加 (非被覆ペレットに基づいて) に被覆した。この製品は、無傷ペレットからのナ
ルトレキソン放出の著しい減少を結果としてもたらし、その一方では、粉砕されたペレッ
トからの放出を高めることを示す。
【0250】
実施例 8 の 非被覆の塩酸ナルトレキソン製剤を下記の表 8A に記載する。
【表42】
実施例 8 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/時の速度で連続供給する。ブレンド物を 85℃ 〜 90℃ のバレル温度で
直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。
押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0251】
実施例 8 によって調製し、表 8A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。本ペレットを、エチルセルロースで10% 重量増加、継い
で、メタクリル共重合体 (非被覆ペレットに基づいて) で15% 重量増加に被覆した。被覆
ペレットは下記の表に記載した。
【表43】
【0252】
実施例 8 における被覆塩酸ナルトレキソン製剤を下記を用いて調製した:
工程
1. エチルセルロースコーティングの分散: Surelease を合計 20% (w/w) 固体分散を達成
するのに十分な量の水と混合する。
2. メタクリルコーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して
15 分間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の
水に分散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
3. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を達成する。
4. エチルセルロースコーティング: 以下のパラメータ指針を用いて Surelease 分散液を
700 g スケールでナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 9.0 〜 9.2 m/秒
* 吸気温度: 50 ℃
* 分散液噴霧速度:10 g/分
5. メタクリルコーティングコーティング: 以下のパラメータ指針を用いて、Sureleaseコ
ーティングの完了時に、Eudragit 分散液をナルトレキソンのペレットに流動床プロセッ
サ (GPCG-1) を用いて噴霧する。
* エアスピード: 9.0 m/秒 .
* 吸気温度: 35 ℃
* 分散液噴霧速度:10.7 g/分
6. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 750 〜 760 CPM
* 吸気温度: 50 ℃
* 分散液噴霧速度: 9.2 g/分
7. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
8. 硬化工程: スクリーニングしたペレットを 45℃ のオーブンに配置し、一部は 24 時
間硬化した後に除去し、残存材料は 48 時間硬化する。
【0253】
In vitro 溶解 (無傷ペレット)
実施例 8 に従って調製した、疎水性コーティング(エチルセルロース及びメタクリル共
重合体コーティング)で被覆した製剤を以下の溶解方法で検査を行い、以下の表 8C に記
載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表44】
【0254】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 8 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。溶解結果は
下記の表 8D に記載した。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表45】
【0255】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比: 42.9
【0256】
3つの 25% 被覆ペレットの比較
実施例 4、7、及び 8 では、ナルトレキソン MEM ペレットを、様々なコーティング材
料または順序を用いて合計 25% のコーティングで被覆した。粉砕/無傷比を次の通りに比
較検討した。
【表46】
【0257】
粉砕 及び無傷 MEM ペレットの in-vitro 溶解データに基づくと、25% の Eudragit RS
コーティングは、組み合わせコーティングと比べてわずかに優れていると思われる。
【実施例9】
【0258】
放出制御塩酸オキシコドンの10 mg 錠剤は、次の通りのこの机上の実施例で調製するこ
とができる。 有機製造の塩酸オキシコドン (10 mg/錠剤) 及び噴霧乾燥ラクトース (71.
25 mg/錠剤) を適当なサイズのミキサーに移動し、およそ 6 分間混合する。Eudragit(
登録商標)RS PM 粉末 (6 mg/錠剤) をエタノールに分散する。 粉末を混合中に、粉末を
分散により顆粒化し、湿性の顆粒物質が得られるまで混合を続ける。追加エタノールを、
造粒エンドポイントに到達するために必要ならば添加する。造粒物を流動床の乾燥機に移
動し、30℃ で乾燥し、次いで 12-メッシュスクリーンを通す。残存 Eudragit(登録商標
)RS PM (9 mg/錠剤) を 90 部のエタノールと 10 部の精製水の溶剤に分散し; 顆粒にお
ける流動床の顆粒機/乾燥機に 30℃ で噴霧する。次には、顆粒を 12-メッシュスクリー
ンに通す。ステアリルアルコール (25 mg/錠剤) をおよそ 60 〜 70℃ で溶解する。温か
い顆粒をミキサーへ戻す。混合中、溶解したステアリルアルコールを添加する。被覆した
顆粒をミキサーから取り除き、冷却する。その後、該顆粒を 12-メッシュスクリーンに通
す。次には、顆粒を実施例 5 のナルトレキソン粒子及び薬剤的に望ましい錠剤賦形剤、
たとえば、滑石及びステアリン酸マグネシウムと好適なブレンダーでと混合し、錠剤に圧
縮する。
【実施例10】
【0259】
疼痛治療の方法
本発明に係る経口剤型を投与すると、患者に疼痛軽減を提供することができる。経口剤
型には、オピオイド作動薬及び実質的に放出不可能なオピオイド拮抗薬の経口有効量を含
有することができる。拮抗薬含有粒子のコーティングは、無傷拮抗薬含有粒子から拮抗薬
が漏出することを有利に低下する役割を果たす。
【0260】
経口剤型が経口で投与され、療法を必要とする患者の胃腸管に送達されると、正常消化
中にオピオイド作動薬は剤型から放出され患者に鎮痛を提供する。しかし、オピオイド拮
抗薬は、実質的に放出不可能になっているので、胃腸管を通過中には実質的に放出されな
い。好ましくは、拮抗薬の実質的に放出不可能な形態は、遅延性の結腸通過または胃酸欠
乏状態を管理するために用いられる緩下薬 (鉱物油) に対して耐性を持つ。経口剤型を改
変 (たとえば、機械的混合、加熱、または溶剤中溶出によって) することなく処方のとお
りに摂取する患者は、形剤摂取期間中にオピオイド作動薬の鎮痛有効性低下を結果として
もたらす量のオピオイド拮抗薬を吸収することはない。すなわち、オピオイド拮抗薬が無
傷剤型から放出され (経口で投与されるとき)、胃腸管から吸収され、患者の体内に蓄積
される量が、剤型に含まれているオピオイド作動薬の用量の鎮痛効力に実質的に影響また
は変化を及ぼすレベルに達することはない。
【実施例11】
【0261】
オピオイド作動薬の乱用を防止する方法
本発明に係る経口剤型を用いると、それに含まれるオピオイド作動薬の乱用を防止する
ことが可能である。本経口剤型はオピオイド拮抗薬と組み合わせたオピオイド作動薬を含
有する。オピオイド拮抗薬は、消化中に実質的に放出不可能な形態で存在する。このよう
に、意図されたように経口投与された経口剤型が 胃腸管に送達されると、形剤が改変さ
れていない限り、拮抗薬は実質的に GI システムに放出されない。ただし経口剤型が改変
される場合、たとえば機械混合 (たとえば、粉砕、せん断、細砕)、熱 (たとえば、45℃
of を超える温度、好ましくは45℃ 〜 50℃)、または剤型を溶剤に溶解 (加熱の有無にか
かわらず) することによって、オピオイド拮抗薬は放出されオピオイド効果を鈍化する。
このように、剤型が改変された際に、経口的、鼻腔内的、非経口的または舌下に投与され
ると、オピオイド作動薬の効果は、オピオイド拮抗薬によって少なくとも部分的に遮断さ
れる。
【実施例12】
【0262】
塩酸ナルトレキソンのペレットを有する塩酸ヒドロモルホンの放出制御カプセルは、次
の通りのこの机上の実施例で調製することができる。 製剤は下記の 12A 表に記載した:
【表47】
実施例 5 のカプセルを以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを衝撃式製粉機にかける。
2. ブレンド工程: 塩酸ヒドロモルホン、Eudragit、エチルセルロース及び製粉したステ
アリルアルコールをツインシェルブレンダー内で混合する。
3. 押出成型工程: ブレンドした材料を2軸押出機に連続供給し、結果として生じるスト
ランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いてペレットに切断する。
6. スクリーニング工程: ペレットをスクリーニングし、所望のふるい部分に回収する。
7. フィルムコーティング工程: Opadry ピンク の水分散体を流動床のオピオイドのペレ
ットに噴霧する。
8. カプセル化工程: 126 mg の被覆し押出成形した塩酸ヒドロモルホンのペレット及び 1
49.7 mg の塩酸ナルトレキソンのペレット(実施例 5 から) をハードゼラチンカプセルに
充填する。
【0263】
当業者であれば本発明の変形形態は明らかであり、特許請求の範囲を逸脱しないものと
する。
【実施例13】
【0264】
実施例 13a
実施例 13a では、実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンの
ペレットをさらに疎水性コーティングで被覆した。ペレットを疎水性コーティングで25%
重量増加に被覆した(Eudragit RS 30D に基づいて)。被覆ペレットは下記の表に記載した
。
【表48】
実施例 13A のペレット調製において利用した工程は以下の通りである。
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 40 ℃
* 分散液噴霧速度: 75 〜 90 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 50 〜 55℃ .
* 分散液噴霧速度: 60 〜 70 g/分
5. スクリーニング: 14 US メッシュスクリーン及び26 US メッシュスクリーンを用いて
振動分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残
留した材料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 164.86 mg でハードゼラチ
ンカプセルに充填する。
【0265】
In vitro 溶解 (無傷ペレット)
実施例 13a において疎水性コーティングで被覆した製剤を、バルクのペレット及びカ
プセル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表
13B に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表49】
【0266】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 13a に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の
溶解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキ
ソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表50】
【0267】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 27.0/0.5 = 54
【0268】
実施例 13b
実施例 13b では、実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンの
ペレットをさらに疎水性コーティングで被覆した。ペレットを疎水性コーティングで30%
重量増加に被覆した (Surelease E-7-10901 に基づいて)。
被覆ペレットは下記の表に記載した。
【表51】
実施例 13A のペレット調製において利用した工程は以下の通りである。
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 400 〜 450 CFM .
* 吸気温度: 40 ℃
* 分散液噴霧速度: 75 〜 90 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 50 〜 55℃ .
* 分散液噴霧速度: 60 〜 70 g/分
5. スクリーニング: 14 US メッシュスクリーン及び26 US メッシュスクリーンを用いて
振動分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残
留した材料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 164.86 mg でハードゼラチ
ンカプセルに充填する。
【0269】
In vitro 溶解 (無傷ペレット)
実施例 13b において疎水性コーティングで被覆した製剤を、バルクのペレット及びカ
プセル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表
13E に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表52】
【0270】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 13b に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の
溶解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキ
ソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
5. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
6. サンプリング時間: 45 分間
7. 培地: 700 ml の SGF
8. 分析方法: 高速液体クロマトグラフィー
結果:
【表53】
【0271】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 26.0/l .6 = 16.3
【0272】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
実施例 13a 及び 13b の上記工程を用いて製造したカプセルを2つの個別の臨床研究に
用いて、異なる条件下の MEM 製剤の薬物動態学/バイオアベイラビリティを決定し、即放
性ナルトレキソン錠剤の薬物動態学/バイオアベイラビリティと比較した。ヒト被検体に
無傷塩酸ナルトレキソン MEM カプセル (1 カプセルまたは 5 カプセル、絶食); 粉砕し
た塩酸ナルトレキソン MEM (1 カプセルの含有量、細砕、絶食); 即放性塩酸ナルトレキ
ソン剤型、錠剤、絶食; または 1 MEM カプセル、無傷、給餌状態のいずれかを投与した
。これらの諸研究は、非盲検、単回投与、5 方向、クロスオーバー研究で健常者において
行った治療法は次の通りに設計した:
A. 1x8 mg のナルトレキソン MEM カプセル、無傷、絶食状態
B. 1x8 mg のナルトレキソン MEM カプセル、カプセル内容物粉砕、絶食状態
C. 1x8 mg のナルトレキソン MEM カプセル、無傷、給餌状態
D.5x8 mg (40 mg) ナルトレキソン MEM カプセル無傷、絶食状態
E. 1 x 1 mg のナルトレキソン即放性錠剤、絶食状態
【0273】
予備的な血漿濃度は、無傷状態で採取すると ナルトレキソン MEM ペレットからナルト
レキソンの無視できる量の放出があることを示す。時間曲線データと対比したナルトレキ
ソン濃度 (pg/ml) は、図3 及び 4 に示してある。粉砕/細砕したナルトレキソンのペレ
ットを絶食状態で経口摂取させたとき、ナルトレキソン血漿レベルが大幅に増加した。平
均 Cmax 粉砕 MEM/無傷 MEM カプセル比は 25% の Eudragit コーティング及び 30% Sure
lease コーティングは、それぞれ 187.91 及び 71.98 であった。 同様に、平均 AUCt 粉
砕 MEM/無傷 MEM カプセル比は 25% の Eudragit コーティング及び 30% Surelease コー
ティングは、それぞれ 66.07 及び 39.27 であった。
実施例 l3a 及び 13b の被覆 MEM の in-vitro 及び In-Vivo の比較は下記の表 13G
に示した。
【表54】
【表55】
【0274】
本明細書中で開示した本発明は特有の実施形態及びアプリケーションを用いて説明した
が、当業者であれば幾多の変更形態及び改変は本発明の精神及び範囲から逸脱することな
く行い得る。そのような変更形態は特許請求の範囲内であるものと見なされる。
【図面の簡単な説明】
【0275】
【図1】無傷の塩酸ナルトレキソン MEM (全体)、粉砕した塩酸ナルトレキソン MEM (細砕) 及び実施例 1 の即放性塩酸ナルトレキソン錠剤 (IR NTX) 剤型に対する血漿濃度時間のグラフ表示である。
【0276】
【図2】実施例 5 の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【0277】
【図3】実施例 13A の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【0278】
【図4】実施例 13B の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【背景技術】
【0001】
医薬製品は時折乱用の対象となる。たとえば、オピオイド作動薬の特定用量は、非経口
的に投与すると、同じ用量を経口で投与した場合と比較してさらに強力になる場合がある
。一部の製剤は、改変する(tampered)ことによって該製剤に含まれるオピオイド作動薬
を違法な使用に供することができる。放出制御型オピオイド作動薬は時折薬物乱用者によ
って細砕され、それに含まれるオピオイドが経口または非経口投与直後に即時放出に供さ
れる。
【0002】
オピオイド拮抗薬がオピオイド作動薬の非経口的な乱用を抑止するために、特定のオピ
オイド作動薬と組み合わされている。 従来の技術では、即放性ペンタゾシンとナロキソ
ンの併用が、米国で Sanofi-Winthrop 社から Talwin(登録商標)Nx (タルウィン) とし
て市販されている錠剤で利用されている。Talwin(登録商標)Nx (タルウィン) には、50
mg 塩基(base)に相当する即放性塩酸ペンタゾシン及び 0.5 mg 塩基(base)に相当す
る塩酸ナロキソンが含まれる。チリジン (50 mg) 及びナロキソン (4 mg) を含有する固
定併用療法(fixed combination therapy)が 1978 年以来、疼痛管理のためにドイツで
提供されている (Valoron(登録商標)N、Goedecke)。ブプレノルフィンとナロキソンの
固定併用は疼痛治療のためにニュージーランド (Temgesic(登録商標)Nx、Reckitt & Co
lman) で 1991 年に導入された。
【0003】
米 Purdue Pharma 社は現在徐放性オキシコドンを 10、20、40 及び 80 mg の塩酸オキ
シコドンを含む剤型でオキシコンチンの商品名で市販している。
【0004】
米国特許番号第 5,266,331 号; 第 5,508,042 号; 第 5,549,912 及び第 5,656,295 号
により徐放性オキシコドン製剤が開示されている。
【0005】
米国特許番号第 5,472,943 号(Crain らによる)では、作動薬をオピオイド拮抗薬と共
に投与することによって二峰性に作用するオピオイド作動薬の鎮痛効力を高める方法が記
述されている。
【0006】
米国特許特許番号第 6,277,384 号; 第 6,475,494 号; 第 及び第 6,375,957 号(Kaiko
らによる); 及び第 6,228,863 号 (Colucci らによる)では、オピオイド鎮痛剤型に関連
する乱用の可能性を少なくすることを狙いとしている。
【0007】
PCT 公開番号 WO 01/58451 の発明の名称「不正使用防止経口オピオイド作動薬製剤 (T
amper Resistant Oral Opioid Agonist Formulations)」では、オピオイド作動薬剤型内
の隔離したオピオイド拮抗薬を抱合することによってオピオイド鎮痛剤型に関連する乱用
の可能性を少なくすることをその狙いとする。
【0008】
乱用の可能性が低いオピオイド作動薬を含有する経口剤型技術の必要性が継続して存在
する。
【0009】
前述を含む本明細書中で引用したすべての参照を引用することを以って本明細書の一部
となす。
【発明の開示】
【発明が解決しようとする課題】
【0010】
本発明の目的と要旨
本発明の目的は、オピオイド拮抗薬の放出を実質的に防止するオピオイド拮抗薬を含有
する経口剤型の提供することである。
【課題を解決するための手段】
【0011】
本発明の特定の実施形態の目的は、オピオイド作動薬の乱用の可能性を少なくすること
に有用なオピオイド拮抗薬の処方を含有する経口剤型提供することである。
【0012】
本発明の特定の実施形態の目的は、オピオイド作動薬の鎮痛効果に影響を及ぼすこと、
または退薬を誘発する可能性を受けることなしに、オピオイド作動薬の乱用の可能性を少
なくするために有用なオピオイド拮抗薬の処方を含有する経口剤型を提供することである
。
【0013】
本発明の特定の実施形態の目的は、剤型が無傷で経口投与されるときにオピオイド作動
薬の鎮痛効力を変化させないか、または実質的に変化させないオピオイド作動薬の有効量
及びオピオイド拮抗薬の一回分量を含む経口剤型を提供することである。但し、剤型が改
変されていない場合、オピオイド拮抗薬は大幅に放出され、オピオイド作動薬の効果を妨
害することによって乱用を防止することができる。
【0014】
本発明の特定の実施形態の目的は、剤型が改変された際に作動薬のすべてが即時放出の
ために遊離されないような放出制御製剤内にオピオイド作動薬の有効量を含む経口剤型を
提供することである。
【0015】
本発明の特定の実施形態の目的は、作動薬粒子と拮抗薬粒子が、たとえば、外観、質感
、臭い、味、硬度、形状、サイズおよび/またはその組み合わせにおいて類似するか、ま
たはこれら特質の1つ以上によって互いを実質的に識別できない、オピオイド作動薬粒子
及び隔離した拮抗薬の粒子を含む経口剤型を提供することである。
【0016】
本発明の特定の実施形態の目的は、隔離された、たとえば、形剤の用量が無傷で投与さ
れると生物によって利用可能でないが、剤型が改変される (たとえば、オピオイド作動薬
の用量の誤用が試みられる) と生物によって利用可能な、オピオイド拮抗薬を剤型内に含
むことによって、オピオイド作動薬を含有する経口剤型の乱用を防止するための方法を提
供することである。
【0017】
本発明の特定の実施形態の目的は、経口、非経口、鼻腔内あるいは舌下経路による誤用
の可能性を少なくしたオピオイド作動薬の経口剤型を投与することを含有するヒト患者に
おける疼痛治療の方法を提供することである。
【0018】
上記目的は、数ある中で、複数の粒子、たとえば押出成形した粒子、マトリックス内に
分散したオピオイド拮抗薬を含有する粒子のそれぞれ; 及び剤型内のオピオイド拮抗薬を
環境流体に晒すと、マトリックス及び層が剤型内のオピオイド拮抗薬を隔離する働きをす
る (すなわち、放出または実質的な放出を防止する) 粒子それぞれの周囲に配置した層を
含有する剤型を部分的にその狙いとする本発明によって達成する。
【0019】
特定の実施形態では、マトリックスは疎水性物質を含有する。特定の他の実施形態では
、層は疎水性物質を含有する。
【0020】
特定の実施形態では、本発明は下記を含有する経口剤型医薬品をその狙いとする。a)
粒子、たとえば、第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した
粒子; 及び b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水
性物質が、たとえば、粒子の重量の約 2% 〜約 30%を含有する層。あるいは、第2疎水性
物質が粒子の重量の約 5% 〜約 25%、約 10% 〜約 20%、約 10% 〜約 25%、約 15% 〜約
25%、約 22% 〜約 28%、または約 5% 〜約 15%を含有する。
【0021】
特定の実施形態では、本発明は下記を含有する経口剤型医薬品をその狙いとする。 a)
複数の粒子、たとえば、第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成
形した粒子、及び粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、
該第2疎水性物質が、たとえば、約 2% 〜約 30% 粒子重量を含有する層; b) 第3疎水性
物質内に分散したオピオイド作動薬を含有する複数の粒子; 及び c) 複数個のオピオイド
作動薬粒子及び複数個のオピオイド受容体拮抗薬の粒子を含むカプセル。あるいは、第2
疎水性物質が粒子の重量の約 5% 〜約 25%、約 10% 〜約 20%、約 10% 〜約 25%、約 15%
〜約 25%、約 22% 〜約 28%、または約 5% 〜約 15%を含有する。
【0022】
特定の実施形態では、本発明では、第1マトリックス及びオピオイド作動薬を含有する
複数の粒子を含有する剤型; 及び第2マトリックス及びオピオイド拮抗薬を含有する複数
の粒子 (たとえば、押出成形した粒子)、及びオピオイド拮抗薬を環境流体に晒すと、第
2マトリックス及び層がオピオイド拮抗薬を隔離する働きをするオピオイド拮抗薬を含有
する粒子それぞれの周囲に配置した層をさらにその狙いとする。
【0023】
特定の実施形態では、オピオイド拮抗薬の粒子のマトリックスは疎水性物質を含有する
。特定の他の実施形態では、オピオイド拮抗薬の粒子上の層は疎水性物質を含有する。特
定の実施形態では、マトリックス及び層の両方が疎水性物質を含有する。
【0024】
特定の実施形態では、層を、オピオイド作動薬粒子の外観をオピオイド拮抗薬に類似さ
せるか、またはオピオイド拮抗薬と実質的に識別できないようにするための粒子を含み、
それによって乱用者が粒子を含む作動薬から粒子を含む拮抗薬を物理的に分離する能力を
低下させる粒子を含むオピオイド作動薬の周囲に配置する。作動薬層は、放出制御を提供
するか、または放出制御を増強するために機能層であり得る。あるいは、作動薬層は、た
とえば、放出制御能力を提供しないフィルムコートのような非機能層でもあり得る。
【0025】
特定の実施形態では、本発明は下記を含有する経口剤型をその狙いとする。
(i) 放出可能型の複数の粒子を含有するオピオイド作動薬及び (ii) 複数の粒子、たとえ
ば、疎水性物質を含有するマトリックスを含有する押出成形した粒子、マトリックス内に
分散したオピオイド拮抗薬、及び剤型が無傷で患者に投与されるときにマトリックス及び
層が拮抗薬の放出を防止または実質的に防止するような粒子の周囲に配置した疎水性物質
を含有する層。
【0026】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の 人工胃液 (SGF) 中で 1 時間の剤型の溶出に基づいた、改変後に剤型から放出さ
れる拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率は約 20:1 以上; 約 50:
1 以上; 約 100:1 以上; 約 150:1 以上; または約 1000:1 以上である。
【0027】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の 人工腸液 (SIF ) に切り替えた条件で、
剤型の 2 時間、4 時間、12 時間、24 時間あるいは 36 時間での溶出に基づいた、改変
後に剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率は約
20:1 以上; 約 50:1 以上; 約 100:1 以上; 約 150:1 以上; または約 1000:1 以上であ
る。
【0028】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で剤型の 1 時間での溶出に基づいた無傷剤型から放出される拮抗薬の重
量パーセントは 1.0% 重量未満; 0.5% 重量未満; 0.2% 重量未満; または0.1 重量% 未満
である。
【0029】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 2 時間
での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 2.0% 未満; 1.0%
未満; 0.5% 未満; または0.25% 未満である。
【0030】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 4 時間
での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 2.2% 未満; 1.5%
未満; 1.0% 未満; または 0.75% 未満である。
【0031】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 12 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 3 .0% 未満; 1
.8% 未満; 1.25%; または0.3% 未満である。
【0032】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 24 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 4.8% 未満; 2.
5% 未満; 1.8% 未満; または0.4% 未満である。
【0033】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた条件で、剤型の 36 時
間での溶出に基づいた無傷剤型から放出される拮抗薬の重量パーセントは 未満7.0% 未満
; 6.5% 未満; 3.0%; または未満1.5%.
【0034】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、1.0% 以下の拮抗薬を 1 時間で、2.0% 以下の拮抗薬を 2 時間で、
2.2% 以下の拮抗薬を 4 時間で、3.0% 以下の拮抗薬を 12 時間で、4.8% 以下の拮抗薬を
24 時間で、及び 7.0% 以下の拮抗薬を 36 時間でそれぞれ放出する。
【0035】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、0.5% 以下の拮抗薬を 1 時間で、1.0% 以下の拮抗薬を 2 時間で、
1.5% 以下の拮抗薬を 4 時間で、1.8% 以下の拮抗薬を 12 時間で、2.5% 以下の拮抗薬 2
4 時間で及び 6.5% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0036】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は 0.2% 以下の拮抗薬を 1 時間で、0.5% 以下の拮抗薬を 2 時間で、1
.0% 以下の拮抗薬を 4 時間で、1.25% 以下の拮抗薬を 12 時間で、1.8% 以下の拮抗薬を
24 時間で、及び 3.0% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0037】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で 1 時間、継いで、900 ml の SIF に切り替えた場合の形剤の溶出に基
づいて、無傷剤型は、0.1% 以下の拮抗薬を 1 時間で、0.25% 以下の拮抗薬を 2 時間で
、0.75% 以下の拮抗薬を 4 時間で、0.3% 以下の拮抗薬を 12 時間で、0.4% 以下の拮抗
薬を 24 時間で、及び 1.5% 以下の拮抗薬 36 時間でそれぞれ放出する。
【0038】
本発明の特定の実施形態では、50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 70
0 ml の SGF 中で剤型の 1 時間での溶出に基づいた、改変後剤型から放出される作動薬
の重量百分率は、50% 未満、40% 未満、または 35% 未満である。
【0039】
本発明の特定の実施形態では、改変剤型(tampered dosage form)を患者集団に単回投
与した後に得られる拮抗薬の平均 Cmax の無傷剤型を患者集団に単回投与した後に得られ
る拮抗薬の平均 Cmax に対する比率は、約 20:1 以上; 約 50:1 以上; 約 75:1 以上; 約
100:1 以上; 約 125:1 以上; 約 150:1 以上; または約 1000:1 以上である。これらの
値は好ましくは絶食状態で得た値である。
【0040】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 Cmax の無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対
する比率は約 20:1 〜約 1000:1; 約 20:1 〜約 150:1; 約 20:1 〜約 125:1; 約 20:1
〜約 100:1; 約 20:1 〜約 75:1; または約 20:1 〜約 50:1 である。他の実施形態では
、その範囲は約 50:1 〜約 1000:1; 約 75:1 〜約 1000:1; 約 100:1 〜約 1000:1; 約 1
25:1 〜約 1000:1; または約 150:1 〜約 1000:1 である。これらの値は好ましくは絶食
状態で得た値である。
【0041】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC に対
する比率は、約 5:1 以上; 約 25:1 以上; 約 75:1 以上; 約 100:1 以上; 約 150:1 以
上; 約 200:1 以上または約 250:1 以上である。これらの値は好ましくは絶食状態で得た
値である。
【0042】
本発明の特定の実施形態では、改変剤型を患者集団に単回投与した後に得られる拮抗薬
の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC に対
する比率は約 5:1 〜約 250:1; 約 5:1 〜約 200:1; 約 5:1 〜約 150:1; 約 5:1to 約 1
00:1;約 5:1 〜約 75:1;または約 5:1to 約 25:1 である。他の実施形態では、その範囲
は約 25:1 〜約 250:1; 約 75:1 〜約 250:1; 約 100:1 〜約 250:1; 約 150: I 〜約 10
00:1; または約 200:1 〜約 250:1 である。これらの値は好ましくは絶食状態で得た値で
ある。
【0043】
特定の実施形態では、本発明は、剤型が改変の対象となり、経口、鼻腔内、非経口ある
いは舌下に投与される場合、オピオイド作動薬の効果がオピオイド受容体拮抗薬の放出に
よって大幅または完全に遮断される本明細書で開示した剤型を利用して、オピオイド作動
薬の乱用を防止する方法をさらにその狙いとする。
【0044】
特定の実施形態では、本発明は、必要とする患者に対する鎮痛薬を含有する、本明細書
で開示した本発明の実施形態の任意の医療製品を、たとえば経口で投与することによる疼
痛治療の方法をさらにその狙いとする。
【0045】
オピオイド作動薬を含有する複数粒子と、オピオイド拮抗薬を含有する複数粒子が類似
しているか、または実質的に互いを識別できない実施形態では、粒子の類似性または実質
的な識別不可能性は、(i) 機能層または非機能層、(ii) 層化する必要のない類似の調製
方法、(iii) 類似、または実質的に識別できない最終製品を結果としてもたら異なる調製
方法、(iv) 異なる最終製品を結果としてもたらし、次いで追加処理工程 (たとえば層化)
の対象となり、類似性または実質的な識別不可能性を提供する異なる調製方法、(v) ま
たは所望の特質 (たとえば、外観、質感、臭い、味、硬度、形状、サイズなど) を結果と
してもたらす任意の他方法のためであり得る。
【0046】
特定の好ましい実施形態では、粒子の平均直径は約 0.1 〜約 12 mm; 約 0.1 〜約 2.5
mm; 約 0.2 〜約 6 mm; 約 0.5 〜約 3 mm; 約 0.5 mm 〜約 2 mm; または約 1 mm 〜約
2 mm である。
【0047】
無傷剤型の投与直後に放出されるオピオイド拮抗薬の量は、存在する場合、剤型が引き
続き鎮痛効果を持ち続けられるような量である。
【0048】
本発明の特定の実施形態では、オピオイド作動薬の隔離したオピオイド拮抗薬に対する
比率は重量で約 1:1 〜約 50:1; 好ましくは重量で約 1: I 〜約 20:1; または重量で約
15:1 〜約 30:1 である。オピオイド作動薬の、活性成分の重量を指すオピオイド拮抗薬
に対する重量比。たとえば、オピオイド拮抗薬の重量には、オピオイド拮抗薬を一緒にな
って隔離する働きをする層及びマトリックスの重量が除かれている。特定の好ましい実施
形態では、作動薬の隔離した拮抗薬に対する重量比は、重量で約 1:1 〜約 10:1 である
。
【0049】
オピオイド拮抗薬の実質的に放出不可能な形態と組み合わせたオピオイド作動薬を含む
本発明の経口剤型には、限定するものではないが、錠剤及びカプセルが含まれる。本発明
の経口剤型には、当業者であれば公知の任意の所望の医薬品賦形剤を含むことができる。
経口剤型は、オピオイド作動薬の即放性および/またはオピオイド作動薬の放出制御を提
供することができる。
【0050】
本発明の乱用防止剤型は、オピオイド作動薬の一回分量を含有し、長時間にわたる放出
を意図した放出制御剤型と関連して有用である。薬物乱用者は、そのような放出制御型の
製品を利用し、粉砕、細砕、抽出あるいは別の方法で、剤型の全含有量を放出させ即時吸
収する目的で製品を損傷する。本発明に係る剤型を改変すると、結果としてオピオイド拮
抗薬も同時に吸収に提供されるようになるので、本発明はそのような乱用を妨げる手段を
提供する。
【0051】
また、本発明は、本明細書で開示した剤型を用いた疼痛治療の方法もその狙いとする。
本方法には、開放可能な形態でオピオイド作動薬及び本明細書で開示したとおりの隔離し
た拮抗薬を含む経口剤型を提供すること、及び無傷経口剤型をそのような治療が必要な哺
乳動物 (たとえば、ヒト) に経口で投与することを含有することができる。
【0052】
特定の実施形態では、本発明は、本明細書で開示した経口剤型の調製方法をさらにその
狙いとする。特定の実施形態では、本発明は、押出成形により疎水性物質を含有するマト
リックス内に分散したオピオイド拮抗薬を含有する複数の粒子を調製すること; 及び剤型
が無傷で投与されるときにマトリックス及び層が拮抗薬を隔離する働きをする疎水性物質
を含有する層を押出成形した粒子の周辺に配置することを含有する経口剤型の調製方法を
含有する。本方法には、隔離した拮抗薬を隔離した拮抗薬の完全性を維持するような方法
でオピオイド作動薬の放出可能形態 (たとえば放出制御) と組み合わせることをさらに含
有することができる。本発明のすべての実施形態では、マトリックスの疎水性物質は層の
疎水性物質と同じであっても同じでなくともよい。
【0053】
本発明の好ましい実施形態では完全にオピオイド拮抗薬の放出を防止する形態でオピオ
イド拮抗薬を含有するが、本発明は拮抗薬も実質的に放出不可能な形態で含まれる。用語
「実質的に放出されない」及び「実質的に放出不可能」は、剤型が意図されたように経口
でヒト投与されるとき、放出量が鎮痛効力に影響を及ぼさない、または実質的に影響を及
ぼさない限りにおいて、少量で放出され得る拮抗薬を参照する。
【0054】
隔離された拮抗薬の粒子では、本発明に従っていくつかの可能性が存在する。第1の可
能性は、マトリックスには層なしで拮抗薬を隔離する能力がいくらかあり、層がその隔離
機能を高めること。第2の可能性は、層にはマトリックスなしで拮抗薬を隔離する能力が
いくらかあり、マトリックスがその隔離機能を高めること。第3の可能性は、マトリック
スには層なしで拮抗薬を隔離する能力がなく、層にはマトリックスなしで拮抗薬を隔離す
る能力がなく、マトリックスと層共には拮抗薬を隔離する能力を持つ(たとえば、マトリ
ックス及び層のそれぞれには、拮抗薬の放出制御を個々に提供する能力があるが、同一剤
型内のマトリックスと層共は拮抗薬を隔離する) ことである。第1及び第2の可能性では
、マトリックスおよび/または層は、拮抗薬の放出制御を個々に提供する能力を持つこと
によって隔離機能を高める。
【0055】
本発明の特定の好ましい実施形態では、拮抗薬の実質的に放出不可能な形態は、遅延性
の結腸通過を管理するために用いられる緩下薬 (たとえば鉱物油) に対して耐性を示し、
そして胃酸欠乏状態に対しても耐性を示す。
【0056】
本発明の好ましい実施形態では、オピオイド拮抗薬の実質的に放出不可能な形態は、経
口剤型の機械的、熱的あるいは化学的改変、たとえば、粉砕、せん断、細砕、咀嚼あるい
は加熱 (たとえば約 45℃ を超える) と組み合わせた溶剤溶出による改変に対して無防備
である。改変がなされると、オピオイド拮抗薬の実質的に放出不可能な形態の完全性が損
なわれ、オピオイド拮抗薬が即時放出に供され、それによって少なくとも部分的に及び好
ましくは実質的にオピオイド作動薬の効果が遮断される。このように、オピオイド作動薬
及びオピオイド拮抗薬を含有する経口剤型が、咀嚼、粉砕、細砕または溶剤溶解及び加熱
され、及び経口、鼻腔内、非経口あるいは舌下に投与されると、オピオイドの鎮痛あるい
は陶酔効果は低下または除去される。
【0057】
本発明は、経口剤型内のオピオイド作動薬の乱用の可能性を少なくする方法をさらにそ
の狙いとする。 本方法は、本明細書に記載したように経口剤型内のオピオイド作動薬を
提供することを含有する。
【0058】
用語「鎮痛有効性」は、本発明では、ヒト患者による決定を基にした副作用の許容レベ
ルと共に、疼痛の満足できる軽減または除去として定義する。
【0059】
句「オピオイド作動薬の鎮痛効果を実質的に遮断していない」は、本発明の目的上、鎮
痛を提供する上で治療上の効果が弱い剤型を与えること応じて、オピオイド拮抗薬が十分
な度合いでオピオイド作動薬の効果を遮断しないことを意味する。
【0060】
用語「改変」は、無傷剤型の特質を変える機械的、急速な放出または即時放出のために
少なくともオピオイド作動薬の部分を遊離するため、またはオピオイド作動薬を不正投与
に利用できるようにするための (たとえば非経口的な投与)、熱的あるいは化学的手段に
よる任意の操作を意味する。無傷剤型を改変する方法は、たとえば、粉砕、せん断、細砕
、咀嚼、溶剤溶出、加熱 (たとえば約 45℃ を超える温度で)、またはこの目的を達成す
るそれらの任意の組み合わせであり得る。
【0061】
特定の実施形態では、剤型の改変を乳鉢と乳棒を用いて粉末に粉砕して行う。他の実施
形態では、改変はをクリューキャップ付き錠剤粉砕器によるか、または2つのステンレス
製大さじを用いることによって行う。
【0062】
特定の実施形態では、乳鉢と乳棒を利用して粉砕を実施し、咀嚼をシミュレートする。
たとえば、乳棒の 3 ストロークは軽度咀嚼をシミュレートし、乳棒のストロークは中度
咀嚼を シミュレートし、及び乳棒のストロークは十分な咀嚼をシミュレートする。特定
の実施形態では、乳鉢と乳棒を用いて、たとえば、乳棒の 24、50、500 または 600 スト
ロークで剤型を粉砕して粉末する。
【0063】
特定の実施形態では、スクリューキャップ付き錠剤粉砕器を利用し、剤型を粉砕器に配
置し、スクリューキャップを回転して剤型を粉砕する。次いで、キャップをゆるめ、粉砕
器を硬面上で軽くたたき、粉砕をさらに 2 回繰り返す。
【0064】
特定の実施形態では、ステンレス製大さじを利用し、剤型を第1スプーンに載置し、第
2スプーンを第1スプーン上に配置し、手圧力を用いて剤型をスプーン間で粉砕する。
【0065】
用語「層は実質的に拮抗薬を欠いている」は、原則的として層にはオピオイド拮抗薬が
含まれていないが、押出成形した成分から移動する少量を含む場合があることを意味する
。
【0066】
用語「少なくとも部分的にオピオイド効果を遮断する」は、本発明の目的上、オピオイ
ド拮抗薬は少なくとも実質的にオピオイド作動薬の陶酔効果を遮断することを意味する。
【0067】
用語「放出制御」は、オピオイド作動薬に適用する場合、本発明の目的上、通常 (すな
わち即放性) 製剤の単回投与に比べてより長い作用期間を提供する放出速度で薬物が製剤
から放出されることを意味する。たとえば、典型的な即放性経口製剤は薬物を、たとえば
、1 時間間隔にわたって放出するが、放出制御型の経口製剤は薬物を、たとえば、4 〜 2
4 時間間隔にわたって放出する。
【0068】
本発明の目的上、用語「オピオイド作動薬」は用語「オピオイド」または「オピオイド
鎮痛薬」と互換可能であるものとし、1つの作動薬または1つ以上のオピオイド作動薬の
組み合わせを含み、さらに、オピオイド塩基、混合した作動薬と拮抗薬、部分的作動薬、
その薬剤的に許容できる塩類、その立体異性体、そのエーテル、そのエステル、及び前述
した物質のいずれかからなる混合物の使用が含まれる
【0069】
本発明の目的上、用語「オピオイド拮抗薬」には、1つの拮抗薬及び1つ以上の拮抗薬
の組み合わせを含めるものとし、さらに、塩基、その薬剤的に許容できる塩類、その立体
異性体、そのエーテル、そのエステル、及び前述した物質のいずれかからなる混合物の使
用も含まれる。
【0070】
本発明の明細書では、開示したオピオイド作動薬及び拮抗薬のすべての薬剤的に許容で
きる塩類の使用も包含することを意図する。薬剤的に許容できる塩類には、限定するもの
ではないが、ナトリウム塩類、カリウム塩類、セシウム (secium) 塩類などのような金属
塩類; カルシウム塩類、マグネシウム塩類などのようなアルカリ土類金属; トリエチルア
ミン塩類、ピリジン塩類、ピコリン塩類、エタノールアミン塩類、トリエタノールアミン
塩類、ジシクロヘキシルアミン塩類、N,N'-ジベンジルエチレンジアミン塩類などのよう
な有機アミン塩類; 塩酸塩、ヒドロブロミド、硫酸塩、リン酸塩などのような無機酸塩類
; ホルマート、アセテート、トリフルオロアセテート、マレアート、酒石酸ラートなどの
ような有機酸塩類; メタンスルホナート、ベンゼンスルホナート、p-トルエンスルホナー
トなどのようなスルホナート; アルギネート、アスパルギネート (asparginate)、グルタ
マートなどのようなアミノ酸塩類が含まれる。
【0071】
本発明に従って用いられる一部のオピオイド作動薬及び拮抗薬には、1つ以上の不斉中
心が含まれ、そして鏡像異性体、ジアステレオマー、または他の立体異性体を作り出す。
また、本発明は、そのようなすべての可能形態の使用、さらには、それらのラセミ形態及
び分解形態及びその混合物を包含することも意図する。本明細書に記載した化合物にオレ
フィン二重結合または他の幾何学的な非対称性の中心が含まれるときには、E 及び Z 幾
何異性体の両方を含むことを意図する。また、すべて互変異性体も本発明によって包含す
ることを意図する。
【0072】
用語「層」は、たとえば、コーティングとしても適用できる、粒子の周囲に配置した材
料を意味する(粒子自体及び、たとえば、シールコートのような1つ以上の任意中間層も
含み得る)。マトリックスの層化は、当分野で周知の工程、たとえば、噴霧コーティング
、浸漬またはエンロービングによって実施することができる。
【0073】
用語「の周囲に配置した」は、中間層または該物質と粒子間の層の有無にかかわらず、
粒子の周囲に配置した材料が少なくとも粒子の一部分を覆うことを意味する。特定の実施
形態では、粒子は材料によって完全に覆われる。
【0074】
本明細書中では、用語「立体異性体」は、空間における原子配向のみが異なる個々分子
のすべて異性体に対する一般用語であり、相互の鏡像ではない1つ以上のキラル中心を有
する化合物の鏡像異性体 及び異性体が含まれる (ジアステレオマー)。
【0075】
用語「キラル中心」は、4 つの異なる群が結合されている炭素原子を指す。
【0076】
用語「鏡像異性体」または「鏡像異性」は、その鏡像で重畳不可能であり、従って光学
的に活性である分子を指すものであり、ここでは、鏡像異性体が 偏光の平面を一方向に
回転し、その鏡像が偏光の平面を逆方向に回転するものとする。
【0077】
用語「ラセミ」は等量の鏡像異性体の混合物を指し、光学的に不活性である。
【0078】
用語「分解能」は、分子の2鏡像異性形態のうちの1つの分離または濃度または消耗を
指す。
【0079】
本発明の粒子の周囲に配置した疎水性物質に関する用語「粒子重量の X% 」または「X%
重量増加」は、疎水性物質が層状粒子合計の重量% ではなく、粒子の重量% として測定
されることを意味する。たとえば、100 mg の非層状粒子は、引き続いて層化して10% 重
量増加すると、層内に 10 mg の疎水性物質を持つ。
【0080】
用語「直径」は、粒子の断面直径を意味し、押出成形工程で用いるオリフィス直径によ
って大きく左右される。
【0081】
用語「長さ」は、押出成形した粒子の長さを意味し、押出成形したストランドの切断間
隔によって大きく左右される。
【0082】
用語「医薬製品」は、投与に好適な剤型または剤型の成分を意味する。
【発明を実施するための最良の形態】
【0083】
本発明は、隔離したオピオイド拮抗薬の粒子が、環境流体に晒されると拮抗薬が無傷形
態から「漏出」することをさらに低下する被覆剤を用いて、押出成形したオピオイド拮抗
薬の粒子にコーティングを施すことによって改善できるという観察に基づくものである。
本発明の効力によって、隔離した拮抗薬がオピオイド作動薬と結合すると、好ましくは無
視できるほどに僅かな量の拮抗薬 (すなわち、作動薬によって提供される鎮痛に影響を及
ぼさない量) が所定の使用条件下で放出される。所定の使用条件下で放出される拮抗薬の
量は、皆無であるか、または測定できないほどに少ないことが最も好ましい。
【0084】
特定の実施形態では、本発明には、剤型が改変されていない場合、オピオイド作動薬の
効果を少なくとも実質的に遮断するための量でオピオイド拮抗薬を含有する、複数の押出
成形し、隔離した粒子と共に、経口で治療学的に効率的な量のオピオイド作動薬を含有す
る複数の粒子を含有する経口剤型が含まれる。好ましくは、オピオイド拮抗薬を含有する
複数個の薬剤的に許容できる粒子、及びオピオイド作動薬を含有する複数個の薬剤的に許
容できる粒子は視覚的に類似し、最も好ましくは、それらが視覚的に識別できないものと
する。
【0085】
特定の実施形態では、オピオイド作動薬のオピオイド拮抗薬に対する比率を、経口剤型
が改変されてオピオイド拮抗薬を含有する粒子の完全性が損なわれた場合、放出される拮
抗薬の量が、被検体に経口、非経口、鼻腔内あるいは舌下に投与されたとき、オピオイド
作動薬の陶酔効果を実質的に低下または除去するように設定する。
【0086】
たとえば、本発明の特定の好ましい実施形態では、剤型が非経口的あるいは舌下に誤用
されると、オピオイド拮抗薬によってオピオイド作動薬の陶酔効果は実質的に低下または
除去される。特定の実施形態では、剤型が咀嚼、粉砕、または溶剤溶解及び加熱され、経
口、鼻腔内、非経口あるいは舌下に投与されるとき、オピオイドの鎮痛または陶酔効果は
オピオイド拮抗薬の放出に起因して実質的に低下または除去される。特定の実施形態では
、オピオイド薬物の効果はオピオイド拮抗薬によって少なくとも部分的に遮断される。特
定の他の実施形態では、オピオイド薬物の効果はオピオイド拮抗薬によって実質的に遮断
される。特定の他の実施形態では、オピオイド薬物の効果はオピオイド拮抗薬によって完
全に遮断される。
【0087】
本発明の無傷経口剤型は、意図されたように正しく投与されるとオピオイド拮抗薬を実
質的に放出しないので、経口投与時にオピオイド拮抗薬が GI システムに放出されるよう
に提供される場合、拮抗薬量の変動の範囲はより大きくなる。
【0088】
マトリックス及び層が拮抗薬を実質的に放出不可能にする隔離形態でのオピオイド拮抗
薬は、層が該粒子のそれぞれの周囲に配置された状態で、マトリックス内に分散したオピ
オイド拮抗薬を含有する複数の押出成形した粒子を含有する。一実施形態では、層は薬剤
的に許容できる疎水性物質を含有する。別の実施形態では、マトリックスは薬剤的に許容
できる疎水性物質を含有する。別の実施形態では、マトリックス及び層の両方が薬剤的に
許容できる疎水性物質を含有する。マトリックスの疎水性物質は層の疎水性物質と同じで
あってもよいし、異なるものであってもよい。疎水性物質は、好ましくは、拮抗薬が被覆
されたマトリックスから放出されないか、または実質的に放出されないような量であって
、したがって、GI システムを介した経口剤型の転移中に吸収されるように使用できない
か、または実質的に使用することができない。
【0089】
本発明の特定の好ましい実施形態では、オピオイド拮抗薬を溶解押出成形によってマト
リックス内に分散する。マトリックスが1つまたはそれ以上の薬剤的に許容できる疎水性
物質を含有する。
【0090】
本発明の特定の実施形態では、オピオイド作動薬を含む粒子は放出制御型の押出成形マ
トリックスの複数微粒子である。特定の実施形態では、放出制御型押出成形マトリックス
の複数微粒子がオピオイド作動薬を即時放出に供するように改変されると、作動薬の一部
分のみが即時放出のために遊離されることが見出されている。特定の実施形態では、50 r
pm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で押出成形された剤型
の 1 時間での溶出に基づいた、改変後剤型から放出される作動薬の重量百分率は 50% 未
満、40% 未満、または 35% 未満である。
【0091】
改変直後にオピオイド拮抗薬の一部分のみが即時放出のためにマトリックスの複数微粒
子から遊離させることができるので、改変直後に本発明の所定の目的に必要な量の拮抗薬
が遊離されることを確実にするため、拮抗薬を高添加することができる。たとえば、本発
明の実施形態で改変直後に拮抗薬の 50% が遊離される場合、もし改変直後に 2 mg の拮
抗薬放出を遊離させる必要があれば、剤型は 4 mg の拮抗薬を添加して製剤することがで
きる。本発明の経口剤型では、無傷剤型の投与直後に拮抗薬が放出または実質的に放出さ
れることがないので、拮抗薬の高添加によっても作動薬の鎮痛効力を妨げる量の拮抗薬が
無傷剤型から放出されるようなことは生じない。
【0092】
本発明の押出成形マトリックスに使用する材料には、たとえば、限定するものではない
が、ゴム、セルロースエーテル、アクリル樹脂、タンパク質由来の材料のような親水性あ
るいは疎水性物質; 脂肪酸、脂肪アルコール、脂肪酸のグリセリルエステル、無機質及び
野菜油類及びワックス (天然の及び合成)、及びステアリルアルコールのような消化性の
、長鎖 (C8-C50、特にC12-C40)の、置換または非置換炭化水素; 及びポリアルキレングリ
コールが含まれる。マトリックスには、1% 〜 80% (重量で) の少なくとも1つの親水性
または好ましくは少なくとも1つの疎水性物質を含むことができる。
【0093】
押出成形マトリックスが疎水性物質を含有する場合、疎水性物質はこの目的に役立つ任
意の疎水性物質であってもよいが、好ましくはアルキルセルロース、アクリル及びメタク
リル酸重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、またはその
混合物からなる群より選択されるものとする。本発明の特定の好ましい実施形態では、疎
水性物質は薬剤的に許容できるアクリル重合体であって、アクリル酸及びメタクリル酸共
重合体、メチル メタクリラート、メチル メタクリラート共重合体、エトキシエチル (et
hoxyethyl) メタクリラート、シアノエチル メタクリラート、アミノアルキル メタクリ
ラート共重合体、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタクリル酸アルキルアミン
共重合体、ポリ(メチル メタクリラート)、ポリ(メタクリル酸)(水素化物)、ポリメタク
リラート、ポリアクリルアミド、ポリ(メタクリル酸 水素化物)、及びグリシジル メタク
リラート共重合体のいずれかが含まれるが、これらに限定されるものではない。
【0094】
本発明で有用なアクリル重合体には、限定するものではないが、使用したアクリル及び
メタクリルモノマーのモル毎に約 0.02 〜 0.03 モルのトリ (低級アルキル) アンモニウ
ム基を含むアクリル及びメタクリル酸エステル (たとえば、アクリル酸の低級アルキルエ
ステル及びメタクリル酸の低級アルキルエステルの共重合体) から合成した共重合体を含
有するアクリル樹脂が含まれる。好適なアクリル樹脂の例は、ローム製薬(Rohm Pharma
)有限責任会社によって製造された重合体であり、商標 Eudragit(登録商標)RS で販売
されている。Eudragit(登録商標)RS30D は好ましいものである。Eudragit(登録商標)
RS は、エチル アクリラート(EA)、メチル メタクリラート (MM) 及びトリエチルアンモ
ニウムエチル メタクリラート塩化物 (TAM) の水不溶性共重合体であって、他成分(EA 及
びMM) に対する TAM のモル比は 1:40 である。Eudragit(登録商標)RS のようなアクリ
ル樹脂は水溶性の懸濁液の形で使用することが可能である。
【0095】
他の実施形態では、疎水性物質はハイドロキシプロピルメチルセルロースのような1つ
またはそれ以上のヒドロキシアルキルセルロースのような材料より選択される。
【0096】
特定の実施形態では、本発明で有用な疎水性物質は約 30℃ 〜約 200℃、または約 45
℃ 〜約 90℃の融点を持つ。
【0097】
特定の実施形態では、疎水性物質は、限定はされないが、脂肪酸エステル、脂肪酸グリ
セリド (モノ-、ジ-、及びトリ-グリセリド)、硬化脂肪、炭化水素、正常ワックス、ステ
アリン酸、ステアリルアルコール及び炭化水素主鎖を有する疎水性と親水性の材料を含む
天然または合成ワックス、脂肪アルコール(例: ラウリル、ミリスチル、ステアリル、セ
チルまたはセトステアリルアルコール)、脂肪酸を含有する。好適なワックスには、たと
えば、蜜ロウ、糖ロウ、キャスターロウ及びカルナウバワックスが含まれる。本発明の目
的上、ワックス様物質は、通常は室温で固体であり、約 30 〜約 100 ℃の融点を持つ任
意の材料として定義する。
【0098】
特定の実施形態では、疎水性物質はエチルセルロース、セルロースアセテート、セルロ
ースプロピオナート(低、中または高の分子量)、セルロースアセテートプロピオナート、
セルロースアセテートブチラート、セルロースアセテートフタラート及びセルローストリ
アセテートからなる群より選択されるセルロースポリマーを含有する。エチルセルロース
の例としては、44 〜 55% のエトキシル基含有量を持つものが挙げられる。エチルセルロ
ースはアルコール溶液の形で使用することが可能である。特定の他の実施形態では、疎水
性物質はポリ乳酸、ポリグリコール酸またはポリ乳酸及びポリグリコール酸の共重合体を
含有する。
【0099】
特定の実施形態では、疎水性物質はセルロースエーテル、セルロースエステル、セルロ
ースエステルエーテル、及びセルロースからなる群より選択されるセルロースポリマーを
含有する。特定の実施形態では、セルロースポリマーはアンヒドログルコース ユニット
に対してゼロから最大で 3 までの置換度 (D.S.) を持つ。置換度は、セルロースポリマ
ーを含有するアンヒドログルコース ユニット上に存在している水酸基の平均数が置換群
によって交換されることを意味する。代表的な材料には、セルロースアシレート、セルロ
ースジアシレート、セルローストリアシレート (triacylate)、セルロースアセテート、
セルロースジアセテート、セルローストリアセテート、モノ/ジ/トリセルロース アルカ
ニレート (alkanylate)、モノ/ジ/トリセルロース アロイレート (aroylates)、及びモノ
/ジ/トリセルロースアルケニレート (alkenylate) からなる群より選択される重合体が含
まれる。例示的重合体には、最大で 1 までの D.S. 及び最大で 21% までのアセチル含有
量を有するセルロースアセテート; 最大で 32 〜 39.8% までのアセチル含有量を有する
セルロースアセテート; 1 〜 2 の D.S. 及び 21 〜 35% のアセチル含有量を有するセ
ルロースアセテート; 及び 2 〜 3 の D.S. 及び 35 〜 44.8% のアセチル含有量を有す
るセルロースアセテートが含まれる。
【0100】
特定のセルロースポリマーには、1.8 の D.S.、39.2 〜 45% のプロピル含有量及び 2.
8 〜 5.4% の水酸含有量を有するセルロースプロピオナート; 1.8 の D.S.、13 〜 15%
のアセチル含有量及び 34 〜 39% のブチリル含有量を有するセルロースアセテートブチ
ラート; 2 〜 29% のアセチル含有量、17 〜 53% のブチリル含有量及び 0.5 〜 4.7%
の水酸含有量を有するセルロースアセテートブチラート; 2.9 〜 3 の D.S. を有するセ
ルローストリアセテート、セルロース トリバレラート (tivalerate)、セルローストリラ
ウラート、セルローストリパルミチン酸、セルローストリスクシナート、及びセルロース
トリオクタノアートのようなセルローストリアクレート (triacylate); 2.2 〜 2.6 の
D.S. を有し、セルロースジスクシナート、セルロースジパルミチン酸、セルロースジオ
クタノアート、セルロースジペンタノアートのようなセルロースジアシレート、及びセル
ロースアセテートブチラート、セルロースアセテートオクタノアートブチラート及びセル
ロースアセテートプロピオナートのようなセルロースコエステル (coester)が含まれる。
【0101】
追加セルロースポリマーには、アセトアルデヒドジメチル セルロースアセテート、セ
ルロースアセテートエチルカルバマート、セルロースアセテートメチルカルバマート、及
びセルロースアセテートジメチルアミノセルロースアセテートが含まれる。
【0102】
特定の実施形態では、薬剤的に許容できる疎水性物質には、乳酸及びグリコール酸 (「
PLGA」) の共重合体を含有する生分解性重合体、ポリ乳酸、ポリグリコリド、ポリ無水物
、ポリオルトエステル、ポリカプロラクトン、ポリホスファゼン、多糖類、タンパク質重
合体、ポリエステル、ポリジオキサノン、ポリグルコナート、ポリ乳酸-酸-ポリエチレン
酸化物共重合体、ポリ(ヒドロキシブチラート)、ポリホスホエステル (polyphosphoesthe
r) または前述の任意物質の混合物またはブレンド物が含まれる。
【0103】
特定の実施形態では、PLGA を含有する生分解性重合体は約 2,000 〜約 500,000 ドル
トンの分子量を有する。乳酸のグリコール酸に対する比率は約 100:0 〜約 25:75 である
が、乳酸のグリコール酸に対する比率が約 65:35 であることが好ましい。
【0104】
PLGA は米国特許特許番号第 4,293,539 号 (Ludwig らによる) に示された工程によっ
て調製することが可能であり、その開示を引用することを以って本明細書の一部となす。
手短に言えば、Ludwig は、容易に取り外し可能な重合触媒 (たとえば、Dowex HCR-W2-H
のような強力な酸イオン交換樹脂)の存在下で乳酸酸及びグリコール酸を縮合することに
よって共重合体を調製する。触媒の量は重合にとって重要ではないが、一般には、乳酸と
グリコール酸を組み合わせた総重量に比べて約 0.01 〜約 20 重量部 である。重合反応
は、溶剤を用いずに、約 100℃ 〜約 250℃ の温度で約 48 〜約 96 時間、水分及び副産
物の除去を容易にするために好ましくは低圧下で実行することができる。次いで、PLGA
は、ジクロロメタンまたはアセトンのような有機溶剤中で溶融した反応混合物をろ過する
ことによって回収し、次いでろ過により触媒を除去する。
【0105】
特定の好ましい実施形態では、2つまたはそれ以上の疎水性物質の組み合わせを押出成
形マトリックス内に含む。2つまたはそれ以上の疎水性物質を含む場合、少なくとも1つ
の疎水性物質を、好ましくは天然及び合成ワックス、脂肪酸、脂肪アルコール、及び同物
質の混合物から選択する。その例としては、限定するものではないが、蜜ロウ、カルナウ
バワックス、ステアリン酸及びステアリルアルコールが挙げられる。
【0106】
疎水性物質が炭化水素であるとき、炭化水素は好ましくは 25℃ 〜 90℃ 間の融点を持
つ。長鎖炭化水素材料中、脂肪(脂肪族) アルコールが好ましい。本マトリックスには、
最大で 60% (重量で) までの少なくとも1つの消化性長鎖炭化水素を含むことが可能であ
る。
【0107】
特定の好ましい実施形態では、押出成形マトリックスには最大で 60% (重量で) までの
少なくとも1つのポリアルキレングリコールを含む。
【0108】
1つの好適な押出成形マトリックスは、少なくとも1つの水溶性ヒドロキシアルキルセ
ルロース、少なくとも1つのC12-C36、好ましくは C14-C22、脂肪アルコール及び、任意
選択で、少なくとも1つのポリアルキレングリコールを含有する。ヒドロキシアルキルセ
ルロースは、好ましくはヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセル
ロースまたはヒドロキシエチルセルロースのようなヒドロキシ(C1 〜 C6) アルキルセル
ロースである。経口剤型内のヒドロキシアルキルセルロース量は、特に、所要活性剤の正
確な放出速度によって決定する。脂肪アルコールは、たとえば、ラウリル アルコール、
ミリスチル アルコールまたはステアリル アルコールであってもよい。特に好適な実施形
態では、但し、脂肪アルコールはセチル アルコールまたはセトステアリルアルコールで
ある。経口剤型内の脂肪アルコール量は、特に、所要活性剤の正確な放出速度によって決
定する。これはまた、ポリアルキレングリコールが経口剤型内に存在しているかどうかに
応じても異なる。ポリアルキレングリコールの不在時には、経口剤型は好ましくは脂肪ア
ルコールの約 20% 〜約 50% (重量で) を含む。少なくとも1つのポリアルキレングリコ
ールが経口剤型内に存在するとき、脂肪アルコールとポリアルキレングリコールを組み合
わせた重量は好ましくは全剤型の 20% 〜 50% (重量で) を構成する。
【0109】
一実施形態では、ヒドロキシアルキルセルロースまたはアクリル樹脂の、脂肪アルコー
ル/ ポリアルキレングリコールに対する比率により、活性剤の製剤からの放出速度がかな
りの程度まで決定される。ヒドロキシアルキルセルロースの脂肪アルコール/ポリアルキ
レングリコールに対する比率は 1:2 及び 1:4 が好ましいが、1:3 及び 1:4 の比率は特
に好適である。
【0110】
ポリアルキレングリコールは、たとえば、ポリプロピレングリコールまたはポリエチレ
ングリコールであってよい。ポリアルキレングリコールの平均分子量は、好ましくは約 1
,000 〜約 15,000 であるが、約 1,500 〜約 12,000 が特に好ましい。
【0111】
別の好適な押出成形マトリックスは、アルキルセルロース (特にエチルセルロース)、C
12 〜 C36 脂肪アルコール及び、任意選択で、ポリアルキレングリコールを含有する。
【0112】
別の好適な押出成形マトリックスは、アクリル重合体 (特にEudragit(登録商標)RSPO
)、C12to C36 脂肪アルコール及び、任意選択で、ポリアルキレングリコールを含有する
。
【0113】
特定の好ましい実施形態では、マトリックスには少なくとも2つの薬剤的に許容できる
疎水性材料の組み合わせが含まれる。
【0114】
上記開示したように、オピオイド拮抗薬を含有する薬剤的に許容できるマトリックスを
含有する複数個の押出成形した粒子は、マトリックス材料に加えて、オピオイド拮抗薬を
隔離することができる1つまたはそれ以上の疎水性物質と重層させる。コーティングの疎
水性物質は任意の上述した物質から選択することが可能である。特定の好ましい実施形態
では、疎水性物質はセルロース材料または重合体、アクリル重合体、またはその組み合わ
せである。用語「第1疎水性物質」、「第2疎水性物質」及び「第3疎水性物質」はそれ
ぞれ、少なくとも部分的分散または層状配列 (arraignment) において1つまたはそれ以
上の疎水性物質を包含することを意図する。第1、第2及び第3疎水性物質は同じであっ
ても異なっていてもよい。特定の実施形態では、第1及び第2疎水性物質が同じであり得
; 第1及び第3疎水性物質が同じであり得; 第2及び第3疎水性物質が同じであり得; ま
たは第1、第2及び第3疎水性物質が同じであり得る。
【0115】
実施形態では、層内に1つ以上の疎水性物質がある場合、疎水性物質を相互分散または
部分的に相互分散することができる。あるいは、疎水性物質は層状配列にあってもよい。
たとえば、粒子重量の 25% の量にあたる層は、重量で粒子の15% にあたるエチルセルロ
ース層及び重量で粒子の 10% にあたるエチルセルロース層の周囲に配置したアクリル重
合体層を持つことができる。
【0116】
コーティング組成物は、当分野で周知の任意の好適な噴霧器を用いて噴霧によって複数
個の押出成形した粒子に塗布することができる。たとえば、下部から注入する噴出空気が
被覆材料を流動化し、コーティングを噴霧中に乾燥させる Wurster 流動層システムを使
用してもよい。コーティング厚は、使用する特定のコーティング組成物の特性によって異
なる。
【0117】
本発明の押出成形した粒子を層化するために好適な疎水性物質には、アルキルセルロー
スを始めとするセルロース材料及び重合体が含まれる。好ましいアルキルセルロースポリ
マーの1つとしてエチルセルロースが挙げられるが、他のセルロースあるいはアルキルセ
ルロースポリマーも、本発明に係る疎水性コーティングのすべてまたは一部として単独ま
たは組み合わせて容易に利用することができる。
【0118】
市販されているエチルセルロースの水分散系の1つとして、Aquacoat (登録商標)(FM
C 社、米国ペンシルバニア州フィラデルフィア) が挙げられるが、これは水不混和性有機
溶剤中のエチルセルロースを溶解し、次いで同セルロースを界面活性剤及び安定剤存在下
で水中で乳化することによって作成される。均質化後、サブミクロンの溶滴を作成するに
は、有機溶剤を真空下で蒸発させ、擬似ラテックスを形成する。
【0119】
エチルセルロースの別の水分散系は、Surelease (登録商標)(Colorcon 社、米国ペン
シルバニア州ウェストポイント) として市販されている。この製品は、製造工程中に可塑
剤を分散系内に取り込むことによって作成される。重合体、可塑剤 (ジブチルセバカート
)、及び安定剤 (オレイン酸) の熱溶融を均一な混合物として調製し、次いでアルカリ溶
液で希釈して複数粒子に直接的に塗布できる水分散系を得る。
【0120】
本発明の他の好ましい実施形態では、層の疎水性物質は、アクリル酸及びメタクリル酸
共重合体、メチル メタクリラート共重合体、エトキシエチル (ethoxyethyl) メタクリラ
ート、シアノエチル メタクリラート、ポリ(アクリル酸)、ポリ(メタクリル酸)、メタク
リル酸アルキルアミド共重合体、ポリ(メチル メタクリラート)、ポリメタクリラート、
ポリ(メチル メタクリラート) 共重合体、ポリアクリルアミド、アミノアルキルメタクリ
ラート共重合体、ポリ(メタクリル酸酸無水物)、グリシジル メタクリラート共重合体、
及びその組み合わせを含むが、これらに限定されるものではない薬剤的に許容できるアク
リル重合体である。
【0121】
特定の好ましい実施形態では、アクリル重合体は1つまたはそれ以上のアンモニオ メ
タクリラート共重合体からなる。アンモニオ メタクリラート共重合体は当分野で周知の
ものであり、低含有量の四級アンモニウム基を有するアクリル及びメタクリル酸エステル
の完全重合の共重合体として NF XVII に記載されている。
【0122】
特定の好ましい実施形態では、四級アンモニウム基の中性 (メタクリルエステル) アク
リルエステルに対するモル比が異なるような異なる物性を有する2つ以上のアンモニオ
メタクリラート共重合体取り込むことが必要である場合がある。
【0123】
特定の実施形態では、アクリル層は Eudragit(登録商標)RL30D 及び Eudragit(登録
商標)RS30D の商品名で ローム製薬(Rohm Pharma) (ダルムシュタット(Darnastadt)
、ドイツ) から市販されている2つのアクリル樹脂ラッカーの混合物を含有する。Eudrag
it(登録商標)RL30D 及び Eudragit(登録商標)RS30D は低含有量の四級アンモニウム
基を有するアクリル及びメタクリルエステルの共重合体であって、アンモニウム基の残存
中性 (メタクリルエステル) アクリルエステルに対するモル比は Eudragit(登録商標)
RL30D では 1:20 及び Eudragit(登録商標)RS30D では 1:40 である。平均分子量は約
150,000 である。コード記号表示 RL (高透過性) 及び RS (低透過性) はこれら薬剤の透
過性特質を参照する。特定の実施形態では、本発明における Eudragit(登録商標)RS は
、Eudragit(登録商標)RSPM、Eudragit(登録商標)RSPO、Eudragit(登録商標)RS100
、Eudragit(登録商標)RS12.5、及びその混合物からなる群より選択される。用語 「Eud
ragit(登録商標)RSPM」は Eudragit(登録商標)RS の通常の未細砕粉末を表し、用語
「Eudragit(登録商標)RSPO」は Eudragit(登録商標)RS の細砕微粉末を表し、用語
「Eudragit(登録商標)RS100」は Eudragit(登録商標)RS の顆粒を表し、その一方、
用語 「Eudragit(登録商標)RS12.5」は、Eudragit(登録商標)RS が有機溶剤中に溶解
されている Eudragit(登録商標)RS の溶液製品を表す。特定の実施形態では、本発明に
おいて使用した Eudragit(登録商標)RL は、Eudragit(登録商標)RLPM、Eudragit(登
録商標)RLPO、Eudragit(登録商標)RL100、Eudragit(登録商標)RL12.5、及びその混
合物からなる群より選択される。用語 「PM」、「PO」、「100」、及び「12.5」は Eudra
git(登録商標)RS に関して上記のように定義される。また、Eudragit(登録商標)RS
シリーズ及び Eudragit(登録商標)RL シリーズの混合物も、任意の比率で、本発明のア
ンモニオ メタクリラート共重合体として使用する。
【0124】
本発明の Eudragit(登録商標)RL/RS 分散系は望ましい溶解特性を有する隔離製剤を
最終的に得るために互いに所望の比率で混合することができる。たとえば、望ましい製剤
は、現に、100% の Eudragit(登録商標)RL、50% の Eudragit(登録商標)RL 及び 50%
の Eudragit(登録商標)RS、及び 10% の Eudragit(登録商標)RL、Eudragit(登録商
標)90% RS に由来するコーティングから得ることができる。 もとより、当業者であれば
、たとえば、Eudragit(登録商標)L のような他のアクリル重合体を使用することも可能
であることを認識するであろう。
【0125】
層は、有機または水溶液または分散系の形式で適用することができる。所望の隔離を得
るため、層を適用して、オピオイド拮抗薬を含有する複数個の薬剤的に許容できる粒子の
約 2 〜約 25% の重量増加を達成することが可能である。水分散系に由来するコーティン
グは、たとえば、米国特許特許番号第 5,273,760 号及び第 5,286,493 号に詳しく記載さ
れている。本発明に従って使用できるコーティングの他の例としては、米国特許特許番号
第 5,324,351 号; 第5,356,467 号、及び第 5,472,712 号が挙げられる。
【0126】
オピオイド拮抗薬を含有する複数個の押出成形した粒子が疎水性物質の水分散系と重層
する特定の実施形態では、 疎水性物質の水分散系には好ましくは可塑剤有効量が含まれ
る。
【0127】
層を疎水性物質の水分散系から調製する本発明の実施形態では、水分散系中の可塑剤有
効量を包含すると、層の物性をさらに改善することができる。たとえば、エチルセルロー
スは比較的高いガラス転移温度を持ち、コーティング標準条件下で柔軟な膜を形成しない
ので、可塑剤をエチルセルロースのコーティング溶液に取り込むことが好ましい。一般的
に、溶液に含まれる可塑剤量は、フィルム形成物濃度、たとえば、ほとんどの場合、フィ
ルム形成物の重量の約 1 〜約 50 パーセントに基づく。
【0128】
好適なエチルセルロースに対する可塑剤の例には、ジブチルセバカート、ジエチル フ
タラート、クエン酸トリエチル、クエン酸トリブチル、及びトリアセチンのような不水溶
性可塑剤が含まれるが、他の水不溶性可塑剤 (例: アセチル化モノグリセリド、フタラー
トエステル、ヒマシ油など) を使用することも可能である。クエン酸トリエチルは、エチ
ルセルロースの水分散系に対して特に好ましい可塑剤である。
【0129】
本発明のアクリル重合体に潜在的に好適な可塑剤の例としては、限定するものではない
が、クエン酸トリエチル NF XVI、クエン酸トリブチル、ジブチルフタラート、及びプロ
ピレングリコールのようなクエン酸エステルが挙げられる。Eudragit(登録商標)RL/RS
ラッカー液のようなアクリルフィルムから形成し膜弾性を高めるのに好適であると実証さ
れている他の可塑剤には、ポリエチレングリコール、ジエチル フタラート、ヒマシ油、
及びトリアセチンが含まれる。クエン酸トリエチルはアクリル重合体の水分散系に特に好
ましい可塑剤である。
【0130】
可塑化した疎水性物質は、当分野で既知の任意の好適な噴霧器を用いた噴霧によって、
オピオイド拮抗薬含有する複数個の薬剤的に許容できる粒子に適用することができる。好
ましい方法では、下部から注入する噴出空気が被覆材料を流動化し、コーティングを噴霧
中に乾燥させる Wurster 流動層システムを使用する。
【0131】
本発明のコーティング溶液は、疎水性物質、可塑剤、及び溶剤システム (たとえば、水
) に加えて、エレガンスと製品識別を提供するために着色剤をさらに含有することができ
る。好適な着色剤には、アルコールまたはプロピレングリコール系色分散剤、摩砕したア
ルミニウムレーキ及び二酸化チタン及び鉄酸化物色素のような乳白剤が含まれる。着色剤
は、コーティング工程中に疎水性物質の分散系に添加することができる。あるいは、本発
明の製剤に色を提供する他の任意の好適な方法を使用することもできる。たとえば、Opad
ry(登録商標)のような色塗を薬剤的に許容できる被覆粒子に適用することができる。
【0132】
特定の実施形態では、水分散系が処理中に固着する傾向、あるいは研磨剤としての役割
を果たす傾向を低下させるために少量の滑石を使用することが可能である。
【0133】
複数個の薬剤的に許容できる粒子は、たとえば、摂取し胃液に接触し、次いで腸液に接
触するときに一定の期間にわたって制御下でオピオイド作動薬緩慢に放出する放出制御マ
トリックスに分散したオピオイド作動薬を含有することができる。粒子のマトリックスは
、好ましくは約 8 〜約 24 時間、好ましくは約 12 〜約 24 時間にわたって作動薬の放
出制御を提供する。 オピオイド作動薬を含有する粒子に使用する放出制御マトリックス
には、親水性物質あるいは疎水性物質に関して上記に記述した材料 (たとえば、ゴム、セ
ルロースエーテル、アクリル樹脂、タンパク質由来の材料; 消化性の長鎖 (C8-C50 特にC
12-C40)、置換または非置換炭化水素、例: 脂肪酸、脂肪アルコール、脂肪酸のグリセリ
ルエステル、無機質及び野菜油類及びワックス (天然の及び合成)、及びステアリルアル
コール; 及びポリアルキレングリコール) を含むことができる。
【0134】
特定の実施形態では、オピオイド作動薬含有する粒子は、その表面に配置した放出制御
層を有する即放性マトリックスを含有することができる。放出制御層には、上記に記載し
た1つまたはそれ以上の疎水性物質を含むことができる。
【0135】
特定の実施形態では、オピオイド作動薬を含有する複数個の薬剤的に許容できる粒子を
、任意選択で: (i) オピオイド作動薬放出の調節; (ii) 製剤の保護または (iii) 拮抗薬
を含有する被覆粒子のコーティングと実質的に識別できないコーティング提供すること;
または (i)、(ii) または (iii) の組み合わせに好適な1つまたはそれ以上の材料と重層
させる。たとえば、一実施形態では、コーティングを提供して、たとえば、胃腸液と接触
するときに pH-依存型放出または pH-非依存型放出のいずれかを許容する。pH-依存型コ
ーティングは、オピオイドを胃腸管、たとえば、胃腸または小腸の所望領域に放出する役
割を果たし、したがって、少なくとも約 8 時間及び好ましくは約 12 時間、最大で 約 2
4 時間までの鎮痛を患者に提供する能力がある吸収特性を提供することができる。pH-非
依存性層が所望される場合は、層を環境流体、たとえば、胃腸管における pH 変化などに
拘わらずオピオイドの放出を達成することを目的として設計する。また、胃腸管、たとえ
ば、胃腸の 1 所望領域に用量の一部を放出し、胃腸管の別領域、たとえば、小腸に用量
の残存部を放出する組成物を調剤することも可能である。
【0136】
特定の実施形態では、オピオイド作動薬またはオピオイド拮抗薬を含む複数個の薬剤的
に許容できる粒子を硬化させる。好ましくは、粒子は、複数個の薬剤的に許容できる粒子
が安定した溶解 (または無溶解) を提供する終点に至るまで硬化させる。硬化の終点は、
硬化直後の剤型の溶解特性 (曲線) を、たとえば、温度 40℃ 及び相対湿度 75% で少な
くとも 1 ヶ月間の加速した貯蔵条件に晒した後の剤型の溶解特性 (曲線) に対して比較
することによって決定することが可能である。硬化した製剤は、たとえば、米国特許特許
番号第 5,273,760 号; 第 5,286,493 号; 第 5,500,227 号; 第 5,580,578 号; 第 5,639
,476 号; 第 5,681,585 号; 及び第 6,024,982 号に詳しく記載されている。本発明に従
って使用することが可能である放出制御製剤及びコーティングの他の例には、米国特許特
許番号第 5,324,351 号; 第 5,356,467; 及び 5,472,712 に記載された製剤及びコーティ
ングが含まれる。
【0137】
特定の実施形態では、オピオイド作動薬を含有する複数個の薬剤的に許容できる粒子お
よび/またはオピオイド拮抗薬を含有する薬剤的に許容できる粒子には、薬剤的に許容で
きる粒子からのオピオイド作動薬あるいはオピオイド拮抗薬の放出に実質的に影響を及ぼ
さない材料でフィルムコートを施す。特定の実施形態では、Opadry(登録商標)のような
フィルムコートを複数個の薬剤的に許容できる粒子に適用する。フィルムコートは、該当
する場合、好ましくは粒子の凝集を実質的に低下させるため、または作動薬及び拮抗薬を
含む粒子を互いに識別困難にする助けとなるために提供する。好ましくは、本発明のフィ
ルムコーティングには、スムーズでエレガンであって、色素及び他のコーティング添加物
を支持する能力があり、非毒性、不活性、及び不粘着である強力な連続フィルムを作る能
力のあることが望ましい。
【0138】
上記成分に加えて、オピオイド作動薬含有する粒子及び拮抗薬を含有する粒子のいずれ
かまたは両方には、好適な量の他材料、たとえば、医薬技術では慣習的な希釈液、潤滑剤
、結合剤、造粒剤、偏球化剤、着色剤、加味剤 (flavorants) 及び流動促進剤を含むこと
もできる。これらの追加材料の量は所望の効果を所望の製剤に対して提供するために十分
であろう。
【0139】
潤滑剤の例には、限定するものではないが、ステアリン酸マグネシウム、ステアリン酸
ナトリウム、ステアリン酸、ステアリン酸カルシウム、オレイン酸マグネシウム、オレイ
ン酸、オレイン酸カリウム、カプリル酸、フマル酸ステアリルナトリウム、及びパルミチ
ン酸マグネシウムが含まれる。
【0140】
低粘性の水溶性重合体のような好適な結合剤は、医薬品技術における当業者には周知で
あろう。但し、ヒドロキシプロピルセルロースのような水溶性ヒドロキシ低級アルキルセ
ルロースが好ましい。
【0141】
着色剤には、二酸化チタンおよび/またはF. D. & C として知られている食物に好適な
色素、及び葡萄皮エキス、ビーツの赤粉末、β カロテン、べにの木、カルミン、ウコン
、パプリカ、のような天然着色剤及び前述の任意物質の組み合わせを含むことができる。
【0142】
組成物に組み込む風味は、合成風味油類及び香味料芳香化合物および/または天然の油
類、植物葉、花、および果実の抽出物、及び前述の任意物質の組み合わせから選択するこ
とが可能である。
【0143】
経口剤型調剤するために使用することが可能である薬剤的に許容できる担体、希釈液、
造粒剤、球化剤、流動促進剤及び他の賦形剤の具体例は、医薬品賦形剤ハンドブック (Ha
ndbook of Pharmaceutical Excipients) (米国薬学会) (1986) に記載されている。
【0144】
いくつかのプロセスを用いて、使用する技術が損傷の隔離した拮抗薬の完全性を損なわ
ない限りは本発明の剤型を調製することが可能である (たとえば、拮抗薬の粒子を作動薬
粒子との組み合わせるときに)。隔離した拮抗薬の粒子の完全性を損なうと、作動薬の効
力を損なう量のオピオイド拮抗薬が、無傷剤型の投与直後に放出されることを結果として
もたらすことがある。
【0145】
本発明の粒子を調製するための好ましい工程は、溶融押出成形または溶融造粒技術を介
した工程である。一般的に、溶融造粒技術には、通常は固体の疎水性物質、たとえばワッ
クスを溶融または軟化すること、および散剤をその中に取り込むことが含まれる。特定の
実施形態では、追加の疎水性物質、たとえば、エチルセルロースまたは水不溶性アクリル
重合体を、溶融または軟化した疎水性物質に追加することができる。
【0146】
追加疎水性物質は1つまたはそれ以上のワックス様の熱可塑性物質を含有することがで
きる。特定の実施形態では、製剤内の個々のワックス様の物質は、実質的に分解不可能で
あり、初期放出相中に 胃腸液に不溶性であることが望ましい。有用なワックス様物質は
、約 1:5,000 以下の水溶性を有する物質であってもよい (w/w)。
【0147】
特定の実施形態では、本発明に係る好適な溶解・押出成形マトリックスの調製には、オ
ピオイド作動薬またはオピオイド拮抗薬を、少なくとも1つまたはそれ以上の疎水性物質
と共にブレンドし、均一な混合物を得る工程を含むことができる。次いで、その均一な混
合物を少なくとも押出成形するために十分に軟化するように十分な温度に加熱する。結果
として得られた均一な混合物を次いで押出成型して、たとえば、細長いストランドを形成
する。押出品は、好ましくは冷却し、当分野で周知の任意の手段によって複数微粒子(た
とえば、複数の粒子) に切断する。押出品は、好ましくは約 0.1 〜約 12 mm、約 0.1 〜
約 2.5 mm、約 0.2 〜約 6 mm、0.5 〜約 3 mm; 約 0.5 mm 〜約 2 mm、または約 1 mm
〜約 2 mm の平均直径を持つ。
【0148】
溶解・押出成形マトリックスの調製に有用な好適な疎水性物質には、限定するものでは
ないが、上記のようにアクリル重合体、セルロースポリマー及び脂肪アルコールが含まれ
る。
【0149】
本発明の溶融押出成形を調製するための任意工程では、直接的に疎水性物質、治療活性
剤、及び任意の結合剤を押出機にメータリングし; 成分をブレンド及び加熱して、均一混
合物を形成し; 均一混合物を押出成型して、細長いストランドを形成し; 均一な混合物を
含むストランドを冷却して; ストランドを約 0.1 mm 〜約 12 mm のサイズを有する粒子
に切断する必要がある。本発明のこの実施態様では、比較的に連続的な製造工程が実現す
る。
【0150】
押出機口径または出口の直径は、押出成形したストランドのさまざまな厚みに合わせて
調節することができる。その上、押出機出口は円形である必要はなく、長円形、長方形な
どであってもよい。得られたストランドは、熱線カッター、裁断機などを用いて粒子に細
断することができる。
【0151】
溶融押出成形したマトリックスは、押出機出口の形状により、たとえば、顆粒、偏球ま
たはペレット形状にすることができる。本発明の目的上、用語「溶融押出成形したマトリ
ックス,」及び「溶融押出成形マトリックスシステム」「溶融押出成形した複数微粒子」
及び「溶融押出成形した粒子」はすべて複数単位の粒子を意味し、好ましくは類似サイズ
あるいは類似形状であって、1つまたはそれ以上の活性剤及び1つまたはそれ以上の賦形
剤を含み、好ましくは本明細書に記載したように疎水性物質を含む粒子をいう。この関連
から、溶融押出成形したマトリックスのサイズ範囲は直径あるいは長さにおいて約 0.1
〜約 12 mm; 約 0.1 〜約 2.5 mm; 約 0.2 〜約 6 mm; 約 0.5 〜約 3 mm; 約 0.5 mm 〜
約 2 mm; または約 1 mm 〜約 2 mm である。さらに、溶融押出成形したマトリックスは
、このサイズ範囲内で任意の幾何学的形状であり得ることを理解されるべきである。特定
の実施形態では、押出品は所望の長さに切断し、偏球化工程を必要とせずにオピオイド拮
抗薬またはオピオイド作動薬の単位用量に分割することができる。
【0152】
本発明の他の実施形態では、溶融押出成形した材料は、オピオイド作動薬あるいはオピ
オイド拮抗薬を包含することなく調製し、その後にそれらの薬剤を押出品に対して添加す
る。そのような製剤によると、一般に押出成形マトリックス材料とブレンドした薬物が得
られ、次いで混合物を当分野で周知の方法によって複数微粒子に形成する。そのような製
剤は、たとえば、製剤に含まれているオピオイド作動薬またはオピオイド拮抗薬が、1つ
以上の疎水性物質を軟化するために必要な温度に対して敏感であるとき、有益であり得る
。
【0153】
特定の実施形態では、複数個の薬剤的に許容できる粒子を製造するための工程は押出成
形/偏球化工程である。この工程のため、オピオイド作動薬またはオピオイド拮抗薬は結
合剤で湿潤し、穿孔したプレートまたはダイを介して押出成形し、回転円板に載置する。
押出品は、好ましくは砕けて破片となり、回転円板上で球体、偏球体、または円形棒に丸
める。この方法の好ましい工程及び組成物には、水を用いて、たとえば約 80% 〜 25% の
オピオイド作動薬またはオピオイド拮抗薬とブレンドした、たとえば約 20% 〜 75% のセ
ルロース誘導体を含有するブレンド物を湿潤することが含まれる。
【0154】
特定の実施形態では、複数の薬剤的に許容できる粒子を製造するための工程で有機溶剤
を用いてオピオイド拮抗薬または作動薬をマトリックス材料と混合する補助を行う必要が
ある。この技術は、マトリックス材料を利用しなければ、不適当に高い融点となり、材料
を溶融した状態に利用した場合、薬物またはマトリックス材料の分解を生じるか、または
容認できない溶融粘度が結果としてもたらされ、それによって薬物 (たとえば、オピオイ
ド作動薬またはオピオイド拮抗薬) のマトリックス材料との混合を防止する状態になる場
合に使用することが望ましい。薬物及びマトリックス材料は中程度の量の溶剤と組み合わ
せてペーストを形成することができ、次いで強制的にスクリーンを通して顆粒を形成し、
次いで顆粒から溶剤を除去する。あるいは、薬物及びマトリックス材料を十分な量の溶剤
と組み合わせて、完全にマトリックス材料を溶解し、結果として得られた溶液 (固体薬物
粒子が含まれている可能性がある) を噴霧乾燥して複数個の薬剤的に許容できる粒子を形
成することができる。この技術は、マトリックス材料がセルロースエーテルまたはセルロ
ースエステルのような高分子量の合成重合体のときに好ましい。本工程に典型的に利用さ
れる溶剤にはアセトン、エタノール、アイソプロパノール、エチル アセテート、及びそ
の混合物が含まれる。
【0155】
上述のように、オピオイド拮抗薬を含有する複数個の押出成形した粒子は、粒子のそれ
ぞれの周囲に配置した疎水性物質を好ましくは含有する層を持つ。好ましくは、オピオイ
ド拮抗薬を含有する層状粒子は、オピオイド拮抗薬の放出を実質的に低下またはを防止し
、一方では、オピオイド作動薬を含有する薬剤的に許容できる粒子は、好ましくはオピオ
イド作動薬の放出制御を約 8 〜約 24 時間以上にわたって、最も好ましくは約 12 〜約
24 時間にわたって提供する。
【0156】
好ましい実施形態では、拮抗薬含有マトリックスの周囲に配置した層は、拮抗薬に対し
て不浸透性または実質的に不浸透性であり、GI システム中に不溶性または実質的に不溶
性である。好ましくは、本発明の無傷剤型をヒトに経口投与するとき、オピオイド拮抗薬
は、実質的に放出されず、よって体内吸収に利用できない。このように、オピオイド拮抗
薬は剤型内に存在するが、オピオイド作動薬の鎮痛有効性を実質的に遮断しない。但し、
本発明の経口剤型が改変される場合、それに含まれるオピオイド拮抗薬は放出され、少な
くとも部分的にオピオイド作動薬の効果を遮断する。本発明のこの実施態様は経口剤型内
のオピオイド作動薬の乱用 または転換の可能性を減少することができる。たとえば、個
人が、たとえば、咀嚼、粉砕、細砕または溶剤を加熱溶解 (たとえば約 45℃ 〜約 5O℃
を超える温度で) することによって、本発明の経口剤型に含まれた薬物乱用を試みる場合
、層及びマトリックスは両方とも損傷され、オピオイド拮抗薬を隔離する働きをしなくな
る。改変剤型の投与時、オピオイド拮抗薬は放出され、好ましくはオピオイド作動薬の陶
酔効果を実質的に遮断する。
【0157】
本発明の複数個の薬剤的に許容できる粒子(すなわち、層状オピオイド拮抗薬の押出成
形した粒子及びオピオイド作動薬粒子) は、任意選択で、当分野で周知の従来の賦形剤を
用いて経口剤型にさらに組み込まれる。
【0158】
1つの好ましい実施形態では、経口剤型を調製して、オピオイド作動薬含有粒子及びオ
ピオイド拮抗薬含有粒子の有効量をカプセル内に含ませる。たとえば、複数の薬剤的に許
容できる粒子を、摂取されると有効な持続放出用量を提供するために十分な量でゼラチン
カプセル内に入れることができる。カプセルは密閉型にしてもよいし、粒子を散布できる
ように非密閉型にしてもよい。
【0159】
別の実施形態では、好適な量の層状拮抗薬含有粒子をオピオイド作動薬含有粒子と組み
合わせて、複数個の薬剤的に許容できる粒子の完全性を実質的に崩壊することがないよう
に経口錠剤を作るために圧縮する。
【0160】
別の実施形態では、拮抗薬含有粒子が複数個の薬剤的に許容できる粒子の完全性を崩壊
することなく作動薬マトリックス内に埋め込まれている好適な量の層状拮抗薬含有粒子を
オピオイド製剤 (たとえば、徐放性造粒) と組み合わせて錠剤を作るために圧縮する。
【0161】
錠剤 (圧縮及び成型された) を作成するための技術及び組成物、カプセル (ハードゼラ
チン及びソフトゼラチン) 及び錠剤は、Remington's Pharmaceutical Sciences、(Arthur
Osol 編), 1553-1593 (1980) に記載されている。
【0162】
特定の実施形態では、経口剤型には、迅速な治療効果を提供する量の即放性オピオイド
作動薬が含まれる。特定の実施形態では、即放性オピオイド作動薬を、たとえば、ゼラチ
ンカプセル内に別のペレットとして組み込むか、または剤型の調製後に作動薬含有粒子の
表面を被覆することができる。
【0163】
本発明の放出制御製剤は、たとえば、摂取され、胃液と接触し、次いで腸液と接触する
ときには、好ましくはオピオイド作動薬を緩慢に放出するものである。本発明の製剤の放
出制御特性は、たとえば、遅延剤、すなわち、疎水性物質の量を変えることによって、疎
水性物質に相対して可塑剤の量を変えることによって、追加成分または賦形剤を抱合する
ことによって、製造などの方法を変えることによって改変することができる。
【0164】
好ましい実施形態では、本発明に有用なオピオイド作動薬には、限定するものではない
が、アルフェンタニル、アリルプロジン、アルファプロジン、アニレリジン、ベンジルモ
ルヒネ、ベジトラミド、ブプレノルフィン、ブトルファノール、クロニタゼン、コデイン
、デソモルヒネ、デキストロモルアミド、デゾシン、ジアンプロミド、ジアモルホン、ジ
ヒドロコデイン、ジヒドロモルヒネ、ジメノキサドール、ジメフェプタノール、ジメチル
チアンブテン、ジオキサフェチル ブチラート、ジピパノン、エプタゾシン、エトヘプタ
ジン、エチルメチルチアンブテン、エチルモルヒネ、エトニタゼン、エトルフィン、ジヒ
ドロエトルフィン、フェンタニール及び誘導体、ヘロイン、ヒドロコドン、ヒドロモルホ
ン、ヒドロキシペチジン、イソメタドン、ケトベミドン、レボルファノール、レボフェナ
シルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタゾシン、メサドン
、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノルレボルファノール
、ノルメサドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピパノン、アヘン、
オキシコドン、オキシモルホン、パパベレタム、ペンタゾシン、フェナドキソン、フェノ
モルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミド、プロpヘプタ
ジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニル、チリジン、ト
ラマドル、それらの薬剤的に許容できる塩類、及び前述した物質のいずれかの混合物が含
まれる。特定の実施形態では、剤型内のオピオイド作動薬の量は約 75 ng 〜 750 mg で
あってもよい。
【0165】
好ましい実施形態では、本発明のオピオイド拮抗薬は、ナルトレキソン、ナロキソン、
ナルメフェン、シクラザシン、レバロルファン、それらの薬剤的に許容できる塩類、及び
前述した物質のいずれかの混合物より選択される。特定の好ましい実施形態では、オピオ
イド拮抗薬は、ナルトレキソンまたはその薬剤的に許容できる塩類 (たとえば、塩酸ナル
トレキソン) である。特定の実施形態では、実質的に放出不可能な形態で存在しているオ
ピオイド拮抗薬の量は、約 0.5 mg 〜約 50 mg、約 1 mg 〜約 25 mg、約 2 mg 〜約 20
mg、約 5 mg 〜約 15 mg、約 2 mg 〜約 10 mg、または約 4 mg 〜約 10 mg または約 6
mg 〜約 8 mg であってよい。
【0166】
ナロキソンは、作動薬効果をほとんど無効にするオピオイド拮抗薬である。最大で 12
mg までのナロキソンの皮下投与は識別可能の自覚効果を生ぜず、24 mg のナロキソンは
軽微な傾眠のみを引き起こす。男性の筋肉内または静脈内に与えた少量 (0.4 〜 0.8 mg)
のナロキソンは、モルヒネ様オピオイド作動薬の効果を防止するか、または即座に逆転
する。1 mg のナロキソンの静脈注射により、25 mg のヘロイン効果を完全に遮断するこ
とが報告されている。ナロキソンの効果は静脈内投与のほとんど直後に見られる。経口投
与後薬物は吸収されるが、その肝臓を介した第1通路で代謝されて急速に不活性形態とな
ることが報告されており、非経口的に投与される場合より大幅に低い作用効力を持つこと
が報告されている。1g 以上の経口投薬は、24 時間未満にほとんど完全に代謝されことが
報告されている。また、舌下投与された 25% のナロキソンが吸収されることも報告され
ている。Weinberg らの、Sublingual Absorption of selected Opioid Analgesics, Clin
Pharmacol Ther. (1988); 44:335-340。参照。
【0167】
他のオピオイド拮抗薬、たとえば、窒素のシクロプロピルメチル置換基をそれぞれに持
つシクラゾシン及びナルトレキソンは、経口経路による効力の多くを維持し、その作用時
間ははるかに長く、経口による服用後 24 時間に達する。
【0168】
本発明の好ましい実施形態では、オピオイド作動薬は、オキシコドン、ヒドロコドン、
ヒドロモルホン、モルヒネ、オキシモルホン、コデインまたはそれらの薬剤的に許容でき
る塩類を含有し、オピオイド拮抗薬は、ナルトレキソンまたはその薬剤的に許容できる塩
類を含有し、約 2 mg 〜約 15 mg、約 5 mg 〜約 10 mg、または約 6 mg 〜約 5 mg の量
で存在する。
【0169】
オピオイド作動薬がヒドロコドンまたはその薬剤的に許容できる塩類を含有する実施形
態では、徐放性経口剤型に、鎮痛用量約 5 mg 〜 約 50 mg の ヒドロコドンまたはその
塩類を用量単位毎に含むことができる。ヒドロモルホンまたはその薬剤的に許容できる塩
類が治療学的に活性なオピオイドである徐放性経口剤型では、それは約 2 mg 〜約 64 mg
の量のヒドロモルホンまたはその塩類に含まれる。別の実施形態では、オピオイド作動
薬は、モルヒネまたはその薬剤的に許容できる塩類を含有し、本発明の放出制御経口剤型
には約 2.5 mg 〜約 800 mg のモルヒネまたはその塩類が含まれる。さらに別の実施形態
では、オピオイド作動薬は、オキシコドンまたはその薬剤的に許容できる塩類を含有し、
放出制御経口剤型には約 2.5 mg 〜約 800 mg のオキシコドンまたはその塩類が含まれる
。特定の好ましい実施形態では、徐放性経口剤型には約 5mg、10mg、20mg、40mg、60mg、
80 mg、160 mg または 320 mg の塩酸オキシコドンが含まれる。放出制御オキシコドン製
剤は当分野で周知である。特定の実施形態では、オピオイド作動薬は、トラマドルまたは
その薬剤的に許容できる塩類を含有し、放出制御経口剤型には、用量単位毎に約 25 mg
〜 800 mg のトラマドルを含むことができる。剤型には、1つ以上のオピオイド作動薬を
含むことができ、1つの作動薬製品によって達成される治療効果と比較して等価な治療効
果を提供することが可能である。あるいは、剤型には、本発明で有用なオピオイド作動薬
の他の塩類と等価のモル濃度を含むことが可能である。
【0170】
特定の実施形態では、安定剤を剤型内に含ませ、オピオイド拮抗薬の劣化を防止する。
特定の実施形態では、剤型で使用する安定剤には、たとえば、限定するものではないが、
有機酸、カルボン酸、アミノ酸の酸塩類 (たとえば、システイン、L-システイン、システ
イン塩酸塩、グリシン塩酸塩またはシステイン二塩酸塩)、メタ重亜硫酸ソーダ、アスコ
ルビン酸及びその誘導体、リンゴ酸酸、イソアスコルビン酸、クエン酸、酒石酸酸、パル
ミチン酸、炭酸ナトリウム、炭酸水素ナトリウム、炭酸カルシウム、リン酸水素カルシウ
ム、二酸化硫黄、亜硫酸ナトリウム、重硫酸ナトリウム、トコフェロール、さらには、そ
の水溶性及び脂溶性の誘導体 (たとえばtocofersolan または酢酸トコフェロールのよう
な)、亜硫酸塩剤、重亜硫酸及び亜硫酸水素またはアルカリ金属、アルカリ土類金属及び
他の金属、PHB エステル、ガラート、ブチル化ヒドロキシアニソール (BHA) またはブチ
ル化ヒドロキシトルエン (BHT)、及び2,6-ジ-t-ブチル-アルファ-ジメチルアミノ-p-クレ
ゾール、t-ブチルヒドロキノン、ジ-t-アミルヒドロキノン、ジ-t-ブチルヒドロキノン、
ブチルヒドロキシトルエン、ブチルヒドロキシアニソール、ピロカテコール、ピロガロー
ル、プロピル/ガラート、及びノルジヒドログアヤレト酸、さらには、低級脂肪酸、フル
ーツ酸、リン酸、ソルビン酸及び安息香酸、さらには、それらの塩類、エステル、誘導体
及び異性化合物、アスコルビン酸パルミテート、レシチン、モノ- 及びポリヒドロキシル
化ベンゼン誘導体、エチレンジアミン四酢酸及びその塩類、シトラコン酸、コンアイデン
ドリン(conidendrine)、ジエチル カルボナート、メチレンジオキシフェノール、ケフ
ァリン、β,β'-ジチオプロピオン酸、ビフェニル及び他のフェニル誘導体、それらの薬
剤的に許容できる塩類、及びその混合物が含まれる。
【0171】
本発明の経口剤型には、オピオイド作動薬及び拮抗薬に加えて、それとともに相乗的作
用してもよいし、相乗的作用しなくてもよい1つまたはそれ以上の薬物がさらに含まれる
。このように、特定の実施形態では、オピオイド拮抗薬に加えて、剤型内に2つのオピオ
イド作動薬の組み合わせを含むことができる。たとえば、剤型には、半減期、溶解度、作
用効力のような異なる特質を有する2つのオピオイド作動薬、及び前述の任意物質の組み
合わせを含むことができる。なお、さらなる実施形態では、オピオイド拮抗薬に加えて、
1つまたはそれ以上のオピオイド作動薬を含み、さらに非オピオイド薬物もまた含む。そ
のような非オピオイド薬物は、好ましくは追加鎮痛を提供し、たとえば、アスピリン、ア
セトアミノフェン; 非ステロイド性抗炎症性薬 (「NSAID」)、たとえば、イブプロフェン
、ケトプロフェンなど; N-メチル-D-アスパルタート(NMDA) 受容体拮抗薬、たとえば、モ
ルフィナンのようなデキストロメトルファンまたはデキストロルファン、またはケタミン
; シクロオキシゲナーゼ-II 阻害剤 (「COX-II 阻害剤」); あるいはグリシン受容体拮
抗薬を含む。追加薬剤を、第1作動薬として同じ粒子内または異なる粒子内に含むことが
できる。
【0172】
本発明の特定の好ましい実施形態では、本発明は、NSAID または COX-2 阻害剤のよう
な付加的な非オピオイド作動薬を包含することによって、低用量のオピオイド鎮痛薬の使
用を可能にする。いずれかまたは両方の薬物を低量で用いることによって、ヒトにおける
効果的な疼痛管理に関連する副作用を抑えることが可能である。
【0173】
好適な非ステロイド性抗炎症薬には、イブプロフェン、ジクロフェナク、ナプロキセン
、ベノクサプロフェン (benoxaprofen)、フルルビプロフェン、フェノプロフェン、フ
ルブフェン (flubufen)、ケトプロフェン、インドプロフェン、ピロプロフェン、カル
プロフェン、オキサプロジン、プラモプロフェン (pramoprofen)、ムロプロフェン (m
uroprofen)、トリオキサプロフェン、スプロフェン、アミノプロフェン、チアプロフェ
ン酸、フルプロフェン、ブクロクス酸、インドメタシン、スリンダク、トルメチン、ゾメ
ピラク、チオピナク、ジドメタシン、アセメタシン、フェンチアザク、クリダナク、オク
スピナク (oxpinac)、メフェナム酸、メクロフェナム酸、フルフェナム酸、ニフルム酸
、トルフェナム酸、ジフルリサール (diflurisal)、フルフェニサール、ピロキシカム
、スドキシカムまたはイソキシカム、それらの薬剤的に許容できる塩類、その混合物など
が含まれる。これらの薬物の有用な用量は当業者には公知である。
【0174】
N-メチル-D-アスパルタート(NMDA) 受容体拮抗薬は当分野で周知であり、たとえば、デ
キストロメトルファンまたはデキストロルファンのようなモルフィナン、ケタミン、d-メ
サドンまたはそれらの薬剤的に許容できる塩類を包含する。本発明の目的上、用語「NMDA
拮抗薬」もまた、NMDA-受容体活性化、たとえば、GM1 またはGT1b のようなガングリオ
シド、トリフルオペラジンのようなフェノチアジンまたはN(6-アミノテキシル)-5-クロロ
-l-ナフタレンスルホンアミドのようなナフタレンスルホンアミドの主な細胞内の結果物
を遮断する薬物を包含すると見なされる。これらの薬物は、 米国特許番号第 5,321,0
12 号及び第 5,556,838 号 (両方とも Mayer らによる) では添加薬物、たとえば、モル
ヒネ、コデインなどのような麻薬性鎮痛薬に対する耐性あるいは依存性を抑制することを
趣旨としており、米国特許特許番号第 5,502,058 号(Mayer らによる) では慢性疼痛を治
療することを趣旨としている。
【0175】
グリシン受容体拮抗薬の使用を介した慢性疼痛の治療、及びその薬物の同定は、米国特
許特許番号第 5,514,680 号 (Weber らによる) に記載されている。
【0176】
CQX-2 阻害剤は当技術分野で報告されており、多くの化学構造がシクロオキシゲナーゼ
-2 の抑制を実現することが知られている。COX-2 阻害剤は、たとえば米国特許特許番号
第 5,616,601 号; 第 5,604,260 号; 第 5,593,994 号; 第 5,550,142 号; 第 5,536,752
号; 第 5,521,213 号; 第 5,475,995 号; 第 5,639,780 号; 第 5,604,253 号; 第 5,55
2,422 号; 第 5,510,368 号; 第 5,436,265 号; 第 5,409,944 号; 及び第 5,130,311 号
に記載されている。特定の好ましい COX-2 阻害剤には、セレコクシブ (SC-58635)、DUP-
697、フロスリド (CGP-28238)、メロキシカム、6-メトキシ-2 ナフチル酢酸 (6-MNA)、MK
-966 (またバイオックスとしても知られている)、ナブメトン (6-MNA 用のプロドラッグ)
、ニメスリド、NS-398、SC-5766、SC-58215、T-614; それらの薬剤的に許容できる塩類、
及びその組み合わせが含まれる。1日当たり、体重 1 キログラム当たり約 0.005 mg 〜
約 140 mg の COX-2 阻害剤の投与量レベルは、オピオイド鎮痛薬と組み合わせた場合に
治療効果がある。あるいは、 1 日当たり、患者当たり約 0.25 mg 〜約 7 g の COX-2 阻
害剤をオピオイド鎮痛薬と組み合わせて投与する。オピオイド作動薬と COX-2 阻害剤の
組み合わせは、WO 99/13799 に開示されている。
【0177】
なお、さらならる実施形態では、鎮痛薬以外の、たとえば、鎮咳薬、排痰剤、制吐薬、
充血除去剤、抗ヒスタミン薬、局所麻酔などの所望の効果を提供する非オピオイド薬物を
含むことができる。
【0178】
また、本発明は、活性剤の乱用を抑止するために、異なる活性剤/拮抗薬の組み合わせ
(すなわち非オピオイド) を利用する本明細書で開示した剤型をその狙いとするをする。
たとえば、ベンゾジアゼピンを本発明の剤型内で活性剤として用いると、隔離したベンゾ
ジアゼピン拮抗薬を剤型内で製剤することができる。バルビツール酸塩を本発明の剤型内
で活性剤として用いると、隔離したバルビツール酸塩拮抗薬を剤型内で製剤することがで
きる。アンフェタミンを本発明の剤型内で活性剤として用いると、隔離したアンフェタミ
ン拮抗薬を剤型内で製剤することができる。
【0179】
用語「ベンゾジアゼピン」は中枢神経系を抑圧することができるベンゾジアゼピン、及
びベンゾジアゼピンの誘導体である薬物を指す。ベンゾジアゼピンには、限定するもので
はないが、、アルプラゾラム、ブロマゼパム、クロルジアゼポキシド、クロラゼプ酸、ジ
アゼパム、エスタゾラム、フルラゼパム、ハラゼパム、ケタゾラム、ロラゼパム、ニトラ
ゼパム、オキサゼパム、プラゼパム、クァゼパム、テマゼパム、トリアゾラム、メチルフ
ェニデート及びその混合物が含まれる。
【0180】
本発明で使用することができるベンゾジアゼピン拮抗薬には、限定するものではないが
、フルマゼニルが含まれる。
【0181】
バルビツール酸塩はバルビツール酸 (2、4、6、-トリオキソヘキサヒドロピリミジン)
に由来する鎮静薬・催眠薬を意味する。バルビツール酸塩には、限定するものではないが
、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタルビタール、メトヘ
キシタール、メホバルビタール、メタルビタール、ペントバルビタール、フェノバルビタ
ール、セコバルビタール及びその混合物が含まれる。
【0182】
本発明で使用することができるバルビツール酸塩拮抗薬には、限定するものではないが
、本明細書に記載したようにアンフェタミンが含まれる。
【0183】
刺激薬は中枢神経系を刺激する薬物を意味する。刺激薬には、限定するものではないが
、アンフェタミン、デキストロアンフェタミン樹脂複合体、デキストロアンフェタミン、
塩酸メタンフェタミン、メチルフェニデートのようなアンフェタミン類、及びその混合物
が含まれる。
【0184】
本発明で使用することができる刺激拮抗薬には、限定するものではないが本明細書に記
載したようにベンゾジアゼピンが含まれる。
【0185】
本発明はまた、活性剤の乱用を抑止するするために拮抗薬以外の逆作用剤を利用する本
明細書で開示した剤型をその狙いとする。用語「逆作用剤」は非隔離形態で投与して不快
な効果を作り出すことができる任意の薬剤を指す。拮抗薬以外の逆作用剤の例には、催吐
薬、刺激剤及び苦味剤が含まれる。
【0186】
催吐薬には、限定するものではないが、トコン及びアポモルヒネが含まれる。
【0187】
刺激剤には、限定するものではないが、カプサイチン、カプサイチン類似体、及びその
混合物が含まれる。カプサイチン類似体には、レシニフェラトキシン、ティニアトキシン
(tinyatoxin)、ヘプタノイルイソブチルアミド(heptanoylisobutylamide)、ヘプタノ
イルグアイアシルアミド(heptanoyl guaiacylamide)、他のイソブチルアミド類または
グアイアシルアミド(guaiacylamides)類、ジヒドロカプサイチン、ホモバニリル オク
チルエステル、ノナノイル バニリルアミド、及びその混合物が含まれる。
【0188】
苦味剤には、限定するものではないが、風味油類; 香味料芳香化合物; 含油樹脂; 植物
、葉、花に由来するエキス; フルーツ風味; サッカロース誘導体; クロロサッカロース誘
導体; 硫酸キニーネ; 安息香酸デナトニウム; 及びその組み合わせが含まれる。
【0189】
以下に、添付実施例を参照して本発明をより詳細に説明する。ただし、以下の説明は単
なる例示目的にすぎず、いかようにも本発明を限定するものではないことを理解されるべ
きである。
【実施例1】
【0190】
塩酸ナルトレキソン 2 mg カプセル
本実施例は、隔離製品を産生するために溶融押出成形した複数微粒子 (以下「MEM」と
する) として製剤したオピオイド拮抗薬である塩酸ナルトレキソンの比較実施例である。
選択した重合体及び賦形剤に基づいて、ペレットを無傷で分析すると MEM ペレットは極
めて僅かなナルトレキソンを放出するが、改変(粉砕) されると著しい量のナルトレキソ
ンを放出する。この実施例は、実施例 1 に続く実施例においてコーティングがどのよう
に隔離特質を高めることができるかを示すために参照として含むものである。実施例 1
の 塩酸ナルトレキソン製剤を下記の表に記載する。
【表1】
実施例 1 の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機に1.7 kg/時 〜 2.6 kg/時
の速度で連続供給し、押出成形物を回収する(Leistritz ZSE-27)。ブレンド物を 75℃ 〜
100℃ のバレル温度で直径約 1 mm の ストランドに押出成型する。押出成形したストラ
ンドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
7. カプセル化工程: スクリーニングしたペレットを目標重量 121 mg でハードゼラチン
カプセルに充填する。
【0191】
In vitro 溶解工程:
実施例 1 に従って調製した製剤を以下の in vitro 溶解検査方法で検査を行い、以下
の表 1B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 75 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF (人工胃液) で 1 時間、その後、900 ml の SIF (人工腸液) に
切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表2】
【0192】
模擬改変のプロセス及び溶解:
実施例 1 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の in
-vitro 溶解検査方法で検査した。1 時間の溶解結果を表1 C に記載した。改変プロセス
では、この溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (600 ストローク)
を用いて細砕し、粉末にした。
溶解方法: 上記と同じ
結果:
【表3】
【0193】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 1 時間で溶解した % と無傷ペレットが 36 時
間で溶解した % との比率である。
粉砕/無傷比の結果: 33.5% / 6.2% = 5.4:1
【0194】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
上記の工程と製法を用いて製造したカプセル (MEM) を臨床研究に用いて、即放性ナル
トレキソン錠剤と比較した薬物動態学/バイオアベイラビリティを決定した。ヒト被検体
に無傷塩酸ナルトレキソン MEM (全体)、粉砕した塩酸ナルトレキソン MEM (細砕) また
は即放性塩酸ナルトレキソン錠剤 (IR NTX) 剤型のいずれかを投与した。結果は図1 のグ
ラフ表示に示す。即放性ナルトレキソン (IR NTX) 及び粉砕 (細砕) 及び無傷 (全体) の
IR NTX の用量調節 Cmax と比較した無傷 (全体) 及び粉砕 (細砕) の用量調節 (1 mg
の IR NTX 錠剤に) 暴露度 (AUCt) を下記の表 1D に記載する。
【表4】
【0195】
用量調節した血漿濃度は、無傷状態で採取すると MEM 剤型からナルトレキソンの最小
放出があることを示す。ナルトレキソンのレベルは、MEM を粉砕 (細砕) 状態で採取する
と増加する。平均 Cmax に基づくと、粉砕 MEM/無傷 MEM カプセルの比率は、約 8 であ
る。同様に、平均 AUCt、粉砕 MEM/無傷 MEM カプセルの比率は、約 4.4 である。これは
合計及びピーク時の暴露の比率が粉砕後に大幅に増加することを示す。
【実施例2】
【0196】
エチルセルロース被覆の塩酸ナルトレキソン 2 mg ペレット
実施例 2 では、ナルトレキソン MEM を実施例 1 と同様に調製し、次いで MEM をエチ
ルセルロース (Surelease) でさまざまなレベル (5%、10%、15%、及び20% 重量増加) に
被覆した。実施例 2 の 非被覆の塩酸ナルトレキソン製剤を下記の表 2A に記載する。
【表5】
【0197】
実施例 2 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機に 2.9 kg/時 〜 4.8 kg/時
の速度で連続供給する (Leistritz ZSE-27)。ブレンド物を 95℃ 〜 105℃ のバレル温度
で直径約 1 mm の ストランドに押出成型する。押出成形したストランドをコンベア上に
回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収した。
【0198】
In vitro 溶解工程:
実施例 2 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 2B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表6】
【0199】
模擬改変; プロセス及び溶解:
実施例 2 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2C に記載した。改変プロセスでは、この
溶解研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を
用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表7】
【0200】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比の結果: 31% / 5.3% = 5.8:1
【0201】
実施例 2 によって調製し、表 2A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング(Surelease)で被覆して
、5%、10%、15% 及び20% 重量増加し; 及び疎水性コーティング(Surelease) とカラーコ
ーティング(Opadry)で被覆して20% の重量増加を得た。20% の重量増加を得た製剤例のコ
ーティング及びカラーコーティングを以下の表に記載する。
【表8】
実施例 2 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: 水と混合することによって Surelease 懸濁液を15% (w/w)
固体に希釈する。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Surelease 分散液を 700 g スケ
ール以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 7.0 〜 9.0 m/秒
* 吸気温度: 40 〜 50℃
* 分散液噴霧速度: 8 〜 11 g/分
5%、10%、15% 及び20% の重量増加に対する分散の理論量を噴霧したとき、サンプルを採
取した。
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 7.0 m/秒
* 吸気温度: 50℃
* 分散液噴霧速度: 8.5 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。上記の 20% 製法及び工程に従って 5%、10% 及び 15% の重量増加に被
覆したペレットを、単位当たり 6.05、12.1 及び 18.15 mg の Surelease をそれぞれ用
いて調製した。
【0202】
In vitro 溶解工程:
実施例 2 に従って調製した被覆した製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 2E に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表9】
溶解結果から分かるように、ナルトレキソンのペレットの溶解度は一般的に重合体コーテ
ィングレベルの増加に伴って減少した。
【0203】
模擬改変のプロセス及び溶解:
実施例 2 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表10】
【0204】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 2G に記載した。
粉砕/無傷比の結果:
【表11】
溶解結果から分かるように、コーティングレベルが増加するにつれ、粉砕/無傷比も増加
した。
【0205】
実施例 1 と比較した実施例 2 の結果
このように、実施例 1 における非被覆のMEM と同じ製剤であるMEM を実施例 2 で過剰
コーティングすることによって、薬物の放出は 36 時間で 5% 以上から およそ 2% まで
に下落した。結果として、実施例 1 における非被覆の MEM からの拮抗薬の「漏出」も、
機能コーティングを用いることによって大幅に低下した。粉砕/無傷比は、およそ 5:1 か
ら 10:1 に増加し得る。
【実施例3】
【0206】
エチルセルロース被覆の塩酸ナルトレキソン 8 mg ペレット
実施例 3 では、8mg のナルトレキソンを含むペレットを調製し、次いでエチルセルロ
ース (Surelease) でさまざまなレベル (5%、10%、15%、及び20%、25% 及び 30% 重量増
加) に被覆した。実施例 1 における 非被覆の塩酸ナルトレキソン製剤を下記の表に記載
する。
【表12】
実施例 3 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 3.9
kg/時の速度で連続供給する。ブレンド物を 95℃ 〜 100℃ のバレル温度で直径約 1 mm
の ストランドに押出成型する。押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0207】
In vitro 溶解工程:
実施例 3 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 3B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表13】
【0208】
模擬改変のプロセス及び溶解:
実施例 3 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の結果を表 3C に記載した。改変プロセスでは、この溶解
研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用い
て細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表14】
【0209】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比の結果: 57% / 18.7% = 3.0
【0210】
実施例 3 によって調製し、表 3A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング(Surelease)で被覆して
、5%、10%、15%、20% 及び25% 重量増加し; 及び疎水性コーティング(Surelease) とカラ
ーコーティング(Opadry)で被覆して30% の重量増加を得た。30% の重量増加を得た製剤例
の疎水性コーティング及びカラーコーティングを以下の表に記載する。
【表15】
実施例 3 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: 水と混合することによって Surelease 懸濁液を15% (w/w)
固体に希釈する。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Surelease 分散液を 700 g スケ
ール以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 8.6 〜 9.6 m/秒
* 吸気温度: 40 〜 50℃
* 分散液噴霧速度: 9 〜 14.8 g/分
5%、10%、15%、20%、25%、30% の重量増加に対する分散の理論量を噴霧したときに、サ
ンプルを採取した (単位当たり、それぞれおよそ 6.05、12.1、18.15、24.2 及び 30.25
mg の Surelease)
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 8.6 〜 9.0 m/秒
* 吸気温度: 47 ℃
* 分散液噴霧速度: 9.0 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。
【0211】
In vitro 溶解
実施例 3 に従って調製した、疎水性コーティング (Surelease) 及びカラーコーティン
グ (Opadry) で被覆した製剤を以下の in vitro 溶解検査方法で検査を行い、以下の表 3
E に記載した結果を得た。
方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表16】
溶解結果から分かるように、ナルトレキソンのペレットの溶解は一般的に重合体コーティ
ングレベルの増加に伴って減少した。
【0212】
模擬改変のプロセス及び溶解:
実施例 3 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 2F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表17】
【0213】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 3G に記載した。
粉砕/無傷比の結果:
【表18】
【0214】
上記の溶解結果から分かるように、コーティングレベルが増加するにつれ、無傷ペレッ
トから放出されるナルトレキソンの量が大幅に減少する (36 時間で 18% 以上から 2% 未
満に減少する) が、粉砕後には、拮抗薬のおよそ 50% が放出され、粉砕/無傷比は大幅に
増加する。
【0215】
コーティング後、8 mg の無傷ペレットでは、ナルトレキソンの放出が非被覆の無傷ペ
レットと比較して著しい減少を示した。但し、粉砕した被覆の 8 mg ペレットからの放出
は、粉砕した非被覆の 2 mg ペレットと比較して高い。
【実施例4】
【0216】
メタクリル共重合体の被覆塩酸ナルトレキソン 8 mg ペレット
実施例 4 では、8mg のナルトレキソンを含むペレットを実施例 3 と同様に調製したが
、メタクリル共重合体 (Eudragit RS 30D) でさまざまなレベル (5%、10%、15%、及び20%
、及び25% 重量増加) に被覆した。実施例 4 の 非被覆の塩酸ナルトレキソン製剤を下記
の表 4A に記載する。
【表19】
実施例 1 の非被覆塩酸ナルトレキソン製剤を
以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 3.9
kg/時の速度で連続供給する。ブレンド物を 95℃ 〜 100℃ のバレル温度で直径約 1 mm
の ストランドに押出成型する。押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0217】
In vitro 溶解工程:
実施例 4 に従って調製した非被覆の製剤を以下の in vitro 溶解検査方法で検査を行
い、以下の表 4B に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表20】
【0218】
模擬改変; プロセス及び溶解:
実施例 4 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 4C に記載した。改変プロセスでは、この
溶解研究のため、非被覆のナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を
用いて細砕し、粉末にした。
溶解方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表21】
【0219】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分での溶解分と無傷ペレットが 36 時間で
の溶解分との比率である。
粉砕/無傷比の結果: 57% / 18.7% = 3.0
【0220】
実施例 4 によって調製し、表 4A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティング (Eudragit に基づいて)
で被覆して、5%、10%、15% 及び20% 重量増加し; 及び疎水性コーティング (Eudragit
に基づいて) とカラーコーティング (Opadry) で被覆して25% の重量増加を得た。25% の
重量増加を得た製剤例の疎水性コーティング及びカラーコーティングを以下の表に記載す
る。
【表22】
実施例 4 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 700 g スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴霧
する。
* エアスピード: 8.5 〜 9.5 m/秒
* 吸気温度: 35℃
* 分散液噴霧速度: 14 g/分
5%、10%、15%、20%、及び25% の重量増加に対する分散の理論量を噴霧したときに、サン
プルを採取した。
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 8.5 m/秒
* 吸気温度: 35 ℃
* 分散液噴霧速度: 8.5 g/分
5. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
6. 硬化工程: スクリーニングしたペレット及びサンプルをオーブンに配置し、45℃ で 2
4 時間硬化する。
上記の 20% 製法及び工程に従って 5%、10%、15% 及び20% の重量増加に被覆したペレッ
トを、単位当たり 6.05、12.1、18.15 及び 24.2 mg の Eudragit RS30D (固体) をそれ
ぞれを用いて調製した。
【0221】
In vitro 溶解工程:
実施例 4 に従って調製した、疎水性コーティングで被覆した製剤を以下の in vitro
溶解検査方法で検査を行い、以下の表 4E に記載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、6、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表23】
溶解結果から分かるように、ナルトレキソンのペレットの溶解は一般的に重合体コーテ
ィングレベルの増加に伴って減少した。
【0222】
模擬改変のプロセス及び溶解:
実施例 4 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。45 分の溶解結果を表 4F に記載した。改変プロセスでは、この
溶解研究のため、被覆及び非被覆のナルトレキソンのペレットをそれぞれ個別に乳鉢と乳
棒 (24 ストローク) を用いて細砕し、粉末にした。
【0223】
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表24】
【0224】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記の表 4G に記載した。
結果:
【表25】
【0225】
上記の溶解結果から分かるように、コーティングレベルが増加するにつれ、無傷ペレッ
トから放出されるナルトレキソンの量が大幅に減少する (36 時間で 18% 以上から約 1%
以下に減少する) が、粉砕後には、拮抗薬のおよそ 50% が依然として放出され、粉砕/無
傷比は増加する。
【0226】
この製品は、コーティングの添加が無傷ペレットからのナルトレキソン放出の著しい減
少を結果としてもたらすが、粉砕されたペレットからはかなりの量の拮抗薬を放出する能
力を保持することを示す。
【実施例5】
【0227】
実施例 5 では、実施例 4 の製剤を GMP 条件下でパイロット規模で繰り返し、In-Vivo
評価に使用した。
【0228】
8 mg の ナルトレキソンを含むペレットを実施例 4 と同様に調製し、疎水性コーティ
ングで 15% 重量増加 (Eudragit RS 30D に基づいて) に被覆した。これらのペレットを
次いでサイズ #2 カプセルに充填した。実施例 5 における 非被覆の塩酸ナルトレキソン
製剤を下記の表に記載する。
【表26】
実施例 5 の非被覆ペレットを以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/分の速度で連続供給する。ブレンド物を 80℃ 〜 100℃ のバレル温度
で直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。押出成形したストランドをコ
ンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0229】
実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティングで15% 重量増加に被覆し
た(Eudragit RS 30D に基づいて)。被覆ペレットは下記の表に記載した。
【表27】
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 700 〜 780 CFM
* 吸気温度: 35 ℃
* 分散液噴霧速度:115 〜 135 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 750 〜 760 CFM
* 吸気温度: 35 ℃
* 分散液噴霧速度: 75 〜 95 g/分
5. スクリーニング: 14 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動分離
器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留した材
料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 149.7 mg でハードゼラチ
ンカプセルに充填する。
【0230】
In vitro 溶解 (無傷ペレット)
実施例 5 において疎水性コーティングで被覆した製剤を、バルクのペレット及びカプ
セル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表 5
C に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表28】
【0231】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 5 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表29】
【0232】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 46.4/0.8 = 58.0
【0233】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
この実施例の上記工程と製法 を用いて製造したカプセルを臨床研究に用いて、異なる
条件下の MEM 製剤の薬物動態学/バイオアベイラビリティを決定し、即放性ナルトレキソ
ン錠剤の薬物動態学/バイオアベイラビリティと比較した。ヒト被検体に無傷塩酸ナルト
レキソン MEM カプセル (1 カプセルまたは 5 カプセル、絶食); 粉砕した塩酸ナルトレ
キソン MEM (1 カプセルの含有量、細砕、絶食); 即放性塩酸ナルトレキソン剤型、錠剤
、絶食; または 1 MEM カプセル、無傷、給餌状態のいずれかを投与した。本研究は、非
盲検、単回投与、5 方向で、クロスオーバー研究を 15 健常者に対して、治療間 14 日間
の洗い出しで行った。治療法は次の通りに設計した:
A. 1x8 mg のナルトレキソン MEM カプセル、無傷、絶食状態
B. 1x8 mg のナルトレキソン MEM カプセル、カプセル内容物粉砕、絶食状態
C. 1x8 mg のナルトレキソン MEM カプセル、無傷、給餌状態
D. 5x8 mg (40 mg) ナルトレキソン MEM カプセル無傷、絶食状態
E. 2x0.5 mg (1 mg) ナルトレキソン即放性錠剤、絶食状態
【0234】
血漿濃度は、無傷状態で採取すると ナルトレキソン MEM ペレットからナルトレキソン
の非常に少量の放出があることを示す。時間曲線データと対比したナルトレキソン濃度 (
pg/ml) は、図2 に示してある。粉砕/細砕したナルトレキソンのペレットを絶食状態で経
口摂取させたとき、ナルトレキソン血漿レベルが大幅に増加した。
平均 Cmax 粉砕した MEM (N=14)/無傷 MEM (N=15) カプセルの比率は112.34 である。
同様に、平均 AUCt 粉砕した MEM(N=14)/無傷 MEM (N=15) カプセルの比率は 31.55 であ
る。
実施例 1 の非被覆 MEM 及び実施例 5 の被覆 MEM の in-vitro 及び in-vivo 比較は
下記の表 5E、及び 5F に示した。
【表30】
【表31】
【実施例6】
【0235】
メタクリル共重合体被覆の 2 mg の塩酸ナルトレキソン MEM
実施例 6 の 非被覆の塩酸ナルトレキソン製剤を下記の表 6A に記載する。
【表32】
実施例 6 の塩酸ナルトレキソン製剤を以下を用いて調製した:
工程:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27に 2.9 k
g/時 〜 4.8 kg/時の速度で連続供給する)。ブレンド物を 95℃ 〜 105℃ のバレル温度
で直径約 1 mm の ストランドに押出成型する。押出成形したストランドをコンベア上に
回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 1 mm のペレット
に切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0236】
In vitro 溶解 (無傷ペレット)
実施例 6 に従って調製した製剤を以下の in vitro 溶解検査方法で検査を行い、以下
の表 6B に記載した溶解結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、18、24、36 時間
3. 培地: 700 ml の SGF で 1 時間 / その後 900 ml SIF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表33】
【0237】
模擬改変及び溶解プロセス (粉砕したペレット):
実施例 6 に従って調製した製剤に対して模擬改変プロセスを実施し、以下の in vitro
溶解検査方法で検査した。1 時間の溶解結果を表 6C に記載した。改変プロセスでは、
この溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用い
て細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表34】
【0238】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。
粉砕/無傷比: 31% / 5.3% = 5.8:1
【0239】
実施例 6 によって調製し、表 6A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットを疎水性コーティングで15% 重量増加に被覆し
た(Eudragit RS 30D に基づいて)。15% の重量増加を得た製剤を以下の表に記載する。
【表35】
実施例 6 の被覆した塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 700 g スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴霧
する。
* エアスピード: 9.0 m/秒 .
* 吸気温度: 35 ℃
* 分散液噴霧速度: 8.8 g/分
3. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
【0240】
In vitro 溶解 (無傷ペレット)
実施例 6 に従って調製した、疎水性コーティングで被覆した製剤を以下の in vitro
溶解検査方法で検査を行い、以下の表 6E に記載した結果を得た。
方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー結果:
【表36】
【0241】
模擬改変及び溶解プロセス (粉砕したペレット):
実施例 6 に従って調製した被覆製剤に対して模擬改変プロセスを実施し、次いで以下
の溶解方法で検査した。1 時間の溶解結果を表 6F に記載した。改変プロセスでは、この
溶解研究のため、ナルトレキソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細
砕し、粉末にした。
溶解方法: 上記結果と同じ
【表37】
【0242】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 1 時間で溶解した % と無傷ペレットが 36 時
間で溶解した % との比率である。
【0243】
粉砕/無傷比: 7% / 0.6% = 12 (注: 検出可能なナルトレキソンが無傷ペレットから観
察されなかったので、粉砕/無傷比 12 をはるかに超える可能性があるが確認していない
。
【実施例7】
【0244】
メタクリル共重合体、次いで Surelease 被覆の 8 mg の塩酸ナルトレキソン MEM
実施例 7 では、MEM の2段階の順次コーティング、最初のコーティングは Eudragit R
S 30D により、重量増加は 15%、次のコーティングは Surelease による 10% の重量増加
(非被覆の押出成形したペレットに基づいて) を調製した。8mg のナルトレキソンを含む
ペレットを実施例 5 と同様に調製し、メタクリル共重合体 (Eudragit RS 30D) で 15%
の重量増加、継いで、エチルセルロース (Surelease) で10% の重量増加に被覆した。こ
の製品は、無傷ペレットからのナルトレキソン放出の著しい減少を結果としてもたらし、
その一方では、粉砕されたペレットからの放出を高めることを示す。実施例 7 の 非被覆
の塩酸ナルトレキソン製剤を下記の表 7A に記載する。
【表38】
実施例 7 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/時の速度で連続供給する。ブレンド物を 85℃ 〜 90℃ のバレル温度で
直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。押出成形したストランドをコン
ベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0245】
実施例 7 によって調製し、表 7A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。ペレットをメタクリル共重合体で10% 重量増加に被覆し
、継いでエチルセルロースで 15% 重量増加 (非被覆ペレットに基づいて) に被覆した。
被覆ペレットは下記の表に記載した。
【表39】
被覆塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. メタクリルコーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して
15 分間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の
水に分散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. エチルセルロースコーティングの分散: Surelease を合計 20% (w/w) 固体分散を達成
するのに十分な量の水と混合する。
3. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を達成する。
4. メタクリルコーティングコーティング: 以下のパラメータ指針を用いて Eudragit 分
散液を 700 g スケール以上で調製したナルトレキソンのペレットに流動床プロセッサ (G
PCG-1) を用いて噴霧する。
* エアスピード: 8.8 〜 9.0 m/秒
* 吸気温度: 35 ℃
* 分散液噴霧速度: 9.6 g/分
5. エチルセルロースコーティング: Eudragit コーティングの完了時点で、以下のパラメ
ータ指針を用いて Surelease 分散液を被覆したペレットに噴霧した。
* エアスピード: 9.0 m/秒
* 吸気温度: 40 〜 45℃
* 分散液噴霧速度: 9.2 〜 9.6 g/分
6. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 8.8 〜 9.0 m/秒
* 吸気温度: 50℃
* 分散液噴霧速度: 9.3 g/分
7. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
【0246】
In vitro 溶解 (無傷ペレット)
疎水性コーティング (メタクリル共重合体コーティング及びエチルセルロースコーティ
ング) で被覆し、実施例 7 に従って調製した製剤からは、
以下の in vitro 溶解方法で検査したところ、下記の表 7C に記載の結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表40】
【0247】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 7 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。溶解結果は
下記の表 7D に記載した。
溶解方法:
1. 装置- Apparatus-USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表41】
【0248】
粉砕/無傷比:
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 34 時間
で溶解した % との比率である。
粉砕/無傷比: 92.5
【実施例8】
【0249】
Surelease 次いで、メタクリル共重合体被覆の 8 mg の塩酸ナルトレキソン MEM
8mg のナルトレキソンを含むペレットを実施例 5 と同様に調製し、エチルセルロース
(Surelease) で10% 重量増加、継いで、メタクリル共重合体 (Eudragit RS 30D) で15%
重量増加 (非被覆ペレットに基づいて) に被覆した。この製品は、無傷ペレットからのナ
ルトレキソン放出の著しい減少を結果としてもたらし、その一方では、粉砕されたペレッ
トからの放出を高めることを示す。
【0250】
実施例 8 の 非被覆の塩酸ナルトレキソン製剤を下記の表 8A に記載する。
【表42】
実施例 8 における非被覆の塩酸ナルトレキソン製剤を以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを 16 メッシュのスクリーンを装備した
振動式製粉機にかけて、容易にブレンド可能な粉末を作る。
2. ブレンド工程: 塩酸ナルトレキソン、Eudragit RSPO、製粉したステアリルアルコール
、ステアリン酸及びBHT をツインシェルブレンダー内で混合する。
3. 押出成型工程: 第2工程でブレンドした材料を2軸押出機(Leistritz ZSE-27)に 4.0
kg/時 〜 4.8 kg/時の速度で連続供給する。ブレンド物を 85℃ 〜 90℃ のバレル温度で
直径約 0.8mm 〜 1.2mm の ストランドに押出成型する。
押出成形したストランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いて長さ約 0.8mm 〜 1.4mm
のペレットに切断する。
6. スクリーニング工程: 16 TBC メッシュ及び 26 TBC メッシュスクリーンを用いて振動
分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残留し
た材料を所望の製品として回収する。
【0251】
実施例 8 によって調製し、表 8A に記載した塩酸ナルトレキソンのペレットをさらに
疎水性コーティングで被覆した。本ペレットを、エチルセルロースで10% 重量増加、継い
で、メタクリル共重合体 (非被覆ペレットに基づいて) で15% 重量増加に被覆した。被覆
ペレットは下記の表に記載した。
【表43】
【0252】
実施例 8 における被覆塩酸ナルトレキソン製剤を下記を用いて調製した:
工程
1. エチルセルロースコーティングの分散: Surelease を合計 20% (w/w) 固体分散を達成
するのに十分な量の水と混合する。
2. メタクリルコーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して
15 分間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の
水に分散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
3. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を達成する。
4. エチルセルロースコーティング: 以下のパラメータ指針を用いて Surelease 分散液を
700 g スケールでナルトレキソンのペレットに流動床プロセッサ (GPCG-1) を用いて噴
霧する。
* エアスピード: 9.0 〜 9.2 m/秒
* 吸気温度: 50 ℃
* 分散液噴霧速度:10 g/分
5. メタクリルコーティングコーティング: 以下のパラメータ指針を用いて、Sureleaseコ
ーティングの完了時に、Eudragit 分散液をナルトレキソンのペレットに流動床プロセッ
サ (GPCG-1) を用いて噴霧する。
* エアスピード: 9.0 m/秒 .
* 吸気温度: 35 ℃
* 分散液噴霧速度:10.7 g/分
6. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアスピード: 750 〜 760 CPM
* 吸気温度: 50 ℃
* 分散液噴霧速度: 9.2 g/分
7. スクリーニング工程: 14 US メッシュスクリーン及び20 US メッシュスクリーンを通
してペレットをスクリーニングする。20 US メッシュスクリーン上に残留した材料を所望
の製品として回収する。
8. 硬化工程: スクリーニングしたペレットを 45℃ のオーブンに配置し、一部は 24 時
間硬化した後に除去し、残存材料は 48 時間硬化する。
【0253】
In vitro 溶解 (無傷ペレット)
実施例 8 に従って調製した、疎水性コーティング(エチルセルロース及びメタクリル共
重合体コーティング)で被覆した製剤を以下の溶解方法で検査を行い、以下の表 8C に記
載した結果を得た。
方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表44】
【0254】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 8 に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の溶
解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキソ
ンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。溶解結果は
下記の表 8D に記載した。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表45】
【0255】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比: 42.9
【0256】
3つの 25% 被覆ペレットの比較
実施例 4、7、及び 8 では、ナルトレキソン MEM ペレットを、様々なコーティング材
料または順序を用いて合計 25% のコーティングで被覆した。粉砕/無傷比を次の通りに比
較検討した。
【表46】
【0257】
粉砕 及び無傷 MEM ペレットの in-vitro 溶解データに基づくと、25% の Eudragit RS
コーティングは、組み合わせコーティングと比べてわずかに優れていると思われる。
【実施例9】
【0258】
放出制御塩酸オキシコドンの10 mg 錠剤は、次の通りのこの机上の実施例で調製するこ
とができる。 有機製造の塩酸オキシコドン (10 mg/錠剤) 及び噴霧乾燥ラクトース (71.
25 mg/錠剤) を適当なサイズのミキサーに移動し、およそ 6 分間混合する。Eudragit(
登録商標)RS PM 粉末 (6 mg/錠剤) をエタノールに分散する。 粉末を混合中に、粉末を
分散により顆粒化し、湿性の顆粒物質が得られるまで混合を続ける。追加エタノールを、
造粒エンドポイントに到達するために必要ならば添加する。造粒物を流動床の乾燥機に移
動し、30℃ で乾燥し、次いで 12-メッシュスクリーンを通す。残存 Eudragit(登録商標
)RS PM (9 mg/錠剤) を 90 部のエタノールと 10 部の精製水の溶剤に分散し; 顆粒にお
ける流動床の顆粒機/乾燥機に 30℃ で噴霧する。次には、顆粒を 12-メッシュスクリー
ンに通す。ステアリルアルコール (25 mg/錠剤) をおよそ 60 〜 70℃ で溶解する。温か
い顆粒をミキサーへ戻す。混合中、溶解したステアリルアルコールを添加する。被覆した
顆粒をミキサーから取り除き、冷却する。その後、該顆粒を 12-メッシュスクリーンに通
す。次には、顆粒を実施例 5 のナルトレキソン粒子及び薬剤的に望ましい錠剤賦形剤、
たとえば、滑石及びステアリン酸マグネシウムと好適なブレンダーでと混合し、錠剤に圧
縮する。
【実施例10】
【0259】
疼痛治療の方法
本発明に係る経口剤型を投与すると、患者に疼痛軽減を提供することができる。経口剤
型には、オピオイド作動薬及び実質的に放出不可能なオピオイド拮抗薬の経口有効量を含
有することができる。拮抗薬含有粒子のコーティングは、無傷拮抗薬含有粒子から拮抗薬
が漏出することを有利に低下する役割を果たす。
【0260】
経口剤型が経口で投与され、療法を必要とする患者の胃腸管に送達されると、正常消化
中にオピオイド作動薬は剤型から放出され患者に鎮痛を提供する。しかし、オピオイド拮
抗薬は、実質的に放出不可能になっているので、胃腸管を通過中には実質的に放出されな
い。好ましくは、拮抗薬の実質的に放出不可能な形態は、遅延性の結腸通過または胃酸欠
乏状態を管理するために用いられる緩下薬 (鉱物油) に対して耐性を持つ。経口剤型を改
変 (たとえば、機械的混合、加熱、または溶剤中溶出によって) することなく処方のとお
りに摂取する患者は、形剤摂取期間中にオピオイド作動薬の鎮痛有効性低下を結果として
もたらす量のオピオイド拮抗薬を吸収することはない。すなわち、オピオイド拮抗薬が無
傷剤型から放出され (経口で投与されるとき)、胃腸管から吸収され、患者の体内に蓄積
される量が、剤型に含まれているオピオイド作動薬の用量の鎮痛効力に実質的に影響また
は変化を及ぼすレベルに達することはない。
【実施例11】
【0261】
オピオイド作動薬の乱用を防止する方法
本発明に係る経口剤型を用いると、それに含まれるオピオイド作動薬の乱用を防止する
ことが可能である。本経口剤型はオピオイド拮抗薬と組み合わせたオピオイド作動薬を含
有する。オピオイド拮抗薬は、消化中に実質的に放出不可能な形態で存在する。このよう
に、意図されたように経口投与された経口剤型が 胃腸管に送達されると、形剤が改変さ
れていない限り、拮抗薬は実質的に GI システムに放出されない。ただし経口剤型が改変
される場合、たとえば機械混合 (たとえば、粉砕、せん断、細砕)、熱 (たとえば、45℃
of を超える温度、好ましくは45℃ 〜 50℃)、または剤型を溶剤に溶解 (加熱の有無にか
かわらず) することによって、オピオイド拮抗薬は放出されオピオイド効果を鈍化する。
このように、剤型が改変された際に、経口的、鼻腔内的、非経口的または舌下に投与され
ると、オピオイド作動薬の効果は、オピオイド拮抗薬によって少なくとも部分的に遮断さ
れる。
【実施例12】
【0262】
塩酸ナルトレキソンのペレットを有する塩酸ヒドロモルホンの放出制御カプセルは、次
の通りのこの机上の実施例で調製することができる。 製剤は下記の 12A 表に記載した:
【表47】
実施例 5 のカプセルを以下の工程を用いて調製した:
工程
1. 製粉工程: ステアリルアルコールのフレークを衝撃式製粉機にかける。
2. ブレンド工程: 塩酸ヒドロモルホン、Eudragit、エチルセルロース及び製粉したステ
アリルアルコールをツインシェルブレンダー内で混合する。
3. 押出成型工程: ブレンドした材料を2軸押出機に連続供給し、結果として生じるスト
ランドをコンベア上に回収する。
4. 冷却工程: ストランドをコンベア上で冷却する。
5. ペレット化工程: 冷却したストランドをペレタイザを用いてペレットに切断する。
6. スクリーニング工程: ペレットをスクリーニングし、所望のふるい部分に回収する。
7. フィルムコーティング工程: Opadry ピンク の水分散体を流動床のオピオイドのペレ
ットに噴霧する。
8. カプセル化工程: 126 mg の被覆し押出成形した塩酸ヒドロモルホンのペレット及び 1
49.7 mg の塩酸ナルトレキソンのペレット(実施例 5 から) をハードゼラチンカプセルに
充填する。
【0263】
当業者であれば本発明の変形形態は明らかであり、特許請求の範囲を逸脱しないものと
する。
【実施例13】
【0264】
実施例 13a
実施例 13a では、実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンの
ペレットをさらに疎水性コーティングで被覆した。ペレットを疎水性コーティングで25%
重量増加に被覆した(Eudragit RS 30D に基づいて)。被覆ペレットは下記の表に記載した
。
【表48】
実施例 13A のペレット調製において利用した工程は以下の通りである。
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 40 ℃
* 分散液噴霧速度: 75 〜 90 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 50 〜 55℃ .
* 分散液噴霧速度: 60 〜 70 g/分
5. スクリーニング: 14 US メッシュスクリーン及び26 US メッシュスクリーンを用いて
振動分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残
留した材料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 164.86 mg でハードゼラチ
ンカプセルに充填する。
【0265】
In vitro 溶解 (無傷ペレット)
実施例 13a において疎水性コーティングで被覆した製剤を、バルクのペレット及びカ
プセル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表
13B に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表49】
【0266】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 13a に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の
溶解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキ
ソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 45 分間
3. 培地: 700 ml の SGF
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表50】
【0267】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 27.0/0.5 = 54
【0268】
実施例 13b
実施例 13b では、実施例 5 によって調製し、表 5A に記載した塩酸ナルトレキソンの
ペレットをさらに疎水性コーティングで被覆した。ペレットを疎水性コーティングで30%
重量増加に被覆した (Surelease E-7-10901 に基づいて)。
被覆ペレットは下記の表に記載した。
【表51】
実施例 13A のペレット調製において利用した工程は以下の通りである。
工程
1. 機能コーティングの分散: Eudragit RS 30D をクエン酸トリエチル と混合して 15 分
間可塑化する。Cab-O-Sil を合計 20% (w/w) 固体分散を達成するのに十分な量の水に分
散する。Cab-O-Sil 分散液 を Eudragit 混合物に加える。
2. カラーコーティングの分散: Opadry を水分と混合して10% (w/w) 分散液を得る。
3. 機能コーティング: 以下のパラメータ指針を用いて Eudragit 分散液を 9 kg スケー
ル以上で調製したナルトレキソンのペレットに流動床プロセッサ (GPCG-15) を用いて噴
霧する。
* エアフロー: 400 〜 450 CFM .
* 吸気温度: 40 ℃
* 分散液噴霧速度: 75 〜 90 g/分
4. カラーコーティング: 機能コーティングの完了時点で、以下のパラメータ指針を用い
て Opadry 分散液を被覆したペレットに噴霧した。
* エアフロー: 400 〜 450 CFM
* 吸気温度: 50 〜 55℃ .
* 分散液噴霧速度: 60 〜 70 g/分
5. スクリーニング: 14 US メッシュスクリーン及び26 US メッシュスクリーンを用いて
振動分離器を介してペレットをスクリーニングする。26 TBC メッシュスクリーン上に残
留した材料を所望の製品として回収する。
6. カプセル化工程: スクリーニングしたペレットを目標重量 164.86 mg でハードゼラチ
ンカプセルに充填する。
【0269】
In vitro 溶解 (無傷ペレット)
実施例 13b において疎水性コーティングで被覆した製剤を、バルクのペレット及びカ
プセル化したペレットの形式で、以下の in vitro 溶解検査方法で検査を行い、以下の表
13E に記載した結果を得た。
溶解方法:
1. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
2. サンプリング時間: 1、2、4、8、12、24、36 時間
3. 培地: 700 ml の SGF で 1 時間、その後、900 ml の SIF に切り替える
4. 分析方法: 高速液体クロマトグラフィー
結果:
【表52】
【0270】
模擬改変; プロセス及び溶解 (粉砕したペレット):
実施例 13b に従って調製した製剤に対して模擬改変プロセスを実施し、次いで以下の
溶解検査方法で検査した。改変プロセスでは、この溶解研究のため、被覆したナルトレキ
ソンのペレットを乳鉢と乳棒 (24 ストローク) を用いて細砕し、粉末にした。
溶解方法:
5. 装置- USP 2型 (パドル)、回転数 50 rpm、温度 37℃
6. サンプリング時間: 45 分間
7. 培地: 700 ml の SGF
8. 分析方法: 高速液体クロマトグラフィー
結果:
【表53】
【0271】
粉砕/無傷比
粉砕/無傷の比率とは、粉砕ペレットが 45 分で溶解した % と無傷ペレットが 36 時間
で溶解した % との比率である。結果は下記に記載した。
粉砕/無傷比の結果: 26.0/l .6 = 16.3
【0272】
In Vivo ヒト薬物動態学/バイオアベイラビリティの研究
実施例 13a 及び 13b の上記工程を用いて製造したカプセルを2つの個別の臨床研究に
用いて、異なる条件下の MEM 製剤の薬物動態学/バイオアベイラビリティを決定し、即放
性ナルトレキソン錠剤の薬物動態学/バイオアベイラビリティと比較した。ヒト被検体に
無傷塩酸ナルトレキソン MEM カプセル (1 カプセルまたは 5 カプセル、絶食); 粉砕し
た塩酸ナルトレキソン MEM (1 カプセルの含有量、細砕、絶食); 即放性塩酸ナルトレキ
ソン剤型、錠剤、絶食; または 1 MEM カプセル、無傷、給餌状態のいずれかを投与した
。これらの諸研究は、非盲検、単回投与、5 方向、クロスオーバー研究で健常者において
行った治療法は次の通りに設計した:
A. 1x8 mg のナルトレキソン MEM カプセル、無傷、絶食状態
B. 1x8 mg のナルトレキソン MEM カプセル、カプセル内容物粉砕、絶食状態
C. 1x8 mg のナルトレキソン MEM カプセル、無傷、給餌状態
D.5x8 mg (40 mg) ナルトレキソン MEM カプセル無傷、絶食状態
E. 1 x 1 mg のナルトレキソン即放性錠剤、絶食状態
【0273】
予備的な血漿濃度は、無傷状態で採取すると ナルトレキソン MEM ペレットからナルト
レキソンの無視できる量の放出があることを示す。時間曲線データと対比したナルトレキ
ソン濃度 (pg/ml) は、図3 及び 4 に示してある。粉砕/細砕したナルトレキソンのペレ
ットを絶食状態で経口摂取させたとき、ナルトレキソン血漿レベルが大幅に増加した。平
均 Cmax 粉砕 MEM/無傷 MEM カプセル比は 25% の Eudragit コーティング及び 30% Sure
lease コーティングは、それぞれ 187.91 及び 71.98 であった。 同様に、平均 AUCt 粉
砕 MEM/無傷 MEM カプセル比は 25% の Eudragit コーティング及び 30% Surelease コー
ティングは、それぞれ 66.07 及び 39.27 であった。
実施例 l3a 及び 13b の被覆 MEM の in-vitro 及び In-Vivo の比較は下記の表 13G
に示した。
【表54】
【表55】
【0274】
本明細書中で開示した本発明は特有の実施形態及びアプリケーションを用いて説明した
が、当業者であれば幾多の変更形態及び改変は本発明の精神及び範囲から逸脱することな
く行い得る。そのような変更形態は特許請求の範囲内であるものと見なされる。
【図面の簡単な説明】
【0275】
【図1】無傷の塩酸ナルトレキソン MEM (全体)、粉砕した塩酸ナルトレキソン MEM (細砕) 及び実施例 1 の即放性塩酸ナルトレキソン錠剤 (IR NTX) 剤型に対する血漿濃度時間のグラフ表示である。
【0276】
【図2】実施例 5 の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【0277】
【図3】実施例 13A の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【0278】
【図4】実施例 13B の時間曲線データと対比したナルトレキソン濃度 (pg/ml) のグラフ表示である。
【特許請求の範囲】
【請求項1】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が押
出成型された粒子の約5%〜約30%の重量を有する層。
【請求項2】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子、及び押
出成形した粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2
疎水性物質が、たとえば、押出成形した粒子重量の約5%〜約30%を含有する複数の粒
子;
b) 第3疎水性物質内に分散したオピオイド作動薬を含有する複数の粒子;及び
c) 複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の押出成形した粒子を
含むカプセル。
【請求項3】
その粒子のそれぞれにマトリックス内に分散したオピオイド拮抗薬を含有する複数の押
出成形した粒子;及び押出成形した粒子の周囲に配置した層;無傷剤型内のオピオイド拮
抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項4】
オピオイド作動薬粒子が押出成形によって形成された請求項2の医薬製品。
【請求項5】
オピオイド拮抗薬の粒子が下記の工程によって形成される請求項1〜3の任意の一項に
係る医薬製品。
a) オピオイド拮抗薬と第1疎水性物質をブレンドしてブレンド物を形成する工程;
b) 少なくとも混合物を軟化するのに充分な温度にブレンド物を加熱する工程;
c) 混合物を押出成形してストランドを形成する工程;及び
d) ストランドを粒子に切断する工程。
【請求項6】
オピオイド拮抗薬の粒子が平均直径約0.1〜約6.0mmである請求項5の医薬製品
。
【請求項7】
オピオイド作動薬粒子が下記の工程によって形成される請求項4の医薬製品。
a) オピオイド作動薬及び第3疎水性物質ををブレンドし、混合物を形成する工程;
b) 混合物を少なくとも混合物を軟化するのに充分な温度に加熱する工程;
c) 混合物を押出成形してストランドを形成する工程;及び
d) ストランドを粒子に切断する工程。
【請求項8】
マトリックスが第1疎水性物質を含有する請求項3の医薬製品。
【請求項9】
層が第2疎水性物質を含有する請求項8の医薬製品。
【請求項10】
第1疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項1、2または8の医薬製品。
【請求項11】
第2疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項9の医薬製品。
【請求項12】
第1疎水性物質及び第2疎水性物質が同じである請求項9の医薬製品。
【請求項13】
第3疎水性物質内に分散したオピオイド作動薬をそのそれぞれに含有する薬剤的に許容
できる第2の複数個の粒子をさらに含有する請求項9の医薬製品。
【請求項14】
第3疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項13の医薬製品。
【請求項15】
第1疎水性物質、第2疎水性物質及び第3疎水性物質が同じである請求項13の医薬製
品。
【請求項16】
第1疎水性物質及び第3疎水性物質が同じである請求項13の医薬製品。
【請求項17】
第2疎水性物質及び第3疎水性物質が同じである請求項13の医薬製品。
【請求項18】
改変された剤型の摂取後に放出されるオピオイド拮抗薬量がオピオイド作動薬の陶酔効
果を遮断するのに効果的である請求項2の医薬製品。
【請求項19】
改変された剤型の摂取後に放出されるオピオイド拮抗薬量がオピオイド作動薬の陶酔効
果を遮断するのに効果的である請求項13の医薬製品。
【請求項20】
複数粒子が平均直径約0.1〜約3mmを持つ請求項1〜3の任意の一項の医薬製品。
【請求項21】
オピオイド作動薬が、アルフェンタニル、アリルプロジン、アルファプロジン、アニレ
リジン、ベンジルモルヒネ、ベジトラミド、ブプレノルフィン、ブトルファノール、クロ
ニタゼン、コデイン、デソモルヒネ、デキシトロモルアミド、デゾシン、ジアンプロミド
、ジアモルホン、ジヒドロコデイン、ジヒドロモルヒネ、ジメノキサドール、ジメフェプ
タノール、ジメチルチアンブテン、ジオキサフェチルブチラート、ジピパノン、エプタゾ
シン、エトヘプタジン、エチルメチルチアンブテン、エチルモルヒネ、エトニタゼン、エ
トルフィン、ジヒドロエトルフィン、フェンタニール及び誘導体、ヘロイン、ヒドロコド
ン、ヒドロモルホン、ヒドロキシペチジン、イソメタドン、ケトベミドン、レボルファノ
ール、レボフェナシルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタ
ゾシン、メサドン、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノル
レボルファノール、ノルメサドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピ
パノン、アヘン、オキシコドン、オキシモルホン、パパベレタム、ペンタゾシン、フェナ
ドキソン、フェノモルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミ
ド、プロフェプタジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニ
ル、チリジン、トラマドル、これらの薬剤的に許容できる塩類、及び前述した物質のいず
れかの混合物からなる群より選択される請求項2または13の医薬製品。
【請求項22】
オピオイド作動薬が、ヒドロコドン、モルヒネ、ヒドロモルホン、オキシコドン、コデ
イン、レボルファノール、メペリジン、メサドン、オキシモルホン、ブプレノルフィン、
フェンタニール及びその誘導体、ジピパノン、ヘロイン、トラマドル、エトルフィン、ジ
ヒドロエトルフィン、ブトルファノール、レボルファノール、それらの薬剤的に許容でき
る塩類、前述した物質のいずれかの混合物からなる群より選択される請求項2または13
の医薬製品。
【請求項23】
オピオイド拮抗薬が、ナルトレキソン、ナロキソン、ナルメフェン、シクラザシン、レ
バロルファン、それらの薬剤的に許容できる塩類、及び前述した物質のいずれかの混合物
からなる群より選択される請求項1〜3の任意の一項の医薬製品。
【請求項24】
オピオイド拮抗薬がナルトレキソンまたはその薬剤的に許容できる塩類である請求項1
〜3の任意の一項の医薬製品。
【請求項25】
マトリックスが層なしで拮抗薬を隔離する能力を持ち、その層が隔離機能を高める請求
項3の医薬製品。
【請求項26】
層がマトリックスなしで拮抗薬を隔離する能力を持ち、そのマトリックスが隔離機能を
高める請求項3の医薬製品。
【請求項27】
マトリックスが層なしで拮抗薬を隔離する能力を持たず、層がマトリックスなしで拮抗
薬を隔離する能力を持たず、マトリックス及び層が組み合わさると拮抗薬を隔離する能力
を持つ請求項3の医薬製品。
【請求項28】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の 1 時間での溶出に基づいて、改変後に剤型から放出
される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が 約 20:1 以上;約
50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上となるように剤型内
のオピオイド拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項29】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 2 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項 28 の医薬製品。
【請求項30】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 4 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項 28 の医薬製品。
【請求項31】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間 で 9
00 ml の SIF に切り替える条件で、剤型の 12 時間での溶出に基づいた、改変後に剤型
から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 20:1
以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である請求
項28の医薬製品。
【請求項32】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 24 時間での溶出に基づいた、改変後
に剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約
20:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上であ
る請求項28の医薬製品。
【請求項33】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる700 ml の SGF 中で 1 時間、その
後 900 ml の SIF に切り替える条件で、剤型の 36 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項28の医薬製品。
【請求項34】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の 1 時間での溶出に基づいた無傷剤型から放出される
拮抗薬の重量百分率が1.0 重量%未満;0.5 重量%未満;0.2 重量%;または0.1 重量%
未満となるように剤型内のオピオイド拮抗薬を隔離するマトリックス及び層を含有する医
薬製品。
【請求項35】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 2 時間での溶出に基づいた無傷剤型か
ら放出される拮抗薬の重量百分率が、2.0 重量%未満;1.0 重量%未満;0.5 重量%未満
;または 0.25 重量%未満である請求項34の医薬製品。
【請求項36】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、そ
の後に 900 ml の SIF に切り替える条件で、剤型の 4 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、2.2 重量%未満;1.5 重量%未満;1.0 重量%未
満;または0.75 重量%未満である請求項34の医薬製品。
【請求項37】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 12 時間で 9
00 ml の SIF に切り替える条件で、剤型の 12 時間での溶出に基づいた無傷剤型から放
出される拮抗薬の重量百分率が、3.0 重量%未満;1.8 重量%未満;1.25 重量%未満;
または0.3 重量%未満である請求項34の医薬製品。
【請求項38】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 24 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、4.8 重量%未満;2.5 重量%未満;1.8 重量%未
満;または0.4 重量%未満である請求項34の医薬製品。
【請求項39】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 36 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、7.0 重量%未満;6.5 重量%未満;3.0 重量%;
または未満1.5 重量%である請求項34の医薬製品。
【請求項40】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の溶出に基づいて 1 時間、継いで、900 ml の SIF に
切り替えた条件で無傷剤型が、1.0% 以下の拮抗薬を 1 時間で、2.0%以下の拮抗薬を2
時間で、2.2%以下の拮抗薬を4時間で、3.0%以下の拮抗薬を12時間で、4.8%以下の
拮抗薬を24時間で、及び 7.0%以下の拮抗薬を36時間で放出するように剤型内のオピ
オイド拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項41】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 0.5% 以下
の拮抗薬を 1 時間で、1.0%以下の拮抗薬を 2 時間で、1.5%以下の拮抗薬を 4 時間で
、1.8%以下の拮抗薬を 12 時間で、2.5%以下の拮抗薬を 24 時間で及び 6.5%以下の拮
抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項42】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 02%以下の
拮抗薬を 1 時間で、0.5%以下の拮抗薬を 2 時間で、1.0%以下の拮抗薬を 4 時間で、1
.25%以下の拮抗薬を 12 時間で、及び 1.8%以下の拮抗薬を 24 時間で、及び 3.0%以
下の拮抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項43】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 0.1%以下
の拮抗薬を 1 時間で、0.25%以下の拮抗薬を 2 時間で、0.75%以下の拮抗薬を 4 時間
で、0.3%以下の拮抗薬を 12 時間で、0.4%以下の拮抗薬を 24 時間で、及び 1.5%以下
の拮抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項44】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項2の医薬製品。
【請求項45】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項13の医薬製品。
【請求項46】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項22の医薬製品。
【請求項47】
オピオイド拮抗薬の粒子の平均直径約0.1〜約6.0mmである請求項7及び請求項
28〜46の任意の一項の医薬製品。
【請求項48】
オピオイド拮抗薬の粒子が a) オピオイド拮抗薬と第1疎水性物質をブレンドして混合
物を形成すること;b) 少なくとも混合物を軟化するのに充分な温度に混合物を加熱する
こと;c) 混合物を押出成形してストランドを形成すること;及びd) ストランドを粒子に
切断することによって形成される請求項28〜46の任意の一項の医薬製品。
【請求項49】
オピオイド拮抗薬の粒子の平均直径が約0.1〜約6.0mmである請求項48の医薬
製品。
【請求項50】
作動薬粒子と拮抗薬粒子が、外観、質感、臭い、味、硬度、形状、サイズまたはその組
み合わせからなる群より選択される特質において類似しているような、オピオイド作動薬
及びオピオイド作動薬の周囲に配置した層を含有する複数の粒子;及び複数のオピオイド
拮抗薬の粒子及びオピオイド拮抗薬の粒子の周囲に配置した層を含有する医薬製品。
【請求項51】
作動薬粒子と拮抗薬粒子が、外観、質感、臭い、味、硬度、形状、サイズまたはその組
み合わせからなる群より選択される特質において実質的に識別できないような、オピオイ
ド作動薬及びオピオイド作動薬の周囲に配置した層を含有する複数の粒子;及び複数のオ
ピオイド拮抗薬の粒子及びオピオイド拮抗薬の粒子の周囲に配置した層を含有する医薬製
品。
【請求項52】
オピオイド作動薬を含有する複数の粒子を調製すること;オピオイド拮抗薬を含有する
複数の粒子を調製すること;オピオイド作動薬粒子とオピオイド拮抗薬の粒子が類似する
外観を持つように、層をオピオイド作動薬粒子及びオピオイド拮抗薬の粒子に適用するこ
とを含有する医薬製品の調製方法。
【請求項53】
オピオイド作動薬を含有する複数の粒子を調製すること;オピオイド拮抗薬を含有する
複数の粒子を調製すること;オピオイド作動薬粒子及びオピオイド拮抗薬の粒子の外観が
実質的に識別できないように、層をオピオイド作動薬粒子及びオピオイド拮抗薬の粒子に
適用することを含有する医薬製品の調製方法。
【請求項54】
下記を含有する医薬製品の調製方法。
a) 押出成形で粒子を形成することによってオピオイド拮抗薬を第1疎水性物質内に分散
させること;及び
b) 粒子重量の約5%〜約30%を持つ第2疎水性物質を含有する層を粒子の周辺に配置
すること。
【請求項55】
下記を含有する医薬製品の調製方法。
a) 押出成型で複数の粒子を形成することによってオピオイド拮抗薬を第1疎水性物質内
に分散させること、及び粒子重量の約5%〜約30%の量を持つ第2疎水性物質を含有す
る層を粒子のそれぞれのの周辺に配置すること;
b) 第3疎水性物質内のオピオイド作動薬を分散させ、複数の粒子を形成すること;及び
c) カプセル内に複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の粒子を
含むこと。
【請求項56】
下記を含有する医薬製品の調製方法。
押出成型で複数の薬剤的に許容できる粒子を形成することによって、マトリックス中にオ
ピオイド拮抗薬を分散させること及び該マトリックス及び層が無傷剤型内のオピオイド拮
抗薬を隔離するようにそれぞれの粒子に該層を配置すること。
【請求項57】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び該粒子の周囲に配置した層;無傷剤型内のオピオイド拮抗薬を隔離する
マトリックスを含有する医薬製品。
【請求項58】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 20:1 以上であ
る請求項1〜3の任意の一項の医薬製品。
【請求項59】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 100:1 以上で
ある請求項58の医薬製品。
【請求項60】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 125:1 以上で
ある請求項58の医薬製品。
【請求項61】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 150:1 以上で
ある請求項58の医薬製品。
【請求項62】
改変が粉砕による粉末化によって行われる、請求項28〜33、44〜46または58
〜61の任意の一項の医薬製品。
【請求項63】
粉砕が乳鉢及び乳棒によって行われる、請求項62の医薬製品。
【請求項64】
マトリックス内に分散した約 2 mg のナルトレキソンまたは薬剤的に許容できる塩類を
含有する複数の押出成形した粒子;及び該粒子周囲に配置した層;50 rpm、37 ℃ で USP
2型 (パドル) 装置を用いる剤型の溶出に基づいて、700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF 中で無傷剤型が 0.065 mg 以下の拮抗薬が 36 時間で放出するように
剤型内のナルトレキソンまたはその塩類を隔離するマトリックス及び層を含有する医薬製
品。
【請求項65】
50 rpm、37 ℃ でUSP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF に切り替える条件で、無傷剤型が 0.04 mg 以下の拮抗薬を 36 時間で
放出する、請求項64の医薬製品。
【請求項66】
マトリックス内に分散した約 8 mg のナルトレキソンまたは薬剤的に許容できる塩類を
含有する複数の押出成形した粒子; 及び該粒子周囲に配置した層; 50 rpm、37 ℃ で USP
2型 (パドル) 装置を用いる剤型の溶出に基づいて、700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF 中で無傷剤型が 0.08 mg 以下の拮抗薬が 36 時間で放出するように剤
型内のナルトレキソンまたはその塩類を隔離するマトリックス及び層を含有する医薬製品
。
【請求項67】
50 rpm、37 ℃ でUSP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF に切り替える条件で、無傷剤型が 0.12 mg 以下の拮抗薬を 36 時間で
放出する、請求項64の医薬製品。
【請求項68】
下記を含有する医薬製品:
a) アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合物からなる群より
選択される第1疎水性物質に分散する塩酸ナルトレキソンを含有する押出成形した粒子;
及び
b) 該粒子の周囲に配置した第2疎水性物質で、アルキルセルロース、アクリル樹脂、及
びその混合物からなる群より選択される第2疎水性物質を含有する層。
【請求項69】
a) アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合物からなる群よ
り選択される第1疎水性物質に分散する塩酸ナルトレキソンを含有する複数の押出成形し
た粒子及びアルキルセルロース、アクリル樹脂、及びその混合物からなる群より選択され
る第2疎水性物質を含有し、それぞれの粒子の周囲に配置された層;b) アクリル樹脂、
ステアリルアルコール、ステアリン酸及びその混合物からなる群より選択される第3疎水
性物質内に分散したオキシコドン、ヒドロコドン、ヒドロモルホン及びその薬剤的に許容
できる塩類からなる群より選択されるオピオイド作動薬を含有する複数の粒子;及び c)
複数個のオピオイド作動薬粒子及び複数個の塩酸ナルトレキソン粒子を含むカプセルを含
有する医薬製品。
【請求項70】
それぞれの粒子が、アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合
物からなる群より選択される疎水性物質を含有するマトリックス内に分散した塩酸ナルト
レキソンを含有する、複数の押出成形した粒子;及びアルキルセルロース、アクリル樹脂
及び粒子の周囲に配置したその混合物からなる群より選択される材料を含有する層;無傷
剤型内の塩酸ナルトレキソンを隔離するマトリックス及び層を含有する医薬製品。
【請求項71】
塩酸ナルトレキソンの量が約2mg〜約12mgである請求項68〜70の任意の一項
の医薬製品。
【請求項72】
塩酸ナルトレキソンの量が約2mg〜約8mgである請求項71の医薬製品。
【請求項73】
塩酸ナルトレキソン粒子が90%を超える疎水性物質を含有する請求項68〜70の任
意の一項の医薬製品。
【請求項74】
塩酸ナルトレキソン粒子が95%超える疎水性物質を含有する請求項73の医薬製品。
【請求項75】
層の量が塩酸ナルトレキソン粒子の重量の約5%〜約30%である請求項68〜70の
任意の一項の医薬製品。
【請求項76】
粒子のそれぞれにマトリックス内に分散したオピオイド拮抗薬を含有する複数の押出成
形した粒子;及び該粒子の周囲に配置した層;無傷剤型内のオピオイド拮抗薬を隔離する
マトリックス及び層を含有し、層が二層配列 (bilaminar arraignment) にアクリル重合
体及びセルロースポリマーを含有する、医薬製品。
【請求項77】
アクリル重合体が拮抗薬の粒子の周囲に配置され、セルロースポリマーがアクリル重合
体層状拮抗薬の粒子の周囲に配置された請求項76の医薬製品。
【請求項78】
第2疎水性物質の量が粒子重量の約16%〜約30%である請求項1または2の医薬製
品。
【請求項79】
第2疎水性物質の量が粒子重量の約20%〜約29%である請求項1または2の医薬製
品。
【請求項80】
第2疎水性物質の量が粒子重量の約22%〜約28%である請求項1または2の医薬製
品。
【請求項81】
層が実質的に拮抗薬を欠いている請求項1〜3の任意の一項の医薬製品。
【請求項82】
剤型が即放性拮抗薬を欠いている請求項1〜3の任意の一項の医薬製品。
【請求項83】
前記粉砕が24ストロークの乳鉢及び乳棒で行われる請求項63の医薬製品。
【請求項84】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散した有害薬剤または拮抗薬を含有する押出成形した粒子;及
び
b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が粒
子の約5%〜約30%の重量を有する層。
【請求項85】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散した有害薬剤または拮抗薬を含有する押出成形した粒子、及
び粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物
質が、たとえば、粒子重量の約5%〜約30%を含有する複数の粒子;
b) 第3疎水性物質内に分散した作用薬を含有する複数の粒子;及び
c) 複数個の作用薬粒子及び複数個の有害薬剤または拮抗薬の粒子を含むカプセル。
【請求項86】
複数の押出成形した粒子であって、そのそれぞれにマトリックス内に分散した有害薬剤
または拮抗薬を含有する粒子;及び粒子の周囲に配置した層;無傷剤型内の有害薬剤また
は拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項87】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項6の医薬製品。
【請求項88】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項47の医薬製品
。
【請求項89】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項49の医薬製品
。
【請求項90】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 5:1 以上である
請求項1〜3の医薬製品。
【請求項91】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 25:1 以上であ
る請求項90の医薬製品。
【請求項92】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 75:1 以上であ
る請求項90の医薬製品。
【請求項93】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 200:1 以上であ
る請求項90の医薬製品。
【請求項94】
オピオイド作動薬がオキシコドンまたはその薬剤的に許容できる塩類である請求項2の
医薬製品。
【請求項95】
オピオイド作動薬がヒドロモルホンまたはその薬剤的に許容できる塩類である請求項2
の医薬製品。
【請求項96】
オピオイド作動薬がヒドロコドンまたは薬剤的にその許容できる塩類である請求項2の
医薬製品。
【請求項97】
オピオイド作動薬がオキシモルホンまたはその薬剤的に許容できる塩類である請求項2
の医薬製品。
【請求項98】
オピオイド作動薬がモルヒネまたはその薬剤的に許容できる塩類である請求項2の医薬
製品。
【請求項99】
請求項2〜13の任意の一項の医薬製品をそれを必要とする患者に経口で投与する工程
を含有する疼痛治療の方法。
【請求項100】
請求項1または2の任意の一項の医薬製品をそれを必要とする患者に経口投与すること
を含有する投与の方法。
【請求項101】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有し、改変剤型を患者集団に単回投与した
後に得られる拮抗薬の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮
抗薬の平均 AUC に対する比率が約 5:1 以上である層。
【請求項102】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有し、改変剤型を患者集団に単回投与した
後に得られる拮抗薬の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮
抗薬の平均 AUC に対する比率が約 20:1 以上である層。
【請求項1】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が押
出成型された粒子の約5%〜約30%の重量を有する層。
【請求項2】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する押出成形した粒子、及び押
出成形した粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2
疎水性物質が、たとえば、押出成形した粒子重量の約5%〜約30%を含有する複数の粒
子;
b) 第3疎水性物質内に分散したオピオイド作動薬を含有する複数の粒子;及び
c) 複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の押出成形した粒子を
含むカプセル。
【請求項3】
その粒子のそれぞれにマトリックス内に分散したオピオイド拮抗薬を含有する複数の押
出成形した粒子;及び押出成形した粒子の周囲に配置した層;無傷剤型内のオピオイド拮
抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項4】
オピオイド作動薬粒子が押出成形によって形成された請求項2の医薬製品。
【請求項5】
オピオイド拮抗薬の粒子が下記の工程によって形成される請求項1〜3の任意の一項に
係る医薬製品。
a) オピオイド拮抗薬と第1疎水性物質をブレンドしてブレンド物を形成する工程;
b) 少なくとも混合物を軟化するのに充分な温度にブレンド物を加熱する工程;
c) 混合物を押出成形してストランドを形成する工程;及び
d) ストランドを粒子に切断する工程。
【請求項6】
オピオイド拮抗薬の粒子が平均直径約0.1〜約6.0mmである請求項5の医薬製品
。
【請求項7】
オピオイド作動薬粒子が下記の工程によって形成される請求項4の医薬製品。
a) オピオイド作動薬及び第3疎水性物質ををブレンドし、混合物を形成する工程;
b) 混合物を少なくとも混合物を軟化するのに充分な温度に加熱する工程;
c) 混合物を押出成形してストランドを形成する工程;及び
d) ストランドを粒子に切断する工程。
【請求項8】
マトリックスが第1疎水性物質を含有する請求項3の医薬製品。
【請求項9】
層が第2疎水性物質を含有する請求項8の医薬製品。
【請求項10】
第1疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項1、2または8の医薬製品。
【請求項11】
第2疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項9の医薬製品。
【請求項12】
第1疎水性物質及び第2疎水性物質が同じである請求項9の医薬製品。
【請求項13】
第3疎水性物質内に分散したオピオイド作動薬をそのそれぞれに含有する薬剤的に許容
できる第2の複数個の粒子をさらに含有する請求項9の医薬製品。
【請求項14】
第3疎水性物質が、セルロースポリマー、アクリル重合体及び共重合体、メタクリル酸
重合体及び共重合体、セラック、ゼイン、硬化ヒマシ油、硬化植物油、及び前述した物質
のいずれかの混合物からなる群より選択される請求項13の医薬製品。
【請求項15】
第1疎水性物質、第2疎水性物質及び第3疎水性物質が同じである請求項13の医薬製
品。
【請求項16】
第1疎水性物質及び第3疎水性物質が同じである請求項13の医薬製品。
【請求項17】
第2疎水性物質及び第3疎水性物質が同じである請求項13の医薬製品。
【請求項18】
改変された剤型の摂取後に放出されるオピオイド拮抗薬量がオピオイド作動薬の陶酔効
果を遮断するのに効果的である請求項2の医薬製品。
【請求項19】
改変された剤型の摂取後に放出されるオピオイド拮抗薬量がオピオイド作動薬の陶酔効
果を遮断するのに効果的である請求項13の医薬製品。
【請求項20】
複数粒子が平均直径約0.1〜約3mmを持つ請求項1〜3の任意の一項の医薬製品。
【請求項21】
オピオイド作動薬が、アルフェンタニル、アリルプロジン、アルファプロジン、アニレ
リジン、ベンジルモルヒネ、ベジトラミド、ブプレノルフィン、ブトルファノール、クロ
ニタゼン、コデイン、デソモルヒネ、デキシトロモルアミド、デゾシン、ジアンプロミド
、ジアモルホン、ジヒドロコデイン、ジヒドロモルヒネ、ジメノキサドール、ジメフェプ
タノール、ジメチルチアンブテン、ジオキサフェチルブチラート、ジピパノン、エプタゾ
シン、エトヘプタジン、エチルメチルチアンブテン、エチルモルヒネ、エトニタゼン、エ
トルフィン、ジヒドロエトルフィン、フェンタニール及び誘導体、ヘロイン、ヒドロコド
ン、ヒドロモルホン、ヒドロキシペチジン、イソメタドン、ケトベミドン、レボルファノ
ール、レボフェナシルモルファン、ロフェンタニル、メペリジン、メプタジノール、メタ
ゾシン、メサドン、メトポン、モルヒネ、ミロフィン、ナルセイン、ニコモルヒネ、ノル
レボルファノール、ノルメサドン、ナロルフィン、ナルブフェン、ノルモルヒネ、ノルピ
パノン、アヘン、オキシコドン、オキシモルホン、パパベレタム、ペンタゾシン、フェナ
ドキソン、フェノモルファン、フェナゾシン、フェノペリジン、ピミノジン、ピリトラミ
ド、プロフェプタジン、プロメドール、プロペリジン、プロポキシフェン、スフェンタニ
ル、チリジン、トラマドル、これらの薬剤的に許容できる塩類、及び前述した物質のいず
れかの混合物からなる群より選択される請求項2または13の医薬製品。
【請求項22】
オピオイド作動薬が、ヒドロコドン、モルヒネ、ヒドロモルホン、オキシコドン、コデ
イン、レボルファノール、メペリジン、メサドン、オキシモルホン、ブプレノルフィン、
フェンタニール及びその誘導体、ジピパノン、ヘロイン、トラマドル、エトルフィン、ジ
ヒドロエトルフィン、ブトルファノール、レボルファノール、それらの薬剤的に許容でき
る塩類、前述した物質のいずれかの混合物からなる群より選択される請求項2または13
の医薬製品。
【請求項23】
オピオイド拮抗薬が、ナルトレキソン、ナロキソン、ナルメフェン、シクラザシン、レ
バロルファン、それらの薬剤的に許容できる塩類、及び前述した物質のいずれかの混合物
からなる群より選択される請求項1〜3の任意の一項の医薬製品。
【請求項24】
オピオイド拮抗薬がナルトレキソンまたはその薬剤的に許容できる塩類である請求項1
〜3の任意の一項の医薬製品。
【請求項25】
マトリックスが層なしで拮抗薬を隔離する能力を持ち、その層が隔離機能を高める請求
項3の医薬製品。
【請求項26】
層がマトリックスなしで拮抗薬を隔離する能力を持ち、そのマトリックスが隔離機能を
高める請求項3の医薬製品。
【請求項27】
マトリックスが層なしで拮抗薬を隔離する能力を持たず、層がマトリックスなしで拮抗
薬を隔離する能力を持たず、マトリックス及び層が組み合わさると拮抗薬を隔離する能力
を持つ請求項3の医薬製品。
【請求項28】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の 1 時間での溶出に基づいて、改変後に剤型から放出
される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が 約 20:1 以上;約
50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上となるように剤型内
のオピオイド拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項29】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 2 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項 28 の医薬製品。
【請求項30】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 4 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項 28 の医薬製品。
【請求項31】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間 で 9
00 ml の SIF に切り替える条件で、剤型の 12 時間での溶出に基づいた、改変後に剤型
から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 20:1
以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である請求
項28の医薬製品。
【請求項32】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 24 時間での溶出に基づいた、改変後
に剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約
20:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上であ
る請求項28の医薬製品。
【請求項33】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる700 ml の SGF 中で 1 時間、その
後 900 ml の SIF に切り替える条件で、剤型の 36 時間での溶出に基づいた、改変後に
剤型から放出される拮抗薬量の、無傷剤型から放出される拮抗薬量に対する比率が、約 2
0:1 以上;約 50:1 以上;約 100:1 以上;約 150:1 以上;または約 1000:1 以上である
請求項28の医薬製品。
【請求項34】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の 1 時間での溶出に基づいた無傷剤型から放出される
拮抗薬の重量百分率が1.0 重量%未満;0.5 重量%未満;0.2 重量%;または0.1 重量%
未満となるように剤型内のオピオイド拮抗薬を隔離するマトリックス及び層を含有する医
薬製品。
【請求項35】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 2 時間での溶出に基づいた無傷剤型か
ら放出される拮抗薬の重量百分率が、2.0 重量%未満;1.0 重量%未満;0.5 重量%未満
;または 0.25 重量%未満である請求項34の医薬製品。
【請求項36】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、そ
の後に 900 ml の SIF に切り替える条件で、剤型の 4 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、2.2 重量%未満;1.5 重量%未満;1.0 重量%未
満;または0.75 重量%未満である請求項34の医薬製品。
【請求項37】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 12 時間で 9
00 ml の SIF に切り替える条件で、剤型の 12 時間での溶出に基づいた無傷剤型から放
出される拮抗薬の重量百分率が、3.0 重量%未満;1.8 重量%未満;1.25 重量%未満;
または0.3 重量%未満である請求項34の医薬製品。
【請求項38】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 24 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、4.8 重量%未満;2.5 重量%未満;1.8 重量%未
満;または0.4 重量%未満である請求項34の医薬製品。
【請求項39】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で1 時間、その
後に 900 ml の SIF に切り替える条件で、剤型の 36 時間での溶出に基づいた無傷剤型
から放出される拮抗薬の重量百分率が、7.0 重量%未満;6.5 重量%未満;3.0 重量%;
または未満1.5 重量%である請求項34の医薬製品。
【請求項40】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び粒子の周囲に配置した層;50 rpm、37 ℃ で USP 2型 (パドル) 装置
を用いる 700 ml の SGF 中で剤型の溶出に基づいて 1 時間、継いで、900 ml の SIF に
切り替えた条件で無傷剤型が、1.0% 以下の拮抗薬を 1 時間で、2.0%以下の拮抗薬を2
時間で、2.2%以下の拮抗薬を4時間で、3.0%以下の拮抗薬を12時間で、4.8%以下の
拮抗薬を24時間で、及び 7.0%以下の拮抗薬を36時間で放出するように剤型内のオピ
オイド拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項41】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 0.5% 以下
の拮抗薬を 1 時間で、1.0%以下の拮抗薬を 2 時間で、1.5%以下の拮抗薬を 4 時間で
、1.8%以下の拮抗薬を 12 時間で、2.5%以下の拮抗薬を 24 時間で及び 6.5%以下の拮
抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項42】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 02%以下の
拮抗薬を 1 時間で、0.5%以下の拮抗薬を 2 時間で、1.0%以下の拮抗薬を 4 時間で、1
.25%以下の拮抗薬を 12 時間で、及び 1.8%以下の拮抗薬を 24 時間で、及び 3.0%以
下の拮抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項43】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の溶出に
基づいて 1 時間、継いで、900 ml の SIF に切り替えた条件で、無傷剤型が 0.1%以下
の拮抗薬を 1 時間で、0.25%以下の拮抗薬を 2 時間で、0.75%以下の拮抗薬を 4 時間
で、0.3%以下の拮抗薬を 12 時間で、0.4%以下の拮抗薬を 24 時間で、及び 1.5%以下
の拮抗薬を 36 時間で放出する請求項40の医薬製品。
【請求項44】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項2の医薬製品。
【請求項45】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項13の医薬製品。
【請求項46】
50 rpm、37 ℃ で USP 2型 (パドル) 装置を用いる 700 ml の SGF 中で剤型の 1 時
間での溶出に基づいて、改変後剤型から放出される作動薬の重量百分率が、50 重量%未
満;40 重量%未満;または35 重量%未満である請求項22の医薬製品。
【請求項47】
オピオイド拮抗薬の粒子の平均直径約0.1〜約6.0mmである請求項7及び請求項
28〜46の任意の一項の医薬製品。
【請求項48】
オピオイド拮抗薬の粒子が a) オピオイド拮抗薬と第1疎水性物質をブレンドして混合
物を形成すること;b) 少なくとも混合物を軟化するのに充分な温度に混合物を加熱する
こと;c) 混合物を押出成形してストランドを形成すること;及びd) ストランドを粒子に
切断することによって形成される請求項28〜46の任意の一項の医薬製品。
【請求項49】
オピオイド拮抗薬の粒子の平均直径が約0.1〜約6.0mmである請求項48の医薬
製品。
【請求項50】
作動薬粒子と拮抗薬粒子が、外観、質感、臭い、味、硬度、形状、サイズまたはその組
み合わせからなる群より選択される特質において類似しているような、オピオイド作動薬
及びオピオイド作動薬の周囲に配置した層を含有する複数の粒子;及び複数のオピオイド
拮抗薬の粒子及びオピオイド拮抗薬の粒子の周囲に配置した層を含有する医薬製品。
【請求項51】
作動薬粒子と拮抗薬粒子が、外観、質感、臭い、味、硬度、形状、サイズまたはその組
み合わせからなる群より選択される特質において実質的に識別できないような、オピオイ
ド作動薬及びオピオイド作動薬の周囲に配置した層を含有する複数の粒子;及び複数のオ
ピオイド拮抗薬の粒子及びオピオイド拮抗薬の粒子の周囲に配置した層を含有する医薬製
品。
【請求項52】
オピオイド作動薬を含有する複数の粒子を調製すること;オピオイド拮抗薬を含有する
複数の粒子を調製すること;オピオイド作動薬粒子とオピオイド拮抗薬の粒子が類似する
外観を持つように、層をオピオイド作動薬粒子及びオピオイド拮抗薬の粒子に適用するこ
とを含有する医薬製品の調製方法。
【請求項53】
オピオイド作動薬を含有する複数の粒子を調製すること;オピオイド拮抗薬を含有する
複数の粒子を調製すること;オピオイド作動薬粒子及びオピオイド拮抗薬の粒子の外観が
実質的に識別できないように、層をオピオイド作動薬粒子及びオピオイド拮抗薬の粒子に
適用することを含有する医薬製品の調製方法。
【請求項54】
下記を含有する医薬製品の調製方法。
a) 押出成形で粒子を形成することによってオピオイド拮抗薬を第1疎水性物質内に分散
させること;及び
b) 粒子重量の約5%〜約30%を持つ第2疎水性物質を含有する層を粒子の周辺に配置
すること。
【請求項55】
下記を含有する医薬製品の調製方法。
a) 押出成型で複数の粒子を形成することによってオピオイド拮抗薬を第1疎水性物質内
に分散させること、及び粒子重量の約5%〜約30%の量を持つ第2疎水性物質を含有す
る層を粒子のそれぞれのの周辺に配置すること;
b) 第3疎水性物質内のオピオイド作動薬を分散させ、複数の粒子を形成すること;及び
c) カプセル内に複数個のオピオイド作動薬粒子及び複数個のオピオイド拮抗薬の粒子を
含むこと。
【請求項56】
下記を含有する医薬製品の調製方法。
押出成型で複数の薬剤的に許容できる粒子を形成することによって、マトリックス中にオ
ピオイド拮抗薬を分散させること及び該マトリックス及び層が無傷剤型内のオピオイド拮
抗薬を隔離するようにそれぞれの粒子に該層を配置すること。
【請求項57】
マトリックス内に分散したオピオイド拮抗薬をそれぞれの粒子に含有する複数の押出成
形した粒子;及び該粒子の周囲に配置した層;無傷剤型内のオピオイド拮抗薬を隔離する
マトリックスを含有する医薬製品。
【請求項58】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 20:1 以上であ
る請求項1〜3の任意の一項の医薬製品。
【請求項59】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 100:1 以上で
ある請求項58の医薬製品。
【請求項60】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 125:1 以上で
ある請求項58の医薬製品。
【請求項61】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 Cmax の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 Cmax に対する比率が約 150:1 以上で
ある請求項58の医薬製品。
【請求項62】
改変が粉砕による粉末化によって行われる、請求項28〜33、44〜46または58
〜61の任意の一項の医薬製品。
【請求項63】
粉砕が乳鉢及び乳棒によって行われる、請求項62の医薬製品。
【請求項64】
マトリックス内に分散した約 2 mg のナルトレキソンまたは薬剤的に許容できる塩類を
含有する複数の押出成形した粒子;及び該粒子周囲に配置した層;50 rpm、37 ℃ で USP
2型 (パドル) 装置を用いる剤型の溶出に基づいて、700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF 中で無傷剤型が 0.065 mg 以下の拮抗薬が 36 時間で放出するように
剤型内のナルトレキソンまたはその塩類を隔離するマトリックス及び層を含有する医薬製
品。
【請求項65】
50 rpm、37 ℃ でUSP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF に切り替える条件で、無傷剤型が 0.04 mg 以下の拮抗薬を 36 時間で
放出する、請求項64の医薬製品。
【請求項66】
マトリックス内に分散した約 8 mg のナルトレキソンまたは薬剤的に許容できる塩類を
含有する複数の押出成形した粒子; 及び該粒子周囲に配置した層; 50 rpm、37 ℃ で USP
2型 (パドル) 装置を用いる剤型の溶出に基づいて、700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF 中で無傷剤型が 0.08 mg 以下の拮抗薬が 36 時間で放出するように剤
型内のナルトレキソンまたはその塩類を隔離するマトリックス及び層を含有する医薬製品
。
【請求項67】
50 rpm、37 ℃ でUSP 2型 (パドル) 装置を用いる 700 ml の SGF 中で 1 時間、次い
で 900 ml の SIF に切り替える条件で、無傷剤型が 0.12 mg 以下の拮抗薬を 36 時間で
放出する、請求項64の医薬製品。
【請求項68】
下記を含有する医薬製品:
a) アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合物からなる群より
選択される第1疎水性物質に分散する塩酸ナルトレキソンを含有する押出成形した粒子;
及び
b) 該粒子の周囲に配置した第2疎水性物質で、アルキルセルロース、アクリル樹脂、及
びその混合物からなる群より選択される第2疎水性物質を含有する層。
【請求項69】
a) アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合物からなる群よ
り選択される第1疎水性物質に分散する塩酸ナルトレキソンを含有する複数の押出成形し
た粒子及びアルキルセルロース、アクリル樹脂、及びその混合物からなる群より選択され
る第2疎水性物質を含有し、それぞれの粒子の周囲に配置された層;b) アクリル樹脂、
ステアリルアルコール、ステアリン酸及びその混合物からなる群より選択される第3疎水
性物質内に分散したオキシコドン、ヒドロコドン、ヒドロモルホン及びその薬剤的に許容
できる塩類からなる群より選択されるオピオイド作動薬を含有する複数の粒子;及び c)
複数個のオピオイド作動薬粒子及び複数個の塩酸ナルトレキソン粒子を含むカプセルを含
有する医薬製品。
【請求項70】
それぞれの粒子が、アクリル樹脂、ステアリルアルコール、ステアリン酸及びその混合
物からなる群より選択される疎水性物質を含有するマトリックス内に分散した塩酸ナルト
レキソンを含有する、複数の押出成形した粒子;及びアルキルセルロース、アクリル樹脂
及び粒子の周囲に配置したその混合物からなる群より選択される材料を含有する層;無傷
剤型内の塩酸ナルトレキソンを隔離するマトリックス及び層を含有する医薬製品。
【請求項71】
塩酸ナルトレキソンの量が約2mg〜約12mgである請求項68〜70の任意の一項
の医薬製品。
【請求項72】
塩酸ナルトレキソンの量が約2mg〜約8mgである請求項71の医薬製品。
【請求項73】
塩酸ナルトレキソン粒子が90%を超える疎水性物質を含有する請求項68〜70の任
意の一項の医薬製品。
【請求項74】
塩酸ナルトレキソン粒子が95%超える疎水性物質を含有する請求項73の医薬製品。
【請求項75】
層の量が塩酸ナルトレキソン粒子の重量の約5%〜約30%である請求項68〜70の
任意の一項の医薬製品。
【請求項76】
粒子のそれぞれにマトリックス内に分散したオピオイド拮抗薬を含有する複数の押出成
形した粒子;及び該粒子の周囲に配置した層;無傷剤型内のオピオイド拮抗薬を隔離する
マトリックス及び層を含有し、層が二層配列 (bilaminar arraignment) にアクリル重合
体及びセルロースポリマーを含有する、医薬製品。
【請求項77】
アクリル重合体が拮抗薬の粒子の周囲に配置され、セルロースポリマーがアクリル重合
体層状拮抗薬の粒子の周囲に配置された請求項76の医薬製品。
【請求項78】
第2疎水性物質の量が粒子重量の約16%〜約30%である請求項1または2の医薬製
品。
【請求項79】
第2疎水性物質の量が粒子重量の約20%〜約29%である請求項1または2の医薬製
品。
【請求項80】
第2疎水性物質の量が粒子重量の約22%〜約28%である請求項1または2の医薬製
品。
【請求項81】
層が実質的に拮抗薬を欠いている請求項1〜3の任意の一項の医薬製品。
【請求項82】
剤型が即放性拮抗薬を欠いている請求項1〜3の任意の一項の医薬製品。
【請求項83】
前記粉砕が24ストロークの乳鉢及び乳棒で行われる請求項63の医薬製品。
【請求項84】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散した有害薬剤または拮抗薬を含有する押出成形した粒子;及
び
b) 粒子の周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物質が粒
子の約5%〜約30%の重量を有する層。
【請求項85】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散した有害薬剤または拮抗薬を含有する押出成形した粒子、及
び粒子それぞれの周囲に配置した第2疎水性物質を含有する層であって、該第2疎水性物
質が、たとえば、粒子重量の約5%〜約30%を含有する複数の粒子;
b) 第3疎水性物質内に分散した作用薬を含有する複数の粒子;及び
c) 複数個の作用薬粒子及び複数個の有害薬剤または拮抗薬の粒子を含むカプセル。
【請求項86】
複数の押出成形した粒子であって、そのそれぞれにマトリックス内に分散した有害薬剤
または拮抗薬を含有する粒子;及び粒子の周囲に配置した層;無傷剤型内の有害薬剤また
は拮抗薬を隔離するマトリックス及び層を含有する医薬製品。
【請求項87】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項6の医薬製品。
【請求項88】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項47の医薬製品
。
【請求項89】
オピオイド拮抗薬の粒子の長さが約0.1〜約6.0mmである請求項49の医薬製品
。
【請求項90】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 5:1 以上である
請求項1〜3の医薬製品。
【請求項91】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 25:1 以上であ
る請求項90の医薬製品。
【請求項92】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 75:1 以上であ
る請求項90の医薬製品。
【請求項93】
改変剤型を患者集団に単回投与した後に得られる拮抗薬の平均 AUC の、無傷剤型を患
者集団に単回投与した後に得られる拮抗薬の平均 AUC に対する比率が約 200:1 以上であ
る請求項90の医薬製品。
【請求項94】
オピオイド作動薬がオキシコドンまたはその薬剤的に許容できる塩類である請求項2の
医薬製品。
【請求項95】
オピオイド作動薬がヒドロモルホンまたはその薬剤的に許容できる塩類である請求項2
の医薬製品。
【請求項96】
オピオイド作動薬がヒドロコドンまたは薬剤的にその許容できる塩類である請求項2の
医薬製品。
【請求項97】
オピオイド作動薬がオキシモルホンまたはその薬剤的に許容できる塩類である請求項2
の医薬製品。
【請求項98】
オピオイド作動薬がモルヒネまたはその薬剤的に許容できる塩類である請求項2の医薬
製品。
【請求項99】
請求項2〜13の任意の一項の医薬製品をそれを必要とする患者に経口で投与する工程
を含有する疼痛治療の方法。
【請求項100】
請求項1または2の任意の一項の医薬製品をそれを必要とする患者に経口投与すること
を含有する投与の方法。
【請求項101】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有し、改変剤型を患者集団に単回投与した
後に得られる拮抗薬の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮
抗薬の平均 AUC に対する比率が約 5:1 以上である層。
【請求項102】
下記を含有する医薬製品:
a) 第1疎水性物質内に分散したオピオイド拮抗薬を含有する粒子;及び
b) 粒子の周囲に配置した第2疎水性物質を含有し、改変剤型を患者集団に単回投与した
後に得られる拮抗薬の平均 AUC の、無傷剤型を患者集団に単回投与した後に得られる拮
抗薬の平均 AUC に対する比率が約 20:1 以上である層。
【図1】

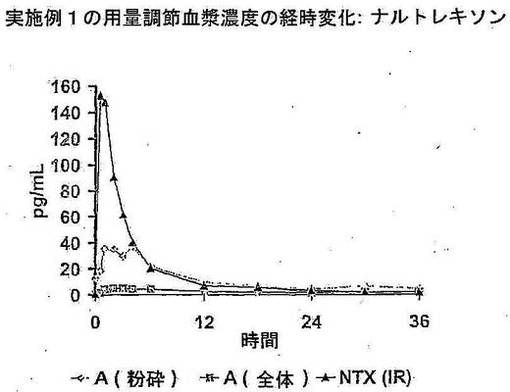
【図2】
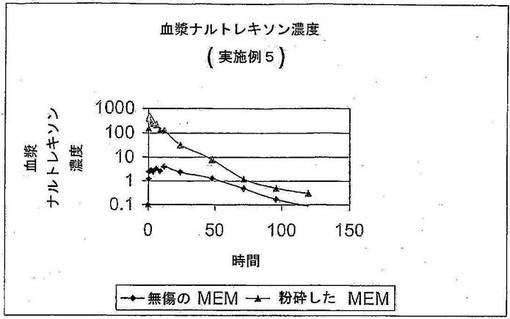
【図3】
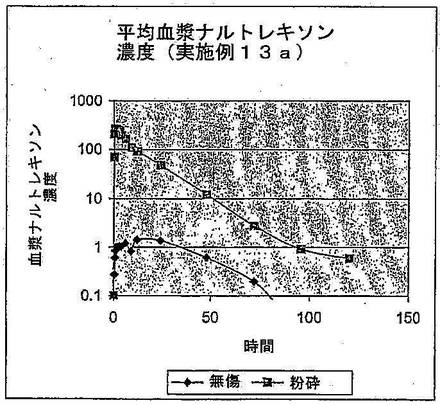
【図4】
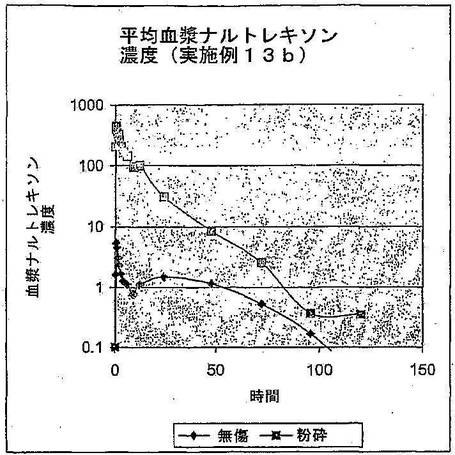
【図2】
【図3】
【図4】
【公開番号】特開2012−67113(P2012−67113A)
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願番号】特願2011−237465(P2011−237465)
【出願日】平成23年10月28日(2011.10.28)
【分割の表示】特願2006−513103(P2006−513103)の分割
【原出願日】平成16年4月19日(2004.4.19)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
【公開日】平成24年4月5日(2012.4.5)
【国際特許分類】
【出願日】平成23年10月28日(2011.10.28)
【分割の表示】特願2006−513103(P2006−513103)の分割
【原出願日】平成16年4月19日(2004.4.19)
【出願人】(599108792)ユーロ−セルティーク エス.エイ. (134)
【Fターム(参考)】
[ Back to top ]