
合成胆汁酸組成物、その方法およびその調製

【課題】胆汁酸組成物、方法および調製を提供すること
【解決手段】
定義された医薬組成物として十分な量の適切な胆汁酸およびその合成方法がここで提供される。そのように提供される胆汁酸組成物および方法は、胆汁酸を自然に産生する哺乳動物または微生物生命体から単離されるものではない。一態様において、動物起源のおよび哺乳動物および/または細菌パイロジェンのすべての部分がない特定のデオキシコール酸医薬組成物ならびに生成および使用のための関連方法が提供される。別の態様において、定義された医薬組成物として十分な量の適切なデオキシコール酸が提供され、これは、関連組成物、製造方法および使用方法とともに、局所的な脂肪除去のための注射用医薬組成物として使用できる。
【解決手段】
定義された医薬組成物として十分な量の適切な胆汁酸およびその合成方法がここで提供される。そのように提供される胆汁酸組成物および方法は、胆汁酸を自然に産生する哺乳動物または微生物生命体から単離されるものではない。一態様において、動物起源のおよび哺乳動物および/または細菌パイロジェンのすべての部分がない特定のデオキシコール酸医薬組成物ならびに生成および使用のための関連方法が提供される。別の態様において、定義された医薬組成物として十分な量の適切なデオキシコール酸が提供され、これは、関連組成物、製造方法および使用方法とともに、局所的な脂肪除去のための注射用医薬組成物として使用できる。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願への相互参照)
本出願は、2007年6月19日に出願された米国仮特許出願第60/945,035号および2007年8月20日に出願された同第60/956,875号;2008年2月21日に出願された米国通常特許出願第12/035,339号および2008年5月16日に出願された同第12/153,446号;ならびに2008年4月25日に出願された英国特許出願第0807615.0号に対する優先権を主張し、これらの各々は、その全体を本明細書において参考として援用される。
【0002】
(発明の分野)
本発明は、広く胆汁酸ならびに関連組成物および方法に関する。一態様において、本発明は、デオキシコール酸および関連組成物、有用な中間体ならびにその合成方法に関する。別の態様において、本発明は、本組成物および方法の、医薬組成物およびその製造方法としての使用に関する。重要なことに、本発明の胆汁酸は、これらの酸を自然に産生する哺乳動物および微生物生命体から単離されるものではなく、故に、そのような生命体に関連するいかなる毒素および汚染物質もない。別の態様において、本発明は、デオキシコール酸ならびに薬学的に許容されるその塩および中間体の合成に関する。
【背景技術】
【0003】
(発明の背景)
コラノロジー(Cholanology)という胆汁酸の研究、特に胆汁酸化学は、世紀の大半にわたって関心を集めてきた。多くは既知であるが、胆汁酸化学には、多くが驚くべき特性を有する多種多様な化学的実体が関与する。総説は、例えば、参照により本明細書に組み込まれる、Mukhopadhyay, S.およびU. Maitra.、Current Science、87巻:1666〜1683頁(2004年)(「Chemistry and biology of bile acids」)を参照されたい。
【0004】
胆汁酸は、2つの連結ユニット、剛性のステロイド核および短い脂肪族側鎖を特徴とする(本出願の図1を参照)。Hofmann, A. F.らを参照。提案された胆汁酸の命名法については、J. Lipid Res.、33巻:599〜604頁(1992年)を参照されたい。核および側鎖はいずれも、多数の可能な立体配置を有する。核は、個々の環の拡張または収縮によって変化させることができ、側鎖は、短縮または伸長できる。加えて、胆汁酸分子の両部分は、多数の可能な極性置換基を有する。イオン化基は、核または側鎖上に存在し得る。最後に、共役基は、核上(例えば、サルフェート、グルクロネート、ホスフェート)または側鎖上(グリシンもしくはタウリンまたは他のアミノ酸、さらには糖類)に存在し得る。側鎖構造は、化合物(胆汁酸または胆汁塩)のクラスを決定する。
【0005】
胆汁酸は、両親媒性(amphiphilic)および両親媒性(amphipathic)の「面」の両方を有する両親媒性物質:
【0006】
【化1】

である。Hofman, A.F.、News Physiol. Sci.、14巻:24〜29頁(1999年)(「Bile Acids: The good, the
Bad, and the Ugly」、25頁、図1)。
【0007】
慣例により、疎水性表面は「β面」と呼ばれ、親水性表面は「α面」と呼ばれる。概して、β面は脂溶性であり、α面は比較的極性である。疎水面上および親水面上に極性基(自然発生的な胆汁酸中のヒドロキシル基)を有するもの、例えばウルソデオキシコール酸等の胆汁酸がある。分子の両親媒性の性質は、ホスファチジルコリン等、両親媒性であるが不水溶性の脂質との混合ミセルの形成を司る。胆汁酸は、胆汁酸が、臨界ミセル化濃度と称される臨界濃度を超えない限り、混合ミセルの形態の食物中の脂質を可溶化しない。
【0008】
ヒトにおいて最大の割合で見られる胆汁酸は、ケノデオキシコール酸およびデオキシコール酸である。デオキシコール酸は、デオキシコレート、コラン酸および3α,12α−ジヒドロキシ−5β−コラネート(cholanate)としても知られている。ヒト体内において、デオキシコール酸は、腸管において吸収のための脂肪の乳化に使用される。研究において、デオキシコール酸は、膜結合タンパク質単離のための低刺激洗浄剤として使用される。実質的に純粋である場合、デオキシコール酸は白色〜オフホワイトの結晶性粉末形態である。デオキシコール酸は、肝臓によって産生される4つの主な酸の1つである。これは、アルコールおよび酢酸に可溶である。デオキシコール酸のCAS番号は[83−44−3]である。
【0009】
体脂肪の迅速な除去は長年の理想であり、多くの物質がそのような結果を達成すると主張されてきたが、結果を示したものはわずかである。「メソセラピー」、つまり脂肪の除去のための注射剤の使用は、1950年代からホメオパシーおよび美容学的主張が為されているが、安全性および有効性の懸念により、医療従事者の間で広く受け入れられていない。メソセラピーは、元来、局所的な医学的および美容学的状態の治療のために化合物の混合物を含有する皮膚注射を利用する方法として欧州で想到された。メソセラピーは、疼痛緩和のために伝統的に用いられていたが、最近ではその美容学的用途、特に脂肪およびセルライトの除去が米国で注目を集めている。局所的な脂肪減少のための1つのそのような報告されている治療は、ブラジルにおいて普及し、ホスファチジルコリンの注射を使用するものであり、メソセラピーと同義であると誤ってみなされてきた。「脂肪を溶解する」とされている注射としてのその魅力にもかかわらず、これらの美容学的治療の安全性および有効性は、ほとんどの患者および医師にとって曖昧なままである。Rotunda,
A.M.およびM. Kolodney、Dermatologic Surgery、32巻:465〜480頁(2006年)(「Mesotherapy and Phosphatidylcholine Injections: Historical
Clarification and Review」)を参照されたい。
【0010】
国際公開第2006/133160号(参照により図面を含むその全体が本明細書に組み込まれる)は、リポモデリング(lipomodeling)のための方法、例えば、神経ペプチドY受容体アンタゴニストを脂肪貯蔵部位に投与することによる脂肪貯蔵物の減少を記載している。Kolonin M. G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)は、強力な脂肪細胞死滅効果を有する脂肪選択的プロアポトーシスペプチドを記載している。記載されているプロアポトーシスペプチドは、死滅させるためには血管系にアクセスする必要がある。
【0011】
最近刊行された文献は、デオキシコール酸がインビボで脂肪性沈着物に注射される場合、脂肪除去特性を有することを報告している。すべて参照により図面を含むその全体が本明細書に組み込まれる、特許文献1および特許文献2、ならびに特許文献3、特許文献4、特許文献5および特許文献6を参照されたい。脂肪組織中に注射されたデオキシコレートは、1)細胞傷害機序によって脂肪細胞を死滅させる、2)皮膚引き締めを引き起こすという2つの効果を有する。これらの効果はいずれも、所望の審美的修正(すなわち、体形矯正)を媒介するために必要である。脂肪中に注射されたデオキシコレートは、タンパク質への曝露によって迅速に不活性化され、その後、迅速に腸の内容物中に戻るため、その効果は空間的に封じ込められる。臨床的安全性を与えるこの減弱作用の結果として、脂肪除去療法は、典型的に4〜6セッションを必要とする。外科手術を必要としないこの局所的な脂肪除去は、病理学的な局所的脂肪沈着物に関する治療的処置(例えば、HIVの治療における医学的介入に伴う脂質異常症)だけでなく、外科手術に固有の付帯リスクがない美容学的脂肪除去(例えば、脂肪吸引術)にも有益である。いずれも参照により本明細書に組み込まれる、Rotundaら、Dermatol. Surgery、30巻:1001〜1008頁(2004年)(「Detergent effects of
sodium deoxycholate are a major feature
of an injectable phosphatidylcholine formulation used for localized fat dissolution」)およびRotundaら、J. Am. Acad. Dermatol.、2005年:973〜978頁(「Lipomas treated with subcutaneous deoxycholate injections」)を参照されたい。
【0012】
医薬品等級の胆汁酸製剤は、比較的低価格で市販されている。この低価格は、胆汁酸が、動物の死体、特に雌ウシおよびヒツジ等の大型動物から得られることによるものである。重要なことに、動物源由来のすべての薬剤と同様に、動物由来胆汁酸製品は、動物病原体、ならびにパイロジェン等の細菌毒素を含む動物または微生物代謝産物および毒素等、他の有害作用物質を含有し得るという懸念がある。
【0013】
そのような動物病原体は、プリオン病を引き起こし得る感染性病原タンパク質の一種であると考えられているプリオンを含み得る。プリオン病は、神経系の変性障害である。1つのそのような疾患である「狂牛」病(クロイツフェルト・ヤコブ病(CJD)の変異型であると考えられている)は、罹患した雌ウシ由来の食用牛肉中に存在するプリオンによって引き起こされると考えられている。ほとんどの場合は未知の伝染様式による散発性なものであり、いくつかの場合は遺伝的なものであり、少数が医療処置によって伝染したものである。感染材料の消費によるヒトプリオン病の蔓延は、歴史的にはクールーに、また最近では変異型CJDに関与するとされている。他の動物プリオン病(ヒツジのスクレピー、伝染性ミンク脳症、シカの慢性消耗病および牛海綿状脳症)はすべて、感染動物との接触によってまたは感染飼料の消費によって横方向に伝染するようである。プリオン病に関するリスク評価および未来事象の予測は、異なる伝染様式、予測不可能な種の障壁、組織中における感染力の可変分布およびいくつかの疾患において見られる系統変動により、解明することが困難である。
【0014】
概して、動物性製品は、パイロジェン(発熱物質)を産生する微生物生命体に曝露され得る。食物および/または医薬品の細菌汚染物質も、腸管出血性E.coliによる食料品の汚染によって証明されている通り、重要課題である。雌ウシに由来する食肉等の製品およびリンゴ、ホウレンソウ等の農産物は、そのような汚染に関与するとされている。そのような場合、ヒトにおいて有害作用を発生させるのは、(細菌自体よりもむしろ)細菌によって産生される毒素である。そのような有害作用は、重度の下痢、腎不全および極限状況では死を含む。パイロジェンの一種である細菌内毒素は、すべての医薬組成物から実質的に排除されなくてはならない。
【0015】
動物性製品は、一般に排除過程によって精製される、すなわち、混合から最終産物を選択するのではなく、最終産物は不純物の排除後に残っている材料である。また、病原体等の潜在的な動物部分に加えて、動物源からの精製の別のアーチファクトは、最終産物が1つまたは複数の胆汁酸の混合物であることである。例えば、デオキシコール酸の市販製剤は、若干のケノデオキシコール酸、ならびに哺乳動物の胆汁酸合成においてデオキシコール酸およびケノデオキシコール酸の両方の前駆体であるコール酸を含有する。デオキシ/ケノ/コールの正確な割合は事前選択されないため、これは、多量の胆汁酸の製造を企図する場合にロット間変動をもたらし得る。そのようなロット間変動は、問題のあるものとなり得、規制当局の承認または品質管理を獲得する際、特に医薬組成物を生成するための取り組みにおいて、追加ステップを生じさせる場合がある。生産者は明らかに、胆汁酸医薬組成物の製造においてロット間予測可能性を望むであろう。
【0016】
現在、動物病原体および他の有害作用物質を含有する動物由来製品に関する懸念は、隔離および検査された動物から調達することによって対処されている。例えば、ニュージーランドの動物に由来するデオキシコール酸は、動物が隔離される等で観察可能な病原体がないままであり続ける限り、米国の規制制度下でヒト使用のための胆汁酸の源である。
【0017】
暗黙的に、そのような政府により管理される規制制度のために必要なのは、動物由来の薬剤が注射される場合における動物病原体の伝染の内在的リスクの認識である。非動物性薬剤代替物が利用可能になれば、政府による規制制度はもはや必要ない。そのような代替物(動物由来の薬剤に置き換わる非動物性薬剤)および関連する利点の例は、ヒト使用のためのインスリンである。米国でのウシインスリンの製造は1998年に中止され、ヒト使用のためのブタインスリンは2006年1月に中止された。動物インスリンはBSEを引き起こす因子または他の病原因子に曝露されていないと分かっている群から得ることができるが、製造施設または過程は、動物性原料を、病原体に曝露された動物に曝露し得る。病原因子のヒトへの伝染のリスクは、組換え的にまたは合成的に製造されるインスリンの使用によって解消することができる。消費者にとって、インスリンの状況はためになるものであり、合成材料が自由に利用可能であれば、動物病原体の伝染のリスクは理論上は解消される。生産者にとって、動物病原体の材料が実質的にない純粋な化学的実体を生成するための能力は、安全性、品質および規制の目的に有利である。さらに、合成過程は、典型的に、生物源に由来するものよりも再現性の高い製品を提供する。
【0018】
現在、動物の死体由来の胆汁酸が比較的豊富であることにより、業界は、胆汁酸を完全に化学的に合成すること、または植物ステロールもしくは微生物出発材料を使用して胆汁酸を調製することのいずれにも踏み出していない。また、胆汁酸誘導体は合成されたが、動物性材料の低価格および入手しやすさにより、この作業には当初、ステロイド化学のための出発材料として、やはり動物由来胆汁酸が関与していた。植物ステロール研究における歴史的に活発な取り組みにもかかわらず、容易に商業的に入手可能な植物ステロール由来の胆汁酸医薬品グレード組成物はない。例えば、非特許文献1を参照されたい(1981年の参考文献、非特許文献2(「First Total Synthesis of (+)−Chenodeoxycholic Acid」)を前提として、いかなる胆汁酸の全合成も実施されなかったということに注目されたい)。細菌等の微生物によって産生された胆汁酸は、細菌産物として、例えば海洋での油流出の浄化のためにインシチュで使用されてきた。非特許文献3(「Bile acids are new products of a mariene bacterium, Myroides sp. Strain SM1」)を参照されたい。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第2005/117900号パンフレット
【特許文献2】国際公開第2005/112942号パンフレット
【特許文献3】米国特許第2005/0261258号明細書
【特許文献4】米国特許第2005/0267080号明細書
【特許文献5】米国特許第2006/127468号明細書
【特許文献6】米国特許第2006/0154906号明細書
【非特許文献】
【0020】
【非特許文献1】Mukhopadhyay, S.およびU. Maitra.、Current Science、87巻:1666〜1683、1670頁(2004年)
【非特許文献2】Kametaniら、J. Am. Chem. Soc.、103巻:2890頁(1981年)
【非特許文献3】Maneeratら、Appl. Microbiol. Biotechnol.、76巻:679〜683頁(2004年)
【発明の概要】
【発明が解決しようとする課題】
【0021】
脂肪除去のためのデオキシコール酸の全潜在能力を現実化するために、動物由来製品の使用に対する懸念にさらに対処することが必須である。明らかに、動物起源の部分(または、動物、特に哺乳動物において作用し得、ヒト使用の場合にはヒトに悪影響を及ぼし得る病原部分)および動物または微生物代謝産物、パイロジェン等の細菌毒素を含む毒素等の他の有害作用物質がないことが冒頭から分かる、ヒトにおいて薬剤として使用するための、適量の有効な胆汁酸およびデオキシコール酸等の関連組成物が必要である。本発明は、動物病原体および他の有害作用物質の潜在的リスクがない合成的に調製された胆汁酸組成物を提供することにより、この懸念に対処するものである。開示されている胆汁酸組成物は、脂肪分解(adipolytic)療法において使用でき、局所的な脂肪除去の領域における研究および開発努力をさらに進める役割を果たす。
【課題を解決するための手段】
【0022】
(発明の要旨)
本明細書では、定義された医薬組成物として十分な量の適切な胆汁酸およびその合成方法が提供される。そのように提供される胆汁酸組成物および方法は、胆汁酸を自然に産生する哺乳動物または微生物生命体から単離されるものではない。一態様において、動物起源のおよび哺乳動物および/または細菌パイロジェンのすべての部分がない特定のデオキシコール酸医薬組成物ならびに生成および使用のための関連方法が提供される。別の態様において、定義された医薬組成物として十分な量の適切なデオキシコール酸が提供され、これは、関連組成物、製造方法および使用方法とともに、局所的な脂肪除去のための注射用医薬組成物として使用できる。本発明の定義されたデオキシコレート注射液を、直交機序(orthogonal mechanism)によって脂肪を消滅させる分子、例えばNPYアンタゴニストおよび/または脂肪選択的プロアポトーシスペプチドと組み合わせ、より少ない治療セッションでの体形矯正を媒介するためのより強力な手段を作成するために使用される作用物質を提供することができる。別の態様において、本発明は、デオキシコール酸および薬学的に許容されるその塩の合成に関する方法および中間体を提供する。合成的に調製されたデオキシコール酸は、脂肪除去のための脂肪分解療法において使用できる。
【図面の簡単な説明】
【0023】
【図1】胆汁酸骨格の炭素のナンバリングシステムを含む、胆汁酸の構造を表す図である。
【図2】ウシ由来デオキシコール酸ナトリウム(Sigma)と比較した、本発明の合成デオキシコール酸ナトリウムによる処理時の初代ヒト脂肪細胞の細胞生存の用量依存的な減少における類似性を示す図である。
【発明を実施するための形態】
【0024】
(参照による組み込み)
本明細書中で言及されるすべての刊行物および特許出願は、各個々の刊行物または特許出願が参照により組み込まれることを具体的かつ個別に指示されている場合と同程度まで、参照により本明細書に組み込まれる。
【0025】
(発明の詳細な説明)
定義
本開示全体を通して、種々の刊行物、特許および公開された特許明細書は、識別引用によって参照される。これらの刊行物、特許および公開された特許明細書の開示は、参照により本開示に組み込まれ、本発明が属する分野の状況をより詳細に記載する。
【0026】
本明細書において使用される場合、いくつかの用語は、下記に定義される意味を有する。本明細書および請求項において使用される場合、単数形「a」、「an」および「the」は、文脈上明らかに別段の定めをした場合を除き、単数および複数の参照物を含む。
【0027】
別段の指示がない限り、本明細書および請求項において使用される原料、反応条件等の数量を表現するすべての数は、すべての場合において用語「約」により修飾されるものとして理解すべきである。したがって、反対の指示がない限り、下記の明細書および添付の請求項において説明される数値パラメーターは近似値である。各数値パラメーターは、少なくとも、報告された有効桁の数を考慮し、通常の丸め技術を適用することによって解釈されるべきである。
【0028】
用語「アセチル化試薬」は、アセチル基CH3C(O)−を分子に添加することができる試薬を指す。
【0029】
用語「酸」は、プロトン供与体を指し、有機および無機酸の両方を含む。
【0030】
用語「アルキル」は、1〜10個の炭素原子、好ましくは1〜6個の炭素原子を有する一価の飽和脂肪族ヒドロカルビル基を指す。この用語は、例として、メチル(CH3−)、エチル(CH3CH2−)、n−プロピル(CH3CH2CH2−)、イソプロピル((CH3)2CH−)、n−ブチル(CH3CH2CH2CH2−)、イソブチル((CH3)2CHCH2−)、sec−ブチル((CH3)(CH3CH2)CH−)、t−ブチル((CH3)3C−)、n−ペンチル(CH3CH2CH2CH2CH2−)およびネオペンチル((CH3)3CCH2−)等の直鎖および分岐鎖のヒドロカルビル基を含む。
【0031】
用語「アリール」は、単環(例えば、フェニル)または多重縮合環(例えば、ナフチル)を有する6〜12個の炭素原子の一価の芳香族炭素環基を指す。
【0032】
用語「動物起源」は、多細胞生命体および単細胞生命体を含む生物の界(動物界)のいずれかに起因していることを指す。
【0033】
用語「脱水試薬」は、水と反応することができる試薬を指す。一態様において、脱水試薬は、分子から除去される水と反応することができる。
【0034】
用語「脱硫試薬」は、硫化物と反応することができる試薬を指す。一態様において、脱硫試薬は、硫化物含有分子と反応して、分子からスルフィド基を除去することができる。
【0035】
用語「エタンジチオールまたはジチアン前駆体」は、カルボニル基との反応によって、エタンジチオールまたはジチアン基を形成する試薬を指す。
【0036】
用語「求電子性アセチル基」は、求電子としてのアセチル基であって、電子に引き付けられ、電子を受け取る傾向がある基を指す。
【0037】
用語「水素化試薬」は、水素を分子に付与することができる試薬を指す。
【0038】
用語「ルイス酸」は、電子対受容体を指す。ルイス酸は、アルキルアルミニウムハライド(例えば、Et2AlClおよびMeAlCl2)等の有機金属試薬を含む。
【0039】
用語「哺乳動物起源」は、任意の哺乳類の生命体に起因していることを指す。用語「哺乳類の生命体」は、乳腺によって分泌される乳でその幼体に栄養を与え、通常は程度の差はあるが体毛で覆われた皮膚を有し、ヒトを含む、温血高等脊椎動物の綱(哺乳綱)(有胎盤類、有袋類または単孔類等)を指す。
【0040】
用語「微生物起源」は、任意の微生物生命体に起因していることを指す。用語「微生物生命体」は、細胞壁を欠いてよいかまたは細胞壁を有する場合にはグラム陽性もしくはグラム陰性であり、多くの場合コロニーに凝集されるかまたは鞭毛を用いて運動性となり、典型的には、土壌、水、有機物中または植物および動物の体内に生息し、通常は、栄養上、独立栄養性、腐生性または寄生性であり、その生化学的作用および病原性で有名な、原核生物の円状、らせん状または棒状の単細胞微生物のドメイン(細菌)を指す。
【0041】
用語「オレフィン化試薬」は、ケトンと反応して、対応するオレフィンを形成する試薬を指す。用語「オレフィン形成条件」は、そのような転換を行うのに適した条件を指す。そのような試薬の例は、ウィッティヒ試薬およびウィッティヒオレフィン化条件を含む。
【0042】
用語「酸化剤」は、酸化還元反応において電子を受け取ることができる試薬を指す。このようにして、ハロゲンまたは酸素を分子に添加することができ、または水素を分子から除去することができる。
【0043】
用語「病原体」は、疾患の特異的な原因因子を指す。
【0044】
「薬学的に許容される塩」は、当該技術分野において既知である多様な有機および無機対イオンに由来する薬学的に許容される塩を指し、ほんの一例として、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、アンモニウムおよびテトラアルキルアンモニウムを含む。適切な塩は、P. Heinrich Stahl、Camille G. Wermuth(編)、Handbook of Pharmaceutical Salts Properties, Selection, and Use、2002年に記載されているものを含む。これらの薬学的に許容される塩は、DCAを適切な塩基と反応させることによって調製できる。例示を目的として、そのような塩基の例は、水酸化ナトリウム、水酸化カリウムまたは水酸化リチウムを含む。代替として、塩は、塩基によるDCAのエステルの加水分解によって、DCAを導き得る任意の酸性ワークアップを省略して調製できる。
【0045】
用語「還元剤」は、酸化還元反応において電子を付与することができる試薬を指す。このようにして、ハロゲンまたは酸素を分子から除去することができ、または水素を分子に添加することができる。
【0046】
組成物および使用方法
本明細書に記載されている種々の態様において、本発明は、薬学的使用のための組成物(および有用な中間体)、その合成方法および本医薬組成物の使用方法を提供する。
【0047】
重要なことに、本胆汁酸組成物は、動物性出発材料から得られた材料に固有のリスクがなく、したがって、動物由来材料の詳細な検査および規定を必要としない。故に、一態様において、本発明は、哺乳動物病原体等の動物起源の材料を含まず、パイロジェン等の細菌起源の毒素を実質的に含まない、胆汁酸医薬組成物を目的としている。
【0048】
デオキシコール酸ナトリウムは、消化管において食物中の脂質を可溶化する、自然に産生される胆汁塩である。これは、コレステロールを出発材料として利用し、ヒトおよび細菌酵素の両方が関与する複合生合成経路を介して、インビボで産生される。デオキシコレートの主要機能は、吸収を容易にするために食物中の脂質を可溶化することによって、消化過程を支援することである。体内において、デオキシコレート生合成は、肝臓内のコレステロールの酵素的酸化、異性化および還元から始まり、そのコレステロール親と構造的に類似した胆汁酸であるコール酸を形成する(Stryer L、27章:Biosynthesis of Membrane Lipids and Steroids、Biochemistry、1995年、W. H. Freeman and Company: New York、691〜707頁)。その後、肝臓内において、コール酸は2つのアミノ酸(タウリンまたはグリシン)の1つと化学的に結合し、「共役」コール酸(すなわち、L−グリココレートおよびタウロコレート)を形成する。その後、これらの共役コール酸は、食物消費まで胆嚢中に保存される。食物消費後、胆汁液は、胆嚢から腸管内に放出され、ここで共役コール酸分子は、腸管微生物叢によって産生された酵素によって媒介される2つの追加の化学修飾に付される(Ridlon J.M.、Kang
D.J.およびHylemon P.B.、Bile salt biotransformations by human intestinal bacteria、J. Lipid Res.、47巻(2号):241〜59頁(2006年))。最初に、共役コール酸は脱ヒドロキシル化されて共役デオキシコレートを形成する。その後、共役デオキシコレートは脱共役されて遊離デオキシコレートを形成し、これが、他の胆汁酸とともに食物中の脂質の可溶化に関与する。デオキシコレートはコール酸合成より下流にあるため、コール酸はデオキシコレートの自然源中に存在する不純物であり得る。
【0049】
デオキシコレートは、333mg/mLまで水に可溶であり、アルコールに難溶であり、アセトンおよび氷酢酸にさらに難溶である。ミセルの可逆的形成は、約2.4mg/mLの臨界ミセル濃度を超過するデオキシコール酸ナトリウム濃度および中性pHで発生し得る(Matsuoka K, M.Y.、Micelle formation of
sodium deoxycholate and sodium ursodeoxycholate (1部)、Biochim. Biophys. Acta.、1580巻(2〜3号):189〜99頁(2002年))。2.4mg/mLの臨界ミセル濃度を超過する濃度において、デオキシコレートはミセルを形成し、細胞、脂質およびタンパク質を可溶化する能力を有する。0.4mg/mL(絶食状態に相当する)等のより低濃度において、デオキシコレートは、26mg/mLのアルブミン(35〜50mg/mLの血清生理学的濃度に近い)の存在下、アルブミンに98%結合する(Roda A.ら、Quantitative aspects of the interaction of bile acids with human serum albumin、J. Lipid Res.、23巻(3号):490〜5頁(1982年))。
【0050】
好ましい実施形態は、デオキシコール酸(DCA)もしくはそのプロドラッグ、または前記化合物または前記プロドラッグの薬学的に許容される塩、ならびに関連組成物および方法を目的としており、ここで、デオキシコール酸(DCA)は、
【0051】
【化2】
であり、ここで、前記化合物は、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない。
【0052】
他の好ましい実施形態は、DCAおよび薬学的に許容されるその塩の立体異性体、DCAならびにその立体異性体および塩の合成における中間体、ならびに関連組成物および方法も目的としている。
【0053】
本胆汁酸医薬組成物は、場合によって塩形態であり、さらに場合によって、薬学的に許容される希釈剤、賦形剤または担体を含有する。一態様において、本発明は、デオキシコール酸(DCA)を自然に産生する哺乳動物または微生物生命体から単離されるものではない、DCAである化合物もしくはそのプロドラッグ、または前記化合物もしくは前記プロドラッグの薬学的に許容される塩:
【0054】
【化3】
および薬学的に許容される賦形剤を目的としている。
【0055】
塩調製のための好ましいカチオンは、ナトリウム(Na+)、カリウム(K+)、リチウム(Li+)、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)、ストロンチウム(Sr2+)およびアンモニウム(NH4+)からなる群から選択できる。塩は、アルカリ金属またはアルカリ土類金属から調製することもできる。アルカリ金属は、ナトリウム(Na+)、カリウム(K+)およびリチウム(Li+)の中から選択できる。アルカリ土類金属は、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)およびストロンチウム(Sr2+)からなる群から選択できる。脂肪の局所的除去のための医薬組成物としての使用には、胆汁塩がデオキシコール酸ナトリウムであることが好ましい。
【0056】
本実施形態の化合物のプロドラッグも企図されている。プロドラッグは、患者へのプロドラッグの投与後、加水分解、代謝等のインビボ生理的作用によって本実施形態の化合物に化学的に修飾される活性または不活性化合物である。例えば、本デオキシコール酸またはその誘導体のC1〜C10エステルまたはアミドを調製することができ、その結果、細胞膜の破壊およびエステラーゼの放出により、デオキシコール酸またはその誘導体の放出が誘発される。エステラーゼの放出によってエステル保護基が開裂されるため、デオキシコール酸活性型またはその誘導体がインシチュの所望の位置に存在する。そのようなC1〜C10エステルは、酸素、硫黄または窒素から選択される1〜4個のヘテロ原子;酸素、硫黄または窒素から選択される1〜4個のヘテロ原子を場合によって有するメチル、エチル、イソプロピル、ブチル、ヘキシル等のアルキル基;任意の許容される置換点に1〜4個のヘテロ原子を場合によって有するベンジルまたはエチルフェニル基等、合計最大10個の炭素原子を有するアルキルフェニル基;ならびにフェニル基等のアリール基を場合によって含み得る。アミドの例は、ヒドロキサメートを含むがこれに限定されない。エステルを含むプロドラッグの一般的議論については、参照により全体が本明細書に組み込まれる、SvenssonおよびTunek、Drug Metabolism Reviews、165巻(1988年)ならびにBundgaard Design of Prodrugs、Elsevier(1985年)を参照されたい。デオキシコール酸のC1〜C10エステルまたはアミドの合成は、当該技術分野において既知である。例えば、エステルは、エステル化反応において、鉱酸の存在下でデオキシコール酸のアルコールとの反応によって合成できる。
【0057】
デオキシコール酸およびケノデオキシコール酸の自然構造は、以下のように示される。4つの環(A、B、C、D)およびD環から伸展するカルボン酸側鎖が示されている。
【0058】
【化4】
【0059】
【化5】
限定されないが、デオキシコール酸およびケノデオキシコール酸(CDCA)またはその誘導体のエステル、ヒドロキサメートおよびヒドロキシアミドの例のいくつかは、以下のように記載される。D環側鎖上のカルボキシル基のエステル化により、異なる官能基がデオキシコール酸またはケノデオキシコール酸に結合してプロドラッグを生成し得る。限定されないが、これらのD環側鎖エステルのいくつかの例が表1に示されている。
【0060】
【表1】
デオキシコール酸またはケノデオキシコール酸およびその誘導体の放出は、細胞膜の破壊およびエステラーゼの放出によって誘発され得る。エステラーゼの放出によってエステル保護基が開裂され得るため、デオキシコール酸のもしくはケノデオキシコール酸の活性型またはその誘導体がインシチュの所望の位置に存在する。
【0061】
デオキシコール酸、ケノデオキシコール酸およびそれらの誘導体のプロドラッグは、自然分子と反対の立体化学を有し得るエピマーも含む。これらのエピマー分子の例は、表2に示されている。
【0062】
【表2】
本実施形態の胆汁酸組成物の注射時に局所刺激がある場合があり、故に、局所麻酔薬を同時または順次に投与することが望ましい場合がある。例えば、リドカインは、ヒトにおいて頻繁に使用され、同時製剤(同じ容器中で同時に注射される)または同時注射(異なる容器から注射される)のいずれとして投与してもよい。リドカイン等の麻酔薬は、パッチまたは軟膏等の局所製剤によって投与してよい。
【0063】
深部組織の場合、麻酔薬を対象組織中により深く注射するか、または全身的に投与してよい(例えば、全身麻酔、硬膜外または他の既知の方法)。
【0064】
本発明の溶液中の胆汁酸(複数可)または胆汁塩(複数可)は、約0.001〜10、0.01〜5または0.1〜2%w/w、w/vまたはv/vの濃度であってよい。好ましくは、上記溶液中の胆汁酸(複数可)または胆汁塩(複数可)は、約0.1〜5%w/wまたはより好ましくは約1%w/wの濃度であってよい。いくつかの実施形態において、脂肪溶解液は、最大100、50、20、10、5、2、1、0.5、0.2、0.05、0.02または0.01グラムの1つまたは複数の洗浄剤、胆汁酸および/または胆汁塩、例えば、デオキシコール酸もしくはその塩またはデオキシコール酸ナトリウムを含む。
【0065】
好ましい実施形態において、本明細書における溶液は、脂質、リン脂質またはホスファチジルコリンを含まない。いくつかの実施形態において、本明細書における溶液は、最大5%w/w、w/vまたはv/vの脂質、リン脂質またはホスファチジルコリンを含む。
【0066】
いくつかの実施形態において、上記溶液は、抗菌剤、血管収縮薬、抗血栓剤、抗凝固剤、発泡抑制剤(suds−depressant)、抗炎症剤、鎮痛剤、分散剤、抗分散剤、浸透促進剤、ステロイド、精神安定薬、筋弛緩薬および下痢止め剤からなる群から選択される第2の治療剤をさらに含み得る。いくつかの実施形態において、溶液は、最大500mLの溶液を収納する容器内にあってよい。そのような容器は、シリンジまたはシリンジ充填可能な容器であってよい。
【0067】
いくつかの実施形態において、組成物および方法は、直交機序によって脂肪を消滅させることが知られている分子をさらに含む。そのような分子は、BIBP−3226(Amgen)、BIBO−3304(Boehringer Ingleheim)、BMS−192548およびAR−H040922(Bristol−Myers Squibb)、LY−357897(Eli Lilly)、1229U91およびGW438014S(GlaxoSmithKline)、JNJ−5207787(Johnson&Johnson)、Lu−AA−44608(Lundbeck)、MK−0557(Merck NPY)、NGD−95−1(Neurgogen)、NLX−E201(Neurologix)、CGP−71683(Novartis)、PD−160170(Pfizer)、SR−120819A、BIIE0246およびS.A.0204(Sanofi Aventis)、S−2367(shiongli)等のNPY受容体アンタゴニスト、NPY受容体アンタゴニストであるジヒドロピリジンおよびジヒドロピリジン誘導体、NPY受容体アンタゴニストである二環化合物、カルバゾール系NPY受容体アンタゴニストならびにNPY受容体アンタゴニストである三環化合物を含むがこれらに限定されない神経ペプチドY(NPY)受容体アンタゴニストを含む。例えば、国際公開第2006/133160号および米国特許第6,313,128号(参照により図面を含むその全体が本明細書に組み込まれる)を参照されたい。白色脂肪血管系にあるCKGGRAKDCペプチド等の脂肪選択的プロアポトーシスペプチドも企図されている。Kolonin M.G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)を参照されたい。
【0068】
一態様において、本発明は、対象における皮下脂肪沈着物を減少させる方法に関する。そのような方法は、(i)脂肪溶解有効量の1つまたは複数の薬理学的に活性な洗浄剤または胆汁酸(複数可)および/もしくは胆汁塩(複数可)またはデオキシコール酸もしくはその塩またはデオキシコール酸ナトリウム;(ii)薬学的、獣医学的または美容学的賦形剤;ならびに(iii)場合によって脂質を含み、ここで、脂質と胆汁酸または胆汁塩との比率が最大1%w/wである組成物であって、リパーゼもコリパーゼも含まない組成物を、対象における皮下脂肪沈着物に局所的に投与するステップを含む。いくつかの実施形態において、脂肪沈着物は、肥満、脂肪再分布症候群、眼瞼脂肪ヘルニア、脂肪腫、ダーカム病、脂肪異栄養症、野牛肩脂肪異栄養症、後頸部(dorsocervical)脂肪、内臓脂肪症、乳房肥大、脂肪過剰症、体幹および腕周囲に散在する体脂肪およびセルライトを伴う脂肪沈着物からなる群から選択される状態に関連する。好ましい実施形態において、上記方法は、前記対象に対して外科手術を実施するステップを含まない。
【0069】
一態様において、本発明は、哺乳動物における選択された位置からの脂肪沈着物の除去方法であって、それを必要とする哺乳動物に、治療有効量のDCAもしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0070】
【化6】
であり、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない前記化合物を投与するステップを含む方法を目的としている。
【0071】
一実施形態において、本発明の方法は、哺乳動物に、神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分を投与するステップをさらに含み得る。一実施形態において、神経ペプチドY(NPY)受容体アンタゴニストは、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、NPY受容体アンタゴニストであるジヒドロピリジン、NPY受容体アンタゴニストであるジヒドロピリジン誘導体、NPY受容体アンタゴニストである二環化合物、カルバゾール系NPY受容体アンタゴニストならびにNPY受容体アンタゴニストである三環化合物からなる群から選択される。例えば、国際公開第2006/133160号および米国特許第6,313,128号(参照により図面を含むその全体が本明細書に組み込まれる)を参照されたい。一実施形態において、脂肪選択的プロアポトーシスペプチドは、白色脂肪血管系にあるCKGGRAKDCペプチドである。Kolonin M.G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)を参照されたい。
【0072】
一態様において、本発明は、対象の皮膚領域における皮膚状態の出現を減少させるための方法に関する。そのような方法は、(i)皮膚引き締め有効量の1つまたは複数の薬理学的に活性な洗浄剤または胆汁酸(複数可)および/もしくは胆汁塩(複数可)またはデオキシコール酸もしくはその塩またはデオキシコール酸ナトリウム;(ii)薬学的、獣医学的または美容学的賦形剤;ならびに(iii)場合によって脂質を含む組成物を、前記皮膚領域に局所的に投与するステップを含む。いくつかの実施形態において、投与するステップは、本明細書における組成物を皮下または経皮注射によって送達することを含む。いくつかの実施形態において、治療または改善される皮膚状態は、弛緩性皮膚、皮膚の老化、皮膚の凹凸およびシワからなる群から選択される。いくつかの実施形態において、治療される皮膚の領域は、目の下、顎の下、腋の下、臀部、頬、額、ふくらはぎ、背部、大腿、足首または腹である。
【0073】
いくつかの実施形態において、皮膚領域における皮膚状態の出現を減少させるために使用される組成物は、皮膚引き締め溶液への製剤である。そのような皮膚引き締め溶液は、抗菌剤、血管収縮薬、抗血栓剤、抗凝固剤、発泡抑制剤、抗炎症剤、鎮痛剤、分散剤、抗分散剤、浸透促進剤、ステロイド、精神安定薬、筋弛緩薬および下痢止め剤からなる群から選択される第2の治療剤をさらに含み得る。
【0074】
好ましい実施形態において、洗浄剤は、デオキシコール酸、コール酸、ケノデオキシコール酸、7−アルファ−デヒドロキシレート(dehydroxylate)ケノデオキシコール酸、リトコール酸、ウルソデオキシコール酸、ジヒドロキシタウリン酸、トリヒドロキシタウリン酸および上記のいずれかのグリシン共役物からなる群から選択される胆汁酸を含む。いくつかの実施形態において、洗浄剤は、ナトリウム(Na+)、カリウム(K+)、リチウム(Li+)、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)、ストロンチウム(Sr2+)およびアンモニウム(NH4+)からなる群から選択されるカチオンを含む胆汁塩を含む。いくつかの実施形態において、洗浄剤は、アルカリ金属またはアルカリ土類金属であるカチオンとの胆汁塩を含む。好ましくは、アルカリ金属は、ナトリウム(Na+)、カリウム(K+)またはリチウム(Li+)であり、アルカリ土類金属は、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)またはストロンチウム(Sr2+)である。より好ましくは、胆汁塩はデオキシコール酸ナトリウムである。
【0075】
別の実施形態は、哺乳動物において脂肪を乳化する方法であって、それを必要とする哺乳動物に、治療有効量のDCAである化合物もしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0076】
【化7】
を投与するステップを含む方法であり、前記化合物はDCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない方法を提供する。
【0077】
別の実施形態は、ホスファチジルコリンを可溶化する方法であって、ホスファチジルコリンと、有効量のDCAである化合物もしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0078】
【化8】
であり、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない前記化合物とを混合するステップを提供する。
【0079】
本発明の別の態様は、デオキシコール酸(DCA)等の脂肪切除胆汁酸を、脂肪細胞を死滅させる作用物質と混合することに関する。一態様において、本発明は、直交機序によって脂肪を消滅させる分子をデオキシコレート注射液中に混合することにより、デオキシコレート注射の審美的作用を増強するための手段を企図している。そのような候補分子の例は、本明細書に記載されている通り、神経ペプチドY(NPY)アンタゴニストおよび脂肪選択的プロアポトーシスペプチドを含むがこれらに限定されない。脂肪細胞死滅および皮膚引き締めはいずれも、所望の作用を媒介するために必要となり得るため、脂肪死滅能力および強力な皮膚引き締め作用を有する作用物質(デオキシコレート等)の作用は、強力な脂肪細胞死滅効果を有する分子の添加によって増強できる。加えて、死滅させるために血管系にアクセスする必要がある分子(毛細血管の腔側に発現したタンパク質と結合するいくつかのプロアポトーシスペプチド等)は、デオキシコレートが血管漏出を引き起こし得るため、これらのタンパク質へのアクセスを得ることができる。故に、そのような作用物質は、デオキシコレートと相乗して、より少ない治療セッションでの体形矯正を媒介するためのより強力な手段を潜在的に作成することができる。
【0080】
合成方法
他の実施形態において、本発明は、DCAの化合物、塩およびプロドラッグならびにそれらの関連中間体の合成方法を提供する。
【0081】
本明細書において使用される場合、ステロイド骨格のナンバリングは、図1に示されている通りの一般的慣例に従う。
【0082】
したがって、提供されるのは、デオキシコール酸(DCA)もしくはそのエステルまたは薬学的に許容されるその塩:
【0083】
【化9】
を調製するための方法であって、
(a)9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1を、水素化条件下、H2と反応させて、化合物2
【0084】
【化10】
を形成するステップと、
(b)化合物2を酸と反応させて、化合物3
【0085】
【化11】
を形成するステップと、
(c)化合物3を還元剤と反応させて、化合物4を4および5
【0086】
【化12】
の混合物として形成するステップと、
(d)化合物4をオレフィン形成条件下で二炭素オレフィン化試薬と反応させて、化合物6
【0087】
【化13】
を形成するステップと、
(e)化合物6を、式7の化合物[式中、Pは保護基である]
【0088】
【化14】
に変換するステップと、
(f)式7の化合物をルイス酸の存在下でアルキルプロピオレートCH2CH2C(O)ORまたはアルキルアクリレートCH2=CHC(O)OR[式中、Rはアルキルである]と反応させて、式8の化合物[式中、Pは保護基であり、Rはアルキルであり、破線
【0089】
【化15−1】
は単結合または二重結合である]
【0090】
【化15−2】
を形成するステップと、
(g)式8の化合物を水素化条件下でH2と反応させて、式9の化合物[式中、Pは保護基であり、Rはアルキルである]
【0091】
【化16】
を形成するステップと、
(h)式9の化合物を酸化剤と反応させて、式10の化合物[式中、Pは保護基であり、Rはアルキルである]
【0092】
【化17】
を形成するステップと、
(i)式10の化合物を水素化条件下でH2と反応させて、式11の化合物[式中、Pは保護基であり、Rはアルキルである]
【0093】
【化18】
を形成するステップと、
(j)式11の化合物を還元剤と反応させて、式12の化合物[式中、Pは保護基であり、Rはアルキルである]
【0094】
【化19】
を形成するステップと、
(k)式12の化合物を脱保護条件に曝露してそのエステルを形成し、場合によって、適切な加水分解条件に曝露して、デオキシコール酸または薬学的に許容されるその塩を形成するステップと
を含む方法である。
【0095】
本発明は、以下のスキーム1に示される下記中間体[式中、PおよびRは上記で定義された通りである]も提供する。
【0096】
スキーム1.デオキシコール酸(DCA)の合成
【0097】
【化20】
一実施形態において、パート(a)の水素化条件はPd/C触媒を含む。
【0098】
一実施形態において、パート(b)の酸は鉱酸である。いくつかの態様において、鉱酸はH2SO4である。
【0099】
一実施形態において、パート(c)の還元剤はLiAl(OtBu)3Hである。
【0100】
一実施形態において、パート(d)の二炭素オレフィン化試薬はPh3PCH2CH3+Br−等のウィッティヒ剤である。
【0101】
一実施形態において、化合物7〜12の保護基Pは−C(O)CH3である。いくつかの態様において、化合物6は、無水酢酸、ならびにEt3N、ピリジンおよび/またはジメチルアミノピリジン等の有機塩基による6の処理等により、アシル化条件に曝露されて7aを形成する。
【0102】
一実施形態において、パート(f)のルイス酸はEtAlCl2である。
【0103】
一実施形態において、パート(f)のアルキルプロピオレートはメチルプロピオレートである。
【0104】
一実施形態において、パート(f)のアルキルアクリレートはメチルアクリレートである。
【0105】
一実施形態において、パート(g)の水素化条件はPtO2またはPd/C触媒を含む。
【0106】
一実施形態において、パート(h)の酸化剤はCrO3である。
【0107】
一実施形態において、パート(i)の水素化条件はPd/C触媒を含む。
【0108】
一実施形態において、パート(j)の還元剤はLiAl(OtBu)3Hである。
【0109】
一実施形態において、Pが−C(O)CH3である場合、パート(k)の脱保護および加水分解条件は、化合物12を、アルカリ土類金属水酸化物、アルカリ土類金属アルコキシドまたは両方の混合物と反応させることを含む。いくつかの態様において、加水分解条件は、デオキシコール酸を得るための酸性ワークアップを含む。他の態様において、酸性ワークアップは、対応する塩を得るために省略される。
【0110】
一実施形態において、アルカリ土類金属アルコキシドはLiOHである。
【0111】
一実施形態において、デオキシコール酸の塩は、アルカリ土類金属アルコキシドまたは水酸化物との反応によって調製できる。デオキシコール酸の塩は、ナトリウム(Na+)、カリウム(K+)およびリチウム(Li+)塩を含む。
【0112】
一実施形態において、提供されるのは、
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2);
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3);
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6);
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a);
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a);
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b);
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a);および
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
からなる群から選択される中間化合物である。
【0113】
医薬組成物および投与様式
組成物は、開示されている化合物を、少なくとも1つの薬学的に許容される賦形剤と組み合わせて構成され得る。許容される賦形剤は、非毒性であり、投与を補助し、開示されている化合物の治療的有用性に悪影響を及ぼさない。そのような賦形剤は、任意の固体、液体、半固体であってよく、またはエアロゾル組成物の場合、当業者に一般に利用可能なガス状賦形剤であってよい。
【0114】
固体医薬品賦形剤は、デンプン、セルロース、タルク、グルコース、ラクトース、スクロース、ゼラチン、麦芽、米、小麦粉、石粉、シリカゲル、ステアリン酸マグネシウム、ステアリン酸ナトリウム、グリセロールモノステアレート、塩化ナトリウム、乾燥脱脂粉乳等を含む。液体および半固体賦形剤は、グリセロール、プロピレングリコール、水、エタノール、ならびに石油、植物または合成起源のもの、例えば、ピーナッツ油、大豆油、鉱油、ゴマ油等を含む種々の油から選択できる。好ましい液体担体は、特に注射用液の場合、水、生理食塩水、ブドウ糖水溶液およびグリコールを含む。
【0115】
概して、好ましい実施形態の化合物は、類似の効用を果たす作用物質の許容されている投与様式のいずれかによって治療有効量で投与される。好ましい実施形態の化合物、すなわち活性成分の実際の量は、治療される疾患の重症度、対象の年齢および相対的健康、使用される化合物の効力、投与の経路および形態ならびに他の要因等の数々の要因によって決まる。薬物は、1日当たり複数回、好ましくは1日当たり1回または2回投与され得る。これらの要因のすべては、担当臨床医の技量の範囲内である。
【0116】
製剤中の化合物の量は、当業者によって用いられる全範囲内で変動し得る。本明細書に記載されている通りの種々の態様における本組成物が調製でき、ここで、デオキシコール酸部分は、水1体積当たりの重量に基づいてまたは水の密度を前提としたw/w(すなわち、重量と体積との1対1対応)に基づいて約0.5%〜10%の範囲である。別の態様において、本実施形態は、最大で希釈剤の飽和点の濃度である現在記載されている医薬組成物に関する。濃度およびpH等の条件に基づき、チキソトロピー粘度を選択することができる。例えば、Mukhopadhyay, S.およびU. Maitra、Current Science、87巻:1666〜1683頁(2004年)の1680頁を参照されたい。
【0117】
いくつかの実施形態において、投与するステップは、本明細書における組成物を、皮膚パッチ、ポンプまたは真皮下デポー剤によって送達することを含む。いくつかの実施形態において、投与するステップは、本明細書における組成物を局所的にまたは皮下に送達することを含む。特定の実施形態において、投与するステップは、前記対象の目の下、顎の下、腋の下、臀部、ふくらはぎ、背部、大腿または腹の領域に局所的に(例えば、皮下または真皮下に)投与することを含む。投与は、皮下または経皮注射によって為され得る。
【0118】
合成スキーム
胆汁酸医薬組成物の完全な化学合成方法および有用な中間体の他の例が以下に提供される。
【0119】
これらの下記の記述および例は、本化合物を自然に産生する哺乳動物または微生物生命体からのDCAの抽出の代替を提供する。合成経路1〜6は、本発明におけるデオキシコール酸(DCA)を合成するための使用が企図されている。合成経路1Bおよび実施例1〜11は、ヒドロコルチゾンからのDCAの合成を示す。
【0120】
1.アドレノステロンからの9(11)−エンまたは11,12−エンを介する合成経路#1A
スキーム1A(以下)のコルチゾン(化合物1.1)は、完全な合成材料として広く利用可能である。クロロクロム酸ピリジニウム(PCC)を使用してこれを効率的に開裂し、C17ケトン化合物を形成することができる。このアドレノステロン(化合物1.2)への開裂は、HIO4またはビスマス酸ナトリウム(NaBiO3)を使用して実施することもできる。化合物1.2を化合物1.3に変換する反応は、既知の化学的プロセスである。化合物1.3の化合物1.4への変換には、モノケタール化が関与する。後続ステップは、3−ケト−4−エンの再生、C5β配置を得るための4,5−エン(H2/Pt/DMF)の選択的還元および化合物1.5を得るためのC3カルボニル基の所望の3α配置への選択的還元である。化合物1.5を化合物1.6に変換する際における保護基の添加およびそれに続く生成物の還元により、C11β−オール(アキシャル配置)、すなわち化合物1.7が得られ、これは、主要な9(11)−エンへの位置選択的脱離(すなわち、化合物1.7の化合物1.8への変換)に適している。
【0121】
ここで、合成スキームは、化合物1.7が化合物1.8または化合物1.9のいずれへの変換のための出発材料として使用され得るかという点で分岐する。C11ヒドロキシル基とC9水素原子との間のトランスジアキシャルの関係により、化合物1.7を化合物1.8に変換するために使用される脱離反応は位置選択的である。化合物1.9の異性体C11−C12オレフィンを得るための脱離の代替様式は、シス−熱脱離(すなわち、化合物1.7の化合物1.9への変換)を含み、同様に位置選択的である。
【0122】
スキーム1A.DCAのC12ヒドロキシル基の2つのC環前駆体の合成
【0123】
【化21】
化合物1.8のアリル酸化(CrO3および3,5ジメチルピラゾールによる処理を介する)により、エノン含有化合物1.10が得られる。化合物1.9の過酸酸化は、ステロイドのアルファ面から立体選択的に進み、C11−12エポキシド化合物1.11が得られる(上記スキーム1Aを参照)。これらの化学転換により、C12ヒドロキシル基官能性の2つの主要な前駆体、すなわち、化合物1.10および化合物2.1が得られる(スキーム1Aおよび2)。
【0124】
当業者であれば、上記コルチゾン経路は、代わりに、同じ炭素骨格および同じ酸素原子の相対的配置を有するヒドロコルチゾンから始まるように変更してよく、ヒドロコルチゾンは、炭素原子を有するC−11酸素の酸化状態のみがコルチゾンと異なっていることを理解するであろう。ヒドロコルチゾンは市販されており、全化学合成(Woodward
R. B.ら、J. Am. Chem. Soc.、74巻:4223頁(1952年))を含むこの化合物の種々の合成が既知である(Szczebaraら、Nature Biotechnology、21巻:143〜149頁(2003年2月))。ケトン1.13は、α,β−不飽和二重結合の水素化分解によってヒドロコルチゾン1.12(スキーム1B)から開始して合成され、続いて、NaIO4を使用する1,2−ジオール開裂を可能にするための水素化ホウ素ナトリウムを使用して完全なケトン還元を行い、それにより、ステロイド環系のD環上にC17ケトンを形成する。それに続くクロロクロム酸ピリジニウム(PCC)による酸化により、1.13が得られる。K−selectride(登録商標)による1.13の処理、続いて無水酢酸/ピリジンによるアセチル化により、保護されたアルコール1.15が得られる。それに続くウィッティヒ試薬による1.15のオレフィン化によってアルケン1.16を得て、その後、これをメチルプロピオレートおよびエチルアルミニウムジクロリドで処理し、ジエン1.17を形成する。両方の二重結合の水素化に続いて、ケトン1.18を還元し、得られたアルコール中間体をピリジン中のSOCl2による処理により脱離し、アルケン1.19を得る。CrO3によるアルケン1.19のアリル酸化および水素化条件下での二重結合の還元により、ケトン1.21が得られる。アセテート保護基の除去および得られたアルコールの酸化により、ジケトン1.22が得られる。LiAlH(O−tBu)3による1.22の還元およびメチルエステルの加水分解により、DCAが得られる。
【0125】
スキーム1B.ヒドロコルチゾンからのDCAの合成
【0126】
【化22】
化合物1.10および2.1のさらなる転換は、スキーム2に示されている。最初に、化合物1.10を、DCAのものと同一の適切に官能基化されたC環系を含有するように修飾する(スキーム2)。C12カルボニル基の立体選択的還元により化合物2.1が得られ、化合物2.1中に存在する9(11)二重結合の触媒水素化により化合物2.2が得られる。
【0127】
スキーム2.アリル酸化経路を使用するC12ヒドロキシル基の導入
【0128】
【化23】
スキーム3は、エポキシド含有化合物1.11の、スキーム2の類似体C12α−ヒドロキシステロイド化合物2.2への転換を提示する。
【0129】
スキーム3.C11−C12エポキシドの立体選択的還元
【0130】
【化24】
これらの経路の両方において上述した通り、共通の中間化合物2.2が形成される。
【0131】
DCAの合成における次のステップは、DCAのカルボキシル側鎖置換D環を含有するような化合物2.2中に存在するD環の修飾である(スキーム4およびスキーム5)。
【0132】
スキーム4.脱保護およびウィッティヒ反応
【0133】
【化25】
最初に、化合物2.2のC17ケタールおよびC3シリルエーテル基を加水分解する。その後、ウィッティヒ反応を実施し、化合物4.2を得る。化合物4.2の化合物5.1への変換がエン反応によって行われる。それに続く化合物5.1の触媒還元およびエステルの加水分解により、DCAが得られる(スキーム5)。
【0134】
スキーム5.DCA側鎖の導入のためのエン反応および触媒還元
【0135】
【化26】
2.コルチゾンからのアドレノステロン(i−ステロイド、3,5−シクロステロール経路)を介する合成経路#2
C17におけるアドレノステロン(化合物1.2、スキーム6)の選択的ケタール化、ホウ化水素還元、メシル化および緩衝化された加水分解により、i−ステロイド(3,5−シクロステロール)含有化合物6.1が得られる。化合物6.1に、9(11)−エン形成(化合物6.1の化合物6.2への変換、スキーム6)およびアリル酸化(化合物6.2の化合物6.3への変換、スキーム6)、続いてカルボニル基還元を受けさせ、化合物6.4を得る。i−ステロールの加水分解および水素化により化合物6.5が得られ、合成経路#1において上記で提示した合成方法によってこれをDCAに変換することができる。
【0136】
スキーム6.i−ステロイド(すなわち、3,5−シクロステロール)の形成によるA−B−環系の保護
【0137】
【化27】
3.ヘコゲニンからの合成経路#3
ヘコゲニン(化合物7.1、スキーム7)は、メキシカンヤムおよびAgave種の他の植物において豊富に見られる植物ステロールである。DCA合成のための出発材料としてのヘコゲニンの主な利点は、DCAに存在するC12酸素官能基をそのまま有することである。
【0138】
ヘコゲニンから開始する合成経路における最初のステップは、ヘコゲニン(化合物7.1)中のC12カルボニル基の、必須のC12−α配置への立体選択的還元(化合物7.1の化合物7.2への変換)である。その後、3−β−オール、5α−AB環系は、3α−オール、5β−AB環系に変換(化合物7.1の、化合物7.2へ、化合物7.3への変換)される(スキーム7)。よく知られているマーカー分解(Marker, R.E.、Rohrmann, E.、Sterols. LXIX. Oxidation Products of Sarsasapogenin. Sarsasapogenoic Acid and Related Substances、J. Am. Chem. Soc.、61巻(8号):2072〜2077頁(1939年))を、化合物7.2の化合物7.3への変換に続いて行い、化合物7.4を得る。D環側鎖(スキーム8)の化合物8.2への導入は、スキーム4および5に示されている方法によって実現される。化合物8.2中の必須のC17ケトンは、化合物8.1の酢酸エノールのオゾン分解によって形成される(スキーム8)。その後、オレフィン化およびエン反応順序を使用するスキーム5と類似の方式で、8.2からDCAが調製される。ヘコゲニンから開始する代替経路は、スキーム9および10に示されている。
【0139】
スキーム7.C12−ヒドロキシル基導入、AB環修飾および側鎖開裂
【0140】
【化28】
スキーム8.側鎖導入
【0141】
【化29】
スキーム9.3−ケト−4−エンへのジチオエタン経路
【0142】
【化30】
スキーム10A.3−ケト−4−エンを介するDCA形成
【0143】
【化31】
4.サポゲニンからの合成経路#4
サポゲニンは、サポニン(すなわち、ステロイド配糖体)のC3ヒドロキシル基に結合している単糖類および二糖類の加水分解に由来する。これらは広く発生する植物生成物である。サポニンは、本来、以下に示される通りのスピロケタール構造として発生する。化合物10.3を、チゴゲニン、ジオスゲニン、クロロゲニン、スミラゲニンおよびヘコゲニン(化合物7.1)から形成することもできる。我々は、DCAが、これらの、すなわち、チゴゲニン、ジオスゲニン、クロロゲニン、スミラゲニンおよびヘコゲニン(化合物7.1)のそれぞれから合成され得ると考えている(Y. Mazur、N. DanieliおよびFranz Sondheimer、J. Am. Chem. Soc.;82巻、5809頁(1960年))。
【0144】
我々は、ヘコゲニンからDCAを合成できたため、上記サポゲニンのいずれも、同様にDCA合成のための出発材料としての役割を果たし得ると認識している。
【0145】
スキーム10B.サポニンからの合成経路
【0146】
【化32】
5.スチグマステロールからの合成経路#5
スチグマステロール(化合物11.1)は、広く利用可能な植物ステロールである。これは、DCA合成のための出発材料として、官能基化されたAB環系および容易に開裂可能な側鎖部分を含有するという利点を有する。スチグマステロールは、DCA合成に不可欠の、C環において必要な官能性を欠くという不利点を有する。
【0147】
この合成経路において、スチグマステロール(化合物11.1)AB環は、i−ステロイド形成によって保護され、続いてオゾン分解によってC17に導入された側鎖が得られ、それはマスクされた形態のカルボキシル基としてのC24−オールに還元される(スキーム11)。後続ステップは、C12にアリル位を生成する(スキーム12)。B環ジエン形成および酢酸第二水銀酸化は既知の過程であり、B環系の触媒還元により、上述した先の経路と共通の中間体が得られる。しかしながら、他の経路とは対照的に、側鎖が既に存在する。アリル酸化(化合物12.4の化合物1.20への変換)および立体選択的還元(化合物1.20の化合物1.21への変換)、続いて前述のステップにより生成物が得られ、これをDCAに変換する。
【0148】
スキーム11.i−ステロイドのオゾン分解および側鎖還元
【0149】
【化33】
スキーム12.トリエン形成およびアリル酸化
【0150】
【化34】
スチグマステロール経路の変形形態では、B環ジエンのディールス・アルダー保護を使用する。これは、9(11)二重結合を単離してアリル酸化ステップ中の考えられる干渉を防止するため、有利である(スキーム13)。
【0151】
スキーム13.トリエン形成および環Bジエンのディールス・アルダー保護
【0152】
【化35】
6.エルゴステロールからの合成経路#6
エルゴステロール(化合物14.1)は、容易に利用可能な出発材料であり、本明細書で説明されている手順を適合することによってDCAを調製するために使用できる。アリル酸化は、C12酸素官能性への簡易経路を提供する(スキーム14)。この経路は、環Bジエンから開始するという利点を有する。これはスチグマステロール経路と収束する。
【0153】
スキーム14.エルゴステロールからのトリエン形成
【0154】
【化36】
好ましい実施形態の化合物は、下記の一般的方法および手順を使用して、容易に利用可能な出発材料から調製できる。典型的なまたは好ましい処理条件(すなわち、反応温度、時間、反応物質のモル比、溶媒、圧力等)が与えられている場合、特に明記しない限り、他の処理条件を使用してもよいことが理解される。最適反応条件は、使用される特定の反応物質または溶媒によって異なり得るが、そのような条件は、当業者が日常的な最適化手順によって決定できる。
【0155】
加えて、当業者には明らかなように、いくつかの官能基が望ましくない反応を受けるのを防止するために、従来の保護基が必要となり得る。種々の官能基に適切な保護基ならびに特定の官能基を保護および脱保護するための適切な条件は、当該技術分野において既知である。例えば、数々の保護基は、T. W. GreeneおよびG. M. Wuts、Protecting Groups in Organic Synthesis、第3版、Wiley、New York、1999年ならびにその中で引用されている参考文献に記載されている。
【0156】
本明細書に記載されている反応のための出発材料および試薬は、概して既知の化合物であるか、または既知の手順もしくはその明白な修正形態によって調製できる。例えば、出発材料および試薬の多くは、Aldrich Chemical Co.(Milwaukee、Wisconsin、USA)、Bachem(Torrance、California、USA)、Emka−ChemまたはSigma(St.Louis、Missouri、USA)等の商業的供給業者から入手可能である。その他は、FieserおよびFieserのReagents for Organic Synthesis、1〜15巻(John Wiley and Sons、1991年)、RoddのChemistry of Carbon Compounds、1〜5巻および補足(Elsevier Science Publishers、1989年)、Organic Reactions、1〜40巻(John Wiley and Sons、1991年)、MarchのAdvanced Organic Chemistry(John Wiley and Sons、第4版)ならびにLarockのComprehensive Organic Transformations(VCH Publishers Inc.、1989年)等の標準的な参考教科書に記載されている手順またはその明白な修正形態によって調製できる。
【実施例】
【0157】
好ましい実施形態の種々の出発材料、中間体および化合物は、適切な場合、沈殿、ろ過、結晶化、蒸発、蒸留およびクロマトグラフィー等の従来の技術を使用して単離および精製できる。これらの化合物の特徴付けは、融点、質量スペクトル、核磁気共鳴および種々の他の分光分析によって等、従来の方法を使用して実施できる。
【0158】
合成経路#1、スキーム1Bにおける生成物の合成を実施するためのステップの例示的な実施形態は、以下でさらに詳細に記載される。表3は、下記の実施例におよび本明細書全体を通して記載されている例示的な反応スキームおよび合成経路において種々の化合物/部分/装置/手順/特性を表現するために使用される略語を記載している。
【0159】
【表3−1】
【0160】
【表3−2】
概要:酸素および水分感受性材料の操作は、アルゴン雰囲気下、二口フレームドライフラスコで行う。カラムクロマトグラフィーは、SE−Makeシリカゲル(60〜120メッシュ)、Spectrochemシリカゲル(230〜400メッシュ)または酸化アルミニウム90中性(SD−Fine Chem.Ltd.、India)を使用して実施する。分析用薄層クロマトグラフィー(TLC)は、Merck Kieselgel 60F254(0.25mm)プレート(Merck&Co.、Whitehouse Station、NJ)で実施した。スポットの可視化は、UV光(254nm)ランプによってまたはエタノール中の硫酸(5%)およびp−アニスアルデヒド(3%)溶液で炭化させることによってのいずれかで検出した。
【0161】
装置:本明細書に記載されている反応スキームおよび合成経路の化合物および生成物の分析は、以下に記載される装置および設備で実行できる。
【0162】
核磁気共鳴(NMR)
プロトンおよび炭素の核磁気共鳴スペクトル(1H NMRおよび13C NMR)は、内部標準として溶媒共鳴(1H NMR、7.26ppmのCHCl3または2.5ppmのDMSOおよび3.33ppmのDMSO−H2O;13C NMR、77.0ppmのCDCl3または39.5ppmのDMSO)を用い、Varian Mercury−Gemini200(1H NMR、200MHz;13C NMR、50MHz)またはVarian Mercury−Inova500(1H NMR、500MHz;13C NMR、125MHz)(Varian,Inc.、Palo Alto、CA)分光計で記録される。1H NMRデータは、下記の通りに報告される:化学シフト(δ、ppm)、多重度(s=一重項、d=二重項、t=三重項、q=四重項、br=広幅、m=多重項)、カップリング定数(Hz)および積分。
【0163】
赤外線分光法
赤外線スペクトル(FT−IR)は、JASCO−460+モデル(Jasco,Inc.、Easton、MD)で動作させる。質量スペクトルは、Perkin ElmerのAPI−2000分光計(Perkin Elmer,Inc.、Waltham、MA)でES+モードを使用して得られる。
【0164】
融点
融点は、LAB−INDIA融点計測装置(Labindia Instruments Pvt.Ltd.、India)を使用して測定したものであり、訂正されない。
【0165】
高圧液体クロマトグラフィー
HPLCクロマトグラムは、PDA検出器を有するSHIMADZU−2010モデル(株式会社島津製作所、日本)を使用して記録した。
【0166】
光学活性
比旋光度([α]D)は、589nmのJASCO−1020(Jasco,Inc.、Easton、MD)を用いて測定し、訂正されない。
【0167】
化学物質:特に断りのない限り、市販の試薬を精製することなく使用する。ジエチルエーテルおよびTHFは、ナトリウム/ベンゾフェノンケチルから蒸留される。無水DMF、DCM、ペンタンおよびヘキサンは、CaH2からの蒸留によって得られる。
【0168】
(実施例1)
アンドロスタン−3,11,17−トリオン(1.13)の調製
10%のPd/C(2.5g、5wt%)を、DMF(250mL)中のヒドロコルチゾン(化合物1.12)(50.0g、138.12mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で12時間水素化する。TLCによって証明される通り、出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去する。粗生成物(48.0g)が無色固体として得られる。
【0169】
NaBH4(2.1g、55.3mmol)を、EtOH(500mL)およびCH2Cl2(500mL)中の上記粗生成物(48.0g、131.86mmol)の溶液に添加する。1時間後、アセトン(50mL)および水(150mL)、続いてNaIO4(70.5g、329.6mmol)を添加する。混合物を室温で終夜撹拌する。
【0170】
蒸留水(500mL)を添加し、混合物を酢酸エチル(3×250mL)で抽出する。酢酸エチル層をシリカゲルプラグに通して洗い流し、溶媒を蒸発させて38gを無色固体として得る。粗生成物を精製することなくさらに酸化させる。
【0171】
PCC(40.4g、187.5mmol)を、CH2Cl2(400mL)中の上記粗生成物の溶液に3等分ずつ30分間かけて添加する。得られた反応混合物を室温で約3〜4時間撹拌する。TLCによってモニターされる通り、反応の完了時に、粗反応混合物をセライトおよびシリカゲルのパッドに通して連続的にろ過し、粗材料を、酢酸エチル/ヘキサン(3:10)[50mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:1)中、化合物1.13のRf=0.37および化合物1.12のRf=0.05]で溶離するカラムクロマトグラフィー[59(W)×700(L)mm、60〜120メッシュシリカ、150g]によって精製し、ジアステレオマー化合物1.13(33.0g、79%収率)を無色固体として得る。
【0172】
得られた粗材料を、Phenomenex Lunov C18カラム(250×30.0mm、10μ)を使用する分取HPLCおよびCH3CN:H2O(12:13)による定組成溶離によって、15mLの画分中、25mL/分流速で精製する。分取HPLCは、精製にのみ使用し、分析には使用しない。表4は、生成物の計測された特性を記載している。
【0173】
【表4】
(実施例2)
3β−ヒドロキシ−アンドロスタン−11,17−ジオン(1.14)
K−selectride(登録商標)(98.39mL、98.01mmol、THF中1M溶液)を、THF(330mL)中の化合物1.13(33.0g、109.27mmol)の溶液に、不活性雰囲気下、−78℃で15分間かけて添加し、−78℃で約3〜4時間撹拌する。反応混合物をNaOH水溶液(2M、70mL)でクエンチする。粗反応混合物を酢酸エチル(500mL)で希釈し、有機層を水(3×75mL)および飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空下で除去し、33gの粗材料を得る。粗生成物を精製することなくアセチル化に付す。
【0174】
粗材料の精製
粗材料を、酢酸エチル/ヘキサン(1:4)[25mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:1)中、化合物1.14のRf=0.3および化合物1.13のRf=0.37]で溶離するカラムクロマトグラフィー[29(W)×600(L)mm、230〜400メッシュシリカ、200g]によって精製し、化合物1.14を得る。表5は、生成物の計測された特性を記載している。
【0175】
【表5】
(実施例3)
3β−ヒドロキシアンドロスタン−11,17−ジオンアセテート(1.15)
無水酢酸(16.6g、162.8mmol)を、ピリジン(150mL)中の化合物1.14(33.0g、108.55mmol)の溶液に、不活性雰囲気下、0℃で添加する。得られた反応混合物を周囲温度で終夜撹拌する。TLCによって証明される通り、反応の完了時に、ピリジンおよび残っている無水酢酸を真空下で除去する。粗残留物を酢酸エチル(500mL)で希釈し、水(3×150mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空蒸発させ、粗材料を、酢酸エチル/ヘキサン(1:10)[25mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(3:7)中、化合物1.15のRf=0.38および化合物1.14のRf=0.1]で溶離するカラムクロマトグラフィー[59(W)×800(L)mm、60〜120メッシュシリカ、150g]によって精製し、化合物1.15(19.0g、66.4%収率)を無色固体として得る。表6は、生成物の計測された特性を記載している。
【0176】
【表6】
(実施例4)
(Z)−3β−ヒドロキシ−5β−プレグ−17(20)−エン−11−オンアセテート(1.16)
カリウムtert−ブトキシド(159.28mL、159.2mmol、THF中1M溶液)を、THF(150mL)中のエチルトリフェニルホスホニウムブロミド(61.16g、164.8mmol)の溶液に、不活性雰囲気下、−5℃で1時間かけて滴下添加する。得られた濃桃色の反応混合物を10〜15℃まで加温し、同じ温度でさらに1時間撹拌する。THF(50mL)中の化合物55(19.0g、54.9mmol)の溶液を、上記ウィッティヒイリド懸濁液に−5℃でゆっくり導入する。溶液をさらに10〜20分間撹拌し、反応混合物を周囲温度までゆっくり加温する。撹拌を約3〜4時間続ける。TLCによって証明される通り、出発材料の完全消失時に、反応混合物を飽和NH4Cl水溶液(75mL)でクエンチする。水層をEtOAc(2×150mL)で抽出し、合わせた有機抽出物を飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:20)[25mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:6)中、化合物1.16のRf=0.54および化合物1.15のRf=0.06]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、300g]によって精製し、化合物1.16(15.5g、78.8%収率)を濃厚な無色液体として得て、これを1〜2日後、0℃でゆっくり凝固させる。表7は、生成物の計測された特性を記載している。
【0177】
【表7】
(実施例5)
メチル(E)−3β−ヒドロキシ−5β−コーラ−16(17),22(23)−ジエン−24−オエートアセテート(1.17)
メチルプロピオレート(9.68g、114.95mmol)を、0℃のCH2Cl2(220mL)中の化合物1.16(16.5g、46mmol)の溶液に添加する。反応混合物を周囲温度まで加温し、不活性雰囲気下、1時間撹拌する。エチルアルミニウムジクロリド(17.5g、137.8mmol)を、0℃の上記混合物に滴下導入し、得られた反応塊を再度周囲温度まで加温し、終夜撹拌する。TLCによって証明される通り、反応の完了時に、粗反応混合物を氷水(100mL)でクエンチし、水層をEtOAc(3×150mL)で抽出する。合わせた有機層を飽和ブライン溶液(100mL)で洗浄し、MgSO4(50g)で乾燥させる。溶媒を真空下で除去し、粗材料を酢酸エチル/ヘキサン(1:7)[15mL画分、10mL/分溶離、TLCによってモニターし、UV光(254nm)ランプまたはp−アニスアルデヒド炭化のいずれかによって検出する;EtOAc/ヘキサン(1:6)中、化合物1.17のRf=0.36および化合物1.16のRf=0.54]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、300g]によって精製し、化合物1.17(16g、79%収率)を無色半固体として得る。表8は、生成物の計測された特性を記載している。
【0178】
【表8】
(実施例6)
メチル3β−ヒドロキシ−5β−コラン−11−オン−24−オエートアセテート(1.18)
10%Pd/C(2.9g、20wt%)を、EtOAc(150mL)中の化合物1.17(14.5g、32.8mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で12時間水素化する。TLC[EtOAc/ヘキサン(1:3)中、化合物1.18のRf=0.43および化合物1.17のRf=0.43;しかしながら、化合物1.17だけは共役エステル発色団によりUV活性である]によって証明される通り、出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去し、化合物1.18(14g、95.7%収率)を無色固体として得る。表9は、生成物の計測された特性を記載している。
【0179】
【表9】
(実施例7)
メチル3β−ヒドロキシ−5β−コール−9(11)−エン−24−オエートアセテート(1.19)
PtO2(5.0g、100wt%)を、触媒量のAcOH(2.0mL)の存在下、EtOAc(75mL)中の1.18(5.0g、11.2mmol)の溶液に添加する。得られたスラリーをParr装置(70psi)内で約14〜16時間水素化する。反応の完了時に、粗混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去する。粗生成物をさらに精製することなく脱離反応に使用する。
【0180】
SOCl2(1.98g、16.78mmol)を、ピリジン(100mL)中の上記粗混合物の溶液に、0℃で滴下導入する。得られた反応混合物を周囲温度まで加温し、約1時間撹拌する。TLCによって証明される通り、反応の完了時に、ピリジンを真空下で除去する。粗残留物を酢酸エチル(100mL)で希釈し、水(2×50mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(40g)で乾燥させる。溶媒を真空蒸発させ、粗材料を、酢酸エチル/ヘキサン(1:10)[10mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:6)中、化合物1.19のRf=0.51および化合物1.18のRf=0.22]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、120g]によって精製し、化合物1.19(4.1g、85.4%収率)を無色固体として得る。表10は、生成物の計測された特性を記載している。
【0181】
【表10】
(実施例8)
メチル3β−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエートアセテート(1.20)
CrO3(8.0g、100wt%、80.0mmol)を、AcOH(150mL)中の化合物1.19(8.0g、18.6mmol)の溶液に添加する。得られた反応混合物を60℃で約24〜36時間加熱する。前駆体の完全消失時に、酢酸を真空蒸発させ、粗材料をジエチルエーテル(400mL)に溶解する。有機層を水(2×100mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(40g)で乾燥させる。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:5)[10mL画分、3mL/分溶離、TLCによってモニターし、UV光(254nm)ランプによって検出する;EtOAc/ヘキサン(1:4)中、化合物1.20のRf=0.28および化合物1.19のRf=0.61]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、120g]によって精製し、化合物1.20(5g、60.5%収率)を無色固体として得る。表11は、生成物の計測された特性を記載している。
【0182】
【表11】
(実施例9)
メチル3β−ヒドロキシ−5β−コラン−12−オン−24−オエートアセテート(1.21)
10%Pd/C(30mg、10wt%)を、EtOAc(30mL)中の化合物1.20(300mg、0.675mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で約16時間水素化する。TLC[EtOAc/ヘキサン(3:7)中、化合物1.21のRf=0.44および化合物1.20のRf=0.44;しかしながら、化合物1.20だけはそのエノン発色団によりUV活性であり;加えて、化合物1.20の炭化は不鮮明であるが化合物1.21は鮮明である]による出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去し、化合物1.21(270mg、90%収率)を無色固体として得る。表12は、生成物の計測された特性を記載している。
【0183】
【表12】
(実施例10)
メチル5β−コーラ−3,12−ジオン−24−オエート(1.22)
NaOH(73mg、1.8mmol)を、MeOH(10mL)中の化合物1.21(270mg、0.6mmol)の溶液に添加する。得られた反応混合物を周囲温度で約2時間撹拌する。TLCによって証明される通り、反応の完了時に、MeOHを真空下で除去し、粗生成物を酢酸エチル(20mL)で希釈する。有機層を飽和ブライン溶液(10mL)で洗浄し、MgSO4(5.0g)で乾燥させる。溶媒を真空下で除去し、粗材料を精製することなくエステル化反応において使用する。
【0184】
SOCl2(0.1mL、1.35mmol)を、0℃のMeOH(10mL)中の上記粗材料の溶液に滴下添加する。得られた反応混合物を周囲温度で約1時間撹拌する。反応の完了時に、MeOHを真空下で除去する。粗反応混合物をEtOAc(30mL)で希釈し、有機層を水(3×10mL)、飽和ブライン溶液(15mL)で洗浄し、MgSO4(5g)で乾燥させる。溶媒を真空蒸発させ、粗生成物を精製することなく酸化反応に使用する。
【0185】
PCC(1.0g、4.6mmol)を、CH2Cl2(25mL)中の得られたエステルの溶液に3等分ずつ約5分間かけて導入する。得られた反応混合物を周囲温度で約3〜4時間撹拌する。TLCによって証明される通り、反応の完了時に、粗反応混合物をセライトのパッドに通してろ過する。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:6)[10mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(2:3)中、化合物1.22のRf=0.57および化合物1.21のRf=0.71]で溶離するカラムクロマトグラフィー[19(W)×400(L)mm、60〜120メッシュシリカ、45g]によって精製し、化合物1.22(170mg、70.8%収率)を無色固体として得る。表13は、生成物の計測された特性を記載している。
【0186】
【表13】
(実施例11)
デオキシコール酸メチル(1.22−エステル)
LiAlH(O−tBu)(332mg、1.3mmol)を、THF(10mL)中の化合物1.22(150mg、0.37mmol)の溶液に、不活性雰囲気下、周囲温度で滴下導入する。約4〜5時間撹拌した後、反応混合物をHCl水溶液(2mL、1N)でクエンチし、粗混合物をEtOAc(30mL)で希釈し、水(15mL)、飽和ブライン溶液(10mL)で洗浄し、MgSO4(3g)で乾燥させる。溶媒を真空下で除去し、粗塊を、MeOH/CH2Cl2(1:20)[5mL画分、3mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;MeOH/CH2Cl2(1:9)中、化合物1.22−エステルのRf=0.42および化合物1.22のRf=0.85]で溶離するカラムクロマトグラフィー[29(W)×500(L)mm、230〜400メッシュシリカ、50g]によって精製し、デオキシコール酸メチル(化合物1.22−エステル)(110mg、72.8%収率)を無色固体として得る。表14は、生成物の計測された特性を記載している。
【0187】
【表14】
(実施例11)
デオキシコール酸
H2O(2.0mL)中のLiOH(23mg、0.55mmol)の溶液を、THF(4mL)中の1.22−エステル(110mg、0.27mmol)の溶液に添加する。得られた反応混合物を周囲温度で約2〜3時間撹拌する。TLC[MeOH/CH2Cl2(1:9)中、化合物DCAのRf=0.35および化合物1.22−エステルのRf=0.42]によるエステルの消失時に、粗反応混合物を酢酸エチル(10mL)で希釈し、飽和ブライン溶液で粉砕し、有機層を完全に分離させる。有機層を飽和NH4Cl溶液(10mL)で洗浄し、MgSO4(3.0g)で乾燥させる。溶媒を真空下で除去し、トルエン(3×5mL)と共沸させることによっていかなる微量の水も除去し、デオキシコール酸(DCA)(100mg、94.3%収率)を無色固体として得る。表15は、生成物の計測された特性を記載している。
【0188】
【表15】
実施例1〜11に記載されている例示的な過程の生成物の収率は、表16に記載されている。
【0189】
【表16】
(実施例12)
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2)
【0190】
【化37】
DMF(150mL)中の9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1(30.0g、99.3mol)の溶液に、10%のPd/C(2.1g)を添加し、得られたスラリーをParr装置(60psi)内で12時間水素化した。TLCによって証明される通り、出発材料の完全消失時に、粗反応混合物をCelite(登録商標)の小型パッドに通してろ過し、溶媒を真空下で除去し、無色固体(30.0g)を得た。この固体を0℃のアセトン(90mL)と合わせ、得られたスラリーを1時間撹拌した。その後、これをろ過し、冷却(0℃)アセトン(30mL)で洗浄し、真空下、同じろ過漏斗中、室温で乾燥させ、化合物2(26.0g、86%)を得た。表17は、生成物の計測された特性を記載している。
【0191】
【表17】
(実施例13)
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3)
【0192】
【化38】
DCM(520mL)中の化合物2(26.0g、85.4mmol)の溶液に、濃硫酸(7.53g、76.8mmol)を、不活性雰囲気下、10℃で15分間かけて添加した。温度を25℃まで上昇させ、得られた溶液を2時間撹拌した。この時点で、TLCによって証明される通り、出発材料は残っていなかった。反応物を10%NaHCO3水溶液(200mL)の添加によってクエンチした。層を分離し、水層をDCM(2×100mL)で2回抽出した。有機層を合わせ、水(100mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、有機相をNa2SO4(75g)で乾燥させ、ろ過した。ろ液を真空蒸発させ、化合物3(23.0g、94%)をオフホワイトの固体として得た。この生成物をさらに精製することなく次のステップにおいて「そのまま」使用した。表18は、生成物の計測された特性を記載している。
【0193】
【表18】
(実施例14)
3α−ヒドロキシ−5β−アンドロスタ−9(11)−エン−17−オン(4)
【0194】
【化39】
リチウムトリ−tert−ブトキシアルミニウムヒドリドのTHF溶液(1.0M、84.4mL、84.4mmol)を、不活性雰囲気下、THF(230mL)中の化合物3(23.0g、80.4mmol)の冷(−40℃)溶液に添加した。得られた反応混合物を2時間撹拌した。この時点で、TLCによって証明される通り、反応は完了したと決定され、1N HCl(200mL)および酢酸エチル(230mL)の混合物を添加することにより、反応混合物をクエンチした。得られた二相混合物を分離し、水層を酢酸エチル(2×100mL)で2回抽出した。有機相を合わせ、水(150mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、有機相をNa2SO4(75g)で乾燥させ、ろ過した。ろ過を真空蒸発させ、化合物4(23.0g)をオフホワイトの固体として得た。上記粗生成物を精製することなく次のステップにおいて「そのまま」使用した。表19は、生成物の計測された特性を記載している。
【0195】
【表19】
(実施例15)
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6)
【0196】
【化40】
THF中のカリウムtert−ブトキシドの溶液(1M、230mL、231mmol)を、THF(150mL)中のエチルトリフェニルホスホニウムブロミド(88.7g、239mmol)の懸濁液に、25℃で1時間かけて滴下添加した。得られた暗赤色混合物を25℃でさらに1時間撹拌した。THF(230mL)中の化合物4(23.0g、79.7mmol)の溶液を、赤色混合物に25℃でゆっくり添加した。得られた混合物を3〜4時間撹拌し、この時点で、TLCにより完了したと決定された。飽和NH4Cl水溶液(75mL)を添加することにより、反応物をクエンチした。相を分離し、水層をEtOAc(3×150mL)で3回抽出した。有機画分を合わせ、飽和ブライン溶液(100mL)で洗浄し、Na2SO4(75g)で乾燥させ、ろ過した。ろ液を真空濃縮し、粗固体を、酢酸エチル/ヘキサン(1:9)で溶離するカラムクロマトグラフィー[49mm(W)×600mm(L)、60〜120メッシュシリカ、300g]によって精製した。生成物を含有する画分を合わせ、濃縮し、化合物6(19.1g、80.0%)を白色固体として得た。表20は、生成物の計測された特性を記載している。
【0197】
【表20】
(実施例16)
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a)
【0198】
【化41】
化合物6(19.0g、63mmol)をCH2Cl2(380mL)に溶解した。トリエチルアミン(17.6mL、126.6mmol)、DMAP(0.772g、6mmol)および無水酢酸(8.98mL、94mmol)を、窒素雰囲気下、25℃で連続的に添加した。得られた溶液を25℃で2時間撹拌し、この時点で、TLCによって反応は完了したと決定された。氷水(100mL)の添加によって反応物をクエンチし、相を分離した。水層をDCM(3×150mL)で3回抽出した。有機画分を合わせ、飽和ブライン溶液(100mL)で洗浄し、無水Na2SO4(50g)で乾燥させ、ろ過した。ろ液を真空濃縮し、化合物7a(22.0g、95%収率)をオフホワイトの固体として得た。表21は、生成物の計測された特性を記載している。
【0199】
【表21】
(実施例17)
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a)
【0200】
【化42】
エチルアルミニウムジクロリド(104.5mL、192mmol、トルエン中1.8M)を、不活性雰囲気下、0℃でDCM(100mL)中のメチルプロピオレート(13.58mL、153mmol)の溶液に添加した。得られた溶液を15分間撹拌し、その後、化合物7a(22g、64.3mmol)を添加した。0℃でさらに20分間撹拌後、温度を25℃まで上昇させ、そこでさらに18時間維持した。この時点で、TLCによって反応は完了したと決定され、混合物を冷(0℃)水(200mL)中に注いだ。相を分離し、水層をDCM(150mL)で抽出した。有機層を合わせ、水(200mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、これを無水Na2SO4(40g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体をメタノール(280mL)中でスラリー化することによって精製し、化合物8a(17.5g、68%)を白色固体として得た。表22は、生成物の計測された特性を記載している。
【0201】
【表22】
(実施例18)
メチル3α−アセトキシ−5β−コール−9(11)−エン−24−オエート(9a)
【0202】
【化43】
EtOAc(350mL)中の化合物8a(17.5g、41mmol)の溶液にPtO2(4.37g)を添加し、得られたスラリーをParr装置(70psi)内で14〜16時間水素化した。この時点で、TLCによって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物9a(17.0g、96.0%)を白色固体として得た。上記生成物をさらに精製することなく次のステップにおいて使用した。表23は、生成物の計測された特性を記載している。
【0203】
【表23】
(実施例19)
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b)
【0204】
【化44】
エチルアルミニウムジクロリド(14.2mL、25mmol、トルエン中1.8M)を、不活性雰囲気下、0℃でDCM(60mL)中のメチルアクリレート(1.89mL、20mmol)の溶液に添加した。得られた溶液を15分間撹拌し、その後、化合物7a(3g、8.7mmol)を添加した。0℃でさらに20分間撹拌後、温度を25℃まで上昇させ、そこでさらに18時間維持した。この時点で、TLCによって反応は完了したと決定され、その後、混合物を冷(0℃)水(60mL)中に注いだ。相を分離し、水層をDCM(60mL)で抽出した。有機層を合わせ、水(50mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、これを無水Na2SO4(5g)で乾燥させ、ろ過した。ろ液を真空濃縮し、化合物8b(2.6g、70%)を得た。表24は、生成物の計測された特性を記載している。
【0205】
【表24】
(実施例20)
メチル3α−アセトキシ−5β−コール−9(11)−エン−24−オエート(9a)
【0206】
【化45】
EtOAc(60mL)中の化合物8a(3g、7mmol)の溶液に10%Pd/C(300mg、10%wt/wt)を添加し、得られたスラリーをParr装置(70psi)内で14〜16時間水素化した。この時点で、TLC(ヘキサン中10%酢酸エチル)によって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物9a(2.6g、86%)を白色固体として得た。表25は、生成物の計測された特性を記載している。
【0207】
【表25】
(実施例21)
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a)
【0208】
【化46】
CrO3(17.0g、170mmol)を、AcOH(270mL)中の化合物9a(17g、39.5mmol)の溶液に添加した。得られた混合物を50℃で24〜36時間加熱した。TLCによる出発材料の完全消失時に、溶媒を真空蒸発させ、粗材料を酢酸エチル(400mL)および水(200mL)に溶解した。2つの相を分離し、有機層を、水(2×100mL)で2回、その後、飽和ブライン溶液(100mL)で1回洗浄した。有機相を無水Na2SO4(40g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体を、酢酸エチル/ヘキサン(1:5)[10mL画分、3mL/分溶離、TLCによってモニターし、UV光(254nm)ランプによって検出する]で溶離するカラムクロマトグラフィー[49mm(W)×600mm(L)、60〜120メッシュシリカ、120g]によって精製した。生成物含有画分を合わせ、真空濃縮し、化合物10a(8.8g、50%収率)を白色固体として得た。表26は、生成物の計測された特性を記載している。
【0209】
【表26】
(実施例22)
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
【0210】
【化47】
10%Pd/C(900mg)を、EtOAc(150mL)中の化合物10a(2.0g、4.5mmol)の溶液に添加し、得られたスラリーをParr装置(50psi)内、50℃で16時間水素化した。この時点で、TLCによって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物11a(1.6g、80%収率)を白色固体として得た。表27は、生成物の計測された特性を記載している。
【0211】
【表27】
(実施例23)
メチル3α−アセトキシ−12α−ヒドロキシ−5β−コラン−24−オエート(12a)
【0212】
【化48】
リチウムトリ−tert−ブトキシアルミニウムヒドリドのTHF溶液(1M、22.4mL、22.4mmol)を、周囲温度でTHF(25mL)中の化合物11a(2.5g、5.6mmol)の溶液に滴下添加した。さらに4〜5時間撹拌後、TLCによって反応は完了したと決定された。HCl水溶液(1M、10mL)を添加することによって反応物をクエンチし、混合物をEtOAc(30mL)で希釈した。相を分離し、有機相を水(15mL)および飽和ブライン溶液(10mL)で連続的に洗浄した。その後、有機相を無水Na2SO4(3g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体を、EtOAc/ヘキサン(2:8)[5mL画分、p−アニスアルデヒド炭化を用いるTLCによってモニターする]で溶離するカラムクロマトグラフィー[29mm(W)×500mm(L)、60〜120メッシュシリカ、50g]によって精製した。生成物を含有する画分を合わせ、真空濃縮し、化合物12a(2.3g、91%)を白色固体として得た。表28は、生成物の計測された特性を記載している。
【0213】
【表28】
(実施例24)
デオキシコール酸(DCA)
【0214】
【化49】
H2O(2.0mL)中のLiOH(187mg、4.4mmol)の溶液を、THF(8mL)およびMeOH(8mL)中の化合物12a(500mg、1.11mmol)の溶液に添加した。得られた混合物を50℃で3〜4時間撹拌した。TLCによる出発材料の完全消失時に、反応混合物を真空濃縮した。水(10mL)および3N HCl(1mL)の混合物を合わせ、0℃まで冷却し、その後、粗生成物に添加した。0℃で1時間撹拌後、沈殿した固体をろ過し、その後、水(10mL)およびヘキサン(20mL)で洗浄した。室温での真空乾燥により、デオキシコール酸(DCA、400mg、91%収率)を白色固体として得た。表29は、生成物の計測された特性を記載している。
【0215】
【表29】
(実施例25)
初代ヒト脂肪細胞を、出発材料として9−HADを使用して合成された様々な濃度の合成デオキシコール酸ナトリウムまたは以下に記載される通りのSigmaから得られたウシ由来デオキシコール酸ナトリウムとともにインキュベートした。
【0216】
材料
脂肪細胞(Zen−Bioカタログ番号SA−1096)
96ウェルプレート(US Scientificカタログ番号セルスター655180番)
無血清RPMI培地(Mediatechカタログ番号17−105−CV)
デオキシコール酸ナトリウム(DC)(Sigmaカタログ番号D6750)
合成グリコデオキシコール酸ナトリウム(Kythera)
PBS(1×)
MTSアッセイキット(Promegaカタログ番号G3580)
到着した脂肪細胞は分化しており、96ウェルプレート中、1ウェル当たり13,000細胞の密度であった。2枚のプレートを入手し、それぞれ同じ試料で処理した。細胞を、37℃、5%CO2で24時間インキュベートした。20mgを2mL培地(無血清)に溶解して各胆汁酸(合成および非合成DCA)の1%原液を作製した。この1%原液を使用して、希釈により下記の11種の溶液を調製した:0.005%、0.01%、0.015%、0.02%、0.025%、0.03%、0.035%、0.04%、0.05%、0.06%および0.1%、ならびに0%(培地のみ)。
【0217】
細胞を150μLの室温1×PBS(リン酸緩衝生理食塩水)で2回洗浄した。プレートを上下逆にし、液体を容器にデカントすることにより、培地、その後PBSを96ウェルプレート中のウェルから除去した。最後のPBS洗浄後、1ウェル当たり80μLの試料を添加した。各濃度の特定の胆汁酸を8つのウェルに添加し、37℃、5%CO2で1時間インキュベートした。その後、プレートをインキュベーターから取り出し、溶液をデカントした。希釈した(1mLのRPMI中40μL)MTS試薬(3−(4,5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム、内塩)の100μL溶液を、各ウェルに直接添加した。対照(胆汁酸なし)ウェルが橙褐色に変色するまで、プレートを37℃、5%CO2でインキュベートし、その後、96ウェルプレートを分析する分光光度計に充填した。試料は、490nm波長設定で操作した。
【0218】
細胞の生存は、Promega製の比色分析アッセイ(MTS)キットを使用して評価した。結果は、合成NaDCまたはSigma製NaDCのいずれも処理時における、細胞生存の用量依存的な減少を示す(図2を参照)。両分子は、本実験において類似の細胞溶解挙動を実証し、合成NaDCおよびウシ由来のSigma製NaDCが脂肪細胞を死滅させるそれらの能力に関して、機能的に同一であることを示している。
【0219】
上述した実施形態および実施例は、本発明を限定することを意図するものではない。本発明の原理に従って、数々の修正形態および変形形態が可能であることを理解すべきである。
本発明の好ましい実施形態においては、以下が提供される。
(項目1)
デオキシコール酸(DCA)を自然に産生する哺乳動物または微生物生命体から単離されるものではない、DCAである化合物もしくはそのプロドラッグ、または前記化合物もしくは前記プロドラッグの薬学的に許容される塩:
【化50】
。
(項目2)
項目1に記載の化合物および薬学的に許容される賦形剤を含む、組成物。
(項目3)
神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分をさらに含む、項目2に記載の組成物。
(項目4)
前記神経ペプチドY(NPY)受容体アンタゴニストが、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、ジヒドロピリジン系NPY受容体アンタゴニスト、ジヒドロピリジン誘導体NPY受容体アンタゴニスト、二環化合物NPY受容体アンタゴニスト、カルバゾール系NPY受容体アンタゴニストおよび三環化合物NPY受容体アンタゴニストからなる群から選択される、項目3に記載の組成物。
(項目5)
前記脂肪選択的プロアポトーシスペプチドが、白色脂肪血管系にあるCKGGRAKDCペプチドである、項目3に記載の組成物。
(項目6)
哺乳動物において選択した位置から脂肪沈着物を除去する方法であって、それを必要とする前記哺乳動物に、治療有効量のDCAもしくはそのプロドラッグ、または前記DCAもしくは前記プロドラッグの薬学的に許容される塩:
【化51】
を投与するステップを含む方法であり、前記化合物はDCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない方法。
(項目7)
前記哺乳動物に、神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分を投与するステップをさらに含む、項目6に記載の方法。
(項目8)
前記神経ペプチドY(NPY)受容体アンタゴニストが、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、ジヒドロピリジン系NPY受容体アンタゴニスト、ジヒドロピリジン誘導体NPY受容体アンタゴニスト、二環化合物NPY受容体アンタゴニスト、カルバゾール系NPY受容体アンタゴニストおよび三環化合物NPY受容体アンタゴニストからなる群から選択される、項目7に記載の方法。
(項目9)
脂肪選択的プロアポトーシスペプチドが、白色脂肪血管系にあるCKGGRAKDCペプチドである、項目7に記載の方法。
(項目10)
デオキシコール酸(DCA)もしくはそのエステル、または薬学的に許容されるその塩:
【化52】
を調製するための方法であって、
(a)9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1を、水素化条件下、H2と反応させて、化合物2
【化53】
を形成するステップと、
(b)化合物2を酸と反応させて、化合物3
【化54】
を形成するステップと、
(c)化合物3を還元剤と反応させて、化合物4を4および5
【化55】
の混合物として形成するステップと、
(d)化合物4をオレフィン形成条件下で二炭素オレフィン化試薬と反応させて、化合物6
【化56】
を形成するステップと、
(e)化合物6を、式7の化合物[式中、Pは保護基である]
【化57】
に変換するステップと、
(f)式7の化合物をルイス酸の存在下でアルキルプロピオレートCH2CH2C(O)ORまたはアルキルアクリレートCH2=CHC(O)OR[式中、Rはアルキルである]と反応させて、式8の化合物[式中、Pは保護基であり、Rはアルキルであり、破線
【化58−1】
は単結合または二重結合である]
【化58−2】
を形成するステップと、
(g)式8の化合物を水素化条件下でH2と反応させて、式9の化合物[式中、Pは保護基であり、Rはアルキルである]
【化59】
を形成するステップと、
(h)式9の化合物を酸化剤と反応させて、式10の化合物[式中、Pは保護基であり、Rはアルキルである]
【化60】
を形成するステップと、
(i)式10の化合物を水素化条件下でH2と反応させて、式11の化合物[式中、Pは保護基であり、Rはアルキルである]
【化61】
を形成するステップと、
(j)式11の化合物を還元剤と反応させて、式12の化合物[式中、Pは保護基であり、Rはアルキルである]
【化62】
を形成するステップと、
(k)式12の化合物を脱保護条件に曝露してそのエステルを形成し、場合によって、適切な加水分解条件に曝露して、デオキシコール酸または薬学的に許容されるその塩を形成するステップと
を含む方法。
(項目11)
パート(a)の前記水素化条件がPd/C触媒を含む、項目10に記載の方法。
(項目12)
パート(b)の前記酸が鉱酸である、項目10に記載の方法。
(項目13)
前記鉱酸がH2SO4である、項目12に記載の方法。
(項目14)
パート(c)の前記還元剤がLiAl(OtBu)3Hである、項目10に記載の方法。
(項目15)
パート(d)の前記二炭素オレフィン化試薬がPh3PCH2CH3+Br−である、項目10に記載の方法。
(項目16)
化合物7〜12の前記保護基Pが−C(O)CH3である、項目10に記載の方法。
(項目17)
パート(f)の前記ルイス酸がEtAlCl2である、項目10に記載の方法。
(項目18)
前記アルキルプロピオレートまたはアルキルアクリレートがメチルプロピオレートまたはメチルアクリレートである、項目10に記載の方法。
(項目19)
パート(g)の前記水素化条件がPtO2触媒を含む、項目10に記載の方法。
(項目20)
パート(h)の前記酸化剤がCrO3である、項目10に記載の方法。
(項目21)
パート(i)の前記水素化条件がPd/C触媒を含む、項目10に記載の方法。
(項目22)
パート(j)の前記還元剤がLiAl(OtBu)3Hである、項目10に記載の方法。
(項目23)
Pが−C(O)CH3である場合、パート(k)の前記脱保護および加水分解条件が、化合物12を、アルカリ土類水酸化物、アルカリ土類金属アルコキシドまたは両方の混合物と反応させることを含む、項目10に記載の方法。
(項目24)
前記アルカリ土類金属アルコキシドがLiOHである、項目23に記載の方法。
(項目25)
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2);
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3);
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6);
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a);
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a);
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b);
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a);および
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
からなる群から選択される、中間化合物。
【技術分野】
【0001】
(関連出願への相互参照)
本出願は、2007年6月19日に出願された米国仮特許出願第60/945,035号および2007年8月20日に出願された同第60/956,875号;2008年2月21日に出願された米国通常特許出願第12/035,339号および2008年5月16日に出願された同第12/153,446号;ならびに2008年4月25日に出願された英国特許出願第0807615.0号に対する優先権を主張し、これらの各々は、その全体を本明細書において参考として援用される。
【0002】
(発明の分野)
本発明は、広く胆汁酸ならびに関連組成物および方法に関する。一態様において、本発明は、デオキシコール酸および関連組成物、有用な中間体ならびにその合成方法に関する。別の態様において、本発明は、本組成物および方法の、医薬組成物およびその製造方法としての使用に関する。重要なことに、本発明の胆汁酸は、これらの酸を自然に産生する哺乳動物および微生物生命体から単離されるものではなく、故に、そのような生命体に関連するいかなる毒素および汚染物質もない。別の態様において、本発明は、デオキシコール酸ならびに薬学的に許容されるその塩および中間体の合成に関する。
【背景技術】
【0003】
(発明の背景)
コラノロジー(Cholanology)という胆汁酸の研究、特に胆汁酸化学は、世紀の大半にわたって関心を集めてきた。多くは既知であるが、胆汁酸化学には、多くが驚くべき特性を有する多種多様な化学的実体が関与する。総説は、例えば、参照により本明細書に組み込まれる、Mukhopadhyay, S.およびU. Maitra.、Current Science、87巻:1666〜1683頁(2004年)(「Chemistry and biology of bile acids」)を参照されたい。
【0004】
胆汁酸は、2つの連結ユニット、剛性のステロイド核および短い脂肪族側鎖を特徴とする(本出願の図1を参照)。Hofmann, A. F.らを参照。提案された胆汁酸の命名法については、J. Lipid Res.、33巻:599〜604頁(1992年)を参照されたい。核および側鎖はいずれも、多数の可能な立体配置を有する。核は、個々の環の拡張または収縮によって変化させることができ、側鎖は、短縮または伸長できる。加えて、胆汁酸分子の両部分は、多数の可能な極性置換基を有する。イオン化基は、核または側鎖上に存在し得る。最後に、共役基は、核上(例えば、サルフェート、グルクロネート、ホスフェート)または側鎖上(グリシンもしくはタウリンまたは他のアミノ酸、さらには糖類)に存在し得る。側鎖構造は、化合物(胆汁酸または胆汁塩)のクラスを決定する。
【0005】
胆汁酸は、両親媒性(amphiphilic)および両親媒性(amphipathic)の「面」の両方を有する両親媒性物質:
【0006】
【化1】
である。Hofman, A.F.、News Physiol. Sci.、14巻:24〜29頁(1999年)(「Bile Acids: The good, the
Bad, and the Ugly」、25頁、図1)。
【0007】
慣例により、疎水性表面は「β面」と呼ばれ、親水性表面は「α面」と呼ばれる。概して、β面は脂溶性であり、α面は比較的極性である。疎水面上および親水面上に極性基(自然発生的な胆汁酸中のヒドロキシル基)を有するもの、例えばウルソデオキシコール酸等の胆汁酸がある。分子の両親媒性の性質は、ホスファチジルコリン等、両親媒性であるが不水溶性の脂質との混合ミセルの形成を司る。胆汁酸は、胆汁酸が、臨界ミセル化濃度と称される臨界濃度を超えない限り、混合ミセルの形態の食物中の脂質を可溶化しない。
【0008】
ヒトにおいて最大の割合で見られる胆汁酸は、ケノデオキシコール酸およびデオキシコール酸である。デオキシコール酸は、デオキシコレート、コラン酸および3α,12α−ジヒドロキシ−5β−コラネート(cholanate)としても知られている。ヒト体内において、デオキシコール酸は、腸管において吸収のための脂肪の乳化に使用される。研究において、デオキシコール酸は、膜結合タンパク質単離のための低刺激洗浄剤として使用される。実質的に純粋である場合、デオキシコール酸は白色〜オフホワイトの結晶性粉末形態である。デオキシコール酸は、肝臓によって産生される4つの主な酸の1つである。これは、アルコールおよび酢酸に可溶である。デオキシコール酸のCAS番号は[83−44−3]である。
【0009】
体脂肪の迅速な除去は長年の理想であり、多くの物質がそのような結果を達成すると主張されてきたが、結果を示したものはわずかである。「メソセラピー」、つまり脂肪の除去のための注射剤の使用は、1950年代からホメオパシーおよび美容学的主張が為されているが、安全性および有効性の懸念により、医療従事者の間で広く受け入れられていない。メソセラピーは、元来、局所的な医学的および美容学的状態の治療のために化合物の混合物を含有する皮膚注射を利用する方法として欧州で想到された。メソセラピーは、疼痛緩和のために伝統的に用いられていたが、最近ではその美容学的用途、特に脂肪およびセルライトの除去が米国で注目を集めている。局所的な脂肪減少のための1つのそのような報告されている治療は、ブラジルにおいて普及し、ホスファチジルコリンの注射を使用するものであり、メソセラピーと同義であると誤ってみなされてきた。「脂肪を溶解する」とされている注射としてのその魅力にもかかわらず、これらの美容学的治療の安全性および有効性は、ほとんどの患者および医師にとって曖昧なままである。Rotunda,
A.M.およびM. Kolodney、Dermatologic Surgery、32巻:465〜480頁(2006年)(「Mesotherapy and Phosphatidylcholine Injections: Historical
Clarification and Review」)を参照されたい。
【0010】
国際公開第2006/133160号(参照により図面を含むその全体が本明細書に組み込まれる)は、リポモデリング(lipomodeling)のための方法、例えば、神経ペプチドY受容体アンタゴニストを脂肪貯蔵部位に投与することによる脂肪貯蔵物の減少を記載している。Kolonin M. G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)は、強力な脂肪細胞死滅効果を有する脂肪選択的プロアポトーシスペプチドを記載している。記載されているプロアポトーシスペプチドは、死滅させるためには血管系にアクセスする必要がある。
【0011】
最近刊行された文献は、デオキシコール酸がインビボで脂肪性沈着物に注射される場合、脂肪除去特性を有することを報告している。すべて参照により図面を含むその全体が本明細書に組み込まれる、特許文献1および特許文献2、ならびに特許文献3、特許文献4、特許文献5および特許文献6を参照されたい。脂肪組織中に注射されたデオキシコレートは、1)細胞傷害機序によって脂肪細胞を死滅させる、2)皮膚引き締めを引き起こすという2つの効果を有する。これらの効果はいずれも、所望の審美的修正(すなわち、体形矯正)を媒介するために必要である。脂肪中に注射されたデオキシコレートは、タンパク質への曝露によって迅速に不活性化され、その後、迅速に腸の内容物中に戻るため、その効果は空間的に封じ込められる。臨床的安全性を与えるこの減弱作用の結果として、脂肪除去療法は、典型的に4〜6セッションを必要とする。外科手術を必要としないこの局所的な脂肪除去は、病理学的な局所的脂肪沈着物に関する治療的処置(例えば、HIVの治療における医学的介入に伴う脂質異常症)だけでなく、外科手術に固有の付帯リスクがない美容学的脂肪除去(例えば、脂肪吸引術)にも有益である。いずれも参照により本明細書に組み込まれる、Rotundaら、Dermatol. Surgery、30巻:1001〜1008頁(2004年)(「Detergent effects of
sodium deoxycholate are a major feature
of an injectable phosphatidylcholine formulation used for localized fat dissolution」)およびRotundaら、J. Am. Acad. Dermatol.、2005年:973〜978頁(「Lipomas treated with subcutaneous deoxycholate injections」)を参照されたい。
【0012】
医薬品等級の胆汁酸製剤は、比較的低価格で市販されている。この低価格は、胆汁酸が、動物の死体、特に雌ウシおよびヒツジ等の大型動物から得られることによるものである。重要なことに、動物源由来のすべての薬剤と同様に、動物由来胆汁酸製品は、動物病原体、ならびにパイロジェン等の細菌毒素を含む動物または微生物代謝産物および毒素等、他の有害作用物質を含有し得るという懸念がある。
【0013】
そのような動物病原体は、プリオン病を引き起こし得る感染性病原タンパク質の一種であると考えられているプリオンを含み得る。プリオン病は、神経系の変性障害である。1つのそのような疾患である「狂牛」病(クロイツフェルト・ヤコブ病(CJD)の変異型であると考えられている)は、罹患した雌ウシ由来の食用牛肉中に存在するプリオンによって引き起こされると考えられている。ほとんどの場合は未知の伝染様式による散発性なものであり、いくつかの場合は遺伝的なものであり、少数が医療処置によって伝染したものである。感染材料の消費によるヒトプリオン病の蔓延は、歴史的にはクールーに、また最近では変異型CJDに関与するとされている。他の動物プリオン病(ヒツジのスクレピー、伝染性ミンク脳症、シカの慢性消耗病および牛海綿状脳症)はすべて、感染動物との接触によってまたは感染飼料の消費によって横方向に伝染するようである。プリオン病に関するリスク評価および未来事象の予測は、異なる伝染様式、予測不可能な種の障壁、組織中における感染力の可変分布およびいくつかの疾患において見られる系統変動により、解明することが困難である。
【0014】
概して、動物性製品は、パイロジェン(発熱物質)を産生する微生物生命体に曝露され得る。食物および/または医薬品の細菌汚染物質も、腸管出血性E.coliによる食料品の汚染によって証明されている通り、重要課題である。雌ウシに由来する食肉等の製品およびリンゴ、ホウレンソウ等の農産物は、そのような汚染に関与するとされている。そのような場合、ヒトにおいて有害作用を発生させるのは、(細菌自体よりもむしろ)細菌によって産生される毒素である。そのような有害作用は、重度の下痢、腎不全および極限状況では死を含む。パイロジェンの一種である細菌内毒素は、すべての医薬組成物から実質的に排除されなくてはならない。
【0015】
動物性製品は、一般に排除過程によって精製される、すなわち、混合から最終産物を選択するのではなく、最終産物は不純物の排除後に残っている材料である。また、病原体等の潜在的な動物部分に加えて、動物源からの精製の別のアーチファクトは、最終産物が1つまたは複数の胆汁酸の混合物であることである。例えば、デオキシコール酸の市販製剤は、若干のケノデオキシコール酸、ならびに哺乳動物の胆汁酸合成においてデオキシコール酸およびケノデオキシコール酸の両方の前駆体であるコール酸を含有する。デオキシ/ケノ/コールの正確な割合は事前選択されないため、これは、多量の胆汁酸の製造を企図する場合にロット間変動をもたらし得る。そのようなロット間変動は、問題のあるものとなり得、規制当局の承認または品質管理を獲得する際、特に医薬組成物を生成するための取り組みにおいて、追加ステップを生じさせる場合がある。生産者は明らかに、胆汁酸医薬組成物の製造においてロット間予測可能性を望むであろう。
【0016】
現在、動物病原体および他の有害作用物質を含有する動物由来製品に関する懸念は、隔離および検査された動物から調達することによって対処されている。例えば、ニュージーランドの動物に由来するデオキシコール酸は、動物が隔離される等で観察可能な病原体がないままであり続ける限り、米国の規制制度下でヒト使用のための胆汁酸の源である。
【0017】
暗黙的に、そのような政府により管理される規制制度のために必要なのは、動物由来の薬剤が注射される場合における動物病原体の伝染の内在的リスクの認識である。非動物性薬剤代替物が利用可能になれば、政府による規制制度はもはや必要ない。そのような代替物(動物由来の薬剤に置き換わる非動物性薬剤)および関連する利点の例は、ヒト使用のためのインスリンである。米国でのウシインスリンの製造は1998年に中止され、ヒト使用のためのブタインスリンは2006年1月に中止された。動物インスリンはBSEを引き起こす因子または他の病原因子に曝露されていないと分かっている群から得ることができるが、製造施設または過程は、動物性原料を、病原体に曝露された動物に曝露し得る。病原因子のヒトへの伝染のリスクは、組換え的にまたは合成的に製造されるインスリンの使用によって解消することができる。消費者にとって、インスリンの状況はためになるものであり、合成材料が自由に利用可能であれば、動物病原体の伝染のリスクは理論上は解消される。生産者にとって、動物病原体の材料が実質的にない純粋な化学的実体を生成するための能力は、安全性、品質および規制の目的に有利である。さらに、合成過程は、典型的に、生物源に由来するものよりも再現性の高い製品を提供する。
【0018】
現在、動物の死体由来の胆汁酸が比較的豊富であることにより、業界は、胆汁酸を完全に化学的に合成すること、または植物ステロールもしくは微生物出発材料を使用して胆汁酸を調製することのいずれにも踏み出していない。また、胆汁酸誘導体は合成されたが、動物性材料の低価格および入手しやすさにより、この作業には当初、ステロイド化学のための出発材料として、やはり動物由来胆汁酸が関与していた。植物ステロール研究における歴史的に活発な取り組みにもかかわらず、容易に商業的に入手可能な植物ステロール由来の胆汁酸医薬品グレード組成物はない。例えば、非特許文献1を参照されたい(1981年の参考文献、非特許文献2(「First Total Synthesis of (+)−Chenodeoxycholic Acid」)を前提として、いかなる胆汁酸の全合成も実施されなかったということに注目されたい)。細菌等の微生物によって産生された胆汁酸は、細菌産物として、例えば海洋での油流出の浄化のためにインシチュで使用されてきた。非特許文献3(「Bile acids are new products of a mariene bacterium, Myroides sp. Strain SM1」)を参照されたい。
【先行技術文献】
【特許文献】
【0019】
【特許文献1】国際公開第2005/117900号パンフレット
【特許文献2】国際公開第2005/112942号パンフレット
【特許文献3】米国特許第2005/0261258号明細書
【特許文献4】米国特許第2005/0267080号明細書
【特許文献5】米国特許第2006/127468号明細書
【特許文献6】米国特許第2006/0154906号明細書
【非特許文献】
【0020】
【非特許文献1】Mukhopadhyay, S.およびU. Maitra.、Current Science、87巻:1666〜1683、1670頁(2004年)
【非特許文献2】Kametaniら、J. Am. Chem. Soc.、103巻:2890頁(1981年)
【非特許文献3】Maneeratら、Appl. Microbiol. Biotechnol.、76巻:679〜683頁(2004年)
【発明の概要】
【発明が解決しようとする課題】
【0021】
脂肪除去のためのデオキシコール酸の全潜在能力を現実化するために、動物由来製品の使用に対する懸念にさらに対処することが必須である。明らかに、動物起源の部分(または、動物、特に哺乳動物において作用し得、ヒト使用の場合にはヒトに悪影響を及ぼし得る病原部分)および動物または微生物代謝産物、パイロジェン等の細菌毒素を含む毒素等の他の有害作用物質がないことが冒頭から分かる、ヒトにおいて薬剤として使用するための、適量の有効な胆汁酸およびデオキシコール酸等の関連組成物が必要である。本発明は、動物病原体および他の有害作用物質の潜在的リスクがない合成的に調製された胆汁酸組成物を提供することにより、この懸念に対処するものである。開示されている胆汁酸組成物は、脂肪分解(adipolytic)療法において使用でき、局所的な脂肪除去の領域における研究および開発努力をさらに進める役割を果たす。
【課題を解決するための手段】
【0022】
(発明の要旨)
本明細書では、定義された医薬組成物として十分な量の適切な胆汁酸およびその合成方法が提供される。そのように提供される胆汁酸組成物および方法は、胆汁酸を自然に産生する哺乳動物または微生物生命体から単離されるものではない。一態様において、動物起源のおよび哺乳動物および/または細菌パイロジェンのすべての部分がない特定のデオキシコール酸医薬組成物ならびに生成および使用のための関連方法が提供される。別の態様において、定義された医薬組成物として十分な量の適切なデオキシコール酸が提供され、これは、関連組成物、製造方法および使用方法とともに、局所的な脂肪除去のための注射用医薬組成物として使用できる。本発明の定義されたデオキシコレート注射液を、直交機序(orthogonal mechanism)によって脂肪を消滅させる分子、例えばNPYアンタゴニストおよび/または脂肪選択的プロアポトーシスペプチドと組み合わせ、より少ない治療セッションでの体形矯正を媒介するためのより強力な手段を作成するために使用される作用物質を提供することができる。別の態様において、本発明は、デオキシコール酸および薬学的に許容されるその塩の合成に関する方法および中間体を提供する。合成的に調製されたデオキシコール酸は、脂肪除去のための脂肪分解療法において使用できる。
【図面の簡単な説明】
【0023】
【図1】胆汁酸骨格の炭素のナンバリングシステムを含む、胆汁酸の構造を表す図である。
【図2】ウシ由来デオキシコール酸ナトリウム(Sigma)と比較した、本発明の合成デオキシコール酸ナトリウムによる処理時の初代ヒト脂肪細胞の細胞生存の用量依存的な減少における類似性を示す図である。
【発明を実施するための形態】
【0024】
(参照による組み込み)
本明細書中で言及されるすべての刊行物および特許出願は、各個々の刊行物または特許出願が参照により組み込まれることを具体的かつ個別に指示されている場合と同程度まで、参照により本明細書に組み込まれる。
【0025】
(発明の詳細な説明)
定義
本開示全体を通して、種々の刊行物、特許および公開された特許明細書は、識別引用によって参照される。これらの刊行物、特許および公開された特許明細書の開示は、参照により本開示に組み込まれ、本発明が属する分野の状況をより詳細に記載する。
【0026】
本明細書において使用される場合、いくつかの用語は、下記に定義される意味を有する。本明細書および請求項において使用される場合、単数形「a」、「an」および「the」は、文脈上明らかに別段の定めをした場合を除き、単数および複数の参照物を含む。
【0027】
別段の指示がない限り、本明細書および請求項において使用される原料、反応条件等の数量を表現するすべての数は、すべての場合において用語「約」により修飾されるものとして理解すべきである。したがって、反対の指示がない限り、下記の明細書および添付の請求項において説明される数値パラメーターは近似値である。各数値パラメーターは、少なくとも、報告された有効桁の数を考慮し、通常の丸め技術を適用することによって解釈されるべきである。
【0028】
用語「アセチル化試薬」は、アセチル基CH3C(O)−を分子に添加することができる試薬を指す。
【0029】
用語「酸」は、プロトン供与体を指し、有機および無機酸の両方を含む。
【0030】
用語「アルキル」は、1〜10個の炭素原子、好ましくは1〜6個の炭素原子を有する一価の飽和脂肪族ヒドロカルビル基を指す。この用語は、例として、メチル(CH3−)、エチル(CH3CH2−)、n−プロピル(CH3CH2CH2−)、イソプロピル((CH3)2CH−)、n−ブチル(CH3CH2CH2CH2−)、イソブチル((CH3)2CHCH2−)、sec−ブチル((CH3)(CH3CH2)CH−)、t−ブチル((CH3)3C−)、n−ペンチル(CH3CH2CH2CH2CH2−)およびネオペンチル((CH3)3CCH2−)等の直鎖および分岐鎖のヒドロカルビル基を含む。
【0031】
用語「アリール」は、単環(例えば、フェニル)または多重縮合環(例えば、ナフチル)を有する6〜12個の炭素原子の一価の芳香族炭素環基を指す。
【0032】
用語「動物起源」は、多細胞生命体および単細胞生命体を含む生物の界(動物界)のいずれかに起因していることを指す。
【0033】
用語「脱水試薬」は、水と反応することができる試薬を指す。一態様において、脱水試薬は、分子から除去される水と反応することができる。
【0034】
用語「脱硫試薬」は、硫化物と反応することができる試薬を指す。一態様において、脱硫試薬は、硫化物含有分子と反応して、分子からスルフィド基を除去することができる。
【0035】
用語「エタンジチオールまたはジチアン前駆体」は、カルボニル基との反応によって、エタンジチオールまたはジチアン基を形成する試薬を指す。
【0036】
用語「求電子性アセチル基」は、求電子としてのアセチル基であって、電子に引き付けられ、電子を受け取る傾向がある基を指す。
【0037】
用語「水素化試薬」は、水素を分子に付与することができる試薬を指す。
【0038】
用語「ルイス酸」は、電子対受容体を指す。ルイス酸は、アルキルアルミニウムハライド(例えば、Et2AlClおよびMeAlCl2)等の有機金属試薬を含む。
【0039】
用語「哺乳動物起源」は、任意の哺乳類の生命体に起因していることを指す。用語「哺乳類の生命体」は、乳腺によって分泌される乳でその幼体に栄養を与え、通常は程度の差はあるが体毛で覆われた皮膚を有し、ヒトを含む、温血高等脊椎動物の綱(哺乳綱)(有胎盤類、有袋類または単孔類等)を指す。
【0040】
用語「微生物起源」は、任意の微生物生命体に起因していることを指す。用語「微生物生命体」は、細胞壁を欠いてよいかまたは細胞壁を有する場合にはグラム陽性もしくはグラム陰性であり、多くの場合コロニーに凝集されるかまたは鞭毛を用いて運動性となり、典型的には、土壌、水、有機物中または植物および動物の体内に生息し、通常は、栄養上、独立栄養性、腐生性または寄生性であり、その生化学的作用および病原性で有名な、原核生物の円状、らせん状または棒状の単細胞微生物のドメイン(細菌)を指す。
【0041】
用語「オレフィン化試薬」は、ケトンと反応して、対応するオレフィンを形成する試薬を指す。用語「オレフィン形成条件」は、そのような転換を行うのに適した条件を指す。そのような試薬の例は、ウィッティヒ試薬およびウィッティヒオレフィン化条件を含む。
【0042】
用語「酸化剤」は、酸化還元反応において電子を受け取ることができる試薬を指す。このようにして、ハロゲンまたは酸素を分子に添加することができ、または水素を分子から除去することができる。
【0043】
用語「病原体」は、疾患の特異的な原因因子を指す。
【0044】
「薬学的に許容される塩」は、当該技術分野において既知である多様な有機および無機対イオンに由来する薬学的に許容される塩を指し、ほんの一例として、ナトリウム、カリウム、リチウム、カルシウム、マグネシウム、アンモニウムおよびテトラアルキルアンモニウムを含む。適切な塩は、P. Heinrich Stahl、Camille G. Wermuth(編)、Handbook of Pharmaceutical Salts Properties, Selection, and Use、2002年に記載されているものを含む。これらの薬学的に許容される塩は、DCAを適切な塩基と反応させることによって調製できる。例示を目的として、そのような塩基の例は、水酸化ナトリウム、水酸化カリウムまたは水酸化リチウムを含む。代替として、塩は、塩基によるDCAのエステルの加水分解によって、DCAを導き得る任意の酸性ワークアップを省略して調製できる。
【0045】
用語「還元剤」は、酸化還元反応において電子を付与することができる試薬を指す。このようにして、ハロゲンまたは酸素を分子から除去することができ、または水素を分子に添加することができる。
【0046】
組成物および使用方法
本明細書に記載されている種々の態様において、本発明は、薬学的使用のための組成物(および有用な中間体)、その合成方法および本医薬組成物の使用方法を提供する。
【0047】
重要なことに、本胆汁酸組成物は、動物性出発材料から得られた材料に固有のリスクがなく、したがって、動物由来材料の詳細な検査および規定を必要としない。故に、一態様において、本発明は、哺乳動物病原体等の動物起源の材料を含まず、パイロジェン等の細菌起源の毒素を実質的に含まない、胆汁酸医薬組成物を目的としている。
【0048】
デオキシコール酸ナトリウムは、消化管において食物中の脂質を可溶化する、自然に産生される胆汁塩である。これは、コレステロールを出発材料として利用し、ヒトおよび細菌酵素の両方が関与する複合生合成経路を介して、インビボで産生される。デオキシコレートの主要機能は、吸収を容易にするために食物中の脂質を可溶化することによって、消化過程を支援することである。体内において、デオキシコレート生合成は、肝臓内のコレステロールの酵素的酸化、異性化および還元から始まり、そのコレステロール親と構造的に類似した胆汁酸であるコール酸を形成する(Stryer L、27章:Biosynthesis of Membrane Lipids and Steroids、Biochemistry、1995年、W. H. Freeman and Company: New York、691〜707頁)。その後、肝臓内において、コール酸は2つのアミノ酸(タウリンまたはグリシン)の1つと化学的に結合し、「共役」コール酸(すなわち、L−グリココレートおよびタウロコレート)を形成する。その後、これらの共役コール酸は、食物消費まで胆嚢中に保存される。食物消費後、胆汁液は、胆嚢から腸管内に放出され、ここで共役コール酸分子は、腸管微生物叢によって産生された酵素によって媒介される2つの追加の化学修飾に付される(Ridlon J.M.、Kang
D.J.およびHylemon P.B.、Bile salt biotransformations by human intestinal bacteria、J. Lipid Res.、47巻(2号):241〜59頁(2006年))。最初に、共役コール酸は脱ヒドロキシル化されて共役デオキシコレートを形成する。その後、共役デオキシコレートは脱共役されて遊離デオキシコレートを形成し、これが、他の胆汁酸とともに食物中の脂質の可溶化に関与する。デオキシコレートはコール酸合成より下流にあるため、コール酸はデオキシコレートの自然源中に存在する不純物であり得る。
【0049】
デオキシコレートは、333mg/mLまで水に可溶であり、アルコールに難溶であり、アセトンおよび氷酢酸にさらに難溶である。ミセルの可逆的形成は、約2.4mg/mLの臨界ミセル濃度を超過するデオキシコール酸ナトリウム濃度および中性pHで発生し得る(Matsuoka K, M.Y.、Micelle formation of
sodium deoxycholate and sodium ursodeoxycholate (1部)、Biochim. Biophys. Acta.、1580巻(2〜3号):189〜99頁(2002年))。2.4mg/mLの臨界ミセル濃度を超過する濃度において、デオキシコレートはミセルを形成し、細胞、脂質およびタンパク質を可溶化する能力を有する。0.4mg/mL(絶食状態に相当する)等のより低濃度において、デオキシコレートは、26mg/mLのアルブミン(35〜50mg/mLの血清生理学的濃度に近い)の存在下、アルブミンに98%結合する(Roda A.ら、Quantitative aspects of the interaction of bile acids with human serum albumin、J. Lipid Res.、23巻(3号):490〜5頁(1982年))。
【0050】
好ましい実施形態は、デオキシコール酸(DCA)もしくはそのプロドラッグ、または前記化合物または前記プロドラッグの薬学的に許容される塩、ならびに関連組成物および方法を目的としており、ここで、デオキシコール酸(DCA)は、
【0051】
【化2】
であり、ここで、前記化合物は、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない。
【0052】
他の好ましい実施形態は、DCAおよび薬学的に許容されるその塩の立体異性体、DCAならびにその立体異性体および塩の合成における中間体、ならびに関連組成物および方法も目的としている。
【0053】
本胆汁酸医薬組成物は、場合によって塩形態であり、さらに場合によって、薬学的に許容される希釈剤、賦形剤または担体を含有する。一態様において、本発明は、デオキシコール酸(DCA)を自然に産生する哺乳動物または微生物生命体から単離されるものではない、DCAである化合物もしくはそのプロドラッグ、または前記化合物もしくは前記プロドラッグの薬学的に許容される塩:
【0054】
【化3】
および薬学的に許容される賦形剤を目的としている。
【0055】
塩調製のための好ましいカチオンは、ナトリウム(Na+)、カリウム(K+)、リチウム(Li+)、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)、ストロンチウム(Sr2+)およびアンモニウム(NH4+)からなる群から選択できる。塩は、アルカリ金属またはアルカリ土類金属から調製することもできる。アルカリ金属は、ナトリウム(Na+)、カリウム(K+)およびリチウム(Li+)の中から選択できる。アルカリ土類金属は、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)およびストロンチウム(Sr2+)からなる群から選択できる。脂肪の局所的除去のための医薬組成物としての使用には、胆汁塩がデオキシコール酸ナトリウムであることが好ましい。
【0056】
本実施形態の化合物のプロドラッグも企図されている。プロドラッグは、患者へのプロドラッグの投与後、加水分解、代謝等のインビボ生理的作用によって本実施形態の化合物に化学的に修飾される活性または不活性化合物である。例えば、本デオキシコール酸またはその誘導体のC1〜C10エステルまたはアミドを調製することができ、その結果、細胞膜の破壊およびエステラーゼの放出により、デオキシコール酸またはその誘導体の放出が誘発される。エステラーゼの放出によってエステル保護基が開裂されるため、デオキシコール酸活性型またはその誘導体がインシチュの所望の位置に存在する。そのようなC1〜C10エステルは、酸素、硫黄または窒素から選択される1〜4個のヘテロ原子;酸素、硫黄または窒素から選択される1〜4個のヘテロ原子を場合によって有するメチル、エチル、イソプロピル、ブチル、ヘキシル等のアルキル基;任意の許容される置換点に1〜4個のヘテロ原子を場合によって有するベンジルまたはエチルフェニル基等、合計最大10個の炭素原子を有するアルキルフェニル基;ならびにフェニル基等のアリール基を場合によって含み得る。アミドの例は、ヒドロキサメートを含むがこれに限定されない。エステルを含むプロドラッグの一般的議論については、参照により全体が本明細書に組み込まれる、SvenssonおよびTunek、Drug Metabolism Reviews、165巻(1988年)ならびにBundgaard Design of Prodrugs、Elsevier(1985年)を参照されたい。デオキシコール酸のC1〜C10エステルまたはアミドの合成は、当該技術分野において既知である。例えば、エステルは、エステル化反応において、鉱酸の存在下でデオキシコール酸のアルコールとの反応によって合成できる。
【0057】
デオキシコール酸およびケノデオキシコール酸の自然構造は、以下のように示される。4つの環(A、B、C、D)およびD環から伸展するカルボン酸側鎖が示されている。
【0058】
【化4】
【0059】
【化5】
限定されないが、デオキシコール酸およびケノデオキシコール酸(CDCA)またはその誘導体のエステル、ヒドロキサメートおよびヒドロキシアミドの例のいくつかは、以下のように記載される。D環側鎖上のカルボキシル基のエステル化により、異なる官能基がデオキシコール酸またはケノデオキシコール酸に結合してプロドラッグを生成し得る。限定されないが、これらのD環側鎖エステルのいくつかの例が表1に示されている。
【0060】
【表1】
デオキシコール酸またはケノデオキシコール酸およびその誘導体の放出は、細胞膜の破壊およびエステラーゼの放出によって誘発され得る。エステラーゼの放出によってエステル保護基が開裂され得るため、デオキシコール酸のもしくはケノデオキシコール酸の活性型またはその誘導体がインシチュの所望の位置に存在する。
【0061】
デオキシコール酸、ケノデオキシコール酸およびそれらの誘導体のプロドラッグは、自然分子と反対の立体化学を有し得るエピマーも含む。これらのエピマー分子の例は、表2に示されている。
【0062】
【表2】
本実施形態の胆汁酸組成物の注射時に局所刺激がある場合があり、故に、局所麻酔薬を同時または順次に投与することが望ましい場合がある。例えば、リドカインは、ヒトにおいて頻繁に使用され、同時製剤(同じ容器中で同時に注射される)または同時注射(異なる容器から注射される)のいずれとして投与してもよい。リドカイン等の麻酔薬は、パッチまたは軟膏等の局所製剤によって投与してよい。
【0063】
深部組織の場合、麻酔薬を対象組織中により深く注射するか、または全身的に投与してよい(例えば、全身麻酔、硬膜外または他の既知の方法)。
【0064】
本発明の溶液中の胆汁酸(複数可)または胆汁塩(複数可)は、約0.001〜10、0.01〜5または0.1〜2%w/w、w/vまたはv/vの濃度であってよい。好ましくは、上記溶液中の胆汁酸(複数可)または胆汁塩(複数可)は、約0.1〜5%w/wまたはより好ましくは約1%w/wの濃度であってよい。いくつかの実施形態において、脂肪溶解液は、最大100、50、20、10、5、2、1、0.5、0.2、0.05、0.02または0.01グラムの1つまたは複数の洗浄剤、胆汁酸および/または胆汁塩、例えば、デオキシコール酸もしくはその塩またはデオキシコール酸ナトリウムを含む。
【0065】
好ましい実施形態において、本明細書における溶液は、脂質、リン脂質またはホスファチジルコリンを含まない。いくつかの実施形態において、本明細書における溶液は、最大5%w/w、w/vまたはv/vの脂質、リン脂質またはホスファチジルコリンを含む。
【0066】
いくつかの実施形態において、上記溶液は、抗菌剤、血管収縮薬、抗血栓剤、抗凝固剤、発泡抑制剤(suds−depressant)、抗炎症剤、鎮痛剤、分散剤、抗分散剤、浸透促進剤、ステロイド、精神安定薬、筋弛緩薬および下痢止め剤からなる群から選択される第2の治療剤をさらに含み得る。いくつかの実施形態において、溶液は、最大500mLの溶液を収納する容器内にあってよい。そのような容器は、シリンジまたはシリンジ充填可能な容器であってよい。
【0067】
いくつかの実施形態において、組成物および方法は、直交機序によって脂肪を消滅させることが知られている分子をさらに含む。そのような分子は、BIBP−3226(Amgen)、BIBO−3304(Boehringer Ingleheim)、BMS−192548およびAR−H040922(Bristol−Myers Squibb)、LY−357897(Eli Lilly)、1229U91およびGW438014S(GlaxoSmithKline)、JNJ−5207787(Johnson&Johnson)、Lu−AA−44608(Lundbeck)、MK−0557(Merck NPY)、NGD−95−1(Neurgogen)、NLX−E201(Neurologix)、CGP−71683(Novartis)、PD−160170(Pfizer)、SR−120819A、BIIE0246およびS.A.0204(Sanofi Aventis)、S−2367(shiongli)等のNPY受容体アンタゴニスト、NPY受容体アンタゴニストであるジヒドロピリジンおよびジヒドロピリジン誘導体、NPY受容体アンタゴニストである二環化合物、カルバゾール系NPY受容体アンタゴニストならびにNPY受容体アンタゴニストである三環化合物を含むがこれらに限定されない神経ペプチドY(NPY)受容体アンタゴニストを含む。例えば、国際公開第2006/133160号および米国特許第6,313,128号(参照により図面を含むその全体が本明細書に組み込まれる)を参照されたい。白色脂肪血管系にあるCKGGRAKDCペプチド等の脂肪選択的プロアポトーシスペプチドも企図されている。Kolonin M.G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)を参照されたい。
【0068】
一態様において、本発明は、対象における皮下脂肪沈着物を減少させる方法に関する。そのような方法は、(i)脂肪溶解有効量の1つまたは複数の薬理学的に活性な洗浄剤または胆汁酸(複数可)および/もしくは胆汁塩(複数可)またはデオキシコール酸もしくはその塩またはデオキシコール酸ナトリウム;(ii)薬学的、獣医学的または美容学的賦形剤;ならびに(iii)場合によって脂質を含み、ここで、脂質と胆汁酸または胆汁塩との比率が最大1%w/wである組成物であって、リパーゼもコリパーゼも含まない組成物を、対象における皮下脂肪沈着物に局所的に投与するステップを含む。いくつかの実施形態において、脂肪沈着物は、肥満、脂肪再分布症候群、眼瞼脂肪ヘルニア、脂肪腫、ダーカム病、脂肪異栄養症、野牛肩脂肪異栄養症、後頸部(dorsocervical)脂肪、内臓脂肪症、乳房肥大、脂肪過剰症、体幹および腕周囲に散在する体脂肪およびセルライトを伴う脂肪沈着物からなる群から選択される状態に関連する。好ましい実施形態において、上記方法は、前記対象に対して外科手術を実施するステップを含まない。
【0069】
一態様において、本発明は、哺乳動物における選択された位置からの脂肪沈着物の除去方法であって、それを必要とする哺乳動物に、治療有効量のDCAもしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0070】
【化6】
であり、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない前記化合物を投与するステップを含む方法を目的としている。
【0071】
一実施形態において、本発明の方法は、哺乳動物に、神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分を投与するステップをさらに含み得る。一実施形態において、神経ペプチドY(NPY)受容体アンタゴニストは、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、NPY受容体アンタゴニストであるジヒドロピリジン、NPY受容体アンタゴニストであるジヒドロピリジン誘導体、NPY受容体アンタゴニストである二環化合物、カルバゾール系NPY受容体アンタゴニストならびにNPY受容体アンタゴニストである三環化合物からなる群から選択される。例えば、国際公開第2006/133160号および米国特許第6,313,128号(参照により図面を含むその全体が本明細書に組み込まれる)を参照されたい。一実施形態において、脂肪選択的プロアポトーシスペプチドは、白色脂肪血管系にあるCKGGRAKDCペプチドである。Kolonin M.G.ら、Nat. Med.、6月、10巻(6号):625〜32頁(2004年)を参照されたい。
【0072】
一態様において、本発明は、対象の皮膚領域における皮膚状態の出現を減少させるための方法に関する。そのような方法は、(i)皮膚引き締め有効量の1つまたは複数の薬理学的に活性な洗浄剤または胆汁酸(複数可)および/もしくは胆汁塩(複数可)またはデオキシコール酸もしくはその塩またはデオキシコール酸ナトリウム;(ii)薬学的、獣医学的または美容学的賦形剤;ならびに(iii)場合によって脂質を含む組成物を、前記皮膚領域に局所的に投与するステップを含む。いくつかの実施形態において、投与するステップは、本明細書における組成物を皮下または経皮注射によって送達することを含む。いくつかの実施形態において、治療または改善される皮膚状態は、弛緩性皮膚、皮膚の老化、皮膚の凹凸およびシワからなる群から選択される。いくつかの実施形態において、治療される皮膚の領域は、目の下、顎の下、腋の下、臀部、頬、額、ふくらはぎ、背部、大腿、足首または腹である。
【0073】
いくつかの実施形態において、皮膚領域における皮膚状態の出現を減少させるために使用される組成物は、皮膚引き締め溶液への製剤である。そのような皮膚引き締め溶液は、抗菌剤、血管収縮薬、抗血栓剤、抗凝固剤、発泡抑制剤、抗炎症剤、鎮痛剤、分散剤、抗分散剤、浸透促進剤、ステロイド、精神安定薬、筋弛緩薬および下痢止め剤からなる群から選択される第2の治療剤をさらに含み得る。
【0074】
好ましい実施形態において、洗浄剤は、デオキシコール酸、コール酸、ケノデオキシコール酸、7−アルファ−デヒドロキシレート(dehydroxylate)ケノデオキシコール酸、リトコール酸、ウルソデオキシコール酸、ジヒドロキシタウリン酸、トリヒドロキシタウリン酸および上記のいずれかのグリシン共役物からなる群から選択される胆汁酸を含む。いくつかの実施形態において、洗浄剤は、ナトリウム(Na+)、カリウム(K+)、リチウム(Li+)、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)、ストロンチウム(Sr2+)およびアンモニウム(NH4+)からなる群から選択されるカチオンを含む胆汁塩を含む。いくつかの実施形態において、洗浄剤は、アルカリ金属またはアルカリ土類金属であるカチオンとの胆汁塩を含む。好ましくは、アルカリ金属は、ナトリウム(Na+)、カリウム(K+)またはリチウム(Li+)であり、アルカリ土類金属は、マグネシウム(Mg2+)、カルシウム(Ca2+)、バリウム(Ba2+)またはストロンチウム(Sr2+)である。より好ましくは、胆汁塩はデオキシコール酸ナトリウムである。
【0075】
別の実施形態は、哺乳動物において脂肪を乳化する方法であって、それを必要とする哺乳動物に、治療有効量のDCAである化合物もしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0076】
【化7】
を投与するステップを含む方法であり、前記化合物はDCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない方法を提供する。
【0077】
別の実施形態は、ホスファチジルコリンを可溶化する方法であって、ホスファチジルコリンと、有効量のDCAである化合物もしくはそのプロドラッグ、またはDCAもしくはプロドラッグの薬学的に許容される塩:
【0078】
【化8】
であり、DCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない前記化合物とを混合するステップを提供する。
【0079】
本発明の別の態様は、デオキシコール酸(DCA)等の脂肪切除胆汁酸を、脂肪細胞を死滅させる作用物質と混合することに関する。一態様において、本発明は、直交機序によって脂肪を消滅させる分子をデオキシコレート注射液中に混合することにより、デオキシコレート注射の審美的作用を増強するための手段を企図している。そのような候補分子の例は、本明細書に記載されている通り、神経ペプチドY(NPY)アンタゴニストおよび脂肪選択的プロアポトーシスペプチドを含むがこれらに限定されない。脂肪細胞死滅および皮膚引き締めはいずれも、所望の作用を媒介するために必要となり得るため、脂肪死滅能力および強力な皮膚引き締め作用を有する作用物質(デオキシコレート等)の作用は、強力な脂肪細胞死滅効果を有する分子の添加によって増強できる。加えて、死滅させるために血管系にアクセスする必要がある分子(毛細血管の腔側に発現したタンパク質と結合するいくつかのプロアポトーシスペプチド等)は、デオキシコレートが血管漏出を引き起こし得るため、これらのタンパク質へのアクセスを得ることができる。故に、そのような作用物質は、デオキシコレートと相乗して、より少ない治療セッションでの体形矯正を媒介するためのより強力な手段を潜在的に作成することができる。
【0080】
合成方法
他の実施形態において、本発明は、DCAの化合物、塩およびプロドラッグならびにそれらの関連中間体の合成方法を提供する。
【0081】
本明細書において使用される場合、ステロイド骨格のナンバリングは、図1に示されている通りの一般的慣例に従う。
【0082】
したがって、提供されるのは、デオキシコール酸(DCA)もしくはそのエステルまたは薬学的に許容されるその塩:
【0083】
【化9】
を調製するための方法であって、
(a)9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1を、水素化条件下、H2と反応させて、化合物2
【0084】
【化10】
を形成するステップと、
(b)化合物2を酸と反応させて、化合物3
【0085】
【化11】
を形成するステップと、
(c)化合物3を還元剤と反応させて、化合物4を4および5
【0086】
【化12】
の混合物として形成するステップと、
(d)化合物4をオレフィン形成条件下で二炭素オレフィン化試薬と反応させて、化合物6
【0087】
【化13】
を形成するステップと、
(e)化合物6を、式7の化合物[式中、Pは保護基である]
【0088】
【化14】
に変換するステップと、
(f)式7の化合物をルイス酸の存在下でアルキルプロピオレートCH2CH2C(O)ORまたはアルキルアクリレートCH2=CHC(O)OR[式中、Rはアルキルである]と反応させて、式8の化合物[式中、Pは保護基であり、Rはアルキルであり、破線
【0089】
【化15−1】
は単結合または二重結合である]
【0090】
【化15−2】
を形成するステップと、
(g)式8の化合物を水素化条件下でH2と反応させて、式9の化合物[式中、Pは保護基であり、Rはアルキルである]
【0091】
【化16】
を形成するステップと、
(h)式9の化合物を酸化剤と反応させて、式10の化合物[式中、Pは保護基であり、Rはアルキルである]
【0092】
【化17】
を形成するステップと、
(i)式10の化合物を水素化条件下でH2と反応させて、式11の化合物[式中、Pは保護基であり、Rはアルキルである]
【0093】
【化18】
を形成するステップと、
(j)式11の化合物を還元剤と反応させて、式12の化合物[式中、Pは保護基であり、Rはアルキルである]
【0094】
【化19】
を形成するステップと、
(k)式12の化合物を脱保護条件に曝露してそのエステルを形成し、場合によって、適切な加水分解条件に曝露して、デオキシコール酸または薬学的に許容されるその塩を形成するステップと
を含む方法である。
【0095】
本発明は、以下のスキーム1に示される下記中間体[式中、PおよびRは上記で定義された通りである]も提供する。
【0096】
スキーム1.デオキシコール酸(DCA)の合成
【0097】
【化20】
一実施形態において、パート(a)の水素化条件はPd/C触媒を含む。
【0098】
一実施形態において、パート(b)の酸は鉱酸である。いくつかの態様において、鉱酸はH2SO4である。
【0099】
一実施形態において、パート(c)の還元剤はLiAl(OtBu)3Hである。
【0100】
一実施形態において、パート(d)の二炭素オレフィン化試薬はPh3PCH2CH3+Br−等のウィッティヒ剤である。
【0101】
一実施形態において、化合物7〜12の保護基Pは−C(O)CH3である。いくつかの態様において、化合物6は、無水酢酸、ならびにEt3N、ピリジンおよび/またはジメチルアミノピリジン等の有機塩基による6の処理等により、アシル化条件に曝露されて7aを形成する。
【0102】
一実施形態において、パート(f)のルイス酸はEtAlCl2である。
【0103】
一実施形態において、パート(f)のアルキルプロピオレートはメチルプロピオレートである。
【0104】
一実施形態において、パート(f)のアルキルアクリレートはメチルアクリレートである。
【0105】
一実施形態において、パート(g)の水素化条件はPtO2またはPd/C触媒を含む。
【0106】
一実施形態において、パート(h)の酸化剤はCrO3である。
【0107】
一実施形態において、パート(i)の水素化条件はPd/C触媒を含む。
【0108】
一実施形態において、パート(j)の還元剤はLiAl(OtBu)3Hである。
【0109】
一実施形態において、Pが−C(O)CH3である場合、パート(k)の脱保護および加水分解条件は、化合物12を、アルカリ土類金属水酸化物、アルカリ土類金属アルコキシドまたは両方の混合物と反応させることを含む。いくつかの態様において、加水分解条件は、デオキシコール酸を得るための酸性ワークアップを含む。他の態様において、酸性ワークアップは、対応する塩を得るために省略される。
【0110】
一実施形態において、アルカリ土類金属アルコキシドはLiOHである。
【0111】
一実施形態において、デオキシコール酸の塩は、アルカリ土類金属アルコキシドまたは水酸化物との反応によって調製できる。デオキシコール酸の塩は、ナトリウム(Na+)、カリウム(K+)およびリチウム(Li+)塩を含む。
【0112】
一実施形態において、提供されるのは、
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2);
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3);
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6);
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a);
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a);
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b);
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a);および
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
からなる群から選択される中間化合物である。
【0113】
医薬組成物および投与様式
組成物は、開示されている化合物を、少なくとも1つの薬学的に許容される賦形剤と組み合わせて構成され得る。許容される賦形剤は、非毒性であり、投与を補助し、開示されている化合物の治療的有用性に悪影響を及ぼさない。そのような賦形剤は、任意の固体、液体、半固体であってよく、またはエアロゾル組成物の場合、当業者に一般に利用可能なガス状賦形剤であってよい。
【0114】
固体医薬品賦形剤は、デンプン、セルロース、タルク、グルコース、ラクトース、スクロース、ゼラチン、麦芽、米、小麦粉、石粉、シリカゲル、ステアリン酸マグネシウム、ステアリン酸ナトリウム、グリセロールモノステアレート、塩化ナトリウム、乾燥脱脂粉乳等を含む。液体および半固体賦形剤は、グリセロール、プロピレングリコール、水、エタノール、ならびに石油、植物または合成起源のもの、例えば、ピーナッツ油、大豆油、鉱油、ゴマ油等を含む種々の油から選択できる。好ましい液体担体は、特に注射用液の場合、水、生理食塩水、ブドウ糖水溶液およびグリコールを含む。
【0115】
概して、好ましい実施形態の化合物は、類似の効用を果たす作用物質の許容されている投与様式のいずれかによって治療有効量で投与される。好ましい実施形態の化合物、すなわち活性成分の実際の量は、治療される疾患の重症度、対象の年齢および相対的健康、使用される化合物の効力、投与の経路および形態ならびに他の要因等の数々の要因によって決まる。薬物は、1日当たり複数回、好ましくは1日当たり1回または2回投与され得る。これらの要因のすべては、担当臨床医の技量の範囲内である。
【0116】
製剤中の化合物の量は、当業者によって用いられる全範囲内で変動し得る。本明細書に記載されている通りの種々の態様における本組成物が調製でき、ここで、デオキシコール酸部分は、水1体積当たりの重量に基づいてまたは水の密度を前提としたw/w(すなわち、重量と体積との1対1対応)に基づいて約0.5%〜10%の範囲である。別の態様において、本実施形態は、最大で希釈剤の飽和点の濃度である現在記載されている医薬組成物に関する。濃度およびpH等の条件に基づき、チキソトロピー粘度を選択することができる。例えば、Mukhopadhyay, S.およびU. Maitra、Current Science、87巻:1666〜1683頁(2004年)の1680頁を参照されたい。
【0117】
いくつかの実施形態において、投与するステップは、本明細書における組成物を、皮膚パッチ、ポンプまたは真皮下デポー剤によって送達することを含む。いくつかの実施形態において、投与するステップは、本明細書における組成物を局所的にまたは皮下に送達することを含む。特定の実施形態において、投与するステップは、前記対象の目の下、顎の下、腋の下、臀部、ふくらはぎ、背部、大腿または腹の領域に局所的に(例えば、皮下または真皮下に)投与することを含む。投与は、皮下または経皮注射によって為され得る。
【0118】
合成スキーム
胆汁酸医薬組成物の完全な化学合成方法および有用な中間体の他の例が以下に提供される。
【0119】
これらの下記の記述および例は、本化合物を自然に産生する哺乳動物または微生物生命体からのDCAの抽出の代替を提供する。合成経路1〜6は、本発明におけるデオキシコール酸(DCA)を合成するための使用が企図されている。合成経路1Bおよび実施例1〜11は、ヒドロコルチゾンからのDCAの合成を示す。
【0120】
1.アドレノステロンからの9(11)−エンまたは11,12−エンを介する合成経路#1A
スキーム1A(以下)のコルチゾン(化合物1.1)は、完全な合成材料として広く利用可能である。クロロクロム酸ピリジニウム(PCC)を使用してこれを効率的に開裂し、C17ケトン化合物を形成することができる。このアドレノステロン(化合物1.2)への開裂は、HIO4またはビスマス酸ナトリウム(NaBiO3)を使用して実施することもできる。化合物1.2を化合物1.3に変換する反応は、既知の化学的プロセスである。化合物1.3の化合物1.4への変換には、モノケタール化が関与する。後続ステップは、3−ケト−4−エンの再生、C5β配置を得るための4,5−エン(H2/Pt/DMF)の選択的還元および化合物1.5を得るためのC3カルボニル基の所望の3α配置への選択的還元である。化合物1.5を化合物1.6に変換する際における保護基の添加およびそれに続く生成物の還元により、C11β−オール(アキシャル配置)、すなわち化合物1.7が得られ、これは、主要な9(11)−エンへの位置選択的脱離(すなわち、化合物1.7の化合物1.8への変換)に適している。
【0121】
ここで、合成スキームは、化合物1.7が化合物1.8または化合物1.9のいずれへの変換のための出発材料として使用され得るかという点で分岐する。C11ヒドロキシル基とC9水素原子との間のトランスジアキシャルの関係により、化合物1.7を化合物1.8に変換するために使用される脱離反応は位置選択的である。化合物1.9の異性体C11−C12オレフィンを得るための脱離の代替様式は、シス−熱脱離(すなわち、化合物1.7の化合物1.9への変換)を含み、同様に位置選択的である。
【0122】
スキーム1A.DCAのC12ヒドロキシル基の2つのC環前駆体の合成
【0123】
【化21】
化合物1.8のアリル酸化(CrO3および3,5ジメチルピラゾールによる処理を介する)により、エノン含有化合物1.10が得られる。化合物1.9の過酸酸化は、ステロイドのアルファ面から立体選択的に進み、C11−12エポキシド化合物1.11が得られる(上記スキーム1Aを参照)。これらの化学転換により、C12ヒドロキシル基官能性の2つの主要な前駆体、すなわち、化合物1.10および化合物2.1が得られる(スキーム1Aおよび2)。
【0124】
当業者であれば、上記コルチゾン経路は、代わりに、同じ炭素骨格および同じ酸素原子の相対的配置を有するヒドロコルチゾンから始まるように変更してよく、ヒドロコルチゾンは、炭素原子を有するC−11酸素の酸化状態のみがコルチゾンと異なっていることを理解するであろう。ヒドロコルチゾンは市販されており、全化学合成(Woodward
R. B.ら、J. Am. Chem. Soc.、74巻:4223頁(1952年))を含むこの化合物の種々の合成が既知である(Szczebaraら、Nature Biotechnology、21巻:143〜149頁(2003年2月))。ケトン1.13は、α,β−不飽和二重結合の水素化分解によってヒドロコルチゾン1.12(スキーム1B)から開始して合成され、続いて、NaIO4を使用する1,2−ジオール開裂を可能にするための水素化ホウ素ナトリウムを使用して完全なケトン還元を行い、それにより、ステロイド環系のD環上にC17ケトンを形成する。それに続くクロロクロム酸ピリジニウム(PCC)による酸化により、1.13が得られる。K−selectride(登録商標)による1.13の処理、続いて無水酢酸/ピリジンによるアセチル化により、保護されたアルコール1.15が得られる。それに続くウィッティヒ試薬による1.15のオレフィン化によってアルケン1.16を得て、その後、これをメチルプロピオレートおよびエチルアルミニウムジクロリドで処理し、ジエン1.17を形成する。両方の二重結合の水素化に続いて、ケトン1.18を還元し、得られたアルコール中間体をピリジン中のSOCl2による処理により脱離し、アルケン1.19を得る。CrO3によるアルケン1.19のアリル酸化および水素化条件下での二重結合の還元により、ケトン1.21が得られる。アセテート保護基の除去および得られたアルコールの酸化により、ジケトン1.22が得られる。LiAlH(O−tBu)3による1.22の還元およびメチルエステルの加水分解により、DCAが得られる。
【0125】
スキーム1B.ヒドロコルチゾンからのDCAの合成
【0126】
【化22】
化合物1.10および2.1のさらなる転換は、スキーム2に示されている。最初に、化合物1.10を、DCAのものと同一の適切に官能基化されたC環系を含有するように修飾する(スキーム2)。C12カルボニル基の立体選択的還元により化合物2.1が得られ、化合物2.1中に存在する9(11)二重結合の触媒水素化により化合物2.2が得られる。
【0127】
スキーム2.アリル酸化経路を使用するC12ヒドロキシル基の導入
【0128】
【化23】
スキーム3は、エポキシド含有化合物1.11の、スキーム2の類似体C12α−ヒドロキシステロイド化合物2.2への転換を提示する。
【0129】
スキーム3.C11−C12エポキシドの立体選択的還元
【0130】
【化24】
これらの経路の両方において上述した通り、共通の中間化合物2.2が形成される。
【0131】
DCAの合成における次のステップは、DCAのカルボキシル側鎖置換D環を含有するような化合物2.2中に存在するD環の修飾である(スキーム4およびスキーム5)。
【0132】
スキーム4.脱保護およびウィッティヒ反応
【0133】
【化25】
最初に、化合物2.2のC17ケタールおよびC3シリルエーテル基を加水分解する。その後、ウィッティヒ反応を実施し、化合物4.2を得る。化合物4.2の化合物5.1への変換がエン反応によって行われる。それに続く化合物5.1の触媒還元およびエステルの加水分解により、DCAが得られる(スキーム5)。
【0134】
スキーム5.DCA側鎖の導入のためのエン反応および触媒還元
【0135】
【化26】
2.コルチゾンからのアドレノステロン(i−ステロイド、3,5−シクロステロール経路)を介する合成経路#2
C17におけるアドレノステロン(化合物1.2、スキーム6)の選択的ケタール化、ホウ化水素還元、メシル化および緩衝化された加水分解により、i−ステロイド(3,5−シクロステロール)含有化合物6.1が得られる。化合物6.1に、9(11)−エン形成(化合物6.1の化合物6.2への変換、スキーム6)およびアリル酸化(化合物6.2の化合物6.3への変換、スキーム6)、続いてカルボニル基還元を受けさせ、化合物6.4を得る。i−ステロールの加水分解および水素化により化合物6.5が得られ、合成経路#1において上記で提示した合成方法によってこれをDCAに変換することができる。
【0136】
スキーム6.i−ステロイド(すなわち、3,5−シクロステロール)の形成によるA−B−環系の保護
【0137】
【化27】
3.ヘコゲニンからの合成経路#3
ヘコゲニン(化合物7.1、スキーム7)は、メキシカンヤムおよびAgave種の他の植物において豊富に見られる植物ステロールである。DCA合成のための出発材料としてのヘコゲニンの主な利点は、DCAに存在するC12酸素官能基をそのまま有することである。
【0138】
ヘコゲニンから開始する合成経路における最初のステップは、ヘコゲニン(化合物7.1)中のC12カルボニル基の、必須のC12−α配置への立体選択的還元(化合物7.1の化合物7.2への変換)である。その後、3−β−オール、5α−AB環系は、3α−オール、5β−AB環系に変換(化合物7.1の、化合物7.2へ、化合物7.3への変換)される(スキーム7)。よく知られているマーカー分解(Marker, R.E.、Rohrmann, E.、Sterols. LXIX. Oxidation Products of Sarsasapogenin. Sarsasapogenoic Acid and Related Substances、J. Am. Chem. Soc.、61巻(8号):2072〜2077頁(1939年))を、化合物7.2の化合物7.3への変換に続いて行い、化合物7.4を得る。D環側鎖(スキーム8)の化合物8.2への導入は、スキーム4および5に示されている方法によって実現される。化合物8.2中の必須のC17ケトンは、化合物8.1の酢酸エノールのオゾン分解によって形成される(スキーム8)。その後、オレフィン化およびエン反応順序を使用するスキーム5と類似の方式で、8.2からDCAが調製される。ヘコゲニンから開始する代替経路は、スキーム9および10に示されている。
【0139】
スキーム7.C12−ヒドロキシル基導入、AB環修飾および側鎖開裂
【0140】
【化28】
スキーム8.側鎖導入
【0141】
【化29】
スキーム9.3−ケト−4−エンへのジチオエタン経路
【0142】
【化30】
スキーム10A.3−ケト−4−エンを介するDCA形成
【0143】
【化31】
4.サポゲニンからの合成経路#4
サポゲニンは、サポニン(すなわち、ステロイド配糖体)のC3ヒドロキシル基に結合している単糖類および二糖類の加水分解に由来する。これらは広く発生する植物生成物である。サポニンは、本来、以下に示される通りのスピロケタール構造として発生する。化合物10.3を、チゴゲニン、ジオスゲニン、クロロゲニン、スミラゲニンおよびヘコゲニン(化合物7.1)から形成することもできる。我々は、DCAが、これらの、すなわち、チゴゲニン、ジオスゲニン、クロロゲニン、スミラゲニンおよびヘコゲニン(化合物7.1)のそれぞれから合成され得ると考えている(Y. Mazur、N. DanieliおよびFranz Sondheimer、J. Am. Chem. Soc.;82巻、5809頁(1960年))。
【0144】
我々は、ヘコゲニンからDCAを合成できたため、上記サポゲニンのいずれも、同様にDCA合成のための出発材料としての役割を果たし得ると認識している。
【0145】
スキーム10B.サポニンからの合成経路
【0146】
【化32】
5.スチグマステロールからの合成経路#5
スチグマステロール(化合物11.1)は、広く利用可能な植物ステロールである。これは、DCA合成のための出発材料として、官能基化されたAB環系および容易に開裂可能な側鎖部分を含有するという利点を有する。スチグマステロールは、DCA合成に不可欠の、C環において必要な官能性を欠くという不利点を有する。
【0147】
この合成経路において、スチグマステロール(化合物11.1)AB環は、i−ステロイド形成によって保護され、続いてオゾン分解によってC17に導入された側鎖が得られ、それはマスクされた形態のカルボキシル基としてのC24−オールに還元される(スキーム11)。後続ステップは、C12にアリル位を生成する(スキーム12)。B環ジエン形成および酢酸第二水銀酸化は既知の過程であり、B環系の触媒還元により、上述した先の経路と共通の中間体が得られる。しかしながら、他の経路とは対照的に、側鎖が既に存在する。アリル酸化(化合物12.4の化合物1.20への変換)および立体選択的還元(化合物1.20の化合物1.21への変換)、続いて前述のステップにより生成物が得られ、これをDCAに変換する。
【0148】
スキーム11.i−ステロイドのオゾン分解および側鎖還元
【0149】
【化33】
スキーム12.トリエン形成およびアリル酸化
【0150】
【化34】
スチグマステロール経路の変形形態では、B環ジエンのディールス・アルダー保護を使用する。これは、9(11)二重結合を単離してアリル酸化ステップ中の考えられる干渉を防止するため、有利である(スキーム13)。
【0151】
スキーム13.トリエン形成および環Bジエンのディールス・アルダー保護
【0152】
【化35】
6.エルゴステロールからの合成経路#6
エルゴステロール(化合物14.1)は、容易に利用可能な出発材料であり、本明細書で説明されている手順を適合することによってDCAを調製するために使用できる。アリル酸化は、C12酸素官能性への簡易経路を提供する(スキーム14)。この経路は、環Bジエンから開始するという利点を有する。これはスチグマステロール経路と収束する。
【0153】
スキーム14.エルゴステロールからのトリエン形成
【0154】
【化36】
好ましい実施形態の化合物は、下記の一般的方法および手順を使用して、容易に利用可能な出発材料から調製できる。典型的なまたは好ましい処理条件(すなわち、反応温度、時間、反応物質のモル比、溶媒、圧力等)が与えられている場合、特に明記しない限り、他の処理条件を使用してもよいことが理解される。最適反応条件は、使用される特定の反応物質または溶媒によって異なり得るが、そのような条件は、当業者が日常的な最適化手順によって決定できる。
【0155】
加えて、当業者には明らかなように、いくつかの官能基が望ましくない反応を受けるのを防止するために、従来の保護基が必要となり得る。種々の官能基に適切な保護基ならびに特定の官能基を保護および脱保護するための適切な条件は、当該技術分野において既知である。例えば、数々の保護基は、T. W. GreeneおよびG. M. Wuts、Protecting Groups in Organic Synthesis、第3版、Wiley、New York、1999年ならびにその中で引用されている参考文献に記載されている。
【0156】
本明細書に記載されている反応のための出発材料および試薬は、概して既知の化合物であるか、または既知の手順もしくはその明白な修正形態によって調製できる。例えば、出発材料および試薬の多くは、Aldrich Chemical Co.(Milwaukee、Wisconsin、USA)、Bachem(Torrance、California、USA)、Emka−ChemまたはSigma(St.Louis、Missouri、USA)等の商業的供給業者から入手可能である。その他は、FieserおよびFieserのReagents for Organic Synthesis、1〜15巻(John Wiley and Sons、1991年)、RoddのChemistry of Carbon Compounds、1〜5巻および補足(Elsevier Science Publishers、1989年)、Organic Reactions、1〜40巻(John Wiley and Sons、1991年)、MarchのAdvanced Organic Chemistry(John Wiley and Sons、第4版)ならびにLarockのComprehensive Organic Transformations(VCH Publishers Inc.、1989年)等の標準的な参考教科書に記載されている手順またはその明白な修正形態によって調製できる。
【実施例】
【0157】
好ましい実施形態の種々の出発材料、中間体および化合物は、適切な場合、沈殿、ろ過、結晶化、蒸発、蒸留およびクロマトグラフィー等の従来の技術を使用して単離および精製できる。これらの化合物の特徴付けは、融点、質量スペクトル、核磁気共鳴および種々の他の分光分析によって等、従来の方法を使用して実施できる。
【0158】
合成経路#1、スキーム1Bにおける生成物の合成を実施するためのステップの例示的な実施形態は、以下でさらに詳細に記載される。表3は、下記の実施例におよび本明細書全体を通して記載されている例示的な反応スキームおよび合成経路において種々の化合物/部分/装置/手順/特性を表現するために使用される略語を記載している。
【0159】
【表3−1】
【0160】
【表3−2】
概要:酸素および水分感受性材料の操作は、アルゴン雰囲気下、二口フレームドライフラスコで行う。カラムクロマトグラフィーは、SE−Makeシリカゲル(60〜120メッシュ)、Spectrochemシリカゲル(230〜400メッシュ)または酸化アルミニウム90中性(SD−Fine Chem.Ltd.、India)を使用して実施する。分析用薄層クロマトグラフィー(TLC)は、Merck Kieselgel 60F254(0.25mm)プレート(Merck&Co.、Whitehouse Station、NJ)で実施した。スポットの可視化は、UV光(254nm)ランプによってまたはエタノール中の硫酸(5%)およびp−アニスアルデヒド(3%)溶液で炭化させることによってのいずれかで検出した。
【0161】
装置:本明細書に記載されている反応スキームおよび合成経路の化合物および生成物の分析は、以下に記載される装置および設備で実行できる。
【0162】
核磁気共鳴(NMR)
プロトンおよび炭素の核磁気共鳴スペクトル(1H NMRおよび13C NMR)は、内部標準として溶媒共鳴(1H NMR、7.26ppmのCHCl3または2.5ppmのDMSOおよび3.33ppmのDMSO−H2O;13C NMR、77.0ppmのCDCl3または39.5ppmのDMSO)を用い、Varian Mercury−Gemini200(1H NMR、200MHz;13C NMR、50MHz)またはVarian Mercury−Inova500(1H NMR、500MHz;13C NMR、125MHz)(Varian,Inc.、Palo Alto、CA)分光計で記録される。1H NMRデータは、下記の通りに報告される:化学シフト(δ、ppm)、多重度(s=一重項、d=二重項、t=三重項、q=四重項、br=広幅、m=多重項)、カップリング定数(Hz)および積分。
【0163】
赤外線分光法
赤外線スペクトル(FT−IR)は、JASCO−460+モデル(Jasco,Inc.、Easton、MD)で動作させる。質量スペクトルは、Perkin ElmerのAPI−2000分光計(Perkin Elmer,Inc.、Waltham、MA)でES+モードを使用して得られる。
【0164】
融点
融点は、LAB−INDIA融点計測装置(Labindia Instruments Pvt.Ltd.、India)を使用して測定したものであり、訂正されない。
【0165】
高圧液体クロマトグラフィー
HPLCクロマトグラムは、PDA検出器を有するSHIMADZU−2010モデル(株式会社島津製作所、日本)を使用して記録した。
【0166】
光学活性
比旋光度([α]D)は、589nmのJASCO−1020(Jasco,Inc.、Easton、MD)を用いて測定し、訂正されない。
【0167】
化学物質:特に断りのない限り、市販の試薬を精製することなく使用する。ジエチルエーテルおよびTHFは、ナトリウム/ベンゾフェノンケチルから蒸留される。無水DMF、DCM、ペンタンおよびヘキサンは、CaH2からの蒸留によって得られる。
【0168】
(実施例1)
アンドロスタン−3,11,17−トリオン(1.13)の調製
10%のPd/C(2.5g、5wt%)を、DMF(250mL)中のヒドロコルチゾン(化合物1.12)(50.0g、138.12mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で12時間水素化する。TLCによって証明される通り、出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去する。粗生成物(48.0g)が無色固体として得られる。
【0169】
NaBH4(2.1g、55.3mmol)を、EtOH(500mL)およびCH2Cl2(500mL)中の上記粗生成物(48.0g、131.86mmol)の溶液に添加する。1時間後、アセトン(50mL)および水(150mL)、続いてNaIO4(70.5g、329.6mmol)を添加する。混合物を室温で終夜撹拌する。
【0170】
蒸留水(500mL)を添加し、混合物を酢酸エチル(3×250mL)で抽出する。酢酸エチル層をシリカゲルプラグに通して洗い流し、溶媒を蒸発させて38gを無色固体として得る。粗生成物を精製することなくさらに酸化させる。
【0171】
PCC(40.4g、187.5mmol)を、CH2Cl2(400mL)中の上記粗生成物の溶液に3等分ずつ30分間かけて添加する。得られた反応混合物を室温で約3〜4時間撹拌する。TLCによってモニターされる通り、反応の完了時に、粗反応混合物をセライトおよびシリカゲルのパッドに通して連続的にろ過し、粗材料を、酢酸エチル/ヘキサン(3:10)[50mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:1)中、化合物1.13のRf=0.37および化合物1.12のRf=0.05]で溶離するカラムクロマトグラフィー[59(W)×700(L)mm、60〜120メッシュシリカ、150g]によって精製し、ジアステレオマー化合物1.13(33.0g、79%収率)を無色固体として得る。
【0172】
得られた粗材料を、Phenomenex Lunov C18カラム(250×30.0mm、10μ)を使用する分取HPLCおよびCH3CN:H2O(12:13)による定組成溶離によって、15mLの画分中、25mL/分流速で精製する。分取HPLCは、精製にのみ使用し、分析には使用しない。表4は、生成物の計測された特性を記載している。
【0173】
【表4】
(実施例2)
3β−ヒドロキシ−アンドロスタン−11,17−ジオン(1.14)
K−selectride(登録商標)(98.39mL、98.01mmol、THF中1M溶液)を、THF(330mL)中の化合物1.13(33.0g、109.27mmol)の溶液に、不活性雰囲気下、−78℃で15分間かけて添加し、−78℃で約3〜4時間撹拌する。反応混合物をNaOH水溶液(2M、70mL)でクエンチする。粗反応混合物を酢酸エチル(500mL)で希釈し、有機層を水(3×75mL)および飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空下で除去し、33gの粗材料を得る。粗生成物を精製することなくアセチル化に付す。
【0174】
粗材料の精製
粗材料を、酢酸エチル/ヘキサン(1:4)[25mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:1)中、化合物1.14のRf=0.3および化合物1.13のRf=0.37]で溶離するカラムクロマトグラフィー[29(W)×600(L)mm、230〜400メッシュシリカ、200g]によって精製し、化合物1.14を得る。表5は、生成物の計測された特性を記載している。
【0175】
【表5】
(実施例3)
3β−ヒドロキシアンドロスタン−11,17−ジオンアセテート(1.15)
無水酢酸(16.6g、162.8mmol)を、ピリジン(150mL)中の化合物1.14(33.0g、108.55mmol)の溶液に、不活性雰囲気下、0℃で添加する。得られた反応混合物を周囲温度で終夜撹拌する。TLCによって証明される通り、反応の完了時に、ピリジンおよび残っている無水酢酸を真空下で除去する。粗残留物を酢酸エチル(500mL)で希釈し、水(3×150mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空蒸発させ、粗材料を、酢酸エチル/ヘキサン(1:10)[25mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(3:7)中、化合物1.15のRf=0.38および化合物1.14のRf=0.1]で溶離するカラムクロマトグラフィー[59(W)×800(L)mm、60〜120メッシュシリカ、150g]によって精製し、化合物1.15(19.0g、66.4%収率)を無色固体として得る。表6は、生成物の計測された特性を記載している。
【0176】
【表6】
(実施例4)
(Z)−3β−ヒドロキシ−5β−プレグ−17(20)−エン−11−オンアセテート(1.16)
カリウムtert−ブトキシド(159.28mL、159.2mmol、THF中1M溶液)を、THF(150mL)中のエチルトリフェニルホスホニウムブロミド(61.16g、164.8mmol)の溶液に、不活性雰囲気下、−5℃で1時間かけて滴下添加する。得られた濃桃色の反応混合物を10〜15℃まで加温し、同じ温度でさらに1時間撹拌する。THF(50mL)中の化合物55(19.0g、54.9mmol)の溶液を、上記ウィッティヒイリド懸濁液に−5℃でゆっくり導入する。溶液をさらに10〜20分間撹拌し、反応混合物を周囲温度までゆっくり加温する。撹拌を約3〜4時間続ける。TLCによって証明される通り、出発材料の完全消失時に、反応混合物を飽和NH4Cl水溶液(75mL)でクエンチする。水層をEtOAc(2×150mL)で抽出し、合わせた有機抽出物を飽和ブライン溶液(100mL)で洗浄し、MgSO4(75g)で乾燥させる。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:20)[25mL画分、10mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:6)中、化合物1.16のRf=0.54および化合物1.15のRf=0.06]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、300g]によって精製し、化合物1.16(15.5g、78.8%収率)を濃厚な無色液体として得て、これを1〜2日後、0℃でゆっくり凝固させる。表7は、生成物の計測された特性を記載している。
【0177】
【表7】
(実施例5)
メチル(E)−3β−ヒドロキシ−5β−コーラ−16(17),22(23)−ジエン−24−オエートアセテート(1.17)
メチルプロピオレート(9.68g、114.95mmol)を、0℃のCH2Cl2(220mL)中の化合物1.16(16.5g、46mmol)の溶液に添加する。反応混合物を周囲温度まで加温し、不活性雰囲気下、1時間撹拌する。エチルアルミニウムジクロリド(17.5g、137.8mmol)を、0℃の上記混合物に滴下導入し、得られた反応塊を再度周囲温度まで加温し、終夜撹拌する。TLCによって証明される通り、反応の完了時に、粗反応混合物を氷水(100mL)でクエンチし、水層をEtOAc(3×150mL)で抽出する。合わせた有機層を飽和ブライン溶液(100mL)で洗浄し、MgSO4(50g)で乾燥させる。溶媒を真空下で除去し、粗材料を酢酸エチル/ヘキサン(1:7)[15mL画分、10mL/分溶離、TLCによってモニターし、UV光(254nm)ランプまたはp−アニスアルデヒド炭化のいずれかによって検出する;EtOAc/ヘキサン(1:6)中、化合物1.17のRf=0.36および化合物1.16のRf=0.54]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、300g]によって精製し、化合物1.17(16g、79%収率)を無色半固体として得る。表8は、生成物の計測された特性を記載している。
【0178】
【表8】
(実施例6)
メチル3β−ヒドロキシ−5β−コラン−11−オン−24−オエートアセテート(1.18)
10%Pd/C(2.9g、20wt%)を、EtOAc(150mL)中の化合物1.17(14.5g、32.8mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で12時間水素化する。TLC[EtOAc/ヘキサン(1:3)中、化合物1.18のRf=0.43および化合物1.17のRf=0.43;しかしながら、化合物1.17だけは共役エステル発色団によりUV活性である]によって証明される通り、出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去し、化合物1.18(14g、95.7%収率)を無色固体として得る。表9は、生成物の計測された特性を記載している。
【0179】
【表9】
(実施例7)
メチル3β−ヒドロキシ−5β−コール−9(11)−エン−24−オエートアセテート(1.19)
PtO2(5.0g、100wt%)を、触媒量のAcOH(2.0mL)の存在下、EtOAc(75mL)中の1.18(5.0g、11.2mmol)の溶液に添加する。得られたスラリーをParr装置(70psi)内で約14〜16時間水素化する。反応の完了時に、粗混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去する。粗生成物をさらに精製することなく脱離反応に使用する。
【0180】
SOCl2(1.98g、16.78mmol)を、ピリジン(100mL)中の上記粗混合物の溶液に、0℃で滴下導入する。得られた反応混合物を周囲温度まで加温し、約1時間撹拌する。TLCによって証明される通り、反応の完了時に、ピリジンを真空下で除去する。粗残留物を酢酸エチル(100mL)で希釈し、水(2×50mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(40g)で乾燥させる。溶媒を真空蒸発させ、粗材料を、酢酸エチル/ヘキサン(1:10)[10mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(1:6)中、化合物1.19のRf=0.51および化合物1.18のRf=0.22]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、120g]によって精製し、化合物1.19(4.1g、85.4%収率)を無色固体として得る。表10は、生成物の計測された特性を記載している。
【0181】
【表10】
(実施例8)
メチル3β−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエートアセテート(1.20)
CrO3(8.0g、100wt%、80.0mmol)を、AcOH(150mL)中の化合物1.19(8.0g、18.6mmol)の溶液に添加する。得られた反応混合物を60℃で約24〜36時間加熱する。前駆体の完全消失時に、酢酸を真空蒸発させ、粗材料をジエチルエーテル(400mL)に溶解する。有機層を水(2×100mL)、飽和ブライン溶液(100mL)で洗浄し、MgSO4(40g)で乾燥させる。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:5)[10mL画分、3mL/分溶離、TLCによってモニターし、UV光(254nm)ランプによって検出する;EtOAc/ヘキサン(1:4)中、化合物1.20のRf=0.28および化合物1.19のRf=0.61]で溶離するカラムクロマトグラフィー[49(W)×600(L)mm、60〜120メッシュシリカ、120g]によって精製し、化合物1.20(5g、60.5%収率)を無色固体として得る。表11は、生成物の計測された特性を記載している。
【0182】
【表11】
(実施例9)
メチル3β−ヒドロキシ−5β−コラン−12−オン−24−オエートアセテート(1.21)
10%Pd/C(30mg、10wt%)を、EtOAc(30mL)中の化合物1.20(300mg、0.675mmol)の溶液に添加する。得られたスラリーをParr装置(50psi)内で約16時間水素化する。TLC[EtOAc/ヘキサン(3:7)中、化合物1.21のRf=0.44および化合物1.20のRf=0.44;しかしながら、化合物1.20だけはそのエノン発色団によりUV活性であり;加えて、化合物1.20の炭化は不鮮明であるが化合物1.21は鮮明である]による出発材料の完全消失時に、粗反応混合物をセライトの小型プラグに通してろ過し、溶媒を真空下で除去し、化合物1.21(270mg、90%収率)を無色固体として得る。表12は、生成物の計測された特性を記載している。
【0183】
【表12】
(実施例10)
メチル5β−コーラ−3,12−ジオン−24−オエート(1.22)
NaOH(73mg、1.8mmol)を、MeOH(10mL)中の化合物1.21(270mg、0.6mmol)の溶液に添加する。得られた反応混合物を周囲温度で約2時間撹拌する。TLCによって証明される通り、反応の完了時に、MeOHを真空下で除去し、粗生成物を酢酸エチル(20mL)で希釈する。有機層を飽和ブライン溶液(10mL)で洗浄し、MgSO4(5.0g)で乾燥させる。溶媒を真空下で除去し、粗材料を精製することなくエステル化反応において使用する。
【0184】
SOCl2(0.1mL、1.35mmol)を、0℃のMeOH(10mL)中の上記粗材料の溶液に滴下添加する。得られた反応混合物を周囲温度で約1時間撹拌する。反応の完了時に、MeOHを真空下で除去する。粗反応混合物をEtOAc(30mL)で希釈し、有機層を水(3×10mL)、飽和ブライン溶液(15mL)で洗浄し、MgSO4(5g)で乾燥させる。溶媒を真空蒸発させ、粗生成物を精製することなく酸化反応に使用する。
【0185】
PCC(1.0g、4.6mmol)を、CH2Cl2(25mL)中の得られたエステルの溶液に3等分ずつ約5分間かけて導入する。得られた反応混合物を周囲温度で約3〜4時間撹拌する。TLCによって証明される通り、反応の完了時に、粗反応混合物をセライトのパッドに通してろ過する。溶媒を真空下で除去し、粗材料を、酢酸エチル/ヘキサン(1:6)[10mL画分、5mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;EtOAc/ヘキサン(2:3)中、化合物1.22のRf=0.57および化合物1.21のRf=0.71]で溶離するカラムクロマトグラフィー[19(W)×400(L)mm、60〜120メッシュシリカ、45g]によって精製し、化合物1.22(170mg、70.8%収率)を無色固体として得る。表13は、生成物の計測された特性を記載している。
【0186】
【表13】
(実施例11)
デオキシコール酸メチル(1.22−エステル)
LiAlH(O−tBu)(332mg、1.3mmol)を、THF(10mL)中の化合物1.22(150mg、0.37mmol)の溶液に、不活性雰囲気下、周囲温度で滴下導入する。約4〜5時間撹拌した後、反応混合物をHCl水溶液(2mL、1N)でクエンチし、粗混合物をEtOAc(30mL)で希釈し、水(15mL)、飽和ブライン溶液(10mL)で洗浄し、MgSO4(3g)で乾燥させる。溶媒を真空下で除去し、粗塊を、MeOH/CH2Cl2(1:20)[5mL画分、3mL/分溶離、p−アニスアルデヒド炭化を用いるTLCによってモニターする;MeOH/CH2Cl2(1:9)中、化合物1.22−エステルのRf=0.42および化合物1.22のRf=0.85]で溶離するカラムクロマトグラフィー[29(W)×500(L)mm、230〜400メッシュシリカ、50g]によって精製し、デオキシコール酸メチル(化合物1.22−エステル)(110mg、72.8%収率)を無色固体として得る。表14は、生成物の計測された特性を記載している。
【0187】
【表14】
(実施例11)
デオキシコール酸
H2O(2.0mL)中のLiOH(23mg、0.55mmol)の溶液を、THF(4mL)中の1.22−エステル(110mg、0.27mmol)の溶液に添加する。得られた反応混合物を周囲温度で約2〜3時間撹拌する。TLC[MeOH/CH2Cl2(1:9)中、化合物DCAのRf=0.35および化合物1.22−エステルのRf=0.42]によるエステルの消失時に、粗反応混合物を酢酸エチル(10mL)で希釈し、飽和ブライン溶液で粉砕し、有機層を完全に分離させる。有機層を飽和NH4Cl溶液(10mL)で洗浄し、MgSO4(3.0g)で乾燥させる。溶媒を真空下で除去し、トルエン(3×5mL)と共沸させることによっていかなる微量の水も除去し、デオキシコール酸(DCA)(100mg、94.3%収率)を無色固体として得る。表15は、生成物の計測された特性を記載している。
【0188】
【表15】
実施例1〜11に記載されている例示的な過程の生成物の収率は、表16に記載されている。
【0189】
【表16】
(実施例12)
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2)
【0190】
【化37】
DMF(150mL)中の9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1(30.0g、99.3mol)の溶液に、10%のPd/C(2.1g)を添加し、得られたスラリーをParr装置(60psi)内で12時間水素化した。TLCによって証明される通り、出発材料の完全消失時に、粗反応混合物をCelite(登録商標)の小型パッドに通してろ過し、溶媒を真空下で除去し、無色固体(30.0g)を得た。この固体を0℃のアセトン(90mL)と合わせ、得られたスラリーを1時間撹拌した。その後、これをろ過し、冷却(0℃)アセトン(30mL)で洗浄し、真空下、同じろ過漏斗中、室温で乾燥させ、化合物2(26.0g、86%)を得た。表17は、生成物の計測された特性を記載している。
【0191】
【表17】
(実施例13)
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3)
【0192】
【化38】
DCM(520mL)中の化合物2(26.0g、85.4mmol)の溶液に、濃硫酸(7.53g、76.8mmol)を、不活性雰囲気下、10℃で15分間かけて添加した。温度を25℃まで上昇させ、得られた溶液を2時間撹拌した。この時点で、TLCによって証明される通り、出発材料は残っていなかった。反応物を10%NaHCO3水溶液(200mL)の添加によってクエンチした。層を分離し、水層をDCM(2×100mL)で2回抽出した。有機層を合わせ、水(100mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、有機相をNa2SO4(75g)で乾燥させ、ろ過した。ろ液を真空蒸発させ、化合物3(23.0g、94%)をオフホワイトの固体として得た。この生成物をさらに精製することなく次のステップにおいて「そのまま」使用した。表18は、生成物の計測された特性を記載している。
【0193】
【表18】
(実施例14)
3α−ヒドロキシ−5β−アンドロスタ−9(11)−エン−17−オン(4)
【0194】
【化39】
リチウムトリ−tert−ブトキシアルミニウムヒドリドのTHF溶液(1.0M、84.4mL、84.4mmol)を、不活性雰囲気下、THF(230mL)中の化合物3(23.0g、80.4mmol)の冷(−40℃)溶液に添加した。得られた反応混合物を2時間撹拌した。この時点で、TLCによって証明される通り、反応は完了したと決定され、1N HCl(200mL)および酢酸エチル(230mL)の混合物を添加することにより、反応混合物をクエンチした。得られた二相混合物を分離し、水層を酢酸エチル(2×100mL)で2回抽出した。有機相を合わせ、水(150mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、有機相をNa2SO4(75g)で乾燥させ、ろ過した。ろ過を真空蒸発させ、化合物4(23.0g)をオフホワイトの固体として得た。上記粗生成物を精製することなく次のステップにおいて「そのまま」使用した。表19は、生成物の計測された特性を記載している。
【0195】
【表19】
(実施例15)
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6)
【0196】
【化40】
THF中のカリウムtert−ブトキシドの溶液(1M、230mL、231mmol)を、THF(150mL)中のエチルトリフェニルホスホニウムブロミド(88.7g、239mmol)の懸濁液に、25℃で1時間かけて滴下添加した。得られた暗赤色混合物を25℃でさらに1時間撹拌した。THF(230mL)中の化合物4(23.0g、79.7mmol)の溶液を、赤色混合物に25℃でゆっくり添加した。得られた混合物を3〜4時間撹拌し、この時点で、TLCにより完了したと決定された。飽和NH4Cl水溶液(75mL)を添加することにより、反応物をクエンチした。相を分離し、水層をEtOAc(3×150mL)で3回抽出した。有機画分を合わせ、飽和ブライン溶液(100mL)で洗浄し、Na2SO4(75g)で乾燥させ、ろ過した。ろ液を真空濃縮し、粗固体を、酢酸エチル/ヘキサン(1:9)で溶離するカラムクロマトグラフィー[49mm(W)×600mm(L)、60〜120メッシュシリカ、300g]によって精製した。生成物を含有する画分を合わせ、濃縮し、化合物6(19.1g、80.0%)を白色固体として得た。表20は、生成物の計測された特性を記載している。
【0197】
【表20】
(実施例16)
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a)
【0198】
【化41】
化合物6(19.0g、63mmol)をCH2Cl2(380mL)に溶解した。トリエチルアミン(17.6mL、126.6mmol)、DMAP(0.772g、6mmol)および無水酢酸(8.98mL、94mmol)を、窒素雰囲気下、25℃で連続的に添加した。得られた溶液を25℃で2時間撹拌し、この時点で、TLCによって反応は完了したと決定された。氷水(100mL)の添加によって反応物をクエンチし、相を分離した。水層をDCM(3×150mL)で3回抽出した。有機画分を合わせ、飽和ブライン溶液(100mL)で洗浄し、無水Na2SO4(50g)で乾燥させ、ろ過した。ろ液を真空濃縮し、化合物7a(22.0g、95%収率)をオフホワイトの固体として得た。表21は、生成物の計測された特性を記載している。
【0199】
【表21】
(実施例17)
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a)
【0200】
【化42】
エチルアルミニウムジクロリド(104.5mL、192mmol、トルエン中1.8M)を、不活性雰囲気下、0℃でDCM(100mL)中のメチルプロピオレート(13.58mL、153mmol)の溶液に添加した。得られた溶液を15分間撹拌し、その後、化合物7a(22g、64.3mmol)を添加した。0℃でさらに20分間撹拌後、温度を25℃まで上昇させ、そこでさらに18時間維持した。この時点で、TLCによって反応は完了したと決定され、混合物を冷(0℃)水(200mL)中に注いだ。相を分離し、水層をDCM(150mL)で抽出した。有機層を合わせ、水(200mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、これを無水Na2SO4(40g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体をメタノール(280mL)中でスラリー化することによって精製し、化合物8a(17.5g、68%)を白色固体として得た。表22は、生成物の計測された特性を記載している。
【0201】
【表22】
(実施例18)
メチル3α−アセトキシ−5β−コール−9(11)−エン−24−オエート(9a)
【0202】
【化43】
EtOAc(350mL)中の化合物8a(17.5g、41mmol)の溶液にPtO2(4.37g)を添加し、得られたスラリーをParr装置(70psi)内で14〜16時間水素化した。この時点で、TLCによって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物9a(17.0g、96.0%)を白色固体として得た。上記生成物をさらに精製することなく次のステップにおいて使用した。表23は、生成物の計測された特性を記載している。
【0203】
【表23】
(実施例19)
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b)
【0204】
【化44】
エチルアルミニウムジクロリド(14.2mL、25mmol、トルエン中1.8M)を、不活性雰囲気下、0℃でDCM(60mL)中のメチルアクリレート(1.89mL、20mmol)の溶液に添加した。得られた溶液を15分間撹拌し、その後、化合物7a(3g、8.7mmol)を添加した。0℃でさらに20分間撹拌後、温度を25℃まで上昇させ、そこでさらに18時間維持した。この時点で、TLCによって反応は完了したと決定され、その後、混合物を冷(0℃)水(60mL)中に注いだ。相を分離し、水層をDCM(60mL)で抽出した。有機層を合わせ、水(50mL)および飽和ブライン溶液(100mL)で連続的に洗浄した。その後、これを無水Na2SO4(5g)で乾燥させ、ろ過した。ろ液を真空濃縮し、化合物8b(2.6g、70%)を得た。表24は、生成物の計測された特性を記載している。
【0205】
【表24】
(実施例20)
メチル3α−アセトキシ−5β−コール−9(11)−エン−24−オエート(9a)
【0206】
【化45】
EtOAc(60mL)中の化合物8a(3g、7mmol)の溶液に10%Pd/C(300mg、10%wt/wt)を添加し、得られたスラリーをParr装置(70psi)内で14〜16時間水素化した。この時点で、TLC(ヘキサン中10%酢酸エチル)によって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物9a(2.6g、86%)を白色固体として得た。表25は、生成物の計測された特性を記載している。
【0207】
【表25】
(実施例21)
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a)
【0208】
【化46】
CrO3(17.0g、170mmol)を、AcOH(270mL)中の化合物9a(17g、39.5mmol)の溶液に添加した。得られた混合物を50℃で24〜36時間加熱した。TLCによる出発材料の完全消失時に、溶媒を真空蒸発させ、粗材料を酢酸エチル(400mL)および水(200mL)に溶解した。2つの相を分離し、有機層を、水(2×100mL)で2回、その後、飽和ブライン溶液(100mL)で1回洗浄した。有機相を無水Na2SO4(40g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体を、酢酸エチル/ヘキサン(1:5)[10mL画分、3mL/分溶離、TLCによってモニターし、UV光(254nm)ランプによって検出する]で溶離するカラムクロマトグラフィー[49mm(W)×600mm(L)、60〜120メッシュシリカ、120g]によって精製した。生成物含有画分を合わせ、真空濃縮し、化合物10a(8.8g、50%収率)を白色固体として得た。表26は、生成物の計測された特性を記載している。
【0209】
【表26】
(実施例22)
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
【0210】
【化47】
10%Pd/C(900mg)を、EtOAc(150mL)中の化合物10a(2.0g、4.5mmol)の溶液に添加し、得られたスラリーをParr装置(50psi)内、50℃で16時間水素化した。この時点で、TLCによって反応は完了したと決定された。混合物をCelite(登録商標)の小型プラグに通してろ過し、溶媒を真空下で除去し、化合物11a(1.6g、80%収率)を白色固体として得た。表27は、生成物の計測された特性を記載している。
【0211】
【表27】
(実施例23)
メチル3α−アセトキシ−12α−ヒドロキシ−5β−コラン−24−オエート(12a)
【0212】
【化48】
リチウムトリ−tert−ブトキシアルミニウムヒドリドのTHF溶液(1M、22.4mL、22.4mmol)を、周囲温度でTHF(25mL)中の化合物11a(2.5g、5.6mmol)の溶液に滴下添加した。さらに4〜5時間撹拌後、TLCによって反応は完了したと決定された。HCl水溶液(1M、10mL)を添加することによって反応物をクエンチし、混合物をEtOAc(30mL)で希釈した。相を分離し、有機相を水(15mL)および飽和ブライン溶液(10mL)で連続的に洗浄した。その後、有機相を無水Na2SO4(3g)で乾燥させ、ろ過した。ろ液を真空濃縮し、得られた固体を、EtOAc/ヘキサン(2:8)[5mL画分、p−アニスアルデヒド炭化を用いるTLCによってモニターする]で溶離するカラムクロマトグラフィー[29mm(W)×500mm(L)、60〜120メッシュシリカ、50g]によって精製した。生成物を含有する画分を合わせ、真空濃縮し、化合物12a(2.3g、91%)を白色固体として得た。表28は、生成物の計測された特性を記載している。
【0213】
【表28】
(実施例24)
デオキシコール酸(DCA)
【0214】
【化49】
H2O(2.0mL)中のLiOH(187mg、4.4mmol)の溶液を、THF(8mL)およびMeOH(8mL)中の化合物12a(500mg、1.11mmol)の溶液に添加した。得られた混合物を50℃で3〜4時間撹拌した。TLCによる出発材料の完全消失時に、反応混合物を真空濃縮した。水(10mL)および3N HCl(1mL)の混合物を合わせ、0℃まで冷却し、その後、粗生成物に添加した。0℃で1時間撹拌後、沈殿した固体をろ過し、その後、水(10mL)およびヘキサン(20mL)で洗浄した。室温での真空乾燥により、デオキシコール酸(DCA、400mg、91%収率)を白色固体として得た。表29は、生成物の計測された特性を記載している。
【0215】
【表29】
(実施例25)
初代ヒト脂肪細胞を、出発材料として9−HADを使用して合成された様々な濃度の合成デオキシコール酸ナトリウムまたは以下に記載される通りのSigmaから得られたウシ由来デオキシコール酸ナトリウムとともにインキュベートした。
【0216】
材料
脂肪細胞(Zen−Bioカタログ番号SA−1096)
96ウェルプレート(US Scientificカタログ番号セルスター655180番)
無血清RPMI培地(Mediatechカタログ番号17−105−CV)
デオキシコール酸ナトリウム(DC)(Sigmaカタログ番号D6750)
合成グリコデオキシコール酸ナトリウム(Kythera)
PBS(1×)
MTSアッセイキット(Promegaカタログ番号G3580)
到着した脂肪細胞は分化しており、96ウェルプレート中、1ウェル当たり13,000細胞の密度であった。2枚のプレートを入手し、それぞれ同じ試料で処理した。細胞を、37℃、5%CO2で24時間インキュベートした。20mgを2mL培地(無血清)に溶解して各胆汁酸(合成および非合成DCA)の1%原液を作製した。この1%原液を使用して、希釈により下記の11種の溶液を調製した:0.005%、0.01%、0.015%、0.02%、0.025%、0.03%、0.035%、0.04%、0.05%、0.06%および0.1%、ならびに0%(培地のみ)。
【0217】
細胞を150μLの室温1×PBS(リン酸緩衝生理食塩水)で2回洗浄した。プレートを上下逆にし、液体を容器にデカントすることにより、培地、その後PBSを96ウェルプレート中のウェルから除去した。最後のPBS洗浄後、1ウェル当たり80μLの試料を添加した。各濃度の特定の胆汁酸を8つのウェルに添加し、37℃、5%CO2で1時間インキュベートした。その後、プレートをインキュベーターから取り出し、溶液をデカントした。希釈した(1mLのRPMI中40μL)MTS試薬(3−(4,5−ジメチルチアゾール−2−イル)−5−(3−カルボキシメトキシフェニル)−2−(4−スルホフェニル)−2H−テトラゾリウム、内塩)の100μL溶液を、各ウェルに直接添加した。対照(胆汁酸なし)ウェルが橙褐色に変色するまで、プレートを37℃、5%CO2でインキュベートし、その後、96ウェルプレートを分析する分光光度計に充填した。試料は、490nm波長設定で操作した。
【0218】
細胞の生存は、Promega製の比色分析アッセイ(MTS)キットを使用して評価した。結果は、合成NaDCまたはSigma製NaDCのいずれも処理時における、細胞生存の用量依存的な減少を示す(図2を参照)。両分子は、本実験において類似の細胞溶解挙動を実証し、合成NaDCおよびウシ由来のSigma製NaDCが脂肪細胞を死滅させるそれらの能力に関して、機能的に同一であることを示している。
【0219】
上述した実施形態および実施例は、本発明を限定することを意図するものではない。本発明の原理に従って、数々の修正形態および変形形態が可能であることを理解すべきである。
本発明の好ましい実施形態においては、以下が提供される。
(項目1)
デオキシコール酸(DCA)を自然に産生する哺乳動物または微生物生命体から単離されるものではない、DCAである化合物もしくはそのプロドラッグ、または前記化合物もしくは前記プロドラッグの薬学的に許容される塩:
【化50】
。
(項目2)
項目1に記載の化合物および薬学的に許容される賦形剤を含む、組成物。
(項目3)
神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分をさらに含む、項目2に記載の組成物。
(項目4)
前記神経ペプチドY(NPY)受容体アンタゴニストが、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、ジヒドロピリジン系NPY受容体アンタゴニスト、ジヒドロピリジン誘導体NPY受容体アンタゴニスト、二環化合物NPY受容体アンタゴニスト、カルバゾール系NPY受容体アンタゴニストおよび三環化合物NPY受容体アンタゴニストからなる群から選択される、項目3に記載の組成物。
(項目5)
前記脂肪選択的プロアポトーシスペプチドが、白色脂肪血管系にあるCKGGRAKDCペプチドである、項目3に記載の組成物。
(項目6)
哺乳動物において選択した位置から脂肪沈着物を除去する方法であって、それを必要とする前記哺乳動物に、治療有効量のDCAもしくはそのプロドラッグ、または前記DCAもしくは前記プロドラッグの薬学的に許容される塩:
【化51】
を投与するステップを含む方法であり、前記化合物はDCAを自然に産生する哺乳動物または微生物生命体から単離されるものではない方法。
(項目7)
前記哺乳動物に、神経ペプチドY(NPY)受容体アンタゴニストおよび脂肪選択的プロアポトーシスペプチドからなる群から選択される少なくとも1つの追加の活性成分を投与するステップをさらに含む、項目6に記載の方法。
(項目8)
前記神経ペプチドY(NPY)受容体アンタゴニストが、BIBP−3226、神経ペプチドY5アンタゴニスト(AmgenのNPY受容体アンタゴニスト)、BIBO−3304(Boehringer IngleheimのNPY受容体アンタゴニスト)、BMS−192548(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、AR−H040922(Bristol−Myers SquibbのNPY受容体アンタゴニスト)、LY−357897(Eli LillyのNPY受容体アンタゴニスト)、Esteve NPY−Y5受容体アンタゴニスト、1229U91(GlaxoSmithKlineのNPY受容体アンタゴニスト)、GW438014S(GlaxoSmithKlineのNPY受容体アンタゴニスト)、JNJ−5207787(Johnson&JohnsonのNPY受容体アンタゴニスト)、Lu−AA−44608(LundbeckのNPY受容体アンタゴニスト)、MK−0557(MerckのNPY受容体アンタゴニスト)、NGD−95−1(NeurgogenのNPY受容体アンタゴニスト)、NLX−E201(NeurologixのNPY受容体アンタゴニスト)、CGP−71683(NovartisのNPY受容体アンタゴニスト)、PD−160170(PfizerのNPY受容体アンタゴニスト)、SR−120819A(Sanofi AventisのNPY受容体アンタゴニスト)、BIIE0246(Sanofi AventisのNPY受容体アンタゴニスト)、S.A.0204(Sanofi AventisのNPY受容体アンタゴニスト)、S−2367(ShiongliのNPY受容体アンタゴニスト)、ジヒドロピリジン系NPY受容体アンタゴニスト、ジヒドロピリジン誘導体NPY受容体アンタゴニスト、二環化合物NPY受容体アンタゴニスト、カルバゾール系NPY受容体アンタゴニストおよび三環化合物NPY受容体アンタゴニストからなる群から選択される、項目7に記載の方法。
(項目9)
脂肪選択的プロアポトーシスペプチドが、白色脂肪血管系にあるCKGGRAKDCペプチドである、項目7に記載の方法。
(項目10)
デオキシコール酸(DCA)もしくはそのエステル、または薬学的に許容されるその塩:
【化52】
を調製するための方法であって、
(a)9α−ヒドロキシアンドロスタ−4−エン−3,17−ジオン1を、水素化条件下、H2と反応させて、化合物2
【化53】
を形成するステップと、
(b)化合物2を酸と反応させて、化合物3
【化54】
を形成するステップと、
(c)化合物3を還元剤と反応させて、化合物4を4および5
【化55】
の混合物として形成するステップと、
(d)化合物4をオレフィン形成条件下で二炭素オレフィン化試薬と反応させて、化合物6
【化56】
を形成するステップと、
(e)化合物6を、式7の化合物[式中、Pは保護基である]
【化57】
に変換するステップと、
(f)式7の化合物をルイス酸の存在下でアルキルプロピオレートCH2CH2C(O)ORまたはアルキルアクリレートCH2=CHC(O)OR[式中、Rはアルキルである]と反応させて、式8の化合物[式中、Pは保護基であり、Rはアルキルであり、破線
【化58−1】
は単結合または二重結合である]
【化58−2】
を形成するステップと、
(g)式8の化合物を水素化条件下でH2と反応させて、式9の化合物[式中、Pは保護基であり、Rはアルキルである]
【化59】
を形成するステップと、
(h)式9の化合物を酸化剤と反応させて、式10の化合物[式中、Pは保護基であり、Rはアルキルである]
【化60】
を形成するステップと、
(i)式10の化合物を水素化条件下でH2と反応させて、式11の化合物[式中、Pは保護基であり、Rはアルキルである]
【化61】
を形成するステップと、
(j)式11の化合物を還元剤と反応させて、式12の化合物[式中、Pは保護基であり、Rはアルキルである]
【化62】
を形成するステップと、
(k)式12の化合物を脱保護条件に曝露してそのエステルを形成し、場合によって、適切な加水分解条件に曝露して、デオキシコール酸または薬学的に許容されるその塩を形成するステップと
を含む方法。
(項目11)
パート(a)の前記水素化条件がPd/C触媒を含む、項目10に記載の方法。
(項目12)
パート(b)の前記酸が鉱酸である、項目10に記載の方法。
(項目13)
前記鉱酸がH2SO4である、項目12に記載の方法。
(項目14)
パート(c)の前記還元剤がLiAl(OtBu)3Hである、項目10に記載の方法。
(項目15)
パート(d)の前記二炭素オレフィン化試薬がPh3PCH2CH3+Br−である、項目10に記載の方法。
(項目16)
化合物7〜12の前記保護基Pが−C(O)CH3である、項目10に記載の方法。
(項目17)
パート(f)の前記ルイス酸がEtAlCl2である、項目10に記載の方法。
(項目18)
前記アルキルプロピオレートまたはアルキルアクリレートがメチルプロピオレートまたはメチルアクリレートである、項目10に記載の方法。
(項目19)
パート(g)の前記水素化条件がPtO2触媒を含む、項目10に記載の方法。
(項目20)
パート(h)の前記酸化剤がCrO3である、項目10に記載の方法。
(項目21)
パート(i)の前記水素化条件がPd/C触媒を含む、項目10に記載の方法。
(項目22)
パート(j)の前記還元剤がLiAl(OtBu)3Hである、項目10に記載の方法。
(項目23)
Pが−C(O)CH3である場合、パート(k)の前記脱保護および加水分解条件が、化合物12を、アルカリ土類水酸化物、アルカリ土類金属アルコキシドまたは両方の混合物と反応させることを含む、項目10に記載の方法。
(項目24)
前記アルカリ土類金属アルコキシドがLiOHである、項目23に記載の方法。
(項目25)
9α−ヒドロキシ−5β−アンドロスタン−3,17−ジオン(2);
5β−アンドロスタ−9(11)−エン−3,17−ジオン(3);
(Z)−3α−ヒドロキシ−5β−プレグナ−9(11),17(20)−ジエン(6);
(Z)−3α−アセトキシ−5β−プレグナ−9(11),17(20)−ジエン(7a);
(E)−メチル3α−アセトキシ−5β−コール−9(11),16,22−トリエン−24−オエート(8a);
メチル3α−アセトキシ−5β−コール−9(11),16−ジエン−24−オエート(8b);
メチル3α−ヒドロキシ−5β−コール−9(11)−エン−12−オン−24−オエート(10a);および
メチル3α−アセトキシ−5β−コラン−12−オン−24−オエート(11a)
からなる群から選択される、中間化合物。
【特許請求の範囲】
【請求項1】
本願明細書または図面に記載の発明。
【請求項1】
本願明細書または図面に記載の発明。
【図1】

【図2】
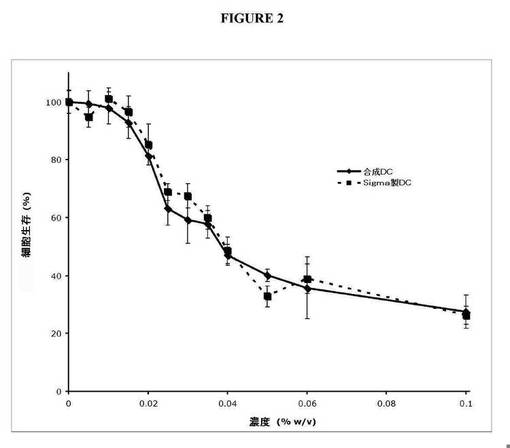
【図2】
【公開番号】特開2013−75918(P2013−75918A)
【公開日】平成25年4月25日(2013.4.25)
【国際特許分類】
【外国語出願】
【出願番号】特願2013−7521(P2013−7521)
【出願日】平成25年1月18日(2013.1.18)
【分割の表示】特願2010−513387(P2010−513387)の分割
【原出願日】平成20年6月18日(2008.6.18)
【出願人】(508276419)カイセラ バイオファーマシューティカルズ, インコーポレイテッド (4)
【Fターム(参考)】
【公開日】平成25年4月25日(2013.4.25)
【国際特許分類】
【出願番号】特願2013−7521(P2013−7521)
【出願日】平成25年1月18日(2013.1.18)
【分割の表示】特願2010−513387(P2010−513387)の分割
【原出願日】平成20年6月18日(2008.6.18)
【出願人】(508276419)カイセラ バイオファーマシューティカルズ, インコーポレイテッド (4)
【Fターム(参考)】
[ Back to top ]