
抗CD37抗体
【課題】B細胞悪性疾患及びCD37陽性B細胞の枯渇に応答する他の疾患の治療を目的とする新規なCD37アンタゴニスト、また、改善されたエフェクター機能を有する抗CD37抗体を提供する。
【解決手段】ヒトCD37と結合し、a)i)特定のアミノ酸配列を含む可変重鎖、及びii)特定のアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、又はb)i)特定のアミノ酸配列を可変重鎖内に含むCDR;ii)特定のアミノ酸配列を可変軽鎖内に含むCDR;iii)ヒト抗体から誘導される、前記CDRを支持するフレームワーク;iv)ヒト抗体に由来する定常重鎖及び軽鎖、によって規定されるヒト化抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体。
【解決手段】ヒトCD37と結合し、a)i)特定のアミノ酸配列を含む可変重鎖、及びii)特定のアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、又はb)i)特定のアミノ酸配列を可変重鎖内に含むCDR;ii)特定のアミノ酸配列を可変軽鎖内に含むCDR;iii)ヒト抗体から誘導される、前記CDRを支持するフレームワーク;iv)ヒト抗体に由来する定常重鎖及び軽鎖、によって規定されるヒト化抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、B細胞枯渇を基礎とする免疫療法に関する。特に本発明は、そのような療法、例えばB細胞悪性疾患及び自己免疫症状の治療で使用される抗CD37抗体分子に関する。
【背景技術】
【0002】
モノクローナル抗体(mAb)を用いる免疫療法は、B細胞枯渇を原理とする、癌及び他の疾患の治療を目的とする安全で選択性を示す方法として出現した。特にB細胞枯渇を原理とする療法、例えばB細胞悪性疾患の治療におけるモノクローナル抗体の役割は、リツキシマブ(Rituxan(商標))(B細胞表面のCD20抗原に対して誘導された抗体)の導入以来拡大した。多くの研究によって、単剤療法及び併用療法としてのリツキシマブの有効性が以下の疾患で確認された:低度NHL(Hiddemann et al. 2005a;Hiddemann et al. 2005b;Hainsworth 2004;McLaughlin et al. 1998)、マントル細胞リンパ腫(Forstpointner et al. 2004;Kahl et al. 2006;Foran et al. 2000;Howard et al. 2002;Romaguera et al. 2005)、びまん性大細胞型リンパ腫(DLCL)(Coiffier et al. 1998;Feugier et al. 2005)、及びバーキット白血病/リンパ腫(Thomas et al. 2006)。しかしながら一部分の患者のみが治療に応答し、大半の患者はリツキシマブ治療後に最終的には再発する。したがって、B細胞悪性疾患の治療に向けてCD20よりも潜在的に有効なB細胞上の新規な治療薬標的が探索されてきた(Zhao et al. 2007)。CD37抗原は細胞表面抗原であり、今日までB細胞悪性疾患のための標的としてB細胞抗原CD20と同じ程度では考慮されてこなかった。
CD37(テトラスパニンスーパーファミリーのメンバーである)は、高度にグリコシル化された細胞表面分子であり、4つのトランスメンブレンドメイン及び2つの細胞外ループを有する。CD37はほぼ例外なく成熟B細胞上で発現され、抹消血B細胞では最高レベルで、プラズマ細胞では低レベルで、さらに骨髄中のCD10+ B前駆細胞では検出不能レベルで発現される。CD37の低レベル発現はまた、休止及び活性化T細胞、顆粒球並びに単球で報告されている。B細胞新形成では、CD37発現は、主として急速進行性非ホジキンリンパ腫(NHL)及び慢性リンパ性白血病(CCL)で観察される。高レベルのCD37発現はまたマントル細胞リンパ腫(MCL)で見出される。この発現パターンによりCD37は抗体仲介癌療法の有望な標的となっている。
【0003】
CD37は1986年に初めて記載され、ネズミのモノクローナル抗体MB-1によって性状が決定された(Link et al. 1986)。
CD37の生理学的役割は不明である。CD37欠損マウスは、リンパ性器官の発育及び細胞構成に変化を示さないが、IgG1レベルを低下させ、T細胞媒介免疫反応を弱める(Knobeloch et al. 2000)。CD37-/-T細胞に関する研究によって、T細胞増殖におけるCD37の役割が示唆されている(van Spriel et al. 2004)。
種々の疾患で悪性B細胞上におけるCD37の発現が報告されている。CD37は、バーキットリンパ腫、濾胞性リンパ腫及びリンパ球性リンパ腫のような大半の成熟B細胞性悪性疾患で発現される(Link et al. 1986)。高レベルのCD37発現が、毛様細胞白血病において並びに慢性リンパ球性白血病(CLL)及び種々の非ホジキン型リンパ腫(NHL)(マントル細胞リンパ腫(MCL)を含む)患者のサンプルにおいて観察されている(Schwartz-Albiez et al. 1988;Barrena et al. 2005)。免疫表現型について抗体マイクロアレイを利用したある報告では、CD37は、悪性CCL細胞(CD37高発現)と正常抹消血(PB)リンパ球(CD37低発現)との間の良好な識別物質であると主張されている(Beloz et al. 2001)。
CD37特異的mAbと癌細胞との結合は多様な作用メカニズムの引き金となりえる:第一に、抗体がCD37抗原の細胞外ドメインと結合した後、補体カスケードを活性化し、標的細胞を溶解させることができる。第二に、抗CD37抗体は、標的細胞に対する抗体依存細胞媒介性細胞傷害(ADCC)を仲介することができる(ADCCは免疫系の細胞傷害性細胞上の適切なレセプターによる結合抗体のFc部分の認識後に生じる)。
第三に、抗体は、抗原又は他の刺激に対して応答するB細胞の能力を変化させることができる。最後に抗CD37抗体はプログラムされた細胞死(アポトーシス)を開始させることができる。
【0004】
抗CD37 mAbのMB-1は、B-NHL(B細胞非ホジキンリンパ腫)患者について2つの放射線免疫療法の臨床試験で評価された(Press et al. 1989;Kaminski et al. 1992)。一方の臨床試験で、治療用量の131I-MB-1が6人のNHL再発患者に投与され、6人の患者全員が、平均期間が7ヶ月の臨床的に完全な緩解(CR)を達成した。重要なことには、6人の患者のうち2人が、ほんの微量用量のMB-1の投与後に既に臨床徴候の沈静を示し、抗体自体の直接的な抗腫瘍作用を示唆した。第二の臨床試験では、放射能標識MB-1を難治性NHL患者の治療に適用し、9人の評価可能な患者のうち3人が限定的期間において客観的応答を示した(Kaminski et al. 1992)。両臨床試験において、微量標識MB-1抗体の注射後に急速で一過性の抹消B細胞の枯渇が報告された。これらの観察は、MB-1は細胞傷害性活性をそれ自体有するという結論を支持した。要約すれば、これらの臨床試験は、B細胞性悪性疾患に対するCD37ターゲティングの有効性を強調し、抗CD37療法の潜在的な臨床応用性を指摘している。
CD37特異抗体類似単鎖分子(“小モジュール免疫薬(Small Modular ImmunoPharmaceutical, SMIP)”)を用いて、当該分子による治療はin vitroでアポトーシスを誘発し、さらにin vivoの異種移植モデルでバーキットリンパ腫の増殖を遅らせるという実験的証拠が得られている。Trubion社の組換え抗CD37 SMIP Tru16.4の抗アポトーシス活性が最近報告された(Zhao et al. 2004)。Tru 16.4は、腫瘍患者由来の原発性CLL細胞に対してカスパーゼ非依存性アポトーシスを誘発した。これら細胞におけるアポトーシスの誘導はリツキシマブのそれよりも強く、さらにアレムツズマブ(CD52アンタゴニスト)のアポトーシス誘発に匹敵した。アポトーシス誘発の程度はCD37の細胞表面発現に正比例し、抗ヒトIgG抗体による架橋によってさらに強化することができた。CD37発現とADCCとの相関性が細胞株においてin vitroで示された。バーキットリンパ腫マウスモデル(Raji)で、抗CD37 scFvによる処置は治療有効性を示した(Zhao et al. 2007)。これらのデータは、CD37ターゲティングは、アポトーシス及びADCCの誘導による標的誘導抗腫瘍療法のための有望なアプローチであるという最初の証拠を提供する。
結論として、CD37抗原は、いくつかのヒトB細胞悪性疾患の腫瘍細胞及び正常な成熟B細胞で頻繁に発現され、さらに抗CD37系療法はB細胞悪性疾患の治療のための有望なアプローチでありえることが示された。CD37陽性正常B細胞の枯渇は重大であるとは考えられない。なぜならば、多数の患者の臨床データが、抗CD20 mAbによる6ヶ月までのB細胞の長期的枯渇でさえ、IgG血清レベルを顕著には低下させず、感染リスクを増大させることもないことを示しているからである(Van der Kolk et al. 2002)。
上記に記載の抗CD37抗体又は抗体様分子(MB-1及びSMIP Tru16.4)は、B細胞悪性疾患における抗腫瘍有効性及びCD37を標的とする潜在能力を示したが、B細胞枯渇に基づく療法を改善するためにまた別の抗CD37インヒビターが希求される。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、B細胞悪性疾患及びCD37陽性B細胞の枯渇に応答する他の疾患の治療を目的とする新規なCD37アンタゴニストを提供することであった。
さらにまた本発明の目的は、改善されたエフェクター機能を有する抗CD37抗体を提供することであった。特に、本発明者らは、抗体依存細胞媒介性細胞傷害(ADCC)を有する抗CD37 mAbを提供しようとした。
【課題を解決するための手段】
【0006】
本発明の根幹に横たわる問題を解決するために、出発抗体として、ネズミモノクローナル抗CD37抗体をヒトの治療に有用なキメラ及びヒト化抗CD37抗体を作製するために用いた。
第一の特徴では、本発明は、ヒトCD37と結合し、さらに
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定されるネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体を提供する。
【図面の簡単な説明】
【0007】
【図1】キメラ抗体A0はCD37抗原を特異的に認識する(FACS競合アッセイによって決定)。
【図2】細胞CD37抗原とヒト化型A0との結合(FACSによって決定)。
【図3】細胞CD37抗原とヒト化型A0との結合(FACSによって決定)。
【図4】細胞CD37抗原に対するヒト化型A0の親和性(FACSスキャチャードによって決定)。
【図5】ヒト化型A0のRamos細胞に対するADCC活性。
【図6】ヒト化型A0のRamos細胞に対するアポトーシス促進活性。
【図7】Fc-操作型mAb A0のRamos細胞に対するADCC活性。
【図8】Fc-操作型mAb B0のRamos細胞に対するADCC活性。
【図9】mAb A0及びB0のアポトーシス促進活性。
【図10】Fc-操作型mAb A0のアポトーシス促進活性。
【図11A】リツキシマブと比較した、全血アッセイにおけるFc-操作抗体A2及びB2による正常ヒトB細胞の枯渇。
【図11B】リツキシマブと比較した、Fc-操作後の抗体の優れたB細胞枯渇活性。
【図11C】抗体A2及びB2は、全血アッセイでT細胞及び単球を枯渇させない。
【図12】リツキシマブと比較した、Fc-操作後の優れたADCC活性。
【図13】リツキシマブと比較した、全血アッセイにおけるFc-操作抗体A2及びB2によるRamosバーキットリンパ腫細胞の枯渇。
【図14】ヌードマウスにおけるFc-操作抗体A2及びB2によるRamos異種移植腫瘍のin vivo腫瘍増殖阻害。
【図15】多発性ミエローマ細胞におけるCD37の発現。
【図16】抗体A2及びB2の多発性ミエローマ細胞に対するADCC活性。
【図17】抗体A2及びB2の患者由来CLL細胞に対するアポトーシス促進活性。
【発明を実施するための形態】
【0008】
上述したように、本発明の根幹に横たわる問題を解決するために、出発抗体として、ネズミモノクローナル抗CD37抗体をヒトの治療に有用なキメラ及びヒト化抗CD37抗体を作製するために用いた。
第一の特徴では、本発明は、ヒトCD37と結合し、さらに
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定されるネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体を提供する。
以下から理解されるように、別の抗体(すなわち出発抗体)から“誘導された”抗体とは、抗体が、以下に記載するように出発抗体を改変することにより作製されることを意味する。
好ましい実施態様では、抗体分子は、a)に規定した出発抗体から誘導されたキメラ又はヒト化抗体分子である。関連配列を有する抗体はG28.1と称され、WO2005/017148に開示された。
【0009】
カテゴリーb)の出発抗体は、例えば、第三回HLDAワークショップでCD37抗原を性状決定したG28.1のようなCD37特異抗体から選択することができる。これらの抗体はHD28、HH1、BI14、F97-3G6と称された(Ling and MacLennam, 1987)。報告されている他のCD37特異抗体には、RFB-7、Y29/55、MB-1、M-B371、M-B372及びIPO-24が含まれる。Moldenhauer(2000)及びSchwartz-Albiezら(1988)によれば、これらの抗体はいずれも(G28.1を含む)、同じ又は近接するか若しくはオーバーラップするCD37エピトープを認識する。Schwartz-Albiezら(1988)は、このエピトープはCD37の炭水化物部分に位置することを指摘している。上記の多数の抗体が市場で入手可能である(例えばHH1(SantaCruz)、RFB-7(Biodesign)、Y29/55(Biogenesis)、M-B371(BD Biosciences)、M-B372(SantaCruz)及びIPO-24(AbCam))。
他のCD37特異抗体は、S-B3(Biosys)、NMN46(Chemicon)及びICO-66(Bioprobe)である。抗体がG28.1と同じエピトープを認識するか否かは、競合結合アッセイによって、又は交差阻害放射性免疫アッセイ(Moldenhauerら(1987)及びMoldenhauer(2000)により記載された)によって決定することができる。
例示すれば、競合結合は、CD37タンパク質若しくはCD37ペプチドを被覆したプレート、又はCD37陽性細胞を用い、競合候補抗体の存在下でビオチン化抗体の結合を測定することによってELISAで決定することができる。競合性抗体又は抗体誘導フラグメントの存在下では、ビオチン化G28.1(又は同じエピトープを認識することが判明している別の抗体)の結合は、抗体が共通エピトープを認識する場合には低下する。G28.1エピトープペプチドを識別するために、CD37配列から誘導されたフラグメント若しくは短いポリペプチド又は組換えタンパク質を合成するか又は生成し、G28.1と前記ペプチド/ポリペプチドとの結合をELISAアッセイで測定することができる。競合結合はまた、実施例で述べるようにFACS分析によって決定することができる。
【0010】
キメラ又はヒト化抗体分子の生成のための出発抗体としてb)に規定する抗体を、G28.1と同様な態様で用いることができる。
カテゴリーb)の出発抗体はまた、対応するエピトープを含むペプチド又はタンパク質フラグメント又はそのようなペプチド/フラグメントをコードするDNA分子をそれぞれ免疫に用い、G28.1と同じエピトープと反応する抗体を得ることによって、de novoに作製することができる。
出発抗体b)はまた、関連エピトープを保持する全細胞で免疫することによって得ることができる。このようにして得たハイブリドーマ細胞を続いて分泌抗体の競合性結合についてスクリーニングする。
“抗CD37抗体分子”という用語は、抗CD37抗体及び抗CD37抗体フラグメントとともに抗体分子とのコンジュゲートを包含する。本発明の意味では、抗体にはキメラモノクローナル抗体及びヒト化抗体が含まれる。“抗体”という用語(前記は“抗体分子”と相互に用いられる)は、完全な免疫グロブリン(リンパ球によって生成され、例えば血清中に存在する)、ハイブリドーマ細胞株によって分泌されるモノクローナル抗体、宿主細胞で組換え発現によって生成されるポリペプチド(免疫グロブリン又はモノクローナル抗体の結合特異性を有する)、並びに改変及び更なるプロセッシングによって前記のような抗体から誘導され、なおそれらの結合特異性を維持している分子を包含するであろう。
本発明のある実施態様では、抗CD37抗体分子は、
i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、
ii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖、
iii)ヒト起源である定常重鎖及び軽鎖、
によって規定されるキメラ抗体である。
【0011】
キメラマウス/ヒト抗体の構築及び生成は当分野では周知である(Boulianne et al. 1984)。非ヒト抗体の可変領域は、典型的にはヒト免疫グロブリンの免疫グロブリン定常領域の少なくとも一部分(Fc)と連結される。ヒト定常領域DNA配列は、周知の方法にしたがって多様なヒト細胞、好ましくは不朽化B細胞から単離することができる(以下を参照されたい:Kabat et al. 1991;及びWO87/02671)。抗体分子は、定常領域の全部又は一部分のみを、それら分子がCD37抗原及びFcレセプターとの特異的結合を示すかぎり含むことができる。定常領域の型及び長さの選択は、補体結合又は抗体依存細胞媒介性細胞傷害のようなエフェクター機能が所望されるか否か、及び抗体分子の所望される薬理学的特性に左右される。
ある種の実施態様では、本発明の抗体分子は、ヒト重鎖定常領域IgG1と融合したa)又はb)に規定される非ヒト抗体の重鎖可変領域、及びヒト軽鎖定常領域カッパと融合したa)又はb)に規定される非ヒト抗体の軽鎖可変領域を有するキメラCD37特異抗体である。
さらに別の実施態様では、本抗体分子は、ヒト重鎖定常領域IgG1(配列番号:24(コードDNA配列は配列番号:23)に示される配列を有するIgG1分子)又は前記から誘導された変異IgG1と融合した配列番号:2に示される重鎖可変領域を有し、さらに配列番号:26(コードDNA配列は配列番号:25)に示されるヒト軽鎖定常領域カッパと融合した配列番号:4に示される軽鎖可変領域を有するキメラCD37特異抗体である。
a)又はb)に規定される非ヒト出発抗体をキメラ化するための他のヒト定常領域も当業者には利用可能であり、例えばIgG2、IgG3、IgG4、IgA、IgE若しくはIgM(IgG1の代わり)又はラムダ(カッパの代わり)である。定常領域もまたキメラであってもよく、例えば重鎖IgG1/IgG2又はIgG1/IgG3キメラである。
【0012】
本発明のある種の実施態様では、抗CD37抗体分子は、
i)配列番号:2に示される可変重鎖内に含まれるCDR、
ii)配列番号:4に示される可変軽鎖内に含まれるCDR、
iii)ヒト抗体から誘導される、前記CDRを支えるフレームワーク、
iv)ヒト抗体に由来する定常重鎖及び軽鎖、
によって規定されるヒト化抗体である。
非ヒト抗体(例えばネズミ、ラット、又はウサギ抗体)のヒト化型は、非ヒト免疫グロブリンから誘導された最小限の配列を含む免疫グロブリン、免疫グロブリン鎖又はそのフラグメント(例えばFv、Fab、Fab’、F(ab’)2、又は抗体の部分配列を有する他の抗原結合分子)である。
ヒト化抗体は、レシピエント抗体の相補性決定領域が、望ましい特異性、親和性及び性能を有する非ヒト種(ドナー抗体)、例えばマウス、ラット又はウサギのCDR由来残基によって置き換えられた(レシピエント抗体由来)ヒト免疫グロブリンを含む。いくつかの事例では、ヒト免疫グロブリンのFvフレームワーク残基は対応する非ヒト残基によって置き換えられる。
本発明のヒト化抗体では、a)又はb)に規定される非ヒト出発抗体のCDRをコードする配列は、ヒト免疫グロブリンの重鎖及び軽鎖の対応する遺伝子に移植された。
モノクローナル抗体の“相補性決定領域”(CDR)は、Chothia and Lesk(1987)の論文に関連するKabatら(1991)の論文にしたがえば、特異的な抗原結合に中心的に関与するアミノ酸配列であると理解される。配列の特徴についてKabatの配列データベースを検索することによって、配列番号:2及び配列番号:4に示される可変領域の配列からCDR配列を日常的に決定することができる。
ヒト化抗体を得るための技術は当業者には日常的に利用可能であり、それら技術はとりわけUS5,225,539、US6,548,640及びUS6,982,321に記載されている。
【0013】
CDR移植抗体の適切なフレームワーク残基をネズミ残基に戻して、結合親和性を改善することができる。上記に記載したように、当業者は、与えられた非ヒト抗体からCDRの入手の仕方、適切なヒト免疫グロブリン遺伝子の選択及び入手の仕方、これら遺伝子へのCDRの移植の仕方、選択したフレームワークの改変の仕方、CDR移植抗体の適切な宿主細胞(例えばチャイニーズハムスター卵巣(CHO)細胞)での発現の仕方、及び得られた組換え抗体の結合親和性及び特異性の試験の仕方を当分野の関係する方法から知ることができる。
ヒト化抗体を得るために、抗原結合部位(重鎖のCDR及び軽鎖のCDRによって形成される)を、げっ歯類(ネズミ)のモノクローナル抗体を分泌する細胞のDNAから切り出し、ヒト抗体のフレームワークをコードするDNAに移植する。
CDR移植の代わりに、非ヒト(特にネズミ)抗CD37抗体は、いわゆる“表面再仕上げ(resurfacing)”技術によってヒト化することができる。前記技術によって、げっ歯類フレームワーク領域は、US5,639,641に記載されたように、表面露出残基を除き変化を受けない。
さらに別の特徴では、本発明は、配列番号:6に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
別の特徴では、本発明は、配列番号:8に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
別の特徴では、本発明は、配列番号:10に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
上記に規定したヒト化抗体は表1に示される。
【0014】
ある種の実施態様では、ヒト化抗体は、ヒト重鎖定常領域IgG1及びヒト軽鎖定常領域カッパを有する。キメラ抗体について上記で述べたように、定常領域は他のクラス及びサブクラスから選択してもよい。
ある種の実施態様では、本発明のヒト化抗体では、ヒト定常重鎖IgG1は配列番号:24に示される配列を有するIgG1分子又は前記から誘導された変異IgG1分子であり、ヒト軽鎖定常領域カッパは配列番号:26に示される配列を有する。
本発明の抗CD37抗体分子はまた、配列表に示されるアミノ酸配列によって規定される抗体の変種であってもよい。日常的に利用可能な技術を用いて、当業者は、上記に規定の抗体の機能的変種を調製、試験及び利用することができるであろう。前記の例は、CDR及び/又はフレームワーク内の少なくとも1つの位置が変異した変種抗体、フレームワーク領域内に生殖系列配列から逸脱したただ1つのアミノ酸置換を有する変種抗体、保存的アミノ酸置換を有する抗体、配列表に提示されている可変鎖をコードするDNA分子とストリンジェントな条件下でハイブリダイズするDNA分子によってコードされる抗体である。
個々のアミノ酸の特性が提供されるならば、合理的な置換を実施して、出発抗体の全体的な分子構造を保存した抗体変種を得ることができる。アミノ酸置換、すなわち“保存的置換”は、対応するアミノ酸の例えば極性、荷電、可溶性、疎水性、親水性及び/又は両親媒性を基準にして実施することができる。当業者は、一般的に実施されるアミノ酸置換(例えばWO2007/042309に記載されている)及びそのように改変された抗体を得る方法を熟知していよう。遺伝暗号並びに組換え及び合成DNA技術を提供されるならば、1つ又は2つ以上の保存的アミノ酸交換を有する変種抗体をコードするDNA分子を日常的に設計して、対応する抗体を容易に得ることができる。
【0015】
配列表に示されるその可変鎖によって規定される抗体と比較して、本発明に包含される抗体変種は、CDR領域に少なくとも60%、より好ましくは少なくとも70%又は80%、さらに好ましくは少なくとも90%、もっとも好ましくは少なくとも95%の配列同一性を有する。好ましい抗体はまた、CDR領域に少なくとも80%、より好ましくは90%、もっとも好ましくは95%の配列類似性を有する。好ましい抗体変種は、可変領域に少なくとも60%、より好ましくは少なくとも70%又は80%、さらに好ましくは少なくとも90%、もっとも好ましくは少なくとも95%の配列同一性を有する。好ましい抗体はまた、可変領域に少なくとも80%、より好ましくは90%、もっとも好ましくは95%の配列類似性を有する。
2つのポリペプチド間における“配列同一性”は、当該配列間で同一であるアミノ酸のパーセンテージを示す。“配列類似性”は、同一であるか又は保存的アミノ酸置換を示すアミノ酸のパーセンテージを示す。
変種はまた、配列表に示される規定の配列を有する抗体を最適化のための出発点として用い、1つ又は2つ以上のアミノ酸残基(好ましくは1つ又は2つ以上のCDR内のアミノ酸残基)を多様化させることによって、さらに得られた抗体変種の収集物を改善された特性を有する変種についてスクリーニングすることによって入手することができる。可変軽鎖のCDR3、可変重鎖のCDR3、可変軽鎖のCDR1及び/又は可変重鎖のCDR2内の1つ又は2つ以上のアミノ酸残基の多様化が有用であることが実証された。多様化は、当分野で公知の方法、WO2007/042309に言及されている例えばいわゆるTRIM技術によって実施することができる。
【0016】
さらに別の実施態様では、本発明の抗CD37抗体分子は“アフィニティ成熟”抗体である。
“アフィニティ成熟”抗CD37抗体は、配列表に示された配列を有する抗体から誘導される抗CD37抗体であり、1つ又は2つ以上のCDR内に1つ又は2つ以上の変異を有し、それにより対応する原型の非成熟抗体と比較して抗原に対する親和性の改善がもたらされる。そのような抗体の変異体を作製する方法の1つはファージディスプレイを必要とする(Hawkins et al. 1992;及びLowman et al. 1991)。略記すれば、いくつかの超可変領域部位(例えば6−7部位)を変異させて、各部位で全ての可能なアミノ酸置換を作製する。このようにして作製した抗体の変異体を、繊維状ファージから各粒子内にパッケージされたM13の遺伝子III生成物との融合物として一価の態様でディスプレイさせる。続いてその生物学的活性(例えば結合親和性)について、このファージディスプレイ変異体を本明細書に開示するようにスクリーニングする。
アフィニティ成熟抗体はまた、例えばMarksら(1992)(可変重鎖(VH)及び可変軽鎖(VL)ドメインシャッフリングによるアフィニティ成熟)、又はBarbasら(1994)、Shierら(1995)、Yeltonら(1995)、Jacksonら(1995)及びHawkinsら(1992)(CDR及び/又はフレームワーク残基のランダム変異導入)が記載した方法によって作製することができる。好ましいアフィニティ成熟抗体は標的抗原に対してナノモルレベル又は場合によってピコモルレベルの親和性を有するであろう。さらに別の実施態様では、本発明の抗CD37抗体分子は“脱免疫化”抗体である。
“脱免疫化”抗CD37抗体は、配列表に示された配列を有するヒト化又はキメラ抗体から誘導される抗体であり、そのアミノ酸配列内に1つ又は2つ以上の変異を有し、それにより対応する原型の脱免疫されていない抗体と比較して抗体の免疫原性の低下がもたらされる。そのような抗体変異体を作製する方法の1つは、抗体分子のT細胞エピトープの識別及び除去を必要とする(Baker and Jones, 2007)。第一の工程では、抗体の免疫原性をいくつかの方法によって、例えばin vitro T細胞エピトープ決定又はそのようなエピトープのin silico予想によって、文献に記載されているように(Jones et al. 2004;Jones et al. 2005;Reche et al. 2004;Hertz et al. 2006)決定することができる。いったんT細胞エピトープ機能に決定的な残基を識別したら、免疫原性を除去し抗体活性を維持するために変異を導入することができる(Jones et al. 2005;Tangri et al. 2005)。タンパク質における変異導入方法は当分野で周知であり、例えばオーバーラップPCR技術によって実施される。
【0017】
抗体のFc領域は多数のFcレセプターと相互作用するので(その相互作用の結果、多数の重要な機能的能力(“エフェクター機能”と呼ばれる)が生じる)、ある種の実施態様では、抗体は、完全長抗体又はFc領域の一部分を含む抗体である(ただし後者は抗原の対応する部分及びFcレセプターとの特異的な結合を示す必要がある)。定常領域のタイプ及び長さの選択は、補体の結合又は抗体依存細胞媒介性細胞傷害のようなエフェクター機能が所望されるか否か及び抗体タンパク質の所望される薬理学的特性に左右される。
本発明のある実施態様では、抗CD37抗体は、Fc領域又はその対応する断片を含むキメラ又はヒト化抗体であり、前記Fc領域は、エフェクター機能を調節するため、特に1つ又は2つ以上のFcレセプターと抗体との結合を強化して、それによりADCCエフェクター機能を強化するために操作されている。Fc領域の操作は、Fc領域が操作されていない親抗体の場合よりもエフェクター細胞の存在下で抗体のエフェクター機能をより効率的に仲介する。ある実施態様では、そのような抗体変種は、親抗体によって仲介されるADCCよりも強力なADCCを仲介する(以下では、特段の記載がなければ、抗体分子の関係又はIgG若しくはFc領域の関係で“親”という用語は、それぞれ操作されていない抗体分子、Fc領域又はIgG(それらから変異した(操作された)分子が誘導される)を意味する。
Fc領域の多様な改変が、当分野で学術文献及び特許文書の両方で、例えばEP0307434、WO9304173、WO9734631、WO9744362、WO9805787、WO9943713、WO9951642、WO9958572、WO02060919、WO03074679、WO2004016750、WO2004029207、WO2004063351、WO2004074455、WO2004035752、WO2004099249、WO2005077981、WO2005092925、WO2006019447、WO2006031994、WO2006047350、WO2006053301、WO2006088494及びWO2007041635で提唱されている。
【0018】
好ましい実施態様では、本発明の抗体は、332位及び/又は239位及び/又は236位にアミノ酸置換を有するFc変種である。好ましい実施態様では、本発明の抗体は、以下の群から選択されるFcドメインの変異を有する:
i)332位におけるただ1つの置換、好ましくはI332E;
ii)239位及び332位における置換の組合せ、好ましくはS239D/I332E;
iii)236位及び332位における置換の組合せ、好ましくはG236A/I332E;
iv)236位、239位及び332位における置換の組合せ、好ましくはG236A/S239D/I332E。
上記に規定した置換は、Lazarら(2006)の文献、WO2004029207及びWO2007041635に記載されている。
本発明の抗体におけるFc変種は、それらを含むアミノ酸改変にしたがって規定される。したがって、例えばI332Eは、親のFcポリペプチドに対して置換I332Eを有するFc変種である。同様に、S239D/I332Eは、親のFcポリペプチドに対して置換S239D及びI332Eを有するFc変種を規定し、S239D/I332E/G236Aは、置換S239D、I332E及びG236Aを有するFc変種を規定する。
番号付与はEU番号付与体系(Kabat et al. 1991)にしたがい、前記はEU抗体(Edelman et al. 1969)の番号付与に該当する。当業者には理解されるところであるが、これらの取り決めは、免疫グロブリン配列の特別な領域における非連続的番号付与から成り、これによって免疫グロブリンファミリーにおける保存的な位置に対し標準化された参照が可能である。
上記に規定した抗体で、置換された236位、239位及び332位は、配列番号:24に示されるIgG1重鎖のそれぞれ119位、122位及び215位に対応する(配列番号:28、32、36及び40に示される抗体A2、A4、B2及びB4の重鎖の完全長配列では、置換されたアミノ酸は235位、238位及び331位に存在する)。
【0019】
ある種の実施態様では、本発明のFc領域はヒトIgG配列を土台にしており、したがってヒトIgG配列が、他の配列を比較する“基準”配列として用いられる。本発明の抗体のために、操作されるFc領域は好ましくはIgG、特にIgG1であるが、Fc領域はまた、IgG2若しくは他の免疫グロブリンクラス(例えばIgA、IgE、IgGD、IgM)由来の変種配列、又は2つ以上の免疫グロブリンクラスのキメラ型(例えばIgG2/IgG1)などであってもよい。本発明のFc変種は1つの親IgGにおいて操作されるが、この変種は別の第二の親IgGにおいて操作されても、又は前記第二の親IgGに“移され”てもよい。これは、“等価である”又は“対応する”残基を決定し、典型的には第一及び第二のIgGの配列間の配列相同性又は構造相同性を基準にしてこの第一及び第二のIgGとの間で置換を実施することによって達成される。相同性を確立するために、本明細書に概略する第一のIgGのアミノ酸配列を第二のIgGの配列と直接比較する。当分野で公知の1つ又は2つ以上の相同性アラインメントプログラムを用い、アラインメントを維持するため(すなわち任意の欠失及び挿入による保存残基の排除を回避するため)に必要な挿入及び欠失を可能にして配列のアラインメントを実施した後、第一のFc変種の一次配列における具体的なアミノ酸と等価の残基が規定される。決定された等価又は対応する残基が何であれ、さらにIgGが作製される親IgGの実体が何であれ、言わんとすることは、本発明で用いられるFc変種を操作して、Fc変種と顕著な配列又は構造相同性を有する任意の第二の親IgGを得ることができるということである。したがって、例えば、親抗体がヒトIgG1である変種抗体が、上記に記載の方法又は等価残基を決定する他の方法を用いることによって作製される場合、変種抗体は、例えばヒトIgG2親抗体、ヒトIgA親抗体で操作されえる(WO2007041635を参照されたい)。
【0020】
本発明の抗体は抗原CD37を標的とし、これは、CD37発現レベルがCD20の発現レベルより高い疾患(例えば慢性リンパ球性白血病、この疾患ではサンプルはCD20 mRNAの低発現レベルと比較してCD37 mRNAの高発現レベルを示す)でCD20を標的とするよりも有利でありえる。
本発明の抗体は、Ramos細胞に対するADCC活性、全血における正常B細胞枯渇及びRamosバーキットリンパ腫細胞枯渇という点についてリツキシマブ(公認された抗CD20抗体)よりも優れていることが示された。本発明の実験で示しえたように、本発明の抗体(Fc未操作及びFc操作の両抗体)は、リツキシマブのB細胞枯渇活性を超える活性を有する。変異Fc領域を有する抗体は、リツキシマブと比較してほぼ10倍高いB細胞枯渇活性を示す(図11B)。
本発明の代表的CD37抗体は、架橋することなく強力なアポトーシス促進活性を示す。これに関しては、この特性を有する抗体は、抗CD37 SMIP Tru16.4(架橋無しにはアポトーシスを示さない(Zhao et al. 2007))よりも優れている。架橋無しでのアポトーシスの誘発(本発明の抗体(Fc操作及びFc未操作の両抗体)はこれを示すことができた)は、in vivoの架橋因子(例えばFcγレセプターを保持するエフェクター細胞)の非存在下で、又は標的抗原CD37の低濃度下で(例えばCD37発現が低レベルの腫瘍細胞)有利である。架橋無しにアポトーシスを誘導する抗体はなお細胞死を引き起こすことができるが、一方、架橋依存抗体は細胞死を引き起こさない。
【0021】
さらに別の特徴では、本発明の抗CD37抗体分子は、本発明のヒト化又はキメラCD37特異抗体から誘導される抗体フラグメントである。抗体フラグメント、例えばFabフラグメントを得るために、日常的技術による手段によって、例えばパパインを用いて消化を実施することができる。パパイン消化の例はWO94/29348及びUS4,342,566に記載されている。抗体のパパイン消化によって典型的には2つの同一の抗原結合フラグメント(いわゆるFabフラグメント、各々はただ1つの抗原結合部位を有する)及び残余のFc領域が生成される。ペプシン処理によって2つの抗原結合部位を有し、なお抗原と架橋することができるF(ab’)2フラグメントが得られる。
抗体の消化によって得られたFabフラグメントはまた、軽鎖の定常ドメイン及び重鎖の第一の定常ドメイン(CH1)を含む。Fab’フラグメントとFabとは、Fab’フラグメントは、抗体ヒンジ領域由来の1つ以上のシステインを含む追加の残基を重鎖CH1ドメインのカルボキシ末端に含むという点で異なる。Fab’-SHは定常ドメインのシステイン残基が遊離チオール基を保持するFab’の本明細書における記述である。F(ab’)2抗体フラグメントは最初、Fab’間のシステイン残基を有するFab’フラグメント対として生成された。抗体フラグメントはまた、それぞれのコードDNAフラグメントを作製する分子生物学的方法によって生成することができる。
抗体分子は典型的には、2つの軽鎖/重鎖対から成るテトラマーであるが、又はダイマーであってもよく(すなわち1つの軽鎖/重鎖対(例えばFab又はFvフラグメント)から成る)、又はモノマー単鎖抗体(scFv;Johnson and Bird, 1991)、ミニボディ又はジアボディであってもよい。
【0022】
抗CD37抗体分子はまた、コンジュゲートの形態(すなわち細胞傷害性物質、特に腫瘍細胞の細胞傷害(例えばアポトーシス又は有糸分裂停止)を誘発する細胞傷害性物質と化学的に結合された抗体分子)であってもよい。正常な薬理学的クリアランス機構の結果として、医薬コンジュゲート(イムノコンジュゲート)で用いられる抗体は、限定的な量でのみ標的細胞と接触しこれと結合する。したがって、コンジュゲートで用いられる細胞傷害性物質は、十分な細胞殺滅を引き起こし治療効果を誘引することができるように高度に細胞傷害性でなければならない。US2004/0241174に記載されているように、そのような細胞傷害性物質の例には、タキサン(例えばWO01/38318及びWO03/097625を参照されたい)、DNAアルキル化剤(例えばCC-1065アナローグ)、アントラサイクリン、チュブリシンアナローグ、デュオカルマイシンアナローグ、ドキソルビシン、アウリスタチンE、リシンA毒素、及び反応性ポリエチレングリコール部分を含む細胞傷害性物質(例えば以下を参照されたい:Sasse et al. 2000;Suzawa et al. 2000;Ichimura et al. 1991;Francisco et al. 2003;US5,475,092;US6,340,701;US6,372,738及びUS6,436,931;US2001/0036923;US2004/0001838;US2003/0199519;及びWO01/49698)が含まれる。
好ましい実施態様では、細胞傷害性物質はメイタンシノイド、すなわちメイタンシン(CAS35846538)の誘導体である。当分野ではメイタンシノイドにはメイタンシン、メイタンシノール、メイタンシノールのC-3エステル、及び他のメイタンシノールアナローグ及び誘導体が含まれることが知られている(例えばUS5,208,020及びUS6,411,163を参照されたい)。
抗CD37抗体イムノコンジュゲートは、抗FAPイムノコンジュゲートについてWO2007/077173に記載されているように、設計及び合成することができる。
【0023】
さらに別の実施態様では、本発明の抗CD37分子は放射能標識して、放射性イムノコンジュゲート(抗CD37抗体MB-1について提唱されたアプローチである(Buchsbaum et al. 1992、上記を参照されたい))を生成することができる。有利な放射線特性を有する放射性核種は当分野で公知であり、その例はリン-32、ストロンチウム-89、イットリウム-90、ヨウ素-131、サマリウム-153、エルビウム-169、イッテリビウム-175、レニウム-188であり、これらは良好にかつ安定的にMAbと結合した。本発明の抗CD37抗体分子は、US6,241,961に記載されているように、当分野で公知の直接標識又は間接標識方法を用いて多様な放射性核種で標識することができる。本発明で有用な新規な放射能標識抗体コンジュゲートを作製しこれを適用する技術に関する概論は以下の文献で提供されている:Goldenberg and Shakey, 2007。
本発明の抗体分子(Fcが操作されていようといまいと)はまた二重特異性であることができる。二重特異性抗体は、すなわち2つの異なる標的と結合し、そのうちの1つはCD37であり、他方は、例えばT細胞によって発現される表面抗原、例えばCD3、CD16及びCD56である。
【0024】
本発明はまた、本発明のキメラ及びヒト化抗CD37抗体分子をコードするDNA分子に関する。本発明の抗体分子の可変重鎖をコードする配列は、配列番号:1、配列番号:5、配列番号:7及び配列番号:9に示される。本発明の抗体分子の可変軽鎖をコードする配列は、配列番号:3、配列番号:11、配列番号:13、配列番号:15、配列番号:17、配列番号:19、配列番号:21に示される。
軽鎖及び重鎖をコードする核酸分子は、標準的な方法により化学的及び酵素的に(PCR増幅)合成することができる。第一に、当分野で公知の方法(例えばGait, 1984)を用いて適切なオリゴヌクレオチドを合成し、このオリゴヌクレオチドを用いて合成遺伝子を生成することができる。オリゴヌクレオチドから合成遺伝子を生成する方法は当分野で公知である(例えば、Stemmer et al. 1995;Ye et al. 1992;Hayden et Mandecki, 1988;Frank et al. 1987)。
本発明のDNA分子には、配列表に示されるDNA分子が含まれるが、ただしこれらに限定されない。したがって、本発明はまた、WO2007/042309で規定する高ストリンジェンシーの結合及び洗浄条件下で、配列表に示されるDNA分子とハイブリダイズする核酸分子に関し、そのような核酸分子は、配列表に示される配列によってコードされる抗体と等価であるか又はそれらより優れた特性を有する抗体又はその機能的フラグメントをコードする。好ましい分子(mRNAの観点から)は、本明細書に記載のDNA分子の1つと少なくとも75%又は80%(好ましくは少なくとも85%、より好ましくは少なくとも90%、もっとも好ましくは少なくとも95%)の相同性又は配列同一性を有するものである。
本発明の範囲内のさらに別のDNA変種クラスは、それらがコードするポリペプチドに照らして規定することができる。これらのDNA分子は、配列表に示されるものからその配列に関して逸脱するが、それらは、遺伝暗号の縮合のために同一のアミノ酸配列をもつ抗体をコードする。例示すれば、真核細胞で当該抗体を発現させるという見地から、配列表に示されるDNA配列は、真核細胞でのコドン使用頻度に適合するように設計されている。抗体を大腸菌(E. coli)で発現させることを所望するならば、これらの配列は大腸菌のコドン使用頻度に適合するように変更することができる。本発明のDNA分子の変種は、例えばWO2007/042309に記載されたようないくつかの異なる方法で構築することができる。
【0025】
本発明の組換え抗CD37抗体分子を作製するために、完全長の軽鎖及び重鎖をコードするDNA分子又はそのフラグメントを、それら配列が転写及び翻訳制御配列に機能的に連結されるように発現ベクターに挿入する。本発明の抗体を製造するために、当業者は、当分野で周知の極めて多様な発現系から、概説論文(例えばKipriyanow and Le Gall, 2004)に記載の発現系を選択することができる。
発現ベクターには、プラスミド、レトロウイルス、コスミド、EBV由来エピソームなどが含まれる。発現ベクター及び発現制御配列は、宿主細胞と適合するように選択される。抗体軽鎖遺伝子及び抗体重鎖遺伝子は別々のベクターに挿入してもよい。ある種の実施態様では、両DNA配列は同じ発現ベクターに挿入される。便利なベクターは機能的に完全なヒトCH又はCL免疫グロブリン配列をコードするベクターであり、前記は、任意のVH又はVL配列を上記に記載したように容易に挿入及び発現することができるように、適切な制限部位が操作されている。定常鎖は通常、抗体軽鎖についてはカッパ又はラムダであり、抗体重鎖については、任意のIgGアイソタイプ(IgG1、IgG2、IgG3、IgG4)又は他の免疫グロブリン(対立遺伝子変種を含む)でありえるが、ただしこれらに限定されない。
組換え発現ベクターはまた、抗体鎖の宿主細胞からの分泌を容易にするシグナルペプチドを含むことができる。抗体鎖をコードするDNAは、シグナルペプチドが成熟抗体鎖のアミノ末端にインフレームで連結されえるようにベクターでクローニングすることができる。シグナルペプチドは、免疫グロブリンシグナルペプチドであっても、又は非免疫グロブリンタンパク質由来の異種ペプチドであってもよい。あるいは、抗体鎖をコードするDNA配列は既にシグナルペプチド配列を含んでいてもよい。
【0026】
抗体鎖をコードするDNA配列に加えて、組換え発現ベクターは、プロモータ、エンハンサー、転写終結及びポリアデニル化シグナル、並びに宿主細胞で抗体鎖の発現を制御する他の発現制御エレメントを含む調節配列を保持する。プロモータ配列の例(哺乳動物細胞で発現を達成する)は、CMV(例えばCMVサルウイルス40(SV40))(例えばSV40プロモータ/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモータ(AdMLP))、ポリオーマ及び強力な哺乳動物プロモータ(例えば天然の免疫グロブリン及びアクチンプロモータ)から誘導されるプロモータ及び/又はエンハンサーである。ポリアデニル化シグナルの例は、BGH polyA、SV40後期又は初期polyA;あるいは免疫グロブリン遺伝子の3’UTRなどを用いることができる。
組換え発現ベクターはまた、宿主細胞でのベクターの複製を調節する配列(例えば複製起点)及び選別可能マーカー遺伝子を保持することができる。抗CD37抗体の重鎖又はその抗原結合部分及び/又は軽鎖又はその抗原結合部分をコードする核酸分子、及びこれらのDNA分子を含むベクターを、宿主細胞、例えば細菌細胞又は高等真核細胞、例えば哺乳動物細胞に当分野で周知のトランスフェクション方法にしたがって導入することができる。前記トランスフェクション方法には、リポソーム仲介トランスフェクション、ポリカチオンイオン仲介トランスフェクション、プロトプラスト融合、マイクロインジェクション、リン酸カルシウム沈殿、エレクトロポレーション又はウイルスベクターによる移転が含まれる。
好ましくは、重鎖及び軽鎖をコードするDNA分子は2つのベクターに存在し、それらベクターは宿主細胞(好ましくは哺乳動物細胞)に同時トランスフェクトされる。
【0027】
発現用宿主として利用可能な哺乳動物細胞株は当分野では周知であり、とりわけチャイニーズハムスター卵巣(CHO、CHO-DG44)細胞、NSO、SP2/0細胞、HeLa細胞、ベビーハムスター腎(BHK)細胞、サル腎細胞(COS)、ヒト癌腫細胞(例えばHep G2)、A549細胞、3T3細胞、又はそのような任意の細胞株の誘導細胞/子孫細胞が含まれる。他の哺乳動物細胞もまた用いることができる。前記にはヒト、マウス、ラット、サル及びげっ歯類細胞株を含まれるが、ただしこれらに限定されない。又は他の真核細胞(酵母、昆虫細胞及び植物細胞を含むが、ただしこれらに限定されない)又は原核細胞、例えば細菌を用いてもよい。本発明の抗CD37抗体分子は、宿主細胞で抗体分子を発現させるために十分な期間当該宿主細胞を培養することによって製造される。
抗体分子は、好ましくは分泌されたポリペプチドとして培養媒体から回収するか、又は前記抗体分子は、例えば分泌シグナル無しに発現させた場合は宿主細胞溶解物から回収することができる。組換えタンパク質及び宿主細胞タンパク質について用いられる標準的なタンパク質精製方法を、実質的に均質な抗体調製物が得られる態様で用いて抗体分子を精製することが必要である。例示すれば、本発明の抗CD37抗体分子得るために有用な現在の精製技術は、第一の工程として、培養媒体又は溶解物から細胞及び/又は粒子状細胞屑の除去を含む。続いて可溶性夾雑タンパク質、ポリペプチド及び核酸から、例えばイムノアフィニティ又はイオン交換カラム、エタノール沈殿、逆相HPLC、セファデックスクロマトグラフィー、シリカゲル若しくはイオン交換樹脂クロマトグラフィーによって抗体を精製する。抗CD37抗体分子調製物を得るための最終工程として、治療薬としての利用のために下記に記載するように、精製抗体分子を乾燥(例えば凍結乾燥)させることができる。
さらに別の特徴では、本発明は、活性成分として本発明の抗CD37抗体分子を含む医薬組成物に関する。
【0028】
治療に用いられるためには、抗CD37抗体は、動物又はヒトへの投与を容易にするために適切な医薬組成物に混合される。抗CD37抗体分子の典型的な処方物は、抗CD37抗体分子を生理学的に許容できる担体、賦形剤又は安定化剤と混合することによって、凍結乾燥あるいは乾燥処方物又は水溶液又は水性若しくは非水性懸濁物の形態で調製することができる。担体、賦形剤、改変剤又は安定化剤は、用いられる用量及び濃度で非毒性である。前記には、緩衝系(例えばリン酸、クエン酸、酢酸及び他の無機若しくは有機酸及びそれらの塩);抗酸化剤(アスコルビン酸及びメチオニンが含まれる);保存料(例えばオクタデシルジメチルベンジルアンモニウムクロリド;ヘキサメトニウムクロリド;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチル又はベンジルアルコール;アルキルパラベン、例えばメチル又はプロピルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;及びm-クレゾール);タンパク質、例えば血清アルブミン、ゼラチン又は免疫グロブリン;親水性ポリマー、例えばポリビニルピロリドン又はポリエチレングリコール(PEG);アミノ酸、例えばグリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン又はリジン;単糖類、二糖類、オリゴ糖又は多糖類及び他の炭水化物(グルコース、マンノース、シュクロース、トレハロース、デキストリン又はデキストランを含む);キレート剤(例えばEDTA);糖アルコール(例えばマンニトール又はソルビトール);塩形成対イオン(例えばナトリウム);金属複合体(例えば亜鉛-タンパク質複合体);及び/又はイオン性又は非イオン性表面活性剤、例えばTWEENTM(ポリソルベート)、PLURONICSTM又は脂肪酸エステル、脂肪酸エーテル又は糖エステルが含まれる。さらに有機溶媒、例えばエタノール又はイソプロパノールもまた抗体処方物に含まれえる。賦形剤はまた放出改変機能又は吸収改変機能を有することができる。
【0029】
抗CD37抗体分子はまた、乾燥させるか(凍結乾燥、噴霧乾燥、噴霧凍結乾燥、近臨界若しくは超臨界ガスによる乾燥、真空乾燥、通気乾燥)、沈殿させるか、又は結晶化させるか、又はマイクロカプセル中に被包化することができる。マイクロカプセルは、例えばコアセルベーション技術によって又は界面重合(それぞれ例えばヒドロキシメチルセルロース又はゼラチン及びポリ-(メチルメタクリレート)を用いる)によって、コロイド性薬剤デリバリー系(例えばリポソーム、アルブミン微小球、マイクロエマルジョン、ナノ粒子及びナノカプセル)で、担体若しくは表面でのマクロエマルジョン又は沈殿又は固定化(例えばpcmc技術(タンパク質被覆微結晶(protein coated microcrystal))により調製することができる。そのような技術は成書(Remington: The Science and Practice of Pharmacy, 21st edition, Hendrickson R. Ed.)に記載されている。
もちろんのこと、in vivo投与に用いられる処方物は無菌的でなければならない。滅菌は、通常の技術、例えば無菌的ろ過膜を通過させるろ過によって実施することができる。
いわゆる高濃度液体処方物(HCLF)を得るために抗CD37抗体の濃度を高めることは有用でありえる。そのようなHCLFを生成するための多様な方法が記載されている。
【0030】
抗CD37抗体分子はまた徐放性調製物に含まれえる。そのような調製物は、疎水性又は親水性ポリマーの固体、半固体、又は液体マトリックスを含み、成形品、例えばフィルム、スティック又はマイクロカプセルの形態を有し、適用装置を介して適用することができる。徐放性マトリックスの例には、ポリエステル、ヒドロゲル(例えばポリ(2-ヒドロキシエチル-メタクリレート)又はシュクロースアセテートブチレート)又はポリ(ビニルアルコール)、ポリラクチド(US3,773,919)、L-グルタミン酸とγエチル-L-グルタメートとのコポリマー、非分解性エチレン-ビニルアセテート、分解性乳酸-グリコール酸コポリマー(例えばLUPRON DEPOTTM(乳酸-グリコール酸コポリマー及び酢酸ロイプロリドを含む注射可能な微小球)及びポリ-D-(-)-3-ヒドロキシ酪酸が含まれる。ポリマー(例えばエチレン-酢酸ビニル及び乳酸-グリコール酸)は100日間にわたって分子の放出を許容するが、一方、ある種のヒドロゲルはより短い期間の間タンパク質を放出する。被包化された抗体が体内で長期間存続するとき、それらは、37℃で水分に暴露される結果として変性又は凝集し、生物学的活性の低下及び免疫原性における変化を生じる可能性がある。関与するメカニズムに応じて安定化のために合理的な対策を講じることができる。例えば、凝集メカニズムがチオ-ジスルフィド交換による分子間S-S結合形成であると判明したら、安定化は、スルフヒドリル残基の改変、酸性溶液からの凍結乾燥(例えばWO89/011297に記載)、水分含有量の管理、適切な添加剤の使用、および特別なポリマーマトリックス組成の開発によって達成することができる。
本発明の抗CD37抗体分子のためにまた用いることができる処方物は、US7,060,268及びUS6,991,790に記載されている。
【0031】
CD37抗体分子はまた他の適用形(例えば分散剤、懸濁剤又はリポソーム、錠剤、カプセル、散剤、スプレー、経皮若しくは皮内パッチ又はクリーム(浸透強化仕掛け付き又は仕掛け無し)、ウェファース、鼻用、頬腔用又は肺用処方物)に取り入れることができる。CD37抗体分子はまた、移植細胞によって、又は遺伝子治療後に個体自身の細胞によって生成させることができる。
抗CD37抗体分子はまた、化学基、例えばポリエチレングリコール(PEG)、メチル若しくはエチル基、又は炭水化物基を用いて誘導体化することができる。これらの基は、本発明の抗体の生物学的特徴の改善、例えば血中半減期の増加又は組織結合の増加に有用でありえる。
適用の好ましい態様は非経口(輸液又は注射(静脈内、筋肉内、皮下、腹腔内、皮内))であるが、他の適用態様(例えば吸入、経皮、鼻内、頬腔、経口適用)もまた利用することができる。
疾患の予防又は治療の場合、抗体の適切な投与量は、治療されるべき疾患のタイプ、当該疾患の重篤度及び経過、抗体が予防目的又は治療目的のために投与されるのか否か、これまでの治療、患者の病歴及び抗体に対する応答、並びに主治医の判断に左右されるであろう。抗体は、一度に又は一連の治療の間適切に患者に投与されるであろう。
疾患のタイプ及び重篤度に応じて、約0.01μg/kgから40mg/kg(例えば0.1−20mg/kg)の抗体が、例えば1回又は2回以上の別々の投与にせよ、又は連続輸液投与にせよ、患者への最初の候補投与量である。症状に応じて数日又はそれより長い期間にわたる反復投与の場合、治療は、症状の所望の抑制が生じるまで維持される。しかしながら、他の投与計画も有用でありえる。本治療法の進行は、通常の技術及びアッセイ(例えばフローサイトメトリーの使用)、例えばB細胞枯渇の程度を決定することによって容易にモニターされる。
投与される抗体の“治療的に有効な量”は、疾患又は異常の予防、緩和又は治療のために必要な最少量である。
【0032】
本発明の抗CD37抗体分子及び前記を含む医薬組成物は、その表面にCD37を発現し、癌性又は自己免疫/炎症疾患を引き起こすB細胞の枯渇に有用である。
第一の特徴では、本発明の医薬組成物は、癌(特に任意のCD37陽性悪性疾患)の治療に有用である。
B細胞悪性疾患には以下が含まれる(ただしこれらに限定されない):B細胞リンパ腫(例えばホジキン症の種々の型、B細胞非ホジキンリンパ腫(NHL)及び関連リンパ腫(例えばヴァルデンストレームマクログロブリン血症(リンパ形質細胞性リンパ腫又は免疫細胞腫とも称される)又は中枢神経系リンパ腫)、白血病(例えば急性リンパ芽球性白血病(ALL)、慢性リンパ球性白血病(CLL、B細胞慢性リンパ球性白血病BCLLとも称される)、毛状細胞白血病及び慢性骨髄芽球性白血病)及びミエローマ(例えば多発性ミエローマ)。さらに別のB細胞悪性疾患には、小リンパ球型リンパ腫、B細胞前リンパ球性白血病、リンパ形質細胞性リンパ腫、脾臓辺縁層リンパ腫、形質細胞ミエローマ、骨の孤立形質細胞腫、骨外形質細胞腫、粘膜結合類リンパ組織(MALT)の外リンパ節辺縁層B細胞リンパ腫、リンパ節辺縁層B細胞リンパ腫、濾胞性リンパ腫、マントル細胞リンパ腫、びまん性大B細胞型リンパ腫、縦隔(胸腺)大B細胞型リンパ腫、血管内大B細胞型リンパ腫、原発性滲出型リンパ腫、バーキットリンパ腫/白血病、灰白色層リンパ腫(grey zone lymphoma)、悪性性が未確認のB細胞増殖、類リンパ腫、肉芽腫症及び移植後のリンパ球増殖性異常が含まれる。
【0033】
さらに別の特徴では、抗CD37抗体を含む医薬組成物は、その病理においてB細胞が中心的に関与する自己免疫及び炎症性疾患の治療に有用である。
そのような疾患には以下が含まれる(ただしこれらに限定されない):関節炎、慢性関節リウマチ、若年性慢性関節リウマチ、変形性関節症、多発性軟骨炎、乾癬性関節炎、乾癬、皮膚炎、多発性筋炎/皮膚筋炎、封入体性筋炎、炎症性筋炎、中毒性表皮壊死症、全身性皮膚硬化症及び硬化症、CREST症候群、炎症性大腸疾患付随応答、クローン病、潰瘍性結腸炎、呼吸窮迫症候群、成人呼吸窮迫症候群(ARDS)、髄膜炎、脳炎、ブドウ膜炎、結腸炎、糸球体腎炎、アレルギー疾患、湿疹、喘息、T細胞浸潤及び慢性炎症性応答を随伴する症状、アテローム性硬化症、自己免疫性心筋症、白血球接着不全、全身性紅斑性狼瘡(SLE)、亜急性皮膚性エリテマトーデス、円板状狼瘡、狼瘡性筋炎、狼瘡性脳炎、若年発症性糖尿病、多発性硬化症、アレルギー性脳脊髄炎、視神経脊髄炎、リウマチ熱、シドナム舞踏病、サイトカイン及びTリンパ球により媒介される急性及び遅延型過敏症に付随する免疫応答、結核、サルコイドーシス、ヴェーゲナー肉芽腫症及びチャーグ-ストラウス病を含む肉芽腫症、顆粒球減少症、脈管炎(過敏性脈管炎/血管炎、ANCA及びリウマチ性脈管炎を含む)、再生不良性貧血、ダイアモンド-ブラックファン貧血、免疫性溶血性貧血(自己免疫性溶血性貧血(AIHA)を含む)、悪性貧血、赤芽球ろう(PRCA)、第VIII因子欠損、血友病A、自己免疫好中球減少症、汎血球減少症、白血球減少症、白血球漏出を伴う疾患、中枢神経系(CNS)の炎症性疾患、多器官損傷症候群、重症筋無力症、抗原抗体複合物介在疾患、抗糸球体基底膜病、抗リン脂質抗体症候群、アレルギー性神経炎、ベーチェット病、キャッスルマン症候群、グッドパスチャー症候群、ランバート-イートン筋無力症症候群、レイノー症候群、シェーグレン症候群、スティーヴェンズ-ジョンソン症候群、固体器官移植拒絶、対宿主性移植片病(GVHD)、水疱性類天疱瘡、天疱瘡、自己免疫性多発性内分泌障害、血清陰性脊椎関節症、ライター病、スティッフマン症候群、巨細胞動脈炎、免疫複合体性腎炎、IgA腎症、IgM多発性腎症又はIgM介在腎症、特発性血小板減少性紫斑病(ITP)、血栓性血小板減少性紫斑病(TTP)、ヘノッホ-シェーンライン紫斑病、自己免疫性血小板減少症、精巣及び卵巣の自己免疫疾患(自己免疫性精巣炎及び卵巣炎を含む)、原発性甲状腺機能低下症;自己免疫性内分泌疾患(自己免疫性甲状腺炎、慢性甲状腺炎(橋本甲状腺炎)、亜急性甲状腺炎、特発性甲状腺機能低下症、アジソン病、グレーヴズ病、自己免疫性多発性腺症候群(又は多発性腺内分泌障害)、I型糖尿病(インスリン依存真性糖尿病(IDDM)とも称される)及びシーハン症候群;自己免疫性肝炎、類リンパ球性間質肺炎(HIV)、NSIPに対する閉塞性細気管支炎(非移植性)(bronchiolitis obliterans (non-transplant) vs NSIP)、ギラン-バレー症候群、大血管性血管炎(リウマチ性多発性筋肉痛及び巨細胞性(タカヤス)動脈炎を含む)、中血管性血管炎(川崎病及び結節性多発性動脈炎を含む)、結節性多発性動脈炎(PAN)強直性脊椎炎、バージャー病(IgA腎症)、急速に進行する糸球体腎炎、原発性胆汁性肝硬変、腹腔スプルー(グルテン腸症)クリオグロブリン血症、肝炎に付随するクリオグロブリン血症、筋萎縮性側索硬化症(ALS)、冠状動脈疾患、家族性地中海熱、微細多発性血管炎、コーガン症候群、ウィスコット-アルドリッチ症候群及び閉塞性血栓性血管炎(WO2007/014278を参照されたい)。
【0034】
治療されるべき疾患に応じて、本発明の抗CD37抗体分子は、そのもの単独で、又は1つ又は2つ以上の追加の治療薬剤と併用して用いることができる。前記追加の治療薬剤は、特に、DNA損傷薬剤若しくはチュブリン結合薬剤、又は癌細胞における血管形成、シグナルトランスダクション経路又は有糸分裂チェックポイントを阻害する治療的に活性を有する化合物から選択される。
前記追加の治療薬剤は、場合によって同じ医薬調製物の成分として同時に投与するか、又は抗CD37抗体分子の投与の前後に投与することができる。
ある種の実施態様では、追加の治療薬剤は、EGFRファミリー、VEGFRファミリー、IGF-1R、インスリンレセプター、オーロラA、オーロラB、PLK及びPI3キナーゼ、FGFR、PDGFR、Raf、KSP又はPDK1の阻害剤の群から選択される1つ又は2つ以上の阻害剤でありえるが、ただしこれらに限定されない。
追加薬剤のさらに別の例は、CDK、Akt、Src、Bcr-Ab1、cKit、cMet/HGF、c-Myc、Flt3、HSP90、ヘッジホッグアンタゴニストの阻害剤、JAK/STAT、Mek、mTor、NFkappaB、プロテアソーム、Rhoの阻害剤、Wntシグナリング又はNotchシグナリングの阻害剤、又はユビキチン化経路の阻害剤である。
Aurora阻害剤の例は、PHA-739358、AZD-1152、AT-9283、CYC-116、R-763、VX-667、MLN-8045、PF-3814735、SNS-314、VX-689、GSK-1070916、TTP-607、PHA-680626、MLN-8237及びENMD-2076であるが、ただしこれらに限定されない。
PLK阻害剤の例はGSK-461364である。
raf阻害剤の例は、BAY-73-4506(VEGFR阻害剤でもある)、PLX-4032、RAF-265(VEGFR阻害剤でもある)、RAF-265(VEGFR阻害剤でもある)、ソラフェニブ(sorafenib)(VEGFR阻害剤でもある)、XL-281及びNevavar(VEGFR阻害剤でもある)である。
【0035】
KSP阻害剤の例は、イスピネシブ(ispinesib)、ARRY-520、AZD-4877、CK-1122697、GSK-246053A、GSK-923295、MK-0731、SB-743921、LY-2523355、及びEMD-534085である。
src及び/又はbcr-ab1阻害剤の例は、ダサチニビ(dasatinib)、AZD-0530、ボスチニブ(bostinib)、XL-228(IGF-1R阻害剤でもある)、ニロチニブ(nilotinib)(PDGFR及びcKit阻害剤でもある)、イマチニブ(imatinib)(cKit阻害剤でもある)、NS-187、KX2-391、AP-24534(EGFR、FGFR、Tie2、Flt3阻害剤でもある)、KM-80及びLS-104(Flt3、Jak2阻害剤でもある)である。
PDK1阻害剤の例はAR-12である。Rho阻害剤の例はBA-210である。
PI3キナーゼ阻害剤の例はPX-866、PX-867、BEZ-235(mTor阻害剤でもある)、XL-147及びXL-765(mTor阻害剤でもある)、BGT-226、CDC-0941である。
cMet又はHGFの阻害剤の例はXL-184(VEGFR、cKit、Flt3阻害剤でもある)、PF-2341066、MK-2461、XL-880(VEGFR阻害剤でもある)、MGCD-265(VEGFR、Ron、Tie2阻害剤でもある)、SU-11274、PHA-665752、AMG-102、AV-299、ARQ-197、MetMAb、CGEN-241、BMS-777607、JNJ-38877605、PF-4217903、SGX-126、CEP-17940、AMG-458、INCB-028060及びE-7050である。
c-Myc阻害剤の例はCX-3543である。
Flt3阻害剤の例は、AC-220(cKit及びPDGFR阻害剤でもある)、KW-2449、LS-104(bcr-ab1及びJak2阻害剤でもある)、MC-2002、SB-1317、レスタウルチニブ(lestaurtinib)(VEGFR、PDGFR、PKC阻害剤でもある)、TG-101348(JAK2阻害剤でもある)、XL-999(cKit、FGFR、PDGFR及びVEGFR阻害剤でもある)、スニチニブ(sunitinib)(PDGFR、VEGFR及びcKit阻害剤でもある)及びタンヅチニブ(tandutinib)(PDGFR及びcKit阻害剤でもある)である。
【0036】
HSP90阻害剤の例はタネスピマイシン、アルヴェスピマイシン、IPI-504、STA-9090、MEDI-561、AUY-922、CNF-2024及びSNX-5422である。
JAK/STAT阻害剤の例は、CYT-997(またチューブリンと相互作用する)、TG-101348(Flt3阻害剤でもある)及びXL-019である。
Mek阻害剤の例は、ARRY-142886、AS-703026、PD-325901、AZD-8330、ARRY-704、RDEA-119及びXL-518である。
mTor阻害剤の例は、テムシロリムス(temsirolimus)、デフォロリムス(deforolimus)(VEGF阻害剤としても作用する)、エヴェロリムス(everolimus)(さらにVEGF阻害剤)、XL-765(PI3キナーゼ阻害剤でもある)及びBEZ-235(PI3キナーゼ阻害剤でもある)である。
Akt阻害剤の例は、ペリフォシン、GSK-690693、RX-0201及びトリシリビンである。
cKit阻害剤の例は、マスチニブ(mastinib)、OSI-930(VEGFR阻害剤としてもまた作用する)、AC-220(Flt3及びPDGFR阻害剤でもある)、タンヅチニブ(Flt3及びPDGFR阻害剤でもある)、アキシチニブ(axitinib)(VEGFR及びPDGFRの阻害剤でもある)、スニチニブ(Flt3、PDGFR、VEGFR阻害剤でもある)及びXL-820(VEGFR及びPDGFR阻害剤としてもまた作用する)、イマチニブ(bcr-ab1阻害剤でもある)、ニロチニブ(bcr-ab1及びPDGFR阻害剤でもある)である。
ヘッジホッグアンタゴニストの例は、IPI-609、CUR-61414、GDC-0449、IPI-926及びXL-139である。
CDK阻害剤の例は、セリシクリブ(seliciclib)、AT-7519、P-276、ZK-CDK(VEGFR2及びPDGFRもまた阻害する)、PD-332991、R-547、SNS-032、PHA-690509、PHA-848125及びSCH-727965である。
【0037】
プロテアソーム阻害剤の例は、ボルテゾミブ(bortezomib)、カルフィルゾミブ(carfilzomib)及びNPI-0052(NFkappaBの阻害剤でもある)である。
プロテアソーム阻害剤/NFkappaB経路阻害剤の例は、ボルテゾミブ、カルフィルゾミブ、NPI-0052、CEP-18770、MLN-2238、PR-047、PR-957、AVE-8680及びSPC-839である。
ユビキチン化経路の阻害剤の例はHBX-41108である。
抗血管形成剤の例は、FGFR、PDGFR及びVEGF(R)の阻害剤、並びにサリドマイドであり、そのような薬剤は以下から選択される(ただしこれらに限定されない):ベヴァシズマブ、モテサニブ、CDP-791、SU-14813、テラチニブ、KRN-951、ZK-CDK(CDKの阻害剤でもある)、ABT-869、BMS-690514、RAF-265、IMC-KDR、IMC-18F1、IMiD、サリドマイド、CC-4047、レナリドマイド、ENMD-0995、IMC-D11、Ki-23057、ブリヴァニブ(brivanib)、セディラニブ(cediranib)、1B3、CP-868596、IMC-3G3、R-1530(Flt3の阻害剤でもある)、スニチニブ(cKit及びFlt3の阻害剤でもある)、アキシチニブ(cKitの阻害剤でもある)、レスタウルチニブ(Flt3及びPKCの阻害剤でもある)、ヴァタラニブ、タンヅチニブ(Flt3及びcKitの阻害剤でもある)、パゾパニブ(pazopanib)、PF-337210、アフリベルセプト(aflibercept)、E-7080、CHIR-258、ソラフェニブ(sorafenib)トシレート(Rafの阻害剤でもある)、ヴァンデタニブ、CP-547632、OSI-930、AEE-788(EGFR及びHer2阻害剤でもある)、BAY-57-9352(Rafの阻害剤でもある)、BAY-73-4506(Rafの阻害剤でもある)、XL-880(cMetの阻害剤でもある)、XL-647(EGFR及びEphB4の阻害剤でもある)、XL-820(cKitの阻害剤でもある)、ニロチニブ(cKit及びbrc-ab1の阻害剤でもある)、CYT-116、PTC-299、BMS-584622、CEP-11981、どドヴィチニブ(dovitinib)、CY-2401401及びENMD-2976.
【0038】
追加の治療薬剤はまたEGFR阻害剤から選択することができる。前記は小分子EGFR阻害剤であっても、又は抗EGFR抗体であってもよい。抗EGFR抗体の例は、セツキシマブ、パニツムマブ、ニモツズマブ、ザルツムマブであるが、ただしこれらに限定されない。小分子EGFR阻害剤の例は、ゲフィチニブ、エルロチニブ(erlotinib)及びヴァンデタニブ(VEGFRの阻害剤でもある)である。EGFR調整物質の別の例はEGF融合毒素である。
本発明の抗CD37抗体分子との併用に有用なさらに別のEGFR及び/又はHer2阻害剤は、ラパチニブ、トラスツズマブ、ペルツズマブ(pertuzumab)、XL-647、ネラチニブ、BMS-599626、ARRY-334543、AV-412、mAB-806、BMS-690514、JNJ-26483327、AEE-788(VEGFRの阻害剤でもある)、AZD-8931、ARRY-380、ARRY-333786、IMC-11F8、Zemab、TAK-285、AZD-4769である。
追加される薬剤はまた、IGF-1R及びインスリンレセプター経路を標的とする薬剤から選択することができる。そのような薬剤には、IGF-1R(例えばCP-751871、AMG-479、IMC-A12、MK-0646、AVE-1642、R-1507、BIIB-022、SCH-717454、rhu Mab IGFR)と結合する抗体、及びIGF1-Rのキナーゼドメインを標的とする新規な化学物質(例えばOSI-906又はBMS-554417、XL-228、BMS-754807)が含まれる。
本発明の抗CD37抗体分子を用いる治療法で有利に併用することができる他の薬剤はCD20を標的とする分子であり、CD20特異的抗体(例えばリツキシマブ、LY-2469298、オクレリズマブ、MEDI-552、IMMU-106、GA-101(=R7159)、XmAb-0367、オフアツムマブ)、放射能標識CD20抗体(例えばトシツムマブ及びイブリツモマブチウキセタン)、又は他のCD20誘導タンパク質(例えばSMIP Tru015、PRO-131921、FBT-A05、ヴェルツズマブ、R-7159)が含まれる。
【0039】
CD37抗体は、白血球で発現される他の表面抗原の阻害剤と併用することができる。前記阻害剤は、特に抗体又は抗体様分子であり、例えば抗CD20(シプリズマブ)、抗CD4(ザノリムマブ)、抗CD19(MT-103、MDX-1342、SAR-3419、XmAb-5574)、抗CD22(エプラツズマブ)、抗CD23(ルミリキシマブ)、抗CD30(イラツムマブ)、抗CD32B(MGA-321)、抗CD38(HuMax-CD38)、抗CD40(SGN40)、抗CD52(アレムツズマブ)、抗CD80(ガリキシマブ)である。本発明の抗体はまた別のCD37アンタゴニスト、例えばTRU-016と併用することができる。
CD37抗体と併用できる他の薬剤は、イムノトキシン(例えばBL-22(抗CD22イムノトキシン)、イノツズマブオゾガミシン(抗CD23抗体-カリケアミシンコンジュゲート)、RFT5.dgA(抗CD25リシン毒素A-鎖)、SGN-35(抗CD30-アウリスタチンEコンジュゲート)及びゲムツズマブオゾガミシン(抗CD33カリケアミシンコンジュゲート)、MDX-1411(抗CD70コンジュゲート))、又は放射能標識抗体(例えば90Y-エプラツズマブ(抗CD22放射性イムノコンジュゲート))である。
さらにまた抗CD37抗体は以下と併用することができる:免疫調節剤、例えばアポトーシスを誘発するか又はシグナルトランスダクション経路を改変する抗体、例えばTRAILレセプター調節物質マパツムマブ(TRAIL-1レセプターアゴニスト)、レキサツムマブ(TRAIL-2レセプターアゴニスト)、チガツズマブ、Apomab、AMG-951及びAMG-655;抗HLA-DR抗体(例えば1D09C3)、抗CD74、破骨細胞分化因子リガンド阻害剤(例えばデノスマブ)、BAFFアンタゴニスト(例えばAMG-623a)、又はToll様レセプターのアゴニスト(例えばTLR-4又はTLR-9)。
【0040】
本発明の抗CD37抗体と一緒に用いることができる他の薬剤は以下から選択される(ただしこれらに限定されない):ホルモン、ホルモン類似体及び抗ホルモナール(例えばタモキシフェン、トレミフェン、ラロキシフェン、フルヴェストラント、酢酸メゲストロール、フルタミド、ニルタミド、ビカルタミド、酢酸シプロテロン、フィナステリド、酢酸ブセレリン、フルドロコルチゾン、フルオキシメステロン、メドロキシプロジェステロン、カプロン酸ヒドロキシプロジェステロン、ジエチルスチルベストロール、プロピオン酸テストステロン、フルオキシメステロン/等価物、オクトレオチド、アルゾキシフェン、パシレオチド、ヴァプレオチド、アドレノコルチコステロイド/アンタゴニスト、プレドニソン、デキサメタゾン、アイノグルテチミド(ainoglutethimide))、アロマターゼ阻害剤(例えばアナストロゾール、レトロゾール、リアロゾール、エクセメスタン、アタメスタン、フォルメスタン)、LHRHアゴニスト及びアンタゴニスト(例えば酢酸ゴセレリン、ロイプロリド、アバレリクス、セトロレリクス、デスロレリン、ヒストレリン、トリプテレリン)、抗代謝薬(例えばアンチフォレート、例えばメトトレキセート、トリメトトレキセート、ペメトレキセート(pemetrexed)、ピリミジン類似体、例えば5-フルオロウラシル、フルオロデオキシウリジン、カペシタビン、デシタビン、ネララビン、5-アザシチジン及びゲミシタビン、プリン及びアデノシン類似体、例えばメルカプトプリン、チオグアニン、アザチオプリン、クラドリビン及びペントスタチン、シタラビン、フルダラビン、クロファラビン);抗腫瘍抗生物質(例えばアントラサイクリン、例えばドキソルビシン、ダウノルビシン、エピルビシン及びイダルビシン、マイトマイシンC、ブレオマイシン、ダクチノマイシン、プリカマイシン、スプリカマイシン、アクチノマイシンD、ミトキサントロン、ミトキサントロンイダルビシン、ピキサントロン、ストレプトゾシン、アフィジコリン);白金誘導体(例えばシスプラチン、オキサリプラチン、カルボプラチン、ロバプラチン、サトラプラチン);アルキル化剤(例えばエストラムスチン、セムスチン、メクロレタミン、メルファラン、クロラムブシル、ブスルファン、ダカルバジン、シクロホスファミド、イフォスファミド、ヒドロキシウレア、テモゾロミド、ニトロソウレア(例えばカルムスチン及びロムスチン)、チオテパ);抗有糸分裂剤(例えばヴィンカアルカロイド、例えばヴィンブラスチン、ヴィンデシン、ヴィノレルビン、ヴィンフルニン);及びタキサン、例えばパクリタキセル、ドセタキセル及びそれらの処方物、ラロタキセル;シモタキセル、及びエポチロン、例えばイキサベピロン、パツピロン、ZK-EPO;トポイソメラーゼ阻害剤(例えばエピポドフィロトキシン、例えばエトポシド及びエトポフォス、テニポシド、アムサクリン、トポテカン、イリノテカン、バノキサントロン、カンプトテシン)及び種々雑多な化学療法剤、例えばレチン酸誘導体、アミフォスチン、アナグレリド、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン-2、プロカルバジン、N-メチルヒドラジン、ミトタン、及びポルフィマー、ベキサロテン、セレコキシブ、エチルエネミン/メチルメラミン、トリエチレンメラミン、トリエチレンチオホスホラミド、ヘキサメチルメラミン、及び酵素、L-アスパラギナーゼ、L-アルギナーゼ及びメトロニダゾール、ミソニダゾール、デスメチルミソニダゾール、ピモニダゾール、エタニダゾール、ニモラゾール、RSU1069、EO9、RB6145、SR4233、ニコチンアミド、5-ブロモデオジウリジン、5-ヨードデオキシウリジン、ブロモデオキシシチジン、エリスロヒドロキシノニル-アデニン、アントラセネジオン、GRN-163L(競合性テロメアーゼ鋳型アンタゴニスト)、SDX-101(PPARアゴニスト)、タラボスタット(DPP阻害剤)、フォロデシン(PNP阻害剤)、アタシセプト(TNFファミリーメンバーBLyS及びAPRILを標的とする可溶性レセプター)、TNF-アルファ中和剤(Enbrel、Humira、Remicade)、XL-844(CHK1/2阻害剤)、VNP-40101M(DNAアルキル化剤)、SPC-2996(アンチセンスbcl2阻害剤)、オバトクラクス(bcl2阻害剤)、エンザスタウリン(PKCベータ調節剤)、ヴォリニスター(HDAC阻害剤)、ロミデプシン(HDAC阻害剤)、AT-101(Bcl-2/Bcl-xL阻害剤)、プリチデプシン(多機能性デプシペプチド)、SL-11047(ポリアミン代謝調節剤)。
【0041】
ある種の実施態様では、抗CD37抗体分子は、“CHOP”(シクロホスファミド、ドキソルビシン、ヴィンクリスチン及びプレドニソンの組合せ)と一緒に適用される。
本発明の抗CD37抗体分子はまた、他の治療法(外科手術、放射線療法、内分泌療法、生物学的応答改変療法、高温療法及び低温療法を含む)及びいずれかの副作用を緩和するための薬剤(例えば鎮吐剤)と一緒に用いることができる。前記は、G-CSF、GM-CSF、光感作剤、例えばヘマトポルフィリン誘導体、フォトフリン(商標)、ベンゾポルフィリン誘導体、Npe6、錫エチオポルフィリン、フェオボリド-a、バクテリオクロロフィル-a、ナフタロシアニン、フタロシアニン、亜鉛フタロシアニンである。
モノクローナル抗体は完璧な抗原特異性を示し、しばしばヒトの標的抗原とのみ反応し、動物種由来の同族タンパク質とは反応しない。治療用抗体の開発を促進するために、in vivo毒性及び動的薬理変化を評価するための適切な動物モデルが要求される。in vivoモデルの1つの可能性はトランスジェニックマウスであり、このマウスでは内因性標的抗原はそのヒト同族体によって置き換えられている(“ノックアウト/ノックインマウス”)。特に、治療用抗CD37抗体の開発のために、ネズミCD37遺伝子をヒトCD37遺伝子によって置き換えることができる。これは、非翻訳配列によってフランキングされているヒトCD37遺伝子のコードゲノム配列を含むターゲティングベクターを構築することにより達成することができる。マウスES細胞を用いた相同組換えにこのターゲティングベクターを用いることができる。ヒトCD37発現についてホモ接合性であるトランスジェニック動物を用いて、ヒトCD37に対して作製した抗体の動的薬理作用を、例えば抗体適用後に抹消B細胞数をモニターすることによって評価することができる。あるいは、それらのマウスを用いて、静脈内適用後のヒトCD37特異抗体の潜在的な毒性作用を解明することができる。
モノクローナル抗体の動物交差反応性が欠如する場合の別の可能性は、いわゆる代用抗体の生成である。代用抗体は、動的薬理作用及び毒性作用の解明のために有用な関連する動物種(例えばマウス又はカニクイザル)の同族タンパク質と反応する抗体である。CD37の場合、理想的にはそれぞれマカクCD37又はマウスCD37に特異的であるモノクローナル抗体が生成され、そのような代用抗体は当該生成抗体と類似の結合及び機能特性を有するはずである。これは、マカク又はマウスCD37発現細胞を標的細胞として利用するアッセイ系、例えば結合については、FACSスキャチャード分析、ADCC及びアポトーシス分析を用いることにより精査することができる。最終的には、代用抗体は、マカク又はマウス血液におけるin vivoにおけるB細胞枯渇活性により選択することができる。
【0042】
本発明の実施態様の例を以下にまとめる。
(1)ヒトCD37と結合し、
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体
から誘導される抗体分子であって、キメラ抗体又はヒト化抗体である前記抗体分子。
(2)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、ii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖、iii)ヒト起源である定常重鎖及び軽鎖によって規定されるキメラ抗体である、(1)に記載の抗体分子。
(3)i)定常重鎖がIgG1鎖であり、さらにii)定常軽鎖がカッパ鎖である、(2)に記載の抗体。
(4)定常重鎖i)が配列番号:24に示されるアミノ酸配列を含み、さらに定常軽鎖ii)が配列番号:26に示されるアミノ酸配列を含む、(3)に記載の抗体。
(5)下記a)−d)によって規定されるヒト化抗体である、(1)に記載の抗体:
a)配列番号:2に示される可変重鎖内に含まれるCDR、
b)配列番号:4に示される可変軽鎖内に含まれるCDR、
c)ヒト抗体から誘導される、前記CDRを支持するフレームワーク、
d)ヒト抗体に由来する定常重鎖及び軽鎖。
(6)配列番号:6に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(7)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(6)に記載の抗体。
(8)配列番号:8に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(9)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(8)に記載の抗体。
(10)配列番号:10に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(11)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(10)に記載の抗体。
(12)抗体が、Fcドメイン内に1つ又は2つ以上のエフェクター機能を調節する1つ又は2つ以上の変異を有する、(1)から(11)のいずれかに記載の抗体。
(13)エフェクター機能の調節が抗体依存細胞媒介性細胞傷害の増加である、(12)に記載の抗体。
(14)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき332位の単一置換である、(12)または(13)に記載の抗体。
(15)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、239位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(16)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、236位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(17)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、236位、239位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(18)置換がI332E、S239D及びG236Aである、(14)から(17)のいずれかに記載の抗体。
(19)ヒトCD37と結合し、さらに配列番号:28のアミノ酸配列を含む重鎖を有する抗体。
(20)配列番号:30のアミノ酸配列を含む軽鎖を有する、(19)に記載の抗体。
(21)ヒトCD37と結合し、さらに配列番号:36のアミノ酸配列を含む重鎖を有する抗体。
(22)配列番号:38のアミノ酸配列を含む軽鎖を有する、(21)に記載の抗体。
(23)ヒトCD37と結合し、さらに配列番号:32のアミノ酸配列を含む重鎖を有する抗体。
(24)配列番号:34のアミノ酸配列を含む軽鎖を有する、(23)に記載の抗体。
(25)ヒトCD37と結合し、さらに配列番号:40のアミノ酸配列を含む重鎖を有する抗体。
(26)配列番号:42のアミノ酸配列を含む軽鎖を有する、(25)に記載の抗体。
(27)(1)から(26)のいずれかに記載の抗体の可変重鎖をコードする領域を含むDNA分子。
(28)可変重鎖コード領域がヒト起源の定常重鎖をコードする領域と融合される、(27)に記載のDNA分子。
(29)ヒト定常重鎖がIgG1である、(28)に記載のDNA分子。
(30)IgG1が配列番号:23に示される配列によってコードされる、(29)に記載のDNA分子。
(31)ヒト定常重鎖が、(14)から(18)のいずれかに記載の1つ又は2つ以上の置換をFc領域内に有する、(28)から(30)のいずれかに記載のDNA分子。
(32)(1)から(26)のいずれかの項に記載の抗体の可変軽鎖をコードする領域を含むDNA分子。
(33)可変軽鎖コード領域がヒト起源の定常軽鎖をコードする領域と融合される、(32)に記載のDNA分子。
(34)定常軽鎖がカッパ鎖である、(33)に記載のDNA分子。
(35)カッパ軽鎖が配列番号:25に示される配列によってコードされる、(34)に記載のDNA分子。
(36)(27)から(31)のいずれかに記載のDNA分子及び/又は(32)から(35)のいずれかに記載のDNA分子を含む発現ベクター。
(37)(36)に記載の1つ又は2つ以上のベクターを保有する宿主細胞。
(38)(27)から(31)のいずれかに記載のDNA分子を含む発現ベクター、及び(32)から(35)のいずれかの項に記載のDNA分子を含む第二の発現ベクターを保有する、(37)に記載の宿主細胞。
(39)哺乳動物細胞である、(37)に記載の宿主細胞。
(40)(36)に記載の1つ又は2つ以上のベクターを哺乳動物宿主細胞にトランスフェクトする工程、前記宿主細胞を培養する工程、並びに前記抗体分子を回収及び精製する工程を含む、(1)から(26)のいずれかに記載の抗体を製造する方法。
(41)活性成分として、(1)から(26)のいずれかの項に記載の1つ又は2つ以上の抗CD37抗体分子及び医薬的に許容できる担体を含む、医薬組成物。
(42)さらに1つ又は2つ以上の追加の治療薬剤を含む、(41)に記載の医薬組成物。
(43)1つ又は2つ以上の追加の治療薬剤が、CD37以外のB細胞抗原を標的とする薬剤から選択される、(42)に記載の医薬組成物。
(44)B細胞抗原がCD20である、(43)に記載の医薬組成物。
(45)1つ又は2つ以上の追加の治療薬剤が、アポトーシスを誘発する薬剤から選択される、(42)に記載の医薬組成物。
(46)薬剤がTRAILレセプターの調節物質である、(45)に記載の医薬組成物。
(47)表面にCD37を発現するB細胞を枯渇させることを目的とする、(41)から(46)のいずれかに記載の医薬組成物。
(48)B細胞悪性疾患の治療を目的とする、(47)に記載の医薬組成物。
(49)B細胞悪性疾患が、B細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択される、(48)に記載の医薬組成物。
(50)病理にB細胞が中心的に関与する自己免疫疾患又は炎症性疾患の治療を目的とする、(47)に記載の医薬組成物。
(51)細胞集団に(1)から(26)のいずれかに記載の抗体分子又はそのような抗体分子を含む医薬組成物を投与する工程を含む、細胞集団からCD37発現細胞を枯渇させる方法。
(52)in vitroで実施される、(51)に記載の方法。
(53)(41)から(46)のいずれかに記載の医薬組成物の有効量を前記患者に投与する工程を含む、B細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択されるB細胞悪性疾患に罹患している患者を治療する方法。
【実施例1】
【0043】
キメラ及びヒト化抗CD37抗体の作製
a)キメラ抗体A0の作製
配列番号:2及び配列番号:4に示す可変重鎖及び可変軽鎖アミノ酸配列(GeneArt, Regenburg, Germany)を基にして、哺乳動物細胞用最適化コドン使用頻度を適用しつつ対応するDNA配列を合成し、5’末端にHindIIIクローニング部位及び3’末端にBamHIクローニング部位を追加する。この合成DNA分子をHindIII及びBamHIで消化し、得られたDNAフラグメント(配列番号:1及び配列番号:3プラス制限部位)を、ヒトIgG1定常領域及びヒトカッパ軽鎖定常領域(それぞれ配列番号:24及び配列番号:26)をコードするpcDNA3.1系発現ベクターにてクローニングする。エンドフリープラスミド調製物(Qiagen)を作製し、各プラスミド1mg/Lの濃度で供給業者のプロトコルにしたがって、重鎖及び軽鎖プラスミドをHEK293フリースタイル細胞(Invitrogen)に同時トランスフェクトする。72時間後に、上清を採集し、ELISAによってIgG濃度を決定する。得られたキメラ抗CD37抗体(A0と称する)を改変プロテインAカラム(GE Healthcare)で精製し、クエン酸緩衝液に溶出し、続いてPBSで透析する。
【0044】
b)キメラ抗体A0のヒト化型の作製
a)で得たキメラmAb A0のヒト化は、例えばUS5,225,539、US6,548,640、US6,982,321に記載されたCDR移植方法を用いて実施する。
mAb A0のVLドメインの構造モデルを確立するために、構造の鋳型をBrookhaven National Laboratoryのタンパク質データバンク(PDB)から選択する。ネズミモノクローナル抗体エントリー“1KB5”のVLドメインが、88%配列同一性/81%類似性及び2.5Å解像により選択される。mAb A0のVHドメインのためには、90%配列同一性及び91%類似性を有する同じマウスモノクローナル抗体構造“1KB5”が主要な造形鋳型として選択される。ヒトコンセンサスフレームワークに対する最高の一致は、ヒトVカッパ1(hVK1)及びヒトVH1(hVH1)のタイプであることが見出された。また別の設計として、もっとも安定なヒトコンセンサスドメインhVK3及びhVH3への移植が選択される。移植のためには、mAb A0_VL及びmAb A0_VHモデルをヒトコンセンサスドメインモデルhVK1、hVK3、hVH1A及びhVH3と結合させ、Fv-モデルを作製する。ループ移植は、ネズミmAb A0 CDR領域をヒト抗体フレームワークに埋め込むことによって実施し、ヒト化された鎖の構築物のDNA分子を合成する。
それぞれのヒト化可変領域を合成し、免疫グロブリン発現ベクターでクローニングし、さらに、表1に示す重鎖及び軽鎖配列を一緒に、a)で述べたようにHEK293フリースタイル発現系(Invitrogen)で一過性に発現させ、プロテインAカラムで精製する。
【0045】
表1:実施例で用いられるキメラ及びヒト化抗CD37抗体の可変重鎖及び軽鎖の配列

【0046】
c)Fc操作キメラ及びヒト化抗CD37抗体の作製
Fc変異体の作製は、Lazarら(2006)の記載のように実施する。得られたFc操作重鎖配列を発現ベクターpAD-CMV1(EP393438に記載)に導入し、軽鎖コード配列を含むプラスミドと一緒にCHO-DG44細胞に同時トランスフェクトする。トランスフェクション後5から7日で細胞培養液から抗体を採集し、改変プロテインAクロマトグラフィーで精製し、クエン酸緩衝液に溶出し、続いてPBSで透析する。サンプルのタンパク質含有量はプロテインA HPLCで決定し、エンドトキシン含有量はKinetic-QCL色原体動力学アッセイ(Lonza)で決定する。サンプルのモノマー含有量はHP-SECで決定し、機能試験に用いた全てのサンプルが95%を超えるモノマー含有量を示していた。
【0047】
表2:キメラ及びヒト化抗CD37抗体の可変重鎖及び軽鎖配列(列III及びIV)並びに変異(列II)(抗体A0、B0、C0などは、表1の抗体A、B、Cなどと同一である)
重鎖及び軽鎖の完全長配列は列VおよびVIに記載される(星印を付した列Vの配列は、列IIに対応する置換(及びコードDNAに対応する変異)を含むように改変した配列番号:24及び23(野生型)のIgG1配列に該当する)
【実施例2】
【0048】
キメラmAb A0は特異的にCD37抗原を認識する
細胞CD37に対するMAb A0の特異性をFACS競合アッセイによりRamosバーキットリンパ腫細胞(ATCC#CRL−1596)で試験する。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。新しい培養液で1:6の比にて週に2−3回、この培養を継代培養により3x105から1.8x106細胞/mLの細胞濃度で維持する。FACS競合アッセイのためには、フィコエリスリン(PE)で直接標識したCD37特異的mAb HH1(Santa Cruz)を1μg/mLの濃度で用いる。抗体を非標識競合抗体A0と表示のモル比で4℃にて10分間プレインキュベートする。その後、1x105のRamos細胞を抗体混合物と氷上で30分インキュベートする。その後で、細胞をリン酸緩衝食塩水(PBS)で2回洗浄し、FACS緩衝液に再懸濁し、BD FACS Cantoで測定する。そのようなアッセイの結果は図1に示されている。20倍モル過剰のコントロールヒトIgG1抗体(Sigma IgG1カッパ)の添加は、Ramos細胞の平均蛍光強度(MFI)を有意には低下させない。20倍モル過剰の非標識HH1抗体又はA0抗体の添加は、直接標識HH1抗体の結合をほぼ完全に抑制する。このことは、A0及びHH1抗体は、Ramos細胞上の同一又は類似のエピトープを認識し、細胞CD37抗原との結合に競合することを示している。
【実施例3】
【0049】
MAb A0ヒト化型と細胞CD37抗原との結合
A0のヒト化型をそれらの細胞CD37抗原結合についてFACS分析によって試験する。抗体をRamos細胞に表示の濃度で添加し、4℃で30分結合させる。その後、PE-標識ヤギ抗ヒトIgG抗体(Sigma)を用い結合抗体を検出する。細胞をPBSで2回洗浄し、その後、FACS緩衝液に細胞を再懸濁し、BD FACS CantoでFACSにより分析する。例は図2及び3に示されている(それぞれ抗体A、B、C、D、I又はA、H、I、J、K、L及びM,表1参照)。A0のヒト化型のいくつかは、親抗体A0と同様なRamosとの結合を示し、ヒト化は細胞CD37抗原との結合を低下させないことを示している。
【実施例4】
【0050】
キメラMAb A0ヒト化型のFACSスキャチャード分析
抗体A0ヒト化型(B、C、D、H、I及びKと称される、表1参照)の細胞CD37抗原に対する親和性を、別のところに記載されている(Brockhoff et al. 1994)FACSスキャチャード分析によって決定する。略記すれば、抗体希釈を96ウェルプレートで第一のウェル(80μL)の100−400nMから始め、それに続く11希釈工程により調製する(1:2、40+40μL)。50μLのmAb希釈物をFACSチューブに添加し、150μLの細胞(0.8x106/mL=1.2x105細胞/チューブ)を各FACSチューブに加える。細胞を穏やかに混合し、1時間氷上でインキュベートする。その後、50μLのFITC結合二次抗体(濃度、15μg/mL;マウスmAb抗hu IgG全サブクラス、Zymed 05-4211)を添加し、混合し、氷上で30分間インキュベートする。その後、0.02%の酸を含む4mLのPBS(pH7.2)を添加し、細胞を沈殿させ、300μLのPBS(pH7.2)に再懸濁し、BD FACS Cantoを用いFACS分析に付す。全ての実験工程は氷水上で実施し、全ての抗体希釈物はPBS/0.5%BSA+0.02%の酸で作成する。FACSの目盛り定めはQuantum FITC MESF(Premix)High Level Beads(Bangs Laboratories)を用いて実施する。全サンプルを同じFACSパラメータを用いて測定する。結合IgG対遊離IgGの比を種々の抗体濃度でのMFI値から算出し、スキャチャードブロットとして表示する。図4は、A0のいくつかのヒト化変種におけるMFI/抗体濃度の関係を示す。これらの結果は、出発抗体に類似する、いくつかのヒト化型のRamos細胞結合を示し、解離定数(Kd)は2.15から4.90ナノモル/Lの範囲である。
【実施例5】
【0051】
キメラmAb A0のヒト化型のADCC活性
抗体依存細胞媒介性細胞傷害(ADCC)を仲介するA0ヒト化型(B、C、D、H、J、Kと称される、表1参照)の能力を、標的細胞としてRamos細胞及びエフェクター細胞としてIL2-刺激ヒトPBMCを用いて評価する。Ramos細胞(バーキットリンパ腫、ATCC#CRL−1596)をATCCから購入した。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。この培養を、新しい培養液で1:6の比にて週に2−3回、継代培養により3x105から1.8x106/mLの細胞濃度で維持する。1.5x106から1.8x106/mLの細胞濃度及びログ期増殖にある細胞培養のアリコットを10分間遠心する(200xg、すなわち1000rpm)。細胞を洗浄培養液(L-グルタミン非含有RPMI1640)で1回洗浄し、ペレットにする(200xg、すなわち1000rpm)。細胞ペレットをアッセイ培養液(L-グルタミン非含有RPMI1640に1%BSA)に再懸濁し、細胞数を決定する。細胞濃度を2x105に調整する。
健常ドナーから採取した約50−80mLの全血をPBMCの単離に使用する。50mLチューブで10mLの全血を26mLのHBSS(カルシウム及びマグネシウム非含有Hanksバランス塩類溶液)により1:3.6に希釈する。18mLの希釈全血を、50mLチューブ中の12mLのLymphoprep(Nycomed Pharma)の最上部に重層し、370xg(1400rpm)で35分遠心する。接触面から単核細胞を吸引し、1回目の洗浄をHBSS(750xg、すなわち1900rpm、10分)で、続いて2回目の洗浄をHBSS(300xg、すなわち1200rpm、10分)で、最後にHBSS(160xg、すなわち900rpm、10分)で洗浄する。ペレットにした細胞を培養液/アッセイ培養液(L-グルタミン非含有RPMI1640中に10%熱不活化ヒトAB血清)に、ピペットを用い穏やかに再懸濁させ、細胞数をセルカウンターで決定する。PBMC濃度を1x107/mLに調整する。新しく単離したPBMC(5x105/mL)を培養液(10%ヒトAB血清を補充したL-グルタミン非含有RPMI1640)中で組織培養フラスコ(75cm2)にて37℃で一晩、CO2インキュベーターで維持する。次の日、細胞を最終濃度1U/mLのhIL-2でさらに3日間刺激する。IL-2刺激PBMCをLymphoprepグラディエントで細胞屑から分離する。精製したIL-2刺激PBMCを、1x107/mLの濃度で培養液/アッセイ培養液に懸濁させる。
特異的又は非特異的抗体の存在下でのエフェクター細胞と標的細胞との同時培養を、96ウェル丸底マイクロタイタープレートによりデュープリケート又はトリプリケートで実施する。各ウェルに付き最終体積200μLのアッセイ培養液は、1:1比の10%ヒトAB血清及びRPMI中の1%BSAから成る。最初にエフェクター細胞(各ウェルに付き10%ヒトAB血清含有RPMIの100μL中に新しく単離したPBMC)をプレートし、続いて標的細胞及び1%BSA RPMI 50μL中で希釈した抗体溶液をプレートする。コントロールとして、エフェクター細胞をアッセイ培養液のみで培養し(エフェクター細胞コントロール)、さらに標的細胞をアッセイ培養液のみ(自発的溶解)又は1%トリトンX-100補充アッセイ培養液(最大溶解)で培養する。前記同時培養を湿潤CO2インキュベーターにて3時間37℃でインキュベートする。インキュベーション終了時に、室温で遠心して(200xg、すなわち1000rpm、10分)細胞を培養液から取り除く。細胞を含まない上清(100μL/ウェル)を96ウェル平底プレートの対応するウェルに移す。これらの上清100μL中のLDH活性を決定するために、反応混合物(新しく混合した250μL触媒と11.25mLの色素溶液)を各ウェルに添加し、暗所にて室温で30分インキュベートする。続いて下記に記載するように吸収を測定する。
細胞傷害検出キット(LDH、Roche)を用いてADCC活性を測定する。細胞傷害の検出は、形質膜損傷細胞から遊離されるLDH酵素活性の測定を基準にする。培養上清中に放出されるLDHは、キットのテトラゾリウム塩をフォルマザンに還元する。フォルマザン色素の最大吸収は、ELISAプレートリーダーで650nmの参照波長に対して490nmで測定される。パーセント細胞媒介細胞傷害を算出するために、各実験設定で5つのコントロールを実施する。
バックグラウンドコントロールI(1):アッセイ培養液に含まれるLDH活性、これを値(3)及び(5)から差し引く。
バックグラウンドコントロールII(2):1%トリトンX100アッセイ培養液に含まれるLDH活性、これを最大LDH放出値(4)から差し引く。
自発的LDH放出(3):標的細胞単独から放出されるLDH活性。
最大LDH放出(4):標的細胞の放出可能最大LDH活性。
エフェクター細胞コントロール(5):エフェクター細胞単独から放出されるLDH活性。
細胞媒介性細胞傷害のパーセンテージを決定するために、トリプリケート又はデュープリケートの平均吸収を算出し、製造業者の指示にしたがってバックグラウンドを差し引く。図5では、E:T比25:1及びRamos標的細胞を用いたADCCアッセイの結果が示されている。抗体は30ng/mLの濃度で添加される。出発mAb及びそのヒト化型の両方がRamos細胞に対して類似するADCC活性を示す。結論すれば、抗CD37 mAb Aのヒト化はそのADCC誘発能力を有意には変化させない。
【実施例6】
【0052】
mAB A0のヒト化型のアポトーシス促進活性
mAb A0(=A)及びそのヒト化型(B、C、D及びI;表1参照)のアポトーシス促進活性を、Ramos細胞とmAbとのインキュベーション後のアネキシンV/PI陽性細胞を測定することによって評価する。Ramos細胞(バーキットリンパ腫;ATCC#CRL-1596)をATCCから入手する。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。この培養を、新しい培養液で1:6の比にて週に2−3回、継代培養により3x105から1.8x106/mLの細胞濃度で維持する。1.5x106から1.8x106/mLの細胞濃度及びログ期増殖にある細胞培養のアリコットを10分間遠心する(200xg、すなわち1000rpm)。細胞を洗浄培養液(L-グルタミン非含有RPMI1640)で1回洗浄し、ペレットにする(200xg、すなわち1000rpm)。細胞ペレットを培養液に再懸濁し、細胞数を決定する。細胞濃度を1x106に調整する。96ウェルの丸底プレートにウェル当たり100μLの細胞懸濁液をプレートする。10%FBSを含む細胞培養液で抗体を希釈し、ウェル当たり100μLの抗体溶液を添加する。細胞をCO2インキュベーターで37℃にて20から24時間インキュベートし、その後Vybrantアポトーシスアッセイキット#2を用いて染色する。Alexa Fluor488標識アネキシンV及びヨウ化プロピジウム溶液を前記細胞に添加し、暗所で15分間インキュベートする。その後、細胞を400μLのアネキシンV結合緩衝液に再懸濁させ、BD FACS Cantoを用いてFACS分析に付す。アネキシンV陽性/PI陰性細胞及びアネキシンV/PI陽性細胞のパーセンテージを、FL1/FL2チャンネルを用いて二次元ドットブロットで決定する。アイソタイプ合致非結合抗体(Sigma human IgG1)を陰性コントロールとして用いる。
図6では、Ramos細胞に対するmAb Aの種々のヒト化型のアポトーシス促進作用が示されている。細胞を10μg/mLの抗体とともに24時間インキュベートし、アネキシンV陽性細胞の全パーセンテージ(PI陽性及びPI陰性)が示されている。親のmAb Aは強力なアポトーシス促進活性を示す。驚くべきことに、ヒト化型は、親mAb Aと比較して有意に低いアネキシンV陽性細胞数を示し、ヒト化抗体のアポトーシス促進活性の変化が指摘される。結論すれば、この実験設定ではmAb Aのヒト化はアポトーシス促進活性の低下をもたらす。
【実施例7】
【0053】
キメラmAB A0のFc操作型のADCC活性
mAB A0のFc操作型(A1、A2、A3、A4、表2参照)のADCC活性を、標的細胞としてRamos細胞を用いて評価する。ADCCアッセイは上記記載(実施例5)のように実施する。実験結果は図7に示されている。A0のFc操作型は、親のmAb A0と比較して明瞭に改善された潜在能力及び有効性を示している。ある種のFc変種は、親mAbと比較して100%に達する最大溶解の改善、さらに親mAbと比較して10倍に達するEC50の改善を示す。結論すれば、特定のFc変異の導入はキメラmAb A0のADCC活性を大きく増進させる。
【実施例8】
【0054】
mAB B0のFc操作型のADCC活性
mAB B0のFc操作型(B1、B2、B3、B4、表2参照)のADCC活性を、標的細胞としてRamos細胞を用いて評価する。ADCCアッセイは上記記載(実施例5)のように実施する。B0のFc操作型は、親のmAb B0と比較して明瞭に改善された潜在能力及び有効性を示す。ある種のFc変種は、親mAbと比較して80%に達する最大溶解の改善、さらに親mAbと比較して20倍に達するEC50の改善を示す。結論すれば、特定のFc変異の導入はヒト化mAb B0のADCC活性を大きく増進させる。この実験結果は図8に示されている。
【実施例9】
【0055】
mAb A0及びB0のアポトーシス促進活性
抗IgG mAbによる架橋の前及び後におけるRamos細胞に対するmAb A0及びB0のアポトーシス促進活性を図9に示す。アポトーシスアッセイは実施例6に記載したように実施し、抗体架橋のためには、抗ヒトIgG抗体(γ鎖特異的、Sigma)を1:1の比で抗体に添加し、標的細胞への添加前に37℃で15分インキュベートする。図9では、CD37特異mAbは、1μg/mLの濃度で架橋された状態又は架橋されていない状態で添加される。キメラmAb A0はたとえ架橋されてなくても強力なアポトーシス誘発因子であり、この作用はmAbの架橋後に顕著に強化される。驚くべきことに、架橋されない状態で、ヒト化mAb B0はアポトーシス促進活性を完全に欠くが、抗IgG Abによる架橋後に強力なアポトーシス促進活性を示す。結論すれば、この実験は、mAb A0のヒト化型のアポトーシス促進活性は、抗体架橋後に回復されえることを示している。
【実施例10】
【0056】
mAB A0のFc操作型のアポトーシス促進活性
Ramos細胞に対するキメラmAb A0のFc操作型のアポトーシス促進活性を、実施例6に記載したようにアネキシンV/PI染色によって評価する。親抗体A0並びにFc操作変種A2及びA4を0.1から10,000ng/mLの濃度範囲にわたって力価を調べる。図10で分かるように、3抗体はいずれも同様なアポトーシス促進活性を示している。結論すれば、この実験は、mAb A0のFc操作はそのアポトーシス促進活性を変化させない。
【実施例11】
【0057】
a)Fc操作抗体A2およびB2の全血アッセイにおけるB細胞枯渇活性
全血アッセイを用いて、正常B細胞のヒト血液からの枯渇の効能及び潜在能力を評価する。このアッセイ様式では、ヒト健常個体血液のEDTA処理サンプルに被検抗体を添加し、続いて37℃での3から4時間のインキュベーション後に、4色FACSアッセイによって定量的に測定される。緩衝液又はIgGコントロールとの比較によって、被検物質によるB細胞枯渇の程度を算出することができる。in vivoでの人間の状況と類似するヒトIgGレベル及びエフェクター細胞の存在により、本アッセイタイプは被検抗体のin vivoにおける作用の予測のために高い相関性を有すると考えられる。
定量的FACSアッセイを用いて、健常個体由来の血液サンプルのB細胞及び/又はスパイクされたRamos細胞の数を決定する。定量は、既知数の蛍光ビーズを含むBD Trucountチューブを用いて実施する(前記ビーズは対象の細胞集団の定量のために内部標準として機能する)。B細胞は、FSC/SSC分析を組み合わせ4つの異なるCDマーカー(CD3/CD14/CD19/CD45)を用いて4色分析により識別する。
48ウェルプレートのウェル当たり270μLの新鮮血を、30μLの抗体希釈物(PBSにて)又はPBSとともにデュープリケートでインキュベートする。サンプルを37℃で4時間インキュベートし、その後直ちに氷上に置く。33μLのCDマーカーマスターミックスをTrucountチューブに加え、50μLの血液-抗体混合物を添加する。サンプルをヴォルテックスミキサーで混合し、15分間室温でインキュベートする。その後、450μLの溶解緩衝液を加え、ヴォルテックスミキサーで混合し、さらに15分間室温でインキュベートする。サンプルを氷上に置き、直ちにBD FACS CantoTMフローサイトメーターを用いてFACS分析に付す。データの判定はBD FACSDivaソフト(ヴァージョン5.0.2)を用いて実施する。
Fc操作キメラ及びヒト化mAb A2及びB2は、正常B細胞枯渇に優れた潜在能力を示し、EC50値は0.15から0.35nMの範囲である。正常B細胞枯渇の程度は57%から65%の範囲である。リツキシマブ(B-NHLの治療に用いられる公認抗体)を並行して試験し、このアッセイ様式では極めて弱いB細胞枯渇を示す(図11A)。
b)A0及びB0のFc操作はリツキシマブと比較して優れたB細胞枯渇を導入する
健常個体に由来するヒト血液のB細胞枯渇に対するmAbの作用をa)に記載したように評価する。非Fc操作mAb A0及びB0は、リツキシマブに類似する13%から26%の範囲のB細胞枯渇活性を示す。Fc操作は、両mAbのB細胞枯渇活性の劇的な増加をもたらし、平均枯渇パーセンテージは75%である。これは、リツキシマブと比較してA2及びB2が優れていることを明瞭に示している(図11B)。
c)抗体A2及びB2は全血アッセイでT細胞及び単球を枯渇させない
Tリンパ球(CD3陽性)及び単球(CD14陽性)に対するA2及びB2の作用を、Bリンパ球に対する作用と並行して評価する。T細胞数又は単球数のいずれにも有意な変化は観察されないが、一方、B細胞数には顕著な減少が認められる(図11C)。これは、A2及びB2はヒト血液から特異的にB細胞を枯渇させることを示している。
【実施例12】
【0058】
Fc操作はリツキシマブと比較して優れたADCC活性を導入する
Ramos細胞を標的細胞として用いてmAb A0のFc操作型A2のADCC活性を評価する。ADCCアッセイは上記(実施例5)に記載したように実施する。非Fc操作抗体A0は、リツキシマブより弱いRamos標的細胞最大溶解を示す(リツキシマブは、B細胞リンパ腫罹患患者のための承認治療であるCD20特異的抗体である)。驚くべきことに、A0のFc操作は、リツキシマブを超える明瞭に改善されたA2の潜在能力及び有効性をもたらす。これは、Ramos細胞のCD20及びCD37の同様な抗原濃度で、Fc操作抗 CD37mAb A2はリツキシマブより明瞭に改善されたADCC活性を示すことを指摘している(図12)。
【実施例13】
【0059】
全血アッセイにおけるFc操作抗体A2及びB2のリンパ腫細胞枯渇活性
Ramos細胞(ヒト血液から得られたバーキットリンパ腫由来細胞株)枯渇の有効性及び潜在能力を、実施例11に記載した全血アッセイを用いて評価する。アッセイの改変として、内在性B細胞と比べて約10倍過剰のRamos腫瘍細胞を全血マトリックスにスパイクし、それらの枯渇をFACS分析でモニターする。Fc操作キメラ及びヒト化mAb A2及びB2はRamos細胞枯渇に対し良好な潜在能力を示し、EC50値は0.35から0.54nMの範囲である。Ramos細胞枯渇の程度は36%から55%の範囲である。リツキシマブ(B-NHLの治療に用いられる公認抗体)を並行して試験し、このアッセイ様式では有意に低いRamos細胞の枯渇が認められる(図13)。
【実施例14】
【0060】
疾患関連モデルにおけるFc操作抗体A2及びB2のin vivo有効性
mAb A2及びB2のin vivo抗腫瘍有効性をヌードマウスのRamosバーキットリンパ腫モデルを用いて評価する。この動物の大腿部外側の皮下にCD37陽性Ramos細胞を注射し、腫瘍が樹立されたときに動物の静脈内処置を開始する。週に2回の処置スケジュール(q3/4d)を選択し、2つの異なる用量(8mg/kg及び25mg/kg)を並行して試験する。両mAbは顕著な抗腫瘍有効性を示し、T/C値は0.2%から26%の範囲である。2つの用量レベル及び2種の抗体間に有意な相違は観察されない。しかしながら、高用量のA2処置マウスはより優れた有効性傾向を示し、T/Cは0.2%であり5/10で完全な腫瘍の退縮がある。いずれの処置も良好な治療寛容を示し、見かけの体重低下は認められない。結論すれば、mAb A2及びB2はRamosバーキットリンパ腫モデルで顕著な抗腫瘍有効性を示し、8mg/kgの用量レベルで最大活性が既に得られていた。この活性は、並行して試験したリツキシマブの活性に匹敵する。Fc操作抗体A2及びB2で観察されたin vivo活性は、これらのmAbは、ネズミのエフェクター細胞ではなくヒトの細胞との相互作用のために最適化されているが故に過小評価されている可能性があることは特記される必要がある。この最適化された相互作用(前記はヒトエフェクター細胞を用いたとき極めて改善されたin vivoのADCC活性をもたらす(実施例8))は、用いられたマウスモデルには反映されない。しかしながら、この実験で得られたデータ(図14に提示)はCD37ターゲティングの概念のin vivoでの証明を提供し、したがってヒトでの治療用量の概算に用いることができる。
【実施例15】
【0061】
ヒトにおける治療用量の概算のための、マウスにおけるA2及びB2の薬物動態作用及び薬力学作用の相関性
A2及びB2の血清濃度とそれらの薬力学作用との間の相関性はRamos腫瘍異種移植モデルを用いてマウスで確立された。これらの実験によって、8mg/kgのA2及びB2(クエン酸緩衝液(25mMのクエン酸ナトリウム、115mMのNaCl、0.04%トゥイーン80、pH6.0)中で処方)の用量が、この侵襲性の皮下腫瘍モデルの顕著な腫瘍増殖の遅延を引き起こすことが、標準的なマウスのq3又は4dの抗体投与スケジュールにより示され、したがって、投与休止期間を通して継続的な活性が指摘された。さらにまた、薬物動態データも同じ用量について確立された。
マウスにおけるこのPK/PD関係により、ヒト化抗体のヒトでのクリアランス(CL)についての報告されたデータ(Lobo et al. 2004)を用い、ヒトのための概算用量を算出することができる。
A2のための完全な計算:
−8mg/kgの1回投与後の平均AUC(0−∞)=6099μg・h/mL;
−マウスの与えられたAUC(0−∞)=マウスのAUC(ss,τ)、及びAUC(ss,τ)/τ=C(ave,ss);
−マウスのC(ave,ss)(τ=84時間の場合)=73μg/mL、これは、おそらくマウスのC(ave,ss)(τ=168時間の場合)と等価である;
−ヒトのAUC(ss,τ)=D/CLであるので、さらにLoboら(2004)が報告した、ヒトにおけるヒト化抗体クリアランス(CL)範囲であるCL=7mL/h/70kgから15mL/h/70kgを用いると;
−7mL/h/70kgの場合:168時間x7=1176mLx73μg=86mg;
−15mL/h/70kgの場合:168時間x15=2520mLx73μg=184mg。
したがって、A2の場合、70kgのヒトの場合、1週間の概算用量は86から184mgの範囲である。上記に記載した同じ仮定を用いるならば、B2の場合に算出される70kgのヒトの1週間の概算用量は189から404mgである。
【実施例16】
【0062】
抗体A2及びB2は多発性ミエローマ細胞に対しADCC活性を示す
多発性ミエローマ細胞株群におけるCD37発現を、CD37に特異的抗体を用いてFACS分析により評価する。細胞を直接蛍光標識抗CD37抗体でインキュベートするか、又は非標識CD37特異的抗体でインキュベートし、続いて一次抗体に対して誘導した第二の蛍光標識抗体とインキュベートする。標識された細胞の蛍光活性をFACS Cantoフローサイトメーター(BD Bioscience)により測定し、FACS Divaソフトを用いMFIとして蛍光強度を記録する。11の被検多発性ミエローマのうち6つがCD37の細胞表面発現を示した(図15)。続いて1つの細胞株(RPMI8226)を、CD37特異抗体A2及びB2を用いて、実施例5に記載したようにADCCアッセイで試験する。両抗体はRPMI8226に対して強力なADCC活性を示し、EC50値は25ng/mLの範囲にあり、最大細胞溶解は約20%である(図16)。本実施例は、CD37陽性多発性ミエローマ細胞は、CD37特異的mAb A2及びB2を用いたADCCによる細胞溶解に感受性であることを示している。
図15は、CD37発現についての6つの多発性ミエローマ由来細胞株のFACS分析を示す。白地曲線はCD37特異抗体との反応性を示し、黒塗り曲線は陰性コントロール抗体を表している。
【実施例17】
【0063】
患者由来CLL細胞に対する抗体A2及びB2のアポトーシス促進活性
患者由来の慢性リンパ球性白血病(CCL)細胞に対するA2及びB2のアポトーシス促進活性を評価する。CLLと診断された患者から抹消血単核細胞(PBMC)を、ヘルシンキ宣言にしたがってインフォームド・コンセントの後で調製した。Ficoll-Paque(商標)プラス工程(StemCell Technologies, Meylan, France)にしたがって、新しく採集した血液から原発性CLL細胞を精製し、RPMI1640培養液(10%熱不活化ヒトAB血清(Sigma, France)を含む)で使用まで4℃で保存する。原発性CLL細胞用培養液は、2mMのL-グルタミン及び10%熱不活化ヒトAB血清を補充したRPMI1640である。実験に使用するために、原発性CLL細胞を血球計算板で計測し、その生存率は0.25%トリパンブルー排除により評価する。CLLサンプルの生存率は90%を超える。細胞を30μg/mLの抗体とともに37℃で24時間インキュベートし、その後アネキシンV陽性細胞のパーセンテージを実施例6に記載したように決定する。図17に示すように、Fc操作抗体A2及びB2は、原発性CLL細胞に対し強力なアポトーシス促進活性を示し、アネキシンV陽性細胞は約90%(A2)及び40%(B2)である。両mAb抗体は明らかにリツキシマブより優れている(リツキシマブはB-NHL治療について承認されたB細胞特異的抗体である)。mAb A2はまた、アレムツズマブ(B-CLL治療について承認された抗体)と比較しても明瞭に優れた活性を示す。
【実施例18】
【0064】
内因性CD37遺伝子がそのヒト同族体によって置き換えられているトランスジェニックマウスモデルの作製
非翻訳配列によってフランキングされているヒトCD37コード配列(BAC(細菌人工染色体)ID:RP11-433N13、RP11-50I11)を含むターゲティングベクターを構築する。続いて、このターゲティングベクター(さらにまたエクソン3−4にフランキングするloxP部位及びfrt部位によってフランキングされているneo選別マーカーを含む)を相同性組換えに用い、マウスES細胞及び標準的技術により、マウスゲノム配列のエクソン1−8をヒトの対応する配列と置き換える。最後に、有糸分裂を不活化させたマウス胚線維芽細胞(MEF)で構成されたフィーダー層上で、C57BL/6N ES細胞株をDMEM高グルコース培養液(20%FBS(PAN)及び1200U/mLの白血病阻害因子(Millipore ESG 1107)を含む)で増殖させる。1x107細胞及び30gの直鎖化DNAベクターを240V及び500Fでエレクトロポレートする(Biorad Gene Pulser)。G418選別(200g/mL)は2日目に開始する。ガンシクロビル(2M)によるカウンター選別はエレクトロポレーション後5日目に開始する。ESクローンを8日目に単離し、増殖させさらに液体窒素でクローンを凍結した後で、標準的方法によるサザンブロッティング、例えば標的遺伝子に特異的な放射能標識DNAプローブを使用することによって分析する。続いてトランスジェニック動物を、当分野で公知の標準的な方法によって、例えば胚盤胞注入とそれに続くキメラ動物の発生によって作製する。ヒトCD37についてヘテロ接合及びホモ接合の動物を、それぞれキメラ動物及びヘテロ接合動物の通常交配によって入手する。ネズミCD37遺伝子のノックアウト及びヒトCD37遺伝子のノックインの成功は、標準的方法、例えば抹消血リンパ球のFACS分析又は組織切片の免疫組織化学的分析を用いてタンパク質レベルでモニターする。
【実施例19】
【0065】
代用抗体の作製
マカクCD37に特異的なモノクローナル抗体を、マカクCD37抗原の完全なコード配列(Acc No. ENSMMUT00000020744)を用いるマウス及びウサギの遺伝子免疫によって生成する。特異抗体は、マカクCD37抗原を発現する組換えHEK293又はCHO細胞を用い標準的なELISA又はFACS技術によって選別する。これら抗体の可変重鎖及び軽鎖コード配列をPCRクローニングによって回収し、ネズミ又はウサギの出発抗体に由来するVH及びVL領域を保有し、Fc部分は、本発明の抗体、例えばA2又はB2のそれと同一であるキメラ抗体の作製に用いる(実施例1に記載したとおり)。結合特性及び機能特性は、標的細胞としてマカクCD37発現細胞を用いるアッセイ系を使用することによって、例えば結合、FACS、スキャチャード分析、ADCC及びアポトーシスアッセイで精査することができる。最終的に、代用抗体は、カニクイザルの血液でそのB細胞枯渇活性によりin vitroで選別される。
【実施例20】
【0066】
抗体産生クローンの調製
本発明の抗体(例えば抗体A2、A4、B2又はB4)を産生するクローンを調製するために、完全な重鎖をコードするDNA分子(例えば配列番号:27、31、35又は39に示す配列をそれぞれ有する)を、pBI-26と称される真核細胞発現ベクター(ハムスター由来ジヒドロフォレートレダクターゼ選別マーカーもまた別にコードする)に挿入する。
配列番号:29、33、37及び41にそれぞれ示される完全な軽鎖をコードするDNA分子を、ネオマイシンホスホトランスフェラーゼ選別マーカーをまた別にコードするpBI-49と称される真核細胞発現ベクターに挿入する。前記完全な重鎖及び軽鎖のDNA配列を完全に配列決定する。
化学的に規定した培養液にて懸濁状態で増殖させたハムスター細胞株CHO-DG44に、上記に記載の重鎖及び軽鎖のための真核細胞発現ベクターを同時トランスフェクトする。トランスフェクト細胞を、ヒポキサンチン及びチミジンを含まず、抗生物質G418を含む培養液で選別する。続いて、細胞を段階的選別、及びメトトレキセート(MTX)の濃度を増加させる増幅に付す。800nMのMTX増幅工程から、単一細胞クローンを増殖パフォーマンス及び抗体産生を基準にスピナー操作で選別し、Safty Cell Bank(SCB)で凍結保存する。
【0067】
参考文献
American Cancer Society (Cancer Facts & Figures 2005).
Baker and Jones, Curr Opinion in Drug Discovery & Development, 10, 219-227, 2007.
Barbas, et al., Proc. Nat. Acad. Sci, USA 91:3809-3813, 1994.
Barrena et al., Leukemia 19: 1376-1383, 2005.
Belov et al., Cancer Research 61: 4483-4489, 2001.
Boulianne G. L., Hozumi N. and Shulman, M .J., Production of functional chimeric mouse/human antibody. Nature 312: 643, 1984.
Brockhoff G, Hofstaedter F, Knuechel R., Cytometry 1994, 17(1):75-83.
Buchsbaum et al., Cancer Research 52: 6476-6481, 1992.
Chothia and Lesk., J. Mol. Biol. 196: 901-917, 1987.
Coiffier B, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood 1998; 92: 1927-1932.
Coiffier, JCO, 23, 6387-93, 2005.
Edelman et al. Proc Natl Acad Sci USA 63: 78-85, 1969.
Feugier P, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2005; 23: 4117-4126.
Foran JM, et al. European phase II study of rituximab (chimeric anti-CD20 monoclonal antibody) for patients with newly diagnosed mantle-cell lymphoma and previously treated mantle-cell lymphoma, immunocytoma, and small B-cell lymphocytic lymphoma. J Clin Oncol 2000; 18: 317-324.
Forstpointner R, et al. The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood, 2004; 104: 3064-3071.
Francisco et al., Blood, 2003 Aug 15;102(4):1458-65.
Frank, et al., Methods Enzymol. 154: 221-249, 1987.
Gait, M.J., Oligonucleotide Synthesis. A Practical Approach. IRL Press, Oxford, UK (1984).
Goldenberg DM and Sharkey RM, Oncogene (2007) 26, 3734-3744. Novel radiolabeled antibody conjugates.
Hainsworth JD. Prolonging remission with rituximab maintenance therapy. Semin Oncol 2004; 31: 17-21.
Hawkins et al., J. Mol. Biol. 254:889-896, 1992.
Hayden and Mandecki. Gene synthesis by serial cloning of oligonucleotides. DNA 7(8): 571-7, 1988.
Hertz T, Yanover C: PepDist: A new framework for protein-peptide binding prediction based on learning peptide distance functions. BMC Bioinform (2006) 7 (Suppl 1):S3-S17.
Hiddemann W, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study. Group. Blood 2005; 106: 3725-3732 (2005a).
Hiddemann W, et al. Treatment strategies in follicular lymphomas: current status and future perspectives. J Clin Oncol 2005b: 23; 6394-6399.
Howard OM, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002; 20: 1288-1294.
Ichimura et al., J. Antibiot. (Tokyo), 44, 1045-53, 1991.
Jackson et al., 1995, J. Immunol. 154(7):3310-9.
Johnson S, Bird R E. Construction of single-chain derivatives of monoclonal antibodies and their production in Escherichia coli. Methods Enzymol. 203: 88-98, 1991.
Jones TD, Hanlon M, Smith BJ, Heise CT, Nayee PD, Sanders DA, Hamilton A, Sweet C, Unitt E, Alexander G, Lo KM et al: The development of a modified human IFN-α2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res (2004) 24(9):560-572.
Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JS, Baker MP: Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of Factor VIII. J Thromb Haemost (2005) 3(5): 991 1000.
Kabat E. A., Wu T. T., Perry H. M., Gottesman K. S. and Foeller C. Sequences of Proteins of Immunological Interest (5th Ed.). NIH Publication No. 91-3242. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda, MD 1991.
Kahl B, et al. Maintenance rituximab following induction chemoimmunotherapy may prolong progression-free survival in mantle cell lymphoma: a pilot study from the Wisconsin Oncology Network. Ann Oncol, 2006; 17: 1418-1423.
Kaminski et al., JCO 10: 1696-1711, 1992.
Kipriyanow and Le Gall, Molecular Biotechnology 26: 39- 60, 2004.
Knobeloch et al., MolCellBiol 20: 5363-5369, 2000.
van der Kolk LE, Baars JW, Prins MH, van Oers MH. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood, 2002, Sept 15;100(6):2257-9.
McLaughlin P, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16: 2825-2833.
Lazar et al., Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4005-10, 2006.
Ling and MacLennan, pp. 302-335 in Leucocyte Typing III. White Cell Differentiation Antigens, Oxford University Press, 1987.
Link et al., Journal Immunol 137: 3013-3018, 1986.
Lobo et al., J Pharm Sci 2004;93(11):2645-2668.
Lowman et al., Biochemistry 30(45): 10832-10837, 1991.
Marks et al., Biotechnology 10:779-783, 1992.
Moldenhauer G., et al., 1987. Biochemical characterization and epitope analysis of B lymphocyte-specific surface antigens defined by clustering workshop monoclonal antibodies. In Leukocyte Typing 111. A. McMichael. ed. Oxford University Press, Oxford. p. 378.
Moldenhauer G.: CD37. J. Biol Regul Homeost Agents 2000; 14: 281-83.
Press et al., JCO 7, 1989.
Press OW, et al. Radiolabeled-antibody therapy of B-cell lymphoma with autologous bone marrow support. N Engl J Med 1993; 329: 1219-1224.
Reche PA, Glutting JP, Zhang H, Reinherz EL: Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics (2004), 56(6):405-419.
Remington: The Science and Practice of Pharmacy, 21st edition, Hendrickson R. Ed.
Romaguera JE, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol, 2005; 23: 7013-7023.
Sasse et al., J. Antibiot. (Tokyo), 53, 879-85, 2000.
Shier et al., 1995, Gene 169:147-155.
Schwartz-Albiez et al, Journal Immunol 140: 905-914, 1988.
Stemmer et al. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides, Gene 164(1): 49-53, 1995.
van Spriel et al., Journal Immunol 172: 2953-2961, 2004.
Suzawa et al., Bioorg. Med. Chem., 8, 2175-84, 2000.
Tangri S, Mothe BR, Eisenbraun J, Sidney J, Southwood S, Briggs K, Zinckgraf J, Bilsel P, Newman M, Chesnut R, LiCalsi C, Sette A: Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol (2005) 174(6):3187-3196.
Thomas DA, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 2006; 106: 1569-1580.
Ye et al. Gene synthesis and expression in E. coli for pump, a human matrix metalloproteinase. Biochem Biophys Res Commun 186(1):143-9, 1992.
Yelton et al., 1995, Immunol. 155:1994-2004.
Zhao et al., Blood 104: Abstract 2515, 2004.
Zhao XB, Lapalombella R, Joshi T, Cheney C, Gowda A, Hayden-Ledbetter MS, Baum PR, Lin TS, Jarjoura D, Lehman A, Kussewitt D, Lee RJ, Caligiuri MA, Tridandapani S, Muthusamy N, Byrd JC. Targeting CD37+ lymphoid malignancies with a novel engineered small modular immunopharmaceutical 2007; BLOOD, 1 OCTOBER 2007, VOLUME 110, NUMBER 7 (Epub ahead of print . Blood. 2007 Apr 17)
【技術分野】
【0001】
本発明は、B細胞枯渇を基礎とする免疫療法に関する。特に本発明は、そのような療法、例えばB細胞悪性疾患及び自己免疫症状の治療で使用される抗CD37抗体分子に関する。
【背景技術】
【0002】
モノクローナル抗体(mAb)を用いる免疫療法は、B細胞枯渇を原理とする、癌及び他の疾患の治療を目的とする安全で選択性を示す方法として出現した。特にB細胞枯渇を原理とする療法、例えばB細胞悪性疾患の治療におけるモノクローナル抗体の役割は、リツキシマブ(Rituxan(商標))(B細胞表面のCD20抗原に対して誘導された抗体)の導入以来拡大した。多くの研究によって、単剤療法及び併用療法としてのリツキシマブの有効性が以下の疾患で確認された:低度NHL(Hiddemann et al. 2005a;Hiddemann et al. 2005b;Hainsworth 2004;McLaughlin et al. 1998)、マントル細胞リンパ腫(Forstpointner et al. 2004;Kahl et al. 2006;Foran et al. 2000;Howard et al. 2002;Romaguera et al. 2005)、びまん性大細胞型リンパ腫(DLCL)(Coiffier et al. 1998;Feugier et al. 2005)、及びバーキット白血病/リンパ腫(Thomas et al. 2006)。しかしながら一部分の患者のみが治療に応答し、大半の患者はリツキシマブ治療後に最終的には再発する。したがって、B細胞悪性疾患の治療に向けてCD20よりも潜在的に有効なB細胞上の新規な治療薬標的が探索されてきた(Zhao et al. 2007)。CD37抗原は細胞表面抗原であり、今日までB細胞悪性疾患のための標的としてB細胞抗原CD20と同じ程度では考慮されてこなかった。
CD37(テトラスパニンスーパーファミリーのメンバーである)は、高度にグリコシル化された細胞表面分子であり、4つのトランスメンブレンドメイン及び2つの細胞外ループを有する。CD37はほぼ例外なく成熟B細胞上で発現され、抹消血B細胞では最高レベルで、プラズマ細胞では低レベルで、さらに骨髄中のCD10+ B前駆細胞では検出不能レベルで発現される。CD37の低レベル発現はまた、休止及び活性化T細胞、顆粒球並びに単球で報告されている。B細胞新形成では、CD37発現は、主として急速進行性非ホジキンリンパ腫(NHL)及び慢性リンパ性白血病(CCL)で観察される。高レベルのCD37発現はまたマントル細胞リンパ腫(MCL)で見出される。この発現パターンによりCD37は抗体仲介癌療法の有望な標的となっている。
【0003】
CD37は1986年に初めて記載され、ネズミのモノクローナル抗体MB-1によって性状が決定された(Link et al. 1986)。
CD37の生理学的役割は不明である。CD37欠損マウスは、リンパ性器官の発育及び細胞構成に変化を示さないが、IgG1レベルを低下させ、T細胞媒介免疫反応を弱める(Knobeloch et al. 2000)。CD37-/-T細胞に関する研究によって、T細胞増殖におけるCD37の役割が示唆されている(van Spriel et al. 2004)。
種々の疾患で悪性B細胞上におけるCD37の発現が報告されている。CD37は、バーキットリンパ腫、濾胞性リンパ腫及びリンパ球性リンパ腫のような大半の成熟B細胞性悪性疾患で発現される(Link et al. 1986)。高レベルのCD37発現が、毛様細胞白血病において並びに慢性リンパ球性白血病(CLL)及び種々の非ホジキン型リンパ腫(NHL)(マントル細胞リンパ腫(MCL)を含む)患者のサンプルにおいて観察されている(Schwartz-Albiez et al. 1988;Barrena et al. 2005)。免疫表現型について抗体マイクロアレイを利用したある報告では、CD37は、悪性CCL細胞(CD37高発現)と正常抹消血(PB)リンパ球(CD37低発現)との間の良好な識別物質であると主張されている(Beloz et al. 2001)。
CD37特異的mAbと癌細胞との結合は多様な作用メカニズムの引き金となりえる:第一に、抗体がCD37抗原の細胞外ドメインと結合した後、補体カスケードを活性化し、標的細胞を溶解させることができる。第二に、抗CD37抗体は、標的細胞に対する抗体依存細胞媒介性細胞傷害(ADCC)を仲介することができる(ADCCは免疫系の細胞傷害性細胞上の適切なレセプターによる結合抗体のFc部分の認識後に生じる)。
第三に、抗体は、抗原又は他の刺激に対して応答するB細胞の能力を変化させることができる。最後に抗CD37抗体はプログラムされた細胞死(アポトーシス)を開始させることができる。
【0004】
抗CD37 mAbのMB-1は、B-NHL(B細胞非ホジキンリンパ腫)患者について2つの放射線免疫療法の臨床試験で評価された(Press et al. 1989;Kaminski et al. 1992)。一方の臨床試験で、治療用量の131I-MB-1が6人のNHL再発患者に投与され、6人の患者全員が、平均期間が7ヶ月の臨床的に完全な緩解(CR)を達成した。重要なことには、6人の患者のうち2人が、ほんの微量用量のMB-1の投与後に既に臨床徴候の沈静を示し、抗体自体の直接的な抗腫瘍作用を示唆した。第二の臨床試験では、放射能標識MB-1を難治性NHL患者の治療に適用し、9人の評価可能な患者のうち3人が限定的期間において客観的応答を示した(Kaminski et al. 1992)。両臨床試験において、微量標識MB-1抗体の注射後に急速で一過性の抹消B細胞の枯渇が報告された。これらの観察は、MB-1は細胞傷害性活性をそれ自体有するという結論を支持した。要約すれば、これらの臨床試験は、B細胞性悪性疾患に対するCD37ターゲティングの有効性を強調し、抗CD37療法の潜在的な臨床応用性を指摘している。
CD37特異抗体類似単鎖分子(“小モジュール免疫薬(Small Modular ImmunoPharmaceutical, SMIP)”)を用いて、当該分子による治療はin vitroでアポトーシスを誘発し、さらにin vivoの異種移植モデルでバーキットリンパ腫の増殖を遅らせるという実験的証拠が得られている。Trubion社の組換え抗CD37 SMIP Tru16.4の抗アポトーシス活性が最近報告された(Zhao et al. 2004)。Tru 16.4は、腫瘍患者由来の原発性CLL細胞に対してカスパーゼ非依存性アポトーシスを誘発した。これら細胞におけるアポトーシスの誘導はリツキシマブのそれよりも強く、さらにアレムツズマブ(CD52アンタゴニスト)のアポトーシス誘発に匹敵した。アポトーシス誘発の程度はCD37の細胞表面発現に正比例し、抗ヒトIgG抗体による架橋によってさらに強化することができた。CD37発現とADCCとの相関性が細胞株においてin vitroで示された。バーキットリンパ腫マウスモデル(Raji)で、抗CD37 scFvによる処置は治療有効性を示した(Zhao et al. 2007)。これらのデータは、CD37ターゲティングは、アポトーシス及びADCCの誘導による標的誘導抗腫瘍療法のための有望なアプローチであるという最初の証拠を提供する。
結論として、CD37抗原は、いくつかのヒトB細胞悪性疾患の腫瘍細胞及び正常な成熟B細胞で頻繁に発現され、さらに抗CD37系療法はB細胞悪性疾患の治療のための有望なアプローチでありえることが示された。CD37陽性正常B細胞の枯渇は重大であるとは考えられない。なぜならば、多数の患者の臨床データが、抗CD20 mAbによる6ヶ月までのB細胞の長期的枯渇でさえ、IgG血清レベルを顕著には低下させず、感染リスクを増大させることもないことを示しているからである(Van der Kolk et al. 2002)。
上記に記載の抗CD37抗体又は抗体様分子(MB-1及びSMIP Tru16.4)は、B細胞悪性疾患における抗腫瘍有効性及びCD37を標的とする潜在能力を示したが、B細胞枯渇に基づく療法を改善するためにまた別の抗CD37インヒビターが希求される。
【発明の概要】
【発明が解決しようとする課題】
【0005】
本発明の目的は、B細胞悪性疾患及びCD37陽性B細胞の枯渇に応答する他の疾患の治療を目的とする新規なCD37アンタゴニストを提供することであった。
さらにまた本発明の目的は、改善されたエフェクター機能を有する抗CD37抗体を提供することであった。特に、本発明者らは、抗体依存細胞媒介性細胞傷害(ADCC)を有する抗CD37 mAbを提供しようとした。
【課題を解決するための手段】
【0006】
本発明の根幹に横たわる問題を解決するために、出発抗体として、ネズミモノクローナル抗CD37抗体をヒトの治療に有用なキメラ及びヒト化抗CD37抗体を作製するために用いた。
第一の特徴では、本発明は、ヒトCD37と結合し、さらに
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定されるネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体を提供する。
【図面の簡単な説明】
【0007】
【図1】キメラ抗体A0はCD37抗原を特異的に認識する(FACS競合アッセイによって決定)。
【図2】細胞CD37抗原とヒト化型A0との結合(FACSによって決定)。
【図3】細胞CD37抗原とヒト化型A0との結合(FACSによって決定)。
【図4】細胞CD37抗原に対するヒト化型A0の親和性(FACSスキャチャードによって決定)。
【図5】ヒト化型A0のRamos細胞に対するADCC活性。
【図6】ヒト化型A0のRamos細胞に対するアポトーシス促進活性。
【図7】Fc-操作型mAb A0のRamos細胞に対するADCC活性。
【図8】Fc-操作型mAb B0のRamos細胞に対するADCC活性。
【図9】mAb A0及びB0のアポトーシス促進活性。
【図10】Fc-操作型mAb A0のアポトーシス促進活性。
【図11A】リツキシマブと比較した、全血アッセイにおけるFc-操作抗体A2及びB2による正常ヒトB細胞の枯渇。
【図11B】リツキシマブと比較した、Fc-操作後の抗体の優れたB細胞枯渇活性。
【図11C】抗体A2及びB2は、全血アッセイでT細胞及び単球を枯渇させない。
【図12】リツキシマブと比較した、Fc-操作後の優れたADCC活性。
【図13】リツキシマブと比較した、全血アッセイにおけるFc-操作抗体A2及びB2によるRamosバーキットリンパ腫細胞の枯渇。
【図14】ヌードマウスにおけるFc-操作抗体A2及びB2によるRamos異種移植腫瘍のin vivo腫瘍増殖阻害。
【図15】多発性ミエローマ細胞におけるCD37の発現。
【図16】抗体A2及びB2の多発性ミエローマ細胞に対するADCC活性。
【図17】抗体A2及びB2の患者由来CLL細胞に対するアポトーシス促進活性。
【発明を実施するための形態】
【0008】
上述したように、本発明の根幹に横たわる問題を解決するために、出発抗体として、ネズミモノクローナル抗CD37抗体をヒトの治療に有用なキメラ及びヒト化抗CD37抗体を作製するために用いた。
第一の特徴では、本発明は、ヒトCD37と結合し、さらに
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定されるネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体を提供する。
以下から理解されるように、別の抗体(すなわち出発抗体)から“誘導された”抗体とは、抗体が、以下に記載するように出発抗体を改変することにより作製されることを意味する。
好ましい実施態様では、抗体分子は、a)に規定した出発抗体から誘導されたキメラ又はヒト化抗体分子である。関連配列を有する抗体はG28.1と称され、WO2005/017148に開示された。
【0009】
カテゴリーb)の出発抗体は、例えば、第三回HLDAワークショップでCD37抗原を性状決定したG28.1のようなCD37特異抗体から選択することができる。これらの抗体はHD28、HH1、BI14、F97-3G6と称された(Ling and MacLennam, 1987)。報告されている他のCD37特異抗体には、RFB-7、Y29/55、MB-1、M-B371、M-B372及びIPO-24が含まれる。Moldenhauer(2000)及びSchwartz-Albiezら(1988)によれば、これらの抗体はいずれも(G28.1を含む)、同じ又は近接するか若しくはオーバーラップするCD37エピトープを認識する。Schwartz-Albiezら(1988)は、このエピトープはCD37の炭水化物部分に位置することを指摘している。上記の多数の抗体が市場で入手可能である(例えばHH1(SantaCruz)、RFB-7(Biodesign)、Y29/55(Biogenesis)、M-B371(BD Biosciences)、M-B372(SantaCruz)及びIPO-24(AbCam))。
他のCD37特異抗体は、S-B3(Biosys)、NMN46(Chemicon)及びICO-66(Bioprobe)である。抗体がG28.1と同じエピトープを認識するか否かは、競合結合アッセイによって、又は交差阻害放射性免疫アッセイ(Moldenhauerら(1987)及びMoldenhauer(2000)により記載された)によって決定することができる。
例示すれば、競合結合は、CD37タンパク質若しくはCD37ペプチドを被覆したプレート、又はCD37陽性細胞を用い、競合候補抗体の存在下でビオチン化抗体の結合を測定することによってELISAで決定することができる。競合性抗体又は抗体誘導フラグメントの存在下では、ビオチン化G28.1(又は同じエピトープを認識することが判明している別の抗体)の結合は、抗体が共通エピトープを認識する場合には低下する。G28.1エピトープペプチドを識別するために、CD37配列から誘導されたフラグメント若しくは短いポリペプチド又は組換えタンパク質を合成するか又は生成し、G28.1と前記ペプチド/ポリペプチドとの結合をELISAアッセイで測定することができる。競合結合はまた、実施例で述べるようにFACS分析によって決定することができる。
【0010】
キメラ又はヒト化抗体分子の生成のための出発抗体としてb)に規定する抗体を、G28.1と同様な態様で用いることができる。
カテゴリーb)の出発抗体はまた、対応するエピトープを含むペプチド又はタンパク質フラグメント又はそのようなペプチド/フラグメントをコードするDNA分子をそれぞれ免疫に用い、G28.1と同じエピトープと反応する抗体を得ることによって、de novoに作製することができる。
出発抗体b)はまた、関連エピトープを保持する全細胞で免疫することによって得ることができる。このようにして得たハイブリドーマ細胞を続いて分泌抗体の競合性結合についてスクリーニングする。
“抗CD37抗体分子”という用語は、抗CD37抗体及び抗CD37抗体フラグメントとともに抗体分子とのコンジュゲートを包含する。本発明の意味では、抗体にはキメラモノクローナル抗体及びヒト化抗体が含まれる。“抗体”という用語(前記は“抗体分子”と相互に用いられる)は、完全な免疫グロブリン(リンパ球によって生成され、例えば血清中に存在する)、ハイブリドーマ細胞株によって分泌されるモノクローナル抗体、宿主細胞で組換え発現によって生成されるポリペプチド(免疫グロブリン又はモノクローナル抗体の結合特異性を有する)、並びに改変及び更なるプロセッシングによって前記のような抗体から誘導され、なおそれらの結合特異性を維持している分子を包含するであろう。
本発明のある実施態様では、抗CD37抗体分子は、
i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、
ii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖、
iii)ヒト起源である定常重鎖及び軽鎖、
によって規定されるキメラ抗体である。
【0011】
キメラマウス/ヒト抗体の構築及び生成は当分野では周知である(Boulianne et al. 1984)。非ヒト抗体の可変領域は、典型的にはヒト免疫グロブリンの免疫グロブリン定常領域の少なくとも一部分(Fc)と連結される。ヒト定常領域DNA配列は、周知の方法にしたがって多様なヒト細胞、好ましくは不朽化B細胞から単離することができる(以下を参照されたい:Kabat et al. 1991;及びWO87/02671)。抗体分子は、定常領域の全部又は一部分のみを、それら分子がCD37抗原及びFcレセプターとの特異的結合を示すかぎり含むことができる。定常領域の型及び長さの選択は、補体結合又は抗体依存細胞媒介性細胞傷害のようなエフェクター機能が所望されるか否か、及び抗体分子の所望される薬理学的特性に左右される。
ある種の実施態様では、本発明の抗体分子は、ヒト重鎖定常領域IgG1と融合したa)又はb)に規定される非ヒト抗体の重鎖可変領域、及びヒト軽鎖定常領域カッパと融合したa)又はb)に規定される非ヒト抗体の軽鎖可変領域を有するキメラCD37特異抗体である。
さらに別の実施態様では、本抗体分子は、ヒト重鎖定常領域IgG1(配列番号:24(コードDNA配列は配列番号:23)に示される配列を有するIgG1分子)又は前記から誘導された変異IgG1と融合した配列番号:2に示される重鎖可変領域を有し、さらに配列番号:26(コードDNA配列は配列番号:25)に示されるヒト軽鎖定常領域カッパと融合した配列番号:4に示される軽鎖可変領域を有するキメラCD37特異抗体である。
a)又はb)に規定される非ヒト出発抗体をキメラ化するための他のヒト定常領域も当業者には利用可能であり、例えばIgG2、IgG3、IgG4、IgA、IgE若しくはIgM(IgG1の代わり)又はラムダ(カッパの代わり)である。定常領域もまたキメラであってもよく、例えば重鎖IgG1/IgG2又はIgG1/IgG3キメラである。
【0012】
本発明のある種の実施態様では、抗CD37抗体分子は、
i)配列番号:2に示される可変重鎖内に含まれるCDR、
ii)配列番号:4に示される可変軽鎖内に含まれるCDR、
iii)ヒト抗体から誘導される、前記CDRを支えるフレームワーク、
iv)ヒト抗体に由来する定常重鎖及び軽鎖、
によって規定されるヒト化抗体である。
非ヒト抗体(例えばネズミ、ラット、又はウサギ抗体)のヒト化型は、非ヒト免疫グロブリンから誘導された最小限の配列を含む免疫グロブリン、免疫グロブリン鎖又はそのフラグメント(例えばFv、Fab、Fab’、F(ab’)2、又は抗体の部分配列を有する他の抗原結合分子)である。
ヒト化抗体は、レシピエント抗体の相補性決定領域が、望ましい特異性、親和性及び性能を有する非ヒト種(ドナー抗体)、例えばマウス、ラット又はウサギのCDR由来残基によって置き換えられた(レシピエント抗体由来)ヒト免疫グロブリンを含む。いくつかの事例では、ヒト免疫グロブリンのFvフレームワーク残基は対応する非ヒト残基によって置き換えられる。
本発明のヒト化抗体では、a)又はb)に規定される非ヒト出発抗体のCDRをコードする配列は、ヒト免疫グロブリンの重鎖及び軽鎖の対応する遺伝子に移植された。
モノクローナル抗体の“相補性決定領域”(CDR)は、Chothia and Lesk(1987)の論文に関連するKabatら(1991)の論文にしたがえば、特異的な抗原結合に中心的に関与するアミノ酸配列であると理解される。配列の特徴についてKabatの配列データベースを検索することによって、配列番号:2及び配列番号:4に示される可変領域の配列からCDR配列を日常的に決定することができる。
ヒト化抗体を得るための技術は当業者には日常的に利用可能であり、それら技術はとりわけUS5,225,539、US6,548,640及びUS6,982,321に記載されている。
【0013】
CDR移植抗体の適切なフレームワーク残基をネズミ残基に戻して、結合親和性を改善することができる。上記に記載したように、当業者は、与えられた非ヒト抗体からCDRの入手の仕方、適切なヒト免疫グロブリン遺伝子の選択及び入手の仕方、これら遺伝子へのCDRの移植の仕方、選択したフレームワークの改変の仕方、CDR移植抗体の適切な宿主細胞(例えばチャイニーズハムスター卵巣(CHO)細胞)での発現の仕方、及び得られた組換え抗体の結合親和性及び特異性の試験の仕方を当分野の関係する方法から知ることができる。
ヒト化抗体を得るために、抗原結合部位(重鎖のCDR及び軽鎖のCDRによって形成される)を、げっ歯類(ネズミ)のモノクローナル抗体を分泌する細胞のDNAから切り出し、ヒト抗体のフレームワークをコードするDNAに移植する。
CDR移植の代わりに、非ヒト(特にネズミ)抗CD37抗体は、いわゆる“表面再仕上げ(resurfacing)”技術によってヒト化することができる。前記技術によって、げっ歯類フレームワーク領域は、US5,639,641に記載されたように、表面露出残基を除き変化を受けない。
さらに別の特徴では、本発明は、配列番号:6に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
別の特徴では、本発明は、配列番号:8に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
別の特徴では、本発明は、配列番号:10に示される配列を有する可変重鎖、及び配列番号:12、配列番号:14、配列番号:16、配列番号:18、配列番号:20及び配列番号:22に示される配列から選択される配列を有する可変軽鎖をもつヒト化抗体に関する。
上記に規定したヒト化抗体は表1に示される。
【0014】
ある種の実施態様では、ヒト化抗体は、ヒト重鎖定常領域IgG1及びヒト軽鎖定常領域カッパを有する。キメラ抗体について上記で述べたように、定常領域は他のクラス及びサブクラスから選択してもよい。
ある種の実施態様では、本発明のヒト化抗体では、ヒト定常重鎖IgG1は配列番号:24に示される配列を有するIgG1分子又は前記から誘導された変異IgG1分子であり、ヒト軽鎖定常領域カッパは配列番号:26に示される配列を有する。
本発明の抗CD37抗体分子はまた、配列表に示されるアミノ酸配列によって規定される抗体の変種であってもよい。日常的に利用可能な技術を用いて、当業者は、上記に規定の抗体の機能的変種を調製、試験及び利用することができるであろう。前記の例は、CDR及び/又はフレームワーク内の少なくとも1つの位置が変異した変種抗体、フレームワーク領域内に生殖系列配列から逸脱したただ1つのアミノ酸置換を有する変種抗体、保存的アミノ酸置換を有する抗体、配列表に提示されている可変鎖をコードするDNA分子とストリンジェントな条件下でハイブリダイズするDNA分子によってコードされる抗体である。
個々のアミノ酸の特性が提供されるならば、合理的な置換を実施して、出発抗体の全体的な分子構造を保存した抗体変種を得ることができる。アミノ酸置換、すなわち“保存的置換”は、対応するアミノ酸の例えば極性、荷電、可溶性、疎水性、親水性及び/又は両親媒性を基準にして実施することができる。当業者は、一般的に実施されるアミノ酸置換(例えばWO2007/042309に記載されている)及びそのように改変された抗体を得る方法を熟知していよう。遺伝暗号並びに組換え及び合成DNA技術を提供されるならば、1つ又は2つ以上の保存的アミノ酸交換を有する変種抗体をコードするDNA分子を日常的に設計して、対応する抗体を容易に得ることができる。
【0015】
配列表に示されるその可変鎖によって規定される抗体と比較して、本発明に包含される抗体変種は、CDR領域に少なくとも60%、より好ましくは少なくとも70%又は80%、さらに好ましくは少なくとも90%、もっとも好ましくは少なくとも95%の配列同一性を有する。好ましい抗体はまた、CDR領域に少なくとも80%、より好ましくは90%、もっとも好ましくは95%の配列類似性を有する。好ましい抗体変種は、可変領域に少なくとも60%、より好ましくは少なくとも70%又は80%、さらに好ましくは少なくとも90%、もっとも好ましくは少なくとも95%の配列同一性を有する。好ましい抗体はまた、可変領域に少なくとも80%、より好ましくは90%、もっとも好ましくは95%の配列類似性を有する。
2つのポリペプチド間における“配列同一性”は、当該配列間で同一であるアミノ酸のパーセンテージを示す。“配列類似性”は、同一であるか又は保存的アミノ酸置換を示すアミノ酸のパーセンテージを示す。
変種はまた、配列表に示される規定の配列を有する抗体を最適化のための出発点として用い、1つ又は2つ以上のアミノ酸残基(好ましくは1つ又は2つ以上のCDR内のアミノ酸残基)を多様化させることによって、さらに得られた抗体変種の収集物を改善された特性を有する変種についてスクリーニングすることによって入手することができる。可変軽鎖のCDR3、可変重鎖のCDR3、可変軽鎖のCDR1及び/又は可変重鎖のCDR2内の1つ又は2つ以上のアミノ酸残基の多様化が有用であることが実証された。多様化は、当分野で公知の方法、WO2007/042309に言及されている例えばいわゆるTRIM技術によって実施することができる。
【0016】
さらに別の実施態様では、本発明の抗CD37抗体分子は“アフィニティ成熟”抗体である。
“アフィニティ成熟”抗CD37抗体は、配列表に示された配列を有する抗体から誘導される抗CD37抗体であり、1つ又は2つ以上のCDR内に1つ又は2つ以上の変異を有し、それにより対応する原型の非成熟抗体と比較して抗原に対する親和性の改善がもたらされる。そのような抗体の変異体を作製する方法の1つはファージディスプレイを必要とする(Hawkins et al. 1992;及びLowman et al. 1991)。略記すれば、いくつかの超可変領域部位(例えば6−7部位)を変異させて、各部位で全ての可能なアミノ酸置換を作製する。このようにして作製した抗体の変異体を、繊維状ファージから各粒子内にパッケージされたM13の遺伝子III生成物との融合物として一価の態様でディスプレイさせる。続いてその生物学的活性(例えば結合親和性)について、このファージディスプレイ変異体を本明細書に開示するようにスクリーニングする。
アフィニティ成熟抗体はまた、例えばMarksら(1992)(可変重鎖(VH)及び可変軽鎖(VL)ドメインシャッフリングによるアフィニティ成熟)、又はBarbasら(1994)、Shierら(1995)、Yeltonら(1995)、Jacksonら(1995)及びHawkinsら(1992)(CDR及び/又はフレームワーク残基のランダム変異導入)が記載した方法によって作製することができる。好ましいアフィニティ成熟抗体は標的抗原に対してナノモルレベル又は場合によってピコモルレベルの親和性を有するであろう。さらに別の実施態様では、本発明の抗CD37抗体分子は“脱免疫化”抗体である。
“脱免疫化”抗CD37抗体は、配列表に示された配列を有するヒト化又はキメラ抗体から誘導される抗体であり、そのアミノ酸配列内に1つ又は2つ以上の変異を有し、それにより対応する原型の脱免疫されていない抗体と比較して抗体の免疫原性の低下がもたらされる。そのような抗体変異体を作製する方法の1つは、抗体分子のT細胞エピトープの識別及び除去を必要とする(Baker and Jones, 2007)。第一の工程では、抗体の免疫原性をいくつかの方法によって、例えばin vitro T細胞エピトープ決定又はそのようなエピトープのin silico予想によって、文献に記載されているように(Jones et al. 2004;Jones et al. 2005;Reche et al. 2004;Hertz et al. 2006)決定することができる。いったんT細胞エピトープ機能に決定的な残基を識別したら、免疫原性を除去し抗体活性を維持するために変異を導入することができる(Jones et al. 2005;Tangri et al. 2005)。タンパク質における変異導入方法は当分野で周知であり、例えばオーバーラップPCR技術によって実施される。
【0017】
抗体のFc領域は多数のFcレセプターと相互作用するので(その相互作用の結果、多数の重要な機能的能力(“エフェクター機能”と呼ばれる)が生じる)、ある種の実施態様では、抗体は、完全長抗体又はFc領域の一部分を含む抗体である(ただし後者は抗原の対応する部分及びFcレセプターとの特異的な結合を示す必要がある)。定常領域のタイプ及び長さの選択は、補体の結合又は抗体依存細胞媒介性細胞傷害のようなエフェクター機能が所望されるか否か及び抗体タンパク質の所望される薬理学的特性に左右される。
本発明のある実施態様では、抗CD37抗体は、Fc領域又はその対応する断片を含むキメラ又はヒト化抗体であり、前記Fc領域は、エフェクター機能を調節するため、特に1つ又は2つ以上のFcレセプターと抗体との結合を強化して、それによりADCCエフェクター機能を強化するために操作されている。Fc領域の操作は、Fc領域が操作されていない親抗体の場合よりもエフェクター細胞の存在下で抗体のエフェクター機能をより効率的に仲介する。ある実施態様では、そのような抗体変種は、親抗体によって仲介されるADCCよりも強力なADCCを仲介する(以下では、特段の記載がなければ、抗体分子の関係又はIgG若しくはFc領域の関係で“親”という用語は、それぞれ操作されていない抗体分子、Fc領域又はIgG(それらから変異した(操作された)分子が誘導される)を意味する。
Fc領域の多様な改変が、当分野で学術文献及び特許文書の両方で、例えばEP0307434、WO9304173、WO9734631、WO9744362、WO9805787、WO9943713、WO9951642、WO9958572、WO02060919、WO03074679、WO2004016750、WO2004029207、WO2004063351、WO2004074455、WO2004035752、WO2004099249、WO2005077981、WO2005092925、WO2006019447、WO2006031994、WO2006047350、WO2006053301、WO2006088494及びWO2007041635で提唱されている。
【0018】
好ましい実施態様では、本発明の抗体は、332位及び/又は239位及び/又は236位にアミノ酸置換を有するFc変種である。好ましい実施態様では、本発明の抗体は、以下の群から選択されるFcドメインの変異を有する:
i)332位におけるただ1つの置換、好ましくはI332E;
ii)239位及び332位における置換の組合せ、好ましくはS239D/I332E;
iii)236位及び332位における置換の組合せ、好ましくはG236A/I332E;
iv)236位、239位及び332位における置換の組合せ、好ましくはG236A/S239D/I332E。
上記に規定した置換は、Lazarら(2006)の文献、WO2004029207及びWO2007041635に記載されている。
本発明の抗体におけるFc変種は、それらを含むアミノ酸改変にしたがって規定される。したがって、例えばI332Eは、親のFcポリペプチドに対して置換I332Eを有するFc変種である。同様に、S239D/I332Eは、親のFcポリペプチドに対して置換S239D及びI332Eを有するFc変種を規定し、S239D/I332E/G236Aは、置換S239D、I332E及びG236Aを有するFc変種を規定する。
番号付与はEU番号付与体系(Kabat et al. 1991)にしたがい、前記はEU抗体(Edelman et al. 1969)の番号付与に該当する。当業者には理解されるところであるが、これらの取り決めは、免疫グロブリン配列の特別な領域における非連続的番号付与から成り、これによって免疫グロブリンファミリーにおける保存的な位置に対し標準化された参照が可能である。
上記に規定した抗体で、置換された236位、239位及び332位は、配列番号:24に示されるIgG1重鎖のそれぞれ119位、122位及び215位に対応する(配列番号:28、32、36及び40に示される抗体A2、A4、B2及びB4の重鎖の完全長配列では、置換されたアミノ酸は235位、238位及び331位に存在する)。
【0019】
ある種の実施態様では、本発明のFc領域はヒトIgG配列を土台にしており、したがってヒトIgG配列が、他の配列を比較する“基準”配列として用いられる。本発明の抗体のために、操作されるFc領域は好ましくはIgG、特にIgG1であるが、Fc領域はまた、IgG2若しくは他の免疫グロブリンクラス(例えばIgA、IgE、IgGD、IgM)由来の変種配列、又は2つ以上の免疫グロブリンクラスのキメラ型(例えばIgG2/IgG1)などであってもよい。本発明のFc変種は1つの親IgGにおいて操作されるが、この変種は別の第二の親IgGにおいて操作されても、又は前記第二の親IgGに“移され”てもよい。これは、“等価である”又は“対応する”残基を決定し、典型的には第一及び第二のIgGの配列間の配列相同性又は構造相同性を基準にしてこの第一及び第二のIgGとの間で置換を実施することによって達成される。相同性を確立するために、本明細書に概略する第一のIgGのアミノ酸配列を第二のIgGの配列と直接比較する。当分野で公知の1つ又は2つ以上の相同性アラインメントプログラムを用い、アラインメントを維持するため(すなわち任意の欠失及び挿入による保存残基の排除を回避するため)に必要な挿入及び欠失を可能にして配列のアラインメントを実施した後、第一のFc変種の一次配列における具体的なアミノ酸と等価の残基が規定される。決定された等価又は対応する残基が何であれ、さらにIgGが作製される親IgGの実体が何であれ、言わんとすることは、本発明で用いられるFc変種を操作して、Fc変種と顕著な配列又は構造相同性を有する任意の第二の親IgGを得ることができるということである。したがって、例えば、親抗体がヒトIgG1である変種抗体が、上記に記載の方法又は等価残基を決定する他の方法を用いることによって作製される場合、変種抗体は、例えばヒトIgG2親抗体、ヒトIgA親抗体で操作されえる(WO2007041635を参照されたい)。
【0020】
本発明の抗体は抗原CD37を標的とし、これは、CD37発現レベルがCD20の発現レベルより高い疾患(例えば慢性リンパ球性白血病、この疾患ではサンプルはCD20 mRNAの低発現レベルと比較してCD37 mRNAの高発現レベルを示す)でCD20を標的とするよりも有利でありえる。
本発明の抗体は、Ramos細胞に対するADCC活性、全血における正常B細胞枯渇及びRamosバーキットリンパ腫細胞枯渇という点についてリツキシマブ(公認された抗CD20抗体)よりも優れていることが示された。本発明の実験で示しえたように、本発明の抗体(Fc未操作及びFc操作の両抗体)は、リツキシマブのB細胞枯渇活性を超える活性を有する。変異Fc領域を有する抗体は、リツキシマブと比較してほぼ10倍高いB細胞枯渇活性を示す(図11B)。
本発明の代表的CD37抗体は、架橋することなく強力なアポトーシス促進活性を示す。これに関しては、この特性を有する抗体は、抗CD37 SMIP Tru16.4(架橋無しにはアポトーシスを示さない(Zhao et al. 2007))よりも優れている。架橋無しでのアポトーシスの誘発(本発明の抗体(Fc操作及びFc未操作の両抗体)はこれを示すことができた)は、in vivoの架橋因子(例えばFcγレセプターを保持するエフェクター細胞)の非存在下で、又は標的抗原CD37の低濃度下で(例えばCD37発現が低レベルの腫瘍細胞)有利である。架橋無しにアポトーシスを誘導する抗体はなお細胞死を引き起こすことができるが、一方、架橋依存抗体は細胞死を引き起こさない。
【0021】
さらに別の特徴では、本発明の抗CD37抗体分子は、本発明のヒト化又はキメラCD37特異抗体から誘導される抗体フラグメントである。抗体フラグメント、例えばFabフラグメントを得るために、日常的技術による手段によって、例えばパパインを用いて消化を実施することができる。パパイン消化の例はWO94/29348及びUS4,342,566に記載されている。抗体のパパイン消化によって典型的には2つの同一の抗原結合フラグメント(いわゆるFabフラグメント、各々はただ1つの抗原結合部位を有する)及び残余のFc領域が生成される。ペプシン処理によって2つの抗原結合部位を有し、なお抗原と架橋することができるF(ab’)2フラグメントが得られる。
抗体の消化によって得られたFabフラグメントはまた、軽鎖の定常ドメイン及び重鎖の第一の定常ドメイン(CH1)を含む。Fab’フラグメントとFabとは、Fab’フラグメントは、抗体ヒンジ領域由来の1つ以上のシステインを含む追加の残基を重鎖CH1ドメインのカルボキシ末端に含むという点で異なる。Fab’-SHは定常ドメインのシステイン残基が遊離チオール基を保持するFab’の本明細書における記述である。F(ab’)2抗体フラグメントは最初、Fab’間のシステイン残基を有するFab’フラグメント対として生成された。抗体フラグメントはまた、それぞれのコードDNAフラグメントを作製する分子生物学的方法によって生成することができる。
抗体分子は典型的には、2つの軽鎖/重鎖対から成るテトラマーであるが、又はダイマーであってもよく(すなわち1つの軽鎖/重鎖対(例えばFab又はFvフラグメント)から成る)、又はモノマー単鎖抗体(scFv;Johnson and Bird, 1991)、ミニボディ又はジアボディであってもよい。
【0022】
抗CD37抗体分子はまた、コンジュゲートの形態(すなわち細胞傷害性物質、特に腫瘍細胞の細胞傷害(例えばアポトーシス又は有糸分裂停止)を誘発する細胞傷害性物質と化学的に結合された抗体分子)であってもよい。正常な薬理学的クリアランス機構の結果として、医薬コンジュゲート(イムノコンジュゲート)で用いられる抗体は、限定的な量でのみ標的細胞と接触しこれと結合する。したがって、コンジュゲートで用いられる細胞傷害性物質は、十分な細胞殺滅を引き起こし治療効果を誘引することができるように高度に細胞傷害性でなければならない。US2004/0241174に記載されているように、そのような細胞傷害性物質の例には、タキサン(例えばWO01/38318及びWO03/097625を参照されたい)、DNAアルキル化剤(例えばCC-1065アナローグ)、アントラサイクリン、チュブリシンアナローグ、デュオカルマイシンアナローグ、ドキソルビシン、アウリスタチンE、リシンA毒素、及び反応性ポリエチレングリコール部分を含む細胞傷害性物質(例えば以下を参照されたい:Sasse et al. 2000;Suzawa et al. 2000;Ichimura et al. 1991;Francisco et al. 2003;US5,475,092;US6,340,701;US6,372,738及びUS6,436,931;US2001/0036923;US2004/0001838;US2003/0199519;及びWO01/49698)が含まれる。
好ましい実施態様では、細胞傷害性物質はメイタンシノイド、すなわちメイタンシン(CAS35846538)の誘導体である。当分野ではメイタンシノイドにはメイタンシン、メイタンシノール、メイタンシノールのC-3エステル、及び他のメイタンシノールアナローグ及び誘導体が含まれることが知られている(例えばUS5,208,020及びUS6,411,163を参照されたい)。
抗CD37抗体イムノコンジュゲートは、抗FAPイムノコンジュゲートについてWO2007/077173に記載されているように、設計及び合成することができる。
【0023】
さらに別の実施態様では、本発明の抗CD37分子は放射能標識して、放射性イムノコンジュゲート(抗CD37抗体MB-1について提唱されたアプローチである(Buchsbaum et al. 1992、上記を参照されたい))を生成することができる。有利な放射線特性を有する放射性核種は当分野で公知であり、その例はリン-32、ストロンチウム-89、イットリウム-90、ヨウ素-131、サマリウム-153、エルビウム-169、イッテリビウム-175、レニウム-188であり、これらは良好にかつ安定的にMAbと結合した。本発明の抗CD37抗体分子は、US6,241,961に記載されているように、当分野で公知の直接標識又は間接標識方法を用いて多様な放射性核種で標識することができる。本発明で有用な新規な放射能標識抗体コンジュゲートを作製しこれを適用する技術に関する概論は以下の文献で提供されている:Goldenberg and Shakey, 2007。
本発明の抗体分子(Fcが操作されていようといまいと)はまた二重特異性であることができる。二重特異性抗体は、すなわち2つの異なる標的と結合し、そのうちの1つはCD37であり、他方は、例えばT細胞によって発現される表面抗原、例えばCD3、CD16及びCD56である。
【0024】
本発明はまた、本発明のキメラ及びヒト化抗CD37抗体分子をコードするDNA分子に関する。本発明の抗体分子の可変重鎖をコードする配列は、配列番号:1、配列番号:5、配列番号:7及び配列番号:9に示される。本発明の抗体分子の可変軽鎖をコードする配列は、配列番号:3、配列番号:11、配列番号:13、配列番号:15、配列番号:17、配列番号:19、配列番号:21に示される。
軽鎖及び重鎖をコードする核酸分子は、標準的な方法により化学的及び酵素的に(PCR増幅)合成することができる。第一に、当分野で公知の方法(例えばGait, 1984)を用いて適切なオリゴヌクレオチドを合成し、このオリゴヌクレオチドを用いて合成遺伝子を生成することができる。オリゴヌクレオチドから合成遺伝子を生成する方法は当分野で公知である(例えば、Stemmer et al. 1995;Ye et al. 1992;Hayden et Mandecki, 1988;Frank et al. 1987)。
本発明のDNA分子には、配列表に示されるDNA分子が含まれるが、ただしこれらに限定されない。したがって、本発明はまた、WO2007/042309で規定する高ストリンジェンシーの結合及び洗浄条件下で、配列表に示されるDNA分子とハイブリダイズする核酸分子に関し、そのような核酸分子は、配列表に示される配列によってコードされる抗体と等価であるか又はそれらより優れた特性を有する抗体又はその機能的フラグメントをコードする。好ましい分子(mRNAの観点から)は、本明細書に記載のDNA分子の1つと少なくとも75%又は80%(好ましくは少なくとも85%、より好ましくは少なくとも90%、もっとも好ましくは少なくとも95%)の相同性又は配列同一性を有するものである。
本発明の範囲内のさらに別のDNA変種クラスは、それらがコードするポリペプチドに照らして規定することができる。これらのDNA分子は、配列表に示されるものからその配列に関して逸脱するが、それらは、遺伝暗号の縮合のために同一のアミノ酸配列をもつ抗体をコードする。例示すれば、真核細胞で当該抗体を発現させるという見地から、配列表に示されるDNA配列は、真核細胞でのコドン使用頻度に適合するように設計されている。抗体を大腸菌(E. coli)で発現させることを所望するならば、これらの配列は大腸菌のコドン使用頻度に適合するように変更することができる。本発明のDNA分子の変種は、例えばWO2007/042309に記載されたようないくつかの異なる方法で構築することができる。
【0025】
本発明の組換え抗CD37抗体分子を作製するために、完全長の軽鎖及び重鎖をコードするDNA分子又はそのフラグメントを、それら配列が転写及び翻訳制御配列に機能的に連結されるように発現ベクターに挿入する。本発明の抗体を製造するために、当業者は、当分野で周知の極めて多様な発現系から、概説論文(例えばKipriyanow and Le Gall, 2004)に記載の発現系を選択することができる。
発現ベクターには、プラスミド、レトロウイルス、コスミド、EBV由来エピソームなどが含まれる。発現ベクター及び発現制御配列は、宿主細胞と適合するように選択される。抗体軽鎖遺伝子及び抗体重鎖遺伝子は別々のベクターに挿入してもよい。ある種の実施態様では、両DNA配列は同じ発現ベクターに挿入される。便利なベクターは機能的に完全なヒトCH又はCL免疫グロブリン配列をコードするベクターであり、前記は、任意のVH又はVL配列を上記に記載したように容易に挿入及び発現することができるように、適切な制限部位が操作されている。定常鎖は通常、抗体軽鎖についてはカッパ又はラムダであり、抗体重鎖については、任意のIgGアイソタイプ(IgG1、IgG2、IgG3、IgG4)又は他の免疫グロブリン(対立遺伝子変種を含む)でありえるが、ただしこれらに限定されない。
組換え発現ベクターはまた、抗体鎖の宿主細胞からの分泌を容易にするシグナルペプチドを含むことができる。抗体鎖をコードするDNAは、シグナルペプチドが成熟抗体鎖のアミノ末端にインフレームで連結されえるようにベクターでクローニングすることができる。シグナルペプチドは、免疫グロブリンシグナルペプチドであっても、又は非免疫グロブリンタンパク質由来の異種ペプチドであってもよい。あるいは、抗体鎖をコードするDNA配列は既にシグナルペプチド配列を含んでいてもよい。
【0026】
抗体鎖をコードするDNA配列に加えて、組換え発現ベクターは、プロモータ、エンハンサー、転写終結及びポリアデニル化シグナル、並びに宿主細胞で抗体鎖の発現を制御する他の発現制御エレメントを含む調節配列を保持する。プロモータ配列の例(哺乳動物細胞で発現を達成する)は、CMV(例えばCMVサルウイルス40(SV40))(例えばSV40プロモータ/エンハンサー)、アデノウイルス(例えばアデノウイルス主要後期プロモータ(AdMLP))、ポリオーマ及び強力な哺乳動物プロモータ(例えば天然の免疫グロブリン及びアクチンプロモータ)から誘導されるプロモータ及び/又はエンハンサーである。ポリアデニル化シグナルの例は、BGH polyA、SV40後期又は初期polyA;あるいは免疫グロブリン遺伝子の3’UTRなどを用いることができる。
組換え発現ベクターはまた、宿主細胞でのベクターの複製を調節する配列(例えば複製起点)及び選別可能マーカー遺伝子を保持することができる。抗CD37抗体の重鎖又はその抗原結合部分及び/又は軽鎖又はその抗原結合部分をコードする核酸分子、及びこれらのDNA分子を含むベクターを、宿主細胞、例えば細菌細胞又は高等真核細胞、例えば哺乳動物細胞に当分野で周知のトランスフェクション方法にしたがって導入することができる。前記トランスフェクション方法には、リポソーム仲介トランスフェクション、ポリカチオンイオン仲介トランスフェクション、プロトプラスト融合、マイクロインジェクション、リン酸カルシウム沈殿、エレクトロポレーション又はウイルスベクターによる移転が含まれる。
好ましくは、重鎖及び軽鎖をコードするDNA分子は2つのベクターに存在し、それらベクターは宿主細胞(好ましくは哺乳動物細胞)に同時トランスフェクトされる。
【0027】
発現用宿主として利用可能な哺乳動物細胞株は当分野では周知であり、とりわけチャイニーズハムスター卵巣(CHO、CHO-DG44)細胞、NSO、SP2/0細胞、HeLa細胞、ベビーハムスター腎(BHK)細胞、サル腎細胞(COS)、ヒト癌腫細胞(例えばHep G2)、A549細胞、3T3細胞、又はそのような任意の細胞株の誘導細胞/子孫細胞が含まれる。他の哺乳動物細胞もまた用いることができる。前記にはヒト、マウス、ラット、サル及びげっ歯類細胞株を含まれるが、ただしこれらに限定されない。又は他の真核細胞(酵母、昆虫細胞及び植物細胞を含むが、ただしこれらに限定されない)又は原核細胞、例えば細菌を用いてもよい。本発明の抗CD37抗体分子は、宿主細胞で抗体分子を発現させるために十分な期間当該宿主細胞を培養することによって製造される。
抗体分子は、好ましくは分泌されたポリペプチドとして培養媒体から回収するか、又は前記抗体分子は、例えば分泌シグナル無しに発現させた場合は宿主細胞溶解物から回収することができる。組換えタンパク質及び宿主細胞タンパク質について用いられる標準的なタンパク質精製方法を、実質的に均質な抗体調製物が得られる態様で用いて抗体分子を精製することが必要である。例示すれば、本発明の抗CD37抗体分子得るために有用な現在の精製技術は、第一の工程として、培養媒体又は溶解物から細胞及び/又は粒子状細胞屑の除去を含む。続いて可溶性夾雑タンパク質、ポリペプチド及び核酸から、例えばイムノアフィニティ又はイオン交換カラム、エタノール沈殿、逆相HPLC、セファデックスクロマトグラフィー、シリカゲル若しくはイオン交換樹脂クロマトグラフィーによって抗体を精製する。抗CD37抗体分子調製物を得るための最終工程として、治療薬としての利用のために下記に記載するように、精製抗体分子を乾燥(例えば凍結乾燥)させることができる。
さらに別の特徴では、本発明は、活性成分として本発明の抗CD37抗体分子を含む医薬組成物に関する。
【0028】
治療に用いられるためには、抗CD37抗体は、動物又はヒトへの投与を容易にするために適切な医薬組成物に混合される。抗CD37抗体分子の典型的な処方物は、抗CD37抗体分子を生理学的に許容できる担体、賦形剤又は安定化剤と混合することによって、凍結乾燥あるいは乾燥処方物又は水溶液又は水性若しくは非水性懸濁物の形態で調製することができる。担体、賦形剤、改変剤又は安定化剤は、用いられる用量及び濃度で非毒性である。前記には、緩衝系(例えばリン酸、クエン酸、酢酸及び他の無機若しくは有機酸及びそれらの塩);抗酸化剤(アスコルビン酸及びメチオニンが含まれる);保存料(例えばオクタデシルジメチルベンジルアンモニウムクロリド;ヘキサメトニウムクロリド;塩化ベンザルコニウム、塩化ベンゼトニウム;フェノール、ブチル又はベンジルアルコール;アルキルパラベン、例えばメチル又はプロピルパラベン;カテコール;レゾルシノール;シクロヘキサノール;3-ペンタノール;及びm-クレゾール);タンパク質、例えば血清アルブミン、ゼラチン又は免疫グロブリン;親水性ポリマー、例えばポリビニルピロリドン又はポリエチレングリコール(PEG);アミノ酸、例えばグリシン、グルタミン、アスパラギン、ヒスチジン、アルギニン又はリジン;単糖類、二糖類、オリゴ糖又は多糖類及び他の炭水化物(グルコース、マンノース、シュクロース、トレハロース、デキストリン又はデキストランを含む);キレート剤(例えばEDTA);糖アルコール(例えばマンニトール又はソルビトール);塩形成対イオン(例えばナトリウム);金属複合体(例えば亜鉛-タンパク質複合体);及び/又はイオン性又は非イオン性表面活性剤、例えばTWEENTM(ポリソルベート)、PLURONICSTM又は脂肪酸エステル、脂肪酸エーテル又は糖エステルが含まれる。さらに有機溶媒、例えばエタノール又はイソプロパノールもまた抗体処方物に含まれえる。賦形剤はまた放出改変機能又は吸収改変機能を有することができる。
【0029】
抗CD37抗体分子はまた、乾燥させるか(凍結乾燥、噴霧乾燥、噴霧凍結乾燥、近臨界若しくは超臨界ガスによる乾燥、真空乾燥、通気乾燥)、沈殿させるか、又は結晶化させるか、又はマイクロカプセル中に被包化することができる。マイクロカプセルは、例えばコアセルベーション技術によって又は界面重合(それぞれ例えばヒドロキシメチルセルロース又はゼラチン及びポリ-(メチルメタクリレート)を用いる)によって、コロイド性薬剤デリバリー系(例えばリポソーム、アルブミン微小球、マイクロエマルジョン、ナノ粒子及びナノカプセル)で、担体若しくは表面でのマクロエマルジョン又は沈殿又は固定化(例えばpcmc技術(タンパク質被覆微結晶(protein coated microcrystal))により調製することができる。そのような技術は成書(Remington: The Science and Practice of Pharmacy, 21st edition, Hendrickson R. Ed.)に記載されている。
もちろんのこと、in vivo投与に用いられる処方物は無菌的でなければならない。滅菌は、通常の技術、例えば無菌的ろ過膜を通過させるろ過によって実施することができる。
いわゆる高濃度液体処方物(HCLF)を得るために抗CD37抗体の濃度を高めることは有用でありえる。そのようなHCLFを生成するための多様な方法が記載されている。
【0030】
抗CD37抗体分子はまた徐放性調製物に含まれえる。そのような調製物は、疎水性又は親水性ポリマーの固体、半固体、又は液体マトリックスを含み、成形品、例えばフィルム、スティック又はマイクロカプセルの形態を有し、適用装置を介して適用することができる。徐放性マトリックスの例には、ポリエステル、ヒドロゲル(例えばポリ(2-ヒドロキシエチル-メタクリレート)又はシュクロースアセテートブチレート)又はポリ(ビニルアルコール)、ポリラクチド(US3,773,919)、L-グルタミン酸とγエチル-L-グルタメートとのコポリマー、非分解性エチレン-ビニルアセテート、分解性乳酸-グリコール酸コポリマー(例えばLUPRON DEPOTTM(乳酸-グリコール酸コポリマー及び酢酸ロイプロリドを含む注射可能な微小球)及びポリ-D-(-)-3-ヒドロキシ酪酸が含まれる。ポリマー(例えばエチレン-酢酸ビニル及び乳酸-グリコール酸)は100日間にわたって分子の放出を許容するが、一方、ある種のヒドロゲルはより短い期間の間タンパク質を放出する。被包化された抗体が体内で長期間存続するとき、それらは、37℃で水分に暴露される結果として変性又は凝集し、生物学的活性の低下及び免疫原性における変化を生じる可能性がある。関与するメカニズムに応じて安定化のために合理的な対策を講じることができる。例えば、凝集メカニズムがチオ-ジスルフィド交換による分子間S-S結合形成であると判明したら、安定化は、スルフヒドリル残基の改変、酸性溶液からの凍結乾燥(例えばWO89/011297に記載)、水分含有量の管理、適切な添加剤の使用、および特別なポリマーマトリックス組成の開発によって達成することができる。
本発明の抗CD37抗体分子のためにまた用いることができる処方物は、US7,060,268及びUS6,991,790に記載されている。
【0031】
CD37抗体分子はまた他の適用形(例えば分散剤、懸濁剤又はリポソーム、錠剤、カプセル、散剤、スプレー、経皮若しくは皮内パッチ又はクリーム(浸透強化仕掛け付き又は仕掛け無し)、ウェファース、鼻用、頬腔用又は肺用処方物)に取り入れることができる。CD37抗体分子はまた、移植細胞によって、又は遺伝子治療後に個体自身の細胞によって生成させることができる。
抗CD37抗体分子はまた、化学基、例えばポリエチレングリコール(PEG)、メチル若しくはエチル基、又は炭水化物基を用いて誘導体化することができる。これらの基は、本発明の抗体の生物学的特徴の改善、例えば血中半減期の増加又は組織結合の増加に有用でありえる。
適用の好ましい態様は非経口(輸液又は注射(静脈内、筋肉内、皮下、腹腔内、皮内))であるが、他の適用態様(例えば吸入、経皮、鼻内、頬腔、経口適用)もまた利用することができる。
疾患の予防又は治療の場合、抗体の適切な投与量は、治療されるべき疾患のタイプ、当該疾患の重篤度及び経過、抗体が予防目的又は治療目的のために投与されるのか否か、これまでの治療、患者の病歴及び抗体に対する応答、並びに主治医の判断に左右されるであろう。抗体は、一度に又は一連の治療の間適切に患者に投与されるであろう。
疾患のタイプ及び重篤度に応じて、約0.01μg/kgから40mg/kg(例えば0.1−20mg/kg)の抗体が、例えば1回又は2回以上の別々の投与にせよ、又は連続輸液投与にせよ、患者への最初の候補投与量である。症状に応じて数日又はそれより長い期間にわたる反復投与の場合、治療は、症状の所望の抑制が生じるまで維持される。しかしながら、他の投与計画も有用でありえる。本治療法の進行は、通常の技術及びアッセイ(例えばフローサイトメトリーの使用)、例えばB細胞枯渇の程度を決定することによって容易にモニターされる。
投与される抗体の“治療的に有効な量”は、疾患又は異常の予防、緩和又は治療のために必要な最少量である。
【0032】
本発明の抗CD37抗体分子及び前記を含む医薬組成物は、その表面にCD37を発現し、癌性又は自己免疫/炎症疾患を引き起こすB細胞の枯渇に有用である。
第一の特徴では、本発明の医薬組成物は、癌(特に任意のCD37陽性悪性疾患)の治療に有用である。
B細胞悪性疾患には以下が含まれる(ただしこれらに限定されない):B細胞リンパ腫(例えばホジキン症の種々の型、B細胞非ホジキンリンパ腫(NHL)及び関連リンパ腫(例えばヴァルデンストレームマクログロブリン血症(リンパ形質細胞性リンパ腫又は免疫細胞腫とも称される)又は中枢神経系リンパ腫)、白血病(例えば急性リンパ芽球性白血病(ALL)、慢性リンパ球性白血病(CLL、B細胞慢性リンパ球性白血病BCLLとも称される)、毛状細胞白血病及び慢性骨髄芽球性白血病)及びミエローマ(例えば多発性ミエローマ)。さらに別のB細胞悪性疾患には、小リンパ球型リンパ腫、B細胞前リンパ球性白血病、リンパ形質細胞性リンパ腫、脾臓辺縁層リンパ腫、形質細胞ミエローマ、骨の孤立形質細胞腫、骨外形質細胞腫、粘膜結合類リンパ組織(MALT)の外リンパ節辺縁層B細胞リンパ腫、リンパ節辺縁層B細胞リンパ腫、濾胞性リンパ腫、マントル細胞リンパ腫、びまん性大B細胞型リンパ腫、縦隔(胸腺)大B細胞型リンパ腫、血管内大B細胞型リンパ腫、原発性滲出型リンパ腫、バーキットリンパ腫/白血病、灰白色層リンパ腫(grey zone lymphoma)、悪性性が未確認のB細胞増殖、類リンパ腫、肉芽腫症及び移植後のリンパ球増殖性異常が含まれる。
【0033】
さらに別の特徴では、抗CD37抗体を含む医薬組成物は、その病理においてB細胞が中心的に関与する自己免疫及び炎症性疾患の治療に有用である。
そのような疾患には以下が含まれる(ただしこれらに限定されない):関節炎、慢性関節リウマチ、若年性慢性関節リウマチ、変形性関節症、多発性軟骨炎、乾癬性関節炎、乾癬、皮膚炎、多発性筋炎/皮膚筋炎、封入体性筋炎、炎症性筋炎、中毒性表皮壊死症、全身性皮膚硬化症及び硬化症、CREST症候群、炎症性大腸疾患付随応答、クローン病、潰瘍性結腸炎、呼吸窮迫症候群、成人呼吸窮迫症候群(ARDS)、髄膜炎、脳炎、ブドウ膜炎、結腸炎、糸球体腎炎、アレルギー疾患、湿疹、喘息、T細胞浸潤及び慢性炎症性応答を随伴する症状、アテローム性硬化症、自己免疫性心筋症、白血球接着不全、全身性紅斑性狼瘡(SLE)、亜急性皮膚性エリテマトーデス、円板状狼瘡、狼瘡性筋炎、狼瘡性脳炎、若年発症性糖尿病、多発性硬化症、アレルギー性脳脊髄炎、視神経脊髄炎、リウマチ熱、シドナム舞踏病、サイトカイン及びTリンパ球により媒介される急性及び遅延型過敏症に付随する免疫応答、結核、サルコイドーシス、ヴェーゲナー肉芽腫症及びチャーグ-ストラウス病を含む肉芽腫症、顆粒球減少症、脈管炎(過敏性脈管炎/血管炎、ANCA及びリウマチ性脈管炎を含む)、再生不良性貧血、ダイアモンド-ブラックファン貧血、免疫性溶血性貧血(自己免疫性溶血性貧血(AIHA)を含む)、悪性貧血、赤芽球ろう(PRCA)、第VIII因子欠損、血友病A、自己免疫好中球減少症、汎血球減少症、白血球減少症、白血球漏出を伴う疾患、中枢神経系(CNS)の炎症性疾患、多器官損傷症候群、重症筋無力症、抗原抗体複合物介在疾患、抗糸球体基底膜病、抗リン脂質抗体症候群、アレルギー性神経炎、ベーチェット病、キャッスルマン症候群、グッドパスチャー症候群、ランバート-イートン筋無力症症候群、レイノー症候群、シェーグレン症候群、スティーヴェンズ-ジョンソン症候群、固体器官移植拒絶、対宿主性移植片病(GVHD)、水疱性類天疱瘡、天疱瘡、自己免疫性多発性内分泌障害、血清陰性脊椎関節症、ライター病、スティッフマン症候群、巨細胞動脈炎、免疫複合体性腎炎、IgA腎症、IgM多発性腎症又はIgM介在腎症、特発性血小板減少性紫斑病(ITP)、血栓性血小板減少性紫斑病(TTP)、ヘノッホ-シェーンライン紫斑病、自己免疫性血小板減少症、精巣及び卵巣の自己免疫疾患(自己免疫性精巣炎及び卵巣炎を含む)、原発性甲状腺機能低下症;自己免疫性内分泌疾患(自己免疫性甲状腺炎、慢性甲状腺炎(橋本甲状腺炎)、亜急性甲状腺炎、特発性甲状腺機能低下症、アジソン病、グレーヴズ病、自己免疫性多発性腺症候群(又は多発性腺内分泌障害)、I型糖尿病(インスリン依存真性糖尿病(IDDM)とも称される)及びシーハン症候群;自己免疫性肝炎、類リンパ球性間質肺炎(HIV)、NSIPに対する閉塞性細気管支炎(非移植性)(bronchiolitis obliterans (non-transplant) vs NSIP)、ギラン-バレー症候群、大血管性血管炎(リウマチ性多発性筋肉痛及び巨細胞性(タカヤス)動脈炎を含む)、中血管性血管炎(川崎病及び結節性多発性動脈炎を含む)、結節性多発性動脈炎(PAN)強直性脊椎炎、バージャー病(IgA腎症)、急速に進行する糸球体腎炎、原発性胆汁性肝硬変、腹腔スプルー(グルテン腸症)クリオグロブリン血症、肝炎に付随するクリオグロブリン血症、筋萎縮性側索硬化症(ALS)、冠状動脈疾患、家族性地中海熱、微細多発性血管炎、コーガン症候群、ウィスコット-アルドリッチ症候群及び閉塞性血栓性血管炎(WO2007/014278を参照されたい)。
【0034】
治療されるべき疾患に応じて、本発明の抗CD37抗体分子は、そのもの単独で、又は1つ又は2つ以上の追加の治療薬剤と併用して用いることができる。前記追加の治療薬剤は、特に、DNA損傷薬剤若しくはチュブリン結合薬剤、又は癌細胞における血管形成、シグナルトランスダクション経路又は有糸分裂チェックポイントを阻害する治療的に活性を有する化合物から選択される。
前記追加の治療薬剤は、場合によって同じ医薬調製物の成分として同時に投与するか、又は抗CD37抗体分子の投与の前後に投与することができる。
ある種の実施態様では、追加の治療薬剤は、EGFRファミリー、VEGFRファミリー、IGF-1R、インスリンレセプター、オーロラA、オーロラB、PLK及びPI3キナーゼ、FGFR、PDGFR、Raf、KSP又はPDK1の阻害剤の群から選択される1つ又は2つ以上の阻害剤でありえるが、ただしこれらに限定されない。
追加薬剤のさらに別の例は、CDK、Akt、Src、Bcr-Ab1、cKit、cMet/HGF、c-Myc、Flt3、HSP90、ヘッジホッグアンタゴニストの阻害剤、JAK/STAT、Mek、mTor、NFkappaB、プロテアソーム、Rhoの阻害剤、Wntシグナリング又はNotchシグナリングの阻害剤、又はユビキチン化経路の阻害剤である。
Aurora阻害剤の例は、PHA-739358、AZD-1152、AT-9283、CYC-116、R-763、VX-667、MLN-8045、PF-3814735、SNS-314、VX-689、GSK-1070916、TTP-607、PHA-680626、MLN-8237及びENMD-2076であるが、ただしこれらに限定されない。
PLK阻害剤の例はGSK-461364である。
raf阻害剤の例は、BAY-73-4506(VEGFR阻害剤でもある)、PLX-4032、RAF-265(VEGFR阻害剤でもある)、RAF-265(VEGFR阻害剤でもある)、ソラフェニブ(sorafenib)(VEGFR阻害剤でもある)、XL-281及びNevavar(VEGFR阻害剤でもある)である。
【0035】
KSP阻害剤の例は、イスピネシブ(ispinesib)、ARRY-520、AZD-4877、CK-1122697、GSK-246053A、GSK-923295、MK-0731、SB-743921、LY-2523355、及びEMD-534085である。
src及び/又はbcr-ab1阻害剤の例は、ダサチニビ(dasatinib)、AZD-0530、ボスチニブ(bostinib)、XL-228(IGF-1R阻害剤でもある)、ニロチニブ(nilotinib)(PDGFR及びcKit阻害剤でもある)、イマチニブ(imatinib)(cKit阻害剤でもある)、NS-187、KX2-391、AP-24534(EGFR、FGFR、Tie2、Flt3阻害剤でもある)、KM-80及びLS-104(Flt3、Jak2阻害剤でもある)である。
PDK1阻害剤の例はAR-12である。Rho阻害剤の例はBA-210である。
PI3キナーゼ阻害剤の例はPX-866、PX-867、BEZ-235(mTor阻害剤でもある)、XL-147及びXL-765(mTor阻害剤でもある)、BGT-226、CDC-0941である。
cMet又はHGFの阻害剤の例はXL-184(VEGFR、cKit、Flt3阻害剤でもある)、PF-2341066、MK-2461、XL-880(VEGFR阻害剤でもある)、MGCD-265(VEGFR、Ron、Tie2阻害剤でもある)、SU-11274、PHA-665752、AMG-102、AV-299、ARQ-197、MetMAb、CGEN-241、BMS-777607、JNJ-38877605、PF-4217903、SGX-126、CEP-17940、AMG-458、INCB-028060及びE-7050である。
c-Myc阻害剤の例はCX-3543である。
Flt3阻害剤の例は、AC-220(cKit及びPDGFR阻害剤でもある)、KW-2449、LS-104(bcr-ab1及びJak2阻害剤でもある)、MC-2002、SB-1317、レスタウルチニブ(lestaurtinib)(VEGFR、PDGFR、PKC阻害剤でもある)、TG-101348(JAK2阻害剤でもある)、XL-999(cKit、FGFR、PDGFR及びVEGFR阻害剤でもある)、スニチニブ(sunitinib)(PDGFR、VEGFR及びcKit阻害剤でもある)及びタンヅチニブ(tandutinib)(PDGFR及びcKit阻害剤でもある)である。
【0036】
HSP90阻害剤の例はタネスピマイシン、アルヴェスピマイシン、IPI-504、STA-9090、MEDI-561、AUY-922、CNF-2024及びSNX-5422である。
JAK/STAT阻害剤の例は、CYT-997(またチューブリンと相互作用する)、TG-101348(Flt3阻害剤でもある)及びXL-019である。
Mek阻害剤の例は、ARRY-142886、AS-703026、PD-325901、AZD-8330、ARRY-704、RDEA-119及びXL-518である。
mTor阻害剤の例は、テムシロリムス(temsirolimus)、デフォロリムス(deforolimus)(VEGF阻害剤としても作用する)、エヴェロリムス(everolimus)(さらにVEGF阻害剤)、XL-765(PI3キナーゼ阻害剤でもある)及びBEZ-235(PI3キナーゼ阻害剤でもある)である。
Akt阻害剤の例は、ペリフォシン、GSK-690693、RX-0201及びトリシリビンである。
cKit阻害剤の例は、マスチニブ(mastinib)、OSI-930(VEGFR阻害剤としてもまた作用する)、AC-220(Flt3及びPDGFR阻害剤でもある)、タンヅチニブ(Flt3及びPDGFR阻害剤でもある)、アキシチニブ(axitinib)(VEGFR及びPDGFRの阻害剤でもある)、スニチニブ(Flt3、PDGFR、VEGFR阻害剤でもある)及びXL-820(VEGFR及びPDGFR阻害剤としてもまた作用する)、イマチニブ(bcr-ab1阻害剤でもある)、ニロチニブ(bcr-ab1及びPDGFR阻害剤でもある)である。
ヘッジホッグアンタゴニストの例は、IPI-609、CUR-61414、GDC-0449、IPI-926及びXL-139である。
CDK阻害剤の例は、セリシクリブ(seliciclib)、AT-7519、P-276、ZK-CDK(VEGFR2及びPDGFRもまた阻害する)、PD-332991、R-547、SNS-032、PHA-690509、PHA-848125及びSCH-727965である。
【0037】
プロテアソーム阻害剤の例は、ボルテゾミブ(bortezomib)、カルフィルゾミブ(carfilzomib)及びNPI-0052(NFkappaBの阻害剤でもある)である。
プロテアソーム阻害剤/NFkappaB経路阻害剤の例は、ボルテゾミブ、カルフィルゾミブ、NPI-0052、CEP-18770、MLN-2238、PR-047、PR-957、AVE-8680及びSPC-839である。
ユビキチン化経路の阻害剤の例はHBX-41108である。
抗血管形成剤の例は、FGFR、PDGFR及びVEGF(R)の阻害剤、並びにサリドマイドであり、そのような薬剤は以下から選択される(ただしこれらに限定されない):ベヴァシズマブ、モテサニブ、CDP-791、SU-14813、テラチニブ、KRN-951、ZK-CDK(CDKの阻害剤でもある)、ABT-869、BMS-690514、RAF-265、IMC-KDR、IMC-18F1、IMiD、サリドマイド、CC-4047、レナリドマイド、ENMD-0995、IMC-D11、Ki-23057、ブリヴァニブ(brivanib)、セディラニブ(cediranib)、1B3、CP-868596、IMC-3G3、R-1530(Flt3の阻害剤でもある)、スニチニブ(cKit及びFlt3の阻害剤でもある)、アキシチニブ(cKitの阻害剤でもある)、レスタウルチニブ(Flt3及びPKCの阻害剤でもある)、ヴァタラニブ、タンヅチニブ(Flt3及びcKitの阻害剤でもある)、パゾパニブ(pazopanib)、PF-337210、アフリベルセプト(aflibercept)、E-7080、CHIR-258、ソラフェニブ(sorafenib)トシレート(Rafの阻害剤でもある)、ヴァンデタニブ、CP-547632、OSI-930、AEE-788(EGFR及びHer2阻害剤でもある)、BAY-57-9352(Rafの阻害剤でもある)、BAY-73-4506(Rafの阻害剤でもある)、XL-880(cMetの阻害剤でもある)、XL-647(EGFR及びEphB4の阻害剤でもある)、XL-820(cKitの阻害剤でもある)、ニロチニブ(cKit及びbrc-ab1の阻害剤でもある)、CYT-116、PTC-299、BMS-584622、CEP-11981、どドヴィチニブ(dovitinib)、CY-2401401及びENMD-2976.
【0038】
追加の治療薬剤はまたEGFR阻害剤から選択することができる。前記は小分子EGFR阻害剤であっても、又は抗EGFR抗体であってもよい。抗EGFR抗体の例は、セツキシマブ、パニツムマブ、ニモツズマブ、ザルツムマブであるが、ただしこれらに限定されない。小分子EGFR阻害剤の例は、ゲフィチニブ、エルロチニブ(erlotinib)及びヴァンデタニブ(VEGFRの阻害剤でもある)である。EGFR調整物質の別の例はEGF融合毒素である。
本発明の抗CD37抗体分子との併用に有用なさらに別のEGFR及び/又はHer2阻害剤は、ラパチニブ、トラスツズマブ、ペルツズマブ(pertuzumab)、XL-647、ネラチニブ、BMS-599626、ARRY-334543、AV-412、mAB-806、BMS-690514、JNJ-26483327、AEE-788(VEGFRの阻害剤でもある)、AZD-8931、ARRY-380、ARRY-333786、IMC-11F8、Zemab、TAK-285、AZD-4769である。
追加される薬剤はまた、IGF-1R及びインスリンレセプター経路を標的とする薬剤から選択することができる。そのような薬剤には、IGF-1R(例えばCP-751871、AMG-479、IMC-A12、MK-0646、AVE-1642、R-1507、BIIB-022、SCH-717454、rhu Mab IGFR)と結合する抗体、及びIGF1-Rのキナーゼドメインを標的とする新規な化学物質(例えばOSI-906又はBMS-554417、XL-228、BMS-754807)が含まれる。
本発明の抗CD37抗体分子を用いる治療法で有利に併用することができる他の薬剤はCD20を標的とする分子であり、CD20特異的抗体(例えばリツキシマブ、LY-2469298、オクレリズマブ、MEDI-552、IMMU-106、GA-101(=R7159)、XmAb-0367、オフアツムマブ)、放射能標識CD20抗体(例えばトシツムマブ及びイブリツモマブチウキセタン)、又は他のCD20誘導タンパク質(例えばSMIP Tru015、PRO-131921、FBT-A05、ヴェルツズマブ、R-7159)が含まれる。
【0039】
CD37抗体は、白血球で発現される他の表面抗原の阻害剤と併用することができる。前記阻害剤は、特に抗体又は抗体様分子であり、例えば抗CD20(シプリズマブ)、抗CD4(ザノリムマブ)、抗CD19(MT-103、MDX-1342、SAR-3419、XmAb-5574)、抗CD22(エプラツズマブ)、抗CD23(ルミリキシマブ)、抗CD30(イラツムマブ)、抗CD32B(MGA-321)、抗CD38(HuMax-CD38)、抗CD40(SGN40)、抗CD52(アレムツズマブ)、抗CD80(ガリキシマブ)である。本発明の抗体はまた別のCD37アンタゴニスト、例えばTRU-016と併用することができる。
CD37抗体と併用できる他の薬剤は、イムノトキシン(例えばBL-22(抗CD22イムノトキシン)、イノツズマブオゾガミシン(抗CD23抗体-カリケアミシンコンジュゲート)、RFT5.dgA(抗CD25リシン毒素A-鎖)、SGN-35(抗CD30-アウリスタチンEコンジュゲート)及びゲムツズマブオゾガミシン(抗CD33カリケアミシンコンジュゲート)、MDX-1411(抗CD70コンジュゲート))、又は放射能標識抗体(例えば90Y-エプラツズマブ(抗CD22放射性イムノコンジュゲート))である。
さらにまた抗CD37抗体は以下と併用することができる:免疫調節剤、例えばアポトーシスを誘発するか又はシグナルトランスダクション経路を改変する抗体、例えばTRAILレセプター調節物質マパツムマブ(TRAIL-1レセプターアゴニスト)、レキサツムマブ(TRAIL-2レセプターアゴニスト)、チガツズマブ、Apomab、AMG-951及びAMG-655;抗HLA-DR抗体(例えば1D09C3)、抗CD74、破骨細胞分化因子リガンド阻害剤(例えばデノスマブ)、BAFFアンタゴニスト(例えばAMG-623a)、又はToll様レセプターのアゴニスト(例えばTLR-4又はTLR-9)。
【0040】
本発明の抗CD37抗体と一緒に用いることができる他の薬剤は以下から選択される(ただしこれらに限定されない):ホルモン、ホルモン類似体及び抗ホルモナール(例えばタモキシフェン、トレミフェン、ラロキシフェン、フルヴェストラント、酢酸メゲストロール、フルタミド、ニルタミド、ビカルタミド、酢酸シプロテロン、フィナステリド、酢酸ブセレリン、フルドロコルチゾン、フルオキシメステロン、メドロキシプロジェステロン、カプロン酸ヒドロキシプロジェステロン、ジエチルスチルベストロール、プロピオン酸テストステロン、フルオキシメステロン/等価物、オクトレオチド、アルゾキシフェン、パシレオチド、ヴァプレオチド、アドレノコルチコステロイド/アンタゴニスト、プレドニソン、デキサメタゾン、アイノグルテチミド(ainoglutethimide))、アロマターゼ阻害剤(例えばアナストロゾール、レトロゾール、リアロゾール、エクセメスタン、アタメスタン、フォルメスタン)、LHRHアゴニスト及びアンタゴニスト(例えば酢酸ゴセレリン、ロイプロリド、アバレリクス、セトロレリクス、デスロレリン、ヒストレリン、トリプテレリン)、抗代謝薬(例えばアンチフォレート、例えばメトトレキセート、トリメトトレキセート、ペメトレキセート(pemetrexed)、ピリミジン類似体、例えば5-フルオロウラシル、フルオロデオキシウリジン、カペシタビン、デシタビン、ネララビン、5-アザシチジン及びゲミシタビン、プリン及びアデノシン類似体、例えばメルカプトプリン、チオグアニン、アザチオプリン、クラドリビン及びペントスタチン、シタラビン、フルダラビン、クロファラビン);抗腫瘍抗生物質(例えばアントラサイクリン、例えばドキソルビシン、ダウノルビシン、エピルビシン及びイダルビシン、マイトマイシンC、ブレオマイシン、ダクチノマイシン、プリカマイシン、スプリカマイシン、アクチノマイシンD、ミトキサントロン、ミトキサントロンイダルビシン、ピキサントロン、ストレプトゾシン、アフィジコリン);白金誘導体(例えばシスプラチン、オキサリプラチン、カルボプラチン、ロバプラチン、サトラプラチン);アルキル化剤(例えばエストラムスチン、セムスチン、メクロレタミン、メルファラン、クロラムブシル、ブスルファン、ダカルバジン、シクロホスファミド、イフォスファミド、ヒドロキシウレア、テモゾロミド、ニトロソウレア(例えばカルムスチン及びロムスチン)、チオテパ);抗有糸分裂剤(例えばヴィンカアルカロイド、例えばヴィンブラスチン、ヴィンデシン、ヴィノレルビン、ヴィンフルニン);及びタキサン、例えばパクリタキセル、ドセタキセル及びそれらの処方物、ラロタキセル;シモタキセル、及びエポチロン、例えばイキサベピロン、パツピロン、ZK-EPO;トポイソメラーゼ阻害剤(例えばエピポドフィロトキシン、例えばエトポシド及びエトポフォス、テニポシド、アムサクリン、トポテカン、イリノテカン、バノキサントロン、カンプトテシン)及び種々雑多な化学療法剤、例えばレチン酸誘導体、アミフォスチン、アナグレリド、インターフェロンアルファ、インターフェロンベータ、インターフェロンガンマ、インターロイキン-2、プロカルバジン、N-メチルヒドラジン、ミトタン、及びポルフィマー、ベキサロテン、セレコキシブ、エチルエネミン/メチルメラミン、トリエチレンメラミン、トリエチレンチオホスホラミド、ヘキサメチルメラミン、及び酵素、L-アスパラギナーゼ、L-アルギナーゼ及びメトロニダゾール、ミソニダゾール、デスメチルミソニダゾール、ピモニダゾール、エタニダゾール、ニモラゾール、RSU1069、EO9、RB6145、SR4233、ニコチンアミド、5-ブロモデオジウリジン、5-ヨードデオキシウリジン、ブロモデオキシシチジン、エリスロヒドロキシノニル-アデニン、アントラセネジオン、GRN-163L(競合性テロメアーゼ鋳型アンタゴニスト)、SDX-101(PPARアゴニスト)、タラボスタット(DPP阻害剤)、フォロデシン(PNP阻害剤)、アタシセプト(TNFファミリーメンバーBLyS及びAPRILを標的とする可溶性レセプター)、TNF-アルファ中和剤(Enbrel、Humira、Remicade)、XL-844(CHK1/2阻害剤)、VNP-40101M(DNAアルキル化剤)、SPC-2996(アンチセンスbcl2阻害剤)、オバトクラクス(bcl2阻害剤)、エンザスタウリン(PKCベータ調節剤)、ヴォリニスター(HDAC阻害剤)、ロミデプシン(HDAC阻害剤)、AT-101(Bcl-2/Bcl-xL阻害剤)、プリチデプシン(多機能性デプシペプチド)、SL-11047(ポリアミン代謝調節剤)。
【0041】
ある種の実施態様では、抗CD37抗体分子は、“CHOP”(シクロホスファミド、ドキソルビシン、ヴィンクリスチン及びプレドニソンの組合せ)と一緒に適用される。
本発明の抗CD37抗体分子はまた、他の治療法(外科手術、放射線療法、内分泌療法、生物学的応答改変療法、高温療法及び低温療法を含む)及びいずれかの副作用を緩和するための薬剤(例えば鎮吐剤)と一緒に用いることができる。前記は、G-CSF、GM-CSF、光感作剤、例えばヘマトポルフィリン誘導体、フォトフリン(商標)、ベンゾポルフィリン誘導体、Npe6、錫エチオポルフィリン、フェオボリド-a、バクテリオクロロフィル-a、ナフタロシアニン、フタロシアニン、亜鉛フタロシアニンである。
モノクローナル抗体は完璧な抗原特異性を示し、しばしばヒトの標的抗原とのみ反応し、動物種由来の同族タンパク質とは反応しない。治療用抗体の開発を促進するために、in vivo毒性及び動的薬理変化を評価するための適切な動物モデルが要求される。in vivoモデルの1つの可能性はトランスジェニックマウスであり、このマウスでは内因性標的抗原はそのヒト同族体によって置き換えられている(“ノックアウト/ノックインマウス”)。特に、治療用抗CD37抗体の開発のために、ネズミCD37遺伝子をヒトCD37遺伝子によって置き換えることができる。これは、非翻訳配列によってフランキングされているヒトCD37遺伝子のコードゲノム配列を含むターゲティングベクターを構築することにより達成することができる。マウスES細胞を用いた相同組換えにこのターゲティングベクターを用いることができる。ヒトCD37発現についてホモ接合性であるトランスジェニック動物を用いて、ヒトCD37に対して作製した抗体の動的薬理作用を、例えば抗体適用後に抹消B細胞数をモニターすることによって評価することができる。あるいは、それらのマウスを用いて、静脈内適用後のヒトCD37特異抗体の潜在的な毒性作用を解明することができる。
モノクローナル抗体の動物交差反応性が欠如する場合の別の可能性は、いわゆる代用抗体の生成である。代用抗体は、動的薬理作用及び毒性作用の解明のために有用な関連する動物種(例えばマウス又はカニクイザル)の同族タンパク質と反応する抗体である。CD37の場合、理想的にはそれぞれマカクCD37又はマウスCD37に特異的であるモノクローナル抗体が生成され、そのような代用抗体は当該生成抗体と類似の結合及び機能特性を有するはずである。これは、マカク又はマウスCD37発現細胞を標的細胞として利用するアッセイ系、例えば結合については、FACSスキャチャード分析、ADCC及びアポトーシス分析を用いることにより精査することができる。最終的には、代用抗体は、マカク又はマウス血液におけるin vivoにおけるB細胞枯渇活性により選択することができる。
【0042】
本発明の実施態様の例を以下にまとめる。
(1)ヒトCD37と結合し、
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、
又は
b)a)に規定した抗体と同じヒトCD37のエピトープを認識するか、又は前記エピトープと近接するか若しくはオーバーラップするエピトープを認識する非ヒト抗体
から誘導される抗体分子であって、キメラ抗体又はヒト化抗体である前記抗体分子。
(2)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、ii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖、iii)ヒト起源である定常重鎖及び軽鎖によって規定されるキメラ抗体である、(1)に記載の抗体分子。
(3)i)定常重鎖がIgG1鎖であり、さらにii)定常軽鎖がカッパ鎖である、(2)に記載の抗体。
(4)定常重鎖i)が配列番号:24に示されるアミノ酸配列を含み、さらに定常軽鎖ii)が配列番号:26に示されるアミノ酸配列を含む、(3)に記載の抗体。
(5)下記a)−d)によって規定されるヒト化抗体である、(1)に記載の抗体:
a)配列番号:2に示される可変重鎖内に含まれるCDR、
b)配列番号:4に示される可変軽鎖内に含まれるCDR、
c)ヒト抗体から誘導される、前記CDRを支持するフレームワーク、
d)ヒト抗体に由来する定常重鎖及び軽鎖。
(6)配列番号:6に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(7)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(6)に記載の抗体。
(8)配列番号:8に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(9)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(8)に記載の抗体。
(10)配列番号:10に示される配列を有する可変重鎖を含む、(5)に記載の抗体。
(11)配列番号:12、14、16、18、20又は22に示される配列を有する可変軽鎖を含む、(10)に記載の抗体。
(12)抗体が、Fcドメイン内に1つ又は2つ以上のエフェクター機能を調節する1つ又は2つ以上の変異を有する、(1)から(11)のいずれかに記載の抗体。
(13)エフェクター機能の調節が抗体依存細胞媒介性細胞傷害の増加である、(12)に記載の抗体。
(14)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき332位の単一置換である、(12)または(13)に記載の抗体。
(15)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、239位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(16)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、236位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(17)Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき、236位、239位及び332位の置換の組合せである、(12)または(13)に記載の抗体。
(18)置換がI332E、S239D及びG236Aである、(14)から(17)のいずれかに記載の抗体。
(19)ヒトCD37と結合し、さらに配列番号:28のアミノ酸配列を含む重鎖を有する抗体。
(20)配列番号:30のアミノ酸配列を含む軽鎖を有する、(19)に記載の抗体。
(21)ヒトCD37と結合し、さらに配列番号:36のアミノ酸配列を含む重鎖を有する抗体。
(22)配列番号:38のアミノ酸配列を含む軽鎖を有する、(21)に記載の抗体。
(23)ヒトCD37と結合し、さらに配列番号:32のアミノ酸配列を含む重鎖を有する抗体。
(24)配列番号:34のアミノ酸配列を含む軽鎖を有する、(23)に記載の抗体。
(25)ヒトCD37と結合し、さらに配列番号:40のアミノ酸配列を含む重鎖を有する抗体。
(26)配列番号:42のアミノ酸配列を含む軽鎖を有する、(25)に記載の抗体。
(27)(1)から(26)のいずれかに記載の抗体の可変重鎖をコードする領域を含むDNA分子。
(28)可変重鎖コード領域がヒト起源の定常重鎖をコードする領域と融合される、(27)に記載のDNA分子。
(29)ヒト定常重鎖がIgG1である、(28)に記載のDNA分子。
(30)IgG1が配列番号:23に示される配列によってコードされる、(29)に記載のDNA分子。
(31)ヒト定常重鎖が、(14)から(18)のいずれかに記載の1つ又は2つ以上の置換をFc領域内に有する、(28)から(30)のいずれかに記載のDNA分子。
(32)(1)から(26)のいずれかの項に記載の抗体の可変軽鎖をコードする領域を含むDNA分子。
(33)可変軽鎖コード領域がヒト起源の定常軽鎖をコードする領域と融合される、(32)に記載のDNA分子。
(34)定常軽鎖がカッパ鎖である、(33)に記載のDNA分子。
(35)カッパ軽鎖が配列番号:25に示される配列によってコードされる、(34)に記載のDNA分子。
(36)(27)から(31)のいずれかに記載のDNA分子及び/又は(32)から(35)のいずれかに記載のDNA分子を含む発現ベクター。
(37)(36)に記載の1つ又は2つ以上のベクターを保有する宿主細胞。
(38)(27)から(31)のいずれかに記載のDNA分子を含む発現ベクター、及び(32)から(35)のいずれかの項に記載のDNA分子を含む第二の発現ベクターを保有する、(37)に記載の宿主細胞。
(39)哺乳動物細胞である、(37)に記載の宿主細胞。
(40)(36)に記載の1つ又は2つ以上のベクターを哺乳動物宿主細胞にトランスフェクトする工程、前記宿主細胞を培養する工程、並びに前記抗体分子を回収及び精製する工程を含む、(1)から(26)のいずれかに記載の抗体を製造する方法。
(41)活性成分として、(1)から(26)のいずれかの項に記載の1つ又は2つ以上の抗CD37抗体分子及び医薬的に許容できる担体を含む、医薬組成物。
(42)さらに1つ又は2つ以上の追加の治療薬剤を含む、(41)に記載の医薬組成物。
(43)1つ又は2つ以上の追加の治療薬剤が、CD37以外のB細胞抗原を標的とする薬剤から選択される、(42)に記載の医薬組成物。
(44)B細胞抗原がCD20である、(43)に記載の医薬組成物。
(45)1つ又は2つ以上の追加の治療薬剤が、アポトーシスを誘発する薬剤から選択される、(42)に記載の医薬組成物。
(46)薬剤がTRAILレセプターの調節物質である、(45)に記載の医薬組成物。
(47)表面にCD37を発現するB細胞を枯渇させることを目的とする、(41)から(46)のいずれかに記載の医薬組成物。
(48)B細胞悪性疾患の治療を目的とする、(47)に記載の医薬組成物。
(49)B細胞悪性疾患が、B細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択される、(48)に記載の医薬組成物。
(50)病理にB細胞が中心的に関与する自己免疫疾患又は炎症性疾患の治療を目的とする、(47)に記載の医薬組成物。
(51)細胞集団に(1)から(26)のいずれかに記載の抗体分子又はそのような抗体分子を含む医薬組成物を投与する工程を含む、細胞集団からCD37発現細胞を枯渇させる方法。
(52)in vitroで実施される、(51)に記載の方法。
(53)(41)から(46)のいずれかに記載の医薬組成物の有効量を前記患者に投与する工程を含む、B細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択されるB細胞悪性疾患に罹患している患者を治療する方法。
【実施例1】
【0043】
キメラ及びヒト化抗CD37抗体の作製
a)キメラ抗体A0の作製
配列番号:2及び配列番号:4に示す可変重鎖及び可変軽鎖アミノ酸配列(GeneArt, Regenburg, Germany)を基にして、哺乳動物細胞用最適化コドン使用頻度を適用しつつ対応するDNA配列を合成し、5’末端にHindIIIクローニング部位及び3’末端にBamHIクローニング部位を追加する。この合成DNA分子をHindIII及びBamHIで消化し、得られたDNAフラグメント(配列番号:1及び配列番号:3プラス制限部位)を、ヒトIgG1定常領域及びヒトカッパ軽鎖定常領域(それぞれ配列番号:24及び配列番号:26)をコードするpcDNA3.1系発現ベクターにてクローニングする。エンドフリープラスミド調製物(Qiagen)を作製し、各プラスミド1mg/Lの濃度で供給業者のプロトコルにしたがって、重鎖及び軽鎖プラスミドをHEK293フリースタイル細胞(Invitrogen)に同時トランスフェクトする。72時間後に、上清を採集し、ELISAによってIgG濃度を決定する。得られたキメラ抗CD37抗体(A0と称する)を改変プロテインAカラム(GE Healthcare)で精製し、クエン酸緩衝液に溶出し、続いてPBSで透析する。
【0044】
b)キメラ抗体A0のヒト化型の作製
a)で得たキメラmAb A0のヒト化は、例えばUS5,225,539、US6,548,640、US6,982,321に記載されたCDR移植方法を用いて実施する。
mAb A0のVLドメインの構造モデルを確立するために、構造の鋳型をBrookhaven National Laboratoryのタンパク質データバンク(PDB)から選択する。ネズミモノクローナル抗体エントリー“1KB5”のVLドメインが、88%配列同一性/81%類似性及び2.5Å解像により選択される。mAb A0のVHドメインのためには、90%配列同一性及び91%類似性を有する同じマウスモノクローナル抗体構造“1KB5”が主要な造形鋳型として選択される。ヒトコンセンサスフレームワークに対する最高の一致は、ヒトVカッパ1(hVK1)及びヒトVH1(hVH1)のタイプであることが見出された。また別の設計として、もっとも安定なヒトコンセンサスドメインhVK3及びhVH3への移植が選択される。移植のためには、mAb A0_VL及びmAb A0_VHモデルをヒトコンセンサスドメインモデルhVK1、hVK3、hVH1A及びhVH3と結合させ、Fv-モデルを作製する。ループ移植は、ネズミmAb A0 CDR領域をヒト抗体フレームワークに埋め込むことによって実施し、ヒト化された鎖の構築物のDNA分子を合成する。
それぞれのヒト化可変領域を合成し、免疫グロブリン発現ベクターでクローニングし、さらに、表1に示す重鎖及び軽鎖配列を一緒に、a)で述べたようにHEK293フリースタイル発現系(Invitrogen)で一過性に発現させ、プロテインAカラムで精製する。
【0045】
表1:実施例で用いられるキメラ及びヒト化抗CD37抗体の可変重鎖及び軽鎖の配列
【0046】
c)Fc操作キメラ及びヒト化抗CD37抗体の作製
Fc変異体の作製は、Lazarら(2006)の記載のように実施する。得られたFc操作重鎖配列を発現ベクターpAD-CMV1(EP393438に記載)に導入し、軽鎖コード配列を含むプラスミドと一緒にCHO-DG44細胞に同時トランスフェクトする。トランスフェクション後5から7日で細胞培養液から抗体を採集し、改変プロテインAクロマトグラフィーで精製し、クエン酸緩衝液に溶出し、続いてPBSで透析する。サンプルのタンパク質含有量はプロテインA HPLCで決定し、エンドトキシン含有量はKinetic-QCL色原体動力学アッセイ(Lonza)で決定する。サンプルのモノマー含有量はHP-SECで決定し、機能試験に用いた全てのサンプルが95%を超えるモノマー含有量を示していた。
【0047】
表2:キメラ及びヒト化抗CD37抗体の可変重鎖及び軽鎖配列(列III及びIV)並びに変異(列II)(抗体A0、B0、C0などは、表1の抗体A、B、Cなどと同一である)
重鎖及び軽鎖の完全長配列は列VおよびVIに記載される(星印を付した列Vの配列は、列IIに対応する置換(及びコードDNAに対応する変異)を含むように改変した配列番号:24及び23(野生型)のIgG1配列に該当する)
【実施例2】
【0048】
キメラmAb A0は特異的にCD37抗原を認識する
細胞CD37に対するMAb A0の特異性をFACS競合アッセイによりRamosバーキットリンパ腫細胞(ATCC#CRL−1596)で試験する。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。新しい培養液で1:6の比にて週に2−3回、この培養を継代培養により3x105から1.8x106細胞/mLの細胞濃度で維持する。FACS競合アッセイのためには、フィコエリスリン(PE)で直接標識したCD37特異的mAb HH1(Santa Cruz)を1μg/mLの濃度で用いる。抗体を非標識競合抗体A0と表示のモル比で4℃にて10分間プレインキュベートする。その後、1x105のRamos細胞を抗体混合物と氷上で30分インキュベートする。その後で、細胞をリン酸緩衝食塩水(PBS)で2回洗浄し、FACS緩衝液に再懸濁し、BD FACS Cantoで測定する。そのようなアッセイの結果は図1に示されている。20倍モル過剰のコントロールヒトIgG1抗体(Sigma IgG1カッパ)の添加は、Ramos細胞の平均蛍光強度(MFI)を有意には低下させない。20倍モル過剰の非標識HH1抗体又はA0抗体の添加は、直接標識HH1抗体の結合をほぼ完全に抑制する。このことは、A0及びHH1抗体は、Ramos細胞上の同一又は類似のエピトープを認識し、細胞CD37抗原との結合に競合することを示している。
【実施例3】
【0049】
MAb A0ヒト化型と細胞CD37抗原との結合
A0のヒト化型をそれらの細胞CD37抗原結合についてFACS分析によって試験する。抗体をRamos細胞に表示の濃度で添加し、4℃で30分結合させる。その後、PE-標識ヤギ抗ヒトIgG抗体(Sigma)を用い結合抗体を検出する。細胞をPBSで2回洗浄し、その後、FACS緩衝液に細胞を再懸濁し、BD FACS CantoでFACSにより分析する。例は図2及び3に示されている(それぞれ抗体A、B、C、D、I又はA、H、I、J、K、L及びM,表1参照)。A0のヒト化型のいくつかは、親抗体A0と同様なRamosとの結合を示し、ヒト化は細胞CD37抗原との結合を低下させないことを示している。
【実施例4】
【0050】
キメラMAb A0ヒト化型のFACSスキャチャード分析
抗体A0ヒト化型(B、C、D、H、I及びKと称される、表1参照)の細胞CD37抗原に対する親和性を、別のところに記載されている(Brockhoff et al. 1994)FACSスキャチャード分析によって決定する。略記すれば、抗体希釈を96ウェルプレートで第一のウェル(80μL)の100−400nMから始め、それに続く11希釈工程により調製する(1:2、40+40μL)。50μLのmAb希釈物をFACSチューブに添加し、150μLの細胞(0.8x106/mL=1.2x105細胞/チューブ)を各FACSチューブに加える。細胞を穏やかに混合し、1時間氷上でインキュベートする。その後、50μLのFITC結合二次抗体(濃度、15μg/mL;マウスmAb抗hu IgG全サブクラス、Zymed 05-4211)を添加し、混合し、氷上で30分間インキュベートする。その後、0.02%の酸を含む4mLのPBS(pH7.2)を添加し、細胞を沈殿させ、300μLのPBS(pH7.2)に再懸濁し、BD FACS Cantoを用いFACS分析に付す。全ての実験工程は氷水上で実施し、全ての抗体希釈物はPBS/0.5%BSA+0.02%の酸で作成する。FACSの目盛り定めはQuantum FITC MESF(Premix)High Level Beads(Bangs Laboratories)を用いて実施する。全サンプルを同じFACSパラメータを用いて測定する。結合IgG対遊離IgGの比を種々の抗体濃度でのMFI値から算出し、スキャチャードブロットとして表示する。図4は、A0のいくつかのヒト化変種におけるMFI/抗体濃度の関係を示す。これらの結果は、出発抗体に類似する、いくつかのヒト化型のRamos細胞結合を示し、解離定数(Kd)は2.15から4.90ナノモル/Lの範囲である。
【実施例5】
【0051】
キメラmAb A0のヒト化型のADCC活性
抗体依存細胞媒介性細胞傷害(ADCC)を仲介するA0ヒト化型(B、C、D、H、J、Kと称される、表1参照)の能力を、標的細胞としてRamos細胞及びエフェクター細胞としてIL2-刺激ヒトPBMCを用いて評価する。Ramos細胞(バーキットリンパ腫、ATCC#CRL−1596)をATCCから購入した。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。この培養を、新しい培養液で1:6の比にて週に2−3回、継代培養により3x105から1.8x106/mLの細胞濃度で維持する。1.5x106から1.8x106/mLの細胞濃度及びログ期増殖にある細胞培養のアリコットを10分間遠心する(200xg、すなわち1000rpm)。細胞を洗浄培養液(L-グルタミン非含有RPMI1640)で1回洗浄し、ペレットにする(200xg、すなわち1000rpm)。細胞ペレットをアッセイ培養液(L-グルタミン非含有RPMI1640に1%BSA)に再懸濁し、細胞数を決定する。細胞濃度を2x105に調整する。
健常ドナーから採取した約50−80mLの全血をPBMCの単離に使用する。50mLチューブで10mLの全血を26mLのHBSS(カルシウム及びマグネシウム非含有Hanksバランス塩類溶液)により1:3.6に希釈する。18mLの希釈全血を、50mLチューブ中の12mLのLymphoprep(Nycomed Pharma)の最上部に重層し、370xg(1400rpm)で35分遠心する。接触面から単核細胞を吸引し、1回目の洗浄をHBSS(750xg、すなわち1900rpm、10分)で、続いて2回目の洗浄をHBSS(300xg、すなわち1200rpm、10分)で、最後にHBSS(160xg、すなわち900rpm、10分)で洗浄する。ペレットにした細胞を培養液/アッセイ培養液(L-グルタミン非含有RPMI1640中に10%熱不活化ヒトAB血清)に、ピペットを用い穏やかに再懸濁させ、細胞数をセルカウンターで決定する。PBMC濃度を1x107/mLに調整する。新しく単離したPBMC(5x105/mL)を培養液(10%ヒトAB血清を補充したL-グルタミン非含有RPMI1640)中で組織培養フラスコ(75cm2)にて37℃で一晩、CO2インキュベーターで維持する。次の日、細胞を最終濃度1U/mLのhIL-2でさらに3日間刺激する。IL-2刺激PBMCをLymphoprepグラディエントで細胞屑から分離する。精製したIL-2刺激PBMCを、1x107/mLの濃度で培養液/アッセイ培養液に懸濁させる。
特異的又は非特異的抗体の存在下でのエフェクター細胞と標的細胞との同時培養を、96ウェル丸底マイクロタイタープレートによりデュープリケート又はトリプリケートで実施する。各ウェルに付き最終体積200μLのアッセイ培養液は、1:1比の10%ヒトAB血清及びRPMI中の1%BSAから成る。最初にエフェクター細胞(各ウェルに付き10%ヒトAB血清含有RPMIの100μL中に新しく単離したPBMC)をプレートし、続いて標的細胞及び1%BSA RPMI 50μL中で希釈した抗体溶液をプレートする。コントロールとして、エフェクター細胞をアッセイ培養液のみで培養し(エフェクター細胞コントロール)、さらに標的細胞をアッセイ培養液のみ(自発的溶解)又は1%トリトンX-100補充アッセイ培養液(最大溶解)で培養する。前記同時培養を湿潤CO2インキュベーターにて3時間37℃でインキュベートする。インキュベーション終了時に、室温で遠心して(200xg、すなわち1000rpm、10分)細胞を培養液から取り除く。細胞を含まない上清(100μL/ウェル)を96ウェル平底プレートの対応するウェルに移す。これらの上清100μL中のLDH活性を決定するために、反応混合物(新しく混合した250μL触媒と11.25mLの色素溶液)を各ウェルに添加し、暗所にて室温で30分インキュベートする。続いて下記に記載するように吸収を測定する。
細胞傷害検出キット(LDH、Roche)を用いてADCC活性を測定する。細胞傷害の検出は、形質膜損傷細胞から遊離されるLDH酵素活性の測定を基準にする。培養上清中に放出されるLDHは、キットのテトラゾリウム塩をフォルマザンに還元する。フォルマザン色素の最大吸収は、ELISAプレートリーダーで650nmの参照波長に対して490nmで測定される。パーセント細胞媒介細胞傷害を算出するために、各実験設定で5つのコントロールを実施する。
バックグラウンドコントロールI(1):アッセイ培養液に含まれるLDH活性、これを値(3)及び(5)から差し引く。
バックグラウンドコントロールII(2):1%トリトンX100アッセイ培養液に含まれるLDH活性、これを最大LDH放出値(4)から差し引く。
自発的LDH放出(3):標的細胞単独から放出されるLDH活性。
最大LDH放出(4):標的細胞の放出可能最大LDH活性。
エフェクター細胞コントロール(5):エフェクター細胞単独から放出されるLDH活性。
細胞媒介性細胞傷害のパーセンテージを決定するために、トリプリケート又はデュープリケートの平均吸収を算出し、製造業者の指示にしたがってバックグラウンドを差し引く。図5では、E:T比25:1及びRamos標的細胞を用いたADCCアッセイの結果が示されている。抗体は30ng/mLの濃度で添加される。出発mAb及びそのヒト化型の両方がRamos細胞に対して類似するADCC活性を示す。結論すれば、抗CD37 mAb Aのヒト化はそのADCC誘発能力を有意には変化させない。
【実施例6】
【0052】
mAB A0のヒト化型のアポトーシス促進活性
mAb A0(=A)及びそのヒト化型(B、C、D及びI;表1参照)のアポトーシス促進活性を、Ramos細胞とmAbとのインキュベーション後のアネキシンV/PI陽性細胞を測定することによって評価する。Ramos細胞(バーキットリンパ腫;ATCC#CRL-1596)をATCCから入手する。10%の熱不活化ウシ胎児血清、12.5mMのHEPES、1mMピルビン酸ナトリウム、1%MEM非必須アミノ酸補充RPMI-1640+GlutaMAXを培養液として用いて、細胞を組織培養フラスコ(175cm2)で増殖させる。初期濃度3x105細胞/mL、37℃及び5%CO2湿潤雰囲気下で細胞を3日間培養する。この培養を、新しい培養液で1:6の比にて週に2−3回、継代培養により3x105から1.8x106/mLの細胞濃度で維持する。1.5x106から1.8x106/mLの細胞濃度及びログ期増殖にある細胞培養のアリコットを10分間遠心する(200xg、すなわち1000rpm)。細胞を洗浄培養液(L-グルタミン非含有RPMI1640)で1回洗浄し、ペレットにする(200xg、すなわち1000rpm)。細胞ペレットを培養液に再懸濁し、細胞数を決定する。細胞濃度を1x106に調整する。96ウェルの丸底プレートにウェル当たり100μLの細胞懸濁液をプレートする。10%FBSを含む細胞培養液で抗体を希釈し、ウェル当たり100μLの抗体溶液を添加する。細胞をCO2インキュベーターで37℃にて20から24時間インキュベートし、その後Vybrantアポトーシスアッセイキット#2を用いて染色する。Alexa Fluor488標識アネキシンV及びヨウ化プロピジウム溶液を前記細胞に添加し、暗所で15分間インキュベートする。その後、細胞を400μLのアネキシンV結合緩衝液に再懸濁させ、BD FACS Cantoを用いてFACS分析に付す。アネキシンV陽性/PI陰性細胞及びアネキシンV/PI陽性細胞のパーセンテージを、FL1/FL2チャンネルを用いて二次元ドットブロットで決定する。アイソタイプ合致非結合抗体(Sigma human IgG1)を陰性コントロールとして用いる。
図6では、Ramos細胞に対するmAb Aの種々のヒト化型のアポトーシス促進作用が示されている。細胞を10μg/mLの抗体とともに24時間インキュベートし、アネキシンV陽性細胞の全パーセンテージ(PI陽性及びPI陰性)が示されている。親のmAb Aは強力なアポトーシス促進活性を示す。驚くべきことに、ヒト化型は、親mAb Aと比較して有意に低いアネキシンV陽性細胞数を示し、ヒト化抗体のアポトーシス促進活性の変化が指摘される。結論すれば、この実験設定ではmAb Aのヒト化はアポトーシス促進活性の低下をもたらす。
【実施例7】
【0053】
キメラmAB A0のFc操作型のADCC活性
mAB A0のFc操作型(A1、A2、A3、A4、表2参照)のADCC活性を、標的細胞としてRamos細胞を用いて評価する。ADCCアッセイは上記記載(実施例5)のように実施する。実験結果は図7に示されている。A0のFc操作型は、親のmAb A0と比較して明瞭に改善された潜在能力及び有効性を示している。ある種のFc変種は、親mAbと比較して100%に達する最大溶解の改善、さらに親mAbと比較して10倍に達するEC50の改善を示す。結論すれば、特定のFc変異の導入はキメラmAb A0のADCC活性を大きく増進させる。
【実施例8】
【0054】
mAB B0のFc操作型のADCC活性
mAB B0のFc操作型(B1、B2、B3、B4、表2参照)のADCC活性を、標的細胞としてRamos細胞を用いて評価する。ADCCアッセイは上記記載(実施例5)のように実施する。B0のFc操作型は、親のmAb B0と比較して明瞭に改善された潜在能力及び有効性を示す。ある種のFc変種は、親mAbと比較して80%に達する最大溶解の改善、さらに親mAbと比較して20倍に達するEC50の改善を示す。結論すれば、特定のFc変異の導入はヒト化mAb B0のADCC活性を大きく増進させる。この実験結果は図8に示されている。
【実施例9】
【0055】
mAb A0及びB0のアポトーシス促進活性
抗IgG mAbによる架橋の前及び後におけるRamos細胞に対するmAb A0及びB0のアポトーシス促進活性を図9に示す。アポトーシスアッセイは実施例6に記載したように実施し、抗体架橋のためには、抗ヒトIgG抗体(γ鎖特異的、Sigma)を1:1の比で抗体に添加し、標的細胞への添加前に37℃で15分インキュベートする。図9では、CD37特異mAbは、1μg/mLの濃度で架橋された状態又は架橋されていない状態で添加される。キメラmAb A0はたとえ架橋されてなくても強力なアポトーシス誘発因子であり、この作用はmAbの架橋後に顕著に強化される。驚くべきことに、架橋されない状態で、ヒト化mAb B0はアポトーシス促進活性を完全に欠くが、抗IgG Abによる架橋後に強力なアポトーシス促進活性を示す。結論すれば、この実験は、mAb A0のヒト化型のアポトーシス促進活性は、抗体架橋後に回復されえることを示している。
【実施例10】
【0056】
mAB A0のFc操作型のアポトーシス促進活性
Ramos細胞に対するキメラmAb A0のFc操作型のアポトーシス促進活性を、実施例6に記載したようにアネキシンV/PI染色によって評価する。親抗体A0並びにFc操作変種A2及びA4を0.1から10,000ng/mLの濃度範囲にわたって力価を調べる。図10で分かるように、3抗体はいずれも同様なアポトーシス促進活性を示している。結論すれば、この実験は、mAb A0のFc操作はそのアポトーシス促進活性を変化させない。
【実施例11】
【0057】
a)Fc操作抗体A2およびB2の全血アッセイにおけるB細胞枯渇活性
全血アッセイを用いて、正常B細胞のヒト血液からの枯渇の効能及び潜在能力を評価する。このアッセイ様式では、ヒト健常個体血液のEDTA処理サンプルに被検抗体を添加し、続いて37℃での3から4時間のインキュベーション後に、4色FACSアッセイによって定量的に測定される。緩衝液又はIgGコントロールとの比較によって、被検物質によるB細胞枯渇の程度を算出することができる。in vivoでの人間の状況と類似するヒトIgGレベル及びエフェクター細胞の存在により、本アッセイタイプは被検抗体のin vivoにおける作用の予測のために高い相関性を有すると考えられる。
定量的FACSアッセイを用いて、健常個体由来の血液サンプルのB細胞及び/又はスパイクされたRamos細胞の数を決定する。定量は、既知数の蛍光ビーズを含むBD Trucountチューブを用いて実施する(前記ビーズは対象の細胞集団の定量のために内部標準として機能する)。B細胞は、FSC/SSC分析を組み合わせ4つの異なるCDマーカー(CD3/CD14/CD19/CD45)を用いて4色分析により識別する。
48ウェルプレートのウェル当たり270μLの新鮮血を、30μLの抗体希釈物(PBSにて)又はPBSとともにデュープリケートでインキュベートする。サンプルを37℃で4時間インキュベートし、その後直ちに氷上に置く。33μLのCDマーカーマスターミックスをTrucountチューブに加え、50μLの血液-抗体混合物を添加する。サンプルをヴォルテックスミキサーで混合し、15分間室温でインキュベートする。その後、450μLの溶解緩衝液を加え、ヴォルテックスミキサーで混合し、さらに15分間室温でインキュベートする。サンプルを氷上に置き、直ちにBD FACS CantoTMフローサイトメーターを用いてFACS分析に付す。データの判定はBD FACSDivaソフト(ヴァージョン5.0.2)を用いて実施する。
Fc操作キメラ及びヒト化mAb A2及びB2は、正常B細胞枯渇に優れた潜在能力を示し、EC50値は0.15から0.35nMの範囲である。正常B細胞枯渇の程度は57%から65%の範囲である。リツキシマブ(B-NHLの治療に用いられる公認抗体)を並行して試験し、このアッセイ様式では極めて弱いB細胞枯渇を示す(図11A)。
b)A0及びB0のFc操作はリツキシマブと比較して優れたB細胞枯渇を導入する
健常個体に由来するヒト血液のB細胞枯渇に対するmAbの作用をa)に記載したように評価する。非Fc操作mAb A0及びB0は、リツキシマブに類似する13%から26%の範囲のB細胞枯渇活性を示す。Fc操作は、両mAbのB細胞枯渇活性の劇的な増加をもたらし、平均枯渇パーセンテージは75%である。これは、リツキシマブと比較してA2及びB2が優れていることを明瞭に示している(図11B)。
c)抗体A2及びB2は全血アッセイでT細胞及び単球を枯渇させない
Tリンパ球(CD3陽性)及び単球(CD14陽性)に対するA2及びB2の作用を、Bリンパ球に対する作用と並行して評価する。T細胞数又は単球数のいずれにも有意な変化は観察されないが、一方、B細胞数には顕著な減少が認められる(図11C)。これは、A2及びB2はヒト血液から特異的にB細胞を枯渇させることを示している。
【実施例12】
【0058】
Fc操作はリツキシマブと比較して優れたADCC活性を導入する
Ramos細胞を標的細胞として用いてmAb A0のFc操作型A2のADCC活性を評価する。ADCCアッセイは上記(実施例5)に記載したように実施する。非Fc操作抗体A0は、リツキシマブより弱いRamos標的細胞最大溶解を示す(リツキシマブは、B細胞リンパ腫罹患患者のための承認治療であるCD20特異的抗体である)。驚くべきことに、A0のFc操作は、リツキシマブを超える明瞭に改善されたA2の潜在能力及び有効性をもたらす。これは、Ramos細胞のCD20及びCD37の同様な抗原濃度で、Fc操作抗 CD37mAb A2はリツキシマブより明瞭に改善されたADCC活性を示すことを指摘している(図12)。
【実施例13】
【0059】
全血アッセイにおけるFc操作抗体A2及びB2のリンパ腫細胞枯渇活性
Ramos細胞(ヒト血液から得られたバーキットリンパ腫由来細胞株)枯渇の有効性及び潜在能力を、実施例11に記載した全血アッセイを用いて評価する。アッセイの改変として、内在性B細胞と比べて約10倍過剰のRamos腫瘍細胞を全血マトリックスにスパイクし、それらの枯渇をFACS分析でモニターする。Fc操作キメラ及びヒト化mAb A2及びB2はRamos細胞枯渇に対し良好な潜在能力を示し、EC50値は0.35から0.54nMの範囲である。Ramos細胞枯渇の程度は36%から55%の範囲である。リツキシマブ(B-NHLの治療に用いられる公認抗体)を並行して試験し、このアッセイ様式では有意に低いRamos細胞の枯渇が認められる(図13)。
【実施例14】
【0060】
疾患関連モデルにおけるFc操作抗体A2及びB2のin vivo有効性
mAb A2及びB2のin vivo抗腫瘍有効性をヌードマウスのRamosバーキットリンパ腫モデルを用いて評価する。この動物の大腿部外側の皮下にCD37陽性Ramos細胞を注射し、腫瘍が樹立されたときに動物の静脈内処置を開始する。週に2回の処置スケジュール(q3/4d)を選択し、2つの異なる用量(8mg/kg及び25mg/kg)を並行して試験する。両mAbは顕著な抗腫瘍有効性を示し、T/C値は0.2%から26%の範囲である。2つの用量レベル及び2種の抗体間に有意な相違は観察されない。しかしながら、高用量のA2処置マウスはより優れた有効性傾向を示し、T/Cは0.2%であり5/10で完全な腫瘍の退縮がある。いずれの処置も良好な治療寛容を示し、見かけの体重低下は認められない。結論すれば、mAb A2及びB2はRamosバーキットリンパ腫モデルで顕著な抗腫瘍有効性を示し、8mg/kgの用量レベルで最大活性が既に得られていた。この活性は、並行して試験したリツキシマブの活性に匹敵する。Fc操作抗体A2及びB2で観察されたin vivo活性は、これらのmAbは、ネズミのエフェクター細胞ではなくヒトの細胞との相互作用のために最適化されているが故に過小評価されている可能性があることは特記される必要がある。この最適化された相互作用(前記はヒトエフェクター細胞を用いたとき極めて改善されたin vivoのADCC活性をもたらす(実施例8))は、用いられたマウスモデルには反映されない。しかしながら、この実験で得られたデータ(図14に提示)はCD37ターゲティングの概念のin vivoでの証明を提供し、したがってヒトでの治療用量の概算に用いることができる。
【実施例15】
【0061】
ヒトにおける治療用量の概算のための、マウスにおけるA2及びB2の薬物動態作用及び薬力学作用の相関性
A2及びB2の血清濃度とそれらの薬力学作用との間の相関性はRamos腫瘍異種移植モデルを用いてマウスで確立された。これらの実験によって、8mg/kgのA2及びB2(クエン酸緩衝液(25mMのクエン酸ナトリウム、115mMのNaCl、0.04%トゥイーン80、pH6.0)中で処方)の用量が、この侵襲性の皮下腫瘍モデルの顕著な腫瘍増殖の遅延を引き起こすことが、標準的なマウスのq3又は4dの抗体投与スケジュールにより示され、したがって、投与休止期間を通して継続的な活性が指摘された。さらにまた、薬物動態データも同じ用量について確立された。
マウスにおけるこのPK/PD関係により、ヒト化抗体のヒトでのクリアランス(CL)についての報告されたデータ(Lobo et al. 2004)を用い、ヒトのための概算用量を算出することができる。
A2のための完全な計算:
−8mg/kgの1回投与後の平均AUC(0−∞)=6099μg・h/mL;
−マウスの与えられたAUC(0−∞)=マウスのAUC(ss,τ)、及びAUC(ss,τ)/τ=C(ave,ss);
−マウスのC(ave,ss)(τ=84時間の場合)=73μg/mL、これは、おそらくマウスのC(ave,ss)(τ=168時間の場合)と等価である;
−ヒトのAUC(ss,τ)=D/CLであるので、さらにLoboら(2004)が報告した、ヒトにおけるヒト化抗体クリアランス(CL)範囲であるCL=7mL/h/70kgから15mL/h/70kgを用いると;
−7mL/h/70kgの場合:168時間x7=1176mLx73μg=86mg;
−15mL/h/70kgの場合:168時間x15=2520mLx73μg=184mg。
したがって、A2の場合、70kgのヒトの場合、1週間の概算用量は86から184mgの範囲である。上記に記載した同じ仮定を用いるならば、B2の場合に算出される70kgのヒトの1週間の概算用量は189から404mgである。
【実施例16】
【0062】
抗体A2及びB2は多発性ミエローマ細胞に対しADCC活性を示す
多発性ミエローマ細胞株群におけるCD37発現を、CD37に特異的抗体を用いてFACS分析により評価する。細胞を直接蛍光標識抗CD37抗体でインキュベートするか、又は非標識CD37特異的抗体でインキュベートし、続いて一次抗体に対して誘導した第二の蛍光標識抗体とインキュベートする。標識された細胞の蛍光活性をFACS Cantoフローサイトメーター(BD Bioscience)により測定し、FACS Divaソフトを用いMFIとして蛍光強度を記録する。11の被検多発性ミエローマのうち6つがCD37の細胞表面発現を示した(図15)。続いて1つの細胞株(RPMI8226)を、CD37特異抗体A2及びB2を用いて、実施例5に記載したようにADCCアッセイで試験する。両抗体はRPMI8226に対して強力なADCC活性を示し、EC50値は25ng/mLの範囲にあり、最大細胞溶解は約20%である(図16)。本実施例は、CD37陽性多発性ミエローマ細胞は、CD37特異的mAb A2及びB2を用いたADCCによる細胞溶解に感受性であることを示している。
図15は、CD37発現についての6つの多発性ミエローマ由来細胞株のFACS分析を示す。白地曲線はCD37特異抗体との反応性を示し、黒塗り曲線は陰性コントロール抗体を表している。
【実施例17】
【0063】
患者由来CLL細胞に対する抗体A2及びB2のアポトーシス促進活性
患者由来の慢性リンパ球性白血病(CCL)細胞に対するA2及びB2のアポトーシス促進活性を評価する。CLLと診断された患者から抹消血単核細胞(PBMC)を、ヘルシンキ宣言にしたがってインフォームド・コンセントの後で調製した。Ficoll-Paque(商標)プラス工程(StemCell Technologies, Meylan, France)にしたがって、新しく採集した血液から原発性CLL細胞を精製し、RPMI1640培養液(10%熱不活化ヒトAB血清(Sigma, France)を含む)で使用まで4℃で保存する。原発性CLL細胞用培養液は、2mMのL-グルタミン及び10%熱不活化ヒトAB血清を補充したRPMI1640である。実験に使用するために、原発性CLL細胞を血球計算板で計測し、その生存率は0.25%トリパンブルー排除により評価する。CLLサンプルの生存率は90%を超える。細胞を30μg/mLの抗体とともに37℃で24時間インキュベートし、その後アネキシンV陽性細胞のパーセンテージを実施例6に記載したように決定する。図17に示すように、Fc操作抗体A2及びB2は、原発性CLL細胞に対し強力なアポトーシス促進活性を示し、アネキシンV陽性細胞は約90%(A2)及び40%(B2)である。両mAb抗体は明らかにリツキシマブより優れている(リツキシマブはB-NHL治療について承認されたB細胞特異的抗体である)。mAb A2はまた、アレムツズマブ(B-CLL治療について承認された抗体)と比較しても明瞭に優れた活性を示す。
【実施例18】
【0064】
内因性CD37遺伝子がそのヒト同族体によって置き換えられているトランスジェニックマウスモデルの作製
非翻訳配列によってフランキングされているヒトCD37コード配列(BAC(細菌人工染色体)ID:RP11-433N13、RP11-50I11)を含むターゲティングベクターを構築する。続いて、このターゲティングベクター(さらにまたエクソン3−4にフランキングするloxP部位及びfrt部位によってフランキングされているneo選別マーカーを含む)を相同性組換えに用い、マウスES細胞及び標準的技術により、マウスゲノム配列のエクソン1−8をヒトの対応する配列と置き換える。最後に、有糸分裂を不活化させたマウス胚線維芽細胞(MEF)で構成されたフィーダー層上で、C57BL/6N ES細胞株をDMEM高グルコース培養液(20%FBS(PAN)及び1200U/mLの白血病阻害因子(Millipore ESG 1107)を含む)で増殖させる。1x107細胞及び30gの直鎖化DNAベクターを240V及び500Fでエレクトロポレートする(Biorad Gene Pulser)。G418選別(200g/mL)は2日目に開始する。ガンシクロビル(2M)によるカウンター選別はエレクトロポレーション後5日目に開始する。ESクローンを8日目に単離し、増殖させさらに液体窒素でクローンを凍結した後で、標準的方法によるサザンブロッティング、例えば標的遺伝子に特異的な放射能標識DNAプローブを使用することによって分析する。続いてトランスジェニック動物を、当分野で公知の標準的な方法によって、例えば胚盤胞注入とそれに続くキメラ動物の発生によって作製する。ヒトCD37についてヘテロ接合及びホモ接合の動物を、それぞれキメラ動物及びヘテロ接合動物の通常交配によって入手する。ネズミCD37遺伝子のノックアウト及びヒトCD37遺伝子のノックインの成功は、標準的方法、例えば抹消血リンパ球のFACS分析又は組織切片の免疫組織化学的分析を用いてタンパク質レベルでモニターする。
【実施例19】
【0065】
代用抗体の作製
マカクCD37に特異的なモノクローナル抗体を、マカクCD37抗原の完全なコード配列(Acc No. ENSMMUT00000020744)を用いるマウス及びウサギの遺伝子免疫によって生成する。特異抗体は、マカクCD37抗原を発現する組換えHEK293又はCHO細胞を用い標準的なELISA又はFACS技術によって選別する。これら抗体の可変重鎖及び軽鎖コード配列をPCRクローニングによって回収し、ネズミ又はウサギの出発抗体に由来するVH及びVL領域を保有し、Fc部分は、本発明の抗体、例えばA2又はB2のそれと同一であるキメラ抗体の作製に用いる(実施例1に記載したとおり)。結合特性及び機能特性は、標的細胞としてマカクCD37発現細胞を用いるアッセイ系を使用することによって、例えば結合、FACS、スキャチャード分析、ADCC及びアポトーシスアッセイで精査することができる。最終的に、代用抗体は、カニクイザルの血液でそのB細胞枯渇活性によりin vitroで選別される。
【実施例20】
【0066】
抗体産生クローンの調製
本発明の抗体(例えば抗体A2、A4、B2又はB4)を産生するクローンを調製するために、完全な重鎖をコードするDNA分子(例えば配列番号:27、31、35又は39に示す配列をそれぞれ有する)を、pBI-26と称される真核細胞発現ベクター(ハムスター由来ジヒドロフォレートレダクターゼ選別マーカーもまた別にコードする)に挿入する。
配列番号:29、33、37及び41にそれぞれ示される完全な軽鎖をコードするDNA分子を、ネオマイシンホスホトランスフェラーゼ選別マーカーをまた別にコードするpBI-49と称される真核細胞発現ベクターに挿入する。前記完全な重鎖及び軽鎖のDNA配列を完全に配列決定する。
化学的に規定した培養液にて懸濁状態で増殖させたハムスター細胞株CHO-DG44に、上記に記載の重鎖及び軽鎖のための真核細胞発現ベクターを同時トランスフェクトする。トランスフェクト細胞を、ヒポキサンチン及びチミジンを含まず、抗生物質G418を含む培養液で選別する。続いて、細胞を段階的選別、及びメトトレキセート(MTX)の濃度を増加させる増幅に付す。800nMのMTX増幅工程から、単一細胞クローンを増殖パフォーマンス及び抗体産生を基準にスピナー操作で選別し、Safty Cell Bank(SCB)で凍結保存する。
【0067】
参考文献
American Cancer Society (Cancer Facts & Figures 2005).
Baker and Jones, Curr Opinion in Drug Discovery & Development, 10, 219-227, 2007.
Barbas, et al., Proc. Nat. Acad. Sci, USA 91:3809-3813, 1994.
Barrena et al., Leukemia 19: 1376-1383, 2005.
Belov et al., Cancer Research 61: 4483-4489, 2001.
Boulianne G. L., Hozumi N. and Shulman, M .J., Production of functional chimeric mouse/human antibody. Nature 312: 643, 1984.
Brockhoff G, Hofstaedter F, Knuechel R., Cytometry 1994, 17(1):75-83.
Buchsbaum et al., Cancer Research 52: 6476-6481, 1992.
Chothia and Lesk., J. Mol. Biol. 196: 901-917, 1987.
Coiffier B, et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: a multicenter phase II study. Blood 1998; 92: 1927-1932.
Coiffier, JCO, 23, 6387-93, 2005.
Edelman et al. Proc Natl Acad Sci USA 63: 78-85, 1969.
Feugier P, et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d'Etude des Lymphomes de l'Adulte. J Clin Oncol 2005; 23: 4117-4126.
Foran JM, et al. European phase II study of rituximab (chimeric anti-CD20 monoclonal antibody) for patients with newly diagnosed mantle-cell lymphoma and previously treated mantle-cell lymphoma, immunocytoma, and small B-cell lymphocytic lymphoma. J Clin Oncol 2000; 18: 317-324.
Forstpointner R, et al. The addition of rituximab to a combination of fludarabine, cyclophosphamide, mitoxantrone (FCM) significantly increases the response rate and prolongs survival as compared with FCM alone in patients with relapsed and refractory follicular and mantle cell lymphomas: results of a prospective randomized study of the German Low-Grade Lymphoma Study Group. Blood, 2004; 104: 3064-3071.
Francisco et al., Blood, 2003 Aug 15;102(4):1458-65.
Frank, et al., Methods Enzymol. 154: 221-249, 1987.
Gait, M.J., Oligonucleotide Synthesis. A Practical Approach. IRL Press, Oxford, UK (1984).
Goldenberg DM and Sharkey RM, Oncogene (2007) 26, 3734-3744. Novel radiolabeled antibody conjugates.
Hainsworth JD. Prolonging remission with rituximab maintenance therapy. Semin Oncol 2004; 31: 17-21.
Hawkins et al., J. Mol. Biol. 254:889-896, 1992.
Hayden and Mandecki. Gene synthesis by serial cloning of oligonucleotides. DNA 7(8): 571-7, 1988.
Hertz T, Yanover C: PepDist: A new framework for protein-peptide binding prediction based on learning peptide distance functions. BMC Bioinform (2006) 7 (Suppl 1):S3-S17.
Hiddemann W, et al. Frontline therapy with rituximab added to the combination of cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) significantly improves the outcome for patients with advanced-stage follicular lymphoma compared with therapy with CHOP alone: results of a prospective randomized study of the German Low-Grade Lymphoma Study. Group. Blood 2005; 106: 3725-3732 (2005a).
Hiddemann W, et al. Treatment strategies in follicular lymphomas: current status and future perspectives. J Clin Oncol 2005b: 23; 6394-6399.
Howard OM, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol 2002; 20: 1288-1294.
Ichimura et al., J. Antibiot. (Tokyo), 44, 1045-53, 1991.
Jackson et al., 1995, J. Immunol. 154(7):3310-9.
Johnson S, Bird R E. Construction of single-chain derivatives of monoclonal antibodies and their production in Escherichia coli. Methods Enzymol. 203: 88-98, 1991.
Jones TD, Hanlon M, Smith BJ, Heise CT, Nayee PD, Sanders DA, Hamilton A, Sweet C, Unitt E, Alexander G, Lo KM et al: The development of a modified human IFN-α2b linked to the Fc portion of human IgG1 as a novel potential therapeutic for the treatment of hepatitis C virus infection. J Interferon Cytokine Res (2004) 24(9):560-572.
Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, Gaston JS, Baker MP: Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of Factor VIII. J Thromb Haemost (2005) 3(5): 991 1000.
Kabat E. A., Wu T. T., Perry H. M., Gottesman K. S. and Foeller C. Sequences of Proteins of Immunological Interest (5th Ed.). NIH Publication No. 91-3242. U.S. Department of Health and Human Services, Public Health Service, National Institutes of Health, Bethesda, MD 1991.
Kahl B, et al. Maintenance rituximab following induction chemoimmunotherapy may prolong progression-free survival in mantle cell lymphoma: a pilot study from the Wisconsin Oncology Network. Ann Oncol, 2006; 17: 1418-1423.
Kaminski et al., JCO 10: 1696-1711, 1992.
Kipriyanow and Le Gall, Molecular Biotechnology 26: 39- 60, 2004.
Knobeloch et al., MolCellBiol 20: 5363-5369, 2000.
van der Kolk LE, Baars JW, Prins MH, van Oers MH. Rituximab treatment results in impaired secondary humoral immune responsiveness. Blood, 2002, Sept 15;100(6):2257-9.
McLaughlin P, et al. Rituximab chimeric anti-CD20 monoclonal antibody therapy for relapsed indolent lymphoma: half of patients respond to a four-dose treatment program. J Clin Oncol 1998; 16: 2825-2833.
Lazar et al., Proc Natl Acad Sci U S A. 2006 Mar 14;103(11):4005-10, 2006.
Ling and MacLennan, pp. 302-335 in Leucocyte Typing III. White Cell Differentiation Antigens, Oxford University Press, 1987.
Link et al., Journal Immunol 137: 3013-3018, 1986.
Lobo et al., J Pharm Sci 2004;93(11):2645-2668.
Lowman et al., Biochemistry 30(45): 10832-10837, 1991.
Marks et al., Biotechnology 10:779-783, 1992.
Moldenhauer G., et al., 1987. Biochemical characterization and epitope analysis of B lymphocyte-specific surface antigens defined by clustering workshop monoclonal antibodies. In Leukocyte Typing 111. A. McMichael. ed. Oxford University Press, Oxford. p. 378.
Moldenhauer G.: CD37. J. Biol Regul Homeost Agents 2000; 14: 281-83.
Press et al., JCO 7, 1989.
Press OW, et al. Radiolabeled-antibody therapy of B-cell lymphoma with autologous bone marrow support. N Engl J Med 1993; 329: 1219-1224.
Reche PA, Glutting JP, Zhang H, Reinherz EL: Enhancement to the RANKPEP resource for the prediction of peptide binding to MHC molecules using profiles. Immunogenetics (2004), 56(6):405-419.
Remington: The Science and Practice of Pharmacy, 21st edition, Hendrickson R. Ed.
Romaguera JE, et al. High rate of durable remissions after treatment of newly diagnosed aggressive mantle-cell lymphoma with rituximab plus hyper-CVAD alternating with rituximab plus high-dose methotrexate and cytarabine. J Clin Oncol, 2005; 23: 7013-7023.
Sasse et al., J. Antibiot. (Tokyo), 53, 879-85, 2000.
Shier et al., 1995, Gene 169:147-155.
Schwartz-Albiez et al, Journal Immunol 140: 905-914, 1988.
Stemmer et al. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides, Gene 164(1): 49-53, 1995.
van Spriel et al., Journal Immunol 172: 2953-2961, 2004.
Suzawa et al., Bioorg. Med. Chem., 8, 2175-84, 2000.
Tangri S, Mothe BR, Eisenbraun J, Sidney J, Southwood S, Briggs K, Zinckgraf J, Bilsel P, Newman M, Chesnut R, LiCalsi C, Sette A: Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol (2005) 174(6):3187-3196.
Thomas DA, et al. Chemoimmunotherapy with hyper-CVAD plus rituximab for the treatment of adult Burkitt and Burkitt-type lymphoma or acute lymphoblastic leukemia. Cancer 2006; 106: 1569-1580.
Ye et al. Gene synthesis and expression in E. coli for pump, a human matrix metalloproteinase. Biochem Biophys Res Commun 186(1):143-9, 1992.
Yelton et al., 1995, Immunol. 155:1994-2004.
Zhao et al., Blood 104: Abstract 2515, 2004.
Zhao XB, Lapalombella R, Joshi T, Cheney C, Gowda A, Hayden-Ledbetter MS, Baum PR, Lin TS, Jarjoura D, Lehman A, Kussewitt D, Lee RJ, Caligiuri MA, Tridandapani S, Muthusamy N, Byrd JC. Targeting CD37+ lymphoid malignancies with a novel engineered small modular immunopharmaceutical 2007; BLOOD, 1 OCTOBER 2007, VOLUME 110, NUMBER 7 (Epub ahead of print . Blood. 2007 Apr 17)
【特許請求の範囲】
【請求項1】
ヒトCD37と結合し、
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、
又は
b)i)配列番号:2に示される可変重鎖内に含まれるCDR;
ii)配列番号:4に示される可変軽鎖内に含まれるCDR;
iii)ヒト抗体から誘導される、前記CDRを支持するフレームワーク;
iv)ヒト抗体に由来する定常重鎖及び軽鎖、
によって規定されるヒト化抗体
から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体。
【請求項2】
配列番号:6に示される配列を有する可変重鎖を含む、請求項1記載の抗体。
【請求項3】
配列番号:12に示される配列を有する可変軽鎖を含む、請求項2記載の抗体。
【請求項4】
Fcドメイン内に1つ又は2つ以上のエフェクター機能を調節する1つ又は2つ以上の変異を有する、請求項1〜3のいずれか1項記載の抗体。
【請求項5】
エフェクター機能の調節が抗体依存細胞媒介性細胞傷害の増加である、請求項4記載の抗体。
【請求項6】
Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき239位および332の置換の組合せ、または、Kabat EU番号インデックスにしたがって番号付与したとき236位及び332位の置換の組合せ、または、Kabat EU番号インデックスにしたがって番号付与したとき236位、239位および332位の置換の組合である、請求項4または5記載の抗体。
【請求項7】
置換が332位の置換がI332Eであり、239位の置換がS239Dであり、236位の置換がG236Aである、請求項6記載の抗体。
【請求項8】
ヒトCD37と結合し、さらに配列番号:36のアミノ酸配列を含む重鎖を有する抗体。
【請求項9】
配列番号:38のアミノ酸配列を含む軽鎖を有する、請求項8記載の抗体。
【請求項10】
ヒトCD37と結合し、配列番号:40のアミノ酸配列を含む重鎖を有する抗体。
【請求項11】
配列番号:42のアミノ酸配列を含む軽鎖を有する、請求項10記載の抗体。
【請求項12】
請求項1から11のいずれか1項記載の抗体の可変重鎖をコードする領域を含むDNA分子。
【請求項13】
可変重鎖コード領域がヒト起源の定常重鎖をコードする領域と融合された、請求項12記載のDNA分子。
【請求項14】
ヒト定常重鎖がIgG1である、請求項12記載のDNA分子。
【請求項15】
ヒト定常重鎖がFc領域に請求項6または7に示した1以上の置換を有する、請求項13または14記載のDNA分子。
【請求項16】
IgG1が配列番号:23、または配列番号:29または配列番号:35に示される配列によってコードされる、請求項14または15記載のDNA分子。
【請求項17】
請求項1から11のいずれか1項記載の抗体の可変軽鎖をコードする領域を含むDNA分子。
【請求項18】
可変軽鎖コード領域がヒト起源の定常軽鎖をコードする領域と融合された、請求項17記載のDNA分子。
【請求項19】
定常軽鎖がカッパ鎖である、請求項18記載のDNA分子。
【請求項20】
カッパ軽鎖が配列番号:25、または配列番号:37または配列番号:41に示される配列によってコードされる、請求項19記載のDNA分子。
【請求項21】
請求項12から16のいずれか1項記載のDNA分子及び/又は請求項17から20のいずれか1項記載のDNA分子を含む発現ベクター。
【請求項22】
請求項16記載のDNA分子及び/又は請求項20記載のDNA分子を含む請求項21記載の発現ベクター。
【請求項23】
請求項21または22記載の1つ又は2つ以上の発現ベクターを保有する宿主細胞。
【請求項24】
請求項16記載のDNA分子を含む発現ベクターおよび請求項20記載のDNA分子を含む第2の発現ベクターを含む、請求項23記載の宿主細胞。
【請求項25】
哺乳動物細胞である請求項23または24記載の宿主細胞。
【請求項26】
請求項21または22記載の1つ又は2つ以上のベクターを哺乳動物宿主細胞にトランスフェクトする工程、前記宿主細胞を培養する工程、並びに抗体を回収及び精製する工程を含む、請求項1から11のいずれか1項記載の抗体を製造する方法。
【請求項27】
活性成分として、請求項1から11のいずれか1項に記載の1つ又は2つ以上の抗CD37抗体及び医薬的に許容できる担体を含む、医薬組成物。
【請求項28】
さらに1つ又は2つ以上の追加の治療薬剤を含む、請求項27記載の医薬組成物。
【請求項29】
1つ又は2つ以上の追加の治療薬剤がCD37以外のB細胞抗原を標的とする薬剤から選択される、請求項28記載の医薬組成物。
【請求項30】
B細胞抗原がCD20である、請求項29記載の医薬組成物。
【請求項31】
表面にCD37を発現するB細胞を枯渇させるための、請求項27〜30のいずれか1項記載の医薬組成物。
【請求項32】
B細胞悪性疾患、病理にB細胞が関与する自己免疫疾患又は炎症性疾患の治療を目的とする、請求項31記載の医薬組成物。
【請求項33】
B細胞悪性疾患がB細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択される、請求項32記載の医薬組成物。
【請求項34】
細胞集団に請求項1から11のいずれか1項記載の抗体又はそのような抗体を含む医薬組成物を投与する工程を含む、細胞集団からCD37を発現するB細胞を枯渇させるin vitroで行われる方法。
【請求項1】
ヒトCD37と結合し、
a)i)配列番号:2に示されるアミノ酸配列を含む可変重鎖、及びii)配列番号:4に示されるアミノ酸配列を含む可変軽鎖によって規定される、ネズミモノクローナル抗体、
又は
b)i)配列番号:2に示される可変重鎖内に含まれるCDR;
ii)配列番号:4に示される可変軽鎖内に含まれるCDR;
iii)ヒト抗体から誘導される、前記CDRを支持するフレームワーク;
iv)ヒト抗体に由来する定常重鎖及び軽鎖、
によって規定されるヒト化抗体
から誘導される抗体であって、キメラ抗体又はヒト化抗体である前記抗体。
【請求項2】
配列番号:6に示される配列を有する可変重鎖を含む、請求項1記載の抗体。
【請求項3】
配列番号:12に示される配列を有する可変軽鎖を含む、請求項2記載の抗体。
【請求項4】
Fcドメイン内に1つ又は2つ以上のエフェクター機能を調節する1つ又は2つ以上の変異を有する、請求項1〜3のいずれか1項記載の抗体。
【請求項5】
エフェクター機能の調節が抗体依存細胞媒介性細胞傷害の増加である、請求項4記載の抗体。
【請求項6】
Fcドメイン内の1つ又は2つ以上の変異が、Kabat EU番号インデックスにしたがって番号付与したとき239位および332の置換の組合せ、または、Kabat EU番号インデックスにしたがって番号付与したとき236位及び332位の置換の組合せ、または、Kabat EU番号インデックスにしたがって番号付与したとき236位、239位および332位の置換の組合である、請求項4または5記載の抗体。
【請求項7】
置換が332位の置換がI332Eであり、239位の置換がS239Dであり、236位の置換がG236Aである、請求項6記載の抗体。
【請求項8】
ヒトCD37と結合し、さらに配列番号:36のアミノ酸配列を含む重鎖を有する抗体。
【請求項9】
配列番号:38のアミノ酸配列を含む軽鎖を有する、請求項8記載の抗体。
【請求項10】
ヒトCD37と結合し、配列番号:40のアミノ酸配列を含む重鎖を有する抗体。
【請求項11】
配列番号:42のアミノ酸配列を含む軽鎖を有する、請求項10記載の抗体。
【請求項12】
請求項1から11のいずれか1項記載の抗体の可変重鎖をコードする領域を含むDNA分子。
【請求項13】
可変重鎖コード領域がヒト起源の定常重鎖をコードする領域と融合された、請求項12記載のDNA分子。
【請求項14】
ヒト定常重鎖がIgG1である、請求項12記載のDNA分子。
【請求項15】
ヒト定常重鎖がFc領域に請求項6または7に示した1以上の置換を有する、請求項13または14記載のDNA分子。
【請求項16】
IgG1が配列番号:23、または配列番号:29または配列番号:35に示される配列によってコードされる、請求項14または15記載のDNA分子。
【請求項17】
請求項1から11のいずれか1項記載の抗体の可変軽鎖をコードする領域を含むDNA分子。
【請求項18】
可変軽鎖コード領域がヒト起源の定常軽鎖をコードする領域と融合された、請求項17記載のDNA分子。
【請求項19】
定常軽鎖がカッパ鎖である、請求項18記載のDNA分子。
【請求項20】
カッパ軽鎖が配列番号:25、または配列番号:37または配列番号:41に示される配列によってコードされる、請求項19記載のDNA分子。
【請求項21】
請求項12から16のいずれか1項記載のDNA分子及び/又は請求項17から20のいずれか1項記載のDNA分子を含む発現ベクター。
【請求項22】
請求項16記載のDNA分子及び/又は請求項20記載のDNA分子を含む請求項21記載の発現ベクター。
【請求項23】
請求項21または22記載の1つ又は2つ以上の発現ベクターを保有する宿主細胞。
【請求項24】
請求項16記載のDNA分子を含む発現ベクターおよび請求項20記載のDNA分子を含む第2の発現ベクターを含む、請求項23記載の宿主細胞。
【請求項25】
哺乳動物細胞である請求項23または24記載の宿主細胞。
【請求項26】
請求項21または22記載の1つ又は2つ以上のベクターを哺乳動物宿主細胞にトランスフェクトする工程、前記宿主細胞を培養する工程、並びに抗体を回収及び精製する工程を含む、請求項1から11のいずれか1項記載の抗体を製造する方法。
【請求項27】
活性成分として、請求項1から11のいずれか1項に記載の1つ又は2つ以上の抗CD37抗体及び医薬的に許容できる担体を含む、医薬組成物。
【請求項28】
さらに1つ又は2つ以上の追加の治療薬剤を含む、請求項27記載の医薬組成物。
【請求項29】
1つ又は2つ以上の追加の治療薬剤がCD37以外のB細胞抗原を標的とする薬剤から選択される、請求項28記載の医薬組成物。
【請求項30】
B細胞抗原がCD20である、請求項29記載の医薬組成物。
【請求項31】
表面にCD37を発現するB細胞を枯渇させるための、請求項27〜30のいずれか1項記載の医薬組成物。
【請求項32】
B細胞悪性疾患、病理にB細胞が関与する自己免疫疾患又は炎症性疾患の治療を目的とする、請求項31記載の医薬組成物。
【請求項33】
B細胞悪性疾患がB細胞非ホジキンリンパ腫、B細胞慢性リンパ球性白血病及び多発性ミエローマから選択される、請求項32記載の医薬組成物。
【請求項34】
細胞集団に請求項1から11のいずれか1項記載の抗体又はそのような抗体を含む医薬組成物を投与する工程を含む、細胞集団からCD37を発現するB細胞を枯渇させるin vitroで行われる方法。
【図1】


【図2】
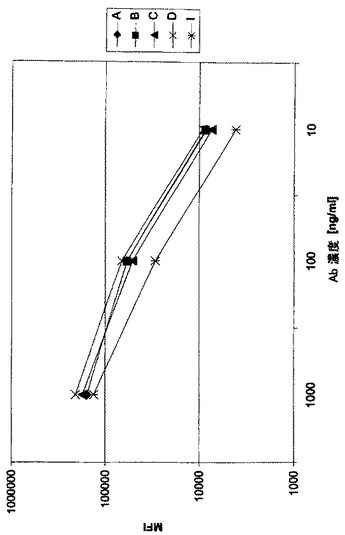
【図3】
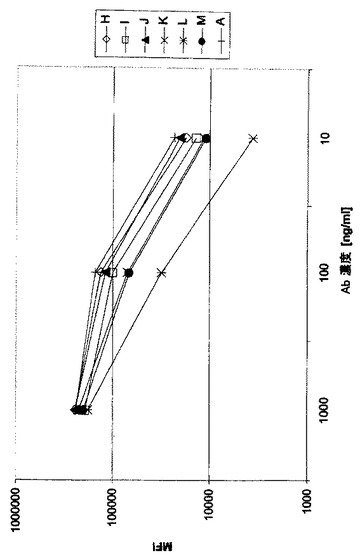
【図4】
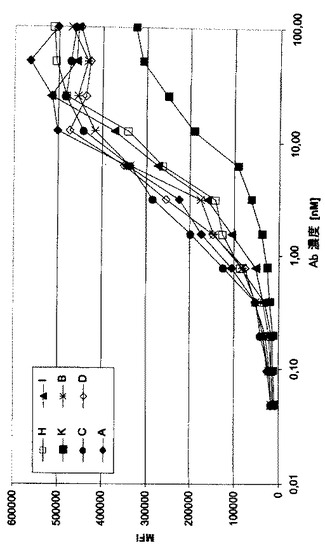
【図5】

【図6】

【図7】
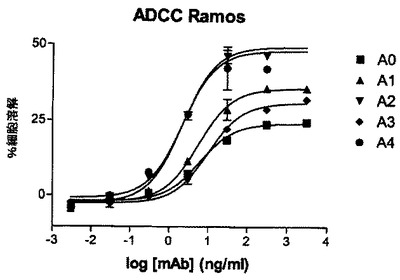
【図8】
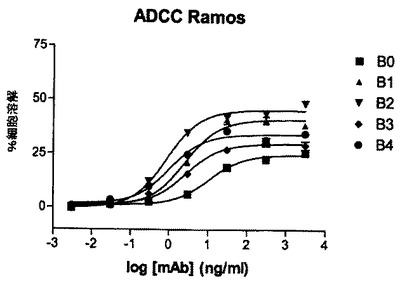
【図9】
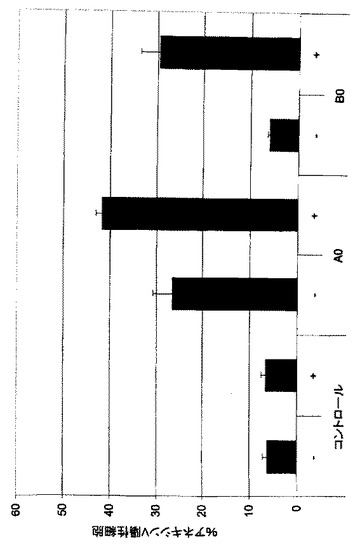
【図10】
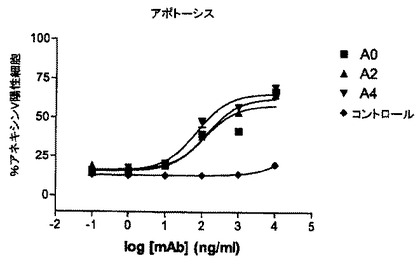
【図11A】
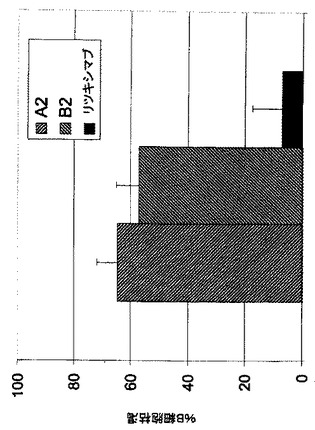
【図11B】
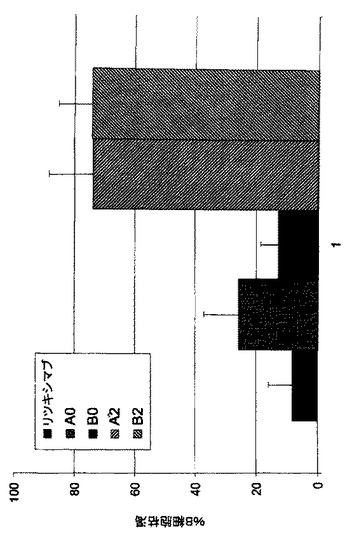
【図11C】

【図12】
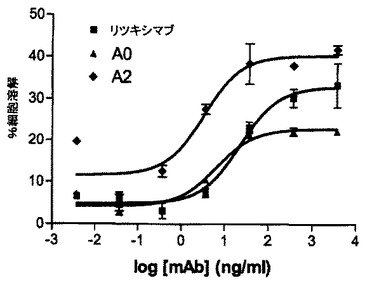
【図13】
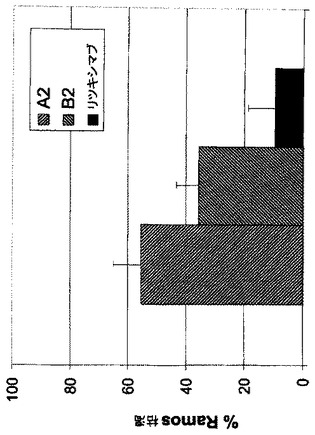
【図14】
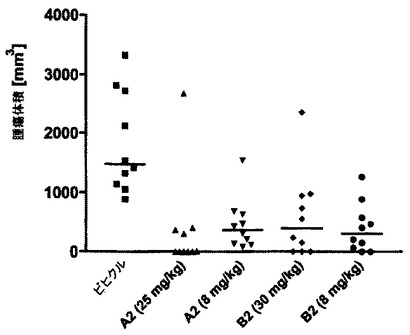
【図15】
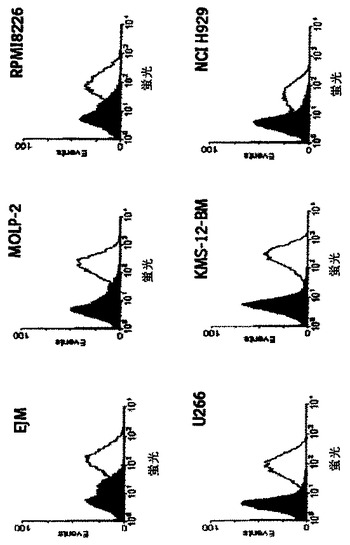
【図16】
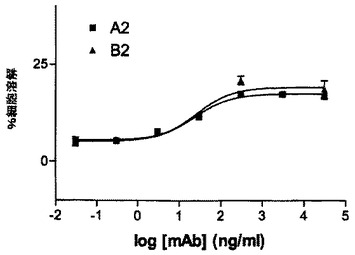
【図17】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11A】
【図11B】
【図11C】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【公開番号】特開2011−4758(P2011−4758A)
【公開日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願番号】特願2010−183075(P2010−183075)
【出願日】平成22年8月18日(2010.8.18)
【分割の表示】特願2010−519473(P2010−519473)の分割
【原出願日】平成20年8月8日(2008.8.8)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
【公開日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願日】平成22年8月18日(2010.8.18)
【分割の表示】特願2010−519473(P2010−519473)の分割
【原出願日】平成20年8月8日(2008.8.8)
【出願人】(503385923)ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング (976)
【Fターム(参考)】
[ Back to top ]