
拡大タグを使用したシークエンシング方法
【課題】位置的情報を伴って、または伴わないで使用することが可能な新しいシークエンシング方法の提供。
【解決手段】核酸分子の一部の配列に加えて前記一部の位置に関係する情報をも決定することによって、標的核酸分子の全部また一部をシークエンシングする方法を提供し、特に塩基の1個または複数の塩基を拡大して同定を促進することを含む新規のシークエンシング方法、特に、配列部分の配列情報およびその配列内のその部分の位置情報を合わせ持つ長い核酸分子のシークエンシングに適した方法、およびこのような方法を実施するキットに関する。
【解決手段】核酸分子の一部の配列に加えて前記一部の位置に関係する情報をも決定することによって、標的核酸分子の全部また一部をシークエンシングする方法を提供し、特に塩基の1個または複数の塩基を拡大して同定を促進することを含む新規のシークエンシング方法、特に、配列部分の配列情報およびその配列内のその部分の位置情報を合わせ持つ長い核酸分子のシークエンシングに適した方法、およびこのような方法を実施するキットに関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、各塩基によって表される情報を効果的に増幅する新規のシークエンシング方法、および特に、配列部分の配列情報およびその配列内のその部分の位置情報を合わせ持つ長い核酸分子のシークエンシングに適した方法、およびこのような方法を実施するキットに関するものである。
【背景技術】
【0002】
WatsonおよびCrickが1953年にDNA分子の構造を明らかにして以来、遺伝子研究者等は個々のDNA分子の配列を決定する迅速かつ安価な方法を求めてきた。Sanger/BarrellおよびMaxam/Gilbertは、シークエンシング技術において飛躍的な進歩を示した2種の新しいDNAシークエンシング方法を1975年と1977年との間に開発した。今日広く使用されている方法は全て、Sanger/Barrell法に基づいており、過去23年間に開発されたDNAシークエンシング法は多かれ少なかれこの方法を変更したものである。
【0003】
しかし、1988年に、DNAシークエンシング技術は全く新しい目的を得た。合衆国を筆頭として、18カ国が共に科学史上最大の単一プロジェクトにおいて、数種の他のより小さなゲノムに加えて3×109bpの全ヒトゲノムのシークエンシング(Human Genome Project、HGPとも称する)に参加した。現在、この目的は2003年中に完了することになっている。このプロジェクトは多大な科学的資源を占有し、莫大な費用がかさむという事実にもかかわらず、このプロジェクトの利益はこの費用に見合うほど充分に重要であると考えられている。
【0004】
このプロジェクトの要となるのは、現在の技術よりも手頃な値段で、かつ迅速な新しいDNAシークエンシング方法の開発である。基本的に、これらはゲルをベースとした手法(主として新規のSanger/Barrell変法)およびゲルをベースとしない手法に分けることができる。ゲルをベースとしない手法は、おそらく大きな可能性を秘めており、質量分析法、フローサイトメトリー、および小さなDNA分子をハイブリダイズする遺伝子チップの使用が、目下試みられている手法のいくつかである。現行の方法よりも実質的に優れた方法があれば、多くの患者に遺伝子試験の機会を提供し、薬剤の同定および開発に重要な役割を果たすことが可能であるため、遺伝子研究だけでなく、現代医学にも変革をもたらすであろう。このような方法の経済的将来性も当然非常に大きい。
【0005】
現在公知のシークエンシング技術を使用すると、各シークエンシング反応で読みとれる配列の長さを延長することは困難であることが明らかになっており、現在使用されている方法のほとんどは、シークエンシング反応当たり約7〜800塩基対に限定されている。今日広く使用されている方法では、シークエンシング反応当たり複数の配列をシークエンシングすることは不可能である。
【0006】
多くの、または長い配列を決定するためには、一般に多くのシークエンシング反応を並行して実施することが必要である(例えば、60億塩基対の2倍体ヒトゲノムをシークエンシングするためには、数百万のシークエンシング反応を並行することが必要であろう)。行程の総数、酵素および試薬の使用、必要とされる独特なプライマーの数などが、実施するシークエンシング反応数に直接比例することが多いため、このことは大きな関門である。さらに、重複した配列を決定するためにしばしば資力を費やさなければならない。加えて、異なった種類の組織的な仕事、例えばDNAライブラリの構築およびソーティングを実施しなければならない。標的となる可能性のある配列が他の配列の中にある場合は、それを単離するために資力を費やすこともまた必要である。
【0007】
シークエンシング反応の長さを制限する基本的問題を説明するために、現在使用中および開発中のシークエンシング方法を2種の群に大別することが適切である(この分別から外れる個々の方法もあるが、少数である)。第1の群では、ポリヌクレオチドの大きさの範囲に基づく方法がある。出発点は、分子全部が1個の共通末端および1個の任意末端を有する1種以上のポリヌクレオチドの「梯子」を形成することである。例えば、SangerおよびMaxam−Gilbertの古典的なシークエンシング方法は、4個の塩基A、C、GおよびTの各々を表す4種の配列梯子に基づいている。
【0008】
読み取ることができるシークエンシング反応の長さに関して制限となる要素は、1個のモノマーだけが変化したポリヌクレオチド類を区別することができなければならないことである。配列梯子中のポリヌクレオチドが長くなればなるほど、ポリヌクレオチド間の大きさの相対的差違は小さくなる。したがって、分子の大きさを決定する方法のほとんどは、すぐに2種の近接したポリヌクレオチド間を区別することができないという限界にすぐに達する。
【0009】
もう1つの群は、異なる原理に基づく方法である。標的分子中に存在する短い配列断片を同定することによって、配列断片間の重複を利用して標的配列を再構築することができる。
【0010】
したがって、多くのシークエンシング方法では、標的分子はより小さい断片に切断され、各断片の組成が論理的に導き出され、重複した配列を発見することによって元の配列が構成される。例えば、ミクロアレイ(microarrays)では65,536のアドレスが生じ、各アドレスは特有の8量体を含む。したがって、8量体(48=65536)の順列が網羅されている。次いで、この標的分子を蛍光で標識し、8量体とハイブリダイズさせる場合、標的配列にどんな配列断片が存在するかについての情報は、蛍光で標識したアドレスを記録することによって得ることができる。
【0011】
読み取ることができるシークエンシング反応の長さに関して重大な制限となる要素は、以下の組み合わさった問題である。実施するシークエンシング反応が長くなればなるほど、標的配列の再構成を可能にするための配列断片が長くなる。しかし、検討されなければならない順列の数は、同定すべき配列断片の長さとともに指数関数的に増大する。これによって、対応する方式ではミクロアレイでの特有のアドレスの必要性が増加する。
【0012】
ミクロアレイの他の用途は、例えば集団中の遺伝子変異のスクリーニングによる公知の配列の再配列である。この目的のため、必要なアドレスの数を減少させ、同定する配列断片の長さを増大させることができるように、オリゴヌクレオチドを公知の配列に適合させることができる。しかし、特定の目的のためのミクロアレイを考案することは高くつき、資源が必要で、現在あるのは2、3のDNA配列用のミクロアレイだけである。ヒトゲノムは約100,000〜140,000遺伝子から成り立っているため、この方法でヒトゲノムの大量シークエンシングをすることは非常に多くの資源が要求されるであろう。
【0013】
ミクロアレイを使用する他の欠点は、現在の構築技術(例えば、フォトリソグラフィ)の限界のため、約10×10マイクロメートル未満の画素(pixels)を生じることが不可能なことである。したがって、蛍光スキャナの解像能力のごく一部しか利用されない。現在の蛍光スキャナは0.1×0.1マイクロメートルの画素を区別することができ、ミクロアレイは現在含む情報よりも10,000倍の情報を含むことができることを意味する。
【0014】
したがって、前述の組み合わさった問題を回避できる長い配列断片を同定する新しい方法/原理を開発することが有利であろう。同様に、同定しなければならない配列断片の長さが、標的配列の長さに対して指数関数的に増加することのないように長い標的配列の決定を可能にする新しい方法/原理の開発が有利であろう。
【0015】
配列断片の同定に基づく(例えば、米国特許出願第5,714,330号で実施されているような)別のシークエンシング方法は、読み取りプレート上に断片化した標的DNAを分配することから成る。その後、1個または数個の最初の塩基対を表す蛍光シグナルを標的DNAに固定するようにこの標的DNAを処理する。各位置の蛍光シグナルは、標的DNAの次の塩基対についてこの手順を繰り返す前に読み取る。DNA分子を読み取りプレート上の位置に固定されたとき、いくつかのサイクルを行うことによって配列情報を有する長い断片の構築が可能になる。
【0016】
サイクル当たり数個の塩基対を読み取る能力は、必要とされる特有の蛍光シグナル数が塩基対の数に対して指数関数的に増加するために限界がある。1塩基対を読み取るために、4色が必要で、2個には16色、3個には64色などとなる。現在の技術で64色の異なる蛍光色を区別することが可能であるかどうかはよくわからない。とにかく、必要とされる読み取り時間および費用は多数の色を使用することでかなり増加するであろう。したがって、解決策は多くのサイクルを実施することであろう。これは、次には酵素的段階および蛍光読み取りの数が増大することを意味する。
【0017】
前述の戦略によって比較的長い配列断片の同定が可能であっても、再構成には重要な問題が待ち受けている。生物学的DNAは組成において非常に偏りがある。短い配列、および長い配列が、「巨視的」および「微視的」レベルにおいてにいくつかの箇所で繰り返されていることが多い。繰り返しDNA配列の領域では特に再構成は困難である。これらは、生物学的に興味深い可能性が多く、例としてはトリヌクレオチドの長さが「繰り返されている」。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】米国特許出願第5,714,330号
【発明の概要】
【発明が解決しようとする課題】
【0019】
前記の問題を克服する新しい試みが現在開発されている。驚くべきことに、位置情報(すなわち、標的配列内の該配列位置の情報)に関連付けられた配列情報が得られたならば、長い配列を正確に同定することができる。さらに、本発明は、位置的情報を伴って、または伴わないで使用することが可能な新しいシークエンシング方法を提供し、本明細書では拡大と称した、1種または複数の塩基に関連したシグナルの増幅が行われる。
【課題を解決するための手段】
【0020】
したがって最初の観点からは、第1態様によれば、本発明は少なくとも標的核酸分子の全部または一部のシークエンシング方法において
a)前記核酸分子の部分の配列を決定する段階と、
b)前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階とを含む方法を提供する。
【0021】
好都合には、複数の部分配列およびその位置が決定され、この情報が組み合わされる。
【図面の簡単な説明】
【0022】
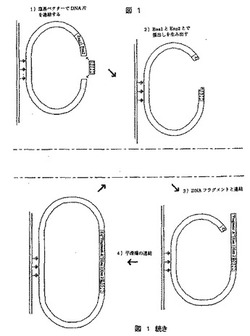
【図1】2種類の制限酵素、ハイブリダイゼーションおよび連結を使用して標的配列に拡大タグを導入する方法を示す図である。
【図2】2つの制限部位を運ぶベクターを使用して、標的配列に拡大タグを導入する方法を示す図である。この部位は両方とも張出し部、およびこれもまたそのような制限部位を含むアダプターになる。図2Aは標的DNAを含む塩基ベクターを示し、図2Bは使用することができるアダプターを示し、図2Cは各塩基を拡大するために使用される4つの酵素段階を示す。
【図3】プルーフリーディングのメカニズムによって、サイクルあたり9個の塩基をシークエンシングする方法を示す図である。図3Aは使用されるアダプター、図3Bは各塩基を拡大するために使用される酵素段階を示す。
【図4】標的分子の末端塩基を同定するために制限部位を完成させることを含むシークエンシングの方法を示す図である。A)はこのプロセスで使用されるアダプターを示し、B)は制限部位の一部を含むリンカー分子を示し、C)は標的DNAに結合し制限部位を完成させるリンカー分子を示す。
【図5】ハイブリダイゼーションおよび連結と結びつくKlenow修復反応を使用するシークエンシングの方法を示す図である。A)はその内部に標的分子が連結した塩基ベクターを示し、B)はサイクル1、3、5などに使用されるアダプターAの構造を示し、C)は大きさが同等な張出し部が形成されたかどうかに応じた、失敗した連結および成功した連結を示す。
【図6】一本鎖標的分子に結合するときにプライマーとして機能するアダプターを使用するシークエンシングの方法を示す図である。A)は標的分子に結合するためにプライマーとして使用されるアダプターを示し、B)は標的分子に結合するアダプターを示す。
【図7】拡大タグを保有し標的にハイブリダイズしセルフ−ハイブリダイズする、隣接して一直線に並べられたアダプターを示す図である。
【図8】複数の塩基に対応する拡大タグを含むアダプターの作成方法を示す図である。
【図9】ヘアピン型アダプターの使用に基づく転換方法を示す図である。拡大タグは標的分子の両末端にコピーされる。
【図10】転換されたDNA断片をお互いに連結させることに基づく転換の方法を示す図である。
【図11】核酸分子を2倍にする方法を示す図である。
【図12】プライマー依存のソーティング方策を使用するDNAシークエンシングのための方法を示す図である。A)は一般的な手順を例示し、この手順ではシークエンシング用プライマーが固形担体に結合され(段階1)、ここに標的分子が結合され(段階2)、ポリメラーゼ伸長によって伸長され(段階3)、次いで異なるシグナルを担持するプライマーが結合され(段階4)、鎖が延長され(段階5)、その結果生じた伸長生成物は次いでソーティング用に切り離される(段階6)。B)は、異なるシグナルが付着したウェル1において使用するための代表的なプライマーを示し、C)は16ウェルそれぞれについて得られる蛍光情報を示す。
【図13】ハイブリダイゼーションおよび得られた結果に基づくソーティング方策を使用する、DNAシークエンシングのための手順を示す図である。A)は次いで伸張される複数のアドレスを有する固形担体上での、一本鎖の蛍光標識された標的分子とオクタマープローブの結合を示し、B)はスキャン表面からの部分を示し、これは蛍光シグナルが各アドレスにおいて異なる長さに対応して直線状にどのように分配されているかを例示し、C)は1つのアドレスにおける蛍光の強度を示す。
【図14】標的DNA分子が固定リファレンスポイントに付着されるDNAシークエンシングの方法を示す図である。A)はPNAおよび対応する組成を有する一鎖の拡大タグ(ここではシグナル鎖として同定された)からなる転換アダプターを示し、B)は標的DNAの固形担体への結合、PNAアダプターとの接着、および伸張を示し、C)は分子が伸張された後の走査表面の外観を示す。
【図15】ナノポアシークエンシングの前のバイナリー末端転換の方法を示す図である。この中で転換アダプターを含む標的分子を生成するための手順、および方向タグに加え位置および配列の情報からなる生成したシグナルを示す。
【図16】塩基配列を表すシグナルを発生させるために、細胞がどのように利用されているかを示す図である。A)はエンハンサーがどのようにしてレポーター遺伝子型の拡大タグを保有する標的分子に加えられ、その後プライマーが付着されるかを示し、B)はシグナル分布を例示するヒストグラムを示す。
【図17】3つの拡大塩基各々と異なるシークエンシング梯子を作成することによるシークエンシングの方法を示す図である。この方法では結合標的分子が非特異的に切断され、拡大タグを付着させるための張出し部を発生させるために切断されるアダプターに結合する。
【図18】シークエンシングの方法を示す図である。固体担体に付着されるDNA分子の長さに沿って固定点を作成し、断片にされたDNAの末端が拡大される。
【図19】シークエンシングの方法を示す図である。この方法では拡大タグの鎖が固形担体に連続的に付着され、DNAリンカーを含むアダプターが使用され、DNAリンカーはシークエンシングされているDNAからのシグナルに一定間隔を置き、次のサイクルでシークエンシングされている分子へのアクセスを可能にするように取り除くことができる。
【図20】シークエンシングされる断片を含む分子に位置マーカーを導入する方法を示す図である。断片はアダプターによってシークエンシングされる。
【図21】ソーティングの方法を示す図である。この方法では固形担体上に存在するアダプターに標的分子が結合される。次いで分子の一末端が固形担体に結合され、他端が切り離され隣接するDNA鎖の転換を可能にする。(4)は、伸張後に生成したDNAを示す。
【図22】マッピング手順用の標的分子の調製方法を示す図である。この手順では数種の制限酵素が使用され、長さおよび方向の両方が異なる張出し部(3’または5’張出し部)を生成し、これは次いで拡大タグの鎖に連結される。
【図23】拡大の一般原則を示す図である。この原則では標的DNA分子の4つの最外部塩基が、拡大タグの鎖を用いた連結反応によって拡大される。拡大されない標的DNA分子の部分は、図示されるような位置情報を得るために、この場合光学マッピングに基づく方策を用いて読み取ることによって、使用することができる。
【図24】本明細書に記載されるソーティング方法を示す図である。この方法はミクロアレイで行われ、標的DNA中の4塩基の張出し部が256のアドレスを有するミクロアレイと混合され連結される。アドレス1はAAAA張出し部を含み、したがってTTTT張出し部を有する標的分子と結合する。
【図25】配列情報(左)および位置情報(右)の両方を得るために、シグナル鎖がどのように使用されるかを表す例を示す図である。A)は、特有なパターンで標的分子に結合する蛍光標識されたプローブを使用する、DIRVISHベースの方法であり、B)は光学マッピングに基づく方法を示し、この方法では配列の位置を与えるために制限パターンが使用され、C)はDNA結合タンパク質がミクロ/ナノ−ポアを通過する際に、その特有なパターンが記録される方法を示し、D)は、蛍光検出器を通過する際に記録される蛍光標識プローブ、タンパク質などを使用する方法を示す。
【発明を実施するための形態】
【0023】
本明細書では、標的核酸分子は、天然に生じるか、または合成したポリヌクレオチド分子、例えばDNAであり、これはゲノムまたはcDNA、RNA、例えば、mRNA、PNAおよびそれらの類縁体のことであり、適切ならば1本鎖、2本鎖および3本鎖であることが可能である。シークエンシングする部分には、標的分子全てが含まれることが好ましいが、例えば完全な分子より小さい、例えば4塩基から1kb、例えば4から100塩基であることも可能である。
【0024】
好ましくは、シークエンシングされる部分は、4個以上の塩基を有し、および/または前記部分の該標的分子内の前記部分の位置が1kb未満(すなわち1kb未満の解像度で)、特に好ましくは100塩基未満、さらに特に好ましくは10塩基未満の精度で決定される。数kb以上の解像度では、本明細書で記載した方法によって容易に実現される8〜10塩基より長い断片についての配列情報を得ることは通常不必要である。
【0025】
配列情報は、任意の便利な方法で得ることが可能で、1個または複数の塩基、特に好ましくは2個以上の塩基、例えば2から20塩基、好都合には4から10塩基について適切に得られる。理解されるように、配列部分の標的分子内の配置に基づいている前述したシークエンシング技法には、配列情報と同時にまたは別々に評価することが可能な位置情報を保持することは絶対必要である。いくつかの適切な技法を以下に説明する。
【0026】
位置情報は、いくつかの便利な方法で同様に得ることが可能で、これらの方法もまた以下に説明する。
【0027】
前述のように、本発明のこの観点では、得られる配列は、情報的に標的配列中のその位置にリンクしていなければならない。これはいくつかの方法、例えば核酸分子の末端領域または内部領域をシークエンシングすること、および位置についての、例えば分子の大きさ(例えば、長さまたは量)、発生したシグナルの強度または位置マーカーまたはアンカーまでの距離を参考にしてその位置を確立することによって実現することが可能である。配列情報は、1種または複数のシークエンシング反応サイクルを実施することによって得ることが可能である。
【0028】
前述したように、長い分子のシークエンシングにおける難点の1つは、長くなるにつれて、1塩基だけが変化した分子の大きさの相対的な違いを区別することが次第に困難になることである。一実施形態では、本発明は分子間の大きさ、強さ、長さまたはシグナルの違いを「拡大する」ことによってこの特定の問題を解決する。したがって、好ましい態様では、本発明はシークエンシングのサイクル当たり、2個以上の塩基(例えば、3個以上、好ましくは4個以上)の配列を決定し、および/または各塩基に関連したシグナルを拡大する標的核酸分子の配列の全部または一部を決定する方法を提供する。
【0029】
本明細書では、シークエンシングの「サイクル」とは、例えばシグナルを発生させること、または読み取ることによって、処理し配列情報を得ることが可能な最終産物を生じる一連の段階を実行することである。本明細書で記載した拡大方法およびシークエンシング反応では、複数のサイクル、例えば2個以上のサイクル、特に好ましくは4個以上、具体的には10個までのサイクルを実施することが好ましい。
【0030】
塩基に関連したシグナルの「拡大」とは、単一の塩基に関連する、または起着することが可能なシグナルの増強のことである。これは、例えば(このシグナルが塩基の大きさの場合)大きさの増加あるいは新しいシグナルの発生、例えば該塩基に標識もしくは他のシグナル手段を付加または結合させることであってもよい。
【0031】
同定された配列部分の長さを増加すると、大きさを決定する際の精度の低さを補うことができる。それによって、現在では精度が十分ではない大きさの決定法、例えば、フローサイトメトリ、DNA伸長などの方法を使用できると同時に、質量分析、ゲルによるソーティングおよび同様の方法の潜在力を利用することができる。
【0032】
分子間の違いを拡大することは、いくつかの方法で実現することができる。まず、得られた分子の長さが塩基2個以上異なり、したがって例えば、同時に位置情報をもたらすシークエンシング梯子における区別が可能になるように、サイクル当たり数個(例えば、4個以上)の塩基のシークエンシングを行ってもよい。(例えば実施例17参照)。あるいは、各塩基によって表される情報は、拡大することができてより容易に区別が可能になる。これらの異なる技法の例を以下に説明する。
【0033】
サイクル当たり2個以上の塩基(例えば4個)の配列を決定することは、任意の便利な技法によって実施することができる。位置情報に依存するシークエンシング方法では、得られる配列情報を標的分子内のこれらの塩基位置に再び関係付けることができるとの条件付きで、どんな技法を使用してもよい。このような場合、例えば相補的プローブ(例えば、固相担体に担持されている)へのハイブリダイゼーションを使用することが可能で、標的分子が結合するプローブを同定することによって、標的分子の末端配列が示される。例えば、2塩基の順列全てに相補的なプローブを、担持した、すなわち16個の異なるプローブを担持した固相担体を使用することが可能である。同様に、4個の塩基の順列全てに対するプローブ、すなわち256個の異なるプローブを相補的配列で標的分子を捕獲するために固相担体に付着させることが可能である。
【0034】
例示的手法では、(プローブはTTTTで終わるとき)AAAAで終わらない標的分子全ては結合せず、除去されるだろう。同様に、他のアドレスでは、特定の末端配列を有する標的分子が選択的に結合するであろう。この標的分子は、配列がそれぞれ末端または内部でも結合でき、さらに同定することが可能であるように(1本鎖が張り出した)2本鎖または1本鎖であることが可能である。PNAを相補的プローブとして使用する場合、このような分子は2本鎖形態に結合することが可能なので、2本鎖形態の内部配列もまた結合することが可能である。一般的用語では、この技法は本明細書では1個または数個の末端塩基対に基づいてソーティングするものとされ、1回または複数回のサイクルで実施することが可能である。この技法は、本明細書で記載した様な他の技法と組み合わせることが可能である。
【0035】
前記の逆の技法として、1本鎖標的DNAを固形基質に固定することによってシークエンシングを実施することが可能である。次いで、この標的DNAを、例えば16断片アダプターと共に混合しハイブリダイズさせることが可能である。アダプターは以下に説明するが、通常標的配列をシグナル増強または拡大された標的配列に適合させる分子のことである。次いで、ハイブリダイズしなかったアダプターは、溶液から洗浄される。これによって1本鎖DNAに相補的な張出しを有するアダプターだけが残る。例えば本明細書で説明した分析方法を使用して、溶液中にどのアダプターが残り、その結果16塩基対のどんな配列断片がこのDNAに含まれるかを確認することができる。
【0036】
このシークエンシング技法は、位置情報と組み合わせて実施するとき、またはシークエンシング技法だけで使用するとき、本発明の好ましい特性を示す。この方法で、単一の塩基によって伝えられる情報が、具体的にはその塩基を増幅すること、またはこの塩基をシグナルを発生させるために使用することができる「拡大」標識で置換することまたは増強することによって拡大される。(本明細書で意味する拡大はまた、いくつかの例では「転換」と称される。)
標的核酸分子の拡大は、増幅、例えば標的分子、またはシークエンシングする部分を含むそれらの部分を1倍または複数倍に倍加することによって実施することが可能である。分子間の、例えば10および11塩基の差を検出することは、拡大された分子、320および352塩基(5倍に倍加)間の差を検出することよりも困難であることは理解されよう。したがって、倍加の原理を使用して、例えばほとんどのDNA分析方法を改善することができる。これを実施するための適切な一技法を実施例11に説明する。他の適切な技法もまた使用することが可能である。
【0037】
したがって、この方法は、核酸分子間の大きさの違いを検出することに基づくほとんどの方法、例えばゲルをベースとするまたはベースとしない技法を改善するために使用することができる。この戦略によってまた、数個の塩基対の違いを区別するために感度が十分でない技法を使用して核酸物質を分析することが可能になる。例えば、改良したMaxam−Gilbert法が考えられ、そこでは1本鎖核酸分子(例えば5’−ビオチンを有する)を、ストレプトアビジンを結合したプレートに付着させる。次いで、シークエンシングを実施し、このプレートを洗浄し、その後得た核酸分子を例えば10倍に倍加して、1024塩基対の段階が得られる。これらの長さは、以下に説明した分析方法の1種によって測定することが可能である。
【0038】
したがって、他の観点から考えると、本発明は本明細書で記載したような標的分子の全部または一部をシークエンシングする方法を提供し、各塩基(または複数の塩基)に関連したシグナルは、前記塩基が前記配列に表れる回数の増加によって拡大する。
【0039】
本明細書では、特定の塩基(または1個以上の塩基)による「シグナル」とは、直接的にまたは間接的にその特性によって塩基(または塩基の集団)を検出する可能性のことを意味する。したがって、これは、直接的または間接的にあるいは1個または複数の他分子の結合によって、例えば前記塩基とシグナルが直接的または間接的に生じることが可能な標識部分との結合によって、検出することが可能な、大きさ、電荷または空間的配置といったその特性のことを意味することが可能である。したがって、直接的に検出可能なシグナルが与えられてもよく、またはシグナルを生じることが可能なシグナル伝達手段が提供されてもよい。このシグナルは、複数の塩基に特有であることが可能で、すなわちこのシグナルは1対の塩基対を示すか、または表すことが可能で、例えば、ATなどに使用するシグナルとは異なるAAに対するシグナルを使用することが可能である。このような分子および生じることが可能なシグナルに関連する、異なった機構については、以下に詳述する。
【0040】
さらに好ましい拡大技法には、1個または複数の特有のシグナル(またはこのようなシグナルを発生する手段)を配列中の1個または複数の塩基に関連させることが含まれる。前記シグナルが、複数の塩基に関連するとき、それぞれが1個または複数の塩基に対応する一連のシグナル(またはシグナル伝達手段)、または2個以上の塩基に特有な単一のシグナル(またはシグナル伝達手段)を使用することによってこれを実現することが可能である。これらのシグナルは、アダプター分子によって配列に付着することになるかもしれない拡大標識上にある。本明細書では、「関連(association)」とは、前記塩基(または複数の塩基)を前記シグナル(またはシグナル伝達手段)と置き換えること、および前記シグナル(またはシグナル伝達手段)を共存するように前記塩基(または複数の塩基)に付加することの両方を意味する。このシグナル(またはシグナル伝達手段)は、関連する塩基(または複数の塩基)に直接的に付着(または特異的に置換)している必要性はなく、この関連は間接的に、例えば1個または複数の分子が介在していることが可能である。関連は適切な化学反応、例えば、疎水性、イオン性、共有結合性などによることが可能であるが、標的核酸分子または関与分子との共有結合性相互作用によることが好ましい。
【0041】
本明細書では、「対応する」とは、塩基とシグナル、例えば、特定の塩基の存在を示すものとして読み取ることが可能な拡大標識によってもたらされるシグナルとの間の関係のことである。あるいは、マッピング手順の場合では、これはヌクレアーゼとこのヌクレアーゼの切断を示すマーカーとして使用されるシグナルとの関係のことである。
【0042】
本明細書では、「拡大タグ」とは、単一の分子あるいは1個または複数のシグナルを生じるための手段をもたらすタグ部分を含む、例えば標識を担うかまたは標識が結合することが可能な部位を有する分子複合体のことである。1個または複数のシグナルを生じる手段は、配列情報以外の情報が必要とされる場合、例えば標的分子または使用した切断方法に関係する情報の指示体として、取り込ませることが可能である。例えば拡大タグがポリヌクレオチドのときには、この拡大タグは、本来、1個または複数のヌクレオチド塩基に特異的に結合する他の部分を本来有することが可能である。この場合、このタグはさらに本明細書で前述したアダプターを含むものと考えられる。あるいは、この拡大タグは、標的配列に結合することを可能とするアダプターに付着するか、または付着するための手段を含むことが可能である。
【0043】
一般的に言って、この方法の例は以下に説明することが可能である。標的核酸物質中の塩基対は、4塩基、アデニン、シトシン、グアニン、およびチミンのそれぞれを表す4種の異なるタグ(以後拡大タグと称する)と結合する。したがって、A−T塩基対の場合は、「拡大タグA」が伴い、C−Gには「拡大タグC」が伴うなどである。それによって、元の塩基の順番、例えばACGTTが、「拡大タグA」−「拡大タグC」−「拡大タグG」などで増幅される場合、新しいDNA分子が生じる。それぞれの拡大タグは、シグナルを発生する手段を提供し、好ましい場合ではポリヌクレオチド分子であってもよい。この場合、必要に応じて、4個のタグの長さは2塩基対から数百kbp(所望するならばそれ以上)で変化させることが可能である。対応的に、DNA断片はリポーター遺伝子および他の生物学的情報を含むことができるか、または公知の生物学的機能のない配列だけから成ることができる。
【0044】
任意の便利な拡大タグを使用することが可能であるが、もちろん少なくとも4個、すなわち各塩基用の特有のタグが存在することは、配列を決定する目的のためには、不可欠である。もちろん、使用するタグは、シークエンシング技法、およびこれを実施する場合は、位置情報を引き出すために使用する方法に依存する。
【0045】
タグは、いくつかの他の形態でもたらすことが可能である。このタグは、特定のシグナルを生じることによって、直接的または間接的に検出するための手段を有する、すなわち、このタグは1個または複数のシグナル伝達手段を含む。蛍光、放射線、磁気、常磁性、電荷、大きさ、および量が特性の例で、拡大タグの粒子に、それらを検出し、かつ互いの分離を可能にするために付けることができる。これらの特性は、拡大タグ上に存在する1個または複数の標識に存在することが可能で、そこから出るシグナルは直接的または間接的に検出することが可能である。適切な標識は、シグナルの発生によって直接的または間接的に拡大タグを検出および/または測定させるものである。このような標識には、例えば、放射標識、化学標識(例えば、EtBr、TOTO、YOYOおよび他の色素)、発色団または蛍光体(例えば、フルオレセインおよびローダミンなどの色素)、またはフェリチン、ヘモシアニンまたは金コロイドなど高い電子密度の試薬が含まれる。あるいは、この標識は、酵素、例えば過酸化酵素またはアルカリホスファターゼであることが可能で、酵素の存在は適切な実体、例えば基質との相互作用によって視覚化される。この標識はまた、シグナル伝達対の一部を形成することが可能で、この対の他の因子はすぐ近傍に導入することが可能で、例えば蛍光化合物およびクエンチ蛍光基質を使用することが可能である。
【0046】
標識はまた、異なる実体、少なくとも拡大タグの一領域、例えば拡大タグのペプチド部分を認識する抗体上に与えることが可能である。この拡大タグがポリヌクレオチドであるならば、標識を導入する一方法は、例えば標識を担う適切な結合相手、例えば蛍光標識プローブまたはDNA結合蛋白質を結合することである。したがって、あるいはこのタグは、例えばその配列によって、標識が付着することが可能な分子を有するか、またはそれ自身が、その分子であることが可能である。標識は、単一の分子または微粒子、ナノ粒子(nanoparticles)、リポゾームまたは他の適切な形態の担体として付着することが可能である。
【0047】
好ましい態様では、この拡大タグは少なくとも2塩基、例えば30から1000塩基、好ましくは6から100塩基、特に好ましくは10から30塩基の長さの核酸配列自身である。これらの配列は、例えばその配列に相補的な蛍光プローブ、蛋白質などの使用によって、それらに付着した1個または複数の標識を有することが可能で、それから1個または複数のシグナルが生じることが可能である。あるいは、蛋白分子は、タグを含むか、またはタグに付着することが可能で、例えば免疫試薬または他の適切な結合相手、例えばDNA、DNA結合蛋白質によって認識されてもよい。このようなタグ分子の他の特性はまた、(制限酵素または蛋白分解酵素による)切断パターン、電荷、大きさ、形状などを調べることが可能である。
【0048】
拡大タグはまた、シグナルを生じるために使用することができる配列によって、情報を含むことが可能である。したがって、別の戦略はリポーター遺伝子、cis−調節因子など含む鎖を創出することである。その後これらは、細胞中にトランスフェクト/形質転換することができ、例えばリポーター遺伝子またはcis−調節因子の構成物は1個または数個のシグナルに転換される。この技法は、形質転換/トランスフェクション段階を必要とするが、細胞は転換段階(すなわち拡大タグの付加)を含む完全なシークエンシング反応を実施するようにプログラムされていてもよい。蛍光蛋白質または蛍光標識することができる膜蛋白質を発現する遺伝子、抗生物質抵抗性などを発現する遺伝子を使用するなど、極めて広い範囲のシグナルが生じることが可能である。時間による変化および配列中の特定塩基の存在を示すその他の特性に加えて、シグナルの質、量および位置も使用することができる。
【0049】
多細胞生物または構造体もまた使用することができるが、この方法では単細胞を使用することが都合がよい。生きていない細胞の同等物はまた、例えばナノテクノロジー(nanotechnology)を使用して、標識またはシグナルの発生に使用することができる。適切であれば、生じたシグナルは、例えば異なるプロモーターを使用することによって、同定のために他の場所に向けることが可能である。この技法をどのように実施するかの実例を実施例18に示す。
【0050】
ヌクレオチド塩基の各々に特異的な4個の拡大タグを好都合に使用することが可能である一方、前述のように、適切ならば複数の塩基に特有のシグナルを生じるために使用することが可能な拡大タグを使用してもよい。したがって、例えば16個の異なる蛍光体を使用することができる読み取り方法では、2塩基対の全ての順列を表す16個の異なるシグナルを発生するために使用する16個の異なるタグを使用することが適切であろう。
【0051】
他の状況では、4個よりも少ない異なるタグを使用することが適切であろう。例えば、1個がA/Tのとき、他方はC/Gである2個だけの拡大タグである。あるいは、4個の拡大タグを作り出す4個未満の特別のシグナル事象を使用することであり、これらのシグナル事象の特定の組み合わせによって4個の特有のシグナルが発生する(この場合個々の塩基にタグが付いている)。例えば、シークエンシング情報は2進法に転換することが可能である。この方法では、アデニンは一連のシグナル事象「0」+「0」に転換することが可能で、シトシンは「0」+「1」に、グアニンは「1」+「0」に、およびチミンは「1」+「1」に転換することが可能である。基本的には、配列情報を読み取るための1種またはおそらく2種の色または特有のシグナルがあれば十分である。これは、次に、いくつかのシグナルを使用した場合よりも読み取りが速いと同時に、より費用のかからない蛍光スキャナを使用することができることを意味するだろう。少なくとも4種の特有の拡大タグをもたらすために、例えば2進法タイプの読み出し形態を作り出すために、空間的に配置された単一のシグナル伝達手段を使用することは、本発明の好ましい態様であり、すなわち、前記シグナルは前記拡大タグ上に特有のシグナルを発生する単一のシグナル事象から成るパターンを含む。この場合、シグナル事象とは、測定可能なシグナル、例えば単一分子または他のこのような標識からの蛍光のことである。複数の拡大タグ、好ましくは20から100個のタグを使用するとき、これらは、必要なとき、位置情報を保存するために、直線的に、例えば長いDNA断片として連結していることが好ましい。
【0052】
タグ(直接である必要はない、例えばアダプターを介して生じてもよい。)とそれが表す塩基(または複数の塩基)との関係は、例えば塩基−塩基相補性による特定の塩基認識に依存する。しかし、本明細書での相補性には、Watson−Crick塩基対におけるヌクレオチドの対合に加えて、ヌクレオシド類縁体、例えば標的核酸分子中の塩基に特異的にハイブリダイズすることが可能なデオキシイノシン、および特異的なハイブリダイゼーションをもたらす他の類縁体、例えばPNA、RNA、DNAおよびそれらの類縁体の対合が含まれる。
【0053】
したがって、例えばDNA、RNAまたはPNA配列、またはそれらのハイブリッド、例えば4から20塩基、好ましくは6から12塩基の長さのオリゴヌクレオチドから成るプローブであって、(相補配列が存在する)標的分子の特定領域に結合し、拡大タグ、または一連のタグを付着し、各タグが、プローブが結合する1個または複数のヌクレオチド塩基を表すプローブを使用することができる。この場合、このプローブは標的配列へのタグの結合を促進するアダプター分子として作用する。あるいは、縮重したプローブの混合物は、特定の部位、例えばNNNNAAで1個または複数の特定の不変塩基のみと共に使用することが可能である。存在する拡大タグの数は、それらの結合した、結合することになるプローブの特定塩基の数に対応することが好ましい。しかし、特有のタグが2個以上の塩基の順列に対応して形成される場合、必要とされるタグはより少ない。
【0054】
この技法は、標的分子の不連続的な領域または部分を同定したり、または標的分子の配列の全てまたは本質的に全ての配列を得たりすることに使用してもよい。
【0055】
しかし、より精密なシークエンシングの別法は、配列を得るために読みとることができるシグナルを出す隣接した拡大タグ鎖の生成によってもたらされる。この効果を実現するためには他の方法があるが、最も便利な技法には、拡大タグを標的分子に挿入することが含まれる。この反応は、転換およびその後の一連の塩基の配列読み取りを行いつつ繰り返して実施することが特に好ましい。
【0056】
拡大タグを、拡大すべき塩基(または複数の塩基)に関連した標的分子に挿入するために、その塩基(または複数の塩基)および周辺の塩基に対する相補性、または認識性を利用することが必要である。この相補性を使用して、直接拡大タグを導入することが可能で、または最終的にその塩基に対応するタグを導入する方法を開始することに使用してもよい(実施例4参照)。
【0057】
これは、拡大タグと結合することができる張出し(すなわち、1本鎖である領域)を標的核酸分子に生成することによって実現することが便利である。(しかし、このような張出しはタグ分子またはその中間体、例えばそのアダプターが2本鎖型、例えばPNAを認識しかつ結合する場合は必要ない。)一方法は、標的分子の末端を、認識配列の外側で切断する制限酵素、例えばIPまたはIIS型の制限酵素の結合部位を含む短いDNA分子とを結合することである。これらの酵素は、切断される配列に対して特異性を示さず、したがって全ての種類の塩基構成を有する張出しを形成することができる。実際の標的分子、例えばDNA分子を問題の制限酵素とインキュベートする場合は、張出しがDNAの内部に形成されるように、結合部位を位置させることが可能である。実際に、3〜4塩基対の張出しを生成する酵素を選択することは好ましいと思われる(増幅し、固相に付着した標的分子にこのような張出しを生成するための一般的手順を示した実施例19を参照のこと)。
【0058】
70種類以上のIIS制限エンドヌクレアーゼが同定されており、基質特異性および切断パターンに関しては多くの変化がある。さらに、これらの酵素は、特定の必要性のために新しい酵素を創造することができるような「モジュールスワッピング」実験に非常に適していることがわかった(Huang−B 他;J−Protein−Chem.1996,15(5):481−9,Bickle,T.A.;1993 in Nucleases(2nd edn),Kim−YG他;PNAS 1994,91:883−887)。したがって、これらの酵素の非常に多くの組み合わせおよび変異体が、本明細書に記載した原理にしたがって使用できる。
【0059】
IIS型制限エンドヌクレアーゼは、いくつかの異なる目的のために使用されてきた。例えば、所定部位のほとんどの部位で1本鎖基質を切断することができる万能の制限エンドヌクレアーゼとしてである(Podhajska,A.J.,Szybalski,W.;Gene 1985,40:175−182,Podhajska,A.J.,Kim,S.C.,Szybalski,W.;Methods in Enzymology;1992,216:303−309,Szybalski,W.;Gene 1985,40:169−173)。
【0060】
シークエンシングの状況において、それらは米国特許出願第5714330号に記載されている前述の方法に対して用いられてきた。しかしながらこれらの場合、標的分子に連結したままである複数の拡大タグの導入は考慮されていなかった。
【0061】
IIS酵素での開裂は、多様な長さ、例えば−5から+6塩基の長さの張出し部をもたらす。ひとたび張出し部が作られると、張出し部の1つまたは複数の塩基に対応して、アダプターに担われ得る拡大タグを張出し部に取り付けることができる。
【0062】
IIS系または類似の系を用いて拡大タグを組み込むことのできるいくつかの異なる方法を以下に記載する。
【0063】
記載する第1の技法は、1つまたは複数の拡大タグを担い、一本鎖領域、すなわち張出し部を生成するために変性された標的核酸分子に相補的な張出し部を有するアダプターを使用することを含む。アダプター自体もまた、張出し部を生成するために用いた酵素と同一でも異なっていてもよいもう一つのIIS酵素のための認識部位を担っている。この技法の例は、実施例1に例示する。
【0064】
簡潔には、標的配列は、挿入点の近くにそれ自体がIIS部位を担うベクターに連結されるか、または標的配列は、そのような部位を含有するように設計される。適切なIIS酵素は次に、適切に配置されたとき標的配列に張出し部をもたらすIIS部位の開裂のために用いられる。一実施形態において、切断ベクターの少なくとも1つの末端が、例えば、IISに隣接するもう一つの制限酵素部位の使用によって平滑にされる。
【0065】
適切なアダプターはその後、張出し部の1つまたは複数の塩基に結合し、それによって拡大を可能にするために用いることができる。単一塩基の拡大の場合には、例えば4塩基張出し部、ANNN、TNNN、CNNN、およびGNNNに対するその形態の一本鎖部分、および拡大タグA、T、C、およびGをそれぞれ有する縮重アダプターを用いることができる。別法として、このアダプターは複数の張出し部塩基に対応して複数の拡大タグ、例えばATGCの張出し部を、当該の場合、線状に付された1つまたは複数の塩基に対応する拡大タグとともに、担うことができる。
【0066】
アダプターの張出し部および開裂ベクターがハイブリダイズされると、これらの分子は連結することができる。これは、張出し部の全長にわたる完全な相補性が実現され、反応の特異性を助ける場合にのみ得られる。次に平滑末端連結を達成し、アダプターの他の末端をベクターに接合することができる。前に用いた酵素と同一でも異なっていてもよい、もう一つのIIS部位(または他の適切な制限酵素部位)の適切な配置によって、最初のアダプターが向けられた配列の下流にある標的配列に張出し部が作られるように開裂を達成することができる。このようにして、隣接または重複する配列は拡大タグを担う配列に連続的に転換することができ、続いてそこからのシグナルを読み取り、本明細書の以下に記載する方法によって配列を決定することができる。この重複配列のシークエンシングは、前サイクルにおいて読み取られた配列を効果的にプルーフリーディング(proof-reading)し、これにより確認を可能にする。
【0067】
平滑末端が生成されず、その代わりにベクターが生成され、IISまたは類似の酵素で開裂が行われた後、もう一つの制限酵素を用い、ベクター内に挿入されるすべてのアダプターの末端に対し普遍的に相補的である張出し部を作るこの技法のわずかな変更を実施例2に示す。この方法は同様にベクターへのアダプターの連結、したがって拡大タグの連結を可能にする。
【0068】
類似しているが、より複雑な実施例を実施例3に例示する。この場合、隣接するDNAストレッチに対応して非相補的な張出し部が作られる。これらはともに適切な拡大タグを付したアダプターにハイブリダイズされる。シークエンシングが一方向に起こるように、アダプターのただ1つだけが、次のサイクルのための制限酵素部位を含有する。明らかにこれらの異なる性質を備えたアダプターの結合を可能にするために、隣接するDNAストレッチの張出し部は、識別可能な、例えば異なる長さでなければならない。これは、異なる長さの張出し部をもたらす異なる制限酵素を用いることによって達成できる。アダプターの2つの異なる型の末端は意図的に相補的であり、それゆえハイブリダイズし、連結されてベクターを形成することができる。それを含有するアダプターの制限部位は、開裂部位がさらに標的配列内に移され、隣接部位のシークエンシングが可能となるように適切に配置される。
【0069】
したがって、例えば5+4の張出し部が作られ、開裂部位が標的配列内へ4塩基に移される場合、次の9塩基が張出し部に転換され、その後、拡大タグに連結されるとき、それらの塩基のうちの5つが前サイクルの拡大タグと連結する。これによって配列の読み取り時に、前の5塩基の同一性を確認することが可能になり、したがってプルーフリーディング機構が導入される。
【0070】
IIS系を用いる他の技法は、DNAポリメラーゼのKlenow断片を使用することを含み、ほとんどのDNAリガーゼが異なる大きさの張出し部を連結できないという事実を利用している。これは、例えば実施例5に示す。この技法において、アダプターの張出し部よりも長い張出し部が作られる。この標的張出し部は、1つのタイプのヌクレオチド存在下でKlenowによって減ぜられる。1つの塩基だけ適切に伸長された標的だけがアダプターに結合し、これによりそのアダプターに付された対応する拡大タグによって導入された塩基の同定を可能にする。
【0071】
実施例4〜7に例示した他の技法は、拡大タグを担うアダプターの一本鎖標的へのハイブリダイゼーションを含み、それは次に標的に連結される。次にアダプターを、二本鎖分子を形成するためのポリメラーゼ伸長反応のプライマーとして用いる。さらなる別法は、ソーティングアダプター(sorting adapter)(この場合、必ずしも拡大タグに連結している必要はなく、単にソーティングのために用いることができる)を用い、アダプターは標的分子上に作られた張出し部より多い張出し部を有する固形担体に取り付けられる。したがって、例えばこのアダプターは8〜10塩基の張出し部を有していてもよい。例えばDNA片(二本鎖形態)が4塩基の張出し部を有するならば、張出し部のもっとも内側の塩基において塩基が互いに相補する場合にのみこれらの分子は連結するであろう。その後、ポリメラーゼ伸長が行われる。ポリメラーゼ伸長反応を成功させる要件は、プライマーとして機能できるように、アダプターの張出し部の残りの部分がDNA片と相補的なことである。このように、ポリメラーゼ伸長は、標的分子の末端配列がアダプターの張出し部に相補的である場合にのみ起こる。
【0072】
別法として、ハイブリダイゼーション単独で用いることができ、隣接する配列のストレッチに連結する拡大タグを合わせて、都合よく連結することができる。
【0073】
さらなる別法は、代謝酵素の認識部位に対する代謝酵素の特異性を利用する。そのような技法を、制限酵素を用いる実施例に例示する。しかしながら、いくつかの代替酵素、例えばトランスウポゼース(transposase)なども用いることができる。この方法において、シークエンシングされる標的分子は、4つの異なる標準的制限酵素で開裂されて平滑末端を生成し、(開裂すると張出し部を生成する)4つの異なる制限酵素の1つに対する制限部位の部分でそれぞれが終わる4つの異なるDNA分子に連結される。
これらは次に、標的分子上に連結される。標的分子が制限部位の残りの塩基を与える塩基で終わっている場合、制限認識部位が生成される。これは、その制限酵素を用いる開裂によって決定することができる。完全な形態で認識部位を有する分子だけが開裂される。開裂された分子を認識するために、張出し部と相補的なアダプターを用いることができる。これらのアダプターは次に、制限部位を完成するために標的分子によって提供された塩基の数に応じて、1つまたは複数の適切な拡大タグを担うことができる。この分子は次に環状化されることができ、サイクリングの繰返しが可能となる。好都合には、アダプターはその配列内に、平滑末端および張出し部の両方を生成する制限酵素のために適切に置かれた制限部位を有し、それにより隣接または重複する標的配列領域に対応する拡大タグを導入させることで繰り返しサイクルを行うことができる。
【0074】
本発明はしたがって、一態様において、標的核酸分子の一部分を同定する方法に関連し、この方法において前記部分を認識し結合する部分、および1つまたは複数の拡大タグを含む部分、好ましくは前記部分内の塩基を表す前記タグの鎖を含むアダプター分子は前記部分に取り付けられる、あるいは前記部分を置換する。
【0075】
したがって好ましい観点から見ると、本発明は、標的核酸分子の配列のすべてまたは一部を拡大する方法を提供し、この方法において、1つまたは複数の拡大タグは標的配列の1つまたは複数の塩基に連結しており、前記タグは前記標識配列の1つまたは複数の塩基に対応している。好ましくは、前記拡大タグはともに、少なくとも2つ、好ましくは少なくとも4つの塩基に対応する。好ましくは、前記拡大タグはそれぞれ、少なくとも2つ、好ましくは少なくとも4つの塩基に対応している。代替の実施形態において、それぞれの拡大タグは1つの塩基に対応し、ともに少なくとも4つの塩基、例えば8から20個の塩基に対応する拡大タグの鎖が用いられる。これは、例えば各サイクルに単一の拡大タグを加えて複数のサイクルを行うことによって、または単一のサイクルにおいて連結されたタグの鎖を用いることによって達成することができる。
【0076】
好ましくは、
前記方法は少なくとも、
a)前記標的配列の少なくとも一部分を、アダプター分子を結合するのに適した形態、好ましくは一本鎖形態に転換する段階と、
b)段階a)で作られたアダプター分子を結合するのに適した前記領域、好ましくは一本鎖領域の少なくとも一部分に、1つまたは複数の拡大タグを含む、または1種または複数の拡大タグを付着するための手段を含むアダプター分子を結合する段階であって、前記タグが前記標的配列の1つまたは複数の塩基に対応しており、好ましくは、前記アダプター分子を結合するのに適した前記領域、好ましくは前記アダプター分子が結合する前記一本鎖領域、または前記領域の近傍の1つまたは複数の塩基に対応する段階と、
c)所望により、少なくとも前記拡大タグが前記標的分子に結合したままであるように、前記標的分子を前記アダプター分子に連結する段階と、
d)所望により段階a)を繰り返す段階であって、作られた前記アダプターを結合するのに適した前記領域、好ましくは前記一本鎖領域が、段階b)による拡大タグに結合していない1つまたは複数の塩基を含む段階と、
e)所望により段階b)からd)を繰り返す段階であって、前サイクルのアダプター分子が結合した領域に関係する前記標的分子の隣接、または重複する領域に前記アダプター分子が結合する段階とを含む。
【0077】
段階e)は、いくつかの技法において省略することができ、例えば、拡大およびソーティングを連結することによってシークエンシングが実行され、そのため1回だけ拡大サイクルが行われる場合である。
【0078】
アダプター分子を結合するのに適した形態への「転換」は、標的分子が適切な形態でない場合にのみ必要である。したがって、PNA分子を結合するために、二本鎖標的分子の転換は必要でない。同様に、分子が一本鎖である場合、オリゴヌクレオチドであるアダプターを結合するために転換は必要でない。しかしながら、いくつかの場合において、アダプターの特異的、選択的結合を可能にするために、例えばDNA断片を溶融することによる転換が必要とされる可能性がある。分子全体を異なる形態に転換する必要はなく、適切な場合においては、一部のみが転換される。この部分は、少なくともアダプターの結合部分の長さ、したがって好ましくは4から500塩基、例えば6から30塩基の長さを含むべきである。この文脈における一形態から他の形態への転換の言及は、拡大に関して用いられるときの転換という言葉の使用と混同されるべきではない。
【0079】
本明細書において「アダプター分子」とは、標的配列をシグナルが強化されたまたは拡大された標的配列に適合させる分子である。本発明で用いられるアダプター分子は、単一分子、あるいは同一の種類でも異なっていてもよい分子の複合体である。アダプター配列は、前記標的配列に結合する結合部分を含み、例えば、標的配列の1つまたは複数の塩基に相補的な特定の塩基配列、より好ましくはポリヌクレオチド配列を認識するタンパク質である。好ましくは、結合配列は3から30塩基、好ましくは4から10塩基の長さである。アダプター分子は、1つまたは複数の拡大タグ、またはそのようなタグを付するための手段を追加的に含み、例えば、相補的である配列、または結合パートナーである。好ましくは、アダプターは、1つまたは複数のヌクレアーゼ認識部位、特に好ましくはその認識部位の外側を開裂するヌクレアーゼのための制限部位(または、少なくとも認識部位)、特に好ましくは、IIS酵素またはその類似体、特にFokI、および本明細書に記載する他の酵素の制限部位を含有する。好ましくは、他の制限酵素に関する部位は、このアダプターから除かれる。
【0080】
好都合には、アダプター分子は専ら核酸分子からなり、アダプターの様々な性質はアダプターの異なる領域によって与えられる。しかしながら、前述したように拡大タグは多様な形態を取ることができ、例えばタンパク質などの標識が含まれる。アダプターはこのように、拡大タグが結合することのできる分子を提供することができ、例えば、標的に結合するための領域に加えて、適切な結合パートナーを提供する。
【0081】
段階c)において、「少なくとも」前記拡大タグが連結したままであることが示される。したがって、アダプター、またはその部分は除去できることが考察される。
【0082】
本明細書において拡大タグの「鎖」は、拡大サイクルの前に結合させ、1つのアダプターに取り付けるか、または各サイクルの終わりに合わせて結合させたタグ、あるいは両方の組み合わせを指す。この結合は、任意の適切な手段によることができるが、しかしながら共有結合の手段による取り付けが好ましい。
【0083】
好ましくは、上述の方法は、前記標的配列に取り付けられた拡大タグから発生したシグナルを同定することによって前記標的分子の配列を決定することに加えて、上述の段階を含む本発明のシークエンシング法において用いられる。拡大タグを同定するために、読み取り可能なシグナルは拡大タグから発生されなければならない。これは、例えばタグがある種の性質を備えた標識(例えば、放射性標識)を担う場合のように、本来的に存在してもよく、あるいは、例えばさらなる分子の付加(例えば、それ自体が標識を担う結合パートナー)、または拡大タグの読み取り可能な形態への処理のような(例えば読み取り可能シグナルへの転換、具体的には読み取られるシグナルが発現タンパク質であるようなレポーター遺伝子の発現による)シグナル発生のためのさらなる段階を必要としてもよい。
【0084】
したがって好ましい態様において、本発明は標的核酸分子のすべて、または一部分をシークエンシングする方法を提供し、この方法において、好ましくは標的配列の1つまたは複数の塩基に連結した1つまたは複数の拡大タグの使用によって、前記標的核酸分子の配列の少なくとも一部分は拡大され、前記拡大配列は所望により読み取り可能なシグナルに転換され、前記配列は発生するシグナルの評価によって決定される。
【0085】
本明細書において「評価」は、絶対的または相対的に決定することのできる定量評価、および定性評価の両方を指す。
【0086】
連結は、化学的に、あるいは適切な天然にあるリガーゼまたはその変異体を使用することによって達成することができる。連結は本発明の好ましい特徴のみを表すものであるが、これは特異性を高めるために都合よく用いられる。ハイブリダイゼーションと比べて、連結がT4DNAリガーゼに基づく場合には、特異性は10倍に増大する。多くの場合ハイブリダイゼーションに基づくシークエンシング法は、許容できない高いエラー率を伴うためにこのことは重要である。さらに、耐熱性リガーゼ、例えばPfu、Taq、およびTTH DNAリガーゼなどを使用することによって特異性が向上し、一方で劇的に効率が上昇し、結果としてインキュベーションの時間が低減される。
【0087】
拡大タグを用いるこのシークエンシング法は、既知のシークエンシング法に対していくつかの利点を提供する。各サイクルにおいて複数の塩基を転換、または拡大することができ、したがって標的分子の特定の長さをシークエンシングするために必要なサイクルの数が低減される。拡大タグの選択、およびそれらが生成するシグナルに応じて、単純化された読み出しシグナルを生成することができ、例えばこのシグナルは二進法の読み出し形態であってもよく、すなわち単一のシグナル事象、例えば蛍光の適切な組み合わせ、例えば線状もしくは位置レイアウトによって1つまたは複数の塩基に対して特定のシグナルが発生される。これは、必要とされる特定のシグナル事象の数を低減する。したがって、例えば2塩基の組み合わせのそれぞれに16個の異なる標識、あるいは3塩基の組み合わせのそれぞれに64個の異なる標識を必要とする代わりに、本発明においては、各拡大タグ上に単一のシグナル事象を生成する手段のパターン、例えば、蛍光プローブを結合するための部位のパターンを提供することによって、16、または64またはそれ以上の特定のシグナルを発生させることができる。
【0088】
このシグナル情報は、緊密にパックされていることが可能である。タグは標識ヌクレオチドのみに限定されず、使用できる拡大タグおよび発生できるシグナルの種類において、より大きな融通性が与えられる。いくつかの実施形態において、サイクリング(cycling)が行われないときでも、配列の大部分を、それらの部分に拡大タグの鎖を用いることによってシークエンシングすることができ、したがって繰返しサイクリングに含まれる複雑な反応、および、シグナルを読み取ることのできる方法を限定する(例えば、ミクロ/ナノポア、またはフローサイトメーターを用いることはできなかった)ような、例えば読み取りプレートに固定された標的分子を用いて各サイクルからの情報を特定の標的配列に関連づける必要性が回避される。
【0089】
本発明の好ましい態様において、少なくとも部分的に一本鎖である形態への標的分子の転換は、一本鎖分子を用いて、あるいは例えば認識部位から離れた開裂部位を有する適切なヌクレアーゼ、例えばIIS酵素などを用いて張出し部を作ることによって達成される。
【0090】
好ましくは、この反応がサイクル的に行われるとき、各サイクルの拡大タグは、例えば会合もしくは連結によって、例えばそれらを含有する一本鎖の生成によってともに接合される。さらに、前記標的分子の前記アダプター分子への連結後、結果として生じた前記分子は、好ましくは環状化される。好都合には、これは標的分子をベクターに導入すること(または、標的分子の一部分を担体に取り付け、分子内で開裂後の自由な相互作用を可能にすることによって、実施例22を参照)、および前記アダプター分子が導入された後に、開裂および連結の適切な工程を用いることによって達成される。別法としては、生成される拡大タグの鎖を、有効な環状化を必要とせずに、標的分子の遠隔部位に転移または複写することができる。これを実行するための適切なプロトコルを実施例9に例示する。
【0091】
過剰なサイクリングの必要性を回避する別の好都合な技法は、より小さな転換断片、すなわち拡大タグが取り付けられた核酸分子のハイブリダイゼーションを含む。これらの断片は、それら自体が1つまたは複数の転換サイクルに供することが可能であったし、その後、非転換配列または拡大タグに担われた情報、例えばタグのヌクレオチド配列に、相補性によって結合することができる(実施例10を参照)。
【0092】
この反応のサイクリングを達成するために、反応に用いられる特定の酵素の制御が必要とされる。これは、用いられた酵素に応じて、異なった方法で達成することができる。したがって、結合および/または制限部位における開裂を防ぐためにメチル化を用いることができる。末端塩基のリン酸化状態を制御することによって、例えば、キナーゼ、またはホスファターゼの適切な使用によって、連結を防止する、または可能とすることができる。分子間連結を回避するために、適切に大容量を用いることもできる。効率を上げるために、制限反応の間、好ましくは少容量が用いられる。
【0093】
好ましくは、拡大の各サイクル(または本明細書に記載のシークエンシング)において、サイクル当たり少なくとも2つの塩基、好ましくは3と100個の間、特に好ましくは4から20個の塩基が転換される。好都合には、複数の拡大タグが、各サイクルにおいて1つまたは複数の塩基に連結される。例えば、好ましい実施形態において、各々が1つまたは複数の塩基に対応し、集合的に前記配列の一部分に対応するタグの集合(例えば、線状の列または鎖)を導入し、例えば4から12個の隣接塩基に対応する、具体的には複数のタグ、例えば4つを超えるタグである。好都合には、これは、それ自体が複数の塩基に向けられているタグ、例えば各塩基対に対する特定のタグと結合させることができる。
【0094】
実施例1に示すように、好ましい実施形態において、張出し部を形成するために上述の性質を有するヌクレアーゼが用いられる。さらに、前記ベクターは追加的に、張出し部を生成するためにヌクレアーゼ開裂から生じる末端の1つにおいて平滑末端開裂を生成するための制限酵素部位を含有する。別法として、最初の張出し部を作るために用いた酵素とは異なる制限酵素を用いることができ、この反応に用いたすべてのアダプターの一末端に対し厳密な相補性を有する張出し部を生成する。
【0095】
実施例3の方法を行うために、好都合には張出し部の隣接もしくは重複の領域を生成するヌクレアーゼ部位を用いる。これらの部位は、好ましくは用いられるアダプター内に位置する。各サイクルにおいて2つのアダプターが用いられ、それらは都合よく、末端にある相補的張出し部を用いることによって、標的配列の一本鎖部分に結合している領域に、ともに連結することを可能にする。したがって本発明の好ましい態様において、特にプルーフリーディングを可能にするために、用いられるアダプターは、認識部位から離れた開裂部位を有する2つ以上のヌクレアーゼに対する認識部位を含み、前記ヌクレアーゼでの開裂は、隣接し、または重複している一本鎖領域を生成する。本明細書において「重複」は、共通して塩基を有する配列、またはそのような配列に相補的である、すなわち対応する鎖上の配列を指す。したがって、重複領域を得るために二本鎖の標的の各鎖を利用することができ、重複しているが、相補的な領域をシークエンシングすることができる。好都合には、この作用を得るために各サイクルにおいて複数のアダプターを標的分子に結合される。この方法は、重複領域がシークエンシングされる場合、特定の塩基または塩基の集合体に対応に複数の拡大タグが取り付けられ、その塩基に対する繰返しシグナルを発生させるため、プルーフリーディングが可能となる。本発明によって、塩基当たり1つのタグが必要とされず、それゆえ塩基対などに対するタグを繰り返すことができることが理解されるであろう。
【0096】
Klenow断片の使用を包含する実施態様の実行において、段階a)において作られる前記一本鎖領域は、アダプター上に存在する核酸の一本鎖領域よりも長い1つまたは複数の塩基である。さらに、段階b)の後に追加の段階が必要であり、そこまでは標的分子の一本鎖領域の長さはポリマー伸長反応によって短縮される。
【0097】
一本鎖の標的分子を含む技法を実施するために、好都合には、サイクリングは、好ましくはポリメラーゼ伸長反応におけるプライマーとしてアダプターを使用することによって、二本鎖分子を生成することを含む。
【0098】
認識部位が完成され、その部位を完成するために必要な末端塩基を有する分子が同定される方法は、上に一般的表現で記載したものとはわずかに異なる技法を提供する。アダプターは張出し部に結合するが、アダプター分子が結合する一本鎖領域の1つまたは複数の塩基に必ずしも対応しなくてもよいタグを担うからである。この一本鎖領域は、標的配列のいくつかの塩基を含む制限部位の開裂によって作られた張出し部から構成される。しかしながら、開裂部位に応じて、それらの塩基は一本鎖形態であっても、そうでなくてもよく、例えば、張出し部はすべて非標的分子の塩基からなることができる。その代わりに、この適切なタグの付加は、制限部位が完成している場所にのみアダプターが結合するという事実を利用している。したがって段階b)は、前記一本鎖領域、または前記領域の近く、具体的には前記領域に隣接する1つまたは複数の塩基に対応するタグの参照を含む。さらにこの方法において、段階a)に先立って、代謝酵素認識部位の一部を含むリンカーDNA片を前記標的分子に取り付け、次いで前記酵素、例えばヌクレアーゼを使用し、段階a)の一本鎖形態を生成する。
【0099】
前述のとおり、シークエンシングはソーティングに基づいて実行することができる。この方法は、独立して、または上に記載した拡大技法と組み合わせて用いることができる。例えば、シークエンシングプロトコルは、4つの塩基対に基づいて標的核酸分子をソーティングすることによって実行することができ、その後、隣接する塩基対を転換して、その配列を決定することができる。例えば、ソーティング戦略は、前に述べたように、標的核酸分子に4塩基を備えた張出し部を作ることから構成され得る。次にそれを、短鎖DNA分子、ソーティングアダプターによってすべて覆われた256ウェル(wells)の中に分配される(これらのアダプターは必ずしも拡大タグを担っている必要はない)。このソーティングアダプターはウェルの壁に固定され、標的DNA上に作られた張出し部を相補することのできる4塩基を備えた張出し部を有する。さらに、ソーティングアダプターは、IIS酵素、または他の適切なヌクレアーゼのための結合部位を含有することができる。この結合部位は、標的DNAに作られた最初の張出し部の近くに位置する塩基対を備えた張出し部を各々のIIS酵素が作ることができるように置かれる。ソーティングアダプターを備えた表面積を増大するために、別法は常磁性ビーズ、または類似物などの固形担体にそれらを固定することである。
【0100】
例えば、ウェル1内のDNA分子は、AAAA張出し部を有し、ウェル2のDNA分子は、AAAC張出し部を有する。これにより256ウェルは、4塩基の張出し部の、すべての順列をカバーする。標的DNAをリガーゼとともにウェルに添加するとき、例えば、TTTT張出し部を備えたDNA分子はそれ自体がウェル1に取り付き、TTTG張出し部を備えた標的DNAはウェル2に取り付く。ソーティングアダプターに連結しなかった標的DNA分子を洗い落とした後、IIS酵素を添加し、それにより標的DNA分子が遊離し、同時に標的配列の次の4塩基対を表す新しい張出し部が作られる。この張出し部はその後、新しいソーティング過程の出発点として用いることができ、あるいは転換/拡大を続けることができる。
【0101】
DNA分子を洗い流すソーティング戦略は、DNA分子の比較的大きな損失を含む。しかしながら、本特許出願において提案されるほとんどのシークエンシングプロトコルは個々の分子の分析に基づいており、これは非常に少ないDNA分子が必要とされることを意味する。したがって、99.9%以上の損失であってもほとんど問題はない。
【0102】
異なるウェルを用いる代わりに、別法は「マイクロアレイ」上の異なる位置を用いることであろう。例えば、アドレス1においては、固定されたTTTTで終わるDNA分子だけであり、アドレス2においては、固定されたTTTG末端を備えたDNA分子である。他の別法は、異なる末端を備えたDNA分子を異なる時点で取り付け/転換させること、ゲルソーティングを使用することなどである。
【0103】
例えば、「マイクロアレイ」上の256区画に分布された256の異なるソーティングアダプターが存在する方式を用いることができる。例えば、区画1に、AAAA張出し部を備えたソーティングアダプターがあり、区画2において、ソーティングアダプターはAAAC張出し部を有する。したがって、標的DNA分子は、例えばTTTT張出し部を備えたものがスクエア1に取り付けられ、GTTT張出し部がスクエア2に取り付けられるようにソートされる。DNA片の他の末端を、例えばビオチン/ストレプトアビジンを用いて基質に固定することによっても、DNA分子を読み取りプレート上のその位置から離すことなく、次の転換/拡大段階を続けることができる。DNA分子がその位置から離れることを防ぐ他の方法は、256ウェル/スペースに分けられた読み取りプレートを用いることである。
【0104】
ソーティングは当然のことながら、256よりも少ない、または多い順列を用いて行えることも指摘しなければならない。ソーティングは、いくつかの過程で行うこともできる。例えば、65536の異なる区画を備えた「マイクロアレイ」を用いる場合、ハイブリダイゼーション単独によるソーティングによって8bpを同定することが可能であろう。これは多くの適用例に関して、うまく再構成を行うために十分である。したがって、転換または拡大を使用することなく、ソーティングはそれだけでシークエンシング法として機能できる。
【0105】
ソーティングはさらに、非リガーゼベースの戦略を用いて行うこともできる。原則的に、拡大に関して挙げられたすべての方法を含めて、塩基対を認識するのに適した任意の方法を用いることができる。
【0106】
ソーティング法の特異性は、同じソーティング手順を1回または数回繰り返すことによって、ほとんどの目的に適合され得ることも指摘されるべきである。特異性を高めるために、競合プローブ/張出し部を用いることも適切である。
【0107】
このように、好ましい態様において本発明は、本明細書に記載したとおり標的分子をシークエンシングする方法を提供し、前記配列は少なくとも、
a)前記標的配列の少なくとも一部分を、固形担体に取り付けられた、または固形担体に取り付けるための手段を担う相補的プローブを結合するのに適した形態、好ましくは一本鎖形態に転換する段階と、
b)前記相補的プローブを、段階a)で作られた相補的プローブを結合するのに適した前記領域の、好ましくは一本鎖領域の少なくとも一部分、好ましくは4から12塩基の長さに結合する段階と、
c)所望により、段階a)およびb)を繰り返す段階であって、前サイクルの相補的プローブが結合した領域に対して、前記標的分子の隣接する、または重複する領域に前記相補的プローブが結合する段階と、
d)前記標的配列が結合した相補的プローブを同定することによって前記標的配列の配列を決定する段階
を含むプロセスにより、前記分子の一部分の相補性を評価する段階とによって決定される。
【0108】
本明細書において「プローブ」とは、適切な核酸分子、例えばオリゴヌクレオチド、またはPNA分子を指す。
【0109】
追加的な段階を含むこともでき、例えば相補的プローブはプライマーの役割を果たすことができ、この場合、必要に応じてポリメラーゼ反応を行うこともできる。
【0110】
上述のとおり、このソーティング技法は、好ましくは複数の相補的プローブを用いて行われ、サイクル当たり好ましくは2と8個の間、特に好ましくは4個の塩基が同定可能であるが、この情報はシークエンシング反応の完了時に集められるだけであってよい。2と8個の間、好ましくは4個の特有の不変塩基を備えた特に好ましい相補的プローブは、前記固形担体の異なる離散する部位に取り付けられる。第2の引き続いて起こるサイクルにおいて、前記標的分子の隣接または重複領域をシークエンシングするために、前記プローブに結合した標的分子は、相補的プローブを有する1つまたは複数のさらなる固形担体に移される。これを達成するために、段階a)は、拡大プロセスのために記載したものと類似の方法で行うことができ、すなわちプローブは、適切な張出し部が形成されるように認識部位の外側を開裂するヌクレアーゼ、例えばIIS酵素のための制限部位をプローブ自体が含有することができる。
【0111】
上述の手順は拡大の手順と連結することができ、それにより、シークエンシングを、例えば段階b)の後で、ソーティングと拡大の組合わせによって行うことができ、張出し部を記載のとおり生成することができ、適切な拡大タグを担うアダプターを前記張出し部に結合するために用いることができる。その後、拡大タグの読み取り、標的分子が結合したプローブの同定の組み合わせによって配列を決定することができる。したがって好ましい特徴において、本発明は、前記配列の一部分が本明細書に記載の拡大方法によって決定され、隣接するまたは重複する部分が本明細書に記載の相補的プローブの使用によって決定されるといった、本明細書で記載のシークエンシング法を提供する。
【0112】
ほとんどの場合、配列部分の位置決定のために採用される技法は、シークエンシングのために標的DNAが生成される方法によって決まり、例えば、共通の点から始まっているか、または様々な点から始まる標的分子をもたらす断片化によって生成されているかである。
【0113】
シークエンシングのための核酸分子は、様々な方法で生成することができる。少量のDNAを、DNアーゼ、超音波破砕、ボルテックス(vortexing)、または類似の技法で処理することによって、核酸分子を細片に断片化することができる。そのような技法は当業分野でよく知られており、例えばランダムサブクローン生成のプロトコルを記載するhttp://dnal.chem.ou.edu/protocol_book/protocol_partII.htmlを参照されたい。これらの技法のパラメーターを調整することによって、標的DNA断片の平均の大きさを調整することが可能である(一般に、最適であるのは数百塩基対の平均の大きさを有することである)。この方法はまた、元の配列におけるほとんどの場所で切断/破壊されたDNA片が統計的に得られるように、DNA分子を切断/破壊する場所に関して比較的に非特異的であるべきであろう。
【0114】
断片化DNA分子の末端は、平滑末端、および1〜2塩基の短い張出し部の両方からなることを、研究は示している。所望するなら、張出し部は平滑末端となるように処理することができる(Klenow充填など)。
【0115】
好都合には、一本鎖張出し部の形成を利用する本発明の好ましい方法を実行するために、核酸分子はそのような張出し部を形成する手順によって断片化することができる。前述のように、超音波破砕、ボルテックス、およびDNアーゼIは、短い張出し部を作る。非特異的に開裂する制限酵素も用いることができる。IIS酵素は特に、DNA結合ドメインを交換することができる「ドメインスワッピングテスト」に十分適していることをいくつかの研究が示している。したがって、DNAを非特異的に結合するDNA結合ドメインに切断ドメインが結合している場合、新しいIIS酵素を作ることができる。
【0116】
既知のIIS酵素によって形成される張出し部は、−5から+6塩基まで多様である。6個を超える塩基の張出し部が所望であるならば、他の系/方策を用いるのが適切である可能性がある。実行可能な1つの手段は、それ自体の結合部位の外側で二本鎖DNAに切れ目(nick)を形成するニッキング酵素(nicking enzymes)を用いることである。6個を超える塩基対の内部距離を有し、二重らせんのいずれかの側に置かれたそのようなニッキング酵素の2つの結合部位は、6個を超える塩基対の張出し部を形成するであろう。現存のニッキング酵素に加え、例えばIPおよびIIS制限酵素の突然変異によって、新しいニッキング酵素を作ることも可能である。
【0117】
断片化の代替法として、標的配列の断片がPCRまたは類似の方法の助力で生成される戦略を選ぶことも可能である。例えば、標的DNA上の既知の配列でもって開始し、次にこの領域をポリメラーゼ伸長におけるプライマーに対するテンプレートとして用いることができる。任意の部位でポリメラーゼ伸長反応を停止させる方法を用いる場合、「DNA梯子」が作られ、そこには多くの異なる長さのDNA分子が存在するが、すべて共通する一末端を有する。別法として、断片のすべての可能な組み合わせが標的配列から生成されるように、短いランダム化プライマーを用いることができる。しかしながら、ポリメラーゼ伸長を用いるとき、限定要因は多様なポリメラーゼの伸長の長さである。
【0118】
拡大技法は、本明細書に記載のソーティングおよび転換技法と合わせることができる。例えば、一本鎖の標的に結合するとき、アダプターをプライマーとして用いることができる。ポリメラーゼ反応は、アダプターと標的配列との間の、相補性の存在を確立する手段をさらに提供する。
【0119】
本発明の好ましい一実施形態において、標的分子は固形担体に固定される。これはいくつかの異なる方法で達成することができる。標的分子は1つまたは複数の部分に取り付けられているように設計することができ、それが分子の固形担体への結合を可能にし、例えば、末端(またはいくつかの内部の部位)は結合の対の1つのパートナー、例えばビオチンを備えていることができ、次にそれをストレプトアビジン担持固形担体に取り付けることが可能である。
【0120】
標的分子は、いくつかの知られている方法でそのような結合部分を担うように設計することができる。例えば、PCR反応は上記結合部分を導入するように、例えば適切に標識されたプライマーを用いることによって実施することができる(例えば実施例17を参照のこと)。別法として、標的核酸は、例えば標的核酸分子を制限酵素で開裂して、次に末端が結合部分で標識されたアダプター/リンカーにその分子を連結することによって、結合部分に連結することができる。そのような方法は、IIS制限酵素を用い、非パリンドローム性張出し部を形成する場合、特に適しているであろう。他の別法は、すでに結合部分を担っている、またはそのような結合部分の導入を促進するような配列を含有するベクターに標的分子をクローンすることである。そのような方法は、以下により詳しく述べる位置マーカーを導入するために、同じく用いることができた。
【0121】
別法として、核酸分子自体が結合対の1つのパートナーである限り、結合部分を取り付ける必要はなく核酸分子を固形担体に取り付けてもよい。したがって、例えば固形担体に取り付ける短いPNA分子を用いることができる。PNA分子は、ハイブリダイズし、二本鎖DNAに結合する能力を有し、この非溶解核酸材料はそれゆえこの方法で固形担体に取り付けることが可能である。同様に、オリゴヌクレオチドプローブは、相補的配列を固形担体に結合するために用いることができる。そのような技法はまた、以下に記載するように特定の核酸分子を固形担体上の特定の位置に結合することによって、シークエンシングを開始するために用いることもできる。
【0122】
標的分子を取り付けるための固定化部分として適した適切な固形担体は、当分野でよく知られ、文献に広く記載されている。一般的に言ってこの固形担体は、化学的または生化学的手順において固定化、分離などのために現在広く用いられている、または提案されている任意のよく知られた担体または材料であってもよい。したがって、例えばこの固定化部分は、例えばポリマー材料、例えばアガロース、セルロース、アルギン酸塩、テフロン、ラテックス、またはポリスチレンからなる、ビーズ、粒子、シート、ゲル、フィルタ、膜、微小繊維ストリップ、チューブまたはプレート、繊維または毛細管の形態を取ることができる。粒状材料、例えばビーズが一般に好ましい。好都合には、この固定化部分は、超常磁性体粒子などの磁性粒子を含むことができる。さらに好ましい実施形態において、線状配置にある分子の固定を可能にするためにプレート、またはシートが用いられる。このプレートはまた、分子を取り付けることのできるプレートと直角をなす壁を含むことができる。
【0123】
固形担体への取り付けは、直接的に、または間接的に実施することができ、用いられる技法は、取り付けられる分子が標的分子を同定するためのプローブであるか、標的分子自体であるかによって決まる。標的分子を取り付けるために、好都合には、核酸分子および/または固形担体に担持された付着部分を使用することによって間接的に行うことができる。したがって、例えば親和性結合パートナー対を用いることができ、アビジン、ストレプトアビジン、またはビオチン、DNA、またはDNA結合タンパク質(例えば、lacIリプレッサタンパク質、またはそれが結合するlacオペレータ配列)、抗体(モノクロナールであっても、ポリクロナールであってもよい)、抗体断片、あるいは抗体のエピトープまたはハプテンなどである。これらの場合、結合対の1つのパートナーが固形担体に取り付けられ(または本質的にその一部にである)、もう一方のパートナーが核酸分子に取り付けられる(または本質的にその一部にである)。直接に取り付ける他の技法を用いることができ、例えばフィルタが用いられる場合、取り付けはUV誘導架橋反応によって行うことができる。DNA断片を取り付けるとき、ガラスに密着するDNAの固有の傾向を用いることもできる。
【0124】
固形担体への適切な官能基の取り付けは、当分野でよく知られている方法によって行うことができ、例えば、ヒドロキシル、カルボキシル、アルデヒド、またはアミノ基による取り付けが含まれ、それらは適した表面被覆がもたらされるように固形担体を処理することによって提供することができる。本発明の核酸分子への適切な官能基の取り付けは、連結によって実施することができ、あるいは合成または増幅の間に導入することができ、例えばビオチン、または捕捉するための特定の配列など適切な部分を担うプライマーが用いられる。
【0125】
本明細書に記載のとおり、標的分子は、固形担体に取り付けられる相補的プローブに都合よく取り付けられる。
【0126】
複数であるが離散した相補的プローブを使用する技法において、これらの異なるプローブが取り付けられる固形担体は、都合よく物理的に取り付けられるが、各プローブへの標的分子の付着によって発生するシグナルは独立して決定可能でなければならない。したがって、例えば複数のウェルを備えたプレートを、異なるウェルにおいて異なるプローブを備えた固形担体として用いることができ、あるいは固形担体の領域は異なるアドレスを含むことができ、例えば、異なるプローブは離散した部位においてフィルタに結合していてもよい。
【0127】
固形担体への取り付けは、核酸分子断片が生成される前、または後に行うことができる。例えば、結合部分を担う標的核酸分子を固形担体に取り付け、その後、DNアーゼIまたは類似体で処理することができる。別法として、開裂を行ってもよく、次にその断片を担体に取り付けることができる。
【0128】
多くの状況において、目的は他の配列の内部、または他の配列と共に存在する1つまたはいくつかの配列をシークエンシングすることである。例えば、わずか5〜10%のヒトゲノム配列が、直接に生物学的重要性があると想定されている。したがってヒトゲノムのマススクリーニングに関して、生物学的重要性の低い領域のシークエンシングを回避できることが有用であろう。
【0129】
したがって、用いることのできる1つの戦略は、単離される標的配列を相補するポリヌクレオチドを固形担体に固定することである(ウェル内部、単分散球、マイクロアレイなど)。配列プール中のポリヌクレオチドを固形担体上のポリマーと共にハイブリダイズすることによって、所望でないポリヌクレオチドを洗い流し、その後シークエンシングの段階に進むことができる。所望であれば、ハイブリダイゼーションと洗浄のサイクルを数回実行することによって、特異性を高めることができる。個々の適用例にとっては有利である可能性があっても、相補的ポリヌクレオチドが規則的なパターンで固定されているかどうかには依存しない。連結、PNAハイブリダイゼーションなどに基づく類似の方法も可能である。
【0130】
例えば、特定のmRNA/cDNA分子を単離するために、相補的cDNA/mRNA分子を常磁性球などに固定することができる。次に該球体は、標的配列を含有する溶液と共にチューブ内で混合することができる。mRNA/cDNA分子が球に固定されたmRNA/cDNAとハイブリダイズされているとき、所望でない分子は、磁石または類似体を用いて球体をチューブ内に保持するのと同時に洗い流すことができる。所望の標的分子を次に、温度の上昇、pHの変化、またはハイブリダイズ化分子を溶解する他の方法を用いることによって遊離することができる。
読み取りプレート上で行われるシークエンシングプロトコルのために用いることのできる類似の方法は、特定の標的配列を所定のアドレスに固定することである。例えば、一本鎖標的DNAを、異なるアドレスに固定されるプライマーにハイブリダイズすることができる。所望であれば、このプライマーは次に、ポリマー伸長のテンプレートとして用いることができる。プライマーを標的配列に適合させることによって、所望どおりアドレスすることができる。
【0131】
類似の方法は、PNA分子を異なるアドレスに固定することであり得る。PNA分子は二本鎖DNAの特定配列を認識する能力を有することが知られており、そのような方法はしたがって、固定したい配列を認識するPNA分子を用いて二本鎖DNAをアドレスするために用いることができる。
【0132】
上述のとおり、シークエンシングされる分子は、2つの種類に分けることができる。1つの共通な末端と1つの任意の末端を有するもの、および2つの任意の末端を有するものである。位置情報は種々の方法で、これらの異なる種類の分子から得ることができる。
【0133】
すべての標的分子が共通の末端を有する場合、各標的分子の長さは、共通の末端と他の任意の末端との間の距離に比例するであろう。同様に、標的分子の特定の部分に起因する配列情報は、共通の末端から配列情報の部位までの距離を算出することによって位置を定めることができる。好都合には、配列情報が標的分子の末端に関係する場合、分子全体の長さ/大きさからその位置を決定することができる。
【0134】
核酸断片が共通の末端から始まっていない場合には、位置情報は種々の方法で得ることができる。1つの別法は、配列ごとに変わる特有のフィンガープリントを作る、または同定することである。したがって、配列片の位置は、どのフィンガープリントにそれが連結しているか、および場合によってはフィンガープリントのどこにそれが位置しているかを記録することによって引き出すことができる。特有のパターンを作るのに使用される、非常に多くの技法が考慮され得る。DNA配列における制限酵素の開裂パターンは、例えば「光学マッピング」(optical mapping)、または類似の方法の助力によって記録することができる。
【0135】
知られている「光学マッピング」法の1つの欠点は、用いられる制限酵素のための切断部位が必ずしも切断されないことである。さらに、不正確な開裂が起こることがあり、DNA断片の長さの測定に関連していくらか不確定性である可能性がある。したがって、多くの同一DNA分子の分析に基づいた各地図片の平均図を作製する必要がある。問題は、どのDNA分子が同一であるのかを知るのが困難であり得ることである。
【0136】
光学マッピングの現行方法に伴う他の問題は、DNA断片の内部配置を観察できるようにするためにDNA分子を真っすぐに整えた後に、制限酵素などを用いた処理を行わなければならないことである。これは、酵素的調製などといったことのためDNA分子の利用可能性を低減する。位置配置に加えて末端シークエンシングを提供する本発明は、そのような問題を克服することを可能にする。例えば、実施例23に記載の技法を参照されたい。
【0137】
特有のパターンを作る蛍光性プローブ/タグを用いることもできる。これは、いわゆる「DIRVISH」技法の背後にある原理である。類似の方法は、原子間力顕微鏡法(atomic force microscopy,AFM)、ミクロ/ナノポア、または特有のパターンで結合したタンパク質の大きさと位置を記録するための他の方法などを用いることである。
【0138】
前に述べたように、細胞性アダプターを用いることもできる。例えば、拡大した標的DNAを細胞に形質転換/トランスフェクトする場合、レポーター遺伝子の転写頻度はシス調節因子への距離によって変わるという事実を利用することができる。レポーター遺伝子を構成する1つの末端にエンハンサーがあり、もう一方に1つまたは複数の拡大タグがある場合、レポータータンパク質の相対量は位置価(position value)を算出するために用いることができる。
【0139】
さらに、位置価を引き出すために用いる要素で標的配列を標識し、または組み込むことも可能である。そのような方法は、例えば非常に類似した2つの配列のフィンガープリントを互いに識別することが困難である場合に有利であり得る。例えば、姉妹染色体をシークエンシングしたい場合、任意に組み込まれる多数の挿入要素(トランスポゾンなど)を組み込むことができる。次に染色体を増幅し、位置マーカーとして挿入要素を使用すれば、各姉妹染色体に関して1つまたはいくつかの特有のパターンができる。
【0140】
位置マーカーを導入することができ、しかもその部位で配列の同定を可能にする、使用可能な別の方法は、PCR反応にプライマーとしてアダプターを使用することを含む。各PCR反応は結果として連結した2つのアダプターをもたらし、2つのアダプター間の距離は標的DNA上のアダプター配列の距離に対応し、同時に位置情報を提供する。
【0141】
標的分子の配列は、拡大することなく位置マーカーを生成するために必要な手段を提供することができる。例えば、いくらかの配列情報が既知である場合、プローブをその配列にハイブリダイズするために用いることができ、その後それは位置マーカーを提供する。別法として、適切な位置マーカーを標的分子内に置くことができ、例えば異なる位置マーカーをゲノム内に規則的な間隔で置くことができる。異なる位置マーカー間の識別を可能にするために、それらのマーカーによって異なるシグナルが提供され、例えば、それらはプローブすることのできる異なる配列または長さを有する。実施例21は、位置マーカーを使用する1つの方法を記載する。
【0142】
したがって、好ましい態様から見ると、本発明は核酸分子を(完全に、または部分的に)シークエンシングする方法を提供し、少なくとも
a)前記核酸分子の一部分の配列を決定する段階と、
b)位置インジケーター、好ましくは位置マーカーの参照により前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階を含む。
【0143】
前述のとおり、好ましくは複数の配列およびその位置が決定される。
【0144】
本明細書において、位置インジケーターは前述のとおり、分子の大きさ、発生したシグナルの強度、あるいは位置マーカー、アンカー、またはフィンガープリントへの距離であってもよい。
【0145】
これらの方法を行うためのいくつかの異なる技術を、本発明を説明するためにここに記載する。
【0146】
原則として、ポリマーの大きさを決定するための各方法を使用することができる。しかしながら、同定されている配列片の長さを大きさの決定の精度に合わせなければならない。精度が低いほど、配列片が長いに違いない。
【0147】
大きさをソーティングするためのいくつかの方法が、以下の分野で存在する。ゲルソーティング、微毛細管ソーティング、読み取りプレート上に伸張しているポリマーの長さの測定、非特異的にタグを付されているポリマーの蛍光強度(または他の)の測定(フローサイトメーター、蛍光顕微鏡などによって)、質量分析法、ポリマーがミクロ−またはナノポアをブロックするために使用する時間などである。このような手順は、配列の決定のためにシグナルが読み取られる前または後に行うことができる。例えばゲル電気泳動法が使用されるとき、ゲル上で分離されているサンプル上で、またはゲルから溶出されたサンプル上で読み取りを行うことができる。
【0148】
DNA分子が切断される(例えばDNアーゼI、超音波処理などによって)可能性はDNA分子の長さに正比例するという原理に基づいて、核酸分子の長さを決定することもできる。例えば200個の塩基対を有するDNA分子は、限られた量のDNアーゼIを含む溶液中で、100個の塩基対を有するDNA分子と比べて2倍の頻度で切断されるはずである。このことは、例えば異なる分子を末端標識しそれらの分子を切断し、次いで単一におよび二重に標識された分子の量を、同様に標識された既知の長さの標準物と比較して調べることによって実施ができるはずである。
【0149】
DNA分子の長さまたは固定点までの距離を決定するためには、DNA分子を延長または伸張させるのが好都合である。DNA分子を伸張させるための1つの方法は、ガラスの小ビーズとDNA分子が1:1の比で結合するように、DNA分子と大余剰のガラスの小ビーズと混合させることである(ビーズは自然にDNA分子と結合する)。DNA分子は液体流中ではガラスビーズよりも抵抗力が弱いはずであり、その結果DNA分子は伸張されるまでお互い同士離れる傾向がある。その液体流が強力であるか、またはガラスビーズが大きくDNA分子とガラスビーズの間の抵抗力の違いが大きいならば、DNA分子は裂けてしまう可能性がある。しかしながら、この問題は液体の流速を下げることによって、またはより小さなガラスビーズを使用することによって避けることができる。単位面積当たりの配列情報の量が増加するようにDNA分子が通常の様式で配置されているなら、この方法は特に効率的なものになる。これを行う1つの方法は、DNA分子をビオチンで標識し、次いで該分子を通常のストレプトアビジン配列を有するプレートに固定する。代替方法としてレーザービーム、いわゆるレーザートラッピングを使用することによって固定を得ることができる。
【0150】
液体流を使用してDNA分子を真直ぐにするかわりに、一方向に負に荷電したDNA分子を引っ張る正電荷を使用することができる。この方策を使用することによって、読み取り効率が増大するようである。原則として最も簡単な方法は、正または負のスポット電荷を読み取りプレートの前に置くことである。クーロンの法則によると、DNA分子上の電荷の力は距離に反比例する。従って電荷に最も近いDNA分子は、さらに下部にあるDNA分子よりもより大きな力によって伸長させられるはずである。読み取りの瞬間にすべてのDNA分子に平等に影響を与えるためには、読み取り単位を有する段階においてスポット電荷を動かすことがしたがって必要であるはずである。プレート上の力の差異を減らすように、スポット電荷を読み取りプレートのはるか下部に置くこともできる。代替方法として、力のベクトルが円弧の中心で直線は同じ大きさであるように、電荷を円弧状に配置することが可能である。したがって、読み取り単位が側路で動かされるときは、電荷を動かしさえすればよい。
【0151】
代替方法として、分子のアンカー上の力を減少させるために、ある異なる技術を使用してよい。電気的に帯電した2枚のプレートを読み取りプレートの下に置くことができ、その読み取りプレート上で標的分子は伸長させられる。トッププレートは弱い負電荷を有し、一方ボトムプレートは相対的に強い正電荷を有する。負に荷電した粒子(例えばDNA)が負のプレートのすぐ上に置かれるならば、そのプレートからの斥力は正のプレートからの引力よりも大きいはずである。次いでその粒子は上方に押し出されるはずである。しかしながら、このプレート状態から動かすことは逆の結果になる。正のプレートの引力は負のプレートの斥力よりも大きい。プレートの電荷を調整することによって、読み取りプレートの上の所与の高さにおいて、斥力と引力の間の平衡が生まれるはずである。標的分子はこの平衡面に押しやられるはずである。この方法では、DNA分子が平衡面にある限り、DNA分子にかかる正味の力はほぼ0に等しい。これによって破裂の可能性が減少する。
【0152】
この2枚の帯電したプレートに加えて、読み取りプレートの左の正電荷も使用することができる。これによって、この方向の正味の力が生み出されるはずである。2枚の帯電したプレートをお互いに関して、および読み取りプレートに関して傾けることによって、同じことを実現することができる。
【0153】
標的分子がフローサイトメーターまたは同等の装置を介して伸張させられながら動かされるならば、負に帯電した管を使用することができる。このような技術を使用することによって、標的分子は斥力が最も弱い管中央に向かって押されるはずである。
【0154】
さらなる代替の伸張技術が、機械的伸張によって提供される。例えばこの方法では、標的分子のいずれかの末端と相補的なオリゴヌクレオチドが接着されるときには、2枚の隣接するプレートを使用してもよい。標的分子がひとたびこれらのプローブにハイブリダイズされると、分子がプレートの間で伸張されるまでプレートを分離することができる。
【0155】
前述の方法において発生したシグナルは、発生したシグナルに応じて、および位置情報がどのようにして得られるかに応じて、いくつかの異なる方法によって読み取ることができる。例えば、標的DNAに付着した蛍光DNAプローブの位置を特定するために、このDNAを前述のように伸張させてよい。例えば、モレキュラーコーミング(molecular combing)として知られている、Weier他によって開発された方法(Hum.Mol.Gen.,1995,Vol.4(10),p1903−1910)を使用することができる。この方法では標的DNAを含む溶液を、DNA分子がガラスプレートにその一末端を付着するように作製した平らなガラス表面上に置いた。次いでこのDNA分子を、液体流を使用することによって真直ぐにした。次いで蛍光顕微鏡によって、伸張させたDNA分子に接着したプローブの相対的な位置を観察できるはずである。
【0156】
本発明では、例えば異なる蛍光体で標識された4つのプローブおよびDNAの特異的な伸張体である拡大タグを使用することによって、そのプローブは、A、C、G、およびTを表す4つの拡大タグにハイブリダイズするようにこれらのタグに向けられてもよい。すなわち、前に記載したDIRVISH技術を使用する。次いで配列順序を、蛍光顕微鏡を用いて直接読み取ることができる。前述のように、拡大タグがどのようにして構築されるかによって、多数または少数のプローブを使用することができる。具体的には、特異的なシグナル、例えばバイナリーコードの発生を生み出す各拡大タグにプローブが結合する方式においては、単一のプローブを使用することができる。代替方法として、拡大タグが2個以上の塩基に対応する場合は、5個以上のプローブを使用してよい。顕微鏡にガラスプレートをスキャンさせるソフトウェアを開発し、一方、同時に自動的に配列順序を分析することによって、塩基対を非常に早く読み取ることが可能なはずである。
【0157】
さらなる代替方法として、早い読み取り用にフローサイトメーターを使用して蛍光プローブを読み取ることができる。このための必要条件は、DNA分子がA、C、G、およびTを表す拡大タグが順番に通過するように伸張した型で、フローサイトメーターの読み取り単位を通過することである。これは前述の技術を使用することによって行うことができる。この特別な実施形態の代替方法として、電界または磁界を液体流の代わりに使用して、蛍光検出器を通過する粒子を引っ張ることができる。これは、DNA分子が負に荷電している一方でガラスビーズが正電荷を有している事実を使用することによって、またはガラスの代わりに超常磁性ビーズを使用することによって達成される。ついでこのビーズは、背後で長い撚り糸のようにDNA分子を引っ張るはずである。
【0158】
この方策における重要なパラメーターの1つは、フローサイトメーターの蛍光検出限界が低いことである。いくつかのグループは、流速を下げることによって個々の蛍光体分子をなんとか検出した。しかしながら、1秒あたり20〜30,000粒子の分析スピードを持つ従来型のフローサイトメーターを使用するためには、多くの蛍光体を各プローブに固定することができるように、より長いプローブが使用されなければならない。
【0159】
最速のフローサイトメーターは、現在1秒当たり約200,000の蛍光粒子を分析するための容量を有するが、このようなフローサイトメーターは市販されていない。さらに、DNA分子が壊れる前に伸張した型で高速耐性のDNA分子が何であるかは確かではない。しかしながら、DNA分子は非常に早い読み取りを可能にする速度に耐えるはずであると考えるのが現実的である。
【0160】
さらなる代替方法は、通常の方法で固形担体上、例えばストレプトアビジンで覆われたプレートにDNA分子を固定することである。配列(例えば一続きの拡大タグによって発生したシグナル)は、読み取りプレート内に挿入された小さな検出器を有することによって読み取られる。これらの検出器は、例えば断片に固定された拡大タグ上のレポーター分子によって、例えば固形担体上にセンサを結合させることによって電気回路を遮断または導通することによって、非活性化または活性化される。例えば、読み取りプレート上のレポーター分子とモジュールの間で強い結合が形成される可能性がある。後者の場合、DNA分子が引き剥がされるならば、モジュールを読み取りプレートから引き剥がすことができるような方法でモジュールを形作ることができる。モジュールが緩んでくると、読み取りプレートからどのモジュールが取り除かれたかを記録するような方式で、モジュールは電流回路を遮断する。首尾よく結合する可能性を増大させるためには、断片上の同じ位置にいくつかのレポーター分子を固定してよい。各塩基A、C、G、およびT用に4つの異なるレポーター分子を使用することができるが、または断片上の4つの異なる場所に位置する同じレポーター分子を使用できる。マルチプレックス、パラレルコンピュータ・インプットおよび現代の他の電子機器によって、素早いシークエンシングを可能にするような1秒当たり数百万のシグナルを記録することが可能であると考えられる。
【0161】
好ましい実施形態では、これらの方法は本明細書に記載されている拡大技術と組み合わせて適切に使用される。すなわち複数の、好ましくは1つの鎖の拡大タグの存在によって前記標的核酸分子の部分が決定される。しかしながらこれらの方法は、拡大が行われないときでも使用することができる。DNA分子を拡大する代わりに、塩基にセンサーが記録することができる異なる付着物を取り込ませることができる。
【0162】
ひとたびシグナル情報が蓄積されると、コンピュータプログラムを使用して配列片を最終配列に組み立てる。この段階においてエラーが発生する可能性は、主に5つのパラメーターに依存する。シークエンシングされるDNA分子の長さ、DNA配列の塩基対組成がどの程度ランダムであるか、読み取られるDNA片の長さ、読み取られるDNA片の数、およびシークエンシング反応におけるエラー率である。
【0163】
本発明者は、前述のパラメーターの重要性を分析するためのコンピュータプログラムをすでに作製している。すでにシークエンシングされているヒトゲノムDNAに基づいて、この分析は、30断片からなる長さのDNA、6×108個のDNA片を読み取り、およびシークエンシング反応におけるエラー率が10%であると(点突然変異を考慮する)、1つのヒトゲノムは1つのシークエンス反応中で読み取られ、点突然変異/欠失がほとんどないであろうことを示している。しかしながら1つの例外は、DNA片の長さが増加しているに違いない非常に非ランダムな領域(サテライトDNAおよび他の繰り返し領域)である。しかしながら、これらの領域の生物学的情報は、コード配列およびシス−調節要素と比べるとそれほど重要ではない。
【0164】
DNA片が何回も読み取られるときは、シークエンシング反応におけるエラー率が非常に高くても、それは埋め合わせられることもデータ分析は示す。例えば、配列の長さの10倍もの多くの塩基対を読むことによって、シークエンシング反応におけるエラー率が高くても、たいていの欠失および点突然変異は無くなるはずである。
【0165】
シークエンシングに使用される技術に応じて、不均一のサンプルのシークエンシングを行うこと、例えばパラレル・シークエンシングを行うことがある状況においては可能である。これを可能にする手順によって、本発明の好ましい態様が形成される。このような技術では、異なる標的分子からのシグナルが識別できることが必要である。これはいくつかの方法によって、例えば特定の標的分子などを同定するマーカーの特定の位置、含有、同定を制限することによって達成される。例えば、標的核酸分子の領域に相補的である固形担体を使用して、特定の分子を単離し保持することができる。このことは、シークエンスの少なくとも部分的な知識があれば、すなわち特定の分子を特定の部位に結合させるために行うことができる。あるいはこうした知識なしに、異なる分子が結合し次いで平行してシークエンシングすることができる、本質的にランダムに結合するプローブを使用することによって、配列をその分子を関連付ける複数の技術を利用することによって、例えばアドレスまたは位置マーカーによって行うことができる。本明細書に記載されている技術は特に有利である。なぜなら、本技術によって個々の分子がシークエンシングされ、したがってパラレル・シークエンシング反応がさらに促進されるからである。
【0166】
本明細書に記載されているいくつかの技術は、標的分子の一部分だけをシークエンシングするために、またはフィンガープリンティング、プロフィール分析またはマッピングのために使用することができる。すなわち分子の不連続おかつ特有部分を、例えばRNA発現(最初に分析用のためDNAに転換することができる)の分析用に同定するため使用することができる。例えば実施例23に記載されるように、拡大タグ(一つの鎖の拡大タグが好ましい)が付着することができる特定の張出し部を生成する制限酵素を用いて、標的サンプルを消化することができる。このようなタグは配列に関する情報を保持するだけでなく、切断をもたらす酵素に関する、すなわちそうした切断から生じる起因する断片のマーカーとしての情報を追加的に保持することができる。制限酵素が、異なるアダプターが結合できる、異なる長さの張出し部を生成するならば、複数の制限酵素を同時に使用してもよい。代替方法として、連続的なサイクルにおいて異なる制限酵素を使用してもよい。
【0167】
したがって、例えば相補的な張出し部に付した増幅タグによって、例えばその相補性を示すタグ、例えばタグ自体がヌクレオチド塩基でできているタグの使用によって、結果として生じる断片を一列に並べることができる。このことから、実施例23に記載されるように制限地図を作成することができる。したがって本発明のさらなる態様では、本明細書に記載されるような前記部分の位置情報に加えて、本明細書に記載されるような前記配列の不連続部分についての配列情報を得ることを含む標的配列の地図を生成する方法を提供する。
【0168】
好ましい特徴はにおいて前期地図は、前記配列の不連続部分の配列情報を得ることによって生み出され、前記部分は複数のヌクレアーゼの開裂部位のすべてまたは一部分、および/または前記ヌクレアーゼの制限部位のすべてまたは一部分を含み、前記配列の位置が前記ヌクレアーゼで消化した後の前記標的核酸分子の断片の末端における配列の比較によって決定される。
【0169】
配列情報は、本明細書に記載されるような複数のヌクレアーゼによる前記標的分子の切断によって得られるのが好ましく、相補的な一本鎖領域およびアダプター分子と前記標的分子の領域の結合(開裂部位に、または開裂部位に隣接しているのが好ましい)を生成するために得られるのが好ましい。前記アダプター分子は本明細書に記載されるような複数の拡大タグを保持し、前記タグは前記領域の複数の塩基に対応するシグナル部分を含み、前記アダプター分子はそこに結合し、切断用に使用されるヌクレアーゼに対応するさらなるシグナル部分を追加的に含む。充分なヌクレアーゼが使用されるいくつかの例では、この方法をシークエンシングの方法として使用してもよい。
【0170】
例えば、バクテリアは以下の方法によって同定することができる。バクテリアは溶解させることができ、DNAを単離することができる。次いでクラスII制限エンドヌクレアーゼまたは本明細書に記載されるような他のこのようなヌクレアーゼを用いて、分子を切断することができる。次いでDNA分子をアダプターに結合させ、張出し部を同定することができる。次いでDNA分子を読み取りプレートに固定し伸張させることができる。蛍光スキャナを用いて読み取りプレートをスキャンすることによって、制限の長さについて情報または特有なパターンを、およびこれらの分子の末端に由来する配列情報を得ることができる。このような拡大が含まれる技術は、その末端配列により同じ大きさの分子の識別を可能とする。したがって本発明のさらなる特徴では、本発明は、標的DNA分子のフィンガープリントを得る方法を提供する。この方法は、本明細書に記載されるよういに位置情報を得ることに加えて本明細書に記載される複数のシークエンシング技術の使用を含む。
【0171】
本発明の好ましい特徴では、前記標的分子の特有な制限地図を参照することによって位置情報が得られる。さらに好ましい特徴には、複数の拡大タグを同定するための制限地図の使用、およびフローサイトメーターまたはナノ/ミクロポア分析を使って効果的に読み取ることができるタグの使用がある。
【0172】
本特許出願に導入されている原理およびプロトコルを使用することによって、シークエンシング/ソーティング反応中およびシグナルを読み取るときに、プルーフリーディングを使用してエラー率を減少させることができる。
【0173】
ソーティングが使用されるならば、同じ配列片を数回ソーティングすることが可能である。例えばAAAAで始まる標的DNAはすべて、ウェル1にソーティングされる。次いで同じ手順が繰り返され、そこで誤ってソーティングされたAAAAで終わらないDNA分子が洗い流される。原則としてこの手順は、望ましいエラー百分率が得られるまで繰り返すことができる。
【0174】
拡大/転換が使われるならば、拡大タグの繰り返し鎖(シグナル鎖)が得られるように、標的分子の同じ配列片を数回転換することが可能である。したがって、繰り返される転換生成物が一様でないときは、たいていのエラー転換は発見することができる。位置情報を引き出すために使用される標的分子の部分も、同じ方法によってコピーすることができる。
【0175】
さらに、各配列片は何回も読み取られる可能性がある。なぜなら、分析される標的分子の数が非常に多い可能性があるためである。
【0176】
本明細書に記載されているシークエンシングまたは拡大方法を行うためのキットは、本発明の好ましい態様を形成する。したがってさらなる観点から見ると、本発明は標的核酸分子の複数の塩基を拡大するためのキットを提供する。この標的核酸分子は前に記載されるような少なくとも1つまたは複数のアダプターを含み、所望するなら複数の固形担体に付着し、好ましくは1つまたはそれ自身がシグナル手段を含む拡大タグを有する。
【0177】
所望するならば、キットは以下のものを含むリストから選択される他の適切な成分を含んでもよい。それは反応中に使用するための制限酵素、標的分子がその内部に連結され得るベクター、リガーゼ、制限または連結部位の不活性化および活性化に必要な酵素、増幅および/または適切な酵素用のプライマー、緩衝液および溶液である。本明細書に記載されているシークエンシング反応の他の態様を行うためのキットも、本発明の範囲内に含まれる。したがって、例えばソーティング反応を行うためのキットは、少なくとも1つの固形担体を含んでよく、この固形担体は複数の相補的なプローブ、好ましくは固形担体上、または分離した固形担体上の異なるアドレスで複数の塩基によりお互いのプローブにミスマッチした一連の不連続プローブを担持する。このようなキットには、適切な標識手段が含まれてもよい。
【0178】
標的核酸分子の拡大用またはシークエンシング用にこのようなキットを使用することによって、本発明のさらなる態様が形成される。
【実施例】
【0179】
説明のためのみに以下の実施例が与えられ、その中で参照される図は下記の通りである。
【0180】
[実施例1]:1方は張出し部を形成し他方は平滑末端を形成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
《方法》
1.シークエンシングされるDNA配列からなる純粋なDNAの集団を、元の配列の細片(以後はDNA片と呼ぶ)からなるDNA分子の集団が形成されるように、非特異的な方法で切断/破壊する。
【0181】
2.DNA片中の塩基対を、4つの各塩基アデニン、シトシン、グアニン、およびチミンを表す4つの異なるDNA配列(以後は拡大タグに対応するDNA断片と呼ぶ)に置き換える。したがって、例えばA−Tの塩基対があったところには「断片A」が挿入され、C−Gは「断片C」で置き換えられる。そのため元の塩基の順序、例えばACGTTが断片A、断片C、断片Gなどに置き換えられる場所では新しいDNA分子が発生する。原則として、これら4つのDNA断片の長さは2塩基対から数百kbp(望むならばこれより多く)まで要件に従って変化する可能性がある。これに対応して、DNA断片はレポーター遺伝子および他の生物学的情報を含む可能性があり、または既知の生物学的機能を持たない配列のみからなる可能性がある。
【0182】
3.個々のDNA分子用の4タイプのDNA断片の順序が読み取られる。これにより、元のDNA片の塩基の順序が決定される。
【0183】
4.コンピュータプログラムによってDNA片の間の重複を使用し、出発時点で使用されたDNA配列の配列のために段階3からの情報を一緒にする。
【0184】
図1では、自らのDNA結合部位の外側を切断する制限酵素に基づいている段階2を行う1つの方法を例示する。この方法は以下のように行われる。
【0185】
1)段階1からのDNA片をプラスミドに連結させる。このプラスミドは平滑末端片切断を発生させ、自らのDNA結合部位の外側を切断して、一塩基対の張出し部(Enz2)を発生させる制限酵素(Enz1)用の結合部位を有する。さらに、プラスミドがストレプトアビジン処理した試薬チューブに粘着しそこで反応が起こるように、ビオチン塩基をプラスミドに取り込ませる。
【0186】
2)試薬チューブを洗浄し、Enz1およびEnz2を含む新しい反応混合物を加え、1つの平滑末端およびDNA片の第1の塩基が張出し部を構成する1つの末端が形成されるようにインキュベートする。
【0187】
3)試薬チューブを再び洗浄し、4つの異なるDNA断片および耐熱性DNAリガーゼ(例えばPfuまたはTaqDNAリガーゼ)を含む反応混合物を加えてインキュベートする。耐熱性DNAリガーゼの有利点は、非常に特異的に連結する一方、同時に平滑末端を連結させないことである。それによって、例えば「断片A」はアデノシン張出し部のある場所に連結されるはずであり、「断片C」はシトシン張出し部のある場所に連結されるはずである。
【0188】
4)試薬チューブをさらにもう1度洗浄し、平滑末端が連結されるようにT4リガーゼを有する反応混合物を加えてインキュベートする。挿入される断片はプラスミドが元来有していたようなEnz1およびEnz2用の部位を有しているため、次の塩基を新しいサイクルにおいてDNA断片に置き換えることができるように出発点に戻る。
【0189】
[実施例2]:両方ともに張出し部を形成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
この方法の出発点は、すべてのBspMI部位が不活性になるように、転換すべき標的DNA断片はすべてBspMIメチラーゼで処理されるということである。次いでDNA断片は、図2aで示されるように塩基ベクターに連結され、この図はDNA片を有する塩基ベクターがそれに連結されストレプトアビジン基質に固定したことを示す。この塩基ベクターは、実線で示されるDNA断片を切断するために使用されるBspMI部位を含む。この実施例で作成される張出し部は内部に「T」を有するはずであり、この張出し部に連結することができるのはAアダプターのみである。AatII部位は、アダプターAがBspMIで作成された張出し部に連結された後で、塩基ベクターを環状にするために使用される。この塩基ベクターはビオチンでタグされた塩基も含むため、ストレプトアビジンで覆われた基質に固定され得る。
【0190】
使用されるアダプターは、図2bに示される。アダプターA1(トップ)とA2(ボトム)の間の唯一の違いは、AatII部位/張出し部がPstI部位/張出し部とともに場所を変えていることである。A1およびA2アダプターは、例えばA1がサイクル1、3、5で使用され、A2がサイクル2、4、6で使用されるように2回に1回のサイクルで使用されるはずである。5’張出し部は5’に沿って3つの共通なヌクレオチドおよびアデニンからなる。そのためこれらの張出し部は、張出し部(3’に沿った)の内側でチミンを有する他の5’張出し部と連結するはずである。下のボトムアダプターの5’張出し部の外側の太線は、アダプターがDNA断片に連結された後に、BspMIによる切断によって形成される張出し部を示す。Aアダプターに加えて、C、G、およびT用にアダプターが作成されなければならない。
【0191】
塩基ベクターに連結されると、DNA断片はストレプトアビジン基質、例えばパラメーター領域に固定される。Dynabeadのkilobase BINDER kitを使用することによって、非常に強いビオチン−ストレプトアビジン結合が得られ、したがってサイクル数が非常に多くてもDNAの損失を最小限にして、反応溶液を素早く効率的に変化させることができる(Biomagnetic Techniques in Molecular Biology,3rd Edition,pp.158−60,Dynal ASによって販売された)。この手順の残りは4つの酵素反応の1サイクルからなり、1サイクルあたり1つの塩基が転換される(図2C)。
【0192】
この手順では、5’張出し部の内側にDNA断片の第1の塩基があるように、BspMIによる切断によってサイクルが開始される。この場合、それはチミンである。その後、かなり過剰なアダプターA1、C1、G1、およびT1が加えられる。これらは例えばアダプターA1が内部にチミジンを持つ張出し部に連結し、C1が内部にグアニンを持つ張出し部に連結するように設計されている。またアダプターはホスファターゼ(例えばウシ小腸由来のアルカリホスファターゼ、AP、Promega)で処理されており、したがってアダプター間の連結が避けられる。耐熱性リガーゼを使用することによって、この段階において高度の特異性が得られる。第3の段階では、最終段階においてベクターを環状にするために使用される張出し部が形成されるように、AatIIによって切断を行う。これによってこの手順は完了し、出発点に戻る。唯一の違いは、内部のDNA断片の第2の塩基によって張出し部が作成されるように、BspMI部位がさらに1塩基対先に位置していることである。またAatII部位はPstI部位に置き換えられるので、次のサイクルではPstI張出し部を有するアダプターが使用されなければならない。(AatIi/PstIアダプターを2回に1回使用する理由は、ベクターを環状にする前にアダプターが再び切断されるのを防ぐためである。)
[実施例3]:プルーフリーディングを可能にする標的DNA分子の隣接領域に張出し部を作成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
この変異体のための主発点は、4つの塩基につき張出し部のすべての組み合わせである256のアダプター、および5つの塩基につき張出し部のすべての組み合わせである1024のアダプターである(図3A)。トップアダプター型の1024の変異体を作成しなければならず、一方ではボトムアダプター型の256の変異体を作成する。断片の相対的な大きさは、図が示すよりも大きい。両アダプターともにPstI張出し部を有し、それによってお互いに連結するのが可能になっていることに留意されたい。しかしながら、キナーゼで処理されるまでアダプター間の連結が起こらないように、張出し部はホスファターゼで処理されてしまっているはずである。
【0193】
塩基ベクターでは、第1の変異体で使用される塩基ベクターと比べて、BspMIおよびPstI部位がHgaIおよびSfaI部位に置き換えられる(図2)。
【0194】
この手順の残りは4つの酵素反応の1サイクルからなり、1サイクルあたり9つの塩基が転換される(図3B)。転換の終わりには、反応を出発点に戻しながらベクターが環状にされる。唯一の違いは、HgaIおよびSfaNI部位が押しのけられ、4つの塩基対がさらにDNA断片に入ることである。したがって次のサイクルでは4つの新しい塩基対、およびこのサイクルで転換された5つの塩基対が作成される。前述の4つの塩基対が両方のサイクルで同じようにして転換されたことを検証することによって、1つまたは複数の不適切な転換が起こったかどうかを調べることができる。
【0195】
《方法》
1)標的DNA分子を、数百の塩基対の断片が形成されるように、DNase1またはその類似物によって断片にする。これらは、HgaIおよびSfaNI部位をメチル化するために処理される。次いで断片を、常磁性ビーズに付着している塩基ベクターに連結させる。
【0196】
2)HgaI切断を行う。
【0197】
3)SfaNIを行う。
【0198】
4)HgaIおよびSfaNIメチラーゼ、またはHgaIおよびSfaNI部位を不活性化させる他のメチラーゼでメチル化を行う。
【0199】
5)かなり過剰なアダプターを加え、例えばPfuまたはTaqを使用して、段階2)および3)でHgaIおよびSfaNIによって形成された張出し部に連結させる。この段階ではPstI張出し部は連結された状態にはならない。なぜなら、それらはすでにホスファターゼで処理されているためである。
【0200】
6)アダプターのPstI張出し部をリン酸化し、次いで例えばT4DNAリガーゼで連結させ、ベクターを環状にする。
【0201】
7)このサイクルを段階2)で再び開始することによって望ましい回数だけ繰り返す。
【0202】
8)転換された標的分子を連結した標的分子の側面にある塩基ベクター内の制限開裂部位を使用することにより切断によって塩基ベクターから切り離す。拡大タグ内にありうるいかなる切断部位も事前に不活性化しておかなければならない。
【0203】
9)転換されたDNA分子を一本鎖にし、蛍光プローブとハイブリダイズさせる。
【0204】
10)転換されたDNA分子をスキャン表面に固定し伸張させ、蛍光プローブを蛍光スキャナまたはその類似品によってスキャンする(DIRVISHのように)。
【0205】
11)適切なソフトウェアを標的配列のイメージ認識および再構築のために使用する。
【0206】
これを行うことができる1つの方法は以下のようなものであり、この方法では体積はすべてビーズの体積を除いて計算する。
【0207】
1)ランダムな断片を、前に記載したような常磁性ビーズに付着した塩基ベクターにクローン化する。
【0208】
2)ビーズを沈降させるために磁石を使用し、管を約100μlの1× NE緩衝液1で洗浄する。
【0209】
3)10μlの10×NE緩衝液1、DNA1μgあたり4ユニットのHga、および水を最終体積100μlまで加える。これを37℃で1時間かけてインキュベートする。
【0210】
4)HgaI酵素を65℃で20分間かけて不活性化させる。
【0211】
5)ビーズを沈降させるために磁石を使用し、管を1×NE緩衝液3で洗浄する。
【0212】
6)10μlの10×NE緩衝液3、DNA1μgあたり2ユニットのSfaNI、および水を最終体積100μlまで加える。これを37℃で1時間かけてインキュベートする。
【0213】
7)SfaNIおよびHgaI部位をメチル化する。
【0214】
8)ビーズを沈降させるために磁石を使用し、管を1×リガーゼ緩衝液で洗浄する。
【0215】
9)転換アダプターを含む溶液を加える。標的DNA分子と転換アダプターの比は1:50であってよい。100μlの10×リガーゼ緩衝液、10μlのT4DNAリガーゼ(400U/μl、NEB #202)および水を最終体積1mlまで加える。これを16℃で12〜16時間かけてインキュベートする。
【0216】
10)ビーズを沈降させるために磁石を使用し、管を1×キナーゼ緩衝液で洗浄する。
【0217】
11)2μlの10mM rATP、10μlの10×キナーゼ緩衝液、2μlのT4ポリヌクレオチドキナーゼ(30U/μl)および水を最終体積100μlまで加える。これを37℃で10〜30分かけてインキュベートする。(United States BiochemicalsからのT4ポリヌクレオチドキナーゼ(70031)。)
12)ビーズを沈降させるために磁石を使用し、管を1×リガーゼ緩衝液で洗浄する。
【0218】
13)100μlの10×リガーゼ緩衝液、10μlのT4DNAリガーゼ(400U/μl、NEB #202)および水を最終体積1mlまで加える。これを16℃で12〜16時間かけてインキュベートする。
【0219】
14)段階2)〜13)を1回または数回繰り返す。
【0220】
例えばDNA分子を伸張させるためにBensimonの方法(Michalet他.,1997,Science,277,p1518−1523)が使用されるならば、スキャン表面当たり500kbの約100万のDNA分子を伸張させることができる。各シグナルが約5kbであるならば、これは500kbの各DNA分子が、100塩基対の配列に関する情報を提供することを意味する。このことは1つのスキャン表面が約1億の塩基対の情報を提供するであろうことを意味する。しかしながら、標的配列の首尾良い再構築は、多くの塩基対が少なくとも2回はスキャンされなければならないような配列片の重複に依存するはずである。
【0221】
[実施例4]:標的分子の末端塩基を同定するために制限部位を完成させることを含むシークエンシングの方法
この方法は、DNA代謝において活性のある多くの酵素が基質中の認識において有している高度な特異性に基づく。この方法は制限酵素とともに以下のように例示されるが、部位特異的な制限酵素、トランスポゼースなどのような他のいくつかのDNA代謝酵素も使用することができる。大部分の制限酵素にとって、塩基対の1つが開裂部位で突然変異すると、それは一般に酵素によるさらなる切断を防ぐのに充分である。この方法では、制限部位の一部のみを含むリンカーに標的分子が連結されている。標的DNAによってこの部位が完成している場所では、切断が影響される可能性があり、この後に、相補的アダプターが結合され、部位を完成させそれゆえ特定の末端塩基を表すこれらの分子を示している。アデニン用のこの方法を図4に示す。
【0222】
《方法》
1)シークエンシングすべきDNA分子を4つの異なる標準制限酵素(EnzA、EnzC、EnzG、およびEnzT)で切断する。
【0223】
2)次いでこれらの分子を4つの異なるDNAリンカー分子(分子A、C、G、およびT)に連結する。これらの分子の1つ1つは、それぞれEnzA、C、G、およびT用にほぼ完全な部位を有し、完全な塩基対を得るために末端では各分子は1つの塩基対のみ欠いている(それぞれA、C、G、およびT)。このようなリンカー分子の一例を図4Bに示す。この分子にはA/T塩基対を欠いたHindIII部位がある。このリンカーがA/T塩基対を持たないDNA片と連結するならば、DNA片から該分子を取り除くためにMnIIを使用することができる。図4Cは、末端にA/T塩基対を有するDNA片に連結されたリンカー分子Aを示し、したがって完全なHindIII部位が作成された。HindIIIが切断用に使用される次のステップでは、アダプターAに連結される可能性があるHindIII張出し部が作成されるであろう。
【0224】
3)切断を可能にするために4つの制限酵素を溶液に加える。完全な切断部位のみが存在するはずであり、各リンカー分子A、C、G、およびTは末端で塩基対を欠くDNA分子に連結する(分子A、C、G、またはT用にそれぞれA、C、G、またはT)。
【0225】
4)アダプターを張出し部に加える。この張出し部は、制限酵素によって発生しアダプターが正しいDNA片に固定されるように連結された張出し部を補う。適切なアダプターを図4Aに示す。トップアダプターをサイクル1、3、5などのために使用し、一方ボトムアダプターをサイクル2、4、6などのために使用する。これらのアダプターは、HindIIIによって生成される張出し部に相補的な張出し部を有する。トップアダプター上のAatII張出し部は、塩基ベクターが環状化されるように、アダプターの一方の末端を塩基ベクターに連結させるために使用されるはずである。MnII部位は、新しいサイクルを開始することができるように、DNA片上に平滑末端を発生させるであろう。PstI部位は次のサイクルで新しいアダプターを連結させるために使用されるはずであり、PstI張出し部を有するアダプターが使用される。
【0226】
5)アダプターの一方の末端を塩基ベクターに連結させることができるように、例えばAatIIで切断することによって、DNA片/アダプターを用いて塩基ベクターを環状にする。HindIIIが形成されない場合は、PstI部位を有する小さな断片を塩基ベクターのAatII張出し部に連結させる。
【0227】
6)新しいサイクルを開始させることができるように、DNA片上に新しい平滑末端を発生させる制限酵素によって切断を行う。
【0228】
[実施例5]:ハイブリダイゼーションおよび連結と結びついたKlenow修復反応を使用するシークエンシングの方法
この方法は、DNAポリメラーゼのKlenow部分がヌクレオチドの取り込みのための非常に高度な特異性と、大部分のDNAリガーゼには異なる大きさの張出し部を連結させる能力がないという事実に基づく。この方法は図5に示され、標的分子内で張出し部が作成され、それはアダプター分子内の張出し部より長い。張出し部を減少させるため適切に伸長し、さらなる正しい塩基を含む標的分子のみがアダプターに連結されるであろう。図5にアデニン用の方法を例示する。
【0229】
《方法》
1)図5Aに示すようにDNA片を塩基ベクターに連結させる。ストレプトアビジン基質(例えばDynalのM280ストレプトアビジン被覆磁性球)に該分子を固定するのを可能にするビオチンとは別に、塩基ベクターはポリヌクレオチド内を切断する制限酵素(例えばHgaI)のための部位、および標準的な制限酵素(例えばEcoRI)のための部位を含む。
【0230】
2)ポリヌクレオチドから5つの塩基対を有する張出し部が形成されるように、HgaIでベクターを切断する。
【0231】
3)その後、「T」で始まる張出し部が4塩基対の張出し部に短縮されるように、例えば塩基AおよびKlenowを加える。
【0232】
4)次いで反応溶液を、リガーゼおよび4塩基対の張出し部を有する遺伝子断片(アダプター)の溶液に置き換える。この張出し部は共通(universal)のヌクレオチド、および4塩基対を有する張出し部のあらゆる可能な組成物の組み合わせのいずれかを含むことができる。共通ヌクレオチドを有する張出し部は、あらゆる組み合わせの4塩基の5’張出し部に連結する能力がある。遺伝子断片は、ポリヌクレオチド内で切断を行う制限酵素(例えばHgaI)のための部位、および標準的な制限酵素(例えばEcoRI)のための部位、およびシグナル「T」を含む配列(プローブとして使用することができる配列など)も含む(図5B)。AatII張出し部およびPstI部位には実施例4の1と同じ機能がある。5塩基対の張出し部を有するポリヌクレオチドを4塩基対の張出し部を有する遺伝子断片に連結させることはできないため遺伝子断片に連結されるのは元来最も内側で「T」を有していたポリヌクレオチドだけである。成功した連結および失敗した連結を実施例5Cに示す。張出し部の内側には塩基は取り込まれなかったことから、上側のDNA片はアダプターAに連結することができない。内部に「A」を有する張出し部のみが塩基を取り込んだので、下部で見られるようにアダプターAに連結されるのはこれらの張出し部だけである。DNA片の実線は、HgaI切断を介して形成される張出し部を示す。次いで同じプロセスを塩基C、G、およびTを用いて繰り返す。
【0233】
5)最後に、塩基ベクターをDNA片/アダプターを用いて切断によって、例えばEcoRIを用いて連結によって環状にする。
【0234】
6)次いでHgaIを用いた切断を行う。これによってDNA片中に新しい張出し部が発生し、したがって反応サイクルが再び始めることができる。
【0235】
連続する塩基が同一である例では、異なる大きさの張出し部、例えば2塩基のKlenw修復を可能にする3塩基の張出し部を有するアダプターを使用することができる。
【0236】
[実施例6]:1本鎖標的分子に結合する際、プライマーとして機能するアダプターを用いるシークエンシング方法
《方法》
1)断片組成物に相当する張出し部を有し、本法に用いられるアダプターは、実施例6Aに例示される。この張出し部中の塩基組成物に対応する断片組成物に注意されたい。断片の全ての組み合わせに相当するDNAアダプターを、構築する必要がある。
【0237】
2)標的DNA片を1本鎖片にし、RNAリガーゼの助けでアダプターの3’末端およびプライマー鋳型の5’末端に連結される(図6B)。アダプターとDNA片との連結を成功させる要件は、アダプターの張出し部が、DNA片の5’末端と相補的であることである。これにより、正しい断片が、DNA片に結合されることが保証される。
【0238】
3)分子が、互いに連結する前に、標的DNA片をアダプターとハイブリダイズする。次いで1回または数回のPCRサイクルが行われる。PCRサイクルを成功させる必要条件は、ssDNA片をハイブリダイズして、アダプターに連結しておくことである。
【0239】
4)EcoRI部位が、アダプターとDNA片との連結後、形成されるDNA分子を環状化させるために用いられる。次にHgaI部位は、DNA片に新たな切断を生じるように用いられ、その結果、次のサイクルが開始できる。EcoRIで切断後の分子間連結を減じるために、この分子を、基質、例えばストレプトアビジン被覆球に固定することが有利と思われる。
【0240】
[実施例7]:自己ハイブリダイズするアダプターを用いるシークエンシング方法
本法の出発点は、シークエンシングされるDNA分子とハイブリダイズする能力を有し、同時に自己ハイブリダイズする領域に加えて、それらが、ハイブリダイズする塩基に対応する断片(拡大タグ)を担持する1本鎖DNAまたはRNAアダプターである(図7)。全ての可能な断片の組み合わせでこのようなアダプターを作成し、シークエンシングされるDNA分子とそれらをハイブリダイズすることにより、複合数アダプターを並列に並べられてもよい(図7を参照)。アダプターを正しく、すなわち、最初の2つのアダプターに誤った組み合わせがないように並べると、それらを互いに連結することが可能となり、より長い鎖を形成することができる。
【0241】
[実施例8]:1つ以上の塩基に対応するアダプターの構築方法
アダプターを構築する1つの方策は、DNAチップを構築するのに用いられる原理と同様の原理を利用する。この方法において、種々のオリゴヌクレオチド類が、DNAチップの構築と同じ方法で種々のアドレスに合成される。次いで、この同じ原理は、塩基対を伸長するオリゴヌクレオチド類に固定するのと同じ方法で、DNA片をオリゴヌクレオチド類に固定するのに用いられる。最後に、DNA分子が解かれ、アダプター溶液を得る。
【0242】
本実施例ではまた、アダプターを調製する別法を提供する。これは、図8に例示されている。図8に例示されるように、各々1つのタグを有する8種類のアダプターを、各々2つのタグを有する16種類のアダプターを合成するのに用いる。他の8種類の1タグを有するアダプターとともに出発して、16個の新たな2タグアダプターを合成でき、これは順次最初の16個と結合して、256種類の4タグアダプターを生成することができる。この方法で、アダプター混合物を生成することが可能であり、ここでは、解答に合う異なる分子の数のみが、順列の数を限定する。最初に必要とされる、異なる1タグアダプターの数は、各アダプターにおけるタグ数の4倍に等しい。例えば、我々が、16タグのアダプター類(4.29×109順列)を作成しようとする場合、16×4種類の1タグアダプターが、最初に必要とされる。
【0243】
《方法》
1)図8に示すように8種類の1−タグアダプターが、用いられる。左側のアダプター類は、EcoRI張出し部、分子上の極く右側にある塩基に特異的なタグ、およびBseMIのための開裂部位から成る。BseMIは、それ自身の部位の外側を切断し、分子右側の塩基のすぐ隣で平滑な切断を生じさせる。アダプターはまた、ホスファターゼで処理されて、これらのアダプター間の連結を減じる。右側のアダプターは、分子の極く左側の塩基に対応するタグおよびEcoRIのための切断部位から成る。アダプターはまた、基質に固定され、これらのアダプター間の連結を防止する。
【0244】
2)2つのアダプター集団を混合し、連結させることにより工程が開始される。これにより、2つのタグを有する全順列に対応する16種類のDNA分子を生成する。
【0245】
3)EcoRIは、開裂に用いられる。次いで、これらの分子を環状化させるために、連結が実施される。
【0246】
4)最後に、BseMIにより開裂を実施すると、16種の2−タグアダプターを有する集団が生成される。
【0247】
[実施例9]:ヘアピン型アダプターに基づく転換方法
前記のいくつかの転換代替方法における主要な点は、塩基対の転換後、DNA断片を、転換されつつあるDNA分子の他の末端に移動させることである。これにより、転換されつつある末端を開放し、DNA断片を蓄えると同時に、次のサイクルに進むことができる。DNA断片をDNA片の他の末端に移動させる他の方法は、以下に示す。
【0248】
出発点は、T4DNAリガーゼなどの多くのリガーゼ類が、dsDNAの張出し部をssDNAの末端に連結できることである。これは、図9に例示した方法に利用され得る。
【0249】
《方法》
1)標的DNAを、1本鎖形に転換する。
【0250】
2)転換アダプターを添加し、DNA片の3’末端に連結する。
【0251】
3)ポリメラーゼ伸長を実施する。
【0252】
4)ヘアピンア型アダプターを添加し、連結する。転換アダプターの末端を予め処理すると、ヘアピン型アダプターに連結しない(例えば、ホスファターゼで処理)。
【0253】
5)DNA分子を融解する。
【0254】
6)1本鎖DNA分子を、断片と相補的なDNA分子とハイブリダイズする。相補的DNA分子はまた、標的DNA中の第1の塩基とハイブリダイズする共通(ユニバーサル)塩基の付いた張出し部を有する。
【0255】
7)自身の認識配列の外側に張出し部を形成する酵素に対する開裂部位の助けにより、次の転換サイクルのためにDNAが作成される。
【0256】
上記の各サイクルにおいて、拡大タグは、各サイクルにおいて複製される。これにより、プルーフリーディングを可能にする。
【0257】
[実施例10]:転換されたDNA分子の連結に基づく転換法
前記の多くの転換法は、サイクル工程で生じる転換に基づいている。それにより拡大タグ(またはシグナル鎖)の1本鎖当たりの転換塩基対数は、サイクル数と共に直線的に増加する。別の方法としては、転換DNA片を長鎖中に結合することである。この原理に基づく方法としては、非常に多くの可能な方法があるが、以下に1つの提案を示し、図10に例示する。
【0258】
《方法》
本法は、大きさに従って標的DNAを切断し、ソーティングすることから始まる。次に、特定の長さ、例えば30塩基対のDNA片を、この手順から除かれる。
【0259】
1)DNA片の末端を、前記方法を用いて転換する。
【0260】
2)転換DNA分子を環状化させる。
【0261】
3)転換アダプターの末端に位置する開裂部位を用いるIIS酵素を添加する。これは、図10に示すようにDNA片を切断する。
【0262】
4)このDNA分子を融解し、断片、例えば蛍光体で標識したプローブに相補的なDNA分子とハイブリダイズする。
【0263】
5)最後に、転換DNA片をハイブリダイズし連結するが、必要なら溶液中で行う。
【0264】
前記実施例において標的DNAを有する張出し部は、相補的な張出し部を捜し求めることから、各転換DNA片は、遭遇したDNA片とハイブリダイズ/連結する。これにより、22の未知塩基(例えば、AGCTGTGA N22 AGTCTGCA N22 TGAC)により遮断される8塩基対の配列片についての情報を提供する拡大タグ鎖(シグナル鎖)を生み出す。未知の塩基対数は、DNA片の当初の長さから、1DNA片当たり転換した塩基対の数を差し引くことにより決定される。シグナル鎖間の重複に基づいて、次に、繰返しの配列の領域においても標的配列を再構築することが可能である。
【0265】
[実施例11]:DNAのダブリング化(doubling)法
1本鎖DNA分子は、図11に示されるように2回のダブリングサイクルに供される。ダブリング(doubling)は、ヘアピン型アダプターを分子の3’末端に連結させることにより開始する。例えば、逆転写酵素が、プライマーとして3’ヘアピンループを用いるのと同じ方法で、アダプターを、分子を伸長するポリメラーゼのためのプライマーとして使用することができる。最後に、DNA分子を溶解して出発点に戻る。長さy bpであるアダプターの助けによりx bpのDNA分子をn倍にダブリングする本法を用いると、DNAの長さは:
・ x・2n+(2n−1)y
となる。次に、ダブリング前、各々xおよびx+1 bpであった2つのDNA分子間の差は:
2)(x+1)・2n+(2n−1)y−x・2n+(2n−1)y=2n
となる。
【0266】
したがって、2つのダブリングされたDNA分子間の長さの差は、増幅前のそれらの絶対的な長さの差によってのみ決定され、相対的な長さの差によってではない。
【0267】
[実施例12]:プライマーに基づくソーティング法を利用するDNAシークエンシング方法
本実施例は、16種類の異なるゲル分離および16種類の標識、例えば蛍光団を用いて、互いに分離できる256種類のシークエンシング梯子の作成法を説明する。シークエンス反応の長さは、4種類のシークエンシング梯子のみを用いる方法と比較してかなり増加できる。したがって、ソーティング作業量、長い配列をシークエンシングする際などに必要なプライマー数をとりわけ減じることが可能である。16種類のシークエンシング梯子並びに16種類の発蛍光団が、本実施例で使用されるが、明らかにこのシークエンシング梯子数、および蛍光団数は、大部分の要件および利用できる装置に適合できる。シークエンシング梯子および蛍光団をより多く使用するほど、シークエンス反応はより長くなる可能性がある。
【0268】
《方法》
使用する本法の概要を、図12Aに示す。
【0269】
1)標的DNA溶液を、ウェルの基質に固着されたシークエンシング・プライマーを含有する16個のウェルに分配する。これらのプライマーは、ポリメラーゼ反応のための一定の出発点を決定することから、この共通の起点により、生成する最終生成物のサイズは、その起点から最終配列の距離を示すものとされる。
【0270】
2)ポリメラーゼ伸長反応を実施し、DNA分子を加熱して融解し、次いでウェルを洗浄する。
【0271】
3)次に、図12Bに示すように、16種のプライマー類を、16個のウェルの各々に添加する(全部で256種のプライマー)。各ウェルに添加された全てのプライマーは、3’末端での塩基3および4以外は同一である。この位置にAAを有するプライマー類は、シグナル1に連結され、この位置にACを有するプライマー類は、シグナル2などに連結される。ウェル2のプライマー類は、それらが3’末端にAAの替わりに、例えば、ACで開始すること以外は同一であるが、ウェル3のプライマー類は、AGなどで開始する。このように全部で、3’末端に256種の4塩基順列の全て包含する256種のプライマー類がある。特定の蛍光シグナルを、各16種類のプライマーに付加する。次いで、さらなるポリメラーゼの伸長反応を実施する。
【0272】
4)次いでウェルを洗浄してからDNA分子を融解する。次に、これにより解かれた1本鎖DNA分子を、16の異なるゲル分離(各ウェルにつき1つ)を用いてサイズに従ってソーティングする。
【0273】
5)蛍光シグナルを記録し、標的配列を、適当なソフトウェアで再構築する。
【0274】
《結果》
この結果を図12Cに示す。各蛍光シグナルは、4塩基配列断片についての情報を提供する。最初の2つの塩基についての情報は、蛍光シグナルが読取られるウェルの参照により引き出し得るが、最後の2つの塩基は、存在する特定のシグナルに基づいて決定できる。
【0275】
[実施例13]:ハイブリダイゼーションに基づくソーティング方法を利用するDNAシークエンシング方法
本法では標的DNA分子を、走査表面にあるオクタマーとのハイブリダイゼーションによって走査表面の異なる部位にアドレスする。次いで、この分子を引き伸ばし、蛍光シグナルから垂直保留線までの距離が評価され、標的配列中のオクタマーの位置についての情報が提供される。一般手順を、図13に示す。
【0276】
《方法》
1)出発点は、65,536のアドレスから成る走査表面である。1本鎖オクタマーを有する垂直保留線を各アドレスに付される。AAAAAAAAオクタマーを、アドレス1のプレートに固定し、AAAAAAACオクタマーは、アドレス2のプレートに固定するなどの結果、65,536の各オクタマー順列全てが、それ自身のアドレスを有することになる。
【0277】
2)次に、一末端または両末端に蛍光標識を有する1本鎖標的DNA分子を、走査表面上に混合し、それらをオクタマーにハイブリダイズさせることができる(図13A)。
【0278】
3)所望するなら、分子をUV照射に曝露することにより、オクタマー/標的DNA結合を強化し、オクタマーをプライマーとして、または他の手段によりポリメラーゼ伸長を実施してもよい。
【0279】
4)次に、走査表面を洗浄し、DNA分子を伸展する(図13B)。
【0280】
5)蛍光スキャナを用いて表面を走査し、保留線に対する距離の関数として、各アドレスにおける蛍光強度を記録し、標的配列を適当なソフトウェアを用いて
再構築する。
【0281】
《結果》
得られた結果を図13Cに示す。例示したアドレスにおいて、約150kb、約300kb、約500kb、約550kb、約780kb、約870kbおよび約1040kb(DNA分子が1マイクロメートル当たり2kb伸長する場合)と7種類のDNA分子長がある。
【0282】
[実施例14]:リガーゼに基づくソーティング法を利用するDNAシークエンシング法
本実施例は、リガーゼに基づくソーティング法であり、65,536のシークエンシング梯子が、65,536のアドレスにソーティングされる。各々が塩基を表す4個のシークエンシング梯子を利用する他の方法とは対照的に、本法における65,536のシークエンシング梯子の各々は、8個の塩基の配列片を表すことになる。これは、4個の梯子のみを用いる方法と比較して、サイズによるソーティングに対する精密要件を減ずる。反応の長さは、このように増加でき、サイズによってポリマー類をソーティングするための広範囲の方法を利用することもまた可能である。
【0283】
本実施例においては、サイズによるソーティングは、伸長したDNA分子の長さを測定する方法により示される。しかし、サイズによるソーティングを走査表面上で直接実施するなど他の変法が考えられる−例えば、DNA分子のシグナル強度が長さに比例するといった標識方法を使用してから、DNA分子のシグナル強度を測定するなどの方法がある。また、シークエンシング梯子を、物理的に分離した状態において時間などを違えて基質から解放し、フローサイトメーター、質量分析、ナノポア分析、ゲルソーティング法などを用いて各65,536のシークエンシング梯子を個々に分析することを可能にさせる変法もまた考えられる。
【0284】
《方法》
1)例えば1Mbの標的配列から出発して、ここに記載されるように調製し、シークエンシング梯子を生成する。
【0285】
2)標的DNAをメチル化して、段階3)および6)で用いられる制限酵素の開裂部位を不活化させる。
【0286】
3)4塩基の張出し部を、ここに記載しているように標的DNAの任意の末端に作成する。DNA分子の任意の末端を、例えば、4塩基対の張出し部を作るIIS酵素の結合部位を含有するDNAリンカーに連結してもよい。結合部位を、実際の標的DNAに張出し部を作るために配置する。次いで分子をIIS酵素で開裂する。
【0287】
4)次に標的DNAは、256のウェル間に溶液を分配することにより、実施例12に記載のとおりソーティングする。ウェル壁を、段階3)で作成された張出し部と相補的であり得る4塩基張出し部を有するソーティングアダプターで覆う。ソーティングアダプターはまた、IIS制限酵素、例えば、FokIに対する結合部位を含み、この結合部位は、張出し部が、段階5で切断されたもののそばにある4塩基対を含んで形成できるように配置される。
【0288】
5)標的DNAをソーティングアダプターと連結し、次に管を洗浄して、結合しなかったDNAを除く。
【0289】
6)IIS酵素による開裂を実施し、標的DNAを解放して新たに4塩基張出し部を形成する。
【0290】
7)実施例12に記載しているように、標的DNAを256のウェルから256のミクロアレイ間に分配する。全ミクロアレイは等しく、段階6)で作成された張出し部と相補的であり得る4塩基張出し部を有するソーティングアダプターの付いた256のアドレスから成る。アドレス1において、ソーティングアダプターは、AAAA張出し部を有し、アドレス2では、AAAC張出し部を有するなどとなる。
【0291】
8)リガーゼを添加し、この混合物をインキュベートする。アドレス1において、TTTT張出し部を有する標的DNAが存在し、アドレス2ではTTTG張出し部などを有する標的DNAが存在することとなるであろう。
【0292】
9)走査面を洗浄し、DNA分子をアドレス化し、TOTO−1,YOYO−1または同様のもので着色する。
【0293】
10)CCDカメラまたは同様のものが、アドレスを撮影するために使用される。CCDカメラを、例えば、1アドレス当たり1枚の写真を撮るために設定してよい。
【0294】
11)蛍光DNA分子を認識し、それらの長さを測定し、次いで実際の標的配列を再構築するために適当なソフトウェアを使用する。
【0295】
これを実施できる1つの方法は、以下のとおりである:
1)各ウェルに、任意の4塩基張出し部を含む標的DNA分子の一定分量、10μlの10×リガーゼ緩衝液、1μlのT4DNAリガーゼ(400U/μl、NEB#202)を加え、最終容量が100μlになるように水を加える。16℃で12〜16時間インキュベートする。
【0296】
2)液体を除去し、1×NE緩衝液4でウェルを1回または数回洗浄する。
【0297】
3)10μlの10×NE緩衝液4、DNA1μg当たり4単位のFokI(New England Biolabs、#109)を加え、最終容量が100μlになるように水を加える。37℃で1時間インキュベートする。
【0298】
4)65℃で20分間不活化する。
【0299】
5)EtOHにより個々の管の各ウェルからDNA分子を沈殿させる。
【0300】
6)ペレットを溶解し、10μlの10×リガーゼ緩衝液、1μlのT4DNAリガーゼ(400U/μl、NEB#202)を加え、最終容量が100μlになるように水を加える。16℃で12〜16時間マイクロアレイでインキュベートする。
【0301】
7)分子を伸長し、標識して解析する。
【0302】
《結果》
特定のアドレスにおける分子の有無およびサイズは、配列情報およびその位置の双方を示す。したがって、マイクロアレイ1のアドレス1が、100マイクロメートルのDNA分子を含む場合、これは、その分子を結合するために用いたオクタマーに相当する配列が(たとえ2段階ソーティングによっても)、+200kb(例えば、TTTTTTTT)で存在することを示す。同様に、2つの異なる大きさの分子の存在は、特定配列の反復を示すと思われる。特定アドレスにおける任意の分子の欠如は、標的配列において固定化オクタマーに相補的な配列の欠如を示すものと思われる。
【0303】
上記実施例において、誤ったソーティングの潜在的な原因としては、粗ソーティングアダプター類がまた、精ソーティングアダプター類として働く可能性が考えられる。しかしながら、この問題は、粗ソーティングアダプター類に別の制限エンドヌクレアーゼのための切断部位を持たせることにより避けることができ、これにより、走査する前に粗ソーティングアダプター類を切り離すことができる。上記実施例で記載しなかったが、粗ソーティングアダプターに付加していないDNA片の先を末端となすこともまた重要である。これは、例えばKlenow充填により行い得る。
【0304】
Bensimon法(Michaletら、1997年、上記)を、DNA分子を伸長するため上記の方法において用いると、100万〜200万のDNA分子を、1.28×1.28cmを測定する走査面上で伸長できる。256のアドレスの各々は、約4、000〜8、000の伸長DNA分子を含むであろう。8塩基対を有する配列片が、65,536番目の塩基対ごとに反復されるため、標的配列が1Mb(1,000,000/65,536=15.2)であると、各アドレスに平均して15種類の長さがあることになる。したがって、各長さは、平均して260〜520回(4,000〜8,000/15.2=260〜520)測定されることになる。
【0305】
[実施例15]:標的DNA分子を固定参照点に固定されるDNAシークエンシング法
本法においては、直線状配列の拡大タグ(シグナル鎖)を担持するPNAオクタマー類を、走査面上に固定化された標的DNAにハイブリダイズし、次にそれがスキャンされ、標的配列内のオクタマー類に相補的な領域の位置が決定される。一般的手順を図14に示す。
【0306】
《方法》
1)このプロトコルの出発点は、本明細書記載されるように走査面上の固定された参照点、例えば走査板に垂直な保留線に標的2本鎖DNA分子を固着することである。
【0307】
2)上記オクタマーに対応する組成物を有するシグナル鎖に付加されたPNAオクタマーカーら成る転換アダプター類(すなわち、拡大タグを担持するアダプター類)の65,536の順列を、次に添加する。シグナルは、蛍光標識球、ビーズであってよく、または他の適当な標識を担持してよい。このように、PNA分子を標的DNA分子とハイブリダイズする。
【0308】
3)分子を伸長し、それらの位置およびシグナル鎖の組成を記録した。
【0309】
4)適当なソフトウェアを、標的配列を再構築するために使用する。
【0310】
《結果》
この結果を図14に示す。シグナル鎖と固定した保留点との間の距離は、標的配列の各配列片の位置について情報を提供する。
【0311】
[実施例16]:ソーティングまたは転換と組み合わせたソーティングに基づくシークエンシングおよび光学的マッピング法
本法により、特別の長さのDNA配列、例えば、ゲノム類をマッピングまたはシークエンス反応においてマッピングまたはシークエンシングすることが可能になる。この方法は、光学的マッピング単独にまたはマッピングに加えてシークエンシングに用いることができる。この方法が、同一のシークエンス反応において多数の異なる標的配列類のシークエンシングを可能にすることに注目することが重要である。
【0312】
《方法(ソーティング単独)》
実施例14の方法に従うが、段階1の替わりに標的DNAをDNアーゼIまたは同様のもので切断して、数百の塩基の断片類を生成する。段階2から段階8を、実施例14に記載されるとおり実施する。次いで、走査面を洗浄し、DNA分子を伸長する。光学的マッピング法または同様の方法を、次に実施する。走査面を、蛍光スキャナまたは同様のもので走査し、配列を再構築するために適当なソフトウェアを使用する。
【0313】
《方法(転換を伴うソーティング)》
段階1から段階6を、上記の光学的マッピング法のとおり実施する、その後、
7)256の転換アダプターを添加し、段階6)で形成された張出し部と連結する。転換アダプターは、例えば、1のシグナルが、特定の制限酵素に係る多数の開裂部位を含むDNA配列類であり、0のシグナルは、そのような部位を含まないDNA配列類である2進法シグナル鎖を有してもよい。
【0314】
8)各ウェルからの転換DNA分子を、それ自体の走査表面に移し、1シグナルにおいて切断部位を有する制限酵素を用い光学的マッピング法を実施する。
【0315】
9)走査面を、蛍光スキャナまたは同様のもので走査し、適当なソフトウェアを、配列を再構築するために使用する。
【0316】
[実施例17]:2進末端転換およびナノポア分析に基づくDNA塩基シークエンシング法
電場により、脂質膜のイオンチャネルを介して1本鎖RNAおよびDNA分子を駆動できることが示された。分子の移動は、イオン電流の一過性の減少として検出できる。大きさの違いのために、プリン類とピリミジン類とを識別することが可能であることが示された。したがって、この方法は、高速シークエンシングに利用できることが示唆された。しかしながら、大きさの相異が小さいために、異なるプリン類(アデニンまたはグアニン)間および異なるピリミジン類(シトシン、チミンまたはウラシル)間を識別することが困難であることが示された。本実施例においては、この問題を、標的のDNAをプリン/ピリミジンシグナルから成る2成分コードに転換することにより如何に解決できるかが示される。
【0317】
《方法》
1)標的DNAの断片は、DNアーゼIなどで開裂することにより生成し、平滑末端を作成するために処理された。
【0318】
2)標的DNA分子を、IIS制限酵素(例えばFokI)の1つまたは複数の結合部位を含むリンカー類と連結する。
【0319】
3)張出し部を、IIS制限酵素での開裂により標的DNA中に生成させる。
【0320】
4)張出し部を、ホスファターゼ酵素で処理する。
【0321】
5)張出し部を、転換アダプターと連結させる。アダプターにはまた、方向タグも含んでいて、ソフトウェア分析をより容易にする。
【0322】
6)プリン類/ピリミジン類の組成を、ナノポア分析により読取る。転換されなかった標的DNA部分を、位置情報を得るために使用してもよい。
【0323】
7)適当なソフトウェアプログラムを、標的配列を再構築するのに使用する。転換アダプターと標的DNAとの間の張出し領域は、プルーフリーディングとして配列片情報と比較できる。
【0324】
《結果》
シグナルは、A=プリン+プリン;C=プリン+ピリミジンなどの2成分プリン/ピリミジンコードから成る。これを図15に示す。
【0325】
[実施例18]:シークエンシング反応のシグナル生成における細胞の利用
本実施例は、シークエンシング反応においてシグナル生成および特定の塩基を表示する拡大タグとしてそれ自体の作用双方のため細胞の利用を例示する。
【0326】
《方法》
A)本法において、レポーター遺伝子が、拡大タグとして使用され、発現による相対的シグナル強度が、配列中の特定塩基の相対位置の指標として使用される。使用される方法および得られる結果を図16に示す。
【0327】
1)ポリメラーゼ伸長反応は、鋳型としての標的DNA、および図16に例示するように、エンハンサーおよびレポーター遺伝子を有する1本鎖または2本鎖に付加されるシークエンシングプライマーを用いて実施する。シークエンシングプライマーは、シークエンシングしようとする配列の開始を表示する標的DNA中の既知配列に結合するために使用される。
【0328】
2)ポリメラーゼ伸長を、4つの異なるプライマーから成るプライマー混合物により実施する。各プライマーは、A、C、GまたはTの何れかであるたいていの3’塩基を除き、ユニバーサル塩基(U)またはランダム塩基(N)から成る。プライマーは、4つの異なるレポーター遺伝子に付加する。ほとんどの3’位にAを有するプライマー類は、レポーター遺伝子Aに付加し、他も同様にする。この段階で使用される転換プライマー類は、ポリメラーゼ伸長の成功に重要な大部分の3’塩基を除いて、無作為に標的DNAに結合する。
【0329】
3)1つまたは複数のポリメラーゼ伸長反応を、段階1および段階2に使用されるプライマー類の5’末端に相補的なプライマーを用いて実施する。
【0330】
4)転換されたDNA分子を、好適な細胞に形質転換/形質移入する。
【0331】
5)細胞を、レポーター遺伝子が発現できる条件下で増殖させる。
【0332】
6)レポーター遺伝子の発現は、フローサイトメーターで分析され、好適なソフトウェアを配列を再構築するために使用する。
【0333】
B)本法において、異なる塩基と関連するシグナルが、細胞内の異なる位置またはシグナル鎖内それらの位置について他の構造表示に向けられる。
【0334】
1)標的DNAを、DNアーゼIまたは同様の技術により断片化する。
【0335】
2)1標的DNA分子あたり16の塩基対を1つのシグナル鎖に転換する。4つのシグナルが、塩基A、C、GまたはTの各々を表示するのに用いられる。各シグナルは、各シグナルについて異なる位置に発現するプロモーターに結合したレポーター遺伝子A、C、GまたはTから成る、すなわち、16塩基対では、シグナルは16種のプロモーターにより16の異なる位置に向けられる。この位置としては、多細胞生物中の細胞または細胞群であってもよい。それはまた、細胞上(例えば、外膜の一部)の位置であってもよい。シグナル鎖を、生物体/構造体のもとなる細胞に形質転換/形質移入する。
【0336】
3)細胞を、生物体/構造体が発育できる条件下で増殖させる。
【0337】
4)各生物体/構造体における4つの異なるシグナルの分布を、異なる位置に記録し、シグナル鎖を伸展させるために用いられる配列に沿って、いずれの塩基がどの位置に出現するかの写真を作成する。
【0338】
《結果》
A)特定の位置に生成したシグナル強度が調査できる。これを図16に示す。シグナル強度は、エンハンサーとレポーター遺伝子との間の距離に逆比例することから、出発塩基に関して特定シグナル(したがって塩基)の位置を確定できる。
【0339】
理想的には、創製される異種分子間の識別を助けるために、標的核酸分子を、伸長生成物が複数塩基だけ異なるように、末端配列に従って最初にソーティングする。
【0340】
[実施例19]:各々3つの拡大塩基だけ異なるシークエンシング梯子を創製することによるシークエンシング法
本法は、転換(すなわち拡大)および配列の読み取りが同一の固形担体上で実施されるシークエンシング梯子の形成を記載する。これらの手順における重要な点は、短い領域(3塩基増すごとに6〜9bp以上)の塩基組成物を獲得することに加えて、より大きなDNA分子(数kbまで)上のそれらの内部位置についての情報もまた得ることである。これは、配列情報の再構築にとって重要であり、この方法は、例えば、前記の変法を介して引き出された配列情報を補足するのに用いることができる。この原理を、9塩基対の長さのポリヌクレオチドのシークエンシングに関して図17に例示する。
【0341】
《方法》
1)シークエンシングしようとするDNA配列は、PCRにより増幅される。プライマーの1つの一端をビオチンで標識し、その結果DNA分子をストレプトアビジン基質に固定できる。ストレプトアビジンは、細い線に並べて、DNA分子を互いに隣接した1列に固定する。
【0342】
2)この分子を、DNアーゼI(または同様のもの)で処理し、無作為な切断を生じさせる(図17の段階1)。
【0343】
3)切断末端を、それ自体の結合部位の外側を切断するクラスIIの制限エンドヌクレアーゼ(この場合EarI)に対する結合部位を含むポリヌクレオチドに連結させる(図17の段階2)。
【0344】
4)次に、制限エンドヌクレアーゼを加えて、該ポリヌクレオチドに張出し部を創製する(図17の段階3)。
【0345】
5)ポリヌクレオチド張出し部を認識し、特異的に連結するアダプターを加える(図17の段階4)。それにより、AGC張出しを有する上部のポリヌクレオチドを、AGC断片の組み合わせなどを有するアダプターに連結させる。
【0346】
6)DNA分子は、液体流、電界または同様のものの助けにより引き伸ばされて、蛍光標識されたアダプター類を、蛍光スキャナで読取ることができる。
【0347】
7)この配列を、配列情報を有する断片を並べることにより再構築する。
【0348】
各アダプターについての相対位置が、ポリヌクレオチドのDNアーゼIにより切断される場所により変化することに注意されたい。この方法において、配列情報を有する各断片が、ポリヌクレオチド上に相対的位置を与えられ、これにより、配列の再構築を、より容易にする。
【0349】
最後に、同一の読み取りプレート上(図17)で異なる数種のDNA配列読取ることにより、読み取りの可能性を増大できることを強調する必要がある。例えば、PCRの助けにより、遺伝子特異的プライマーを使って多数の遺伝子を増幅することができる。次いで、特定の張出し部が、増幅遺伝子配列上で作成される。これにより、例えば、最後のサイクルにおいて特別に長いプライマー類を用いることにより実施でき、めったに切断しない制限エンドヌクレアーゼなどの切断部位を挿入できる。それにより、各区画が、種々の遺伝子に特異的なオリゴヌクレオチドから成る、DNAチップに遺伝子をハイブリダイズすることができる。それにより、遺伝子Aに対応するDNA分子は、区画Aにハイブリダイズし、遺伝子Bは、区画Bにハイブリダイズするなどとなる。医療などで関心のあるゲノムにおいて、これらの特異的な領域を選び出すことが可能な場合、この方法は、人々のゲノム類のマススクリーニングにとって特に適切である。
【0350】
異なる遺伝子の平行シークエンシングはまた、遺伝子特異的プライマー類を用いる増幅により達成できる。次に、各遺伝子に特異的な張出し部を生成できてから、遺伝子類は、DNAチップ上のオリゴヌクレオチド類にハイブリダイズされる。このDNAチップは、異なる遺伝子類に相補的なオリゴヌクレオチド類が、異なる部位を有するように構築される。実際に、同一の読み取りプレート上に数千の異なるアドレスを創製することが可能であり、その結果平行して数千の遺伝子をシークエンシングすることが可能である。
【0351】
図18に示すように、さらなる変法は、2次元で位置情報を得ることである。
【0352】
1)この手順の出発点は、シークエンシングされるDNA分子を、2、3kb以上の長さの分子に切断することである。次いで、ビオチンを、DNA分子に取り込ませると、例えば、平均して二、三百の塩基(必要とされるものに依っては、それ以上または以下)の間隔でビオチンを有する塩基があることになる。次に、このDNA分子は、ストレプトアビジンで被覆されるプレートに一端が固定される。この末端についての固定化機構は、ストレプトアビジン/ビオチンとは違うものである必要がある。
【0353】
2)この分子は、液体流、電界または他の手段の助けにより引き伸ばされる。このDNA分子を、ビオチン−ストレプトアビジン結合を創製する反応溶液を添加することにより基質に保留する。
【0354】
3)次にこのDNA分子を、DNアーゼIまたは他の手段で切断してから、遊離末端を、IIS型制限エンドヌクレアーゼ(示していないが)の結合部位を含有するプレアダプターに連結する。次いで、それぞれのエンドヌクレアーゼで切断することにより、配列情報を有するアダプター類に連結する張出し部が生じる。
【0355】
4)次に、ACGTの断片組合せをもつアダプターを、ACGT張出し部などに連結する。
【0356】
5)液体流、電界または同様の工程が、シークエンシングされるDNA分子の方向に対して、90°の方向にDNAアダプター類を引き伸ばすために使用されてから、それらを、ストレプトアビジン/ビオチン系で固着する。全てのDNA分子が、基質に固着されたら、この工程を、塩基対の所望する数が、転換拡大されるまで、繰り返すことができる。また、アダプター間の相対距離が、シークエンシングされるDNA分子中の配列片の内部距離に対応することを注目されたい。1枚の読み取りプレート上に、極めて多数のDNA分子を平行してシークエンシングすることが、勿論可能であることも言及するべきだろう。
【0357】
[実施例20]:標的分子から拡大部分の間隔を空けるリンカーの付いたアダプターを利用するシークエンシング法
以下に述べられた方法は、シークエンシングの1サイクル以上が達成できる方法を示している。この方法では、創製される拡大タグが、固形担体に付けられる。続いて、対応する配列からタグの間隔を空けるリンカーが取り除かれ、次に標的塩基配列の隣接部分が拡大される。この手順を図19に示す。
【0358】
《方法》
1)シークエンシングしようとするDNA分子、ACGTGAGCTの一端をストレプトアビジン被覆のプレートに固定する。その固定化機構は、ストレプトアビジン/ビオチン以外の機構である必要がある。
【0359】
2)結合部位の外側に切断部位を有するタイプIIの制限エンドヌクレアーゼ(例えば、図19に示すようなBspMI)に係る結合部位を含むポリヌクレオチドにDNA分子を連結する。
【0360】
3)次の段階で、制限エンドヌクレアーゼを加え、開裂が、シークエンシングしようとするDNA分子からの塩基を有する張出し部を形成する。
【0361】
4)次に、種々のアダプターとリガーゼを有する溶液を加える。図19は、ACGT張出し部を認識し、これと結合したアダプターを示す。ACGT張出し部に対応する蛍光標識した断片に加えて、2個以上のビオチン分子がアダプターに組込まれた。
【0362】
5)DNA分子を、液体流または電界の助けによって引き伸ばす。断片は図のように基質に固定することができる。(DNAリンカー領域の機能は、シークエンシングするDNA分子から断片の間隔を空けることである。これにより、次の段階における新たなアダプターのための余地が残る。)
6)SmaIおよびBspMIによって開裂が行われ、DNAの結合が取り除かれると同時に、DNA分子上に次の4塩基対から成る新たな張出し部が形成される。このことにより、新たなアダプターは、蛍光標識された断片と連結することが可能となる。唯一の違いは、このアダプターがDNAの結合を含まないことである。次に、蛍光標識したアダプターは、ストレプトアビジン基質上の新たな位置に固定されことになる。種々の長さのDNA結合を使って、複数の連続的な転換サイクルを実施することが可能である。
【0363】
[実施例21]:シークエンシング法における位置マーカーの利用
この方法では、シークエンシングされる分子に、位置マーカーが付され、得られる配列情報の位置を定める助けとなる。
【0364】
《方法》
用いられる方法を図20に示す。出発点は、例えば、100kbの環状標的DNA分子である。この分子は、明るい灰色および暗い灰色で印を付した2種の配列を含んでおり(図20)、これは、位置マーカーとして利用されることになる。
【0365】
1)DNA分子をBst71Iメチラーゼでメチル化する。
【0366】
2)上記分子をDNアーゼIまたは同様のもので線状化し、その後Bst71Iに対する開裂部位を含むアダプターを連結反応により付加する(標的DNA分子内の最初の4塩基を有する張出し部を作るために利用できるように、切断部位を配置する。それによって2つの4bp張出し部は、8bp上の連続的配列についての情報を提供できるようになる)。
【0367】
3)Bst71Iによって開裂を行い、断片アダプターを付加して連結する。
【0368】
4)DNA分子を1本鎖形に転換してから、モレキュラーコーミング(molecular combing)電界または同様の手段で、スライド上に固定し、引き伸ばし、同時に断片と位置マーカーを認識する蛍光プローブとハイブリダイズする。また、DNA分子をYOYO−1または同様のもので染色するのも適切であると思われる。
【0369】
5)次に、蛍光顕微鏡/スキャナを使って、配列片を走査し、位置マーカーに付いていたプローブまでの距離を測定する。これにより、シークエンシングするDNA分子上の8bpの配列片各々におおよその位置を割り当てることができる。
【0370】
[実施例22]:ソーティング後の拡大を含むシークエンシング法
この方法では、断片は、それらの末端の4塩基によって固形担体上にソーティングされた後、分子の終末端が固形担体に付着される。次いで、隣接する4塩基が、拡大により評価される。この方法の例を図21に示す。
【0371】
《方法》
256のアドレスに分けたDNAチップを使う。各アドレスは、ソーティングアダプター、張出し部、クラスIISの制限エンドヌクレアーゼに係る結合部位および平滑末端切断を行う制限エンドヌクレアーゼに係る結合部位を含む。張出し部は、アドレスによって変化するため、アドレス1は、AAAA張出し部の付いたソーティングアダプターを有し、アドレス2は、AAAC張出し部を有するものとなる。さらに、全てのアドレスは、結合性を有する分子、例えば、ストレプトアビジンで被覆される。
【0372】
1)標的DNAを断片に切断し、4塩基の張出し部を形成するようDNA断片の末端を処理してから、ソーティングが開始される。
【0373】
2)断片が連結されるソーティングアダプターを担持する固形担体に導入される。TTTT張出し部を有するDNA片は、ソーティングアダプターが、相補的な張出し部AAAAを有するアドレス1に結合し、GTTT張出し部を有するDNA片は、アドレス2に結合するなどとなる。
【0374】
3)DNA片上の他の張出し部は、その末端が下敷に固定できるように処理される。例えば、これは、Klenow充填反応により達成でき、該末端をビオチンで標識したり、ビオチン標識したユニバーサルアダプターに末端結合したりする。
【0375】
4)IISおよび平滑末端酵素による開裂を実施する(この場合は、FokIおよびDraIを用いて示される)。このようにして、DNA片内に次の4塩基を表す新たな張出し部が得られる。
【0376】
5)転換アダプターを加え、これらの塩基をシグナル鎖へと転換する。
【0377】
6)DNA分子が引き伸ばされ、例えば、蛍光スキャナを用いて走査する。DNA分子の一端の位置は、我々に4塩基についての情報を提供し、他の末端のシグナル鎖は、次の4塩基についての情報を提供する。
【0378】
[実施例23]:マッピング手順、プロフィル解析などのための標的分子調製方法
このプロトコルの背景となる原理は、自身の認識配列外側の切断を優先的に行う1種またはいくつかのヌクレアーゼ、例えばIIS酵素による標的DNAを分解することであるが、他の種類のヌクレアーゼもまた、例えば−5から+5の範囲の張出し部を作るために用いることができる。次に張出し部は、配列情報を含む一部分、および張出し部の性質についての情報を含む一部分(すなわち、どの制限酵素がその張出し部を作ったか)から成るシグナル鎖に連結される。分解された分子の各々は、これにより、末端のシグナル組成および両末端間の長さの異なる記号へと転換される。両端を相補的配列で並べることにより(例えば、温浸で創製された相補的張出し部に付着する1つまたは複数の拡大タグの整列)、この情報が制限マップを作るために利用することができる。この記号はまた、混合DNA集団における標的配列の同定に利用することもできる。FokIを利用する原理を図22に示す。
【0379】
水に溶解した100kbBACクローンである標的DNAにおけるFokI部位のマップを作りたい場合、以下のプロトコルが利用できる。
【0380】
1)FokI(New England Biolabs、#109)1ユニット、2.5μlの10×NE緩衝液4、1μgのBAC DNAに、水を加えて最終容量25μlとする。
【0381】
2)37℃で1時間インキュベートする。
【0382】
3)65℃で20分間加熱に不活化する。
【0383】
4)EtOHでDNAを沈殿させる。
【0384】
5)ペレットを水に溶かし、ホスファターゼ処理した転換アダプターを加え(転換アダプターと標的DNAとのモル比は、少なくとも50:1である必要がある)、200ユニットのT4DNAリガーゼ(New England Biolabs、#202)、2.5μlの10×T4 DNAリガーゼ反応緩衝液を加える。最終容量:25μl。
【0385】
6)16℃で4〜16時間インキュベートする。
【0386】
これで分析を行うことができる。しかし、非連結のアダプター除去の手順を実施することが好ましいと思われる。例えば、
7)5’の4塩基張出し部の全ての順列を含むアダプターで被覆した1.5〜2×108のダイナビーズM−280スレプトアビジン(Dynal、#112.05または#112.06)、2000ユニットのT4DNAリガーゼ(New England Biolabs、#202)、22.5μlの10×T4DNAリガーゼ反応緩衝液と水を加え、最終容量を250μlとする。
【0387】
8)16℃で4〜16時間インキュベートする。
【0388】
9)「Biomagnetic Techniques in Molecular Biology」第3版(ノルウェー、Dynal ASより配布)で説明されているように、磁石でビーズを沈殿させ、上澄液を除去する。
【0389】
10)上澄液中のDNAをEtOHで沈殿させる。
【0390】
11)DNA分子を適切な溶液と容量に溶解する。
【0391】
このプロトコルに使用される転換アダプターは、張出し部の256の全順列を表してもよいし、また、256の張出し部の部分集合、または1つ以上の縮退塩基を有する張出し部を表してもよい。分子を光学的マッピング法で解析しようとする場合、シグナルは、例えば、EcoRI部位のない0−シグナルおよび非常に多数のEcoRI部位を含む1−シグナルから成ってもよい。
【0392】
上記の手順を、分子を分析する前に他の制限酵素およびシグナル鎖のセットで1回または複数回反復してもよい。しかし、1回目のラウンドで生成した張出し部と連結しているシグナル鎖を、2回目に使用される酵素の分解から守る必要があることを指摘しなければならない。これは、該当部位をメチル化するなど2回目に使用される制限酵素に対する結合部位のないシグナル鎖を用いることにより達成できる。
【0393】
この手順はまた、ソーティング手順または各末端で得られる配列長を増加させる他の手順と関連して実施してもよい。
【0394】
一部は、標的DNAの生成された断片の他の末端に存在する制限部位を同定することにより、また、一部は、これらの断片が結合している相補的な張出し部および制限部位を含む他の標的断片を同定することにより、いずれの制限部位が特定の制限部位の横に位置するかを決めることにより、標的配列内の制限部位の位置を決定することができる。十分な制限酵素を使用することにより、非常に正確に該位置を決定することができるため、各断片の長さを決定する必要はない。
【0395】
光学的マッピングに関する従来の方法に比べて、この方法はいくつかの重要で有利な特徴を有する。
【0396】
1)分解能がはるかによい:わずか2〜3塩基対離れている制限部位間の識別が可能である。一方、従来の方法では、少なくとも二、三百塩基対離れていることが必要である。
【0397】
2)複数の制限酵素の使用によって制限マップの作成がより容易である。
【0398】
3)再構築に伴う統計学的問題が激減する。というのは、配列が、部分的にのみ切断されている制限部位間のDNA長についての不確かな測定よりもむしろ、シグナル鎖の組成に基づいているからである。
【0399】
4)自身の認識部位の外側で開裂する酵素を用いる場合、マップ内の各位置は、結合部位の長さに加えて、張出し部の長さ(通常9個以上の塩基)に基づいている。このように制限部位は、結合部位の配列および創製された張出し部に基づいて同定することができる。これは、希少な開裂(8塩基)の酵素によって作成された従来の遺伝子マップに比べて有利である。
【技術分野】
【0001】
本発明は、各塩基によって表される情報を効果的に増幅する新規のシークエンシング方法、および特に、配列部分の配列情報およびその配列内のその部分の位置情報を合わせ持つ長い核酸分子のシークエンシングに適した方法、およびこのような方法を実施するキットに関するものである。
【背景技術】
【0002】
WatsonおよびCrickが1953年にDNA分子の構造を明らかにして以来、遺伝子研究者等は個々のDNA分子の配列を決定する迅速かつ安価な方法を求めてきた。Sanger/BarrellおよびMaxam/Gilbertは、シークエンシング技術において飛躍的な進歩を示した2種の新しいDNAシークエンシング方法を1975年と1977年との間に開発した。今日広く使用されている方法は全て、Sanger/Barrell法に基づいており、過去23年間に開発されたDNAシークエンシング法は多かれ少なかれこの方法を変更したものである。
【0003】
しかし、1988年に、DNAシークエンシング技術は全く新しい目的を得た。合衆国を筆頭として、18カ国が共に科学史上最大の単一プロジェクトにおいて、数種の他のより小さなゲノムに加えて3×109bpの全ヒトゲノムのシークエンシング(Human Genome Project、HGPとも称する)に参加した。現在、この目的は2003年中に完了することになっている。このプロジェクトは多大な科学的資源を占有し、莫大な費用がかさむという事実にもかかわらず、このプロジェクトの利益はこの費用に見合うほど充分に重要であると考えられている。
【0004】
このプロジェクトの要となるのは、現在の技術よりも手頃な値段で、かつ迅速な新しいDNAシークエンシング方法の開発である。基本的に、これらはゲルをベースとした手法(主として新規のSanger/Barrell変法)およびゲルをベースとしない手法に分けることができる。ゲルをベースとしない手法は、おそらく大きな可能性を秘めており、質量分析法、フローサイトメトリー、および小さなDNA分子をハイブリダイズする遺伝子チップの使用が、目下試みられている手法のいくつかである。現行の方法よりも実質的に優れた方法があれば、多くの患者に遺伝子試験の機会を提供し、薬剤の同定および開発に重要な役割を果たすことが可能であるため、遺伝子研究だけでなく、現代医学にも変革をもたらすであろう。このような方法の経済的将来性も当然非常に大きい。
【0005】
現在公知のシークエンシング技術を使用すると、各シークエンシング反応で読みとれる配列の長さを延長することは困難であることが明らかになっており、現在使用されている方法のほとんどは、シークエンシング反応当たり約7〜800塩基対に限定されている。今日広く使用されている方法では、シークエンシング反応当たり複数の配列をシークエンシングすることは不可能である。
【0006】
多くの、または長い配列を決定するためには、一般に多くのシークエンシング反応を並行して実施することが必要である(例えば、60億塩基対の2倍体ヒトゲノムをシークエンシングするためには、数百万のシークエンシング反応を並行することが必要であろう)。行程の総数、酵素および試薬の使用、必要とされる独特なプライマーの数などが、実施するシークエンシング反応数に直接比例することが多いため、このことは大きな関門である。さらに、重複した配列を決定するためにしばしば資力を費やさなければならない。加えて、異なった種類の組織的な仕事、例えばDNAライブラリの構築およびソーティングを実施しなければならない。標的となる可能性のある配列が他の配列の中にある場合は、それを単離するために資力を費やすこともまた必要である。
【0007】
シークエンシング反応の長さを制限する基本的問題を説明するために、現在使用中および開発中のシークエンシング方法を2種の群に大別することが適切である(この分別から外れる個々の方法もあるが、少数である)。第1の群では、ポリヌクレオチドの大きさの範囲に基づく方法がある。出発点は、分子全部が1個の共通末端および1個の任意末端を有する1種以上のポリヌクレオチドの「梯子」を形成することである。例えば、SangerおよびMaxam−Gilbertの古典的なシークエンシング方法は、4個の塩基A、C、GおよびTの各々を表す4種の配列梯子に基づいている。
【0008】
読み取ることができるシークエンシング反応の長さに関して制限となる要素は、1個のモノマーだけが変化したポリヌクレオチド類を区別することができなければならないことである。配列梯子中のポリヌクレオチドが長くなればなるほど、ポリヌクレオチド間の大きさの相対的差違は小さくなる。したがって、分子の大きさを決定する方法のほとんどは、すぐに2種の近接したポリヌクレオチド間を区別することができないという限界にすぐに達する。
【0009】
もう1つの群は、異なる原理に基づく方法である。標的分子中に存在する短い配列断片を同定することによって、配列断片間の重複を利用して標的配列を再構築することができる。
【0010】
したがって、多くのシークエンシング方法では、標的分子はより小さい断片に切断され、各断片の組成が論理的に導き出され、重複した配列を発見することによって元の配列が構成される。例えば、ミクロアレイ(microarrays)では65,536のアドレスが生じ、各アドレスは特有の8量体を含む。したがって、8量体(48=65536)の順列が網羅されている。次いで、この標的分子を蛍光で標識し、8量体とハイブリダイズさせる場合、標的配列にどんな配列断片が存在するかについての情報は、蛍光で標識したアドレスを記録することによって得ることができる。
【0011】
読み取ることができるシークエンシング反応の長さに関して重大な制限となる要素は、以下の組み合わさった問題である。実施するシークエンシング反応が長くなればなるほど、標的配列の再構成を可能にするための配列断片が長くなる。しかし、検討されなければならない順列の数は、同定すべき配列断片の長さとともに指数関数的に増大する。これによって、対応する方式ではミクロアレイでの特有のアドレスの必要性が増加する。
【0012】
ミクロアレイの他の用途は、例えば集団中の遺伝子変異のスクリーニングによる公知の配列の再配列である。この目的のため、必要なアドレスの数を減少させ、同定する配列断片の長さを増大させることができるように、オリゴヌクレオチドを公知の配列に適合させることができる。しかし、特定の目的のためのミクロアレイを考案することは高くつき、資源が必要で、現在あるのは2、3のDNA配列用のミクロアレイだけである。ヒトゲノムは約100,000〜140,000遺伝子から成り立っているため、この方法でヒトゲノムの大量シークエンシングをすることは非常に多くの資源が要求されるであろう。
【0013】
ミクロアレイを使用する他の欠点は、現在の構築技術(例えば、フォトリソグラフィ)の限界のため、約10×10マイクロメートル未満の画素(pixels)を生じることが不可能なことである。したがって、蛍光スキャナの解像能力のごく一部しか利用されない。現在の蛍光スキャナは0.1×0.1マイクロメートルの画素を区別することができ、ミクロアレイは現在含む情報よりも10,000倍の情報を含むことができることを意味する。
【0014】
したがって、前述の組み合わさった問題を回避できる長い配列断片を同定する新しい方法/原理を開発することが有利であろう。同様に、同定しなければならない配列断片の長さが、標的配列の長さに対して指数関数的に増加することのないように長い標的配列の決定を可能にする新しい方法/原理の開発が有利であろう。
【0015】
配列断片の同定に基づく(例えば、米国特許出願第5,714,330号で実施されているような)別のシークエンシング方法は、読み取りプレート上に断片化した標的DNAを分配することから成る。その後、1個または数個の最初の塩基対を表す蛍光シグナルを標的DNAに固定するようにこの標的DNAを処理する。各位置の蛍光シグナルは、標的DNAの次の塩基対についてこの手順を繰り返す前に読み取る。DNA分子を読み取りプレート上の位置に固定されたとき、いくつかのサイクルを行うことによって配列情報を有する長い断片の構築が可能になる。
【0016】
サイクル当たり数個の塩基対を読み取る能力は、必要とされる特有の蛍光シグナル数が塩基対の数に対して指数関数的に増加するために限界がある。1塩基対を読み取るために、4色が必要で、2個には16色、3個には64色などとなる。現在の技術で64色の異なる蛍光色を区別することが可能であるかどうかはよくわからない。とにかく、必要とされる読み取り時間および費用は多数の色を使用することでかなり増加するであろう。したがって、解決策は多くのサイクルを実施することであろう。これは、次には酵素的段階および蛍光読み取りの数が増大することを意味する。
【0017】
前述の戦略によって比較的長い配列断片の同定が可能であっても、再構成には重要な問題が待ち受けている。生物学的DNAは組成において非常に偏りがある。短い配列、および長い配列が、「巨視的」および「微視的」レベルにおいてにいくつかの箇所で繰り返されていることが多い。繰り返しDNA配列の領域では特に再構成は困難である。これらは、生物学的に興味深い可能性が多く、例としてはトリヌクレオチドの長さが「繰り返されている」。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】米国特許出願第5,714,330号
【発明の概要】
【発明が解決しようとする課題】
【0019】
前記の問題を克服する新しい試みが現在開発されている。驚くべきことに、位置情報(すなわち、標的配列内の該配列位置の情報)に関連付けられた配列情報が得られたならば、長い配列を正確に同定することができる。さらに、本発明は、位置的情報を伴って、または伴わないで使用することが可能な新しいシークエンシング方法を提供し、本明細書では拡大と称した、1種または複数の塩基に関連したシグナルの増幅が行われる。
【課題を解決するための手段】
【0020】
したがって最初の観点からは、第1態様によれば、本発明は少なくとも標的核酸分子の全部または一部のシークエンシング方法において
a)前記核酸分子の部分の配列を決定する段階と、
b)前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階とを含む方法を提供する。
【0021】
好都合には、複数の部分配列およびその位置が決定され、この情報が組み合わされる。
【図面の簡単な説明】
【0022】
【図1】2種類の制限酵素、ハイブリダイゼーションおよび連結を使用して標的配列に拡大タグを導入する方法を示す図である。
【図2】2つの制限部位を運ぶベクターを使用して、標的配列に拡大タグを導入する方法を示す図である。この部位は両方とも張出し部、およびこれもまたそのような制限部位を含むアダプターになる。図2Aは標的DNAを含む塩基ベクターを示し、図2Bは使用することができるアダプターを示し、図2Cは各塩基を拡大するために使用される4つの酵素段階を示す。
【図3】プルーフリーディングのメカニズムによって、サイクルあたり9個の塩基をシークエンシングする方法を示す図である。図3Aは使用されるアダプター、図3Bは各塩基を拡大するために使用される酵素段階を示す。
【図4】標的分子の末端塩基を同定するために制限部位を完成させることを含むシークエンシングの方法を示す図である。A)はこのプロセスで使用されるアダプターを示し、B)は制限部位の一部を含むリンカー分子を示し、C)は標的DNAに結合し制限部位を完成させるリンカー分子を示す。
【図5】ハイブリダイゼーションおよび連結と結びつくKlenow修復反応を使用するシークエンシングの方法を示す図である。A)はその内部に標的分子が連結した塩基ベクターを示し、B)はサイクル1、3、5などに使用されるアダプターAの構造を示し、C)は大きさが同等な張出し部が形成されたかどうかに応じた、失敗した連結および成功した連結を示す。
【図6】一本鎖標的分子に結合するときにプライマーとして機能するアダプターを使用するシークエンシングの方法を示す図である。A)は標的分子に結合するためにプライマーとして使用されるアダプターを示し、B)は標的分子に結合するアダプターを示す。
【図7】拡大タグを保有し標的にハイブリダイズしセルフ−ハイブリダイズする、隣接して一直線に並べられたアダプターを示す図である。
【図8】複数の塩基に対応する拡大タグを含むアダプターの作成方法を示す図である。
【図9】ヘアピン型アダプターの使用に基づく転換方法を示す図である。拡大タグは標的分子の両末端にコピーされる。
【図10】転換されたDNA断片をお互いに連結させることに基づく転換の方法を示す図である。
【図11】核酸分子を2倍にする方法を示す図である。
【図12】プライマー依存のソーティング方策を使用するDNAシークエンシングのための方法を示す図である。A)は一般的な手順を例示し、この手順ではシークエンシング用プライマーが固形担体に結合され(段階1)、ここに標的分子が結合され(段階2)、ポリメラーゼ伸長によって伸長され(段階3)、次いで異なるシグナルを担持するプライマーが結合され(段階4)、鎖が延長され(段階5)、その結果生じた伸長生成物は次いでソーティング用に切り離される(段階6)。B)は、異なるシグナルが付着したウェル1において使用するための代表的なプライマーを示し、C)は16ウェルそれぞれについて得られる蛍光情報を示す。
【図13】ハイブリダイゼーションおよび得られた結果に基づくソーティング方策を使用する、DNAシークエンシングのための手順を示す図である。A)は次いで伸張される複数のアドレスを有する固形担体上での、一本鎖の蛍光標識された標的分子とオクタマープローブの結合を示し、B)はスキャン表面からの部分を示し、これは蛍光シグナルが各アドレスにおいて異なる長さに対応して直線状にどのように分配されているかを例示し、C)は1つのアドレスにおける蛍光の強度を示す。
【図14】標的DNA分子が固定リファレンスポイントに付着されるDNAシークエンシングの方法を示す図である。A)はPNAおよび対応する組成を有する一鎖の拡大タグ(ここではシグナル鎖として同定された)からなる転換アダプターを示し、B)は標的DNAの固形担体への結合、PNAアダプターとの接着、および伸張を示し、C)は分子が伸張された後の走査表面の外観を示す。
【図15】ナノポアシークエンシングの前のバイナリー末端転換の方法を示す図である。この中で転換アダプターを含む標的分子を生成するための手順、および方向タグに加え位置および配列の情報からなる生成したシグナルを示す。
【図16】塩基配列を表すシグナルを発生させるために、細胞がどのように利用されているかを示す図である。A)はエンハンサーがどのようにしてレポーター遺伝子型の拡大タグを保有する標的分子に加えられ、その後プライマーが付着されるかを示し、B)はシグナル分布を例示するヒストグラムを示す。
【図17】3つの拡大塩基各々と異なるシークエンシング梯子を作成することによるシークエンシングの方法を示す図である。この方法では結合標的分子が非特異的に切断され、拡大タグを付着させるための張出し部を発生させるために切断されるアダプターに結合する。
【図18】シークエンシングの方法を示す図である。固体担体に付着されるDNA分子の長さに沿って固定点を作成し、断片にされたDNAの末端が拡大される。
【図19】シークエンシングの方法を示す図である。この方法では拡大タグの鎖が固形担体に連続的に付着され、DNAリンカーを含むアダプターが使用され、DNAリンカーはシークエンシングされているDNAからのシグナルに一定間隔を置き、次のサイクルでシークエンシングされている分子へのアクセスを可能にするように取り除くことができる。
【図20】シークエンシングされる断片を含む分子に位置マーカーを導入する方法を示す図である。断片はアダプターによってシークエンシングされる。
【図21】ソーティングの方法を示す図である。この方法では固形担体上に存在するアダプターに標的分子が結合される。次いで分子の一末端が固形担体に結合され、他端が切り離され隣接するDNA鎖の転換を可能にする。(4)は、伸張後に生成したDNAを示す。
【図22】マッピング手順用の標的分子の調製方法を示す図である。この手順では数種の制限酵素が使用され、長さおよび方向の両方が異なる張出し部(3’または5’張出し部)を生成し、これは次いで拡大タグの鎖に連結される。
【図23】拡大の一般原則を示す図である。この原則では標的DNA分子の4つの最外部塩基が、拡大タグの鎖を用いた連結反応によって拡大される。拡大されない標的DNA分子の部分は、図示されるような位置情報を得るために、この場合光学マッピングに基づく方策を用いて読み取ることによって、使用することができる。
【図24】本明細書に記載されるソーティング方法を示す図である。この方法はミクロアレイで行われ、標的DNA中の4塩基の張出し部が256のアドレスを有するミクロアレイと混合され連結される。アドレス1はAAAA張出し部を含み、したがってTTTT張出し部を有する標的分子と結合する。
【図25】配列情報(左)および位置情報(右)の両方を得るために、シグナル鎖がどのように使用されるかを表す例を示す図である。A)は、特有なパターンで標的分子に結合する蛍光標識されたプローブを使用する、DIRVISHベースの方法であり、B)は光学マッピングに基づく方法を示し、この方法では配列の位置を与えるために制限パターンが使用され、C)はDNA結合タンパク質がミクロ/ナノ−ポアを通過する際に、その特有なパターンが記録される方法を示し、D)は、蛍光検出器を通過する際に記録される蛍光標識プローブ、タンパク質などを使用する方法を示す。
【発明を実施するための形態】
【0023】
本明細書では、標的核酸分子は、天然に生じるか、または合成したポリヌクレオチド分子、例えばDNAであり、これはゲノムまたはcDNA、RNA、例えば、mRNA、PNAおよびそれらの類縁体のことであり、適切ならば1本鎖、2本鎖および3本鎖であることが可能である。シークエンシングする部分には、標的分子全てが含まれることが好ましいが、例えば完全な分子より小さい、例えば4塩基から1kb、例えば4から100塩基であることも可能である。
【0024】
好ましくは、シークエンシングされる部分は、4個以上の塩基を有し、および/または前記部分の該標的分子内の前記部分の位置が1kb未満(すなわち1kb未満の解像度で)、特に好ましくは100塩基未満、さらに特に好ましくは10塩基未満の精度で決定される。数kb以上の解像度では、本明細書で記載した方法によって容易に実現される8〜10塩基より長い断片についての配列情報を得ることは通常不必要である。
【0025】
配列情報は、任意の便利な方法で得ることが可能で、1個または複数の塩基、特に好ましくは2個以上の塩基、例えば2から20塩基、好都合には4から10塩基について適切に得られる。理解されるように、配列部分の標的分子内の配置に基づいている前述したシークエンシング技法には、配列情報と同時にまたは別々に評価することが可能な位置情報を保持することは絶対必要である。いくつかの適切な技法を以下に説明する。
【0026】
位置情報は、いくつかの便利な方法で同様に得ることが可能で、これらの方法もまた以下に説明する。
【0027】
前述のように、本発明のこの観点では、得られる配列は、情報的に標的配列中のその位置にリンクしていなければならない。これはいくつかの方法、例えば核酸分子の末端領域または内部領域をシークエンシングすること、および位置についての、例えば分子の大きさ(例えば、長さまたは量)、発生したシグナルの強度または位置マーカーまたはアンカーまでの距離を参考にしてその位置を確立することによって実現することが可能である。配列情報は、1種または複数のシークエンシング反応サイクルを実施することによって得ることが可能である。
【0028】
前述したように、長い分子のシークエンシングにおける難点の1つは、長くなるにつれて、1塩基だけが変化した分子の大きさの相対的な違いを区別することが次第に困難になることである。一実施形態では、本発明は分子間の大きさ、強さ、長さまたはシグナルの違いを「拡大する」ことによってこの特定の問題を解決する。したがって、好ましい態様では、本発明はシークエンシングのサイクル当たり、2個以上の塩基(例えば、3個以上、好ましくは4個以上)の配列を決定し、および/または各塩基に関連したシグナルを拡大する標的核酸分子の配列の全部または一部を決定する方法を提供する。
【0029】
本明細書では、シークエンシングの「サイクル」とは、例えばシグナルを発生させること、または読み取ることによって、処理し配列情報を得ることが可能な最終産物を生じる一連の段階を実行することである。本明細書で記載した拡大方法およびシークエンシング反応では、複数のサイクル、例えば2個以上のサイクル、特に好ましくは4個以上、具体的には10個までのサイクルを実施することが好ましい。
【0030】
塩基に関連したシグナルの「拡大」とは、単一の塩基に関連する、または起着することが可能なシグナルの増強のことである。これは、例えば(このシグナルが塩基の大きさの場合)大きさの増加あるいは新しいシグナルの発生、例えば該塩基に標識もしくは他のシグナル手段を付加または結合させることであってもよい。
【0031】
同定された配列部分の長さを増加すると、大きさを決定する際の精度の低さを補うことができる。それによって、現在では精度が十分ではない大きさの決定法、例えば、フローサイトメトリ、DNA伸長などの方法を使用できると同時に、質量分析、ゲルによるソーティングおよび同様の方法の潜在力を利用することができる。
【0032】
分子間の違いを拡大することは、いくつかの方法で実現することができる。まず、得られた分子の長さが塩基2個以上異なり、したがって例えば、同時に位置情報をもたらすシークエンシング梯子における区別が可能になるように、サイクル当たり数個(例えば、4個以上)の塩基のシークエンシングを行ってもよい。(例えば実施例17参照)。あるいは、各塩基によって表される情報は、拡大することができてより容易に区別が可能になる。これらの異なる技法の例を以下に説明する。
【0033】
サイクル当たり2個以上の塩基(例えば4個)の配列を決定することは、任意の便利な技法によって実施することができる。位置情報に依存するシークエンシング方法では、得られる配列情報を標的分子内のこれらの塩基位置に再び関係付けることができるとの条件付きで、どんな技法を使用してもよい。このような場合、例えば相補的プローブ(例えば、固相担体に担持されている)へのハイブリダイゼーションを使用することが可能で、標的分子が結合するプローブを同定することによって、標的分子の末端配列が示される。例えば、2塩基の順列全てに相補的なプローブを、担持した、すなわち16個の異なるプローブを担持した固相担体を使用することが可能である。同様に、4個の塩基の順列全てに対するプローブ、すなわち256個の異なるプローブを相補的配列で標的分子を捕獲するために固相担体に付着させることが可能である。
【0034】
例示的手法では、(プローブはTTTTで終わるとき)AAAAで終わらない標的分子全ては結合せず、除去されるだろう。同様に、他のアドレスでは、特定の末端配列を有する標的分子が選択的に結合するであろう。この標的分子は、配列がそれぞれ末端または内部でも結合でき、さらに同定することが可能であるように(1本鎖が張り出した)2本鎖または1本鎖であることが可能である。PNAを相補的プローブとして使用する場合、このような分子は2本鎖形態に結合することが可能なので、2本鎖形態の内部配列もまた結合することが可能である。一般的用語では、この技法は本明細書では1個または数個の末端塩基対に基づいてソーティングするものとされ、1回または複数回のサイクルで実施することが可能である。この技法は、本明細書で記載した様な他の技法と組み合わせることが可能である。
【0035】
前記の逆の技法として、1本鎖標的DNAを固形基質に固定することによってシークエンシングを実施することが可能である。次いで、この標的DNAを、例えば16断片アダプターと共に混合しハイブリダイズさせることが可能である。アダプターは以下に説明するが、通常標的配列をシグナル増強または拡大された標的配列に適合させる分子のことである。次いで、ハイブリダイズしなかったアダプターは、溶液から洗浄される。これによって1本鎖DNAに相補的な張出しを有するアダプターだけが残る。例えば本明細書で説明した分析方法を使用して、溶液中にどのアダプターが残り、その結果16塩基対のどんな配列断片がこのDNAに含まれるかを確認することができる。
【0036】
このシークエンシング技法は、位置情報と組み合わせて実施するとき、またはシークエンシング技法だけで使用するとき、本発明の好ましい特性を示す。この方法で、単一の塩基によって伝えられる情報が、具体的にはその塩基を増幅すること、またはこの塩基をシグナルを発生させるために使用することができる「拡大」標識で置換することまたは増強することによって拡大される。(本明細書で意味する拡大はまた、いくつかの例では「転換」と称される。)
標的核酸分子の拡大は、増幅、例えば標的分子、またはシークエンシングする部分を含むそれらの部分を1倍または複数倍に倍加することによって実施することが可能である。分子間の、例えば10および11塩基の差を検出することは、拡大された分子、320および352塩基(5倍に倍加)間の差を検出することよりも困難であることは理解されよう。したがって、倍加の原理を使用して、例えばほとんどのDNA分析方法を改善することができる。これを実施するための適切な一技法を実施例11に説明する。他の適切な技法もまた使用することが可能である。
【0037】
したがって、この方法は、核酸分子間の大きさの違いを検出することに基づくほとんどの方法、例えばゲルをベースとするまたはベースとしない技法を改善するために使用することができる。この戦略によってまた、数個の塩基対の違いを区別するために感度が十分でない技法を使用して核酸物質を分析することが可能になる。例えば、改良したMaxam−Gilbert法が考えられ、そこでは1本鎖核酸分子(例えば5’−ビオチンを有する)を、ストレプトアビジンを結合したプレートに付着させる。次いで、シークエンシングを実施し、このプレートを洗浄し、その後得た核酸分子を例えば10倍に倍加して、1024塩基対の段階が得られる。これらの長さは、以下に説明した分析方法の1種によって測定することが可能である。
【0038】
したがって、他の観点から考えると、本発明は本明細書で記載したような標的分子の全部または一部をシークエンシングする方法を提供し、各塩基(または複数の塩基)に関連したシグナルは、前記塩基が前記配列に表れる回数の増加によって拡大する。
【0039】
本明細書では、特定の塩基(または1個以上の塩基)による「シグナル」とは、直接的にまたは間接的にその特性によって塩基(または塩基の集団)を検出する可能性のことを意味する。したがって、これは、直接的または間接的にあるいは1個または複数の他分子の結合によって、例えば前記塩基とシグナルが直接的または間接的に生じることが可能な標識部分との結合によって、検出することが可能な、大きさ、電荷または空間的配置といったその特性のことを意味することが可能である。したがって、直接的に検出可能なシグナルが与えられてもよく、またはシグナルを生じることが可能なシグナル伝達手段が提供されてもよい。このシグナルは、複数の塩基に特有であることが可能で、すなわちこのシグナルは1対の塩基対を示すか、または表すことが可能で、例えば、ATなどに使用するシグナルとは異なるAAに対するシグナルを使用することが可能である。このような分子および生じることが可能なシグナルに関連する、異なった機構については、以下に詳述する。
【0040】
さらに好ましい拡大技法には、1個または複数の特有のシグナル(またはこのようなシグナルを発生する手段)を配列中の1個または複数の塩基に関連させることが含まれる。前記シグナルが、複数の塩基に関連するとき、それぞれが1個または複数の塩基に対応する一連のシグナル(またはシグナル伝達手段)、または2個以上の塩基に特有な単一のシグナル(またはシグナル伝達手段)を使用することによってこれを実現することが可能である。これらのシグナルは、アダプター分子によって配列に付着することになるかもしれない拡大標識上にある。本明細書では、「関連(association)」とは、前記塩基(または複数の塩基)を前記シグナル(またはシグナル伝達手段)と置き換えること、および前記シグナル(またはシグナル伝達手段)を共存するように前記塩基(または複数の塩基)に付加することの両方を意味する。このシグナル(またはシグナル伝達手段)は、関連する塩基(または複数の塩基)に直接的に付着(または特異的に置換)している必要性はなく、この関連は間接的に、例えば1個または複数の分子が介在していることが可能である。関連は適切な化学反応、例えば、疎水性、イオン性、共有結合性などによることが可能であるが、標的核酸分子または関与分子との共有結合性相互作用によることが好ましい。
【0041】
本明細書では、「対応する」とは、塩基とシグナル、例えば、特定の塩基の存在を示すものとして読み取ることが可能な拡大標識によってもたらされるシグナルとの間の関係のことである。あるいは、マッピング手順の場合では、これはヌクレアーゼとこのヌクレアーゼの切断を示すマーカーとして使用されるシグナルとの関係のことである。
【0042】
本明細書では、「拡大タグ」とは、単一の分子あるいは1個または複数のシグナルを生じるための手段をもたらすタグ部分を含む、例えば標識を担うかまたは標識が結合することが可能な部位を有する分子複合体のことである。1個または複数のシグナルを生じる手段は、配列情報以外の情報が必要とされる場合、例えば標的分子または使用した切断方法に関係する情報の指示体として、取り込ませることが可能である。例えば拡大タグがポリヌクレオチドのときには、この拡大タグは、本来、1個または複数のヌクレオチド塩基に特異的に結合する他の部分を本来有することが可能である。この場合、このタグはさらに本明細書で前述したアダプターを含むものと考えられる。あるいは、この拡大タグは、標的配列に結合することを可能とするアダプターに付着するか、または付着するための手段を含むことが可能である。
【0043】
一般的に言って、この方法の例は以下に説明することが可能である。標的核酸物質中の塩基対は、4塩基、アデニン、シトシン、グアニン、およびチミンのそれぞれを表す4種の異なるタグ(以後拡大タグと称する)と結合する。したがって、A−T塩基対の場合は、「拡大タグA」が伴い、C−Gには「拡大タグC」が伴うなどである。それによって、元の塩基の順番、例えばACGTTが、「拡大タグA」−「拡大タグC」−「拡大タグG」などで増幅される場合、新しいDNA分子が生じる。それぞれの拡大タグは、シグナルを発生する手段を提供し、好ましい場合ではポリヌクレオチド分子であってもよい。この場合、必要に応じて、4個のタグの長さは2塩基対から数百kbp(所望するならばそれ以上)で変化させることが可能である。対応的に、DNA断片はリポーター遺伝子および他の生物学的情報を含むことができるか、または公知の生物学的機能のない配列だけから成ることができる。
【0044】
任意の便利な拡大タグを使用することが可能であるが、もちろん少なくとも4個、すなわち各塩基用の特有のタグが存在することは、配列を決定する目的のためには、不可欠である。もちろん、使用するタグは、シークエンシング技法、およびこれを実施する場合は、位置情報を引き出すために使用する方法に依存する。
【0045】
タグは、いくつかの他の形態でもたらすことが可能である。このタグは、特定のシグナルを生じることによって、直接的または間接的に検出するための手段を有する、すなわち、このタグは1個または複数のシグナル伝達手段を含む。蛍光、放射線、磁気、常磁性、電荷、大きさ、および量が特性の例で、拡大タグの粒子に、それらを検出し、かつ互いの分離を可能にするために付けることができる。これらの特性は、拡大タグ上に存在する1個または複数の標識に存在することが可能で、そこから出るシグナルは直接的または間接的に検出することが可能である。適切な標識は、シグナルの発生によって直接的または間接的に拡大タグを検出および/または測定させるものである。このような標識には、例えば、放射標識、化学標識(例えば、EtBr、TOTO、YOYOおよび他の色素)、発色団または蛍光体(例えば、フルオレセインおよびローダミンなどの色素)、またはフェリチン、ヘモシアニンまたは金コロイドなど高い電子密度の試薬が含まれる。あるいは、この標識は、酵素、例えば過酸化酵素またはアルカリホスファターゼであることが可能で、酵素の存在は適切な実体、例えば基質との相互作用によって視覚化される。この標識はまた、シグナル伝達対の一部を形成することが可能で、この対の他の因子はすぐ近傍に導入することが可能で、例えば蛍光化合物およびクエンチ蛍光基質を使用することが可能である。
【0046】
標識はまた、異なる実体、少なくとも拡大タグの一領域、例えば拡大タグのペプチド部分を認識する抗体上に与えることが可能である。この拡大タグがポリヌクレオチドであるならば、標識を導入する一方法は、例えば標識を担う適切な結合相手、例えば蛍光標識プローブまたはDNA結合蛋白質を結合することである。したがって、あるいはこのタグは、例えばその配列によって、標識が付着することが可能な分子を有するか、またはそれ自身が、その分子であることが可能である。標識は、単一の分子または微粒子、ナノ粒子(nanoparticles)、リポゾームまたは他の適切な形態の担体として付着することが可能である。
【0047】
好ましい態様では、この拡大タグは少なくとも2塩基、例えば30から1000塩基、好ましくは6から100塩基、特に好ましくは10から30塩基の長さの核酸配列自身である。これらの配列は、例えばその配列に相補的な蛍光プローブ、蛋白質などの使用によって、それらに付着した1個または複数の標識を有することが可能で、それから1個または複数のシグナルが生じることが可能である。あるいは、蛋白分子は、タグを含むか、またはタグに付着することが可能で、例えば免疫試薬または他の適切な結合相手、例えばDNA、DNA結合蛋白質によって認識されてもよい。このようなタグ分子の他の特性はまた、(制限酵素または蛋白分解酵素による)切断パターン、電荷、大きさ、形状などを調べることが可能である。
【0048】
拡大タグはまた、シグナルを生じるために使用することができる配列によって、情報を含むことが可能である。したがって、別の戦略はリポーター遺伝子、cis−調節因子など含む鎖を創出することである。その後これらは、細胞中にトランスフェクト/形質転換することができ、例えばリポーター遺伝子またはcis−調節因子の構成物は1個または数個のシグナルに転換される。この技法は、形質転換/トランスフェクション段階を必要とするが、細胞は転換段階(すなわち拡大タグの付加)を含む完全なシークエンシング反応を実施するようにプログラムされていてもよい。蛍光蛋白質または蛍光標識することができる膜蛋白質を発現する遺伝子、抗生物質抵抗性などを発現する遺伝子を使用するなど、極めて広い範囲のシグナルが生じることが可能である。時間による変化および配列中の特定塩基の存在を示すその他の特性に加えて、シグナルの質、量および位置も使用することができる。
【0049】
多細胞生物または構造体もまた使用することができるが、この方法では単細胞を使用することが都合がよい。生きていない細胞の同等物はまた、例えばナノテクノロジー(nanotechnology)を使用して、標識またはシグナルの発生に使用することができる。適切であれば、生じたシグナルは、例えば異なるプロモーターを使用することによって、同定のために他の場所に向けることが可能である。この技法をどのように実施するかの実例を実施例18に示す。
【0050】
ヌクレオチド塩基の各々に特異的な4個の拡大タグを好都合に使用することが可能である一方、前述のように、適切ならば複数の塩基に特有のシグナルを生じるために使用することが可能な拡大タグを使用してもよい。したがって、例えば16個の異なる蛍光体を使用することができる読み取り方法では、2塩基対の全ての順列を表す16個の異なるシグナルを発生するために使用する16個の異なるタグを使用することが適切であろう。
【0051】
他の状況では、4個よりも少ない異なるタグを使用することが適切であろう。例えば、1個がA/Tのとき、他方はC/Gである2個だけの拡大タグである。あるいは、4個の拡大タグを作り出す4個未満の特別のシグナル事象を使用することであり、これらのシグナル事象の特定の組み合わせによって4個の特有のシグナルが発生する(この場合個々の塩基にタグが付いている)。例えば、シークエンシング情報は2進法に転換することが可能である。この方法では、アデニンは一連のシグナル事象「0」+「0」に転換することが可能で、シトシンは「0」+「1」に、グアニンは「1」+「0」に、およびチミンは「1」+「1」に転換することが可能である。基本的には、配列情報を読み取るための1種またはおそらく2種の色または特有のシグナルがあれば十分である。これは、次に、いくつかのシグナルを使用した場合よりも読み取りが速いと同時に、より費用のかからない蛍光スキャナを使用することができることを意味するだろう。少なくとも4種の特有の拡大タグをもたらすために、例えば2進法タイプの読み出し形態を作り出すために、空間的に配置された単一のシグナル伝達手段を使用することは、本発明の好ましい態様であり、すなわち、前記シグナルは前記拡大タグ上に特有のシグナルを発生する単一のシグナル事象から成るパターンを含む。この場合、シグナル事象とは、測定可能なシグナル、例えば単一分子または他のこのような標識からの蛍光のことである。複数の拡大タグ、好ましくは20から100個のタグを使用するとき、これらは、必要なとき、位置情報を保存するために、直線的に、例えば長いDNA断片として連結していることが好ましい。
【0052】
タグ(直接である必要はない、例えばアダプターを介して生じてもよい。)とそれが表す塩基(または複数の塩基)との関係は、例えば塩基−塩基相補性による特定の塩基認識に依存する。しかし、本明細書での相補性には、Watson−Crick塩基対におけるヌクレオチドの対合に加えて、ヌクレオシド類縁体、例えば標的核酸分子中の塩基に特異的にハイブリダイズすることが可能なデオキシイノシン、および特異的なハイブリダイゼーションをもたらす他の類縁体、例えばPNA、RNA、DNAおよびそれらの類縁体の対合が含まれる。
【0053】
したがって、例えばDNA、RNAまたはPNA配列、またはそれらのハイブリッド、例えば4から20塩基、好ましくは6から12塩基の長さのオリゴヌクレオチドから成るプローブであって、(相補配列が存在する)標的分子の特定領域に結合し、拡大タグ、または一連のタグを付着し、各タグが、プローブが結合する1個または複数のヌクレオチド塩基を表すプローブを使用することができる。この場合、このプローブは標的配列へのタグの結合を促進するアダプター分子として作用する。あるいは、縮重したプローブの混合物は、特定の部位、例えばNNNNAAで1個または複数の特定の不変塩基のみと共に使用することが可能である。存在する拡大タグの数は、それらの結合した、結合することになるプローブの特定塩基の数に対応することが好ましい。しかし、特有のタグが2個以上の塩基の順列に対応して形成される場合、必要とされるタグはより少ない。
【0054】
この技法は、標的分子の不連続的な領域または部分を同定したり、または標的分子の配列の全てまたは本質的に全ての配列を得たりすることに使用してもよい。
【0055】
しかし、より精密なシークエンシングの別法は、配列を得るために読みとることができるシグナルを出す隣接した拡大タグ鎖の生成によってもたらされる。この効果を実現するためには他の方法があるが、最も便利な技法には、拡大タグを標的分子に挿入することが含まれる。この反応は、転換およびその後の一連の塩基の配列読み取りを行いつつ繰り返して実施することが特に好ましい。
【0056】
拡大タグを、拡大すべき塩基(または複数の塩基)に関連した標的分子に挿入するために、その塩基(または複数の塩基)および周辺の塩基に対する相補性、または認識性を利用することが必要である。この相補性を使用して、直接拡大タグを導入することが可能で、または最終的にその塩基に対応するタグを導入する方法を開始することに使用してもよい(実施例4参照)。
【0057】
これは、拡大タグと結合することができる張出し(すなわち、1本鎖である領域)を標的核酸分子に生成することによって実現することが便利である。(しかし、このような張出しはタグ分子またはその中間体、例えばそのアダプターが2本鎖型、例えばPNAを認識しかつ結合する場合は必要ない。)一方法は、標的分子の末端を、認識配列の外側で切断する制限酵素、例えばIPまたはIIS型の制限酵素の結合部位を含む短いDNA分子とを結合することである。これらの酵素は、切断される配列に対して特異性を示さず、したがって全ての種類の塩基構成を有する張出しを形成することができる。実際の標的分子、例えばDNA分子を問題の制限酵素とインキュベートする場合は、張出しがDNAの内部に形成されるように、結合部位を位置させることが可能である。実際に、3〜4塩基対の張出しを生成する酵素を選択することは好ましいと思われる(増幅し、固相に付着した標的分子にこのような張出しを生成するための一般的手順を示した実施例19を参照のこと)。
【0058】
70種類以上のIIS制限エンドヌクレアーゼが同定されており、基質特異性および切断パターンに関しては多くの変化がある。さらに、これらの酵素は、特定の必要性のために新しい酵素を創造することができるような「モジュールスワッピング」実験に非常に適していることがわかった(Huang−B 他;J−Protein−Chem.1996,15(5):481−9,Bickle,T.A.;1993 in Nucleases(2nd edn),Kim−YG他;PNAS 1994,91:883−887)。したがって、これらの酵素の非常に多くの組み合わせおよび変異体が、本明細書に記載した原理にしたがって使用できる。
【0059】
IIS型制限エンドヌクレアーゼは、いくつかの異なる目的のために使用されてきた。例えば、所定部位のほとんどの部位で1本鎖基質を切断することができる万能の制限エンドヌクレアーゼとしてである(Podhajska,A.J.,Szybalski,W.;Gene 1985,40:175−182,Podhajska,A.J.,Kim,S.C.,Szybalski,W.;Methods in Enzymology;1992,216:303−309,Szybalski,W.;Gene 1985,40:169−173)。
【0060】
シークエンシングの状況において、それらは米国特許出願第5714330号に記載されている前述の方法に対して用いられてきた。しかしながらこれらの場合、標的分子に連結したままである複数の拡大タグの導入は考慮されていなかった。
【0061】
IIS酵素での開裂は、多様な長さ、例えば−5から+6塩基の長さの張出し部をもたらす。ひとたび張出し部が作られると、張出し部の1つまたは複数の塩基に対応して、アダプターに担われ得る拡大タグを張出し部に取り付けることができる。
【0062】
IIS系または類似の系を用いて拡大タグを組み込むことのできるいくつかの異なる方法を以下に記載する。
【0063】
記載する第1の技法は、1つまたは複数の拡大タグを担い、一本鎖領域、すなわち張出し部を生成するために変性された標的核酸分子に相補的な張出し部を有するアダプターを使用することを含む。アダプター自体もまた、張出し部を生成するために用いた酵素と同一でも異なっていてもよいもう一つのIIS酵素のための認識部位を担っている。この技法の例は、実施例1に例示する。
【0064】
簡潔には、標的配列は、挿入点の近くにそれ自体がIIS部位を担うベクターに連結されるか、または標的配列は、そのような部位を含有するように設計される。適切なIIS酵素は次に、適切に配置されたとき標的配列に張出し部をもたらすIIS部位の開裂のために用いられる。一実施形態において、切断ベクターの少なくとも1つの末端が、例えば、IISに隣接するもう一つの制限酵素部位の使用によって平滑にされる。
【0065】
適切なアダプターはその後、張出し部の1つまたは複数の塩基に結合し、それによって拡大を可能にするために用いることができる。単一塩基の拡大の場合には、例えば4塩基張出し部、ANNN、TNNN、CNNN、およびGNNNに対するその形態の一本鎖部分、および拡大タグA、T、C、およびGをそれぞれ有する縮重アダプターを用いることができる。別法として、このアダプターは複数の張出し部塩基に対応して複数の拡大タグ、例えばATGCの張出し部を、当該の場合、線状に付された1つまたは複数の塩基に対応する拡大タグとともに、担うことができる。
【0066】
アダプターの張出し部および開裂ベクターがハイブリダイズされると、これらの分子は連結することができる。これは、張出し部の全長にわたる完全な相補性が実現され、反応の特異性を助ける場合にのみ得られる。次に平滑末端連結を達成し、アダプターの他の末端をベクターに接合することができる。前に用いた酵素と同一でも異なっていてもよい、もう一つのIIS部位(または他の適切な制限酵素部位)の適切な配置によって、最初のアダプターが向けられた配列の下流にある標的配列に張出し部が作られるように開裂を達成することができる。このようにして、隣接または重複する配列は拡大タグを担う配列に連続的に転換することができ、続いてそこからのシグナルを読み取り、本明細書の以下に記載する方法によって配列を決定することができる。この重複配列のシークエンシングは、前サイクルにおいて読み取られた配列を効果的にプルーフリーディング(proof-reading)し、これにより確認を可能にする。
【0067】
平滑末端が生成されず、その代わりにベクターが生成され、IISまたは類似の酵素で開裂が行われた後、もう一つの制限酵素を用い、ベクター内に挿入されるすべてのアダプターの末端に対し普遍的に相補的である張出し部を作るこの技法のわずかな変更を実施例2に示す。この方法は同様にベクターへのアダプターの連結、したがって拡大タグの連結を可能にする。
【0068】
類似しているが、より複雑な実施例を実施例3に例示する。この場合、隣接するDNAストレッチに対応して非相補的な張出し部が作られる。これらはともに適切な拡大タグを付したアダプターにハイブリダイズされる。シークエンシングが一方向に起こるように、アダプターのただ1つだけが、次のサイクルのための制限酵素部位を含有する。明らかにこれらの異なる性質を備えたアダプターの結合を可能にするために、隣接するDNAストレッチの張出し部は、識別可能な、例えば異なる長さでなければならない。これは、異なる長さの張出し部をもたらす異なる制限酵素を用いることによって達成できる。アダプターの2つの異なる型の末端は意図的に相補的であり、それゆえハイブリダイズし、連結されてベクターを形成することができる。それを含有するアダプターの制限部位は、開裂部位がさらに標的配列内に移され、隣接部位のシークエンシングが可能となるように適切に配置される。
【0069】
したがって、例えば5+4の張出し部が作られ、開裂部位が標的配列内へ4塩基に移される場合、次の9塩基が張出し部に転換され、その後、拡大タグに連結されるとき、それらの塩基のうちの5つが前サイクルの拡大タグと連結する。これによって配列の読み取り時に、前の5塩基の同一性を確認することが可能になり、したがってプルーフリーディング機構が導入される。
【0070】
IIS系を用いる他の技法は、DNAポリメラーゼのKlenow断片を使用することを含み、ほとんどのDNAリガーゼが異なる大きさの張出し部を連結できないという事実を利用している。これは、例えば実施例5に示す。この技法において、アダプターの張出し部よりも長い張出し部が作られる。この標的張出し部は、1つのタイプのヌクレオチド存在下でKlenowによって減ぜられる。1つの塩基だけ適切に伸長された標的だけがアダプターに結合し、これによりそのアダプターに付された対応する拡大タグによって導入された塩基の同定を可能にする。
【0071】
実施例4〜7に例示した他の技法は、拡大タグを担うアダプターの一本鎖標的へのハイブリダイゼーションを含み、それは次に標的に連結される。次にアダプターを、二本鎖分子を形成するためのポリメラーゼ伸長反応のプライマーとして用いる。さらなる別法は、ソーティングアダプター(sorting adapter)(この場合、必ずしも拡大タグに連結している必要はなく、単にソーティングのために用いることができる)を用い、アダプターは標的分子上に作られた張出し部より多い張出し部を有する固形担体に取り付けられる。したがって、例えばこのアダプターは8〜10塩基の張出し部を有していてもよい。例えばDNA片(二本鎖形態)が4塩基の張出し部を有するならば、張出し部のもっとも内側の塩基において塩基が互いに相補する場合にのみこれらの分子は連結するであろう。その後、ポリメラーゼ伸長が行われる。ポリメラーゼ伸長反応を成功させる要件は、プライマーとして機能できるように、アダプターの張出し部の残りの部分がDNA片と相補的なことである。このように、ポリメラーゼ伸長は、標的分子の末端配列がアダプターの張出し部に相補的である場合にのみ起こる。
【0072】
別法として、ハイブリダイゼーション単独で用いることができ、隣接する配列のストレッチに連結する拡大タグを合わせて、都合よく連結することができる。
【0073】
さらなる別法は、代謝酵素の認識部位に対する代謝酵素の特異性を利用する。そのような技法を、制限酵素を用いる実施例に例示する。しかしながら、いくつかの代替酵素、例えばトランスウポゼース(transposase)なども用いることができる。この方法において、シークエンシングされる標的分子は、4つの異なる標準的制限酵素で開裂されて平滑末端を生成し、(開裂すると張出し部を生成する)4つの異なる制限酵素の1つに対する制限部位の部分でそれぞれが終わる4つの異なるDNA分子に連結される。
これらは次に、標的分子上に連結される。標的分子が制限部位の残りの塩基を与える塩基で終わっている場合、制限認識部位が生成される。これは、その制限酵素を用いる開裂によって決定することができる。完全な形態で認識部位を有する分子だけが開裂される。開裂された分子を認識するために、張出し部と相補的なアダプターを用いることができる。これらのアダプターは次に、制限部位を完成するために標的分子によって提供された塩基の数に応じて、1つまたは複数の適切な拡大タグを担うことができる。この分子は次に環状化されることができ、サイクリングの繰返しが可能となる。好都合には、アダプターはその配列内に、平滑末端および張出し部の両方を生成する制限酵素のために適切に置かれた制限部位を有し、それにより隣接または重複する標的配列領域に対応する拡大タグを導入させることで繰り返しサイクルを行うことができる。
【0074】
本発明はしたがって、一態様において、標的核酸分子の一部分を同定する方法に関連し、この方法において前記部分を認識し結合する部分、および1つまたは複数の拡大タグを含む部分、好ましくは前記部分内の塩基を表す前記タグの鎖を含むアダプター分子は前記部分に取り付けられる、あるいは前記部分を置換する。
【0075】
したがって好ましい観点から見ると、本発明は、標的核酸分子の配列のすべてまたは一部を拡大する方法を提供し、この方法において、1つまたは複数の拡大タグは標的配列の1つまたは複数の塩基に連結しており、前記タグは前記標識配列の1つまたは複数の塩基に対応している。好ましくは、前記拡大タグはともに、少なくとも2つ、好ましくは少なくとも4つの塩基に対応する。好ましくは、前記拡大タグはそれぞれ、少なくとも2つ、好ましくは少なくとも4つの塩基に対応している。代替の実施形態において、それぞれの拡大タグは1つの塩基に対応し、ともに少なくとも4つの塩基、例えば8から20個の塩基に対応する拡大タグの鎖が用いられる。これは、例えば各サイクルに単一の拡大タグを加えて複数のサイクルを行うことによって、または単一のサイクルにおいて連結されたタグの鎖を用いることによって達成することができる。
【0076】
好ましくは、
前記方法は少なくとも、
a)前記標的配列の少なくとも一部分を、アダプター分子を結合するのに適した形態、好ましくは一本鎖形態に転換する段階と、
b)段階a)で作られたアダプター分子を結合するのに適した前記領域、好ましくは一本鎖領域の少なくとも一部分に、1つまたは複数の拡大タグを含む、または1種または複数の拡大タグを付着するための手段を含むアダプター分子を結合する段階であって、前記タグが前記標的配列の1つまたは複数の塩基に対応しており、好ましくは、前記アダプター分子を結合するのに適した前記領域、好ましくは前記アダプター分子が結合する前記一本鎖領域、または前記領域の近傍の1つまたは複数の塩基に対応する段階と、
c)所望により、少なくとも前記拡大タグが前記標的分子に結合したままであるように、前記標的分子を前記アダプター分子に連結する段階と、
d)所望により段階a)を繰り返す段階であって、作られた前記アダプターを結合するのに適した前記領域、好ましくは前記一本鎖領域が、段階b)による拡大タグに結合していない1つまたは複数の塩基を含む段階と、
e)所望により段階b)からd)を繰り返す段階であって、前サイクルのアダプター分子が結合した領域に関係する前記標的分子の隣接、または重複する領域に前記アダプター分子が結合する段階とを含む。
【0077】
段階e)は、いくつかの技法において省略することができ、例えば、拡大およびソーティングを連結することによってシークエンシングが実行され、そのため1回だけ拡大サイクルが行われる場合である。
【0078】
アダプター分子を結合するのに適した形態への「転換」は、標的分子が適切な形態でない場合にのみ必要である。したがって、PNA分子を結合するために、二本鎖標的分子の転換は必要でない。同様に、分子が一本鎖である場合、オリゴヌクレオチドであるアダプターを結合するために転換は必要でない。しかしながら、いくつかの場合において、アダプターの特異的、選択的結合を可能にするために、例えばDNA断片を溶融することによる転換が必要とされる可能性がある。分子全体を異なる形態に転換する必要はなく、適切な場合においては、一部のみが転換される。この部分は、少なくともアダプターの結合部分の長さ、したがって好ましくは4から500塩基、例えば6から30塩基の長さを含むべきである。この文脈における一形態から他の形態への転換の言及は、拡大に関して用いられるときの転換という言葉の使用と混同されるべきではない。
【0079】
本明細書において「アダプター分子」とは、標的配列をシグナルが強化されたまたは拡大された標的配列に適合させる分子である。本発明で用いられるアダプター分子は、単一分子、あるいは同一の種類でも異なっていてもよい分子の複合体である。アダプター配列は、前記標的配列に結合する結合部分を含み、例えば、標的配列の1つまたは複数の塩基に相補的な特定の塩基配列、より好ましくはポリヌクレオチド配列を認識するタンパク質である。好ましくは、結合配列は3から30塩基、好ましくは4から10塩基の長さである。アダプター分子は、1つまたは複数の拡大タグ、またはそのようなタグを付するための手段を追加的に含み、例えば、相補的である配列、または結合パートナーである。好ましくは、アダプターは、1つまたは複数のヌクレアーゼ認識部位、特に好ましくはその認識部位の外側を開裂するヌクレアーゼのための制限部位(または、少なくとも認識部位)、特に好ましくは、IIS酵素またはその類似体、特にFokI、および本明細書に記載する他の酵素の制限部位を含有する。好ましくは、他の制限酵素に関する部位は、このアダプターから除かれる。
【0080】
好都合には、アダプター分子は専ら核酸分子からなり、アダプターの様々な性質はアダプターの異なる領域によって与えられる。しかしながら、前述したように拡大タグは多様な形態を取ることができ、例えばタンパク質などの標識が含まれる。アダプターはこのように、拡大タグが結合することのできる分子を提供することができ、例えば、標的に結合するための領域に加えて、適切な結合パートナーを提供する。
【0081】
段階c)において、「少なくとも」前記拡大タグが連結したままであることが示される。したがって、アダプター、またはその部分は除去できることが考察される。
【0082】
本明細書において拡大タグの「鎖」は、拡大サイクルの前に結合させ、1つのアダプターに取り付けるか、または各サイクルの終わりに合わせて結合させたタグ、あるいは両方の組み合わせを指す。この結合は、任意の適切な手段によることができるが、しかしながら共有結合の手段による取り付けが好ましい。
【0083】
好ましくは、上述の方法は、前記標的配列に取り付けられた拡大タグから発生したシグナルを同定することによって前記標的分子の配列を決定することに加えて、上述の段階を含む本発明のシークエンシング法において用いられる。拡大タグを同定するために、読み取り可能なシグナルは拡大タグから発生されなければならない。これは、例えばタグがある種の性質を備えた標識(例えば、放射性標識)を担う場合のように、本来的に存在してもよく、あるいは、例えばさらなる分子の付加(例えば、それ自体が標識を担う結合パートナー)、または拡大タグの読み取り可能な形態への処理のような(例えば読み取り可能シグナルへの転換、具体的には読み取られるシグナルが発現タンパク質であるようなレポーター遺伝子の発現による)シグナル発生のためのさらなる段階を必要としてもよい。
【0084】
したがって好ましい態様において、本発明は標的核酸分子のすべて、または一部分をシークエンシングする方法を提供し、この方法において、好ましくは標的配列の1つまたは複数の塩基に連結した1つまたは複数の拡大タグの使用によって、前記標的核酸分子の配列の少なくとも一部分は拡大され、前記拡大配列は所望により読み取り可能なシグナルに転換され、前記配列は発生するシグナルの評価によって決定される。
【0085】
本明細書において「評価」は、絶対的または相対的に決定することのできる定量評価、および定性評価の両方を指す。
【0086】
連結は、化学的に、あるいは適切な天然にあるリガーゼまたはその変異体を使用することによって達成することができる。連結は本発明の好ましい特徴のみを表すものであるが、これは特異性を高めるために都合よく用いられる。ハイブリダイゼーションと比べて、連結がT4DNAリガーゼに基づく場合には、特異性は10倍に増大する。多くの場合ハイブリダイゼーションに基づくシークエンシング法は、許容できない高いエラー率を伴うためにこのことは重要である。さらに、耐熱性リガーゼ、例えばPfu、Taq、およびTTH DNAリガーゼなどを使用することによって特異性が向上し、一方で劇的に効率が上昇し、結果としてインキュベーションの時間が低減される。
【0087】
拡大タグを用いるこのシークエンシング法は、既知のシークエンシング法に対していくつかの利点を提供する。各サイクルにおいて複数の塩基を転換、または拡大することができ、したがって標的分子の特定の長さをシークエンシングするために必要なサイクルの数が低減される。拡大タグの選択、およびそれらが生成するシグナルに応じて、単純化された読み出しシグナルを生成することができ、例えばこのシグナルは二進法の読み出し形態であってもよく、すなわち単一のシグナル事象、例えば蛍光の適切な組み合わせ、例えば線状もしくは位置レイアウトによって1つまたは複数の塩基に対して特定のシグナルが発生される。これは、必要とされる特定のシグナル事象の数を低減する。したがって、例えば2塩基の組み合わせのそれぞれに16個の異なる標識、あるいは3塩基の組み合わせのそれぞれに64個の異なる標識を必要とする代わりに、本発明においては、各拡大タグ上に単一のシグナル事象を生成する手段のパターン、例えば、蛍光プローブを結合するための部位のパターンを提供することによって、16、または64またはそれ以上の特定のシグナルを発生させることができる。
【0088】
このシグナル情報は、緊密にパックされていることが可能である。タグは標識ヌクレオチドのみに限定されず、使用できる拡大タグおよび発生できるシグナルの種類において、より大きな融通性が与えられる。いくつかの実施形態において、サイクリング(cycling)が行われないときでも、配列の大部分を、それらの部分に拡大タグの鎖を用いることによってシークエンシングすることができ、したがって繰返しサイクリングに含まれる複雑な反応、および、シグナルを読み取ることのできる方法を限定する(例えば、ミクロ/ナノポア、またはフローサイトメーターを用いることはできなかった)ような、例えば読み取りプレートに固定された標的分子を用いて各サイクルからの情報を特定の標的配列に関連づける必要性が回避される。
【0089】
本発明の好ましい態様において、少なくとも部分的に一本鎖である形態への標的分子の転換は、一本鎖分子を用いて、あるいは例えば認識部位から離れた開裂部位を有する適切なヌクレアーゼ、例えばIIS酵素などを用いて張出し部を作ることによって達成される。
【0090】
好ましくは、この反応がサイクル的に行われるとき、各サイクルの拡大タグは、例えば会合もしくは連結によって、例えばそれらを含有する一本鎖の生成によってともに接合される。さらに、前記標的分子の前記アダプター分子への連結後、結果として生じた前記分子は、好ましくは環状化される。好都合には、これは標的分子をベクターに導入すること(または、標的分子の一部分を担体に取り付け、分子内で開裂後の自由な相互作用を可能にすることによって、実施例22を参照)、および前記アダプター分子が導入された後に、開裂および連結の適切な工程を用いることによって達成される。別法としては、生成される拡大タグの鎖を、有効な環状化を必要とせずに、標的分子の遠隔部位に転移または複写することができる。これを実行するための適切なプロトコルを実施例9に例示する。
【0091】
過剰なサイクリングの必要性を回避する別の好都合な技法は、より小さな転換断片、すなわち拡大タグが取り付けられた核酸分子のハイブリダイゼーションを含む。これらの断片は、それら自体が1つまたは複数の転換サイクルに供することが可能であったし、その後、非転換配列または拡大タグに担われた情報、例えばタグのヌクレオチド配列に、相補性によって結合することができる(実施例10を参照)。
【0092】
この反応のサイクリングを達成するために、反応に用いられる特定の酵素の制御が必要とされる。これは、用いられた酵素に応じて、異なった方法で達成することができる。したがって、結合および/または制限部位における開裂を防ぐためにメチル化を用いることができる。末端塩基のリン酸化状態を制御することによって、例えば、キナーゼ、またはホスファターゼの適切な使用によって、連結を防止する、または可能とすることができる。分子間連結を回避するために、適切に大容量を用いることもできる。効率を上げるために、制限反応の間、好ましくは少容量が用いられる。
【0093】
好ましくは、拡大の各サイクル(または本明細書に記載のシークエンシング)において、サイクル当たり少なくとも2つの塩基、好ましくは3と100個の間、特に好ましくは4から20個の塩基が転換される。好都合には、複数の拡大タグが、各サイクルにおいて1つまたは複数の塩基に連結される。例えば、好ましい実施形態において、各々が1つまたは複数の塩基に対応し、集合的に前記配列の一部分に対応するタグの集合(例えば、線状の列または鎖)を導入し、例えば4から12個の隣接塩基に対応する、具体的には複数のタグ、例えば4つを超えるタグである。好都合には、これは、それ自体が複数の塩基に向けられているタグ、例えば各塩基対に対する特定のタグと結合させることができる。
【0094】
実施例1に示すように、好ましい実施形態において、張出し部を形成するために上述の性質を有するヌクレアーゼが用いられる。さらに、前記ベクターは追加的に、張出し部を生成するためにヌクレアーゼ開裂から生じる末端の1つにおいて平滑末端開裂を生成するための制限酵素部位を含有する。別法として、最初の張出し部を作るために用いた酵素とは異なる制限酵素を用いることができ、この反応に用いたすべてのアダプターの一末端に対し厳密な相補性を有する張出し部を生成する。
【0095】
実施例3の方法を行うために、好都合には張出し部の隣接もしくは重複の領域を生成するヌクレアーゼ部位を用いる。これらの部位は、好ましくは用いられるアダプター内に位置する。各サイクルにおいて2つのアダプターが用いられ、それらは都合よく、末端にある相補的張出し部を用いることによって、標的配列の一本鎖部分に結合している領域に、ともに連結することを可能にする。したがって本発明の好ましい態様において、特にプルーフリーディングを可能にするために、用いられるアダプターは、認識部位から離れた開裂部位を有する2つ以上のヌクレアーゼに対する認識部位を含み、前記ヌクレアーゼでの開裂は、隣接し、または重複している一本鎖領域を生成する。本明細書において「重複」は、共通して塩基を有する配列、またはそのような配列に相補的である、すなわち対応する鎖上の配列を指す。したがって、重複領域を得るために二本鎖の標的の各鎖を利用することができ、重複しているが、相補的な領域をシークエンシングすることができる。好都合には、この作用を得るために各サイクルにおいて複数のアダプターを標的分子に結合される。この方法は、重複領域がシークエンシングされる場合、特定の塩基または塩基の集合体に対応に複数の拡大タグが取り付けられ、その塩基に対する繰返しシグナルを発生させるため、プルーフリーディングが可能となる。本発明によって、塩基当たり1つのタグが必要とされず、それゆえ塩基対などに対するタグを繰り返すことができることが理解されるであろう。
【0096】
Klenow断片の使用を包含する実施態様の実行において、段階a)において作られる前記一本鎖領域は、アダプター上に存在する核酸の一本鎖領域よりも長い1つまたは複数の塩基である。さらに、段階b)の後に追加の段階が必要であり、そこまでは標的分子の一本鎖領域の長さはポリマー伸長反応によって短縮される。
【0097】
一本鎖の標的分子を含む技法を実施するために、好都合には、サイクリングは、好ましくはポリメラーゼ伸長反応におけるプライマーとしてアダプターを使用することによって、二本鎖分子を生成することを含む。
【0098】
認識部位が完成され、その部位を完成するために必要な末端塩基を有する分子が同定される方法は、上に一般的表現で記載したものとはわずかに異なる技法を提供する。アダプターは張出し部に結合するが、アダプター分子が結合する一本鎖領域の1つまたは複数の塩基に必ずしも対応しなくてもよいタグを担うからである。この一本鎖領域は、標的配列のいくつかの塩基を含む制限部位の開裂によって作られた張出し部から構成される。しかしながら、開裂部位に応じて、それらの塩基は一本鎖形態であっても、そうでなくてもよく、例えば、張出し部はすべて非標的分子の塩基からなることができる。その代わりに、この適切なタグの付加は、制限部位が完成している場所にのみアダプターが結合するという事実を利用している。したがって段階b)は、前記一本鎖領域、または前記領域の近く、具体的には前記領域に隣接する1つまたは複数の塩基に対応するタグの参照を含む。さらにこの方法において、段階a)に先立って、代謝酵素認識部位の一部を含むリンカーDNA片を前記標的分子に取り付け、次いで前記酵素、例えばヌクレアーゼを使用し、段階a)の一本鎖形態を生成する。
【0099】
前述のとおり、シークエンシングはソーティングに基づいて実行することができる。この方法は、独立して、または上に記載した拡大技法と組み合わせて用いることができる。例えば、シークエンシングプロトコルは、4つの塩基対に基づいて標的核酸分子をソーティングすることによって実行することができ、その後、隣接する塩基対を転換して、その配列を決定することができる。例えば、ソーティング戦略は、前に述べたように、標的核酸分子に4塩基を備えた張出し部を作ることから構成され得る。次にそれを、短鎖DNA分子、ソーティングアダプターによってすべて覆われた256ウェル(wells)の中に分配される(これらのアダプターは必ずしも拡大タグを担っている必要はない)。このソーティングアダプターはウェルの壁に固定され、標的DNA上に作られた張出し部を相補することのできる4塩基を備えた張出し部を有する。さらに、ソーティングアダプターは、IIS酵素、または他の適切なヌクレアーゼのための結合部位を含有することができる。この結合部位は、標的DNAに作られた最初の張出し部の近くに位置する塩基対を備えた張出し部を各々のIIS酵素が作ることができるように置かれる。ソーティングアダプターを備えた表面積を増大するために、別法は常磁性ビーズ、または類似物などの固形担体にそれらを固定することである。
【0100】
例えば、ウェル1内のDNA分子は、AAAA張出し部を有し、ウェル2のDNA分子は、AAAC張出し部を有する。これにより256ウェルは、4塩基の張出し部の、すべての順列をカバーする。標的DNAをリガーゼとともにウェルに添加するとき、例えば、TTTT張出し部を備えたDNA分子はそれ自体がウェル1に取り付き、TTTG張出し部を備えた標的DNAはウェル2に取り付く。ソーティングアダプターに連結しなかった標的DNA分子を洗い落とした後、IIS酵素を添加し、それにより標的DNA分子が遊離し、同時に標的配列の次の4塩基対を表す新しい張出し部が作られる。この張出し部はその後、新しいソーティング過程の出発点として用いることができ、あるいは転換/拡大を続けることができる。
【0101】
DNA分子を洗い流すソーティング戦略は、DNA分子の比較的大きな損失を含む。しかしながら、本特許出願において提案されるほとんどのシークエンシングプロトコルは個々の分子の分析に基づいており、これは非常に少ないDNA分子が必要とされることを意味する。したがって、99.9%以上の損失であってもほとんど問題はない。
【0102】
異なるウェルを用いる代わりに、別法は「マイクロアレイ」上の異なる位置を用いることであろう。例えば、アドレス1においては、固定されたTTTTで終わるDNA分子だけであり、アドレス2においては、固定されたTTTG末端を備えたDNA分子である。他の別法は、異なる末端を備えたDNA分子を異なる時点で取り付け/転換させること、ゲルソーティングを使用することなどである。
【0103】
例えば、「マイクロアレイ」上の256区画に分布された256の異なるソーティングアダプターが存在する方式を用いることができる。例えば、区画1に、AAAA張出し部を備えたソーティングアダプターがあり、区画2において、ソーティングアダプターはAAAC張出し部を有する。したがって、標的DNA分子は、例えばTTTT張出し部を備えたものがスクエア1に取り付けられ、GTTT張出し部がスクエア2に取り付けられるようにソートされる。DNA片の他の末端を、例えばビオチン/ストレプトアビジンを用いて基質に固定することによっても、DNA分子を読み取りプレート上のその位置から離すことなく、次の転換/拡大段階を続けることができる。DNA分子がその位置から離れることを防ぐ他の方法は、256ウェル/スペースに分けられた読み取りプレートを用いることである。
【0104】
ソーティングは当然のことながら、256よりも少ない、または多い順列を用いて行えることも指摘しなければならない。ソーティングは、いくつかの過程で行うこともできる。例えば、65536の異なる区画を備えた「マイクロアレイ」を用いる場合、ハイブリダイゼーション単独によるソーティングによって8bpを同定することが可能であろう。これは多くの適用例に関して、うまく再構成を行うために十分である。したがって、転換または拡大を使用することなく、ソーティングはそれだけでシークエンシング法として機能できる。
【0105】
ソーティングはさらに、非リガーゼベースの戦略を用いて行うこともできる。原則的に、拡大に関して挙げられたすべての方法を含めて、塩基対を認識するのに適した任意の方法を用いることができる。
【0106】
ソーティング法の特異性は、同じソーティング手順を1回または数回繰り返すことによって、ほとんどの目的に適合され得ることも指摘されるべきである。特異性を高めるために、競合プローブ/張出し部を用いることも適切である。
【0107】
このように、好ましい態様において本発明は、本明細書に記載したとおり標的分子をシークエンシングする方法を提供し、前記配列は少なくとも、
a)前記標的配列の少なくとも一部分を、固形担体に取り付けられた、または固形担体に取り付けるための手段を担う相補的プローブを結合するのに適した形態、好ましくは一本鎖形態に転換する段階と、
b)前記相補的プローブを、段階a)で作られた相補的プローブを結合するのに適した前記領域の、好ましくは一本鎖領域の少なくとも一部分、好ましくは4から12塩基の長さに結合する段階と、
c)所望により、段階a)およびb)を繰り返す段階であって、前サイクルの相補的プローブが結合した領域に対して、前記標的分子の隣接する、または重複する領域に前記相補的プローブが結合する段階と、
d)前記標的配列が結合した相補的プローブを同定することによって前記標的配列の配列を決定する段階
を含むプロセスにより、前記分子の一部分の相補性を評価する段階とによって決定される。
【0108】
本明細書において「プローブ」とは、適切な核酸分子、例えばオリゴヌクレオチド、またはPNA分子を指す。
【0109】
追加的な段階を含むこともでき、例えば相補的プローブはプライマーの役割を果たすことができ、この場合、必要に応じてポリメラーゼ反応を行うこともできる。
【0110】
上述のとおり、このソーティング技法は、好ましくは複数の相補的プローブを用いて行われ、サイクル当たり好ましくは2と8個の間、特に好ましくは4個の塩基が同定可能であるが、この情報はシークエンシング反応の完了時に集められるだけであってよい。2と8個の間、好ましくは4個の特有の不変塩基を備えた特に好ましい相補的プローブは、前記固形担体の異なる離散する部位に取り付けられる。第2の引き続いて起こるサイクルにおいて、前記標的分子の隣接または重複領域をシークエンシングするために、前記プローブに結合した標的分子は、相補的プローブを有する1つまたは複数のさらなる固形担体に移される。これを達成するために、段階a)は、拡大プロセスのために記載したものと類似の方法で行うことができ、すなわちプローブは、適切な張出し部が形成されるように認識部位の外側を開裂するヌクレアーゼ、例えばIIS酵素のための制限部位をプローブ自体が含有することができる。
【0111】
上述の手順は拡大の手順と連結することができ、それにより、シークエンシングを、例えば段階b)の後で、ソーティングと拡大の組合わせによって行うことができ、張出し部を記載のとおり生成することができ、適切な拡大タグを担うアダプターを前記張出し部に結合するために用いることができる。その後、拡大タグの読み取り、標的分子が結合したプローブの同定の組み合わせによって配列を決定することができる。したがって好ましい特徴において、本発明は、前記配列の一部分が本明細書に記載の拡大方法によって決定され、隣接するまたは重複する部分が本明細書に記載の相補的プローブの使用によって決定されるといった、本明細書で記載のシークエンシング法を提供する。
【0112】
ほとんどの場合、配列部分の位置決定のために採用される技法は、シークエンシングのために標的DNAが生成される方法によって決まり、例えば、共通の点から始まっているか、または様々な点から始まる標的分子をもたらす断片化によって生成されているかである。
【0113】
シークエンシングのための核酸分子は、様々な方法で生成することができる。少量のDNAを、DNアーゼ、超音波破砕、ボルテックス(vortexing)、または類似の技法で処理することによって、核酸分子を細片に断片化することができる。そのような技法は当業分野でよく知られており、例えばランダムサブクローン生成のプロトコルを記載するhttp://dnal.chem.ou.edu/protocol_book/protocol_partII.htmlを参照されたい。これらの技法のパラメーターを調整することによって、標的DNA断片の平均の大きさを調整することが可能である(一般に、最適であるのは数百塩基対の平均の大きさを有することである)。この方法はまた、元の配列におけるほとんどの場所で切断/破壊されたDNA片が統計的に得られるように、DNA分子を切断/破壊する場所に関して比較的に非特異的であるべきであろう。
【0114】
断片化DNA分子の末端は、平滑末端、および1〜2塩基の短い張出し部の両方からなることを、研究は示している。所望するなら、張出し部は平滑末端となるように処理することができる(Klenow充填など)。
【0115】
好都合には、一本鎖張出し部の形成を利用する本発明の好ましい方法を実行するために、核酸分子はそのような張出し部を形成する手順によって断片化することができる。前述のように、超音波破砕、ボルテックス、およびDNアーゼIは、短い張出し部を作る。非特異的に開裂する制限酵素も用いることができる。IIS酵素は特に、DNA結合ドメインを交換することができる「ドメインスワッピングテスト」に十分適していることをいくつかの研究が示している。したがって、DNAを非特異的に結合するDNA結合ドメインに切断ドメインが結合している場合、新しいIIS酵素を作ることができる。
【0116】
既知のIIS酵素によって形成される張出し部は、−5から+6塩基まで多様である。6個を超える塩基の張出し部が所望であるならば、他の系/方策を用いるのが適切である可能性がある。実行可能な1つの手段は、それ自体の結合部位の外側で二本鎖DNAに切れ目(nick)を形成するニッキング酵素(nicking enzymes)を用いることである。6個を超える塩基対の内部距離を有し、二重らせんのいずれかの側に置かれたそのようなニッキング酵素の2つの結合部位は、6個を超える塩基対の張出し部を形成するであろう。現存のニッキング酵素に加え、例えばIPおよびIIS制限酵素の突然変異によって、新しいニッキング酵素を作ることも可能である。
【0117】
断片化の代替法として、標的配列の断片がPCRまたは類似の方法の助力で生成される戦略を選ぶことも可能である。例えば、標的DNA上の既知の配列でもって開始し、次にこの領域をポリメラーゼ伸長におけるプライマーに対するテンプレートとして用いることができる。任意の部位でポリメラーゼ伸長反応を停止させる方法を用いる場合、「DNA梯子」が作られ、そこには多くの異なる長さのDNA分子が存在するが、すべて共通する一末端を有する。別法として、断片のすべての可能な組み合わせが標的配列から生成されるように、短いランダム化プライマーを用いることができる。しかしながら、ポリメラーゼ伸長を用いるとき、限定要因は多様なポリメラーゼの伸長の長さである。
【0118】
拡大技法は、本明細書に記載のソーティングおよび転換技法と合わせることができる。例えば、一本鎖の標的に結合するとき、アダプターをプライマーとして用いることができる。ポリメラーゼ反応は、アダプターと標的配列との間の、相補性の存在を確立する手段をさらに提供する。
【0119】
本発明の好ましい一実施形態において、標的分子は固形担体に固定される。これはいくつかの異なる方法で達成することができる。標的分子は1つまたは複数の部分に取り付けられているように設計することができ、それが分子の固形担体への結合を可能にし、例えば、末端(またはいくつかの内部の部位)は結合の対の1つのパートナー、例えばビオチンを備えていることができ、次にそれをストレプトアビジン担持固形担体に取り付けることが可能である。
【0120】
標的分子は、いくつかの知られている方法でそのような結合部分を担うように設計することができる。例えば、PCR反応は上記結合部分を導入するように、例えば適切に標識されたプライマーを用いることによって実施することができる(例えば実施例17を参照のこと)。別法として、標的核酸は、例えば標的核酸分子を制限酵素で開裂して、次に末端が結合部分で標識されたアダプター/リンカーにその分子を連結することによって、結合部分に連結することができる。そのような方法は、IIS制限酵素を用い、非パリンドローム性張出し部を形成する場合、特に適しているであろう。他の別法は、すでに結合部分を担っている、またはそのような結合部分の導入を促進するような配列を含有するベクターに標的分子をクローンすることである。そのような方法は、以下により詳しく述べる位置マーカーを導入するために、同じく用いることができた。
【0121】
別法として、核酸分子自体が結合対の1つのパートナーである限り、結合部分を取り付ける必要はなく核酸分子を固形担体に取り付けてもよい。したがって、例えば固形担体に取り付ける短いPNA分子を用いることができる。PNA分子は、ハイブリダイズし、二本鎖DNAに結合する能力を有し、この非溶解核酸材料はそれゆえこの方法で固形担体に取り付けることが可能である。同様に、オリゴヌクレオチドプローブは、相補的配列を固形担体に結合するために用いることができる。そのような技法はまた、以下に記載するように特定の核酸分子を固形担体上の特定の位置に結合することによって、シークエンシングを開始するために用いることもできる。
【0122】
標的分子を取り付けるための固定化部分として適した適切な固形担体は、当分野でよく知られ、文献に広く記載されている。一般的に言ってこの固形担体は、化学的または生化学的手順において固定化、分離などのために現在広く用いられている、または提案されている任意のよく知られた担体または材料であってもよい。したがって、例えばこの固定化部分は、例えばポリマー材料、例えばアガロース、セルロース、アルギン酸塩、テフロン、ラテックス、またはポリスチレンからなる、ビーズ、粒子、シート、ゲル、フィルタ、膜、微小繊維ストリップ、チューブまたはプレート、繊維または毛細管の形態を取ることができる。粒状材料、例えばビーズが一般に好ましい。好都合には、この固定化部分は、超常磁性体粒子などの磁性粒子を含むことができる。さらに好ましい実施形態において、線状配置にある分子の固定を可能にするためにプレート、またはシートが用いられる。このプレートはまた、分子を取り付けることのできるプレートと直角をなす壁を含むことができる。
【0123】
固形担体への取り付けは、直接的に、または間接的に実施することができ、用いられる技法は、取り付けられる分子が標的分子を同定するためのプローブであるか、標的分子自体であるかによって決まる。標的分子を取り付けるために、好都合には、核酸分子および/または固形担体に担持された付着部分を使用することによって間接的に行うことができる。したがって、例えば親和性結合パートナー対を用いることができ、アビジン、ストレプトアビジン、またはビオチン、DNA、またはDNA結合タンパク質(例えば、lacIリプレッサタンパク質、またはそれが結合するlacオペレータ配列)、抗体(モノクロナールであっても、ポリクロナールであってもよい)、抗体断片、あるいは抗体のエピトープまたはハプテンなどである。これらの場合、結合対の1つのパートナーが固形担体に取り付けられ(または本質的にその一部にである)、もう一方のパートナーが核酸分子に取り付けられる(または本質的にその一部にである)。直接に取り付ける他の技法を用いることができ、例えばフィルタが用いられる場合、取り付けはUV誘導架橋反応によって行うことができる。DNA断片を取り付けるとき、ガラスに密着するDNAの固有の傾向を用いることもできる。
【0124】
固形担体への適切な官能基の取り付けは、当分野でよく知られている方法によって行うことができ、例えば、ヒドロキシル、カルボキシル、アルデヒド、またはアミノ基による取り付けが含まれ、それらは適した表面被覆がもたらされるように固形担体を処理することによって提供することができる。本発明の核酸分子への適切な官能基の取り付けは、連結によって実施することができ、あるいは合成または増幅の間に導入することができ、例えばビオチン、または捕捉するための特定の配列など適切な部分を担うプライマーが用いられる。
【0125】
本明細書に記載のとおり、標的分子は、固形担体に取り付けられる相補的プローブに都合よく取り付けられる。
【0126】
複数であるが離散した相補的プローブを使用する技法において、これらの異なるプローブが取り付けられる固形担体は、都合よく物理的に取り付けられるが、各プローブへの標的分子の付着によって発生するシグナルは独立して決定可能でなければならない。したがって、例えば複数のウェルを備えたプレートを、異なるウェルにおいて異なるプローブを備えた固形担体として用いることができ、あるいは固形担体の領域は異なるアドレスを含むことができ、例えば、異なるプローブは離散した部位においてフィルタに結合していてもよい。
【0127】
固形担体への取り付けは、核酸分子断片が生成される前、または後に行うことができる。例えば、結合部分を担う標的核酸分子を固形担体に取り付け、その後、DNアーゼIまたは類似体で処理することができる。別法として、開裂を行ってもよく、次にその断片を担体に取り付けることができる。
【0128】
多くの状況において、目的は他の配列の内部、または他の配列と共に存在する1つまたはいくつかの配列をシークエンシングすることである。例えば、わずか5〜10%のヒトゲノム配列が、直接に生物学的重要性があると想定されている。したがってヒトゲノムのマススクリーニングに関して、生物学的重要性の低い領域のシークエンシングを回避できることが有用であろう。
【0129】
したがって、用いることのできる1つの戦略は、単離される標的配列を相補するポリヌクレオチドを固形担体に固定することである(ウェル内部、単分散球、マイクロアレイなど)。配列プール中のポリヌクレオチドを固形担体上のポリマーと共にハイブリダイズすることによって、所望でないポリヌクレオチドを洗い流し、その後シークエンシングの段階に進むことができる。所望であれば、ハイブリダイゼーションと洗浄のサイクルを数回実行することによって、特異性を高めることができる。個々の適用例にとっては有利である可能性があっても、相補的ポリヌクレオチドが規則的なパターンで固定されているかどうかには依存しない。連結、PNAハイブリダイゼーションなどに基づく類似の方法も可能である。
【0130】
例えば、特定のmRNA/cDNA分子を単離するために、相補的cDNA/mRNA分子を常磁性球などに固定することができる。次に該球体は、標的配列を含有する溶液と共にチューブ内で混合することができる。mRNA/cDNA分子が球に固定されたmRNA/cDNAとハイブリダイズされているとき、所望でない分子は、磁石または類似体を用いて球体をチューブ内に保持するのと同時に洗い流すことができる。所望の標的分子を次に、温度の上昇、pHの変化、またはハイブリダイズ化分子を溶解する他の方法を用いることによって遊離することができる。
読み取りプレート上で行われるシークエンシングプロトコルのために用いることのできる類似の方法は、特定の標的配列を所定のアドレスに固定することである。例えば、一本鎖標的DNAを、異なるアドレスに固定されるプライマーにハイブリダイズすることができる。所望であれば、このプライマーは次に、ポリマー伸長のテンプレートとして用いることができる。プライマーを標的配列に適合させることによって、所望どおりアドレスすることができる。
【0131】
類似の方法は、PNA分子を異なるアドレスに固定することであり得る。PNA分子は二本鎖DNAの特定配列を認識する能力を有することが知られており、そのような方法はしたがって、固定したい配列を認識するPNA分子を用いて二本鎖DNAをアドレスするために用いることができる。
【0132】
上述のとおり、シークエンシングされる分子は、2つの種類に分けることができる。1つの共通な末端と1つの任意の末端を有するもの、および2つの任意の末端を有するものである。位置情報は種々の方法で、これらの異なる種類の分子から得ることができる。
【0133】
すべての標的分子が共通の末端を有する場合、各標的分子の長さは、共通の末端と他の任意の末端との間の距離に比例するであろう。同様に、標的分子の特定の部分に起因する配列情報は、共通の末端から配列情報の部位までの距離を算出することによって位置を定めることができる。好都合には、配列情報が標的分子の末端に関係する場合、分子全体の長さ/大きさからその位置を決定することができる。
【0134】
核酸断片が共通の末端から始まっていない場合には、位置情報は種々の方法で得ることができる。1つの別法は、配列ごとに変わる特有のフィンガープリントを作る、または同定することである。したがって、配列片の位置は、どのフィンガープリントにそれが連結しているか、および場合によってはフィンガープリントのどこにそれが位置しているかを記録することによって引き出すことができる。特有のパターンを作るのに使用される、非常に多くの技法が考慮され得る。DNA配列における制限酵素の開裂パターンは、例えば「光学マッピング」(optical mapping)、または類似の方法の助力によって記録することができる。
【0135】
知られている「光学マッピング」法の1つの欠点は、用いられる制限酵素のための切断部位が必ずしも切断されないことである。さらに、不正確な開裂が起こることがあり、DNA断片の長さの測定に関連していくらか不確定性である可能性がある。したがって、多くの同一DNA分子の分析に基づいた各地図片の平均図を作製する必要がある。問題は、どのDNA分子が同一であるのかを知るのが困難であり得ることである。
【0136】
光学マッピングの現行方法に伴う他の問題は、DNA断片の内部配置を観察できるようにするためにDNA分子を真っすぐに整えた後に、制限酵素などを用いた処理を行わなければならないことである。これは、酵素的調製などといったことのためDNA分子の利用可能性を低減する。位置配置に加えて末端シークエンシングを提供する本発明は、そのような問題を克服することを可能にする。例えば、実施例23に記載の技法を参照されたい。
【0137】
特有のパターンを作る蛍光性プローブ/タグを用いることもできる。これは、いわゆる「DIRVISH」技法の背後にある原理である。類似の方法は、原子間力顕微鏡法(atomic force microscopy,AFM)、ミクロ/ナノポア、または特有のパターンで結合したタンパク質の大きさと位置を記録するための他の方法などを用いることである。
【0138】
前に述べたように、細胞性アダプターを用いることもできる。例えば、拡大した標的DNAを細胞に形質転換/トランスフェクトする場合、レポーター遺伝子の転写頻度はシス調節因子への距離によって変わるという事実を利用することができる。レポーター遺伝子を構成する1つの末端にエンハンサーがあり、もう一方に1つまたは複数の拡大タグがある場合、レポータータンパク質の相対量は位置価(position value)を算出するために用いることができる。
【0139】
さらに、位置価を引き出すために用いる要素で標的配列を標識し、または組み込むことも可能である。そのような方法は、例えば非常に類似した2つの配列のフィンガープリントを互いに識別することが困難である場合に有利であり得る。例えば、姉妹染色体をシークエンシングしたい場合、任意に組み込まれる多数の挿入要素(トランスポゾンなど)を組み込むことができる。次に染色体を増幅し、位置マーカーとして挿入要素を使用すれば、各姉妹染色体に関して1つまたはいくつかの特有のパターンができる。
【0140】
位置マーカーを導入することができ、しかもその部位で配列の同定を可能にする、使用可能な別の方法は、PCR反応にプライマーとしてアダプターを使用することを含む。各PCR反応は結果として連結した2つのアダプターをもたらし、2つのアダプター間の距離は標的DNA上のアダプター配列の距離に対応し、同時に位置情報を提供する。
【0141】
標的分子の配列は、拡大することなく位置マーカーを生成するために必要な手段を提供することができる。例えば、いくらかの配列情報が既知である場合、プローブをその配列にハイブリダイズするために用いることができ、その後それは位置マーカーを提供する。別法として、適切な位置マーカーを標的分子内に置くことができ、例えば異なる位置マーカーをゲノム内に規則的な間隔で置くことができる。異なる位置マーカー間の識別を可能にするために、それらのマーカーによって異なるシグナルが提供され、例えば、それらはプローブすることのできる異なる配列または長さを有する。実施例21は、位置マーカーを使用する1つの方法を記載する。
【0142】
したがって、好ましい態様から見ると、本発明は核酸分子を(完全に、または部分的に)シークエンシングする方法を提供し、少なくとも
a)前記核酸分子の一部分の配列を決定する段階と、
b)位置インジケーター、好ましくは位置マーカーの参照により前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階を含む。
【0143】
前述のとおり、好ましくは複数の配列およびその位置が決定される。
【0144】
本明細書において、位置インジケーターは前述のとおり、分子の大きさ、発生したシグナルの強度、あるいは位置マーカー、アンカー、またはフィンガープリントへの距離であってもよい。
【0145】
これらの方法を行うためのいくつかの異なる技術を、本発明を説明するためにここに記載する。
【0146】
原則として、ポリマーの大きさを決定するための各方法を使用することができる。しかしながら、同定されている配列片の長さを大きさの決定の精度に合わせなければならない。精度が低いほど、配列片が長いに違いない。
【0147】
大きさをソーティングするためのいくつかの方法が、以下の分野で存在する。ゲルソーティング、微毛細管ソーティング、読み取りプレート上に伸張しているポリマーの長さの測定、非特異的にタグを付されているポリマーの蛍光強度(または他の)の測定(フローサイトメーター、蛍光顕微鏡などによって)、質量分析法、ポリマーがミクロ−またはナノポアをブロックするために使用する時間などである。このような手順は、配列の決定のためにシグナルが読み取られる前または後に行うことができる。例えばゲル電気泳動法が使用されるとき、ゲル上で分離されているサンプル上で、またはゲルから溶出されたサンプル上で読み取りを行うことができる。
【0148】
DNA分子が切断される(例えばDNアーゼI、超音波処理などによって)可能性はDNA分子の長さに正比例するという原理に基づいて、核酸分子の長さを決定することもできる。例えば200個の塩基対を有するDNA分子は、限られた量のDNアーゼIを含む溶液中で、100個の塩基対を有するDNA分子と比べて2倍の頻度で切断されるはずである。このことは、例えば異なる分子を末端標識しそれらの分子を切断し、次いで単一におよび二重に標識された分子の量を、同様に標識された既知の長さの標準物と比較して調べることによって実施ができるはずである。
【0149】
DNA分子の長さまたは固定点までの距離を決定するためには、DNA分子を延長または伸張させるのが好都合である。DNA分子を伸張させるための1つの方法は、ガラスの小ビーズとDNA分子が1:1の比で結合するように、DNA分子と大余剰のガラスの小ビーズと混合させることである(ビーズは自然にDNA分子と結合する)。DNA分子は液体流中ではガラスビーズよりも抵抗力が弱いはずであり、その結果DNA分子は伸張されるまでお互い同士離れる傾向がある。その液体流が強力であるか、またはガラスビーズが大きくDNA分子とガラスビーズの間の抵抗力の違いが大きいならば、DNA分子は裂けてしまう可能性がある。しかしながら、この問題は液体の流速を下げることによって、またはより小さなガラスビーズを使用することによって避けることができる。単位面積当たりの配列情報の量が増加するようにDNA分子が通常の様式で配置されているなら、この方法は特に効率的なものになる。これを行う1つの方法は、DNA分子をビオチンで標識し、次いで該分子を通常のストレプトアビジン配列を有するプレートに固定する。代替方法としてレーザービーム、いわゆるレーザートラッピングを使用することによって固定を得ることができる。
【0150】
液体流を使用してDNA分子を真直ぐにするかわりに、一方向に負に荷電したDNA分子を引っ張る正電荷を使用することができる。この方策を使用することによって、読み取り効率が増大するようである。原則として最も簡単な方法は、正または負のスポット電荷を読み取りプレートの前に置くことである。クーロンの法則によると、DNA分子上の電荷の力は距離に反比例する。従って電荷に最も近いDNA分子は、さらに下部にあるDNA分子よりもより大きな力によって伸長させられるはずである。読み取りの瞬間にすべてのDNA分子に平等に影響を与えるためには、読み取り単位を有する段階においてスポット電荷を動かすことがしたがって必要であるはずである。プレート上の力の差異を減らすように、スポット電荷を読み取りプレートのはるか下部に置くこともできる。代替方法として、力のベクトルが円弧の中心で直線は同じ大きさであるように、電荷を円弧状に配置することが可能である。したがって、読み取り単位が側路で動かされるときは、電荷を動かしさえすればよい。
【0151】
代替方法として、分子のアンカー上の力を減少させるために、ある異なる技術を使用してよい。電気的に帯電した2枚のプレートを読み取りプレートの下に置くことができ、その読み取りプレート上で標的分子は伸長させられる。トッププレートは弱い負電荷を有し、一方ボトムプレートは相対的に強い正電荷を有する。負に荷電した粒子(例えばDNA)が負のプレートのすぐ上に置かれるならば、そのプレートからの斥力は正のプレートからの引力よりも大きいはずである。次いでその粒子は上方に押し出されるはずである。しかしながら、このプレート状態から動かすことは逆の結果になる。正のプレートの引力は負のプレートの斥力よりも大きい。プレートの電荷を調整することによって、読み取りプレートの上の所与の高さにおいて、斥力と引力の間の平衡が生まれるはずである。標的分子はこの平衡面に押しやられるはずである。この方法では、DNA分子が平衡面にある限り、DNA分子にかかる正味の力はほぼ0に等しい。これによって破裂の可能性が減少する。
【0152】
この2枚の帯電したプレートに加えて、読み取りプレートの左の正電荷も使用することができる。これによって、この方向の正味の力が生み出されるはずである。2枚の帯電したプレートをお互いに関して、および読み取りプレートに関して傾けることによって、同じことを実現することができる。
【0153】
標的分子がフローサイトメーターまたは同等の装置を介して伸張させられながら動かされるならば、負に帯電した管を使用することができる。このような技術を使用することによって、標的分子は斥力が最も弱い管中央に向かって押されるはずである。
【0154】
さらなる代替の伸張技術が、機械的伸張によって提供される。例えばこの方法では、標的分子のいずれかの末端と相補的なオリゴヌクレオチドが接着されるときには、2枚の隣接するプレートを使用してもよい。標的分子がひとたびこれらのプローブにハイブリダイズされると、分子がプレートの間で伸張されるまでプレートを分離することができる。
【0155】
前述の方法において発生したシグナルは、発生したシグナルに応じて、および位置情報がどのようにして得られるかに応じて、いくつかの異なる方法によって読み取ることができる。例えば、標的DNAに付着した蛍光DNAプローブの位置を特定するために、このDNAを前述のように伸張させてよい。例えば、モレキュラーコーミング(molecular combing)として知られている、Weier他によって開発された方法(Hum.Mol.Gen.,1995,Vol.4(10),p1903−1910)を使用することができる。この方法では標的DNAを含む溶液を、DNA分子がガラスプレートにその一末端を付着するように作製した平らなガラス表面上に置いた。次いでこのDNA分子を、液体流を使用することによって真直ぐにした。次いで蛍光顕微鏡によって、伸張させたDNA分子に接着したプローブの相対的な位置を観察できるはずである。
【0156】
本発明では、例えば異なる蛍光体で標識された4つのプローブおよびDNAの特異的な伸張体である拡大タグを使用することによって、そのプローブは、A、C、G、およびTを表す4つの拡大タグにハイブリダイズするようにこれらのタグに向けられてもよい。すなわち、前に記載したDIRVISH技術を使用する。次いで配列順序を、蛍光顕微鏡を用いて直接読み取ることができる。前述のように、拡大タグがどのようにして構築されるかによって、多数または少数のプローブを使用することができる。具体的には、特異的なシグナル、例えばバイナリーコードの発生を生み出す各拡大タグにプローブが結合する方式においては、単一のプローブを使用することができる。代替方法として、拡大タグが2個以上の塩基に対応する場合は、5個以上のプローブを使用してよい。顕微鏡にガラスプレートをスキャンさせるソフトウェアを開発し、一方、同時に自動的に配列順序を分析することによって、塩基対を非常に早く読み取ることが可能なはずである。
【0157】
さらなる代替方法として、早い読み取り用にフローサイトメーターを使用して蛍光プローブを読み取ることができる。このための必要条件は、DNA分子がA、C、G、およびTを表す拡大タグが順番に通過するように伸張した型で、フローサイトメーターの読み取り単位を通過することである。これは前述の技術を使用することによって行うことができる。この特別な実施形態の代替方法として、電界または磁界を液体流の代わりに使用して、蛍光検出器を通過する粒子を引っ張ることができる。これは、DNA分子が負に荷電している一方でガラスビーズが正電荷を有している事実を使用することによって、またはガラスの代わりに超常磁性ビーズを使用することによって達成される。ついでこのビーズは、背後で長い撚り糸のようにDNA分子を引っ張るはずである。
【0158】
この方策における重要なパラメーターの1つは、フローサイトメーターの蛍光検出限界が低いことである。いくつかのグループは、流速を下げることによって個々の蛍光体分子をなんとか検出した。しかしながら、1秒あたり20〜30,000粒子の分析スピードを持つ従来型のフローサイトメーターを使用するためには、多くの蛍光体を各プローブに固定することができるように、より長いプローブが使用されなければならない。
【0159】
最速のフローサイトメーターは、現在1秒当たり約200,000の蛍光粒子を分析するための容量を有するが、このようなフローサイトメーターは市販されていない。さらに、DNA分子が壊れる前に伸張した型で高速耐性のDNA分子が何であるかは確かではない。しかしながら、DNA分子は非常に早い読み取りを可能にする速度に耐えるはずであると考えるのが現実的である。
【0160】
さらなる代替方法は、通常の方法で固形担体上、例えばストレプトアビジンで覆われたプレートにDNA分子を固定することである。配列(例えば一続きの拡大タグによって発生したシグナル)は、読み取りプレート内に挿入された小さな検出器を有することによって読み取られる。これらの検出器は、例えば断片に固定された拡大タグ上のレポーター分子によって、例えば固形担体上にセンサを結合させることによって電気回路を遮断または導通することによって、非活性化または活性化される。例えば、読み取りプレート上のレポーター分子とモジュールの間で強い結合が形成される可能性がある。後者の場合、DNA分子が引き剥がされるならば、モジュールを読み取りプレートから引き剥がすことができるような方法でモジュールを形作ることができる。モジュールが緩んでくると、読み取りプレートからどのモジュールが取り除かれたかを記録するような方式で、モジュールは電流回路を遮断する。首尾よく結合する可能性を増大させるためには、断片上の同じ位置にいくつかのレポーター分子を固定してよい。各塩基A、C、G、およびT用に4つの異なるレポーター分子を使用することができるが、または断片上の4つの異なる場所に位置する同じレポーター分子を使用できる。マルチプレックス、パラレルコンピュータ・インプットおよび現代の他の電子機器によって、素早いシークエンシングを可能にするような1秒当たり数百万のシグナルを記録することが可能であると考えられる。
【0161】
好ましい実施形態では、これらの方法は本明細書に記載されている拡大技術と組み合わせて適切に使用される。すなわち複数の、好ましくは1つの鎖の拡大タグの存在によって前記標的核酸分子の部分が決定される。しかしながらこれらの方法は、拡大が行われないときでも使用することができる。DNA分子を拡大する代わりに、塩基にセンサーが記録することができる異なる付着物を取り込ませることができる。
【0162】
ひとたびシグナル情報が蓄積されると、コンピュータプログラムを使用して配列片を最終配列に組み立てる。この段階においてエラーが発生する可能性は、主に5つのパラメーターに依存する。シークエンシングされるDNA分子の長さ、DNA配列の塩基対組成がどの程度ランダムであるか、読み取られるDNA片の長さ、読み取られるDNA片の数、およびシークエンシング反応におけるエラー率である。
【0163】
本発明者は、前述のパラメーターの重要性を分析するためのコンピュータプログラムをすでに作製している。すでにシークエンシングされているヒトゲノムDNAに基づいて、この分析は、30断片からなる長さのDNA、6×108個のDNA片を読み取り、およびシークエンシング反応におけるエラー率が10%であると(点突然変異を考慮する)、1つのヒトゲノムは1つのシークエンス反応中で読み取られ、点突然変異/欠失がほとんどないであろうことを示している。しかしながら1つの例外は、DNA片の長さが増加しているに違いない非常に非ランダムな領域(サテライトDNAおよび他の繰り返し領域)である。しかしながら、これらの領域の生物学的情報は、コード配列およびシス−調節要素と比べるとそれほど重要ではない。
【0164】
DNA片が何回も読み取られるときは、シークエンシング反応におけるエラー率が非常に高くても、それは埋め合わせられることもデータ分析は示す。例えば、配列の長さの10倍もの多くの塩基対を読むことによって、シークエンシング反応におけるエラー率が高くても、たいていの欠失および点突然変異は無くなるはずである。
【0165】
シークエンシングに使用される技術に応じて、不均一のサンプルのシークエンシングを行うこと、例えばパラレル・シークエンシングを行うことがある状況においては可能である。これを可能にする手順によって、本発明の好ましい態様が形成される。このような技術では、異なる標的分子からのシグナルが識別できることが必要である。これはいくつかの方法によって、例えば特定の標的分子などを同定するマーカーの特定の位置、含有、同定を制限することによって達成される。例えば、標的核酸分子の領域に相補的である固形担体を使用して、特定の分子を単離し保持することができる。このことは、シークエンスの少なくとも部分的な知識があれば、すなわち特定の分子を特定の部位に結合させるために行うことができる。あるいはこうした知識なしに、異なる分子が結合し次いで平行してシークエンシングすることができる、本質的にランダムに結合するプローブを使用することによって、配列をその分子を関連付ける複数の技術を利用することによって、例えばアドレスまたは位置マーカーによって行うことができる。本明細書に記載されている技術は特に有利である。なぜなら、本技術によって個々の分子がシークエンシングされ、したがってパラレル・シークエンシング反応がさらに促進されるからである。
【0166】
本明細書に記載されているいくつかの技術は、標的分子の一部分だけをシークエンシングするために、またはフィンガープリンティング、プロフィール分析またはマッピングのために使用することができる。すなわち分子の不連続おかつ特有部分を、例えばRNA発現(最初に分析用のためDNAに転換することができる)の分析用に同定するため使用することができる。例えば実施例23に記載されるように、拡大タグ(一つの鎖の拡大タグが好ましい)が付着することができる特定の張出し部を生成する制限酵素を用いて、標的サンプルを消化することができる。このようなタグは配列に関する情報を保持するだけでなく、切断をもたらす酵素に関する、すなわちそうした切断から生じる起因する断片のマーカーとしての情報を追加的に保持することができる。制限酵素が、異なるアダプターが結合できる、異なる長さの張出し部を生成するならば、複数の制限酵素を同時に使用してもよい。代替方法として、連続的なサイクルにおいて異なる制限酵素を使用してもよい。
【0167】
したがって、例えば相補的な張出し部に付した増幅タグによって、例えばその相補性を示すタグ、例えばタグ自体がヌクレオチド塩基でできているタグの使用によって、結果として生じる断片を一列に並べることができる。このことから、実施例23に記載されるように制限地図を作成することができる。したがって本発明のさらなる態様では、本明細書に記載されるような前記部分の位置情報に加えて、本明細書に記載されるような前記配列の不連続部分についての配列情報を得ることを含む標的配列の地図を生成する方法を提供する。
【0168】
好ましい特徴はにおいて前期地図は、前記配列の不連続部分の配列情報を得ることによって生み出され、前記部分は複数のヌクレアーゼの開裂部位のすべてまたは一部分、および/または前記ヌクレアーゼの制限部位のすべてまたは一部分を含み、前記配列の位置が前記ヌクレアーゼで消化した後の前記標的核酸分子の断片の末端における配列の比較によって決定される。
【0169】
配列情報は、本明細書に記載されるような複数のヌクレアーゼによる前記標的分子の切断によって得られるのが好ましく、相補的な一本鎖領域およびアダプター分子と前記標的分子の領域の結合(開裂部位に、または開裂部位に隣接しているのが好ましい)を生成するために得られるのが好ましい。前記アダプター分子は本明細書に記載されるような複数の拡大タグを保持し、前記タグは前記領域の複数の塩基に対応するシグナル部分を含み、前記アダプター分子はそこに結合し、切断用に使用されるヌクレアーゼに対応するさらなるシグナル部分を追加的に含む。充分なヌクレアーゼが使用されるいくつかの例では、この方法をシークエンシングの方法として使用してもよい。
【0170】
例えば、バクテリアは以下の方法によって同定することができる。バクテリアは溶解させることができ、DNAを単離することができる。次いでクラスII制限エンドヌクレアーゼまたは本明細書に記載されるような他のこのようなヌクレアーゼを用いて、分子を切断することができる。次いでDNA分子をアダプターに結合させ、張出し部を同定することができる。次いでDNA分子を読み取りプレートに固定し伸張させることができる。蛍光スキャナを用いて読み取りプレートをスキャンすることによって、制限の長さについて情報または特有なパターンを、およびこれらの分子の末端に由来する配列情報を得ることができる。このような拡大が含まれる技術は、その末端配列により同じ大きさの分子の識別を可能とする。したがって本発明のさらなる特徴では、本発明は、標的DNA分子のフィンガープリントを得る方法を提供する。この方法は、本明細書に記載されるよういに位置情報を得ることに加えて本明細書に記載される複数のシークエンシング技術の使用を含む。
【0171】
本発明の好ましい特徴では、前記標的分子の特有な制限地図を参照することによって位置情報が得られる。さらに好ましい特徴には、複数の拡大タグを同定するための制限地図の使用、およびフローサイトメーターまたはナノ/ミクロポア分析を使って効果的に読み取ることができるタグの使用がある。
【0172】
本特許出願に導入されている原理およびプロトコルを使用することによって、シークエンシング/ソーティング反応中およびシグナルを読み取るときに、プルーフリーディングを使用してエラー率を減少させることができる。
【0173】
ソーティングが使用されるならば、同じ配列片を数回ソーティングすることが可能である。例えばAAAAで始まる標的DNAはすべて、ウェル1にソーティングされる。次いで同じ手順が繰り返され、そこで誤ってソーティングされたAAAAで終わらないDNA分子が洗い流される。原則としてこの手順は、望ましいエラー百分率が得られるまで繰り返すことができる。
【0174】
拡大/転換が使われるならば、拡大タグの繰り返し鎖(シグナル鎖)が得られるように、標的分子の同じ配列片を数回転換することが可能である。したがって、繰り返される転換生成物が一様でないときは、たいていのエラー転換は発見することができる。位置情報を引き出すために使用される標的分子の部分も、同じ方法によってコピーすることができる。
【0175】
さらに、各配列片は何回も読み取られる可能性がある。なぜなら、分析される標的分子の数が非常に多い可能性があるためである。
【0176】
本明細書に記載されているシークエンシングまたは拡大方法を行うためのキットは、本発明の好ましい態様を形成する。したがってさらなる観点から見ると、本発明は標的核酸分子の複数の塩基を拡大するためのキットを提供する。この標的核酸分子は前に記載されるような少なくとも1つまたは複数のアダプターを含み、所望するなら複数の固形担体に付着し、好ましくは1つまたはそれ自身がシグナル手段を含む拡大タグを有する。
【0177】
所望するならば、キットは以下のものを含むリストから選択される他の適切な成分を含んでもよい。それは反応中に使用するための制限酵素、標的分子がその内部に連結され得るベクター、リガーゼ、制限または連結部位の不活性化および活性化に必要な酵素、増幅および/または適切な酵素用のプライマー、緩衝液および溶液である。本明細書に記載されているシークエンシング反応の他の態様を行うためのキットも、本発明の範囲内に含まれる。したがって、例えばソーティング反応を行うためのキットは、少なくとも1つの固形担体を含んでよく、この固形担体は複数の相補的なプローブ、好ましくは固形担体上、または分離した固形担体上の異なるアドレスで複数の塩基によりお互いのプローブにミスマッチした一連の不連続プローブを担持する。このようなキットには、適切な標識手段が含まれてもよい。
【0178】
標的核酸分子の拡大用またはシークエンシング用にこのようなキットを使用することによって、本発明のさらなる態様が形成される。
【実施例】
【0179】
説明のためのみに以下の実施例が与えられ、その中で参照される図は下記の通りである。
【0180】
[実施例1]:1方は張出し部を形成し他方は平滑末端を形成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
《方法》
1.シークエンシングされるDNA配列からなる純粋なDNAの集団を、元の配列の細片(以後はDNA片と呼ぶ)からなるDNA分子の集団が形成されるように、非特異的な方法で切断/破壊する。
【0181】
2.DNA片中の塩基対を、4つの各塩基アデニン、シトシン、グアニン、およびチミンを表す4つの異なるDNA配列(以後は拡大タグに対応するDNA断片と呼ぶ)に置き換える。したがって、例えばA−Tの塩基対があったところには「断片A」が挿入され、C−Gは「断片C」で置き換えられる。そのため元の塩基の順序、例えばACGTTが断片A、断片C、断片Gなどに置き換えられる場所では新しいDNA分子が発生する。原則として、これら4つのDNA断片の長さは2塩基対から数百kbp(望むならばこれより多く)まで要件に従って変化する可能性がある。これに対応して、DNA断片はレポーター遺伝子および他の生物学的情報を含む可能性があり、または既知の生物学的機能を持たない配列のみからなる可能性がある。
【0182】
3.個々のDNA分子用の4タイプのDNA断片の順序が読み取られる。これにより、元のDNA片の塩基の順序が決定される。
【0183】
4.コンピュータプログラムによってDNA片の間の重複を使用し、出発時点で使用されたDNA配列の配列のために段階3からの情報を一緒にする。
【0184】
図1では、自らのDNA結合部位の外側を切断する制限酵素に基づいている段階2を行う1つの方法を例示する。この方法は以下のように行われる。
【0185】
1)段階1からのDNA片をプラスミドに連結させる。このプラスミドは平滑末端片切断を発生させ、自らのDNA結合部位の外側を切断して、一塩基対の張出し部(Enz2)を発生させる制限酵素(Enz1)用の結合部位を有する。さらに、プラスミドがストレプトアビジン処理した試薬チューブに粘着しそこで反応が起こるように、ビオチン塩基をプラスミドに取り込ませる。
【0186】
2)試薬チューブを洗浄し、Enz1およびEnz2を含む新しい反応混合物を加え、1つの平滑末端およびDNA片の第1の塩基が張出し部を構成する1つの末端が形成されるようにインキュベートする。
【0187】
3)試薬チューブを再び洗浄し、4つの異なるDNA断片および耐熱性DNAリガーゼ(例えばPfuまたはTaqDNAリガーゼ)を含む反応混合物を加えてインキュベートする。耐熱性DNAリガーゼの有利点は、非常に特異的に連結する一方、同時に平滑末端を連結させないことである。それによって、例えば「断片A」はアデノシン張出し部のある場所に連結されるはずであり、「断片C」はシトシン張出し部のある場所に連結されるはずである。
【0188】
4)試薬チューブをさらにもう1度洗浄し、平滑末端が連結されるようにT4リガーゼを有する反応混合物を加えてインキュベートする。挿入される断片はプラスミドが元来有していたようなEnz1およびEnz2用の部位を有しているため、次の塩基を新しいサイクルにおいてDNA断片に置き換えることができるように出発点に戻る。
【0189】
[実施例2]:両方ともに張出し部を形成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
この方法の出発点は、すべてのBspMI部位が不活性になるように、転換すべき標的DNA断片はすべてBspMIメチラーゼで処理されるということである。次いでDNA断片は、図2aで示されるように塩基ベクターに連結され、この図はDNA片を有する塩基ベクターがそれに連結されストレプトアビジン基質に固定したことを示す。この塩基ベクターは、実線で示されるDNA断片を切断するために使用されるBspMI部位を含む。この実施例で作成される張出し部は内部に「T」を有するはずであり、この張出し部に連結することができるのはAアダプターのみである。AatII部位は、アダプターAがBspMIで作成された張出し部に連結された後で、塩基ベクターを環状にするために使用される。この塩基ベクターはビオチンでタグされた塩基も含むため、ストレプトアビジンで覆われた基質に固定され得る。
【0190】
使用されるアダプターは、図2bに示される。アダプターA1(トップ)とA2(ボトム)の間の唯一の違いは、AatII部位/張出し部がPstI部位/張出し部とともに場所を変えていることである。A1およびA2アダプターは、例えばA1がサイクル1、3、5で使用され、A2がサイクル2、4、6で使用されるように2回に1回のサイクルで使用されるはずである。5’張出し部は5’に沿って3つの共通なヌクレオチドおよびアデニンからなる。そのためこれらの張出し部は、張出し部(3’に沿った)の内側でチミンを有する他の5’張出し部と連結するはずである。下のボトムアダプターの5’張出し部の外側の太線は、アダプターがDNA断片に連結された後に、BspMIによる切断によって形成される張出し部を示す。Aアダプターに加えて、C、G、およびT用にアダプターが作成されなければならない。
【0191】
塩基ベクターに連結されると、DNA断片はストレプトアビジン基質、例えばパラメーター領域に固定される。Dynabeadのkilobase BINDER kitを使用することによって、非常に強いビオチン−ストレプトアビジン結合が得られ、したがってサイクル数が非常に多くてもDNAの損失を最小限にして、反応溶液を素早く効率的に変化させることができる(Biomagnetic Techniques in Molecular Biology,3rd Edition,pp.158−60,Dynal ASによって販売された)。この手順の残りは4つの酵素反応の1サイクルからなり、1サイクルあたり1つの塩基が転換される(図2C)。
【0192】
この手順では、5’張出し部の内側にDNA断片の第1の塩基があるように、BspMIによる切断によってサイクルが開始される。この場合、それはチミンである。その後、かなり過剰なアダプターA1、C1、G1、およびT1が加えられる。これらは例えばアダプターA1が内部にチミジンを持つ張出し部に連結し、C1が内部にグアニンを持つ張出し部に連結するように設計されている。またアダプターはホスファターゼ(例えばウシ小腸由来のアルカリホスファターゼ、AP、Promega)で処理されており、したがってアダプター間の連結が避けられる。耐熱性リガーゼを使用することによって、この段階において高度の特異性が得られる。第3の段階では、最終段階においてベクターを環状にするために使用される張出し部が形成されるように、AatIIによって切断を行う。これによってこの手順は完了し、出発点に戻る。唯一の違いは、内部のDNA断片の第2の塩基によって張出し部が作成されるように、BspMI部位がさらに1塩基対先に位置していることである。またAatII部位はPstI部位に置き換えられるので、次のサイクルではPstI張出し部を有するアダプターが使用されなければならない。(AatIi/PstIアダプターを2回に1回使用する理由は、ベクターを環状にする前にアダプターが再び切断されるのを防ぐためである。)
[実施例3]:プルーフリーディングを可能にする標的DNA分子の隣接領域に張出し部を作成する、2種類の制限酵素を使用することによって拡大タグを導入するシークエンシング
この変異体のための主発点は、4つの塩基につき張出し部のすべての組み合わせである256のアダプター、および5つの塩基につき張出し部のすべての組み合わせである1024のアダプターである(図3A)。トップアダプター型の1024の変異体を作成しなければならず、一方ではボトムアダプター型の256の変異体を作成する。断片の相対的な大きさは、図が示すよりも大きい。両アダプターともにPstI張出し部を有し、それによってお互いに連結するのが可能になっていることに留意されたい。しかしながら、キナーゼで処理されるまでアダプター間の連結が起こらないように、張出し部はホスファターゼで処理されてしまっているはずである。
【0193】
塩基ベクターでは、第1の変異体で使用される塩基ベクターと比べて、BspMIおよびPstI部位がHgaIおよびSfaI部位に置き換えられる(図2)。
【0194】
この手順の残りは4つの酵素反応の1サイクルからなり、1サイクルあたり9つの塩基が転換される(図3B)。転換の終わりには、反応を出発点に戻しながらベクターが環状にされる。唯一の違いは、HgaIおよびSfaNI部位が押しのけられ、4つの塩基対がさらにDNA断片に入ることである。したがって次のサイクルでは4つの新しい塩基対、およびこのサイクルで転換された5つの塩基対が作成される。前述の4つの塩基対が両方のサイクルで同じようにして転換されたことを検証することによって、1つまたは複数の不適切な転換が起こったかどうかを調べることができる。
【0195】
《方法》
1)標的DNA分子を、数百の塩基対の断片が形成されるように、DNase1またはその類似物によって断片にする。これらは、HgaIおよびSfaNI部位をメチル化するために処理される。次いで断片を、常磁性ビーズに付着している塩基ベクターに連結させる。
【0196】
2)HgaI切断を行う。
【0197】
3)SfaNIを行う。
【0198】
4)HgaIおよびSfaNIメチラーゼ、またはHgaIおよびSfaNI部位を不活性化させる他のメチラーゼでメチル化を行う。
【0199】
5)かなり過剰なアダプターを加え、例えばPfuまたはTaqを使用して、段階2)および3)でHgaIおよびSfaNIによって形成された張出し部に連結させる。この段階ではPstI張出し部は連結された状態にはならない。なぜなら、それらはすでにホスファターゼで処理されているためである。
【0200】
6)アダプターのPstI張出し部をリン酸化し、次いで例えばT4DNAリガーゼで連結させ、ベクターを環状にする。
【0201】
7)このサイクルを段階2)で再び開始することによって望ましい回数だけ繰り返す。
【0202】
8)転換された標的分子を連結した標的分子の側面にある塩基ベクター内の制限開裂部位を使用することにより切断によって塩基ベクターから切り離す。拡大タグ内にありうるいかなる切断部位も事前に不活性化しておかなければならない。
【0203】
9)転換されたDNA分子を一本鎖にし、蛍光プローブとハイブリダイズさせる。
【0204】
10)転換されたDNA分子をスキャン表面に固定し伸張させ、蛍光プローブを蛍光スキャナまたはその類似品によってスキャンする(DIRVISHのように)。
【0205】
11)適切なソフトウェアを標的配列のイメージ認識および再構築のために使用する。
【0206】
これを行うことができる1つの方法は以下のようなものであり、この方法では体積はすべてビーズの体積を除いて計算する。
【0207】
1)ランダムな断片を、前に記載したような常磁性ビーズに付着した塩基ベクターにクローン化する。
【0208】
2)ビーズを沈降させるために磁石を使用し、管を約100μlの1× NE緩衝液1で洗浄する。
【0209】
3)10μlの10×NE緩衝液1、DNA1μgあたり4ユニットのHga、および水を最終体積100μlまで加える。これを37℃で1時間かけてインキュベートする。
【0210】
4)HgaI酵素を65℃で20分間かけて不活性化させる。
【0211】
5)ビーズを沈降させるために磁石を使用し、管を1×NE緩衝液3で洗浄する。
【0212】
6)10μlの10×NE緩衝液3、DNA1μgあたり2ユニットのSfaNI、および水を最終体積100μlまで加える。これを37℃で1時間かけてインキュベートする。
【0213】
7)SfaNIおよびHgaI部位をメチル化する。
【0214】
8)ビーズを沈降させるために磁石を使用し、管を1×リガーゼ緩衝液で洗浄する。
【0215】
9)転換アダプターを含む溶液を加える。標的DNA分子と転換アダプターの比は1:50であってよい。100μlの10×リガーゼ緩衝液、10μlのT4DNAリガーゼ(400U/μl、NEB #202)および水を最終体積1mlまで加える。これを16℃で12〜16時間かけてインキュベートする。
【0216】
10)ビーズを沈降させるために磁石を使用し、管を1×キナーゼ緩衝液で洗浄する。
【0217】
11)2μlの10mM rATP、10μlの10×キナーゼ緩衝液、2μlのT4ポリヌクレオチドキナーゼ(30U/μl)および水を最終体積100μlまで加える。これを37℃で10〜30分かけてインキュベートする。(United States BiochemicalsからのT4ポリヌクレオチドキナーゼ(70031)。)
12)ビーズを沈降させるために磁石を使用し、管を1×リガーゼ緩衝液で洗浄する。
【0218】
13)100μlの10×リガーゼ緩衝液、10μlのT4DNAリガーゼ(400U/μl、NEB #202)および水を最終体積1mlまで加える。これを16℃で12〜16時間かけてインキュベートする。
【0219】
14)段階2)〜13)を1回または数回繰り返す。
【0220】
例えばDNA分子を伸張させるためにBensimonの方法(Michalet他.,1997,Science,277,p1518−1523)が使用されるならば、スキャン表面当たり500kbの約100万のDNA分子を伸張させることができる。各シグナルが約5kbであるならば、これは500kbの各DNA分子が、100塩基対の配列に関する情報を提供することを意味する。このことは1つのスキャン表面が約1億の塩基対の情報を提供するであろうことを意味する。しかしながら、標的配列の首尾良い再構築は、多くの塩基対が少なくとも2回はスキャンされなければならないような配列片の重複に依存するはずである。
【0221】
[実施例4]:標的分子の末端塩基を同定するために制限部位を完成させることを含むシークエンシングの方法
この方法は、DNA代謝において活性のある多くの酵素が基質中の認識において有している高度な特異性に基づく。この方法は制限酵素とともに以下のように例示されるが、部位特異的な制限酵素、トランスポゼースなどのような他のいくつかのDNA代謝酵素も使用することができる。大部分の制限酵素にとって、塩基対の1つが開裂部位で突然変異すると、それは一般に酵素によるさらなる切断を防ぐのに充分である。この方法では、制限部位の一部のみを含むリンカーに標的分子が連結されている。標的DNAによってこの部位が完成している場所では、切断が影響される可能性があり、この後に、相補的アダプターが結合され、部位を完成させそれゆえ特定の末端塩基を表すこれらの分子を示している。アデニン用のこの方法を図4に示す。
【0222】
《方法》
1)シークエンシングすべきDNA分子を4つの異なる標準制限酵素(EnzA、EnzC、EnzG、およびEnzT)で切断する。
【0223】
2)次いでこれらの分子を4つの異なるDNAリンカー分子(分子A、C、G、およびT)に連結する。これらの分子の1つ1つは、それぞれEnzA、C、G、およびT用にほぼ完全な部位を有し、完全な塩基対を得るために末端では各分子は1つの塩基対のみ欠いている(それぞれA、C、G、およびT)。このようなリンカー分子の一例を図4Bに示す。この分子にはA/T塩基対を欠いたHindIII部位がある。このリンカーがA/T塩基対を持たないDNA片と連結するならば、DNA片から該分子を取り除くためにMnIIを使用することができる。図4Cは、末端にA/T塩基対を有するDNA片に連結されたリンカー分子Aを示し、したがって完全なHindIII部位が作成された。HindIIIが切断用に使用される次のステップでは、アダプターAに連結される可能性があるHindIII張出し部が作成されるであろう。
【0224】
3)切断を可能にするために4つの制限酵素を溶液に加える。完全な切断部位のみが存在するはずであり、各リンカー分子A、C、G、およびTは末端で塩基対を欠くDNA分子に連結する(分子A、C、G、またはT用にそれぞれA、C、G、またはT)。
【0225】
4)アダプターを張出し部に加える。この張出し部は、制限酵素によって発生しアダプターが正しいDNA片に固定されるように連結された張出し部を補う。適切なアダプターを図4Aに示す。トップアダプターをサイクル1、3、5などのために使用し、一方ボトムアダプターをサイクル2、4、6などのために使用する。これらのアダプターは、HindIIIによって生成される張出し部に相補的な張出し部を有する。トップアダプター上のAatII張出し部は、塩基ベクターが環状化されるように、アダプターの一方の末端を塩基ベクターに連結させるために使用されるはずである。MnII部位は、新しいサイクルを開始することができるように、DNA片上に平滑末端を発生させるであろう。PstI部位は次のサイクルで新しいアダプターを連結させるために使用されるはずであり、PstI張出し部を有するアダプターが使用される。
【0226】
5)アダプターの一方の末端を塩基ベクターに連結させることができるように、例えばAatIIで切断することによって、DNA片/アダプターを用いて塩基ベクターを環状にする。HindIIIが形成されない場合は、PstI部位を有する小さな断片を塩基ベクターのAatII張出し部に連結させる。
【0227】
6)新しいサイクルを開始させることができるように、DNA片上に新しい平滑末端を発生させる制限酵素によって切断を行う。
【0228】
[実施例5]:ハイブリダイゼーションおよび連結と結びついたKlenow修復反応を使用するシークエンシングの方法
この方法は、DNAポリメラーゼのKlenow部分がヌクレオチドの取り込みのための非常に高度な特異性と、大部分のDNAリガーゼには異なる大きさの張出し部を連結させる能力がないという事実に基づく。この方法は図5に示され、標的分子内で張出し部が作成され、それはアダプター分子内の張出し部より長い。張出し部を減少させるため適切に伸長し、さらなる正しい塩基を含む標的分子のみがアダプターに連結されるであろう。図5にアデニン用の方法を例示する。
【0229】
《方法》
1)図5Aに示すようにDNA片を塩基ベクターに連結させる。ストレプトアビジン基質(例えばDynalのM280ストレプトアビジン被覆磁性球)に該分子を固定するのを可能にするビオチンとは別に、塩基ベクターはポリヌクレオチド内を切断する制限酵素(例えばHgaI)のための部位、および標準的な制限酵素(例えばEcoRI)のための部位を含む。
【0230】
2)ポリヌクレオチドから5つの塩基対を有する張出し部が形成されるように、HgaIでベクターを切断する。
【0231】
3)その後、「T」で始まる張出し部が4塩基対の張出し部に短縮されるように、例えば塩基AおよびKlenowを加える。
【0232】
4)次いで反応溶液を、リガーゼおよび4塩基対の張出し部を有する遺伝子断片(アダプター)の溶液に置き換える。この張出し部は共通(universal)のヌクレオチド、および4塩基対を有する張出し部のあらゆる可能な組成物の組み合わせのいずれかを含むことができる。共通ヌクレオチドを有する張出し部は、あらゆる組み合わせの4塩基の5’張出し部に連結する能力がある。遺伝子断片は、ポリヌクレオチド内で切断を行う制限酵素(例えばHgaI)のための部位、および標準的な制限酵素(例えばEcoRI)のための部位、およびシグナル「T」を含む配列(プローブとして使用することができる配列など)も含む(図5B)。AatII張出し部およびPstI部位には実施例4の1と同じ機能がある。5塩基対の張出し部を有するポリヌクレオチドを4塩基対の張出し部を有する遺伝子断片に連結させることはできないため遺伝子断片に連結されるのは元来最も内側で「T」を有していたポリヌクレオチドだけである。成功した連結および失敗した連結を実施例5Cに示す。張出し部の内側には塩基は取り込まれなかったことから、上側のDNA片はアダプターAに連結することができない。内部に「A」を有する張出し部のみが塩基を取り込んだので、下部で見られるようにアダプターAに連結されるのはこれらの張出し部だけである。DNA片の実線は、HgaI切断を介して形成される張出し部を示す。次いで同じプロセスを塩基C、G、およびTを用いて繰り返す。
【0233】
5)最後に、塩基ベクターをDNA片/アダプターを用いて切断によって、例えばEcoRIを用いて連結によって環状にする。
【0234】
6)次いでHgaIを用いた切断を行う。これによってDNA片中に新しい張出し部が発生し、したがって反応サイクルが再び始めることができる。
【0235】
連続する塩基が同一である例では、異なる大きさの張出し部、例えば2塩基のKlenw修復を可能にする3塩基の張出し部を有するアダプターを使用することができる。
【0236】
[実施例6]:1本鎖標的分子に結合する際、プライマーとして機能するアダプターを用いるシークエンシング方法
《方法》
1)断片組成物に相当する張出し部を有し、本法に用いられるアダプターは、実施例6Aに例示される。この張出し部中の塩基組成物に対応する断片組成物に注意されたい。断片の全ての組み合わせに相当するDNAアダプターを、構築する必要がある。
【0237】
2)標的DNA片を1本鎖片にし、RNAリガーゼの助けでアダプターの3’末端およびプライマー鋳型の5’末端に連結される(図6B)。アダプターとDNA片との連結を成功させる要件は、アダプターの張出し部が、DNA片の5’末端と相補的であることである。これにより、正しい断片が、DNA片に結合されることが保証される。
【0238】
3)分子が、互いに連結する前に、標的DNA片をアダプターとハイブリダイズする。次いで1回または数回のPCRサイクルが行われる。PCRサイクルを成功させる必要条件は、ssDNA片をハイブリダイズして、アダプターに連結しておくことである。
【0239】
4)EcoRI部位が、アダプターとDNA片との連結後、形成されるDNA分子を環状化させるために用いられる。次にHgaI部位は、DNA片に新たな切断を生じるように用いられ、その結果、次のサイクルが開始できる。EcoRIで切断後の分子間連結を減じるために、この分子を、基質、例えばストレプトアビジン被覆球に固定することが有利と思われる。
【0240】
[実施例7]:自己ハイブリダイズするアダプターを用いるシークエンシング方法
本法の出発点は、シークエンシングされるDNA分子とハイブリダイズする能力を有し、同時に自己ハイブリダイズする領域に加えて、それらが、ハイブリダイズする塩基に対応する断片(拡大タグ)を担持する1本鎖DNAまたはRNAアダプターである(図7)。全ての可能な断片の組み合わせでこのようなアダプターを作成し、シークエンシングされるDNA分子とそれらをハイブリダイズすることにより、複合数アダプターを並列に並べられてもよい(図7を参照)。アダプターを正しく、すなわち、最初の2つのアダプターに誤った組み合わせがないように並べると、それらを互いに連結することが可能となり、より長い鎖を形成することができる。
【0241】
[実施例8]:1つ以上の塩基に対応するアダプターの構築方法
アダプターを構築する1つの方策は、DNAチップを構築するのに用いられる原理と同様の原理を利用する。この方法において、種々のオリゴヌクレオチド類が、DNAチップの構築と同じ方法で種々のアドレスに合成される。次いで、この同じ原理は、塩基対を伸長するオリゴヌクレオチド類に固定するのと同じ方法で、DNA片をオリゴヌクレオチド類に固定するのに用いられる。最後に、DNA分子が解かれ、アダプター溶液を得る。
【0242】
本実施例ではまた、アダプターを調製する別法を提供する。これは、図8に例示されている。図8に例示されるように、各々1つのタグを有する8種類のアダプターを、各々2つのタグを有する16種類のアダプターを合成するのに用いる。他の8種類の1タグを有するアダプターとともに出発して、16個の新たな2タグアダプターを合成でき、これは順次最初の16個と結合して、256種類の4タグアダプターを生成することができる。この方法で、アダプター混合物を生成することが可能であり、ここでは、解答に合う異なる分子の数のみが、順列の数を限定する。最初に必要とされる、異なる1タグアダプターの数は、各アダプターにおけるタグ数の4倍に等しい。例えば、我々が、16タグのアダプター類(4.29×109順列)を作成しようとする場合、16×4種類の1タグアダプターが、最初に必要とされる。
【0243】
《方法》
1)図8に示すように8種類の1−タグアダプターが、用いられる。左側のアダプター類は、EcoRI張出し部、分子上の極く右側にある塩基に特異的なタグ、およびBseMIのための開裂部位から成る。BseMIは、それ自身の部位の外側を切断し、分子右側の塩基のすぐ隣で平滑な切断を生じさせる。アダプターはまた、ホスファターゼで処理されて、これらのアダプター間の連結を減じる。右側のアダプターは、分子の極く左側の塩基に対応するタグおよびEcoRIのための切断部位から成る。アダプターはまた、基質に固定され、これらのアダプター間の連結を防止する。
【0244】
2)2つのアダプター集団を混合し、連結させることにより工程が開始される。これにより、2つのタグを有する全順列に対応する16種類のDNA分子を生成する。
【0245】
3)EcoRIは、開裂に用いられる。次いで、これらの分子を環状化させるために、連結が実施される。
【0246】
4)最後に、BseMIにより開裂を実施すると、16種の2−タグアダプターを有する集団が生成される。
【0247】
[実施例9]:ヘアピン型アダプターに基づく転換方法
前記のいくつかの転換代替方法における主要な点は、塩基対の転換後、DNA断片を、転換されつつあるDNA分子の他の末端に移動させることである。これにより、転換されつつある末端を開放し、DNA断片を蓄えると同時に、次のサイクルに進むことができる。DNA断片をDNA片の他の末端に移動させる他の方法は、以下に示す。
【0248】
出発点は、T4DNAリガーゼなどの多くのリガーゼ類が、dsDNAの張出し部をssDNAの末端に連結できることである。これは、図9に例示した方法に利用され得る。
【0249】
《方法》
1)標的DNAを、1本鎖形に転換する。
【0250】
2)転換アダプターを添加し、DNA片の3’末端に連結する。
【0251】
3)ポリメラーゼ伸長を実施する。
【0252】
4)ヘアピンア型アダプターを添加し、連結する。転換アダプターの末端を予め処理すると、ヘアピン型アダプターに連結しない(例えば、ホスファターゼで処理)。
【0253】
5)DNA分子を融解する。
【0254】
6)1本鎖DNA分子を、断片と相補的なDNA分子とハイブリダイズする。相補的DNA分子はまた、標的DNA中の第1の塩基とハイブリダイズする共通(ユニバーサル)塩基の付いた張出し部を有する。
【0255】
7)自身の認識配列の外側に張出し部を形成する酵素に対する開裂部位の助けにより、次の転換サイクルのためにDNAが作成される。
【0256】
上記の各サイクルにおいて、拡大タグは、各サイクルにおいて複製される。これにより、プルーフリーディングを可能にする。
【0257】
[実施例10]:転換されたDNA分子の連結に基づく転換法
前記の多くの転換法は、サイクル工程で生じる転換に基づいている。それにより拡大タグ(またはシグナル鎖)の1本鎖当たりの転換塩基対数は、サイクル数と共に直線的に増加する。別の方法としては、転換DNA片を長鎖中に結合することである。この原理に基づく方法としては、非常に多くの可能な方法があるが、以下に1つの提案を示し、図10に例示する。
【0258】
《方法》
本法は、大きさに従って標的DNAを切断し、ソーティングすることから始まる。次に、特定の長さ、例えば30塩基対のDNA片を、この手順から除かれる。
【0259】
1)DNA片の末端を、前記方法を用いて転換する。
【0260】
2)転換DNA分子を環状化させる。
【0261】
3)転換アダプターの末端に位置する開裂部位を用いるIIS酵素を添加する。これは、図10に示すようにDNA片を切断する。
【0262】
4)このDNA分子を融解し、断片、例えば蛍光体で標識したプローブに相補的なDNA分子とハイブリダイズする。
【0263】
5)最後に、転換DNA片をハイブリダイズし連結するが、必要なら溶液中で行う。
【0264】
前記実施例において標的DNAを有する張出し部は、相補的な張出し部を捜し求めることから、各転換DNA片は、遭遇したDNA片とハイブリダイズ/連結する。これにより、22の未知塩基(例えば、AGCTGTGA N22 AGTCTGCA N22 TGAC)により遮断される8塩基対の配列片についての情報を提供する拡大タグ鎖(シグナル鎖)を生み出す。未知の塩基対数は、DNA片の当初の長さから、1DNA片当たり転換した塩基対の数を差し引くことにより決定される。シグナル鎖間の重複に基づいて、次に、繰返しの配列の領域においても標的配列を再構築することが可能である。
【0265】
[実施例11]:DNAのダブリング化(doubling)法
1本鎖DNA分子は、図11に示されるように2回のダブリングサイクルに供される。ダブリング(doubling)は、ヘアピン型アダプターを分子の3’末端に連結させることにより開始する。例えば、逆転写酵素が、プライマーとして3’ヘアピンループを用いるのと同じ方法で、アダプターを、分子を伸長するポリメラーゼのためのプライマーとして使用することができる。最後に、DNA分子を溶解して出発点に戻る。長さy bpであるアダプターの助けによりx bpのDNA分子をn倍にダブリングする本法を用いると、DNAの長さは:
・ x・2n+(2n−1)y
となる。次に、ダブリング前、各々xおよびx+1 bpであった2つのDNA分子間の差は:
2)(x+1)・2n+(2n−1)y−x・2n+(2n−1)y=2n
となる。
【0266】
したがって、2つのダブリングされたDNA分子間の長さの差は、増幅前のそれらの絶対的な長さの差によってのみ決定され、相対的な長さの差によってではない。
【0267】
[実施例12]:プライマーに基づくソーティング法を利用するDNAシークエンシング方法
本実施例は、16種類の異なるゲル分離および16種類の標識、例えば蛍光団を用いて、互いに分離できる256種類のシークエンシング梯子の作成法を説明する。シークエンス反応の長さは、4種類のシークエンシング梯子のみを用いる方法と比較してかなり増加できる。したがって、ソーティング作業量、長い配列をシークエンシングする際などに必要なプライマー数をとりわけ減じることが可能である。16種類のシークエンシング梯子並びに16種類の発蛍光団が、本実施例で使用されるが、明らかにこのシークエンシング梯子数、および蛍光団数は、大部分の要件および利用できる装置に適合できる。シークエンシング梯子および蛍光団をより多く使用するほど、シークエンス反応はより長くなる可能性がある。
【0268】
《方法》
使用する本法の概要を、図12Aに示す。
【0269】
1)標的DNA溶液を、ウェルの基質に固着されたシークエンシング・プライマーを含有する16個のウェルに分配する。これらのプライマーは、ポリメラーゼ反応のための一定の出発点を決定することから、この共通の起点により、生成する最終生成物のサイズは、その起点から最終配列の距離を示すものとされる。
【0270】
2)ポリメラーゼ伸長反応を実施し、DNA分子を加熱して融解し、次いでウェルを洗浄する。
【0271】
3)次に、図12Bに示すように、16種のプライマー類を、16個のウェルの各々に添加する(全部で256種のプライマー)。各ウェルに添加された全てのプライマーは、3’末端での塩基3および4以外は同一である。この位置にAAを有するプライマー類は、シグナル1に連結され、この位置にACを有するプライマー類は、シグナル2などに連結される。ウェル2のプライマー類は、それらが3’末端にAAの替わりに、例えば、ACで開始すること以外は同一であるが、ウェル3のプライマー類は、AGなどで開始する。このように全部で、3’末端に256種の4塩基順列の全て包含する256種のプライマー類がある。特定の蛍光シグナルを、各16種類のプライマーに付加する。次いで、さらなるポリメラーゼの伸長反応を実施する。
【0272】
4)次いでウェルを洗浄してからDNA分子を融解する。次に、これにより解かれた1本鎖DNA分子を、16の異なるゲル分離(各ウェルにつき1つ)を用いてサイズに従ってソーティングする。
【0273】
5)蛍光シグナルを記録し、標的配列を、適当なソフトウェアで再構築する。
【0274】
《結果》
この結果を図12Cに示す。各蛍光シグナルは、4塩基配列断片についての情報を提供する。最初の2つの塩基についての情報は、蛍光シグナルが読取られるウェルの参照により引き出し得るが、最後の2つの塩基は、存在する特定のシグナルに基づいて決定できる。
【0275】
[実施例13]:ハイブリダイゼーションに基づくソーティング方法を利用するDNAシークエンシング方法
本法では標的DNA分子を、走査表面にあるオクタマーとのハイブリダイゼーションによって走査表面の異なる部位にアドレスする。次いで、この分子を引き伸ばし、蛍光シグナルから垂直保留線までの距離が評価され、標的配列中のオクタマーの位置についての情報が提供される。一般手順を、図13に示す。
【0276】
《方法》
1)出発点は、65,536のアドレスから成る走査表面である。1本鎖オクタマーを有する垂直保留線を各アドレスに付される。AAAAAAAAオクタマーを、アドレス1のプレートに固定し、AAAAAAACオクタマーは、アドレス2のプレートに固定するなどの結果、65,536の各オクタマー順列全てが、それ自身のアドレスを有することになる。
【0277】
2)次に、一末端または両末端に蛍光標識を有する1本鎖標的DNA分子を、走査表面上に混合し、それらをオクタマーにハイブリダイズさせることができる(図13A)。
【0278】
3)所望するなら、分子をUV照射に曝露することにより、オクタマー/標的DNA結合を強化し、オクタマーをプライマーとして、または他の手段によりポリメラーゼ伸長を実施してもよい。
【0279】
4)次に、走査表面を洗浄し、DNA分子を伸展する(図13B)。
【0280】
5)蛍光スキャナを用いて表面を走査し、保留線に対する距離の関数として、各アドレスにおける蛍光強度を記録し、標的配列を適当なソフトウェアを用いて
再構築する。
【0281】
《結果》
得られた結果を図13Cに示す。例示したアドレスにおいて、約150kb、約300kb、約500kb、約550kb、約780kb、約870kbおよび約1040kb(DNA分子が1マイクロメートル当たり2kb伸長する場合)と7種類のDNA分子長がある。
【0282】
[実施例14]:リガーゼに基づくソーティング法を利用するDNAシークエンシング法
本実施例は、リガーゼに基づくソーティング法であり、65,536のシークエンシング梯子が、65,536のアドレスにソーティングされる。各々が塩基を表す4個のシークエンシング梯子を利用する他の方法とは対照的に、本法における65,536のシークエンシング梯子の各々は、8個の塩基の配列片を表すことになる。これは、4個の梯子のみを用いる方法と比較して、サイズによるソーティングに対する精密要件を減ずる。反応の長さは、このように増加でき、サイズによってポリマー類をソーティングするための広範囲の方法を利用することもまた可能である。
【0283】
本実施例においては、サイズによるソーティングは、伸長したDNA分子の長さを測定する方法により示される。しかし、サイズによるソーティングを走査表面上で直接実施するなど他の変法が考えられる−例えば、DNA分子のシグナル強度が長さに比例するといった標識方法を使用してから、DNA分子のシグナル強度を測定するなどの方法がある。また、シークエンシング梯子を、物理的に分離した状態において時間などを違えて基質から解放し、フローサイトメーター、質量分析、ナノポア分析、ゲルソーティング法などを用いて各65,536のシークエンシング梯子を個々に分析することを可能にさせる変法もまた考えられる。
【0284】
《方法》
1)例えば1Mbの標的配列から出発して、ここに記載されるように調製し、シークエンシング梯子を生成する。
【0285】
2)標的DNAをメチル化して、段階3)および6)で用いられる制限酵素の開裂部位を不活化させる。
【0286】
3)4塩基の張出し部を、ここに記載しているように標的DNAの任意の末端に作成する。DNA分子の任意の末端を、例えば、4塩基対の張出し部を作るIIS酵素の結合部位を含有するDNAリンカーに連結してもよい。結合部位を、実際の標的DNAに張出し部を作るために配置する。次いで分子をIIS酵素で開裂する。
【0287】
4)次に標的DNAは、256のウェル間に溶液を分配することにより、実施例12に記載のとおりソーティングする。ウェル壁を、段階3)で作成された張出し部と相補的であり得る4塩基張出し部を有するソーティングアダプターで覆う。ソーティングアダプターはまた、IIS制限酵素、例えば、FokIに対する結合部位を含み、この結合部位は、張出し部が、段階5で切断されたもののそばにある4塩基対を含んで形成できるように配置される。
【0288】
5)標的DNAをソーティングアダプターと連結し、次に管を洗浄して、結合しなかったDNAを除く。
【0289】
6)IIS酵素による開裂を実施し、標的DNAを解放して新たに4塩基張出し部を形成する。
【0290】
7)実施例12に記載しているように、標的DNAを256のウェルから256のミクロアレイ間に分配する。全ミクロアレイは等しく、段階6)で作成された張出し部と相補的であり得る4塩基張出し部を有するソーティングアダプターの付いた256のアドレスから成る。アドレス1において、ソーティングアダプターは、AAAA張出し部を有し、アドレス2では、AAAC張出し部を有するなどとなる。
【0291】
8)リガーゼを添加し、この混合物をインキュベートする。アドレス1において、TTTT張出し部を有する標的DNAが存在し、アドレス2ではTTTG張出し部などを有する標的DNAが存在することとなるであろう。
【0292】
9)走査面を洗浄し、DNA分子をアドレス化し、TOTO−1,YOYO−1または同様のもので着色する。
【0293】
10)CCDカメラまたは同様のものが、アドレスを撮影するために使用される。CCDカメラを、例えば、1アドレス当たり1枚の写真を撮るために設定してよい。
【0294】
11)蛍光DNA分子を認識し、それらの長さを測定し、次いで実際の標的配列を再構築するために適当なソフトウェアを使用する。
【0295】
これを実施できる1つの方法は、以下のとおりである:
1)各ウェルに、任意の4塩基張出し部を含む標的DNA分子の一定分量、10μlの10×リガーゼ緩衝液、1μlのT4DNAリガーゼ(400U/μl、NEB#202)を加え、最終容量が100μlになるように水を加える。16℃で12〜16時間インキュベートする。
【0296】
2)液体を除去し、1×NE緩衝液4でウェルを1回または数回洗浄する。
【0297】
3)10μlの10×NE緩衝液4、DNA1μg当たり4単位のFokI(New England Biolabs、#109)を加え、最終容量が100μlになるように水を加える。37℃で1時間インキュベートする。
【0298】
4)65℃で20分間不活化する。
【0299】
5)EtOHにより個々の管の各ウェルからDNA分子を沈殿させる。
【0300】
6)ペレットを溶解し、10μlの10×リガーゼ緩衝液、1μlのT4DNAリガーゼ(400U/μl、NEB#202)を加え、最終容量が100μlになるように水を加える。16℃で12〜16時間マイクロアレイでインキュベートする。
【0301】
7)分子を伸長し、標識して解析する。
【0302】
《結果》
特定のアドレスにおける分子の有無およびサイズは、配列情報およびその位置の双方を示す。したがって、マイクロアレイ1のアドレス1が、100マイクロメートルのDNA分子を含む場合、これは、その分子を結合するために用いたオクタマーに相当する配列が(たとえ2段階ソーティングによっても)、+200kb(例えば、TTTTTTTT)で存在することを示す。同様に、2つの異なる大きさの分子の存在は、特定配列の反復を示すと思われる。特定アドレスにおける任意の分子の欠如は、標的配列において固定化オクタマーに相補的な配列の欠如を示すものと思われる。
【0303】
上記実施例において、誤ったソーティングの潜在的な原因としては、粗ソーティングアダプター類がまた、精ソーティングアダプター類として働く可能性が考えられる。しかしながら、この問題は、粗ソーティングアダプター類に別の制限エンドヌクレアーゼのための切断部位を持たせることにより避けることができ、これにより、走査する前に粗ソーティングアダプター類を切り離すことができる。上記実施例で記載しなかったが、粗ソーティングアダプターに付加していないDNA片の先を末端となすこともまた重要である。これは、例えばKlenow充填により行い得る。
【0304】
Bensimon法(Michaletら、1997年、上記)を、DNA分子を伸長するため上記の方法において用いると、100万〜200万のDNA分子を、1.28×1.28cmを測定する走査面上で伸長できる。256のアドレスの各々は、約4、000〜8、000の伸長DNA分子を含むであろう。8塩基対を有する配列片が、65,536番目の塩基対ごとに反復されるため、標的配列が1Mb(1,000,000/65,536=15.2)であると、各アドレスに平均して15種類の長さがあることになる。したがって、各長さは、平均して260〜520回(4,000〜8,000/15.2=260〜520)測定されることになる。
【0305】
[実施例15]:標的DNA分子を固定参照点に固定されるDNAシークエンシング法
本法においては、直線状配列の拡大タグ(シグナル鎖)を担持するPNAオクタマー類を、走査面上に固定化された標的DNAにハイブリダイズし、次にそれがスキャンされ、標的配列内のオクタマー類に相補的な領域の位置が決定される。一般的手順を図14に示す。
【0306】
《方法》
1)このプロトコルの出発点は、本明細書記載されるように走査面上の固定された参照点、例えば走査板に垂直な保留線に標的2本鎖DNA分子を固着することである。
【0307】
2)上記オクタマーに対応する組成物を有するシグナル鎖に付加されたPNAオクタマーカーら成る転換アダプター類(すなわち、拡大タグを担持するアダプター類)の65,536の順列を、次に添加する。シグナルは、蛍光標識球、ビーズであってよく、または他の適当な標識を担持してよい。このように、PNA分子を標的DNA分子とハイブリダイズする。
【0308】
3)分子を伸長し、それらの位置およびシグナル鎖の組成を記録した。
【0309】
4)適当なソフトウェアを、標的配列を再構築するために使用する。
【0310】
《結果》
この結果を図14に示す。シグナル鎖と固定した保留点との間の距離は、標的配列の各配列片の位置について情報を提供する。
【0311】
[実施例16]:ソーティングまたは転換と組み合わせたソーティングに基づくシークエンシングおよび光学的マッピング法
本法により、特別の長さのDNA配列、例えば、ゲノム類をマッピングまたはシークエンス反応においてマッピングまたはシークエンシングすることが可能になる。この方法は、光学的マッピング単独にまたはマッピングに加えてシークエンシングに用いることができる。この方法が、同一のシークエンス反応において多数の異なる標的配列類のシークエンシングを可能にすることに注目することが重要である。
【0312】
《方法(ソーティング単独)》
実施例14の方法に従うが、段階1の替わりに標的DNAをDNアーゼIまたは同様のもので切断して、数百の塩基の断片類を生成する。段階2から段階8を、実施例14に記載されるとおり実施する。次いで、走査面を洗浄し、DNA分子を伸長する。光学的マッピング法または同様の方法を、次に実施する。走査面を、蛍光スキャナまたは同様のもので走査し、配列を再構築するために適当なソフトウェアを使用する。
【0313】
《方法(転換を伴うソーティング)》
段階1から段階6を、上記の光学的マッピング法のとおり実施する、その後、
7)256の転換アダプターを添加し、段階6)で形成された張出し部と連結する。転換アダプターは、例えば、1のシグナルが、特定の制限酵素に係る多数の開裂部位を含むDNA配列類であり、0のシグナルは、そのような部位を含まないDNA配列類である2進法シグナル鎖を有してもよい。
【0314】
8)各ウェルからの転換DNA分子を、それ自体の走査表面に移し、1シグナルにおいて切断部位を有する制限酵素を用い光学的マッピング法を実施する。
【0315】
9)走査面を、蛍光スキャナまたは同様のもので走査し、適当なソフトウェアを、配列を再構築するために使用する。
【0316】
[実施例17]:2進末端転換およびナノポア分析に基づくDNA塩基シークエンシング法
電場により、脂質膜のイオンチャネルを介して1本鎖RNAおよびDNA分子を駆動できることが示された。分子の移動は、イオン電流の一過性の減少として検出できる。大きさの違いのために、プリン類とピリミジン類とを識別することが可能であることが示された。したがって、この方法は、高速シークエンシングに利用できることが示唆された。しかしながら、大きさの相異が小さいために、異なるプリン類(アデニンまたはグアニン)間および異なるピリミジン類(シトシン、チミンまたはウラシル)間を識別することが困難であることが示された。本実施例においては、この問題を、標的のDNAをプリン/ピリミジンシグナルから成る2成分コードに転換することにより如何に解決できるかが示される。
【0317】
《方法》
1)標的DNAの断片は、DNアーゼIなどで開裂することにより生成し、平滑末端を作成するために処理された。
【0318】
2)標的DNA分子を、IIS制限酵素(例えばFokI)の1つまたは複数の結合部位を含むリンカー類と連結する。
【0319】
3)張出し部を、IIS制限酵素での開裂により標的DNA中に生成させる。
【0320】
4)張出し部を、ホスファターゼ酵素で処理する。
【0321】
5)張出し部を、転換アダプターと連結させる。アダプターにはまた、方向タグも含んでいて、ソフトウェア分析をより容易にする。
【0322】
6)プリン類/ピリミジン類の組成を、ナノポア分析により読取る。転換されなかった標的DNA部分を、位置情報を得るために使用してもよい。
【0323】
7)適当なソフトウェアプログラムを、標的配列を再構築するのに使用する。転換アダプターと標的DNAとの間の張出し領域は、プルーフリーディングとして配列片情報と比較できる。
【0324】
《結果》
シグナルは、A=プリン+プリン;C=プリン+ピリミジンなどの2成分プリン/ピリミジンコードから成る。これを図15に示す。
【0325】
[実施例18]:シークエンシング反応のシグナル生成における細胞の利用
本実施例は、シークエンシング反応においてシグナル生成および特定の塩基を表示する拡大タグとしてそれ自体の作用双方のため細胞の利用を例示する。
【0326】
《方法》
A)本法において、レポーター遺伝子が、拡大タグとして使用され、発現による相対的シグナル強度が、配列中の特定塩基の相対位置の指標として使用される。使用される方法および得られる結果を図16に示す。
【0327】
1)ポリメラーゼ伸長反応は、鋳型としての標的DNA、および図16に例示するように、エンハンサーおよびレポーター遺伝子を有する1本鎖または2本鎖に付加されるシークエンシングプライマーを用いて実施する。シークエンシングプライマーは、シークエンシングしようとする配列の開始を表示する標的DNA中の既知配列に結合するために使用される。
【0328】
2)ポリメラーゼ伸長を、4つの異なるプライマーから成るプライマー混合物により実施する。各プライマーは、A、C、GまたはTの何れかであるたいていの3’塩基を除き、ユニバーサル塩基(U)またはランダム塩基(N)から成る。プライマーは、4つの異なるレポーター遺伝子に付加する。ほとんどの3’位にAを有するプライマー類は、レポーター遺伝子Aに付加し、他も同様にする。この段階で使用される転換プライマー類は、ポリメラーゼ伸長の成功に重要な大部分の3’塩基を除いて、無作為に標的DNAに結合する。
【0329】
3)1つまたは複数のポリメラーゼ伸長反応を、段階1および段階2に使用されるプライマー類の5’末端に相補的なプライマーを用いて実施する。
【0330】
4)転換されたDNA分子を、好適な細胞に形質転換/形質移入する。
【0331】
5)細胞を、レポーター遺伝子が発現できる条件下で増殖させる。
【0332】
6)レポーター遺伝子の発現は、フローサイトメーターで分析され、好適なソフトウェアを配列を再構築するために使用する。
【0333】
B)本法において、異なる塩基と関連するシグナルが、細胞内の異なる位置またはシグナル鎖内それらの位置について他の構造表示に向けられる。
【0334】
1)標的DNAを、DNアーゼIまたは同様の技術により断片化する。
【0335】
2)1標的DNA分子あたり16の塩基対を1つのシグナル鎖に転換する。4つのシグナルが、塩基A、C、GまたはTの各々を表示するのに用いられる。各シグナルは、各シグナルについて異なる位置に発現するプロモーターに結合したレポーター遺伝子A、C、GまたはTから成る、すなわち、16塩基対では、シグナルは16種のプロモーターにより16の異なる位置に向けられる。この位置としては、多細胞生物中の細胞または細胞群であってもよい。それはまた、細胞上(例えば、外膜の一部)の位置であってもよい。シグナル鎖を、生物体/構造体のもとなる細胞に形質転換/形質移入する。
【0336】
3)細胞を、生物体/構造体が発育できる条件下で増殖させる。
【0337】
4)各生物体/構造体における4つの異なるシグナルの分布を、異なる位置に記録し、シグナル鎖を伸展させるために用いられる配列に沿って、いずれの塩基がどの位置に出現するかの写真を作成する。
【0338】
《結果》
A)特定の位置に生成したシグナル強度が調査できる。これを図16に示す。シグナル強度は、エンハンサーとレポーター遺伝子との間の距離に逆比例することから、出発塩基に関して特定シグナル(したがって塩基)の位置を確定できる。
【0339】
理想的には、創製される異種分子間の識別を助けるために、標的核酸分子を、伸長生成物が複数塩基だけ異なるように、末端配列に従って最初にソーティングする。
【0340】
[実施例19]:各々3つの拡大塩基だけ異なるシークエンシング梯子を創製することによるシークエンシング法
本法は、転換(すなわち拡大)および配列の読み取りが同一の固形担体上で実施されるシークエンシング梯子の形成を記載する。これらの手順における重要な点は、短い領域(3塩基増すごとに6〜9bp以上)の塩基組成物を獲得することに加えて、より大きなDNA分子(数kbまで)上のそれらの内部位置についての情報もまた得ることである。これは、配列情報の再構築にとって重要であり、この方法は、例えば、前記の変法を介して引き出された配列情報を補足するのに用いることができる。この原理を、9塩基対の長さのポリヌクレオチドのシークエンシングに関して図17に例示する。
【0341】
《方法》
1)シークエンシングしようとするDNA配列は、PCRにより増幅される。プライマーの1つの一端をビオチンで標識し、その結果DNA分子をストレプトアビジン基質に固定できる。ストレプトアビジンは、細い線に並べて、DNA分子を互いに隣接した1列に固定する。
【0342】
2)この分子を、DNアーゼI(または同様のもの)で処理し、無作為な切断を生じさせる(図17の段階1)。
【0343】
3)切断末端を、それ自体の結合部位の外側を切断するクラスIIの制限エンドヌクレアーゼ(この場合EarI)に対する結合部位を含むポリヌクレオチドに連結させる(図17の段階2)。
【0344】
4)次に、制限エンドヌクレアーゼを加えて、該ポリヌクレオチドに張出し部を創製する(図17の段階3)。
【0345】
5)ポリヌクレオチド張出し部を認識し、特異的に連結するアダプターを加える(図17の段階4)。それにより、AGC張出しを有する上部のポリヌクレオチドを、AGC断片の組み合わせなどを有するアダプターに連結させる。
【0346】
6)DNA分子は、液体流、電界または同様のものの助けにより引き伸ばされて、蛍光標識されたアダプター類を、蛍光スキャナで読取ることができる。
【0347】
7)この配列を、配列情報を有する断片を並べることにより再構築する。
【0348】
各アダプターについての相対位置が、ポリヌクレオチドのDNアーゼIにより切断される場所により変化することに注意されたい。この方法において、配列情報を有する各断片が、ポリヌクレオチド上に相対的位置を与えられ、これにより、配列の再構築を、より容易にする。
【0349】
最後に、同一の読み取りプレート上(図17)で異なる数種のDNA配列読取ることにより、読み取りの可能性を増大できることを強調する必要がある。例えば、PCRの助けにより、遺伝子特異的プライマーを使って多数の遺伝子を増幅することができる。次いで、特定の張出し部が、増幅遺伝子配列上で作成される。これにより、例えば、最後のサイクルにおいて特別に長いプライマー類を用いることにより実施でき、めったに切断しない制限エンドヌクレアーゼなどの切断部位を挿入できる。それにより、各区画が、種々の遺伝子に特異的なオリゴヌクレオチドから成る、DNAチップに遺伝子をハイブリダイズすることができる。それにより、遺伝子Aに対応するDNA分子は、区画Aにハイブリダイズし、遺伝子Bは、区画Bにハイブリダイズするなどとなる。医療などで関心のあるゲノムにおいて、これらの特異的な領域を選び出すことが可能な場合、この方法は、人々のゲノム類のマススクリーニングにとって特に適切である。
【0350】
異なる遺伝子の平行シークエンシングはまた、遺伝子特異的プライマー類を用いる増幅により達成できる。次に、各遺伝子に特異的な張出し部を生成できてから、遺伝子類は、DNAチップ上のオリゴヌクレオチド類にハイブリダイズされる。このDNAチップは、異なる遺伝子類に相補的なオリゴヌクレオチド類が、異なる部位を有するように構築される。実際に、同一の読み取りプレート上に数千の異なるアドレスを創製することが可能であり、その結果平行して数千の遺伝子をシークエンシングすることが可能である。
【0351】
図18に示すように、さらなる変法は、2次元で位置情報を得ることである。
【0352】
1)この手順の出発点は、シークエンシングされるDNA分子を、2、3kb以上の長さの分子に切断することである。次いで、ビオチンを、DNA分子に取り込ませると、例えば、平均して二、三百の塩基(必要とされるものに依っては、それ以上または以下)の間隔でビオチンを有する塩基があることになる。次に、このDNA分子は、ストレプトアビジンで被覆されるプレートに一端が固定される。この末端についての固定化機構は、ストレプトアビジン/ビオチンとは違うものである必要がある。
【0353】
2)この分子は、液体流、電界または他の手段の助けにより引き伸ばされる。このDNA分子を、ビオチン−ストレプトアビジン結合を創製する反応溶液を添加することにより基質に保留する。
【0354】
3)次にこのDNA分子を、DNアーゼIまたは他の手段で切断してから、遊離末端を、IIS型制限エンドヌクレアーゼ(示していないが)の結合部位を含有するプレアダプターに連結する。次いで、それぞれのエンドヌクレアーゼで切断することにより、配列情報を有するアダプター類に連結する張出し部が生じる。
【0355】
4)次に、ACGTの断片組合せをもつアダプターを、ACGT張出し部などに連結する。
【0356】
5)液体流、電界または同様の工程が、シークエンシングされるDNA分子の方向に対して、90°の方向にDNAアダプター類を引き伸ばすために使用されてから、それらを、ストレプトアビジン/ビオチン系で固着する。全てのDNA分子が、基質に固着されたら、この工程を、塩基対の所望する数が、転換拡大されるまで、繰り返すことができる。また、アダプター間の相対距離が、シークエンシングされるDNA分子中の配列片の内部距離に対応することを注目されたい。1枚の読み取りプレート上に、極めて多数のDNA分子を平行してシークエンシングすることが、勿論可能であることも言及するべきだろう。
【0357】
[実施例20]:標的分子から拡大部分の間隔を空けるリンカーの付いたアダプターを利用するシークエンシング法
以下に述べられた方法は、シークエンシングの1サイクル以上が達成できる方法を示している。この方法では、創製される拡大タグが、固形担体に付けられる。続いて、対応する配列からタグの間隔を空けるリンカーが取り除かれ、次に標的塩基配列の隣接部分が拡大される。この手順を図19に示す。
【0358】
《方法》
1)シークエンシングしようとするDNA分子、ACGTGAGCTの一端をストレプトアビジン被覆のプレートに固定する。その固定化機構は、ストレプトアビジン/ビオチン以外の機構である必要がある。
【0359】
2)結合部位の外側に切断部位を有するタイプIIの制限エンドヌクレアーゼ(例えば、図19に示すようなBspMI)に係る結合部位を含むポリヌクレオチドにDNA分子を連結する。
【0360】
3)次の段階で、制限エンドヌクレアーゼを加え、開裂が、シークエンシングしようとするDNA分子からの塩基を有する張出し部を形成する。
【0361】
4)次に、種々のアダプターとリガーゼを有する溶液を加える。図19は、ACGT張出し部を認識し、これと結合したアダプターを示す。ACGT張出し部に対応する蛍光標識した断片に加えて、2個以上のビオチン分子がアダプターに組込まれた。
【0362】
5)DNA分子を、液体流または電界の助けによって引き伸ばす。断片は図のように基質に固定することができる。(DNAリンカー領域の機能は、シークエンシングするDNA分子から断片の間隔を空けることである。これにより、次の段階における新たなアダプターのための余地が残る。)
6)SmaIおよびBspMIによって開裂が行われ、DNAの結合が取り除かれると同時に、DNA分子上に次の4塩基対から成る新たな張出し部が形成される。このことにより、新たなアダプターは、蛍光標識された断片と連結することが可能となる。唯一の違いは、このアダプターがDNAの結合を含まないことである。次に、蛍光標識したアダプターは、ストレプトアビジン基質上の新たな位置に固定されことになる。種々の長さのDNA結合を使って、複数の連続的な転換サイクルを実施することが可能である。
【0363】
[実施例21]:シークエンシング法における位置マーカーの利用
この方法では、シークエンシングされる分子に、位置マーカーが付され、得られる配列情報の位置を定める助けとなる。
【0364】
《方法》
用いられる方法を図20に示す。出発点は、例えば、100kbの環状標的DNA分子である。この分子は、明るい灰色および暗い灰色で印を付した2種の配列を含んでおり(図20)、これは、位置マーカーとして利用されることになる。
【0365】
1)DNA分子をBst71Iメチラーゼでメチル化する。
【0366】
2)上記分子をDNアーゼIまたは同様のもので線状化し、その後Bst71Iに対する開裂部位を含むアダプターを連結反応により付加する(標的DNA分子内の最初の4塩基を有する張出し部を作るために利用できるように、切断部位を配置する。それによって2つの4bp張出し部は、8bp上の連続的配列についての情報を提供できるようになる)。
【0367】
3)Bst71Iによって開裂を行い、断片アダプターを付加して連結する。
【0368】
4)DNA分子を1本鎖形に転換してから、モレキュラーコーミング(molecular combing)電界または同様の手段で、スライド上に固定し、引き伸ばし、同時に断片と位置マーカーを認識する蛍光プローブとハイブリダイズする。また、DNA分子をYOYO−1または同様のもので染色するのも適切であると思われる。
【0369】
5)次に、蛍光顕微鏡/スキャナを使って、配列片を走査し、位置マーカーに付いていたプローブまでの距離を測定する。これにより、シークエンシングするDNA分子上の8bpの配列片各々におおよその位置を割り当てることができる。
【0370】
[実施例22]:ソーティング後の拡大を含むシークエンシング法
この方法では、断片は、それらの末端の4塩基によって固形担体上にソーティングされた後、分子の終末端が固形担体に付着される。次いで、隣接する4塩基が、拡大により評価される。この方法の例を図21に示す。
【0371】
《方法》
256のアドレスに分けたDNAチップを使う。各アドレスは、ソーティングアダプター、張出し部、クラスIISの制限エンドヌクレアーゼに係る結合部位および平滑末端切断を行う制限エンドヌクレアーゼに係る結合部位を含む。張出し部は、アドレスによって変化するため、アドレス1は、AAAA張出し部の付いたソーティングアダプターを有し、アドレス2は、AAAC張出し部を有するものとなる。さらに、全てのアドレスは、結合性を有する分子、例えば、ストレプトアビジンで被覆される。
【0372】
1)標的DNAを断片に切断し、4塩基の張出し部を形成するようDNA断片の末端を処理してから、ソーティングが開始される。
【0373】
2)断片が連結されるソーティングアダプターを担持する固形担体に導入される。TTTT張出し部を有するDNA片は、ソーティングアダプターが、相補的な張出し部AAAAを有するアドレス1に結合し、GTTT張出し部を有するDNA片は、アドレス2に結合するなどとなる。
【0374】
3)DNA片上の他の張出し部は、その末端が下敷に固定できるように処理される。例えば、これは、Klenow充填反応により達成でき、該末端をビオチンで標識したり、ビオチン標識したユニバーサルアダプターに末端結合したりする。
【0375】
4)IISおよび平滑末端酵素による開裂を実施する(この場合は、FokIおよびDraIを用いて示される)。このようにして、DNA片内に次の4塩基を表す新たな張出し部が得られる。
【0376】
5)転換アダプターを加え、これらの塩基をシグナル鎖へと転換する。
【0377】
6)DNA分子が引き伸ばされ、例えば、蛍光スキャナを用いて走査する。DNA分子の一端の位置は、我々に4塩基についての情報を提供し、他の末端のシグナル鎖は、次の4塩基についての情報を提供する。
【0378】
[実施例23]:マッピング手順、プロフィル解析などのための標的分子調製方法
このプロトコルの背景となる原理は、自身の認識配列外側の切断を優先的に行う1種またはいくつかのヌクレアーゼ、例えばIIS酵素による標的DNAを分解することであるが、他の種類のヌクレアーゼもまた、例えば−5から+5の範囲の張出し部を作るために用いることができる。次に張出し部は、配列情報を含む一部分、および張出し部の性質についての情報を含む一部分(すなわち、どの制限酵素がその張出し部を作ったか)から成るシグナル鎖に連結される。分解された分子の各々は、これにより、末端のシグナル組成および両末端間の長さの異なる記号へと転換される。両端を相補的配列で並べることにより(例えば、温浸で創製された相補的張出し部に付着する1つまたは複数の拡大タグの整列)、この情報が制限マップを作るために利用することができる。この記号はまた、混合DNA集団における標的配列の同定に利用することもできる。FokIを利用する原理を図22に示す。
【0379】
水に溶解した100kbBACクローンである標的DNAにおけるFokI部位のマップを作りたい場合、以下のプロトコルが利用できる。
【0380】
1)FokI(New England Biolabs、#109)1ユニット、2.5μlの10×NE緩衝液4、1μgのBAC DNAに、水を加えて最終容量25μlとする。
【0381】
2)37℃で1時間インキュベートする。
【0382】
3)65℃で20分間加熱に不活化する。
【0383】
4)EtOHでDNAを沈殿させる。
【0384】
5)ペレットを水に溶かし、ホスファターゼ処理した転換アダプターを加え(転換アダプターと標的DNAとのモル比は、少なくとも50:1である必要がある)、200ユニットのT4DNAリガーゼ(New England Biolabs、#202)、2.5μlの10×T4 DNAリガーゼ反応緩衝液を加える。最終容量:25μl。
【0385】
6)16℃で4〜16時間インキュベートする。
【0386】
これで分析を行うことができる。しかし、非連結のアダプター除去の手順を実施することが好ましいと思われる。例えば、
7)5’の4塩基張出し部の全ての順列を含むアダプターで被覆した1.5〜2×108のダイナビーズM−280スレプトアビジン(Dynal、#112.05または#112.06)、2000ユニットのT4DNAリガーゼ(New England Biolabs、#202)、22.5μlの10×T4DNAリガーゼ反応緩衝液と水を加え、最終容量を250μlとする。
【0387】
8)16℃で4〜16時間インキュベートする。
【0388】
9)「Biomagnetic Techniques in Molecular Biology」第3版(ノルウェー、Dynal ASより配布)で説明されているように、磁石でビーズを沈殿させ、上澄液を除去する。
【0389】
10)上澄液中のDNAをEtOHで沈殿させる。
【0390】
11)DNA分子を適切な溶液と容量に溶解する。
【0391】
このプロトコルに使用される転換アダプターは、張出し部の256の全順列を表してもよいし、また、256の張出し部の部分集合、または1つ以上の縮退塩基を有する張出し部を表してもよい。分子を光学的マッピング法で解析しようとする場合、シグナルは、例えば、EcoRI部位のない0−シグナルおよび非常に多数のEcoRI部位を含む1−シグナルから成ってもよい。
【0392】
上記の手順を、分子を分析する前に他の制限酵素およびシグナル鎖のセットで1回または複数回反復してもよい。しかし、1回目のラウンドで生成した張出し部と連結しているシグナル鎖を、2回目に使用される酵素の分解から守る必要があることを指摘しなければならない。これは、該当部位をメチル化するなど2回目に使用される制限酵素に対する結合部位のないシグナル鎖を用いることにより達成できる。
【0393】
この手順はまた、ソーティング手順または各末端で得られる配列長を増加させる他の手順と関連して実施してもよい。
【0394】
一部は、標的DNAの生成された断片の他の末端に存在する制限部位を同定することにより、また、一部は、これらの断片が結合している相補的な張出し部および制限部位を含む他の標的断片を同定することにより、いずれの制限部位が特定の制限部位の横に位置するかを決めることにより、標的配列内の制限部位の位置を決定することができる。十分な制限酵素を使用することにより、非常に正確に該位置を決定することができるため、各断片の長さを決定する必要はない。
【0395】
光学的マッピングに関する従来の方法に比べて、この方法はいくつかの重要で有利な特徴を有する。
【0396】
1)分解能がはるかによい:わずか2〜3塩基対離れている制限部位間の識別が可能である。一方、従来の方法では、少なくとも二、三百塩基対離れていることが必要である。
【0397】
2)複数の制限酵素の使用によって制限マップの作成がより容易である。
【0398】
3)再構築に伴う統計学的問題が激減する。というのは、配列が、部分的にのみ切断されている制限部位間のDNA長についての不確かな測定よりもむしろ、シグナル鎖の組成に基づいているからである。
【0399】
4)自身の認識部位の外側で開裂する酵素を用いる場合、マップ内の各位置は、結合部位の長さに加えて、張出し部の長さ(通常9個以上の塩基)に基づいている。このように制限部位は、結合部位の配列および創製された張出し部に基づいて同定することができる。これは、希少な開裂(8塩基)の酵素によって作成された従来の遺伝子マップに比べて有利である。
【特許請求の範囲】
【請求項1】
標的核酸分子の配列の全部または一部を拡大する方法であって、1種または複数の拡大タグが標的配列の1つまたは複数の塩基に結合し、前記タグが前記標的分子中の1つまたは複数の塩基に対応する方法。
【請求項2】
少なくとも、
a)前記標的配列の少なくとも一部分を、アダプター分子を結合するのに適する形態、好ましくは1本鎖形態に転換する段階と、
b)段階a)で生成したアダプター分子の結合に適した前記領域、好ましくは前記1本鎖領域の少なくとも一部分に、1種または複数の拡大タグを含み、または1種または複数の拡大タグを付着する手段を含むアダプターを結合する段階であって、前記タグが前記標的配列の1つまたは複数の塩基に対応しており、好ましくは前記アダプター分子の結合に適した前記領域、好ましくは、前記アダプター分子が結合するか、または前記領域の付近にある前記1本鎖領域の1種または複数の塩基に対応している段階と、
c)所望により、少なくとも前記拡大タグが前記標的分子に結合したままであるように、前記標的分子を前記アダプター分子に連結する段階と、
d)所望により段階a)を繰り返す段階であって、前記アダプターの結合に適した前記領域、好ましくは生成された前記1本鎖領域が、段階b)により拡大タグに結合していない1つまたは複数の塩基を含む段階と、
e)所望により段階b)からd)の段階を繰り返す段階であって、前記アダプター分子が前サイクルの前記アダプター分子が結合した領域に関係する前記標的分子の隣接領域または重複領域に結合する段階と
を含む請求項1に記載の方法。
【請求項3】
前記拡大タグそれぞれが少なくとも2個の塩基に対応する請求項1または2に記載の方法。
【請求項4】
前記拡大タグが一緒に、少なくとも2個の塩基、好ましくは少なくとも4個の塩基に対応する請求項1〜3のいずれか一項に記載の方法。
【請求項5】
拡大タグの鎖が前記分子に結合し、好ましくは少なくとも4個の隣接した塩基に対応する4個以上の拡大タグを含む請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記拡大タグが少なくとも2個の塩基、好ましくは10から30個の塩基の長さの核酸配列である請求項1〜5のいずれか一項に記載の方法。
【請求項7】
段階a)からc)の各サイクルの前記拡大タグが一緒に結合している請求項2〜6のいずれか一項に記載の方法。
【請求項8】
前記アダプターが、認識部位から離れた開裂部位を有するヌクレアーゼの該認識部位を含む請求項2〜7のいずれか一項に記載の方法。
【請求項9】
前記アダプターが認識部位から離れた開裂部位を有する2個以上のヌクレアーゼの認識部位を含み、前記ヌクレアーゼによる開裂が隣接した、または重複した1本鎖領域を生じる請求項2〜8のいずれか一項に記載の方法。
【請求項10】
複数のアダプターが段階b)で、好ましくは前記部分の重複領域または隣接領域に結合する請求項2〜9のいずれか一項に記載の方法。
【請求項11】
前記アダプターが前記配列の重複領域に結合し、それによって複数の拡大タグが各塩基に結合できる請求項10に記載の方法。
【請求項12】
段階c)およびe)が実施される請求項2〜11のいずれか一項に記載の方法。
【請求項13】
シークエンシングのサイクル当たり2個以上、好ましくは4個以上の塩基がシークエンシングされ、および/または各塩基、または複数の塩基に関連したシグナルが拡大される標的核酸分子の全部または一部をシークエンシングする方法。
【請求項14】
各塩基に関連した前記シグナルが、前記塩基が前記配列に現れる回数の増加によって拡大される請求項13に記載のシークエンシング方法。
【請求項15】
前記標的核酸分子の該配列の少なくとも一部分が、請求項1〜12のいずれか一項で定義されたように拡大され、前記拡大配列が所望により読み取り可能なシグナルに転換され、該配列が生じたシグナルの評価によって決定される請求項13または14に記載のシークエンシング方法。
【請求項16】
前記シグナルのそれぞれが、各拡大タグ上に特有のシグナルを生じる単一のシグナル事象から成るパターンを含む請求項15に記載の方法。
【請求項17】
少なくとも、
a)前記核酸分子の一部分の配列を決定する段階と、
b)前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階と
を含む標的核酸分子の全部または一部のシークエンシング方法。
【請求項18】
前記位置が位置マーカーを参照することによって決定される請求項17に記載の方法。
【請求項19】
前記位置が前記標的分子の制限地図を参考することによって決定される請求項17または18に記載の方法。
【請求項20】
シークエンシングする部分が、4個以上の塩基を有し、および/または前記標的分子内にある前記部分の前記位置が1kb未満の精度で決定される請求項17〜19のいずれか一項に記載のシークエンシング方法。
【請求項21】
前記部分が請求項13〜16のいずれか一項で定義された方法によってシークエンシングされる請求項17〜20のいずれか一項に記載の方法。
【請求項22】
少なくとも、
a)前記標的配列の少なくとも一部分を固形担体に付着した、または固形担体に付着する手段を有する相補的プローブの結合に適した形態、好ましくは1本鎖形態に転換する段階と、
b)前記相補的プローブを、段階a)で生成した、相補的プローブの結合に適した前記領域の、好ましくは前記1本鎖領域の少なくとも一部分、好ましくは4から12塩基の長さである一部分に結合させる段階と、
c)所望により、前記相補的プローブが、前サイクルの前記相補的プローブが結合した領域に関係する前記標的分子の隣接領域または重複領域に結合する段階aおよびb)を繰り返す段階と、
d)前記標的配列が結合した相補的プローブ(類)を同定することによって前記標的配列の配列を決定する段階と
を含むプロセスによって、前記分子の一部分の相補性を評価することによって前記配列を決定する、請求項17〜20のいずれか一項に記載の方法。
【請求項23】
前記配列の一部分が請求項13から16のいずれかに定義された拡大に基づくシークエンシング方法によって決定され、隣接部分または重複部分が請求項22で記載されたように決定される請求項17〜22のいずれか一項に記載のシークエンシング方法。
【請求項24】
所望により1種または複数の固状担体に付着した、請求項2から12のいずれか定義された少なくとも1種または複数のアダプターを含む標的核酸分子の1個または複数の塩基を拡大するためのキット。
【請求項25】
前記方法が不均質な試料に関して実施される請求項1〜24のいずれか一項に記載の拡大またはシークエンシング方法。
【請求項26】
前記標的分子を1種または複数のヌクレアーゼ、好ましくはその認識部位から離れた開裂部位を有するヌクレアーゼで切断し、好ましくは相補的な1本鎖領域を生成することによって前記配列の部分に関する配列情報を得ること、およびアダプター分子を前記標的分子の領域に結合させること、ここで、前記アダプター分子は、請求項2から11のいずれか一項で記載した1個または複数の拡大タグを有し(前記タグは前記アダプター分子が結合する前記領域の1個または複数の塩基に対応するシグナル伝達部分を含む。)さらに開裂に使用したヌクレアーゼに対応したシグナル伝達部分を含むものであり(前記部分は前記ヌクレアーゼの開裂部位の全部または一部および/または前記ヌクレアーゼの開裂部位の全部または一部を含む。)、および前記標的配列内の前記部分の位置を決定することを含む標的核酸分子の地図の作成方法。
【請求項1】
標的核酸分子の配列の全部または一部を拡大する方法であって、1種または複数の拡大タグが標的配列の1つまたは複数の塩基に結合し、前記タグが前記標的分子中の1つまたは複数の塩基に対応する方法。
【請求項2】
少なくとも、
a)前記標的配列の少なくとも一部分を、アダプター分子を結合するのに適する形態、好ましくは1本鎖形態に転換する段階と、
b)段階a)で生成したアダプター分子の結合に適した前記領域、好ましくは前記1本鎖領域の少なくとも一部分に、1種または複数の拡大タグを含み、または1種または複数の拡大タグを付着する手段を含むアダプターを結合する段階であって、前記タグが前記標的配列の1つまたは複数の塩基に対応しており、好ましくは前記アダプター分子の結合に適した前記領域、好ましくは、前記アダプター分子が結合するか、または前記領域の付近にある前記1本鎖領域の1種または複数の塩基に対応している段階と、
c)所望により、少なくとも前記拡大タグが前記標的分子に結合したままであるように、前記標的分子を前記アダプター分子に連結する段階と、
d)所望により段階a)を繰り返す段階であって、前記アダプターの結合に適した前記領域、好ましくは生成された前記1本鎖領域が、段階b)により拡大タグに結合していない1つまたは複数の塩基を含む段階と、
e)所望により段階b)からd)の段階を繰り返す段階であって、前記アダプター分子が前サイクルの前記アダプター分子が結合した領域に関係する前記標的分子の隣接領域または重複領域に結合する段階と
を含む請求項1に記載の方法。
【請求項3】
前記拡大タグそれぞれが少なくとも2個の塩基に対応する請求項1または2に記載の方法。
【請求項4】
前記拡大タグが一緒に、少なくとも2個の塩基、好ましくは少なくとも4個の塩基に対応する請求項1〜3のいずれか一項に記載の方法。
【請求項5】
拡大タグの鎖が前記分子に結合し、好ましくは少なくとも4個の隣接した塩基に対応する4個以上の拡大タグを含む請求項1〜4のいずれか一項に記載の方法。
【請求項6】
前記拡大タグが少なくとも2個の塩基、好ましくは10から30個の塩基の長さの核酸配列である請求項1〜5のいずれか一項に記載の方法。
【請求項7】
段階a)からc)の各サイクルの前記拡大タグが一緒に結合している請求項2〜6のいずれか一項に記載の方法。
【請求項8】
前記アダプターが、認識部位から離れた開裂部位を有するヌクレアーゼの該認識部位を含む請求項2〜7のいずれか一項に記載の方法。
【請求項9】
前記アダプターが認識部位から離れた開裂部位を有する2個以上のヌクレアーゼの認識部位を含み、前記ヌクレアーゼによる開裂が隣接した、または重複した1本鎖領域を生じる請求項2〜8のいずれか一項に記載の方法。
【請求項10】
複数のアダプターが段階b)で、好ましくは前記部分の重複領域または隣接領域に結合する請求項2〜9のいずれか一項に記載の方法。
【請求項11】
前記アダプターが前記配列の重複領域に結合し、それによって複数の拡大タグが各塩基に結合できる請求項10に記載の方法。
【請求項12】
段階c)およびe)が実施される請求項2〜11のいずれか一項に記載の方法。
【請求項13】
シークエンシングのサイクル当たり2個以上、好ましくは4個以上の塩基がシークエンシングされ、および/または各塩基、または複数の塩基に関連したシグナルが拡大される標的核酸分子の全部または一部をシークエンシングする方法。
【請求項14】
各塩基に関連した前記シグナルが、前記塩基が前記配列に現れる回数の増加によって拡大される請求項13に記載のシークエンシング方法。
【請求項15】
前記標的核酸分子の該配列の少なくとも一部分が、請求項1〜12のいずれか一項で定義されたように拡大され、前記拡大配列が所望により読み取り可能なシグナルに転換され、該配列が生じたシグナルの評価によって決定される請求項13または14に記載のシークエンシング方法。
【請求項16】
前記シグナルのそれぞれが、各拡大タグ上に特有のシグナルを生じる単一のシグナル事象から成るパターンを含む請求項15に記載の方法。
【請求項17】
少なくとも、
a)前記核酸分子の一部分の配列を決定する段階と、
b)前記核酸分子内の前記部分の位置を決定する段階と、
c)段階a)およびb)で得られた情報を組み合わせて、前記分子の配列を得る段階と
を含む標的核酸分子の全部または一部のシークエンシング方法。
【請求項18】
前記位置が位置マーカーを参照することによって決定される請求項17に記載の方法。
【請求項19】
前記位置が前記標的分子の制限地図を参考することによって決定される請求項17または18に記載の方法。
【請求項20】
シークエンシングする部分が、4個以上の塩基を有し、および/または前記標的分子内にある前記部分の前記位置が1kb未満の精度で決定される請求項17〜19のいずれか一項に記載のシークエンシング方法。
【請求項21】
前記部分が請求項13〜16のいずれか一項で定義された方法によってシークエンシングされる請求項17〜20のいずれか一項に記載の方法。
【請求項22】
少なくとも、
a)前記標的配列の少なくとも一部分を固形担体に付着した、または固形担体に付着する手段を有する相補的プローブの結合に適した形態、好ましくは1本鎖形態に転換する段階と、
b)前記相補的プローブを、段階a)で生成した、相補的プローブの結合に適した前記領域の、好ましくは前記1本鎖領域の少なくとも一部分、好ましくは4から12塩基の長さである一部分に結合させる段階と、
c)所望により、前記相補的プローブが、前サイクルの前記相補的プローブが結合した領域に関係する前記標的分子の隣接領域または重複領域に結合する段階aおよびb)を繰り返す段階と、
d)前記標的配列が結合した相補的プローブ(類)を同定することによって前記標的配列の配列を決定する段階と
を含むプロセスによって、前記分子の一部分の相補性を評価することによって前記配列を決定する、請求項17〜20のいずれか一項に記載の方法。
【請求項23】
前記配列の一部分が請求項13から16のいずれかに定義された拡大に基づくシークエンシング方法によって決定され、隣接部分または重複部分が請求項22で記載されたように決定される請求項17〜22のいずれか一項に記載のシークエンシング方法。
【請求項24】
所望により1種または複数の固状担体に付着した、請求項2から12のいずれか定義された少なくとも1種または複数のアダプターを含む標的核酸分子の1個または複数の塩基を拡大するためのキット。
【請求項25】
前記方法が不均質な試料に関して実施される請求項1〜24のいずれか一項に記載の拡大またはシークエンシング方法。
【請求項26】
前記標的分子を1種または複数のヌクレアーゼ、好ましくはその認識部位から離れた開裂部位を有するヌクレアーゼで切断し、好ましくは相補的な1本鎖領域を生成することによって前記配列の部分に関する配列情報を得ること、およびアダプター分子を前記標的分子の領域に結合させること、ここで、前記アダプター分子は、請求項2から11のいずれか一項で記載した1個または複数の拡大タグを有し(前記タグは前記アダプター分子が結合する前記領域の1個または複数の塩基に対応するシグナル伝達部分を含む。)さらに開裂に使用したヌクレアーゼに対応したシグナル伝達部分を含むものであり(前記部分は前記ヌクレアーゼの開裂部位の全部または一部および/または前記ヌクレアーゼの開裂部位の全部または一部を含む。)、および前記標的配列内の前記部分の位置を決定することを含む標的核酸分子の地図の作成方法。
【図1】


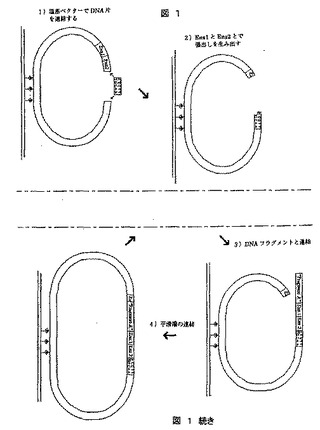
【図2】
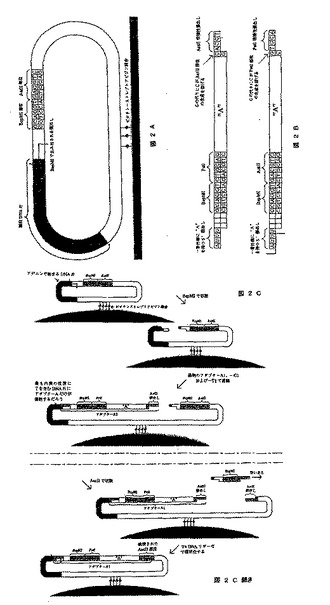
【図3】
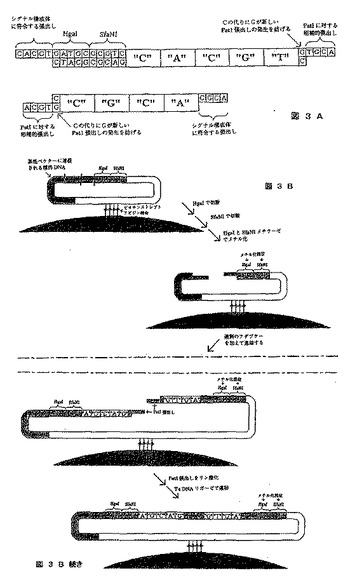
【図4】
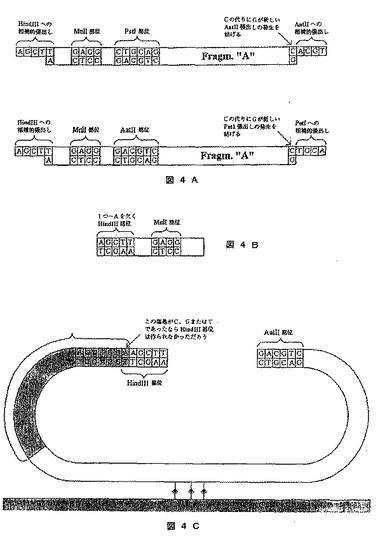
【図5】
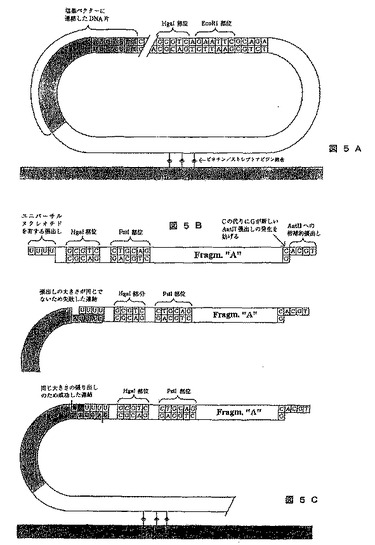
【図6】
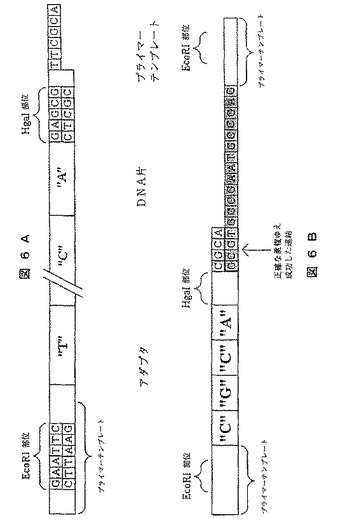
【図7】
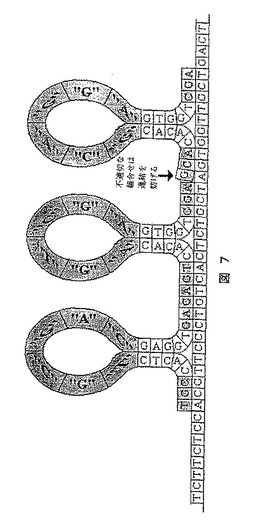
【図8】
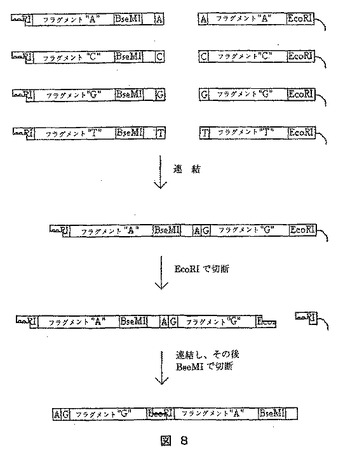
【図9】
【図10】
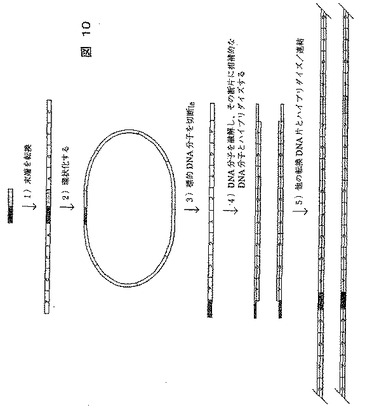
【図11】
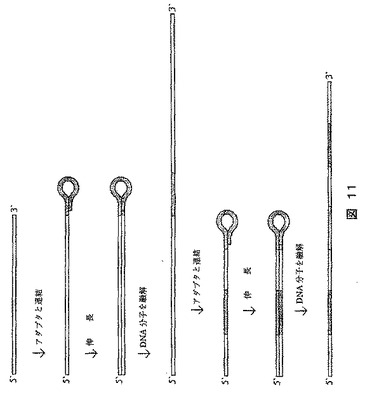
【図12】
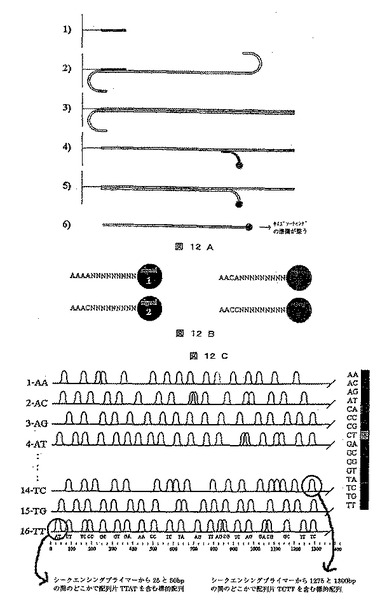
【図13】
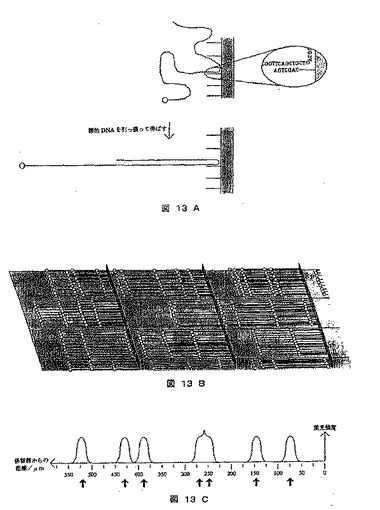
【図14】
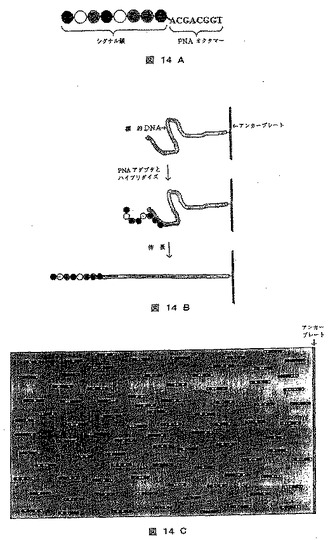
【図15】

【図16】
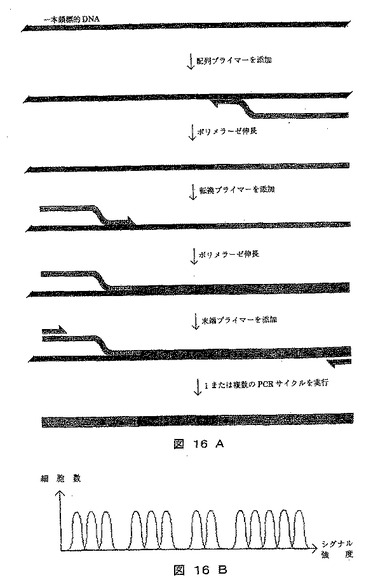
【図17】
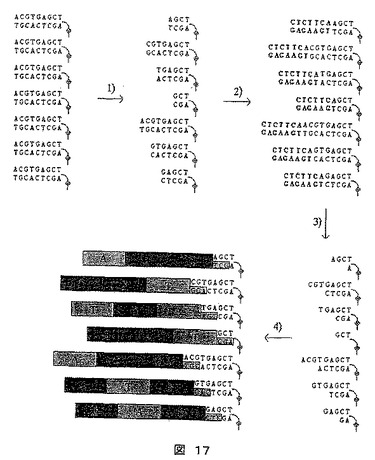
【図18】
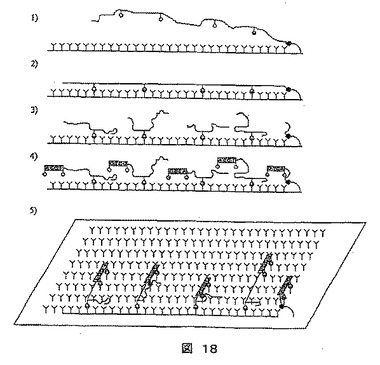
【図19】
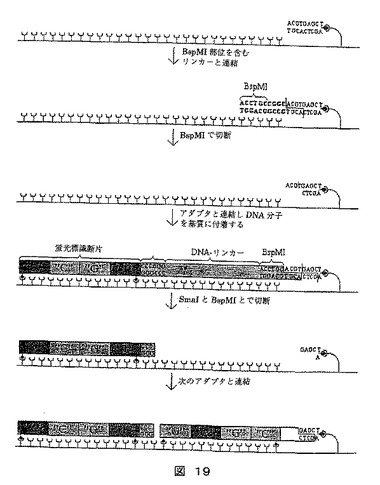
【図20】
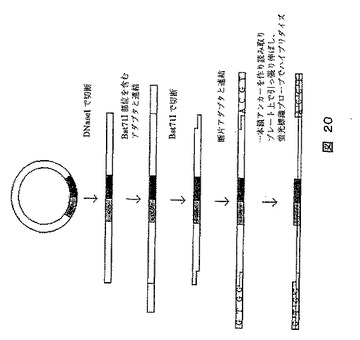
【図21】
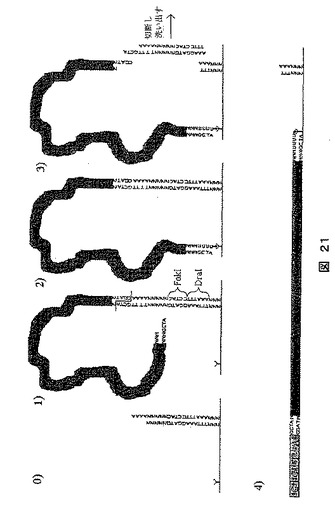
【図22】
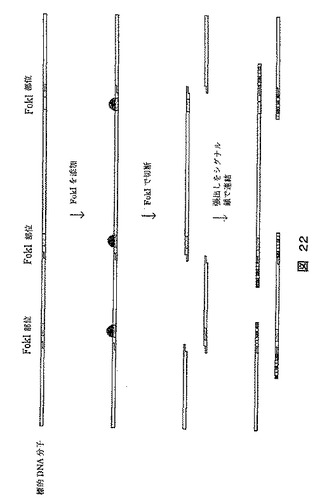
【図23】
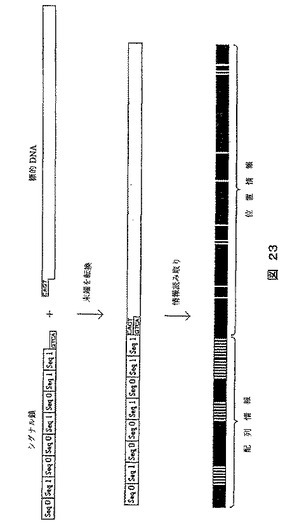
【図24】
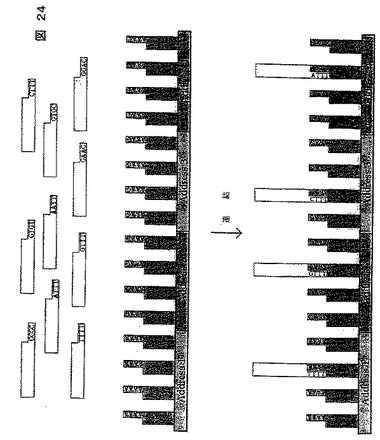
【図25】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【公開番号】特開2011−172573(P2011−172573A)
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願番号】特願2011−54130(P2011−54130)
【出願日】平成23年3月11日(2011.3.11)
【分割の表示】特願2000−591221(P2000−591221)の分割
【原出願日】平成11年12月23日(1999.12.23)
【出願人】(501253279)
【Fターム(参考)】
【公開日】平成23年9月8日(2011.9.8)
【国際特許分類】
【出願日】平成23年3月11日(2011.3.11)
【分割の表示】特願2000−591221(P2000−591221)の分割
【原出願日】平成11年12月23日(1999.12.23)
【出願人】(501253279)
【Fターム(参考)】
[ Back to top ]