
標識インテグリン結合剤
本発明は、被験体で発現されるαvβ6の検出で使用するのに適した代替インビボイメージング剤を提供する。本発明はまた、前記インビボイメージング剤を得るための方法、及び被験体で発現されるαvβ6の測定におけるインビボイメージング剤の使用も提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インビボイメージング、特に新規なインビボイメージング剤に関する。本発明はまた、本発明のインビボイメージング剤の製造方法及び前記方法で有用な前駆体化合物も提供する。本発明のインビボイメージング剤は、インテグリンαvβ6の発現について常態からの逸脱が存在する状態の診断に有用である。
【背景技術】
【0002】
インテグリンは、α及びβサブユニットから構成される膜結合糖タンパク質のファミリーである。インテグリンαvβ6は比較的希少であって(Busk et al 1992 J Biol Chem; 267(9): 5790)、これは修復過程中に上皮組織で生成され、天然のマトリックス分子であるフィブロネクチン(Weinacker et al 1994 J Biol Chem; 269: 6940-6948)及びテネイシン(Wang et al, 1996, Am J Respir Cell Mol Biol; 15(5): 664)と優先的に結合する。ビトロネクチンもまたαvβ6に結合し(Huang et al 1998 J Cell Sci; 111(15): 2189-2195)、TGF−βはαvβ6との相互作用によって活性化される(Massague and Chen 2000 Genes & Dev; 14: 627-644)。
【0003】
インテグリンαvβ6は、創傷治癒、炎症及び癌のような病理学的過程においてアップレギュレートされることが判明している。αvβ6が遊走及び浸潤を促進する能力を有することは、癌の攻撃性の指標としての潜在的な役割を示している(Bates et al J Clin Invest 2005; 115(2): 339-347)。Popov et al, J Hepatol 2008; 48: 453-64には、肝線維症の経過中にαvβ6が活性化胆管上皮及び肝細胞上で発現されることが実証されており、αvβ6が肝線維症の治療のための標的として提唱されている。
【0004】
国際公開第2002/074730号及び同第2005/039547号は、αvβ6インテグリンレセプターを優先的に阻害し、肝線維症のような線維症性疾患状態を含む一連の疾患状態で薬剤として使用できる非標識ビフェニル化合物を開示している。
【0005】
ビフェニル化合物EMD527040は、αvβ6のアンタゴニストとして報告されている(Popov et al J Hepatol 2008; 48: 453-464)。EMD527040の化学構造は下記の通りである。
【0006】
【化1】

国際公開第2005/039547号は、活性化された線維芽細胞又は筋線維芽細胞が関係する状態の治療におけるEMD527040の使用を教示している。Patsenker et al, Gastroenterology 2008; 135: 660-670には、EMD527040が特異的αvβ6アンタゴニストであることが報告されている。Popov et al, Gastroenterology 2008; 134(4) SI : A-827には、肝線維形成のインビボイメージングで使用するための99mTcで標識されたEMD527040の三量体について言及されている。99mTc標識三量体のバックグラウンド肝臓取込みは注射量の1.5%であると報告されているが、これは肝臓内又はその付近における病変の検出を可能にするためには問題となることがある。
【0007】
αvβ6の異常発現に関連する一連の疾患状態の診断で使用するためにαvβ6を標的化する代替インビボイメージング剤に対する余地が存在している。
【先行技術文献】
【特許文献】
【0008】
国際公開第02/074730号パンフレット
【発明の概要】
【0009】
本発明は、被験体で発現されるαvβ6の検出で使用するのに適した代替インビボイメージング剤を提供する。本発明はまた、前記インビボイメージング剤を得るための方法、及び被験体で発現されるαvβ6の測定におけるインビボイメージング剤の使用も提供する。先行技術に比べて、本発明のインビボイメージング剤は比較的小さく、これは例えば薬物動態学的性質を調整するために修飾するのが容易であることを意味している。本発明のインビボイメージング剤はまた、既知のαvβ6アンタゴニストEMD527040と同様な生物学的性質を示す。
【図面の簡単な説明】
【0010】
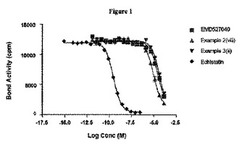
【図1】図1は、3種の試験化合物に関し、インテグリンレセプターαvβ3に対する親和性をインビトロで評価した結果を示している。
【発明を実施するための形態】
【0011】
一態様では、本発明は次の式Iのインビボイメージング剤又はその塩もしくは溶媒和物を提供する。
【0012】
【化2】
式中、
nは0〜6の整数であり、
mは0〜8の整数であり、
X1はO、S又はNR’(式中、R’は水素又はC1-4アルキルである。)であり、
R1は−C(=NH)−NHR’(式中、R’はX1に関して定義した通りである。)であるか、或いはR1は1又は2個の窒素ヘテロ原子を有するC3-5含窒素ヘテロアリール環であり、
R2〜R4の各々は独立に水素、C1-4アルキル又はハロゲンであるか、或いはインビボイメージング成分を含む置換基であり、或いはR2及びR3又はR3及びR4はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-5ヘテロアリール環を形成し、
R5は水素又はR6R7基であり、R6はアミノ酸残基、炭水化物残基、−C(OH)−、−(CR'2)−、−C(=O)−C(CR'2)−、−C(=O)−NR'−、−(CR'2−O−CR'2)−、−CR'2−NR'−、−CR'2−S(O2)−CR'2−及び−(CR'2)−O−N=CR'−(式中、R’はX1に関して定義した通りである。)から選択される1〜50の二価リンカー単位を有する二価リンカー基であり、R7は水素であるか、或いはインビボイメージング成分を含む置換基であり、
R2〜R4の1以上がインビボイメージング成分を含む置換基であるか、或いはR5がR6R7(式中、R7はインビボイメージング成分を含む置換基である。)である。
【0013】
「インビボイメージング剤」という用語は、被験体における特定の生理学的又は病態生理学的状態を標的化するように設計され、被験体へのインビボ投与後に検出できる化合物を意味する。
【0014】
「その塩又は溶媒和物」という用語に係る好適な塩には、(i)鉱酸(例えば、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸及び硫酸)から導かれるもの並びに有機酸(例えば、酒石酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、グリコール酸、グルコン酸、コハク酸、メタンスルホン酸及びp−トルエンスルホン酸)から導かれるもののような生理学的に許容される酸付加塩、並びに(ii)アンモニウム塩、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、有機塩基(例えば、トリエタノールアミン、N−メチル−D−グルカミン、ピペリジン、ピリジン、ピペラジン及びモルホリン)との塩、及びアミノ酸(例えば、アルギニン及びリシン)との塩のような生理学的に許容される塩基塩がある。「その塩又は溶媒和物」という用語に係る好適な溶媒和物には、エタノール、水、食塩水、生理的緩衝液及びグリコールと共に生成されるものがある。
【0015】
「アルキル」という用語は、好ましくは1〜4の炭素原子を含む直鎖又は枝分れアルキル基を意味する。かかる基の例には、メチル、エチル及びプロピルがある。
【0016】
「アリール」という用語は、環系中に5〜6の炭素原子を有する環式芳香族基(例えば、フェニル又はナフチル)を意味する。
【0017】
「ヘテロアリール」置換基は、上記に定義したアリールにおいて、環の炭素原子の1以上がN、S及びOから選択される「ヘテロ原子」で置き換えられたものである。
【0018】
本明細書中で「含窒素ヘテロアリール環」という用語は、上記に定義したアリールにおいて、環が1又は2個の窒素ヘテロ原子を含むものを意味する。
【0019】
「ハロゲン」という用語は、置換基としてのヨウ素、臭素、塩素及びフッ素並びにインビボイメージングに適したその同位体を包含する。
【0020】
「インビボイメージング成分」という用語は、前記被験体に投与した後に前記被験体の体外で検出できる原子又は原子団を意味する。
【0021】
「インビボイメージング成分を含む置換基」という用語は、それ自体がインビボイメージング成分である置換基、又は前記インビボイメージング成分を含む化学基を意味する。さらなる詳細は、下記において特定のインビボイメージング成分を論議する際に示す。
【0022】
「アミノ酸残基」という用語は、L−又はD−アミノ酸、アミノ酸類似体(例えば、ナフチルアラニン)或いはアミノ酸模倣体を意味し、これらは天然のもの又は純粋に合成由来のものであってよく、光学的に純粋なもの(即ち、単一の鏡像異性体、したがってキラルなもの)であるか、或いは鏡像異性体の混合物であってよい。好ましくは、本発明のアミノ酸は光学的に純粋なものである。
【0023】
「炭水化物残基」という用語は、多価アルコールの二価アルデヒド又はケトン誘導体を意味する。それはフルクトース又はグルコースのようなモノマー(単糖)であってもよいし、或いは2つの糖が互いに結合して二糖を形成していてもよい。二糖には、グルコース及びフルクトースから構成されるスクロースのような糖がある。「糖」という用語は、置換糖及び非置換糖の両方並びに糖の誘導体を包含する。好ましくは、糖はグルコース、グルコサミン、ガラクトース、ガラクトサミン、マンノース、ラクトース、フコース及びこれらの誘導体(例えば、シアル酸及びグルコサミンの誘導体)から選択される。糖は好ましくはα又はβである。糖は特にマンノピラノシド又はガラクトースピラノシドであり得る。糖上のヒドロキシル基は、例えば1以上のアセチル基で保護することができる。糖部分は、好ましくはN−アセチル化されている。かかる糖の好ましい例には、N−アセチルガラクトサミン、シアル酸、ノイラミン酸、N−アセチルガラクトース及びN−アセチルグルコサミンがある。
【0024】
本発明の特定のインビボイメージング剤がキラル中心を含む場合、本発明の技術的範囲は、2種の鏡像異性体のラセミ混合物並びに他方の鏡像異性体を実質的に含まずに各鏡像異性体を個別に含む組成物も包含する。即ち本発明では、例えば、R鏡像体を実質的に含まずにS鏡像体を含む組成物、又はS鏡像体を実質的に含まずにR鏡像体を含む組成物も想定されている。「実質的に含まずに」とは、組成物が10%未満又は8%未満又は5%未満又は3%未満又は1%未満の少量鏡像体を含むことを意味する。特定のインビボイメージング剤が2以上のキラル中心を含む場合、本発明の技術的範囲は、各種のジアステレオマーの混合物を含む組成物並びに他のジアステレオマーを実質的に含まずに各ジアステレオマーを含む組成物も包含する。特定のジアステレオマーのいずれかに言及せずに記載されたインビボイメージング剤は、4種のジアステレオマーのすべてを含む組成物、R,R及びS,S異性体のラセミ混合物を含む組成物、R,S及びS,R異性体のラセミ混合物を含む組成物、他のジアステレオマーを実質的に含まずにR,R鏡像体を含む組成物、他のジアステレオマーを実質的に含まずにS,S鏡像体を含む組成物、他のジアステレオマーを実質的に含まずにR,S鏡像体を含む組成物、及び他のジアステレオマーを実質的に含まずにS,R鏡像体を含む組成物を包含する。
【0025】
好ましくは、式Iのnは1〜4であり、最も好ましくは3である。
【0026】
好ましくは、式Iのmは0〜3であり、最も好ましくは1である。
【0027】
好ましくは、式IのX1はNR’であり、最も好ましくはNHである。
【0028】
好ましくは、式IのR1は五員又は六員含窒素ヘテロアリール環であり、最も好ましくはピリジル又はイミダゾリルである。R1がピリジルである場合、それは好ましくは2−ピリジルである。
【0029】
好ましくは、式IのR2〜R4の各々は独立に水素、クロロ、ヨード又はフルオロであり、最も好ましくは水素、ヨード又はクロロである。
【0030】
好ましくは、式IのR5はR6R7基である。X1がNHである場合、R6R7は好ましくは−C(=O)−R6aR7a基(式中、R6aは1〜20の二価リンカー単位を有する二価リンカー基であって、前記二価リンカー単位はR6に関して上記に定義した通りであり、R7aはR7に関して上記に定義した通りである。)である。
【0031】
式Iに関する好ましいインビボイメージング成分は、
(i)放射性金属イオン、
(ii)常磁性金属イオン、
(iii)γ放出型放射性ハロゲン、
(iv)陽電子放出型放射性非金属、及び
(v)インビボ光学イメージングに適したレポーター
から選択される。
【0032】
イメージング成分が放射性金属イオン(即ち、放射性金属)である場合、好適な放射性金属は、64Cu、48V、52Fe、55Co、94mTc又は68Gaのような陽電子放射体、及び99mTc、111In、113mIn又は67Gaのようなγ放射体であり得る。好ましい放射性金属は99mTc、64Cu、68Ga及び111Inである。最も好ましい放射性金属はγ放射体、特に99mTcである。
【0033】
イメージング成分が常磁性金属イオンである場合、好適なかかる金属イオンには、Gd(III)、Mn(II)、Cu(II)、Cr(III)、Fe(III)、Co(II)、Er(II)、Ni(II)、Eu(III)及びDy(III)がある。好ましい常磁性金属イオンはGd(III)、Mn(II)及びFe(III)であり、Gd(III)が特に好ましい。
【0034】
イメージング成分がγ放出型放射性ハロゲンである場合、放射性ハロゲンは好適には123I、131I及び77Brから選択される。125Iは、診断イメージング用のイメージング成分として使用するのに適さないので、特別に除外される。好ましいγ放出型放射性ハロゲンは123Iである。
【0035】
イメージング成分が陽電子放出型放射性非金属である場合、好適なかかる陽電子放射体には、11C、13N、15O、17F、18F、75Br、76Br及び124Iがある。好ましい陽電子放出型放射性非金属は11C、13N、18F及び124Iであり、特に好ましくは11C及び18Fであり、最も特に好ましくは18Fである。
【0036】
イメージング成分がインビボ光学イメージングに適したレポーターである場合、レポーターは光学イメージング方法で直接又は間接に検出できる任意の成分である。レポーターは、光散乱剤(例えば、着色又は無着色粒子)、吸光剤又は発光剤であり得る。さらに好ましくは、レポーターは発色団又は蛍光化合物のような色素である。色素は、紫外域乃至近赤外域の波長をもった電磁スペクトル中の光と相互作用する任意の色素であり得る。最も好ましくは、レポーターは蛍光特性を有する。好ましい有機発色性及び発蛍光性レポーターには、広範な非局在化電子系を有する群、例えばシアニン類、メロシアニン類、インドシアニン類、フタロシアニン類、ナフタロシアニン類、トリフェニルメチン類、ポルフィリン類、ピリリウム色素、チアピリリウム色素、スクアリリウム色素、クロコニウム色素、アズレニウム色素、インドアニリン類、ベンゾフェノキサジニウム色素、ベンゾチアフェノチアジニウム色素、アントラキノン類、ナフトキノン類、インダスレン類、フタロイルアクリドン類、トリスフェノキノン類、アゾ色素、分子内及び分子間電荷移動色素及び色素錯体、トロポン類、テトラジン類、ビス(ジチオレン)錯体、ビス(ベンゼン−ジチオレート)錯体、ヨードアニリン色素、ビス(S,O−ジチオレン)錯体がある。緑色蛍光タンパク質(GFP)及び異なる吸光/発光特性を有するGFPの変種のような蛍光タンパク質も有用である。特定の状況においてはある種の希土類金属(例えば、ユウロピウム、サマリウム、テルビウム又はジスプロシウム)の錯体が使用され、蛍光ナノ結晶(量子ドット)についても同様である。使用できる発色団の具体例には、フルオレセイン、スルホローダミン101(Texas Red)、ローダミンB、ローダミン6G、ローダミン19、インドシアニングリーン、Cy2、Cy3、Cy3.5、Cy5、Cy5.5、Cy7、Marina Blue、Pacific Blue、Oregon Green 88、Oregon Green 514、テトラメチルローダミン、Alexa Fluor 350、Alexa Fluor 430、Alexa Fluor 532、Alexa Fluor 546、Alexa Fluor 555、Alexa Fluor 568、Alexa Fluor 594、Alexa Fluor 633、Alexa Fluor 647、Alexa Fluor 660、Alexa Fluor 680、Alexa Fluor 700及びAlexa Fluor 750がある。特に好ましいのは、400nm乃至3μm、特に600〜1300nmの範囲内の可視又は近赤外(NIR)領域内に吸収極大を有する色素である。光学イメージングモダリティー及び測定技法には、特に限定されないが、ルミネセンスイメージング、内視鏡検査、蛍光内視鏡検査、光学コヒーレンス断層撮影、透過率イメージング、時間分解透過率イメージング、共焦点イメージング、非線形顕微鏡検査、光音響イメージング、音響光学イメージング、スペクトル分析、反射スペクトル分析、干渉分析、コヒーレンス干渉分析、拡散光学断層撮影及び蛍光媒介拡散光学断層撮影(連続波、時間ドメイン及び周波数ドメインシステム)、並びに光散乱、吸光、偏光、ルミネセンス、蛍光寿命、量子収量及び消光の測定がある。インビボ光学イメージングに適した好ましいレポーターはCy色素である。
【0037】
最も好ましいインビボイメージング成分は、好適なもの及び好ましいものとして上記にそれぞれ定義したような、放射性金属イオン、γ放出型放射性ハロゲン、陽電子放出型放射性非金属、又はインビボ光学イメージングに適したレポーターである。
【0038】
式Iのインビボイメージング剤に関して好ましくは、R2〜R4の1つは18F又は123Iである。別法として、式Iのインビボイメージング剤に関して好ましくは、R5はR6R70基である。式中、R7は、18F、放射性金属イオン又は常磁性金属イオンを含む金属錯体、及び光学イメージングに適したレポーターから選択されるイメージング成分を含んでいる。本明細書中に前述した通り、X1がNHである場合、R6R7は好ましくは−C(=O)−R6aR7aである。この実施形態に関しては、R7aは好ましくは前記イメージング成分を含んでいる。
【0039】
「金属錯体」という用語は、金属イオンと1以上のリガンドとの配位錯体を意味する。金属錯体は、「キレート交換に耐える」こと(即ち、金属配位座に関して他の潜在的に競合するリガンドとのリガンド交換を受けにくいこと)が極めて好ましい。潜在的に競合するリガンドは、インビトロでは前駆体化合物自体又は製剤中の他の賦形剤(例えば、製剤中に使用される放射線防護剤又は抗菌防腐剤)中に存在することがあり、インビボでは内因性化合物(例えば、グルタチオン、トランスフェリン又は血漿タンパク質)であり得る。
【0040】
式Iの二価リンカー基は、好ましくは10〜50原子の鎖であり、最も好ましくは10〜30原子の鎖である。最も好ましくは、二価リンカー基はバイオモディファイアー部分として作用する。「バイオモディファイアー部分」は、式Iのインビボイメージング剤の薬物動態及び血中クリアランス速度を調整する機能を有する。好適なバイオモディファイアー部分の例は、単分散PEG構成単位に基づいていて、1〜20の前記構成単位を含むものである。さらに、前記バイオモディファイアー部分は1〜10のアミノ酸残基を表すこともできる。前記バイオモディファイアー部分用の好ましいアミノ酸残基は、リシン及びグルタミン酸のような帯電アミノ酸、或いはシステイン酸及びホスホノアラニンのような帯電非天然アミノ酸である。加えて、アミノ酸であるグリシン、アスパラギン酸及びセリンも含めることができる。
【0041】
式Iの好ましいインビボイメージング剤は、次の式Iaの化合物又はその塩もしくは溶媒和物である。
【0042】
【化3】
式中、
R1aはR1に関して上記に好適なもの及び好ましいものとして定義した通りであり、
R2a〜R4aはR2〜R4に関して上記に好適なもの及び好ましいものとして定義した通りであり、
R5aは水素であるか、或いは−C(=O)−R6aR7a基(式中、R6a及びR7aは上記に好適なもの及び好ましいものとして定義した通りである。)である。
【0043】
本発明に係る好ましいインビボイメージング剤の非限定的な例は下記の通りである。
【0044】
【化4】
【0045】
【化5】
別の態様では、本発明は、本明細書中に好適なもの及び好ましいものとして定義した式Iのインビボイメージング剤の製造方法であって、インビボイメージング成分の適当な供給源を次の式IIの前駆体化合物と反応させる段階を含んでなる方法を提供する。
【0046】
【化6】
式中、
R11は式IのR1に関して好適なもの及び好ましいものとして定義した通りであり、
R12〜R14は水素、C1-4アルキル及びハロゲン並びに前駆体基から独立に選択され、
R15は水素又は前駆体基であるか、或いはR16R17基(式中、R16はR6に関して本明細書中に好適なもの及び好ましいものとして定義した二価リンカー基であり、R17は水素又は前駆体基である。)であり、
X11は式IのX1に関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
m’は式Iのmに関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
n’は式Iのnに関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
R12〜R14の1以上が前駆体基であるか、或いはR15がR16R17(式中、R17は前駆体基である。)であり、
前記前駆体基以外の反応基は化学的に保護されている。
【0047】
概して言えば、前駆体化合物をインビボイメージング成分の適当な供給源と「反応させる」段階は、できるだけ高い収率で所望のインビボイメージング剤を生成するのに適した反応条件下で2種の反応体を合わせることを含んでいる。
【0048】
「インビボイメージング成分の適当な供給源」とは、インビボイメージング成分が共有結合することで本発明のインビボイメージング剤を生成するようにして前駆体化合物の前駆体基と反応し得る化学形態のインビボイメージング成分を意味する。
【0049】
「前駆体化合物」は、好都合な化学形態のインビボイメージング成分との化学反応が部位特異的に起こり、最小数の段階(理想的にはただ1つの段階)で反応を実施でき、かつ格別の精製の必要なしに(理想的にはいかなる追加の精製も必要なしに)所望のインビボイメージング剤が得られるように設計された、インビボイメージング剤の誘導体からなる。かかる前駆体化合物は合成品であり、良好な化学純度で簡便に得ることができる。前駆体化合物は、不要の反応を回避するため、ある種の官能基又は「反応基」に関して1以上の保護基を任意に含むことができる。
【0050】
「保護基」という用語は、望ましくない化学反応を阻止又は抑制するが、分子の残部を変質させない十分に温和な条件下で問題の官能基から脱離させ得るのに十分な反応性を有するように設計された基を意味する。脱保護後には所望の生成物が得られる。保護基は当業者に公知であり、アミン基に関してはBoc(ここでBocはtert−ブチルオキシカルボニルである)、Fmoc(ここでFmocはフルオレニルメトキシカルボニルである)、トリフルオロアセチル、アリルオキシカルボニル、Dde(1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル)及びNpys(3−ニトロ−2−ピリジンスルフェニル)から適宜に選択され、カルボキシル基に関してはメチルエステル、tert−ブチルエステル及びベンジルエステルから適宜に選択される。ヒドロキシル基に関しては、好適な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)、又はテトラブチルジメチルシリルのようなトリアルキルシリルである。チオール基に関しては、好適な保護基はトリチル及び4−メトキシベンジルである。本明細書中では、保護基を含む置換基は「化学的に保護されている」。保護基の使用は、'Protective Groups in Organic Synthesis', Theorodora W. Greene and Peter G. M. Wuts, (Fourth Edition, John Wiley & Sons, 2006)に記載されている。
【0051】
「前駆体基」とは、インビボイメージング剤を得るため、インビボイメージング成分の適当な供給源と優先的に反応する化学基である。
【0052】
式Iaの好ましいインビボイメージング剤に関しては、前記式IIの前駆体化合物は次の式IIaを有している。
【0053】
【化7】
式中、R11a〜R15aは式IIのR11〜R15に関して上記に定義した通りである。
【0054】
下記のスキーム1は、Goodman et al, 2002 J Med Chem; 45: 1045-1051に記載された方法に基づいており、上記に好適なもの及び好ましいものとして定義した式IIの前駆体化合物の製造における初期段階を記載している。
【0055】
【化8】
式中、
R21は式IのR1に関して上記に定義した通りであり、
R22〜R24は水素、C1-4アルキル及びハロゲンから独立に選択され、或いはR22及びR23又はR23及びR24はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-6ヘテロアリール環を形成し、
Y21は−X21−R25(式中、X21は式IのX1に関して上記に定義した通りであり、R25は水素又は保護基である。)であり、
m”は式Iのmに関して定義した通りであり、
n”は式Iのnに関して定義した通りである。
【0056】
上記のスキーム1では、商業的に入手可能なアルデヒド1を段階(a)でマロン酸及び酢酸アンモニウムと反応させて中間体2を得る。段階(b)で中間体2をメタノール及びSOCl2と反応させれば、中間体3が得られる。標準的なペプチドカップリング試薬及び方法を用いて、中間体3を段階(c)でBoc−NH−CH((CH2)m"−Y21)−C(=O)OHと反応させる(このタイプの各種試薬がNovabiochem社、Bachem社及びAdvanced Chemtech社のような販売業者から商業的に入手可能であり、例えばBoc−Lys(Fmoc)−OH、Boc−Lys(Z)−OH又は対応するジアミノプロピオン酸(例えば、Boc−Dpr(Fmoc)−OH)がある)。標準的な方法(例えば、ジオキサン中の2M HCl)を用いてBoc保護基を段階(d)で除去することで、中間体5が得られる。段階(e)では、中間体5をR21−NH−(CH2)n1−C(=O)−OHと反応させるが、この反応体はGoodman et al, 2002 J Med Chem; 45: 1045-1051に記載の方法に従うか又はそれを改変することで製造される。段階(f)において、得られた中間体6を適当な溶媒中でNaOHと反応させることでエステルを対応するカルボン酸に転化させ、したがって中間体7に到達する。次いで、当業者にとって公知の方法を用いて中間体7を修飾することで、R22〜R24のいずれか又はY21の位置に前駆体基を導入し、本明細書中に定義した式IIの前駆体化合物に到達することができる。かかる方法は、特定のインビボイメージング成分の論議に際して下記に一層詳しく記載される。
【0057】
式IIに関して好ましくは、R12〜R14の1つが前記前駆体基である。
【0058】
別法として、式IIに関して好ましくは、R15がR16R17(式中、R17は前記前駆体基である。)である。
【0059】
式IIの好ましい前駆体基は、
(i)金属イメージング成分を錯体化できる1種以上のリガンド、
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体、
(iii)求核置換用のアルキルハライド、アルキルトシレート又はアルキルメシレートを含む誘導体、
(iv)求核又は求電子置換に向けて活性化された芳香環を含む誘導体、
(v)アシル化を受けやすい官能基を含む誘導体、
(vi)ベンズアルデヒドと反応させた場合にオキシム生成に参加する官能基を含む誘導体、
(vii)ビニルスルホン官能基を含む誘導体、
(viii)容易にアルキル化を受ける官能基を含む誘導体、及び
(ix)チオール含有化合物をアルキル化してチオエーテル含有生成物を与える誘導体
から選択される。
【0060】
式IIの前駆体化合物の前駆体基が(i)又は(ii)である場合、式IIの前駆体化合物は本発明の別の態様をなす。
【0061】
インビボイメージング成分の特定の供給源と反応させるためにいかなる前駆体基を選択すべきかは、インビボイメージング剤の技術分野において公知である。これは、本発明の特定のインビボイメージング剤を得るための方法について読者を手引きするため、以下に一層詳しく記載される。
【0062】
インビボイメージング成分が金属イオンである場合、前駆体基は金属イメージング成分を錯体化できる1種以上のリガンドを含む。キレート交換に耐える金属錯体を形成するために本発明で使用するのに適したリガンドには、2〜6(好ましくは2〜4)の金属ドナー原子が(金属ドナー原子を結合する炭素原子又は非配位ヘテロ原子の非配位主鎖を有することで)五員又は六員キレート環を生じるように配列されたキレート剤、或いは金属イオンと強く結合するドナー原子を含む単座リガンド(例えば、イソニトリル、ホスフィン、チオール又はジアゼニド)がある。キレート剤の一部として金属とよく結合するドナー原子種の例は、アミン、チオール、アミド、オキシム及びホスフィンである。ホスフィンは非常に強い金属錯体を形成するので、単座又は二座のホスフィンでも好適な金属錯体を形成する。イソニトリル及びジアゼニドの直線形状はキレート剤中に組み込みにくいものであり、したがって通例は単座リガンドとして使用される。好適なイソニトリルの例には、tert−ブチルイソニトリルのような単純アルキルイソニトリル、及びMIBI(即ち、1−イソシアノ−2−メトキシ−2−メチルプロパン)のようなエーテル置換イソニトリルがある。好適なホスフィンの例には、テトロフォスミン(Tetrofosmin)、及びトリス(3−メトキシプロピル)ホスフィンのような単座ホスフィンがある。好適なジアゼニドの例には、HYNIC系列のリガンド(即ち、ヒドラジン置換ピリジン又はニコチンアミド)がある。
【0063】
キレート交換に耐える金属錯体を形成する好適なキレート剤には、特に限定されないが、下記のものがある。
(i)ジアミンジオキシム、
(ii)MAG3(メルカプトアセチルトリグリシン)のようなチオールトリアミドドナーセット又はPicaのようなジアミドピリジンチオールドナーセットを有するN3Sリガンド及び関連するリガンド、
(iii)BAT又はECD(即ち、エチルシステイネート二量体)のようなジアミンジチオールドナーセット或いはMAMAのようなアミドアミンジチオールドナーセットを有するN2S2リガンド、
(iv)シクラム、モノオキソシクラム又はジオキソシクラムのようなテトラミン、アミドトリアミン又はジアミドジアミンドナーセットを有する開鎖又は大環状リガンドであるN4リガンド、並びに
(v)ジアミンジフェノールドナーセットを有するN2O2リガンド。
【0064】
上述のリガンドはテクネチウム(例えば、94mTc又は99mTc)を錯体化するために特に適しており、Jurisson et al, 1999 Chem Rev; 99: 2205-2218にさらに詳細に記載されている。かかるリガンドは、銅(64Cu又は67Cu)、バナジウム(例えば、48V)、鉄(例えば、52Fe)或いはコバルト(例えば、55Co)のような他の金属に対しても有用である。他の好適なリガンドは国際公開第91/01144号に記載されており、その中にはインジウム、イットリウム及びガドリニウムに対して特に好適なリガンド(特に大環状アミノカルボキシレート及びアミノホスホン酸リガンド)が含まれる。ガドリニウムの非イオン性(即ち、中性)金属錯体を形成するリガンドが知られており、米国特許第4,885,363号に記載されている。ガドリニウムに対して特に好ましいのは、DTPA、エチレンジアミン四酢酸(EDTA)、トリエチレンテトラアミン六酢酸(TTHA)、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸(DOTA)、10−(2−ヒドロキシプロピル)−1,4,7,10−テトラアザシクロドデカン−1,4,7−三酢酸(DO3A)及びこれらの誘導体を含むキレートである。
【0065】
上記のスキーム1について述べれば、前駆体基がキレート剤である前駆体化合物は、Y21が−X21−R25(式中、R25は保護基である。)である化合物7から出発して得ることができる。次いで、スキーム1の段階(f)後にR25保護基を除去することができる。キレート剤の反応性誘導体を脱保護化合物7と反応させることで前駆体化合物が得られる。反応性誘導体は、例えば、脱保護化合物7中のNH2との反応のためには活性エステル誘導体又はカルボン酸誘導体であり得る。前駆体基がキレート剤である場合に好ましくは、R15はR16R17基(式中、R17はキレート剤である。)である。この場合には、キレート剤の添加前に、脱保護化合物7を二価リンカー基R16の反応性誘導体と反応させる。
【0066】
適当なキレート剤を含む式IIの前駆体化合物の99mTc標識のためには、通常のテクネチウム出発原料は、テクネチウムがTc(VII)酸化状態にある過テクネチウム酸塩(即ち、TcO4-)である。過テクネチウム酸塩自体は容易に金属錯体を形成せず、したがってテクネチウム錯体の調製には、第一スズイオンのような適当な還元剤を添加してテクネチウムの酸化状態を低い酸化状態(通常はTc(I)乃至Tc(V))に還元することで錯体化を容易にすることが通常必要である。溶媒は、有機溶媒又は水性溶媒或いはこれらの混合物であり得る。溶媒が有機溶媒を含む場合、有機溶媒は好ましくはエタノール又はDMSOのような生体適合性溶媒である。好ましくは、溶媒は水性溶媒であり、最も好ましくは等張食塩水である。
【0067】
式Iの放射性ヨウ素化インビボイメージング剤を製造するのに適した式IIの前駆体化合物は、求電子又は求核放射性ヨウ素化を受ける誘導体或いは標識アルデヒド又はケトンとの縮合を受ける誘導体を含んでいる。第1のカテゴリーの例は下記の(a)〜(c)である。
(a)トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えば、トリメチルシリル)或いは有機ホウ素化合物(例えば、ボロネートエステル又はオルガノトリフルオロボレート)のような有機金属誘導体、
(b)ハロゲン交換用の非放射性アルキルブロミド、或いは求核ヨウ素化用のアルキルトシレート、メシレート又はトリフレート、並びに
(c)求核ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0068】
好ましいかかる前駆体化合物は、(放射性ヨウ素交換を可能にするための)アリールヨージド又はブロミドのような非放射性ハロゲン原子、有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、或いはトリアゼンのような有機前駆体又は求核置換のための良好な脱離基(例えば、ヨードニウム塩)からなっている。放射性ヨウ素化のために好ましくは、前駆体化合物は有機金属前駆体化合物からなり、最も好ましくはトリアルキルスズからなる。
【0069】
有機分子中に放射性ヨウ素を導入するための前駆体化合物及び方法は、Bolton, 2002 J Lab Comp Radiopharm: 45: 485-528に記載されている。好適なボロネートエステル有機ホウ素化合物及びその製法は、Kabalka et al, 2002 Nucl Med Biol; 29: 841-843; and 2003 Nuc Med Biol; 30: 369-373に記載されている。好適なオルガノトリフルオロボレート及びその製法は、Kabalka et al, 2004 Nucl Med Biol; 31 : 935-938に記載されている。
【0070】
放射性ヨウ素が結合できるアリール基の例を以下に示す。
【0071】
【化9】
いずれも、芳香環上への容易な放射性ヨウ素置換を可能にする置換基を含んでいる。チロシン残基は、その固有のフェノール基を用いて放射性ヨウ素化を実施することを可能にする。
【0072】
放射性ヨウ素を含む別の置換基は、例えば次式のように、放射性ハロゲン交換による直接ヨウ素化で合成することができる。
【0073】
【化10】
飽和脂肪族系に結合したヨウ素原子はインビボでの代謝を受けやすく、したがって放射性ヨウ素の損失を招きやすいことが知られているので、放射性ヨウ素原子は好ましくはベンゼン環のような芳香環への直接共有結合により又はビニル基を介して結合される。
【0074】
上記のスキーム1について述べれば、放射性ヨウ素化に適した前駆体化合物は、R22〜R24の1つがブロモ基である化合物7から出発して得ることができる。この化合物を適当なスタンナンと反応させてトリアルキルスズ前駆体化合物を得、これを放射性ヨウ素と反応させて式Iの放射性ヨウ素化インビボイメージング剤を得ることができる。別法として、化合物7のR22〜R24の1つが127Iであり得るが、これは放射性ハロゲン交換による放射性ヨウ素化に適した前駆体化合物である。
【0075】
放射性フッ素化に適した前駆体化合物は、[18F]−フッ化物で直接に標識されるか、或いは18F含有補欠分子族との反応性を有するように設計できる。
【0076】
放射性フッ素化は、[18F]−フッ化物と式IIの前駆体化合物中の適当な前駆体基との反応を用いる直接標識によって実施できる。前駆体基は、アルキルブロミド、アルキルメシレート又はアルキルトシレートのような良好な脱離基であり得る。[18F]−フッ化物による直接放射性フッ素化はまた、求核芳香族置換によっても実施できる。アリール系に関しては、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。別法として、前駆体化合物はクロロニコチンアミド前駆体基を含み得る。この場合、クロロの位置における18F−フッ化物求核置換によって[18F]フルオロニコチンアミド化合物が得られる(Greguric et al 2009 J Med Chem; 52: 5299-5302)。[18F]−フッ化物での直接標識が実施される場合に好ましくは、前駆体基は式IIのR15の位置に存在する。かかる前駆体化合物は、Y21が−X21−R25(式中、X21はNであり、R25は保護基である。)であるスキーム1の化合物7から出発して得ることができる。このR25保護基の除去後、遊離アミノ基の位置でカップリング反応を実施できる。例えば、前駆体基R15の反応性誘導体を用いてR15前駆体基を導入できる。別法として、R16リンカーの任意に保護された反応性誘導体をカップリングした後、脱保護し、R17前駆体基の反応性誘導体と反応させることができる。
【0077】
18Fはまた、前駆体化合物と18F標識補欠分子族との反応によっても導入できる。例えば、N−ハロアセチル前駆体基を補欠分子族18F(CH2)3OHでアルキル化して−NH(CO)CH2O(CH2)318F誘導体を得ることができる。本発明の18F標識化合物はまた、18F−フルオロジアルキルアミンを生成させ、次いで18F−フルオロジアルキルアミンを例えば塩素、P(O)Ph3及び活性化エステルから選択した前駆体基を含む前駆体化合物と反応させた場合のアミド生成によっても得ることができる。[18F]−N−メチルアミノオキシ含有補欠分子族(Olberg et al Bioconjugate Chem 2008; 19: 1301-1308)を前駆体化合物中のビニルスルホニル前駆体基と反応させて18F標識インビボイメージング剤を得ることもできる。別の好適な補欠分子族は、国際公開第2004/080492号に記載の通り得られる4−[18F]フルオロベンズアルデヒドであり、これはNH2と反応して−N=CH−[18F]フルオロフェニル誘導体を生成する。前段に記載した[18F]フルオロニコチンアミド化合物はまた、同時係属英国特許出願第0905438.8号に記載されているように、遊離アミノ基を有する前駆体化合物を[18F]−6−フルオロニコチン酸テトラフルオロフェニルエステルと反応させて得ることもできる。そこに記載されているように、この反応は、適当な溶媒(例えば、2〜11、好適には3〜11のpH範囲内の水性緩衝液)中において5〜70℃の極端でない温度(好ましくは周囲温度)で実施できる。
【0078】
18F標識誘導体の合成経路のさらなる詳細は、Bolton, 2002 J Lab Comp Radiopharm; 45: 485-528に記載されている。
【0079】
光学イメージングに適したレポーターを含む本発明のインビボイメージング剤を得ることに関しては、Licha et al, 2002 Topics Curr Chem; 222: 1-29; 2005 Adv Drug Deliv Rev; 57: 1087-1108に記載の方法を参照されたい。蛍光色素標識試薬を用いる標識の総説及び例に関しては、"Non-Radioactive Labelling, a Practical Introduction", Garman, A.J. Academic Press, 1997; "Bioconjugation - Protein Coupling Techniques for the Biomedical Sciences", Aslam, M. and Dent, A., Macmillan Reference Ltd, (1998)を参照されたい。コンジュゲーションに適した官能化シアニン色素(CyD)は、GE Healthcare社、Atto−Tec社、Dyomics社、Molecular Probes社などから商業的に入手できる。かかる色素の大部分はNHSエステルとして入手でき、これらは本明細書中に定義された式IIの前駆体化合物中のアミンと反応して所望のインビボイメージング剤を生成し得る。ヒドラジド、マレイミド又はスクシンイミジルエステル基で官能化されたAlexa Fluor(商標)647は、Molecular Probes社から商業的に入手できる。カルボキシル又はマレイミド基で官能化されたCyDは、欧州特許出願公開第1816475号に記載された方法に従って製造できる。これらの官能化CyD化合物は、ヒドロキシ又はアミン前駆体基を含む前駆体化合物と反応させて式Iの所望インビボイメージング剤を得ることができる。CyDにとって好ましい位置は、式IのR5である。
【0080】
式Iのインビボイメージング剤の製造方法はまた、固相合成によっても実施できる。「固相」は架橋された不溶性ポリマー材料であって、合成条件に対して化学的に不活性である。固相は通例は球状粒子(例えば、直径0.04〜0.15mmのビーズ)の形態を取るが、シート、ピン状粒子及び円盤状粒子も使用される。
【0081】
【化11】
R22〜R24、Y21及びm”は、スキーム1に関して上記に好適なもの及び好ましいものとして定義した通りである。固相は、スキーム2中にクロスハッチングを施した円で示されている。出発化合物はスキーム1からの化合物2であり、これを段階(a)でFmoc保護し、次いで段階(b)で固相に結合し、続いてFmoc保護基を段階(c)で(例えば、ジメチルホルムアミド又はNMP中の20%ピペリジンにより)除去する。段階(d)では、固相化合物2をFmoc−NH−CH(CH2)m"−Y21と反応させる。上記に記載したようにしてFmocを開裂させ、次いでスキーム1の(d)〜(f)に類似した段階をスキーム1の化合物5の固相バージョンであるものに関して実施する。前駆体化合物全体を固相上で合成し、標準的な方法(例えば、ジクロロメタン中のトリフルオロ酢酸)で開裂除去することができる。別法として、関係する化学に応じ、適当な段階で中間体を開裂除去し、以後の段階を溶液中で実施することもできる。
【0082】
好ましい実施形態では、本発明のインビボイメージング剤の製造方法は自動化される。自動化合成は、自動化合成装置、例えばTracerlab(商標)及びFastlab(商標)(いずれもGE Healthcare社から入手可能)によって簡便に実施できる。Fastlab(商標)は自動化陽電子放出断層撮影(PET)ラジオトレーサー合成プラットホームに関する技術の現状を表すものであり、新しいPETラジオトレーサーの開発に当たっては、その合成がFastlab(商標)に適合していることが望ましい。放射化学は、「カセット」を装置に取り付けることにより、自動化合成装置上で実施される。通常、かかるカセットは流体通路、反応器、及び試薬バイアル並びに放射合成後の清掃段階で使用される任意の固相抽出カートリッジを受け入れるためのポートを含んでいる。
【0083】
したがって、本発明のさらに別の態様では、本発明の自動化方法を実施するためのカセットであって、
(i)本発明の方法に関して上記に好適なもの及び好ましいものとして定義した前駆体化合物を含む容器、及び
(ii)本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング成分の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセットが提供される。
【0084】
カセットはまた、過剰のインビボイメージング成分を除去するためのイオン交換カートリッジを含み得る。自動化合成のために必要な試薬、溶媒及び他の消耗品もまた、濃度、体積、送出時間などに関する最終ユーザーの要求条件を満たすように自動化合成装置を運転させるソフトウェアを保持したコンパクトディスクのようなデータ媒体と共に含めることができる。本発明のカセットは、インビボイメージング成分が18Fである本発明のインビボイメージング剤を製造するために特に適している。
【0085】
さらに別の態様では、本発明は、本明細書中に定義したインビボイメージング剤を哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる組成物として定義される「医薬組成物」を提供する。「生体適合性キャリヤー」は、組成物が「哺乳動物への投与に適する」ようにして(即ち、毒性又は過度の不快感なしに哺乳動物体に投与できるようにして)インビボイメージング剤を懸濁又は溶解するための流体(特に液体)である。生体適合性キャリヤー媒質は、好適には、無菌のパイロジェンフリー注射用水、(有利には注射用の最終生成物が等張性又は非低張性になるように平衡させ得る)食塩水のような水溶液、或いは1種以上の張度調整物質(例えば、血漿陽イオンと生体適合性対イオンとの塩)、糖(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール物質(例えば、ポリエチレングリコール、プロピレングリコールなど)の水溶液のような注射可能なキャリヤー液体である。生体適合性キャリヤー媒質はまた、エタノールのような生体適合性有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は配合物を可溶化するために有用である。好ましくは、生体適合性キャリヤー媒質はパイロジェンフリー注射用水、等張食塩水又はエタノール水溶液である。静脈内注射用生体適合性キャリヤー媒質のpHは好適には4.0〜10.5の範囲内にある。
【0086】
本発明の医薬組成物は、好適には、無菌保全性を維持しながら皮下注射針による1回又は数回の穿刺に適したシール(例えば、クリンプ加工した隔壁シールクロージャー)を備えた容器に入れた状態で供給される。かかる容器は1回分又は複数回分の患者用量を含み得る。好ましい複数用量容器は、複数の患者用量を含む(例えば、容積10〜30cm3の)単一のバルクバイアルからなり、したがって臨床的状況に合わせて製剤の実用寿命中に様々な時間間隔で1回分の患者用量を臨床グレードの注射器に抜き取ることができる。予備充填注射器は1回分のヒト用量又は「単位用量」を含むように設計され、したがって好ましくは臨床用に適した使い捨て注射器又は他の注射器である。医薬組成物が放射性医薬組成物である場合、予備充填注射器には、施術者を放射線量から防護するための注射器シールドを任意に設けることができる。好適なかかる放射性医薬品注射器シールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなっている。
【0087】
本発明の医薬組成物はキットから製造できる。別法として、医薬組成物を無菌製造条件下で製造することで所望の無菌生成物を得ることができる。かかる医薬組成物を非無菌条件下で製造し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いて終末滅菌を施すこともできる。好ましくは、本発明の医薬組成物はキットから製造される。
【0088】
かかるキットは、本発明の好適な前駆体、好ましくは無菌で非発熱性の形態にある前駆体であって、インビボイメージング成分の無菌供給源との反応によって最小数の操作で所望の医薬組成物を与えるような前駆体を含んでいる。
キットが本発明の前駆体化合物を含む場合、前記キット自体は本発明のさらに別の態様をなす。かかる考慮事項は、放射性医薬品(特に、放射性同位体が比較的短い半減期を有する場合)にとって特に重要であると共に、取扱いを容易にして放射線薬剤師に対する放射線量を低減させるためにも特に重要である。したがって、かかるキットの再構成用の反応媒質は好ましくは上記に定義したような生体適合性キャリヤーであり、最も好ましくは水性のものである。キット中に使用するための前駆体を無菌製造条件下で使用すれば、所望の無菌で非発熱性の材料を得ることができる。また、前駆体を非無菌条件下で使用し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いる終末滅菌を施すこともできる。好ましくは、前駆体は無菌で非発熱性の形態で使用される。
【0089】
キットはさらに、放射線防護剤、抗菌防腐剤、pH調整剤又はフィラーのような追加成分を任意に含むことができる。
【0090】
「放射線防護剤」という用語は、水の放射線分解から生じる酸素含有フリーラジカルのような高反応性フリーラジカルを捕捉することで分解反応(例えば、レドックス過程)を阻止する化合物を意味する。好適な放射線防護剤は、アスコルビン酸、p−アミノ安息香酸(即ち、4−アミノ安息香酸)、ゲンチシン酸(即ち、2,5−ジヒドロキシ安息香酸)、及び生体適合性陽イオンを有するこれらの塩から選択される。
【0091】
「抗菌防腐剤」という用語は、潜在的に有害な微生物(例えば、細菌、酵母又はかび)の増殖を阻止する薬剤を意味する。抗菌防腐剤はまた、用量に応じ、多少の殺菌性を示すこともある。本発明の抗菌防腐剤の主な役割は、再構成後の医薬組成物(即ち、イメージング剤そのもの)中におけるこのような微生物の増殖を阻止することである。しかし、抗菌防腐剤は、再構成前のキットの1種以上の成分中における潜在的に有害な微生物の増殖を阻止するためにも任意に使用できる。好適な抗菌防腐剤には、パラベン類(即ち、メチル、エチル、プロピル又はブチルパラベン或いはこれらの混合物)、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオメルサールがある。好ましい抗菌防腐剤はパラベン類である。
【0092】
「pH調整剤」という用語は、再構成されたキットのpHがヒト又は哺乳動物への投与のために許容し得る範囲(およそpH4.0〜10.5)内にあることを保証するために有用な化合物又は化合物の混合物を意味する。好適なかかるpH調整剤には、トリシン、リン酸塩又はTRIS(即ち、トリス(ヒドロキシルメチル)アミノメタン)のような薬学的に許容される緩衝剤、及び炭酸ナトリウム、重炭酸ナトリウム又はこれらの混合物のような薬学的に許容される塩基がある。
【0093】
「フィラー」という用語は、製造及び凍結乾燥中における材料の取扱いを容易にすることができる薬学的に許容される増量剤を意味する。好適なフィラーには、塩化ナトリウムのような無機塩、及びスクロース、マルトース、マンニトール又はトレハロースのような水溶性糖又は糖アルコールがある。
【0094】
さらに別の態様では、本発明は、被験体で発現されるαvβ6の量及び/又は位置を決定するためのインビボイメージング方法であって、
(i)本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング剤を前記被験体を投与する段階、
(ii)段階(i)で投与した前記インビボイメージング剤を前記被験体で発現されるαvβ6に結合させる段階、
(iii)段階(ii)で結合させた前記インビボイメージング剤に含まれるインビボイメージング成分から放出される信号を検出する段階、並びに
(iv)前記信号の位置及び量を表す画像を生成する段階であって、前記信号が前記被験体で発現されるαvβ6の量及び/又は位置を表す段階
を含んでなるインビボイメージング方法を提供する。
【0095】
本発明の「被験体」は、任意のヒト又は動物被験体であり得る。好ましくは、本発明の被験体は哺乳動物である。最も好ましくは、前記被験体はインタクトな哺乳動物生体である。特に好ましい実施形態では、本発明の被験体はヒトである。
【0096】
インビボイメージング剤を「投与する」段階は、好ましくは非経口的に実施され、最も好ましくは静脈内に実施される。静脈内経路は、インビボイメージング剤を被験体の身体全域に送達し、前記被験体で発現されたαvβ6に接触させるための最も効率的な方法である。本発明のインビボイメージング剤は、好ましくは本明細書中に定義した本発明の医薬組成物として投与される。
【0097】
投与段階後かつ検出段階前に、インビボイメージング剤をαvβ6に結合させる。インビボイメージング剤は哺乳動物の身体を通って動的に移動し、体内の様々な組織に接触する。ひとたびインビボイメージング剤がαvβ6に接触すれば、特異的な相互作用が起こる結果、αvβ6をもった組織からのインビボイメージング剤のクリアランスは、αvβ6をもたない組織又は少ないαvβ6を発現する組織よりも長い時間がかかる。一定の時点に達すれば、αvβ6をもった組織に結合したインビボイメージング剤と少ないαvβ6を発現する組織(又はαvβ6を発現しない組織)に結合したインビボイメージング剤との比の結果として、αvβ6に特異的に結合したインビボイメージング剤の検出が可能となる。
【0098】
「信号を検出する」段階は、インビボイメージング成分から放出される信号を、前記信号に対して感受性を有する検出器によって検出することを含んでいる。かかる検出器は当技術分野で公知である。例えば、インビボイメージング成分がγ放射体である場合、検出は単光子放出コンピューター断層撮影(SPECT)カメラを用いて実施でき、インビボイメージング成分が常磁性金属イオンである場合、検出は磁気共鳴イメージング(MRI)カメラを用いて実施できる。
【0099】
「画像を生成する」段階は、取得された信号データに再構築アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作することで、前記インビボイメージング成分から放出される信号の位置及び/又は量を示す画像が生成される。
【0100】
「αvβ6状態」とは、αvβ6の異常発現(通例はαvβ6の過剰発現)によって特徴づけられる病的状態を意味する。本発明のインビボイメージング方法が有用なかかる状態の例には、炎症、癌及び線維症がある。本発明のインビボイメージング方法は、好ましくは前記被験体がαvβ6状態を有することが知られ又は疑われている場合に実施され、最も好ましくは前記被験体がαvβ6状態を有することが知られている場合に実施される。被験体がαvβ6状態を有することが知られている場合、前記インビボイメージング方法は好ましくは前記被験体に関する治療計画の進行中に繰り返して実施され、前記治療計画は前記αvβ6状態と戦うための薬物の投与を含んでいる。
【0101】
好ましい実施形態では、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング方法はさらに、αvβ6レセプターの量及び/又は位置を特定の臨床像に帰因させる段階(v)を含んでいる。
【0102】
さらに別の態様では、本発明は、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング方法で使用するための、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング剤を提供する。
【実施例】
【0103】
次に、以下の非限定的な実施例によって本発明を説明する。
【0104】
実施例の簡単な説明
実施例1は、非放射性イメージング剤1の合成法を記載している。
【0105】
実施例2は、イメージング剤2の合成法を記載している。
【0106】
実施例3は、イメージング剤3の合成法を記載している。
【0107】
実施例4は、非放射性イメージング剤4の合成法を記載している。
【0108】
実施例5は、イメージング剤4の合成法を記載している。
【0109】
実施例6は、イメージング剤5の合成法を記載している。
【0110】
実施例7は、イメージング剤6の合成法を記載している。
【0111】
実施例8は、αvβ3結合を評価するためのインビトロ千葉県香取郡神崎町新アッセイを記載している。
【0112】
実施例9は、イメージング剤7の合成法を記載している。
【0113】
実施例10は、イメージング剤7のフローサイトメトリー評価を記載している。
【0114】
実施例中で使用される略語のリスト
aq 水性
Boc t−ブチルオキシカルボニル
Bu ブチル
Bzl ベンジル
Dpr ジアミノプロピオン酸
ESI−MS エレクトロスプレーイオン化質量分析法
Fmoc 9−フルオレニルメチルオキシムカルボニル
h 時間
LC−MS 液体クロマトグラフィー質量分析法
MDP メチレンジホスホン酸
Me メチル
MH+ 分子イオン
min 分
m/z 質量/電荷比
PEG ポリエチレングリコール
Ser セリン
TFA トリフルオロ酢酸
tR 保持時間
trityl トリメチルフェニル
UV 紫外。
【0115】
実施例1:3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパン酸(非放射性イメージング剤1)の合成
実施例1(i):3−(Fmoc−アミノ)−3−(3−ブロモ−5−クロロフェニル)プロパン酸の合成
この化合物は、下記の実施例2(i)に記載されたようにして、しかし3−ブロモ−5−クロロベンズアルデヒド(ABCR、3.0g、14mmol)から出発して合成した。収量1.3g(2段階を通して19%)。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm、溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸、勾配:5分かけて05〜80%B、流量:0.6mL/分、UV検出:214及び254nm、ESI−MS)、tR=4.1分、m/z期待値500.0、実測値500.0。
【0116】
実施例1(ii):3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパン酸の合成
この化合物は、下記の実施例2(ii)、2(iii)、2(vi)及び2(vii)に記載されたものと同じ方法を用いて、しかし3−(Fmoc−アミノ)−3−(3−ブロモ−5−クロロフェニル)プロパン酸(実施例1(i)に記載、0.6g、1.2mmol)をトリチルクロリド樹脂(Novabiochem社、置換度1.6mmol/g、0.5g)上に結合することから出発し、続いてそれぞれFmoc−Ser(Bzl)−OH(Aldrich社、0.5g、1.2mmol)及び5−((tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸(下記実施例2(v)に記載、0.21g、0.71mmol)をカップリングすることにより固体担体上で合成した。Boc脱保護及び固体担体からの切断によって0.211gの粗物質を得た。
【0117】
実施例1(iii):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパノエートの合成
3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパン酸(実施例1(ii)に記載、0.167g、0.264mmol)をメタノール(3mL)に溶解した溶液に、塩化チオニル(Sigma Aldrich社、79mg、0.66mmol)を0℃(氷/水浴)でゆっくりと添加した。混合物を室温まで加温し、45分後に水で奪活した。溶液を蒸発させ、生成物をメタノールから再結晶して0.155g(91%)を得た。
【0118】
実施例1(iv):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−(トリメチルスタンニル)フェニル)プロパノエートの合成
この反応はマイクロ波装置(Biotage社)内で実施した。メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパノエート(実施例1(iii)に記載、10mg、15μmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(Strem chemicals社、1mg、0.8μmol)及びヘキサメチル二スズ(Sigma Aldrich社、40.0mg、122μmol)をトルエン(2mL)中に含む混合物を窒素でパージし、140℃で30分間加熱した。フラッシュクロマトグラフィー(シリカ、1%トリエチルアミンを含むジクロロメタン/メタノール(9:1))によって精製することで、5mgの半純粋生成物を得た。
【0119】
実施例1(v):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパノエートの合成
ヨウ素(Fluka社、2mg、74μmol)及びメチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−(トリメチルスタンニル)フェニル)プロパノエート(実施例1(iv)に記載、5mg、7μmol)をジクロロメタン(2mL)に溶解した溶液を室温で2時間撹拌した。反応混合物をチオ硫酸ナトリウム溶液(0.3M、1mL)で処理した。相の分離後、ジクロロメタン溶液を水及びブラインで洗浄し、(硫酸ナトリウムで)乾燥し、濃縮して4mgの粗物質を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値693.1(MH+)、実測値693.3。
【0120】
実施例1(vi):3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパン酸の合成
メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパノエート(実施例1(v)に記載、4mg、6μmol)を、メタノール(1mL)と水酸化ナトリウム水溶液(1M、0.2mL)との混合物に溶解した。反応混合物を室温で1時間撹拌した。溶液をジクロロメタン(2×2mL)で抽出した。水性相を塩酸(1M)でpH6に調整し、次いでジクロロメタン(2×2mL)及び酢酸エチル(2×2mL)で抽出した。有機相を合わせ、(硫酸ナトリウムで)乾燥し、濃縮して0.5mgを得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値679.1(MH+)、実測値679.2。
【0121】
実施例2:(6S,Z)−3−(3,5−ジクロロフェニル)−22−(ヒドロキシイミノ)−17−(((Z)−3−(ヒドロキシイミノ)−2−メチルブタン−2−イル)アミノ)エチル)−21,21−ジメチル−5,9,13−トリオキソ−6−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−4,8,14,20−テトラアザトリコサン−1−酸(イメージング剤2)の合成
実施例2(i):Fmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸の合成
3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸(100mg、0.43mmol)(J Med Chem; 45: 1045-1051及びTetrahedron Asym; 19: 2072-2077に記載の通り合成)をクロロホルム/メタノール/水(65:25:4、4.5mL)中に懸濁した撹拌懸濁液に、ジイソプロピルエチルアミン(Fluka社、227μL、1.30mmol)を添加した。この溶液に9−フルオレニルメトキシカルボニル−N−ヒドロキシスクシンイミド(Novabiochem社、169mg、0.50mmol)を添加した。1時間後、混合物を(ロタベイパー(rotavapor)で)濃縮し、残留物を0.1%トリフルオロ酢酸含有アセトニトリル(10mL)中に取り、5分間撹拌し、濾過した。溶液を濃縮し、残留物をフラッシュクロマトグラフィーに付した。固体物質をカラム上に直接適用し、ヘキサン/酢酸エチル(1:1)で溶出して不純物を除去し、続いて生成物をクロロホルム/メタノール(9:1)で溶出することで、177mg(91%)の生成物を無色油状物として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=4.1分、m/z期待値456.1(MH+)、実測値456.1。
【0122】
実施例2(ii):固体担体へのFmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸の結合
トリチルクロリド樹脂(Novabiochem社、置換度1.6mmol/g、0.406g)に、Fmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸(実施例2(i)に記載、0.30g、0.65mmol)をジクロロメタンとN−メチルピロリドンとの混合物(8:1、9mL)に溶解した溶液を添加し、続いてジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、2.6mL、5.2mmol)を添加した。混合物をローラーテーブル上に5時間保ち、次いで手動窒素バブラー装置に移した。樹脂を排液し、ジクロロメタン/メタノール/ジイソプロピルエチルアミン溶液(17:2:1、6mL)で3回処理し、ジクロロメタンで洗浄し、乾燥した。
【0123】
実施例2(iii):Fmoc−Dpr(ivDde)−OHのカップリング
この反応は、実施例2(ii)に記載した樹脂(0.65mmol)上において手動窒素バブラー装置内で実施した。標準的な処理(N−メチルピロリドン中20%ピペリジン)によってFmoc基を除去した。Fmoc−Dpr(ivDde)−OH(Novabiochem社、0.69g、1.3mmol)を、N−メチルピロリドン(7mL)中においてHATU((N−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシド、Genscript社、0.49g、1.3mmol)及びジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、1.3mL、2.6mmol)で5分間予備活性化し、樹脂に添加した。2時間後、樹脂を排液し、N−メチルピロリドンで洗浄した。標準的なカイザー試験は反応が完了していないことを示し、カップリングを繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=4.4分、m/z期待値748.3(MH+)、実測値748.2。
【0124】
実施例2(iv):5−(tert−ブトキシカルボニル−ピリジン−2−イル−アミノ)ペンタン酸メチルエステル
2−(N−(tert−ブトキシカルボニル)アミノ)ピリジン(1.0g、5.2mmol、J Org Chem;67:4965−4967に記載の通り合成した)を無水ジメチルホルムアミド(15mL)に溶解した溶液を氷/水浴中で5℃に冷却した。水素化ナトリウム(Aldrich社、鉱油中60%、0.25g、6.3mmol)を少量ずつ添加した。冷却浴を取り除き、混合物を20分間激しく撹拌した。次いで、混合物を(氷/水浴で)冷却し、5−ブロモ吉草酸メチル(Aldrich社、0.89mL、6.3mmol)を滴下した。10分後、冷却浴を取り除き、混合物を2時間撹拌し、次いで(ロタベイパーで)濃縮した。残留物をジクロロメタン(30mL)と塩酸(0.02N、20mL)との混合物中に取り、激しく振盪した。相を分離し、有機相を飽和重炭酸ナトリウム水溶液(20mL)及びブライン(20mL)で抽出し、(Na2SO4で)乾燥した。(ロタベイパーで)濃縮した後、粗物質を無色油状物(1.52g)として得た。粗物質のアリコート(0.5g)をフラッシュクロマトグラフィー(ヘキサン/酢酸エチル、1:1)によって精製することで、生成物を無色油状物(収量490mg(93%))として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.6分、m/z期待値309.2(MH+)、実測値309.4。
【0125】
実施例2(v):5−(tert−ブトキシカルボニル−ピリジン−2−イル−アミノ)ペンタン酸
実施例2(iv)に記載したエステル(0.69g、2.3mmol)をメタノール(10mL)に溶解した撹拌溶液に、水酸化ナトリウム(4N、2mL)を周囲温度で添加した。反応を薄層クロマトグラフィー(シリカ、ヘキサン/酢酸エチル、1:1)によってモニターした。3時間後、溶媒を(ロタベイパーで)蒸発させた。水(8mL)を残留物に添加し、水溶液をエーテル(2×10mL)で抽出し、次いで塩酸(2N)を用いてやや酸性(pH約5)にした後、ジクロロメタン(3×10mL)で抽出した。ジクロロメタン相を合わせて(Na2SO4で)乾燥し、濾過し、蒸発させることで、生成物を無色油状物(収量594mg(90%))として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.0分、m/z期待値295.2(MH+)、実測値295.4。
【0126】
実施例2(vi):5−(tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸のカップリング
この反応は手動窒素バブラー装置内で実施した。標準的な処理(N−メチルピロリドン中20%ピペリジン)により、実施例2(iii)に記載した樹脂のFmoc基を除去した。5−(tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸(実施例2(v)に記載、0.25g、0.85mmol)を、N−メチルピロリドン(6mL)中においてHATU(Genscript社、0.32g、0.85mmol)及びジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、0.845mL、1.69mmol)で5分間予備活性化し、樹脂に添加した。3時間後、樹脂を排液し、洗浄した。カイザー試験は反応が完了していないことを示したので、カップリングを繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=3.5分、m/z期待値802.3(MH+)、実測値802.3。
【0127】
実施例2(vii):保護基の除去及び固体担体からの開裂
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.325mmol)に、2%ヒドラジンをN−メチルピロリドン(5mL)に溶解した溶液を添加した。2分後、樹脂を排液し、それぞれ5分及び10分の反応時間で処理を2回繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄した。トリフルオロ酢酸/ジクロロメタン/トリイソプロピルシラン溶液(49:49:2、6.2mL)中において、Boc基の除去及び樹脂からの生成物の開裂を同時に行った。2.5時間後、樹脂を濾別し、溶液を(ロタベイパーで)濃縮乾固した。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)による分析:tR=1.5分、m/z期待値496.1(MH+)、実測値496.1。
【0128】
実施例2(viii):cPn216−グルタル酸とのコンジュゲーション
(国際公開第2003/006070号に記載の通り合成した)cPn216−グルタル酸(0.26g、0.57mmol)を、ジメチルホルムアミド(3mL)中においてPyAOP(7−アザベンゾトリアゾール−1−イルオキシ−トリス(ピロリジノ)ホスホニウムヘキサフルオロホスフェート、Applied Biosystems社、0.222g、0.426mmol)及びジイソプロピルエチルアミン(Aldrich社、148μL、0.852mmol)で10分間予備活性化した。実施例2(vii)からの化合物(0.141g、0.284mmol)をジメチルホルムアミド(6mL)に溶解し、予備活性化したキレートに添加した。反応の進行をLC−MS分析によってモニターした。1時間後、ジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M、0.4mL、0.4mmol)を添加し、混合物を1時間撹拌した。予備活性化したcPn216−グルタル酸の第2部分(0.143g、0.312mmol)を添加した(同じカップリング試薬)。1時間後、反応は完了し、混合物を(ロタベイパーで)濃縮した。化合物を分取HPLC(カラム:Phenomenex Luna C18(2)10μm 250×50.0mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜40%B;流量:50mL/分;214nm及び254nmでUV検出)によって精製し、凍結乾燥して56mg(21%)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値935.5(MH+)、実測値935.5。
【0129】
実施例2(ix):イメージング剤2を得るための99mTc標識
実施例2(viii)で得た前駆体化合物を99mTcで標識するためには、(実施例2(viii)に記載したようにして得た)100μgの前駆体化合物を、メタノール中において0.5mlのNa2CO3/NaHCO3緩衝液(pH9.2)、99mTcジェネレーターから得た0.5mlのTcO4-及び0.1mlのSnCl2/MDP溶液と共に室温でインキュベートすればよい。
【0130】
実施例3:3−((S)−3−(4−(2−アミノエチルアミノ)−3−((2−アミノエチルアミノ)メチル)ブタンアミド)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド−3−(3,5−ジクロロフェニル)プロパン酸(イメージング剤3)の合成
実施例3(i):テトラ−Boc−テトラアミン−NHSエステルのカップリング
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.074mmol)に、2%ヒドラジンをN−メチルピロリドン(5mL)に溶解した溶液を添加した。2分後、樹脂を排液し、それぞれ5分及び10分の反応時間で処理を2回繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄し、排液した。樹脂をN−メチルピロリドン中に懸濁し、次いで(国際公開第2006/008496号に記載の通り合成した)テトラ−Boc−テトラアミン−NHSエステル(0.053g、0.074mmol)及びN−メチルモルホリン(Merck社、9.7μL、0.088mmol)を添加した。2時間後、樹脂を排液し、洗浄した。カップリングを標準的なカイザー試験で追跡し、さらに3回繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で0〜60%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=3.2分、m/z期待値348.7((M−Boc5)H2)2+、実測値348.6。
【0131】
実施例3(ii):Boc脱保護及び固体担体からの開裂
実施例3(i)に記載した樹脂(0.074mmol)をトリフルオロ酢酸/トリイソプロピルシラン/水(1.9mL、5μL、5μL)溶液で2時間処理することで、Boc保護基の除去及び固体担体からの生成物の開裂を同時に行った。溶液を樹脂から濾別し、(ロタベイパーで)濃縮した。生成物を分取HPLC(カラム:Phenomenex Luna C18(2)5μm 250×21.2mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で5〜50%B;流量:10.0mL/分;214nm及び254nmでUV検出)によって精製し、凍結乾燥した。収量8.1mg(16%)。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.3分、m/z期待値696.3(MH+)、実測値696.3。
【0132】
実施例3(iii):イメージング剤3を得るための99mTc標識
実施例3(ii)で得た前駆体化合物は、実施例2(ix)に記載した方法を用いて99mTcで標識することができる。
【0133】
実施例4:(48S,E)−51−(3,5−ジクロロフェニル)−1−(4−フルオレニルフェニル)−5,45,49−トリオキソ−48−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−3,9,12,15,18,21,24,27,30,33,36,39,42−トリデカオキサ−2,6,46,50−テトラアザトリペンタコント−1−エン−53−酸(非放射性イメージング剤4)
実施例4(i):Fmoc−PEG(12)プロピオン酸とのコンジュゲーション
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.17mmol)に、N−メチルピロリドン(10mL)中の2%ヒドラジンを添加した。5分後、樹脂を排液し、5分の反応時間で処理を繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄した。1−(9H−フルオレン−9−イル)−3−オキソ−2,7,10,13,16,19,22,25,28,31,34,37,40−トリデカオキサ−4−アザトリテトラコンタン−43−酸(Polypure社、0.244g、0.290mmol)を、N−メチルピロリドン(2mL)中においてHATU(Applied Biosystems社、0.11g、0.29mmol)及びジイソプロピルエチルアミン(Fluka社、50μL、0.29mmol)で5分間予備活性化し、樹脂に添加した。カップリングを標準的なカイザー試験で追跡した。2時間後、樹脂を排液し、N−メチルピロリドン(2mL)中で5分間予備活性化したFmoc−アミノPEG(12)プロピオン酸(0.134mg、0.160mmol)、HATU(0.070g、0.18mmol)及びジイソプロピルエチルアミン(80μL、0.46mmol)の溶液を用いてカップリングを繰り返した。3時間後、混合物を排液し、N−メチルピロリドン及びジクロロメタンで洗浄した。標準的なカイザー試験は反応が完了したことを示した。
【0134】
実施例4(ii):Fmoc−アミノオキシ酢酸とのコンジュゲーション
この反応は手動窒素バブラー装置内で実施した。標準的なFmoc基除去のため、実施例4(i)に記載した樹脂(0.17mmol)をN−メチルピロリドン中の20%ピペリジンで処理した。Fmoc−アミノオキシ酢酸(Iris Biotech GmbH、0.050g、0.16mmol)を、
N−メチルピロリドン(2mL)中においてHATU(Applied Biosystems社、0.070g、0.18mmol)及びジイソプロピルエチルアミン(Fluka社、80μL、0.46mmol)で5分間予備活性化し、次いで樹脂に添加した。2時間後、樹脂を排液し、洗浄した。カイザー試験は転化が不完全であることを示し、同じ条件を用いてカップリングを2回繰り返した。
【0135】
実施例4(iii):保護基の除去及び固体担体からの開裂
実施例4(ii)からの樹脂(0.17mmol)を、手動窒素バブラー装置内において、N−メチルピロリドン中の20%ピペリジンを用いる標準的なFmoc脱保護に付した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄し、排液した。トリフルオロ酢酸(3mL)、ジクロロメタン(3mL)及びトリイソプロピルシラン(150μL)の混合物を用いて、Boc保護基の除去及び樹脂からの物質の開裂を同時に行った。2.5時間後、溶液を濾過し、(ロタベイパーで)濃縮した。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=3.5分、m/z期待値584.8(MH2)2+、実測値584.3。
【0136】
実施例4(iv):4−フルオロベンズアルデヒドとのオキシム形成(非放射性イメージング剤4)
実施例4(iii)に記載した生成物(0.17mmol)を水/アセトニトリル(4:1、3mL)に溶解した溶液に、4−フルオロベンズアルデヒド(Fluka社、4μL、0.04mmol)を添加した。反応混合物を70℃で1時間撹拌した。混合物を濃縮し、生成物を分取HPLC(カラム:Phenomenex Luna C18(2)5μm 21.2×250mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜80%B;流量:10.0mL/分;214nm及び254nmでUV検出)によって精製することで、凍結乾燥後に収量11mg(5%)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.3分、m/z期待値637.8(MH2)2+、実測値637.4。
【0137】
実施例5:イメージング剤4の合成
Goodman et al(上述)の方法に従い、商業的に入手可能なベンズアルデヒド(Sigma−Aldrich社から入手可能)を段階(a)でマロン酸及び酢酸アンモニウムと反応させ、次いで段階(b)でメタノール及びSOCl2と反応させる。標準的なペプチドカップリング試薬及び方法を用いて、Boc−Dpr(Fmoc)−OHを段階(c)で段階(b)の生成物と反応させ、次いでジオキサン中の2M HClを用いて段階(d)で脱保護することでBoc保護基を除去する。次いで、Boc脱保護化合物を段階(e)で(Goodman他(上掲)に記載の通り得られる)5−(ピリジン−2−イル)アミノペンタン酸と反応させる。段階(f)では、エステル基をけん化し、NaOH(水溶液)/ジオキサンを用いてFmoc側鎖を開裂する。段階(f)で得られた化合物を段階(g)で、標準的なペプチドカップリング試薬で予備活性化したBoc−アミノ−PEG−カルボン酸(Polypure社)と反応させる。段階(h)では、段階(g)で得られた化合物を(i)ジオキサン中のHCl又はジクロロメタン中のTFAと反応させ、(ii)予備活性化したBoc−アミノオキシ酢酸と反応させ、(iii)ジオキサン中のHCl又はジクロロメタン中のTFAと反応させて前駆体化合物を得る。この前駆体化合物を4−[18F]フルオロベンズアルデヒド(その合成法は国際公開第2004/080492号に記載されている)との反応によって18Fで標識することで、インビボイメージング剤4を得ることができる。
【0138】
実施例6:イメージング剤5の合成
実施例5に記載したようにして得られた出発化合物を、段階(a)でビニルスルホニル酢酸/カップリング試薬/塩基と反応させる(Scoberl and Biederman 1968 Liebigs Ann Chem; 716: 37-46; Olberg et al 2009 J Labelled Compd Radiopharm; published online 2009)。段階(b)では、ジエチレングリコールを3つの段階で18F標識補欠分子族に転化させ、次いでOlberg et al, 2008 Bioconjugate Chem; 19: 1301-1308及びOlberg et al, 2009 J Labelled Compd Radiopharm; published onlineに記載の方法に従って段階(c)でコンジュゲーションを行う。
【0139】
実施例7:イメージング剤6の合成
実施例5に記載したようにして得られた出発化合物を、段階(a)でBoc保護アミノ−PEGと反応させ、次いで例えばジクロロメタン中のTFA又はジオキサン中のHClを用いて段階(b)で脱保護して前駆体化合物を得る。18F標識は、段階(c)及び段階(d)からなる2段階法を用いて実施できる。即ち、Hocke et al 2005 Bioorg Med Chem Lett; 15: 4819及びGreguric et al 2009 J Med Chem; 52: 5299に記載の方法に従って段階(c)で6−クロロニコチン酸と反応させ、次いで段階(d)で[18F]−フッ化物と反応させる。別法として、18F標識は1つの段階(e)でも実施できる。この場合には、同時係属英国特許出願第0905438.8号に記載の通り、前駆体化合物を[18F]−6−フルオロニコチン酸テトラフルオロフェニルエステルと反応させる。そこに記載されているように、この反応は2〜11のpH範囲内の水性緩衝液中において周囲温度で実施できる。
【0140】
実施例8:αvβ3に対するインビトロ親和性評価
インテグリンレセプターαvβ3に対する親和性を、ホモジナイザーを用いてヒト内皮腺癌細胞株EA−Hy926から調製されかつ超遠心分離によって単離された膜画分に関する[125I]−エキスタチンとの競合実験によって測定した。試験化合物の希釈液をラジオトレーサー及び膜画分と混合した後、37℃で1時間インキュベートした。洗浄手順後、Skatronマイクロハーベスターを用いてフィルター上に膜を回収した。最後にフィルタースポットを切り取り、Packard γ−カウンターでカウントした。Prismソフトウェアを用いて[125I]−エキスタチンのKdを計算した。コールドエキスタチンを参照標準として使用し、Kiを0.14nMと測定した。使用した化合物は、EMD527040(ベンチマーく)、実施例2(viii)のcPn216キレート化化合物、及び実施例3(ii)のテトラアミンキレート化化合物であった。3種の化合物のすべてが同じ挙動を示した(図1)。試験した最高濃度は144μMであったが、この濃度でも放射性標識エキスタチンの結合を完全に競合排除することは不可能であった。
【0141】
この実験はαvβ3に対する低い親和性を実証しており、ベンチマーク化合物EMD527040に比べてキレート化バージョンが同様な結合曲線を有することを表している。
【0142】
実施例9:イメージング剤7の合成
この反応は手動窒素バブラー装置内で実施した。Fmoc基の標準的な除去のため、実施例4(i)に記載した樹脂(0.08mmol)をN−メチルピロリドン中の20%ピペリジンで処理した。(国際公開第2008/139206号に記載された方法に従って製造した)Cy5**(40mg、0.080mmol)、PyAOP(42mg、0.080mmol)及びジイソプロピルエチルアミン(40μl、0.24mmol)をN−メチルピロリドン(2mL)に溶解した溶液を5分間予備活性化し、樹脂に添加した。2時間後、HPLC分析は出発原料が部分的にしか転化していないことを示し、カップリングを繰り返した(反応時間20時間)。トリフルオロ酢酸(3mL)、ジクロロメタン(3mL)及びトリイソプロピルシラン(150μL)の混合物を用いて、Boc保護基の除去及び樹脂からの物質の開裂を同時に行った。2.5時間後、溶液を濾過し、(ロタベイパーで)濃縮した。残留物を、分取HPLC(カラム:Phenomenex Luna C18(2)5μm 21.2×250mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜50%B;流量:10.0mL/分;214nmでUV検出)によって精製することで、5mgのイメージング剤7(1−(Cy5**−アミド)−45−(3,5−ジクロロフェニル)−39,43−ジオキソ−42−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−3,6,9,12,15,18,21,24,27,30,33,36−ドデカオキサ−40,44−ジアザヘプタテトラコンタン−47−酸)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=3.1分、m/z期待値982.4(MH)2+、実測値982.8。
【0143】
実施例10:イメージング剤7のフローサイトメトリー評価
(実施例9に記載したようにして製造した)イメージング剤7の肺腺癌細胞株H2009(Cancer Res; 67: 5889-5895)に対する結合を、フローサイトメトリックアナライザー(Beckman Coulter FC500MPL)を用いて調べた。100nMの濃度で(37℃で1時間のインキュベーション)、約40%の細胞において陽性の取込みが証明され、これは分子中のこの位置への置換基の導入が許容されることを示している。
【技術分野】
【0001】
本発明は、インビボイメージング、特に新規なインビボイメージング剤に関する。本発明はまた、本発明のインビボイメージング剤の製造方法及び前記方法で有用な前駆体化合物も提供する。本発明のインビボイメージング剤は、インテグリンαvβ6の発現について常態からの逸脱が存在する状態の診断に有用である。
【背景技術】
【0002】
インテグリンは、α及びβサブユニットから構成される膜結合糖タンパク質のファミリーである。インテグリンαvβ6は比較的希少であって(Busk et al 1992 J Biol Chem; 267(9): 5790)、これは修復過程中に上皮組織で生成され、天然のマトリックス分子であるフィブロネクチン(Weinacker et al 1994 J Biol Chem; 269: 6940-6948)及びテネイシン(Wang et al, 1996, Am J Respir Cell Mol Biol; 15(5): 664)と優先的に結合する。ビトロネクチンもまたαvβ6に結合し(Huang et al 1998 J Cell Sci; 111(15): 2189-2195)、TGF−βはαvβ6との相互作用によって活性化される(Massague and Chen 2000 Genes & Dev; 14: 627-644)。
【0003】
インテグリンαvβ6は、創傷治癒、炎症及び癌のような病理学的過程においてアップレギュレートされることが判明している。αvβ6が遊走及び浸潤を促進する能力を有することは、癌の攻撃性の指標としての潜在的な役割を示している(Bates et al J Clin Invest 2005; 115(2): 339-347)。Popov et al, J Hepatol 2008; 48: 453-64には、肝線維症の経過中にαvβ6が活性化胆管上皮及び肝細胞上で発現されることが実証されており、αvβ6が肝線維症の治療のための標的として提唱されている。
【0004】
国際公開第2002/074730号及び同第2005/039547号は、αvβ6インテグリンレセプターを優先的に阻害し、肝線維症のような線維症性疾患状態を含む一連の疾患状態で薬剤として使用できる非標識ビフェニル化合物を開示している。
【0005】
ビフェニル化合物EMD527040は、αvβ6のアンタゴニストとして報告されている(Popov et al J Hepatol 2008; 48: 453-464)。EMD527040の化学構造は下記の通りである。
【0006】
【化1】
国際公開第2005/039547号は、活性化された線維芽細胞又は筋線維芽細胞が関係する状態の治療におけるEMD527040の使用を教示している。Patsenker et al, Gastroenterology 2008; 135: 660-670には、EMD527040が特異的αvβ6アンタゴニストであることが報告されている。Popov et al, Gastroenterology 2008; 134(4) SI : A-827には、肝線維形成のインビボイメージングで使用するための99mTcで標識されたEMD527040の三量体について言及されている。99mTc標識三量体のバックグラウンド肝臓取込みは注射量の1.5%であると報告されているが、これは肝臓内又はその付近における病変の検出を可能にするためには問題となることがある。
【0007】
αvβ6の異常発現に関連する一連の疾患状態の診断で使用するためにαvβ6を標的化する代替インビボイメージング剤に対する余地が存在している。
【先行技術文献】
【特許文献】
【0008】
国際公開第02/074730号パンフレット
【発明の概要】
【0009】
本発明は、被験体で発現されるαvβ6の検出で使用するのに適した代替インビボイメージング剤を提供する。本発明はまた、前記インビボイメージング剤を得るための方法、及び被験体で発現されるαvβ6の測定におけるインビボイメージング剤の使用も提供する。先行技術に比べて、本発明のインビボイメージング剤は比較的小さく、これは例えば薬物動態学的性質を調整するために修飾するのが容易であることを意味している。本発明のインビボイメージング剤はまた、既知のαvβ6アンタゴニストEMD527040と同様な生物学的性質を示す。
【図面の簡単な説明】
【0010】
【図1】図1は、3種の試験化合物に関し、インテグリンレセプターαvβ3に対する親和性をインビトロで評価した結果を示している。
【発明を実施するための形態】
【0011】
一態様では、本発明は次の式Iのインビボイメージング剤又はその塩もしくは溶媒和物を提供する。
【0012】
【化2】
式中、
nは0〜6の整数であり、
mは0〜8の整数であり、
X1はO、S又はNR’(式中、R’は水素又はC1-4アルキルである。)であり、
R1は−C(=NH)−NHR’(式中、R’はX1に関して定義した通りである。)であるか、或いはR1は1又は2個の窒素ヘテロ原子を有するC3-5含窒素ヘテロアリール環であり、
R2〜R4の各々は独立に水素、C1-4アルキル又はハロゲンであるか、或いはインビボイメージング成分を含む置換基であり、或いはR2及びR3又はR3及びR4はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-5ヘテロアリール環を形成し、
R5は水素又はR6R7基であり、R6はアミノ酸残基、炭水化物残基、−C(OH)−、−(CR'2)−、−C(=O)−C(CR'2)−、−C(=O)−NR'−、−(CR'2−O−CR'2)−、−CR'2−NR'−、−CR'2−S(O2)−CR'2−及び−(CR'2)−O−N=CR'−(式中、R’はX1に関して定義した通りである。)から選択される1〜50の二価リンカー単位を有する二価リンカー基であり、R7は水素であるか、或いはインビボイメージング成分を含む置換基であり、
R2〜R4の1以上がインビボイメージング成分を含む置換基であるか、或いはR5がR6R7(式中、R7はインビボイメージング成分を含む置換基である。)である。
【0013】
「インビボイメージング剤」という用語は、被験体における特定の生理学的又は病態生理学的状態を標的化するように設計され、被験体へのインビボ投与後に検出できる化合物を意味する。
【0014】
「その塩又は溶媒和物」という用語に係る好適な塩には、(i)鉱酸(例えば、塩酸、臭化水素酸、リン酸、メタリン酸、硝酸及び硫酸)から導かれるもの並びに有機酸(例えば、酒石酸、トリフルオロ酢酸、クエン酸、リンゴ酸、乳酸、フマル酸、安息香酸、グリコール酸、グルコン酸、コハク酸、メタンスルホン酸及びp−トルエンスルホン酸)から導かれるもののような生理学的に許容される酸付加塩、並びに(ii)アンモニウム塩、アルカリ金属塩(例えば、ナトリウム塩及びカリウム塩)、アルカリ土類金属塩(例えば、カルシウム塩及びマグネシウム塩)、有機塩基(例えば、トリエタノールアミン、N−メチル−D−グルカミン、ピペリジン、ピリジン、ピペラジン及びモルホリン)との塩、及びアミノ酸(例えば、アルギニン及びリシン)との塩のような生理学的に許容される塩基塩がある。「その塩又は溶媒和物」という用語に係る好適な溶媒和物には、エタノール、水、食塩水、生理的緩衝液及びグリコールと共に生成されるものがある。
【0015】
「アルキル」という用語は、好ましくは1〜4の炭素原子を含む直鎖又は枝分れアルキル基を意味する。かかる基の例には、メチル、エチル及びプロピルがある。
【0016】
「アリール」という用語は、環系中に5〜6の炭素原子を有する環式芳香族基(例えば、フェニル又はナフチル)を意味する。
【0017】
「ヘテロアリール」置換基は、上記に定義したアリールにおいて、環の炭素原子の1以上がN、S及びOから選択される「ヘテロ原子」で置き換えられたものである。
【0018】
本明細書中で「含窒素ヘテロアリール環」という用語は、上記に定義したアリールにおいて、環が1又は2個の窒素ヘテロ原子を含むものを意味する。
【0019】
「ハロゲン」という用語は、置換基としてのヨウ素、臭素、塩素及びフッ素並びにインビボイメージングに適したその同位体を包含する。
【0020】
「インビボイメージング成分」という用語は、前記被験体に投与した後に前記被験体の体外で検出できる原子又は原子団を意味する。
【0021】
「インビボイメージング成分を含む置換基」という用語は、それ自体がインビボイメージング成分である置換基、又は前記インビボイメージング成分を含む化学基を意味する。さらなる詳細は、下記において特定のインビボイメージング成分を論議する際に示す。
【0022】
「アミノ酸残基」という用語は、L−又はD−アミノ酸、アミノ酸類似体(例えば、ナフチルアラニン)或いはアミノ酸模倣体を意味し、これらは天然のもの又は純粋に合成由来のものであってよく、光学的に純粋なもの(即ち、単一の鏡像異性体、したがってキラルなもの)であるか、或いは鏡像異性体の混合物であってよい。好ましくは、本発明のアミノ酸は光学的に純粋なものである。
【0023】
「炭水化物残基」という用語は、多価アルコールの二価アルデヒド又はケトン誘導体を意味する。それはフルクトース又はグルコースのようなモノマー(単糖)であってもよいし、或いは2つの糖が互いに結合して二糖を形成していてもよい。二糖には、グルコース及びフルクトースから構成されるスクロースのような糖がある。「糖」という用語は、置換糖及び非置換糖の両方並びに糖の誘導体を包含する。好ましくは、糖はグルコース、グルコサミン、ガラクトース、ガラクトサミン、マンノース、ラクトース、フコース及びこれらの誘導体(例えば、シアル酸及びグルコサミンの誘導体)から選択される。糖は好ましくはα又はβである。糖は特にマンノピラノシド又はガラクトースピラノシドであり得る。糖上のヒドロキシル基は、例えば1以上のアセチル基で保護することができる。糖部分は、好ましくはN−アセチル化されている。かかる糖の好ましい例には、N−アセチルガラクトサミン、シアル酸、ノイラミン酸、N−アセチルガラクトース及びN−アセチルグルコサミンがある。
【0024】
本発明の特定のインビボイメージング剤がキラル中心を含む場合、本発明の技術的範囲は、2種の鏡像異性体のラセミ混合物並びに他方の鏡像異性体を実質的に含まずに各鏡像異性体を個別に含む組成物も包含する。即ち本発明では、例えば、R鏡像体を実質的に含まずにS鏡像体を含む組成物、又はS鏡像体を実質的に含まずにR鏡像体を含む組成物も想定されている。「実質的に含まずに」とは、組成物が10%未満又は8%未満又は5%未満又は3%未満又は1%未満の少量鏡像体を含むことを意味する。特定のインビボイメージング剤が2以上のキラル中心を含む場合、本発明の技術的範囲は、各種のジアステレオマーの混合物を含む組成物並びに他のジアステレオマーを実質的に含まずに各ジアステレオマーを含む組成物も包含する。特定のジアステレオマーのいずれかに言及せずに記載されたインビボイメージング剤は、4種のジアステレオマーのすべてを含む組成物、R,R及びS,S異性体のラセミ混合物を含む組成物、R,S及びS,R異性体のラセミ混合物を含む組成物、他のジアステレオマーを実質的に含まずにR,R鏡像体を含む組成物、他のジアステレオマーを実質的に含まずにS,S鏡像体を含む組成物、他のジアステレオマーを実質的に含まずにR,S鏡像体を含む組成物、及び他のジアステレオマーを実質的に含まずにS,R鏡像体を含む組成物を包含する。
【0025】
好ましくは、式Iのnは1〜4であり、最も好ましくは3である。
【0026】
好ましくは、式Iのmは0〜3であり、最も好ましくは1である。
【0027】
好ましくは、式IのX1はNR’であり、最も好ましくはNHである。
【0028】
好ましくは、式IのR1は五員又は六員含窒素ヘテロアリール環であり、最も好ましくはピリジル又はイミダゾリルである。R1がピリジルである場合、それは好ましくは2−ピリジルである。
【0029】
好ましくは、式IのR2〜R4の各々は独立に水素、クロロ、ヨード又はフルオロであり、最も好ましくは水素、ヨード又はクロロである。
【0030】
好ましくは、式IのR5はR6R7基である。X1がNHである場合、R6R7は好ましくは−C(=O)−R6aR7a基(式中、R6aは1〜20の二価リンカー単位を有する二価リンカー基であって、前記二価リンカー単位はR6に関して上記に定義した通りであり、R7aはR7に関して上記に定義した通りである。)である。
【0031】
式Iに関する好ましいインビボイメージング成分は、
(i)放射性金属イオン、
(ii)常磁性金属イオン、
(iii)γ放出型放射性ハロゲン、
(iv)陽電子放出型放射性非金属、及び
(v)インビボ光学イメージングに適したレポーター
から選択される。
【0032】
イメージング成分が放射性金属イオン(即ち、放射性金属)である場合、好適な放射性金属は、64Cu、48V、52Fe、55Co、94mTc又は68Gaのような陽電子放射体、及び99mTc、111In、113mIn又は67Gaのようなγ放射体であり得る。好ましい放射性金属は99mTc、64Cu、68Ga及び111Inである。最も好ましい放射性金属はγ放射体、特に99mTcである。
【0033】
イメージング成分が常磁性金属イオンである場合、好適なかかる金属イオンには、Gd(III)、Mn(II)、Cu(II)、Cr(III)、Fe(III)、Co(II)、Er(II)、Ni(II)、Eu(III)及びDy(III)がある。好ましい常磁性金属イオンはGd(III)、Mn(II)及びFe(III)であり、Gd(III)が特に好ましい。
【0034】
イメージング成分がγ放出型放射性ハロゲンである場合、放射性ハロゲンは好適には123I、131I及び77Brから選択される。125Iは、診断イメージング用のイメージング成分として使用するのに適さないので、特別に除外される。好ましいγ放出型放射性ハロゲンは123Iである。
【0035】
イメージング成分が陽電子放出型放射性非金属である場合、好適なかかる陽電子放射体には、11C、13N、15O、17F、18F、75Br、76Br及び124Iがある。好ましい陽電子放出型放射性非金属は11C、13N、18F及び124Iであり、特に好ましくは11C及び18Fであり、最も特に好ましくは18Fである。
【0036】
イメージング成分がインビボ光学イメージングに適したレポーターである場合、レポーターは光学イメージング方法で直接又は間接に検出できる任意の成分である。レポーターは、光散乱剤(例えば、着色又は無着色粒子)、吸光剤又は発光剤であり得る。さらに好ましくは、レポーターは発色団又は蛍光化合物のような色素である。色素は、紫外域乃至近赤外域の波長をもった電磁スペクトル中の光と相互作用する任意の色素であり得る。最も好ましくは、レポーターは蛍光特性を有する。好ましい有機発色性及び発蛍光性レポーターには、広範な非局在化電子系を有する群、例えばシアニン類、メロシアニン類、インドシアニン類、フタロシアニン類、ナフタロシアニン類、トリフェニルメチン類、ポルフィリン類、ピリリウム色素、チアピリリウム色素、スクアリリウム色素、クロコニウム色素、アズレニウム色素、インドアニリン類、ベンゾフェノキサジニウム色素、ベンゾチアフェノチアジニウム色素、アントラキノン類、ナフトキノン類、インダスレン類、フタロイルアクリドン類、トリスフェノキノン類、アゾ色素、分子内及び分子間電荷移動色素及び色素錯体、トロポン類、テトラジン類、ビス(ジチオレン)錯体、ビス(ベンゼン−ジチオレート)錯体、ヨードアニリン色素、ビス(S,O−ジチオレン)錯体がある。緑色蛍光タンパク質(GFP)及び異なる吸光/発光特性を有するGFPの変種のような蛍光タンパク質も有用である。特定の状況においてはある種の希土類金属(例えば、ユウロピウム、サマリウム、テルビウム又はジスプロシウム)の錯体が使用され、蛍光ナノ結晶(量子ドット)についても同様である。使用できる発色団の具体例には、フルオレセイン、スルホローダミン101(Texas Red)、ローダミンB、ローダミン6G、ローダミン19、インドシアニングリーン、Cy2、Cy3、Cy3.5、Cy5、Cy5.5、Cy7、Marina Blue、Pacific Blue、Oregon Green 88、Oregon Green 514、テトラメチルローダミン、Alexa Fluor 350、Alexa Fluor 430、Alexa Fluor 532、Alexa Fluor 546、Alexa Fluor 555、Alexa Fluor 568、Alexa Fluor 594、Alexa Fluor 633、Alexa Fluor 647、Alexa Fluor 660、Alexa Fluor 680、Alexa Fluor 700及びAlexa Fluor 750がある。特に好ましいのは、400nm乃至3μm、特に600〜1300nmの範囲内の可視又は近赤外(NIR)領域内に吸収極大を有する色素である。光学イメージングモダリティー及び測定技法には、特に限定されないが、ルミネセンスイメージング、内視鏡検査、蛍光内視鏡検査、光学コヒーレンス断層撮影、透過率イメージング、時間分解透過率イメージング、共焦点イメージング、非線形顕微鏡検査、光音響イメージング、音響光学イメージング、スペクトル分析、反射スペクトル分析、干渉分析、コヒーレンス干渉分析、拡散光学断層撮影及び蛍光媒介拡散光学断層撮影(連続波、時間ドメイン及び周波数ドメインシステム)、並びに光散乱、吸光、偏光、ルミネセンス、蛍光寿命、量子収量及び消光の測定がある。インビボ光学イメージングに適した好ましいレポーターはCy色素である。
【0037】
最も好ましいインビボイメージング成分は、好適なもの及び好ましいものとして上記にそれぞれ定義したような、放射性金属イオン、γ放出型放射性ハロゲン、陽電子放出型放射性非金属、又はインビボ光学イメージングに適したレポーターである。
【0038】
式Iのインビボイメージング剤に関して好ましくは、R2〜R4の1つは18F又は123Iである。別法として、式Iのインビボイメージング剤に関して好ましくは、R5はR6R70基である。式中、R7は、18F、放射性金属イオン又は常磁性金属イオンを含む金属錯体、及び光学イメージングに適したレポーターから選択されるイメージング成分を含んでいる。本明細書中に前述した通り、X1がNHである場合、R6R7は好ましくは−C(=O)−R6aR7aである。この実施形態に関しては、R7aは好ましくは前記イメージング成分を含んでいる。
【0039】
「金属錯体」という用語は、金属イオンと1以上のリガンドとの配位錯体を意味する。金属錯体は、「キレート交換に耐える」こと(即ち、金属配位座に関して他の潜在的に競合するリガンドとのリガンド交換を受けにくいこと)が極めて好ましい。潜在的に競合するリガンドは、インビトロでは前駆体化合物自体又は製剤中の他の賦形剤(例えば、製剤中に使用される放射線防護剤又は抗菌防腐剤)中に存在することがあり、インビボでは内因性化合物(例えば、グルタチオン、トランスフェリン又は血漿タンパク質)であり得る。
【0040】
式Iの二価リンカー基は、好ましくは10〜50原子の鎖であり、最も好ましくは10〜30原子の鎖である。最も好ましくは、二価リンカー基はバイオモディファイアー部分として作用する。「バイオモディファイアー部分」は、式Iのインビボイメージング剤の薬物動態及び血中クリアランス速度を調整する機能を有する。好適なバイオモディファイアー部分の例は、単分散PEG構成単位に基づいていて、1〜20の前記構成単位を含むものである。さらに、前記バイオモディファイアー部分は1〜10のアミノ酸残基を表すこともできる。前記バイオモディファイアー部分用の好ましいアミノ酸残基は、リシン及びグルタミン酸のような帯電アミノ酸、或いはシステイン酸及びホスホノアラニンのような帯電非天然アミノ酸である。加えて、アミノ酸であるグリシン、アスパラギン酸及びセリンも含めることができる。
【0041】
式Iの好ましいインビボイメージング剤は、次の式Iaの化合物又はその塩もしくは溶媒和物である。
【0042】
【化3】
式中、
R1aはR1に関して上記に好適なもの及び好ましいものとして定義した通りであり、
R2a〜R4aはR2〜R4に関して上記に好適なもの及び好ましいものとして定義した通りであり、
R5aは水素であるか、或いは−C(=O)−R6aR7a基(式中、R6a及びR7aは上記に好適なもの及び好ましいものとして定義した通りである。)である。
【0043】
本発明に係る好ましいインビボイメージング剤の非限定的な例は下記の通りである。
【0044】
【化4】
【0045】
【化5】
別の態様では、本発明は、本明細書中に好適なもの及び好ましいものとして定義した式Iのインビボイメージング剤の製造方法であって、インビボイメージング成分の適当な供給源を次の式IIの前駆体化合物と反応させる段階を含んでなる方法を提供する。
【0046】
【化6】
式中、
R11は式IのR1に関して好適なもの及び好ましいものとして定義した通りであり、
R12〜R14は水素、C1-4アルキル及びハロゲン並びに前駆体基から独立に選択され、
R15は水素又は前駆体基であるか、或いはR16R17基(式中、R16はR6に関して本明細書中に好適なもの及び好ましいものとして定義した二価リンカー基であり、R17は水素又は前駆体基である。)であり、
X11は式IのX1に関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
m’は式Iのmに関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
n’は式Iのnに関して本明細書中に好適なもの及び好ましいものとして定義した通りであり、
R12〜R14の1以上が前駆体基であるか、或いはR15がR16R17(式中、R17は前駆体基である。)であり、
前記前駆体基以外の反応基は化学的に保護されている。
【0047】
概して言えば、前駆体化合物をインビボイメージング成分の適当な供給源と「反応させる」段階は、できるだけ高い収率で所望のインビボイメージング剤を生成するのに適した反応条件下で2種の反応体を合わせることを含んでいる。
【0048】
「インビボイメージング成分の適当な供給源」とは、インビボイメージング成分が共有結合することで本発明のインビボイメージング剤を生成するようにして前駆体化合物の前駆体基と反応し得る化学形態のインビボイメージング成分を意味する。
【0049】
「前駆体化合物」は、好都合な化学形態のインビボイメージング成分との化学反応が部位特異的に起こり、最小数の段階(理想的にはただ1つの段階)で反応を実施でき、かつ格別の精製の必要なしに(理想的にはいかなる追加の精製も必要なしに)所望のインビボイメージング剤が得られるように設計された、インビボイメージング剤の誘導体からなる。かかる前駆体化合物は合成品であり、良好な化学純度で簡便に得ることができる。前駆体化合物は、不要の反応を回避するため、ある種の官能基又は「反応基」に関して1以上の保護基を任意に含むことができる。
【0050】
「保護基」という用語は、望ましくない化学反応を阻止又は抑制するが、分子の残部を変質させない十分に温和な条件下で問題の官能基から脱離させ得るのに十分な反応性を有するように設計された基を意味する。脱保護後には所望の生成物が得られる。保護基は当業者に公知であり、アミン基に関してはBoc(ここでBocはtert−ブチルオキシカルボニルである)、Fmoc(ここでFmocはフルオレニルメトキシカルボニルである)、トリフルオロアセチル、アリルオキシカルボニル、Dde(1−(4,4−ジメチル−2,6−ジオキソシクロヘキシリデン)エチル)及びNpys(3−ニトロ−2−ピリジンスルフェニル)から適宜に選択され、カルボキシル基に関してはメチルエステル、tert−ブチルエステル及びベンジルエステルから適宜に選択される。ヒドロキシル基に関しては、好適な保護基は、メチル、エチル又はtert−ブチル、アルコキシメチル又はアルコキシエチル、ベンジル、アセチル、ベンゾイル、トリチル(Trt)、又はテトラブチルジメチルシリルのようなトリアルキルシリルである。チオール基に関しては、好適な保護基はトリチル及び4−メトキシベンジルである。本明細書中では、保護基を含む置換基は「化学的に保護されている」。保護基の使用は、'Protective Groups in Organic Synthesis', Theorodora W. Greene and Peter G. M. Wuts, (Fourth Edition, John Wiley & Sons, 2006)に記載されている。
【0051】
「前駆体基」とは、インビボイメージング剤を得るため、インビボイメージング成分の適当な供給源と優先的に反応する化学基である。
【0052】
式Iaの好ましいインビボイメージング剤に関しては、前記式IIの前駆体化合物は次の式IIaを有している。
【0053】
【化7】
式中、R11a〜R15aは式IIのR11〜R15に関して上記に定義した通りである。
【0054】
下記のスキーム1は、Goodman et al, 2002 J Med Chem; 45: 1045-1051に記載された方法に基づいており、上記に好適なもの及び好ましいものとして定義した式IIの前駆体化合物の製造における初期段階を記載している。
【0055】
【化8】
式中、
R21は式IのR1に関して上記に定義した通りであり、
R22〜R24は水素、C1-4アルキル及びハロゲンから独立に選択され、或いはR22及びR23又はR23及びR24はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-6ヘテロアリール環を形成し、
Y21は−X21−R25(式中、X21は式IのX1に関して上記に定義した通りであり、R25は水素又は保護基である。)であり、
m”は式Iのmに関して定義した通りであり、
n”は式Iのnに関して定義した通りである。
【0056】
上記のスキーム1では、商業的に入手可能なアルデヒド1を段階(a)でマロン酸及び酢酸アンモニウムと反応させて中間体2を得る。段階(b)で中間体2をメタノール及びSOCl2と反応させれば、中間体3が得られる。標準的なペプチドカップリング試薬及び方法を用いて、中間体3を段階(c)でBoc−NH−CH((CH2)m"−Y21)−C(=O)OHと反応させる(このタイプの各種試薬がNovabiochem社、Bachem社及びAdvanced Chemtech社のような販売業者から商業的に入手可能であり、例えばBoc−Lys(Fmoc)−OH、Boc−Lys(Z)−OH又は対応するジアミノプロピオン酸(例えば、Boc−Dpr(Fmoc)−OH)がある)。標準的な方法(例えば、ジオキサン中の2M HCl)を用いてBoc保護基を段階(d)で除去することで、中間体5が得られる。段階(e)では、中間体5をR21−NH−(CH2)n1−C(=O)−OHと反応させるが、この反応体はGoodman et al, 2002 J Med Chem; 45: 1045-1051に記載の方法に従うか又はそれを改変することで製造される。段階(f)において、得られた中間体6を適当な溶媒中でNaOHと反応させることでエステルを対応するカルボン酸に転化させ、したがって中間体7に到達する。次いで、当業者にとって公知の方法を用いて中間体7を修飾することで、R22〜R24のいずれか又はY21の位置に前駆体基を導入し、本明細書中に定義した式IIの前駆体化合物に到達することができる。かかる方法は、特定のインビボイメージング成分の論議に際して下記に一層詳しく記載される。
【0057】
式IIに関して好ましくは、R12〜R14の1つが前記前駆体基である。
【0058】
別法として、式IIに関して好ましくは、R15がR16R17(式中、R17は前記前駆体基である。)である。
【0059】
式IIの好ましい前駆体基は、
(i)金属イメージング成分を錯体化できる1種以上のリガンド、
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体、
(iii)求核置換用のアルキルハライド、アルキルトシレート又はアルキルメシレートを含む誘導体、
(iv)求核又は求電子置換に向けて活性化された芳香環を含む誘導体、
(v)アシル化を受けやすい官能基を含む誘導体、
(vi)ベンズアルデヒドと反応させた場合にオキシム生成に参加する官能基を含む誘導体、
(vii)ビニルスルホン官能基を含む誘導体、
(viii)容易にアルキル化を受ける官能基を含む誘導体、及び
(ix)チオール含有化合物をアルキル化してチオエーテル含有生成物を与える誘導体
から選択される。
【0060】
式IIの前駆体化合物の前駆体基が(i)又は(ii)である場合、式IIの前駆体化合物は本発明の別の態様をなす。
【0061】
インビボイメージング成分の特定の供給源と反応させるためにいかなる前駆体基を選択すべきかは、インビボイメージング剤の技術分野において公知である。これは、本発明の特定のインビボイメージング剤を得るための方法について読者を手引きするため、以下に一層詳しく記載される。
【0062】
インビボイメージング成分が金属イオンである場合、前駆体基は金属イメージング成分を錯体化できる1種以上のリガンドを含む。キレート交換に耐える金属錯体を形成するために本発明で使用するのに適したリガンドには、2〜6(好ましくは2〜4)の金属ドナー原子が(金属ドナー原子を結合する炭素原子又は非配位ヘテロ原子の非配位主鎖を有することで)五員又は六員キレート環を生じるように配列されたキレート剤、或いは金属イオンと強く結合するドナー原子を含む単座リガンド(例えば、イソニトリル、ホスフィン、チオール又はジアゼニド)がある。キレート剤の一部として金属とよく結合するドナー原子種の例は、アミン、チオール、アミド、オキシム及びホスフィンである。ホスフィンは非常に強い金属錯体を形成するので、単座又は二座のホスフィンでも好適な金属錯体を形成する。イソニトリル及びジアゼニドの直線形状はキレート剤中に組み込みにくいものであり、したがって通例は単座リガンドとして使用される。好適なイソニトリルの例には、tert−ブチルイソニトリルのような単純アルキルイソニトリル、及びMIBI(即ち、1−イソシアノ−2−メトキシ−2−メチルプロパン)のようなエーテル置換イソニトリルがある。好適なホスフィンの例には、テトロフォスミン(Tetrofosmin)、及びトリス(3−メトキシプロピル)ホスフィンのような単座ホスフィンがある。好適なジアゼニドの例には、HYNIC系列のリガンド(即ち、ヒドラジン置換ピリジン又はニコチンアミド)がある。
【0063】
キレート交換に耐える金属錯体を形成する好適なキレート剤には、特に限定されないが、下記のものがある。
(i)ジアミンジオキシム、
(ii)MAG3(メルカプトアセチルトリグリシン)のようなチオールトリアミドドナーセット又はPicaのようなジアミドピリジンチオールドナーセットを有するN3Sリガンド及び関連するリガンド、
(iii)BAT又はECD(即ち、エチルシステイネート二量体)のようなジアミンジチオールドナーセット或いはMAMAのようなアミドアミンジチオールドナーセットを有するN2S2リガンド、
(iv)シクラム、モノオキソシクラム又はジオキソシクラムのようなテトラミン、アミドトリアミン又はジアミドジアミンドナーセットを有する開鎖又は大環状リガンドであるN4リガンド、並びに
(v)ジアミンジフェノールドナーセットを有するN2O2リガンド。
【0064】
上述のリガンドはテクネチウム(例えば、94mTc又は99mTc)を錯体化するために特に適しており、Jurisson et al, 1999 Chem Rev; 99: 2205-2218にさらに詳細に記載されている。かかるリガンドは、銅(64Cu又は67Cu)、バナジウム(例えば、48V)、鉄(例えば、52Fe)或いはコバルト(例えば、55Co)のような他の金属に対しても有用である。他の好適なリガンドは国際公開第91/01144号に記載されており、その中にはインジウム、イットリウム及びガドリニウムに対して特に好適なリガンド(特に大環状アミノカルボキシレート及びアミノホスホン酸リガンド)が含まれる。ガドリニウムの非イオン性(即ち、中性)金属錯体を形成するリガンドが知られており、米国特許第4,885,363号に記載されている。ガドリニウムに対して特に好ましいのは、DTPA、エチレンジアミン四酢酸(EDTA)、トリエチレンテトラアミン六酢酸(TTHA)、1,4,7,10−テトラアザシクロドデカン−1,4,7,10−四酢酸(DOTA)、10−(2−ヒドロキシプロピル)−1,4,7,10−テトラアザシクロドデカン−1,4,7−三酢酸(DO3A)及びこれらの誘導体を含むキレートである。
【0065】
上記のスキーム1について述べれば、前駆体基がキレート剤である前駆体化合物は、Y21が−X21−R25(式中、R25は保護基である。)である化合物7から出発して得ることができる。次いで、スキーム1の段階(f)後にR25保護基を除去することができる。キレート剤の反応性誘導体を脱保護化合物7と反応させることで前駆体化合物が得られる。反応性誘導体は、例えば、脱保護化合物7中のNH2との反応のためには活性エステル誘導体又はカルボン酸誘導体であり得る。前駆体基がキレート剤である場合に好ましくは、R15はR16R17基(式中、R17はキレート剤である。)である。この場合には、キレート剤の添加前に、脱保護化合物7を二価リンカー基R16の反応性誘導体と反応させる。
【0066】
適当なキレート剤を含む式IIの前駆体化合物の99mTc標識のためには、通常のテクネチウム出発原料は、テクネチウムがTc(VII)酸化状態にある過テクネチウム酸塩(即ち、TcO4-)である。過テクネチウム酸塩自体は容易に金属錯体を形成せず、したがってテクネチウム錯体の調製には、第一スズイオンのような適当な還元剤を添加してテクネチウムの酸化状態を低い酸化状態(通常はTc(I)乃至Tc(V))に還元することで錯体化を容易にすることが通常必要である。溶媒は、有機溶媒又は水性溶媒或いはこれらの混合物であり得る。溶媒が有機溶媒を含む場合、有機溶媒は好ましくはエタノール又はDMSOのような生体適合性溶媒である。好ましくは、溶媒は水性溶媒であり、最も好ましくは等張食塩水である。
【0067】
式Iの放射性ヨウ素化インビボイメージング剤を製造するのに適した式IIの前駆体化合物は、求電子又は求核放射性ヨウ素化を受ける誘導体或いは標識アルデヒド又はケトンとの縮合を受ける誘導体を含んでいる。第1のカテゴリーの例は下記の(a)〜(c)である。
(a)トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)、トリアルキルシラン(例えば、トリメチルシリル)或いは有機ホウ素化合物(例えば、ボロネートエステル又はオルガノトリフルオロボレート)のような有機金属誘導体、
(b)ハロゲン交換用の非放射性アルキルブロミド、或いは求核ヨウ素化用のアルキルトシレート、メシレート又はトリフレート、並びに
(c)求核ヨウ素化に向けて活性化された芳香環(例えば、アリールヨードニウム塩、アリールジアゾニウム塩、アリールトリアルキルアンモニウム塩又はニトロアリール誘導体)。
【0068】
好ましいかかる前駆体化合物は、(放射性ヨウ素交換を可能にするための)アリールヨージド又はブロミドのような非放射性ハロゲン原子、有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)、或いはトリアゼンのような有機前駆体又は求核置換のための良好な脱離基(例えば、ヨードニウム塩)からなっている。放射性ヨウ素化のために好ましくは、前駆体化合物は有機金属前駆体化合物からなり、最も好ましくはトリアルキルスズからなる。
【0069】
有機分子中に放射性ヨウ素を導入するための前駆体化合物及び方法は、Bolton, 2002 J Lab Comp Radiopharm: 45: 485-528に記載されている。好適なボロネートエステル有機ホウ素化合物及びその製法は、Kabalka et al, 2002 Nucl Med Biol; 29: 841-843; and 2003 Nuc Med Biol; 30: 369-373に記載されている。好適なオルガノトリフルオロボレート及びその製法は、Kabalka et al, 2004 Nucl Med Biol; 31 : 935-938に記載されている。
【0070】
放射性ヨウ素が結合できるアリール基の例を以下に示す。
【0071】
【化9】
いずれも、芳香環上への容易な放射性ヨウ素置換を可能にする置換基を含んでいる。チロシン残基は、その固有のフェノール基を用いて放射性ヨウ素化を実施することを可能にする。
【0072】
放射性ヨウ素を含む別の置換基は、例えば次式のように、放射性ハロゲン交換による直接ヨウ素化で合成することができる。
【0073】
【化10】
飽和脂肪族系に結合したヨウ素原子はインビボでの代謝を受けやすく、したがって放射性ヨウ素の損失を招きやすいことが知られているので、放射性ヨウ素原子は好ましくはベンゼン環のような芳香環への直接共有結合により又はビニル基を介して結合される。
【0074】
上記のスキーム1について述べれば、放射性ヨウ素化に適した前駆体化合物は、R22〜R24の1つがブロモ基である化合物7から出発して得ることができる。この化合物を適当なスタンナンと反応させてトリアルキルスズ前駆体化合物を得、これを放射性ヨウ素と反応させて式Iの放射性ヨウ素化インビボイメージング剤を得ることができる。別法として、化合物7のR22〜R24の1つが127Iであり得るが、これは放射性ハロゲン交換による放射性ヨウ素化に適した前駆体化合物である。
【0075】
放射性フッ素化に適した前駆体化合物は、[18F]−フッ化物で直接に標識されるか、或いは18F含有補欠分子族との反応性を有するように設計できる。
【0076】
放射性フッ素化は、[18F]−フッ化物と式IIの前駆体化合物中の適当な前駆体基との反応を用いる直接標識によって実施できる。前駆体基は、アルキルブロミド、アルキルメシレート又はアルキルトシレートのような良好な脱離基であり得る。[18F]−フッ化物による直接放射性フッ素化はまた、求核芳香族置換によっても実施できる。アリール系に関しては、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第四級アンモニウム塩からの18F−フッ化物求核置換が、アリール−18F誘導体への好適な経路である。別法として、前駆体化合物はクロロニコチンアミド前駆体基を含み得る。この場合、クロロの位置における18F−フッ化物求核置換によって[18F]フルオロニコチンアミド化合物が得られる(Greguric et al 2009 J Med Chem; 52: 5299-5302)。[18F]−フッ化物での直接標識が実施される場合に好ましくは、前駆体基は式IIのR15の位置に存在する。かかる前駆体化合物は、Y21が−X21−R25(式中、X21はNであり、R25は保護基である。)であるスキーム1の化合物7から出発して得ることができる。このR25保護基の除去後、遊離アミノ基の位置でカップリング反応を実施できる。例えば、前駆体基R15の反応性誘導体を用いてR15前駆体基を導入できる。別法として、R16リンカーの任意に保護された反応性誘導体をカップリングした後、脱保護し、R17前駆体基の反応性誘導体と反応させることができる。
【0077】
18Fはまた、前駆体化合物と18F標識補欠分子族との反応によっても導入できる。例えば、N−ハロアセチル前駆体基を補欠分子族18F(CH2)3OHでアルキル化して−NH(CO)CH2O(CH2)318F誘導体を得ることができる。本発明の18F標識化合物はまた、18F−フルオロジアルキルアミンを生成させ、次いで18F−フルオロジアルキルアミンを例えば塩素、P(O)Ph3及び活性化エステルから選択した前駆体基を含む前駆体化合物と反応させた場合のアミド生成によっても得ることができる。[18F]−N−メチルアミノオキシ含有補欠分子族(Olberg et al Bioconjugate Chem 2008; 19: 1301-1308)を前駆体化合物中のビニルスルホニル前駆体基と反応させて18F標識インビボイメージング剤を得ることもできる。別の好適な補欠分子族は、国際公開第2004/080492号に記載の通り得られる4−[18F]フルオロベンズアルデヒドであり、これはNH2と反応して−N=CH−[18F]フルオロフェニル誘導体を生成する。前段に記載した[18F]フルオロニコチンアミド化合物はまた、同時係属英国特許出願第0905438.8号に記載されているように、遊離アミノ基を有する前駆体化合物を[18F]−6−フルオロニコチン酸テトラフルオロフェニルエステルと反応させて得ることもできる。そこに記載されているように、この反応は、適当な溶媒(例えば、2〜11、好適には3〜11のpH範囲内の水性緩衝液)中において5〜70℃の極端でない温度(好ましくは周囲温度)で実施できる。
【0078】
18F標識誘導体の合成経路のさらなる詳細は、Bolton, 2002 J Lab Comp Radiopharm; 45: 485-528に記載されている。
【0079】
光学イメージングに適したレポーターを含む本発明のインビボイメージング剤を得ることに関しては、Licha et al, 2002 Topics Curr Chem; 222: 1-29; 2005 Adv Drug Deliv Rev; 57: 1087-1108に記載の方法を参照されたい。蛍光色素標識試薬を用いる標識の総説及び例に関しては、"Non-Radioactive Labelling, a Practical Introduction", Garman, A.J. Academic Press, 1997; "Bioconjugation - Protein Coupling Techniques for the Biomedical Sciences", Aslam, M. and Dent, A., Macmillan Reference Ltd, (1998)を参照されたい。コンジュゲーションに適した官能化シアニン色素(CyD)は、GE Healthcare社、Atto−Tec社、Dyomics社、Molecular Probes社などから商業的に入手できる。かかる色素の大部分はNHSエステルとして入手でき、これらは本明細書中に定義された式IIの前駆体化合物中のアミンと反応して所望のインビボイメージング剤を生成し得る。ヒドラジド、マレイミド又はスクシンイミジルエステル基で官能化されたAlexa Fluor(商標)647は、Molecular Probes社から商業的に入手できる。カルボキシル又はマレイミド基で官能化されたCyDは、欧州特許出願公開第1816475号に記載された方法に従って製造できる。これらの官能化CyD化合物は、ヒドロキシ又はアミン前駆体基を含む前駆体化合物と反応させて式Iの所望インビボイメージング剤を得ることができる。CyDにとって好ましい位置は、式IのR5である。
【0080】
式Iのインビボイメージング剤の製造方法はまた、固相合成によっても実施できる。「固相」は架橋された不溶性ポリマー材料であって、合成条件に対して化学的に不活性である。固相は通例は球状粒子(例えば、直径0.04〜0.15mmのビーズ)の形態を取るが、シート、ピン状粒子及び円盤状粒子も使用される。
【0081】
【化11】
R22〜R24、Y21及びm”は、スキーム1に関して上記に好適なもの及び好ましいものとして定義した通りである。固相は、スキーム2中にクロスハッチングを施した円で示されている。出発化合物はスキーム1からの化合物2であり、これを段階(a)でFmoc保護し、次いで段階(b)で固相に結合し、続いてFmoc保護基を段階(c)で(例えば、ジメチルホルムアミド又はNMP中の20%ピペリジンにより)除去する。段階(d)では、固相化合物2をFmoc−NH−CH(CH2)m"−Y21と反応させる。上記に記載したようにしてFmocを開裂させ、次いでスキーム1の(d)〜(f)に類似した段階をスキーム1の化合物5の固相バージョンであるものに関して実施する。前駆体化合物全体を固相上で合成し、標準的な方法(例えば、ジクロロメタン中のトリフルオロ酢酸)で開裂除去することができる。別法として、関係する化学に応じ、適当な段階で中間体を開裂除去し、以後の段階を溶液中で実施することもできる。
【0082】
好ましい実施形態では、本発明のインビボイメージング剤の製造方法は自動化される。自動化合成は、自動化合成装置、例えばTracerlab(商標)及びFastlab(商標)(いずれもGE Healthcare社から入手可能)によって簡便に実施できる。Fastlab(商標)は自動化陽電子放出断層撮影(PET)ラジオトレーサー合成プラットホームに関する技術の現状を表すものであり、新しいPETラジオトレーサーの開発に当たっては、その合成がFastlab(商標)に適合していることが望ましい。放射化学は、「カセット」を装置に取り付けることにより、自動化合成装置上で実施される。通常、かかるカセットは流体通路、反応器、及び試薬バイアル並びに放射合成後の清掃段階で使用される任意の固相抽出カートリッジを受け入れるためのポートを含んでいる。
【0083】
したがって、本発明のさらに別の態様では、本発明の自動化方法を実施するためのカセットであって、
(i)本発明の方法に関して上記に好適なもの及び好ましいものとして定義した前駆体化合物を含む容器、及び
(ii)本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング成分の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセットが提供される。
【0084】
カセットはまた、過剰のインビボイメージング成分を除去するためのイオン交換カートリッジを含み得る。自動化合成のために必要な試薬、溶媒及び他の消耗品もまた、濃度、体積、送出時間などに関する最終ユーザーの要求条件を満たすように自動化合成装置を運転させるソフトウェアを保持したコンパクトディスクのようなデータ媒体と共に含めることができる。本発明のカセットは、インビボイメージング成分が18Fである本発明のインビボイメージング剤を製造するために特に適している。
【0085】
さらに別の態様では、本発明は、本明細書中に定義したインビボイメージング剤を哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる組成物として定義される「医薬組成物」を提供する。「生体適合性キャリヤー」は、組成物が「哺乳動物への投与に適する」ようにして(即ち、毒性又は過度の不快感なしに哺乳動物体に投与できるようにして)インビボイメージング剤を懸濁又は溶解するための流体(特に液体)である。生体適合性キャリヤー媒質は、好適には、無菌のパイロジェンフリー注射用水、(有利には注射用の最終生成物が等張性又は非低張性になるように平衡させ得る)食塩水のような水溶液、或いは1種以上の張度調整物質(例えば、血漿陽イオンと生体適合性対イオンとの塩)、糖(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール物質(例えば、ポリエチレングリコール、プロピレングリコールなど)の水溶液のような注射可能なキャリヤー液体である。生体適合性キャリヤー媒質はまた、エタノールのような生体適合性有機溶媒を含んでいてもよい。かかる有機溶媒は、親油性の高い化合物又は配合物を可溶化するために有用である。好ましくは、生体適合性キャリヤー媒質はパイロジェンフリー注射用水、等張食塩水又はエタノール水溶液である。静脈内注射用生体適合性キャリヤー媒質のpHは好適には4.0〜10.5の範囲内にある。
【0086】
本発明の医薬組成物は、好適には、無菌保全性を維持しながら皮下注射針による1回又は数回の穿刺に適したシール(例えば、クリンプ加工した隔壁シールクロージャー)を備えた容器に入れた状態で供給される。かかる容器は1回分又は複数回分の患者用量を含み得る。好ましい複数用量容器は、複数の患者用量を含む(例えば、容積10〜30cm3の)単一のバルクバイアルからなり、したがって臨床的状況に合わせて製剤の実用寿命中に様々な時間間隔で1回分の患者用量を臨床グレードの注射器に抜き取ることができる。予備充填注射器は1回分のヒト用量又は「単位用量」を含むように設計され、したがって好ましくは臨床用に適した使い捨て注射器又は他の注射器である。医薬組成物が放射性医薬組成物である場合、予備充填注射器には、施術者を放射線量から防護するための注射器シールドを任意に設けることができる。好適なかかる放射性医薬品注射器シールドは当技術分野で公知であり、好ましくは鉛又はタングステンからなっている。
【0087】
本発明の医薬組成物はキットから製造できる。別法として、医薬組成物を無菌製造条件下で製造することで所望の無菌生成物を得ることができる。かかる医薬組成物を非無菌条件下で製造し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いて終末滅菌を施すこともできる。好ましくは、本発明の医薬組成物はキットから製造される。
【0088】
かかるキットは、本発明の好適な前駆体、好ましくは無菌で非発熱性の形態にある前駆体であって、インビボイメージング成分の無菌供給源との反応によって最小数の操作で所望の医薬組成物を与えるような前駆体を含んでいる。
キットが本発明の前駆体化合物を含む場合、前記キット自体は本発明のさらに別の態様をなす。かかる考慮事項は、放射性医薬品(特に、放射性同位体が比較的短い半減期を有する場合)にとって特に重要であると共に、取扱いを容易にして放射線薬剤師に対する放射線量を低減させるためにも特に重要である。したがって、かかるキットの再構成用の反応媒質は好ましくは上記に定義したような生体適合性キャリヤーであり、最も好ましくは水性のものである。キット中に使用するための前駆体を無菌製造条件下で使用すれば、所望の無菌で非発熱性の材料を得ることができる。また、前駆体を非無菌条件下で使用し、次いで例えばγ線照射、オートクレーブ処理、乾熱又は(例えば、エチレンオキシドによる)化学処理を用いる終末滅菌を施すこともできる。好ましくは、前駆体は無菌で非発熱性の形態で使用される。
【0089】
キットはさらに、放射線防護剤、抗菌防腐剤、pH調整剤又はフィラーのような追加成分を任意に含むことができる。
【0090】
「放射線防護剤」という用語は、水の放射線分解から生じる酸素含有フリーラジカルのような高反応性フリーラジカルを捕捉することで分解反応(例えば、レドックス過程)を阻止する化合物を意味する。好適な放射線防護剤は、アスコルビン酸、p−アミノ安息香酸(即ち、4−アミノ安息香酸)、ゲンチシン酸(即ち、2,5−ジヒドロキシ安息香酸)、及び生体適合性陽イオンを有するこれらの塩から選択される。
【0091】
「抗菌防腐剤」という用語は、潜在的に有害な微生物(例えば、細菌、酵母又はかび)の増殖を阻止する薬剤を意味する。抗菌防腐剤はまた、用量に応じ、多少の殺菌性を示すこともある。本発明の抗菌防腐剤の主な役割は、再構成後の医薬組成物(即ち、イメージング剤そのもの)中におけるこのような微生物の増殖を阻止することである。しかし、抗菌防腐剤は、再構成前のキットの1種以上の成分中における潜在的に有害な微生物の増殖を阻止するためにも任意に使用できる。好適な抗菌防腐剤には、パラベン類(即ち、メチル、エチル、プロピル又はブチルパラベン或いはこれらの混合物)、ベンジルアルコール、フェノール、クレゾール、セトリミド及びチオメルサールがある。好ましい抗菌防腐剤はパラベン類である。
【0092】
「pH調整剤」という用語は、再構成されたキットのpHがヒト又は哺乳動物への投与のために許容し得る範囲(およそpH4.0〜10.5)内にあることを保証するために有用な化合物又は化合物の混合物を意味する。好適なかかるpH調整剤には、トリシン、リン酸塩又はTRIS(即ち、トリス(ヒドロキシルメチル)アミノメタン)のような薬学的に許容される緩衝剤、及び炭酸ナトリウム、重炭酸ナトリウム又はこれらの混合物のような薬学的に許容される塩基がある。
【0093】
「フィラー」という用語は、製造及び凍結乾燥中における材料の取扱いを容易にすることができる薬学的に許容される増量剤を意味する。好適なフィラーには、塩化ナトリウムのような無機塩、及びスクロース、マルトース、マンニトール又はトレハロースのような水溶性糖又は糖アルコールがある。
【0094】
さらに別の態様では、本発明は、被験体で発現されるαvβ6の量及び/又は位置を決定するためのインビボイメージング方法であって、
(i)本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング剤を前記被験体を投与する段階、
(ii)段階(i)で投与した前記インビボイメージング剤を前記被験体で発現されるαvβ6に結合させる段階、
(iii)段階(ii)で結合させた前記インビボイメージング剤に含まれるインビボイメージング成分から放出される信号を検出する段階、並びに
(iv)前記信号の位置及び量を表す画像を生成する段階であって、前記信号が前記被験体で発現されるαvβ6の量及び/又は位置を表す段階
を含んでなるインビボイメージング方法を提供する。
【0095】
本発明の「被験体」は、任意のヒト又は動物被験体であり得る。好ましくは、本発明の被験体は哺乳動物である。最も好ましくは、前記被験体はインタクトな哺乳動物生体である。特に好ましい実施形態では、本発明の被験体はヒトである。
【0096】
インビボイメージング剤を「投与する」段階は、好ましくは非経口的に実施され、最も好ましくは静脈内に実施される。静脈内経路は、インビボイメージング剤を被験体の身体全域に送達し、前記被験体で発現されたαvβ6に接触させるための最も効率的な方法である。本発明のインビボイメージング剤は、好ましくは本明細書中に定義した本発明の医薬組成物として投与される。
【0097】
投与段階後かつ検出段階前に、インビボイメージング剤をαvβ6に結合させる。インビボイメージング剤は哺乳動物の身体を通って動的に移動し、体内の様々な組織に接触する。ひとたびインビボイメージング剤がαvβ6に接触すれば、特異的な相互作用が起こる結果、αvβ6をもった組織からのインビボイメージング剤のクリアランスは、αvβ6をもたない組織又は少ないαvβ6を発現する組織よりも長い時間がかかる。一定の時点に達すれば、αvβ6をもった組織に結合したインビボイメージング剤と少ないαvβ6を発現する組織(又はαvβ6を発現しない組織)に結合したインビボイメージング剤との比の結果として、αvβ6に特異的に結合したインビボイメージング剤の検出が可能となる。
【0098】
「信号を検出する」段階は、インビボイメージング成分から放出される信号を、前記信号に対して感受性を有する検出器によって検出することを含んでいる。かかる検出器は当技術分野で公知である。例えば、インビボイメージング成分がγ放射体である場合、検出は単光子放出コンピューター断層撮影(SPECT)カメラを用いて実施でき、インビボイメージング成分が常磁性金属イオンである場合、検出は磁気共鳴イメージング(MRI)カメラを用いて実施できる。
【0099】
「画像を生成する」段階は、取得された信号データに再構築アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作することで、前記インビボイメージング成分から放出される信号の位置及び/又は量を示す画像が生成される。
【0100】
「αvβ6状態」とは、αvβ6の異常発現(通例はαvβ6の過剰発現)によって特徴づけられる病的状態を意味する。本発明のインビボイメージング方法が有用なかかる状態の例には、炎症、癌及び線維症がある。本発明のインビボイメージング方法は、好ましくは前記被験体がαvβ6状態を有することが知られ又は疑われている場合に実施され、最も好ましくは前記被験体がαvβ6状態を有することが知られている場合に実施される。被験体がαvβ6状態を有することが知られている場合、前記インビボイメージング方法は好ましくは前記被験体に関する治療計画の進行中に繰り返して実施され、前記治療計画は前記αvβ6状態と戦うための薬物の投与を含んでいる。
【0101】
好ましい実施形態では、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング方法はさらに、αvβ6レセプターの量及び/又は位置を特定の臨床像に帰因させる段階(v)を含んでいる。
【0102】
さらに別の態様では、本発明は、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング方法で使用するための、本明細書中に好適なもの及び好ましいものとして定義したインビボイメージング剤を提供する。
【実施例】
【0103】
次に、以下の非限定的な実施例によって本発明を説明する。
【0104】
実施例の簡単な説明
実施例1は、非放射性イメージング剤1の合成法を記載している。
【0105】
実施例2は、イメージング剤2の合成法を記載している。
【0106】
実施例3は、イメージング剤3の合成法を記載している。
【0107】
実施例4は、非放射性イメージング剤4の合成法を記載している。
【0108】
実施例5は、イメージング剤4の合成法を記載している。
【0109】
実施例6は、イメージング剤5の合成法を記載している。
【0110】
実施例7は、イメージング剤6の合成法を記載している。
【0111】
実施例8は、αvβ3結合を評価するためのインビトロ千葉県香取郡神崎町新アッセイを記載している。
【0112】
実施例9は、イメージング剤7の合成法を記載している。
【0113】
実施例10は、イメージング剤7のフローサイトメトリー評価を記載している。
【0114】
実施例中で使用される略語のリスト
aq 水性
Boc t−ブチルオキシカルボニル
Bu ブチル
Bzl ベンジル
Dpr ジアミノプロピオン酸
ESI−MS エレクトロスプレーイオン化質量分析法
Fmoc 9−フルオレニルメチルオキシムカルボニル
h 時間
LC−MS 液体クロマトグラフィー質量分析法
MDP メチレンジホスホン酸
Me メチル
MH+ 分子イオン
min 分
m/z 質量/電荷比
PEG ポリエチレングリコール
Ser セリン
TFA トリフルオロ酢酸
tR 保持時間
trityl トリメチルフェニル
UV 紫外。
【0115】
実施例1:3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパン酸(非放射性イメージング剤1)の合成
実施例1(i):3−(Fmoc−アミノ)−3−(3−ブロモ−5−クロロフェニル)プロパン酸の合成
この化合物は、下記の実施例2(i)に記載されたようにして、しかし3−ブロモ−5−クロロベンズアルデヒド(ABCR、3.0g、14mmol)から出発して合成した。収量1.3g(2段階を通して19%)。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm、溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸、勾配:5分かけて05〜80%B、流量:0.6mL/分、UV検出:214及び254nm、ESI−MS)、tR=4.1分、m/z期待値500.0、実測値500.0。
【0116】
実施例1(ii):3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパン酸の合成
この化合物は、下記の実施例2(ii)、2(iii)、2(vi)及び2(vii)に記載されたものと同じ方法を用いて、しかし3−(Fmoc−アミノ)−3−(3−ブロモ−5−クロロフェニル)プロパン酸(実施例1(i)に記載、0.6g、1.2mmol)をトリチルクロリド樹脂(Novabiochem社、置換度1.6mmol/g、0.5g)上に結合することから出発し、続いてそれぞれFmoc−Ser(Bzl)−OH(Aldrich社、0.5g、1.2mmol)及び5−((tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸(下記実施例2(v)に記載、0.21g、0.71mmol)をカップリングすることにより固体担体上で合成した。Boc脱保護及び固体担体からの切断によって0.211gの粗物質を得た。
【0117】
実施例1(iii):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパノエートの合成
3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパン酸(実施例1(ii)に記載、0.167g、0.264mmol)をメタノール(3mL)に溶解した溶液に、塩化チオニル(Sigma Aldrich社、79mg、0.66mmol)を0℃(氷/水浴)でゆっくりと添加した。混合物を室温まで加温し、45分後に水で奪活した。溶液を蒸発させ、生成物をメタノールから再結晶して0.155g(91%)を得た。
【0118】
実施例1(iv):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−(トリメチルスタンニル)フェニル)プロパノエートの合成
この反応はマイクロ波装置(Biotage社)内で実施した。メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−ブロモ−5−クロロフェニル)プロパノエート(実施例1(iii)に記載、10mg、15μmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(Strem chemicals社、1mg、0.8μmol)及びヘキサメチル二スズ(Sigma Aldrich社、40.0mg、122μmol)をトルエン(2mL)中に含む混合物を窒素でパージし、140℃で30分間加熱した。フラッシュクロマトグラフィー(シリカ、1%トリエチルアミンを含むジクロロメタン/メタノール(9:1))によって精製することで、5mgの半純粋生成物を得た。
【0119】
実施例1(v):メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパノエートの合成
ヨウ素(Fluka社、2mg、74μmol)及びメチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−(トリメチルスタンニル)フェニル)プロパノエート(実施例1(iv)に記載、5mg、7μmol)をジクロロメタン(2mL)に溶解した溶液を室温で2時間撹拌した。反応混合物をチオ硫酸ナトリウム溶液(0.3M、1mL)で処理した。相の分離後、ジクロロメタン溶液を水及びブラインで洗浄し、(硫酸ナトリウムで)乾燥し、濃縮して4mgの粗物質を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値693.1(MH+)、実測値693.3。
【0120】
実施例1(vi):3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパン酸の合成
メチル3−(3−(ベンジルオキシ)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド)−3−(3−クロロ−5−ヨードフェニル)プロパノエート(実施例1(v)に記載、4mg、6μmol)を、メタノール(1mL)と水酸化ナトリウム水溶液(1M、0.2mL)との混合物に溶解した。反応混合物を室温で1時間撹拌した。溶液をジクロロメタン(2×2mL)で抽出した。水性相を塩酸(1M)でpH6に調整し、次いでジクロロメタン(2×2mL)及び酢酸エチル(2×2mL)で抽出した。有機相を合わせ、(硫酸ナトリウムで)乾燥し、濃縮して0.5mgを得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値679.1(MH+)、実測値679.2。
【0121】
実施例2:(6S,Z)−3−(3,5−ジクロロフェニル)−22−(ヒドロキシイミノ)−17−(((Z)−3−(ヒドロキシイミノ)−2−メチルブタン−2−イル)アミノ)エチル)−21,21−ジメチル−5,9,13−トリオキソ−6−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−4,8,14,20−テトラアザトリコサン−1−酸(イメージング剤2)の合成
実施例2(i):Fmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸の合成
3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸(100mg、0.43mmol)(J Med Chem; 45: 1045-1051及びTetrahedron Asym; 19: 2072-2077に記載の通り合成)をクロロホルム/メタノール/水(65:25:4、4.5mL)中に懸濁した撹拌懸濁液に、ジイソプロピルエチルアミン(Fluka社、227μL、1.30mmol)を添加した。この溶液に9−フルオレニルメトキシカルボニル−N−ヒドロキシスクシンイミド(Novabiochem社、169mg、0.50mmol)を添加した。1時間後、混合物を(ロタベイパー(rotavapor)で)濃縮し、残留物を0.1%トリフルオロ酢酸含有アセトニトリル(10mL)中に取り、5分間撹拌し、濾過した。溶液を濃縮し、残留物をフラッシュクロマトグラフィーに付した。固体物質をカラム上に直接適用し、ヘキサン/酢酸エチル(1:1)で溶出して不純物を除去し、続いて生成物をクロロホルム/メタノール(9:1)で溶出することで、177mg(91%)の生成物を無色油状物として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=4.1分、m/z期待値456.1(MH+)、実測値456.1。
【0122】
実施例2(ii):固体担体へのFmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸の結合
トリチルクロリド樹脂(Novabiochem社、置換度1.6mmol/g、0.406g)に、Fmoc−3−アミノ−3−(3,5−ジクロロフェニル)プロパン酸(実施例2(i)に記載、0.30g、0.65mmol)をジクロロメタンとN−メチルピロリドンとの混合物(8:1、9mL)に溶解した溶液を添加し、続いてジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、2.6mL、5.2mmol)を添加した。混合物をローラーテーブル上に5時間保ち、次いで手動窒素バブラー装置に移した。樹脂を排液し、ジクロロメタン/メタノール/ジイソプロピルエチルアミン溶液(17:2:1、6mL)で3回処理し、ジクロロメタンで洗浄し、乾燥した。
【0123】
実施例2(iii):Fmoc−Dpr(ivDde)−OHのカップリング
この反応は、実施例2(ii)に記載した樹脂(0.65mmol)上において手動窒素バブラー装置内で実施した。標準的な処理(N−メチルピロリドン中20%ピペリジン)によってFmoc基を除去した。Fmoc−Dpr(ivDde)−OH(Novabiochem社、0.69g、1.3mmol)を、N−メチルピロリドン(7mL)中においてHATU((N−[(ジメチルアミノ)−1H−1,2,3−トリアゾロ[4,5−b]ピリジン−1−イルメチレン]−N−メチルメタンアミニウムヘキサフルオロホスフェートN−オキシド、Genscript社、0.49g、1.3mmol)及びジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、1.3mL、2.6mmol)で5分間予備活性化し、樹脂に添加した。2時間後、樹脂を排液し、N−メチルピロリドンで洗浄した。標準的なカイザー試験は反応が完了していないことを示し、カップリングを繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=4.4分、m/z期待値748.3(MH+)、実測値748.2。
【0124】
実施例2(iv):5−(tert−ブトキシカルボニル−ピリジン−2−イル−アミノ)ペンタン酸メチルエステル
2−(N−(tert−ブトキシカルボニル)アミノ)ピリジン(1.0g、5.2mmol、J Org Chem;67:4965−4967に記載の通り合成した)を無水ジメチルホルムアミド(15mL)に溶解した溶液を氷/水浴中で5℃に冷却した。水素化ナトリウム(Aldrich社、鉱油中60%、0.25g、6.3mmol)を少量ずつ添加した。冷却浴を取り除き、混合物を20分間激しく撹拌した。次いで、混合物を(氷/水浴で)冷却し、5−ブロモ吉草酸メチル(Aldrich社、0.89mL、6.3mmol)を滴下した。10分後、冷却浴を取り除き、混合物を2時間撹拌し、次いで(ロタベイパーで)濃縮した。残留物をジクロロメタン(30mL)と塩酸(0.02N、20mL)との混合物中に取り、激しく振盪した。相を分離し、有機相を飽和重炭酸ナトリウム水溶液(20mL)及びブライン(20mL)で抽出し、(Na2SO4で)乾燥した。(ロタベイパーで)濃縮した後、粗物質を無色油状物(1.52g)として得た。粗物質のアリコート(0.5g)をフラッシュクロマトグラフィー(ヘキサン/酢酸エチル、1:1)によって精製することで、生成物を無色油状物(収量490mg(93%))として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.6分、m/z期待値309.2(MH+)、実測値309.4。
【0125】
実施例2(v):5−(tert−ブトキシカルボニル−ピリジン−2−イル−アミノ)ペンタン酸
実施例2(iv)に記載したエステル(0.69g、2.3mmol)をメタノール(10mL)に溶解した撹拌溶液に、水酸化ナトリウム(4N、2mL)を周囲温度で添加した。反応を薄層クロマトグラフィー(シリカ、ヘキサン/酢酸エチル、1:1)によってモニターした。3時間後、溶媒を(ロタベイパーで)蒸発させた。水(8mL)を残留物に添加し、水溶液をエーテル(2×10mL)で抽出し、次いで塩酸(2N)を用いてやや酸性(pH約5)にした後、ジクロロメタン(3×10mL)で抽出した。ジクロロメタン相を合わせて(Na2SO4で)乾燥し、濾過し、蒸発させることで、生成物を無色油状物(収量594mg(90%))として得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.0分、m/z期待値295.2(MH+)、実測値295.4。
【0126】
実施例2(vi):5−(tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸のカップリング
この反応は手動窒素バブラー装置内で実施した。標準的な処理(N−メチルピロリドン中20%ピペリジン)により、実施例2(iii)に記載した樹脂のFmoc基を除去した。5−(tert−ブトキシカルボニル(ピリジン−2−イル)アミノ)ペンタン酸(実施例2(v)に記載、0.25g、0.85mmol)を、N−メチルピロリドン(6mL)中においてHATU(Genscript社、0.32g、0.85mmol)及びジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M溶液、0.845mL、1.69mmol)で5分間予備活性化し、樹脂に添加した。3時間後、樹脂を排液し、洗浄した。カイザー試験は反応が完了していないことを示したので、カップリングを繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=3.5分、m/z期待値802.3(MH+)、実測値802.3。
【0127】
実施例2(vii):保護基の除去及び固体担体からの開裂
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.325mmol)に、2%ヒドラジンをN−メチルピロリドン(5mL)に溶解した溶液を添加した。2分後、樹脂を排液し、それぞれ5分及び10分の反応時間で処理を2回繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄した。トリフルオロ酢酸/ジクロロメタン/トリイソプロピルシラン溶液(49:49:2、6.2mL)中において、Boc基の除去及び樹脂からの生成物の開裂を同時に行った。2.5時間後、樹脂を濾別し、溶液を(ロタベイパーで)濃縮乾固した。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)による分析:tR=1.5分、m/z期待値496.1(MH+)、実測値496.1。
【0128】
実施例2(viii):cPn216−グルタル酸とのコンジュゲーション
(国際公開第2003/006070号に記載の通り合成した)cPn216−グルタル酸(0.26g、0.57mmol)を、ジメチルホルムアミド(3mL)中においてPyAOP(7−アザベンゾトリアゾール−1−イルオキシ−トリス(ピロリジノ)ホスホニウムヘキサフルオロホスフェート、Applied Biosystems社、0.222g、0.426mmol)及びジイソプロピルエチルアミン(Aldrich社、148μL、0.852mmol)で10分間予備活性化した。実施例2(vii)からの化合物(0.141g、0.284mmol)をジメチルホルムアミド(6mL)に溶解し、予備活性化したキレートに添加した。反応の進行をLC−MS分析によってモニターした。1時間後、ジイソプロピルエチルアミン(Fluka社、N−メチルピロリドン中2M、0.4mL、0.4mmol)を添加し、混合物を1時間撹拌した。予備活性化したcPn216−グルタル酸の第2部分(0.143g、0.312mmol)を添加した(同じカップリング試薬)。1時間後、反応は完了し、混合物を(ロタベイパーで)濃縮した。化合物を分取HPLC(カラム:Phenomenex Luna C18(2)10μm 250×50.0mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜40%B;流量:50mL/分;214nm及び254nmでUV検出)によって精製し、凍結乾燥して56mg(21%)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.8分、m/z期待値935.5(MH+)、実測値935.5。
【0129】
実施例2(ix):イメージング剤2を得るための99mTc標識
実施例2(viii)で得た前駆体化合物を99mTcで標識するためには、(実施例2(viii)に記載したようにして得た)100μgの前駆体化合物を、メタノール中において0.5mlのNa2CO3/NaHCO3緩衝液(pH9.2)、99mTcジェネレーターから得た0.5mlのTcO4-及び0.1mlのSnCl2/MDP溶液と共に室温でインキュベートすればよい。
【0130】
実施例3:3−((S)−3−(4−(2−アミノエチルアミノ)−3−((2−アミノエチルアミノ)メチル)ブタンアミド)−2−(5−(ピリジン−2−イルアミノ)ペンタンアミド)プロパンアミド−3−(3,5−ジクロロフェニル)プロパン酸(イメージング剤3)の合成
実施例3(i):テトラ−Boc−テトラアミン−NHSエステルのカップリング
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.074mmol)に、2%ヒドラジンをN−メチルピロリドン(5mL)に溶解した溶液を添加した。2分後、樹脂を排液し、それぞれ5分及び10分の反応時間で処理を2回繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄し、排液した。樹脂をN−メチルピロリドン中に懸濁し、次いで(国際公開第2006/008496号に記載の通り合成した)テトラ−Boc−テトラアミン−NHSエステル(0.053g、0.074mmol)及びN−メチルモルホリン(Merck社、9.7μL、0.088mmol)を添加した。2時間後、樹脂を排液し、洗浄した。カップリングを標準的なカイザー試験で追跡し、さらに3回繰り返した。樹脂のアリコートを開裂し(ジクロロメタン中5%トリフルオロ酢酸、5分間)、LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で0〜60%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS)によって分析した。tR=3.2分、m/z期待値348.7((M−Boc5)H2)2+、実測値348.6。
【0131】
実施例3(ii):Boc脱保護及び固体担体からの開裂
実施例3(i)に記載した樹脂(0.074mmol)をトリフルオロ酢酸/トリイソプロピルシラン/水(1.9mL、5μL、5μL)溶液で2時間処理することで、Boc保護基の除去及び固体担体からの生成物の開裂を同時に行った。溶液を樹脂から濾別し、(ロタベイパーで)濃縮した。生成物を分取HPLC(カラム:Phenomenex Luna C18(2)5μm 250×21.2mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で5〜50%B;流量:10.0mL/分;214nm及び254nmでUV検出)によって精製し、凍結乾燥した。収量8.1mg(16%)。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.3分、m/z期待値696.3(MH+)、実測値696.3。
【0132】
実施例3(iii):イメージング剤3を得るための99mTc標識
実施例3(ii)で得た前駆体化合物は、実施例2(ix)に記載した方法を用いて99mTcで標識することができる。
【0133】
実施例4:(48S,E)−51−(3,5−ジクロロフェニル)−1−(4−フルオレニルフェニル)−5,45,49−トリオキソ−48−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−3,9,12,15,18,21,24,27,30,33,36,39,42−トリデカオキサ−2,6,46,50−テトラアザトリペンタコント−1−エン−53−酸(非放射性イメージング剤4)
実施例4(i):Fmoc−PEG(12)プロピオン酸とのコンジュゲーション
この反応は手動窒素バブラー装置内で実施した。実施例2(vi)に記載した樹脂(0.17mmol)に、N−メチルピロリドン(10mL)中の2%ヒドラジンを添加した。5分後、樹脂を排液し、5分の反応時間で処理を繰り返した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄した。1−(9H−フルオレン−9−イル)−3−オキソ−2,7,10,13,16,19,22,25,28,31,34,37,40−トリデカオキサ−4−アザトリテトラコンタン−43−酸(Polypure社、0.244g、0.290mmol)を、N−メチルピロリドン(2mL)中においてHATU(Applied Biosystems社、0.11g、0.29mmol)及びジイソプロピルエチルアミン(Fluka社、50μL、0.29mmol)で5分間予備活性化し、樹脂に添加した。カップリングを標準的なカイザー試験で追跡した。2時間後、樹脂を排液し、N−メチルピロリドン(2mL)中で5分間予備活性化したFmoc−アミノPEG(12)プロピオン酸(0.134mg、0.160mmol)、HATU(0.070g、0.18mmol)及びジイソプロピルエチルアミン(80μL、0.46mmol)の溶液を用いてカップリングを繰り返した。3時間後、混合物を排液し、N−メチルピロリドン及びジクロロメタンで洗浄した。標準的なカイザー試験は反応が完了したことを示した。
【0134】
実施例4(ii):Fmoc−アミノオキシ酢酸とのコンジュゲーション
この反応は手動窒素バブラー装置内で実施した。標準的なFmoc基除去のため、実施例4(i)に記載した樹脂(0.17mmol)をN−メチルピロリドン中の20%ピペリジンで処理した。Fmoc−アミノオキシ酢酸(Iris Biotech GmbH、0.050g、0.16mmol)を、
N−メチルピロリドン(2mL)中においてHATU(Applied Biosystems社、0.070g、0.18mmol)及びジイソプロピルエチルアミン(Fluka社、80μL、0.46mmol)で5分間予備活性化し、次いで樹脂に添加した。2時間後、樹脂を排液し、洗浄した。カイザー試験は転化が不完全であることを示し、同じ条件を用いてカップリングを2回繰り返した。
【0135】
実施例4(iii):保護基の除去及び固体担体からの開裂
実施例4(ii)からの樹脂(0.17mmol)を、手動窒素バブラー装置内において、N−メチルピロリドン中の20%ピペリジンを用いる標準的なFmoc脱保護に付した。樹脂をN−メチルピロリドン及びジクロロメタンで洗浄し、排液した。トリフルオロ酢酸(3mL)、ジクロロメタン(3mL)及びトリイソプロピルシラン(150μL)の混合物を用いて、Boc保護基の除去及び樹脂からの物質の開裂を同時に行った。2.5時間後、溶液を濾過し、(ロタベイパーで)濃縮した。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で5〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=3.5分、m/z期待値584.8(MH2)2+、実測値584.3。
【0136】
実施例4(iv):4−フルオロベンズアルデヒドとのオキシム形成(非放射性イメージング剤4)
実施例4(iii)に記載した生成物(0.17mmol)を水/アセトニトリル(4:1、3mL)に溶解した溶液に、4−フルオロベンズアルデヒド(Fluka社、4μL、0.04mmol)を添加した。反応混合物を70℃で1時間撹拌した。混合物を濃縮し、生成物を分取HPLC(カラム:Phenomenex Luna C18(2)5μm 21.2×250mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜80%B;流量:10.0mL/分;214nm及び254nmでUV検出)によって精製することで、凍結乾燥後に収量11mg(5%)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜80%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=2.3分、m/z期待値637.8(MH2)2+、実測値637.4。
【0137】
実施例5:イメージング剤4の合成
Goodman et al(上述)の方法に従い、商業的に入手可能なベンズアルデヒド(Sigma−Aldrich社から入手可能)を段階(a)でマロン酸及び酢酸アンモニウムと反応させ、次いで段階(b)でメタノール及びSOCl2と反応させる。標準的なペプチドカップリング試薬及び方法を用いて、Boc−Dpr(Fmoc)−OHを段階(c)で段階(b)の生成物と反応させ、次いでジオキサン中の2M HClを用いて段階(d)で脱保護することでBoc保護基を除去する。次いで、Boc脱保護化合物を段階(e)で(Goodman他(上掲)に記載の通り得られる)5−(ピリジン−2−イル)アミノペンタン酸と反応させる。段階(f)では、エステル基をけん化し、NaOH(水溶液)/ジオキサンを用いてFmoc側鎖を開裂する。段階(f)で得られた化合物を段階(g)で、標準的なペプチドカップリング試薬で予備活性化したBoc−アミノ−PEG−カルボン酸(Polypure社)と反応させる。段階(h)では、段階(g)で得られた化合物を(i)ジオキサン中のHCl又はジクロロメタン中のTFAと反応させ、(ii)予備活性化したBoc−アミノオキシ酢酸と反応させ、(iii)ジオキサン中のHCl又はジクロロメタン中のTFAと反応させて前駆体化合物を得る。この前駆体化合物を4−[18F]フルオロベンズアルデヒド(その合成法は国際公開第2004/080492号に記載されている)との反応によって18Fで標識することで、インビボイメージング剤4を得ることができる。
【0138】
実施例6:イメージング剤5の合成
実施例5に記載したようにして得られた出発化合物を、段階(a)でビニルスルホニル酢酸/カップリング試薬/塩基と反応させる(Scoberl and Biederman 1968 Liebigs Ann Chem; 716: 37-46; Olberg et al 2009 J Labelled Compd Radiopharm; published online 2009)。段階(b)では、ジエチレングリコールを3つの段階で18F標識補欠分子族に転化させ、次いでOlberg et al, 2008 Bioconjugate Chem; 19: 1301-1308及びOlberg et al, 2009 J Labelled Compd Radiopharm; published onlineに記載の方法に従って段階(c)でコンジュゲーションを行う。
【0139】
実施例7:イメージング剤6の合成
実施例5に記載したようにして得られた出発化合物を、段階(a)でBoc保護アミノ−PEGと反応させ、次いで例えばジクロロメタン中のTFA又はジオキサン中のHClを用いて段階(b)で脱保護して前駆体化合物を得る。18F標識は、段階(c)及び段階(d)からなる2段階法を用いて実施できる。即ち、Hocke et al 2005 Bioorg Med Chem Lett; 15: 4819及びGreguric et al 2009 J Med Chem; 52: 5299に記載の方法に従って段階(c)で6−クロロニコチン酸と反応させ、次いで段階(d)で[18F]−フッ化物と反応させる。別法として、18F標識は1つの段階(e)でも実施できる。この場合には、同時係属英国特許出願第0905438.8号に記載の通り、前駆体化合物を[18F]−6−フルオロニコチン酸テトラフルオロフェニルエステルと反応させる。そこに記載されているように、この反応は2〜11のpH範囲内の水性緩衝液中において周囲温度で実施できる。
【0140】
実施例8:αvβ3に対するインビトロ親和性評価
インテグリンレセプターαvβ3に対する親和性を、ホモジナイザーを用いてヒト内皮腺癌細胞株EA−Hy926から調製されかつ超遠心分離によって単離された膜画分に関する[125I]−エキスタチンとの競合実験によって測定した。試験化合物の希釈液をラジオトレーサー及び膜画分と混合した後、37℃で1時間インキュベートした。洗浄手順後、Skatronマイクロハーベスターを用いてフィルター上に膜を回収した。最後にフィルタースポットを切り取り、Packard γ−カウンターでカウントした。Prismソフトウェアを用いて[125I]−エキスタチンのKdを計算した。コールドエキスタチンを参照標準として使用し、Kiを0.14nMと測定した。使用した化合物は、EMD527040(ベンチマーく)、実施例2(viii)のcPn216キレート化化合物、及び実施例3(ii)のテトラアミンキレート化化合物であった。3種の化合物のすべてが同じ挙動を示した(図1)。試験した最高濃度は144μMであったが、この濃度でも放射性標識エキスタチンの結合を完全に競合排除することは不可能であった。
【0141】
この実験はαvβ3に対する低い親和性を実証しており、ベンチマーク化合物EMD527040に比べてキレート化バージョンが同様な結合曲線を有することを表している。
【0142】
実施例9:イメージング剤7の合成
この反応は手動窒素バブラー装置内で実施した。Fmoc基の標準的な除去のため、実施例4(i)に記載した樹脂(0.08mmol)をN−メチルピロリドン中の20%ピペリジンで処理した。(国際公開第2008/139206号に記載された方法に従って製造した)Cy5**(40mg、0.080mmol)、PyAOP(42mg、0.080mmol)及びジイソプロピルエチルアミン(40μl、0.24mmol)をN−メチルピロリドン(2mL)に溶解した溶液を5分間予備活性化し、樹脂に添加した。2時間後、HPLC分析は出発原料が部分的にしか転化していないことを示し、カップリングを繰り返した(反応時間20時間)。トリフルオロ酢酸(3mL)、ジクロロメタン(3mL)及びトリイソプロピルシラン(150μL)の混合物を用いて、Boc保護基の除去及び樹脂からの物質の開裂を同時に行った。2.5時間後、溶液を濾過し、(ロタベイパーで)濃縮した。残留物を、分取HPLC(カラム:Phenomenex Luna C18(2)5μm 21.2×250mm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:60分で10〜50%B;流量:10.0mL/分;214nmでUV検出)によって精製することで、5mgのイメージング剤7(1−(Cy5**−アミド)−45−(3,5−ジクロロフェニル)−39,43−ジオキソ−42−(5−(ピリジン−2−イルアミノ)ペンタンアミド)−3,6,9,12,15,18,21,24,27,30,33,36−ドデカオキサ−40,44−ジアザヘプタテトラコンタン−47−酸)を得た。LC−MS(カラム:Phenomenex Luna C18(2)20×2mm,3μm;溶媒:A=水/0.1%トリフルオロ酢酸及びB=アセトニトリル/0.1%トリフルオロ酢酸;勾配:5分で10〜40%B;流量:0.6mL/分;214nm及び254nmでUV検出;ESI−MS):tR=3.1分、m/z期待値982.4(MH)2+、実測値982.8。
【0143】
実施例10:イメージング剤7のフローサイトメトリー評価
(実施例9に記載したようにして製造した)イメージング剤7の肺腺癌細胞株H2009(Cancer Res; 67: 5889-5895)に対する結合を、フローサイトメトリックアナライザー(Beckman Coulter FC500MPL)を用いて調べた。100nMの濃度で(37℃で1時間のインキュベーション)、約40%の細胞において陽性の取込みが証明され、これは分子中のこの位置への置換基の導入が許容されることを示している。
【特許請求の範囲】
【請求項1】
次の式Iのインビボイメージング剤又はその塩もしくは溶媒和物。
【化1】
式中、
nは0〜6の整数であり、
mは0〜8の整数であり、
X1はO、S又はNR’(式中、R’は水素又はC1-4アルキルである。)であり、
R1は−C(=NH)−NHR’(式中、R’はX1に関して定義した通りである。)であるか、或いはR1は1又は2個の窒素ヘテロ原子を有するC3-5含窒素ヘテロアリール環であり、
R2〜R4の各々は独立に水素、C1-4アルキル又はハロゲンであるか、或いはインビボイメージング成分を含む置換基であり、或いはR2及びR3又はR3及びR4はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-5ヘテロアリール環を形成し、
R5は水素又はR6R7基であり、R6はアミノ酸残基、炭水化物残基、−C(OH)−、−(CR'2)−、−C(=O)−C(CR'2)−、−C(=O)−NR'−、−(CR'2−O−CR'2)−、−CR'2−NR'−、−CR'2−S(O2)−CR'2−及び−(CR'2)−O−N=CR'−(式中、R’はX1に関して定義した通りである。)から選択される1〜50の二価リンカー単位を有する二価リンカー基であり、R7は水素であるか、或いはインビボイメージング成分を含む置換基であり、
R2〜R4の1以上がインビボイメージング成分を含む置換基であるか、或いはR5がR6R7(式中、R7はインビボイメージング成分を含む置換基である。)である。
【請求項2】
前記インビボイメージング成分が
(i)放射性金属イオン、
(ii)常磁性金属イオン、
(iii)γ放出型放射性ハロゲン、
(iv)陽電子放出型放射性非金属、及び
(v)インビボ光学イメージングに適したレポーター
から選択される、請求項1記載のインビボイメージング剤。
【請求項3】
nが1〜4である、請求項1又は請求項2記載のインビボイメージング剤。
【請求項4】
mが0〜3である、請求項1乃至請求項3のいずれか1項記載のインビボイメージング剤。
【請求項5】
X1がNR’である、請求項1乃至請求項4のいずれか1項記載のインビボイメージング剤。
【請求項6】
R1が五員又は六員含窒素ヘテロアリール環である、請求項1乃至請求項5のいずれか1項記載のインビボイメージング剤。
【請求項7】
R2〜R4の各々が独立に水素、クロロ、ヨード又はフルオロである、請求項1乃至請求項6のいずれか1項記載のインビボイメージング剤。
【請求項8】
R2〜R4の各々が18F又は123Iである、請求項1乃至請求項7のいずれか1項記載のインビボイメージング剤。
【請求項9】
R5がR6R7基である、請求項1乃至請求項8のいずれか1項記載のインビボイメージング剤。
【請求項10】
R6R7が−C(=O)−R6aR7a基(式中、R6aは1〜20の二価リンカー単位を有する二価リンカー基であって、前記二価リンカー単位はR6に関して請求項1で定義した通りであり、R7aはR7に関して請求項1で定義した通りである。)である、請求項9記載のインビボイメージング剤。
【請求項11】
R7aが18Fを含む、請求項10記載のインビボイメージング剤。
【請求項12】
R7aが金属錯体であり、前記金属錯体が放射性金属イオン又は常磁性金属イオンを含む、請求項10記載のインビボイメージング剤。
【請求項13】
次の式Iaの化合物又はその塩もしくは溶媒和物である、請求項1記載の式Iのインビボイメージング剤。
【化2】
式中、
R1aはR1に関して請求項1又は請求項5で定義した通りであり、
R2a〜R4aはR2〜R4に関して請求項1、請求項6又は請求項7で定義した通りであり、
R5aは水素であるか、或いは請求項9乃至請求項11のいずれかで定義したR6aR7a基である。
【請求項14】
請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤の製造方法であって、インビボイメージング成分の適当な供給源を次の式IIの前駆体化合物と反応させる段階を含んでなる方法。
【化3】
式中、
R11はR1に関して請求項1又は請求項6で定義した通りであり、
R12〜R14は水素、C1-4アルキル及びハロゲン並びに前駆体基から独立に選択され、
R15は水素又は前駆体基であるか、或いはR16R17基(式中、R16はR6に関して請求項1で定義した二価リンカー基であり、R17は水素又は前駆体基である。)であり、
X11はX1に関して請求項1又は請求項5で定義した通りであり、
m’はmに関して請求項1又は請求項4で定義した通りであり、
n’はnに関して請求項1又は請求項3で定義した通りであり、
R12〜R14の1以上が前駆体基であるか、或いはR15がR16R17(式中、R17は前駆体基である。)であり、
前記前駆体基以外の反応基は化学的に保護されている。
【請求項15】
前記式IIの前駆体化合物が次の式IIaを有する、請求項14記載の方法。
【化4】
式中、R11a〜R15aは式IIのR11〜R15に関して請求項14で定義した通りである。
【請求項16】
前記前駆体基が
(i)金属イメージング成分を錯体化できる1種以上のリガンド、
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体、
(iii)求核置換用のアルキルハライド、アルキルトシレート又はアルキルメシレートを含む誘導体、
(iv)求核又は求電子置換に向けて活性化された芳香環を含む誘導体、
(v)アシル化を受けやすい官能基を含む誘導体、
(vi)ベンズアルデヒドと反応させた場合にオキシム生成に参加する官能基を含む誘導体、
(vii)ビニルスルホン官能基を含む誘導体、
(viii)容易にアルキル化を受ける官能基を含む誘導体、及び
(ix)チオール含有化合物をアルキル化してチオエーテル含有生成物を与える誘導体
から選択される、請求項14又は請求項15記載の方法。
【請求項17】
R12〜R14の1つが前記前駆体基である、請求項14乃至請求項16のいずれか1項記載の方法。
【請求項18】
R15がR16R17(式中、R17は前記前駆体基である。)である、請求項14乃至請求項16のいずれか1項記載の方法。
【請求項19】
固相上で実施される、請求項14乃至請求項18のいずれか1項記載の方法。
【請求項20】
自動化される、請求項14乃至請求項19のいずれか1項記載の方法。
【請求項21】
請求項14乃至請求項18のいずれか1項記載の方法で定義した式IIの前駆体化合物であって、前記前駆体基が
(i)金属イメージング成分を錯体化できるキレーター、及び
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体
から選択される、前駆体化合物。
【請求項22】
請求項20記載の方法を実施するためのカセットであって、
(i)請求項14乃至請求項18のいずれか1項記載の方法で定義した前駆体化合物、又は請求項21記載の前駆体化合物を含む容器、及び
(ii)請求項1又は請求項2で定義したインビボイメージング成分の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセット。
【請求項23】
さらに、(iii)過剰のインビボイメージング成分を除去するためのイオン交換カートリッジを含む、請求項22記載のカセット。
【請求項24】
請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる医薬組成物。
【請求項25】
請求項24記載の医薬組成物を製造するためのキットであって、請求項21記載の前駆体化合物を含んでなるキット。
【請求項26】
被験体で発現されるαvβ6の量及び/又は位置を決定するためのインビボイメージング方法であって、
(i)請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤を前記被験体を投与する段階、
(ii)段階(i)で投与した前記インビボイメージング剤を前記被験体で発現されるαvβ6に結合させる段階、
(iii)段階(ii)で結合させた前記インビボイメージング剤に含まれるインビボイメージング成分から放出される信号を検出する段階、並びに
(iv)前記信号の位置及び量を表す画像を生成する段階であって、前記信号が前記被験体で発現されるαvβ6の量及び/又は位置を表す段階
を含んでなるインビボイメージング方法。
【請求項27】
前記インビボイメージング剤が、段階(i)で請求項24記載の医薬組成物として投与される、請求項26記載のインビボイメージング方法。
【請求項28】
前記被験体が哺乳動物である、請求項27記載のインビボイメージング方法。
【請求項29】
前記被験体がαvβ6状態を有することが知られ又は疑われている、請求項28記載のインビボイメージング方法。
【請求項30】
前記被験体がαvβ6状態を有することが知られ又は疑われており、当該方法が前記被験体に関する治療計画の進行中に繰り返して実施され、前記治療計画がαvβ6状態と戦うための薬物の投与を含む、請求項29記載のインビボイメージング方法。
【請求項31】
さらに、発現されるαvβ6の量及び/又は位置を特定の臨床像に帰因させる段階(v)を含む、請求項26乃至請求項30のいずれか1項記載のインビボイメージング方法。
【請求項32】
請求項26乃至請求項31のいずれか1項記載のインビボイメージング方法で使用するための、請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤。
【請求項1】
次の式Iのインビボイメージング剤又はその塩もしくは溶媒和物。
【化1】
式中、
nは0〜6の整数であり、
mは0〜8の整数であり、
X1はO、S又はNR’(式中、R’は水素又はC1-4アルキルである。)であり、
R1は−C(=NH)−NHR’(式中、R’はX1に関して定義した通りである。)であるか、或いはR1は1又は2個の窒素ヘテロ原子を有するC3-5含窒素ヘテロアリール環であり、
R2〜R4の各々は独立に水素、C1-4アルキル又はハロゲンであるか、或いはインビボイメージング成分を含む置換基であり、或いはR2及びR3又はR3及びR4はそれらが結合した炭素原子と共にC5-6アリール環又は1若しくは2個のヘテロ原子を有するC3-5ヘテロアリール環を形成し、
R5は水素又はR6R7基であり、R6はアミノ酸残基、炭水化物残基、−C(OH)−、−(CR'2)−、−C(=O)−C(CR'2)−、−C(=O)−NR'−、−(CR'2−O−CR'2)−、−CR'2−NR'−、−CR'2−S(O2)−CR'2−及び−(CR'2)−O−N=CR'−(式中、R’はX1に関して定義した通りである。)から選択される1〜50の二価リンカー単位を有する二価リンカー基であり、R7は水素であるか、或いはインビボイメージング成分を含む置換基であり、
R2〜R4の1以上がインビボイメージング成分を含む置換基であるか、或いはR5がR6R7(式中、R7はインビボイメージング成分を含む置換基である。)である。
【請求項2】
前記インビボイメージング成分が
(i)放射性金属イオン、
(ii)常磁性金属イオン、
(iii)γ放出型放射性ハロゲン、
(iv)陽電子放出型放射性非金属、及び
(v)インビボ光学イメージングに適したレポーター
から選択される、請求項1記載のインビボイメージング剤。
【請求項3】
nが1〜4である、請求項1又は請求項2記載のインビボイメージング剤。
【請求項4】
mが0〜3である、請求項1乃至請求項3のいずれか1項記載のインビボイメージング剤。
【請求項5】
X1がNR’である、請求項1乃至請求項4のいずれか1項記載のインビボイメージング剤。
【請求項6】
R1が五員又は六員含窒素ヘテロアリール環である、請求項1乃至請求項5のいずれか1項記載のインビボイメージング剤。
【請求項7】
R2〜R4の各々が独立に水素、クロロ、ヨード又はフルオロである、請求項1乃至請求項6のいずれか1項記載のインビボイメージング剤。
【請求項8】
R2〜R4の各々が18F又は123Iである、請求項1乃至請求項7のいずれか1項記載のインビボイメージング剤。
【請求項9】
R5がR6R7基である、請求項1乃至請求項8のいずれか1項記載のインビボイメージング剤。
【請求項10】
R6R7が−C(=O)−R6aR7a基(式中、R6aは1〜20の二価リンカー単位を有する二価リンカー基であって、前記二価リンカー単位はR6に関して請求項1で定義した通りであり、R7aはR7に関して請求項1で定義した通りである。)である、請求項9記載のインビボイメージング剤。
【請求項11】
R7aが18Fを含む、請求項10記載のインビボイメージング剤。
【請求項12】
R7aが金属錯体であり、前記金属錯体が放射性金属イオン又は常磁性金属イオンを含む、請求項10記載のインビボイメージング剤。
【請求項13】
次の式Iaの化合物又はその塩もしくは溶媒和物である、請求項1記載の式Iのインビボイメージング剤。
【化2】
式中、
R1aはR1に関して請求項1又は請求項5で定義した通りであり、
R2a〜R4aはR2〜R4に関して請求項1、請求項6又は請求項7で定義した通りであり、
R5aは水素であるか、或いは請求項9乃至請求項11のいずれかで定義したR6aR7a基である。
【請求項14】
請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤の製造方法であって、インビボイメージング成分の適当な供給源を次の式IIの前駆体化合物と反応させる段階を含んでなる方法。
【化3】
式中、
R11はR1に関して請求項1又は請求項6で定義した通りであり、
R12〜R14は水素、C1-4アルキル及びハロゲン並びに前駆体基から独立に選択され、
R15は水素又は前駆体基であるか、或いはR16R17基(式中、R16はR6に関して請求項1で定義した二価リンカー基であり、R17は水素又は前駆体基である。)であり、
X11はX1に関して請求項1又は請求項5で定義した通りであり、
m’はmに関して請求項1又は請求項4で定義した通りであり、
n’はnに関して請求項1又は請求項3で定義した通りであり、
R12〜R14の1以上が前駆体基であるか、或いはR15がR16R17(式中、R17は前駆体基である。)であり、
前記前駆体基以外の反応基は化学的に保護されている。
【請求項15】
前記式IIの前駆体化合物が次の式IIaを有する、請求項14記載の方法。
【化4】
式中、R11a〜R15aは式IIのR11〜R15に関して請求項14で定義した通りである。
【請求項16】
前記前駆体基が
(i)金属イメージング成分を錯体化できる1種以上のリガンド、
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体、
(iii)求核置換用のアルキルハライド、アルキルトシレート又はアルキルメシレートを含む誘導体、
(iv)求核又は求電子置換に向けて活性化された芳香環を含む誘導体、
(v)アシル化を受けやすい官能基を含む誘導体、
(vi)ベンズアルデヒドと反応させた場合にオキシム生成に参加する官能基を含む誘導体、
(vii)ビニルスルホン官能基を含む誘導体、
(viii)容易にアルキル化を受ける官能基を含む誘導体、及び
(ix)チオール含有化合物をアルキル化してチオエーテル含有生成物を与える誘導体
から選択される、請求項14又は請求項15記載の方法。
【請求項17】
R12〜R14の1つが前記前駆体基である、請求項14乃至請求項16のいずれか1項記載の方法。
【請求項18】
R15がR16R17(式中、R17は前記前駆体基である。)である、請求項14乃至請求項16のいずれか1項記載の方法。
【請求項19】
固相上で実施される、請求項14乃至請求項18のいずれか1項記載の方法。
【請求項20】
自動化される、請求項14乃至請求項19のいずれか1項記載の方法。
【請求項21】
請求項14乃至請求項18のいずれか1項記載の方法で定義した式IIの前駆体化合物であって、前記前駆体基が
(i)金属イメージング成分を錯体化できるキレーター、及び
(ii)トリアルキルスタンナン又はトリアルキルシランのような有機金属誘導体
から選択される、前駆体化合物。
【請求項22】
請求項20記載の方法を実施するためのカセットであって、
(i)請求項14乃至請求項18のいずれか1項記載の方法で定義した前駆体化合物、又は請求項21記載の前駆体化合物を含む容器、及び
(ii)請求項1又は請求項2で定義したインビボイメージング成分の適当な供給源を用いて容器を溶出するための手段
を含んでなるカセット。
【請求項23】
さらに、(iii)過剰のインビボイメージング成分を除去するためのイオン交換カートリッジを含む、請求項22記載のカセット。
【請求項24】
請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤を、哺乳動物への投与に適した形態の生体適合性キャリヤーと共に含んでなる医薬組成物。
【請求項25】
請求項24記載の医薬組成物を製造するためのキットであって、請求項21記載の前駆体化合物を含んでなるキット。
【請求項26】
被験体で発現されるαvβ6の量及び/又は位置を決定するためのインビボイメージング方法であって、
(i)請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤を前記被験体を投与する段階、
(ii)段階(i)で投与した前記インビボイメージング剤を前記被験体で発現されるαvβ6に結合させる段階、
(iii)段階(ii)で結合させた前記インビボイメージング剤に含まれるインビボイメージング成分から放出される信号を検出する段階、並びに
(iv)前記信号の位置及び量を表す画像を生成する段階であって、前記信号が前記被験体で発現されるαvβ6の量及び/又は位置を表す段階
を含んでなるインビボイメージング方法。
【請求項27】
前記インビボイメージング剤が、段階(i)で請求項24記載の医薬組成物として投与される、請求項26記載のインビボイメージング方法。
【請求項28】
前記被験体が哺乳動物である、請求項27記載のインビボイメージング方法。
【請求項29】
前記被験体がαvβ6状態を有することが知られ又は疑われている、請求項28記載のインビボイメージング方法。
【請求項30】
前記被験体がαvβ6状態を有することが知られ又は疑われており、当該方法が前記被験体に関する治療計画の進行中に繰り返して実施され、前記治療計画がαvβ6状態と戦うための薬物の投与を含む、請求項29記載のインビボイメージング方法。
【請求項31】
さらに、発現されるαvβ6の量及び/又は位置を特定の臨床像に帰因させる段階(v)を含む、請求項26乃至請求項30のいずれか1項記載のインビボイメージング方法。
【請求項32】
請求項26乃至請求項31のいずれか1項記載のインビボイメージング方法で使用するための、請求項1乃至請求項13のいずれか1項記載のインビボイメージング剤。
【図1】

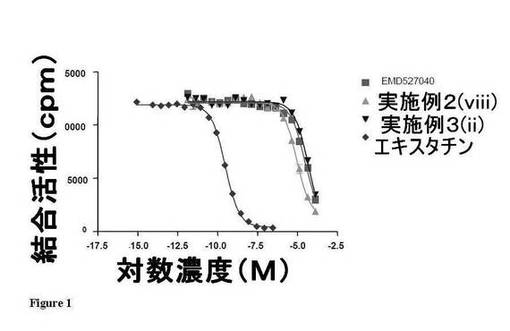
【公表番号】特表2013−514326(P2013−514326A)
【公表日】平成25年4月25日(2013.4.25)
【国際特許分類】
【出願番号】特願2012−543763(P2012−543763)
【出願日】平成22年12月16日(2010.12.16)
【国際出願番号】PCT/EP2010/069958
【国際公開番号】WO2011/073340
【国際公開日】平成23年6月23日(2011.6.23)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成25年4月25日(2013.4.25)
【国際特許分類】
【出願日】平成22年12月16日(2010.12.16)
【国際出願番号】PCT/EP2010/069958
【国際公開番号】WO2011/073340
【国際公開日】平成23年6月23日(2011.6.23)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]