
毛細血管拡張性失調症治療薬及びその用途
【課題】毛細血管拡張性失調症治療薬および、毛細血管拡張性失調症等の神経変性の治療ないし予防に有効な化合物のスクリーニング法を提供する。
【解決手段】標的神経細胞内で機能的ジンクフィンガーホメオボックス3タンパク質を増大又は安定化する化合物を含む毛細血管拡張性失調症治療薬。以下のステップ、(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ、(2)試験群に対して、カイニン酸とともに被験物質を投与するステップ、(3)ステップ(2)後の試験群と、被験物質を投与しない対照群について、脳内でのオートファジー又は神経変性を検定するステップ、(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップ、を含む、毛細血管拡張性失調症等の神経変性の治療ないし予防に有効な化合物のスクリーニング法。
【解決手段】標的神経細胞内で機能的ジンクフィンガーホメオボックス3タンパク質を増大又は安定化する化合物を含む毛細血管拡張性失調症治療薬。以下のステップ、(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ、(2)試験群に対して、カイニン酸とともに被験物質を投与するステップ、(3)ステップ(2)後の試験群と、被験物質を投与しない対照群について、脳内でのオートファジー又は神経変性を検定するステップ、(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップ、を含む、毛細血管拡張性失調症等の神経変性の治療ないし予防に有効な化合物のスクリーニング法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は毛細血管拡張性失調症治療薬及びその用途、並びに毛細血管拡張性失調症等の神経変性を予防し或いはその治療にに有効な化合物をスクリーニングする方法に関する。
【背景技術】
【0002】
毛細血管拡張性失調症(Ataxia telangiectasia, A-T)は神経変性を伴う遺伝性疾患である(非特許文献1)。原因遺伝子はDNAの二重らせんの切断に反応する、セリン・スレオニンリン酸化酵素の一つであるATM(ataxia telangiectasia mutated)の障害による(非特許文献2、3)。疾患名であるA-Tは、合併する臨床兆候である小脳変性に由来する運動失調症(Ataxia)と毛細血管拡張(Telangiectasia)を合体させた名称で構成されている。
【0003】
A-T患者の臨床所見としてアルファフェトプロテイン(AFP)の血中異常上昇が知られている。AFP遺伝子はアルブミン遺伝子と並んでヒトの第4染色体上にコードされている(非特許文献4、5)。AFPは生理的には胎児の肝臓で発現し、成体の肝臓ではほぼ完全に発現が抑制されている。ただし成体に肝細胞癌が発生するとその腫瘍細胞が再びAFPを産生するようになる。この病態反応を利用することにより、血中AFPを肝臓癌の診断用バイオマーカとして臨床応用することができる。ところが、不思議なことにA-T患者では肝臓癌の発生とは無関係に血中AFPの異常上昇が認められる。AFPの異常上昇は、A-T患者が持つAFP遺伝子の発現抑制系に異常が起こっていることを示唆している。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S. et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749-1753.
【非特許文献2】Khanna, K. K. and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247-254.
【非特許文献3】Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155-168.
【非特許文献4】Kao, F. T., Hawkins, J. W., Law, M. L. and Dugaiczyk, A. (1982). Assignment of the structural gene coding for albumin to human chromosome 4. Hum. Genet. 62, 337-341.
【非特許文献5】Urano, Y., Sakai, M., Watanabe, K. and Tamaoki, T. (1984). Tandem arrangement of the albumin and alpha-fetoprotein genes in the human genome. Gene 32, 255-261.
【非特許文献6】Morinaga, T., Yasuda, H., Hashimoto, T., Higashio, K. and Tamaoki, T. (1991). A human alpha-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Mol. Cell. Biol. 11, 6041-6049.
【非特許文献7】Miura, Y., Tam, T., Ido, A., Morinaga, T., Miki, T., Hashimoto, T. and Tamaoki, T. (1995). Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J. Biol. Chem. 270, 26840-26848.
【非特許文献8】Jung, C. G., Kim, H. J., Kawaguchi, M., Khanna, K. K., Hida, H., Asai, K., Nishino, H. and Miura, Y. (2005). Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development 132, 5137-5145.
【非特許文献9】Yasuda, H., Mizuno, A., Tamaoki, T. and Morinaga, T. (1994). ATBF1, a multiplehomeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol. Cell. Biol. 14, 1395-1401.
【非特許文献10】Wilkinson, D. S., Tsai, W. W., Schumacher, M. A. and Barton, M. C. (2008). Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol. Cell. Biol. 28, 1988-1998.
【非特許文献11】Miura, Y., Kataoka, H., Joh, T., Tada, T., Asai, K., Nakanishi, M., Okada, N. and Okada, H. (2004). Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Waf1/Cip1) promoter. Microbiol. Immunol. 48, 137-145.
【非特許文献12】Kim, C. J., Song, J. H., Cho, Y. G., Cao, Z., Lee, Y. S., Nam, S. W., Lee, J. Y. and Park, W. S. (2008). Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology 52, 552-559.
【非特許文献13】Kataoka, H., Miura, Y., Joh, T., Seno, K., Tada, T., Tamaoki, T., Nakabayashi, H., Kawaguchi, M., Asai, K., Kato, T. et al. (2001). Alpha-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene 20, 869-873.
【非特許文献14】Cho, Y. G., Song, J. H., Kim, C. J., Lee, Y. S., Kim, S. Y., Nam, S. W., Lee, J. Y. and Park, W. S. (2007). Genetic alterations of the ATBF1 gene in gastric cancer. Clin. Cancer Res. 13, 4355-4359.
【非特許文献15】Alterman, N., Fattal-Valevski, A., Moyal, L., Crawford, T. O., Lederman, H. M., Ziv, Y. and Shiloh, Y. (2007). Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am. J. Med. Genet 143A, 1827-1834.
【非特許文献16】Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., 3rd, Hurov, K. E., Luo, J., Bakalarski, C. E., Zhao, Z., Solimini, N., Lerenthal, Y. et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160-1166.
【非特許文献17】Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., Smorodinsky, N. I., Prives, C., Reiss, Y., Shiloh, Y. et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674-1677.
【非特許文献18】Shieh, S. Y., Ikeda, M., Taya, Y. and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334.
【非特許文献19】Burgner, D., Davila, S., Breunis, W. B., Ng, S. B., Li, Y., Bonnard, C., Ling, L., Wright, V. J., Thalamuthu, A., Odam, M. et al. (2009). A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 5, e1000319.
【発明の概要】
【発明が解決しようとする課題】
【0005】
ATMというリン酸化酵素をコードしている遺伝子の変異がA-Tの原因であることが解明されたものの、神経変性のメカニズムは不明であり、A-Tに対する適切な治療法/予防法の確立が切望されている。このような状況下、本発明は、A-Tの治療又は予防に有効な手段を提供することを目的とする。また、A-T等の神経変性疾患の治療法ないし予防法の確立に資するツールを提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らはA-T患者に特徴的な症状と遺伝子転写調節因子ZFHX3(ATBF1)の機能の間に深い関係があると考えて研究を進めた。ZFHX3はAFP遺伝子プロモータ上流の発現調節領域にあるA(アデニン)とT(チミン)に富む(AT-rich)DNA配列に結合するDNA結合タンパク質(AT-rich binding factor 1)として1991年にクローニングされた(非特許文献6)。最初に9-kbのATBF1 mRNAがクローニングされ、1995年には主要な遺伝子転写産物として全長12-kbのATBF1 mRNAがATBF1-Aと命名された。ATBF1-Aについては神経細胞の分化に関連する機能が明らかにされた(非特許文献7、8)。ごく最近になってATBF1の名称はヒトゲノム国際機構(Human Genome Organization)によりZFHX3(zinc finger homeobox 3)と改名された。ZFHX3はAFP遺伝子の発現調節領域にあるATに富む(AT-rich)領域に結合してその活性を抑制する作用がある(非特許文献6、9)。最近、p53とTGF-βシグナル伝達因子とが複合体を形成してAFP遺伝子の発現を抑制するという新しいメカニズムが明らかになった(非特許文献10)。p53はZFHX3とタンパク質相互作用する因子であり、p53とZFHX3の複合体がp21waf1/cip1のプロモータを活性化する(非特許文献11)。これらの事実から、AFP遺伝子に関しても、p53、TGF-βシグナル伝達因子及びZFHX3が複合体形成しするタンパク質相互作用を通じて転写抑制をしている可能性がある。
【0007】
A-T患者以外では、成人の血中AFPが異常上昇する臨床状態は肝細胞癌(Hepatocellular carcinoma, HCC)やAFP産生胃癌の患者に認められる。その背景としてHCCの癌細胞でZFHX3発現量が著しく減少していることが明らかにされた。また、AFP産生胃癌の癌細胞ではZFHX3の発現がほとんど消失していることが示された(非特許文献12)。AFP産生胃癌は、AFPの異常な発現上昇と同時に、原発巣が小さい段階から高率に遠隔転移を起こす特徴を示し、極めて悪性度の高い癌である(非特許文献13、14)。癌の転移の問題は細胞接着性と深く関連する問題であり、ZFHX3の機能不全との関連が推察されている。
【0008】
A-T患者にとって最も深刻な問題は神経変性であり、神経変性は病態早期から進行する。A-T患者に神経変性が起こるメカニズムとしてDNAの二重らせん切断(double strand breaks, DSBs)の修復異常が想定されてきた。しかしA-Tと診断された症例の中には、臨床的な観察結果および定量的な神経学的検査結果から神経症状が軽微であるにも関わらず、遺伝子検査結果からはATM遺伝子に重大な中途欠損があって、ATM機能に著しい障害を持つことが予想される例がみつかる。それらの患者から採取された試料を用いた細胞機能テストではDNAの二重らせん切断に対するDNA修復反応の著しい損傷を確認できることから、典型的なATM症状を呈することを期待できる。もしも神経変性がDNA修復障害のみに由来するのであれば、当然著しい神経症状が出ると期待される。しかし、DNA修復反応が障害されていても、必ずしも神経変性の兆候が重篤になるとは限らない。この事実関係はA-T患者における神経変性を起こすメカニズムは単純にATMのDNA修復障害では説明できないことを示唆しているものであり、従来から知られているATMのDNA修復シグナル系とは独立した別のメカニズムが神経変性過程に関わっている可能性がある(非特許文献15)。
【0009】
ATMキナーゼの下流の標的として数百の因子が推定されているが、その一つとしてZFHX3が検出されている(非特許文献16)。ZFHX3がATMによってリン酸化を受ける機能的な意味は、ATMによってリン酸化されるp53の機能調節と類似している可能性がある。p53はATMによってリン酸化されることでタンパク質が安定化し転写因子としての機能を発揮するようになる。p53のリン酸化は分解を促進する因子であるMDM2とp53の結合を抑制する作用がある(非特許文献17、18)。ZFHX3もATMによってリン酸化されることでタンパク質として安定化している可能性がある。本発明者らは、A-T患者が示す様々な臨床症状の背景としてZFHX3の機能障害が関連していると考えた。最近の論文報告によって、ZFHX3の異常は川崎病の遺伝的背景因子であることが解明された。川崎病は小児急性熱性皮膚粘膜リンパ節症候群として知られる病気であり、心臓冠動脈瘤の合併症を引き起こす(非特許文献19)。この事実はA-T患者の臨床兆候の一つである毛細血管拡張症状と関連して、ZFHX3が血管の保護的役割を持っていることを示唆するものであり、ZFHX3に動脈瘤が発生することを防御する役割があることを示している。
【0010】
以上の考察を踏まえて本発明者らは、ZFHX3の機能とA-T患者が持つ多彩な臨床症状との密接な結びつきに着目し、数々の実験を行った。その結果、特に重要な成果として、レチノイン酸刺激がATMの活性化を誘導し、活性化したATMがCREBを活性化し、続いてCREBがZFHX3の遺伝子プロモータ上にあるCRE(cAMP Responsive Element)のコンセンサスDNA配列に結合し、標的遺伝子であるZFHX3遺伝子の転写を活性化するという、シグナル伝達経路を明らかにした。また、酸化ストレスに反応して神経細胞の細胞質内でATMが活性化する事実及びこのATMの活性化にPDGFRBの活性化が必須であることを解明した。これらの成果によって、A-Tの原因遺伝子であるATMの活性化に関するシグナル伝達経路の全体像が明らかになった。その結果、ATMの活性化においてZFHX3が重要な分子であり、ZFHX3が良好に作用する環境を形成することがA-Tの治療に有効であるとの知見がもたらされた。「ZFHX3が良好に作用する環境」を形成するための具体的な手段は、機能的ZFHX3(即ちリン酸化状態のZFHX3)の量を増大させること、或いは機能的ZFHX3の安定化(維持)である。前者は、例えば、本研究で使用したレチノイン酸によって達成可能である。一方、本発明者らの検討の結果、後者に関連して重要かつ興味深い実験結果が得られた。即ち、モデル神経細胞を用いた実験によって、免疫抑制剤として知られるタクロリムス(FK506)が神経細胞死を抑制できること、酸化ストレス下の神経細胞ではZFHX3の分断化が生ずること、タクロリムスの投与によってこのZFHX3の分断化を抑制できること、及びタクロリムスの投与によってZFHX3の脱リン酸化が抑制できること、が明らかとなった。ここで、タクロリムスがプロテインフォスファターゼ2B(略称PP2B。カルシニューリンとも呼ばれる)に対する阻害活性を示すことを踏まえて上記結果を考察すれば、酸化ストレス下の神経細胞ではZFHX3がPP2Bによる脱リン酸化を受けて分断化することがわかるとともに、タクロリムスによれば当該分断化の抑制を通して神経細胞死を抑制できるといえる。この知見は、タクロリムス自体が機能的ZFHX3の安定化という作用によってA-Tの治療/予防に有効であることを示すとともに、タクロリムスと同様の作用、即ち、ZFHX3を脱リン酸化する酵素(PP2B)に対する阻害活性を示す物質がA-Tの治療/予防に有効であること、更には、シグナル伝達経路においてPP2Bの上流に位置するカルパインの活性を阻害する物質にもA-Tに対する治療/予防効果を期待できることを示唆する。
【0011】
一方、カイニン酸刺激を負荷した動物の小脳深部核等の反応性を検定するという、本研究で使用した実験系は、上記の如き重要且つ有意義な事実の発見に寄与したことからも明らかな通り、小脳の変性を伴う疾患に有効な薬物をスクリーニングするためのツールとしては当然のこと、グルタミン酸作動性ニューロンでの神経過興奮に由来する酸化ストレスがもたらす各種神経変性に有効な薬物をスクリーニングするツールとしても、それ自体が大きな価値を有する。
【0012】
以下に示す本発明は主として上記成果に基づく。
[1]標的神経細胞内で機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を含む、毛細血管拡張性失調症治療薬。
[2]前記化合物が、機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質の発現増強作用を示す化合物である、[1]に記載の毛細血管拡張性失調症治療薬。
[3]前記化合物がレチノイン酸である、[2]に記載の毛細血管拡張性失調症治療薬。
[4]前記化合物がプロテインフォスファターゼ2B(PP2B)阻害剤である、[1]に記載の毛細血管拡張性失調症治療薬。
[5]前記プロテインフォスファターゼ2B(PP2B)阻害剤がタクロリムスである、[4]に記載の毛細血管拡張性失調症治療薬。
[6]前記化合物がカルパイン阻害剤である、[1]に記載の毛細血管拡張性失調症治療薬。
[7]機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を毛細血管拡張性失調症患者に対して治療上有効量、投与するステップを含む、毛細血管拡張性失調症の治療法。
[8]前記化合物がレチノイン酸又はタクロリムスである、[7]に記載の治療法。
[9]以下のステップ(1)〜(4)を含む、毛細血管拡張性失調症に起因又は付随する神経変性或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物のスクリーニング法:
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
[10]ステップ(3)において、小脳でのオートファジー又は神経変性が検定される、[9]に記載のスクリーニング法。
[11]ステップ(3)において、小脳深部核でのオートファジー又は神経変性が検定される、[9]に記載のスクリーニング法。
[12]
非ヒト哺乳動物が野生型動物である、[9]〜[11]のいずれか一項に記載のスクリーニング法。
[13]非ヒト哺乳動物がATM遺伝子欠損動物である、[9]〜[11]のいずれか一項に記載のスクリーニング法。
[14]非ヒト哺乳動物がマウスである、[9]〜[13]のいずれか一項に記載のスクリーニング法。
【図面の簡単な説明】
【0013】
【図1】レチノイン酸(RA)刺激を用いた実験の結果。レチノイン酸の刺激はATMキナーゼの活性化を介してCREB(cAMP Responsive Element)の活性化を誘導する。この活性化したCREBがZFHX3遺伝子プロモータ上流に存在するCREに結合することでZFHX3遺伝子が活性化する。(A) ヒトZFHX3ゲノムの遺伝子構造の概略図を示す。四角形の部分はエクソン(1〜10)をコードしている領域を示す。選択的スプライシング1によって2種類のmRNAが作り出される。長型mRNAをATBF1-A、短型mRANをATBF1-Bと呼ぶ。ZFHX3から転写されるATBF1-AとATBF1-Bの二種類のmRNAはそれぞれ2カ所の転写開始点(PAとPB)からの転写産物である。神経細胞で発現しているZFHX3は長型mRNAであるATBF1-Aタイプであるので、この転写制御メカニズムを解明するために転写開始点(PA)から5’上流の5.6-kbまでの領域に着目して転写制御メカニズムを詳細に解析した。(B) CREのコンセンサス配列は転写開始点(PA)の上流域約5.5-kbの位置に存在する。その位置を白抜き円で示した。ルシフェラーゼ解析で用いたDNA断片には以下の名称を付けた。5.6は転写開始点(PA)から5’上流側の全長5.6-kb (ゲノムからSalI-BamHで切り出されるDNA断片)を含むDNA配列である。5.6Δは5.6-kbから途中の3.8-kb (HindII-StuIで切り出されるDNA断片)を取り除いた、転写開始点の177-bpと連結した配列である。4.0は5.6-kbの上流側の1.6-kb部分を取り除いた部分配列である。0.42は転写開始点直近の0.42-kbの配列である。これらのDNA断片を使ったルシフェラーゼレポータ解析を実施し、少なくとも3回の独立した測定をし、有意性(P<0.01)をボンフェローニ補正マンホイットニー検定で評価した。
【図2】PDGFRB及びVEGFR-2に対する特異的阻害剤AG1433を用いた実験の結果。AG1433は酸化ストレスに反応するATMの活性化を抑制した。P19細胞(マウス奇形腫瘍由来の胎児性癌細胞株)を神経分化誘導刺激し、その5日目の細胞を神経細胞のモデル細胞として以下の実験を行った。(A) 過酸化水素水(100μM)で処理する方法で酸化ストレス(0, 5, 15, 30, 60分)を与えた時に観察されるATMキナーゼの活性化(Ser1981のリン酸化抗体)を検定した。(B)刺激後30分の過酸化水素水濃度を各種(1, 10, 100, 500μM)用意して、ATMキナーゼの活性化を調べた。尚、Xで示したレーンはX線照射(10Gy)後1時間のサンプルを用いてATMの活性化を調べた結果を示したものであり、ATMキナーゼ活性化のポジティブコントロールとなる。(C) 過酸化水素水(100μM)処理後30分の時点のATMの活性化量(レーン3〜7)を示している。AG1433はPDGFRBとVEGFR-2に対する特異的な阻害剤であり、その濃度依存性にATMの活性化が抑制された。AG1433を投与する際の溶媒としてDMSOを使用した。溶媒であるDMSOの効果を差し引くために、DMSOの単独投与(0.1%)の影響も検討した。その投与量はAD1433 (5μM)投与実験で使ったDMSO濃度に相当する(レーン3)。(D) 以上の実験を3回繰り返してpS-ATM(Ser1981がリン酸化した活性型ATM)量とATM量の相対比率を計算し、その相対比率の変化について有意検定を行い、結果を棒グラフに纏めた。(E) 10 GyのX-線照射後1時間のATMの活性化量(レーン3〜7)についてはAG1433を投与しても有意な変化が起こらなかった。この実験で使用したDMSOによる効果を差し引くために、DMSO単独投与(0.1%)の比較実験を行った(レーン3)。(F) 以上の実験を3回繰り返してpS-ATM(Ser1981がリン酸化した活性型ATM)量とATM量の比率を計算し、その相対比率の変化について有意検定を行って棒グラフとして示した。有意性(P<0.01)をボンフェローニ補正マンホイットニー検定で評価した。
【図3】ATMの活性化に関する実験の結果。PDGFRBの抑制及び/又はVEGFR-2シグナルの抑制は、DNA損傷ストレスではなく酸化ストレスに反応したATMの活性化(pS-ATM)を減弱する。培養5日目のP19由来神経細胞(レチノイン酸によって4日間、神経細胞へ分化誘導したもの)を、PDGFRB及びVEGFR-2の特異的阻害剤であるAG1433(5μM)で前処理し、その30分後に100μMの過酸化水素水又はX線(10Gy)で処理した。この処理から30分後に細胞をパラホルムアルデヒドで固定した。活性化ATM(pS-ATM、緑)、DAN二重らせん切断(γ-H2AX、赤)及びDNA(DAPI、青)を染色した。スケールバーは10μm。
【図4】カイニン酸刺激が小脳神経細胞へ及ぼす影響に関する実験の結果。カイニン酸刺激によって生ずる、小脳深部核神経細胞での血管形成。pS-ATM(茶色)の免疫組織化学。カイニン酸刺激に反応して細胞質内でATMが活性化する(B1,B2,B3)。対照的に、X線照射に反応してATMは核内で活性化する(C1,C2,C3)。マウスの脳切片をヘマトキシリン・エオジン染色した(A,B,C)。図中の番号は大脳皮質(1)、海馬(2)及び小脳深部核(DCN)(3)を示す。マウスをカイニン酸(30mg/kg)による処理(B)、又はX線照射(C)に供し、4時間後に屠殺した。続いて、大脳皮質(A1,B1,C1)、海馬(A2,B2,C2)及びDCN(A3,B3,C3)について脳切片を作製した。注目すべきことにDCNの神経細胞では血管形成を認める(B3、矢印)。ヘマトキシリン(青)で対比染色した。スケールバーは10μm。
【図5】ATMの活性化が起こる細胞内の特異的領域(細胞質/核)の鑑別結果を示す図。成体マウスの腹腔内にカイニン酸(A1, A2; 30 mg/kg)を注射投与し、脳神経の過剰興奮を誘発させた実験条件と、X線を照射(B1, B2; 10 Gy)してDNA二重らせん切断を誘発した実験条件を比較してATMの活性化を評価した。カイニン酸投与後4時間で神経細胞の細胞質に活性型ATM(pS-ATM)が有意に検出された(A1)。X線照射刺激後4時間の神経細胞の核に活性型ATM(pS-ATM)が有意に集積していた。X線照射群ではDNA損傷のマーカーであるγ-H2AXが核に強く検出されるが、カイニン酸投与細胞ではγ-H2AXはまったく検出されない。拡大率を示すバーは10μm。
【図6】カイニン酸刺激による細胞質内でのATMの活性化を示す図。カイニン酸刺激から5時間後にマウス大脳皮質の脳切片を固定した。同様にX線照射(10Gy)から1時間後にマウス大脳皮質の脳切片を固定した。X線照射の場合、活性化ATM(pS-ATM、緑)は核に局在するが、カイニン酸刺激の場合は細胞質に局在する。DAN二重らせん切断(γ-H2AX、赤)、DNA(DAPI、青)の染色像も示した。スケールバーは10μm。
【図7】PDGFRBの発現に関する実験の結果。PDGFRBは小脳深部核(DCN)の神経細胞に有意に強く発現している。(A) P19細胞を神経系細胞に分化誘導する処理過程の7日目(神経分化の最終段階)に至るとPDGFRBとZFHX3が共にもっとも強く発現する。分化途中の段階で観察できる発現量の有意差(P<0.01)はマンホイットニーのU検定で検定した。(B)ラット胎仔(E14)の脊髄では未分化神経前細胞群が増殖している領域においてDNA合成を示すBrdUの取り込みが観察できる(2i)。神経への分化が終了して、BrdUの取り込みが観察できない領域でZFHX3 (4ii)とPDGFRB(6ii)の発現が同時に認められる。拡大率を示すバーをそれぞれの図の中に示した。(C) Atmノックアウトモデルマウスとコントロールマウスの脳を正中線近傍で切断した標本の免疫組織化学検査。それぞれの切断部位は白色の破線で示し(1及び8)それらの切断面のヘマトキシリン・エオジン染色像(2及び9)を示した。小脳組織の拡大部分を抗GFAP抗体で染色し、ヘマトキシリン染色液で核を対比染色した(3及び10)。画像中にあるDCN領域を白色破線で囲んで示した。ZFHX3の発現量(4及び5; 11及び12)とPDGFRBの発現量(6及び7; 13及びと14)を解析した結果、Atmノックアウトモデルマウスの小脳のDCNの神経細胞で観察できるZFHX3とPDGFRBの発現量は、コントロールマウスの小脳DCNの神経細胞と比較すると、いずれの発現量も有意に減少していることが判明した。画像の拡大率を示すバーはパネル3と10では500μm、パネル6,11,13では50μm、パネル5, 7, 12, 14では20μmである。
【図8】DCNにおけるオートファジー関連タンパク質LC3の発現を示す図。マウスを4%パラホルムアルデヒドで灌流固定した。ホルマリン固定、パラフィン包埋組織から切り出した4μmの組織切片を使用した。組織切片を脱パラフィン・再水和処理した後、電子レンジを使用して2回、賦活処理した(98℃、10分間、10mMクエン酸緩衝液(pH6.3))。コントロールマウスの脳切片(1)、カイニン酸処理(20mg/kg、腹腔内注射)から4時間後のマウスの脳切片(2)、2回のカイニン酸処理(10mg/kg、腹腔内注射。24時間の間隔)の後、24時間後のマウスの脳切片(3)をウサギ抗LC3抗体(1000倍希釈:株式会社医学生物学研究所)に1時間反応させ、その後、ホースラディッシュペルオキシダーゼ標識抗ウサギIgG抗体(2次抗体)を反応させた。ジアミノベンジジンを用いて可視化した。ヘマトキシリンを用いて組織切片を対比染色した。スケールバーは50μm。
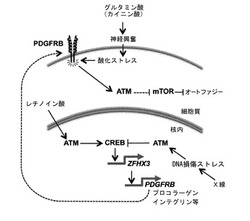
【図9】ATMキナーゼ活性化とZFHX3による転写制御が相互依存的に関わる細胞内シグナル伝達系の概念図。レチノイン酸の刺激で誘導されるATMの活性化によってCREBのSer133のリン酸化反応が進む。一方、X線照射によって誘導されるATMの活性化はDNAの二重らせん切断(DSBs, DNA double strand breaks)部分で起こり、CREBのThe100、Ser111及びSer121のリン酸化の反応が進むことになる。ATMの活性化がどのような原因によって誘導されるかに応じて下流のCREBのリン酸化部位に変化が生じ、その下流へのシグナル伝達に機能的な変化が起こる。レチノイン酸刺激に誘導されるCREBの活性化によって特異的にZFHX3の転写が活性化する。ZFHX3のプロモータの上流5.5-kbにはCRE(5’-TGACGTCA-3’)配列が存在し、そこにCREBが結合して転写を活性化していると考えられる。転写されたZFHX3 mRNAはZFHX3タンパク質に翻訳され、転写因子としてさまざまな標的遺伝子の発現制御に関わる。マイクロアレイによるZHFX3の標的遺伝子のスクリーニング結果から、細胞接着因子群(プロコラーゲン類、インテグリン類)及びPDGFRB(血小板由来増殖因子受容体β)遺伝子群が同定された。神経興奮によって引き起こされる代謝性酸化ストレスに対する自己防御シグナルとしてATMが細胞質内で活性化することは重要である。この自己防御シグナルを活性化するためにPDGFRBの活性化がその上流因子として必須であることが今回の研究で始めて明らかになった。細胞質内で活性化したATMのリン酸化シグナルはLKB1、AMPK、TSC2のカスケードに伝えられてmTORを抑制する制御系として作用する。活性型のmTORはオートファジー(ミトコンドリアなどの細胞内小器官の自己貪食機構)の抑制因子として知られており、mTORを抑制するATMからのシグナルカスケードはオートファジーを誘導するシグナルとして機能する。
【図10】神経細胞死に対するFK506(タクロリムス)の効果。FK506の投与によって神経細胞死を抑制することができる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスを与えるモデル神経細胞をレチノイン酸(RA)刺激(5μM、4日間処理)で前処理しておくと、カスパーゼ3の活性化が有意に上昇することが分かる(レーン3)。FK506処理をするとカスパーゼ3の活性化が抑制できる(レーン4)。この神経細胞モデル系でThapsigarginを作用させることは神経過興奮のモデルとして捉えることができることから、神経の過興奮で神経細胞死が誘導されるメカニズムとしてカスパーゼ3が活性化することが想定できる(レーン5)。この神経細胞死シグナルの誘導はFK506の投与によって抑制できる(レーン6)。尚、FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。Thapsgargin処理については、最終濃度2μMで1時間の処理とした。
【図11】ZFHX3の分断化に対するFK506(タクロリムス)の効果。FK506の投与によってZFHX3の分断化が抑制できる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスがZFHX3タンパク質に与える影響を調べるため、抗ATBF1抗体(AT-6)で免疫沈降したタンパク質をAT-6抗体でウエスタンブロット解析した。その結果、レチノイン酸(RA)刺激で神経分化誘導した細胞では、Thapsigargin処理によってZFHX3タンパク質の分断化が有意に強く起きた(レーン5)。未分化P19細胞に由来するZFHX3ではこのような変化は認められなかった。このZFHX3の分断化現象はFK506の投与で抑制できる(レーン6)。ZFHX3の分断化現象はThapsigargin処理をしないP19由来神経細胞内でも弱いながら観察でき(レーン3)、この分断化もFK506の投与で抑制できる(レーン4)。尚、FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。Thapsgargin処理については、最終濃度2μMで1時間の処理とした。
【図12】ZFHX3の脱リン酸化に対するFK506(タクロリムス)の効果。FK506の投与によってZFHX3の脱リン酸化が抑制できる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスがZFHX3のリン酸化に与える影響をPro-Q Diamond phosphoprotein stain kit (Invitrogen社、カタログ番号 P-33356)を用いて検定した。P19細胞の溶解液を抗ATBF1抗体(AT-6)で免疫沈降して得られたタンパク質をSDS-PAGEで展開し、上記の試薬に反応させた。レチノイン酸(RA)刺激(5μM、4日間処理)で前処理をして神経分化誘導した細胞ではZFHX3のリン酸化が有意に強くなっていることが分かる(レーン7, 8, 13, 14)。Thapsigargin処理によるCa2+ストレスが加わらなければ、FK506による処理の有無によってZFHX3のリン酸化レベルには大きな差が検出できないが(レーン7, 8)、Thapsigargin処理をしてCa2+ストレスを細胞に加えた系では、FK506による処理細胞群でZFHX3のリン酸化状態が有意に高く保たれている(レーン13, 14)。図中の矢印は404-kDaの全長ZFHX3がリン酸化していることを示している。尚、レーン1、4、6、10、12、15は分子量マーカーである。
【図13】ZFHX3タンパク質量の検定結果。図12の解析に使ったタンパク質をSYPRO Ruby staining(Invitrogen社、カタログ番号 No.S12001)で検定した結果。いずれの条件においても、矢印で示した404-kDaの位置に等量のタンパク質量として全長ZFHX3タンパク質が検出されている(レーン2, 3, 7, 8, 13, 14)。尚、レーン1、4、6、10、12、15は分子量マーカーである。
【発明を実施するための形態】
【0014】
1.毛細血管拡張性失調症用の治療薬
本発明の第1の局面は毛細血管拡張性失調症(以下、「A-T」を略称する)の治療薬に関する。本発明における「治療薬」とは、標的の疾病ないし病態であるA-Tに対する治療的又は予防的効果を示す医薬のことをいう。治療的効果には、A-Tに特徴的な症状又は随伴症状を緩和すること(軽症化)、症状の悪化を阻止ないし遅延すること等が含まれる。後者については、重症化を予防するという点において予防的効果の一つと捉えることができる。このように、治療的効果と予防的効果は一部において重複する概念であることから、明確に区別して捉えることは困難であり、またそうすることの実益は少ない。尚、予防的効果の典型的なものは、A-Tに特徴的な症状の再発を阻止ないし遅延することである。尚、A-Tに対して何らかの治療的効果又は予防的効果、或いはこの両者を示す限り、A-T治療薬に該当する。
【0015】
本発明の治療薬は有効成分として、標的神経細胞内で機能的ジンクフィンガーホメオボックス3(以下、慣例に従い「ZFHX3」と略称する)タンパク質を増大又は安定化する化合物を含む。換言すれば、本発明の治療薬における有効成分は、標的細胞内において機能的ZFHX3タンパク質を増大させる効果、或いは標的細胞内において機能的ZFHX3タンパク質を安定化させる効果を発揮する。本発明における用語「標的神経細胞」とは、本発明の治療薬の標的となる神経細胞である。本発明の治療薬は神経細胞内でその作用を発揮する。典型的には、小脳に存在する神経細胞(小脳深部核の神経細胞やプルキンエ細胞など)が標的となり、A-Tに特徴的な小脳神経変性に対する抑制的ないし阻害的治療効果をもたらす。
【0016】
本発明における用語「機能的ZFHX3」とは、その生理機能を発揮できる状態のZFHX3タンパク質をいう。生理的条件下ではZFHX3タンパク質はリン酸化状態にあり、その機能を発揮する。一方、ZFHX3タンパク質の脱リン酸化はZFHX3タンパク質の分断化・分解を引き起こす。そこで本明細書では、用語「機能的ZFHX3タンパク質」を用語「リン酸化状態のZFHX3タンパク質」と交換可能に使用する。
【0017】
ZFHX3はAFP遺伝子プロモータ上流の発現調節領域にあるA(アデニン)とT(チミン)に富む(AT-rich)DNA配列に結合するDNA結合タンパク質であり、ATBF1と呼ばれていた。ATBF1については、異なったプロモータの使用及び選択的スプライシングにより形成される、分子量の異なった2つのアイソフォームATBF1-A(404kDa、アミノ酸配列を配列番号1に示す。また、ATBF1-Aをコードする塩基配列(GenBank accession number:L32832を参照)を配列番号2に示す。)とATBF1-B(306kDa、アミノ酸配列を配列番号3に示す。また、ATBF1-Bをコードする塩基配列(GenBank accession number:L32833を参照)を配列番号4に示す。)の存在が知られている。
【0018】
「機能的ZFHX3タンパク質を増大させる効果」とは、直接又は間接的にZFHX3遺伝子を発現制御し、機能的ZFHX3タンパク質の発現量の増大をもたらす効果をいう。機能的ZFHX3タンパク質を増大させる効果を発揮する化合物として、ZFHX3タンパク質の発現を増強する化合物を用いることができる。該当する化合物の具体例としてレチノイン酸を挙げることができる。レチノイン酸はATM及びCREBを介してZFHX3遺伝子の転写活性を高める。ZFHX3遺伝子に対するこのような作用を示す化合物である限り、この例に限定することなく本発明の治療薬の有効成分として使用可能である。
【0019】
一方、「機能的ZFHX3タンパク質を安定化させる効果」とは、機能的ZFHX3タンパク質のリン酸化状態を維持する効果をいう。リン酸化状態が維持されることによってZFHX3は分断化・分解を回避でき、その本来の生理機能を発揮できる。ZFHX3の脱リン酸化状態の維持には、ZFHX3を標的とした脱リン酸化酵素の活性阻害が有効である。そこで、本発明の治療薬の有効成分としてプロテインフォスファターゼ2B(以下、「PP2B」と略称する)阻害剤を使用するとよい。PP2B阻害剤の具体例としてタクロリムス(別名FK506、CAS登録番号104987-11-3)、シクロスポリンAを挙げることができる。タクロリムスは土壌細菌より単離された化合物であり、移植時の免疫抑制、アトピー性皮膚炎、重症筋無力症、関節リウマチ、ループス腎炎等に使用されている。プログラフ(登録商標)(タクロリムス水和物)、グラセプター(登録商標)(タクロリムス水和物)、プロトピック(登録商標)(タクロリムス水和物)等の商品名でタクロリムス製剤がアステラス製薬株式会社によって販売されている。また、シクロスポリンはタクロリムスと類似の薬理作用を持ち、免疫拒絶反応の抑制や自己免疫疾患の治療に使用されている。ネオーラル(登録商標)、サンディミュン(登録商標)の商品名でシクロスポリン製剤がノバルティスファーマ株式会社によって販売されている。
【0020】
PP2B阻害剤に代えて、シグナル伝達経路においてPP2Bの上流に位置するカルパインに対する阻害剤を用いることにしてもよい。カルパイン阻害剤の例はカルパスタチン、ロイペプチン、E-64、E-64d(E-64の誘導体)、カルペプチンである。
【0021】
ZFHX3の脱リン酸化を抑制し得る限りにおいて、以上の例に限定することなく本発明の治療薬の有効成分として使用可能である。尚、後述の実施例に示したアッセイ系或いは参考文献に記載されるアッセイ系を利用すれば、公知の化合物群或いは新規に合成又は単離した化合物の中から、機能的ZFHX3タンパク質を増大又は安定化する化合物をルーチン的操作によって同定することが可能である。
【0022】
後述の実施例に示す通り、本発明者らは、神経細胞においてA-Tの原因遺伝子とされるATMとZFHX3が密接な関わりをもつことを見出した。詳しくは、ZFHX3が血小板由来成長因子受容体β(以下、「PDGFRB」と略称する)の活性化を介してATMの活性化に関与し、オートファジーの誘導ないし活性化が生ずるという、酸化ストレス下でのシグナル伝達経路の存在を明らかにした。理論に拘泥する訳ではないが、標的神経細胞における機能的ZFHX3タンパク質の増大は、本発明者らが見出したシグナル経路を活性化(正に制御)し、オートファジーの誘導ないし活性化を引き起こす。機能的ZFHX3タンパク質の安定化も同様にオートファジーの誘導ないし活性化に寄与する。
【0023】
本発明の治療薬の製剤化は常法で行うことができる。製剤化する場合には製剤上許容される他の成分(例えば、担体、賦形剤、崩壊剤、緩衝剤、乳化剤、懸濁剤、無痛化剤、安定剤、保存剤、防腐剤、生理食塩水など)を含有させることができる。賦形剤としては乳糖、デンプン、ソルビトール、D-マンニトール、白糖等を用いることができる。崩壊剤としてはデンプン、カルボキシメチルセルロース、炭酸カルシウム等を用いることができる。緩衝剤としてはリン酸塩、クエン酸塩、酢酸塩等を用いることができる。乳化剤としてはアラビアゴム、アルギン酸ナトリウム、トラガント等を用いることができる。懸濁剤としてはモノステアリン酸グリセリン、モノステアリン酸アルミニウム、メチルセルロース、カルボキシメチルセルロース、ヒドロキシメチルセルロース、ラウリル硫酸ナトリウム等を用いることができる。無痛化剤としてはベンジルアルコール、クロロブタノール、ソルビトール等を用いることができる。安定剤としてはプロピレングリコール、アスコルビン酸等を用いることができる。保存剤としてはフェノール、塩化ベンザルコニウム、ベンジルアルコール、クロロブタノール、メチルパラベン等を用いることができる。防腐剤としては塩化ベンザルコニウム、パラオキシ安息香酸、クロロブタノール等と用いることができる。
【0024】
製剤化する場合の剤形も特に限定されない。剤形の例は錠剤、散剤、細粒剤、顆粒剤、カプセル剤、シロップ剤、注射剤及び吸入剤である。本発明の治療薬には、期待される治療効果(又は予防効果)を得るために必要な量(即ち治療上有効量)の有効成分が含有される。本発明の治療薬中の有効成分量は一般に剤形によって異なるが、所望の投与量を達成できるように有効成分量を例えば約0.1重量%〜約95重量%の範囲内で設定する。
【0025】
本発明の治療薬はその剤形に応じて経口投与又は非経口投与(静脈内、動脈内、皮下、皮内、筋肉内、又は腹腔内注射など)によって患者に適用される。これらの投与経路は互いに排他的なものではなく、任意に選択される二つ以上を併用することもできる(例えば、経口投与と同時に又は所定時間経過後に静脈注射等を行う等)。全身投与によらず、局所投与することにしてもよい。局所投与として、目的の組織への直接注入又は塗布を例示することができる。
【0026】
本発明の治療薬の投与量及び投与スケジュールは、期待される治療効果が得られるように設定される。治療上有効な投与量の設定においては一般に患者の症状、年齢、性別、及び体重などが考慮される。尚、当業者であればこれらの事項を考慮して適当な投与量を設定することが可能である。例えば、成人(体重約60kg)を対象として一日当たりの有効成分量が約0.01mg〜約100mg、好ましくは約0.2mg〜約10mgとなるよう投与量を設定することができる。投与スケジュールとしては例えば1日1回〜数回、2日に1回、或いは3日に1回などを採用できる。投与スケジュールの作成においては、患者の症状や有効成分の効果持続時間などを考慮することができる。
【0027】
以上の記述から明らかな通り、本出願は、機能的ZFHX3タンパク質を増大又は安定化する化合物をA-T患者に対して治療上有効量、投与するステップを含む、A-Tの治療法も提供する。本発明の治療法によれば、A-T患者の小脳神経細胞内における機能的ZFHX3タンパク質が増大及び/又は安定化し、小脳での神経変性が抑制される。機能的ZFHX3タンパク質を増大する化合物としては例えばレチノイン酸が採用される。また、機能的ZFHX3タンパク質を安定化する化合物としては、PP2B阻害剤やカルパイン阻害剤等を使用できる。PP2B阻害剤の具体例はタクロリムスである。
【0028】
2.神経変性の予防又は治療に有効な化合物のスクリーニング法
本発明の第2の局面は、A-Tに起因又は付随する神経変性、或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物をスクリーニングする方法に関する。本発明のスクリーニング法で選抜される化合物は、それ自体又は必要な改変を加えた上で、A-T等の神経変性疾患に対する治療薬の有効成分として、或いはA-T等の神経変性疾患に対する治療薬を開発するためのリード化合物等として使用される。A-Tに起因又は付随するものであるか、又は神経過興奮に由来した酸化ストレスによるものである限り、本発明のスクリーニング法が標的とする神経変性に該当する。好ましい一態様では、A-Tの治療に有効な化合物を見出す手段として本発明のスクリーニング法は構成される。
【0029】
本発明のスクリーニング法では以下の(1)〜(4)のステップを行う。
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
【0030】
ステップ(1)では複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分ける。同種同系非ヒト哺乳動物としてマウス、ラット、モルモット、ハムスターを例示することができる。好ましくは、入手及び取り扱いが比較的容易なマウス又はラットを使用する。試験群及び対照群に含まれる個体数は特に限定されない。一般に使用する個体数が多くなるほど信頼性の高い結果が得られるが、多数の個体を同時に取り扱うことは使用する個体の確保や操作(飼育を含む)の面で困難を伴う。そこで例えば各群に含まれる個体数を1〜50、好ましくは2〜30、さらに好ましくは3〜20とする。通常は試験群と対照群の個体数を等しくする。
【0031】
本発明の一態様では、野生型動物を用いて本発明のスクリーニング法を実施する。ここでの「野生型」とはATM遺伝子に関して野生型であること、即ち、正常なATM遺伝子を保有していることを意味する。慣例に従い、当該野生型動物をATM(+/+)と表現することができる。他の態様として、ATM遺伝子欠損動物(ATMノックアウト動物)を用いたスクリーニング法が提供される。この態様はA-Tの原因・病態を反映させた動物を用いたものであり、A-Tに対する有効性や特異性が一層高い化合物の同定に有利である。ATM遺伝子欠損動物はホモ型でもヘテロ型でもよい。A-Tに対する特異性が高い化合物を同定するという目的の下では、ホモ型のATM遺伝子欠損動物を採用することが特に好ましい。尚、慣例に従い、ホモ型ATM遺伝子欠損動物及びヘテロ型ATM遺伝子欠損動物はそれぞれATM(-/-)及びATM(+/-)と表現することができる。
【0032】
ATM遺伝子欠損動物は、例えば、ジーンターゲティングを利用してATM遺伝子に点変異を導入すること又は点変異を含む変異型ATM遺伝子を導入することによって作製できる。ジーンターゲティングは遺伝子の相同組換えを利用してゲノムに改変を加える技術である。ジーンターゲティングを利用すれば、特定の遺伝子が欠損した動物(ノックアウト動物)を作製することができる。ジーンターゲティングを利用したノックアウト動物の作製法は、マウスの場合を例にとれば、大別して(a)ターゲティングベクターの構築、(b)ES細胞へのターゲティングベクターの導入及び相同組換え体の同定、(c)ブラストシストへの相同組み換え体の注入、(d)仮親の子宮内への胚移植及び出産(キメラ仔マウス)、(e)キメラ仔マウスと野生型マウスの交配(F1世代であるヘテロ接合体マウスの産出)、及び(f)F1世代同士の交配(F2世代であるホモ接合体マウスの産出)からなる。ラットなど、他の齧歯類を用いた場合も同様の手順で遺伝子改変動物を得ることができる。尚、ジーンターゲティングによる遺伝子改変動物の作製法については例えばJoyner, A. L.:Gene Targeting (IRL press)、Capeccchi, M. R., Science, 244, 1288-1292 (1989)、Hua Gu, et al., Science, 265, 103-106 (1994)、McHugh, T. J. et al., Cell, 87, 1339-1349 (1996)、Shibata, H. et al., Science, 278, 120-123 (1997)等が参考になる。
【0033】
ステップ(2)では試験群に対してカイニン酸を投与し、併せて被験物質を投与する。カイニン酸及び被験物質の投与態様は特に限定されないが、カイニン酸については例えば腹腔内注射、静脈内注射、脳質内投与等によって投与するとよい。操作の簡便性と安全性の点から、腹腔内注射による投与が特に好ましい。神経興奮に由来する神経障害ストレスを生じさせるためにカイニン酸が投与される。当該目的を達成できる限りにおいて、カイニン酸の投与量や投与回数は特に問わない。カイニン酸の投与回数及び投与量は使用する動物の種、体重等を考慮して設定することができる。投与回数の例は1〜5回である。また、投与量の一例を示せば、マウスを使用した場合において短期間投与で30mg/kg、1回/1日で3日まで、長期間投与で30mg/kg、2日ごと2ヶ月間まで、である。尚、適切な投与回数、投与量は予備実験を通して決定することができる。
【0034】
試験群全体に同量のカイニン酸を投与するのではなく、投与量の異なる動物個体を用意することにしてもよい(例えばカイニン酸の投与量が基準量の個体、2倍量の個体、5倍量の個体を用意する)。この態様の場合、カイニン酸の投与量(即ちカイニン酸刺激のレベル)を考慮しつつ、後述のステップ(3)の検定及びステップ(4)の判定が行われる。
【0035】
被験物質の投与にあたっては、例えば被験物質を含む餌又は飲料水を用意し、これを摂取させる。或いは、被験物質又は被験物質を含む溶液を用意し、これを経口、経鼻、経気管、注射(静脈内、動脈内、皮下、筋肉内、腹腔内等)によって投与する。被験物質を複数回投与することにしてもよい。その場合には各回の投与量は同一であっても異なっていても良い。継続的に投与することにしてもよい。被験物質は、カイニン酸の投与の前、カイニン酸の投与と同時、又はカイニン酸の投与の後に投与される。被験物質を複数回投与する場合についても、各回の投与の時期とカイニン酸の投与の時期との関係に特段の制約はない。例えば、カイニン酸の投与後、被験物質を所定の間隔をおいて複数回投与する。尚、対照群については被験物質を投与しないこと以外は同一の条件下で処理・飼育する。
【0036】
被験物質としては様々な分子サイズの有機化合物又は無機化合物を用いることができる。有機化合物の例として、核酸、ペプチド、タンパク質、脂質(単純脂質、複合脂質(ホスホグリセリド、スフィンゴ脂質、グリコシルグリセリド、セレブロシド等)、プロスタグランジン、イソプレノイド、テルペン、ステロイド、ポリフェノール、カテキン、ビタミン(B1、B2、B3、B5、B6、B7、B9、B12、C、A、D、E等)を例示できる。被験物質は天然物由来であっても、或いは合成によるものであってもよい。後者の場合には例えばコンビナトリアル合成の手法を利用して効率的なスクリーニング系を構築することができる。尚、植物抽出液、細胞抽出液、培養上清などを被験物質として用いてもよい。また、既存の薬剤を被験物質としてもよい。2種類以上の被験物質を同時に添加することにより、被験物質間の相互作用、相乗作用などを調べることにしてもよい。
【0037】
ステップ(3)では、ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定する。このように本発明では、脳内でのオートファジー又は神経変性(例えば小脳におけるプルキンエ細胞や小脳深部核神経細胞等の変性・脱落)が検定される。オートファジーと神経変性の両者を検定することにしてもよい。検定に供される脳部位の具体例は、小脳、大脳皮質、海馬である。好ましい一態様では小脳を検定に供する。また、カイニン酸による刺激に対して小脳深部核が顕著な反応性を示したという知見を考慮し(後述の実施例を参照)、好ましくは、小脳深部核に関してオートファジー又は神経変性の検定を行う。ここで「オートファジー」とは、プロテアソーム系と並ぶ主要な細胞内分解機構である。オートファジーは生体の恒常性維持に関与する。また、感染防御や細胞死、老化、癌化、神経変性などにもオートファジーが関与していることが明らかになってきている。一方、「神経変性」は、神経細胞の変性・脱落と同義である。
【0038】
オートファジーを検定するためには、例えばオートファジー関連因子の量、ATMの活性化量又は空胞形成の程度を検出すればよい。これらの検出対象はいずれもオートファジーのマーカーとなり、オートファジーの促進ないし誘導を反映してその量ないし程度が増大する。オートファジー関連因子として微小管関連タンパク質軽鎖3(LC3)、GABAA受容体結合タンパク質(GABARAP)及びはゴルジ関連ATPアーゼエンハンサー(GATE-16)を例示することができる。検出対象に応じて、免疫組織化学、ウエスタンブロット解析が利用される。尚、ホモ型のATM遺伝子欠損動物を用いて本発明のスクリーニング法を構築した場合には、ATM遺伝子の発現が見られないことから、オートファジー関連因子の量又は空胞形成の程度を検出することになる。
【0039】
神経変性の検定には、例えばTUNEL染色や活性型カスペース3(Caspase)染色等の免疫組織化学を利用できる。TUNEL染色及び活性型カスペース3(Caspase)染色については専用のキットが市販されている。当該キットを利用すれば神経変性を定量的に検定・評価することも可能である。
【0040】
ステップ(4)では、試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定する。本発明では、判定の際、「オートファジーの促進ないし誘導を認めること(指標1)」、又は「神経変性の抑制を認めること(指標2)」を、被験物質の有効性の指標とする。換言すれば、オートファジーの促進ないし誘導の程度が対照群よりも試験群で高いとき(指標1に基づく判定)又は神経変性の抑制の程度が対象群よりも試験群で高いとき(指標2に基づく判定)、被験物質が有効であると判定する。ステップ(3)において、脳内でのオートファジーを検定することにした場合には指標1が採用される。他方、ステップ(3)において、脳内での神経変性を検定することにした場合には指標2が採用される。
【0041】
有効性を認めた複数の被験化合物を用いて再度ステップ(1)〜(4)を行い、有効性の高い物質の絞り込みを行うことにしてもよい。本発明のスクリーニング法によって選択された物質が十分な薬効を有する場合には、当該物質をそのまま治療薬等の有効成分として使用することができる。一方で十分な薬効を有しない場合には化学的修飾などの改変を施してその薬効を高めた上で、治療薬等の有効成分として使用することができる。勿論、十分な薬効を有する場合であっても、更なる薬効の増大を目的として同様の改変を施してもよい。
【0042】
ところで、これまでにもATM遺伝子欠損マウス(ATM(-/-)マウス)を用いて神経変性を誘導する実験・研究が行われている。但し、従来の方法では、X線照射によってDNA損傷刺激を与え、神経変性を誘導している。このような誘導法は、神経興奮という神経本来の機能に付随する神経変性メカニズムとは無関係な刺激であり、治療薬の開発には結びつかない。換言すれば、ATM遺伝子欠損マウスを用いた従来のアッセイ系は治療薬の開発に有用とはいえない。対照的に、本発明で採用するカイニン酸刺激は、DNA損傷と独立した(即ち、DNA損傷とは関係のない)本来の「神経興奮」に由来する神経障害ストレスであるといえることから、神経変性の評価系として、治療薬の開発などに有用である。特に、小脳機能に特化した神経変性の評価系として、小脳での神経変性を伴う疾病に対する治療薬の開発などにおいて有用である。このように本発明のスクリーニング法は有用性及び実用性の点で従来の技術とは比較にならないほど優れたものである。尚、カイニン酸刺激が海馬神経に神経障害を与えることは既知であり(Science 262:689-695, 1993)、それ自体は新しいことではない。本発明において重要なことは、カイニン酸が小脳深部核(DCN)に存在するニューロンの過剰興奮を引き起こすという、カイニン酸刺激に対する小脳の反応を明らかにしたことであり、当該成果があったからこそ、有用性及び実用性に優れた本発明のスクリーニング法を完成することができた。小脳の出力系のニューロンに着目することで完成し得た本発明のスクリーニング法は、小脳機能に特化した神経変性の定性的或いは定量的な評価に特に有用であり、小脳での神経変性の抑制ないし阻害に有効な物質の探索・同定を可能とする。
【実施例】
【0043】
ZFHX3の機能とA-T患者が持つ多彩な臨床症状との密接な結びつきに着目し、ZFHX3の発現制御及びその標的遺伝子群に焦点をあてた網羅的な解析を行った。
【0044】
1.方法
(1)細胞培養
P19マウス胎児性癌細胞株をα-MEMで維持した(Rudnicki and McBurney, 1987)。神経細胞への分化を誘導するために、P19細胞を凝集体の状態で、0.5μMのオールトランス型レチノイン酸(RA)(Sigma社)及び10%ウシ胎仔血清(FBS)を添加した培養液で4日間培養した。続いて、1%のFBS含有α-MEM(RAは含まない)を培養液として、0.01% ポリ-L-リジン(P8920, Sigma社)をコートしたスライドガラス上で3時間、そして1μg/mlウシ胎仔血漿フィブロネクチン(Invitrogen社)をコートしたスライドガラス上で終夜、室温にて細胞を培養した。必要な場合には、ATMキナーゼ阻害剤のKU-55933(2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one; 2-morpholino-6-(thianthren-1-yl)-4H-pyran-4-one (Calbiochem社))(10 μM)を毎日培養液に添加し、そして、PDGFRB及びVEGFR-2の特異的阻害剤であるAG1433(2-(3,4-dihydroxyphenyl)-6,7-dimethylquinoxaline(Calbiochem社))を過酸化水素水の添加30分前に所定の濃度(0.05, 0.5, 1.0, 5.0μM)で添加した。
【0045】
(2)マウス
野生型及びATMノックアウトマウス(ハーバード・メディカル・スクールのP. Leder氏より供与)を周囲条件下(23℃、湿度55%)、所定の明暗サイクルで飼育した。ゲノムDNAを尾先端より採取し、PCRによってAtm遺伝子を増幅させた。PCRには以下のプライマーを使用した。
KO-1F: 5'-TGGTCAGTGTAACAGTCATTGTGC-3'(配列番号5)
KO-1R: 5'-AAGGTTGTAGATAGGTCAGCATTG-3'(配列番号6)
KO-2R: 5'-AACGAGATCAGCAGCCTCTGTTCC-3'(配列番号7)
【0046】
プライマーKO-1Fはエクソン34の220bp上流(イントロン34)に位置し、プライマーKO-1Rはエクソン34内の123bpに位置し、プライマーKO-2RはPGKneoのポリA領域内の87bpに位置する。これらのプライマーを使用した場合のPCR産物の長さは、野生型マウスの場合は342bpであり、Atm(-/-)マウスの場合は406bpとなる。カイニン酸(2-carboxy-3-carboxymethyl-4-isopropenyl-pyrrolidine (Enzo Life Science社))をオスDDYマウスに投与した(30 mg/kg、腹腔内投与(i.p.))。4%パラホルムアルデヒドを使用した灌流固定後に3月齢のマウスから脳全体を摘出した。妊娠ラット(Std Wister ST)にBrdU(5-bromo-2'-deoxy-uridine)(50mg/kg)を3時間投与し、E14.5の胚を標識化した。続いて胚を摘出し、その脳を4%パラホルムアルデヒドで固定した後、パラフィン包埋した。
【0047】
(3)X線照射
X線装置CAX-210(株式会社中部メディカル)を用い、細胞、マウスにX線(10 Gy)を照射した(210 kV、10 mA、4分45秒、銅シールド)。
【0048】
(4)トランスフェクション及びプラスミド
一連のルシフェラーゼリポータープラスミドを構築するため、SalI及びBamHIを使用し、様々な長さのヒトZFHX3プロモータ領域をpBluescriptIIKS(+)(Agilent Technologies社、Stratagene社製品)にクローニングした。ZFHX3プロモータ領域を含むBssHII/BamHI断片をpGV-B基本ベクター(東洋ビーネット株式会社)のMluIサイトとBglIIサイトの間、ルシフェラーゼ遺伝子の5'プライム位置にサブクローニングした。以下のプライマーセットを用いたPCRによる部位特異的変異導入法により、1.0 kbpのZFHX3プロモータ内のCREサイトに変異を導入した。
pGV-F(pGV-B内ルシフェラーゼ遺伝子の5'隣接配列を含むフォワードプライマー): 5'-CAATGTATCTTATGGTACTG-3'(配列番号8)
mCRE-R(CREサイトへの変異導入用のリバースプライマー): 5'-CGGAAATGACCACAGCAAAG-3'(配列番号9)
mCRE-F(CREサイトへの変異導入用のフォワードプライマー):5'-CTTTGCTGTGGTCATTTCCG-3'(配列番号10)
pGV-R1(pGV-B内ルシフェラーゼ遺伝子の最初のATGコドンを含むリバースプライマー): 5'-CTTTATGTTTTTGGCGTCTTCC-3'(配列番号11)
pGV-R2(pGV-B内ルシフェラーゼ遺伝子のpGV-R1プライマーよりさらに35bp下流に位置するリバースプライマー):5'-CCATCCTCTAGAGGATAGAATG-3'(配列番号12)
【0049】
pGV-F及びmCRE-Rを使用したPCR産物とmCRE-F及びpVG-R2を使用したPCR産物を得た。一部で重複する、これら一次産物を一緒に変性させ、pGV-F及びpGVR1を用いて再び増幅させた。その結果得られたPCR産物をMluI及びHindIIIで処理した後、pGV-B基本ベクターの同一クローニングサイトにサブクローニングした。CREBドミナントネガティブ発現ベクターpCMVCREB133はClontech社(カタログ番号631925)から購入した。
【0050】
(5)免疫組織化学
4%パラホルムアルデヒド(PFA)固定・パラフィン包埋組織から切り出した4μm断片を使用した。組織断片を脱パラフィン及び再水和処理した後、クエン酸バッファー(0.01 M、pH 6.0)内で加熱処理した(ZFHX3免疫染色用では圧力釜使用、110℃。ATM免疫染色用では電子レンジ使用、98℃)。PDFGRBの免疫染色については、熱賦活処理を行わなかった。染色は室温で行った。内在性ペルオキシダーゼ活性を阻害するため、全ての組織断片を0.3% 過酸化水素及び1.0%アジ化ナトリウムを含むメタノールでインキュベートした。続いて、一部の組織断片について、マウス抗ATM Ser1981リン酸部位(pSATM)モノクローナル抗体(1200倍希釈、7C10D8; Rockland社)とともにインキュベートした後、ホースラディッシュペルオキシダーゼ(HRP)標識抗マウスIgG抗体(Envision labeled polymer,Dako Cytomation社)とともにインキュベートした。同様に、一部の組織断片については、ウサギ抗ZFHX3ポリクローナル抗体(50倍希釈、D1-120; 株式会社医学生物学研究所)とともにインキュベートした後、HRP標識抗ウサギIgG抗体(Envision labeled polymer, Dako Cytomation社)とともにインキュベートした。また、一部の組織断片については、ヤギ抗マウスPDGFRBポリクローナル抗体(100倍希釈、R&D Systems社)とともにインキュベートした後、HRP結合抗ヤギIgG抗体(MAX-PO (G),株式会社ニチレイ)とともにインキュベートした。インキュベート条件は、一次抗体及び二次抗体いずれについても、室温、1時間とした。免疫反応産物をジアミノベンジジンで可視化した。BrdU標識及び検出にはロッシュ社のキットを用いた。組織断片をヘマトキシリンで対比染色した。
【0051】
(6)免疫細胞化学
4% PFAを含むPBSに細胞を室温にて20分間浸漬し、固定した。その後、0.25% Triton-Xを含むPBSで洗浄し、10%正常ヤギ血清でブロッキングした。続いて、細胞を室温にて1時間、一次抗体(ウサギ抗ZFHX3ポリクローナル抗体(100倍希釈)、マウス抗β-チューブリンアイソタイプIIIモノクローナル抗体(500倍希釈、3D10; Sigma社)、抗ATM Ser1981リン酸化サイト(pS-ATM)モノクローナル抗体(1:100希釈、10H11.E12;Rockland社)、ウサギ抗γH2AXポリクローナル抗体(1:100希釈、BETHYL社))と反応させた。0.25% Triton-Xを含むPBSで3回洗浄した後、二次抗体(マウスモノクローナル抗体用にAlexa-Fluor-488標識ヤギ抗マウス抗体、ウサギポリクローナル抗体用にAlexa-Fluor-546標識ヤギ抗ウサギ抗体)を1時間反応させ、細胞を可視化した。その後、PBSで2回洗浄した後、2.0μg/ml DAPI(1 mg/ml溶液を500倍希釈、和光純薬株式会社)で5分間、核染色した。
【0052】
(7)ウエスタンブロット
細胞を洗浄バッファー(10 mM 0.1 Mリン酸バッファー、250 mMシュークロース及び50 mM NaFを含む)で洗浄し、TNEバッファー(20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% Nonident-P40, 50 mM NaF)を用いて全細胞ライセートを調製した。タンパク質を15分間氷上でインキュベートした後、15,000 g、4℃で30分間、遠心処理した。上清を回収し、-80℃で保存した。ブラッドフォード法(Bio-Rad社)にて全タンパク質を定量した。タンパク質の検出のため、サンプル中のタンパク質を5〜20%の濃度勾配を有するポリアクリルアミドゲルで分離し、PVDF膜(Millipore社)に転写した。次に、転写後の膜を3% BSAを含むトリス緩衝液(0.05% Tween 20含有:TBS-T)で1時間ブロッキングした後、TBS-Tで洗浄した。続いて、室温にて1時間、一次抗体(マウス抗α-チューブリンモノクローナル抗体(8000倍希釈、B-5-1-2; Sigma社)、ウサギ抗ZFHX3抗体(2000倍希釈、AT-6; 株式会社医学生物学研究所)、マウス抗HAタグモノクローナル抗体(500倍希釈、5D8; 株式会社医学生物学研究所)、マウス抗ATMモノクローナル抗体(200倍希釈、5C2; Santa Cruz社)、マウス抗pS-ATMモノクローナル抗体(1000倍希釈、10H11.E12; Rockland社)、ヤギ抗PDGFRBポリクローナル抗体(1000倍希釈、R&D Systems社)、ウサギ抗カスパーゼ3ポリクローナル抗体(1000倍希釈、AAP-113; Assay Designs社))と反応させた。TBS-Tで洗浄した後、ペルオキシダーゼ標識二次抗体(マウスモノクローナル抗体用に抗マウスIgG抗体(10,000倍希釈、抗マウスIgG (H+L chain)-HRP; 株式会社医学生物学研究所)、ウサギポリクローナル抗体用に抗ウサギIgG抗体(5000倍希釈、抗ウサギIgG(H+L chain)-HRP, 株式会社医学生物学研究所)、ヤギポリクローナル抗体用に抗ヤギIgG抗体(5000倍希釈、抗ヤギIgG (H+L chain)-HRP;株式会社医学生物学研究所))を室温にて1時間、反応させた。TBS-Tで洗浄後、免疫反応をAmersham ECL Plus western blotting detection reagents(GE Healthcare社)で可視化した。
【0053】
(8)RNA抽出及びRT-PCR分析
Trizol試薬(Invitrogen社)を用いて全RNAを細胞から単離した。Ready-To-Go You-Prime First-Strand Beads kit(GE Healthcare社)を用い、1μgの全RNAからcDNAを調製した。qPCR MasterMix Plus for SYBR Green (Eurogentec社)を用いてリアルタイムPCRを行った。操作方法は添付の説明書に従い、95℃で10分間のインキュベーションの後、40サイクルの増幅(95℃で15秒間、60℃で1分間、72℃で45秒間、80℃で15秒間)とし、SYBR Greenを検出した。使用したプライマーは以下の通りである。
Gapdh:5'-TGTGTCCGTCGTGGATCTGA-3'(配列番号13)及び5'-CCTGCTTCACCACCTTCTTGA-3'(配列番号14)
Zfhx3:5'-TTCTTTTCCTCCTCTCTCCTCATC-3'(配列番号15)及び5'-CGGTCCGTCGGACTTTTG-3'(配列番号16)
Col3a1:5'-GCACAGCAGTCCAACGTAGA-3'(配列番号17)及び5'-TCTCCAAATGGGATCTCTGG-3'(配列番号18)
Itga8:5'-AGTGGGAGGACCTGGAAGTT-3'(配列番号19)及び5'-AGTGGGAGGACCTGGAAGTT-3'(配列番号20)
Pdfgrb: 5'-AACCCCCTTACAGCTGTCCT-3'(配列番号21)及び5'-TAATCCCGTCAGCATCTTCC-3'(配列番号22)
【0054】
Gapdh mRNAの発現量を用いて各遺伝子の発現量を補正した。ABI Prism 7500 fast Sequence Detection System (Applied Biosystemsa社)のユーザーガイド(User Bulletin #2)を用い、比較CT(2-ΔΔCT)法で各増幅産物の相対量を求めた。
【0055】
(9)統計
ルシフェラーゼアッセイ及びウエスタンブロットの結果については、ボンフェローニ補正マンホイットニー検定によって統計解析した。少なくとも3回の独立した実験を行い、平均±s.e.m.(標準誤差)で全ての数値を表した。P値が0.01未満の場合に有意差ありとした。RT-PCRの結果は、Gapdhの発現レベルで補正し、コントロールの発現レベルと比較した。スチューデントのt検定で統計的有意差(P<0.001)を評価した。
【0056】
(10)Thapsigargin処理による神経過興奮モデル
神経細胞への分化を誘導したP19細胞をThapsigargin(2μM、1時間)で処理し、Ca2+濃度を上昇させるストレス(神経過興奮)を与えた。この神経過興奮モデルを用い、FK506(タクロリムス)の効果を検討した。FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。
【0057】
2.結果
<ZFHX3プロモータはATMの活性化に反応してCREBにより活性化される>
レチノイン酸(RA)はHoxホメオティック遺伝子の発現に影響する。マウス胚の後脳及び鰓部の神経幹細胞におけるHox遺伝子の発現は特にRAに感受性が高い(Marshall et al., 1996)。ヒト細胞やマウス胎仔性癌細胞を用いた、RAによる神経への分化誘導法が確立されている(Andrews et al., (1984))。RA処理による遺伝子発現制御には、標的遺伝子内の特定の配列モチーフ(レチノイン酸応答エレメント(RAREs)と呼ばれる)を認識する核内レチノイン酸レセプター(RARs又はRXRと呼ばれる)が関与する(Marshall et al., 1996)。ZFHX3遺伝子は、胎児脳における分裂後ニューロンで発現される非クラスターホメオティック因子である(Jung et al., 2005)。重要なことは、プロモータがRAREsを含まないにも拘わらず、ZFHX3遺伝子の発現はRAで誘導されることである(Miura et al., 1995)。最近になって、神経分化に関与する新たなRA経路、即ちATM及びcAMP応答エレメント結合タンパク質(CREB)に依存する経路が同定された(Fernandes et al., 2007)。注目すべきことに、本発明者らの検討の結果、ヒトZFHX3遺伝子プロモータの5'隣接配列内にCREB結合エレメント(CRE)があることが明らかとなった。この事実から、ATM及びCREBの活性化を介したRAからのシグナルが、ZFHX3遺伝子の発現開始に重要な役割を果たしていると予想される。
【0058】
本発明者らは様々な長さのヒトZFHX3遺伝子プロモータをリポーター遺伝子(ルシフェラーゼ遺伝子)の上流にクローニングした(図1B)。そして、これらのコンストラクトでマウス胎仔性癌細胞株P19を一過性にトランスフェクトし、RAによる刺激の存在下及び非存在下でレポーター活性を測定した。その結果、RAは濃度依存的にニューロン特異的5.6-kbプロモータを活性化した(図1Bの-5.6)。一方、5'側1.6-kbを欠失させると基礎レベル(RAに反応しない0.42-kbのプロモータの場合の活性レベル。図1Bの-0.42)までプロモータ活性が低下した(図1の-4.0)。CREコンセンサス配列を含む1.6-kb配列を0.42-kbプロモータ配列の5'側に付加するとRAへの反応が顕著に増加した(図1Bの-5.6Δ)。ATM特異的キナーゼ阻害剤KU-55933で前処理すると、この反応は認められない(図1C)。ニューロンの分化に関して重要な生存因子であるCREBの関与を確認するために、133番セリンがアラニンに変異された変異CREBタンパク質を発現するpCMV-CREB133ベクターを用い、ドミナントネガティブCREB(dn-CREB)を過剰発現させた。CREBの当該変異はプロテインキナーゼAによるCREBのリン酸化を阻害し、cAMPの活性化を抑制する。dn-CREBを発現する細胞ではRA処理後のRA応答プロモータコンストラクトのトランス活性化が有意に抑制された(図1Dの-5.6Δ)。また、プロモータ内のCREB結合サイトの変異によって、dn-CREBの発現の有無に関係なく、RAに反応したプロモータの活性化が阻害された(図1Dの-5.6Δmut)。これらの結果は、ATM及びCREB経路の活性化が、RAによるニューロン特異的ZFHX3遺伝子プロモータのトランス活性化のメカニズムであることを示唆する。
【0059】
CREに加え、活性因子のマルチプルコンセンサスエレメントを含むことから、5.6-kbプロモータ配列に最大のプロモータ活性が認められるものと予想された。しかしながら、5.6-kbプロモータの活性は、短いCRE含有フラグメント(5.6Δ)の活性よりも低かった。この事実は負の調節エレメントがプロモータ内に存在することを示唆する。
【0060】
<酸化ストレス応答性のATM活性化には膜受容体が必要である>
Pdgfrb遺伝子変異マウスは、明らかな解剖学的異常を示すことなく成長するが、その脳はN-メチル-D-アスパラギン酸(NMDA)又は凍傷に対して脆弱である(Ishii et al., 2006)。このことはグルタミン酸作動性興奮毒性からニューロンを保護するためにPDGFRBの発現が重要であることを示唆する。PDGF受容体とインテグリンによって受容体のクラスタリングが生じ、PDGFの結合量が増加し、PDGF受容体が活性化する(Zemskov et al., 2009)。インテグリン及びPDGFRBの発現誘導において、ZFHX3が重要な役割を果たすかもしれない。
【0061】
酸化ストレスは、いくつかの成人神経変性疾患や脳卒中、トラウマ、発作などの原因因子或いは少なくとも補助的因子である(Coyle and Puttfarcken 1993)。また、酸化ストレスはA-T変異マウスのプルキンエニューロンの不完全な生存の主要原因の一つと考えられている(Chen et al., 2003)。酸化ストレスは様々な細胞においてPDGFRB介在性のATMキナーゼ活性化を引き起こす(Chen et al., 2003)。新たに同定されたPDGFRB介在性シグナル経路は興奮毒性のメカニズムを理解する上で重要と考えられる。そこで、過酸化水素による酸化ストレスの存在下でP19由来ニューロン様細胞においてPDGFRBがATMの活性化に関与するか否かを調べることにした。Ser1981(pS-ATM)のリン酸化を指標に評価した結果、ATMの速やかな活性化を認めた(100μMの過酸化水素添加後15分以内)(図2A)。過酸化水素の添加量(1-500μM)に応じてATMの活性化量は上昇した(図2B)。また、酸化ストレスに反応した当該ATMの活性化はPDGFRB及びVEGFR-2(vascular endothelial growth factor receptor 2; also known as FLK-1 and KDR)の特異的阻害剤であるAG1433によって顕著に抑制された。(図2C,D)。対照的に、X線照射に反応したATMの活性化はAG1433の添加によって影響を受けない(図2E,F)。以上の結果は、神経細胞における酸化ストレス下でのATMの活性化にこれらの膜受容体が重要な役割を果たしていることを示す。
【0062】
<神経興奮に反応してATMは細胞質内で活性化される>
DNA損傷ストレスに反応して活性化されたATMは核点状構造(nuclear foci)に局在する。しかしながら、低張刺激やクロロキン処理(Bakkenist and Kastan, 2003)或いはRA刺激(Fernandes et al., 2007)に反応して活性化したATMは、びまん性に広がるパターンを示す。本発明者らの検討したところ、酸化ストレス誘導性DNA二重らせん切断(DSB)に反応して活性化したATMの核点状構造を認め、それはDNA二重らせん切断のマーカーであるγ-H2AXとともに局在していた(図3)。最近になって、酸化ストレスに反応して細胞質内でATMが活性化されることが報告され、この活性化は核での活性化と独立したものであることが示された(Alexander et al., 2010)。本発明者らの検討では、当初、負荷する酸化ストレスの量に問題があり、核でのATMの活性化と細胞質でのATMの活性化を区別することはできなかった(図3)。そこで、細胞質内でのATMの活性化を検討するため、より生理的条件に近い酸化ストレス条件を設定した。ニューロンでは様々な因子が酸化ストレスを引き起こすが、脳での興奮毒性を誘導する因子としては神経伝達物質のグルタミン酸が最も重要である。カイニン酸(KA)は、非NMDA型グルタミン酸受容体選択的なグルタミン酸受容体アゴニストである(Olney et al., 1974)。そこで、マウスニューロン(Coyle and Puttfarcken, 1993; Zhang et al., 2002)における代謝性酸化ストレスを誘導するため、KAによる刺激を使用することにした。KA刺激はマウスに2種類の発作を引き起こす。一つは頭部を繰り返し曲げる動作である。もう一つは異常な筋収縮であり、マウスの大脳皮質、海馬及び小脳深部核(DCN)でのATMの活性化を引き起こす(図4)。γ-H2AXの誘導がないと(図5A2、図6)活性化ATMは主に細胞質に存在することが明らかとなった(図5A1、図6)。対照的に、X線照射はDNA損傷ストレスを引き起こし、その場合の活性化ATMはγ-H2AXの点状構造(図5B2、図6)を伴い核に局在した(図5B1、図6)。KA刺激によって、DCN内のニューロンに明確な液胞の形成を認めた。
【0063】
<ATMノックアウトマウスのDCNでのPDGFRBの抑制>
神経への分化前及び分化後のP19細胞における、ZFHX3とPDGFRBの発現との関係をより詳細に調べることにした。ニューロンへ終末分化したP19ニューロン様細胞(7日目)においてZFHX3及びPDGFRBタンパク質の発現が顕著に上昇していた(図7A)。in situハイブリダイゼーションによるスクリーニング解析の結果、PDGFRBはDCNで高発現していた。免疫組織化学の結果では、成体コントロールマウス脳DCNにおける大型ニューロンにZFHX3(図7C4,C5)及びPDGFRB(図7C6,C7)の明瞭な染色を認めた。一方で、ATMノックアウトマウスではいずれのタンパク質の発現も強力に抑制された(図7C11-C14)。ラット胚において、増殖細胞(図7B2i)と分裂後細胞(図7B2ii)を識別するためにBrdU染色を行った。分裂後ニューロンにおける、ZFHX3の発現とPDGFRBの発現の相関性は、神経分化モデルを使用したマイクロアレイ解析の結果に符合した。ATMノックアウトマウスのDCNにおいてPDGFRBが抑制されていた事実は、小脳での特定神経変性のメカニズムを説明する上で重要といえる。
【0064】
<FK506の投与によって神経細胞死を抑制することができる>
神経細胞へ分化誘導させたP19細胞をThapsigarginで処理し、Ca2+濃度を上昇させるストレス(神経過興奮)を与えた。FK506を投与すると、当該ストレスによって誘導されるカスパーゼ3の活性化が抑制された(図10、レーン4)。即ち、FK506の投与によって神経細胞死が抑制された。
【0065】
<FK506の投与によってZFHX3の分断化が抑制できる>
Thapsigargin処理によるストレスがZFHX3タンパク質に与える影響を調べるため、抗ATBF1抗体(AT-6)で免疫沈降したタンパク質をAT-6抗体でウエスタンブロット解析した。その結果、Thapsigargin処理によって生ずるZFHX3タンパク質の分断化をFK506が有意に抑制した(図11、レーン6)。
【0066】
<FK506の投与によってZFHX3の脱リン酸化が抑制できる>
Thapsigargin処理によるストレスがZFHX3タンパク質のリン酸化に与える影響を調べるため、Pro-Q Diamond phosphoprotein stain kit (Invitrogen社、カタログ番号 P-33356)を用いた検定を行った。その結果、Thapsigargin処理によって生ずるZFHX3タンパク質の脱リン酸化をFK506が抑制した(図12、レーン14)。
【0067】
以上の結果より、免疫抑制剤として知られるFK506が、神経過興奮による神経細胞死を抑制できることが明らかになるとともに、その作用機序は「脱リン酸化の抑制(リン酸化状態の維持)を通してZFHX3の分断化を防ぐことにある」との知見が得られた。
【0068】
3.考察
ATMが細胞質内で活性化する現象は、神経細胞が酸化ストレスを受けた場合の防御シグナルとして重要な意味を持つことを明らかにした。以前にも神経細胞を含む各種細胞の観察結果として、核ではなく細胞質内でATMが活性化する現象が報告されている(Kuljis et al., 1999;Barlow et al., 1996;Boehrs et al., 2007;Gorodetsky et al., 2007)。神経細胞で観察された、細胞質内でのATMの活性化はシナプスの機能と関係している可能性が示唆されている(Li et al., 2009)。シナプスに存在する小胞ではATMがVAM2やシナプシン-1と結合した状態で存在する。ATMが核内タンパク質としての機能以外に細胞質タンパク質として機能していることを示す数々の研究論文が出されるようになり、細胞質内で活性化するATMは細胞の防御機構として機能している可能性が予想されるようになった。Alexanderらは細胞が酸化ストレスを受けた際にATMが活性化してmTORの抑制シグナルとして機能することを示した(Alexander et al., 2010)。mRORC1複合体の機能を抑制することによりオートファジー反応が誘導される。この細胞質内のシグナル伝達系は、核で進むDNAの二重鎖切断反応からのシグナルとは関わりのないことが示されている。Alexanderらは酸化ストレスに反応して、どのようにATMが活性化するのか、その未知のメカニズムについて実証的な根拠を与えることができなかったが、強い酸化反応でATMを構成するタンパク質のスルホニル基が修飾を受けることで、ATM自身の高次構造が変化することが、ATM自身の活性化の引き金になっている可能性を提唱した。
【0069】
本発明者らは、P19細胞をレチノン酸(RA)刺激で神経分化誘導する実験モデル系を使い、ZFHX3(ATBF1)の遺伝子発現誘導されるシグナル伝達系を解明した。RA刺激はATMの活性化を誘導し、そして、活性化したATMがCREBを活性化する。CREBはZFHX3の遺伝子プロモータ上にあるCRE(cAMP Responsive Element)のコンセンサスDNA配列に結合して、標的遺伝子であるZFHX3遺伝子の転写を活性化する。本発明者らはZFHX3の標的遺伝子の網羅的検索実験も行い、細胞接着因子の遺伝子群(プロコラーゲンIII型α1、インテグリンα8)を同定した。ZFHX3の標的遺伝子の中にはPDGFRB(血小板由来増殖因子受容体β)も含まれている。PDGFRBは結合組織間の細胞接着因子との相互作用で重要な受容体であるばかりでなく、神経細胞がグルタミン酸刺激による興奮等に由来する酸化ストレスを受けた状況下での神経保護作用のためのシグナル伝達系において重要な役割を演じる受容体である(Ishii et al., 2006)。本発明者らはPFGFRBまたはVEGFR-2からのシグナルが神経細胞の酸化ストレス反応の際、ATMの活性化の必須因子になることを実験的に証明した。本研究で用いた、P19細胞をレチノン酸(RA)刺激で神経分化誘導する実験モデル系において、ZFHX3の発現誘導に同期してPdgfrbとVegfr-2のmRNAの発現上昇を確認している。神経細胞中はその中に抗酸化物質が少ないにもかかわらず、神経興奮に由来する酸化ストレスに暴露され続ける(Brooks et al., 2000)。従って、十分な防御系を持たなければ酸化ストレスのために長期間生存することが難しい。酸化ストレスが生じた場合に迅速に反応する必要があり、細胞膜上の受容体から細胞内に情報を伝達して防御反応を誘導することは、神経細胞が長期間生存し続けるために非常に大切なメカニズムになっている。
【0070】
本発明者らは酸化ストレスを受けた神経細胞の細胞質内でATMが活性化する事実を明らかにした。神経細胞の興奮に起因する生化学反応産物が必然的に酸化ストレスを引き起こす。繰り返される神経興奮よって引き起こされる酸化ストレスは慢性的に進行する神経変性の主因となり、或いは少なくともその副次的な原因になっている(Coyle and Puttfarcken 1993)。本発明者らは、酸化ストレスが細胞質でATMを活性化するシグナル伝達系において、PDGFRBまたはVEGFR-2の活性化が必須であること解明した。神経興奮よって細胞内に酸化ストレスが生じても、ATMノックアウトマウスでは小脳深部核(DCN)にPDGFRBが発現していないために、細胞内で酸化ストレスに対応するためのシグナル伝達系が作動しない状況にあると考えられる。本発明者らは、マウスを使用したカイニン酸刺激実験で神経細胞に強い酸化ストレスを誘導した結果、小脳深部核のニューロンに顕著な空胞形成が生じることを見出した(図4B3)。小脳深部核にあるニューロン群にはPDGFRBが強く発現している(図5C6,C7)。小脳深部核のニューロンは主要な小脳の出力系中継場所として重要な役割を担っている。プルキンエ細胞と直接的にシナプスで繋がり、抑制シグナルを受け取っている。もしも小脳深部核ニューロンが障害を受けて消失すると、直結していたプルキンエ細胞との繋がりが障害され、その結果プルキンエ細胞は小脳深部核のニューロンから神経栄養因子を受け取れなくなり、プルキンエ細胞の変性が進む要因になる。プルキンエ細胞と小脳深部核との機能的な繋がりがA-Tの病態に密接に関与すると考えられる。カイニン酸刺激によって生じた、DCNにおける細胞質内のATM活性化は、過剰な神経興奮によってニューロン内に生じた酸化ストレスに対する保護的な応答反応と思われる。DCNに認められた空胞形成は、細胞質でのATMの活性化がmTORの抑制を介してオートファジーを誘導するという事実(Alexander et al., 2010)に関連しているであろう。DCNでは、カイニン酸処理に反応して、オートファジーの指標となる微小管関連タンパク質軽鎖3(LC3)の顕著な蓄積を認めた(図8)。オートファジー反応は障害を受けた細胞内小器官などを小胞に包んで酵素分解する反応であるので、かつては急性の神経細胞死の反応であると考えられていた(Wang et al., 2008)。しかし最近の研究によると、オートファジー反応は神経細胞死を導く反応というよりも、むしろ障害した細胞内小器官をきれいに分解/清掃して、細胞を生存維持するために必要な反応であると考えられるようになった。パーキンソン病の神経細胞では、オートファジー反応が障害され、障害を受けたままのミトコンドリアが蓄積することが神経細胞死を引き起こす原因になっていることが知られている(Narendra et al., 2008)。
【0071】
マイクロアレイによる網羅的発現解析によって、ZFHX3の標的遺伝子群としてプロコラーゲン遺伝子群(プロコラーゲンII型を除く)を同定した。ZFHX3は細胞外マトリックスの産生をコントロールして組織障害修復に関連する転写調節因子である。尚、プロコラーゲンII型は関節軟骨に特異的な因子であるので(Cheah et al., 1985)、今回用いた神経分化誘導の実験系モデル系ではプロコラーゲンII型の発現量に全く変化がなかったことは当然といえる。PDGFRBは外科的な損傷を受けた組織周囲にある血管で発現量が顕著に上昇する(Reuterdahl et al., 1993)。PDGFRBは組織修復過程で重要な機能を担っていると考えられている。ZFHX3はプロコラーゲン類とPDGFRBの両方の遺伝子を同時に転写活性化する機能を有し、組織損傷治癒過程できわめて重要な遺伝子調節因子であると言える。ZFHX3が機能障害に陥ると、血管系の疾患が惹起される可能性がある。事実、最近の研究報告によると、ZFHX3の変異は川崎病や心房細動や虚血性心疾患を起こす遺伝的背景になっている(Burgner et al., 2009;Benjamin et al., 2009;Gudbjartsson et al., 2009)。大規模な疫学調査結果から考察すると、ZFHX3は障害を受けた血管系の治癒過程に必要な重要な転写因子であることが分かる。コントロールマウスの小脳ではPDGFRBが強く発現しているにも関わらず(Lein et al., 2007)、ATMノックアウトマウスの小脳ではその発現量が低下していることは、A-T患者の小脳では組織損傷治癒反応が起こりにくいために神経変性に陥る危険性が生じていることを示している。
【0072】
以上の知見を図9として纏める。ATMの活性化経路として3種類の異なるシグナル伝達系がある。第一はDNAのDSBsに由来する核内のDNA二重鎖切断が起こす局所のATM活性化である。第二はレチノイン酸刺激(Fernandes et al., 2007)に誘導されて、核内にびまん性に広がるATMの活性化(Bakkenist and Kastan 2003)である。第三は酸化ストレスに誘導されて、細胞質で起こるATMの活性化である(Alexander et al., 2010)。酸化ストレス反応で起こるATMの活性化は、神経興奮で必然的に誘導される酸化ストレスとの関連において非常に重要である。本発明者らは細胞質で起こるATMの活性化反応にとって膜受容体型のチロシンリン酸化酵素のシグナルが必須であることを発見した。細胞質で起こるATMの活性化シグナルは、従来知られていた核内のDNA損傷ストレスに起因するATMの活性化シグナルとは独立したシグナルである。酸化ストレスに起因するATMの活性化シグナルは、mTORの抑制を介してオートファジー反応を誘導する。オートファジー反応は障害を受けた細胞内小器官を清掃して細胞機能を生理的に維持するために必要な反応である。以上を総合すると、細胞質で活性化するATMのシグナル伝達系は、酸化ストレスを受けた神経細胞を慢性的な神経変性から保護するためのメカニズムを起動するために重要な役割を演じていると言える。
【0073】
<参考論文>
Alexander, A., Cai, S. L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., Inoki, K., Guan, K. L., Shen, J., Person, M. D. et al. (2010). ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 107, 4153-4158.
Alterman, N., Fattal-Valevski, A., Moyal, L., Crawford, T. O., Lederman, H. M., Ziv, Y. and Shiloh, Y. (2007). Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am. J. Med. Genet 143A, 1827-1834.
Andrews, P. W. (1984). Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 103, 285-293.
Andrews, P. W., Damjanov, I., Simon, D., Banting, G. S., Carlin, C., Dracopoli, N. C. and Fogh, J. (1984). Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Invest. 50, 147-162.
Bakkenist, C. J. and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499-506.
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., Smorodinsky, N. I., Prives, C., Reiss, Y., Shiloh, Y. et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674-1677.
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley, J. N., Ried, T., Tagle, D. et al. (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159-171.
Benjamin, E. J., Rice, K. M., Arking, D. E., Pfeufer, A., van Noord, C., Smith, A. V., Schnabel, R. B., Bis, J. C., Boerwinkle, E., Sinner, M. F. et al. (2009). Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat. Genet. 41, 879-881.
Boehrs, J. K., He, J., Halaby, M. J. and Yang, D. Q. (2007). Constitutive expression and cytoplasmic compartmentalization of ATM protein in differentiated human neuronlike SH-SY5Y cells. J. Neurochem. 100, 337-345.
Brooks, P. J., Wise, D. S., Berry, D. A., Kosmoski, J. V., Smerdon, M. J., Somers, R. L., Mackie, H., Spoonde, A. Y., Ackerman, E. J., Coleman, K. et al. (2000). The oxidative DNA lesion 8,5'-(S)-cyclo-2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 275, 22355-22362.
Burgner, D., Davila, S., Breunis, W. B., Ng, S. B., Li, Y., Bonnard, C., Ling, L., Wright, V. J., Thalamuthu, A., Odam, M. et al. (2009). A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 5, e1000319.
Cheah, K. S., Stoker, N. G., Griffin, J. R., Grosveld, F. G. and Solomon, E. (1985). Identification and characterization of the human type II collagen gene (COL2A1). Proc. Natl. Acad. Sci. USA 82, 2555-2559.
Chen, K., Albano, A., Ho, A. and Keaney, J. F., Jr (2003). Activation of p53 by oxidative stress involves platelet-derived growth factor-beta receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J. Biol. Chem. 278, 39527-39533.
Chen, P., Peng, C., Luff, J., Spring, K., Watters, D., Bottle, S., Furuya, S. and Lavin, M. F. (2003). Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J. Neurosci. 23, 11453-11460.
Cho, Y. G., Song, J. H., Kim, C. J., Lee, Y. S., Kim, S. Y., Nam, S. W., Lee, J. Y. and Park, W. S. (2007). Genetic alterations of the ATBF1 gene in gastric cancer. Clin. Cancer Res. 13, 4355-4359.
Coyle, J. T. and Puttfarcken, P. (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689-695.
Dupre, A., Boyer-Chatenet, L. and Gautier, J. (2006). Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 13, 451-457.
Fernandes, N. D., Sun, Y. and Price, B. D. (2007). Activation of the kinase activity of ATM by retinoic acid is required for CREB-dependent differentiation of neuroblastoma cells. J. Biol. Chem. 282, 16577-16584.
Gorodetsky, E., Calkins, S., Ahn, J. and Brooks, P. J. (2007). ATM, the Mre11/Rad50/Nbs1 complex, and topoisomerase I are concentrated in the nucleus of Purkinje neurons in the juvenile human brain. DNA Rep. 6, 1698-1707.
Gudbjartsson, D. F., Holm, H., Gretarsdottir, S., Thorleifsson, G., Walters, G. B., Thorgeirsson, G., Gulcher, J., Mathiesen, E. B., Njolstad, I., Nyrnes, A. et al. (2009). A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 41, 876-878.
Ishii, Y., Oya, T., Zheng, L., Gao, Z., Kawaguchi, M., Sabit, H., Matsushima, T., Tokunaga, A., Ishizawa, S., Hori, E. et al. (2006). Mouse brains deficient in neuronal PDGF receptor-beta develop normally but are vulnerable to injury. J. Neurochem. 98, 588-600.
Jung, C. G., Kim, H. J., Kawaguchi, M., Khanna, K. K., Hida, H., Asai, K., Nishino, H. and Miura, Y. (2005). Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development 132, 5137-5145.
Kao, F. T., Hawkins, J. W., Law, M. L. and Dugaiczyk, A. (1982). Assignment of the structural gene coding for albumin to human chromosome 4. Hum. Genet. 62, 337-341.
Kataoka, H., Miura, Y., Joh, T., Seno, K., Tada, T., Tamaoki, T., Nakabayashi, H., Kawaguchi, M., Asai, K., Kato, T. et al. (2001). Alpha-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene 20, 869-873.
Khanna, K. K. and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247-254.
Kim, C. J., Song, J. H., Cho, Y. G., Cao, Z., Lee, Y. S., Nam, S. W., Lee, J. Y. and Park, W. S. (2008). Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology 52, 552-559.
Kuljis, R. O., Chen, G., Lee, E. Y., Aguila, M. C. and Xu, Y. (1999). ATM immunolocalization in mouse neuronal endosomes: implications for ataxiatelangiectasia. Brain Res. 842, 351-358.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A. F., Boguski, M. S., Brockway, K. S., Byrnes, E. J. et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168-176.
Li, J., Han, Y. R., Plummer, M. R. and Herrup, K. (2009). Cytoplasmic ATM in neurons modulates synaptic function. Curr. Biol. 19, 2091-2096.
Marshall, H., Morrison, A., Studer, M., Popperl, H. and Krumlauf, R. (1996). Retinoids and Hox genes. FASEB J. 10, 969-978.
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., 3rd, Hurov, K. E., Luo, J., Bakalarski, C. E., Zhao, Z., Solimini, N., Lerenthal, Y. et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160-1166.
Miura, Y., Tam, T., Ido, A., Morinaga, T., Miki, T., Hashimoto, T. and Tamaoki, T. (1995). Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J. Biol. Chem. 270, 26840-26848.
Miura, Y., Kataoka, H., Joh, T., Tada, T., Asai, K., Nakanishi, M., Okada, N. and Okada, H. (2004). Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Waf1/Cip1) promoter. Microbiol. Immunol. 48, 137-145.
Morinaga, T., Yasuda, H., Hashimoto, T., Higashio, K. and Tamaoki, T. (1991). A human alpha-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Mol. Cell. Biol. 11, 6041-6049.
Narendra, D., Tanaka, A., Suen, D. F. and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795-803.
Olney, J. W., Rhee, V. and Ho, O. L. (1974). Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 77, 507-512.
Reuterdahl, C., Sundberg, C., Rubin, K., Funa, K. and Gerdin, B. (1993). Tissue localization of beta receptors for platelet-derived growth factor and platelet-derived growth factor B chain during wound repair in humans. J. Clin. Invest. 91, 2065-2075.
Rudnicki, M. A. and McBurney, M. W. (1987). Cell culture methods and induction of differentiatin of embryonal carcinoma cell lines. In Teratocarcinomas and Embryonic Stem Cells a Practical Approach (ed. E. J. Robertson), pp. 14-49. Oxford, Washington DC: IRL Press.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S. et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749-1753.
Shi, Y., Venkataraman, S. L., Dodson, G. E., Mabb, A. M., LeBlanc, S. and Tibbetts, R. S. (2004). Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 101, 5898-5903.
Shieh, S. Y., Ikeda, M., Taya, Y. and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334.
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155-168.
Sun, X., Frierson, H. F., Chen, C., Li, C., Ran, Q., Otto, K. B., Cantarel, B. L., Vessella, R. L., Gao, A. C., Petros, J. et al. (2005). Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat. Genet. 37, 407-412.
Urano, Y., Sakai, M., Watanabe, K. and Tamaoki, T. (1984). Tandem arrangement of the albumin and alpha-fetoprotein genes in the human genome. Gene 32, 255-261.
Wang, Y., Han, R., Liang, Z. Q., Wu, J. C., Zhang, X. D., Gu, Z. L. and Qin, Z. H. (2008). An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy 4, 214-226.
Wilkinson, D. S., Tsai, W. W., Schumacher, M. A. and Barton, M. C. (2008). Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol. Cell. Biol. 28, 1988-1998.
Yasuda, H., Mizuno, A., Tamaoki, T. and Morinaga, T. (1994). ATBF1, a multiplehomeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol. Cell. Biol. 14, 1395-1401.
Zemskov, E. A., Loukinova, E., Mikhailenko, I., Coleman, R. A., Strickland, D. K. and Belkin, A. M. (2009). Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J. Biol. Chem. 284, 16693-16703.
Zhang, J., Zhang, D., McQuade, J. S., Behbehani, M., Tsien, J. Z. and Xu, M. (2002). c-fos regulates neuronal excitability and survival. Nat. Genet. 30, 416-420.
【産業上の利用可能性】
【0074】
毛細血管拡張性失調症の原因遺伝子とされるATM遺伝子が関わる新たなシグナル伝達経路が解明された。本発明の治療薬は当該シグナル伝達経路において重要な役割を果たすZFHX3を標的としたものであり、毛細血管拡張性失調症の治療に有効である。本発明の一態様では有効成分としてタクロリムス(免疫抑制剤として知られている)を使用する。タクロリムスの新たな用途を提供するという点においても本発明の意義は大きい。一方、本願が提供するスクリーニング法には、毛細血管拡張性失調症等における神経変性に対して有効な新規化合物の創出ないし同定手段として活用されることが期待される。
【0075】
この発明は、上記発明の実施の形態及び実施例の説明に何ら限定されるものではない。特許請求の範囲の記載を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
本明細書の中で明示した論文、公開特許公報、及び特許公報などの内容は、その全ての内容を援用によって引用することとする。
【配列表フリーテキスト】
【0076】
配列番号5:人工配列の説明:プライマーKO-1F;配列番号6:人工配列の説明:プライマーKO-1R;配列番号7:人工配列の説明:プライマーKO-2R;配列番号8:人工配列の説明:プライマーpGV-F;配列番号9:人工配列の説明:プライマーmCRE-R;配列番号10:人工配列の説明:プライマーmCRE-F;配列番号11:人工配列の説明:プライマーpGV-R1;配列番号12:人工配列の説明:プライマーpGV-R2;配列番号13:人工配列の説明:プライマーGapdh;配列番号14:人工配列の説明:プライマーGapdh;配列番号15:人工配列の説明:プライマーZfhx3;配列番号16:人工配列の説明:プライマーZfhx3;配列番号17:人工配列の説明:プライマーCol3a1;配列番号18:人工配列の説明:プライマーCol3a1;配列番号19:人工配列の説明:プライマーItga8;配列番号20:人工配列の説明:プライマーItga8;配列番号21:人工配列の説明:プライマーPdfgrb;配列番号22:人工配列の説明:プライマーPdfgrb
【技術分野】
【0001】
本発明は毛細血管拡張性失調症治療薬及びその用途、並びに毛細血管拡張性失調症等の神経変性を予防し或いはその治療にに有効な化合物をスクリーニングする方法に関する。
【背景技術】
【0002】
毛細血管拡張性失調症(Ataxia telangiectasia, A-T)は神経変性を伴う遺伝性疾患である(非特許文献1)。原因遺伝子はDNAの二重らせんの切断に反応する、セリン・スレオニンリン酸化酵素の一つであるATM(ataxia telangiectasia mutated)の障害による(非特許文献2、3)。疾患名であるA-Tは、合併する臨床兆候である小脳変性に由来する運動失調症(Ataxia)と毛細血管拡張(Telangiectasia)を合体させた名称で構成されている。
【0003】
A-T患者の臨床所見としてアルファフェトプロテイン(AFP)の血中異常上昇が知られている。AFP遺伝子はアルブミン遺伝子と並んでヒトの第4染色体上にコードされている(非特許文献4、5)。AFPは生理的には胎児の肝臓で発現し、成体の肝臓ではほぼ完全に発現が抑制されている。ただし成体に肝細胞癌が発生するとその腫瘍細胞が再びAFPを産生するようになる。この病態反応を利用することにより、血中AFPを肝臓癌の診断用バイオマーカとして臨床応用することができる。ところが、不思議なことにA-T患者では肝臓癌の発生とは無関係に血中AFPの異常上昇が認められる。AFPの異常上昇は、A-T患者が持つAFP遺伝子の発現抑制系に異常が起こっていることを示唆している。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S. et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749-1753.
【非特許文献2】Khanna, K. K. and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247-254.
【非特許文献3】Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155-168.
【非特許文献4】Kao, F. T., Hawkins, J. W., Law, M. L. and Dugaiczyk, A. (1982). Assignment of the structural gene coding for albumin to human chromosome 4. Hum. Genet. 62, 337-341.
【非特許文献5】Urano, Y., Sakai, M., Watanabe, K. and Tamaoki, T. (1984). Tandem arrangement of the albumin and alpha-fetoprotein genes in the human genome. Gene 32, 255-261.
【非特許文献6】Morinaga, T., Yasuda, H., Hashimoto, T., Higashio, K. and Tamaoki, T. (1991). A human alpha-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Mol. Cell. Biol. 11, 6041-6049.
【非特許文献7】Miura, Y., Tam, T., Ido, A., Morinaga, T., Miki, T., Hashimoto, T. and Tamaoki, T. (1995). Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J. Biol. Chem. 270, 26840-26848.
【非特許文献8】Jung, C. G., Kim, H. J., Kawaguchi, M., Khanna, K. K., Hida, H., Asai, K., Nishino, H. and Miura, Y. (2005). Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development 132, 5137-5145.
【非特許文献9】Yasuda, H., Mizuno, A., Tamaoki, T. and Morinaga, T. (1994). ATBF1, a multiplehomeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol. Cell. Biol. 14, 1395-1401.
【非特許文献10】Wilkinson, D. S., Tsai, W. W., Schumacher, M. A. and Barton, M. C. (2008). Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol. Cell. Biol. 28, 1988-1998.
【非特許文献11】Miura, Y., Kataoka, H., Joh, T., Tada, T., Asai, K., Nakanishi, M., Okada, N. and Okada, H. (2004). Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Waf1/Cip1) promoter. Microbiol. Immunol. 48, 137-145.
【非特許文献12】Kim, C. J., Song, J. H., Cho, Y. G., Cao, Z., Lee, Y. S., Nam, S. W., Lee, J. Y. and Park, W. S. (2008). Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology 52, 552-559.
【非特許文献13】Kataoka, H., Miura, Y., Joh, T., Seno, K., Tada, T., Tamaoki, T., Nakabayashi, H., Kawaguchi, M., Asai, K., Kato, T. et al. (2001). Alpha-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene 20, 869-873.
【非特許文献14】Cho, Y. G., Song, J. H., Kim, C. J., Lee, Y. S., Kim, S. Y., Nam, S. W., Lee, J. Y. and Park, W. S. (2007). Genetic alterations of the ATBF1 gene in gastric cancer. Clin. Cancer Res. 13, 4355-4359.
【非特許文献15】Alterman, N., Fattal-Valevski, A., Moyal, L., Crawford, T. O., Lederman, H. M., Ziv, Y. and Shiloh, Y. (2007). Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am. J. Med. Genet 143A, 1827-1834.
【非特許文献16】Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., 3rd, Hurov, K. E., Luo, J., Bakalarski, C. E., Zhao, Z., Solimini, N., Lerenthal, Y. et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160-1166.
【非特許文献17】Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., Smorodinsky, N. I., Prives, C., Reiss, Y., Shiloh, Y. et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674-1677.
【非特許文献18】Shieh, S. Y., Ikeda, M., Taya, Y. and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334.
【非特許文献19】Burgner, D., Davila, S., Breunis, W. B., Ng, S. B., Li, Y., Bonnard, C., Ling, L., Wright, V. J., Thalamuthu, A., Odam, M. et al. (2009). A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 5, e1000319.
【発明の概要】
【発明が解決しようとする課題】
【0005】
ATMというリン酸化酵素をコードしている遺伝子の変異がA-Tの原因であることが解明されたものの、神経変性のメカニズムは不明であり、A-Tに対する適切な治療法/予防法の確立が切望されている。このような状況下、本発明は、A-Tの治療又は予防に有効な手段を提供することを目的とする。また、A-T等の神経変性疾患の治療法ないし予防法の確立に資するツールを提供することを目的とする。
【課題を解決するための手段】
【0006】
本発明者らはA-T患者に特徴的な症状と遺伝子転写調節因子ZFHX3(ATBF1)の機能の間に深い関係があると考えて研究を進めた。ZFHX3はAFP遺伝子プロモータ上流の発現調節領域にあるA(アデニン)とT(チミン)に富む(AT-rich)DNA配列に結合するDNA結合タンパク質(AT-rich binding factor 1)として1991年にクローニングされた(非特許文献6)。最初に9-kbのATBF1 mRNAがクローニングされ、1995年には主要な遺伝子転写産物として全長12-kbのATBF1 mRNAがATBF1-Aと命名された。ATBF1-Aについては神経細胞の分化に関連する機能が明らかにされた(非特許文献7、8)。ごく最近になってATBF1の名称はヒトゲノム国際機構(Human Genome Organization)によりZFHX3(zinc finger homeobox 3)と改名された。ZFHX3はAFP遺伝子の発現調節領域にあるATに富む(AT-rich)領域に結合してその活性を抑制する作用がある(非特許文献6、9)。最近、p53とTGF-βシグナル伝達因子とが複合体を形成してAFP遺伝子の発現を抑制するという新しいメカニズムが明らかになった(非特許文献10)。p53はZFHX3とタンパク質相互作用する因子であり、p53とZFHX3の複合体がp21waf1/cip1のプロモータを活性化する(非特許文献11)。これらの事実から、AFP遺伝子に関しても、p53、TGF-βシグナル伝達因子及びZFHX3が複合体形成しするタンパク質相互作用を通じて転写抑制をしている可能性がある。
【0007】
A-T患者以外では、成人の血中AFPが異常上昇する臨床状態は肝細胞癌(Hepatocellular carcinoma, HCC)やAFP産生胃癌の患者に認められる。その背景としてHCCの癌細胞でZFHX3発現量が著しく減少していることが明らかにされた。また、AFP産生胃癌の癌細胞ではZFHX3の発現がほとんど消失していることが示された(非特許文献12)。AFP産生胃癌は、AFPの異常な発現上昇と同時に、原発巣が小さい段階から高率に遠隔転移を起こす特徴を示し、極めて悪性度の高い癌である(非特許文献13、14)。癌の転移の問題は細胞接着性と深く関連する問題であり、ZFHX3の機能不全との関連が推察されている。
【0008】
A-T患者にとって最も深刻な問題は神経変性であり、神経変性は病態早期から進行する。A-T患者に神経変性が起こるメカニズムとしてDNAの二重らせん切断(double strand breaks, DSBs)の修復異常が想定されてきた。しかしA-Tと診断された症例の中には、臨床的な観察結果および定量的な神経学的検査結果から神経症状が軽微であるにも関わらず、遺伝子検査結果からはATM遺伝子に重大な中途欠損があって、ATM機能に著しい障害を持つことが予想される例がみつかる。それらの患者から採取された試料を用いた細胞機能テストではDNAの二重らせん切断に対するDNA修復反応の著しい損傷を確認できることから、典型的なATM症状を呈することを期待できる。もしも神経変性がDNA修復障害のみに由来するのであれば、当然著しい神経症状が出ると期待される。しかし、DNA修復反応が障害されていても、必ずしも神経変性の兆候が重篤になるとは限らない。この事実関係はA-T患者における神経変性を起こすメカニズムは単純にATMのDNA修復障害では説明できないことを示唆しているものであり、従来から知られているATMのDNA修復シグナル系とは独立した別のメカニズムが神経変性過程に関わっている可能性がある(非特許文献15)。
【0009】
ATMキナーゼの下流の標的として数百の因子が推定されているが、その一つとしてZFHX3が検出されている(非特許文献16)。ZFHX3がATMによってリン酸化を受ける機能的な意味は、ATMによってリン酸化されるp53の機能調節と類似している可能性がある。p53はATMによってリン酸化されることでタンパク質が安定化し転写因子としての機能を発揮するようになる。p53のリン酸化は分解を促進する因子であるMDM2とp53の結合を抑制する作用がある(非特許文献17、18)。ZFHX3もATMによってリン酸化されることでタンパク質として安定化している可能性がある。本発明者らは、A-T患者が示す様々な臨床症状の背景としてZFHX3の機能障害が関連していると考えた。最近の論文報告によって、ZFHX3の異常は川崎病の遺伝的背景因子であることが解明された。川崎病は小児急性熱性皮膚粘膜リンパ節症候群として知られる病気であり、心臓冠動脈瘤の合併症を引き起こす(非特許文献19)。この事実はA-T患者の臨床兆候の一つである毛細血管拡張症状と関連して、ZFHX3が血管の保護的役割を持っていることを示唆するものであり、ZFHX3に動脈瘤が発生することを防御する役割があることを示している。
【0010】
以上の考察を踏まえて本発明者らは、ZFHX3の機能とA-T患者が持つ多彩な臨床症状との密接な結びつきに着目し、数々の実験を行った。その結果、特に重要な成果として、レチノイン酸刺激がATMの活性化を誘導し、活性化したATMがCREBを活性化し、続いてCREBがZFHX3の遺伝子プロモータ上にあるCRE(cAMP Responsive Element)のコンセンサスDNA配列に結合し、標的遺伝子であるZFHX3遺伝子の転写を活性化するという、シグナル伝達経路を明らかにした。また、酸化ストレスに反応して神経細胞の細胞質内でATMが活性化する事実及びこのATMの活性化にPDGFRBの活性化が必須であることを解明した。これらの成果によって、A-Tの原因遺伝子であるATMの活性化に関するシグナル伝達経路の全体像が明らかになった。その結果、ATMの活性化においてZFHX3が重要な分子であり、ZFHX3が良好に作用する環境を形成することがA-Tの治療に有効であるとの知見がもたらされた。「ZFHX3が良好に作用する環境」を形成するための具体的な手段は、機能的ZFHX3(即ちリン酸化状態のZFHX3)の量を増大させること、或いは機能的ZFHX3の安定化(維持)である。前者は、例えば、本研究で使用したレチノイン酸によって達成可能である。一方、本発明者らの検討の結果、後者に関連して重要かつ興味深い実験結果が得られた。即ち、モデル神経細胞を用いた実験によって、免疫抑制剤として知られるタクロリムス(FK506)が神経細胞死を抑制できること、酸化ストレス下の神経細胞ではZFHX3の分断化が生ずること、タクロリムスの投与によってこのZFHX3の分断化を抑制できること、及びタクロリムスの投与によってZFHX3の脱リン酸化が抑制できること、が明らかとなった。ここで、タクロリムスがプロテインフォスファターゼ2B(略称PP2B。カルシニューリンとも呼ばれる)に対する阻害活性を示すことを踏まえて上記結果を考察すれば、酸化ストレス下の神経細胞ではZFHX3がPP2Bによる脱リン酸化を受けて分断化することがわかるとともに、タクロリムスによれば当該分断化の抑制を通して神経細胞死を抑制できるといえる。この知見は、タクロリムス自体が機能的ZFHX3の安定化という作用によってA-Tの治療/予防に有効であることを示すとともに、タクロリムスと同様の作用、即ち、ZFHX3を脱リン酸化する酵素(PP2B)に対する阻害活性を示す物質がA-Tの治療/予防に有効であること、更には、シグナル伝達経路においてPP2Bの上流に位置するカルパインの活性を阻害する物質にもA-Tに対する治療/予防効果を期待できることを示唆する。
【0011】
一方、カイニン酸刺激を負荷した動物の小脳深部核等の反応性を検定するという、本研究で使用した実験系は、上記の如き重要且つ有意義な事実の発見に寄与したことからも明らかな通り、小脳の変性を伴う疾患に有効な薬物をスクリーニングするためのツールとしては当然のこと、グルタミン酸作動性ニューロンでの神経過興奮に由来する酸化ストレスがもたらす各種神経変性に有効な薬物をスクリーニングするツールとしても、それ自体が大きな価値を有する。
【0012】
以下に示す本発明は主として上記成果に基づく。
[1]標的神経細胞内で機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を含む、毛細血管拡張性失調症治療薬。
[2]前記化合物が、機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質の発現増強作用を示す化合物である、[1]に記載の毛細血管拡張性失調症治療薬。
[3]前記化合物がレチノイン酸である、[2]に記載の毛細血管拡張性失調症治療薬。
[4]前記化合物がプロテインフォスファターゼ2B(PP2B)阻害剤である、[1]に記載の毛細血管拡張性失調症治療薬。
[5]前記プロテインフォスファターゼ2B(PP2B)阻害剤がタクロリムスである、[4]に記載の毛細血管拡張性失調症治療薬。
[6]前記化合物がカルパイン阻害剤である、[1]に記載の毛細血管拡張性失調症治療薬。
[7]機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を毛細血管拡張性失調症患者に対して治療上有効量、投与するステップを含む、毛細血管拡張性失調症の治療法。
[8]前記化合物がレチノイン酸又はタクロリムスである、[7]に記載の治療法。
[9]以下のステップ(1)〜(4)を含む、毛細血管拡張性失調症に起因又は付随する神経変性或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物のスクリーニング法:
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
[10]ステップ(3)において、小脳でのオートファジー又は神経変性が検定される、[9]に記載のスクリーニング法。
[11]ステップ(3)において、小脳深部核でのオートファジー又は神経変性が検定される、[9]に記載のスクリーニング法。
[12]
非ヒト哺乳動物が野生型動物である、[9]〜[11]のいずれか一項に記載のスクリーニング法。
[13]非ヒト哺乳動物がATM遺伝子欠損動物である、[9]〜[11]のいずれか一項に記載のスクリーニング法。
[14]非ヒト哺乳動物がマウスである、[9]〜[13]のいずれか一項に記載のスクリーニング法。
【図面の簡単な説明】
【0013】
【図1】レチノイン酸(RA)刺激を用いた実験の結果。レチノイン酸の刺激はATMキナーゼの活性化を介してCREB(cAMP Responsive Element)の活性化を誘導する。この活性化したCREBがZFHX3遺伝子プロモータ上流に存在するCREに結合することでZFHX3遺伝子が活性化する。(A) ヒトZFHX3ゲノムの遺伝子構造の概略図を示す。四角形の部分はエクソン(1〜10)をコードしている領域を示す。選択的スプライシング1によって2種類のmRNAが作り出される。長型mRNAをATBF1-A、短型mRANをATBF1-Bと呼ぶ。ZFHX3から転写されるATBF1-AとATBF1-Bの二種類のmRNAはそれぞれ2カ所の転写開始点(PAとPB)からの転写産物である。神経細胞で発現しているZFHX3は長型mRNAであるATBF1-Aタイプであるので、この転写制御メカニズムを解明するために転写開始点(PA)から5’上流の5.6-kbまでの領域に着目して転写制御メカニズムを詳細に解析した。(B) CREのコンセンサス配列は転写開始点(PA)の上流域約5.5-kbの位置に存在する。その位置を白抜き円で示した。ルシフェラーゼ解析で用いたDNA断片には以下の名称を付けた。5.6は転写開始点(PA)から5’上流側の全長5.6-kb (ゲノムからSalI-BamHで切り出されるDNA断片)を含むDNA配列である。5.6Δは5.6-kbから途中の3.8-kb (HindII-StuIで切り出されるDNA断片)を取り除いた、転写開始点の177-bpと連結した配列である。4.0は5.6-kbの上流側の1.6-kb部分を取り除いた部分配列である。0.42は転写開始点直近の0.42-kbの配列である。これらのDNA断片を使ったルシフェラーゼレポータ解析を実施し、少なくとも3回の独立した測定をし、有意性(P<0.01)をボンフェローニ補正マンホイットニー検定で評価した。
【図2】PDGFRB及びVEGFR-2に対する特異的阻害剤AG1433を用いた実験の結果。AG1433は酸化ストレスに反応するATMの活性化を抑制した。P19細胞(マウス奇形腫瘍由来の胎児性癌細胞株)を神経分化誘導刺激し、その5日目の細胞を神経細胞のモデル細胞として以下の実験を行った。(A) 過酸化水素水(100μM)で処理する方法で酸化ストレス(0, 5, 15, 30, 60分)を与えた時に観察されるATMキナーゼの活性化(Ser1981のリン酸化抗体)を検定した。(B)刺激後30分の過酸化水素水濃度を各種(1, 10, 100, 500μM)用意して、ATMキナーゼの活性化を調べた。尚、Xで示したレーンはX線照射(10Gy)後1時間のサンプルを用いてATMの活性化を調べた結果を示したものであり、ATMキナーゼ活性化のポジティブコントロールとなる。(C) 過酸化水素水(100μM)処理後30分の時点のATMの活性化量(レーン3〜7)を示している。AG1433はPDGFRBとVEGFR-2に対する特異的な阻害剤であり、その濃度依存性にATMの活性化が抑制された。AG1433を投与する際の溶媒としてDMSOを使用した。溶媒であるDMSOの効果を差し引くために、DMSOの単独投与(0.1%)の影響も検討した。その投与量はAD1433 (5μM)投与実験で使ったDMSO濃度に相当する(レーン3)。(D) 以上の実験を3回繰り返してpS-ATM(Ser1981がリン酸化した活性型ATM)量とATM量の相対比率を計算し、その相対比率の変化について有意検定を行い、結果を棒グラフに纏めた。(E) 10 GyのX-線照射後1時間のATMの活性化量(レーン3〜7)についてはAG1433を投与しても有意な変化が起こらなかった。この実験で使用したDMSOによる効果を差し引くために、DMSO単独投与(0.1%)の比較実験を行った(レーン3)。(F) 以上の実験を3回繰り返してpS-ATM(Ser1981がリン酸化した活性型ATM)量とATM量の比率を計算し、その相対比率の変化について有意検定を行って棒グラフとして示した。有意性(P<0.01)をボンフェローニ補正マンホイットニー検定で評価した。
【図3】ATMの活性化に関する実験の結果。PDGFRBの抑制及び/又はVEGFR-2シグナルの抑制は、DNA損傷ストレスではなく酸化ストレスに反応したATMの活性化(pS-ATM)を減弱する。培養5日目のP19由来神経細胞(レチノイン酸によって4日間、神経細胞へ分化誘導したもの)を、PDGFRB及びVEGFR-2の特異的阻害剤であるAG1433(5μM)で前処理し、その30分後に100μMの過酸化水素水又はX線(10Gy)で処理した。この処理から30分後に細胞をパラホルムアルデヒドで固定した。活性化ATM(pS-ATM、緑)、DAN二重らせん切断(γ-H2AX、赤)及びDNA(DAPI、青)を染色した。スケールバーは10μm。
【図4】カイニン酸刺激が小脳神経細胞へ及ぼす影響に関する実験の結果。カイニン酸刺激によって生ずる、小脳深部核神経細胞での血管形成。pS-ATM(茶色)の免疫組織化学。カイニン酸刺激に反応して細胞質内でATMが活性化する(B1,B2,B3)。対照的に、X線照射に反応してATMは核内で活性化する(C1,C2,C3)。マウスの脳切片をヘマトキシリン・エオジン染色した(A,B,C)。図中の番号は大脳皮質(1)、海馬(2)及び小脳深部核(DCN)(3)を示す。マウスをカイニン酸(30mg/kg)による処理(B)、又はX線照射(C)に供し、4時間後に屠殺した。続いて、大脳皮質(A1,B1,C1)、海馬(A2,B2,C2)及びDCN(A3,B3,C3)について脳切片を作製した。注目すべきことにDCNの神経細胞では血管形成を認める(B3、矢印)。ヘマトキシリン(青)で対比染色した。スケールバーは10μm。
【図5】ATMの活性化が起こる細胞内の特異的領域(細胞質/核)の鑑別結果を示す図。成体マウスの腹腔内にカイニン酸(A1, A2; 30 mg/kg)を注射投与し、脳神経の過剰興奮を誘発させた実験条件と、X線を照射(B1, B2; 10 Gy)してDNA二重らせん切断を誘発した実験条件を比較してATMの活性化を評価した。カイニン酸投与後4時間で神経細胞の細胞質に活性型ATM(pS-ATM)が有意に検出された(A1)。X線照射刺激後4時間の神経細胞の核に活性型ATM(pS-ATM)が有意に集積していた。X線照射群ではDNA損傷のマーカーであるγ-H2AXが核に強く検出されるが、カイニン酸投与細胞ではγ-H2AXはまったく検出されない。拡大率を示すバーは10μm。
【図6】カイニン酸刺激による細胞質内でのATMの活性化を示す図。カイニン酸刺激から5時間後にマウス大脳皮質の脳切片を固定した。同様にX線照射(10Gy)から1時間後にマウス大脳皮質の脳切片を固定した。X線照射の場合、活性化ATM(pS-ATM、緑)は核に局在するが、カイニン酸刺激の場合は細胞質に局在する。DAN二重らせん切断(γ-H2AX、赤)、DNA(DAPI、青)の染色像も示した。スケールバーは10μm。
【図7】PDGFRBの発現に関する実験の結果。PDGFRBは小脳深部核(DCN)の神経細胞に有意に強く発現している。(A) P19細胞を神経系細胞に分化誘導する処理過程の7日目(神経分化の最終段階)に至るとPDGFRBとZFHX3が共にもっとも強く発現する。分化途中の段階で観察できる発現量の有意差(P<0.01)はマンホイットニーのU検定で検定した。(B)ラット胎仔(E14)の脊髄では未分化神経前細胞群が増殖している領域においてDNA合成を示すBrdUの取り込みが観察できる(2i)。神経への分化が終了して、BrdUの取り込みが観察できない領域でZFHX3 (4ii)とPDGFRB(6ii)の発現が同時に認められる。拡大率を示すバーをそれぞれの図の中に示した。(C) Atmノックアウトモデルマウスとコントロールマウスの脳を正中線近傍で切断した標本の免疫組織化学検査。それぞれの切断部位は白色の破線で示し(1及び8)それらの切断面のヘマトキシリン・エオジン染色像(2及び9)を示した。小脳組織の拡大部分を抗GFAP抗体で染色し、ヘマトキシリン染色液で核を対比染色した(3及び10)。画像中にあるDCN領域を白色破線で囲んで示した。ZFHX3の発現量(4及び5; 11及び12)とPDGFRBの発現量(6及び7; 13及びと14)を解析した結果、Atmノックアウトモデルマウスの小脳のDCNの神経細胞で観察できるZFHX3とPDGFRBの発現量は、コントロールマウスの小脳DCNの神経細胞と比較すると、いずれの発現量も有意に減少していることが判明した。画像の拡大率を示すバーはパネル3と10では500μm、パネル6,11,13では50μm、パネル5, 7, 12, 14では20μmである。
【図8】DCNにおけるオートファジー関連タンパク質LC3の発現を示す図。マウスを4%パラホルムアルデヒドで灌流固定した。ホルマリン固定、パラフィン包埋組織から切り出した4μmの組織切片を使用した。組織切片を脱パラフィン・再水和処理した後、電子レンジを使用して2回、賦活処理した(98℃、10分間、10mMクエン酸緩衝液(pH6.3))。コントロールマウスの脳切片(1)、カイニン酸処理(20mg/kg、腹腔内注射)から4時間後のマウスの脳切片(2)、2回のカイニン酸処理(10mg/kg、腹腔内注射。24時間の間隔)の後、24時間後のマウスの脳切片(3)をウサギ抗LC3抗体(1000倍希釈:株式会社医学生物学研究所)に1時間反応させ、その後、ホースラディッシュペルオキシダーゼ標識抗ウサギIgG抗体(2次抗体)を反応させた。ジアミノベンジジンを用いて可視化した。ヘマトキシリンを用いて組織切片を対比染色した。スケールバーは50μm。
【図9】ATMキナーゼ活性化とZFHX3による転写制御が相互依存的に関わる細胞内シグナル伝達系の概念図。レチノイン酸の刺激で誘導されるATMの活性化によってCREBのSer133のリン酸化反応が進む。一方、X線照射によって誘導されるATMの活性化はDNAの二重らせん切断(DSBs, DNA double strand breaks)部分で起こり、CREBのThe100、Ser111及びSer121のリン酸化の反応が進むことになる。ATMの活性化がどのような原因によって誘導されるかに応じて下流のCREBのリン酸化部位に変化が生じ、その下流へのシグナル伝達に機能的な変化が起こる。レチノイン酸刺激に誘導されるCREBの活性化によって特異的にZFHX3の転写が活性化する。ZFHX3のプロモータの上流5.5-kbにはCRE(5’-TGACGTCA-3’)配列が存在し、そこにCREBが結合して転写を活性化していると考えられる。転写されたZFHX3 mRNAはZFHX3タンパク質に翻訳され、転写因子としてさまざまな標的遺伝子の発現制御に関わる。マイクロアレイによるZHFX3の標的遺伝子のスクリーニング結果から、細胞接着因子群(プロコラーゲン類、インテグリン類)及びPDGFRB(血小板由来増殖因子受容体β)遺伝子群が同定された。神経興奮によって引き起こされる代謝性酸化ストレスに対する自己防御シグナルとしてATMが細胞質内で活性化することは重要である。この自己防御シグナルを活性化するためにPDGFRBの活性化がその上流因子として必須であることが今回の研究で始めて明らかになった。細胞質内で活性化したATMのリン酸化シグナルはLKB1、AMPK、TSC2のカスケードに伝えられてmTORを抑制する制御系として作用する。活性型のmTORはオートファジー(ミトコンドリアなどの細胞内小器官の自己貪食機構)の抑制因子として知られており、mTORを抑制するATMからのシグナルカスケードはオートファジーを誘導するシグナルとして機能する。
【図10】神経細胞死に対するFK506(タクロリムス)の効果。FK506の投与によって神経細胞死を抑制することができる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスを与えるモデル神経細胞をレチノイン酸(RA)刺激(5μM、4日間処理)で前処理しておくと、カスパーゼ3の活性化が有意に上昇することが分かる(レーン3)。FK506処理をするとカスパーゼ3の活性化が抑制できる(レーン4)。この神経細胞モデル系でThapsigarginを作用させることは神経過興奮のモデルとして捉えることができることから、神経の過興奮で神経細胞死が誘導されるメカニズムとしてカスパーゼ3が活性化することが想定できる(レーン5)。この神経細胞死シグナルの誘導はFK506の投与によって抑制できる(レーン6)。尚、FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。Thapsgargin処理については、最終濃度2μMで1時間の処理とした。
【図11】ZFHX3の分断化に対するFK506(タクロリムス)の効果。FK506の投与によってZFHX3の分断化が抑制できる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスがZFHX3タンパク質に与える影響を調べるため、抗ATBF1抗体(AT-6)で免疫沈降したタンパク質をAT-6抗体でウエスタンブロット解析した。その結果、レチノイン酸(RA)刺激で神経分化誘導した細胞では、Thapsigargin処理によってZFHX3タンパク質の分断化が有意に強く起きた(レーン5)。未分化P19細胞に由来するZFHX3ではこのような変化は認められなかった。このZFHX3の分断化現象はFK506の投与で抑制できる(レーン6)。ZFHX3の分断化現象はThapsigargin処理をしないP19由来神経細胞内でも弱いながら観察でき(レーン3)、この分断化もFK506の投与で抑制できる(レーン4)。尚、FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。Thapsgargin処理については、最終濃度2μMで1時間の処理とした。
【図12】ZFHX3の脱リン酸化に対するFK506(タクロリムス)の効果。FK506の投与によってZFHX3の脱リン酸化が抑制できる。モデル神経細胞(P19細胞の神経分化誘導)に対して、Thapsigargin処理により細胞内のCa2+濃度を上昇させるストレスを与えた。このストレスがZFHX3のリン酸化に与える影響をPro-Q Diamond phosphoprotein stain kit (Invitrogen社、カタログ番号 P-33356)を用いて検定した。P19細胞の溶解液を抗ATBF1抗体(AT-6)で免疫沈降して得られたタンパク質をSDS-PAGEで展開し、上記の試薬に反応させた。レチノイン酸(RA)刺激(5μM、4日間処理)で前処理をして神経分化誘導した細胞ではZFHX3のリン酸化が有意に強くなっていることが分かる(レーン7, 8, 13, 14)。Thapsigargin処理によるCa2+ストレスが加わらなければ、FK506による処理の有無によってZFHX3のリン酸化レベルには大きな差が検出できないが(レーン7, 8)、Thapsigargin処理をしてCa2+ストレスを細胞に加えた系では、FK506による処理細胞群でZFHX3のリン酸化状態が有意に高く保たれている(レーン13, 14)。図中の矢印は404-kDaの全長ZFHX3がリン酸化していることを示している。尚、レーン1、4、6、10、12、15は分子量マーカーである。
【図13】ZFHX3タンパク質量の検定結果。図12の解析に使ったタンパク質をSYPRO Ruby staining(Invitrogen社、カタログ番号 No.S12001)で検定した結果。いずれの条件においても、矢印で示した404-kDaの位置に等量のタンパク質量として全長ZFHX3タンパク質が検出されている(レーン2, 3, 7, 8, 13, 14)。尚、レーン1、4、6、10、12、15は分子量マーカーである。
【発明を実施するための形態】
【0014】
1.毛細血管拡張性失調症用の治療薬
本発明の第1の局面は毛細血管拡張性失調症(以下、「A-T」を略称する)の治療薬に関する。本発明における「治療薬」とは、標的の疾病ないし病態であるA-Tに対する治療的又は予防的効果を示す医薬のことをいう。治療的効果には、A-Tに特徴的な症状又は随伴症状を緩和すること(軽症化)、症状の悪化を阻止ないし遅延すること等が含まれる。後者については、重症化を予防するという点において予防的効果の一つと捉えることができる。このように、治療的効果と予防的効果は一部において重複する概念であることから、明確に区別して捉えることは困難であり、またそうすることの実益は少ない。尚、予防的効果の典型的なものは、A-Tに特徴的な症状の再発を阻止ないし遅延することである。尚、A-Tに対して何らかの治療的効果又は予防的効果、或いはこの両者を示す限り、A-T治療薬に該当する。
【0015】
本発明の治療薬は有効成分として、標的神経細胞内で機能的ジンクフィンガーホメオボックス3(以下、慣例に従い「ZFHX3」と略称する)タンパク質を増大又は安定化する化合物を含む。換言すれば、本発明の治療薬における有効成分は、標的細胞内において機能的ZFHX3タンパク質を増大させる効果、或いは標的細胞内において機能的ZFHX3タンパク質を安定化させる効果を発揮する。本発明における用語「標的神経細胞」とは、本発明の治療薬の標的となる神経細胞である。本発明の治療薬は神経細胞内でその作用を発揮する。典型的には、小脳に存在する神経細胞(小脳深部核の神経細胞やプルキンエ細胞など)が標的となり、A-Tに特徴的な小脳神経変性に対する抑制的ないし阻害的治療効果をもたらす。
【0016】
本発明における用語「機能的ZFHX3」とは、その生理機能を発揮できる状態のZFHX3タンパク質をいう。生理的条件下ではZFHX3タンパク質はリン酸化状態にあり、その機能を発揮する。一方、ZFHX3タンパク質の脱リン酸化はZFHX3タンパク質の分断化・分解を引き起こす。そこで本明細書では、用語「機能的ZFHX3タンパク質」を用語「リン酸化状態のZFHX3タンパク質」と交換可能に使用する。
【0017】
ZFHX3はAFP遺伝子プロモータ上流の発現調節領域にあるA(アデニン)とT(チミン)に富む(AT-rich)DNA配列に結合するDNA結合タンパク質であり、ATBF1と呼ばれていた。ATBF1については、異なったプロモータの使用及び選択的スプライシングにより形成される、分子量の異なった2つのアイソフォームATBF1-A(404kDa、アミノ酸配列を配列番号1に示す。また、ATBF1-Aをコードする塩基配列(GenBank accession number:L32832を参照)を配列番号2に示す。)とATBF1-B(306kDa、アミノ酸配列を配列番号3に示す。また、ATBF1-Bをコードする塩基配列(GenBank accession number:L32833を参照)を配列番号4に示す。)の存在が知られている。
【0018】
「機能的ZFHX3タンパク質を増大させる効果」とは、直接又は間接的にZFHX3遺伝子を発現制御し、機能的ZFHX3タンパク質の発現量の増大をもたらす効果をいう。機能的ZFHX3タンパク質を増大させる効果を発揮する化合物として、ZFHX3タンパク質の発現を増強する化合物を用いることができる。該当する化合物の具体例としてレチノイン酸を挙げることができる。レチノイン酸はATM及びCREBを介してZFHX3遺伝子の転写活性を高める。ZFHX3遺伝子に対するこのような作用を示す化合物である限り、この例に限定することなく本発明の治療薬の有効成分として使用可能である。
【0019】
一方、「機能的ZFHX3タンパク質を安定化させる効果」とは、機能的ZFHX3タンパク質のリン酸化状態を維持する効果をいう。リン酸化状態が維持されることによってZFHX3は分断化・分解を回避でき、その本来の生理機能を発揮できる。ZFHX3の脱リン酸化状態の維持には、ZFHX3を標的とした脱リン酸化酵素の活性阻害が有効である。そこで、本発明の治療薬の有効成分としてプロテインフォスファターゼ2B(以下、「PP2B」と略称する)阻害剤を使用するとよい。PP2B阻害剤の具体例としてタクロリムス(別名FK506、CAS登録番号104987-11-3)、シクロスポリンAを挙げることができる。タクロリムスは土壌細菌より単離された化合物であり、移植時の免疫抑制、アトピー性皮膚炎、重症筋無力症、関節リウマチ、ループス腎炎等に使用されている。プログラフ(登録商標)(タクロリムス水和物)、グラセプター(登録商標)(タクロリムス水和物)、プロトピック(登録商標)(タクロリムス水和物)等の商品名でタクロリムス製剤がアステラス製薬株式会社によって販売されている。また、シクロスポリンはタクロリムスと類似の薬理作用を持ち、免疫拒絶反応の抑制や自己免疫疾患の治療に使用されている。ネオーラル(登録商標)、サンディミュン(登録商標)の商品名でシクロスポリン製剤がノバルティスファーマ株式会社によって販売されている。
【0020】
PP2B阻害剤に代えて、シグナル伝達経路においてPP2Bの上流に位置するカルパインに対する阻害剤を用いることにしてもよい。カルパイン阻害剤の例はカルパスタチン、ロイペプチン、E-64、E-64d(E-64の誘導体)、カルペプチンである。
【0021】
ZFHX3の脱リン酸化を抑制し得る限りにおいて、以上の例に限定することなく本発明の治療薬の有効成分として使用可能である。尚、後述の実施例に示したアッセイ系或いは参考文献に記載されるアッセイ系を利用すれば、公知の化合物群或いは新規に合成又は単離した化合物の中から、機能的ZFHX3タンパク質を増大又は安定化する化合物をルーチン的操作によって同定することが可能である。
【0022】
後述の実施例に示す通り、本発明者らは、神経細胞においてA-Tの原因遺伝子とされるATMとZFHX3が密接な関わりをもつことを見出した。詳しくは、ZFHX3が血小板由来成長因子受容体β(以下、「PDGFRB」と略称する)の活性化を介してATMの活性化に関与し、オートファジーの誘導ないし活性化が生ずるという、酸化ストレス下でのシグナル伝達経路の存在を明らかにした。理論に拘泥する訳ではないが、標的神経細胞における機能的ZFHX3タンパク質の増大は、本発明者らが見出したシグナル経路を活性化(正に制御)し、オートファジーの誘導ないし活性化を引き起こす。機能的ZFHX3タンパク質の安定化も同様にオートファジーの誘導ないし活性化に寄与する。
【0023】
本発明の治療薬の製剤化は常法で行うことができる。製剤化する場合には製剤上許容される他の成分(例えば、担体、賦形剤、崩壊剤、緩衝剤、乳化剤、懸濁剤、無痛化剤、安定剤、保存剤、防腐剤、生理食塩水など)を含有させることができる。賦形剤としては乳糖、デンプン、ソルビトール、D-マンニトール、白糖等を用いることができる。崩壊剤としてはデンプン、カルボキシメチルセルロース、炭酸カルシウム等を用いることができる。緩衝剤としてはリン酸塩、クエン酸塩、酢酸塩等を用いることができる。乳化剤としてはアラビアゴム、アルギン酸ナトリウム、トラガント等を用いることができる。懸濁剤としてはモノステアリン酸グリセリン、モノステアリン酸アルミニウム、メチルセルロース、カルボキシメチルセルロース、ヒドロキシメチルセルロース、ラウリル硫酸ナトリウム等を用いることができる。無痛化剤としてはベンジルアルコール、クロロブタノール、ソルビトール等を用いることができる。安定剤としてはプロピレングリコール、アスコルビン酸等を用いることができる。保存剤としてはフェノール、塩化ベンザルコニウム、ベンジルアルコール、クロロブタノール、メチルパラベン等を用いることができる。防腐剤としては塩化ベンザルコニウム、パラオキシ安息香酸、クロロブタノール等と用いることができる。
【0024】
製剤化する場合の剤形も特に限定されない。剤形の例は錠剤、散剤、細粒剤、顆粒剤、カプセル剤、シロップ剤、注射剤及び吸入剤である。本発明の治療薬には、期待される治療効果(又は予防効果)を得るために必要な量(即ち治療上有効量)の有効成分が含有される。本発明の治療薬中の有効成分量は一般に剤形によって異なるが、所望の投与量を達成できるように有効成分量を例えば約0.1重量%〜約95重量%の範囲内で設定する。
【0025】
本発明の治療薬はその剤形に応じて経口投与又は非経口投与(静脈内、動脈内、皮下、皮内、筋肉内、又は腹腔内注射など)によって患者に適用される。これらの投与経路は互いに排他的なものではなく、任意に選択される二つ以上を併用することもできる(例えば、経口投与と同時に又は所定時間経過後に静脈注射等を行う等)。全身投与によらず、局所投与することにしてもよい。局所投与として、目的の組織への直接注入又は塗布を例示することができる。
【0026】
本発明の治療薬の投与量及び投与スケジュールは、期待される治療効果が得られるように設定される。治療上有効な投与量の設定においては一般に患者の症状、年齢、性別、及び体重などが考慮される。尚、当業者であればこれらの事項を考慮して適当な投与量を設定することが可能である。例えば、成人(体重約60kg)を対象として一日当たりの有効成分量が約0.01mg〜約100mg、好ましくは約0.2mg〜約10mgとなるよう投与量を設定することができる。投与スケジュールとしては例えば1日1回〜数回、2日に1回、或いは3日に1回などを採用できる。投与スケジュールの作成においては、患者の症状や有効成分の効果持続時間などを考慮することができる。
【0027】
以上の記述から明らかな通り、本出願は、機能的ZFHX3タンパク質を増大又は安定化する化合物をA-T患者に対して治療上有効量、投与するステップを含む、A-Tの治療法も提供する。本発明の治療法によれば、A-T患者の小脳神経細胞内における機能的ZFHX3タンパク質が増大及び/又は安定化し、小脳での神経変性が抑制される。機能的ZFHX3タンパク質を増大する化合物としては例えばレチノイン酸が採用される。また、機能的ZFHX3タンパク質を安定化する化合物としては、PP2B阻害剤やカルパイン阻害剤等を使用できる。PP2B阻害剤の具体例はタクロリムスである。
【0028】
2.神経変性の予防又は治療に有効な化合物のスクリーニング法
本発明の第2の局面は、A-Tに起因又は付随する神経変性、或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物をスクリーニングする方法に関する。本発明のスクリーニング法で選抜される化合物は、それ自体又は必要な改変を加えた上で、A-T等の神経変性疾患に対する治療薬の有効成分として、或いはA-T等の神経変性疾患に対する治療薬を開発するためのリード化合物等として使用される。A-Tに起因又は付随するものであるか、又は神経過興奮に由来した酸化ストレスによるものである限り、本発明のスクリーニング法が標的とする神経変性に該当する。好ましい一態様では、A-Tの治療に有効な化合物を見出す手段として本発明のスクリーニング法は構成される。
【0029】
本発明のスクリーニング法では以下の(1)〜(4)のステップを行う。
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
【0030】
ステップ(1)では複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分ける。同種同系非ヒト哺乳動物としてマウス、ラット、モルモット、ハムスターを例示することができる。好ましくは、入手及び取り扱いが比較的容易なマウス又はラットを使用する。試験群及び対照群に含まれる個体数は特に限定されない。一般に使用する個体数が多くなるほど信頼性の高い結果が得られるが、多数の個体を同時に取り扱うことは使用する個体の確保や操作(飼育を含む)の面で困難を伴う。そこで例えば各群に含まれる個体数を1〜50、好ましくは2〜30、さらに好ましくは3〜20とする。通常は試験群と対照群の個体数を等しくする。
【0031】
本発明の一態様では、野生型動物を用いて本発明のスクリーニング法を実施する。ここでの「野生型」とはATM遺伝子に関して野生型であること、即ち、正常なATM遺伝子を保有していることを意味する。慣例に従い、当該野生型動物をATM(+/+)と表現することができる。他の態様として、ATM遺伝子欠損動物(ATMノックアウト動物)を用いたスクリーニング法が提供される。この態様はA-Tの原因・病態を反映させた動物を用いたものであり、A-Tに対する有効性や特異性が一層高い化合物の同定に有利である。ATM遺伝子欠損動物はホモ型でもヘテロ型でもよい。A-Tに対する特異性が高い化合物を同定するという目的の下では、ホモ型のATM遺伝子欠損動物を採用することが特に好ましい。尚、慣例に従い、ホモ型ATM遺伝子欠損動物及びヘテロ型ATM遺伝子欠損動物はそれぞれATM(-/-)及びATM(+/-)と表現することができる。
【0032】
ATM遺伝子欠損動物は、例えば、ジーンターゲティングを利用してATM遺伝子に点変異を導入すること又は点変異を含む変異型ATM遺伝子を導入することによって作製できる。ジーンターゲティングは遺伝子の相同組換えを利用してゲノムに改変を加える技術である。ジーンターゲティングを利用すれば、特定の遺伝子が欠損した動物(ノックアウト動物)を作製することができる。ジーンターゲティングを利用したノックアウト動物の作製法は、マウスの場合を例にとれば、大別して(a)ターゲティングベクターの構築、(b)ES細胞へのターゲティングベクターの導入及び相同組換え体の同定、(c)ブラストシストへの相同組み換え体の注入、(d)仮親の子宮内への胚移植及び出産(キメラ仔マウス)、(e)キメラ仔マウスと野生型マウスの交配(F1世代であるヘテロ接合体マウスの産出)、及び(f)F1世代同士の交配(F2世代であるホモ接合体マウスの産出)からなる。ラットなど、他の齧歯類を用いた場合も同様の手順で遺伝子改変動物を得ることができる。尚、ジーンターゲティングによる遺伝子改変動物の作製法については例えばJoyner, A. L.:Gene Targeting (IRL press)、Capeccchi, M. R., Science, 244, 1288-1292 (1989)、Hua Gu, et al., Science, 265, 103-106 (1994)、McHugh, T. J. et al., Cell, 87, 1339-1349 (1996)、Shibata, H. et al., Science, 278, 120-123 (1997)等が参考になる。
【0033】
ステップ(2)では試験群に対してカイニン酸を投与し、併せて被験物質を投与する。カイニン酸及び被験物質の投与態様は特に限定されないが、カイニン酸については例えば腹腔内注射、静脈内注射、脳質内投与等によって投与するとよい。操作の簡便性と安全性の点から、腹腔内注射による投与が特に好ましい。神経興奮に由来する神経障害ストレスを生じさせるためにカイニン酸が投与される。当該目的を達成できる限りにおいて、カイニン酸の投与量や投与回数は特に問わない。カイニン酸の投与回数及び投与量は使用する動物の種、体重等を考慮して設定することができる。投与回数の例は1〜5回である。また、投与量の一例を示せば、マウスを使用した場合において短期間投与で30mg/kg、1回/1日で3日まで、長期間投与で30mg/kg、2日ごと2ヶ月間まで、である。尚、適切な投与回数、投与量は予備実験を通して決定することができる。
【0034】
試験群全体に同量のカイニン酸を投与するのではなく、投与量の異なる動物個体を用意することにしてもよい(例えばカイニン酸の投与量が基準量の個体、2倍量の個体、5倍量の個体を用意する)。この態様の場合、カイニン酸の投与量(即ちカイニン酸刺激のレベル)を考慮しつつ、後述のステップ(3)の検定及びステップ(4)の判定が行われる。
【0035】
被験物質の投与にあたっては、例えば被験物質を含む餌又は飲料水を用意し、これを摂取させる。或いは、被験物質又は被験物質を含む溶液を用意し、これを経口、経鼻、経気管、注射(静脈内、動脈内、皮下、筋肉内、腹腔内等)によって投与する。被験物質を複数回投与することにしてもよい。その場合には各回の投与量は同一であっても異なっていても良い。継続的に投与することにしてもよい。被験物質は、カイニン酸の投与の前、カイニン酸の投与と同時、又はカイニン酸の投与の後に投与される。被験物質を複数回投与する場合についても、各回の投与の時期とカイニン酸の投与の時期との関係に特段の制約はない。例えば、カイニン酸の投与後、被験物質を所定の間隔をおいて複数回投与する。尚、対照群については被験物質を投与しないこと以外は同一の条件下で処理・飼育する。
【0036】
被験物質としては様々な分子サイズの有機化合物又は無機化合物を用いることができる。有機化合物の例として、核酸、ペプチド、タンパク質、脂質(単純脂質、複合脂質(ホスホグリセリド、スフィンゴ脂質、グリコシルグリセリド、セレブロシド等)、プロスタグランジン、イソプレノイド、テルペン、ステロイド、ポリフェノール、カテキン、ビタミン(B1、B2、B3、B5、B6、B7、B9、B12、C、A、D、E等)を例示できる。被験物質は天然物由来であっても、或いは合成によるものであってもよい。後者の場合には例えばコンビナトリアル合成の手法を利用して効率的なスクリーニング系を構築することができる。尚、植物抽出液、細胞抽出液、培養上清などを被験物質として用いてもよい。また、既存の薬剤を被験物質としてもよい。2種類以上の被験物質を同時に添加することにより、被験物質間の相互作用、相乗作用などを調べることにしてもよい。
【0037】
ステップ(3)では、ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定する。このように本発明では、脳内でのオートファジー又は神経変性(例えば小脳におけるプルキンエ細胞や小脳深部核神経細胞等の変性・脱落)が検定される。オートファジーと神経変性の両者を検定することにしてもよい。検定に供される脳部位の具体例は、小脳、大脳皮質、海馬である。好ましい一態様では小脳を検定に供する。また、カイニン酸による刺激に対して小脳深部核が顕著な反応性を示したという知見を考慮し(後述の実施例を参照)、好ましくは、小脳深部核に関してオートファジー又は神経変性の検定を行う。ここで「オートファジー」とは、プロテアソーム系と並ぶ主要な細胞内分解機構である。オートファジーは生体の恒常性維持に関与する。また、感染防御や細胞死、老化、癌化、神経変性などにもオートファジーが関与していることが明らかになってきている。一方、「神経変性」は、神経細胞の変性・脱落と同義である。
【0038】
オートファジーを検定するためには、例えばオートファジー関連因子の量、ATMの活性化量又は空胞形成の程度を検出すればよい。これらの検出対象はいずれもオートファジーのマーカーとなり、オートファジーの促進ないし誘導を反映してその量ないし程度が増大する。オートファジー関連因子として微小管関連タンパク質軽鎖3(LC3)、GABAA受容体結合タンパク質(GABARAP)及びはゴルジ関連ATPアーゼエンハンサー(GATE-16)を例示することができる。検出対象に応じて、免疫組織化学、ウエスタンブロット解析が利用される。尚、ホモ型のATM遺伝子欠損動物を用いて本発明のスクリーニング法を構築した場合には、ATM遺伝子の発現が見られないことから、オートファジー関連因子の量又は空胞形成の程度を検出することになる。
【0039】
神経変性の検定には、例えばTUNEL染色や活性型カスペース3(Caspase)染色等の免疫組織化学を利用できる。TUNEL染色及び活性型カスペース3(Caspase)染色については専用のキットが市販されている。当該キットを利用すれば神経変性を定量的に検定・評価することも可能である。
【0040】
ステップ(4)では、試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定する。本発明では、判定の際、「オートファジーの促進ないし誘導を認めること(指標1)」、又は「神経変性の抑制を認めること(指標2)」を、被験物質の有効性の指標とする。換言すれば、オートファジーの促進ないし誘導の程度が対照群よりも試験群で高いとき(指標1に基づく判定)又は神経変性の抑制の程度が対象群よりも試験群で高いとき(指標2に基づく判定)、被験物質が有効であると判定する。ステップ(3)において、脳内でのオートファジーを検定することにした場合には指標1が採用される。他方、ステップ(3)において、脳内での神経変性を検定することにした場合には指標2が採用される。
【0041】
有効性を認めた複数の被験化合物を用いて再度ステップ(1)〜(4)を行い、有効性の高い物質の絞り込みを行うことにしてもよい。本発明のスクリーニング法によって選択された物質が十分な薬効を有する場合には、当該物質をそのまま治療薬等の有効成分として使用することができる。一方で十分な薬効を有しない場合には化学的修飾などの改変を施してその薬効を高めた上で、治療薬等の有効成分として使用することができる。勿論、十分な薬効を有する場合であっても、更なる薬効の増大を目的として同様の改変を施してもよい。
【0042】
ところで、これまでにもATM遺伝子欠損マウス(ATM(-/-)マウス)を用いて神経変性を誘導する実験・研究が行われている。但し、従来の方法では、X線照射によってDNA損傷刺激を与え、神経変性を誘導している。このような誘導法は、神経興奮という神経本来の機能に付随する神経変性メカニズムとは無関係な刺激であり、治療薬の開発には結びつかない。換言すれば、ATM遺伝子欠損マウスを用いた従来のアッセイ系は治療薬の開発に有用とはいえない。対照的に、本発明で採用するカイニン酸刺激は、DNA損傷と独立した(即ち、DNA損傷とは関係のない)本来の「神経興奮」に由来する神経障害ストレスであるといえることから、神経変性の評価系として、治療薬の開発などに有用である。特に、小脳機能に特化した神経変性の評価系として、小脳での神経変性を伴う疾病に対する治療薬の開発などにおいて有用である。このように本発明のスクリーニング法は有用性及び実用性の点で従来の技術とは比較にならないほど優れたものである。尚、カイニン酸刺激が海馬神経に神経障害を与えることは既知であり(Science 262:689-695, 1993)、それ自体は新しいことではない。本発明において重要なことは、カイニン酸が小脳深部核(DCN)に存在するニューロンの過剰興奮を引き起こすという、カイニン酸刺激に対する小脳の反応を明らかにしたことであり、当該成果があったからこそ、有用性及び実用性に優れた本発明のスクリーニング法を完成することができた。小脳の出力系のニューロンに着目することで完成し得た本発明のスクリーニング法は、小脳機能に特化した神経変性の定性的或いは定量的な評価に特に有用であり、小脳での神経変性の抑制ないし阻害に有効な物質の探索・同定を可能とする。
【実施例】
【0043】
ZFHX3の機能とA-T患者が持つ多彩な臨床症状との密接な結びつきに着目し、ZFHX3の発現制御及びその標的遺伝子群に焦点をあてた網羅的な解析を行った。
【0044】
1.方法
(1)細胞培養
P19マウス胎児性癌細胞株をα-MEMで維持した(Rudnicki and McBurney, 1987)。神経細胞への分化を誘導するために、P19細胞を凝集体の状態で、0.5μMのオールトランス型レチノイン酸(RA)(Sigma社)及び10%ウシ胎仔血清(FBS)を添加した培養液で4日間培養した。続いて、1%のFBS含有α-MEM(RAは含まない)を培養液として、0.01% ポリ-L-リジン(P8920, Sigma社)をコートしたスライドガラス上で3時間、そして1μg/mlウシ胎仔血漿フィブロネクチン(Invitrogen社)をコートしたスライドガラス上で終夜、室温にて細胞を培養した。必要な場合には、ATMキナーゼ阻害剤のKU-55933(2-morpholin-4-yl-6-thianthren-1-yl-pyran-4-one; 2-morpholino-6-(thianthren-1-yl)-4H-pyran-4-one (Calbiochem社))(10 μM)を毎日培養液に添加し、そして、PDGFRB及びVEGFR-2の特異的阻害剤であるAG1433(2-(3,4-dihydroxyphenyl)-6,7-dimethylquinoxaline(Calbiochem社))を過酸化水素水の添加30分前に所定の濃度(0.05, 0.5, 1.0, 5.0μM)で添加した。
【0045】
(2)マウス
野生型及びATMノックアウトマウス(ハーバード・メディカル・スクールのP. Leder氏より供与)を周囲条件下(23℃、湿度55%)、所定の明暗サイクルで飼育した。ゲノムDNAを尾先端より採取し、PCRによってAtm遺伝子を増幅させた。PCRには以下のプライマーを使用した。
KO-1F: 5'-TGGTCAGTGTAACAGTCATTGTGC-3'(配列番号5)
KO-1R: 5'-AAGGTTGTAGATAGGTCAGCATTG-3'(配列番号6)
KO-2R: 5'-AACGAGATCAGCAGCCTCTGTTCC-3'(配列番号7)
【0046】
プライマーKO-1Fはエクソン34の220bp上流(イントロン34)に位置し、プライマーKO-1Rはエクソン34内の123bpに位置し、プライマーKO-2RはPGKneoのポリA領域内の87bpに位置する。これらのプライマーを使用した場合のPCR産物の長さは、野生型マウスの場合は342bpであり、Atm(-/-)マウスの場合は406bpとなる。カイニン酸(2-carboxy-3-carboxymethyl-4-isopropenyl-pyrrolidine (Enzo Life Science社))をオスDDYマウスに投与した(30 mg/kg、腹腔内投与(i.p.))。4%パラホルムアルデヒドを使用した灌流固定後に3月齢のマウスから脳全体を摘出した。妊娠ラット(Std Wister ST)にBrdU(5-bromo-2'-deoxy-uridine)(50mg/kg)を3時間投与し、E14.5の胚を標識化した。続いて胚を摘出し、その脳を4%パラホルムアルデヒドで固定した後、パラフィン包埋した。
【0047】
(3)X線照射
X線装置CAX-210(株式会社中部メディカル)を用い、細胞、マウスにX線(10 Gy)を照射した(210 kV、10 mA、4分45秒、銅シールド)。
【0048】
(4)トランスフェクション及びプラスミド
一連のルシフェラーゼリポータープラスミドを構築するため、SalI及びBamHIを使用し、様々な長さのヒトZFHX3プロモータ領域をpBluescriptIIKS(+)(Agilent Technologies社、Stratagene社製品)にクローニングした。ZFHX3プロモータ領域を含むBssHII/BamHI断片をpGV-B基本ベクター(東洋ビーネット株式会社)のMluIサイトとBglIIサイトの間、ルシフェラーゼ遺伝子の5'プライム位置にサブクローニングした。以下のプライマーセットを用いたPCRによる部位特異的変異導入法により、1.0 kbpのZFHX3プロモータ内のCREサイトに変異を導入した。
pGV-F(pGV-B内ルシフェラーゼ遺伝子の5'隣接配列を含むフォワードプライマー): 5'-CAATGTATCTTATGGTACTG-3'(配列番号8)
mCRE-R(CREサイトへの変異導入用のリバースプライマー): 5'-CGGAAATGACCACAGCAAAG-3'(配列番号9)
mCRE-F(CREサイトへの変異導入用のフォワードプライマー):5'-CTTTGCTGTGGTCATTTCCG-3'(配列番号10)
pGV-R1(pGV-B内ルシフェラーゼ遺伝子の最初のATGコドンを含むリバースプライマー): 5'-CTTTATGTTTTTGGCGTCTTCC-3'(配列番号11)
pGV-R2(pGV-B内ルシフェラーゼ遺伝子のpGV-R1プライマーよりさらに35bp下流に位置するリバースプライマー):5'-CCATCCTCTAGAGGATAGAATG-3'(配列番号12)
【0049】
pGV-F及びmCRE-Rを使用したPCR産物とmCRE-F及びpVG-R2を使用したPCR産物を得た。一部で重複する、これら一次産物を一緒に変性させ、pGV-F及びpGVR1を用いて再び増幅させた。その結果得られたPCR産物をMluI及びHindIIIで処理した後、pGV-B基本ベクターの同一クローニングサイトにサブクローニングした。CREBドミナントネガティブ発現ベクターpCMVCREB133はClontech社(カタログ番号631925)から購入した。
【0050】
(5)免疫組織化学
4%パラホルムアルデヒド(PFA)固定・パラフィン包埋組織から切り出した4μm断片を使用した。組織断片を脱パラフィン及び再水和処理した後、クエン酸バッファー(0.01 M、pH 6.0)内で加熱処理した(ZFHX3免疫染色用では圧力釜使用、110℃。ATM免疫染色用では電子レンジ使用、98℃)。PDFGRBの免疫染色については、熱賦活処理を行わなかった。染色は室温で行った。内在性ペルオキシダーゼ活性を阻害するため、全ての組織断片を0.3% 過酸化水素及び1.0%アジ化ナトリウムを含むメタノールでインキュベートした。続いて、一部の組織断片について、マウス抗ATM Ser1981リン酸部位(pSATM)モノクローナル抗体(1200倍希釈、7C10D8; Rockland社)とともにインキュベートした後、ホースラディッシュペルオキシダーゼ(HRP)標識抗マウスIgG抗体(Envision labeled polymer,Dako Cytomation社)とともにインキュベートした。同様に、一部の組織断片については、ウサギ抗ZFHX3ポリクローナル抗体(50倍希釈、D1-120; 株式会社医学生物学研究所)とともにインキュベートした後、HRP標識抗ウサギIgG抗体(Envision labeled polymer, Dako Cytomation社)とともにインキュベートした。また、一部の組織断片については、ヤギ抗マウスPDGFRBポリクローナル抗体(100倍希釈、R&D Systems社)とともにインキュベートした後、HRP結合抗ヤギIgG抗体(MAX-PO (G),株式会社ニチレイ)とともにインキュベートした。インキュベート条件は、一次抗体及び二次抗体いずれについても、室温、1時間とした。免疫反応産物をジアミノベンジジンで可視化した。BrdU標識及び検出にはロッシュ社のキットを用いた。組織断片をヘマトキシリンで対比染色した。
【0051】
(6)免疫細胞化学
4% PFAを含むPBSに細胞を室温にて20分間浸漬し、固定した。その後、0.25% Triton-Xを含むPBSで洗浄し、10%正常ヤギ血清でブロッキングした。続いて、細胞を室温にて1時間、一次抗体(ウサギ抗ZFHX3ポリクローナル抗体(100倍希釈)、マウス抗β-チューブリンアイソタイプIIIモノクローナル抗体(500倍希釈、3D10; Sigma社)、抗ATM Ser1981リン酸化サイト(pS-ATM)モノクローナル抗体(1:100希釈、10H11.E12;Rockland社)、ウサギ抗γH2AXポリクローナル抗体(1:100希釈、BETHYL社))と反応させた。0.25% Triton-Xを含むPBSで3回洗浄した後、二次抗体(マウスモノクローナル抗体用にAlexa-Fluor-488標識ヤギ抗マウス抗体、ウサギポリクローナル抗体用にAlexa-Fluor-546標識ヤギ抗ウサギ抗体)を1時間反応させ、細胞を可視化した。その後、PBSで2回洗浄した後、2.0μg/ml DAPI(1 mg/ml溶液を500倍希釈、和光純薬株式会社)で5分間、核染色した。
【0052】
(7)ウエスタンブロット
細胞を洗浄バッファー(10 mM 0.1 Mリン酸バッファー、250 mMシュークロース及び50 mM NaFを含む)で洗浄し、TNEバッファー(20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 2 mM EDTA, 1% Nonident-P40, 50 mM NaF)を用いて全細胞ライセートを調製した。タンパク質を15分間氷上でインキュベートした後、15,000 g、4℃で30分間、遠心処理した。上清を回収し、-80℃で保存した。ブラッドフォード法(Bio-Rad社)にて全タンパク質を定量した。タンパク質の検出のため、サンプル中のタンパク質を5〜20%の濃度勾配を有するポリアクリルアミドゲルで分離し、PVDF膜(Millipore社)に転写した。次に、転写後の膜を3% BSAを含むトリス緩衝液(0.05% Tween 20含有:TBS-T)で1時間ブロッキングした後、TBS-Tで洗浄した。続いて、室温にて1時間、一次抗体(マウス抗α-チューブリンモノクローナル抗体(8000倍希釈、B-5-1-2; Sigma社)、ウサギ抗ZFHX3抗体(2000倍希釈、AT-6; 株式会社医学生物学研究所)、マウス抗HAタグモノクローナル抗体(500倍希釈、5D8; 株式会社医学生物学研究所)、マウス抗ATMモノクローナル抗体(200倍希釈、5C2; Santa Cruz社)、マウス抗pS-ATMモノクローナル抗体(1000倍希釈、10H11.E12; Rockland社)、ヤギ抗PDGFRBポリクローナル抗体(1000倍希釈、R&D Systems社)、ウサギ抗カスパーゼ3ポリクローナル抗体(1000倍希釈、AAP-113; Assay Designs社))と反応させた。TBS-Tで洗浄した後、ペルオキシダーゼ標識二次抗体(マウスモノクローナル抗体用に抗マウスIgG抗体(10,000倍希釈、抗マウスIgG (H+L chain)-HRP; 株式会社医学生物学研究所)、ウサギポリクローナル抗体用に抗ウサギIgG抗体(5000倍希釈、抗ウサギIgG(H+L chain)-HRP, 株式会社医学生物学研究所)、ヤギポリクローナル抗体用に抗ヤギIgG抗体(5000倍希釈、抗ヤギIgG (H+L chain)-HRP;株式会社医学生物学研究所))を室温にて1時間、反応させた。TBS-Tで洗浄後、免疫反応をAmersham ECL Plus western blotting detection reagents(GE Healthcare社)で可視化した。
【0053】
(8)RNA抽出及びRT-PCR分析
Trizol試薬(Invitrogen社)を用いて全RNAを細胞から単離した。Ready-To-Go You-Prime First-Strand Beads kit(GE Healthcare社)を用い、1μgの全RNAからcDNAを調製した。qPCR MasterMix Plus for SYBR Green (Eurogentec社)を用いてリアルタイムPCRを行った。操作方法は添付の説明書に従い、95℃で10分間のインキュベーションの後、40サイクルの増幅(95℃で15秒間、60℃で1分間、72℃で45秒間、80℃で15秒間)とし、SYBR Greenを検出した。使用したプライマーは以下の通りである。
Gapdh:5'-TGTGTCCGTCGTGGATCTGA-3'(配列番号13)及び5'-CCTGCTTCACCACCTTCTTGA-3'(配列番号14)
Zfhx3:5'-TTCTTTTCCTCCTCTCTCCTCATC-3'(配列番号15)及び5'-CGGTCCGTCGGACTTTTG-3'(配列番号16)
Col3a1:5'-GCACAGCAGTCCAACGTAGA-3'(配列番号17)及び5'-TCTCCAAATGGGATCTCTGG-3'(配列番号18)
Itga8:5'-AGTGGGAGGACCTGGAAGTT-3'(配列番号19)及び5'-AGTGGGAGGACCTGGAAGTT-3'(配列番号20)
Pdfgrb: 5'-AACCCCCTTACAGCTGTCCT-3'(配列番号21)及び5'-TAATCCCGTCAGCATCTTCC-3'(配列番号22)
【0054】
Gapdh mRNAの発現量を用いて各遺伝子の発現量を補正した。ABI Prism 7500 fast Sequence Detection System (Applied Biosystemsa社)のユーザーガイド(User Bulletin #2)を用い、比較CT(2-ΔΔCT)法で各増幅産物の相対量を求めた。
【0055】
(9)統計
ルシフェラーゼアッセイ及びウエスタンブロットの結果については、ボンフェローニ補正マンホイットニー検定によって統計解析した。少なくとも3回の独立した実験を行い、平均±s.e.m.(標準誤差)で全ての数値を表した。P値が0.01未満の場合に有意差ありとした。RT-PCRの結果は、Gapdhの発現レベルで補正し、コントロールの発現レベルと比較した。スチューデントのt検定で統計的有意差(P<0.001)を評価した。
【0056】
(10)Thapsigargin処理による神経過興奮モデル
神経細胞への分化を誘導したP19細胞をThapsigargin(2μM、1時間)で処理し、Ca2+濃度を上昇させるストレス(神経過興奮)を与えた。この神経過興奮モデルを用い、FK506(タクロリムス)の効果を検討した。FK506による処理の方法は次の通りとした。即ち、P19細胞の培養中の7日間にわたって毎日、培地交換時に最終濃度20 nMとなるようにFK506を培地に添加した。
【0057】
2.結果
<ZFHX3プロモータはATMの活性化に反応してCREBにより活性化される>
レチノイン酸(RA)はHoxホメオティック遺伝子の発現に影響する。マウス胚の後脳及び鰓部の神経幹細胞におけるHox遺伝子の発現は特にRAに感受性が高い(Marshall et al., 1996)。ヒト細胞やマウス胎仔性癌細胞を用いた、RAによる神経への分化誘導法が確立されている(Andrews et al., (1984))。RA処理による遺伝子発現制御には、標的遺伝子内の特定の配列モチーフ(レチノイン酸応答エレメント(RAREs)と呼ばれる)を認識する核内レチノイン酸レセプター(RARs又はRXRと呼ばれる)が関与する(Marshall et al., 1996)。ZFHX3遺伝子は、胎児脳における分裂後ニューロンで発現される非クラスターホメオティック因子である(Jung et al., 2005)。重要なことは、プロモータがRAREsを含まないにも拘わらず、ZFHX3遺伝子の発現はRAで誘導されることである(Miura et al., 1995)。最近になって、神経分化に関与する新たなRA経路、即ちATM及びcAMP応答エレメント結合タンパク質(CREB)に依存する経路が同定された(Fernandes et al., 2007)。注目すべきことに、本発明者らの検討の結果、ヒトZFHX3遺伝子プロモータの5'隣接配列内にCREB結合エレメント(CRE)があることが明らかとなった。この事実から、ATM及びCREBの活性化を介したRAからのシグナルが、ZFHX3遺伝子の発現開始に重要な役割を果たしていると予想される。
【0058】
本発明者らは様々な長さのヒトZFHX3遺伝子プロモータをリポーター遺伝子(ルシフェラーゼ遺伝子)の上流にクローニングした(図1B)。そして、これらのコンストラクトでマウス胎仔性癌細胞株P19を一過性にトランスフェクトし、RAによる刺激の存在下及び非存在下でレポーター活性を測定した。その結果、RAは濃度依存的にニューロン特異的5.6-kbプロモータを活性化した(図1Bの-5.6)。一方、5'側1.6-kbを欠失させると基礎レベル(RAに反応しない0.42-kbのプロモータの場合の活性レベル。図1Bの-0.42)までプロモータ活性が低下した(図1の-4.0)。CREコンセンサス配列を含む1.6-kb配列を0.42-kbプロモータ配列の5'側に付加するとRAへの反応が顕著に増加した(図1Bの-5.6Δ)。ATM特異的キナーゼ阻害剤KU-55933で前処理すると、この反応は認められない(図1C)。ニューロンの分化に関して重要な生存因子であるCREBの関与を確認するために、133番セリンがアラニンに変異された変異CREBタンパク質を発現するpCMV-CREB133ベクターを用い、ドミナントネガティブCREB(dn-CREB)を過剰発現させた。CREBの当該変異はプロテインキナーゼAによるCREBのリン酸化を阻害し、cAMPの活性化を抑制する。dn-CREBを発現する細胞ではRA処理後のRA応答プロモータコンストラクトのトランス活性化が有意に抑制された(図1Dの-5.6Δ)。また、プロモータ内のCREB結合サイトの変異によって、dn-CREBの発現の有無に関係なく、RAに反応したプロモータの活性化が阻害された(図1Dの-5.6Δmut)。これらの結果は、ATM及びCREB経路の活性化が、RAによるニューロン特異的ZFHX3遺伝子プロモータのトランス活性化のメカニズムであることを示唆する。
【0059】
CREに加え、活性因子のマルチプルコンセンサスエレメントを含むことから、5.6-kbプロモータ配列に最大のプロモータ活性が認められるものと予想された。しかしながら、5.6-kbプロモータの活性は、短いCRE含有フラグメント(5.6Δ)の活性よりも低かった。この事実は負の調節エレメントがプロモータ内に存在することを示唆する。
【0060】
<酸化ストレス応答性のATM活性化には膜受容体が必要である>
Pdgfrb遺伝子変異マウスは、明らかな解剖学的異常を示すことなく成長するが、その脳はN-メチル-D-アスパラギン酸(NMDA)又は凍傷に対して脆弱である(Ishii et al., 2006)。このことはグルタミン酸作動性興奮毒性からニューロンを保護するためにPDGFRBの発現が重要であることを示唆する。PDGF受容体とインテグリンによって受容体のクラスタリングが生じ、PDGFの結合量が増加し、PDGF受容体が活性化する(Zemskov et al., 2009)。インテグリン及びPDGFRBの発現誘導において、ZFHX3が重要な役割を果たすかもしれない。
【0061】
酸化ストレスは、いくつかの成人神経変性疾患や脳卒中、トラウマ、発作などの原因因子或いは少なくとも補助的因子である(Coyle and Puttfarcken 1993)。また、酸化ストレスはA-T変異マウスのプルキンエニューロンの不完全な生存の主要原因の一つと考えられている(Chen et al., 2003)。酸化ストレスは様々な細胞においてPDGFRB介在性のATMキナーゼ活性化を引き起こす(Chen et al., 2003)。新たに同定されたPDGFRB介在性シグナル経路は興奮毒性のメカニズムを理解する上で重要と考えられる。そこで、過酸化水素による酸化ストレスの存在下でP19由来ニューロン様細胞においてPDGFRBがATMの活性化に関与するか否かを調べることにした。Ser1981(pS-ATM)のリン酸化を指標に評価した結果、ATMの速やかな活性化を認めた(100μMの過酸化水素添加後15分以内)(図2A)。過酸化水素の添加量(1-500μM)に応じてATMの活性化量は上昇した(図2B)。また、酸化ストレスに反応した当該ATMの活性化はPDGFRB及びVEGFR-2(vascular endothelial growth factor receptor 2; also known as FLK-1 and KDR)の特異的阻害剤であるAG1433によって顕著に抑制された。(図2C,D)。対照的に、X線照射に反応したATMの活性化はAG1433の添加によって影響を受けない(図2E,F)。以上の結果は、神経細胞における酸化ストレス下でのATMの活性化にこれらの膜受容体が重要な役割を果たしていることを示す。
【0062】
<神経興奮に反応してATMは細胞質内で活性化される>
DNA損傷ストレスに反応して活性化されたATMは核点状構造(nuclear foci)に局在する。しかしながら、低張刺激やクロロキン処理(Bakkenist and Kastan, 2003)或いはRA刺激(Fernandes et al., 2007)に反応して活性化したATMは、びまん性に広がるパターンを示す。本発明者らの検討したところ、酸化ストレス誘導性DNA二重らせん切断(DSB)に反応して活性化したATMの核点状構造を認め、それはDNA二重らせん切断のマーカーであるγ-H2AXとともに局在していた(図3)。最近になって、酸化ストレスに反応して細胞質内でATMが活性化されることが報告され、この活性化は核での活性化と独立したものであることが示された(Alexander et al., 2010)。本発明者らの検討では、当初、負荷する酸化ストレスの量に問題があり、核でのATMの活性化と細胞質でのATMの活性化を区別することはできなかった(図3)。そこで、細胞質内でのATMの活性化を検討するため、より生理的条件に近い酸化ストレス条件を設定した。ニューロンでは様々な因子が酸化ストレスを引き起こすが、脳での興奮毒性を誘導する因子としては神経伝達物質のグルタミン酸が最も重要である。カイニン酸(KA)は、非NMDA型グルタミン酸受容体選択的なグルタミン酸受容体アゴニストである(Olney et al., 1974)。そこで、マウスニューロン(Coyle and Puttfarcken, 1993; Zhang et al., 2002)における代謝性酸化ストレスを誘導するため、KAによる刺激を使用することにした。KA刺激はマウスに2種類の発作を引き起こす。一つは頭部を繰り返し曲げる動作である。もう一つは異常な筋収縮であり、マウスの大脳皮質、海馬及び小脳深部核(DCN)でのATMの活性化を引き起こす(図4)。γ-H2AXの誘導がないと(図5A2、図6)活性化ATMは主に細胞質に存在することが明らかとなった(図5A1、図6)。対照的に、X線照射はDNA損傷ストレスを引き起こし、その場合の活性化ATMはγ-H2AXの点状構造(図5B2、図6)を伴い核に局在した(図5B1、図6)。KA刺激によって、DCN内のニューロンに明確な液胞の形成を認めた。
【0063】
<ATMノックアウトマウスのDCNでのPDGFRBの抑制>
神経への分化前及び分化後のP19細胞における、ZFHX3とPDGFRBの発現との関係をより詳細に調べることにした。ニューロンへ終末分化したP19ニューロン様細胞(7日目)においてZFHX3及びPDGFRBタンパク質の発現が顕著に上昇していた(図7A)。in situハイブリダイゼーションによるスクリーニング解析の結果、PDGFRBはDCNで高発現していた。免疫組織化学の結果では、成体コントロールマウス脳DCNにおける大型ニューロンにZFHX3(図7C4,C5)及びPDGFRB(図7C6,C7)の明瞭な染色を認めた。一方で、ATMノックアウトマウスではいずれのタンパク質の発現も強力に抑制された(図7C11-C14)。ラット胚において、増殖細胞(図7B2i)と分裂後細胞(図7B2ii)を識別するためにBrdU染色を行った。分裂後ニューロンにおける、ZFHX3の発現とPDGFRBの発現の相関性は、神経分化モデルを使用したマイクロアレイ解析の結果に符合した。ATMノックアウトマウスのDCNにおいてPDGFRBが抑制されていた事実は、小脳での特定神経変性のメカニズムを説明する上で重要といえる。
【0064】
<FK506の投与によって神経細胞死を抑制することができる>
神経細胞へ分化誘導させたP19細胞をThapsigarginで処理し、Ca2+濃度を上昇させるストレス(神経過興奮)を与えた。FK506を投与すると、当該ストレスによって誘導されるカスパーゼ3の活性化が抑制された(図10、レーン4)。即ち、FK506の投与によって神経細胞死が抑制された。
【0065】
<FK506の投与によってZFHX3の分断化が抑制できる>
Thapsigargin処理によるストレスがZFHX3タンパク質に与える影響を調べるため、抗ATBF1抗体(AT-6)で免疫沈降したタンパク質をAT-6抗体でウエスタンブロット解析した。その結果、Thapsigargin処理によって生ずるZFHX3タンパク質の分断化をFK506が有意に抑制した(図11、レーン6)。
【0066】
<FK506の投与によってZFHX3の脱リン酸化が抑制できる>
Thapsigargin処理によるストレスがZFHX3タンパク質のリン酸化に与える影響を調べるため、Pro-Q Diamond phosphoprotein stain kit (Invitrogen社、カタログ番号 P-33356)を用いた検定を行った。その結果、Thapsigargin処理によって生ずるZFHX3タンパク質の脱リン酸化をFK506が抑制した(図12、レーン14)。
【0067】
以上の結果より、免疫抑制剤として知られるFK506が、神経過興奮による神経細胞死を抑制できることが明らかになるとともに、その作用機序は「脱リン酸化の抑制(リン酸化状態の維持)を通してZFHX3の分断化を防ぐことにある」との知見が得られた。
【0068】
3.考察
ATMが細胞質内で活性化する現象は、神経細胞が酸化ストレスを受けた場合の防御シグナルとして重要な意味を持つことを明らかにした。以前にも神経細胞を含む各種細胞の観察結果として、核ではなく細胞質内でATMが活性化する現象が報告されている(Kuljis et al., 1999;Barlow et al., 1996;Boehrs et al., 2007;Gorodetsky et al., 2007)。神経細胞で観察された、細胞質内でのATMの活性化はシナプスの機能と関係している可能性が示唆されている(Li et al., 2009)。シナプスに存在する小胞ではATMがVAM2やシナプシン-1と結合した状態で存在する。ATMが核内タンパク質としての機能以外に細胞質タンパク質として機能していることを示す数々の研究論文が出されるようになり、細胞質内で活性化するATMは細胞の防御機構として機能している可能性が予想されるようになった。Alexanderらは細胞が酸化ストレスを受けた際にATMが活性化してmTORの抑制シグナルとして機能することを示した(Alexander et al., 2010)。mRORC1複合体の機能を抑制することによりオートファジー反応が誘導される。この細胞質内のシグナル伝達系は、核で進むDNAの二重鎖切断反応からのシグナルとは関わりのないことが示されている。Alexanderらは酸化ストレスに反応して、どのようにATMが活性化するのか、その未知のメカニズムについて実証的な根拠を与えることができなかったが、強い酸化反応でATMを構成するタンパク質のスルホニル基が修飾を受けることで、ATM自身の高次構造が変化することが、ATM自身の活性化の引き金になっている可能性を提唱した。
【0069】
本発明者らは、P19細胞をレチノン酸(RA)刺激で神経分化誘導する実験モデル系を使い、ZFHX3(ATBF1)の遺伝子発現誘導されるシグナル伝達系を解明した。RA刺激はATMの活性化を誘導し、そして、活性化したATMがCREBを活性化する。CREBはZFHX3の遺伝子プロモータ上にあるCRE(cAMP Responsive Element)のコンセンサスDNA配列に結合して、標的遺伝子であるZFHX3遺伝子の転写を活性化する。本発明者らはZFHX3の標的遺伝子の網羅的検索実験も行い、細胞接着因子の遺伝子群(プロコラーゲンIII型α1、インテグリンα8)を同定した。ZFHX3の標的遺伝子の中にはPDGFRB(血小板由来増殖因子受容体β)も含まれている。PDGFRBは結合組織間の細胞接着因子との相互作用で重要な受容体であるばかりでなく、神経細胞がグルタミン酸刺激による興奮等に由来する酸化ストレスを受けた状況下での神経保護作用のためのシグナル伝達系において重要な役割を演じる受容体である(Ishii et al., 2006)。本発明者らはPFGFRBまたはVEGFR-2からのシグナルが神経細胞の酸化ストレス反応の際、ATMの活性化の必須因子になることを実験的に証明した。本研究で用いた、P19細胞をレチノン酸(RA)刺激で神経分化誘導する実験モデル系において、ZFHX3の発現誘導に同期してPdgfrbとVegfr-2のmRNAの発現上昇を確認している。神経細胞中はその中に抗酸化物質が少ないにもかかわらず、神経興奮に由来する酸化ストレスに暴露され続ける(Brooks et al., 2000)。従って、十分な防御系を持たなければ酸化ストレスのために長期間生存することが難しい。酸化ストレスが生じた場合に迅速に反応する必要があり、細胞膜上の受容体から細胞内に情報を伝達して防御反応を誘導することは、神経細胞が長期間生存し続けるために非常に大切なメカニズムになっている。
【0070】
本発明者らは酸化ストレスを受けた神経細胞の細胞質内でATMが活性化する事実を明らかにした。神経細胞の興奮に起因する生化学反応産物が必然的に酸化ストレスを引き起こす。繰り返される神経興奮よって引き起こされる酸化ストレスは慢性的に進行する神経変性の主因となり、或いは少なくともその副次的な原因になっている(Coyle and Puttfarcken 1993)。本発明者らは、酸化ストレスが細胞質でATMを活性化するシグナル伝達系において、PDGFRBまたはVEGFR-2の活性化が必須であること解明した。神経興奮よって細胞内に酸化ストレスが生じても、ATMノックアウトマウスでは小脳深部核(DCN)にPDGFRBが発現していないために、細胞内で酸化ストレスに対応するためのシグナル伝達系が作動しない状況にあると考えられる。本発明者らは、マウスを使用したカイニン酸刺激実験で神経細胞に強い酸化ストレスを誘導した結果、小脳深部核のニューロンに顕著な空胞形成が生じることを見出した(図4B3)。小脳深部核にあるニューロン群にはPDGFRBが強く発現している(図5C6,C7)。小脳深部核のニューロンは主要な小脳の出力系中継場所として重要な役割を担っている。プルキンエ細胞と直接的にシナプスで繋がり、抑制シグナルを受け取っている。もしも小脳深部核ニューロンが障害を受けて消失すると、直結していたプルキンエ細胞との繋がりが障害され、その結果プルキンエ細胞は小脳深部核のニューロンから神経栄養因子を受け取れなくなり、プルキンエ細胞の変性が進む要因になる。プルキンエ細胞と小脳深部核との機能的な繋がりがA-Tの病態に密接に関与すると考えられる。カイニン酸刺激によって生じた、DCNにおける細胞質内のATM活性化は、過剰な神経興奮によってニューロン内に生じた酸化ストレスに対する保護的な応答反応と思われる。DCNに認められた空胞形成は、細胞質でのATMの活性化がmTORの抑制を介してオートファジーを誘導するという事実(Alexander et al., 2010)に関連しているであろう。DCNでは、カイニン酸処理に反応して、オートファジーの指標となる微小管関連タンパク質軽鎖3(LC3)の顕著な蓄積を認めた(図8)。オートファジー反応は障害を受けた細胞内小器官などを小胞に包んで酵素分解する反応であるので、かつては急性の神経細胞死の反応であると考えられていた(Wang et al., 2008)。しかし最近の研究によると、オートファジー反応は神経細胞死を導く反応というよりも、むしろ障害した細胞内小器官をきれいに分解/清掃して、細胞を生存維持するために必要な反応であると考えられるようになった。パーキンソン病の神経細胞では、オートファジー反応が障害され、障害を受けたままのミトコンドリアが蓄積することが神経細胞死を引き起こす原因になっていることが知られている(Narendra et al., 2008)。
【0071】
マイクロアレイによる網羅的発現解析によって、ZFHX3の標的遺伝子群としてプロコラーゲン遺伝子群(プロコラーゲンII型を除く)を同定した。ZFHX3は細胞外マトリックスの産生をコントロールして組織障害修復に関連する転写調節因子である。尚、プロコラーゲンII型は関節軟骨に特異的な因子であるので(Cheah et al., 1985)、今回用いた神経分化誘導の実験系モデル系ではプロコラーゲンII型の発現量に全く変化がなかったことは当然といえる。PDGFRBは外科的な損傷を受けた組織周囲にある血管で発現量が顕著に上昇する(Reuterdahl et al., 1993)。PDGFRBは組織修復過程で重要な機能を担っていると考えられている。ZFHX3はプロコラーゲン類とPDGFRBの両方の遺伝子を同時に転写活性化する機能を有し、組織損傷治癒過程できわめて重要な遺伝子調節因子であると言える。ZFHX3が機能障害に陥ると、血管系の疾患が惹起される可能性がある。事実、最近の研究報告によると、ZFHX3の変異は川崎病や心房細動や虚血性心疾患を起こす遺伝的背景になっている(Burgner et al., 2009;Benjamin et al., 2009;Gudbjartsson et al., 2009)。大規模な疫学調査結果から考察すると、ZFHX3は障害を受けた血管系の治癒過程に必要な重要な転写因子であることが分かる。コントロールマウスの小脳ではPDGFRBが強く発現しているにも関わらず(Lein et al., 2007)、ATMノックアウトマウスの小脳ではその発現量が低下していることは、A-T患者の小脳では組織損傷治癒反応が起こりにくいために神経変性に陥る危険性が生じていることを示している。
【0072】
以上の知見を図9として纏める。ATMの活性化経路として3種類の異なるシグナル伝達系がある。第一はDNAのDSBsに由来する核内のDNA二重鎖切断が起こす局所のATM活性化である。第二はレチノイン酸刺激(Fernandes et al., 2007)に誘導されて、核内にびまん性に広がるATMの活性化(Bakkenist and Kastan 2003)である。第三は酸化ストレスに誘導されて、細胞質で起こるATMの活性化である(Alexander et al., 2010)。酸化ストレス反応で起こるATMの活性化は、神経興奮で必然的に誘導される酸化ストレスとの関連において非常に重要である。本発明者らは細胞質で起こるATMの活性化反応にとって膜受容体型のチロシンリン酸化酵素のシグナルが必須であることを発見した。細胞質で起こるATMの活性化シグナルは、従来知られていた核内のDNA損傷ストレスに起因するATMの活性化シグナルとは独立したシグナルである。酸化ストレスに起因するATMの活性化シグナルは、mTORの抑制を介してオートファジー反応を誘導する。オートファジー反応は障害を受けた細胞内小器官を清掃して細胞機能を生理的に維持するために必要な反応である。以上を総合すると、細胞質で活性化するATMのシグナル伝達系は、酸化ストレスを受けた神経細胞を慢性的な神経変性から保護するためのメカニズムを起動するために重要な役割を演じていると言える。
【0073】
<参考論文>
Alexander, A., Cai, S. L., Kim, J., Nanez, A., Sahin, M., MacLean, K. H., Inoki, K., Guan, K. L., Shen, J., Person, M. D. et al. (2010). ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc. Natl. Acad. Sci. USA 107, 4153-4158.
Alterman, N., Fattal-Valevski, A., Moyal, L., Crawford, T. O., Lederman, H. M., Ziv, Y. and Shiloh, Y. (2007). Ataxia-telangiectasia: mild neurological presentation despite null ATM mutation and severe cellular phenotype. Am. J. Med. Genet 143A, 1827-1834.
Andrews, P. W. (1984). Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 103, 285-293.
Andrews, P. W., Damjanov, I., Simon, D., Banting, G. S., Carlin, C., Dracopoli, N. C. and Fogh, J. (1984). Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Invest. 50, 147-162.
Bakkenist, C. J. and Kastan, M. B. (2003). DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499-506.
Banin, S., Moyal, L., Shieh, S., Taya, Y., Anderson, C. W., Chessa, L., Smorodinsky, N. I., Prives, C., Reiss, Y., Shiloh, Y. et al. (1998). Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674-1677.
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley, J. N., Ried, T., Tagle, D. et al. (1996). Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159-171.
Benjamin, E. J., Rice, K. M., Arking, D. E., Pfeufer, A., van Noord, C., Smith, A. V., Schnabel, R. B., Bis, J. C., Boerwinkle, E., Sinner, M. F. et al. (2009). Variants in ZFHX3 are associated with atrial fibrillation in individuals of European ancestry. Nat. Genet. 41, 879-881.
Boehrs, J. K., He, J., Halaby, M. J. and Yang, D. Q. (2007). Constitutive expression and cytoplasmic compartmentalization of ATM protein in differentiated human neuronlike SH-SY5Y cells. J. Neurochem. 100, 337-345.
Brooks, P. J., Wise, D. S., Berry, D. A., Kosmoski, J. V., Smerdon, M. J., Somers, R. L., Mackie, H., Spoonde, A. Y., Ackerman, E. J., Coleman, K. et al. (2000). The oxidative DNA lesion 8,5'-(S)-cyclo-2’-deoxyadenosine is repaired by the nucleotide excision repair pathway and blocks gene expression in mammalian cells. J. Biol. Chem. 275, 22355-22362.
Burgner, D., Davila, S., Breunis, W. B., Ng, S. B., Li, Y., Bonnard, C., Ling, L., Wright, V. J., Thalamuthu, A., Odam, M. et al. (2009). A genome-wide association study identifies novel and functionally related susceptibility Loci for Kawasaki disease. PLoS Genet. 5, e1000319.
Cheah, K. S., Stoker, N. G., Griffin, J. R., Grosveld, F. G. and Solomon, E. (1985). Identification and characterization of the human type II collagen gene (COL2A1). Proc. Natl. Acad. Sci. USA 82, 2555-2559.
Chen, K., Albano, A., Ho, A. and Keaney, J. F., Jr (2003). Activation of p53 by oxidative stress involves platelet-derived growth factor-beta receptor-mediated ataxia telangiectasia mutated (ATM) kinase activation. J. Biol. Chem. 278, 39527-39533.
Chen, P., Peng, C., Luff, J., Spring, K., Watters, D., Bottle, S., Furuya, S. and Lavin, M. F. (2003). Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J. Neurosci. 23, 11453-11460.
Cho, Y. G., Song, J. H., Kim, C. J., Lee, Y. S., Kim, S. Y., Nam, S. W., Lee, J. Y. and Park, W. S. (2007). Genetic alterations of the ATBF1 gene in gastric cancer. Clin. Cancer Res. 13, 4355-4359.
Coyle, J. T. and Puttfarcken, P. (1993). Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689-695.
Dupre, A., Boyer-Chatenet, L. and Gautier, J. (2006). Two-step activation of ATM by DNA and the Mre11-Rad50-Nbs1 complex. Nat. Struct. Mol. Biol. 13, 451-457.
Fernandes, N. D., Sun, Y. and Price, B. D. (2007). Activation of the kinase activity of ATM by retinoic acid is required for CREB-dependent differentiation of neuroblastoma cells. J. Biol. Chem. 282, 16577-16584.
Gorodetsky, E., Calkins, S., Ahn, J. and Brooks, P. J. (2007). ATM, the Mre11/Rad50/Nbs1 complex, and topoisomerase I are concentrated in the nucleus of Purkinje neurons in the juvenile human brain. DNA Rep. 6, 1698-1707.
Gudbjartsson, D. F., Holm, H., Gretarsdottir, S., Thorleifsson, G., Walters, G. B., Thorgeirsson, G., Gulcher, J., Mathiesen, E. B., Njolstad, I., Nyrnes, A. et al. (2009). A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 41, 876-878.
Ishii, Y., Oya, T., Zheng, L., Gao, Z., Kawaguchi, M., Sabit, H., Matsushima, T., Tokunaga, A., Ishizawa, S., Hori, E. et al. (2006). Mouse brains deficient in neuronal PDGF receptor-beta develop normally but are vulnerable to injury. J. Neurochem. 98, 588-600.
Jung, C. G., Kim, H. J., Kawaguchi, M., Khanna, K. K., Hida, H., Asai, K., Nishino, H. and Miura, Y. (2005). Homeotic factor ATBF1 induces the cell cycle arrest associated with neuronal differentiation. Development 132, 5137-5145.
Kao, F. T., Hawkins, J. W., Law, M. L. and Dugaiczyk, A. (1982). Assignment of the structural gene coding for albumin to human chromosome 4. Hum. Genet. 62, 337-341.
Kataoka, H., Miura, Y., Joh, T., Seno, K., Tada, T., Tamaoki, T., Nakabayashi, H., Kawaguchi, M., Asai, K., Kato, T. et al. (2001). Alpha-fetoprotein producing gastric cancer lacks transcription factor ATBF1. Oncogene 20, 869-873.
Khanna, K. K. and Jackson, S. P. (2001). DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247-254.
Kim, C. J., Song, J. H., Cho, Y. G., Cao, Z., Lee, Y. S., Nam, S. W., Lee, J. Y. and Park, W. S. (2008). Down-regulation of ATBF1 is a major inactivating mechanism in hepatocellular carcinoma. Histopathology 52, 552-559.
Kuljis, R. O., Chen, G., Lee, E. Y., Aguila, M. C. and Xu, Y. (1999). ATM immunolocalization in mouse neuronal endosomes: implications for ataxiatelangiectasia. Brain Res. 842, 351-358.
Lein, E. S., Hawrylycz, M. J., Ao, N., Ayres, M., Bensinger, A., Bernard, A., Boe, A. F., Boguski, M. S., Brockway, K. S., Byrnes, E. J. et al. (2007). Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168-176.
Li, J., Han, Y. R., Plummer, M. R. and Herrup, K. (2009). Cytoplasmic ATM in neurons modulates synaptic function. Curr. Biol. 19, 2091-2096.
Marshall, H., Morrison, A., Studer, M., Popperl, H. and Krumlauf, R. (1996). Retinoids and Hox genes. FASEB J. 10, 969-978.
Matsuoka, S., Ballif, B. A., Smogorzewska, A., McDonald, E. R., 3rd, Hurov, K. E., Luo, J., Bakalarski, C. E., Zhao, Z., Solimini, N., Lerenthal, Y. et al. (2007). ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316, 1160-1166.
Miura, Y., Tam, T., Ido, A., Morinaga, T., Miki, T., Hashimoto, T. and Tamaoki, T. (1995). Cloning and characterization of an ATBF1 isoform that expresses in a neuronal differentiation-dependent manner. J. Biol. Chem. 270, 26840-26848.
Miura, Y., Kataoka, H., Joh, T., Tada, T., Asai, K., Nakanishi, M., Okada, N. and Okada, H. (2004). Susceptibility to killer T cells of gastric cancer cells enhanced by Mitomycin-C involves induction of ATBF1 and activation of p21 (Waf1/Cip1) promoter. Microbiol. Immunol. 48, 137-145.
Morinaga, T., Yasuda, H., Hashimoto, T., Higashio, K. and Tamaoki, T. (1991). A human alpha-fetoprotein enhancer-binding protein, ATBF1, contains four homeodomains and seventeen zinc fingers. Mol. Cell. Biol. 11, 6041-6049.
Narendra, D., Tanaka, A., Suen, D. F. and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183, 795-803.
Olney, J. W., Rhee, V. and Ho, O. L. (1974). Kainic acid: a powerful neurotoxic analogue of glutamate. Brain Res. 77, 507-512.
Reuterdahl, C., Sundberg, C., Rubin, K., Funa, K. and Gerdin, B. (1993). Tissue localization of beta receptors for platelet-derived growth factor and platelet-derived growth factor B chain during wound repair in humans. J. Clin. Invest. 91, 2065-2075.
Rudnicki, M. A. and McBurney, M. W. (1987). Cell culture methods and induction of differentiatin of embryonal carcinoma cell lines. In Teratocarcinomas and Embryonic Stem Cells a Practical Approach (ed. E. J. Robertson), pp. 14-49. Oxford, Washington DC: IRL Press.
Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L., Tagle, D. A., Smith, S., Uziel, T., Sfez, S. et al. (1995). A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749-1753.
Shi, Y., Venkataraman, S. L., Dodson, G. E., Mabb, A. M., LeBlanc, S. and Tibbetts, R. S. (2004). Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 101, 5898-5903.
Shieh, S. Y., Ikeda, M., Taya, Y. and Prives, C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334.
Shiloh, Y. (2003). ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 3, 155-168.
Sun, X., Frierson, H. F., Chen, C., Li, C., Ran, Q., Otto, K. B., Cantarel, B. L., Vessella, R. L., Gao, A. C., Petros, J. et al. (2005). Frequent somatic mutations of the transcription factor ATBF1 in human prostate cancer. Nat. Genet. 37, 407-412.
Urano, Y., Sakai, M., Watanabe, K. and Tamaoki, T. (1984). Tandem arrangement of the albumin and alpha-fetoprotein genes in the human genome. Gene 32, 255-261.
Wang, Y., Han, R., Liang, Z. Q., Wu, J. C., Zhang, X. D., Gu, Z. L. and Qin, Z. H. (2008). An autophagic mechanism is involved in apoptotic death of rat striatal neurons induced by the non-N-methyl-D-aspartate receptor agonist kainic acid. Autophagy 4, 214-226.
Wilkinson, D. S., Tsai, W. W., Schumacher, M. A. and Barton, M. C. (2008). Chromatin-bound p53 anchors activated Smads and the mSin3A corepressor to confer transforming-growth-factor-beta-mediated transcription repression. Mol. Cell. Biol. 28, 1988-1998.
Yasuda, H., Mizuno, A., Tamaoki, T. and Morinaga, T. (1994). ATBF1, a multiplehomeodomain zinc finger protein, selectively down-regulates AT-rich elements of the human alpha-fetoprotein gene. Mol. Cell. Biol. 14, 1395-1401.
Zemskov, E. A., Loukinova, E., Mikhailenko, I., Coleman, R. A., Strickland, D. K. and Belkin, A. M. (2009). Regulation of platelet-derived growth factor receptor function by integrin-associated cell surface transglutaminase. J. Biol. Chem. 284, 16693-16703.
Zhang, J., Zhang, D., McQuade, J. S., Behbehani, M., Tsien, J. Z. and Xu, M. (2002). c-fos regulates neuronal excitability and survival. Nat. Genet. 30, 416-420.
【産業上の利用可能性】
【0074】
毛細血管拡張性失調症の原因遺伝子とされるATM遺伝子が関わる新たなシグナル伝達経路が解明された。本発明の治療薬は当該シグナル伝達経路において重要な役割を果たすZFHX3を標的としたものであり、毛細血管拡張性失調症の治療に有効である。本発明の一態様では有効成分としてタクロリムス(免疫抑制剤として知られている)を使用する。タクロリムスの新たな用途を提供するという点においても本発明の意義は大きい。一方、本願が提供するスクリーニング法には、毛細血管拡張性失調症等における神経変性に対して有効な新規化合物の創出ないし同定手段として活用されることが期待される。
【0075】
この発明は、上記発明の実施の形態及び実施例の説明に何ら限定されるものではない。特許請求の範囲の記載を逸脱せず、当業者が容易に想到できる範囲で種々の変形態様もこの発明に含まれる。
本明細書の中で明示した論文、公開特許公報、及び特許公報などの内容は、その全ての内容を援用によって引用することとする。
【配列表フリーテキスト】
【0076】
配列番号5:人工配列の説明:プライマーKO-1F;配列番号6:人工配列の説明:プライマーKO-1R;配列番号7:人工配列の説明:プライマーKO-2R;配列番号8:人工配列の説明:プライマーpGV-F;配列番号9:人工配列の説明:プライマーmCRE-R;配列番号10:人工配列の説明:プライマーmCRE-F;配列番号11:人工配列の説明:プライマーpGV-R1;配列番号12:人工配列の説明:プライマーpGV-R2;配列番号13:人工配列の説明:プライマーGapdh;配列番号14:人工配列の説明:プライマーGapdh;配列番号15:人工配列の説明:プライマーZfhx3;配列番号16:人工配列の説明:プライマーZfhx3;配列番号17:人工配列の説明:プライマーCol3a1;配列番号18:人工配列の説明:プライマーCol3a1;配列番号19:人工配列の説明:プライマーItga8;配列番号20:人工配列の説明:プライマーItga8;配列番号21:人工配列の説明:プライマーPdfgrb;配列番号22:人工配列の説明:プライマーPdfgrb
【特許請求の範囲】
【請求項1】
標的神経細胞内で機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を含む、毛細血管拡張性失調症治療薬。
【請求項2】
前記化合物が、機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質の発現増強作用を示す化合物である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項3】
前記化合物がレチノイン酸である、請求項2に記載の毛細血管拡張性失調症治療薬。
【請求項4】
前記化合物がプロテインフォスファターゼ2B(PP2B)阻害剤である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項5】
前記プロテインフォスファターゼ2B(PP2B)阻害剤がタクロリムスである、請求項4に記載の毛細血管拡張性失調症治療薬。
【請求項6】
前記化合物がカルパイン阻害剤である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項7】
機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を毛細血管拡張性失調症患者に対して治療上有効量、投与するステップを含む、毛細血管拡張性失調症の治療法。
【請求項8】
前記化合物がレチノイン酸又はタクロリムスである、請求項7に記載の治療法。
【請求項9】
以下のステップ(1)〜(4)を含む、毛細血管拡張性失調症に起因又は付随する神経変性或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物のスクリーニング法:
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
【請求項10】
ステップ(3)において、小脳でのオートファジー又は神経変性が検定される、請求項9に記載のスクリーニング法。
【請求項11】
ステップ(3)において、小脳深部核でのオートファジー又は神経変性が検定される、請求項9に記載のスクリーニング法。
【請求項12】
非ヒト哺乳動物が野生型動物である、請求項9〜11のいずれか一項に記載のスクリーニング法。
【請求項13】
非ヒト哺乳動物がATM遺伝子欠損動物である、請求項9〜11のいずれか一項に記載のスクリーニング法。
【請求項14】
非ヒト哺乳動物がマウスである、請求項9〜13のいずれか一項に記載のスクリーニング法。
【請求項1】
標的神経細胞内で機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を含む、毛細血管拡張性失調症治療薬。
【請求項2】
前記化合物が、機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質の発現増強作用を示す化合物である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項3】
前記化合物がレチノイン酸である、請求項2に記載の毛細血管拡張性失調症治療薬。
【請求項4】
前記化合物がプロテインフォスファターゼ2B(PP2B)阻害剤である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項5】
前記プロテインフォスファターゼ2B(PP2B)阻害剤がタクロリムスである、請求項4に記載の毛細血管拡張性失調症治療薬。
【請求項6】
前記化合物がカルパイン阻害剤である、請求項1に記載の毛細血管拡張性失調症治療薬。
【請求項7】
機能的ジンクフィンガーホメオボックス3(ZFHX3)タンパク質を増大又は安定化する化合物を毛細血管拡張性失調症患者に対して治療上有効量、投与するステップを含む、毛細血管拡張性失調症の治療法。
【請求項8】
前記化合物がレチノイン酸又はタクロリムスである、請求項7に記載の治療法。
【請求項9】
以下のステップ(1)〜(4)を含む、毛細血管拡張性失調症に起因又は付随する神経変性或いは神経過興奮に由来した酸化ストレスによる神経変性の予防又は治療に有効な化合物のスクリーニング法:
(1)複数匹の同種同系非ヒト哺乳動物を用意し、試験群と対照群に分けるステップ;
(2)試験群に対して、カイニン酸を投与するとともに被験物質を投与するステップ;
(3)ステップ(2)後の試験群と、被験物質を投与しないこと以外は試験群と同様の処置を施した対照群について、脳内でのオートファジー又は神経変性を検定するステップ;
(4)試験群と対照群の間で検定結果を比較し、比較結果に基づき被験物質の有効性を判定するステップであって、オートファジーの促進ないし誘導を認めること、又は神経変性の抑制を認めること、が被験物質の有効性の指標となる、ステップ。
【請求項10】
ステップ(3)において、小脳でのオートファジー又は神経変性が検定される、請求項9に記載のスクリーニング法。
【請求項11】
ステップ(3)において、小脳深部核でのオートファジー又は神経変性が検定される、請求項9に記載のスクリーニング法。
【請求項12】
非ヒト哺乳動物が野生型動物である、請求項9〜11のいずれか一項に記載のスクリーニング法。
【請求項13】
非ヒト哺乳動物がATM遺伝子欠損動物である、請求項9〜11のいずれか一項に記載のスクリーニング法。
【請求項14】
非ヒト哺乳動物がマウスである、請求項9〜13のいずれか一項に記載のスクリーニング法。
【図1】


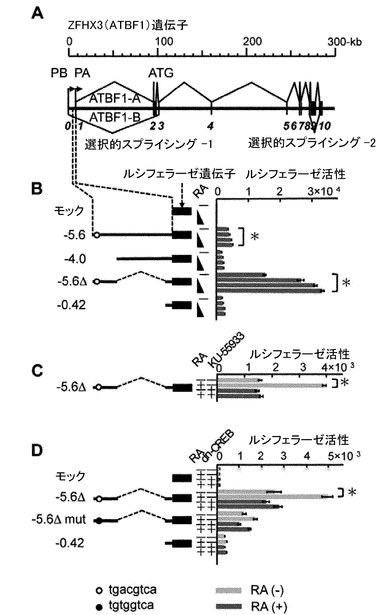
【図2】
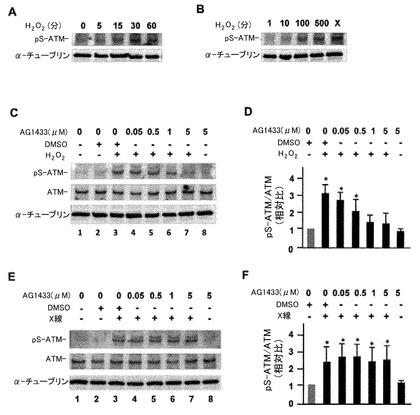
【図3】
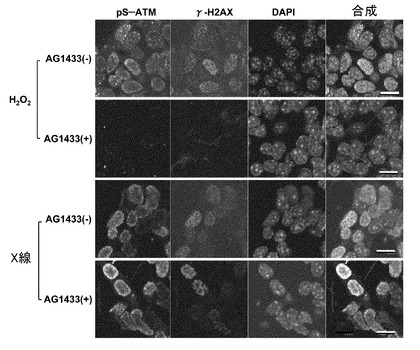
【図4】
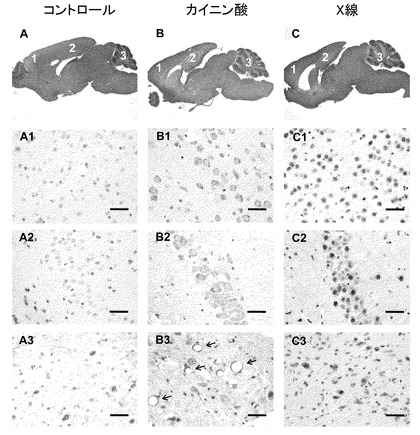
【図5】
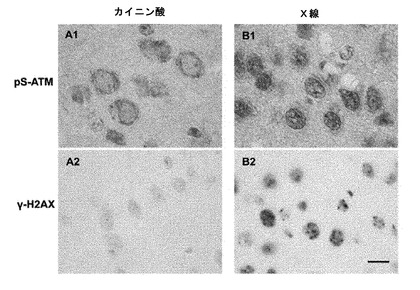
【図6】
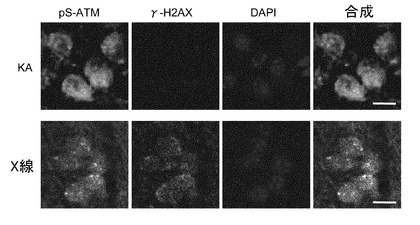
【図7】
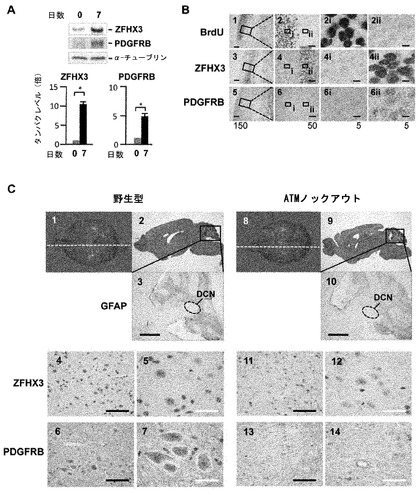
【図8】
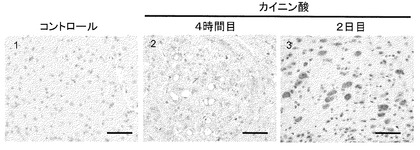
【図9】
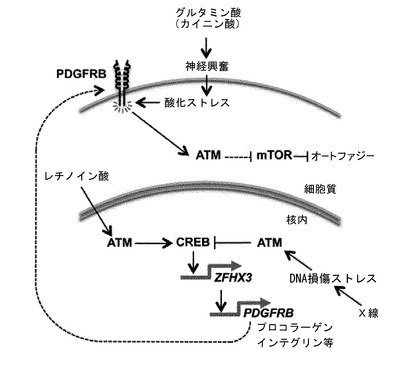
【図10】
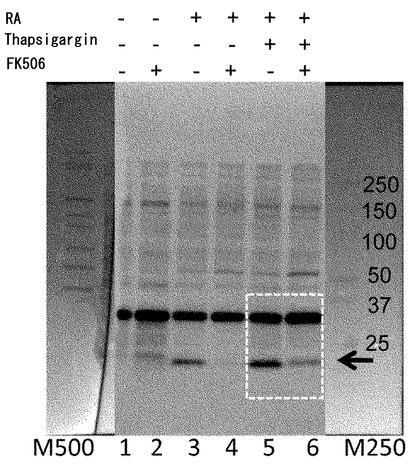
【図11】
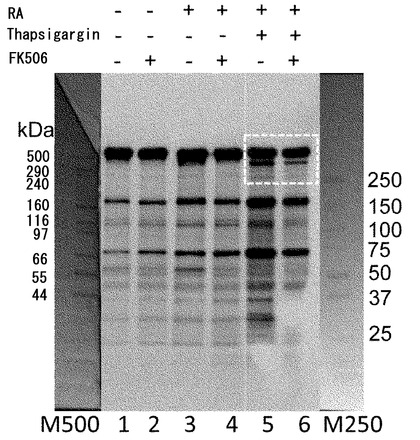
【図12】
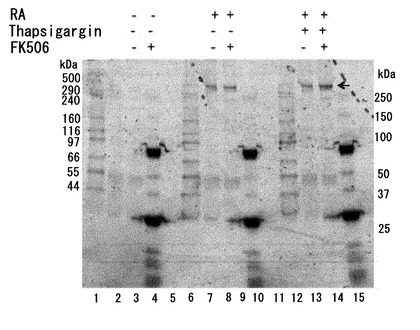
【図13】
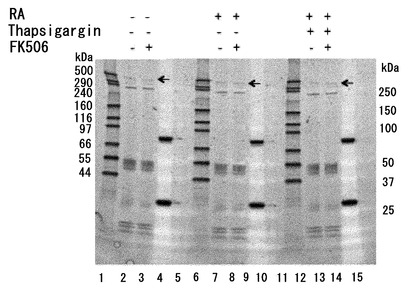
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2012−171895(P2012−171895A)
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願番号】特願2011−33855(P2011−33855)
【出願日】平成23年2月18日(2011.2.18)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 掲載アドレス:http://share.dynacom.jp/bmb2010abst/index.php?p_no=3P−0377&btn_syousai=on 掲載日 :平成22年11月19日
【出願人】(506218664)公立大学法人名古屋市立大学 (48)
【Fターム(参考)】
【公開日】平成24年9月10日(2012.9.10)
【国際特許分類】
【出願日】平成23年2月18日(2011.2.18)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 掲載アドレス:http://share.dynacom.jp/bmb2010abst/index.php?p_no=3P−0377&btn_syousai=on 掲載日 :平成22年11月19日
【出願人】(506218664)公立大学法人名古屋市立大学 (48)
【Fターム(参考)】
[ Back to top ]