
特定遺伝子の検出方法
【課題】被検試料における核酸の分離・精製等の前処理を必要とせず、簡易に高感度で検出し得る特定遺伝子、具体的には、特定ウイルスまたは特定微生物の検出方法を提供することである。
【解決手段】表面処理された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、被検試料のなかで、被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、DNA鎖断片またはRNA鎖断片を分離する第3工程と、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片を増幅する第4工程とを備える特定遺伝子の検出方法に関する。
【解決手段】表面処理された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、被検試料のなかで、被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、DNA鎖断片またはRNA鎖断片を分離する第3工程と、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片を増幅する第4工程とを備える特定遺伝子の検出方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定遺伝子の検出方法に関する。より詳細には、生体試料中に存在するウイルス、または、食品や臨床試料中に含まれる微生物を特異的に検出することのできる特定遺伝子の検出方法に関する。
【背景技術】
【0002】
ウイルス病の診断を確実に行なうこと、又未知の流行病を新しいウイルス病として同定することは、当該ウイルス病の地域分布と流行時期を知り、当該疾患に対する予防と治療対策を立てる上で重要なことである。しかも、ウイルス病は速やかにしかも正確に診断されないと、適切な対策処理が手遅れとなることが多い。
【0003】
臨床検体中の特定ウイルスを直接検出する方法としては、たとえば検体中に存在するウイルス抗原を蛍光等で標識した抗体と反応させて検出する方法が挙げられる。当該方法は2〜3時間程度で結果が得られるという利点はあるが、検体中の夾雑物質との非特異反応による偽陽性反応の可能性や検体中に一定以上のウイルス抗原がないと検出できないという検出感度の問題が指摘されている。また最近では、生体試料から、生体に潜在しているウイルス核酸やmRNAの存在を証明して診断する方法が開発されている。当該方法によると、PCR法(Science,230,1350(1985))やRT−PCR法を利用することによって生体試料中に微量存在するウイルス遺伝子でも高感度に検出できるが、この場合検査結果の取得に1日以上要する。
【0004】
また、たとえば、輸血用血液や血液製剤の安全性を確保するため、献血血液に対して、通常、ウイルス検査が行なわれる。このような検査としては、血液中のウイルス抗原やウイルス関連抗体の有無を調べる血清学的検査が主に行われている。1999年から、血清学的検査で陰性となった血液に対して、2次検査として核酸増幅検査(Nucleic Acid Amplification Testing: NAT)が行われている。NATとは、ウイルス遺伝子の一部の核酸を取り出し(抽出)、その核酸を増やし(増幅)、増えた核酸を検出することでウイルス遺伝子の有無を確認する検査法のことである。NATを使用したウイルス検査法は、ウイルスの持つ遺伝子を数万倍以上に増幅して検出するため、従来の血清学的な検査法に比べ、極めて感度の高い検査法である。この検査で再度陰性になった献血血液が、輸血用血液および血液製剤の原料などとして使用される。
【0005】
このようなNATの導入により、献血血液を原料とした輸血用血液や血液製剤等の安全性は以前よりも向上してきたが、輸血後の感染症発生率は依然として0%に達していない。通常、NATを用いた2次検査では1人からたとえば約4μLといった微少量の血液を採取し、約50人分を1つにまとめて分析を行なっている。このため、感染直後のようにウイルス量が極端に少ない場合などには、検査に回した血液約4μL中にウイルスが含まれない確率が高い。その場合、献血によって集められた血液中にウイルスが存在していても検出されずに輸血感染する恐れがある。
【0006】
また、食品や臨床試料、または環境中などの特定微生物の有無を調べる場合には、従来、被検試料中の微生物を分離培養し、コロニーについて視覚的観察(コロニーの色素反応、コロニー形態等)、顕微鏡観察、グラム染色、生化学的性状の検査等を行なっていた。特定微生物の有無が判明するまでに少なくとも2日間を要する。したがって、この方法は、微生物が検出されたとしてとしてもそれに対する対応が遅くなることから、食品メーカーによる食品出荷前の自主検査には採用し難い。
【0007】
食品の殺菌技術や加工技術が向上したことにより、微量であっても被検試料中に存在する微生物の生死の状態を識別するニーズが高まっている。特に、食品衛生検査や臨床検査領域においては、微生物の迅速検出法として、PCR法により各微生物の特異遺伝子を視覚的に捉えられる量まで増幅し、各微生物の存在の有無を判別および定量する試みがなされている。しかし、微生物のDNAにターゲットを当てた場合、被検試料に元来含まれている死菌のバックグラウンドまで検出されるため、PCR法で陽性判定がでた場合、必ずしも生きた微生物の存在を示唆しているとは限らなかった。
【0008】
一方、生きた微生物(生菌)の存在を確認するためには、食品から微生物を分離培養し、得られたコロニーについて、特定微生物のDNAに対応したプライマーを用いたPCR法により特定微生物のDNAを迅速に検出する方法が開発されている。しかし、この方法によっても、食品から微生物を分離培養するのに少なくとも1日間を要する。そのために、食品衛生や臨床検査の分野では、PCR法は高感度・迅速でありながら普及していないのが現状であった。
【0009】
ここで、特許文献1、特許文献2および非特許文献1には、ホスホリルコリン基を有する第一単位と電子求引性の置換基がカルボニル基に結合してなるカルボン酸誘導基を有する第二単位とを含む高分子物質を表面にDNA鎖を固定化した不溶性担体に検出対象遺伝子のDNA断片またはRNA断片を含有する溶液を添加し、該DNA鎖とハイブリダイズさせ、溶液中の遺伝子断片の含有状況を判定することを特徴とする遺伝子の検出方法が開示されている。
【0010】
また、最近では、mRNAをターゲットにして、逆転写酵素によりcDNAを作製後、各微生物の特異プライマーを用いてPCRを行い、被検試料中の生菌のみを検出・定量する試みがなされている。しかし、この方法では、死菌のバックグラウンドを検出してしまうため、生死の判別法としては十分なものとは言えなかった。具体的には、PCR法を利用した微生物等の生死を判別する方法としては、特許文献4に記載の方法が開示されている。
【0011】
また、非特許文献2には、ノロウイルスの検出法が開示されており、特許文献3には微生物等の検出等に利用されるLAMP(Loop−mediated Isothermal Amplification)法が開示されており、非特許文献3には、微生物等の検出等に利用されるRT−LAMP法が開示されている。
【特許文献1】特開2006−187270号公報
【特許文献2】特開2006−322739号公報
【特許文献3】特開2004−154008号公報
【特許文献4】特表2003−530118号公報
【非特許文献1】Kinoshita, K. et al., Nucleic Acids Res. 2007,Vol.35,No.1 e3
【非特許文献2】ノロウイルスの検出法(平成15年11月5日付け食安監発第1105001号)
【非特許文献3】Fukuda, S. et al.,J. Clinical Microbiology Vol.44,No.4: 1376-1381 (2006) (RT−LAMP)
【発明の開示】
【発明が解決しようとする課題】
【0012】
上記のように、特定ウイルスの検出および特定微生物の検出には、さまざまな問題が未
だ解決されていない。そして、特許文献1、特許文献2および非特許文献1には、微量の核酸を迅速にかつ簡便に測定することができる方法については開示されていない。
【0013】
そしてさらに、患者から採取した生体試料を対象として、該生体試料中に含まれるウイルス遺伝子の検出を目的としてハイブリダイゼーションが広く行われてきているが、1個の不溶性担体内でウイルス遺伝子を捕捉し、洗浄後、分離した遺伝子を増幅してウイルスを検出する方法は知られていなかった。
【0014】
また、試料中の微量の核酸を増幅するためにはPCR法が広く普及している。しかしながら、PCR法では試料中に含まれる夾雑物により遺伝子の増幅反応を阻害してしまう場合がある。したがって、PCR法を用いた場合には、増幅反応の前に分離・精製等の煩雑な前処理を行なう必要がある。
【0015】
このような背景から、本発明者らは、特定ウイルスの検出において、血液中に含まれるウイルス数が極端に少ない場合でも、簡便かつ短時間にウイルスを検出できる検査法の開発を行なってきた。また、本発明者らは、特定微生物の検出において、食品や臨床試料中に含まれる特定の複数の微生物または微生物群の生菌の有無を死菌に比べて選択的、簡単かつ迅速に検出する方法の開発を行なってきた。
【0016】
したがって、本発明の目的は、被検試料における核酸の分離・精製等の前処理を必要とせず、簡易に高感度で検出し得る特定遺伝子、具体的には、特定ウイルスまたは特定微生物の検出方法を提供することである。
【課題を解決するための手段】
【0017】
本発明者は、上記課題の解決を目的として種々検討していたところ、ウイルスに感染した患者の生体試料を公知のグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液でウイルスを溶解させ、次いでDNA鎖を固定化した不溶性担体にハイブリダイゼーションを行なうことにより、生体試料中に存在するウイルス遺伝子の捕捉、分離を簡便かつ迅速に行なうことができ、しかも遺伝子断片をポリメラーゼ連鎖反応(PCR)、LAMP法、鎖置換増幅(SDA)、逆転写酵素鎖置換増幅(RT−SDA)、逆転写酵素ポリメラーゼ連鎖反応(RT−PCR)、逆転写LAMP法(RT−LAMP)、核酸配列に基づく増幅(NASBA)、転写媒介性増幅(TMA)、ローリングサークル型増幅などの遺伝子増幅法を用いて特異的に検出することができることを見出し、増幅することによって得られた結果が患者検体の臨床状態を的確に反映した結果であることを確認した。本発明は、かかる知見に基づいて開発されたものである。
【0018】
ウイルスを含み得る被検試料中のウイルス遺伝子の存在の有無を判定することを特徴とするウイルス感染の診断方法である。なお、当該診断方法には、ウイルス感染成立を診断する方法だけでなく、ウイルス感染の経過や治療経過を診断する方法もまた含まれる。
【0019】
つまり、本発明は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する第3工程と、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する第4工程とを備える特定遺伝子の検出方法に関する。
【0020】
また、本発明は、ホスホリルコリン基またはアルキレングリコール基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、抗体、レクチンおよび複合糖質のいずれかで微生物またはウイルスを捕捉する工程と、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、生体高分子固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程とを備える特定遺伝子の検出方法に関する。
【0021】
また、本発明においては、被検試料が、鼻汁、鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液、細胞組織または食品であることが好ましい。
【0022】
また、本発明においては、第3工程において、4〜70℃で5分〜90分間、ハイブリダイズすることが好ましい。
【0023】
また、本発明においては、第4工程において、RNA鎖断片を逆転写するための逆転写反応のプライマーとして、ランダムプライマー、dTプライマーまたは1個以上の配列特異的なスペシフィックプライマーから合成されたcDNAを使用することが好ましい。
【0024】
また、本発明においては、第4工程、または、DNA鎖断片もしくはRNA鎖断片で増幅する工程の後に増幅したDNA鎖断片を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する第5工程をさらに備えることが好ましい。
【0025】
また、本発明においては、キャプチャーDNA鎖は、10〜50塩基からなり、23S rRNA遺伝子配列または16S rRNA遺伝子配列に相補的な配列から選択されることが好ましい。
【0026】
また、本発明においては、キャプチャーDNA鎖のターゲット配列領域がさらに、トポイソメラーゼ、または、DNAジャイレースを含む微生物特異的な配列であることが好ましい。
【0027】
また、本発明においては、微生物がグラム陽性菌またはグラム陰性菌であることが好ましい。
【0028】
また、本発明においてはウイルスがC型肝炎ウイルス(HCV)、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス(黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス)、トガウイルス(アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス)、ペスチウイルス(ブタコレラウイルス、ウシ下痢ウイルス)、パラミクソウイルス(パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス)、オルソクソウイルス(ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス)、ラブドウイルス(狂犬病ウイルス、水泡性口内炎ウイルス)、ピコルナウイルス(ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス)、コロナウイルス(ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス)、アレナウイルス(リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス)、レトロウイルス(HTLV:ヒト成人白血病ウイルス、HIV:エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス)、レオウイルス(ロタウイルス)、カリシウイルス(ノーウオークウイルス)、ノロウイルス、ブンヤウイルス(腎症候性出血熱ウイルス)、フィロウイルス(エボラウイルス、マールブルグウイルス)、B型肝炎ウイルス(HBV)、ポックスウイルス(ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス)、パルボウイルス(ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス)、パポーバウイルス(パピローマウイルス、ポリオーマウイルス)、アデノウイルス、ヘルペスウイルス(単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルス)およびアフリカ豚コレラウイルスよりなる群から選択される少なくとも1種であることが好ましい。
【発明の効果】
【0029】
本発明の方法によると、被検試料における核酸の分離・精製等の前処理を必要とせず、簡易に高感度で検出し得る特定遺伝子、具体的には、特定ウイルスまたは特定微生物の検出をすることができる。
【発明を実施するための最良の形態】
【0030】
本発明の特定遺伝子の検出方法においては、少なくとも第1工程〜第4工程を備える。第1工程は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する工程である。第2工程は、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する工程である。第3工程は、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する工程である。第4工程は、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程である。
【0031】
また、本発明の別の形態における特定遺伝子の検出方法においては、抗体、レクチンおよび複合糖質のいずれかが固定化されたチューブ状の生体高分子固定化担体を用いる。そして、該特定遺伝子の検出方法においても4つの工程を備える。これらの工程は、ホスホリルコリン基またはアルキレングリコール基等の生体適合性の高い官能基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、抗体、レクチンおよび複合糖質のいずれかで微生物またはウイルスを捕捉する工程と、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、生体高分子固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程とからなる。なお、ホスホリルコリン基またはアルキレングリコール基等の生体適合性の高い官能基を有する高分子物質については、特開2008−128854号公報に記載されているものを挙げることができる。
【0032】
[第1実施形態]
以下、各工程について詳細に説明する。
【0033】
<第1工程>
本工程においては、チューブ状のDNA固定化担体を作製する。より具体的には、チューブ状の不溶性担体の少なくとも内側を高分子物質で被覆して、その上にキャプチャーDNA鎖を固定化させることによってチューブ状のDNA固定化担体を作製する。
【0034】
まず、本実施形態の本工程における「高分子物質」は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含み、DNA鎖およびRNA鎖の非特異的吸着を抑制する性質とDNA鎖を固定化する性質とを併せ持つポリマーを示す。特に、第一単位に含まれるホスホリルコリン基は鋳型RNA断片の非特異的吸着を抑制する役割を果たし、第二単位に含まれるカルボン酸誘導基はキャプチャーDNA鎖を化学的に固定化する役割を果たす。すなわち、キャプチャーDNA鎖は、このコーティング層の活性エステル基の部位で共有結合して、当該不溶性担体の表面に固定化される。具体的には、住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブを例として挙げることができる。なお、チューブ状のDNA固定化担体の作製方法については、特許文献2に詳細が記載されている。
【0035】
次に、本発明における「不溶性担体」は、プラスチック、ガラス等からなるチューブのほか、96穴ウェル等を挙げることができる。
【0036】
チューブ状とは、中空状態のものをいい、底があるPCRチューブや、エッペンドルフチューブのような形状であってもよい。
【0037】
そして、本発明における「キャプチャーDNA鎖」は、後述する被検試料に含まれる微生物由来遺伝子またはウイルス由来遺伝子におけるターゲット配列領域の一部と相補的な配列を有するDNA鎖をいう。なお、キャプチャーDNA鎖は、ターゲット配列領域の相補的な配列と相同性が50%以上であればよい。キャプチャーDNA鎖には、塩基数が100以下より好ましくは80以下のDNA鎖を用いることが好ましい。これは、キャプチャーDNAの長さが100以下である場合には、合成が容易であり、また、後述するハイブリダイゼーションを安定して行なうことができるためである。また、キャプチャーDNA鎖には、修飾されたオリゴDNAを用いることが好ましいが、無修飾のオリゴDNAを用いても固定化が可能である。さらには、キャプチャーDNA鎖がアミノ基が導入されたオリゴDNAである場合には、固定化効率が高くなる。アミノ基は活性エステル基との反応性に優れるため、キャプチャーDNA鎖として、アミノ基が導入されたオリゴDNAを用いることにより、該キャプチャーDNA鎖は、効率よくかつ強固に該高分子物質上にプライマーを固定化することができる。アミノ基の導入位置はキャプチャーDNA鎖の分子鎖末端あるいは側鎖であってもよいが、分子鎖末端に導入されていることが、相補的なDNA断片またはRNA断片とのハイブリダイズをより一層効率よく行なうことができるという観点からは、好ましい。
【0038】
本発明において「ターゲット配列領域」とは、微生物またはウイルスにおける全遺伝子のうち、本発明に用いるプライマーを用いたPCRにより増幅され得る配列領域であり、検出対象のウイルスまたは微生物を検出することができるものであれば特に制限されず、目的に応じて適宜設定することができる。
【0039】
たとえば、被検試料に検出対象のウイルスまたは微生物と異なる種類の細胞が含まれる場合には、ターゲット配列領域は、検出対象のウイルスまたは微生物に特異的な配列を有することが好ましい。また、目的によっては、複数種のウイルスまたは微生物に共通する配列を有するものであってもよい。さらに、ターゲット配列領域は単一であっても、複数であってもよい。
【0040】
キャプチャーDNA鎖のターゲット配列領域がさらに、トポイソメラーゼ、または、DNAジャイレースを含む微生物特異的な配列であってもよい。
【0041】
ターゲット配列領域の長さとしては、通常20〜数十万塩基程度まで適用できるが、500〜3000塩基であることが好ましい。検出対象が微生物である場合には、具体的には、ターゲット配列領域は、16S rRNA遺伝子および23S rRNA遺伝子が挙げられる。これは、広汎な微生物に応用して使用することができるためである。
【0042】
<第2工程>
本工程は、特定遺伝子の検出がされるかどうか検査したい被検試料、具体的には、微生物の特定遺伝子またはウイルスの特定遺伝子の検出がされるかどうか検査したい被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する工程である。
【0043】
本発明において、「被検試料」が「ウイルスを含む被検試料」である場合においては、該被検試料は、ウイルス感染によってウイルスが存在しえる生体試料であれば特に制限されることなく任意に使用することができる。
【0044】
ウイルスの特定遺伝子の検出がされるかどうか検査したい被検試料は、対象とする被験者から、たとえば滅菌綿棒などの滅菌器具および容器を用いることにより無菌的に採取することが好ましい。得られた該被検試料は、採取後、なるべく速やかにグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液でウイルスを溶解させることができる。このような操作によって、遺伝子溶解溶液を得ることができる。
【0045】
また、後述する第3工程でDNA鎖断片またはRNA鎖断片を捕捉するために、希NaOH溶液で被検試料を溶解した場合には、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えることが好ましい。また、該遺伝子溶解溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0046】
本発明において、「被検試料」が「微生物を含む被検試料」である場合においては、該被検試料は、その中に存在する微生物の生菌を検出する対象であり、PCR法による染色体DNAの特定領域の増幅によって存在を検出することが可能なものであれば特に制限されない。
【0047】
微生物の特定遺伝子の検出がされるかどうか検査したい被検試料は、食品原材料付着の製品または生体試料そのものであってもよく、これを希釈もしくは濃縮したもの、または本発明の方法による処理以外の前処理をしたものであってもよい。前処理としては、加熱処理、濾過、遠心分離等が挙げられる。得られた被検試料は、採取後、なるべく速やかにグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液で微生物を溶解させることができるが、次工程のハイブリダイゼーションで対象RNA遺伝子を捕捉するためには、希NaOH溶液で生体試料を、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えたほう好ましい。溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0048】
また、被検試料は、鼻汁、粘膜ぬぐい液(たとえば鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液等)、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液等の各種体液、または各種組織(細胞組織)または食品を例示することができる。本発明方法の対象となる食品は、特に制限されない。本発明方法は、飲料、麺製品、食肉、魚介類、野菜、穀類、乳製品およびこれらの加工物等の広い範囲の食品を対象にすることができる。
【0049】
また、被検試料としての食品からRNAを抽出する場合において、食品から分離培養した微生物からRNAを抽出してもよいが、食品から直接RNAを抽出することもできる。また、微生物を含むかもしれない食品を遠心処理することにより、微生物を含むかもしれない画分を食品と分離した後に、微生物を含むかもしれない画分からRNAを抽出することもできる。
【0050】
<第3工程>
本工程は、キャプチャーDNA鎖と相補的な配列を有する遺伝子溶解溶液中のDNA鎖断片またはRNA鎖断片を分離する工程である。具体的には、チューブ状のDNA固定化担体の内側(内部)に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせ、DNA鎖断片またはRNA鎖断片を捕捉する。以上の工程によって、遺伝子溶解溶液中の遺伝子のなかからキャプチャーDNA鎖と相補的な配列を有する遺伝子溶解溶液中のDNA鎖断片またはRNA鎖断片を分離することができる。
【0051】
本工程においては、4〜70℃、より好ましくは15〜37℃(室温)で静置または攪拌状態でキャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせる。本工程は、5分〜90分間で完了できる。90分間超過の時間をかけてハイブリダイズさせることもできるが、本発明においては、1時間程度のハイブリダイズで十分であり、結果として迅速に特定遺伝子の検出を行なうことができる。
【0052】
<第4工程>
本工程は、キャプチャーDNA鎖に捕捉されたDNA鎖断片またはRNA鎖断片を増幅する工程である。本工程においては、上述のチューブ状のDNA固定化担体に対して直接、ポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施すことができる。具体的な操作は以下のとおりである。
【0053】
まず、第3工程の後のチューブ状のDNA固定化担体から被検試料を除去する。そして、該DNA固定化担体の表面をPBSにて1〜数回洗浄することが好ましい。本発明において、被検試料中に存在する微生物や夾雑物質等による検出誤差を回避するためである。これにより、チューブ状のDNA固定化担体の内部における核酸はキャプチャーDNA鎖とキャプチャーDNA鎖に捕捉されたDNA鎖断片またはRNA鎖断片が残る。
【0054】
次に、DNA固定化担体の内部に所定の物質(dNTP等)を添加して核酸の増幅を行なう。
【0055】
キャプチャーDNA鎖に捕捉された核酸がDNA鎖断片の場合においては、直接PCR法またはLAMP法等を行なうことができる。また、キャプチャーDNA鎖に捕捉された核酸がRNA断片の場合においては、RNA鎖断片の場合は逆転写反応を行なった後、ポリメラーゼ連鎖反応(PCR)、LAMP法、鎖置換増幅(SDA)、逆転写酵素鎖置換増幅(RT−SDA)、逆転写酵素ポリメラーゼ連鎖反応(RT−PCR)、逆転写LAMP法(RT−LAMP)、核酸配列に基づく増幅(NASBA)、転写媒介性増幅(TMA)、ローリングサークル型増幅などの遺伝子増幅法を用いて増幅することができる。
【0056】
検出対象の微生物に特異的なターゲット配列領域に対応するプライマーセットと、広汎なウイルスまたは微生物のDNAに対応するプライマーセットを用いると、検出対象の微生物の生菌量と、多数種の微生物の生菌量を、同時に測定することができる。該ターゲット配列領域の長さとしては、通常20〜数十万塩基程度まで適用できるが、500〜3000塩基であることが好ましい。検出対象が微生物である場合には、具体的には、ターゲット配列領域は、16S rRNA遺伝子および23S rRNA遺伝子が挙げられる。
【0057】
本発明においてPCR法に用いるプライマーは、上記ターゲット配列領域を特異的に増幅することができるものであれば特に制限されない。検出対象が微生物である場合に、具体的には、23S rRNA遺伝子に対応するプライマーとしては、配列番号1、2および3に示すプライマーセット(Journal of Clinical Microbiology,42 1048-1057(2004))が挙げられる。また、16S rRNA遺伝子に対応するプライマーとしては、配列番号4および5に示すプライマー(MicroSeq500)セット、配列番号6および7に示すプライマーセット(Journal of Clinical Microbiology,32:335−351(1994)、Journal of Clinical Microbiology,43:3390−33397(2005)、 Journal of Molecular Diagnostics,7:575581−33397(2005))、および配列番号8および9に示すプライマーセット(Experimental Biology and Medicine 230:587−591(2005))が挙げられる。
配列番号1:5’−GACAGCCAGGATGTTGGCTTAGAAGCAGC−3’(43a2)
配列番号2:5’−GGAATTTCGCTACCTTAGGACCGTTATAGTTACG−3’(69ar2)
配列番号3:5’−GGAATTTCGCTACCTTAGGATGGTTATAGTTACC−3’(69arrh)
配列番号4:5’−GAGTTTGATCCTGGCTCAG−3’(16S−9F)
配列番号5:5’−GTATTACCGCGGCTGCTG−3’(16S−536R)
配列番号6:5’−AACTGGAGGAAGGTGGGGAY−3’(RW01&RDR080、1170−1189)
配列番号7:5’−AGGAGGTGATCCAACCGCA−3’(DG74、1522−1540)
配列番号8:5’−ACTCCTACGGGAGGCAGCAGT−3’(Pba1、358-378)
配列番号9:5’−TCACCGGCCGTGTGTACAAG−3’(Pba2、1444-1425)
LAMP法は、栄研化学(株)社が開発した核酸増幅法である。LAMP法は、標的遺伝子の6箇所の領域に対して4種類のプライマーを設定して、鎖置換反応を利用し一定温度で反応させることを特徴とする。反応はサンプルとなる遺伝子、LAMPプライマー、鎖置換型DNA合成酵素、基質核酸等を同一の反応チューブに混合し、一定温度(65℃付近)で保温することにより、遺伝子の増幅から検出までを1ステップの工程で行なうことができる。増幅効率が高く、DNAを15分〜1時間で109〜1010倍に増幅することが可能である。その極めて高い特異性から、増幅産物の有無で目的とする標的遺伝子配列の有無を判定することができる。また、増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無によっても標的遺伝子配列の有無を判別できる。また、過剰のインターカレーターSYBR Green IをLAMP反応液の核酸増幅産物に添加することによって、標的核酸の有無を特別な光源や検出装置を用いることなく目視確認するする方法でも標的遺伝子配列の有無を判別できる。さらには、電気泳動によって、アガロースゲルのウェルから尾を引いたようなバンドが確認できれば、遺伝子が増幅されたことを示している(特許文献3参照)。
【0058】
RT−LAMP(Reverse Transcription−Loop−mediated Isothermal Amplification)法は、LAMP法を応用し、ノロウイルスのように標的遺伝子であるRNAから逆転写酵素反応よりcDNAを合成して増幅して検出する方法である。サンプルとなるRNAに対して、DNAを標的遺伝子とする場合と同様の試薬(プライマー、鎖置換型DNA合成酵素、基質核酸等)に逆転写酵素を追加して混合することによって、一定温度(60〜65℃)下で増幅から検出までの工程を1ステップで行なうことができる。増幅から検出までを1ステップで行なうことができる特徴を有する(非特許文献3参照)。
【0059】
<第5工程>
本工程は、第4工程の後にさらに加えられることが好ましい工程である。本工程では、第4工程の後に増幅したDNA鎖断片(PCR増幅産物)を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する。
【0060】
本工程における解析法は、PCR増幅産物の検出または定量が可能なものであれば特に制限されず、電気泳動法、リアルタイムPCR法等が挙げられる。電気泳動法によれば、PCR増幅産物の量、およびその大きさを評価することができる。また、リアルタイムPCR法によれば、迅速にPCR増幅産物の定量を行なうことができる。リアルタイムPCR法を採用する場合、一般に増幅サイクル数1〜10までは蛍光強度の変化はノイズレベルでありゼロに等しいので、それらを増幅産物ゼロのサンプルブランクと見なし、それらの標準偏差SDを算出しその10を乗じた蛍光値をスレッショード値とし、そのスレッショード値を最初に上回るPCRサイクル数をサイクルスレッショード値(Ct値)という。したがって、PCR反応溶液に初期のDNA鋳型量が多い程、Ct値は小さな値となり、鋳型DNA量が少ない程、Ct値は大きな値となる。また、鋳型DNA量が同じでも、その鋳型内のPCRのターゲット配列領域に切断が生じている割合が多くなる程、同領域のPCR反応のCt値は大きな値となる。
【0061】
また、複数種の微生物に共通するプライマーを用いると、被検試料中の複数種の微生物の生菌を検出することができる。また、特定の微生物に特異的なプライマーを用いると、被検試料中の特定の菌種の生菌を検出することができる。
【0062】
PCRの条件は、PCRの原理にのっとった特異的な増幅が起る限り特に制限されず、適宜設定することができる。
【0063】
本発明の方法によって生菌を検出する場合、PCR増幅産物の解析は、同定されている微生物の標準試料を用いて作成された微生物量および増幅産物との関連を示す標準曲線を用いると、生菌の有無または定量の精度を高めることができる。標準曲線は予め作成しておいたものを用いることができるが、被検試料と同時に標準試料について本発明の各ステップを行なって作成した標準曲線を用いることが好ましい。また、予め微生物量とRNA量との相関を調べておけば、その微生物から単離されたDNAを標準試料として用いることもできる。
【0064】
<各用語>
本発明においては、「ウイルス」という用語は、たとえば、C型肝炎ウイルス(HCV)、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス(黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス)、トガウイルス(アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス)、ペスチウイルス(ブタコレラウイルス、ウシ下痢ウイルス)、パラミクソウイルス(パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス)、オルソクソウイルス(ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス)、ラブドウイルス(狂犬病ウイルス、水泡性口内炎ウイルス)、ピコルナウイルス(ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス)、コロナウイルス(ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス)、アレナウイルス(リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス)、レトロウイルス(HTLV:ヒト成人白血病ウイルス、HIV:エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス)、レオウイルス(ロタウイルス)、カリシウイルス(ノーウオークウイルス)、ノロウイルス、ブンヤウイルス(腎症候性出血熱ウイルス)、フィロウイルス(エボラウイルス、マールブルグウイルス)、B型肝炎ウイルス(HBV)、ポックスウイルス(ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス)、パルボウイルス(ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス)、パポーバウイルス(パピローマウイルス、ポリオーマウイルス)、アデノウイルス、ヘルペスウイルス(単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルス)およびアフリカ豚コレラウイルスよりなる群から選択される。
【0065】
本発明においては、「微生物」という用語は、原核生物、細菌、真菌、寄生生物、または原生動物を意味するのに使用する。「微生物」としては、グラム陽性菌およびグラム陰性菌のいずれもが含まれ、ブドウ球菌(スタフィロコッカス・エピダーミディス(Staphylococcus epidermidis))のスタフィロコッカス属細菌、肺炎連鎖球菌(Streptococcus pneumoniae)等のストレプトコッカス属細菌、リステリア・モノサイトゲネス(Listeria monocytogenes)等のリステリア属細菌、バチラス・セレウス(Bacillus cereus)等のバチラス属細菌、黄色ブドウ球菌(Staphylococcus aureus)、マイコバクテリウム属細菌、大腸菌(Escherichia coli)、表皮ブドウ球菌(Staphylococcus epidermidis)、肺炎桿菌(Klebsiella pneumoniae)、エンテロコッカスフェカリス(Enterococcus faecalis)、緑膿菌(Pseudomonas aeruginosa)、エンテロバクター・サカザキ(Enterobacter sakazakii)等のエンテロバクター属細菌、シトロバクター・コーセリ(Citrobacter koseri)等のシトロバクター属細菌、クレブシェラ・オキシトカ(Klebsiella oxytoca)等のクレブシェラ属細菌に代表される腸内細菌群、サルモネラ属細菌、ビブリオ属細菌、シュードモナス属細菌等ミュータンス連鎖球菌(Streptococcus mutans)、ストレプトコッカスゴルドニイ(Streptococcus gordonii)、ウェルシュ菌(Clostridium perfringens)、ボツリヌス菌(Clostridium botulinum)、インフルエンザ菌(Haemophilus influenzae)、エンテロコッカスデュランス(Enterococcus durans)、化膿連鎖球菌(Streptococcus pyogenes)、ストレプトコッカスアガラクティエ(Streptococcus agalacticae)、クロストリジウムディフィシレ(Clostridium difficile)およびエンテロコッカスフェシウム(Enterococcus faecium)などが挙げられる。
【0066】
本発明において、微生物における「生菌」の検出とは、被検試料中の生菌の有無の判別および生菌の量の決定のいずれをも含む。また、生菌の量とは、絶対的な量に限られず、対照試料に対する相対的な量であってもよい。得られる菌数が少なすぎるために十分量のRNAを得ることができない場合や、死菌がPCR反応の結果に影響を与えると考えられる場合には、被験試料に滅菌した微生物用液体培地を添加し、30〜37℃程度で培養することにより菌数を増加させればよい。これによりRNA検出感度を向上させることができ、また死菌の影響を抑えることができる。液体培地の種類は特に制限されず、ブレインハートインフィージョン培地、トリプトソーヤ培地等の一般微生物用培地を広い範囲から適宜選択して使用することができる。ビブリオ属細菌の場合は、これらの培地において食塩濃度を1〜3%程度に調製したものまたはアルカリ性ペプトン培地を使用すればよい。
【0067】
また、検出対象の微生物が病原性微生物である場合には、ターゲット配列領域としては病原遺伝子が挙げられる。病原遺伝子としては、リステリア属細菌のリステリオリシンO(hlyA)遺伝子、サルモネラ属細菌のエンテロトキシン遺伝子やinvA遺伝子、病原性大腸菌O−157のベロ毒素遺伝子、エンテロバクター属細菌のMMS遺伝子(エンテロバクター・サカザキ菌)、黄色ブドウ球菌エンテロトキシン遺伝子、バチルス・セレウス菌のセレウリド(嘔吐毒素)遺伝子やエンテロトキシン遺伝子、ボツリヌス菌の各種毒素遺伝子等が挙げられる。
【0068】
[第2実施形態]
<チューブ状の生体高分子固定化担体を作製する工程>
本工程においては、チューブ状の生体高分子固定化担体を作製する。より具体的には、チューブ状の不溶性担体の少なくとも内側を高分子物質で被覆して、その上に抗体、レクチンおよび複合糖質のいずれかを固定化させることによってチューブ状の生体高分子固定化担体を作製する。
【0069】
まず、本実施形態の本工程における「高分子物質」は、ホスホリルコリン基またはアルキレングリコール基など生体適合性の高い官能基を有するような高分子物質であり、生体成分の非特異的吸着抑制するモノクローナル抗体、タンパク質、レクチン、複合糖質などの生体分子を固定する性質を持つポリマーを示す。具体的には、住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブ、ビーズ単体、プレートを例として挙げることができる。なお、固定化担体の作製方法については、特開2008−128854号公報に記載されている。
【0070】
次に、本発明における「不溶性担体」は、プラスチック、ガラス等からなるチューブのほか、96穴ウェル等を挙げることができる。
【0071】
チューブ状とは、中空状態のものをいい、底があるPCRチューブや、エッペンドルフチューブのような形状であってもよい。
【0072】
そして、本発明における「抗体」、「レクチン」、「複合糖質」は、後述する被検試料に含まれる微生物またはウイルスの一部をエピトープ部位または認識部位として感知できるものであれば特に限定されない。抗体には、免疫グロブリンのようなものを用いることができる。
【0073】
本発明において「エピトープ部位」または「認識部位」には、微生物の表面構造(鞭毛、夾膜、細胞壁糖鎖など)またはウイルスのカプシドおよびエンベロープ部分を設定することができる。
【0074】
たとえば、被検試料に検出対象のウイルスまたは微生物と異なる種類の細胞が含まれる場合には、ターゲット配列領域は、検出対象のウイルスまたは微生物に特異的な配列を有することが好ましい。また、目的によっては、複数種のウイルスまたは微生物に共通する配列を有するものであってもよい。さらに、ターゲット配列領域は単一であっても、複数であってもよい。
【0075】
<微生物またはウイルスを捕捉する工程>
本工程は、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、該微生物またはウイルスを分離する工程である。具体的には、チューブ状の生体高分子固定化担体の内側(内部)に被検試料を添加し、該生体高分子固定化担体を2時間程度、静置することで、該微生物またはウイルスを捕捉する。
【0076】
<遺伝子溶解溶液を作製する工程>
本工程は、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する。
【0077】
上記微生物またはウイルスを捕捉した生体高分子固定化担体を前処理する。該生体高分子固定化担体に、グアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液で微生物を溶解させることができるが、次工程のハイブリダイゼーションで対象RNA遺伝子を捕捉するためには、希NaOH溶液で生体試料を、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えたほう好ましい。溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0078】
<DNA鎖断片もしくはRNA鎖断片で増幅する工程>
本工程は、DNA鎖断片またはRNA鎖断片を増幅する工程である。本工程においては、上述のチューブ状のDNA固定化担体に対して直接、ポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施すことができる。具体的な操作は上述した方法をとることができる。
【0079】
その他、被検試料等については、上述のものと同様のものを応用することができる。
[応用]
本発明は、特に、食品衛生検査や臨床検査において、穏和な加熱処理や抗生物質投与により、損傷菌の状態を呈した微生物の検出が注目されており、本発明においては、生菌の検出のみならず、生菌または死菌との識別も可能な微生物の検出方法を提供するものである。
【0080】
本発明により、ウイルスの感染判定または微生物(細菌)の感染判定を迅速・簡便に行なうことが可能なキットを提供される。試験に要する時間は、患者からの被検試料受領後約2時間であり、従来技術とは完全に区別される迅速診断法である。本発明のキットを用いれば、検査担当者の2次感染を防止でき、ウイルス感染の伝播防止と感染者への迅速な対応と効果的な治療が可能になり、公衆衛生上大きく寄与するものと考えられる。
【0081】
そして、本発明の特定遺伝子の検出方法を用いることによって、ウイルス感染症および細菌感染の診断に関して、患者の臨床状態を迅速にしかも的確に反映することのできる治療法を提供することができる。該特定遺伝子の検出方法は、ウイルス感染症のまたは細菌感染の診断および治療に有効であるだけでなく、他者への感染のリスクの予測、それによる院内感染の防止、治療効果の追跡による現行処置の適否判断やさらなる治療対策の検討が可能となる。
【0082】
そして、本発明の特定遺伝子の検出方法では、生体試料権対中のウイルス溶解処理から遺伝子増幅および検出までに要する時間はわずか2時間程度で実施可能である。この短時間の操作により迅速かつ簡便にウィルスを検出することができる。
【0083】
すなわち、本発明の特定遺伝子の検出方法によると、短時間で且つ高感度にウイルスまたは微生物を検出できるため、ウイルス感染症患者の病態および細菌感染症患者の病態を的確に反映することができる。さらに、本発明は、従来の分離および精製等の前処理を必要とせず、簡易に高感度で検出し得るウイルスの検出方法を提供することで遺伝子検査作業者の2次感染予防の効果が期待できる。
【0084】
さらに本発明の特定遺伝子の検出方法によると、主要な食中毒細菌の食品中への混入の可能性を簡単かつ高感度に調べることができる。食品メーカーによる製品出荷前の自主細菌検査に好適に利用できる。また、本発明の細菌の検出方法において、食品の破砕工程と遠心分離操作とを行なう場合には、細菌を食品から効率よく分離採取できるため、細菌を食品から分離培養する必要がなくなり、その分検査時間が短縮される。
【0085】
以下、実施例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0086】
<実施例1>
≪第1工程(a):ノロウイルスG1型検出用PCRチューブの作製≫
非特許文献2(ノロウイルスの検出法)記載の公知のノロウイルスG1型特異的DNA塩基配列5’−CTTAGACGCCATCATCATTYAC−3’(22塩基)(配列番号10)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製した。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの内部底面に添加し、室温で1時間静置することで、キャプチャーDNA鎖を固定化させてDNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNaseフリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0087】
≪第1工程(b):ノロウイルスG2型検出用PCRチューブの作製≫
非特許文献2(ノロウイルスの検出法)記載の公知のノロウイルスG2型特異的DNA塩基配列5’−TCGACGCCATCTTCATTCACA−3’(21塩基)(配列番号11)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの内部底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させてDNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNase フリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0088】
≪第2工程:ノロウイルス感染患者糞便検体の処理≫
ノロウイルス感染患者糞便検体にPBSを加え、10%乳剤を作製し、この10%乳剤を激しく攪拌した後、12,000rpmで20分間冷却遠心分離して得られた遠心上清をノロウイルスを含有するサンプルとして使用した。
【0089】
≪第3工程:ノロウイルス遺伝子の分離≫
15μLのサンプルを、作製したノロウイルスG1型およびG2型検出用PCRチューブそれぞれに添加し、5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))をさらに加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で該チューブを30分間静置してノロウイルス遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内の「ノロウイルスを含有するサンプル」を、マイクロピペットで完全に除去後、PCRチューブ内をPBS150μLで洗浄した。上記操作にて、ノロウイルス遺伝子を分離したPCRチューブを使用して以下のRT−LAMP法による遺伝子検出法に供した。
【0090】
≪第4工程、第5工程:RT−LAMP法によるノロウイルス遺伝子の増幅および検出≫
ノロウイルス遺伝子の検出は、栄研化学(株)社製LoopampノロウイルスGI検出試薬キットおよびLoopampノロウイルスGII検出試薬キットを使用して行なった。反応条件は添付のプロトコールにしたがって実施した。具体的には、上述したようにノロウイルス感染患者糞便検体からノロウイルス遺伝子をオリゴDNAが固定化されたロウイルスG1およびG2検出用PCRチューブでそれぞれ分離し、ノロウイルスG1検出用PCRチューブには、LoopampノロウイルスGI検出試薬キットより調整された反応液を添加し、ノロウイルスG2検出用PCRチューブには、LoopampノロウイルスGII検出試薬キットより調整された反応液を添加した。そして、それぞれのPCRチューブに対してRT−LAMP法による遺伝子増幅反応を63℃、60分間で行ない、遺伝子増幅反応終了後、80℃、5分間にて酵素を失活させた。
【0091】
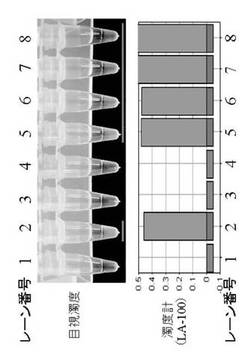
図1は、実施例1のLAMP法反応の増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無による標的遺伝子配列の有無を判別した図である((上):目視濁度結果、(下):栄研化学(株)社製Loopampエンドポイント濁度測定装置(LA−100)による濁度測定結果。レーン番号1および2は、水またはコントロールRNAを栄研化学(株)社製LoopampノロウイルスGI検出試薬キットを用いて行なったLAMP法のコントロール反応)。なお、図1の濁度測定結果の縦軸は濁度(単位:OD660値(透過率))を示す。
【0092】
図2は、実施例1のLAMP法反応液に約1μLの1000×の濃度のSYBR Green I添加(最終濃度:100×)によるLAMP法反応液の核酸増幅産物の目視確認結果を示す図である。
【0093】
図1、図2におけるレーン番号1〜8は、以下のとおりである。
1:ネガティブコントロール(水添加による通常LAMP法)
2:ポジティブコントロール(G1コントロールRNA添加による通常LAMP法)
3:ノロウイルスG1型検出用PCRチューブ(水添加)
4:ノロウイルスG2型検出用PCRチューブ(水添加)
5:ノロウイルスG1型検出用PCRチューブ(G1コントロールRNA添加)
6:ノロウイルスG2型検出用PCRチューブ(G2コントロールRNA添加)
7:ノロウイルスG1型検出用PCRチューブ(G1糞便検体処理液添加)
8:ノロウイルスG2型検出用PCRチューブ(G2糞便検体処理液添加)
[結果]
ポジティブコントロールと同様にレーン番号5〜8においてもノロウイルスを検出することができた。
【0094】
<確認実験1>
TaqMan RT−qPCR法、並びにNASBA(Nucleic Acid Sequence−Based Amplification)法のスイフトジーンノロウイルスGI/GII「カイノス」(株式会社カイノス製)、TRC (Transcription Reverse transcription Concerted reaction)法のノロウイルスRNA検出試薬TRCRtest Noro (東ソー株式会社製)の各キットを用いた増幅反応を同一のノロウイルス感染患者由来の糞便サンプルを用いて行なった。いずれの結果も、LAMP法と同等の結果を得ることができた。
【0095】
<確認実験2:LAMP法の感度>
本発明の検出方法にLAMP法を導入したことにより、まずLAMP法の遺伝子増幅の限界を確認する必要がある。
【0096】
そこで、10000〜10コピー/チューブのノロウイルスG2コントロールRNA並びにノロウイルスG2感染患者糞便サンプルから通知法(非特許文献2)に従い抽出した3種類のRNA(S1,S2,S3)を用いてLAMP法の遺伝子増幅の限界を確認した。ノロウイルスG2RNAをノロウイルスG2型検出用PCRチューブに添加してノロウイルス遺伝子を分離後、LAMP法の増幅反応液に添加し、63℃、60分の増幅反応後、アガロースゲル電気泳動を行なった。コントロールとして、ノロウイルスG2 RNAをLAMP法の増幅反応液に添加し、添付プロトコールに従い実施した。結果は図3に示した。図3は、実施例1のコントロールのノロウイルスRNAとノロウイルス感染患者糞便サンプル(S1〜3)を用いたLAMP法反応結果を示す図であり、図3Aは、ノロウイルスG2型検出用PCRチューブを用いてノロウイルス遺伝子を分離後、LoopampノロウイルスG2検出試薬キットを用いて行なったLAMP法遺伝子増幅反応産物を電気泳動した図である。図3Bは、ノロウイルス遺伝子を直接LoopampノロウイルスGI検出試薬キットに添加して行なったLAMP法のコントロールの結果である。
【0097】
図3の各レーン番号は以下のとおりとなっている。
M:マーカー
1:10コピー/チューブ (G2コントロールRNA添加)
2:100コピー/チューブ (G2コントロールRNA添加)
3:1000コピー/チューブ (G2コントロールRNA添加)
4:10000コピー/チューブ (G2コントロールRNA添加)
5:ノロウイルス感染患者糞便サンプル(S1)
6:ノロウイルス感染患者糞便サンプル(S2)
7:ノロウイルス感染患者糞便サンプル(S3)
B:ネガティブコントロール(水添加)
なお、電気泳動によって、アガロースゲルのウェルから尾を引いたようなバンドが確認できれば、遺伝子が増幅されたことを示している。
【0098】
図3に示すようにノロウイルスG2型検出用PCRチューブを使用してウイルス遺伝子を分離後、LAMP法を実施した増幅限界は10000コピー/チューブまでであった。この検出限界は、一般的に広く用いられているPCR法と同程度以下ではある。また、ノロウイルス感染患者糞便サンプル(S1,S2,S3)から抽出したRNAを用いた場合には、いずれも検出可能であった。コントロールの通常のLAMP反応液に直接ノロウイルスRNAを添加したLAMP法の結果は、非特許文献3の結果とほぼ同等の結果であった。この非特許文献3においては、LAMP法の増幅時間はPCR法が約2時間かかるのに対し、本実施例においてLAMP法は1時間であった。これより、核酸増幅にLAMP法を用いることで検査時間の短縮が可能であると考えられた。また、ノロウイルスG1に関しても同様の実験を行い、同等の検出感度であることを確認した。
【0099】
<確認実験3:再現性について>
繰り返して100回、上記同様の測定した結果、毎回ほぼ同程度の結果が検出された。これにより、LAMP法ではほぼ決まった長さの生成物が同程度増幅されることが確認された。
【0100】
<実施例2>
≪第1工程:HIV−1検出用PCRチューブの作製≫
公知のHIV−1特異的DNA塩基配列5’−TACTAGTAGTTCCTGCTATGTCACTTCC−3’(28塩基)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させることで、DNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNase フリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0101】
≪第2工程:HIV−1コントロールRNAの作製≫
HIV−1より作製された公知のプラスミドDNA(pNL432)のgag遺伝子領域をPCR法で遺伝子増幅し、pSTBlue−1 AccepTor(TM) Vector Kitsを使用して常法によりクローニングを行い、in VitroでHIV−1コントロールRNAを作成した。その遺伝子配列は、以下の通りである。
cagcagctgacacaggaaacagcaacaaggtcagccaaaattaccctatagtgcagaacatccaggggcaaatggtacatcaggccatatcacctagaactttaaatgcatgggtaaaagtagtagaagagaaggcttttagcccagaagtaatacccatgttttcagcattatcagaaggagccaccccacaagatttaaacaccatgctaaacacagtggggggacatcaagcagccatgcaaatgttaaaagagaccatcaatgaggaagctgcagaatgggatagattgcatccagtgcaggcagggcctgtcgcaccaggccagatgagagaaccaaggggaagtgacatagcaggaactactagtacccttcaggaacaaataggatggatgacaaataatccacctatcccagtaggagaaatctataaaagatggataatcctgggattaaataaaatagtaaggatgtatagccctgccagcattctggacataagacaaggaccaaaggaaccctttagagattatgtagaccggttctataaaactctaagagccgagcaagcttcaca(配列番号12)
≪第3工程:市販ヒト血清へコントロールHIV−1 RNAの添加およびHIV−1遺伝子の分離回収)
作製したHIV−1検出用PCRチューブに10μLの市販ヒト血清を4μLのPBS希釈して添加し、HIV−1コントロールRNAを106〜102コピー/チューブ加えた。このヒト血清とHIV−1コントロールRNAとの混合物からなるサンプルは、実質的にHIV感染患者の血清と同様なものとして利用した。さらに、該サンプルに5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))を加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機でスピンダウン後、室温で30分間静置してHIV−1遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のHIV−1溶解液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。操作にてHIV−1遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による増幅に供した。
【0102】
≪第4工程:RT−PCR法による増幅≫
RT−PCR法の条件設定を行なった。これは、目的のRNAを増幅するための条件設定のために行なった。
【0103】
(TaqMan RT−qPCR条件調整)
ReverTra Ace(東洋紡績)を100U/μlの原液を専用の希釈バッファーで10U/μl(10倍希釈)希釈する。この希釈液を下記の表に従い、逆転写反応液試薬(RT反応液)を調整した。
【0104】
【表1】

【0105】
次に下記のプロトコールに従い、ワンステップのTaqMan RT−qPCR反応液を調整した。
【0106】
【表2】
【0107】
SK145(Forward):5’−AGTGGGGGGACATCAAGCAGCCATGCAAAT−3’(配列番号13)
SKCC1B(Reverse):5’−TACTAGTAGTTCCTGCTATGTCACTTCC−3’(配列番号14)
SK102−TP(TaqMan Probe):5’−GAGACCATCAATGAGGAAGCTGCAGAATGGGAT−3’(配列番号15)
(RT−PCRプログラム)
48℃,20分→95℃,5分→94℃,15秒⇔54℃,45秒、45サイクル
(結果)
TaqMan RT−qPCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行なった。表3に示すようにHIV−1検出用PCRチューブを使用してウイルス遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は100コピー/チューブまでであることが分かった。
【0108】
【表3】
【0109】
(RT−PCR条件)
TaqMan RT−qPCR反応液に調整したRT反応液を使用して、次に下記のプロトコールに従い、RT−PCR反応液の調整を行なった。
【0110】
【表4】
【0111】
G60(Forward):5’−CAGCCAAAATTACCCTATAGTGCAG−3’(配列番号16)
SK39(Reverse):5’−TTTGGTCCTTGTCTTATGTCCAGAATGCTG−3’(配列番号17)
(RT−PCRプログラム)
48℃,20分→95℃,1分→94℃,15秒⇔54℃,1分、40サイクル
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図4は、コントロールHIV−1 RNAを添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動(458bp)法で検出した結果を示す図である。そして、具体的には、図4Aは、上述したヒトヒト血清とHIV−1コントロールRNAとの混合物からなるサンプルを用いて第1工程〜第4工程までした結果を示し、図4Bは、HIVコントロールRNAのみを第1工程〜第4工程までした結果を示す。
【0112】
図4における各レーン番号は以下のとおりとなっている。
M:マーカー
1:100コピー/チューブ (コントロールHIV−1 RNA添加)
2:1000コピー/チューブ (コントロールHIV−1 RNA添加)
3:10000コピー/チューブ (コントロールHIV−1 RNA添加)
B:ネガティブコントロール(水添加)
図4に示すようにHIV−1検出用PCRチューブを使用してウイルス遺伝子を分離後、RT−PCR法を実施した増幅限界は10000コピー/チューブまでであった。
【0113】
[まとめ]
以上から、このヒト血清とHIV−1コントロールRNAとの混合物からなるサンプル(実質的にHIV感染患者の血清と同様なもの)に対して、本発明のDNA固定化担体を用いた特定遺伝子の検出方法を有効に利用することができることが分かった。
【0114】
<実施例3>
≪第3工程:市販ヒト血清へHIV−1 LAI株の添加およびHIV−1遺伝子の分離回収≫
実施例2で作製したHIV−1検出用PCRチューブに10μLの市販ヒト血清を4μLのPBS希釈して添加し、HIV−1 LAI株 ウイルスを105コピー/チューブ加えた。さらに、5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))を加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機でスピンダウン後、室温で30分間静置してHIV−1遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のHIV−1溶解液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。操作にてHIV−1遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子増幅に供した。
【0115】
≪第4工程:RT−PCR法による増幅≫
(RT−PCR条件)
TaqMan RT−qPCR反応液に調整したRT反応液を使用して、次に下記のプロトコールに従い、RT−PCR反応液の調整を行なった。
【0116】
【表5】
【0117】
G60(Forward):5’−CAGCCAAAATTACCCTATAGTGCAG−3’(配列番号18)
SK39(Reverse):5’−TTTGGTCCTTGTCTTATGTCCAGAATGCTG−3’(配列番号19)
(RT−PCRプログラム)
48℃,20分→95℃,1分→94℃,15秒⇔54℃,1分、40サイクル
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。実施例3のHIV−1 LAI株を添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動(458bp)法で検出した結果を示す図である。なお、図5の各レーンは、以下のとおりである。
レーン番号1と3:ネガティブコントロール
レーン番号2:ポジティブコントロール(コントロールHIV−1 LAI RNA添加)
レーン番号4:HIV−1検出用PCRチューブ使用したRT−PCR法の結果
[まとめ]
図5から、HIV−1 LAI株 ウイルス(市販)に対しても本発明のDNA固定化担体を用いた特定遺伝子の検出方法を有効に利用することができることが分かった
[考察]
表1、図4、5に示すようにHIV−1検出用PCRチューブを使用して血清中に浮遊するウイルス粒子を溶解し、溶出するウイルス遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は100コピー/チューブまでであった。この検出限界は、一般的に広く用いられているPCR法と同程度であり、HIV−1感染患者の血清中ウイルス(約106コピー/mL)を直接遺伝子検査できる範囲にあることを確認した。これより、血清からウイルス遺伝子を抽出する操作が省略でき、検査時間の短縮が可能であると考えられた。
【0118】
<実施例4:大腸菌全RNA標準液による反応性の確認>
≪第1工程:細菌16S rRNA検出用PCRチューブの作製≫
公知の大腸菌ゲノムDNAの16S rRNAをコードする配列中の細菌全般に保存されているDNA塩基配列5’−GTATTACCGCGGCTGCTG−3’(18塩基、519−536)(配列番号5)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させることで、DNA固定化担体とてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNaseフリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0119】
≪第2工程および第3工程:大腸菌全RNAからの16S rRNA遺伝子の分離≫
大腸菌全RNA標準液(ロシュ社製)を水で希釈して1,10,100,1000pg/μLの各溶液を調整した。16S rRNA検出用PCRチューブに各濃度RNA溶液1μLにおよびPBS19μLを加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0120】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0121】
(RT−PCR条件)
RNA−direct SYBR Green Realtime PCR Master Mix (TOYOBO)添付書類に従い、ワンステップのRT−PCR反応液の調整を行なった。
【0122】
【表6】
【0123】
(RT−PCRプログラム)
【0124】
【表7】
【0125】
16S−9F (Forward):5’− GAGTTTGATCCTGGCTCAG−3’(配列番号20)
16S−536R (Reverse):5’−GTATTACCGCGGCTGCTG−3’(配列番号21)
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図6は、大腸菌全RNA標準液(ロシュ社製)をチューブ当り1,10,100,1000pg/μL添加して、RT−PCRを行なった結果を示す図(RT−PCR電気泳動(528bp))である。図6に示すように細菌16S rRNA検出用PCRチューブを使用して細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。増幅限界は1ピコグラム/チューブまでであった。なお、図6の各レーンは以下のとおりである。
レーン0:ネガティブコントロール
レーン番号1:1pg/μL
レーン番号10:10pg/μL
レーン番号100:100pg/μL
レーン番号1000:1000pg/μL
以上から、本発明においては、微生物特異的な配列キャプチャーDNAが高感度で微生物を検出できることが分かった。
【0126】
<実施例5:複数の逆転写プライマーによる効果の確認>
≪第2工程および第3工程:大腸菌全RNAからの16S rRNA遺伝子の分離≫
本実施例においては、実施例4で作製したPCRチューブを使用した(第1工程)。水で希釈しての各溶液を調整する。16S rRNA検出用PCRチューブに濃度1pg/μLの大腸菌全RNA標準液(ロシュ社製)を1μLおよびPBS19μLを加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0127】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0128】
(RT−PCR条件)
RNA−direct SYBR Green Realtime PCR Master Mix (TOYOBO)添付書類に従い、ワンステップのRT−PCR反応液の調整を行なった。
【0129】
【表8】
【0130】
16S−9F(Forward):5‘− GAGTTTGATCCTGGCTCAG−3’(配列番号22)
16S−536R(Reverse):5’−GTATTACCGCGGCTGCTG−3’(配列番号23)
(逆転写プライマー(RT Primer))
Pba2:5’−TCACCGGCCGTGTGTACAAG−3’(配列番号24)
Microbio:5’−GGACTACCAGGGTATCTAATCCTGTT−3’(Microbiology, 148: 257−266 (2002))(配列番号25)
(RT−PCRプログラム):実施例4と同一条件で行なった。
【0131】
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図1に示すように細菌16S rRNA検出用PCRチューブを使用して細菌16S rRNA遺伝子を分離後、逆転写プライマーを追加した条件下でRT−PCR法を実施した。図7は、複数の逆転写プライマーによる効果の確認結果を示す図(RT−PCR 電気泳動 (528bp))である。2種類のプライマーを用いたときにバンドが濃くなることから、プライマーが1つより2つの方が検出感度が上がることことから、逆転写プライマーを追加の効果は明白に確認できた。なお、図7の各レーンは以下のとおりである。
Normal Tube:対象となるコントロール実験結果
GeneTube:本発明の実験結果
レーン番号1:逆転写プライマー添加なし
レーン番号2:Pba2
レーン番号3:Microbio
レーン番号4:Pba2+Microbio
<実施例6:標準菌株培養における反応性の確認>
本実施例においては、実施例4の第1工程で作製したPCRチューブを用いた。
【0132】
≪第2工程(a):大腸菌標準菌株の培養および溶菌≫
培養は通常液体培地あるいは普通寒天培地を用い、37℃、一昼夜(14−18時間)行なう。培地はインスタント培地である「普通ブイヨン栄研」を用い、液体培地は「普通ブイヨン栄研」の指示量を脱塩水に溶かし、寒天培地はそれに1.6%の寒天末を加え、それぞれオートクレーブ後、使用した。
【0133】
1Lあたりの培地組成は以下の通りとした。
肉エキス3g、ペプトン10g、NaCl5g(pH7.0)
そして、大腸菌(Escherichia coli ATCC25922)被検リボゾームRNAを以下のようにして調製した。被検菌を白金耳などで上記液体培地に懸濁し、37℃で一晩培養した。なお、生菌数は10μlの懸濁液をとり適当に希釈して栄養寒天培地に塗抹し、37℃で一晩培養し、出現したコロニー数と希釈率とから求め、生菌数は約109cfu/mlであった。菌体培養液20μlを10000×gで5分間遠心し、沈殿を採取した。沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5% SDS,5%ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0134】
≪第2工程(b):大腸菌標準菌株の添加食品の培養操作後にRT−PCR反応を行なう方法≫
大腸菌(Escherichia coli ATCC25922)を小麦粉に1g当たり10個程度となるように添加した。菌が添加された小麦粉重量の9倍量の通常液体培地(普通ブイヨン栄研)を加え、37℃で一晩培養した。培養前の小麦懸濁中の菌密度は、1個/mlであったが、4時間培養後には約1×103個/ml増殖した。培養液1mlを採取し、100×gで1分間遠心し、上清を10000×gで5分間遠心し、沈殿を採取した。この沈殿を洗うためTE緩衝液(5mM Tris 0.1mM EDTA)に懸濁し、10000×gで5分間遠心し、沈殿を採取した。沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5%SDS,5%ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0135】
≪第3工程:被検RNA溶液からの16S rRNA遺伝子の分離>
16S rRNA検出用PCRチューブに被検RNA溶液をそれぞれ加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0136】
≪第4工程:RT−PCR法によるDNAの増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0137】
(RT−PCR条件)
ワンステップRT−PCR反応液は実施例4と同一条件で行なった。
【0138】
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図8(RT−PCR電気泳動(528bp))に示すように細菌16S rRNA検出用PCRチューブを使用して、大腸菌標準株培養から得た被検RNA溶液から細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。大腸菌の場合には、小麦1g当たり10個程度の細菌が存在している場合には、前述の操作による培養時間を4時間以上にすれば、PCR反応によりその16S rRNAを検出できることが分かる。また、黄色ブドウ球菌(Staphylococcus aureus ATCC25923)、緑脳菌(Pseudomonas aeruginosa ATCC27853)およびサルモネラ菌(Salmonella enterica serovar Typhimurium ATCC14028)から選ばれる微生物の各被検リボゾームRNAからも同一のプライマーの組み合わせを用いることにより同様の結果が得られた。なお、図8における各レーンは以下のとおりである。
レーン番号1:大腸菌標準株培養から得た被検RNA溶液
レーン番号2:大腸菌標準菌株の添加食品の培養から得た被検RNA溶液
<実施例7:大腸菌DH5α株の培養経過における16S rRNAの確認>
≪第2工程:被検RNA溶液の作製≫
大腸菌DH5α(Bethesda Research Laborotories製)102〜103cfuをLB培地10mLに懸濁し、37℃で培養した。培養液50μlを1、2、4時間経過後にそれぞれサンプリングし、10000×gで5分間遠心し、沈殿を採取した。各沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5% SDS,5% ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0139】
≪第3工程:被検RNA溶液からの16S rRNA遺伝子の分離≫
16S rRNA検出用PCRチューブ(実施例4で作製したPCRチューブ)に被検RNA溶液を加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0140】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0141】
(RT−PCR条件)
逆転写反応:PrimeScript RT−PCR Kit (Takara, PR037A)を使用して以下の条件で行なった。
【0142】
【表9】
【0143】
【表10】
【0144】
PCR反応は以下の条件で実施した。
【0145】
【表11】
【0146】
【表12】
【0147】
(結果)
RT−PCR測定は、Applied Biosystems 9700PCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図9は、大腸菌DH5α株の培養時間(h)経過における16S rRNAの確認した結果を示す図(RT−PCR 電気泳動 (528bp))である。細菌16S rRNA検出用PCRチューブを使用して、大腸菌標準株培養から得た被検RNA溶液から細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。2時間経過した培養液中から明白な16S rRNA遺伝子増幅産物が確認できた。
【0148】
[考察]
図6〜9に示すように16S rRNA検出用PCRチューブを使用して、様々なサンプル形態に応じて細菌の16S rRNA遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は細菌10個/チューブであった。この検出限界は、一般的に広く用いられているPCR法と同程度であることを確認した。これより、サンプルから16S rRNA遺伝子を抽出する操作が省略でき、検査時間の短縮が可能であると考えられる。本発明の微生物検出法では、被検試料検体中の微生物溶解処理から遺伝子増幅・検出までに要する時間はわずか2時間程度で実施可能である。この短時間の操作により迅速・簡便に微生物を検出することができ、公衆衛生上大きく寄与するものと考えられた。
【0149】
今回開示された実施の形態および実施例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【図面の簡単な説明】
【0150】
【図1】実施例1のLAMP法反応の増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無による標的遺伝子配列の有無を判別した図である((上):目視濁度結果、(下):栄研化学(株)社製Loopampエンドポイント濁度測定装置(LA−100)による濁度測定結果。レーン番号1および2は、水またはコントロールRNAを栄研化学(株)社製LoopampノロウイルスGI検出試薬キットを用いて行なったLAMP法のコントロール反応)。
【図2】実施例1のLAMP法反応液に約1μLの1000×の濃度のSYBR Green I添加(最終濃度:100×)によるLAMP法反応液の核酸増幅産物の目視確認結果を示す図である。
【図3】実施例1のコントロールのノロウイルスRNAとノロウイルス感染患者糞便サンプル(S1〜3)を用いたLAMP法反応結果を示す図である(A:ノロウイルスG2型検出用PCRチューブを用いてノロウイルス遺伝子を分離後、LoopampノロウイルスG2検出試薬キットを用いて行なったLAMP法遺伝子増幅反応産物を電気泳動。B:ノロウイルス遺伝子を直接LoopampノロウイルスGI検出試薬キットに添加して行なったLAMP法のコントロールの結果)。
【図4】実施例2のコントロールHIV−1 RNAを添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動法で検出した結果を示す図である。
【図5】実施例3のHIV−1 LAI株を添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動法で検出した結果を示す図である。
【図6】実施例4の大腸菌全RNA標準液(ロシュ社製)をチューブ当り1,10,100,1000pg/μL添加して、RT−PCRを行なった結果を示す図である。
【図7】実施例5の複数の逆転写プライマーによる効果の確認結果を示す図である(Normal Tube:対象となるコントロール実験結果、GeneTube:本発明の実験結果。レーン番号1:逆転写プライマー添加なし、レーン番号2:Pba2、レーン3:Microbio、レーン4:Pba2+Microbio)。
【図8】実施例6の標準菌株培養における反応性の確認結果を示す図である(レーン番号1:大腸菌標準株培養から得た被検RNA溶液、レーン番号2:大腸菌標準菌株の添加食品の培養から得た被検RNA溶液)。
【図9】実施例7の大腸菌DH5α株の培養時間経過における16S rRNAの確認した結果を示す図である。
【技術分野】
【0001】
本発明は、特定遺伝子の検出方法に関する。より詳細には、生体試料中に存在するウイルス、または、食品や臨床試料中に含まれる微生物を特異的に検出することのできる特定遺伝子の検出方法に関する。
【背景技術】
【0002】
ウイルス病の診断を確実に行なうこと、又未知の流行病を新しいウイルス病として同定することは、当該ウイルス病の地域分布と流行時期を知り、当該疾患に対する予防と治療対策を立てる上で重要なことである。しかも、ウイルス病は速やかにしかも正確に診断されないと、適切な対策処理が手遅れとなることが多い。
【0003】
臨床検体中の特定ウイルスを直接検出する方法としては、たとえば検体中に存在するウイルス抗原を蛍光等で標識した抗体と反応させて検出する方法が挙げられる。当該方法は2〜3時間程度で結果が得られるという利点はあるが、検体中の夾雑物質との非特異反応による偽陽性反応の可能性や検体中に一定以上のウイルス抗原がないと検出できないという検出感度の問題が指摘されている。また最近では、生体試料から、生体に潜在しているウイルス核酸やmRNAの存在を証明して診断する方法が開発されている。当該方法によると、PCR法(Science,230,1350(1985))やRT−PCR法を利用することによって生体試料中に微量存在するウイルス遺伝子でも高感度に検出できるが、この場合検査結果の取得に1日以上要する。
【0004】
また、たとえば、輸血用血液や血液製剤の安全性を確保するため、献血血液に対して、通常、ウイルス検査が行なわれる。このような検査としては、血液中のウイルス抗原やウイルス関連抗体の有無を調べる血清学的検査が主に行われている。1999年から、血清学的検査で陰性となった血液に対して、2次検査として核酸増幅検査(Nucleic Acid Amplification Testing: NAT)が行われている。NATとは、ウイルス遺伝子の一部の核酸を取り出し(抽出)、その核酸を増やし(増幅)、増えた核酸を検出することでウイルス遺伝子の有無を確認する検査法のことである。NATを使用したウイルス検査法は、ウイルスの持つ遺伝子を数万倍以上に増幅して検出するため、従来の血清学的な検査法に比べ、極めて感度の高い検査法である。この検査で再度陰性になった献血血液が、輸血用血液および血液製剤の原料などとして使用される。
【0005】
このようなNATの導入により、献血血液を原料とした輸血用血液や血液製剤等の安全性は以前よりも向上してきたが、輸血後の感染症発生率は依然として0%に達していない。通常、NATを用いた2次検査では1人からたとえば約4μLといった微少量の血液を採取し、約50人分を1つにまとめて分析を行なっている。このため、感染直後のようにウイルス量が極端に少ない場合などには、検査に回した血液約4μL中にウイルスが含まれない確率が高い。その場合、献血によって集められた血液中にウイルスが存在していても検出されずに輸血感染する恐れがある。
【0006】
また、食品や臨床試料、または環境中などの特定微生物の有無を調べる場合には、従来、被検試料中の微生物を分離培養し、コロニーについて視覚的観察(コロニーの色素反応、コロニー形態等)、顕微鏡観察、グラム染色、生化学的性状の検査等を行なっていた。特定微生物の有無が判明するまでに少なくとも2日間を要する。したがって、この方法は、微生物が検出されたとしてとしてもそれに対する対応が遅くなることから、食品メーカーによる食品出荷前の自主検査には採用し難い。
【0007】
食品の殺菌技術や加工技術が向上したことにより、微量であっても被検試料中に存在する微生物の生死の状態を識別するニーズが高まっている。特に、食品衛生検査や臨床検査領域においては、微生物の迅速検出法として、PCR法により各微生物の特異遺伝子を視覚的に捉えられる量まで増幅し、各微生物の存在の有無を判別および定量する試みがなされている。しかし、微生物のDNAにターゲットを当てた場合、被検試料に元来含まれている死菌のバックグラウンドまで検出されるため、PCR法で陽性判定がでた場合、必ずしも生きた微生物の存在を示唆しているとは限らなかった。
【0008】
一方、生きた微生物(生菌)の存在を確認するためには、食品から微生物を分離培養し、得られたコロニーについて、特定微生物のDNAに対応したプライマーを用いたPCR法により特定微生物のDNAを迅速に検出する方法が開発されている。しかし、この方法によっても、食品から微生物を分離培養するのに少なくとも1日間を要する。そのために、食品衛生や臨床検査の分野では、PCR法は高感度・迅速でありながら普及していないのが現状であった。
【0009】
ここで、特許文献1、特許文献2および非特許文献1には、ホスホリルコリン基を有する第一単位と電子求引性の置換基がカルボニル基に結合してなるカルボン酸誘導基を有する第二単位とを含む高分子物質を表面にDNA鎖を固定化した不溶性担体に検出対象遺伝子のDNA断片またはRNA断片を含有する溶液を添加し、該DNA鎖とハイブリダイズさせ、溶液中の遺伝子断片の含有状況を判定することを特徴とする遺伝子の検出方法が開示されている。
【0010】
また、最近では、mRNAをターゲットにして、逆転写酵素によりcDNAを作製後、各微生物の特異プライマーを用いてPCRを行い、被検試料中の生菌のみを検出・定量する試みがなされている。しかし、この方法では、死菌のバックグラウンドを検出してしまうため、生死の判別法としては十分なものとは言えなかった。具体的には、PCR法を利用した微生物等の生死を判別する方法としては、特許文献4に記載の方法が開示されている。
【0011】
また、非特許文献2には、ノロウイルスの検出法が開示されており、特許文献3には微生物等の検出等に利用されるLAMP(Loop−mediated Isothermal Amplification)法が開示されており、非特許文献3には、微生物等の検出等に利用されるRT−LAMP法が開示されている。
【特許文献1】特開2006−187270号公報
【特許文献2】特開2006−322739号公報
【特許文献3】特開2004−154008号公報
【特許文献4】特表2003−530118号公報
【非特許文献1】Kinoshita, K. et al., Nucleic Acids Res. 2007,Vol.35,No.1 e3
【非特許文献2】ノロウイルスの検出法(平成15年11月5日付け食安監発第1105001号)
【非特許文献3】Fukuda, S. et al.,J. Clinical Microbiology Vol.44,No.4: 1376-1381 (2006) (RT−LAMP)
【発明の開示】
【発明が解決しようとする課題】
【0012】
上記のように、特定ウイルスの検出および特定微生物の検出には、さまざまな問題が未
だ解決されていない。そして、特許文献1、特許文献2および非特許文献1には、微量の核酸を迅速にかつ簡便に測定することができる方法については開示されていない。
【0013】
そしてさらに、患者から採取した生体試料を対象として、該生体試料中に含まれるウイルス遺伝子の検出を目的としてハイブリダイゼーションが広く行われてきているが、1個の不溶性担体内でウイルス遺伝子を捕捉し、洗浄後、分離した遺伝子を増幅してウイルスを検出する方法は知られていなかった。
【0014】
また、試料中の微量の核酸を増幅するためにはPCR法が広く普及している。しかしながら、PCR法では試料中に含まれる夾雑物により遺伝子の増幅反応を阻害してしまう場合がある。したがって、PCR法を用いた場合には、増幅反応の前に分離・精製等の煩雑な前処理を行なう必要がある。
【0015】
このような背景から、本発明者らは、特定ウイルスの検出において、血液中に含まれるウイルス数が極端に少ない場合でも、簡便かつ短時間にウイルスを検出できる検査法の開発を行なってきた。また、本発明者らは、特定微生物の検出において、食品や臨床試料中に含まれる特定の複数の微生物または微生物群の生菌の有無を死菌に比べて選択的、簡単かつ迅速に検出する方法の開発を行なってきた。
【0016】
したがって、本発明の目的は、被検試料における核酸の分離・精製等の前処理を必要とせず、簡易に高感度で検出し得る特定遺伝子、具体的には、特定ウイルスまたは特定微生物の検出方法を提供することである。
【課題を解決するための手段】
【0017】
本発明者は、上記課題の解決を目的として種々検討していたところ、ウイルスに感染した患者の生体試料を公知のグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液でウイルスを溶解させ、次いでDNA鎖を固定化した不溶性担体にハイブリダイゼーションを行なうことにより、生体試料中に存在するウイルス遺伝子の捕捉、分離を簡便かつ迅速に行なうことができ、しかも遺伝子断片をポリメラーゼ連鎖反応(PCR)、LAMP法、鎖置換増幅(SDA)、逆転写酵素鎖置換増幅(RT−SDA)、逆転写酵素ポリメラーゼ連鎖反応(RT−PCR)、逆転写LAMP法(RT−LAMP)、核酸配列に基づく増幅(NASBA)、転写媒介性増幅(TMA)、ローリングサークル型増幅などの遺伝子増幅法を用いて特異的に検出することができることを見出し、増幅することによって得られた結果が患者検体の臨床状態を的確に反映した結果であることを確認した。本発明は、かかる知見に基づいて開発されたものである。
【0018】
ウイルスを含み得る被検試料中のウイルス遺伝子の存在の有無を判定することを特徴とするウイルス感染の診断方法である。なお、当該診断方法には、ウイルス感染成立を診断する方法だけでなく、ウイルス感染の経過や治療経過を診断する方法もまた含まれる。
【0019】
つまり、本発明は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する第3工程と、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する第4工程とを備える特定遺伝子の検出方法に関する。
【0020】
また、本発明は、ホスホリルコリン基またはアルキレングリコール基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、抗体、レクチンおよび複合糖質のいずれかで微生物またはウイルスを捕捉する工程と、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、生体高分子固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程とを備える特定遺伝子の検出方法に関する。
【0021】
また、本発明においては、被検試料が、鼻汁、鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液、細胞組織または食品であることが好ましい。
【0022】
また、本発明においては、第3工程において、4〜70℃で5分〜90分間、ハイブリダイズすることが好ましい。
【0023】
また、本発明においては、第4工程において、RNA鎖断片を逆転写するための逆転写反応のプライマーとして、ランダムプライマー、dTプライマーまたは1個以上の配列特異的なスペシフィックプライマーから合成されたcDNAを使用することが好ましい。
【0024】
また、本発明においては、第4工程、または、DNA鎖断片もしくはRNA鎖断片で増幅する工程の後に増幅したDNA鎖断片を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する第5工程をさらに備えることが好ましい。
【0025】
また、本発明においては、キャプチャーDNA鎖は、10〜50塩基からなり、23S rRNA遺伝子配列または16S rRNA遺伝子配列に相補的な配列から選択されることが好ましい。
【0026】
また、本発明においては、キャプチャーDNA鎖のターゲット配列領域がさらに、トポイソメラーゼ、または、DNAジャイレースを含む微生物特異的な配列であることが好ましい。
【0027】
また、本発明においては、微生物がグラム陽性菌またはグラム陰性菌であることが好ましい。
【0028】
また、本発明においてはウイルスがC型肝炎ウイルス(HCV)、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス(黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス)、トガウイルス(アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス)、ペスチウイルス(ブタコレラウイルス、ウシ下痢ウイルス)、パラミクソウイルス(パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス)、オルソクソウイルス(ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス)、ラブドウイルス(狂犬病ウイルス、水泡性口内炎ウイルス)、ピコルナウイルス(ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス)、コロナウイルス(ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス)、アレナウイルス(リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス)、レトロウイルス(HTLV:ヒト成人白血病ウイルス、HIV:エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス)、レオウイルス(ロタウイルス)、カリシウイルス(ノーウオークウイルス)、ノロウイルス、ブンヤウイルス(腎症候性出血熱ウイルス)、フィロウイルス(エボラウイルス、マールブルグウイルス)、B型肝炎ウイルス(HBV)、ポックスウイルス(ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス)、パルボウイルス(ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス)、パポーバウイルス(パピローマウイルス、ポリオーマウイルス)、アデノウイルス、ヘルペスウイルス(単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルス)およびアフリカ豚コレラウイルスよりなる群から選択される少なくとも1種であることが好ましい。
【発明の効果】
【0029】
本発明の方法によると、被検試料における核酸の分離・精製等の前処理を必要とせず、簡易に高感度で検出し得る特定遺伝子、具体的には、特定ウイルスまたは特定微生物の検出をすることができる。
【発明を実施するための最良の形態】
【0030】
本発明の特定遺伝子の検出方法においては、少なくとも第1工程〜第4工程を備える。第1工程は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する工程である。第2工程は、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する工程である。第3工程は、DNA固定化担体の上に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する工程である。第4工程は、DNA固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程である。
【0031】
また、本発明の別の形態における特定遺伝子の検出方法においては、抗体、レクチンおよび複合糖質のいずれかが固定化されたチューブ状の生体高分子固定化担体を用いる。そして、該特定遺伝子の検出方法においても4つの工程を備える。これらの工程は、ホスホリルコリン基またはアルキレングリコール基等の生体適合性の高い官能基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、抗体、レクチンおよび複合糖質のいずれかで微生物またはウイルスを捕捉する工程と、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、生体高分子固定化担体に対して直接、DNA鎖断片またはRNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施してDNA鎖断片またはRNA鎖断片で増幅する工程とからなる。なお、ホスホリルコリン基またはアルキレングリコール基等の生体適合性の高い官能基を有する高分子物質については、特開2008−128854号公報に記載されているものを挙げることができる。
【0032】
[第1実施形態]
以下、各工程について詳細に説明する。
【0033】
<第1工程>
本工程においては、チューブ状のDNA固定化担体を作製する。より具体的には、チューブ状の不溶性担体の少なくとも内側を高分子物質で被覆して、その上にキャプチャーDNA鎖を固定化させることによってチューブ状のDNA固定化担体を作製する。
【0034】
まず、本実施形態の本工程における「高分子物質」は、ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含み、DNA鎖およびRNA鎖の非特異的吸着を抑制する性質とDNA鎖を固定化する性質とを併せ持つポリマーを示す。特に、第一単位に含まれるホスホリルコリン基は鋳型RNA断片の非特異的吸着を抑制する役割を果たし、第二単位に含まれるカルボン酸誘導基はキャプチャーDNA鎖を化学的に固定化する役割を果たす。すなわち、キャプチャーDNA鎖は、このコーティング層の活性エステル基の部位で共有結合して、当該不溶性担体の表面に固定化される。具体的には、住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブを例として挙げることができる。なお、チューブ状のDNA固定化担体の作製方法については、特許文献2に詳細が記載されている。
【0035】
次に、本発明における「不溶性担体」は、プラスチック、ガラス等からなるチューブのほか、96穴ウェル等を挙げることができる。
【0036】
チューブ状とは、中空状態のものをいい、底があるPCRチューブや、エッペンドルフチューブのような形状であってもよい。
【0037】
そして、本発明における「キャプチャーDNA鎖」は、後述する被検試料に含まれる微生物由来遺伝子またはウイルス由来遺伝子におけるターゲット配列領域の一部と相補的な配列を有するDNA鎖をいう。なお、キャプチャーDNA鎖は、ターゲット配列領域の相補的な配列と相同性が50%以上であればよい。キャプチャーDNA鎖には、塩基数が100以下より好ましくは80以下のDNA鎖を用いることが好ましい。これは、キャプチャーDNAの長さが100以下である場合には、合成が容易であり、また、後述するハイブリダイゼーションを安定して行なうことができるためである。また、キャプチャーDNA鎖には、修飾されたオリゴDNAを用いることが好ましいが、無修飾のオリゴDNAを用いても固定化が可能である。さらには、キャプチャーDNA鎖がアミノ基が導入されたオリゴDNAである場合には、固定化効率が高くなる。アミノ基は活性エステル基との反応性に優れるため、キャプチャーDNA鎖として、アミノ基が導入されたオリゴDNAを用いることにより、該キャプチャーDNA鎖は、効率よくかつ強固に該高分子物質上にプライマーを固定化することができる。アミノ基の導入位置はキャプチャーDNA鎖の分子鎖末端あるいは側鎖であってもよいが、分子鎖末端に導入されていることが、相補的なDNA断片またはRNA断片とのハイブリダイズをより一層効率よく行なうことができるという観点からは、好ましい。
【0038】
本発明において「ターゲット配列領域」とは、微生物またはウイルスにおける全遺伝子のうち、本発明に用いるプライマーを用いたPCRにより増幅され得る配列領域であり、検出対象のウイルスまたは微生物を検出することができるものであれば特に制限されず、目的に応じて適宜設定することができる。
【0039】
たとえば、被検試料に検出対象のウイルスまたは微生物と異なる種類の細胞が含まれる場合には、ターゲット配列領域は、検出対象のウイルスまたは微生物に特異的な配列を有することが好ましい。また、目的によっては、複数種のウイルスまたは微生物に共通する配列を有するものであってもよい。さらに、ターゲット配列領域は単一であっても、複数であってもよい。
【0040】
キャプチャーDNA鎖のターゲット配列領域がさらに、トポイソメラーゼ、または、DNAジャイレースを含む微生物特異的な配列であってもよい。
【0041】
ターゲット配列領域の長さとしては、通常20〜数十万塩基程度まで適用できるが、500〜3000塩基であることが好ましい。検出対象が微生物である場合には、具体的には、ターゲット配列領域は、16S rRNA遺伝子および23S rRNA遺伝子が挙げられる。これは、広汎な微生物に応用して使用することができるためである。
【0042】
<第2工程>
本工程は、特定遺伝子の検出がされるかどうか検査したい被検試料、具体的には、微生物の特定遺伝子またはウイルスの特定遺伝子の検出がされるかどうか検査したい被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する工程である。
【0043】
本発明において、「被検試料」が「ウイルスを含む被検試料」である場合においては、該被検試料は、ウイルス感染によってウイルスが存在しえる生体試料であれば特に制限されることなく任意に使用することができる。
【0044】
ウイルスの特定遺伝子の検出がされるかどうか検査したい被検試料は、対象とする被験者から、たとえば滅菌綿棒などの滅菌器具および容器を用いることにより無菌的に採取することが好ましい。得られた該被検試料は、採取後、なるべく速やかにグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液でウイルスを溶解させることができる。このような操作によって、遺伝子溶解溶液を得ることができる。
【0045】
また、後述する第3工程でDNA鎖断片またはRNA鎖断片を捕捉するために、希NaOH溶液で被検試料を溶解した場合には、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えることが好ましい。また、該遺伝子溶解溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0046】
本発明において、「被検試料」が「微生物を含む被検試料」である場合においては、該被検試料は、その中に存在する微生物の生菌を検出する対象であり、PCR法による染色体DNAの特定領域の増幅によって存在を検出することが可能なものであれば特に制限されない。
【0047】
微生物の特定遺伝子の検出がされるかどうか検査したい被検試料は、食品原材料付着の製品または生体試料そのものであってもよく、これを希釈もしくは濃縮したもの、または本発明の方法による処理以外の前処理をしたものであってもよい。前処理としては、加熱処理、濾過、遠心分離等が挙げられる。得られた被検試料は、採取後、なるべく速やかにグアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液で微生物を溶解させることができるが、次工程のハイブリダイゼーションで対象RNA遺伝子を捕捉するためには、希NaOH溶液で生体試料を、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えたほう好ましい。溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0048】
また、被検試料は、鼻汁、粘膜ぬぐい液(たとえば鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液等)、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液等の各種体液、または各種組織(細胞組織)または食品を例示することができる。本発明方法の対象となる食品は、特に制限されない。本発明方法は、飲料、麺製品、食肉、魚介類、野菜、穀類、乳製品およびこれらの加工物等の広い範囲の食品を対象にすることができる。
【0049】
また、被検試料としての食品からRNAを抽出する場合において、食品から分離培養した微生物からRNAを抽出してもよいが、食品から直接RNAを抽出することもできる。また、微生物を含むかもしれない食品を遠心処理することにより、微生物を含むかもしれない画分を食品と分離した後に、微生物を含むかもしれない画分からRNAを抽出することもできる。
【0050】
<第3工程>
本工程は、キャプチャーDNA鎖と相補的な配列を有する遺伝子溶解溶液中のDNA鎖断片またはRNA鎖断片を分離する工程である。具体的には、チューブ状のDNA固定化担体の内側(内部)に遺伝子溶解溶液を添加し、キャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせ、DNA鎖断片またはRNA鎖断片を捕捉する。以上の工程によって、遺伝子溶解溶液中の遺伝子のなかからキャプチャーDNA鎖と相補的な配列を有する遺伝子溶解溶液中のDNA鎖断片またはRNA鎖断片を分離することができる。
【0051】
本工程においては、4〜70℃、より好ましくは15〜37℃(室温)で静置または攪拌状態でキャプチャーDNA鎖と遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせる。本工程は、5分〜90分間で完了できる。90分間超過の時間をかけてハイブリダイズさせることもできるが、本発明においては、1時間程度のハイブリダイズで十分であり、結果として迅速に特定遺伝子の検出を行なうことができる。
【0052】
<第4工程>
本工程は、キャプチャーDNA鎖に捕捉されたDNA鎖断片またはRNA鎖断片を増幅する工程である。本工程においては、上述のチューブ状のDNA固定化担体に対して直接、ポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施すことができる。具体的な操作は以下のとおりである。
【0053】
まず、第3工程の後のチューブ状のDNA固定化担体から被検試料を除去する。そして、該DNA固定化担体の表面をPBSにて1〜数回洗浄することが好ましい。本発明において、被検試料中に存在する微生物や夾雑物質等による検出誤差を回避するためである。これにより、チューブ状のDNA固定化担体の内部における核酸はキャプチャーDNA鎖とキャプチャーDNA鎖に捕捉されたDNA鎖断片またはRNA鎖断片が残る。
【0054】
次に、DNA固定化担体の内部に所定の物質(dNTP等)を添加して核酸の増幅を行なう。
【0055】
キャプチャーDNA鎖に捕捉された核酸がDNA鎖断片の場合においては、直接PCR法またはLAMP法等を行なうことができる。また、キャプチャーDNA鎖に捕捉された核酸がRNA断片の場合においては、RNA鎖断片の場合は逆転写反応を行なった後、ポリメラーゼ連鎖反応(PCR)、LAMP法、鎖置換増幅(SDA)、逆転写酵素鎖置換増幅(RT−SDA)、逆転写酵素ポリメラーゼ連鎖反応(RT−PCR)、逆転写LAMP法(RT−LAMP)、核酸配列に基づく増幅(NASBA)、転写媒介性増幅(TMA)、ローリングサークル型増幅などの遺伝子増幅法を用いて増幅することができる。
【0056】
検出対象の微生物に特異的なターゲット配列領域に対応するプライマーセットと、広汎なウイルスまたは微生物のDNAに対応するプライマーセットを用いると、検出対象の微生物の生菌量と、多数種の微生物の生菌量を、同時に測定することができる。該ターゲット配列領域の長さとしては、通常20〜数十万塩基程度まで適用できるが、500〜3000塩基であることが好ましい。検出対象が微生物である場合には、具体的には、ターゲット配列領域は、16S rRNA遺伝子および23S rRNA遺伝子が挙げられる。
【0057】
本発明においてPCR法に用いるプライマーは、上記ターゲット配列領域を特異的に増幅することができるものであれば特に制限されない。検出対象が微生物である場合に、具体的には、23S rRNA遺伝子に対応するプライマーとしては、配列番号1、2および3に示すプライマーセット(Journal of Clinical Microbiology,42 1048-1057(2004))が挙げられる。また、16S rRNA遺伝子に対応するプライマーとしては、配列番号4および5に示すプライマー(MicroSeq500)セット、配列番号6および7に示すプライマーセット(Journal of Clinical Microbiology,32:335−351(1994)、Journal of Clinical Microbiology,43:3390−33397(2005)、 Journal of Molecular Diagnostics,7:575581−33397(2005))、および配列番号8および9に示すプライマーセット(Experimental Biology and Medicine 230:587−591(2005))が挙げられる。
配列番号1:5’−GACAGCCAGGATGTTGGCTTAGAAGCAGC−3’(43a2)
配列番号2:5’−GGAATTTCGCTACCTTAGGACCGTTATAGTTACG−3’(69ar2)
配列番号3:5’−GGAATTTCGCTACCTTAGGATGGTTATAGTTACC−3’(69arrh)
配列番号4:5’−GAGTTTGATCCTGGCTCAG−3’(16S−9F)
配列番号5:5’−GTATTACCGCGGCTGCTG−3’(16S−536R)
配列番号6:5’−AACTGGAGGAAGGTGGGGAY−3’(RW01&RDR080、1170−1189)
配列番号7:5’−AGGAGGTGATCCAACCGCA−3’(DG74、1522−1540)
配列番号8:5’−ACTCCTACGGGAGGCAGCAGT−3’(Pba1、358-378)
配列番号9:5’−TCACCGGCCGTGTGTACAAG−3’(Pba2、1444-1425)
LAMP法は、栄研化学(株)社が開発した核酸増幅法である。LAMP法は、標的遺伝子の6箇所の領域に対して4種類のプライマーを設定して、鎖置換反応を利用し一定温度で反応させることを特徴とする。反応はサンプルとなる遺伝子、LAMPプライマー、鎖置換型DNA合成酵素、基質核酸等を同一の反応チューブに混合し、一定温度(65℃付近)で保温することにより、遺伝子の増幅から検出までを1ステップの工程で行なうことができる。増幅効率が高く、DNAを15分〜1時間で109〜1010倍に増幅することが可能である。その極めて高い特異性から、増幅産物の有無で目的とする標的遺伝子配列の有無を判定することができる。また、増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無によっても標的遺伝子配列の有無を判別できる。また、過剰のインターカレーターSYBR Green IをLAMP反応液の核酸増幅産物に添加することによって、標的核酸の有無を特別な光源や検出装置を用いることなく目視確認するする方法でも標的遺伝子配列の有無を判別できる。さらには、電気泳動によって、アガロースゲルのウェルから尾を引いたようなバンドが確認できれば、遺伝子が増幅されたことを示している(特許文献3参照)。
【0058】
RT−LAMP(Reverse Transcription−Loop−mediated Isothermal Amplification)法は、LAMP法を応用し、ノロウイルスのように標的遺伝子であるRNAから逆転写酵素反応よりcDNAを合成して増幅して検出する方法である。サンプルとなるRNAに対して、DNAを標的遺伝子とする場合と同様の試薬(プライマー、鎖置換型DNA合成酵素、基質核酸等)に逆転写酵素を追加して混合することによって、一定温度(60〜65℃)下で増幅から検出までの工程を1ステップで行なうことができる。増幅から検出までを1ステップで行なうことができる特徴を有する(非特許文献3参照)。
【0059】
<第5工程>
本工程は、第4工程の後にさらに加えられることが好ましい工程である。本工程では、第4工程の後に増幅したDNA鎖断片(PCR増幅産物)を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する。
【0060】
本工程における解析法は、PCR増幅産物の検出または定量が可能なものであれば特に制限されず、電気泳動法、リアルタイムPCR法等が挙げられる。電気泳動法によれば、PCR増幅産物の量、およびその大きさを評価することができる。また、リアルタイムPCR法によれば、迅速にPCR増幅産物の定量を行なうことができる。リアルタイムPCR法を採用する場合、一般に増幅サイクル数1〜10までは蛍光強度の変化はノイズレベルでありゼロに等しいので、それらを増幅産物ゼロのサンプルブランクと見なし、それらの標準偏差SDを算出しその10を乗じた蛍光値をスレッショード値とし、そのスレッショード値を最初に上回るPCRサイクル数をサイクルスレッショード値(Ct値)という。したがって、PCR反応溶液に初期のDNA鋳型量が多い程、Ct値は小さな値となり、鋳型DNA量が少ない程、Ct値は大きな値となる。また、鋳型DNA量が同じでも、その鋳型内のPCRのターゲット配列領域に切断が生じている割合が多くなる程、同領域のPCR反応のCt値は大きな値となる。
【0061】
また、複数種の微生物に共通するプライマーを用いると、被検試料中の複数種の微生物の生菌を検出することができる。また、特定の微生物に特異的なプライマーを用いると、被検試料中の特定の菌種の生菌を検出することができる。
【0062】
PCRの条件は、PCRの原理にのっとった特異的な増幅が起る限り特に制限されず、適宜設定することができる。
【0063】
本発明の方法によって生菌を検出する場合、PCR増幅産物の解析は、同定されている微生物の標準試料を用いて作成された微生物量および増幅産物との関連を示す標準曲線を用いると、生菌の有無または定量の精度を高めることができる。標準曲線は予め作成しておいたものを用いることができるが、被検試料と同時に標準試料について本発明の各ステップを行なって作成した標準曲線を用いることが好ましい。また、予め微生物量とRNA量との相関を調べておけば、その微生物から単離されたDNAを標準試料として用いることもできる。
【0064】
<各用語>
本発明においては、「ウイルス」という用語は、たとえば、C型肝炎ウイルス(HCV)、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス(黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス)、トガウイルス(アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス)、ペスチウイルス(ブタコレラウイルス、ウシ下痢ウイルス)、パラミクソウイルス(パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス)、オルソクソウイルス(ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス)、ラブドウイルス(狂犬病ウイルス、水泡性口内炎ウイルス)、ピコルナウイルス(ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス)、コロナウイルス(ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス)、アレナウイルス(リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス)、レトロウイルス(HTLV:ヒト成人白血病ウイルス、HIV:エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス)、レオウイルス(ロタウイルス)、カリシウイルス(ノーウオークウイルス)、ノロウイルス、ブンヤウイルス(腎症候性出血熱ウイルス)、フィロウイルス(エボラウイルス、マールブルグウイルス)、B型肝炎ウイルス(HBV)、ポックスウイルス(ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス)、パルボウイルス(ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス)、パポーバウイルス(パピローマウイルス、ポリオーマウイルス)、アデノウイルス、ヘルペスウイルス(単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルス)およびアフリカ豚コレラウイルスよりなる群から選択される。
【0065】
本発明においては、「微生物」という用語は、原核生物、細菌、真菌、寄生生物、または原生動物を意味するのに使用する。「微生物」としては、グラム陽性菌およびグラム陰性菌のいずれもが含まれ、ブドウ球菌(スタフィロコッカス・エピダーミディス(Staphylococcus epidermidis))のスタフィロコッカス属細菌、肺炎連鎖球菌(Streptococcus pneumoniae)等のストレプトコッカス属細菌、リステリア・モノサイトゲネス(Listeria monocytogenes)等のリステリア属細菌、バチラス・セレウス(Bacillus cereus)等のバチラス属細菌、黄色ブドウ球菌(Staphylococcus aureus)、マイコバクテリウム属細菌、大腸菌(Escherichia coli)、表皮ブドウ球菌(Staphylococcus epidermidis)、肺炎桿菌(Klebsiella pneumoniae)、エンテロコッカスフェカリス(Enterococcus faecalis)、緑膿菌(Pseudomonas aeruginosa)、エンテロバクター・サカザキ(Enterobacter sakazakii)等のエンテロバクター属細菌、シトロバクター・コーセリ(Citrobacter koseri)等のシトロバクター属細菌、クレブシェラ・オキシトカ(Klebsiella oxytoca)等のクレブシェラ属細菌に代表される腸内細菌群、サルモネラ属細菌、ビブリオ属細菌、シュードモナス属細菌等ミュータンス連鎖球菌(Streptococcus mutans)、ストレプトコッカスゴルドニイ(Streptococcus gordonii)、ウェルシュ菌(Clostridium perfringens)、ボツリヌス菌(Clostridium botulinum)、インフルエンザ菌(Haemophilus influenzae)、エンテロコッカスデュランス(Enterococcus durans)、化膿連鎖球菌(Streptococcus pyogenes)、ストレプトコッカスアガラクティエ(Streptococcus agalacticae)、クロストリジウムディフィシレ(Clostridium difficile)およびエンテロコッカスフェシウム(Enterococcus faecium)などが挙げられる。
【0066】
本発明において、微生物における「生菌」の検出とは、被検試料中の生菌の有無の判別および生菌の量の決定のいずれをも含む。また、生菌の量とは、絶対的な量に限られず、対照試料に対する相対的な量であってもよい。得られる菌数が少なすぎるために十分量のRNAを得ることができない場合や、死菌がPCR反応の結果に影響を与えると考えられる場合には、被験試料に滅菌した微生物用液体培地を添加し、30〜37℃程度で培養することにより菌数を増加させればよい。これによりRNA検出感度を向上させることができ、また死菌の影響を抑えることができる。液体培地の種類は特に制限されず、ブレインハートインフィージョン培地、トリプトソーヤ培地等の一般微生物用培地を広い範囲から適宜選択して使用することができる。ビブリオ属細菌の場合は、これらの培地において食塩濃度を1〜3%程度に調製したものまたはアルカリ性ペプトン培地を使用すればよい。
【0067】
また、検出対象の微生物が病原性微生物である場合には、ターゲット配列領域としては病原遺伝子が挙げられる。病原遺伝子としては、リステリア属細菌のリステリオリシンO(hlyA)遺伝子、サルモネラ属細菌のエンテロトキシン遺伝子やinvA遺伝子、病原性大腸菌O−157のベロ毒素遺伝子、エンテロバクター属細菌のMMS遺伝子(エンテロバクター・サカザキ菌)、黄色ブドウ球菌エンテロトキシン遺伝子、バチルス・セレウス菌のセレウリド(嘔吐毒素)遺伝子やエンテロトキシン遺伝子、ボツリヌス菌の各種毒素遺伝子等が挙げられる。
【0068】
[第2実施形態]
<チューブ状の生体高分子固定化担体を作製する工程>
本工程においては、チューブ状の生体高分子固定化担体を作製する。より具体的には、チューブ状の不溶性担体の少なくとも内側を高分子物質で被覆して、その上に抗体、レクチンおよび複合糖質のいずれかを固定化させることによってチューブ状の生体高分子固定化担体を作製する。
【0069】
まず、本実施形態の本工程における「高分子物質」は、ホスホリルコリン基またはアルキレングリコール基など生体適合性の高い官能基を有するような高分子物質であり、生体成分の非特異的吸着抑制するモノクローナル抗体、タンパク質、レクチン、複合糖質などの生体分子を固定する性質を持つポリマーを示す。具体的には、住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブ、ビーズ単体、プレートを例として挙げることができる。なお、固定化担体の作製方法については、特開2008−128854号公報に記載されている。
【0070】
次に、本発明における「不溶性担体」は、プラスチック、ガラス等からなるチューブのほか、96穴ウェル等を挙げることができる。
【0071】
チューブ状とは、中空状態のものをいい、底があるPCRチューブや、エッペンドルフチューブのような形状であってもよい。
【0072】
そして、本発明における「抗体」、「レクチン」、「複合糖質」は、後述する被検試料に含まれる微生物またはウイルスの一部をエピトープ部位または認識部位として感知できるものであれば特に限定されない。抗体には、免疫グロブリンのようなものを用いることができる。
【0073】
本発明において「エピトープ部位」または「認識部位」には、微生物の表面構造(鞭毛、夾膜、細胞壁糖鎖など)またはウイルスのカプシドおよびエンベロープ部分を設定することができる。
【0074】
たとえば、被検試料に検出対象のウイルスまたは微生物と異なる種類の細胞が含まれる場合には、ターゲット配列領域は、検出対象のウイルスまたは微生物に特異的な配列を有することが好ましい。また、目的によっては、複数種のウイルスまたは微生物に共通する配列を有するものであってもよい。さらに、ターゲット配列領域は単一であっても、複数であってもよい。
【0075】
<微生物またはウイルスを捕捉する工程>
本工程は、被検試料のなかで、微生物またはウイルスを含む被検試料を生体高分子固定化担体の上に添加し、該微生物またはウイルスを分離する工程である。具体的には、チューブ状の生体高分子固定化担体の内側(内部)に被検試料を添加し、該生体高分子固定化担体を2時間程度、静置することで、該微生物またはウイルスを捕捉する。
【0076】
<遺伝子溶解溶液を作製する工程>
本工程は、微生物またはウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する。
【0077】
上記微生物またはウイルスを捕捉した生体高分子固定化担体を前処理する。該生体高分子固定化担体に、グアニジンイソチオシアネート、SDS、NaOHなどを含む緩衝溶液で微生物を溶解させることができるが、次工程のハイブリダイゼーションで対象RNA遺伝子を捕捉するためには、希NaOH溶液で生体試料を、希HCl溶液で中和するなど溶解後生体内の組成を大きく変化させないような工夫を加えたほう好ましい。溶解液をPBSなどの緩衝液に希釈して−20℃で長期間保存することも可能である。
【0078】
<DNA鎖断片もしくはRNA鎖断片で増幅する工程>
本工程は、DNA鎖断片またはRNA鎖断片を増幅する工程である。本工程においては、上述のチューブ状のDNA固定化担体に対して直接、ポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施すことができる。具体的な操作は上述した方法をとることができる。
【0079】
その他、被検試料等については、上述のものと同様のものを応用することができる。
[応用]
本発明は、特に、食品衛生検査や臨床検査において、穏和な加熱処理や抗生物質投与により、損傷菌の状態を呈した微生物の検出が注目されており、本発明においては、生菌の検出のみならず、生菌または死菌との識別も可能な微生物の検出方法を提供するものである。
【0080】
本発明により、ウイルスの感染判定または微生物(細菌)の感染判定を迅速・簡便に行なうことが可能なキットを提供される。試験に要する時間は、患者からの被検試料受領後約2時間であり、従来技術とは完全に区別される迅速診断法である。本発明のキットを用いれば、検査担当者の2次感染を防止でき、ウイルス感染の伝播防止と感染者への迅速な対応と効果的な治療が可能になり、公衆衛生上大きく寄与するものと考えられる。
【0081】
そして、本発明の特定遺伝子の検出方法を用いることによって、ウイルス感染症および細菌感染の診断に関して、患者の臨床状態を迅速にしかも的確に反映することのできる治療法を提供することができる。該特定遺伝子の検出方法は、ウイルス感染症のまたは細菌感染の診断および治療に有効であるだけでなく、他者への感染のリスクの予測、それによる院内感染の防止、治療効果の追跡による現行処置の適否判断やさらなる治療対策の検討が可能となる。
【0082】
そして、本発明の特定遺伝子の検出方法では、生体試料権対中のウイルス溶解処理から遺伝子増幅および検出までに要する時間はわずか2時間程度で実施可能である。この短時間の操作により迅速かつ簡便にウィルスを検出することができる。
【0083】
すなわち、本発明の特定遺伝子の検出方法によると、短時間で且つ高感度にウイルスまたは微生物を検出できるため、ウイルス感染症患者の病態および細菌感染症患者の病態を的確に反映することができる。さらに、本発明は、従来の分離および精製等の前処理を必要とせず、簡易に高感度で検出し得るウイルスの検出方法を提供することで遺伝子検査作業者の2次感染予防の効果が期待できる。
【0084】
さらに本発明の特定遺伝子の検出方法によると、主要な食中毒細菌の食品中への混入の可能性を簡単かつ高感度に調べることができる。食品メーカーによる製品出荷前の自主細菌検査に好適に利用できる。また、本発明の細菌の検出方法において、食品の破砕工程と遠心分離操作とを行なう場合には、細菌を食品から効率よく分離採取できるため、細菌を食品から分離培養する必要がなくなり、その分検査時間が短縮される。
【0085】
以下、実施例を挙げて本発明をより詳細に説明するが、本発明はこれらに限定されるものではない。
【実施例】
【0086】
<実施例1>
≪第1工程(a):ノロウイルスG1型検出用PCRチューブの作製≫
非特許文献2(ノロウイルスの検出法)記載の公知のノロウイルスG1型特異的DNA塩基配列5’−CTTAGACGCCATCATCATTYAC−3’(22塩基)(配列番号10)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製した。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの内部底面に添加し、室温で1時間静置することで、キャプチャーDNA鎖を固定化させてDNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNaseフリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0087】
≪第1工程(b):ノロウイルスG2型検出用PCRチューブの作製≫
非特許文献2(ノロウイルスの検出法)記載の公知のノロウイルスG2型特異的DNA塩基配列5’−TCGACGCCATCTTCATTCACA−3’(21塩基)(配列番号11)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの内部底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させてDNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNase フリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0088】
≪第2工程:ノロウイルス感染患者糞便検体の処理≫
ノロウイルス感染患者糞便検体にPBSを加え、10%乳剤を作製し、この10%乳剤を激しく攪拌した後、12,000rpmで20分間冷却遠心分離して得られた遠心上清をノロウイルスを含有するサンプルとして使用した。
【0089】
≪第3工程:ノロウイルス遺伝子の分離≫
15μLのサンプルを、作製したノロウイルスG1型およびG2型検出用PCRチューブそれぞれに添加し、5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))をさらに加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で該チューブを30分間静置してノロウイルス遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内の「ノロウイルスを含有するサンプル」を、マイクロピペットで完全に除去後、PCRチューブ内をPBS150μLで洗浄した。上記操作にて、ノロウイルス遺伝子を分離したPCRチューブを使用して以下のRT−LAMP法による遺伝子検出法に供した。
【0090】
≪第4工程、第5工程:RT−LAMP法によるノロウイルス遺伝子の増幅および検出≫
ノロウイルス遺伝子の検出は、栄研化学(株)社製LoopampノロウイルスGI検出試薬キットおよびLoopampノロウイルスGII検出試薬キットを使用して行なった。反応条件は添付のプロトコールにしたがって実施した。具体的には、上述したようにノロウイルス感染患者糞便検体からノロウイルス遺伝子をオリゴDNAが固定化されたロウイルスG1およびG2検出用PCRチューブでそれぞれ分離し、ノロウイルスG1検出用PCRチューブには、LoopampノロウイルスGI検出試薬キットより調整された反応液を添加し、ノロウイルスG2検出用PCRチューブには、LoopampノロウイルスGII検出試薬キットより調整された反応液を添加した。そして、それぞれのPCRチューブに対してRT−LAMP法による遺伝子増幅反応を63℃、60分間で行ない、遺伝子増幅反応終了後、80℃、5分間にて酵素を失活させた。
【0091】
図1は、実施例1のLAMP法反応の増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無による標的遺伝子配列の有無を判別した図である((上):目視濁度結果、(下):栄研化学(株)社製Loopampエンドポイント濁度測定装置(LA−100)による濁度測定結果。レーン番号1および2は、水またはコントロールRNAを栄研化学(株)社製LoopampノロウイルスGI検出試薬キットを用いて行なったLAMP法のコントロール反応)。なお、図1の濁度測定結果の縦軸は濁度(単位:OD660値(透過率))を示す。
【0092】
図2は、実施例1のLAMP法反応液に約1μLの1000×の濃度のSYBR Green I添加(最終濃度:100×)によるLAMP法反応液の核酸増幅産物の目視確認結果を示す図である。
【0093】
図1、図2におけるレーン番号1〜8は、以下のとおりである。
1:ネガティブコントロール(水添加による通常LAMP法)
2:ポジティブコントロール(G1コントロールRNA添加による通常LAMP法)
3:ノロウイルスG1型検出用PCRチューブ(水添加)
4:ノロウイルスG2型検出用PCRチューブ(水添加)
5:ノロウイルスG1型検出用PCRチューブ(G1コントロールRNA添加)
6:ノロウイルスG2型検出用PCRチューブ(G2コントロールRNA添加)
7:ノロウイルスG1型検出用PCRチューブ(G1糞便検体処理液添加)
8:ノロウイルスG2型検出用PCRチューブ(G2糞便検体処理液添加)
[結果]
ポジティブコントロールと同様にレーン番号5〜8においてもノロウイルスを検出することができた。
【0094】
<確認実験1>
TaqMan RT−qPCR法、並びにNASBA(Nucleic Acid Sequence−Based Amplification)法のスイフトジーンノロウイルスGI/GII「カイノス」(株式会社カイノス製)、TRC (Transcription Reverse transcription Concerted reaction)法のノロウイルスRNA検出試薬TRCRtest Noro (東ソー株式会社製)の各キットを用いた増幅反応を同一のノロウイルス感染患者由来の糞便サンプルを用いて行なった。いずれの結果も、LAMP法と同等の結果を得ることができた。
【0095】
<確認実験2:LAMP法の感度>
本発明の検出方法にLAMP法を導入したことにより、まずLAMP法の遺伝子増幅の限界を確認する必要がある。
【0096】
そこで、10000〜10コピー/チューブのノロウイルスG2コントロールRNA並びにノロウイルスG2感染患者糞便サンプルから通知法(非特許文献2)に従い抽出した3種類のRNA(S1,S2,S3)を用いてLAMP法の遺伝子増幅の限界を確認した。ノロウイルスG2RNAをノロウイルスG2型検出用PCRチューブに添加してノロウイルス遺伝子を分離後、LAMP法の増幅反応液に添加し、63℃、60分の増幅反応後、アガロースゲル電気泳動を行なった。コントロールとして、ノロウイルスG2 RNAをLAMP法の増幅反応液に添加し、添付プロトコールに従い実施した。結果は図3に示した。図3は、実施例1のコントロールのノロウイルスRNAとノロウイルス感染患者糞便サンプル(S1〜3)を用いたLAMP法反応結果を示す図であり、図3Aは、ノロウイルスG2型検出用PCRチューブを用いてノロウイルス遺伝子を分離後、LoopampノロウイルスG2検出試薬キットを用いて行なったLAMP法遺伝子増幅反応産物を電気泳動した図である。図3Bは、ノロウイルス遺伝子を直接LoopampノロウイルスGI検出試薬キットに添加して行なったLAMP法のコントロールの結果である。
【0097】
図3の各レーン番号は以下のとおりとなっている。
M:マーカー
1:10コピー/チューブ (G2コントロールRNA添加)
2:100コピー/チューブ (G2コントロールRNA添加)
3:1000コピー/チューブ (G2コントロールRNA添加)
4:10000コピー/チューブ (G2コントロールRNA添加)
5:ノロウイルス感染患者糞便サンプル(S1)
6:ノロウイルス感染患者糞便サンプル(S2)
7:ノロウイルス感染患者糞便サンプル(S3)
B:ネガティブコントロール(水添加)
なお、電気泳動によって、アガロースゲルのウェルから尾を引いたようなバンドが確認できれば、遺伝子が増幅されたことを示している。
【0098】
図3に示すようにノロウイルスG2型検出用PCRチューブを使用してウイルス遺伝子を分離後、LAMP法を実施した増幅限界は10000コピー/チューブまでであった。この検出限界は、一般的に広く用いられているPCR法と同程度以下ではある。また、ノロウイルス感染患者糞便サンプル(S1,S2,S3)から抽出したRNAを用いた場合には、いずれも検出可能であった。コントロールの通常のLAMP反応液に直接ノロウイルスRNAを添加したLAMP法の結果は、非特許文献3の結果とほぼ同等の結果であった。この非特許文献3においては、LAMP法の増幅時間はPCR法が約2時間かかるのに対し、本実施例においてLAMP法は1時間であった。これより、核酸増幅にLAMP法を用いることで検査時間の短縮が可能であると考えられた。また、ノロウイルスG1に関しても同様の実験を行い、同等の検出感度であることを確認した。
【0099】
<確認実験3:再現性について>
繰り返して100回、上記同様の測定した結果、毎回ほぼ同程度の結果が検出された。これにより、LAMP法ではほぼ決まった長さの生成物が同程度増幅されることが確認された。
【0100】
<実施例2>
≪第1工程:HIV−1検出用PCRチューブの作製≫
公知のHIV−1特異的DNA塩基配列5’−TACTAGTAGTTCCTGCTATGTCACTTCC−3’(28塩基)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させることで、DNA固定化担体としてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNase フリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0101】
≪第2工程:HIV−1コントロールRNAの作製≫
HIV−1より作製された公知のプラスミドDNA(pNL432)のgag遺伝子領域をPCR法で遺伝子増幅し、pSTBlue−1 AccepTor(TM) Vector Kitsを使用して常法によりクローニングを行い、in VitroでHIV−1コントロールRNAを作成した。その遺伝子配列は、以下の通りである。
cagcagctgacacaggaaacagcaacaaggtcagccaaaattaccctatagtgcagaacatccaggggcaaatggtacatcaggccatatcacctagaactttaaatgcatgggtaaaagtagtagaagagaaggcttttagcccagaagtaatacccatgttttcagcattatcagaaggagccaccccacaagatttaaacaccatgctaaacacagtggggggacatcaagcagccatgcaaatgttaaaagagaccatcaatgaggaagctgcagaatgggatagattgcatccagtgcaggcagggcctgtcgcaccaggccagatgagagaaccaaggggaagtgacatagcaggaactactagtacccttcaggaacaaataggatggatgacaaataatccacctatcccagtaggagaaatctataaaagatggataatcctgggattaaataaaatagtaaggatgtatagccctgccagcattctggacataagacaaggaccaaaggaaccctttagagattatgtagaccggttctataaaactctaagagccgagcaagcttcaca(配列番号12)
≪第3工程:市販ヒト血清へコントロールHIV−1 RNAの添加およびHIV−1遺伝子の分離回収)
作製したHIV−1検出用PCRチューブに10μLの市販ヒト血清を4μLのPBS希釈して添加し、HIV−1コントロールRNAを106〜102コピー/チューブ加えた。このヒト血清とHIV−1コントロールRNAとの混合物からなるサンプルは、実質的にHIV感染患者の血清と同様なものとして利用した。さらに、該サンプルに5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))を加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機でスピンダウン後、室温で30分間静置してHIV−1遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のHIV−1溶解液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。操作にてHIV−1遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による増幅に供した。
【0102】
≪第4工程:RT−PCR法による増幅≫
RT−PCR法の条件設定を行なった。これは、目的のRNAを増幅するための条件設定のために行なった。
【0103】
(TaqMan RT−qPCR条件調整)
ReverTra Ace(東洋紡績)を100U/μlの原液を専用の希釈バッファーで10U/μl(10倍希釈)希釈する。この希釈液を下記の表に従い、逆転写反応液試薬(RT反応液)を調整した。
【0104】
【表1】
【0105】
次に下記のプロトコールに従い、ワンステップのTaqMan RT−qPCR反応液を調整した。
【0106】
【表2】
【0107】
SK145(Forward):5’−AGTGGGGGGACATCAAGCAGCCATGCAAAT−3’(配列番号13)
SKCC1B(Reverse):5’−TACTAGTAGTTCCTGCTATGTCACTTCC−3’(配列番号14)
SK102−TP(TaqMan Probe):5’−GAGACCATCAATGAGGAAGCTGCAGAATGGGAT−3’(配列番号15)
(RT−PCRプログラム)
48℃,20分→95℃,5分→94℃,15秒⇔54℃,45秒、45サイクル
(結果)
TaqMan RT−qPCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行なった。表3に示すようにHIV−1検出用PCRチューブを使用してウイルス遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は100コピー/チューブまでであることが分かった。
【0108】
【表3】
【0109】
(RT−PCR条件)
TaqMan RT−qPCR反応液に調整したRT反応液を使用して、次に下記のプロトコールに従い、RT−PCR反応液の調整を行なった。
【0110】
【表4】
【0111】
G60(Forward):5’−CAGCCAAAATTACCCTATAGTGCAG−3’(配列番号16)
SK39(Reverse):5’−TTTGGTCCTTGTCTTATGTCCAGAATGCTG−3’(配列番号17)
(RT−PCRプログラム)
48℃,20分→95℃,1分→94℃,15秒⇔54℃,1分、40サイクル
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図4は、コントロールHIV−1 RNAを添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動(458bp)法で検出した結果を示す図である。そして、具体的には、図4Aは、上述したヒトヒト血清とHIV−1コントロールRNAとの混合物からなるサンプルを用いて第1工程〜第4工程までした結果を示し、図4Bは、HIVコントロールRNAのみを第1工程〜第4工程までした結果を示す。
【0112】
図4における各レーン番号は以下のとおりとなっている。
M:マーカー
1:100コピー/チューブ (コントロールHIV−1 RNA添加)
2:1000コピー/チューブ (コントロールHIV−1 RNA添加)
3:10000コピー/チューブ (コントロールHIV−1 RNA添加)
B:ネガティブコントロール(水添加)
図4に示すようにHIV−1検出用PCRチューブを使用してウイルス遺伝子を分離後、RT−PCR法を実施した増幅限界は10000コピー/チューブまでであった。
【0113】
[まとめ]
以上から、このヒト血清とHIV−1コントロールRNAとの混合物からなるサンプル(実質的にHIV感染患者の血清と同様なもの)に対して、本発明のDNA固定化担体を用いた特定遺伝子の検出方法を有効に利用することができることが分かった。
【0114】
<実施例3>
≪第3工程:市販ヒト血清へHIV−1 LAI株の添加およびHIV−1遺伝子の分離回収≫
実施例2で作製したHIV−1検出用PCRチューブに10μLの市販ヒト血清を4μLのPBS希釈して添加し、HIV−1 LAI株 ウイルスを105コピー/チューブ加えた。さらに、5μLのウイルス溶解バッファー(4M GuSCN、25mM Sodium citrate(pH7.0)、0.5%lauroylsarcosine(w/v))を加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機でスピンダウン後、室温で30分間静置してHIV−1遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のHIV−1溶解液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。操作にてHIV−1遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子増幅に供した。
【0115】
≪第4工程:RT−PCR法による増幅≫
(RT−PCR条件)
TaqMan RT−qPCR反応液に調整したRT反応液を使用して、次に下記のプロトコールに従い、RT−PCR反応液の調整を行なった。
【0116】
【表5】
【0117】
G60(Forward):5’−CAGCCAAAATTACCCTATAGTGCAG−3’(配列番号18)
SK39(Reverse):5’−TTTGGTCCTTGTCTTATGTCCAGAATGCTG−3’(配列番号19)
(RT−PCRプログラム)
48℃,20分→95℃,1分→94℃,15秒⇔54℃,1分、40サイクル
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。実施例3のHIV−1 LAI株を添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動(458bp)法で検出した結果を示す図である。なお、図5の各レーンは、以下のとおりである。
レーン番号1と3:ネガティブコントロール
レーン番号2:ポジティブコントロール(コントロールHIV−1 LAI RNA添加)
レーン番号4:HIV−1検出用PCRチューブ使用したRT−PCR法の結果
[まとめ]
図5から、HIV−1 LAI株 ウイルス(市販)に対しても本発明のDNA固定化担体を用いた特定遺伝子の検出方法を有効に利用することができることが分かった
[考察]
表1、図4、5に示すようにHIV−1検出用PCRチューブを使用して血清中に浮遊するウイルス粒子を溶解し、溶出するウイルス遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は100コピー/チューブまでであった。この検出限界は、一般的に広く用いられているPCR法と同程度であり、HIV−1感染患者の血清中ウイルス(約106コピー/mL)を直接遺伝子検査できる範囲にあることを確認した。これより、血清からウイルス遺伝子を抽出する操作が省略でき、検査時間の短縮が可能であると考えられた。
【0118】
<実施例4:大腸菌全RNA標準液による反応性の確認>
≪第1工程:細菌16S rRNA検出用PCRチューブの作製≫
公知の大腸菌ゲノムDNAの16S rRNAをコードする配列中の細菌全般に保存されているDNA塩基配列5’−GTATTACCGCGGCTGCTG−3’(18塩基、519−536)(配列番号5)を含み、5’末端がアミノ基修飾したキャプチャーDNA鎖としての合成DNAプローブを5M K2HPO4(pH9.0)に溶解し、0.1μMのキャプチャーDNA鎖溶液を調製する。キャプチャーDNA鎖溶液20μLを住友ベークライト株式会社の開発した表面処理(S−BIO PrimeSurface)を施したPCRチューブの底面に添加し、室温で1時間静置してキャプチャーDNA鎖を固定化させることで、DNA固定化担体とてのPCRチューブを作製した。その後、PCRチューブ内を300μLの0.1N NaOHでブロッキング処理を施し、RNaseフリー水300μLで2回洗浄したPCRチューブを以下の実験に使用した。
【0119】
≪第2工程および第3工程:大腸菌全RNAからの16S rRNA遺伝子の分離≫
大腸菌全RNA標準液(ロシュ社製)を水で希釈して1,10,100,1000pg/μLの各溶液を調整した。16S rRNA検出用PCRチューブに各濃度RNA溶液1μLにおよびPBS19μLを加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0120】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0121】
(RT−PCR条件)
RNA−direct SYBR Green Realtime PCR Master Mix (TOYOBO)添付書類に従い、ワンステップのRT−PCR反応液の調整を行なった。
【0122】
【表6】
【0123】
(RT−PCRプログラム)
【0124】
【表7】
【0125】
16S−9F (Forward):5’− GAGTTTGATCCTGGCTCAG−3’(配列番号20)
16S−536R (Reverse):5’−GTATTACCGCGGCTGCTG−3’(配列番号21)
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図6は、大腸菌全RNA標準液(ロシュ社製)をチューブ当り1,10,100,1000pg/μL添加して、RT−PCRを行なった結果を示す図(RT−PCR電気泳動(528bp))である。図6に示すように細菌16S rRNA検出用PCRチューブを使用して細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。増幅限界は1ピコグラム/チューブまでであった。なお、図6の各レーンは以下のとおりである。
レーン0:ネガティブコントロール
レーン番号1:1pg/μL
レーン番号10:10pg/μL
レーン番号100:100pg/μL
レーン番号1000:1000pg/μL
以上から、本発明においては、微生物特異的な配列キャプチャーDNAが高感度で微生物を検出できることが分かった。
【0126】
<実施例5:複数の逆転写プライマーによる効果の確認>
≪第2工程および第3工程:大腸菌全RNAからの16S rRNA遺伝子の分離≫
本実施例においては、実施例4で作製したPCRチューブを使用した(第1工程)。水で希釈しての各溶液を調整する。16S rRNA検出用PCRチューブに濃度1pg/μLの大腸菌全RNA標準液(ロシュ社製)を1μLおよびPBS19μLを加え、ボルテックスミキサーで激しく撹拌して、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0127】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0128】
(RT−PCR条件)
RNA−direct SYBR Green Realtime PCR Master Mix (TOYOBO)添付書類に従い、ワンステップのRT−PCR反応液の調整を行なった。
【0129】
【表8】
【0130】
16S−9F(Forward):5‘− GAGTTTGATCCTGGCTCAG−3’(配列番号22)
16S−536R(Reverse):5’−GTATTACCGCGGCTGCTG−3’(配列番号23)
(逆転写プライマー(RT Primer))
Pba2:5’−TCACCGGCCGTGTGTACAAG−3’(配列番号24)
Microbio:5’−GGACTACCAGGGTATCTAATCCTGTT−3’(Microbiology, 148: 257−266 (2002))(配列番号25)
(RT−PCRプログラム):実施例4と同一条件で行なった。
【0131】
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図1に示すように細菌16S rRNA検出用PCRチューブを使用して細菌16S rRNA遺伝子を分離後、逆転写プライマーを追加した条件下でRT−PCR法を実施した。図7は、複数の逆転写プライマーによる効果の確認結果を示す図(RT−PCR 電気泳動 (528bp))である。2種類のプライマーを用いたときにバンドが濃くなることから、プライマーが1つより2つの方が検出感度が上がることことから、逆転写プライマーを追加の効果は明白に確認できた。なお、図7の各レーンは以下のとおりである。
Normal Tube:対象となるコントロール実験結果
GeneTube:本発明の実験結果
レーン番号1:逆転写プライマー添加なし
レーン番号2:Pba2
レーン番号3:Microbio
レーン番号4:Pba2+Microbio
<実施例6:標準菌株培養における反応性の確認>
本実施例においては、実施例4の第1工程で作製したPCRチューブを用いた。
【0132】
≪第2工程(a):大腸菌標準菌株の培養および溶菌≫
培養は通常液体培地あるいは普通寒天培地を用い、37℃、一昼夜(14−18時間)行なう。培地はインスタント培地である「普通ブイヨン栄研」を用い、液体培地は「普通ブイヨン栄研」の指示量を脱塩水に溶かし、寒天培地はそれに1.6%の寒天末を加え、それぞれオートクレーブ後、使用した。
【0133】
1Lあたりの培地組成は以下の通りとした。
肉エキス3g、ペプトン10g、NaCl5g(pH7.0)
そして、大腸菌(Escherichia coli ATCC25922)被検リボゾームRNAを以下のようにして調製した。被検菌を白金耳などで上記液体培地に懸濁し、37℃で一晩培養した。なお、生菌数は10μlの懸濁液をとり適当に希釈して栄養寒天培地に塗抹し、37℃で一晩培養し、出現したコロニー数と希釈率とから求め、生菌数は約109cfu/mlであった。菌体培養液20μlを10000×gで5分間遠心し、沈殿を採取した。沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5% SDS,5%ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0134】
≪第2工程(b):大腸菌標準菌株の添加食品の培養操作後にRT−PCR反応を行なう方法≫
大腸菌(Escherichia coli ATCC25922)を小麦粉に1g当たり10個程度となるように添加した。菌が添加された小麦粉重量の9倍量の通常液体培地(普通ブイヨン栄研)を加え、37℃で一晩培養した。培養前の小麦懸濁中の菌密度は、1個/mlであったが、4時間培養後には約1×103個/ml増殖した。培養液1mlを採取し、100×gで1分間遠心し、上清を10000×gで5分間遠心し、沈殿を採取した。この沈殿を洗うためTE緩衝液(5mM Tris 0.1mM EDTA)に懸濁し、10000×gで5分間遠心し、沈殿を採取した。沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5%SDS,5%ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0135】
≪第3工程:被検RNA溶液からの16S rRNA遺伝子の分離>
16S rRNA検出用PCRチューブに被検RNA溶液をそれぞれ加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0136】
≪第4工程:RT−PCR法によるDNAの増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0137】
(RT−PCR条件)
ワンステップRT−PCR反応液は実施例4と同一条件で行なった。
【0138】
(結果)
RT−PCR測定は、Applied Biosystems 7300リアルタイムPCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図8(RT−PCR電気泳動(528bp))に示すように細菌16S rRNA検出用PCRチューブを使用して、大腸菌標準株培養から得た被検RNA溶液から細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。大腸菌の場合には、小麦1g当たり10個程度の細菌が存在している場合には、前述の操作による培養時間を4時間以上にすれば、PCR反応によりその16S rRNAを検出できることが分かる。また、黄色ブドウ球菌(Staphylococcus aureus ATCC25923)、緑脳菌(Pseudomonas aeruginosa ATCC27853)およびサルモネラ菌(Salmonella enterica serovar Typhimurium ATCC14028)から選ばれる微生物の各被検リボゾームRNAからも同一のプライマーの組み合わせを用いることにより同様の結果が得られた。なお、図8における各レーンは以下のとおりである。
レーン番号1:大腸菌標準株培養から得た被検RNA溶液
レーン番号2:大腸菌標準菌株の添加食品の培養から得た被検RNA溶液
<実施例7:大腸菌DH5α株の培養経過における16S rRNAの確認>
≪第2工程:被検RNA溶液の作製≫
大腸菌DH5α(Bethesda Research Laborotories製)102〜103cfuをLB培地10mLに懸濁し、37℃で培養した。培養液50μlを1、2、4時間経過後にそれぞれサンプリングし、10000×gで5分間遠心し、沈殿を採取した。各沈殿にPBS18μlを加え、溶菌液(0.5N NaOH,0.5% SDS,5% ポリエチエングリコール1000)1μlを混合し、10分間室温で静置した後、氷冷し、中和液(0.2M リン酸緩衝液(pH7.0)、0.5N HCl)1μlを加えて中和し、これを被検RNA溶液とした。
【0139】
≪第3工程:被検RNA溶液からの16S rRNA遺伝子の分離≫
16S rRNA検出用PCRチューブ(実施例4で作製したPCRチューブ)に被検RNA溶液を加え、ボルテックスミキサーで激しく撹拌し、簡易卓上遠心機で遠心分離後、室温で30分間静置して16S rRNA遺伝子をハイブリダイゼーションで捕捉した。PCRチューブ内のRNA溶液は、マイクロピペットで完全に除去後、PBS150μLで洗浄した。
【0140】
≪第4工程:RT−PCR法によるDNA増幅≫
操作にて16S rRNA遺伝子を分離したPCRチューブを使用して以下のRT−PCR法による遺伝子検出法に供した。
【0141】
(RT−PCR条件)
逆転写反応:PrimeScript RT−PCR Kit (Takara, PR037A)を使用して以下の条件で行なった。
【0142】
【表9】
【0143】
【表10】
【0144】
PCR反応は以下の条件で実施した。
【0145】
【表11】
【0146】
【表12】
【0147】
(結果)
RT−PCR測定は、Applied Biosystems 9700PCRシステムを使用して行い、アガロースゲル電気泳動にて増幅された遺伝子断片の確認を行なった。図9は、大腸菌DH5α株の培養時間(h)経過における16S rRNAの確認した結果を示す図(RT−PCR 電気泳動 (528bp))である。細菌16S rRNA検出用PCRチューブを使用して、大腸菌標準株培養から得た被検RNA溶液から細菌16S rRNA遺伝子を分離後、RT−PCR法を実施した。2時間経過した培養液中から明白な16S rRNA遺伝子増幅産物が確認できた。
【0148】
[考察]
図6〜9に示すように16S rRNA検出用PCRチューブを使用して、様々なサンプル形態に応じて細菌の16S rRNA遺伝子を分離後、リアルタイムRT−qPCR法を実施した増幅限界は細菌10個/チューブであった。この検出限界は、一般的に広く用いられているPCR法と同程度であることを確認した。これより、サンプルから16S rRNA遺伝子を抽出する操作が省略でき、検査時間の短縮が可能であると考えられる。本発明の微生物検出法では、被検試料検体中の微生物溶解処理から遺伝子増幅・検出までに要する時間はわずか2時間程度で実施可能である。この短時間の操作により迅速・簡便に微生物を検出することができ、公衆衛生上大きく寄与するものと考えられた。
【0149】
今回開示された実施の形態および実施例はすべての点で例示であって制限的なものではないと考えられるべきである。本発明の範囲は上記した説明ではなくて特許請求の範囲によって示され、特許請求の範囲と均等の意味および範囲内でのすべての変更が含まれることが意図される。
【図面の簡単な説明】
【0150】
【図1】実施例1のLAMP法反応の増幅副産物であるピロリン酸マグネシウムMg2P2O7(白色沈殿物質)の白濁の有無による標的遺伝子配列の有無を判別した図である((上):目視濁度結果、(下):栄研化学(株)社製Loopampエンドポイント濁度測定装置(LA−100)による濁度測定結果。レーン番号1および2は、水またはコントロールRNAを栄研化学(株)社製LoopampノロウイルスGI検出試薬キットを用いて行なったLAMP法のコントロール反応)。
【図2】実施例1のLAMP法反応液に約1μLの1000×の濃度のSYBR Green I添加(最終濃度:100×)によるLAMP法反応液の核酸増幅産物の目視確認結果を示す図である。
【図3】実施例1のコントロールのノロウイルスRNAとノロウイルス感染患者糞便サンプル(S1〜3)を用いたLAMP法反応結果を示す図である(A:ノロウイルスG2型検出用PCRチューブを用いてノロウイルス遺伝子を分離後、LoopampノロウイルスG2検出試薬キットを用いて行なったLAMP法遺伝子増幅反応産物を電気泳動。B:ノロウイルス遺伝子を直接LoopampノロウイルスGI検出試薬キットに添加して行なったLAMP法のコントロールの結果)。
【図4】実施例2のコントロールHIV−1 RNAを添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動法で検出した結果を示す図である。
【図5】実施例3のHIV−1 LAI株を添加した市販血清から、HIV−1検出用PCRチューブを用いてウイルス遺伝子を分離、RT−PCR法で増幅し、電気泳動法で検出した結果を示す図である。
【図6】実施例4の大腸菌全RNA標準液(ロシュ社製)をチューブ当り1,10,100,1000pg/μL添加して、RT−PCRを行なった結果を示す図である。
【図7】実施例5の複数の逆転写プライマーによる効果の確認結果を示す図である(Normal Tube:対象となるコントロール実験結果、GeneTube:本発明の実験結果。レーン番号1:逆転写プライマー添加なし、レーン番号2:Pba2、レーン3:Microbio、レーン4:Pba2+Microbio)。
【図8】実施例6の標準菌株培養における反応性の確認結果を示す図である(レーン番号1:大腸菌標準株培養から得た被検RNA溶液、レーン番号2:大腸菌標準菌株の添加食品の培養から得た被検RNA溶液)。
【図9】実施例7の大腸菌DH5α株の培養時間経過における16S rRNAの確認した結果を示す図である。
【特許請求の範囲】
【請求項1】
ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、
被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、
前記DNA固定化担体の上に前記遺伝子溶解溶液を添加し、前記キャプチャーDNA鎖と前記遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、前記キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する第3工程と、
前記DNA固定化担体に対して直接、前記DNA鎖断片または前記RNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施して前記DNA鎖断片または前記RNA鎖断片で増幅する第4工程と、
を備える特定遺伝子の検出方法。
【請求項2】
ホスホリルコリン基またはアルキレングリコール基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、
被検試料のなかで、微生物またはウイルスを含む被検試料を前記生体高分子固定化担体の上に添加し、前記抗体、レクチンおよび複合糖質のいずれかで前記微生物または前記ウイルスを捕捉する工程と、
前記微生物または前記ウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、
前記生体高分子固定化担体に対して直接、前記DNA鎖断片または前記RNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施して前記DNA鎖断片もしくは前記RNA鎖断片で増幅する工程と、
を備える特定遺伝子の検出方法。
【請求項3】
被検試料が、鼻汁、鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液、細胞組織または食品である請求項1または2に記載の特定遺伝子の検出方法。
【請求項4】
前記第3工程において、4〜70℃で5分〜90分間、ハイブリダイズする請求項1または3に記載の特定遺伝子の検出方法。
【請求項5】
前記RNA鎖断片を逆転写するための逆転写反応のプライマーとして、ランダムプライマー、dTプライマーまたは1個以上の配列特異的なスペシフィックプライマーから合成されたcDNAを使用する請求項1〜4のいずれかに記載の特定遺伝子の検出方法。
【請求項6】
前記第4工程、または、前記DNA鎖断片もしくは前記RNA鎖断片で増幅する工程の後に増幅したDNA鎖断片を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する第5工程をさらに備える請求項1〜5のいずれかに記載の特定遺伝子の検出方法。
【請求項7】
前記キャプチャーDNA鎖は、23S rRNA遺伝子配列または16S rRNA遺伝子配列に相補的な配列から選択される請求項1または請求項3〜6のいずれかに記載の特定遺伝子の検出方法。
【請求項8】
前記微生物がグラム陽性菌またはグラム陰性菌である請求項1〜7のいずれかに記載の特定遺伝子の検出方法。
【請求項9】
前記ウイルスが、C型肝炎ウイルス、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス、黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス、トガウイルス、アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス、ペスチウイルス、ブタコレラウイルス、ウシ下痢ウイルス、パラミクソウイルス、パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス、オルソクソウイルス、ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス、ラブドウイルス、狂犬病ウイルス、水泡性口内炎ウイルス、ピコルナウイルス、ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス、コロナウイルス、ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス、アレナウイルス、リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス、レトロウイルス、ヒト成人白血病ウイルス、エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス、レオウイルス、ロタウイルス、カリシウイルス、ノーウオークウイルス、ノロウイルス、ブンヤウイルス、腎症候性出血熱ウイルス、フィロウイルス、エボラウイルス、マールブルグウイルス、B型肝炎ウイルス、ポックスウイルス、ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス、パルボウイルス、ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス、パポーバウイルス、パピローマウイルス、ポリオーマウイルス、アデノウイルス、ヘルペスウイルス、単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルスおよびアフリカ豚コレラウイルスよりなる群から選択される少なくとも1種である請求項1〜8のいずれかに記載の特定遺伝子の検出方法。
【請求項1】
ホスホリルコリン基を有する第一単位とカルボン酸誘導基を有する第二単位とを含む高分子物質で被覆された不溶性担体の上にキャプチャーDNA鎖を固定化させてチューブ状のDNA固定化担体を作製する第1工程と、
被検試料のなかで、微生物またはウイルスを含む被検試料から微生物由来遺伝子またはウイルス由来遺伝子を遊離して、遺伝子溶解溶液を作製する第2工程と、
前記DNA固定化担体の上に前記遺伝子溶解溶液を添加し、前記キャプチャーDNA鎖と前記遺伝子溶解溶液中のDNA鎖またはRNA鎖とをハイブリダイズさせて、前記キャプチャーDNA鎖と相補的な配列を有するDNA鎖断片またはRNA鎖断片を分離する第3工程と、
前記DNA固定化担体に対して直接、前記DNA鎖断片または前記RNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施して前記DNA鎖断片または前記RNA鎖断片で増幅する第4工程と、
を備える特定遺伝子の検出方法。
【請求項2】
ホスホリルコリン基またはアルキレングリコール基を有する高分子物質で被覆された不溶性担体の上に抗体、レクチンおよび複合糖質のいずれかを固定化させてチューブ状の生体高分子固定化担体を作製する工程と、
被検試料のなかで、微生物またはウイルスを含む被検試料を前記生体高分子固定化担体の上に添加し、前記抗体、レクチンおよび複合糖質のいずれかで前記微生物または前記ウイルスを捕捉する工程と、
前記微生物または前記ウイルスにおける微生物由来遺伝子またはウイルス由来遺伝子を遊離して、DNA鎖断片またはRNA鎖断片を含む遺伝子溶解溶液を作製する工程と、
前記生体高分子固定化担体に対して直接、前記DNA鎖断片または前記RNA鎖断片をポリメラーゼ連鎖反応、LAMP法、鎖置換増幅、逆転写酵素鎖置換増幅、逆転写酵素ポリメラーゼ連鎖反応、逆転写LAMP法、核酸配列に基づく増幅、転写媒介性増幅およびローリングサークル型増幅法から選ばれる方法を施して前記DNA鎖断片もしくは前記RNA鎖断片で増幅する工程と、
を備える特定遺伝子の検出方法。
【請求項3】
被検試料が、鼻汁、鼻腔ぬぐい液、眼結膜ぬぐい液、咽頭ぬぐい液、喀痰、糞便、全血、血清、血漿、髄液、唾液、尿、汗、精液、細胞組織または食品である請求項1または2に記載の特定遺伝子の検出方法。
【請求項4】
前記第3工程において、4〜70℃で5分〜90分間、ハイブリダイズする請求項1または3に記載の特定遺伝子の検出方法。
【請求項5】
前記RNA鎖断片を逆転写するための逆転写反応のプライマーとして、ランダムプライマー、dTプライマーまたは1個以上の配列特異的なスペシフィックプライマーから合成されたcDNAを使用する請求項1〜4のいずれかに記載の特定遺伝子の検出方法。
【請求項6】
前記第4工程、または、前記DNA鎖断片もしくは前記RNA鎖断片で増幅する工程の後に増幅したDNA鎖断片を検量して、被検試料中の微生物遺伝子またはウイルス遺伝子の存在の有無を判定する第5工程をさらに備える請求項1〜5のいずれかに記載の特定遺伝子の検出方法。
【請求項7】
前記キャプチャーDNA鎖は、23S rRNA遺伝子配列または16S rRNA遺伝子配列に相補的な配列から選択される請求項1または請求項3〜6のいずれかに記載の特定遺伝子の検出方法。
【請求項8】
前記微生物がグラム陽性菌またはグラム陰性菌である請求項1〜7のいずれかに記載の特定遺伝子の検出方法。
【請求項9】
前記ウイルスが、C型肝炎ウイルス、D型肝炎ウイルス、E型肝炎ウイルス、G型肝炎ウイルス、手足口病ウイルス、フラビウイルス、黄熱ウイルス、西ナイルウイルス、日本脳炎ウイルス、デングウイルス、トガウイルス、アルファウイルス、ルビウイルス、アルテリウイルス、ルベラウイルス、ペスチウイルス、ブタコレラウイルス、ウシ下痢ウイルス、パラミクソウイルス、パラインフルエンザウイルス1,2,3,4、イヌジステムパ−ウイルス、ニューカッスル病ウイルス、RSウイルス、リンダペストウイルス、サルパラインフルエンザウイルス、麻疹ウイルス、ムンプスウイルス、オルソクソウイルス、ヒトインフルエンザウイルス、トリインフルエンザウイルス、ウマインフルエンザウイルス、ブタインフルエンザウイルス、ラブドウイルス、狂犬病ウイルス、水泡性口内炎ウイルス、ピコルナウイルス、ポリオウイルス、コクサッキーウイルス、エコーウイルス、ウシエンテロウイルス、ブタエンテロウイルス、サルエンテロウイルス、マウス脳脊髄炎ウイルス、ヒトライノウイルス、ウシライノウイルス、ウマライノウイルス、口蹄疫ウイルス、A型肝炎ウイルス、コロナウイルス、ヒトコロナウイルス、ニワトリ伝染性気管支炎ウイルス、マウス肝炎ウイルス、豚伝染性胃腸炎ウイルス、アレナウイルス、リンパ球性脈絡髄膜炎ウイルス、ラサウイルス、韓国型出血熱ウイルス、レトロウイルス、ヒト成人白血病ウイルス、エイズウイルス、ネコ白血病肉腫ウイルス、牛白血病ウイルス、ラウス肉腫ウイルス、レオウイルス、ロタウイルス、カリシウイルス、ノーウオークウイルス、ノロウイルス、ブンヤウイルス、腎症候性出血熱ウイルス、フィロウイルス、エボラウイルス、マールブルグウイルス、B型肝炎ウイルス、ポックスウイルス、ワクシニアウイルス、アラストリウムウイルス、牛痘ウイルス、天然痘ウイルス、パルボウイルス、ヒトパルボウイルス、豚パルボウイルス、牛パルボウイルス、犬パルボウイルス、ネコ白血球減少症ウイルス、ミンクアリューシャン病ウイルス、パポーバウイルス、パピローマウイルス、ポリオーマウイルス、アデノウイルス、ヘルペスウイルス、単純ヘルペスウイルス、サイトメガロウイルス、水痘帯状疱疹ウイルス、EBウイルス、馬ヘルペスウイルス、ネコヘルペスウイルス、マレック病ウイルスおよびアフリカ豚コレラウイルスよりなる群から選択される少なくとも1種である請求項1〜8のいずれかに記載の特定遺伝子の検出方法。
【図1】

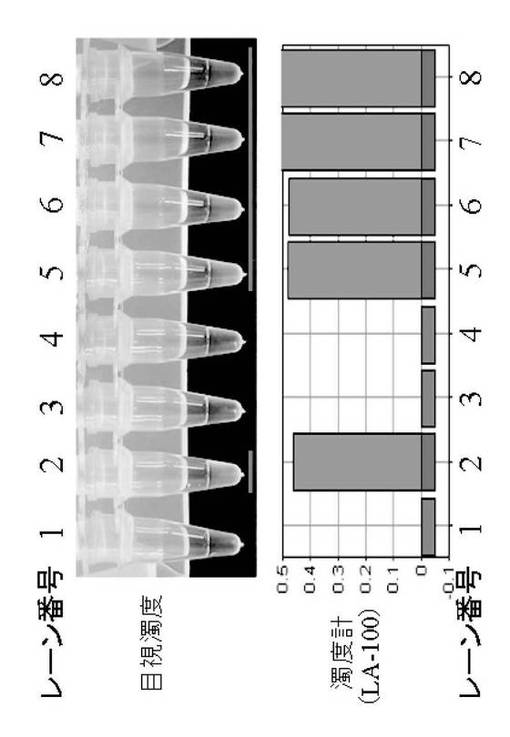
【図2】
【図3】
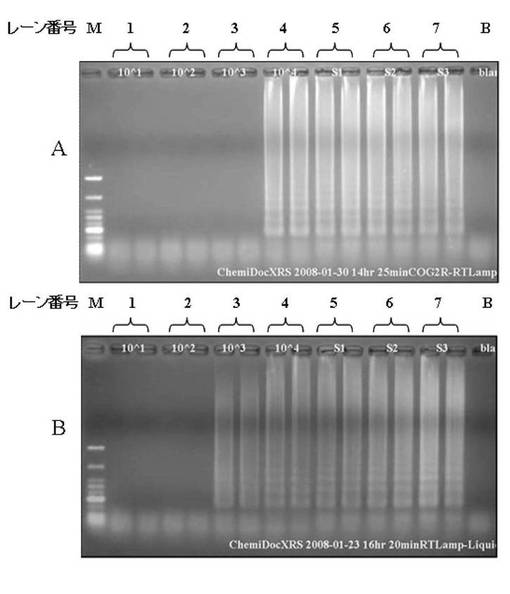
【図4】
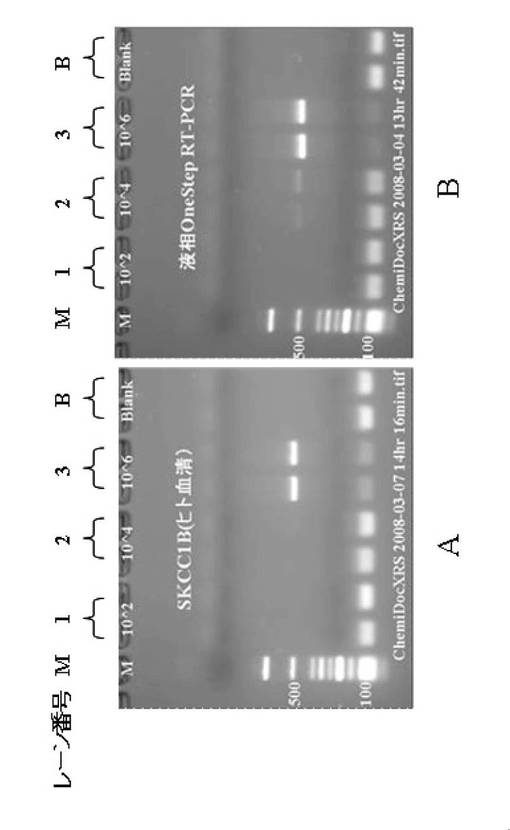
【図5】
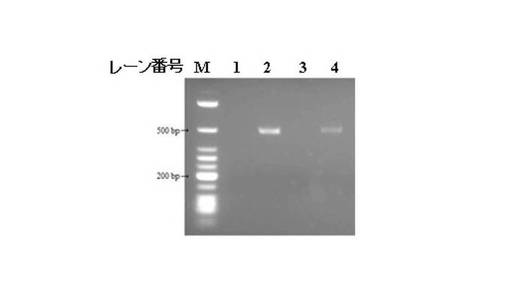
【図6】
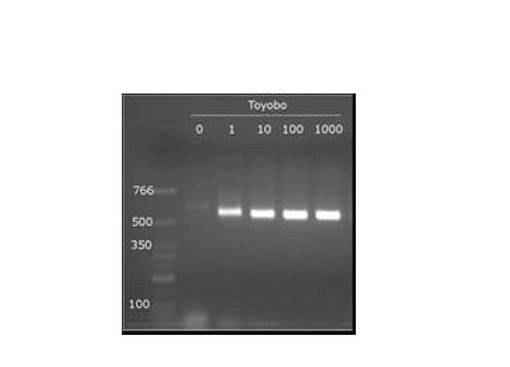
【図7】
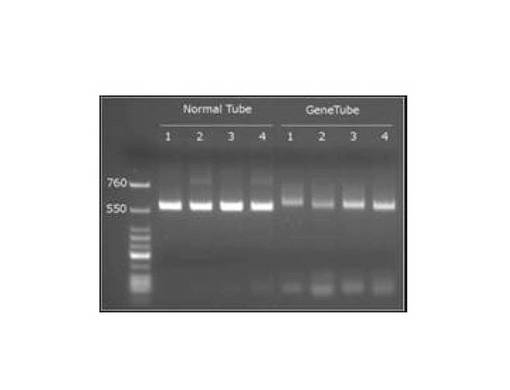
【図8】
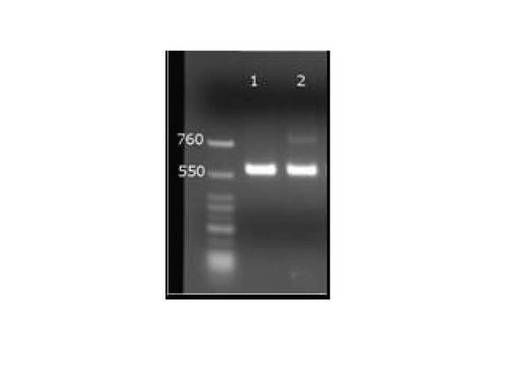
【図9】
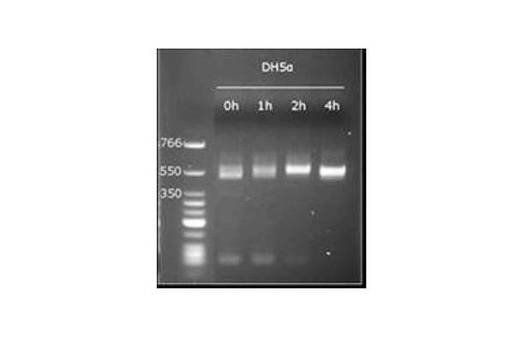
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2010−46042(P2010−46042A)
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願番号】特願2008−215591(P2008−215591)
【出願日】平成20年8月25日(2008.8.25)
【出願人】(599125249)学校法人武庫川学院 (24)
【Fターム(参考)】
【公開日】平成22年3月4日(2010.3.4)
【国際特許分類】
【出願日】平成20年8月25日(2008.8.25)
【出願人】(599125249)学校法人武庫川学院 (24)
【Fターム(参考)】
[ Back to top ]