
癌細胞におけるアポトーシスの誘導のための化合物および方法
【課題】癌の治療方法を提供する。
【解決手段】本発明は、癌細胞と、アポゴシポール、誘導体である化合物とを接触させることを含む哺乳類における癌の治療方法を提供する。
【解決手段】本発明は、癌細胞と、アポゴシポール、誘導体である化合物とを接触させることを含む哺乳類における癌の治療方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願のクロスリファレンス
本出願は2003年6月25日出願の米国仮特許出願第60/482,886号(その内容を引用により本出願に含める)からの優先権を主張する。
政府援助
本明細書に記載の本発明は、the National Institute of Healthにより与えられた認可番号 CA78040-05下、政府の支援によりなされた。米国政府は本発明における一定の権利を有する。
【背景技術】
【0002】
発明の背景
現在、癌、特にメラノーマ、頸癌、または白血病を予防および/または治療するための新規、強力、かつ選択的な物質が望まれている。現在研究中の方法の一つはアポトーシスの選択的誘導である。アポトーシスの選択的誘導に関連する生理過程のさらなる研究のための薬理手段も必要とされている。
【0003】
プログラムされた細胞死 (アポトーシス)は、組織恒常性、発達過程における望ましくない細胞の生理的除去、および宿主の防御機構に必須である(Vaux et al.、Cell、96:245 (1999))。アポトーシスの阻害は既知のヒトの悪性腫瘍のいずれにおいても伴っている。この阻害によって悪性細胞に選択的増殖の利点が与えられ、照射または化学療法によっても生存可能となってしまう(Reed、Curr. Opin. Oncol.、7:541 (1995)、およびKelekar et al.、Trends Cell. Biol.、8:324 (1998)参照)。Bcl-2 ファミリーのタンパク質はアポトーシスの重要な制御因子であると考えられている;このファミリーの生存促進性のメンバー、例えば、Bcl-xLは、表面に疎水性の溝を含み、そこでアポトーシス促進性対応物のBH3 ドメインへの結合が可能となると考えられている (Johnstone et al.、Cell、108:153 (2002))。この結合はアポトーシス制御に重要であると考えられており、実際生存促進性および抗生存タンパク質が二量体化を介して互いの機能を逆転することが出来る。抗アポトーシス性 Bcl-2 ファミリーメンバーは、多くのヒト悪性腫瘍において一般的に過剰発現していると考えられている。かかる観察によりBcl-2 ファミリーの抗アポトーシス性タンパク質、主に、Bcl-xLを標的とする低分子の候補抗癌治療薬としての発見に関する興味が増すことになった (Wang et al.、Proc. Natl. Acad. Sci. U.S.A.、97:7124 (2000))、Degterev et al.、Nat. Cell Biol.、3:173 (2001); Tzung et al.、Nat. Cell Biol.、3:183 (2001); Enyedy et al.、J. Med. Chem.、44:4313 (2001))。しかし現在でもなお、提案された化合物は、インビボ活性が低いか、あるいはインビトロ親和性が低いために、候補抗癌薬としてのBcl-xL 阻害物質の役割を完全に確証されるには至っていない(Kaneko et al.、Bioorg. Med. Chem. Lett.、11:887 (2001); Chin et al.、Angew. Chem. Int. Ed. Engl.、40:3806 (2001); Kutzki et al.、J. Am. Chem. Soc.、124:11838 (2002))。
【0004】
それゆえ、例えば、Bcl-xL、Bcl-2、Mcl-1、Bcl-W、 またはBcl-BなどのBcl-2 ファミリーの受容体を標的化する強力な細胞透過性化合物を同定する必要がある。BH3の Bcl-2 受容体への結合を阻害できるアゴニストが望まれている。
【0005】
さらに特に、癌細胞(例えば、リンパ腫、神経芽腫、乳癌、肺癌、前立腺癌、卵巣癌、白血病等)が、抗アポトーシス性 Bcl-2 ファミリータンパク質、例えばBcl-xL、Bcl-2、Mcl-1または Bcl-Bを過剰生産しているタイプの癌における化学感作物質として有用な化合物が望まれている。
【発明の概要】
【0006】
発明の概要
本発明は、癌細胞と、ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびそれらの混合物からなる群から選択される、癌細胞の生存度を低下させるのに有効な化合物とを接触させることを含む、患者における癌の治療方法を提供する。
【0007】
さらに、本発明は患者におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導方法を提供し、該方法は、標的細胞と、アポトーシス誘導、カスパーゼ活性調節、または標的細胞の細胞死誘導に有効な、以下からなる群から選択される化合物とを接触させることを含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびそれらの混合物。
【0008】
さらに、本発明は、Bcl-2 ファミリータンパク質を過剰発現している細胞におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導方法を提供し、該方法は、標的細胞と、本明細書に開示する本発明の化合物とを接触させることを含む。
【0009】
別の態様において、本発明は患者における癌の治療方法を提供し、該方法は、対象に、抗癌剤と組み合わせて以下からなる群から選択される化学感作物質を投与することを含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびその混合物。
【0010】
さらに、本発明は、Bcl-2 タンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1、Bcl-W、およびBcl-B)の、Bcl-2 ファミリーのアポトーシス促進性メンバーであるタンパク質(例えば、Bid、Bad、Bak、Bax )のBH3 ドメインへの、またはBH3 ドメインのみを含むペプチドへの、結合を調節するのに有効な化合物の同定方法を提供する。
【0011】
本発明は、 Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Bcl-W、Mcl-1、および Bcl-B)に結合する、または Bcl- 2活性を調節する化合物の同定方法を提供する。さらに、本発明は、Bcl-2 ファミリーのアポトーシス促進性メンバーであるタンパク質(例えば、Bid、Bad、Bak、Bax)のBH3 ドメインまたは BH3 ドメインのみを含むペプチドと複合体を形成した場合に、Bcl-2 ファミリータンパク質に結合する、またはBcl-2活性を調節する化合物の同定方法を提供する。
【0012】
本発明は、薬物療法における使用(例えば、アポトーシスの誘導、カスパーゼ活性の調節、細胞死の誘導、または癌の治療における使用)、好ましくは、以下の治療における使用のための本明細書に記載の化合物を提供する:肺癌、乳癌、前立腺癌、その他の形態の癌、および白血病、例えば、急性リンパ球性白血病 (ALL)、急性骨髄性白血病 (AML)、慢性骨髄性白血病 (CML)、およびその他の増殖性疾患。さらに本発明は、哺乳類、例えばヒトにおけるアポトーシスの誘導、カスパーゼ活性の調節、細胞死の誘導または癌の治療(好ましくは肺癌、乳癌、前立腺癌、CML、ALL、AML、その他の形態の癌または白血病およびその他の増殖性疾患の治療)のための医薬の製造における式Iの化合物の使用を提供する。本発明の化合物はまた、アポトーシスが一つの徴候である疾患(例えば、心臓病、パーキンソン病、アルツハイマー病等)の、AHPN アンタゴニスト経路を用いた治療に有用である。
【0013】
本発明はまた、アポトーシスまたは細胞死の誘導方法を提供し、該方法は、細胞と、有効量の (本明細書に記載の) 本発明の化合物をインビトロまたはインビボで接触させることを含む。
【0014】
本発明はまた、治療を必要とする哺乳類におけるアポトーシスの誘導方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0015】
本発明はまた、細胞におけるカスパーゼの活性化方法(例えば、Bcl-2 ファミリーの抗アポトーシス性タンパク質の阻害によるカスパーゼ 3 および 7の活性化方法)を提供し、該方法は細胞を、インビトロまたはインビボで、有効量の(本明細書に記載の)本発明の化合物と接触させることを含む。
【0016】
本発明はまた、ヒトなどの哺乳類におけるカスパーゼ活性化(例えば、Bcl-2 ファミリーの抗アポトーシス性タンパク質の阻害を介するカスパーゼ 3 および 7の活性化)が関連する病状または症状の予防または治療方法を提供し、該方法は、かかる治療を必要とする哺乳類にカスパーゼ-調節に有効な量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0017】
本発明はまた、細胞死を誘導する治療方法を提供し、該方法は細胞と、インビトロまたはインビボで、有効量の(本明細書に記載の)本発明の化合物とを接触させることを含む。
【0018】
本発明はまた、治療を必要とする哺乳類における細胞死の誘導方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0019】
本発明はまた、治療を必要とする哺乳類における癌 (例えば、肺癌、結腸直腸癌、乳癌、前立腺癌、ALL、CLL、AML、固形腫瘍、その他の形態の癌または白血病、例えば、リンパ腫、神経芽腫、およびその他の増殖性疾患)の治療方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0020】
本発明はまた、Bcl-2 ファミリーのタンパク質(例えば、Bcl-xLおよびBcl-2)の抗アポトーシス活性を阻害する物質の同定方法を提供し、該方法は以下の工程を含む: a)標識化 Bcl-xLに結合している選択的 Bcl-xLまたはBcl-2 阻害物質を検出する工程、ここで該 Bcl-xL 阻害物質は以下からなる群から選択されるコア構造を含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-)ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)およびプルプロガリン誘導体; b)結合しているBcl-xLと候補物質とを接触させる工程、ここで該候補物質はBcl-xLを阻害することが出来ると予測されている;および、 c)該Bcl-xL 阻害物質の該標識化 Bcl-xLからの分離を検出する工程、これによって該候補物質はBcl-xLの阻害物質であると同定される。
【図面の簡単な説明】
【0021】
図面の簡単な説明
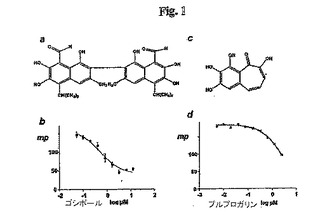
【図1】図1. ゴシポールおよびプルプロガリンがBcl-xLのBH3-結合ポケットについて競合している。ゴシポール (a)およびプルプロガリン (c)の化学構造。フルオレセイン-標識化 Bad ペプチド (NLWAAQRYGRELRRMSD-K(FITC)-FVD) (Synpep Corporation、Dublin、CA)を用いた蛍光偏光に基づく競合結合アッセイ (FPA)の結果を (c)ゴシポールおよび(d)プルプロガリンについて示す。
【0022】
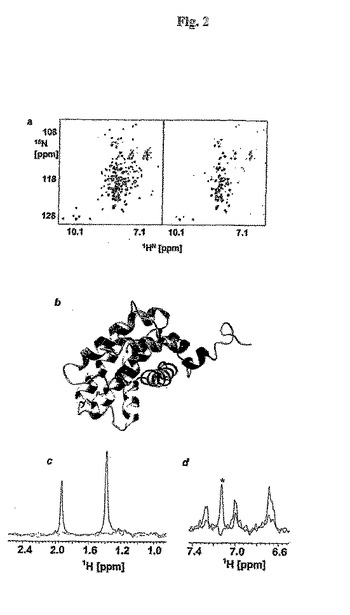
【図2】図 2. NMR 結合研究。(a)アポ(左)およびゴシポール-結合(右)形態におけるBcl-xLについての 2 D [15N,1H]-TROSY スペクトル。(b) Bak ペプチドとの複合体におけるBcl-xLの三次元構造へのゴシポールの化学シフトマッピング(PDB コード1BXL)。ペプチドを黄色で示す。ゴシポールの結合により影響を受けた領域を赤色で示す。(c)および(d) それぞれゴシポールおよびプルプロガリンによるT1ρ 実験(300 ms 緩和時間):青色、タンパク質無し、および赤色、10 μM Bcl-xLの存在下。(c)に示すピークはゴシポールにおけるイソプロピルおよびメチル基を表す。 (d)において、星印で示したピークは、タンパク質調製物において存在する残余のイミダゾールを示す。
【0023】
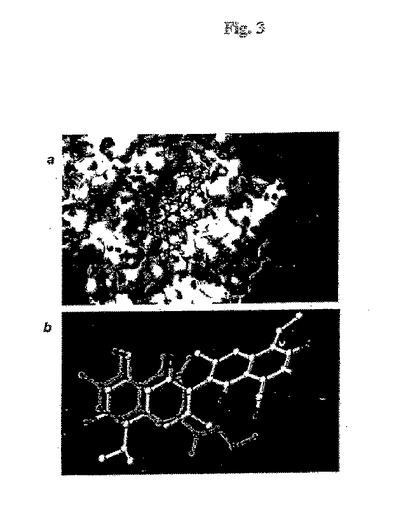
【図3】図 3. 分子モデリング研究。(a) FlexXによって得られたBcl-xLとゴシポールのドッキング構造の表面表示。(b) 5D1 (緑色)およびゴシポール (白色、酸素原子は赤色)の重ね合わせ。
【0024】
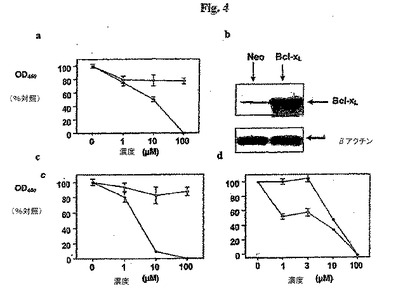
【図4】図 4.化合物の癌細胞生存に対する阻害効果。培養中の腫瘍細胞の生存度に対するゴシポールの効果をXTT アッセイを用いてモニターした。(a) MCF7 および(b) ZR75-1 細胞株 (黒丸)を用いた。陰性対照として、ジェネリック(generic)ポリフェノール化合物も試験した(白丸)。低継代 HeLa 細胞 (継代数 10〜20)をpcDNA3-Bcl-xL (黒丸)または対照pcDNA3 プラスミド(白丸)でトランスフェクトした。 (c) イムノブロット分析により、pcDNA3-対照トランスフェクタントと比較してpcDNA3-Bcl-xL 化合物でトランスフェクトした細胞においてBcl-xL の過剰発現が確認された(細胞溶解液を全タンパク質含量について正規化した; 25 μg/レーン)。(d) HeLa-トランスフェクタントを様々な用量のゴシポール (0、1、3、10 および 100μM)で処理した。示したデータは平均±標準偏差 (n = 4)を表す。
【0025】
【図5】図 5は、以前に処理されず、新たにCLLと診断されたものにおけるアポゴシポール (6C1)およびゴシポールの単剤としての活性を示す。
【0026】
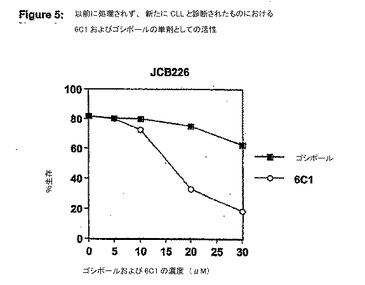
【図6】図 6は、アポゴシポール (6C1)とフルダラビン (F-ara-A)との相加効果を示す。
【0027】
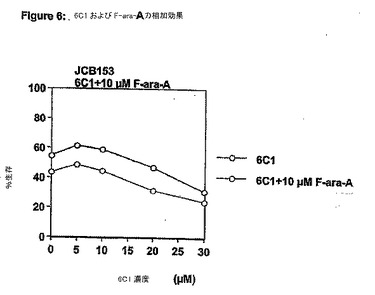
【図7】図 7は、アポゴシポール (6C1)と F-ara-Aとの相乗効果を示す。
【0028】
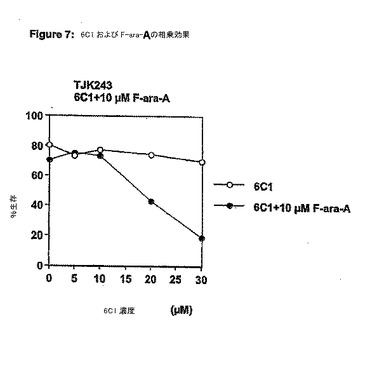
【図8】図 8は、 (-)EGCG のBcl-xL 受容体に対する結合を示す; (a) 10 μM Bcl-xLの添加の前(上側スペクトル)と後(下側スペクトル)の(-)EGCGによるT1ρ実験 (200 ms 緩和時間)。(a)に示すピークはカテキン基(group)のプロトン4αおよび4βを示し、* は DMSOを示す; (b) (-)EGCGについての蛍光偏光に基づく競合結合アッセイの結果;および、 (c) FlexXによって得られたBcl-xL と(-)EGCGのドッキング構造の表面表示。リガンドによって占有された3つのサブポケット (P1、P2およびP3)が示される。
【0029】
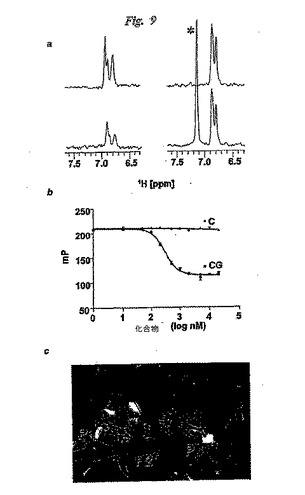
【図9】図 9は(-)CGと(-)Cとの比較を示す; (a) (-)CG (左側)と(-)C (右側)のT1ρ実験(200 ms 緩和時間); タンパク質の非存在下で記録したスペクトルを青色で示す; *はタンパク質バッファーからのイミダゾールを示す; (b) (-)CGと(-)CのFPA結果の重ね合わせ;および、 (c) (-)CG (赤色)と (-)C (青色)のドッキング構造によってくぼみのついた(pocked)Bcl-xL結合の表面表示。
【0030】
【図10】図 10はテアフラビン-3’-ガラートのBcl-xL 受容体への結合を示す: (a)テアフラビン-3’-ガラート (1mM) の添加前(左)および添加後(右)のBcl-xL (0.250 mM)の2D [15N,1H]-TROSY スペクトル; (b)テアフラビン-3’-ガラートについてのFPA結果;および、 (c)テアフラビン-3 '-ガラートのドッキング構造によりくぼみのついたBcl-xL結合の表面表示、リガンドに占有された3つのサブポケット (P1、P2およびP3)を丸で示す。
【0031】
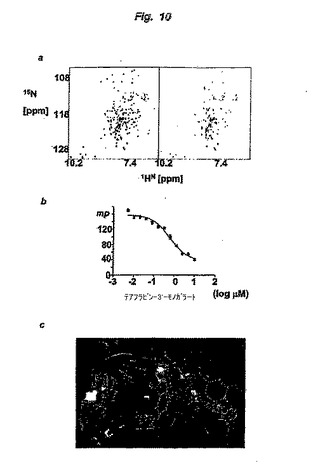
【図11】図 11は、示したテアフラビンを用いるBcl-xLの阻害を示す。
【0032】
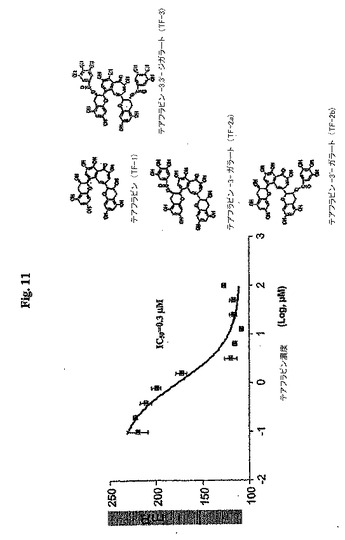
【図12】図 12はテアフラバニンを用いるBcl-xLの阻害を示す。
【0033】
【図13】図 13はXTTアッセイにおけるHela 細胞に対するテアフラバニンの効果を示す。
【発明を実施するための形態】
【0034】
詳細な説明
本発明の化合物は、Bcl-2 タンパク質 (例えば、Bcl-2およびBcl-xL)の受容体の拮抗(antagonizing)に有用である。これらのタンパク質は通常の抗癌治療、例えば、放射療法または化学療法に対して癌細胞をより抵抗性にすることができる。したがって、これら細胞は死滅せず、増殖しうる。本明細書に記載の化合物によるかかるタンパク質の阻害によって、細胞の抗癌治療に対する抵抗性を減少または排除しうる。それゆえ、これらの化合物は化学感作物質として有用である。したがって、本発明の化合物は癌細胞を抗癌治療、例えば、放射療法または化学療法に対してより感受性にしうる。したがって、本発明は、Bcl-2 タンパク質とアポトーシス促進性 Bcl-2 ファミリーメンバーのBH3 ドメインとの複合体の形成を調節する方法を提供し、これら複合体の量または安定性を調節するのに有用な化合物を提供する。
【0035】
本発明は、本発明の化合物が抗アポトーシス性タンパク質 Bcl-xLに結合する能力を測定するためにスペクトル技術を用いて化合物をスクリーニングする方法を提供する。該方法により、その他の抗癌化合物と組み合わせて用いることが出来る化合物が同定される。様々な緑茶および黒茶ポリフェノール化合物を、核磁気共鳴 (NMR) 結合アッセイ、蛍光偏光アッセイ (FPA) およびコンピュータによるドッキング研究を組み合わせて用いることによりスクリーニングした。
【0036】
一つの態様において、本発明は、抗アポトーシス性タンパク質 Bcl-xLに結合し、そしてBcl-2 ファミリーの生存促進性メンバーとのその相互作用を阻害することを介して部分的に抗癌薬として作用しうる茶のポリフェノール類の活性を評価する方法を提供する。
【0037】
別の態様において、本発明はBcl-xLのBH3への結合を有効に調節することが出来る化合物の同定方法を提供する。該方法は以下の工程を含む:(a) Bcl-xL-BH3 複合体の形成に好適な条件下でBcl-xLとBH3とを接触させる工程; (b) Bcl-xL-BH3 複合体と被験化合物とを接触させる工程;および、(c)被験化合物が、Bcl-xLのBH3への結合を調節する能力を判定する工程、ここでBcl-xLのBH3への結合の調節は、被験化合物がBcl-xLのBH3への結合を調節する有効な化合物であることを示す。
【0038】
別の態様において、本発明は、 Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1 Bcl-W、またはBcl-B)の結合を有効に調節しうる物質の同定方法を提供し、該方法は、Bcl-2 阻害物質または標識化 Bcl-2 阻害物質を同定する工程を含み、ここで該阻害物質は表1、2、3に挙げる化合物およびアポゴシポール誘導体からなる群から選択される。該方法は、(a)Bcl-2 阻害物質または標識化 Bcl-2 阻害物質を同定する工程(ここで該阻害物質はゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、およびプルプロガリン誘導体からなる群から選択される)、Bcl-2 ファミリータンパク質と該阻害物質(例えば表1に挙げるもの)とを、Bcl-2 ファミリータンパク質との複合体形成に好適な条件下で接触させる工程; (b)該Bcl-2 阻害物質複合体と被験化合物とを接触させる工程; および (c) Bcl-xLの阻害物質への結合を調節する被験化合物の能力を判定する工程(ここで、Bcl-2 タンパク質の阻害物質化合物への結合の調節は、被験化合物 (物質)がBcl-2 の阻害物質化合物への結合の調節に有効な化合物であることを示す)。
【0039】
別の態様において、本発明は、癌の治療のための伝統的な化学療法の有効性を向上させるために、癌細胞を感作するのに有用なポリフェノール化合物を提供する。
【0040】
別の態様において、本発明は、アポゴシポールまたはそのアナログの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0041】
別の態様において、本発明は、プルプロガリンまたはそのアナログの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0042】
別の態様において、本発明は、黒茶または緑茶由来のポリフェノールの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0043】
別の態様において、本発明はポリフェノールの癌予防薬としての使用を提供する。本発明に記載するポリフェノール化合物は、癌(例えば、肺癌、乳癌、前立腺癌、結腸直腸癌、および白血病)の発症の確立が高いと疑われる患者に投与され、患者がかかる癌を発症する可能性を低減させうる。
【0044】
本発明において、緑茶および黒茶由来のポリフェノールを試験した。緑茶はCamelia Sinensis の未発酵の葉から生産され、ポリフェノール(カテキンとして知られる)はその主な化学成分を構成する。エピカテキン (EC)、エピカテキン-3 ガラート (ECG)、エピガロカテキン (EGC)、およびエピガロカテキン-3 ガラート (表 1)が緑茶に含まれる主なカテキンである(Chu et al.、In: Yamamoto、T.、Juneja、J. R.、Cu、D.C. and Kim、M. Chemistry and Application of Grean Tea、pp. 13-22、New York: CRC Press、1997)。黒茶は、ポリフェノールの、テアフラビン等の重合生成物への高度な酵素酸化によって作られる (Pan et al.)。テアフラビン、テアフラビン-3 ガラート、テアフラビン-3’-ガラート、およびテアフラビン-3-3’-ジガラートが黒茶における主なテアフラビン類である。
【0045】
さらに、本発明は、茶の抗癌活性と、Bcl-2 ファミリーの抗アポトーシス性タンパク質(例えば、Bcl-xL およびBcl-2)とのその相互作用を相関させる方法を提供する。
【0046】
さらに、本発明は、茶の抗癌活性とBcl-2 ファミリーの抗アポトーシス性タンパク質(例えば、Bcl-xLおよび Bcl-2)とのその相互作用を関連させる方法を提供する。
【0047】
別の態様において、本発明は、抗癌活性について化合物をスクリーニングする方法を提供し、そして 、以下に記載のアッセイを用いる薬理学モデルを用いて、本発明の化合物が癌治療を補助しうることを判定する方法を提供する。
【0048】
本発明はまた、本明細書に記載の化合物またはその医薬上許容される塩を、医薬上許容される希釈剤または担体と組み合わせて含む医薬組成物も提供する。さらに、本発明は、その他の既知の抗癌化合物と組み合わせての本明細書に開示の化合物の使用を提供する。
【0049】
本発明は、治療を必要とする哺乳類に、有効量の本明細書に記載の化合物、別の抗癌化合物と組み合わせた本明細書に記載の化合物またはその医薬上許容される塩を投与することを含む癌の治療方法を提供する。
【0050】
さらに、本発明は、癌の予防方法またはかかる癌を患者が発症しうる可能性を低下させる方法を提供し、該方法は、治療を必要とする哺乳類に、有効量の本明細書に記載の化合物またはその医薬上許容される塩を投与することを含む。
【0051】
特に断りのない限り以下の定義を用いる:ハロは、フルオロ、クロロ、ブロモ、またはヨードである。アルキル、アルコキシ、アルケニル、アルキニルなどは、直鎖状と分枝状の基の両方を含む;しかし個々の基についての、例えば、「プロピル」という言及は、直鎖状基のみを含み、分枝状異性体、例えば「イソプロピル」は具体的に言及される。アリールは、フェニル基、または約9から10の環原子を有し少なくとも1つの環が芳香族であるオルト-縮合二環式炭素環基をいう。ヘテロアリールは、炭素および1から4のヘテロ原子(それぞれ非ペルオキシド酸素、硫黄およびN(X)からなる群から選択され、ここで、Xは存在しないか、またはH、O、(C1-C4)アルキル、フェニルまたはベンジル)からなる5または6の環原子を含む単環式芳香族環の環炭素を介して結合した基、およびそれに由来する約8から10の環原子のオルト-縮合二環式-ヘテロ環基であり、特に、ベンズ-誘導体またはプロピレン、トリメチレン、またはテトラメチレンジ基がそれに縮合することにより生ずるものである。
【0052】
具体的には、「アルキル」の語は、炭素原子数が1から6の分枝状または非分枝状飽和炭化水素基であり、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、t-ブチル、ペンチル等である。本明細書において好ましいアルキル基は炭素原子数1から6のもの、例えば、メチル、エチル等である。
【0053】
本明細書において用いる「シクロアルキル」の語は、炭素原子数3から8、好ましくは、3、5または6の環状アルキル基である。本明細書において用いる「シクロアルキレン」の語は、二価環状アルキレン基をいい、典型的には、3-、5-、6-、または8-員環である。
【0054】
本明細書において用いる「アルコキシ」の語は、末端エーテル単結合を介して結合したアルキル基をいい、即ち「アルコキシ」基は、Rが上記のアルキルである- ORと定義できる。「低級アルコキシ」基は、炭素原子数1から6のアルコキシ基をいう。
【0055】
本明細書において用いる「アリール」の語は、典型的には、少なくとも1つの環が芳香族である6-または10- 員環の芳香族炭素環をいう。
【0056】
当業者であれば、不斉中心を有する本発明の化合物は、光学活性形態およびラセミ形態にて存在および単離しうることを理解するであろう。化合物のなかには多形を示すものもあり得る。本発明は、本明細書に記載の有用な性質を有する、あらゆる本発明の化合物のラセミ、光学活性、多型、または幾何異性形態、あるいはその混合物を含むことを理解されたい。当該技術分野において、光学活性形態をいかにして調製するか(例えば、再結晶技術によるラセミ体の分割、光学活性出発物質からの合成、キラル合成またはキラル固定相を用いたクロマトグラフィー分割による)、そして本明細書に記載の標準的試験または当該技術分野において周知の類似の試験を用いて抗癌活性をいかにして判定するかは周知である。
【0057】
基、置換基および範囲について以下に挙げる具体的かつ好ましい値は、単に例示の目的である; それらは基および置換基について規定されるその他の値あるいは規定される範囲内の別の値を排除するものではない。
【0058】
具体的には、アルキルは以下のいずれかであり得る:メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、ペンチル、3-ペンチル、またはヘキシル; シクロアルキルは以下のいずれかであり得る:シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル; -O(C1-C6)アルキル(アルコキシ) は以下のいずれかであり得る: メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソ-ブトキシ、sec-ブトキシ、ペントキシ、3-ペントキシ、またはヘキシルオキシ。
【0059】
本発明の実施に有用な具体的化合物を表 1および表 2に挙げる。
表1
【表1−1】

【表1−2】
【0060】
表 2
【表2】
【0061】
黒茶または緑茶から単離されるポリフェノールの非限定的な例としては以下が含まれる:カテキン、例えば、エピカテキン (EC)、エピカテキン-3 ガラート (ECG)、エピガロカテキン (EGC)、エピガロカテキン-3 ガラート等; テアフラビン、例えば、テアフラビン、テアフラビン-3 ガラート、テアフラビン-3’-ガラート、テアフラビン-3-3’ジガラート等。
【0062】
別の具体的な本発明の化合物にはプルプロガリンおよびその誘導体、例えば、表 3に示す化合物が含まれる。
【0063】
表3
プルプロガリン誘導体
【化1】
【表3】
【0064】
本発明の実施に有用なさらなる化合物には以下が含まれる:例えば、正常細胞に対しては毒性が低いが、癌細胞に対しては、ゴシポールと同様の細胞毒性を有する化合物であるアポゴシポールおよびアポゴシポールのアナログ。かかるアナログは、Bcl-xL/2に対する強度および選択性が増強している。本発明のアナログ化合物は式Iを有するか、またはその医薬上許容される塩である:
【化2】
(I)
[式中:
各R6、R8、R9およびR10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ;
各R7は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【0065】
具体的なR6、R8、R9基は独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである。
【0066】
具体的なR7基は独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、シクロヘキシルである。
【0067】
具体的なR10基は独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである。
【0068】
具体的な本発明の化合物は式(I)を有し、ここでR6、R8、R9はそれぞれアセテート (-OC(O)CH3); R7はi-Pr;およびR10は-CH3 (アポゴシポールヘクス(hex)アセテート)である。この化合物はアポゴシポールの経口投与用のプロドラッグとしても有用である。別の態様において、本発明の化合物は式(I)の化合物であってR6基の1つが水素以外の基であるものも含む。
【0069】
本明細書において用いる、「患者」の語は、本発明の方法によって治療すべき生物をいう。かかる生物としては、これに限定されないが、ヒトが挙げられる。本発明において、「対象」の語は一般に、Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1またはBcl-B)の過剰発現によって特徴づけられる疾患の治療 (例えば、本発明の化合物、および所望により1以上の抗癌薬の投与)を受ける、または治療を受けた個体をいう。
【0070】
本明細書において用いる、「抗癌剤」または「化学療法抗癌剤」の語は、癌の治療に用いられるあらゆる化学療法化合物、放射療法、および外科技術をいう。
【0071】
本発明の実施に有用な抗癌薬としては、緊張性薬剤または癌化学療法薬剤が挙げられる。本発明の実施に有用な緊張性薬剤の非限定的な例としては、低分子、ポリペプチド、ペプチド、ペプチド疑似体(peptidomimetics)、核酸分子、細胞およびウイルスが挙げられる。非限定的な例として、本発明に有用な細胞毒性薬剤としては、細胞毒性低分子、即ち、典型的にはDNAに関連する過程を標的とする化合物、例えば、ドキソルビシン、ドセタキセル、トラスツズマブ、シクロホスファミド、メルファラン、マイトマイシン C、ビゼレシン、シスプラチン、ドキソルビシン、エトポシド、ミトキサントロン、SN 38、Et 743、アクチノマイシン D、ブレオマイシン、TLK286等; 抗菌性ペプチド、例えば以下に記載するもの; アポトーシス促進性ポリペプチド、例えば、カスパーゼおよび毒素、例えば、カスパーゼ 8; ジフテリア毒素 A 鎖、緑膿菌外毒素 A、コレラ毒素、リガンド融合毒素、例えば、DAB389EGF、トウゴマ毒素 (リシン);および細胞毒性細胞、例えば細胞障害性 T 細胞等が挙げられる。例えば、Martin et al.、Cancer Res.、60:3218 (2000); Kreitman et al.、Blood、90:252 (1997); Allam et al.、Cancer Res.、57:2615 (1997);および Osborne et al.、Cancer J. Sci. Am.、2:175 (1996)を参照されたい。
【0072】
本発明での使用に好適なさらなる抗癌化学療法薬剤としては、これらに限定されないが、タキサン、例えば、ドセタキセル(タキソテール、Aventis Pharmaceuticals、Inc.; Parsippany、NJ) および パクリタキセル (タキソール、Bristol Myers Squibb; Princeton、NJ); アントラサイクリン、例えば、ドキソルビシン、イダルビシン、ダウノルビシン等;アルキル化剤;ビンカアルカロイド;代謝拮抗剤;白金薬剤、例えば、シスプラチンまたはカルボプラチン;ステロイド、例えば、メトトレキサート;抗生物質、例えば、アドリアマイシン;イソファミド;または選択的エストロゲン受容体調節因子;抗体、例えば、トラスツズマブ等が挙げられる。
【0073】
ドキソルビシンは一般によく用いられている癌化学療法薬剤であり、例えば、乳癌の治療に有用である(Stewart et al.、In: ”Cancer: Principles and Practice of Oncology” 5th ed.、Chap. 19 (eds. DeVita、Jr.、et al.; J.P. Lippincott 1997); Harris et al.、”Cancer: Principles and Practice of Oncology,”前掲、1997)。さらに、ドキソルビシンは抗血管形成活性を有し(Folkman、Nature Biotechnology、15:510 (1997); Steiner、”Angiogenesis: Key principles Science、technology and medicine,” pp. 449 454 (eds. Steiner et al.; Birkhauser Verlag、1992))、その活性が癌の治療におけるその有効性に寄与し得る。
【0074】
アルキル化剤、例えば、メルファランまたはクロランブシルは本発明の併用治療に有用な癌化学療法薬剤である。同様に、ビンカアルカロイド、例えば、ビンデシン、ビンブラスチンまたはビノレルビン;あるいは代謝拮抗剤、例えば、 5-フルオロウラシル、5-フルオロウリジンまたはそれらの誘導体は本発明の併用療法に有用な癌化学療法薬剤である。
【0075】
白金薬剤は本発明の併用療法に有用な化学療法薬剤である。かかる白金薬剤は例えばCrown、Seminars in Oncol.、28:28 (2001)に記載の、例えば、シスプラチンまたはカルボプラチンであり得る。本発明の併用療法に有用な化学療法薬剤としてはこれらに限定されないが、メトトレキサート、マイトマイシン C、アドリアマイシン、イホスファミドおよびアンサマイシンが挙げられる。
【0076】
乳癌およびその他のホルモン依存性癌の治療に用いられる癌化学療法薬剤は、エストロゲンの作用を拮抗する薬剤としても使用し得、例えば、選択的エストロゲン受容体修飾因子または抗エストロゲンが挙げられる。選択的エストロゲン受容体調節因子である、タモキシフェンは乳癌の治療のための本発明の併用療法に利用できる癌化学療法薬剤である(Fisher et al.、J. Natl. Cancer Instit.、90:1371 (1998))。
【0077】
本発明の併用療法に有用な別のタイプの治療薬は抗体、例えばヒト化モノクローナル抗体である。非限定的な例としては、抗上皮成長因子受容体 2 (HER2) 抗体が挙げられる。トラスツズマブ (ハーセプチン; Genentech、South San Francisco、CA)はHER2/neuを過剰発現する乳癌の治療のために本発明の組み合わせに有用なさらなる治療薬である(White et al.、Annu. Rev. Med.、52:125 (2001))。
【0078】
本発明に有用な別の治療薬は、本明細書において用いる場合、直接または間接的に細胞死を促進するいずれかの分子である細胞毒性薬剤でもありうる。
【0079】
具体的な抗癌薬としては、フラボピリドール、アドリアマイシン(ドキソルビシン)、VP16 (エトポシド)、タキソール (パクリタキセル)、シスプラチン等が挙げられる。
【0080】
化合物が安定な非毒性の酸または塩基塩を形成するのに十分に塩基性または酸性の場合は、化合物の塩としての投与が適当であり得る。医薬上許容される塩の例は、生理的に許容される陰イオンを形成する酸と形成される有機酸付加塩、例えば、トシレート、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α-ケトグルタル酸塩、およびα-グリセロリン酸塩などである。好適な無機塩も形成され得、例えば、塩酸塩、硫酸塩、硝酸塩、炭酸水素塩、および炭酸塩が挙げられる。
【0081】
医薬上許容される塩は当該技術分野において周知の標準的手順を用いて得ることが出来、例えば十分に塩基性の化合物、例えば、アミンを生理的に許容される陰イオンを与える好適な酸と反応させることにより得ることが出来る。カルボン酸のアルカリ金属 (例えば、ナトリウム、カリウムまたはリチウム)あるいはアルカリ土類金属 (例えばカルシウム)塩も作ることが出来る。
【0082】
本発明の実施に有用な化合物は医薬組成物として選択された投与経路、即ち、経口または非経口、静脈内、筋肉内、局所または皮下経路に応じた様々な形態に製剤し、哺乳類宿主、例えば、ヒト患者に投与してもよい。
【0083】
したがって、化合物は全身投与してもよく、例えば、医薬上許容される媒体、例えば、不活性希釈剤または吸収可能な食用担体とともに経口投与してもよい。それらは殻の硬いまたは軟らかいゼラチンカプセルに封入してもよいし、圧縮して錠剤としてもよいし、患者の食事の食品に直接組み込んでもよい。経口治療的投与について、活性化合物は1以上の賦形剤と組み合わせて、消化可能な錠剤、舌下錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、ウエハースなどの形態で用いてもよい。かかる組成物および調製物は少なくとも0.1%の活性化合物を含むべきである。組成物および調製物のパーセンテージはもちろん変動させてもよく、好都合なことに所与の単位剤形の重量の約2から約60%の間であってよい。かかる治療上有用な組成物における活性化合物の量は有効投与量レベルが得られるものである。
【0084】
錠剤、トローチ、丸薬、カプセル等は以下のものを含んでいてもよい: 結合剤、例えば、トラガカントゴム、アカシアゴム、コーンスターチまたはゼラチン; 賦形剤、例えば、第二リン酸カルシウム; 崩壊剤、例えば、コーンスターチ、ジャガイモデンプン、アルギン酸等; 滑沢剤、例えば、ステアリン酸マグネシウム;および甘味料、例えば、スクロース、フルクトース、ラクトースまたはアスパルテームあるいは香味料、例えば、ペパーミント、ウィンターグリーン油、またはチェリーフレーバーを添加してもよい。単位剤形がカプセルの場合、それは上記タイプの物質に加えて、液体担体、例えば、植物油またはポリエチレングリコールを含んでいてもよい。その他の様々な物質が被覆として存在する場合もあるし、固体単位剤形の物理形態を修飾する場合もある。例えば、錠剤、丸薬またはカプセルはゼラチン、ろう、セラックまたは糖等で被覆してもよい。シロップまたはエリキシルは、活性化合物、甘味料としてスクロースまたはフルクトース、保存料としてメチルおよびプロピルパラベン、色素および香料、例えば、チェリーまたはオレンジフレーバーを含んでいてもよい。もちろん、単位剤形の調製にもちいる物質はいずれも使用する量において医薬上許容され、実質的に非毒性のものでなければならない。さらに、活性化合物は徐放調製物および装置に組み込んでもよい。
【0085】
活性化合物は注入または注射によって静脈内または腹腔内に投与してもよい。活性化合物またはその塩の溶液を水中に調製してもよく、所望により非毒性の界面活性剤を添加してもよい。分散液をグリセロール、液体ポリエチレングリコール、トリアセチン、およびその混合物ならびに油中に調製してもよい。貯蔵および使用の通常の条件下では、これら調製物には微生物の増殖の阻害のために保存料を含める。
【0086】
注射または注入に好適な薬理剤形には、無菌水溶液または分散液あるいは無菌注射可能または注入可能溶液または分散液の即時調製に適した活性成分を含む無菌粉末が含まれ、リポソームに封入されたものでもよい。何れの場合も、最終的な剤形は無菌、液体、かつ製造と貯蔵の条件下で安定なものであるべきである。液体の担体または媒体は溶媒または液体分散媒であり得、例えば、水、エタノール、ポリオール (例えば、グリセロール、プロピレングリコール、液体ポリエチレングリコール等)、植物油、非毒性グリセリルエステルおよびそれらの好適な混合物を含みうる。適切な流動性は、例えば、リポソームの形成、分散液の場合は所望の粒径の維持または界面活性剤の使用により維持されうる。微生物の作用の阻止は、様々な抗細菌および抗真菌薬、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等によってもたらされ得る。多くの場合、等張化剤、例えば、糖、バッファーまたは塩化ナトリウムを含むのが好ましい。注射可能組成物の持続的吸収は、吸収遅延剤、例えば、モノステアリン酸アルミニウムおよびゼラチンの組成物中での使用によりもたらされうる。
【0087】
無菌注射可能溶液は、所望の量の活性化合物を適当な溶媒に所望により様々なその他の上記成分とともに組込み、次いでろ過滅菌することにより調製される。無菌注射可能溶液の調製用の無菌粉末の場合、好ましい調製方法は減圧乾燥および凍結乾燥技術であり、それによって前もって無菌ろ過した溶液中に存在する活性成分とさらなる所望の成分の粉末が得られる。
【0088】
局所投与のためには、本発明の化合物を、それが液体の場合は純粋な形態で適用することができる。しかし、皮膚科学的に許容される担体と組み合わせた組成物または製剤として皮膚に投与するのが一般に望ましく、それは固体であっても液体であってもよい。
【0089】
有用な固体担体には、微細な固体、例えば、タルク、クレイ、微晶質セルロース、シリカ、アルミナ等が挙げられる。有用な液体担体には、水、アルコールまたはグリコールまたは水-アルコール/グリコール混合物が挙げられ、そのなかに本発明の化合物が有効レベルで、所望により非毒性界面活性剤の助けにより溶解または分散され得る。補助剤、例えば、香料およびさらなる抗菌剤を添加して所望の用途のための性質を最適にしてもよい。その結果得られる液体組成物は吸着剤パッドから適用されるか、絆創膏およびその他の包帯剤に浸透させて用いられるか、あるいはポンプ型またはエアゾールスプレーを用いて患部に噴霧される。
【0090】
増粘剤、例えば、合成高分子、脂肪酸、脂肪酸塩およびエステル、脂肪アルコール、修飾セルロースまたは修飾ミネラル物質を液体担体とともに用いて、塗布可能なペースト、ゲル、軟膏、ソープ等を形成して、使用者の皮膚に直接塗布してもよい。
【0091】
式Iの化合物を皮膚に送達するのに用いることが出来る有用な皮膚用組成物の例は当該技術分野で知られている; 例えば、Jacquet et al. (米国特許第4608392号)、Geria (米国特許第4992478号)、Smith et al. (米国特許第4559157号) およびWortzman (米国特許第4820508号)を参照されたい。
【0092】
式Iの化合物の有用な用量はインビトロ活性と動物モデルにおけるインビボ活性を比較することにより決定できる。マウスおよびその他の動物の有効用量をヒトに外挿する方法は当該技術分野で知られている; 例えば、米国特許第4938949号参照。
【0093】
一般に、例えば、ローションなどの液体組成物中の式Iの化合物の濃度は、約 0.1-25 重量%、好ましくは約 0.5-10 重量%である。ゲルまたは粉末などの半固体または固体組成物中の濃度は、約 0.1-5 重量%、好ましくは 約 0.5-2.5重量%である。
【0094】
治療での使用に必要な化合物、またはその活性な塩あるいは誘導体の量は、選択した特定の塩によってのみならず、投与経路、治療すべき症状の性質、および患者の年齢と状態によって変動し、最終的にはかかりつけの医師または臨床家の決定によるであろう。
【0095】
しかし一般に、好適な用量は約 0.5〜約 100 mg/kg体重/日の範囲、例えば、約 10〜約 75 mg/kg体重/日の範囲であり、例えば、 3〜約 50 mg /kg(レシピエントの)体重/日、好ましくは6〜90 mg/kg/日の範囲、もっとも好ましくは15〜 60 mg/kg/日の範囲である。
【0096】
本発明の化合物は当該技術分野において公知の方法を用いて標識することができる。好ましい検出可能な基は蛍光基である。蛍光基は典型的には高い信号対雑音比を生じるので、検出手順において高い解像度と感度を提供する。好ましくは、蛍光基は波長が約 300 nmを超える、より好ましくは約 350 nmを超える、もっとも好ましくは約 400 nmを超える光を吸収する。蛍光基によって放射される光の波長は好ましくは 約 310 nmを超え、より好ましくは約 360 nmを超え、もっとも好ましくは約 410 nmを超える。
【0097】
蛍光性の検出可能な部分は様々な構造分類から選択され、以下の非限定的な例を含む: 1-および2-アミノナフタレン、p,p’ジアミノスチルベン、ピレン、四級フェナントリジン塩、9-アミノアクリジン、p,p’-ジアミノベンゾフェノンイミン、アントラセン、オキサカルボシアニン、マロシアニン、3-アミノエキレニン、ペリレン、ビスベンズオキサゾール、ビス-p-オキサゾリルベンゼン、1,2-ベンゾフェナジン、レチノール、ビス-3-アミノピリジニウム(pridinium)塩、ヘレブリゲニン(hellebrigenin)、テトラサイクリン、ステロフェノール、ベンズイミダゾリルフェニルアミン、2-オキソ-3-クロメン、インドール、キサンテン、7-ヒドロキシクマリン、フェノキサジン、サリチレート、ストロファンチジン、ポルフィリン、トリアリールメタン、フラビン、キサンテン色素 (例えば、フルオレセインおよびローダミン色素); シアニン色素; 4,4-ジフルオロ-4-ボラ-3a,4a-ジアザ-s-インダセン色素および蛍光タンパク質 (例えば、緑色蛍光タンパク質、フィコビリタンパク質)。
【0098】
化合物は、自然にシグナルを放射するか、または好適な刺激の導入によってシグナルを生成する標識基によって標識することができる。標識としては、例えば、以下のような原子が挙げられる:例えば、13C、15N、19F、1H 等。
【0099】
化合物は便宜に単位剤形にて投与される;例えば、5 〜 1000 mg、便宜には 10〜750 mg、もっとも便宜には、50〜500 mgの活性成分が単位剤形当たりに含まれる。
【0100】
理想的には、活性成分は、約 0.5〜約 75 μM、好ましくは、約 1 〜 50 μM、もっとも好ましくは、約 2 〜 約 30 μMの活性化合物のピーク血漿濃度を達成するように投与される。これは、例えば、活性成分の、所望により生理食塩水中の0.05 〜 5% 溶液の静脈内注射により、または活性成分を約 1-100 mg含むボーラスとしての経口投与により達成される。所望の血中レベルは、約 0.01-5.0 mg/kg/時をもたらす連続注入または活性成分を約 0.4-15 mg/kg含む間欠的注入によって維持されうる。
【0101】
所望の用量は、単回用量において、または適宜の間隔、例えば、1日あたり分割用量を2、3、4または5以上の複数回用量として便宜に提供すればよい。分割用量自体をさらに分割してもよく、例えば、多数の不連続な緊密でない間隔を開けた投与にしてもよい;例えば、吸入器からの複数回の吸入など。
【0102】
Sybyl (TRIPOS) に備えられたFlexX ソフトウェア (Kramer et al.、Proteins、37:228 (1999))による、Bak-ペプチドとの複合体においてみられるBcl-xLの立体構造を用いたドッキング研究により、通常は複合体においてBak らせん状 BH3 ペプチドによって占有されている疎水性の深い裂け目中にゴシポールの最適位置があることが示された (図3a)。本発明者らはゴシポールの(+)と(-)の両方の立体異性体をドッキングさせた。というのは、これらは以前の細胞に基づくアッセイにおいて異なる活性を示したからであり、細胞毒性薬剤として(-) ゴシポールは(+)ゴシポールより十倍活性が高かった(Qiu et al.、Exp. Biol. Med.、227:398 (2002))。スコアリング関数およびSybylのDOCKルーチンによる最小化後の分子間エネルギーによって測定した適合度(Pervushin et al.、Proc. Natl. Acad. Sci. U.S.A.、94:12366 (1997)は、(-) ゴシポールの方がかなり良好であり((-) ゴシポールは-32.7 Kcal/molであるのに対し、(+) ゴシポールは-25 Kcal/mol)、これら観察と一致した。 (-) ゴシポールの構造を示すが (図3a)、ゴシポールの両方の立体異性体の全体的な位置づけは非常に類似している。
【0103】
ヒト腫瘍細胞に対する本発明の化合物の細胞毒性活性を評価するために、本発明者らはそれらの生理活性を2つの乳癌細胞株を用いてXTT 色素排除 アッセイによって試験した: MCF7 (Bcl-2/Bcl-xL高発現株)およびZR75-1 (Bcl-2/Bcl-xL低発現株)。ゴシポールはMCF7およびZR75-1 細胞に対して細胞毒性薬剤であり(図4 a,b)、細胞生存度を用量依存的に低下させ、それぞれIC50 値は13.2μMおよび8.4μMであった。しかしプルプロガリンはこれらアッセイにおいて感知できる活性を示さず、それはおそらくその疎水性の特徴による (ClogP ~ 0.7)。この観察と一致して、(その ClogPが ~ 2.5であることに基づいて)より良好な細胞膜透過特性を有することが予測されるプルプロガリン誘導体 5D1は細胞生存度を用量依存的に低下させ、ZR75-1 細胞株に対するIC50 値は~ 50μMであった(示さず)。これらの理由のため、本発明者らはさらに、化合物取り込みに対して選択性が低いことが知られているHeLa 細胞における本発明の化合物の細胞活性を評価した(表 3)。HeLa 細胞生存度アッセイにより得られた阻害データはBcl-xLを用いたインビトロ結合データと同等であり(表 3)、相関係数r = 0.9 (p = 0.001)であった。
【0104】
実験セクション
蛍光偏光アッセイ(FPA)
FPA アッセイをLJL Analyst HT (Molecular Devices Co.、Sunnyvale、CA)を用い、フルオレセイン-標識化 Bad ペプチド (NLWAAQRYGRELRRMSD-K(FITC)-FVD) (Synpep Corporation、Dublin、CA)を用いて行った。すべてのストックおよびサンプルについての希釈バッファーは、 50 mM Tris-Bis pH 7.4、0.01% ウシガンマグロブリンであった。ゴシポールの一連の2倍希釈物を調製した(即ち希釈バッファー中、100μM、50μM、0.1μMまで)。各チューブに、30 nM のBcl-xLと4 nM フルオレセイン標識ペプチドを含む溶液を添加した。チューブを5 分間室温でインキュベートし、20μlの各反応混合物を96-ウェルブラック PS、HE マイクロプレート (LJL Biosystems Co.)に移した。すべてのアッセイは四連で行い、ブランクウェルにはゴシポールを入れなかった。次いで、プレートを全強度について読み、偏光 (mP単位)を測定した。対照にはタンパク質の非存在下での用量応答測定を含め、化合物とFITC-BH3 ペプチドとの相互作用を評価した。結果としての効果をサブトラクション(差し引くこと)によって考慮した。
【0105】
NMR 分光法
Bcl-xLの2D [15N,1H]-TROSY (Pervushin et al.、Proc. Natl. Acad. Sci. U.S.A.、94:12366 (1997); Pellecchia et al.、Nat. Rev. Drug Disc.、1:211 (2002)) スペクトルを、15N-標識化 Bcl-xLの0.5 mM サンプルを用いて測定した。15N-標識化および非標識化 Bcl-xLを調製し、Sattler et al.、Science、275:983 (1997)に記載のように精製した。化学シフトマッピングおよびドッキング研究のために、本発明者らは、Bak ペプチド (PDBコード 1BXL)との複合体におけるBcl-xLの三次元構造を用いた。標識化タンパク質による化学シフトマッピングに加えて、T1ρ測定(Hajduk et al.、J. Am. Chem. Soc.、119:12257 (1997))および 飽和移動実験 、例えば、WaterLOGSY 実験(Dalvit et al.、J. Biomol. NMR、18:65 (2000))も行って、被験化合物のBcl-xLに対する結合をさらに確認した。すべての実験は500 MHz Varian Unity+ 分光計または600 MHz Bruker Avance600 分光計を用いて行った。これら分光計はともに、4つの rf チャンネルおよびz-軸パルスフィールドグラジエントを備えていた。 選択的水飽和を連続する選択的 IBURP2パルス(7 msの持続時間、10 msの遅延による間隔)によって行った。用いた全飽和時間は2.5sであった。T1ρシリーズは長さが可変のスピンロックパルスによって行った。測定は1 ms、10 ms、50 ms、150 ms、200 ms、250 msおよび300 ms スピンロック時間で100μM 化合物を用いて10 μM タンパク質の非存在下および存在下で行った。すべての実験において、残余の水シグナルの位相の散逸(de-phasing)が WATERGATE シークエンス(sequence)によって得られた。
【0106】
分子モデリング
分子モデリング研究はソフトウェアパッケージ Sybyl バージョン 6.9 (TRIPOS)によりいくつかのR12000 SGI Octane ワークステーションで行った。Sybylに備えられていたために最初にFlexX (Kramer et al.、Proteins、37:228 (1999))によって、ゴシポールのドッキング構造を得た。2つの計算を行った。まず、すべての結合部位ねじれ角を固定して維持し、次に側鎖ねじれ角を自由に変化できるようにした。30の最適解についての平均スコアリング関数は、側鎖を自由に回転させた場合わずかに低くなるにすぎなかった。モデルにおける側鎖の位置は最初の値から実質的に変化しなかった。(+) ゴシポールのスコアリング関数は(-) ゴシポールよりも劣っていたが、両方の立体異性体の全体的な配置は非常に類似していた。結果として得られた最適スコアリング構造のエネルギーを次いでSYBYL のルーチンDOCKを用いて、部位を固定しながら最小化した。DOCK最小化の後のリガンドのエネルギーはそのエネルギーの全最小値(global minimum of energy)から5 Kcal/molの範囲内であった。化合物の重ね合わせはSYBYLのルーチンMULTIFITにより得た。三次元構造を示すカラー図をプログラム SYBYLおよび MOLMOL (Koradi et al.、J. Mol. Graph.、14:29; 14:51 (1996))を用いて調製した。
【0107】
化合物の癌細胞生存に対する阻害効果
培養中の腫瘍細胞の生存度に対する本発明において研究している化合物の効果をXTT (Weislow et al.、J. Natl. Cancer Inst.、81:577 (1989)) アッセイにより、 MCF7および ZR75-1 細胞株を用いてモニターした。MCF7 細胞を10% 胎児ウシ血清、ペニシリン/ストレプトマイシン、さらに10-10 M インスリン、1 mM ピルビン酸ナトリウムおよびグルタミンを含むDMEMで培養した。ZR75-1 細胞を、10% 胎児ウシ血清、ペニシリン/ストレプトマイシン、さらにHEPES バッファー、1 mM ピルビン酸ナトリウムおよびグルタミンを含むRPMIで培養した。細胞をマイコプラズマによるコンタミネーションについて定期的に試験した。細胞を三連で最初の細胞密度1,000 細胞/ウェルにて播いた。ブランクウェルには細胞を入れなかった。ゴシポール、プルプロガリンおよび5D1を終濃度0、1、10および 100μMにて添加し、3日間インキュベートした。生細胞の相対数をXTT アッセイで判定した。簡単に説明すると、96-ウェルプレートに50 μlの以下の混合物を各ウェルに添加した:1 mg/ml の、0.025 mM PMS (フェナジンメトスルフェート)を含有するXTT (Weislow et al.、J. Natl. Cancer Inst.、81:577 (1989)) (2,3-ビス(2-メトキシ-4-ニトロ-5-スルホフェニル)-5-[(フェニルアミノ)カルボニル]-2H-テトラゾリウムヒドロキシド) (Polysciences、Washington、PA)。96-ウェルプレートをさらに4時間再びインキュベートしてXTT ホルマザンの生成を可能にした。次いで、各プレートの内容物を混合し、吸光度を波長450 nm (OD450)で測定した。正味のOD450をブランクウェルのOD450を差し引いた後に判定した。低継代 HeLa 細胞(継代数 10〜20)をpcDNA3-Bcl-xLまたは対照 pcDNA3 プラスミドにLipofectamine Plus 試薬(Invitrogen)を用いてトランスフェクトし、800 μg/mlのG418を含む培地で選択した。Bcl-xLのイムノブロット分析は(Krajanski 1996 -Cancer Res)に記載のように行った。 HeLa-トランスフェクタントを様々な用量のゴシポール、プルプロガリンおよびその誘導体(0、1、3、10 および100 μM)で処理した。
【0108】
化学物質
純粋なポリフェノールをSIGMA (ゴシポールおよびプルプロガリン)および/またはMicrosource Discovery Systems (プルプロガリン誘導体)から得た。対照化合物をChembridge Corp. (San Diego)から得た。ゴシポールは(+)および(-)異性体のラセミ体として試験した。化合物をDMSOに100 mM 濃度で溶解し、-20℃で保存した。NMR 分析を定期的に質的対照として化合物について行い、その後、さらに結合および置換アッセイのために希釈した。ゴシポールの反応性を、15N-標識化被験タンパク質 (XIAP のBIR3 ドメイン)を用いて試験した。1 mM ゴシポールおよび200μM 15N-標識化 BIR3を含む溶液を2時間インキュベートし、 [15N,1H]- 相関スペクトルを記録し、アポ-Bir3のスペクトルと比較した。スペクトルにおいて感知できる差は観察されなかった。
【0109】
【化3】
【表4】
プルプロガリン誘導体の構造活性相関(SAR)
【0110】
すべての刊行物、特許、および特許文献は、個々に引用により含まれているかのように参考として本明細書に含まれる。本発明は様々な特定の好ましい態様および技術に関して記載した。しかし、本発明の精神と枠内で多くの改変および修飾がなされうることを理解されたい。
【技術分野】
【0001】
関連出願のクロスリファレンス
本出願は2003年6月25日出願の米国仮特許出願第60/482,886号(その内容を引用により本出願に含める)からの優先権を主張する。
政府援助
本明細書に記載の本発明は、the National Institute of Healthにより与えられた認可番号 CA78040-05下、政府の支援によりなされた。米国政府は本発明における一定の権利を有する。
【背景技術】
【0002】
発明の背景
現在、癌、特にメラノーマ、頸癌、または白血病を予防および/または治療するための新規、強力、かつ選択的な物質が望まれている。現在研究中の方法の一つはアポトーシスの選択的誘導である。アポトーシスの選択的誘導に関連する生理過程のさらなる研究のための薬理手段も必要とされている。
【0003】
プログラムされた細胞死 (アポトーシス)は、組織恒常性、発達過程における望ましくない細胞の生理的除去、および宿主の防御機構に必須である(Vaux et al.、Cell、96:245 (1999))。アポトーシスの阻害は既知のヒトの悪性腫瘍のいずれにおいても伴っている。この阻害によって悪性細胞に選択的増殖の利点が与えられ、照射または化学療法によっても生存可能となってしまう(Reed、Curr. Opin. Oncol.、7:541 (1995)、およびKelekar et al.、Trends Cell. Biol.、8:324 (1998)参照)。Bcl-2 ファミリーのタンパク質はアポトーシスの重要な制御因子であると考えられている;このファミリーの生存促進性のメンバー、例えば、Bcl-xLは、表面に疎水性の溝を含み、そこでアポトーシス促進性対応物のBH3 ドメインへの結合が可能となると考えられている (Johnstone et al.、Cell、108:153 (2002))。この結合はアポトーシス制御に重要であると考えられており、実際生存促進性および抗生存タンパク質が二量体化を介して互いの機能を逆転することが出来る。抗アポトーシス性 Bcl-2 ファミリーメンバーは、多くのヒト悪性腫瘍において一般的に過剰発現していると考えられている。かかる観察によりBcl-2 ファミリーの抗アポトーシス性タンパク質、主に、Bcl-xLを標的とする低分子の候補抗癌治療薬としての発見に関する興味が増すことになった (Wang et al.、Proc. Natl. Acad. Sci. U.S.A.、97:7124 (2000))、Degterev et al.、Nat. Cell Biol.、3:173 (2001); Tzung et al.、Nat. Cell Biol.、3:183 (2001); Enyedy et al.、J. Med. Chem.、44:4313 (2001))。しかし現在でもなお、提案された化合物は、インビボ活性が低いか、あるいはインビトロ親和性が低いために、候補抗癌薬としてのBcl-xL 阻害物質の役割を完全に確証されるには至っていない(Kaneko et al.、Bioorg. Med. Chem. Lett.、11:887 (2001); Chin et al.、Angew. Chem. Int. Ed. Engl.、40:3806 (2001); Kutzki et al.、J. Am. Chem. Soc.、124:11838 (2002))。
【0004】
それゆえ、例えば、Bcl-xL、Bcl-2、Mcl-1、Bcl-W、 またはBcl-BなどのBcl-2 ファミリーの受容体を標的化する強力な細胞透過性化合物を同定する必要がある。BH3の Bcl-2 受容体への結合を阻害できるアゴニストが望まれている。
【0005】
さらに特に、癌細胞(例えば、リンパ腫、神経芽腫、乳癌、肺癌、前立腺癌、卵巣癌、白血病等)が、抗アポトーシス性 Bcl-2 ファミリータンパク質、例えばBcl-xL、Bcl-2、Mcl-1または Bcl-Bを過剰生産しているタイプの癌における化学感作物質として有用な化合物が望まれている。
【発明の概要】
【0006】
発明の概要
本発明は、癌細胞と、ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびそれらの混合物からなる群から選択される、癌細胞の生存度を低下させるのに有効な化合物とを接触させることを含む、患者における癌の治療方法を提供する。
【0007】
さらに、本発明は患者におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導方法を提供し、該方法は、標的細胞と、アポトーシス誘導、カスパーゼ活性調節、または標的細胞の細胞死誘導に有効な、以下からなる群から選択される化合物とを接触させることを含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびそれらの混合物。
【0008】
さらに、本発明は、Bcl-2 ファミリータンパク質を過剰発現している細胞におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導方法を提供し、該方法は、標的細胞と、本明細書に開示する本発明の化合物とを接触させることを含む。
【0009】
別の態様において、本発明は患者における癌の治療方法を提供し、該方法は、対象に、抗癌剤と組み合わせて以下からなる群から選択される化学感作物質を投与することを含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびその混合物。
【0010】
さらに、本発明は、Bcl-2 タンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1、Bcl-W、およびBcl-B)の、Bcl-2 ファミリーのアポトーシス促進性メンバーであるタンパク質(例えば、Bid、Bad、Bak、Bax )のBH3 ドメインへの、またはBH3 ドメインのみを含むペプチドへの、結合を調節するのに有効な化合物の同定方法を提供する。
【0011】
本発明は、 Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Bcl-W、Mcl-1、および Bcl-B)に結合する、または Bcl- 2活性を調節する化合物の同定方法を提供する。さらに、本発明は、Bcl-2 ファミリーのアポトーシス促進性メンバーであるタンパク質(例えば、Bid、Bad、Bak、Bax)のBH3 ドメインまたは BH3 ドメインのみを含むペプチドと複合体を形成した場合に、Bcl-2 ファミリータンパク質に結合する、またはBcl-2活性を調節する化合物の同定方法を提供する。
【0012】
本発明は、薬物療法における使用(例えば、アポトーシスの誘導、カスパーゼ活性の調節、細胞死の誘導、または癌の治療における使用)、好ましくは、以下の治療における使用のための本明細書に記載の化合物を提供する:肺癌、乳癌、前立腺癌、その他の形態の癌、および白血病、例えば、急性リンパ球性白血病 (ALL)、急性骨髄性白血病 (AML)、慢性骨髄性白血病 (CML)、およびその他の増殖性疾患。さらに本発明は、哺乳類、例えばヒトにおけるアポトーシスの誘導、カスパーゼ活性の調節、細胞死の誘導または癌の治療(好ましくは肺癌、乳癌、前立腺癌、CML、ALL、AML、その他の形態の癌または白血病およびその他の増殖性疾患の治療)のための医薬の製造における式Iの化合物の使用を提供する。本発明の化合物はまた、アポトーシスが一つの徴候である疾患(例えば、心臓病、パーキンソン病、アルツハイマー病等)の、AHPN アンタゴニスト経路を用いた治療に有用である。
【0013】
本発明はまた、アポトーシスまたは細胞死の誘導方法を提供し、該方法は、細胞と、有効量の (本明細書に記載の) 本発明の化合物をインビトロまたはインビボで接触させることを含む。
【0014】
本発明はまた、治療を必要とする哺乳類におけるアポトーシスの誘導方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0015】
本発明はまた、細胞におけるカスパーゼの活性化方法(例えば、Bcl-2 ファミリーの抗アポトーシス性タンパク質の阻害によるカスパーゼ 3 および 7の活性化方法)を提供し、該方法は細胞を、インビトロまたはインビボで、有効量の(本明細書に記載の)本発明の化合物と接触させることを含む。
【0016】
本発明はまた、ヒトなどの哺乳類におけるカスパーゼ活性化(例えば、Bcl-2 ファミリーの抗アポトーシス性タンパク質の阻害を介するカスパーゼ 3 および 7の活性化)が関連する病状または症状の予防または治療方法を提供し、該方法は、かかる治療を必要とする哺乳類にカスパーゼ-調節に有効な量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0017】
本発明はまた、細胞死を誘導する治療方法を提供し、該方法は細胞と、インビトロまたはインビボで、有効量の(本明細書に記載の)本発明の化合物とを接触させることを含む。
【0018】
本発明はまた、治療を必要とする哺乳類における細胞死の誘導方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0019】
本発明はまた、治療を必要とする哺乳類における癌 (例えば、肺癌、結腸直腸癌、乳癌、前立腺癌、ALL、CLL、AML、固形腫瘍、その他の形態の癌または白血病、例えば、リンパ腫、神経芽腫、およびその他の増殖性疾患)の治療方法を提供し、該方法は、哺乳類に有効量の(本明細書に記載の)本発明の化合物を投与することを含む。
【0020】
本発明はまた、Bcl-2 ファミリーのタンパク質(例えば、Bcl-xLおよびBcl-2)の抗アポトーシス活性を阻害する物質の同定方法を提供し、該方法は以下の工程を含む: a)標識化 Bcl-xLに結合している選択的 Bcl-xLまたはBcl-2 阻害物質を検出する工程、ここで該 Bcl-xL 阻害物質は以下からなる群から選択されるコア構造を含む:ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-)ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)およびプルプロガリン誘導体; b)結合しているBcl-xLと候補物質とを接触させる工程、ここで該候補物質はBcl-xLを阻害することが出来ると予測されている;および、 c)該Bcl-xL 阻害物質の該標識化 Bcl-xLからの分離を検出する工程、これによって該候補物質はBcl-xLの阻害物質であると同定される。
【図面の簡単な説明】
【0021】
図面の簡単な説明
【図1】図1. ゴシポールおよびプルプロガリンがBcl-xLのBH3-結合ポケットについて競合している。ゴシポール (a)およびプルプロガリン (c)の化学構造。フルオレセイン-標識化 Bad ペプチド (NLWAAQRYGRELRRMSD-K(FITC)-FVD) (Synpep Corporation、Dublin、CA)を用いた蛍光偏光に基づく競合結合アッセイ (FPA)の結果を (c)ゴシポールおよび(d)プルプロガリンについて示す。
【0022】
【図2】図 2. NMR 結合研究。(a)アポ(左)およびゴシポール-結合(右)形態におけるBcl-xLについての 2 D [15N,1H]-TROSY スペクトル。(b) Bak ペプチドとの複合体におけるBcl-xLの三次元構造へのゴシポールの化学シフトマッピング(PDB コード1BXL)。ペプチドを黄色で示す。ゴシポールの結合により影響を受けた領域を赤色で示す。(c)および(d) それぞれゴシポールおよびプルプロガリンによるT1ρ 実験(300 ms 緩和時間):青色、タンパク質無し、および赤色、10 μM Bcl-xLの存在下。(c)に示すピークはゴシポールにおけるイソプロピルおよびメチル基を表す。 (d)において、星印で示したピークは、タンパク質調製物において存在する残余のイミダゾールを示す。
【0023】
【図3】図 3. 分子モデリング研究。(a) FlexXによって得られたBcl-xLとゴシポールのドッキング構造の表面表示。(b) 5D1 (緑色)およびゴシポール (白色、酸素原子は赤色)の重ね合わせ。
【0024】
【図4】図 4.化合物の癌細胞生存に対する阻害効果。培養中の腫瘍細胞の生存度に対するゴシポールの効果をXTT アッセイを用いてモニターした。(a) MCF7 および(b) ZR75-1 細胞株 (黒丸)を用いた。陰性対照として、ジェネリック(generic)ポリフェノール化合物も試験した(白丸)。低継代 HeLa 細胞 (継代数 10〜20)をpcDNA3-Bcl-xL (黒丸)または対照pcDNA3 プラスミド(白丸)でトランスフェクトした。 (c) イムノブロット分析により、pcDNA3-対照トランスフェクタントと比較してpcDNA3-Bcl-xL 化合物でトランスフェクトした細胞においてBcl-xL の過剰発現が確認された(細胞溶解液を全タンパク質含量について正規化した; 25 μg/レーン)。(d) HeLa-トランスフェクタントを様々な用量のゴシポール (0、1、3、10 および 100μM)で処理した。示したデータは平均±標準偏差 (n = 4)を表す。
【0025】
【図5】図 5は、以前に処理されず、新たにCLLと診断されたものにおけるアポゴシポール (6C1)およびゴシポールの単剤としての活性を示す。
【0026】
【図6】図 6は、アポゴシポール (6C1)とフルダラビン (F-ara-A)との相加効果を示す。
【0027】
【図7】図 7は、アポゴシポール (6C1)と F-ara-Aとの相乗効果を示す。
【0028】
【図8】図 8は、 (-)EGCG のBcl-xL 受容体に対する結合を示す; (a) 10 μM Bcl-xLの添加の前(上側スペクトル)と後(下側スペクトル)の(-)EGCGによるT1ρ実験 (200 ms 緩和時間)。(a)に示すピークはカテキン基(group)のプロトン4αおよび4βを示し、* は DMSOを示す; (b) (-)EGCGについての蛍光偏光に基づく競合結合アッセイの結果;および、 (c) FlexXによって得られたBcl-xL と(-)EGCGのドッキング構造の表面表示。リガンドによって占有された3つのサブポケット (P1、P2およびP3)が示される。
【0029】
【図9】図 9は(-)CGと(-)Cとの比較を示す; (a) (-)CG (左側)と(-)C (右側)のT1ρ実験(200 ms 緩和時間); タンパク質の非存在下で記録したスペクトルを青色で示す; *はタンパク質バッファーからのイミダゾールを示す; (b) (-)CGと(-)CのFPA結果の重ね合わせ;および、 (c) (-)CG (赤色)と (-)C (青色)のドッキング構造によってくぼみのついた(pocked)Bcl-xL結合の表面表示。
【0030】
【図10】図 10はテアフラビン-3’-ガラートのBcl-xL 受容体への結合を示す: (a)テアフラビン-3’-ガラート (1mM) の添加前(左)および添加後(右)のBcl-xL (0.250 mM)の2D [15N,1H]-TROSY スペクトル; (b)テアフラビン-3’-ガラートについてのFPA結果;および、 (c)テアフラビン-3 '-ガラートのドッキング構造によりくぼみのついたBcl-xL結合の表面表示、リガンドに占有された3つのサブポケット (P1、P2およびP3)を丸で示す。
【0031】
【図11】図 11は、示したテアフラビンを用いるBcl-xLの阻害を示す。
【0032】
【図12】図 12はテアフラバニンを用いるBcl-xLの阻害を示す。
【0033】
【図13】図 13はXTTアッセイにおけるHela 細胞に対するテアフラバニンの効果を示す。
【発明を実施するための形態】
【0034】
詳細な説明
本発明の化合物は、Bcl-2 タンパク質 (例えば、Bcl-2およびBcl-xL)の受容体の拮抗(antagonizing)に有用である。これらのタンパク質は通常の抗癌治療、例えば、放射療法または化学療法に対して癌細胞をより抵抗性にすることができる。したがって、これら細胞は死滅せず、増殖しうる。本明細書に記載の化合物によるかかるタンパク質の阻害によって、細胞の抗癌治療に対する抵抗性を減少または排除しうる。それゆえ、これらの化合物は化学感作物質として有用である。したがって、本発明の化合物は癌細胞を抗癌治療、例えば、放射療法または化学療法に対してより感受性にしうる。したがって、本発明は、Bcl-2 タンパク質とアポトーシス促進性 Bcl-2 ファミリーメンバーのBH3 ドメインとの複合体の形成を調節する方法を提供し、これら複合体の量または安定性を調節するのに有用な化合物を提供する。
【0035】
本発明は、本発明の化合物が抗アポトーシス性タンパク質 Bcl-xLに結合する能力を測定するためにスペクトル技術を用いて化合物をスクリーニングする方法を提供する。該方法により、その他の抗癌化合物と組み合わせて用いることが出来る化合物が同定される。様々な緑茶および黒茶ポリフェノール化合物を、核磁気共鳴 (NMR) 結合アッセイ、蛍光偏光アッセイ (FPA) およびコンピュータによるドッキング研究を組み合わせて用いることによりスクリーニングした。
【0036】
一つの態様において、本発明は、抗アポトーシス性タンパク質 Bcl-xLに結合し、そしてBcl-2 ファミリーの生存促進性メンバーとのその相互作用を阻害することを介して部分的に抗癌薬として作用しうる茶のポリフェノール類の活性を評価する方法を提供する。
【0037】
別の態様において、本発明はBcl-xLのBH3への結合を有効に調節することが出来る化合物の同定方法を提供する。該方法は以下の工程を含む:(a) Bcl-xL-BH3 複合体の形成に好適な条件下でBcl-xLとBH3とを接触させる工程; (b) Bcl-xL-BH3 複合体と被験化合物とを接触させる工程;および、(c)被験化合物が、Bcl-xLのBH3への結合を調節する能力を判定する工程、ここでBcl-xLのBH3への結合の調節は、被験化合物がBcl-xLのBH3への結合を調節する有効な化合物であることを示す。
【0038】
別の態様において、本発明は、 Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1 Bcl-W、またはBcl-B)の結合を有効に調節しうる物質の同定方法を提供し、該方法は、Bcl-2 阻害物質または標識化 Bcl-2 阻害物質を同定する工程を含み、ここで該阻害物質は表1、2、3に挙げる化合物およびアポゴシポール誘導体からなる群から選択される。該方法は、(a)Bcl-2 阻害物質または標識化 Bcl-2 阻害物質を同定する工程(ここで該阻害物質はゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、(-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、およびプルプロガリン誘導体からなる群から選択される)、Bcl-2 ファミリータンパク質と該阻害物質(例えば表1に挙げるもの)とを、Bcl-2 ファミリータンパク質との複合体形成に好適な条件下で接触させる工程; (b)該Bcl-2 阻害物質複合体と被験化合物とを接触させる工程; および (c) Bcl-xLの阻害物質への結合を調節する被験化合物の能力を判定する工程(ここで、Bcl-2 タンパク質の阻害物質化合物への結合の調節は、被験化合物 (物質)がBcl-2 の阻害物質化合物への結合の調節に有効な化合物であることを示す)。
【0039】
別の態様において、本発明は、癌の治療のための伝統的な化学療法の有効性を向上させるために、癌細胞を感作するのに有用なポリフェノール化合物を提供する。
【0040】
別の態様において、本発明は、アポゴシポールまたはそのアナログの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0041】
別の態様において、本発明は、プルプロガリンまたはそのアナログの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0042】
別の態様において、本発明は、黒茶または緑茶由来のポリフェノールの、単独で、または抗癌剤と組み合わせての癌の治療のための使用を提供する。
【0043】
別の態様において、本発明はポリフェノールの癌予防薬としての使用を提供する。本発明に記載するポリフェノール化合物は、癌(例えば、肺癌、乳癌、前立腺癌、結腸直腸癌、および白血病)の発症の確立が高いと疑われる患者に投与され、患者がかかる癌を発症する可能性を低減させうる。
【0044】
本発明において、緑茶および黒茶由来のポリフェノールを試験した。緑茶はCamelia Sinensis の未発酵の葉から生産され、ポリフェノール(カテキンとして知られる)はその主な化学成分を構成する。エピカテキン (EC)、エピカテキン-3 ガラート (ECG)、エピガロカテキン (EGC)、およびエピガロカテキン-3 ガラート (表 1)が緑茶に含まれる主なカテキンである(Chu et al.、In: Yamamoto、T.、Juneja、J. R.、Cu、D.C. and Kim、M. Chemistry and Application of Grean Tea、pp. 13-22、New York: CRC Press、1997)。黒茶は、ポリフェノールの、テアフラビン等の重合生成物への高度な酵素酸化によって作られる (Pan et al.)。テアフラビン、テアフラビン-3 ガラート、テアフラビン-3’-ガラート、およびテアフラビン-3-3’-ジガラートが黒茶における主なテアフラビン類である。
【0045】
さらに、本発明は、茶の抗癌活性と、Bcl-2 ファミリーの抗アポトーシス性タンパク質(例えば、Bcl-xL およびBcl-2)とのその相互作用を相関させる方法を提供する。
【0046】
さらに、本発明は、茶の抗癌活性とBcl-2 ファミリーの抗アポトーシス性タンパク質(例えば、Bcl-xLおよび Bcl-2)とのその相互作用を関連させる方法を提供する。
【0047】
別の態様において、本発明は、抗癌活性について化合物をスクリーニングする方法を提供し、そして 、以下に記載のアッセイを用いる薬理学モデルを用いて、本発明の化合物が癌治療を補助しうることを判定する方法を提供する。
【0048】
本発明はまた、本明細書に記載の化合物またはその医薬上許容される塩を、医薬上許容される希釈剤または担体と組み合わせて含む医薬組成物も提供する。さらに、本発明は、その他の既知の抗癌化合物と組み合わせての本明細書に開示の化合物の使用を提供する。
【0049】
本発明は、治療を必要とする哺乳類に、有効量の本明細書に記載の化合物、別の抗癌化合物と組み合わせた本明細書に記載の化合物またはその医薬上許容される塩を投与することを含む癌の治療方法を提供する。
【0050】
さらに、本発明は、癌の予防方法またはかかる癌を患者が発症しうる可能性を低下させる方法を提供し、該方法は、治療を必要とする哺乳類に、有効量の本明細書に記載の化合物またはその医薬上許容される塩を投与することを含む。
【0051】
特に断りのない限り以下の定義を用いる:ハロは、フルオロ、クロロ、ブロモ、またはヨードである。アルキル、アルコキシ、アルケニル、アルキニルなどは、直鎖状と分枝状の基の両方を含む;しかし個々の基についての、例えば、「プロピル」という言及は、直鎖状基のみを含み、分枝状異性体、例えば「イソプロピル」は具体的に言及される。アリールは、フェニル基、または約9から10の環原子を有し少なくとも1つの環が芳香族であるオルト-縮合二環式炭素環基をいう。ヘテロアリールは、炭素および1から4のヘテロ原子(それぞれ非ペルオキシド酸素、硫黄およびN(X)からなる群から選択され、ここで、Xは存在しないか、またはH、O、(C1-C4)アルキル、フェニルまたはベンジル)からなる5または6の環原子を含む単環式芳香族環の環炭素を介して結合した基、およびそれに由来する約8から10の環原子のオルト-縮合二環式-ヘテロ環基であり、特に、ベンズ-誘導体またはプロピレン、トリメチレン、またはテトラメチレンジ基がそれに縮合することにより生ずるものである。
【0052】
具体的には、「アルキル」の語は、炭素原子数が1から6の分枝状または非分枝状飽和炭化水素基であり、例えば、メチル、エチル、n-プロピル、イソプロピル、n-ブチル、イソブチル、t-ブチル、ペンチル等である。本明細書において好ましいアルキル基は炭素原子数1から6のもの、例えば、メチル、エチル等である。
【0053】
本明細書において用いる「シクロアルキル」の語は、炭素原子数3から8、好ましくは、3、5または6の環状アルキル基である。本明細書において用いる「シクロアルキレン」の語は、二価環状アルキレン基をいい、典型的には、3-、5-、6-、または8-員環である。
【0054】
本明細書において用いる「アルコキシ」の語は、末端エーテル単結合を介して結合したアルキル基をいい、即ち「アルコキシ」基は、Rが上記のアルキルである- ORと定義できる。「低級アルコキシ」基は、炭素原子数1から6のアルコキシ基をいう。
【0055】
本明細書において用いる「アリール」の語は、典型的には、少なくとも1つの環が芳香族である6-または10- 員環の芳香族炭素環をいう。
【0056】
当業者であれば、不斉中心を有する本発明の化合物は、光学活性形態およびラセミ形態にて存在および単離しうることを理解するであろう。化合物のなかには多形を示すものもあり得る。本発明は、本明細書に記載の有用な性質を有する、あらゆる本発明の化合物のラセミ、光学活性、多型、または幾何異性形態、あるいはその混合物を含むことを理解されたい。当該技術分野において、光学活性形態をいかにして調製するか(例えば、再結晶技術によるラセミ体の分割、光学活性出発物質からの合成、キラル合成またはキラル固定相を用いたクロマトグラフィー分割による)、そして本明細書に記載の標準的試験または当該技術分野において周知の類似の試験を用いて抗癌活性をいかにして判定するかは周知である。
【0057】
基、置換基および範囲について以下に挙げる具体的かつ好ましい値は、単に例示の目的である; それらは基および置換基について規定されるその他の値あるいは規定される範囲内の別の値を排除するものではない。
【0058】
具体的には、アルキルは以下のいずれかであり得る:メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、ペンチル、3-ペンチル、またはヘキシル; シクロアルキルは以下のいずれかであり得る:シクロプロピル、シクロブチル、シクロペンチル、またはシクロヘキシル; -O(C1-C6)アルキル(アルコキシ) は以下のいずれかであり得る: メトキシ、エトキシ、プロポキシ、イソプロポキシ、ブトキシ、イソ-ブトキシ、sec-ブトキシ、ペントキシ、3-ペントキシ、またはヘキシルオキシ。
【0059】
本発明の実施に有用な具体的化合物を表 1および表 2に挙げる。
表1
【表1−1】
【表1−2】
【0060】
表 2
【表2】
【0061】
黒茶または緑茶から単離されるポリフェノールの非限定的な例としては以下が含まれる:カテキン、例えば、エピカテキン (EC)、エピカテキン-3 ガラート (ECG)、エピガロカテキン (EGC)、エピガロカテキン-3 ガラート等; テアフラビン、例えば、テアフラビン、テアフラビン-3 ガラート、テアフラビン-3’-ガラート、テアフラビン-3-3’ジガラート等。
【0062】
別の具体的な本発明の化合物にはプルプロガリンおよびその誘導体、例えば、表 3に示す化合物が含まれる。
【0063】
表3
プルプロガリン誘導体
【化1】
【表3】
【0064】
本発明の実施に有用なさらなる化合物には以下が含まれる:例えば、正常細胞に対しては毒性が低いが、癌細胞に対しては、ゴシポールと同様の細胞毒性を有する化合物であるアポゴシポールおよびアポゴシポールのアナログ。かかるアナログは、Bcl-xL/2に対する強度および選択性が増強している。本発明のアナログ化合物は式Iを有するか、またはその医薬上許容される塩である:
【化2】
(I)
[式中:
各R6、R8、R9およびR10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ;
各R7は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【0065】
具体的なR6、R8、R9基は独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである。
【0066】
具体的なR7基は独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、シクロヘキシルである。
【0067】
具体的なR10基は独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである。
【0068】
具体的な本発明の化合物は式(I)を有し、ここでR6、R8、R9はそれぞれアセテート (-OC(O)CH3); R7はi-Pr;およびR10は-CH3 (アポゴシポールヘクス(hex)アセテート)である。この化合物はアポゴシポールの経口投与用のプロドラッグとしても有用である。別の態様において、本発明の化合物は式(I)の化合物であってR6基の1つが水素以外の基であるものも含む。
【0069】
本明細書において用いる、「患者」の語は、本発明の方法によって治療すべき生物をいう。かかる生物としては、これに限定されないが、ヒトが挙げられる。本発明において、「対象」の語は一般に、Bcl-2 ファミリータンパク質(例えば、Bcl-xL、Bcl-2、Mcl-1またはBcl-B)の過剰発現によって特徴づけられる疾患の治療 (例えば、本発明の化合物、および所望により1以上の抗癌薬の投与)を受ける、または治療を受けた個体をいう。
【0070】
本明細書において用いる、「抗癌剤」または「化学療法抗癌剤」の語は、癌の治療に用いられるあらゆる化学療法化合物、放射療法、および外科技術をいう。
【0071】
本発明の実施に有用な抗癌薬としては、緊張性薬剤または癌化学療法薬剤が挙げられる。本発明の実施に有用な緊張性薬剤の非限定的な例としては、低分子、ポリペプチド、ペプチド、ペプチド疑似体(peptidomimetics)、核酸分子、細胞およびウイルスが挙げられる。非限定的な例として、本発明に有用な細胞毒性薬剤としては、細胞毒性低分子、即ち、典型的にはDNAに関連する過程を標的とする化合物、例えば、ドキソルビシン、ドセタキセル、トラスツズマブ、シクロホスファミド、メルファラン、マイトマイシン C、ビゼレシン、シスプラチン、ドキソルビシン、エトポシド、ミトキサントロン、SN 38、Et 743、アクチノマイシン D、ブレオマイシン、TLK286等; 抗菌性ペプチド、例えば以下に記載するもの; アポトーシス促進性ポリペプチド、例えば、カスパーゼおよび毒素、例えば、カスパーゼ 8; ジフテリア毒素 A 鎖、緑膿菌外毒素 A、コレラ毒素、リガンド融合毒素、例えば、DAB389EGF、トウゴマ毒素 (リシン);および細胞毒性細胞、例えば細胞障害性 T 細胞等が挙げられる。例えば、Martin et al.、Cancer Res.、60:3218 (2000); Kreitman et al.、Blood、90:252 (1997); Allam et al.、Cancer Res.、57:2615 (1997);および Osborne et al.、Cancer J. Sci. Am.、2:175 (1996)を参照されたい。
【0072】
本発明での使用に好適なさらなる抗癌化学療法薬剤としては、これらに限定されないが、タキサン、例えば、ドセタキセル(タキソテール、Aventis Pharmaceuticals、Inc.; Parsippany、NJ) および パクリタキセル (タキソール、Bristol Myers Squibb; Princeton、NJ); アントラサイクリン、例えば、ドキソルビシン、イダルビシン、ダウノルビシン等;アルキル化剤;ビンカアルカロイド;代謝拮抗剤;白金薬剤、例えば、シスプラチンまたはカルボプラチン;ステロイド、例えば、メトトレキサート;抗生物質、例えば、アドリアマイシン;イソファミド;または選択的エストロゲン受容体調節因子;抗体、例えば、トラスツズマブ等が挙げられる。
【0073】
ドキソルビシンは一般によく用いられている癌化学療法薬剤であり、例えば、乳癌の治療に有用である(Stewart et al.、In: ”Cancer: Principles and Practice of Oncology” 5th ed.、Chap. 19 (eds. DeVita、Jr.、et al.; J.P. Lippincott 1997); Harris et al.、”Cancer: Principles and Practice of Oncology,”前掲、1997)。さらに、ドキソルビシンは抗血管形成活性を有し(Folkman、Nature Biotechnology、15:510 (1997); Steiner、”Angiogenesis: Key principles Science、technology and medicine,” pp. 449 454 (eds. Steiner et al.; Birkhauser Verlag、1992))、その活性が癌の治療におけるその有効性に寄与し得る。
【0074】
アルキル化剤、例えば、メルファランまたはクロランブシルは本発明の併用治療に有用な癌化学療法薬剤である。同様に、ビンカアルカロイド、例えば、ビンデシン、ビンブラスチンまたはビノレルビン;あるいは代謝拮抗剤、例えば、 5-フルオロウラシル、5-フルオロウリジンまたはそれらの誘導体は本発明の併用療法に有用な癌化学療法薬剤である。
【0075】
白金薬剤は本発明の併用療法に有用な化学療法薬剤である。かかる白金薬剤は例えばCrown、Seminars in Oncol.、28:28 (2001)に記載の、例えば、シスプラチンまたはカルボプラチンであり得る。本発明の併用療法に有用な化学療法薬剤としてはこれらに限定されないが、メトトレキサート、マイトマイシン C、アドリアマイシン、イホスファミドおよびアンサマイシンが挙げられる。
【0076】
乳癌およびその他のホルモン依存性癌の治療に用いられる癌化学療法薬剤は、エストロゲンの作用を拮抗する薬剤としても使用し得、例えば、選択的エストロゲン受容体修飾因子または抗エストロゲンが挙げられる。選択的エストロゲン受容体調節因子である、タモキシフェンは乳癌の治療のための本発明の併用療法に利用できる癌化学療法薬剤である(Fisher et al.、J. Natl. Cancer Instit.、90:1371 (1998))。
【0077】
本発明の併用療法に有用な別のタイプの治療薬は抗体、例えばヒト化モノクローナル抗体である。非限定的な例としては、抗上皮成長因子受容体 2 (HER2) 抗体が挙げられる。トラスツズマブ (ハーセプチン; Genentech、South San Francisco、CA)はHER2/neuを過剰発現する乳癌の治療のために本発明の組み合わせに有用なさらなる治療薬である(White et al.、Annu. Rev. Med.、52:125 (2001))。
【0078】
本発明に有用な別の治療薬は、本明細書において用いる場合、直接または間接的に細胞死を促進するいずれかの分子である細胞毒性薬剤でもありうる。
【0079】
具体的な抗癌薬としては、フラボピリドール、アドリアマイシン(ドキソルビシン)、VP16 (エトポシド)、タキソール (パクリタキセル)、シスプラチン等が挙げられる。
【0080】
化合物が安定な非毒性の酸または塩基塩を形成するのに十分に塩基性または酸性の場合は、化合物の塩としての投与が適当であり得る。医薬上許容される塩の例は、生理的に許容される陰イオンを形成する酸と形成される有機酸付加塩、例えば、トシレート、メタンスルホン酸塩、酢酸塩、クエン酸塩、マロン酸塩、酒石酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、α-ケトグルタル酸塩、およびα-グリセロリン酸塩などである。好適な無機塩も形成され得、例えば、塩酸塩、硫酸塩、硝酸塩、炭酸水素塩、および炭酸塩が挙げられる。
【0081】
医薬上許容される塩は当該技術分野において周知の標準的手順を用いて得ることが出来、例えば十分に塩基性の化合物、例えば、アミンを生理的に許容される陰イオンを与える好適な酸と反応させることにより得ることが出来る。カルボン酸のアルカリ金属 (例えば、ナトリウム、カリウムまたはリチウム)あるいはアルカリ土類金属 (例えばカルシウム)塩も作ることが出来る。
【0082】
本発明の実施に有用な化合物は医薬組成物として選択された投与経路、即ち、経口または非経口、静脈内、筋肉内、局所または皮下経路に応じた様々な形態に製剤し、哺乳類宿主、例えば、ヒト患者に投与してもよい。
【0083】
したがって、化合物は全身投与してもよく、例えば、医薬上許容される媒体、例えば、不活性希釈剤または吸収可能な食用担体とともに経口投与してもよい。それらは殻の硬いまたは軟らかいゼラチンカプセルに封入してもよいし、圧縮して錠剤としてもよいし、患者の食事の食品に直接組み込んでもよい。経口治療的投与について、活性化合物は1以上の賦形剤と組み合わせて、消化可能な錠剤、舌下錠、トローチ、カプセル、エリキシル、懸濁液、シロップ、ウエハースなどの形態で用いてもよい。かかる組成物および調製物は少なくとも0.1%の活性化合物を含むべきである。組成物および調製物のパーセンテージはもちろん変動させてもよく、好都合なことに所与の単位剤形の重量の約2から約60%の間であってよい。かかる治療上有用な組成物における活性化合物の量は有効投与量レベルが得られるものである。
【0084】
錠剤、トローチ、丸薬、カプセル等は以下のものを含んでいてもよい: 結合剤、例えば、トラガカントゴム、アカシアゴム、コーンスターチまたはゼラチン; 賦形剤、例えば、第二リン酸カルシウム; 崩壊剤、例えば、コーンスターチ、ジャガイモデンプン、アルギン酸等; 滑沢剤、例えば、ステアリン酸マグネシウム;および甘味料、例えば、スクロース、フルクトース、ラクトースまたはアスパルテームあるいは香味料、例えば、ペパーミント、ウィンターグリーン油、またはチェリーフレーバーを添加してもよい。単位剤形がカプセルの場合、それは上記タイプの物質に加えて、液体担体、例えば、植物油またはポリエチレングリコールを含んでいてもよい。その他の様々な物質が被覆として存在する場合もあるし、固体単位剤形の物理形態を修飾する場合もある。例えば、錠剤、丸薬またはカプセルはゼラチン、ろう、セラックまたは糖等で被覆してもよい。シロップまたはエリキシルは、活性化合物、甘味料としてスクロースまたはフルクトース、保存料としてメチルおよびプロピルパラベン、色素および香料、例えば、チェリーまたはオレンジフレーバーを含んでいてもよい。もちろん、単位剤形の調製にもちいる物質はいずれも使用する量において医薬上許容され、実質的に非毒性のものでなければならない。さらに、活性化合物は徐放調製物および装置に組み込んでもよい。
【0085】
活性化合物は注入または注射によって静脈内または腹腔内に投与してもよい。活性化合物またはその塩の溶液を水中に調製してもよく、所望により非毒性の界面活性剤を添加してもよい。分散液をグリセロール、液体ポリエチレングリコール、トリアセチン、およびその混合物ならびに油中に調製してもよい。貯蔵および使用の通常の条件下では、これら調製物には微生物の増殖の阻害のために保存料を含める。
【0086】
注射または注入に好適な薬理剤形には、無菌水溶液または分散液あるいは無菌注射可能または注入可能溶液または分散液の即時調製に適した活性成分を含む無菌粉末が含まれ、リポソームに封入されたものでもよい。何れの場合も、最終的な剤形は無菌、液体、かつ製造と貯蔵の条件下で安定なものであるべきである。液体の担体または媒体は溶媒または液体分散媒であり得、例えば、水、エタノール、ポリオール (例えば、グリセロール、プロピレングリコール、液体ポリエチレングリコール等)、植物油、非毒性グリセリルエステルおよびそれらの好適な混合物を含みうる。適切な流動性は、例えば、リポソームの形成、分散液の場合は所望の粒径の維持または界面活性剤の使用により維持されうる。微生物の作用の阻止は、様々な抗細菌および抗真菌薬、例えば、パラベン、クロロブタノール、フェノール、ソルビン酸、チメロサール等によってもたらされ得る。多くの場合、等張化剤、例えば、糖、バッファーまたは塩化ナトリウムを含むのが好ましい。注射可能組成物の持続的吸収は、吸収遅延剤、例えば、モノステアリン酸アルミニウムおよびゼラチンの組成物中での使用によりもたらされうる。
【0087】
無菌注射可能溶液は、所望の量の活性化合物を適当な溶媒に所望により様々なその他の上記成分とともに組込み、次いでろ過滅菌することにより調製される。無菌注射可能溶液の調製用の無菌粉末の場合、好ましい調製方法は減圧乾燥および凍結乾燥技術であり、それによって前もって無菌ろ過した溶液中に存在する活性成分とさらなる所望の成分の粉末が得られる。
【0088】
局所投与のためには、本発明の化合物を、それが液体の場合は純粋な形態で適用することができる。しかし、皮膚科学的に許容される担体と組み合わせた組成物または製剤として皮膚に投与するのが一般に望ましく、それは固体であっても液体であってもよい。
【0089】
有用な固体担体には、微細な固体、例えば、タルク、クレイ、微晶質セルロース、シリカ、アルミナ等が挙げられる。有用な液体担体には、水、アルコールまたはグリコールまたは水-アルコール/グリコール混合物が挙げられ、そのなかに本発明の化合物が有効レベルで、所望により非毒性界面活性剤の助けにより溶解または分散され得る。補助剤、例えば、香料およびさらなる抗菌剤を添加して所望の用途のための性質を最適にしてもよい。その結果得られる液体組成物は吸着剤パッドから適用されるか、絆創膏およびその他の包帯剤に浸透させて用いられるか、あるいはポンプ型またはエアゾールスプレーを用いて患部に噴霧される。
【0090】
増粘剤、例えば、合成高分子、脂肪酸、脂肪酸塩およびエステル、脂肪アルコール、修飾セルロースまたは修飾ミネラル物質を液体担体とともに用いて、塗布可能なペースト、ゲル、軟膏、ソープ等を形成して、使用者の皮膚に直接塗布してもよい。
【0091】
式Iの化合物を皮膚に送達するのに用いることが出来る有用な皮膚用組成物の例は当該技術分野で知られている; 例えば、Jacquet et al. (米国特許第4608392号)、Geria (米国特許第4992478号)、Smith et al. (米国特許第4559157号) およびWortzman (米国特許第4820508号)を参照されたい。
【0092】
式Iの化合物の有用な用量はインビトロ活性と動物モデルにおけるインビボ活性を比較することにより決定できる。マウスおよびその他の動物の有効用量をヒトに外挿する方法は当該技術分野で知られている; 例えば、米国特許第4938949号参照。
【0093】
一般に、例えば、ローションなどの液体組成物中の式Iの化合物の濃度は、約 0.1-25 重量%、好ましくは約 0.5-10 重量%である。ゲルまたは粉末などの半固体または固体組成物中の濃度は、約 0.1-5 重量%、好ましくは 約 0.5-2.5重量%である。
【0094】
治療での使用に必要な化合物、またはその活性な塩あるいは誘導体の量は、選択した特定の塩によってのみならず、投与経路、治療すべき症状の性質、および患者の年齢と状態によって変動し、最終的にはかかりつけの医師または臨床家の決定によるであろう。
【0095】
しかし一般に、好適な用量は約 0.5〜約 100 mg/kg体重/日の範囲、例えば、約 10〜約 75 mg/kg体重/日の範囲であり、例えば、 3〜約 50 mg /kg(レシピエントの)体重/日、好ましくは6〜90 mg/kg/日の範囲、もっとも好ましくは15〜 60 mg/kg/日の範囲である。
【0096】
本発明の化合物は当該技術分野において公知の方法を用いて標識することができる。好ましい検出可能な基は蛍光基である。蛍光基は典型的には高い信号対雑音比を生じるので、検出手順において高い解像度と感度を提供する。好ましくは、蛍光基は波長が約 300 nmを超える、より好ましくは約 350 nmを超える、もっとも好ましくは約 400 nmを超える光を吸収する。蛍光基によって放射される光の波長は好ましくは 約 310 nmを超え、より好ましくは約 360 nmを超え、もっとも好ましくは約 410 nmを超える。
【0097】
蛍光性の検出可能な部分は様々な構造分類から選択され、以下の非限定的な例を含む: 1-および2-アミノナフタレン、p,p’ジアミノスチルベン、ピレン、四級フェナントリジン塩、9-アミノアクリジン、p,p’-ジアミノベンゾフェノンイミン、アントラセン、オキサカルボシアニン、マロシアニン、3-アミノエキレニン、ペリレン、ビスベンズオキサゾール、ビス-p-オキサゾリルベンゼン、1,2-ベンゾフェナジン、レチノール、ビス-3-アミノピリジニウム(pridinium)塩、ヘレブリゲニン(hellebrigenin)、テトラサイクリン、ステロフェノール、ベンズイミダゾリルフェニルアミン、2-オキソ-3-クロメン、インドール、キサンテン、7-ヒドロキシクマリン、フェノキサジン、サリチレート、ストロファンチジン、ポルフィリン、トリアリールメタン、フラビン、キサンテン色素 (例えば、フルオレセインおよびローダミン色素); シアニン色素; 4,4-ジフルオロ-4-ボラ-3a,4a-ジアザ-s-インダセン色素および蛍光タンパク質 (例えば、緑色蛍光タンパク質、フィコビリタンパク質)。
【0098】
化合物は、自然にシグナルを放射するか、または好適な刺激の導入によってシグナルを生成する標識基によって標識することができる。標識としては、例えば、以下のような原子が挙げられる:例えば、13C、15N、19F、1H 等。
【0099】
化合物は便宜に単位剤形にて投与される;例えば、5 〜 1000 mg、便宜には 10〜750 mg、もっとも便宜には、50〜500 mgの活性成分が単位剤形当たりに含まれる。
【0100】
理想的には、活性成分は、約 0.5〜約 75 μM、好ましくは、約 1 〜 50 μM、もっとも好ましくは、約 2 〜 約 30 μMの活性化合物のピーク血漿濃度を達成するように投与される。これは、例えば、活性成分の、所望により生理食塩水中の0.05 〜 5% 溶液の静脈内注射により、または活性成分を約 1-100 mg含むボーラスとしての経口投与により達成される。所望の血中レベルは、約 0.01-5.0 mg/kg/時をもたらす連続注入または活性成分を約 0.4-15 mg/kg含む間欠的注入によって維持されうる。
【0101】
所望の用量は、単回用量において、または適宜の間隔、例えば、1日あたり分割用量を2、3、4または5以上の複数回用量として便宜に提供すればよい。分割用量自体をさらに分割してもよく、例えば、多数の不連続な緊密でない間隔を開けた投与にしてもよい;例えば、吸入器からの複数回の吸入など。
【0102】
Sybyl (TRIPOS) に備えられたFlexX ソフトウェア (Kramer et al.、Proteins、37:228 (1999))による、Bak-ペプチドとの複合体においてみられるBcl-xLの立体構造を用いたドッキング研究により、通常は複合体においてBak らせん状 BH3 ペプチドによって占有されている疎水性の深い裂け目中にゴシポールの最適位置があることが示された (図3a)。本発明者らはゴシポールの(+)と(-)の両方の立体異性体をドッキングさせた。というのは、これらは以前の細胞に基づくアッセイにおいて異なる活性を示したからであり、細胞毒性薬剤として(-) ゴシポールは(+)ゴシポールより十倍活性が高かった(Qiu et al.、Exp. Biol. Med.、227:398 (2002))。スコアリング関数およびSybylのDOCKルーチンによる最小化後の分子間エネルギーによって測定した適合度(Pervushin et al.、Proc. Natl. Acad. Sci. U.S.A.、94:12366 (1997)は、(-) ゴシポールの方がかなり良好であり((-) ゴシポールは-32.7 Kcal/molであるのに対し、(+) ゴシポールは-25 Kcal/mol)、これら観察と一致した。 (-) ゴシポールの構造を示すが (図3a)、ゴシポールの両方の立体異性体の全体的な位置づけは非常に類似している。
【0103】
ヒト腫瘍細胞に対する本発明の化合物の細胞毒性活性を評価するために、本発明者らはそれらの生理活性を2つの乳癌細胞株を用いてXTT 色素排除 アッセイによって試験した: MCF7 (Bcl-2/Bcl-xL高発現株)およびZR75-1 (Bcl-2/Bcl-xL低発現株)。ゴシポールはMCF7およびZR75-1 細胞に対して細胞毒性薬剤であり(図4 a,b)、細胞生存度を用量依存的に低下させ、それぞれIC50 値は13.2μMおよび8.4μMであった。しかしプルプロガリンはこれらアッセイにおいて感知できる活性を示さず、それはおそらくその疎水性の特徴による (ClogP ~ 0.7)。この観察と一致して、(その ClogPが ~ 2.5であることに基づいて)より良好な細胞膜透過特性を有することが予測されるプルプロガリン誘導体 5D1は細胞生存度を用量依存的に低下させ、ZR75-1 細胞株に対するIC50 値は~ 50μMであった(示さず)。これらの理由のため、本発明者らはさらに、化合物取り込みに対して選択性が低いことが知られているHeLa 細胞における本発明の化合物の細胞活性を評価した(表 3)。HeLa 細胞生存度アッセイにより得られた阻害データはBcl-xLを用いたインビトロ結合データと同等であり(表 3)、相関係数r = 0.9 (p = 0.001)であった。
【0104】
実験セクション
蛍光偏光アッセイ(FPA)
FPA アッセイをLJL Analyst HT (Molecular Devices Co.、Sunnyvale、CA)を用い、フルオレセイン-標識化 Bad ペプチド (NLWAAQRYGRELRRMSD-K(FITC)-FVD) (Synpep Corporation、Dublin、CA)を用いて行った。すべてのストックおよびサンプルについての希釈バッファーは、 50 mM Tris-Bis pH 7.4、0.01% ウシガンマグロブリンであった。ゴシポールの一連の2倍希釈物を調製した(即ち希釈バッファー中、100μM、50μM、0.1μMまで)。各チューブに、30 nM のBcl-xLと4 nM フルオレセイン標識ペプチドを含む溶液を添加した。チューブを5 分間室温でインキュベートし、20μlの各反応混合物を96-ウェルブラック PS、HE マイクロプレート (LJL Biosystems Co.)に移した。すべてのアッセイは四連で行い、ブランクウェルにはゴシポールを入れなかった。次いで、プレートを全強度について読み、偏光 (mP単位)を測定した。対照にはタンパク質の非存在下での用量応答測定を含め、化合物とFITC-BH3 ペプチドとの相互作用を評価した。結果としての効果をサブトラクション(差し引くこと)によって考慮した。
【0105】
NMR 分光法
Bcl-xLの2D [15N,1H]-TROSY (Pervushin et al.、Proc. Natl. Acad. Sci. U.S.A.、94:12366 (1997); Pellecchia et al.、Nat. Rev. Drug Disc.、1:211 (2002)) スペクトルを、15N-標識化 Bcl-xLの0.5 mM サンプルを用いて測定した。15N-標識化および非標識化 Bcl-xLを調製し、Sattler et al.、Science、275:983 (1997)に記載のように精製した。化学シフトマッピングおよびドッキング研究のために、本発明者らは、Bak ペプチド (PDBコード 1BXL)との複合体におけるBcl-xLの三次元構造を用いた。標識化タンパク質による化学シフトマッピングに加えて、T1ρ測定(Hajduk et al.、J. Am. Chem. Soc.、119:12257 (1997))および 飽和移動実験 、例えば、WaterLOGSY 実験(Dalvit et al.、J. Biomol. NMR、18:65 (2000))も行って、被験化合物のBcl-xLに対する結合をさらに確認した。すべての実験は500 MHz Varian Unity+ 分光計または600 MHz Bruker Avance600 分光計を用いて行った。これら分光計はともに、4つの rf チャンネルおよびz-軸パルスフィールドグラジエントを備えていた。 選択的水飽和を連続する選択的 IBURP2パルス(7 msの持続時間、10 msの遅延による間隔)によって行った。用いた全飽和時間は2.5sであった。T1ρシリーズは長さが可変のスピンロックパルスによって行った。測定は1 ms、10 ms、50 ms、150 ms、200 ms、250 msおよび300 ms スピンロック時間で100μM 化合物を用いて10 μM タンパク質の非存在下および存在下で行った。すべての実験において、残余の水シグナルの位相の散逸(de-phasing)が WATERGATE シークエンス(sequence)によって得られた。
【0106】
分子モデリング
分子モデリング研究はソフトウェアパッケージ Sybyl バージョン 6.9 (TRIPOS)によりいくつかのR12000 SGI Octane ワークステーションで行った。Sybylに備えられていたために最初にFlexX (Kramer et al.、Proteins、37:228 (1999))によって、ゴシポールのドッキング構造を得た。2つの計算を行った。まず、すべての結合部位ねじれ角を固定して維持し、次に側鎖ねじれ角を自由に変化できるようにした。30の最適解についての平均スコアリング関数は、側鎖を自由に回転させた場合わずかに低くなるにすぎなかった。モデルにおける側鎖の位置は最初の値から実質的に変化しなかった。(+) ゴシポールのスコアリング関数は(-) ゴシポールよりも劣っていたが、両方の立体異性体の全体的な配置は非常に類似していた。結果として得られた最適スコアリング構造のエネルギーを次いでSYBYL のルーチンDOCKを用いて、部位を固定しながら最小化した。DOCK最小化の後のリガンドのエネルギーはそのエネルギーの全最小値(global minimum of energy)から5 Kcal/molの範囲内であった。化合物の重ね合わせはSYBYLのルーチンMULTIFITにより得た。三次元構造を示すカラー図をプログラム SYBYLおよび MOLMOL (Koradi et al.、J. Mol. Graph.、14:29; 14:51 (1996))を用いて調製した。
【0107】
化合物の癌細胞生存に対する阻害効果
培養中の腫瘍細胞の生存度に対する本発明において研究している化合物の効果をXTT (Weislow et al.、J. Natl. Cancer Inst.、81:577 (1989)) アッセイにより、 MCF7および ZR75-1 細胞株を用いてモニターした。MCF7 細胞を10% 胎児ウシ血清、ペニシリン/ストレプトマイシン、さらに10-10 M インスリン、1 mM ピルビン酸ナトリウムおよびグルタミンを含むDMEMで培養した。ZR75-1 細胞を、10% 胎児ウシ血清、ペニシリン/ストレプトマイシン、さらにHEPES バッファー、1 mM ピルビン酸ナトリウムおよびグルタミンを含むRPMIで培養した。細胞をマイコプラズマによるコンタミネーションについて定期的に試験した。細胞を三連で最初の細胞密度1,000 細胞/ウェルにて播いた。ブランクウェルには細胞を入れなかった。ゴシポール、プルプロガリンおよび5D1を終濃度0、1、10および 100μMにて添加し、3日間インキュベートした。生細胞の相対数をXTT アッセイで判定した。簡単に説明すると、96-ウェルプレートに50 μlの以下の混合物を各ウェルに添加した:1 mg/ml の、0.025 mM PMS (フェナジンメトスルフェート)を含有するXTT (Weislow et al.、J. Natl. Cancer Inst.、81:577 (1989)) (2,3-ビス(2-メトキシ-4-ニトロ-5-スルホフェニル)-5-[(フェニルアミノ)カルボニル]-2H-テトラゾリウムヒドロキシド) (Polysciences、Washington、PA)。96-ウェルプレートをさらに4時間再びインキュベートしてXTT ホルマザンの生成を可能にした。次いで、各プレートの内容物を混合し、吸光度を波長450 nm (OD450)で測定した。正味のOD450をブランクウェルのOD450を差し引いた後に判定した。低継代 HeLa 細胞(継代数 10〜20)をpcDNA3-Bcl-xLまたは対照 pcDNA3 プラスミドにLipofectamine Plus 試薬(Invitrogen)を用いてトランスフェクトし、800 μg/mlのG418を含む培地で選択した。Bcl-xLのイムノブロット分析は(Krajanski 1996 -Cancer Res)に記載のように行った。 HeLa-トランスフェクタントを様々な用量のゴシポール、プルプロガリンおよびその誘導体(0、1、3、10 および100 μM)で処理した。
【0108】
化学物質
純粋なポリフェノールをSIGMA (ゴシポールおよびプルプロガリン)および/またはMicrosource Discovery Systems (プルプロガリン誘導体)から得た。対照化合物をChembridge Corp. (San Diego)から得た。ゴシポールは(+)および(-)異性体のラセミ体として試験した。化合物をDMSOに100 mM 濃度で溶解し、-20℃で保存した。NMR 分析を定期的に質的対照として化合物について行い、その後、さらに結合および置換アッセイのために希釈した。ゴシポールの反応性を、15N-標識化被験タンパク質 (XIAP のBIR3 ドメイン)を用いて試験した。1 mM ゴシポールおよび200μM 15N-標識化 BIR3を含む溶液を2時間インキュベートし、 [15N,1H]- 相関スペクトルを記録し、アポ-Bir3のスペクトルと比較した。スペクトルにおいて感知できる差は観察されなかった。
【0109】
【化3】
【表4】
プルプロガリン誘導体の構造活性相関(SAR)
【0110】
すべての刊行物、特許、および特許文献は、個々に引用により含まれているかのように参考として本明細書に含まれる。本発明は様々な特定の好ましい態様および技術に関して記載した。しかし、本発明の精神と枠内で多くの改変および修飾がなされうることを理解されたい。
【特許請求の範囲】
【請求項1】
癌細胞の生存度を低下させるのに有効な式(I)の化合物またはその医薬上許容される塩と、癌細胞とを接触させることを含む哺乳類における癌の治療方法;
【化1】
(I)
[式中:各R6、R8、R9およびR10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ;各R7は独立に水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項2】
各R6、R8、およびR9 基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項1の方法。
【請求項3】
各R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項1または2の方法。
【請求項4】
各R10基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである請求項1-3のいずれかの方法。
【請求項5】
各R6、R8、およびR9が-OC(O)CH3; 各R7がi-プロピル; および 各 R10が-CH3である請求項1-4のいずれかの方法。
【請求項6】
化合物がアポゴシポールのプロドラッグである請求項1-5のいずれかの方法。
【請求項7】
癌が、肺癌、乳癌、前立腺癌、結腸直腸癌、または白血病である請求項1-6のいずれかの方法。
【請求項8】
白血病が、急性リンパ球性白血病、慢性リンパ球性白血病、急性骨髄性白血病、または慢性骨髄性白血病である請求項7の方法。
【請求項9】
ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-)エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびその混合物からなる群から選択される化学感作物質を、抗癌剤と組み合わせて対象に投与することを含む、対象における癌の治療方法。
【請求項10】
アポゴシポール誘導体が式(I)の化合物またはその医薬上許容される塩である請求項9の方法:
【化2】
(I)
[式中: 各 R6、R8、R9 および R10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ; 各R7 は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項11】
各R6、R8、R9、および R10基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項10の方法。
【請求項12】
各 R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項10 または 11の方法。
【請求項13】
各 R6、R8、および R9が-OC(O)CH3; 各 R7がi-プロピル; および 各 R10が-CH3である請求項10-13のいずれかの方法。
【請求項14】
化学感作物質および抗癌剤が同時に投与される請求項9-13のいずれかの方法。
【請求項15】
化学感作物質が抗癌剤よりも先に投与される請求項9-13のいずれかの方法。
【請求項16】
抗癌剤が、フラボピリドール、アドリアマイシン、エトポシド、タキソール、シスプラチンまたはそれらの組み合わせである請求項10-15のいずれかの方法。
【請求項17】
プルプロガリン誘導体が5D1、1163、または1142である請求項9、または 14-16のいずれかの方法。
【請求項18】
癌が、肺癌、乳癌、前立腺癌、結腸直腸癌、または白血病である請求項9-17のいずれかの方法。
【請求項19】
白血病が、急性リンパ球性白血病、慢性リンパ球性白血病、急性骨髄性白血病、または慢性骨髄性白血病である請求項18の方法。
【請求項20】
標的細胞のアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導に有効な式(I)の化合物またはその医薬上許容される塩を、標的細胞と接触させることを含む、哺乳類におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導のための方法:
【化3】
(I)
[式中:
各 R6、R8、R9 および R10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ; 各 R7は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項21】
各 R6、R8、R9、および R10 基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項 20の方法。
【請求項22】
各 R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項 20の方法。
【請求項23】
各 R6、R8、および R9が-OC(O)CH3; 各 R7がi-プロピル; および各 R10が-CH3である請求項 20の方法。
【請求項24】
以下の工程を含むBcl-2 ファミリータンパク質の抗アポトーシス活性を阻害する物質の同定方法:
(a) Bcl-2 阻害物質または標識化Bcl-2阻害物質を同定する工程、ここで、該阻害物質はゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、 (-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、およびプルプロガリン誘導体からなる群から選択される;
(b)結合している阻害物質を候補物質と接触させる工程、ここで該候補物質はBcl-2 ファミリータンパク質の阻害が可能であると予測されるものである; および、
(c)該阻害物質のBcl-2 ファミリータンパク質からの解離を検出する工程、これによって、物質がBcl-2 ファミリータンパク質を阻害する物質であると同定される。
【請求項25】
Bcl-2 ファミリータンパク質がBcl-xL、Bcl-2、Mcl-1、Bcl-W、または Bcl-Bである請求項 24の方法。
【請求項26】
阻害物質が標識化されている請求項 24の方法。
【請求項27】
標識がフルオレセイン、フルオレセイン誘導体、クマリンまたはクマリン誘導体である請求項 25の方法。
【請求項28】
標識がフルオレセインである請求項27の方法。
【請求項29】
化合物が同位体で標識化されている請求項26の方法。
【請求項30】
同位体が13C、15N、19F、または 1Hである請求項29の方法。
【請求項31】
スペクトル技術が核磁気共鳴 (NMR) 結合アッセイを含む請求項 24の方法。
【請求項32】
スペクトル技術が蛍光偏光アッセイ (FPA) を含む請求項 24の方法。
【請求項1】
癌細胞の生存度を低下させるのに有効な式(I)の化合物またはその医薬上許容される塩と、癌細胞とを接触させることを含む哺乳類における癌の治療方法;
【化1】
(I)
[式中:各R6、R8、R9およびR10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ;各R7は独立に水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項2】
各R6、R8、およびR9 基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項1の方法。
【請求項3】
各R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項1または2の方法。
【請求項4】
各R10基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、またはBrである請求項1-3のいずれかの方法。
【請求項5】
各R6、R8、およびR9が-OC(O)CH3; 各R7がi-プロピル; および 各 R10が-CH3である請求項1-4のいずれかの方法。
【請求項6】
化合物がアポゴシポールのプロドラッグである請求項1-5のいずれかの方法。
【請求項7】
癌が、肺癌、乳癌、前立腺癌、結腸直腸癌、または白血病である請求項1-6のいずれかの方法。
【請求項8】
白血病が、急性リンパ球性白血病、慢性リンパ球性白血病、急性骨髄性白血病、または慢性骨髄性白血病である請求項7の方法。
【請求項9】
ゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート(EGCG)、(-)カテキン-3-ガラート (CG)、(-)エピカテキン-3-ガラート (ECG)、プルプロガリン誘導体、およびその混合物からなる群から選択される化学感作物質を、抗癌剤と組み合わせて対象に投与することを含む、対象における癌の治療方法。
【請求項10】
アポゴシポール誘導体が式(I)の化合物またはその医薬上許容される塩である請求項9の方法:
【化2】
(I)
[式中: 各 R6、R8、R9 および R10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ; 各R7 は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項11】
各R6、R8、R9、および R10基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項10の方法。
【請求項12】
各 R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項10 または 11の方法。
【請求項13】
各 R6、R8、および R9が-OC(O)CH3; 各 R7がi-プロピル; および 各 R10が-CH3である請求項10-13のいずれかの方法。
【請求項14】
化学感作物質および抗癌剤が同時に投与される請求項9-13のいずれかの方法。
【請求項15】
化学感作物質が抗癌剤よりも先に投与される請求項9-13のいずれかの方法。
【請求項16】
抗癌剤が、フラボピリドール、アドリアマイシン、エトポシド、タキソール、シスプラチンまたはそれらの組み合わせである請求項10-15のいずれかの方法。
【請求項17】
プルプロガリン誘導体が5D1、1163、または1142である請求項9、または 14-16のいずれかの方法。
【請求項18】
癌が、肺癌、乳癌、前立腺癌、結腸直腸癌、または白血病である請求項9-17のいずれかの方法。
【請求項19】
白血病が、急性リンパ球性白血病、慢性リンパ球性白血病、急性骨髄性白血病、または慢性骨髄性白血病である請求項18の方法。
【請求項20】
標的細胞のアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導に有効な式(I)の化合物またはその医薬上許容される塩を、標的細胞と接触させることを含む、哺乳類におけるアポトーシスの誘導、カスパーゼ活性の調節、または細胞死の誘導のための方法:
【化3】
(I)
[式中:
各 R6、R8、R9 および R10は独立に、水素、ヒドロキシル、-(C1-C6)アルキル、-O(C1-C6)アルキル、-(C1-C6)アルキルハロ、-OC(O)(C1-C6)アルキル、またはハロ; 各 R7は独立に、水素、-(C3-C8)シクロアルキル、-(C6-C10)アリール、または-(C1-C6)アルキル(C6-C10)アリール]。
【請求項21】
各 R6、R8、R9、および R10 基が独立に、水素、-OH、-OCH3、-CF3、-CH3、-OC2H5、-OC(O)CH3、F、Cl、または Brである請求項 20の方法。
【請求項22】
各 R7基が独立に、水素、-C2H5; -i-Pr、n-Pr、n-Bu、t-Bu、i-Bu、s-Bu、またはシクロヘキシルである請求項 20の方法。
【請求項23】
各 R6、R8、および R9が-OC(O)CH3; 各 R7がi-プロピル; および各 R10が-CH3である請求項 20の方法。
【請求項24】
以下の工程を含むBcl-2 ファミリータンパク質の抗アポトーシス活性を阻害する物質の同定方法:
(a) Bcl-2 阻害物質または標識化Bcl-2阻害物質を同定する工程、ここで、該阻害物質はゴシポール、アポゴシポール、アポゴシポール誘導体、テアフラビン、テアフラビン-3’-ガラート、テアフラバニン、(-) ガロカテキン-3-ガラート (GCG)、(-) エピガロカテキン-3-ガラート (EGCG)、 (-)カテキン-3-ガラート (CG)、(-) エピカテキン-3-ガラート (ECG)、およびプルプロガリン誘導体からなる群から選択される;
(b)結合している阻害物質を候補物質と接触させる工程、ここで該候補物質はBcl-2 ファミリータンパク質の阻害が可能であると予測されるものである; および、
(c)該阻害物質のBcl-2 ファミリータンパク質からの解離を検出する工程、これによって、物質がBcl-2 ファミリータンパク質を阻害する物質であると同定される。
【請求項25】
Bcl-2 ファミリータンパク質がBcl-xL、Bcl-2、Mcl-1、Bcl-W、または Bcl-Bである請求項 24の方法。
【請求項26】
阻害物質が標識化されている請求項 24の方法。
【請求項27】
標識がフルオレセイン、フルオレセイン誘導体、クマリンまたはクマリン誘導体である請求項 25の方法。
【請求項28】
標識がフルオレセインである請求項27の方法。
【請求項29】
化合物が同位体で標識化されている請求項26の方法。
【請求項30】
同位体が13C、15N、19F、または 1Hである請求項29の方法。
【請求項31】
スペクトル技術が核磁気共鳴 (NMR) 結合アッセイを含む請求項 24の方法。
【請求項32】
スペクトル技術が蛍光偏光アッセイ (FPA) を含む請求項 24の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】

【図9】
【図10】
【図11】
【図12】
【図13】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公開番号】特開2012−25767(P2012−25767A)
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【外国語出願】
【出願番号】特願2011−215077(P2011−215077)
【出願日】平成23年9月29日(2011.9.29)
【分割の表示】特願2006−517702(P2006−517702)の分割
【原出願日】平成16年6月25日(2004.6.25)
【出願人】(500114586)ザ バーナム インスティチュート (8)
【Fターム(参考)】
【公開日】平成24年2月9日(2012.2.9)
【国際特許分類】
【出願番号】特願2011−215077(P2011−215077)
【出願日】平成23年9月29日(2011.9.29)
【分割の表示】特願2006−517702(P2006−517702)の分割
【原出願日】平成16年6月25日(2004.6.25)
【出願人】(500114586)ザ バーナム インスティチュート (8)
【Fターム(参考)】
[ Back to top ]