
置換ピラゾロピリミジン類の結晶形
本発明は、ピラゾロ−ピリミジン類と共結晶形成剤の新規な共結晶に関し、この共結晶形成剤は有機カルボン酸、好ましくはゲンチシン酸、コハク酸およびキシナホ酸の群より選択される。
【化1】

【化1】
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規の結晶形態、特に置換ピラゾロ−ピリミジン類と有機酸との共結晶に関する。特に興味深いのは、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(以下において、時々「化合物A」と呼ばれる)、および特に該化合物のR異性体、いわゆる6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと有機カルボン酸との共結晶である。
【0002】
さらに、本発明は置換ピラゾロ−ピリミジン類および特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、有機モノカルボン酸およびジカルボン酸との共結晶の調製方法を提供する。
【背景技術】
【0003】
化合物6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンのような典型的な置換ピラゾロ−ピリミジン類(その異性体を含む)、およびそれらの調製方法は、特許文献1に記載されている。この文書は、ピラゾロ−ピリミジン類、特に化合物Aが効果のあるmGluR5モジュレーターであり、急性および慢性の神経障害、特にグルタミン酸誘導性茂樹に関与するCNS(中枢神経系)障害の予防および治療に有用である。
【0004】
化合物AのR−エナンチオマーの化学構造は以下に示される:
【化1】
【0005】
医薬組成物用の原薬(active pharmaceutical ingredients(API))は、種々の異なる形態に調製され得ることが知られている。ほとんどの薬物化合物または原薬(例えば、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン)は、固体として投与される。多くの固体APIは、1つまたはいくつかの結晶形態で存在している。しばしば、このAPIは、それ自身の上には結晶化しないか、さもなくば不利益な物理的および生物薬剤学的特性を呈す結晶形態中に結晶化しない。より良好な医薬品製品を提供するために、代替の結晶形態を探す必要がある。特定の形態の良い例としては、多形体、塩、溶媒和物および水和物が挙げられる。これらの確立された結晶APIの改変(modification)に加え、APIを含む結晶分子複合体としてまた記載され得る医薬品共結晶は、化学者の注目を引いてきた。薬学的有効成分の特定の物理学的形態の選択は、溶解度、溶解速度、吸湿性、物理安定性および化学安定性のような物理特性を最適化するための戦略的なチャンスを示している。
【0006】
いくつかの薬学的に有効な化合物は多型を示す。いくつかの化合物は、10より多い異なる結晶形態変位で存在している。多型は、所定の化合物の固体形態であり、固体状態にある化合物の分子の少なくとも2つの異なる配置の可能性に起因している。この多形の形成は、しばしば結晶化の状態に依存している。所定の化合物の異なる多型は、独特な一連の物理化学的特性を示す。不利な点として、化合物の新規の多型形態は、通常いくつかの例に限定される。
【0007】
薬学的に有効な成分の新規の結晶形態を得るための別の公知のアプローチは、水和物および溶媒和物の形成である。しばしば、結晶化の間に溶媒は結合され結晶構造の一部として取り込まれる。多くの溶媒は生物学的に毒性であり、従って溶媒を含有する結晶はしばしば薬物化合物の固体形態の開発においては避けられる。しかしながら、水和物(これにおいては結晶構造は水を取り込む)は、APIの通常形態であり、医薬品製品において周知である。多くの目的の医薬品分子が、水和物を形成することができる。しかしながら、水和物はしばしば不安定であり、温度、圧力または相対湿度といった貯蔵状態の変化の結果として無水結晶に変化する。この水和物から無水物への変換(例えば貯蔵の間、または製剤工程の間)は、薬物製品の品質を急激に損なう可能性がある。
【0008】
APIの塩の形成は、原薬の特性を変更するための別の公知のプロ−地である。API間の酸−塩基反応として記載することができる塩形成は、塩基性および/または酸性の官能基、および酸性または塩基性の物質を示す。薬物化合物の塩は、その結晶格子中にAPI分子のイオン形態を含んでいる。多くの医薬品化合物は酸性または塩基性の機能を呈しているため、塩形成は、APIの新規の結晶形態を得るための魅力的な方法である。塩の広範囲にわたる用途は、多くの市販されている薬物化合物の結晶塩によって証明されている。
【0009】
共結晶はずいぶん以前に発見されたが、医薬品共結晶は未だなお、特に置換ピラゾロ−ピリミジン類のような複素環式薬物化合物についての開発および最適化の興味深いゴールである。本発明に従う共結晶は、非共有相互作用(しばしば水素結合を含む)を通して結晶格子内で一緒に結合される2つまたはそれ以上の中性分子の結晶複合体として理解することができる。通常、APIとさらなる分子化合物(共結晶形成剤またはカウンター分子)の間のプロトン移動は起こらない。本発明に従う共結晶化技術の適用は、塩形成と比較していくつかの利点を提供する。原則的には、従来塩形成の能力が限定されているかまたはその能力がないために物理特性の最適化という点で高いリスクを示すと考えられている、イオン化力の弱い化合物や非イオン化化合物を含むあらゆるタイプの分子が共結晶を形成することができる。
【0010】
2002年に、その各々が水素結合アクセプターとして作用する、6つの異なるカウンター分子を持つ鎮痛薬パラセタモール(paracetamol(アセトアミノフェン))が販売された。その後すぐにいくつかの水素結合アクセプターを持つ薬物化合物の共結晶、イブプロフェン、フルルビプロフェンおよびアスピリンが記載された。これらの例は、通常の水素結合特徴を持つ一連の共結晶は得ることができることを示している。融点データだけではなく、これらの報告書はこれらの共結晶の機能特性および薬理学特性に目を向けることなしに特に構造の特徴に焦点を置いている。
【0011】
本発明に従う共結晶のさらなる利点は、ソルトスクリーン(salt screen)においては、わずかな数の酸性または塩基性対イオンのみだけが考慮されるにもかかわらず、ピラゾロ−ピリミジン類と共結晶を調製するのに用いられ得る共結晶を形成する可能性のある薬剤(共結晶形成剤またはカウンター分子とも呼ばれる)はいくつも存在するという点である。可能性のある薬剤は、米国食品医薬品局による「一般的に安全であると認められる」物質のリストから、実験用に選択することができる。共結晶の高められた範囲は、薬物の望ましい物理特性プロフィールを達成するより大きい可能性の示唆において有利であるが、スクリーニングの労力という点においてはかなりの困難も示される。共結晶のスクリーニング、特にハイスループットスクリーニング法(改良された合理的な共結晶デザインおよびより効果的な共結晶スクリーニングプロトコルを含む)は、新規の結晶形態の開発において重要なツールである。共結晶の調製のためのいくつかの一般的な方法は文献に記載されている。特定の物理特性を高めるために共結晶を用いる公知の例が存在している。共結晶の調製方法には、従来の結晶化技術および固体の粉砕のようなより特別な方法も含まれる。
【0012】
共結晶の形成は、調査に入る前に研究されており、種々の重要な研究が共結晶デザインの理解を目的としている。初期の研究においては、良好なプロトンドナーとアクセプターが水素結合に用いられること、および最適なドナーは典型的には所定の結晶構造において最良のアクセプターと対となることを含む、いくつかの「水素結合ルール」が提唱された。水素結合ルールと幾何解析との組み合わせて用いることが、多くの新規の超分子構造の合成における合理的な共結晶デザインを実行するために行われた。
【0013】
特許文献1に記載されるような多くのピラゾロ−ピリミジン類、および特に化合物Aは、塩基性官能基を示している。化合物Aは、その低い塩基性能に起因して、約−1.97(ピラゾロ[1,5−a]ピリミジンと相関して算出した)という低いpKa値を示している。さらに、この化合物Aは水または水性溶媒においては溶解性が乏しい(10μg/mL未満)。この物理化学特性のおかげで、化合物Aはいくつかの不利な医薬品特性(例えば、完全でないバイオアベイラビリティー)を示している。上記のように、化合物Aはパートナー間のpKa値の差異が十分大きくないために、鉱物酸または不安定な塩と一緒に簡単に塩を形成することができない。従って、塩の形成は化合物Aの医薬品特性を向上するために困難な方法である。
【0014】
改良された結晶形態である(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンについては、増強された溶解度および融解特性ならびに良好な貯蔵安定性特性を示す医薬品組成物の調製のために高い必要性がある。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】WO2008/015269
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、改良された結晶形態、特にピラゾロ−ピリミジン類、および特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、適切な共結晶形成剤との共結晶を提供することである。この新規の共結晶は、好ましくは改良された薬学的特性および良好な貯蔵安定性(例えば、水へのより高い溶解性、全くないかまたはわずかな吸湿性)を示す化合物Aである。
【課題を解決するための手段】
【0017】
驚いたことに、薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび特にこのR−異性体は、特定の有機カルボン酸と安定な共結晶を形成することが見出された。さらに、多くの有機カルボン酸およびアミノ酸は、通常の結晶化手順を用いて6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびその光学異性体と共結晶を形成しないことが明らかとなった。いくつかの例は、安息香酸、リンゴ酸(1−ヒドロキシブタン二酸)、マンデル酸(2−ヒドロキシ−2−フェニル酢酸)、D/L酒石酸(2,3−ジヒドロキシブタン二酸)、バニリン酸(4−ヒドロキシ−3−メトキシ安息香酸)、またはL−アスパラギン酸(2−アミノブタン二酸)である。
【0018】
本発明は、ピラゾロ−ピリミジン類、特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、および特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、以下に記載されるような少なくとも1種の共結晶形成剤、好ましくは1種、2種または3種の共結晶形成剤との共結晶との共結晶に関する。
【0019】
本発明は、ピラゾロ−ピリミジン類、特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、および特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、共結晶形成剤との共結晶に関し、該共結晶形成剤が、一般式(I):
【化2】
[式中、Rは以下:
a)
【化3】
(式中、nは1、2、3または4である)
または
b)
【化4】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸である。
【0020】
好ましい実施態様において、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、該共結晶形成剤が、式(I):
【化5】
[式中、Rは以下:
a)
【化6】
(式中、nは2または3である)
または
b)
【化7】
(式中、
R1およびR2は、互いに独立して水素またはヒドロキシルであり、
R3およびR4は、水素であるかまたは、
R3およびR4は、それらを保持する炭素原子と一緒になって、非置換の芳香族6員環を形成してもよい)
を表わす]
のカルボン酸である。
【0021】
好ましくは、このカルボン酸共結晶形成剤は、少なくとも2つの水素供与基を含み得、その1つはヒドロキシル基から選択され、またその1つはカルボキシル基から選択され、好ましい強力な水素結合をした2分子の環モチーフの形成が可能である。
【0022】
さらに、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンは、コハク酸、ゲンチシン酸、およびキシナホ酸(xinafoic acid)を含む群より選択される少なくとも1種のカルボン酸と安定な共結晶を形成することが明らかとなった。さらに、コハク酸、ゲンチシン酸、およびキシナホ酸の共結晶は、例えばそれらは吸湿性ではないかまたは化合物A自体よりも吸湿性が少ないという特に有利な特性を有していることも明らかとなった。上述の全ての共結晶は、遊離型の薬物化合物に比べ、より良好な水溶解性を示した。
【0023】
本発明における用語「共結晶」は、室温(20〜25℃)である2個以上の中性分子化合物が、非共有相互作用(しばしば、水素結合、πスタッキング、ゲスト−ホスト錯体、ファンデルワールス相互作用などを含む)によって結晶格子中に一緒に結合されている結晶複合体を意味している。特に、該非共有相互作用は、水素結合を含む。水素結合は、例えば、異なる分子間構造(例えば二量体、線状鎖、または環状構造)をもたらし得る。これらの共結晶の各々は、構造(例えば、PXRDパターンで特徴づけられる)、融点、融解熱のような独特の物理特性を示し、とりわけそれにより特徴づけられ得る。
【0024】
本発明に従う共結晶は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびその化合物におそらく水素結合している共結晶形成剤を含む。上述したような他の相互作用はまた、本発明に従う共結晶の形成にも役割を果たしている。
【0025】
共結晶形成剤をさらに含まない化合物Aの塩および溶媒和物は、本発明に従う共結晶として考慮されない。しかしながら、本発明に従う共結晶は、結晶格子中に1種またはそれ以上の溶媒和物分子を含んでいてもよい。従って、室温で液体である化合物をさらに含む共結晶の溶媒和物は、本発明のより広義の範囲に含まれる。本発明に従う共結晶はまた、化合物Aの塩と共結晶形成剤との共結晶であってもよいが、化合物Aおよび共結晶形成剤は、好ましくは水素結合を介して一緒に構築または結合される。この共結晶形成剤は、化合物Aに直接結合していてもよいし、または化合物Aに結合しているさらなる分子(例えば溶媒和物分子)に結合していてもよい。上に概説したように、本発明に関する共結晶は、典型的な塩および溶媒和物/水和物の意味からは区別され得る。
【0026】
本発明に記載される(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの新規の共結晶は、pKa値により従来の塩としては考えられない。さらに、実験データにより、本発明に従う結晶化合物が共結晶であることが確認されている。
【0027】
本発明に記載される共結晶の有利な特性は、遊離型の化合物Aと比較して、例えば水への良好な溶解性、より高い溶解速度、低いかまたは全くない吸湿特性、および良好な貯蔵安定性である。
【0028】
本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと少なくとも1種の共結晶形成剤との共結晶に関し、該共結晶形成剤は、好ましくはコハク酸(ブタン二酸)、ゲンチシン酸(2,5−ジヒドロキシ安息香酸)、およびキシナホ酸(1−ヒドロキシ−2−ナフトエ酸)からなる群より選択されるカルボン酸である。
【0029】
本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、該共結晶形成剤は、好ましくはコハク酸(ブタン二酸)、ゲンチシン酸(2,5−ジヒドロキシ安息香酸)、およびキシナホ酸(1−ヒドロキシ−2−ナフトエ酸)からなる群より選択されるカルボン酸である。
【0030】
特に、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、複素環(化合物A):共結晶形成剤のモル比は、1:0.1〜1:10の範囲、好ましくは1:1〜1:10の範囲、好ましくは1:1〜1:5の範囲、より好ましくは、約1:1である。この共結晶は、好ましくは結晶性共結晶である。
【0031】
さらなる局面において、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶を提供し、9.3、16.0、20.0、22.9、および26.0°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも2つ、好ましくは少なくとも3つ、より好ましくは少なくとも4つ、さらにより好ましくは少なくとも5つの粉末X線回折(PXRD)ピークによって特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、図1に実質的に従うPXRDパターンによって特徴づけられ得る。
【0032】
さらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、DSC(示差走査熱量測定)で約156.9℃の融解ピークを有している。さらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、実質的に図2に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンダイアグラムによって特徴づけられる。
【0033】
本発明はまた、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶を提供し、この共結晶が、6.0、7.0、14.0、17.6、21.0、23.4、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも4つの粉末X線回折(PXRD)ピークにより特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図3に従うPXRDパターンで特徴づけられ得る。
【0034】
本発明はまた、ゲンチシン酸共結晶の第2の多型を提供する。従って、本発明は(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶に関し、この共結晶が、6.9、12.6、21.2、および27.5°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、より好ましくは4つの粉末X線回折(PXRD)ピークの選択によって特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図4に従うPXRDパターンによって特徴づけられる。
【0035】
別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、DSC(示差走査熱量測定)により約147.4℃の融解ピークで特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(好ましくは2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図5に従うDSC(示差走査熱量測定)によって特徴づけられ得る。
【0036】
本発明はまた、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶を提供し、この共結晶が、3.9、11.6、18.1、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、さらにより好ましくは4つの粉末X線回折(PXRD)ピークによって特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、実質的に図6に従うPXRDパターンによって特徴づけられる。
【0037】
本発明のさらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、DSC(示差走査熱量測定)によって約139.2℃の融解ピークで特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、実質的に図7に従うDSC(示差走査熱量測定)によって特徴づけられ得る。
【0038】
各共結晶は、1つまたはそれ以上の上述の物理特性(PXRDピーク、DSCピーク)によって特徴づけられ得る。従って、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤(好ましくはゲンチシン酸、コハク酸、キシナホ酸より選択される)との共結晶に関し、この共結晶は、1つまたはそれ以上の上述の物理データによって特徴付けられる。
【0039】
本発明はさらに、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法に関し、該共結晶形成剤は、一般式(I):
【化8】
[式中、Rは以下:
a)
【化9】
(式中、nは1、2、3または4である)
または
b)
【化10】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸であり、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、スラリーの状態で分散させる工程、
を包含する。
【0040】
本発明はさらに、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法に関し、該共結晶形成剤が、ゲンチシン酸、コハク酸、およびキシナホ酸からなる群より選択されるカルボン酸であり、
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、スラリーの状態で分散させる工程、
を包含する。
【0041】
好ましい実施態様において、化合物Aと、コハク酸およびキシナホ酸より選択される共結晶形成剤との共結晶の調製方法は、相平衡工程c)を含む。
【0042】
本発明の実施態様において、溶解工程a)は、室温(20〜25℃)および通常圧力(1013.25hPa)下で、1〜60分間実施される。別の好ましい実施態様において、この溶解工程a)は、20℃〜100℃の範囲で温度を上げられた状態で実施される。
【0043】
工程a)は、最初に溶媒S1に化合物Aを溶解し、その後その溶液に共結晶形成剤を加えることによって実施される。さらに、別の実施態様において、この化合物Aおよび共結晶形成剤は、固体として最初に混合され、その後溶媒S1に溶解させてもよい。さらなる実施態様として、工程a)は、溶媒S1に溶解させた化合物Aの溶液と溶媒S1に溶解させた共結晶形成剤の溶液とを混合することにより実施することができ、ここで化合物Aと共結晶形成剤を溶解させるための溶媒は、異なっていても同じでもよい。化合物Aと共結晶形成剤を溶解させるための溶媒は等しいことが好ましい。
【0044】
好ましい実施態様において、蒸発(工程b)は、室温(20〜25℃)および通常圧力(1013.25hPa)下で実施される。しばしば、この工程b)は、空気または窒素流下で、場合によりフロー制御をおこないながら実施される。場合により、溶媒S1の蒸発(工程b))は、減圧下で実施される。
【0045】
相平衡工程c)は、しばしば室温(20〜25℃)下で実施され、別の実施態様において、工程c)は、0〜100℃の範囲に上げられたまたは下げられた温度下で実施される。
【0046】
本発明の1実施態様において、カルボン酸共結晶形成剤および化合物Aは、0.1〜10の範囲のモル比で、好ましくは約1:1のモル比で本発明に従う方法に用いられる。さらなる実施態様において、このカルボン酸共結晶形成剤は、化合物Aに対して1.1〜10のモル過多で用いられる。本発明のさらなる実施態様において、カルボン酸共結晶形成剤は、化合物Aに対して0.1〜0.95のモル比で用いられる。好ましくは、コハク酸は、化合物Aに対して1〜7.5の範囲、より好ましくは約1.2:1のモル比で用いられる。
【0047】
薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、およびカルボン酸共結晶形成剤は、しばしば溶媒S1中に等モル比で溶解される(工程a)。
【0048】
本発明は、上記のような共結晶に関し、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、とカルボン酸共結晶形成剤のモル比は、1:0.1〜1:10の範囲、好ましくは1:1〜1:10の範囲、好ましくは1:1〜1:5の範囲である。より好ましい実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとカルボン酸共結晶形成剤のモル比は、1H−NMR分光法で測定した場合1:1である。
【0049】
好ましくは、本発明に従う共結晶は、化合物Aと共結晶形成剤を溶解し、その後上記のようにその溶媒を蒸発させることによって調製される。別の実施態様において、この共結晶は、他の通常な結晶化手順を用いて調製され得る。例えば、化合物Aは、溶液または固体状態で温度勾配を用いてカルボン酸共結晶形成剤と共結晶化され得る。1つの例はハンギングドロップ拡散法であり、これは少量の共結晶の調製に用いられる方法である。
【0050】
溶媒S1および場合によりS2は、好ましくは以下からなる群より選択される少なくとも1つの有機溶媒である:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)およびN−メチルピロリドン(NMP)。好ましくは、この有機溶媒S1は、アセトン、エタノール、酢酸エチル、テトラヒドロフラン、およびイソプロピルエーテル(IPE)から、より好ましくはアセトン、イソプロピルエーテル(IPE)および酢酸エチルから選択される。
【0051】
溶媒S1は、さらに上述の溶媒の2つ、3つまたはそれ以上の混合物であってもよい。この溶媒はしばしば、有機溶媒(上記のもの)と水の混合物である。典型的な例は、以下の混合物である:エタノール:水(1:1)およびテトラヒドロフラン:水(1:1)。特に、選択された溶媒への共結晶形成剤の溶解度が低い場合、上記のような溶媒混合物が用いられる。
【0052】
溶媒S2は、好ましくは以下からなる群の少なくとも1つの有機または無期溶媒から選択される:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)、およびN−メチルピロリドン(NMP)。好ましくは、溶媒S2は、tert−ブチル−メチルエーテル(TBME)、1−プロパノール、2−プロパノール、トルエン、水、およびイソプロピルエーテル(IPE)から、より好ましくはイソプロピルエーテル(IPE)および2−プロパノールから選択される。
【0053】
溶媒S2は、さらに上述の溶媒の2つ、3つまたはそれ以上の混合物であってもよい。しばしば、この溶媒は、例えばエタノール:水(1:1)混合物およびテトラヒドロフラン:水(1:1)混合物のような有機溶媒と水との混合物である。
【0054】
本発明の好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶の調製方法に関し、ここで溶媒S1は1−プロパノールであり、溶媒S2は、イソプロピルエーテル(IPE)および2−プロパノールから選択される少なくとも1つの溶媒である。
【0055】
好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法に関し、溶媒S1は、アセトンおよびイソプロピルエーテル(IPE)から選択される少なくとも1つの溶媒である。
【0056】
好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)との共結晶の調製方法に関し、溶媒S1は酢酸エチルである。
【0057】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびコハク酸を、2−プロパノール中に1:1〜1:10の範囲のモル比で溶解する工程、
b)好ましくは空気または窒素流下、室温下で、2−プロパノールを蒸発させる工程、
c)工程b)で得られた残留物を、イソプロピルエーテル(IPE)および2−プロパノールから選択される少なくとも1つの溶媒中に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、攪拌しながら分散させる工程(相平衡)。
【0058】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびゲンチシン酸(2,5−ジヒドロキシ安息香酸)を、約1:1〜1:1.25の範囲、好ましくは約1:1.2〜1:1.25の範囲のモル比で、アセトンおよびイソプロピルエーテル(IPE)から選択される溶媒中に溶解する工程、
b)好ましくは窒素流または空気中で室温にてアセトンを蒸発させる工程。
【0059】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびゲンチシン酸(2,5−ジヒドロキシ安息香酸)を、約1:1〜1:1.2.25の範囲のモル比、好ましくは約1:1のモル比でアセトン中に溶解させる工程、
b)好ましくは窒素流下で室温にてアセトンを蒸発させる工程。
【0060】
1実施態様において、上記の工程a)は、最初に化合物Aをアセトンに溶解させ、その後ゲンチシン酸を加えることにより実施される。別の実施態様において、工程b)は、アセトンに溶解させた化合物Aの溶液とアセトンに溶解させたゲンチシン酸の溶液とを混合することによって実施される。
【0061】
本発明の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)との共結晶の焼成方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R )−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)を、約1:1のモル比で酢酸エチルに溶解させる工程、
b)好ましくは窒素流下で室温にて酢酸エチルを蒸発させる工程。
【0062】
さらに、本発明は(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと少なくとも1種の上記共結晶形成剤との共結晶の調製方法に関し、好ましくは1種、2種または3種の共結晶形成剤が上記のような方法に適用される。
【0063】
本発明はまた、本発明に従う少なくとも1種、好ましくは1種、2種または3種の共結晶を含む医薬組成物にも関する。
【0064】
本発明はまた、1種またはそれ以上の薬学的に許容可能な賦形剤と一緒に、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、または薬学的に許容可能なその誘導体もしくはアナログの共結晶および本発明に従う前出の共結晶を含む医薬組成物に関する。このタイプの医薬組成物、賦形剤および調製は、WO2008/015270およびWO2008/015269により詳細に記載されている。
【0065】
本発明はまた、異常なグルタミン酸神経伝達に関連している状態または疾患、好ましくは以下に記載するような状態または疾患の治療および/または予防に用いるための、本発明に記載されるような(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶にも関する。
【0066】
好ましくは、本発明は、アルツハイマー病、統合失調症の陽性および/もしくは陰性症状、認知障害の状態または疾患の治療および/または予防に用いるため、または認知向上および神経保護に用いるための、本発明に記載されるような(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関する。
【0067】
用語「アナログ」または「誘導体」は、本明細書において慣用の製薬的意味で用いられ、参照分子に構造的に類似している分子であるが、参照分子の1つまたは複数の特定の置換基を代替置換基によって置換し、そのことによって、参照分子に構造的に類似した分子を作り出すために標的化され、制御された様式で修飾されている分子のことである。アナログを合成およびスクリーニングして、改善された、または偏りを持たせた特徴(特定の標的受容体タイプにおけるより高い効力および/または選択性、脳血管関門を通過するより高い能力、より少ない副作用など)を有し得る、既知化合物をわずかに修飾した変型を同定することは、製薬化学においてよく知られた薬剤設計法である。また、当業者に知られている方法を用いて、改善された治療有効性、すなわち、特定の標的受容体タイプにおけるより高い効力および/または選択性、脳血管関門を通過するより高い能力またはより低い能力(例えば、より高い、またはより低い脳血管関門通過率)、より少ない副作用などを有する本発明の化合物のアナログおよび誘導体を作り出すことができる。
【0068】
本発明の組成物に関連して用いられる語句「薬学的に許容できる」とは、哺乳動物(例えばヒト)に投与した際、生理学的に許容性であり、一般に有害な反応を生じさせない分子全体およびこのような組成物の他の成分のことである。本明細書に用いられる用語「薬学的に許容できる」は、哺乳動物における使用が、特にヒトにおける使用が、連邦政府または州政府の監督機関によって承認されているか、または米国薬局方もしくは他の一般に認められた薬局方に記載されていることを意味することが好ましい。
【0069】
本発明に従う共結晶は、生きている動物の身体、特にヒトの種々の障害の治療および/または予防における適用を見出し得る。共結晶はまた、特定の状態が存在する必要はないが、特定の生理学的パラメータがインスタント化合物(instant compound)の投与を介して向上され得る(認知エンハンスメントを含む)、生きている動物の身体、特にヒトにおける徴候の治療に用途を見出す。
【0070】
生きている動物の体に存在する選択された病気の進行の阻害または緩和のための、本発明の共結晶によるその治療方法は、緩和されることが所望される特定の病気の緩和において有効である選択された用量を用いて、任意の通常受け入れられる医薬品経路により、以前に述べたように可能である。選択された病気または状態(特に病気または状態が、グループImGluRモジュレーターによる治療に影響を受けやすいもの)の進行の阻害または緩和のための、生きている動物の治療のための医薬の製造における本発明の共結晶の使用は、薬学的に受容可能な希釈剤、賦形剤またはキャリアと一緒に、有効量の本発明の共結晶を投与する工程を包含する通常の方法で実施される。
【0071】
それぞれの医薬組成物は、共結晶成分と、1種またはそれ以上の適切かつ薬学的に受容可能な賦形剤とを組み合わせることにより調製され得る。これらの組成物は経口、経皮、非経口、肺、直腸、経粘膜および鼻孔経路のような異なる経路を介して適用され得る。医薬品投薬形態は、動物および好ましくはヒトへの用途のための医薬を製造するために、例えば、粉末、顆粒、錠剤、フィルムコーティングされた錠剤、放出調整錠剤、ハードカプセル、ソフトカプセル、溶液、懸濁液、エマルジョン、クリーム、軟膏、ゲル、経皮パッチ、エアロゾル製剤、吸入およびマイクロまたはナノ粒子ベースの製剤であり得る。適切な本発明の製剤は、さらに溶媒に溶解させた共結晶の懸濁液である。
【0072】
本発明の医薬組成物において、1日の投与用の投薬単位あたり、本発明に従う共結晶は、例えば、0.1〜4000mg、好ましくは1〜2000mgの前記化合物をが乳する投薬単位として処方される。本発明の全ての局面について、特に医薬の局面においては、化合物または組成物の投与は、投薬レジメを有しており、治療している医師によって最終的に決定され、用いられる化合物、動物タイプ、性別、年齢、体重、症状の重篤度、投与方法、副作用反応および/または他の反徴候のような因子を考慮する。
【0073】
本発明に従う生理学的に許容可能な化合物は、通常例えば0.01mg/kg(処置される哺乳類の体重1kgあたりのmg)〜100mg/kg、好ましくは0.1mg/kg〜75mg/kgの経口用量である。
【0074】
さらに、本発明は、ヒトを含む動物における神経保護を提供するための医薬としての、本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(および/またはそのR−エナンチオマー)の共結晶を含む組成物の使用に関する。
【0075】
さらに、本発明は、異常なグルタミン酸神経伝達に関連している状態の治療のため、またはその状態において治療の利益をもたらすmGluR5の調節のための、本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(および/またはそのR−エナンチオマー)の共結晶の使用に関する。
【0076】
特に、本発明は以下から選択される状態または疾患の予防および/または治療のための医薬の調製のための、本発明に従う共結晶の使用を取り扱う:
アルツハイマー病、クロイツフェルト−ヤコブ症候群/病、ウシの海綿状脳症(BSE)、プリオン関連の感染症、ミトコンドリア機能障害が関与する疾患、β−アミロイドおよび/またはタウオパシーが関与する疾患、ダウン症候群、肝性脳症、ハンチントン病、運動ニューロン疾患、筋萎縮側索硬化症(ALS)、オリーブ橋小脳萎縮、術後認知機能障害(POCD)、全身性エリテマトーデス、全身性硬化症、シェーグレン症候群、神経セロイドリポフスチノーシス、神経変性小脳性運動失調、パーキンソン病、パーキンソン病の認知症、軽度認知障害、軽度認知障害の種々の形態の認知欠陥、認知症の種々の形態の認知欠陥、ボクサー認知症、血管性および前頭葉型認知症、認知障害、学習障害、眼の損傷、眼の疾患、眼の障害、緑内障、網膜症、黄斑変性、頭部もしくは脳または脊髄の損傷、頭部もしくは脳または脊髄の外傷、外傷、低血糖、低酸素症、周産期低酸素症、虚血、心停止もしくは脳卒中もしくはバイパス手術もしくは移植から生じる虚血、痙攣、てんかん様痙攣、てんかん、側頭葉てんかん、間代性筋痙攣、内耳傷害、耳鳴における内耳傷害、耳鳴、音−または薬物−誘発性内耳傷害、音−または薬物−誘発性耳鳴、L−ドーパ−誘発性ジスキネジア、パーキンソン病の治療におけるL−ドーパ誘発性ジスキネジア、ジスキネジア、ハンチントン病におけるジスキネジア、薬物誘発性ジスキネジア、神経遮断薬−誘発性ジスキネジア、ハロペリドール−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、舞踏病、ハンチントン舞踏病、アテトーシス、ジストニア、常同症、バリズム、遅発性ジスキネジア、チック障害、痙性斜頚、眼瞼痙攣、局所性および全身性ジストニア、眼振、遺伝性小脳性運動失調、大脳皮質基底核変性症、振せん、本態性振せん、乱用、嗜癖、ニコチン嗜癖、ニコチン乱用、アルコール嗜癖、アルコール乱用、アヘン嗜癖、アヘン乱用、コカイン嗜癖、コカイン乱用、アンフェタミン嗜癖、アンフェタミン乱用、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、注意欠如多動性障害(ADHD)、注意欠陥症候群(ADS)、レストレスレッグ症候群(RLS)、子供における多動、自閉症、認知症、アルツハイマー病における認知症、コルサコフ症候群における認知症、コルサコフ症候群、血管性認知症、HIV感染症に関連する認知症、HIV−1脳障害、エイズ脳症、エイズ認知症複合、エイズ関連認知症、大うつ病性障害、大うつ病、うつ病、ボルナウイルス感染から生じるうつ病、ボルナウイルス感染から生じる大うつ病、双極性躁うつ病、薬物耐性、オピオイドに対する薬物耐性、運動障害、ぜい弱X症候群、過敏性腸症候群(IBS)、片頭痛、多発性硬化症(MS)、筋痙攣、疼痛、慢性疼痛、急性疼痛、炎症性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、関節リウマチに関連する疼痛、アロディニア、痛覚過敏、侵害受容性疼痛、癌性疼痛、心的外傷後ストレス障害(PTSD)、統合失調症、統合失調症の陽性もしくは認知または陰性症状、痙性、ツレット症候群、尿失禁、嘔吐、そう痒状態、そう痒症、睡眠障害、排尿障害、下部尿路における神経筋障害、胃食道逆流性疾患(GERD)、胃腸障害、下部食道括約筋(LES)疾患、機能性胃腸障害、消化不良、吐出、気道感染症、神経性過食症、慢性喉頭炎、喘息、逆流関連の喘息、肺疾患、摂食障害、肥満、肥満関連の障害、肥満乱用、食品嗜癖、過食症、広場恐怖、全般性不安障害、強迫性障害、パニック障害、心的外傷後ストレス障害、社会恐怖症、恐怖症、物質誘発性不安障害、妄想性障害、統合失調性感情障害、統合失調症様障害、物質誘発性精神病性障害、または、せん妄;末梢組織、末梢神経系およびCNSにおける腫瘍細胞成長、移動、侵入、付着および毒性の阻害;新形成、過形成、異形成、癌、癌腫、肉腫、口腔癌、扁平上皮癌(SCC)、口腔扁平上皮癌(SCC)、肺癌、肺腺癌、乳癌、前立腺癌、胃癌、肝臓癌、大腸癌、結腸直腸癌腫、横紋筋肉腫、脳腫瘍、神経組織の腫瘍、神経膠腫、悪性神経膠腫、星細胞腫、神経膠腫、神経芽腫、グリア芽腫、髄芽腫、皮膚細胞の癌、黒色腫、悪性黒色腫、上皮新生物、リンパ腫、骨髄腫、ホジキン病、バーキットリンパ腫、白血病、胸腺腫および他の腫瘍。
【0077】
治療することができる障害は、すでに上に記載されている。好ましい状態および適応症は、以下の通りである:
a)mGluR5モジュレーターについて:慢性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、癌性疼痛、関節リウマチに関連する疼痛、炎症性疼痛、L−ドーパ−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、パーキンソン病治療におけるL−ドーパ−誘発性ジスキネジア、パーキンソン病治療におけるドーパミン模倣薬−誘発性ジスキネジア、遅発性ジスキネジア、パーキンソン病、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、全般性不安障害、物質誘発性不安障害、摂食障害、肥満、過食症、ハンチントン舞踏病、てんかん、アルツハイマー病、統合失調症の陽性および陰性症状、認知障害、機能性胃腸障害、胃食道逆流性疾患(GERD)、片頭痛、過敏性腸症候群(IBS)、または認知強化および/または神経保護のため。
b)mGluR5の負の調整について:慢性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、癌性疼痛、関節リウマチに関連する疼痛、炎症性疼痛、L−ドーパ−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、パーキンソン病治療におけるL−ドーパ−誘発性ジスキネジア、パーキンソン病治療におけるドーパミン模倣薬−誘発性ジスキネジア、遅発性ジスキネジア、パーキンソン病、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、全般性不安障害、物質誘発性不安障害、摂食障害、肥満、過食症、片頭痛、過敏性腸症候群(IBS)、機能性胃腸障害、胃食道逆流性疾患(GERD)、ハンチントン舞踏病および/またはてんかん。
c)mGluR5の正の調整について:アルツハイマー病、統合失調症の陽性および/または陰性症状、認知障害、または認知強化および/または神経保護のため。
【0078】
本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの共結晶は特に、過食症障害の治療のために用いることができる。
【図面の簡単な説明】
【0079】
【図1】実施例2aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の粉末X線回折チャートである。調製:実施例2aに従って入手されるような粉末、0.1mm Si(シリコン)。
【図2】実施例2aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。(ピーク=158.20℃;ピークkorr.=156.9℃、ピーク高さ=17.6434mW、ΔH=112.6J/g)。
【図3】実施例3aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の粉末X線回折のチャートである。調製:実施例3aに従って入手した粉末、0.1mm Si(シリコン)。
【図4】実施例3cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の粉末X線回折チャートである。調製:実施例3cに従って入手した粉末、0.1mm Si(シリコン)。
【図5】実施例3cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。融解ピーク:ピーク=148.17℃;ピークkorr.=147.4℃、ピーク高さ=4.4950mW、Area=185.679mJ、ΔH=56.8696J/g。吸熱事象:ピーク=105.0℃;ピークkorr.=104.1℃、ピーク高さ=0.1501mW、Area=16.336mJ、ΔH=5.0035J/g。
【図6】実施例4cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸の共結晶の粉末X線回折チャートである。調製:実施例4cに従って入手した粉末、0.1mm Si(シリコン)。
【図7】実施例4cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。融解ピーク:ピーク=140.30℃;ピークkorr.=139.2℃、ピーク高さ=10.2330mW、Area=229.500mJ、ΔH=79.3568J/g.吸熱事象:ピーク=202.9℃;ピークkorr.=202.6℃、ピーク高さ=0.5661mW、Area=67.712mJ、ΔH=23.4134J/g。析出:ピーク=240.70℃;ピークkorr.=241.1℃、ピーク高さ=−8.7764mW、Area=−816.376mJ、ΔH=−282.2879J/g。
【実施例】
【0080】
(実施例1) 出発物質の特徴づけ
出発物質である薬学的に有効な成分(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンをWO 2008/015269に記載されたように調製した。
【0081】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンのpKa値をACD/Labs、pKa DB v10.0を用いて算出した(ピラゾロ(1,5−a)ピリミジンを相関化合物として用いた)。プロトン化形態の算出pKa値は、−1.97± 0.30である。従って、この分子は非常に弱い塩基であり、この低いpKa値は伝統的な塩形成のために適切ではない。
【0082】
さらに、この出発物質を、実施例7に記載されるように特にPXRD、FT−Ramanおよび1H NMR分光法によって特徴づけた。
【0083】
遊離薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの結晶構造をX線結晶学によって測定した。結晶(無色のブロック、0.16×0.32×0.36)をKappa APEX2回折計でT=123Kにてθmax=36.345°でのグラファイト−単色化モリブデン−K−α(Kα)照射(波長 λ=0.71073Å)を用いて測定した。APEX2ソフトウエアパッケージを補正および照射に用いた。この構造を、プログラムSIR92を用いたダイレクトメソッドによって解析した。プログラムCRYSTALSを用いて、全ての非水素原子において、Fに対する最小二乗精密化を実施した。
以下の結晶データが見出された:
F(000)=752、斜方晶系、空間グループP212121、Z=4 算出密度Dca,,=1.605mg・m-3;a=7.5918(2)Å(オングストローム)、b=13.3879(4)Å(オングストローム)、c=15.1119(5)Å(オングストローム)、α(アルファ)=90°、β(ベータ)=90°、γ(ガンマ)=90°、V=1535.95(8)Å3(オングストローム)。
【0084】
共結晶形成剤2,5−ジヒドロキシ安息香酸(ゲンチシン酸、GEN)を、Flukaより購入した(注文番号37550、C7H6O4;Mw154.12g/mol)。1−ヒドロキシ−2−ナフトエ酸(キシナホ酸、XIN)を、Flukaより購入した(注文番号55910;C11H8O3;Mw188.18g/mol)。ブタン二酸(コハク酸、SUC)をFlukaより購入した(注文番号14079、C4H6O4;Mw118.09g/mol)。
【0085】
(実施例2)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の調製
実施例2a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび57.8mgのコハク酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温で24時間攪拌し、最終的に溶媒を空気中で室温にて(解放バイアル)蒸発させた。白色粉末が得られた。
【0086】
得られた粉末をFT Ramanによって特徴づけた。このFT Ramanスペクトルは、遊離有効成分とコハク酸の混合物を示している。
【0087】
2mlのイソプロピルエーテルを得られた粉末の残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し、空気中で乾燥させた。無色の粉末が得られ、実施例7に記載されているようにFT Raman、PXRD、1H−NMR、TG−FTIR、DSC、DVSによって特徴づけた。
【0088】
得られた結晶粉末は、独特のRamanスペクトルを示し、NMRデータにより所定の共結晶構造が確認された。
【0089】
この粉末をPXRDパターンによって特徴付けした(これを図1に示す)。
DVSデータは、得られた結晶粉末は水分を取り込まない(吸湿性ではない)ことを示していた。
【0090】
TG−FTIR測定は、得られた結晶粉末は、微量のイソプロピルエーテル/水を含んでおり、150℃を超えると分解することを示している。さらに、得られた結晶粉末は156.9℃で融解ピークによって特徴づけられた(DSC)。このDSCダイアグラムを図2に示している。
【0091】
実施例2b:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解させた。57.8mgのコハク酸(2mlのアセトンに溶解)を加えた。この溶媒を流量制御をせずに窒素流下で室温にて蒸発させた。無色の粉末が得られた。この得られた粉末を、実施例7に記載したようにFT Ramanによって特徴付けし、このRamanスペクトルは遊離塩基と同様である。
【0092】
2mlの2−プロパノールを得られた残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し空気中で乾燥させた。無色の粉末が得られ、これをFT Ramanで特徴付けした。この共結晶は実施例2aに従う共結晶と同じRamanスペクトルを示している。
【0093】
実施例3:(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと2,5−ジヒドロキシ安息香酸(ゲンチシン酸)の共結晶の調製
実施例3a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解した。75.5mgのゲンチシン酸(2mlのアセトンに溶解させた)を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末が得られた。
【0094】
得られた生成物を実施例7に記載したようにFT Raman、PXRD(図3)、1H−NMRによって特徴付けした。
【0095】
得られた粉末のX線回折パターンを図3に示しており、結晶形態(共結晶)が確認される。1H−NMRスペクトルにより共結晶の所定の構造が確認された。
【0096】
実施例3b:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと75.5mgのゲンチシン酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温で約24時間攪拌し、最終的に溶媒を室温にて空気中で(開口バイアル)蒸発させた。アイボリー色の粉末が得られた。得られた粉末のFT Ramanスペクトルは、実施例3a)のFT Ramanと一致する。
【0097】
実施例3c:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解させた。62.3mgのゲンチシン酸を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末を得た。
【0098】
得られた生成物を実施例7に記載されるようにRaman、PXRD(図4)、1H−NMR、TG−FTIR、DSC(図5)、DVS、およびFT Ramanによって特徴付けした。
【0099】
図4に示されれているようなPXRDパターンは、実施例3aに従う共結晶のX線パターンとは異なる結晶構造を示している。従って、共結晶の別の多型形態が得られた。さらに得られた共結晶は、微量のイソプロピルエーテル/アセトンを含んでいる。NMRデータは、所定の構造と一致している。
【0100】
このTG−FTIR分析は、150℃より高い温度での分解を示しており;DSCダイアグラムは、104℃での吸熱事象、147℃での融解ピークを示している(DSCダイアグラム図6を参照のこと)。
【0101】
DVS測定の結果として、得られた共結晶は最小限の水の取込を示しており、かつ吸湿性がない。
【0102】
実施例4:(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと1−ヒドロキシ−2−ナフトエ酸(キシナホ酸)の共結晶
実施例4a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを、5mlのEtOAcに溶解した。92.1mgの1−ヒドロキシ−2−ナフトエ酸(キシナホ酸)(2mlの酢酸エチルに溶解)を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末が得られ、これを実施例7に記載されているようFT Raman、PXRD、1H−NMRで特徴づけした。
【0103】
得られた粉末のX線回折パターンは、実施例4に従うPXRDパターンと広範囲にわたって同一であり、結晶形態(共結晶)が確認される。1H−NMRスペクトルにより、共結晶の予測された構造と一致した。
【0104】
実施例4b:150mgの6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと92.1mgの1−ヒドロキシ−2−ナフトエ酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温(r.t.)で約24時間攪拌し、最終的に溶媒を空気中(開口バイアル)で室温(r.t.)にて蒸発させた。アイボリー色の粉末が得られ、得られた粉末のRamanスペクトルは、共結晶と有効成分の混合物であった。
【0105】
2mlのイソプロピルエーテルを得られた残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し、空気中で乾燥させた。アイボリー色の粉末が得られた。得られた粉末のFT Ramanは、実施例4aに従って得られた共結晶のスペクトルと一致した。
【0106】
実施例4c:150mgの6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのEtOAcに溶解させた。76mgの1−ヒドロキシ−2−ナフトエ酸(2mlのEtOAcに溶解)を加えた。この溶媒を流量制御なしに窒素下で室温(r.t.)にて蒸発させた。アイボリー色の粉末が得られ、実施例7に記載されるように、FT Raman、PXRD(図6)、1H NMR、TG−FTIR、DSC(図7)、DVSで特徴付けした。
【0107】
得られた粉末のFT Ramanは、実施例4aに従って得られた共結晶のスペクトルと一致した。
【0108】
得られた粉末のX線回折パターンを図6に示しており、結晶形態(共結晶)であることが確認される。1H−NMRスペクトルは、共結晶の構造と一致した。
【0109】
DSC分析(DSCダイアグラムは、図7に示されている)は、得られた共結晶が、139℃で融解ピーク、241℃で吸熱事象を表わしていることを示している。TG−FTIR測定は、キシナホ酸共結晶が微量の酢酸エチルを含有しており、150℃を超える温度で分解を示すことを実証している。
【0110】
DVS測定により、可逆的な水の取込が、80%の相対湿度を超えることを示している。従って、公知の化合物Aとキシナホン酸の得られた共結晶は、わずかに吸湿性である。
【0111】
実施例5:溶解度の測定
各共結晶(実施例2a、3c;および4cに従う)を水中に懸濁させた。このサンプルを、Eppendorf製の温度制御された「Thermomixer comfort」を用いて800rpm(24時間、23℃)で振盪させた。得られた懸濁液を、Millipore Centrifugal Filter Device UFC30VVNB(0.1)およびCentrifuge Hettich EBA 12R(10.000g)を用いて濾過した。得られた固体をFT−Raman分光計によって特徴付けし、溶解性のテスト前のスペクトルと比較すると、形態の変化は観察されなかった。
【0112】
ろ液のpHを測定し、遊離塩基の濃度をHPLCによって測定した(方法は実施例7に記載されている)。
【0113】
同様に(同じ条件で)測定した検出限界付近である遊離薬物化合物の溶解度は、pH8.5にて10μg/ml未満(0.01mg/l未満)を示している。
【0114】
新規の共結晶の測定された溶解度を表1にまとめた。
【0115】
【表1】
【0116】
本発明に従う共結晶は、驚いたことに遊離薬物と比較して2倍の溶解度であった。
【0117】
実施例6:熱分析技術によるコハク酸、ゲンチシン酸、およびキシナホ酸の共結晶の特徴付け
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの以下の共結晶を実施例7に記載されるようにTG−FTIR、DSCおよびDVSによって特徴付けした:
実施例2aに従うコハク酸の共結晶、
実施例3cに従うゲンチシン酸の共結晶、および
実施例4cに従うキシナホン酸の共結晶。
【0118】
実施例6a:TG−FTIRを上記のサンプルに対して実施した。結果を以下の表1にまとめる。
【0119】
【表2】
【0120】
コハク酸共結晶サンプルは、微量のイソプロピルエーテル(調製に用いられる溶媒)を含有している。150℃を超えて分解が観察された。
【0121】
ゲンチシン酸共結晶サンプルは、微量のアセトン(調製に用いられる溶媒)を含有している。180℃を超えて分解が観察された。
【0122】
キシナホン酸共結晶サンプルは、微量の酢酸エチル(調製に用いられる溶媒)を含有している。160℃を超えて分解が観察された。
【0123】
実施例6b:上記のサンプルに対してDSCを実施した。結果を以下の表2にまとめる。
【0124】
【表3】
【0125】
化合物Aは、132〜140℃の範囲で融解する(ピーク132.9℃)。
【0126】
実施例2aに従うコハク酸共結晶のDSC(図2を参照)は、156.9℃で鋭い融解ピークを示している。
【0127】
実施例3cに従うゲンチシン酸共結晶のDSC(図5を参照)は、104℃で広範な吸熱事象を示している(ΔH=5J/g)。おそらくこのサンプルは、微量の出発物質および/または他の不純物を含有している。147℃における第2の吸熱事象は、共結晶の融解に寄与し得る。分解は200℃を超えて観察された。
【0128】
実施例4に従うキシナホン酸共結晶のDSC(図7を参照)は、139.2℃で鋭い融解ピークを示し、200℃を超えて分解を示している。
【0129】
実施例6c:DVS(50% −> 0% −> 95% −> 50% r.h.)を、上述のサンプルに対して実施した。この結果を以下にまとめる:
【0130】
ゲンチシン酸共結晶のDVS(実施例2a)は、試験した湿度範囲にわたって最小かつ可逆的な質量変化のみを示している。約0.1%の質量変化Δm(50〜85%までの相対湿度(r.h.)の変化)が観察され、この共結晶は吸湿性ではない。DVS Ramanスペクトル後は、形態に何の変化も示さない。
【0131】
コハク酸共結晶のDVS(実施例3c)は、試験した湿度範囲にわたって最小かつ可逆的な質量変化のみを示している。約0.1%の質量変化Δm(50〜85%までの相対湿度(r.h.)の変化)が観察され、この共結晶は吸湿性ではない。DVS Ramanスペクトル後は、形態に何の変化も示さない。
【0132】
キシナホン酸共結晶のDVS(実施例4c)は、ヒステリシスで80%の相対湿度を超える可逆的な水の取込を示している。約1%の質量変化Δm(50〜85%までの変化)が観察された。この共結晶はわずかに吸湿性である。湿度が95%の相対湿度まで増加した場合より多くの水が取り込まれた((約2質量%の総水含有量、平衡到達)。相対湿度を再び下げると、水含有量は減少し、元の質量まで戻った。DVS Ramanスペクトル後は形態に何の変化も示さない。
【0133】
実施例7:装置の測定条件
実施例7a:DSC(示差走査熱量測定)/Perkin Elmer DSC 7は、閉じたAu製のるつぼを用いた。加熱速度:10または20℃/分、範囲:−50℃〜250℃。
【0134】
実施例7b:DVS(動的水蒸気吸脱着測定装置)
Surface Measurement Systems Ltd.製のDVS−1水蒸気吸着分析器およびProjekt Messtechnik製のSPS 11−100n マルチサンプル水蒸気吸着分析器を用いた。
【0135】
このサンプルを開始前に50%の相対湿度で平衡にしておいた(例えば、以下の所定の湿度プログラム):
0%の相対湿度で2時間
95%の相対湿度で0〜95%(5%/時)
95%の相対湿度で3時間
95%〜0%(10%/時)
0%の相対湿度で2時間。
【0136】
実施例7c:使用したHPLC(高速液体クロマトグラフィー)システムは、以下のように特徴付けられる:
装置:TSP HPLC(UV3000、AS3000、P4000、SCM1000
Soft. Version 4.1)
カラム:水、XTerra MS C18 4.6x100mm 5μ(CC01)
移動相A:希釈H2O + 0.1%TFA
移動相B:MeCN + 0.1%TFA
参照濃度:ca. 0.04mg/ml
保持時間:13.1分
勾配: 0.0分 95% A / 5% B
20.0分 5% A / 95% B
21.0分 95% A / 5% B
30.0分 95% A / 5% B
流量:1.0ml/分
注入量:10μl
波長:254nm
【0137】
実施例7d:NMR(核磁気共鳴)
1H NMRスペクトルを、Bruker DPX300装置で、300.13MHzで記録した。
【0138】
実施例7e:ラマン顕微
安定化ダイオードレーザー785−nm励起および検出器としてNIR−増強ペルチェ吸熱CCDカメラを用いて、ラマン顕微をRenishaw RM 1000で記録した。測定は長作動距離20倍で実施した。測定範囲は2000〜100cm-1である。
【0139】
実施例7f:FT−ラマン分光法(フーリエ変換ラマン分光法)
FT−ラマンスペクトルを、Nd:YAG 1064nm励起、100mWのレーザーパワーおよびGE検出器、64スキャン、範囲25〜3500cm-1、2cm-1解像度で、Bruker RFS100で記録した。
【0140】
実施例7g:TG−FTIR(フーリエ変換赤外分光法と組み合わせた熱重量計)
TG−FTIRを、Al製のるつぼ(開口または微細孔のあるもの)中、N2雰囲気下、10℃min-1の加熱速度、25〜250℃の範囲でNetzsch Thermo−Microbalance TG 209 with Bruker FT−IR Spectrometer、Vector 22を用いて実施した。
【0141】
実施例7h:溶解度測定
水への共結晶の懸濁液をEppendorf製の温度制御された「Thermomixer comfort」を用いて800rpmで(24時間、23℃)攪拌した。この懸濁液をMillipore Centrifugal Filter Device UFC30VVNB(0.1μ)およびCentrifuge Hettich EBA 12 R(10´000g)で濾過した。
【0142】
実施例7i:PXRD(粉末X線回折)
粉末X線回折パターンを、銅Kα放射(Cu−Kα1、波長λ=1.540598Å(オングストローム)、40kV/40mAでBruker D8にて、およびLynxEye検出器、0.02°2θのステップサイズ、37sのステップ時間で記録した。
【0143】
サンプル調製:このサンプルは、通常平坦な表面を得るためにわずかな圧力をかける以外には何の特別な処理もなく測定した。シリコン単結晶サンプルホルダータイプ:a)多型スクリーニング用の標準ホルダー、0.1mm深さ、20mg未満のサンプル;b)0.5mm深さ、c.40mgのために12mmの孔直径;c)1.0mm深さ、c.80mgのために12mmの孔直径。Bruker D8で測定される全てのサンプルは測定の間に回転される。
【0144】
実施例8:溶解速度の測定
各共結晶(実施例2a、3cおよび4cに従う)について、および遊離薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン)について、圧縮された錠剤の溶解を、胃腸管を通る錠剤の通過をシミュレートするために4つのセクターを通してpHを増加させながら、UV吸収分光分析を用いて0.15Mの塩化カリウム水溶液中でモニターした(セクターIpH2.0;セクターII:pH 3.9;セクターIII:pH 5.4;セクターIV:pH7.3)。
この錠剤を約50000ポンド/inch-2の重さで圧縮し直径を3mmにした、錠剤の一面のみを、酢酸/リン酸緩衝液経を含む溶解培地に曝露し、薬物の溶解に起因する実験的なpHの逸脱を最小にした。溶液の攪拌を一定の速度で続けた。吸収データを事前に測定したpH依存係数、モルモル吸光係数を用いてサンプル重量の絶対値に変換した。適切な波長範囲は、UV光源の彩度に起因した誤った溶解データを避ける、1.3未満の吸光値の分光データが分析されることが保障されるように選択した。溶解速度は、得られた実験データに一次指数方程式をフィッティングすることで算出した。
【0145】
表3に列挙した溶解速度は、pH2.0、3.9、5.4、および7.3(±0.1)、温度23℃(±1℃)で測定した。表3にデータがない場合は、溶解速度は得番目〜3番目および4番目への移動において溶解速度が少しも変化しなかったことがわかる。
【0146】
【表4】
【0147】
例えば、MRZ8456のコハク酸共結晶の場合においては、pH2における遊離薬物MRZ8456と比較して3倍の溶解速度の増加が観察された。
【技術分野】
【0001】
本発明は、新規の結晶形態、特に置換ピラゾロ−ピリミジン類と有機酸との共結晶に関する。特に興味深いのは、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(以下において、時々「化合物A」と呼ばれる)、および特に該化合物のR異性体、いわゆる6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと有機カルボン酸との共結晶である。
【0002】
さらに、本発明は置換ピラゾロ−ピリミジン類および特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、有機モノカルボン酸およびジカルボン酸との共結晶の調製方法を提供する。
【背景技術】
【0003】
化合物6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンのような典型的な置換ピラゾロ−ピリミジン類(その異性体を含む)、およびそれらの調製方法は、特許文献1に記載されている。この文書は、ピラゾロ−ピリミジン類、特に化合物Aが効果のあるmGluR5モジュレーターであり、急性および慢性の神経障害、特にグルタミン酸誘導性茂樹に関与するCNS(中枢神経系)障害の予防および治療に有用である。
【0004】
化合物AのR−エナンチオマーの化学構造は以下に示される:
【化1】
【0005】
医薬組成物用の原薬(active pharmaceutical ingredients(API))は、種々の異なる形態に調製され得ることが知られている。ほとんどの薬物化合物または原薬(例えば、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン)は、固体として投与される。多くの固体APIは、1つまたはいくつかの結晶形態で存在している。しばしば、このAPIは、それ自身の上には結晶化しないか、さもなくば不利益な物理的および生物薬剤学的特性を呈す結晶形態中に結晶化しない。より良好な医薬品製品を提供するために、代替の結晶形態を探す必要がある。特定の形態の良い例としては、多形体、塩、溶媒和物および水和物が挙げられる。これらの確立された結晶APIの改変(modification)に加え、APIを含む結晶分子複合体としてまた記載され得る医薬品共結晶は、化学者の注目を引いてきた。薬学的有効成分の特定の物理学的形態の選択は、溶解度、溶解速度、吸湿性、物理安定性および化学安定性のような物理特性を最適化するための戦略的なチャンスを示している。
【0006】
いくつかの薬学的に有効な化合物は多型を示す。いくつかの化合物は、10より多い異なる結晶形態変位で存在している。多型は、所定の化合物の固体形態であり、固体状態にある化合物の分子の少なくとも2つの異なる配置の可能性に起因している。この多形の形成は、しばしば結晶化の状態に依存している。所定の化合物の異なる多型は、独特な一連の物理化学的特性を示す。不利な点として、化合物の新規の多型形態は、通常いくつかの例に限定される。
【0007】
薬学的に有効な成分の新規の結晶形態を得るための別の公知のアプローチは、水和物および溶媒和物の形成である。しばしば、結晶化の間に溶媒は結合され結晶構造の一部として取り込まれる。多くの溶媒は生物学的に毒性であり、従って溶媒を含有する結晶はしばしば薬物化合物の固体形態の開発においては避けられる。しかしながら、水和物(これにおいては結晶構造は水を取り込む)は、APIの通常形態であり、医薬品製品において周知である。多くの目的の医薬品分子が、水和物を形成することができる。しかしながら、水和物はしばしば不安定であり、温度、圧力または相対湿度といった貯蔵状態の変化の結果として無水結晶に変化する。この水和物から無水物への変換(例えば貯蔵の間、または製剤工程の間)は、薬物製品の品質を急激に損なう可能性がある。
【0008】
APIの塩の形成は、原薬の特性を変更するための別の公知のプロ−地である。API間の酸−塩基反応として記載することができる塩形成は、塩基性および/または酸性の官能基、および酸性または塩基性の物質を示す。薬物化合物の塩は、その結晶格子中にAPI分子のイオン形態を含んでいる。多くの医薬品化合物は酸性または塩基性の機能を呈しているため、塩形成は、APIの新規の結晶形態を得るための魅力的な方法である。塩の広範囲にわたる用途は、多くの市販されている薬物化合物の結晶塩によって証明されている。
【0009】
共結晶はずいぶん以前に発見されたが、医薬品共結晶は未だなお、特に置換ピラゾロ−ピリミジン類のような複素環式薬物化合物についての開発および最適化の興味深いゴールである。本発明に従う共結晶は、非共有相互作用(しばしば水素結合を含む)を通して結晶格子内で一緒に結合される2つまたはそれ以上の中性分子の結晶複合体として理解することができる。通常、APIとさらなる分子化合物(共結晶形成剤またはカウンター分子)の間のプロトン移動は起こらない。本発明に従う共結晶化技術の適用は、塩形成と比較していくつかの利点を提供する。原則的には、従来塩形成の能力が限定されているかまたはその能力がないために物理特性の最適化という点で高いリスクを示すと考えられている、イオン化力の弱い化合物や非イオン化化合物を含むあらゆるタイプの分子が共結晶を形成することができる。
【0010】
2002年に、その各々が水素結合アクセプターとして作用する、6つの異なるカウンター分子を持つ鎮痛薬パラセタモール(paracetamol(アセトアミノフェン))が販売された。その後すぐにいくつかの水素結合アクセプターを持つ薬物化合物の共結晶、イブプロフェン、フルルビプロフェンおよびアスピリンが記載された。これらの例は、通常の水素結合特徴を持つ一連の共結晶は得ることができることを示している。融点データだけではなく、これらの報告書はこれらの共結晶の機能特性および薬理学特性に目を向けることなしに特に構造の特徴に焦点を置いている。
【0011】
本発明に従う共結晶のさらなる利点は、ソルトスクリーン(salt screen)においては、わずかな数の酸性または塩基性対イオンのみだけが考慮されるにもかかわらず、ピラゾロ−ピリミジン類と共結晶を調製するのに用いられ得る共結晶を形成する可能性のある薬剤(共結晶形成剤またはカウンター分子とも呼ばれる)はいくつも存在するという点である。可能性のある薬剤は、米国食品医薬品局による「一般的に安全であると認められる」物質のリストから、実験用に選択することができる。共結晶の高められた範囲は、薬物の望ましい物理特性プロフィールを達成するより大きい可能性の示唆において有利であるが、スクリーニングの労力という点においてはかなりの困難も示される。共結晶のスクリーニング、特にハイスループットスクリーニング法(改良された合理的な共結晶デザインおよびより効果的な共結晶スクリーニングプロトコルを含む)は、新規の結晶形態の開発において重要なツールである。共結晶の調製のためのいくつかの一般的な方法は文献に記載されている。特定の物理特性を高めるために共結晶を用いる公知の例が存在している。共結晶の調製方法には、従来の結晶化技術および固体の粉砕のようなより特別な方法も含まれる。
【0012】
共結晶の形成は、調査に入る前に研究されており、種々の重要な研究が共結晶デザインの理解を目的としている。初期の研究においては、良好なプロトンドナーとアクセプターが水素結合に用いられること、および最適なドナーは典型的には所定の結晶構造において最良のアクセプターと対となることを含む、いくつかの「水素結合ルール」が提唱された。水素結合ルールと幾何解析との組み合わせて用いることが、多くの新規の超分子構造の合成における合理的な共結晶デザインを実行するために行われた。
【0013】
特許文献1に記載されるような多くのピラゾロ−ピリミジン類、および特に化合物Aは、塩基性官能基を示している。化合物Aは、その低い塩基性能に起因して、約−1.97(ピラゾロ[1,5−a]ピリミジンと相関して算出した)という低いpKa値を示している。さらに、この化合物Aは水または水性溶媒においては溶解性が乏しい(10μg/mL未満)。この物理化学特性のおかげで、化合物Aはいくつかの不利な医薬品特性(例えば、完全でないバイオアベイラビリティー)を示している。上記のように、化合物Aはパートナー間のpKa値の差異が十分大きくないために、鉱物酸または不安定な塩と一緒に簡単に塩を形成することができない。従って、塩の形成は化合物Aの医薬品特性を向上するために困難な方法である。
【0014】
改良された結晶形態である(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンについては、増強された溶解度および融解特性ならびに良好な貯蔵安定性特性を示す医薬品組成物の調製のために高い必要性がある。
【先行技術文献】
【特許文献】
【0015】
【特許文献1】WO2008/015269
【発明の概要】
【発明が解決しようとする課題】
【0016】
本発明の目的は、改良された結晶形態、特にピラゾロ−ピリミジン類、および特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、特に6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、適切な共結晶形成剤との共結晶を提供することである。この新規の共結晶は、好ましくは改良された薬学的特性および良好な貯蔵安定性(例えば、水へのより高い溶解性、全くないかまたはわずかな吸湿性)を示す化合物Aである。
【課題を解決するための手段】
【0017】
驚いたことに、薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび特にこのR−異性体は、特定の有機カルボン酸と安定な共結晶を形成することが見出された。さらに、多くの有機カルボン酸およびアミノ酸は、通常の結晶化手順を用いて6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびその光学異性体と共結晶を形成しないことが明らかとなった。いくつかの例は、安息香酸、リンゴ酸(1−ヒドロキシブタン二酸)、マンデル酸(2−ヒドロキシ−2−フェニル酢酸)、D/L酒石酸(2,3−ジヒドロキシブタン二酸)、バニリン酸(4−ヒドロキシ−3−メトキシ安息香酸)、またはL−アスパラギン酸(2−アミノブタン二酸)である。
【0018】
本発明は、ピラゾロ−ピリミジン類、特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、および特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、以下に記載されるような少なくとも1種の共結晶形成剤、好ましくは1種、2種または3種の共結晶形成剤との共結晶との共結晶に関する。
【0019】
本発明は、ピラゾロ−ピリミジン類、特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、および特に(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと、共結晶形成剤との共結晶に関し、該共結晶形成剤が、一般式(I):
【化2】
[式中、Rは以下:
a)
【化3】
(式中、nは1、2、3または4である)
または
b)
【化4】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸である。
【0020】
好ましい実施態様において、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、該共結晶形成剤が、式(I):
【化5】
[式中、Rは以下:
a)
【化6】
(式中、nは2または3である)
または
b)
【化7】
(式中、
R1およびR2は、互いに独立して水素またはヒドロキシルであり、
R3およびR4は、水素であるかまたは、
R3およびR4は、それらを保持する炭素原子と一緒になって、非置換の芳香族6員環を形成してもよい)
を表わす]
のカルボン酸である。
【0021】
好ましくは、このカルボン酸共結晶形成剤は、少なくとも2つの水素供与基を含み得、その1つはヒドロキシル基から選択され、またその1つはカルボキシル基から選択され、好ましい強力な水素結合をした2分子の環モチーフの形成が可能である。
【0022】
さらに、6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンは、コハク酸、ゲンチシン酸、およびキシナホ酸(xinafoic acid)を含む群より選択される少なくとも1種のカルボン酸と安定な共結晶を形成することが明らかとなった。さらに、コハク酸、ゲンチシン酸、およびキシナホ酸の共結晶は、例えばそれらは吸湿性ではないかまたは化合物A自体よりも吸湿性が少ないという特に有利な特性を有していることも明らかとなった。上述の全ての共結晶は、遊離型の薬物化合物に比べ、より良好な水溶解性を示した。
【0023】
本発明における用語「共結晶」は、室温(20〜25℃)である2個以上の中性分子化合物が、非共有相互作用(しばしば、水素結合、πスタッキング、ゲスト−ホスト錯体、ファンデルワールス相互作用などを含む)によって結晶格子中に一緒に結合されている結晶複合体を意味している。特に、該非共有相互作用は、水素結合を含む。水素結合は、例えば、異なる分子間構造(例えば二量体、線状鎖、または環状構造)をもたらし得る。これらの共結晶の各々は、構造(例えば、PXRDパターンで特徴づけられる)、融点、融解熱のような独特の物理特性を示し、とりわけそれにより特徴づけられ得る。
【0024】
本発明に従う共結晶は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびその化合物におそらく水素結合している共結晶形成剤を含む。上述したような他の相互作用はまた、本発明に従う共結晶の形成にも役割を果たしている。
【0025】
共結晶形成剤をさらに含まない化合物Aの塩および溶媒和物は、本発明に従う共結晶として考慮されない。しかしながら、本発明に従う共結晶は、結晶格子中に1種またはそれ以上の溶媒和物分子を含んでいてもよい。従って、室温で液体である化合物をさらに含む共結晶の溶媒和物は、本発明のより広義の範囲に含まれる。本発明に従う共結晶はまた、化合物Aの塩と共結晶形成剤との共結晶であってもよいが、化合物Aおよび共結晶形成剤は、好ましくは水素結合を介して一緒に構築または結合される。この共結晶形成剤は、化合物Aに直接結合していてもよいし、または化合物Aに結合しているさらなる分子(例えば溶媒和物分子)に結合していてもよい。上に概説したように、本発明に関する共結晶は、典型的な塩および溶媒和物/水和物の意味からは区別され得る。
【0026】
本発明に記載される(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの新規の共結晶は、pKa値により従来の塩としては考えられない。さらに、実験データにより、本発明に従う結晶化合物が共結晶であることが確認されている。
【0027】
本発明に記載される共結晶の有利な特性は、遊離型の化合物Aと比較して、例えば水への良好な溶解性、より高い溶解速度、低いかまたは全くない吸湿特性、および良好な貯蔵安定性である。
【0028】
本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと少なくとも1種の共結晶形成剤との共結晶に関し、該共結晶形成剤は、好ましくはコハク酸(ブタン二酸)、ゲンチシン酸(2,5−ジヒドロキシ安息香酸)、およびキシナホ酸(1−ヒドロキシ−2−ナフトエ酸)からなる群より選択されるカルボン酸である。
【0029】
本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、該共結晶形成剤は、好ましくはコハク酸(ブタン二酸)、ゲンチシン酸(2,5−ジヒドロキシ安息香酸)、およびキシナホ酸(1−ヒドロキシ−2−ナフトエ酸)からなる群より選択されるカルボン酸である。
【0030】
特に、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関し、複素環(化合物A):共結晶形成剤のモル比は、1:0.1〜1:10の範囲、好ましくは1:1〜1:10の範囲、好ましくは1:1〜1:5の範囲、より好ましくは、約1:1である。この共結晶は、好ましくは結晶性共結晶である。
【0031】
さらなる局面において、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶を提供し、9.3、16.0、20.0、22.9、および26.0°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも2つ、好ましくは少なくとも3つ、より好ましくは少なくとも4つ、さらにより好ましくは少なくとも5つの粉末X線回折(PXRD)ピークによって特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、図1に実質的に従うPXRDパターンによって特徴づけられ得る。
【0032】
さらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、DSC(示差走査熱量測定)で約156.9℃の融解ピークを有している。さらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶は、実質的に図2に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンダイアグラムによって特徴づけられる。
【0033】
本発明はまた、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶を提供し、この共結晶が、6.0、7.0、14.0、17.6、21.0、23.4、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも4つの粉末X線回折(PXRD)ピークにより特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図3に従うPXRDパターンで特徴づけられ得る。
【0034】
本発明はまた、ゲンチシン酸共結晶の第2の多型を提供する。従って、本発明は(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶に関し、この共結晶が、6.9、12.6、21.2、および27.5°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、より好ましくは4つの粉末X線回折(PXRD)ピークの選択によって特徴づけられる。別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図4に従うPXRDパターンによって特徴づけられる。
【0035】
別の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶は、DSC(示差走査熱量測定)により約147.4℃の融解ピークで特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(好ましくは2,5−ジヒドロキシ安息香酸)との共結晶は、実質的に図5に従うDSC(示差走査熱量測定)によって特徴づけられ得る。
【0036】
本発明はまた、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶を提供し、この共結晶が、3.9、11.6、18.1、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つ、好ましくは少なくとも2つ、より好ましくは少なくとも3つ、さらにより好ましくは4つの粉末X線回折(PXRD)ピークによって特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、実質的に図6に従うPXRDパターンによって特徴づけられる。
【0037】
本発明のさらなる実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、DSC(示差走査熱量測定)によって約139.2℃の融解ピークで特徴づけられる。(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸;1−ヒドロキシ−2−ナフトエ酸)との共結晶は、実質的に図7に従うDSC(示差走査熱量測定)によって特徴づけられ得る。
【0038】
各共結晶は、1つまたはそれ以上の上述の物理特性(PXRDピーク、DSCピーク)によって特徴づけられ得る。従って、本発明は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤(好ましくはゲンチシン酸、コハク酸、キシナホ酸より選択される)との共結晶に関し、この共結晶は、1つまたはそれ以上の上述の物理データによって特徴付けられる。
【0039】
本発明はさらに、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法に関し、該共結晶形成剤は、一般式(I):
【化8】
[式中、Rは以下:
a)
【化9】
(式中、nは1、2、3または4である)
または
b)
【化10】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸であり、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、スラリーの状態で分散させる工程、
を包含する。
【0040】
本発明はさらに、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法に関し、該共結晶形成剤が、ゲンチシン酸、コハク酸、およびキシナホ酸からなる群より選択されるカルボン酸であり、
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、スラリーの状態で分散させる工程、
を包含する。
【0041】
好ましい実施態様において、化合物Aと、コハク酸およびキシナホ酸より選択される共結晶形成剤との共結晶の調製方法は、相平衡工程c)を含む。
【0042】
本発明の実施態様において、溶解工程a)は、室温(20〜25℃)および通常圧力(1013.25hPa)下で、1〜60分間実施される。別の好ましい実施態様において、この溶解工程a)は、20℃〜100℃の範囲で温度を上げられた状態で実施される。
【0043】
工程a)は、最初に溶媒S1に化合物Aを溶解し、その後その溶液に共結晶形成剤を加えることによって実施される。さらに、別の実施態様において、この化合物Aおよび共結晶形成剤は、固体として最初に混合され、その後溶媒S1に溶解させてもよい。さらなる実施態様として、工程a)は、溶媒S1に溶解させた化合物Aの溶液と溶媒S1に溶解させた共結晶形成剤の溶液とを混合することにより実施することができ、ここで化合物Aと共結晶形成剤を溶解させるための溶媒は、異なっていても同じでもよい。化合物Aと共結晶形成剤を溶解させるための溶媒は等しいことが好ましい。
【0044】
好ましい実施態様において、蒸発(工程b)は、室温(20〜25℃)および通常圧力(1013.25hPa)下で実施される。しばしば、この工程b)は、空気または窒素流下で、場合によりフロー制御をおこないながら実施される。場合により、溶媒S1の蒸発(工程b))は、減圧下で実施される。
【0045】
相平衡工程c)は、しばしば室温(20〜25℃)下で実施され、別の実施態様において、工程c)は、0〜100℃の範囲に上げられたまたは下げられた温度下で実施される。
【0046】
本発明の1実施態様において、カルボン酸共結晶形成剤および化合物Aは、0.1〜10の範囲のモル比で、好ましくは約1:1のモル比で本発明に従う方法に用いられる。さらなる実施態様において、このカルボン酸共結晶形成剤は、化合物Aに対して1.1〜10のモル過多で用いられる。本発明のさらなる実施態様において、カルボン酸共結晶形成剤は、化合物Aに対して0.1〜0.95のモル比で用いられる。好ましくは、コハク酸は、化合物Aに対して1〜7.5の範囲、より好ましくは約1.2:1のモル比で用いられる。
【0047】
薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、およびカルボン酸共結晶形成剤は、しばしば溶媒S1中に等モル比で溶解される(工程a)。
【0048】
本発明は、上記のような共結晶に関し、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、とカルボン酸共結晶形成剤のモル比は、1:0.1〜1:10の範囲、好ましくは1:1〜1:10の範囲、好ましくは1:1〜1:5の範囲である。より好ましい実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとカルボン酸共結晶形成剤のモル比は、1H−NMR分光法で測定した場合1:1である。
【0049】
好ましくは、本発明に従う共結晶は、化合物Aと共結晶形成剤を溶解し、その後上記のようにその溶媒を蒸発させることによって調製される。別の実施態様において、この共結晶は、他の通常な結晶化手順を用いて調製され得る。例えば、化合物Aは、溶液または固体状態で温度勾配を用いてカルボン酸共結晶形成剤と共結晶化され得る。1つの例はハンギングドロップ拡散法であり、これは少量の共結晶の調製に用いられる方法である。
【0050】
溶媒S1および場合によりS2は、好ましくは以下からなる群より選択される少なくとも1つの有機溶媒である:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)およびN−メチルピロリドン(NMP)。好ましくは、この有機溶媒S1は、アセトン、エタノール、酢酸エチル、テトラヒドロフラン、およびイソプロピルエーテル(IPE)から、より好ましくはアセトン、イソプロピルエーテル(IPE)および酢酸エチルから選択される。
【0051】
溶媒S1は、さらに上述の溶媒の2つ、3つまたはそれ以上の混合物であってもよい。この溶媒はしばしば、有機溶媒(上記のもの)と水の混合物である。典型的な例は、以下の混合物である:エタノール:水(1:1)およびテトラヒドロフラン:水(1:1)。特に、選択された溶媒への共結晶形成剤の溶解度が低い場合、上記のような溶媒混合物が用いられる。
【0052】
溶媒S2は、好ましくは以下からなる群の少なくとも1つの有機または無期溶媒から選択される:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)、およびN−メチルピロリドン(NMP)。好ましくは、溶媒S2は、tert−ブチル−メチルエーテル(TBME)、1−プロパノール、2−プロパノール、トルエン、水、およびイソプロピルエーテル(IPE)から、より好ましくはイソプロピルエーテル(IPE)および2−プロパノールから選択される。
【0053】
溶媒S2は、さらに上述の溶媒の2つ、3つまたはそれ以上の混合物であってもよい。しばしば、この溶媒は、例えばエタノール:水(1:1)混合物およびテトラヒドロフラン:水(1:1)混合物のような有機溶媒と水との混合物である。
【0054】
本発明の好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶の調製方法に関し、ここで溶媒S1は1−プロパノールであり、溶媒S2は、イソプロピルエーテル(IPE)および2−プロパノールから選択される少なくとも1つの溶媒である。
【0055】
好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法に関し、溶媒S1は、アセトンおよびイソプロピルエーテル(IPE)から選択される少なくとも1つの溶媒である。
【0056】
好ましい実施態様は、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)との共結晶の調製方法に関し、溶媒S1は酢酸エチルである。
【0057】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびコハク酸を、2−プロパノール中に1:1〜1:10の範囲のモル比で溶解する工程、
b)好ましくは空気または窒素流下、室温下で、2−プロパノールを蒸発させる工程、
c)工程b)で得られた残留物を、イソプロピルエーテル(IPE)および2−プロパノールから選択される少なくとも1つの溶媒中に少なくとも10時間、好ましくは少なくとも15時間、より好ましくは少なくとも24時間、攪拌しながら分散させる工程(相平衡)。
【0058】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびゲンチシン酸(2,5−ジヒドロキシ安息香酸)を、約1:1〜1:1.25の範囲、好ましくは約1:1.2〜1:1.25の範囲のモル比で、アセトンおよびイソプロピルエーテル(IPE)から選択される溶媒中に溶解する工程、
b)好ましくは窒素流または空気中で室温にてアセトンを蒸発させる工程。
【0059】
本発明の1実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸(2,5−ジヒドロキシ安息香酸)との共結晶の調製方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびゲンチシン酸(2,5−ジヒドロキシ安息香酸)を、約1:1〜1:1.2.25の範囲のモル比、好ましくは約1:1のモル比でアセトン中に溶解させる工程、
b)好ましくは窒素流下で室温にてアセトンを蒸発させる工程。
【0060】
1実施態様において、上記の工程a)は、最初に化合物Aをアセトンに溶解させ、その後ゲンチシン酸を加えることにより実施される。別の実施態様において、工程b)は、アセトンに溶解させた化合物Aの溶液とアセトンに溶解させたゲンチシン酸の溶液とを混合することによって実施される。
【0061】
本発明の実施態様において、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)との共結晶の焼成方法は、以下の工程を包含する(または以下の工程からなる):
a)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R )−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよびキシナホ酸(1−ヒドロキシ−2−ナフタレン−2−カルボン酸)を、約1:1のモル比で酢酸エチルに溶解させる工程、
b)好ましくは窒素流下で室温にて酢酸エチルを蒸発させる工程。
【0062】
さらに、本発明は(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと少なくとも1種の上記共結晶形成剤との共結晶の調製方法に関し、好ましくは1種、2種または3種の共結晶形成剤が上記のような方法に適用される。
【0063】
本発明はまた、本発明に従う少なくとも1種、好ましくは1種、2種または3種の共結晶を含む医薬組成物にも関する。
【0064】
本発明はまた、1種またはそれ以上の薬学的に許容可能な賦形剤と一緒に、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、好ましくは(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン、または薬学的に許容可能なその誘導体もしくはアナログの共結晶および本発明に従う前出の共結晶を含む医薬組成物に関する。このタイプの医薬組成物、賦形剤および調製は、WO2008/015270およびWO2008/015269により詳細に記載されている。
【0065】
本発明はまた、異常なグルタミン酸神経伝達に関連している状態または疾患、好ましくは以下に記載するような状態または疾患の治療および/または予防に用いるための、本発明に記載されるような(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶にも関する。
【0066】
好ましくは、本発明は、アルツハイマー病、統合失調症の陽性および/もしくは陰性症状、認知障害の状態または疾患の治療および/または予防に用いるため、または認知向上および神経保護に用いるための、本発明に記載されるような(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶に関する。
【0067】
用語「アナログ」または「誘導体」は、本明細書において慣用の製薬的意味で用いられ、参照分子に構造的に類似している分子であるが、参照分子の1つまたは複数の特定の置換基を代替置換基によって置換し、そのことによって、参照分子に構造的に類似した分子を作り出すために標的化され、制御された様式で修飾されている分子のことである。アナログを合成およびスクリーニングして、改善された、または偏りを持たせた特徴(特定の標的受容体タイプにおけるより高い効力および/または選択性、脳血管関門を通過するより高い能力、より少ない副作用など)を有し得る、既知化合物をわずかに修飾した変型を同定することは、製薬化学においてよく知られた薬剤設計法である。また、当業者に知られている方法を用いて、改善された治療有効性、すなわち、特定の標的受容体タイプにおけるより高い効力および/または選択性、脳血管関門を通過するより高い能力またはより低い能力(例えば、より高い、またはより低い脳血管関門通過率)、より少ない副作用などを有する本発明の化合物のアナログおよび誘導体を作り出すことができる。
【0068】
本発明の組成物に関連して用いられる語句「薬学的に許容できる」とは、哺乳動物(例えばヒト)に投与した際、生理学的に許容性であり、一般に有害な反応を生じさせない分子全体およびこのような組成物の他の成分のことである。本明細書に用いられる用語「薬学的に許容できる」は、哺乳動物における使用が、特にヒトにおける使用が、連邦政府または州政府の監督機関によって承認されているか、または米国薬局方もしくは他の一般に認められた薬局方に記載されていることを意味することが好ましい。
【0069】
本発明に従う共結晶は、生きている動物の身体、特にヒトの種々の障害の治療および/または予防における適用を見出し得る。共結晶はまた、特定の状態が存在する必要はないが、特定の生理学的パラメータがインスタント化合物(instant compound)の投与を介して向上され得る(認知エンハンスメントを含む)、生きている動物の身体、特にヒトにおける徴候の治療に用途を見出す。
【0070】
生きている動物の体に存在する選択された病気の進行の阻害または緩和のための、本発明の共結晶によるその治療方法は、緩和されることが所望される特定の病気の緩和において有効である選択された用量を用いて、任意の通常受け入れられる医薬品経路により、以前に述べたように可能である。選択された病気または状態(特に病気または状態が、グループImGluRモジュレーターによる治療に影響を受けやすいもの)の進行の阻害または緩和のための、生きている動物の治療のための医薬の製造における本発明の共結晶の使用は、薬学的に受容可能な希釈剤、賦形剤またはキャリアと一緒に、有効量の本発明の共結晶を投与する工程を包含する通常の方法で実施される。
【0071】
それぞれの医薬組成物は、共結晶成分と、1種またはそれ以上の適切かつ薬学的に受容可能な賦形剤とを組み合わせることにより調製され得る。これらの組成物は経口、経皮、非経口、肺、直腸、経粘膜および鼻孔経路のような異なる経路を介して適用され得る。医薬品投薬形態は、動物および好ましくはヒトへの用途のための医薬を製造するために、例えば、粉末、顆粒、錠剤、フィルムコーティングされた錠剤、放出調整錠剤、ハードカプセル、ソフトカプセル、溶液、懸濁液、エマルジョン、クリーム、軟膏、ゲル、経皮パッチ、エアロゾル製剤、吸入およびマイクロまたはナノ粒子ベースの製剤であり得る。適切な本発明の製剤は、さらに溶媒に溶解させた共結晶の懸濁液である。
【0072】
本発明の医薬組成物において、1日の投与用の投薬単位あたり、本発明に従う共結晶は、例えば、0.1〜4000mg、好ましくは1〜2000mgの前記化合物をが乳する投薬単位として処方される。本発明の全ての局面について、特に医薬の局面においては、化合物または組成物の投与は、投薬レジメを有しており、治療している医師によって最終的に決定され、用いられる化合物、動物タイプ、性別、年齢、体重、症状の重篤度、投与方法、副作用反応および/または他の反徴候のような因子を考慮する。
【0073】
本発明に従う生理学的に許容可能な化合物は、通常例えば0.01mg/kg(処置される哺乳類の体重1kgあたりのmg)〜100mg/kg、好ましくは0.1mg/kg〜75mg/kgの経口用量である。
【0074】
さらに、本発明は、ヒトを含む動物における神経保護を提供するための医薬としての、本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(および/またはそのR−エナンチオマー)の共結晶を含む組成物の使用に関する。
【0075】
さらに、本発明は、異常なグルタミン酸神経伝達に関連している状態の治療のため、またはその状態において治療の利益をもたらすmGluR5の調節のための、本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン(および/またはそのR−エナンチオマー)の共結晶の使用に関する。
【0076】
特に、本発明は以下から選択される状態または疾患の予防および/または治療のための医薬の調製のための、本発明に従う共結晶の使用を取り扱う:
アルツハイマー病、クロイツフェルト−ヤコブ症候群/病、ウシの海綿状脳症(BSE)、プリオン関連の感染症、ミトコンドリア機能障害が関与する疾患、β−アミロイドおよび/またはタウオパシーが関与する疾患、ダウン症候群、肝性脳症、ハンチントン病、運動ニューロン疾患、筋萎縮側索硬化症(ALS)、オリーブ橋小脳萎縮、術後認知機能障害(POCD)、全身性エリテマトーデス、全身性硬化症、シェーグレン症候群、神経セロイドリポフスチノーシス、神経変性小脳性運動失調、パーキンソン病、パーキンソン病の認知症、軽度認知障害、軽度認知障害の種々の形態の認知欠陥、認知症の種々の形態の認知欠陥、ボクサー認知症、血管性および前頭葉型認知症、認知障害、学習障害、眼の損傷、眼の疾患、眼の障害、緑内障、網膜症、黄斑変性、頭部もしくは脳または脊髄の損傷、頭部もしくは脳または脊髄の外傷、外傷、低血糖、低酸素症、周産期低酸素症、虚血、心停止もしくは脳卒中もしくはバイパス手術もしくは移植から生じる虚血、痙攣、てんかん様痙攣、てんかん、側頭葉てんかん、間代性筋痙攣、内耳傷害、耳鳴における内耳傷害、耳鳴、音−または薬物−誘発性内耳傷害、音−または薬物−誘発性耳鳴、L−ドーパ−誘発性ジスキネジア、パーキンソン病の治療におけるL−ドーパ誘発性ジスキネジア、ジスキネジア、ハンチントン病におけるジスキネジア、薬物誘発性ジスキネジア、神経遮断薬−誘発性ジスキネジア、ハロペリドール−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、舞踏病、ハンチントン舞踏病、アテトーシス、ジストニア、常同症、バリズム、遅発性ジスキネジア、チック障害、痙性斜頚、眼瞼痙攣、局所性および全身性ジストニア、眼振、遺伝性小脳性運動失調、大脳皮質基底核変性症、振せん、本態性振せん、乱用、嗜癖、ニコチン嗜癖、ニコチン乱用、アルコール嗜癖、アルコール乱用、アヘン嗜癖、アヘン乱用、コカイン嗜癖、コカイン乱用、アンフェタミン嗜癖、アンフェタミン乱用、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、注意欠如多動性障害(ADHD)、注意欠陥症候群(ADS)、レストレスレッグ症候群(RLS)、子供における多動、自閉症、認知症、アルツハイマー病における認知症、コルサコフ症候群における認知症、コルサコフ症候群、血管性認知症、HIV感染症に関連する認知症、HIV−1脳障害、エイズ脳症、エイズ認知症複合、エイズ関連認知症、大うつ病性障害、大うつ病、うつ病、ボルナウイルス感染から生じるうつ病、ボルナウイルス感染から生じる大うつ病、双極性躁うつ病、薬物耐性、オピオイドに対する薬物耐性、運動障害、ぜい弱X症候群、過敏性腸症候群(IBS)、片頭痛、多発性硬化症(MS)、筋痙攣、疼痛、慢性疼痛、急性疼痛、炎症性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、関節リウマチに関連する疼痛、アロディニア、痛覚過敏、侵害受容性疼痛、癌性疼痛、心的外傷後ストレス障害(PTSD)、統合失調症、統合失調症の陽性もしくは認知または陰性症状、痙性、ツレット症候群、尿失禁、嘔吐、そう痒状態、そう痒症、睡眠障害、排尿障害、下部尿路における神経筋障害、胃食道逆流性疾患(GERD)、胃腸障害、下部食道括約筋(LES)疾患、機能性胃腸障害、消化不良、吐出、気道感染症、神経性過食症、慢性喉頭炎、喘息、逆流関連の喘息、肺疾患、摂食障害、肥満、肥満関連の障害、肥満乱用、食品嗜癖、過食症、広場恐怖、全般性不安障害、強迫性障害、パニック障害、心的外傷後ストレス障害、社会恐怖症、恐怖症、物質誘発性不安障害、妄想性障害、統合失調性感情障害、統合失調症様障害、物質誘発性精神病性障害、または、せん妄;末梢組織、末梢神経系およびCNSにおける腫瘍細胞成長、移動、侵入、付着および毒性の阻害;新形成、過形成、異形成、癌、癌腫、肉腫、口腔癌、扁平上皮癌(SCC)、口腔扁平上皮癌(SCC)、肺癌、肺腺癌、乳癌、前立腺癌、胃癌、肝臓癌、大腸癌、結腸直腸癌腫、横紋筋肉腫、脳腫瘍、神経組織の腫瘍、神経膠腫、悪性神経膠腫、星細胞腫、神経膠腫、神経芽腫、グリア芽腫、髄芽腫、皮膚細胞の癌、黒色腫、悪性黒色腫、上皮新生物、リンパ腫、骨髄腫、ホジキン病、バーキットリンパ腫、白血病、胸腺腫および他の腫瘍。
【0077】
治療することができる障害は、すでに上に記載されている。好ましい状態および適応症は、以下の通りである:
a)mGluR5モジュレーターについて:慢性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、癌性疼痛、関節リウマチに関連する疼痛、炎症性疼痛、L−ドーパ−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、パーキンソン病治療におけるL−ドーパ−誘発性ジスキネジア、パーキンソン病治療におけるドーパミン模倣薬−誘発性ジスキネジア、遅発性ジスキネジア、パーキンソン病、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、全般性不安障害、物質誘発性不安障害、摂食障害、肥満、過食症、ハンチントン舞踏病、てんかん、アルツハイマー病、統合失調症の陽性および陰性症状、認知障害、機能性胃腸障害、胃食道逆流性疾患(GERD)、片頭痛、過敏性腸症候群(IBS)、または認知強化および/または神経保護のため。
b)mGluR5の負の調整について:慢性疼痛、神経因性疼痛、糖尿病性神経因性疼痛(DNP)、癌性疼痛、関節リウマチに関連する疼痛、炎症性疼痛、L−ドーパ−誘発性ジスキネジア、ドーパミン模倣薬−誘発性ジスキネジア、パーキンソン病治療におけるL−ドーパ−誘発性ジスキネジア、パーキンソン病治療におけるドーパミン模倣薬−誘発性ジスキネジア、遅発性ジスキネジア、パーキンソン病、不安障害、パニック障害、不安およびパニック障害、社会不安障害(SAD)、全般性不安障害、物質誘発性不安障害、摂食障害、肥満、過食症、片頭痛、過敏性腸症候群(IBS)、機能性胃腸障害、胃食道逆流性疾患(GERD)、ハンチントン舞踏病および/またはてんかん。
c)mGluR5の正の調整について:アルツハイマー病、統合失調症の陽性および/または陰性症状、認知障害、または認知強化および/または神経保護のため。
【0078】
本発明に従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの共結晶は特に、過食症障害の治療のために用いることができる。
【図面の簡単な説明】
【0079】
【図1】実施例2aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の粉末X線回折チャートである。調製:実施例2aに従って入手されるような粉末、0.1mm Si(シリコン)。
【図2】実施例2aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。(ピーク=158.20℃;ピークkorr.=156.9℃、ピーク高さ=17.6434mW、ΔH=112.6J/g)。
【図3】実施例3aに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の粉末X線回折のチャートである。調製:実施例3aに従って入手した粉末、0.1mm Si(シリコン)。
【図4】実施例3cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の粉末X線回折チャートである。調製:実施例3cに従って入手した粉末、0.1mm Si(シリコン)。
【図5】実施例3cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとゲンチシン酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。融解ピーク:ピーク=148.17℃;ピークkorr.=147.4℃、ピーク高さ=4.4950mW、Area=185.679mJ、ΔH=56.8696J/g。吸熱事象:ピーク=105.0℃;ピークkorr.=104.1℃、ピーク高さ=0.1501mW、Area=16.336mJ、ΔH=5.0035J/g。
【図6】実施例4cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸の共結晶の粉末X線回折チャートである。調製:実施例4cに従って入手した粉末、0.1mm Si(シリコン)。
【図7】実施例4cに従う(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとキシナホ酸の共結晶の示差走査熱量測定チャート(DSC分析チャート)である。DSC測定は、閉じてあるAu製のるつぼ中にて窒素下で、−50.00℃〜240℃まで10.00℃/分の加熱速度で加熱しながら実施した。融解ピーク:ピーク=140.30℃;ピークkorr.=139.2℃、ピーク高さ=10.2330mW、Area=229.500mJ、ΔH=79.3568J/g.吸熱事象:ピーク=202.9℃;ピークkorr.=202.6℃、ピーク高さ=0.5661mW、Area=67.712mJ、ΔH=23.4134J/g。析出:ピーク=240.70℃;ピークkorr.=241.1℃、ピーク高さ=−8.7764mW、Area=−816.376mJ、ΔH=−282.2879J/g。
【実施例】
【0080】
(実施例1) 出発物質の特徴づけ
出発物質である薬学的に有効な成分(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンをWO 2008/015269に記載されたように調製した。
【0081】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンのpKa値をACD/Labs、pKa DB v10.0を用いて算出した(ピラゾロ(1,5−a)ピリミジンを相関化合物として用いた)。プロトン化形態の算出pKa値は、−1.97± 0.30である。従って、この分子は非常に弱い塩基であり、この低いpKa値は伝統的な塩形成のために適切ではない。
【0082】
さらに、この出発物質を、実施例7に記載されるように特にPXRD、FT−Ramanおよび1H NMR分光法によって特徴づけた。
【0083】
遊離薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの結晶構造をX線結晶学によって測定した。結晶(無色のブロック、0.16×0.32×0.36)をKappa APEX2回折計でT=123Kにてθmax=36.345°でのグラファイト−単色化モリブデン−K−α(Kα)照射(波長 λ=0.71073Å)を用いて測定した。APEX2ソフトウエアパッケージを補正および照射に用いた。この構造を、プログラムSIR92を用いたダイレクトメソッドによって解析した。プログラムCRYSTALSを用いて、全ての非水素原子において、Fに対する最小二乗精密化を実施した。
以下の結晶データが見出された:
F(000)=752、斜方晶系、空間グループP212121、Z=4 算出密度Dca,,=1.605mg・m-3;a=7.5918(2)Å(オングストローム)、b=13.3879(4)Å(オングストローム)、c=15.1119(5)Å(オングストローム)、α(アルファ)=90°、β(ベータ)=90°、γ(ガンマ)=90°、V=1535.95(8)Å3(オングストローム)。
【0084】
共結晶形成剤2,5−ジヒドロキシ安息香酸(ゲンチシン酸、GEN)を、Flukaより購入した(注文番号37550、C7H6O4;Mw154.12g/mol)。1−ヒドロキシ−2−ナフトエ酸(キシナホ酸、XIN)を、Flukaより購入した(注文番号55910;C11H8O3;Mw188.18g/mol)。ブタン二酸(コハク酸、SUC)をFlukaより購入した(注文番号14079、C4H6O4;Mw118.09g/mol)。
【0085】
(実施例2)(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンとコハク酸の共結晶の調製
実施例2a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび57.8mgのコハク酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温で24時間攪拌し、最終的に溶媒を空気中で室温にて(解放バイアル)蒸発させた。白色粉末が得られた。
【0086】
得られた粉末をFT Ramanによって特徴づけた。このFT Ramanスペクトルは、遊離有効成分とコハク酸の混合物を示している。
【0087】
2mlのイソプロピルエーテルを得られた粉末の残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し、空気中で乾燥させた。無色の粉末が得られ、実施例7に記載されているようにFT Raman、PXRD、1H−NMR、TG−FTIR、DSC、DVSによって特徴づけた。
【0088】
得られた結晶粉末は、独特のRamanスペクトルを示し、NMRデータにより所定の共結晶構造が確認された。
【0089】
この粉末をPXRDパターンによって特徴付けした(これを図1に示す)。
DVSデータは、得られた結晶粉末は水分を取り込まない(吸湿性ではない)ことを示していた。
【0090】
TG−FTIR測定は、得られた結晶粉末は、微量のイソプロピルエーテル/水を含んでおり、150℃を超えると分解することを示している。さらに、得られた結晶粉末は156.9℃で融解ピークによって特徴づけられた(DSC)。このDSCダイアグラムを図2に示している。
【0091】
実施例2b:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解させた。57.8mgのコハク酸(2mlのアセトンに溶解)を加えた。この溶媒を流量制御をせずに窒素流下で室温にて蒸発させた。無色の粉末が得られた。この得られた粉末を、実施例7に記載したようにFT Ramanによって特徴付けし、このRamanスペクトルは遊離塩基と同様である。
【0092】
2mlの2−プロパノールを得られた残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し空気中で乾燥させた。無色の粉末が得られ、これをFT Ramanで特徴付けした。この共結晶は実施例2aに従う共結晶と同じRamanスペクトルを示している。
【0093】
実施例3:(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと2,5−ジヒドロキシ安息香酸(ゲンチシン酸)の共結晶の調製
実施例3a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解した。75.5mgのゲンチシン酸(2mlのアセトンに溶解させた)を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末が得られた。
【0094】
得られた生成物を実施例7に記載したようにFT Raman、PXRD(図3)、1H−NMRによって特徴付けした。
【0095】
得られた粉末のX線回折パターンを図3に示しており、結晶形態(共結晶)が確認される。1H−NMRスペクトルにより共結晶の所定の構造が確認された。
【0096】
実施例3b:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと75.5mgのゲンチシン酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温で約24時間攪拌し、最終的に溶媒を室温にて空気中で(開口バイアル)蒸発させた。アイボリー色の粉末が得られた。得られた粉末のFT Ramanスペクトルは、実施例3a)のFT Ramanと一致する。
【0097】
実施例3c:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのアセトンに溶解させた。62.3mgのゲンチシン酸を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末を得た。
【0098】
得られた生成物を実施例7に記載されるようにRaman、PXRD(図4)、1H−NMR、TG−FTIR、DSC(図5)、DVS、およびFT Ramanによって特徴付けした。
【0099】
図4に示されれているようなPXRDパターンは、実施例3aに従う共結晶のX線パターンとは異なる結晶構造を示している。従って、共結晶の別の多型形態が得られた。さらに得られた共結晶は、微量のイソプロピルエーテル/アセトンを含んでいる。NMRデータは、所定の構造と一致している。
【0100】
このTG−FTIR分析は、150℃より高い温度での分解を示しており;DSCダイアグラムは、104℃での吸熱事象、147℃での融解ピークを示している(DSCダイアグラム図6を参照のこと)。
【0101】
DVS測定の結果として、得られた共結晶は最小限の水の取込を示しており、かつ吸湿性がない。
【0102】
実施例4:(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと1−ヒドロキシ−2−ナフトエ酸(キシナホ酸)の共結晶
実施例4a:150mgの(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを、5mlのEtOAcに溶解した。92.1mgの1−ヒドロキシ−2−ナフトエ酸(キシナホ酸)(2mlの酢酸エチルに溶解)を加えた。この溶媒を流量制御なしに窒素流下で室温にて蒸発させた。アイボリー色の粉末が得られ、これを実施例7に記載されているようFT Raman、PXRD、1H−NMRで特徴づけした。
【0103】
得られた粉末のX線回折パターンは、実施例4に従うPXRDパターンと広範囲にわたって同一であり、結晶形態(共結晶)が確認される。1H−NMRスペクトルにより、共結晶の予測された構造と一致した。
【0104】
実施例4b:150mgの6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと92.1mgの1−ヒドロキシ−2−ナフトエ酸を混合した。0.15mlのイソプロピルエーテルを加えた。この混合物を室温(r.t.)で約24時間攪拌し、最終的に溶媒を空気中(開口バイアル)で室温(r.t.)にて蒸発させた。アイボリー色の粉末が得られ、得られた粉末のRamanスペクトルは、共結晶と有効成分の混合物であった。
【0105】
2mlのイソプロピルエーテルを得られた残留物に加えた。この混合物を約16時間攪拌した。得られた固体を濾過し、空気中で乾燥させた。アイボリー色の粉末が得られた。得られた粉末のFT Ramanは、実施例4aに従って得られた共結晶のスペクトルと一致した。
【0106】
実施例4c:150mgの6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンを5mlのEtOAcに溶解させた。76mgの1−ヒドロキシ−2−ナフトエ酸(2mlのEtOAcに溶解)を加えた。この溶媒を流量制御なしに窒素下で室温(r.t.)にて蒸発させた。アイボリー色の粉末が得られ、実施例7に記載されるように、FT Raman、PXRD(図6)、1H NMR、TG−FTIR、DSC(図7)、DVSで特徴付けした。
【0107】
得られた粉末のFT Ramanは、実施例4aに従って得られた共結晶のスペクトルと一致した。
【0108】
得られた粉末のX線回折パターンを図6に示しており、結晶形態(共結晶)であることが確認される。1H−NMRスペクトルは、共結晶の構造と一致した。
【0109】
DSC分析(DSCダイアグラムは、図7に示されている)は、得られた共結晶が、139℃で融解ピーク、241℃で吸熱事象を表わしていることを示している。TG−FTIR測定は、キシナホ酸共結晶が微量の酢酸エチルを含有しており、150℃を超える温度で分解を示すことを実証している。
【0110】
DVS測定により、可逆的な水の取込が、80%の相対湿度を超えることを示している。従って、公知の化合物Aとキシナホン酸の得られた共結晶は、わずかに吸湿性である。
【0111】
実施例5:溶解度の測定
各共結晶(実施例2a、3c;および4cに従う)を水中に懸濁させた。このサンプルを、Eppendorf製の温度制御された「Thermomixer comfort」を用いて800rpm(24時間、23℃)で振盪させた。得られた懸濁液を、Millipore Centrifugal Filter Device UFC30VVNB(0.1)およびCentrifuge Hettich EBA 12R(10.000g)を用いて濾過した。得られた固体をFT−Raman分光計によって特徴付けし、溶解性のテスト前のスペクトルと比較すると、形態の変化は観察されなかった。
【0112】
ろ液のpHを測定し、遊離塩基の濃度をHPLCによって測定した(方法は実施例7に記載されている)。
【0113】
同様に(同じ条件で)測定した検出限界付近である遊離薬物化合物の溶解度は、pH8.5にて10μg/ml未満(0.01mg/l未満)を示している。
【0114】
新規の共結晶の測定された溶解度を表1にまとめた。
【0115】
【表1】
【0116】
本発明に従う共結晶は、驚いたことに遊離薬物と比較して2倍の溶解度であった。
【0117】
実施例6:熱分析技術によるコハク酸、ゲンチシン酸、およびキシナホ酸の共結晶の特徴付け
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンの以下の共結晶を実施例7に記載されるようにTG−FTIR、DSCおよびDVSによって特徴付けした:
実施例2aに従うコハク酸の共結晶、
実施例3cに従うゲンチシン酸の共結晶、および
実施例4cに従うキシナホン酸の共結晶。
【0118】
実施例6a:TG−FTIRを上記のサンプルに対して実施した。結果を以下の表1にまとめる。
【0119】
【表2】
【0120】
コハク酸共結晶サンプルは、微量のイソプロピルエーテル(調製に用いられる溶媒)を含有している。150℃を超えて分解が観察された。
【0121】
ゲンチシン酸共結晶サンプルは、微量のアセトン(調製に用いられる溶媒)を含有している。180℃を超えて分解が観察された。
【0122】
キシナホン酸共結晶サンプルは、微量の酢酸エチル(調製に用いられる溶媒)を含有している。160℃を超えて分解が観察された。
【0123】
実施例6b:上記のサンプルに対してDSCを実施した。結果を以下の表2にまとめる。
【0124】
【表3】
【0125】
化合物Aは、132〜140℃の範囲で融解する(ピーク132.9℃)。
【0126】
実施例2aに従うコハク酸共結晶のDSC(図2を参照)は、156.9℃で鋭い融解ピークを示している。
【0127】
実施例3cに従うゲンチシン酸共結晶のDSC(図5を参照)は、104℃で広範な吸熱事象を示している(ΔH=5J/g)。おそらくこのサンプルは、微量の出発物質および/または他の不純物を含有している。147℃における第2の吸熱事象は、共結晶の融解に寄与し得る。分解は200℃を超えて観察された。
【0128】
実施例4に従うキシナホン酸共結晶のDSC(図7を参照)は、139.2℃で鋭い融解ピークを示し、200℃を超えて分解を示している。
【0129】
実施例6c:DVS(50% −> 0% −> 95% −> 50% r.h.)を、上述のサンプルに対して実施した。この結果を以下にまとめる:
【0130】
ゲンチシン酸共結晶のDVS(実施例2a)は、試験した湿度範囲にわたって最小かつ可逆的な質量変化のみを示している。約0.1%の質量変化Δm(50〜85%までの相対湿度(r.h.)の変化)が観察され、この共結晶は吸湿性ではない。DVS Ramanスペクトル後は、形態に何の変化も示さない。
【0131】
コハク酸共結晶のDVS(実施例3c)は、試験した湿度範囲にわたって最小かつ可逆的な質量変化のみを示している。約0.1%の質量変化Δm(50〜85%までの相対湿度(r.h.)の変化)が観察され、この共結晶は吸湿性ではない。DVS Ramanスペクトル後は、形態に何の変化も示さない。
【0132】
キシナホン酸共結晶のDVS(実施例4c)は、ヒステリシスで80%の相対湿度を超える可逆的な水の取込を示している。約1%の質量変化Δm(50〜85%までの変化)が観察された。この共結晶はわずかに吸湿性である。湿度が95%の相対湿度まで増加した場合より多くの水が取り込まれた((約2質量%の総水含有量、平衡到達)。相対湿度を再び下げると、水含有量は減少し、元の質量まで戻った。DVS Ramanスペクトル後は形態に何の変化も示さない。
【0133】
実施例7:装置の測定条件
実施例7a:DSC(示差走査熱量測定)/Perkin Elmer DSC 7は、閉じたAu製のるつぼを用いた。加熱速度:10または20℃/分、範囲:−50℃〜250℃。
【0134】
実施例7b:DVS(動的水蒸気吸脱着測定装置)
Surface Measurement Systems Ltd.製のDVS−1水蒸気吸着分析器およびProjekt Messtechnik製のSPS 11−100n マルチサンプル水蒸気吸着分析器を用いた。
【0135】
このサンプルを開始前に50%の相対湿度で平衡にしておいた(例えば、以下の所定の湿度プログラム):
0%の相対湿度で2時間
95%の相対湿度で0〜95%(5%/時)
95%の相対湿度で3時間
95%〜0%(10%/時)
0%の相対湿度で2時間。
【0136】
実施例7c:使用したHPLC(高速液体クロマトグラフィー)システムは、以下のように特徴付けられる:
装置:TSP HPLC(UV3000、AS3000、P4000、SCM1000
Soft. Version 4.1)
カラム:水、XTerra MS C18 4.6x100mm 5μ(CC01)
移動相A:希釈H2O + 0.1%TFA
移動相B:MeCN + 0.1%TFA
参照濃度:ca. 0.04mg/ml
保持時間:13.1分
勾配: 0.0分 95% A / 5% B
20.0分 5% A / 95% B
21.0分 95% A / 5% B
30.0分 95% A / 5% B
流量:1.0ml/分
注入量:10μl
波長:254nm
【0137】
実施例7d:NMR(核磁気共鳴)
1H NMRスペクトルを、Bruker DPX300装置で、300.13MHzで記録した。
【0138】
実施例7e:ラマン顕微
安定化ダイオードレーザー785−nm励起および検出器としてNIR−増強ペルチェ吸熱CCDカメラを用いて、ラマン顕微をRenishaw RM 1000で記録した。測定は長作動距離20倍で実施した。測定範囲は2000〜100cm-1である。
【0139】
実施例7f:FT−ラマン分光法(フーリエ変換ラマン分光法)
FT−ラマンスペクトルを、Nd:YAG 1064nm励起、100mWのレーザーパワーおよびGE検出器、64スキャン、範囲25〜3500cm-1、2cm-1解像度で、Bruker RFS100で記録した。
【0140】
実施例7g:TG−FTIR(フーリエ変換赤外分光法と組み合わせた熱重量計)
TG−FTIRを、Al製のるつぼ(開口または微細孔のあるもの)中、N2雰囲気下、10℃min-1の加熱速度、25〜250℃の範囲でNetzsch Thermo−Microbalance TG 209 with Bruker FT−IR Spectrometer、Vector 22を用いて実施した。
【0141】
実施例7h:溶解度測定
水への共結晶の懸濁液をEppendorf製の温度制御された「Thermomixer comfort」を用いて800rpmで(24時間、23℃)攪拌した。この懸濁液をMillipore Centrifugal Filter Device UFC30VVNB(0.1μ)およびCentrifuge Hettich EBA 12 R(10´000g)で濾過した。
【0142】
実施例7i:PXRD(粉末X線回折)
粉末X線回折パターンを、銅Kα放射(Cu−Kα1、波長λ=1.540598Å(オングストローム)、40kV/40mAでBruker D8にて、およびLynxEye検出器、0.02°2θのステップサイズ、37sのステップ時間で記録した。
【0143】
サンプル調製:このサンプルは、通常平坦な表面を得るためにわずかな圧力をかける以外には何の特別な処理もなく測定した。シリコン単結晶サンプルホルダータイプ:a)多型スクリーニング用の標準ホルダー、0.1mm深さ、20mg未満のサンプル;b)0.5mm深さ、c.40mgのために12mmの孔直径;c)1.0mm深さ、c.80mgのために12mmの孔直径。Bruker D8で測定される全てのサンプルは測定の間に回転される。
【0144】
実施例8:溶解速度の測定
各共結晶(実施例2a、3cおよび4cに従う)について、および遊離薬物化合物(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン)について、圧縮された錠剤の溶解を、胃腸管を通る錠剤の通過をシミュレートするために4つのセクターを通してpHを増加させながら、UV吸収分光分析を用いて0.15Mの塩化カリウム水溶液中でモニターした(セクターIpH2.0;セクターII:pH 3.9;セクターIII:pH 5.4;セクターIV:pH7.3)。
この錠剤を約50000ポンド/inch-2の重さで圧縮し直径を3mmにした、錠剤の一面のみを、酢酸/リン酸緩衝液経を含む溶解培地に曝露し、薬物の溶解に起因する実験的なpHの逸脱を最小にした。溶液の攪拌を一定の速度で続けた。吸収データを事前に測定したpH依存係数、モルモル吸光係数を用いてサンプル重量の絶対値に変換した。適切な波長範囲は、UV光源の彩度に起因した誤った溶解データを避ける、1.3未満の吸光値の分光データが分析されることが保障されるように選択した。溶解速度は、得られた実験データに一次指数方程式をフィッティングすることで算出した。
【0145】
表3に列挙した溶解速度は、pH2.0、3.9、5.4、および7.3(±0.1)、温度23℃(±1℃)で測定した。表3にデータがない場合は、溶解速度は得番目〜3番目および4番目への移動において溶解速度が少しも変化しなかったことがわかる。
【0146】
【表4】
【0147】
例えば、MRZ8456のコハク酸共結晶の場合においては、pH2における遊離薬物MRZ8456と比較して3倍の溶解速度の増加が観察された。
【特許請求の範囲】
【請求項1】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であって、共結晶形成剤が、一般式(I):
【化1】
[式中、Rは以下:
a)
【化2】
(式中、nは1、2、3または4である)
または
b)
【化3】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸である、上記共結晶。
【請求項2】
請求項1に記載の共結晶であって、共結晶形成剤が、式(I):
【化4】
[式中、Rは以下:
a)
【化5】
(式中、nは2または3である)
または
b)
【化6】
(式中、
R1およびR2は、互いに独立して水素またはヒドロキシルであり、
R3およびR4は、水素であるかまたは、
R3およびR4は、それらを保持する炭素原子と一緒になって、非置換の芳香族6員環を形成してもよい)
を表わす]
のカルボン酸である、上記共結晶。
【請求項3】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、ここで共結晶形成剤が、ゲンチシン酸、コハク酸およびキシナホ酸からなる群より選択されるカルボン酸である、請求項1または2に記載の共結晶。
【請求項4】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン:共結晶形成剤のモル比が、1:0.1〜1:10の範囲内である、請求項1〜3のいずれか1項に記載の共結晶。
【請求項5】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がコハク酸であり、かつ共結晶が、以下:
9.3、16.0、20.0、22.9、および26.0°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも2つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約156.9℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項6】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がコハク酸であり、かつ共結晶が、
6.0、7.0、14.0、17.6、21.0、23.4、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも4つの粉末X線回折(PXRD)ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項7】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がゲンチシン酸であり、かつ共結晶が、以下:
6.9、12.6、21.2、および27.5°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約147.4℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項8】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がキシナホ酸であり、かつ共結晶が、以下:
3.9、11.6、18.1、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約139.2℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項9】
1つまたはそれ以上の薬学的に許容可能な賦形剤と一緒に、有効成分として請求項1〜8のいずれか1項に記載の共結晶を含む、医薬組成物。
【請求項10】
異常なグルタミン酸神経伝達に関連している状態または疾患の治療および/または予防に用いるための、請求項1〜8のいずれか1項に記載の共結晶。
【請求項11】
アルツハイマー病、統合失調症の陽性および/もしくは陰性症状、認知障害の状態または疾患の治療および/または予防に用いるための、または認知向上および/または神経保護に用いるための、請求項10に記載の共結晶。
【請求項12】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法であって、共結晶形成剤が、一般式(I):
【化7】
[式中、Rは以下:
a)
【化8】
(式中、nは1、2、3または4である)
または
b)
【化9】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸であり、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、スラリーの状態で分散させる工程、
を含む、上記方法。
【請求項13】
共結晶形成剤が、ゲンチシン酸、コハク酸、およびキシナホ酸からなる群より選択されるカルボン酸である、請求項12に記載の共結晶の調製方法であって、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、スラリーの状態で分散させる工程、
を含む、上記方法。
【請求項14】
溶媒S1および場合によるS2が、以下からなる群より選択される少なくとも1つの溶媒である、請求項12または13に記載の共結晶の調製方法:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)、およびN−メチルピロリドン(NMP)。
【請求項1】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であって、共結晶形成剤が、一般式(I):
【化1】
[式中、Rは以下:
a)
【化2】
(式中、nは1、2、3または4である)
または
b)
【化3】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸である、上記共結晶。
【請求項2】
請求項1に記載の共結晶であって、共結晶形成剤が、式(I):
【化4】
[式中、Rは以下:
a)
【化5】
(式中、nは2または3である)
または
b)
【化6】
(式中、
R1およびR2は、互いに独立して水素またはヒドロキシルであり、
R3およびR4は、水素であるかまたは、
R3およびR4は、それらを保持する炭素原子と一緒になって、非置換の芳香族6員環を形成してもよい)
を表わす]
のカルボン酸である、上記共結晶。
【請求項3】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、ここで共結晶形成剤が、ゲンチシン酸、コハク酸およびキシナホ酸からなる群より選択されるカルボン酸である、請求項1または2に記載の共結晶。
【請求項4】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノン:共結晶形成剤のモル比が、1:0.1〜1:10の範囲内である、請求項1〜3のいずれか1項に記載の共結晶。
【請求項5】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がコハク酸であり、かつ共結晶が、以下:
9.3、16.0、20.0、22.9、および26.0°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも2つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約156.9℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項6】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がコハク酸であり、かつ共結晶が、
6.0、7.0、14.0、17.6、21.0、23.4、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも4つの粉末X線回折(PXRD)ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項7】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がゲンチシン酸であり、かつ共結晶が、以下:
6.9、12.6、21.2、および27.5°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約147.4℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項8】
共結晶が、(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶であり、共結晶形成剤がキシナホ酸であり、かつ共結晶が、以下:
3.9、11.6、18.1、および27.2°(2θ)+/−0.3°(2θ)からなる群より選択される少なくとも1つの粉末X線回折(PXRD)ピーク;
示差走査熱量測定(DSC)によって測定される、約139.2℃での融解ピーク;
により特徴づけられる、請求項1〜4のいずれか1項に記載の共結晶。
【請求項9】
1つまたはそれ以上の薬学的に許容可能な賦形剤と一緒に、有効成分として請求項1〜8のいずれか1項に記載の共結晶を含む、医薬組成物。
【請求項10】
異常なグルタミン酸神経伝達に関連している状態または疾患の治療および/または予防に用いるための、請求項1〜8のいずれか1項に記載の共結晶。
【請求項11】
アルツハイマー病、統合失調症の陽性および/もしくは陰性症状、認知障害の状態または疾患の治療および/または予防に用いるための、または認知向上および/または神経保護に用いるための、請求項10に記載の共結晶。
【請求項12】
(6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンと共結晶形成剤との共結晶の調製方法であって、共結晶形成剤が、一般式(I):
【化7】
[式中、Rは以下:
a)
【化8】
(式中、nは1、2、3または4である)
または
b)
【化9】
(式中、
R1およびR2は、互いに独立して水素、ヒドロキシルまたはカルボキシルであり、
R3およびR4は、互いに独立して水素、ヒドロキシルもしくはカルボキシルであるか、
またはR3およびR4は、それらを保持する炭素原子と一緒になって、C1−C5アルキル、ヒドロキシル、およびカルボキシルから選択される1〜4つの基で置換されてもよい芳香族6員環を形成する)
を表わす]
のカルボン酸であり、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、スラリーの状態で分散させる工程、
を含む、上記方法。
【請求項13】
共結晶形成剤が、ゲンチシン酸、コハク酸、およびキシナホ酸からなる群より選択されるカルボン酸である、請求項12に記載の共結晶の調製方法であって、以下:
a) 6−ブロモ−ピラゾロ[1,5−a]ピリミジン−2−イル)−(1(R)−メチル−3,4−ジヒドロ−1H−イソキノリン−2−イル)−メタノンおよび共結晶形成剤を溶媒S1に溶解する工程、
b)溶媒S1を蒸発させる工程、
c)場合により、工程b)で得られた残留物を溶媒S2に少なくとも10時間、スラリーの状態で分散させる工程、
を含む、上記方法。
【請求項14】
溶媒S1および場合によるS2が、以下からなる群より選択される少なくとも1つの溶媒である、請求項12または13に記載の共結晶の調製方法:アセトン、1−ブタノール、tert−ブチル−メチルエーテル(TBME)、ジメチルスルホキシド(DMSO)、エタノール、酢酸エチル、メチルエチルケトン(MEK)、1−プロパノール、2−プロパノール、テトラヒドロフラン(THF)、アセトニトリル、ジクロロメタン、N,N−ジメチルホルムアミド(DMF)、1−オクタノール、メタノール、トルエン、水、イソプロピルエーテル(IPE)、およびN−メチルピロリドン(NMP)。
【図1】

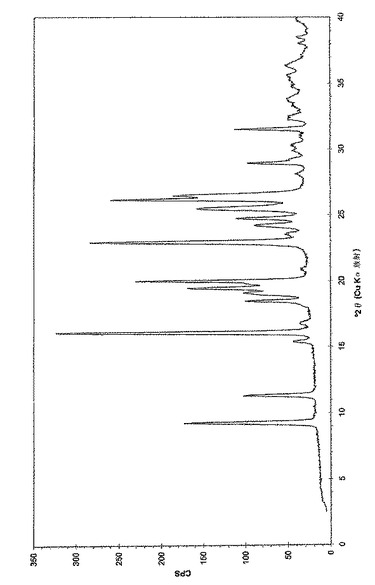
【図2】
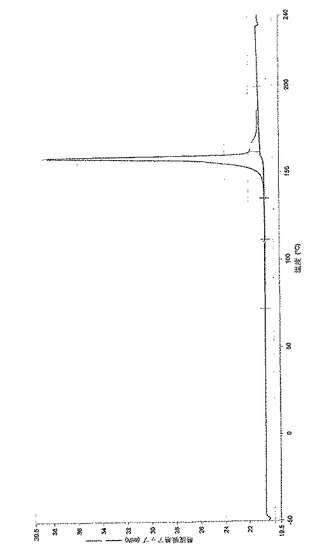
【図3】
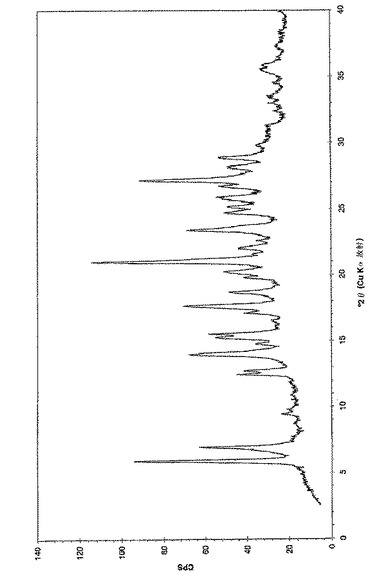
【図4】
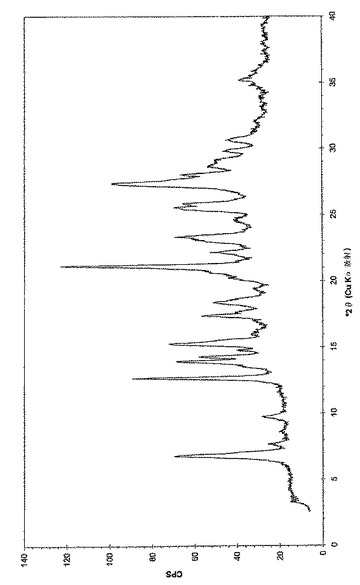
【図5】
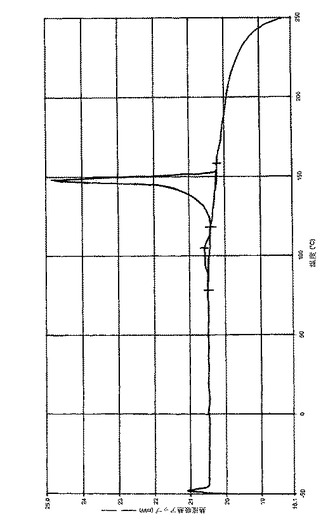
【図6】
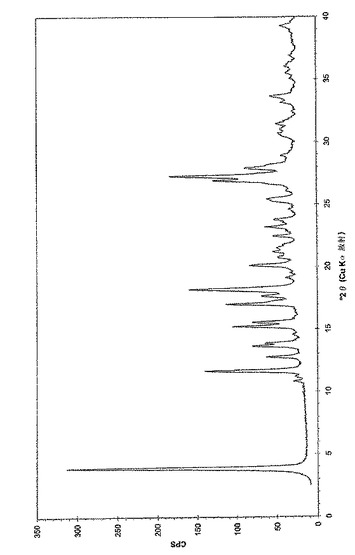
【図7】
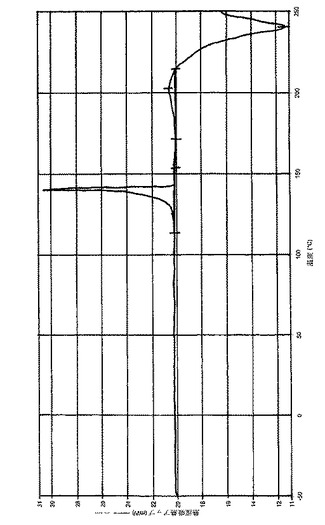
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2013−512216(P2013−512216A)
【公表日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願番号】特願2012−540411(P2012−540411)
【出願日】平成22年11月24日(2010.11.24)
【国際出願番号】PCT/EP2010/068093
【国際公開番号】WO2011/064237
【国際公開日】平成23年6月3日(2011.6.3)
【出願人】(397045220)メルツ・ファルマ・ゲゼルシヤフト・ミト・ベシュレンクテル・ハフツング・ウント・コンパニー・コマンデイトゲゼルシヤフト・アウフ・アクティーン (31)
【Fターム(参考)】
【公表日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願日】平成22年11月24日(2010.11.24)
【国際出願番号】PCT/EP2010/068093
【国際公開番号】WO2011/064237
【国際公開日】平成23年6月3日(2011.6.3)
【出願人】(397045220)メルツ・ファルマ・ゲゼルシヤフト・ミト・ベシュレンクテル・ハフツング・ウント・コンパニー・コマンデイトゲゼルシヤフト・アウフ・アクティーン (31)
【Fターム(参考)】
[ Back to top ]