
試料解析方法及び試料解析プログラム
【課題】試料に含まれる成分を分析するに際して、優れた分析能を達成することができる試料解析方法及び試料解析プログラムを提供する。
【解決手段】本発明に係る試料解析方法は、試料の分析の結果として得られた多次元データにおける、少なくとも1次元のパラメータを補正する工程aと、上記工程aにより得られる補正後のデータを複数の試料について比較する工程bとを含む。
【解決手段】本発明に係る試料解析方法は、試料の分析の結果として得られた多次元データにおける、少なくとも1次元のパラメータを補正する工程aと、上記工程aにより得られる補正後のデータを複数の試料について比較する工程bとを含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、試料の分析の結果として得られた多次元データを用いた試料解析方法及び試料解析プログラムに関する。
【背景技術】
【0002】
例えば、液体クロマトグラフィー(以下LCと略記する)と質量分析(以下MSと略記する)を連結した液体クロマトグラフィー質量分析(以下LC−MSと略記する)の結果として、横軸に質量/電荷比(以下m/zと略記する)、縦軸にイオン強度をとったグラフとして2次元上にスペクトルデータを得ることができる。ここで、LCの役割は、MSの処理能力に適応させるために、試料を単に分画することにある。
【0003】
すなわち、LCによって分画された試料をMSで分析することによって、上述したような2次元のスペクトルデータを得ることができ、試料中の成分分析を行うことができる。ところが、LCの役割を単に分画に限定することにより、従来のLC−MSでは、検体中から検出・識別できるタンパク質の種類が網羅的でなく、分析能・解析能が低いといった問題がある。
【0004】
一方で、クロマトグラフィーを単に分画だけでなく試料の特性を現す情報として利用する目的で、複数のクロマトグラフィー結果を比較できるようにするために、時間軸を補正して重ね合わせる(align)方法がいくつか提案されている。その代表的なものとして、Dynamic Time Warping (以下、DTWと略記する)および Correlation Optimized Warping (以下、COWと略記する)がある。いずれも動的計画法アルゴリズムに基づいた1つの実装形態として、それぞれユークリッド距離、あるいは相関を2つのクロマトグラフィーの距離もしくは類似性の指標とするものである(V. Pravdova, B. Walczak, D.L.Massart, “A comparison of two algorithms for warping of analytical signals”, Anal. Chim. Acta 456: 77-92 (2002))。しかしながら、これらの方法は、クロマトグラフィーの時間軸と信号強度との2次元で表されるクロマトグラムに適用されるものであるため、多次元データにおける少なくとも1次元のパラメータを補正するものではない。
【0005】
さらに、このような重ね合わせ方法は、比較対象となるクロマトグラムあるいはスペクトログラムがある程度類似していることが前提となる。実際、DTWにせよCOWにせよ、比較対象のプロファイル間における距離の最小化あるいは相関の最大化を目指した重ねあわせを行うため、比較対象のプロファイルの共通性が乏しい場合には適切な重ねあわせが得られない可能性が十分にある。このように高い共通性を前提とした方法は、例えば、実際の疾患病態解析や薬剤応答性解析のように、多くの因子の変動が予想され、しかもそれぞれの変動量が微細で、個体差や測定誤差などに紛れそうな場合には不適切である。
【0006】
そこで、本発明は、上述したような実状に鑑み、試料に含まれる成分を分析するに際して、優れた分析能を達成することができる試料解析方法及び試料解析プログラムを提供することを目的とする。
【0007】
【非特許文献1】V. Pravdova, B. Walczak, D.L.Massart, “A comparison of two algorithms for warping of analytical signals”, Anal. Chim. Acta 456: 77-92 (2002)
【発明の開示】
【課題を解決するための手段】
【0008】
上述した目的を達成した本発明は以下を包含する。
(1)試料の分析の結果として得られた多次元データにおける、少なくとも1次元のパラメータを補正する工程aと、上記工程aにより得られる補正後のデータを複数の試料について比較する工程bとを含む試料解析方法。
【0009】
本試料解析方法において、多次元データとしては、クロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを挙げることができる。また、このとき、工程aでは保持時間を示すパラメータを補正することが好ましい。
【0010】
また、ここで、補正対象のパラメータを除いたパラメータに関するプロファイルを参照プロファイルと定義し、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を与えることができる。この場合、工程aでは、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることができる。
【0011】
このとき、評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることが好ましい。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【0012】
また、工程aでは、補正対象のパラメータについて最適解探索問題として評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることができる。この動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることが好ましい。さらに、この場合、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることが好ましい。
【0013】
上記(1)に係る試料解析方法は、特に上記工程aにおいて、あらかじめ添加された標準物質に由来する情報を用いることによって、解析の精度をより向上できるとともに、補正処理の能力をも向上させる。本発明に係る試料解析方法のなかでも、このような特徴を備える方法をinternal standard guided optimal profile alignment(i-OPAL)法と命名する。
【0014】
また、上述した本発明に係る試料解析方法は、各種データを入力する機能を有する入力手段、プログラムに従って演算処理を実行する機能を有する演算処理手段、当該演算処理の結果等を表示する機能を有する表示手段を備えるコンピュータに実行させるためのプログラムとして実現することができる。
【0015】
一方、本発明に係る試料解析方法は、異なった種類の試料の間で量の異なる物質を検出・同定することができる。具体的には、複数の試料において多次元データとして、クロマトグラフィー質量分析の結果として得られる質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを測定し、複数の試料間で3次元データを比較し、イオン強度が有意に異なるシグナルを検出・同定することができる。これらのシグナルの特性、即ち、質量/電荷比および保持時間に十分近接した特性を持つシグナルを生じさせた物質に対して、更なる解析を施すことで、当該物質を同定することが可能である。
【0016】
さらに、検出・同定工程を、例えば疾患由来試料と、健常者あるいは健常組織由来試料とに適用することにより、疾患群と健常群で有意に異なる存在量を示す物質を検出・同定することができる。これによって同定された物質は、バイオマーカーとして利用することができる。バイオマーカー検出・同定結果を利用して、疾患の診断や治療法の選択へ利用することが可能である。
【0017】
さらにまた、検出・同定工程を、例えば特定の治療法あるいは薬物への応答性の差異、あるいは副作用の差異を示す患者集団由来の試料へ適用することで、治療法/薬剤応答性あるいは副作用のマーカーとなる物質の検出・同定へ利用可能である。
【発明を実施するための最良の形態】
【0018】
以下、図面を参照して本発明を詳細に説明する。
1.試料の調整
本発明に係る試料解析方法では、先ず、解析対象の試料を採取する。解析対象の試料としては、特に限定されないが、例えば、動物個体由来の臓器の組織切片や血漿・リンパ液などの体液成分、植物の緑葉や花弁などの器官、環境中の土壌・水成分などがあげられる。これらの試料に含まれる分析対象物質としては、特に以下に限定されないが、例えば、有機化合物、無機化合物、有機金属化合物、金属イオン、ペプチド、蛋白質、金属蛋白質、リン酸化を含む翻訳後修飾を受けたペプチド、リン酸化を含む翻訳後修飾を受けた蛋白質、核酸、糖質、脂質などがあげられるが、特に望ましくはペプチド、蛋白質、金属蛋白質、翻訳後修飾を受けたペプチドもしくは蛋白質である。
【0019】
また、採取した試料は、分析の目的及び採取した試料の特性に合わせて、必要であれば各種処理を施すことが好ましい。例えば、(ア)タンパク質群の分離あるいは分画、(イ)タンパク質群の酵素的及び/又は化学的切断、(ウ)切断によって生じたペプチド混合物の分離あるいは分画、および(エ)標準物質の添加、の全ての要素あるいは一部の要素の組み合わせて行う分析前調製を施すことが好ましい。
【0020】
より具体的に、「(ア)タンパク質群の分離あるいは分画」は、一次元のドデシル硫酸ナトリウム(SDS)電気泳動法、二次元電気泳動法、キヤピラリー電気泳動法、イオン交換クロマトグラフィー、ゲルろ過クロマトグラフィー、順相クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトグラフイー、或いはこれらの組み合わせによる多次元分離・分画等によって行うことができる。
【0021】
また、「(イ)タンパク質群の酵素的及び/又は化学的切断」は、トリプシン消化、キモトリプシン消化、Lys-C消化、Asp-N消化、Glu-C消化、臭化シアン分解或いはこれらの組み合わせによる切断等によって行うことができる。
【0022】
さらに、「(ウ)切断によって生じたペプチド混合物の分離あるいは分画」は、一次元のドデシル硫酸ナトリウム(SDS)電気泳動法、二次元電気泳動法、キャピラリー電気泳動法、イオン交換クロマトグラフィー、ゲルろ過クロマトグラフィー、順相クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトゲラフイー或いはこれらの組み合わせによる多次元分離・分画等によって行うことができる。
【0023】
さらにまた、「(エ)標準物質の添加」において標準物質は、選択したイオン化法にてイオン化できるものであって、測定のLC保持時間の範囲内に溶出するものであって、溶出時間および分子イオン強度の再現性が高いものを選択することが好ましい。このような好ましい標準物質としては、例えば、有機化合物、無機化合物、有機金属化合物、金属イオン、ペプチド、蛋白質、金属蛋白質、リン酸化を含む翻訳後修飾を受けたペプチド、リン酸化を含む翻訳後修飾を含む蛋白質、核酸、糖質、脂質など、より好ましくは、ペプチド・蛋白質で市販品、天然に存在する物質あるいは合成された物質を挙げることができる。
【0024】
以上、(ア)〜(エ)に示した分析前における各種処理は、例えば、「ア、エ、イ、ウの順」、「エ、イ、ウの順」、「イ、エ、ウの順」、「エ、アの順」、「エ、イの順」「イ、エの順」又は「エのみ」で行うことができる。
【0025】
2.試料分析
次に、試料を分析することによって、試料に関する多次元データを取得する。具体的には、試料をLC-MSにより分析し、m/z、イオン強度及び保持時間からなる多次元データを測定する。ここで、LC−MSにより分析するとは、試料をクロマトグラフィーの原理に従って分離又は分画し、その後、分離又は分画された試料に含まれる成分を質量分析の原理で測定することを意味する。なお、保持時間は、試料をクロマトグラフィーの原理に従って分離又は分画する際の時間として測定される。また、m/z及びイオン強度は、質量分析の結果として測定される。
【0026】
また、クロマトグラフィーの原理としては、特に限定されないが、逆相クロマトグラフィー、キヤピラリー電気泳動、アフィニティークロマトグラフイー、クロマトフォーカシング、等電点フォーカシング、ゲルろ過クロマトグラフィー等の各種クロマトグラフィーの原理を適用することができる。特に、本明細書においてLCと表記する場合、液体クロマトグラフィーのみを意味するのではなく、広く一般的なクロマトグラフィーを意味する。
【0027】
LC−MSにおけるクロマトグラフィーでは、再現性の高い溶出プロファイルが得られること、分離能が高いこと、適当なイオン化のインターフェースを介してMSに直接分子イオンを導入することが可能であることが好ましい。
【0028】
より具体的に、液体クロマトグラフィーにおける好ましい条件としては次の通りである。試料中のペプチド群を分析対称とする場合には、水・アセトニトリル溶液に低濃度の蟻酸などの強酸を含む溶離液を用いたC18カラムによる逆相液体クロマトグラフィーが好ましい。また蛋白質群を分析対称とする場合には、水・アセトニトリル溶液に低濃度の蟻酸などの強酸を含む溶離液を用いたC4カラムによる逆相液体クロマトグラフィーが好ましい。
【0029】
質量分析は、特に限定されないが、磁場型質量分析計、飛行時間型質量分析計、四重極質量分析計、イオントラップ質量分析計、フーリエ変換質量分析計またはこれらのハイブリッド及びタンデム質量分析計等により行うことができる。より好ましくは、エレクトロスプレーイオン化またはナノエレクトロスプレーイオン化と結合できる磁場型質量分析計、飛行時間型質量分析計、四重極質量分析計、イオントラップ質量分析計、フーリエ変換質量分析計またはこれらのハイブリッド若しくはタンデム質量分析計を用いて質量分析を行うことが好ましい。
【0030】
LC−MSにおける質量分析では、再現性の高い質量スペクトルが得られること、500ppm以下の高い質量精度を有すること、一定範囲のm/zの分子イオンに対して衝突誘起解離(CID)をかけ、当該分子イオンのフラグメントイオンの質量スペクトルが得られることが好ましい。
【0031】
このように、試料をLC−MSにより分析し、m/z、イオン強度及び保持時間を測定することによって、試料の分析結果を3次元データとして取得することができる。なお、LC−MSによる分析は、保持時間に関するデータ、m/zに関するシグナル及びイオン強度に関するデータを、入力手段を介してコンピュータに入力し、詳細を後述するアルゴリズムに従って演算処理手段によって処理を行うことにより図1に示すような3次元データとして取得することができる。本アルゴリズムは、コンピュータソフトウェアに搭載することができる。当該ソフトウェアをコンピュータにインストールすることによって、CPU等の演算処理手段により本アルゴリズムをコンピュータ上で実現することができる。従って、図1に示すような3次元データは、コンピュータの表示装置に表示することができる。
【0032】
従前のLC−MSによる解析方法においては、単に試料の分画のためにLCを行っているため、保持時間は解析対象パラメータとして使われておらず、試料の分析結果として横軸にm/z、縦軸にイオン強度をとった2次元データが解析対象となりうるに過ぎなかった。これに対して本発明に係る解析方法によれば、試料の分析結果を3次元空間上にプロットされるプロファイルとして取得することができるため、試料の分析能を飛躍的に向上させることができる。具体的には、本発明に係る解析方法によれば、保持時間を示す軸の方向に広がりを持った多数のスペクトルの重ね合わせとしてデータを取得でき、従前の解析方法と比較してより多数の成分に関して、スペクトルに基づく同定を行うことができる。このため、例えば、複数の試料について得られた多次元データを比較することで各試料の成分分析をより厳密に行うことができる。
【0033】
3.データ解析
次に、本発明に係る解析方法においては、以上のように測定した保持時間を、演算処理手段の制御のもとに本発明に係るアルゴリズムによって補正することもできる。ここで、一般に保持時間は、LCにおける移動相の組成、流速、カラム温度等のファクターが時間的に微小な変化を生じることから、非線形的に変動することが多い。したがって、本発明に係る解析方法で取得された3次元データに関しても、複数の試料について解析を行った場合に試料間の保持時間を示す軸が非線形的に変動していることが考えられる。そこで、本発明に係るアルゴリズムにおいては、保持時間の補正(以下、時間軸補正とも言う)を行う。
【0034】
但し、本発明に係るアルゴリズムが対象とする時間軸補正は、従前の方法でDTWアルゴリズム等によるクロマトグラムの時間軸補正のような、保持時間−信号強度の2次元空間で表現される単一次元プロファイルの補正ではない。本発明が対象とするデータは、時間軸に対して補正されるべきプロファイルが少なくとも2次元以上の多次元で表現されるものである。
【0035】
以下、当該アルゴリズムについて説明するが、当該アルゴリズムは保持時間の補正に限定されず、多次元のパラメータが得られた場合に少なくとも1次元のパラメータを補正する場合に広く適用することができる。言い換えると、当該アルゴリズムは、試料の分析の結果として得られた多次元のパラメータ(例えば3次元のパラメータ)における、少なくとも1次元のパラメータを補正する際に適用することができる。従って、以下の説明においては、p+q次元の測定データを取得した場合のアルゴリズムについて説明する。
【0036】
先ず、補正対象とするパラメータを含むp次元の測定値を(x1…xp)とし、補正の際に参照するq次元の測定値を(y1…yq)とすると、データの集合(プロファイル)ZはZ=(x1…xp y1…yq)となる。ここで、x及びyは、データ点の個数Nの次元を持つ列ベクトルである。
【0037】
なお、データ点とは、上記プロファイル行列(Z)の1つの行を構成するp+q次元のベクトルであり、測定対象の1つの要素について、測定パラメータと値の組を表している。特に、
【数1】

番目のデータ点を
【数2】
のようにも表す。
【0038】
また、補正の基準となる測定値を
【数3】
とする。ここで、Sは
【数4】
(Sは基準点の数)を意味する。また、
【数5】
は、いずれも各基準点のとる値が推定可能な範囲に収まらなくてはならない。
【0039】
さらに、本アルゴリズムにおいて補正を行うためには2つ以上のプロファイルデータ
【数6】
が必要となる。
【0040】
以上のような定義の下で、本アルゴリズムにおいては先ず、p個のパラメータ軸x1…xpそれぞれにおいて取りうる値を量子化する。但し、量子化プロセスは、計算精度と計算時間との兼ね合いで行うものであり、値のとりうる場合の数が十分計算可能な範囲にあるならば、必ずしもこの段階で行わなくてもよい。次いで、p個のパラメータ軸x1…xpそれぞれにおいて
【数7】
の各データ点を、順列を保って対応付けを行う。なお、一般に、Z(1)及びZ(2)に含まれるデータ点の個数は異なりうるので、全てのデータ点が1対1に対応するわけではなく、対応する相手のないデータ点も含むことに留意する。
【0041】
このとき、例えば、以下のような評価関数を用いてプロファイル全体での対応付けの評価得点Eを算出する。なおこの評価得点は、類似性を表す尺度として、大きいほどよい「得点」として定義することも、逆に距離を表す尺度として、小さければよい「失点」として定義することも可能である。以下では失点としての定義で説明する。
【0042】
【数8】
ここで
【数9】
は、i番目のデータ点におけるr番目のパラメータの値を表し、N1、N2はそれぞれ1番目と2番目のプロファイルにおけるデータ点の総数である。また、上記評価関数において関数fは、対応する点の類似度合いの距離を与える関数で、例えば以下のような関数を挙げることができる。
【0043】
【数10】
ここで上記式(I)における、右辺第1項目は補正すべきパラメータxrにおいて当該パラメータの測度における差異の程度に応じたペナルティ、第2項目は合わせるべき測定パラメータysが補正後どれだけずれたかを当該パラメータ測度上の距離に応じたペナルティ、第3項目はパラメータ補正によって2つの点が全てのパラメータにおいて一致したことのボーナスとして与えられるスコア、逆に第4項目は補正対象パラメータ軸上での2つの点が一致しなかったことによるペナルティスコアに相当する。また第5項目は、後述するように、標準物質によるシグナルの一致をボーナスとして評価するための項である。
【0044】
また、上記式(I)における、α、β、σ及びπは、それぞれを含む項における係数であり、適宜設定できる値である。一例としては、αを1.0とし、βを0.1とし、パラメータ補正によって点が一致した場合にはσを0とし、一致しなかった場合にはπを100とすることができる。
【0045】
なお、関数δr(i,j)は、着目するパラメータrの値がi,jによって指定されるデータ点において対応した場合は1、しなかった場合は0を与える関数、逆に
【0046】
【数11】
は対応した場合が0、非対応の場合が1となる関数である。
【0047】
上記式(I)において、第2項目は、補正対称のパラメータを除いたパラメータに関するプロファイル(参照プロファイル)について、試料間での配置類似性の尺度を示している。
また、ここでは2点の不一致によるペナルティを与える式として、対応・非対応によって定数となる例を示したが、所定の関数によって算出される値であっても良い。例えば、隣接するデータ点が対応するか否か、対応しないデータ点が出現した列の長さ等を考慮した関数によって第4項目を算出することができる。
【0048】
また、上記式(I)において、ノルム
【数12】
は一般的なベクトル空間上の距離を表し、必ずしもユークリッド距離に限定するものではない。さらに
【数13】
のように2点間の値の差を計算する際に、対応する点がない場合は、値を0(または適当な欠損値の代替値)に置き換えて計算するものとする。
【0049】
なお、本発明において評価関数は、上記式(I)に示す関数に限定されるものではない。例えば、このようなデータ点(i,j)間の補正対象パラメータ或いは参照パラメータの距離の線形結合だけではなく、両者の距離に応じた任意の関数、更には直前若しくはそれまでに連続して対応したデータ点列におけるパラメータ間の距離も勘案した関数を定義することも可能である。また、評価関数としては、上記式(I)に限定されず、試料間において参照プロファイの配置類似性の尺度となるような関数を定義することも可能である。
【0050】
あるいは、ここでは失点としての例を示したが、上記式(I)における右辺各項の符号を逆転させた上で、距離の部分を相関などに置き換えることにより、逆に類似性を表す指標として、大きければ大きいほどよい得点としての評価関数を定義することも容易に可能であり、そのような評価関数も本アルゴリズムに適用することができる。
【0051】
上記式(I)の第5項で例示したように、対応する点が標準物質由来の基準点か否かで、例えば次のような特別な得点を与えるものとする。即ち、対応するデータ点がいずれも標準物質由来のものであった場合は、θ(i,j)=Smとして、はるかに大きな得点を設定することで、評価関数(この場合は距離即ち失点としての定義)が大きな負の値をとりうることで結果的にこのような対応関係が望ましいことを定義する。さらに、一方が標準物質由来であるのに他方がそうでない場合は、逆にθ(i,j)=-Sdとしてはるかに大きな距離となるように定義することも可能である。
【0052】
以上で説明した評価関数を最適化するアルゴリズムによって、上記「2.試料分析」で取得した3次元データに関して、保持時間を示すパラメータの補正を行うことができる。上記「2.試料分析」で取得した3次元データに関して、最適化アルゴリズムを適用する場合、以下の(a)〜(d)の手順に従って説明することができる。
【0053】
(a)保持時間補正の概念
保持時間を補正する操作は、m/z、イオン強度及び補正時間からなる単一の三次元パラメータ集合体を対象とするのではなく、2つの三次元パラメータ集合体の比較によって実現される。三次元パラメータ集合体は、図2に示すように、m/zと保持時間とをそれぞれ行と列にとった行列において、m/z及び保持時間が対応する位置の行列要素にイオン強度が入るような形で表される。保持時間を補正する対象の三次元パラメータ集合体をそれぞれZ(1)及びZ(2)とすると、保持時間の補正操作は、Z(1)及びZ(2)における2つの行列で、保持時間軸に相当する列の対応関係を決める操作(以下、「対応配置の検索」と呼ぶ)に他ならない。例えば、図2に示す行列をZ(1)の行列とし、図3に示す行列をZ(2)の行列とすると、図4のような配置が望ましい対応配置(重ね合わせ配置)である。
【0054】
(b)2つの3次元データにおけるパラメータ集合体間の対応配置の探索
図4に示すような対応配置を探索するためには、可能なすべての保持時間の対応付けを考える。この際、配置の対応関係の良し悪しを評価するスコアを定義し、配置毎にスコアを計算し、その中でもっともスコアがよいものを採用することで目的とする最適な対応配置を得ることができる。図5は、図2と図3に示した三次元パラメータ集合体Z(1)及びZ(2)に関して、保持時間の可能なすべての対応付けを示したものである。横方向にZ(1)の保持時間、縦方向にZ(2)の保持時間が記されているが、(イ)Z(1)及びZ(2)でそれぞれ対応する保持時間のある場合が斜線、(ロ)Z(1)の所定の保持時間に対してZ(2)の方に対応するものがない場合が横線、(ハ)Z(2)の所定の保持時間に対してZ(1)の方に対応するものがない場合が縦線で示されている。Z(1)及びZ(2)の全体的な保持時間の対応付けは、図5の格子の最左上角から最右下角にいたる経路を、これら斜線・横線・縦線をなぞることで求めることに相当する。但し、一度下がったり右に進んだら、進んだ点から逆に上や左に戻るような経路は許されない。なお、図5において太線で示された経路は、図4の対応対置に相当する。
【0055】
(c)保持時間対応配置の良否を判断するためのスコア
保持時間に関する対応配置の良否を判断するスコアは、例えば、次のようにして定義することができる。
i)最左上点におけるスコア、すなわちまだ対応関係がまったく決まっていない点でのスコアを0とする。
ii)前述の(イ)(ロ)及び(ハ)のうちいずれかの場合をとることにより、対応関係が1段階進んだ場合は、その直前のスコアに対して、(イ)(ロ)及び(ハ)毎に決められたスコアを加算することで、新たな対応関係の点におけるスコアとなる。例えば、以下のように(イ)(ロ)及び(ハ)毎にスコアを設定することができる。
【0056】
(イ)の場合(図5において斜線方向に進む場合):
この場合、所定の保持時間に関して、Z(1)及びZ(2)が互いに対応付けられるわけである。したがって、この場合、加算されるスコアとしては、m/zパラメータとイオン強度パラメータがZ(1)及びZ(2)間でどれだけ類似或いは離れているかを反映した値を設定することができる。以下の説明では類似度としてスコアを定義した場合について説明する。例えば、Z(1)において所定のm/zの元でイオン強度が検出されているのにZ(2)には同m/zの元でイオン強度が検出されなかったケース、あるいはその逆のケースであれば、一定の値(ペナルティスコア)を減じるようにスコアを設定することができる。また、所定のm/zにおいてZ(1)及びZ(2)それぞれにイオン強度が得られている場合、例えば両イオン強度の差の絶対値に所定の係数を乗じて算出される値(ペナルティスコア)を減じるようにスコアを設定することができる。さらに、スコアとしては、両イオン強度の違いが大きければ大きいほど、得点が小さくなるような関数で算出されるものであっても良い。
【0057】
一方、Z(1)及びZ(2)における保持時間のずれもスコアに反映させることもできる。例えば、Z(1)及びZ(2)における保持時間の差の絶対値に所定の係数を乗じて算出された値(ペナルティスコア)を減じるようにスコアを設定することができる。スコアとしては、Z(1)及びZ(2)における保持時間の違いが大きければ大きいほど、得点が小さくなるような関数で算出されるものであっても良い。
【0058】
なお、標準物質由来のシグナルがZ(1)及びZ(2)で対応する場合には、後述する計算方法の工夫に加えて、スコア算定上も特別な措置を施すことが好ましい。特に、これらの点がZ(1)及びZ(2)間で一致することは強く求められるので、Z(1)及びZ(2)ともに標準物質由来シグナルとして対応付けられる場合には大きな得点を、逆に一方だけしか標準物質由来シグナルが見つからない場合には大きな失点を与える。
【0059】
(ロ)及び(ハ)の場合(図5において縦又は横方向に進む場合):
この場合、所定の保持時間に関して、Z(1)及びZ(2)において対応する保持時間を見出せなかったわけである。したがって、この場合、所定の値(ペナルティスコア)を減じるようなスコアを設定する。
iii)このようにして図5の格子の最左上角から最右下角にいたるまで、段階的にスコアを求めてゆき、最後に最右下角までいたった時点でのスコアが、得られた対応配置に対応するスコアになる。
【0060】
(d)保持時間に関する最適な対応配置を求める手順
基本的には、可能なすべての対応配置を列挙し、それぞれについてスコアを計算し、その中で最大のスコアを示す対応配置を選択すればよいわけであるが、上述したように、スコアは漸化式で与えられるため、「動的計画法」に適している問題である。すなわち、3次元データZ(1)に含まれるi番目の保持時間とZ(2)に含まれるj番目の保持時間の対応関係を考える際には、(イ)Z(1)に含まれるi−1番目及びZ(2)に含まれるj−1番目に次いでZ(1)及びZ(2)両者ともに対応付けられる場合、(ロ)Z(1)に含まれるi−1番目及びZ(2)に含まれるj番目に次いでZ(1)の保持時間に対応するZ(2)のパラメータがない場合、(ハ)Z(1)に含まれるi番目とZ(2)に含まれるj−1番目に次いでZ(2)の保持時間に対応するZ(1)のパラメータがない場合、の3通りを考えることとなる。いずれの場合も1段階前の状態におけるスコアがわかっていればZ(1)及びZ(2)の(i、j)番目のスコアを算出することが可能となる。
【0061】
そこで(イ)(ロ)及び(ハ)の3通りのうち、最もよいスコアを与えた場合のスコアとそこに至るまでの経路のみを記録しておき、このステップを図5に示した格子の最左上角の出発点から最右下角のゴールに達するまで続ける。そして、記録した経路を最右下角から出発点まで、逆にたどることにより、最適経路、すなわちZ(1)及びZ(2)における保持時間に関して、最適な対応配置を求めることができる。
【0062】
以上、(a)〜(d)の手順として説明した手法は、動的計画法に基づいた最適解探索手法と言い換えることができるが、本発明において適用可能なアルゴリズムは動的計画法に限定されるものではない。すなわち、目的の評価関数を最適化するより一般的な探索問題と捉えることで、他の最適探索アルゴリズムを用いて実装することも可能である。このようなアルゴリズムとしては、例えばA*アルゴリズム、遺伝的アルゴリズム(GA)、シミュレーテッドアニーリング(SA)、最急降下法等による非線形計画法等によって実装することもできる。
【0063】
(a)〜(d)の手順として説明した手法は、いわゆる動的計画法に基づく手法であり、動的計画法に基づいている点では、DTW法やCOW法と類似する部分がある。しかしながら、DTWやCOWは、評価関数の形や計算手法もユークリッド距離あるいは相関を評価関数として、時系列データ点列そのままあるいは一定間隔の時間区切りでセグメント化した上での区間ごとの比較として、さらに2つのプロファイルの時刻0を出発点とし、それぞれのプロファイルの終了時刻をゴールとする同じ大域的制約条件の下に探索を実施する方式に限定されたものである。さらに、DTWやCOWを用いる手法は、基本的に2次元データとして表される時系列プロファイル、即ち時間軸とシグナル強度軸で表されるようなデータセットに対し、時間軸の非線形伸縮を行うことによって強度のプロファイルを重ね合わせるものである。
【0064】
したがって、DTWやCOWを用いる手法では、(1)特定の軸について特定の値をとる1つないし複数個の切断面を用いて、あるいは、(2)特定の軸に沿って全ての値を集約することによって、重ねあわせ操作を行わせることは、これらの方法の自然な拡張として容易に考えられる。例えば、DTWやCOWを用いる手法では、LC-MS分析で得られた保持時間、m/z、イオン強度からなる3次元データについても、いくつか特定のm/zに限定することにより、あるいは、全イオンクロマトグラム(TIC)のように保持時間軸に沿って全てのイオン強度を足し合わせることによって、時間軸補正が可能である。
【0065】
しかしながら、(a)〜(d)の手順として説明した手法では、DTWやCOWを拡張した方法とは異なり、補正対象の次元(保持時間軸)を除いた多次元プロファイルはそのままの形で直接比較することで、補正対象の次元を伸縮させることで、多次元プロファイルの重ねあわせを実現している。DTWやCOWを拡張した方法では、(1)特定の切片に限った手法をとると、精度を保ちつつプロファイル全体を重ね合わせるのと同じ結果が得られる保証がないこと、特定の切片を選択するための汎用性のある有効な手段がないこと、そのような保障なしに限定した重ね合わせ処理を行うことで、結果が恣意的になりうる危険があること、(2)TICのように情報を集約することで、せっかく次元を多くしてより分解能を向上させているメリットが得られなくなってしまう、といった問題がある。これに対して、(a)〜(d)の手順として説明した手法では、上記(1)及び(2)の問題が全くなく、高精度にプロファイルの重ね合わせが可能となり、且つ、多次元データという高分解能を維持したままプロファイルの重ね合わせが可能となる。
【0066】
4.データ解析における標準物質の役割
また、本発明に係る試料測定方法においては、以下に述べるような標準物質由来の情報を、本アルゴリズムを用いた計算手法に取り入れることで高い精度及び計算効率を達成することができる。
【0067】
上記「2.試料の分析」に先立って又は「2.試料の分析」の途中で標準物質を入れることにより、測定及び解析の過程で生じる可能性のあるバイアスを補正すると同時に、これらの情報を用いることによって、上述した最適な対応配置、すなわち、プロファイル同士の重ねあわせをより正確にかつより効率的に行うことが可能となる。すなわち、標準物質を用いることによって以下のメリットを得ることができる。
(1)あらかじめ既知の量の標準物質を入れることで、全体のシグナル強度を補正し、定量的な比較が可能となること
(2)いくつかの標準物質を、補正すべきパラメータ(時間軸等)を合わせる際のランドマークとして使用できること
(3)プロファイル重ねあわせにおいて、ある程度のプロファイル形状の共通性を与えることにより、重ねあわせが容易になること
【0068】
これらメリットを上記「3.データの解析」において最大限発揮させるべく、計算手法を以下のように改変することができる。すなわち、上述したアルゴリズムにおいて、標準物質に由来するシグナルのピーク部分を必ず通らなくてはいけない点として扱うように改変することができる。より詳しく説明すると、上述したアルゴリズムによる最適解探索では、図5に示したような格子状の探索空間上で、左上の出発点から右下の到着点に至る最適経路(太線)を探索するものであるが、いまここで仮に縦列上の保持時間15の点と、横の行で保持時間13の点がそれぞれ標準物質由来であったと仮定すると、解となりうる経路は必ずこの点を通らなければいけないというように拘束条件を設ける。そして、このような拘束条件を設定することによって、列15と行13を通る線で区分される探索空間のうちの左下と右上の部分空間を通る経路は排除されるため、探索しなければいけない空間を削減することができる(図6)。
【0069】
このように、標準物質由来のシグナルについて拘束条件を設定するようにアルゴリズムを改変することによって、本発明に係る試料解析プログラムは、プロファイル同士の重ねあわせをより正確に行うことができるとともに、処理の能率を大幅に向上させることができる。
【0070】
さらに、標準物質由来のシグナルの数が増えるにつれて、検索空間はより限定されることになるため、プロファイル同士の重ねあわせ精度はより向上するとともに、より能率向上が期待できる。実際、図7に示すように、丸印で示した点を標準物質由来シグナルの一致点として拘束条件を設定すると、灰色でマスクした領域は探索空間から削除される。n種類の標準物質由来シグナルで時間軸がn+1個に分割されたとすると、最良のケースとして分割が等間隔で行われたならば、探索空間は最大
【数14】
まで減少する。なお、標準物質由来のシグナルを拘束条件に設定する場合、標準物質由来シグナルが均等に広く分布するように標準物質を選択することで、探索空間削減効果を最も発揮することができる。
【0071】
なお、探索空間を限定して探索効率を上げるためのアルゴリズムに対する改変方法としては、図5に示した探索空間の左上の出発点から右下の到達点に至る対角線の前後に、所定の幅を持った空間に限定するといった拘束条件も考え得る。しかしながら、この場合、どの程度限定すれば良いのかといった確実な前提知識は一般に得られない虞がある。さらに、この場合、出発点や到達点が比較すべき多次元データ毎に大きくずれた場合には、求めるべき最適経路が限定された空間からはみ出す虞がある。例えば、クロマトグラフィーの溶出開始時刻は、大きく変動しうるので、この時刻を確実に観測できなければ、上記対角線の前後に所定の幅を持った空間に探索を限定する方法は適切とは言いがたい。
【0072】
それに比べて、標準物質由来のシグナルを拘束条件に設定するようにアルゴリズムを改変する方法では、検体内部に添加した標準物質の出現した時点を揃えており、標準物質に由来するシグナルは最も確証のある参照点となる。その上、探索空間は最大
【数15】
近くまで減らせるため、確実性及び効率性の両面から見ても優れているといえる。
【0073】
さらに、標準物質由来シグナルで限定される1つないしいくつかの部分空間に限って最適経路探索を行うことで、部分的な最適プロファイル重ね合わせを求めることができる。この際に、前述の評価関数の値を、プロファイル重ね合わせの程度の指標とすることで、プロファイル同士の類似性(あるいは距離)を測ることができる。多くの場合、主要なシグナルは限られた時間領域に集中的に現れるため、部分空間に限って最適経路探索を行って評価関数の値を求めることで、プロファイル同士、さらにはそれらのプロファイルを生じさせた試料同士の類似性(あるいは距離)を効率よく求めることができる。
【0074】
数多くの試料由来のプロファイルを重ね合わせて平均プロファイルを求めなければいけない場合や、あらかじめ試料の属性に関する情報が十分に得られていない場合は、まず部分空間に限って最適経路探索を行って試料間の類似性(または距離)を求めておき、その順番で逐次プロファイル重ねあわせを行い、あるいは試料同士のグルーピングを行うことが可能である。殊に、プロファイルの重ね合わせ処理を行う場合は、重ね合わせる順番に依存して結果が変化しうるので、できるだけ近いものから重ね合わせることが望ましいが、そのような処理を行う際にこの手法は有効である。
【0075】
5.データ解析後の処理
2つの多次元データについて、最適な重ね合わせが得られたならば、補正されたパラメータについて、新たな補正後の値を生成する。特に、クロマトグラムの保持時間を補正した場合は、補正後の保持時間を求める。補正後の保持時間を求める方法としては、重ね合わせる2つの多次元データのうち一方を参照用データとし、他方の多次元データにおける保持時間を当該参照用データにおける保持時間に一致させる方法(非対称型)と、重ね合わせる2つの多次元データの両方を補正する方法(対称型)とを挙げることができる。特に対称型で補正後の保持時間を求めることが好ましい。
【0076】
補正後の保持時間を求めて非対称型で2つの多次元データを重ね合わせる場合には、参照用データの保持時間軸にあわせるべく、一致した点に関しては参照用データの保持時間をそのまま用い、参照データの対応する点が得られなかった場合は、その前後で一致した点を用いて内挿によって補正後の保持時間を決めることができる。
【0077】
しかしながら、非対称型で2つの多次元データを重ね合わせるには、いずれの多次元データを参照用とすべきかが予め決まっている必要がある。例えば、標準物質のみを含むブランクを参照用データとすることも考えられるが、この場合は最初に重ね合わせに用いられた多次元データのプロファイルの影響がかなり大きくなる可能性が高い。
【0078】
これに対して、補正後の保持時間を求めて対称型で2つの多次元データを重ね合わせる場合には、2つの多次元データ間で対応する点が得られたところについては、それぞれの保持時間の算術平均とする。2つの多次元データのいずれか一方だけのデータ点しか得られなかった場合は、その前後の最も近い対応点の補正後の保持時間の組から、内挿によって補正後の保持時間を得る。内挿による補正が不可能な場合は、最も近くにある対応した点の補正後の保持時間をベースにして、データセット全体での平均時間スケールを係数とした外挿によって補正後の保持時間を得ることが可能である。
なお、この場合、前述のようにあらかじめ全ての多次元データ間の類似性(距離)を事前に計算した上で、近いものから順次重ね合わせても良い。
【0079】
6.出力処理
上記「5.データ解析後の処理」で得られたプロファイルの出力方法としては、以下の(1)及び(2)を挙げることができる。
(1)対応する点が得られなかった箇所も含めて、全て出力する。
(2)対応する点に限って出力する。
【0080】
いずれの出力方法を選択するかは、本発明に係る試料解析方法の使用目的に応じて適宜選択することができる。例えば、測定誤差を相殺するため同一試料を複数回に亘って測定した結果から平均を求めることを目的とする場合、又はきわめて類似した条件での複数試料の測定から代表的なプロファイルを得たい場合には上記(2)の出力方法が有効である。上記(2)の出力方法によれば、出力されるプロファイルは共通部分に限られるため、データの大きさを軽減して処理効率を上げることができる。
【0081】
また、例えば異なった試料群の群間差を検出する場合には、上記(1)の出力方法を使用しなくてはならない。上記(1)の出力方法では、データの大きさは一般に増大するが、情報の損失は起こらない。
【0082】
また、上記(1)の出力方法を選択する場合、さらに共通するプロファイルに重きを置いた重ね合わせを行うこともできる。この場合、以前の重ね合わせプロセスで対応する点に対して新たな点を対応させる場合は、評価関数のスコアを良くするような新たな項を当該評価関数に設けて、できるだけ同じ点に重なるように調整することも可能である。すなわち、例えば、上記式(I)で与えられる評価関数の最後に-μ・δm(i,j)のような新たな項を付与した新たな評価関数を用いて評価得点を算出する。そして、このような評価関数を用いる場合、以前の重ね合わせ点に対して対応が取れた場合にはδm(i,j)を1、それ以外は0とする。
【0083】
一方、本発明に係る試料解析プログラムの出力は次のような形式となっている。
・重ね合わせ処理で新たに得られた点の情報
・対応する入力データセット1(一方の多次元データ)の点の情報
・対応する入力データセット2(他方の多次元データ)の点の情報
出力は、これらの情報が重ね合わせ処理の結果として得られたデータ点の数だけ繰り返された形をとる。但し、対応する点がない場合には、それぞれ入力データセット1あるいは2の情報は存在しない。このように、出力には、対応する入力データセットの点の情報も含まれることにより、後述する例のように、最終的に得られた、重ね合わせプロファイルの各点が、元の多次元データのどれに由来するかを求めることが可能となる。なお、以上の情報の他にも必要であれば付加的情報をつけて出力することも可能である。
【0084】
このようにして得られた重ね合わせ後のプロファイルについて、必要に応じてさらにいくつかのパラメータに関して集約あるいは量子化処理を行ってもよい。例えば、特に上記(1)のように全ての点を出力した場合は、時間軸分解能が求めるレベル以上に詳細になりすぎることもある。この場合は時間軸上きわめて近接した点同士はさらに集約して1つにした方が、後の処理上得策である。集約された点の強度は、集約前の個々の点の強度の加算で置き換えることができる。同様にして、m/z軸においても必要分解能以上に近接した点は集約することができる。但しこの操作は重ね合わせ処理を行う都度実行してもよいし、必要な重ね合わせをまず行ってから、最後に1回だけ行ってもよい。
【0085】
7.イオン化強度の規格化と標準物質
本発明に係る試料解析方法においては、測定したイオン強度について、上記「3.データ解析」に先立って規格化を行うことが好ましい。以下にイオン強度の規格化について説明するが、イオン強度の規格化手法は何ら限定されるものではない。
【0086】
具体的には、先ず、LC−MS分析に結果として得られたRAWファイルを、例えばXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換する。次に、C言語およびPerl言語で作成されたプログラムにより、以下の一連のデータ処理を適用する。
(1)ノイズレベルのデータを除去するため、イオン強度が所定の値(例えば、102以下)以下のシグナルを除去する。
(2)必要ならば、処理時間の節約のためにデータ点を集約する。具体的に、例えば、m/zは1刻みに、保持時間は0.2刻みになるよう、元データのm/z値および保持時間の値を丸め、同じ値を持つ(m/z、保持時間)のデータ点は加算集計する。
(3)あらかじめ調べたm/z値と保持時間から標準物質由来のシグナルを同定し、そのイオン強度値をもって測定値全体を除算することで規格化する。この際、1つないし複数の標準物質由来の複数のシグナルの平均値などの代表値を標準物質イオン強度値として用いる方法、予備実験などでシグナルの安定性を事前に検討したうえでもっとも安定なシグナルの値を用いる方法、などがある。
【0087】
より具体的に、例えばニワトリ卵白リゾチームを標準物質とした場合、m/z値715近傍及び877近傍のシグナルを標準シグナルとすることができる。サンプルの測定データに対しては、m/zについては前後±1の範囲で、保持時間に関してはm/z 715(715±1)のシグナルについては早期溶出シグナル群より、m/z 877(877±1)のシグナルについては後期溶出シグナル群より探索することで、標準物質由来シグナルを探索することができる。なお、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正するといった更なる補正を行ってもよい。
【0088】
また、ペプチドそのもの、例えばペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)およびβカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)を標準物質として用いた場合、それぞれm/z値859近傍及び791近傍のシグナルを標準シグナルとすることができる。前者のペプチドは比較的親水性であり、後者は疎水性である。本解析方法で保持時間による分離のために用いられている逆相クロマトグラフィーでは、前者の保持時間の値は低く、後者のそれはより高い。大部分のサンプル由来のペプチドの保持時間はこの2種類のペプチドの保持時間の間にある。サンプルの測定データに対しては、m/zについては前後±1の範囲で、保持時間に関してはあらかじめ標準物質のみを測定して得られたクロマトグラムからおおよその値を見積もってから、その前後一定の範囲で探索することで、標準物質由来シグナルを見出すことができる。
【0089】
本解析方法において標準物質としてペプチドを用いる場合、当該ペプチド以外の物質(例えば、夾雑物)を含まないことがシグナルノイズの検出を最小限に抑えるために重要である。したがって、天然物から抽出・精製して得られるものよりも、化学的に合成されたペプチド分子の方が、望ましい。また、ペプチド分子の性質については、あらかじめ設定された測定条件下で構造が安定なことや不溶化しないことが重要である。ペプチド分子を構成するアミノ酸残基については、メチオニン、トリプトファン、ヒスチジンなど容易に酸化されうるアミノ酸残基を含まないこと、および塩基性の官能基を2つ以上含まないことが望ましい。とくに後者については、測定手段のMSで原理的に多価イオンを生じるエレクトロスプレーイオン化法を用いた場合、ひとつの標準物質から価数の異なる複数のイオンシグナルが検出されることを避けるためである。
【0090】
標準物質として蛋白質から加水分解あるいは化学的切断によって生じるペプチド断片を用いる場合、当該蛋白質に由来するペプチドイオンシグナルのうちで標準物質として採用したもの以外のペプチドイオンシグナルの強度は出来る限り低いことが望ましい。
【0091】
以上(1)〜(3)によって、測定されたイオン強度値を規格化することができ、複数の試料間におけるイオン強度の量的な比較を行うことができる。なお、測定されたイオン強度値の規格化は、上述した保持時間の補正に先立って行われるべきである。
【0092】
8.試料間の比較分析
本発明に係る試料解析方法によってm/z、規格化されたイオン強度及び補正した保持時間からなる3次元データを用いて、試料中に含まれるタンパク質群等の各種成分分析をコンピュータ上で行うことができる。具体的に、成分分析としては、(a)加算の方法、(b)減算の方法を挙げることができる。
a.加算の方法
上述したように、本発明に係る試料解析方法によって取得された複数の3次元データにおいては、保持時間のパラメータをそれぞれ適切に補正しているため、データ点間の対応関係を正確に取ることができる。したがって、複数の3次元データにおいて、データ点同士の規格化されたイオン強度値同士を足し合わせることができる。
b.減算の方法
「a.加算の方法」と同様に、本発明に係る試料解析方法によって取得された複数の3次元データにおいてはデータ点間の対応関係を正確に取ることができるため、データ点同士の規格化されたイオン強度値の差を求めることができる。
【0093】
このように、本発明に係る試料解析方法によって取得された複数の3次元データについて、加算或いは減算することができるため、以下のような、成分分析のアプローチをコンピュータ上で実現することができる。
【0094】
(1)実験データを集計する際への応用:
1つのサンプル由来の試料を、測定の便宜上、複数の分画に分割して測定した場合であっても、当該複数の分画それぞれから取得された3次元データにおいては、データ点間の対応関係を正確に取ることができる。したがって、上述した加算の方法に従って、全ての3次元データを足し合わせることができる。これにより元のサンプル全体に含まれる成分の解析等を行うことが可能となる。
【0095】
なお、このような集計を行う際には、複数の分画全てに亘って足し合わせ、全体で1つのプロファイルにまとめる方法のほかに、あらかじめ指示された個数により、近接する数個のプロファイル毎に集約する方法もある。この場合、全体がn個に分画され、そのうち近接するm個ずつを集約した場合はn-m+1個のプロファイルが得られることになるが、その場合は得られたプロファイルについては、対応する分画同士以外はそれぞれ別個に扱って以下の操作を行うことになる。いずれにせよ、多次元分画法で複数の分画を得た場合も、そのおのおのが完全に重なりなくなることはまれで、多くの場合複数分画に亘るキャリーオーバーがありうるので、このような集計操作は必要である。
【0096】
(2)複数のサンプルの測定結果の代表値を求める際への応用:
異なるサンプルに由来する複数の試料について測定した場合であっても、本発明に係る解析方法によれば、取得された複数の3次元データ間において、データ点間の対応関係を正確に取ることができる。したがって、上述した加算の方法に従って、全ての3次元データを足し合わせることができる。そして、得られた3次元データの総和をサンプル数で除算することで相加平均を求めることができる。なお、必要に応じて、各サンプルに重みを設定し、当該重みを反映させた重みつき平均を算出することもできる。
これによれば、例えば、同じ範暗に属すると考えられる複数のサンプルについて、当該範暗の代表値を求めることができる。
【0097】
(3)2つのサンプル間での測定結果の差分を求める際への応用:
例えば、同一のサンプル由来であるが状態の異なるときに採取した試料について測定した場合であっても、取得された2つの3次元データ間において、データ点間の対応関係を正確に取ることができる。したがって、上述した減算の方法に従って、2つの3次元データ間の差分を求めることができる。これにより、状態の変化に起因する試料中の成分変化を解析することができる。
【0098】
また、例えば、上述した(1)に準じて、複数のサンプルを含む2群についてそれぞれ相加平均等の代表値を求めた上で、これら2群の代表値の差を求めることができる。得られた差については、統計的な検定などによって有意性を検討することで、各群に特異的な成分を同定することができる。
【0099】
以上の(1)〜(3)に示す成分分析アプローチは、本発明に係る試料解析方法によって得られた複数の3次元スペクトルデータを格納したデータベースを用いても良いし、当該データベースに格納されたデータと現実に得られたデータとを用いて行っても良い。いずれの場合であっても、上述した(1)〜(3)に示す成分分析アプローチは、コンピュータを用いて容易に実現することができる。
【0100】
このようにして、本発明に係る試料解析方法によって得られた、例えば群特異的シグナル成分については、得られたシグナル領域に範囲を限定したタンデムMS分析などにより、当該シグナルが由来する蛋白質群を同定することができる。すなわち、本発明に係る試料解析方法においては、試料をLC−MSにより分析したときに、特定のm/z値を持つペプチド分子イオンが検出された場合、当該イオンのCIDスペクトルを測定することができる。
【0101】
そして、得られたCIDスペクトルをコンピュータに入力し、データベース検索ソフトウェアを用いて蛋白質一次構造データベース、ゲノム配列データベースやcDNA配列データベースから得られる蛋白質配列に対して検索する。このデータベース検索によって有意なヒットスコアを示した場合には、データベースに登録されたタンパク質或いはアミノ酸配列等の情報を得ることができ、得られたCIDスペクトルに対して当該情報を関連付けることができる。
【0102】
例えば、上述した(3)の成分分析アプローチにおいて、各群に特異的な成分として同定されたシグナルに関するCIDスペクトルを測定することで、当該シグナルが示すタンパク質群を同定することができる。
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【実施例1】
【0103】
実施例1では、アミノ酸配列が既に知られているタンパク質のプロテアーゼ消化物を混合して得られるペプチド試料をLC-MSによって測定し、この測定によって得られた保持時間、m/z値およびイオン強度からなる三次元プロファイルに対して本発明に係るアルゴリズムを適用し、測定されたペプチド試料を定量的に特性づけた。また、実施例1では、比較定量のためのモデル実験としてアミノ酸配列が既に知られているタンパク質のプロテアーゼ消化物を混合したペプチド試料数種を各々LC−MSによって測定し、本発明の試料解析方法を適用して各三次元プロファイルを比較することによって、各ペプチド試料に含まれるタンパク質の種類の違いが検出されることを示した。
【0104】
ペプチド試料の調製
以下に列挙する24種類のタンパク質のトリプシン消化物を、本実施例におけるペプチド試料として調製した。(1)ウシキモトリプシノーゲン、(2)ウシカタラーゼ、(3)ウシカルボニックアンヒドラーゼ、(4)ウシアポトランスフェリン、(5)ウシカルボキシペプチダーゼA、(6)ウシ血清アルブミン、(7)ウマシトクロムc、(8)ブタガンマ免疫グロブリン、(9)ウシヘモグロビン、(10)ウマミオグロビン、(11)ウシベータラクトグロブリン、(12)ウシデオキシリボヌクレアーゼ、(13)ウサギグリセルアルデヒド3リン酸デヒドロゲナーゼ、(14)トリコンアルブミン、(15)セイヨウワサビペルオキシダーゼ、(16)枯草菌アルファアミラーゼ、(17)ウマグルタチオンS−トランスフェラーゼ、(18)ウシグルタミン酸デヒドロゲナーゼ、(19)ウシラクトペルオキシダーゼ、(20)コウジカビアミログルコシダーゼ、(21)ウサギホスホリラーゼB、(22)ウシベータガラクトシダーゼ、(23)ウサギ乳酸デヒドロゲナーゼ、(24)ニワトリ卵白リゾチーム。これらの消化物はMichrom BioResources社より購入した。
【0105】
これら24種類の各タンパク質のトリプシン消化物を以下に示すように混合し、合計3種類(A群〜C群)のペプチド試料を用意した。
A群:(1)、(2)、(7)〜(24)の20種類のタンパク質のトリプシン消化物。A群を特徴付けるタンパク質は(1)及び(2)である。B群:(3)、(4)、(7)〜(24)の20種類のタンパク質のトリプシン消化物。B群を特徴付けるタンパク質は(3)及び(4)である。C群:(5)〜(24)の20種類の蛋白質のトリプシン消化物。C群を特徴付ける蛋白質は(5)及び(6)である。各群のサンプルは3つずつ調製した。
【0106】
LC-MS解析
各ペプチド試料の三次元プロファイルを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al,Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリルおよび水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
【0107】
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。また、スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。これを各群3回ずつ行うことによって、各試料に対応する三次元パラメータの集合体、3群計9通りを得た。このデータセットをそれぞれAl、A2、A3(A群)、Bl、B2、B3(B群)、Cl、C2、C3(C群)とした。
【0108】
三次元パラメータ集合体を含むファイルはXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した。C言語及びPerl言語で作成されたプログラムにより、以下の(1)〜(5)のデータ処理を実行した。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)処理時間の節約のためにデータ点を集約した。具体的には、m/zは1刻みに、保持時間は0.2刻みになるよう、元データのm/z値および保持時間の値を丸め、同じ値を持つm/zと保持時間の2つ組みで指定されるデータ点は加算集計した。
(3)標準物質であるニワトリ卵白リゾチーム由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、次いでそのデータ点を中心に、イオン強度値が単調減少しかつ0より大きな範囲にあるデータ点を拾い、これらを標準物質由来のシグナルによるデータ点であると見なした。標準物質由来のシグナルの総イオン強度値としては、標準物質由来シグナルと見なされたデータ点のイオン強度の総和をもって当てた。具体的には、ニワトリ卵白リゾチーム由来のm/z値が715近傍及び877近傍のシグナルを標準シグナルとし、サンプルの測定データからこれらの標準物質由来シグナルを探索する際には、m/zについては前後±1の範囲で、保持時間に関してはm/z 715のシグナルについては6〜16分の範囲で、m/z 877のシグナルについては13〜23分の範囲で探索した。
(4)得られた標準物質由来シグナルの総イオン強度値をもって、各シグナルのイオン強度を除算し、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正した。
(5)便宜上、m/z 715のシグナルとm/z 877のシグナルのピーク位置がそれぞれ保持時間に関して10分、20分となるよう、保持時間軸を線型変換した。
【0109】
次に、A、B及びC群それぞれ3例ずつあるサンプルから得られた三次元プロファイルの代表点を求めた。すなわち上述したように、同じ群に属するサンプルを集約した。m/zおよび保持時間の重なる点のイオン強度は加算して集計した。
【0110】
また本例で使用したスコアは高いほどよいとする得点として計算し、算出式の係数は次の通り。イオン強度差については、それぞれの常用対数の差の絶対値に対して係数−1をかけたものを使用した。保持時間の差については、差の絶対値に対して係数−1000をかけたものを使用した。また各群間で対応するデータ点のシグナルがともに標準物質由来であった場合の加算点は50000点とした。一方の群において対応する保持時間の点がなかった場合の失点は5000点とした。本実施例ではこれらを単純に加算してスコアとした。
【0111】
次いで、上述したように、A−B群間、B−C群聞及びC−A群間で差を求めた。得られた差の有意性はt検定によって有意水準0.1%の両側検定で検討した。
【0112】
その結果、保持時間を補正した三次元データを比較することによって、A、B及びCの各群から以下に列挙するm/z値を持つペプチド分子イオンが各群に特異的なシグナルとして検出された。
A群:495,524,546,560,671,696,779,845,871,908,962等。
B群:451,464,509,513,546,555,583,585,626,635,649,653,701,720,723,740,741,753,768,789,819,821,847,873,886,922,928,952,966,973,978,1057,1230等。
C群:636,670,674,679,683,718,734,735,770,824,870,918等。
【0113】
また、本実施例では、特異的なシグナルとして検出されたペプチド分子イオンのCIDスペクトルを得るため、各試料をLC−MS/MS分析にかけた。分析条件は以下に示す操作以外は、上述した通りとした。すなわち、LC−MS/MS分析に際しては、イオントラップ型質量分析計の測定条件を変更し、上に列挙したm/z値を持つペプチド分子イオンが検出された場合に当該イオンのCIDを必ず行うように測定条件を設定して試料の測定を行った。
【0114】
その結果、各ペプチド分子イオンから得られたCIDスペクトルを、Matrix Science社のデータベース検索ソフトウェアであるMASCOTTMを用いて、SWISS−PROT蛋白質配列データベースに対して検索したところ、各群において特異的なタンパク質由来ペプチドとして添加した各群2種類(すなわち、A群においては上記(1)及び(2)のタンパク質、B群においては(3)及び(4)のタンパク質、C群においては(5)及び(6)のタンパク質)、合計6種類はいずれも有意なヒットスコアをもって同定された。このことから、本実施例で行った試料解析方法の妥当性が示された。
【実施例2】
【0115】
実施例2では、所定の濃度組成の蛋白質混合物中に、濃度を変化させた別の蛋白質標品を混合して得られた試料をプロテアーゼ消化し、LC-MSによって測定することで得られた保持時間、m/z値、イオン強度からなる3次元データに対して本発明に係る方法を適用し、異なる濃度の試料の測定で得られた3次元データ同士を比較することで、定量的に変動するシグナルを検出した。これによって、本手法で定量的に変化する物質を検出できることを示したものである。
【0116】
試料とその調製
以下に列挙する6種類のタンパク質のトリプシン消化物を、本実施例におけるペプチド試料として調製した。(1)ウシカタラーゼ、(2) ウシベータラクトグロブリン、(3) ウシラクトペルオキシダーゼ、(4) ウマグルタチオンS−トランスフェラーゼ、(5) セイヨウワサビペルオキシダーゼ、(6) ウシ血清アルブミン。これらのタンパク質はシグマ社より購入した。
これらのタンパク質は水溶液中でブタトリプシン(プロメガ社より購入)と反応させることにより、トリプシン消化物とした。
【0117】
これら6種類の各タンパク質のトリプシン消化物を以下に示すように混合し、合計7種類のペプチド試料を用意した。
(1):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は0フェムトモル。
(2):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は10フェムトモル。
(3):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は50フェムトモル。
(4):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は100フェムトモル。
(5):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は500フェムトモル。
(6):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は1 ピコモル。
(7):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は5 ピコモル。
各群のサンプルは5測定分ずつ調整した。上記各試料に対し、さらにペプチドTおよびβカソモルフィン7をそれぞれ10ピコモルおよび1ピコモルずつ標準物質として混入した。
【0118】
LC-MS解析
各ペプチド試料の三次元データを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al, Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリル及び水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
【0119】
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。また、質量分析計でのスキャン回数を細かく取るためにTurbo Scan方法を適用した。この測定を各群5回ずつ行うことによって、各試料に対応する三次元パラメータの集合体、7群計35通りを得た。図1に得られたプロファイルの例を示す。
【0120】
データ処理
三次元データを含むファイルはXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した。C、C++言語及びPerl言語で作成されたプログラムにより、以下の(1)〜(7)のデータ処理を実行した。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)標準物質であるペプチドTおよびβカソモルフィン7由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、その周囲で規定の範囲内にあるシグナルについて、さらにその点を頂点とするガウス分布で近似される強度範囲内に収まるシグナルを拾い上げ、それらを全て標準物質由来シグナルとみなした。より具体的には、m/zは858.9 および791.0 を中心に前後±2を限度とし、また保持時間は9および25分を中心に前後±6を限度として、上述の手順により探索した。従って使用した標準物質由来シグナルはm/z 858.9, 保持時間9分、およびm/z 791.0, 保持時間25分近傍に存在する2つである。なお、イオン強度補正を行う際には標準物質由来シグナル強度を全て足し合わせ、その値が109となるように規格化した。また時間軸補正の際の動的計画法探索空間上の拘束点としては、上述2つの標準物質シグナルのうち、それぞれ強度ピークを与える1点ずつの計2点を選定した。
(3)BSAの濃度を変えた7種類の試料それぞれにつき、5回の測定結果得られたプロファイルを平均化するため、本発明によるプロファイル重ね合わせプログラムを用い、7群の試料それぞれについての代表プロファイルを求めた。重ね合わせの際の評価関数のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=0.1(但しシグナル強度さは常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=0、不一致点に対するペナルティπ=100、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、一致点のみとした。
【0121】
図8に、BSA濃度が500フェムトモルの試料(5)の5回の測定結果から得られたプロファイルのうち、m/z 620.0 近傍、残留時間15から19分の間のクロマトグラムを示す。時間軸上少しずつずれた灰色の5本の波形が、重ね合わせ処理前の5回の測定結果であり、実線がこれら5つに重ね合わせ処理を施してすべてのシグナルを足し合わせた結果である。図に示したように時間軸上の揺らぎが補正されて1つの大きなピークとして扱えるようになった。
(4)次に、群間で有意に変動しているシグナルを拾い出すため、前述のプロセスで得た7群の代表プロファイルを、さらに重ね合わせ処理によってアライメントをとり、最終的に1つの集約プロファイルを得た。このときの評価関数のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=0.1(但しシグナル強度を常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=0、不一致点に対するペナルティπ=100、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、不一致を含む全ての点とした。
(5)試料間の相互比較に必要十分な分解能を保ちつつデータ点を集約するため、m/z範囲±0.75(絶対範囲±2)、保持時間範囲±1.25(絶対範囲±4)の範囲内にある点について、次の条件を満たしている点については1つに集約した。即ち、シグナル強度の高い順に前述範囲内の全てのデータ点をチェックし、これらがピークシグナルを頂点とするガウス分布で近似される範囲内に入ると判定されたものについてはそれらを集約した。
【0122】
図9に、BSA濃度が異なる7種類の試料ごとの、時間軸補正および集約後のプロファイルの例を示す。図では特定のm/z値(この例では752)で切った断面を、時間軸に沿って強度をプロットしたクロマトグラムとして示している。17分および19分近傍の集約シグナルは、BSA濃度が最も高い06の試料(図中、“DS:Spl 06-Ave”として示す。BSA 5ピコモル)が最も高いピークを示し、順次Spl 05、Spl 04,…と続いているため、これらはBSA由来のシグナルと判定できる。一方、25分近傍の緩やかなピークはどの試料でも同様に出ているため、BSA以外の共通物質あるいはバックグラウンド由来と判定できる。
【0123】
(6)前述の集約プロファイルを出発点として、順次(5),(4),(3)のプロセスを逆にたどることにより、最終的に得られた集約プロファイル上の点が、測定結果の7群×5測定のいずれに由来するかをたどることができる。集約プロファイル上の各点について、測定結果データに戻ることで、群ごとのプロファイルや群間の差プロファイルを得ることができる。図10にBSA濃度が500フェムトモルの試料(5)と、0フェムトモルの試料(1)との差プロファイルを示す。m/z−保持時間平面より上に伸びている線は試料(5)で、逆に下に伸びている線は試料(1)で強く観測されたシグナルである。
【0124】
(7)BSAを入れていない試料(1)から得られた集約プロファイルを基準とし(基準プロファイル)、残りの(2)から(7)までのBSA濃度が異なる6種類の試料から得られた集約プロファイルそれぞれに関して(対象プロファイル)、次のような条件を満たすデータ点を求めた。即ち、(1)対象プロファイル上の点は、5回の測定すべてよりデータが得られていること。(2)集約した対象プロファイル上の点のシグナル強度は106以上であること。(3)対象プロファイル上の各点について、基準プロファイル上の対応する点の強度を引いた差が0以上であること。(4)対象プロファイルの各点について、基準プロファイル上の対応する点とのシグナル強度の差が10000未満であるという帰無仮説が、片側t検定結果によって有意水準0.5%で棄却されること。
【0125】
上記の条件で選別されたシグナルを、試料(5)の場合について図11に示す。ここでは127個のシグナルが前述条件に合致したものとして残った。プロットマークの大きさは、試料(5)のプロファイルにおけるシグナル強度を表す。このうち、プロットマーク○のものは、後述するプロセスでBSAシグナルと対応付けられたもの、×のものはそうでなかったものを示している。
【0126】
これら127個のシグナルについて、別途行ったBSA消化物のMS/MS実験とつき合わせたところ、このうち103個がBSA由来シグナルと一致した。即ち、検出された127個のシグナルのうちの少なくとも81%が真に求めたいものであったといえる。
【0127】
同様にして、他のBSA濃度の試料から検出されたシグナルについて、前述の同じ条件で選別されたシグナルをBSA由来シグナルと付き合わせたところ、対象プロファイル(2)から(7)について、それぞれ65、64、75、81(既述)、76、48%のシグナルがBSA由来シグナルと判定された。なお最後の(7)試料(BSA濃度5ピコモル)の正答率が落ちているが、これは高濃度BSAに由来する強度の強いシグナルが多数存在することによるプロファイルの閾値が変わったことで、偽陽性シグナルが増大したことに起因すると考えられる。実際、他の濃度の試料とほぼ同程度の数のシグナルが選ばれるよう、選定条件の(2)を3×106に調整すると、正答率は75%になった。
【0128】
なお、本発明に係る工夫の1つに、動的計画法の探索空間節減があるが、その効果をCPU時間の実測によって評価したところ、試料(5)の5つの測定結果を重ね合わせる場合を例とすると、CPU時間の比にして43〜45%の削減効果が得られた。
【0129】
ここでは2種類の標準物質由来シグナルを用いているので、もし完全に均等にシグナルが分布していれば1/3の時間短縮が期待できるが、実際多くのシグナルは2つの標準物質シグナルの間に存在するので、探索空間の分割は不均等である。このことを勘案すると、45%程度の削減はおよそ期待通りであり、実用上も十分効果があると考えられる。
【0130】
以上により、本発明に係る試料解析方法が、試料中で量的に変化するシグナルを実用上有用な確度で検出できることを示し、また本発明における計算手法の工夫が有用であることを示した。
【実施例3】
【0131】
実施例3では、実際の患者由来組織サンプルを用いて、いくつかの病態群間で有意に変動している蛋白質由来シグナルを求め、それを元にさらにMS/MS解析を行ってそのうちのいくつかの蛋白質を同定することにより、本方法の有効性、特にバイオマーカーの探索等にも有効であることを示したものである。
【0132】
具体的には、肺の腺癌を対象として、外科的に切除された組織を用い、組織より後述する手法で蛋白質を抽出し、測定した。得られたプロファイルを、後日病理診断によってリンパ節転移ありと判定された群となしと判定された群にわけ、両群間で有意に変動しているシグナルを拾い出し、それらについてMS/MS解析を行って蛋白質を同定した。
【0133】
試料
試料は、36名の異なった肺癌患者より、外科的に切除された肺の組織片を用いた。病理診断により、これらの患者は腫瘍径の大きな群と小さな群、および所属リンパ節への転移が認められる群と認められない群の合計4群に分けられる。
腫瘍径小でリンパ節転移なしの群には10例、腫瘍径大でリンパ節転移なしの群には11例、腫瘍径小でリンパ節転移ありの群には12例、腫瘍径大でリンパ節転移ありの群には3例にぞれぞれ分類された。
【0134】
試料調製および蛋白質分画
ドデシル硫酸ナトリウム(SDS)-ポリアクリルアミドゲル電気泳動(PAGE)用の試料緩衝液中にて各組織片を破砕した。試料緩衝液の組成は以下のとおりである。62.5mMトリス-塩酸(pH6.8)、2%w/v SDS、5%v/v 2-メルカプトエタノール、10%v/v グリセリン、0.0025%w/vブロモフェノールブルー。この懸濁液を室温にて30分間振とうしたのち、上清と沈殿に遠心分離した。上清の蛋白質の濃度をLowry法の変法によって測定した。蛋白質100μg分の試料上清に対して同じ組成の試料緩衝液を加え、総液量を50μLにした。この溶液に対して1Mトリス水溶液を加え、pHを8.8にした。システイン残基の還元アルキル化のため、2μLの400mMジチオトレイトールを加え、60℃にて30分間保温した。つぎに10μLの400mMヨードアセトアミド溶液を加え、室温・暗条件下で60分間放置した。約5μLの1.0N塩酸を加え、pHを6.8に戻した。この溶液をLaemmliのSDS-PAGEにかけた。このときに用いたポリアクリルアミドゲルは不連続の緩衝液系、すなわち上部の濃縮ゲル(pH6.8)と下部の分離ゲル(pH8.8)から成った。ポリアクリルアミドゲルの濃度は各々4%と12.5%であり、全体の大きさは幅14cm、高さ14cm、および厚さ1mmであった。電気泳動時の電流は一定の10mAであった。色素ブロモフェノールの泳動フロントが濃縮ゲルと分離ゲルの界面から分離ゲルの48mmまで達したところで泳動を停止した。ポリアクリルアミドゲルを40%メタノール、10%酢酸水溶液中で振とうし、ポリアクリルアミドゲル内で分離されている蛋白質を固定した。このあと、ポリアクリルアミドゲルを水で2回洗浄した。洗浄したポリアクリルミドゲルを試料当たり24個のゲル片に切り出し分画した。すなわち、泳動方向に対して垂直の向きに2mmの等幅で梯子状に切り出し、各切片をさらに一辺約1mmのサイコロ状に分割した。
【0135】
標準蛋白質の調製
内部標準蛋白質はゲル中に固定された状態で各試料ゲル分画に加えた。最初に12.5%のアクリルアミド、0.1%SDS、375mMトリス塩酸(pH8.8)よりなる水溶液に対して卵白リゾチーム水溶液を混合し、さらにN,N,N’,N’-テトラメチルエチレンジアミンと過硫酸アンモニウムを加えてガラス板にはさまれた1mm幅の間でアクリルアミドを重合した。この内部標準蛋白質を含むゲルを直径1.5mmの円形に繰り抜いた。ゲル切片当たり2.5pmolの卵白リゾチームが含まれるように、重合前の水溶液の蛋白質濃度をあらかじめ計算しておいた。
【0136】
プロテアーゼ消化
上に示した一定量の標準蛋白質を含むゲルの繰り抜き切片を各試料ゲル分画に1個ずつ加えた。つぎに各分画ごとにゲル片を十分量の水で洗浄してからアセトニトリルで脱水した。ゲル片に残った水とアセトニトリルを減圧留去してからトリプシン水溶液をゲル片全部が浸かる程度に加え氷中で45分間放置した。ゲル中に染み込まなかった水溶液を除き、50mM重炭酸アンモニウム水溶液をゲル片全部が浸かる程度に加え、37℃にて16時間保温し消化反応を行った。ゲル片に含まれるペプチド断片の抽出は25mM重炭酸アンモニウム/50%アセトニトリル水溶液で1回、続いて5%蟻酸/50%アセトニトリル水溶液で2回行い、抽出溶液は1個の容器にまとめて減圧濃縮した。
【0137】
LC-MS解析
各ペプチド試料の三次元プロファイルを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al, Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリル及び水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。
【0138】
データ処理
得られたLC-MSプロファイルデータは、36サンプル×24バンド分の、合計864個である。これらのプロファイルデータについて、XcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した後、C、C++言語及びPerl言語で作成されたプログラムにより、以下のような手順で解析を行った。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)処理時間の節約のために、m/zと保持時間を量子化することでデータ点を集約した。具体的には、保持時間が約1刻みになるよう、シグナル強度が大きなものから順次、時間差1を最大限度に近傍を探し、単調減少する範囲までを1つのシグナルとしてまとめられる範囲とした。またm/zは1刻みになるよう、元データのm/z値の値を丸め、前述の時間範囲内でm/zが同じ値を持つデータ点は加算集計した。
(3)標準物質であるニワトリ卵白リゾチーム由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、次いでそのデータ点を中心に、イオン強度値が単調減少しかつ0より大きな範囲にあるデータ点を拾い、これらを標準物質由来のシグナルによるデータ点であると見なした。標準物質由来のシグナルの総イオン強度値としては、標準物質由来シグナルと見なされたデータ点のイオン強度の総和をもって当てた。具体的には、ニワトリ卵白リゾチーム由来のm/z値が715近傍及び877近傍のシグナルを標準シグナルとし、サンプルの測定データからこれらの標準物質由来シグナルを探索する際には、m/zについては前後±1の範囲で、保持時間に関してはm/z 715のシグナルについては10分±5分の範囲で、m/z 877のシグナルについては18分±5分の範囲で探索した。但し、得られた標準物質由来シグナルの絶対強度、全シグナル中での相対強度、および2種類の標準物質由来シグナルの強度比のいずれかが他のものに比べてかけ離れているものについては、個別にプロファイルのプロットを確認のうえ、標準物質由来シグナルと考えられるシグナル群のピークが探索時のパラメータの中心点に来るように調整した上で取り直した。得られた標準物質由来シグナルの総イオン強度値をもって、各シグナルのイオン強度を除算し、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正した。さらに、便宜上、m/z 715のシグナルとm/z 877のシグナルのピーク位置がそれぞれ保持時間に関して10分、20分となるよう、保持時間軸を線型変換した。
【0139】
(4)SDS-PAGEによって分画した24バンド分のプロファイルについて、バンド間にまたがって存在する蛋白質の定量性を保証するため、全バンドについて重ね合わせたプロファイルをもって、各試料のプロファイルとして扱った。具体的には、本発明に係る試料解析プログラムの、プロファイル重ね合わせ機能を用い、隣接バンド間のプロファイルを逐次重ね合わせて加算集計した。即ち、まずはバンド1+2, 2+3, 3+4, … , 23+24 のように、共通バンドを有する形で隣接バンドを重ねあわせ、次段階では1+2と2+3から1~3の重ね合わせ得るという形で、常に最低1バンドは重なるようにして重ねてゆくと、6段階の重ね合わせ操作により、最終的には1~17と9~24の重ね合わせによって、全バンドの重ね合わせを得た。なお、重複して重ね合わせたバンドに関しては、最終段階で重複回数を除算することで定量性を保つよう調整した。
【0140】
重ね合わせの際のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=1.0(但しシグナル強度を常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=100、不一致点に対するペナルティπ=10、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、不一致を含む全ての点とした。さらに重ね合わせの処理が終わるごとに、保持時間とm/zがそれぞれ分解能1.0および1.0となるようデータ点の集約操作を行った。
【0141】
(5)リンパ節転移のある群とない群を特徴付ける蛋白質を探すため、前述の試料の4種類の分類に従って、まずは群内でプロファイル同士を重ね合わせて集約プロファイルを得、次いで群間について同様にプロファイル重ね合わせ操作を行った。このときの重ね合わせ処理の際のパラメータは、前述のバンド間重ね合わせ処理と同じとした。また重ね合わせ順序については、群内については事前に総当りで行っておいた同一パラメータの下での重ね合わせ処理の評価関数スコアをもとに、近いものから順次重ね合わせた。群間については、まずリンパ節転移のある群内の腫瘍径の違いの2群と、リンパ節転移のない群内での腫瘍径の違いの2群を重ねた後、最終的にリンパ節転移のある群とない群を重ねた。
図12に最終的な重ね合わせプロファイルについて、リンパ節転移陽性群に表れたシグナルを上方向に、陰性群に存在したシグナルを下方向にプロットしたものを示す。
【0142】
(6)前述の重ね合わせた順序を逆にたどることで、重ね合わせの出発点となっている36検体×24バンドの元データにたどれるようにし、最終的な全てを重ね合わせたプロファイル上の各点について、元データとの対応を取れるようにした。
(7)最終的な集約プロファイル上の各点について、リンパ節転移ありの検体由来のデータと、なしの検体由来のデータをそれぞれ集計し、これら両群の平均値の差について両側t検定を行い、両群間の平均値の差と検定によるp値を得た。
図13に、上記検定でp値が0.005未満となった点を、図12と同様のプロットで示す。この段階で5,889個のシグナルが得られた。
【0143】
(8)上述のようにして選別されたシグナルについて、このシグナルの情報をもとにターゲットMS/MSを行い、あるいは別途行ったMS/MS解析から、蛋白質同定ソフトMASCOTTMを用いて、シグナルが由来するところの蛋白質同定を行った。図14に、この同定によって蛋白質情報と関連付けることができたシグナルを示す。約半数の2,753個のシグナルについて、何らかの既知蛋白と関連付けることができた。
【0144】
最後に、上述の既知蛋白質と関連付けられたものの中から、癌の転移に何らかのかかわりを持つといわれている蛋白質の一部をリストにして示す(図15)。このように、癌転移に関わると考えられる蛋白質が有効に発見しうることを示せた。
【0145】
以上より、本発明に係る試料解析システムおよびプログラムが、実際の臨床検体を用いた解析に有効であり、殊に病理的・臨床的な差異を蛋白質の量的な差異に結び付けて検出することができ、またその結果を利用して有効に蛋白質同定に繋げられることから、バイオマーカーの探索や新規診断方法の開発にも有用であることを示したものである。
【産業上の利用可能性】
【0146】
以上、詳細に説明したように、本発明に係る試料解析方法及び試料解析プログラムによれば、試料に含まれる成分を分析するに際して、優れた分析能を達成することができる。したがって、本発明によれば、分析対象の試料中に含まれる多数の成分を網羅的に解析する場合に非常に有効且つ有益な試料分析方法及び試料分析プログラムを提供することができる。
【0147】
特に本発明に係る試料解析方法及び試料解析プログラムによれば、実際の臨床検体を用いて、何らかの疾患の病態の違いに関係する物質を探索する目的にたいへん有効であり、これによりバイオマーカーの探索や診断法の開発ができるという点でも有用性はきわめて大である。
【図面の簡単な説明】
【0148】
【図1】本発明に係る試料解析方法及び試料解析プログラムにより取得された3次元のスペクトルデータの一例を示す図である。
【図2】3次元データの一例を示す図である。
【図3】図2に示した3次元データとの対応関係を検索するために設定したもう一つの3次元データの一例を示す図である。
【図4】図2に示した3次元データと図3に示した3次元データとの最適な対応配置を示した図である。
【図5】図2に示した3次元データと図3に示した3次元データとの最適な対応配置を検索する際の概念を示す図である。
【図6】図5に示した最適配置探索において、標準物質由来の情報を利用して経路を限定すると、探索空間のうちの灰色部分はもはや探索する必要がないことを示した図である。
【図7】標準物質由来の情報をより多く利用して経路の拘束条件を増やすと、探索不要な空間が増えて、探索効率がいっそう向上することを示した図である。
【図8】本発明に係る試料解析プログラムによって、同一種類の5つの測定結果で時間軸上揺らいでいる波形を、1つに重ね合わせてシグナルを足し合わせた結果を示した図である。
【図9】本発明に係る試料解析プログラムによって、異なる7種類の試料測定から得られたプロファイルを時間軸をそろえて重ね合わせることにより、時点ごとに異なった試料間での相互比較が行えるようになったことを示した図である。
【図10】本発明に係る試料解析プログラムによって、異なった2種類の試料間の差スペクトルを計算して示した図である。
【図11】本発明に係る試料解析プログラムによって、試料群間に有意な量的変動があるシグナルとして選別されたものを示した図である。
【図12】本発明に係る試料解析プログラムを、実際の臨床検体を用いたマーカー探索に応用し、異なった病理診断結果によるグルーピングに応じてシグナルを分類できることを示した図である。
【図13】図12に示した結果に対し、さらに統計検定を行うことで、異なった病理診断結果に応じて量的に変化しているシグナルを拾い出したものを示した図である。
【図14】図13に示した結果の各シグナルに対し、さらにMS/MSによる蛋白質同定との関連付けを行った結果を示した図である。
【図15】図14に示したような形で既知の蛋白質と関連付けられたもののうち、特に癌の転移に関連することが知られている蛋白について、本発明に係る試料解析システムで見出された結果の一部を示した表である。
【技術分野】
【0001】
本発明は、試料の分析の結果として得られた多次元データを用いた試料解析方法及び試料解析プログラムに関する。
【背景技術】
【0002】
例えば、液体クロマトグラフィー(以下LCと略記する)と質量分析(以下MSと略記する)を連結した液体クロマトグラフィー質量分析(以下LC−MSと略記する)の結果として、横軸に質量/電荷比(以下m/zと略記する)、縦軸にイオン強度をとったグラフとして2次元上にスペクトルデータを得ることができる。ここで、LCの役割は、MSの処理能力に適応させるために、試料を単に分画することにある。
【0003】
すなわち、LCによって分画された試料をMSで分析することによって、上述したような2次元のスペクトルデータを得ることができ、試料中の成分分析を行うことができる。ところが、LCの役割を単に分画に限定することにより、従来のLC−MSでは、検体中から検出・識別できるタンパク質の種類が網羅的でなく、分析能・解析能が低いといった問題がある。
【0004】
一方で、クロマトグラフィーを単に分画だけでなく試料の特性を現す情報として利用する目的で、複数のクロマトグラフィー結果を比較できるようにするために、時間軸を補正して重ね合わせる(align)方法がいくつか提案されている。その代表的なものとして、Dynamic Time Warping (以下、DTWと略記する)および Correlation Optimized Warping (以下、COWと略記する)がある。いずれも動的計画法アルゴリズムに基づいた1つの実装形態として、それぞれユークリッド距離、あるいは相関を2つのクロマトグラフィーの距離もしくは類似性の指標とするものである(V. Pravdova, B. Walczak, D.L.Massart, “A comparison of two algorithms for warping of analytical signals”, Anal. Chim. Acta 456: 77-92 (2002))。しかしながら、これらの方法は、クロマトグラフィーの時間軸と信号強度との2次元で表されるクロマトグラムに適用されるものであるため、多次元データにおける少なくとも1次元のパラメータを補正するものではない。
【0005】
さらに、このような重ね合わせ方法は、比較対象となるクロマトグラムあるいはスペクトログラムがある程度類似していることが前提となる。実際、DTWにせよCOWにせよ、比較対象のプロファイル間における距離の最小化あるいは相関の最大化を目指した重ねあわせを行うため、比較対象のプロファイルの共通性が乏しい場合には適切な重ねあわせが得られない可能性が十分にある。このように高い共通性を前提とした方法は、例えば、実際の疾患病態解析や薬剤応答性解析のように、多くの因子の変動が予想され、しかもそれぞれの変動量が微細で、個体差や測定誤差などに紛れそうな場合には不適切である。
【0006】
そこで、本発明は、上述したような実状に鑑み、試料に含まれる成分を分析するに際して、優れた分析能を達成することができる試料解析方法及び試料解析プログラムを提供することを目的とする。
【0007】
【非特許文献1】V. Pravdova, B. Walczak, D.L.Massart, “A comparison of two algorithms for warping of analytical signals”, Anal. Chim. Acta 456: 77-92 (2002)
【発明の開示】
【課題を解決するための手段】
【0008】
上述した目的を達成した本発明は以下を包含する。
(1)試料の分析の結果として得られた多次元データにおける、少なくとも1次元のパラメータを補正する工程aと、上記工程aにより得られる補正後のデータを複数の試料について比較する工程bとを含む試料解析方法。
【0009】
本試料解析方法において、多次元データとしては、クロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを挙げることができる。また、このとき、工程aでは保持時間を示すパラメータを補正することが好ましい。
【0010】
また、ここで、補正対象のパラメータを除いたパラメータに関するプロファイルを参照プロファイルと定義し、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を与えることができる。この場合、工程aでは、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることができる。
【0011】
このとき、評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることが好ましい。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【0012】
また、工程aでは、補正対象のパラメータについて最適解探索問題として評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることができる。この動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることが好ましい。さらに、この場合、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることが好ましい。
【0013】
上記(1)に係る試料解析方法は、特に上記工程aにおいて、あらかじめ添加された標準物質に由来する情報を用いることによって、解析の精度をより向上できるとともに、補正処理の能力をも向上させる。本発明に係る試料解析方法のなかでも、このような特徴を備える方法をinternal standard guided optimal profile alignment(i-OPAL)法と命名する。
【0014】
また、上述した本発明に係る試料解析方法は、各種データを入力する機能を有する入力手段、プログラムに従って演算処理を実行する機能を有する演算処理手段、当該演算処理の結果等を表示する機能を有する表示手段を備えるコンピュータに実行させるためのプログラムとして実現することができる。
【0015】
一方、本発明に係る試料解析方法は、異なった種類の試料の間で量の異なる物質を検出・同定することができる。具体的には、複数の試料において多次元データとして、クロマトグラフィー質量分析の結果として得られる質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを測定し、複数の試料間で3次元データを比較し、イオン強度が有意に異なるシグナルを検出・同定することができる。これらのシグナルの特性、即ち、質量/電荷比および保持時間に十分近接した特性を持つシグナルを生じさせた物質に対して、更なる解析を施すことで、当該物質を同定することが可能である。
【0016】
さらに、検出・同定工程を、例えば疾患由来試料と、健常者あるいは健常組織由来試料とに適用することにより、疾患群と健常群で有意に異なる存在量を示す物質を検出・同定することができる。これによって同定された物質は、バイオマーカーとして利用することができる。バイオマーカー検出・同定結果を利用して、疾患の診断や治療法の選択へ利用することが可能である。
【0017】
さらにまた、検出・同定工程を、例えば特定の治療法あるいは薬物への応答性の差異、あるいは副作用の差異を示す患者集団由来の試料へ適用することで、治療法/薬剤応答性あるいは副作用のマーカーとなる物質の検出・同定へ利用可能である。
【発明を実施するための最良の形態】
【0018】
以下、図面を参照して本発明を詳細に説明する。
1.試料の調整
本発明に係る試料解析方法では、先ず、解析対象の試料を採取する。解析対象の試料としては、特に限定されないが、例えば、動物個体由来の臓器の組織切片や血漿・リンパ液などの体液成分、植物の緑葉や花弁などの器官、環境中の土壌・水成分などがあげられる。これらの試料に含まれる分析対象物質としては、特に以下に限定されないが、例えば、有機化合物、無機化合物、有機金属化合物、金属イオン、ペプチド、蛋白質、金属蛋白質、リン酸化を含む翻訳後修飾を受けたペプチド、リン酸化を含む翻訳後修飾を受けた蛋白質、核酸、糖質、脂質などがあげられるが、特に望ましくはペプチド、蛋白質、金属蛋白質、翻訳後修飾を受けたペプチドもしくは蛋白質である。
【0019】
また、採取した試料は、分析の目的及び採取した試料の特性に合わせて、必要であれば各種処理を施すことが好ましい。例えば、(ア)タンパク質群の分離あるいは分画、(イ)タンパク質群の酵素的及び/又は化学的切断、(ウ)切断によって生じたペプチド混合物の分離あるいは分画、および(エ)標準物質の添加、の全ての要素あるいは一部の要素の組み合わせて行う分析前調製を施すことが好ましい。
【0020】
より具体的に、「(ア)タンパク質群の分離あるいは分画」は、一次元のドデシル硫酸ナトリウム(SDS)電気泳動法、二次元電気泳動法、キヤピラリー電気泳動法、イオン交換クロマトグラフィー、ゲルろ過クロマトグラフィー、順相クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトグラフイー、或いはこれらの組み合わせによる多次元分離・分画等によって行うことができる。
【0021】
また、「(イ)タンパク質群の酵素的及び/又は化学的切断」は、トリプシン消化、キモトリプシン消化、Lys-C消化、Asp-N消化、Glu-C消化、臭化シアン分解或いはこれらの組み合わせによる切断等によって行うことができる。
【0022】
さらに、「(ウ)切断によって生じたペプチド混合物の分離あるいは分画」は、一次元のドデシル硫酸ナトリウム(SDS)電気泳動法、二次元電気泳動法、キャピラリー電気泳動法、イオン交換クロマトグラフィー、ゲルろ過クロマトグラフィー、順相クロマトグラフィー、逆相クロマトグラフィー、アフィニティークロマトゲラフイー或いはこれらの組み合わせによる多次元分離・分画等によって行うことができる。
【0023】
さらにまた、「(エ)標準物質の添加」において標準物質は、選択したイオン化法にてイオン化できるものであって、測定のLC保持時間の範囲内に溶出するものであって、溶出時間および分子イオン強度の再現性が高いものを選択することが好ましい。このような好ましい標準物質としては、例えば、有機化合物、無機化合物、有機金属化合物、金属イオン、ペプチド、蛋白質、金属蛋白質、リン酸化を含む翻訳後修飾を受けたペプチド、リン酸化を含む翻訳後修飾を含む蛋白質、核酸、糖質、脂質など、より好ましくは、ペプチド・蛋白質で市販品、天然に存在する物質あるいは合成された物質を挙げることができる。
【0024】
以上、(ア)〜(エ)に示した分析前における各種処理は、例えば、「ア、エ、イ、ウの順」、「エ、イ、ウの順」、「イ、エ、ウの順」、「エ、アの順」、「エ、イの順」「イ、エの順」又は「エのみ」で行うことができる。
【0025】
2.試料分析
次に、試料を分析することによって、試料に関する多次元データを取得する。具体的には、試料をLC-MSにより分析し、m/z、イオン強度及び保持時間からなる多次元データを測定する。ここで、LC−MSにより分析するとは、試料をクロマトグラフィーの原理に従って分離又は分画し、その後、分離又は分画された試料に含まれる成分を質量分析の原理で測定することを意味する。なお、保持時間は、試料をクロマトグラフィーの原理に従って分離又は分画する際の時間として測定される。また、m/z及びイオン強度は、質量分析の結果として測定される。
【0026】
また、クロマトグラフィーの原理としては、特に限定されないが、逆相クロマトグラフィー、キヤピラリー電気泳動、アフィニティークロマトグラフイー、クロマトフォーカシング、等電点フォーカシング、ゲルろ過クロマトグラフィー等の各種クロマトグラフィーの原理を適用することができる。特に、本明細書においてLCと表記する場合、液体クロマトグラフィーのみを意味するのではなく、広く一般的なクロマトグラフィーを意味する。
【0027】
LC−MSにおけるクロマトグラフィーでは、再現性の高い溶出プロファイルが得られること、分離能が高いこと、適当なイオン化のインターフェースを介してMSに直接分子イオンを導入することが可能であることが好ましい。
【0028】
より具体的に、液体クロマトグラフィーにおける好ましい条件としては次の通りである。試料中のペプチド群を分析対称とする場合には、水・アセトニトリル溶液に低濃度の蟻酸などの強酸を含む溶離液を用いたC18カラムによる逆相液体クロマトグラフィーが好ましい。また蛋白質群を分析対称とする場合には、水・アセトニトリル溶液に低濃度の蟻酸などの強酸を含む溶離液を用いたC4カラムによる逆相液体クロマトグラフィーが好ましい。
【0029】
質量分析は、特に限定されないが、磁場型質量分析計、飛行時間型質量分析計、四重極質量分析計、イオントラップ質量分析計、フーリエ変換質量分析計またはこれらのハイブリッド及びタンデム質量分析計等により行うことができる。より好ましくは、エレクトロスプレーイオン化またはナノエレクトロスプレーイオン化と結合できる磁場型質量分析計、飛行時間型質量分析計、四重極質量分析計、イオントラップ質量分析計、フーリエ変換質量分析計またはこれらのハイブリッド若しくはタンデム質量分析計を用いて質量分析を行うことが好ましい。
【0030】
LC−MSにおける質量分析では、再現性の高い質量スペクトルが得られること、500ppm以下の高い質量精度を有すること、一定範囲のm/zの分子イオンに対して衝突誘起解離(CID)をかけ、当該分子イオンのフラグメントイオンの質量スペクトルが得られることが好ましい。
【0031】
このように、試料をLC−MSにより分析し、m/z、イオン強度及び保持時間を測定することによって、試料の分析結果を3次元データとして取得することができる。なお、LC−MSによる分析は、保持時間に関するデータ、m/zに関するシグナル及びイオン強度に関するデータを、入力手段を介してコンピュータに入力し、詳細を後述するアルゴリズムに従って演算処理手段によって処理を行うことにより図1に示すような3次元データとして取得することができる。本アルゴリズムは、コンピュータソフトウェアに搭載することができる。当該ソフトウェアをコンピュータにインストールすることによって、CPU等の演算処理手段により本アルゴリズムをコンピュータ上で実現することができる。従って、図1に示すような3次元データは、コンピュータの表示装置に表示することができる。
【0032】
従前のLC−MSによる解析方法においては、単に試料の分画のためにLCを行っているため、保持時間は解析対象パラメータとして使われておらず、試料の分析結果として横軸にm/z、縦軸にイオン強度をとった2次元データが解析対象となりうるに過ぎなかった。これに対して本発明に係る解析方法によれば、試料の分析結果を3次元空間上にプロットされるプロファイルとして取得することができるため、試料の分析能を飛躍的に向上させることができる。具体的には、本発明に係る解析方法によれば、保持時間を示す軸の方向に広がりを持った多数のスペクトルの重ね合わせとしてデータを取得でき、従前の解析方法と比較してより多数の成分に関して、スペクトルに基づく同定を行うことができる。このため、例えば、複数の試料について得られた多次元データを比較することで各試料の成分分析をより厳密に行うことができる。
【0033】
3.データ解析
次に、本発明に係る解析方法においては、以上のように測定した保持時間を、演算処理手段の制御のもとに本発明に係るアルゴリズムによって補正することもできる。ここで、一般に保持時間は、LCにおける移動相の組成、流速、カラム温度等のファクターが時間的に微小な変化を生じることから、非線形的に変動することが多い。したがって、本発明に係る解析方法で取得された3次元データに関しても、複数の試料について解析を行った場合に試料間の保持時間を示す軸が非線形的に変動していることが考えられる。そこで、本発明に係るアルゴリズムにおいては、保持時間の補正(以下、時間軸補正とも言う)を行う。
【0034】
但し、本発明に係るアルゴリズムが対象とする時間軸補正は、従前の方法でDTWアルゴリズム等によるクロマトグラムの時間軸補正のような、保持時間−信号強度の2次元空間で表現される単一次元プロファイルの補正ではない。本発明が対象とするデータは、時間軸に対して補正されるべきプロファイルが少なくとも2次元以上の多次元で表現されるものである。
【0035】
以下、当該アルゴリズムについて説明するが、当該アルゴリズムは保持時間の補正に限定されず、多次元のパラメータが得られた場合に少なくとも1次元のパラメータを補正する場合に広く適用することができる。言い換えると、当該アルゴリズムは、試料の分析の結果として得られた多次元のパラメータ(例えば3次元のパラメータ)における、少なくとも1次元のパラメータを補正する際に適用することができる。従って、以下の説明においては、p+q次元の測定データを取得した場合のアルゴリズムについて説明する。
【0036】
先ず、補正対象とするパラメータを含むp次元の測定値を(x1…xp)とし、補正の際に参照するq次元の測定値を(y1…yq)とすると、データの集合(プロファイル)ZはZ=(x1…xp y1…yq)となる。ここで、x及びyは、データ点の個数Nの次元を持つ列ベクトルである。
【0037】
なお、データ点とは、上記プロファイル行列(Z)の1つの行を構成するp+q次元のベクトルであり、測定対象の1つの要素について、測定パラメータと値の組を表している。特に、
【数1】
番目のデータ点を
【数2】
のようにも表す。
【0038】
また、補正の基準となる測定値を
【数3】
とする。ここで、Sは
【数4】
(Sは基準点の数)を意味する。また、
【数5】
は、いずれも各基準点のとる値が推定可能な範囲に収まらなくてはならない。
【0039】
さらに、本アルゴリズムにおいて補正を行うためには2つ以上のプロファイルデータ
【数6】
が必要となる。
【0040】
以上のような定義の下で、本アルゴリズムにおいては先ず、p個のパラメータ軸x1…xpそれぞれにおいて取りうる値を量子化する。但し、量子化プロセスは、計算精度と計算時間との兼ね合いで行うものであり、値のとりうる場合の数が十分計算可能な範囲にあるならば、必ずしもこの段階で行わなくてもよい。次いで、p個のパラメータ軸x1…xpそれぞれにおいて
【数7】
の各データ点を、順列を保って対応付けを行う。なお、一般に、Z(1)及びZ(2)に含まれるデータ点の個数は異なりうるので、全てのデータ点が1対1に対応するわけではなく、対応する相手のないデータ点も含むことに留意する。
【0041】
このとき、例えば、以下のような評価関数を用いてプロファイル全体での対応付けの評価得点Eを算出する。なおこの評価得点は、類似性を表す尺度として、大きいほどよい「得点」として定義することも、逆に距離を表す尺度として、小さければよい「失点」として定義することも可能である。以下では失点としての定義で説明する。
【0042】
【数8】
ここで
【数9】
は、i番目のデータ点におけるr番目のパラメータの値を表し、N1、N2はそれぞれ1番目と2番目のプロファイルにおけるデータ点の総数である。また、上記評価関数において関数fは、対応する点の類似度合いの距離を与える関数で、例えば以下のような関数を挙げることができる。
【0043】
【数10】
ここで上記式(I)における、右辺第1項目は補正すべきパラメータxrにおいて当該パラメータの測度における差異の程度に応じたペナルティ、第2項目は合わせるべき測定パラメータysが補正後どれだけずれたかを当該パラメータ測度上の距離に応じたペナルティ、第3項目はパラメータ補正によって2つの点が全てのパラメータにおいて一致したことのボーナスとして与えられるスコア、逆に第4項目は補正対象パラメータ軸上での2つの点が一致しなかったことによるペナルティスコアに相当する。また第5項目は、後述するように、標準物質によるシグナルの一致をボーナスとして評価するための項である。
【0044】
また、上記式(I)における、α、β、σ及びπは、それぞれを含む項における係数であり、適宜設定できる値である。一例としては、αを1.0とし、βを0.1とし、パラメータ補正によって点が一致した場合にはσを0とし、一致しなかった場合にはπを100とすることができる。
【0045】
なお、関数δr(i,j)は、着目するパラメータrの値がi,jによって指定されるデータ点において対応した場合は1、しなかった場合は0を与える関数、逆に
【0046】
【数11】
は対応した場合が0、非対応の場合が1となる関数である。
【0047】
上記式(I)において、第2項目は、補正対称のパラメータを除いたパラメータに関するプロファイル(参照プロファイル)について、試料間での配置類似性の尺度を示している。
また、ここでは2点の不一致によるペナルティを与える式として、対応・非対応によって定数となる例を示したが、所定の関数によって算出される値であっても良い。例えば、隣接するデータ点が対応するか否か、対応しないデータ点が出現した列の長さ等を考慮した関数によって第4項目を算出することができる。
【0048】
また、上記式(I)において、ノルム
【数12】
は一般的なベクトル空間上の距離を表し、必ずしもユークリッド距離に限定するものではない。さらに
【数13】
のように2点間の値の差を計算する際に、対応する点がない場合は、値を0(または適当な欠損値の代替値)に置き換えて計算するものとする。
【0049】
なお、本発明において評価関数は、上記式(I)に示す関数に限定されるものではない。例えば、このようなデータ点(i,j)間の補正対象パラメータ或いは参照パラメータの距離の線形結合だけではなく、両者の距離に応じた任意の関数、更には直前若しくはそれまでに連続して対応したデータ点列におけるパラメータ間の距離も勘案した関数を定義することも可能である。また、評価関数としては、上記式(I)に限定されず、試料間において参照プロファイの配置類似性の尺度となるような関数を定義することも可能である。
【0050】
あるいは、ここでは失点としての例を示したが、上記式(I)における右辺各項の符号を逆転させた上で、距離の部分を相関などに置き換えることにより、逆に類似性を表す指標として、大きければ大きいほどよい得点としての評価関数を定義することも容易に可能であり、そのような評価関数も本アルゴリズムに適用することができる。
【0051】
上記式(I)の第5項で例示したように、対応する点が標準物質由来の基準点か否かで、例えば次のような特別な得点を与えるものとする。即ち、対応するデータ点がいずれも標準物質由来のものであった場合は、θ(i,j)=Smとして、はるかに大きな得点を設定することで、評価関数(この場合は距離即ち失点としての定義)が大きな負の値をとりうることで結果的にこのような対応関係が望ましいことを定義する。さらに、一方が標準物質由来であるのに他方がそうでない場合は、逆にθ(i,j)=-Sdとしてはるかに大きな距離となるように定義することも可能である。
【0052】
以上で説明した評価関数を最適化するアルゴリズムによって、上記「2.試料分析」で取得した3次元データに関して、保持時間を示すパラメータの補正を行うことができる。上記「2.試料分析」で取得した3次元データに関して、最適化アルゴリズムを適用する場合、以下の(a)〜(d)の手順に従って説明することができる。
【0053】
(a)保持時間補正の概念
保持時間を補正する操作は、m/z、イオン強度及び補正時間からなる単一の三次元パラメータ集合体を対象とするのではなく、2つの三次元パラメータ集合体の比較によって実現される。三次元パラメータ集合体は、図2に示すように、m/zと保持時間とをそれぞれ行と列にとった行列において、m/z及び保持時間が対応する位置の行列要素にイオン強度が入るような形で表される。保持時間を補正する対象の三次元パラメータ集合体をそれぞれZ(1)及びZ(2)とすると、保持時間の補正操作は、Z(1)及びZ(2)における2つの行列で、保持時間軸に相当する列の対応関係を決める操作(以下、「対応配置の検索」と呼ぶ)に他ならない。例えば、図2に示す行列をZ(1)の行列とし、図3に示す行列をZ(2)の行列とすると、図4のような配置が望ましい対応配置(重ね合わせ配置)である。
【0054】
(b)2つの3次元データにおけるパラメータ集合体間の対応配置の探索
図4に示すような対応配置を探索するためには、可能なすべての保持時間の対応付けを考える。この際、配置の対応関係の良し悪しを評価するスコアを定義し、配置毎にスコアを計算し、その中でもっともスコアがよいものを採用することで目的とする最適な対応配置を得ることができる。図5は、図2と図3に示した三次元パラメータ集合体Z(1)及びZ(2)に関して、保持時間の可能なすべての対応付けを示したものである。横方向にZ(1)の保持時間、縦方向にZ(2)の保持時間が記されているが、(イ)Z(1)及びZ(2)でそれぞれ対応する保持時間のある場合が斜線、(ロ)Z(1)の所定の保持時間に対してZ(2)の方に対応するものがない場合が横線、(ハ)Z(2)の所定の保持時間に対してZ(1)の方に対応するものがない場合が縦線で示されている。Z(1)及びZ(2)の全体的な保持時間の対応付けは、図5の格子の最左上角から最右下角にいたる経路を、これら斜線・横線・縦線をなぞることで求めることに相当する。但し、一度下がったり右に進んだら、進んだ点から逆に上や左に戻るような経路は許されない。なお、図5において太線で示された経路は、図4の対応対置に相当する。
【0055】
(c)保持時間対応配置の良否を判断するためのスコア
保持時間に関する対応配置の良否を判断するスコアは、例えば、次のようにして定義することができる。
i)最左上点におけるスコア、すなわちまだ対応関係がまったく決まっていない点でのスコアを0とする。
ii)前述の(イ)(ロ)及び(ハ)のうちいずれかの場合をとることにより、対応関係が1段階進んだ場合は、その直前のスコアに対して、(イ)(ロ)及び(ハ)毎に決められたスコアを加算することで、新たな対応関係の点におけるスコアとなる。例えば、以下のように(イ)(ロ)及び(ハ)毎にスコアを設定することができる。
【0056】
(イ)の場合(図5において斜線方向に進む場合):
この場合、所定の保持時間に関して、Z(1)及びZ(2)が互いに対応付けられるわけである。したがって、この場合、加算されるスコアとしては、m/zパラメータとイオン強度パラメータがZ(1)及びZ(2)間でどれだけ類似或いは離れているかを反映した値を設定することができる。以下の説明では類似度としてスコアを定義した場合について説明する。例えば、Z(1)において所定のm/zの元でイオン強度が検出されているのにZ(2)には同m/zの元でイオン強度が検出されなかったケース、あるいはその逆のケースであれば、一定の値(ペナルティスコア)を減じるようにスコアを設定することができる。また、所定のm/zにおいてZ(1)及びZ(2)それぞれにイオン強度が得られている場合、例えば両イオン強度の差の絶対値に所定の係数を乗じて算出される値(ペナルティスコア)を減じるようにスコアを設定することができる。さらに、スコアとしては、両イオン強度の違いが大きければ大きいほど、得点が小さくなるような関数で算出されるものであっても良い。
【0057】
一方、Z(1)及びZ(2)における保持時間のずれもスコアに反映させることもできる。例えば、Z(1)及びZ(2)における保持時間の差の絶対値に所定の係数を乗じて算出された値(ペナルティスコア)を減じるようにスコアを設定することができる。スコアとしては、Z(1)及びZ(2)における保持時間の違いが大きければ大きいほど、得点が小さくなるような関数で算出されるものであっても良い。
【0058】
なお、標準物質由来のシグナルがZ(1)及びZ(2)で対応する場合には、後述する計算方法の工夫に加えて、スコア算定上も特別な措置を施すことが好ましい。特に、これらの点がZ(1)及びZ(2)間で一致することは強く求められるので、Z(1)及びZ(2)ともに標準物質由来シグナルとして対応付けられる場合には大きな得点を、逆に一方だけしか標準物質由来シグナルが見つからない場合には大きな失点を与える。
【0059】
(ロ)及び(ハ)の場合(図5において縦又は横方向に進む場合):
この場合、所定の保持時間に関して、Z(1)及びZ(2)において対応する保持時間を見出せなかったわけである。したがって、この場合、所定の値(ペナルティスコア)を減じるようなスコアを設定する。
iii)このようにして図5の格子の最左上角から最右下角にいたるまで、段階的にスコアを求めてゆき、最後に最右下角までいたった時点でのスコアが、得られた対応配置に対応するスコアになる。
【0060】
(d)保持時間に関する最適な対応配置を求める手順
基本的には、可能なすべての対応配置を列挙し、それぞれについてスコアを計算し、その中で最大のスコアを示す対応配置を選択すればよいわけであるが、上述したように、スコアは漸化式で与えられるため、「動的計画法」に適している問題である。すなわち、3次元データZ(1)に含まれるi番目の保持時間とZ(2)に含まれるj番目の保持時間の対応関係を考える際には、(イ)Z(1)に含まれるi−1番目及びZ(2)に含まれるj−1番目に次いでZ(1)及びZ(2)両者ともに対応付けられる場合、(ロ)Z(1)に含まれるi−1番目及びZ(2)に含まれるj番目に次いでZ(1)の保持時間に対応するZ(2)のパラメータがない場合、(ハ)Z(1)に含まれるi番目とZ(2)に含まれるj−1番目に次いでZ(2)の保持時間に対応するZ(1)のパラメータがない場合、の3通りを考えることとなる。いずれの場合も1段階前の状態におけるスコアがわかっていればZ(1)及びZ(2)の(i、j)番目のスコアを算出することが可能となる。
【0061】
そこで(イ)(ロ)及び(ハ)の3通りのうち、最もよいスコアを与えた場合のスコアとそこに至るまでの経路のみを記録しておき、このステップを図5に示した格子の最左上角の出発点から最右下角のゴールに達するまで続ける。そして、記録した経路を最右下角から出発点まで、逆にたどることにより、最適経路、すなわちZ(1)及びZ(2)における保持時間に関して、最適な対応配置を求めることができる。
【0062】
以上、(a)〜(d)の手順として説明した手法は、動的計画法に基づいた最適解探索手法と言い換えることができるが、本発明において適用可能なアルゴリズムは動的計画法に限定されるものではない。すなわち、目的の評価関数を最適化するより一般的な探索問題と捉えることで、他の最適探索アルゴリズムを用いて実装することも可能である。このようなアルゴリズムとしては、例えばA*アルゴリズム、遺伝的アルゴリズム(GA)、シミュレーテッドアニーリング(SA)、最急降下法等による非線形計画法等によって実装することもできる。
【0063】
(a)〜(d)の手順として説明した手法は、いわゆる動的計画法に基づく手法であり、動的計画法に基づいている点では、DTW法やCOW法と類似する部分がある。しかしながら、DTWやCOWは、評価関数の形や計算手法もユークリッド距離あるいは相関を評価関数として、時系列データ点列そのままあるいは一定間隔の時間区切りでセグメント化した上での区間ごとの比較として、さらに2つのプロファイルの時刻0を出発点とし、それぞれのプロファイルの終了時刻をゴールとする同じ大域的制約条件の下に探索を実施する方式に限定されたものである。さらに、DTWやCOWを用いる手法は、基本的に2次元データとして表される時系列プロファイル、即ち時間軸とシグナル強度軸で表されるようなデータセットに対し、時間軸の非線形伸縮を行うことによって強度のプロファイルを重ね合わせるものである。
【0064】
したがって、DTWやCOWを用いる手法では、(1)特定の軸について特定の値をとる1つないし複数個の切断面を用いて、あるいは、(2)特定の軸に沿って全ての値を集約することによって、重ねあわせ操作を行わせることは、これらの方法の自然な拡張として容易に考えられる。例えば、DTWやCOWを用いる手法では、LC-MS分析で得られた保持時間、m/z、イオン強度からなる3次元データについても、いくつか特定のm/zに限定することにより、あるいは、全イオンクロマトグラム(TIC)のように保持時間軸に沿って全てのイオン強度を足し合わせることによって、時間軸補正が可能である。
【0065】
しかしながら、(a)〜(d)の手順として説明した手法では、DTWやCOWを拡張した方法とは異なり、補正対象の次元(保持時間軸)を除いた多次元プロファイルはそのままの形で直接比較することで、補正対象の次元を伸縮させることで、多次元プロファイルの重ねあわせを実現している。DTWやCOWを拡張した方法では、(1)特定の切片に限った手法をとると、精度を保ちつつプロファイル全体を重ね合わせるのと同じ結果が得られる保証がないこと、特定の切片を選択するための汎用性のある有効な手段がないこと、そのような保障なしに限定した重ね合わせ処理を行うことで、結果が恣意的になりうる危険があること、(2)TICのように情報を集約することで、せっかく次元を多くしてより分解能を向上させているメリットが得られなくなってしまう、といった問題がある。これに対して、(a)〜(d)の手順として説明した手法では、上記(1)及び(2)の問題が全くなく、高精度にプロファイルの重ね合わせが可能となり、且つ、多次元データという高分解能を維持したままプロファイルの重ね合わせが可能となる。
【0066】
4.データ解析における標準物質の役割
また、本発明に係る試料測定方法においては、以下に述べるような標準物質由来の情報を、本アルゴリズムを用いた計算手法に取り入れることで高い精度及び計算効率を達成することができる。
【0067】
上記「2.試料の分析」に先立って又は「2.試料の分析」の途中で標準物質を入れることにより、測定及び解析の過程で生じる可能性のあるバイアスを補正すると同時に、これらの情報を用いることによって、上述した最適な対応配置、すなわち、プロファイル同士の重ねあわせをより正確にかつより効率的に行うことが可能となる。すなわち、標準物質を用いることによって以下のメリットを得ることができる。
(1)あらかじめ既知の量の標準物質を入れることで、全体のシグナル強度を補正し、定量的な比較が可能となること
(2)いくつかの標準物質を、補正すべきパラメータ(時間軸等)を合わせる際のランドマークとして使用できること
(3)プロファイル重ねあわせにおいて、ある程度のプロファイル形状の共通性を与えることにより、重ねあわせが容易になること
【0068】
これらメリットを上記「3.データの解析」において最大限発揮させるべく、計算手法を以下のように改変することができる。すなわち、上述したアルゴリズムにおいて、標準物質に由来するシグナルのピーク部分を必ず通らなくてはいけない点として扱うように改変することができる。より詳しく説明すると、上述したアルゴリズムによる最適解探索では、図5に示したような格子状の探索空間上で、左上の出発点から右下の到着点に至る最適経路(太線)を探索するものであるが、いまここで仮に縦列上の保持時間15の点と、横の行で保持時間13の点がそれぞれ標準物質由来であったと仮定すると、解となりうる経路は必ずこの点を通らなければいけないというように拘束条件を設ける。そして、このような拘束条件を設定することによって、列15と行13を通る線で区分される探索空間のうちの左下と右上の部分空間を通る経路は排除されるため、探索しなければいけない空間を削減することができる(図6)。
【0069】
このように、標準物質由来のシグナルについて拘束条件を設定するようにアルゴリズムを改変することによって、本発明に係る試料解析プログラムは、プロファイル同士の重ねあわせをより正確に行うことができるとともに、処理の能率を大幅に向上させることができる。
【0070】
さらに、標準物質由来のシグナルの数が増えるにつれて、検索空間はより限定されることになるため、プロファイル同士の重ねあわせ精度はより向上するとともに、より能率向上が期待できる。実際、図7に示すように、丸印で示した点を標準物質由来シグナルの一致点として拘束条件を設定すると、灰色でマスクした領域は探索空間から削除される。n種類の標準物質由来シグナルで時間軸がn+1個に分割されたとすると、最良のケースとして分割が等間隔で行われたならば、探索空間は最大
【数14】
まで減少する。なお、標準物質由来のシグナルを拘束条件に設定する場合、標準物質由来シグナルが均等に広く分布するように標準物質を選択することで、探索空間削減効果を最も発揮することができる。
【0071】
なお、探索空間を限定して探索効率を上げるためのアルゴリズムに対する改変方法としては、図5に示した探索空間の左上の出発点から右下の到達点に至る対角線の前後に、所定の幅を持った空間に限定するといった拘束条件も考え得る。しかしながら、この場合、どの程度限定すれば良いのかといった確実な前提知識は一般に得られない虞がある。さらに、この場合、出発点や到達点が比較すべき多次元データ毎に大きくずれた場合には、求めるべき最適経路が限定された空間からはみ出す虞がある。例えば、クロマトグラフィーの溶出開始時刻は、大きく変動しうるので、この時刻を確実に観測できなければ、上記対角線の前後に所定の幅を持った空間に探索を限定する方法は適切とは言いがたい。
【0072】
それに比べて、標準物質由来のシグナルを拘束条件に設定するようにアルゴリズムを改変する方法では、検体内部に添加した標準物質の出現した時点を揃えており、標準物質に由来するシグナルは最も確証のある参照点となる。その上、探索空間は最大
【数15】
近くまで減らせるため、確実性及び効率性の両面から見ても優れているといえる。
【0073】
さらに、標準物質由来シグナルで限定される1つないしいくつかの部分空間に限って最適経路探索を行うことで、部分的な最適プロファイル重ね合わせを求めることができる。この際に、前述の評価関数の値を、プロファイル重ね合わせの程度の指標とすることで、プロファイル同士の類似性(あるいは距離)を測ることができる。多くの場合、主要なシグナルは限られた時間領域に集中的に現れるため、部分空間に限って最適経路探索を行って評価関数の値を求めることで、プロファイル同士、さらにはそれらのプロファイルを生じさせた試料同士の類似性(あるいは距離)を効率よく求めることができる。
【0074】
数多くの試料由来のプロファイルを重ね合わせて平均プロファイルを求めなければいけない場合や、あらかじめ試料の属性に関する情報が十分に得られていない場合は、まず部分空間に限って最適経路探索を行って試料間の類似性(または距離)を求めておき、その順番で逐次プロファイル重ねあわせを行い、あるいは試料同士のグルーピングを行うことが可能である。殊に、プロファイルの重ね合わせ処理を行う場合は、重ね合わせる順番に依存して結果が変化しうるので、できるだけ近いものから重ね合わせることが望ましいが、そのような処理を行う際にこの手法は有効である。
【0075】
5.データ解析後の処理
2つの多次元データについて、最適な重ね合わせが得られたならば、補正されたパラメータについて、新たな補正後の値を生成する。特に、クロマトグラムの保持時間を補正した場合は、補正後の保持時間を求める。補正後の保持時間を求める方法としては、重ね合わせる2つの多次元データのうち一方を参照用データとし、他方の多次元データにおける保持時間を当該参照用データにおける保持時間に一致させる方法(非対称型)と、重ね合わせる2つの多次元データの両方を補正する方法(対称型)とを挙げることができる。特に対称型で補正後の保持時間を求めることが好ましい。
【0076】
補正後の保持時間を求めて非対称型で2つの多次元データを重ね合わせる場合には、参照用データの保持時間軸にあわせるべく、一致した点に関しては参照用データの保持時間をそのまま用い、参照データの対応する点が得られなかった場合は、その前後で一致した点を用いて内挿によって補正後の保持時間を決めることができる。
【0077】
しかしながら、非対称型で2つの多次元データを重ね合わせるには、いずれの多次元データを参照用とすべきかが予め決まっている必要がある。例えば、標準物質のみを含むブランクを参照用データとすることも考えられるが、この場合は最初に重ね合わせに用いられた多次元データのプロファイルの影響がかなり大きくなる可能性が高い。
【0078】
これに対して、補正後の保持時間を求めて対称型で2つの多次元データを重ね合わせる場合には、2つの多次元データ間で対応する点が得られたところについては、それぞれの保持時間の算術平均とする。2つの多次元データのいずれか一方だけのデータ点しか得られなかった場合は、その前後の最も近い対応点の補正後の保持時間の組から、内挿によって補正後の保持時間を得る。内挿による補正が不可能な場合は、最も近くにある対応した点の補正後の保持時間をベースにして、データセット全体での平均時間スケールを係数とした外挿によって補正後の保持時間を得ることが可能である。
なお、この場合、前述のようにあらかじめ全ての多次元データ間の類似性(距離)を事前に計算した上で、近いものから順次重ね合わせても良い。
【0079】
6.出力処理
上記「5.データ解析後の処理」で得られたプロファイルの出力方法としては、以下の(1)及び(2)を挙げることができる。
(1)対応する点が得られなかった箇所も含めて、全て出力する。
(2)対応する点に限って出力する。
【0080】
いずれの出力方法を選択するかは、本発明に係る試料解析方法の使用目的に応じて適宜選択することができる。例えば、測定誤差を相殺するため同一試料を複数回に亘って測定した結果から平均を求めることを目的とする場合、又はきわめて類似した条件での複数試料の測定から代表的なプロファイルを得たい場合には上記(2)の出力方法が有効である。上記(2)の出力方法によれば、出力されるプロファイルは共通部分に限られるため、データの大きさを軽減して処理効率を上げることができる。
【0081】
また、例えば異なった試料群の群間差を検出する場合には、上記(1)の出力方法を使用しなくてはならない。上記(1)の出力方法では、データの大きさは一般に増大するが、情報の損失は起こらない。
【0082】
また、上記(1)の出力方法を選択する場合、さらに共通するプロファイルに重きを置いた重ね合わせを行うこともできる。この場合、以前の重ね合わせプロセスで対応する点に対して新たな点を対応させる場合は、評価関数のスコアを良くするような新たな項を当該評価関数に設けて、できるだけ同じ点に重なるように調整することも可能である。すなわち、例えば、上記式(I)で与えられる評価関数の最後に-μ・δm(i,j)のような新たな項を付与した新たな評価関数を用いて評価得点を算出する。そして、このような評価関数を用いる場合、以前の重ね合わせ点に対して対応が取れた場合にはδm(i,j)を1、それ以外は0とする。
【0083】
一方、本発明に係る試料解析プログラムの出力は次のような形式となっている。
・重ね合わせ処理で新たに得られた点の情報
・対応する入力データセット1(一方の多次元データ)の点の情報
・対応する入力データセット2(他方の多次元データ)の点の情報
出力は、これらの情報が重ね合わせ処理の結果として得られたデータ点の数だけ繰り返された形をとる。但し、対応する点がない場合には、それぞれ入力データセット1あるいは2の情報は存在しない。このように、出力には、対応する入力データセットの点の情報も含まれることにより、後述する例のように、最終的に得られた、重ね合わせプロファイルの各点が、元の多次元データのどれに由来するかを求めることが可能となる。なお、以上の情報の他にも必要であれば付加的情報をつけて出力することも可能である。
【0084】
このようにして得られた重ね合わせ後のプロファイルについて、必要に応じてさらにいくつかのパラメータに関して集約あるいは量子化処理を行ってもよい。例えば、特に上記(1)のように全ての点を出力した場合は、時間軸分解能が求めるレベル以上に詳細になりすぎることもある。この場合は時間軸上きわめて近接した点同士はさらに集約して1つにした方が、後の処理上得策である。集約された点の強度は、集約前の個々の点の強度の加算で置き換えることができる。同様にして、m/z軸においても必要分解能以上に近接した点は集約することができる。但しこの操作は重ね合わせ処理を行う都度実行してもよいし、必要な重ね合わせをまず行ってから、最後に1回だけ行ってもよい。
【0085】
7.イオン化強度の規格化と標準物質
本発明に係る試料解析方法においては、測定したイオン強度について、上記「3.データ解析」に先立って規格化を行うことが好ましい。以下にイオン強度の規格化について説明するが、イオン強度の規格化手法は何ら限定されるものではない。
【0086】
具体的には、先ず、LC−MS分析に結果として得られたRAWファイルを、例えばXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換する。次に、C言語およびPerl言語で作成されたプログラムにより、以下の一連のデータ処理を適用する。
(1)ノイズレベルのデータを除去するため、イオン強度が所定の値(例えば、102以下)以下のシグナルを除去する。
(2)必要ならば、処理時間の節約のためにデータ点を集約する。具体的に、例えば、m/zは1刻みに、保持時間は0.2刻みになるよう、元データのm/z値および保持時間の値を丸め、同じ値を持つ(m/z、保持時間)のデータ点は加算集計する。
(3)あらかじめ調べたm/z値と保持時間から標準物質由来のシグナルを同定し、そのイオン強度値をもって測定値全体を除算することで規格化する。この際、1つないし複数の標準物質由来の複数のシグナルの平均値などの代表値を標準物質イオン強度値として用いる方法、予備実験などでシグナルの安定性を事前に検討したうえでもっとも安定なシグナルの値を用いる方法、などがある。
【0087】
より具体的に、例えばニワトリ卵白リゾチームを標準物質とした場合、m/z値715近傍及び877近傍のシグナルを標準シグナルとすることができる。サンプルの測定データに対しては、m/zについては前後±1の範囲で、保持時間に関してはm/z 715(715±1)のシグナルについては早期溶出シグナル群より、m/z 877(877±1)のシグナルについては後期溶出シグナル群より探索することで、標準物質由来シグナルを探索することができる。なお、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正するといった更なる補正を行ってもよい。
【0088】
また、ペプチドそのもの、例えばペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)およびβカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)を標準物質として用いた場合、それぞれm/z値859近傍及び791近傍のシグナルを標準シグナルとすることができる。前者のペプチドは比較的親水性であり、後者は疎水性である。本解析方法で保持時間による分離のために用いられている逆相クロマトグラフィーでは、前者の保持時間の値は低く、後者のそれはより高い。大部分のサンプル由来のペプチドの保持時間はこの2種類のペプチドの保持時間の間にある。サンプルの測定データに対しては、m/zについては前後±1の範囲で、保持時間に関してはあらかじめ標準物質のみを測定して得られたクロマトグラムからおおよその値を見積もってから、その前後一定の範囲で探索することで、標準物質由来シグナルを見出すことができる。
【0089】
本解析方法において標準物質としてペプチドを用いる場合、当該ペプチド以外の物質(例えば、夾雑物)を含まないことがシグナルノイズの検出を最小限に抑えるために重要である。したがって、天然物から抽出・精製して得られるものよりも、化学的に合成されたペプチド分子の方が、望ましい。また、ペプチド分子の性質については、あらかじめ設定された測定条件下で構造が安定なことや不溶化しないことが重要である。ペプチド分子を構成するアミノ酸残基については、メチオニン、トリプトファン、ヒスチジンなど容易に酸化されうるアミノ酸残基を含まないこと、および塩基性の官能基を2つ以上含まないことが望ましい。とくに後者については、測定手段のMSで原理的に多価イオンを生じるエレクトロスプレーイオン化法を用いた場合、ひとつの標準物質から価数の異なる複数のイオンシグナルが検出されることを避けるためである。
【0090】
標準物質として蛋白質から加水分解あるいは化学的切断によって生じるペプチド断片を用いる場合、当該蛋白質に由来するペプチドイオンシグナルのうちで標準物質として採用したもの以外のペプチドイオンシグナルの強度は出来る限り低いことが望ましい。
【0091】
以上(1)〜(3)によって、測定されたイオン強度値を規格化することができ、複数の試料間におけるイオン強度の量的な比較を行うことができる。なお、測定されたイオン強度値の規格化は、上述した保持時間の補正に先立って行われるべきである。
【0092】
8.試料間の比較分析
本発明に係る試料解析方法によってm/z、規格化されたイオン強度及び補正した保持時間からなる3次元データを用いて、試料中に含まれるタンパク質群等の各種成分分析をコンピュータ上で行うことができる。具体的に、成分分析としては、(a)加算の方法、(b)減算の方法を挙げることができる。
a.加算の方法
上述したように、本発明に係る試料解析方法によって取得された複数の3次元データにおいては、保持時間のパラメータをそれぞれ適切に補正しているため、データ点間の対応関係を正確に取ることができる。したがって、複数の3次元データにおいて、データ点同士の規格化されたイオン強度値同士を足し合わせることができる。
b.減算の方法
「a.加算の方法」と同様に、本発明に係る試料解析方法によって取得された複数の3次元データにおいてはデータ点間の対応関係を正確に取ることができるため、データ点同士の規格化されたイオン強度値の差を求めることができる。
【0093】
このように、本発明に係る試料解析方法によって取得された複数の3次元データについて、加算或いは減算することができるため、以下のような、成分分析のアプローチをコンピュータ上で実現することができる。
【0094】
(1)実験データを集計する際への応用:
1つのサンプル由来の試料を、測定の便宜上、複数の分画に分割して測定した場合であっても、当該複数の分画それぞれから取得された3次元データにおいては、データ点間の対応関係を正確に取ることができる。したがって、上述した加算の方法に従って、全ての3次元データを足し合わせることができる。これにより元のサンプル全体に含まれる成分の解析等を行うことが可能となる。
【0095】
なお、このような集計を行う際には、複数の分画全てに亘って足し合わせ、全体で1つのプロファイルにまとめる方法のほかに、あらかじめ指示された個数により、近接する数個のプロファイル毎に集約する方法もある。この場合、全体がn個に分画され、そのうち近接するm個ずつを集約した場合はn-m+1個のプロファイルが得られることになるが、その場合は得られたプロファイルについては、対応する分画同士以外はそれぞれ別個に扱って以下の操作を行うことになる。いずれにせよ、多次元分画法で複数の分画を得た場合も、そのおのおのが完全に重なりなくなることはまれで、多くの場合複数分画に亘るキャリーオーバーがありうるので、このような集計操作は必要である。
【0096】
(2)複数のサンプルの測定結果の代表値を求める際への応用:
異なるサンプルに由来する複数の試料について測定した場合であっても、本発明に係る解析方法によれば、取得された複数の3次元データ間において、データ点間の対応関係を正確に取ることができる。したがって、上述した加算の方法に従って、全ての3次元データを足し合わせることができる。そして、得られた3次元データの総和をサンプル数で除算することで相加平均を求めることができる。なお、必要に応じて、各サンプルに重みを設定し、当該重みを反映させた重みつき平均を算出することもできる。
これによれば、例えば、同じ範暗に属すると考えられる複数のサンプルについて、当該範暗の代表値を求めることができる。
【0097】
(3)2つのサンプル間での測定結果の差分を求める際への応用:
例えば、同一のサンプル由来であるが状態の異なるときに採取した試料について測定した場合であっても、取得された2つの3次元データ間において、データ点間の対応関係を正確に取ることができる。したがって、上述した減算の方法に従って、2つの3次元データ間の差分を求めることができる。これにより、状態の変化に起因する試料中の成分変化を解析することができる。
【0098】
また、例えば、上述した(1)に準じて、複数のサンプルを含む2群についてそれぞれ相加平均等の代表値を求めた上で、これら2群の代表値の差を求めることができる。得られた差については、統計的な検定などによって有意性を検討することで、各群に特異的な成分を同定することができる。
【0099】
以上の(1)〜(3)に示す成分分析アプローチは、本発明に係る試料解析方法によって得られた複数の3次元スペクトルデータを格納したデータベースを用いても良いし、当該データベースに格納されたデータと現実に得られたデータとを用いて行っても良い。いずれの場合であっても、上述した(1)〜(3)に示す成分分析アプローチは、コンピュータを用いて容易に実現することができる。
【0100】
このようにして、本発明に係る試料解析方法によって得られた、例えば群特異的シグナル成分については、得られたシグナル領域に範囲を限定したタンデムMS分析などにより、当該シグナルが由来する蛋白質群を同定することができる。すなわち、本発明に係る試料解析方法においては、試料をLC−MSにより分析したときに、特定のm/z値を持つペプチド分子イオンが検出された場合、当該イオンのCIDスペクトルを測定することができる。
【0101】
そして、得られたCIDスペクトルをコンピュータに入力し、データベース検索ソフトウェアを用いて蛋白質一次構造データベース、ゲノム配列データベースやcDNA配列データベースから得られる蛋白質配列に対して検索する。このデータベース検索によって有意なヒットスコアを示した場合には、データベースに登録されたタンパク質或いはアミノ酸配列等の情報を得ることができ、得られたCIDスペクトルに対して当該情報を関連付けることができる。
【0102】
例えば、上述した(3)の成分分析アプローチにおいて、各群に特異的な成分として同定されたシグナルに関するCIDスペクトルを測定することで、当該シグナルが示すタンパク質群を同定することができる。
以下、実施例を用いて本発明をより詳細に説明するが、本発明の技術的範囲は以下の実施例に限定されるものではない。
【実施例1】
【0103】
実施例1では、アミノ酸配列が既に知られているタンパク質のプロテアーゼ消化物を混合して得られるペプチド試料をLC-MSによって測定し、この測定によって得られた保持時間、m/z値およびイオン強度からなる三次元プロファイルに対して本発明に係るアルゴリズムを適用し、測定されたペプチド試料を定量的に特性づけた。また、実施例1では、比較定量のためのモデル実験としてアミノ酸配列が既に知られているタンパク質のプロテアーゼ消化物を混合したペプチド試料数種を各々LC−MSによって測定し、本発明の試料解析方法を適用して各三次元プロファイルを比較することによって、各ペプチド試料に含まれるタンパク質の種類の違いが検出されることを示した。
【0104】
ペプチド試料の調製
以下に列挙する24種類のタンパク質のトリプシン消化物を、本実施例におけるペプチド試料として調製した。(1)ウシキモトリプシノーゲン、(2)ウシカタラーゼ、(3)ウシカルボニックアンヒドラーゼ、(4)ウシアポトランスフェリン、(5)ウシカルボキシペプチダーゼA、(6)ウシ血清アルブミン、(7)ウマシトクロムc、(8)ブタガンマ免疫グロブリン、(9)ウシヘモグロビン、(10)ウマミオグロビン、(11)ウシベータラクトグロブリン、(12)ウシデオキシリボヌクレアーゼ、(13)ウサギグリセルアルデヒド3リン酸デヒドロゲナーゼ、(14)トリコンアルブミン、(15)セイヨウワサビペルオキシダーゼ、(16)枯草菌アルファアミラーゼ、(17)ウマグルタチオンS−トランスフェラーゼ、(18)ウシグルタミン酸デヒドロゲナーゼ、(19)ウシラクトペルオキシダーゼ、(20)コウジカビアミログルコシダーゼ、(21)ウサギホスホリラーゼB、(22)ウシベータガラクトシダーゼ、(23)ウサギ乳酸デヒドロゲナーゼ、(24)ニワトリ卵白リゾチーム。これらの消化物はMichrom BioResources社より購入した。
【0105】
これら24種類の各タンパク質のトリプシン消化物を以下に示すように混合し、合計3種類(A群〜C群)のペプチド試料を用意した。
A群:(1)、(2)、(7)〜(24)の20種類のタンパク質のトリプシン消化物。A群を特徴付けるタンパク質は(1)及び(2)である。B群:(3)、(4)、(7)〜(24)の20種類のタンパク質のトリプシン消化物。B群を特徴付けるタンパク質は(3)及び(4)である。C群:(5)〜(24)の20種類の蛋白質のトリプシン消化物。C群を特徴付ける蛋白質は(5)及び(6)である。各群のサンプルは3つずつ調製した。
【0106】
LC-MS解析
各ペプチド試料の三次元プロファイルを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al,Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリルおよび水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
【0107】
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。また、スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。これを各群3回ずつ行うことによって、各試料に対応する三次元パラメータの集合体、3群計9通りを得た。このデータセットをそれぞれAl、A2、A3(A群)、Bl、B2、B3(B群)、Cl、C2、C3(C群)とした。
【0108】
三次元パラメータ集合体を含むファイルはXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した。C言語及びPerl言語で作成されたプログラムにより、以下の(1)〜(5)のデータ処理を実行した。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)処理時間の節約のためにデータ点を集約した。具体的には、m/zは1刻みに、保持時間は0.2刻みになるよう、元データのm/z値および保持時間の値を丸め、同じ値を持つm/zと保持時間の2つ組みで指定されるデータ点は加算集計した。
(3)標準物質であるニワトリ卵白リゾチーム由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、次いでそのデータ点を中心に、イオン強度値が単調減少しかつ0より大きな範囲にあるデータ点を拾い、これらを標準物質由来のシグナルによるデータ点であると見なした。標準物質由来のシグナルの総イオン強度値としては、標準物質由来シグナルと見なされたデータ点のイオン強度の総和をもって当てた。具体的には、ニワトリ卵白リゾチーム由来のm/z値が715近傍及び877近傍のシグナルを標準シグナルとし、サンプルの測定データからこれらの標準物質由来シグナルを探索する際には、m/zについては前後±1の範囲で、保持時間に関してはm/z 715のシグナルについては6〜16分の範囲で、m/z 877のシグナルについては13〜23分の範囲で探索した。
(4)得られた標準物質由来シグナルの総イオン強度値をもって、各シグナルのイオン強度を除算し、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正した。
(5)便宜上、m/z 715のシグナルとm/z 877のシグナルのピーク位置がそれぞれ保持時間に関して10分、20分となるよう、保持時間軸を線型変換した。
【0109】
次に、A、B及びC群それぞれ3例ずつあるサンプルから得られた三次元プロファイルの代表点を求めた。すなわち上述したように、同じ群に属するサンプルを集約した。m/zおよび保持時間の重なる点のイオン強度は加算して集計した。
【0110】
また本例で使用したスコアは高いほどよいとする得点として計算し、算出式の係数は次の通り。イオン強度差については、それぞれの常用対数の差の絶対値に対して係数−1をかけたものを使用した。保持時間の差については、差の絶対値に対して係数−1000をかけたものを使用した。また各群間で対応するデータ点のシグナルがともに標準物質由来であった場合の加算点は50000点とした。一方の群において対応する保持時間の点がなかった場合の失点は5000点とした。本実施例ではこれらを単純に加算してスコアとした。
【0111】
次いで、上述したように、A−B群間、B−C群聞及びC−A群間で差を求めた。得られた差の有意性はt検定によって有意水準0.1%の両側検定で検討した。
【0112】
その結果、保持時間を補正した三次元データを比較することによって、A、B及びCの各群から以下に列挙するm/z値を持つペプチド分子イオンが各群に特異的なシグナルとして検出された。
A群:495,524,546,560,671,696,779,845,871,908,962等。
B群:451,464,509,513,546,555,583,585,626,635,649,653,701,720,723,740,741,753,768,789,819,821,847,873,886,922,928,952,966,973,978,1057,1230等。
C群:636,670,674,679,683,718,734,735,770,824,870,918等。
【0113】
また、本実施例では、特異的なシグナルとして検出されたペプチド分子イオンのCIDスペクトルを得るため、各試料をLC−MS/MS分析にかけた。分析条件は以下に示す操作以外は、上述した通りとした。すなわち、LC−MS/MS分析に際しては、イオントラップ型質量分析計の測定条件を変更し、上に列挙したm/z値を持つペプチド分子イオンが検出された場合に当該イオンのCIDを必ず行うように測定条件を設定して試料の測定を行った。
【0114】
その結果、各ペプチド分子イオンから得られたCIDスペクトルを、Matrix Science社のデータベース検索ソフトウェアであるMASCOTTMを用いて、SWISS−PROT蛋白質配列データベースに対して検索したところ、各群において特異的なタンパク質由来ペプチドとして添加した各群2種類(すなわち、A群においては上記(1)及び(2)のタンパク質、B群においては(3)及び(4)のタンパク質、C群においては(5)及び(6)のタンパク質)、合計6種類はいずれも有意なヒットスコアをもって同定された。このことから、本実施例で行った試料解析方法の妥当性が示された。
【実施例2】
【0115】
実施例2では、所定の濃度組成の蛋白質混合物中に、濃度を変化させた別の蛋白質標品を混合して得られた試料をプロテアーゼ消化し、LC-MSによって測定することで得られた保持時間、m/z値、イオン強度からなる3次元データに対して本発明に係る方法を適用し、異なる濃度の試料の測定で得られた3次元データ同士を比較することで、定量的に変動するシグナルを検出した。これによって、本手法で定量的に変化する物質を検出できることを示したものである。
【0116】
試料とその調製
以下に列挙する6種類のタンパク質のトリプシン消化物を、本実施例におけるペプチド試料として調製した。(1)ウシカタラーゼ、(2) ウシベータラクトグロブリン、(3) ウシラクトペルオキシダーゼ、(4) ウマグルタチオンS−トランスフェラーゼ、(5) セイヨウワサビペルオキシダーゼ、(6) ウシ血清アルブミン。これらのタンパク質はシグマ社より購入した。
これらのタンパク質は水溶液中でブタトリプシン(プロメガ社より購入)と反応させることにより、トリプシン消化物とした。
【0117】
これら6種類の各タンパク質のトリプシン消化物を以下に示すように混合し、合計7種類のペプチド試料を用意した。
(1):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は0フェムトモル。
(2):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は10フェムトモル。
(3):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は50フェムトモル。
(4):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は100フェムトモル。
(5):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は500フェムトモル。
(6):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は1 ピコモル。
(7):(1) 〜(5) はそれぞれが一測定当り500フェムトモル、(6)は5 ピコモル。
各群のサンプルは5測定分ずつ調整した。上記各試料に対し、さらにペプチドTおよびβカソモルフィン7をそれぞれ10ピコモルおよび1ピコモルずつ標準物質として混入した。
【0118】
LC-MS解析
各ペプチド試料の三次元データを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al, Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリル及び水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
【0119】
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。また、質量分析計でのスキャン回数を細かく取るためにTurbo Scan方法を適用した。この測定を各群5回ずつ行うことによって、各試料に対応する三次元パラメータの集合体、7群計35通りを得た。図1に得られたプロファイルの例を示す。
【0120】
データ処理
三次元データを含むファイルはXcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した。C、C++言語及びPerl言語で作成されたプログラムにより、以下の(1)〜(7)のデータ処理を実行した。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)標準物質であるペプチドTおよびβカソモルフィン7由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、その周囲で規定の範囲内にあるシグナルについて、さらにその点を頂点とするガウス分布で近似される強度範囲内に収まるシグナルを拾い上げ、それらを全て標準物質由来シグナルとみなした。より具体的には、m/zは858.9 および791.0 を中心に前後±2を限度とし、また保持時間は9および25分を中心に前後±6を限度として、上述の手順により探索した。従って使用した標準物質由来シグナルはm/z 858.9, 保持時間9分、およびm/z 791.0, 保持時間25分近傍に存在する2つである。なお、イオン強度補正を行う際には標準物質由来シグナル強度を全て足し合わせ、その値が109となるように規格化した。また時間軸補正の際の動的計画法探索空間上の拘束点としては、上述2つの標準物質シグナルのうち、それぞれ強度ピークを与える1点ずつの計2点を選定した。
(3)BSAの濃度を変えた7種類の試料それぞれにつき、5回の測定結果得られたプロファイルを平均化するため、本発明によるプロファイル重ね合わせプログラムを用い、7群の試料それぞれについての代表プロファイルを求めた。重ね合わせの際の評価関数のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=0.1(但しシグナル強度さは常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=0、不一致点に対するペナルティπ=100、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、一致点のみとした。
【0121】
図8に、BSA濃度が500フェムトモルの試料(5)の5回の測定結果から得られたプロファイルのうち、m/z 620.0 近傍、残留時間15から19分の間のクロマトグラムを示す。時間軸上少しずつずれた灰色の5本の波形が、重ね合わせ処理前の5回の測定結果であり、実線がこれら5つに重ね合わせ処理を施してすべてのシグナルを足し合わせた結果である。図に示したように時間軸上の揺らぎが補正されて1つの大きなピークとして扱えるようになった。
(4)次に、群間で有意に変動しているシグナルを拾い出すため、前述のプロセスで得た7群の代表プロファイルを、さらに重ね合わせ処理によってアライメントをとり、最終的に1つの集約プロファイルを得た。このときの評価関数のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=0.1(但しシグナル強度を常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=0、不一致点に対するペナルティπ=100、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、不一致を含む全ての点とした。
(5)試料間の相互比較に必要十分な分解能を保ちつつデータ点を集約するため、m/z範囲±0.75(絶対範囲±2)、保持時間範囲±1.25(絶対範囲±4)の範囲内にある点について、次の条件を満たしている点については1つに集約した。即ち、シグナル強度の高い順に前述範囲内の全てのデータ点をチェックし、これらがピークシグナルを頂点とするガウス分布で近似される範囲内に入ると判定されたものについてはそれらを集約した。
【0122】
図9に、BSA濃度が異なる7種類の試料ごとの、時間軸補正および集約後のプロファイルの例を示す。図では特定のm/z値(この例では752)で切った断面を、時間軸に沿って強度をプロットしたクロマトグラムとして示している。17分および19分近傍の集約シグナルは、BSA濃度が最も高い06の試料(図中、“DS:Spl 06-Ave”として示す。BSA 5ピコモル)が最も高いピークを示し、順次Spl 05、Spl 04,…と続いているため、これらはBSA由来のシグナルと判定できる。一方、25分近傍の緩やかなピークはどの試料でも同様に出ているため、BSA以外の共通物質あるいはバックグラウンド由来と判定できる。
【0123】
(6)前述の集約プロファイルを出発点として、順次(5),(4),(3)のプロセスを逆にたどることにより、最終的に得られた集約プロファイル上の点が、測定結果の7群×5測定のいずれに由来するかをたどることができる。集約プロファイル上の各点について、測定結果データに戻ることで、群ごとのプロファイルや群間の差プロファイルを得ることができる。図10にBSA濃度が500フェムトモルの試料(5)と、0フェムトモルの試料(1)との差プロファイルを示す。m/z−保持時間平面より上に伸びている線は試料(5)で、逆に下に伸びている線は試料(1)で強く観測されたシグナルである。
【0124】
(7)BSAを入れていない試料(1)から得られた集約プロファイルを基準とし(基準プロファイル)、残りの(2)から(7)までのBSA濃度が異なる6種類の試料から得られた集約プロファイルそれぞれに関して(対象プロファイル)、次のような条件を満たすデータ点を求めた。即ち、(1)対象プロファイル上の点は、5回の測定すべてよりデータが得られていること。(2)集約した対象プロファイル上の点のシグナル強度は106以上であること。(3)対象プロファイル上の各点について、基準プロファイル上の対応する点の強度を引いた差が0以上であること。(4)対象プロファイルの各点について、基準プロファイル上の対応する点とのシグナル強度の差が10000未満であるという帰無仮説が、片側t検定結果によって有意水準0.5%で棄却されること。
【0125】
上記の条件で選別されたシグナルを、試料(5)の場合について図11に示す。ここでは127個のシグナルが前述条件に合致したものとして残った。プロットマークの大きさは、試料(5)のプロファイルにおけるシグナル強度を表す。このうち、プロットマーク○のものは、後述するプロセスでBSAシグナルと対応付けられたもの、×のものはそうでなかったものを示している。
【0126】
これら127個のシグナルについて、別途行ったBSA消化物のMS/MS実験とつき合わせたところ、このうち103個がBSA由来シグナルと一致した。即ち、検出された127個のシグナルのうちの少なくとも81%が真に求めたいものであったといえる。
【0127】
同様にして、他のBSA濃度の試料から検出されたシグナルについて、前述の同じ条件で選別されたシグナルをBSA由来シグナルと付き合わせたところ、対象プロファイル(2)から(7)について、それぞれ65、64、75、81(既述)、76、48%のシグナルがBSA由来シグナルと判定された。なお最後の(7)試料(BSA濃度5ピコモル)の正答率が落ちているが、これは高濃度BSAに由来する強度の強いシグナルが多数存在することによるプロファイルの閾値が変わったことで、偽陽性シグナルが増大したことに起因すると考えられる。実際、他の濃度の試料とほぼ同程度の数のシグナルが選ばれるよう、選定条件の(2)を3×106に調整すると、正答率は75%になった。
【0128】
なお、本発明に係る工夫の1つに、動的計画法の探索空間節減があるが、その効果をCPU時間の実測によって評価したところ、試料(5)の5つの測定結果を重ね合わせる場合を例とすると、CPU時間の比にして43〜45%の削減効果が得られた。
【0129】
ここでは2種類の標準物質由来シグナルを用いているので、もし完全に均等にシグナルが分布していれば1/3の時間短縮が期待できるが、実際多くのシグナルは2つの標準物質シグナルの間に存在するので、探索空間の分割は不均等である。このことを勘案すると、45%程度の削減はおよそ期待通りであり、実用上も十分効果があると考えられる。
【0130】
以上により、本発明に係る試料解析方法が、試料中で量的に変化するシグナルを実用上有用な確度で検出できることを示し、また本発明における計算手法の工夫が有用であることを示した。
【実施例3】
【0131】
実施例3では、実際の患者由来組織サンプルを用いて、いくつかの病態群間で有意に変動している蛋白質由来シグナルを求め、それを元にさらにMS/MS解析を行ってそのうちのいくつかの蛋白質を同定することにより、本方法の有効性、特にバイオマーカーの探索等にも有効であることを示したものである。
【0132】
具体的には、肺の腺癌を対象として、外科的に切除された組織を用い、組織より後述する手法で蛋白質を抽出し、測定した。得られたプロファイルを、後日病理診断によってリンパ節転移ありと判定された群となしと判定された群にわけ、両群間で有意に変動しているシグナルを拾い出し、それらについてMS/MS解析を行って蛋白質を同定した。
【0133】
試料
試料は、36名の異なった肺癌患者より、外科的に切除された肺の組織片を用いた。病理診断により、これらの患者は腫瘍径の大きな群と小さな群、および所属リンパ節への転移が認められる群と認められない群の合計4群に分けられる。
腫瘍径小でリンパ節転移なしの群には10例、腫瘍径大でリンパ節転移なしの群には11例、腫瘍径小でリンパ節転移ありの群には12例、腫瘍径大でリンパ節転移ありの群には3例にぞれぞれ分類された。
【0134】
試料調製および蛋白質分画
ドデシル硫酸ナトリウム(SDS)-ポリアクリルアミドゲル電気泳動(PAGE)用の試料緩衝液中にて各組織片を破砕した。試料緩衝液の組成は以下のとおりである。62.5mMトリス-塩酸(pH6.8)、2%w/v SDS、5%v/v 2-メルカプトエタノール、10%v/v グリセリン、0.0025%w/vブロモフェノールブルー。この懸濁液を室温にて30分間振とうしたのち、上清と沈殿に遠心分離した。上清の蛋白質の濃度をLowry法の変法によって測定した。蛋白質100μg分の試料上清に対して同じ組成の試料緩衝液を加え、総液量を50μLにした。この溶液に対して1Mトリス水溶液を加え、pHを8.8にした。システイン残基の還元アルキル化のため、2μLの400mMジチオトレイトールを加え、60℃にて30分間保温した。つぎに10μLの400mMヨードアセトアミド溶液を加え、室温・暗条件下で60分間放置した。約5μLの1.0N塩酸を加え、pHを6.8に戻した。この溶液をLaemmliのSDS-PAGEにかけた。このときに用いたポリアクリルアミドゲルは不連続の緩衝液系、すなわち上部の濃縮ゲル(pH6.8)と下部の分離ゲル(pH8.8)から成った。ポリアクリルアミドゲルの濃度は各々4%と12.5%であり、全体の大きさは幅14cm、高さ14cm、および厚さ1mmであった。電気泳動時の電流は一定の10mAであった。色素ブロモフェノールの泳動フロントが濃縮ゲルと分離ゲルの界面から分離ゲルの48mmまで達したところで泳動を停止した。ポリアクリルアミドゲルを40%メタノール、10%酢酸水溶液中で振とうし、ポリアクリルアミドゲル内で分離されている蛋白質を固定した。このあと、ポリアクリルアミドゲルを水で2回洗浄した。洗浄したポリアクリルミドゲルを試料当たり24個のゲル片に切り出し分画した。すなわち、泳動方向に対して垂直の向きに2mmの等幅で梯子状に切り出し、各切片をさらに一辺約1mmのサイコロ状に分割した。
【0135】
標準蛋白質の調製
内部標準蛋白質はゲル中に固定された状態で各試料ゲル分画に加えた。最初に12.5%のアクリルアミド、0.1%SDS、375mMトリス塩酸(pH8.8)よりなる水溶液に対して卵白リゾチーム水溶液を混合し、さらにN,N,N’,N’-テトラメチルエチレンジアミンと過硫酸アンモニウムを加えてガラス板にはさまれた1mm幅の間でアクリルアミドを重合した。この内部標準蛋白質を含むゲルを直径1.5mmの円形に繰り抜いた。ゲル切片当たり2.5pmolの卵白リゾチームが含まれるように、重合前の水溶液の蛋白質濃度をあらかじめ計算しておいた。
【0136】
プロテアーゼ消化
上に示した一定量の標準蛋白質を含むゲルの繰り抜き切片を各試料ゲル分画に1個ずつ加えた。つぎに各分画ごとにゲル片を十分量の水で洗浄してからアセトニトリルで脱水した。ゲル片に残った水とアセトニトリルを減圧留去してからトリプシン水溶液をゲル片全部が浸かる程度に加え氷中で45分間放置した。ゲル中に染み込まなかった水溶液を除き、50mM重炭酸アンモニウム水溶液をゲル片全部が浸かる程度に加え、37℃にて16時間保温し消化反応を行った。ゲル片に含まれるペプチド断片の抽出は25mM重炭酸アンモニウム/50%アセトニトリル水溶液で1回、続いて5%蟻酸/50%アセトニトリル水溶液で2回行い、抽出溶液は1個の容器にまとめて減圧濃縮した。
【0137】
LC-MS解析
各ペプチド試料の三次元プロファイルを得るために、以下に示す装置と操作によってペプチド試料を分析した(Kawakami,T.et al, Jpn.J.Electrophoresis 44: 185-190 (2000))。まず、減圧濃縮したペプチド試料を、トリフルオロ酢酸、アセトニトリル及び水の混合比が0.1:2:98の溶媒45μlに溶解した。これを溶解液とする。
次に、CTC Analytics社製のオートサンプラーPAL LC-1TMを用い、Michrom BioResources社製のMAGIC MSTM C18キャピラリーカラム(内径0.2mm、長さ50mm、粒径5μm、孔径200オングストローム)に溶解液20μlを導入した。ペプチドの溶出はMAGIC 2002TM HPLCシステム(Michrom BioResources社)を用いて行った。このときのHPLC移動相Aは蟻酸、アセトニトリル及び水を0.1:2:98の容積比で混合した溶媒であり、対して移動相Bの混合比は0.1:90:10であった。そして移動相Bの濃度を5%から85%まで直線勾配で上げ、ペプチド断片を連続的に溶出した。このときの流速は約1μl/minとした。LCの溶出液は、New Objective社製のPicoChipTMニードル(内径20μm)を介し、LCQTMイオントラップ型質量分析計(ThermoQuest社)のイオン源に直接導入した。NanoESIニードルの位置は加熱キャピラリーとの距離を微調整できるようになっている。スプレー電圧はニードルではなく、溶離液に直接荷電するようにした。噴霧のためにガスは使用せず、スプレー電流は3.0mAとした。
【0138】
データ処理
得られたLC-MSプロファイルデータは、36サンプル×24バンド分の、合計864個である。これらのプロファイルデータについて、XcaliburTMのユーティリティソフトウェアを用いてテキストファイルに変換した後、C、C++言語及びPerl言語で作成されたプログラムにより、以下のような手順で解析を行った。
(1)ノイズレベルのデータを除去するため、イオン強度が102以下のシグナルを除去した。
(2)処理時間の節約のために、m/zと保持時間を量子化することでデータ点を集約した。具体的には、保持時間が約1刻みになるよう、シグナル強度が大きなものから順次、時間差1を最大限度に近傍を探し、単調減少する範囲までを1つのシグナルとしてまとめられる範囲とした。またm/zは1刻みになるよう、元データのm/z値の値を丸め、前述の時間範囲内でm/zが同じ値を持つデータ点は加算集計した。
(3)標準物質であるニワトリ卵白リゾチーム由来のシグナルを同定した。すなわち、予備実験で実測した標準物質のm/z値および保持時間の値の前後ある範囲内で、最も高いイオン強度を与えるデータ点を探し、次いでそのデータ点を中心に、イオン強度値が単調減少しかつ0より大きな範囲にあるデータ点を拾い、これらを標準物質由来のシグナルによるデータ点であると見なした。標準物質由来のシグナルの総イオン強度値としては、標準物質由来シグナルと見なされたデータ点のイオン強度の総和をもって当てた。具体的には、ニワトリ卵白リゾチーム由来のm/z値が715近傍及び877近傍のシグナルを標準シグナルとし、サンプルの測定データからこれらの標準物質由来シグナルを探索する際には、m/zについては前後±1の範囲で、保持時間に関してはm/z 715のシグナルについては10分±5分の範囲で、m/z 877のシグナルについては18分±5分の範囲で探索した。但し、得られた標準物質由来シグナルの絶対強度、全シグナル中での相対強度、および2種類の標準物質由来シグナルの強度比のいずれかが他のものに比べてかけ離れているものについては、個別にプロファイルのプロットを確認のうえ、標準物質由来シグナルと考えられるシグナル群のピークが探索時のパラメータの中心点に来るように調整した上で取り直した。得られた標準物質由来シグナルの総イオン強度値をもって、各シグナルのイオン強度を除算し、得られた値に107を乗じることで、標準物質由来のシグナル強度を107に補正した。さらに、便宜上、m/z 715のシグナルとm/z 877のシグナルのピーク位置がそれぞれ保持時間に関して10分、20分となるよう、保持時間軸を線型変換した。
【0139】
(4)SDS-PAGEによって分画した24バンド分のプロファイルについて、バンド間にまたがって存在する蛋白質の定量性を保証するため、全バンドについて重ね合わせたプロファイルをもって、各試料のプロファイルとして扱った。具体的には、本発明に係る試料解析プログラムの、プロファイル重ね合わせ機能を用い、隣接バンド間のプロファイルを逐次重ね合わせて加算集計した。即ち、まずはバンド1+2, 2+3, 3+4, … , 23+24 のように、共通バンドを有する形で隣接バンドを重ねあわせ、次段階では1+2と2+3から1~3の重ね合わせ得るという形で、常に最低1バンドは重なるようにして重ねてゆくと、6段階の重ね合わせ操作により、最終的には1~17と9~24の重ね合わせによって、全バンドの重ね合わせを得た。なお、重複して重ね合わせたバンドに関しては、最終段階で重複回数を除算することで定量性を保つよう調整した。
【0140】
重ね合わせの際のパラメータは以下のとおり:
上記式(I)において、時間軸上の差(絶対値)のペナルティα=1.0、シグナル強度の差のペナルティβ=1.0(但しシグナル強度を常用対数に変換後の差の絶対値とした)、点の一致に対するボーナス点σ=100、不一致点に対するペナルティπ=10、標準物質由来シグナルに対する一致のボーナス点θ( i, j ) = Sm = 1000。また重ね合わせプロファイルの出力オプションは、不一致を含む全ての点とした。さらに重ね合わせの処理が終わるごとに、保持時間とm/zがそれぞれ分解能1.0および1.0となるようデータ点の集約操作を行った。
【0141】
(5)リンパ節転移のある群とない群を特徴付ける蛋白質を探すため、前述の試料の4種類の分類に従って、まずは群内でプロファイル同士を重ね合わせて集約プロファイルを得、次いで群間について同様にプロファイル重ね合わせ操作を行った。このときの重ね合わせ処理の際のパラメータは、前述のバンド間重ね合わせ処理と同じとした。また重ね合わせ順序については、群内については事前に総当りで行っておいた同一パラメータの下での重ね合わせ処理の評価関数スコアをもとに、近いものから順次重ね合わせた。群間については、まずリンパ節転移のある群内の腫瘍径の違いの2群と、リンパ節転移のない群内での腫瘍径の違いの2群を重ねた後、最終的にリンパ節転移のある群とない群を重ねた。
図12に最終的な重ね合わせプロファイルについて、リンパ節転移陽性群に表れたシグナルを上方向に、陰性群に存在したシグナルを下方向にプロットしたものを示す。
【0142】
(6)前述の重ね合わせた順序を逆にたどることで、重ね合わせの出発点となっている36検体×24バンドの元データにたどれるようにし、最終的な全てを重ね合わせたプロファイル上の各点について、元データとの対応を取れるようにした。
(7)最終的な集約プロファイル上の各点について、リンパ節転移ありの検体由来のデータと、なしの検体由来のデータをそれぞれ集計し、これら両群の平均値の差について両側t検定を行い、両群間の平均値の差と検定によるp値を得た。
図13に、上記検定でp値が0.005未満となった点を、図12と同様のプロットで示す。この段階で5,889個のシグナルが得られた。
【0143】
(8)上述のようにして選別されたシグナルについて、このシグナルの情報をもとにターゲットMS/MSを行い、あるいは別途行ったMS/MS解析から、蛋白質同定ソフトMASCOTTMを用いて、シグナルが由来するところの蛋白質同定を行った。図14に、この同定によって蛋白質情報と関連付けることができたシグナルを示す。約半数の2,753個のシグナルについて、何らかの既知蛋白と関連付けることができた。
【0144】
最後に、上述の既知蛋白質と関連付けられたものの中から、癌の転移に何らかのかかわりを持つといわれている蛋白質の一部をリストにして示す(図15)。このように、癌転移に関わると考えられる蛋白質が有効に発見しうることを示せた。
【0145】
以上より、本発明に係る試料解析システムおよびプログラムが、実際の臨床検体を用いた解析に有効であり、殊に病理的・臨床的な差異を蛋白質の量的な差異に結び付けて検出することができ、またその結果を利用して有効に蛋白質同定に繋げられることから、バイオマーカーの探索や新規診断方法の開発にも有用であることを示したものである。
【産業上の利用可能性】
【0146】
以上、詳細に説明したように、本発明に係る試料解析方法及び試料解析プログラムによれば、試料に含まれる成分を分析するに際して、優れた分析能を達成することができる。したがって、本発明によれば、分析対象の試料中に含まれる多数の成分を網羅的に解析する場合に非常に有効且つ有益な試料分析方法及び試料分析プログラムを提供することができる。
【0147】
特に本発明に係る試料解析方法及び試料解析プログラムによれば、実際の臨床検体を用いて、何らかの疾患の病態の違いに関係する物質を探索する目的にたいへん有効であり、これによりバイオマーカーの探索や診断法の開発ができるという点でも有用性はきわめて大である。
【図面の簡単な説明】
【0148】
【図1】本発明に係る試料解析方法及び試料解析プログラムにより取得された3次元のスペクトルデータの一例を示す図である。
【図2】3次元データの一例を示す図である。
【図3】図2に示した3次元データとの対応関係を検索するために設定したもう一つの3次元データの一例を示す図である。
【図4】図2に示した3次元データと図3に示した3次元データとの最適な対応配置を示した図である。
【図5】図2に示した3次元データと図3に示した3次元データとの最適な対応配置を検索する際の概念を示す図である。
【図6】図5に示した最適配置探索において、標準物質由来の情報を利用して経路を限定すると、探索空間のうちの灰色部分はもはや探索する必要がないことを示した図である。
【図7】標準物質由来の情報をより多く利用して経路の拘束条件を増やすと、探索不要な空間が増えて、探索効率がいっそう向上することを示した図である。
【図8】本発明に係る試料解析プログラムによって、同一種類の5つの測定結果で時間軸上揺らいでいる波形を、1つに重ね合わせてシグナルを足し合わせた結果を示した図である。
【図9】本発明に係る試料解析プログラムによって、異なる7種類の試料測定から得られたプロファイルを時間軸をそろえて重ね合わせることにより、時点ごとに異なった試料間での相互比較が行えるようになったことを示した図である。
【図10】本発明に係る試料解析プログラムによって、異なった2種類の試料間の差スペクトルを計算して示した図である。
【図11】本発明に係る試料解析プログラムによって、試料群間に有意な量的変動があるシグナルとして選別されたものを示した図である。
【図12】本発明に係る試料解析プログラムを、実際の臨床検体を用いたマーカー探索に応用し、異なった病理診断結果によるグルーピングに応じてシグナルを分類できることを示した図である。
【図13】図12に示した結果に対し、さらに統計検定を行うことで、異なった病理診断結果に応じて量的に変化しているシグナルを拾い出したものを示した図である。
【図14】図13に示した結果の各シグナルに対し、さらにMS/MSによる蛋白質同定との関連付けを行った結果を示した図である。
【図15】図14に示したような形で既知の蛋白質と関連付けられたもののうち、特に癌の転移に関連することが知られている蛋白について、本発明に係る試料解析システムで見出された結果の一部を示した表である。
【特許請求の範囲】
【請求項1】
複数の試料についてクロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データにおける、質量/電荷比を示すパラメータ及びイオン強度を示すパラメータに関するプロファイルを参照プロファイルとし、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を用いて、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることで、保持時間を示すパラメータを補正する工程aと、
上記工程aにより得られる補正後の3次元データを、複数の試料について比較する工程bとを含む試料解析方法。
【請求項2】
上記評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることを特徴とする請求項1記載の試料解析方法。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【請求項3】
上記工程aでは、上記補正対象のパラメータについて最適解探索問題として上記評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることを特徴とする請求項1記載の試料解析方法。
【請求項4】
上記動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることを特徴とする請求項3記載の試料解析方法。
【請求項5】
上記動的計画法のアルゴリズムでは、補正対象のパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることを特徴とする請求項3記載の試料解析方法。
【請求項6】
上記試料は、蛋白質群及び/又はペプチド群を含むことを特徴とする請求項1記載の試料解析方法。
【請求項7】
上記標準物質は、ペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)、βカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)及びこれらの構造類似体からなる群から選ばれる少なくとも1種以上のペプチドであることを特徴とする請求項1記載の試料解析方法。
【請求項8】
上記標準物質をゲル内に固定した状態で上記試料に添加することを特徴とする請求項1記載の試料解析方法。
【請求項9】
複数の試料についてクロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを入力する手順aと、
入力された3次元データにおける、質量/電荷比を示すパラメータ及びイオン強度を示すパラメータに関するプロファイルを参照プロファイルとし、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を用いて、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることで、保持時間を示すパラメータについてデータを補正する手順bと、
上記手順bで補正した後の3次元データを、複数の試料について比較する手順cとをコンピュータに実行させる試料解析プログラム。
【請求項10】
上記評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることを特徴とする請求項9項記載の試料解析プログラム。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【請求項11】
上記手順aでは、上記補正対象のパラメータについて最適解探索問題として上記評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることを特徴とする請求項9記載の試料解析プログラム。
【請求項12】
上記動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることを特徴とする請求項11項記載の試料解析プログラム。
【請求項13】
上記動的計画法のアルゴリズムでは、補正対象のパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることを特徴とする請求項11記載の試料解析プログラム。
【請求項14】
上記試料は、蛋白質群及び/又はペプチド群を含み、当該蛋白質群及び/又はペプチド群に由来する3次元データを解析することを特徴とする請求項9記載の試料解析プログラム。
【請求項15】
上記標準物質は、ペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)、βカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)及びこれらの構造類似体からなる群から選ばれる少なくとも1種以上のペプチドであることを特徴とする請求項9記載の試料解析プログラム。
【請求項16】
上記標準物質をゲル内に固定した状態で上記試料に添加することを特徴とする請求項9記載の試料解析プログラム。
【請求項1】
複数の試料についてクロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データにおける、質量/電荷比を示すパラメータ及びイオン強度を示すパラメータに関するプロファイルを参照プロファイルとし、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を用いて、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることで、保持時間を示すパラメータを補正する工程aと、
上記工程aにより得られる補正後の3次元データを、複数の試料について比較する工程bとを含む試料解析方法。
【請求項2】
上記評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることを特徴とする請求項1記載の試料解析方法。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【請求項3】
上記工程aでは、上記補正対象のパラメータについて最適解探索問題として上記評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることを特徴とする請求項1記載の試料解析方法。
【請求項4】
上記動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることを特徴とする請求項3記載の試料解析方法。
【請求項5】
上記動的計画法のアルゴリズムでは、補正対象のパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることを特徴とする請求項3記載の試料解析方法。
【請求項6】
上記試料は、蛋白質群及び/又はペプチド群を含むことを特徴とする請求項1記載の試料解析方法。
【請求項7】
上記標準物質は、ペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)、βカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)及びこれらの構造類似体からなる群から選ばれる少なくとも1種以上のペプチドであることを特徴とする請求項1記載の試料解析方法。
【請求項8】
上記標準物質をゲル内に固定した状態で上記試料に添加することを特徴とする請求項1記載の試料解析方法。
【請求項9】
複数の試料についてクロマトグラフィー質量分析の結果として得られる、質量/電荷比を示すパラメータと、イオン強度を示すパラメータと、保持時間を示すパラメータとからなる3次元データを入力する手順aと、
入力された3次元データにおける、質量/電荷比を示すパラメータ及びイオン強度を示すパラメータに関するプロファイルを参照プロファイルとし、複数の試料間で複数の参照プロファイルに関する配置類似性の尺度となる評価関数を用いて、当該評価関数の値を最適化する最適解探索問題として各プロファイルの配置を求めることで、保持時間を示すパラメータについてデータを補正する手順bと、
上記手順bで補正した後の3次元データを、複数の試料について比較する手順cとをコンピュータに実行させる試料解析プログラム。
【請求項10】
上記評価関数は、以下の(1)〜(6)からなる群から選ばれる1以上の項で定義されることを特徴とする請求項9項記載の試料解析プログラム。
(1)補正対象のパラメータに関するプロファイル間の類似性及び/又は距離に関する項
(2)参照プロファイルに関するプロファイル間の類似性及び/又は距離に関する項
(3)比較対象となるプロファイル間でのデータ点の一致の程度に関する項
(4)比較対象となるプロファイル間でのデータ点の不一致の程度に関する項
(5)比較対象となるプロファイル間での標準物質由来シグナルの一致又は不一致の程度に関する項
(6)比較操作を繰り返す際に、過去の比較における一致の程度に関する項
【請求項11】
上記手順aでは、上記補正対象のパラメータについて最適解探索問題として上記評価関数の値を最適化する際に動的計画法のアルゴリズムを用いることを特徴とする請求項9記載の試料解析プログラム。
【請求項12】
上記動的計画法のアルゴリズムでは、補正の対象となるパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、当該スコアを良くする設定とすることを特徴とする請求項11項記載の試料解析プログラム。
【請求項13】
上記動的計画法のアルゴリズムでは、補正対象のパラメータに含まれるデータ点の最適な対応関係を、スコアを算出して評価する際に、標準物質に由来するデータ点に関する対応関係については、必ず指定された点で対応することを拘束条件とすることを特徴とする請求項11記載の試料解析プログラム。
【請求項14】
上記試料は、蛋白質群及び/又はペプチド群を含み、当該蛋白質群及び/又はペプチド群に由来する3次元データを解析することを特徴とする請求項9記載の試料解析プログラム。
【請求項15】
上記標準物質は、ペプチドT(Ala-Ser-Thr-Thr-Asn-Tyr-Thr)、βカソモルフィン7(Tyr-Pro-Phe-Pro-Gly-Pro-Ile)及びこれらの構造類似体からなる群から選ばれる少なくとも1種以上のペプチドであることを特徴とする請求項9記載の試料解析プログラム。
【請求項16】
上記標準物質をゲル内に固定した状態で上記試料に添加することを特徴とする請求項9記載の試料解析プログラム。
【図1】

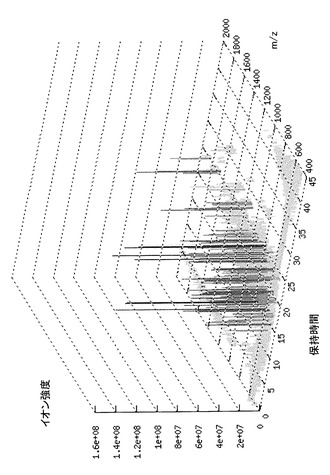
【図2】
【図3】
【図4】
【図5】
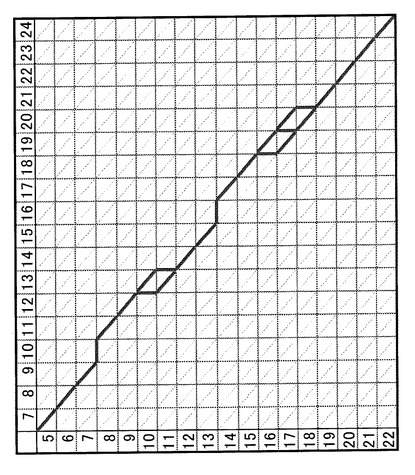
【図6】
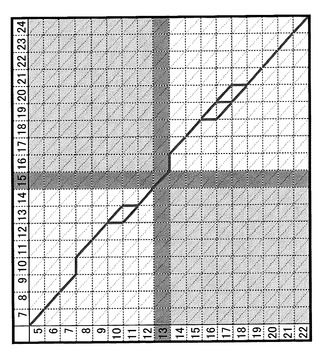
【図7】

【図8】
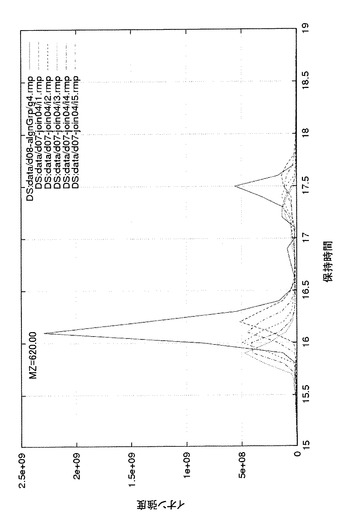
【図9】
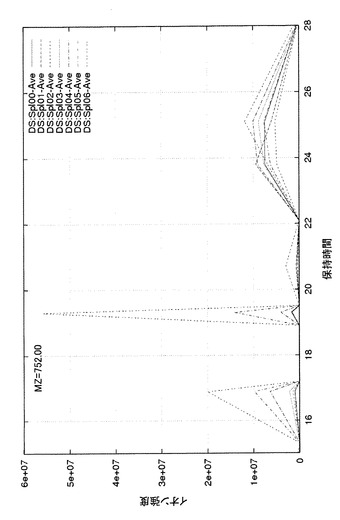
【図10】
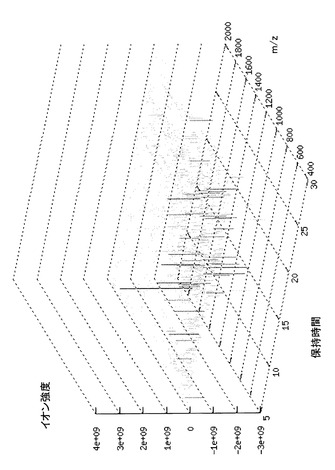
【図11】
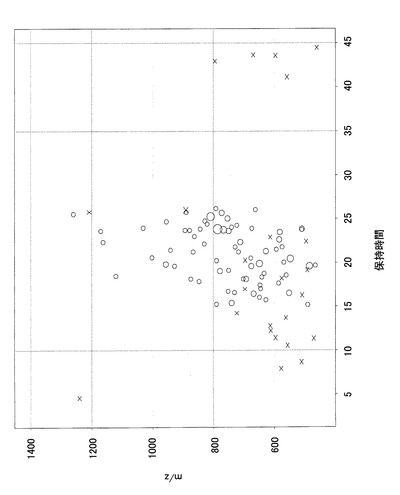
【図12】

【図13】

【図14】
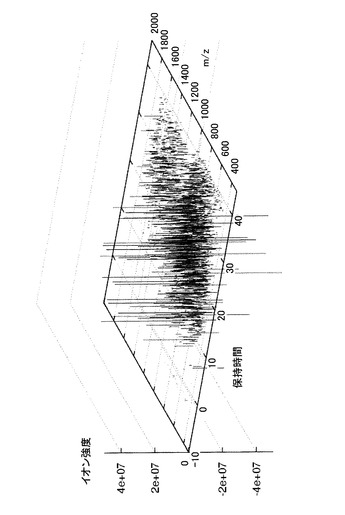
【図15】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2008−241721(P2008−241721A)
【公開日】平成20年10月9日(2008.10.9)
【国際特許分類】
【出願番号】特願2008−136968(P2008−136968)
【出願日】平成20年5月26日(2008.5.26)
【分割の表示】特願2005−505220(P2005−505220)の分割
【原出願日】平成16年3月31日(2004.3.31)
【出願人】(503119030)株式会社メディカル・プロテオスコープ (5)
【Fターム(参考)】
【公開日】平成20年10月9日(2008.10.9)
【国際特許分類】
【出願日】平成20年5月26日(2008.5.26)
【分割の表示】特願2005−505220(P2005−505220)の分割
【原出願日】平成16年3月31日(2004.3.31)
【出願人】(503119030)株式会社メディカル・プロテオスコープ (5)
【Fターム(参考)】
[ Back to top ]