
金属化合物の合成
(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな生成物を残すステップを含むメソポーラスなリチウム遷移金属化合物の合成方法を提供する。前記メソポーラスな生成物は酸化物、リン酸塩、ホウ酸塩またはケイ酸塩であり得、場合によりステップ(i)で追加のリン酸塩、ホウ酸塩またはケイ酸塩試薬を添加してもよい。反応混合物は任意のキレート化剤を含み得、ステップ(i)及び(ii)の反応条件を液晶相の不安定化を防止するようにコントロールすることが好ましい。本発明は、メソポーラスなコバルト酸リチウム及びリン酸鉄リチウムを製造するために特に適している。前記方法は、10m2/gを超える、好ましくは15m2/g以上の比表面積を有するメソポーラスなコバルト酸リチウムを合成するために使用され得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スーパーキャパシタ及び電池を含めたリチウム電池用材料を製造するための改良方法に関する。本発明は特に、リチウム二次電池に適した材料、具体的にはメソポーラスなリチウム遷移金属化合物の製造、より具体的にはメソポーラスなリチウム遷移金属酸化物及びリン酸塩の製造、更により具体的にはメソポーラスなコバルト酸リチウム及びリン酸鉄リチウムの製造に関する。本発明は、この方法により形成される材料にも関する。
【背景技術】
【0002】
リチウム二次電池は、重要な携帯用DC電力源であり、最近燃料電池が開発されたにも関わらず多年にわたりそのようにあり続けているようである。携帯用電子及び電気システムは効果的に作動させるためにハイパワー、コンパクトで永続性な電力源がますます必要とされており、よって改善された携帯用電力源が必要である。既存のシステムのために必要とされる時間の何分の1かで再充電され得、改良された電池サイクル寿命をも有する高エネルギーリチウムイオン電池が特に興味深い。
【0003】
性能を所望するように向上させるための1つの方法は改良された活性正極材料を使用することである。二次リチウム電池用高性能活性正極材料としてリチウム遷移金属酸化物を使用することは公知であり、コバルト酸リチウムは最も一般的に使用されている市販材料の1つである。別の興味ある材料はリン酸鉄リチウムであるが、大量に合成されるリン酸鉄リチウムは高い電気抵抗を有している。このことはハイパワー用途にとっては望ましくなく、導電率を高め、よってレート特性を高めるために電極材料に炭素を配合することが必要である。
【0004】
電池電極は、典型的には活性正極材料、結合剤及び導電性炭素からなるペーストを集電体(例えば、アルミニウムホイル)にコーティングすることにより製作されている。活性材料は必要な電気化学容量を与える電極の主成分である。結合剤は粒子間接続性及び集電体に対する密着性を与え、導電性炭素は電極反応のための電子伝導率のレベルを高める。活性材料粒子のサイズは均一で接着性のコーティングを作成するために、また所望レベルの電極性能のために重要であり、粒子が大きいと電極の厚さが厚くなるだけでなく、不均一なコーティングが生ずることは公知である。電極性能に関して、大きい粒子及び厚いコーティングは、充電/放電中リチウムイオンが電極中に及びそこから拡散し得るレートを限定し、よって電極の電力/レート特性を制限する。
【0005】
リチウムイオン電池の充電/放電中、リチウムイオンは活性正極材料粒子中に及びそこから移動して、前記粒子はプロセス中膨張し、収縮する。連続サイクリングで粒子が繰り返し膨張し、収縮すると、徐々に粒子間の電気接触が失われ、その結果電極の抵抗が増加する。また、抵抗が増加すると、すべての容量が引き出され得る前に時期尚早に充電/放電カットオフ電圧に達するために電極の利用率が低くなる。これは「容量フェード」として公知の現象である。
【0006】
よって、電池が高いレート、長いサイクル寿命及び良好な利用率を発揮するためには、電池電極材料を多孔率の高い小粒子から構成することが望ましい。こうすると、電極の活性表面積が大きくなり、非常に薄い電極コーティングを作成することができる。(比表面積として測定した)表面積が大きいと、速い電極反応も促進され、よってレート性能及び電力特性が高まる。
【0007】
高表面積の電池電極材料を得るための従来方法は、粒子サイズを小さくするためのボールミリング及び多孔質材料を得るための熱分解またはソリッドステートルートを含む。しかしながら、これらの方法は、より小さい粒子が凝集したり、高価であったり、スケールアップに適さないことを含めた多数の欠点を有する傾向にある。電池電極のための既存の製作方法との適合性も問題であり得る。
【0008】
また、リチウム遷移金属酸化物はナノ粒子として直接合成され得る。例えば、リチウム遷移金属酸化物のナノ粒子を製造する1つの公知方法は、単相金属水酸化物または多相混合金属水酸化物を直接溶解沈殿させた後、水酸化物をそれぞれの酸化物または化合物酸化物生成物に変換する焼成ステップによる。リチウム遷移金属酸化物のナノ粒子を製造する別の公知方法は溶解ゾル−ゲル合成により、例えばHahnら(Proc.9th Asia Pacific Physics Conference,Hanoi,Vietnam,October 25−31,2004)はクエン酸反応試薬の存在下で硝酸リチウムを硝酸コバルトと反応させて約100nmの粒度を有するコバルト酸リチウム粉末を製造することを記載している。しかしながら、上記方法で製造したコバルト酸リチウムナノ材料は最高でも約5m2/gの表面積しか達成し得ない傾向にある。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Hahnら(Proc.9th Asia Pacific Physics Conference,Hanoi,Vietnam,October 25−31,2004)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の第1態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな生成物を残すステップを含むメソポーラスなリチウム遷移金属化合物の合成方法が提供される。
【0011】
リチウム塩及び1つ以上の遷移金属塩は、ステップ(ii)で高分子金属酸化物網状構造を形成し得、これによりメソポーラスなリチウム遷移金属酸化物生成物が形成される。
【0012】
或いは、リチウム塩及び1つ以上の遷移金属塩は、ステップ(ii)で高分子金属リン酸素網状構造を形成し得、これによりメソポーラスなリチウム遷移金属リン酸塩生成物が形成される。リン酸塩を主成分とするゾル−ゲルを形成するために追加のリン酸塩試薬、例えばリン酸二水素アンモニウムが必要なことがある。好ましい実施形態では、リチウム塩を鉄塩及びリン酸塩と好ましくは酸性条件下で反応させて、メソポーラスなLiFePO4を製造する。好ましくは、リチウム塩は硝酸リチウムであり、鉄塩はシュウ酸第一鉄二水和物または硝酸鉄(例えば、Fe(NO3)3・9H2O)であり、リン酸塩はリン酸二水素アンモニウムである。或いは、鉄塩(例えば、クエン酸Fe(III))を追加のリン酸塩試薬なしでリン酸リチウム(例えば、LiH2PO4)と反応させて、高分子金属リン酸素網状構造を形成してもよい。場合により、反応混合物は硝酸を含む。有利には、LiFePO4材料にメソ多孔性を付与することにより該材料の導電率が大きく高められ得ることが知見された。
【0013】
本発明は、リチウム遷移金属ホウ酸塩及びケイ酸塩、例えばLiMBO3(ここで、MはFe、CoまたはMnであり得る)及びLi2MSiO4(ここで、MはFeまたはMnであり得る)を合成するためにも使用され得る。この場合、リチウム塩及び1つ以上の遷移金属塩はステップ(ii)で高分子金属ホウ素酸素または高分子金属ケイ素酸素網状構造のいずれかを形成し、これによりメソポーラスなリチウム遷移金属ホウ酸塩またはケイ酸塩生成物が形成される。この場合でも、ゾル−ゲルを形成するために追加のホウ酸塩またはケイ酸塩試薬が必要なことがあり、当業者は適当なゾル−ゲル試薬を認識している。
【0014】
リン酸鉄リチウム(LiFePO4)と同様に、LiMBO3(ここで、MはFe、CoまたはMnであり得る)及びLi2MSiO4(ここで、MはFeまたはMnであり得る)は通常低い導電率を有する潜在的高エネルギー密度正極材料である。材料を本発明の方法に従って合成することにより、その導電率を高め、その材料をリチウム電池中に使用することができる。
【0015】
いずれの場合も、本発明の方法は、ゾル−ゲル合成と液晶テンプレート化を組み合わせることによりメソポーラスなリチウム遷移金属化合物を製造する。結果は、単相のメソポーラスなリチウム遷移金属化合物を製造するために容易に実施される便利な低温方法である。
【0016】
ここで、メソポーラスなコバルト酸リチウムの製造に特に言及して本発明を説明する。しかしながら、教示内容は当業者により一般的には上記したリチウム遷移金属化合物に適用され得る。コバルト酸リチウムは、コバルト酸リチウム(LiCoO2)または1つ以上の追加の遷移金属包含物を含むコバルト酸リチウム(例えば、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2)のいずれかを意味し、以後化合物コバルト酸リチウムと称する。いずれの場合も、1つ以上の遷移金属塩はコバルト塩を含む。LiCoO2を製造するためには、ステップ(i)でリチウム塩をコバルト塩と反応させる。しかしながら、化合物コバルト酸リチウムを製造するためにリチウム塩をコバルト塩及び1つ以上の更なる遷移金属塩と反応させ、1つ以上の更なる塩は好ましくはNi、Mn及びAlからなる群から選択される遷移金属を含む。より好ましくは、試薬をLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される化合物コバルト酸リチウム生成物を形成するように選択する。
【0017】
上に表した化学式は厳密に限定するものと意図されず、非化学量論的バリアントを包含すると理解されたい。
【0018】
メソポーラスなコバルト酸リチウムにより、市販の電池において通常相互に相いれないパラメーターである高いエネルギー及び高い再充電/放電レート特性を有するリチウムイオン電池を開発する可能性がある。メソポーラスなコバルト酸リチウムを主成分とする正極材料を使用する別の効果は、破損の主メカニズム(物理的分解により正極の電子抵抗が高くなる)がメソ多孔性により大きく妨害されるために電池サイクル寿命が向上することである。
【0019】
慣例的に、多孔質材料の孔径範囲は次のように定義される:ミクロポーラス 5nm未満、メソポーラス 5〜50nm、ナノポーラス 50〜100nm、マクロポーラス 100nm以上。しかしながら、範囲及び長距離秩序は非常に僅かに変わり得るのでこれらの定義を厳密に解釈するつもりはない。従って、本発明の方法により製造されるメソポーラスな材料の幾つかの孔は50nmより大きく、幾つかの孔は5nmより小さい。
【0020】
本発明の方法は単相のメソポーラスなリチウム遷移金属酸化物、好ましくはメソポーラスなコバルト酸リチウムを製造し得る。本発明に従って製造されるコバルト酸リチウム粉末をX線回折(XRD)分析、透過型電子顕微鏡(TEM)及びブルナウア−エメット−テラー(BET)表面積分析により特性決定され、メソポーラスであり且つ10m2/gを超える、典型的には15m2/g以上の比表面積を有していることが判明した。1つの実施例では、20m2/gのBET表面積が記録された。
【0021】
液晶テンプレート化は、規則的に離間されているシリンダーの自己組織化アレイからなる秩序化液晶相を形成するために反応混合物中に高濃度の界面活性剤を使用する。シリンダーの間隔、よって最終テンプレート化生成物の孔径は選択した界面活性剤の鎖長によりコントロールされる。本発明の方法にとって好ましい液晶相であるヘキサゴナル相の場合、シリンダーはヘキサゴナルアレイで配置され、界面活性剤の疎水性部分がシリンダーの内部に向かって集中し、親水性、通常水性領域は外部に集中するように構成される。適切にコントロールされると、液晶テンプレート化により高秩序化テンプレート構造が得られ、その周りで材料が合成され得る。典型的には、液晶テンプレート化によりリオトロピック液晶相が使用でき、従って他のよりゆるく秩序化されている界面活性剤を用いる技術(例えば、ミセルテンプレート化)よりもより高い界面活性剤濃度で実施される。
【0022】
ゾル−ゲル方法は、無機及び/または有機金属塩前駆体からリチウム遷移金属酸化物粉末を含めた金属酸化物粉末の合成のために使用され得る低温方法であり、通常水性条件下で行う。ゾル−ゲル方法では、まず前駆体からコロイド状懸濁液(ゾル)を形成した後、ゾルを加水分解及び縮合反応により連続液相中に固相−M−O−M−高分子網状構造(ゲル)を形成することにより金属酸化物網状構造が形成される。その後、ゲルを加熱して酸化物生成物を乾燥し、所要により求められる結晶相を与える。金属塩前駆体の組合せを使用することにより、2、3、4、5またはそれ以上の金属包含物を含む混合金属酸化物ゾルが形成され得る。正極材料用の所望の混合金属酸化物にはLiCoO2、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2が含まれる。
【0023】
ゾル−ゲル方法は他の金属化合物、例えばリン酸塩、ホウ酸塩及びケイ酸塩を合成するためにも使用され得る。この場合、高分子網状構造は金属リン酸素網状構造、金属ホウ素酸素網状構造、金属ケイ素酸素網状構造等である。
【0024】
非テンプレート化ゾル−ゲル合成から製造される金属酸化物粉末は通常約5m2/g未満、より典型的には約1m2/g未満の比表面積を有する個々に分散されている及び/または凝集されているサブミクロン及び/またはナノサイズ粒子からなる。これは、ゾル−ゲル合成によると小さい粒度を有するが限定された多孔性を有する酸化物生成物が製造されるからである。
【0025】
本発明の方法では、反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させる。反応混合物を加熱し、場合によりエージングしてゾル−ゲルを形成し、これにより金属酸化物の合成の場合には液晶相の親水性ドメイン中に高分子リチウム−遷移金属酸化物網状構造が形成される。言い換えれば、反応混合物の親水性領域、典型的には液晶相中のロッドまたはシリンダーの外の領域がゾル−ゲル方法によりリチウム遷移金属酸化物(−Li−O−M−)網状構造の合成を助けるために使用される。こうして、無機高分子金属酸化物網状構造が好ましくはヘキサゴナルアレイで配置されている界面活性剤ロッドの周りに形成し、これにより液晶テンプレートが金属酸化物構造に導入される。次いで、界面活性剤を除去すると、メソポーラスな酸化物生成物が形成される。
【0026】
好ましくは、1つ以上の遷移金属塩には、液晶相がそれぞれ高分子−Li−O−Co−網状構造または−Li−O−Co−O−M−網状構造の形成を助けるLiCoO2、すなわち化合物コバルト酸リチウムを製造するためのコバルト塩が含まれる。
【0027】
界面活性剤は、ゾル−ゲルから適当な方法、例えば溶媒抽出または熱分解により除去され得る。界面活性剤を除去すると、単相の三次元的に結合しているメソポーラス構造を有する酸化物生成物が残る。
【0028】
合成された生成物、好ましくは合成された酸化物生成物は少なくとも部分的に非晶質形態をとるようである。非晶質材料は通常電池材料として使用するためには望ましくなく、従って材料を所望の結晶相に変換するために生成物を焼成することが好ましい。界面活性剤除去及び焼成段階を組み合わせること、例えば界面活性剤を焼成に適した温度で熱分解することにより除去することが好都合である。通常、生成物を適切な結晶相に変換するために約400℃以上の温度が使用され得るが、焼成温度が高くなるとメソポーラスな生成物中の孔が崩壊する可能性が高くなることを本発明者らは知見した。こうなると、達成可能な表面積の拡大が対応して低下する。従って、(孔の一体性を維持するために)できるだけ低いが、正しい結晶性を得るために十分に高い焼成温度を使用することが望ましい。電池グレードLiCoO2を液晶テンプレート化合成するための好ましい焼成温度は400〜700℃、より好ましくは400〜600℃の範囲である。リン酸鉄リチウムを合成するためには、焼成温度は好ましくは450〜700℃の範囲、より好ましくは450〜650℃の範囲である。
【0029】
材料を液晶テンプレート化方法によりうまく合成するためには、通常反応混合物中に所望の液晶相を形成した後この相を合成反応の実質的すべての間維持することが重要である。反応条件を例えば望ましくない副生成物を除去したり及び/またはステップ(i)及び(ii)で界面活性剤濃度を所望範囲に維持することにより注意深くコントロールしないと、液晶相は不安定になることがあり得る。液晶相が不安定化すると、メソポーラスな生成物を形成するのに適していない望ましくないミセル相、すなわちよりゆるく秩序化されている構造が形成される恐れがある。むしろ、生成物は単に全くまたは殆ど相互接続性を持たず、比表面積が小さいゆるく秩序化されているナノ粒子からなる傾向にある。
【0030】
従って、本発明の方法では、ステップ(i)及び(ii)で液晶相の不安定化を防止するように反応条件をコントロールすることが望ましく、理想的にはステップ(i)及び(ii)のすべてまたは実質的にすべてを通して相不安定化を防止するように反応条件をコントロールする。安定な液晶相、好ましくは安定なヘキサゴナル相が維持されるように反応混合物中の界面活性剤の濃度をコントロールすることが特に重要であり得る。
【0031】
安定な液晶相、特に好ましいヘキサゴナル液晶相を製造するための典型的な界面活性剤濃度は40〜60wt%であり、上記にてらしてステップ(i)及び(ii)のすべてまたは実質的にすべてを通して界面活性剤の濃度を上記範囲に維持することが好ましい。より好ましくは、界面活性剤濃度は45〜55wt%の範囲であり、更により好ましくは界面活性剤濃度は約50wt%である。この場合でも、ステップ(i)及び(ii)のすべてまたは実質的にすべてを通して、すなわちゾル−ゲル無機高分子網状構造の形成中、界面活性剤濃度を上記範囲に維持することが好ましい。
【0032】
本発明の方法の作用効果は、反応混合物に試薬を更に添加することを伴わないことである。換言すれば、金属塩、界面活性剤及び任意の試薬(例えば、上記したリン酸塩、ホウ酸塩またはケイ酸塩及び/または下記するキレート化剤)を組み合わせて反応混合物、好ましくは水性反応混合物を形成したら、合成中更なる試薬を添加する必要がなく、その結果界面活性剤濃度はその後希釈されない。こうして、単純で簡単な合成方法が提供されるだけでなく、界面活性剤濃度を変化させる恐れがある主メカニズムがステップ(i)及び(ii)中の水分損失であることを意味する。従って、これらのステップ中の水分損失を最小限とすることにより反応混合物中の界面活性剤濃度(よって、液晶相挙動)を簡単且つ効果的にコントロールすることができる。
【0033】
水分損失を最小限とする1つの好ましい方法は、液晶相が完全に乾燥しないように、よって相不安定化を受けないように反応温度をコントロールすることである。従来の金属酸化物のゾル−ゲル合成は60〜80℃の範囲の温度で進行し得るが、50〜60℃の範囲のより低い温度が通常本発明の方法のために適している。より好ましくは、反応混合物を55〜60℃の範囲の温度に加熱するが、この温度はコバルト酸リチウムの場合の70〜80℃という通常の非テンプレート化反応温度よりも低い。本発明者らは、55〜60℃の範囲の反応温度がゾル−ゲル合成の加水分解及び縮合反応の促進と反応混合物からの水分損失の低減の間の適切なバランスを与えることを知見した。低い反応温度を補うために、本発明のテンプレート化ゾル−ゲル合成を従来の対応する非テンプレート化合成よりも長く進行させることが好ましい。有利には、この任意のエージングプロセスにより、その後の加工ステップ(特に、任意の焼成ステップ)で崩壊しにくいより良好で、より相互接続され、高密度化されている高分子網状構造が作成され得る。
【0034】
反応温度をコントロールする代わりにまたはそれに加えて、ステップ(i)及び(ii)で生ずる水蒸気の逃散をコントロールすることにより水分損失を低減させることができる。従って、水分損失の程度をコントロールするように、より好ましくは水分損失を実質的に防止するようにテンプレート化ゾル−ゲル反応を場合により密封されたまたは部分的に密封された容器または反応容器中で実施する。
【0035】
本発明のために適したテンプレート化剤として作用させるために、ロッドまたはシリンダーからなる液晶相を形成し得る、より好ましくはH1ヘキサゴナル相を形成し得る界面活性剤が通常必要である。多くの界面活性剤が当業者に公知であり、市販業者から容易に入手し得、反応混合物において所望の液晶相挙動を与える界面活性剤が本発明の方法で使用され得る。イオン性または非イオン性界面活性剤が適当であり得る。適しているイオン性界面活性剤には臭化セチルジメチルエチルアンモニウムまたは臭化セチルトリメチルアンモニウム(CTAB)が含まれ、適している非イオン性界面活性剤にはエチレンオキシド及びプロピレンオキシドを主成分とするブロックコポリマー(一般的にPluronic(登録商標)界面活性剤として公知)、ポリオキシエチレンセチルエーテルまたはオクタエチレンエーテル(OEE)が含まれる。しかしながら、化学的に不活性であり、ゾル−ゲル反応に関与しない界面活性剤を選択することが好ましい。この点で、リチオ化界面活性剤(例えば、ラウリルエーテルリチウム酢酸塩)が反応混合物中の液晶相に対する望ましくない不安定化作用のために本発明の方法で使用するのに適さないことを知見した。
【0036】
好ましくは、界面活性剤はテンプレート化ゾル−ゲル合成の温度範囲で必要な液晶相を形成し得なければならない。秩序化テンプレートにおいて約5〜10nm、好ましくは7〜8nmの反復距離を与え得る鎖長(または、鎖長範囲)を有する界面活性剤を選択することも有利である。
【0037】
好ましくは、ゾル−ゲル反応を水性条件下で実施し、理想的には液晶相を不安定化し得る溶媒を排除した条件下で実施する。通常、有機溶媒(例えば、エタノール及びメタノール)は液晶相を壊し、これによりテンプレート構造を完全または部分的に崩壊し、所望のメソ多孔性を有する生成物を得ることができないので、これらの溶媒の使用を避けることが望ましい。
【0038】
場合により、試薬の混合を高め、反応する前駆体の表面積を最大限とし、(望ましくない多相生成物の形成を導く)金属酸化物の時期尚早の沈殿を防止するために反応混合物はキレート化剤を含む。例えば硝酸リチウムを硝酸コバルトと反応させてLi/O/Coを主成分とするゾルを製造する場合、通常反応混合物中に追加のキレート化剤が必要である。
【0039】
しかしながら、別のキレート化剤は必ずしも必要でない。例えば酢酸リチウムを酢酸コバルトと反応させてLi/O/Coを主成分とするゾル−ゲルを製造する場合、酢酸対イオンそれ自体がキレート化剤として作用し得、これにより別の試薬が必要でなくなる(換言すると、酢酸塩自己キレート)。
【0040】
一般的に、ゾル−ゲル試薬それ自体がキレート化特性を有していない場合にはキレート化剤が望ましい傾向にある。材料を本発明のテンプレート化ゾル−ゲル合成においてキレート化剤として作用させるためには、材料はテンプレート内の疎水性ドメインよりもむしろテンプレートのロッドまたはシリンダーの外の親水性ドメインでキレート化し得なければならない。本発明者らは予期せぬことに、クエン酸及びプロピオン酸が本発明の方法においてキレート化剤として作用し得、コストが安く、容易に入手し得るためにクエン酸が好ましいことを知見した。
【0041】
1つ以上の遷移金属塩は有機アニオン(例えば、酢酸塩、カルボン酸塩、クエン酸塩またはシュウ酸塩)または無機アニオン(例えば、硝酸塩、リン酸二水素塩またはリン酸塩)を含み得る。幾つかの場合には、1つ以上の更なる遷移金属塩は異なるアニオンを含み得るが、多くの場合各遷移金属試薬は同一のアニオンを含む。
【0042】
また、リチウム塩は有機アニオン(例えば、酢酸塩、カルボン酸塩、クエン酸塩またはシュウ酸塩)または無機アニオン(例えば、硝酸塩、リン酸二水素塩またはリン酸塩)を含み得る。通常(必須ではないが)、リチウム塩は1つ以上の遷移金属塩の少なくとも1つと同一のアニオンを含み、通常リチウム塩及び1つ以上の遷移金属塩はすべて同一のアニオンを含む。好ましくは、リチウム塩及び1つ以上の遷移金属塩のすべてが同一のアニオンを含み、アニオンは酢酸塩及び硝酸塩、より好ましくは硝酸塩から選択される。よって、LiCoO2を酢酸リチウム及び硝酸コバルト、または硝酸リチウム及び硝酸コバルトのいずれかを含む反応混合物から合成することが好ましく、硝酸リチウム及び硝酸コバルトを含む反応混合物から合成することがより好ましい。通常硝酸塩が水もアルコールも含まない縮合生成物を生ずるので、硝酸塩を含むゾル−ゲル反応が本発明の方法にとって有利である。
【0043】
LiFePO4のための好ましいゾル−ゲル前駆体の2つの例は、
クエン酸Fe(III)+LiH2PO4(H3PO4及びLi3PO4から製造)、
Fe(NO3)3・9H2O+NH4H2PO4+LiNO3+クエン酸
である。
【0044】
本発明の第2態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな酸化物生成物を残すステップを含むメソポーラスなリチウム遷移金属酸化物の合成方法を提供する。
【0045】
本発明の第3態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスなリン酸塩生成物を残すステップを含むリン酸鉄リチウムの合成方法を提供する。好ましくは、リチウム塩はリン酸塩である。或いは、ステップ(i)で追加のリン酸塩を添加してもよい。いずれの場合も、リン酸二水素塩が好ましいリン酸塩である。
【0046】
本発明の第4態様によれば、メソポーラスなリチウム遷移金属酸化物材料、好ましくはコバルト酸リチウム材料を提供する。本発明のメソポーラスな酸化物は好ましくは10m2/gを超える、より好ましくは15m2/g以上のBET表面積を有している。1つの実施形態では、リチウム遷移金属酸化物は化合物コバルト酸リチウムであり、好ましくはLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される。代替実施形態では、リチウム遷移金属酸化物はLiCoO2であり、好ましくは結晶構造を有するLiCoO2である。
【0047】
本発明の第5態様によれば、上記したメソポーラスなリチウム遷移材料を含む電極を提供する。
【0048】
本発明の第6態様によれば、上記したメソポーラスなリチウム遷移材料を含む電池を提供する。好ましくは、電池はリチウム二次電池またはスーパーキャパシタであり得る。
【0049】
本発明の第2、第3、第4、第5及び第6態様の好ましい要件は第1態様に関連して上記した通りである。
【0050】
本発明の具体的実施形態を添付図面を参照して説明する。
【図面の簡単な説明】
【0051】
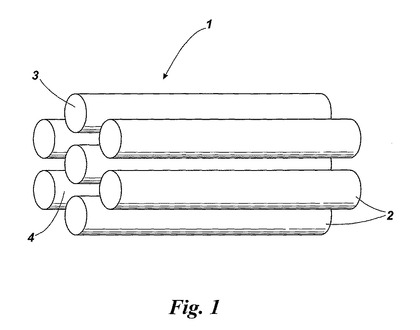
【図1】液晶ヘキサゴナルカラムアレイの概略図である。
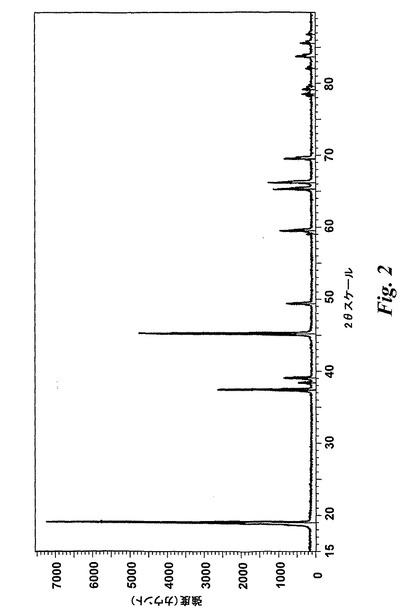
【図2】市販LiCoO2のXRDパターンを示す。
【図3a】市販LiCoO2のTEM顕微鏡写真を示す。
【図3b】市販LiCoO2のTEM顕微鏡写真を示す。
【図4】非テンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
【図5】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のXRDパターンを示す。
【図6a】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
【図6b】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
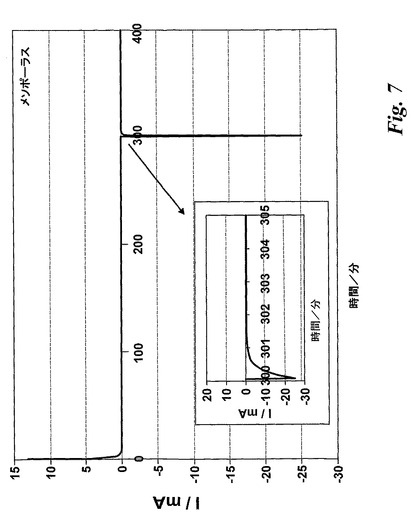
【図7】本発明の方法により製造したメソポーラスなLiCoO2を含む電池についての電位ステップトレースを示す。
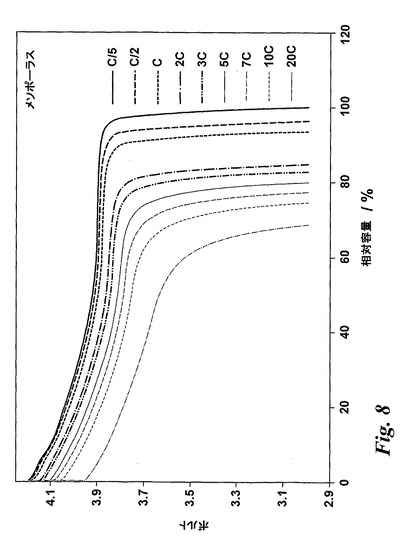
【図8】本発明の方法により製造したメソポーラスなLiCoO2についての異なる放電レートでの定電流放電プロフィールを示す。
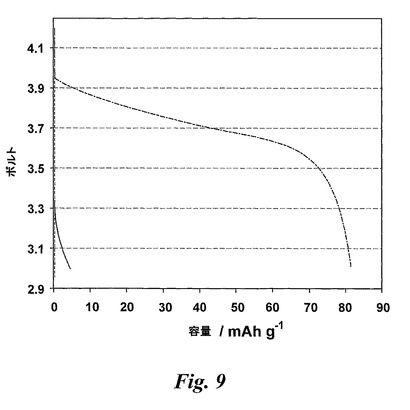
【図9】テンプレート化(真ん中の曲線)、非テンプレート化(上の曲線)及び市販の(下の曲線)LiCoO2電極材料の20Cレートでの定電流放電プロフィールを示す。
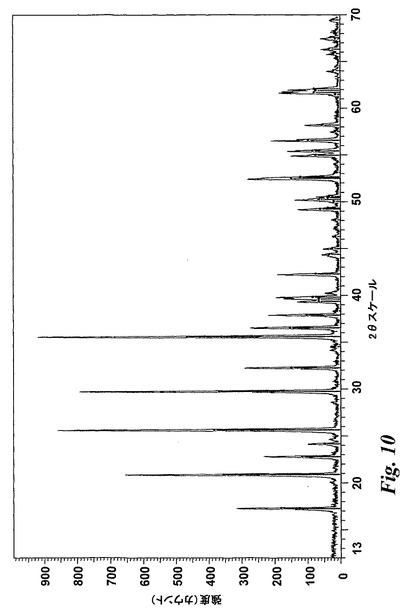
【図10】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiFePO4のXRDパターンを示す。
【発明を実施するための形態】
【0052】
図1は、疎水性内部領域3及び水性ゾル−ゲル反応を助け得る親水性外部領域4を有するシリンダーまたはカラム2からなる液晶ヘキサゴナルカラムアレイ1の概略図である。本発明の方法では、ヘキサゴナルアレイの周りの親水性領域4に高度に相互接続されている高分子金属酸化物網状構造を形成し、界面活性剤を除去するとシリンダー形態が酸化物構造中に残る。
【0053】
液晶相安定性
液晶相がゾル−ゲル合成の実質的にすべての間、すなわち本発明の方法のステップ(i)及び(ii)の間60〜80℃のゾル−ゲル温度範囲で安定のままであることが望ましい。その目的を達するために、好ましいヘキサゴナル(H1)液晶相の安定を2つのゾル−ゲル試薬系(酢酸リチウム/酢酸コバルト及び硝酸リチウム/硝酸コバルト)の存在下で典型的なゾル−ゲル反応温度で2つの適している界面活性剤を用いて研究した。
【0054】
使用した2つの界面活性剤は、高純度で比較的高価な化合物であるオクタエチレングリコールモノヘキサデシルエーテル(OEE)(Fluka)及びより安価な原料であるポリオキシエチレン10セチルエーテル(Brij(登録商標)56)(Aldrich)であった。
【0055】
(ゾル−ゲル反応混合物由来の)金属イオンの存在は相構造を不安定としなかったことを確かめることも重要であり、その目的を達するために金属イオンを含む及び含まない界面活性剤/水溶液の相挙動も研究した。
【0056】
別の実験では、水溶液にキレート化剤としてクエン酸も含有させた。
【0057】
金属酢酸塩または硝酸塩の水溶液とOEEまたはBrij(登録商標)56の混合物は、金属イオン溶液に30、40、50及び60wt%に相当する組成で界面活性剤を添加することにより調製した。界面活性剤が液体になるまで混合物を加熱(約40〜50℃)した後、均質な混合物が得られるまで迅速に混合した。混合物を交差分極光学顕微鏡のホットステージアセンブリに移し、初期のサンプル組織を調べた後、更に95℃の最高温度まで加熱した。95℃を超えて加熱すると、混合物が沸騰して、水分が急速に失われ、その結果相挙動が変化した。
【0058】
30wt% OEEを含む硝酸塩系の場合、ヘキサゴナル相は観察されなかった。40、50及び60wt%では、材料の剪断の結果として見られる断面に応じて2つのH1組織が得られた。1つの組織は古典的な「柱状ディスコチック」扇形組織であり、他の組織はカラミチックなスメクチックI/FまたはB相で観察される層状H1秩序相に類似の小板組織であった。混合物は40、50及び60wt%で(すなわち、金属硝酸塩の非存在下で)ホスト系から構成され、相組織は金属イオン系と同じであると確認された。2つの組織を示す混合物の接触研究から、研究した温度範囲で連続性を示した。
【0059】
有利には、ヘキサゴナル相の安定性が水系に比して硝酸塩系で強化され、更に混合物にクエン酸を添加しても大きく低下しなかったことが知見された。
【0060】
酢酸塩/OEE混合物の場合、ヘキサゴナル相の安定性はホスト系に比して低下したが、当該温度範囲でなお実用的であった。より複雑な相変化が60wt%を超える界面活性剤濃度で明らかであったが、これらは興味あるヘキサゴナル相であったので更に研究されなかった。
【0061】
OEEの代わりにBrij(登録商標)56を用いて調製した一連の混合物(酢酸塩、硝酸塩またはホスト)に関して次のような所見が得られた。これらはヘキサゴナルH1相の温度安定性範囲の拡大及び立方V1相の低下を示し、これらは本発明の目的にとって有益であった。50:50wt% Brij(登録商標)56/水溶液混合物はH1相に対して最も広い温度安定性範囲(室温〜約95℃)を与えることを知見し、従って研究中のすべての実験にとって好ましかった。
【0062】
95℃以下の温度で液晶混合物にクエン酸を添加してもヘキサゴナル相の安定性に影響を与えないことを知見した。しかしながら、酢酸塩混合物のヘキサゴナル相は通常余り熱的に安定でなく、液晶相遷移も余りうまく規定されなかった。
【0063】
要するに、2つの界面活性剤とリチウム及びコバルトの硝酸塩及び酢酸塩の水溶液の液晶混合物のヘキサゴナル相の組成及び温度安定性範囲がうまく決定された。市販されている低価な界面活性剤(Brij(登録商標)56)を含有する溶液は、より高価な代替界面活性剤(OEE)よりも好ましいヘキサゴナル相のより広い温度安定性範囲を有していることが知見された。更に、テンプレート化混合物にクエン酸を添加してもヘキサゴナル相の安定性が影響されないことが知見された。
【0064】
酢酸塩混合物のヘキサゴナル相は通常余り熱的に安定でなく、液晶相遷移も余りうまく規定されなかった。よって、リチウム及びコバルトの硝酸塩を含む混合物は酢酸塩よりも好ましかった。
【0065】
市販のコバルト酸リチウムの特性決定
市販のLiCoO2は供給業者(Pred Materials,電池グレード)から黒色微粉として受け取り、XRD分析、TEM及びBET表面積分析により調べられた。XRD分析用サンプルは、まず乳棒及び乳鉢を用いて微粉に粉砕することにより調製した。TEMサンプルは、電子ビームが透過するのに十分な薄さの断片が得られるようにエタノール中で数時間超音波処理にかけた後粉末断片として調べた。BET測定は供給業者から受け取ったままの粉末に対して実施した。
【0066】
XRDは結晶性無機化合物を同定するための標準技術であり、しばしば化合物のフィンガープリントをとるように作用する。XRDパターンは特定化合物毎に独自であり、結晶面からの反射による回折ピークからなる。市販の電池グレードLiCoO2のXRDパターンを標準の純結晶LiCoO2と比較し、図2に示す。図2では、純結晶LiCoO2標準についてのXRDピークを縦線として表す。標準サンプルからのピークと市販LiCoO2粉末からのピークが(ピークの数及びビークの位置の点で)かなり整合していることが明らかとなり得る。このことから、市販LiCoO2が標準LiCoO2であることが確認される。
【0067】
TEMは、非常に小さい孔(<100nm)を有する高表面積材料中の孔径及び孔構造を同定するために電子ビームを使用する高分解能特性決定技術である。材料がナノスケールで多孔性でないならば、電子ビームは構造を透過し得ないのでTEM顕微鏡写真は暗い像として現れる。しかしながら、メソポーラスな材料の場合、TEM顕微鏡写真は暗い領域及び明るい領域として(明るい領域はビームが孔を透過するためであり、暗い領域は孔壁を透過しないためである)、孔構造の規則性及びビームの透過面に応じて黒い及び白い線または円を有する規則性パターンとして現れる。線間の測定距離は孔の直径または孔反復距離であり、線幅は孔壁の厚さである。図3a及び3bは、市販粉末由来の断片のTEM顕微鏡写真を示す。顕微鏡写真は特色がなくて暗く、メソスケールでの多孔性を欠くことを示している。
【0068】
BET表面積分析は、高多孔性材料の表面積を測定するために使用されている標準技術である。この方法は特定温度での窒素吸着及び脱着に依存している。通常、吸着または脱着された窒素の量は材料の表面積に直接関係している。BET表面積が高ければ、材料の表面積、よって多孔性が大きくなる。市販LiCoO2材料は1m2/g未満のBET値を有していた。
【0069】
コバルト酸リチウムの非テンプレート化ゾル−ゲル方法による合成
LiCoO2の非テンプレート化ゾル−ゲル合成を対照実験として実施した。紫〜ピンク色の外皮のあるゲルが形成されるまで、硝酸リチウム(Suprapur,Merck Chemicals International)、硝酸コバルト(Suprapur,Merck Chemicals International)及びクエン酸(Puriss Grade,Fluka Chemie AG)の水溶液を開放溶液中で連続攪拌しながら70〜80℃に加熱した。次いで、ゲルを75℃で数時間保持した後、放冷した。生成物を微粉に粉砕した後、650〜700℃の温度で16時間焼成した。暗灰色〜黒色粉末が形成された。市販のコバルト酸リチウムの特性決定に関して上に概説した方法を用いてXRD、BET及びTEMにより生成物の特性を決定した。
【0070】
非テンプレート化生成物について標準サンプルとのピークの整合を示したXRDトレースを記録し、非テンプレート化合成の生成物が市販LiCoO2に類似する単相構造を有していることを確認した。
【0071】
非テンプレート化合成により製造したLiCoO2材料のTEM顕微鏡写真を図4に示す。像は特色がなくて暗く(写真の上部領域)、メソスケールの多孔性を欠いていることを示している。
【0072】
非テンプレート化合成由来の粉末は約1m2/gのBET表面積を有していた。
【0073】
コバルト酸リチウムのテンプレート化ゾル−ゲル方法による合成
LiCoO2のテンプレート化ゾル−ゲル合成を本発明に従って実施した。硝酸リチウム、硝酸コバルト及びクエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、100mlのBrij(登録商標)56(Sigma Aldrich Inc.)界面活性剤の溶融溶液を40℃で添加した。次いで、ヘキサゴナル液晶相が形成されるまで混合物を攪拌した。次いで、粘性ペーストのコンシステンシーを有する液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを明紫色から暗紫色に変化するまで60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。次いで、冷却した固体/ゲルを炉に移して600〜650℃で16時間一定加熱した。液晶のヘキサゴナル相を破壊し、よって孔構造形態の崩壊を導く水分損失が観察されたので、テンプレート化ゾル−ゲル合成を密封容器中で実施した。
【0074】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、市販のコバルト酸リチウムの特性決定について上に概説した方法を用いてXRD、BET及びTEMにより特性を決定した。
【0075】
図5は、テンプレート化生成物についてのXRDトレースを示す。標準サンプルとピーク整合していることが明らかであり、このことからテンプレート化合成の生成物はLiCoO2標準及び市販LiCoO2と類似の構造を有していることが確認される。
【0076】
図6a及び6bは、テンプレート化合成により製造した粒子についてのTEM顕微鏡写真を示す。いずれの像も規則的に離間している暗い線及び明るい欄の領域またはクラスターを有するメソ多孔性の証拠を示している。線間隔(すなわち、孔径)及び反復距離は約3nmであり、線幅(すなわち、孔壁厚さ)は約1.5nmである。
【0077】
テンプレート化合成由来の粉末は15.8m2/gのBET表面積を有していた。
【0078】
上記実験を数回繰り返した。いずれの場合も、合成粉末のBET表面積は10mz/g以上であり(記録した最低値は13.0mz/g)、BET表面積は通常15m2/g以上であった。1例で、20.0m2/gのBET表面積を記録した。
【0079】
従って、一般的に、本発明の合成は15m2/g以上のBET表面積を有する粉末を製造したのに対して、非テンプレート化合成由来の粉末及び市販の材料はそれぞれ1m2/g及び1m2/g未満の値を有していた。このことは、テンプレート化合成由来の粉末の表面積が市販LiCoO2に比して少なくとも15倍大きくなることを示している。
【0080】
結論として、XRD分析からテンプレート化ゾル−ゲル合成により製造した粉末がLiCoO2であることが確認される。TEM測定値は、LiCoO2粒子が市販の電池グレードLiCoO2及び同等の非テンプレート化合成により製造した材料よりも少なくとも15倍高いBET表面積を有する高度のメソ多孔性であることを示している。
【0081】
LiNi0.33Co0.33Mn0.33O2のテンプレート化ゾル−ゲル方法による合成
LiNi0.33Co0.33Mn0.33O2のテンプレート化ゾル−ゲル合成を本発明に従って実施した。硝酸リチウム、硝酸コバルト、硝酸ニッケル、硝酸マンガン(1:0.33:0.33:0.33のモル比)及びクエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、40wt%の前駆体溶液を60wt%のCTABと混合した。混合物を攪拌し、安定なヘキサゴナルメソ相が形成された。次いで、液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを明紫色から濃い紫色に変化するまで60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。次いで、冷却した固体/ゲルを炉に移して900〜950℃で10時間一定加熱した。
【0082】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、XRD及びBETにより特性を決定した。
【0083】
XRDトレースは標準LiCoO2についてのXRDトレースと比較してピークの位置がより高い角度に移っていることを示した。このことから、コバルトが構造を変化させることなく部分的にマンガン及びニッケルで置換されている均質な結晶構造が形成されたことが示唆され、予想に一致した。
【0084】
テンプレート化合成由来の粉末は14.3m2/gのBET表面積を有し、これは市販のLiNi0.33Co0.33Mn0.33O2に比して10倍以上の表面積の増加に相当した。
【0085】
電気的性能試験
本発明のテンプレート化ゾル−ゲル方法を用いて製造したLiCoO2材料の電気的性能を確認し、その性能をベンチマークの市販LiCoO2及び非テンプレート化方法由来の材料と比較するために実験を実施した。
【0086】
上記した材料の各々を複合電極コーティングに作成し、リチウムホイル負極を有する実験室半電池において試験した。新しい材料のレート性能を確認するために、電位ステップ分析及び定電流サイクリングの技術を使用した。
【0087】
電位ステップ分析を用いて、メソポーラスな材料のレート性能及び電力特性を確認した。電位ステップ分析は、電圧を開放電圧(OCV)から4.2Vまで充電に相当する5時間で上昇させ、放電に相当する5時間で3Vまで低下させることからなる。材料が短時間、すなわち数秒でその容量の大部分を送出し得るならば、その材料は高レートと分類される。
【0088】
図7は、本発明の方法により製造したメソポーラスなLiCoO2を用いた電池について開放電圧(3.1V)から4.2Vへ及び3Vまでの電位ステップ後の電流の変化を経時的に示す。市販LiCoO2及び非テンプレート化ゾル−ゲル方法を用いて製造したLiCoO2についてもプロフィールを得た。
【0089】
市販の複合電極の場合20分後電流は直ぐに0に低下し、非テンプレート化電極の場合45分後に電流は0に減衰したことが判明した。しかしながら、テンプレート化電極の場合電流はたった2分以内に0に減衰した。
【0090】
重要なパラメーターは個々の電極より送出される電荷の量である。電位ステップ分析から得た電流/時間プロフィールを積分することにより、電池を4.2Vから3Vに低下させたときに得られる電荷の量を計算した。結果を表1にまとめる。ここから、36秒後メソポーラスな電極が示す容量は61mAh/gであるのに対して、非テンプレート化電極の場合8.4mAh/g、市販の電極の場合16mAh/gであることが明らかとなり得る。これらの結果から、約36秒でその容量の11%しか送出しない市販の電極に比してメソポーラスな電極は同じ時間でその容量の少なくとも70%を送出できるであろうことを示唆している。
【0091】
【表1】

【0092】
メソポーラスな電極材料のレート及び電力特性を確認するために定電流サイクリングも使用した。電気化学電池を定電流をC/5(5時間で充電)のレートで用いてOCVから4.2Vまで充電した後、異なるレートで3Vまで放電した。メソポーラスな複合電極の性能を再び非テンプレート化LiCoO2及び市販のLiCoO2と比較した。メソポーラスな電極、非テンプレート化電極及び市販の電極について異なる放電レートでの放電プロフィールを得、図8はメソポーラスな電極についてのプロフィールを示し、異なるレートでの容量はC/5で得た容量に対して正規化されている。図8中、最左のプロフィールは20Cプロフィールであり、最右のプロフィールはC/5プロフィールである。
【0093】
被覆したメソポーラスな材料のレート性能が非テンプレート化電極及び市販の電極よりも特に高いレートで優れていることに関して同じ所見が得られた。例えば、20Cのレートで(図9参照)、テンプレート化電極の容量保持は70%であるのに対して、非テンプレート化電極の場合16%、市販の電極の場合0%であった。これらの結果は、テンプレート化電極の場合36秒で最高の電荷が送出された電位ステップ実験から得た結果と一致している。
【0094】
リン酸鉄リチウムのテンプレート化ゾル−ゲル方法による合成
リン酸鉄リチウム(LiFePO4)のテンプレート化ゾル−ゲル合成を本発明に従って実施した。0.5M リン酸二水素リチウム(Sigma Aldrich Inc.)、0.5M クエン酸第二鉄(Sigma Aldrich)及び1M クエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、100mlのBrij(登録商標)56(Sigma Aldrich Inc.)界面活性剤の溶融溶液を40℃で添加した。次いで、ヘキサゴナル液晶相が形成されるまで混合物を攪拌した。次いで、粘性ペーストのコンシステンシーを有する液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。
【0095】
次いで、冷却した固体/ゲルを炉に移してアルゴン雰囲気下約700℃で16時間一定加熱した。液晶のヘキサゴナル相を破壊し、よって孔構造形態の崩壊を導く水分損失が観察されたので、テンプレート化ゾル−ゲル合成を密封容器中で実施した。
【0096】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、XRDにより特性を決定した。XRDトレース(図10)から、リン酸鉄リチウムの純度が確認された。LiFePO4生成物のメソ多孔性を最適化するために焼成温度を450〜700℃の範囲で変更し得ることが知見された。
【技術分野】
【0001】
本発明は、スーパーキャパシタ及び電池を含めたリチウム電池用材料を製造するための改良方法に関する。本発明は特に、リチウム二次電池に適した材料、具体的にはメソポーラスなリチウム遷移金属化合物の製造、より具体的にはメソポーラスなリチウム遷移金属酸化物及びリン酸塩の製造、更により具体的にはメソポーラスなコバルト酸リチウム及びリン酸鉄リチウムの製造に関する。本発明は、この方法により形成される材料にも関する。
【背景技術】
【0002】
リチウム二次電池は、重要な携帯用DC電力源であり、最近燃料電池が開発されたにも関わらず多年にわたりそのようにあり続けているようである。携帯用電子及び電気システムは効果的に作動させるためにハイパワー、コンパクトで永続性な電力源がますます必要とされており、よって改善された携帯用電力源が必要である。既存のシステムのために必要とされる時間の何分の1かで再充電され得、改良された電池サイクル寿命をも有する高エネルギーリチウムイオン電池が特に興味深い。
【0003】
性能を所望するように向上させるための1つの方法は改良された活性正極材料を使用することである。二次リチウム電池用高性能活性正極材料としてリチウム遷移金属酸化物を使用することは公知であり、コバルト酸リチウムは最も一般的に使用されている市販材料の1つである。別の興味ある材料はリン酸鉄リチウムであるが、大量に合成されるリン酸鉄リチウムは高い電気抵抗を有している。このことはハイパワー用途にとっては望ましくなく、導電率を高め、よってレート特性を高めるために電極材料に炭素を配合することが必要である。
【0004】
電池電極は、典型的には活性正極材料、結合剤及び導電性炭素からなるペーストを集電体(例えば、アルミニウムホイル)にコーティングすることにより製作されている。活性材料は必要な電気化学容量を与える電極の主成分である。結合剤は粒子間接続性及び集電体に対する密着性を与え、導電性炭素は電極反応のための電子伝導率のレベルを高める。活性材料粒子のサイズは均一で接着性のコーティングを作成するために、また所望レベルの電極性能のために重要であり、粒子が大きいと電極の厚さが厚くなるだけでなく、不均一なコーティングが生ずることは公知である。電極性能に関して、大きい粒子及び厚いコーティングは、充電/放電中リチウムイオンが電極中に及びそこから拡散し得るレートを限定し、よって電極の電力/レート特性を制限する。
【0005】
リチウムイオン電池の充電/放電中、リチウムイオンは活性正極材料粒子中に及びそこから移動して、前記粒子はプロセス中膨張し、収縮する。連続サイクリングで粒子が繰り返し膨張し、収縮すると、徐々に粒子間の電気接触が失われ、その結果電極の抵抗が増加する。また、抵抗が増加すると、すべての容量が引き出され得る前に時期尚早に充電/放電カットオフ電圧に達するために電極の利用率が低くなる。これは「容量フェード」として公知の現象である。
【0006】
よって、電池が高いレート、長いサイクル寿命及び良好な利用率を発揮するためには、電池電極材料を多孔率の高い小粒子から構成することが望ましい。こうすると、電極の活性表面積が大きくなり、非常に薄い電極コーティングを作成することができる。(比表面積として測定した)表面積が大きいと、速い電極反応も促進され、よってレート性能及び電力特性が高まる。
【0007】
高表面積の電池電極材料を得るための従来方法は、粒子サイズを小さくするためのボールミリング及び多孔質材料を得るための熱分解またはソリッドステートルートを含む。しかしながら、これらの方法は、より小さい粒子が凝集したり、高価であったり、スケールアップに適さないことを含めた多数の欠点を有する傾向にある。電池電極のための既存の製作方法との適合性も問題であり得る。
【0008】
また、リチウム遷移金属酸化物はナノ粒子として直接合成され得る。例えば、リチウム遷移金属酸化物のナノ粒子を製造する1つの公知方法は、単相金属水酸化物または多相混合金属水酸化物を直接溶解沈殿させた後、水酸化物をそれぞれの酸化物または化合物酸化物生成物に変換する焼成ステップによる。リチウム遷移金属酸化物のナノ粒子を製造する別の公知方法は溶解ゾル−ゲル合成により、例えばHahnら(Proc.9th Asia Pacific Physics Conference,Hanoi,Vietnam,October 25−31,2004)はクエン酸反応試薬の存在下で硝酸リチウムを硝酸コバルトと反応させて約100nmの粒度を有するコバルト酸リチウム粉末を製造することを記載している。しかしながら、上記方法で製造したコバルト酸リチウムナノ材料は最高でも約5m2/gの表面積しか達成し得ない傾向にある。
【先行技術文献】
【非特許文献】
【0009】
【非特許文献1】Hahnら(Proc.9th Asia Pacific Physics Conference,Hanoi,Vietnam,October 25−31,2004)
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の第1態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな生成物を残すステップを含むメソポーラスなリチウム遷移金属化合物の合成方法が提供される。
【0011】
リチウム塩及び1つ以上の遷移金属塩は、ステップ(ii)で高分子金属酸化物網状構造を形成し得、これによりメソポーラスなリチウム遷移金属酸化物生成物が形成される。
【0012】
或いは、リチウム塩及び1つ以上の遷移金属塩は、ステップ(ii)で高分子金属リン酸素網状構造を形成し得、これによりメソポーラスなリチウム遷移金属リン酸塩生成物が形成される。リン酸塩を主成分とするゾル−ゲルを形成するために追加のリン酸塩試薬、例えばリン酸二水素アンモニウムが必要なことがある。好ましい実施形態では、リチウム塩を鉄塩及びリン酸塩と好ましくは酸性条件下で反応させて、メソポーラスなLiFePO4を製造する。好ましくは、リチウム塩は硝酸リチウムであり、鉄塩はシュウ酸第一鉄二水和物または硝酸鉄(例えば、Fe(NO3)3・9H2O)であり、リン酸塩はリン酸二水素アンモニウムである。或いは、鉄塩(例えば、クエン酸Fe(III))を追加のリン酸塩試薬なしでリン酸リチウム(例えば、LiH2PO4)と反応させて、高分子金属リン酸素網状構造を形成してもよい。場合により、反応混合物は硝酸を含む。有利には、LiFePO4材料にメソ多孔性を付与することにより該材料の導電率が大きく高められ得ることが知見された。
【0013】
本発明は、リチウム遷移金属ホウ酸塩及びケイ酸塩、例えばLiMBO3(ここで、MはFe、CoまたはMnであり得る)及びLi2MSiO4(ここで、MはFeまたはMnであり得る)を合成するためにも使用され得る。この場合、リチウム塩及び1つ以上の遷移金属塩はステップ(ii)で高分子金属ホウ素酸素または高分子金属ケイ素酸素網状構造のいずれかを形成し、これによりメソポーラスなリチウム遷移金属ホウ酸塩またはケイ酸塩生成物が形成される。この場合でも、ゾル−ゲルを形成するために追加のホウ酸塩またはケイ酸塩試薬が必要なことがあり、当業者は適当なゾル−ゲル試薬を認識している。
【0014】
リン酸鉄リチウム(LiFePO4)と同様に、LiMBO3(ここで、MはFe、CoまたはMnであり得る)及びLi2MSiO4(ここで、MはFeまたはMnであり得る)は通常低い導電率を有する潜在的高エネルギー密度正極材料である。材料を本発明の方法に従って合成することにより、その導電率を高め、その材料をリチウム電池中に使用することができる。
【0015】
いずれの場合も、本発明の方法は、ゾル−ゲル合成と液晶テンプレート化を組み合わせることによりメソポーラスなリチウム遷移金属化合物を製造する。結果は、単相のメソポーラスなリチウム遷移金属化合物を製造するために容易に実施される便利な低温方法である。
【0016】
ここで、メソポーラスなコバルト酸リチウムの製造に特に言及して本発明を説明する。しかしながら、教示内容は当業者により一般的には上記したリチウム遷移金属化合物に適用され得る。コバルト酸リチウムは、コバルト酸リチウム(LiCoO2)または1つ以上の追加の遷移金属包含物を含むコバルト酸リチウム(例えば、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2)のいずれかを意味し、以後化合物コバルト酸リチウムと称する。いずれの場合も、1つ以上の遷移金属塩はコバルト塩を含む。LiCoO2を製造するためには、ステップ(i)でリチウム塩をコバルト塩と反応させる。しかしながら、化合物コバルト酸リチウムを製造するためにリチウム塩をコバルト塩及び1つ以上の更なる遷移金属塩と反応させ、1つ以上の更なる塩は好ましくはNi、Mn及びAlからなる群から選択される遷移金属を含む。より好ましくは、試薬をLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される化合物コバルト酸リチウム生成物を形成するように選択する。
【0017】
上に表した化学式は厳密に限定するものと意図されず、非化学量論的バリアントを包含すると理解されたい。
【0018】
メソポーラスなコバルト酸リチウムにより、市販の電池において通常相互に相いれないパラメーターである高いエネルギー及び高い再充電/放電レート特性を有するリチウムイオン電池を開発する可能性がある。メソポーラスなコバルト酸リチウムを主成分とする正極材料を使用する別の効果は、破損の主メカニズム(物理的分解により正極の電子抵抗が高くなる)がメソ多孔性により大きく妨害されるために電池サイクル寿命が向上することである。
【0019】
慣例的に、多孔質材料の孔径範囲は次のように定義される:ミクロポーラス 5nm未満、メソポーラス 5〜50nm、ナノポーラス 50〜100nm、マクロポーラス 100nm以上。しかしながら、範囲及び長距離秩序は非常に僅かに変わり得るのでこれらの定義を厳密に解釈するつもりはない。従って、本発明の方法により製造されるメソポーラスな材料の幾つかの孔は50nmより大きく、幾つかの孔は5nmより小さい。
【0020】
本発明の方法は単相のメソポーラスなリチウム遷移金属酸化物、好ましくはメソポーラスなコバルト酸リチウムを製造し得る。本発明に従って製造されるコバルト酸リチウム粉末をX線回折(XRD)分析、透過型電子顕微鏡(TEM)及びブルナウア−エメット−テラー(BET)表面積分析により特性決定され、メソポーラスであり且つ10m2/gを超える、典型的には15m2/g以上の比表面積を有していることが判明した。1つの実施例では、20m2/gのBET表面積が記録された。
【0021】
液晶テンプレート化は、規則的に離間されているシリンダーの自己組織化アレイからなる秩序化液晶相を形成するために反応混合物中に高濃度の界面活性剤を使用する。シリンダーの間隔、よって最終テンプレート化生成物の孔径は選択した界面活性剤の鎖長によりコントロールされる。本発明の方法にとって好ましい液晶相であるヘキサゴナル相の場合、シリンダーはヘキサゴナルアレイで配置され、界面活性剤の疎水性部分がシリンダーの内部に向かって集中し、親水性、通常水性領域は外部に集中するように構成される。適切にコントロールされると、液晶テンプレート化により高秩序化テンプレート構造が得られ、その周りで材料が合成され得る。典型的には、液晶テンプレート化によりリオトロピック液晶相が使用でき、従って他のよりゆるく秩序化されている界面活性剤を用いる技術(例えば、ミセルテンプレート化)よりもより高い界面活性剤濃度で実施される。
【0022】
ゾル−ゲル方法は、無機及び/または有機金属塩前駆体からリチウム遷移金属酸化物粉末を含めた金属酸化物粉末の合成のために使用され得る低温方法であり、通常水性条件下で行う。ゾル−ゲル方法では、まず前駆体からコロイド状懸濁液(ゾル)を形成した後、ゾルを加水分解及び縮合反応により連続液相中に固相−M−O−M−高分子網状構造(ゲル)を形成することにより金属酸化物網状構造が形成される。その後、ゲルを加熱して酸化物生成物を乾燥し、所要により求められる結晶相を与える。金属塩前駆体の組合せを使用することにより、2、3、4、5またはそれ以上の金属包含物を含む混合金属酸化物ゾルが形成され得る。正極材料用の所望の混合金属酸化物にはLiCoO2、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2が含まれる。
【0023】
ゾル−ゲル方法は他の金属化合物、例えばリン酸塩、ホウ酸塩及びケイ酸塩を合成するためにも使用され得る。この場合、高分子網状構造は金属リン酸素網状構造、金属ホウ素酸素網状構造、金属ケイ素酸素網状構造等である。
【0024】
非テンプレート化ゾル−ゲル合成から製造される金属酸化物粉末は通常約5m2/g未満、より典型的には約1m2/g未満の比表面積を有する個々に分散されている及び/または凝集されているサブミクロン及び/またはナノサイズ粒子からなる。これは、ゾル−ゲル合成によると小さい粒度を有するが限定された多孔性を有する酸化物生成物が製造されるからである。
【0025】
本発明の方法では、反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させる。反応混合物を加熱し、場合によりエージングしてゾル−ゲルを形成し、これにより金属酸化物の合成の場合には液晶相の親水性ドメイン中に高分子リチウム−遷移金属酸化物網状構造が形成される。言い換えれば、反応混合物の親水性領域、典型的には液晶相中のロッドまたはシリンダーの外の領域がゾル−ゲル方法によりリチウム遷移金属酸化物(−Li−O−M−)網状構造の合成を助けるために使用される。こうして、無機高分子金属酸化物網状構造が好ましくはヘキサゴナルアレイで配置されている界面活性剤ロッドの周りに形成し、これにより液晶テンプレートが金属酸化物構造に導入される。次いで、界面活性剤を除去すると、メソポーラスな酸化物生成物が形成される。
【0026】
好ましくは、1つ以上の遷移金属塩には、液晶相がそれぞれ高分子−Li−O−Co−網状構造または−Li−O−Co−O−M−網状構造の形成を助けるLiCoO2、すなわち化合物コバルト酸リチウムを製造するためのコバルト塩が含まれる。
【0027】
界面活性剤は、ゾル−ゲルから適当な方法、例えば溶媒抽出または熱分解により除去され得る。界面活性剤を除去すると、単相の三次元的に結合しているメソポーラス構造を有する酸化物生成物が残る。
【0028】
合成された生成物、好ましくは合成された酸化物生成物は少なくとも部分的に非晶質形態をとるようである。非晶質材料は通常電池材料として使用するためには望ましくなく、従って材料を所望の結晶相に変換するために生成物を焼成することが好ましい。界面活性剤除去及び焼成段階を組み合わせること、例えば界面活性剤を焼成に適した温度で熱分解することにより除去することが好都合である。通常、生成物を適切な結晶相に変換するために約400℃以上の温度が使用され得るが、焼成温度が高くなるとメソポーラスな生成物中の孔が崩壊する可能性が高くなることを本発明者らは知見した。こうなると、達成可能な表面積の拡大が対応して低下する。従って、(孔の一体性を維持するために)できるだけ低いが、正しい結晶性を得るために十分に高い焼成温度を使用することが望ましい。電池グレードLiCoO2を液晶テンプレート化合成するための好ましい焼成温度は400〜700℃、より好ましくは400〜600℃の範囲である。リン酸鉄リチウムを合成するためには、焼成温度は好ましくは450〜700℃の範囲、より好ましくは450〜650℃の範囲である。
【0029】
材料を液晶テンプレート化方法によりうまく合成するためには、通常反応混合物中に所望の液晶相を形成した後この相を合成反応の実質的すべての間維持することが重要である。反応条件を例えば望ましくない副生成物を除去したり及び/またはステップ(i)及び(ii)で界面活性剤濃度を所望範囲に維持することにより注意深くコントロールしないと、液晶相は不安定になることがあり得る。液晶相が不安定化すると、メソポーラスな生成物を形成するのに適していない望ましくないミセル相、すなわちよりゆるく秩序化されている構造が形成される恐れがある。むしろ、生成物は単に全くまたは殆ど相互接続性を持たず、比表面積が小さいゆるく秩序化されているナノ粒子からなる傾向にある。
【0030】
従って、本発明の方法では、ステップ(i)及び(ii)で液晶相の不安定化を防止するように反応条件をコントロールすることが望ましく、理想的にはステップ(i)及び(ii)のすべてまたは実質的にすべてを通して相不安定化を防止するように反応条件をコントロールする。安定な液晶相、好ましくは安定なヘキサゴナル相が維持されるように反応混合物中の界面活性剤の濃度をコントロールすることが特に重要であり得る。
【0031】
安定な液晶相、特に好ましいヘキサゴナル液晶相を製造するための典型的な界面活性剤濃度は40〜60wt%であり、上記にてらしてステップ(i)及び(ii)のすべてまたは実質的にすべてを通して界面活性剤の濃度を上記範囲に維持することが好ましい。より好ましくは、界面活性剤濃度は45〜55wt%の範囲であり、更により好ましくは界面活性剤濃度は約50wt%である。この場合でも、ステップ(i)及び(ii)のすべてまたは実質的にすべてを通して、すなわちゾル−ゲル無機高分子網状構造の形成中、界面活性剤濃度を上記範囲に維持することが好ましい。
【0032】
本発明の方法の作用効果は、反応混合物に試薬を更に添加することを伴わないことである。換言すれば、金属塩、界面活性剤及び任意の試薬(例えば、上記したリン酸塩、ホウ酸塩またはケイ酸塩及び/または下記するキレート化剤)を組み合わせて反応混合物、好ましくは水性反応混合物を形成したら、合成中更なる試薬を添加する必要がなく、その結果界面活性剤濃度はその後希釈されない。こうして、単純で簡単な合成方法が提供されるだけでなく、界面活性剤濃度を変化させる恐れがある主メカニズムがステップ(i)及び(ii)中の水分損失であることを意味する。従って、これらのステップ中の水分損失を最小限とすることにより反応混合物中の界面活性剤濃度(よって、液晶相挙動)を簡単且つ効果的にコントロールすることができる。
【0033】
水分損失を最小限とする1つの好ましい方法は、液晶相が完全に乾燥しないように、よって相不安定化を受けないように反応温度をコントロールすることである。従来の金属酸化物のゾル−ゲル合成は60〜80℃の範囲の温度で進行し得るが、50〜60℃の範囲のより低い温度が通常本発明の方法のために適している。より好ましくは、反応混合物を55〜60℃の範囲の温度に加熱するが、この温度はコバルト酸リチウムの場合の70〜80℃という通常の非テンプレート化反応温度よりも低い。本発明者らは、55〜60℃の範囲の反応温度がゾル−ゲル合成の加水分解及び縮合反応の促進と反応混合物からの水分損失の低減の間の適切なバランスを与えることを知見した。低い反応温度を補うために、本発明のテンプレート化ゾル−ゲル合成を従来の対応する非テンプレート化合成よりも長く進行させることが好ましい。有利には、この任意のエージングプロセスにより、その後の加工ステップ(特に、任意の焼成ステップ)で崩壊しにくいより良好で、より相互接続され、高密度化されている高分子網状構造が作成され得る。
【0034】
反応温度をコントロールする代わりにまたはそれに加えて、ステップ(i)及び(ii)で生ずる水蒸気の逃散をコントロールすることにより水分損失を低減させることができる。従って、水分損失の程度をコントロールするように、より好ましくは水分損失を実質的に防止するようにテンプレート化ゾル−ゲル反応を場合により密封されたまたは部分的に密封された容器または反応容器中で実施する。
【0035】
本発明のために適したテンプレート化剤として作用させるために、ロッドまたはシリンダーからなる液晶相を形成し得る、より好ましくはH1ヘキサゴナル相を形成し得る界面活性剤が通常必要である。多くの界面活性剤が当業者に公知であり、市販業者から容易に入手し得、反応混合物において所望の液晶相挙動を与える界面活性剤が本発明の方法で使用され得る。イオン性または非イオン性界面活性剤が適当であり得る。適しているイオン性界面活性剤には臭化セチルジメチルエチルアンモニウムまたは臭化セチルトリメチルアンモニウム(CTAB)が含まれ、適している非イオン性界面活性剤にはエチレンオキシド及びプロピレンオキシドを主成分とするブロックコポリマー(一般的にPluronic(登録商標)界面活性剤として公知)、ポリオキシエチレンセチルエーテルまたはオクタエチレンエーテル(OEE)が含まれる。しかしながら、化学的に不活性であり、ゾル−ゲル反応に関与しない界面活性剤を選択することが好ましい。この点で、リチオ化界面活性剤(例えば、ラウリルエーテルリチウム酢酸塩)が反応混合物中の液晶相に対する望ましくない不安定化作用のために本発明の方法で使用するのに適さないことを知見した。
【0036】
好ましくは、界面活性剤はテンプレート化ゾル−ゲル合成の温度範囲で必要な液晶相を形成し得なければならない。秩序化テンプレートにおいて約5〜10nm、好ましくは7〜8nmの反復距離を与え得る鎖長(または、鎖長範囲)を有する界面活性剤を選択することも有利である。
【0037】
好ましくは、ゾル−ゲル反応を水性条件下で実施し、理想的には液晶相を不安定化し得る溶媒を排除した条件下で実施する。通常、有機溶媒(例えば、エタノール及びメタノール)は液晶相を壊し、これによりテンプレート構造を完全または部分的に崩壊し、所望のメソ多孔性を有する生成物を得ることができないので、これらの溶媒の使用を避けることが望ましい。
【0038】
場合により、試薬の混合を高め、反応する前駆体の表面積を最大限とし、(望ましくない多相生成物の形成を導く)金属酸化物の時期尚早の沈殿を防止するために反応混合物はキレート化剤を含む。例えば硝酸リチウムを硝酸コバルトと反応させてLi/O/Coを主成分とするゾルを製造する場合、通常反応混合物中に追加のキレート化剤が必要である。
【0039】
しかしながら、別のキレート化剤は必ずしも必要でない。例えば酢酸リチウムを酢酸コバルトと反応させてLi/O/Coを主成分とするゾル−ゲルを製造する場合、酢酸対イオンそれ自体がキレート化剤として作用し得、これにより別の試薬が必要でなくなる(換言すると、酢酸塩自己キレート)。
【0040】
一般的に、ゾル−ゲル試薬それ自体がキレート化特性を有していない場合にはキレート化剤が望ましい傾向にある。材料を本発明のテンプレート化ゾル−ゲル合成においてキレート化剤として作用させるためには、材料はテンプレート内の疎水性ドメインよりもむしろテンプレートのロッドまたはシリンダーの外の親水性ドメインでキレート化し得なければならない。本発明者らは予期せぬことに、クエン酸及びプロピオン酸が本発明の方法においてキレート化剤として作用し得、コストが安く、容易に入手し得るためにクエン酸が好ましいことを知見した。
【0041】
1つ以上の遷移金属塩は有機アニオン(例えば、酢酸塩、カルボン酸塩、クエン酸塩またはシュウ酸塩)または無機アニオン(例えば、硝酸塩、リン酸二水素塩またはリン酸塩)を含み得る。幾つかの場合には、1つ以上の更なる遷移金属塩は異なるアニオンを含み得るが、多くの場合各遷移金属試薬は同一のアニオンを含む。
【0042】
また、リチウム塩は有機アニオン(例えば、酢酸塩、カルボン酸塩、クエン酸塩またはシュウ酸塩)または無機アニオン(例えば、硝酸塩、リン酸二水素塩またはリン酸塩)を含み得る。通常(必須ではないが)、リチウム塩は1つ以上の遷移金属塩の少なくとも1つと同一のアニオンを含み、通常リチウム塩及び1つ以上の遷移金属塩はすべて同一のアニオンを含む。好ましくは、リチウム塩及び1つ以上の遷移金属塩のすべてが同一のアニオンを含み、アニオンは酢酸塩及び硝酸塩、より好ましくは硝酸塩から選択される。よって、LiCoO2を酢酸リチウム及び硝酸コバルト、または硝酸リチウム及び硝酸コバルトのいずれかを含む反応混合物から合成することが好ましく、硝酸リチウム及び硝酸コバルトを含む反応混合物から合成することがより好ましい。通常硝酸塩が水もアルコールも含まない縮合生成物を生ずるので、硝酸塩を含むゾル−ゲル反応が本発明の方法にとって有利である。
【0043】
LiFePO4のための好ましいゾル−ゲル前駆体の2つの例は、
クエン酸Fe(III)+LiH2PO4(H3PO4及びLi3PO4から製造)、
Fe(NO3)3・9H2O+NH4H2PO4+LiNO3+クエン酸
である。
【0044】
本発明の第2態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな酸化物生成物を残すステップを含むメソポーラスなリチウム遷移金属酸化物の合成方法を提供する。
【0045】
本発明の第3態様によれば、(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスなリン酸塩生成物を残すステップを含むリン酸鉄リチウムの合成方法を提供する。好ましくは、リチウム塩はリン酸塩である。或いは、ステップ(i)で追加のリン酸塩を添加してもよい。いずれの場合も、リン酸二水素塩が好ましいリン酸塩である。
【0046】
本発明の第4態様によれば、メソポーラスなリチウム遷移金属酸化物材料、好ましくはコバルト酸リチウム材料を提供する。本発明のメソポーラスな酸化物は好ましくは10m2/gを超える、より好ましくは15m2/g以上のBET表面積を有している。1つの実施形態では、リチウム遷移金属酸化物は化合物コバルト酸リチウムであり、好ましくはLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される。代替実施形態では、リチウム遷移金属酸化物はLiCoO2であり、好ましくは結晶構造を有するLiCoO2である。
【0047】
本発明の第5態様によれば、上記したメソポーラスなリチウム遷移材料を含む電極を提供する。
【0048】
本発明の第6態様によれば、上記したメソポーラスなリチウム遷移材料を含む電池を提供する。好ましくは、電池はリチウム二次電池またはスーパーキャパシタであり得る。
【0049】
本発明の第2、第3、第4、第5及び第6態様の好ましい要件は第1態様に関連して上記した通りである。
【0050】
本発明の具体的実施形態を添付図面を参照して説明する。
【図面の簡単な説明】
【0051】
【図1】液晶ヘキサゴナルカラムアレイの概略図である。
【図2】市販LiCoO2のXRDパターンを示す。
【図3a】市販LiCoO2のTEM顕微鏡写真を示す。
【図3b】市販LiCoO2のTEM顕微鏡写真を示す。
【図4】非テンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
【図5】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のXRDパターンを示す。
【図6a】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
【図6b】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiCoO2のTEM顕微鏡写真を示す。
【図7】本発明の方法により製造したメソポーラスなLiCoO2を含む電池についての電位ステップトレースを示す。
【図8】本発明の方法により製造したメソポーラスなLiCoO2についての異なる放電レートでの定電流放電プロフィールを示す。
【図9】テンプレート化(真ん中の曲線)、非テンプレート化(上の曲線)及び市販の(下の曲線)LiCoO2電極材料の20Cレートでの定電流放電プロフィールを示す。
【図10】本発明に従うテンプレート化ゾル−ゲル合成により製造したLiFePO4のXRDパターンを示す。
【発明を実施するための形態】
【0052】
図1は、疎水性内部領域3及び水性ゾル−ゲル反応を助け得る親水性外部領域4を有するシリンダーまたはカラム2からなる液晶ヘキサゴナルカラムアレイ1の概略図である。本発明の方法では、ヘキサゴナルアレイの周りの親水性領域4に高度に相互接続されている高分子金属酸化物網状構造を形成し、界面活性剤を除去するとシリンダー形態が酸化物構造中に残る。
【0053】
液晶相安定性
液晶相がゾル−ゲル合成の実質的にすべての間、すなわち本発明の方法のステップ(i)及び(ii)の間60〜80℃のゾル−ゲル温度範囲で安定のままであることが望ましい。その目的を達するために、好ましいヘキサゴナル(H1)液晶相の安定を2つのゾル−ゲル試薬系(酢酸リチウム/酢酸コバルト及び硝酸リチウム/硝酸コバルト)の存在下で典型的なゾル−ゲル反応温度で2つの適している界面活性剤を用いて研究した。
【0054】
使用した2つの界面活性剤は、高純度で比較的高価な化合物であるオクタエチレングリコールモノヘキサデシルエーテル(OEE)(Fluka)及びより安価な原料であるポリオキシエチレン10セチルエーテル(Brij(登録商標)56)(Aldrich)であった。
【0055】
(ゾル−ゲル反応混合物由来の)金属イオンの存在は相構造を不安定としなかったことを確かめることも重要であり、その目的を達するために金属イオンを含む及び含まない界面活性剤/水溶液の相挙動も研究した。
【0056】
別の実験では、水溶液にキレート化剤としてクエン酸も含有させた。
【0057】
金属酢酸塩または硝酸塩の水溶液とOEEまたはBrij(登録商標)56の混合物は、金属イオン溶液に30、40、50及び60wt%に相当する組成で界面活性剤を添加することにより調製した。界面活性剤が液体になるまで混合物を加熱(約40〜50℃)した後、均質な混合物が得られるまで迅速に混合した。混合物を交差分極光学顕微鏡のホットステージアセンブリに移し、初期のサンプル組織を調べた後、更に95℃の最高温度まで加熱した。95℃を超えて加熱すると、混合物が沸騰して、水分が急速に失われ、その結果相挙動が変化した。
【0058】
30wt% OEEを含む硝酸塩系の場合、ヘキサゴナル相は観察されなかった。40、50及び60wt%では、材料の剪断の結果として見られる断面に応じて2つのH1組織が得られた。1つの組織は古典的な「柱状ディスコチック」扇形組織であり、他の組織はカラミチックなスメクチックI/FまたはB相で観察される層状H1秩序相に類似の小板組織であった。混合物は40、50及び60wt%で(すなわち、金属硝酸塩の非存在下で)ホスト系から構成され、相組織は金属イオン系と同じであると確認された。2つの組織を示す混合物の接触研究から、研究した温度範囲で連続性を示した。
【0059】
有利には、ヘキサゴナル相の安定性が水系に比して硝酸塩系で強化され、更に混合物にクエン酸を添加しても大きく低下しなかったことが知見された。
【0060】
酢酸塩/OEE混合物の場合、ヘキサゴナル相の安定性はホスト系に比して低下したが、当該温度範囲でなお実用的であった。より複雑な相変化が60wt%を超える界面活性剤濃度で明らかであったが、これらは興味あるヘキサゴナル相であったので更に研究されなかった。
【0061】
OEEの代わりにBrij(登録商標)56を用いて調製した一連の混合物(酢酸塩、硝酸塩またはホスト)に関して次のような所見が得られた。これらはヘキサゴナルH1相の温度安定性範囲の拡大及び立方V1相の低下を示し、これらは本発明の目的にとって有益であった。50:50wt% Brij(登録商標)56/水溶液混合物はH1相に対して最も広い温度安定性範囲(室温〜約95℃)を与えることを知見し、従って研究中のすべての実験にとって好ましかった。
【0062】
95℃以下の温度で液晶混合物にクエン酸を添加してもヘキサゴナル相の安定性に影響を与えないことを知見した。しかしながら、酢酸塩混合物のヘキサゴナル相は通常余り熱的に安定でなく、液晶相遷移も余りうまく規定されなかった。
【0063】
要するに、2つの界面活性剤とリチウム及びコバルトの硝酸塩及び酢酸塩の水溶液の液晶混合物のヘキサゴナル相の組成及び温度安定性範囲がうまく決定された。市販されている低価な界面活性剤(Brij(登録商標)56)を含有する溶液は、より高価な代替界面活性剤(OEE)よりも好ましいヘキサゴナル相のより広い温度安定性範囲を有していることが知見された。更に、テンプレート化混合物にクエン酸を添加してもヘキサゴナル相の安定性が影響されないことが知見された。
【0064】
酢酸塩混合物のヘキサゴナル相は通常余り熱的に安定でなく、液晶相遷移も余りうまく規定されなかった。よって、リチウム及びコバルトの硝酸塩を含む混合物は酢酸塩よりも好ましかった。
【0065】
市販のコバルト酸リチウムの特性決定
市販のLiCoO2は供給業者(Pred Materials,電池グレード)から黒色微粉として受け取り、XRD分析、TEM及びBET表面積分析により調べられた。XRD分析用サンプルは、まず乳棒及び乳鉢を用いて微粉に粉砕することにより調製した。TEMサンプルは、電子ビームが透過するのに十分な薄さの断片が得られるようにエタノール中で数時間超音波処理にかけた後粉末断片として調べた。BET測定は供給業者から受け取ったままの粉末に対して実施した。
【0066】
XRDは結晶性無機化合物を同定するための標準技術であり、しばしば化合物のフィンガープリントをとるように作用する。XRDパターンは特定化合物毎に独自であり、結晶面からの反射による回折ピークからなる。市販の電池グレードLiCoO2のXRDパターンを標準の純結晶LiCoO2と比較し、図2に示す。図2では、純結晶LiCoO2標準についてのXRDピークを縦線として表す。標準サンプルからのピークと市販LiCoO2粉末からのピークが(ピークの数及びビークの位置の点で)かなり整合していることが明らかとなり得る。このことから、市販LiCoO2が標準LiCoO2であることが確認される。
【0067】
TEMは、非常に小さい孔(<100nm)を有する高表面積材料中の孔径及び孔構造を同定するために電子ビームを使用する高分解能特性決定技術である。材料がナノスケールで多孔性でないならば、電子ビームは構造を透過し得ないのでTEM顕微鏡写真は暗い像として現れる。しかしながら、メソポーラスな材料の場合、TEM顕微鏡写真は暗い領域及び明るい領域として(明るい領域はビームが孔を透過するためであり、暗い領域は孔壁を透過しないためである)、孔構造の規則性及びビームの透過面に応じて黒い及び白い線または円を有する規則性パターンとして現れる。線間の測定距離は孔の直径または孔反復距離であり、線幅は孔壁の厚さである。図3a及び3bは、市販粉末由来の断片のTEM顕微鏡写真を示す。顕微鏡写真は特色がなくて暗く、メソスケールでの多孔性を欠くことを示している。
【0068】
BET表面積分析は、高多孔性材料の表面積を測定するために使用されている標準技術である。この方法は特定温度での窒素吸着及び脱着に依存している。通常、吸着または脱着された窒素の量は材料の表面積に直接関係している。BET表面積が高ければ、材料の表面積、よって多孔性が大きくなる。市販LiCoO2材料は1m2/g未満のBET値を有していた。
【0069】
コバルト酸リチウムの非テンプレート化ゾル−ゲル方法による合成
LiCoO2の非テンプレート化ゾル−ゲル合成を対照実験として実施した。紫〜ピンク色の外皮のあるゲルが形成されるまで、硝酸リチウム(Suprapur,Merck Chemicals International)、硝酸コバルト(Suprapur,Merck Chemicals International)及びクエン酸(Puriss Grade,Fluka Chemie AG)の水溶液を開放溶液中で連続攪拌しながら70〜80℃に加熱した。次いで、ゲルを75℃で数時間保持した後、放冷した。生成物を微粉に粉砕した後、650〜700℃の温度で16時間焼成した。暗灰色〜黒色粉末が形成された。市販のコバルト酸リチウムの特性決定に関して上に概説した方法を用いてXRD、BET及びTEMにより生成物の特性を決定した。
【0070】
非テンプレート化生成物について標準サンプルとのピークの整合を示したXRDトレースを記録し、非テンプレート化合成の生成物が市販LiCoO2に類似する単相構造を有していることを確認した。
【0071】
非テンプレート化合成により製造したLiCoO2材料のTEM顕微鏡写真を図4に示す。像は特色がなくて暗く(写真の上部領域)、メソスケールの多孔性を欠いていることを示している。
【0072】
非テンプレート化合成由来の粉末は約1m2/gのBET表面積を有していた。
【0073】
コバルト酸リチウムのテンプレート化ゾル−ゲル方法による合成
LiCoO2のテンプレート化ゾル−ゲル合成を本発明に従って実施した。硝酸リチウム、硝酸コバルト及びクエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、100mlのBrij(登録商標)56(Sigma Aldrich Inc.)界面活性剤の溶融溶液を40℃で添加した。次いで、ヘキサゴナル液晶相が形成されるまで混合物を攪拌した。次いで、粘性ペーストのコンシステンシーを有する液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを明紫色から暗紫色に変化するまで60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。次いで、冷却した固体/ゲルを炉に移して600〜650℃で16時間一定加熱した。液晶のヘキサゴナル相を破壊し、よって孔構造形態の崩壊を導く水分損失が観察されたので、テンプレート化ゾル−ゲル合成を密封容器中で実施した。
【0074】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、市販のコバルト酸リチウムの特性決定について上に概説した方法を用いてXRD、BET及びTEMにより特性を決定した。
【0075】
図5は、テンプレート化生成物についてのXRDトレースを示す。標準サンプルとピーク整合していることが明らかであり、このことからテンプレート化合成の生成物はLiCoO2標準及び市販LiCoO2と類似の構造を有していることが確認される。
【0076】
図6a及び6bは、テンプレート化合成により製造した粒子についてのTEM顕微鏡写真を示す。いずれの像も規則的に離間している暗い線及び明るい欄の領域またはクラスターを有するメソ多孔性の証拠を示している。線間隔(すなわち、孔径)及び反復距離は約3nmであり、線幅(すなわち、孔壁厚さ)は約1.5nmである。
【0077】
テンプレート化合成由来の粉末は15.8m2/gのBET表面積を有していた。
【0078】
上記実験を数回繰り返した。いずれの場合も、合成粉末のBET表面積は10mz/g以上であり(記録した最低値は13.0mz/g)、BET表面積は通常15m2/g以上であった。1例で、20.0m2/gのBET表面積を記録した。
【0079】
従って、一般的に、本発明の合成は15m2/g以上のBET表面積を有する粉末を製造したのに対して、非テンプレート化合成由来の粉末及び市販の材料はそれぞれ1m2/g及び1m2/g未満の値を有していた。このことは、テンプレート化合成由来の粉末の表面積が市販LiCoO2に比して少なくとも15倍大きくなることを示している。
【0080】
結論として、XRD分析からテンプレート化ゾル−ゲル合成により製造した粉末がLiCoO2であることが確認される。TEM測定値は、LiCoO2粒子が市販の電池グレードLiCoO2及び同等の非テンプレート化合成により製造した材料よりも少なくとも15倍高いBET表面積を有する高度のメソ多孔性であることを示している。
【0081】
LiNi0.33Co0.33Mn0.33O2のテンプレート化ゾル−ゲル方法による合成
LiNi0.33Co0.33Mn0.33O2のテンプレート化ゾル−ゲル合成を本発明に従って実施した。硝酸リチウム、硝酸コバルト、硝酸ニッケル、硝酸マンガン(1:0.33:0.33:0.33のモル比)及びクエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、40wt%の前駆体溶液を60wt%のCTABと混合した。混合物を攪拌し、安定なヘキサゴナルメソ相が形成された。次いで、液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを明紫色から濃い紫色に変化するまで60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。次いで、冷却した固体/ゲルを炉に移して900〜950℃で10時間一定加熱した。
【0082】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、XRD及びBETにより特性を決定した。
【0083】
XRDトレースは標準LiCoO2についてのXRDトレースと比較してピークの位置がより高い角度に移っていることを示した。このことから、コバルトが構造を変化させることなく部分的にマンガン及びニッケルで置換されている均質な結晶構造が形成されたことが示唆され、予想に一致した。
【0084】
テンプレート化合成由来の粉末は14.3m2/gのBET表面積を有し、これは市販のLiNi0.33Co0.33Mn0.33O2に比して10倍以上の表面積の増加に相当した。
【0085】
電気的性能試験
本発明のテンプレート化ゾル−ゲル方法を用いて製造したLiCoO2材料の電気的性能を確認し、その性能をベンチマークの市販LiCoO2及び非テンプレート化方法由来の材料と比較するために実験を実施した。
【0086】
上記した材料の各々を複合電極コーティングに作成し、リチウムホイル負極を有する実験室半電池において試験した。新しい材料のレート性能を確認するために、電位ステップ分析及び定電流サイクリングの技術を使用した。
【0087】
電位ステップ分析を用いて、メソポーラスな材料のレート性能及び電力特性を確認した。電位ステップ分析は、電圧を開放電圧(OCV)から4.2Vまで充電に相当する5時間で上昇させ、放電に相当する5時間で3Vまで低下させることからなる。材料が短時間、すなわち数秒でその容量の大部分を送出し得るならば、その材料は高レートと分類される。
【0088】
図7は、本発明の方法により製造したメソポーラスなLiCoO2を用いた電池について開放電圧(3.1V)から4.2Vへ及び3Vまでの電位ステップ後の電流の変化を経時的に示す。市販LiCoO2及び非テンプレート化ゾル−ゲル方法を用いて製造したLiCoO2についてもプロフィールを得た。
【0089】
市販の複合電極の場合20分後電流は直ぐに0に低下し、非テンプレート化電極の場合45分後に電流は0に減衰したことが判明した。しかしながら、テンプレート化電極の場合電流はたった2分以内に0に減衰した。
【0090】
重要なパラメーターは個々の電極より送出される電荷の量である。電位ステップ分析から得た電流/時間プロフィールを積分することにより、電池を4.2Vから3Vに低下させたときに得られる電荷の量を計算した。結果を表1にまとめる。ここから、36秒後メソポーラスな電極が示す容量は61mAh/gであるのに対して、非テンプレート化電極の場合8.4mAh/g、市販の電極の場合16mAh/gであることが明らかとなり得る。これらの結果から、約36秒でその容量の11%しか送出しない市販の電極に比してメソポーラスな電極は同じ時間でその容量の少なくとも70%を送出できるであろうことを示唆している。
【0091】
【表1】
【0092】
メソポーラスな電極材料のレート及び電力特性を確認するために定電流サイクリングも使用した。電気化学電池を定電流をC/5(5時間で充電)のレートで用いてOCVから4.2Vまで充電した後、異なるレートで3Vまで放電した。メソポーラスな複合電極の性能を再び非テンプレート化LiCoO2及び市販のLiCoO2と比較した。メソポーラスな電極、非テンプレート化電極及び市販の電極について異なる放電レートでの放電プロフィールを得、図8はメソポーラスな電極についてのプロフィールを示し、異なるレートでの容量はC/5で得た容量に対して正規化されている。図8中、最左のプロフィールは20Cプロフィールであり、最右のプロフィールはC/5プロフィールである。
【0093】
被覆したメソポーラスな材料のレート性能が非テンプレート化電極及び市販の電極よりも特に高いレートで優れていることに関して同じ所見が得られた。例えば、20Cのレートで(図9参照)、テンプレート化電極の容量保持は70%であるのに対して、非テンプレート化電極の場合16%、市販の電極の場合0%であった。これらの結果は、テンプレート化電極の場合36秒で最高の電荷が送出された電位ステップ実験から得た結果と一致している。
【0094】
リン酸鉄リチウムのテンプレート化ゾル−ゲル方法による合成
リン酸鉄リチウム(LiFePO4)のテンプレート化ゾル−ゲル合成を本発明に従って実施した。0.5M リン酸二水素リチウム(Sigma Aldrich Inc.)、0.5M クエン酸第二鉄(Sigma Aldrich)及び1M クエン酸の水溶液を開放容器中で連続攪拌しながら55〜60℃に加熱した。溶液を更に10時間攪拌して均質混合物を得た後、100mlのBrij(登録商標)56(Sigma Aldrich Inc.)界面活性剤の溶融溶液を40℃で添加した。次いで、ヘキサゴナル液晶相が形成されるまで混合物を攪拌した。次いで、粘性ペーストのコンシステンシーを有する液晶相混合物を密封サンプルボトルに移し、60℃に加熱した。ここで、混合物は粘性液体であり、これを60℃で更に24〜36時間攪拌した。次いで、加熱を数時間続け、溶液をアルミナるつぼに移し、そこで放冷した。
【0095】
次いで、冷却した固体/ゲルを炉に移してアルゴン雰囲気下約700℃で16時間一定加熱した。液晶のヘキサゴナル相を破壊し、よって孔構造形態の崩壊を導く水分損失が観察されたので、テンプレート化ゾル−ゲル合成を密封容器中で実施した。
【0096】
生じた暗灰色〜黒色固体を粉砕して微粉とした後、XRDにより特性を決定した。XRDトレース(図10)から、リン酸鉄リチウムの純度が確認された。LiFePO4生成物のメソ多孔性を最適化するために焼成温度を450〜700℃の範囲で変更し得ることが知見された。
【特許請求の範囲】
【請求項1】
(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな生成物を残すステップを含むメソポーラスなリチウム遷移金属化合物の合成方法。
【請求項2】
金属化合物は酸化物、リン酸塩、ホウ酸塩またはケイ酸塩である請求項1に記載の方法。
【請求項3】
金属化合物はリン酸塩、ホウ酸塩またはケイ酸塩であり、ステップ(i)で追加のリン酸塩、ホウ酸塩またはケイ酸塩試薬を反応混合物に添加する請求項2に記載の方法。
【請求項4】
ステップ(ii)でリチウム塩及び1つ以上の遷移金属塩は高分子金属リン酸素網状構造を形成して、メソポーラスなリチウム遷移金属リン酸塩生成物を形成する請求項1〜3のいずれか1項に記載の方法。
【請求項5】
リチウム塩をメソポーラスなLiFePO4を製造するような酸性条件下で鉄塩及びリン酸塩と反応させる請求項4に記載の方法。
【請求項6】
リン酸二水素リチウムを鉄塩と反応させて、メソポーラスなLiFePO4を製造する請求項4に記載の方法。
【請求項7】
ステップ(ii)でリチウム塩及び1つ以上の遷移金属塩は高分子金属酸化物網状構造を形成して、メソポーラスなリチウム遷移金属酸化物生成物を形成する請求項1または2に記載の方法。
【請求項8】
ステップ(i)でリチウム塩をコバルト塩及び1つ以上の追加遷移金属塩と反応させて、化合物コバルト酸リチウムを形成する請求項7に記載の方法。
【請求項9】
1つ以上の更なる塩はNi、Mn及びAlからなる群から選択される遷移金属を含む請求項8に記載の方法。
【請求項10】
メソポーラスな酸化物生成物はLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される化合物コバルト酸リチウムである請求項8または9に記載の方法。
【請求項11】
ステップ(i)でリチウム塩をコバルト塩と反応させて、メソポーラスなLiCoO2を形成する請求項7に記載の方法。
【請求項12】
リチウム塩は硝酸リチウムであり、コバルト塩は硝酸コバルトである請求項11に記載の方法。
【請求項13】
界面活性剤を焼成することにより除去し、焼成温度は400〜700℃の範囲である請求項11または12に記載の方法。
【請求項14】
焼成温度は400〜600℃の範囲である請求項13に記載の方法。
【請求項15】
リチウム塩をコバルト塩と反応させてメソポーラスなLiCoO2生成物を製造し、ステップ(ii)で反応物を50〜60℃、より好ましくは55〜60℃の範囲で加熱する請求項11〜14のいずれか1項に記載の方法。
【請求項16】
界面活性剤を焼成により除去する請求項1〜15のいずれかに記載の方法。
【請求項17】
液晶相はヘキサゴナル相である請求項1〜16のいずれかに記載の方法。
【請求項18】
界面活性剤はポリオキシエチレンセチルエーテル、オクタエチレンエーテル及びCTABからなる群から選択される請求項1〜17のいずれかに記載の方法。
【請求項19】
反応混合物中の界面活性剤濃度は40〜60wt%、好ましくは45〜55wt%、より好ましくは50wt%の範囲である請求項1〜18のいずれかに記載の方法。
【請求項20】
反応を水性溶媒中で行う請求項1〜19のいずれかに記載の方法。
【請求項21】
ステップ(i)及び(ii)の反応条件を液晶相の不安定化を防止するようにコントロールする請求項1〜20のいずれかに記載の方法。
【請求項22】
ステップ(i)及び(ii)で液晶相の不安定化を防止するように水分損失をコントロールする請求項1〜21のいずれかに記載の方法。
【請求項23】
水分損失を実質的に防止する請求項22に記載の方法。
【請求項24】
反応混合物は更にキレート化剤を含む請求項1〜23のいずれかに記載の方法。
【請求項25】
キレート化剤はクエン酸及びプロピオン酸から選択される請求項24に記載の方法。
【請求項26】
リチウム塩は硝酸リチウムである請求項1〜25のいずれかに記載の方法。
【請求項27】
1つ以上の遷移金属塩は硝酸塩である請求項1〜26のいずれかに記載の方法。
【請求項28】
請求項1〜27のいずれかに記載の方法により得られ得るメソポーラスなリチウム遷移金属化合物。
【請求項29】
請求項1または2、或いは請求項7〜27のいずれか1項に記載の方法により得られ得るメソポーラスなコバルト酸リチウム、すなわち化合物コバルト酸リチウム。
【請求項30】
LiCoO2、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される請求項29に記載のメソポーラスなコバルト酸リチウム。
【請求項31】
10m2/g以上、好ましくは15m2/g以上のBET表面積を有するメソポーラスなLiCoO2。
【請求項32】
請求項28〜31のいずれか1項に記載の材料を含む電極。
【請求項33】
請求項28〜31のいずれか1項に記載の材料を含む電池。
【請求項34】
請求項28〜31のいずれか1項に記載の材料を含む二次リチウム電池。
【請求項35】
請求項1〜6、或いは請求項16〜27のいずれかに記載の方法により製造されるメソポーラスなLiFePO4。
【請求項36】
実質的に先に記載されている方法または生成物。
【請求項37】
実質的に先に記載されている新規な特徴または特徴の組合せ。
【請求項1】
(i)反応混合物中に液晶相を形成するのに十分な量の界面活性剤の存在下でリチウム塩を1つ以上の遷移金属塩と反応させるステップ、(ii)反応混合物を加熱してゾル−ゲルを形成するステップ、及び(iii)界面活性剤を除去して、メソポーラスな生成物を残すステップを含むメソポーラスなリチウム遷移金属化合物の合成方法。
【請求項2】
金属化合物は酸化物、リン酸塩、ホウ酸塩またはケイ酸塩である請求項1に記載の方法。
【請求項3】
金属化合物はリン酸塩、ホウ酸塩またはケイ酸塩であり、ステップ(i)で追加のリン酸塩、ホウ酸塩またはケイ酸塩試薬を反応混合物に添加する請求項2に記載の方法。
【請求項4】
ステップ(ii)でリチウム塩及び1つ以上の遷移金属塩は高分子金属リン酸素網状構造を形成して、メソポーラスなリチウム遷移金属リン酸塩生成物を形成する請求項1〜3のいずれか1項に記載の方法。
【請求項5】
リチウム塩をメソポーラスなLiFePO4を製造するような酸性条件下で鉄塩及びリン酸塩と反応させる請求項4に記載の方法。
【請求項6】
リン酸二水素リチウムを鉄塩と反応させて、メソポーラスなLiFePO4を製造する請求項4に記載の方法。
【請求項7】
ステップ(ii)でリチウム塩及び1つ以上の遷移金属塩は高分子金属酸化物網状構造を形成して、メソポーラスなリチウム遷移金属酸化物生成物を形成する請求項1または2に記載の方法。
【請求項8】
ステップ(i)でリチウム塩をコバルト塩及び1つ以上の追加遷移金属塩と反応させて、化合物コバルト酸リチウムを形成する請求項7に記載の方法。
【請求項9】
1つ以上の更なる塩はNi、Mn及びAlからなる群から選択される遷移金属を含む請求項8に記載の方法。
【請求項10】
メソポーラスな酸化物生成物はLiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される化合物コバルト酸リチウムである請求項8または9に記載の方法。
【請求項11】
ステップ(i)でリチウム塩をコバルト塩と反応させて、メソポーラスなLiCoO2を形成する請求項7に記載の方法。
【請求項12】
リチウム塩は硝酸リチウムであり、コバルト塩は硝酸コバルトである請求項11に記載の方法。
【請求項13】
界面活性剤を焼成することにより除去し、焼成温度は400〜700℃の範囲である請求項11または12に記載の方法。
【請求項14】
焼成温度は400〜600℃の範囲である請求項13に記載の方法。
【請求項15】
リチウム塩をコバルト塩と反応させてメソポーラスなLiCoO2生成物を製造し、ステップ(ii)で反応物を50〜60℃、より好ましくは55〜60℃の範囲で加熱する請求項11〜14のいずれか1項に記載の方法。
【請求項16】
界面活性剤を焼成により除去する請求項1〜15のいずれかに記載の方法。
【請求項17】
液晶相はヘキサゴナル相である請求項1〜16のいずれかに記載の方法。
【請求項18】
界面活性剤はポリオキシエチレンセチルエーテル、オクタエチレンエーテル及びCTABからなる群から選択される請求項1〜17のいずれかに記載の方法。
【請求項19】
反応混合物中の界面活性剤濃度は40〜60wt%、好ましくは45〜55wt%、より好ましくは50wt%の範囲である請求項1〜18のいずれかに記載の方法。
【請求項20】
反応を水性溶媒中で行う請求項1〜19のいずれかに記載の方法。
【請求項21】
ステップ(i)及び(ii)の反応条件を液晶相の不安定化を防止するようにコントロールする請求項1〜20のいずれかに記載の方法。
【請求項22】
ステップ(i)及び(ii)で液晶相の不安定化を防止するように水分損失をコントロールする請求項1〜21のいずれかに記載の方法。
【請求項23】
水分損失を実質的に防止する請求項22に記載の方法。
【請求項24】
反応混合物は更にキレート化剤を含む請求項1〜23のいずれかに記載の方法。
【請求項25】
キレート化剤はクエン酸及びプロピオン酸から選択される請求項24に記載の方法。
【請求項26】
リチウム塩は硝酸リチウムである請求項1〜25のいずれかに記載の方法。
【請求項27】
1つ以上の遷移金属塩は硝酸塩である請求項1〜26のいずれかに記載の方法。
【請求項28】
請求項1〜27のいずれかに記載の方法により得られ得るメソポーラスなリチウム遷移金属化合物。
【請求項29】
請求項1または2、或いは請求項7〜27のいずれか1項に記載の方法により得られ得るメソポーラスなコバルト酸リチウム、すなわち化合物コバルト酸リチウム。
【請求項30】
LiCoO2、LiCoMnO2、LiCoNiO、LiCoAlO2及びLiNi0.33Co0.33Mn0.33O2からなる群から選択される請求項29に記載のメソポーラスなコバルト酸リチウム。
【請求項31】
10m2/g以上、好ましくは15m2/g以上のBET表面積を有するメソポーラスなLiCoO2。
【請求項32】
請求項28〜31のいずれか1項に記載の材料を含む電極。
【請求項33】
請求項28〜31のいずれか1項に記載の材料を含む電池。
【請求項34】
請求項28〜31のいずれか1項に記載の材料を含む二次リチウム電池。
【請求項35】
請求項1〜6、或いは請求項16〜27のいずれかに記載の方法により製造されるメソポーラスなLiFePO4。
【請求項36】
実質的に先に記載されている方法または生成物。
【請求項37】
実質的に先に記載されている新規な特徴または特徴の組合せ。
【図1】

【図2】
【図3a】

【図3b】

【図4】

【図5】

【図6a】

【図6b】

【図7】
【図8】
【図9】
【図10】
【図2】
【図3a】
【図3b】
【図4】
【図5】
【図6a】
【図6b】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2012−506354(P2012−506354A)
【公表日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願番号】特願2011−531556(P2011−531556)
【出願日】平成21年10月19日(2009.10.19)
【国際出願番号】PCT/GB2009/002485
【国際公開番号】WO2010/046629
【国際公開日】平成22年4月29日(2010.4.29)
【出願人】(501352882)キネテイツク・リミテツド (93)
【Fターム(参考)】
【公表日】平成24年3月15日(2012.3.15)
【国際特許分類】
【出願日】平成21年10月19日(2009.10.19)
【国際出願番号】PCT/GB2009/002485
【国際公開番号】WO2010/046629
【国際公開日】平成22年4月29日(2010.4.29)
【出願人】(501352882)キネテイツク・リミテツド (93)
【Fターム(参考)】
[ Back to top ]