
非水性液体組成物中の検体の溶出速度測定方法
本発明は非水性液体組成物から水性媒体への検体の移動を特徴づける方法を提供し、特に徐放性製剤からの薬物溶出のインビトロ測定方法を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は非水性溶液から水性媒体への検体の移動を特徴づける方法に関し、具体的には、徐放性製剤からの薬物溶出のインビトロ測定方法に関する。
【背景技術】
【0002】
医薬組成物の製剤化の1つの重要な態様は、薬物の薬物動態学的挙動である。薬物の物理状態(即ち、気体、液体、固体)、結晶形、粒子径、剤形、および用いる賦形剤などの種々の要素によって、体内での薬物の時間に依存した放出は大幅に変化しうる。たとえ、同一薬物が同一剤形で提供された場合であっても、ロット間変動は起こりうる。
【0003】
規制当局の認可を得るために、薬物動態学的挙動は、多くの場合、動物またはヒトに薬物を投与し、投与後のある複数の時点で、例えば血中における薬物またはその代謝物の量を測定することにより測定される。この方法は時間と費用がかかり、一般的には、製造工程中で医薬品の質を制御するためには用いられていない。インビトロ試験において、薬物のインビボでの薬物動態学的挙動を評価するための多くの方法が考案されてきた。これらの試験の一部は標準化され、米国薬局方(USP)などに記載されている。一般的に用いられるUSP法は、バスケット法(USP第1法)およびパドル法(USP第2法)である。これらの標準化法に加えて、特定の個別用途のための多くの方法が記載されてきた。多くの溶出方法に関する概説は、例えば、G. K. Shiu, Drug Information Journal, 30, 1045-1054, (1996)などにおいて見出すことができる。
【0004】
Andonaeguiら(Drug
Development and Industrial Pharmacy, 25 (11), 1199- 1203 (1999))は、絶食条件および高脂肪食条件で投与した徐放性テオフィリンマトリックス錠の、インビボ性能を予測するためのインビトロ法を記載している。3種類の徐放性マトリックス錠中のテオフィリンの溶出挙動が研究された。高脂肪食に関するインビトロ/インビボ相関性を高めるために、USPパドル試験における溶出試験の前に落花生油と混合することにより錠剤が前処理された。
【0005】
日本特許出願JP05−249097は、徐放性錠剤のインビボ放出を予測するための溶出試験を記載している。該錠剤にパドル試験を行い、取り出し、オイルおよび脂質で処理し、次いで水性溶出溶媒中のビーズと共にパドル装置に戻すかあるいはバスケット中に沈める。この方法は、徐放性錠剤の放出制御メカニズムによる影響を受けないで、生体内の血漿中薬物濃度を予測するといわれている。
【0006】
米国特許第6132751号は、眼科用PBS乳濁液組成物中の薬物の溶解性の評価を開示している。
【0007】
微粒子ドラッグデリバリーシステムのための種々のインビトロ溶出法が、Contiら Drug Development and Industrial Pharmacy, 21(10), 1233-1233
(1995)により比較されている。撹拌速度、イオン強度および界面活性剤の存在の影響が研究されている。
【0008】
油状薬物製剤を試験するための溶出方法もまた記載されている。Takahashiら(Chem. Pharm. Bull., 42 (8), 1672-1675, (1994))は、パドル法と回転透析セル法を比較している。回転透析セル法の変法において、オクタノールを外相として用い、一方で内相として酸性溶液を用いた。
【0009】
Machidaら(Chem.
Pharm. Bull., 34 (6), 2637-2641, (1986))は、油性薬物製剤の溶出特性の測定において遭遇する問題を克服するための1つの試みを記載している。水性溶出溶媒の表面を撹拌するための追加の補助翼を用いる、日本薬局法第2法のパドル法の変法を用いることを彼らは提案している。さらに、撹拌を改善するためにビーズを加え、かつ水性溶出溶媒として胆汁酸塩溶液を用いた。
【0010】
薬物が非水性基剤中に溶解し、分散し、懸濁し、または別な風に調製された非水医薬組成物の薬物動態学的挙動は、従来の技術を用いて確実に予測することは困難である。インビトロ測定の精度および信頼度は低いことが多く、インビトロ測定の結果は、インビボでの薬物の性質といつも相関するとは限らない。従って、本発明の目的の1つは、非水性液体組成物中の検体の溶出を特徴づけるための信頼できる方法を提供することである。本発明の他の目的は、低溶解性を有する検体の溶出を特徴づけるための方法を提供することである。本発明の他の目的は、ゆっくりしか溶けない検体の溶出を特徴づけるための方法を提供することである。本発明の他の目的は、インプロセスアッセイとして製造中に利用できる、検体の溶出を特徴づけるための迅速な方法を提供することである。本発明の他の目的は、本出願の明細書および特許請求の範囲を読むことにより容易に明らかとなるであろう。
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は非水性液体組成物中の検体の溶出を特徴づける方法に関し、
(a)検体および非水性基剤を含む非水性液体組成物を用意する段階;
(b)非水性液体組成物を水性溶出溶媒と混合する段階;
(c)非水性液体組成物と水性溶出溶媒を撹拌して乳濁液を形成する段階;および
(d)水性溶出溶媒中の検体の量を測定する段階;
を含む方法に関する。
発明の詳細な説明
【0012】
本発明は、非水性液体組成物中の検体のインビトロ溶出を特徴づける、信頼できる方法を提供する。本方法は、好ましくは、医薬組成物からの医薬活性成分の溶出を定量するために用いられるが、本方法はさらに、例えばオイルから環境への汚染物質の溶出速度の測定、防錆剤などの活性成分の油性基剤からの減少速度の測定または殺虫剤もしくは肥料からの成分の放出速度などの、分析化学の他の分野においても用いることができる。
【0013】
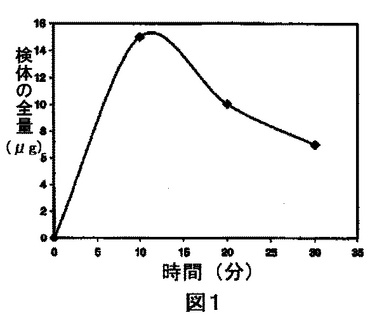
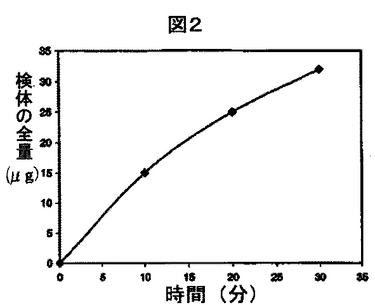
溶出は、種々の方法により特徴づけかつ定量することができる。例えば、水性溶出溶媒中の検体の量が、所定の時間で1回のみ測定される場合もある。例えば、3μgの検体が30分後に溶解したと測定される場合、30分で3μg溶解したと溶出が特徴づけられる。水性溶出溶媒中の検体の量を2回以上測定する場合、溶出はいくつかの異なる方法で説明することができるが、これらの方法は当該技術分野において公知である。一般的な方法の1つは、二次元グラフ中にデータをプロットすることである。x軸は時間軸を表す。y軸はn回目と(n−1)回目の水性溶出溶媒の分析の間に溶解した検体の量を表す。他の一般的な方法の1つは、x軸がここでも時間軸を表す、二次元グラフ中にデータをプロットすることである。y軸は、測定の最初と水性溶出溶媒のn番目の分析の間に溶解した検体の総量を表す。もちろん、上記の二次元グラフ以外にも、同一の情報を表または他の適切な形で表すことができる。以下の一連の実験は、例として使用できる:非水性液体組成物を調べ、10分(n=1)、20分(n=2)、および30分(n=3)の時点で、溶解した検体の量を測定する。総量で、10分後に15μgの検体が溶解し、20分後に25μgの検体が溶解し、そして30分後に32μgの検体が溶解した。第1の場合では、図1に示すようなプロットが得られるであろうし、また第2の場合では、プロットは図2に示すようなものになるであろう。
【0014】
本明細書においては、「非水性液体組成物」という用語は、接触温度で液体でありかつ検体および非水性基剤を含む任意の組成物のことを言う。検体および非水性基剤の混合物は任意の形であることもでき、例えば、溶液、乳濁液または懸濁液であることができる。検体が非水性基剤に懸濁化されている場合、検体の粒子径は、一般的に約50nm〜約200ミクロンの範囲であり、好ましくは約100nm〜約200ミクロンの範囲である。非水性液体組成物中の検体の濃度は特に限定されるわけではない。例えばこれらの濃度は、約0.00001mg/mL〜約5,000mg/mLの間で変化し、好ましくは約0.01mg/mL〜約1,000mg/mLの間で変化することができる。
【0015】
非水性液体組成物は、好ましくは医薬組成物である。本発明の方法において、医薬組成物は一般的に、非経口、経口、舌下、経鼻、気管支内、肺、乳房内、直腸、膣、眼球、または局所投与に適した液体である。しかしながら、医薬組成物がカプセル剤に含まれている医薬組成物中の検体の溶出速度を測定することもまた可能である。この場合、カプセルの殻は水性溶出溶媒と接触して崩壊し、その内容物を放出する。
【0016】
溶出が特徴づけられる検体は、非水性液体組成物の任意の成分であることもできる。検体の例は、汚染物質、活性成分、または不活性成分であるが、これに限定されない。医薬組成物の場合、検体は一般的に医薬活性成分であるが、医薬組成物の賦形剤またはその他の成分であることもできる。本発明の方法は、単一の検体の測定に限定されない。所望であれば、2以上の検体を測定することができる。本発明の方法は、何らかの特定の物理的または化学的性質を有する検体の測定に、限定されない。検体が、本方法のために選択された水性溶出溶媒に少なくとも一部が溶解する限りは、ほとんど全ての検体(有機であれ、無機であれ)を本発明の方法により測定することができる。本発明の方法を用いて測定できる検体の例は、以下の例示的、非限定的なクラス:ACE阻害剤;α−アドレナリンアゴニスト;β−アドレナリンアゴニスト;α−アドレナリン遮断薬;β−アドレナリン遮断薬(β遮断薬);抗酒薬;アルドースレダクターゼ阻害剤;アルドステロンアンタゴニスト;アミノ酸;タンパク同化剤;鎮痛剤(麻薬性および非麻薬性の両方);麻酔剤;摂食障害;制酸剤;駆虫薬;抗にきび剤;抗アレルギー剤;抗男性ホルモン;抗狭心症薬;抗不安薬(antianxiety);抗不整脈薬;抗喘息剤;抗菌剤および抗生物質;脱毛防止薬およびはげ予防薬;抗アメーバ剤;抗体;抗コリン剤;抗凝血物質および血液希釈剤;大腸炎治療剤;鎮痙薬薬;膀胱炎治療剤;抗うつ薬;抗糖尿病薬;下痢止め;抗利尿薬;解毒剤;制吐剤;抗エストロゲン剤;抗膨満薬;抗真菌剤;抗原;緑内障治療薬;抗ヒスタミン剤;過剰行動治療剤;抗高リポタンパク血症剤;降圧剤;甲状腺機能亢進症治療薬;低血圧治療剤;甲状腺機能低下症治療薬;抗感染薬;抗炎症剤(ステロイド性および非ステロイド性の両方);抗マラリア薬;偏頭痛治療薬;抗腫瘍剤;抗肥満薬;抗パーキンソン薬およびジスキネジア治療剤;肺炎治療剤;抗原虫剤;かゆみ止め;抗乾癬剤;抗精神病薬;解熱剤;リウマチ治療剤;分泌抑制薬;抗ショック剤;抗痙攣薬;抗血栓剤;抗癌剤;鎮咳剤;抗潰瘍剤;抗ウイルス剤;抗不安薬(anxiolytics);殺菌素;骨密度増加剤;気管支拡張剤;カルシウムチャンネル遮断薬;炭酸脱水酵素阻害剤;強心薬および強心剤;化学療法剤;胆汁分泌促進物質;コリン作用薬;慢性疲労症候群治療薬;中枢興奮薬;凝固剤;避妊薬;嚢胞性線維症薬;充血除去剤;利尿薬;ドーパミン受容体アゴニスト;ドーパミン受容体アンタゴニスト;酵素;エストロゲン;去痰薬;胃過敏治療剤;グルココルチコイド;止血薬;HMG−CoAレダクターゼ阻害剤;ホルモン;睡眠剤;免疫調節剤;免疫抑制剤;下剤;口腔および歯周疾患用医薬;縮瞳薬;モノアミン酸化酵素阻害剤;粘液溶解剤;多発性硬化症薬;筋弛緩剤;散瞳薬;麻薬拮抗薬;NMDA受容体アンタゴニスト;オリゴヌクレオチド;眼科用薬;子宮収縮薬;ペプチド、ポリペプチドおよびタンパク質;多糖類;プロゲストゲン;プロスタグランジン;プロテアーゼ阻害剤;呼吸促進剤;鎮静剤;セロトニン取り込み阻害剤;アンドロゲンを含む性ホルモン;禁煙薬;平滑筋弛緩薬;平滑筋刺激薬;血栓溶解剤;鎮静剤;尿酸性化剤;尿失禁薬;血管拡張剤;血管保護剤;およびそれらの組み合わせ;を含む。特定の薬剤化合物に対して行った本明細書のいずれの引用も、その化合物の互変異性体、立体異性体、エナンチオマー、塩およびプロドラッグを含み、かつその薬物のいずれか1つの固体状態形に限定されないことが理解されなければならない。
【0017】
本発明の方法は、特に第三世代セファロスポリンなどのセファロスポリンの溶出速度の測定に適している。セファロスポリンの例には、セフチオフル、セフェピム、セフィキシム、セフォペラゾン、セフォタキシム、セフポドキシム、セフタジジム、セフチゾキシム、セフトリアキソン、モクサラクタム(moxalactam)、これらの薬学的に許容される塩および誘導体があるが、これらに限定されない。特に好ましいセファロスポリンはセフチオフル、薬学的に許容される塩およびそれらの誘導体である。
【0018】
セフチオフル(ceftiofur)は、現在、ナクセル(登録商標)(Naxel)およびエクセネル(登録商標)(Excenel)という販売名でファルマシア社より市販されている。セフチオフルのもう1つの好ましい形態は、セフチオフル結晶性遊離酸(ceftiofur
crystalline free acid)(CCFA)である。この化合物およびその医薬製剤は、米国特許第5,721,359号に記載されており、その全体を本願に引用して援用する。
【0019】
非水性液体組成物は、接触温度で一般的に液体であり、かつ水と混和性、部分混和性、あるいは非混和性でありうる、非水性基剤もまた含む。非水性基剤は、水素化または非水素化、飽和、不飽和、またはポリ不飽和の、脂質(脂肪、オイル、ワックス、ステロール、およびグリセリド)であることができ、かつ当該技術分野において公知の技術によりさらに修飾することができる。非水性基剤は、好ましくは、天然または合成の脂肪、ワックス、またはオイルからなる群から選択され、さらに好ましくは、非水性基剤は天然または合成のオイルである。オイルという用語は、植物、動物、鉱物、および海洋起源由来のものを含む、トリグリセリド脂肪およびオイルを含む。非水性基剤として適切な合成オイルの具体例は、6〜24炭素原子を有する飽和または不飽和脂肪酸のトリグリセリド、またはプロピレングリコールジエステルを含む。かかるカルボン酸は、例えば、ヘキサン酸、オクタン酸(カプリル酸)、ノナン酸(ペラルゴン酸)、デカン酸(カプリン酸)、ウンデカン酸、ラウリン酸、トリデカン酸、テトラデカン酸(ミリスチン酸)、ペンタデカン酸、ヘキサデカン酸(パルミチン酸)、ヘプタデカン酸、オクタデカン酸(ステアリン酸)、ノナデカン酸、エイコサン酸、ヘンエイコサン酸、ドコサン酸およびリグノセリン酸などの、6〜24炭素原子を有するカルボン酸を含む。不飽和カルボン酸の例は、オレイン酸、リノール酸およびリノレン酸などを含む。トリグリセリドビヒクルは、脂肪酸のモノ−、ジ−、またはトリグリセリルエステル、またはグリセロールの少なくとも1つの分子が異なる炭素原子長の脂肪酸でエステル化された混合グリセリドおよび/またはプロピレングリコールジエステルを含むことができると考えられている。以下:トリオレイン、トリリノレインおよびトリリノレニンを含むトリ不飽和エステル;トリパルミチン、トリステアリン、およびトリデカノインを含む飽和トリ飽和エステル;はトリグリセリルエステルの例である。トリグリセリルエステルの他の例は、ジ飽和モノ不飽和タイプ:1,2−ジパルミトイル−3−オレオイル−rac−グリセロールまたは1,3−ジパルミトイル−2−オレオイル−rac−グリセロールなどのオレオジ飽和エステル;1,3−ジパルミトイル−2−リノレオイル−rac−グリセロールなどのリノレオジ飽和エステル;を含む。トリグリセリドの他の例は、1−パルミトイル−2−オレオイル−3−リノレオイル−rac−グリセロールおよび1−リノレオイル−2−オレオイル−3−ステアロイル−rac−グリセロールを含むモノ飽和オレオリノレイン酸エステルならびに1,2−ジリノレオイル−3−パルミトイル−rac−グリセロールを含むモノ飽和ジリノレインエステルなどの、モノ飽和ジ不飽和エステルである。
【0020】
ジグリセリルエステルの例は:1,2−ジオレインまたは1,3−ジオレイン、1,2−ジリノレインまたは1,3−ジリノレインおよび1,2−ジリノレニンまたは1,3−ジリノレニンなどのジ不飽和エステル;1,2−ジパルミチンまたは1,3−ジパルミチン、1,2−ジステアリンまたは1,3−ジステアリン、および1,2−ジデカノインまたは1,3−ジデカノインなどの飽和ジ飽和エステル;l−パルミトイル−2−オレオイル−グリセロールまたは1−オレオイル−2−パルミトイル−グリセロール、l−パルミトイル−2−リノレオイル−グリセロールまたは1−リノレオイル−2−パルミトイル−グリセロールなどの飽和不飽和ジグリセリルエステル;を含む。
【0021】
モノグリセリルエステルの例は:1−オレインまたは2−オレイン、1−リノレインまたは2−リノレインおよび1−リノレニンまたは2−リノレニンなどの不飽和エステル;1−パルミチンまたは2−パルミチン、1−ステアリンまたは2−ステアリン、および1−デカノインまたは2−デカノインなどの飽和エステル;を含む。
【0022】
ポリエチレングリコール(PEG)ジエステルの例は:1,2−ジオレインまたは1,3−ジオレイン、1,2−ジリノレインまたは1,3−ジリノレインおよび1,2−ジリノレニンまたは1,3−ジリノレニンなどのジ不飽和エステル;1,2−ジパルミチンまたは1,3−ジパルミチン、1,2−ジステアリンまたは1,3−ジステアリン、および1,2−ジデカノインまたは1,3−ジデカノインなどの飽和ジ飽和エステル、を含む。飽和不飽和ジグリセリルエステルからのPEGジエステルの他の例は、l−パルミトイル−2−オレオイル−グリセロールまたは1−オレオイル−2−パルミトイル−グリセロール、1−パルミトイル−2−リノレオイル−グリセロールまたは1−リノレオイル−2−パルミトイル−グリセロールを含む。
【0023】
天然オイルの具体例は、キャノーラ油、ヤシ油、コーン油、落花生油、ゴマ油、オリーブ油、パーム油、ベニバナ油、大豆油、綿実油、菜種油、ヒマワリ油およびそれらの混合物である。これらの中で綿実油が好ましい。
【0024】
当該技術分野において公知な方法により非水性基剤を改変することができる。例えば過酸化不飽和オイル基剤を用いる実施態様において、改変基剤は約0.1〜約600の過酸化物価を有することができ、一部の実施態様において、約10、約20、約40、または約80あるいはその間の任意の値を有することができる。本明細書においては、過酸化物価は、1000グラムのオイルサンプルに対するミリ当量(mEq)の過酸化物で表す。
【0025】
前記の成分以外に、非水性液体組成物は追加の化合物を含むこともできる。例えば、非水性液体組成物が医薬組成物の場合、該組成物は任意の薬学的に許容される成分を含むことができる。典型的な追加の成分は、例えば、医薬活性成分、賦形剤、添加剤、懸濁化剤、防腐剤、湿潤剤、増粘剤、緩衝液および凝集剤である。ゴム類(例えばアカシアゴム、カラギナン、アルギン酸ナトリウムおよびトラガント)、セルロース誘導体(例えばカルボキシメチルセルロースナトリウム、微結晶セルロース、およびヒドロキシエチルセルロース)、および粘土(例えばベントナイトおよびコロイド状アルミニウムマグネシウム)などの懸濁化剤を含むことができる。メチルおよびプロピルパラベン、ベンジルアルコール、クロロブタノールならびにチメロサールなどの防腐剤を加えることができる。陰イオン界面活性剤(例えばドキュセートナトリウムおよびラウリル硫酸ナトリウム)、非イオン性界面活性剤(例えばポリソルベート、ポリオキサマー、オクトキシノール−9)、および陽イオン界面活性剤(例えば臭化トリメチルテトラデシルアンモニウム、塩化ベンザルコニウム、塩化ベンゼトニウム、ミリスチルγ塩化ピコリニウム)を使用することができる。ゼラチン、天然ゴム類およびセルロース誘導体(懸濁化剤として上に記したものなど)などの増粘剤を加えることができる。クエン酸およびリン酸緩衝化剤などの緩衝液、および塩化ナトリウムおよびマンニトールなどの浸透物質を含むことができる。経口投与される医薬組成物のためには、風味剤、甘味料(例えばマンニトール、ショ糖、ソルビトールおよびデキストロース)、着色剤ならびに芳香剤を使用することができる。医薬組成物において、モノオレイン酸ソルビタン(Span 80(登録商標)としてシグマアルドリッチ社から入手可能)などの賦形剤およびホスファチジルコリン(ホスホリポン(Phospholipon)90HとしてAmerican Lecithin Companyから入手可能)を使用することができる。
【0026】
本発明の水性溶出溶媒は、当該技術分野において公知なすべての水性溶出溶媒であって良い。一般的に用いられる溶出溶媒は、水、塩酸(例えば約0.001M〜約0.1MHC1の範囲の濃度を有する)、ペプシン含有あるいは非含有の人工胃液(simulated gastric fluid)、種々の緩衝液(グリシン、クエン酸、酢酸、リン酸、およびホウ酸緩衝液)、酵素含有あるいは非含有の人工腸液(simulated intestinal fluid)(例えばパンクレアチン含有あるいは非含有の0.05Mリン酸緩衝液(pH7.5))、界面活性剤を含む水、界面活性剤を含む緩衝液および水性アルコール性溶液(例えば補助溶剤の役割を果たす、一般的に5以下の炭素を含む、水に溶ける低分子量アルコール)である。これらの種々の要素を、所定の検体の溶解条件を代えるために調製することができる。実験を繰返すことにより、所望の範囲内にインビトロ薬物放出速度を調節することを実験者に可能にさせる、最適の組成物を経験的に導き出すことが可能である。溶解度条件の調節はまた、インビボで異なる振舞いをするロット間の差異をインビトロで見分けることを実験者に可能にすることもできる。
【0027】
本発明の特定の実施態様において、所望により、界面活性剤を含む緩衝液は水性溶出溶媒として用いられる。緩衝液の種類は特に限定されないが、特定の系次第により選択されなければならない。緩衝液は、薬物放出媒体中の検体の溶解性を調節し、薬物放出挙動を最適化し、そして重要なサンプル間の識別の程度を最適化するように選択することができる。緩衝液の具体例は、pH2〜3の0.05Mグリシン緩衝液、pH3の0.05Mクエン酸緩衝液、pH4〜5の0.05M酢酸緩衝液、通常の生理食塩水中のpH5.5の0.05M酢酸緩衝液、pH6〜8の0.05Mリン酸緩衝液、カリウムを含まないpH6.8の0.05Mリン酸緩衝液、通常の生理食塩中のpH7.4の0.05Mリン酸緩衝液、pH8〜10の0.05Mホウ酸緩衝液である。好ましい緩衝液は、pH6〜7の0.05Mリン酸緩衝液である。緩衝液は、任意の適切なモル濃度を有することができるが、例えば約0.001M〜約0.5M、好ましくは約0.01〜約0.1のモル濃度を有することができる。溶出緩衝液に関する一般情報は、USP 24, pp. 2231-2240, United States Pharmacopeial Convention Inc,
Jan 1, 2000で見出すことができる。
【0028】
他の実施態様において、水性溶出溶媒は、所望により界面活性剤を含む水である。
【0029】
選択的に、水性溶出溶媒は界面活性剤を含むことができるが、界面活性剤は系の溶解性を調節するもう1つの方法である。典型的な有用な界面活性剤は非イオン、陽イオン、陰イオンおよび両性イオン界面活性剤である。本発明における使用に適した界面活性剤の具体例は、ドデシル硫酸ナトリウム、モノオレイン酸ポリオキシエチレンソルビタン(Tween 80(登録商標))、ケノデオキシコール酸、グリココール酸ナトリウム塩、モノラウリン酸ポリ(オキシエチレン)n−ソルビタン(Tween 20(登録商標))、タウロコール酸、オクチルフェノールエチレンオキシド縮合物(Triton X−100(登録商標))、および臭化ヘキサデシルトリメチルアンモニウムである。
【0030】
界面活性剤の種類と量は、検体、非水性液体組成物および水性溶出溶媒の特定の系により決まり、当業者により決定されうる。界面活性剤濃度は、臨界ミセル濃度以上であることも以下であることもできる。界面活性剤の典型的な濃度範囲は、約0.001%〜約1%である。
【0031】
水性溶出溶媒のpHは、研究する特定の系に従って選択されなければならない。一般的に、水性溶出溶媒のpHは、約1〜約10であり、好ましくは約2〜約8である。水性溶出溶媒のpHは検体の溶解性に影響することができ、このpHは実験におけるシンク条件の調節の方法の1つであることが一般的に知られている。水性溶出溶媒のpHを最適化することにより、一部の検体の溶出特性を調節することができる。医薬品の場合、これによりインビトロ薬物放出特性とインビボ薬物動態学的性能の間の相関関係を構築することが可能になる。
【0032】
水性溶出溶媒のpHを最適化することにより、一部の場合において界面活性剤の使用が必要でなくなる場合がある。水性溶出溶媒として特に好ましい系は、最適のpH値を有する水性緩衝液である。
【0033】
最適化したpH値を有する水性溶出溶媒を用い、かつ薬物放出媒体中の界面活性剤の使用を除去することにより、単一の濾過段階を用いて乳濁液混合物の残りの部分から溶液中の検体を分離することが可能となる場合がある。このことにより、大きな孔径を有するフィルターを通過させる濾過よりもより複雑で時間のかかる限外濾過の必要性が回避される。界面活性剤が用いられない場合、関心のある検体の水溶性分画を分離するために単一のフィルターを用いることができ、一方で乳濁液の油相小球体が実質的にフィルターを通過せず、水性溶出溶媒中の検体の量の測定に悪影響を及ぼさないことを確実にするために、実験パラメーター(例えば撹拌速度、ストローク長さ、撹拌時間)を乳濁液の小球体(即ち、小滴またはミセル)サイズが十分大きいように選択することができる。乳濁液のミセルサイズが小さすぎ(例えば0.2ミクロン以下)、単一のフィルターを通過する場合、溶液状態の検体を溶液状態で分離するために限外濾過を用いることができる。機械撹拌パラメーターを、小球体サイズを最適化するために調節することができる。
【0034】
1つの実施態様において、pH、界面活性剤濃度、およびシステムの機械撹拌パラメーターは、約0.2ミクロンの孔径を有するフィルター(Acrodisk(登録商標)シリンジフィルター、製品番号4496、Gelman Laboratory)を通して乳濁液を濾過でき、一方で乳濁液の油相小球体が実質的にフィルターを通過しないように最適化される。溶出系の物理的パラメーター(温度、撹拌速度、ストローク長さ、容器の形状、サンプルサイズおよび溶出溶媒の量)および化学的パラメーター(pH、シンク条件、緩衝液の濃度、界面活性剤、および補助溶剤)は、反復法により最適化できる。所望の孔径を有するフィルターが選択される。本発明の方法は、固定されたpH(例えばpH=7)を有する水性溶出溶媒を用いて実施される。形成された乳濁液は、選択されたフィルターを通して濾過される。乳濁液の小滴が実質的にフィルターを通過しない場合、単一の濾過で十分であり、水層中の検体の濃度は測定できると思われる。しかしながら、乳濁液の小滴がフィルターを通過してしまう場合、検体の水溶性部分の定量に移る前に、限外濾過または他の適切な精製段階(例えば液/液抽出)が必要な場合がある。小滴が実質的にフィルターを通過するか否かは、濾液を視覚的に検査することにより判定できる。濾液が透明でかすんでいない場合、小滴は実質的にフィルターを通過していない。ついで、物理的および化学的パラメーターを反復的に調節し、さらに最適な条件が特定されるまで実験を繰返すことができる。最適と分類されたいずれのpH値も、本発明の好ましい実施態様に使用できる。上記の追加の方法は、いかにして最適のpH範囲、界面活性剤の種類および濃度ならびに機械および熱的撹拌パラメーターを特定できるかを説明している。
【0035】
非水性液体組成物中の検体の溶出を測定するために、組成物を水性溶出溶媒と混合する。水性溶出溶媒と混合される組成物の量は、組成物の性質、溶出溶媒の性質、および用いる溶出溶媒の量などの種々の要素によって大きく変化しうる。適切な分析方法を用いて検出できる水層中の検体濃度として得られるいずれの量も許容できる。従って、水性溶出溶媒に対する非水性液体組成物の比率は場合により大きく変化することができる。一般的に、水性溶出溶媒に対する非水性液体組成物の比率は、約1:100〜約1:2000であり、好ましくは約1:250〜約1:1000である。本発明の他の利点は、少しのサンプル量で溶出を実施できることである。約500mL〜1000mLの水性溶出溶媒の量を必要とする標準的な溶出方法と比較して、本発明の方法の一部の実施態様において、本発明の方法は約10mL〜約100mL、または好ましくは、約20〜約50mLの水性溶出溶媒を用いて実施できる。
【0036】
非水性液体組成物と水性溶出溶媒を混合した後、得られた混合物を乳濁液が形成されるように撹拌する。「乳濁液」という用語は、少なくとも2つの相が不混和性または部分不混和性の液体である、2以上の相を含む分散系を意味する。
【0037】
非水性液体組成物と水性溶出溶媒の混合物は、いずれの適切な撹拌装置を用いても撹拌できる。一般的に、装置は混合物のために少なくとも1つの容器を有する。容器は,その中で混合物を混合でき、かつ撹拌中に混合物の損失がないように作られている任意のうつわ、入れ物、切り込みまたは他の形のことを言う。単数または複数の容器は、撹拌装置の常設部品であっても良く、あるいは撹拌装置と分離可能なものであっても良い。一般的には、再使用する前に洗浄するかあるいは廃棄できるように、容器は撹拌装置と分離可能なものが良いと思われる。典型的な容器は、プラスチック、ゴム、ガラス、金属、または加工紙製で使い捨ての、EPA型バイアル、遠心管、試験管、血清バイアル、ビーカー、エーレンマイヤーフラスコ、反応バイアル、または他の種類の入れ物を含む。好ましい容器は、ガラス製EPA型40mLバイアル、またはゴム栓のついたガラス製血清バイアル(50〜100mLサイズ)のいずれかの使い捨てバイアルである。
【0038】
乳濁液を製造できる装置であれば、任意の撹拌装置を使用できる。適切な撹拌装置の例は、市販の(温度制御つきあるいは温度制御無しの)種々の実験室用シェーカーであり、オービタル式、リニア式(レシプロ式)、または他の方式で撹拌できる。好ましい撹拌装置は、容器に含まれる混合物が激しく撹拌されるシェーカーである。シェーカーは、容器を水平方向に、垂直方向に、シーソー風にまたはそれらの任意の組み合わせで容器を動かすことができる。特に好ましいシェーカーはレシプロシェーカーである。撹拌装置は、特定の環境下では乳濁液を形成できるかも知れないが、一般的には、パドルアセンブリ中などで単に撹拌しただけでは、非水性液体組成物と水性溶出溶媒は乳濁液を形成しない。これらの装置中では、非水性液体組成物は一般的に水性溶出溶媒の表面に浮く。従って、これらの2成分の間の接触面積は、本発明における場合よりも小さく、検体の溶出速度はより小さいと思われる。
【0039】
図3は、本発明の方法に使用できる撹拌装置の概略図である。この装置はシェーカーであり、容器2が2つ取り付けてある水平プレート1を含む。作動中は、水平プレートは矢印で示される方向に水平に動く。容器はシェーカーの表面に横たわるように示してあるが、容器はまた直立の状態であることもできる。これにより、本発明の方法の精度と信頼度をさらに改良することができる。容器の形状が細長い場合、シェーカーのストロークは、例えば図3に矢印で示されているように、容器の延長線に並行であるのが好ましいが、他の任意の適切な方向であることもできる。
【0040】
非水性液体組成物と水性溶出溶媒は所定時間撹拌される。撹拌時間は大きく変化させることができ、例えば、撹拌量、検体、非水性液体組成物、溶出溶媒、温度、検体の量を測定するために用いる検出法の感度、および多くの他の要素によって決まる。さらに、撹拌時間は、短時間、中程度の時間または長時間の溶出速度あるいはそれらの組み合わせに関する情報が所望されているかどうかで決まる。一般的に、撹拌は、水性溶出溶媒中の検体の総量の約1%〜約100%、好ましくは約10%〜約100%が溶解するまで続けられる。一般的に撹拌は約5分〜約24時間、好ましくは約15分〜約60分行われる。
【0041】
撹拌段階中、非水性液体組成物と水性溶出溶媒の混合物は、任意の所望の接触温度に保つことができる。通例、混合物は、例えば室温(即ち約22−25℃)または約37℃などの比較的一定の接触温度に保たれる。しかしながら、溶出速度をあげるために高温を使用することができるし、溶出速度を下げるために低温を使用することができる。混合物の温度は溶出速度に影響するため、2以上の実験結果を比較するためには、各実験に関して同一の温度を用いなければならない。本明細書においては、「同一温度」は、異なる実験の温度間の差異が、多くとも5℃、好ましくは多くとも2℃であることを意味する。好ましくは接触温度は室温(即ち22〜25℃)である。
【0042】
振盪速度などの、接触中の撹拌量は、検体の溶出速度にも影響し、最適の条件は、撹拌容器のサイズおよび形状、非水性液体組成物および水性溶出溶媒などの種々の要素に基づいて決定されなければならない。一般的に、サイクル数は、約100〜約500であり、好ましくは約100〜約300である。ストローク長さは、好ましくは約0.5インチ〜約2インチであり、さらに好ましくは約0.75インチ〜約1.5インチである。
【0043】
非水性液体組成物および水性溶出溶媒の混合物を所定時間撹拌した後、水性溶出溶媒中の検体の量を測定する。一部の検出法を用いれば、溶出試験装置中に水性溶出溶媒が残っていても検体の量は測定できる。しかしながら、一般的に、例えばシリンジまたは常設試料採取管により、溶出試験装置から少なくとも一部の水性溶出溶媒が取り出される。分析のために水性溶出溶媒の全てを使用することが可能であるし、このことは一部の検出法には必要であるが、他の場合には、水性溶出溶媒の一部のみを用いる。検体の量を測定するために取り出すサンプルサイズは、種々の要素、特に用いる検出法によって決まり、約0.1mL〜約25mL、好ましくは約0.5mL〜約15mLであることができる。所望であれば、検体の量を測定するために使用する水性溶出溶媒のサンプルは、溶出試験装置から取り出した後、濾過または遠心分離することができる。これによりオイルまたは固相から水性層を除くことができ、従って検体の測定を妨害し測定を混乱させる可能性のある、検体を含む粒子または乳濁液を水層から取り除くことができる。約0.1〜約50ミクロン、好ましくは約0.1〜約0.3ミクロンの平均孔径を有するフィルターを通して濾過するなどのいずれの適切な方法を用いても濾過を達成できる。一部の場合において、限外濾過が必要な場合がある。その場合、約0.001ミクロン〜約0.1ミクロン、好ましくは約0.001〜約0.01の孔径を有するフィルターがより適切であろう。当該技術分野において公知である適切なフィルター用具の多くの例がある。これらのフィルターは、例えば、Gelman Laboratory製Acrodisk(登録商標)、およびMillipore Corporation製Centriprep50(登録商標)などの販売名で市販されている。所望による濾過段階の後、水性溶出溶媒中の検体の量が測定される。検体の量を測定するために適切な任意の分析方法も使用できる。分析方法の選択は、検体の性質、検体の濃度範囲、溶出溶媒を含む種々のパラメーターによって決まるが、これらの分析方法もまた研究室で利用可能である。分析方法の具体例は、分離技術(例えば高速液体クロマトグラフィー、液体クロマトグラフィー、薄層クロマトグラフィー、キャピラリー電気泳動、ガスクロマトグラフィ−)、光度技術および分光光度技術(例えば紫外・可視(UV−Vis)、フーリエ変換赤外(FTIR)、原子吸光(AA)、原子発光(AE)、質量分析法(MS))である。クロマトグラフ法、特にガスクロマトグラフィ−(GC)および高速液体クロマトグラフィー(HPLC)が好ましい。適切なクロマトグラフ法の例は、当該技術分野に公知な種々の検出技術のいずれかを組み込んだ、逆相高速液体クロマトグラフィー(RP−HPLC)、順層高速液体クロマトグラフィー(NP−HPLC)である。適切なクロマトグラフ法と共に用いることができる検出技術の例は、UV−Vis、屈折率、質量分析法、および光散乱検出を含む。分析方法として、UV−Vis検出法と組み合わせたフローインジェクション分析(FIA)もまた使用できる。リアルタイムで製造システムのインプロセス評価を行う場合など、サンプルのハイスループットが必要とされる時にFIAはとりわけ適切である。
【0044】
溶解した検体の量が単一の所定時間で測定される実施態様に関して、本発明の方法を上記で説明した。多くの場合、検体が一定速度で放出されるか、あるいは速度が時間と共に変化(例えば、溶出試験の開始時に多量、そしてその後、より少量)するかどうか測定するために、一定の時間に亘って、溶出速度をモニターすることは興味の持たれるところである。これらの場合、異なる所定の時間に2以上のサンプルを取り出し、これらのサンプルを個々に分析するために、十分に大きな容器を使用することができる。2以上の同一の実験を用意し、撹拌時間のみを変える以外は同一の条件で撹拌することも可能である。この場合、図3に示すように、同一の撹拌装置を用いて各混合物を同時に撹拌することができる。異なる装置を用いて、混合物を交互に撹拌することもまた可能である。これらの別個の混合物から異なる時点得採取した水性溶出溶媒を個別に分析する。その結果を、次に溶出速度の時間依存性プロファイルを測定するために用いることができる。
【0045】
本発明の方法を用いれば、非水性液体組成物中における検体の溶出速度を信頼度高く正確に測定することができる。薬学的応用において、本発明のインビトロ法により得られる結果は、インビボ薬物動態学的研究の結果と良く相関させることができる。従って、本方法は、適切な生物学的性能およびロットの一貫性を確実にするための、医薬品の製造中における品質管理における厳格で信頼性のある方法として使用できる。本方法は簡単で安価でありかつ迅速であるため、医薬品およびそれらの剤形の開発に有利に使用することもできる。本発明の方法は、従来法よりも迅速であるため、遅い溶出速度を有する薬物または徐放性製剤の溶出速度を測定するために有用である。医薬品の剤形における薬物放出を減じる(徐放性を付与する)ことを意図する製造方法のリアルタイムモニターに本方法はとりわけ有用である。
【0046】
以下の実施例は本発明を説明するために提示される。しかしながら、これらを制限と解釈すべきではない。特記しない限り、実施例中に引用した全てのパーセンテージ、比率、割合などは重量換算で示してある。
【実施例】
【0047】
精度:
本発明の方法の精度は、繰り返し測定の相対標準偏差(RSD)を算出することにより測定できる。低い標準偏差値は、高い精度を示す。一般的に、相対標準偏差は検体の溶出速度を同一の条件で3回以上測定することにより測定される。次いで、以下の式に従って相対標準偏差値を算出する:
【数1】

ここで、s.d.は以下で定義される標準偏差値である:
【数2】
ここで、Xは個別の結果であり;Nはデータポイント数であり;そして
【数3】
は平均値である。本発明の方法により得られる相対標準偏差値は1%未満であることができる。
正確さ:
本発明の方法の正確さは、既知量の検体を加えた非水性液体組成物から水性媒体への検体の移動を測定することにより測定できる。加えた非水性液体組成物は、振盪または撹拌により水性薬物放出媒体と平衡され、しかる後水性溶出溶媒中の検体の量が測定される。次いで水性媒体へ移動した検体の濃度を、検体が理論的に100%移動した場合(例えばピペッティングおよび計量の誤差または損失が無く、検体が100%溶解し検体が100%検出されると仮定する場合)に得られる濃度と比較する。本発明の方法は、約70%〜約100%の正確さを有し、好ましくは約90%〜約100%の正確さを有する。
一般的溶出方法
装置:
プラットフォームシェーカー:レシプロシェーカーモデル5850(Eberbach社から入手可能)、公称ストローク長さおよそ2.54cm、回転数200サイクル/分。バイアルは水平位置に設置し、図3に示すようにストローク方向に並行に並べた。プラットフォームシェーカーを適切に調節した温度環境(例えば22℃)に保つ。
バイアル:40mL(EPA型)テフロン(登録商標)裏打ちスクリューキャップ。Qorpak社から入手可能(部品番号7588T)。
プラスティックシリンジ:BD Disposable 10mLプラスティックシリンジまたは同等物
フィルター:Acrodisk 0.2ミクロン(部品番号4496)
方法:
空の40mLバイアルに、溶出試験のための組成物の適切な量(例えば30−70mg)を分配する。適切な温度(例えば22℃)で平衡させる。溶出溶媒を適切な温度(例えば22℃)で平衡させる。適切な量(例えば30mL)の溶出溶媒を、溶出試験のためのサンプルを含むバイアルに加える。全てのサンプルに関して繰返す。この方法を約2〜3分以内に完了する。撹拌を開始する。所定時点で、定量分析のためにサンプルを取り出す。必要に応じて濾過する。定量分析に進む。
一般的定量分析方法
特記しない限り、以下の一般的方法に従った。
装置:定組成操作が可能なHPLC(例えばAgilent 1100(Agilent Technologiesから入手可能))。
検出器:UV−Vis254nm検出器(例えばダイオードアレイ検出器(Diode array detector)、検出波長:254nm、Agilent Technologiesから入手可能)。
カラム:Waters Symmetry C8, 3.9 x 50 mm、Waters Corporationから入手可能
注入量:10μl
流速:1〜2mL/min
圧力:3000psi
移動層:3.85gの酢酸アンモニウムおよび13.5mLの40%テトラブチル水酸化アンモニウムをMilli−Q水に溶解し、全部で700mLの容量にした。氷酢酸を用いてpHを6.7±0.1に調節した。次いで、溶液を0.45μmのメンブランフィルターを通して濾過した。濾過後、200mLのメタノールおよび110mLのテトラヒドロフランを加え、混合物を減圧下超音波処理し脱気した。
検体の放出量の算出
各時点での放出された検体(例えばセフチオフル)の量は、以下の式に従って算出できる。
【数4】
式中、
Wstd=標準試料の重量(mg)
P=セフチオフル遊離酸としての参照標準の純度
Rstd=標準試料のピーク面積
DISVOL=溶出液の容量(mL)(30)
WSVOL=実用標準の容量(mL)(10)
Rsam=測定試料のピーク面積
Wsam=サンプル懸濁液の重量(mg)
1000=サンプル重量のmgからグラムへの変換
である。
(実施例1)
【0048】
以下に記載した方法を用いて、2つのCCFA懸濁液サンプルを測定した。
【0049】
綿実油をジャケットつき容器に注ぎいれ、115℃に加熱することにより、非水性ビヒクルを調製した。ホスホリポン90H(0.05重量%)(American Lecithin Co. から入手可能)を加え、混合した。溶液を45℃に冷却した。モノオレイン酸ソルビタン(0.15重量%)を加え、混合した。CCFAを100mg/mL加え、懸濁液が均一になるまでトリブレンダーを通して混合した。懸濁液をトリブレンダーを通して再循環させ、タンク撹拌器を作動させながら濾過した。得られた懸濁液を滅菌バイアルに充填し、栓をし、シールした。シールしたバイアルをγ線照射を用いて滅菌した。これらのロットを40,700および40,620とラベルをつけた。1%Tween 80界面活性剤(Sigma−Aldrich社から入手可能)を0.05MpH6.0リン酸緩衝液に溶解することにより、水性溶出溶媒を調製した。21.8グラムの第一リン酸カリウムおよび3.48グラムの第二リン酸カリウムを脱イオン水に加え、200mLに希釈してpH6.0リン酸緩衝原液を調製した。50mLの緩衝原液を脱イオン水を用いて900mLに希釈することにより0.05MpH6.0リン酸緩衝原液を調製した。
【0050】
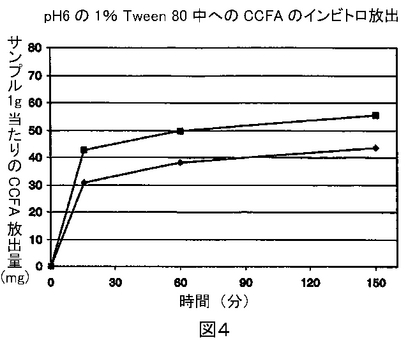
65mgから85mgの間のCCFA懸濁液ロット番号40,700および30mLの水性溶出溶媒を、テフロン(登録商標)裏打ちスクリューキャップ(Qorpak社から入手可能)つきの40mL EPA型ガラスバイアルに入れた。全部で9つの同一のバイアルを調製した。第2のロットである、CCFA懸濁液ロット番号40,620を用いてこの方法を繰り返し、別の9つのバイアルを調製した。これらのバイアルをレシプロシェーカー(Eberbach Model 5850、ストローク長さ=17/16インチ、振盪速度=200サイクル/分)上に水平におき、撹拌を開始した。15分の撹拌の後、各ロットの最初の3つのバイアルをシェーカーから取り除いた。各バイアルから15ミリリットルのサンプルを採取し、限外濾過装置(Centriprep 50限外濾過装置、AmiconまたはMillipore Corporationから入手可能)を用いて限外濾過した。前述の一般的分析方法を用いて濾液中のCCFA量を測定した。この方法を撹拌の60分および120分に繰返した。各ロットの残りのバイアルをそれぞれ同様な方法で取り除き処理した。
【0051】
得られた結果を図4に示す(ロット40,620のデータを菱形で表し、ロット40,700のデータを正方形で表す)。測定された溶出速度は、前回インビボ実験で観察された薬物動態学的性能と相関関係を示した。
【0052】
界面活性剤として、1% Tween 80の代わりに1% Tween 20を用いて、実験を繰返した。結果を図5に示す(ロット40,620のデータを菱形で表し、ロット40,700のデータを正方形で表す)。測定された溶出速度は、前回インビボ実験で観察された薬物動態学的性能と相関関係を示した。
(実施例2)
【0053】
本実施例は、本発明の方法により得られたインビトロ結果と薬物動態学的研究により得られたインビボ結果との間の相関関係を明らかにする。
【0054】
異なるインビボ薬物動態学的性能を示すCCFA S.Supensionの3つのロット(40,620;40,700;JMS−144F)を本評価に用いた。各ロットは同一の組成を有し、かつ前述の実施例1におけると同じ方法を用いて(ただしJMS−144Fはγ照射処理によって滅菌されていない)製造したが、これらのロットは異なるインビボ薬物動態学的性能を示した。薬物動態学的性能における差異は、セフチオフルが動物の血流中で検出される時間数により示される徐放効果の持続時間における差異により明らかにされた。セフチオフルに関する分析検出限界は、血漿1mLあたり0.2マイクログラムのセフチオフルである。徐放効果の持続時間は、一般に、「0.2mcg/mL以上の時間」として表わされる。
【0055】
本発明に記載の方法を用いて3つのロットを試験した。インビトロ薬物放出結果と0.2mcg/mLを超す時間との相関関係を図6に示す。60分(正方形で表されるデータポイント)および150分(三角形により表されるデータポイント)の時点に関するインビトロ結果を示す。実線はデータに適合する、最小二乗法による回帰直線であり、インビトロ放出量とインビボで観察される徐放効果の持続時間との逆相関を説明する例に含まれる。所定時間にインビトロでより多くのCCFAを放出したロットは、インビボ徐放性の持続時間がより短かった。
(実施例3)
【0056】
本実施例は、水性溶出溶媒のpHを最適化することにより、薬物放出媒体における界面活性剤の使用を除去することができ、かつ限外濾過の必要性もまた除去できる。本実施例において、CCFAを検体として用いた。CCFAの溶解性はpHに応じて変化する。pH5以下では、CCFAは比較的不溶性であり;CCFAの溶解性はpHの増加に伴って増加する。この薬物放出実験において、2つの異なる薬物放出媒体:(1)1% Tween 20を含むpH6.0リン酸緩衝液、および(2)外因性界面活性剤を含まないpH6.5リン酸緩衝液、を用いた。緩衝液を以下の方法で調製した:21.8グラムの第一リン酸カリウムおよび3.48グラムの第二リン酸カリウムを脱イオン水に加え希釈して200mLにし、次いで脱イオン水により再度18倍に希釈して(即ち、50mLを脱イオン水により希釈して900mLにする)、0.05MpH6.0リン酸緩衝液を調製した。31.98グラムの第一リン酸カリウムおよび15.39グラムの第二リン酸カリウムを脱イオン水に加え1000mLにし、次いで脱イオン水により再度10倍に希釈して(即ち、100mLの緩衝原液を脱イオン水により1000mLにする)、0.05MpH6.5リン酸緩衝液を調製した。
【0057】
異なるインビボ薬物動態学的性能を示したCCFA S.Suspension(40,620;40,700;JMS−111)の3つのロットを、本評価に用いた。各ロットは同じ組成を有し、かつ前述の実施例1におけると同じ方法(ただしロットJMS−111はγ照射を用いる処理により滅菌されていない)を用いて製造されたが、これらのロットは、最初の薬物放出媒体(1% Tween 20を含むpH6.0リン酸緩衝液)を用いて試験した場合、異なるインビトロ薬物放出速度を示した。ロット間のインビトロ放出の変動は、60分で放出されたCCFAの種々の量に反映されている(表1)。
【0058】
外因性界面活性剤を含まないpH6.5リン酸緩衝液からなる薬物放出媒体を調製し、インビトロ薬物放出測定を繰り返した。pHを6.0から6.5に上昇させ、薬物放出媒体からTween 20を除去することにより、3つのロットに関するインビトロ放出速度の順位関係は同一であり、乳濁液の油相小球体はかなり大きく、かつ単一の濾過により除去できることが見出された。これにより、限外濾過精製段階を単一の濾過に置き換えることができた。さらに、全3サンプルに関するインビトロ放出速度は、pH6.5媒体においてより速いことが見出された。したがって、アッセイ時間は60分から30分に短縮化することができた。最後に、3つのCCFAロット間の適切な絶対分解能が達成され、より速く(60分に対して30分)かつより簡単な(Tween 20を除去し、かつ限外濾過よりも簡単な)方法により、種々のインビトロ放出速度を有するロットを適切に識別することができることが見出された。絶対分解能は、薬物放出媒体中のロットが、その媒体に関して具体的に選択した時点で放出したCCFAの量の間の差異として定義した。
【表1】
(実施例4)
【0059】
CCFA懸濁液のための2つの異なる非水性基剤を調製し、それぞれをCCFAに加えた。添加物は2つのおおよその濃度レベルで3回調整した。正確なCCFA添加量を記録した。
【0060】
非水性基剤である基剤−1は100%ヤシ油(Miglyol 812として入手可能)であり、一方、非水性基剤である基剤−2はヤシ油(Miglyol 812としてHulsAmericaより入手可能)および綿実油(Welch,Home & Clark Companyより入手可能)の1:1(v:v)混合物であった。
【0061】
CCFAを加えた基剤混合物は、40mL EPAバイアル中のおよそ65mgの基剤(1または2)に既知量の薬物を正確に加え混合することにより調整した。基剤混合物の調製物について、レシプロシェーカー上で振盪することによりサンプルを溶出溶媒で4時間抽出した。サンプルを濾過し、濾液中のCCFA濃度をHPLCにより測定した。結果を表2に要約する。98.4〜100.1%の範囲のCCFAの回収率が得られた。
【表2】
【図面の簡単な説明】
【0062】
【図1】検体が2回以上測定される場合の、1つの可能性のある溶出速度のプロットの図である。
【図2】検体が2回以上測定される場合の、他の可能性のある溶出速度のプロットの図である。
【図3】シェーカーの図式を示す図である。
【図4】界面活性剤としてTween 80を使用する実施例1の溶出試験の結果を示す図である。
【図5】界面活性剤としてTween 20を使用する実施例1の溶出試験の結果を示す図である。
【図6】図6は実施例2のインビトロ−インビボ相関関係の結果を示す図である。
【技術分野】
【0001】
本発明は非水性溶液から水性媒体への検体の移動を特徴づける方法に関し、具体的には、徐放性製剤からの薬物溶出のインビトロ測定方法に関する。
【背景技術】
【0002】
医薬組成物の製剤化の1つの重要な態様は、薬物の薬物動態学的挙動である。薬物の物理状態(即ち、気体、液体、固体)、結晶形、粒子径、剤形、および用いる賦形剤などの種々の要素によって、体内での薬物の時間に依存した放出は大幅に変化しうる。たとえ、同一薬物が同一剤形で提供された場合であっても、ロット間変動は起こりうる。
【0003】
規制当局の認可を得るために、薬物動態学的挙動は、多くの場合、動物またはヒトに薬物を投与し、投与後のある複数の時点で、例えば血中における薬物またはその代謝物の量を測定することにより測定される。この方法は時間と費用がかかり、一般的には、製造工程中で医薬品の質を制御するためには用いられていない。インビトロ試験において、薬物のインビボでの薬物動態学的挙動を評価するための多くの方法が考案されてきた。これらの試験の一部は標準化され、米国薬局方(USP)などに記載されている。一般的に用いられるUSP法は、バスケット法(USP第1法)およびパドル法(USP第2法)である。これらの標準化法に加えて、特定の個別用途のための多くの方法が記載されてきた。多くの溶出方法に関する概説は、例えば、G. K. Shiu, Drug Information Journal, 30, 1045-1054, (1996)などにおいて見出すことができる。
【0004】
Andonaeguiら(Drug
Development and Industrial Pharmacy, 25 (11), 1199- 1203 (1999))は、絶食条件および高脂肪食条件で投与した徐放性テオフィリンマトリックス錠の、インビボ性能を予測するためのインビトロ法を記載している。3種類の徐放性マトリックス錠中のテオフィリンの溶出挙動が研究された。高脂肪食に関するインビトロ/インビボ相関性を高めるために、USPパドル試験における溶出試験の前に落花生油と混合することにより錠剤が前処理された。
【0005】
日本特許出願JP05−249097は、徐放性錠剤のインビボ放出を予測するための溶出試験を記載している。該錠剤にパドル試験を行い、取り出し、オイルおよび脂質で処理し、次いで水性溶出溶媒中のビーズと共にパドル装置に戻すかあるいはバスケット中に沈める。この方法は、徐放性錠剤の放出制御メカニズムによる影響を受けないで、生体内の血漿中薬物濃度を予測するといわれている。
【0006】
米国特許第6132751号は、眼科用PBS乳濁液組成物中の薬物の溶解性の評価を開示している。
【0007】
微粒子ドラッグデリバリーシステムのための種々のインビトロ溶出法が、Contiら Drug Development and Industrial Pharmacy, 21(10), 1233-1233
(1995)により比較されている。撹拌速度、イオン強度および界面活性剤の存在の影響が研究されている。
【0008】
油状薬物製剤を試験するための溶出方法もまた記載されている。Takahashiら(Chem. Pharm. Bull., 42 (8), 1672-1675, (1994))は、パドル法と回転透析セル法を比較している。回転透析セル法の変法において、オクタノールを外相として用い、一方で内相として酸性溶液を用いた。
【0009】
Machidaら(Chem.
Pharm. Bull., 34 (6), 2637-2641, (1986))は、油性薬物製剤の溶出特性の測定において遭遇する問題を克服するための1つの試みを記載している。水性溶出溶媒の表面を撹拌するための追加の補助翼を用いる、日本薬局法第2法のパドル法の変法を用いることを彼らは提案している。さらに、撹拌を改善するためにビーズを加え、かつ水性溶出溶媒として胆汁酸塩溶液を用いた。
【0010】
薬物が非水性基剤中に溶解し、分散し、懸濁し、または別な風に調製された非水医薬組成物の薬物動態学的挙動は、従来の技術を用いて確実に予測することは困難である。インビトロ測定の精度および信頼度は低いことが多く、インビトロ測定の結果は、インビボでの薬物の性質といつも相関するとは限らない。従って、本発明の目的の1つは、非水性液体組成物中の検体の溶出を特徴づけるための信頼できる方法を提供することである。本発明の他の目的は、低溶解性を有する検体の溶出を特徴づけるための方法を提供することである。本発明の他の目的は、ゆっくりしか溶けない検体の溶出を特徴づけるための方法を提供することである。本発明の他の目的は、インプロセスアッセイとして製造中に利用できる、検体の溶出を特徴づけるための迅速な方法を提供することである。本発明の他の目的は、本出願の明細書および特許請求の範囲を読むことにより容易に明らかとなるであろう。
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明は非水性液体組成物中の検体の溶出を特徴づける方法に関し、
(a)検体および非水性基剤を含む非水性液体組成物を用意する段階;
(b)非水性液体組成物を水性溶出溶媒と混合する段階;
(c)非水性液体組成物と水性溶出溶媒を撹拌して乳濁液を形成する段階;および
(d)水性溶出溶媒中の検体の量を測定する段階;
を含む方法に関する。
発明の詳細な説明
【0012】
本発明は、非水性液体組成物中の検体のインビトロ溶出を特徴づける、信頼できる方法を提供する。本方法は、好ましくは、医薬組成物からの医薬活性成分の溶出を定量するために用いられるが、本方法はさらに、例えばオイルから環境への汚染物質の溶出速度の測定、防錆剤などの活性成分の油性基剤からの減少速度の測定または殺虫剤もしくは肥料からの成分の放出速度などの、分析化学の他の分野においても用いることができる。
【0013】
溶出は、種々の方法により特徴づけかつ定量することができる。例えば、水性溶出溶媒中の検体の量が、所定の時間で1回のみ測定される場合もある。例えば、3μgの検体が30分後に溶解したと測定される場合、30分で3μg溶解したと溶出が特徴づけられる。水性溶出溶媒中の検体の量を2回以上測定する場合、溶出はいくつかの異なる方法で説明することができるが、これらの方法は当該技術分野において公知である。一般的な方法の1つは、二次元グラフ中にデータをプロットすることである。x軸は時間軸を表す。y軸はn回目と(n−1)回目の水性溶出溶媒の分析の間に溶解した検体の量を表す。他の一般的な方法の1つは、x軸がここでも時間軸を表す、二次元グラフ中にデータをプロットすることである。y軸は、測定の最初と水性溶出溶媒のn番目の分析の間に溶解した検体の総量を表す。もちろん、上記の二次元グラフ以外にも、同一の情報を表または他の適切な形で表すことができる。以下の一連の実験は、例として使用できる:非水性液体組成物を調べ、10分(n=1)、20分(n=2)、および30分(n=3)の時点で、溶解した検体の量を測定する。総量で、10分後に15μgの検体が溶解し、20分後に25μgの検体が溶解し、そして30分後に32μgの検体が溶解した。第1の場合では、図1に示すようなプロットが得られるであろうし、また第2の場合では、プロットは図2に示すようなものになるであろう。
【0014】
本明細書においては、「非水性液体組成物」という用語は、接触温度で液体でありかつ検体および非水性基剤を含む任意の組成物のことを言う。検体および非水性基剤の混合物は任意の形であることもでき、例えば、溶液、乳濁液または懸濁液であることができる。検体が非水性基剤に懸濁化されている場合、検体の粒子径は、一般的に約50nm〜約200ミクロンの範囲であり、好ましくは約100nm〜約200ミクロンの範囲である。非水性液体組成物中の検体の濃度は特に限定されるわけではない。例えばこれらの濃度は、約0.00001mg/mL〜約5,000mg/mLの間で変化し、好ましくは約0.01mg/mL〜約1,000mg/mLの間で変化することができる。
【0015】
非水性液体組成物は、好ましくは医薬組成物である。本発明の方法において、医薬組成物は一般的に、非経口、経口、舌下、経鼻、気管支内、肺、乳房内、直腸、膣、眼球、または局所投与に適した液体である。しかしながら、医薬組成物がカプセル剤に含まれている医薬組成物中の検体の溶出速度を測定することもまた可能である。この場合、カプセルの殻は水性溶出溶媒と接触して崩壊し、その内容物を放出する。
【0016】
溶出が特徴づけられる検体は、非水性液体組成物の任意の成分であることもできる。検体の例は、汚染物質、活性成分、または不活性成分であるが、これに限定されない。医薬組成物の場合、検体は一般的に医薬活性成分であるが、医薬組成物の賦形剤またはその他の成分であることもできる。本発明の方法は、単一の検体の測定に限定されない。所望であれば、2以上の検体を測定することができる。本発明の方法は、何らかの特定の物理的または化学的性質を有する検体の測定に、限定されない。検体が、本方法のために選択された水性溶出溶媒に少なくとも一部が溶解する限りは、ほとんど全ての検体(有機であれ、無機であれ)を本発明の方法により測定することができる。本発明の方法を用いて測定できる検体の例は、以下の例示的、非限定的なクラス:ACE阻害剤;α−アドレナリンアゴニスト;β−アドレナリンアゴニスト;α−アドレナリン遮断薬;β−アドレナリン遮断薬(β遮断薬);抗酒薬;アルドースレダクターゼ阻害剤;アルドステロンアンタゴニスト;アミノ酸;タンパク同化剤;鎮痛剤(麻薬性および非麻薬性の両方);麻酔剤;摂食障害;制酸剤;駆虫薬;抗にきび剤;抗アレルギー剤;抗男性ホルモン;抗狭心症薬;抗不安薬(antianxiety);抗不整脈薬;抗喘息剤;抗菌剤および抗生物質;脱毛防止薬およびはげ予防薬;抗アメーバ剤;抗体;抗コリン剤;抗凝血物質および血液希釈剤;大腸炎治療剤;鎮痙薬薬;膀胱炎治療剤;抗うつ薬;抗糖尿病薬;下痢止め;抗利尿薬;解毒剤;制吐剤;抗エストロゲン剤;抗膨満薬;抗真菌剤;抗原;緑内障治療薬;抗ヒスタミン剤;過剰行動治療剤;抗高リポタンパク血症剤;降圧剤;甲状腺機能亢進症治療薬;低血圧治療剤;甲状腺機能低下症治療薬;抗感染薬;抗炎症剤(ステロイド性および非ステロイド性の両方);抗マラリア薬;偏頭痛治療薬;抗腫瘍剤;抗肥満薬;抗パーキンソン薬およびジスキネジア治療剤;肺炎治療剤;抗原虫剤;かゆみ止め;抗乾癬剤;抗精神病薬;解熱剤;リウマチ治療剤;分泌抑制薬;抗ショック剤;抗痙攣薬;抗血栓剤;抗癌剤;鎮咳剤;抗潰瘍剤;抗ウイルス剤;抗不安薬(anxiolytics);殺菌素;骨密度増加剤;気管支拡張剤;カルシウムチャンネル遮断薬;炭酸脱水酵素阻害剤;強心薬および強心剤;化学療法剤;胆汁分泌促進物質;コリン作用薬;慢性疲労症候群治療薬;中枢興奮薬;凝固剤;避妊薬;嚢胞性線維症薬;充血除去剤;利尿薬;ドーパミン受容体アゴニスト;ドーパミン受容体アンタゴニスト;酵素;エストロゲン;去痰薬;胃過敏治療剤;グルココルチコイド;止血薬;HMG−CoAレダクターゼ阻害剤;ホルモン;睡眠剤;免疫調節剤;免疫抑制剤;下剤;口腔および歯周疾患用医薬;縮瞳薬;モノアミン酸化酵素阻害剤;粘液溶解剤;多発性硬化症薬;筋弛緩剤;散瞳薬;麻薬拮抗薬;NMDA受容体アンタゴニスト;オリゴヌクレオチド;眼科用薬;子宮収縮薬;ペプチド、ポリペプチドおよびタンパク質;多糖類;プロゲストゲン;プロスタグランジン;プロテアーゼ阻害剤;呼吸促進剤;鎮静剤;セロトニン取り込み阻害剤;アンドロゲンを含む性ホルモン;禁煙薬;平滑筋弛緩薬;平滑筋刺激薬;血栓溶解剤;鎮静剤;尿酸性化剤;尿失禁薬;血管拡張剤;血管保護剤;およびそれらの組み合わせ;を含む。特定の薬剤化合物に対して行った本明細書のいずれの引用も、その化合物の互変異性体、立体異性体、エナンチオマー、塩およびプロドラッグを含み、かつその薬物のいずれか1つの固体状態形に限定されないことが理解されなければならない。
【0017】
本発明の方法は、特に第三世代セファロスポリンなどのセファロスポリンの溶出速度の測定に適している。セファロスポリンの例には、セフチオフル、セフェピム、セフィキシム、セフォペラゾン、セフォタキシム、セフポドキシム、セフタジジム、セフチゾキシム、セフトリアキソン、モクサラクタム(moxalactam)、これらの薬学的に許容される塩および誘導体があるが、これらに限定されない。特に好ましいセファロスポリンはセフチオフル、薬学的に許容される塩およびそれらの誘導体である。
【0018】
セフチオフル(ceftiofur)は、現在、ナクセル(登録商標)(Naxel)およびエクセネル(登録商標)(Excenel)という販売名でファルマシア社より市販されている。セフチオフルのもう1つの好ましい形態は、セフチオフル結晶性遊離酸(ceftiofur
crystalline free acid)(CCFA)である。この化合物およびその医薬製剤は、米国特許第5,721,359号に記載されており、その全体を本願に引用して援用する。
【0019】
非水性液体組成物は、接触温度で一般的に液体であり、かつ水と混和性、部分混和性、あるいは非混和性でありうる、非水性基剤もまた含む。非水性基剤は、水素化または非水素化、飽和、不飽和、またはポリ不飽和の、脂質(脂肪、オイル、ワックス、ステロール、およびグリセリド)であることができ、かつ当該技術分野において公知の技術によりさらに修飾することができる。非水性基剤は、好ましくは、天然または合成の脂肪、ワックス、またはオイルからなる群から選択され、さらに好ましくは、非水性基剤は天然または合成のオイルである。オイルという用語は、植物、動物、鉱物、および海洋起源由来のものを含む、トリグリセリド脂肪およびオイルを含む。非水性基剤として適切な合成オイルの具体例は、6〜24炭素原子を有する飽和または不飽和脂肪酸のトリグリセリド、またはプロピレングリコールジエステルを含む。かかるカルボン酸は、例えば、ヘキサン酸、オクタン酸(カプリル酸)、ノナン酸(ペラルゴン酸)、デカン酸(カプリン酸)、ウンデカン酸、ラウリン酸、トリデカン酸、テトラデカン酸(ミリスチン酸)、ペンタデカン酸、ヘキサデカン酸(パルミチン酸)、ヘプタデカン酸、オクタデカン酸(ステアリン酸)、ノナデカン酸、エイコサン酸、ヘンエイコサン酸、ドコサン酸およびリグノセリン酸などの、6〜24炭素原子を有するカルボン酸を含む。不飽和カルボン酸の例は、オレイン酸、リノール酸およびリノレン酸などを含む。トリグリセリドビヒクルは、脂肪酸のモノ−、ジ−、またはトリグリセリルエステル、またはグリセロールの少なくとも1つの分子が異なる炭素原子長の脂肪酸でエステル化された混合グリセリドおよび/またはプロピレングリコールジエステルを含むことができると考えられている。以下:トリオレイン、トリリノレインおよびトリリノレニンを含むトリ不飽和エステル;トリパルミチン、トリステアリン、およびトリデカノインを含む飽和トリ飽和エステル;はトリグリセリルエステルの例である。トリグリセリルエステルの他の例は、ジ飽和モノ不飽和タイプ:1,2−ジパルミトイル−3−オレオイル−rac−グリセロールまたは1,3−ジパルミトイル−2−オレオイル−rac−グリセロールなどのオレオジ飽和エステル;1,3−ジパルミトイル−2−リノレオイル−rac−グリセロールなどのリノレオジ飽和エステル;を含む。トリグリセリドの他の例は、1−パルミトイル−2−オレオイル−3−リノレオイル−rac−グリセロールおよび1−リノレオイル−2−オレオイル−3−ステアロイル−rac−グリセロールを含むモノ飽和オレオリノレイン酸エステルならびに1,2−ジリノレオイル−3−パルミトイル−rac−グリセロールを含むモノ飽和ジリノレインエステルなどの、モノ飽和ジ不飽和エステルである。
【0020】
ジグリセリルエステルの例は:1,2−ジオレインまたは1,3−ジオレイン、1,2−ジリノレインまたは1,3−ジリノレインおよび1,2−ジリノレニンまたは1,3−ジリノレニンなどのジ不飽和エステル;1,2−ジパルミチンまたは1,3−ジパルミチン、1,2−ジステアリンまたは1,3−ジステアリン、および1,2−ジデカノインまたは1,3−ジデカノインなどの飽和ジ飽和エステル;l−パルミトイル−2−オレオイル−グリセロールまたは1−オレオイル−2−パルミトイル−グリセロール、l−パルミトイル−2−リノレオイル−グリセロールまたは1−リノレオイル−2−パルミトイル−グリセロールなどの飽和不飽和ジグリセリルエステル;を含む。
【0021】
モノグリセリルエステルの例は:1−オレインまたは2−オレイン、1−リノレインまたは2−リノレインおよび1−リノレニンまたは2−リノレニンなどの不飽和エステル;1−パルミチンまたは2−パルミチン、1−ステアリンまたは2−ステアリン、および1−デカノインまたは2−デカノインなどの飽和エステル;を含む。
【0022】
ポリエチレングリコール(PEG)ジエステルの例は:1,2−ジオレインまたは1,3−ジオレイン、1,2−ジリノレインまたは1,3−ジリノレインおよび1,2−ジリノレニンまたは1,3−ジリノレニンなどのジ不飽和エステル;1,2−ジパルミチンまたは1,3−ジパルミチン、1,2−ジステアリンまたは1,3−ジステアリン、および1,2−ジデカノインまたは1,3−ジデカノインなどの飽和ジ飽和エステル、を含む。飽和不飽和ジグリセリルエステルからのPEGジエステルの他の例は、l−パルミトイル−2−オレオイル−グリセロールまたは1−オレオイル−2−パルミトイル−グリセロール、1−パルミトイル−2−リノレオイル−グリセロールまたは1−リノレオイル−2−パルミトイル−グリセロールを含む。
【0023】
天然オイルの具体例は、キャノーラ油、ヤシ油、コーン油、落花生油、ゴマ油、オリーブ油、パーム油、ベニバナ油、大豆油、綿実油、菜種油、ヒマワリ油およびそれらの混合物である。これらの中で綿実油が好ましい。
【0024】
当該技術分野において公知な方法により非水性基剤を改変することができる。例えば過酸化不飽和オイル基剤を用いる実施態様において、改変基剤は約0.1〜約600の過酸化物価を有することができ、一部の実施態様において、約10、約20、約40、または約80あるいはその間の任意の値を有することができる。本明細書においては、過酸化物価は、1000グラムのオイルサンプルに対するミリ当量(mEq)の過酸化物で表す。
【0025】
前記の成分以外に、非水性液体組成物は追加の化合物を含むこともできる。例えば、非水性液体組成物が医薬組成物の場合、該組成物は任意の薬学的に許容される成分を含むことができる。典型的な追加の成分は、例えば、医薬活性成分、賦形剤、添加剤、懸濁化剤、防腐剤、湿潤剤、増粘剤、緩衝液および凝集剤である。ゴム類(例えばアカシアゴム、カラギナン、アルギン酸ナトリウムおよびトラガント)、セルロース誘導体(例えばカルボキシメチルセルロースナトリウム、微結晶セルロース、およびヒドロキシエチルセルロース)、および粘土(例えばベントナイトおよびコロイド状アルミニウムマグネシウム)などの懸濁化剤を含むことができる。メチルおよびプロピルパラベン、ベンジルアルコール、クロロブタノールならびにチメロサールなどの防腐剤を加えることができる。陰イオン界面活性剤(例えばドキュセートナトリウムおよびラウリル硫酸ナトリウム)、非イオン性界面活性剤(例えばポリソルベート、ポリオキサマー、オクトキシノール−9)、および陽イオン界面活性剤(例えば臭化トリメチルテトラデシルアンモニウム、塩化ベンザルコニウム、塩化ベンゼトニウム、ミリスチルγ塩化ピコリニウム)を使用することができる。ゼラチン、天然ゴム類およびセルロース誘導体(懸濁化剤として上に記したものなど)などの増粘剤を加えることができる。クエン酸およびリン酸緩衝化剤などの緩衝液、および塩化ナトリウムおよびマンニトールなどの浸透物質を含むことができる。経口投与される医薬組成物のためには、風味剤、甘味料(例えばマンニトール、ショ糖、ソルビトールおよびデキストロース)、着色剤ならびに芳香剤を使用することができる。医薬組成物において、モノオレイン酸ソルビタン(Span 80(登録商標)としてシグマアルドリッチ社から入手可能)などの賦形剤およびホスファチジルコリン(ホスホリポン(Phospholipon)90HとしてAmerican Lecithin Companyから入手可能)を使用することができる。
【0026】
本発明の水性溶出溶媒は、当該技術分野において公知なすべての水性溶出溶媒であって良い。一般的に用いられる溶出溶媒は、水、塩酸(例えば約0.001M〜約0.1MHC1の範囲の濃度を有する)、ペプシン含有あるいは非含有の人工胃液(simulated gastric fluid)、種々の緩衝液(グリシン、クエン酸、酢酸、リン酸、およびホウ酸緩衝液)、酵素含有あるいは非含有の人工腸液(simulated intestinal fluid)(例えばパンクレアチン含有あるいは非含有の0.05Mリン酸緩衝液(pH7.5))、界面活性剤を含む水、界面活性剤を含む緩衝液および水性アルコール性溶液(例えば補助溶剤の役割を果たす、一般的に5以下の炭素を含む、水に溶ける低分子量アルコール)である。これらの種々の要素を、所定の検体の溶解条件を代えるために調製することができる。実験を繰返すことにより、所望の範囲内にインビトロ薬物放出速度を調節することを実験者に可能にさせる、最適の組成物を経験的に導き出すことが可能である。溶解度条件の調節はまた、インビボで異なる振舞いをするロット間の差異をインビトロで見分けることを実験者に可能にすることもできる。
【0027】
本発明の特定の実施態様において、所望により、界面活性剤を含む緩衝液は水性溶出溶媒として用いられる。緩衝液の種類は特に限定されないが、特定の系次第により選択されなければならない。緩衝液は、薬物放出媒体中の検体の溶解性を調節し、薬物放出挙動を最適化し、そして重要なサンプル間の識別の程度を最適化するように選択することができる。緩衝液の具体例は、pH2〜3の0.05Mグリシン緩衝液、pH3の0.05Mクエン酸緩衝液、pH4〜5の0.05M酢酸緩衝液、通常の生理食塩水中のpH5.5の0.05M酢酸緩衝液、pH6〜8の0.05Mリン酸緩衝液、カリウムを含まないpH6.8の0.05Mリン酸緩衝液、通常の生理食塩中のpH7.4の0.05Mリン酸緩衝液、pH8〜10の0.05Mホウ酸緩衝液である。好ましい緩衝液は、pH6〜7の0.05Mリン酸緩衝液である。緩衝液は、任意の適切なモル濃度を有することができるが、例えば約0.001M〜約0.5M、好ましくは約0.01〜約0.1のモル濃度を有することができる。溶出緩衝液に関する一般情報は、USP 24, pp. 2231-2240, United States Pharmacopeial Convention Inc,
Jan 1, 2000で見出すことができる。
【0028】
他の実施態様において、水性溶出溶媒は、所望により界面活性剤を含む水である。
【0029】
選択的に、水性溶出溶媒は界面活性剤を含むことができるが、界面活性剤は系の溶解性を調節するもう1つの方法である。典型的な有用な界面活性剤は非イオン、陽イオン、陰イオンおよび両性イオン界面活性剤である。本発明における使用に適した界面活性剤の具体例は、ドデシル硫酸ナトリウム、モノオレイン酸ポリオキシエチレンソルビタン(Tween 80(登録商標))、ケノデオキシコール酸、グリココール酸ナトリウム塩、モノラウリン酸ポリ(オキシエチレン)n−ソルビタン(Tween 20(登録商標))、タウロコール酸、オクチルフェノールエチレンオキシド縮合物(Triton X−100(登録商標))、および臭化ヘキサデシルトリメチルアンモニウムである。
【0030】
界面活性剤の種類と量は、検体、非水性液体組成物および水性溶出溶媒の特定の系により決まり、当業者により決定されうる。界面活性剤濃度は、臨界ミセル濃度以上であることも以下であることもできる。界面活性剤の典型的な濃度範囲は、約0.001%〜約1%である。
【0031】
水性溶出溶媒のpHは、研究する特定の系に従って選択されなければならない。一般的に、水性溶出溶媒のpHは、約1〜約10であり、好ましくは約2〜約8である。水性溶出溶媒のpHは検体の溶解性に影響することができ、このpHは実験におけるシンク条件の調節の方法の1つであることが一般的に知られている。水性溶出溶媒のpHを最適化することにより、一部の検体の溶出特性を調節することができる。医薬品の場合、これによりインビトロ薬物放出特性とインビボ薬物動態学的性能の間の相関関係を構築することが可能になる。
【0032】
水性溶出溶媒のpHを最適化することにより、一部の場合において界面活性剤の使用が必要でなくなる場合がある。水性溶出溶媒として特に好ましい系は、最適のpH値を有する水性緩衝液である。
【0033】
最適化したpH値を有する水性溶出溶媒を用い、かつ薬物放出媒体中の界面活性剤の使用を除去することにより、単一の濾過段階を用いて乳濁液混合物の残りの部分から溶液中の検体を分離することが可能となる場合がある。このことにより、大きな孔径を有するフィルターを通過させる濾過よりもより複雑で時間のかかる限外濾過の必要性が回避される。界面活性剤が用いられない場合、関心のある検体の水溶性分画を分離するために単一のフィルターを用いることができ、一方で乳濁液の油相小球体が実質的にフィルターを通過せず、水性溶出溶媒中の検体の量の測定に悪影響を及ぼさないことを確実にするために、実験パラメーター(例えば撹拌速度、ストローク長さ、撹拌時間)を乳濁液の小球体(即ち、小滴またはミセル)サイズが十分大きいように選択することができる。乳濁液のミセルサイズが小さすぎ(例えば0.2ミクロン以下)、単一のフィルターを通過する場合、溶液状態の検体を溶液状態で分離するために限外濾過を用いることができる。機械撹拌パラメーターを、小球体サイズを最適化するために調節することができる。
【0034】
1つの実施態様において、pH、界面活性剤濃度、およびシステムの機械撹拌パラメーターは、約0.2ミクロンの孔径を有するフィルター(Acrodisk(登録商標)シリンジフィルター、製品番号4496、Gelman Laboratory)を通して乳濁液を濾過でき、一方で乳濁液の油相小球体が実質的にフィルターを通過しないように最適化される。溶出系の物理的パラメーター(温度、撹拌速度、ストローク長さ、容器の形状、サンプルサイズおよび溶出溶媒の量)および化学的パラメーター(pH、シンク条件、緩衝液の濃度、界面活性剤、および補助溶剤)は、反復法により最適化できる。所望の孔径を有するフィルターが選択される。本発明の方法は、固定されたpH(例えばpH=7)を有する水性溶出溶媒を用いて実施される。形成された乳濁液は、選択されたフィルターを通して濾過される。乳濁液の小滴が実質的にフィルターを通過しない場合、単一の濾過で十分であり、水層中の検体の濃度は測定できると思われる。しかしながら、乳濁液の小滴がフィルターを通過してしまう場合、検体の水溶性部分の定量に移る前に、限外濾過または他の適切な精製段階(例えば液/液抽出)が必要な場合がある。小滴が実質的にフィルターを通過するか否かは、濾液を視覚的に検査することにより判定できる。濾液が透明でかすんでいない場合、小滴は実質的にフィルターを通過していない。ついで、物理的および化学的パラメーターを反復的に調節し、さらに最適な条件が特定されるまで実験を繰返すことができる。最適と分類されたいずれのpH値も、本発明の好ましい実施態様に使用できる。上記の追加の方法は、いかにして最適のpH範囲、界面活性剤の種類および濃度ならびに機械および熱的撹拌パラメーターを特定できるかを説明している。
【0035】
非水性液体組成物中の検体の溶出を測定するために、組成物を水性溶出溶媒と混合する。水性溶出溶媒と混合される組成物の量は、組成物の性質、溶出溶媒の性質、および用いる溶出溶媒の量などの種々の要素によって大きく変化しうる。適切な分析方法を用いて検出できる水層中の検体濃度として得られるいずれの量も許容できる。従って、水性溶出溶媒に対する非水性液体組成物の比率は場合により大きく変化することができる。一般的に、水性溶出溶媒に対する非水性液体組成物の比率は、約1:100〜約1:2000であり、好ましくは約1:250〜約1:1000である。本発明の他の利点は、少しのサンプル量で溶出を実施できることである。約500mL〜1000mLの水性溶出溶媒の量を必要とする標準的な溶出方法と比較して、本発明の方法の一部の実施態様において、本発明の方法は約10mL〜約100mL、または好ましくは、約20〜約50mLの水性溶出溶媒を用いて実施できる。
【0036】
非水性液体組成物と水性溶出溶媒を混合した後、得られた混合物を乳濁液が形成されるように撹拌する。「乳濁液」という用語は、少なくとも2つの相が不混和性または部分不混和性の液体である、2以上の相を含む分散系を意味する。
【0037】
非水性液体組成物と水性溶出溶媒の混合物は、いずれの適切な撹拌装置を用いても撹拌できる。一般的に、装置は混合物のために少なくとも1つの容器を有する。容器は,その中で混合物を混合でき、かつ撹拌中に混合物の損失がないように作られている任意のうつわ、入れ物、切り込みまたは他の形のことを言う。単数または複数の容器は、撹拌装置の常設部品であっても良く、あるいは撹拌装置と分離可能なものであっても良い。一般的には、再使用する前に洗浄するかあるいは廃棄できるように、容器は撹拌装置と分離可能なものが良いと思われる。典型的な容器は、プラスチック、ゴム、ガラス、金属、または加工紙製で使い捨ての、EPA型バイアル、遠心管、試験管、血清バイアル、ビーカー、エーレンマイヤーフラスコ、反応バイアル、または他の種類の入れ物を含む。好ましい容器は、ガラス製EPA型40mLバイアル、またはゴム栓のついたガラス製血清バイアル(50〜100mLサイズ)のいずれかの使い捨てバイアルである。
【0038】
乳濁液を製造できる装置であれば、任意の撹拌装置を使用できる。適切な撹拌装置の例は、市販の(温度制御つきあるいは温度制御無しの)種々の実験室用シェーカーであり、オービタル式、リニア式(レシプロ式)、または他の方式で撹拌できる。好ましい撹拌装置は、容器に含まれる混合物が激しく撹拌されるシェーカーである。シェーカーは、容器を水平方向に、垂直方向に、シーソー風にまたはそれらの任意の組み合わせで容器を動かすことができる。特に好ましいシェーカーはレシプロシェーカーである。撹拌装置は、特定の環境下では乳濁液を形成できるかも知れないが、一般的には、パドルアセンブリ中などで単に撹拌しただけでは、非水性液体組成物と水性溶出溶媒は乳濁液を形成しない。これらの装置中では、非水性液体組成物は一般的に水性溶出溶媒の表面に浮く。従って、これらの2成分の間の接触面積は、本発明における場合よりも小さく、検体の溶出速度はより小さいと思われる。
【0039】
図3は、本発明の方法に使用できる撹拌装置の概略図である。この装置はシェーカーであり、容器2が2つ取り付けてある水平プレート1を含む。作動中は、水平プレートは矢印で示される方向に水平に動く。容器はシェーカーの表面に横たわるように示してあるが、容器はまた直立の状態であることもできる。これにより、本発明の方法の精度と信頼度をさらに改良することができる。容器の形状が細長い場合、シェーカーのストロークは、例えば図3に矢印で示されているように、容器の延長線に並行であるのが好ましいが、他の任意の適切な方向であることもできる。
【0040】
非水性液体組成物と水性溶出溶媒は所定時間撹拌される。撹拌時間は大きく変化させることができ、例えば、撹拌量、検体、非水性液体組成物、溶出溶媒、温度、検体の量を測定するために用いる検出法の感度、および多くの他の要素によって決まる。さらに、撹拌時間は、短時間、中程度の時間または長時間の溶出速度あるいはそれらの組み合わせに関する情報が所望されているかどうかで決まる。一般的に、撹拌は、水性溶出溶媒中の検体の総量の約1%〜約100%、好ましくは約10%〜約100%が溶解するまで続けられる。一般的に撹拌は約5分〜約24時間、好ましくは約15分〜約60分行われる。
【0041】
撹拌段階中、非水性液体組成物と水性溶出溶媒の混合物は、任意の所望の接触温度に保つことができる。通例、混合物は、例えば室温(即ち約22−25℃)または約37℃などの比較的一定の接触温度に保たれる。しかしながら、溶出速度をあげるために高温を使用することができるし、溶出速度を下げるために低温を使用することができる。混合物の温度は溶出速度に影響するため、2以上の実験結果を比較するためには、各実験に関して同一の温度を用いなければならない。本明細書においては、「同一温度」は、異なる実験の温度間の差異が、多くとも5℃、好ましくは多くとも2℃であることを意味する。好ましくは接触温度は室温(即ち22〜25℃)である。
【0042】
振盪速度などの、接触中の撹拌量は、検体の溶出速度にも影響し、最適の条件は、撹拌容器のサイズおよび形状、非水性液体組成物および水性溶出溶媒などの種々の要素に基づいて決定されなければならない。一般的に、サイクル数は、約100〜約500であり、好ましくは約100〜約300である。ストローク長さは、好ましくは約0.5インチ〜約2インチであり、さらに好ましくは約0.75インチ〜約1.5インチである。
【0043】
非水性液体組成物および水性溶出溶媒の混合物を所定時間撹拌した後、水性溶出溶媒中の検体の量を測定する。一部の検出法を用いれば、溶出試験装置中に水性溶出溶媒が残っていても検体の量は測定できる。しかしながら、一般的に、例えばシリンジまたは常設試料採取管により、溶出試験装置から少なくとも一部の水性溶出溶媒が取り出される。分析のために水性溶出溶媒の全てを使用することが可能であるし、このことは一部の検出法には必要であるが、他の場合には、水性溶出溶媒の一部のみを用いる。検体の量を測定するために取り出すサンプルサイズは、種々の要素、特に用いる検出法によって決まり、約0.1mL〜約25mL、好ましくは約0.5mL〜約15mLであることができる。所望であれば、検体の量を測定するために使用する水性溶出溶媒のサンプルは、溶出試験装置から取り出した後、濾過または遠心分離することができる。これによりオイルまたは固相から水性層を除くことができ、従って検体の測定を妨害し測定を混乱させる可能性のある、検体を含む粒子または乳濁液を水層から取り除くことができる。約0.1〜約50ミクロン、好ましくは約0.1〜約0.3ミクロンの平均孔径を有するフィルターを通して濾過するなどのいずれの適切な方法を用いても濾過を達成できる。一部の場合において、限外濾過が必要な場合がある。その場合、約0.001ミクロン〜約0.1ミクロン、好ましくは約0.001〜約0.01の孔径を有するフィルターがより適切であろう。当該技術分野において公知である適切なフィルター用具の多くの例がある。これらのフィルターは、例えば、Gelman Laboratory製Acrodisk(登録商標)、およびMillipore Corporation製Centriprep50(登録商標)などの販売名で市販されている。所望による濾過段階の後、水性溶出溶媒中の検体の量が測定される。検体の量を測定するために適切な任意の分析方法も使用できる。分析方法の選択は、検体の性質、検体の濃度範囲、溶出溶媒を含む種々のパラメーターによって決まるが、これらの分析方法もまた研究室で利用可能である。分析方法の具体例は、分離技術(例えば高速液体クロマトグラフィー、液体クロマトグラフィー、薄層クロマトグラフィー、キャピラリー電気泳動、ガスクロマトグラフィ−)、光度技術および分光光度技術(例えば紫外・可視(UV−Vis)、フーリエ変換赤外(FTIR)、原子吸光(AA)、原子発光(AE)、質量分析法(MS))である。クロマトグラフ法、特にガスクロマトグラフィ−(GC)および高速液体クロマトグラフィー(HPLC)が好ましい。適切なクロマトグラフ法の例は、当該技術分野に公知な種々の検出技術のいずれかを組み込んだ、逆相高速液体クロマトグラフィー(RP−HPLC)、順層高速液体クロマトグラフィー(NP−HPLC)である。適切なクロマトグラフ法と共に用いることができる検出技術の例は、UV−Vis、屈折率、質量分析法、および光散乱検出を含む。分析方法として、UV−Vis検出法と組み合わせたフローインジェクション分析(FIA)もまた使用できる。リアルタイムで製造システムのインプロセス評価を行う場合など、サンプルのハイスループットが必要とされる時にFIAはとりわけ適切である。
【0044】
溶解した検体の量が単一の所定時間で測定される実施態様に関して、本発明の方法を上記で説明した。多くの場合、検体が一定速度で放出されるか、あるいは速度が時間と共に変化(例えば、溶出試験の開始時に多量、そしてその後、より少量)するかどうか測定するために、一定の時間に亘って、溶出速度をモニターすることは興味の持たれるところである。これらの場合、異なる所定の時間に2以上のサンプルを取り出し、これらのサンプルを個々に分析するために、十分に大きな容器を使用することができる。2以上の同一の実験を用意し、撹拌時間のみを変える以外は同一の条件で撹拌することも可能である。この場合、図3に示すように、同一の撹拌装置を用いて各混合物を同時に撹拌することができる。異なる装置を用いて、混合物を交互に撹拌することもまた可能である。これらの別個の混合物から異なる時点得採取した水性溶出溶媒を個別に分析する。その結果を、次に溶出速度の時間依存性プロファイルを測定するために用いることができる。
【0045】
本発明の方法を用いれば、非水性液体組成物中における検体の溶出速度を信頼度高く正確に測定することができる。薬学的応用において、本発明のインビトロ法により得られる結果は、インビボ薬物動態学的研究の結果と良く相関させることができる。従って、本方法は、適切な生物学的性能およびロットの一貫性を確実にするための、医薬品の製造中における品質管理における厳格で信頼性のある方法として使用できる。本方法は簡単で安価でありかつ迅速であるため、医薬品およびそれらの剤形の開発に有利に使用することもできる。本発明の方法は、従来法よりも迅速であるため、遅い溶出速度を有する薬物または徐放性製剤の溶出速度を測定するために有用である。医薬品の剤形における薬物放出を減じる(徐放性を付与する)ことを意図する製造方法のリアルタイムモニターに本方法はとりわけ有用である。
【0046】
以下の実施例は本発明を説明するために提示される。しかしながら、これらを制限と解釈すべきではない。特記しない限り、実施例中に引用した全てのパーセンテージ、比率、割合などは重量換算で示してある。
【実施例】
【0047】
精度:
本発明の方法の精度は、繰り返し測定の相対標準偏差(RSD)を算出することにより測定できる。低い標準偏差値は、高い精度を示す。一般的に、相対標準偏差は検体の溶出速度を同一の条件で3回以上測定することにより測定される。次いで、以下の式に従って相対標準偏差値を算出する:
【数1】
ここで、s.d.は以下で定義される標準偏差値である:
【数2】
ここで、Xは個別の結果であり;Nはデータポイント数であり;そして
【数3】
は平均値である。本発明の方法により得られる相対標準偏差値は1%未満であることができる。
正確さ:
本発明の方法の正確さは、既知量の検体を加えた非水性液体組成物から水性媒体への検体の移動を測定することにより測定できる。加えた非水性液体組成物は、振盪または撹拌により水性薬物放出媒体と平衡され、しかる後水性溶出溶媒中の検体の量が測定される。次いで水性媒体へ移動した検体の濃度を、検体が理論的に100%移動した場合(例えばピペッティングおよび計量の誤差または損失が無く、検体が100%溶解し検体が100%検出されると仮定する場合)に得られる濃度と比較する。本発明の方法は、約70%〜約100%の正確さを有し、好ましくは約90%〜約100%の正確さを有する。
一般的溶出方法
装置:
プラットフォームシェーカー:レシプロシェーカーモデル5850(Eberbach社から入手可能)、公称ストローク長さおよそ2.54cm、回転数200サイクル/分。バイアルは水平位置に設置し、図3に示すようにストローク方向に並行に並べた。プラットフォームシェーカーを適切に調節した温度環境(例えば22℃)に保つ。
バイアル:40mL(EPA型)テフロン(登録商標)裏打ちスクリューキャップ。Qorpak社から入手可能(部品番号7588T)。
プラスティックシリンジ:BD Disposable 10mLプラスティックシリンジまたは同等物
フィルター:Acrodisk 0.2ミクロン(部品番号4496)
方法:
空の40mLバイアルに、溶出試験のための組成物の適切な量(例えば30−70mg)を分配する。適切な温度(例えば22℃)で平衡させる。溶出溶媒を適切な温度(例えば22℃)で平衡させる。適切な量(例えば30mL)の溶出溶媒を、溶出試験のためのサンプルを含むバイアルに加える。全てのサンプルに関して繰返す。この方法を約2〜3分以内に完了する。撹拌を開始する。所定時点で、定量分析のためにサンプルを取り出す。必要に応じて濾過する。定量分析に進む。
一般的定量分析方法
特記しない限り、以下の一般的方法に従った。
装置:定組成操作が可能なHPLC(例えばAgilent 1100(Agilent Technologiesから入手可能))。
検出器:UV−Vis254nm検出器(例えばダイオードアレイ検出器(Diode array detector)、検出波長:254nm、Agilent Technologiesから入手可能)。
カラム:Waters Symmetry C8, 3.9 x 50 mm、Waters Corporationから入手可能
注入量:10μl
流速:1〜2mL/min
圧力:3000psi
移動層:3.85gの酢酸アンモニウムおよび13.5mLの40%テトラブチル水酸化アンモニウムをMilli−Q水に溶解し、全部で700mLの容量にした。氷酢酸を用いてpHを6.7±0.1に調節した。次いで、溶液を0.45μmのメンブランフィルターを通して濾過した。濾過後、200mLのメタノールおよび110mLのテトラヒドロフランを加え、混合物を減圧下超音波処理し脱気した。
検体の放出量の算出
各時点での放出された検体(例えばセフチオフル)の量は、以下の式に従って算出できる。
【数4】
式中、
Wstd=標準試料の重量(mg)
P=セフチオフル遊離酸としての参照標準の純度
Rstd=標準試料のピーク面積
DISVOL=溶出液の容量(mL)(30)
WSVOL=実用標準の容量(mL)(10)
Rsam=測定試料のピーク面積
Wsam=サンプル懸濁液の重量(mg)
1000=サンプル重量のmgからグラムへの変換
である。
(実施例1)
【0048】
以下に記載した方法を用いて、2つのCCFA懸濁液サンプルを測定した。
【0049】
綿実油をジャケットつき容器に注ぎいれ、115℃に加熱することにより、非水性ビヒクルを調製した。ホスホリポン90H(0.05重量%)(American Lecithin Co. から入手可能)を加え、混合した。溶液を45℃に冷却した。モノオレイン酸ソルビタン(0.15重量%)を加え、混合した。CCFAを100mg/mL加え、懸濁液が均一になるまでトリブレンダーを通して混合した。懸濁液をトリブレンダーを通して再循環させ、タンク撹拌器を作動させながら濾過した。得られた懸濁液を滅菌バイアルに充填し、栓をし、シールした。シールしたバイアルをγ線照射を用いて滅菌した。これらのロットを40,700および40,620とラベルをつけた。1%Tween 80界面活性剤(Sigma−Aldrich社から入手可能)を0.05MpH6.0リン酸緩衝液に溶解することにより、水性溶出溶媒を調製した。21.8グラムの第一リン酸カリウムおよび3.48グラムの第二リン酸カリウムを脱イオン水に加え、200mLに希釈してpH6.0リン酸緩衝原液を調製した。50mLの緩衝原液を脱イオン水を用いて900mLに希釈することにより0.05MpH6.0リン酸緩衝原液を調製した。
【0050】
65mgから85mgの間のCCFA懸濁液ロット番号40,700および30mLの水性溶出溶媒を、テフロン(登録商標)裏打ちスクリューキャップ(Qorpak社から入手可能)つきの40mL EPA型ガラスバイアルに入れた。全部で9つの同一のバイアルを調製した。第2のロットである、CCFA懸濁液ロット番号40,620を用いてこの方法を繰り返し、別の9つのバイアルを調製した。これらのバイアルをレシプロシェーカー(Eberbach Model 5850、ストローク長さ=17/16インチ、振盪速度=200サイクル/分)上に水平におき、撹拌を開始した。15分の撹拌の後、各ロットの最初の3つのバイアルをシェーカーから取り除いた。各バイアルから15ミリリットルのサンプルを採取し、限外濾過装置(Centriprep 50限外濾過装置、AmiconまたはMillipore Corporationから入手可能)を用いて限外濾過した。前述の一般的分析方法を用いて濾液中のCCFA量を測定した。この方法を撹拌の60分および120分に繰返した。各ロットの残りのバイアルをそれぞれ同様な方法で取り除き処理した。
【0051】
得られた結果を図4に示す(ロット40,620のデータを菱形で表し、ロット40,700のデータを正方形で表す)。測定された溶出速度は、前回インビボ実験で観察された薬物動態学的性能と相関関係を示した。
【0052】
界面活性剤として、1% Tween 80の代わりに1% Tween 20を用いて、実験を繰返した。結果を図5に示す(ロット40,620のデータを菱形で表し、ロット40,700のデータを正方形で表す)。測定された溶出速度は、前回インビボ実験で観察された薬物動態学的性能と相関関係を示した。
(実施例2)
【0053】
本実施例は、本発明の方法により得られたインビトロ結果と薬物動態学的研究により得られたインビボ結果との間の相関関係を明らかにする。
【0054】
異なるインビボ薬物動態学的性能を示すCCFA S.Supensionの3つのロット(40,620;40,700;JMS−144F)を本評価に用いた。各ロットは同一の組成を有し、かつ前述の実施例1におけると同じ方法を用いて(ただしJMS−144Fはγ照射処理によって滅菌されていない)製造したが、これらのロットは異なるインビボ薬物動態学的性能を示した。薬物動態学的性能における差異は、セフチオフルが動物の血流中で検出される時間数により示される徐放効果の持続時間における差異により明らかにされた。セフチオフルに関する分析検出限界は、血漿1mLあたり0.2マイクログラムのセフチオフルである。徐放効果の持続時間は、一般に、「0.2mcg/mL以上の時間」として表わされる。
【0055】
本発明に記載の方法を用いて3つのロットを試験した。インビトロ薬物放出結果と0.2mcg/mLを超す時間との相関関係を図6に示す。60分(正方形で表されるデータポイント)および150分(三角形により表されるデータポイント)の時点に関するインビトロ結果を示す。実線はデータに適合する、最小二乗法による回帰直線であり、インビトロ放出量とインビボで観察される徐放効果の持続時間との逆相関を説明する例に含まれる。所定時間にインビトロでより多くのCCFAを放出したロットは、インビボ徐放性の持続時間がより短かった。
(実施例3)
【0056】
本実施例は、水性溶出溶媒のpHを最適化することにより、薬物放出媒体における界面活性剤の使用を除去することができ、かつ限外濾過の必要性もまた除去できる。本実施例において、CCFAを検体として用いた。CCFAの溶解性はpHに応じて変化する。pH5以下では、CCFAは比較的不溶性であり;CCFAの溶解性はpHの増加に伴って増加する。この薬物放出実験において、2つの異なる薬物放出媒体:(1)1% Tween 20を含むpH6.0リン酸緩衝液、および(2)外因性界面活性剤を含まないpH6.5リン酸緩衝液、を用いた。緩衝液を以下の方法で調製した:21.8グラムの第一リン酸カリウムおよび3.48グラムの第二リン酸カリウムを脱イオン水に加え希釈して200mLにし、次いで脱イオン水により再度18倍に希釈して(即ち、50mLを脱イオン水により希釈して900mLにする)、0.05MpH6.0リン酸緩衝液を調製した。31.98グラムの第一リン酸カリウムおよび15.39グラムの第二リン酸カリウムを脱イオン水に加え1000mLにし、次いで脱イオン水により再度10倍に希釈して(即ち、100mLの緩衝原液を脱イオン水により1000mLにする)、0.05MpH6.5リン酸緩衝液を調製した。
【0057】
異なるインビボ薬物動態学的性能を示したCCFA S.Suspension(40,620;40,700;JMS−111)の3つのロットを、本評価に用いた。各ロットは同じ組成を有し、かつ前述の実施例1におけると同じ方法(ただしロットJMS−111はγ照射を用いる処理により滅菌されていない)を用いて製造されたが、これらのロットは、最初の薬物放出媒体(1% Tween 20を含むpH6.0リン酸緩衝液)を用いて試験した場合、異なるインビトロ薬物放出速度を示した。ロット間のインビトロ放出の変動は、60分で放出されたCCFAの種々の量に反映されている(表1)。
【0058】
外因性界面活性剤を含まないpH6.5リン酸緩衝液からなる薬物放出媒体を調製し、インビトロ薬物放出測定を繰り返した。pHを6.0から6.5に上昇させ、薬物放出媒体からTween 20を除去することにより、3つのロットに関するインビトロ放出速度の順位関係は同一であり、乳濁液の油相小球体はかなり大きく、かつ単一の濾過により除去できることが見出された。これにより、限外濾過精製段階を単一の濾過に置き換えることができた。さらに、全3サンプルに関するインビトロ放出速度は、pH6.5媒体においてより速いことが見出された。したがって、アッセイ時間は60分から30分に短縮化することができた。最後に、3つのCCFAロット間の適切な絶対分解能が達成され、より速く(60分に対して30分)かつより簡単な(Tween 20を除去し、かつ限外濾過よりも簡単な)方法により、種々のインビトロ放出速度を有するロットを適切に識別することができることが見出された。絶対分解能は、薬物放出媒体中のロットが、その媒体に関して具体的に選択した時点で放出したCCFAの量の間の差異として定義した。
【表1】
(実施例4)
【0059】
CCFA懸濁液のための2つの異なる非水性基剤を調製し、それぞれをCCFAに加えた。添加物は2つのおおよその濃度レベルで3回調整した。正確なCCFA添加量を記録した。
【0060】
非水性基剤である基剤−1は100%ヤシ油(Miglyol 812として入手可能)であり、一方、非水性基剤である基剤−2はヤシ油(Miglyol 812としてHulsAmericaより入手可能)および綿実油(Welch,Home & Clark Companyより入手可能)の1:1(v:v)混合物であった。
【0061】
CCFAを加えた基剤混合物は、40mL EPAバイアル中のおよそ65mgの基剤(1または2)に既知量の薬物を正確に加え混合することにより調整した。基剤混合物の調製物について、レシプロシェーカー上で振盪することによりサンプルを溶出溶媒で4時間抽出した。サンプルを濾過し、濾液中のCCFA濃度をHPLCにより測定した。結果を表2に要約する。98.4〜100.1%の範囲のCCFAの回収率が得られた。
【表2】
【図面の簡単な説明】
【0062】
【図1】検体が2回以上測定される場合の、1つの可能性のある溶出速度のプロットの図である。
【図2】検体が2回以上測定される場合の、他の可能性のある溶出速度のプロットの図である。
【図3】シェーカーの図式を示す図である。
【図4】界面活性剤としてTween 80を使用する実施例1の溶出試験の結果を示す図である。
【図5】界面活性剤としてTween 20を使用する実施例1の溶出試験の結果を示す図である。
【図6】図6は実施例2のインビトロ−インビボ相関関係の結果を示す図である。
【特許請求の範囲】
【請求項1】
非水性液体組成物から水性媒体への検体の移動を特徴づける方法であって、以下の段階:
(a)検体および非水性基剤を含む非水性液体組成物を用意する段階;
(b)非水性液体組成物を水性溶出溶媒と混合する段階;
(c)非水性液体組成物と水性溶出溶媒を撹拌して乳濁液を形成する段階;および
(d)水性溶出溶媒中の検体の量を測定する段階;
を含む方法。
【請求項2】
段階(d)において、水性溶出溶媒中の検体の量が2以上の時点で測定される、請求項1に記載の方法。
【請求項3】
水性溶出溶媒中の検体の量を測定する前に、水性溶出溶媒中の検体の量を測定するために用いられる、水性溶出溶媒をフィルターに通過させる段階をさらに含む、請求項1に記載の方法。
【請求項4】
フィルターの孔径が約0.1〜約50ミクロンの範囲である、請求項3に記載の方法。
【請求項5】
非水性液体組成物が医薬組成物である、請求項1に記載の方法。
【請求項6】
検体が医薬組成物中の医薬活性成分である、請求項5に記載の方法。
【請求項7】
医薬組成物が徐放性製剤である、請求項5に記載の方法。
【請求項8】
医薬組成物が薬学的に許容される成分をさらに含む、請求項5に記載の方法。
【請求項9】
ACE阻害剤;α−アドレナリンアゴニスト;β−アドレナリンアゴニスト;α−アドレナリン遮断薬;β−アドレナリン遮断薬(β遮断薬);抗酒薬;アルドースレダクターゼ阻害剤;アルドステロンアンタゴニスト;アミノ酸;タンパク同化剤;鎮痛剤(麻薬性および非麻薬性の両方);麻酔剤;摂食障害;制酸剤;駆虫薬;抗にきび剤;抗アレルギー剤;抗男性ホルモン;抗狭心症薬;抗不安薬(antianxiety);抗不整脈薬;抗喘息剤;抗菌剤および抗生物質;脱毛防止薬およびはげ予防薬;抗アメーバ剤;抗体;抗コリン剤;抗凝血物質および血液希釈剤;大腸炎治療剤;鎮痙薬;膀胱炎治療剤;抗うつ薬;抗糖尿病薬;下痢止め;抗利尿薬;解毒剤;制吐剤;抗エストロゲン剤;抗膨満薬;抗真菌剤;抗原;緑内障治療薬;抗ヒスタミン剤;過剰行動治療剤;抗高リポタンパク血症剤;降圧剤;甲状腺機能亢進症治療薬;低血圧治療剤;甲状腺機能低下症治療薬;抗感染薬;抗炎症剤(ステロイド性および非ステロイド性の両方);抗マラリア薬;偏頭痛治療薬;抗腫瘍剤;抗肥満薬;抗パーキンソン薬およびジスキネジア治療剤;肺炎治療剤;抗原虫剤;かゆみ止め;抗乾癬剤;抗精神病薬;解熱剤;リウマチ治療剤;分泌抑制薬;抗ショック剤;抗痙攣薬;抗血栓剤;抗癌剤;鎮咳剤;抗潰瘍剤;抗ウイルス剤;抗不安薬(anxiolytics);殺菌素;骨密度増加剤;気管支拡張剤;カルシウムチャンネル遮断薬;炭酸脱水酵素阻害剤;強心薬および強心剤;化学療法剤;胆汁分泌促進物質;コリン作用薬;慢性疲労症候群治療薬;中枢興奮薬;凝固剤;避妊薬;嚢胞性線維症薬;充血除去剤;利尿薬;ドーパミン受容体アゴニスト;ドーパミン受容体アンタゴニスト;酵素;エストロゲン;去痰薬;胃過敏治療剤;グルココルチコイド;止血薬;HMG−CoAレダクターゼ阻害剤;ホルモン;睡眠剤;免疫調節剤;免疫抑制剤;下剤;口腔および歯周疾患用医薬;縮瞳薬;モノアミン酸化酵素阻害剤;粘液溶解剤;多発性硬化症薬;筋弛緩剤;散瞳薬;麻薬拮抗薬;NMDA受容体アンタゴニスト;オリゴヌクレオチド;眼科用薬;子宮収縮薬;ペプチド、ポリペプチドおよびタンパク質;多糖類;プロゲストゲン;プロスタグランジン;プロテアーゼ阻害剤;呼吸促進剤;鎮静剤;セロトニン取り込み阻害剤;アンドロゲンを含む性ホルモン;禁煙薬;平滑筋弛緩薬;平滑筋刺激薬;血栓溶解剤;鎮静剤;尿酸性化剤;尿失禁薬;血管拡張剤;血管保護剤;およびそれらの組み合わせから検体が選択される、請求項1に記載の方法。
【請求項10】
検体がセファロスポリンである、請求項1に記載の方法。
【請求項11】
検体がセフチオフル、その薬学的に許容される塩または誘導体である、請求項1に記載の方法。
【請求項12】
非水性基剤が脂質である、請求項1に記載の方法。
【請求項13】
非水性基剤がオイルである、請求項12に記載の方法。
【請求項14】
キャノーラ油、ヤシ油、コーン油、落花生油、ゴマ油、オリーブ油、パーム油、ベニバナ油、大豆油、綿実油、菜種油、ヒマワリ油およびそれらの混合物からなる群からオイルが選択される、請求項13に記載の方法。
【請求項15】
オイルが綿実油である、請求項14に記載の方法。
【請求項16】
非水性液体組成物が懸濁液、溶液または分散体である、請求項1に記載の方法。
【請求項17】
水性溶出溶媒が緩衝液を含む、請求項1に記載の方法。
【請求項18】
グリシン緩衝液、クエン酸緩衝液、酢酸緩衝液、リン酸緩衝液、およびホウ酸緩衝液からなる群から緩衝液が選択される、請求項17に記載の方法。
【請求項19】
緩衝液が最適のpHを有する、請求項18に記載の方法。
【請求項20】
水性溶出溶媒が最適のpHを有する、請求項1に記載の方法。
【請求項21】
水性溶出溶媒が界面活性剤を含まない、請求項1、21、または22に記載の方法。
【請求項22】
水性溶出溶媒が界面活性剤を含む、請求項1に記載の方法。
【請求項23】
水性溶出溶媒に対する非水性液体組成物の重量比が約1:100〜約1:2000である、請求項1に記載の方法。
【請求項24】
段階(c)がシェーカー上で行われる、請求項1に記載の方法。
【請求項25】
シェーカーがレシプロシェーカーである、請求項24に記載の方法。
【請求項26】
シェーカーが約50〜約400サイクル/分のストローク速度を有する、請求項24に記載の方法。
【請求項27】
段階(c)において、非水性液体組成物と水性溶出溶媒が40mL EPA型バイアルまたは50〜100mL血清型バイアルに含まれる、請求項1または24に記載の方法。
【請求項28】
薬学的に許容される成分が、賦形剤、添加剤、懸濁化剤、防腐剤、湿潤剤、増粘剤、緩衝液、凝集剤、風味剤、甘味料、着色剤および芳香剤からなる群から選択される、請求項8に記載の方法。
【請求項29】
セファロスポリンがセフチオフル、セフェピム、セフィキシム、セフォペラゾン、セフォタキシム、セフポドキシム、セフタジジム、セフチゾキシム、セフトリアキソン、モクサラクタム、およびそれらの薬学的に許容される塩ならびに誘導体からなる群から選択される、請求項10に記載の方法。
【請求項30】
水、塩酸溶液、人工胃液、緩衝液、人工腸液、界面活性剤を含む水、界面活性剤を含む緩衝液、および水性アルコール性溶液から水性溶出溶媒が選択される、請求項1に記載の方法。
【請求項31】
非水性液体組成物が懸濁液、溶液、または分散体である、請求項1に記載の方法。
【請求項32】
非水性液体組成物が乳濁液では無い、請求項1に記載の方法。
【請求項33】
検体を含む非水医薬組成物のインビボ性能を予測する方法であって、請求項1に記載の方法に従った、組成物から水性媒体への検体の移動を特徴づける段階を含む該方法。
【請求項34】
公共利用のために、検体を含む非水医薬組成物を商業的に製造し販売する方法であって、
(a)非水医薬組成物を製造する段階;
(b)請求項1に記載の方法に従って、組成物から水性媒体への検体の移動を特徴づけル段階;および
(c)段階(b)からの結果が所望の標準内に収まることを確認する段階;
を含む該方法。
【請求項1】
非水性液体組成物から水性媒体への検体の移動を特徴づける方法であって、以下の段階:
(a)検体および非水性基剤を含む非水性液体組成物を用意する段階;
(b)非水性液体組成物を水性溶出溶媒と混合する段階;
(c)非水性液体組成物と水性溶出溶媒を撹拌して乳濁液を形成する段階;および
(d)水性溶出溶媒中の検体の量を測定する段階;
を含む方法。
【請求項2】
段階(d)において、水性溶出溶媒中の検体の量が2以上の時点で測定される、請求項1に記載の方法。
【請求項3】
水性溶出溶媒中の検体の量を測定する前に、水性溶出溶媒中の検体の量を測定するために用いられる、水性溶出溶媒をフィルターに通過させる段階をさらに含む、請求項1に記載の方法。
【請求項4】
フィルターの孔径が約0.1〜約50ミクロンの範囲である、請求項3に記載の方法。
【請求項5】
非水性液体組成物が医薬組成物である、請求項1に記載の方法。
【請求項6】
検体が医薬組成物中の医薬活性成分である、請求項5に記載の方法。
【請求項7】
医薬組成物が徐放性製剤である、請求項5に記載の方法。
【請求項8】
医薬組成物が薬学的に許容される成分をさらに含む、請求項5に記載の方法。
【請求項9】
ACE阻害剤;α−アドレナリンアゴニスト;β−アドレナリンアゴニスト;α−アドレナリン遮断薬;β−アドレナリン遮断薬(β遮断薬);抗酒薬;アルドースレダクターゼ阻害剤;アルドステロンアンタゴニスト;アミノ酸;タンパク同化剤;鎮痛剤(麻薬性および非麻薬性の両方);麻酔剤;摂食障害;制酸剤;駆虫薬;抗にきび剤;抗アレルギー剤;抗男性ホルモン;抗狭心症薬;抗不安薬(antianxiety);抗不整脈薬;抗喘息剤;抗菌剤および抗生物質;脱毛防止薬およびはげ予防薬;抗アメーバ剤;抗体;抗コリン剤;抗凝血物質および血液希釈剤;大腸炎治療剤;鎮痙薬;膀胱炎治療剤;抗うつ薬;抗糖尿病薬;下痢止め;抗利尿薬;解毒剤;制吐剤;抗エストロゲン剤;抗膨満薬;抗真菌剤;抗原;緑内障治療薬;抗ヒスタミン剤;過剰行動治療剤;抗高リポタンパク血症剤;降圧剤;甲状腺機能亢進症治療薬;低血圧治療剤;甲状腺機能低下症治療薬;抗感染薬;抗炎症剤(ステロイド性および非ステロイド性の両方);抗マラリア薬;偏頭痛治療薬;抗腫瘍剤;抗肥満薬;抗パーキンソン薬およびジスキネジア治療剤;肺炎治療剤;抗原虫剤;かゆみ止め;抗乾癬剤;抗精神病薬;解熱剤;リウマチ治療剤;分泌抑制薬;抗ショック剤;抗痙攣薬;抗血栓剤;抗癌剤;鎮咳剤;抗潰瘍剤;抗ウイルス剤;抗不安薬(anxiolytics);殺菌素;骨密度増加剤;気管支拡張剤;カルシウムチャンネル遮断薬;炭酸脱水酵素阻害剤;強心薬および強心剤;化学療法剤;胆汁分泌促進物質;コリン作用薬;慢性疲労症候群治療薬;中枢興奮薬;凝固剤;避妊薬;嚢胞性線維症薬;充血除去剤;利尿薬;ドーパミン受容体アゴニスト;ドーパミン受容体アンタゴニスト;酵素;エストロゲン;去痰薬;胃過敏治療剤;グルココルチコイド;止血薬;HMG−CoAレダクターゼ阻害剤;ホルモン;睡眠剤;免疫調節剤;免疫抑制剤;下剤;口腔および歯周疾患用医薬;縮瞳薬;モノアミン酸化酵素阻害剤;粘液溶解剤;多発性硬化症薬;筋弛緩剤;散瞳薬;麻薬拮抗薬;NMDA受容体アンタゴニスト;オリゴヌクレオチド;眼科用薬;子宮収縮薬;ペプチド、ポリペプチドおよびタンパク質;多糖類;プロゲストゲン;プロスタグランジン;プロテアーゼ阻害剤;呼吸促進剤;鎮静剤;セロトニン取り込み阻害剤;アンドロゲンを含む性ホルモン;禁煙薬;平滑筋弛緩薬;平滑筋刺激薬;血栓溶解剤;鎮静剤;尿酸性化剤;尿失禁薬;血管拡張剤;血管保護剤;およびそれらの組み合わせから検体が選択される、請求項1に記載の方法。
【請求項10】
検体がセファロスポリンである、請求項1に記載の方法。
【請求項11】
検体がセフチオフル、その薬学的に許容される塩または誘導体である、請求項1に記載の方法。
【請求項12】
非水性基剤が脂質である、請求項1に記載の方法。
【請求項13】
非水性基剤がオイルである、請求項12に記載の方法。
【請求項14】
キャノーラ油、ヤシ油、コーン油、落花生油、ゴマ油、オリーブ油、パーム油、ベニバナ油、大豆油、綿実油、菜種油、ヒマワリ油およびそれらの混合物からなる群からオイルが選択される、請求項13に記載の方法。
【請求項15】
オイルが綿実油である、請求項14に記載の方法。
【請求項16】
非水性液体組成物が懸濁液、溶液または分散体である、請求項1に記載の方法。
【請求項17】
水性溶出溶媒が緩衝液を含む、請求項1に記載の方法。
【請求項18】
グリシン緩衝液、クエン酸緩衝液、酢酸緩衝液、リン酸緩衝液、およびホウ酸緩衝液からなる群から緩衝液が選択される、請求項17に記載の方法。
【請求項19】
緩衝液が最適のpHを有する、請求項18に記載の方法。
【請求項20】
水性溶出溶媒が最適のpHを有する、請求項1に記載の方法。
【請求項21】
水性溶出溶媒が界面活性剤を含まない、請求項1、21、または22に記載の方法。
【請求項22】
水性溶出溶媒が界面活性剤を含む、請求項1に記載の方法。
【請求項23】
水性溶出溶媒に対する非水性液体組成物の重量比が約1:100〜約1:2000である、請求項1に記載の方法。
【請求項24】
段階(c)がシェーカー上で行われる、請求項1に記載の方法。
【請求項25】
シェーカーがレシプロシェーカーである、請求項24に記載の方法。
【請求項26】
シェーカーが約50〜約400サイクル/分のストローク速度を有する、請求項24に記載の方法。
【請求項27】
段階(c)において、非水性液体組成物と水性溶出溶媒が40mL EPA型バイアルまたは50〜100mL血清型バイアルに含まれる、請求項1または24に記載の方法。
【請求項28】
薬学的に許容される成分が、賦形剤、添加剤、懸濁化剤、防腐剤、湿潤剤、増粘剤、緩衝液、凝集剤、風味剤、甘味料、着色剤および芳香剤からなる群から選択される、請求項8に記載の方法。
【請求項29】
セファロスポリンがセフチオフル、セフェピム、セフィキシム、セフォペラゾン、セフォタキシム、セフポドキシム、セフタジジム、セフチゾキシム、セフトリアキソン、モクサラクタム、およびそれらの薬学的に許容される塩ならびに誘導体からなる群から選択される、請求項10に記載の方法。
【請求項30】
水、塩酸溶液、人工胃液、緩衝液、人工腸液、界面活性剤を含む水、界面活性剤を含む緩衝液、および水性アルコール性溶液から水性溶出溶媒が選択される、請求項1に記載の方法。
【請求項31】
非水性液体組成物が懸濁液、溶液、または分散体である、請求項1に記載の方法。
【請求項32】
非水性液体組成物が乳濁液では無い、請求項1に記載の方法。
【請求項33】
検体を含む非水医薬組成物のインビボ性能を予測する方法であって、請求項1に記載の方法に従った、組成物から水性媒体への検体の移動を特徴づける段階を含む該方法。
【請求項34】
公共利用のために、検体を含む非水医薬組成物を商業的に製造し販売する方法であって、
(a)非水医薬組成物を製造する段階;
(b)請求項1に記載の方法に従って、組成物から水性媒体への検体の移動を特徴づけル段階;および
(c)段階(b)からの結果が所望の標準内に収まることを確認する段階;
を含む該方法。
【図1】

【図2】
【図3】

【図4】
【図5】

【図6】

【図2】
【図3】
【図4】
【図5】
【図6】
【公表番号】特表2006−503269(P2006−503269A)
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願番号】特願2004−536037(P2004−536037)
【出願日】平成15年9月10日(2003.9.10)
【国際出願番号】PCT/US2003/025853
【国際公開番号】WO2004/024121
【国際公開日】平成16年3月25日(2004.3.25)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
【公表日】平成18年1月26日(2006.1.26)
【国際特許分類】
【出願日】平成15年9月10日(2003.9.10)
【国際出願番号】PCT/US2003/025853
【国際公開番号】WO2004/024121
【国際公開日】平成16年3月25日(2004.3.25)
【出願人】(504396379)ファルマシア・アンド・アップジョン・カンパニー・エルエルシー (130)
【Fターム(参考)】
[ Back to top ]