
C型レクチンを介する免疫
本発明は免疫系の調節に関し、特に、CLEC9a分子が、MHCクラスI経路を介して細胞外抗原を交差提示することができる樹状細胞マーカーであるという発見に関する。このためこれらは、細胞傷害性Tリンパ球応答の発生に特に適する。材料および方法は、標的抗原に対する免疫応答の誘導、およびこれらの細胞が関与する望ましくない免疫応答の阻害または抑制の両方のために提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、免疫系の調節に関し、特に、標的抗原に対する免疫応答をプライミングおよび阻害するための、CLEC9a分子に対して親和性を有する分子の使用に関する。
【背景技術】
【0002】
免疫系は、感染性病原体の存在を検出し、自己組織を破壊することなく病原体に対する応答を誘発することができる。病原体の膨大な分子多様性ならびにその複製および突然変異率の高さを考えれば、この現象は驚くべきものである。多細胞生物は、進化の過程にわたっていくつかの性質の異なる免疫認識機構、すなわち「先天性」免疫系および「適応性」免疫系を発達させるよう促されてきた。
【0003】
進化的に古い先天性免疫系は、感染の存在および性質を検出し、宿主防御の第一線を担い、適応性免疫系のエフェクタークラスの開始および決定を制御する。
【0004】
樹状細胞(DC、dendritic cell)は、先天性免疫と抗原特異的適応応答を結び付ける上で極めて重要な役割を果たす。免疫応答を開始するために、DCは、(とりわけ)病原体が示す病原体関連分子パターン(PAMP、pathogen-associated molecular pattern)によってプライミングされる。その後DCは、適応性免疫応答の進行をうまく調整し、これは、抗原特異的T細胞およびB細胞によってさらにいっそう特殊化され駆動される。
【0005】
病原体に対するDCの抗原認識および取り込み機能は、PAMPを識別するパターン認識受容体(PRR、pattern recognition receptor)によって媒介される。DCにより発現されるこれらのPRRはToll様受容体(TLR、Toll like receptor)を含む1。DCはさらに、別のクラスの受容体としてC型レクチンを発現し、その中にはPRRとして機能し2-4、かつ/または細胞間コミュニケーションを媒介する5-8ことができるものがある。C型レクチンの中に、構造的かつ進化的に密接な関係があり、NK遺伝子複合体(NKC、NK gene complex)中にクラスターを形成している、単一の細胞外C型レクチンドメイン(CTLD、C-type lectin domain)を有するII型タンパク質の群がある。これらの受容体は、カルシウム結合部位および典型的な糖認識ドメインを欠くが、それにもかかわらずデクチン1で示されるように糖質に結合することができる。これらの受容体のいくつか(CD94/NKG2、NKG2D)は、MHC-Iまたは関連分子と相互作用し、阻害および活性化シグナルのバランスの結果としてNKおよびT細胞傷害性を阻害または活性化する。しかしながら、NKレクチン受容体の大部分について、NKまたはDC機能における結合特異性および関連性は不明である。したがって、DC上で発現されるC型レクチンは、微生物を認識するように作用する可能があるだけでなく、特異的な細胞の相補的構造を認識することによりDCの他の細胞とのコミュニケーションを調節する可能性もある。
【0006】
個体発生および/またはDCの置かれる微環境は、DCによる表面受容体の特徴的な組合せの発現をもたらし得る。例えば、表現型基準のみによって、マウスリンパ節DCを6つの主な亜集団に分類することができる10。これらのうち、リンパ組織中の通常の非形質細胞様DCは、伝統的にCD8αおよびCD8α+亜集団に細分される。異なるDCサブセットがある特定の病原体の特異的認識に関与および/または異なる免疫応答、例えば、Th1もしくはTh2(免疫)または制御性T細胞(寛容)などを調節している可能性があることが議論されてきた11。しかしながら、DCの表現型および機能的挙動は外部の活性化刺激によっても大きく左右され、顕著な可塑性を示す。DCサブセット間の差異を理解するための最初のアプローチとして、DC亜集団を単離しそのin vitroでの特性を評価した:マウスにおいて、脾臓DCのCD8α+およびCD8αサブセットは、in vitroでIL-12を産生する能力が異なっている12,13。しかしながら、in vitroでの示差的IL-12産生はパターン認識によっても測定され、異なるDCサブセットの機能的柔軟性を示した14。第2のアプローチとして、DCサブセットが単離され、抗原パルスされ、次いでin vivoに再注入された。CD8α+およびCD8α-サブセットは、in vivoでTh1およびTh2応答を別個にプライミングした15,16。免疫療法は、特定のDCサブセットにおいて選択的に発現される分子を測定できれば、実現可能である。こうした分子は、次にこのDCサブセットの機能を改変するための標的とすることができる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】PCT/US92/0996
【特許文献2】WO94/13804
【特許文献3】EP-A-0120694
【特許文献4】EP-A-0125023
【非特許文献】
【0008】
【非特許文献1】Mallat Z.ら、Circulation 108:1232〜7、2003年
【非特許文献2】Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年
【非特許文献3】Altschulら、Methods in Enzymology、266:460〜480(1996年)
【非特許文献4】Viorittoら、(Clin Immunol 122(2)、125〜134(2007年))
【非特許文献5】Tesniereら(Corr Op Immunol 21、1〜8(2008年))
【非特許文献6】Kimら(Immunity 27、321〜333(2007年)
【非特許文献7】Ward,E.S.ら、Nature 341、544〜546(1989年)
【非特許文献8】Birdら、Science、242、423〜426、1988年
【非特許文献9】Hustonら、PNAS USA、85、5879〜5883、1988年
【非特許文献10】P.Holligerら、Proc.Natl.Acad.Sci.USA 90 6444〜6448、1993年
【非特許文献11】Remington's Pharmaceutical Sciences、第20版、2000年、pub. Lippincott、WilliamsおよびWilkins
【非特許文献12】Turnerら、Nature 378、298(1995年)
【非特許文献13】Sanchoら、J Clin Invest.2008、118(6):2098〜110頁
【非特許文献14】Liuら、2002.J.Exp.Med.196:1091〜1097頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明者らは、CLEC9aが、マウスにおいてCD8を発現し、そのためCD8+樹状細胞と呼ばれる樹状細胞のサブセット上で選択的に発現されることを発見した。これは、細胞外由来の抗原を処理することができ、MHCクラスI分子を介してそれらをT細胞に提示することができると考えられているため、重要な細胞型である。これは、MHCクラスII分子を介して細胞外由来抗原を提示する大部分の抗原提示細胞とは対照的である。したがって、この抗原提示機構を「交差提示」と呼ぶこともある。これらの細胞はそのため、細胞内病原体(例えば、ウイルスなど)および癌に対する免疫応答の不可欠な部分である細胞傷害性T細胞(CTL)応答の発生および刺激において重要な役割を果たす。
【0010】
この知見は多くの応用を開拓する。例えば、抗原を交差提示可能な樹状細胞に特異的に標的化することを可能にする。
【課題を解決するための手段】
【0011】
したがって、第1の態様において、本発明は、抗原を抗原提示細胞に標的化する方法であって、抗原提示細胞を、抗原を含む組成物と接触させる段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原提示細胞がCLEC9aを発現する方法を提供する。
【0012】
本方法は、in vitroまたはin vivoで適用することができる。抗原提示細胞は、典型的には樹状細胞とし、好ましくはMHCクラスI分子を介して細胞外抗原を交差提示することができる。「細胞外の」とは、抗原が、細胞外環境から細胞によって、典型的にはエンドサイトーシスまたは食作用によって取り込まれたものであることを意味する。
【0013】
本発明はさらに、対象において抗原に対する免疫応答を刺激する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合している方法を提供する。
【0014】
本方法は、単回投与、または適当に決定された時間間隔を空けた一連の2回以上の投与を含んでもよい。例えば、本方法は、プライミング段階(すなわち、初回投与)に続いて1回または複数回のブースター段階(1回または複数回の追加投与)を含むことができる。例えば、初回投与と第2回投与は、1日以上、1週間以上または1か月以上、好ましくは2週間から1か月の間隔を空けてもよい。追加の投与は、1週間以上または1か月以上の後に提供することができる。
【0015】
CLEC9a標的化によって刺激された免疫応答は、CTLまたはヘルパーT細胞であり得るT細胞の増殖を伴う。CLEC9aを発現する抗原提示細胞(および特に樹状細胞)は、CD8+T細胞およびCD4+T細胞の両方の増殖を誘導することができ、任意の与えられた免疫応答において両方の型のT細胞の増殖を刺激し得る。
【0016】
特定の条件下において、上記細胞は制御性T細胞(Treg)増殖を刺激することができると考えられている。Treg細胞は、Foxp3(Forkhead box p3)転写因子の発現により特徴付けられる。大部分のTreg細胞はCD4+およびCD25+細胞であり、ヘルパーT細胞のサブセットとみなすことができるが、ごく少数はCD8+である可能性がある。したがって、本発明の方法により刺激される免疫応答は、抗原に応答してTreg細胞の増殖を誘導する段階を含み得る。したがって、本方法は、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物を対象に投与する段階を含むことができる。抗原は、Treg細胞の増殖を促進するアジュバントと共に投与されてもよい。
【0017】
本方法が特異的抗原に応答してTreg細胞の増殖および分化を刺激する段階を伴う限り、免疫応答を刺激する方法であるとみなすことができる。しかしながら、Treg細胞が抗原に対して、例えばその活性の阻害または抑制などの別の方法で、免疫系の別の細胞の応答を調整することができ得ると仮定すると、全体としての免疫系に対する影響は、その抗原に対する応答を調整(例えば、抑制または阻害)するものとなる可能性がある。したがって、本発明の本態様の方法は、抗原に対する免疫応答を調整(例えば、阻害または抑制)する方法と同様に呼ぶことができる。
【0018】
したがって実際には、本発明のこれらの方法は、特定の抗原に対する望ましくない免疫応答を阻害または抑制するために、その抗原に対する免疫が既に存在するまたは免疫応答が進行している対象においても、治療的または予防的に使用することができる。このことは、(例えば)自己免疫疾患の治療において特に有用である。
【0019】
特定の条件下において、抗原をCLEC9aを発現する抗原提示細胞に標的化することによって、特定の抗原に対して対象を寛容化することも可能であり得る。したがって本発明は、抗原に対して対象における寛容を誘導する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原がアジュバントの非存在下で投与される方法を提供する。
【0020】
この文脈において寛容は、上記のような免疫細胞において、そうでなければその抗原に対して応答する、または抗原に対する応答性の持続的な低下を誘導することができる免疫細胞の減少を典型的に含む。
【0021】
典型的には、対象は脊椎動物、好ましくは哺乳動物である。対象は、ヒト、別の霊長類、またはマウス、ラット、モルモット、ウサギ目(例えば、ウサギ)、ネコ、イヌ、ブタ、ウシ、ウマ、ヒツジもしくはヤギなどの飼育動物、実験動物もしくは家畜であってもよい。
【0022】
本発明はさらに、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物を提供する。組成物は、抗原およびその結合している結合剤を薬学的に許容される担体と組み合わせて含む医薬組成物、例えば、ワクチンであってもよい。組成物は、静脈、筋肉内、腹腔内、経鼻、皮下、皮内などを含むがこれらに限定されない任意の適した投与経路用に製剤化することができる。
【0023】
本発明は、医療方法において使用するための、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物をさらに提供する。
【0024】
抗原に対する免疫応答を刺激するのに使用するための、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物もまた提供される。
【0025】
抗原に対する免疫応答を刺激するための医薬の調製における、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物の使用もまた提供される。
【0026】
CD8α+樹状細胞は、少なくともTh1、Th2、およびTh17型免疫応答に関係している可能性がある。したがって、本発明の方法は、任意の抗原に対する様々な型の免疫応答の刺激に適用することができる。しかしながら、これらの細胞は、CTL応答の発生において特に重要であると考えられているため、刺激される免疫応答は好ましくはCTL応答である。本方法は、典型的にはCD8を発現するT細胞であり、MHCクラスI分子に関連して同種抗原をディスプレイしている細胞に対して細胞傷害活性能がある、CTLの産生および/または増殖を測定する段階を含んでもよい。
【0027】
それにもかかわらず、抗原のCLEC9a+樹状細胞への標的化は、CTLに加えて、または代わりにヘルパーT細胞の増殖をもたらすことができる。したがって、本方法は追加としてまたは代替として、ヘルパーT細胞の産生および/または増殖を測定する段階を含んでもよい。ヘルパーT細胞はCD4+T細胞であってもよく、Th1、Th2、Th17またはTreg型であってもよい。ヘルパーT細胞はまた、CD4、例えば、CD8+Treg細胞を発現しない他の型のTreg細胞を含むことができる。
【0028】
したがって、上述した方法および組成物は、癌、またはウイルス感染などの細胞内寄生体もしくは病原体による感染症などの、CTL応答を誘導することが望ましい任意の状態の予防および/または治療のために使用することができることが理解されるはずである。
【0029】
最大のCTL刺激および増殖ならびに/または他のT細胞型の刺激および増殖を達成するために、さらなる免疫刺激剤を投与することが望ましい場合もある。これらは、樹状細胞を活性化、およびT細胞活性化を促進する能力を刺激することができる作用物質を含み得る。このような作用物質は、アジュバントと呼ばれることがある。アジュバントは、CD40のアゴニスト(可溶性CD40リガンド、またはCD40特異的アゴニスト抗体など)、CD28、CD27またはOX40のアゴニスト(例えば、これらの分子の1つに特異的なアゴニスト抗体)、CTLA-4アンタゴニスト(例えば、CTLA-4特異的遮断抗体)、および/またはToll様受容体(TLR)アゴニスト、および/または樹状細胞活性化を誘導することができる任意の他の作用物質を含み得る。TLRアゴニストは、Toll様受容体を活性化する物質である。好ましくは、TLRアゴニストはTLR3、TLR4、TLR5、TLR7またはTLR9の活性化剤である。適したTLRアゴニストは、TLR4に結合するMPL(モノホスホリル脂質A)である。使用することができる他のTLRアゴニストは、LTA(TLR2に結合するリポタイコ酸); TLR3に結合するポリI:C(ポリイノシン酸-ポリシチジル酸);TLR5に結合するフラジェリン; TLR7、およびTLR9に結合するCpG(DNA CpGモチーフ)に結合するイミキモドまたはポリU RNA(1-(2-メチルプロピル)-1H-イミダゾ(4,5-c)キノリン-4-アミン);またはTLRに結合および活性化する任意の他の成分である。より詳細には、Reis e Sousa、Toll-like receptors and dendritic cells、Seminars in Immunology 16:27、2004年を参照されたい。TLRを介して作用しない可能性があるアジュバントは、5'三リン酸RNA、ポリI:C、およびカードラン((β-1,3-グルカン))などのβ-グルカンを含む。TNF-αまたはIL-1などの炎症誘発性サイトカインもまた、アジュバントとして使用することができる。
【0030】
CLEC9a活性を有する(以下でより詳細に論じられる、例えば、樹状細胞表面上のCLEC9aと交差結合することができる)結合剤もまた、樹状細胞を活性化することができ得る。そのためこのようなアゴニスト結合剤は、それら自体がアジュバントとみなすことができる。したがって、このような結合剤が使用される場合、(そうすることがやはり望ましい可能性があるが)上述のものなどのさらなるアジュバントを投与する必要はない可能性がある。CLEC9aアゴニストとして作用することができる結合剤は、以下でより詳細に論じられる。
【0031】
理論に拘束されることを望むものではないが、使用されるアジュバントの性質は得られる応答の種類に影響する可能性があると考えられる。CLEC9aを発現する抗原提示細胞は、CD4+T細胞およびCD8+T細胞の両方を刺激することができ、CD4+応答の性質は特に使用されるアジュバントに影響される可能性がある。例えば、ポリI:Cの使用はTh1型CD4+応答の発生に有利であるようである。カードランはTh17型CD4+応答を刺激するようである。
【0032】
特定のアジュバントはTreg細胞の刺激を促進する。これらはレチノイン酸、および特にトレチノインとしても知られるオールトランスレチノイン酸(ATRA、all-trans retinoic acid)を含む。したがって、刺激される免疫応答がTreg応答である(実際には特定の抗原に対する免疫系の他の成分の応答を抑制してもよい)場合、Treg促進アジュバントを使用することが適切である可能性がある。アジュバントを投与せずにTreg細胞刺激を刺激することもまた可能である。
【0033】
本発明の組成物は、同じまたは別個の組成物において順次にまたは同時に、アジュバントと共に投与、または共に投与するために製剤化することができる。したがって、本発明の組成物は、アジュバントを含む必要はないが、含んでもよい。
【0034】
理論に拘束されることを望むものではないが、また上記で説明された通り、アジュバントの非存在下における抗原の投与は抗原に対する寛容の発生をもたらすと考えられる。すなわち、免疫系は同じ抗原のさらなる投与に応答しないように誘導される。これは、応答の能動的抑制が可能なTreg細胞の発生を含む(必要はないが)ことができる。したがって、抗原の、その抗原に対して寛容化された対象へのさらなる投与は、その抗原に対してナイーブな(すなわち、免疫系が以前にその抗原に曝露されていない)対象よりも弱い免疫応答をもたらすべきである。免疫応答の大きさは、炎症の出現、腫脹、細胞増殖(例えば、Th1、Th2もしくはTh17 CD4+T細胞、またはCTLの)、または炎症性サイトカイン産生(例えば、IL-1、IL-4、IL-12、IFN-γ、TNF-α)などの任意の適切な基準によって評価することができる。特定の実施形態において、寛容化された個体はその抗原に対する免疫応答を実質的に示さないはずである。
【0035】
上述した組成物および方法において、抗原は結合剤と物理的に結合しており、これは共有または非共有(例えば、静電またはファンデルワールス)相互作用を介していてもよい。好ましくは、抗原は結合剤に共有結合している。例えば、結合剤は、適したカップリング試薬を介して抗原と結合していてもよい。当業者は、このようなカップリング反応に使用することができる適した方法および試薬をよく知っている。
【0036】
代替として、抗原および結合剤は、同じペプチド鎖の一部であってもよく、すなわち、融合タンパク質として発現されてもよい。融合タンパク質は、抗原および結合剤間にリンカー配列を含有することができる。代替として、抗原および結合剤は、化学結合せずに物理的に近接して、例えば、リポソームに存在することができる。
【0037】
結合剤は、CLEC9aに対する十分に特異的な親和性を有する任意の適した分子であってよい。本明細書の全体にわたってこのような結合剤が言及される場合はいつでも、それはタンパク質、核酸(例えば、アプタマー)、糖質、または小分子であってよい。結合剤は、CLEC9aの生理的リガンドまたはその変異体もしくはアナログであってもよい。しかしながら、CLEC9aに対する抗体およびその機能的断片は特に好ましい。したがって、結合剤はCLEC9aに特異的な抗体結合部位を含んでもよい。結合剤は、以下で詳細に論じる通り多価であってもよい。
【0038】
抗原はペプチド抗原である。用語「ペプチド」は、抗原の性質、すなわち、ペプチド結合によって結合しているアミノ酸から形成されるものをいい、いかなる特定の大きさまたは長さを意味すると解釈されるべきではない。典型的には、ペプチド抗体は少なくとも長さが8アミノ酸であるはずであり、長さ30アミノ酸まで、長さ50アミノ酸まで、100アミノ酸まで、200アミノ酸まで、またはさらに長いものであってもよく、グリカン鎖などのアミノ酸に結合した残基を有してもよい。例えば、長さが25〜35アミノ酸であってもよい。
【0039】
いかなる特定の理論にも拘束されることを望むものではないが、ペプチド抗原は、MHCクラスIIまたはMHCクラスI分子に結合可能であるべきであり、または抗原提示細胞(樹状細胞など)内でプロセシングされてMHCクラスIIまたはMHCクラスI分子に結合できる1つまたは複数のペプチドを生じさせることが可能であるべきである。最近、長さが約8アミノ酸の短鎖エピトープペプチドが、長鎖ペプチド(例えば、長さが約30アミノ酸)よりも短いCTL反応性を誘導し得ることが示唆された(21)。MHCクラスI分子は典型的に、長さが8または9アミノ酸のペプチドに結合するが、MHCクラスII分子は8アミノ酸から20アミノ酸まで、30アミノ酸までの、またはさらに長いペプチドに結合することができる。
【0040】
抗原は、それに対して免疫応答、特にCTL応答だけでなくTh17応答またはTreg応答を生じさせることが望ましい任意のタンパク質またはその断片であってよい。これらは、癌細胞および細胞内病原体または寄生体を含有する細胞を含む、それに対してCTL応答を刺激することが望ましい細胞に結合している、上記細胞によって発現される、上記細胞上でディスプレイされる、または上記細胞によって分泌される抗原を含む。例えば、抗原は、細胞内病原体または寄生体によって発現されるタンパク質(ウイルスタンパク質など)由来の、または癌もしくは腫瘍細胞によって発現されるタンパク質由来のエピトープペプチドであってもよく、または上記エピトープペプチドを含んでもよい。したがって、抗原は腫瘍特異的抗原であってもよい。用語「腫瘍特異的」抗原は、固形腫瘍由来の抗原に限定されると解釈されるべきではなく、任意の癌性、形質転換または悪性細胞によって特異的に発現された抗原を包含すると解釈されるべきである。
【0041】
対象が望ましくない免疫応答を示す、または発生する危険性がある抗原に対するTreg応答を生じさせることが特に望ましい場合がある。例えば、それは自己免疫疾患において免疫応答が起こる自己抗原であり得る。特異的抗原が潜在的に病原的に重要であると確認された自己免疫疾患の例は、多発性硬化症(ミエリン塩基性タンパク質)、インシュリン依存性糖尿病(グルタミン酸脱炭酸酵素)、インシュリン抵抗性糖尿病(インシュリン受容体)、セリアック病(グリアジン)、水疱性類天疱瘡(XVII型コラーゲン)、自己免疫性溶血性貧血(Rhタンパク質)、自己免疫性血小板減少症(GpIIb/IIIa)、重症筋無力症(アセチルコリン受容体)、グレーブズ病(甲状腺刺激ホルモン受容体)、グッドパスチャー病(α3(IV)NC1コラーゲン)などの糸球体腎炎、および悪性貧血(内因子)を含む。代替として、標的抗原は、宿主組織にも損傷を与える応答を刺激する外因性抗原であってもよい。例えば、急性リウマチ熱は、心筋細胞抗原と交差反応する連鎖球菌抗原に対する抗体反応により引き起こされる。したがってこれらの抗原、またはその特定の断片もしくはエピトープは、本発明において使用するために適した抗原であり得る。
【0042】
Treg細胞の減少またはTreg細胞機能の障害は、マウスモデルにおいて自己免疫疾患をもたらすことが示されている。試験動物において引き起こされた疾患は、関節炎(例えば、関節リウマチ)、炎症性腸疾患、胃炎、悪性貧血、甲状腺炎、膵島炎、糖尿病、唾液腺炎、副腎炎、自己免疫性精巣炎/卵巣炎、糸球体腎炎、慢性閉塞性肺疾患ならびに実験的自己免疫性脳炎および多発性硬化症を含む。
【0043】
制御性T細胞1型応答の誘導はまた、マウスモデルにおいてアテローム性動脈硬化症の発生を減少させることも示されている(Mallat Z.ら、Circulation 108:1232〜7、2003年)。
【0044】
Treg活性はまた、同種移植の拒絶率において重要であることも示されている。Treg細胞の減少または機能障害は拒絶率を促進するが、Treg細胞において濃縮された同系リンパ球の試験動物への注入は移植片の生着を延長することが示されている。
【0045】
したがって本発明の方法は、任意のこれらの状態の治療において利用法を見出すことができる。
【0046】
対象に抗原自体を直接投与することなく、対象の体内に存在する抗原に対して、(CTL応答であれTreg応答を含む任意の別の種類のT細胞応答であれ)免疫応答を刺激することもまた可能であり得る。
【0047】
これは、結合剤が対象の体内の抗原と複合体を形成することができ、抗原をCLEC9aを発現する抗原提示細胞に標的化することができるように、抗原に対して親和性を有し、またCLEC9aに対して親和性も有する結合剤を使用して達成することができる。
【0048】
したがって本発明は、CLEC9aに対して親和性を有する第1の結合部位および抗原に対して親和性を有する第2の結合部位を有する結合剤を提供する。このような結合剤は、本明細書において「二重特異的」結合剤と呼ぶことにする。しかしながら、結合剤が、例えば、他の抗原に対する代替の結合親和性を有するさらなる結合部位を含んでもよく、用語「二重特異的」はそれに応じて解釈されるべきであることが理解されるはずである。
【0049】
本発明は、医療方法において、例えば、標的細胞に対する免疫応答の刺激において使用するための、上述の二重特異的結合剤をさらに提供する。標的細胞は、癌細胞または寄生細胞であってもよい。
【0050】
標的細胞に対する免疫応答の刺激のための医薬の製造における、上述の二重特異的結合剤の使用がさらに提供される。標的細胞は、癌細胞または寄生細胞であってもよい。
【0051】
二重特異的結合剤を対象に投与する段階を含む、前記対象において標的細胞に対する免疫応答を刺激する方法がさらに提供される。対象は、(例えば)癌または細胞内寄生体感染症に罹患していてもよい。
【0052】
二重特異的結合剤は、本明細書の他の部分に記載したアジュバントとの併用投与用に処方することができ、またはアジュバントと同じ組成物中に処方することができる。アジュバントの性質は所望の応答の性質に応じて選択することができる。したがって、例えば、ATRAは抗原に対するTreg応答を誘導することが望ましい場合に使用することができ、カードランはTh17応答が所望される場合に使用することができ、任意の別の適したアジュバント(例えば、抗CD40、ポリI:Cなど)はより一般的なCTL応答が必要とされる場合に使用することができる。アジュバントが投与されない場合、Treg細胞の刺激および/または抗原に対する寛容を誘導することが可能であり得る。
【0053】
本発明は、薬学的に許容される担体との混合物中の上述の二重特異的結合剤を含む医薬組成物をさらに提供する。組成物は、本明細書の他の部分に記載の通り、アジュバントをさらに含んでもよく、またはアジュバントとの併用投与用であってもよい。
【0054】
いくつかの実施形態において、一方または両方の結合部位がそれぞれCLEC9aまたは抗原特異的抗体結合部位であってもよい。したがって、結合剤は、少なくともCLEC9aに特異的な第1の抗体結合部位および少なくとも抗原に対して親和性を有する第2の抗体結合部位を含む二重特異的抗体であってもよい。用語「二重特異的抗体」は、二重特異的単鎖Fv二量体および「ダイアボディ」(下記参照)などの、上記2つの結合部位を有する任意の分子または分子複合体を包含すると解釈されるべきである。
【0055】
CLEC9a分子が樹状細胞の特定のサブセットのマーカーであるという発見はまた、CLEC9a分子を望ましくない免疫応答の下方制御の適した治療標的とする。
【0056】
したがって、さらなる態様において本発明は、CLEC9aに対して親和性を有する結合剤を投与する段階を含む、対象において免疫応答を阻害する方法を提供する。
【0057】
本発明の本態様において、結合剤は、結合する樹状細胞において樹状細胞機能を直接的または間接的に阻害することができる。結合剤は、CLEC9aが同種リガンドへ結合するのを阻害することができ、またはアジュバントに応じた成熟または抗原提示などの細胞機能の一面を阻害することができる。代替として、結合剤は細胞を直接的または間接的に死滅させることができ、これは「枯渇」活性とみなすことができる。典型的には、結合剤は、細胞を直接的または間接的に死滅させること、またはそうでなければ抗原提示に影響を与えることに関与する「エフェクター」部分を含む。
【0058】
例えば、結合剤は、対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる。例えば、抗体Fc領域を使用して補体系の成分を動員することができ、それにより補体の溶解経路、または細胞のオプソニン作用およびその後の好中球またはマクロファージなどの免疫系の食細胞による食作用を介して細胞の溶解をもたらすことができる。食細胞はまた、抗体の結合している細胞の食菌を可能にするFc受容体を有している。したがって抗体は、補体を活性化することなく細胞に食作用の印をつけることができる。このように、エフェクター部分は抗体Fc領域、例えば、IgGまたはIgMなどを含むことができる。
【0059】
結合剤は、標的細胞を死滅させるように作用する他の種類のエフェクター部分を含んでもよい。例えば、細胞を死滅させることができる毒素分子を含んでもよい。本発明者らが、CLEC9aに結合する抗体は樹状細胞にエンドサイトーシスによって取り込まれることができ、そのためコンジュゲート毒素分子が高い可能性で細胞に取り込まれるはずであることを示したように、この機構は特に有効であり得る。
【0060】
代替として、エフェクター部分は、細胞の近くでプロドラッグを活性化する、例えば、非毒性分子を毒性分子に変換することができる酵素であってもよい。
【0061】
既述したように、結合剤はCLEC9aに特異的な抗体結合部位を含んでもよい。
【0062】
有用な結合剤は、したがって、CLEC9aに特異的な抗体結合部位を含む分子、および抗体Fc領域、毒素分子、またはプロドラッグを活性化することができる酵素のうち1つまたは複数を含む。
【0063】
結合剤自体が、CLEC9aの機能的アンタゴニストであってもよい。したがって、CLEC9aをその同種リガンドに結合させない、または、例えば、樹状細胞成熟および/または抗原提示における通常のシグナル伝達の役割を発揮させないことによって阻害機能を発揮するために、結合剤はCLEC9に結合するだけで十分であり得る。このような実施形態において、結合剤が1個のみまたは2個のCLEC9aに特異的な結合部位(例えば、抗体結合部位)を含むことが望ましい場合がある。例えば、結合剤は、抗体またはFabもしくはscFv断片などの抗体断片であっても(を含んでも)よい。
【0064】
CLEC9aとリガンドの結合を妨害するアンタゴニストは、自己免疫疾患の治療において特に有用であり得る。いくつかの自己免疫疾患は、異常に高レベルの細胞死により特徴付けられ、これらの細胞に結合している自己抗原に対する免疫応答がこれらの状態の病原性に寄与する可能性があると考えられている。したがって、CLEC9aアンタゴニストを使用してCLEC9aが死細胞または瀕死細胞(特に免疫原性細胞死を起こしている細胞)に曝露されたリガンドに結合するのを妨害することができ、したがってこれらの抗原に対する免疫応答の刺激を阻害または妨害することができる。
【0065】
他の機能的CLEC9aアンタゴニストもまた、記載の方法において有用であり得る。このようなアンタゴニストは、CLEC9aをコードするmRNAまたはDNAにハイブリダイズすることができる核酸またはそのアナログ(例えば、リボザイム、アンチセンスRNAまたはDNA分子、siRNAなど)、CLEC9aの小分子アンタゴニストなどを含む。
【0066】
なおさらなるCLEC9aアンタゴニストは、樹状細胞結合CLEC9aのリガンド上の結合部位を遮断することができ、そのためリガンドが樹状細胞に認識、または結合されるのを妨害することができるCLEC9aリガンドの競合物である。適した競合物は、CLEC9aリガンドに結合するために十分なCLEC9aの細胞外ドメインまたはその一部を含む可溶性分子を含む。
【0067】
CLEC9a細胞外ドメイン(またはその一部)は、非相同部分と結合していてもよく、これはin vivoでの薬物動態特性などのアンタゴニストの特性を若干変える可能性がある。細胞外ドメインは、非相同部分に共有または非共有結合していてもよく、または非相同部分を有する融合タンパク質として発現されてもよい。例えば、血中半減期の増加をもたらし、アンタゴニストとCLEC9aリガンドの複合体の効率的な除去を可能にするために、非相同部分は抗体Fcドメインであってもよい。
【0068】
他の考えられる非相同部分の機能は、CLEC9a細胞外ドメインのオリゴマー形成の媒介、およびアンタゴニストの精製または試料からのCLEC9a単離の簡易化を含む。例えば、適したアンタゴニストは、アビジン単量体に結合しているCLEC9a細胞外ドメイン(またはCLEC9aリガンドに結合するのに十分なその断片)を含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む複合体をもたらす。この構築体は、ビーズなどの固体支持体上で提供することができるビオチンとの接触により容易に単離することができる。
【0069】
結合剤またはアンタゴニストがタンパク質である場合、アンタゴニストをコードする核酸(例えば、DNA)を投与することが可能であり得る。典型的には、核酸は体内の細胞(例えば、筋細胞)によって取り込まれ、発現され、これらの細胞から分泌されるはずである。このアプローチはしばしばDNAワクチン接種と呼ばれる。
【0070】
抗体は、結合剤およびアンタゴニストとして特に適しており、scFv形態で好都合に発現することができる。必要であれば、抗体を、抗原または上述のエフェクター部分と共に融合タンパク質としてコードすることができる。DNAワクチン接種アプローチの例は、Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年に記載されている。
【0071】
核酸は典型的に、核酸を取り込む細胞によるタンパク質の適切な発現および分泌を確実にするために、転写および翻訳調節配列と作動可能に連結して、場合により任意の所望の融合パートナーと共に、結合剤またはアンタゴニストをコードするコード領域を含む。このような配列は、転写開始配列(例えば、プロモーターおよびエンハンサー)、転写終結配列、適切なスプライシングシグナル、翻訳開始および終結配列、ならびに分泌を可能にするシグナルペプチドを含む(がこれらに限定されない)。
【0072】
したがって、本発明は、医療方法において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸(例えば、DNA)をさらに提供する。その方法および治療的使用において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸もまた提供される。
【0073】
結合剤またはアンタゴニストが投与される対象は、炎症または自己免疫状態、特に望ましくないCTL活性を特徴とする状態および/または高レベルの細胞死を特徴とする状態に罹患していてもよい。このような状態は、以下を含む:
- 免疫成分による関節リウマチおよび他の種類の慢性または急性関節炎または関節症、全身性エリテマトーデス(特に高レベルの細胞死を伴うことが知られている)、強皮症、シェーグレン症候群、自己免疫性(特にI型)糖尿病、甲状腺炎、ならびに乾癬を含む他の臓器特異的免疫疾患を含む自己免疫疾患;
- 多発性硬化症、重症筋無力症、および他の免疫介在性神経疾患を含む神経疾患。クローン病、大腸炎、セリアック病、および肝炎を含む胃腸疾患もまた含まれる;
- 現在では重大な免疫介在性の成分を含むと認識されている、アテローム性動脈硬化症、心筋症、リウマチ熱、心内膜炎、血管炎および他の免疫介在性心臓血管疾患を含む心臓血管疾患;
- 肺気腫、呼吸器気道感染症、および他の免疫介在性呼吸器疾患を含む免疫介在性呼吸器疾患;
- 喘息、鼻炎、および他の免疫介在性過敏性反応を含むアレルギープロセスおよび過敏性反応(I、II、III、およびIV型);
- 例えば、臓器移植、組織移植、輸血、骨髄移植中またはその後に起こる移植または移植片拒絶反応および移植片対宿主病;
- 敗血症性ショック症候群を含む感染性病原体に対する免疫病理学的反応;
- 小膠細胞などの免疫担当細胞に影響を与える神経変性過程などの変性過程。
【0074】
本発明は、医療方法において使用するための、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤をさらに提供する。
【0075】
免疫応答の阻害において使用するための、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤もまた提供される。
【0076】
免疫応答の阻害のための医薬の調製における、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤の使用もまた提供される。
【0077】
本発明者らはまた、抗原と物理的に結合していない場合であっても、CLEC9aアンタゴニストが樹状細胞を活性化することができ、したがって免疫機能を刺激するのに有用である可能性があることも発見した。したがって本発明は、CLEC9aに対して親和性を有する結合剤を投与する段階を含む、対象において免疫応答を刺激する方法を提供する。
【0078】
CLEC9aアゴニストは、CLEC9aに対して親和性を有する結合剤であってもよく、CLEC9aに特異的な抗体結合部位を含んでもよい。
【0079】
理論に拘束されることを望むものではないが、2個以上のCLEC9aに特異的な結合部位(例えば、抗体結合部位)を有する結合剤は、おそらく細胞表面上のCLEC9aと交差結合またはその結合もしくは多量体化をもたらすことができるため、CLEC9a活性の特に有効なアゴニストである可能性があると考えられる。結合剤は、2個のこのような結合部位を有する場合、「二価」と、または2個以上のこのような結合部位を有する場合「多価」と呼ばれることがある。したがって、結合剤は好ましくは多価であり、少なくとも3、4、5、10個またはさらにそれ以上の結合部位を含んでもよい。このような結合剤は、(例えば、ラテックスの)ビーズ、リポソームもしくはベシクルなどの粒子、または任意の他の適した粒子中または上に固定化された複数の結合部位を含むことができる。このように、結合部位は微粒子固相上または中に固定化することができる。代替として、多価結合剤は、互いに共有結合または別な方法で結合した2個以上の結合部位を含むのみであってもよい。例えば、抗体全体またはその機能的断片(下記参照)は、融合タンパク質としておよび/または化学的交差結合によって結合していてもよい。当業者は、このような多価結合剤を調製するための適した技術をよく知っている。本方法がin vitroで行われる場合、培養容器の表面上に固定化された結合剤(Clec9aに対する抗体など)をアンタゴニストとして使用することができ、この場合培養容器の被覆された表面自体が多価結合剤であると考えることができる。
【0080】
アゴニストは、免疫機能を広く刺激するために単独で、またはそれに対する免疫応答を誘導することが望ましい標的抗原と併用して投与することができる。アゴニストおよび抗原は、必要に応じて、別々にまたは同じ組成物において同時にまたは順次に投与することができる。アゴニストは、本発明の第1の態様に関して上述した通り抗原と物理的に結合していてもよく、または2個の成分が物理的に独立した別個の存在であってもよい。
【0081】
既述した通り、記載した方法および組成物は、癌、またはウイルス感染などの細胞内寄生体もしくは病原体による感染症などの、CTL応答を誘導することが望ましい任意の状態の予防および/または治療のために特に有用であり得る。
【0082】
記載した方法および組成物はまた、Treg応答を誘導することが望ましい任意の状態、例えば、自己免疫疾患などの特定の状態に対する望ましくないまたは不適切な免疫応答を伴う状態の予防および/または治療のために使用することもできる。
【0083】
上述のさらなる免疫刺激剤またはアジュバントを、アゴニストおよび場合により抗原を伴って投与することもできる。したがってアジュバントは、例えば、CD40アゴニストまたはTLRアゴニストを含むことができる。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0084】
樹状細胞のサブセットのマーカーとしてのCLEC9aの同定は、生体試料からこのような細胞を同定および単離するための手段を提供する。
【0085】
したがって、さらなる態様において本発明は、試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤と1つまたは複数の細胞の結合を測定する段階とを含む、試料中の抗原提示細胞を検出する方法を提供する。
【0086】
本方法はまた、試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤が結合している1つまたは複数の細胞を単離する段階とを含む、試料から抗原提示細胞を単離する方法も提供する。結合剤は、単離を容易にするために固体支持体(磁気ビーズなど)上に固定化されていてもよい。
【0087】
上記の議論から明らかになるように、抗原提示細胞は、典型的には樹状細胞であり、MHCクラスI分子を介して細胞外抗原を交差提示することができ得る。
【0088】
CLEC9a結合剤により同定された細胞が樹状細胞であることを確認するために、または細胞をCLEC9a結合剤と接触させる前に試料の樹状細胞を濃縮するために、本方法は、試料を樹状細胞マーカーに対して親和性を有する第2の結合剤と接触させる段階と、第2の結合剤と1つまたは複数の細胞の結合を測定する段階とを含むことができる。2つの結合剤を、同時にまたは順次に、任意の順序で細胞と接触させてもよい。いくつかの実施形態において、第1および第2の結合剤両方が結合する細胞のみが同定または単離される。
【0089】
樹状細胞マーカーは、CD11などの全樹状細胞マーカー、特に(マウスにおける)CD11cとすることができる。
【0090】
ヒト樹状細胞の試料について、樹状細胞マーカーはHLA-DRであってもよい。細胞がlineage陰性である、すなわちCD3、CD14、CD19またはCD56を発現しないことを確認することが望ましい場合がある。
【0091】
ヒト細胞の試料について、CLEC9a結合剤によって同定された細胞がBDCA-3(CD141またはトロンボモジュリンとしても知られている)も発現することを確認することが望ましい場合がある。したがって本方法は、試料をBDCA-3に対して親和性を有するさらなる結合剤と接触させる段階と、さらなる結合剤と1つまたは複数の細胞の結合を測定する段階および/またはさらなる結合剤が結合する細胞を単離する段階とを含むことができる。CLEC9aおよびBDCA-3の結合剤を、同時にまたは順次に、任意の順序で細胞と接触させてもよい。いくつかの実施形態において、両方の結合剤が結合する細胞のみが同定または単離される。
【0092】
追加としてまたは代替として、1つまたは複数の不要な細胞型の負の選択によって、試料の所望の細胞型を濃縮することが望ましい場合がある。不要な細胞型は、CD123またはLy6Cに対する負の選択によって除外され得る、形質細胞様樹状細胞(pDC)などの樹状細胞の他の亜群を含み得る。負の選択は、CLEC9a発現細胞に対する選択の前に、同時に、または後に行うことができる。負の選択は、CD3、CD14、CD19および/またはCD56に対して行うことができる。
【0093】
細胞上のCLEC9a発現レベルを測定することが望ましい場合もある。このことは、所望のレベルのCLEC9a、例えば、検出可能なレベルでCLEC9aを発現する別の細胞集団よりも高レベルのCLEC9aを発現する細胞を選択することのみを可能にする。例えば、理論に拘束されることを望むものではないが、CD8を発現する(またはヒトにおけるそのサブセットと同等である)DCのサブセットはpDCよりも高レベルのCLEC9aを発現すると考えられる。したがって、特定の閾値レベルを超えるCLEC9aを発現する細胞を選択することのみによって、CD8 DCまたはその同等物を選択することが可能であり得る。
【0094】
結合剤は、例えば、検出可能なシグナル(蛍光または放射性標識など)を発することができる標識によって、または結合パートナーにより特異的に結合されることが可能な親和性タグによって、細胞の検出および/または単離を容易にするために標識されていてもよい。親和性タグと結合パートナーの例は、エピトープと同種抗原、正に荷電したペプチド(例えば、ポリ-His)と金属(例えば、ニッケル)イオン、アビジン/ストレプトアビジンとビオチン、糖質とレクチンなどを含む。当業者であれば、これらの特定の要件に応じて適した系を設計することができるはずである。
【0095】
細胞の同定または単離は、試料を第1および/または第2の結合剤に結合することができる1つまたは複数の検出剤と接触させる段階を含むことができる。検出剤自体が、上述のように標識されていてもよい。
【0096】
単離および検出を容易にするために、結合剤または検出剤は固体支持体上に固定化されていてもよい。
【0097】
本発明は、上述の方法により単離された抗原提示細胞の集団をさらに提供する。
【0098】
これらの方法により単離された細胞は、in vitro試験およびex vivo療法を含む様々な目的のために使用することができる。例えば、単離された細胞にin vitroで所望の抗原をパルスすることができる。その後、抗原に対する免疫応答を刺激するために、細胞を対象に投与することができる。
【0099】
したがって、本発明は、ペプチド抗原に対する免疫応答を刺激する方法であって、上述した方法により単離された抗原提示細胞またはその集団を提供する段階と、前記細胞または細胞集団を前記抗原と接触させる段階とを含む方法をさらに提供する。
【0100】
好ましくは、抗原提示細胞はMHCクラスI分子に関連して前記抗原またはその断片を提示する。
【0101】
前記接触させる段階に続いて、細胞または細胞集団を対象に投与することができる。好ましくは、細胞は、細胞の由来した対象に再投与される。
【0102】
細胞または細胞集団はまた、上述した通り、アジュバントと接触させてもよい。接触は、例えば、抗原との接触と同時にまたはほぼ同時に、または受容対象への投与時または後にin vitroで行うことができる。アジュバントとの接触は、細胞または細胞集団の、抗原に応答したT細胞増殖を活性化または促進する能力を刺激することができる。アジュバントは、細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。免疫刺激アジュバントは、例えば、CD40アゴニストまたはTLRアゴニストであってもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0103】
したがって本発明は、上述した方法によって得られる、プライミングされた抗原提示細胞またはその集団を提供する。「プライミングされた」は、細胞が抗原と接触したこと、MHC分子、好ましくはMHCクラスI分子に関連して、その抗原またはエピトープを提示していること、およびそれに応答してT細胞の増殖およびエフェクター細胞への分化を活性化または刺激することができることを意味する。
【0104】
医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるプライミングされた抗原提示細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、プライミングされた抗原提示細胞またはその集団の使用もまた提供される。
【0105】
代替として、抗原に特異的なT細胞(特にCTLだけでなくTh17およびTreg細胞を含むCD4+T細胞も)を発生させるために、プライミングされた細胞をin vitroでT細胞と接触させることができる。したがって、前記接触させる段階に続いて、本方法は、前記抗原提示細胞を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含むことができる。集団中のT細胞は、抗原に特異的な集団中のT細胞の数または比率を増加させるために、培養で増殖させることができる。その後T細胞を対象に投与することができる。場合により、T細胞は投与前に集団中の他の細胞から分離される。
【0106】
細胞集団はまた、例えば、プライミングされた抗原提示細胞と接触するのと実質的に同時に、アジュバントと接触させてもよい。
【0107】
好ましくは、T細胞および抗原提示細胞は自己由来、すなわち、同じ対象由来または遺伝学的に同一の対象由来である。
【0108】
T細胞は、細胞(またはその祖先)の由来した対象に再投与されてもよい。
【0109】
この場合もやはり、アジュバントは対象に投与されるときにT細胞と共に投与されてもよい。アジュバントは、CD40アゴニスト(CD40に特異的な抗体など)またはTLRアゴニストであってもよい。アジュバントは、T細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。
【0110】
したがって本発明は、上述した方法によって得られるT細胞またはその集団をさらに提供する。医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるT細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、T細胞またはその集団の使用もまた提供される。
【0111】
本発明は、CLEC9aを発現するヒト樹状細胞の単離された集団をさらに提供する。集団は、少なくとも5個、少なくとも10個、少なくとも100個、少なくとも1000個または少なくとも10000個の樹状細胞を含有することができる。好ましくは、集団中の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、または少なくとも95%の樹状細胞がCLEC9aを発現する。この目的のために、樹状細胞はlineage陰性、HLA-DR+細胞であると考えられている。lineage陰性細胞は、CD3、CD14、CD19またはCD56を発現しない。
【0112】
単離された集団は、試料中の樹状細胞の所要の比率がCLEC9aを発現する限り、他の細胞型を含む試料中に含まれてもよい。したがって試料は、リンパ球(例えば、T細胞、および特にCTL)または樹状細胞でない他の種類の抗原提示細胞を含むことができる。
【0113】
CLEC9aを発現する樹状細胞は、典型的にはBDCA-3+であるはずである。また、CD123-、CD34-、CD16-およびCD11b/c-であってもよい。
【0114】
本発明は、ペプチド抗原に対する免疫応答を刺激する方法であって、CLEC9aを発現するヒト樹状細胞の集団を提供する段階と、前記集団を前記抗原と接触させる段階とを含む方法をさらに提供する。
【0115】
前記接触させる段階に続いて、CLEC9aを発現するヒト樹状細胞の集団を自己由来対象に投与する(すなわち、もともと集団が由来した対象に投与される)ことができる。
【0116】
CLEC9aを発現するヒト樹状細胞集団はまた、上述した通り、アジュバントと接触させてもよい。接触は、例えば、抗原との接触と同時にまたはほぼ同時に、または受容対象への投与時または後にin vitroで行うことができる。アジュバントとの接触は、細胞集団の、抗原に応答したT細胞増殖を活性化または促進する能力を刺激することができる。アジュバントは、細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。免疫刺激アジュバントは、例えば、CD40アゴニストまたはTLRアゴニストであってもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0117】
したがって本発明は、抗原によってプライミングされた、CLEC9aを発現するヒト樹状細胞の集団を提供する。「プライミングされた」は、細胞が抗原と接触したこと、MHC分子、好ましくはMHCクラスI分子に関連して、その抗原またはエピトープを提示していること、およびそれに応答してT細胞の増殖およびエフェクター細胞への分化を活性化または刺激することができることを意味する。
【0118】
医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、CLEC9aを発現するヒト樹状細胞のプライミングされた集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、CLEC9aを発現するヒト樹状細胞のプライミングされた集団の使用もまた提供される。
【0119】
代替として、抗原に特異的なT細胞(特にCTLだけでなくヘルパーT細胞またはTregも)を発生させるために、プライミングされた細胞をin vitroでT細胞と接触させることができる。したがって、前記接触させる段階に続いて、本方法は、前記CLEC9aを発現するヒト樹状細胞の集団を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含むことができる。集団中のT細胞は、抗原に特異的な集団中のT細胞の数または比率を増加させるために、培養で増殖させることができる。その後T細胞を対象に投与することができる。場合により、T細胞は投与前に集団中の他の細胞から分離される。本発明の別の態様にあるように、樹状細胞およびT細胞もまた、アジュバントと接触してもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、CTL応答、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0120】
好ましくは、T細胞および樹状細胞は自己由来であり、すなわち、同じ対象由来、または遺伝学的に同一の対象由来である。
【0121】
T細胞は、細胞(またはその祖先)の由来した対象に再投与されてもよい。
【0122】
この場合もやはり、アジュバントはT細胞と共に投与されてもよく、アジュバントはCD40アゴニスト(CD40に特異的な抗体など)またはTLRアゴニストであってもよい。アジュバントは、T細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。
【0123】
したがって本発明は、上述した方法によって得られるT細胞またはその集団をさらに提供する。医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるT細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、T細胞またはその集団の使用もまた提供される。
【0124】
全ての上述の態様において、抗原は、それに対して免疫応答、特にCTL応答、Th17型応答またはTreg応答を刺激することが望ましい任意の抗体であってよい。例えば、抗原は、本明細書の他の部分に記載の通り、細胞内病原体または寄生体によって発現される抗原であってもよく、または癌細胞によって発現されてもよい。代替として、抗原は、それに対して望ましくないまたは不適切な応答が起こる(例えば、自己免疫疾患において)、およびそれに対してTreg応答を刺激することが望ましい抗原であってもよい。
【0125】
CLEC9aが、感染性病原体ではなく哺乳動物細胞上または中にみられるリガンドに結合するという驚くべき発見はまた、このリガンドの同定のためのアッセイを利用可能にもする。
【0126】
したがって本発明は、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合するのに十分なその一部を含む標的物質を、哺乳動物細胞の成分である試験物質と接触させる段階と、標的物質と試験物質の結合を測定する段階とを含む、CLEC9aの生理的リガンドをスクリーニングする方法を提供する。
【0127】
結合により、試験物質がCLEC9aの1つの(または特定の)生理的リガンドであることを示すことができる。
【0128】
結合が起こる場合、本方法は、試験物質を同定する段階をさらに含むことができる。
【0129】
CLEC9aのリガンドは、一部のまたは全ての正常哺乳動物細胞において構成的に発現されるが、細胞が正常であり続ける限りは、CLEC9aと接触して相互作用することができないと考えられている。特定の種類の細胞損傷または細胞死(特に、原発性または二次性壊死などの免疫原性の場合)は、リガンドが樹状細胞上のCLEC9aと接触して相互作用できるように、リガンドを露出させる。
【0130】
したがって、試験物質は、哺乳動物細胞(例えば、細胞内寄生体または病原体に感染していない正常な哺乳動物細胞)の細胞内成分であってもよい。試験物質は、タンパク質、糖質、脂質または核酸を含んでも、またはそれらからなってもよい。試験物質は、これらの成分の2つ以上を含んでもよく、例えば、糖質および脂質成分を含むグリコシル化タンパク質、脂質およびタンパク質(および場合により糖質も)成分を含む脂質アンカータンパク質、または脂質および糖質成分を含む糖脂質であってもよい。
【0131】
試験物質は、生理的条件下で哺乳動物細胞中に存在するであろう分子または物質の全体を含まない可能性があるが、CLEC9aと相互作用するために十分なその一部を含み得ることが理解されるはずである。例えば、試験物質は、細胞タンパク質の単離ドメイン、またはペプチド(例えば、5〜10アミノ酸、20アミノ酸まで、50アミノ酸まで、または100アミノ酸まで)さえも含むことができる。
【0132】
典型的には、試験物質は、標的物質中に存在するCLEC9a(またはその一部)と同じ哺乳動物種由来であるはずである。
【0133】
本方法は、標的物質を、試験物質を含む試料(例えば、水性試料などの液体試料)と接触させる段階を含むことができる。
【0134】
標的物質は溶液中(例えば、水溶液中)に提供されてもよく、または固体支持体上に固定化されてもよい。
【0135】
試料は、透過性哺乳動物細胞を含んでもよい。「透過性」は、(細胞によって能動的に取り込まれずには)通常、原形質膜を通過することができない存在への拡散によって、原形質膜が透過性になったことを意味する。通常の条件下で原形質膜を透過しないヨウ化プロピジウムおよびTO-PRO3などの染料は、原形質膜完全性/透過性を試験するために慣習的に使用されている。したがって、原形質膜はこのような物質に対して透過性であってもよい。例えば、原形質膜は、500Daを超える、1kDaを超える、10kDaを超える、50kDaを超える、100kDaを超える、またはさらに大きい分子量を有する物質に対して透過性であってもよい。用語「透過性」はこの場合、個々の細胞が依然として(例えば、顕微鏡法によって)識別できるように、実質的に細胞構造(原形質膜、および潜在的に細胞内の他の膜の透過性増加は別にして)を保持する細胞のことをいうために使用される。用語「溶解物」、「抽出物」または「ホモジネート」は、個々の細胞がもはや識別できない、または存在しない程度まで細胞の構造が粉砕される調製のために使用することができる。
【0136】
透過性細胞は壊死性であってもよい。壊死は、原発性または二次性壊死であってもよい。原発性壊死は、(例えば)照射(例えば、UV光などの電離放射線による)、血清除去、少なくとも1回の凍結/融解サイクルによって、またはアントラサイクリン(ドキソルビシンおよびダウノルビシンなど)およびアントラセンジオン(ミトキサントロンなど)などの壊死誘導化学物質を用いた処理によって、実験的に誘導することができる。二次性壊死は、アポトーシスに入るよう誘導された細胞が隣接細胞によって食菌されず、原形質膜がその後破裂する場合に起こる。
【0137】
代替として、正常細胞は、原形質膜を破裂させるまたは膜に孔を形成する作用物質を用いて、直接透過性化してもよい。適した透過性化剤は、コレステロールを沈殿させ、したがって膜から除去して透過性を増加させるサポニンなどの孔形成剤(例えば、β-エスシン)、細胞溶解素(例えば、黄色ブドウ球菌のストレプトリシン-O)および黄色ブドウ球菌(Staphylococcus aureus)のα-トキシンなどの様々な細菌毒素、ならびにTriton X-100、Brij-96、Tweenなどの界面活性剤を含む。
【0138】
場合により、細胞は、透過性化の前または後に固定されてもよい。透過性化後にリガンドが(例えば、拡散によって)細胞から失われる機会を減少させるために、透過性化前の固定が好ましい場合がある。固定は、アセトン、メタノール、エタノールまたはそれらの混合物などの有機溶媒(一般に、細胞構造上のタンパク質を沈殿させると同時に、細胞の脂質を除去し脱水する)および/またはホルムアルデヒド(例えば、ホルマリン)もしくはパラホルムアルデヒド(タンパク質などの細胞成分を、アミノ基などのこれらの細胞成分に存在する遊離反応基を介して架橋する)などの架橋試薬を用いて行うことができる。
【0139】
試料が透過性細胞を含む場合、本方法は、標的物質の検出によって、標的物質の結合が起こる細胞内位置を測定する段階を含むことができる。
【0140】
検出は直接的または間接的であってもよい。例えば、標的物質は標識を含むことができ、本方法は標識の細胞内位置を測定する段階を含むことができる。代替として、本方法は、標的物質を、標的物質に結合可能な結合剤であってもよい検出剤と接触させる、さらなる段階を含むことができる。検出剤自体が標識を含んでもよい。検出は、顕微鏡法、例えば、共焦点顕微鏡法などの任意の適した技術によって達成することができる。例えば、標識は蛍光であってもよい。
【0141】
代替として、試料は、哺乳動物細胞の細胞溶解物、抽出物または細胞内画分であっても、または含んでもよい。例えば、試料は、全細胞溶解物、またはインタクトな正常細胞中の外部環境に曝露されていない細胞内画分を含むことができる。例えば、試料は、単離された細胞質画分、単離された核画分、単離された小胞体画分、単離されたゴルジ画分、または単離されたミトコンドリア画分を含むことができる。この文脈において「単離された」は、インタクトな細胞の少なくとも1つの他の正常成分(原形質膜など)から分離されていることを意味する。このような画分中の核またはミトコンドリアなどの細胞内オルガネラは、インタクトであってもまたは破壊されていてもよい。
【0142】
第1の試料の陽性結合反応に続いて、第1の試料は、第1の試料に存在する1つまたは複数の成分を欠く第2の試料を提供するためにさらに分画されてもよい。その後、本方法を繰り返してもよい。これは、標的物質に結合する試験物質の同定において役に立ち得る。このプロセスは、徐々に小さくなる画分を使用して、所望の回数繰り返すことができる。
【0143】
追加としてまたは代替として、本方法は、標的物質および試験物質を含む複合体を単離する段階を含むことができる。これは、標的物質が結合している固体支持体(例えば、ビーズ)を単離することによって達成することができる。代替として、標的物質に結合することができる結合剤を利用してもよい。結合剤は、固体支持体上に固定化することができる。標的物質は特異的結合対のメンバー含むことができ、結合剤は特異的結合対の第2のメンバーを含むことができる。例えば、結合剤は、標的物質に特異的な抗体であってもよい。代替として、結合剤および標的物質のうち1つがアビジン/ストレプトアビジン部分を含み、もう一方がビオチン部分を含むことができる。
【0144】
用語「特異的結合対」は、互いに特別な特異性を有し、通常の条件において他の分子への結合に優先して互いに結合する、特異的結合メンバー(sbm)および結合パートナー(bp)を含む分子の対を説明するために使用される。特異的結合対の例は、抗体とその同種エピトープ/抗原、リガンド(ホルモンなど)と受容体、アビジン/ストレプトアビジンとビオチン、レクチンと糖質、および相補的ヌクレオチド配列である。
【0145】
単離を容易にするために、単一の結合剤が多価、すなわち、1つまたは複数の標的物質に同時に結合することができるものであってもよい。標的物質もまた多価(すなわち、同じ種類の2つ以上の結合剤に同時に結合することができる)である場合、架橋複合体が形成されてもよい。
【0146】
当業者は、利用できる免疫沈降技術などの適した技術をよく知っている。
【0147】
例えば、標的物質は、アビジン単量体に結合している(例えば、コンジュゲートした、または融合タンパク質として)CLEC9a細胞外ドメインを含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む結合剤をもたらす。この構築体はしたがって、本明細書に記載のアッセイにおいて試験物質との複合体を形成し、その後ビーズなどの固体支持体上に存在し得るビオチンとの接触によって単離することができる。
【0148】
代替として、試験物質はメンブレン、マイクロタイタープレート、またはマイクロアレイチップなどの固体支持体上に固定化されてもよい。支持体を標的物質と接触させて、任意の結合している標的物質の位置を測定することができる。
【0149】
標的物質に結合できる物質を実際に含有するかどうかを確かめるために、CLEC9aのリガンドを含有する疑いがある複数の試料(例えば、異なる細胞画分)を試験することが望ましい場合がある。追加としてまたは代替として、標的物質に結合できるかどうかを確かめるために、複数の既知物質(例えば、タンパク質)を試験することが望ましい場合がある。
【0150】
したがって、単一の固体支持体は唯1つの試料または試験物質を含むことができ、または複数の試料または試験物質をそれぞれ支持体上の規定された位置に有することができる。
【0151】
アッセイの形式に応じて、本方法は、陽性結合反応が起こる特定の支持体、または陽性結合反応が起こる支持体上の位置の同定段階を含むことができる。したがってこれにより、陽性反応の原因である試験物質を含む試料の性質を明らかにすることができ、または試験物質の正体を直接明らかにすることができる。
【0152】
代替として、固体支持体から試験物質および標的物質の複合体を単離し、試験物質を同定するためのさらなる分析を行うことも可能であり得る。
【0153】
いかなるアッセイの形式であっても、試験物質がタンパク質である場合、試験物質を同定するためにプロテオミクス技術を使用することが可能であり得る。これは典型的には、質量分析およびデータベース照合と組み合わせるはずである。試験物質(または試験および標的物質の複合体)は、既知のNまたはC末端残基を有するペプチドを得るために、(使用される特定のプロテアーゼに応じて)既知の標的切断配列を有する1つまたは複数のプロテアーゼによる消化に供してもよい。得られたペプチドを、次いで質量分析(例えば、MALDI-TOF)にかけて分子量を測定する。次いで、適したタンパク質配列データベースを、このようなペプチドを生じさせることができるタンパク質を同定するために照合することができる。
【0154】
代替として、試験物質は細胞またはウイルスの表面上にディスプレイされてもよい。例えば、ファージディスプレイ技術を使用して、試験物質またはその断片をバクテリオファージの表面上にディスプレイすることができる。代替として、細胞は、表面上に試験物質をディスプレイするように処理されてもよい。代替として、試験物質と標的物質の間の相互作用が、細胞発現系または無細胞発現系において起こってもよく、レポーター遺伝子の発現などの検出可能な反応を誘導してもよい。このような系の一例は酵母2ハイブリッド系であるが、当業者であれば、2つのタンパク質間の「bait-prey」相互作用に依存してレポーター遺伝子の発現を駆動する他の系をよく知っているはずである。核酸にコードされるタンパク質が細胞発現系または無細胞発現系において発現される場合、このような形式を使用して、(存在する場合)標的物質に結合できる物質をコードする核酸分子を同定するために、核酸分子集団(例えば、cDNAライブラリー)をスクリーニングすることができる。このような形式において、本方法は、陽性結果の原因である核酸を単離する(例えば、クローニング)段階と、核酸によりコードされるタンパク質の正体を特定する段階とを含むことができる。
【0155】
試験物質がタンパク質ではない場合、他の分析技術を利用することができる。
【0156】
本発明を、限定ではなく例として、添付の図面および実施例を参照することによって以下により詳細に説明する。
【図面の簡単な説明】
【0157】
【図1】ヒトCLEC9aタンパク質に関するcDNAおよびアミノ酸配列を示す図である。
【図2】マウスCLEC9aタンパク質に関するcDNAおよびアミノ酸配列を示す図である。
【図3】ヒトPhoenix細胞中で発現されたマウスCLEC9aタンパク質の検出を示す図である。還元(R)および非還元(NR)状態において、CLEC9aを発現するPhoenix細胞(FNX-C9)または親細胞(FNX)からの完全溶解物で抗CLEC9aを使用するウエスタンブロット。
【図4】脾臓DCおよびin vitro培養BMDCにおけるマウスCLEC9a転写産物の分布を示す図である。脾臓DCのサブセット由来のmRNAまたはGMCSFもしくはFlt3L-BMDCの精製サブセット由来のmRNAを、Methodsで示したように、CLEC9a特異的プライマー(上レーン)またはβ-アクチンプライマー(下レーン)を使用するRT-PCRに施した。
【図5】この実験中に作製したCD3ζ-NKRP1-CLEC9aキメラを示す図である。キメラ分子中に存在するドメインおよびCLEC9aの細胞外ドメイン、NKRP1由来の膜貫通ドメインおよびCD3ζ由来の細胞質ドメインを示す。
【図6】初代細胞中でのCLEC9aの発現を示す図である。脾細胞はビオチン抗CLEC9a(太線、10mg/ml)またはビオチン-ラットIgG1(太線、アイソタイプ対照)、次にストレプトアビジン-PE(1:1000)で染色し、かつCD11c、CD4、CD8またはLy6Cで対比染色した。ヒストグラムはCD11c陰性細胞とCD11c陽性細胞に関する染色を示し、次いでそれらは従来型DCのCD4/CD8サブセットまたはpDCに相当するLy6C+サブセットにおいてさらに分析する。
【図7】CLEC9a細胞質ペプチドでのSykの破壊を示すウエスタンブロットの図である。CLEC9a細胞質ドメインまたはデクチン-1対照に相当するビオチン化ペプチドを使用して、Methods中に示したように組換えSykを破壊した。
【図8】抗CLEC9aを用いたキナーゼ活性化を示す図である。CLEC9aを発現するLK細胞をプレート抗体を用いて示した時間活性化させ、次いで溶解し、抗P-Sykまたは抗-P-Erk抗体を用いたSDS-PAGEおよびWBに施してこれらの経路の活性化を検出した。抗Sykまたは抗Erkを用いたタンパク質対照は全体のキナーゼを示す。
【図9】抗CLEC9aハイブリドーマによるサイトカインの誘導を示す図である。CLEC9aを発現するLK細胞(105個細胞/ウエル)は、上清の回収およびIL-2に関する試験前に、示したハイブリドーマと共に培養した。
【図10】プレートした抗CLEC9aが、Sykの存在下でCLEC9aのWT尾部を介して、CLEC9a-CD3ζを発現するY7.B3Z(σ)、またはCLEC9a-WT(ν)またはCLEC9a-Y7F(λ)を発現するB3Z-Sykの存在に依存してNFAT活性を誘導することを示す図である。NFAT活性はMethodsで示したように測定する。X軸はプレートをコーティングするために使用した抗CLEC9a抗体の濃度(μg/ml)を示す。
【図11】プラスチック上に固定化した抗Clec9a抗体は、Flt3L BMDCにおけるIL12-23 p40タンパク質の産生を誘導することを示す図である。Flt3L BMDCは抗CLEC9aモノクローナル抗体をコーティングしたプレート上でインキュベートし、一晩培養後の上清をIL12-23 p40に関して分析した。
【図12AB】抗CLEC9a-S1モノクローナル抗体を使用したin vivoでのCD11c+CD8+の標的化を示す図である。S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体(5μg)を静脈内注射し、Methodsで示したように翌日脾細胞を処理した。AおよびB.CD11c陽性および陰性脾細胞を、CFSE標識OT-I細胞と共に4日間培養した。A.25×103個の標的CD11c-またはCD11c+脾細胞の存在下でのOT-I細胞のCFSEプロファイル。B.左図:ウエル当たりのOT-I細胞の絶対数。右図:増殖OT-I細胞の上清中のIFN-γ産生。
【図12CD】抗CLEC9a-S1モノクローナル抗体を使用したin vivoでのCD11c+CD8+の標的化を示す図である。S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体(5μg)を静脈内注射し、Methodsで示したように翌日脾細胞を処理した。CおよびD.S1抗CLEC9a標的マウス中のCD11c+脾細胞を、CD11c+B220-CD8+(CD8+DC)、CD11c+B220-CD4+(CD4+DC)およびCD11c+B220-CD4-CD8-(DN DC)として分類し、CFSE標識OT-I細胞と共に3日間培養した。C.25×103個の標的CD8+、DN、CD4+DCの存在下でのOT-I細胞のCFSEプロファイル。D.左図:ウエル当たりのOT-I細胞の絶対数。右図:培養の終期における上清中のIFN-γ産生。
【図13A】抗CLEC9aモノクローナル抗体を使用した標的化によるCTL免疫の誘導を示す図である。A:S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体を、Methodsで示したように、抗CD40と一緒に、または抗CD40とは別に皮下注射した。5日後、20nM(0.03μMのCFSE)、200nM(0.3μMのCFSE)のSIINFEKLを導入した、またはペプチド(3μMのCFSE)を導入しなかった標的細胞(コンジェニックCD45.1)を静脈内注射した。結果はin vivo死滅アッセイにおける高用量ペプチドの特異的溶解率の算術平均±SEMとして表す(n=5、対照と比較したDEC205およびCLEC9a群のp<0.001。一元配置ANOVA)。B:抗CD40有りまたは無しで投与した、アイソタイプ対照または抗CLEC9aと結合したS2を使用した同様の実験の結果。
【図13B】抗CLEC9aモノクローナル抗体を使用した標的化によるCTL免疫の誘導を示す図である。B:抗CD40有りまたは無しで投与した、アイソタイプ対照または抗CLEC9aと結合したS2を使用した同様の実験の結果。
【図14】B16メラノーマの治療における抗CLEC9a+抗CD40の治療効果を示す図である。A.B16-OVA-GETメラノーマ細胞を使用した腫瘍治療実験の時間的経過。腫瘍細胞(2×105個)を第0日に静脈内注射し、抗体-S1+抗CD40の治療を第6日に実施し、第18日に肺を摘出し腫瘍を計数した。B.1つの代表的な実験におけるそれぞれのマウス中の腫瘍数を示す。全身腫瘍組織量の減少は、抗CLEC9aまたは抗DBC205および抗CD40を用いて有意である(p<0.001、一元配置ANOVA)。
【図15】ヒトCLEC9aの発現がBDCA-3+血中DCに限られることを示す図である。(A)ヒトPBMCを抗hCLEC9a(8F9)またはアイソタイプ適合対照抗体(マウスIgG2a)で染色し、様々な血液白血球マーカー用に対比染色した。ヒストグラムは、T細胞、B細胞、単球、NK細胞、系統陰性HLA-DR-細胞および系統陰性HLA-DR+細胞において染色したCLEC9aを示す。数字は後者の分画中のhCLEC9a+細胞の割合を示す。(B)(A)からのPBMCを系統陰性HLA-DR+細胞でゲートした。ドットプロットは、様々な血中DCサブセットマーカーに対する抗CLEC9aまたはアイソタイプ適合対照モノクローナル抗体を用いた染色を示す。数字はそれぞれの四分円中の細胞の割合を表す。特異的染色はBDCA-3+DCにおいてのみ見られる。4つの中の1つの代表的な実験。
【図16】CLEC-9aに腫瘍抗原を標的化することによるB16メラノーマの免疫療法を示す図である。(A)腫瘍治療実験を、抗CLEC9aまたはアイソタイプ適合対照抗体と共有結合したメラニン細胞分化内在性抗原(「Endo」:gp100、TRP-1およびTRP-2)の知られているエピトープを含むペプチドを使用して示したように実施した(上左)。ポリI:C+抗CD40はアジュバントとして使用した。左下図は、示したように治療したマウス由来の肺の代表図を示す。右図はそれぞれのマウス中の肺腫瘍の定量化を示す。データは2回の独立した実験(n=9マウス/群)からプールし、それぞれのポイントは1匹のマウスを表す。(B)(A)中の個々のマウス由来の脾細胞を、免疫処置に使用したメラニン細胞分化抗原ペプチド(10μM)を用いてin vitroで再度刺激した。2dの培養後のIFN-γのレベルを示す。データは2回の独立した実験(n=9マウス/群)からプールした。P値はマンホイットニーのU検定を使用して計算した。(C)B16細胞を注入する1日前にワクチンを与えたこと以外、図16中と同様に実験を実施した。データはマウス当たりの肺腫瘍の数を示す。データは2回の独立した実験(n=7マウス/群)からプールし、それぞれのポイントは1匹のマウスを表す。P値はマンホイットニーのU検定を使用して計算した。
【図17】死/死滅細胞上でCLEC9Aがリガンドと結合することを示す図である。(A)BWZ-マウスおよびヒトCLEC9Aζレポーターの基底活性化は、死滅TO-PRO3+細胞の数と相関関係がある。LacZと結合したNFATの受容体を発現し、かつマウスまたはヒトCLEC9Aまたは対照デクチン-1由来の細胞外ドメインおよびCD3ζ由来の細胞内尾部を有するキメラ分子を安定的に発現するBWZ細胞を、CLEC9Aの天然リガンドに関するスクリーニング用に作製した。異なる濃度の細胞を2日間増殖させ、異なる程度の異常増殖を示し、培養中の死滅細胞の増大をTO-PRO3染色によって追跡した。同量の生存BWZ細胞を次いで新たな培地に平板培養し、一晩の培養後、Methodsで示したように、BWZ細胞中のNFAT活性を比色分析アッセイにおいて測定した。 (B)UVC処理死滅細胞をCLEC9Aのリガンドに曝す。細胞死を誘導するためにUVCで処理し24時間放置した生存MEF(Ctrl)またはMEFを、キメラマウスCLEC9A-ζ、ヒトCLEC9A-ζ、およびデクチン-1-ζを発現するBWZ NFATレポーター細胞と共に培養した。示した場合、対照(p21)または抗mCLEC9A(1F6)または抗hCLEC9A(8F9)の一価Fab断片を培養物に加えた。BWZ細胞中のNFAT活性は(A)中と同様に測定した。 (C)UV処理細胞中の用量応答。LK細胞はMethods中で示したように異なる線量のUVCに曝し、mCLEC9A-ζキメラを発現するBWZ細胞または対照BWZとの培養前に24時間放置して細胞死を誘導した。BWZ細胞中のNFAT活性は(A)中と同様に測定した。 (D)CLEC9Aの組換え可溶性C型レクチンドメイン(rsCTLD)は、TO-PRO3+死滅細胞によって曝される分子を選択的に認識する。CLEC9Aまたは(対照として)デクチン-1のrsCTLDのPE-テトラマーは、UV照射した不死化MEFの染色用に使用した(ドットプロット)。ヒストグラムは、デクチン-1のrsCTLDに陽性なザイモサンの染色を示す。 (E)異なる死誘導処理はBWZ-CLEC9Aζレポーター細胞を誘発する。BWZ-CLEC9Aζ細胞は単独で(Ctrl)、または未処理(LK)またはUVC(UV)、ミトキサントロン(Mtx)、血清枯渇(SD)、浸透圧ショック(OS)または凍結および解凍(FT)で処理したLK細胞と共に一晩培養した。BWZレポーターの活性(左側y軸)ならびに同時培養開始時のLK細胞中のto-pro3+およびCLEC9A rsCTLD+の頻度(右側y軸)を示す。 (F)CLEC9AのrsCTLDのPE-テトラマーを使用した死滅MEFの染色。MEFはUVで処理して24時間放置し、または染色前に浸透圧ショック(OS)で処理した。 (A)〜(F)実施した少なくとも3つの中の1つの代表的な実験を示す。
【図18】CLEC9Aがin vitroでのCD8α+DCによる死細胞とのクロスプライミングに関与することを示す図である。 (A)、(B)CLEC9Aの阻害は、死滅細胞関連抗原とのOT-I細胞のクロスプライミングの低下をもたらす。(A)OT-I OVA特異的T細胞を、抗CLEC9AFab(1F6)または対照Fab(p21)の有無の下においてOVA導入死滅bm1脾細胞で刺激したCD8a様Flt3L BMDCと共に培養した。3日後、増殖(OT-I細胞の絶対数)、IL-2およびIFNγ産生(未処理対照に対する産生率)を測定した。6つの中の1つの代表的な実験を示す。(B)実施した6回の独立した実験に関する平均IL-2およびIFNγ産生は、抗CLEC9AFab(1F6)または対照Fab(p21)に関する未処理対照による産生率として示す。 (C)CLEC9A-/-およびWTCD8α様Flt3L BMDCは、死細胞の物質を捕捉する同じ能力を有する。WTまたはCLEC9A-/-CD8α様Flt3L BMDCは、異なる割合でPKH26標識およびUVC処理bm1脾細胞と共に2時間インキュベートした。結合(4℃)および結合+摂取(37℃)を、次いでそれぞれの型のDCに関するフローサイトメトリーにより定量化した。 (D)OVA発現またはOVA導入bm1死滅細胞で刺激したCLEC9A-/-Flt3L BMDCとのインキュベーション後の、エフェクターOT-I細胞に対する増殖および分化の低下。CLEC9A-/-Flt3L BMDCは、UVC処理OVA導入bm1脾細胞またはUVC処理OVA発現bm1 MEFと共に培養した。OVA特異的OT-I T細胞を次いで加え、かつ同時培養の3日後、OT-I細胞(左図)またはIFN-γ(右図)の絶対数を測定した。結果は一群当たり2匹のマウスの平均±SEMを示す。実施した3つの中の1つの代表的な実験を示す。 (E)(D)中と同様にOVA発現UV死滅bm1 MEFで刺激したCLEC9A-/-Flt3L BMDCとのインキュベーション後の、OT-I細胞の分化の低下。同じアッセイを使用して分析した四群中の一群当たりの1匹の代表的なマウスを示す。
【図19】CLEC9Aが免疫細胞の死を感知してin vivoでのクロスプライミングを促進することを示す図である。 (A)、(B)CLEC9Aの阻害はin vivoでの死滅細胞関連抗原とのクロスプライミングを低減する。未治療マウス、または抗CLEC9A(1F6、400μg/マウス)またはアイソタイプ対照(ラットIgG1)で治療したマウスを、切断型OVA-GFP融合タンパク質を発現する0.75×106個のUV照射bm1 MEFを用いた静脈内注射によって免疫処置した。6日後、H2Kb-OVAペプチドテトラマー陽性細胞(A)およびex vivoでのSIINFEKLに応じたIFNγ産生(B)を、内因性レパートリーから生じるCD8+T細胞エフェクター応答の誘導に関する読み出し値として測定した。(3つの中)1つの代表的な実験に関する個々のマウスおよび平均を示す。 (C)〜(E)CLEC9Aの欠失はin vivoでの死滅細胞関連抗原とのクロスプライミングを低減する。CLEC9a-/-マウスまたは対照同腹子は(A)中と同様に免疫処置した。OVA特異的内因性CD8+T細胞(C〜D)の頻度を(A)中と同様に定量化した。(C)それぞれのドットは、6回の独立した実験からプールしMethodsで示したように正規化した個々のマウスを表す。(D)CLEC9a-/-およびCLEC9a+マウスに関するそれぞれの同腹子におけるテトラマー陽性細胞の平均を表す。(E)ex vivoでのSIINFEKLに応じたIFNγ産生を、(B)中と同様に測定した。(3つの中)1つの代表的な実験に関する個々のマウスおよび平均を示す。
【図20】固定/透過処理後にCLEC9aのリガンドを曝したことを示す図である。 LK細胞を2%ホルムアルデヒドまたは2%パラホルムアルデヒドで固定し、Tween(0.5%)およびTx-100(0.5%)で透過処理したか、またはしなかった。細胞は大々的に洗浄し、透過性細胞の割合をTo-pro3を使用して定量化した。固定または透過処理細胞はキメラマウスCLAC9a-CD3ζを発現するBWZ NFATレポーター細胞または対照と同時培養し、NFAT活性を測定した。
【図21】CLEC9a wt細胞質尾部を介した死滅細胞のシグナルを示す図である。 LK細胞はMethods中で示したように異なる線量のUVCに曝し、CLEC9a wtまたはTyr7がPheに突然変異したCLEC9a(Y7F)で安定的にトランスフェクトしたB3Z細胞と共に培養し、Sykと同時発現させるかまたはさせない前に24時間放置して細胞死を誘導した。B3Z細胞中のNFAT活性はMethodsで記載したように測定した。
【図22】Fab一価と完全二価抗CLEC9a抗体の両方がCLEC9aを介した死滅細胞のシグナルを阻害することを示す図である。 細胞死を誘導するためにUVCで処理し24時間放置したLK細胞を、CLEC9a wtまたはTyr7がPheに突然変異したCLEC9a(Y7F)で安定的にトランスフェクトしたB3Z細胞と共に培養し、Sykまたは対照としてB3Z-デクチン-1-ζと同時発現させた。示した場合、対照(p21)または抗CLEC9A(1F6)の一価Fab断片、またはアイソタイプ対照(ラットIgG1、ラットIgG2a)および抗mCLEC9a(1F6、397、7H11)を含めた完全二価抗体を培養物に加えた。B3Z細胞中のNFAT活性はMethodsで記載したように測定した。
【図23】抗CLEC9a-0VA323-339に応じたOVA特異的OT-II CD4 T細胞の増殖を示す図である。未感作CFSE標識OT-II CD4 T細胞をC57BL/6マウスに静脈内注射で移した。1日後、抗CLEC9aまたはアイソタイプ対照抗体のいずれかと結合したOVA323-339ペプチドを脚部に皮下接種した。脾臓および流入領域リンパ節を3〜4日後に回収しOT-II細胞のin vivo増殖をCFSE希釈後に追跡した。サンプル間で正規化したOT-II細胞の絶対数を示す。
【図24】in vivoでの標的化中のアジュバントの添加は、強力なTh1の応答を誘導することを示す図である。アジュバント(OT-II系)の有無の下でのin vivo標的化。マウスは図23中と同様に処理した。上図:(正規化後の)OVA特異的CD4 T細胞の絶対数。下図:割合(左)または絶対数(右、正規化後)として表したOT-II CD4 T細胞集団中のIFN-γ産生。ポリI:Cの存在下で観察した増殖および分化に対する影響はこのアジュバントに限られない。カードランは同様の影響を有する。
【図25】CD4 T細胞応答は、標的化試薬と同時に注射するアジュバント/免疫調節剤の型によって調節することができることを示す図である。アジュバントの有無の下でのin vivo標的化。OT-II系は図24中と同様に実施した。上図:割合(左)または絶対数(右、正規化後)として表したOT-II CD4 T細胞集団中のIL-17産生。カードラン、デクチン-1アゴニストは、標的化抗原と投与すると、Th17分極の強力なアジュバントとして作用した。
【図26】アジュバントの不在下での標的化は、抗原特異的耐性をもたらすことを示す図である。未感作OVA特異的DO.11.10 CD4 T細胞はBALB/cマウスに移した。1日後、抗CLEC9a-OVA 323-339をアジュバント(ポリI:C)有りまたは無しで脚部に皮下接種した。免疫処置後の異なる時間地点で、脾臓DO.11.10区画の大きさを、KJ.126クローンタイプ抗体を使用したフローサイトメトリーによって決定した。結果は(正規化後の)OVA特異的KJ.126細胞の絶対数として、またはCD4 T細胞区画中のKJ.126+細胞の割合として表す。第20日に、半数のマウスをOVA、フロイント完全アジュバント中で皮下攻撃した。5日後、DO.11.10の応答をフローサイトメトリーによってモニターした。第0日にPBSのみを注射したマウス中で、残りのDO.11.10細胞は、再攻撃後のそれらの強力な増殖応答によって示されるように、依然として応答性がある。より強力な応答を抗CLEC9a+アジュバントを事前に注射したマウス中で検出し、この最初の免疫処置がおそらく記憶細胞の生成をもたらしたことが示された。対照的に、抗CLEC9aのみで最初に免疫処置したマウス中では応答を検出せず、残りの細胞は抗原に耐性があったことが示された。
【発明を実施するための形態】
【0158】
CLEC9a
CLEC9aは、樹状細胞上に発現されるC型レクチンである。本明細書において使用される場合、用語「CLEC9a」は、ヒトタンパク質(図1に示す核酸およびタンパク質配列)、マウスタンパク質(図2に示す核酸およびタンパク質配列)、他の種におけるそれらの相同体(特に相同分子種)、ならびにCLEC9a活性を保持するそれらの変異体および誘導体を包含することを意図する。このような変異体および誘導体は、図1に示すヒトタンパク質配列に対して好ましくは少なくとも約30%の配列同一性、より好ましくは少なくとも約35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%または95%の配列同一性、または図1に示すヒトタンパク質配列の細胞外ドメインに対して少なくとも約約35%の同一性、より好ましくは少なくとも約40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%または95%の同一性を有する。
【0159】
特に、CLEC9a配列における保存的置換(参照配列と比較して)は、機能に実質的な影響がなく特に忍容性良好であり得る。
【0160】
保存的置換は、アミノ酸クラス内の置換および/またはBLOSUM62マトリックスにおいて陽性と判定される置換と定義することができる。
【0161】
1つの分類によれば、アミノ酸クラスは酸性、塩基性、非荷電極性および非極性であり、酸性アミノ酸はAspおよびGluであり;塩基性アミノ酸はArg、LysおよびHisであり;非荷電極性アミノ酸はAsn、Gln、Ser、ThrおよびTyrであり;および非極性アミノ酸はAla、Gly、Val、Leu、Ile、Pro、Phe、Met、TrpおよびCysである。
【0162】
別の分類によれば、アミノ酸クラスは、低分子親水性、酸/酸アミド/親水性、塩基性、低分子疎水性および芳香族であり、低分子親水性アミノ酸はSer、Thr、Pro、AlaおよびGlyであり;酸/酸アミド/親水性アミノ酸はAsn、Asp、GluおよびGlnであり;塩基性アミノ酸はHis、ArgおよびLysであり;低分子疎水性アミノ酸はMet、Ile、LeuおよびValであり;および芳香族アミノ酸はPhe、TyrおよびTrpである。
【0163】
BLOSUM62マトリックスにおいて陽性と判定される置換は以下の通りである:
【0164】
【表1】

【0165】
参照配列に対するアミノ酸配列同一性パーセント(%)は、最大配列同一性パーセントを達成するために必要であれば、配列のアラインメントおよびギャップ導入後、保存的置換を配列同一性の一部とみなさずに、参照配列中のアミノ酸残基に同一の候補配列中のアミノ酸残基の割合として定義される。同一性%値は、WU-BLAST-2(Altschulら、Methods in Enzymology、266:460〜480(1996年))によって決定することができる。WU-BLAST-2はいくつかの検索パラメータを使用し、これらのほとんどは初期値に設定される。調整可能なパラメータは以下の値に設定される:オーバーラップスパン=1、オーバーラップフラクション=0.125、ワード閾値(T)=11。アミノ酸配列同一性%値は、WU-BLAST-2によって決定されるマッチングする同一な残基の数を、参照配列(無視されるアラインメントスコアを最大にするために参照配列中にWU-BLAST-2によってギャップを導入した)の全残基数で割って、100をかけることによって決定される。
【0166】
CLEC9aアゴニストは、典型的には、その細胞外ドメイン(ECD)に結合することおよびその細胞内ドメインを介して細胞内シグナル伝達を誘導することによって、CLEC9a活性を誘導することができる作用物質である。シグナル伝達は、Sykの細胞内ドメインへの結合、Sykリン酸化(およびしたがって活性化)、および/またはErkリン酸化および/またはNFAT活性化の1つまたは複数を含み得る。Clec9aおよびSykをトランスフェクトしたB3Z細胞を使用した例示的なアッセイを実施例に記載する。当業者であれば、Clec9aの細胞外ドメインおよび異なるタンパク質由来の細胞内ドメインを有するキメラタンパク質もまた、Clec9aアゴニスト活性をアッセイするために使用できることを理解するはずである。膜貫通ドメインもまた、Clec9a由来、細胞内ドメインと同じタンパク質、または別のタンパク質由来であってもよい。一例は図5に例示したCD3ζ-NKRPl-CLEC9aであり、実施例により詳細を記載する。
【0167】
CLEC9aアンタゴニストは、CLEC9a機能を阻害または遮断することができる作用物質である。例えば、CLEC9aアンタゴニストは、通常の発現、生理的CLEC9aリガンドに結合する能力、結合した分子を吸収する(エンドサイトーシスによって取り込む)能力、細胞内でシグナル伝達する能力(上記参照)、または例えば別の細胞上でそのECDが結合パートナー(リガンドまたは受容体)と相互作用する能力を妨害することができる。アンタゴニストの別の考えられる作用機構は、顕著な細胞内シグナル伝達を誘導することなく、Clec9aの吸収を促進し、そのため天然リガンドまたは別のアゴニストと相互作用するために利用可能な細胞表面上のClec9aのプールを減少させることである可能性がある。アンタゴニストは、有意なアゴニスト活性を欠く抗Clec9a抗体などの、CLEC9aに対して親和性を有する結合剤を含む。これらは「遮断」抗体と呼ばれることがある。アゴニスト活性を有さない一価または二価抗体は、遮断抗体として特に適する可能性がある。別のClec9aアンタゴニストは、CLEC9aをコードするDNAまたはRNAとハイブリダイズすることができる核酸分子またはそのアナログを含む。このような作用物質はリボザイム、RNAi、siRNAなどを含む。
【0168】
さらなるCLEC9aアンタゴニストは、樹状細胞結合CLEC9aのリガンド上の結合部位を遮断することができ、そのためリガンドが樹状細胞に認識、または結合されるのを妨害することができるCLEC9aリガンドの競合物である。適した競合物は、CLEC9aリガンドに結合するために十分なCLEC9aの細胞外ドメインまたはその一部を含む可溶性分子を含む。したがって、この分子は、図1に示すヒトCLEC9a、または図2に示すマウスCLEC9a、またはCLEC9aリガンドに親和性を有するその断片の細胞外ドメイン(CTLD)に対して、少なくとも70%の同一性、少なくとも75%の同一性、少なくとも80%の同一性、少なくとも85%の同一性、少なくとも90%の同一性、または少なくとも95%の同一性を有するアミノ酸配列を含むことができる。断片は、それぞれの細胞外ドメイン配列またはそれに所要のレベルの同一性を有する配列の、少なくとも20、30、40、50、60、70、80、90、100、110個または少なくとも120個のアミノ酸を含むことができる。
【0169】
CLEC9a細胞外ドメイン(またはその一部)は、非相同部分と結合していてもよく、これはin vivoでの薬物動態特性などのアンタゴニストの特性を若干変える可能性がある。細胞外ドメインは、非相同部分に共有または非共有結合していてもよく、または非相同部分を有する融合タンパク質として発現されてもよい。例えば、血中半減期の増加をもたらし、アンタゴニストとCLEC9aリガンドの複合体の効率的な除去を可能にするために、非相同部分は抗体Fcドメインであってもよい。
【0170】
他の考えられる非相同部分の機能は、CLEC9a細胞外ドメインのオリゴマー形成の媒介、およびアンタゴニストの精製または試料からの単離の簡易化を含む。例えば、適したアンタゴニストは、アビジン単量体に結合しているCLEC9a細胞外ドメイン(またはCLEC9aリガンドに結合するのに十分なその断片)を含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む複合体をもたらす。この構築体は、ビーズなどの固体支持体上で提供することができるビオチンとの接触により容易に単離することができる。
【0171】
結合剤またはアンタゴニストがタンパク質である場合、アンタゴニストをコードする核酸(例えば、DNA)を投与することが可能であり得る。典型的には、核酸は体(例えば、筋細胞)内で細胞によって取り込まれ、これらの細胞から発現され、分泌されるはずである。このアプローチはしばしばDNAワクチン接種と呼ばれる。
【0172】
抗体は、結合剤およびアンタゴニストとして特に適しており、scFv形態で好都合に発現することができる。必要であれば、抗体を、抗原または上述のエフェクター部分と共に融合タンパク質としてコードすることができる。DNAワクチン接種アプローチの例は、Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年に記載されている。
【0173】
核酸は典型的に、核酸を取り込む細胞によるタンパク質の適切な発現および分泌を確実にするために、転写および翻訳調節配列と作動可能に連結して、場合により任意の所望の融合パートナーと共に、結合剤またはアンタゴニストをコードするコード領域を含む。このような配列は、転写開始配列(例えば、プロモーターおよびエンハンサー)、転写終結配列、適切なスプライシングシグナル、翻訳開始および終結配列、ならびに分泌を可能にするシグナルペプチドを含む(がこれらに限定されない)。
【0174】
本発明は、医療方法において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸(例えば、DNA)をさらに提供する。その方法および治療的使用において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸もまた提供される。
【0175】
CLEC9aリガンド
本発明者らは、CLEC9aが特定の種類の死または瀕死哺乳動物細胞によりディスプレイされたリガンドを認識することを発見した。特に、特定の種類の細胞死はリガンドのディスプレイを誘発するようである。C型レクチンファミリーの多くの既知のメンバー(CLEC9aに最も密接に関連したタンパク質であるデクチン1を含む)は、病原体関連分子パターンの受容体であり、そのため自己分子よりも病原体によってディスプレイされる構造を認識するので、これは驚くべき発見である。
【0176】
自己細胞死の特定の機構が免疫応答を誘発することができることはよく認識されている。これらは免疫原性細胞死とみなすことができる。アポトーシス(通常、原形質膜の破裂および細胞内容物の放出をもたらさない)による死は非免疫原性であり、一方、壊死(原形質膜の破裂および細胞内容物の放出を伴う)などの他の機構による死は免疫原性であると提唱されている。しかしながら、生理学的状況はこれよりも複雑であるようである。例えば、in vivoにおけるアポトーシス細胞は通常、細胞死のプロセスが完了する前にマクロファージなどの隣接細胞により吸収(食菌)される。しかしながら、細胞が食菌されなければ、いわゆる二次性壊死が起こる可能性があり、原形質膜は破裂し、細胞内容物は放出される可能性がある。この性質の細胞死は、少なくとも一部はアポトーシスであるにもかかわらず免疫原性とすることができる。
【0177】
免疫原性細胞死は、自己免疫疾患の発症、進行または持続において役割を果たしている可能性がある。このことは、例えば、Viorittoら、(Clin Immunol 122(2)、125〜134(2007年))、Tesniereら(Corr Op Immunol 21、1〜8(2008年))およびKimら(Immunity 27、321〜333(2007年))によって概説されている。
【0178】
樹状細胞は、死細胞または瀕死細胞の細胞破片を取り込むこと(またはさらに細胞全体を吸収すること)、およびプロセシングされた断片をT細胞に提示することによって、免疫原性細胞死により引き起こされる任意の免疫応答の誘導において役割を果たしている可能性がある。
【0179】
本発明者らは今回、CLEC9aが死細胞または瀕死細胞によりディスプレイされるリガンドに結合できること、およびCLEC9aシグナル伝達がこの相互作用によって誘発され得ることを発見した。
【0180】
細胞死プロセスの間、リガンドが新たに合成されないこともまた考えられる。むしろ、リガンドは一部のまたは全ての哺乳動物細胞によって構成的に発現されるが、細胞が正常であり続ける限りは、CLEC9aと接触して相互作用することができない可能性がある。特定の種類の細胞死は、CLEC9aと相互作用可能な形態でリガンドの露出および/または細胞からのリガンド放出をもたらす。これは原形質膜の破裂を伴い得る。
【0181】
実験的には、リガンドの露出は、照射(例えば、UV光などの電離放射線による)、血清除去、少なくとも1回の凍結/融解サイクルなどの処理によって、またはアントラサイクリン(ドキソルビシンおよびダウノルビシンなど)およびアントラセンジオン(ミトキサントロンなど)などの化学療法剤を用いた処理によって、引き起こすことができる。しかしながら、浸透圧ショックによる死はリガンドを露出させないようである。
【0182】
したがって、いかなる特定の理論に拘束されることを望むものではないが、CLEC9aは免疫原性細胞死によって引き起こされる免疫応答の発生に関与する可能性があると考えられる。CLEC9aおよびそのリガンド間の相互作用は、CLEC9aアンタゴニストによって阻害することができる。これらは、CLEC9a(の細胞外ドメイン)に結合することができる結合剤を含む。別の例は、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合できるその一部(例えば、細胞外ドメインの少なくとも20アミノ酸、少なくとも50アミノ酸、少なくとも100アミノ酸、少なくとも150アミノ酸、または少なくとも200アミノ酸)を含む可溶性作用物質などの、リガンド上のCLEC9a結合部位の競合物を含む。
【0183】
CLEC9aおよびそのリガンド間の結合を阻害することができるアンタゴニストは、CLEC9aシグナル伝達を誘導することができる適した死細胞または瀕死細胞(例えば、UV照射された哺乳動物細胞)、その溶解物、抽出物または画分と接触させれば、CLEC9aシグナル伝達を阻害することができるはずである。任意の適した試験系を、この能力を評価するために使用することができる。例えば、CLEC9aを発現する樹状細胞においてCLEC9aシグナル伝達は、Sykキナーゼのリン酸化を測定することによって評価することができる。代替として、実施例に記載したCD3zetaキメラなどのレポーター系に機能的に連結したCLEC9a細胞外ドメインを含む人工レポーター系を使用することができる。
【0184】
結合剤
CLEC9aに対する十分に高い親和性および特異性を有する任意の適した分子を結合剤として使用することができる。分子は、タンパク質、核酸(例えば、アプタマー)、糖質(例えば、オリゴ糖または多糖)、小分子などであってもよい。特に好ましい結合剤は、CLEC9aの生理的リガンド、ならびにCLEC9aに対する抗体およびその機能的断片である。
【0185】
結合剤は、好ましくはCLEC9a、特にCLEC9a ECDに対する、少なくとも105M-1、少なくとも106M-1、少なくとも107M-1、好ましくは少なくとも108M-1、より好ましくは少なくとも109M-1の結合親和性(親和性定数)を有する。
【0186】
結合剤は、好ましくは他のC型レクチンを含む任意のCLEC9aではない分子に対して少なくとも2×、および好ましくは少なくとも5×、少なくとも10×、少なくとも50×のまたは少なくとも100×を超える親和性を有する。
【0187】
全抗体の断片が結合抗原の機能を果たすことができることはよく知られている。機能的結合断片の例は、(i)VL、VH、CLおよびCH1からなるFab断片;(ii)VHおよびCH1ドメインからなるFd断片;(iii)単一抗体のVLおよびVHドメインからなるFv断片;(iv)VHドメインからなるdAb断片(Ward,E.S.ら、Nature 341、544〜546(1989年));(v)単離CDR領域;(vi)2つの連結したFab断片を含む二価断片であるF(ab')2断片;(vii)VHドメインおよびVLドメインがペプチドリンカーにより連結しており、2つのドメインが結合して抗原結合部位を形成することが可能である、単鎖Fv分子(scFv)(Birdら、Science、242、423〜426、1988年; Hustonら、PNAS USA、85、5879〜5883、1988年);(viii)二重特異的単鎖Fv二量体(PCT/US92/0996);(ix)遺伝子融合により構築される多価または多重特異的断片、「ダイアボディ」(WO94/13804;P.Holligerら、Proc.Natl.Acad.Sci.USA 90 6444〜6448、1993年)である。
【0188】
抗体は多くの方法で修飾することができるので、用語「抗体」はしたがって、必要な特異性を有する結合ドメインを有する任意の特異的に結合する物質も含むと解釈されるべきである。したがってこの用語は、天然または合成であるかにかかわらず、免疫グロブリン結合ドメインを含む任意のポリペプチドを含む、上述の抗体断片ならびに抗体の誘導体、機能的同等物および相同体を含む。したがって、別のポリペプチドに融合した、免疫グロブリン結合ドメイン、または同等物を含むキメラ分子が含まれる。キメラ抗体のクローニングおよび発現はEP-A-0120694およびEP-A-0125023に記載されている。
【0189】
本明細書中に記載の本方法において使用される結合剤は一般に、所要の作用を発揮するためにCLEC9aの細胞外ドメインに結合することが必要であることが理解されるはずである。CLEC9aに結合することができる結合剤への参照は、文脈上特に明記しない限り、それに応じて解釈されるべきである。
【0190】
本発明の特定の態様において、抗原(例えば、タンパク質またはペプチド抗原)を記載の結合剤と架橋させることが望ましい。当業者は、適した方法および試薬をよく知っている。結合剤がタンパク質である場合、抗原は結合剤のスルフヒドリル基を介して結合することができる。スルフヒドリル基は、通常遊離していてもよく、または通常結合剤の選択的還元によって露出するジスルフィド結合の一部であってもよい。例えば、抗体は、還元剤メルカプトエタンスルホネートを使用してヒンジ領域において選択的に、穏やかに還元することができる。次いで、抗原を、タンパク質の第3級アミンと反応するヘテロ二官能性架橋試薬、スルホ-SMCCを使用して活性化して、遊離スルフヒドリル反応基を生じさせる。次いで、抗体および活性化された抗原を一緒にインキュベートして、一価抗体とコンジュゲートしたタンパク質を得る18。代替として、抗原由来の適した免疫原性ペプチド配列が既知であれば、遊離スルフヒドリルを有するシステインを含有するこのようなペプチドを合成して、スルホ-SMCC活性化抗体に結合させることができ、これは二価で、1抗体分子につきいくつかのペプチドが結合しているままである。
【0191】
医薬組成物
本明細書中に記載のポリペプチド、抗体、ペプチド、核酸および細胞は、医薬組成物に処方することができる。これらの組成物は、上記物質の1つに加えて、薬学的に許容される賦形剤、担体、緩衝液、安定剤または当業者によく知られている他の材料を含むことができる。このような材料は、非毒性であるべきであり、または活性成分の有効性を妨げるべきではない。担体または他の材料の明確な性質は、例えば、経口、静脈内、皮膚または皮下、経鼻、筋肉内、腹腔内経路などの投与経路に依存し得る。
【0192】
経口投与用の医薬組成物は、錠剤、カプセル、粉末または液体形態であってもよい。錠剤は、ゼラチンなどの固体担体またはアジュバントを含み得る。液体医薬組成物は、一般に、水、石油、動物性または植物性油、鉱物油または合成油などの液体担体を含む。生理食塩水、デキストロースもしくは他の糖溶液、またはエチレングリコール、プロピレングリコール、もしくはポリエチレングリコールなどのグリコールが含まれ得る。
【0193】
静脈内、皮膚もしくは皮下注射、または患部への注射用の活性成分は、発熱物質を含まない、適したpH、等張性および安定性を有する、非経口的に許容される水性溶液の形態であるはずである。当業者は、例えば、塩化ナトリウム注射、リンゲル液注射、乳酸加リンゲル液注射などの等張性ビヒクルを使用して、適した溶液を容易に調製することができる。防腐剤、安定剤、緩衝液、酸化防止剤および/または他の添加剤を、必要に応じて含むことができる。
【0194】
投与は、好ましくは「予防的有効量」または「治療的有効量」(場合によっては、予防が治療とも考え得るが)であることが好ましく、これは個体に対する有益性を示すの十分である。実際の投与量ならびに投与率および投与の経過は、治療されるものの性質および重症度に依存するはずである。例えば、用量の決定などの治療の処方箋は一般開業医および他の医師の責任であり、典型的には、治療する疾患、個々の患者の状態、投与部位、投与方法および開業医に既知の他の要因を考慮する。上記の技術および手順の例は、Remington's Pharmaceutical Sciences、第20版、2000年、pub. Lippincott、WilliamsおよびWilkinsで見ることができる。
【0195】
組成物は、単独でまたは他の治療と組み合わせて、治療される状態に応じて同時にまたは順次に、投与することができる。
【0196】
(実施例)
CLEC9aの配列および構造
NCBI遺伝子データベースの検索は、CLEC9aの配列はハツカネズミ(Mus musculus)、チンパンジー(Pan troglodytes)、ホモサピエンス(Homo sapiens)およびアカゲザル(Macaca mu1atta)において既に同定されていることを示す。マウスCLEC9aのタンパク質配列を使用するblast検索も、ドブネズミ(Rattus norvegicus)、ケイネスファミリアリス(Canis familiaris)およびボスタウラス(Bos Taurus)における予想CLEC9aタンパク質を示す。ヒトcDNA配列および関連ドメインを有する注釈付きタンパク質配列は図1中に詳述する。本発明者らがマウスCD8α+DCからクローニングしたマウスcDNA配列およびその注釈付きタンパク質配列は図2中に詳述する。この配列は、分子の末端へのフレームシフトを引き起こし図2中に示すタンパク質より長いタンパク質をもたらす、追加的G残基を含有する公開cDNA配列とは異なる。本発明者らの配列は正確であるようである。それは公開ゲノム配列(NC_000072.4GI:94471533)に適合し、このページ中で見ることができるように、ゲノム(ATTT)の位置13480は本発明者らのcDNA配列に適合するからである。この配列によって、図1および2中で強調する一種保存チロシンを含有する、C型レクチン様ドメイン(CTLD)、茎部、膜貫通領域および細胞質ドメインを有するC型レクチンファミリータンパク質を予想する。本発明者らは、マウスCLEC9aの3つのアイソフォームに関する転写産物を発見しており、本発明者らは、それらを長いアイソフォーム(エクソン1〜7)、二量体化に関与する推定システインを含むエクソン4を欠く短いアイソフォーム、およびエクソン3〜エクソン7と結合し、発現する場合CLEC9aと膜貫通-細胞内ドメインを共有し得るが、短い異なる細胞外ドメインを有し得る膜貫通タンパク質をコードするmRNAを生成する、非常に短いアイソフォームと名付けている。本発明者らは、長いアイソフォームに関するタンパク質発現の証拠のみを有する。
【0197】
マウスCLEC9aの構造を分析した。コアタンパク質は約29.67KDaの予想分子量(Mw)を有する。しかしながら、HEK-293細胞系中で発現されるとき、Mwは非還元状態で約100KDaであり、かつ還元状態でMwは約45KDaである(図3)。これらの結果は、この分子が茎部中のシステインを介して二量体を形成し、ファミリー中の他のレクチンと同様に、モノマーは強くグリコシル化されることを示す。
【0198】
CLEC9aの発現
マウス脾臓CD11c+CD8+およびCD11c+CD8-細胞のサンプル間の代表的な差分解析の結果として、本発明者らの研究室においてCLEC9aを最初に検出した。結果は、ESTクローンAW318446に相当する配列、CLEC9aに相当する配列がCD11c+CD8+転写産物中で選択的に見られたことを示した。
【0199】
脾臓DCの分類したサブセットにおける転写産物の分析によってCD8+サブセット中でのCLEC9aの高発現が明らかになったが、二重陰性(CD4-CD8-)およびB220+脾臓DCもCLEC9aに関するいくつかの転写産物を示した(図4)。GMCSF由来BMDCにおいて有意な発現は見られなかった(図4)。F1t3L(50ng/ml)の存在下で10日間培養したマウス骨髄は、機能上脾臓の従来のDCに相当しCD8様サブセット19を含むCD11b+、または形質細胞様DC(pDC)の機能的同等物であるB220+のいずれかであるCD11c+細胞を生成する。CLEC9aの高発現は分類したCD11b+のサブセットにおいて見られたが、pDCのサブセットはこの分子のいくらかの発現を示した(図4)。
【0200】
RT-PCRは非常に低レベルの転写産物を検出することができる高感度の技法であるので、それはサンプルの精製の質によって制限される。CLEC9aの発現のパターンを明確に決定するために、本発明者らは、Methodsで示したように、B3Z細胞中で発現されるCD3ζ-NKRP1-CLEC9aキメラおよびβ-Gal受容体(図5)を使用して、マウスCLEC9aに対するラットモノクローナル抗体(mAb)を作製した。本発明者らは1F6、397、および7H11という名称の3つのモノクローナル抗体を選択した。これらのモノクローナル抗体を使用して、本発明者らは、マウス脾臓および骨髄中の分子の発現のパターンを試験した。CLEC9aはCD8α+従来型DCにおいて高度に発現され(MFI約350〜400)、pDCにおいて適度な発現(MFI約65〜70)を示した(図6)。B細胞、T細胞、NK細胞、NKT細胞、単球、マクロファージおよび顆粒球を含めた調べた他の細胞型においてこの分子は検出しなかった。
【0201】
in vitroでのCLEC9a機能の分析用のモデルとして、本発明者らはマウスGMCSF-およびF1t3L-由来BMDC中の発現を分析した。本発明者らは、GMCSF-由来BMDC中で分子の発現は検出しなかったが、一方CLEC9aはCD8+脾臓DC19と機能的に相同なFlt3L BMDCのCD11blo、CD24hi、B220-サブセット中、およびpDCと同等なCD11bloB220+のサブセット中でも選択的に発現された(データ示さず)。
【0202】
CLEC9aはSykキナーゼを介してシグナル伝達し、DC活性化を促進する。配列分析によって、推定SH2結合ドメインおよび/またはチロシンベースの分類シグナルとして働き得る、同じ構造EXXYXXLを有する全種で保存されたマウス由来のTyr7が明らかになる。この配列はマウスデクチン-1におけるSykの結合を可能/仲介し、「HemITAM」22と呼ばれる。しかしながら、この推定配列は必要であるが、Sykの結合を予想するには十分ではない。したがって本発明者らは、リン酸化Tyr7を発現するマウスCLEC9a、またはリン酸化無しかまたはさらにPheに突然変異したTyr7を有するマウスCLEC9aの細胞質尾部を有するビオチン化ペプチドを設計した。Tyr7リン酸化ペプチドは、陽性対照23としてリン酸化Tyrをいずれも発現するマウスデクチン-1由来の細胞質尾部と同程度、組換えSykを破壊することができた(図7)。
【0203】
CLEC9aがシグナル伝達受容体として実際作用するかどうかを決定するために、本発明者らは、前に記載したラット抗マウスCLEC9aモノクローナル抗体を使用した。本発明者らは、LK細胞、CLEC9a発現に陰性であるが内因性レベルのSyk23を含有するマウスB細胞系において、CLEC9aを発現するトランスフェクタントを作製した。試験した全3個のプレート抗CLEC9a抗体を用いたCLEC9aの誘発は、LK細胞中でのSykおよびErkのリン酸化をもたらした(図8)。
【0204】
SykがCLEC9aのシグナル伝達に必要であるかどうかを決定するために、本発明者らは、Sykを発現しない受容体T細胞系B3Zの安定したトランスフェクタントを使用した。CLEC9aのシグナル伝達に対するTyr7の貢献を、Pheに突然変異したTyr7を有するCLEC9aの突然変異型(CLEC9a Y7F)を使用して分析した。本発明者らはCLEC9a wtまたはY7FをB3Z細胞に形質導入し、Sykキナーゼを同時形質導入したか、またはしなかった。プレートした抗CLEC9aモノクローナル抗体は、C9のwt細胞質尾部を介してB3Z-C9wt-SykにおいてNFATの活性化を誘導したが、SykまたはY7のいずれかの不在下では誘導しなかった(示さず)。
【0205】
CLEC9aを介した免疫調節性サイトカイン/同時刺激性分子の調節。
CLEC9aを介したシグナル伝達が調節性サイトカインの産生、または発現する場合細胞中での同時刺激性分子の発現に貢献し得るかどうかを決定するために、本発明者らはwt CLEC9またはY7F突然変異体でLKトランスフェクタントを分析した。397抗CLEC9a、および低い程度で7H11を発現するハイブリドーマ細胞は、Y7F突然変異体において無効であったCLEC9a分子による特異的IL-2産生を誘発した(図9)。DEC-205対照ハイブリドーマはいかなる応答も誘発せず、1F6抗CLEC9aは低い応答を誘発し、このことはこれらの抗体がサイトカイン産生に関して差次的に挙動することができ、CLEC9a標的化の展望から非常に魅力的であるという可能性を開く(アゴニスト抗体、397、対遮断抗体、1F6)。
【0206】
CLEC9a WT尾部活性化に関する要件を決定するために、本発明者らは、Sykを発現しない受容体T細胞系B3Zの安定したトランスフェクタントを分析した。CLEC9aのシグナル伝達に対するTyr7の貢献を、Pheに突然変異したTyr7を有するCLEC9aの突然変異型(CLEC9a Y7F)を使用して分析した。本発明者らはCLEC9a wtまたはY7FをB3Z細胞に形質導入し、Sykキナーゼを同時形質導入したか、またはしなかった。プレートした抗CLEC9aモノクローナル抗体は、C9のwt細胞質尾部を介してB3Z-C9wt-SykにおいてNFATの活性化を誘導したが、いずれかのSykの不在下、またはY7突然変異体を使用したときは誘導しなかった(図10)。
【0207】
Flt3L BMDCはIL12-23 p40タンパク質の産生によってプレートした抗CLEC9aモノクローナル抗体に応答する(図11)。
【0208】
これらの結果は、CLEC9aがDCを活性化することができるシグナル伝達分子であることを実証する。
【0209】
CLEC9aはin vivoでCD11c+CD8+DCを選択的に標的化するエンドサイトーシス受容体である
Flt3L BMDCにおいて発現される内因性CLEC9aによって抗CLEC9aモノクローナル抗体が内在化される可能性をFACSによって分析し、CLEC9aがエンドサイトーシス分子であることを明らかした。共焦点分析によって、細胞内区画への抗体の標的化を示した(データ示さず)。これらの結果は、CLEC9aはエンドサイトーシス受容体であり、それは抗原と結合した抗体によって標的化して(腫瘍/ウイルス予防接種)、分子を選択的に発現する細胞サブセットに薬剤を特異的に送達することができることを示唆する。CLEC9aモノクローナル抗体がin vivoで標的化ツールとして働くかどうかを決定するために、本発明者らは7H11-Alexa-488またはアイソタイプ対照を静脈内注射した。16時間後、本発明者らは、全脾細胞および抗体がCD8α+DC(MFI約350〜400)、および低い親和性で、PDC(MFI約65〜70)を選択的に標的化したと分析した。抗ラットCy5を用いた脾細胞の標識は、大部分のラット抗マウスCLEC9aモノクローナル抗体は、それは抗ラット二次試薬と同時染色しなかったので、取り込まれたことを示唆した(示さず)。
【0210】
CLEC9aによる標的化が、in vivoで特異的サブセットによる抗原のプロセシングおよび提示をもたらすかどうかを調べるために、本発明者らは、Methodsで示したように、CTL応答に関する免疫優勢ペプチドのビオチン化誘導体とOVAタンパク質(SIINFEKLC-biot、S1と呼ぶ)、アイソタイプ対照または抗CLEC9aモノクローナル抗体を結合させた。ペプチドのビオチン化によって本発明者らは、全ての場合1抗体当たり1〜1.2のペプチドが存在したと決定することができた。5μgのS1結合抗CLEC9aまたはS1-アイソタイプ対照のいずれかをマウスに静脈内注射した。翌日、脾細胞をCD11c+またはCD11c-サブセットにおいて増大させ、OT-I T細胞増殖およびサイトカイン産生を誘導するそれらの能力に関して試験した(図12aおよびb)。抗CLEC9aのみがCD11c+細胞を標的化し、OT-I細胞による増殖およびIFN-γ産生をもたらし、CLEC9aがCD11c+DCを特異的に標的化し、抗原特異的T細胞に対する提示をもたらすことを示す。
【0211】
樹状細胞のどのサブセットが抗C9によって標的化されるかをさらに決定するために、S1結合抗C9を注射したマウスを、前述のように従来型DCの3つの主要サブセットに分類し、OT-I T細胞を用いて試験した。DCのCD8+サブセットのみがT細胞による増殖およびIFN-γ産生を仲介し、CLEC9はCD11c+CD8+DCを特異的に標的化し、抗原特異的T細胞のプライミングをもたらすことを確認した(図12cおよびd)。
【0212】
抗CLEC9aモノクローナル抗体および抗CD40を使用するin vivo標的化はCTLのプライミングおよび腫瘍拒絶をもたらす
本発明者らは、in vivoでのCLEC9aの標的化が特異的T細胞の活性化を誘導し得るかどうかを調べた。アイソタイプ対照ではなく2μgの抗C9-S1の注射は、抗CD40と同時投与したとき、in vivoにおいて内因性レパートリーからの特異的CTL活性の誘導をもたらす(図13)。この挙動は、in vivo死滅アッセイにより証明した前に記載した24抗DEC205-S1で観察した挙動と同様である(図13A)。
【0213】
対照モノクローナル抗体と結合したS1を与えたマウスは、抗CD40の同時投与と無関係に標的細胞を除去しなかった。対照的に、抗CLEC9aおよび抗CD40と結合したS1を与えたマウスからは、標的細胞は完全に除去された。抗CD40モノクローナル抗体を削除したとき応答は見られなかった。標的細胞の除去と一致して、相当数のテトラマー陽性OVA/H-2 Kb特異的CD8+T細胞を、抗CLEC9a-S1および抗CD40を与えていたマウスの脾臓および血液のみで発見した。in vitroでのSIINFEKLペプチドを用いた同じ細胞の再刺激は、二次増殖、ならびにIFN-γ産生および特異的死滅活性をもたらした。SIINFEKLエピトープを含有するOVAの長鎖ペプチドと結合した抗CLEC9aを使用して同一の結果を得た(「S2」、SIINFEKLTEWTSSNVMEERC;図13B)。
【0214】
特に、遊離S1ペプチドは、抗CLEC9a-S1結合体中に存在する量の100倍過剰で与えたときでさえ、in vivo死滅応答を誘導することまたは相当数のテトラマー陽性細胞を誘発することができなかった(示さず)。本発明者らは、CLEC-9aおよび適切なアジュバントへの外因性抗原の標的化は、CD8+T細胞の有効なクロスプライミングを可能にすると結論付ける。
【0215】
CTL活性のCLEC9aプライミングが腫瘍治療をもたらし得るかどうかを決定するために、本発明者らはB16メラノーマ肺転移のモデルを使用した。本発明者らは2×105個のB16-OVA-GETメラノーマ細胞を静脈内接種し、6日後、S1 SIINFEKL誘導体(10μg)および抗CD40(25μg)と結合した異なる抗体を脚部に皮下注射した(図14)。腫瘍の注入後18日で、肺腫瘍の数を分析した。これらの結果によって、抗CLEC9a+抗CD40は腫瘍治療に有効であることが明らかになった(p<0.001、一元配置ANOVA)(図14)。
【0216】
本発明者らは実験を延長して、抗CLEC9a標的化をさらに使用して、B16腫瘍関連抗原(25〜27)として働くことができる内在性メラニン細胞分化タンパク質に対する免疫応答を誘導することができるかどうかを決定した。本発明者らは、gp100、TRP-1およびTRP-2(25〜27)由来のH-2KbおよびH-2Db拘束性抗原エピトープを含むビオチン化ペプチドを合成し、これらを抗CLEC-9aと共有結合させ、抗体結合体ならびにアジュバントとしてポリI:Cおよび抗CD40でマウスを免疫処置した。図16中に示すように、B16メラノーマ細胞移動の3日後に治療的に与えた1回用量のワクチンは、肺擬似転移のほぼ完全な根絶を誘導した。これはメラノーマ抗原に対する強力なIFN-γ応答の誘導によって実施した(図16)。対照的に、(対照アイソタイプ適合モノクローナル抗体と結合した)非標的型の同じ抗原は、防御またはIFN-γ応答を誘導することはできなかった(図16A、B)。B16攻撃前にワクチンを与えた予防モデルにおいて同様の結果を得た(図16C)。本発明者らは、CLEC-9a標的化による特異的CTLのプライミングは、マウス腫瘍に対する予防的または治療的ワクチン接種に使用することができると結論付ける。
【0217】
ヒトClec9aの発現は血中DCの小サブセットに限られる
これらの発見をヒトに広げるために、本発明者らはhClec9aをクローニングし、それに対するマウスモノクローナル抗体を作製した(Materials and Methodsを参照)。これらのモノクローナル抗体の1つを選択して、ヒト末梢血単球細胞間のClec9a発現のパターンを分析した。ヒトClec9aの発現は、リンパ球、単球、NK細胞および系統陰性HLADR-細胞からはなかった(図15A)。GM-CSFおよびIL-4における培養によって生成した単球由来DC中でもそれは検出しなかった(データ示さず)。しかしながら、Clec9aの発現は、系統陰性HLA-DR+細胞で定義したように、血中DCの別の亜集団において明らかであった(図15A)。
【0218】
CD123+pDCの集団およびCD16、CD1b/c、BDCA-3、およびCD34(22)の発現に基づいて区別可能な推定ミエロイドCD123-DCの異なるサブセットを含めた、血中DCの5つの異なるサブセットを報告している。DCのClec9a+亜集団はCD123に陰性であり、マウス中でのpDCと異なり、ヒトpDCはClec9aを発現しないことを示唆した(図15B)。Clec9a+血中DCはCD34、CD16およびCD1b/cにも陰性であった。しかしながら、Clec9a+DCはBDCA-3に均一に陽性であった(図15B)。したがってヒトClec9aは、BDCA-3+DCの異なる集団を選択的に示す。
【0219】
最後に、本発明者らは、ヒトCLEC-9aは、そのマウスオルソログと同様に、DCにおいてエンドサイトーシス受容体として機能することができるかどうか評価した。BDCA-3+DCはAlexa 488標識抗CLEC-9aを用いて4℃で染色した。1時間後、4℃ではなく37℃において、細胞内区画中の蛍光を発見した(示さず)。したがって、ヒトCLEC-9aはBDCA3+DCにおける結合抗体のエンドサイトーシスを仲介し、それによってヒトCLEC-9aは、ヒト中でのこれらの細胞への抗原標的化に使用することができることを示唆する。
【0220】
死/死滅細胞はCLEC9Aのリガンドを発現する。
CLEC9A-CD3ζキメラを含有するBWZ細胞は短い生成時間を有し、容易に異常増殖し、培養中に相当な割合の死滅細胞を生成する可能性がある。本発明者らは、任意の追加的な刺激無しでCLEC9Aを発現するBWZトランスフェクタントにおいて基底活性化を観察し、BWZ培養中の死滅細胞の数と関連付けた(図17a)。
【0221】
これらの結果を確認するために、本発明者らは、BWZ-CLEC9A-ζトランスフェクタントに加えたときに応答を誘導しない細胞系を使用した。LK細胞はレポーター細胞に曝したときに応答を誘導しない。しかしながら、LK細胞をUV照射して細胞死を誘導すると、それらはレポーターの強力なインデューサーに変わり(図17b)、UV処理後に改変した細胞はCLEC9Aのリガンド(複数可)を発現することを示唆した。二価抗体はB3Z細胞系中で発現される分子の架橋およびレポーターの活性化を誘発するので、相互作用の特異性を実証するためにこの系において遮断抗体を使用することはできない。しかしながら、抗CLEC9A抗体の一価Fab断片はレポーターの活性を誘発せず、かつ種特異的な形式でUV処理細胞による誘導を遮断し(図17b)、抗体はCLEC9A受容体の遮断に特異的に作用してリガンドとの相互作用を回避することを実証した。
【0222】
この結果は、他の独立したUV照射細胞型(ネズミ3T3、LK細胞、MEF、EL-4)、ラットRBL細胞、およびヒトHEK-293において確認した。細胞を(UV誘導型DNA損傷およびアポトーシスを妨げる)カフェインとプレインキュベートし、次いでUV放射に曝すと、リガンドの露出は誘導されない。
【0223】
本発明者らはLK細胞を異なる線量のUVに曝し、かつ本発明者らは、LK細胞中のリガンドの発現は死滅LK細胞の数と相関関係があることを発見し(図17c)、リガンドはこの集団中で選択的に発現されることを示した。
【0224】
これを独立形式で確認するために、本発明者らは、それぞれモノビオチン化用のBirA配列と結合した、マウスCLEC9Aの組換え可溶性細胞外ドメイン(rsCTLD)、および対照としてマウスデクチン-1を作製した。モノビオチン化rsCTLDを使用してPE-テトラマーを作製した。本発明者らは24時間前にUVで処理した細胞を染色し、かつ本発明者らは、CLEC9A rsCTLDテトラマーとTO-PRO3陽性細胞の特異的結合を検出した(図17d、ドットプロット)。対照として、デクチン-1 rsCTLDテトラマーは死滅細胞と結合しなかったが、それらはその特異的リガンドザイモサンとは結合した(図17d、ヒストグラム)。
【0225】
UV処理はDNA損傷および一連の関連ストレスマーカーを誘導するので、本発明者らは、リガンドの誘導はDNA損傷によって引き起こされたか、または大部分は細胞死に関与するプロセスによって引き起こされたかどうか試験した。DNA損傷誘導試薬だけでなく、免疫細胞死をもたらす原発性壊死を促進することが示されている、血清枯渇またはさらに凍結解凍もリガンドの露出をもたらした(図17e)。しかしながら、寛容原性で働くことが示されている細胞の即死を誘導する浸透圧ショックは、CLEC9Aリガンドを露出しなかった(図17eおよびf)。結論として、特定の型の原発性および続発性壊死の後にCLEC9Aリガンドが細胞中で露出される。さらに本発明者らは、細胞の固定(または固定および透過処理)は細胞をTO-PRO3に透過可能にする変化を瞬時に促進し、CLEC9Aのリガンドを「露出する」ことを見出しており(図20)、リガンドの合成はUVに対する損傷応答の結果として誘導されないが、特定の刺激に応答する死のプロセスの結果としてリガンドが露出されることをさらに実証している。
【0226】
CLEC9Aは死細胞により樹状細胞に送達されるアジュバントシグナルを仲介する。
UV死滅細胞はSykおよびY7依存形式でCLEC9Aの細胞質ドメインを介してシグナル伝達する(図21)。この系によって本発明者らは、抗CLEC9A抗体がこの相互作用の特異的遮断試薬として働き得るかどうかを調べることができ、かつ本発明者らは、可溶形のFab抗体と完全抗体の両方が強力な遮断試薬であったことを見出した(図22)。樹状細胞における死細胞の影響およびin vitroにおけるそれらのエフェクター機能を試験するために、本発明者らは交差提示アッセイを設計し、その中ではOVAタンパク質を導入したUV処理bm-1細胞を、遮断抗CLEC9Aの有無の下でFlt3L BMDCと相互作用させることが可能であった。bm-1細胞はB6ハプロタイプ由来であるが、OVAの免疫優勢クラスIペプチド(SIINFEKL)と結合しない突然変異H2Kbを発現する。クロスプライミングに関するDCの能力の読み出しとして、特異的OT-I細胞をアッセイに加えた。交差提示された抗原の量に関する読み出しとしてのOT-I細胞の増殖は、大きく影響は受けなかった(図18a)。しかしながら、アジュバント効果により活性化されるDCによって促進される助力に依存するOT-I細胞によるサイトカイン産生は、遮断CLEC9A抗体を使用したとき大幅に阻害された(図18aおよびb)。
【0227】
これらの結果を確認するために、本発明者らは、Clec9aプロモーター(Clec9aegfp/-を)の調節下でEGFPを発現するCLEC9A欠損マウスを作製した。本発明者らはCLEC9A欠損または非欠損Flt3L BMDCを作製し、かつ本発明者らは、死細胞の摂取が影響を受けたかどうか分析した。図18cは、CLEC9A+とCLEC9A- Flt3L BMDCの間の死細胞の摂取に関する能力の間に差がないことを示す。次いで本発明者らは、UV処理bm-1細胞中に導入したかまたはUV処理したbm-1MEF中の非分泌型OVA-GFP融合タンパク質において細胞内で発現されたいずれかのOVAタンパク質との交差提示における、Flt3L BMDC欠損CLEC9Aの影響をアッセイした(図18d)。OT-I増殖は影響を受け、抗体遮断より交差提示の遮断において大きな影響を示唆した。しかしながら、bm1MEFにより発現されたOVAを含めたOVAの高用量では、増殖は影響を受けず、IFNγ産生は大幅に阻害された(図18dおよびe)。これらの結果は、DC中のCLEC9Aの不在下におけるin vitroでのクロスプライミング中に死細胞と関係があるアジュバント効果の遮断があることを示唆する。
【0228】
CLEC9Aが免疫細胞の死を感知してin vivoでのクロスプライミングを促進する
CLEC9Aを発現するCD8α+DCは、in vivoでの死滅細胞関連抗原とのクロスプライミングを促進することを特徴とする主な細胞型であるので、本発明者らは、この機能におけるCLEC9A遮断の影響を試験した。OVAを発現するUV処理bm1 MEFを与えたマウスは、6日後内因性レパートリーからのOVAに対してCD8+T細胞の増大を示した(図19a、左図)。これらのCD8+T細胞はSIINFEKLに応答してIFN-γを産生することができ、それらがエフェクター細胞であること、および死滅細胞のアジュバント活性と関係がある抗原の免疫原性担体として死滅細胞が働いたことを示した(図19a、右図)。アイソタイプ対照ではなく抗CLEC9A遮断抗体を用いた予備処理は、特異的CD8応答とそのエフェクター活性の両方を遮断した(図19a)。
【0229】
死滅細胞関連抗原とのクロスプライミングと関連がある死滅細胞の免疫原性のプロセスにおけるCLEC9Aの正確な役割を決定するために、本発明者らはCLEC9aegfp/-マウスにおいてアッセイを利用した。その結果は、CLEC9Aの不在下でin vivoでの死滅細胞関連抗原とのクロスプライミングの非常に有意で部分的な阻害を示した(図19b、左図)。ノックアウトマウスを混合型C57BL/6-129バックグラウンドで作製し、C57BL/6(N3)で戻し交配しているので、本発明者らはその個々を図19b、左図においてプールした12匹のメス同腹子を分類し、かつ本発明者らはCLEC9A+マウスとCLEC9A-マウスの間の平均を比較し、12匹の同腹子のうち12匹が、平均30.05%の阻害で低減を示したことを示した(p<0.0001、スチューデントのt検定)(図19b、右図)。図19b、右図中に示すように、異なる同腹子間のバックグラウンド浸透度の違いは、同腹子間のクロスプライミング能力の有意な変動性を説明し、一部分のみではあるが依然として非常に有意である、CLEC9A+マウスとCLEC9A-マウスの間の真の違いを抑えることができた。さらに、ex vivoでのSIINFEKLに応じたIFNγ産生は大幅に影響を受け、CLEC9Aの不在下での死滅細胞関連抗原とのクロスプライミングによって生成するエフェクター応答の欠損を示した(図19c)。結論として、CLEC9A欠損は死滅細胞関連抗原の低下したアジュバント性をもたらし、これは死滅細胞関連抗原に対する特異的T細胞エフェクター応答の遮断をもたらす。
【0230】
方法
マウス
C57BL/6マウス、Rag-/-C57BL/6バックグラウンドのOT-IマウスおよびB6.SJLバックグラウンドのマウス(コンジェニックCD45.1+)を、特定病原体未感染条件で英国癌研究所において飼育した。Kbm-1マウスはJackson laboratoryから購入し(Bar Harbor、Maine;ストック番号001060)、C57BL/6マウス、Rag-/-C57BL/6バックグラウンドのOT-Iマウス、MyD88-TRIF二重ノックアウト、Clec9a+、およびClec9a-/-マウスと一緒に特定病原体未感染条件で英国癌研究所において飼育した。前に記載されたように(Turnerら、Nature 378、298(1995年))骨髄キメラをSyk欠損胎児肝細胞から作製した。全ての動物実験は、動物ケアに関する国内および機関ガイドラインに従って実施した。
【0231】
試薬
培養培地はグルタミン、ペニシリン、ストレプトマイシン、2-メルカプトエタノール(いずれもInvitrogenから)および10%熱不活性化ウシ胎児血清(Bioclear)を補充したRPMI1640(Invitrogen)であった。フローサイトメトリー分析実験に使用した抗体はBD Pharmingenからのものであり、CD11c(クローンHL3、ハムスターIgG1)、CD24、CD11b、B220、Ly6C、CD4(RM4-5、ラットIgG2a)、CD8に特異的な抗体を含んでいた。ELISAに使用した抗体は捕捉IFN-γ(XMG1.2、ラットIgG1)検出/IL12-23 p40であった。精製した2.4G2(抗FcgRIII/II、ラットIgG2b、非特異的抗体結合を遮断するために使用した)は、英国癌研究所の抗体産生サービスからのものであった。フローサイトメトリー用に、2mMのEDTA、1%のFCSおよび0.02%のアジ化ナトリウムを補充した氷冷PBSで細胞懸濁液を染色した。データをFACSCalibur(BD Biosciences)で得て、FlowJoソフトウェア(Treestar、San Carlos、CA)を使用して分析した。
【0232】
細胞
マウス骨髄由来のDC(BMDC)をGM-CSFを使用して生成し、抗CD1cマイクロビーズを用いた磁気選択によってバルク培養物から精製した(GM-CSF BMDC)。あるいは、10日間100ng/mlのFlt3L(R&D)の存在下で骨髄細胞を培養することによってBMDCを生成し、この時間まで全ての生存細胞はCD11cに陽性であった(Flt3L BMDC)。脾細胞はリベラーゼ/DNAse消化によって調製し、抗CD11cマイクロビーズを用いた正の選択によってDCを増大させた。OT-IT細胞(リンパ節および脾臓由来)は、ビオチン化抗体(抗CD11c、CD11b、B220、FcγR、Gr-1、およびCD4)のカクテル、次にストレプトアビジンマイクロビーズを使用した負の選択によって精製した。ヒト末梢血単球細胞(PBMC)は、Ficoll-Hypaque(GE-Healthcare)での沈降作用によって1のドナー白血球軟膜(National Blood Transfusion Service)から調製した。
【0233】
PCR/RT-PCR
全RNAは抗CD11cマイクロビーズ(Miltenyi)を用いてCD11cで増大した脾臓DCのサブセットからトリゾール(Invitrogen)を使用して抽出し、FacsAriaソート(BD)でCD4およびCD8発現に関して分類した。さらに、GMCSFおよびFlt3Lin vitro由来BMDCの分類したサブセットからRNAを抽出した。RNAをDNAse消化(DNAフリー、Ambion)によって調製し、SuperscriptII逆転写酵素(Gibco)、1μMのdNTPおよび10μMのランダムヘキサヌクレオチド(Gibco)を使用して逆転写した。cDNAは30秒間94℃、30秒間55℃、1分間72℃からなる35PCRサイクルを使用して増幅した。プライマーの配列は:mCLEC9a順方向5'AGACTGCTTCACCACTCCAA;mCLEC9a逆方向Rv:5'CTTGGCACAATGGACAAGGT;b-アクチン順方向:5'GTTTGAGACCTTCAACACCCC、b-アクチン逆方向:5'GTGGCCATCTCCTGCTCGAAGTC;hClec9a正方向:5'CCCAAGTCTCATTTGGAGGA;hClec9a-1逆方向:5'AAATCTGGACGGTGTGGAAGであった。
【0234】
抗CLEC9aモノクローナル抗体の作製
ネズミClec9aに対するモノクローナル抗体
ウイスターラットをHAエピトープと融合したCLEC9aでトランスフェクトしたRBL-2H3細胞で免疫処置し、過免疫したラット由来の脾細胞とラットメラノーマ細胞系Y3の融合は、標準的な手順に従い実施した。陽性の検出用に、本発明者らは、CLEC9aの細胞外ドメイン、NKRP1由来の膜貫通領域およびCD3ζの細胞内尾部および次にIRES-GFPを有するキメラを発現するB3Z細胞系25、記載されている戦略21を使用した。mCLEC9aに関する融合キメラのcDNA配列は図3中に示す。B3Z細胞系はβ-Gal活性と関係があるNFATに関するレポーターを含有し、このキメラ分子の任意の関与がNFATおよびレポーターの活性化をもたらし、したがってこれはb-Gal活性に関する標準的なアッセイに従い明らかにすることができる。抗体に関するスクリーニングは、親細胞系と比較したキメラを発現するB3Zの機能活性化によるものであった。陽性として選択した抗体は、CLEC9aキメラ発現細胞(EGFP+)と比較して親細胞系(EGFP-)においてFAGS分析によって確認した。この方法は、図4中に示した1F6、397、および7H11という名称の3つのモノクローナル抗体の選択を可能にした。次いでモノクローナル抗体を染色用のビオチンまたはAlexa488(Invitrogen)と結合させ、またはS1ペプチドとの結合用に使用した。
【0235】
ヒトClec9aに対するモノクローナル抗体
HAエピトープと融合したヒトClec9aを発現するRBL-2H3細胞を用いて3〜4回BALB/cマウスを免疫処置した。脾細胞とマウスメラノーマ系SP2/0の融合は、標準的な手順を使用して実施した。ハイブリドーマスクリーニング用に、本発明者らは、NFATに関するβ-galレポーターを発現するB3Z細胞系を使用した(23)。NKRP1B由来の膜貫通領域と融合したヒトClec9aの細胞外ドメイン、およびCD3ζの細胞内尾部、次にIRES配列およびGFP遺伝子のキメラをコードするレトロウイルスを、この細胞系に形質導入した(24)。ハイブリドーマ上清を、Clec9aキメラと結合しNFATレポーターの活性化およびβ-gal活性の誘導をもたらす能力に関してスクリーニングした(24)。このアッセイ中で陽性反応を示した上清を、キメラClec9aを発現するB3Z細胞(GFP+)と親B3Z細胞(GFP-)の混合物を使用したフローサイトメトリーによって、さらにスクリーニングした。この方法は、hClec9aに特異的な1つのマウスモノクローナル抗体の選択を可能にした(8F9(IgG2a))。
【0236】
フローサイトメトリー
マウスCD11c、CD24、CD11b、B220、Ly6C、CD4およびCD8αに特異的なフルオロクロムまたはビオチン標識抗体は、BD Pharmingenからのものであった。精製2.4G2(抗FcγRIII/II)は、英国癌研究所の抗体産生サービスからのものであった。マウス細胞懸濁液を10μg/mlの2.4G2モノクローナル抗体と共にインキュベートしてFcγ受容体を遮断し、次いで2mMのEDTA、1%のFCSおよび0.02%のアジ化ナトリウムを補充した氷冷PBSで染色した。エンドサイトーシス試験用に、4℃において30分間5μg/mlのビオチン化抗Clec9aモノクローナル抗体でFcγ遮断細胞を標識した。次いで細胞を二回洗浄し、氷に移しストレプトアビジンPEを加える前に、4℃または37℃において異なる回数インキュベートした。in vivo標識試験用に、Alexa-488結合抗Clec9aまたはアイソタイプ適合対照モノクローナル抗体を示した用量で静脈内注射し、組織を調製し、16時間後に分析した。ヒトCD3、CD14、CD19、CD56、HLA-DR、CD34、CD123およびCD16に特異的な抗体はBDから購入し、かつCD1b/c、およびBDCA-3はAbCam(ケンブリッジ、英国)から購入した。ヒト単球細胞は100μg/mlのヒトIgG(Sigma-Aldrich)で遮断し、前述のように染色した。データをFACSCalibur(BD Biosciences)で得て、FlowJoソフトウェア(Treestar、San Carlos、CA)を使用して分析した。
【0237】
BM-DCの培養および刺激
GM-CSF BM-DCを記載したように生成し26、使用前に抗CD11cマイクロビーズを用いてバルク培養物からDCを精製した(Miltenyi Biotec)。BM-DCの純度はFACSによって調べ、常に98%を超えていた(データ示さず)。F1t3LBMDCは、10日間75ng/mlまたは50ng/mlのF1t3L(R&D)の存在下で培養した骨髄から生成した。サイトカイン産生および表面マーカー発現の分析用に、ウエル当たり5〜10×104個のFlt3L BM-DCを、アイソタイプ対照または抗CLEC9aモノクローナル抗体で事前にコーティングした96ウエル平底プレート中のFlt3Lを含有する200mlの培養培地中で18〜24時間培養した。上清中のサイトカインレベルを、いずれもBDからの捕捉用抗IL-12 p40-p70(C15.6)および検出用ビオチン抗IL12p40-p70(C17.8)を使用してサンドウィッチELISAによって測定した。F1t3LBMDCにおけるエンドサイトーシス用に、FcγRは10μg/mlの2.4G2モノクローナル抗体で遮断し、次いで細胞を5μg/mlのビオチン化モノクローナル抗体と共に30分間4℃または37℃において培養し、洗浄し、4℃において二次試薬(ストレプトアビジンPE)で同時に洗浄および添加する前に、アッセイ温度で1.5時間または0.5時間放置した。共焦点顕微鏡による分析用に、Alexa 488結合モノクローナル抗体を4℃または37℃において30分間5mg/mlでFcγR遮断F1t3L-BMDCに加え、次いで洗浄し、ポリ-L-リシンコーティングカバースリップに接着させ室温において20分間2%PFA中で固定する前に、アッセイ温度でさらに1.5時間放置した。Fluoromount(Southern Biotech、Birmingham、AL)でスライドにサンプルを載せた。共焦点系の微分干渉コントラストおよび蛍光画像を、63°-Plan-ApochromatNA1.4油浸対物レンズを有するレーザー走査共焦点顕微鏡(Axioplan2、Zeiss、ドイツ)を用いて同時に得た。画像解析はLSM510ソフトウェア(Zeiss、ドイツ)で実施した。
【0238】
ペプチドの破壊およびウエスタンブロッティング
ペプチド破壊のために、溶解バッファー中に希釈する前に、ビオチン結合ペプチドを40%のDMSO中に溶かした。溶解バッファー中に希釈した組換えヒトSyk(Upstate)は、CLEC9aおよびデクチン-1細胞内尾部に相当する示したビオチン化ペプチド(英国癌研究所ペプチド合成研究室)およびストレプトアビジン-セファロース(Sigma Biosciences AB、Uppsala、スウェーデン)と共にインキュベートした。親和性による精製後、セファロースビーズを溶解バッファー中で1回洗浄し、10%のβ-メルカプトエタノールを含有するSDSゲル導入バッファー中で煮沸した。タンパク質はドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)によって分離し、Immobilon PVDF膜(Millipore Corporation、Bedford、MA)に移し、ウサギ抗Syk(ネズミSyk20のアミノ酸318〜330に相当する合成ペプチドに対して産生した2131血清と、ヒトSykのC末端に相当する合成ペプチドに対して産生したCell Signaling Technology、Inc.からの抗Syk、カタログ番号2712の組合せ)でプローブ処理し、次に化学発光を検出した。
【0239】
LK細胞の活性化アッセイでは、示した時間抗C9またはアイソタイプ対照でコーティングした6ウエルプレートにLK細胞を平板培養した。細胞抽出物を溶解バッファー(50mMのHEPBS[pH7.4]、150mMの塩化ナトリウム、100mMのフッ化ナトリウム、10mMのピロリン酸四ナトリウム、1mMのオルトバナジン酸ナトリウム[pH10.0]、1mMのBDTA[pH8.0]、1.5mMの塩化マグネシウム、10%のグリセロール、1%のTritonX-100、1mMのPMSF、および「完全」プロテアーゼ阻害剤カクテル錠剤[Roche])中で調製し、不溶性物質を廃棄し、一定量の溶解物を前に記載したSDS-PAGEによって調べた。ウエスタンブロッティング用の前述のウサギ抗Sykおよび抗P-Syk、抗Erk、抗P-ErkはCell Signalingからのものであった。
【0240】
B3ZおよびBWZ細胞におけるNFATレポーターアッセイ
LacZ活性と関係があるNFATのレポータープラスミドを含有するB3Z細胞は以前に記載されている25。CLEC9a WTまたは突然変異型Y7F(Stratagene)を、wtまたはSyk形質導入B3Z細胞に形質導入した。細胞はアイソタイプ対照または抗CLEC9aモノクローナル抗体でコーティングした96ウエルプレート中に平板培養し、一晩培養した後、PBS中で洗浄し、記載したように21CPRG含有バッファー中に溶かした。4時間後、参照としてOD655を使用してA595を測定した。
【0241】
CLEC9Aリガンドを検出するために、NKRP1B由来の膜貫通領域と融合したマウスまたはヒトCLEC9Aの細胞外ドメイン、およびCD3ζの細胞内尾部、次にIRES配列およびGFP遺伝子のキメラをコードするレトロウイルスを、BWZ細胞系に形質導入した(24)。CLEC9Aキメラと結合したリガンドは、NFATレポーターの活性化およびβ-gal活性の誘導をもたらす可能性がある。マウスおよびヒトCLEC9A-CD3ζキメラ受容体を発現するBWZ細胞の基底活性化をアッセイするために、4、2、1、または0.5×106個の細胞/mlを6ウエルプレート中の3mlにおいて培養して2日間(異常)増殖させた。死滅細胞の頻度はTO-PR03色素(Invitrogen)を使用して決定し、生存細胞は96ウエル平底プレートにおいて2×105個/ウエルで新たな培地中に平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0242】
異なる細胞型におけるリガンドの露出を決定するために、LK(B細胞系)、標準的なプロトコルを使用してbm-1マウス{Ref}から誘導したSV-40不死化マウス胎児線維芽細胞(MEF)、RBLラット白血病細胞、3T3線維芽細胞、およびHEK-293ヒト胎児腎臓細胞をUV照射し(240mJ/cm2)、対照Fabまたは抗マウスCLEC9A(1F6)および抗ヒトCLEC9A(8F9)Fab(Sanchoら、J Clin Invest.2008、118(6):2098〜110頁)の有無の下において1:1の比で96ウエル平底プレート中の2×105個のBWZトランスフェクタントに加える前に、24時間放置して細胞死を誘導した。一晩培養した後、LacZの活性を前述のように測定した。いくつかのアッセイ中で、CLEC9A wtでトランスフェクタントしたBWZ細胞を使用して、リガンドが細胞質尾部を介してシグナル伝達するかどうか決定し、Fabに匹敵する遮断試薬として完全抗体を評価した。
【0243】
以下の線量のUV(mJ/cm2):0、0.5、1.5、5、15、50、240でLK細胞を照射することによってUV線量反応を実施した。死滅細胞の頻度はto-pro3色素を使用して24時間後に決定し、細胞は新たな培地中にBWZトランスフェクタント(2×105個/ウエル)と共に1:1で平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0244】
LK細胞およびbm-1不死化MEFに関する異なる処理をリガンドの露出において評価した。細胞はミトキサントロン(1μM)と共に、または血清の不在下で(血清枯渇)24時間培養した。浸透圧ショックは以前に記載されたように実施した(Liuら、2002.J.Exp.Med.196:1091〜1097頁)。3サイクルの凍結解凍を細胞のペレットで実施し、液体N2中に凍結し37℃で解凍した。これらの処理後、死滅細胞の頻度はto-pro3色素を使用して決定し、細胞は以下に示すようにrsCTLDに関して染色した。細胞は新たな培地中にBWZトランスフェクタント(2×105個/ウエル)と共に1:1で平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0245】
SIINFEKLC-biot(S1)とモノクローナル抗体のカップリング
免疫優勢OVAペプチドSIINFEKLと二価抗体を結合させるために、遊離スルフヒドリル基を生成するための付加システイン(C)および標識を追跡するためのビオチンを含有する、S1と呼ぶSIINFEKLペプチドの誘導体が、英国癌研究所において合成され高性能液体クロマトグラフィーによって精製された。モノクローナル抗体は室温において30分間スルホ-SMCCで処理し、第三級アミンにおいてスルホ反応基を生成し、次に分子サイズ排除クロマトグラフィーカラム(Pierce)中で活性化抗体を精製した。次いで、S1ペプチドを新たに調製し、放置して37℃で1時間等モル量で活性化抗体と反応させ、クロマトグラフィーカラム中で精製した。モノクローナル抗体のビオチン化の程度によって、本発明者らは、Fluoreporterキット(Invitrogen)を使用し製造者の説明書に従い、生成した全ての抗体結合体中でモノクローナル抗体分子当たり1〜1.2の結合ペプチドを定量化することができた。
【0246】
S2ペプチドおよびメラニン細胞分化抗原ペプチドとモノクローナル抗体のカップリング
S2ペプチド(SIINFEKLTEWTSSNVMEERC-ビオチン)は、英国癌研究所において合成されHPLCによって精製された。PBS中のモノクローナル抗体は室温において30分間スルホ-SMCCで処理して、第三級アミンにおいてスルホ反応基を生成した。活性化抗体は分子サイズ排除クロマトグラフィーカラム(Pierce)によって精製し、S2ペプチドを加え(2:1のモル比)、反応は37℃で1時間進行させた。結合体はImmunobindセファロースカラムを使用して精製した。モノクローナル抗体のビオチン化の程度は、製造者の説明書に従いFluoreporterキット(Invitrogen)を使用して評価した。
【0247】
同じ戦略を使用して、メラニン細胞分化抗原、gp100(EGSRNQDWLおよびKVPRNQDWL;H-2Db-拘束性(25))、TRP-1(TWHRYHLLおよびTAYRYHLL;H-2Kb-拘束性(26))およびTRP-2(SVYDFFVWL;H-2Kb拘束性(27))からペプチドを合成し、C末端にシステイン-eahx-ビオチンを付加することによってそれぞれを修飾した。
【0248】
抗CLEC9aモノクローナル抗体を用いたin vivo標的化。
抗CLEC9a、アイソタイプ対照、または抗DEC-205をAlexa488(Invitrogen)またはS1ペプチドと結合させた。Alexa488モノクローナル抗体を使用する標的化の実験用に、モノクローナル抗体-Alexa488を静脈内注射し(20μg)、脾細胞を抽出し、16時間後に分析した。
【0249】
in vivoでの特異的サブセットへの抗原の標的化の実験では、S1-抗体を静脈内注射し(5μg)、脾細胞を抽出し、抗CD11cマイクロビーズ(Miltenyi Biotec)を用いてCD11c陽性および陰性分画に精製した。OT-I細胞をOT-IRag-/-マウスのリンパ節および脾細胞から得て、ビオチン化抗体(抗CD11c、CD11b、B220、FcgR、Gr-1、およびCD4)のカクテル、次にストレプトアビジンマイクロビーズ(Miltenyi Biotec)を使用した負の選択によって精製した。図の凡例に示すように、異なる量のin vivo標的化DCを、U底プレートにおいて2μMのCFSE(Invitrogen)で標識した105個のOT-I細胞と共に培養した。3日後、Vβ5.1およびCD8陽性細胞ならびにTO-PRO3陰性細胞におけるCFSE希釈によって増殖を決定した。細胞は実際のビーズ数(...)で得て細胞の絶対数を定量化した。上清中のIFN-γはサンドウィッチELISAによって測定した。
【0250】
特異的CTL応答を標的化するためのin vivo標的化の実験では、モノクローナル抗体-S1を25μgの抗CD40(3/23、BD Pharmingen)有りまたは無しで後脚に皮下注射し(2μg)、5日後、in vivo死滅アッセイを記載したように実施した27。簡単に言うと、B6.SJLバックグラウンド(コンジェニックCD45.1+)由来の標的脾細胞に20nM、200nMまたは0nMのSIINFEKLペプチドを導入し、37℃において20分間、0.03μM、0.3μMまたは3μMのCFSEでそれぞれを標識した。標識した脾細胞(107)は静脈内注射した。翌日脾細胞を抽出し、CD45.1陽性集団をCFSEに関して分析した。さらに、5×105個の脾細胞を24時間1μMのSIINFEKLの有無の下で培養し、上清はELISAによりIFNγに関して定量化した。血液および脾細胞もSIINFEKL-H2Kbテトラマー(Beckman Coulter)、抗CD8および抗Thy1.2で標識し、CD8+T細胞集団中のテトラマー+細胞に関して分析した。
【0251】
B16メラノーマ腫瘍治療モデル。
B16メラノーマ細胞にOVA-GFP融合タンパク質を形質導入し、GFP発現に関して分類した。腫瘍細胞(2×105個)をコンジェニックB6マウスの尾部に静脈内注射し、6日後、抗体-S1+抗CD40からなる療法治療剤を脚部に皮下注射した。腫瘍注射後第18日で、肺腫瘍を計数した。腫瘍治療実験は、抗体治療3日前にマウスにB16-OVAを与えたこと以外は類似の形式で行った。
【0252】
腫瘍治療および予防実験も非形質導入親B16細胞を用いて実施した。これらをgpl00、TRP-1およびTRP-2由来の5ペプチドの混合物と共有結合した抗CLEC-9aまたは対照抗体(1μg/脚部)、ならびに抗CD40(12.5μg/脚部)およびポリI:C(5μg/脚部)を用いた免疫処置の3日前(治療)または1日後(予防)のいずれかに静脈内注射した(5×105個/マウス)。全身腫瘍組織量は肺病巣を計数することによって評価した。これらが数えるのに多すぎるほど(マウス当たり250を超えて)存在したとき、それらは250として示す。CTL応答は前に記載したようにモニターした。
【0253】
CLEC9a-/-マウスの生成。
Red/ET組換え(Gene Bridges、Heidelberg、ドイツ)を使用して遺伝子の領域を直接捕捉しBACクローンRP-23 248-K14(InvitrogenからのC57BL/6BACクローン)から改変してマウスを生成した。相同的組換えES細胞クローンの単離用に、陽性(ネオマイシン耐性遺伝子)と陰性(単純ヘルペスウイルスチミジンキナーゼ遺伝子)選択の両方の戦略を使用する、従来の遺伝子標的置換ベクター:pFloxRI+tkを、ベクターと対形成した20ヌクレオチドおよび捕捉対象のClec9aの領域と対形成した70ヌクレオチドを有する示したプライマーを使用してPCRによって増幅した。使用したプライマーは順方向3アーム24330pFlox5'ATAATATCATATTTCTATAATATCATTGTAATGACAAAACCACTGAACTA GTGCCTGTAAAGGCAGGAGGGGTACCGAGCTCGAATTCTACCG3';逆方向pFlox5アーム5'TGCTATATTACAGATTTTCAAGTGGGGTAGCCTGGAGTAACAAGATGGCAGGGCATAATCACTAGTGCGGCCGCCACCGCGGTGGAGCTCCAGCTTT3'であった。
【0254】
修飾するための領域をAmp耐性ベクター中に含めた後、ファルネシル化EGFP、およびPGK-gb2プロモーター次にKan/Neoを含むカセットは、Kanに関する選択で組換え相同的組換えステップの反復を可能にした。EGFP-FKan-Neo耐性カセットの増幅に使用したプライマーは、逆方向NeoKan3アーム5'TGCTTTTGTACTTACACTTGATGCCCAAGAAAATGGACGTTGCTAACAAGCCCATACAGACCACACCTCGAGATAACTTCGTATAATGTAT3'および順方向5アームEGFP-F5'TTTGTGCCAGGCTCCTATGTAGACTGCTTCACCACTCCAAGCGCCTTCAGCATGCATGTCGACATGGTGAGCAAGGGCGAGGAG3'であった。
【0255】
標的化ベクターを調製して、CLEC9A由来の最初の2アミノ酸のすぐ下流に強力なポリAを有するEGFP-Fを発現させ、エクソン1および2を破壊し、強力なポリAで転写を終了させた。標的化ベクターはNotIを使用したトランスフェクション前に線状化した。S6B6ハイブリッド129S6/C57BL/6F1由来胚幹(ES)細胞のトランスフェクションはエレクトロポレーションによって実施し、組換え体クローンはG418およびガンシクロビル中での培養後に単離した。淘汰を生存したES細胞は、2つの独立したプライマー対を使用したPCRによってスクリーニングし、そのプライマーの1つは短いアームの外側に存在した。使用したプライマー対は、Scr順方向1 5'GATCTGTGTGTTGGTTTTTGTGTGC3';Scr逆方向1、5'TAGCATGGCACTTCTCCATTACCTT3'アンプリコン順方向1逆方向1:2138bp.Scr順方向2、5'GCGAATTCGGTACCAATAAA AGAGC3';Scr逆方向2、5'CAGAAGCTTCCTGGTTTTGGTTTTT3'アンプリコン順方向2逆方向2:2352であった。
【0256】
正確に標的化したので、核典型的な正倍数体ESクローンを3.5日後のcoitumC57BL/6肺盤胞にマイクロインジェクションし、毛色がキメラ状態である生成した子孫はC57BL/6のメスと交配させて生殖細胞系の伝達を確認した。生殖細胞系が伝達したキメラを後にC57BL/6のメスと交配させて、純粋なC57BL/6バックグラウンドで遺伝子標的となる対立遺伝子を固定した。ヘテロ接合型動物を異種交配させて、ホモ接合型欠損動物および適合同腹子対照を生成した。ノックアウトマウスにおけるClec9aと連結したNK1.1C57BL/6遺伝子の発現は、標的化C57BL/6BACクローンの相同的組換えがS6B6 ES細胞におけるF1のC57BL/6コピー中に組み込まれたことを示す。
【0257】
この実験で使用したClec9a-/-マウスは、C57BL/6に戻し交配した第三世代の混合型129/Sv×C57BL/6の遺伝的背景であった。
【0258】
組換え可溶性CTLDの生成。
マウスCLEC9Aおよびデクチン-1に関するCTLDを、付加BirAモノビオチン化配列を有するSigmaからのp3xFlag-CMV-9発現ベクターにおいて、インフレームで独立にクローニングした。CTLD増幅に使用したプライマーは、mCLEC9A順方向5'GGATCC
mCLEC9A逆方向5'GGATCC
mデクチン順方向5'GGATCC3'mデクチン逆方向5'GGATCC3'であった。
CHO細胞をトランスフェクトしG418(1mg/ml)で選択した。安定したトランスフェクタントを限界希釈法によって二回クローニングし、上清にデクチン-1またはCLEC9A由来のrsCTLDを分泌するクローンを選択し、捕捉抗体として抗flag M2(Sigma)および検出用にビオチン-2A11抗デクチン-1またはビオチン-1F6抗CLEC9Aを使用したサンドウィッチELISAによって検出した。CHOクローンから濃縮した上清をCELLineバイオリアクター(Integra Biosciences、Chur、スイス)において生成し、抗flag M2アガロース(Sigma)を使用して精製した。次いでモノビオチン化を標準的な手順を使用して実施し{Ref}、PE-ストレプトアビジン(Sigma)を使用してテトラマーを作製した。次いでrsCTLDのPE-テトラマーを、通常のFACSバッファー中での4℃における30分間の死滅細胞またはザイモサンの染色に使用した。サンプルはto-pro3で対比染色し、フローサイトメトリーによって得た。
【0259】
in vitroでの交差提示。
in vitroでの交差提示アッセイにおいて、本発明者らは、3つの細胞型:OVA導入またはOVA発現死滅細胞、CLEC9Aおよび読み出しを阻害する抗体の有無の下の異なる起源に由来するFlt3L BMDC、OT-I OVA特異的トランスジェニックT細胞を同時培養した。本発明者らは、CLEC9a-/-またはCLEC9a+同腹子由来のFlt3L BMDCを試験した。さらに、C57BL/6由来のFlt3L BMDCを、対照Fab、または抗CLEC9A(1F6)Fab(10μg/ml)、およびCLEC9Aまたはアイソタイプ対照に対する完全抗体(20μg/ml)の有無の下で培養した。示した場合、CD11blo同およびCD24hi同を有するCD8α様Flt3L BMDCを記載したように(19)分類した。交差提示に関してOVA抗原と関係がある死滅細胞の供給源として、本発明者らはbm-1脾細胞、SIINFEKL、H2Kbの免疫優勢クラスI OVAペプチドの結合を妨げるH2Kb分子において突然変異体を発現するC57BL/6コンジェニックマウスを使用した。そのように、OVAを導入した細胞はOVAペプチドを直接提示することができないはずであり、プロセシングおよび交差提示して応答を生成しなければならない。吸着によりOVAを導入するために、本発明者らは、PBS中で五回洗浄する前に、20分間PBS中で低レベルのエンドトキシンおよびbm-1脾細胞と共に、示した用量の可溶性OVA(Calbiochem)をインキュベートした。あるいは、本発明者らはbm-1マウス胎児線維芽細胞(MEF)を作製し、かつ本発明者らは、SV-40 Tラージ抗原を用いてそれらを不死化した。次いで、本発明者らは、切断型非分泌OVA-GFP融合タンパク質を次いで形質導入し、OVA-GFPの相同的発現を分類した。OVA導入脾細胞とOVA発現MEFの両方を次いでUV照射し(240mJ/cm2)、完全培地中で一晩培養した。翌日、脾細胞は5:1の比でFlt3L BMDCと同時培養し(105個細胞/ウエル、96U底)、OVA-MEFは1:1の比でFlt3L BMDCと培養した。MACSビーズ(Miltenyi)を使用して負の選択をし(80%を超える純度)CFSE(2μM)で標識したOT-I細胞を、アッセイに加えた(105個細胞/ウエル)。3日後、上清を採取してサンドウィッチELISAによってIL-2およびIFN-γを検出し、細胞はPMA(10ng/ml)およびイオノマイシン(500ng/ml)で4時間刺激し、培養の最後の3時間中にブレフェルジンA(10μg/ml、Sigma)を加えた。次いで細胞を細胞内染色によってVβ5、CD8およびIFN-γに関して染色し、フローサイトメトリーによって得て、絶対数、一定数のキャリブレーションビーズ(BD)を導入したサンプル、および細胞内染色によるIFN-γ産生を決定した。
【0260】
死滅細胞の摂取
WTまたはCLEC9A-/-CD8α様Flt3L BMDCは、24時間前にUVC(240mJ/cm2)で処理しPKH26(Sigma)で標識した異なる割合の脾細胞と共に2時間インキュベートした。Flt3L BMDCはCD24に関して標識したので、PKH26およびCD24に関する二重陽性は死細胞の結合(4℃)または結合+摂取(37℃)が原因である可能性があり、頻度はそれぞれの型のDCに関するフローサイトメトリーにより定量化した。
【0261】
in vivoでの交差提示
前に示したように作製したOVA発現不死化bm-1 MEFをUVC処理し(240mJ/cm2)、静脈内注射(0.75×106個細胞/マウス)する前に前に一晩培養した。マウス(C57BL/6)は、静脈内注射の30分前に、PBSまたは400μgのアイソタイプ対照(AFRO-MAC49、ラットIgG1)または1F6抗CLEC9Aの腹腔内注射で前治療した。あるいは、Clec9a-/-または同腹子Clec9a+を使用した。6日後、内因性レパートリーから生じるCD8+T細胞エフェクター応答の誘導を、H2Kb-OVAペプチドテトラマー陽性細胞の数およびCD8サブセットにおけるex vivoのSIINFEKLに応じたIFNγ産生によって追跡した。独立した実験中の数匹の同腹子からデータをプールした図19cおよび19dにおいて、本発明者らはプール前にデータの正規化を実施した。それぞれの同腹子(メス)は独立した実験で考え、それぞれの同腹子における生データは、その同腹子中のClec9a+マウスの平均に正規化し、独立した実験における全同腹子中のClec9a+マウスの集団全体に関して得た算術平均を掛けた。同腹子の倍数変化の分析のために、Clec9a+マウスまたはClec9a-/-マウスに関する算術平均をそれぞれの同腹子において計算した。分析した12匹のうち12匹全ての同腹子が、Clec9a+と比較してClec9a-/-マウスのCD8 T細胞サブセット中のテトラマー陽性細胞の算術平均の数倍の低下を示した。
【0262】
統計
統計分析は、群間の差に関してスチューデントの両側t検定、またはデータの正規性を推測できなかったときはマンホイットニーのU検定を用いて実施した。p<0.05を統計上有意であると考えた。他に言及しない限り定量データは平均±SEMとして表す。
【0263】
上述の具体例としての実施形態と共に本発明を説明してきたが、この開示を与えられた場合多くの同等な修正形態および変形形態が、当業者には明らかなはずである。したがって、記載した本発明の具体例としての実施形態は、例示であって限定ではないとみなされるべきである。本発明の精神および範囲を逸脱することなく、記載の実施例の様々な変更がなされ得る。本明細書中の全ての引用文書は、参照により明確に援用される。
【0264】
(参考文献)
【技術分野】
【0001】
本発明は、免疫系の調節に関し、特に、標的抗原に対する免疫応答をプライミングおよび阻害するための、CLEC9a分子に対して親和性を有する分子の使用に関する。
【背景技術】
【0002】
免疫系は、感染性病原体の存在を検出し、自己組織を破壊することなく病原体に対する応答を誘発することができる。病原体の膨大な分子多様性ならびにその複製および突然変異率の高さを考えれば、この現象は驚くべきものである。多細胞生物は、進化の過程にわたっていくつかの性質の異なる免疫認識機構、すなわち「先天性」免疫系および「適応性」免疫系を発達させるよう促されてきた。
【0003】
進化的に古い先天性免疫系は、感染の存在および性質を検出し、宿主防御の第一線を担い、適応性免疫系のエフェクタークラスの開始および決定を制御する。
【0004】
樹状細胞(DC、dendritic cell)は、先天性免疫と抗原特異的適応応答を結び付ける上で極めて重要な役割を果たす。免疫応答を開始するために、DCは、(とりわけ)病原体が示す病原体関連分子パターン(PAMP、pathogen-associated molecular pattern)によってプライミングされる。その後DCは、適応性免疫応答の進行をうまく調整し、これは、抗原特異的T細胞およびB細胞によってさらにいっそう特殊化され駆動される。
【0005】
病原体に対するDCの抗原認識および取り込み機能は、PAMPを識別するパターン認識受容体(PRR、pattern recognition receptor)によって媒介される。DCにより発現されるこれらのPRRはToll様受容体(TLR、Toll like receptor)を含む1。DCはさらに、別のクラスの受容体としてC型レクチンを発現し、その中にはPRRとして機能し2-4、かつ/または細胞間コミュニケーションを媒介する5-8ことができるものがある。C型レクチンの中に、構造的かつ進化的に密接な関係があり、NK遺伝子複合体(NKC、NK gene complex)中にクラスターを形成している、単一の細胞外C型レクチンドメイン(CTLD、C-type lectin domain)を有するII型タンパク質の群がある。これらの受容体は、カルシウム結合部位および典型的な糖認識ドメインを欠くが、それにもかかわらずデクチン1で示されるように糖質に結合することができる。これらの受容体のいくつか(CD94/NKG2、NKG2D)は、MHC-Iまたは関連分子と相互作用し、阻害および活性化シグナルのバランスの結果としてNKおよびT細胞傷害性を阻害または活性化する。しかしながら、NKレクチン受容体の大部分について、NKまたはDC機能における結合特異性および関連性は不明である。したがって、DC上で発現されるC型レクチンは、微生物を認識するように作用する可能があるだけでなく、特異的な細胞の相補的構造を認識することによりDCの他の細胞とのコミュニケーションを調節する可能性もある。
【0006】
個体発生および/またはDCの置かれる微環境は、DCによる表面受容体の特徴的な組合せの発現をもたらし得る。例えば、表現型基準のみによって、マウスリンパ節DCを6つの主な亜集団に分類することができる10。これらのうち、リンパ組織中の通常の非形質細胞様DCは、伝統的にCD8αおよびCD8α+亜集団に細分される。異なるDCサブセットがある特定の病原体の特異的認識に関与および/または異なる免疫応答、例えば、Th1もしくはTh2(免疫)または制御性T細胞(寛容)などを調節している可能性があることが議論されてきた11。しかしながら、DCの表現型および機能的挙動は外部の活性化刺激によっても大きく左右され、顕著な可塑性を示す。DCサブセット間の差異を理解するための最初のアプローチとして、DC亜集団を単離しそのin vitroでの特性を評価した:マウスにおいて、脾臓DCのCD8α+およびCD8αサブセットは、in vitroでIL-12を産生する能力が異なっている12,13。しかしながら、in vitroでの示差的IL-12産生はパターン認識によっても測定され、異なるDCサブセットの機能的柔軟性を示した14。第2のアプローチとして、DCサブセットが単離され、抗原パルスされ、次いでin vivoに再注入された。CD8α+およびCD8α-サブセットは、in vivoでTh1およびTh2応答を別個にプライミングした15,16。免疫療法は、特定のDCサブセットにおいて選択的に発現される分子を測定できれば、実現可能である。こうした分子は、次にこのDCサブセットの機能を改変するための標的とすることができる。
【先行技術文献】
【特許文献】
【0007】
【特許文献1】PCT/US92/0996
【特許文献2】WO94/13804
【特許文献3】EP-A-0120694
【特許文献4】EP-A-0125023
【非特許文献】
【0008】
【非特許文献1】Mallat Z.ら、Circulation 108:1232〜7、2003年
【非特許文献2】Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年
【非特許文献3】Altschulら、Methods in Enzymology、266:460〜480(1996年)
【非特許文献4】Viorittoら、(Clin Immunol 122(2)、125〜134(2007年))
【非特許文献5】Tesniereら(Corr Op Immunol 21、1〜8(2008年))
【非特許文献6】Kimら(Immunity 27、321〜333(2007年)
【非特許文献7】Ward,E.S.ら、Nature 341、544〜546(1989年)
【非特許文献8】Birdら、Science、242、423〜426、1988年
【非特許文献9】Hustonら、PNAS USA、85、5879〜5883、1988年
【非特許文献10】P.Holligerら、Proc.Natl.Acad.Sci.USA 90 6444〜6448、1993年
【非特許文献11】Remington's Pharmaceutical Sciences、第20版、2000年、pub. Lippincott、WilliamsおよびWilkins
【非特許文献12】Turnerら、Nature 378、298(1995年)
【非特許文献13】Sanchoら、J Clin Invest.2008、118(6):2098〜110頁
【非特許文献14】Liuら、2002.J.Exp.Med.196:1091〜1097頁
【発明の概要】
【発明が解決しようとする課題】
【0009】
本発明者らは、CLEC9aが、マウスにおいてCD8を発現し、そのためCD8+樹状細胞と呼ばれる樹状細胞のサブセット上で選択的に発現されることを発見した。これは、細胞外由来の抗原を処理することができ、MHCクラスI分子を介してそれらをT細胞に提示することができると考えられているため、重要な細胞型である。これは、MHCクラスII分子を介して細胞外由来抗原を提示する大部分の抗原提示細胞とは対照的である。したがって、この抗原提示機構を「交差提示」と呼ぶこともある。これらの細胞はそのため、細胞内病原体(例えば、ウイルスなど)および癌に対する免疫応答の不可欠な部分である細胞傷害性T細胞(CTL)応答の発生および刺激において重要な役割を果たす。
【0010】
この知見は多くの応用を開拓する。例えば、抗原を交差提示可能な樹状細胞に特異的に標的化することを可能にする。
【課題を解決するための手段】
【0011】
したがって、第1の態様において、本発明は、抗原を抗原提示細胞に標的化する方法であって、抗原提示細胞を、抗原を含む組成物と接触させる段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原提示細胞がCLEC9aを発現する方法を提供する。
【0012】
本方法は、in vitroまたはin vivoで適用することができる。抗原提示細胞は、典型的には樹状細胞とし、好ましくはMHCクラスI分子を介して細胞外抗原を交差提示することができる。「細胞外の」とは、抗原が、細胞外環境から細胞によって、典型的にはエンドサイトーシスまたは食作用によって取り込まれたものであることを意味する。
【0013】
本発明はさらに、対象において抗原に対する免疫応答を刺激する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合している方法を提供する。
【0014】
本方法は、単回投与、または適当に決定された時間間隔を空けた一連の2回以上の投与を含んでもよい。例えば、本方法は、プライミング段階(すなわち、初回投与)に続いて1回または複数回のブースター段階(1回または複数回の追加投与)を含むことができる。例えば、初回投与と第2回投与は、1日以上、1週間以上または1か月以上、好ましくは2週間から1か月の間隔を空けてもよい。追加の投与は、1週間以上または1か月以上の後に提供することができる。
【0015】
CLEC9a標的化によって刺激された免疫応答は、CTLまたはヘルパーT細胞であり得るT細胞の増殖を伴う。CLEC9aを発現する抗原提示細胞(および特に樹状細胞)は、CD8+T細胞およびCD4+T細胞の両方の増殖を誘導することができ、任意の与えられた免疫応答において両方の型のT細胞の増殖を刺激し得る。
【0016】
特定の条件下において、上記細胞は制御性T細胞(Treg)増殖を刺激することができると考えられている。Treg細胞は、Foxp3(Forkhead box p3)転写因子の発現により特徴付けられる。大部分のTreg細胞はCD4+およびCD25+細胞であり、ヘルパーT細胞のサブセットとみなすことができるが、ごく少数はCD8+である可能性がある。したがって、本発明の方法により刺激される免疫応答は、抗原に応答してTreg細胞の増殖を誘導する段階を含み得る。したがって、本方法は、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物を対象に投与する段階を含むことができる。抗原は、Treg細胞の増殖を促進するアジュバントと共に投与されてもよい。
【0017】
本方法が特異的抗原に応答してTreg細胞の増殖および分化を刺激する段階を伴う限り、免疫応答を刺激する方法であるとみなすことができる。しかしながら、Treg細胞が抗原に対して、例えばその活性の阻害または抑制などの別の方法で、免疫系の別の細胞の応答を調整することができ得ると仮定すると、全体としての免疫系に対する影響は、その抗原に対する応答を調整(例えば、抑制または阻害)するものとなる可能性がある。したがって、本発明の本態様の方法は、抗原に対する免疫応答を調整(例えば、阻害または抑制)する方法と同様に呼ぶことができる。
【0018】
したがって実際には、本発明のこれらの方法は、特定の抗原に対する望ましくない免疫応答を阻害または抑制するために、その抗原に対する免疫が既に存在するまたは免疫応答が進行している対象においても、治療的または予防的に使用することができる。このことは、(例えば)自己免疫疾患の治療において特に有用である。
【0019】
特定の条件下において、抗原をCLEC9aを発現する抗原提示細胞に標的化することによって、特定の抗原に対して対象を寛容化することも可能であり得る。したがって本発明は、抗原に対して対象における寛容を誘導する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原がアジュバントの非存在下で投与される方法を提供する。
【0020】
この文脈において寛容は、上記のような免疫細胞において、そうでなければその抗原に対して応答する、または抗原に対する応答性の持続的な低下を誘導することができる免疫細胞の減少を典型的に含む。
【0021】
典型的には、対象は脊椎動物、好ましくは哺乳動物である。対象は、ヒト、別の霊長類、またはマウス、ラット、モルモット、ウサギ目(例えば、ウサギ)、ネコ、イヌ、ブタ、ウシ、ウマ、ヒツジもしくはヤギなどの飼育動物、実験動物もしくは家畜であってもよい。
【0022】
本発明はさらに、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物を提供する。組成物は、抗原およびその結合している結合剤を薬学的に許容される担体と組み合わせて含む医薬組成物、例えば、ワクチンであってもよい。組成物は、静脈、筋肉内、腹腔内、経鼻、皮下、皮内などを含むがこれらに限定されない任意の適した投与経路用に製剤化することができる。
【0023】
本発明は、医療方法において使用するための、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物をさらに提供する。
【0024】
抗原に対する免疫応答を刺激するのに使用するための、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物もまた提供される。
【0025】
抗原に対する免疫応答を刺激するための医薬の調製における、CLEC9aに対して親和性を有する結合剤と結合している抗原を含む組成物の使用もまた提供される。
【0026】
CD8α+樹状細胞は、少なくともTh1、Th2、およびTh17型免疫応答に関係している可能性がある。したがって、本発明の方法は、任意の抗原に対する様々な型の免疫応答の刺激に適用することができる。しかしながら、これらの細胞は、CTL応答の発生において特に重要であると考えられているため、刺激される免疫応答は好ましくはCTL応答である。本方法は、典型的にはCD8を発現するT細胞であり、MHCクラスI分子に関連して同種抗原をディスプレイしている細胞に対して細胞傷害活性能がある、CTLの産生および/または増殖を測定する段階を含んでもよい。
【0027】
それにもかかわらず、抗原のCLEC9a+樹状細胞への標的化は、CTLに加えて、または代わりにヘルパーT細胞の増殖をもたらすことができる。したがって、本方法は追加としてまたは代替として、ヘルパーT細胞の産生および/または増殖を測定する段階を含んでもよい。ヘルパーT細胞はCD4+T細胞であってもよく、Th1、Th2、Th17またはTreg型であってもよい。ヘルパーT細胞はまた、CD4、例えば、CD8+Treg細胞を発現しない他の型のTreg細胞を含むことができる。
【0028】
したがって、上述した方法および組成物は、癌、またはウイルス感染などの細胞内寄生体もしくは病原体による感染症などの、CTL応答を誘導することが望ましい任意の状態の予防および/または治療のために使用することができることが理解されるはずである。
【0029】
最大のCTL刺激および増殖ならびに/または他のT細胞型の刺激および増殖を達成するために、さらなる免疫刺激剤を投与することが望ましい場合もある。これらは、樹状細胞を活性化、およびT細胞活性化を促進する能力を刺激することができる作用物質を含み得る。このような作用物質は、アジュバントと呼ばれることがある。アジュバントは、CD40のアゴニスト(可溶性CD40リガンド、またはCD40特異的アゴニスト抗体など)、CD28、CD27またはOX40のアゴニスト(例えば、これらの分子の1つに特異的なアゴニスト抗体)、CTLA-4アンタゴニスト(例えば、CTLA-4特異的遮断抗体)、および/またはToll様受容体(TLR)アゴニスト、および/または樹状細胞活性化を誘導することができる任意の他の作用物質を含み得る。TLRアゴニストは、Toll様受容体を活性化する物質である。好ましくは、TLRアゴニストはTLR3、TLR4、TLR5、TLR7またはTLR9の活性化剤である。適したTLRアゴニストは、TLR4に結合するMPL(モノホスホリル脂質A)である。使用することができる他のTLRアゴニストは、LTA(TLR2に結合するリポタイコ酸); TLR3に結合するポリI:C(ポリイノシン酸-ポリシチジル酸);TLR5に結合するフラジェリン; TLR7、およびTLR9に結合するCpG(DNA CpGモチーフ)に結合するイミキモドまたはポリU RNA(1-(2-メチルプロピル)-1H-イミダゾ(4,5-c)キノリン-4-アミン);またはTLRに結合および活性化する任意の他の成分である。より詳細には、Reis e Sousa、Toll-like receptors and dendritic cells、Seminars in Immunology 16:27、2004年を参照されたい。TLRを介して作用しない可能性があるアジュバントは、5'三リン酸RNA、ポリI:C、およびカードラン((β-1,3-グルカン))などのβ-グルカンを含む。TNF-αまたはIL-1などの炎症誘発性サイトカインもまた、アジュバントとして使用することができる。
【0030】
CLEC9a活性を有する(以下でより詳細に論じられる、例えば、樹状細胞表面上のCLEC9aと交差結合することができる)結合剤もまた、樹状細胞を活性化することができ得る。そのためこのようなアゴニスト結合剤は、それら自体がアジュバントとみなすことができる。したがって、このような結合剤が使用される場合、(そうすることがやはり望ましい可能性があるが)上述のものなどのさらなるアジュバントを投与する必要はない可能性がある。CLEC9aアゴニストとして作用することができる結合剤は、以下でより詳細に論じられる。
【0031】
理論に拘束されることを望むものではないが、使用されるアジュバントの性質は得られる応答の種類に影響する可能性があると考えられる。CLEC9aを発現する抗原提示細胞は、CD4+T細胞およびCD8+T細胞の両方を刺激することができ、CD4+応答の性質は特に使用されるアジュバントに影響される可能性がある。例えば、ポリI:Cの使用はTh1型CD4+応答の発生に有利であるようである。カードランはTh17型CD4+応答を刺激するようである。
【0032】
特定のアジュバントはTreg細胞の刺激を促進する。これらはレチノイン酸、および特にトレチノインとしても知られるオールトランスレチノイン酸(ATRA、all-trans retinoic acid)を含む。したがって、刺激される免疫応答がTreg応答である(実際には特定の抗原に対する免疫系の他の成分の応答を抑制してもよい)場合、Treg促進アジュバントを使用することが適切である可能性がある。アジュバントを投与せずにTreg細胞刺激を刺激することもまた可能である。
【0033】
本発明の組成物は、同じまたは別個の組成物において順次にまたは同時に、アジュバントと共に投与、または共に投与するために製剤化することができる。したがって、本発明の組成物は、アジュバントを含む必要はないが、含んでもよい。
【0034】
理論に拘束されることを望むものではないが、また上記で説明された通り、アジュバントの非存在下における抗原の投与は抗原に対する寛容の発生をもたらすと考えられる。すなわち、免疫系は同じ抗原のさらなる投与に応答しないように誘導される。これは、応答の能動的抑制が可能なTreg細胞の発生を含む(必要はないが)ことができる。したがって、抗原の、その抗原に対して寛容化された対象へのさらなる投与は、その抗原に対してナイーブな(すなわち、免疫系が以前にその抗原に曝露されていない)対象よりも弱い免疫応答をもたらすべきである。免疫応答の大きさは、炎症の出現、腫脹、細胞増殖(例えば、Th1、Th2もしくはTh17 CD4+T細胞、またはCTLの)、または炎症性サイトカイン産生(例えば、IL-1、IL-4、IL-12、IFN-γ、TNF-α)などの任意の適切な基準によって評価することができる。特定の実施形態において、寛容化された個体はその抗原に対する免疫応答を実質的に示さないはずである。
【0035】
上述した組成物および方法において、抗原は結合剤と物理的に結合しており、これは共有または非共有(例えば、静電またはファンデルワールス)相互作用を介していてもよい。好ましくは、抗原は結合剤に共有結合している。例えば、結合剤は、適したカップリング試薬を介して抗原と結合していてもよい。当業者は、このようなカップリング反応に使用することができる適した方法および試薬をよく知っている。
【0036】
代替として、抗原および結合剤は、同じペプチド鎖の一部であってもよく、すなわち、融合タンパク質として発現されてもよい。融合タンパク質は、抗原および結合剤間にリンカー配列を含有することができる。代替として、抗原および結合剤は、化学結合せずに物理的に近接して、例えば、リポソームに存在することができる。
【0037】
結合剤は、CLEC9aに対する十分に特異的な親和性を有する任意の適した分子であってよい。本明細書の全体にわたってこのような結合剤が言及される場合はいつでも、それはタンパク質、核酸(例えば、アプタマー)、糖質、または小分子であってよい。結合剤は、CLEC9aの生理的リガンドまたはその変異体もしくはアナログであってもよい。しかしながら、CLEC9aに対する抗体およびその機能的断片は特に好ましい。したがって、結合剤はCLEC9aに特異的な抗体結合部位を含んでもよい。結合剤は、以下で詳細に論じる通り多価であってもよい。
【0038】
抗原はペプチド抗原である。用語「ペプチド」は、抗原の性質、すなわち、ペプチド結合によって結合しているアミノ酸から形成されるものをいい、いかなる特定の大きさまたは長さを意味すると解釈されるべきではない。典型的には、ペプチド抗体は少なくとも長さが8アミノ酸であるはずであり、長さ30アミノ酸まで、長さ50アミノ酸まで、100アミノ酸まで、200アミノ酸まで、またはさらに長いものであってもよく、グリカン鎖などのアミノ酸に結合した残基を有してもよい。例えば、長さが25〜35アミノ酸であってもよい。
【0039】
いかなる特定の理論にも拘束されることを望むものではないが、ペプチド抗原は、MHCクラスIIまたはMHCクラスI分子に結合可能であるべきであり、または抗原提示細胞(樹状細胞など)内でプロセシングされてMHCクラスIIまたはMHCクラスI分子に結合できる1つまたは複数のペプチドを生じさせることが可能であるべきである。最近、長さが約8アミノ酸の短鎖エピトープペプチドが、長鎖ペプチド(例えば、長さが約30アミノ酸)よりも短いCTL反応性を誘導し得ることが示唆された(21)。MHCクラスI分子は典型的に、長さが8または9アミノ酸のペプチドに結合するが、MHCクラスII分子は8アミノ酸から20アミノ酸まで、30アミノ酸までの、またはさらに長いペプチドに結合することができる。
【0040】
抗原は、それに対して免疫応答、特にCTL応答だけでなくTh17応答またはTreg応答を生じさせることが望ましい任意のタンパク質またはその断片であってよい。これらは、癌細胞および細胞内病原体または寄生体を含有する細胞を含む、それに対してCTL応答を刺激することが望ましい細胞に結合している、上記細胞によって発現される、上記細胞上でディスプレイされる、または上記細胞によって分泌される抗原を含む。例えば、抗原は、細胞内病原体または寄生体によって発現されるタンパク質(ウイルスタンパク質など)由来の、または癌もしくは腫瘍細胞によって発現されるタンパク質由来のエピトープペプチドであってもよく、または上記エピトープペプチドを含んでもよい。したがって、抗原は腫瘍特異的抗原であってもよい。用語「腫瘍特異的」抗原は、固形腫瘍由来の抗原に限定されると解釈されるべきではなく、任意の癌性、形質転換または悪性細胞によって特異的に発現された抗原を包含すると解釈されるべきである。
【0041】
対象が望ましくない免疫応答を示す、または発生する危険性がある抗原に対するTreg応答を生じさせることが特に望ましい場合がある。例えば、それは自己免疫疾患において免疫応答が起こる自己抗原であり得る。特異的抗原が潜在的に病原的に重要であると確認された自己免疫疾患の例は、多発性硬化症(ミエリン塩基性タンパク質)、インシュリン依存性糖尿病(グルタミン酸脱炭酸酵素)、インシュリン抵抗性糖尿病(インシュリン受容体)、セリアック病(グリアジン)、水疱性類天疱瘡(XVII型コラーゲン)、自己免疫性溶血性貧血(Rhタンパク質)、自己免疫性血小板減少症(GpIIb/IIIa)、重症筋無力症(アセチルコリン受容体)、グレーブズ病(甲状腺刺激ホルモン受容体)、グッドパスチャー病(α3(IV)NC1コラーゲン)などの糸球体腎炎、および悪性貧血(内因子)を含む。代替として、標的抗原は、宿主組織にも損傷を与える応答を刺激する外因性抗原であってもよい。例えば、急性リウマチ熱は、心筋細胞抗原と交差反応する連鎖球菌抗原に対する抗体反応により引き起こされる。したがってこれらの抗原、またはその特定の断片もしくはエピトープは、本発明において使用するために適した抗原であり得る。
【0042】
Treg細胞の減少またはTreg細胞機能の障害は、マウスモデルにおいて自己免疫疾患をもたらすことが示されている。試験動物において引き起こされた疾患は、関節炎(例えば、関節リウマチ)、炎症性腸疾患、胃炎、悪性貧血、甲状腺炎、膵島炎、糖尿病、唾液腺炎、副腎炎、自己免疫性精巣炎/卵巣炎、糸球体腎炎、慢性閉塞性肺疾患ならびに実験的自己免疫性脳炎および多発性硬化症を含む。
【0043】
制御性T細胞1型応答の誘導はまた、マウスモデルにおいてアテローム性動脈硬化症の発生を減少させることも示されている(Mallat Z.ら、Circulation 108:1232〜7、2003年)。
【0044】
Treg活性はまた、同種移植の拒絶率において重要であることも示されている。Treg細胞の減少または機能障害は拒絶率を促進するが、Treg細胞において濃縮された同系リンパ球の試験動物への注入は移植片の生着を延長することが示されている。
【0045】
したがって本発明の方法は、任意のこれらの状態の治療において利用法を見出すことができる。
【0046】
対象に抗原自体を直接投与することなく、対象の体内に存在する抗原に対して、(CTL応答であれTreg応答を含む任意の別の種類のT細胞応答であれ)免疫応答を刺激することもまた可能であり得る。
【0047】
これは、結合剤が対象の体内の抗原と複合体を形成することができ、抗原をCLEC9aを発現する抗原提示細胞に標的化することができるように、抗原に対して親和性を有し、またCLEC9aに対して親和性も有する結合剤を使用して達成することができる。
【0048】
したがって本発明は、CLEC9aに対して親和性を有する第1の結合部位および抗原に対して親和性を有する第2の結合部位を有する結合剤を提供する。このような結合剤は、本明細書において「二重特異的」結合剤と呼ぶことにする。しかしながら、結合剤が、例えば、他の抗原に対する代替の結合親和性を有するさらなる結合部位を含んでもよく、用語「二重特異的」はそれに応じて解釈されるべきであることが理解されるはずである。
【0049】
本発明は、医療方法において、例えば、標的細胞に対する免疫応答の刺激において使用するための、上述の二重特異的結合剤をさらに提供する。標的細胞は、癌細胞または寄生細胞であってもよい。
【0050】
標的細胞に対する免疫応答の刺激のための医薬の製造における、上述の二重特異的結合剤の使用がさらに提供される。標的細胞は、癌細胞または寄生細胞であってもよい。
【0051】
二重特異的結合剤を対象に投与する段階を含む、前記対象において標的細胞に対する免疫応答を刺激する方法がさらに提供される。対象は、(例えば)癌または細胞内寄生体感染症に罹患していてもよい。
【0052】
二重特異的結合剤は、本明細書の他の部分に記載したアジュバントとの併用投与用に処方することができ、またはアジュバントと同じ組成物中に処方することができる。アジュバントの性質は所望の応答の性質に応じて選択することができる。したがって、例えば、ATRAは抗原に対するTreg応答を誘導することが望ましい場合に使用することができ、カードランはTh17応答が所望される場合に使用することができ、任意の別の適したアジュバント(例えば、抗CD40、ポリI:Cなど)はより一般的なCTL応答が必要とされる場合に使用することができる。アジュバントが投与されない場合、Treg細胞の刺激および/または抗原に対する寛容を誘導することが可能であり得る。
【0053】
本発明は、薬学的に許容される担体との混合物中の上述の二重特異的結合剤を含む医薬組成物をさらに提供する。組成物は、本明細書の他の部分に記載の通り、アジュバントをさらに含んでもよく、またはアジュバントとの併用投与用であってもよい。
【0054】
いくつかの実施形態において、一方または両方の結合部位がそれぞれCLEC9aまたは抗原特異的抗体結合部位であってもよい。したがって、結合剤は、少なくともCLEC9aに特異的な第1の抗体結合部位および少なくとも抗原に対して親和性を有する第2の抗体結合部位を含む二重特異的抗体であってもよい。用語「二重特異的抗体」は、二重特異的単鎖Fv二量体および「ダイアボディ」(下記参照)などの、上記2つの結合部位を有する任意の分子または分子複合体を包含すると解釈されるべきである。
【0055】
CLEC9a分子が樹状細胞の特定のサブセットのマーカーであるという発見はまた、CLEC9a分子を望ましくない免疫応答の下方制御の適した治療標的とする。
【0056】
したがって、さらなる態様において本発明は、CLEC9aに対して親和性を有する結合剤を投与する段階を含む、対象において免疫応答を阻害する方法を提供する。
【0057】
本発明の本態様において、結合剤は、結合する樹状細胞において樹状細胞機能を直接的または間接的に阻害することができる。結合剤は、CLEC9aが同種リガンドへ結合するのを阻害することができ、またはアジュバントに応じた成熟または抗原提示などの細胞機能の一面を阻害することができる。代替として、結合剤は細胞を直接的または間接的に死滅させることができ、これは「枯渇」活性とみなすことができる。典型的には、結合剤は、細胞を直接的または間接的に死滅させること、またはそうでなければ抗原提示に影響を与えることに関与する「エフェクター」部分を含む。
【0058】
例えば、結合剤は、対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる。例えば、抗体Fc領域を使用して補体系の成分を動員することができ、それにより補体の溶解経路、または細胞のオプソニン作用およびその後の好中球またはマクロファージなどの免疫系の食細胞による食作用を介して細胞の溶解をもたらすことができる。食細胞はまた、抗体の結合している細胞の食菌を可能にするFc受容体を有している。したがって抗体は、補体を活性化することなく細胞に食作用の印をつけることができる。このように、エフェクター部分は抗体Fc領域、例えば、IgGまたはIgMなどを含むことができる。
【0059】
結合剤は、標的細胞を死滅させるように作用する他の種類のエフェクター部分を含んでもよい。例えば、細胞を死滅させることができる毒素分子を含んでもよい。本発明者らが、CLEC9aに結合する抗体は樹状細胞にエンドサイトーシスによって取り込まれることができ、そのためコンジュゲート毒素分子が高い可能性で細胞に取り込まれるはずであることを示したように、この機構は特に有効であり得る。
【0060】
代替として、エフェクター部分は、細胞の近くでプロドラッグを活性化する、例えば、非毒性分子を毒性分子に変換することができる酵素であってもよい。
【0061】
既述したように、結合剤はCLEC9aに特異的な抗体結合部位を含んでもよい。
【0062】
有用な結合剤は、したがって、CLEC9aに特異的な抗体結合部位を含む分子、および抗体Fc領域、毒素分子、またはプロドラッグを活性化することができる酵素のうち1つまたは複数を含む。
【0063】
結合剤自体が、CLEC9aの機能的アンタゴニストであってもよい。したがって、CLEC9aをその同種リガンドに結合させない、または、例えば、樹状細胞成熟および/または抗原提示における通常のシグナル伝達の役割を発揮させないことによって阻害機能を発揮するために、結合剤はCLEC9に結合するだけで十分であり得る。このような実施形態において、結合剤が1個のみまたは2個のCLEC9aに特異的な結合部位(例えば、抗体結合部位)を含むことが望ましい場合がある。例えば、結合剤は、抗体またはFabもしくはscFv断片などの抗体断片であっても(を含んでも)よい。
【0064】
CLEC9aとリガンドの結合を妨害するアンタゴニストは、自己免疫疾患の治療において特に有用であり得る。いくつかの自己免疫疾患は、異常に高レベルの細胞死により特徴付けられ、これらの細胞に結合している自己抗原に対する免疫応答がこれらの状態の病原性に寄与する可能性があると考えられている。したがって、CLEC9aアンタゴニストを使用してCLEC9aが死細胞または瀕死細胞(特に免疫原性細胞死を起こしている細胞)に曝露されたリガンドに結合するのを妨害することができ、したがってこれらの抗原に対する免疫応答の刺激を阻害または妨害することができる。
【0065】
他の機能的CLEC9aアンタゴニストもまた、記載の方法において有用であり得る。このようなアンタゴニストは、CLEC9aをコードするmRNAまたはDNAにハイブリダイズすることができる核酸またはそのアナログ(例えば、リボザイム、アンチセンスRNAまたはDNA分子、siRNAなど)、CLEC9aの小分子アンタゴニストなどを含む。
【0066】
なおさらなるCLEC9aアンタゴニストは、樹状細胞結合CLEC9aのリガンド上の結合部位を遮断することができ、そのためリガンドが樹状細胞に認識、または結合されるのを妨害することができるCLEC9aリガンドの競合物である。適した競合物は、CLEC9aリガンドに結合するために十分なCLEC9aの細胞外ドメインまたはその一部を含む可溶性分子を含む。
【0067】
CLEC9a細胞外ドメイン(またはその一部)は、非相同部分と結合していてもよく、これはin vivoでの薬物動態特性などのアンタゴニストの特性を若干変える可能性がある。細胞外ドメインは、非相同部分に共有または非共有結合していてもよく、または非相同部分を有する融合タンパク質として発現されてもよい。例えば、血中半減期の増加をもたらし、アンタゴニストとCLEC9aリガンドの複合体の効率的な除去を可能にするために、非相同部分は抗体Fcドメインであってもよい。
【0068】
他の考えられる非相同部分の機能は、CLEC9a細胞外ドメインのオリゴマー形成の媒介、およびアンタゴニストの精製または試料からのCLEC9a単離の簡易化を含む。例えば、適したアンタゴニストは、アビジン単量体に結合しているCLEC9a細胞外ドメイン(またはCLEC9aリガンドに結合するのに十分なその断片)を含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む複合体をもたらす。この構築体は、ビーズなどの固体支持体上で提供することができるビオチンとの接触により容易に単離することができる。
【0069】
結合剤またはアンタゴニストがタンパク質である場合、アンタゴニストをコードする核酸(例えば、DNA)を投与することが可能であり得る。典型的には、核酸は体内の細胞(例えば、筋細胞)によって取り込まれ、発現され、これらの細胞から分泌されるはずである。このアプローチはしばしばDNAワクチン接種と呼ばれる。
【0070】
抗体は、結合剤およびアンタゴニストとして特に適しており、scFv形態で好都合に発現することができる。必要であれば、抗体を、抗原または上述のエフェクター部分と共に融合タンパク質としてコードすることができる。DNAワクチン接種アプローチの例は、Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年に記載されている。
【0071】
核酸は典型的に、核酸を取り込む細胞によるタンパク質の適切な発現および分泌を確実にするために、転写および翻訳調節配列と作動可能に連結して、場合により任意の所望の融合パートナーと共に、結合剤またはアンタゴニストをコードするコード領域を含む。このような配列は、転写開始配列(例えば、プロモーターおよびエンハンサー)、転写終結配列、適切なスプライシングシグナル、翻訳開始および終結配列、ならびに分泌を可能にするシグナルペプチドを含む(がこれらに限定されない)。
【0072】
したがって、本発明は、医療方法において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸(例えば、DNA)をさらに提供する。その方法および治療的使用において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸もまた提供される。
【0073】
結合剤またはアンタゴニストが投与される対象は、炎症または自己免疫状態、特に望ましくないCTL活性を特徴とする状態および/または高レベルの細胞死を特徴とする状態に罹患していてもよい。このような状態は、以下を含む:
- 免疫成分による関節リウマチおよび他の種類の慢性または急性関節炎または関節症、全身性エリテマトーデス(特に高レベルの細胞死を伴うことが知られている)、強皮症、シェーグレン症候群、自己免疫性(特にI型)糖尿病、甲状腺炎、ならびに乾癬を含む他の臓器特異的免疫疾患を含む自己免疫疾患;
- 多発性硬化症、重症筋無力症、および他の免疫介在性神経疾患を含む神経疾患。クローン病、大腸炎、セリアック病、および肝炎を含む胃腸疾患もまた含まれる;
- 現在では重大な免疫介在性の成分を含むと認識されている、アテローム性動脈硬化症、心筋症、リウマチ熱、心内膜炎、血管炎および他の免疫介在性心臓血管疾患を含む心臓血管疾患;
- 肺気腫、呼吸器気道感染症、および他の免疫介在性呼吸器疾患を含む免疫介在性呼吸器疾患;
- 喘息、鼻炎、および他の免疫介在性過敏性反応を含むアレルギープロセスおよび過敏性反応(I、II、III、およびIV型);
- 例えば、臓器移植、組織移植、輸血、骨髄移植中またはその後に起こる移植または移植片拒絶反応および移植片対宿主病;
- 敗血症性ショック症候群を含む感染性病原体に対する免疫病理学的反応;
- 小膠細胞などの免疫担当細胞に影響を与える神経変性過程などの変性過程。
【0074】
本発明は、医療方法において使用するための、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤をさらに提供する。
【0075】
免疫応答の阻害において使用するための、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤もまた提供される。
【0076】
免疫応答の阻害のための医薬の調製における、CLEC9aまたはCLEC9aアンタゴニストに対して親和性を有する結合剤の使用もまた提供される。
【0077】
本発明者らはまた、抗原と物理的に結合していない場合であっても、CLEC9aアンタゴニストが樹状細胞を活性化することができ、したがって免疫機能を刺激するのに有用である可能性があることも発見した。したがって本発明は、CLEC9aに対して親和性を有する結合剤を投与する段階を含む、対象において免疫応答を刺激する方法を提供する。
【0078】
CLEC9aアゴニストは、CLEC9aに対して親和性を有する結合剤であってもよく、CLEC9aに特異的な抗体結合部位を含んでもよい。
【0079】
理論に拘束されることを望むものではないが、2個以上のCLEC9aに特異的な結合部位(例えば、抗体結合部位)を有する結合剤は、おそらく細胞表面上のCLEC9aと交差結合またはその結合もしくは多量体化をもたらすことができるため、CLEC9a活性の特に有効なアゴニストである可能性があると考えられる。結合剤は、2個のこのような結合部位を有する場合、「二価」と、または2個以上のこのような結合部位を有する場合「多価」と呼ばれることがある。したがって、結合剤は好ましくは多価であり、少なくとも3、4、5、10個またはさらにそれ以上の結合部位を含んでもよい。このような結合剤は、(例えば、ラテックスの)ビーズ、リポソームもしくはベシクルなどの粒子、または任意の他の適した粒子中または上に固定化された複数の結合部位を含むことができる。このように、結合部位は微粒子固相上または中に固定化することができる。代替として、多価結合剤は、互いに共有結合または別な方法で結合した2個以上の結合部位を含むのみであってもよい。例えば、抗体全体またはその機能的断片(下記参照)は、融合タンパク質としておよび/または化学的交差結合によって結合していてもよい。当業者は、このような多価結合剤を調製するための適した技術をよく知っている。本方法がin vitroで行われる場合、培養容器の表面上に固定化された結合剤(Clec9aに対する抗体など)をアンタゴニストとして使用することができ、この場合培養容器の被覆された表面自体が多価結合剤であると考えることができる。
【0080】
アゴニストは、免疫機能を広く刺激するために単独で、またはそれに対する免疫応答を誘導することが望ましい標的抗原と併用して投与することができる。アゴニストおよび抗原は、必要に応じて、別々にまたは同じ組成物において同時にまたは順次に投与することができる。アゴニストは、本発明の第1の態様に関して上述した通り抗原と物理的に結合していてもよく、または2個の成分が物理的に独立した別個の存在であってもよい。
【0081】
既述した通り、記載した方法および組成物は、癌、またはウイルス感染などの細胞内寄生体もしくは病原体による感染症などの、CTL応答を誘導することが望ましい任意の状態の予防および/または治療のために特に有用であり得る。
【0082】
記載した方法および組成物はまた、Treg応答を誘導することが望ましい任意の状態、例えば、自己免疫疾患などの特定の状態に対する望ましくないまたは不適切な免疫応答を伴う状態の予防および/または治療のために使用することもできる。
【0083】
上述のさらなる免疫刺激剤またはアジュバントを、アゴニストおよび場合により抗原を伴って投与することもできる。したがってアジュバントは、例えば、CD40アゴニストまたはTLRアゴニストを含むことができる。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0084】
樹状細胞のサブセットのマーカーとしてのCLEC9aの同定は、生体試料からこのような細胞を同定および単離するための手段を提供する。
【0085】
したがって、さらなる態様において本発明は、試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤と1つまたは複数の細胞の結合を測定する段階とを含む、試料中の抗原提示細胞を検出する方法を提供する。
【0086】
本方法はまた、試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤が結合している1つまたは複数の細胞を単離する段階とを含む、試料から抗原提示細胞を単離する方法も提供する。結合剤は、単離を容易にするために固体支持体(磁気ビーズなど)上に固定化されていてもよい。
【0087】
上記の議論から明らかになるように、抗原提示細胞は、典型的には樹状細胞であり、MHCクラスI分子を介して細胞外抗原を交差提示することができ得る。
【0088】
CLEC9a結合剤により同定された細胞が樹状細胞であることを確認するために、または細胞をCLEC9a結合剤と接触させる前に試料の樹状細胞を濃縮するために、本方法は、試料を樹状細胞マーカーに対して親和性を有する第2の結合剤と接触させる段階と、第2の結合剤と1つまたは複数の細胞の結合を測定する段階とを含むことができる。2つの結合剤を、同時にまたは順次に、任意の順序で細胞と接触させてもよい。いくつかの実施形態において、第1および第2の結合剤両方が結合する細胞のみが同定または単離される。
【0089】
樹状細胞マーカーは、CD11などの全樹状細胞マーカー、特に(マウスにおける)CD11cとすることができる。
【0090】
ヒト樹状細胞の試料について、樹状細胞マーカーはHLA-DRであってもよい。細胞がlineage陰性である、すなわちCD3、CD14、CD19またはCD56を発現しないことを確認することが望ましい場合がある。
【0091】
ヒト細胞の試料について、CLEC9a結合剤によって同定された細胞がBDCA-3(CD141またはトロンボモジュリンとしても知られている)も発現することを確認することが望ましい場合がある。したがって本方法は、試料をBDCA-3に対して親和性を有するさらなる結合剤と接触させる段階と、さらなる結合剤と1つまたは複数の細胞の結合を測定する段階および/またはさらなる結合剤が結合する細胞を単離する段階とを含むことができる。CLEC9aおよびBDCA-3の結合剤を、同時にまたは順次に、任意の順序で細胞と接触させてもよい。いくつかの実施形態において、両方の結合剤が結合する細胞のみが同定または単離される。
【0092】
追加としてまたは代替として、1つまたは複数の不要な細胞型の負の選択によって、試料の所望の細胞型を濃縮することが望ましい場合がある。不要な細胞型は、CD123またはLy6Cに対する負の選択によって除外され得る、形質細胞様樹状細胞(pDC)などの樹状細胞の他の亜群を含み得る。負の選択は、CLEC9a発現細胞に対する選択の前に、同時に、または後に行うことができる。負の選択は、CD3、CD14、CD19および/またはCD56に対して行うことができる。
【0093】
細胞上のCLEC9a発現レベルを測定することが望ましい場合もある。このことは、所望のレベルのCLEC9a、例えば、検出可能なレベルでCLEC9aを発現する別の細胞集団よりも高レベルのCLEC9aを発現する細胞を選択することのみを可能にする。例えば、理論に拘束されることを望むものではないが、CD8を発現する(またはヒトにおけるそのサブセットと同等である)DCのサブセットはpDCよりも高レベルのCLEC9aを発現すると考えられる。したがって、特定の閾値レベルを超えるCLEC9aを発現する細胞を選択することのみによって、CD8 DCまたはその同等物を選択することが可能であり得る。
【0094】
結合剤は、例えば、検出可能なシグナル(蛍光または放射性標識など)を発することができる標識によって、または結合パートナーにより特異的に結合されることが可能な親和性タグによって、細胞の検出および/または単離を容易にするために標識されていてもよい。親和性タグと結合パートナーの例は、エピトープと同種抗原、正に荷電したペプチド(例えば、ポリ-His)と金属(例えば、ニッケル)イオン、アビジン/ストレプトアビジンとビオチン、糖質とレクチンなどを含む。当業者であれば、これらの特定の要件に応じて適した系を設計することができるはずである。
【0095】
細胞の同定または単離は、試料を第1および/または第2の結合剤に結合することができる1つまたは複数の検出剤と接触させる段階を含むことができる。検出剤自体が、上述のように標識されていてもよい。
【0096】
単離および検出を容易にするために、結合剤または検出剤は固体支持体上に固定化されていてもよい。
【0097】
本発明は、上述の方法により単離された抗原提示細胞の集団をさらに提供する。
【0098】
これらの方法により単離された細胞は、in vitro試験およびex vivo療法を含む様々な目的のために使用することができる。例えば、単離された細胞にin vitroで所望の抗原をパルスすることができる。その後、抗原に対する免疫応答を刺激するために、細胞を対象に投与することができる。
【0099】
したがって、本発明は、ペプチド抗原に対する免疫応答を刺激する方法であって、上述した方法により単離された抗原提示細胞またはその集団を提供する段階と、前記細胞または細胞集団を前記抗原と接触させる段階とを含む方法をさらに提供する。
【0100】
好ましくは、抗原提示細胞はMHCクラスI分子に関連して前記抗原またはその断片を提示する。
【0101】
前記接触させる段階に続いて、細胞または細胞集団を対象に投与することができる。好ましくは、細胞は、細胞の由来した対象に再投与される。
【0102】
細胞または細胞集団はまた、上述した通り、アジュバントと接触させてもよい。接触は、例えば、抗原との接触と同時にまたはほぼ同時に、または受容対象への投与時または後にin vitroで行うことができる。アジュバントとの接触は、細胞または細胞集団の、抗原に応答したT細胞増殖を活性化または促進する能力を刺激することができる。アジュバントは、細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。免疫刺激アジュバントは、例えば、CD40アゴニストまたはTLRアゴニストであってもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0103】
したがって本発明は、上述した方法によって得られる、プライミングされた抗原提示細胞またはその集団を提供する。「プライミングされた」は、細胞が抗原と接触したこと、MHC分子、好ましくはMHCクラスI分子に関連して、その抗原またはエピトープを提示していること、およびそれに応答してT細胞の増殖およびエフェクター細胞への分化を活性化または刺激することができることを意味する。
【0104】
医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるプライミングされた抗原提示細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、プライミングされた抗原提示細胞またはその集団の使用もまた提供される。
【0105】
代替として、抗原に特異的なT細胞(特にCTLだけでなくTh17およびTreg細胞を含むCD4+T細胞も)を発生させるために、プライミングされた細胞をin vitroでT細胞と接触させることができる。したがって、前記接触させる段階に続いて、本方法は、前記抗原提示細胞を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含むことができる。集団中のT細胞は、抗原に特異的な集団中のT細胞の数または比率を増加させるために、培養で増殖させることができる。その後T細胞を対象に投与することができる。場合により、T細胞は投与前に集団中の他の細胞から分離される。
【0106】
細胞集団はまた、例えば、プライミングされた抗原提示細胞と接触するのと実質的に同時に、アジュバントと接触させてもよい。
【0107】
好ましくは、T細胞および抗原提示細胞は自己由来、すなわち、同じ対象由来または遺伝学的に同一の対象由来である。
【0108】
T細胞は、細胞(またはその祖先)の由来した対象に再投与されてもよい。
【0109】
この場合もやはり、アジュバントは対象に投与されるときにT細胞と共に投与されてもよい。アジュバントは、CD40アゴニスト(CD40に特異的な抗体など)またはTLRアゴニストであってもよい。アジュバントは、T細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。
【0110】
したがって本発明は、上述した方法によって得られるT細胞またはその集団をさらに提供する。医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるT細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、T細胞またはその集団の使用もまた提供される。
【0111】
本発明は、CLEC9aを発現するヒト樹状細胞の単離された集団をさらに提供する。集団は、少なくとも5個、少なくとも10個、少なくとも100個、少なくとも1000個または少なくとも10000個の樹状細胞を含有することができる。好ましくは、集団中の少なくとも50%、少なくとも60%、少なくとも70%、少なくとも80%、少なくとも90%、または少なくとも95%の樹状細胞がCLEC9aを発現する。この目的のために、樹状細胞はlineage陰性、HLA-DR+細胞であると考えられている。lineage陰性細胞は、CD3、CD14、CD19またはCD56を発現しない。
【0112】
単離された集団は、試料中の樹状細胞の所要の比率がCLEC9aを発現する限り、他の細胞型を含む試料中に含まれてもよい。したがって試料は、リンパ球(例えば、T細胞、および特にCTL)または樹状細胞でない他の種類の抗原提示細胞を含むことができる。
【0113】
CLEC9aを発現する樹状細胞は、典型的にはBDCA-3+であるはずである。また、CD123-、CD34-、CD16-およびCD11b/c-であってもよい。
【0114】
本発明は、ペプチド抗原に対する免疫応答を刺激する方法であって、CLEC9aを発現するヒト樹状細胞の集団を提供する段階と、前記集団を前記抗原と接触させる段階とを含む方法をさらに提供する。
【0115】
前記接触させる段階に続いて、CLEC9aを発現するヒト樹状細胞の集団を自己由来対象に投与する(すなわち、もともと集団が由来した対象に投与される)ことができる。
【0116】
CLEC9aを発現するヒト樹状細胞集団はまた、上述した通り、アジュバントと接触させてもよい。接触は、例えば、抗原との接触と同時にまたはほぼ同時に、または受容対象への投与時または後にin vitroで行うことができる。アジュバントとの接触は、細胞集団の、抗原に応答したT細胞増殖を活性化または促進する能力を刺激することができる。アジュバントは、細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。免疫刺激アジュバントは、例えば、CD40アゴニストまたはTLRアゴニストであってもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0117】
したがって本発明は、抗原によってプライミングされた、CLEC9aを発現するヒト樹状細胞の集団を提供する。「プライミングされた」は、細胞が抗原と接触したこと、MHC分子、好ましくはMHCクラスI分子に関連して、その抗原またはエピトープを提示していること、およびそれに応答してT細胞の増殖およびエフェクター細胞への分化を活性化または刺激することができることを意味する。
【0118】
医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、CLEC9aを発現するヒト樹状細胞のプライミングされた集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、CLEC9aを発現するヒト樹状細胞のプライミングされた集団の使用もまた提供される。
【0119】
代替として、抗原に特異的なT細胞(特にCTLだけでなくヘルパーT細胞またはTregも)を発生させるために、プライミングされた細胞をin vitroでT細胞と接触させることができる。したがって、前記接触させる段階に続いて、本方法は、前記CLEC9aを発現するヒト樹状細胞の集団を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含むことができる。集団中のT細胞は、抗原に特異的な集団中のT細胞の数または比率を増加させるために、培養で増殖させることができる。その後T細胞を対象に投与することができる。場合により、T細胞は投与前に集団中の他の細胞から分離される。本発明の別の態様にあるように、樹状細胞およびT細胞もまた、アジュバントと接触してもよい。アジュバントの性質は所望の免疫応答の性質に応じて選択することができる。したがって、例えば、CTL応答、Th17型CD4 T細胞応答またはTreg応答が望ましい場合、アジュバントはそれに応じて選択することができる。
【0120】
好ましくは、T細胞および樹状細胞は自己由来であり、すなわち、同じ対象由来、または遺伝学的に同一の対象由来である。
【0121】
T細胞は、細胞(またはその祖先)の由来した対象に再投与されてもよい。
【0122】
この場合もやはり、アジュバントはT細胞と共に投与されてもよく、アジュバントはCD40アゴニスト(CD40に特異的な抗体など)またはTLRアゴニストであってもよい。アジュバントは、T細胞と共に同時に、または同じまたは異なる組成物において順次に投与することができる。
【0123】
したがって本発明は、上述した方法によって得られるT細胞またはその集団をさらに提供する。医療方法において使用するための、および特に標的抗原に対する免疫応答を刺激する方法において使用するための、上述した方法によって得られるT細胞またはその集団もまた提供される。標的抗原に対する免疫応答の刺激のための医薬の調製における、上述した方法により得られる、T細胞またはその集団の使用もまた提供される。
【0124】
全ての上述の態様において、抗原は、それに対して免疫応答、特にCTL応答、Th17型応答またはTreg応答を刺激することが望ましい任意の抗体であってよい。例えば、抗原は、本明細書の他の部分に記載の通り、細胞内病原体または寄生体によって発現される抗原であってもよく、または癌細胞によって発現されてもよい。代替として、抗原は、それに対して望ましくないまたは不適切な応答が起こる(例えば、自己免疫疾患において)、およびそれに対してTreg応答を刺激することが望ましい抗原であってもよい。
【0125】
CLEC9aが、感染性病原体ではなく哺乳動物細胞上または中にみられるリガンドに結合するという驚くべき発見はまた、このリガンドの同定のためのアッセイを利用可能にもする。
【0126】
したがって本発明は、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合するのに十分なその一部を含む標的物質を、哺乳動物細胞の成分である試験物質と接触させる段階と、標的物質と試験物質の結合を測定する段階とを含む、CLEC9aの生理的リガンドをスクリーニングする方法を提供する。
【0127】
結合により、試験物質がCLEC9aの1つの(または特定の)生理的リガンドであることを示すことができる。
【0128】
結合が起こる場合、本方法は、試験物質を同定する段階をさらに含むことができる。
【0129】
CLEC9aのリガンドは、一部のまたは全ての正常哺乳動物細胞において構成的に発現されるが、細胞が正常であり続ける限りは、CLEC9aと接触して相互作用することができないと考えられている。特定の種類の細胞損傷または細胞死(特に、原発性または二次性壊死などの免疫原性の場合)は、リガンドが樹状細胞上のCLEC9aと接触して相互作用できるように、リガンドを露出させる。
【0130】
したがって、試験物質は、哺乳動物細胞(例えば、細胞内寄生体または病原体に感染していない正常な哺乳動物細胞)の細胞内成分であってもよい。試験物質は、タンパク質、糖質、脂質または核酸を含んでも、またはそれらからなってもよい。試験物質は、これらの成分の2つ以上を含んでもよく、例えば、糖質および脂質成分を含むグリコシル化タンパク質、脂質およびタンパク質(および場合により糖質も)成分を含む脂質アンカータンパク質、または脂質および糖質成分を含む糖脂質であってもよい。
【0131】
試験物質は、生理的条件下で哺乳動物細胞中に存在するであろう分子または物質の全体を含まない可能性があるが、CLEC9aと相互作用するために十分なその一部を含み得ることが理解されるはずである。例えば、試験物質は、細胞タンパク質の単離ドメイン、またはペプチド(例えば、5〜10アミノ酸、20アミノ酸まで、50アミノ酸まで、または100アミノ酸まで)さえも含むことができる。
【0132】
典型的には、試験物質は、標的物質中に存在するCLEC9a(またはその一部)と同じ哺乳動物種由来であるはずである。
【0133】
本方法は、標的物質を、試験物質を含む試料(例えば、水性試料などの液体試料)と接触させる段階を含むことができる。
【0134】
標的物質は溶液中(例えば、水溶液中)に提供されてもよく、または固体支持体上に固定化されてもよい。
【0135】
試料は、透過性哺乳動物細胞を含んでもよい。「透過性」は、(細胞によって能動的に取り込まれずには)通常、原形質膜を通過することができない存在への拡散によって、原形質膜が透過性になったことを意味する。通常の条件下で原形質膜を透過しないヨウ化プロピジウムおよびTO-PRO3などの染料は、原形質膜完全性/透過性を試験するために慣習的に使用されている。したがって、原形質膜はこのような物質に対して透過性であってもよい。例えば、原形質膜は、500Daを超える、1kDaを超える、10kDaを超える、50kDaを超える、100kDaを超える、またはさらに大きい分子量を有する物質に対して透過性であってもよい。用語「透過性」はこの場合、個々の細胞が依然として(例えば、顕微鏡法によって)識別できるように、実質的に細胞構造(原形質膜、および潜在的に細胞内の他の膜の透過性増加は別にして)を保持する細胞のことをいうために使用される。用語「溶解物」、「抽出物」または「ホモジネート」は、個々の細胞がもはや識別できない、または存在しない程度まで細胞の構造が粉砕される調製のために使用することができる。
【0136】
透過性細胞は壊死性であってもよい。壊死は、原発性または二次性壊死であってもよい。原発性壊死は、(例えば)照射(例えば、UV光などの電離放射線による)、血清除去、少なくとも1回の凍結/融解サイクルによって、またはアントラサイクリン(ドキソルビシンおよびダウノルビシンなど)およびアントラセンジオン(ミトキサントロンなど)などの壊死誘導化学物質を用いた処理によって、実験的に誘導することができる。二次性壊死は、アポトーシスに入るよう誘導された細胞が隣接細胞によって食菌されず、原形質膜がその後破裂する場合に起こる。
【0137】
代替として、正常細胞は、原形質膜を破裂させるまたは膜に孔を形成する作用物質を用いて、直接透過性化してもよい。適した透過性化剤は、コレステロールを沈殿させ、したがって膜から除去して透過性を増加させるサポニンなどの孔形成剤(例えば、β-エスシン)、細胞溶解素(例えば、黄色ブドウ球菌のストレプトリシン-O)および黄色ブドウ球菌(Staphylococcus aureus)のα-トキシンなどの様々な細菌毒素、ならびにTriton X-100、Brij-96、Tweenなどの界面活性剤を含む。
【0138】
場合により、細胞は、透過性化の前または後に固定されてもよい。透過性化後にリガンドが(例えば、拡散によって)細胞から失われる機会を減少させるために、透過性化前の固定が好ましい場合がある。固定は、アセトン、メタノール、エタノールまたはそれらの混合物などの有機溶媒(一般に、細胞構造上のタンパク質を沈殿させると同時に、細胞の脂質を除去し脱水する)および/またはホルムアルデヒド(例えば、ホルマリン)もしくはパラホルムアルデヒド(タンパク質などの細胞成分を、アミノ基などのこれらの細胞成分に存在する遊離反応基を介して架橋する)などの架橋試薬を用いて行うことができる。
【0139】
試料が透過性細胞を含む場合、本方法は、標的物質の検出によって、標的物質の結合が起こる細胞内位置を測定する段階を含むことができる。
【0140】
検出は直接的または間接的であってもよい。例えば、標的物質は標識を含むことができ、本方法は標識の細胞内位置を測定する段階を含むことができる。代替として、本方法は、標的物質を、標的物質に結合可能な結合剤であってもよい検出剤と接触させる、さらなる段階を含むことができる。検出剤自体が標識を含んでもよい。検出は、顕微鏡法、例えば、共焦点顕微鏡法などの任意の適した技術によって達成することができる。例えば、標識は蛍光であってもよい。
【0141】
代替として、試料は、哺乳動物細胞の細胞溶解物、抽出物または細胞内画分であっても、または含んでもよい。例えば、試料は、全細胞溶解物、またはインタクトな正常細胞中の外部環境に曝露されていない細胞内画分を含むことができる。例えば、試料は、単離された細胞質画分、単離された核画分、単離された小胞体画分、単離されたゴルジ画分、または単離されたミトコンドリア画分を含むことができる。この文脈において「単離された」は、インタクトな細胞の少なくとも1つの他の正常成分(原形質膜など)から分離されていることを意味する。このような画分中の核またはミトコンドリアなどの細胞内オルガネラは、インタクトであってもまたは破壊されていてもよい。
【0142】
第1の試料の陽性結合反応に続いて、第1の試料は、第1の試料に存在する1つまたは複数の成分を欠く第2の試料を提供するためにさらに分画されてもよい。その後、本方法を繰り返してもよい。これは、標的物質に結合する試験物質の同定において役に立ち得る。このプロセスは、徐々に小さくなる画分を使用して、所望の回数繰り返すことができる。
【0143】
追加としてまたは代替として、本方法は、標的物質および試験物質を含む複合体を単離する段階を含むことができる。これは、標的物質が結合している固体支持体(例えば、ビーズ)を単離することによって達成することができる。代替として、標的物質に結合することができる結合剤を利用してもよい。結合剤は、固体支持体上に固定化することができる。標的物質は特異的結合対のメンバー含むことができ、結合剤は特異的結合対の第2のメンバーを含むことができる。例えば、結合剤は、標的物質に特異的な抗体であってもよい。代替として、結合剤および標的物質のうち1つがアビジン/ストレプトアビジン部分を含み、もう一方がビオチン部分を含むことができる。
【0144】
用語「特異的結合対」は、互いに特別な特異性を有し、通常の条件において他の分子への結合に優先して互いに結合する、特異的結合メンバー(sbm)および結合パートナー(bp)を含む分子の対を説明するために使用される。特異的結合対の例は、抗体とその同種エピトープ/抗原、リガンド(ホルモンなど)と受容体、アビジン/ストレプトアビジンとビオチン、レクチンと糖質、および相補的ヌクレオチド配列である。
【0145】
単離を容易にするために、単一の結合剤が多価、すなわち、1つまたは複数の標的物質に同時に結合することができるものであってもよい。標的物質もまた多価(すなわち、同じ種類の2つ以上の結合剤に同時に結合することができる)である場合、架橋複合体が形成されてもよい。
【0146】
当業者は、利用できる免疫沈降技術などの適した技術をよく知っている。
【0147】
例えば、標的物質は、アビジン単量体に結合している(例えば、コンジュゲートした、または融合タンパク質として)CLEC9a細胞外ドメインを含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む結合剤をもたらす。この構築体はしたがって、本明細書に記載のアッセイにおいて試験物質との複合体を形成し、その後ビーズなどの固体支持体上に存在し得るビオチンとの接触によって単離することができる。
【0148】
代替として、試験物質はメンブレン、マイクロタイタープレート、またはマイクロアレイチップなどの固体支持体上に固定化されてもよい。支持体を標的物質と接触させて、任意の結合している標的物質の位置を測定することができる。
【0149】
標的物質に結合できる物質を実際に含有するかどうかを確かめるために、CLEC9aのリガンドを含有する疑いがある複数の試料(例えば、異なる細胞画分)を試験することが望ましい場合がある。追加としてまたは代替として、標的物質に結合できるかどうかを確かめるために、複数の既知物質(例えば、タンパク質)を試験することが望ましい場合がある。
【0150】
したがって、単一の固体支持体は唯1つの試料または試験物質を含むことができ、または複数の試料または試験物質をそれぞれ支持体上の規定された位置に有することができる。
【0151】
アッセイの形式に応じて、本方法は、陽性結合反応が起こる特定の支持体、または陽性結合反応が起こる支持体上の位置の同定段階を含むことができる。したがってこれにより、陽性反応の原因である試験物質を含む試料の性質を明らかにすることができ、または試験物質の正体を直接明らかにすることができる。
【0152】
代替として、固体支持体から試験物質および標的物質の複合体を単離し、試験物質を同定するためのさらなる分析を行うことも可能であり得る。
【0153】
いかなるアッセイの形式であっても、試験物質がタンパク質である場合、試験物質を同定するためにプロテオミクス技術を使用することが可能であり得る。これは典型的には、質量分析およびデータベース照合と組み合わせるはずである。試験物質(または試験および標的物質の複合体)は、既知のNまたはC末端残基を有するペプチドを得るために、(使用される特定のプロテアーゼに応じて)既知の標的切断配列を有する1つまたは複数のプロテアーゼによる消化に供してもよい。得られたペプチドを、次いで質量分析(例えば、MALDI-TOF)にかけて分子量を測定する。次いで、適したタンパク質配列データベースを、このようなペプチドを生じさせることができるタンパク質を同定するために照合することができる。
【0154】
代替として、試験物質は細胞またはウイルスの表面上にディスプレイされてもよい。例えば、ファージディスプレイ技術を使用して、試験物質またはその断片をバクテリオファージの表面上にディスプレイすることができる。代替として、細胞は、表面上に試験物質をディスプレイするように処理されてもよい。代替として、試験物質と標的物質の間の相互作用が、細胞発現系または無細胞発現系において起こってもよく、レポーター遺伝子の発現などの検出可能な反応を誘導してもよい。このような系の一例は酵母2ハイブリッド系であるが、当業者であれば、2つのタンパク質間の「bait-prey」相互作用に依存してレポーター遺伝子の発現を駆動する他の系をよく知っているはずである。核酸にコードされるタンパク質が細胞発現系または無細胞発現系において発現される場合、このような形式を使用して、(存在する場合)標的物質に結合できる物質をコードする核酸分子を同定するために、核酸分子集団(例えば、cDNAライブラリー)をスクリーニングすることができる。このような形式において、本方法は、陽性結果の原因である核酸を単離する(例えば、クローニング)段階と、核酸によりコードされるタンパク質の正体を特定する段階とを含むことができる。
【0155】
試験物質がタンパク質ではない場合、他の分析技術を利用することができる。
【0156】
本発明を、限定ではなく例として、添付の図面および実施例を参照することによって以下により詳細に説明する。
【図面の簡単な説明】
【0157】
【図1】ヒトCLEC9aタンパク質に関するcDNAおよびアミノ酸配列を示す図である。
【図2】マウスCLEC9aタンパク質に関するcDNAおよびアミノ酸配列を示す図である。
【図3】ヒトPhoenix細胞中で発現されたマウスCLEC9aタンパク質の検出を示す図である。還元(R)および非還元(NR)状態において、CLEC9aを発現するPhoenix細胞(FNX-C9)または親細胞(FNX)からの完全溶解物で抗CLEC9aを使用するウエスタンブロット。
【図4】脾臓DCおよびin vitro培養BMDCにおけるマウスCLEC9a転写産物の分布を示す図である。脾臓DCのサブセット由来のmRNAまたはGMCSFもしくはFlt3L-BMDCの精製サブセット由来のmRNAを、Methodsで示したように、CLEC9a特異的プライマー(上レーン)またはβ-アクチンプライマー(下レーン)を使用するRT-PCRに施した。
【図5】この実験中に作製したCD3ζ-NKRP1-CLEC9aキメラを示す図である。キメラ分子中に存在するドメインおよびCLEC9aの細胞外ドメイン、NKRP1由来の膜貫通ドメインおよびCD3ζ由来の細胞質ドメインを示す。
【図6】初代細胞中でのCLEC9aの発現を示す図である。脾細胞はビオチン抗CLEC9a(太線、10mg/ml)またはビオチン-ラットIgG1(太線、アイソタイプ対照)、次にストレプトアビジン-PE(1:1000)で染色し、かつCD11c、CD4、CD8またはLy6Cで対比染色した。ヒストグラムはCD11c陰性細胞とCD11c陽性細胞に関する染色を示し、次いでそれらは従来型DCのCD4/CD8サブセットまたはpDCに相当するLy6C+サブセットにおいてさらに分析する。
【図7】CLEC9a細胞質ペプチドでのSykの破壊を示すウエスタンブロットの図である。CLEC9a細胞質ドメインまたはデクチン-1対照に相当するビオチン化ペプチドを使用して、Methods中に示したように組換えSykを破壊した。
【図8】抗CLEC9aを用いたキナーゼ活性化を示す図である。CLEC9aを発現するLK細胞をプレート抗体を用いて示した時間活性化させ、次いで溶解し、抗P-Sykまたは抗-P-Erk抗体を用いたSDS-PAGEおよびWBに施してこれらの経路の活性化を検出した。抗Sykまたは抗Erkを用いたタンパク質対照は全体のキナーゼを示す。
【図9】抗CLEC9aハイブリドーマによるサイトカインの誘導を示す図である。CLEC9aを発現するLK細胞(105個細胞/ウエル)は、上清の回収およびIL-2に関する試験前に、示したハイブリドーマと共に培養した。
【図10】プレートした抗CLEC9aが、Sykの存在下でCLEC9aのWT尾部を介して、CLEC9a-CD3ζを発現するY7.B3Z(σ)、またはCLEC9a-WT(ν)またはCLEC9a-Y7F(λ)を発現するB3Z-Sykの存在に依存してNFAT活性を誘導することを示す図である。NFAT活性はMethodsで示したように測定する。X軸はプレートをコーティングするために使用した抗CLEC9a抗体の濃度(μg/ml)を示す。
【図11】プラスチック上に固定化した抗Clec9a抗体は、Flt3L BMDCにおけるIL12-23 p40タンパク質の産生を誘導することを示す図である。Flt3L BMDCは抗CLEC9aモノクローナル抗体をコーティングしたプレート上でインキュベートし、一晩培養後の上清をIL12-23 p40に関して分析した。
【図12AB】抗CLEC9a-S1モノクローナル抗体を使用したin vivoでのCD11c+CD8+の標的化を示す図である。S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体(5μg)を静脈内注射し、Methodsで示したように翌日脾細胞を処理した。AおよびB.CD11c陽性および陰性脾細胞を、CFSE標識OT-I細胞と共に4日間培養した。A.25×103個の標的CD11c-またはCD11c+脾細胞の存在下でのOT-I細胞のCFSEプロファイル。B.左図:ウエル当たりのOT-I細胞の絶対数。右図:増殖OT-I細胞の上清中のIFN-γ産生。
【図12CD】抗CLEC9a-S1モノクローナル抗体を使用したin vivoでのCD11c+CD8+の標的化を示す図である。S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体(5μg)を静脈内注射し、Methodsで示したように翌日脾細胞を処理した。CおよびD.S1抗CLEC9a標的マウス中のCD11c+脾細胞を、CD11c+B220-CD8+(CD8+DC)、CD11c+B220-CD4+(CD4+DC)およびCD11c+B220-CD4-CD8-(DN DC)として分類し、CFSE標識OT-I細胞と共に3日間培養した。C.25×103個の標的CD8+、DN、CD4+DCの存在下でのOT-I細胞のCFSEプロファイル。D.左図:ウエル当たりのOT-I細胞の絶対数。右図:培養の終期における上清中のIFN-γ産生。
【図13A】抗CLEC9aモノクローナル抗体を使用した標的化によるCTL免疫の誘導を示す図である。A:S1結合抗CLEC9aまたはアイソタイプ対照モノクローナル抗体を、Methodsで示したように、抗CD40と一緒に、または抗CD40とは別に皮下注射した。5日後、20nM(0.03μMのCFSE)、200nM(0.3μMのCFSE)のSIINFEKLを導入した、またはペプチド(3μMのCFSE)を導入しなかった標的細胞(コンジェニックCD45.1)を静脈内注射した。結果はin vivo死滅アッセイにおける高用量ペプチドの特異的溶解率の算術平均±SEMとして表す(n=5、対照と比較したDEC205およびCLEC9a群のp<0.001。一元配置ANOVA)。B:抗CD40有りまたは無しで投与した、アイソタイプ対照または抗CLEC9aと結合したS2を使用した同様の実験の結果。
【図13B】抗CLEC9aモノクローナル抗体を使用した標的化によるCTL免疫の誘導を示す図である。B:抗CD40有りまたは無しで投与した、アイソタイプ対照または抗CLEC9aと結合したS2を使用した同様の実験の結果。
【図14】B16メラノーマの治療における抗CLEC9a+抗CD40の治療効果を示す図である。A.B16-OVA-GETメラノーマ細胞を使用した腫瘍治療実験の時間的経過。腫瘍細胞(2×105個)を第0日に静脈内注射し、抗体-S1+抗CD40の治療を第6日に実施し、第18日に肺を摘出し腫瘍を計数した。B.1つの代表的な実験におけるそれぞれのマウス中の腫瘍数を示す。全身腫瘍組織量の減少は、抗CLEC9aまたは抗DBC205および抗CD40を用いて有意である(p<0.001、一元配置ANOVA)。
【図15】ヒトCLEC9aの発現がBDCA-3+血中DCに限られることを示す図である。(A)ヒトPBMCを抗hCLEC9a(8F9)またはアイソタイプ適合対照抗体(マウスIgG2a)で染色し、様々な血液白血球マーカー用に対比染色した。ヒストグラムは、T細胞、B細胞、単球、NK細胞、系統陰性HLA-DR-細胞および系統陰性HLA-DR+細胞において染色したCLEC9aを示す。数字は後者の分画中のhCLEC9a+細胞の割合を示す。(B)(A)からのPBMCを系統陰性HLA-DR+細胞でゲートした。ドットプロットは、様々な血中DCサブセットマーカーに対する抗CLEC9aまたはアイソタイプ適合対照モノクローナル抗体を用いた染色を示す。数字はそれぞれの四分円中の細胞の割合を表す。特異的染色はBDCA-3+DCにおいてのみ見られる。4つの中の1つの代表的な実験。
【図16】CLEC-9aに腫瘍抗原を標的化することによるB16メラノーマの免疫療法を示す図である。(A)腫瘍治療実験を、抗CLEC9aまたはアイソタイプ適合対照抗体と共有結合したメラニン細胞分化内在性抗原(「Endo」:gp100、TRP-1およびTRP-2)の知られているエピトープを含むペプチドを使用して示したように実施した(上左)。ポリI:C+抗CD40はアジュバントとして使用した。左下図は、示したように治療したマウス由来の肺の代表図を示す。右図はそれぞれのマウス中の肺腫瘍の定量化を示す。データは2回の独立した実験(n=9マウス/群)からプールし、それぞれのポイントは1匹のマウスを表す。(B)(A)中の個々のマウス由来の脾細胞を、免疫処置に使用したメラニン細胞分化抗原ペプチド(10μM)を用いてin vitroで再度刺激した。2dの培養後のIFN-γのレベルを示す。データは2回の独立した実験(n=9マウス/群)からプールした。P値はマンホイットニーのU検定を使用して計算した。(C)B16細胞を注入する1日前にワクチンを与えたこと以外、図16中と同様に実験を実施した。データはマウス当たりの肺腫瘍の数を示す。データは2回の独立した実験(n=7マウス/群)からプールし、それぞれのポイントは1匹のマウスを表す。P値はマンホイットニーのU検定を使用して計算した。
【図17】死/死滅細胞上でCLEC9Aがリガンドと結合することを示す図である。(A)BWZ-マウスおよびヒトCLEC9Aζレポーターの基底活性化は、死滅TO-PRO3+細胞の数と相関関係がある。LacZと結合したNFATの受容体を発現し、かつマウスまたはヒトCLEC9Aまたは対照デクチン-1由来の細胞外ドメインおよびCD3ζ由来の細胞内尾部を有するキメラ分子を安定的に発現するBWZ細胞を、CLEC9Aの天然リガンドに関するスクリーニング用に作製した。異なる濃度の細胞を2日間増殖させ、異なる程度の異常増殖を示し、培養中の死滅細胞の増大をTO-PRO3染色によって追跡した。同量の生存BWZ細胞を次いで新たな培地に平板培養し、一晩の培養後、Methodsで示したように、BWZ細胞中のNFAT活性を比色分析アッセイにおいて測定した。 (B)UVC処理死滅細胞をCLEC9Aのリガンドに曝す。細胞死を誘導するためにUVCで処理し24時間放置した生存MEF(Ctrl)またはMEFを、キメラマウスCLEC9A-ζ、ヒトCLEC9A-ζ、およびデクチン-1-ζを発現するBWZ NFATレポーター細胞と共に培養した。示した場合、対照(p21)または抗mCLEC9A(1F6)または抗hCLEC9A(8F9)の一価Fab断片を培養物に加えた。BWZ細胞中のNFAT活性は(A)中と同様に測定した。 (C)UV処理細胞中の用量応答。LK細胞はMethods中で示したように異なる線量のUVCに曝し、mCLEC9A-ζキメラを発現するBWZ細胞または対照BWZとの培養前に24時間放置して細胞死を誘導した。BWZ細胞中のNFAT活性は(A)中と同様に測定した。 (D)CLEC9Aの組換え可溶性C型レクチンドメイン(rsCTLD)は、TO-PRO3+死滅細胞によって曝される分子を選択的に認識する。CLEC9Aまたは(対照として)デクチン-1のrsCTLDのPE-テトラマーは、UV照射した不死化MEFの染色用に使用した(ドットプロット)。ヒストグラムは、デクチン-1のrsCTLDに陽性なザイモサンの染色を示す。 (E)異なる死誘導処理はBWZ-CLEC9Aζレポーター細胞を誘発する。BWZ-CLEC9Aζ細胞は単独で(Ctrl)、または未処理(LK)またはUVC(UV)、ミトキサントロン(Mtx)、血清枯渇(SD)、浸透圧ショック(OS)または凍結および解凍(FT)で処理したLK細胞と共に一晩培養した。BWZレポーターの活性(左側y軸)ならびに同時培養開始時のLK細胞中のto-pro3+およびCLEC9A rsCTLD+の頻度(右側y軸)を示す。 (F)CLEC9AのrsCTLDのPE-テトラマーを使用した死滅MEFの染色。MEFはUVで処理して24時間放置し、または染色前に浸透圧ショック(OS)で処理した。 (A)〜(F)実施した少なくとも3つの中の1つの代表的な実験を示す。
【図18】CLEC9Aがin vitroでのCD8α+DCによる死細胞とのクロスプライミングに関与することを示す図である。 (A)、(B)CLEC9Aの阻害は、死滅細胞関連抗原とのOT-I細胞のクロスプライミングの低下をもたらす。(A)OT-I OVA特異的T細胞を、抗CLEC9AFab(1F6)または対照Fab(p21)の有無の下においてOVA導入死滅bm1脾細胞で刺激したCD8a様Flt3L BMDCと共に培養した。3日後、増殖(OT-I細胞の絶対数)、IL-2およびIFNγ産生(未処理対照に対する産生率)を測定した。6つの中の1つの代表的な実験を示す。(B)実施した6回の独立した実験に関する平均IL-2およびIFNγ産生は、抗CLEC9AFab(1F6)または対照Fab(p21)に関する未処理対照による産生率として示す。 (C)CLEC9A-/-およびWTCD8α様Flt3L BMDCは、死細胞の物質を捕捉する同じ能力を有する。WTまたはCLEC9A-/-CD8α様Flt3L BMDCは、異なる割合でPKH26標識およびUVC処理bm1脾細胞と共に2時間インキュベートした。結合(4℃)および結合+摂取(37℃)を、次いでそれぞれの型のDCに関するフローサイトメトリーにより定量化した。 (D)OVA発現またはOVA導入bm1死滅細胞で刺激したCLEC9A-/-Flt3L BMDCとのインキュベーション後の、エフェクターOT-I細胞に対する増殖および分化の低下。CLEC9A-/-Flt3L BMDCは、UVC処理OVA導入bm1脾細胞またはUVC処理OVA発現bm1 MEFと共に培養した。OVA特異的OT-I T細胞を次いで加え、かつ同時培養の3日後、OT-I細胞(左図)またはIFN-γ(右図)の絶対数を測定した。結果は一群当たり2匹のマウスの平均±SEMを示す。実施した3つの中の1つの代表的な実験を示す。 (E)(D)中と同様にOVA発現UV死滅bm1 MEFで刺激したCLEC9A-/-Flt3L BMDCとのインキュベーション後の、OT-I細胞の分化の低下。同じアッセイを使用して分析した四群中の一群当たりの1匹の代表的なマウスを示す。
【図19】CLEC9Aが免疫細胞の死を感知してin vivoでのクロスプライミングを促進することを示す図である。 (A)、(B)CLEC9Aの阻害はin vivoでの死滅細胞関連抗原とのクロスプライミングを低減する。未治療マウス、または抗CLEC9A(1F6、400μg/マウス)またはアイソタイプ対照(ラットIgG1)で治療したマウスを、切断型OVA-GFP融合タンパク質を発現する0.75×106個のUV照射bm1 MEFを用いた静脈内注射によって免疫処置した。6日後、H2Kb-OVAペプチドテトラマー陽性細胞(A)およびex vivoでのSIINFEKLに応じたIFNγ産生(B)を、内因性レパートリーから生じるCD8+T細胞エフェクター応答の誘導に関する読み出し値として測定した。(3つの中)1つの代表的な実験に関する個々のマウスおよび平均を示す。 (C)〜(E)CLEC9Aの欠失はin vivoでの死滅細胞関連抗原とのクロスプライミングを低減する。CLEC9a-/-マウスまたは対照同腹子は(A)中と同様に免疫処置した。OVA特異的内因性CD8+T細胞(C〜D)の頻度を(A)中と同様に定量化した。(C)それぞれのドットは、6回の独立した実験からプールしMethodsで示したように正規化した個々のマウスを表す。(D)CLEC9a-/-およびCLEC9a+マウスに関するそれぞれの同腹子におけるテトラマー陽性細胞の平均を表す。(E)ex vivoでのSIINFEKLに応じたIFNγ産生を、(B)中と同様に測定した。(3つの中)1つの代表的な実験に関する個々のマウスおよび平均を示す。
【図20】固定/透過処理後にCLEC9aのリガンドを曝したことを示す図である。 LK細胞を2%ホルムアルデヒドまたは2%パラホルムアルデヒドで固定し、Tween(0.5%)およびTx-100(0.5%)で透過処理したか、またはしなかった。細胞は大々的に洗浄し、透過性細胞の割合をTo-pro3を使用して定量化した。固定または透過処理細胞はキメラマウスCLAC9a-CD3ζを発現するBWZ NFATレポーター細胞または対照と同時培養し、NFAT活性を測定した。
【図21】CLEC9a wt細胞質尾部を介した死滅細胞のシグナルを示す図である。 LK細胞はMethods中で示したように異なる線量のUVCに曝し、CLEC9a wtまたはTyr7がPheに突然変異したCLEC9a(Y7F)で安定的にトランスフェクトしたB3Z細胞と共に培養し、Sykと同時発現させるかまたはさせない前に24時間放置して細胞死を誘導した。B3Z細胞中のNFAT活性はMethodsで記載したように測定した。
【図22】Fab一価と完全二価抗CLEC9a抗体の両方がCLEC9aを介した死滅細胞のシグナルを阻害することを示す図である。 細胞死を誘導するためにUVCで処理し24時間放置したLK細胞を、CLEC9a wtまたはTyr7がPheに突然変異したCLEC9a(Y7F)で安定的にトランスフェクトしたB3Z細胞と共に培養し、Sykまたは対照としてB3Z-デクチン-1-ζと同時発現させた。示した場合、対照(p21)または抗CLEC9A(1F6)の一価Fab断片、またはアイソタイプ対照(ラットIgG1、ラットIgG2a)および抗mCLEC9a(1F6、397、7H11)を含めた完全二価抗体を培養物に加えた。B3Z細胞中のNFAT活性はMethodsで記載したように測定した。
【図23】抗CLEC9a-0VA323-339に応じたOVA特異的OT-II CD4 T細胞の増殖を示す図である。未感作CFSE標識OT-II CD4 T細胞をC57BL/6マウスに静脈内注射で移した。1日後、抗CLEC9aまたはアイソタイプ対照抗体のいずれかと結合したOVA323-339ペプチドを脚部に皮下接種した。脾臓および流入領域リンパ節を3〜4日後に回収しOT-II細胞のin vivo増殖をCFSE希釈後に追跡した。サンプル間で正規化したOT-II細胞の絶対数を示す。
【図24】in vivoでの標的化中のアジュバントの添加は、強力なTh1の応答を誘導することを示す図である。アジュバント(OT-II系)の有無の下でのin vivo標的化。マウスは図23中と同様に処理した。上図:(正規化後の)OVA特異的CD4 T細胞の絶対数。下図:割合(左)または絶対数(右、正規化後)として表したOT-II CD4 T細胞集団中のIFN-γ産生。ポリI:Cの存在下で観察した増殖および分化に対する影響はこのアジュバントに限られない。カードランは同様の影響を有する。
【図25】CD4 T細胞応答は、標的化試薬と同時に注射するアジュバント/免疫調節剤の型によって調節することができることを示す図である。アジュバントの有無の下でのin vivo標的化。OT-II系は図24中と同様に実施した。上図:割合(左)または絶対数(右、正規化後)として表したOT-II CD4 T細胞集団中のIL-17産生。カードラン、デクチン-1アゴニストは、標的化抗原と投与すると、Th17分極の強力なアジュバントとして作用した。
【図26】アジュバントの不在下での標的化は、抗原特異的耐性をもたらすことを示す図である。未感作OVA特異的DO.11.10 CD4 T細胞はBALB/cマウスに移した。1日後、抗CLEC9a-OVA 323-339をアジュバント(ポリI:C)有りまたは無しで脚部に皮下接種した。免疫処置後の異なる時間地点で、脾臓DO.11.10区画の大きさを、KJ.126クローンタイプ抗体を使用したフローサイトメトリーによって決定した。結果は(正規化後の)OVA特異的KJ.126細胞の絶対数として、またはCD4 T細胞区画中のKJ.126+細胞の割合として表す。第20日に、半数のマウスをOVA、フロイント完全アジュバント中で皮下攻撃した。5日後、DO.11.10の応答をフローサイトメトリーによってモニターした。第0日にPBSのみを注射したマウス中で、残りのDO.11.10細胞は、再攻撃後のそれらの強力な増殖応答によって示されるように、依然として応答性がある。より強力な応答を抗CLEC9a+アジュバントを事前に注射したマウス中で検出し、この最初の免疫処置がおそらく記憶細胞の生成をもたらしたことが示された。対照的に、抗CLEC9aのみで最初に免疫処置したマウス中では応答を検出せず、残りの細胞は抗原に耐性があったことが示された。
【発明を実施するための形態】
【0158】
CLEC9a
CLEC9aは、樹状細胞上に発現されるC型レクチンである。本明細書において使用される場合、用語「CLEC9a」は、ヒトタンパク質(図1に示す核酸およびタンパク質配列)、マウスタンパク質(図2に示す核酸およびタンパク質配列)、他の種におけるそれらの相同体(特に相同分子種)、ならびにCLEC9a活性を保持するそれらの変異体および誘導体を包含することを意図する。このような変異体および誘導体は、図1に示すヒトタンパク質配列に対して好ましくは少なくとも約30%の配列同一性、より好ましくは少なくとも約35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%または95%の配列同一性、または図1に示すヒトタンパク質配列の細胞外ドメインに対して少なくとも約約35%の同一性、より好ましくは少なくとも約40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%または95%の同一性を有する。
【0159】
特に、CLEC9a配列における保存的置換(参照配列と比較して)は、機能に実質的な影響がなく特に忍容性良好であり得る。
【0160】
保存的置換は、アミノ酸クラス内の置換および/またはBLOSUM62マトリックスにおいて陽性と判定される置換と定義することができる。
【0161】
1つの分類によれば、アミノ酸クラスは酸性、塩基性、非荷電極性および非極性であり、酸性アミノ酸はAspおよびGluであり;塩基性アミノ酸はArg、LysおよびHisであり;非荷電極性アミノ酸はAsn、Gln、Ser、ThrおよびTyrであり;および非極性アミノ酸はAla、Gly、Val、Leu、Ile、Pro、Phe、Met、TrpおよびCysである。
【0162】
別の分類によれば、アミノ酸クラスは、低分子親水性、酸/酸アミド/親水性、塩基性、低分子疎水性および芳香族であり、低分子親水性アミノ酸はSer、Thr、Pro、AlaおよびGlyであり;酸/酸アミド/親水性アミノ酸はAsn、Asp、GluおよびGlnであり;塩基性アミノ酸はHis、ArgおよびLysであり;低分子疎水性アミノ酸はMet、Ile、LeuおよびValであり;および芳香族アミノ酸はPhe、TyrおよびTrpである。
【0163】
BLOSUM62マトリックスにおいて陽性と判定される置換は以下の通りである:
【0164】
【表1】
【0165】
参照配列に対するアミノ酸配列同一性パーセント(%)は、最大配列同一性パーセントを達成するために必要であれば、配列のアラインメントおよびギャップ導入後、保存的置換を配列同一性の一部とみなさずに、参照配列中のアミノ酸残基に同一の候補配列中のアミノ酸残基の割合として定義される。同一性%値は、WU-BLAST-2(Altschulら、Methods in Enzymology、266:460〜480(1996年))によって決定することができる。WU-BLAST-2はいくつかの検索パラメータを使用し、これらのほとんどは初期値に設定される。調整可能なパラメータは以下の値に設定される:オーバーラップスパン=1、オーバーラップフラクション=0.125、ワード閾値(T)=11。アミノ酸配列同一性%値は、WU-BLAST-2によって決定されるマッチングする同一な残基の数を、参照配列(無視されるアラインメントスコアを最大にするために参照配列中にWU-BLAST-2によってギャップを導入した)の全残基数で割って、100をかけることによって決定される。
【0166】
CLEC9aアゴニストは、典型的には、その細胞外ドメイン(ECD)に結合することおよびその細胞内ドメインを介して細胞内シグナル伝達を誘導することによって、CLEC9a活性を誘導することができる作用物質である。シグナル伝達は、Sykの細胞内ドメインへの結合、Sykリン酸化(およびしたがって活性化)、および/またはErkリン酸化および/またはNFAT活性化の1つまたは複数を含み得る。Clec9aおよびSykをトランスフェクトしたB3Z細胞を使用した例示的なアッセイを実施例に記載する。当業者であれば、Clec9aの細胞外ドメインおよび異なるタンパク質由来の細胞内ドメインを有するキメラタンパク質もまた、Clec9aアゴニスト活性をアッセイするために使用できることを理解するはずである。膜貫通ドメインもまた、Clec9a由来、細胞内ドメインと同じタンパク質、または別のタンパク質由来であってもよい。一例は図5に例示したCD3ζ-NKRPl-CLEC9aであり、実施例により詳細を記載する。
【0167】
CLEC9aアンタゴニストは、CLEC9a機能を阻害または遮断することができる作用物質である。例えば、CLEC9aアンタゴニストは、通常の発現、生理的CLEC9aリガンドに結合する能力、結合した分子を吸収する(エンドサイトーシスによって取り込む)能力、細胞内でシグナル伝達する能力(上記参照)、または例えば別の細胞上でそのECDが結合パートナー(リガンドまたは受容体)と相互作用する能力を妨害することができる。アンタゴニストの別の考えられる作用機構は、顕著な細胞内シグナル伝達を誘導することなく、Clec9aの吸収を促進し、そのため天然リガンドまたは別のアゴニストと相互作用するために利用可能な細胞表面上のClec9aのプールを減少させることである可能性がある。アンタゴニストは、有意なアゴニスト活性を欠く抗Clec9a抗体などの、CLEC9aに対して親和性を有する結合剤を含む。これらは「遮断」抗体と呼ばれることがある。アゴニスト活性を有さない一価または二価抗体は、遮断抗体として特に適する可能性がある。別のClec9aアンタゴニストは、CLEC9aをコードするDNAまたはRNAとハイブリダイズすることができる核酸分子またはそのアナログを含む。このような作用物質はリボザイム、RNAi、siRNAなどを含む。
【0168】
さらなるCLEC9aアンタゴニストは、樹状細胞結合CLEC9aのリガンド上の結合部位を遮断することができ、そのためリガンドが樹状細胞に認識、または結合されるのを妨害することができるCLEC9aリガンドの競合物である。適した競合物は、CLEC9aリガンドに結合するために十分なCLEC9aの細胞外ドメインまたはその一部を含む可溶性分子を含む。したがって、この分子は、図1に示すヒトCLEC9a、または図2に示すマウスCLEC9a、またはCLEC9aリガンドに親和性を有するその断片の細胞外ドメイン(CTLD)に対して、少なくとも70%の同一性、少なくとも75%の同一性、少なくとも80%の同一性、少なくとも85%の同一性、少なくとも90%の同一性、または少なくとも95%の同一性を有するアミノ酸配列を含むことができる。断片は、それぞれの細胞外ドメイン配列またはそれに所要のレベルの同一性を有する配列の、少なくとも20、30、40、50、60、70、80、90、100、110個または少なくとも120個のアミノ酸を含むことができる。
【0169】
CLEC9a細胞外ドメイン(またはその一部)は、非相同部分と結合していてもよく、これはin vivoでの薬物動態特性などのアンタゴニストの特性を若干変える可能性がある。細胞外ドメインは、非相同部分に共有または非共有結合していてもよく、または非相同部分を有する融合タンパク質として発現されてもよい。例えば、血中半減期の増加をもたらし、アンタゴニストとCLEC9aリガンドの複合体の効率的な除去を可能にするために、非相同部分は抗体Fcドメインであってもよい。
【0170】
他の考えられる非相同部分の機能は、CLEC9a細胞外ドメインのオリゴマー形成の媒介、およびアンタゴニストの精製または試料からの単離の簡易化を含む。例えば、適したアンタゴニストは、アビジン単量体に結合しているCLEC9a細胞外ドメイン(またはCLEC9aリガンドに結合するのに十分なその断片)を含む、またはからなる可溶性分子であってもよい。アビジン単量体は、四量体に結合する傾向があるはずであり、4個のCLEC9aドメインおよび4個のアビジンサブユニットを含む複合体をもたらす。この構築体は、ビーズなどの固体支持体上で提供することができるビオチンとの接触により容易に単離することができる。
【0171】
結合剤またはアンタゴニストがタンパク質である場合、アンタゴニストをコードする核酸(例えば、DNA)を投与することが可能であり得る。典型的には、核酸は体(例えば、筋細胞)内で細胞によって取り込まれ、これらの細胞から発現され、分泌されるはずである。このアプローチはしばしばDNAワクチン接種と呼ばれる。
【0172】
抗体は、結合剤およびアンタゴニストとして特に適しており、scFv形態で好都合に発現することができる。必要であれば、抗体を、抗原または上述のエフェクター部分と共に融合タンパク質としてコードすることができる。DNAワクチン接種アプローチの例は、Nchindaら、J.Clin.Invest.118(4)、1427〜36、2008年に記載されている。
【0173】
核酸は典型的に、核酸を取り込む細胞によるタンパク質の適切な発現および分泌を確実にするために、転写および翻訳調節配列と作動可能に連結して、場合により任意の所望の融合パートナーと共に、結合剤またはアンタゴニストをコードするコード領域を含む。このような配列は、転写開始配列(例えば、プロモーターおよびエンハンサー)、転写終結配列、適切なスプライシングシグナル、翻訳開始および終結配列、ならびに分泌を可能にするシグナルペプチドを含む(がこれらに限定されない)。
【0174】
本発明は、医療方法において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸(例えば、DNA)をさらに提供する。その方法および治療的使用において使用するための、CLEC9aアンタゴニストまたは結合剤をコードする核酸もまた提供される。
【0175】
CLEC9aリガンド
本発明者らは、CLEC9aが特定の種類の死または瀕死哺乳動物細胞によりディスプレイされたリガンドを認識することを発見した。特に、特定の種類の細胞死はリガンドのディスプレイを誘発するようである。C型レクチンファミリーの多くの既知のメンバー(CLEC9aに最も密接に関連したタンパク質であるデクチン1を含む)は、病原体関連分子パターンの受容体であり、そのため自己分子よりも病原体によってディスプレイされる構造を認識するので、これは驚くべき発見である。
【0176】
自己細胞死の特定の機構が免疫応答を誘発することができることはよく認識されている。これらは免疫原性細胞死とみなすことができる。アポトーシス(通常、原形質膜の破裂および細胞内容物の放出をもたらさない)による死は非免疫原性であり、一方、壊死(原形質膜の破裂および細胞内容物の放出を伴う)などの他の機構による死は免疫原性であると提唱されている。しかしながら、生理学的状況はこれよりも複雑であるようである。例えば、in vivoにおけるアポトーシス細胞は通常、細胞死のプロセスが完了する前にマクロファージなどの隣接細胞により吸収(食菌)される。しかしながら、細胞が食菌されなければ、いわゆる二次性壊死が起こる可能性があり、原形質膜は破裂し、細胞内容物は放出される可能性がある。この性質の細胞死は、少なくとも一部はアポトーシスであるにもかかわらず免疫原性とすることができる。
【0177】
免疫原性細胞死は、自己免疫疾患の発症、進行または持続において役割を果たしている可能性がある。このことは、例えば、Viorittoら、(Clin Immunol 122(2)、125〜134(2007年))、Tesniereら(Corr Op Immunol 21、1〜8(2008年))およびKimら(Immunity 27、321〜333(2007年))によって概説されている。
【0178】
樹状細胞は、死細胞または瀕死細胞の細胞破片を取り込むこと(またはさらに細胞全体を吸収すること)、およびプロセシングされた断片をT細胞に提示することによって、免疫原性細胞死により引き起こされる任意の免疫応答の誘導において役割を果たしている可能性がある。
【0179】
本発明者らは今回、CLEC9aが死細胞または瀕死細胞によりディスプレイされるリガンドに結合できること、およびCLEC9aシグナル伝達がこの相互作用によって誘発され得ることを発見した。
【0180】
細胞死プロセスの間、リガンドが新たに合成されないこともまた考えられる。むしろ、リガンドは一部のまたは全ての哺乳動物細胞によって構成的に発現されるが、細胞が正常であり続ける限りは、CLEC9aと接触して相互作用することができない可能性がある。特定の種類の細胞死は、CLEC9aと相互作用可能な形態でリガンドの露出および/または細胞からのリガンド放出をもたらす。これは原形質膜の破裂を伴い得る。
【0181】
実験的には、リガンドの露出は、照射(例えば、UV光などの電離放射線による)、血清除去、少なくとも1回の凍結/融解サイクルなどの処理によって、またはアントラサイクリン(ドキソルビシンおよびダウノルビシンなど)およびアントラセンジオン(ミトキサントロンなど)などの化学療法剤を用いた処理によって、引き起こすことができる。しかしながら、浸透圧ショックによる死はリガンドを露出させないようである。
【0182】
したがって、いかなる特定の理論に拘束されることを望むものではないが、CLEC9aは免疫原性細胞死によって引き起こされる免疫応答の発生に関与する可能性があると考えられる。CLEC9aおよびそのリガンド間の相互作用は、CLEC9aアンタゴニストによって阻害することができる。これらは、CLEC9a(の細胞外ドメイン)に結合することができる結合剤を含む。別の例は、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合できるその一部(例えば、細胞外ドメインの少なくとも20アミノ酸、少なくとも50アミノ酸、少なくとも100アミノ酸、少なくとも150アミノ酸、または少なくとも200アミノ酸)を含む可溶性作用物質などの、リガンド上のCLEC9a結合部位の競合物を含む。
【0183】
CLEC9aおよびそのリガンド間の結合を阻害することができるアンタゴニストは、CLEC9aシグナル伝達を誘導することができる適した死細胞または瀕死細胞(例えば、UV照射された哺乳動物細胞)、その溶解物、抽出物または画分と接触させれば、CLEC9aシグナル伝達を阻害することができるはずである。任意の適した試験系を、この能力を評価するために使用することができる。例えば、CLEC9aを発現する樹状細胞においてCLEC9aシグナル伝達は、Sykキナーゼのリン酸化を測定することによって評価することができる。代替として、実施例に記載したCD3zetaキメラなどのレポーター系に機能的に連結したCLEC9a細胞外ドメインを含む人工レポーター系を使用することができる。
【0184】
結合剤
CLEC9aに対する十分に高い親和性および特異性を有する任意の適した分子を結合剤として使用することができる。分子は、タンパク質、核酸(例えば、アプタマー)、糖質(例えば、オリゴ糖または多糖)、小分子などであってもよい。特に好ましい結合剤は、CLEC9aの生理的リガンド、ならびにCLEC9aに対する抗体およびその機能的断片である。
【0185】
結合剤は、好ましくはCLEC9a、特にCLEC9a ECDに対する、少なくとも105M-1、少なくとも106M-1、少なくとも107M-1、好ましくは少なくとも108M-1、より好ましくは少なくとも109M-1の結合親和性(親和性定数)を有する。
【0186】
結合剤は、好ましくは他のC型レクチンを含む任意のCLEC9aではない分子に対して少なくとも2×、および好ましくは少なくとも5×、少なくとも10×、少なくとも50×のまたは少なくとも100×を超える親和性を有する。
【0187】
全抗体の断片が結合抗原の機能を果たすことができることはよく知られている。機能的結合断片の例は、(i)VL、VH、CLおよびCH1からなるFab断片;(ii)VHおよびCH1ドメインからなるFd断片;(iii)単一抗体のVLおよびVHドメインからなるFv断片;(iv)VHドメインからなるdAb断片(Ward,E.S.ら、Nature 341、544〜546(1989年));(v)単離CDR領域;(vi)2つの連結したFab断片を含む二価断片であるF(ab')2断片;(vii)VHドメインおよびVLドメインがペプチドリンカーにより連結しており、2つのドメインが結合して抗原結合部位を形成することが可能である、単鎖Fv分子(scFv)(Birdら、Science、242、423〜426、1988年; Hustonら、PNAS USA、85、5879〜5883、1988年);(viii)二重特異的単鎖Fv二量体(PCT/US92/0996);(ix)遺伝子融合により構築される多価または多重特異的断片、「ダイアボディ」(WO94/13804;P.Holligerら、Proc.Natl.Acad.Sci.USA 90 6444〜6448、1993年)である。
【0188】
抗体は多くの方法で修飾することができるので、用語「抗体」はしたがって、必要な特異性を有する結合ドメインを有する任意の特異的に結合する物質も含むと解釈されるべきである。したがってこの用語は、天然または合成であるかにかかわらず、免疫グロブリン結合ドメインを含む任意のポリペプチドを含む、上述の抗体断片ならびに抗体の誘導体、機能的同等物および相同体を含む。したがって、別のポリペプチドに融合した、免疫グロブリン結合ドメイン、または同等物を含むキメラ分子が含まれる。キメラ抗体のクローニングおよび発現はEP-A-0120694およびEP-A-0125023に記載されている。
【0189】
本明細書中に記載の本方法において使用される結合剤は一般に、所要の作用を発揮するためにCLEC9aの細胞外ドメインに結合することが必要であることが理解されるはずである。CLEC9aに結合することができる結合剤への参照は、文脈上特に明記しない限り、それに応じて解釈されるべきである。
【0190】
本発明の特定の態様において、抗原(例えば、タンパク質またはペプチド抗原)を記載の結合剤と架橋させることが望ましい。当業者は、適した方法および試薬をよく知っている。結合剤がタンパク質である場合、抗原は結合剤のスルフヒドリル基を介して結合することができる。スルフヒドリル基は、通常遊離していてもよく、または通常結合剤の選択的還元によって露出するジスルフィド結合の一部であってもよい。例えば、抗体は、還元剤メルカプトエタンスルホネートを使用してヒンジ領域において選択的に、穏やかに還元することができる。次いで、抗原を、タンパク質の第3級アミンと反応するヘテロ二官能性架橋試薬、スルホ-SMCCを使用して活性化して、遊離スルフヒドリル反応基を生じさせる。次いで、抗体および活性化された抗原を一緒にインキュベートして、一価抗体とコンジュゲートしたタンパク質を得る18。代替として、抗原由来の適した免疫原性ペプチド配列が既知であれば、遊離スルフヒドリルを有するシステインを含有するこのようなペプチドを合成して、スルホ-SMCC活性化抗体に結合させることができ、これは二価で、1抗体分子につきいくつかのペプチドが結合しているままである。
【0191】
医薬組成物
本明細書中に記載のポリペプチド、抗体、ペプチド、核酸および細胞は、医薬組成物に処方することができる。これらの組成物は、上記物質の1つに加えて、薬学的に許容される賦形剤、担体、緩衝液、安定剤または当業者によく知られている他の材料を含むことができる。このような材料は、非毒性であるべきであり、または活性成分の有効性を妨げるべきではない。担体または他の材料の明確な性質は、例えば、経口、静脈内、皮膚または皮下、経鼻、筋肉内、腹腔内経路などの投与経路に依存し得る。
【0192】
経口投与用の医薬組成物は、錠剤、カプセル、粉末または液体形態であってもよい。錠剤は、ゼラチンなどの固体担体またはアジュバントを含み得る。液体医薬組成物は、一般に、水、石油、動物性または植物性油、鉱物油または合成油などの液体担体を含む。生理食塩水、デキストロースもしくは他の糖溶液、またはエチレングリコール、プロピレングリコール、もしくはポリエチレングリコールなどのグリコールが含まれ得る。
【0193】
静脈内、皮膚もしくは皮下注射、または患部への注射用の活性成分は、発熱物質を含まない、適したpH、等張性および安定性を有する、非経口的に許容される水性溶液の形態であるはずである。当業者は、例えば、塩化ナトリウム注射、リンゲル液注射、乳酸加リンゲル液注射などの等張性ビヒクルを使用して、適した溶液を容易に調製することができる。防腐剤、安定剤、緩衝液、酸化防止剤および/または他の添加剤を、必要に応じて含むことができる。
【0194】
投与は、好ましくは「予防的有効量」または「治療的有効量」(場合によっては、予防が治療とも考え得るが)であることが好ましく、これは個体に対する有益性を示すの十分である。実際の投与量ならびに投与率および投与の経過は、治療されるものの性質および重症度に依存するはずである。例えば、用量の決定などの治療の処方箋は一般開業医および他の医師の責任であり、典型的には、治療する疾患、個々の患者の状態、投与部位、投与方法および開業医に既知の他の要因を考慮する。上記の技術および手順の例は、Remington's Pharmaceutical Sciences、第20版、2000年、pub. Lippincott、WilliamsおよびWilkinsで見ることができる。
【0195】
組成物は、単独でまたは他の治療と組み合わせて、治療される状態に応じて同時にまたは順次に、投与することができる。
【0196】
(実施例)
CLEC9aの配列および構造
NCBI遺伝子データベースの検索は、CLEC9aの配列はハツカネズミ(Mus musculus)、チンパンジー(Pan troglodytes)、ホモサピエンス(Homo sapiens)およびアカゲザル(Macaca mu1atta)において既に同定されていることを示す。マウスCLEC9aのタンパク質配列を使用するblast検索も、ドブネズミ(Rattus norvegicus)、ケイネスファミリアリス(Canis familiaris)およびボスタウラス(Bos Taurus)における予想CLEC9aタンパク質を示す。ヒトcDNA配列および関連ドメインを有する注釈付きタンパク質配列は図1中に詳述する。本発明者らがマウスCD8α+DCからクローニングしたマウスcDNA配列およびその注釈付きタンパク質配列は図2中に詳述する。この配列は、分子の末端へのフレームシフトを引き起こし図2中に示すタンパク質より長いタンパク質をもたらす、追加的G残基を含有する公開cDNA配列とは異なる。本発明者らの配列は正確であるようである。それは公開ゲノム配列(NC_000072.4GI:94471533)に適合し、このページ中で見ることができるように、ゲノム(ATTT)の位置13480は本発明者らのcDNA配列に適合するからである。この配列によって、図1および2中で強調する一種保存チロシンを含有する、C型レクチン様ドメイン(CTLD)、茎部、膜貫通領域および細胞質ドメインを有するC型レクチンファミリータンパク質を予想する。本発明者らは、マウスCLEC9aの3つのアイソフォームに関する転写産物を発見しており、本発明者らは、それらを長いアイソフォーム(エクソン1〜7)、二量体化に関与する推定システインを含むエクソン4を欠く短いアイソフォーム、およびエクソン3〜エクソン7と結合し、発現する場合CLEC9aと膜貫通-細胞内ドメインを共有し得るが、短い異なる細胞外ドメインを有し得る膜貫通タンパク質をコードするmRNAを生成する、非常に短いアイソフォームと名付けている。本発明者らは、長いアイソフォームに関するタンパク質発現の証拠のみを有する。
【0197】
マウスCLEC9aの構造を分析した。コアタンパク質は約29.67KDaの予想分子量(Mw)を有する。しかしながら、HEK-293細胞系中で発現されるとき、Mwは非還元状態で約100KDaであり、かつ還元状態でMwは約45KDaである(図3)。これらの結果は、この分子が茎部中のシステインを介して二量体を形成し、ファミリー中の他のレクチンと同様に、モノマーは強くグリコシル化されることを示す。
【0198】
CLEC9aの発現
マウス脾臓CD11c+CD8+およびCD11c+CD8-細胞のサンプル間の代表的な差分解析の結果として、本発明者らの研究室においてCLEC9aを最初に検出した。結果は、ESTクローンAW318446に相当する配列、CLEC9aに相当する配列がCD11c+CD8+転写産物中で選択的に見られたことを示した。
【0199】
脾臓DCの分類したサブセットにおける転写産物の分析によってCD8+サブセット中でのCLEC9aの高発現が明らかになったが、二重陰性(CD4-CD8-)およびB220+脾臓DCもCLEC9aに関するいくつかの転写産物を示した(図4)。GMCSF由来BMDCにおいて有意な発現は見られなかった(図4)。F1t3L(50ng/ml)の存在下で10日間培養したマウス骨髄は、機能上脾臓の従来のDCに相当しCD8様サブセット19を含むCD11b+、または形質細胞様DC(pDC)の機能的同等物であるB220+のいずれかであるCD11c+細胞を生成する。CLEC9aの高発現は分類したCD11b+のサブセットにおいて見られたが、pDCのサブセットはこの分子のいくらかの発現を示した(図4)。
【0200】
RT-PCRは非常に低レベルの転写産物を検出することができる高感度の技法であるので、それはサンプルの精製の質によって制限される。CLEC9aの発現のパターンを明確に決定するために、本発明者らは、Methodsで示したように、B3Z細胞中で発現されるCD3ζ-NKRP1-CLEC9aキメラおよびβ-Gal受容体(図5)を使用して、マウスCLEC9aに対するラットモノクローナル抗体(mAb)を作製した。本発明者らは1F6、397、および7H11という名称の3つのモノクローナル抗体を選択した。これらのモノクローナル抗体を使用して、本発明者らは、マウス脾臓および骨髄中の分子の発現のパターンを試験した。CLEC9aはCD8α+従来型DCにおいて高度に発現され(MFI約350〜400)、pDCにおいて適度な発現(MFI約65〜70)を示した(図6)。B細胞、T細胞、NK細胞、NKT細胞、単球、マクロファージおよび顆粒球を含めた調べた他の細胞型においてこの分子は検出しなかった。
【0201】
in vitroでのCLEC9a機能の分析用のモデルとして、本発明者らはマウスGMCSF-およびF1t3L-由来BMDC中の発現を分析した。本発明者らは、GMCSF-由来BMDC中で分子の発現は検出しなかったが、一方CLEC9aはCD8+脾臓DC19と機能的に相同なFlt3L BMDCのCD11blo、CD24hi、B220-サブセット中、およびpDCと同等なCD11bloB220+のサブセット中でも選択的に発現された(データ示さず)。
【0202】
CLEC9aはSykキナーゼを介してシグナル伝達し、DC活性化を促進する。配列分析によって、推定SH2結合ドメインおよび/またはチロシンベースの分類シグナルとして働き得る、同じ構造EXXYXXLを有する全種で保存されたマウス由来のTyr7が明らかになる。この配列はマウスデクチン-1におけるSykの結合を可能/仲介し、「HemITAM」22と呼ばれる。しかしながら、この推定配列は必要であるが、Sykの結合を予想するには十分ではない。したがって本発明者らは、リン酸化Tyr7を発現するマウスCLEC9a、またはリン酸化無しかまたはさらにPheに突然変異したTyr7を有するマウスCLEC9aの細胞質尾部を有するビオチン化ペプチドを設計した。Tyr7リン酸化ペプチドは、陽性対照23としてリン酸化Tyrをいずれも発現するマウスデクチン-1由来の細胞質尾部と同程度、組換えSykを破壊することができた(図7)。
【0203】
CLEC9aがシグナル伝達受容体として実際作用するかどうかを決定するために、本発明者らは、前に記載したラット抗マウスCLEC9aモノクローナル抗体を使用した。本発明者らは、LK細胞、CLEC9a発現に陰性であるが内因性レベルのSyk23を含有するマウスB細胞系において、CLEC9aを発現するトランスフェクタントを作製した。試験した全3個のプレート抗CLEC9a抗体を用いたCLEC9aの誘発は、LK細胞中でのSykおよびErkのリン酸化をもたらした(図8)。
【0204】
SykがCLEC9aのシグナル伝達に必要であるかどうかを決定するために、本発明者らは、Sykを発現しない受容体T細胞系B3Zの安定したトランスフェクタントを使用した。CLEC9aのシグナル伝達に対するTyr7の貢献を、Pheに突然変異したTyr7を有するCLEC9aの突然変異型(CLEC9a Y7F)を使用して分析した。本発明者らはCLEC9a wtまたはY7FをB3Z細胞に形質導入し、Sykキナーゼを同時形質導入したか、またはしなかった。プレートした抗CLEC9aモノクローナル抗体は、C9のwt細胞質尾部を介してB3Z-C9wt-SykにおいてNFATの活性化を誘導したが、SykまたはY7のいずれかの不在下では誘導しなかった(示さず)。
【0205】
CLEC9aを介した免疫調節性サイトカイン/同時刺激性分子の調節。
CLEC9aを介したシグナル伝達が調節性サイトカインの産生、または発現する場合細胞中での同時刺激性分子の発現に貢献し得るかどうかを決定するために、本発明者らはwt CLEC9またはY7F突然変異体でLKトランスフェクタントを分析した。397抗CLEC9a、および低い程度で7H11を発現するハイブリドーマ細胞は、Y7F突然変異体において無効であったCLEC9a分子による特異的IL-2産生を誘発した(図9)。DEC-205対照ハイブリドーマはいかなる応答も誘発せず、1F6抗CLEC9aは低い応答を誘発し、このことはこれらの抗体がサイトカイン産生に関して差次的に挙動することができ、CLEC9a標的化の展望から非常に魅力的であるという可能性を開く(アゴニスト抗体、397、対遮断抗体、1F6)。
【0206】
CLEC9a WT尾部活性化に関する要件を決定するために、本発明者らは、Sykを発現しない受容体T細胞系B3Zの安定したトランスフェクタントを分析した。CLEC9aのシグナル伝達に対するTyr7の貢献を、Pheに突然変異したTyr7を有するCLEC9aの突然変異型(CLEC9a Y7F)を使用して分析した。本発明者らはCLEC9a wtまたはY7FをB3Z細胞に形質導入し、Sykキナーゼを同時形質導入したか、またはしなかった。プレートした抗CLEC9aモノクローナル抗体は、C9のwt細胞質尾部を介してB3Z-C9wt-SykにおいてNFATの活性化を誘導したが、いずれかのSykの不在下、またはY7突然変異体を使用したときは誘導しなかった(図10)。
【0207】
Flt3L BMDCはIL12-23 p40タンパク質の産生によってプレートした抗CLEC9aモノクローナル抗体に応答する(図11)。
【0208】
これらの結果は、CLEC9aがDCを活性化することができるシグナル伝達分子であることを実証する。
【0209】
CLEC9aはin vivoでCD11c+CD8+DCを選択的に標的化するエンドサイトーシス受容体である
Flt3L BMDCにおいて発現される内因性CLEC9aによって抗CLEC9aモノクローナル抗体が内在化される可能性をFACSによって分析し、CLEC9aがエンドサイトーシス分子であることを明らかした。共焦点分析によって、細胞内区画への抗体の標的化を示した(データ示さず)。これらの結果は、CLEC9aはエンドサイトーシス受容体であり、それは抗原と結合した抗体によって標的化して(腫瘍/ウイルス予防接種)、分子を選択的に発現する細胞サブセットに薬剤を特異的に送達することができることを示唆する。CLEC9aモノクローナル抗体がin vivoで標的化ツールとして働くかどうかを決定するために、本発明者らは7H11-Alexa-488またはアイソタイプ対照を静脈内注射した。16時間後、本発明者らは、全脾細胞および抗体がCD8α+DC(MFI約350〜400)、および低い親和性で、PDC(MFI約65〜70)を選択的に標的化したと分析した。抗ラットCy5を用いた脾細胞の標識は、大部分のラット抗マウスCLEC9aモノクローナル抗体は、それは抗ラット二次試薬と同時染色しなかったので、取り込まれたことを示唆した(示さず)。
【0210】
CLEC9aによる標的化が、in vivoで特異的サブセットによる抗原のプロセシングおよび提示をもたらすかどうかを調べるために、本発明者らは、Methodsで示したように、CTL応答に関する免疫優勢ペプチドのビオチン化誘導体とOVAタンパク質(SIINFEKLC-biot、S1と呼ぶ)、アイソタイプ対照または抗CLEC9aモノクローナル抗体を結合させた。ペプチドのビオチン化によって本発明者らは、全ての場合1抗体当たり1〜1.2のペプチドが存在したと決定することができた。5μgのS1結合抗CLEC9aまたはS1-アイソタイプ対照のいずれかをマウスに静脈内注射した。翌日、脾細胞をCD11c+またはCD11c-サブセットにおいて増大させ、OT-I T細胞増殖およびサイトカイン産生を誘導するそれらの能力に関して試験した(図12aおよびb)。抗CLEC9aのみがCD11c+細胞を標的化し、OT-I細胞による増殖およびIFN-γ産生をもたらし、CLEC9aがCD11c+DCを特異的に標的化し、抗原特異的T細胞に対する提示をもたらすことを示す。
【0211】
樹状細胞のどのサブセットが抗C9によって標的化されるかをさらに決定するために、S1結合抗C9を注射したマウスを、前述のように従来型DCの3つの主要サブセットに分類し、OT-I T細胞を用いて試験した。DCのCD8+サブセットのみがT細胞による増殖およびIFN-γ産生を仲介し、CLEC9はCD11c+CD8+DCを特異的に標的化し、抗原特異的T細胞のプライミングをもたらすことを確認した(図12cおよびd)。
【0212】
抗CLEC9aモノクローナル抗体および抗CD40を使用するin vivo標的化はCTLのプライミングおよび腫瘍拒絶をもたらす
本発明者らは、in vivoでのCLEC9aの標的化が特異的T細胞の活性化を誘導し得るかどうかを調べた。アイソタイプ対照ではなく2μgの抗C9-S1の注射は、抗CD40と同時投与したとき、in vivoにおいて内因性レパートリーからの特異的CTL活性の誘導をもたらす(図13)。この挙動は、in vivo死滅アッセイにより証明した前に記載した24抗DEC205-S1で観察した挙動と同様である(図13A)。
【0213】
対照モノクローナル抗体と結合したS1を与えたマウスは、抗CD40の同時投与と無関係に標的細胞を除去しなかった。対照的に、抗CLEC9aおよび抗CD40と結合したS1を与えたマウスからは、標的細胞は完全に除去された。抗CD40モノクローナル抗体を削除したとき応答は見られなかった。標的細胞の除去と一致して、相当数のテトラマー陽性OVA/H-2 Kb特異的CD8+T細胞を、抗CLEC9a-S1および抗CD40を与えていたマウスの脾臓および血液のみで発見した。in vitroでのSIINFEKLペプチドを用いた同じ細胞の再刺激は、二次増殖、ならびにIFN-γ産生および特異的死滅活性をもたらした。SIINFEKLエピトープを含有するOVAの長鎖ペプチドと結合した抗CLEC9aを使用して同一の結果を得た(「S2」、SIINFEKLTEWTSSNVMEERC;図13B)。
【0214】
特に、遊離S1ペプチドは、抗CLEC9a-S1結合体中に存在する量の100倍過剰で与えたときでさえ、in vivo死滅応答を誘導することまたは相当数のテトラマー陽性細胞を誘発することができなかった(示さず)。本発明者らは、CLEC-9aおよび適切なアジュバントへの外因性抗原の標的化は、CD8+T細胞の有効なクロスプライミングを可能にすると結論付ける。
【0215】
CTL活性のCLEC9aプライミングが腫瘍治療をもたらし得るかどうかを決定するために、本発明者らはB16メラノーマ肺転移のモデルを使用した。本発明者らは2×105個のB16-OVA-GETメラノーマ細胞を静脈内接種し、6日後、S1 SIINFEKL誘導体(10μg)および抗CD40(25μg)と結合した異なる抗体を脚部に皮下注射した(図14)。腫瘍の注入後18日で、肺腫瘍の数を分析した。これらの結果によって、抗CLEC9a+抗CD40は腫瘍治療に有効であることが明らかになった(p<0.001、一元配置ANOVA)(図14)。
【0216】
本発明者らは実験を延長して、抗CLEC9a標的化をさらに使用して、B16腫瘍関連抗原(25〜27)として働くことができる内在性メラニン細胞分化タンパク質に対する免疫応答を誘導することができるかどうかを決定した。本発明者らは、gp100、TRP-1およびTRP-2(25〜27)由来のH-2KbおよびH-2Db拘束性抗原エピトープを含むビオチン化ペプチドを合成し、これらを抗CLEC-9aと共有結合させ、抗体結合体ならびにアジュバントとしてポリI:Cおよび抗CD40でマウスを免疫処置した。図16中に示すように、B16メラノーマ細胞移動の3日後に治療的に与えた1回用量のワクチンは、肺擬似転移のほぼ完全な根絶を誘導した。これはメラノーマ抗原に対する強力なIFN-γ応答の誘導によって実施した(図16)。対照的に、(対照アイソタイプ適合モノクローナル抗体と結合した)非標的型の同じ抗原は、防御またはIFN-γ応答を誘導することはできなかった(図16A、B)。B16攻撃前にワクチンを与えた予防モデルにおいて同様の結果を得た(図16C)。本発明者らは、CLEC-9a標的化による特異的CTLのプライミングは、マウス腫瘍に対する予防的または治療的ワクチン接種に使用することができると結論付ける。
【0217】
ヒトClec9aの発現は血中DCの小サブセットに限られる
これらの発見をヒトに広げるために、本発明者らはhClec9aをクローニングし、それに対するマウスモノクローナル抗体を作製した(Materials and Methodsを参照)。これらのモノクローナル抗体の1つを選択して、ヒト末梢血単球細胞間のClec9a発現のパターンを分析した。ヒトClec9aの発現は、リンパ球、単球、NK細胞および系統陰性HLADR-細胞からはなかった(図15A)。GM-CSFおよびIL-4における培養によって生成した単球由来DC中でもそれは検出しなかった(データ示さず)。しかしながら、Clec9aの発現は、系統陰性HLA-DR+細胞で定義したように、血中DCの別の亜集団において明らかであった(図15A)。
【0218】
CD123+pDCの集団およびCD16、CD1b/c、BDCA-3、およびCD34(22)の発現に基づいて区別可能な推定ミエロイドCD123-DCの異なるサブセットを含めた、血中DCの5つの異なるサブセットを報告している。DCのClec9a+亜集団はCD123に陰性であり、マウス中でのpDCと異なり、ヒトpDCはClec9aを発現しないことを示唆した(図15B)。Clec9a+血中DCはCD34、CD16およびCD1b/cにも陰性であった。しかしながら、Clec9a+DCはBDCA-3に均一に陽性であった(図15B)。したがってヒトClec9aは、BDCA-3+DCの異なる集団を選択的に示す。
【0219】
最後に、本発明者らは、ヒトCLEC-9aは、そのマウスオルソログと同様に、DCにおいてエンドサイトーシス受容体として機能することができるかどうか評価した。BDCA-3+DCはAlexa 488標識抗CLEC-9aを用いて4℃で染色した。1時間後、4℃ではなく37℃において、細胞内区画中の蛍光を発見した(示さず)。したがって、ヒトCLEC-9aはBDCA3+DCにおける結合抗体のエンドサイトーシスを仲介し、それによってヒトCLEC-9aは、ヒト中でのこれらの細胞への抗原標的化に使用することができることを示唆する。
【0220】
死/死滅細胞はCLEC9Aのリガンドを発現する。
CLEC9A-CD3ζキメラを含有するBWZ細胞は短い生成時間を有し、容易に異常増殖し、培養中に相当な割合の死滅細胞を生成する可能性がある。本発明者らは、任意の追加的な刺激無しでCLEC9Aを発現するBWZトランスフェクタントにおいて基底活性化を観察し、BWZ培養中の死滅細胞の数と関連付けた(図17a)。
【0221】
これらの結果を確認するために、本発明者らは、BWZ-CLEC9A-ζトランスフェクタントに加えたときに応答を誘導しない細胞系を使用した。LK細胞はレポーター細胞に曝したときに応答を誘導しない。しかしながら、LK細胞をUV照射して細胞死を誘導すると、それらはレポーターの強力なインデューサーに変わり(図17b)、UV処理後に改変した細胞はCLEC9Aのリガンド(複数可)を発現することを示唆した。二価抗体はB3Z細胞系中で発現される分子の架橋およびレポーターの活性化を誘発するので、相互作用の特異性を実証するためにこの系において遮断抗体を使用することはできない。しかしながら、抗CLEC9A抗体の一価Fab断片はレポーターの活性を誘発せず、かつ種特異的な形式でUV処理細胞による誘導を遮断し(図17b)、抗体はCLEC9A受容体の遮断に特異的に作用してリガンドとの相互作用を回避することを実証した。
【0222】
この結果は、他の独立したUV照射細胞型(ネズミ3T3、LK細胞、MEF、EL-4)、ラットRBL細胞、およびヒトHEK-293において確認した。細胞を(UV誘導型DNA損傷およびアポトーシスを妨げる)カフェインとプレインキュベートし、次いでUV放射に曝すと、リガンドの露出は誘導されない。
【0223】
本発明者らはLK細胞を異なる線量のUVに曝し、かつ本発明者らは、LK細胞中のリガンドの発現は死滅LK細胞の数と相関関係があることを発見し(図17c)、リガンドはこの集団中で選択的に発現されることを示した。
【0224】
これを独立形式で確認するために、本発明者らは、それぞれモノビオチン化用のBirA配列と結合した、マウスCLEC9Aの組換え可溶性細胞外ドメイン(rsCTLD)、および対照としてマウスデクチン-1を作製した。モノビオチン化rsCTLDを使用してPE-テトラマーを作製した。本発明者らは24時間前にUVで処理した細胞を染色し、かつ本発明者らは、CLEC9A rsCTLDテトラマーとTO-PRO3陽性細胞の特異的結合を検出した(図17d、ドットプロット)。対照として、デクチン-1 rsCTLDテトラマーは死滅細胞と結合しなかったが、それらはその特異的リガンドザイモサンとは結合した(図17d、ヒストグラム)。
【0225】
UV処理はDNA損傷および一連の関連ストレスマーカーを誘導するので、本発明者らは、リガンドの誘導はDNA損傷によって引き起こされたか、または大部分は細胞死に関与するプロセスによって引き起こされたかどうか試験した。DNA損傷誘導試薬だけでなく、免疫細胞死をもたらす原発性壊死を促進することが示されている、血清枯渇またはさらに凍結解凍もリガンドの露出をもたらした(図17e)。しかしながら、寛容原性で働くことが示されている細胞の即死を誘導する浸透圧ショックは、CLEC9Aリガンドを露出しなかった(図17eおよびf)。結論として、特定の型の原発性および続発性壊死の後にCLEC9Aリガンドが細胞中で露出される。さらに本発明者らは、細胞の固定(または固定および透過処理)は細胞をTO-PRO3に透過可能にする変化を瞬時に促進し、CLEC9Aのリガンドを「露出する」ことを見出しており(図20)、リガンドの合成はUVに対する損傷応答の結果として誘導されないが、特定の刺激に応答する死のプロセスの結果としてリガンドが露出されることをさらに実証している。
【0226】
CLEC9Aは死細胞により樹状細胞に送達されるアジュバントシグナルを仲介する。
UV死滅細胞はSykおよびY7依存形式でCLEC9Aの細胞質ドメインを介してシグナル伝達する(図21)。この系によって本発明者らは、抗CLEC9A抗体がこの相互作用の特異的遮断試薬として働き得るかどうかを調べることができ、かつ本発明者らは、可溶形のFab抗体と完全抗体の両方が強力な遮断試薬であったことを見出した(図22)。樹状細胞における死細胞の影響およびin vitroにおけるそれらのエフェクター機能を試験するために、本発明者らは交差提示アッセイを設計し、その中ではOVAタンパク質を導入したUV処理bm-1細胞を、遮断抗CLEC9Aの有無の下でFlt3L BMDCと相互作用させることが可能であった。bm-1細胞はB6ハプロタイプ由来であるが、OVAの免疫優勢クラスIペプチド(SIINFEKL)と結合しない突然変異H2Kbを発現する。クロスプライミングに関するDCの能力の読み出しとして、特異的OT-I細胞をアッセイに加えた。交差提示された抗原の量に関する読み出しとしてのOT-I細胞の増殖は、大きく影響は受けなかった(図18a)。しかしながら、アジュバント効果により活性化されるDCによって促進される助力に依存するOT-I細胞によるサイトカイン産生は、遮断CLEC9A抗体を使用したとき大幅に阻害された(図18aおよびb)。
【0227】
これらの結果を確認するために、本発明者らは、Clec9aプロモーター(Clec9aegfp/-を)の調節下でEGFPを発現するCLEC9A欠損マウスを作製した。本発明者らはCLEC9A欠損または非欠損Flt3L BMDCを作製し、かつ本発明者らは、死細胞の摂取が影響を受けたかどうか分析した。図18cは、CLEC9A+とCLEC9A- Flt3L BMDCの間の死細胞の摂取に関する能力の間に差がないことを示す。次いで本発明者らは、UV処理bm-1細胞中に導入したかまたはUV処理したbm-1MEF中の非分泌型OVA-GFP融合タンパク質において細胞内で発現されたいずれかのOVAタンパク質との交差提示における、Flt3L BMDC欠損CLEC9Aの影響をアッセイした(図18d)。OT-I増殖は影響を受け、抗体遮断より交差提示の遮断において大きな影響を示唆した。しかしながら、bm1MEFにより発現されたOVAを含めたOVAの高用量では、増殖は影響を受けず、IFNγ産生は大幅に阻害された(図18dおよびe)。これらの結果は、DC中のCLEC9Aの不在下におけるin vitroでのクロスプライミング中に死細胞と関係があるアジュバント効果の遮断があることを示唆する。
【0228】
CLEC9Aが免疫細胞の死を感知してin vivoでのクロスプライミングを促進する
CLEC9Aを発現するCD8α+DCは、in vivoでの死滅細胞関連抗原とのクロスプライミングを促進することを特徴とする主な細胞型であるので、本発明者らは、この機能におけるCLEC9A遮断の影響を試験した。OVAを発現するUV処理bm1 MEFを与えたマウスは、6日後内因性レパートリーからのOVAに対してCD8+T細胞の増大を示した(図19a、左図)。これらのCD8+T細胞はSIINFEKLに応答してIFN-γを産生することができ、それらがエフェクター細胞であること、および死滅細胞のアジュバント活性と関係がある抗原の免疫原性担体として死滅細胞が働いたことを示した(図19a、右図)。アイソタイプ対照ではなく抗CLEC9A遮断抗体を用いた予備処理は、特異的CD8応答とそのエフェクター活性の両方を遮断した(図19a)。
【0229】
死滅細胞関連抗原とのクロスプライミングと関連がある死滅細胞の免疫原性のプロセスにおけるCLEC9Aの正確な役割を決定するために、本発明者らはCLEC9aegfp/-マウスにおいてアッセイを利用した。その結果は、CLEC9Aの不在下でin vivoでの死滅細胞関連抗原とのクロスプライミングの非常に有意で部分的な阻害を示した(図19b、左図)。ノックアウトマウスを混合型C57BL/6-129バックグラウンドで作製し、C57BL/6(N3)で戻し交配しているので、本発明者らはその個々を図19b、左図においてプールした12匹のメス同腹子を分類し、かつ本発明者らはCLEC9A+マウスとCLEC9A-マウスの間の平均を比較し、12匹の同腹子のうち12匹が、平均30.05%の阻害で低減を示したことを示した(p<0.0001、スチューデントのt検定)(図19b、右図)。図19b、右図中に示すように、異なる同腹子間のバックグラウンド浸透度の違いは、同腹子間のクロスプライミング能力の有意な変動性を説明し、一部分のみではあるが依然として非常に有意である、CLEC9A+マウスとCLEC9A-マウスの間の真の違いを抑えることができた。さらに、ex vivoでのSIINFEKLに応じたIFNγ産生は大幅に影響を受け、CLEC9Aの不在下での死滅細胞関連抗原とのクロスプライミングによって生成するエフェクター応答の欠損を示した(図19c)。結論として、CLEC9A欠損は死滅細胞関連抗原の低下したアジュバント性をもたらし、これは死滅細胞関連抗原に対する特異的T細胞エフェクター応答の遮断をもたらす。
【0230】
方法
マウス
C57BL/6マウス、Rag-/-C57BL/6バックグラウンドのOT-IマウスおよびB6.SJLバックグラウンドのマウス(コンジェニックCD45.1+)を、特定病原体未感染条件で英国癌研究所において飼育した。Kbm-1マウスはJackson laboratoryから購入し(Bar Harbor、Maine;ストック番号001060)、C57BL/6マウス、Rag-/-C57BL/6バックグラウンドのOT-Iマウス、MyD88-TRIF二重ノックアウト、Clec9a+、およびClec9a-/-マウスと一緒に特定病原体未感染条件で英国癌研究所において飼育した。前に記載されたように(Turnerら、Nature 378、298(1995年))骨髄キメラをSyk欠損胎児肝細胞から作製した。全ての動物実験は、動物ケアに関する国内および機関ガイドラインに従って実施した。
【0231】
試薬
培養培地はグルタミン、ペニシリン、ストレプトマイシン、2-メルカプトエタノール(いずれもInvitrogenから)および10%熱不活性化ウシ胎児血清(Bioclear)を補充したRPMI1640(Invitrogen)であった。フローサイトメトリー分析実験に使用した抗体はBD Pharmingenからのものであり、CD11c(クローンHL3、ハムスターIgG1)、CD24、CD11b、B220、Ly6C、CD4(RM4-5、ラットIgG2a)、CD8に特異的な抗体を含んでいた。ELISAに使用した抗体は捕捉IFN-γ(XMG1.2、ラットIgG1)検出/IL12-23 p40であった。精製した2.4G2(抗FcgRIII/II、ラットIgG2b、非特異的抗体結合を遮断するために使用した)は、英国癌研究所の抗体産生サービスからのものであった。フローサイトメトリー用に、2mMのEDTA、1%のFCSおよび0.02%のアジ化ナトリウムを補充した氷冷PBSで細胞懸濁液を染色した。データをFACSCalibur(BD Biosciences)で得て、FlowJoソフトウェア(Treestar、San Carlos、CA)を使用して分析した。
【0232】
細胞
マウス骨髄由来のDC(BMDC)をGM-CSFを使用して生成し、抗CD1cマイクロビーズを用いた磁気選択によってバルク培養物から精製した(GM-CSF BMDC)。あるいは、10日間100ng/mlのFlt3L(R&D)の存在下で骨髄細胞を培養することによってBMDCを生成し、この時間まで全ての生存細胞はCD11cに陽性であった(Flt3L BMDC)。脾細胞はリベラーゼ/DNAse消化によって調製し、抗CD11cマイクロビーズを用いた正の選択によってDCを増大させた。OT-IT細胞(リンパ節および脾臓由来)は、ビオチン化抗体(抗CD11c、CD11b、B220、FcγR、Gr-1、およびCD4)のカクテル、次にストレプトアビジンマイクロビーズを使用した負の選択によって精製した。ヒト末梢血単球細胞(PBMC)は、Ficoll-Hypaque(GE-Healthcare)での沈降作用によって1のドナー白血球軟膜(National Blood Transfusion Service)から調製した。
【0233】
PCR/RT-PCR
全RNAは抗CD11cマイクロビーズ(Miltenyi)を用いてCD11cで増大した脾臓DCのサブセットからトリゾール(Invitrogen)を使用して抽出し、FacsAriaソート(BD)でCD4およびCD8発現に関して分類した。さらに、GMCSFおよびFlt3Lin vitro由来BMDCの分類したサブセットからRNAを抽出した。RNAをDNAse消化(DNAフリー、Ambion)によって調製し、SuperscriptII逆転写酵素(Gibco)、1μMのdNTPおよび10μMのランダムヘキサヌクレオチド(Gibco)を使用して逆転写した。cDNAは30秒間94℃、30秒間55℃、1分間72℃からなる35PCRサイクルを使用して増幅した。プライマーの配列は:mCLEC9a順方向5'AGACTGCTTCACCACTCCAA;mCLEC9a逆方向Rv:5'CTTGGCACAATGGACAAGGT;b-アクチン順方向:5'GTTTGAGACCTTCAACACCCC、b-アクチン逆方向:5'GTGGCCATCTCCTGCTCGAAGTC;hClec9a正方向:5'CCCAAGTCTCATTTGGAGGA;hClec9a-1逆方向:5'AAATCTGGACGGTGTGGAAGであった。
【0234】
抗CLEC9aモノクローナル抗体の作製
ネズミClec9aに対するモノクローナル抗体
ウイスターラットをHAエピトープと融合したCLEC9aでトランスフェクトしたRBL-2H3細胞で免疫処置し、過免疫したラット由来の脾細胞とラットメラノーマ細胞系Y3の融合は、標準的な手順に従い実施した。陽性の検出用に、本発明者らは、CLEC9aの細胞外ドメイン、NKRP1由来の膜貫通領域およびCD3ζの細胞内尾部および次にIRES-GFPを有するキメラを発現するB3Z細胞系25、記載されている戦略21を使用した。mCLEC9aに関する融合キメラのcDNA配列は図3中に示す。B3Z細胞系はβ-Gal活性と関係があるNFATに関するレポーターを含有し、このキメラ分子の任意の関与がNFATおよびレポーターの活性化をもたらし、したがってこれはb-Gal活性に関する標準的なアッセイに従い明らかにすることができる。抗体に関するスクリーニングは、親細胞系と比較したキメラを発現するB3Zの機能活性化によるものであった。陽性として選択した抗体は、CLEC9aキメラ発現細胞(EGFP+)と比較して親細胞系(EGFP-)においてFAGS分析によって確認した。この方法は、図4中に示した1F6、397、および7H11という名称の3つのモノクローナル抗体の選択を可能にした。次いでモノクローナル抗体を染色用のビオチンまたはAlexa488(Invitrogen)と結合させ、またはS1ペプチドとの結合用に使用した。
【0235】
ヒトClec9aに対するモノクローナル抗体
HAエピトープと融合したヒトClec9aを発現するRBL-2H3細胞を用いて3〜4回BALB/cマウスを免疫処置した。脾細胞とマウスメラノーマ系SP2/0の融合は、標準的な手順を使用して実施した。ハイブリドーマスクリーニング用に、本発明者らは、NFATに関するβ-galレポーターを発現するB3Z細胞系を使用した(23)。NKRP1B由来の膜貫通領域と融合したヒトClec9aの細胞外ドメイン、およびCD3ζの細胞内尾部、次にIRES配列およびGFP遺伝子のキメラをコードするレトロウイルスを、この細胞系に形質導入した(24)。ハイブリドーマ上清を、Clec9aキメラと結合しNFATレポーターの活性化およびβ-gal活性の誘導をもたらす能力に関してスクリーニングした(24)。このアッセイ中で陽性反応を示した上清を、キメラClec9aを発現するB3Z細胞(GFP+)と親B3Z細胞(GFP-)の混合物を使用したフローサイトメトリーによって、さらにスクリーニングした。この方法は、hClec9aに特異的な1つのマウスモノクローナル抗体の選択を可能にした(8F9(IgG2a))。
【0236】
フローサイトメトリー
マウスCD11c、CD24、CD11b、B220、Ly6C、CD4およびCD8αに特異的なフルオロクロムまたはビオチン標識抗体は、BD Pharmingenからのものであった。精製2.4G2(抗FcγRIII/II)は、英国癌研究所の抗体産生サービスからのものであった。マウス細胞懸濁液を10μg/mlの2.4G2モノクローナル抗体と共にインキュベートしてFcγ受容体を遮断し、次いで2mMのEDTA、1%のFCSおよび0.02%のアジ化ナトリウムを補充した氷冷PBSで染色した。エンドサイトーシス試験用に、4℃において30分間5μg/mlのビオチン化抗Clec9aモノクローナル抗体でFcγ遮断細胞を標識した。次いで細胞を二回洗浄し、氷に移しストレプトアビジンPEを加える前に、4℃または37℃において異なる回数インキュベートした。in vivo標識試験用に、Alexa-488結合抗Clec9aまたはアイソタイプ適合対照モノクローナル抗体を示した用量で静脈内注射し、組織を調製し、16時間後に分析した。ヒトCD3、CD14、CD19、CD56、HLA-DR、CD34、CD123およびCD16に特異的な抗体はBDから購入し、かつCD1b/c、およびBDCA-3はAbCam(ケンブリッジ、英国)から購入した。ヒト単球細胞は100μg/mlのヒトIgG(Sigma-Aldrich)で遮断し、前述のように染色した。データをFACSCalibur(BD Biosciences)で得て、FlowJoソフトウェア(Treestar、San Carlos、CA)を使用して分析した。
【0237】
BM-DCの培養および刺激
GM-CSF BM-DCを記載したように生成し26、使用前に抗CD11cマイクロビーズを用いてバルク培養物からDCを精製した(Miltenyi Biotec)。BM-DCの純度はFACSによって調べ、常に98%を超えていた(データ示さず)。F1t3LBMDCは、10日間75ng/mlまたは50ng/mlのF1t3L(R&D)の存在下で培養した骨髄から生成した。サイトカイン産生および表面マーカー発現の分析用に、ウエル当たり5〜10×104個のFlt3L BM-DCを、アイソタイプ対照または抗CLEC9aモノクローナル抗体で事前にコーティングした96ウエル平底プレート中のFlt3Lを含有する200mlの培養培地中で18〜24時間培養した。上清中のサイトカインレベルを、いずれもBDからの捕捉用抗IL-12 p40-p70(C15.6)および検出用ビオチン抗IL12p40-p70(C17.8)を使用してサンドウィッチELISAによって測定した。F1t3LBMDCにおけるエンドサイトーシス用に、FcγRは10μg/mlの2.4G2モノクローナル抗体で遮断し、次いで細胞を5μg/mlのビオチン化モノクローナル抗体と共に30分間4℃または37℃において培養し、洗浄し、4℃において二次試薬(ストレプトアビジンPE)で同時に洗浄および添加する前に、アッセイ温度で1.5時間または0.5時間放置した。共焦点顕微鏡による分析用に、Alexa 488結合モノクローナル抗体を4℃または37℃において30分間5mg/mlでFcγR遮断F1t3L-BMDCに加え、次いで洗浄し、ポリ-L-リシンコーティングカバースリップに接着させ室温において20分間2%PFA中で固定する前に、アッセイ温度でさらに1.5時間放置した。Fluoromount(Southern Biotech、Birmingham、AL)でスライドにサンプルを載せた。共焦点系の微分干渉コントラストおよび蛍光画像を、63°-Plan-ApochromatNA1.4油浸対物レンズを有するレーザー走査共焦点顕微鏡(Axioplan2、Zeiss、ドイツ)を用いて同時に得た。画像解析はLSM510ソフトウェア(Zeiss、ドイツ)で実施した。
【0238】
ペプチドの破壊およびウエスタンブロッティング
ペプチド破壊のために、溶解バッファー中に希釈する前に、ビオチン結合ペプチドを40%のDMSO中に溶かした。溶解バッファー中に希釈した組換えヒトSyk(Upstate)は、CLEC9aおよびデクチン-1細胞内尾部に相当する示したビオチン化ペプチド(英国癌研究所ペプチド合成研究室)およびストレプトアビジン-セファロース(Sigma Biosciences AB、Uppsala、スウェーデン)と共にインキュベートした。親和性による精製後、セファロースビーズを溶解バッファー中で1回洗浄し、10%のβ-メルカプトエタノールを含有するSDSゲル導入バッファー中で煮沸した。タンパク質はドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動(SDS-PAGE)によって分離し、Immobilon PVDF膜(Millipore Corporation、Bedford、MA)に移し、ウサギ抗Syk(ネズミSyk20のアミノ酸318〜330に相当する合成ペプチドに対して産生した2131血清と、ヒトSykのC末端に相当する合成ペプチドに対して産生したCell Signaling Technology、Inc.からの抗Syk、カタログ番号2712の組合せ)でプローブ処理し、次に化学発光を検出した。
【0239】
LK細胞の活性化アッセイでは、示した時間抗C9またはアイソタイプ対照でコーティングした6ウエルプレートにLK細胞を平板培養した。細胞抽出物を溶解バッファー(50mMのHEPBS[pH7.4]、150mMの塩化ナトリウム、100mMのフッ化ナトリウム、10mMのピロリン酸四ナトリウム、1mMのオルトバナジン酸ナトリウム[pH10.0]、1mMのBDTA[pH8.0]、1.5mMの塩化マグネシウム、10%のグリセロール、1%のTritonX-100、1mMのPMSF、および「完全」プロテアーゼ阻害剤カクテル錠剤[Roche])中で調製し、不溶性物質を廃棄し、一定量の溶解物を前に記載したSDS-PAGEによって調べた。ウエスタンブロッティング用の前述のウサギ抗Sykおよび抗P-Syk、抗Erk、抗P-ErkはCell Signalingからのものであった。
【0240】
B3ZおよびBWZ細胞におけるNFATレポーターアッセイ
LacZ活性と関係があるNFATのレポータープラスミドを含有するB3Z細胞は以前に記載されている25。CLEC9a WTまたは突然変異型Y7F(Stratagene)を、wtまたはSyk形質導入B3Z細胞に形質導入した。細胞はアイソタイプ対照または抗CLEC9aモノクローナル抗体でコーティングした96ウエルプレート中に平板培養し、一晩培養した後、PBS中で洗浄し、記載したように21CPRG含有バッファー中に溶かした。4時間後、参照としてOD655を使用してA595を測定した。
【0241】
CLEC9Aリガンドを検出するために、NKRP1B由来の膜貫通領域と融合したマウスまたはヒトCLEC9Aの細胞外ドメイン、およびCD3ζの細胞内尾部、次にIRES配列およびGFP遺伝子のキメラをコードするレトロウイルスを、BWZ細胞系に形質導入した(24)。CLEC9Aキメラと結合したリガンドは、NFATレポーターの活性化およびβ-gal活性の誘導をもたらす可能性がある。マウスおよびヒトCLEC9A-CD3ζキメラ受容体を発現するBWZ細胞の基底活性化をアッセイするために、4、2、1、または0.5×106個の細胞/mlを6ウエルプレート中の3mlにおいて培養して2日間(異常)増殖させた。死滅細胞の頻度はTO-PR03色素(Invitrogen)を使用して決定し、生存細胞は96ウエル平底プレートにおいて2×105個/ウエルで新たな培地中に平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0242】
異なる細胞型におけるリガンドの露出を決定するために、LK(B細胞系)、標準的なプロトコルを使用してbm-1マウス{Ref}から誘導したSV-40不死化マウス胎児線維芽細胞(MEF)、RBLラット白血病細胞、3T3線維芽細胞、およびHEK-293ヒト胎児腎臓細胞をUV照射し(240mJ/cm2)、対照Fabまたは抗マウスCLEC9A(1F6)および抗ヒトCLEC9A(8F9)Fab(Sanchoら、J Clin Invest.2008、118(6):2098〜110頁)の有無の下において1:1の比で96ウエル平底プレート中の2×105個のBWZトランスフェクタントに加える前に、24時間放置して細胞死を誘導した。一晩培養した後、LacZの活性を前述のように測定した。いくつかのアッセイ中で、CLEC9A wtでトランスフェクタントしたBWZ細胞を使用して、リガンドが細胞質尾部を介してシグナル伝達するかどうか決定し、Fabに匹敵する遮断試薬として完全抗体を評価した。
【0243】
以下の線量のUV(mJ/cm2):0、0.5、1.5、5、15、50、240でLK細胞を照射することによってUV線量反応を実施した。死滅細胞の頻度はto-pro3色素を使用して24時間後に決定し、細胞は新たな培地中にBWZトランスフェクタント(2×105個/ウエル)と共に1:1で平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0244】
LK細胞およびbm-1不死化MEFに関する異なる処理をリガンドの露出において評価した。細胞はミトキサントロン(1μM)と共に、または血清の不在下で(血清枯渇)24時間培養した。浸透圧ショックは以前に記載されたように実施した(Liuら、2002.J.Exp.Med.196:1091〜1097頁)。3サイクルの凍結解凍を細胞のペレットで実施し、液体N2中に凍結し37℃で解凍した。これらの処理後、死滅細胞の頻度はto-pro3色素を使用して決定し、細胞は以下に示すようにrsCTLDに関して染色した。細胞は新たな培地中にBWZトランスフェクタント(2×105個/ウエル)と共に1:1で平板培養した。一晩培養した後、LacZの活性を前述のように測定した。
【0245】
SIINFEKLC-biot(S1)とモノクローナル抗体のカップリング
免疫優勢OVAペプチドSIINFEKLと二価抗体を結合させるために、遊離スルフヒドリル基を生成するための付加システイン(C)および標識を追跡するためのビオチンを含有する、S1と呼ぶSIINFEKLペプチドの誘導体が、英国癌研究所において合成され高性能液体クロマトグラフィーによって精製された。モノクローナル抗体は室温において30分間スルホ-SMCCで処理し、第三級アミンにおいてスルホ反応基を生成し、次に分子サイズ排除クロマトグラフィーカラム(Pierce)中で活性化抗体を精製した。次いで、S1ペプチドを新たに調製し、放置して37℃で1時間等モル量で活性化抗体と反応させ、クロマトグラフィーカラム中で精製した。モノクローナル抗体のビオチン化の程度によって、本発明者らは、Fluoreporterキット(Invitrogen)を使用し製造者の説明書に従い、生成した全ての抗体結合体中でモノクローナル抗体分子当たり1〜1.2の結合ペプチドを定量化することができた。
【0246】
S2ペプチドおよびメラニン細胞分化抗原ペプチドとモノクローナル抗体のカップリング
S2ペプチド(SIINFEKLTEWTSSNVMEERC-ビオチン)は、英国癌研究所において合成されHPLCによって精製された。PBS中のモノクローナル抗体は室温において30分間スルホ-SMCCで処理して、第三級アミンにおいてスルホ反応基を生成した。活性化抗体は分子サイズ排除クロマトグラフィーカラム(Pierce)によって精製し、S2ペプチドを加え(2:1のモル比)、反応は37℃で1時間進行させた。結合体はImmunobindセファロースカラムを使用して精製した。モノクローナル抗体のビオチン化の程度は、製造者の説明書に従いFluoreporterキット(Invitrogen)を使用して評価した。
【0247】
同じ戦略を使用して、メラニン細胞分化抗原、gp100(EGSRNQDWLおよびKVPRNQDWL;H-2Db-拘束性(25))、TRP-1(TWHRYHLLおよびTAYRYHLL;H-2Kb-拘束性(26))およびTRP-2(SVYDFFVWL;H-2Kb拘束性(27))からペプチドを合成し、C末端にシステイン-eahx-ビオチンを付加することによってそれぞれを修飾した。
【0248】
抗CLEC9aモノクローナル抗体を用いたin vivo標的化。
抗CLEC9a、アイソタイプ対照、または抗DEC-205をAlexa488(Invitrogen)またはS1ペプチドと結合させた。Alexa488モノクローナル抗体を使用する標的化の実験用に、モノクローナル抗体-Alexa488を静脈内注射し(20μg)、脾細胞を抽出し、16時間後に分析した。
【0249】
in vivoでの特異的サブセットへの抗原の標的化の実験では、S1-抗体を静脈内注射し(5μg)、脾細胞を抽出し、抗CD11cマイクロビーズ(Miltenyi Biotec)を用いてCD11c陽性および陰性分画に精製した。OT-I細胞をOT-IRag-/-マウスのリンパ節および脾細胞から得て、ビオチン化抗体(抗CD11c、CD11b、B220、FcgR、Gr-1、およびCD4)のカクテル、次にストレプトアビジンマイクロビーズ(Miltenyi Biotec)を使用した負の選択によって精製した。図の凡例に示すように、異なる量のin vivo標的化DCを、U底プレートにおいて2μMのCFSE(Invitrogen)で標識した105個のOT-I細胞と共に培養した。3日後、Vβ5.1およびCD8陽性細胞ならびにTO-PRO3陰性細胞におけるCFSE希釈によって増殖を決定した。細胞は実際のビーズ数(...)で得て細胞の絶対数を定量化した。上清中のIFN-γはサンドウィッチELISAによって測定した。
【0250】
特異的CTL応答を標的化するためのin vivo標的化の実験では、モノクローナル抗体-S1を25μgの抗CD40(3/23、BD Pharmingen)有りまたは無しで後脚に皮下注射し(2μg)、5日後、in vivo死滅アッセイを記載したように実施した27。簡単に言うと、B6.SJLバックグラウンド(コンジェニックCD45.1+)由来の標的脾細胞に20nM、200nMまたは0nMのSIINFEKLペプチドを導入し、37℃において20分間、0.03μM、0.3μMまたは3μMのCFSEでそれぞれを標識した。標識した脾細胞(107)は静脈内注射した。翌日脾細胞を抽出し、CD45.1陽性集団をCFSEに関して分析した。さらに、5×105個の脾細胞を24時間1μMのSIINFEKLの有無の下で培養し、上清はELISAによりIFNγに関して定量化した。血液および脾細胞もSIINFEKL-H2Kbテトラマー(Beckman Coulter)、抗CD8および抗Thy1.2で標識し、CD8+T細胞集団中のテトラマー+細胞に関して分析した。
【0251】
B16メラノーマ腫瘍治療モデル。
B16メラノーマ細胞にOVA-GFP融合タンパク質を形質導入し、GFP発現に関して分類した。腫瘍細胞(2×105個)をコンジェニックB6マウスの尾部に静脈内注射し、6日後、抗体-S1+抗CD40からなる療法治療剤を脚部に皮下注射した。腫瘍注射後第18日で、肺腫瘍を計数した。腫瘍治療実験は、抗体治療3日前にマウスにB16-OVAを与えたこと以外は類似の形式で行った。
【0252】
腫瘍治療および予防実験も非形質導入親B16細胞を用いて実施した。これらをgpl00、TRP-1およびTRP-2由来の5ペプチドの混合物と共有結合した抗CLEC-9aまたは対照抗体(1μg/脚部)、ならびに抗CD40(12.5μg/脚部)およびポリI:C(5μg/脚部)を用いた免疫処置の3日前(治療)または1日後(予防)のいずれかに静脈内注射した(5×105個/マウス)。全身腫瘍組織量は肺病巣を計数することによって評価した。これらが数えるのに多すぎるほど(マウス当たり250を超えて)存在したとき、それらは250として示す。CTL応答は前に記載したようにモニターした。
【0253】
CLEC9a-/-マウスの生成。
Red/ET組換え(Gene Bridges、Heidelberg、ドイツ)を使用して遺伝子の領域を直接捕捉しBACクローンRP-23 248-K14(InvitrogenからのC57BL/6BACクローン)から改変してマウスを生成した。相同的組換えES細胞クローンの単離用に、陽性(ネオマイシン耐性遺伝子)と陰性(単純ヘルペスウイルスチミジンキナーゼ遺伝子)選択の両方の戦略を使用する、従来の遺伝子標的置換ベクター:pFloxRI+tkを、ベクターと対形成した20ヌクレオチドおよび捕捉対象のClec9aの領域と対形成した70ヌクレオチドを有する示したプライマーを使用してPCRによって増幅した。使用したプライマーは順方向3アーム24330pFlox5'ATAATATCATATTTCTATAATATCATTGTAATGACAAAACCACTGAACTA GTGCCTGTAAAGGCAGGAGGGGTACCGAGCTCGAATTCTACCG3';逆方向pFlox5アーム5'TGCTATATTACAGATTTTCAAGTGGGGTAGCCTGGAGTAACAAGATGGCAGGGCATAATCACTAGTGCGGCCGCCACCGCGGTGGAGCTCCAGCTTT3'であった。
【0254】
修飾するための領域をAmp耐性ベクター中に含めた後、ファルネシル化EGFP、およびPGK-gb2プロモーター次にKan/Neoを含むカセットは、Kanに関する選択で組換え相同的組換えステップの反復を可能にした。EGFP-FKan-Neo耐性カセットの増幅に使用したプライマーは、逆方向NeoKan3アーム5'TGCTTTTGTACTTACACTTGATGCCCAAGAAAATGGACGTTGCTAACAAGCCCATACAGACCACACCTCGAGATAACTTCGTATAATGTAT3'および順方向5アームEGFP-F5'TTTGTGCCAGGCTCCTATGTAGACTGCTTCACCACTCCAAGCGCCTTCAGCATGCATGTCGACATGGTGAGCAAGGGCGAGGAG3'であった。
【0255】
標的化ベクターを調製して、CLEC9A由来の最初の2アミノ酸のすぐ下流に強力なポリAを有するEGFP-Fを発現させ、エクソン1および2を破壊し、強力なポリAで転写を終了させた。標的化ベクターはNotIを使用したトランスフェクション前に線状化した。S6B6ハイブリッド129S6/C57BL/6F1由来胚幹(ES)細胞のトランスフェクションはエレクトロポレーションによって実施し、組換え体クローンはG418およびガンシクロビル中での培養後に単離した。淘汰を生存したES細胞は、2つの独立したプライマー対を使用したPCRによってスクリーニングし、そのプライマーの1つは短いアームの外側に存在した。使用したプライマー対は、Scr順方向1 5'GATCTGTGTGTTGGTTTTTGTGTGC3';Scr逆方向1、5'TAGCATGGCACTTCTCCATTACCTT3'アンプリコン順方向1逆方向1:2138bp.Scr順方向2、5'GCGAATTCGGTACCAATAAA AGAGC3';Scr逆方向2、5'CAGAAGCTTCCTGGTTTTGGTTTTT3'アンプリコン順方向2逆方向2:2352であった。
【0256】
正確に標的化したので、核典型的な正倍数体ESクローンを3.5日後のcoitumC57BL/6肺盤胞にマイクロインジェクションし、毛色がキメラ状態である生成した子孫はC57BL/6のメスと交配させて生殖細胞系の伝達を確認した。生殖細胞系が伝達したキメラを後にC57BL/6のメスと交配させて、純粋なC57BL/6バックグラウンドで遺伝子標的となる対立遺伝子を固定した。ヘテロ接合型動物を異種交配させて、ホモ接合型欠損動物および適合同腹子対照を生成した。ノックアウトマウスにおけるClec9aと連結したNK1.1C57BL/6遺伝子の発現は、標的化C57BL/6BACクローンの相同的組換えがS6B6 ES細胞におけるF1のC57BL/6コピー中に組み込まれたことを示す。
【0257】
この実験で使用したClec9a-/-マウスは、C57BL/6に戻し交配した第三世代の混合型129/Sv×C57BL/6の遺伝的背景であった。
【0258】
組換え可溶性CTLDの生成。
マウスCLEC9Aおよびデクチン-1に関するCTLDを、付加BirAモノビオチン化配列を有するSigmaからのp3xFlag-CMV-9発現ベクターにおいて、インフレームで独立にクローニングした。CTLD増幅に使用したプライマーは、mCLEC9A順方向5'GGATCC
mCLEC9A逆方向5'GGATCC
mデクチン順方向5'GGATCC3'mデクチン逆方向5'GGATCC3'であった。
CHO細胞をトランスフェクトしG418(1mg/ml)で選択した。安定したトランスフェクタントを限界希釈法によって二回クローニングし、上清にデクチン-1またはCLEC9A由来のrsCTLDを分泌するクローンを選択し、捕捉抗体として抗flag M2(Sigma)および検出用にビオチン-2A11抗デクチン-1またはビオチン-1F6抗CLEC9Aを使用したサンドウィッチELISAによって検出した。CHOクローンから濃縮した上清をCELLineバイオリアクター(Integra Biosciences、Chur、スイス)において生成し、抗flag M2アガロース(Sigma)を使用して精製した。次いでモノビオチン化を標準的な手順を使用して実施し{Ref}、PE-ストレプトアビジン(Sigma)を使用してテトラマーを作製した。次いでrsCTLDのPE-テトラマーを、通常のFACSバッファー中での4℃における30分間の死滅細胞またはザイモサンの染色に使用した。サンプルはto-pro3で対比染色し、フローサイトメトリーによって得た。
【0259】
in vitroでの交差提示。
in vitroでの交差提示アッセイにおいて、本発明者らは、3つの細胞型:OVA導入またはOVA発現死滅細胞、CLEC9Aおよび読み出しを阻害する抗体の有無の下の異なる起源に由来するFlt3L BMDC、OT-I OVA特異的トランスジェニックT細胞を同時培養した。本発明者らは、CLEC9a-/-またはCLEC9a+同腹子由来のFlt3L BMDCを試験した。さらに、C57BL/6由来のFlt3L BMDCを、対照Fab、または抗CLEC9A(1F6)Fab(10μg/ml)、およびCLEC9Aまたはアイソタイプ対照に対する完全抗体(20μg/ml)の有無の下で培養した。示した場合、CD11blo同およびCD24hi同を有するCD8α様Flt3L BMDCを記載したように(19)分類した。交差提示に関してOVA抗原と関係がある死滅細胞の供給源として、本発明者らはbm-1脾細胞、SIINFEKL、H2Kbの免疫優勢クラスI OVAペプチドの結合を妨げるH2Kb分子において突然変異体を発現するC57BL/6コンジェニックマウスを使用した。そのように、OVAを導入した細胞はOVAペプチドを直接提示することができないはずであり、プロセシングおよび交差提示して応答を生成しなければならない。吸着によりOVAを導入するために、本発明者らは、PBS中で五回洗浄する前に、20分間PBS中で低レベルのエンドトキシンおよびbm-1脾細胞と共に、示した用量の可溶性OVA(Calbiochem)をインキュベートした。あるいは、本発明者らはbm-1マウス胎児線維芽細胞(MEF)を作製し、かつ本発明者らは、SV-40 Tラージ抗原を用いてそれらを不死化した。次いで、本発明者らは、切断型非分泌OVA-GFP融合タンパク質を次いで形質導入し、OVA-GFPの相同的発現を分類した。OVA導入脾細胞とOVA発現MEFの両方を次いでUV照射し(240mJ/cm2)、完全培地中で一晩培養した。翌日、脾細胞は5:1の比でFlt3L BMDCと同時培養し(105個細胞/ウエル、96U底)、OVA-MEFは1:1の比でFlt3L BMDCと培養した。MACSビーズ(Miltenyi)を使用して負の選択をし(80%を超える純度)CFSE(2μM)で標識したOT-I細胞を、アッセイに加えた(105個細胞/ウエル)。3日後、上清を採取してサンドウィッチELISAによってIL-2およびIFN-γを検出し、細胞はPMA(10ng/ml)およびイオノマイシン(500ng/ml)で4時間刺激し、培養の最後の3時間中にブレフェルジンA(10μg/ml、Sigma)を加えた。次いで細胞を細胞内染色によってVβ5、CD8およびIFN-γに関して染色し、フローサイトメトリーによって得て、絶対数、一定数のキャリブレーションビーズ(BD)を導入したサンプル、および細胞内染色によるIFN-γ産生を決定した。
【0260】
死滅細胞の摂取
WTまたはCLEC9A-/-CD8α様Flt3L BMDCは、24時間前にUVC(240mJ/cm2)で処理しPKH26(Sigma)で標識した異なる割合の脾細胞と共に2時間インキュベートした。Flt3L BMDCはCD24に関して標識したので、PKH26およびCD24に関する二重陽性は死細胞の結合(4℃)または結合+摂取(37℃)が原因である可能性があり、頻度はそれぞれの型のDCに関するフローサイトメトリーにより定量化した。
【0261】
in vivoでの交差提示
前に示したように作製したOVA発現不死化bm-1 MEFをUVC処理し(240mJ/cm2)、静脈内注射(0.75×106個細胞/マウス)する前に前に一晩培養した。マウス(C57BL/6)は、静脈内注射の30分前に、PBSまたは400μgのアイソタイプ対照(AFRO-MAC49、ラットIgG1)または1F6抗CLEC9Aの腹腔内注射で前治療した。あるいは、Clec9a-/-または同腹子Clec9a+を使用した。6日後、内因性レパートリーから生じるCD8+T細胞エフェクター応答の誘導を、H2Kb-OVAペプチドテトラマー陽性細胞の数およびCD8サブセットにおけるex vivoのSIINFEKLに応じたIFNγ産生によって追跡した。独立した実験中の数匹の同腹子からデータをプールした図19cおよび19dにおいて、本発明者らはプール前にデータの正規化を実施した。それぞれの同腹子(メス)は独立した実験で考え、それぞれの同腹子における生データは、その同腹子中のClec9a+マウスの平均に正規化し、独立した実験における全同腹子中のClec9a+マウスの集団全体に関して得た算術平均を掛けた。同腹子の倍数変化の分析のために、Clec9a+マウスまたはClec9a-/-マウスに関する算術平均をそれぞれの同腹子において計算した。分析した12匹のうち12匹全ての同腹子が、Clec9a+と比較してClec9a-/-マウスのCD8 T細胞サブセット中のテトラマー陽性細胞の算術平均の数倍の低下を示した。
【0262】
統計
統計分析は、群間の差に関してスチューデントの両側t検定、またはデータの正規性を推測できなかったときはマンホイットニーのU検定を用いて実施した。p<0.05を統計上有意であると考えた。他に言及しない限り定量データは平均±SEMとして表す。
【0263】
上述の具体例としての実施形態と共に本発明を説明してきたが、この開示を与えられた場合多くの同等な修正形態および変形形態が、当業者には明らかなはずである。したがって、記載した本発明の具体例としての実施形態は、例示であって限定ではないとみなされるべきである。本発明の精神および範囲を逸脱することなく、記載の実施例の様々な変更がなされ得る。本明細書中の全ての引用文書は、参照により明確に援用される。
【0264】
(参考文献)
【特許請求の範囲】
【請求項1】
ペプチド抗原を抗原提示細胞に標的化する方法であって、抗原提示細胞を、前記抗原を含む組成物と接触させる段階を含み、前記抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原提示細胞がCLEC9aを発現する、方法。
【請求項2】
抗原提示細胞が樹状細胞である、請求項1に記載の方法。
【請求項3】
樹状細胞が、MHCクラスI分子を介して細胞外抗原を交差提示することができる、請求項2に記載の方法。
【請求項4】
in vitroまたはin vivoで行われる、請求項1から3のいずれか一項に記載の方法。
【請求項5】
対象においてペプチド抗原に対する免疫応答を刺激する方法であって、対象に抗原を含む組成物を投与する段階を含み、前記抗原は、CLEC9aに対して親和性を有する結合剤と結合している、方法。
【請求項6】
前記組成物の複数回投与を含む、請求項5に記載の方法。
【請求項7】
CLEC9aに対して親和性を有する結合剤と結合しているペプチド抗原を含む組成物。
【請求項8】
前記抗原および前記結合剤を薬学的に許容される担体と組み合わせて含む、請求項7に記載の組成物。
【請求項9】
静脈、筋肉内、腹腔内、経鼻、皮下または皮内投与用に処方される、請求項7または請求項8に記載の組成物。
【請求項10】
医療方法において使用するための、請求項7から9のいずれか一項に記載の組成物。
【請求項11】
前記抗原に対する免疫応答を刺激するのに使用するための、請求項7から10のいずれか一項に記載の組成物。
【請求項12】
前記抗原に対する免疫応答を刺激するための医薬の調製における、請求項7から11のいずれか一項に記載の組成物の使用。
【請求項13】
刺激される免疫応答がCTL応答またはTreg応答である、請求項1から12のいずれか一項に記載の方法、組成物または使用。
【請求項14】
前記組成物がアジュバントと共に投与される、またはアジュバントと共に投与するために処方される、請求項1から13のいずれか一項に記載の方法、組成物または使用。
【請求項15】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニスト、または別の免疫刺激剤である、請求項14に記載の方法、組成物または使用。
【請求項16】
前記抗原が結合剤と共有結合している、請求項1から15のいずれか一項に記載の方法、組成物または使用。
【請求項17】
前記抗原および結合剤が同じペプチド鎖の一部である、請求項16に記載の方法、組成物または使用。
【請求項18】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項1から17のいずれか一項に記載の方法、組成物または使用。
【請求項19】
結合剤が抗体またはその機能的断片である、請求項18に記載の方法、組成物または使用。
【請求項20】
結合剤がCLEC9aのアゴニストである、請求項1から19のいずれか一項に記載の方法、組成物または使用。
【請求項21】
結合剤が少なくとも2個のCLEC9a結合部位を有する、請求項1から20のいずれか一項に記載の方法、組成物または使用。
【請求項22】
前記抗原が、病原体または寄生体により発現されるタンパク質、または癌細胞のタンパク質のペプチドである、またはこれを含む、請求項1から21のいずれか一項に記載の方法、組成物または使用。
【請求項23】
前記抗原がウイルスタンパク質または腫瘍特異的抗原である、請求項22に記載の方法、組成物または使用。
【請求項24】
前記応答がTreg応答であり、前記抗原が、それに対する望ましくない免疫応答を阻害または抑制することが望ましいものである、請求項13に記載の方法、組成物または使用。
【請求項25】
CLEC9aアンタゴニストを対象に投与する段階を含む、対象において免疫応答を阻害する方法。
【請求項26】
前記アンタゴニストが、CLEC9aに対して親和性を有する結合剤、またはCLEC9aに対して親和性を有する結合剤をコードする核酸である、請求項25に記載の方法。
【請求項27】
結合剤が、結合する細胞の機能を直接的または間接的に阻害することができる、請求項26に記載の方法。
【請求項28】
結合剤が、前記細胞を直接的または間接的に死滅させることができる、請求項27に記載の方法。
【請求項29】
結合剤が対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる、請求項27または請求項28に記載の方法。
【請求項30】
結合剤が抗体Fc領域を含む、請求項26から29のいずれか一項に記載の方法。
【請求項31】
結合剤が、前記細胞を死滅させることができる毒素分子を含む、請求項28に記載の方法。
【請求項32】
結合剤が、前記細胞の近くでプロドラッグを活性化することができる酵素を含む、請求項28に記載の方法。
【請求項33】
酵素が非毒性分子を毒性分子に変換する、請求項32に記載の方法。
【請求項34】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項26から33のいずれか一項に記載の方法。
【請求項35】
結合剤がCLEC9aに特異的な結合部位を1個のみ有する、請求項26から34のいずれか一項に記載の方法。
【請求項36】
CLEC9aアンタゴニストが、CLEC9aとリガンドの結合を阻害することができる、請求項25に記載の方法。
【請求項37】
CLEC9aアンタゴニストが、CLEC9aリガンドに結合することができるCLEC9aの細胞外ドメインまたはその断片を含む可溶性分子である、請求項36に記載の方法。
【請求項38】
アンタゴニストが投与される対象が、炎症または自己免疫状態、特に望ましくないCTL活性を特徴とする状態に罹患している、請求項25から37のいずれか一項に記載の方法。
【請求項39】
前記状態が、
免疫成分による関節リウマチおよび他の種類の慢性または急性関節炎または関節症、全身性エリテマトーデス、強皮症、シェーグレン症候群、自己免疫性(特にI型)糖尿病、甲状腺炎、ならびに乾癬を含む他の臓器特異的免疫疾患を含む自己免疫疾患;
多発性硬化症、重症筋無力症、および他の免疫介在性神経疾患を含む神経疾患;
クローン病、大腸炎、セリアック病、および肝炎を含む胃腸疾患;
アテローム性動脈硬化症、心筋症、リウマチ熱、心内膜炎、血管炎および他の免疫介在性心臓血管疾患を含む心臓血管疾患;
肺気腫、呼吸器気道感染症、および他の免疫介在性呼吸器疾患を含む免疫介在性呼吸器疾患;
喘息、鼻炎、および他の免疫介在性過敏性反応を含むアレルギー作用および過敏性反応(I、II、III、およびIV型);
例えば、臓器移植、組織移植、輸血、骨髄移植中またはその後に起こる移植または移植片拒絶反応および移植片対宿主病;
敗血症性ショック症候群を含む感染性病原体に対する免疫病理学的反応;
小膠細胞などの免疫担当細胞に影響を与える神経変性過程などの変性過程
から選択される、請求項38に記載の方法。
【請求項40】
医療方法において使用するための、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤。
【請求項41】
免疫応答の阻害において使用するための、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤。
【請求項42】
免疫応答を阻害するための医薬の調製における、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤の使用。
【請求項43】
CLEC9aに特異的な抗体結合部位を含む、請求項40から42のいずれか一項に記載の結合剤または使用。
【請求項44】
結合剤が、結合する細胞による抗原提示を直接的または間接的に阻害することができる、請求項40から43のいずれか一項に記載の結合剤または使用。
【請求項45】
結合剤が、前記細胞を直接的または間接的に死滅させることができる、請求項44に記載の結合剤または使用。
【請求項46】
結合剤が対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる、請求項44または請求項45に記載の結合剤または使用。
【請求項47】
結合剤が抗体Fc領域を含む、請求項46に記載の結合剤または使用。
【請求項48】
結合剤が、前記細胞を死滅させることができる毒素分子を含む、請求項45に記載の結合剤または使用。
【請求項49】
結合剤が、前記細胞の近くでプロドラッグを活性化することができる酵素を含む、請求項45に記載の結合剤または使用。
【請求項50】
酵素が非毒性分子を毒性分子に変換する、請求項49に記載の結合剤または使用。
【請求項51】
CLEC9aアンタゴニストが、CLEC9aとリガンドの結合を阻害することができる、請求項40から42のいずれか一項に記載のCLEC9aアンタゴニストまたは使用。
【請求項52】
CLEC9aアンタゴニストが、CLEC9aリガンドに結合することができるCLEC9aの細胞外ドメインまたはその断片を含む可溶性分子である、請求項51に記載のCLEC9aアンタゴニストまたは使用。
【請求項53】
CLEC9aアゴニストを対象に投与する段階を含む、対象において免疫応答を刺激する方法。
【請求項54】
アゴニストがCLEC9aに対して親和性を有する結合剤である、請求項53に記載の方法。
【請求項55】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項54に記載の方法。
【請求項56】
結合剤がCLEC9aに特異的な結合部位を2個より多く含む、請求項54または請求項55に記載の方法。
【請求項57】
試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤と1つまたは複数の細胞の結合を測定する段階とを含む、試料中の細胞を検出する方法。
【請求項58】
試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤が結合している1つまたは複数の細胞を単離する段階とを含む、試料から細胞を単離する方法。
【請求項59】
結合剤が固体支持体上に固定化される、請求項57または請求項58に記載の方法。
【請求項60】
抗原提示細胞が樹状細胞である、請求項57から59のいずれか一項に記載の方法。
【請求項61】
樹状細胞が、MHCクラスI分子を介して細胞外抗原を交差提示することができる、請求項60に記載の方法。
【請求項62】
試料を、樹状細胞マーカーに対して親和性を有する第2の結合剤と接触させる段階を含む、請求項57から61のいずれか一項に記載の方法。
【請求項63】
樹状細胞マーカーが、CD11、好ましくはCD11c、もしくはHLA-DR、またはBDCA-3である、は請求項62に記載の方法。
【請求項64】
第1および第2の結合剤両方が結合する細胞のみが同定または単離される、請求項62または請求項63に記載の方法。
【請求項65】
1つまたは複数の不要な細胞型の負の選択を含む、請求項57から64のいずれか一項に記載の方法。
【請求項66】
不要な細胞型が樹状細胞の亜群である、請求項65に記載の方法。
【請求項67】
不要な細胞型が形質細胞様樹状細胞(pDC、plasmacytoid dendritic cell)である、請求項65に記載の方法。
【請求項68】
細胞上のCLEC9a発現レベルを測定する段階を含む、請求項57から67のいずれか一項に記載の方法。
【請求項69】
請求項58から68のいずれか一項に記載の方法により単離された、抗原提示細胞またはその集団。
【請求項70】
ペプチド抗原に対する免疫応答を刺激する方法であって、請求項69に記載の抗原提示細胞またはその集団を提供する段階と、前記細胞または細胞集団を前記抗原と接触させる段階とを含む、方法。
【請求項71】
細胞または細胞集団を対象に投与する段階を含む、請求項70に記載の方法。
【請求項72】
細胞が、細胞の由来した対象に再投与される、請求項71に記載の方法。
【請求項73】
前記細胞または細胞集団をアジュバントと接触させる段階をさらに含む、請求項71または請求項72に記載の方法。
【請求項74】
細胞または細胞集団をアジュバントおよび抗原と実質的に同時に接触させる、請求項73に記載の方法。
【請求項75】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニスト、または別の免疫刺激剤である、請求項73または請求項74に記載の方法。
【請求項76】
医療方法において使用するための、請求項69に記載の抗原提示細胞またはその集団。
【請求項77】
標的抗原に対する免疫応答を刺激する方法において使用するための、請求項69に記載の抗原提示細胞またはその集団。
【請求項78】
標的抗原に対する免疫応答の刺激のための医薬の調製における、請求項70に記載の方法により得られる、プライミングされた抗原提示細胞またはその集団の使用。
【請求項79】
前記接触段階に続いて、前記抗原提示細胞を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含む、請求項70に記載の方法。
【請求項80】
集団中のT細胞を増殖させる段階を含む、請求項79に記載の方法。
【請求項81】
T細胞を対象に投与する段階を含む、請求項79または請求項80に記載の方法。
【請求項82】
T細胞および抗原提示細胞が自己由来である、請求項79から81のいずれか一項に記載の方法。
【請求項83】
T細胞および抗原提示細胞が同じ対象に由来する、請求項82に記載の方法。
【請求項84】
T細胞が、T細胞の由来した対象に投与される、請求項83に記載の方法。
【請求項85】
アジュバントを投与する段階を含む、請求項81から84のいずれか一項に記載の方法。
【請求項86】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニストである、請求項85に記載の方法。
【請求項87】
T細胞が、CTLまたはTreg細胞もしくはTh17細胞を含むヘルパーT細胞を含む、請求項79から86のいずれか一項に記載の方法。
【請求項88】
請求項79または請求項80に記載の方法により得られるT細胞またはその集団。
【請求項89】
医療方法において使用するための、請求項88に記載のT細胞またはその集団。
【請求項90】
標的抗原に対する免疫応答を刺激する方法において使用するための、請求項88に記載のT細胞またはその集団。
【請求項91】
標的抗原に対する免疫応答の刺激のための医薬の調製における、請求項88に記載のT細胞またはその集団の使用。
【請求項92】
抗原に対する対象の寛容を誘導する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原がアジュバントの非存在下で投与される方法。
【請求項93】
前記抗原に対する対象の寛容を誘導するのに使用するための、請求項7から9のいずれか一項に記載の組成物。
【請求項94】
前記抗原に対する対象の寛容を誘導する方法において使用するための医薬の調製における、請求項7から9のいずれか一項に記載の組成物の使用。
【請求項95】
CLEC9aの生理的リガンドをスクリーニングする方法であって、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合するのに十分なその一部を含む標的物質を、哺乳動物細胞の成分である試験物質と接触させる段階と、標的物質と試験物質の結合を測定する段階とを含む方法。
【請求項96】
試験物質を同定する段階をさらに含む、請求項95に記載の方法。
【請求項97】
試験物質が哺乳動物細胞の細胞内成分である、請求項95または請求項96に記載の方法。
【請求項98】
試験物質および標的物質中に存在するCLEC9aまたはその一部が同種由来である、請求項95から97のいずれか一項に記載の方法。
【請求項99】
標的物質を、試験物質を含む試料と接触させる段階を含む、請求項95から98のいずれか一項に記載の方法。
【請求項100】
試料が透過性哺乳動物細胞を含む、請求項99に記載の方法。
【請求項101】
細胞が固定される、請求項100に記載の方法。
【請求項102】
標的物質の検出によって、標的物質の結合が起こる細胞内位置を測定する段階を含む、請求項100または請求項101に記載の方法。
【請求項103】
試料が哺乳動物細胞の細胞溶解物、抽出物または細胞内画分を含む、請求項99に記載の方法。
【請求項104】
標的物質および試験物質を含む複合体を単離する段階を含む、請求項95から103のいずれか一項に記載の方法。
【請求項105】
試験物質が固体支持体上に固定化される、請求項95から103のいずれか一項に記載の方法。
【請求項106】
プロテオミクス技術による試験物質の同定を含む、請求項95から105のいずれか一項に記載の方法。
【請求項107】
試験物質と標的物質の間の相互作用が、細胞発現系または無細胞発現系において起こり、レポーター遺伝子の発現などの検出可能な反応を誘導する、請求項95に記載の方法。
【請求項1】
ペプチド抗原を抗原提示細胞に標的化する方法であって、抗原提示細胞を、前記抗原を含む組成物と接触させる段階を含み、前記抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原提示細胞がCLEC9aを発現する、方法。
【請求項2】
抗原提示細胞が樹状細胞である、請求項1に記載の方法。
【請求項3】
樹状細胞が、MHCクラスI分子を介して細胞外抗原を交差提示することができる、請求項2に記載の方法。
【請求項4】
in vitroまたはin vivoで行われる、請求項1から3のいずれか一項に記載の方法。
【請求項5】
対象においてペプチド抗原に対する免疫応答を刺激する方法であって、対象に抗原を含む組成物を投与する段階を含み、前記抗原は、CLEC9aに対して親和性を有する結合剤と結合している、方法。
【請求項6】
前記組成物の複数回投与を含む、請求項5に記載の方法。
【請求項7】
CLEC9aに対して親和性を有する結合剤と結合しているペプチド抗原を含む組成物。
【請求項8】
前記抗原および前記結合剤を薬学的に許容される担体と組み合わせて含む、請求項7に記載の組成物。
【請求項9】
静脈、筋肉内、腹腔内、経鼻、皮下または皮内投与用に処方される、請求項7または請求項8に記載の組成物。
【請求項10】
医療方法において使用するための、請求項7から9のいずれか一項に記載の組成物。
【請求項11】
前記抗原に対する免疫応答を刺激するのに使用するための、請求項7から10のいずれか一項に記載の組成物。
【請求項12】
前記抗原に対する免疫応答を刺激するための医薬の調製における、請求項7から11のいずれか一項に記載の組成物の使用。
【請求項13】
刺激される免疫応答がCTL応答またはTreg応答である、請求項1から12のいずれか一項に記載の方法、組成物または使用。
【請求項14】
前記組成物がアジュバントと共に投与される、またはアジュバントと共に投与するために処方される、請求項1から13のいずれか一項に記載の方法、組成物または使用。
【請求項15】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニスト、または別の免疫刺激剤である、請求項14に記載の方法、組成物または使用。
【請求項16】
前記抗原が結合剤と共有結合している、請求項1から15のいずれか一項に記載の方法、組成物または使用。
【請求項17】
前記抗原および結合剤が同じペプチド鎖の一部である、請求項16に記載の方法、組成物または使用。
【請求項18】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項1から17のいずれか一項に記載の方法、組成物または使用。
【請求項19】
結合剤が抗体またはその機能的断片である、請求項18に記載の方法、組成物または使用。
【請求項20】
結合剤がCLEC9aのアゴニストである、請求項1から19のいずれか一項に記載の方法、組成物または使用。
【請求項21】
結合剤が少なくとも2個のCLEC9a結合部位を有する、請求項1から20のいずれか一項に記載の方法、組成物または使用。
【請求項22】
前記抗原が、病原体または寄生体により発現されるタンパク質、または癌細胞のタンパク質のペプチドである、またはこれを含む、請求項1から21のいずれか一項に記載の方法、組成物または使用。
【請求項23】
前記抗原がウイルスタンパク質または腫瘍特異的抗原である、請求項22に記載の方法、組成物または使用。
【請求項24】
前記応答がTreg応答であり、前記抗原が、それに対する望ましくない免疫応答を阻害または抑制することが望ましいものである、請求項13に記載の方法、組成物または使用。
【請求項25】
CLEC9aアンタゴニストを対象に投与する段階を含む、対象において免疫応答を阻害する方法。
【請求項26】
前記アンタゴニストが、CLEC9aに対して親和性を有する結合剤、またはCLEC9aに対して親和性を有する結合剤をコードする核酸である、請求項25に記載の方法。
【請求項27】
結合剤が、結合する細胞の機能を直接的または間接的に阻害することができる、請求項26に記載の方法。
【請求項28】
結合剤が、前記細胞を直接的または間接的に死滅させることができる、請求項27に記載の方法。
【請求項29】
結合剤が対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる、請求項27または請求項28に記載の方法。
【請求項30】
結合剤が抗体Fc領域を含む、請求項26から29のいずれか一項に記載の方法。
【請求項31】
結合剤が、前記細胞を死滅させることができる毒素分子を含む、請求項28に記載の方法。
【請求項32】
結合剤が、前記細胞の近くでプロドラッグを活性化することができる酵素を含む、請求項28に記載の方法。
【請求項33】
酵素が非毒性分子を毒性分子に変換する、請求項32に記載の方法。
【請求項34】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項26から33のいずれか一項に記載の方法。
【請求項35】
結合剤がCLEC9aに特異的な結合部位を1個のみ有する、請求項26から34のいずれか一項に記載の方法。
【請求項36】
CLEC9aアンタゴニストが、CLEC9aとリガンドの結合を阻害することができる、請求項25に記載の方法。
【請求項37】
CLEC9aアンタゴニストが、CLEC9aリガンドに結合することができるCLEC9aの細胞外ドメインまたはその断片を含む可溶性分子である、請求項36に記載の方法。
【請求項38】
アンタゴニストが投与される対象が、炎症または自己免疫状態、特に望ましくないCTL活性を特徴とする状態に罹患している、請求項25から37のいずれか一項に記載の方法。
【請求項39】
前記状態が、
免疫成分による関節リウマチおよび他の種類の慢性または急性関節炎または関節症、全身性エリテマトーデス、強皮症、シェーグレン症候群、自己免疫性(特にI型)糖尿病、甲状腺炎、ならびに乾癬を含む他の臓器特異的免疫疾患を含む自己免疫疾患;
多発性硬化症、重症筋無力症、および他の免疫介在性神経疾患を含む神経疾患;
クローン病、大腸炎、セリアック病、および肝炎を含む胃腸疾患;
アテローム性動脈硬化症、心筋症、リウマチ熱、心内膜炎、血管炎および他の免疫介在性心臓血管疾患を含む心臓血管疾患;
肺気腫、呼吸器気道感染症、および他の免疫介在性呼吸器疾患を含む免疫介在性呼吸器疾患;
喘息、鼻炎、および他の免疫介在性過敏性反応を含むアレルギー作用および過敏性反応(I、II、III、およびIV型);
例えば、臓器移植、組織移植、輸血、骨髄移植中またはその後に起こる移植または移植片拒絶反応および移植片対宿主病;
敗血症性ショック症候群を含む感染性病原体に対する免疫病理学的反応;
小膠細胞などの免疫担当細胞に影響を与える神経変性過程などの変性過程
から選択される、請求項38に記載の方法。
【請求項40】
医療方法において使用するための、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤。
【請求項41】
免疫応答の阻害において使用するための、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤。
【請求項42】
免疫応答を阻害するための医薬の調製における、CLEC9aアンタゴニストまたはCLEC9aに対して親和性を有する結合剤の使用。
【請求項43】
CLEC9aに特異的な抗体結合部位を含む、請求項40から42のいずれか一項に記載の結合剤または使用。
【請求項44】
結合剤が、結合する細胞による抗原提示を直接的または間接的に阻害することができる、請求項40から43のいずれか一項に記載の結合剤または使用。
【請求項45】
結合剤が、前記細胞を直接的または間接的に死滅させることができる、請求項44に記載の結合剤または使用。
【請求項46】
結合剤が対象の免疫系の成分を動員し、したがって細胞への免疫攻撃を刺激することができる、請求項44または請求項45に記載の結合剤または使用。
【請求項47】
結合剤が抗体Fc領域を含む、請求項46に記載の結合剤または使用。
【請求項48】
結合剤が、前記細胞を死滅させることができる毒素分子を含む、請求項45に記載の結合剤または使用。
【請求項49】
結合剤が、前記細胞の近くでプロドラッグを活性化することができる酵素を含む、請求項45に記載の結合剤または使用。
【請求項50】
酵素が非毒性分子を毒性分子に変換する、請求項49に記載の結合剤または使用。
【請求項51】
CLEC9aアンタゴニストが、CLEC9aとリガンドの結合を阻害することができる、請求項40から42のいずれか一項に記載のCLEC9aアンタゴニストまたは使用。
【請求項52】
CLEC9aアンタゴニストが、CLEC9aリガンドに結合することができるCLEC9aの細胞外ドメインまたはその断片を含む可溶性分子である、請求項51に記載のCLEC9aアンタゴニストまたは使用。
【請求項53】
CLEC9aアゴニストを対象に投与する段階を含む、対象において免疫応答を刺激する方法。
【請求項54】
アゴニストがCLEC9aに対して親和性を有する結合剤である、請求項53に記載の方法。
【請求項55】
結合剤がCLEC9aに特異的な抗体結合部位を含む、請求項54に記載の方法。
【請求項56】
結合剤がCLEC9aに特異的な結合部位を2個より多く含む、請求項54または請求項55に記載の方法。
【請求項57】
試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤と1つまたは複数の細胞の結合を測定する段階とを含む、試料中の細胞を検出する方法。
【請求項58】
試料をCLEC9aに対して親和性を有する結合剤と接触させる段階と、結合剤が結合している1つまたは複数の細胞を単離する段階とを含む、試料から細胞を単離する方法。
【請求項59】
結合剤が固体支持体上に固定化される、請求項57または請求項58に記載の方法。
【請求項60】
抗原提示細胞が樹状細胞である、請求項57から59のいずれか一項に記載の方法。
【請求項61】
樹状細胞が、MHCクラスI分子を介して細胞外抗原を交差提示することができる、請求項60に記載の方法。
【請求項62】
試料を、樹状細胞マーカーに対して親和性を有する第2の結合剤と接触させる段階を含む、請求項57から61のいずれか一項に記載の方法。
【請求項63】
樹状細胞マーカーが、CD11、好ましくはCD11c、もしくはHLA-DR、またはBDCA-3である、は請求項62に記載の方法。
【請求項64】
第1および第2の結合剤両方が結合する細胞のみが同定または単離される、請求項62または請求項63に記載の方法。
【請求項65】
1つまたは複数の不要な細胞型の負の選択を含む、請求項57から64のいずれか一項に記載の方法。
【請求項66】
不要な細胞型が樹状細胞の亜群である、請求項65に記載の方法。
【請求項67】
不要な細胞型が形質細胞様樹状細胞(pDC、plasmacytoid dendritic cell)である、請求項65に記載の方法。
【請求項68】
細胞上のCLEC9a発現レベルを測定する段階を含む、請求項57から67のいずれか一項に記載の方法。
【請求項69】
請求項58から68のいずれか一項に記載の方法により単離された、抗原提示細胞またはその集団。
【請求項70】
ペプチド抗原に対する免疫応答を刺激する方法であって、請求項69に記載の抗原提示細胞またはその集団を提供する段階と、前記細胞または細胞集団を前記抗原と接触させる段階とを含む、方法。
【請求項71】
細胞または細胞集団を対象に投与する段階を含む、請求項70に記載の方法。
【請求項72】
細胞が、細胞の由来した対象に再投与される、請求項71に記載の方法。
【請求項73】
前記細胞または細胞集団をアジュバントと接触させる段階をさらに含む、請求項71または請求項72に記載の方法。
【請求項74】
細胞または細胞集団をアジュバントおよび抗原と実質的に同時に接触させる、請求項73に記載の方法。
【請求項75】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニスト、または別の免疫刺激剤である、請求項73または請求項74に記載の方法。
【請求項76】
医療方法において使用するための、請求項69に記載の抗原提示細胞またはその集団。
【請求項77】
標的抗原に対する免疫応答を刺激する方法において使用するための、請求項69に記載の抗原提示細胞またはその集団。
【請求項78】
標的抗原に対する免疫応答の刺激のための医薬の調製における、請求項70に記載の方法により得られる、プライミングされた抗原提示細胞またはその集団の使用。
【請求項79】
前記接触段階に続いて、前記抗原提示細胞を1つまたは複数のT細胞を含む細胞集団と接触させる段階を含む、請求項70に記載の方法。
【請求項80】
集団中のT細胞を増殖させる段階を含む、請求項79に記載の方法。
【請求項81】
T細胞を対象に投与する段階を含む、請求項79または請求項80に記載の方法。
【請求項82】
T細胞および抗原提示細胞が自己由来である、請求項79から81のいずれか一項に記載の方法。
【請求項83】
T細胞および抗原提示細胞が同じ対象に由来する、請求項82に記載の方法。
【請求項84】
T細胞が、T細胞の由来した対象に投与される、請求項83に記載の方法。
【請求項85】
アジュバントを投与する段階を含む、請求項81から84のいずれか一項に記載の方法。
【請求項86】
アジュバントがレチノイン酸、CD40アゴニストもしくはTLRアゴニストである、請求項85に記載の方法。
【請求項87】
T細胞が、CTLまたはTreg細胞もしくはTh17細胞を含むヘルパーT細胞を含む、請求項79から86のいずれか一項に記載の方法。
【請求項88】
請求項79または請求項80に記載の方法により得られるT細胞またはその集団。
【請求項89】
医療方法において使用するための、請求項88に記載のT細胞またはその集団。
【請求項90】
標的抗原に対する免疫応答を刺激する方法において使用するための、請求項88に記載のT細胞またはその集団。
【請求項91】
標的抗原に対する免疫応答の刺激のための医薬の調製における、請求項88に記載のT細胞またはその集団の使用。
【請求項92】
抗原に対する対象の寛容を誘導する方法であって、対象に抗原を含む組成物を投与する段階を含み、抗原がCLEC9aに対して親和性を有する結合剤と結合しており、抗原がアジュバントの非存在下で投与される方法。
【請求項93】
前記抗原に対する対象の寛容を誘導するのに使用するための、請求項7から9のいずれか一項に記載の組成物。
【請求項94】
前記抗原に対する対象の寛容を誘導する方法において使用するための医薬の調製における、請求項7から9のいずれか一項に記載の組成物の使用。
【請求項95】
CLEC9aの生理的リガンドをスクリーニングする方法であって、CLEC9aの細胞外ドメインまたはCLEC9aリガンドに結合するのに十分なその一部を含む標的物質を、哺乳動物細胞の成分である試験物質と接触させる段階と、標的物質と試験物質の結合を測定する段階とを含む方法。
【請求項96】
試験物質を同定する段階をさらに含む、請求項95に記載の方法。
【請求項97】
試験物質が哺乳動物細胞の細胞内成分である、請求項95または請求項96に記載の方法。
【請求項98】
試験物質および標的物質中に存在するCLEC9aまたはその一部が同種由来である、請求項95から97のいずれか一項に記載の方法。
【請求項99】
標的物質を、試験物質を含む試料と接触させる段階を含む、請求項95から98のいずれか一項に記載の方法。
【請求項100】
試料が透過性哺乳動物細胞を含む、請求項99に記載の方法。
【請求項101】
細胞が固定される、請求項100に記載の方法。
【請求項102】
標的物質の検出によって、標的物質の結合が起こる細胞内位置を測定する段階を含む、請求項100または請求項101に記載の方法。
【請求項103】
試料が哺乳動物細胞の細胞溶解物、抽出物または細胞内画分を含む、請求項99に記載の方法。
【請求項104】
標的物質および試験物質を含む複合体を単離する段階を含む、請求項95から103のいずれか一項に記載の方法。
【請求項105】
試験物質が固体支持体上に固定化される、請求項95から103のいずれか一項に記載の方法。
【請求項106】
プロテオミクス技術による試験物質の同定を含む、請求項95から105のいずれか一項に記載の方法。
【請求項107】
試験物質と標的物質の間の相互作用が、細胞発現系または無細胞発現系において起こり、レポーター遺伝子の発現などの検出可能な反応を誘導する、請求項95に記載の方法。
【図1】


【図2】

【図3】
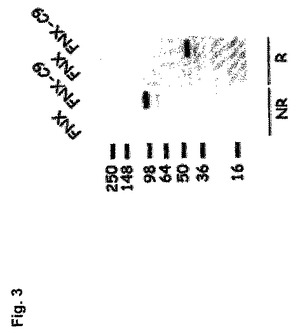
【図9】

【図10】
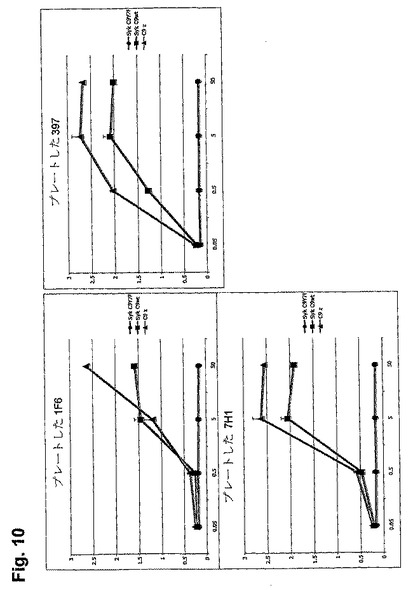
【図13A】
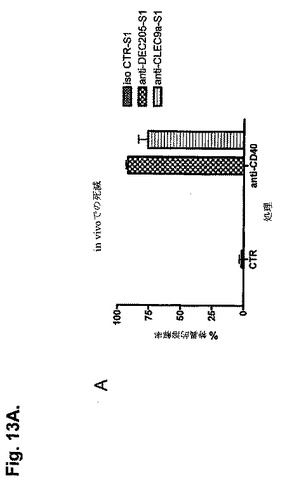
【図13B】
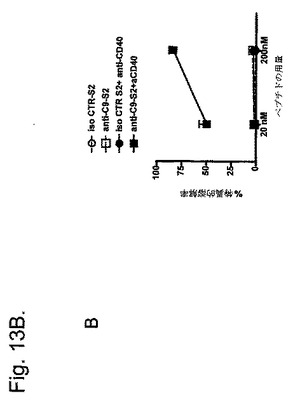
【図14】
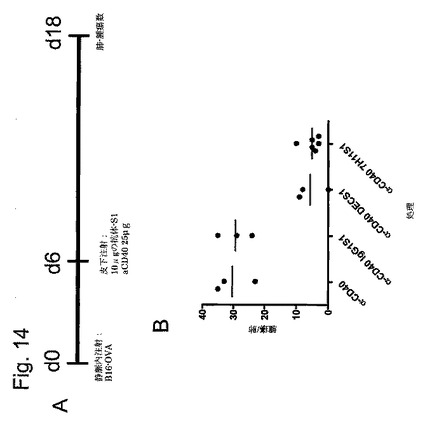
【図21】
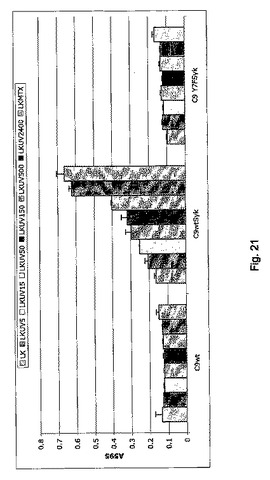
【図23】
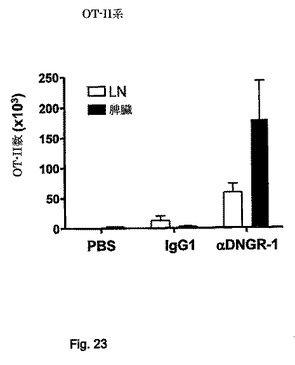
【図24】
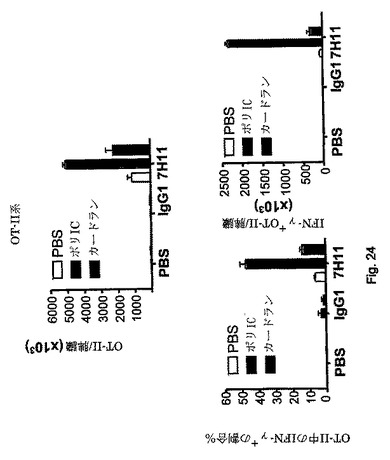
【図25】
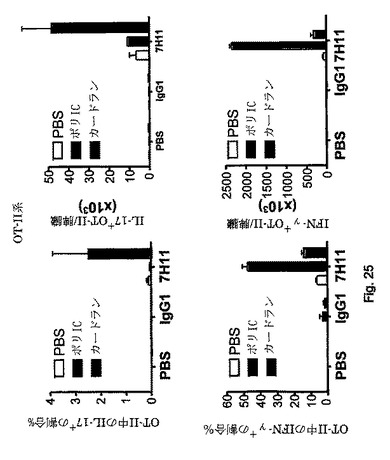
【図26】
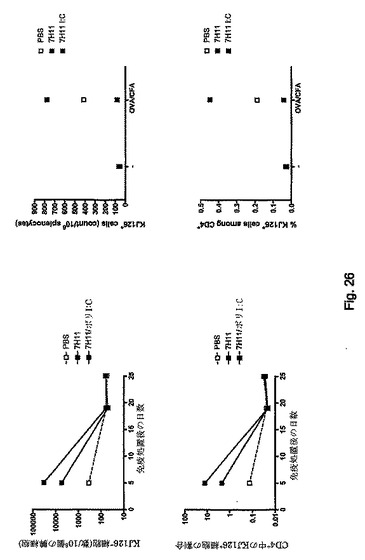
【図4】
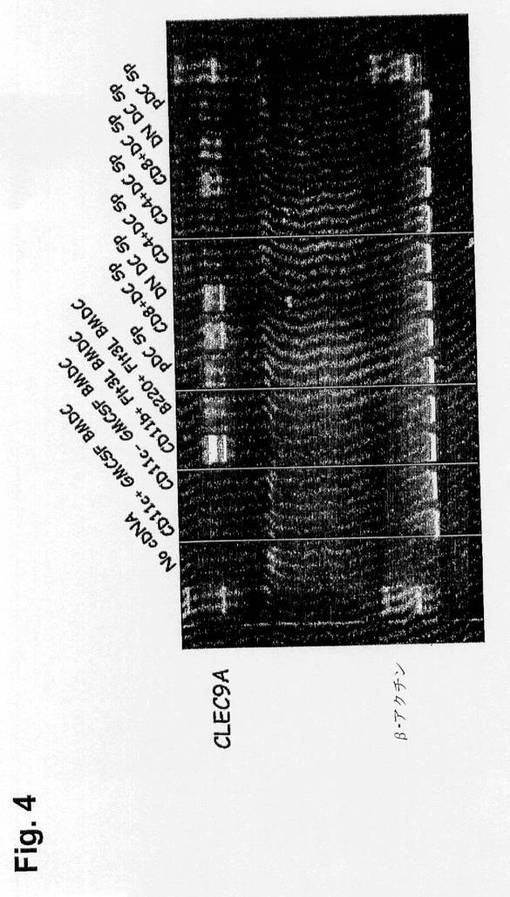
【図5】

【図6】
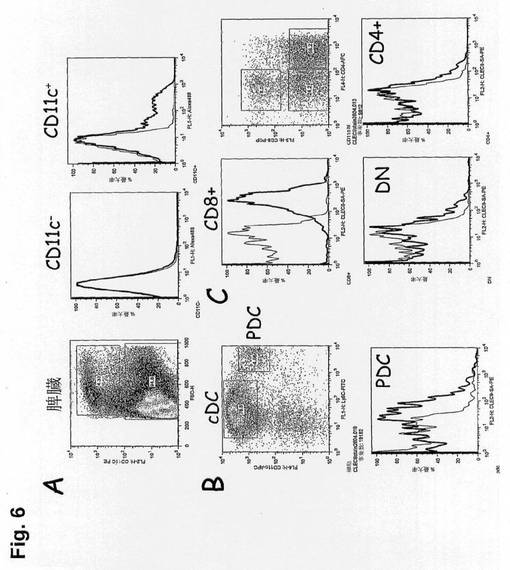
【図7】

【図8】
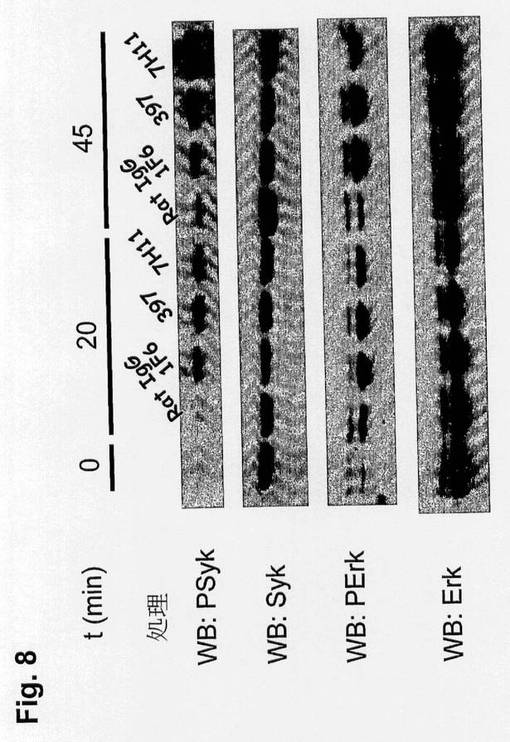
【図11】
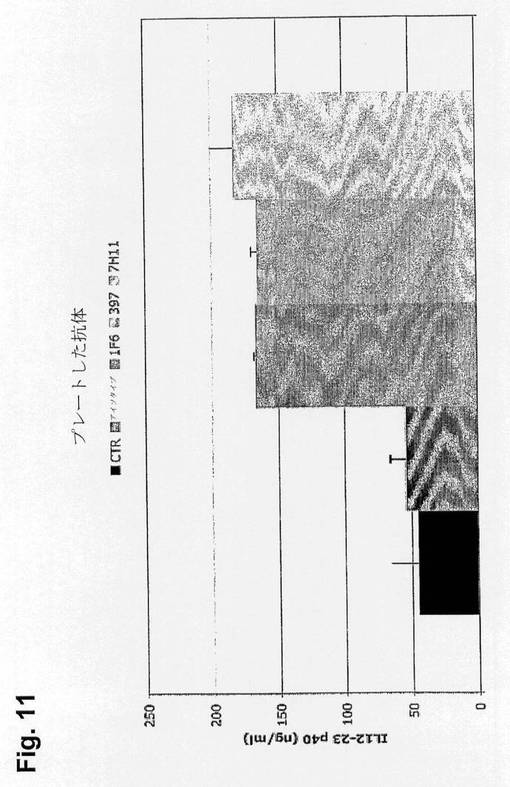
【図12AB】
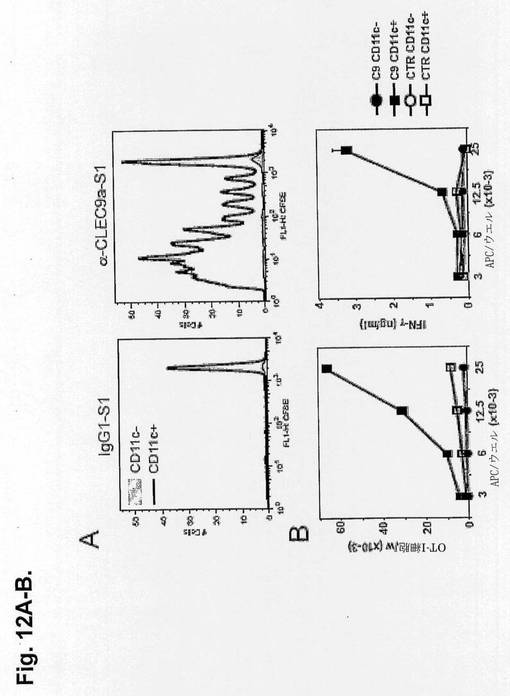
【図12CD】
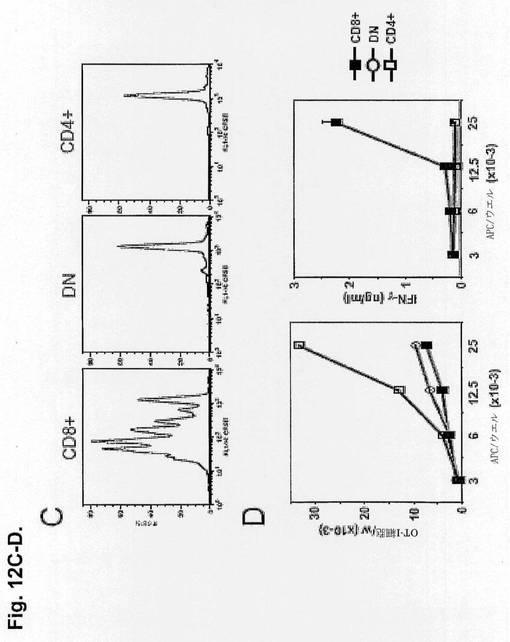
【図15】
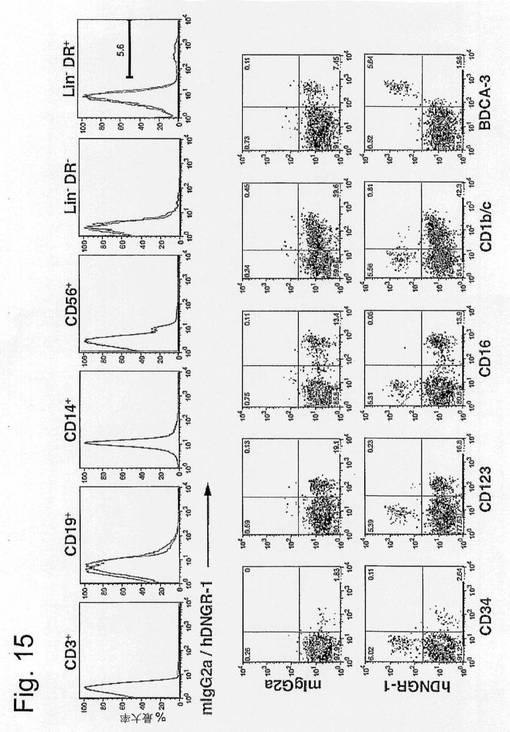
【図16】
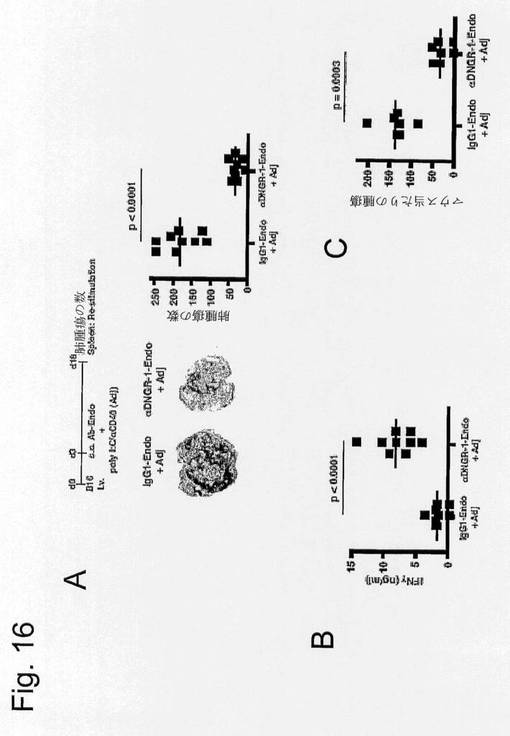
【図17】
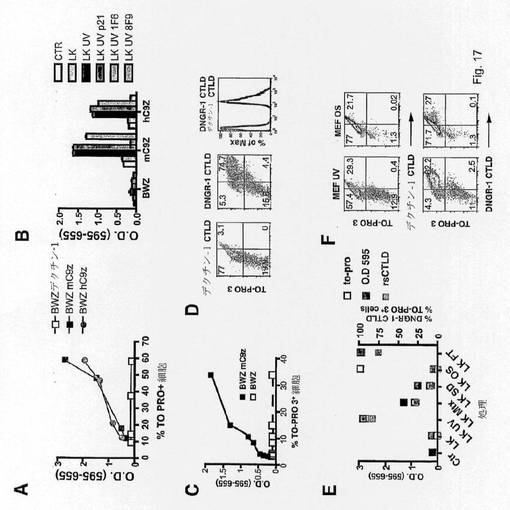
【図18】
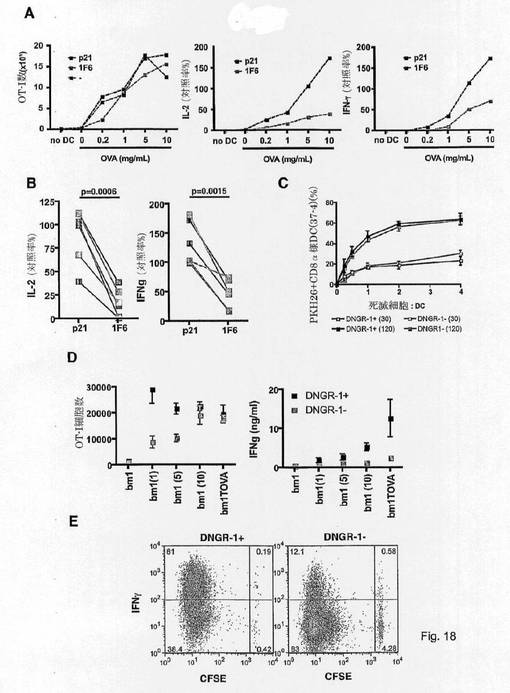
【図19】
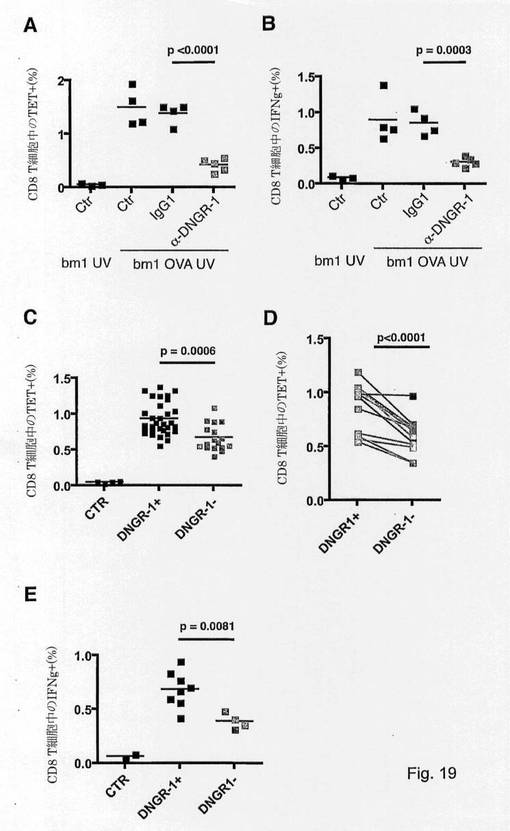
【図20】

【図22】
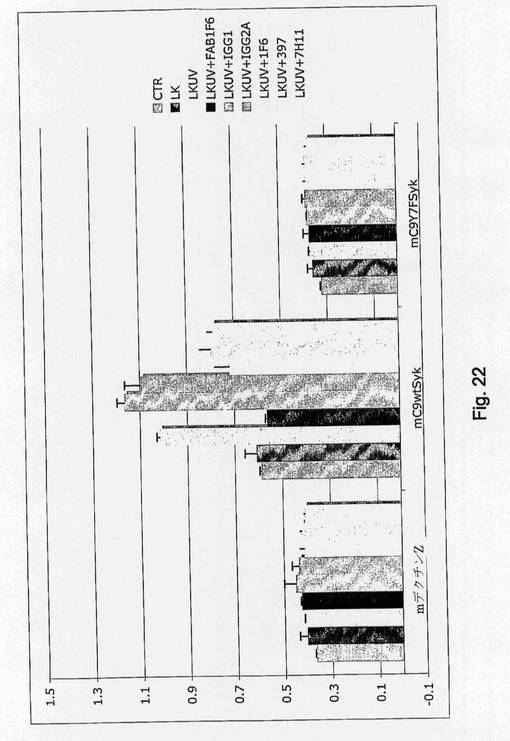
【図2】
【図3】
【図9】
【図10】
【図13A】
【図13B】
【図14】
【図21】
【図23】
【図24】
【図25】
【図26】
【図4】
【図5】
【図6】
【図7】
【図8】
【図11】
【図12AB】
【図12CD】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図22】
【公表番号】特表2010−534200(P2010−534200A)
【公表日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願番号】特願2010−516587(P2010−516587)
【出願日】平成20年7月21日(2008.7.21)
【国際出願番号】PCT/GB2008/002504
【国際公開番号】WO2009/013484
【国際公開日】平成21年1月29日(2009.1.29)
【出願人】(598176569)キャンサー・リサーチ・テクノロジー・リミテッド (57)
【氏名又は名称原語表記】CANCER RESEARCH TECHNOLOGY LIMITED
【Fターム(参考)】
【公表日】平成22年11月4日(2010.11.4)
【国際特許分類】
【出願日】平成20年7月21日(2008.7.21)
【国際出願番号】PCT/GB2008/002504
【国際公開番号】WO2009/013484
【国際公開日】平成21年1月29日(2009.1.29)
【出願人】(598176569)キャンサー・リサーチ・テクノロジー・リミテッド (57)
【氏名又は名称原語表記】CANCER RESEARCH TECHNOLOGY LIMITED
【Fターム(参考)】
[ Back to top ]