
CETP阻害剤を合成するための方法
【課題】CETP(コレステロールエステル転移タンパク質)の阻害剤である化合物を調製するための効率的な方法の提供。
【解決手段】該方法の最終段階は、式(I)の化合物を提供するための解決手段、オキサゾリジノン誘導体とビフェニル部分とのカップリングである。この合成の具体的な実施形態において、非溶媒和結晶性多形体を特徴とする結晶性生成物が製造される。

【解決手段】該方法の最終段階は、式(I)の化合物を提供するための解決手段、オキサゾリジノン誘導体とビフェニル部分とのカップリングである。この合成の具体的な実施形態において、非溶媒和結晶性多形体を特徴とする結晶性生成物が製造される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、コレステロールエステル転移タンパク質(CETP)を阻害する化合物を合成するための方法、およびこの方法により作成された特定の化合物の結晶性多形に関する。該方法の生成物は、哺乳動物におけるHDLコレステロールを上昇させ、アテローム性動脈硬化の治療および/または予防における、ならびにアテローム性動脈硬化の進行を遅延させることにおける有用性を有すると考えられる。
【背景技術】
【0002】
アテローム性動脈硬化および臨床的結果である冠動脈性心疾患(CHD)、卒中および末梢血管疾患は、産業化世界の保健医療制度にとって非常に大きな負担になっている。米国単独において、約1300万人の患者がCHDと診断されており、毎年50万人を超える死亡は、CHDが原因である。更に、人口の平均年齢が増加し、肥満および糖尿病における流行が広がり続けているので、この犠牲は、次の25年にわたって増えると予想される。
【0003】
CETPの阻害は、アテローム性動脈硬化の罹患率を減少させる、有望な新しいアプローチである。スタチンは、LDLコレステロール(「悪玉コレステロール」)を減少させることによるCHDの罹患率を減少させることにおいて重要であったが、HDLコレステロール(「善玉コレステロール」)を増加させることでは、比較的効果がない。CETP阻害剤は、HDLコレステロールを増加させ、および一般集団におけるCHDおよびアテローム性動脈硬化を減少させる、効力のある新たなツールを提供する可能性がある。CETP阻害剤およびスタチンの両方の投与は、アテローム性動脈硬化を治療するおよび予防するために、特に価値がある可能性がある。CETP阻害剤を含む医薬は、現在利用可能でない。Pfizerのトルセトラピブ(torcetrapib)は、現在、臨床試験の第III相中のCETP阻害剤である。
【発明の概要】
【0004】
(発明の概要)
本発明は、式Iを有する化合物を調製するための方法を提供する。これらの新規の化合物は、効力のあるCETP阻害剤:
【0005】
【化1】
である。
【0006】
該方法は、化合物Iの収束的合成を提供する。完全な方法には、2つの重要な中間体IIおよびIIIの合成が含まれる。該重要な中間体は、次いで方法の最終段階において結合され、このことは、以下に示される。
【0007】
【化2】
【0008】
式I、IIおよびIIIを有する化合物において、
R1は、HまたはC1〜4アルキルであり、このC1〜4アルキルは、1から5個のF基で場合により置換されており、
R2、R4、およびR5は、それぞれ独立して、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルからなる群から選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
R3は、H、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルから選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
aおよびbは、それぞれ独立して、1〜4からの整数から選択され、
cは、0〜2からの整数であり、
Xは、HまたはI族金属カチオン(例えば、Na、K、Li、またはCs)であり、ならびに
Yは、脱離基(すなわち、容易に置換される基)である。脱離基の例は、ハロゲン、C1〜3アルカノアート(例えば、アセテート)、トリフルオロアセテート、およびトリフレートを含む。
【0009】
XがHである場合、塩基もまた、該反応において含まれ、ここで、該塩基は、強塩基のアルカリ金属塩である。該アルカリ金属は、Na、K、Li、またはCsであり得、およびサブグループにおいて、該アルカリ金属は、NaまたはKであり得、他のサブグループにおいて、該アルカリ金属は、Naであり得、他のサブグループにおいて、該アルカリ金属は、Kであり得る。強塩基のアルカリ金属塩の例は、ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムを含む。該塩基の使用は、XがNa、K、Li、またはCsである中間化合物IIをもたらす。
【0010】
上記の反応の実施形態において、R1は、H、または1から5個のFで場合により置換されているC1〜3アルキルである。他の実施形態におけるR1は、1から3個のFで場合により置換されているC1〜2アルキルである。好ましい実施形態において、R1は、CH3である。
【0011】
該反応の実施形態において、R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、または1から5個のFで場合により置換されている−OC1〜3アルキルである。他の実施形態において、R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、または1から5個のFで場合により置換されている−OC1〜2アルキルである。他の実施形態において、R2、R4およびR5は、それぞれ独立して、C1〜3アルキル、CF3、−OCH3、−OCF3、およびFから選択される。
【0012】
他の実施形態において、それぞれのR2は、CH3またはCF3である。
【0013】
他の実施形態において、R2は、CF3である。
【0014】
他の実施形態において、R3は、C1〜3アルキル、−OC1〜3アルキル、またはFであり、ここで、C1〜3アルキルおよびOC1〜3アルキルは、1から5個のFで場合により置換されている。
【0015】
他の実施形態において、R3は、CH3、CF3またはFである。
【0016】
他の実施形態において、R3は、CF3である。
【0017】
いくつかの実施形態において、aは、1または2であり、および他の実施形態において、aは、2である。
【0018】
いくつかの実施形態において、bは、1〜3である。他の実施形態において、bは、2または3である。他の実施形態において、bは、3である。
【0019】
いくつかの実施形態において、cは、0または1である。他の実施形態において、cは、0である。
【0020】
いくつかの実施形態において、Xは、H、Na、またはKである。他の実施形態において、Xは、Na、またはKである。
【0021】
いくつかの実施形態において、Yは、ハロゲンである。他の実施形態において、Yは、Cl、BrまたはIである。他の実施形態において、Yは、ClまたはBrである。他の実施形態において、Yは、Clである。
【0022】
別の様に示していない限り、アルキル基は、直鎖または分枝鎖状のいずれでもよい。
【図面の簡単な説明】
【0023】
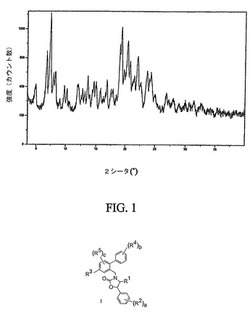
【図1】化合物12の結晶性非溶媒和形態の特徴的なX線粉末回折パターンを示す図である。
【図2】化合物12の結晶性非溶媒和形態の通常のDSC曲線を示す図である。
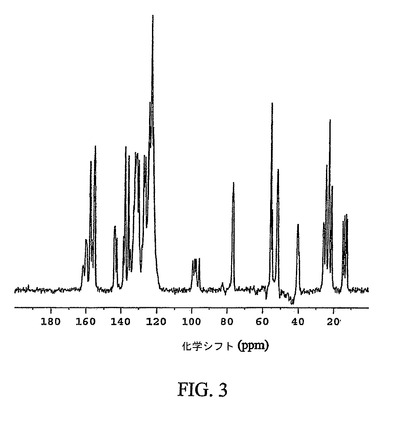
【図3】化合物12の結晶性非溶媒和形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
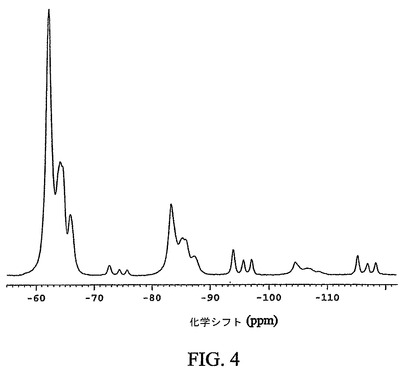
【図4】化合物12の結晶性非溶媒和形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
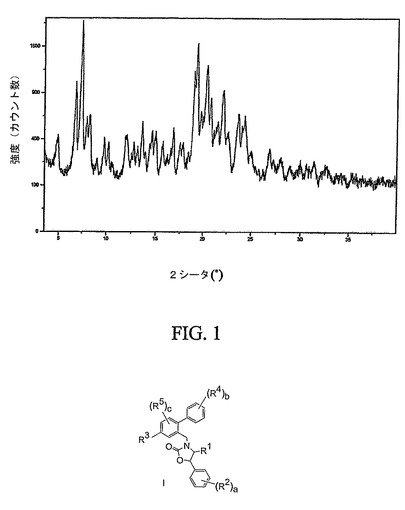
【図5】化合物12の結晶性ヘプタン溶媒和形態の特徴的なX線粉末回折パターンを示す図である。
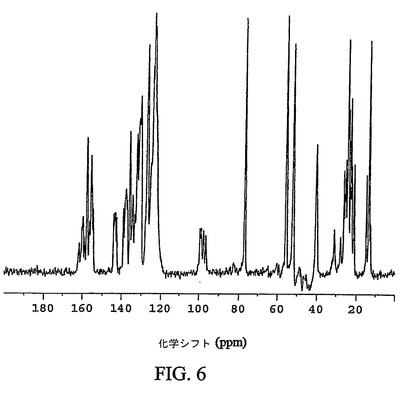
【図6】化合物12の結晶性ヘプタン溶媒和形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図7】化合物12の結晶性ヘプタン溶媒和形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
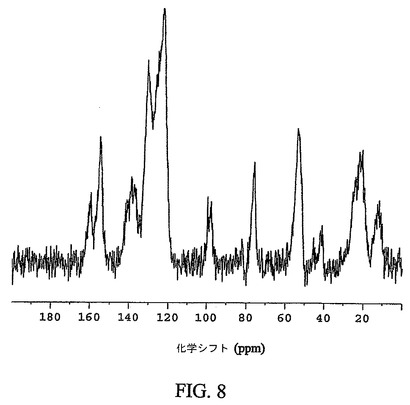
【図8】化合物12の非晶質形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
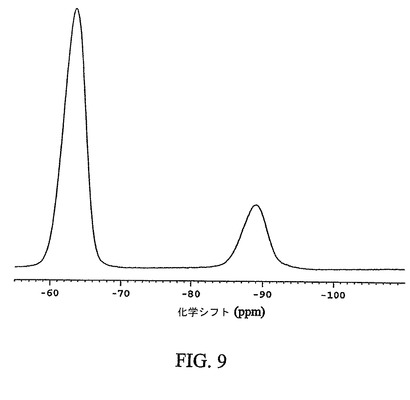
【図9】化合物12の非晶質形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
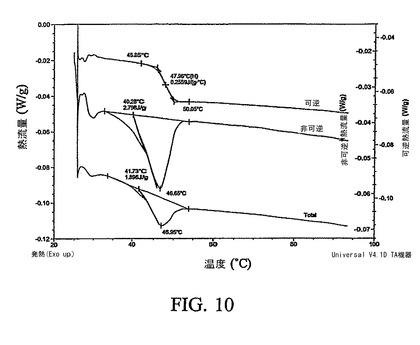
【図10】化合物12の非晶質形態の通常の調節されたDSC(MDSC)曲線を示す図である。
【0024】
(発明の詳細な説明)
更なる実施形態が、以下に記載される。
【0025】
中間体IIにおいて、Xは、HまたはI族アルカリ金属(例えば、Na、K、Li、またはCs)である。該I族金属カチオンは、TMEDAなどのリガンドと、またはエーテルもしくはポリエーテル(IIのオキサゾリジノン群の負に荷電されたNの反応性を増加させる、クラウンエーテルなど)と錯体を形成され得る。Xはまた、Hであってよい。Xが金属カチオン(例えば、Na、K、Li、またはCs)である場合、中間体IIは、オキサゾリジノン(Xは、Hである。)と、金属ヒドリド、アルキル金属化合物、または反応性アルカリ金属アミドとの反応により作成され得る。実施例は、オキサゾリジノン(X=H)と、塩基(ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムなど)との反応を含む。XがHである場合、塩基もまた反応において含まれ、ここで、該塩基は、オキサゾリジノンを反応性化合物(ここで、Xは、Na、K、Li、またはCsである。)に変換するために使用される塩基の1つである。X基は、多くの実施形態において、KおよびNaから選択される。実施例1においてX基は、Naである。
【0026】
中間体IIIにおいて、Yは、脱離基(すなわち、容易に置換される基)である。該脱離基が置換された後、該脱離基は、通常陰イオンである。最も一般的な脱離基は、ハロゲン(Cl、Br、IまたはFなど)である。脱離は、有機酸(トリフレートまたはトリフルオロアセテートなど)の脱プロトン化された形態であってもよい。多くの実施形態において、脱離基Yは、Br、ClおよびIから選択される。多くの実施形態において、脱離基Yは、BrおよびClから選択される。Y基は、実施例1における化合物12の合成に関し、Clである。
【0027】
上記の反応において、中間体IIは、しばしば、中性のオキサゾリジノン(X=H)として反応槽に充填され、次いで、アルカリ金属塩に変換され(ここで、Xは、アルカリ金属である。)、および次いで単離されることなく、中間体IIIと反応する。この適用の目的のために、オキサゾリジノン(XはHである。)は、反応槽に充填される開始物質であるが、これは、オキサゾリジノン(Xは、アルカリ金属である。)のアルカリ金属塩と、中間体IIIとの反応である。あるいは、中間体IIおよびIIIは、最初に反応槽に充填され得、およびXがHである場合、次いで十分な塩基が加えられ、カップリング反応を引き起こす。これはまた、オキサゾリジノン(ここで、Xは、アルカリ金属である。)と中間体IIIとの反応である。
【0028】
該反応は、極性非プロトン性溶媒(HMPA、DMF、またはDMACなど)中で一般に実施される。多くの実施形態において、DMFは、溶媒として使用される。該反応は、温度の穏やかな条件下で進行する。代表的な穏やかな条件は、−20℃、−10℃、0℃、10℃、20℃、30℃、および40℃である。該反応、および特にオキサゾリジノンと塩基との脱プロトン化は、しばしば、引き下げた温度(例えば、−20℃、−10℃、または0℃)で該塩基を加えることおよび次いで該混合物を室温まで温めることにより開始する。
【0029】
好ましい実施形態において、化合物Iは、式12の構造:
【0030】
【化3】
を有し、および以下に示されるように、化合物11および7の反応により作成される。
【0031】
【化4】
【0032】
12の完全な合成を、実施例1に示す。実施例1中の化合物11において、XはNaであり、これは、中性オキサゾリジノン(X=H)からの単離せずに作成され、および単離せずに、中間体7(ここで、Yは、Clである。)と反応される。実施例は、本発明を更に説明するために提供され、いずれかの方法において本発明を限定するものとして扱われるべきではない。本発明の範囲は、特許請求の範囲により定義される。
【実施例1】
【0033】
化合物12(式I)を合成するための完全な方法は、図式Iにおいて要約され、および続いて、段階的に、詳細に開示される。以下に示されるこの方法において、X基はHであり、Hは、単離されない中間体においてナトリウムに変換され、これは、カップリング反応を経て、およびY基はClである。
【0034】
【化5】
【0035】
定義
この適用を通して、および特に実施例において使用される用語は、製法研究の分野において作業する化学者に対して一般に周知である。これらのいくつかはまた、以下に定義される。
「EDC」は、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドである。
「DIPEA」は、ジイソプロピルエチルアミンである。
「DMAC」は、ジメチルアセトアミドである。
「DMSO」は、ジメチルスルホキシドである。
「DMF」は、ジメチルホルムアミドである。
「ハロゲン」は、フッ素、塩素、臭素、およびヨウ素を含む。
「HMPA」は、ヘキサメチルリン酸トリアミドである。
「HOBT」は、1−ヒドロキシベンゾトリアゾールである。
「IPAC」は、イソプロピルアセテートである。
「Me」は、メチルを表す。
「NaHMDS」は、ヘキサメチルジシラジド(disilazide)ナトリウムである。
「TMEDA」は、テトラメチルエチレンジアミンである。
「ワインレブ(Weinreb)アミン」は、N、O−ジメチルヒドロキシルアミンである。
【0036】
中間体7の合成
中間体7は、容易に入手可能な物質から、6段階において作成される。該合成は、ボロン酸中間体5の4段階合成として以下に要約され、このボロン酸中間体5は、固体物質として単離される。ボロン酸は、次いで、2つの更なる段階を経て、重要な中間体7(これはまた、固体生成物として単離される。)になる。
【0037】
ボロン酸中間体は、以下に示される、および図式2において要約される4段階において合成される。
【0038】
【化6】
【0039】
2への1の変換
THF(24L)を、室温で100Lの円筒形槽に加えた。これに、2.75kgのCeCl3を加えた。得られたスラリーを、1.5時間、室温で熟成した。試料を、次いで顕微鏡下で検査し、所望の形態変化が起こったことを確かめた。該スラリーを、9℃に冷却し、およびMeMgClを加えた。添加の速さを調整して、内部温度を19℃未満に維持した。混合物を−11℃に冷却し、内部温度を0℃未満に維持しながらアセトフェノン1の溶液(4.0kg、THFで10Lに希釈された)を滴下した。該反応混合物を、次いで、1時間、0℃未満の温度で熟成した。該反応を、5.7Lの3N HClを、滴下して停止し、内部温度は15℃未満に維持した。該停止された反応混合物を、次いで1.5時間、5〜10℃で熟成し、Solka Flocのプラグを通して濾過した。
【0040】
3への2の水素化
2のTHF溶液は、エタノール(約18Lの容量)に転換された溶媒であり、および1.9LのHClを加え、次いで190gmの10%Pd/C(50%水)を加えた。反応がHPLC分析に基づいて完了するまで、該混合物を、40℃で15psi水素下に置いた。該混合物を、室温まで冷却した。フィルターの補助としてSolka−Flokを用いて、濾過により、触媒を取り除いた。エタノール中のアニソール生成物を、次いで、次の段階のために溶媒をアセトニトリルに転換した。
【0041】
4への3の臭素化
アニソール3を、アセトニトリル(1.72L、4mL MeCN/mMol 3)中に希釈する。この混合物を、35℃に温め、およびNBS(1.1当量、84g)を、1回の固体添加において加える。該反応を、35℃で維持し、および2から4時間内に完了する。該溶液を、400mLの総容量まで濃縮し、1Lのトルエンで希釈する。該溶液を、次いで、チオ硫酸ナトリウムおよび水で洗浄し、副産物であるスクシンイミドを取り除く。有機層を、次いで濃縮し、溶媒をトルエンに転換する。
【0042】
ボロン酸5への臭化アリール4の変換
75Lのガラス反応槽を、1.87kgの臭化アリール4(7.6Mol)で充填し、これは、6.4kgの、トルエン中の4の29.1重量%溶液として加えられた。この溶液を、5.6LのTHFで希釈した。該槽に窒素を流し、トリイソプロピルホウ酸塩(1.35当量、2.35L、10.3Mol)を加えた。該混合物を、―70℃未満に冷却した。次いで、ヘキサン(9.5Mol)中の5.9Lの1.6M n−BuLiを、4時間に亘ってゆっくり加え、温度を55℃未満に維持した。n−BuLi添加の完了後30分に、該反応は、LC分析により完了した。該反応物を、−35℃まで温め、3.0M H2SO4溶液(5.6L)中に反応停止した。該反応停止後の水相は、酸性であるべきである(pH約2)。MTBE(7.5L)を、該混合物に加え、有機層を希釈した。該混合物を攪拌し(15分)、水層を取り出した。有機層を、もう1つの5.6Lの3.0M H2SO4溶液で洗浄した(15分)。層を再び分けた後、有機MTBE/トルエン層を、1M KOHで2回抽出した(初めに15.1Lおよび次いで7.6L)。2つのKOH抽出物を合わせ、2−プロパノール(6.4L)で希釈し、および15℃に冷却した。次いで、温度を15〜20℃に維持しながら、該溶液を、3.0M硫酸(約7.6L)を用いてpH約2にゆっくり酸性化した。得られたスラリーを、1時間攪拌し、次いで濾過した。濾過ケークを水で洗浄し(2×6L)、気流下で1日間乾燥した。濾過した固体を、真空下50℃で、2から3日間、炉中に置き、ジアーリル不純物を分解し、該固体を乾燥した。オフホワイト色の結晶性固体を単離して、ボロン酸5を得た。
【0043】
ボロン酸5を、次いで2段階(この段階は、以下の図式3において要約されおよび続く手順において詳細に記載される。)においてビアリール中間体7に変換する。
【0044】
【化7】
【0045】
段階1:6を得るためのボロン酸5および塩化アリール13のスズキカップリング反応
3M K2CO3溶液を、4.71kgの固体K2CO3を10.3Lの水に加えることにより調製する。冷却を適用し、溶液を20から25℃に保つ。THF(12L)、塩化アリール13(2.69kg)、およびボロン酸5(2.74kg)を、K2CO3に加え、次いで1L THFで濯ぐ。HPLC分析を使用して、5/13の1.00/1.00比を確認する。窒素ガスで70分間散布することにより、該溶液を、脱気する。触媒である、1,1ビス(ジ−tert−ブチルホスフィノ)フェロセンパラジウムジクロリド(42g)を、固体として加え、および次いで、脱気THF(1.5L)で濯ぐ。有機層は、すぐに暗褐色に変わる。2相の混合物を、活発に攪拌しながら36°から40℃で熟成する。HPLCが変換の完了を明らかにした後(15から18時間)、該混合物を、室温に冷却し、および水層を取り除く。有機層にヘプタン(25.6L)および水(25.6L)を加え、該層を取り出す。該有機層を、水(19L)で洗浄する。該有機層を、680gのDarco KB−Bで、室温で60分間処理し、10%THF/ヘプタン(約15L)で濯いでsolca−flocを通して濾過する。0.5v%未満のTHFが残るまで、約45から50℃で、該溶媒を、ヘプタン(約35L)に転換する。更なるヘプタンを加えて、約45から50Lの総容量をもたらす。種晶ベッドが全く形成しない場合、より早い操作から得られる結晶を種晶として入れる。該スラリーをゆっくり室温に冷却し、次いで−15℃に冷却する。−15℃で、1〜2時間熟成させた後、上澄み液のLC後のものは、上澄み液中の生成物の約2g/lの損失が存在することを示し、該スラリーを濾過し、および生成物を冷ヘプタン(約25L)で洗浄し、化合物6を提供する。
【0046】
段階2:7への6の塩素化
10℃で維持された、DMF(17L)中のビアリール化合物6(3.4kg)の溶液に、塩化チオニル(940ml)を加え、および次いで混合物を、室温まで温めた。99.8%を超える変換がHPLCにより測定されるまで、該混合物を熟成した。水(3.4L)を次いで加えた。より早いバッチから得られた種結晶(1重量%)を加え、約1時間に亘って5.1Lの更なる水をゆっくり加える前に、該混合物を、更に30分熟成した。該固体を濾過し、および初めに20Lの1:1DMF:水で洗浄し、次いで、3×20Lの水で洗浄した。0.1重量%未満の水が残るまで、固体生成物7を、20℃で乾燥した。
【0047】
(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジン−2−オン(11)のキラル合成
オキサゾリジノン中間体11を、以下に示される3段階の経路により、キラル開始物質CBZ−L−アラニン(8)から直接作成する。この化合物(4R,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジン−2−オンの鏡像異性体は、CBZ−D−アラニンから開始する類似した経路により作成され得る。
【0048】
段階1:9への8の変換
【0049】
【化8】
【0050】
CBZ−L−アラニン(6.5kg、28.5mol)、HOBT−水和物(4.8kg、34.8mol)、ワインレブアミンHCl塩(3.4kg、36.2mol)およびTHF(32L)を、窒素下で清潔なフラスコに充填する。該混合物を、0〜10℃に冷却し、および次いでDIPEA(12.4L)を、25℃未満の温度で、ゆっくりと加える。EDC−HCl(7Kg、36.2mol)を、次いで、15〜25℃で冷却しながらゆっくりと加える。該スラリーを、20°から25℃で一晩熟成する。該混合物を、次いで、0°〜10℃に冷却し、および3N HCl(12L)を、ゆっくり加える。次いで、IPAC(32L)を加え、および該層を分離する。有機層を、HCl(13L)で一回洗浄し、8%NaHCO3(13L)(注意:発泡)で2回洗浄する。該有機層を、次いで、真空下で、50℃で約15Lに濃縮する。清潔な溶液を、室温にゆっくり冷却し、該生成物を結晶化させる。ヘプタン(約70L)を、次いでゆっくり加える。該スラリーを濾過し、ヘプタン(18L)で洗浄し、および濾過ポット上で室温で乾燥する。生成物は、キラルHPLCにより測定して、99.9%eeを超えて得られる。
【0051】
段階2:10への9の変換
【0052】
【化9】
【0053】
前述の段階からのワインレブアミン9(6kg、22.5mol)および3,5−ビス(トリフルオロメチル)ブロモベンゼン(4.85L、28.1mol)を、無水THF(24L)中に溶解する。該溶液を、窒素でパージし、酸素を取り除く。水含量は、この地点で500ppm未満であるべきである。常圧蒸留が、実施され得、必要であれば、水を共沸的に取り除く。該溶液を、−10℃に冷却し、THF中のイソ−PrMgCl(56.4mol)を、ゆっくり反応物に、添加漏斗を介して加え(2時間)、反応温度を−5℃以下に維持する。アミドが0.5LCAPより小さくなるまで、該溶液を、20℃に温め、20℃で一晩熟成する。該反応物を、次いで窒素下で−10℃に冷却して、0から5℃で維持される5N HCl(14L)中に、2時間に亘ってゆっくり反応を停止する。MTBE(12L)を加えて、および2相性混合物を、5分間攪拌する。20から25℃に温めた後、30分間落ち着かせ、次いで該層を分ける。該有機層を、水で2回(12L)洗浄する。
【0054】
有機層を、1−ミクロンインラインPTFEフィルターを通して蒸留フラスコ中に真空供給し、次いで真空化で約12L(内部温度40℃未満)から最小の攪拌された容量に濃縮する。次いで、溶液を、トルエンで共沸的に乾燥し、再び最小の攪拌された容量にする。ケトン10を含む溶液を、次の段階において直接使用する。
【0055】
段階3:キラルオキサゾリジノン11へのケトン10の還元
【0056】
【化10】
【0057】
ケトン10(6kg)を、50℃で、12L IPA中の0.3当量のAl(O−i−Pr)3(790g)および18Lのトルエンと15.5時間加熱する。該溶液を、周囲温度に冷却し、温度を25℃未満に保ったまま、激しく攪拌した状態で、固体KOHペレット(13.5kg)をゆっくり加える。約2時間後、HPLCが99.5%より大きい環化を示す場合、33Lの1N HCl溶液を加えて反応を停止し、これを、25℃未満に保つ。固体の断片層が形成する場合、それを、濾過するべきである。該断片層は、ラセミオキサゾリジノンであり、除去は、鏡像体過剰率を増加させる。有機層を、次いで36Lの0.5N HClで初めに洗浄し、次いで45Lの水を合わせた6LのIPAで洗浄し、最後に36Lの水を合わせた6LのIPAで洗浄する。該有機層を、インラインフィルターを介して移す。2v%未満のトルエンが残るまで、該溶媒を、約40℃でヘプタン(標的容量は、約42Lである。)に転換する。室温で2時間熟成させ、固体生成物11を得る。
【0058】
7とのオキサゾリジノン11のアルキル化
オキサゾリジノン11を7とアルキル化し、所望の生成物である、(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−3−{[4’−フルオロ−5’−イソプロピル−2’−メトキシ−4−(トリフルオロメチル)ビフェニル−2−イル]メチル}−4−メチル−1,3−オキサゾリジノン−2−オン(12)を得る。
【0059】
【化11】
【0060】
上述で作成された、キラル中間体(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジノン−2−オン(11)を、DMF(32.7L中2.8kg)中に溶解し、−15℃に冷却する。2.0M NaHMDS(39.2L、1.05当量)を、次いで1.5時間に亘って加え、次いでDMF中の塩化ビアリール7(2.8kg)を加えた。該混合物を、+12℃に温め、および完全な変換が起こるまで、熟成した。次いで、5N HCl(3.4L)を加え、次いで16Lの10%IPAC/ヘプタンおよび34Lの水を加え、温度を10℃から20℃の間の全体にわたって保った。該層を取り出し、有機層を、14Lの1:1DMF:水で2回洗浄し、次いで14Lの水で2回洗浄した。有機層を、生成のために分析し、次いで2.4kgのシリカゲルを通して濾過し、0.5%未満までの過剰のオキサゾリジノンを取り除いた。シリカゲルを、5%IPAC/ヘプタンで洗浄した。合わせた有機溶液を蒸留し、1%未満までのIPACを取り除いた。温めたヘプタン溶液を、次いで、10重量%種晶を含む20℃ヘプタン溶液にゆっくり移した。該種結晶を、同じ反応のより早いバッチから最初に得た。該スラリーを次いで−20℃に冷却し、濾過した。濾過ケークを、冷ヘプタンで洗浄し、次いで乾燥し、4.4kg(88%)の所望の生成物12を得た。
【0061】
化合物12の多形
ヘプタンから濾過により上記のように単離される濾過ケークは、初めは結晶性ヘプタン溶媒和物である。濾過および乾燥の間、ヘプタンは蒸発し、無水非溶媒和結晶性生成物を生じる。ヘプタンは、窒素もしくは大気の流れの下または真空下で、室温で脱溶媒する(desolvates)。結晶性生成物は、約69℃で融解する(図2)。化合物12の結晶性非溶媒和形態は、含水せず、湿ったまたは乾燥した大気において水和物に変換しない。化合物12の結晶性非溶媒和形態は、室温で、継続的に他の結晶性形態に変換しないが、長時間、継続的に非晶質形態にゆっくり変換し、および高温で非晶質形態に、より急速に変換する。化合物12の非晶質形態はまた、粉砕により結晶性形態から、およびスプレー乾燥によりまたは貧溶媒(antisolvent)として水を使用する析出により、有機溶媒中の溶液から得られ得る。
【0062】
上述の方法により得られる該結晶性生成物12は、医薬調合物を作成するために使用され得る。化合物12は水中にほとんど溶解しないので、生物学的利用能を増やす形態において化合物12を調合することは、一般に有益である。結晶性生成物12が使用され、もう1つの形態(例えば溶液として、ポリマー中の非晶質懸濁液として、またはプレコンセントレート(preconcentrate)が嚥下されまたは水と混合された後にマイクロエマルジョンを生じるプレコンセントレートの一部として)に有効成分が変化する、医薬調合物を作成することが出来る。化合物12の結晶性非溶媒和形態は、これらの調合物を作成するのに有用な中間体であるが、なぜなら、この形態は、精製しやすくおよび取り扱い易く、含水ではなく、および非晶質形態に対する変化に対して適度な期間、室温で安定であるからである。
【0063】
化合物12を含む医薬調合物は、検出可能な量における化合物12の結晶性非溶媒和形態を含み得る。固体における化合物12の結晶性非溶媒和形態の量は、物理的方法(X線粉末回析法(XRPD)、固体フッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴分光学、固体炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴分光学、固体フーリエ変換赤外分光法、およびラマン分光法など)の使用により定量化することが出来る。化合物12を含む医薬調合物は、約5重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る(調合物中の化合物12の量の%として)。化合物12を含む医薬調合物は、約10重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約25重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約50重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約75重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。調合物中の固体化合物12が、実質的に相純粋結晶性非溶媒和形態であるように、化合物12を含む医薬調合物は、約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。
【0064】
該調合物が、非晶質(例えば、非晶質化合物12の調合物、水溶性ポリマー(例えば、ポリビニルピロリジノン、ポリビニルピロリジノンコポリマー、またはHPMCASなどの水溶性セルロースポリマー)中の化合物12の非晶質懸濁液を含む調合物、またはマイクロエマルジョンプレコンセントレートなどの溶液中の化合物12を含む調合物など)になるために作成される場合でさえ、化合物12を含む医薬調合物は、検出可能な量における結晶性非溶媒和形態中の化合物12を含み得る。結晶性化合物が完全に非結晶性形態に変化しないまたは完全に溶解しないゆえ、または化合物12が長期間継続的に、結晶性非溶媒和形態に段階的に変換するゆえなどの、多くの理由のために、結晶性化合物12は、これらの調合物中に少量において存在し得る。化合物12を含む、このような医薬調合物において、該医薬調合物は、測定可能な量において結晶性非溶媒和形態中の化合物12を含み得、これは、調合物中の化合物12の総量の少なくとも0.1%を表す可能性があり、または調合物中の化合物12の総量の少なくとも0.5%を表す可能性があり、または調合物中の化合物12の総量の少なくとも1%を表す可能性があり、または調合物中の化合物12の総量の少なくとも5%を表す可能性があり、または調合物中の化合物12の総量の少なくとも10%を表す可能性があり、または調合物中の化合物12の総量の少なくとも25%を表す可能性があり、または調合物中の化合物12の総量の少なくとも50%を表す可能性がある。
【0065】
結晶性生成物は、以下で特徴付けられる。
【0066】
特徴付け方法
X線粉末回析検査は、分子の構造、結晶性、および多形性を特徴付けるために、広く使用される。該X線粉末回折パターンは、PW3040/60コンソールを備えるPhilips Analytical X’Pert PRO X線回折システム上で得られる。PW3373/00セラミックCu LEF X線管K−アルファ放射を、源として使用する。
【0067】
上記のX線粉末回折パターンに加えて、化合物の結晶性形態は、固体炭素−13およびフッ素−19核磁気共鳴(NMR)スペクトルによりさらに特徴付けられ得る。固体炭素−13NMRスペクトルは、Bruker4mm二重共鳴CPMASプローブを使用するBrukerDSX400WB上で得られる。該炭素−13NMRスペクトルは、可変振幅交差分極を有する、プロトン/炭素−13交差分極マジック角回転を利用する。該試料を、15.0kHzで回転させ、1024スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、40Hzの線幅拡大を該スペクトルに適用する。第2の基準としてグリシンのカルボニル炭素を用いるTMSスケール(176.03p.p.m)上で、化学シフトを報告する。
【0068】
該固体炭素−13NMRスペクトルをまた、Bruker4mmH/X/YCPMASプローブを用いるBrukerDSX500WB NMRシステム上で得る。該炭素−13NMRスペクトルは、可変振幅交差分極、総側波帯抑制、および100kHzで分断するSPINALを利用する。該試料を、10.0kHzで回転させ、1024スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、10Hzの線幅拡大を該スペクトルに適用する。第2の基準としてグリシンのカルボニル炭素を用いるTMSスケール(176.03p.p.m)上で、化学シフトを報告する。
【0069】
該固体フッ素−19NMRスペクトルを、Bruker4mmCRAMPSプローブを用いるBrukerDSX400WB NMRシステム上で得る。該NMRスペクトルは、単純なパルス獲得パルスプログラムを利用する。該試料を、15.0kHzで回転させ、128スキャンの全てを、5秒の再循環遅延で収集する。べスペル(vespel)エンドキャップを利用して、フッ素バックグラウンドを最小限にする。FTを行う前に、100Hzの線幅拡大を該スペクトルに適用する。−122ppmの化学シフトに割り当てられる外部の第2の基準としてポリ(テトラフルオロエチレン)(Teflon(登録商標))を用いて、化学シフトを報告する。
【0070】
該固体フッ素−19NMRスペクトルをまた、Bruker4mmH/F/X CPMASプローブを用いるBrukerDSX500WB NMRシステム上で得る。該フッ素−19NMRスペクトルは、可変振幅交差分極を有する、プロトン/フッ素−19交差分極マジック角回転、および62.5kHzで分断するTPPMを利用する。該試料を、15.0kHzで回転させ、256スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、10Hzの線幅拡大を該スペクトルに適用する。−122ppmの化学シフトに割り当てられる外部の第2の基準としてポリ(テトラフルオロエチレン)(Teflon(登録商標))を用いて、化学シフトを報告する。
【0071】
DSCデータをまた、TA InstrumentsDSC2910または同等の計測手段を用いて得る。2から6mgの間の重量を有する試料を、開いているはかり皿に量り分ける。このはかり皿を、次いで圧着し、熱量計セル中の試料位置中に置く。空のはかり皿を、基準位置に置く。熱量計セルを閉じ、窒素の流れを、セル中に通過させる。該加熱プログラムを設定し、約100℃の温度に10℃/分の加熱割合で、試料を加熱する。操作が完了する場合、システムソフトウェアにおけるDSC分析プログラムを用いて、データを分析する。吸熱が観察される温度範囲より上およびより下である基線温度点の間で、融解吸熱を、積分する。報告されたデータは、開始温度、ピーク温度およびエンタルピーである。
【0072】
化合物12の非晶質形態が、いくつかの試料において表される可能性があるので、更なる吸熱が、非晶質相存在のエンタルピー(enthalpic)緩和のゆえであり得るDSC曲線において観察される場合、調節されたDSC(MDSC)を使用し、この余分な吸熱が不純物の融解のゆえでないことを確かめる。MDSCは、単線的加熱割合の代わりの加熱割合における正弦のまたは調節された変化を使用する。これは、熱流量を可逆および非可逆成分に分けることを可能にする。非晶質物質のガラス転移は、試料の熱容量の変化のゆえに、基線における変化としての可逆熱流量曲線において検出される。
【0073】
DSCデータを、TA InstrumentsDSCQ1000を用いて得る。2から6mgの間の試料を、開いているはかり皿に量り分ける。このはかり皿を、次いで圧着し、熱量計セル中の試料位置中に置く。空のはかり皿を、基準位置に置く。熱量計セルを閉じ、窒素の流れを、セル中に通過させる。該加熱プログラムを設定し、60秒の調節期間および±1℃の振幅変調での3℃/分の加熱割合で、試料を加熱する。100℃になるように、最終温度を選択する。操作が完了する場合、システムソフトウェアにおけるDSC分析プログラムを用いて、データを分析する。吸熱が観察される温度範囲より上およびより下である基線温度点の間で、全熱流量曲線における融解吸熱を、積分する。報告されたデータは、開始温度、ピーク温度およびエンタルピーである。ガラス転移のゆえに可逆熱流量曲線の基線の変化が観察される場合、報告されたデータは、開始温度、中間点温度、終了温度および熱容量変化である。
【0074】
特徴付けデータ
図1は、結晶性非溶媒和形態の通常のX線粉末回折パターンを示す。結晶性非溶媒和形態は、4.66、4.59および4.36オングストロームの格子面間隔に相当する特徴的な回折ピークを示す。結晶性非溶媒和形態は、11.89、4.02および3.76オングストロームの格子面間隔により更に特徴付けられる。結晶性非溶媒和形態は、12.95、7.41および6.51オングストロームの格子面間隔により、なお更に特徴付けられる。
【0075】
図2は、化合物12の結晶性非溶媒和形態の通常のDSC曲線である。図2における69.62℃の外挿開始温度を有する吸熱は、融解(または結晶性非晶質相転移)のゆえである。
【0076】
図3は、化合物12の結晶性非溶媒和形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。結晶性非溶媒和形態は、123.4、55.8および23.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性非溶媒和形態の更なる特徴は、124.5、155.3および137.7p.p.mの化学シフト値を有するシグナルである。結晶性非溶媒和形態は、24.8、13.1および132.3p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0077】
図4は、化合物12の結晶性非溶媒和形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。結晶性非溶媒和形態は、−62.1、−63.9および−66.0p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性非溶媒和形態は、−115.2、−116.9および−118.3p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0078】
図5は、結晶性ヘプタン溶媒和形態の通常のX線粉末回折パターンを示す。ヘプタン溶媒和形態は、4.79、4.62および4.43オングストロームの格子面間隔に相当する特徴的な回折ピークを示す。結晶性ヘプタン溶媒和形態は、4.20、4.05および3.84オングストロームにより更に特徴付けられる。結晶性ヘプタン溶媒和形態は、13.12、11.99および5.52オングストロームにより、なお更に特徴付けられる。
【0079】
図6は、化合物12の結晶性ヘプタン溶媒和形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。結晶性ヘプタン溶媒和形態は、123.6、55.9および77.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性ヘプタン溶媒和形態の更なる特徴は、24.6、13.6および126.8p.p.mの化学シフト値を有するシグナルである。結晶性ヘプタン溶媒和形態は、52.3、130.5および23.2p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0080】
図7は、化合物12の結晶性ヘプタン溶媒和形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。結晶性ヘプタン溶媒和形態は、−61.8、−62.9および−65.2p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性ヘプタン溶媒和形態は、−114.8、−117.9および−116.7p.p.mの化学シフト値を有するシグナルにより、更に特徴付けられる。
【0081】
図8は、化合物12の非晶質形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。非晶質形態は、54.3、123.5および155.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。非晶質形態の更なる特徴は、22.3、76.6および138.1p.p.mの化学シフト値を有するシグナルである。非晶質形態は、159.8、12.3および98.9p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0082】
図9は、化合物12の非晶質形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。非晶質形態は、−63.3p.p.mの化学シフト値を有する特徴的なシグナルを示す。
【0083】
図10は、化合物12の非晶質形態の通常のMDSC曲線である。47.96℃の中間点温度を有する可逆熱流量曲線において観察される熱容量変化は、非晶質化合物12のガラス転移に相当する。
【0084】
有用性
本明細書に開示される方法により作られる化合物および結晶性多形体は、CETPの阻害剤であり、HDL−コレステロールの量を増加させる、および患者、好ましくはヒト患者におけるLDL−コレステロールの量を減少させることにおける有用性を有する。HDLの増加およびLDLの減少は、アテローム性動脈硬化および伴う疾患において有益な医薬の分野における医師に公知である。
【0085】
本明細書の方法により合成された化合物は、水性の環境において非常に低い可溶性を有し、錠剤を作成するための固体有効成分および賦形剤を用いて従来作られている調合物と比較して、経口の生物学的利用能を増加させる調合物に作成されると可能性がある。これらの調製方法において得られる結晶性生成物は、容易に精製され、およびそれらを油中およびまたは界面活性剤中に溶解させるまたはポリ(ビニルピロリジノン)などの水溶性ポリマー中の非結晶性懸濁液としてそれらを分散させることにより調合され得る。
【0086】
化合物12の結晶性非溶媒和物の典型的な調合物は、ゼラチンカプセルにおける使用のための565mgの溶液を作成するために、十分な油または油および表面活性剤との混合物中に溶解された5mg、10mg、50mg、100mgまたは150mgの用量を含む。このような用量は、1日に1度または2度投与されるであろう。このような調合物は、医薬調合物の当業者に周知である。
【技術分野】
【0001】
本発明は、コレステロールエステル転移タンパク質(CETP)を阻害する化合物を合成するための方法、およびこの方法により作成された特定の化合物の結晶性多形に関する。該方法の生成物は、哺乳動物におけるHDLコレステロールを上昇させ、アテローム性動脈硬化の治療および/または予防における、ならびにアテローム性動脈硬化の進行を遅延させることにおける有用性を有すると考えられる。
【背景技術】
【0002】
アテローム性動脈硬化および臨床的結果である冠動脈性心疾患(CHD)、卒中および末梢血管疾患は、産業化世界の保健医療制度にとって非常に大きな負担になっている。米国単独において、約1300万人の患者がCHDと診断されており、毎年50万人を超える死亡は、CHDが原因である。更に、人口の平均年齢が増加し、肥満および糖尿病における流行が広がり続けているので、この犠牲は、次の25年にわたって増えると予想される。
【0003】
CETPの阻害は、アテローム性動脈硬化の罹患率を減少させる、有望な新しいアプローチである。スタチンは、LDLコレステロール(「悪玉コレステロール」)を減少させることによるCHDの罹患率を減少させることにおいて重要であったが、HDLコレステロール(「善玉コレステロール」)を増加させることでは、比較的効果がない。CETP阻害剤は、HDLコレステロールを増加させ、および一般集団におけるCHDおよびアテローム性動脈硬化を減少させる、効力のある新たなツールを提供する可能性がある。CETP阻害剤およびスタチンの両方の投与は、アテローム性動脈硬化を治療するおよび予防するために、特に価値がある可能性がある。CETP阻害剤を含む医薬は、現在利用可能でない。Pfizerのトルセトラピブ(torcetrapib)は、現在、臨床試験の第III相中のCETP阻害剤である。
【発明の概要】
【0004】
(発明の概要)
本発明は、式Iを有する化合物を調製するための方法を提供する。これらの新規の化合物は、効力のあるCETP阻害剤:
【0005】
【化1】
である。
【0006】
該方法は、化合物Iの収束的合成を提供する。完全な方法には、2つの重要な中間体IIおよびIIIの合成が含まれる。該重要な中間体は、次いで方法の最終段階において結合され、このことは、以下に示される。
【0007】
【化2】
【0008】
式I、IIおよびIIIを有する化合物において、
R1は、HまたはC1〜4アルキルであり、このC1〜4アルキルは、1から5個のF基で場合により置換されており、
R2、R4、およびR5は、それぞれ独立して、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルからなる群から選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
R3は、H、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルから選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
aおよびbは、それぞれ独立して、1〜4からの整数から選択され、
cは、0〜2からの整数であり、
Xは、HまたはI族金属カチオン(例えば、Na、K、Li、またはCs)であり、ならびに
Yは、脱離基(すなわち、容易に置換される基)である。脱離基の例は、ハロゲン、C1〜3アルカノアート(例えば、アセテート)、トリフルオロアセテート、およびトリフレートを含む。
【0009】
XがHである場合、塩基もまた、該反応において含まれ、ここで、該塩基は、強塩基のアルカリ金属塩である。該アルカリ金属は、Na、K、Li、またはCsであり得、およびサブグループにおいて、該アルカリ金属は、NaまたはKであり得、他のサブグループにおいて、該アルカリ金属は、Naであり得、他のサブグループにおいて、該アルカリ金属は、Kであり得る。強塩基のアルカリ金属塩の例は、ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムを含む。該塩基の使用は、XがNa、K、Li、またはCsである中間化合物IIをもたらす。
【0010】
上記の反応の実施形態において、R1は、H、または1から5個のFで場合により置換されているC1〜3アルキルである。他の実施形態におけるR1は、1から3個のFで場合により置換されているC1〜2アルキルである。好ましい実施形態において、R1は、CH3である。
【0011】
該反応の実施形態において、R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、または1から5個のFで場合により置換されている−OC1〜3アルキルである。他の実施形態において、R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、または1から5個のFで場合により置換されている−OC1〜2アルキルである。他の実施形態において、R2、R4およびR5は、それぞれ独立して、C1〜3アルキル、CF3、−OCH3、−OCF3、およびFから選択される。
【0012】
他の実施形態において、それぞれのR2は、CH3またはCF3である。
【0013】
他の実施形態において、R2は、CF3である。
【0014】
他の実施形態において、R3は、C1〜3アルキル、−OC1〜3アルキル、またはFであり、ここで、C1〜3アルキルおよびOC1〜3アルキルは、1から5個のFで場合により置換されている。
【0015】
他の実施形態において、R3は、CH3、CF3またはFである。
【0016】
他の実施形態において、R3は、CF3である。
【0017】
いくつかの実施形態において、aは、1または2であり、および他の実施形態において、aは、2である。
【0018】
いくつかの実施形態において、bは、1〜3である。他の実施形態において、bは、2または3である。他の実施形態において、bは、3である。
【0019】
いくつかの実施形態において、cは、0または1である。他の実施形態において、cは、0である。
【0020】
いくつかの実施形態において、Xは、H、Na、またはKである。他の実施形態において、Xは、Na、またはKである。
【0021】
いくつかの実施形態において、Yは、ハロゲンである。他の実施形態において、Yは、Cl、BrまたはIである。他の実施形態において、Yは、ClまたはBrである。他の実施形態において、Yは、Clである。
【0022】
別の様に示していない限り、アルキル基は、直鎖または分枝鎖状のいずれでもよい。
【図面の簡単な説明】
【0023】
【図1】化合物12の結晶性非溶媒和形態の特徴的なX線粉末回折パターンを示す図である。
【図2】化合物12の結晶性非溶媒和形態の通常のDSC曲線を示す図である。
【図3】化合物12の結晶性非溶媒和形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図4】化合物12の結晶性非溶媒和形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図5】化合物12の結晶性ヘプタン溶媒和形態の特徴的なX線粉末回折パターンを示す図である。
【図6】化合物12の結晶性ヘプタン溶媒和形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図7】化合物12の結晶性ヘプタン溶媒和形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図8】化合物12の非晶質形態の通常の炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図9】化合物12の非晶質形態の通常のフッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴(NMR)スペクトルを示す図である。
【図10】化合物12の非晶質形態の通常の調節されたDSC(MDSC)曲線を示す図である。
【0024】
(発明の詳細な説明)
更なる実施形態が、以下に記載される。
【0025】
中間体IIにおいて、Xは、HまたはI族アルカリ金属(例えば、Na、K、Li、またはCs)である。該I族金属カチオンは、TMEDAなどのリガンドと、またはエーテルもしくはポリエーテル(IIのオキサゾリジノン群の負に荷電されたNの反応性を増加させる、クラウンエーテルなど)と錯体を形成され得る。Xはまた、Hであってよい。Xが金属カチオン(例えば、Na、K、Li、またはCs)である場合、中間体IIは、オキサゾリジノン(Xは、Hである。)と、金属ヒドリド、アルキル金属化合物、または反応性アルカリ金属アミドとの反応により作成され得る。実施例は、オキサゾリジノン(X=H)と、塩基(ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムなど)との反応を含む。XがHである場合、塩基もまた反応において含まれ、ここで、該塩基は、オキサゾリジノンを反応性化合物(ここで、Xは、Na、K、Li、またはCsである。)に変換するために使用される塩基の1つである。X基は、多くの実施形態において、KおよびNaから選択される。実施例1においてX基は、Naである。
【0026】
中間体IIIにおいて、Yは、脱離基(すなわち、容易に置換される基)である。該脱離基が置換された後、該脱離基は、通常陰イオンである。最も一般的な脱離基は、ハロゲン(Cl、Br、IまたはFなど)である。脱離は、有機酸(トリフレートまたはトリフルオロアセテートなど)の脱プロトン化された形態であってもよい。多くの実施形態において、脱離基Yは、Br、ClおよびIから選択される。多くの実施形態において、脱離基Yは、BrおよびClから選択される。Y基は、実施例1における化合物12の合成に関し、Clである。
【0027】
上記の反応において、中間体IIは、しばしば、中性のオキサゾリジノン(X=H)として反応槽に充填され、次いで、アルカリ金属塩に変換され(ここで、Xは、アルカリ金属である。)、および次いで単離されることなく、中間体IIIと反応する。この適用の目的のために、オキサゾリジノン(XはHである。)は、反応槽に充填される開始物質であるが、これは、オキサゾリジノン(Xは、アルカリ金属である。)のアルカリ金属塩と、中間体IIIとの反応である。あるいは、中間体IIおよびIIIは、最初に反応槽に充填され得、およびXがHである場合、次いで十分な塩基が加えられ、カップリング反応を引き起こす。これはまた、オキサゾリジノン(ここで、Xは、アルカリ金属である。)と中間体IIIとの反応である。
【0028】
該反応は、極性非プロトン性溶媒(HMPA、DMF、またはDMACなど)中で一般に実施される。多くの実施形態において、DMFは、溶媒として使用される。該反応は、温度の穏やかな条件下で進行する。代表的な穏やかな条件は、−20℃、−10℃、0℃、10℃、20℃、30℃、および40℃である。該反応、および特にオキサゾリジノンと塩基との脱プロトン化は、しばしば、引き下げた温度(例えば、−20℃、−10℃、または0℃)で該塩基を加えることおよび次いで該混合物を室温まで温めることにより開始する。
【0029】
好ましい実施形態において、化合物Iは、式12の構造:
【0030】
【化3】
を有し、および以下に示されるように、化合物11および7の反応により作成される。
【0031】
【化4】
【0032】
12の完全な合成を、実施例1に示す。実施例1中の化合物11において、XはNaであり、これは、中性オキサゾリジノン(X=H)からの単離せずに作成され、および単離せずに、中間体7(ここで、Yは、Clである。)と反応される。実施例は、本発明を更に説明するために提供され、いずれかの方法において本発明を限定するものとして扱われるべきではない。本発明の範囲は、特許請求の範囲により定義される。
【実施例1】
【0033】
化合物12(式I)を合成するための完全な方法は、図式Iにおいて要約され、および続いて、段階的に、詳細に開示される。以下に示されるこの方法において、X基はHであり、Hは、単離されない中間体においてナトリウムに変換され、これは、カップリング反応を経て、およびY基はClである。
【0034】
【化5】
【0035】
定義
この適用を通して、および特に実施例において使用される用語は、製法研究の分野において作業する化学者に対して一般に周知である。これらのいくつかはまた、以下に定義される。
「EDC」は、1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミドである。
「DIPEA」は、ジイソプロピルエチルアミンである。
「DMAC」は、ジメチルアセトアミドである。
「DMSO」は、ジメチルスルホキシドである。
「DMF」は、ジメチルホルムアミドである。
「ハロゲン」は、フッ素、塩素、臭素、およびヨウ素を含む。
「HMPA」は、ヘキサメチルリン酸トリアミドである。
「HOBT」は、1−ヒドロキシベンゾトリアゾールである。
「IPAC」は、イソプロピルアセテートである。
「Me」は、メチルを表す。
「NaHMDS」は、ヘキサメチルジシラジド(disilazide)ナトリウムである。
「TMEDA」は、テトラメチルエチレンジアミンである。
「ワインレブ(Weinreb)アミン」は、N、O−ジメチルヒドロキシルアミンである。
【0036】
中間体7の合成
中間体7は、容易に入手可能な物質から、6段階において作成される。該合成は、ボロン酸中間体5の4段階合成として以下に要約され、このボロン酸中間体5は、固体物質として単離される。ボロン酸は、次いで、2つの更なる段階を経て、重要な中間体7(これはまた、固体生成物として単離される。)になる。
【0037】
ボロン酸中間体は、以下に示される、および図式2において要約される4段階において合成される。
【0038】
【化6】
【0039】
2への1の変換
THF(24L)を、室温で100Lの円筒形槽に加えた。これに、2.75kgのCeCl3を加えた。得られたスラリーを、1.5時間、室温で熟成した。試料を、次いで顕微鏡下で検査し、所望の形態変化が起こったことを確かめた。該スラリーを、9℃に冷却し、およびMeMgClを加えた。添加の速さを調整して、内部温度を19℃未満に維持した。混合物を−11℃に冷却し、内部温度を0℃未満に維持しながらアセトフェノン1の溶液(4.0kg、THFで10Lに希釈された)を滴下した。該反応混合物を、次いで、1時間、0℃未満の温度で熟成した。該反応を、5.7Lの3N HClを、滴下して停止し、内部温度は15℃未満に維持した。該停止された反応混合物を、次いで1.5時間、5〜10℃で熟成し、Solka Flocのプラグを通して濾過した。
【0040】
3への2の水素化
2のTHF溶液は、エタノール(約18Lの容量)に転換された溶媒であり、および1.9LのHClを加え、次いで190gmの10%Pd/C(50%水)を加えた。反応がHPLC分析に基づいて完了するまで、該混合物を、40℃で15psi水素下に置いた。該混合物を、室温まで冷却した。フィルターの補助としてSolka−Flokを用いて、濾過により、触媒を取り除いた。エタノール中のアニソール生成物を、次いで、次の段階のために溶媒をアセトニトリルに転換した。
【0041】
4への3の臭素化
アニソール3を、アセトニトリル(1.72L、4mL MeCN/mMol 3)中に希釈する。この混合物を、35℃に温め、およびNBS(1.1当量、84g)を、1回の固体添加において加える。該反応を、35℃で維持し、および2から4時間内に完了する。該溶液を、400mLの総容量まで濃縮し、1Lのトルエンで希釈する。該溶液を、次いで、チオ硫酸ナトリウムおよび水で洗浄し、副産物であるスクシンイミドを取り除く。有機層を、次いで濃縮し、溶媒をトルエンに転換する。
【0042】
ボロン酸5への臭化アリール4の変換
75Lのガラス反応槽を、1.87kgの臭化アリール4(7.6Mol)で充填し、これは、6.4kgの、トルエン中の4の29.1重量%溶液として加えられた。この溶液を、5.6LのTHFで希釈した。該槽に窒素を流し、トリイソプロピルホウ酸塩(1.35当量、2.35L、10.3Mol)を加えた。該混合物を、―70℃未満に冷却した。次いで、ヘキサン(9.5Mol)中の5.9Lの1.6M n−BuLiを、4時間に亘ってゆっくり加え、温度を55℃未満に維持した。n−BuLi添加の完了後30分に、該反応は、LC分析により完了した。該反応物を、−35℃まで温め、3.0M H2SO4溶液(5.6L)中に反応停止した。該反応停止後の水相は、酸性であるべきである(pH約2)。MTBE(7.5L)を、該混合物に加え、有機層を希釈した。該混合物を攪拌し(15分)、水層を取り出した。有機層を、もう1つの5.6Lの3.0M H2SO4溶液で洗浄した(15分)。層を再び分けた後、有機MTBE/トルエン層を、1M KOHで2回抽出した(初めに15.1Lおよび次いで7.6L)。2つのKOH抽出物を合わせ、2−プロパノール(6.4L)で希釈し、および15℃に冷却した。次いで、温度を15〜20℃に維持しながら、該溶液を、3.0M硫酸(約7.6L)を用いてpH約2にゆっくり酸性化した。得られたスラリーを、1時間攪拌し、次いで濾過した。濾過ケークを水で洗浄し(2×6L)、気流下で1日間乾燥した。濾過した固体を、真空下50℃で、2から3日間、炉中に置き、ジアーリル不純物を分解し、該固体を乾燥した。オフホワイト色の結晶性固体を単離して、ボロン酸5を得た。
【0043】
ボロン酸5を、次いで2段階(この段階は、以下の図式3において要約されおよび続く手順において詳細に記載される。)においてビアリール中間体7に変換する。
【0044】
【化7】
【0045】
段階1:6を得るためのボロン酸5および塩化アリール13のスズキカップリング反応
3M K2CO3溶液を、4.71kgの固体K2CO3を10.3Lの水に加えることにより調製する。冷却を適用し、溶液を20から25℃に保つ。THF(12L)、塩化アリール13(2.69kg)、およびボロン酸5(2.74kg)を、K2CO3に加え、次いで1L THFで濯ぐ。HPLC分析を使用して、5/13の1.00/1.00比を確認する。窒素ガスで70分間散布することにより、該溶液を、脱気する。触媒である、1,1ビス(ジ−tert−ブチルホスフィノ)フェロセンパラジウムジクロリド(42g)を、固体として加え、および次いで、脱気THF(1.5L)で濯ぐ。有機層は、すぐに暗褐色に変わる。2相の混合物を、活発に攪拌しながら36°から40℃で熟成する。HPLCが変換の完了を明らかにした後(15から18時間)、該混合物を、室温に冷却し、および水層を取り除く。有機層にヘプタン(25.6L)および水(25.6L)を加え、該層を取り出す。該有機層を、水(19L)で洗浄する。該有機層を、680gのDarco KB−Bで、室温で60分間処理し、10%THF/ヘプタン(約15L)で濯いでsolca−flocを通して濾過する。0.5v%未満のTHFが残るまで、約45から50℃で、該溶媒を、ヘプタン(約35L)に転換する。更なるヘプタンを加えて、約45から50Lの総容量をもたらす。種晶ベッドが全く形成しない場合、より早い操作から得られる結晶を種晶として入れる。該スラリーをゆっくり室温に冷却し、次いで−15℃に冷却する。−15℃で、1〜2時間熟成させた後、上澄み液のLC後のものは、上澄み液中の生成物の約2g/lの損失が存在することを示し、該スラリーを濾過し、および生成物を冷ヘプタン(約25L)で洗浄し、化合物6を提供する。
【0046】
段階2:7への6の塩素化
10℃で維持された、DMF(17L)中のビアリール化合物6(3.4kg)の溶液に、塩化チオニル(940ml)を加え、および次いで混合物を、室温まで温めた。99.8%を超える変換がHPLCにより測定されるまで、該混合物を熟成した。水(3.4L)を次いで加えた。より早いバッチから得られた種結晶(1重量%)を加え、約1時間に亘って5.1Lの更なる水をゆっくり加える前に、該混合物を、更に30分熟成した。該固体を濾過し、および初めに20Lの1:1DMF:水で洗浄し、次いで、3×20Lの水で洗浄した。0.1重量%未満の水が残るまで、固体生成物7を、20℃で乾燥した。
【0047】
(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジン−2−オン(11)のキラル合成
オキサゾリジノン中間体11を、以下に示される3段階の経路により、キラル開始物質CBZ−L−アラニン(8)から直接作成する。この化合物(4R,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジン−2−オンの鏡像異性体は、CBZ−D−アラニンから開始する類似した経路により作成され得る。
【0048】
段階1:9への8の変換
【0049】
【化8】
【0050】
CBZ−L−アラニン(6.5kg、28.5mol)、HOBT−水和物(4.8kg、34.8mol)、ワインレブアミンHCl塩(3.4kg、36.2mol)およびTHF(32L)を、窒素下で清潔なフラスコに充填する。該混合物を、0〜10℃に冷却し、および次いでDIPEA(12.4L)を、25℃未満の温度で、ゆっくりと加える。EDC−HCl(7Kg、36.2mol)を、次いで、15〜25℃で冷却しながらゆっくりと加える。該スラリーを、20°から25℃で一晩熟成する。該混合物を、次いで、0°〜10℃に冷却し、および3N HCl(12L)を、ゆっくり加える。次いで、IPAC(32L)を加え、および該層を分離する。有機層を、HCl(13L)で一回洗浄し、8%NaHCO3(13L)(注意:発泡)で2回洗浄する。該有機層を、次いで、真空下で、50℃で約15Lに濃縮する。清潔な溶液を、室温にゆっくり冷却し、該生成物を結晶化させる。ヘプタン(約70L)を、次いでゆっくり加える。該スラリーを濾過し、ヘプタン(18L)で洗浄し、および濾過ポット上で室温で乾燥する。生成物は、キラルHPLCにより測定して、99.9%eeを超えて得られる。
【0051】
段階2:10への9の変換
【0052】
【化9】
【0053】
前述の段階からのワインレブアミン9(6kg、22.5mol)および3,5−ビス(トリフルオロメチル)ブロモベンゼン(4.85L、28.1mol)を、無水THF(24L)中に溶解する。該溶液を、窒素でパージし、酸素を取り除く。水含量は、この地点で500ppm未満であるべきである。常圧蒸留が、実施され得、必要であれば、水を共沸的に取り除く。該溶液を、−10℃に冷却し、THF中のイソ−PrMgCl(56.4mol)を、ゆっくり反応物に、添加漏斗を介して加え(2時間)、反応温度を−5℃以下に維持する。アミドが0.5LCAPより小さくなるまで、該溶液を、20℃に温め、20℃で一晩熟成する。該反応物を、次いで窒素下で−10℃に冷却して、0から5℃で維持される5N HCl(14L)中に、2時間に亘ってゆっくり反応を停止する。MTBE(12L)を加えて、および2相性混合物を、5分間攪拌する。20から25℃に温めた後、30分間落ち着かせ、次いで該層を分ける。該有機層を、水で2回(12L)洗浄する。
【0054】
有機層を、1−ミクロンインラインPTFEフィルターを通して蒸留フラスコ中に真空供給し、次いで真空化で約12L(内部温度40℃未満)から最小の攪拌された容量に濃縮する。次いで、溶液を、トルエンで共沸的に乾燥し、再び最小の攪拌された容量にする。ケトン10を含む溶液を、次の段階において直接使用する。
【0055】
段階3:キラルオキサゾリジノン11へのケトン10の還元
【0056】
【化10】
【0057】
ケトン10(6kg)を、50℃で、12L IPA中の0.3当量のAl(O−i−Pr)3(790g)および18Lのトルエンと15.5時間加熱する。該溶液を、周囲温度に冷却し、温度を25℃未満に保ったまま、激しく攪拌した状態で、固体KOHペレット(13.5kg)をゆっくり加える。約2時間後、HPLCが99.5%より大きい環化を示す場合、33Lの1N HCl溶液を加えて反応を停止し、これを、25℃未満に保つ。固体の断片層が形成する場合、それを、濾過するべきである。該断片層は、ラセミオキサゾリジノンであり、除去は、鏡像体過剰率を増加させる。有機層を、次いで36Lの0.5N HClで初めに洗浄し、次いで45Lの水を合わせた6LのIPAで洗浄し、最後に36Lの水を合わせた6LのIPAで洗浄する。該有機層を、インラインフィルターを介して移す。2v%未満のトルエンが残るまで、該溶媒を、約40℃でヘプタン(標的容量は、約42Lである。)に転換する。室温で2時間熟成させ、固体生成物11を得る。
【0058】
7とのオキサゾリジノン11のアルキル化
オキサゾリジノン11を7とアルキル化し、所望の生成物である、(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−3−{[4’−フルオロ−5’−イソプロピル−2’−メトキシ−4−(トリフルオロメチル)ビフェニル−2−イル]メチル}−4−メチル−1,3−オキサゾリジノン−2−オン(12)を得る。
【0059】
【化11】
【0060】
上述で作成された、キラル中間体(4S,5R)−5−[3,5−ビス(トリフルオロメチル)フェニル]−4−メチル−1,3−オキサゾリジノン−2−オン(11)を、DMF(32.7L中2.8kg)中に溶解し、−15℃に冷却する。2.0M NaHMDS(39.2L、1.05当量)を、次いで1.5時間に亘って加え、次いでDMF中の塩化ビアリール7(2.8kg)を加えた。該混合物を、+12℃に温め、および完全な変換が起こるまで、熟成した。次いで、5N HCl(3.4L)を加え、次いで16Lの10%IPAC/ヘプタンおよび34Lの水を加え、温度を10℃から20℃の間の全体にわたって保った。該層を取り出し、有機層を、14Lの1:1DMF:水で2回洗浄し、次いで14Lの水で2回洗浄した。有機層を、生成のために分析し、次いで2.4kgのシリカゲルを通して濾過し、0.5%未満までの過剰のオキサゾリジノンを取り除いた。シリカゲルを、5%IPAC/ヘプタンで洗浄した。合わせた有機溶液を蒸留し、1%未満までのIPACを取り除いた。温めたヘプタン溶液を、次いで、10重量%種晶を含む20℃ヘプタン溶液にゆっくり移した。該種結晶を、同じ反応のより早いバッチから最初に得た。該スラリーを次いで−20℃に冷却し、濾過した。濾過ケークを、冷ヘプタンで洗浄し、次いで乾燥し、4.4kg(88%)の所望の生成物12を得た。
【0061】
化合物12の多形
ヘプタンから濾過により上記のように単離される濾過ケークは、初めは結晶性ヘプタン溶媒和物である。濾過および乾燥の間、ヘプタンは蒸発し、無水非溶媒和結晶性生成物を生じる。ヘプタンは、窒素もしくは大気の流れの下または真空下で、室温で脱溶媒する(desolvates)。結晶性生成物は、約69℃で融解する(図2)。化合物12の結晶性非溶媒和形態は、含水せず、湿ったまたは乾燥した大気において水和物に変換しない。化合物12の結晶性非溶媒和形態は、室温で、継続的に他の結晶性形態に変換しないが、長時間、継続的に非晶質形態にゆっくり変換し、および高温で非晶質形態に、より急速に変換する。化合物12の非晶質形態はまた、粉砕により結晶性形態から、およびスプレー乾燥によりまたは貧溶媒(antisolvent)として水を使用する析出により、有機溶媒中の溶液から得られ得る。
【0062】
上述の方法により得られる該結晶性生成物12は、医薬調合物を作成するために使用され得る。化合物12は水中にほとんど溶解しないので、生物学的利用能を増やす形態において化合物12を調合することは、一般に有益である。結晶性生成物12が使用され、もう1つの形態(例えば溶液として、ポリマー中の非晶質懸濁液として、またはプレコンセントレート(preconcentrate)が嚥下されまたは水と混合された後にマイクロエマルジョンを生じるプレコンセントレートの一部として)に有効成分が変化する、医薬調合物を作成することが出来る。化合物12の結晶性非溶媒和形態は、これらの調合物を作成するのに有用な中間体であるが、なぜなら、この形態は、精製しやすくおよび取り扱い易く、含水ではなく、および非晶質形態に対する変化に対して適度な期間、室温で安定であるからである。
【0063】
化合物12を含む医薬調合物は、検出可能な量における化合物12の結晶性非溶媒和形態を含み得る。固体における化合物12の結晶性非溶媒和形態の量は、物理的方法(X線粉末回析法(XRPD)、固体フッ素−19交差分極マジック角回転(CPMAS)核磁気共鳴分光学、固体炭素−13交差分極マジック角回転(CPMAS)核磁気共鳴分光学、固体フーリエ変換赤外分光法、およびラマン分光法など)の使用により定量化することが出来る。化合物12を含む医薬調合物は、約5重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る(調合物中の化合物12の量の%として)。化合物12を含む医薬調合物は、約10重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約25重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約50重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。化合物12を含む医薬調合物は、約75重量%から約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。調合物中の固体化合物12が、実質的に相純粋結晶性非溶媒和形態であるように、化合物12を含む医薬調合物は、約100重量%の、化合物12の結晶性非溶媒和形態を含み得る。
【0064】
該調合物が、非晶質(例えば、非晶質化合物12の調合物、水溶性ポリマー(例えば、ポリビニルピロリジノン、ポリビニルピロリジノンコポリマー、またはHPMCASなどの水溶性セルロースポリマー)中の化合物12の非晶質懸濁液を含む調合物、またはマイクロエマルジョンプレコンセントレートなどの溶液中の化合物12を含む調合物など)になるために作成される場合でさえ、化合物12を含む医薬調合物は、検出可能な量における結晶性非溶媒和形態中の化合物12を含み得る。結晶性化合物が完全に非結晶性形態に変化しないまたは完全に溶解しないゆえ、または化合物12が長期間継続的に、結晶性非溶媒和形態に段階的に変換するゆえなどの、多くの理由のために、結晶性化合物12は、これらの調合物中に少量において存在し得る。化合物12を含む、このような医薬調合物において、該医薬調合物は、測定可能な量において結晶性非溶媒和形態中の化合物12を含み得、これは、調合物中の化合物12の総量の少なくとも0.1%を表す可能性があり、または調合物中の化合物12の総量の少なくとも0.5%を表す可能性があり、または調合物中の化合物12の総量の少なくとも1%を表す可能性があり、または調合物中の化合物12の総量の少なくとも5%を表す可能性があり、または調合物中の化合物12の総量の少なくとも10%を表す可能性があり、または調合物中の化合物12の総量の少なくとも25%を表す可能性があり、または調合物中の化合物12の総量の少なくとも50%を表す可能性がある。
【0065】
結晶性生成物は、以下で特徴付けられる。
【0066】
特徴付け方法
X線粉末回析検査は、分子の構造、結晶性、および多形性を特徴付けるために、広く使用される。該X線粉末回折パターンは、PW3040/60コンソールを備えるPhilips Analytical X’Pert PRO X線回折システム上で得られる。PW3373/00セラミックCu LEF X線管K−アルファ放射を、源として使用する。
【0067】
上記のX線粉末回折パターンに加えて、化合物の結晶性形態は、固体炭素−13およびフッ素−19核磁気共鳴(NMR)スペクトルによりさらに特徴付けられ得る。固体炭素−13NMRスペクトルは、Bruker4mm二重共鳴CPMASプローブを使用するBrukerDSX400WB上で得られる。該炭素−13NMRスペクトルは、可変振幅交差分極を有する、プロトン/炭素−13交差分極マジック角回転を利用する。該試料を、15.0kHzで回転させ、1024スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、40Hzの線幅拡大を該スペクトルに適用する。第2の基準としてグリシンのカルボニル炭素を用いるTMSスケール(176.03p.p.m)上で、化学シフトを報告する。
【0068】
該固体炭素−13NMRスペクトルをまた、Bruker4mmH/X/YCPMASプローブを用いるBrukerDSX500WB NMRシステム上で得る。該炭素−13NMRスペクトルは、可変振幅交差分極、総側波帯抑制、および100kHzで分断するSPINALを利用する。該試料を、10.0kHzで回転させ、1024スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、10Hzの線幅拡大を該スペクトルに適用する。第2の基準としてグリシンのカルボニル炭素を用いるTMSスケール(176.03p.p.m)上で、化学シフトを報告する。
【0069】
該固体フッ素−19NMRスペクトルを、Bruker4mmCRAMPSプローブを用いるBrukerDSX400WB NMRシステム上で得る。該NMRスペクトルは、単純なパルス獲得パルスプログラムを利用する。該試料を、15.0kHzで回転させ、128スキャンの全てを、5秒の再循環遅延で収集する。べスペル(vespel)エンドキャップを利用して、フッ素バックグラウンドを最小限にする。FTを行う前に、100Hzの線幅拡大を該スペクトルに適用する。−122ppmの化学シフトに割り当てられる外部の第2の基準としてポリ(テトラフルオロエチレン)(Teflon(登録商標))を用いて、化学シフトを報告する。
【0070】
該固体フッ素−19NMRスペクトルをまた、Bruker4mmH/F/X CPMASプローブを用いるBrukerDSX500WB NMRシステム上で得る。該フッ素−19NMRスペクトルは、可変振幅交差分極を有する、プロトン/フッ素−19交差分極マジック角回転、および62.5kHzで分断するTPPMを利用する。該試料を、15.0kHzで回転させ、256スキャンの全てを、5秒の再循環遅延で収集する。FTを行う前に、10Hzの線幅拡大を該スペクトルに適用する。−122ppmの化学シフトに割り当てられる外部の第2の基準としてポリ(テトラフルオロエチレン)(Teflon(登録商標))を用いて、化学シフトを報告する。
【0071】
DSCデータをまた、TA InstrumentsDSC2910または同等の計測手段を用いて得る。2から6mgの間の重量を有する試料を、開いているはかり皿に量り分ける。このはかり皿を、次いで圧着し、熱量計セル中の試料位置中に置く。空のはかり皿を、基準位置に置く。熱量計セルを閉じ、窒素の流れを、セル中に通過させる。該加熱プログラムを設定し、約100℃の温度に10℃/分の加熱割合で、試料を加熱する。操作が完了する場合、システムソフトウェアにおけるDSC分析プログラムを用いて、データを分析する。吸熱が観察される温度範囲より上およびより下である基線温度点の間で、融解吸熱を、積分する。報告されたデータは、開始温度、ピーク温度およびエンタルピーである。
【0072】
化合物12の非晶質形態が、いくつかの試料において表される可能性があるので、更なる吸熱が、非晶質相存在のエンタルピー(enthalpic)緩和のゆえであり得るDSC曲線において観察される場合、調節されたDSC(MDSC)を使用し、この余分な吸熱が不純物の融解のゆえでないことを確かめる。MDSCは、単線的加熱割合の代わりの加熱割合における正弦のまたは調節された変化を使用する。これは、熱流量を可逆および非可逆成分に分けることを可能にする。非晶質物質のガラス転移は、試料の熱容量の変化のゆえに、基線における変化としての可逆熱流量曲線において検出される。
【0073】
DSCデータを、TA InstrumentsDSCQ1000を用いて得る。2から6mgの間の試料を、開いているはかり皿に量り分ける。このはかり皿を、次いで圧着し、熱量計セル中の試料位置中に置く。空のはかり皿を、基準位置に置く。熱量計セルを閉じ、窒素の流れを、セル中に通過させる。該加熱プログラムを設定し、60秒の調節期間および±1℃の振幅変調での3℃/分の加熱割合で、試料を加熱する。100℃になるように、最終温度を選択する。操作が完了する場合、システムソフトウェアにおけるDSC分析プログラムを用いて、データを分析する。吸熱が観察される温度範囲より上およびより下である基線温度点の間で、全熱流量曲線における融解吸熱を、積分する。報告されたデータは、開始温度、ピーク温度およびエンタルピーである。ガラス転移のゆえに可逆熱流量曲線の基線の変化が観察される場合、報告されたデータは、開始温度、中間点温度、終了温度および熱容量変化である。
【0074】
特徴付けデータ
図1は、結晶性非溶媒和形態の通常のX線粉末回折パターンを示す。結晶性非溶媒和形態は、4.66、4.59および4.36オングストロームの格子面間隔に相当する特徴的な回折ピークを示す。結晶性非溶媒和形態は、11.89、4.02および3.76オングストロームの格子面間隔により更に特徴付けられる。結晶性非溶媒和形態は、12.95、7.41および6.51オングストロームの格子面間隔により、なお更に特徴付けられる。
【0075】
図2は、化合物12の結晶性非溶媒和形態の通常のDSC曲線である。図2における69.62℃の外挿開始温度を有する吸熱は、融解(または結晶性非晶質相転移)のゆえである。
【0076】
図3は、化合物12の結晶性非溶媒和形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。結晶性非溶媒和形態は、123.4、55.8および23.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性非溶媒和形態の更なる特徴は、124.5、155.3および137.7p.p.mの化学シフト値を有するシグナルである。結晶性非溶媒和形態は、24.8、13.1および132.3p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0077】
図4は、化合物12の結晶性非溶媒和形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。結晶性非溶媒和形態は、−62.1、−63.9および−66.0p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性非溶媒和形態は、−115.2、−116.9および−118.3p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0078】
図5は、結晶性ヘプタン溶媒和形態の通常のX線粉末回折パターンを示す。ヘプタン溶媒和形態は、4.79、4.62および4.43オングストロームの格子面間隔に相当する特徴的な回折ピークを示す。結晶性ヘプタン溶媒和形態は、4.20、4.05および3.84オングストロームにより更に特徴付けられる。結晶性ヘプタン溶媒和形態は、13.12、11.99および5.52オングストロームにより、なお更に特徴付けられる。
【0079】
図6は、化合物12の結晶性ヘプタン溶媒和形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。結晶性ヘプタン溶媒和形態は、123.6、55.9および77.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性ヘプタン溶媒和形態の更なる特徴は、24.6、13.6および126.8p.p.mの化学シフト値を有するシグナルである。結晶性ヘプタン溶媒和形態は、52.3、130.5および23.2p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0080】
図7は、化合物12の結晶性ヘプタン溶媒和形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。結晶性ヘプタン溶媒和形態は、−61.8、−62.9および−65.2p.p.mの化学シフト値を有する特徴的なシグナルを示す。結晶性ヘプタン溶媒和形態は、−114.8、−117.9および−116.7p.p.mの化学シフト値を有するシグナルにより、更に特徴付けられる。
【0081】
図8は、化合物12の非晶質形態の通常の固体炭素−13CPMAS NMRスペクトルを示す。非晶質形態は、54.3、123.5および155.1p.p.mの化学シフト値を有する特徴的なシグナルを示す。非晶質形態の更なる特徴は、22.3、76.6および138.1p.p.mの化学シフト値を有するシグナルである。非晶質形態は、159.8、12.3および98.9p.p.mの化学シフト値を有するシグナルにより、なお更に特徴付けられる。
【0082】
図9は、化合物12の非晶質形態の通常の固体フッ素−19CPMAS NMRスペクトルを示す。非晶質形態は、−63.3p.p.mの化学シフト値を有する特徴的なシグナルを示す。
【0083】
図10は、化合物12の非晶質形態の通常のMDSC曲線である。47.96℃の中間点温度を有する可逆熱流量曲線において観察される熱容量変化は、非晶質化合物12のガラス転移に相当する。
【0084】
有用性
本明細書に開示される方法により作られる化合物および結晶性多形体は、CETPの阻害剤であり、HDL−コレステロールの量を増加させる、および患者、好ましくはヒト患者におけるLDL−コレステロールの量を減少させることにおける有用性を有する。HDLの増加およびLDLの減少は、アテローム性動脈硬化および伴う疾患において有益な医薬の分野における医師に公知である。
【0085】
本明細書の方法により合成された化合物は、水性の環境において非常に低い可溶性を有し、錠剤を作成するための固体有効成分および賦形剤を用いて従来作られている調合物と比較して、経口の生物学的利用能を増加させる調合物に作成されると可能性がある。これらの調製方法において得られる結晶性生成物は、容易に精製され、およびそれらを油中およびまたは界面活性剤中に溶解させるまたはポリ(ビニルピロリジノン)などの水溶性ポリマー中の非結晶性懸濁液としてそれらを分散させることにより調合され得る。
【0086】
化合物12の結晶性非溶媒和物の典型的な調合物は、ゼラチンカプセルにおける使用のための565mgの溶液を作成するために、十分な油または油および表面活性剤との混合物中に溶解された5mg、10mg、50mg、100mgまたは150mgの用量を含む。このような用量は、1日に1度または2度投与されるであろう。このような調合物は、医薬調合物の当業者に周知である。
【特許請求の範囲】
【請求項1】
式I:
【化1】
の化合物を合成するための方法であり、
式IIと式IIIとの反応を含み、
【化2】
[式中、R1は、HまたはC1〜4アルキルであり、このC1〜4アルキルは、1から5個のF基で場合により置換されており、
R2、R4、およびR5は、それぞれ独立して、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルからなる群から選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
R3は、H、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルから選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
Xは、H、またはNa、K、Li、およびCsから選択されるI族アルカリ金属カチオンであり、
Yは、オキサゾリジノン環IIの−NX−基により置換され得る脱離基であり、
aおよびbは、それぞれ独立して、1から4の整数から選択され、ならびに
cは、0から2の整数である。]
ここで、IIおよびIIIの反応が、溶媒中で、およびIIのオキサゾリジノン環の−NX−基によるYの置換のために適した温度で実施され、
ならびにXがHである場合、反応が、塩基を更に含む、
方法。
【請求項2】
R1が、HまたはC1〜3アルキルであり、このC1〜3アルキルは、1から5個のF基で場合により置換されており、
R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、および1から5個のFで場合により置換されている−OC1〜3アルキルから選択され、
R3は、C1〜3アルキル、−OC1〜3アルキル、およびFから成る群から選択され、ここで、
C1〜3アルキル、および−OC1〜3アルキルが、1から5個のFで場合により置換されており、
Xが、Na、K、Li、およびCsからなる群から選択され、
Yが、Br、ClおよびIからなる群から選択され、
aが、1から2の整数であり、
bが、1から3の整数であり、ならびに
cが、0または1であり、
ここで、溶媒が、DMF、DMAC、HMPA、DMSO、またはこの混合物を含み、および反応が、40℃未満で実施される、請求項1の方法。
【請求項3】
R1が、1から3個のF基で場合により置換されているC1〜2アルキルであり、
R2およびR4が、それぞれ独立して、C1〜3アルキル、CF3、−OCH3、−OCF3、およびFから選択され、
R3が、CH3、CF3またはFであり、
Xが、Na、K、およびLiからなる群から選択され、
Yが、Br、Cl、およびIからなる群から選択され、
aが、2であり、
bが、2から3の整数であり、ならびに
cが、0であり、
ここで、溶媒が、DMFを含み、および反応が、30℃未満で実施される、請求項1の方法。
【請求項4】
R1が、CH3であり、
R2が、CF3であり、
R3が、CF3であり、
R4が、C1〜3アルキル、−OCH3、およびFからなる群から選択され、
Xが、NaまたはKであり、
Yが、ClまたはBrであり、
aが、2であり、
bが、2または3であり、ならびに
cが、0であり、
ここで、溶媒が、DMFを含み、および反応が、30℃未満で実施される、請求項1の方法。
【請求項5】
Xが、Hであり、ならびに塩基が、ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムからなる群から選択される、請求項1の方法。
【請求項6】
式12:
【化3】
の化合物を合成するための方法であり、
化合物11および化合物7との反応を含み、
【化4】
[式中、Xは、H、Cs、Na、K、およびLiからなる群から選択され、
ならびにYは、Br、Cl、およびIからなる群から選択される。]
ここで、11および7との反応が、溶媒中で、および11のオキサゾリジノン環の−NX−基によるYの置換のために適した温度で実施され、
ならびにXがHである場合、反応が、塩基を更に含む、
方法。
【請求項7】
溶媒がDMFを含み、および反応が30℃未満の温度で実施される、請求項6の方法。
【請求項8】
Xが、Naであり、およびYが、BrまたはClである、請求項7の方法。
【請求項9】
結晶性非溶媒和物または結晶性ヘプタン溶媒和物であることを特徴とする、式12を有する化合物。
【化5】
【請求項10】
結晶性非溶媒和物であることを特徴とする、式12を有する、請求項9の化合物。
【請求項11】
4.66、4.59および4.36オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項12】
11.89、4.02および3.76オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項13】
12.95、7.41および6.51オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項14】
4.66、4.59、4.36、11.89、4.02、3.76、12.95、7.41および6.51オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項15】
123.4、55.8、23.1、124.5、155.3、137.7、24.8、13.1、および132.3ppmの化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項16】
−62.1、−63.9、−66.0、−115.2、−116.9および−118.3ppmの化学シフト値を有する固体フッ素−19CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項17】
69.92℃の外挿開始温度を有する吸熱を有するDSC曲線により特徴付けられる、式12を有する請求項10の化合物。
【請求項18】
結晶性ヘプタン溶媒和物であることを特徴とする、式12を有する請求項9の化合物。
【請求項19】
4.79、4.62および4.43オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項20】
4.20、4.05および3.84オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項21】
13.12、11.99および5.52オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項22】
4.79、4.62、4.43、4.20、4.05、3.84、13.12、11.99および5.52オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項23】
123.6、55.9、77.1、24.6、13.6、126.8、52.3、130.5、および23.2ppmの化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項24】
−61.8.−62.9、−65.2、−114.8、−117.9および−116.7ppmの化学シフト値を有する固体フッ素−19CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項25】
式12を有する請求項9の化合物の結晶性非溶媒和形態を含む、医薬組成物。
【請求項26】
結晶性非溶媒和形態を特徴とする、式12を有する請求項9の化合物の検出可能な量を含む、請求項25の医薬組成物。
【請求項27】
結晶性非溶媒和形態を特徴とする、式12を有する請求項9の化合物の測定可能な量を含む、請求項25の医薬組成物。
【請求項28】
結晶性非溶媒和物を特徴とする、式12を有する請求項9の化合物の治療有効量を投与することを含む、治療の必要な患者におけるアテローム性動脈硬化を治療または予防する方法。
【請求項29】
アテローム性動脈硬化を治療または予防するための医薬の製造における、結晶性非溶媒和物であることを特徴とする、式12を有する請求項9の化合物の使用。
【請求項30】
(a)式12を有する請求項9の化合物の結晶性非溶媒和形態、および(b)スタチン、DPP−IV阻害剤、選択性PPAR−ガンマ部分アゴニスト、およびCB−1逆アゴニストからなる群から選択される第2の有効医薬成分を含む、医薬組成物。
【請求項1】
式I:
【化1】
の化合物を合成するための方法であり、
式IIと式IIIとの反応を含み、
【化2】
[式中、R1は、HまたはC1〜4アルキルであり、このC1〜4アルキルは、1から5個のF基で場合により置換されており、
R2、R4、およびR5は、それぞれ独立して、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルからなる群から選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
R3は、H、ハロゲン、C1〜4アルキル、および−OC1〜4アルキルから選択され、ここで、C1〜4アルキルおよび−OC1〜4アルキルは、1から5個のハロゲンで場合により置換されており、
Xは、H、またはNa、K、Li、およびCsから選択されるI族アルカリ金属カチオンであり、
Yは、オキサゾリジノン環IIの−NX−基により置換され得る脱離基であり、
aおよびbは、それぞれ独立して、1から4の整数から選択され、ならびに
cは、0から2の整数である。]
ここで、IIおよびIIIの反応が、溶媒中で、およびIIのオキサゾリジノン環の−NX−基によるYの置換のために適した温度で実施され、
ならびにXがHである場合、反応が、塩基を更に含む、
方法。
【請求項2】
R1が、HまたはC1〜3アルキルであり、このC1〜3アルキルは、1から5個のF基で場合により置換されており、
R2、R4およびR5は、それぞれ独立して、F、1から5個のFで場合により置換されているC1〜3アルキル、および1から5個のFで場合により置換されている−OC1〜3アルキルから選択され、
R3は、C1〜3アルキル、−OC1〜3アルキル、およびFから成る群から選択され、ここで、
C1〜3アルキル、および−OC1〜3アルキルが、1から5個のFで場合により置換されており、
Xが、Na、K、Li、およびCsからなる群から選択され、
Yが、Br、ClおよびIからなる群から選択され、
aが、1から2の整数であり、
bが、1から3の整数であり、ならびに
cが、0または1であり、
ここで、溶媒が、DMF、DMAC、HMPA、DMSO、またはこの混合物を含み、および反応が、40℃未満で実施される、請求項1の方法。
【請求項3】
R1が、1から3個のF基で場合により置換されているC1〜2アルキルであり、
R2およびR4が、それぞれ独立して、C1〜3アルキル、CF3、−OCH3、−OCF3、およびFから選択され、
R3が、CH3、CF3またはFであり、
Xが、Na、K、およびLiからなる群から選択され、
Yが、Br、Cl、およびIからなる群から選択され、
aが、2であり、
bが、2から3の整数であり、ならびに
cが、0であり、
ここで、溶媒が、DMFを含み、および反応が、30℃未満で実施される、請求項1の方法。
【請求項4】
R1が、CH3であり、
R2が、CF3であり、
R3が、CF3であり、
R4が、C1〜3アルキル、−OCH3、およびFからなる群から選択され、
Xが、NaまたはKであり、
Yが、ClまたはBrであり、
aが、2であり、
bが、2または3であり、ならびに
cが、0であり、
ここで、溶媒が、DMFを含み、および反応が、30℃未満で実施される、請求項1の方法。
【請求項5】
Xが、Hであり、ならびに塩基が、ナトリウムアミド、カリウムアミド、NaHMDS、KHMDS、n−ブチルリチウム、およびt−ブチルリチウムからなる群から選択される、請求項1の方法。
【請求項6】
式12:
【化3】
の化合物を合成するための方法であり、
化合物11および化合物7との反応を含み、
【化4】
[式中、Xは、H、Cs、Na、K、およびLiからなる群から選択され、
ならびにYは、Br、Cl、およびIからなる群から選択される。]
ここで、11および7との反応が、溶媒中で、および11のオキサゾリジノン環の−NX−基によるYの置換のために適した温度で実施され、
ならびにXがHである場合、反応が、塩基を更に含む、
方法。
【請求項7】
溶媒がDMFを含み、および反応が30℃未満の温度で実施される、請求項6の方法。
【請求項8】
Xが、Naであり、およびYが、BrまたはClである、請求項7の方法。
【請求項9】
結晶性非溶媒和物または結晶性ヘプタン溶媒和物であることを特徴とする、式12を有する化合物。
【化5】
【請求項10】
結晶性非溶媒和物であることを特徴とする、式12を有する、請求項9の化合物。
【請求項11】
4.66、4.59および4.36オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項12】
11.89、4.02および3.76オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項13】
12.95、7.41および6.51オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項14】
4.66、4.59、4.36、11.89、4.02、3.76、12.95、7.41および6.51オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項15】
123.4、55.8、23.1、124.5、155.3、137.7、24.8、13.1、および132.3ppmの化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項16】
−62.1、−63.9、−66.0、−115.2、−116.9および−118.3ppmの化学シフト値を有する固体フッ素−19CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項10の化合物。
【請求項17】
69.92℃の外挿開始温度を有する吸熱を有するDSC曲線により特徴付けられる、式12を有する請求項10の化合物。
【請求項18】
結晶性ヘプタン溶媒和物であることを特徴とする、式12を有する請求項9の化合物。
【請求項19】
4.79、4.62および4.43オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項20】
4.20、4.05および3.84オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項21】
13.12、11.99および5.52オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項22】
4.79、4.62、4.43、4.20、4.05、3.84、13.12、11.99および5.52オングストロームの格子面間隔に相当するXRPD回折ピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項23】
123.6、55.9、77.1、24.6、13.6、126.8、52.3、130.5、および23.2ppmの化学シフト値を有する固体炭素−13CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項24】
−61.8.−62.9、−65.2、−114.8、−117.9および−116.7ppmの化学シフト値を有する固体フッ素−19CPMAS NMRスペクトルにおけるピークにより特徴付けられる、式12を有する請求項18の化合物。
【請求項25】
式12を有する請求項9の化合物の結晶性非溶媒和形態を含む、医薬組成物。
【請求項26】
結晶性非溶媒和形態を特徴とする、式12を有する請求項9の化合物の検出可能な量を含む、請求項25の医薬組成物。
【請求項27】
結晶性非溶媒和形態を特徴とする、式12を有する請求項9の化合物の測定可能な量を含む、請求項25の医薬組成物。
【請求項28】
結晶性非溶媒和物を特徴とする、式12を有する請求項9の化合物の治療有効量を投与することを含む、治療の必要な患者におけるアテローム性動脈硬化を治療または予防する方法。
【請求項29】
アテローム性動脈硬化を治療または予防するための医薬の製造における、結晶性非溶媒和物であることを特徴とする、式12を有する請求項9の化合物の使用。
【請求項30】
(a)式12を有する請求項9の化合物の結晶性非溶媒和形態、および(b)スタチン、DPP−IV阻害剤、選択性PPAR−ガンマ部分アゴニスト、およびCB−1逆アゴニストからなる群から選択される第2の有効医薬成分を含む、医薬組成物。
【図1】

【図2】

【図3】
【図4】
【図5】

【図6】
【図7】

【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2013−14594(P2013−14594A)
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−181807(P2012−181807)
【出願日】平成24年8月20日(2012.8.20)
【分割の表示】特願2008−519594(P2008−519594)の分割
【原出願日】平成18年6月29日(2006.6.29)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
【公開日】平成25年1月24日(2013.1.24)
【国際特許分類】
【出願番号】特願2012−181807(P2012−181807)
【出願日】平成24年8月20日(2012.8.20)
【分割の表示】特願2008−519594(P2008−519594)の分割
【原出願日】平成18年6月29日(2006.6.29)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
[ Back to top ]