
PDGFRベータ阻害剤の新たな使用
本発明は、T細胞リンパ腫、特にNHL、ALCL及びPTCLの抗増殖治療における使用のためのPDGFRベータ阻害剤に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、PDGFRベータ阻害剤に関する新たな示唆に関する。
【背景技術】
【0002】
血小板由来増殖因子受容体ベータポリペプチド(別称、PDGFRB、PDGFRベータ、PDGF-Rβ又はCD140B)は、血小板由来増殖因子ファミリーのメンバーに関する細胞表面チロシンキナーゼ受容体をコードするヒト遺伝子である。これらの増殖因子は、間葉系起源の細胞に対するマイトジェンである。受容体単量体に結合する増殖因子の同一性により、機能性受容体がホモ二量体であるか、血小板由来増殖因子受容体のアルファポリペプチド及びベータポリペプチドの双方から成るヘテロ二量体であるかが決定される。この遺伝子は、5番染色体上で顆粒球マクロファージコロニー刺激因子及びマクロファージコロニー刺激因子受容体に関する遺伝子のそばに隣接され、3つの遺伝子は全て、5-q症候群と関係し得る。5番染色体と12番染色体との間の転座であって、5番染色体の遺伝子を転座の遺伝子、ETV6、白血病遺伝子に融合する転座は、好酸球増加症と共に慢性骨髄増殖性疾患を生じる。
【0003】
国際公開第2005/075454号(A2)は、白血病を含む様々な癌疾患の治療のためのイマチニブの使用を開示する。国際公開第2008/037716号(A2)は、慢性骨髄白血病及び消化管間葉性腫瘍の治療のためのニロチニブの使用を開示する。Verbeek等(Nat.Rev.Clin.Onc. 7 (2010) 116-119) は、消化管間葉性腫瘍に罹患する患者におけるイマチニブ誘発性T細胞増殖性疾患を開示する。Rassidakis等(Mod. Pathol 17 (2004) 946-953; 及びBlood 102 (2003), 4619-4620) は、c-kitはホジキン病及び未分化リンパ腫キナーゼ(ALK-) 陽性未分化大細胞リンパ腫(ALCL)株化細胞中に発現されず、従ってc-kitはイマチニブ等の治療剤の適した標的ではない、ことと報告する。Pullarkat等(Leuk. Res. 32 (2008), 1770-1775)は、巨核球性急性転化に罹患する患者の呈する慢性骨髄性発作が、イマチニブでの治療では効果がなかったことを記載する。
【0004】
国際公開第2007/053573号(A2)は、数百もの多くの腫瘍疾患、特にALCLを、有効量のソラフェニブで治療するための組成物及び方法を開示する。
【0005】
Lansigan等(J. Clin. Oncol. 28 (2010) TPS 300)は、再発性又は治療抵抗性T細胞リンパ腫におけるソラフェニブのパイロット研究を開示する。一方、Ambrosini等は、ソラフェニブは、ヒトACLC/HD-株化細胞においてアポトーシスを誘発することができたが、HD及びALCLのインビボ(in vivo)での治療におては効果がないことを記載する (Nguyen et al., Leuk. Res. 34 (2010), 379-386においても記載される)。
【0006】
米国特許出願第2005/0119352号は、例えば、発癌性キナーゼ修飾因子、好ましくはイマチニブと併用した、細胞周期チェックポイント活性化因子、好ましくはベータラパコン、又はその誘導体若しくは類似体の投与によってHDの治療が可能であることを開示する。
【0007】
先行技術を考慮すると、T細胞起源のリンパ腫の治療候補に関する報告は完全に矛盾していた。従って、このような患者、特にALCL患者のための信頼性があり且つ有効な治療が依然として必要である。
【発明の概要】
【課題を解決するための手段】
【0008】
従って、本発明は、細胞増殖性疾患感受性患者におけるT細胞リンパ腫の抗増殖治療のためのPDGFRB阻害剤を供する。それにより、腫瘍増殖を効果的に予防することができる。本発明の阻害剤は、全リンパ腫の治療のために使用することができる。好ましくは、リンパ腫は、NHL、ALCL及びPTCLから成る群から選択される。
【0009】
本発明の阻害剤は、特に再発患者の治療に有用である。従って、特に好ましい治療は、薬剤抵抗性患者の第二選択治療である。
【0010】
他の実施形態において、本発明の阻害剤は患者の第一選択治療のために使用される。
【0011】
本発明の阻害剤は、好ましくは、例えばポリペプチド又は小分子等の、PDGFRB拮抗薬から選択される。
【0012】
特定の実施形態において、本発明の阻害剤は、ニロチニブ、イマチニブ、ダサチニブ、ソラフェニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される。
【0013】
本発明の阻害剤を利用する治療方法は、好ましくは予防又は治療使用のための方法である。
【0014】
好ましくは、本発明の阻害剤は、局所又は全身使用のために、より好ましくは経口使用のために製剤される。
【0015】
好ましい治療レジメントは、0.001 mg/kg/日〜約100 mg/kg/日の用量でイマチニブ等の阻害剤を供する。従って、本発明の好ましい阻害剤は、そのような用量の投与を供するよう製剤される。
【0016】
本発明の好ましい実施形態は、CHOP療法又はこの治療の変更を含む、化学療法及び/又は放射線療法と併用の阻害剤の使用に関する(8)。
【0017】
本発明はさらに、T細胞リンパ腫の治療のためのPDGFRベータ阻害剤を含む医薬製剤の調製方法を供する。PDGF(血小板由来増殖因子)は、血管形成、腫瘍細胞の増殖刺激、腫瘍血管新生及び腫瘍線維芽細胞の動員及び制御に必須である。ホモ又はヘテロ二量体を形成し、2つの異なる受容体 (PDGFRA及びPDGFRB)に結合することができる4つの異なるリガンド (PDGF A-D)が存在する。
【0018】
活性化チロシンキナーゼは、慢性及び急性白血病の発病と関係し、阻害剤治療のための有力な標的を意味する。一部の阻害剤は、癌治療において使用される。
【0019】
イマチニブ(4-[(4-メチルピペラジン-1-イル)メチル]-N-[4-メチル-3-[(4-ピリジン-3-イルピリミジン-2-イル)アミノ]フェニル]ベンズアミド)は、特定のタイプの治療に使用される薬剤である。それは、NovartisからGleevec(登録商標)(アメリカ)又はそのメシル酸塩としてのGlivec(登録商標)(ヨーロッパ/オーストラリア)、イマチニブメシル酸塩(INN)との名称で現在市販される。それは、慢性骨髄性白血病(CML)、消化管間葉性腫瘍(GISTs)及び多数の他の悪性腫瘍の治療において使用される。イマチニブは、肺高血圧症の治療においてもまた機能する。それは、門脈肺高血圧症を含む様々な疾患過程において肺脈管構造の平滑筋肥大及び過形成の双方を減少することが示されている。
【0020】
イマチニブは、多数のチロシンキナーゼ (TK) 酵素の特異的阻害剤として機能する。それは、TK活性部位を占有し、活性の低下につながる。体内には、インスリン受容体を含む多数のTK酵素が存在する。イマチニブは、abl (エーベルソン癌原遺伝子)、c-kit及びPDGF-R (血小板由来増殖因子受容体)におけるTKドメインに特異的である。
【0021】
研究室において、イマチニブは、その受容体PDGFRBを阻害することによって、血小板由来増殖因子を抑制する実験薬剤として使用される。その効果の1つは、糖尿病マウスにおいて粥状動脈硬化を遅延させることである。
【0022】
慢性骨髄性白血病において、フィラデルフィア染色体は、bcr-ablと呼ばれるablのbcr (易切断領域)との融合タンパク質につながる。これは連続活性があるチロシンキナーゼであるので、bcr-abl活性を低下させるためにイマチニブが使用される。
【0023】
チロシンキナーゼの活性部位はそれぞれ、ATPの結合部位を有する。チロシンキナーゼによって触媒される酵素活性は、ATP由来の末端のリン酸のその基質上のチロシン残基への転移であり、プロテインチロシンリン酸化反応として周知の工程である。イマチニブは、bcr-ablのATP結合部位に結合し、タンパク質の酵素活性を競合的に抑制することによって機能する。
【0024】
イマチニブは、bcr-ablに非常に選択性があり、上記の他の標的(c-kit及びPDGF-R)も阻害するが、他にその選択性があるチロシンキナーゼは分かっていない。
【0025】
塩酸一水和物塩形態の、ニロチニブ (4-メチル-N-[3-(4-メチル-1H-イミダゾール-1-イル)-5-(トリフルオロメチル)フェニル]-3-[(4-ピリジン-3-イルピリミジン-2-イル) アミノ]ベンズアミド)は、同様にbcr-ablを阻害するチロシンキナーゼ阻害剤である。
【0026】
それは米国及び欧州において、第二選択治療薬剤抵抗性、フィラデルフィア染色体陽性慢性骨髄性白血病(CML)用の、Tasigna(登録商標)として認可された。ニロチニブは、イマチニブでの治療抵抗性のあるCMLの症例において活性を示し、第一選択治療として現在使用される。
【0027】
抗腫瘍性剤として使用される他のbcr-abl阻害剤は、ダサチニブ(N-(2-クロロ-6-メチルフェニル)-2-[[6-[4-(2-ヒドロキシエチル)-1-ピペラジニル]-2-メチル-4-ピリミジニル]アミノ]-5-チアゾールカルボキサミド一水和物であり、Bristol-Myers Squibbから名称SPRYCEL(登録商標) で販売され、慢性期CMLにおいて活性を示す。
【0028】
名称Nexavar(登録商標)でBayerから販売される、ソラフェニブ(4-[4-[[4-クロロ-3-(トリフルオロメチル)フェニル]カルバモイルアミノ]フェノキシ]-N-メチル-ピリジン-2-カルボキサミドは、腎細胞癌の治療に使用される小分子B-RAF阻害剤であり、血小板由来増殖因子受容体 (PDGFR)及び血管内皮増殖因子受容体(VEGFR)ファミリー由来の受容体チロシンキナーゼに対して活性を有することが示されている。国際公開第2007/053573号(A2)において、ソラフェニブは、数百もの多くの腫瘍疾患、特にALCLにおいて使用されることが示された。しかしながら、当該公報は当業者が実施可能なように記載しておらず、これは推測上の性質にすぎない。従って、ソラフェニブは本発明の好ましいPDGFRベータ阻害剤とみなされず、少なくとも通常のALCLの治療に関する限り、本化合物群に含まれないとみなすことができる。
【0029】
アキシチニブ(N-メチル-2-[[3-[(E)-2-ピリジン-2-イルエチニル]-1H-インダゾール-6-イル]スルファニル]ベンズアミド(別称、AG013736)は、小分子チロシンキナーゼ阻害剤であり、Pfizerにより開発中である。それは、VEGFR-1、VEGFR-2、VEGFR-3、血小板由来増殖因子受容体 (PDGFR)、及びc-Kit (CD117)を含む、複数の標的を阻害する。それは、異種移植モデルにおいて癌の成長を顕著に阻害することが示されており、腎細胞癌 (RCC) 及び複数の他の腫瘍型での試験において成功している。
【0030】
名称Sutent(登録商標)でPfizerから販売される、スニチニブ (N-[2-(ジエチルアミノ)エチル]-5-[(Z)-(5-フルオロ-1,2-ジヒドロ-2-オキソ-3H-インドール-3-イリデン)メチル]-2,4-ジメチル-1H-ピロール-3-カルボキサミドは、腫瘍血管新生及び腫瘍細胞増殖の双方において関与する血小板由来増殖因子のためのそれらの受容体及び血管内皮増殖因子受容体(VEGFRs)を含む、複数の受容体チロシンキナーゼ(RTKs)を標的にすることによって細胞内シグナル伝達を阻害する。従って、これらの標的の同時阻害は、腫瘍血管新生の減少及び癌細胞死の双方を導き、そして最終的に腫瘍の萎縮を導く。腫瘍がc-kitにおいて突然変異を生じこれによりイマチニブに抵抗性が生じている患者、又は薬剤に不耐性を生じている患者のための第二選択治療として、それは推奨されている。
【0031】
トセラニブ ((Z)-5-[(5-フルオロ-2-オキソ-1,2-ジヒドロ-3H-インドール-3-イリデン)メチル]-2,4-ジメチル-N-(2-ピロリジン-1-イルエチル)-1H-ピロール-3-カルボキサミド)は、受容体チロシンキナーゼ阻害剤であり、イヌマスト細胞腫瘍(別称、マスト細胞腫)の治療において使用される。それは、米国食品医薬品局に認可された唯一のイヌ特異性抗癌剤である。そのリン酸塩、リン酸トセラニブ (INN) はPalladia(登録商標) としてPfizerから販売される。トセラニブは、チロシンキナーゼ阻害剤の種類に属する。トセラニブは、イヌマスト細胞腫瘍の25-50%において変異しているc-Kitと呼ばれる特異的腫瘍細胞受容体、及び腫瘍血管新生に関与する2つの他の血管細胞受容体、PDGFR及びVEGFRを標的とするようデザインされる。
【0032】
未分化大細胞リンパ腫(ALCL) は、非ホジキンリンパ腫、主にT細胞起源である悪性の高いタイプである。全非ホジキンリンパ腫症例の約5%に含まれる。ALCL患者の約50%は、転座t(2;5)(p23;q35)を有し、発癌性融合タンパク質NPM-ALK (ヌクレオフォスミン未分化リンパ腫キナーゼ) を生じる。非常に低い頻度であるが、例えば、ATIC、CLTCL、MSN、RanBP2、TFG及びTPM3等の他のタンパク質との融合が観察されている。
【0033】
ALKは、インスリン受容体スーパーファミリーの膜貫通受容体チロシンキナーゼ (RTK) である。内在性ALK発現は、神経系に制限される。それは新生児脳において最も豊富であり、低レベルであるが成人脳において維持される。プレイオトロフィン受容体に関するALKについて強力な証拠が示されているが、ALKの正確な機能に関して研究は進行中である。発癌性ALK融合タンパク質と反対に、内在性ALKは造血組織において発現されない。
【0034】
NPM (ヌクレオフォスミン) は、リボソーム生合成及び中心体複製に関与する核小体タンパク質であり、ストレス刺激への応答において上方制御され、複数の腫瘍抑制遺伝子の機能を調整する。
【0035】
NPM-ALK融合タンパク質 [t(2:5)(p23;q35)] の恒常的過剰発現及び活性化は、ALCLの生存及び増殖を促進する重要な発癌性事象である。
【0036】
受容体チロシンキナーゼALKの触媒部分は、RNA-結合核小体リン酸化タンパク質NPMのオリゴマー形成ドメインと融合する。これは、リンパ系ALCL細胞においてNPM-ALKのホモ二量体形成及び自己リン酸化反応及びALKの発現を通して、ALKの恒常的活性化を導く。ALKの異常発現は、直接ALCLの形成に関与する。
【0037】
Galkin等 (PNAS 104 (2007), 270-275)は、非常に強力且つ選択的な小分子ALK阻害剤、NVP-TAE684を同定した。斯かる阻害剤は、ALCL由来及びALK依存性株化細胞の成長をブロックし、IC50値は2〜10 nMでであった。NVP-TAE684は、インビボ(in vivo)において、ALK-陽性ALCLの2つの独立モデルにおいてリンパ腫発生を抑制することが示され、樹立Karpas-299リンパ腫の退行を誘導した。NVP-TAE684はまた、CD30発現の下方制御を誘導し、これはCD30が治療上のNPM-ALKキナーゼ活性阻害のバイオマーカーとして使用され得ることを示している。
【0038】
NPM-ALKは、インビトロ(in vitro)及びインビボ(in vivo)で形質転換活性を有し、広範囲の発癌性因子及び例えばJak/Stat又はPI3K/Akt等のシグナル伝達ネットワークと相互作用している。ALK陽性(ALK+) ALCLはまた、サイトカイン受容体CD30及び活性化タンパク質 (AP-1) 転写因子ファミリーメンバーJunB及びcJunの高発現を特徴とする。
【0039】
AP-1はDNA結合転写因子であり、細胞増殖、分化及びアポトーシス、並びに発癌性形質転換を含む多くのプロセスにおいて重要な因子である。それは、Fos又はATFファミリーメンバーとホモ二量体又はヘテロ二量体を形成するJunファミリーメンバー (cJun, JunB, JunD) から成る。JunB及びcJunは、度々拮抗薬機能を有する。JunBは、アポトーシス促進性又は抗アポトーシス性のあることが報告され、それは細胞状況次第である。ALCLにおいて、JunB及びcJunが過剰発現されることが確認されており、CD30を活性化するJunBの役割が記載される。従って、cJun及びJunBは、腫瘍増殖と関係付けられる (Mathas et al., EMBO J. 21 (2002), 4104-4113; Kenner et al., J. Cell Biol. 164 (2004), 613-623; Watanabe et al., Can. Res. 65 (2005), 7628-7634; Jacobsen, The Oncologist 11 (2006), 831-840)。
【0040】
全身ALCLは、最終的に死に至る可能性のある進行性疾患である。
【0041】
全身ALCLと診断された患者は、最初の治療手段として度々CHOPで治療を受ける。CHOPは、複数の化学療法剤(シクロホスファミド、ヒドロキシダウノマイシン (ドキソルビシン) 、オンコビン(登録商標) (ビンクリスチン) 、及びプレドニゾン)の組み合わせである。他の化学療法の組み合わせもまた第一の選択として使用することができる。
【0042】
さらに、局在部位に特に大量のリンパ腫が存在する場合又は局所性大リンパ節が正常な器官又は構造を圧迫又は侵襲し、化学療法ではその問題を制御できない場合、放射線療法は治療の重要な補助となり得る。
【0043】
第一選択の組み合わせ治療後の治療選択には、放射線療法と併用した別のより高用量の化学療法を含むことができる。このような高用量の組み合わせ治療は、腫瘍中のリンパ腫細胞及び骨髄中に位置し得るリンパ腫細胞を標的とする。しかしながら、骨髄中の正常な造血細胞もまた死滅され得る。このため、患者自身の血液から回収された循環血液幹細胞又は組織適合血縁又は非血縁ドナー由来の循環血液幹細胞の骨髄移植又は移植が必要とされる。このような治療に耐性を示す又は治療後に再発する患者は、治療選択が制限されている。
【図面の簡単な説明】
【0044】
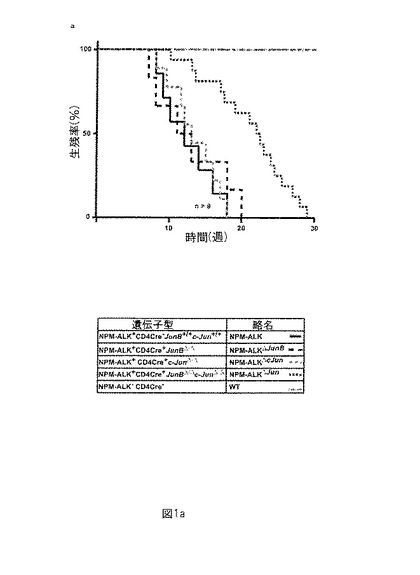
【図1−1】図1aは、JunB及びcJunのT細胞特異的ノックアウトが、NPM-ALKトランスジェニックマウスの生存期間を顕著に増加することを示す(G. Inghirami; University of Turin)。(a) マウスの5つの群、NPM-ALK、NPM-ALKΔJunB(E.F. Wagner)、NPM-ALKΔcJun(E.F. Wagner)、NPM-ALKΔJun及び野生型マウスを使用した。NPM-ALKΔJunマウスはNPM-ALKマウスより顕著に長く生存したが、NPM-ALKΔJunB及びNPM-ALKΔcJunマウスの生存期間に違いはなかった。
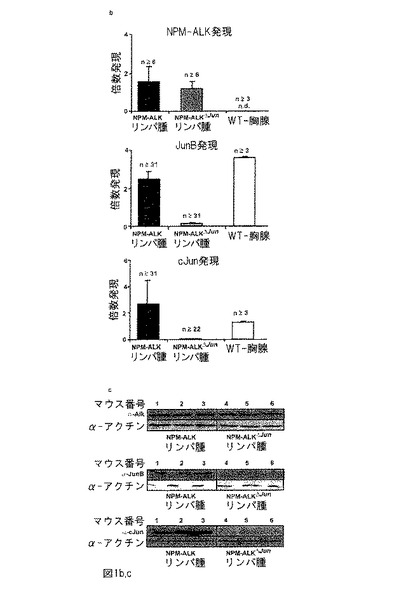
【図1−2】図1bは、qRT-PCRによるNPM-ALK、JunB及びcJun mRNAレベルの分析を表す。NPM-ALK mRNA発現レベルは、NPM-ALK及びNPM-ALKΔJun腫瘍において同様であるが、野生型胸腺では発現は欠如している。JunB発現は野生型胸腺及びNPM-ALK腫瘍において高く、NPM-ALKΔJun腫瘍においては低い。cJun発現はNPM-ALKリンパ腫及び野生型脾臓において高いが、NPM-ALKΔJun腫瘍において低い。図1cは、ウエスタンブロットによるNPM-ALK、JunB及びcJunタンパク質レベルの分析を表す。ALKレベルは、NPM-ALK及びNPM-ALKΔJun腫瘍で同等であり、JunB及びcJunはNPM-ALKに存在するが、NPM-ALKΔJun腫瘍中に存在しない。
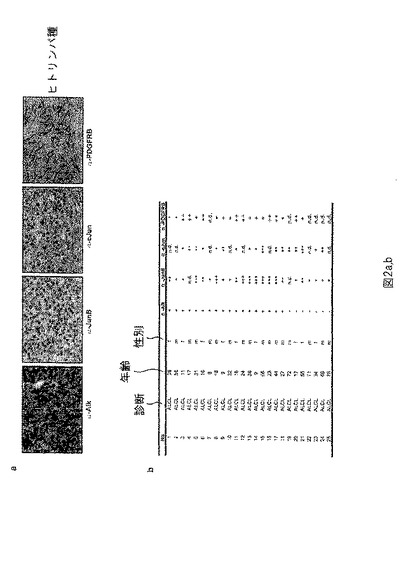
【図2】図2は、NPM-ALK陽性とNPM-ALK陰性ヒトALCL患者試料の免疫組織化学分析を表す。(a) ALCL患者由来の組織マイクロアレイの免疫組織化学分析(TMA) は、NPM-ALK陽性結節及び皮膚リンパ腫におけるALK、JunB、cJun及びPDGFRBの同時発現を示す(n=29)。(b)NPM-ALK陽性とNPM-ALK陰性ヒトALCL患者試料の表である。PDGFRB、cJun及びJunB及びALKに対するAbsを使用してIHC分析を行った。
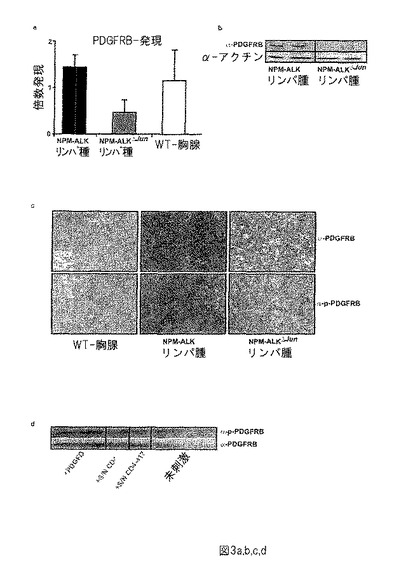
【図3−1】図3−1は、PDGFRB mRNAレベルをqRT-PCRによって分析し、NPM-ALK腫瘍と比較してNPM-ALKΔJun腫瘍及び野生型胸腺において減少されることを示す。(a) PDGFRBは、マウスNPM-ALK腫瘍において高く発現されるが、NPM-ALKΔJun腫瘍及び野生型胸腺においては高くない。(b) タンパク質免疫ブロット定量化は、PDGFRBは、NPM-ALKΔJun腫瘍ではなく、大抵のNPM-ALKにおいて活発に発現されることを明らかにした。(c) は、NPM-ALKマウス腫瘍細胞はマウス線維芽細胞においてPDGFを産生し、PDGFRB発現及びリン酸化反応を刺激できることを示す。(d) 糖タンパク質のIPを行い、ウエスタンブロットによってPDGFRB及びp-PDGFRBのタンパク質レベルを評価した。NPM-ALK誘導リンパ腫は、cJun及びJunBの喪失により増殖を減少しアポトーシス速度を低下することを示す。
【図3−2】図3−2は、PDGFRB mRNAレベルをqRT-PCRによって分析し、NPM-ALK腫瘍と比較してNPM-ALKΔJun腫瘍及び野生型胸腺において減少されることを示す。(e) NPM-ALK及びNPM-ALKΔJunリンパ腫のKi-67陽性増殖速度及びタネル陽性アポトーシス速度を、蛍光免疫染色によって測定し(右)、10 HPFのカウントによって定量化した(左)。さらに、Ki-67陽性増殖速度を、各リンパ腫の10 HPF(強拡大視野)のIHC染色のカウントによって分析した(下)。NPM-ALK腫瘍と比較して、NPM-ALKΔJun腫瘍の増殖速度は顕著に低下したが、アポトーシス速度は増加した。(f)FACSによる細胞周期染色は、NPM-ALKマウスの腫瘍よりNPM-ALKΔJunマウスの腫瘍がG1において顕著に多くの細胞を有し、S期において少ないことを示す。
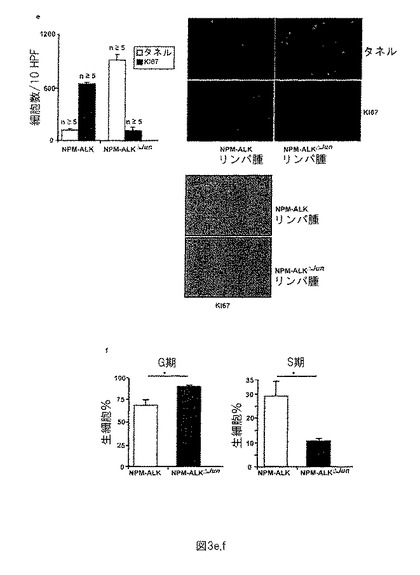
【図4−1】図4−1は、PDGFRBがJunB及びcJunの直接標的であることを示す。(a)は、複数の種におけるPDGFRBの高度の保存AP-1部位を示す。(b)は、マウスPDGFRB-プロモーターのChIP分析及び図を示す。図において、ChIP生成物の部位及びAP-1、c/EBP、SP1、AP-2及びNF-Yの結合部位を示す。NPM-ALKマウス腫瘍株化細胞でChIP分析を行った。抗体なしの細胞抽出物は、負の対照として機能する。陽性対照として、ヒストンH3 Absを使用した。PDGFRBプロモーターのAP-1部位に特異的なプライマーでのQRT PCRを、ChIP後抽出したRNAで行った。JunB及びcJunは共にPDGFRBプロモーター配列に結合した。
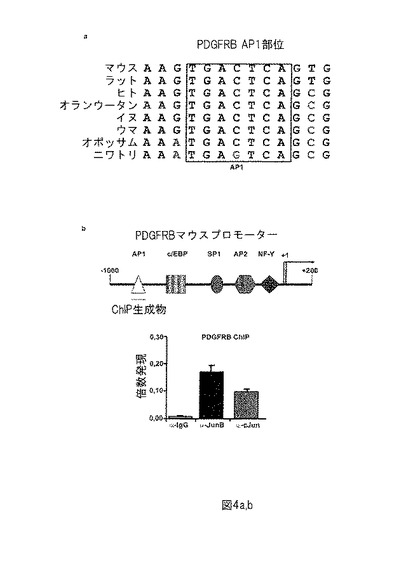
【図4−2】図4−2は、PDGFRBがJunB及びcJunの直接標的であることを示す。(c) (PDGFRB-luc)を含む又は(PDGFRB(w/o AP-1部位)-luc) AP-1部位を欠如する、作成したルシフェラーゼコンストラクトを示す。P-GL3-lucは、ホタルルシフェラーゼを含むベクターであるが、p-vec及びp-PDGFRB-lucはルシフェラーゼを含まないベクター及びPDGFRBプロモーターである。P-cJun、p-JunB及びp-PDGFRB-lucは、ルシフェラーゼを有するcJun及びJunB cDNA (P. Vesely)及びPDGFRBプロモーターを含むベクターをそれぞれ示す。P-vec + p-PDGFRB(w/o AP-1部位)-lucは、欠失AP-1部位を含まないベクター及びPDGFRBプロモーターを示す。p-cJun、p-JunB及びp-PDGFRB-lucのみが共に、強いシグナルを作り、cJun及びJunBのPDGFRB AP-1部位への特異的結合が確認される。(d) 交換された2つの塩基を有する正確なAP-1配列又は変異版 (標識「mut」)を含むEMSAプローブをデザインした。NPM-ALK陽性腫瘍細胞抽出物、及び対照AP-1部位、PDGFRB AP-1部位及びこれらの配列の変異版用のプローブを使用してEMSAを行った。対照AP-1部位及びPDGFRB AP-1両方のプローブであり、変異されていないプローブは、強いシグナルを生じる。さらに、PDGFRB AP-1部位及びJunB及びcJunに特異的な抗体を利用するスーパーシフトアッセイを行った。AP-1バンドは、Ab結合上で明らかに変化し、それぞれのJun-抗体の特異的結合が確認される。
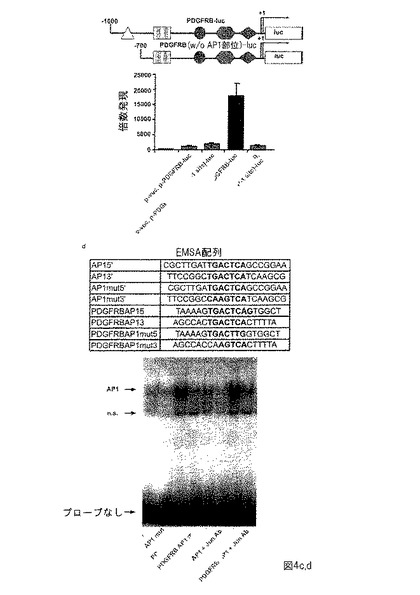
【図5】補足図1は、ヒト及びマウスリンパ腫及びそれぞれの対照におけるNPM-ALK、JunB及びcJunのタンパク質レベルのIHCによる分析を示す。NPM-ALK レベルはヒトNPM-ALK陽性リンパ腫、NPM-ALK及びNPM-ALKΔJunマウス腫瘍で同様であるが、NPM-ALK発現は野生型リンパ節において検出されなかった。JunBはヒトリンパ腫及びNPM-ALK陽性マウスリンパ腫において発現されるが、NPM-ALKΔJun腫瘍及び野生型リンパ節で発現されない。NPM-ALKΔJun腫瘍以外の全組織において、cJunは発現される。
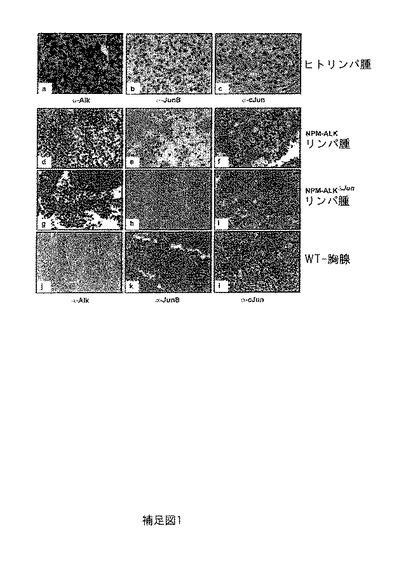
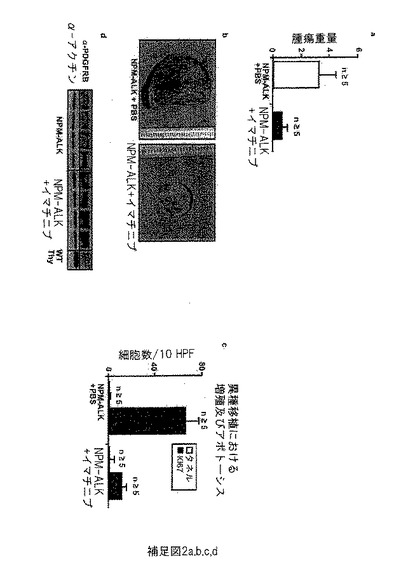
【図6】補足図2は、イマチニブでの特異的阻害の使用によるPDGFRBの阻害が、インビトロ(in vitro)での細胞数の減少及びアポトーシス速度の増加、インビボ(in vivo)での腫瘍サイズの減少及び増殖速度の低下を導き、90%以上の腫瘍サイズの減少が得られることを示す。(a) イマチニブでの処理の5日後、未処理対照腫瘍と比べ腫瘍重量は大幅に減少した。(b) 処理マウスと未処理マウスの腫瘍の直接のサイズ比較は、イマチニブ処理マウス腫瘍サイズで激しい減少を示す。(c) 異種移植マウスの腫瘍における増殖及びアポトーシス速度をそれぞれ、IHC染色のKi-67及びタネルによって評価した。増殖はイマチニブ処理マウスで大きく減少したが、アポトーシス速度で顕著な違いはなかった。(d) ウエスタンブロット解析は、未処理対照と比較して、イマチニブ処理の異種移植マウスの腫瘍の高PDGFRB-発現を明らかにする。
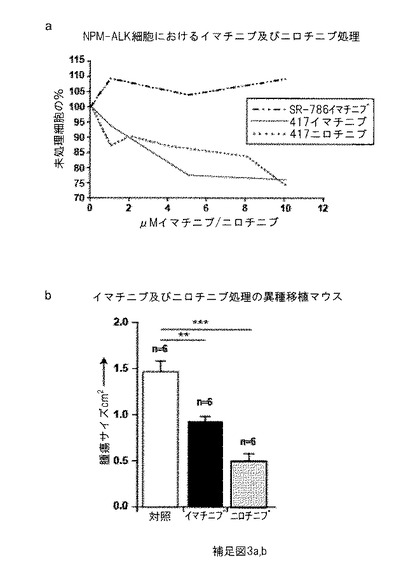
【図7】補足図3は、ニロチニブでの特異的阻害を使用したPDGFRBの阻害が、インビトロ(in vitro)での細胞数の減少及びインビボ(in vivo)での腫瘍サイズの減少を導き、59%以上の腫瘍サイズ減少が得られることを示す。(a) 抗増殖効果: ニロチニブ及びイマチニブでの治療は、PDGFRB発現NPM-ALK発現細胞 (株化細胞417; G. Inghirami; University of Turin) の細胞数の用量依存性減少を導くが、PDGFRB陰性SR-786 (G. Egger; MUW) 株化細胞の増殖速度は影響を受けない。(b) ニロチニブでの処置の6日後、未処理の対照腫瘍と比較して腫瘍重量は大幅に減少した。
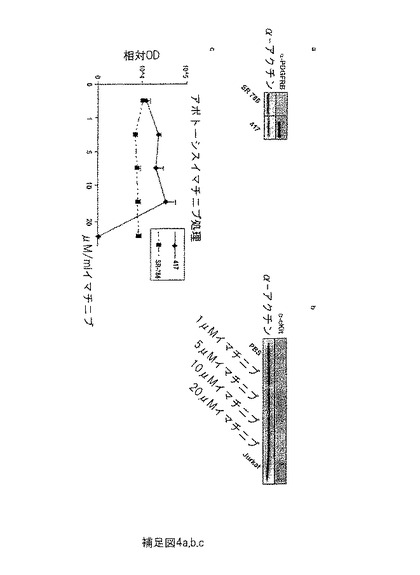
【図8】補足図4は、SR-786及び417株化細胞におけるPDGFRBタンパク質発現のウエスタンブロット解析を示す。(a) 株化細胞 417のみ、PDGFRB陽性である。(b) ウエスタンブロット解析は、c-kitは分析したNPM-ALKリンパ腫試料のいずれにおいても発現されないことを明らかにする。(c)インビトロ(in vitro)でのイマチニブでの処理は、417細胞において用量依存性アポトーシスを導くが、SR-786細胞は影響を受けなかった。アラマーブルーアッセイによってアポトーシスを測定した。株化細胞417は20 μMのイマチニブでの処理の3日後に高アポトーシスを示すが、株化細胞SR-786は影響を受けなかった。
【発明を実施するための形態】
【0045】
分析方法
リアルタイムPCR
リアルタイムPCRを、Chromo 4 cycler (Biorad)上で製造者の標準プロトコールに従って、ABI Prism (Invitrogen)のSybrGreen PCR qPCR Supermixで行った。1ウェルあたり50 ngのcDNAを使用し、試料ごとに3通りに分析し、規準化用にGAPDHを使用した。結果を2-ΔΔCt法で分析した。プライマー: NPM-ALK FW: GTG GTC TTA AGG TTG AAG TGT GGT T (配列番号1); NPM-ALK Rev: GCT TCC GGC GGT ACA CTA CTA A (配列番号2); JunB FW: GGC TTT GCG GAC GGT TT (配列番号3); JunB Rev: GGC GTC ACG TGG TTC ATC T (配列番号4) ; c-jun FW: TGA CTG CAA AGA TGG AAA CG (配列番号5); c-jun Rev: GCT CTC GGA CTG GAG GAA C (配列番号6); PDGFRB FW: TGC CAG TTC CAC CTT GAA TGA A (配列番号7); PDGFRB Rev: AGT TGT GCC TCA GGC TCT GCT T (配列番号8)。
【0046】
ウエスタンブロット
約100 mgの腫瘍組織を、氷上でDounceを使用して、50 mM のトリス-HCl pH 7,4、150 mMのNaCl、0.1% トリトン X-100、5 mM EDTA、1x 完全プロテイナーゼ阻害剤(Roche)、1x HALT ホスファターゼ阻害剤 (Thermo scientific) から成る1 mlの溶解緩衝液中で裁断した。続いて、得られる懸濁液を5分間1200 rpmで遠心分離し、上清を新たなチューブに移し、タンパク質濃度をQubit蛍光光度計(Invitrogen)で測定した。総タンパク質溶解物(50μg)を、ドデシル硫酸ナトリウム-ポリアクリルアミド電気泳動 (SDS-PAGE) に供し、続いて、ニトロセルロース膜に移した。
【0047】
膜を1%ミルク及び1% ポリビニルピロリドン (PVP) を加えたTBS-T中で1時間ブロックし、その後一次抗体(1%ミルク+ 1% PVP (ポリビニルピロリドン及び0.02% NaN3)を加えたTBS-T中で、適した濃度に希釈) で4°Cで一晩インキュベートした。その後、TBS-Tで膜を3回洗浄し、TBS-T中に希釈した適切な二次抗体で1時間インキュベートした。TBSでの最終洗浄工程の後、ECL Plus溶液(Amersham Biosciences) で1分間膜をインキュベートし、続いてルミノメーター (Roche)上で写真を撮った。次の一次抗体を使用した: 抗JunB sc-73X、抗c-jun sc-1694X (Santa Cruz) 、抗PDGFRB #3169 (細胞情報伝達) 、抗ベータアクチン#4967 (細胞情報伝達) 、抗Alk 51-3900 Zymed; 抗phospho-Alk (#33415、細胞情報伝達)、Santa Cruz)、抗cKit (sc-168、Santa Cruz)。
【0048】
免疫組織化学及び免疫蛍光
ホルマリン固定パラフィン包埋 (FFPE) 腫瘍試料の切片を作成し、スライドガラスに貼り付け、脱脂し、再水和した。エピトープをトリス-EDTA中で加熱処理によって回収した。内在性ペルオキシダーゼを10分間3 % H2O2 中でブロックした。アビジン/ビオチンブロック(Vector)、Universal Mouse HRP-Kit (IDLabs) 由来のSuperblock及びマウスブロック中で切片をブロックした。一次抗体染色を一晩4°Cで行った。AEC及びヘマトキシリンを対比染色として使用した。抗PDGFRB (細胞情報伝達、#3169) 、抗JunB (Santa Cruz、sc-46) 、抗c-jun (Santa Cruz、sc-1694) 、抗Alk Zymed (51-3900)抗体を、製造者説明書に従って使用した。ヘマトキシリン/エオシン及びPAS染色をDAKOから購入した染色キットで行った。
【0049】
本発明者は今回、PDGFRB活性化の阻害によってT細胞リンパ腫に対応する治療法を見出した。このような阻害は、特に特定のAP-1陽性癌型における細胞増殖及びアポトーシスの調節に関連する。本発明は、少なくとも一部において、PDGFRB活性化媒介プロセスは、T細胞リンパ腫腫瘍治療、特に高悪性度NHL又はALCLの治療用の、(例えば、選択的)標的として有用であることを見出したことに関する。
【0050】
ALCL及びNPM-ALKとの関係に関して、先行技術に証拠が存在する。しかしながら、NPM-ALK陽性ALCLにおいてPDGFRBが活性化されるという今回の発見は意外であった。PDGFRBはNPM-ALK誘導リンパ腫において過剰発現されることが分かったが、複数の研究はイマチニブ等のPDGFRB阻害剤の使用が、ALCL株化細胞に有益な効果を有しない可能性を示していた(Rassidakis et al., Blood 105 (2005), 827-829; Ergin et al., Exp. Hematol. 29 (2001): 1082-1090)。NPM-ALKはまた、イマチニブでの治療に非感受性であることが分かっていた(Gunby et al., J. Med. Chem. 49 (2006), 5759-5768)。
【0051】
よって、今回のAP-1発現リンパ腫の動物モデルにおけるPDGFRB活性化因子阻害剤の特有の効果は、意外であった。
【0052】
用語「投与」とは、所望の目的の機能が行われるように本発明の阻害剤を必要とする対象に導入される経路を含む。使用できる投与の経路の例は、経口投与を含む。阻害剤は、任意の他の都合の良い経路、例えば、継続点滴又はボーラス投与によって、上皮又は皮膚粘膜(mucocutaneous lining) (例えば、経口、直腸、腟、及び腸管粘膜等)を介した吸収によってもまた投与することができ、他の治療剤と共に投与することができる。投与は、全身又は局所的とすることができる。リポソーム、微小粒子、マイクロカプセル及びカプセル中のカプセル化を含む様々な周知の送達系を、使用することができる。本発明の阻害剤の投与方法は、皮内、筋肉内、腹腔内、静脈内、皮下、鼻腔内、硬膜外、経口、舌下、脳内、腟内、経皮、直腸、吸入、又は局所投与を含むがこれらに限定されない。本発明の阻害剤は、単独、又は他のPDGFRB阻害剤又はT細胞リンパ腫の治療において使用される任意の他の治療剤、或いはこれらの両方と併用して、好ましくは医薬的に許容される担体と共に、投与することができる。本発明の阻害剤は、他の薬剤の投与前に、他の薬剤の投与と同時に、又は他の薬剤の投与後に投与することができる。さらに、本発明の阻害剤は、プロドラッグ形態で投与することもでき、それはインビボ(in vivo)で活性代謝物、又はより活性のある代謝物に変換される。
【0053】
用語「PDGFRB阻害剤」とは、PDGFRBに結合しそれによりその活性化を阻害し、及び/又はPDGFRBの活性化の機能阻害を直接又は間接的に示すリガンド(例えば、キナーゼドメインの全長、N末端、C末端、カルボキシ末端、ATP結合ポケット)に関する。活性の抑制は、HTScan(登録商標) PDGF受容体βキナーゼアッセイキット#7770; 細胞情報伝達等の標準試験によって試験することができる。
【0054】
用語「予防上有効量」とは、患者への単一又は複数用量投与において、細胞増殖性疾患の予防又は治療で効果のある本発明の阻害剤の量に関する。
【0055】
用語「対象」とは、細胞増殖性疾患罹患の可能性がある、又は本発明の化合物の投与から利益を得る可能性のある、ヒト及び非ヒト動物等の生物を含む。好ましいヒトは、本明細書中に記載されるように、細胞増殖性疾患又は関連症状に罹患する又は罹患する傾向のあるヒト患者を含む。用語本発明の「非ヒト動物」とは、あらゆる脊椎動物、例えば、哺乳類、マウス等のげっ歯類、及び非人類霊長類、非哺乳類、例えば、好ましくはヒトT細胞リンパ腫の動物モデルとして使用されるヒツジ、イヌ、ウシ、ニワトリ等を含む。
【0056】
用語「細胞増殖性疾患感受性」とは、例えば癌等の細胞増殖疾患を発症する危険性のある対象、すなわち、癌ウイルスでのウイルス感染症に罹患する対象、電離放射線又は発癌性化合物に曝露した対象、癌家系又は病歴を有する対象等を含むことを意味する。
【0057】
本明細書中で使用される用語「治療」又は「処理」は、細胞増殖性疾患感受性患者を治療するための予防又は治療の両方の手段を含む。
【0058】
特定の実施形態において、本発明は、T細胞増殖性疾患に関係する対象に、T細胞リンパ腫細胞の増殖を抑制することができるPDGFRB阻害剤の有効量を投与することによって、対象を治療する方法を供する。T細胞増殖性疾患は、癌、特に後期癌を含む。
【0059】
本発明の阻害剤は好ましくは、非ホジキンリンパ腫(別称、NHL)、特に高悪性度NHL等のNPM-ALKリンパ腫において、例えば、リンパ節又はリンパ節外部位、又は皮膚結節を含む原発性皮膚ALCLを含む、全身ALCL等のALCLにおける使用のために使用される。さらに、本発明の阻害剤は、例えば、末梢性T細胞リンパ腫 (PTCL)を治療するために、他の高悪性度成熟T-/NK-細胞リンパ腫における、例えば、リンパ節等の成熟胸腺起源のリンパ系組織における使用が示唆される。特に、PDGFRBの過剰発現が判明した場合、それらのT細胞リンパ腫の患者は本発明の阻害剤で治療を受ける。過剰発現は好ましくは、生物試料中の発現の増加によって測定され、その増加はT細胞リンパ腫非易発性対象又は非罹患の対象の基準値の少なくとも1.2倍、より好ましくは少なくとも1.5倍である。
【0060】
本発明の最も好ましい実施形態は、ALK+ T細胞リンパ腫、特にALK+-ALCL、すなわち、腫瘍細胞がALK陽性(特にNPM-ALK+)であるALCLの治療である。これらの腫瘍においてイマチニブ等の物質の標的であるc-kitが認められないことが確認された先行技術を考慮すると、本発明のPDGFRベータ阻害剤、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブ、特にイマチニブ又はニロチニブでの治療が効果のあることは非常に驚きであった。従って、この特異的治療の過程において、まずALCL患者のALK状態を確認し、続いて、最も効果のある治療を決定することが好ましい。これは、当技術分野で周知の方法、例えば、患者由来の腫瘍細胞の試料 (例えば、患者の血液、血清又は血漿試料)を用意し、続いて、これらの細胞がALK+又はALK-であるかを確認することによって行うことができる(例えば、Ergin et al., 2001; Fornari et al., Hematol. Oncol. 27 (2009), 161-170)。イマチニブはヒトALK陽性ALCL株化細胞においてはALKを結合することができず、効果を示すことはできないが (Ergin et al., 2001を参照されたい)、ヒトALCL患者においては、ALK陽性(特にNPM-ALK陽性) ALCLはPDGFRベータ阻害剤、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブ、特にイマチニブ又はニロチニブで良好に治療することができる。この好ましい実施形態に関してもまた、AP-1(の発現)の存在が必要である。
【0061】
従って、ALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療の本発明の好ましい方法において、まずリンパ腫を有する患者由来で腫瘍細胞を含む試料を分析して、腫瘍細胞がALKを発現するか判別し、試料の腫瘍細胞がALKを発現する場合、患者はPDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで治療する。
【0062】
特定の実施形態において、対象は、哺乳類、例えば、ヒト又は非ヒト霊長類を含む霊長類である。本実施形態において、本発明の阻害剤は、PDGFRB、又はその特異的ドメインと直接又は間接的に相互作用し得る。本発明の阻害剤は無制御の増殖を生じている細胞と接触し、細胞増殖を抑制する又はアポトーシスを誘発することができる。本発明の阻害剤との細胞の接触又は本発明の阻害剤の対象への投与は、細胞増殖性疾患に罹患する又は感受性のある細胞又は対象の治療の1つの方法である。
【0063】
特定の実施形態によれば、T細胞リンパ腫疾患に罹患する又は感受性のある対象の治療方法は、それを必要とする対象に、PDGFRBの阻害剤の予防又は治療上有効量を投与する工程を含み、これによりAP-1発現リンパ腫細胞を抑制する。好適な阻害剤は、PDGFRBを結合する少なくとも部分特異性を有するそれらのTK阻害剤、標準的なスクリーニング方法を介して同定された医薬的に許容されるその塩又は類似体を含む。
【0064】
本発明によれば、好ましい小分子阻害剤は、異なる目的で抗癌治療において使用されている、ニロチニブ、イマチニブ、ダサチニブ、ソラフェニブ、アキシチニブ、スニチニブ及びトセラニブ又は証明された抑制活性を有するその機能誘導体等の市販の化合物である。
【0065】
さらに、PDGFRの阻害剤は、活性を抑制するための試験を利用する、適切なスクリーニング技術によって同定することができる。
【0066】
天然のPDGF結合パートナーの構造を模倣するポリペプチド阻害剤、又は拮抗モノクローナル抗体又は抗体断片等のPDGFRB化合物への特異的免疫リガンドであるポリペプチド阻害剤もまた好ましい。
【0067】
それらの阻害剤は、高親和性を有してPDGFRBに結合することが好ましく、Kdが10-6 M未満であることが好ましい。
【0068】
特定の実施形態において、本発明の方法は、他の薬剤上活性な薬剤又は従来の治療方法と併用した、対象に本発明の阻害剤の治療上有効量を投与する工程を含む。薬剤活性化合物の例は、他のPDGFRB阻害剤及び/又はT細胞増殖性疾患を治療するための周知の薬剤、例えば、抗癌剤、抗増殖剤又は化学療法剤を含む。本発明の阻害剤と容易に併用されるT細胞リンパ腫の従来の治療方法は、標準的な化学療法及び/又は放射線治療であり、短パルス化学療法又はCHOP療法の使用を含む(Seidemann et al., Blood 97 (2001), 3699-3706)。本発明はさらに、本発明の阻害剤及び組み合わせ治療に使用される薬剤を含むキットに関する。
【0069】
本発明の阻害剤及び薬剤活性化合物は、同一の医薬組成物又は異なる医薬組成物で、例えば、同時に又は別の時間に対象に投与することができる。
【0070】
本発明の阻害剤の治療上有効量又は予防上有効量は、周知技術の使用及び類似環境下で得られた結果の観察によって、当業者によって容易に決定することができる。インビトロ(in vitro)又はインビボ(in vivo)でのアッセイを任意に利用することは、最適用量範囲の特定の手助けとなり得る。利用される正確な用量はまた、投与経路、状態、治療される状態の重症度、及び治療される個体に関する様々な物理的要因に依存し、当業者の判断に従って決定することができる。
【0071】
治療上又は予防上使用される本発明の阻害剤の有効量は、体重1キログラムあたり1日約0.001 ミリグラム (mg/kg/日)〜約100 mg/kg/日であることが期待される。好ましい用量は、0.1〜10 mg/kg/日の範囲である。さらに好ましい用量は、0.01〜10 g/日、より好ましくは 0.1〜1 g/日、さらに好ましくは0.1〜0.5 g/日、特に200〜400 mg/日である。
【0072】
本発明のさらなる他の態様において、本発明の方法は、T細胞リンパ腫の治療においてPDGFRB阻害に適当な阻害剤を特定するために供される。機能アッセイは、そのような疾患に罹患しPDGFRB過剰発現が判明した対象由来のT細胞リンパ腫株化細胞のエクスビボ(ex vivo)での使用を含む。
【0073】
インビトロ(in vitro)でのPDGFRB抑制活性を測定する方法は、試験化合物の存在下又は不存在下でPDGFRB又はその特異的ドメインの結晶構造を得る工程を含む。続いて化合物は、結晶構造の結合部位中又は上にコンピューターでモデル化する(computer modeled)ことができる。候補調節化合物が同定されると、化合物は、インビトロ(in vitro)、インビボ(in vivo)、又はエクスビボ(ex vivo)での細胞アッセイを使用してスクリーニングすることができる。本態様で同定した化合物は、本発明の好ましい阻害剤の類似体として有用である。
【0074】
本発明の阻害剤は、医薬的に許容される担体又は希釈剤と共に有効量で製剤することができる。例えば、有効量は、医薬的に許容される製剤中に供され、医薬的に許容される製剤の対象への投与後、対象に対して少なくとも12時間、24時間、36時間、48時間、1週間、2週間、3週間、又は4週間、本発明の化合物の輸送を維持する。
【0075】
特定の実施形態において、これらの医薬組成物は対象への局所又は経口投与に適切であり、錠剤、トローチ剤、バッカル剤、トローチ、水性又は油性懸濁剤又は液剤、顆粒、粉末、ペースト、エマルション、カプセル、シロップ又はエリキシル剤を含む。
【0076】
非経口投与に使用される好適な製剤は、例えば、無菌溶液又は懸濁液等の、皮下、筋肉内又は静脈内注射を含む。局所投与用製剤は、クリーム又は軟膏、ペースト及びゲル等の多数の形態を含む。
【0077】
好ましい医薬的に許容される担体は、ビヒクル、ラクトース、グルコース及びスクロース等の糖、コーンスターチ及びポテトスターチ等のデンプン、カルボキシルメチルセルロースナトリウム、エチルセルロース及び酢酸セルロース等のセルロース及びその誘導体、又はグリセリン、ソルビトール、マンニトール及びポリエチレングリコール等のポリオール、或いは医薬製剤において利用される当技術分野で周知の他の希釈剤及び賦形剤を含む。経口及び非経口投与のための液体担体の適切な例は、特に上記添加剤、例えば、カルボキシルメチルセルロースナトリウム溶液を含む、セルロース誘導体を含む水、一価アルコール及び多価アルコール及びその誘導体を含むアルコール、及び油を含む。生理的に許容される賦形剤は、生理食塩水、ゼラチン、デンプン、滑石、ケラチン、コロイドシリカ、尿素等とすることができる。さらに、補助剤、安定化剤、増粘剤、潤滑剤、及び着色剤を使用することができる。
【0078】
それらはまた、例えば、ヒドロキシプロピルメチルセルロース、他のポリマーマトリックス、ゲル、透過性膜、浸透圧系、多層コーティング、微小粒子、リポソーム、微粒子、又はそれらの組み合わせを使用して、それらの活性成分の維持又は制御された放出を供するように製剤され、様々な割合の目的の放出特性を供することができる。阻害剤または、1又は2以上の上記賦形剤と共にマイクロカプセル化形態とすることができる。
【0079】
以下の定義の対象は、本発明の実施形態である:
1.細胞増殖性疾患感受性患者における、T細胞リンパ腫の抗増殖治療のための、PDGFRベータ阻害剤。
2.AP-1発現リンパ腫の治療のための、請求項1に記載の阻害剤。
3.NHL、ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCL、及びPTCLから成る群から選択されるリンパ腫の治療のための、請求項1又は2に記載の阻害剤。
4.再発患者を治療するための、請求項1〜3のいずれか1項に記載の阻害剤。
5.患者の第一選択治療のための、請求項1〜4のいずれか1項に記載の阻害剤。
6.例えばポリペプチド又は小分子等のPDGFRベータ拮抗薬から選択される、請求項1〜5のいずれか1項に記載の阻害剤。
7.ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される、特にイマチニブ又はニロチニブから成る群から選択される、請求項6に記載の阻害剤。
8.予防又は治療使用のための、請求項1〜7のいずれか1項に記載の阻害剤。
9.局所又は全身使用のために製剤される、請求項1〜8のいずれか1項に記載の阻害剤。
10.経口使用のために製剤される、請求項1〜9のいずれか1項に記載の阻害剤。
11.0.001 mg/kg/日〜約100 mg/kg/日の用量で投与される、請求項1〜10のいずれか1項に記載の阻害剤。
12.化学療法及び/又は放射線療法との併用使用のための、請求項1〜11のいずれか1項に記載の阻害剤。
13.T細胞リンパ腫、特にAP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択される、特にイマチニブ又はニロチニブから選択される、PDGFRベータ阻害剤。
14.T細胞リンパ腫の抗増殖治療のためのPDGFRベータ阻害剤を含有する医薬製剤の、調製方法。
15.T細胞リンパ腫、特にAP-1発現及びALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療方法であって、リンパ腫を有する患者の腫瘍細胞を含む試料を分析し、腫瘍細胞がAP-1及び/又はALKを発現するか判別するとともに、試料の腫瘍細胞がAP-1及びALKを発現する場合、PDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで患者を治療する、抗増殖治療方法。
16.AP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、PDGFRベータ阻害剤。
本発明はさらに、次の実施例によってそれらに限定することなく説明する。
【実施例】
【0080】
実施例1: cJunとJunBとのT細胞特異的欠失は、マウスの発達NPM-ALK陽性リンパ腫の生存期間を顕著に増加する。
インビボ(in vivo)でのNPM-ALK陽性リンパ腫の形成におけるJunB及びcJunの役割を検討するために、マウス、T細胞特異的、CD4-プロモーターの制御の下でヒトNPM-ALK融合-チロシンキナーゼ (FTK) を保有するトランスジェニックマウスを、JunB遺伝子又はcJun 遺伝子のいずかのloxP導入版(JunBf/f又はcJunf/f)を保有する、CREリコンビナーゼ遺伝子(CD4-CRE)を独立して発現している同一バックグラウンドのマウスと交配した。(Chiarle et al., Blood 101 (2003): 1919-1927; Wolfer et al., Nat. Immunol. 2 (2001), 235-241; Behrens et al., EMBO J. 21 (2002), 1782-1790)。この手段を使用して、NPM-ALKのT細胞特異的発現を有しJunB及び/又はcJunがT細胞特異的にノックアウトされたマウスを作製した。目的の4つの系統のマウスを樹立した。導入遺伝子を有しJunB 又は cJunがコンディショナルノックアウトされたマウスを、NPM-ALKΔc-Jun又はNPM-ALKΔJunBとそれぞれ命名した。導入遺伝子を保有しJunB及びcJunが二重ノックアウトのマウスをNPM-ALKΔJunと命名した。カプラン・マイヤープロットを使用して異なるマウス系統の生存期間を分析した(図1a)。NPM-ALKΔJunはノックアウトなしの同腹子より顕著に長く生存したが(平均生存期間24 +/- 4,8 vs. 13,5 +/- 3,9 週間)、NPM-ALKΔJunB又はNPM-ALKΔc-Junの生存期間においては顕著な差はなかった(12,8 +/- 4,7及び11,3 +/- 3,2週間 vs. 13,5 +/- 3,9 週間) (図1a)。従って、さらに、NPM-ALKとNPM-ALKΔJunマウスとの違いを解明することに決定した。
【0081】
CREリコンビナーゼ媒介遺伝子欠失が十分に機能し、NPM-ALK mRNA発現レベルは影響を受けないことを確証するために、上記の2つの遺伝子型のマウスのリンパ腫及び対照マウス(胸腺)由来のリンパ組織においてqRT-PCRによって、NPM-ALK、JunB及びcJun mRNAのレベルを検討した(図1b)。腫瘍は胸腺において最も頻繁に生じるので、胸腺が対照組織として適した。NPM-ALK及びNPM-ALKΔJunリンパ腫は、NPM-ALKの同様の発現レベルを示したが、NPM-ALKは野生型マウスの胸腺中で発現されなかった。
【0082】
JunB mRNA発現は、CD4由来のNPM-ALK発現動物のリンパ腫よりも野生型マウスの胸腺においてわずかに高かったが、NPM-ALKΔJunマウスのリンパ腫においてjunB発現を検出することがほとんどできなかった。CJun mRNA発現は、NPM-ALKマウスのリンパ腫及び野生型マウスの胸腺と同程度であったが、期待通りNPM-ALKΔJunリンパ腫においてcjun発現は全くなかった。これらのデータから、JunB及びcJun のCREリコンビナーゼに基づくノックアウト戦略によって、対応するmRNAレベルの効率的な抑止が得られることが確認された。
【0083】
続いて、NPM-ALK及びNPM-ALKΔJunマウスのリンパ腫においてNPM-ALK、JunB及びcJunのタンパク質発現レベルを分析した(図1c)。NPM-ALKタンパク質発現はNPM-ALK及びNPM-ALKΔJunマウスにおいて同程度であり、ノックアウト戦略はNPM-ALKレベルに影響しなかったことが確認された。NPM-ALKマウスと比較した場合、JunB及びcJunタンパク質は、NPM-ALKΔJunリンパ腫において検出できず、再度CD-4 Cre媒介ノックアウト戦略の効果が確認された。
【0084】
実施例2: ヒトALCL患者試料における遺伝子発現プロファイリング
組織マイクロアレイ(TMA)技術を使用して(NPM-ALK陽性及び陰性ALCL患者のパラフィン包埋ヒト患者試料を使用した組織マイクロアレイを、Dr. Ana Schmatz; Medical University Viennaから得た)、ALCL患者試料のタンパク質発現を分析した。JunB (Santa Cruz; sc-46)、cJun (Santa Cruz; sc-1694) 及びPDGFRB (Cell Signaling; Cat.Nr.3169)タンパク質発現レベルは、ALK(Dako; N1614)発現レベルから独立して上昇した(図2a及びb)。
【0085】
PDGFRBは、NPM-ALKにおいて高発現されるが、NPM-ALKΔJun腫瘍においては高発現されない。
マウス及びヒトNPM-ALKリンパ腫の腫瘍試料をPDGFRBのRNA発現に関して分析した。qRT-PCRを使用して、PDGFRB発現がNPM-ALKΔJunマウスのリンパ腫においてNPM-ALKマウスと同様であることが分かった。野生型胸腺において、発現は腫瘍よりも50 %低い(図3a)。NPM-ALKマウスの腫瘍はPDGFRB陽性であるが、PDGFRBタンパク質発現はNPM-ALKΔJunマウスにおいて完全に消失されることを示す(図3b)。我々はまたIHCによって、NPMΔJunリンパ腫及び肝臓と比較してNPM-ALKのPDGFRB発現を分析した。PDGFRB発現は、NPM-ALKリンパ腫において高いが、NPM-ALKΔJunマウスはPDGFRBを発現しない(図3c及び未開示データ)。線維芽細胞を一晩血清非含有培地で血清欠乏させ、続いて、PDGFD(正の対照) 、NPM-ALK陽性マウス腫瘍株化細胞の血清(CD4-4又はCD4-417それぞれ、G. Inghirami; University of Turin) 又は血清非含有培地を、線維芽細胞に添加した。糖タンパク質に関して免疫沈降法を行い、PDGFRB及びp-PDGFRB(Cell Signaling; Cat.Nr.3124) のタンパク質レベルを、マウスNPM-ALKリンパ腫における自己刺激ループのウエスタンブロット解析によって評価した。NPM-ALKマウス腫瘍細胞は、自己刺激態様でマウス線維芽細胞において、PDGFを産生し、PDGFRB発現及びリン酸化反応を刺激することができる(図3d)。
【0086】
実施例3: NPM-ALKΔJunマウスの腫瘍は、増殖の減少及びアポトーシスマーカーの増加を示す。
マウスにおけるNPM-ALKリンパ腫は非常に活動的なので、腫瘍形成のこれらの初期段階の分析は直接は不可能であり、窒息死の数時間前までマウスは疾患の兆候を示さず、生存期間は約8〜約30週間と非常に広範である(図1aを参照されたい)。従って、我々は死亡時に、Ki-67 (Novocastra; NLC-Ki67p) 及びタネル(Chemicon; S7111) アッセイ、それぞれに関して免疫蛍光及び免疫組織化学染色を介してリンパ腫において増殖及びアポトーシス速度を分析した(図2e)。二重ノックアウト動物由来の腫瘍における増殖マーカーKi-67を使用した染色は非常に減少したが、タネル染色によって測定したアポトーシス速度はNPM-ALKΔJunマウスにおいて増加したことが分かった。フローサイトメトリーを使用して、NPM-ALKの細胞周期パラメータをNPM-ALKΔJunリンパ腫細胞に対して分析した。NPM-ALKΔJunリンパ腫細胞は、G期が顕著に長い状態であったが、S期は非常に短かった(図3f)。
【0087】
実施例4: PDGFRBは、JunB及びcJunの新規標的である。
次に、我々はJunB及びcJunによるPDGFRBの制御を分析した。「UCSC Genome Browser on Mouse July 2007 Assembly」及びMathInspectorを使用して(Kent et al., Genome Res. 12(2002): 996-1006; Cartharius et al., Bioinformatics 21 (2005), 2933-2942)、転写開始点から-269bp 〜-263 bp 上流のPDGFRプロモーター内にAP-1コンセンサス配列を同定することができ、かかる配列は複数の哺乳類種に渡って高度に保存されている(図4a)。このインシリコの発見から、PDGFRBプロモーターはAP-1因子を介して制御することができ、PDGFRBはおそらく新規であり未だ知られていないAP-1標的遺伝子であるという仮説が導びかれる。この仮説を試験するために、マウスCD4-NPM-ALK株化細胞 (417系統)を使用してクロマチン免疫沈降 (ChIP) 分析を行った (G. Inghirami, University of Turin)。この株化細胞は、我々が使用する同一のNPM-ALKマウスの原発腫瘍由来であった。ChIPを抗JunB及び抗cJun抗体で行い、得られる物質をPDGFRB-プロモーターの推定AP-1結合部位に結合したプライマーのqRT-PCRによって分析した(図の説明文を参照されたい)。JunB及びcJunがこのAP-1部位に結合することを示すことができ、PDGFRBがAP-1標的遺伝子であることを示唆している(図4b)。
【0088】
これらの結果をさらに確認するために、2つの異なるPDGFRBプロモータールシフェラーゼコンストラクトを使用して、ルシフェラーゼレポーター遺伝子アッセイを行った。両方の場合で、マウスPDGFRBプロモーター部分を、pGL3塩基プロモーターの複数のクローニング部位中にクローン化した。第一の場合(PDGFRB-Luc)において、転写開始点から-269bp〜-263bp上流のAP-1コンセンサス部位を含む断片を使用し、第二の場合(PDGFRBΔ-Luc)、この領域を排除した(図の説明文を参照されたい)。AP-1結合部位を含むPDGFRB-LucプロモーターコンストラクトとのcJun及びJunB発現ベクターの同時導入は強力なルシフェラーゼ活性化を生じ、一方AP-1結合部位欠如のPDGFRBΔ-Lucコンストラクトは、cJun及びJunBによって誘導されなかった。これらのデータは、cJun及びJunBの組み合わせは、AP-1部位を介してPDGFRBプロモーターを強力に誘導することを示す(図4c)。
【0089】
最終的に、EMSAによってNPM-ALKマウス腫瘍タンパク質抽出物におけるPDGFRB AP-1部位を分析した。腫瘍抽出物をTPA (1 2-O-テトラデカノイルホルボール-13-酢酸)-応答エレメント(TRE)を含むプローブでインキュベートした場合、EMSAによって大きなバンド変化が観察された。しかしながら、変異TRE配列はこの効果を消失した(図4d)。さらに、cJun及びJunBに特異的な抗体での腫瘍抽出物のプレインキュベーションは、変化したバンドのほぼ完全な欠如につながる。しかしながらこれらの発見は、PDGFRBが新規AP-1標的遺伝子であることを示す。
【0090】
免疫組織化学 (IHC)を使用して、ヒトALCL、NPM-ALK及びNPM-ALKΔJunマウスリンパ腫及び同系野生型動物由来のリンパ節において、NPM-ALK、JunB及びcJunの発現レベルを評価した。NPM-ALK染色は、全リンパ腫試料において同程度であったが、野生型リンパ節において検出できなかった。ヒト及びマウスNPM-ALK陽性ALCLリンパ腫は、JunB及びcJunの高発現を示すが、NPM-ALKΔJunマウスはJunBもcJunも発現しない。しかしながら、野生型のリンパ組織、すなわち、胸腺はJunB及びcJunを低レベルで発現する(補足図1)。
【0091】
実施例5: イマチニブ又はニロチニブでにマウスNPM-ALK腫瘍細胞の治療は、インビトロ(in vitro)での増殖の減少及びアポトーシスの増加を導く。
NPM-ALK腫瘍形成に対するPDGFRBの役割を検討するために、マウスNPM-ALK細胞及びPDGFRB陽性株化細胞417及びヒトNPM-ALK陽性及びPDGFRB陰性株化細胞SR-786 (補足図3a) [G. Egger, Medical University Vienna] を、異なる濃度のチロシンキナーゼ阻害剤イマチニブ-メシル酸塩 (イマチニブ)及びニロチニブで処理した。細胞を3日間RPMI培地中で培養し、続いて細胞数及びアポトーシス速度に関して分析した。イマチニブ濃度が増加するにつれ、細胞数は株化細胞417において次第に減少した。しかしながらSR-786細胞は、全く影響を受けなかった。
【0092】
実施例6: インビボ(in vivo)でのイマチニブ及びニロチニブでのマウスNPM-ALK腫瘍の治療は、腫瘍サイズの減少につながる。
同様にインビボ(in vivo)でのイマチニブ及びニロチニブの効果を試験するために、マウスNPM-ALK陽性株化細胞CD4-417の細胞を6週齢のSCID(重度の複合免疫不全)マウスの右側腹部の中に注入することによって、マウス異種移植モデルを樹立した。注入の4週間後に、固形の腫瘍が発生し、マウスを経管栄養によって300 mg/kg/日のイマチニブ又は75 mg/kg/日のニロチニブでそれぞれ毎日処置した。7日のコース後、イマチニブで処置したマウスは腫瘍の完全な寛解又は大きな減少を示した(未処置マウス: 2.7 g、処置マウス 0.5 g)(補足図2a及びb; 補足図3b)。さらに、NPM-ALKΔJunマウスにおいて観察された腫瘍表現型に一致して、移植腫瘍のイマチニブでの治療は、Ki-67及びタネル蛍光免疫染色によって測定されるように、増殖の減少及びアポトーシス速度のわずかな増大につながる(補足図2c)。PDGFRBタンパク質発現は、イマチニブ処理マウスにおいて上方制御される。これはおそらく、イマチニブによるPDGFRB-リン酸化反応の阻害への反作用である (補足図2d)。これらのデータは、イマチニブ及びニロチニブがALCLにおいてPDGFRBをブロックすることを支持している。
【0093】
実施例7: マウスNPM-ALK腫瘍細胞の、インビトロ(in vitro)でのイマチニブ又はニロチニブでの治療は、増殖の減少及びアポトーシスの増加につながる。
アポトーシス速度をアラマーブルーアッセイによって測定した。アラマーブルーの活性成分は、非蛍光性濃青色指示色素であるレサズリンである。それは細胞に浸透することができ、生(代謝上活性のある) 細胞による還元反応において、蛍光、鮮赤色色素のレゾルフィンに変換される。生じた蛍光は、代謝上活性のある細胞の数と比例する。
【0094】
アラマーブルーの0-20 μM イマチニブで処理したSR-786 及び417細胞を、摂氏37度で1時間インキュベートした。SR-786細胞は、培地中のイマチニブ濃度にかかわらず、同程度にレサズリンをレゾルフィンに変換することができた。0〜10μMで処理した417細胞の応答は、SR-786細胞と同様であった。20 μM イマチニブで処理した417細胞は濃青色から赤色への色調変化を誘発できなかったが、これはほとんど生細胞が存在しなかったことを示す。増殖率と同様に、SR-786細胞においてアポトーシスは影響を受けなかったが、417細胞においてアポトーシスは20 μM/ml イマチニブで非常に増加した(補足図4c)。これらの結果は、マウス細胞417はイマチニブ及びニロチニブに強く反応するが、ヒトSR-786細胞は影響を受けないままであったことを示し、417細胞の反応はイマチニブの中毒量が原因でないことを証明している。株化細胞の異なる応答の理由は、おそらくSR-786細胞におけるPDGFRB発現の欠如が原因である。ほとんどのヒトNPM-ALK陽性リンパ腫、及びマウス腫瘍及び株化細胞417はPDGFRBに関して陽性なので、ヒト株化細胞はそもそもPDGFRBを発現し、長時間の培養によってそれを失ったと思われる。培養下の細胞は、栄養分の供給のための新たな脈管構造(neovasculature)を生じる必要はなく、よって長時間の培養の細胞に関しては、主に血管の結合に関与するPDGFRB等の因子の発現を当然減少させるであろう。従って、イマチニブはNPM-ALK自体を阻害せずヒトNPM-ALK陽性細胞に影響しないので、ヒトにおいてイマチニブ治療は機能しないという仮説は、妥当でない。
【0095】
c-kitの阻害を介して作用しているイマチニブの可能性を排除するために、c-kitに関するウエスタンブロット解析(Dako, A4502)を行った。予想通り、株化細胞417においてc-kitは発現されなかった (補足図4a及びb)。
【技術分野】
【0001】
本発明は、PDGFRベータ阻害剤に関する新たな示唆に関する。
【背景技術】
【0002】
血小板由来増殖因子受容体ベータポリペプチド(別称、PDGFRB、PDGFRベータ、PDGF-Rβ又はCD140B)は、血小板由来増殖因子ファミリーのメンバーに関する細胞表面チロシンキナーゼ受容体をコードするヒト遺伝子である。これらの増殖因子は、間葉系起源の細胞に対するマイトジェンである。受容体単量体に結合する増殖因子の同一性により、機能性受容体がホモ二量体であるか、血小板由来増殖因子受容体のアルファポリペプチド及びベータポリペプチドの双方から成るヘテロ二量体であるかが決定される。この遺伝子は、5番染色体上で顆粒球マクロファージコロニー刺激因子及びマクロファージコロニー刺激因子受容体に関する遺伝子のそばに隣接され、3つの遺伝子は全て、5-q症候群と関係し得る。5番染色体と12番染色体との間の転座であって、5番染色体の遺伝子を転座の遺伝子、ETV6、白血病遺伝子に融合する転座は、好酸球増加症と共に慢性骨髄増殖性疾患を生じる。
【0003】
国際公開第2005/075454号(A2)は、白血病を含む様々な癌疾患の治療のためのイマチニブの使用を開示する。国際公開第2008/037716号(A2)は、慢性骨髄白血病及び消化管間葉性腫瘍の治療のためのニロチニブの使用を開示する。Verbeek等(Nat.Rev.Clin.Onc. 7 (2010) 116-119) は、消化管間葉性腫瘍に罹患する患者におけるイマチニブ誘発性T細胞増殖性疾患を開示する。Rassidakis等(Mod. Pathol 17 (2004) 946-953; 及びBlood 102 (2003), 4619-4620) は、c-kitはホジキン病及び未分化リンパ腫キナーゼ(ALK-) 陽性未分化大細胞リンパ腫(ALCL)株化細胞中に発現されず、従ってc-kitはイマチニブ等の治療剤の適した標的ではない、ことと報告する。Pullarkat等(Leuk. Res. 32 (2008), 1770-1775)は、巨核球性急性転化に罹患する患者の呈する慢性骨髄性発作が、イマチニブでの治療では効果がなかったことを記載する。
【0004】
国際公開第2007/053573号(A2)は、数百もの多くの腫瘍疾患、特にALCLを、有効量のソラフェニブで治療するための組成物及び方法を開示する。
【0005】
Lansigan等(J. Clin. Oncol. 28 (2010) TPS 300)は、再発性又は治療抵抗性T細胞リンパ腫におけるソラフェニブのパイロット研究を開示する。一方、Ambrosini等は、ソラフェニブは、ヒトACLC/HD-株化細胞においてアポトーシスを誘発することができたが、HD及びALCLのインビボ(in vivo)での治療におては効果がないことを記載する (Nguyen et al., Leuk. Res. 34 (2010), 379-386においても記載される)。
【0006】
米国特許出願第2005/0119352号は、例えば、発癌性キナーゼ修飾因子、好ましくはイマチニブと併用した、細胞周期チェックポイント活性化因子、好ましくはベータラパコン、又はその誘導体若しくは類似体の投与によってHDの治療が可能であることを開示する。
【0007】
先行技術を考慮すると、T細胞起源のリンパ腫の治療候補に関する報告は完全に矛盾していた。従って、このような患者、特にALCL患者のための信頼性があり且つ有効な治療が依然として必要である。
【発明の概要】
【課題を解決するための手段】
【0008】
従って、本発明は、細胞増殖性疾患感受性患者におけるT細胞リンパ腫の抗増殖治療のためのPDGFRB阻害剤を供する。それにより、腫瘍増殖を効果的に予防することができる。本発明の阻害剤は、全リンパ腫の治療のために使用することができる。好ましくは、リンパ腫は、NHL、ALCL及びPTCLから成る群から選択される。
【0009】
本発明の阻害剤は、特に再発患者の治療に有用である。従って、特に好ましい治療は、薬剤抵抗性患者の第二選択治療である。
【0010】
他の実施形態において、本発明の阻害剤は患者の第一選択治療のために使用される。
【0011】
本発明の阻害剤は、好ましくは、例えばポリペプチド又は小分子等の、PDGFRB拮抗薬から選択される。
【0012】
特定の実施形態において、本発明の阻害剤は、ニロチニブ、イマチニブ、ダサチニブ、ソラフェニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される。
【0013】
本発明の阻害剤を利用する治療方法は、好ましくは予防又は治療使用のための方法である。
【0014】
好ましくは、本発明の阻害剤は、局所又は全身使用のために、より好ましくは経口使用のために製剤される。
【0015】
好ましい治療レジメントは、0.001 mg/kg/日〜約100 mg/kg/日の用量でイマチニブ等の阻害剤を供する。従って、本発明の好ましい阻害剤は、そのような用量の投与を供するよう製剤される。
【0016】
本発明の好ましい実施形態は、CHOP療法又はこの治療の変更を含む、化学療法及び/又は放射線療法と併用の阻害剤の使用に関する(8)。
【0017】
本発明はさらに、T細胞リンパ腫の治療のためのPDGFRベータ阻害剤を含む医薬製剤の調製方法を供する。PDGF(血小板由来増殖因子)は、血管形成、腫瘍細胞の増殖刺激、腫瘍血管新生及び腫瘍線維芽細胞の動員及び制御に必須である。ホモ又はヘテロ二量体を形成し、2つの異なる受容体 (PDGFRA及びPDGFRB)に結合することができる4つの異なるリガンド (PDGF A-D)が存在する。
【0018】
活性化チロシンキナーゼは、慢性及び急性白血病の発病と関係し、阻害剤治療のための有力な標的を意味する。一部の阻害剤は、癌治療において使用される。
【0019】
イマチニブ(4-[(4-メチルピペラジン-1-イル)メチル]-N-[4-メチル-3-[(4-ピリジン-3-イルピリミジン-2-イル)アミノ]フェニル]ベンズアミド)は、特定のタイプの治療に使用される薬剤である。それは、NovartisからGleevec(登録商標)(アメリカ)又はそのメシル酸塩としてのGlivec(登録商標)(ヨーロッパ/オーストラリア)、イマチニブメシル酸塩(INN)との名称で現在市販される。それは、慢性骨髄性白血病(CML)、消化管間葉性腫瘍(GISTs)及び多数の他の悪性腫瘍の治療において使用される。イマチニブは、肺高血圧症の治療においてもまた機能する。それは、門脈肺高血圧症を含む様々な疾患過程において肺脈管構造の平滑筋肥大及び過形成の双方を減少することが示されている。
【0020】
イマチニブは、多数のチロシンキナーゼ (TK) 酵素の特異的阻害剤として機能する。それは、TK活性部位を占有し、活性の低下につながる。体内には、インスリン受容体を含む多数のTK酵素が存在する。イマチニブは、abl (エーベルソン癌原遺伝子)、c-kit及びPDGF-R (血小板由来増殖因子受容体)におけるTKドメインに特異的である。
【0021】
研究室において、イマチニブは、その受容体PDGFRBを阻害することによって、血小板由来増殖因子を抑制する実験薬剤として使用される。その効果の1つは、糖尿病マウスにおいて粥状動脈硬化を遅延させることである。
【0022】
慢性骨髄性白血病において、フィラデルフィア染色体は、bcr-ablと呼ばれるablのbcr (易切断領域)との融合タンパク質につながる。これは連続活性があるチロシンキナーゼであるので、bcr-abl活性を低下させるためにイマチニブが使用される。
【0023】
チロシンキナーゼの活性部位はそれぞれ、ATPの結合部位を有する。チロシンキナーゼによって触媒される酵素活性は、ATP由来の末端のリン酸のその基質上のチロシン残基への転移であり、プロテインチロシンリン酸化反応として周知の工程である。イマチニブは、bcr-ablのATP結合部位に結合し、タンパク質の酵素活性を競合的に抑制することによって機能する。
【0024】
イマチニブは、bcr-ablに非常に選択性があり、上記の他の標的(c-kit及びPDGF-R)も阻害するが、他にその選択性があるチロシンキナーゼは分かっていない。
【0025】
塩酸一水和物塩形態の、ニロチニブ (4-メチル-N-[3-(4-メチル-1H-イミダゾール-1-イル)-5-(トリフルオロメチル)フェニル]-3-[(4-ピリジン-3-イルピリミジン-2-イル) アミノ]ベンズアミド)は、同様にbcr-ablを阻害するチロシンキナーゼ阻害剤である。
【0026】
それは米国及び欧州において、第二選択治療薬剤抵抗性、フィラデルフィア染色体陽性慢性骨髄性白血病(CML)用の、Tasigna(登録商標)として認可された。ニロチニブは、イマチニブでの治療抵抗性のあるCMLの症例において活性を示し、第一選択治療として現在使用される。
【0027】
抗腫瘍性剤として使用される他のbcr-abl阻害剤は、ダサチニブ(N-(2-クロロ-6-メチルフェニル)-2-[[6-[4-(2-ヒドロキシエチル)-1-ピペラジニル]-2-メチル-4-ピリミジニル]アミノ]-5-チアゾールカルボキサミド一水和物であり、Bristol-Myers Squibbから名称SPRYCEL(登録商標) で販売され、慢性期CMLにおいて活性を示す。
【0028】
名称Nexavar(登録商標)でBayerから販売される、ソラフェニブ(4-[4-[[4-クロロ-3-(トリフルオロメチル)フェニル]カルバモイルアミノ]フェノキシ]-N-メチル-ピリジン-2-カルボキサミドは、腎細胞癌の治療に使用される小分子B-RAF阻害剤であり、血小板由来増殖因子受容体 (PDGFR)及び血管内皮増殖因子受容体(VEGFR)ファミリー由来の受容体チロシンキナーゼに対して活性を有することが示されている。国際公開第2007/053573号(A2)において、ソラフェニブは、数百もの多くの腫瘍疾患、特にALCLにおいて使用されることが示された。しかしながら、当該公報は当業者が実施可能なように記載しておらず、これは推測上の性質にすぎない。従って、ソラフェニブは本発明の好ましいPDGFRベータ阻害剤とみなされず、少なくとも通常のALCLの治療に関する限り、本化合物群に含まれないとみなすことができる。
【0029】
アキシチニブ(N-メチル-2-[[3-[(E)-2-ピリジン-2-イルエチニル]-1H-インダゾール-6-イル]スルファニル]ベンズアミド(別称、AG013736)は、小分子チロシンキナーゼ阻害剤であり、Pfizerにより開発中である。それは、VEGFR-1、VEGFR-2、VEGFR-3、血小板由来増殖因子受容体 (PDGFR)、及びc-Kit (CD117)を含む、複数の標的を阻害する。それは、異種移植モデルにおいて癌の成長を顕著に阻害することが示されており、腎細胞癌 (RCC) 及び複数の他の腫瘍型での試験において成功している。
【0030】
名称Sutent(登録商標)でPfizerから販売される、スニチニブ (N-[2-(ジエチルアミノ)エチル]-5-[(Z)-(5-フルオロ-1,2-ジヒドロ-2-オキソ-3H-インドール-3-イリデン)メチル]-2,4-ジメチル-1H-ピロール-3-カルボキサミドは、腫瘍血管新生及び腫瘍細胞増殖の双方において関与する血小板由来増殖因子のためのそれらの受容体及び血管内皮増殖因子受容体(VEGFRs)を含む、複数の受容体チロシンキナーゼ(RTKs)を標的にすることによって細胞内シグナル伝達を阻害する。従って、これらの標的の同時阻害は、腫瘍血管新生の減少及び癌細胞死の双方を導き、そして最終的に腫瘍の萎縮を導く。腫瘍がc-kitにおいて突然変異を生じこれによりイマチニブに抵抗性が生じている患者、又は薬剤に不耐性を生じている患者のための第二選択治療として、それは推奨されている。
【0031】
トセラニブ ((Z)-5-[(5-フルオロ-2-オキソ-1,2-ジヒドロ-3H-インドール-3-イリデン)メチル]-2,4-ジメチル-N-(2-ピロリジン-1-イルエチル)-1H-ピロール-3-カルボキサミド)は、受容体チロシンキナーゼ阻害剤であり、イヌマスト細胞腫瘍(別称、マスト細胞腫)の治療において使用される。それは、米国食品医薬品局に認可された唯一のイヌ特異性抗癌剤である。そのリン酸塩、リン酸トセラニブ (INN) はPalladia(登録商標) としてPfizerから販売される。トセラニブは、チロシンキナーゼ阻害剤の種類に属する。トセラニブは、イヌマスト細胞腫瘍の25-50%において変異しているc-Kitと呼ばれる特異的腫瘍細胞受容体、及び腫瘍血管新生に関与する2つの他の血管細胞受容体、PDGFR及びVEGFRを標的とするようデザインされる。
【0032】
未分化大細胞リンパ腫(ALCL) は、非ホジキンリンパ腫、主にT細胞起源である悪性の高いタイプである。全非ホジキンリンパ腫症例の約5%に含まれる。ALCL患者の約50%は、転座t(2;5)(p23;q35)を有し、発癌性融合タンパク質NPM-ALK (ヌクレオフォスミン未分化リンパ腫キナーゼ) を生じる。非常に低い頻度であるが、例えば、ATIC、CLTCL、MSN、RanBP2、TFG及びTPM3等の他のタンパク質との融合が観察されている。
【0033】
ALKは、インスリン受容体スーパーファミリーの膜貫通受容体チロシンキナーゼ (RTK) である。内在性ALK発現は、神経系に制限される。それは新生児脳において最も豊富であり、低レベルであるが成人脳において維持される。プレイオトロフィン受容体に関するALKについて強力な証拠が示されているが、ALKの正確な機能に関して研究は進行中である。発癌性ALK融合タンパク質と反対に、内在性ALKは造血組織において発現されない。
【0034】
NPM (ヌクレオフォスミン) は、リボソーム生合成及び中心体複製に関与する核小体タンパク質であり、ストレス刺激への応答において上方制御され、複数の腫瘍抑制遺伝子の機能を調整する。
【0035】
NPM-ALK融合タンパク質 [t(2:5)(p23;q35)] の恒常的過剰発現及び活性化は、ALCLの生存及び増殖を促進する重要な発癌性事象である。
【0036】
受容体チロシンキナーゼALKの触媒部分は、RNA-結合核小体リン酸化タンパク質NPMのオリゴマー形成ドメインと融合する。これは、リンパ系ALCL細胞においてNPM-ALKのホモ二量体形成及び自己リン酸化反応及びALKの発現を通して、ALKの恒常的活性化を導く。ALKの異常発現は、直接ALCLの形成に関与する。
【0037】
Galkin等 (PNAS 104 (2007), 270-275)は、非常に強力且つ選択的な小分子ALK阻害剤、NVP-TAE684を同定した。斯かる阻害剤は、ALCL由来及びALK依存性株化細胞の成長をブロックし、IC50値は2〜10 nMでであった。NVP-TAE684は、インビボ(in vivo)において、ALK-陽性ALCLの2つの独立モデルにおいてリンパ腫発生を抑制することが示され、樹立Karpas-299リンパ腫の退行を誘導した。NVP-TAE684はまた、CD30発現の下方制御を誘導し、これはCD30が治療上のNPM-ALKキナーゼ活性阻害のバイオマーカーとして使用され得ることを示している。
【0038】
NPM-ALKは、インビトロ(in vitro)及びインビボ(in vivo)で形質転換活性を有し、広範囲の発癌性因子及び例えばJak/Stat又はPI3K/Akt等のシグナル伝達ネットワークと相互作用している。ALK陽性(ALK+) ALCLはまた、サイトカイン受容体CD30及び活性化タンパク質 (AP-1) 転写因子ファミリーメンバーJunB及びcJunの高発現を特徴とする。
【0039】
AP-1はDNA結合転写因子であり、細胞増殖、分化及びアポトーシス、並びに発癌性形質転換を含む多くのプロセスにおいて重要な因子である。それは、Fos又はATFファミリーメンバーとホモ二量体又はヘテロ二量体を形成するJunファミリーメンバー (cJun, JunB, JunD) から成る。JunB及びcJunは、度々拮抗薬機能を有する。JunBは、アポトーシス促進性又は抗アポトーシス性のあることが報告され、それは細胞状況次第である。ALCLにおいて、JunB及びcJunが過剰発現されることが確認されており、CD30を活性化するJunBの役割が記載される。従って、cJun及びJunBは、腫瘍増殖と関係付けられる (Mathas et al., EMBO J. 21 (2002), 4104-4113; Kenner et al., J. Cell Biol. 164 (2004), 613-623; Watanabe et al., Can. Res. 65 (2005), 7628-7634; Jacobsen, The Oncologist 11 (2006), 831-840)。
【0040】
全身ALCLは、最終的に死に至る可能性のある進行性疾患である。
【0041】
全身ALCLと診断された患者は、最初の治療手段として度々CHOPで治療を受ける。CHOPは、複数の化学療法剤(シクロホスファミド、ヒドロキシダウノマイシン (ドキソルビシン) 、オンコビン(登録商標) (ビンクリスチン) 、及びプレドニゾン)の組み合わせである。他の化学療法の組み合わせもまた第一の選択として使用することができる。
【0042】
さらに、局在部位に特に大量のリンパ腫が存在する場合又は局所性大リンパ節が正常な器官又は構造を圧迫又は侵襲し、化学療法ではその問題を制御できない場合、放射線療法は治療の重要な補助となり得る。
【0043】
第一選択の組み合わせ治療後の治療選択には、放射線療法と併用した別のより高用量の化学療法を含むことができる。このような高用量の組み合わせ治療は、腫瘍中のリンパ腫細胞及び骨髄中に位置し得るリンパ腫細胞を標的とする。しかしながら、骨髄中の正常な造血細胞もまた死滅され得る。このため、患者自身の血液から回収された循環血液幹細胞又は組織適合血縁又は非血縁ドナー由来の循環血液幹細胞の骨髄移植又は移植が必要とされる。このような治療に耐性を示す又は治療後に再発する患者は、治療選択が制限されている。
【図面の簡単な説明】
【0044】
【図1−1】図1aは、JunB及びcJunのT細胞特異的ノックアウトが、NPM-ALKトランスジェニックマウスの生存期間を顕著に増加することを示す(G. Inghirami; University of Turin)。(a) マウスの5つの群、NPM-ALK、NPM-ALKΔJunB(E.F. Wagner)、NPM-ALKΔcJun(E.F. Wagner)、NPM-ALKΔJun及び野生型マウスを使用した。NPM-ALKΔJunマウスはNPM-ALKマウスより顕著に長く生存したが、NPM-ALKΔJunB及びNPM-ALKΔcJunマウスの生存期間に違いはなかった。
【図1−2】図1bは、qRT-PCRによるNPM-ALK、JunB及びcJun mRNAレベルの分析を表す。NPM-ALK mRNA発現レベルは、NPM-ALK及びNPM-ALKΔJun腫瘍において同様であるが、野生型胸腺では発現は欠如している。JunB発現は野生型胸腺及びNPM-ALK腫瘍において高く、NPM-ALKΔJun腫瘍においては低い。cJun発現はNPM-ALKリンパ腫及び野生型脾臓において高いが、NPM-ALKΔJun腫瘍において低い。図1cは、ウエスタンブロットによるNPM-ALK、JunB及びcJunタンパク質レベルの分析を表す。ALKレベルは、NPM-ALK及びNPM-ALKΔJun腫瘍で同等であり、JunB及びcJunはNPM-ALKに存在するが、NPM-ALKΔJun腫瘍中に存在しない。
【図2】図2は、NPM-ALK陽性とNPM-ALK陰性ヒトALCL患者試料の免疫組織化学分析を表す。(a) ALCL患者由来の組織マイクロアレイの免疫組織化学分析(TMA) は、NPM-ALK陽性結節及び皮膚リンパ腫におけるALK、JunB、cJun及びPDGFRBの同時発現を示す(n=29)。(b)NPM-ALK陽性とNPM-ALK陰性ヒトALCL患者試料の表である。PDGFRB、cJun及びJunB及びALKに対するAbsを使用してIHC分析を行った。
【図3−1】図3−1は、PDGFRB mRNAレベルをqRT-PCRによって分析し、NPM-ALK腫瘍と比較してNPM-ALKΔJun腫瘍及び野生型胸腺において減少されることを示す。(a) PDGFRBは、マウスNPM-ALK腫瘍において高く発現されるが、NPM-ALKΔJun腫瘍及び野生型胸腺においては高くない。(b) タンパク質免疫ブロット定量化は、PDGFRBは、NPM-ALKΔJun腫瘍ではなく、大抵のNPM-ALKにおいて活発に発現されることを明らかにした。(c) は、NPM-ALKマウス腫瘍細胞はマウス線維芽細胞においてPDGFを産生し、PDGFRB発現及びリン酸化反応を刺激できることを示す。(d) 糖タンパク質のIPを行い、ウエスタンブロットによってPDGFRB及びp-PDGFRBのタンパク質レベルを評価した。NPM-ALK誘導リンパ腫は、cJun及びJunBの喪失により増殖を減少しアポトーシス速度を低下することを示す。
【図3−2】図3−2は、PDGFRB mRNAレベルをqRT-PCRによって分析し、NPM-ALK腫瘍と比較してNPM-ALKΔJun腫瘍及び野生型胸腺において減少されることを示す。(e) NPM-ALK及びNPM-ALKΔJunリンパ腫のKi-67陽性増殖速度及びタネル陽性アポトーシス速度を、蛍光免疫染色によって測定し(右)、10 HPFのカウントによって定量化した(左)。さらに、Ki-67陽性増殖速度を、各リンパ腫の10 HPF(強拡大視野)のIHC染色のカウントによって分析した(下)。NPM-ALK腫瘍と比較して、NPM-ALKΔJun腫瘍の増殖速度は顕著に低下したが、アポトーシス速度は増加した。(f)FACSによる細胞周期染色は、NPM-ALKマウスの腫瘍よりNPM-ALKΔJunマウスの腫瘍がG1において顕著に多くの細胞を有し、S期において少ないことを示す。
【図4−1】図4−1は、PDGFRBがJunB及びcJunの直接標的であることを示す。(a)は、複数の種におけるPDGFRBの高度の保存AP-1部位を示す。(b)は、マウスPDGFRB-プロモーターのChIP分析及び図を示す。図において、ChIP生成物の部位及びAP-1、c/EBP、SP1、AP-2及びNF-Yの結合部位を示す。NPM-ALKマウス腫瘍株化細胞でChIP分析を行った。抗体なしの細胞抽出物は、負の対照として機能する。陽性対照として、ヒストンH3 Absを使用した。PDGFRBプロモーターのAP-1部位に特異的なプライマーでのQRT PCRを、ChIP後抽出したRNAで行った。JunB及びcJunは共にPDGFRBプロモーター配列に結合した。
【図4−2】図4−2は、PDGFRBがJunB及びcJunの直接標的であることを示す。(c) (PDGFRB-luc)を含む又は(PDGFRB(w/o AP-1部位)-luc) AP-1部位を欠如する、作成したルシフェラーゼコンストラクトを示す。P-GL3-lucは、ホタルルシフェラーゼを含むベクターであるが、p-vec及びp-PDGFRB-lucはルシフェラーゼを含まないベクター及びPDGFRBプロモーターである。P-cJun、p-JunB及びp-PDGFRB-lucは、ルシフェラーゼを有するcJun及びJunB cDNA (P. Vesely)及びPDGFRBプロモーターを含むベクターをそれぞれ示す。P-vec + p-PDGFRB(w/o AP-1部位)-lucは、欠失AP-1部位を含まないベクター及びPDGFRBプロモーターを示す。p-cJun、p-JunB及びp-PDGFRB-lucのみが共に、強いシグナルを作り、cJun及びJunBのPDGFRB AP-1部位への特異的結合が確認される。(d) 交換された2つの塩基を有する正確なAP-1配列又は変異版 (標識「mut」)を含むEMSAプローブをデザインした。NPM-ALK陽性腫瘍細胞抽出物、及び対照AP-1部位、PDGFRB AP-1部位及びこれらの配列の変異版用のプローブを使用してEMSAを行った。対照AP-1部位及びPDGFRB AP-1両方のプローブであり、変異されていないプローブは、強いシグナルを生じる。さらに、PDGFRB AP-1部位及びJunB及びcJunに特異的な抗体を利用するスーパーシフトアッセイを行った。AP-1バンドは、Ab結合上で明らかに変化し、それぞれのJun-抗体の特異的結合が確認される。
【図5】補足図1は、ヒト及びマウスリンパ腫及びそれぞれの対照におけるNPM-ALK、JunB及びcJunのタンパク質レベルのIHCによる分析を示す。NPM-ALK レベルはヒトNPM-ALK陽性リンパ腫、NPM-ALK及びNPM-ALKΔJunマウス腫瘍で同様であるが、NPM-ALK発現は野生型リンパ節において検出されなかった。JunBはヒトリンパ腫及びNPM-ALK陽性マウスリンパ腫において発現されるが、NPM-ALKΔJun腫瘍及び野生型リンパ節で発現されない。NPM-ALKΔJun腫瘍以外の全組織において、cJunは発現される。
【図6】補足図2は、イマチニブでの特異的阻害の使用によるPDGFRBの阻害が、インビトロ(in vitro)での細胞数の減少及びアポトーシス速度の増加、インビボ(in vivo)での腫瘍サイズの減少及び増殖速度の低下を導き、90%以上の腫瘍サイズの減少が得られることを示す。(a) イマチニブでの処理の5日後、未処理対照腫瘍と比べ腫瘍重量は大幅に減少した。(b) 処理マウスと未処理マウスの腫瘍の直接のサイズ比較は、イマチニブ処理マウス腫瘍サイズで激しい減少を示す。(c) 異種移植マウスの腫瘍における増殖及びアポトーシス速度をそれぞれ、IHC染色のKi-67及びタネルによって評価した。増殖はイマチニブ処理マウスで大きく減少したが、アポトーシス速度で顕著な違いはなかった。(d) ウエスタンブロット解析は、未処理対照と比較して、イマチニブ処理の異種移植マウスの腫瘍の高PDGFRB-発現を明らかにする。
【図7】補足図3は、ニロチニブでの特異的阻害を使用したPDGFRBの阻害が、インビトロ(in vitro)での細胞数の減少及びインビボ(in vivo)での腫瘍サイズの減少を導き、59%以上の腫瘍サイズ減少が得られることを示す。(a) 抗増殖効果: ニロチニブ及びイマチニブでの治療は、PDGFRB発現NPM-ALK発現細胞 (株化細胞417; G. Inghirami; University of Turin) の細胞数の用量依存性減少を導くが、PDGFRB陰性SR-786 (G. Egger; MUW) 株化細胞の増殖速度は影響を受けない。(b) ニロチニブでの処置の6日後、未処理の対照腫瘍と比較して腫瘍重量は大幅に減少した。
【図8】補足図4は、SR-786及び417株化細胞におけるPDGFRBタンパク質発現のウエスタンブロット解析を示す。(a) 株化細胞 417のみ、PDGFRB陽性である。(b) ウエスタンブロット解析は、c-kitは分析したNPM-ALKリンパ腫試料のいずれにおいても発現されないことを明らかにする。(c)インビトロ(in vitro)でのイマチニブでの処理は、417細胞において用量依存性アポトーシスを導くが、SR-786細胞は影響を受けなかった。アラマーブルーアッセイによってアポトーシスを測定した。株化細胞417は20 μMのイマチニブでの処理の3日後に高アポトーシスを示すが、株化細胞SR-786は影響を受けなかった。
【発明を実施するための形態】
【0045】
分析方法
リアルタイムPCR
リアルタイムPCRを、Chromo 4 cycler (Biorad)上で製造者の標準プロトコールに従って、ABI Prism (Invitrogen)のSybrGreen PCR qPCR Supermixで行った。1ウェルあたり50 ngのcDNAを使用し、試料ごとに3通りに分析し、規準化用にGAPDHを使用した。結果を2-ΔΔCt法で分析した。プライマー: NPM-ALK FW: GTG GTC TTA AGG TTG AAG TGT GGT T (配列番号1); NPM-ALK Rev: GCT TCC GGC GGT ACA CTA CTA A (配列番号2); JunB FW: GGC TTT GCG GAC GGT TT (配列番号3); JunB Rev: GGC GTC ACG TGG TTC ATC T (配列番号4) ; c-jun FW: TGA CTG CAA AGA TGG AAA CG (配列番号5); c-jun Rev: GCT CTC GGA CTG GAG GAA C (配列番号6); PDGFRB FW: TGC CAG TTC CAC CTT GAA TGA A (配列番号7); PDGFRB Rev: AGT TGT GCC TCA GGC TCT GCT T (配列番号8)。
【0046】
ウエスタンブロット
約100 mgの腫瘍組織を、氷上でDounceを使用して、50 mM のトリス-HCl pH 7,4、150 mMのNaCl、0.1% トリトン X-100、5 mM EDTA、1x 完全プロテイナーゼ阻害剤(Roche)、1x HALT ホスファターゼ阻害剤 (Thermo scientific) から成る1 mlの溶解緩衝液中で裁断した。続いて、得られる懸濁液を5分間1200 rpmで遠心分離し、上清を新たなチューブに移し、タンパク質濃度をQubit蛍光光度計(Invitrogen)で測定した。総タンパク質溶解物(50μg)を、ドデシル硫酸ナトリウム-ポリアクリルアミド電気泳動 (SDS-PAGE) に供し、続いて、ニトロセルロース膜に移した。
【0047】
膜を1%ミルク及び1% ポリビニルピロリドン (PVP) を加えたTBS-T中で1時間ブロックし、その後一次抗体(1%ミルク+ 1% PVP (ポリビニルピロリドン及び0.02% NaN3)を加えたTBS-T中で、適した濃度に希釈) で4°Cで一晩インキュベートした。その後、TBS-Tで膜を3回洗浄し、TBS-T中に希釈した適切な二次抗体で1時間インキュベートした。TBSでの最終洗浄工程の後、ECL Plus溶液(Amersham Biosciences) で1分間膜をインキュベートし、続いてルミノメーター (Roche)上で写真を撮った。次の一次抗体を使用した: 抗JunB sc-73X、抗c-jun sc-1694X (Santa Cruz) 、抗PDGFRB #3169 (細胞情報伝達) 、抗ベータアクチン#4967 (細胞情報伝達) 、抗Alk 51-3900 Zymed; 抗phospho-Alk (#33415、細胞情報伝達)、Santa Cruz)、抗cKit (sc-168、Santa Cruz)。
【0048】
免疫組織化学及び免疫蛍光
ホルマリン固定パラフィン包埋 (FFPE) 腫瘍試料の切片を作成し、スライドガラスに貼り付け、脱脂し、再水和した。エピトープをトリス-EDTA中で加熱処理によって回収した。内在性ペルオキシダーゼを10分間3 % H2O2 中でブロックした。アビジン/ビオチンブロック(Vector)、Universal Mouse HRP-Kit (IDLabs) 由来のSuperblock及びマウスブロック中で切片をブロックした。一次抗体染色を一晩4°Cで行った。AEC及びヘマトキシリンを対比染色として使用した。抗PDGFRB (細胞情報伝達、#3169) 、抗JunB (Santa Cruz、sc-46) 、抗c-jun (Santa Cruz、sc-1694) 、抗Alk Zymed (51-3900)抗体を、製造者説明書に従って使用した。ヘマトキシリン/エオシン及びPAS染色をDAKOから購入した染色キットで行った。
【0049】
本発明者は今回、PDGFRB活性化の阻害によってT細胞リンパ腫に対応する治療法を見出した。このような阻害は、特に特定のAP-1陽性癌型における細胞増殖及びアポトーシスの調節に関連する。本発明は、少なくとも一部において、PDGFRB活性化媒介プロセスは、T細胞リンパ腫腫瘍治療、特に高悪性度NHL又はALCLの治療用の、(例えば、選択的)標的として有用であることを見出したことに関する。
【0050】
ALCL及びNPM-ALKとの関係に関して、先行技術に証拠が存在する。しかしながら、NPM-ALK陽性ALCLにおいてPDGFRBが活性化されるという今回の発見は意外であった。PDGFRBはNPM-ALK誘導リンパ腫において過剰発現されることが分かったが、複数の研究はイマチニブ等のPDGFRB阻害剤の使用が、ALCL株化細胞に有益な効果を有しない可能性を示していた(Rassidakis et al., Blood 105 (2005), 827-829; Ergin et al., Exp. Hematol. 29 (2001): 1082-1090)。NPM-ALKはまた、イマチニブでの治療に非感受性であることが分かっていた(Gunby et al., J. Med. Chem. 49 (2006), 5759-5768)。
【0051】
よって、今回のAP-1発現リンパ腫の動物モデルにおけるPDGFRB活性化因子阻害剤の特有の効果は、意外であった。
【0052】
用語「投与」とは、所望の目的の機能が行われるように本発明の阻害剤を必要とする対象に導入される経路を含む。使用できる投与の経路の例は、経口投与を含む。阻害剤は、任意の他の都合の良い経路、例えば、継続点滴又はボーラス投与によって、上皮又は皮膚粘膜(mucocutaneous lining) (例えば、経口、直腸、腟、及び腸管粘膜等)を介した吸収によってもまた投与することができ、他の治療剤と共に投与することができる。投与は、全身又は局所的とすることができる。リポソーム、微小粒子、マイクロカプセル及びカプセル中のカプセル化を含む様々な周知の送達系を、使用することができる。本発明の阻害剤の投与方法は、皮内、筋肉内、腹腔内、静脈内、皮下、鼻腔内、硬膜外、経口、舌下、脳内、腟内、経皮、直腸、吸入、又は局所投与を含むがこれらに限定されない。本発明の阻害剤は、単独、又は他のPDGFRB阻害剤又はT細胞リンパ腫の治療において使用される任意の他の治療剤、或いはこれらの両方と併用して、好ましくは医薬的に許容される担体と共に、投与することができる。本発明の阻害剤は、他の薬剤の投与前に、他の薬剤の投与と同時に、又は他の薬剤の投与後に投与することができる。さらに、本発明の阻害剤は、プロドラッグ形態で投与することもでき、それはインビボ(in vivo)で活性代謝物、又はより活性のある代謝物に変換される。
【0053】
用語「PDGFRB阻害剤」とは、PDGFRBに結合しそれによりその活性化を阻害し、及び/又はPDGFRBの活性化の機能阻害を直接又は間接的に示すリガンド(例えば、キナーゼドメインの全長、N末端、C末端、カルボキシ末端、ATP結合ポケット)に関する。活性の抑制は、HTScan(登録商標) PDGF受容体βキナーゼアッセイキット#7770; 細胞情報伝達等の標準試験によって試験することができる。
【0054】
用語「予防上有効量」とは、患者への単一又は複数用量投与において、細胞増殖性疾患の予防又は治療で効果のある本発明の阻害剤の量に関する。
【0055】
用語「対象」とは、細胞増殖性疾患罹患の可能性がある、又は本発明の化合物の投与から利益を得る可能性のある、ヒト及び非ヒト動物等の生物を含む。好ましいヒトは、本明細書中に記載されるように、細胞増殖性疾患又は関連症状に罹患する又は罹患する傾向のあるヒト患者を含む。用語本発明の「非ヒト動物」とは、あらゆる脊椎動物、例えば、哺乳類、マウス等のげっ歯類、及び非人類霊長類、非哺乳類、例えば、好ましくはヒトT細胞リンパ腫の動物モデルとして使用されるヒツジ、イヌ、ウシ、ニワトリ等を含む。
【0056】
用語「細胞増殖性疾患感受性」とは、例えば癌等の細胞増殖疾患を発症する危険性のある対象、すなわち、癌ウイルスでのウイルス感染症に罹患する対象、電離放射線又は発癌性化合物に曝露した対象、癌家系又は病歴を有する対象等を含むことを意味する。
【0057】
本明細書中で使用される用語「治療」又は「処理」は、細胞増殖性疾患感受性患者を治療するための予防又は治療の両方の手段を含む。
【0058】
特定の実施形態において、本発明は、T細胞増殖性疾患に関係する対象に、T細胞リンパ腫細胞の増殖を抑制することができるPDGFRB阻害剤の有効量を投与することによって、対象を治療する方法を供する。T細胞増殖性疾患は、癌、特に後期癌を含む。
【0059】
本発明の阻害剤は好ましくは、非ホジキンリンパ腫(別称、NHL)、特に高悪性度NHL等のNPM-ALKリンパ腫において、例えば、リンパ節又はリンパ節外部位、又は皮膚結節を含む原発性皮膚ALCLを含む、全身ALCL等のALCLにおける使用のために使用される。さらに、本発明の阻害剤は、例えば、末梢性T細胞リンパ腫 (PTCL)を治療するために、他の高悪性度成熟T-/NK-細胞リンパ腫における、例えば、リンパ節等の成熟胸腺起源のリンパ系組織における使用が示唆される。特に、PDGFRBの過剰発現が判明した場合、それらのT細胞リンパ腫の患者は本発明の阻害剤で治療を受ける。過剰発現は好ましくは、生物試料中の発現の増加によって測定され、その増加はT細胞リンパ腫非易発性対象又は非罹患の対象の基準値の少なくとも1.2倍、より好ましくは少なくとも1.5倍である。
【0060】
本発明の最も好ましい実施形態は、ALK+ T細胞リンパ腫、特にALK+-ALCL、すなわち、腫瘍細胞がALK陽性(特にNPM-ALK+)であるALCLの治療である。これらの腫瘍においてイマチニブ等の物質の標的であるc-kitが認められないことが確認された先行技術を考慮すると、本発明のPDGFRベータ阻害剤、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブ、特にイマチニブ又はニロチニブでの治療が効果のあることは非常に驚きであった。従って、この特異的治療の過程において、まずALCL患者のALK状態を確認し、続いて、最も効果のある治療を決定することが好ましい。これは、当技術分野で周知の方法、例えば、患者由来の腫瘍細胞の試料 (例えば、患者の血液、血清又は血漿試料)を用意し、続いて、これらの細胞がALK+又はALK-であるかを確認することによって行うことができる(例えば、Ergin et al., 2001; Fornari et al., Hematol. Oncol. 27 (2009), 161-170)。イマチニブはヒトALK陽性ALCL株化細胞においてはALKを結合することができず、効果を示すことはできないが (Ergin et al., 2001を参照されたい)、ヒトALCL患者においては、ALK陽性(特にNPM-ALK陽性) ALCLはPDGFRベータ阻害剤、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブ、特にイマチニブ又はニロチニブで良好に治療することができる。この好ましい実施形態に関してもまた、AP-1(の発現)の存在が必要である。
【0061】
従って、ALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療の本発明の好ましい方法において、まずリンパ腫を有する患者由来で腫瘍細胞を含む試料を分析して、腫瘍細胞がALKを発現するか判別し、試料の腫瘍細胞がALKを発現する場合、患者はPDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで治療する。
【0062】
特定の実施形態において、対象は、哺乳類、例えば、ヒト又は非ヒト霊長類を含む霊長類である。本実施形態において、本発明の阻害剤は、PDGFRB、又はその特異的ドメインと直接又は間接的に相互作用し得る。本発明の阻害剤は無制御の増殖を生じている細胞と接触し、細胞増殖を抑制する又はアポトーシスを誘発することができる。本発明の阻害剤との細胞の接触又は本発明の阻害剤の対象への投与は、細胞増殖性疾患に罹患する又は感受性のある細胞又は対象の治療の1つの方法である。
【0063】
特定の実施形態によれば、T細胞リンパ腫疾患に罹患する又は感受性のある対象の治療方法は、それを必要とする対象に、PDGFRBの阻害剤の予防又は治療上有効量を投与する工程を含み、これによりAP-1発現リンパ腫細胞を抑制する。好適な阻害剤は、PDGFRBを結合する少なくとも部分特異性を有するそれらのTK阻害剤、標準的なスクリーニング方法を介して同定された医薬的に許容されるその塩又は類似体を含む。
【0064】
本発明によれば、好ましい小分子阻害剤は、異なる目的で抗癌治療において使用されている、ニロチニブ、イマチニブ、ダサチニブ、ソラフェニブ、アキシチニブ、スニチニブ及びトセラニブ又は証明された抑制活性を有するその機能誘導体等の市販の化合物である。
【0065】
さらに、PDGFRの阻害剤は、活性を抑制するための試験を利用する、適切なスクリーニング技術によって同定することができる。
【0066】
天然のPDGF結合パートナーの構造を模倣するポリペプチド阻害剤、又は拮抗モノクローナル抗体又は抗体断片等のPDGFRB化合物への特異的免疫リガンドであるポリペプチド阻害剤もまた好ましい。
【0067】
それらの阻害剤は、高親和性を有してPDGFRBに結合することが好ましく、Kdが10-6 M未満であることが好ましい。
【0068】
特定の実施形態において、本発明の方法は、他の薬剤上活性な薬剤又は従来の治療方法と併用した、対象に本発明の阻害剤の治療上有効量を投与する工程を含む。薬剤活性化合物の例は、他のPDGFRB阻害剤及び/又はT細胞増殖性疾患を治療するための周知の薬剤、例えば、抗癌剤、抗増殖剤又は化学療法剤を含む。本発明の阻害剤と容易に併用されるT細胞リンパ腫の従来の治療方法は、標準的な化学療法及び/又は放射線治療であり、短パルス化学療法又はCHOP療法の使用を含む(Seidemann et al., Blood 97 (2001), 3699-3706)。本発明はさらに、本発明の阻害剤及び組み合わせ治療に使用される薬剤を含むキットに関する。
【0069】
本発明の阻害剤及び薬剤活性化合物は、同一の医薬組成物又は異なる医薬組成物で、例えば、同時に又は別の時間に対象に投与することができる。
【0070】
本発明の阻害剤の治療上有効量又は予防上有効量は、周知技術の使用及び類似環境下で得られた結果の観察によって、当業者によって容易に決定することができる。インビトロ(in vitro)又はインビボ(in vivo)でのアッセイを任意に利用することは、最適用量範囲の特定の手助けとなり得る。利用される正確な用量はまた、投与経路、状態、治療される状態の重症度、及び治療される個体に関する様々な物理的要因に依存し、当業者の判断に従って決定することができる。
【0071】
治療上又は予防上使用される本発明の阻害剤の有効量は、体重1キログラムあたり1日約0.001 ミリグラム (mg/kg/日)〜約100 mg/kg/日であることが期待される。好ましい用量は、0.1〜10 mg/kg/日の範囲である。さらに好ましい用量は、0.01〜10 g/日、より好ましくは 0.1〜1 g/日、さらに好ましくは0.1〜0.5 g/日、特に200〜400 mg/日である。
【0072】
本発明のさらなる他の態様において、本発明の方法は、T細胞リンパ腫の治療においてPDGFRB阻害に適当な阻害剤を特定するために供される。機能アッセイは、そのような疾患に罹患しPDGFRB過剰発現が判明した対象由来のT細胞リンパ腫株化細胞のエクスビボ(ex vivo)での使用を含む。
【0073】
インビトロ(in vitro)でのPDGFRB抑制活性を測定する方法は、試験化合物の存在下又は不存在下でPDGFRB又はその特異的ドメインの結晶構造を得る工程を含む。続いて化合物は、結晶構造の結合部位中又は上にコンピューターでモデル化する(computer modeled)ことができる。候補調節化合物が同定されると、化合物は、インビトロ(in vitro)、インビボ(in vivo)、又はエクスビボ(ex vivo)での細胞アッセイを使用してスクリーニングすることができる。本態様で同定した化合物は、本発明の好ましい阻害剤の類似体として有用である。
【0074】
本発明の阻害剤は、医薬的に許容される担体又は希釈剤と共に有効量で製剤することができる。例えば、有効量は、医薬的に許容される製剤中に供され、医薬的に許容される製剤の対象への投与後、対象に対して少なくとも12時間、24時間、36時間、48時間、1週間、2週間、3週間、又は4週間、本発明の化合物の輸送を維持する。
【0075】
特定の実施形態において、これらの医薬組成物は対象への局所又は経口投与に適切であり、錠剤、トローチ剤、バッカル剤、トローチ、水性又は油性懸濁剤又は液剤、顆粒、粉末、ペースト、エマルション、カプセル、シロップ又はエリキシル剤を含む。
【0076】
非経口投与に使用される好適な製剤は、例えば、無菌溶液又は懸濁液等の、皮下、筋肉内又は静脈内注射を含む。局所投与用製剤は、クリーム又は軟膏、ペースト及びゲル等の多数の形態を含む。
【0077】
好ましい医薬的に許容される担体は、ビヒクル、ラクトース、グルコース及びスクロース等の糖、コーンスターチ及びポテトスターチ等のデンプン、カルボキシルメチルセルロースナトリウム、エチルセルロース及び酢酸セルロース等のセルロース及びその誘導体、又はグリセリン、ソルビトール、マンニトール及びポリエチレングリコール等のポリオール、或いは医薬製剤において利用される当技術分野で周知の他の希釈剤及び賦形剤を含む。経口及び非経口投与のための液体担体の適切な例は、特に上記添加剤、例えば、カルボキシルメチルセルロースナトリウム溶液を含む、セルロース誘導体を含む水、一価アルコール及び多価アルコール及びその誘導体を含むアルコール、及び油を含む。生理的に許容される賦形剤は、生理食塩水、ゼラチン、デンプン、滑石、ケラチン、コロイドシリカ、尿素等とすることができる。さらに、補助剤、安定化剤、増粘剤、潤滑剤、及び着色剤を使用することができる。
【0078】
それらはまた、例えば、ヒドロキシプロピルメチルセルロース、他のポリマーマトリックス、ゲル、透過性膜、浸透圧系、多層コーティング、微小粒子、リポソーム、微粒子、又はそれらの組み合わせを使用して、それらの活性成分の維持又は制御された放出を供するように製剤され、様々な割合の目的の放出特性を供することができる。阻害剤または、1又は2以上の上記賦形剤と共にマイクロカプセル化形態とすることができる。
【0079】
以下の定義の対象は、本発明の実施形態である:
1.細胞増殖性疾患感受性患者における、T細胞リンパ腫の抗増殖治療のための、PDGFRベータ阻害剤。
2.AP-1発現リンパ腫の治療のための、請求項1に記載の阻害剤。
3.NHL、ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCL、及びPTCLから成る群から選択されるリンパ腫の治療のための、請求項1又は2に記載の阻害剤。
4.再発患者を治療するための、請求項1〜3のいずれか1項に記載の阻害剤。
5.患者の第一選択治療のための、請求項1〜4のいずれか1項に記載の阻害剤。
6.例えばポリペプチド又は小分子等のPDGFRベータ拮抗薬から選択される、請求項1〜5のいずれか1項に記載の阻害剤。
7.ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される、特にイマチニブ又はニロチニブから成る群から選択される、請求項6に記載の阻害剤。
8.予防又は治療使用のための、請求項1〜7のいずれか1項に記載の阻害剤。
9.局所又は全身使用のために製剤される、請求項1〜8のいずれか1項に記載の阻害剤。
10.経口使用のために製剤される、請求項1〜9のいずれか1項に記載の阻害剤。
11.0.001 mg/kg/日〜約100 mg/kg/日の用量で投与される、請求項1〜10のいずれか1項に記載の阻害剤。
12.化学療法及び/又は放射線療法との併用使用のための、請求項1〜11のいずれか1項に記載の阻害剤。
13.T細胞リンパ腫、特にAP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択される、特にイマチニブ又はニロチニブから選択される、PDGFRベータ阻害剤。
14.T細胞リンパ腫の抗増殖治療のためのPDGFRベータ阻害剤を含有する医薬製剤の、調製方法。
15.T細胞リンパ腫、特にAP-1発現及びALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療方法であって、リンパ腫を有する患者の腫瘍細胞を含む試料を分析し、腫瘍細胞がAP-1及び/又はALKを発現するか判別するとともに、試料の腫瘍細胞がAP-1及びALKを発現する場合、PDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで患者を治療する、抗増殖治療方法。
16.AP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、PDGFRベータ阻害剤。
本発明はさらに、次の実施例によってそれらに限定することなく説明する。
【実施例】
【0080】
実施例1: cJunとJunBとのT細胞特異的欠失は、マウスの発達NPM-ALK陽性リンパ腫の生存期間を顕著に増加する。
インビボ(in vivo)でのNPM-ALK陽性リンパ腫の形成におけるJunB及びcJunの役割を検討するために、マウス、T細胞特異的、CD4-プロモーターの制御の下でヒトNPM-ALK融合-チロシンキナーゼ (FTK) を保有するトランスジェニックマウスを、JunB遺伝子又はcJun 遺伝子のいずかのloxP導入版(JunBf/f又はcJunf/f)を保有する、CREリコンビナーゼ遺伝子(CD4-CRE)を独立して発現している同一バックグラウンドのマウスと交配した。(Chiarle et al., Blood 101 (2003): 1919-1927; Wolfer et al., Nat. Immunol. 2 (2001), 235-241; Behrens et al., EMBO J. 21 (2002), 1782-1790)。この手段を使用して、NPM-ALKのT細胞特異的発現を有しJunB及び/又はcJunがT細胞特異的にノックアウトされたマウスを作製した。目的の4つの系統のマウスを樹立した。導入遺伝子を有しJunB 又は cJunがコンディショナルノックアウトされたマウスを、NPM-ALKΔc-Jun又はNPM-ALKΔJunBとそれぞれ命名した。導入遺伝子を保有しJunB及びcJunが二重ノックアウトのマウスをNPM-ALKΔJunと命名した。カプラン・マイヤープロットを使用して異なるマウス系統の生存期間を分析した(図1a)。NPM-ALKΔJunはノックアウトなしの同腹子より顕著に長く生存したが(平均生存期間24 +/- 4,8 vs. 13,5 +/- 3,9 週間)、NPM-ALKΔJunB又はNPM-ALKΔc-Junの生存期間においては顕著な差はなかった(12,8 +/- 4,7及び11,3 +/- 3,2週間 vs. 13,5 +/- 3,9 週間) (図1a)。従って、さらに、NPM-ALKとNPM-ALKΔJunマウスとの違いを解明することに決定した。
【0081】
CREリコンビナーゼ媒介遺伝子欠失が十分に機能し、NPM-ALK mRNA発現レベルは影響を受けないことを確証するために、上記の2つの遺伝子型のマウスのリンパ腫及び対照マウス(胸腺)由来のリンパ組織においてqRT-PCRによって、NPM-ALK、JunB及びcJun mRNAのレベルを検討した(図1b)。腫瘍は胸腺において最も頻繁に生じるので、胸腺が対照組織として適した。NPM-ALK及びNPM-ALKΔJunリンパ腫は、NPM-ALKの同様の発現レベルを示したが、NPM-ALKは野生型マウスの胸腺中で発現されなかった。
【0082】
JunB mRNA発現は、CD4由来のNPM-ALK発現動物のリンパ腫よりも野生型マウスの胸腺においてわずかに高かったが、NPM-ALKΔJunマウスのリンパ腫においてjunB発現を検出することがほとんどできなかった。CJun mRNA発現は、NPM-ALKマウスのリンパ腫及び野生型マウスの胸腺と同程度であったが、期待通りNPM-ALKΔJunリンパ腫においてcjun発現は全くなかった。これらのデータから、JunB及びcJun のCREリコンビナーゼに基づくノックアウト戦略によって、対応するmRNAレベルの効率的な抑止が得られることが確認された。
【0083】
続いて、NPM-ALK及びNPM-ALKΔJunマウスのリンパ腫においてNPM-ALK、JunB及びcJunのタンパク質発現レベルを分析した(図1c)。NPM-ALKタンパク質発現はNPM-ALK及びNPM-ALKΔJunマウスにおいて同程度であり、ノックアウト戦略はNPM-ALKレベルに影響しなかったことが確認された。NPM-ALKマウスと比較した場合、JunB及びcJunタンパク質は、NPM-ALKΔJunリンパ腫において検出できず、再度CD-4 Cre媒介ノックアウト戦略の効果が確認された。
【0084】
実施例2: ヒトALCL患者試料における遺伝子発現プロファイリング
組織マイクロアレイ(TMA)技術を使用して(NPM-ALK陽性及び陰性ALCL患者のパラフィン包埋ヒト患者試料を使用した組織マイクロアレイを、Dr. Ana Schmatz; Medical University Viennaから得た)、ALCL患者試料のタンパク質発現を分析した。JunB (Santa Cruz; sc-46)、cJun (Santa Cruz; sc-1694) 及びPDGFRB (Cell Signaling; Cat.Nr.3169)タンパク質発現レベルは、ALK(Dako; N1614)発現レベルから独立して上昇した(図2a及びb)。
【0085】
PDGFRBは、NPM-ALKにおいて高発現されるが、NPM-ALKΔJun腫瘍においては高発現されない。
マウス及びヒトNPM-ALKリンパ腫の腫瘍試料をPDGFRBのRNA発現に関して分析した。qRT-PCRを使用して、PDGFRB発現がNPM-ALKΔJunマウスのリンパ腫においてNPM-ALKマウスと同様であることが分かった。野生型胸腺において、発現は腫瘍よりも50 %低い(図3a)。NPM-ALKマウスの腫瘍はPDGFRB陽性であるが、PDGFRBタンパク質発現はNPM-ALKΔJunマウスにおいて完全に消失されることを示す(図3b)。我々はまたIHCによって、NPMΔJunリンパ腫及び肝臓と比較してNPM-ALKのPDGFRB発現を分析した。PDGFRB発現は、NPM-ALKリンパ腫において高いが、NPM-ALKΔJunマウスはPDGFRBを発現しない(図3c及び未開示データ)。線維芽細胞を一晩血清非含有培地で血清欠乏させ、続いて、PDGFD(正の対照) 、NPM-ALK陽性マウス腫瘍株化細胞の血清(CD4-4又はCD4-417それぞれ、G. Inghirami; University of Turin) 又は血清非含有培地を、線維芽細胞に添加した。糖タンパク質に関して免疫沈降法を行い、PDGFRB及びp-PDGFRB(Cell Signaling; Cat.Nr.3124) のタンパク質レベルを、マウスNPM-ALKリンパ腫における自己刺激ループのウエスタンブロット解析によって評価した。NPM-ALKマウス腫瘍細胞は、自己刺激態様でマウス線維芽細胞において、PDGFを産生し、PDGFRB発現及びリン酸化反応を刺激することができる(図3d)。
【0086】
実施例3: NPM-ALKΔJunマウスの腫瘍は、増殖の減少及びアポトーシスマーカーの増加を示す。
マウスにおけるNPM-ALKリンパ腫は非常に活動的なので、腫瘍形成のこれらの初期段階の分析は直接は不可能であり、窒息死の数時間前までマウスは疾患の兆候を示さず、生存期間は約8〜約30週間と非常に広範である(図1aを参照されたい)。従って、我々は死亡時に、Ki-67 (Novocastra; NLC-Ki67p) 及びタネル(Chemicon; S7111) アッセイ、それぞれに関して免疫蛍光及び免疫組織化学染色を介してリンパ腫において増殖及びアポトーシス速度を分析した(図2e)。二重ノックアウト動物由来の腫瘍における増殖マーカーKi-67を使用した染色は非常に減少したが、タネル染色によって測定したアポトーシス速度はNPM-ALKΔJunマウスにおいて増加したことが分かった。フローサイトメトリーを使用して、NPM-ALKの細胞周期パラメータをNPM-ALKΔJunリンパ腫細胞に対して分析した。NPM-ALKΔJunリンパ腫細胞は、G期が顕著に長い状態であったが、S期は非常に短かった(図3f)。
【0087】
実施例4: PDGFRBは、JunB及びcJunの新規標的である。
次に、我々はJunB及びcJunによるPDGFRBの制御を分析した。「UCSC Genome Browser on Mouse July 2007 Assembly」及びMathInspectorを使用して(Kent et al., Genome Res. 12(2002): 996-1006; Cartharius et al., Bioinformatics 21 (2005), 2933-2942)、転写開始点から-269bp 〜-263 bp 上流のPDGFRプロモーター内にAP-1コンセンサス配列を同定することができ、かかる配列は複数の哺乳類種に渡って高度に保存されている(図4a)。このインシリコの発見から、PDGFRBプロモーターはAP-1因子を介して制御することができ、PDGFRBはおそらく新規であり未だ知られていないAP-1標的遺伝子であるという仮説が導びかれる。この仮説を試験するために、マウスCD4-NPM-ALK株化細胞 (417系統)を使用してクロマチン免疫沈降 (ChIP) 分析を行った (G. Inghirami, University of Turin)。この株化細胞は、我々が使用する同一のNPM-ALKマウスの原発腫瘍由来であった。ChIPを抗JunB及び抗cJun抗体で行い、得られる物質をPDGFRB-プロモーターの推定AP-1結合部位に結合したプライマーのqRT-PCRによって分析した(図の説明文を参照されたい)。JunB及びcJunがこのAP-1部位に結合することを示すことができ、PDGFRBがAP-1標的遺伝子であることを示唆している(図4b)。
【0088】
これらの結果をさらに確認するために、2つの異なるPDGFRBプロモータールシフェラーゼコンストラクトを使用して、ルシフェラーゼレポーター遺伝子アッセイを行った。両方の場合で、マウスPDGFRBプロモーター部分を、pGL3塩基プロモーターの複数のクローニング部位中にクローン化した。第一の場合(PDGFRB-Luc)において、転写開始点から-269bp〜-263bp上流のAP-1コンセンサス部位を含む断片を使用し、第二の場合(PDGFRBΔ-Luc)、この領域を排除した(図の説明文を参照されたい)。AP-1結合部位を含むPDGFRB-LucプロモーターコンストラクトとのcJun及びJunB発現ベクターの同時導入は強力なルシフェラーゼ活性化を生じ、一方AP-1結合部位欠如のPDGFRBΔ-Lucコンストラクトは、cJun及びJunBによって誘導されなかった。これらのデータは、cJun及びJunBの組み合わせは、AP-1部位を介してPDGFRBプロモーターを強力に誘導することを示す(図4c)。
【0089】
最終的に、EMSAによってNPM-ALKマウス腫瘍タンパク質抽出物におけるPDGFRB AP-1部位を分析した。腫瘍抽出物をTPA (1 2-O-テトラデカノイルホルボール-13-酢酸)-応答エレメント(TRE)を含むプローブでインキュベートした場合、EMSAによって大きなバンド変化が観察された。しかしながら、変異TRE配列はこの効果を消失した(図4d)。さらに、cJun及びJunBに特異的な抗体での腫瘍抽出物のプレインキュベーションは、変化したバンドのほぼ完全な欠如につながる。しかしながらこれらの発見は、PDGFRBが新規AP-1標的遺伝子であることを示す。
【0090】
免疫組織化学 (IHC)を使用して、ヒトALCL、NPM-ALK及びNPM-ALKΔJunマウスリンパ腫及び同系野生型動物由来のリンパ節において、NPM-ALK、JunB及びcJunの発現レベルを評価した。NPM-ALK染色は、全リンパ腫試料において同程度であったが、野生型リンパ節において検出できなかった。ヒト及びマウスNPM-ALK陽性ALCLリンパ腫は、JunB及びcJunの高発現を示すが、NPM-ALKΔJunマウスはJunBもcJunも発現しない。しかしながら、野生型のリンパ組織、すなわち、胸腺はJunB及びcJunを低レベルで発現する(補足図1)。
【0091】
実施例5: イマチニブ又はニロチニブでにマウスNPM-ALK腫瘍細胞の治療は、インビトロ(in vitro)での増殖の減少及びアポトーシスの増加を導く。
NPM-ALK腫瘍形成に対するPDGFRBの役割を検討するために、マウスNPM-ALK細胞及びPDGFRB陽性株化細胞417及びヒトNPM-ALK陽性及びPDGFRB陰性株化細胞SR-786 (補足図3a) [G. Egger, Medical University Vienna] を、異なる濃度のチロシンキナーゼ阻害剤イマチニブ-メシル酸塩 (イマチニブ)及びニロチニブで処理した。細胞を3日間RPMI培地中で培養し、続いて細胞数及びアポトーシス速度に関して分析した。イマチニブ濃度が増加するにつれ、細胞数は株化細胞417において次第に減少した。しかしながらSR-786細胞は、全く影響を受けなかった。
【0092】
実施例6: インビボ(in vivo)でのイマチニブ及びニロチニブでのマウスNPM-ALK腫瘍の治療は、腫瘍サイズの減少につながる。
同様にインビボ(in vivo)でのイマチニブ及びニロチニブの効果を試験するために、マウスNPM-ALK陽性株化細胞CD4-417の細胞を6週齢のSCID(重度の複合免疫不全)マウスの右側腹部の中に注入することによって、マウス異種移植モデルを樹立した。注入の4週間後に、固形の腫瘍が発生し、マウスを経管栄養によって300 mg/kg/日のイマチニブ又は75 mg/kg/日のニロチニブでそれぞれ毎日処置した。7日のコース後、イマチニブで処置したマウスは腫瘍の完全な寛解又は大きな減少を示した(未処置マウス: 2.7 g、処置マウス 0.5 g)(補足図2a及びb; 補足図3b)。さらに、NPM-ALKΔJunマウスにおいて観察された腫瘍表現型に一致して、移植腫瘍のイマチニブでの治療は、Ki-67及びタネル蛍光免疫染色によって測定されるように、増殖の減少及びアポトーシス速度のわずかな増大につながる(補足図2c)。PDGFRBタンパク質発現は、イマチニブ処理マウスにおいて上方制御される。これはおそらく、イマチニブによるPDGFRB-リン酸化反応の阻害への反作用である (補足図2d)。これらのデータは、イマチニブ及びニロチニブがALCLにおいてPDGFRBをブロックすることを支持している。
【0093】
実施例7: マウスNPM-ALK腫瘍細胞の、インビトロ(in vitro)でのイマチニブ又はニロチニブでの治療は、増殖の減少及びアポトーシスの増加につながる。
アポトーシス速度をアラマーブルーアッセイによって測定した。アラマーブルーの活性成分は、非蛍光性濃青色指示色素であるレサズリンである。それは細胞に浸透することができ、生(代謝上活性のある) 細胞による還元反応において、蛍光、鮮赤色色素のレゾルフィンに変換される。生じた蛍光は、代謝上活性のある細胞の数と比例する。
【0094】
アラマーブルーの0-20 μM イマチニブで処理したSR-786 及び417細胞を、摂氏37度で1時間インキュベートした。SR-786細胞は、培地中のイマチニブ濃度にかかわらず、同程度にレサズリンをレゾルフィンに変換することができた。0〜10μMで処理した417細胞の応答は、SR-786細胞と同様であった。20 μM イマチニブで処理した417細胞は濃青色から赤色への色調変化を誘発できなかったが、これはほとんど生細胞が存在しなかったことを示す。増殖率と同様に、SR-786細胞においてアポトーシスは影響を受けなかったが、417細胞においてアポトーシスは20 μM/ml イマチニブで非常に増加した(補足図4c)。これらの結果は、マウス細胞417はイマチニブ及びニロチニブに強く反応するが、ヒトSR-786細胞は影響を受けないままであったことを示し、417細胞の反応はイマチニブの中毒量が原因でないことを証明している。株化細胞の異なる応答の理由は、おそらくSR-786細胞におけるPDGFRB発現の欠如が原因である。ほとんどのヒトNPM-ALK陽性リンパ腫、及びマウス腫瘍及び株化細胞417はPDGFRBに関して陽性なので、ヒト株化細胞はそもそもPDGFRBを発現し、長時間の培養によってそれを失ったと思われる。培養下の細胞は、栄養分の供給のための新たな脈管構造(neovasculature)を生じる必要はなく、よって長時間の培養の細胞に関しては、主に血管の結合に関与するPDGFRB等の因子の発現を当然減少させるであろう。従って、イマチニブはNPM-ALK自体を阻害せずヒトNPM-ALK陽性細胞に影響しないので、ヒトにおいてイマチニブ治療は機能しないという仮説は、妥当でない。
【0095】
c-kitの阻害を介して作用しているイマチニブの可能性を排除するために、c-kitに関するウエスタンブロット解析(Dako, A4502)を行った。予想通り、株化細胞417においてc-kitは発現されなかった (補足図4a及びb)。
【特許請求の範囲】
【請求項1】
細胞増殖性疾患感受性患者における、T細胞リンパ腫の抗増殖治療のための、PDGFRベータ阻害剤。
【請求項2】
AP-1発現リンパ腫の治療のための、請求項1に記載の阻害剤。
【請求項3】
NHL、ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCL、及びPTCLから成る群から選択されるリンパ腫の治療のための、請求項1又は2に記載の阻害剤。
【請求項4】
再発患者を治療するための、請求項1〜3のいずれか1項に記載の阻害剤。
【請求項5】
患者の第一選択治療のための、請求項1〜4のいずれか1項に記載の阻害剤。
【請求項6】
例えばポリペプチド又は小分子等のPDGFRベータ拮抗薬から選択される、請求項1〜5のいずれか1項に記載の阻害剤。
【請求項7】
ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される、特にイマチニブ又はニロチニブから成る群から選択される、請求項6に記載の阻害剤。
【請求項8】
予防又は治療使用のための、請求項1〜7のいずれか1項に記載の阻害剤。
【請求項9】
局所又は全身使用のために製剤される、請求項1〜8のいずれか1項に記載の阻害剤。
【請求項10】
経口使用のために製剤される、請求項1〜9のいずれか1項に記載の阻害剤。
【請求項11】
0.001 mg/kg/日〜約100 mg/kg/日の用量で投与される、請求項1〜10のいずれか1項に記載の阻害剤。
【請求項12】
化学療法及び/又は放射線療法との併用使用のための、請求項1〜11のいずれか1項に記載の阻害剤。
【請求項13】
T細胞リンパ腫、特にAP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択される、特にイマチニブ又はニロチニブから選択される、PDGFRベータ阻害剤。
【請求項14】
T細胞リンパ腫の抗増殖治療のためのPDGFRベータ阻害剤を含有する医薬製剤の、調製方法。
【請求項15】
T細胞リンパ腫、特にAP-1発現及びALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療方法であって、リンパ腫を有する患者の腫瘍細胞を含む試料を分析し、腫瘍細胞がAP-1及び/又はALKを発現するか判別するとともに、試料の腫瘍細胞がAP-1及びALKを発現する場合、PDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで患者を治療する、抗増殖治療方法。
【請求項1】
細胞増殖性疾患感受性患者における、T細胞リンパ腫の抗増殖治療のための、PDGFRベータ阻害剤。
【請求項2】
AP-1発現リンパ腫の治療のための、請求項1に記載の阻害剤。
【請求項3】
NHL、ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCL、及びPTCLから成る群から選択されるリンパ腫の治療のための、請求項1又は2に記載の阻害剤。
【請求項4】
再発患者を治療するための、請求項1〜3のいずれか1項に記載の阻害剤。
【請求項5】
患者の第一選択治療のための、請求項1〜4のいずれか1項に記載の阻害剤。
【請求項6】
例えばポリペプチド又は小分子等のPDGFRベータ拮抗薬から選択される、請求項1〜5のいずれか1項に記載の阻害剤。
【請求項7】
ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ及びトセラニブから成る群から選択される、特にイマチニブ又はニロチニブから成る群から選択される、請求項6に記載の阻害剤。
【請求項8】
予防又は治療使用のための、請求項1〜7のいずれか1項に記載の阻害剤。
【請求項9】
局所又は全身使用のために製剤される、請求項1〜8のいずれか1項に記載の阻害剤。
【請求項10】
経口使用のために製剤される、請求項1〜9のいずれか1項に記載の阻害剤。
【請求項11】
0.001 mg/kg/日〜約100 mg/kg/日の用量で投与される、請求項1〜10のいずれか1項に記載の阻害剤。
【請求項12】
化学療法及び/又は放射線療法との併用使用のための、請求項1〜11のいずれか1項に記載の阻害剤。
【請求項13】
T細胞リンパ腫、特にAP-1発現ALCL、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療における使用のための、ニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択される、特にイマチニブ又はニロチニブから選択される、PDGFRベータ阻害剤。
【請求項14】
T細胞リンパ腫の抗増殖治療のためのPDGFRベータ阻害剤を含有する医薬製剤の、調製方法。
【請求項15】
T細胞リンパ腫、特にAP-1発現及びALK発現リンパ腫、好ましくはALK+-ALCL、特にNPM-ALK+-ALCLの抗増殖治療方法であって、リンパ腫を有する患者の腫瘍細胞を含む試料を分析し、腫瘍細胞がAP-1及び/又はALKを発現するか判別するとともに、試料の腫瘍細胞がAP-1及びALKを発現する場合、PDGFRベータ阻害剤の有効量で、好ましくはニロチニブ、イマチニブ、ダサチニブ、アキシチニブ、スニチニブ又はトセラニブから選択されるPDGFRベータ阻害剤で、特にイマチニブ又はニロチニブで患者を治療する、抗増殖治療方法。
【図1−1】


【図1−2】
【図2】
【図3−1】
【図3−2】
【図4−1】
【図4−2】
【図5】
【図6】
【図7】
【図8】
【図1−2】
【図2】
【図3−1】
【図3−2】
【図4−1】
【図4−2】
【図5】
【図6】
【図7】
【図8】
【公表番号】特表2013−505979(P2013−505979A)
【公表日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願番号】特願2012−531363(P2012−531363)
【出願日】平成22年9月28日(2010.9.28)
【国際出願番号】PCT/EP2010/064367
【国際公開番号】WO2011/036305
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(508317424)メディツィニシェ ウニベルジテート ウィーン (2)
【Fターム(参考)】
【公表日】平成25年2月21日(2013.2.21)
【国際特許分類】
【出願日】平成22年9月28日(2010.9.28)
【国際出願番号】PCT/EP2010/064367
【国際公開番号】WO2011/036305
【国際公開日】平成23年3月31日(2011.3.31)
【出願人】(508317424)メディツィニシェ ウニベルジテート ウィーン (2)
【Fターム(参考)】
[ Back to top ]