
R−バクロフェンプロドラッグの持効性放出経口投与形
R−バクロフェンプロドラッグの持効性放出経口投与形が開示される。

【発明の詳細な説明】
【技術分野】
【0001】
本開示により提供される方法は、(R)−バクロフェンプロドラッグの持効性放出経口投与形に関する。
【背景技術】
【0002】
下記式(1):
【化1】
【0003】
で表される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸(1)、すなわちGABABアゴニスト、R−バクロフェン((±)−4−アミノ−3−(4−クロロフェニル)ブタン酸)のプロドラッグは、経口投与されるか、又は哺乳類の結腸中に直接投与される場合、R−バクロフェンとして高い生物学的利用能を示す(Gallop et al., US 7,109,239 and US 7,227,028; 及び Lal et al., J Pharmacology Experimental Therapeutics 2009, 330(3), 911-921)。
【0004】
化合物(1)の投与に続く高いR−バクロフェンの経口生物学的利用能は、経口投与形、例えば持効性経口投与形での化合物(1)の使用、及び疾病、例えば痙攣及び胃食道逆流病の処理のための(van Herwaarden et al., Aliment. Pharmacol. Ther. 2002, 16(9), 1655-62; Ciccaglione and Marzio, Gut 2003, 52(4), 464-70; Andrews et al., US 6,117,908;及び Fara et al., WO 02/096404号);禁煙の促進への(Gessa et al., WO 01/08675号);麻酔剤の中毒傾向の低減への(Robson et al., US 4,126,684号);嘔吐の処理への(Bountra et al., US 5,719,185号);咳嗽の処理のための鎮咳剤として(Kreutner et al., US 5,006,560号);及び神経障害及び筋骨格痛(Benson et al., US 2009/0118365号)、尿失禁(Wun and Wustrow,アメリカ仮出願 番号61/309,336号, March 1, 2010に出願された)、運動障害、例えば失調症及びしゃっくり;末梢神経障害、例えば筋肉刺激障害;脊髄障害、例えば痙攣性対性不全麻痺;脳神経障害、例えば舌咽頭神経痛及び三叉神経痛;多発性硬化症;及び脳性麻痺の処理のためへのそのような経口投与形の使用を好む。
【0005】
化合物(1)の合成は、Gallop et al., US 7,109,239号; Gallop et al., US 7,227,028号; Gallop et al., US 2009/0192325号; Raillard et al., アメリカ出願番号2/537,798号 、August 7, 2009年出願された; 及びRaillard et al., アメリカ出願番号12/537,764 合、August 7, 2009年出願された(それらの個々は引用により本明細書に組込まれる)により記載されている。
【0006】
化合物(1)を含んで成る経口投与は、Kidney et al., US 2008/0206332号; 及びSastry et al., US 2009/0197958号に開示されている。Kidneyなどは、高剪断湿式造粒法を用いて調製された、化合物(1)及び放出速度−制御ポリマーを含んで成る持効性錠剤投与形を示す。Sastryなどは、化合物(1)を含んで成る持効性粒状投与形を開示する。
【0007】
直接的圧縮錠剤製造工程においては、錠剤成分が乾式ブレンドにより組み合わされ、そして得られる環式ブレンドが続いて回転錠剤プレス上でパンチ工具により錠剤に組合される。化合物(1)のこれまでのマトリックス錠剤配合物は、自由に流動する粒状物を形成するために、高剪断湿式造粒工程の間、有機溶媒、例えばアルコールの使用を要した(Kidney et al., US 2008/0206332号)。自由に流動する粒状物は、回転錠剤プレス上で許容できる重量制御を維持することを製造工程において必要とした。高剪断湿式造粒工程に使用される溶媒は続いて、高温での乾燥により許容できるレベルに除去されるべきであり、これは製造費用を高め、そして製造速度を遅くする。
【発明の概要】
【0008】
従って、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の経口投与形を調製するための改良された方法は有用である。
【0009】
乾燥粉末から調製される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸を含んで成る経口錠剤投与形、及びそのような投与形の調製方法は開示されている。乾燥処理は、化学的分離を引起すことができる、水又は溶媒への薬物の暴露を排除する。さらに、乾燥処理は、室温で実施され得る。この良好な温度条件は、薬物が、典型的には湿式造粒化工程の間、導入される残留溶媒を除去するために使用される高温に長時間、暴露される場合に発生する熱分解を最小にするか又は妨げることができる。
【0010】
第1の観点においては、経口錠剤投与形の合計重量に基づいて、約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る経口錠剤投与形が開示される。
【0011】
第2の観点においては、経口錠剤投与形の合計重量に基づいて、約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る混合物を乾式ブレンドし;そして前記ブレンドされた混合物を圧縮し、経口錠剤投与形を供給することを含んで成る経口錠剤投与形の調製方法が開示される。
【0012】
第3の観点においては、リン酸ニカルシウム及び(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩をブレンドし、第1ブレンドを供給し;前記第1ブレンドを円錐型ミルに通し;ヒドロキシプロピルメチルセルロース及びコロイド状ニ酸化珪素を乾燥ブレンドし、第2部位レンドを形成し;前記第2ブレンドを円錐形ミルに通し;前記第1ブレンド、前記第2ブレンド、微晶性セルロース、及び放出速度−制御ポリマーを、高剪断ブレンダーにおいてブレンドし、第3ブレンドを形成し;前記第3ブレンドと共にステアリン酸マグネシウムをブレンドし;そして前記第3ブレンドを圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法が開示される。
【0013】
第4の観点においては、少なくとも1つの本開示により供給される経口投与形を、処理の必要な患者に経口投与することを含んで成る、痙攣、胃食道逆流病、嘔吐、咳嗽、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、神経因性疼痛、及び筋骨格痛から選択された、患者における疾病の処理方法が開示される。
【図面の簡単な説明】
【0014】
当業者は、本明細書に記載される図面が単なる例示目的のためであることを理解しているであろう。図面は本発明の開示の範囲を制限するものではない。
【図1】図1は、500倍率での化合物(1)の次の3種のロットの走査電子顕微鏡(SEM)マイクログラフを示す:ロット4(1A);ロット70(JB);ロット71(1C)。
【図2】図2は、10,000倍率での化合物(1)の次の3種のロットのSEMマイクログラフを示す:ロット4(2A);ロット70(2B);ロット71(2C)。
【0015】
【図3】図3は異なったロットの化合物(1)を含む錠剤についての溶解プロフィールを示す。
【図4】図4は、異なったグレードのEUDRAGIT(商標)を含む錠剤についての溶解プロフィールを示す。
【図5】図5は、20mgの錠剤の重量均一性を示す。
【図6】図6は、20mgの錠剤の含有物均一性を示す。
【図7】図7は、20mgの錠剤の溶解プロフィールを示す。
【0016】
【図8】図8は、30mgの錠剤の重量均一性を示す。
【図9】図9は、30mgの錠剤の含有物均一性を示す。
【図10】図10は、30mgの錠剤の溶解プロフィールを示す。
【図11】図11は、SR3及びSR4投与形の投与に続いての健康なボランティアの血液における定常状態R−バクロフェン薬物力学プロフィールを示す。
【図12】図12は、不統一性粉末組成物を用いて調製されたSR4錠剤における(A)化合物(1)及び(B)DI-TAB(商標)についての錠剤均一性パラメーターを示す。
【図13】図13は、整構造性粉末組成物を用いて調製されたSR4における化合物(1)についての錠剤均一性パラメーターを示す。
【0017】
【図14】図14は、試験規模での不統一性粉末組成物を用いて調製されたSR4錠剤における化合物(1)についての錠剤均一性パラメーターを示す。
【図15】図15は、試験規模での不統一性粉末組成物を用いて調製されたSR4錠剤における化合物(1)についての錠剤均一性パラメーターを示す。
【発明を実施するための形態】
【0018】
定義:
“薬物副作用”とは、所望しない、不快な、有害で又は潜在的に有害である薬物効果を言及する。薬物副作用は、軽い、中位い又は重度であり得る。軽い薬物副作用の例は、消化不良、頭痛、疲労、はっきりしない筋痛、倦怠感及び睡眠パターンにおける変化を包含するが、但しそれらだけには限定されない。中位の薬物副作用は、それらを経験する個人が厄介で、苦しく又は耐え難く思われる反応、例えば皮疹、視力障害、筋肉の震え、排尿困難、ムード又は精神的機能における知覚可能な変化、及び血液組成における一定の変化を表す。重度の薬物副作用の例は、持続的又は有意な障害又は入院をもたらし、そして先天性異常を引起す、生命の脅威である反応を包含する。バクロフィン療法に関連することが知られている副作用の例は、鎮静、認識的な機能の減失、精神錯乱、記憶喪失、眩暈感、衰弱、失調症、ぼけるか又は複視、嘔気、息切れ、痙攣及び体位性低血圧を包含する。
【0019】
“AUC”とは、患者への化合物の投与に続いて、時間の関数としての血液における化合物又はその代謝物の濃度を示す曲線下の領域である。例えば、投与される化合物は、R−バクロフェンプロドラッグ(1)及びその対応する代謝物R−バクロフェンであり得る。AUCは、種々の間隔で、測定するための標準方法、例えば液体クロマトグラフィー−タンデム質量分光法(LC/MS/MS)を用いて、血液中の化合物又はその代謝物の濃度を測定し、そして血液濃度−対−時間の曲線下での領域を計算することにより決定され得る。濃度−対−時間の曲線はまた、薬物力学プロフィールとしても言及される。
【0020】
薬物濃度−対−時間の曲線からAUCを計算するための適切な方法は、当業界において良く知られている。例えば、R−バクロフェンについてのAUCは、患者へのR−バクロフィンプロドラッグ、例えば化合物(1)の投与に続いて、患者の血液におけるR−バクロフェンの濃度を測定することにより決定され得る。AUC0-24は、投与に続いて、投与から24時間までの曲線下の領域である。AUCss, 24は、一定期間(定常状態)にわたって投与される投与レジメに続いて、24時間にわたっての曲線下の領域である。
【0021】
“生物学的利用能”とは、患者への化合物(1)の投与に続いて患者の全身性循環に達するR−バクロフェンの割合及び量を言及し、そしてR−バクロフェンの血液濃度−対−時間のプロフィールを評価することにより決定され得る。血液濃度−対−時間の曲線を特徴づけることにおいて有用なパラメーターは、曲線下の領域(AUC)、ピーク濃度までの時間(Tmax)及び最大R−バクロフェン濃度(Cmax)を包含し、そこでCmaxは、患者への一定用量の化合物(1)の投与に続いて、患者の血液における薬物の最大濃度であり、そしてTmaxは、患者への一定用量の化合物(1)の投与に続いて、患者の血液におけるR−バクロフェンの最大濃度(Cmax)への時間である。
【0022】
絶対的経口生物学的利用能は、等量の化合物又は代謝物の静脈内投与に続いての生物学的利用能に比較して、経口投与に続いての化合物又はその代謝物の生物学的利用能である。化合物又はその代謝物の相対的経口生物学的利用能は、他の投与形及び/又は投与路での等量の化合物又はその代謝の投与に対する、化合物又はその代謝物の経口投与に続いての生物学的利用能である。例えば、一定の態様においては、%Frelとして表される相対的経口生物学的利用能は、持効性投与形としての20mgの化合物(1)の経口投与に続いてのR−バクロフェンの生物学的利用能に対する、患者への化合物(1)の経口投与に続いてのAUC0-24により決定されるR−バクロフェンの生物学的利用能である。
【0023】
“生物学的同等”とは、患者への等用量のR−バクロフェン又は化合物(1)の投与の後、R−バクロフェンの吸収速度及び程度の同等性を言及する。本明細書において使用される場合、2種の薬物学力プロフィールは、それらの2種のプロフィールの平均応答の比率についての90%信頼区間が0.8及び1.25の限界内にある場合、生物学的同等である。前記平均応答は、プロフィールの特徴的パラメーター、例えばCmax、 Tmax及びAUCの少なくとも1つを包含する。
【0024】
“化合物(1)”は、R−バルコフェンプロドラッグ化合物(1)、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸、医薬的に許容できるその塩、前記のいずれかの医薬的に許容できる溶媒化合物、及び前記のいずれかの結晶形を包含する。化合物(1)は、R−バクロフェンプロドラッグ(1)と交換可能的に使用される。一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、遊離酸である。
【0025】
一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、遊離酸であり、そして結晶性である。一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、塩酸塩である。IUPAC命名法を用いれば、化合物(1)はまた、(R)-4-クロロ-β-[[[[2-メチル-1-(S)-(2-メチル-1-オキソプロポキシ)プロポキシ]カルボニル]アミノ]メチル]-ベンゼンプロパン酸としても言及される。
【0026】
化合物(1)はまた、いくつかの互変異形、例えばエノール形、ケト形及びそれらの混合物で存在することもできる。従って、本明細書に示される化学構造体は、例示される化合物のすべての可能な互変異形を包含する。本開示の化合物はまた、同位体標識化合物も包含し、ここで1又は複数の原子が、従来天然において見出される原子質量とは異なった原子量を有する。本明細書に記載される化合物中に組込まれ得る同位体の例は、2H、3H、 11C、 13C、14C、15N、18O、17O、などを包含するが、但しそれらだけには限定されない。
【0027】
化合物(1)は、非溶媒和形及び溶媒和形、例えば水和化形で及びN−酸化物として存在することができる。一般的に、本明細書に言及されるような化合物は、塩、遊離酸、水和化された、N−酸化物、又は前記のいずれかの組合せであり得る。化合物(1)は、多重結晶形、共結晶形又は非結晶形で存在することができる。化合物(1)は、医薬的に許容できるその塩又は前記いずれかの遊離酸形の医薬的に許容できる溶媒和化合物、及び前記いずれかの結晶形を包含する。
【0028】
化合物(1)はまた、溶媒和化合物も包含する。溶媒和化合物とは、化学量論量又は非化学量論量で1又は複数の溶媒分子を有する化合物の分子複合体を言及する。そのような溶媒分子は、患者に対して無害であることが知られている、医薬業界において通常使用されるそれら、例えば水、エタノール及び同様のものである。化合物又は化合物の成分及び溶媒の分子複合体は、非共有分子内力、例えば静電力、ファンデルワース力又は結合により安定化され得る。用語“水和物”とは、1又は複数の溶媒分子が水である溶媒化合物を言及する。
【0029】
本開示の“化合物”は、本明細書に開示される式内のいずれかの特定の化合物を包含する。化合物は、それらの化合物構造及び/又は化学名称のいずれかにより同定され得る。化学構造及び化学名称が相反する場合、化学構造は化合物の識別の決定因子である。本明細書に記載される化合物は、1又は複数のキラル中心、及び/又は二重結合を含んで成り、そして、従って、立体異性体、例えば二重結合異性体(すなわち、幾何学的異性体)、鏡像異性体又はジアステレオーマとして存在することができる。従って、特にことわらない限り、相対的形状で完全に又は部分的に示される、明細書の範囲内のいずれかの化学構造は、例示される化合物のすべての可能性ある鏡像異性体及び立体異性体、例えば立体異性体的に純粋な形(例えば、幾何学的に純粋、鏡像異性体的に純粋又はジアステレオマー的に純粋)、及び鏡像異性体及び立体異性体混合物を包含する。
【0030】
鏡像異性体及び立体異性体混合物は、当業者に良く知られている分離技術法又はキラル合成技法を用いて、それらの成分鏡像異性体又は立体異性体に分解され得る。例えば、鏡像異性体又はジアステレオマーの分解は、例えば従来の方法、例えば分解剤の存在下での結晶化により、又は例えば、キラル高圧液体クロマトグラフィー(HPLC)カラムを用いてのクロマトグラフィーにより達成され得る。
【0031】
“Cmax”は、患者への一定用量の化合物(1)の投与に続いて、患者の血液において観察される最大R−バクロフィン濃度である。Css, maxは、一定期間(定常状態)にわたって投与される投与レジメの間、化合物(1)の投与に続く最大定常状態濃度である。Cass、minは、定常状態での最小濃度である。
【0032】
“C1/2”は、患者への化合物(1)の投与後12時間で患者の血液において観察されるR−バクロフィン濃度である。Css, 12は、一定期間(定常状態)投与される用量レジメの間、化合物(1)の投与後12時間での濃度である。
【0033】
“Tmax”は、患者への一定用量の化合物(1)の投与に続いて患者の血液におけるR−バクロフィンの最大濃度(Cmax)までの時間である。Tss, maxは一定期間(定常状態)にわたって投与される用量レジメの間、化合物(1)の投与に続いての最大濃度までの時間である。 “T1/2”は、患者の血液中のR−バクロフィン濃度が最大薬物濃度の半分に低下した時間とTmaxとの間の時間間隔である。Tss, 1/2は、一定期間(定常状態)にわたって投与される用量レジメの間、化合物(1)の投与に続いて、患者の血液中のR−バックフェン濃度が最大薬物濃度の半分に低下した時間とTmaxとの間の時間間隔である。
【0034】
“投与形”とは、治療効果を達成するために患者に投与され得る。一定量の活性剤又は活性剤のプロドラッグ、すなわちR−バクロフィンプロドラッグ(1)を含む配合物の形を言及する。経口投与形は、口を通して患者に投与されるか又は飲み込まれることが意図される。一定用量の薬物は、同時に又は一定期間にわたって投与される1又は複数の投与形を含むことができる。
【0035】
“絶食された患者”とは、一定用量が患者に投与される時間で及び投与後少なくとも4時間、胃が食物を実質的に有さない患者を言及する。患者の胃が食事に続いて実質的に食物を有さなくなる時間は、多くの因子、例えば食事のサイズ、例えばカロリー数、食事の内容物、例えば、脂肪含有率、患者の健康性、及び患者の胃腸管の状態に依存することができる。健康なヒト対象の胃は、食物の摂取に続いて、約4〜約8時間後、典型的に実質的に食物を有さない。ある態様においては、絶食された患者は、投与の前約10時間、投与の後約4時間まで、いずれの食物を食べず(しかし、いずれかの量の水又は透明な液体を摂取することができる)、投与の前約2時間及び約1時間、約250mlの水、及び投与の後約2時間、約250mlの水を飲み、投与後約4時間、昼食を取り、そして投与後約10時間で夕食を取る。
【0036】
“食事した患者”とは、胃が食物を含む患者を言及する。ある態様においては、食事した患者は、投与の前約30分で試験食事を食べ始め、そして投与の前約5分で試験食事を完結し、投与の後4時間で昼食を食べ、そして投与の後約10時間で夕食を食べる。試験食事は、高脂肪(試験食事における合計カロリー数の約50%)及び高カロリー(約1000合計カロリー)の朝食、例えばバターであげられた2個の卵、2枚のベーコン、バターを塗られた2枚の小麦トースト、4オンスのハッシュブラウンポテト、及び8オンスの全乳を含むことができる。試験食事は、いずれかのカロリー数を含むことができ、そしていくつかの態様においては、約150カロリーのタンパク質、250カロリーの炭水化物及び約500〜600カロリーの脂肪を含む。
【0037】
“最小悪影響濃度”とは、許容できない薬物副作用を生成しない、患者の血液又は血漿における治療化合物の最小濃度を言及する。薬物副作用の受容不可能性は例えば、患者に投与される化合物の治療有益性の観点から、薬物副作用及び/又は知覚される危険性の重症度に少なくとも一部基づいて、患者及び/又は処方する医者により決定され得る。最小悪影響濃度はまた、処理される患者の年齢、体重及び健康性、症状の頻度及び重症度、及び処方する医者の判断に少なくとも一部、依存する。
【0038】
“最小治療有効濃度”とは、意図される治療効果を生成する患者の血液又は血漿における治療化合物の最小濃度を言及する。
“患者”とは、哺乳類、例えばヒトを包含する。
“医薬的に許容できる”とは、U.S.薬局方に列挙されるか、又は哺乳類、例えばヒトへの使用のために他の一般的に認識される薬局方に列挙される、連邦又は州政府の規制局により許可されるか又は許可できることを言及する。
【0039】
“医薬的に許容できる塩”とは、1つの化合物、例えば医薬的に許容でき、そして本発明の化合物の所望する薬理学的活性を有する化合物(1)の塩を言及する。そのような塩は、次のものを包含するが、但しそれらだけには限定されない:(a)無機酸、例えば塩酸、臭酸、硫酸、硝酸、燐酸、及び同様のものにより形成されるか、又は有機酸、例えば酢酸、プロピオン酸、 ヘキサノン酸、 シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、琥珀酸、 リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、 3-(4-ヒドロキソベンゾイル) 安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、 1、2-エタン-二スルホン酸、 2-ヒドロキエタンスルホン酸、 ベンゼンスルホン酸、 4-クロロベンゼンスルホン酸、 2-ナフタレンスルホン酸、 4-トルエンスルホン酸、樟脳スルホン酸、 4-メチルビシクロ[2.2.2]-オクト-2-エン-1-カルボン酸、 グルコヘプトン酸、 3-フェニルプロピオン酸、 トリメチル酢酸、 第三ブチル酢酸、ラウリル硫酸、グルコン酸、 グルタミン酸、 ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸、 及び同様のものにより形成される酸付加塩;及び(b)本発明の化合物に存在する酸性プロトンが金属イオン、例えばアルカリ金属イオン、アルカリ土類金属イオン又はアルミニウムイオンにより置換されるか;又は有機塩基、例えばエタノールアミン、ジエタノールアミン、トリエタノールアミン、N−メチルグルカミン及び同様のものと調和する場合に形成される塩。ある態様においては、化合物(1)の塩は、塩酸塩、及びある態様においては、ナトリウム塩である。
【0040】
“医薬的に許容できるビークル”又は“医薬的に許容できる賦形剤”とは、医薬的に許容できる希釈剤、医薬的に許容できるアジュバント、医薬的に許容できる賦形剤、医薬的に許容できるビークル、医薬的に許容できるキャリヤー、又は前記いずれかの組合せを言及し、それらと共に、その薬理学的活性を破壊せず、そして治療的有効量の化合物、例えばR−バクロフェンプロドラッグ又はR−バクロフェン代謝物を供給するのに十分な用量で投与される場合、非毒性である、化合物、例えばR−バクロフェンプロドラッグ、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸(1)が投与され得る。
【0041】
“医薬組成物”とは、R−バクロフェンプロドラッグ(1)又は医薬的に許容できるその塩、及び少なくとも1つの医薬的に許容できるビークル(これと共に、プロドラッグが患者に投与される)を含んで成る組成物を言及する。
【0042】
“プロドラッグ”とは、活性薬物を開放するために、使用条件下で、例えば身体内で転換を受ける活性化合物の誘導体を言及する。プロドラッグは時折、但し必ずしも必要ではないが、活性薬物に転換されるまで、薬理学的不活性である。プロドラッグは、典型的には官能基を通して薬物にプロ成分(本明細書において定義される)を結合することにより得られる。例えば、R−バクロフェンプロドラッグ(1)は、親薬物R−バクロフェンを供給するために患者の身体内で代謝される。
【0043】
“プロ成分”とは、1つの薬物に、典型的には使用の特定条件下で分解できる結合を通して、薬物の官能基に結合される基を言及する。薬物とプロ成分との間の結合は、酵素又は非酵素手段により分解され得る。使用の条件下で、例えば患者への投与に続いて、薬物とプロ成分との結合は、親薬物を開放するために分解され得る。プロ成分の分解は、例えば加水分解反応を通して、自発的に進行するか、又はそれは他の剤、例えば酵素、光、酸又は物理的又は環境パラメーターの変化又はそれらへの暴露、例えば温度、pH、等の変化により触媒されるか又は誘発され得る。前記剤は、使用の条件、例えばプロドラッグが投与される患者の全身性循環に存在する酵素、又は胃の酸性条件に対して内因性であるか、又は前記剤は外因的に供給され得る。例えば、R−バクロフェンプロドラッグ(1)に関しては、薬物はR−バクロフェンであり、そしてプロ成分は下記構造を有する:
【0044】
【化2】
【0045】
“鎮静”とは、本明細書において使用される場合、最小鎮静及び/又は中位の鎮静を言及する(例えば、American Society of Anesthesiologists, Anesthesiology 2002, 96, 1004-17を参照のこと)。抗不安剤としても言及される最小鎮静は、気道を独立して及び連続的に維持する患者の能力、及び薬理学的又は非薬理学的方法又はその組合せにより生成される物理的刺激又は口頭の指令に対して適切に応答する患者の能力を維持する、最小に低められたレベルの意識である。認識機能及び同調はひかえめに損なわれるが、呼吸及び心血管機能は影響されない。目的が成人における最小鎮静である場合、適切な用量は、モニターされていない自家使用のために処方され得る最大の推薦される用量、例えば最大の推薦される治療用量に過ぎない。
【0046】
中位の鎮静は、患者が単独で又は光触知刺激により達成される、口頭の指令に対して意図的に応答する間、意識の薬物−誘発された低下である。患者の気道を維持するためには介入は必要とされない。鎮静は連続的であり、そして個々の患者がいかに応答するかを予測することは常に可能であるわけではない。鎮静用量は、所望する効果が達成されるまで、薬物、例えばR−バクロフェンプロドラッグ(1)の複数用量を投与するインクリメンタル投与により決定され得る。種々の目盛り、例えばラムゼー目盛り及び他のものが、鎮静を評価するために使用され得る。鎮静の客観的処理は、脳波図パラメーター、例えばBispectral IndexバージョンXP及びPatient State Analyzerの測定を包含する。ある態様においては、鎮静は、最小鎮静、及びある態様においては、中位の鎮静を言及する。
【0047】
“持効性”とは、化合物の直接的な配合物の経口投与により達成される時間に比較して、延長された時間にわたって全身性血液循環において化合物、又はその活性代謝物の治療量を達成するのに効果的な速度で、投与形からの化合物の放出を言及する。いくつかの態様においては、化合物のインビボ放出は、少なくとも約4時間にわたって、いくつかの態様においては、少なくとも約8時間にわたって、いくつかの態様においては、少なくとも約12時間にわたって、いくつかの態様においては、少なくとも約16時間にわたって、いくつかの態様においては、少なくとも約20時間にわたって、及びいくつかの態様においては、少なくとも約24時間にわたって、生じる。
【0048】
ある態様においては、本発明の開示により供給される持効性投与形は、特定速度、例えば75rpmで回転するパドル(USP, タイプII)により撹拌される、pH6.8及び37℃での50mMのリン酸ナトリウム一塩基性緩衝液において、約18時間以内に投与形における約45%〜約55%の化合物(1);約18時間以内に約40〜約45%の化合物(1);及び約18時間以内に約34%〜約44%の化合物(1)を放出する。
【0049】
“治療的行有効量”とは、疾病又は障害、又は疾病又は障害の臨床学的症状の少なくとも1つを処理するために対象に投与される場合、前記疾病、障害又は症状のそのような処理に影響を及ぼすために十分である化合物の量を言及する。治療的有効量は、例えば化合物、疾病、障害、及び/又は疾病の症状、疾病又は障害の重症度、及び/又は疾病又は障害の症状、処理される患者の年齢、体重及び/又は健康、及び処方する医者の判断に依存して変化することができる。治療的有効量は、当業者に確認され得るか、又は通常の実験により決定され得る。
【0050】
いずれかの疾病の“処理する(treating)”又は“処理(treatment)”とは、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つを阻止するか又は改善し、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの獲得の危険性を低め、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの進行を遅くし、又は疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの進行の危険性を低めることを言及する。“処理する”又は“処理”はまた、疾病を、物理的に(例えば、認識できる症状の安定化)、生理学的に(例えば、物理的パラメーターの安定化)、又は両者的に阻害し、そして患者に認識できるか又はできない少なくとも1つの物理的パラメーターを阻害することを言及する。ある態様においては、“処理する”又は“処理”とは、疾病、又は患者が疾病をまだ経験していないか又はその症状をまだ示していない場合でさえ、疾病に暴露されるか又は疾病の傾向を有する患者における疾病又は1又は複数のその症状の開始を遅延することである。
【0051】
投与形及び方法のある態様が詳細に言及される。開示される態様は請求項を制限するものではない。対照的に、請求項はすべての変更、修飾及び同等のことを包含する。
【0052】
1つの観点いおいては、本発明の開示は、次の構造:
【化3】
【0053】
を有する化合物(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸に関する。
化合物(1)は、R−バクロフェンのプロドラッグである。
【0054】
組成物:
もう1つの観点においては、本発明の開示は、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸及び医薬的に許容できる賦形剤を含んで成る持効性経口投与形を供給する。化合物(1)は、Gallop et al., アメリカ特許第7,109,239号; Gallop et al., アメリカ特許第7,227,028号; Gallop et al., US 2009/0192325号; Raillard et al., 2009年8月7日に出願されたアメリカ特許出願番号12/537,798号;及び/又は Raillard et al., 2009年8月7日に出願されたアメリカ特許出願番号12/537,764号(それらの全内容物は引用により本明細書に組込まれる)により記載される方法を用いて調製され得る。
【0055】
もう1つの観点においては、化合物(1)の結晶化は、溶媒の組成及び血漿化速度に少なくとも一部、依存して、異なった形態学を有する生成物を供給する。異なった結晶化速度でアセトン/ヘキサンの混合物から結晶化された化合物(1)の異なった形態学のいくつかが、図1及び2に示されている。中間速度の結晶化で得られる形態学(図1A及び2A)は、化合物(1)の初晶の凝集体を含んで成るものとして特徴づけられ得、それらの凝集体は主に丸くされ、そして約25〜約ミクロンの直径を伴って圧縮し、そして前記初晶は数ミクロン以下の寸法を有する。
【0056】
遅い速度の結晶化で得られる態様学(図1B及び2B)は、約100ミクロンの全体的寸法及び約100μmの長さの線状物を有する、ゆるい線維状の又は線状の塊状物を含んで成るものとして特徴づけられる。早い速度の結晶化で得られる化合物(1)の形態学(図1C及び2C)は、初晶の不規則的に形状化された凝集体により特徴づけられ、ここで前記凝集体は約25〜約50ミクロンの寸法(長さ/幅)を有し、そして初晶は数ミクロン以下の寸法を有する。
【0057】
もう1つの観点においては、化合物(1)の他に、本発明の開示により供給される持効性経口投与形は、医薬的に許容できる賦形剤、例えば微晶性セルロース、ヒドロキシプロピルメチルセルロース、放出速度制御のポリマー、二塩基性リン酸カルシウム二水和物、コロイド状ニ酸化珪素及びステアリン酸マグネシウムを含むことができる。
【0058】
もう1つの観点においては、化合物(1)の他に、本発明の開示により供給される持効性経口投与形は、医薬的に許容できる賦形剤、例えば微晶性セルロース、ヒドロキシプロピルメチルセルロース、放出速度制御のポリマー、二塩基性無水リン酸カルシウム、コロイド状二酸化珪素及びステアリン酸マグネシウムを含むことができる。
【0059】
ある態様においては、本発明の開示により供給される投与形に使用される微晶性セルロースは、約180ミクロンの呼称粒子サイズ、約2〜約5%の湿分、及び約0.29g/cc〜約0.36g/ccのゆるい嵩密度により特徴づけられ、そして例えばAVICEL(商標) PH200 (AVICEL(商標), FMC Biopolymer)を包含する。ある態様においては、微晶性セルロース又は、約12mmのFlodexを示す。
【0060】
ある態様においては、本発明の開示により供給される投与形に使用されるヒドロキシプロピルメチルセルロースは、19〜24%のメトキシル含有率、7〜12%のヒドロキシプロピル含有率及び2%水溶液における3,000〜5,600cPの粘度を有するヒプロメロース2208、例えばMETHOCELTM K4M SP (標準のプレミアム) or METHOCELTM K4M CR (制御された放出)であり得る。ある態様においては、本発明の開示により供給される投与形に使用されるヒドロキシプロピルメチルセルロースは、19〜24%のメトキシル含有率、7〜12%のヒドロキシプロピル含有率、ヒプロメロース2208置換型、2,663〜4,970cPの粘度、0.1〜0.2g/ccの嵩密度、及び最大約5%の湿分を有し、そして例えばMETHOCELTM K4M DC(直接的圧縮)を包含する。ある態様においては、ヒドロキシプロピルメチルセルロースは、約28mm〜約30mmのFlodexを示す。
【0061】
ある態様においては、本発明の開示により供給される投与形に使用される放出速度制御ポリマーは、エチルアクリレート、メチルメタクリレート、及び第四アンモニウム基を有する低含有率のメタクリル酸エステルのコポリマー、例えばトリメチルアンモニオエチルメタクリレートクロライドであり得る。ある態様においては、前記コポリマーは、約150,000ダルトンの平均分子量を有する。ある態様においては、放出速度制御コポリマーは、乾燥物質に基づいて約8.9〜約12.3%のアンモニオメタクリレート単位を含み、そしてある態様においては、EUDRAGIT(商標) RLPO (Evonik Industries AG, Darmstadt, DE)であり得る。ある態様においては、放出速度制御コポリマーは、乾燥物質に基づいて約4.5〜約7.0%のアンモニオメタクリレート単位を含み、そしてある態様においては、EUDRAGIT(商標) RSPO (Evonik Industries AG, Darmstadt, DE)であり得る。ある態様においては、放出速度制御ポリマーは約22mmのFlodexを示す。
【0062】
ある態様においては、本発明の開示により供給される投与形に使用される二塩基性リン酸カルシウム二水和物は、粉砕されていないDI−TAB(商標)であり得る。ある態様においては、二塩基性リン酸カルシウム二水和物は、4mm以下か又はそれに等しいFlodexを示す。
【0063】
ある態様においては、本発明の開示により供給される投与形に使用される二塩基性無水リン酸カルシウムは、粉砕されていないA-TAB(商標)であり得る。ある態様においては、二塩基性無水リン酸カルシウムは、4mm以下か又はそれに等しいFlodexを示す。
【0064】
ある態様においては、本発明の開示により供給される投与形に使用されるコロイド状二酸化珪素又は未処理の非晶性ヒュームドシリカは、CAB-O-SILTM M-5P (Cabot Corporation, Bilerica, MA)であり得る。
【0065】
ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブラン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成るを含んで成る。ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約5重量%〜約15重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約17重量%〜約33重量%の微晶性セルロース;約20重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び約5重量%〜約20重量%の放出速度−制御ポリマーを含んで成る。
【0066】
ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約8重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約17重量%〜約33重量%の微晶性セルロース;約21重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び約5重量%〜約22重量%の放出速度−制御ポリマーを含んで成る。ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約5重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約18重量%〜約22重量%の微晶性セルロース;約33重量%のヒドロキシプロピルメチルセルロース;及び約17重量%の放出速度−制御ポリマーを含んで成る。
【0067】
ある態様においては、持効性投与形は、希釈剤、充填剤及び滑剤から選択された、1又は複数の医薬的に許容できる賦形剤を含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約23重量%〜約33重量%の二塩基性リン酸カルシウム二水和物;約0.1重量%〜約2重量%のコロイド状二酸化珪素;及び約0.1重量%〜約2重量%のステアリン酸マグネシウムを含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約19重量%〜約22重量%の二塩基性リン酸カルシウム二水和物;約1重量%のコロイド状二酸化珪素;及び約1重量%のステアリン酸マグネシウムを含んで成る。
【0068】
ある態様においては、本発明の開示により供給される持効性経口投与形は、約23重量%〜約33重量%の二塩基性無水リン酸カルシウム;約0.1重量%〜約2重量%のコロイド状二酸化珪素;及び約0.1重量%〜約2重量%のステアリン酸マグネシウムを含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約19重量%〜約22重量%の二塩基性無水リン酸カルシウム;約1重量%のコロイド状二酸化珪素;及び約1重量%のステアリン酸マグネシウムを含んで成る。
【0069】
ある態様においては、微晶性セルロースはAVICEL(商標) PH200であり、ヒドロキシプロピルメチルセルロースはMETHOCELTM K4M DC 及びMETHOCELTM K4M CRから選択され、放出速度制御ポリマーはEUDRAGIT(商標) RLPOである。
【0070】
本発明の開示により供給される持効性経口投与形は、約24〜26mmのFlodexを有する(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約12mmのFlodexを有する微晶性セルロース;約28〜20mmのFlodexを有するヒドロキシプロピルメチルセルロース;及び約22mmのFlodexを有する放出速度制御ポリマーを含んで成る。
【0071】
ある態様においては、本発明の開示により供給される錠剤投与形は、約5重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸;約18重量%〜約32重量%のAVICEL(商標) PH200; 約22重量%〜約33重量%の METHOCELTM K4M DC; 約重量%〜約18重量%のEUDRAGIT(商標) RLPO; 約19重量%〜約28重量%のA-TAB(商標);約0.7重量%〜約1.5重量%のステアリン酸マグネシウム;及び約0.9重量%〜約1.1重量%のコロイド状二酸化珪素を含んで成り;そして錠剤投与形はそれぞれ、約150mg〜約400mg;及びある態様においては、約50mg〜約600mgの重量である。
【0072】
ある態様においては、本発明の開示により供給される投与形は、約100mg〜約600mgの合計重量を有する。
【0073】
化合物及びブレンドの特徴化:
種々の観点においては、乾燥粉末ブレンドからの錠剤製造を促進するためには、組合された化合物(1)及び医薬的に許容される賦形剤から形成される乾燥粉末が許容できる流動性質を示すことが所望される。乾燥粉末の流れは、多くのパラメーター、例えば粒度、粒度分布、粒子形状、粒子粗さ、、嵩密度、多孔度、粉末を通しての空気透過性、静電荷、湿度、沈殿効果及び粘着力、例えばLondon分散力及び水素結合力により影響され得る。
【0074】
本発明者は、賦形剤のある組合せ又はブレンドが、個々の賦形剤の紛体流性質により予測されない、直接的圧縮錠剤製造のために有用な改良された粉体流性質を思いがけなく提供することを見出した。粉体流に対する予測されない相乗効果が市販の錠剤操作において有用なブレンドを提供する。さらに、本発明者は、親水性アクリレートポリマーが、同じタイプの疎水性ポリマーからより親水性マトリックス錠剤からの化合物(1)の放出を遅め、そして制御することにおいてより効果的であり得ることを見出した。
【0075】
乾燥粉末ブレンドの流動性質は、Fledexにより特徴づけられ得る。低いFlodexを示す乾燥粉末ブレンドは一般的に例えば製造速度、錠剤重量均一性、薬物含有均一性、硬度均一性、錠剤外観、及び薬物放出プロフィールにおいて影響されるような錠剤製造工程を受けやすい。約22mm又はそれ以下、又は約15mm又はそれ以下のFlodexを示す乾燥粉末ブレンドが有用である。
【0076】
ある態様においては、化合物(1)は、約22mm〜約28mmのFlodexを示す。
ある態様においては、投与形に使用される微晶性セルロースは、約10mmへの約14mmのFlodexを示す。
ある態様においては、投与形に使用されるヒドロキシプロピルメチルセルロースは、約26mmへの約32mmのFlodexを示す。
【0077】
ある態様においては、投与形に使用される放出速度−制御ポリマーは、約20mmへの約24mmのFlodexを示す。
ある態様においては、投与形に使用されるニ塩基性リン酸カルシウム二水和物は、約4mm以下のFlodexを示す。
ある態様においては、投与形に使用されるニ塩基性無水リン酸カルシウムは、約4mm以下のFlodexを示す。
【0078】
製造:
種々の観点においては、本発明の開示により供給される持効性経口投与形は錠剤として供給され得る。本発明の開示により供給される配合物は一般的に、直接的な圧縮により経口錠剤投与形を形成することにおいて有用である。
【0079】
ある態様においては、投与形は、化合物(1)を含んで成る錠剤形で存在することができる。錠剤投与形は、薬物の経口投与のために適切ないずれかの形状、例えば球状、立法体状、卵形又は楕円形状のものであり得る。ある態様においては、本発明の開示により供給される錠剤投与形、例えば錠剤形での経口投与は、R−バクロフェンプロドラッグ(1)が少なくとも1つの放出速度変性化合物を含んで成るマトリックスに分散されているマトリックスシステムである。マトリックスシステムは、例えば“Handbook of Pharmaceutical Controlled Release Technology,” ed. Wise, Marcel Dekker, Inc. (2000) 及び“Treatise on Controlled Drug Delivery, Fundamentals, Optimization, and Applications,” ed. Kydonieus, Marcel Dekker, Inc. (1992)に記載されるように、当業界において良く知られている。
【0080】
ある態様においては、本発明の開示により供給される投与形における化合物(1)の量は、約0.1mg〜約200mg;ある態様においては、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;及びある態様においては、約5mg〜約50mgの範囲である。化合物(1)の医薬的に許容できる塩及び/又は溶媒化合物を含んでいる投与形に関しては、投与形における化合物(1)の量は、その塩及び/又は水和物を含んで成る化合物(1)の等質量を言及する。ある態様においては、錠剤投与形は、治療的有効量の化合物(1)を含んで成る。
【0081】
治療的有効量の化合物(1)は、約1mg−当量〜約100mg−当量のR−バクロフィン;約2mg−当量〜約80mg−当量のR−バクロフィン;約2mg−当量〜約40mg−当量のR−バクロフィン;又は約5mg−当量〜約20mg−当量のR−バクロフィン;を含んで成ることができる。1mgの化合物(1)は、約0.535mg−当量のR−バクロフェンを含んで成る。ある態様においては、本発明の開示により供給される投与形における化合物(1)の量は、患者において中位の鎮静及び運動活動の障害を引起す量以下である。ある態様においては、化合物(1)の治療的有効量は、患者において中位の鎮静及び運動活動の障害を引起す量以下である。
【0082】
錠剤投与形が治療的有効量以下の化合物(1)を含んで成る、ある態様においては、多重錠剤投与形が、治療的有効量の化合物(1)を供給するために、同時に又は一定期間にわたって患者に投与され得る。
【0083】
本明細書に開示される化合物(1)及び放出速度変性化合物の他に、錠剤投与形はまた、1又は複数の医薬的に許容できる賦形剤、例えば界面活性剤、滑剤、可塑剤、結合剤、希釈剤、 抗−付着剤、滑沢剤、 緩衝剤、 顔料、 湿潤剤、 乳化剤、 pH 緩衝剤、安定化剤、増粘剤、崩壊剤、 風味剤、 味覚隠蔽剤及び着色剤を含んで成る。
【0084】
希釈剤又は充填剤は、投与形を圧縮のための粒度にするために嵩密度を高めるために添加され得る。本発明の開示により供給される錠剤投与形において有用な希釈剤の例は次のものを包含する:二塩基性無水リン酸カルシウム、二塩基性リン酸カルシウム二水和物、硫酸カルシウム、 リン酸ニカルシウム、リン酸三カルシウム、ラクトース、セルロース、例えば 微晶性セルロース、カオリン、マンニトール、塩化ナトリウム、 乾燥澱粉、アルファ澱粉、圧縮性糖、 及び前記いずれかの組合せ。ある態様においては、希釈剤は二塩基性リン酸カルシウム及び微晶性セルロースから選択される。充填剤は水不溶性、水溶性、又はその組合せであり得る。
【0085】
有用な水不溶性充填剤の例は、次のものを包含する:二酸化珪素、二酸化チタン、タルク、アルミナ、澱粉、カオリン、ポラクリリンカリウム、粉末セルロース、微晶性セルロース、ヒュームドシリカ、モノステアリン酸グリセリン、ステアリン酸マグネシウム、ステアリン酸カルシウム、コロイド状シリカ、微粒シリカ、三珪酸マグネシウム、石膏、及び前記いずれかの組合せ。
【0086】
有用な水溶性充填剤の例は、水溶性糖アルコール、例えば、ラクトース、グルコース、フルクトース、 スクロース、マンノース、 デキストロース、 ガラクトース、 対応する糖アルコール及び 他の糖アルコール、例えばマンニトール、ソルビトール、キシリトール、及び前記いずれかの組合せを包含する。希釈剤が微晶性セルロースである態様においては、錠剤投与形は、約15重量%〜約35重量%、及びある態様においては、約18重量%〜約20重量%の範囲の量の希釈剤を含んで成ることができる。ある態様においては、希釈剤又は充填剤は、二塩基性無水リン酸カルシウム及びある態様においては、二塩基性リン酸カルシウム二水和物である。
【0087】
錠剤化滑剤は、加工、フィルム形成及び/又は乾燥の間、粘着効果を低めるために、本発明の開示により供給される投与形に含まれ得る。有用な滑剤の例は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、グリセロールモノステアレート、及び前記いずれかの組合せを包含する。
【0088】
滑沢剤は、粉体流を改良するために、本発明の開示により供給される投与形に含まれ得る。有用な滑沢剤の例は、タルク、コロイド状二酸化珪素、沈殿された二酸化珪素、 ヒュームド二酸化珪素、及び前記いずれかの組合せを包含する。ある態様におおいては、滑沢剤は、コロイド状二酸化珪素である。錠剤投与形は、約2重量%以下の滑沢剤、及びある態様においては、約1重量%以下の滑沢剤を含むことができる。ある態様においては、滑沢剤はコロイド状二酸化珪素である。
【0089】
結合剤は、構成成分の付着を促進するために投与形に包含され得る。本発明の開示により供給される錠剤投与形において有用な結合剤の例は、ポリビニルアセテートフタレート、モラッセ、メチルセルロース、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC)、ナトリウムカルボキシメチルセルロース、微晶性セルロース(HCC)及びポリビニルピロリドンを包含する、本発明の開示により供給されるある態様においては、結合剤は、微晶性セルロース、例えばAVICEL(商標) PH200 (FMC Corporation)である。
【0090】
可塑剤は、本発明の開示により供給される錠剤投与形に含まれ得る。本発明の開示により供給される錠剤投与形において有用な可塑剤の例は次のものを包含する:クエン酸アルキル、例えばクエン酸トリエチル、クエン酸アセチルトリエチル、 クエン酸トリブチル、クエン酸アセチルトリエチル、 及びクエン酸アセチルトリブチル; スクロース脂肪酸エステル;グリセリンモノ-、ジ- 及びトリ-脂肪酸エステル、 例えばトリアセチン、 グリセリンモノ-脂肪酸エステル、グリセリンモノステアレート及びアセチル化されたモノグリセリド; ポリグリセリン脂肪酸エステル; ポリエチレングリコール、例えばマクロゴール400、マクロゴール600、マクロゴール1500、マクロゴール4000、マクロゴール6000、マクロゴール20、000、及びマクロゴール35、000; セバシン酸ジブチル; トリブチルセバケート; ビニールピロリドン; プロピレングリコール; 胡麻油; ひまし油; グリセリン; シリコーン樹脂; D-ソルビトール; フィトステロール;アルキルフタレート、例えばフタル酸ジエチル、フタル酸ジブチル及びジオクチルフタレート; アジペートポリエステル; イソプロピルミリステート; 中鎖トリグリセリド; ブチルフタリルブチルグリコレート; ポリオキシエチレンポリオキシプロピレングリコール; 及び前記いずれかの組合せ。
【0091】
錠剤投与形は、約0.1重量%〜約10重量%、約1重量%〜約8重量%、及びある態様においては、約2重量%〜約6重量%の範囲の量の可塑剤を含んで成る。本発明の開示により供給される投与形のある態様においては、投与形は、約2重量%〜約6重量%の可塑剤を含んで成り、前記可塑剤はトリエチルシトレート及びアセチルトリエチルシトレートから選択される。
【0092】
滑剤及び抗−付着剤は、加工目的のために本発明の開示により供給される錠剤投与形に含まれ得る。本発明の開示により供給される錠剤投与形において有用な滑剤及び/又は抗−付着剤の例は、次のも斧を包含する:ステアリン酸カルシウム、グリセリルベヘネート、モノステアリン酸グリセリン、ステアリン酸マグネシウム、鉱油、ポリエチレングリコール、ナトリウムステアリルフマレート、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ステアリン酸、 タルク、 水素化された植物油、ステアリン酸亜鉛、 及び前記いずれかの組合せ。ある態様においては、滑剤は、グリセリルモノステアレートである。ある態様においては、滑剤は、ステアリン酸マグネシウムである。ある態様においては、錠剤投与形は、約0.1重量%〜約5重量%、ある態様においては、約0.1重量%〜約1重量%及びある態様においては、約1重量%の量の滑剤はステアリン酸をマグネシウムである。
【0093】
本発明の開示により供給される錠剤投与形において有用な界面活性剤の例は、次のものを包含する:医薬的に許容できるアニオン性界面活性剤、カチオン性 界面活性剤、双性イオン性、両性(親媒性/両親媒性) 界面活性剤、 非イオン性界面活性剤、ポリエチレングリコールエステル又はエーテル、及び前記いずれかの組合せ。
【0094】
有用な医薬的に許容できるアニオン性界面活性剤の例は、一価のアルキルカルボキシレート、アシルラクチレート、アルキルエーテルカルボキシレート、N−アシルサルコシネート、多価のアルキルカーボネート、N−アシルグルタメート、脂肪酸−ポリペプチド縮合物、硫酸エステル、アルキルスルフェート、例えばラウリル硫酸ナトリウム及びドデシル硫酸ナトリウム、エトキシル化されたアルキルスルフェート、エステル結合されたスルホネート、例えばドキュセートナトリウム及びジオクチルナトリウムスクシネート、α−オレフィン性スルホネート、又はリン酸化されたエトキシル化されたアルコールを包含する。
【0095】
有用な医薬的に許容できるカチオン性界面活性剤の例は、モノアルキル四級アンモニウム塩、ジアルキル四級アンモニア化合物、アミドアミン及びアミンイミドを包含する。有用な医薬的に許容できる両性界面活性剤の例は、N−置換されたアルキルアミド、N−アルキルベタイン、スルホベタイン及びN−アルキル−6−アミノプロピオネートを包含する。
【0096】
有用な医薬的に許容できる非イオン性界面活性剤の例は、ポリエチレンオキシド、ポリプロピレンオキシド、ポリオキシエチレン(20)ソルビタンモノオレエート、及びポリエチレングリコールエステル又はエーテル、例えばポリエトキシル化されたヒマシ油、ポリエトキシル化された水素化ヒマシ油及び水素化ヒマシ油の二ブロック及び三ブロックコポリマーを包含する。ある態様においては、界面活性剤は、ラウリル硫酸ナトリウム及びドデシル硫酸ナトリウムから選択される。ある態様においては、錠剤投与形は、約3重量%以下の界面活性剤、及びある態様においては、約2重量%以下の界面活性剤を含んで成る。
【0097】
崩壊剤は、水に暴露される場合、崩壊剤の膨潤により錠剤の分解を引起すよう錠剤配合物に含まれ得る。有用な崩壊剤の例は、水膨張性物質、例えば低−置換されたヒドロキシプロピルセルロース、架橋されたナトリウムカルボキシメチルセルロース (ナトリウムクロスカルメロース)、ナトリウム澱粉グリコレート、ナトリウムカルボキシメチルセルロース、澱粉グリコール酸ナトリウム、イオン交換樹脂、微結晶セルロース、架橋されたポリビニルピロリドン、澱粉及びアルファ澱粉、ホルマリン−カゼイン、アルギン酸、 一定の複合シリケート、及び前記いずれかの組合せを包含する。
【0098】
本発明の開示により供給される錠剤投与形はさらに、錠剤を部分的に又は完全に被覆することができる1又は複数の被膜を含んで残ることができる。一定の被膜が胃腸管における錠剤投与形からの化合物(1)の放出を変性するか又は影響を及ぼすよう適用され得るが、他はそのような効果を有することはできない。例えば、1又は複数の追加の被膜が錠剤の物質的保護、エステ、容易な飲み込みのために、及び/又は追加の加工を促進するために存在することができる。
【0099】
被膜は、湿気に対して不透過性であるか、又は湿気透過性であり得る。湿気不透過性外部錠剤被膜は、乾燥剤の存在下で包装される投与形において低水分率を維持するために有用であり得、そしてそれにより、例えば錠剤投与形の貯蔵安定性を増強することができる。物理的保護のための被膜において有用な材料の例は、透過性又は溶解材料、例えばヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ラクトース、ヒドロキシプロピルエチルセルロース、ヒドロキシエチルセルロース及びキサンタンガムを包含する。
【0100】
追加の加工を促進するための外部錠剤被膜において有用な材料の例は、タルク、コロイド状シリカ、ポリビニルアルコール、二酸化チタン、微小化されたシリカ、ヒュームドシリカ、グリセロールモノステアレート、マグネシウムトリシリケート及びステアリン酸マグネシウムを包含する。外部錠剤被膜はさらに、1又は複数のビークル、例えば可塑剤、結合剤、充填剤、圧縮助剤及び前記いずれかの組合せを含む。前記1又は複数の追加の被膜は、単一の材料、又は本明細書に開示されるそれらの材料のいずれかを包含する1つよりも多くの材料の組合せを含んで成る。それらの追加の被膜は、当業者に知られている方法により錠剤投与形に適用され得る。
【0101】
ある態様においては、本発明の開示による供給される投与形は、化合物(1)及び/又はR−バクロフェンの分子内環化により形成されるラクタム副生成物を実質的に有さない。投与形は、実質的なラクタム形成、例えば約0.5重量%以下のラクタム、約0.2重量%以下ンラクタム、又は約0.1重量%以下のラクタムの形成を伴わないで、例えば1年以上の延長した貯蔵に対して安定することができる。
【0102】
投与形の溶解プロフィール:
化合物(1)を含んで成る、本発明の開示により供給される投与形の放出特徴は、インビトロ溶解プロフィールにより一部特徴づけられ得る。投与形の溶解プロフィールを決定するための方法は、医薬業界の人々に良く知られている。米国薬局方に示される標準方法論が使用され得る。例えば、溶解プロフィールは、米国薬局方タイプI装置(バスケット)又は米国薬局方タイプII装置(パドル)のいずれかにおいて測定され得る。
【0103】
後者の方法を用いる場合、ある態様においては、本発明の開示により供給される投与形の溶解又は放出プロフィールは、37℃の温度で、pH6.8で50mMの一塩基性リン酸ナトリウム緩衝液(NaH2PO4)に投与形を含浸することにより決定され得る。溶解媒体は75rpmでパドルにより撹拌される(USP, タイプII)。サンプルが、一定間隔で溶解媒体から取り出され、そして溶解媒体における化合物(1)及び/又はR−バクロフェンの含有率が逆相高圧液体クロマトグラフィー(HPLC)を用いて決定される。
【0104】
ある態様においては、本発明の開示により供給される錠剤投与形からの化合物(1)の放出は、75rpmで撹拌される(USP, タイプII)、pH6.8及び37℃での50mMの一塩基性リン酸ナトリウム緩衝液におけるインビトロ溶解プロフィールを示し、ここで約10%〜約30%の化合物が約4時間以内に放出され;約20%〜約50%の化合物が約8時間以内に放出され;約30%〜約65%の化合物が約12時間以内に放出され;約40%〜約80%の化合物が約18時間以内に放出される。
【0105】
ある態様においては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約10%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0106】
約15%〜約35%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約20%〜約50%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約30%〜約80%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0107】
ある態様のおいては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約10%〜約20%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0108】
約20%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約25%〜約45%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約35%〜約55%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0109】
そのような態様のある態様においては、前記放出プロフィールのいずれかを示す錠剤投与形は、約300mg又は約300mgの重量であり、そしてそれぞれ、例9又は10に記載のようにして調製される。
【0110】
ある態様においては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約15.5%〜約21.5%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0111】
約26%〜約32%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約35%〜約41%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約46%〜約51%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0112】
そのような態様のある態様においては、前記放出プロフィールを示す錠剤投与形は、前記方法のいずれかにより調製され、そして構成された粉末ブレンドから調製される錠剤を記載する、例16, 18及び/又は19に記載される組成を含む。
【0113】
ある態様においては、錠剤投与形は、例19、表14に記載されるプロフィールのいずれかに類似する放出プロフィールを示す。
【0114】
“Dissolution Testing of Immediate Release Solid Oral Dosage Forms - Guidance for Industry”, FDA-CDER, August 1997と一致して、溶解プロフィールは、差異因子(f1)及び類似性因子(f2)に基づいて類似すると思われる。類似すると思われる溶解プロフィールに関しては、f1値は0に接近し、そしてf2値は100に近づくべきである。一般的に15(0〜15)までのf2値及び50以上(50〜100)のf2値は、2種の溶解プロフィールの同等性を保証する。f1及びf2を計算するための方法は、前記引例に示されている。ある態様においては、本発明の開示により供給される経口錠剤投与形は、前記溶解プロフィールのいずれか1つと、又は表12又は14に記載される溶解プロフィールのいずれかと比較される場合、15以下のf1差異因子及び50〜100のf2類似性因子を生成する溶解プロフィールを示す。
【0115】
市販の許容できる錠剤は、USP Test No. 1216に従って決定される場合、約1重量%以下の破砕性を有することが一般的に認められる。ある態様においては、本発明の開示により供給される錠剤は、約1重量%以下、ある態様においては、約0.5重量%以下、ある態様においては、約0.3重量%以下、及びある態様においては、約0.2重量%以下の破砕性を有する。
【0116】
薬物力学及びインビボ放出プロフィール:
化合物(1)を含んで成る持効性投与形は、R−バクロフェン及び/又はラセミ体の等用量形で投与される場合、R−バクロフェンの経口生物学的利用能に比較してR−バクロフェンとしての増強された経口生物学的利用能を示す。化合物(1)の増強された経口生物学的利用能は、受動的及び/又は活性輸送機能を通して、結腸を包含する胃腸管じゅうでの化合物(1)の効果的吸収のためであると思われる。本発明の開示により供給される投与形は、胃腸管を通しての投与形の通過の間、投与形からの化合物(1)の放出を提供する。
【0117】
患者、すなわち本発明の開示により供給される投与形を飲み込む患者に経口投与される場合、その投与形は、連続期間、患者の血液にR−バクロフェンの持続した治療的有効濃度を提供する。ある態様においては、投与形は、最小の治療的有効濃度以上であり、そして患者の血液におけるR−バクロフェンの最小影響濃度以下である患者の血液におけるR−バクロフェンの濃度を提供する。ある態様においては、本発明の開示により供給される投与形は、R−バクロフェンの最小悪影響濃度を越えないで、連続した期間、患者の血液において治療的有効濃度のR−バクロフェンを提供する。ある態様においては、患者の血液におけるR−バクロフェンの濃度は、投与形が患者に経口投与された後、いずれの時点でも最小悪影響濃度を越えない。
【0118】
本発明の開示により供給される投与形は、例えばR−バクロフェンを含んで成る経口投与形の投与に続いて観察される、最小悪影響濃度以上の濃度で、高血液濃度のR−バクロフェンに関連する悪影響薬物効果を低めるか又は排除しながら、連続した期間、患者の血液において治療的有効濃度のR−バクロフェンを提供することができる。化合物(1)を含んで成る投与形を用いて達成できるR−バクロフェンの高い生物学的利用能は、R−バクロフェンを含んで成る経口投与形におけるR−バクロフェンの量に比較して、患者の血液においてR−バクロフェンの持続した治療的有効濃度を達成するためへの一定用量での低等価質量のR−バクロフェンの使用を促進することができる。
【0119】
本発明の開示により供給される持効性投与形は、経口投与に続いて、患者の血液に持続した治療的有効濃度のR−バクロフェンを提供することができる。例えば、ある態様においては、投与形は、患者への経口投与の後、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間、少なくとも約16時間、少なくとも約20時間、又は少なくとも約24時間から選択された連続した期間、患者の血液において持続した治療的有効濃度のR−バクロフェンを提供することができる。ある態様においては、、患者の血液中のR−バクロフェンの濃度は、投与形が患者に経口投与された後、いずれの時間でも最小悪影響濃度を越えないであろう、例えば患者において悪影響を及ぼす濃度に達しないであろう。
【0120】
ある態様においては、患者の血液中のR−バクロフェンの治療的有効濃度は、約50ng/ml〜約1,000ng/ml、及びある態様においては、約100ng/ml〜約500ng/mlの範囲であり得る。血液R−バクロフェン濃度の薬物力学的プロフィールは、類似するR−バクロフェン血液AUCを提供する、R−バクロフェンを含んで成る即時放出及び持効性経口配合物に比較して、より低いCmax/C12比及びより低いCmax/用量により特徴づけられ得る。
【0121】
ある態様においては、本発明の開示により供給される経口投与形の反復された1日1度(QD)の投与は、表1に示されるように、血液中のR−バクロフェンの定常状態濃度を提供する。ある態様においては、16人の健康な成人ボランティアへの本発明の開示により経口投与は、約202±56ng/mlのCss, max;約3.9±1.0時間のTss, max;約63 ng/mLのC12;約8〜約15のCss,max/Css,12;約19ng/mlのCss, 12;約10.9±3.8時間のTss, 1/2;及び1803±420ng・時/mlのAUCss, 24により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールを提供する。ある態様においては、本発明の開示により供給される投与形は、約8〜約15のCss,max/Css,12を提供する。
【0122】
表1.SR3錠剤配合物としての60mg(6×10mg)の化合物(1);又はSR4錠剤配合物としての60mg(6×10mg)、60mg(3×20mg)又は60mg(2×30mg)の化合物(1)の健康な成人ボランティアへの4日間、毎日1度(QD)の経口投与の後、定常状態で決定された血液中のR−バクロフェンについての平均(SD)薬物力学的パラメーター:
【0123】
【表1】
【0124】
ある態様においては、少なくとも1つの経口投与形は、約5mg〜約140mg、及びある態様においては、約10mg〜約80mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量で人患者に投与される。前記態様のある態様においては、投与される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量は、患者において中位の鎮静及び運動活動の障害を引起す(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量よりも少ない。
【0125】
本発明の開示により供給される投与形の経口投与を用いる投与レジメが、長期間、最小の治療的有効濃度よりも高く、そして最小の悪影響濃度よりも低い、患者の血液における一定濃度のR−バクロフェンを維持するために開発され得る。ある態様においては、R−バクロフェンの最小の治療的有効濃度は、約1ng/ml〜約200ng/ml、及びある態様においては、約10ng/ml〜約100ng/mlの範囲であり得る。ある態様においては、最小悪影響濃度は、約200ng/ml〜mlの範囲であり得る。ある態様においては、最小悪影響濃度は、約200ng/ml〜約2,000ng/ml, 及びある態様においては、約500ng/ml〜約1,000ng/mlの範囲であり得る。
【0126】
最小治療濃度及び最小悪影響濃度は、多くの因子、例えば処理される疾病、疾病の重症度、意図される臨床学的結果、処理される患者の状態、等に依存するであろう。そのようなレジメは、本発明の開示により供給される1又は複数の投与形の反復した投与を用いることができる。投与の適切な間隔は例えば、投与形における化合物(1)の量、投与形中の組成、投与形からの化合物(1)の放出特徴、処理される疾病、患者の状態、可能性ある悪影響、及び処理医者の判断に依存することができる。投与形レジメは、個々の間隔で同じ投与形又は異なった間隔で異なった投与形の反復された投与を包含する。例えば、毎日2回の投与レジメは、朝での第1の投与形、及び夕方での第2の投与形の投与を包含する。
【0127】
本発明の開示により供給される投与形はさらに、U.S. Food and Drug Administration and discussed in “Guidance for Industry - Bioavailability and Bioequivalence Studies for Orally Administered Drug Products” (2003)により定義されるように、吸収速度及び程度の両者に関して、本明細書に開示される投与形に対して生物学的同等である投与形を包含する。
【0128】
投与量:
R−バクロフェンの持続した全身性濃度を提供する錠剤投与形は、現在、1日当たり3度、投与される即時放出性非プロドラッグ形、すなわち患者のために不便であり、そして患者にとって覚えるのが困難であるレジメに比較して、患者のコンプライアンスを増強するであろうと思われる。さらに、本発明の開示により供給される経口投与形錠剤の使用は、低められた副作用、例えば傾眠状態、衰弱、頭痛、発作、嘔気、嘔吐、低血圧、便秘、精神錯乱、呼吸抑制、不眠、及び頻尿又は尿閉を伴って、増強された効能を提供するであろうと思われる。
【0129】
本明細書に開示される特定の疾病の処理において効果的であろう化合物(1)の量は、疾病の性質に、少なくとも一部、依存し、そして当業界において知られている標準の臨床学的技法により決定され得る。さらに、インビトロ又はインビボアッセイは、最適な投与量範囲の同定を助けるために使用され得る。投与レジメ及び投与間隔はまた、当業者に知られている方法により決定され得る。投与される化合物(1)の量は、他の因子の中でも、処理される対象、対象の体重、疾病の重症者、投与路、及び処方医者の判断に依存することができる。
【0130】
全身性投与に関しては、治療的有効用量は、最初、インビトロアッセイから評価され得る。初期用量はまた、インビボデータ、例えば動物モデルから、当業界において知られている技法を用いて評価され得る。そのような情報は、ヒトにおける有用な用量を、より正確に決定するために使用され得る。当業者は、動物データに基づいて、ヒトへの投与を最適化することができる。
【0131】
化合物(1)の用量は、等モル量又は等価質量のR−バクロフェンを供給するよう調節され得る。用量は、本発明の開示により供給される、複数用量形を含んで成ることができる。ある態様においては、R−バクロフェンの治療的有効用量は一般的に、1日当たり約0.03mg〜約1mg/kg体重である。ある態様においては、毎日の用量は、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;ある態様においては、約5mg〜約60mg;及びある態様においては、約10mg〜約40mgの範囲の等価質量のR−バクロフェンを含んで成ることができる。ある態様においては、化合物(1)の用量は、患者において、中位の鎮静及び運動活動の障害を引起す用量以下である。化合物(1)の用量及び適切な投与間隔は、患者におけるR−バクロフェンの持続した治療的有効濃度を、ある態様においては、最小悪影響濃度を越えないで、維持するよう選択され得る。
【0132】
ある態様においては、本発明の開示により供給される投与形は、1日1度、1日2度、及びある態様においては、1日1度以上の間隔で投与され得る。投与は、単独で、及び他の薬物と組合して提供され、そして疾病の効果的処理のために必要とされる限り、続けることができる。投与は、哺乳類、例えばヒトに、食物を食べた状態又は絶食された状態で投与形を投与することを包含する。
【0133】
用量は、単一投与形で、又は複数の投与形で投与され得る。複数の投与形が使用される場合、その複数投与形の個々内に含まれる化合物(1)の量は同じであっても、又は異なっていても良い。
【0134】
ある態様においては、投与される用量は、毒性用量以下である。本明細書に記載される組成物の毒性は、細胞培養物又は実験運動において標準の医薬方法により、例えばLD50(集団の50%が致死する用量)又はLD100(集団の100%が致死する用量)により決定され得る。毒性効果:治療効果の用量比は、治療指数を示す。それらの細胞培養アッセイ及び動物研究から得られるデータは、ヒトへの使用のために毒性でない投与範囲の配合に使用され得る。化合物(1)の用量は、治療的に有効であり、鎮静用量以下であり、そして/又は毒性をほとんどか又はまったく示さない、血液、血漿又は中枢神経系における一定範囲内の循環濃度であり得る。
【0135】
処理の間、用量及び投与スケジュールは、疾病を処理するために十分な又は定常状態の全身性濃度のR−バクロフェンを提供することができる。ある態様においては、徐々に増大する用量が投与され得る。
【0136】
治療用途:
本発明の開示により供給される持効性経口投与形は、親薬物、すなわちR−バクロフェンが治療的有効であることが知られているか、治療的に有効であると思われるか、又はこの後、治療的有効であることが決定される、いずれかの疾病又は障害を有する患者に投与され得る。R−バクロフェンが処方されており、そして本発明の開示により供給される投与形がまた、効果的である徴候は、痙攣、胃食道逆流病、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、嘔吐、咳嗽、神経因性疼痛、筋骨格痛及び尿失禁を包含する。
【0137】
上記に列挙される疾病の処理における本発明の開示により供給される投与形の適合性は、当業界において記載されている方法により決定され得る。
R−バクロフェン治療の必要な患者に投与される化合物(1)の適切な用量は、等価質量のR−バクロフェン、及び化合物(1)により供給されるR−バクロフェンの増強された経口生物学的利用能に基づいて評価され得る。
【0138】
痙攣:
痙攣は、延伸に対する無意識の速度−依存性の高められた耐性である。痙攣は、上昇する延伸速度による外部的に付加された運動に対して高められた耐性が存在する筋肉緊張過渡により特徴づけられる。痙攣は、誕生の前、間又は後、脳への酸素の欠乏(脳性麻痺);物理的な損傷(脳又は脊髄損傷);脳血管からの出血の遮断(脳卒中); 一定の代謝性疾患; アドレノロイコジストロフィ; フェニルケトン尿症; 神経変性疾病、例えばパーキンソン病及び筋萎縮性側索硬化症;及び神経障害、例えば多発性硬化症により引起され得る。
【0139】
痙攣は、皮質脊髄路に対する損傷に関連し、そして神経学的疾患の通常の合併症である。痙攣が顕著な症状である疾病及び状態は、脳性麻痺、多発性硬化症、脳卒中、頭部及び脊髄損傷、外傷性脳損傷、血液酸素欠乏、 及び神経変性疾病を包含する。痙攣を有する患者は、硬直、無意識の発作及び苦痛の不平をいう。それらの苦痛を伴う発作は、自発的であるか、又は低感覚刺激、例えば患者の接触により引起され得る。
【0140】
痙攣の症状は、高張緊張過度(筋緊張亢進)、クローヌス(一連の急速な筋肉の収縮)、大過度の深部腱反射、痙攣性疾患、シザリング(脚の不本意な交差)、固定接合を有する奇形、硬直、 及び/又は四肢が正常に移動するよう試みることにより引起される疲労を包含する。他の合併症は、尿路感染、慢性便秘、発熱又は他の全身系病気、及び/又は褥瘡を包含する。痙攣の程度は、軽い筋肉硬直から、重度で、苦痛を伴い、且つ不快な筋肉発作まで種々であり得る。
【0141】
痙攣は、他の状態と同時に存在するが、しかし硬直(運動に対する不本意な両方向の非速度依存性耐性)、クローヌス(高張につづく自己持続性振動運動)、ジストニー(ねん回姿勢異常をもたらす不本意な持続性収縮)、アテトーゼ様運動(不本意な不規則的な併合性苦悩動作)、舞踏病(不本意で、突然の、急速で、不規則な、持続しない運動)、バリズム(四肢又は身体の不本意な突進運動)、及び身震い(非自己持続性性の不本意でリズミカルな反復性発振)とは区別される。
【0142】
痙攣は、整形外科的変形、例えば股関節脱臼、拘縮、又は脊柱変形; 毎日の生存活動、例えば衣服の脱着、入浴及びトイレの障害; 移動性の障害、例えば歩くか、回転するか、又は座ることの不能性; ボジショニング困難性及び剪断圧力に続く皮膚の損傷; 疼痛又は異常感覚フィードバック; 高カロリー消費に続く不良な体重増加; 睡眠妨害; 及び/又は機能的独立性の欠乏に続く鬱病を導くことができる。
【0143】
痙攣の処理は、物理的及び作業療法、例えば機能的な基本療法、リハビリテーション、促進作用、例えば神経−発達学的治療、固有受容体神経筋促進法、及び感覚統合;バイオフィードバック;電気的刺激;及び他の方法を包含する。痙攣の処理において有用な経口薬剤は、バクロフェン、ベンゾジアゼピン、例えばジアゼパム、ダントロレンナトリウム;イミダゾリン、例えばクロニジン及びチザニジン;及びガバペンチンを包含する。痙攣の処理においては、有用な鞘内薬剤はバクロフェンを包含する。局部麻酔薬、例えばリドカイン及びキシロカインによる化学脱神経;タイプAポツリヌス毒素及びタイプBボツリス毒素;フェノール及びアルコール注入がまた、痙攣の処理において有用であり得る。痙攣の処理において有用な外科処理は、神経外科、例えば選択的背部神経根切断法;及び整形外科的手術、例えば痙縮解放、腱又は筋肉延伸、腱移動、骨切断、及び関節固定を包含する。
【0144】
哺乳類におけるバクロフェンの主要薬理学的効果は、筋張力の低下であり、そして従って、薬物は時折、痙攣の処理に使用される。
【0145】
痙攣の処理のための本発明の開示により供給される投与形の効能は、痙攣の動物モデルを用いて、及び異なった病因の痙攣の臨床学的に適切な研究において評価され得る。痙攣の動物モデルは知られており、そして(a)痙攣性変異マウス;(b)急性/慢性の脊髄切断されたラット及び急性除脳ラット;(c)ラットにおける一次観察Irwin試験;及び(d)ラット及びマウスにおけるRotard試験を包含する。他の動物モデルは、一時的な脊髄虚血に従ってラットにおいて誘発された痙攣、多発性硬化症のマウスも出るにおける痙攣; 及び脳性麻痺のラットモデルにおける痙攣を包含する。齧歯動物における最大電気ショック痙攣(MES)限界試験が、可能性ある抗痙攣薬剤性質を検出するために敏感である。
【0146】
痙攣の処理のための本発明の開示により供給される投与形の効能は、ニ重盲検プラシーボ−制御された臨床試験を用いて、ヒトにおいて評価され得る。痙攣についての臨床試験結果測定は、Ashworth Scale、改良されたAshworth Scale、筋肉延長反射、間代痙攣の存在、及び有害な刺激に対する反射応答を包含する。痙攣は、当業界において知られている方法、例えば臨床試験、評価尺度、例えばAshworth Scale、改良されたAshworth Scale、発作頻度尺度及び反射評点、生化学的研究、例えばペンダラム試験、電気生理学的研究、例えば筋電図、及び機能的測定、例えばFugl-Meyer Assessment of Sensorimotor Impairment尺度の組合せを用いて評価され得る。他の測定法は、特定の障害、例えば多発性硬化症性痙攣尺度に関連する痙攣を評価するために使用され得る。
【0147】
胃食道逆流病:
胃食道逆流病(GERD)は、食道において異常逆流により生成される慢性症状又は粘膜損傷として定義される。GERDの症状は、胸焼け、食道炎、狭窄症、えん下障害、慢性的な胸の疼痛、咳嗽、 しわがれ声、声の変化、慢性的な耳痛、燃えるような胸痛、嘔気、及び副鼻腔炎を包含する。
【0148】
下部食道括約筋の強い収縮が、食道中への胃内容物の逆流を妨げる主要要因である。一時的な下部食道括約筋弛緩(TLESR)は、正常な対象及びGERDを有する患者における逆流を受ける主要機構である。GABABアゴニスト、例えばR−バクロフェンは、ヒトにおいてTLESRを低めることが示されている(Lidums et al., Gastroenterology 2000, 118(1), 7-13; Vela et al., Aliment Pharmacol Ther 2003, 17(2), 243-51; Ciccaglione and Marzio, Gut 2003, 52(4), 464-70; 及びZhang et al., Gut 2002, 50(1), 19-24)。
【0149】
バクロフェンによるTLESRの頻度の低下は、迷走神経導入、孤束核と迷走神経背側運動核その間の情報移行、及び迷走神経延伸性流出のためであると思われる(Hornby et al., Gastroenterol Clin N Am 2002, 31(4 Suppl), S11-S20)。より特定には、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸、すなわち化合物(1)は、臨床試験において逆流エピソードを低めることが示されている(Gerson et al., Am J Gastroenterol 2009, online publication 29 December 2009; doi: 10.1038/ajg.2009.718)。
【0150】
GERDの処理についての効能は、動物モデルを用いて、及び臨床試験において評価され得る。
【0151】
嘔吐:
悪心、嘔吐及び吐き気は、毒素の吸収に対する基本的なヒトの保護反射及び一定の刺激に対する応答である。悪心は、蒼白又は潮紅、頻拍、及び嘔吐に対する心拍の意識に関連する咽頭又は上胃部の背部における主観的に不快な波状感覚である。発汗、過度の唾液分泌、及び寒さ又は暑さの感覚がまた生じることができる。嘔吐は、腹筋の吸縮、融膜の降下、及び胃噴門の開口により特徴づけられ、口から胃内容物の強力な排除がもたらされる。吐き気は、胃内容物の排除を伴わないで、融膜、及び胸部及び腹筋の筋肉の痙攣性収縮を包含する。嘔吐は、悪心、嘔吐、及び/又は吐き気を言及するために本明細書において使用される。
【0152】
バクロフェンは、モルホリンにより誘発される吐気及び嘔吐を抑制することが示されており、それにより、嘔吐制御経路におけるGABAB受容体の関与を示唆する(Suzuki et al., Neuropharmacology 2005, 49(8), 1121-31)。バクロフェンはまた、動物モデルにおいてニコチン及び運動により誘発される嘔吐を拮抗することが示されている(Chan et al., Eur J Pharmacology 2007, 559(2-3), 196-201)。
【0153】
嘔吐の処理における効能は、適切な動物モデル及び臨床試験を用いて評価され得る。例えば、化学療法剤により誘発される嘔吐の処理における効能は、嘔吐を示す影響、及び異食症、異うっ血、及びラット、マウス又はフェレットにおける低められた食物摂取に基づいて決定され得る。臨床試験いおいては、評価装置、例えばDuke Descriptive Scale、isual Analog Scales、Morrow Assessment of Nausea and Emesis、Rhodes Index of Nausea and Vomiting Form-2、及びFunctional Living Index Emesisが効能を測定するために使用され得る。一般的に、適切に制御された、二重盲検プラシーボ制御される試験が、ヒトにおける効能を評価するために使用され得る。
【0154】
咳嗽:
呼吸気管に位置する咳受容体の活性化により誘発される咳反射は、呼吸気管からの吸入された刺激物及び外来性物質を清浄し、そして粘膜繊毛システムに関連して、呼吸気管からの異常状態下で生成される過剰の気道分泌物を排除することができる。咳は、軽い急性の上部呼吸気管感染、アレルギー、喘息、慢性閉塞性肺疾患、肺癌、胃食道逆流病、後鼻腔滴下、及び心臓又は耳疾患により引起され得る。しかしながら、同定できる原因を有さない慢性非生産性咳は、咳をする患者の数%である。慢性咳は喘息性徴候の増悪、肋骨骨折、息苦しさ、破裂脳動脈瘤腹部筋肉、気胸症、失神、 第2及び第3程度の心臓ブロック、及び意識喪失に関連している。永続的で且つ制御できない咳は、羅病率を導き、そしてそれらの患者の生命の質を重度に障害を与える。
【0155】
咳は、いずれかのタイプの病因学又は病原論の急性及び慢性咳、及び特に、喉頭の感覚神経障害に関連する咳を包含する。
【0156】
バクロフェンの鎮咳効果は良く知られている(Dicpinigaitis and Dobkin, Chest 1997, 111(4), 996-9; Dicpinigaitis and Rauf, Respiration 1998, 65(1), 86-8; Dicpinigaitis et al., J Clin Pharmacol 1998, 38(4), 364-7; 及びKreutner et al., US 5,006,560号及び WO 91/08740号)。
咳の処理における効能は、適切な動物モデル及び臨床試験を用いて評価され得る。
【0157】
物質中毒又は乱用:
臨床試験においては、バクロフェンは、コケイン中毒(Brebner et al., Alcohol 2002, 37(5), 478-84; 及び Haney et al., Neuropsychopharmacology 2006, 31, 1814-21);メトアンフェタミン依存性(Heinzerling et al., Drug Alcohol Depend 2006, 85(3), 177-84);オピオイド依存性(Assadi et al., BMC Psychiatry 2003, November 18, 3(16); 及び Ahmadi-Abhari et al., J Clin Pharm Therapeutics 2001, 26(1), 67-71);アルコール欲求及び摂取(Addolorato et al., Alcohol 2002, 37(5), 504-8; 及びFlannery et al., Alcohol Clin Exp Res 2004, 28(10), 1517-23);ニコチン使用(Markou et al., Ann N.Y. Acad Sci 2004, 1025, 491-503);及び薬物乱用(Cousins et al., Drug Alcohol Dependence 2002, 65(3), 209-20)の処理において効果的であることが示されている。
【0158】
物質中毒及び乱用の処理についての効能は、動物モデルを用いて、及び臨床試験において評価され得る。物質中毒障害の動物モデルは知られている。
【0159】
神経因性疼痛:
神経因性疼痛は、通常、神経組織への直接的な外傷又は損傷の後に生じる感覚入力の異常工程を包含する。神経因性疼痛は、異なった病因学、例えば感染、炎症、 疾病、例えば糖尿病及び多発性硬化症、主要末梢神経に対する損傷又は圧縮、及び化学物質又は照射-誘発された神経損傷により特徴づけられる障害の集積である。新規因性疼痛は典型的には、組織損傷が解決された後、長く持続する。
【0160】
化合物(1)は、神経因性疼痛の処理に使用され得る。ある態様においては、化合物(1)は、神経因性疼痛、例えば帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症の処理に使用され得る。
【0161】
神経因性疼痛の研究についての国際協会(The International Association for the Study of Neuropathic Pain)は、通常の条件下で、中枢神経系への有害情報を伝達する神経系の損傷又は機能不全により特徴づけられる障害として神経因性疼痛を定義する。神経因性疼痛状態の根底にある機構は高く不均等であるが、しかしながら、すべてのタイプの神経因性疼痛は中枢及び/又は末梢神経系における体感覚プロセッシングにおいて神経損傷及び一定の共通する異常を包含することが推定される。神経因性疼痛の可能性ある原因は、物理的損傷、感染及び化学物質暴露を包含する。
【0162】
神経因性疼痛は一般的に、末梢神経系の局部/多病巣性損傷、例えば帯状疱疹後神経痛、末梢神経系の一般化された外傷、 例えば、苦痛な糖尿病性神経障害、 HIV-関連NP、中枢神経系の外傷、 又はより複雑な障害として分類され得る。末梢神経因性疼痛は、外傷及び手術関連の外傷、例えば腕神経叢損傷; 絞扼性神経障害、例えば腰椎圧縮、手根管症候群; 疾病関連の神経障害、例えば糖尿病及びHIV-AIDS; 神経根疾患; 複雑な局所疼痛症候群; 及び/又は神経圧迫か浸潤を導く腫瘍増殖の結果として生じる。中枢神経因性疼痛は、脳卒中、多発性硬化症、後−虚血性脊髄障害; 後−超神経痛; 及び/又は後−外傷性脊髄損傷の結果であり得る。
【0163】
神経因性疼痛は、導入の感覚機能の一部又は完全な損失、及び痛い領域における一定の超現象の矛盾した存在として特徴づけられ得る。神経組織損傷は、脳、脊髄又は末梢神経に見出され得る。症状は、その状態に依存して変化し、そして痛感鋭敏(苦痛域値の低下、及び有害刺激への増加反応)、異痛(非有害刺激、例えば冷感、熱感、又は接触による痛みの喚起)、痛覚過敏(刺激強度が知覚の閾値を超える場合、増強された知覚検出閾値を伴って、皮膚領域から突然喚起される爆発的疼痛反応)、痙攣(自発的に、又は無毒の触覚の刺激又はありのままの圧力による刺激に続いて生じるシューテング、電気的、ショック−様又は速い痛みにより特徴づけられるタイプの喚起された疼痛)、錯感覚(自発的であるか又は喚起され、しばしばピン及び針として記載される、異常であるば、しかし、非苦痛性感覚)、知覚異常(自発的であるか又は外部刺激により引き起こされ得る、異常で不快であるが、しかし必ずしも苦痛な感覚ではない)、 関連痛及び異常疼痛輻射(疼痛の異常な蔓延)、 及びワインド・アップ様疼痛及び残感覚(苦痛な刺激の終結の後、長い疼痛の持続)として明白である。
【0164】
神経因性疼痛を有する患者の典型的には、燃えるような、電撃的な、突き刺すような、締め付けるような、痛み、 及び/又は時々、しっかりと締めつけているような疼痛を表す。その痛みは、発作性であるが又は一定であり得る。末梢神経、脊髄及び脳への病理学的変化は、慢性神経因性疼痛の誘発及び維持に関係されて来た。神経因性疼痛を有する患者は典型的には、現在の薬物療法に対して難治性であり、そして彼らの生命の質に深く影響を及ぼす慢性的衰弱性エピソードに耐えている。三環状抗うつ剤及びガバペンチルを包含する、神経因性疼痛のための現在入手できる処理は、典型的には、大部分の患者において制限された効能を示す。
【0165】
いくつかのタイプの神経因性疼痛が存在する。苦痛を伴った神経障害を引起す損傷のタイプ又は関連する病理生理学に関連する分類は、次のものを包含する:機械的な神経損傷に関連する神経障害、例えば手根管症候群、脊椎骨の椎間板ヘルニア、絞扼性神経障害、尺骨の神経障害、及び神経原性胸郭口症候群; 神経障害に関連する代謝性疾患、例えば糖尿病性多発神経障害; 神経向性のウイルス性疾病に関連する神経障害、例えば帯状疱疹及びヒト免疫不全ウィルス(HIV) 疾患; 神経毒性作用に関連する神経障害、例えば癌腫又は結核の化学療法、放射線療法、薬物誘発性神経障害、及びアルコール神経炎; 炎症及び/又は免疫機構に関連する神経障害、例えば多発性硬化症、抗スルファチド抗体神経障害、モノクローナル高ガンマグロブリン血症に関連する神経障害、シェーグレン病、狼蒼、脈管性神経障害、ポリクローナル炎症性神経障害、ギラン・バレー症候群、慢性炎症性脱髄性神経障害、多病巣性運動神経障害、傍腫瘍自律性ニューロパシー、神経節アセチルコリン受容体抗体自律性ニューロパシー、ランバート=イートン筋無力症症候群及び重症筋無力症; 神経系巣状虚血に関連する神経障害、例えば視床症候群(有痛知覚消失); 多発神経伝達物質系機能不全に関連する神経障害、例えば複雑な局所疼痛症候群(CRPS); 慢性的な/神経因性疼痛に関連する神経障害、例えば変形性関節症、腰痛、線維筋痛症、癌腫骨の疼痛、慢性的断端痛、幻肢痛、及び腫瘍随伴性神経障害;
【0166】
毒性神経炎(例えば、 化学物質への暴露、例えばアクリルアミド、3- クロロフェン、カルバメート、二硫化炭素、 酸化エチレン、 n-ヘキサン、 メチルn-ブチルケトン、臭化メチル、有機リン酸塩、ポリ塩化ビフェニール、 ピリミニル、トリクロロエチレン、 又はジクロロアセチレンへの暴露)、焦点の外傷性の神経障害、幻肢痛及び断端痛、単-神経根疾患、及び三叉神経痛; 中枢神経障害、例えば虚血性脳血管性損傷(脳卒中)、多発性硬化症、脊髄損傷、パーキンソン病、筋萎縮性側索硬化症、脊髄空洞症、腫瘍、クモ膜炎、及び術後痛; 混合された神経障害、例えば糖尿病性神経障害(左右対称の多発神経病変、例えば知覚又は知覚運動性多発神経病変、選択的小ファイバ多発神経病変、及び自律性ニューロパシー; 及び焦点の及び多病巣性の神経障害、例えば頭側神経障害、手足モノニューロパシー、 トランクモノニューロパシー(trunk mononeuropathy)、多発性単神経炎、及び非対称の下周辺運動神経障害)及び 好意的に維持される疼痛。他の神経障害は、焦点の神経障害; 舌咽神経痛; 虚血性疼痛; 三叉神経痛; ファブリー病、小児脂肪便症、遺伝性感覚性ニューロパシー又はB12-欠陥に関連する非定型顔面痛; モノニューロパシー; 多発神経病変; 遺伝性末梢性ニューロパシー、例えばCarcot-Marie-歯疾患、レフサム病、シュトルンペル-ローライン病、及び網膜色素変性症; 急性多発神経根筋障害; 及び慢性的多発神経根筋障害を包含する。
【0167】
腫瘍随伴性神経障害は、腫瘍随伴性亜急性知覚神経障害、 腫瘍随伴性運動神経疾患、腫瘍随伴性神経筋緊張症、腫瘍随伴性脱髄性神経障害、腫瘍随伴性 血管炎性神経障害、 及び 腫瘍随伴性自律神経機能不全症を包含する。本発明の開示により供給されるGABABアゴニストのプロドラッグは、前記タイプの神経因性疼痛のいずれかを処理するために使用され得る。ある態様においては、神経因性疼痛は、帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症から選択される。ある態様においては、神経因性疼痛は、帯状疱疹後神経痛及び三叉神経痛から選択される。
【0168】
臨床研究においては、鞘内バクロフェン投与は、脊髄損傷及び多発性硬化症に関連する神経因性疼痛(Herman et al., Clin J Pain 1992, 8(4), 338-345;及びTaira et al., Stereotactic Funct Neurosurg 1995, 65, 101-105)、痛みを伴った末梢異常(Gatscher et al., Acta Neurochir Suppl 2002, 79, 75-76)、及び交感神経的に維持される痛み(Van Hilten et al., N Engl J Med 2000, 343, 625-630; Becker et al., J Clin Neurosci 2000, 7, 316-319;及びZuniga et al., Reg Anesth Pain Med 2002, 27, 90-93)の処理において効果的であることが示されている。
【0169】
バクロフェンはまた、三叉神経、舌咽、迷走舌咽神経、及び眼-後ヘルペス性神経痛(Bowsher, Br Med Bull 1991, 47, 644-66; Fromm et al., Neurology 1981, 31, 683-687;及び Ringel and Roy, Ann Neurol 1987, 21, 514-515)において、及び糖尿病性神経障害を有する患者(Anghinah et al., Muscle Nerve 1994, 958-59)において有効であることも示されている。約50mg/日〜約60mg/日のバクロフェンの用量が三又神経痛の処理において有効であることが示されている(Fromm et al., Ann Neurol 1984, 15, 240-244)。
【0170】
種々のタイプの神経因性疼痛を処理するための化合物(1)の効能は、当業界に知られている技法、例えばランダム二重盲目プラシーボ制御された方法を用いて、臨床試験において評価され得る。神経因性疼痛のための臨床試験に使用されるエンドポイントは、確認された神経因性疼痛基準、例えばthe Brief Pain Inventory、 Categorical Scale、 Gracely Pain Scale、 Likert Scale、Neuropathic Pain Scale、 Numerical Pain Scale、 Short Form McGill Pain Questionnaire、Verbal Pain Scale、Visual Analog Scale (VAS)、 VAS Pain Intensity Scale及び/又はVAS Pain Relief Scaleを用いて決定され得る。
【0171】
筋骨格痛:
圧痛及び筋肉痙攣を引き起こす筋骨格状態は、線維筋痛症、緊張型頭痛、筋筋膜疼痛症候群、咬合局面関節痛、 内部椎間板離開、体性機能不全、脊椎骨折、化膿性脊椎炎、リウマチ性多発筋痛、環軸関節の不安定性、環椎後頭関節疼痛、骨粗鬆症の脊椎骨の圧縮破壊、ショイエルマン病、 脊椎分離症、脊椎辷り症、棘突起間関節形成症、仙腸関節疼痛、仙椎の圧力骨折、尾骨痛、 衰えた逆症候群、 及び機械的腰痛又は頸痛を包含する(Meleger and Krivickas, Neurol Clin 2007, 25, 419-438)。
【0172】
それらの状態においては、筋肉痙攣は、痙攣の特徴的な高められた調子又は反射を伴わないで、影響された筋肉グループを包含する局部因子に関連する。筋肉、腱、靭帯、椎間板、関節軟骨及び骨は、筋骨格痛に関与する。首及び背部痛を生成できる障害は、肉離れ、靭帯捻挫、 筋膜痛、線維筋痛症、咬合局面関節痛、内部椎間板離開、体性機能不全、脊椎骨折、 脊椎骨髄炎、及びリウマチ性多発筋痛、環軸関節の不安定性及び環椎後頭関節疼痛を包含する。
【0173】
バクロフェンは、全身的に又は中枢的に投与される場合、筋肉−弛緩効果を誘発することが知られている(Malcangio and Bowery, Trends Pharmacol Sci 1996, 17, 457-462)。従って、上位運動ニューロン症候群に関連する痙攣の処理のためへのバクロフェンの使用は十分に確立されている。バクロフェンはまた、筋肉痛及び/又は末梢筋肉骨格状態に関連する痙攣の処理のにおいて有効である研究がまた示されている。
【0174】
例えば、バクロフィンは、片頭痛(Hering-Hanit, Cephalalgia 1999, 19, 589-591; and Hering-Hanit及びGadoth, Headache 2000, 40, 48-51);及び特に、引張型頭痛(Freitag, CNS Drugs 2003, 17(6), 373-381);並びに腰痛及び神経根症(Zuniga et al., Anesthesiology 2000, 92, 876-880; Vatine et al., Pain Clin 1989, 2, 207-217; Dapas et al., Spine 1985, 10(4), 345-349; Raphael et al., BMC Musculoskeletal Disorders 2002, June 20, 3(17);及びMagora et al., Pain Clin 1988, 2, 81-85)の処理において効果的であることが示されている。
【0175】
1又は複数のタイプの筋骨格痛の処理のために化合物(1)のプロドラッグの効能は、神経因性疼痛の動物モデル及び臨床試験において評価され得る。
【0176】
背痛
化合物(1)は、頸部、胸部及び/又は腰背骨領域における背痛を包含する背痛みの処理に使用される。背痛は急性又は慢性であり得る。急性背痛は、4週よりも少ない間、存在する低背痛みとして定義され、時々、3ヶ月よりも少ない間、存在する症状を伴って、亜急性背痛により分類される。慢性低背痛は、3ヶ月以上の間、存在する低背痛として定義される。
【0177】
腰痛:
腰痛は一般的に、腰椎L1−L5の位置における背部のコシ領域において発生する。腰痛は、捻挫、緊張、 又は筋、腱、面関節、 及び/又は背部における仙腸関節の1つに対する痙攣; 背骨捻挫又は過剰圧縮; 又は脊椎板破壊又は湾曲部により引起され得る。腰痛はまた、神経又は筋肉刺激又は骨損傷に影響を及ぼすことができる。ほとんどの腰痛は背部への損傷又は外傷を従えるが、しかし疼痛はまた、変性状態、例えば関節炎又は脊椎板疾患、骨粗鬆症、又は他の骨疾患、ウイルス感染,関節及び脊椎板に対する刺激、又は背骨における先天性異常により引起され得る。
【0178】
肥満、喫煙、妊娠の間の体重増、 ストレス、 不良な物理的状態、 実施される活動のために不適切な姿勢、及び不十分な睡眠位置がまた、腰痛にも寄与している。さらに、外傷の背部がそれ自体治癒する場合に創造される瘢痕組織は、正常組織の強さ又は柔軟性を有さない。反復された外傷からの瘢痕組織の再生は結果的には、背部を弱くし、そしてより重度の外傷を導く。時折、腰痛はより重度の医学的問題を示す。発熱又は腸管又は膀胱制御の損失に伴う疼痛、咳する場合の疼痛、及び脚における進行性衰弱は、狭まれた神経又は他の重度の状態を示すことができる。糖尿病を有する人々は、重度の腰痛、又は神経障害に関連する脚の下側まで広がる疼痛を有する。
【0179】
腰痛は、隆起した脊椎板 (例えば、突出した、ヘルニアを起こした、又は破裂した脊椎板)、坐骨神経痛、背骨の変性、頚椎以外の脊柱管狭窄症、骨粗鬆症、変形性関節症、圧縮破壊、骨格の不規則、線維筋痛症、椎弓分離症及び/又は脊椎辷り症により引起され得る。腰痛を引起すことができるまれな脊椎状態は、強直性脊椎炎、細菌感染、頭蓋骨骨髄炎、背骨の腫瘍、パジェット病、及びショイエルマン病を包含する。臨床結果は、GABABアゴニスト、例えばバクロフェンが腰痛の処理において効果的であり得ることを示唆する(Dapas et al., Spine 1985, 10(4), 345-349; and Raphael et al., BMC Musculoskeletal Disorders 2002, June 20, 3(17))。例えば、約20mg/日〜約80mg/日の用量のバクロフェンが、急性腰痛の処理において有効であることが示されている(Dapas et al., Spine 1985, 10(4), 345-9)。
【0180】
ある態様においては、本発明の開示により提供される、腰痛の処理方法は、腰痛、例えば筋肉痙攣に関連する障害、状態及び/又は症状を処理することを含んで成る。腰痛の症状は、その原因に依存する。例えば、逆捻挫又は背部緊張の症状は、背部及び臀部痙攣、痙縮、硬直及び疼痛を包含する。神経根圧の症状は、坐骨神経痛として言及される脚痛、及び神経関連の集積、例えば1本の脚又は足、下脚、又は両脚における刺痛、しびれ感又は衰弱を包含する。背骨の関節炎の症状は、背部及び股関節において悪化する疼痛及び硬直を包含する。
【0181】
苦痛を伴う急性筋骨格状態に関連する筋肉痙攣:
筋肉痙攣は、苦痛を伴う急性筋骨格状態に関連している。腰痛及び頸痛はそのような状態の共通する表れである。背部の急性筋骨格痙攣は、局在化された疼痛、硬直、低められた移動性、毎日の生活の活動消出、及び睡眠障害を引起す共通する障害である。急性腰痛又は頚痛のほとんどのエピソードは、非特異的である。ほとんどの対象は、腰痛及び頸痛、例えば有意な痙攣、癌、感染又は運動衰弱を示す基準を満たさない。非特異的な腰痛は、機械的背痛、個眼関節痛、変形性関節炎、筋捻挫及び筋痙攣として定義される。腰痛は、傍脊椎筋群における反射性痙攣により引起され得る。急性背痛は不本意であり、そしてしばしば、頸部、胸部及び/又は腰髄部を包含する背部の筋肉の苦痛を伴った収縮である。腰椎に関連する痙攣はまた、下背痙攣としても言及される。
【0182】
急性頸痛及び腰痛についての典型的な薬理学的処理剤は、NSAIDS、アセトアミノフェン及び筋肉弛緩薬である。最近のプラシーボ制御された研究は、バクロフェンが、2週間以下の傍背柱筋痙攣及び機能的不能性の現象を有する急性下背症候群の処理において、効果的で、安全で且つ十分に許容されていることを結論づけた。従って、化合物(1)は、苦痛を伴う筋骨格状態、例えば急性背部痙攣及びより特定には急性腰痙攣に関係する筋肉痙攣の処理に使用され得る。
【0183】
線維筋痛:
線維筋痛は、身体じゅう、但し特に背骨を通して、筋肉、腱及び関節における痛み及び疼痛により特徴づけられる状態である。身体はまた、圧点又はトリガー点として言及される特定領域において接触しにくい。線維筋痛の他の症状は、睡眠妨害、鬱病、昼間の疲労、頭痛、下痢便秘交代症、手足の麻痺及び刺痛、脱力感、 記憶困難、及び眩暈感を包含する。線維筋痛の病因学は知られていないが、ストレス、乱れた睡眼パターン、神経系における疼痛関連の化学物質の異常生成、及び/又は低レベルの成長ホルモンが、線維筋痛の開始に寄与していると思われる。
【0184】
線維筋痛の現在の処理は、疼痛の緩和、睡眼の回復、及び生活の一般的質の改善する目的を伴っての症状に基づかれる。いくつかの非薬理学的処理は、運動、教育、行動的及び物理的治療を包含する。薬理学的処理は、三環状化合物、セロトニン再摂取インヒビター、鎮痛薬、筋弛緩薬、及びACEインヒビターを包含する。バクロフェンが線維筋痛症状の改善において有用であることを示す証拠が存在する(Taylor-Gjevre and Gjevre, Lupis 2005, 14(6), 486-8)。
【0185】
線維筋痛の処理のための本発明の開示により供給される化合物の投与の効能は、線維筋痛の動物及びヒトモデルを用いて、及び臨床試験において評価され得る。神経因性疼痛の動物モデル、又は異なったタイプの神経因性疼痛の臨床学的に適切な研究が、線維筋痛の処理のための治療活動の評価においてい有用であることが見出された。
【0186】
神経因性疼痛、筋骨格痛、腰痛、急性の苦痛を伴った筋骨格状態に関連する筋肉痙攣、及び線維筋症の処理のためへの化合物(1)及び他のR−バクロフェンプロドラッグの使用は、Bensonなど., (US 2009/0118365号)(その全内容の引用により本明細書に組込まれる)に開示されている。
【0187】
尿失禁:
尿失禁は、尿のいずれかの不本意な漏れであり、そして切迫性尿失禁、ストレス性失禁、奇異性尿失禁、機能的な失禁、及び混合汚泥失禁を包含する状態のパターンに基づいて5種のタイプに分類され得る(Abrams et al., Neurology and Urodynamics 2002, 21, 167-178)。
【0188】
切迫性尿失禁は、制御され得ない、排尿する突然で且つ激しい行動であり、続いて制御できない排尿が存在する。切迫性尿失禁は、排尿を阻害する前頭葉における脳の部分の変化のために一部、膀胱筋肉の不良なスクイージング能力と共に、膀胱における筋肉の過剰活性の組合せにより引起され得る。膀胱筋肉の不本意な作用は、膀胱の神経、脊髄及び脳を包含する神経系、又は筋肉自体への損傷のために生じる。筋肉及び神経への損傷は、脳卒中、 手術、又は脳障害、例えば多発性硬化症、パーキンソン病、アルツハイマー病の結果として生じる。
【0189】
ストレス性失禁は、咳をし、りきみ、くしゃみをし、重いものを持ち上げ、又は腹内圧迫を突然、高め、そして一般的に、直腸床筋の不十分な強さにより引起されるいずれかの操作を実施する場合、少量の尿の制御できない排尿である。前立腺手術に続く失禁は、男性におけるストレス性失禁の最も共通する形である。女性においては、ストレス性失禁は、妊娠、出産、閉経又は骨盤の手術に関連する物理的変化に起因する。
【0190】
奇異性尿失禁は、あるタイプの閉塞、又は膀胱筋肉の弱い収縮により通常、引起される少量の尿の制御できない漏れである。奇異性尿失禁は、前立腺外科、前立腺肥大症、便秘、神経損傷、神経メッセージを妨害する脳又は脊髄に影響を及ぼす薬物、糖尿病、 多発性硬化症、腫瘍、脊髄損傷、神経系障害、 及び疾病 、例えば 膀胱からの神経シグナル又は排尿筋による尿の圧出を低めることができる多発性硬化症により引起され得る。
【0191】
機能的失禁は、制限された移動性のために物理的に又は不本意には、トレイに行けないことに起因する排尿を言及する。機能的失禁の原因は、次のものを包含する:混乱、痴呆、 不良な視力、不良な移動性、 不良な機敏性、鬱病によるトイレへの気がすすまないこと、不安感、怒り、泥酔、 又は物理的不可能性 、例えば車椅子の人。非可動性を引起す状態は、脳卒中、重度の関節炎、 及び精神機能を妨害する論争、例えばアルツハイマー病及び重度の鬱病による痴呆を包含する。
【0192】
混合汚尿失禁は、1つよりも多くのタイプの失禁を包含する。
尿失禁はまた、夜尿症又は遺尿を包含する。
【0193】
尿失禁はまた、活動亢進性膀胱を包含する。活動亢進性膀胱は、緊急の失禁を伴って又は伴わないで、通常、頻度及び夜間多尿症を伴って、緊急である(Abrams, Urology 2003, 62 (Suppl 5B), 28-37; Ouslander N Engl J Med 2004, 350, 786-99; and Wein and Rovner, Urology 2002 (Suppl 5A), 7-12)。緊急は、排泄の突然の強制的所望の不満であり;頻度は、男性/女性が日中あまりにも頻繁に排泄すると思われる患者による不満であり;そして夜間多尿症は、個人が排泄のために夜間、約1度よりも多く目をさますべきである不満である。
【0194】
活動亢進性膀胱を有する患者は典型的には、延期するのに困難である排尿の突然の及び強制的な必要性(緊急)、緊急を感じる尿の不本意な漏れ(尿失禁の促進)、頻度(24時間で8回以上の排尿)及び夜間多尿症(排尿するために夜、1度以上の目覚め)の症状を提供する。活動亢進性膀胱の症状は、排尿サイクルの充満期の間、排尿筋の付随意的収縮のためである。それらの付随意的収縮は、排尿筋過剰活性と呼ばれ、そして膀胱ムスカリン作動受容体のアセチルコリン−誘発された刺激により仲介される(Andersson, Urology 1997, 50(Suppl. 6A), 74-84)。排尿筋過剰活性は、自発的であるか又は引起され得る充満期の間、不本意な排尿筋収縮により特徴づけられる尿力学的観察である。排尿筋過剰活性は、適切な神経学的状態が存在する、定義された根本にある原因及び排尿筋過剰活性が存在しない、特発性排尿筋過剰活性として特徴づけられ得る。
【0195】
苦痛を伴った膀胱症候群とも呼ばれる間質性膀胱炎は、尿失禁に関連する障害である。間質性膀胱炎は、多くの因子、例えば自己免疫、アレルギー及び感染症病因学により引起されると思われる膀胱の慢性炎症状態である。間質性膀胱炎の症状は、患者が排尿した後でさえ、排尿する過度の緊急性、1日当たり平均16又はそれ以上の尿頻度、夜間での排尿、恥骨結合上(膀胱/骨盤/会陰)疼痛、及び/又は性交疼痛症を包含する。ある状態においては、本発明の開示により供給される投与形は、間質性膀胱炎の処理に使用され得る。
【0196】
二重盲検クロスオーバー試験においては、1日当たり4度、5mgの用量で投与されるバクロフェンは、不安定な膀胱症候群を有する患者において昼間及び夜間頻尿、及び失禁の重症度を有意に改善することが示されている(Taylor and Bates, British J Urology 1979, 51, 504-505)。従って、化合物(1)は、尿失禁、例えば活動亢進性膀胱及び/又は排尿筋過剰活性の処理において有用であることが予測される。
【0197】
投与量:
R−バクロフェンの持続した全身性濃度を提供する錠剤投与形は、現在、1日当たり3度、投与される即時放出性非プロドラッグ形、すなわち患者のために不便であり、そして患者にとって覚えるのが困難であるレジメに比較して、患者のコンプライアンスを増強するであろうと思われる。さらに、本発明の開示により供給される経口投与形錠剤の使用は、低められた副作用、例えば傾眠状態、衰弱、頭痛、発作、嘔気、嘔吐、低血圧、便秘、精神錯乱、呼吸抑制、不眠、及び頻尿又は尿閉を伴って、増強された効能を提供するであろうと思われる。
【0198】
本明細書に開示される特定の疾病の処理において効果的であろう化合物(1)の量は、疾病の性質に、少なくとも一部、依存し、そして当業界において知られている標準の臨床学的技法により決定され得る。さらに、インビトロ又はインビボアッセイは、最適な投与量範囲の同定を助けるために使用され得る。投与レジメ及び投与間隔はまた、当業者に知られている方法により決定され得る。投与される化合物(1)の量は、他の因子の中でも、処理される対象、対象の体重、疾病の重症者、投与路、及び処方医者の判断に依存することができる。
【0199】
全身性投与に関しては、治療的有効用量は、最初、インビトロアッセイから評価され得る。初期用量はまた、インビボデータ、例えば動物モデルから、当業界において知られている技法を用いて評価され得る。そのような情報は、ヒトにおける有用な用量を、より正確に決定するために使用され得る。当業者は、動物データに基づいて、ヒトへの投与を最適化することができる。
【0200】
化合物(1)の用量は、等モル量又は等価質量のR−バクロフェンを供給するよう調節され得る。用量は、本発明の開示により供給される、複数用量形を含んで成ることができる。ある態様においては、R−バクロフェンの治療的有効用量は一般的に、1日当たり約0.03mg〜約1mg/kg体重である。ある態様においては、毎日の用量は、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;ある態様においては、約5mg〜約60mg;及びある態様においては、約10mg〜約40mgの範囲の等価質量のR−バクロフェンを含んで成ることができる。ある態様においては、化合物(1)の用量は、患者において、中位の鎮静及び運動活動の障害を引起す用量以下である。化合物(1)の用量及び適切な投与間隔は、患者におけるR−バクロフェンの持続した治療的有効濃度を、ある態様においては、最小悪影響濃度を越えないで、維持するよう選択され得る。
【0201】
ある態様においては、本発明の開示により供給される投与形は、1日1度、1日2度、及びある態様においては、1日1度以上の間隔で投与され得る。投与は、単独で、及び他の薬物と組合して提供され、そして疾病の効果的処理のために必要とされる限り、続けることができる。投与は、哺乳類、例えばヒトに、食物を食べた状態又は絶食された状態で投与形を投与することを包含する。
【0202】
用量は、単一投与形で、又は複数の投与形で投与され得る。複数の投与形が使用される場合、その複数投与形の個々内に含まれる化合物(1)の量は同じであっても、又は異なっていても良い。
【0203】
ある態様においては、投与される用量は、毒性用量以下である。本明細書に記載される組成物の毒性は、細胞培養物又は実験運動において標準の医薬方法により、例えばLD50(集団の50%が致死する用量)又はLD100(集団の100%が致死する用量)により決定され得る。毒性効果:治療効果の用量比は、治療指数を示す。それらの細胞培養アッセイ及び動物研究から得られるデータは、ヒトへの使用のために毒性でない投与範囲の配合に使用され得る。化合物(1)の用量は、治療的に有効であり、鎮静用量以下であり、そして/又は毒性をほとんどか又はまったく示さない、血液、血漿又は中枢神経系における一定範囲内の循環濃度であり得る。
【0204】
処理の間、用量及び投与スケジュールは、疾病を処理するために十分な又は定常状態の全身性濃度のR−バクロフェンを提供することができる。ある態様においては、徐々に増大する用量が投与され得る。
【0205】
組合せ療法:
本発明の開示により供給される投与形はさらに、化合物(1)の他に、1又は複数の医薬的化合物を含んで成ることができる。そのような化合物は、化合物(1)により処理される疾病と同じ疾病又は異なった疾病を処理するために供給され得る。
【0206】
ある態様においては、化合物(1)は、少なくとも1つの他の治療剤と組合して使用され得る。ある態様においては、化合物(1)は、移動障害、例えば痙攣、消化障害、例えば胃食道逆流病及び嘔吐、 又は中毒又は乱用障害、 例えば ニコチン中毒又は乱用、 アルコール 中毒又は乱用、麻薬中毒又は乱用、 咳、神経因性疼痛、筋骨格痛、又は尿失禁を処理するために、他の化合物と一緒に患者に投与され得る。ある態様においては、少なくとも1つの他の治療剤は、異なったR−バクロフェンプロドラッグであり得る。種々の観点においては化合物(1)及び少なくとも1つの他の治療剤は、加算的に、及びある態様においては、相乗的に作用することができる。
【0207】
少なくとも1つの追加の治療剤は、化合物(1)を含んで成る同じ投与形に含まれ得るか、又は別の投与形に存在することができる。従って、本発明の開示により提供される方法はさらに、化合物(1)を投与する他に、化合物(1)により処理される疾病と同じか又は異なった疾病を処理するために有効な1又は複数の治療剤を投与することを包含する。本発明の開示により提供される方法は、化合物(1)、及び1又は複数の他の治療剤の投与を包含し、但し組合された投与は化合物(1)の治療効能を阻害せず、そして/又は副組合せ作用を生成しない。
【0208】
ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)を含んで成る投与形と同じ投与形の一部であるか、又は異なった投与形に存在する、他の治療剤の投与と同時に投与され得る。化合物(1)は、他の治療剤の投与の前又は続いて投与され得る。組合せ療法のある態様においては、組合せ療法は、例えば特定薬物に関連する薬物副作用を最少にするために、化合物(1)と他の治療剤を含んで成る組成物との間の投与を交換することを含んで成る。化合物(1)が毒性(但し、それだけには限定されない)を包含する薬物副作用を実質的に生成する他の治療剤と同時に投与され得る場合、前記他の治療剤は好都合には、有害薬物反応が誘発される閾値以下に低下する用量で投与され得る。
【0209】
ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)の放出性、生物学的利用能、治療効能、治療能力、安定性及び同様のものを増強するか、調節するか、そして/又は制御するために1又は複数の物質と共に投与され得る。例えば、化合物(1)又はその代謝物の治療効能を増強するために、R−バクロフェン及び化合物(1)は、同時に投与され得るか、又は化合物(1)を含んで成る投与形は、胃腸管から全身循環への化合物(1)又はR−バクロフェンの吸収又は拡散を高めるために、又は患者の血液における化合物(1)又はR−バクロフェンの分解を阻止するために、1又は複数の剤を含んで成ることができる。ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)の治療効能を増強する薬理学的効果を有する活性剤と共に同時投与され得る。
【0210】
さらに、本発明の開示により供給される投与形は、副作用として、痙攣、胃食道逆流病、嘔吐、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、嘔吐、咳、神経因性疼痛、及び/又は筋骨格痛を、それら自体引起すことが知られている他の薬物と組合して使用され得、これにより、その副作用の発生を妨げるか、又は低めることができる。
【0211】
ある態様においては、本発明の開示により供給される投与形は、移動障害、例えば痙攣を処理するために、治療剤、又は移動障害、例えば痙攣の処理において効果的であることが知られているか、又は思われる他の治療剤と組合して、患者に投与され得る。移動障害、例えば痙攣の処理のための、及び化合物の(1)と共に投与され得る薬物の例は、次のものを包含する:レボドパ、軽い鎮静剤、例えばベンゾジアゼピン、例えばアルプラゾラム、クロルジアゼポキシド、クロナゼパム、 クロラゼパート、ジアゼパム、ロラゼパム、及びオキザゼパム; 筋弛緩剤、例えばバクロフェン、抗コリン作動薬、例えばトリヘキシフェニジル、アトロピン、スコポラミン、及びジフェンヒドラミン; 抗精神病薬、例えばクロルプロマジン、フルフェナジン、ハロペリドール、ロキサピン、メソリダジン、モリンドン、ペルフェナジン、ピモジド、チオリダジン、チオチキセン、トリフルオロペラジン、 アリピラゾール、クロザピン、オランザピン、 クエチアピン、 リスペリドン、及びジプラシドン;及び抗うつ剤 、例えばアミトリプチン。
【0212】
ある態様においては、本発明の開示により供給される投与形は、胃腸障害、例えば胃食道逆流病を処理するために、治療剤、又は胃腸障害、例えば胃食道逆流病の処理において効果的であることが知られているか、又は思われる他の治療剤と組合して、患者に投与され得る。胃腸障害、例えば胃食道逆流病の処理のための、及び化合物の(1)と共に投与され得る薬物の例は、次のものを包含する:H2 インヒビター、例えばシメチジン、ファモチジン、 ニザチジン、及び ラニチジン; プロトンポンプインヒビター、例えばオメプラゾール、 ランソプラゾール、 パントプラゾール、 ラベプラゾール、 及びエキソメプラゾール; 及び消化管運動促進剤、例えばシサプリド、ベタンコール、及びメトクロプラミド。
【0213】
ある態様においては、本発明の開示により供給される投与形は、嘔吐を処理するために、治療剤、又は嘔吐の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。嘔吐(悪心及び吐気)を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ベンズアミン、例えばメトクロプラミド; フェノチアジン、例えばプロクロルペラジン、ペルフェナジン、クロルプロマジン、プロメタジン、及びトリエチルペラジン; ブチロフェノン、例えばドロペリドール及びハロペリドール;ドーパミン2 アンタゴニスト、例えば メトクロルパミド; 5-HT3 アンタゴニスト、例えばオンダンセトロン、グラニセトロン、 ドラセトロン、パロノセトロン; NK-1 受容体アンタゴニスト、例えばアプレピタント、コルチコステロイド、例えばデキサメタゾン; 抗ヒスタミン、例えばジフェンヒドラミン及びヒドロキシジン; カンナビノイド、例えばドロナビノール; 及びベンゾジアゼピン、例えばロラゼパム、ミダゾラム、アルプラゾラム、及びオランザピン。
【0214】
ある態様においては、本発明の開示により供給される投与形は、アルコール中毒症又は乱用を処理するために、治療剤、又はアルコール中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。アルコール中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:アンタビュース、ナルトレキソン、クロニジン、メサドン、 1-α-アセチルメタドール、ブプレノルフィン、及びブプロピオン。
【0215】
ある態様においては、本発明の開示により供給される投与形は、麻薬中毒症又は乱用を処理するために、治療剤、又は麻薬中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。麻薬中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ブプレノルフィン、トラマドール、メサドン、及びナルトレキソン。
【0216】
ある態様においては、本発明の開示により供給される投与形は、ニコチン中毒症又は乱用を処理するために、治療剤、又はニコチン中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。ニコチン中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ブプロピオン、クロニジン、及びニコチン。
【0217】
ある態様においては、本発明の開示により供給される投与形は、咳を処理するために、治療剤、又は咳の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。咳を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:デクストロメトルファン、グアイフェネシン、ヒドロコドン、ベンゾナタート、ジフェンヒドラミン、プソイドエフェドリン、パラセタモール、及びカルビノキサミン。
【0218】
ある態様においては、本発明の開示により供給される投与形は、神経因性疼痛を処理するために、治療剤、又は神経因性疼痛の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。神経因性疼痛を処理するために有用な薬物の例は、次のものを包含する:オピオイド鎮痛薬、例えばモルフィン、コデイン、フェンタニール、メペリディン、メサドン、プロポキシフェン、 レボルフェノール、ヒドロモルフォン、オキシコドン、オキシモルフォン、トラマドール及びペンタゾシン; 非オピオイド鎮痛薬、例えばアスピリン、イブプロフェン、ケトプロフェン、ナプロキセン、及びパラセタモール; 非ステロイド性抗炎症薬、例えばアスペリン、コリンサリチル酸マグネシウム、ジフルニサール、サルサレート、 セレコキシブ、 ロフェコキシブ、バルデコキシブ、ジクロフェナック、エトドラク、フェノプロフェン、フルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、 ケトロラック、 メクロファナメート、メフェナム酸、 メロキシカム、ナブメトン、オキサプロジン、ピロキシカム、スリンダク、及びトメチン;
【0219】
抗癲癇薬、例えばガバペンチン、 プレガバリン、カルバマゼピン、フェニトイン、 ラモトリジン、 及びトピラメート; 抗抑うつ剤、例えばジュロキセチン、アミトリプチン、 ベンラファキシン、 ノルトリプチリン、イミプラミン、及びデシプラミン; 局部麻酔剤、例えばリドカイン、及びメキシレチン; NMDA 受容体アンタゴニスト、例えばデキストロメトルファン、メマンチン、及びケタミン; N-タイプカルシウムチャネル遮断薬、例えばジコノチド; バニロイド受容体-1 モジュレーター、例えばカプサイシン; カンナビノイド受容体モジュレーター、例えばサチベックス; ノイロキニン受容体アンタゴニスト 、例えばラネピタント; 他の痛み止め、例えばノイロトロピン;及び他の薬物、例えばデシプラミン、クロナゼパム、ジバルプロエクス、オキスカルバゼピン、ジバルプロエクス、ブトルファノール、バルデコキシブ、ビコプロフェン、ペンタゾシン、プロポキシヘン、フェノプロフェン、ピロキシカム、 インドメタシン、ヒドロキシジン、ブプレノルフィン、ベンゾカイン、クロニジン、フルルビプロフェン、メペリディン、ラコサミド、 デスベンラファキシン、及び ビシファジン。
【0220】
ある態様においては、神経因性疼痛の処理に有効な薬物は、プロポキシフェン、メペリディン、ヒドロモルフォン、ヒドロコドン、モルフィン、コデイン、 2-ピペリジノール-1-アルカノール、 エリプロジル、イフェンプロジル、 ロフェコキシブ、 セロコキシブ、サリチル酸、ジクロフェナック、ピロキシカムインドメタシン、イブプロフェン、ナプロキセン、 ガバペンチン、 カルベマゼピン、 プレガバリン、トピラメート、バルプロン酸、スマトリプタン、 エリトリプタン、リザトリプタン、ゾルミトリプタン、 ナラトリプタン、フレクセリル、カリソプロドール、ロバキシサル、 ノルゲシック(norgesic)、ダントリウム、ジアゼパム、クロルジアゼポキシド、アルプラゾラム、ロラゼパム、パラセタモール、亜酸化窒素、ハロセン、リドカイン、エチドカイン、ロピバカイン、塩化プロカイン、サラピン、ブピバカイン、カプシシン、デシプラミン、アミトリプチン、ドキセピン、ペルフェナジン、プロトリプチリン、トラニルシプロミン、バクロフェン、クロニジン、メキセリチン( mexelitine)、ジフェンヒドラミン、ヒドロキシジン、カフェイン、プレドニソン、 メチル-プレドニソン、デカドロン、セルトラリン、パロキセチン、フルオキセチン、トラマドール、レボドパ、デクストロメトルファン、 サブスタンス Pアンタゴニスト、及びボツリヌス毒素から選択される。
【0221】
ある態様においては、神経因性疾病の処理にお有用な薬物は、ニコチン受容体部分作動薬及び鎮痛薬から選択され得る。
神経因性疼痛の処理のための非薬理学的療法は、経皮的電気刺激、経皮の電気的神経刺激、及び刺鍼術を包含する。
【0222】
ある態様においては、本発明の開示により供給される投与形は、線維筋痛症を処理するために、治療剤、又は線維筋痛症、又はある態様においては、線維筋痛症に関連する疾病、障害又は状態の処理において効果的であることが知られているいるか、又は思われる他の治療剤と組合して患者に投与され得る。線維筋痛症についての薬物療法は、線維筋痛症の重症度及び頻度に適応され得る。時折のエピソードに関しては、急性処理が示され得る。1月あたり複数回、発生する線維筋痛症エピソードに関しては、又は攻撃が患者の毎日の生活に非常に影響を及ぼす場合、進行に基づいて慢性療法が適切である。
【0223】
エピソードの頻度を低める、繊維筋痛症のための処理は、非ステロイド性抗炎症薬(NSAIDs)、アドレナリン性β遮断剤、カルシウムチャンネル阻害剤、三環系抗うつ薬、選択的セロトニン再取込み阻害薬、抗痙攣薬、 NMDA 受容体アンタゴニスト、ドーパミンアゴニスト、選択的5-HT3受容体アゴニスト、 オピオイド、 筋弛緩薬、催眠鎮静薬、 及び他の治療剤を包含する。線維筋痛症の処理に有用なNSAIDの例は、アスピリン、イブプロフェン、フェノプロフェン、フルルビプロフェン、ケトプロフェン、メフェナム酸、及びナプロキセンを包含する。線維筋痛症の処理に有用なアドレナリン性β遮断剤の例は、アセブトロール、アテノロール、イミロール、メトプロロール、ナドロール、ピンドロール、プロプラノロール、及びチモロールを包含する。
【0224】
線維筋痛症の処理に有用なチャンネル遮断剤の例は、アムロジピン、カルシウム拮抗物質ジルチアゼム、ドタリジン、フェロジピン、フルナリジン、ニカルジピン、ニフェジピン、ニモジピン、ニソルジピン、及びベラパミルを包含する。線維筋痛症の処理のために有用な三環式抗うつ薬の例は、アミトリプチン、デシプラミン、ドキセピン、イミプラミン、ノルトリプチリン、シクロベンザプリン、及びプロトリプチリンを包含する。線維筋痛症の処理のために有用な選択的セロトニン再摂取インヒビターの例は、フルオキセチン、メチセルギド、ネファゾドン、パロキセチン、セルトラリン、及びシタロプラムを包含する。線維筋痛症の処理のために有用な他の抗うつ薬の例は、ブプロピオン、ネファゾドン、ノルエピネフリン、バラファキシン、ジュロキセチン、及びトラゾドンを包含する。
【0225】
線維筋痛症の処理のために有用な抗痙攣薬(抗癲癇薬)の例は、ジバルプロックス・ナトリウム、フェルバメート、 ガバペンチン、 ラモトリジン、 レベチラセタム、オキスカルバゼピン、チアガビン、トピラメート、バルプロエート、及びゾニサミドを包含する。線維筋痛症の処理のために有用なNMDA受容体アンタゴニストの例は、デクストロメトルファン、マグネシウム及びケタミンを包含する。線維筋痛症の処理のために有用なドーパミンアゴニストの例は、α−ジヒドロエルゴクリプチンを包含する。線維筋痛症を妨げるために有用なオピオイドの例は、トラマドール、オキシコドン及びメサドンである。
【0226】
線維筋痛症の処理のために有用な筋弛緩薬の例は、シクロベンザプリンである。線維筋痛症の処理のために有用な治療薬の例は、運動、インターフェロン、成長ホルモン、 ホルモン療法、 動物脂肪が低くそして繊維に富んでいる食物、 及び相補療法、例えばカウンセリング/精神療法、リラクゼーション・トレーニング、段階的筋弛緩法、誘導されたイメージ、横隔膜の呼吸、バイオフィードバック、刺鍼術、及び物理的及びマッサージ療法を包含する。
【0227】
筋肉/骨格痛の重症度及びいずれかの関連する症状を排除するか又は低めるための急性線維筋痛症処理は、セロトニン受容体作動薬、例えば トリプタンス (5-ヒドロキシトリプトファン(5-HT) アゴニスト)、 例えば、アルモトリプタン、 エレトリプタン、 フロバトリプタン、 ナラトリプタン、 リザトリプタン、 スマトリプタン、及びザルミトリプタン; エルゴタミン-に基づく化合物、例えばジヒドロエルゴタミン及びエルゴタミン; 鎮吐薬、例えばメトクロプラミド及びプロクロルペラジン;及び鎮痛効果を提供する化合物を包含する。
【0228】
線維筋痛症の処理において有用な薬物の他の例は、パラセタモール、アスピリン、カフェイン、シプロヘプタジン、メチセルギド、バルプロン酸、 NSAIDs、例えばジクロフェナック、フルルビプロフェン、ケタプロフェン、 ケトロラック、イブプロフェン、インドメタシン、メクロフェナメート 、及びナプロキセンナトリウム; オピオイド、例えばコデイン、メペリディン、及びオキシコドン;及びグルココルチコイ、例えばデキサメサゾン、プレドニソン、及びメチルプレドニゾロンを包含する。
【0229】
ある態様においては、本発明の開示により供給される投与形は筋骨格痛の処理のために、治療剤、又は筋骨格痛の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。筋骨格痛の処理のために有用な薬物の例は、シクロベンザプリン、ダントロレン、メトカルバモール、オーフェナドリン、 トリザニドリン、メタキサロン、カリソプロドール、クロルフェネシン、クロルゾキサゾン、アルプラゾラム、ブロマゼパム、クロルジアゼポキシド、クロラゼプ酸、ジアゼパム、フルニトリアゼパム、ロラゼパム、メダゼパム、ミダゾラム、オキザゼパム、プラゼパム、トリアゾラム、テマゼパム、及びボツリヌス毒素を包含する。ある態様においては、神経因性疼痛の処理のために有用ないずれかの薬物が、筋骨格痛の処理のために化合物(1)と共に同時投与され得る。
【0230】
ある態様においては、本発明の開示により供給される投与形は腰痛の処理のために、治療剤、又は腰痛の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。腰痛の処理のために有用な薬物の例は、NSAIDs、例えばアスピリン、ナプロキセン、及びイブプロフェン; 抗痙攣薬、抗抑うつ薬、例えばアミトリプチン及びデシプラミン; 及びオピオイド、例えばコデイン、オキシコドン、ヒドロコドン、及びモルフィンを包含する。
【0231】
ある態様においては、神経因性疼痛の処理のために有用ないずれかの薬物が、腰痛の処理のためにGABAB アゴニストのプロドラッグと共に同時投与され得る。腰痛のための治療は、冷たくて熱い湿布の使用、床上安静、 運動、背骨の操作、刺鍼術、バイオフィードバック、介入療法、牽引、経皮的電気刺激、超音波、椎体療法、椎骨形成術、椎間板切除、椎間孔拡大術、 椎間板内電気療法、経皮的高周波椎間板減圧術、ラジオ周波外傷、脊椎固定、 及び背骨ラミネクトミーを包含する。
【0232】
ある態様においては、本発明の開示により供給される投与形は、腰痛の処理のために、治療剤、又は筋肉痙攣、例えば腰痛に関連する筋肉痙攣を処理するための他の治療剤、例えば筋肉弛緩剤を組合して患者に投与され得る。筋肉痙攣を処理するための筋肉弛緩剤として有用な薬物の例は、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、ジアゼパム、メタキサロン、メトカルバモール、オーフェナドリン、及びチザニジンを包含する。
【0233】
ある態様においては、本発明の開示により供給される投与形は尿失禁の処理のために、治療剤、又は尿失禁の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。尿失禁の処理のために有用な薬物の例はアミトリプチン、ベラドンナ、darifenacin、デスモプレッシン、ジュロキセチン、発情物質、 フェソテロジン、フラボキサート、ヒヨスチアミン、イミダフェナシン、イミプラミン、ニトロフラントイン、オキシブチニン、 プロピベリン、 ソラベロン、ソリフェナシン、タムスロシン塩酸塩、タムスロシン、トルテロジン、トロスピウム、 タイプ Aボツリヌス毒素、 及びバルデナフィル塩酸塩を包含する。
【0234】
尿失禁及び特に活動亢進性膀胱の処理のために可能性を示す他の薬物は、K+ チャネルに対して作用する薬物、例えば NS-8、KW-7158、 ZD-0947; 5-HT3 アンタゴニスト; 5-HT1aアンタゴニスト、例えばREC-0545; P2X アンタゴニスト; NK1 受容体アンタゴニスト、例えばSSR-240600、TA-5538、及びアプレピタント; β3-アゴニスト、例えばGW-427353及びKUC-7483、 YM-178; 及び 他のもの、例えばDDP-200 (オキシブチニン及びガバペンチン)、ニトロフルルビプロフェン、エロカルシトール、 NCX-2111、及びビピリジンを包含する(Colli et al., Expert Opin Investig Drugs 2007, 16(7), 999-1007)。
【0235】
β3−アドレノ受容体アゴニストは、活動亢進性膀胱の処理のために最近提案されて来た(Tyagi et al., Drugs of the Future 2009, 34(8), 635-640)。尿失禁の処理のために有用な他の薬物は、Robinson and Cardozo, Maturitas 2010 doi:10.1016/j.maturitas.2009.12.022に開示されている。
【実施例】
【0236】
次の例は、化合物(1)を含んで成る経口錠剤投与形を詳細に記載する。材料及び方法に関する多くの修飾は本発明の開示の範囲内で実施され得ることは当業者に明らかであろう。
【0237】
例1:化合物(1)乾燥粉末の調製及び特徴化:
化合物(1)ロット70を、次の方法を用いて結晶化した。機械撹拌機、テフロン(登録商標)被覆されたサーモカップル、還流冷却器及び窒素入口を備えた、5Lの三ツ首丸底フラスコに、化合物(1)(72g)、アセトン(186ml)及びヘキサン(672ml)を添加した。その混合物を、撹拌し(60%の速度)、そして50℃に1時間、加熱した。すべての材料は溶解した。追加のヘキサン(1081ml)を、50℃で温度を維持しながら、1時間にわたって添加した。その溶液を45℃に冷却し、そして45℃で5時間、維持し、この時点で固形物が形成した。溶液をさらに、40℃に12時間、次に35℃に12時間、冷却した。次に、その溶液を22〜25℃に2時間、冷却した。生成物を焼結ガラス漏斗を通しての濾過(早い)により集め、そしてアセトン(100ml)及びヘキサン(900ml)の溶液によりすすいだ。湿ったフィルターケークを真空オーブンに移し、そして40℃で12時間、乾燥し、白色固形物として結晶性化合物(1)(ロット70、70g、97%)を得た。
【0238】
化合物(1)ロット71を、次の方法を用いて結晶化した。機械撹拌機、テフロン(登録商標)被覆されたサーモカップル、還流冷却器及び窒素入口を備えた、5Lの三ツ首丸底フラスコに、化合物(1)(72g)、アセトン(186ml)及びヘキサン(672ml)を添加した。その混合物を、撹拌し(60%の速度)、そして50℃に1時間、加熱した。すべての材料は溶解した。追加のヘキサン(1081ml)を、2分間にわたって添加し、そして温度を39℃に下げ、固形物を形成した。その溶液を22〜25℃に1時間にわたって冷却し、そして次に、0〜5℃に1時間、冷却した。生成物を焼結ガラス漏斗を通しての濾過(遅い)により集め、そしてアセトン(100ml)及びヘキサン(900ml)の溶液によりすすいだ。湿ったフィルターケークを真空オーブンに移し、そして40℃で12時間、乾燥し、白色固形物として結晶性化合物(1)(ロット71、70g、97%)を得た。
【0239】
結晶性化合物(1)ロット4を、同じ濃度及び溶媒を用いて調製した(但し、2.5時間にわたって50℃〜0℃の線状除熱傾斜を用いた)。
遅い結晶化(ロット70)、早い結晶化(ロット71)、及び中間速度の結晶化(ロット4)により調製された結晶性化合物(1)の種々の性質が表2に示されている。
【0240】
【表2】
【0241】
化合物(1)サンプルの粒子サイズ及び形状を、走査電子顕微鏡(SEM)分析により特徴づけた。3種のロットの個々からのサンプルを両面炭素テープ上に取り付け、白金の薄層によりスパター被覆し、そして次に、Hitachi−4700SEMを用いて試験した。500倍率での3種のロットのSEMマイクログラフが図1に示される。10,000倍率での3種のロットの対応する像が図2に示される。
【0242】
ロット4(図1A及び2A)は、約25〜50ミクロンの直径の丸い凝集体を含んで成る。対照的に、ロット70(図1B及び2B)は約100ミクロンの長さの線状体を含んで成る。ロット71(図1C及び2C)は、約20〜50ミクロンの長/幅の不規則な形状の凝集体により特徴づけられるさらにもう1つの形態学を含んで成る。
【0243】
例2. 乾燥粉末の流動特性:
乾燥粉末の流れを、FLODEX(商標) Powder Flowability Index Test Instrument (Hanson Research Corporation, Chatsworth, CA)を用いて特徴づけた。その器具は、流動試験の前、試験粉末を保持する円柱状の金属貯蔵物を備えた。その円柱状貯蔵物は、5.7cmの内径及び7.4mmの長さを有する。貯蔵物の底端は、取りはずせる金属ディスクにより密封され得る。個々のディスクは、そのディスクの中心に丸型開口部を有する。開口部の直径は、1mmの増分で4mm〜10mmの範囲であり、そして2mmの増分で10mm〜34mmの範囲である。流動試験の前、その開口部は閉じられる。
【0244】
次に、粉末を、その閉じられた開口部上に配置する。開口部が閉じられていない場合、粉末は、開口部の直径が十分に大きい場合、重力下でその開口部を通して流動することができる。小さな開口部を通して流れる粉末は、錠剤のために有用な流動性質を有すると思われる。例えば、約24mm以下のFlodex測定(Flodex)が典型的には、商業的規模で高速錠剤化操作のために使用される。約20mm以下のFlodexは、高速錠剤化操作のために有用である。18mm又はそれ以下のFlodexは、特に高速錠剤化操作のために有用であると思われる。
【0245】
重度の積層を回避し、そして粉末層を振動又はタッピングしないで、底での開口部を閉鎖しないで、Flodexを、約70ccの試験粉末により保存物をまず軽く満たすことにより決定する。次に、開口部を開放する。これは、前記装置により供給されるシャッターを開口することにより達成され得る。他方では、シャッターが使用されない場合、ディスクにより固定された粉末充填保存物を、開口部を閉じるために乾燥した平らな表面上に設置する。次に、ゆっくり且つ均等に、貯蔵物を持ち上げ、粉末の流動を可能にする。
【0246】
いずれかの方法においては、粉末が開口部を通して流れる場合、透明なチャネルが粉末層内に放置される。粉末が開口部を通して流れない場合、粉末層内のアーチ形状の腔が開口部上に形成され、そしてアーチとして言及される。流動試験は、良好な流れのための最小開口部サイズが同定されるまで(Flodexとして言及される)、種々の開口部サイズで実施される。Flodexは、粉末が少なくとも3回の測定試験において開口部を通して流れないよりも多く開口部を通して流れる最小開口部直径である。
【0247】
例3.化合物(1)及び賦形剤の流動性質:
純粋化合物(1)及び種々の純粋錠剤化賦形剤の3種のロットについてのFlodexが表3に示される。純粋化合物(1)の3種のロットは化学的に同等であるが、しかし個々のロットの粒子サイズ及び形状により異なる(例1、図1及び2を参照のこと)。表3に示されるように、ロット4は最低のFlodexを示す。低いFlodexは丸型の粒子形状に寄与し、それは、もつれるロット70の特徴的な繊維状粒子塊状物、又は運動下で高い粒子間摩擦力を付与するロット71の特徴的な不規則形状の粒子よりも、より滑らかに流れる。それらの測定は、Flodexが、異なった形状学を有する粒子を含んで成る粉末の流動性質における差異を検出し、そして定量化するために十分に異なることを示す。
【0248】
【表3】
【0249】
表3に列挙されるAVICEL(商標) PH200は、190ミクロンの平均サイズを有する微晶性セルロースである(FMC Biopolymer Corporation, Philadelphia, PA)。METHOCELTM K4M SPは、ヒドロキシプロピルメチルセルロース(METHOCELTM K4M Standard Premium, Dow Chemical Company, Midland, MI)である。この置換されたセルロースポリマーは、約8重量%のヒドロキシプロピル含有物、約22重量%のメトキシル含有物、2%濃度で約4,000センチポアズの水中、呼称粘度、及び少なくとも75重量%が149ミクロ以下になるような粒子サイズを有する。METHOCELTM K4M (DC) (Dow Wolff Cellulosics, Midland, MI)は、METHOCELTM K4M SPと化学的に同一であるが、しかし約250ミクロンの大きな粒子サイズを有する。
【0250】
EUDRAGIT(商標) RLPO (Evonik Industries AG, Darmstadt, DE)は、ポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)(1:2:0.2)を含んで成るコポリマーである(USP/NFアッセイ、乾燥物質に基づいて8.85〜11.96%のアンモニオメタクリレート)。このアクリレートポリマーは、1モル当たり約150,000gの平均分子量、及び少なくとも90重量%が315ミクロン以下であるような粒子サイズを有する。EUDRAGIT(商標) RSPO (Evonik Industries AG, Darmstadt, DE)は、エチルアクリレート、メチルメタクリレート及び低含有率のメタクリル酸エステルのコポリマー(四級アンモニウム基を有する)である(USP/NFアッセイ、乾燥物質に基づいて4.48〜6.77%のアンモニオメタクリレート単位)。DI-TAB(商標)は、180ミクロンの呼称粒子サイズを有する二塩基リン酸カルシウム・二水和物(Innophos, Inc., Cranbury, NJ)である。
【0251】
表3に示されるように、錠剤配合物に使用される賦形剤のFlodexは、有意に異なった流動性質を示すことができる。低い〜高いFlodexの順位は、DI-TAB(商標)、AVICEL(商標) PH200、EUDRAGIT(商標) RLPO、METHOCELTM K4M SP及び METHOCELTM K4M DCである。
【0252】
例4.乾燥粉末ブレンドの流れ:
錠剤化賦形剤の2種の乾燥ブレンドを調製し、そして流動性質を決定した。ブレンドA08−011を、1インチ当たり20のワイヤを有する篩を通して、42.0 gの AVICEL(商標) PH200、43.4 g のMETHOCELTM K4M (SP)及び25.7 gのEUDRAGIT(商標) RLPOを容器中に連続的に通すことにより調製した。36gのDI−TAB(商標)及び1.5gのコロイド状二酸化珪素(CAB-O-SILTM M-5P, Cabot Corporation, Billerica, MA)を予備混合し、そして次に、同じスクリーンを通して容器中に通した。コロイド状二酸化珪素は、流動促進剤としてブレンドに含まれた。
【0253】
サイズ分けされた粉末を、5分間、V−ブレンダーにおいてタンブル−混合した。最終的に、40ワイア/インチを有する篩を前もって通された、1.5gのステアリン酸マグネシウム(Univar)を、粉末混合物に添加し、そして全組成を、5分間、V−ブレンドした。ステアリン酸マグネシウムは、錠剤圧縮の間、摩擦を低め、そして錠剤の滑らかな射出を促進するために錠剤化滑剤として作用した。得られるブレンド(A08−011)の流れは、20mmのFladexを示した。
【0254】
ブレンドA08−016を、ブレンドA08−011を調節するために使用される方法に類似する方法を用いて調製した。ブレンドA08−016は、ブレンドA08−011と組成において同等であるが、但しMETHOCELTM K4M SPがMETHOCELTM K4M DCにより置換された。得られるブレンドA08−016の流れを測定した。
【0255】
2種のブレンドの組成及びそれぞれのFlodexは、表4に提供される。純粋なMETHOCELTM K4M SPは28mmの高いFlodexを示し、そして純粋なMETHOCELTM K4M DCの流れは30mmのさらに高いFlodexを示すが(表3)、組合わされた賦形剤のブレンドは、それぞれ20及び15mmの値を示した。この結果は、表4に列挙される個々の成分のFlodexに基づいて予測されなかった。
【0256】
【表4】
【0257】
例5.化合物(1)を含む乾燥粉末ブレンドの流れ:
賦形剤及び単一ロットの化合物(1)により配合された2種の乾燥粉末ブレンドを調製し、そして流動性質を決定した。30.0gの化合物(1)、69.0 g の AVICEL(商標) PH200、86.7 g のMETHOCELTM K4M SP、及び51.3 g のEUDRAGIT(商標) RLPOを、20メッシュの篩を通してボウル中に連続的に通した。57.0gのDI-TAB(商標)及び3.0gの二酸化珪素を予備混合し、そして同じ20メッシュの篩を通してボウル中に通した。得られるサイズ分けされた粉末を、2クオートのV−ブレンダにおいて5分間タンブル−ブレンドした。最終的に、40メッシュの篩を通して前もってサイズ分けされた、3.0gのステアリン酸マグネシウムを、前記混合された粉末に添加し、そして5分間タンブル混合した。得られるブレンドA08−020のFlodexは22mmであった。
【0258】
第2ブレンドAO8−017を、ブレンドA08-020を調製するために使用される方法に類似する方法を用いて調製した。ブレンドA08-017の組成は、A08-020の組成と同等であったが、但しMETHOCELTM K4M DCをMETHOCELTM K4M SPにより置換した。2種のブレンドの組成及び流動性質が表5に要約されている。METHOCELTM K4M DCにより配合される化合物(1)を含むブレンドの流れは、22mmでMETHOCELTM K4M SPにより配合された同じブレンドに比較して、16mmのFlodexで有意に良好であった。この結果は、METHOCELTM K4M SP and METHOCELTM K4M DCがそれぞれ28mm及び30mmの類似する流動性質を有することを示す、表3に列挙される個々の成分の流れの結果に基づいて単に予測されなかった。
【0259】
【表5】
【0260】
例6.異なったロットからの化合物(1)を含む乾燥粉末ブレンドの流れ:
異なったロットの化合物(1)によりそれぞれ配合された、3種の乾燥粉末ブレンドを調製し、そして得られるブレンドの流動性質を比較した。ブレンドを調製するために使用されるサイズ分け及び混合方法は、例5に記載されるのと同じであった。組成及び対応するFlodex数が表6に要約されている。ブレンドB08-019及びBO8-020はそれぞれ、21mm及び13mmのFlodex数を示した。化合物(1)の個々のロットについてのFlodex数に基づいて(表3)、化合物(1)ロット70により調製されたブレンドが、化合物(1)ロット71により調製されたブレンドのFlodexに相当するFlodexを示したことが予測される。しかしながら、ロット71により配合されたブレンドのFlodexは、ロット70により配合されたブレンドのFlodexよりも有意に良好であり;それぞれ13mm対21mmであった。
【0261】
【表6】
【0262】
例7.化合物(1)及びEUDRAGIT(商標) RLPO又は EUDRAGIT(商標) RSPOを含む乾燥粉末ブレンドの流れ:
異なった品質のEUDRAGIT(商標)によりそれぞれ配合された2種の乾燥粉末ブレンドを調製した。16.5gのAVICEL(商標) PH200、32.9 g の METHOCELTM K4M DC、及び22.1 gのEUDRAGIT(商標) RLPOを、20メッシュの篩を通して通常の容器中に連続的に通した。16.5gのDI-TAB(商標)及び1.0gのコロイド状二酸化珪素を予備混合し、20メッシュの篩を通してサイズ分けし、そしてそのサイズ分けされた粉末に添加した。
【0263】
粉末をV−ブレンダーにおいて5分間タンブル混合した。次に、混合され、そしてサイズ分けされた粉末の半分を除いた。20%メッシュの篩を通して前もってサイズ分けされた10.0gの化合物(1)を、前記半分の粉末上に均一層として広げた。除かれた半分の粉末層を、化合物(1)の層上に適用し、そして得られる三層組成物を5分間タンブル混合した。最終的に、40メッシュの篩を通して前もってサイズ分けされた1.0gのステアリン酸マグネシウムを、前記混合物に添加し、そしてV−ブレンダーにおいて3.5分間タンブル混合し、ブレンドE08-007を形成した。
【0264】
第2ブレンドを、類似する混合方法を用いて調製した。ブレンドE08-013は、組成において同一であったが、但し、EUDRAGIT(商標) RSPOがEUDRAGIT(商標) RLPOにより置換されている。両ブレンドの組成が表7に提供される。
【0265】
【表7】
【0266】
例8.錠剤配合物の溶解プロフィール:
例5及び6に記載されるブレンドを、1/4−インチの丸型標準量凸錠剤型押し及びダイスを用いて、100mgの重量の錠剤に圧縮した。個々のタイプの錠剤を、37℃の温度で、50mMの一塩基性リン酸ナトリウム(pH6.8)900mlにおけるUSA櫂装置(タイプII)における薬物の溶解性について試験した。櫂撹拌速度は75回転/分であった。溶解試験の間、錠剤は、個々の容器の底で錠剤を位置決定するためにステンレス鋼製カゴ内に含まれた。
【0267】
EUDRAGIT(商標) RLPOを含んで成る錠剤(すなわち、 Blend E08-007)はまた、SR4−10錠剤配合物としても言及される。
【0268】
例6に記載されるブレンドを用いて調製された錠剤に関して、一定時間にわたって錠剤から放出される累積%化合物(1)を示す溶解プロフィールが図3に示されている。
【0269】
例7に記載されるブレンドを用いて調製された錠剤に関して、一定時間にわたって錠剤から放出される累積%化合物(1)を示す溶解プロフィールが図4に示されている。EUDRAGIT(商標) RSPOにより配合される錠剤の溶解プロフィールは、そのプロフィールにおいて三角形の記号により示され、そしてEUDRAGIT(商標) RLPOにより配合される錠剤の溶解プロフィールにおいて円形の記号により示される。
【0270】
例9.20mgの化合物(1)錠剤(SR4-20)の調製及び特徴化:
錠剤バッチを、パイロット規模で製造し、ブレンド及び錠剤化操作において許容できる錠剤重量及び薬物含有率の維持の実行可能性を評価した。粉末ブレンドを、20メッシュのスクリーンを備えたRussell Finex 篩 (Russell Finex, Pineville, NC)を通して、順に1,140 g の AVICEL(商標) PH200、1,974 g のMETHOCELTM K4M DC、1,026 g のEUDRAGIT(商標) RLPO、1,140 gのDI-TAB(商標)、及び60gのコロイド状二酸化珪素を通すことにより調製した。
【0271】
サイズ分けされた粉末を、1立方フィートのV−ブレンダーに移し、そして5分間タンブル混合した。約2,670gのブレンドを除き、そしてそばに置いた。20メッシュの篩を通して前もって手動的に通された、600gの化合物(1)(ロット4)を、ブレンダーに残る半分の粉末上に均等層を広げた。2,670gの前もって除かれた粉末をブレンダーに戻し、そしてその三層混合物を5分間ブレンドした。最終的に、40メッシュスクリーンを通され、前もってふるいにかけられた60gのステアリン酸マグネシウムをブレンドに添加し、そしてその混合物を4分間ブレンドし、6,000gのバッチの混合及びブレンド操作を完結した。
【0272】
2,400gの得られるブレンドを、5/16−インチの標準の丸型凹パンチ用具を備えたKilian T100 回転錠剤プレス(Kilian & Co., Inc., Horsham, PA)のホッパーに移した。錠剤を、1分当たり180個の錠剤の速度で圧縮し、それらは錠剤当たり200mgの呼称重量を有する。錠剤のサンプルを約5分ごとに集め、そして錠剤の重量を測定した。10個の錠剤の平均重量を個々の時点で決定した。図5に示される錠剤重量ヒストグラムは、錠剤重量が十分に維持されていることを示す。平均重量は、200mgの錠剤重量(錠剤当たり20mgの化合物(1)の±5%以内に維持された。
【0273】
錠剤における化合物(1)の含有率を決定した。9の点の個々で集められた3個の錠剤を、1.2ml/分の流速で35℃で、Alltech Platinum EPS C18 カラム (4.6 × 150 mm, 3 μm, 100Å) (Alltech Associates, Inc., Deerfield, IL)を用いて、高圧液体クロマトグラフィーにより化合物(1)について分析した。移動相は、0.02 M KH2PO4, pH 2.5/水/アセトニトリル, 461/44/495 (v/v/v)であり、そして検出波長は220nmであった。錠剤を、アセトニトリル:水(80:20)中、1%(w/v)ドデシル硫酸ナトリウム(SDS)に溶解し、そして30分間、音波処理した(標的濃度0.2mg/mlの化合物(1))。すべてのサンプルを、分析の前、0.45μmのナイロンフィルターにより濾過した。少量の界面活性剤(すなわち、ドデシル硫酸ナトリウム(SDS))添加は、薬物の不溶性賦形剤への結合を最少にすることが示されており、改良された薬物抽出効率をもたらした。
【0274】
錠剤についての化合物(1)含有率ヒストグラムが図6に示されており、そして20mgの標的化合物(1)含有量に基づかれている。錠剤サンプルを、錠剤化実施の間、一定間隔で入手し、ここでその間隔は合計錠剤化実施(100%)の%として同定される。錠剤当たり20mgの化合物(1)含有量は、グラフの縦座標に基づいて100%に等しい。錠剤当たりの平均化合物(1)含有量は、すべての時点で標的用量の±5%以内に維持された。個々の錠剤における化合物(1)含有率の範囲は、20mgの標的化合物(1)含有率の92.6%低値〜105.7%の高値の範囲であった。
【0275】
錠剤の厚さをダイヤルキャリパにより測定し、約4.02mmであることがわかった。錠剤の圧縮強さを、錠剤硬度計上で測定し、約8.1kポンドであることがわかった。錠剤を例6に記載される方法を用いて、溶解について試験した。例8に従って決定される、18時間で20mgの錠剤からの化合物(1)の平均累積放出性は、図7に示されるように約45%であった。
【0276】
例10.30mg化合物(1)錠剤(SR4-30)の調製及び特徴化:
例9に記載されるブレンドの 3,600gを、3/8−インチの標準の丸型凹パンチ用具を備えたKilian T100 回転錠剤プレスのホッパーに移した。錠剤を、1分当たり180個の錠剤の速度で圧縮し、それらは錠剤当たり300mg(錠剤当たり30mgの化合物(1))の呼称重量を有する。錠剤のサンプルを約5分ごとに集め、そして錠剤の重量を測定した。10個の錠剤の平均重量を個々の時点で決定した。図8における錠剤重量ヒストグラムは、錠剤重量が300mgの標的重量の5%以内に維持されたことを示す。
【0277】
錠剤における化合物(1)の含有量をまたモニターした。12時点の個々で集められた3個の錠剤を、例9に記載されるようにHPLCを用いて化合物(1)について分析した。化合物(1)含有率のヒストグラムが図9に示されている。錠剤当たり30mgの化合物(1)含有率は、グラフ縦軸上で100%の標的含有率に等しい。錠剤当たりの平均薬物含有率を、錠剤化実施を通して30mgの標的量の±5%以内に維持した。個々の錠剤における薬物含有率の範囲は、30mgの標的重量の95.0%の低値〜103.6%の高値の範囲であった。
【0278】
錠剤の厚さをダイヤルキャリパにより測定し、約4.18mmであることがわかった。錠剤の圧縮強さを、錠剤硬度計上で測定し、約9.5kポンドであることがわかった。錠剤を例8に記載される方法を用いて、溶解について試験した。例8に従って決定される、18時間で30mgの錠剤からの化合物(1)の平均累積放出性は、図10に示されるように約39%であった。
【0279】
例11.10mgの化合物(1)の錠剤(SR4-10)の調製及び特徴化:
10mgの化合物(1)を含んで成り、そして約175.1mgの重量の持効性放出錠剤を例9及び10に記載される方法に類似する方法を用いて調製した。SR4錠剤投与形の組成物は、表8に要約されている。
【0280】
【表8】
【0281】
例12.10mgの化合物(1)SR3錠剤の調製:
化合物(1)及びアンモニオアルキルメタクリレートポリマー、EUDRAGIT(商標) RL 30D, (SR3)を含んで成る持効性放出錠剤を、300gのバッチサイズで調製した。
【0282】
次の量の成分を用いて、300gのバッチを調製した:12.0 g の化合物(1)、128.7 g の 微晶性セルロース (AVICEL(商標) PH200, FMC Corp., Philadelphia, PA)、106.5 gの ヒドロキシプロピルメチルセルロースMETHOCELTM K4M (METHOCELTM K4M, Dow Chemical)、51.3 g のアンモニオアルキルメタクリレートコポリマー(EUDRAGIT(商標) RL 30D, Degussa)、及び1.5 g のステアリン酸マグネシウム (HYQUAL(商標)植物源, Mallinckrodt, Phillipsburg, NJ)。10mgの化合物(1)及びアンモニオアルキルメタクリレートポリマーを含んで成る持効性放出錠剤(SR3)における成分の量が表9に提供される。
【0283】
【表9】
【0284】
化合物 (1) (12 g)、微晶性セルロース(AVICEL(商標)PH200, FMC Biopolymer) (127.8 g)、 及びヒドロキシプロピルメチルセルロース(METHOCELTM K4M SP, Dow Chemical Co.) (106.5 g)を計量し、#20%メッシュのステンレス鋼製スクリーンを通してスクリーンし、そしてV−ブレンダー(2 quart, Model MB-1, Globepharma, New Brunswick, NJ)において5分間、混合した。
【0285】
ブレンドを放し、そして1 Lのボウルを有する KG-5 Mixer/Granulator (Key International, Englishtown, NJ)を用いて、高剪断で湿式粒状化した。湿式粒状化は、100mlの水、1mmの管直径、250rpmのインペラー速度、及び1500rpmのチョッパー速度を用いて実施された。
【0286】
湿った粒状物を、25 SCFMの入口、45℃の入口空気温度、30℃以下の出口空気温度、及び200〜900mmH2Oのフィルター圧力を用いて、Fluid Bed Model 0002 (Fluid Air, Aurora, IL)グラニュレーター/ドライヤーにおいて乾燥した。乾燥後の標的重量損失は、約3%以下であった。
【0287】
乾燥生成物を、0.079インチのガータ型スクリーン(識別番号7L079G03123-(2007)0503)及びステンレス鋼、150粗粒(Ra1.06)表面仕上げを用いて、2500rpmの操作速度でComil Model U5ミル (Quadro Engineering, Inc., Millburn, NJ)に通し、さらなる圧縮のための微粉砕された材料を得た。
【0288】
粒状物を、KG-5 ミキサー/グラニュレーターに戻し、そして250rpmのインペラ速度及び1500rpmのチョッパー速度を混合しながら、2.4ml/分で、171gのEUDRAGIT(商標)RL 30D(Degussaから入手できる、約125,000ドルトン〜約175,000ドルトンの分子量により特徴づけられる、タイプA、アンモニオアルキルメタクリレートコポリマー30%水性分散体)を添加することにより、アンモニオアルキルメタクリレートコポリマー及び賦形剤を含んで成るブレンドにより被覆した。次に粒状物を乾燥した。
【0289】
ステアリン酸マグネシウム(1.5g)(HYQUAL(商標)植物源)を計量し、そして#40メッシュスクリーンに通した。微粉砕された材料及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして25rpmで5分間ブレンドした。
【0290】
ブレンドされた材料を放し、そして圧縮し、250mgの合計重量及び10mg(4重量%)の化合物(1)を有する錠剤を形成した。5/16−インチ直径のIPT標準同心の上下パンチ、及び5/16−インチ(ID)×1.1875ODストレート穴鉄鋼ダイを備えた10ステーション、Mini Press-IIBD (Globepharma, New Brunswick, NJ)を用いて、錠剤を圧縮した。錠剤は、約6kp〜約9kp(59〜88ニュートン)の最終平均硬度を有した。
【0291】
例13.錠剤配合物における化合物(1)の化学的安定性:
温度及び湿度の種々の条件下での化合物(1)の開放皿化学安定性を、SR3-10及びSR4-10錠剤配合物について決定した。錠剤を、温度及び湿度に対して3ヶ月までの間、暴露し、そしてR−バクロフェンの量及びラクタム分解(R−(4−クロロ−フェニル)−ピロリジン−2−オン)を決定した。結果は表10に示される。SR4-10配合物は、3ヶ月での個々の貯蔵条件で一定した低いラクタムレベルにより示されるように、SR3-10配合物に比較して、卓越した化学安定性を示した。
【0292】
【表10】
【0293】
例14.化合物(1)を含んで成る錠剤投与形の投与に続くヒト患者におけるR−バクロフェンの定常状態薬物力学:
健康な成人ボランティアにおける10mgのSR3及び10mg、20mg及び30mgのSR4錠剤配合物の定常状態薬物力学を比較する、ランダム化された、多重投与、4−処理、4−期間クロスオーバー研究を実施した。
【0294】
研究1日目での投与の前、患者を4系列の1つにランダム化した。1日目で、すべての患者は朝食中の10分以内にSR3錠剤として20mg(2×10mg)の用量の化合物(1)を受けた。2日目で、すべての患者は、朝食の完結の後10分以内に、SR3錠剤として30mg(3×10mg)の用量の化合物(1)を受けた。3日目で、すべての患者は、朝食の完結の後10分以内にSR3錠剤として40mg(4×10mg)の用量の化合物を受けた。
【0295】
患者は、期間1(4日目〜7日目)、期間2(8日目〜11日目)、期間3(12日目〜15日目)及び期間4(16日目〜19日目)の間、ランダム化された態様で次の4種の処理の1つを受けた:
【0296】
処理A:朝食の完結後10分以内に1日1度(QD)、4日間、6×10mgの化合物(1)SR3錠剤;
処理B:朝食の完結後10分以内に1日1度(QD)、4日間、6×10mgの化合物(1)SR4-10錠剤;
処理C:朝食の完結後10分以内に1日1度(QD)、4日間、3×20mgの化合物(1)SR4-20錠剤;及び
処理D:朝食の完結後10分以内に1日1度(QD)、4日間、2×30mgの化合物(1)SR4-30錠剤。
個々の処理グループは、16人の健康な成人ボランティアを含んだ。
【0297】
血液サンプル(約4ml)を、投与の前、及びK2EDTAを含む管中への投与後、一定間隔で患者から集めた。血液サンプルアリコートを、メタノールによりすぐに急冷し、化合物(1)のさらなる加水分解を妨げた。2つのアリコート(それぞれ1ml)をすぐに、Nalgene管に移し、そして3mlのメタノールにより急冷した。血液サンプルアリコートを、−80℃で冷凍庫に貯蔵した。血液サンプルアリコートを、敏感で且つ特異的なLC-MS/MS方法を用いて、完全な血液上清液におけるR−バクロフェン及び化合物(1)について分析した。
【0298】
血液中のR−バクロフェン及び化合物(1)についての濃度データを、WINNONLIN(商標) Software バージョン4.1 (Pharsight Corporation, Mountain View, CA)を用いて、非コンパートメント方法により分析した。濃度データ及び薬物力学パラメーターを、SIGMAPLOTTM バージョン9.0 (Systat Software Inc., Point Richmond, CA)を用いてプロットした。実時間点を、薬物力学パラメーターの計算のために使用した。
【0299】
最大の観察される薬物の濃度(Cmax)及びCmaxまでの時間(Tmax)を観察により入手した。見かけ排除半減期(T1/2)を、末端過程における3又はそれ以上の対数−転換されたデータ点の直線回帰法により決定した(In(2)/Kelとして計算され、ここでKelは、対数濃度対時間曲線の末端直線部分の直線回帰により計算される末端排除速度定数である)。直線回帰モデル下の領域が、Windows(登録商標) (SAS Institute, Cary, NC)についてのSASTMバージョン9.1を用いて、AUC対用量及びCmax対用量のために適合された。両モデルにおいては、用量効果が、不等間隔値のための直交多項式係数を用いてパラメーター化された。
【0300】
血液サンプルを、7, 11, 15及び19日目に入手した。期間4に続いて、患者は、4日間にわたって養生法から除外された。研究の間、患者は、約2000kカロリーの毎日の合計カロリー量を有する標準化された臨床食事(脂質からのエネルギーの約30%)を供給された。
4種の処理の間、定常状態で決定される、血液中のR−バクロフェンについての平均薬物力学的パラメーターが表1に要約され、そして薬物力学的パラメーターが図11に示される。3種のSR4配合物についてのAUCss, 24値は、SR3配合物について約1500ng×時/mlのAUCss, 24に比較して、約1750ng×時/ml〜約1850ng×時/mlの範囲であった。
【0301】
例15. 不統一粉末組成物を用いて調製されたSR4錠剤(ベンチスケール):
500gの粉末ブレンドを調製した。最初に、28.6gの化合物(1)中、ヘラにより20−メッシュ篩に通し、そして得られるサイズ分けされた薬物をそばに置いた。次に、105.7 g のAVICEL(商標) PH200、164.5 gのMETHOCELTM K4M 直接圧縮グレード(DC)、85.4 g のEUDRAGIT(商標) RLPO、 5.2 g の二酸化珪素グレード M5P、 及び105.7 g のDI-TAB(商標)としての二塩基性リン酸カルシウム・二水和物を、ヘラを用いて、20−メッシュ篩に連続的に通した。次に、サイズ分けされた賦形剤粉末を、2クォーツのツイン−シェルブレンダーに移し、そして25回転/分の回転速度で5分間タンブル混合した。
【0302】
233gの得られるサイズ分けされ、そして混合された賦形剤粉末を除いた。次に、サイズ分けされた化合物(1)を、ミキサーにおける半分の粉末層上に均一の厚さの層として広げた。233gの除去された粉末を、ブレンダーのシェルに戻し、そして薄層組成物を25rpmで5分間ブレンドした。次に、5.2gのステアリン酸マグネシウムを、60メッシュの篩にヘラにより通し、そして粉末層に添加した。最終的に、粉末の層を、25rpmで4分間タンブル混合した。これは、不統一粉末ブレンド形成した。
【0303】
得られる不統一粉末ブレンドを、5/16インチの丸型標準凹形型押しを備えた2つのステーションを有するKorsch XL 100錠剤プレス(Korsch America Inc., South Easton, MA)のホッパーに移した。粉末を、個々の錠剤が10mgの呼称含有量の化合物(1)を含むよう、175mgの呼称重量を有する錠剤に圧縮した。開始で及び圧縮工程の間、錠剤のサンプルを集めた。得られる錠剤サンプルを、高性能液体クロマトグラフィーにより化合物(1)の含有率について分析した。さらに、錠剤サンプルを、イオンクロマトグラフィーを用いて、DI-TAB(商標)含有率について分析した。
【0304】
得られる錠剤含有率データは図12にプロットされている。図12Aは、錠剤化実施の間、錠剤における化合物(1)の含有率を示す。個々のデータ点は、錠剤化実施の間、特定点でサンプリングされ、そして完全な錠剤化実施(100%)の%として表される10.0mgの化合物(1)の標的量に対する、3個の錠剤における化合物(1)の平均含有率を表す。個々のデータ点についての誤差バーは、個々の時点での3種の測定される値の相対的偏差を表す。化合物(1)含有率データへの直線回帰適合の傾斜は、+0.121であった。その実施を通して化合物(1)の含有率の相対的標準偏差は6.34%であった。図12Bは、同じ錠剤化実施の間、個々の錠剤内のDI−TAB(商標)の含有率を示す。DI-TAB(商標)含有率データに対する直線回帰適合の傾斜は、-0.099であった。その実施を通してのDI-TAB(商標)含有率の相対的標準偏差は、5.60%であった。
【0305】
それらのヒストグラムは、錠剤における化合物(1)の含有率が錠剤化実施の開始で低いことを示す。さらに、錠剤内化合物(1)の含有率は、錠剤化実施の間、上昇する。化合物(1)の含有率はその実施の最後で高いことはまた明白である。さらに、錠剤内のDI-TAB(商標)の含有率は、反対の傾向に従う。DI-TAB(商標)の含有率はその実施の開始で高く、そして錠剤化実施を通して着実に低下する。
【0306】
この現象は、それらの錠剤を調製するために使用される粉末ブレンドの不統一性質及びブレンドを含んで成る主要成分の嵩密度により説明される。この不統一ブレンド内の主要成分の嵩密度値が表11に要約されている。
【0307】
【表11】
【0308】
DI-TAG(商標)の密度は、いずれかの他の成分の密度を越える。それは、ブレンド内のいずれか他の成分の密度の2倍以上である。従って、不統一粉末ブレンド内で、そのブレンドの単純な取り扱い及び加工が、ブレンドの底への重力下での密なDI-TAB(商標)成分の沈降を引起すのに十分な振動を誘発する。そのようにする際、低い密度の成分、例えばバクロフェンプロドラッグ化合物(1)が、ブレンドの上部に置換される。ブレンドが錠剤プレスのホッパーに供給される場合、比較的高い画分のDI-TAB(商標)及び比較的低い画分の化合物(1)を含む、不統一粉末層の最低部分がまず、錠剤に転換される。従って、実施の開始での錠剤は、低い薬分含有率及び高いDI-TAB(商標)含有率を有する。対照的に、錠剤化実施の最後での錠剤は、比較的高い薬物含有率及び比較的低いDI-TAB(商標)含有率を有する。従って、図12におけるデータの直接回帰分析は、化合物(1)の含有率を表す曲線の正の傾斜、及びDI-TAB(商標)の含有率を表す曲線について類似する大きさの対応する負の傾斜に影響を及ぼす。
【0309】
例16.整構造の粉末組成物を用いて調製されるSR4錠剤(ベンチスケール):
整構造の粉末ブレンドを調製した。最初に、33.0gのバクロフェンプロドラッグ化合物(1)、5.94gの二酸化珪素M5P、及び122.1gの DI-TAB(商標)を、ポリエチレンバッグに配置し、そして2分間タンブル混合した。次に、予備混合された粉末を、円錐状ミル(Quadro Comil Model U5, Quadro Engineering Corp., Waterloo, Ontario, Canada)に通した。その円錐上ミルは、457ミクロンの直径を有する丸型メッシュ開口部を有するスクリーンを固定され、そして3000rpmのインペラ速度で作動した。
【0310】
この微紛化工程は、バクロフェンプロドラッグ化合物(1)により被覆され、そして整構造の粉末ブレンドを調製するために使用されるDI-TAB(商標)粒子をもたらした。
【0311】
整構造の粉末ブレンドの調製を続けるために、172.8gのMETHOCELTM K4M DCを、20メッシュの篩に通し、そして2−クォーツの双シェルブレンダー中に充填した。次に、146.4gの円錐状ミル粉砕された化合物(1)/DI-TAB(商標)/二酸化珪素混合物を、ヒドロキシプロピルメチルセルロース上に均等な厚い層として双シェル混合物中に充填した。次に、89.7 g of EUDRAGIT(商標)RLPO 及び 111.0 g のAVICEL(商標)PH200を、20メッシュの篩を通してサイズ分けし、そして双シェルミキサー中に充填した。次に、組成物を25rpmで10分間タンブル混合した。次に、100gのその混合物を除いた。5.4gのステアリン酸マグネシウムを、40メッシュの篩に通し、そして100gのブレンド中にヘラにより撹拌した。最終的に、ステアリン酸マグネシウムを含むサンプル100gをブレンドに添加し、そして全バッチを25rpmで4分間タンブル混合した。これは整構造の粉末ブレンドを形成した。
【0312】
得られる整構造の粉末ブレンドを、Korsch XL100錠剤プレスのホッパーに移した。プレスは、5/16インチの丸型標準凹型パンチ及び2ステーションで設定されるダイを適合された。ブレンドを、10mgの化合物(1)のユニット用量を供給するために、175mgの呼称標的重量を用いて錠剤に圧縮した。錠剤サンプルを、例15に記載される方法に従って、集め、そして分析した。得られるデータは、図13に示される。図13は、全錠剤化実施(100%)の間、一定間隔で、化合物(1)の標的含有率(±SD)に標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰適合の傾斜は、+0.028であった。バッチを通しての化合物(1)の含有率の相対的標準偏差は1.00%であった。
【0313】
図12における不統一ブレンドの含有率結果と図13における整構造ブレンドの結果との比較は、整構造粉末ブレンドにより提供される化合物(1)含有率均質性において有意な改良性を示す。整構造ブレンドから製造された錠剤中の化合物(1)含有率についての直線回帰の傾斜は、+0.121の不統一ブレンドから製造された錠剤中の化合物(1)含有率についての傾斜に比較して、+0.028である。当業者は、傾斜がゼロに近いほど、バッチを通しての平均含有率がより均等になることを理解しているであろう。同様に、整構造粉末ブレンドから製造された錠剤についての含有率の相対的標準偏差(RSD)は、1.00%であった。これは、6.34%の相対的標準偏差を有した不統一ブレンドから製造された錠剤についてよりも、錠剤内の化合物(1)含有率における、より良好な含有率均一性を示唆する。
【0314】
例17.不統一粉末ブレンドを用いて調製されたSR4錠剤(パイロット製造スケール):
次の工程を用いて、6kgの不統一粉末ブレンドを調製した。最初に、0.340kgの化合物(1)を、1インチ当たり20のワイヤのメッシュを有する篩を通してスクリーンした。次に、1.258 kg のAVICEL(商標)PH200、0.061 kgのコロイド状二酸化珪素、 1.958 kgのMETHOCELTM K4M (DC)、1.258 kgのDI-TAB(商標)、及び 1.017 kgのEUDRAGIT(商標)RLPOを、20メッシュ篩を備えたFinex電気シフター(Russell Finex Inc.)に通した。サイズ分けされた賦形剤を、1立方フィートのV−ブレンダー中に充填し、そして25rpmの回転速度で5分間タンブル混合した。得られる賦形剤ブレンドの半分を除いた。
【0315】
次に、サイズ分けされたプロドラッグ化合物(1)を、前記半分の粉末層上に積層した。残りの半分の賦形剤層を化合物(1)上に積層した。次に、得られる3層組成物を、25rpmの回転速度で5分間V−ブレンダーにおいてタンブル混合した。ステアリン酸マグネシウム(0.061kg)を、1インチ当たり40のワイヤを有するメッシュに通し、そして次に、粉末層に添加した。得られる組成物を、25rpmで4分間タンブル混合した。これは、高い剪断混合を伴わないで調製されたSR4ブレンドを形成した。
【0316】
得られる6kgのブレンドを、5/16インチの標準凹型丸形パンチ及びダイの9ステーションを備えたKilian T100回転錠剤プレス(IMA Kilian GmbH & Co. KG, Koeln, DE)に供給した。約175mgの重量及び約7kNの硬度を有する錠剤を、25rpmの速度で圧縮した。個々の錠剤は、10mgの呼称含有量の化合物(1)を含んだ。錠剤サンプルを、約37,000個の錠剤ごとに集めた。化合物(1)の含有率を、個々のサンプリング点で3個の錠剤において高性能液体クロマトグラフィーを用いて決定した。
【0317】
得られるヒストグラムが図14に示されている。図14は、合計の錠剤化実施(100%)の間、一定間隔で化合物(1)の標的含有率(±SD)に対して標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰適合の傾斜は、+0.051であった。約340,000個の錠剤のバッチを通しての化合物(1)含有率の相対的標準偏差は、4.94%であった。
【0318】
例18.整構造粉末ブレンドを用いて調製されたSR4錠剤(パイロット製造スケール):
次の工程を用いて、6kgの整構造粉末ブレンドを調製した。A−TAB(商標)の粒子を、次の方法を用いて、化合物(1)により被覆した。最初に、0.600kgのA−TAB(商標)を、8クオートのV−ブレンダーに充填した。次に、0.350kgの化合物(1)を、A-TAB(商標)上に積層した。0.695kgの追加のA-TAB(商標)を、化合物(1)上に1つの層として適用した。次に、3層組成物を、25rpmで4分間、ブレンダーによりタンブル混合した。得られる混合物を、813ミクロンの直径を有する丸形開口部を有するスクリーン、長方形のインペラー及び0.225インチのインペラースペーサーを備えたQuadro Comil Model 197円錐形ミルに通した。インペラーは、2000rpmの速度で回転した。混合物を、1分当たり約0.3kgの速度でミルにすくい取り、A−TAB(商標)により被覆された化合物(1)を生成した。
【0319】
次に、1.000 kg のMETHOCELTM K4M DC及び 0.063 kg の二酸化珪素を、8クォーツのV−ブレンダーに添加し、そして25rpmで2分間、混合した。得られるヒドロキシプロピルメチルセルロース(HPMC)混合物を、A−TAB(商標)により被覆された化合物(1)を調製するために使用されたのと同じパラメーターを用いて、円錐形ミルに通した。
【0320】
HPMC混合物を、1 ft3 Loedige 高-剪断ブレンダー(Gebrueder Loedige Maschinenbau GmbH, Paderborn, DE)に移した。次に化合物(1)を含む混合物を、HPMC混合物上に1つの層として適用した。次に、1.309 kgのAVICEL(商標)PH200、 及び1.046 kgの EUDRAGIT(商標)RLPOを、ブレンダーに層として添加した。得られる4層混合物を、高い剪断で5分間ブレンドした。
【0321】
得られる混合物を、1ft3のV−ブレンダーに移した。40メッシュの篩を前もって通されたステアリン酸マグネシウム(0.046kg)を混合物に添加し、そして25rpmで4分間ブレンドした。その混合物をドラムに放し、整構造粉末ブレンドの形成を完成した。
【0322】
得られる整構造粉末ブレンドを、フィードフレーム中、5/16標準凹型丸形パンチ及びダイ及び2−櫂フィダーの9ステーションを備えたkilian T100を用いて、錠剤に圧縮した。プレスの砲塔は 25rpmで回転し、そして櫂供給装置は6rpmで作動した。ブレンドをプレスのホッパーにすくい取った。175mgの呼称標的重量及び10.0mgの呼称標的化合物(1)含有量を有する錠剤を圧縮した。錠剤の硬度は約7kpであり、そして錠剤の厚さは約3.6mmであった。錠剤の破砕性は0.3%であった。
【0323】
錠剤サンプルを、圧縮の実施の間、約3,375個の錠剤の間隔で集めた。3個の錠剤を、集められた間隔の個々で化合物(1)含有率について分析した。得られるデータを、図15に表されるヒストグラムにプロットした。図15は、合計の錠剤化実施(100%)の間、一定間隔での化合物(1)の標的含有率(±SD)に対して標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰の傾斜は+0.033であり、そして相対的標準偏差は1.95%であった。
【0324】
例18に記載される整構造粉末ブレンドから製造された錠剤の均一性に比較して、例17に記載されるような不統一の粉末ブレンドにより製造された錠剤における含有物均一性が表12に要約されている。ヒストグラムの傾斜がゼロに近づくほど、バッチの開始から最後までの薬物含有率はより均一である。相対的標準偏差が小さいほど、バッチ内の薬物含有率はより均一である。整構造粉末ブレンドから製造された錠剤は、両測定によれば、化合物(1)の有意に良好な均一性を示す。
【0325】
円錐形ミルへの化合物(1)及びA−TAB(商標)のブレンドの通過は、化合物(1)により被覆されたA−TAB(商標)コアー粒子を生成した。それらの被覆された粒子は、ブレンドの他の成分に付着し、A−TAB(商標)の分離を低めた三次元構造体を形成した。高い剪断ブレンドはさらに、粒子サイズを低め、そしてブレンドを均質化した。
【0326】
【表12】
【0327】
例19.整構造粉末ブレンドを用いて調製されたSR4錠剤の溶解:
錠剤を、丸形標準凹形装置を備えたKorsch XL100錠剤プレスを用いて、10kN〜20kNの圧力で調製した。10mg、20mg、30mg及び40mgの化合物(1)を含むSR4錠剤の組成が表13に要約されている。SR4錠剤についての溶解プロフィールが表14に要約されている。
【0328】
【表13】
【0329】
【表14】
【0330】
最終的に、本明細書に開示される態様を実現するための他の手段が存在することは注目されるべきである。従って、本発明の態様は、例示的であって、制限的ではない。さらに、請求項は本明細書に与えられる詳細に制限されるものではなく、そしてそれらの十分な範囲及びその同等物に権利が与えられる。
【図1A】
【図1B】
【図1C】
【図2A】
【図2B】
【図2C】
【技術分野】
【0001】
本開示により提供される方法は、(R)−バクロフェンプロドラッグの持効性放出経口投与形に関する。
【背景技術】
【0002】
下記式(1):
【化1】
【0003】
で表される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸(1)、すなわちGABABアゴニスト、R−バクロフェン((±)−4−アミノ−3−(4−クロロフェニル)ブタン酸)のプロドラッグは、経口投与されるか、又は哺乳類の結腸中に直接投与される場合、R−バクロフェンとして高い生物学的利用能を示す(Gallop et al., US 7,109,239 and US 7,227,028; 及び Lal et al., J Pharmacology Experimental Therapeutics 2009, 330(3), 911-921)。
【0004】
化合物(1)の投与に続く高いR−バクロフェンの経口生物学的利用能は、経口投与形、例えば持効性経口投与形での化合物(1)の使用、及び疾病、例えば痙攣及び胃食道逆流病の処理のための(van Herwaarden et al., Aliment. Pharmacol. Ther. 2002, 16(9), 1655-62; Ciccaglione and Marzio, Gut 2003, 52(4), 464-70; Andrews et al., US 6,117,908;及び Fara et al., WO 02/096404号);禁煙の促進への(Gessa et al., WO 01/08675号);麻酔剤の中毒傾向の低減への(Robson et al., US 4,126,684号);嘔吐の処理への(Bountra et al., US 5,719,185号);咳嗽の処理のための鎮咳剤として(Kreutner et al., US 5,006,560号);及び神経障害及び筋骨格痛(Benson et al., US 2009/0118365号)、尿失禁(Wun and Wustrow,アメリカ仮出願 番号61/309,336号, March 1, 2010に出願された)、運動障害、例えば失調症及びしゃっくり;末梢神経障害、例えば筋肉刺激障害;脊髄障害、例えば痙攣性対性不全麻痺;脳神経障害、例えば舌咽頭神経痛及び三叉神経痛;多発性硬化症;及び脳性麻痺の処理のためへのそのような経口投与形の使用を好む。
【0005】
化合物(1)の合成は、Gallop et al., US 7,109,239号; Gallop et al., US 7,227,028号; Gallop et al., US 2009/0192325号; Raillard et al., アメリカ出願番号2/537,798号 、August 7, 2009年出願された; 及びRaillard et al., アメリカ出願番号12/537,764 合、August 7, 2009年出願された(それらの個々は引用により本明細書に組込まれる)により記載されている。
【0006】
化合物(1)を含んで成る経口投与は、Kidney et al., US 2008/0206332号; 及びSastry et al., US 2009/0197958号に開示されている。Kidneyなどは、高剪断湿式造粒法を用いて調製された、化合物(1)及び放出速度−制御ポリマーを含んで成る持効性錠剤投与形を示す。Sastryなどは、化合物(1)を含んで成る持効性粒状投与形を開示する。
【0007】
直接的圧縮錠剤製造工程においては、錠剤成分が乾式ブレンドにより組み合わされ、そして得られる環式ブレンドが続いて回転錠剤プレス上でパンチ工具により錠剤に組合される。化合物(1)のこれまでのマトリックス錠剤配合物は、自由に流動する粒状物を形成するために、高剪断湿式造粒工程の間、有機溶媒、例えばアルコールの使用を要した(Kidney et al., US 2008/0206332号)。自由に流動する粒状物は、回転錠剤プレス上で許容できる重量制御を維持することを製造工程において必要とした。高剪断湿式造粒工程に使用される溶媒は続いて、高温での乾燥により許容できるレベルに除去されるべきであり、これは製造費用を高め、そして製造速度を遅くする。
【発明の概要】
【0008】
従って、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の経口投与形を調製するための改良された方法は有用である。
【0009】
乾燥粉末から調製される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸を含んで成る経口錠剤投与形、及びそのような投与形の調製方法は開示されている。乾燥処理は、化学的分離を引起すことができる、水又は溶媒への薬物の暴露を排除する。さらに、乾燥処理は、室温で実施され得る。この良好な温度条件は、薬物が、典型的には湿式造粒化工程の間、導入される残留溶媒を除去するために使用される高温に長時間、暴露される場合に発生する熱分解を最小にするか又は妨げることができる。
【0010】
第1の観点においては、経口錠剤投与形の合計重量に基づいて、約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る経口錠剤投与形が開示される。
【0011】
第2の観点においては、経口錠剤投与形の合計重量に基づいて、約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る混合物を乾式ブレンドし;そして前記ブレンドされた混合物を圧縮し、経口錠剤投与形を供給することを含んで成る経口錠剤投与形の調製方法が開示される。
【0012】
第3の観点においては、リン酸ニカルシウム及び(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩をブレンドし、第1ブレンドを供給し;前記第1ブレンドを円錐型ミルに通し;ヒドロキシプロピルメチルセルロース及びコロイド状ニ酸化珪素を乾燥ブレンドし、第2部位レンドを形成し;前記第2ブレンドを円錐形ミルに通し;前記第1ブレンド、前記第2ブレンド、微晶性セルロース、及び放出速度−制御ポリマーを、高剪断ブレンダーにおいてブレンドし、第3ブレンドを形成し;前記第3ブレンドと共にステアリン酸マグネシウムをブレンドし;そして前記第3ブレンドを圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法が開示される。
【0013】
第4の観点においては、少なくとも1つの本開示により供給される経口投与形を、処理の必要な患者に経口投与することを含んで成る、痙攣、胃食道逆流病、嘔吐、咳嗽、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、神経因性疼痛、及び筋骨格痛から選択された、患者における疾病の処理方法が開示される。
【図面の簡単な説明】
【0014】
当業者は、本明細書に記載される図面が単なる例示目的のためであることを理解しているであろう。図面は本発明の開示の範囲を制限するものではない。
【図1】図1は、500倍率での化合物(1)の次の3種のロットの走査電子顕微鏡(SEM)マイクログラフを示す:ロット4(1A);ロット70(JB);ロット71(1C)。
【図2】図2は、10,000倍率での化合物(1)の次の3種のロットのSEMマイクログラフを示す:ロット4(2A);ロット70(2B);ロット71(2C)。
【0015】
【図3】図3は異なったロットの化合物(1)を含む錠剤についての溶解プロフィールを示す。
【図4】図4は、異なったグレードのEUDRAGIT(商標)を含む錠剤についての溶解プロフィールを示す。
【図5】図5は、20mgの錠剤の重量均一性を示す。
【図6】図6は、20mgの錠剤の含有物均一性を示す。
【図7】図7は、20mgの錠剤の溶解プロフィールを示す。
【0016】
【図8】図8は、30mgの錠剤の重量均一性を示す。
【図9】図9は、30mgの錠剤の含有物均一性を示す。
【図10】図10は、30mgの錠剤の溶解プロフィールを示す。
【図11】図11は、SR3及びSR4投与形の投与に続いての健康なボランティアの血液における定常状態R−バクロフェン薬物力学プロフィールを示す。
【図12】図12は、不統一性粉末組成物を用いて調製されたSR4錠剤における(A)化合物(1)及び(B)DI-TAB(商標)についての錠剤均一性パラメーターを示す。
【図13】図13は、整構造性粉末組成物を用いて調製されたSR4における化合物(1)についての錠剤均一性パラメーターを示す。
【0017】
【図14】図14は、試験規模での不統一性粉末組成物を用いて調製されたSR4錠剤における化合物(1)についての錠剤均一性パラメーターを示す。
【図15】図15は、試験規模での不統一性粉末組成物を用いて調製されたSR4錠剤における化合物(1)についての錠剤均一性パラメーターを示す。
【発明を実施するための形態】
【0018】
定義:
“薬物副作用”とは、所望しない、不快な、有害で又は潜在的に有害である薬物効果を言及する。薬物副作用は、軽い、中位い又は重度であり得る。軽い薬物副作用の例は、消化不良、頭痛、疲労、はっきりしない筋痛、倦怠感及び睡眠パターンにおける変化を包含するが、但しそれらだけには限定されない。中位の薬物副作用は、それらを経験する個人が厄介で、苦しく又は耐え難く思われる反応、例えば皮疹、視力障害、筋肉の震え、排尿困難、ムード又は精神的機能における知覚可能な変化、及び血液組成における一定の変化を表す。重度の薬物副作用の例は、持続的又は有意な障害又は入院をもたらし、そして先天性異常を引起す、生命の脅威である反応を包含する。バクロフィン療法に関連することが知られている副作用の例は、鎮静、認識的な機能の減失、精神錯乱、記憶喪失、眩暈感、衰弱、失調症、ぼけるか又は複視、嘔気、息切れ、痙攣及び体位性低血圧を包含する。
【0019】
“AUC”とは、患者への化合物の投与に続いて、時間の関数としての血液における化合物又はその代謝物の濃度を示す曲線下の領域である。例えば、投与される化合物は、R−バクロフェンプロドラッグ(1)及びその対応する代謝物R−バクロフェンであり得る。AUCは、種々の間隔で、測定するための標準方法、例えば液体クロマトグラフィー−タンデム質量分光法(LC/MS/MS)を用いて、血液中の化合物又はその代謝物の濃度を測定し、そして血液濃度−対−時間の曲線下での領域を計算することにより決定され得る。濃度−対−時間の曲線はまた、薬物力学プロフィールとしても言及される。
【0020】
薬物濃度−対−時間の曲線からAUCを計算するための適切な方法は、当業界において良く知られている。例えば、R−バクロフェンについてのAUCは、患者へのR−バクロフィンプロドラッグ、例えば化合物(1)の投与に続いて、患者の血液におけるR−バクロフェンの濃度を測定することにより決定され得る。AUC0-24は、投与に続いて、投与から24時間までの曲線下の領域である。AUCss, 24は、一定期間(定常状態)にわたって投与される投与レジメに続いて、24時間にわたっての曲線下の領域である。
【0021】
“生物学的利用能”とは、患者への化合物(1)の投与に続いて患者の全身性循環に達するR−バクロフェンの割合及び量を言及し、そしてR−バクロフェンの血液濃度−対−時間のプロフィールを評価することにより決定され得る。血液濃度−対−時間の曲線を特徴づけることにおいて有用なパラメーターは、曲線下の領域(AUC)、ピーク濃度までの時間(Tmax)及び最大R−バクロフェン濃度(Cmax)を包含し、そこでCmaxは、患者への一定用量の化合物(1)の投与に続いて、患者の血液における薬物の最大濃度であり、そしてTmaxは、患者への一定用量の化合物(1)の投与に続いて、患者の血液におけるR−バクロフェンの最大濃度(Cmax)への時間である。
【0022】
絶対的経口生物学的利用能は、等量の化合物又は代謝物の静脈内投与に続いての生物学的利用能に比較して、経口投与に続いての化合物又はその代謝物の生物学的利用能である。化合物又はその代謝物の相対的経口生物学的利用能は、他の投与形及び/又は投与路での等量の化合物又はその代謝の投与に対する、化合物又はその代謝物の経口投与に続いての生物学的利用能である。例えば、一定の態様においては、%Frelとして表される相対的経口生物学的利用能は、持効性投与形としての20mgの化合物(1)の経口投与に続いてのR−バクロフェンの生物学的利用能に対する、患者への化合物(1)の経口投与に続いてのAUC0-24により決定されるR−バクロフェンの生物学的利用能である。
【0023】
“生物学的同等”とは、患者への等用量のR−バクロフェン又は化合物(1)の投与の後、R−バクロフェンの吸収速度及び程度の同等性を言及する。本明細書において使用される場合、2種の薬物学力プロフィールは、それらの2種のプロフィールの平均応答の比率についての90%信頼区間が0.8及び1.25の限界内にある場合、生物学的同等である。前記平均応答は、プロフィールの特徴的パラメーター、例えばCmax、 Tmax及びAUCの少なくとも1つを包含する。
【0024】
“化合物(1)”は、R−バルコフェンプロドラッグ化合物(1)、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸、医薬的に許容できるその塩、前記のいずれかの医薬的に許容できる溶媒化合物、及び前記のいずれかの結晶形を包含する。化合物(1)は、R−バクロフェンプロドラッグ(1)と交換可能的に使用される。一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、遊離酸である。
【0025】
一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、遊離酸であり、そして結晶性である。一定の態様においては、R−バクロフェンプロドラッグ化合物(1)、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸は、塩酸塩である。IUPAC命名法を用いれば、化合物(1)はまた、(R)-4-クロロ-β-[[[[2-メチル-1-(S)-(2-メチル-1-オキソプロポキシ)プロポキシ]カルボニル]アミノ]メチル]-ベンゼンプロパン酸としても言及される。
【0026】
化合物(1)はまた、いくつかの互変異形、例えばエノール形、ケト形及びそれらの混合物で存在することもできる。従って、本明細書に示される化学構造体は、例示される化合物のすべての可能な互変異形を包含する。本開示の化合物はまた、同位体標識化合物も包含し、ここで1又は複数の原子が、従来天然において見出される原子質量とは異なった原子量を有する。本明細書に記載される化合物中に組込まれ得る同位体の例は、2H、3H、 11C、 13C、14C、15N、18O、17O、などを包含するが、但しそれらだけには限定されない。
【0027】
化合物(1)は、非溶媒和形及び溶媒和形、例えば水和化形で及びN−酸化物として存在することができる。一般的に、本明細書に言及されるような化合物は、塩、遊離酸、水和化された、N−酸化物、又は前記のいずれかの組合せであり得る。化合物(1)は、多重結晶形、共結晶形又は非結晶形で存在することができる。化合物(1)は、医薬的に許容できるその塩又は前記いずれかの遊離酸形の医薬的に許容できる溶媒和化合物、及び前記いずれかの結晶形を包含する。
【0028】
化合物(1)はまた、溶媒和化合物も包含する。溶媒和化合物とは、化学量論量又は非化学量論量で1又は複数の溶媒分子を有する化合物の分子複合体を言及する。そのような溶媒分子は、患者に対して無害であることが知られている、医薬業界において通常使用されるそれら、例えば水、エタノール及び同様のものである。化合物又は化合物の成分及び溶媒の分子複合体は、非共有分子内力、例えば静電力、ファンデルワース力又は結合により安定化され得る。用語“水和物”とは、1又は複数の溶媒分子が水である溶媒化合物を言及する。
【0029】
本開示の“化合物”は、本明細書に開示される式内のいずれかの特定の化合物を包含する。化合物は、それらの化合物構造及び/又は化学名称のいずれかにより同定され得る。化学構造及び化学名称が相反する場合、化学構造は化合物の識別の決定因子である。本明細書に記載される化合物は、1又は複数のキラル中心、及び/又は二重結合を含んで成り、そして、従って、立体異性体、例えば二重結合異性体(すなわち、幾何学的異性体)、鏡像異性体又はジアステレオーマとして存在することができる。従って、特にことわらない限り、相対的形状で完全に又は部分的に示される、明細書の範囲内のいずれかの化学構造は、例示される化合物のすべての可能性ある鏡像異性体及び立体異性体、例えば立体異性体的に純粋な形(例えば、幾何学的に純粋、鏡像異性体的に純粋又はジアステレオマー的に純粋)、及び鏡像異性体及び立体異性体混合物を包含する。
【0030】
鏡像異性体及び立体異性体混合物は、当業者に良く知られている分離技術法又はキラル合成技法を用いて、それらの成分鏡像異性体又は立体異性体に分解され得る。例えば、鏡像異性体又はジアステレオマーの分解は、例えば従来の方法、例えば分解剤の存在下での結晶化により、又は例えば、キラル高圧液体クロマトグラフィー(HPLC)カラムを用いてのクロマトグラフィーにより達成され得る。
【0031】
“Cmax”は、患者への一定用量の化合物(1)の投与に続いて、患者の血液において観察される最大R−バクロフィン濃度である。Css, maxは、一定期間(定常状態)にわたって投与される投与レジメの間、化合物(1)の投与に続く最大定常状態濃度である。Cass、minは、定常状態での最小濃度である。
【0032】
“C1/2”は、患者への化合物(1)の投与後12時間で患者の血液において観察されるR−バクロフィン濃度である。Css, 12は、一定期間(定常状態)投与される用量レジメの間、化合物(1)の投与後12時間での濃度である。
【0033】
“Tmax”は、患者への一定用量の化合物(1)の投与に続いて患者の血液におけるR−バクロフィンの最大濃度(Cmax)までの時間である。Tss, maxは一定期間(定常状態)にわたって投与される用量レジメの間、化合物(1)の投与に続いての最大濃度までの時間である。 “T1/2”は、患者の血液中のR−バクロフィン濃度が最大薬物濃度の半分に低下した時間とTmaxとの間の時間間隔である。Tss, 1/2は、一定期間(定常状態)にわたって投与される用量レジメの間、化合物(1)の投与に続いて、患者の血液中のR−バックフェン濃度が最大薬物濃度の半分に低下した時間とTmaxとの間の時間間隔である。
【0034】
“投与形”とは、治療効果を達成するために患者に投与され得る。一定量の活性剤又は活性剤のプロドラッグ、すなわちR−バクロフィンプロドラッグ(1)を含む配合物の形を言及する。経口投与形は、口を通して患者に投与されるか又は飲み込まれることが意図される。一定用量の薬物は、同時に又は一定期間にわたって投与される1又は複数の投与形を含むことができる。
【0035】
“絶食された患者”とは、一定用量が患者に投与される時間で及び投与後少なくとも4時間、胃が食物を実質的に有さない患者を言及する。患者の胃が食事に続いて実質的に食物を有さなくなる時間は、多くの因子、例えば食事のサイズ、例えばカロリー数、食事の内容物、例えば、脂肪含有率、患者の健康性、及び患者の胃腸管の状態に依存することができる。健康なヒト対象の胃は、食物の摂取に続いて、約4〜約8時間後、典型的に実質的に食物を有さない。ある態様においては、絶食された患者は、投与の前約10時間、投与の後約4時間まで、いずれの食物を食べず(しかし、いずれかの量の水又は透明な液体を摂取することができる)、投与の前約2時間及び約1時間、約250mlの水、及び投与の後約2時間、約250mlの水を飲み、投与後約4時間、昼食を取り、そして投与後約10時間で夕食を取る。
【0036】
“食事した患者”とは、胃が食物を含む患者を言及する。ある態様においては、食事した患者は、投与の前約30分で試験食事を食べ始め、そして投与の前約5分で試験食事を完結し、投与の後4時間で昼食を食べ、そして投与の後約10時間で夕食を食べる。試験食事は、高脂肪(試験食事における合計カロリー数の約50%)及び高カロリー(約1000合計カロリー)の朝食、例えばバターであげられた2個の卵、2枚のベーコン、バターを塗られた2枚の小麦トースト、4オンスのハッシュブラウンポテト、及び8オンスの全乳を含むことができる。試験食事は、いずれかのカロリー数を含むことができ、そしていくつかの態様においては、約150カロリーのタンパク質、250カロリーの炭水化物及び約500〜600カロリーの脂肪を含む。
【0037】
“最小悪影響濃度”とは、許容できない薬物副作用を生成しない、患者の血液又は血漿における治療化合物の最小濃度を言及する。薬物副作用の受容不可能性は例えば、患者に投与される化合物の治療有益性の観点から、薬物副作用及び/又は知覚される危険性の重症度に少なくとも一部基づいて、患者及び/又は処方する医者により決定され得る。最小悪影響濃度はまた、処理される患者の年齢、体重及び健康性、症状の頻度及び重症度、及び処方する医者の判断に少なくとも一部、依存する。
【0038】
“最小治療有効濃度”とは、意図される治療効果を生成する患者の血液又は血漿における治療化合物の最小濃度を言及する。
“患者”とは、哺乳類、例えばヒトを包含する。
“医薬的に許容できる”とは、U.S.薬局方に列挙されるか、又は哺乳類、例えばヒトへの使用のために他の一般的に認識される薬局方に列挙される、連邦又は州政府の規制局により許可されるか又は許可できることを言及する。
【0039】
“医薬的に許容できる塩”とは、1つの化合物、例えば医薬的に許容でき、そして本発明の化合物の所望する薬理学的活性を有する化合物(1)の塩を言及する。そのような塩は、次のものを包含するが、但しそれらだけには限定されない:(a)無機酸、例えば塩酸、臭酸、硫酸、硝酸、燐酸、及び同様のものにより形成されるか、又は有機酸、例えば酢酸、プロピオン酸、 ヘキサノン酸、 シクロペンタンプロピオン酸、グリコール酸、ピルビン酸、乳酸、マロン酸、琥珀酸、 リンゴ酸、マレイン酸、フマル酸、酒石酸、クエン酸、安息香酸、 3-(4-ヒドロキソベンゾイル) 安息香酸、桂皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、 1、2-エタン-二スルホン酸、 2-ヒドロキエタンスルホン酸、 ベンゼンスルホン酸、 4-クロロベンゼンスルホン酸、 2-ナフタレンスルホン酸、 4-トルエンスルホン酸、樟脳スルホン酸、 4-メチルビシクロ[2.2.2]-オクト-2-エン-1-カルボン酸、 グルコヘプトン酸、 3-フェニルプロピオン酸、 トリメチル酢酸、 第三ブチル酢酸、ラウリル硫酸、グルコン酸、 グルタミン酸、 ヒドロキシナフトエ酸、サリチル酸、ステアリン酸、ムコン酸、 及び同様のものにより形成される酸付加塩;及び(b)本発明の化合物に存在する酸性プロトンが金属イオン、例えばアルカリ金属イオン、アルカリ土類金属イオン又はアルミニウムイオンにより置換されるか;又は有機塩基、例えばエタノールアミン、ジエタノールアミン、トリエタノールアミン、N−メチルグルカミン及び同様のものと調和する場合に形成される塩。ある態様においては、化合物(1)の塩は、塩酸塩、及びある態様においては、ナトリウム塩である。
【0040】
“医薬的に許容できるビークル”又は“医薬的に許容できる賦形剤”とは、医薬的に許容できる希釈剤、医薬的に許容できるアジュバント、医薬的に許容できる賦形剤、医薬的に許容できるビークル、医薬的に許容できるキャリヤー、又は前記いずれかの組合せを言及し、それらと共に、その薬理学的活性を破壊せず、そして治療的有効量の化合物、例えばR−バクロフェンプロドラッグ又はR−バクロフェン代謝物を供給するのに十分な用量で投与される場合、非毒性である、化合物、例えばR−バクロフェンプロドラッグ、すなわち(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸(1)が投与され得る。
【0041】
“医薬組成物”とは、R−バクロフェンプロドラッグ(1)又は医薬的に許容できるその塩、及び少なくとも1つの医薬的に許容できるビークル(これと共に、プロドラッグが患者に投与される)を含んで成る組成物を言及する。
【0042】
“プロドラッグ”とは、活性薬物を開放するために、使用条件下で、例えば身体内で転換を受ける活性化合物の誘導体を言及する。プロドラッグは時折、但し必ずしも必要ではないが、活性薬物に転換されるまで、薬理学的不活性である。プロドラッグは、典型的には官能基を通して薬物にプロ成分(本明細書において定義される)を結合することにより得られる。例えば、R−バクロフェンプロドラッグ(1)は、親薬物R−バクロフェンを供給するために患者の身体内で代謝される。
【0043】
“プロ成分”とは、1つの薬物に、典型的には使用の特定条件下で分解できる結合を通して、薬物の官能基に結合される基を言及する。薬物とプロ成分との間の結合は、酵素又は非酵素手段により分解され得る。使用の条件下で、例えば患者への投与に続いて、薬物とプロ成分との結合は、親薬物を開放するために分解され得る。プロ成分の分解は、例えば加水分解反応を通して、自発的に進行するか、又はそれは他の剤、例えば酵素、光、酸又は物理的又は環境パラメーターの変化又はそれらへの暴露、例えば温度、pH、等の変化により触媒されるか又は誘発され得る。前記剤は、使用の条件、例えばプロドラッグが投与される患者の全身性循環に存在する酵素、又は胃の酸性条件に対して内因性であるか、又は前記剤は外因的に供給され得る。例えば、R−バクロフェンプロドラッグ(1)に関しては、薬物はR−バクロフェンであり、そしてプロ成分は下記構造を有する:
【0044】
【化2】
【0045】
“鎮静”とは、本明細書において使用される場合、最小鎮静及び/又は中位の鎮静を言及する(例えば、American Society of Anesthesiologists, Anesthesiology 2002, 96, 1004-17を参照のこと)。抗不安剤としても言及される最小鎮静は、気道を独立して及び連続的に維持する患者の能力、及び薬理学的又は非薬理学的方法又はその組合せにより生成される物理的刺激又は口頭の指令に対して適切に応答する患者の能力を維持する、最小に低められたレベルの意識である。認識機能及び同調はひかえめに損なわれるが、呼吸及び心血管機能は影響されない。目的が成人における最小鎮静である場合、適切な用量は、モニターされていない自家使用のために処方され得る最大の推薦される用量、例えば最大の推薦される治療用量に過ぎない。
【0046】
中位の鎮静は、患者が単独で又は光触知刺激により達成される、口頭の指令に対して意図的に応答する間、意識の薬物−誘発された低下である。患者の気道を維持するためには介入は必要とされない。鎮静は連続的であり、そして個々の患者がいかに応答するかを予測することは常に可能であるわけではない。鎮静用量は、所望する効果が達成されるまで、薬物、例えばR−バクロフェンプロドラッグ(1)の複数用量を投与するインクリメンタル投与により決定され得る。種々の目盛り、例えばラムゼー目盛り及び他のものが、鎮静を評価するために使用され得る。鎮静の客観的処理は、脳波図パラメーター、例えばBispectral IndexバージョンXP及びPatient State Analyzerの測定を包含する。ある態様においては、鎮静は、最小鎮静、及びある態様においては、中位の鎮静を言及する。
【0047】
“持効性”とは、化合物の直接的な配合物の経口投与により達成される時間に比較して、延長された時間にわたって全身性血液循環において化合物、又はその活性代謝物の治療量を達成するのに効果的な速度で、投与形からの化合物の放出を言及する。いくつかの態様においては、化合物のインビボ放出は、少なくとも約4時間にわたって、いくつかの態様においては、少なくとも約8時間にわたって、いくつかの態様においては、少なくとも約12時間にわたって、いくつかの態様においては、少なくとも約16時間にわたって、いくつかの態様においては、少なくとも約20時間にわたって、及びいくつかの態様においては、少なくとも約24時間にわたって、生じる。
【0048】
ある態様においては、本発明の開示により供給される持効性投与形は、特定速度、例えば75rpmで回転するパドル(USP, タイプII)により撹拌される、pH6.8及び37℃での50mMのリン酸ナトリウム一塩基性緩衝液において、約18時間以内に投与形における約45%〜約55%の化合物(1);約18時間以内に約40〜約45%の化合物(1);及び約18時間以内に約34%〜約44%の化合物(1)を放出する。
【0049】
“治療的行有効量”とは、疾病又は障害、又は疾病又は障害の臨床学的症状の少なくとも1つを処理するために対象に投与される場合、前記疾病、障害又は症状のそのような処理に影響を及ぼすために十分である化合物の量を言及する。治療的有効量は、例えば化合物、疾病、障害、及び/又は疾病の症状、疾病又は障害の重症度、及び/又は疾病又は障害の症状、処理される患者の年齢、体重及び/又は健康、及び処方する医者の判断に依存して変化することができる。治療的有効量は、当業者に確認され得るか、又は通常の実験により決定され得る。
【0050】
いずれかの疾病の“処理する(treating)”又は“処理(treatment)”とは、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つを阻止するか又は改善し、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの獲得の危険性を低め、疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの進行を遅くし、又は疾病、又は疾病又は障害の臨床学的症状の少なくとも1つの進行の危険性を低めることを言及する。“処理する”又は“処理”はまた、疾病を、物理的に(例えば、認識できる症状の安定化)、生理学的に(例えば、物理的パラメーターの安定化)、又は両者的に阻害し、そして患者に認識できるか又はできない少なくとも1つの物理的パラメーターを阻害することを言及する。ある態様においては、“処理する”又は“処理”とは、疾病、又は患者が疾病をまだ経験していないか又はその症状をまだ示していない場合でさえ、疾病に暴露されるか又は疾病の傾向を有する患者における疾病又は1又は複数のその症状の開始を遅延することである。
【0051】
投与形及び方法のある態様が詳細に言及される。開示される態様は請求項を制限するものではない。対照的に、請求項はすべての変更、修飾及び同等のことを包含する。
【0052】
1つの観点いおいては、本発明の開示は、次の構造:
【化3】
【0053】
を有する化合物(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸に関する。
化合物(1)は、R−バクロフェンのプロドラッグである。
【0054】
組成物:
もう1つの観点においては、本発明の開示は、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸及び医薬的に許容できる賦形剤を含んで成る持効性経口投与形を供給する。化合物(1)は、Gallop et al., アメリカ特許第7,109,239号; Gallop et al., アメリカ特許第7,227,028号; Gallop et al., US 2009/0192325号; Raillard et al., 2009年8月7日に出願されたアメリカ特許出願番号12/537,798号;及び/又は Raillard et al., 2009年8月7日に出願されたアメリカ特許出願番号12/537,764号(それらの全内容物は引用により本明細書に組込まれる)により記載される方法を用いて調製され得る。
【0055】
もう1つの観点においては、化合物(1)の結晶化は、溶媒の組成及び血漿化速度に少なくとも一部、依存して、異なった形態学を有する生成物を供給する。異なった結晶化速度でアセトン/ヘキサンの混合物から結晶化された化合物(1)の異なった形態学のいくつかが、図1及び2に示されている。中間速度の結晶化で得られる形態学(図1A及び2A)は、化合物(1)の初晶の凝集体を含んで成るものとして特徴づけられ得、それらの凝集体は主に丸くされ、そして約25〜約ミクロンの直径を伴って圧縮し、そして前記初晶は数ミクロン以下の寸法を有する。
【0056】
遅い速度の結晶化で得られる態様学(図1B及び2B)は、約100ミクロンの全体的寸法及び約100μmの長さの線状物を有する、ゆるい線維状の又は線状の塊状物を含んで成るものとして特徴づけられる。早い速度の結晶化で得られる化合物(1)の形態学(図1C及び2C)は、初晶の不規則的に形状化された凝集体により特徴づけられ、ここで前記凝集体は約25〜約50ミクロンの寸法(長さ/幅)を有し、そして初晶は数ミクロン以下の寸法を有する。
【0057】
もう1つの観点においては、化合物(1)の他に、本発明の開示により供給される持効性経口投与形は、医薬的に許容できる賦形剤、例えば微晶性セルロース、ヒドロキシプロピルメチルセルロース、放出速度制御のポリマー、二塩基性リン酸カルシウム二水和物、コロイド状ニ酸化珪素及びステアリン酸マグネシウムを含むことができる。
【0058】
もう1つの観点においては、化合物(1)の他に、本発明の開示により供給される持効性経口投与形は、医薬的に許容できる賦形剤、例えば微晶性セルロース、ヒドロキシプロピルメチルセルロース、放出速度制御のポリマー、二塩基性無水リン酸カルシウム、コロイド状二酸化珪素及びステアリン酸マグネシウムを含むことができる。
【0059】
ある態様においては、本発明の開示により供給される投与形に使用される微晶性セルロースは、約180ミクロンの呼称粒子サイズ、約2〜約5%の湿分、及び約0.29g/cc〜約0.36g/ccのゆるい嵩密度により特徴づけられ、そして例えばAVICEL(商標) PH200 (AVICEL(商標), FMC Biopolymer)を包含する。ある態様においては、微晶性セルロース又は、約12mmのFlodexを示す。
【0060】
ある態様においては、本発明の開示により供給される投与形に使用されるヒドロキシプロピルメチルセルロースは、19〜24%のメトキシル含有率、7〜12%のヒドロキシプロピル含有率及び2%水溶液における3,000〜5,600cPの粘度を有するヒプロメロース2208、例えばMETHOCELTM K4M SP (標準のプレミアム) or METHOCELTM K4M CR (制御された放出)であり得る。ある態様においては、本発明の開示により供給される投与形に使用されるヒドロキシプロピルメチルセルロースは、19〜24%のメトキシル含有率、7〜12%のヒドロキシプロピル含有率、ヒプロメロース2208置換型、2,663〜4,970cPの粘度、0.1〜0.2g/ccの嵩密度、及び最大約5%の湿分を有し、そして例えばMETHOCELTM K4M DC(直接的圧縮)を包含する。ある態様においては、ヒドロキシプロピルメチルセルロースは、約28mm〜約30mmのFlodexを示す。
【0061】
ある態様においては、本発明の開示により供給される投与形に使用される放出速度制御ポリマーは、エチルアクリレート、メチルメタクリレート、及び第四アンモニウム基を有する低含有率のメタクリル酸エステルのコポリマー、例えばトリメチルアンモニオエチルメタクリレートクロライドであり得る。ある態様においては、前記コポリマーは、約150,000ダルトンの平均分子量を有する。ある態様においては、放出速度制御コポリマーは、乾燥物質に基づいて約8.9〜約12.3%のアンモニオメタクリレート単位を含み、そしてある態様においては、EUDRAGIT(商標) RLPO (Evonik Industries AG, Darmstadt, DE)であり得る。ある態様においては、放出速度制御コポリマーは、乾燥物質に基づいて約4.5〜約7.0%のアンモニオメタクリレート単位を含み、そしてある態様においては、EUDRAGIT(商標) RSPO (Evonik Industries AG, Darmstadt, DE)であり得る。ある態様においては、放出速度制御ポリマーは約22mmのFlodexを示す。
【0062】
ある態様においては、本発明の開示により供給される投与形に使用される二塩基性リン酸カルシウム二水和物は、粉砕されていないDI−TAB(商標)であり得る。ある態様においては、二塩基性リン酸カルシウム二水和物は、4mm以下か又はそれに等しいFlodexを示す。
【0063】
ある態様においては、本発明の開示により供給される投与形に使用される二塩基性無水リン酸カルシウムは、粉砕されていないA-TAB(商標)であり得る。ある態様においては、二塩基性無水リン酸カルシウムは、4mm以下か又はそれに等しいFlodexを示す。
【0064】
ある態様においては、本発明の開示により供給される投与形に使用されるコロイド状二酸化珪素又は未処理の非晶性ヒュームドシリカは、CAB-O-SILTM M-5P (Cabot Corporation, Bilerica, MA)であり得る。
【0065】
ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブラン酸又は医薬的に許容できるその塩;約15重量%〜約40重量%の微晶性セルロース;約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成るを含んで成る。ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約5重量%〜約15重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約17重量%〜約33重量%の微晶性セルロース;約20重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び約5重量%〜約20重量%の放出速度−制御ポリマーを含んで成る。
【0066】
ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約8重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約17重量%〜約33重量%の微晶性セルロース;約21重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び約5重量%〜約22重量%の放出速度−制御ポリマーを含んで成る。ある態様においては、持効性経口投与形は、経口錠剤投与形の合計重量に基づいて;約5重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約18重量%〜約22重量%の微晶性セルロース;約33重量%のヒドロキシプロピルメチルセルロース;及び約17重量%の放出速度−制御ポリマーを含んで成る。
【0067】
ある態様においては、持効性投与形は、希釈剤、充填剤及び滑剤から選択された、1又は複数の医薬的に許容できる賦形剤を含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約23重量%〜約33重量%の二塩基性リン酸カルシウム二水和物;約0.1重量%〜約2重量%のコロイド状二酸化珪素;及び約0.1重量%〜約2重量%のステアリン酸マグネシウムを含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約19重量%〜約22重量%の二塩基性リン酸カルシウム二水和物;約1重量%のコロイド状二酸化珪素;及び約1重量%のステアリン酸マグネシウムを含んで成る。
【0068】
ある態様においては、本発明の開示により供給される持効性経口投与形は、約23重量%〜約33重量%の二塩基性無水リン酸カルシウム;約0.1重量%〜約2重量%のコロイド状二酸化珪素;及び約0.1重量%〜約2重量%のステアリン酸マグネシウムを含んで成る。ある態様においては、本発明の開示により供給される持効性経口投与形は、約19重量%〜約22重量%の二塩基性無水リン酸カルシウム;約1重量%のコロイド状二酸化珪素;及び約1重量%のステアリン酸マグネシウムを含んで成る。
【0069】
ある態様においては、微晶性セルロースはAVICEL(商標) PH200であり、ヒドロキシプロピルメチルセルロースはMETHOCELTM K4M DC 及びMETHOCELTM K4M CRから選択され、放出速度制御ポリマーはEUDRAGIT(商標) RLPOである。
【0070】
本発明の開示により供給される持効性経口投与形は、約24〜26mmのFlodexを有する(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;約12mmのFlodexを有する微晶性セルロース;約28〜20mmのFlodexを有するヒドロキシプロピルメチルセルロース;及び約22mmのFlodexを有する放出速度制御ポリマーを含んで成る。
【0071】
ある態様においては、本発明の開示により供給される錠剤投与形は、約5重量%〜約12重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸;約18重量%〜約32重量%のAVICEL(商標) PH200; 約22重量%〜約33重量%の METHOCELTM K4M DC; 約重量%〜約18重量%のEUDRAGIT(商標) RLPO; 約19重量%〜約28重量%のA-TAB(商標);約0.7重量%〜約1.5重量%のステアリン酸マグネシウム;及び約0.9重量%〜約1.1重量%のコロイド状二酸化珪素を含んで成り;そして錠剤投与形はそれぞれ、約150mg〜約400mg;及びある態様においては、約50mg〜約600mgの重量である。
【0072】
ある態様においては、本発明の開示により供給される投与形は、約100mg〜約600mgの合計重量を有する。
【0073】
化合物及びブレンドの特徴化:
種々の観点においては、乾燥粉末ブレンドからの錠剤製造を促進するためには、組合された化合物(1)及び医薬的に許容される賦形剤から形成される乾燥粉末が許容できる流動性質を示すことが所望される。乾燥粉末の流れは、多くのパラメーター、例えば粒度、粒度分布、粒子形状、粒子粗さ、、嵩密度、多孔度、粉末を通しての空気透過性、静電荷、湿度、沈殿効果及び粘着力、例えばLondon分散力及び水素結合力により影響され得る。
【0074】
本発明者は、賦形剤のある組合せ又はブレンドが、個々の賦形剤の紛体流性質により予測されない、直接的圧縮錠剤製造のために有用な改良された粉体流性質を思いがけなく提供することを見出した。粉体流に対する予測されない相乗効果が市販の錠剤操作において有用なブレンドを提供する。さらに、本発明者は、親水性アクリレートポリマーが、同じタイプの疎水性ポリマーからより親水性マトリックス錠剤からの化合物(1)の放出を遅め、そして制御することにおいてより効果的であり得ることを見出した。
【0075】
乾燥粉末ブレンドの流動性質は、Fledexにより特徴づけられ得る。低いFlodexを示す乾燥粉末ブレンドは一般的に例えば製造速度、錠剤重量均一性、薬物含有均一性、硬度均一性、錠剤外観、及び薬物放出プロフィールにおいて影響されるような錠剤製造工程を受けやすい。約22mm又はそれ以下、又は約15mm又はそれ以下のFlodexを示す乾燥粉末ブレンドが有用である。
【0076】
ある態様においては、化合物(1)は、約22mm〜約28mmのFlodexを示す。
ある態様においては、投与形に使用される微晶性セルロースは、約10mmへの約14mmのFlodexを示す。
ある態様においては、投与形に使用されるヒドロキシプロピルメチルセルロースは、約26mmへの約32mmのFlodexを示す。
【0077】
ある態様においては、投与形に使用される放出速度−制御ポリマーは、約20mmへの約24mmのFlodexを示す。
ある態様においては、投与形に使用されるニ塩基性リン酸カルシウム二水和物は、約4mm以下のFlodexを示す。
ある態様においては、投与形に使用されるニ塩基性無水リン酸カルシウムは、約4mm以下のFlodexを示す。
【0078】
製造:
種々の観点においては、本発明の開示により供給される持効性経口投与形は錠剤として供給され得る。本発明の開示により供給される配合物は一般的に、直接的な圧縮により経口錠剤投与形を形成することにおいて有用である。
【0079】
ある態様においては、投与形は、化合物(1)を含んで成る錠剤形で存在することができる。錠剤投与形は、薬物の経口投与のために適切ないずれかの形状、例えば球状、立法体状、卵形又は楕円形状のものであり得る。ある態様においては、本発明の開示により供給される錠剤投与形、例えば錠剤形での経口投与は、R−バクロフェンプロドラッグ(1)が少なくとも1つの放出速度変性化合物を含んで成るマトリックスに分散されているマトリックスシステムである。マトリックスシステムは、例えば“Handbook of Pharmaceutical Controlled Release Technology,” ed. Wise, Marcel Dekker, Inc. (2000) 及び“Treatise on Controlled Drug Delivery, Fundamentals, Optimization, and Applications,” ed. Kydonieus, Marcel Dekker, Inc. (1992)に記載されるように、当業界において良く知られている。
【0080】
ある態様においては、本発明の開示により供給される投与形における化合物(1)の量は、約0.1mg〜約200mg;ある態様においては、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;及びある態様においては、約5mg〜約50mgの範囲である。化合物(1)の医薬的に許容できる塩及び/又は溶媒化合物を含んでいる投与形に関しては、投与形における化合物(1)の量は、その塩及び/又は水和物を含んで成る化合物(1)の等質量を言及する。ある態様においては、錠剤投与形は、治療的有効量の化合物(1)を含んで成る。
【0081】
治療的有効量の化合物(1)は、約1mg−当量〜約100mg−当量のR−バクロフィン;約2mg−当量〜約80mg−当量のR−バクロフィン;約2mg−当量〜約40mg−当量のR−バクロフィン;又は約5mg−当量〜約20mg−当量のR−バクロフィン;を含んで成ることができる。1mgの化合物(1)は、約0.535mg−当量のR−バクロフェンを含んで成る。ある態様においては、本発明の開示により供給される投与形における化合物(1)の量は、患者において中位の鎮静及び運動活動の障害を引起す量以下である。ある態様においては、化合物(1)の治療的有効量は、患者において中位の鎮静及び運動活動の障害を引起す量以下である。
【0082】
錠剤投与形が治療的有効量以下の化合物(1)を含んで成る、ある態様においては、多重錠剤投与形が、治療的有効量の化合物(1)を供給するために、同時に又は一定期間にわたって患者に投与され得る。
【0083】
本明細書に開示される化合物(1)及び放出速度変性化合物の他に、錠剤投与形はまた、1又は複数の医薬的に許容できる賦形剤、例えば界面活性剤、滑剤、可塑剤、結合剤、希釈剤、 抗−付着剤、滑沢剤、 緩衝剤、 顔料、 湿潤剤、 乳化剤、 pH 緩衝剤、安定化剤、増粘剤、崩壊剤、 風味剤、 味覚隠蔽剤及び着色剤を含んで成る。
【0084】
希釈剤又は充填剤は、投与形を圧縮のための粒度にするために嵩密度を高めるために添加され得る。本発明の開示により供給される錠剤投与形において有用な希釈剤の例は次のものを包含する:二塩基性無水リン酸カルシウム、二塩基性リン酸カルシウム二水和物、硫酸カルシウム、 リン酸ニカルシウム、リン酸三カルシウム、ラクトース、セルロース、例えば 微晶性セルロース、カオリン、マンニトール、塩化ナトリウム、 乾燥澱粉、アルファ澱粉、圧縮性糖、 及び前記いずれかの組合せ。ある態様においては、希釈剤は二塩基性リン酸カルシウム及び微晶性セルロースから選択される。充填剤は水不溶性、水溶性、又はその組合せであり得る。
【0085】
有用な水不溶性充填剤の例は、次のものを包含する:二酸化珪素、二酸化チタン、タルク、アルミナ、澱粉、カオリン、ポラクリリンカリウム、粉末セルロース、微晶性セルロース、ヒュームドシリカ、モノステアリン酸グリセリン、ステアリン酸マグネシウム、ステアリン酸カルシウム、コロイド状シリカ、微粒シリカ、三珪酸マグネシウム、石膏、及び前記いずれかの組合せ。
【0086】
有用な水溶性充填剤の例は、水溶性糖アルコール、例えば、ラクトース、グルコース、フルクトース、 スクロース、マンノース、 デキストロース、 ガラクトース、 対応する糖アルコール及び 他の糖アルコール、例えばマンニトール、ソルビトール、キシリトール、及び前記いずれかの組合せを包含する。希釈剤が微晶性セルロースである態様においては、錠剤投与形は、約15重量%〜約35重量%、及びある態様においては、約18重量%〜約20重量%の範囲の量の希釈剤を含んで成ることができる。ある態様においては、希釈剤又は充填剤は、二塩基性無水リン酸カルシウム及びある態様においては、二塩基性リン酸カルシウム二水和物である。
【0087】
錠剤化滑剤は、加工、フィルム形成及び/又は乾燥の間、粘着効果を低めるために、本発明の開示により供給される投与形に含まれ得る。有用な滑剤の例は、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸、グリセロールモノステアレート、及び前記いずれかの組合せを包含する。
【0088】
滑沢剤は、粉体流を改良するために、本発明の開示により供給される投与形に含まれ得る。有用な滑沢剤の例は、タルク、コロイド状二酸化珪素、沈殿された二酸化珪素、 ヒュームド二酸化珪素、及び前記いずれかの組合せを包含する。ある態様におおいては、滑沢剤は、コロイド状二酸化珪素である。錠剤投与形は、約2重量%以下の滑沢剤、及びある態様においては、約1重量%以下の滑沢剤を含むことができる。ある態様においては、滑沢剤はコロイド状二酸化珪素である。
【0089】
結合剤は、構成成分の付着を促進するために投与形に包含され得る。本発明の開示により供給される錠剤投与形において有用な結合剤の例は、ポリビニルアセテートフタレート、モラッセ、メチルセルロース、ヒドロキシプロピルセルロース(HPC)、ヒドロキシプロピルメチルセルロース(HPMC)、ナトリウムカルボキシメチルセルロース、微晶性セルロース(HCC)及びポリビニルピロリドンを包含する、本発明の開示により供給されるある態様においては、結合剤は、微晶性セルロース、例えばAVICEL(商標) PH200 (FMC Corporation)である。
【0090】
可塑剤は、本発明の開示により供給される錠剤投与形に含まれ得る。本発明の開示により供給される錠剤投与形において有用な可塑剤の例は次のものを包含する:クエン酸アルキル、例えばクエン酸トリエチル、クエン酸アセチルトリエチル、 クエン酸トリブチル、クエン酸アセチルトリエチル、 及びクエン酸アセチルトリブチル; スクロース脂肪酸エステル;グリセリンモノ-、ジ- 及びトリ-脂肪酸エステル、 例えばトリアセチン、 グリセリンモノ-脂肪酸エステル、グリセリンモノステアレート及びアセチル化されたモノグリセリド; ポリグリセリン脂肪酸エステル; ポリエチレングリコール、例えばマクロゴール400、マクロゴール600、マクロゴール1500、マクロゴール4000、マクロゴール6000、マクロゴール20、000、及びマクロゴール35、000; セバシン酸ジブチル; トリブチルセバケート; ビニールピロリドン; プロピレングリコール; 胡麻油; ひまし油; グリセリン; シリコーン樹脂; D-ソルビトール; フィトステロール;アルキルフタレート、例えばフタル酸ジエチル、フタル酸ジブチル及びジオクチルフタレート; アジペートポリエステル; イソプロピルミリステート; 中鎖トリグリセリド; ブチルフタリルブチルグリコレート; ポリオキシエチレンポリオキシプロピレングリコール; 及び前記いずれかの組合せ。
【0091】
錠剤投与形は、約0.1重量%〜約10重量%、約1重量%〜約8重量%、及びある態様においては、約2重量%〜約6重量%の範囲の量の可塑剤を含んで成る。本発明の開示により供給される投与形のある態様においては、投与形は、約2重量%〜約6重量%の可塑剤を含んで成り、前記可塑剤はトリエチルシトレート及びアセチルトリエチルシトレートから選択される。
【0092】
滑剤及び抗−付着剤は、加工目的のために本発明の開示により供給される錠剤投与形に含まれ得る。本発明の開示により供給される錠剤投与形において有用な滑剤及び/又は抗−付着剤の例は、次のも斧を包含する:ステアリン酸カルシウム、グリセリルベヘネート、モノステアリン酸グリセリン、ステアリン酸マグネシウム、鉱油、ポリエチレングリコール、ナトリウムステアリルフマレート、ラウリル硫酸ナトリウム、ドデシル硫酸ナトリウム、ステアリン酸、 タルク、 水素化された植物油、ステアリン酸亜鉛、 及び前記いずれかの組合せ。ある態様においては、滑剤は、グリセリルモノステアレートである。ある態様においては、滑剤は、ステアリン酸マグネシウムである。ある態様においては、錠剤投与形は、約0.1重量%〜約5重量%、ある態様においては、約0.1重量%〜約1重量%及びある態様においては、約1重量%の量の滑剤はステアリン酸をマグネシウムである。
【0093】
本発明の開示により供給される錠剤投与形において有用な界面活性剤の例は、次のものを包含する:医薬的に許容できるアニオン性界面活性剤、カチオン性 界面活性剤、双性イオン性、両性(親媒性/両親媒性) 界面活性剤、 非イオン性界面活性剤、ポリエチレングリコールエステル又はエーテル、及び前記いずれかの組合せ。
【0094】
有用な医薬的に許容できるアニオン性界面活性剤の例は、一価のアルキルカルボキシレート、アシルラクチレート、アルキルエーテルカルボキシレート、N−アシルサルコシネート、多価のアルキルカーボネート、N−アシルグルタメート、脂肪酸−ポリペプチド縮合物、硫酸エステル、アルキルスルフェート、例えばラウリル硫酸ナトリウム及びドデシル硫酸ナトリウム、エトキシル化されたアルキルスルフェート、エステル結合されたスルホネート、例えばドキュセートナトリウム及びジオクチルナトリウムスクシネート、α−オレフィン性スルホネート、又はリン酸化されたエトキシル化されたアルコールを包含する。
【0095】
有用な医薬的に許容できるカチオン性界面活性剤の例は、モノアルキル四級アンモニウム塩、ジアルキル四級アンモニア化合物、アミドアミン及びアミンイミドを包含する。有用な医薬的に許容できる両性界面活性剤の例は、N−置換されたアルキルアミド、N−アルキルベタイン、スルホベタイン及びN−アルキル−6−アミノプロピオネートを包含する。
【0096】
有用な医薬的に許容できる非イオン性界面活性剤の例は、ポリエチレンオキシド、ポリプロピレンオキシド、ポリオキシエチレン(20)ソルビタンモノオレエート、及びポリエチレングリコールエステル又はエーテル、例えばポリエトキシル化されたヒマシ油、ポリエトキシル化された水素化ヒマシ油及び水素化ヒマシ油の二ブロック及び三ブロックコポリマーを包含する。ある態様においては、界面活性剤は、ラウリル硫酸ナトリウム及びドデシル硫酸ナトリウムから選択される。ある態様においては、錠剤投与形は、約3重量%以下の界面活性剤、及びある態様においては、約2重量%以下の界面活性剤を含んで成る。
【0097】
崩壊剤は、水に暴露される場合、崩壊剤の膨潤により錠剤の分解を引起すよう錠剤配合物に含まれ得る。有用な崩壊剤の例は、水膨張性物質、例えば低−置換されたヒドロキシプロピルセルロース、架橋されたナトリウムカルボキシメチルセルロース (ナトリウムクロスカルメロース)、ナトリウム澱粉グリコレート、ナトリウムカルボキシメチルセルロース、澱粉グリコール酸ナトリウム、イオン交換樹脂、微結晶セルロース、架橋されたポリビニルピロリドン、澱粉及びアルファ澱粉、ホルマリン−カゼイン、アルギン酸、 一定の複合シリケート、及び前記いずれかの組合せを包含する。
【0098】
本発明の開示により供給される錠剤投与形はさらに、錠剤を部分的に又は完全に被覆することができる1又は複数の被膜を含んで残ることができる。一定の被膜が胃腸管における錠剤投与形からの化合物(1)の放出を変性するか又は影響を及ぼすよう適用され得るが、他はそのような効果を有することはできない。例えば、1又は複数の追加の被膜が錠剤の物質的保護、エステ、容易な飲み込みのために、及び/又は追加の加工を促進するために存在することができる。
【0099】
被膜は、湿気に対して不透過性であるか、又は湿気透過性であり得る。湿気不透過性外部錠剤被膜は、乾燥剤の存在下で包装される投与形において低水分率を維持するために有用であり得、そしてそれにより、例えば錠剤投与形の貯蔵安定性を増強することができる。物理的保護のための被膜において有用な材料の例は、透過性又は溶解材料、例えばヒドロキシプロピルメチルセルロース、ヒドロキシプロピルセルロース、ラクトース、ヒドロキシプロピルエチルセルロース、ヒドロキシエチルセルロース及びキサンタンガムを包含する。
【0100】
追加の加工を促進するための外部錠剤被膜において有用な材料の例は、タルク、コロイド状シリカ、ポリビニルアルコール、二酸化チタン、微小化されたシリカ、ヒュームドシリカ、グリセロールモノステアレート、マグネシウムトリシリケート及びステアリン酸マグネシウムを包含する。外部錠剤被膜はさらに、1又は複数のビークル、例えば可塑剤、結合剤、充填剤、圧縮助剤及び前記いずれかの組合せを含む。前記1又は複数の追加の被膜は、単一の材料、又は本明細書に開示されるそれらの材料のいずれかを包含する1つよりも多くの材料の組合せを含んで成る。それらの追加の被膜は、当業者に知られている方法により錠剤投与形に適用され得る。
【0101】
ある態様においては、本発明の開示による供給される投与形は、化合物(1)及び/又はR−バクロフェンの分子内環化により形成されるラクタム副生成物を実質的に有さない。投与形は、実質的なラクタム形成、例えば約0.5重量%以下のラクタム、約0.2重量%以下ンラクタム、又は約0.1重量%以下のラクタムの形成を伴わないで、例えば1年以上の延長した貯蔵に対して安定することができる。
【0102】
投与形の溶解プロフィール:
化合物(1)を含んで成る、本発明の開示により供給される投与形の放出特徴は、インビトロ溶解プロフィールにより一部特徴づけられ得る。投与形の溶解プロフィールを決定するための方法は、医薬業界の人々に良く知られている。米国薬局方に示される標準方法論が使用され得る。例えば、溶解プロフィールは、米国薬局方タイプI装置(バスケット)又は米国薬局方タイプII装置(パドル)のいずれかにおいて測定され得る。
【0103】
後者の方法を用いる場合、ある態様においては、本発明の開示により供給される投与形の溶解又は放出プロフィールは、37℃の温度で、pH6.8で50mMの一塩基性リン酸ナトリウム緩衝液(NaH2PO4)に投与形を含浸することにより決定され得る。溶解媒体は75rpmでパドルにより撹拌される(USP, タイプII)。サンプルが、一定間隔で溶解媒体から取り出され、そして溶解媒体における化合物(1)及び/又はR−バクロフェンの含有率が逆相高圧液体クロマトグラフィー(HPLC)を用いて決定される。
【0104】
ある態様においては、本発明の開示により供給される錠剤投与形からの化合物(1)の放出は、75rpmで撹拌される(USP, タイプII)、pH6.8及び37℃での50mMの一塩基性リン酸ナトリウム緩衝液におけるインビトロ溶解プロフィールを示し、ここで約10%〜約30%の化合物が約4時間以内に放出され;約20%〜約50%の化合物が約8時間以内に放出され;約30%〜約65%の化合物が約12時間以内に放出され;約40%〜約80%の化合物が約18時間以内に放出される。
【0105】
ある態様においては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約10%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0106】
約15%〜約35%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約20%〜約50%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約30%〜約80%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0107】
ある態様のおいては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約10%〜約20%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0108】
約20%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約25%〜約45%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約35%〜約55%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0109】
そのような態様のある態様においては、前記放出プロフィールのいずれかを示す錠剤投与形は、約300mg又は約300mgの重量であり、そしてそれぞれ、例9又は10に記載のようにして調製される。
【0110】
ある態様においては、前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, TypeIII )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:約15.5%〜約21.5%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
【0111】
約26%〜約32%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;約35%〜約41%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして約46%〜約51%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される。
【0112】
そのような態様のある態様においては、前記放出プロフィールを示す錠剤投与形は、前記方法のいずれかにより調製され、そして構成された粉末ブレンドから調製される錠剤を記載する、例16, 18及び/又は19に記載される組成を含む。
【0113】
ある態様においては、錠剤投与形は、例19、表14に記載されるプロフィールのいずれかに類似する放出プロフィールを示す。
【0114】
“Dissolution Testing of Immediate Release Solid Oral Dosage Forms - Guidance for Industry”, FDA-CDER, August 1997と一致して、溶解プロフィールは、差異因子(f1)及び類似性因子(f2)に基づいて類似すると思われる。類似すると思われる溶解プロフィールに関しては、f1値は0に接近し、そしてf2値は100に近づくべきである。一般的に15(0〜15)までのf2値及び50以上(50〜100)のf2値は、2種の溶解プロフィールの同等性を保証する。f1及びf2を計算するための方法は、前記引例に示されている。ある態様においては、本発明の開示により供給される経口錠剤投与形は、前記溶解プロフィールのいずれか1つと、又は表12又は14に記載される溶解プロフィールのいずれかと比較される場合、15以下のf1差異因子及び50〜100のf2類似性因子を生成する溶解プロフィールを示す。
【0115】
市販の許容できる錠剤は、USP Test No. 1216に従って決定される場合、約1重量%以下の破砕性を有することが一般的に認められる。ある態様においては、本発明の開示により供給される錠剤は、約1重量%以下、ある態様においては、約0.5重量%以下、ある態様においては、約0.3重量%以下、及びある態様においては、約0.2重量%以下の破砕性を有する。
【0116】
薬物力学及びインビボ放出プロフィール:
化合物(1)を含んで成る持効性投与形は、R−バクロフェン及び/又はラセミ体の等用量形で投与される場合、R−バクロフェンの経口生物学的利用能に比較してR−バクロフェンとしての増強された経口生物学的利用能を示す。化合物(1)の増強された経口生物学的利用能は、受動的及び/又は活性輸送機能を通して、結腸を包含する胃腸管じゅうでの化合物(1)の効果的吸収のためであると思われる。本発明の開示により供給される投与形は、胃腸管を通しての投与形の通過の間、投与形からの化合物(1)の放出を提供する。
【0117】
患者、すなわち本発明の開示により供給される投与形を飲み込む患者に経口投与される場合、その投与形は、連続期間、患者の血液にR−バクロフェンの持続した治療的有効濃度を提供する。ある態様においては、投与形は、最小の治療的有効濃度以上であり、そして患者の血液におけるR−バクロフェンの最小影響濃度以下である患者の血液におけるR−バクロフェンの濃度を提供する。ある態様においては、本発明の開示により供給される投与形は、R−バクロフェンの最小悪影響濃度を越えないで、連続した期間、患者の血液において治療的有効濃度のR−バクロフェンを提供する。ある態様においては、患者の血液におけるR−バクロフェンの濃度は、投与形が患者に経口投与された後、いずれの時点でも最小悪影響濃度を越えない。
【0118】
本発明の開示により供給される投与形は、例えばR−バクロフェンを含んで成る経口投与形の投与に続いて観察される、最小悪影響濃度以上の濃度で、高血液濃度のR−バクロフェンに関連する悪影響薬物効果を低めるか又は排除しながら、連続した期間、患者の血液において治療的有効濃度のR−バクロフェンを提供することができる。化合物(1)を含んで成る投与形を用いて達成できるR−バクロフェンの高い生物学的利用能は、R−バクロフェンを含んで成る経口投与形におけるR−バクロフェンの量に比較して、患者の血液においてR−バクロフェンの持続した治療的有効濃度を達成するためへの一定用量での低等価質量のR−バクロフェンの使用を促進することができる。
【0119】
本発明の開示により供給される持効性投与形は、経口投与に続いて、患者の血液に持続した治療的有効濃度のR−バクロフェンを提供することができる。例えば、ある態様においては、投与形は、患者への経口投与の後、少なくとも約4時間、少なくとも約8時間、少なくとも約12時間、少なくとも約16時間、少なくとも約20時間、又は少なくとも約24時間から選択された連続した期間、患者の血液において持続した治療的有効濃度のR−バクロフェンを提供することができる。ある態様においては、、患者の血液中のR−バクロフェンの濃度は、投与形が患者に経口投与された後、いずれの時間でも最小悪影響濃度を越えないであろう、例えば患者において悪影響を及ぼす濃度に達しないであろう。
【0120】
ある態様においては、患者の血液中のR−バクロフェンの治療的有効濃度は、約50ng/ml〜約1,000ng/ml、及びある態様においては、約100ng/ml〜約500ng/mlの範囲であり得る。血液R−バクロフェン濃度の薬物力学的プロフィールは、類似するR−バクロフェン血液AUCを提供する、R−バクロフェンを含んで成る即時放出及び持効性経口配合物に比較して、より低いCmax/C12比及びより低いCmax/用量により特徴づけられ得る。
【0121】
ある態様においては、本発明の開示により供給される経口投与形の反復された1日1度(QD)の投与は、表1に示されるように、血液中のR−バクロフェンの定常状態濃度を提供する。ある態様においては、16人の健康な成人ボランティアへの本発明の開示により経口投与は、約202±56ng/mlのCss, max;約3.9±1.0時間のTss, max;約63 ng/mLのC12;約8〜約15のCss,max/Css,12;約19ng/mlのCss, 12;約10.9±3.8時間のTss, 1/2;及び1803±420ng・時/mlのAUCss, 24により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールを提供する。ある態様においては、本発明の開示により供給される投与形は、約8〜約15のCss,max/Css,12を提供する。
【0122】
表1.SR3錠剤配合物としての60mg(6×10mg)の化合物(1);又はSR4錠剤配合物としての60mg(6×10mg)、60mg(3×20mg)又は60mg(2×30mg)の化合物(1)の健康な成人ボランティアへの4日間、毎日1度(QD)の経口投与の後、定常状態で決定された血液中のR−バクロフェンについての平均(SD)薬物力学的パラメーター:
【0123】
【表1】
【0124】
ある態様においては、少なくとも1つの経口投与形は、約5mg〜約140mg、及びある態様においては、約10mg〜約80mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量で人患者に投与される。前記態様のある態様においては、投与される(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量は、患者において中位の鎮静及び運動活動の障害を引起す(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量よりも少ない。
【0125】
本発明の開示により供給される投与形の経口投与を用いる投与レジメが、長期間、最小の治療的有効濃度よりも高く、そして最小の悪影響濃度よりも低い、患者の血液における一定濃度のR−バクロフェンを維持するために開発され得る。ある態様においては、R−バクロフェンの最小の治療的有効濃度は、約1ng/ml〜約200ng/ml、及びある態様においては、約10ng/ml〜約100ng/mlの範囲であり得る。ある態様においては、最小悪影響濃度は、約200ng/ml〜mlの範囲であり得る。ある態様においては、最小悪影響濃度は、約200ng/ml〜約2,000ng/ml, 及びある態様においては、約500ng/ml〜約1,000ng/mlの範囲であり得る。
【0126】
最小治療濃度及び最小悪影響濃度は、多くの因子、例えば処理される疾病、疾病の重症度、意図される臨床学的結果、処理される患者の状態、等に依存するであろう。そのようなレジメは、本発明の開示により供給される1又は複数の投与形の反復した投与を用いることができる。投与の適切な間隔は例えば、投与形における化合物(1)の量、投与形中の組成、投与形からの化合物(1)の放出特徴、処理される疾病、患者の状態、可能性ある悪影響、及び処理医者の判断に依存することができる。投与形レジメは、個々の間隔で同じ投与形又は異なった間隔で異なった投与形の反復された投与を包含する。例えば、毎日2回の投与レジメは、朝での第1の投与形、及び夕方での第2の投与形の投与を包含する。
【0127】
本発明の開示により供給される投与形はさらに、U.S. Food and Drug Administration and discussed in “Guidance for Industry - Bioavailability and Bioequivalence Studies for Orally Administered Drug Products” (2003)により定義されるように、吸収速度及び程度の両者に関して、本明細書に開示される投与形に対して生物学的同等である投与形を包含する。
【0128】
投与量:
R−バクロフェンの持続した全身性濃度を提供する錠剤投与形は、現在、1日当たり3度、投与される即時放出性非プロドラッグ形、すなわち患者のために不便であり、そして患者にとって覚えるのが困難であるレジメに比較して、患者のコンプライアンスを増強するであろうと思われる。さらに、本発明の開示により供給される経口投与形錠剤の使用は、低められた副作用、例えば傾眠状態、衰弱、頭痛、発作、嘔気、嘔吐、低血圧、便秘、精神錯乱、呼吸抑制、不眠、及び頻尿又は尿閉を伴って、増強された効能を提供するであろうと思われる。
【0129】
本明細書に開示される特定の疾病の処理において効果的であろう化合物(1)の量は、疾病の性質に、少なくとも一部、依存し、そして当業界において知られている標準の臨床学的技法により決定され得る。さらに、インビトロ又はインビボアッセイは、最適な投与量範囲の同定を助けるために使用され得る。投与レジメ及び投与間隔はまた、当業者に知られている方法により決定され得る。投与される化合物(1)の量は、他の因子の中でも、処理される対象、対象の体重、疾病の重症者、投与路、及び処方医者の判断に依存することができる。
【0130】
全身性投与に関しては、治療的有効用量は、最初、インビトロアッセイから評価され得る。初期用量はまた、インビボデータ、例えば動物モデルから、当業界において知られている技法を用いて評価され得る。そのような情報は、ヒトにおける有用な用量を、より正確に決定するために使用され得る。当業者は、動物データに基づいて、ヒトへの投与を最適化することができる。
【0131】
化合物(1)の用量は、等モル量又は等価質量のR−バクロフェンを供給するよう調節され得る。用量は、本発明の開示により供給される、複数用量形を含んで成ることができる。ある態様においては、R−バクロフェンの治療的有効用量は一般的に、1日当たり約0.03mg〜約1mg/kg体重である。ある態様においては、毎日の用量は、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;ある態様においては、約5mg〜約60mg;及びある態様においては、約10mg〜約40mgの範囲の等価質量のR−バクロフェンを含んで成ることができる。ある態様においては、化合物(1)の用量は、患者において、中位の鎮静及び運動活動の障害を引起す用量以下である。化合物(1)の用量及び適切な投与間隔は、患者におけるR−バクロフェンの持続した治療的有効濃度を、ある態様においては、最小悪影響濃度を越えないで、維持するよう選択され得る。
【0132】
ある態様においては、本発明の開示により供給される投与形は、1日1度、1日2度、及びある態様においては、1日1度以上の間隔で投与され得る。投与は、単独で、及び他の薬物と組合して提供され、そして疾病の効果的処理のために必要とされる限り、続けることができる。投与は、哺乳類、例えばヒトに、食物を食べた状態又は絶食された状態で投与形を投与することを包含する。
【0133】
用量は、単一投与形で、又は複数の投与形で投与され得る。複数の投与形が使用される場合、その複数投与形の個々内に含まれる化合物(1)の量は同じであっても、又は異なっていても良い。
【0134】
ある態様においては、投与される用量は、毒性用量以下である。本明細書に記載される組成物の毒性は、細胞培養物又は実験運動において標準の医薬方法により、例えばLD50(集団の50%が致死する用量)又はLD100(集団の100%が致死する用量)により決定され得る。毒性効果:治療効果の用量比は、治療指数を示す。それらの細胞培養アッセイ及び動物研究から得られるデータは、ヒトへの使用のために毒性でない投与範囲の配合に使用され得る。化合物(1)の用量は、治療的に有効であり、鎮静用量以下であり、そして/又は毒性をほとんどか又はまったく示さない、血液、血漿又は中枢神経系における一定範囲内の循環濃度であり得る。
【0135】
処理の間、用量及び投与スケジュールは、疾病を処理するために十分な又は定常状態の全身性濃度のR−バクロフェンを提供することができる。ある態様においては、徐々に増大する用量が投与され得る。
【0136】
治療用途:
本発明の開示により供給される持効性経口投与形は、親薬物、すなわちR−バクロフェンが治療的有効であることが知られているか、治療的に有効であると思われるか、又はこの後、治療的有効であることが決定される、いずれかの疾病又は障害を有する患者に投与され得る。R−バクロフェンが処方されており、そして本発明の開示により供給される投与形がまた、効果的である徴候は、痙攣、胃食道逆流病、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、嘔吐、咳嗽、神経因性疼痛、筋骨格痛及び尿失禁を包含する。
【0137】
上記に列挙される疾病の処理における本発明の開示により供給される投与形の適合性は、当業界において記載されている方法により決定され得る。
R−バクロフェン治療の必要な患者に投与される化合物(1)の適切な用量は、等価質量のR−バクロフェン、及び化合物(1)により供給されるR−バクロフェンの増強された経口生物学的利用能に基づいて評価され得る。
【0138】
痙攣:
痙攣は、延伸に対する無意識の速度−依存性の高められた耐性である。痙攣は、上昇する延伸速度による外部的に付加された運動に対して高められた耐性が存在する筋肉緊張過渡により特徴づけられる。痙攣は、誕生の前、間又は後、脳への酸素の欠乏(脳性麻痺);物理的な損傷(脳又は脊髄損傷);脳血管からの出血の遮断(脳卒中); 一定の代謝性疾患; アドレノロイコジストロフィ; フェニルケトン尿症; 神経変性疾病、例えばパーキンソン病及び筋萎縮性側索硬化症;及び神経障害、例えば多発性硬化症により引起され得る。
【0139】
痙攣は、皮質脊髄路に対する損傷に関連し、そして神経学的疾患の通常の合併症である。痙攣が顕著な症状である疾病及び状態は、脳性麻痺、多発性硬化症、脳卒中、頭部及び脊髄損傷、外傷性脳損傷、血液酸素欠乏、 及び神経変性疾病を包含する。痙攣を有する患者は、硬直、無意識の発作及び苦痛の不平をいう。それらの苦痛を伴う発作は、自発的であるか、又は低感覚刺激、例えば患者の接触により引起され得る。
【0140】
痙攣の症状は、高張緊張過度(筋緊張亢進)、クローヌス(一連の急速な筋肉の収縮)、大過度の深部腱反射、痙攣性疾患、シザリング(脚の不本意な交差)、固定接合を有する奇形、硬直、 及び/又は四肢が正常に移動するよう試みることにより引起される疲労を包含する。他の合併症は、尿路感染、慢性便秘、発熱又は他の全身系病気、及び/又は褥瘡を包含する。痙攣の程度は、軽い筋肉硬直から、重度で、苦痛を伴い、且つ不快な筋肉発作まで種々であり得る。
【0141】
痙攣は、他の状態と同時に存在するが、しかし硬直(運動に対する不本意な両方向の非速度依存性耐性)、クローヌス(高張につづく自己持続性振動運動)、ジストニー(ねん回姿勢異常をもたらす不本意な持続性収縮)、アテトーゼ様運動(不本意な不規則的な併合性苦悩動作)、舞踏病(不本意で、突然の、急速で、不規則な、持続しない運動)、バリズム(四肢又は身体の不本意な突進運動)、及び身震い(非自己持続性性の不本意でリズミカルな反復性発振)とは区別される。
【0142】
痙攣は、整形外科的変形、例えば股関節脱臼、拘縮、又は脊柱変形; 毎日の生存活動、例えば衣服の脱着、入浴及びトイレの障害; 移動性の障害、例えば歩くか、回転するか、又は座ることの不能性; ボジショニング困難性及び剪断圧力に続く皮膚の損傷; 疼痛又は異常感覚フィードバック; 高カロリー消費に続く不良な体重増加; 睡眠妨害; 及び/又は機能的独立性の欠乏に続く鬱病を導くことができる。
【0143】
痙攣の処理は、物理的及び作業療法、例えば機能的な基本療法、リハビリテーション、促進作用、例えば神経−発達学的治療、固有受容体神経筋促進法、及び感覚統合;バイオフィードバック;電気的刺激;及び他の方法を包含する。痙攣の処理において有用な経口薬剤は、バクロフェン、ベンゾジアゼピン、例えばジアゼパム、ダントロレンナトリウム;イミダゾリン、例えばクロニジン及びチザニジン;及びガバペンチンを包含する。痙攣の処理においては、有用な鞘内薬剤はバクロフェンを包含する。局部麻酔薬、例えばリドカイン及びキシロカインによる化学脱神経;タイプAポツリヌス毒素及びタイプBボツリス毒素;フェノール及びアルコール注入がまた、痙攣の処理において有用であり得る。痙攣の処理において有用な外科処理は、神経外科、例えば選択的背部神経根切断法;及び整形外科的手術、例えば痙縮解放、腱又は筋肉延伸、腱移動、骨切断、及び関節固定を包含する。
【0144】
哺乳類におけるバクロフェンの主要薬理学的効果は、筋張力の低下であり、そして従って、薬物は時折、痙攣の処理に使用される。
【0145】
痙攣の処理のための本発明の開示により供給される投与形の効能は、痙攣の動物モデルを用いて、及び異なった病因の痙攣の臨床学的に適切な研究において評価され得る。痙攣の動物モデルは知られており、そして(a)痙攣性変異マウス;(b)急性/慢性の脊髄切断されたラット及び急性除脳ラット;(c)ラットにおける一次観察Irwin試験;及び(d)ラット及びマウスにおけるRotard試験を包含する。他の動物モデルは、一時的な脊髄虚血に従ってラットにおいて誘発された痙攣、多発性硬化症のマウスも出るにおける痙攣; 及び脳性麻痺のラットモデルにおける痙攣を包含する。齧歯動物における最大電気ショック痙攣(MES)限界試験が、可能性ある抗痙攣薬剤性質を検出するために敏感である。
【0146】
痙攣の処理のための本発明の開示により供給される投与形の効能は、ニ重盲検プラシーボ−制御された臨床試験を用いて、ヒトにおいて評価され得る。痙攣についての臨床試験結果測定は、Ashworth Scale、改良されたAshworth Scale、筋肉延長反射、間代痙攣の存在、及び有害な刺激に対する反射応答を包含する。痙攣は、当業界において知られている方法、例えば臨床試験、評価尺度、例えばAshworth Scale、改良されたAshworth Scale、発作頻度尺度及び反射評点、生化学的研究、例えばペンダラム試験、電気生理学的研究、例えば筋電図、及び機能的測定、例えばFugl-Meyer Assessment of Sensorimotor Impairment尺度の組合せを用いて評価され得る。他の測定法は、特定の障害、例えば多発性硬化症性痙攣尺度に関連する痙攣を評価するために使用され得る。
【0147】
胃食道逆流病:
胃食道逆流病(GERD)は、食道において異常逆流により生成される慢性症状又は粘膜損傷として定義される。GERDの症状は、胸焼け、食道炎、狭窄症、えん下障害、慢性的な胸の疼痛、咳嗽、 しわがれ声、声の変化、慢性的な耳痛、燃えるような胸痛、嘔気、及び副鼻腔炎を包含する。
【0148】
下部食道括約筋の強い収縮が、食道中への胃内容物の逆流を妨げる主要要因である。一時的な下部食道括約筋弛緩(TLESR)は、正常な対象及びGERDを有する患者における逆流を受ける主要機構である。GABABアゴニスト、例えばR−バクロフェンは、ヒトにおいてTLESRを低めることが示されている(Lidums et al., Gastroenterology 2000, 118(1), 7-13; Vela et al., Aliment Pharmacol Ther 2003, 17(2), 243-51; Ciccaglione and Marzio, Gut 2003, 52(4), 464-70; 及びZhang et al., Gut 2002, 50(1), 19-24)。
【0149】
バクロフェンによるTLESRの頻度の低下は、迷走神経導入、孤束核と迷走神経背側運動核その間の情報移行、及び迷走神経延伸性流出のためであると思われる(Hornby et al., Gastroenterol Clin N Am 2002, 31(4 Suppl), S11-S20)。より特定には、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸、すなわち化合物(1)は、臨床試験において逆流エピソードを低めることが示されている(Gerson et al., Am J Gastroenterol 2009, online publication 29 December 2009; doi: 10.1038/ajg.2009.718)。
【0150】
GERDの処理についての効能は、動物モデルを用いて、及び臨床試験において評価され得る。
【0151】
嘔吐:
悪心、嘔吐及び吐き気は、毒素の吸収に対する基本的なヒトの保護反射及び一定の刺激に対する応答である。悪心は、蒼白又は潮紅、頻拍、及び嘔吐に対する心拍の意識に関連する咽頭又は上胃部の背部における主観的に不快な波状感覚である。発汗、過度の唾液分泌、及び寒さ又は暑さの感覚がまた生じることができる。嘔吐は、腹筋の吸縮、融膜の降下、及び胃噴門の開口により特徴づけられ、口から胃内容物の強力な排除がもたらされる。吐き気は、胃内容物の排除を伴わないで、融膜、及び胸部及び腹筋の筋肉の痙攣性収縮を包含する。嘔吐は、悪心、嘔吐、及び/又は吐き気を言及するために本明細書において使用される。
【0152】
バクロフェンは、モルホリンにより誘発される吐気及び嘔吐を抑制することが示されており、それにより、嘔吐制御経路におけるGABAB受容体の関与を示唆する(Suzuki et al., Neuropharmacology 2005, 49(8), 1121-31)。バクロフェンはまた、動物モデルにおいてニコチン及び運動により誘発される嘔吐を拮抗することが示されている(Chan et al., Eur J Pharmacology 2007, 559(2-3), 196-201)。
【0153】
嘔吐の処理における効能は、適切な動物モデル及び臨床試験を用いて評価され得る。例えば、化学療法剤により誘発される嘔吐の処理における効能は、嘔吐を示す影響、及び異食症、異うっ血、及びラット、マウス又はフェレットにおける低められた食物摂取に基づいて決定され得る。臨床試験いおいては、評価装置、例えばDuke Descriptive Scale、isual Analog Scales、Morrow Assessment of Nausea and Emesis、Rhodes Index of Nausea and Vomiting Form-2、及びFunctional Living Index Emesisが効能を測定するために使用され得る。一般的に、適切に制御された、二重盲検プラシーボ制御される試験が、ヒトにおける効能を評価するために使用され得る。
【0154】
咳嗽:
呼吸気管に位置する咳受容体の活性化により誘発される咳反射は、呼吸気管からの吸入された刺激物及び外来性物質を清浄し、そして粘膜繊毛システムに関連して、呼吸気管からの異常状態下で生成される過剰の気道分泌物を排除することができる。咳は、軽い急性の上部呼吸気管感染、アレルギー、喘息、慢性閉塞性肺疾患、肺癌、胃食道逆流病、後鼻腔滴下、及び心臓又は耳疾患により引起され得る。しかしながら、同定できる原因を有さない慢性非生産性咳は、咳をする患者の数%である。慢性咳は喘息性徴候の増悪、肋骨骨折、息苦しさ、破裂脳動脈瘤腹部筋肉、気胸症、失神、 第2及び第3程度の心臓ブロック、及び意識喪失に関連している。永続的で且つ制御できない咳は、羅病率を導き、そしてそれらの患者の生命の質を重度に障害を与える。
【0155】
咳は、いずれかのタイプの病因学又は病原論の急性及び慢性咳、及び特に、喉頭の感覚神経障害に関連する咳を包含する。
【0156】
バクロフェンの鎮咳効果は良く知られている(Dicpinigaitis and Dobkin, Chest 1997, 111(4), 996-9; Dicpinigaitis and Rauf, Respiration 1998, 65(1), 86-8; Dicpinigaitis et al., J Clin Pharmacol 1998, 38(4), 364-7; 及びKreutner et al., US 5,006,560号及び WO 91/08740号)。
咳の処理における効能は、適切な動物モデル及び臨床試験を用いて評価され得る。
【0157】
物質中毒又は乱用:
臨床試験においては、バクロフェンは、コケイン中毒(Brebner et al., Alcohol 2002, 37(5), 478-84; 及び Haney et al., Neuropsychopharmacology 2006, 31, 1814-21);メトアンフェタミン依存性(Heinzerling et al., Drug Alcohol Depend 2006, 85(3), 177-84);オピオイド依存性(Assadi et al., BMC Psychiatry 2003, November 18, 3(16); 及び Ahmadi-Abhari et al., J Clin Pharm Therapeutics 2001, 26(1), 67-71);アルコール欲求及び摂取(Addolorato et al., Alcohol 2002, 37(5), 504-8; 及びFlannery et al., Alcohol Clin Exp Res 2004, 28(10), 1517-23);ニコチン使用(Markou et al., Ann N.Y. Acad Sci 2004, 1025, 491-503);及び薬物乱用(Cousins et al., Drug Alcohol Dependence 2002, 65(3), 209-20)の処理において効果的であることが示されている。
【0158】
物質中毒及び乱用の処理についての効能は、動物モデルを用いて、及び臨床試験において評価され得る。物質中毒障害の動物モデルは知られている。
【0159】
神経因性疼痛:
神経因性疼痛は、通常、神経組織への直接的な外傷又は損傷の後に生じる感覚入力の異常工程を包含する。神経因性疼痛は、異なった病因学、例えば感染、炎症、 疾病、例えば糖尿病及び多発性硬化症、主要末梢神経に対する損傷又は圧縮、及び化学物質又は照射-誘発された神経損傷により特徴づけられる障害の集積である。新規因性疼痛は典型的には、組織損傷が解決された後、長く持続する。
【0160】
化合物(1)は、神経因性疼痛の処理に使用され得る。ある態様においては、化合物(1)は、神経因性疼痛、例えば帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症の処理に使用され得る。
【0161】
神経因性疼痛の研究についての国際協会(The International Association for the Study of Neuropathic Pain)は、通常の条件下で、中枢神経系への有害情報を伝達する神経系の損傷又は機能不全により特徴づけられる障害として神経因性疼痛を定義する。神経因性疼痛状態の根底にある機構は高く不均等であるが、しかしながら、すべてのタイプの神経因性疼痛は中枢及び/又は末梢神経系における体感覚プロセッシングにおいて神経損傷及び一定の共通する異常を包含することが推定される。神経因性疼痛の可能性ある原因は、物理的損傷、感染及び化学物質暴露を包含する。
【0162】
神経因性疼痛は一般的に、末梢神経系の局部/多病巣性損傷、例えば帯状疱疹後神経痛、末梢神経系の一般化された外傷、 例えば、苦痛な糖尿病性神経障害、 HIV-関連NP、中枢神経系の外傷、 又はより複雑な障害として分類され得る。末梢神経因性疼痛は、外傷及び手術関連の外傷、例えば腕神経叢損傷; 絞扼性神経障害、例えば腰椎圧縮、手根管症候群; 疾病関連の神経障害、例えば糖尿病及びHIV-AIDS; 神経根疾患; 複雑な局所疼痛症候群; 及び/又は神経圧迫か浸潤を導く腫瘍増殖の結果として生じる。中枢神経因性疼痛は、脳卒中、多発性硬化症、後−虚血性脊髄障害; 後−超神経痛; 及び/又は後−外傷性脊髄損傷の結果であり得る。
【0163】
神経因性疼痛は、導入の感覚機能の一部又は完全な損失、及び痛い領域における一定の超現象の矛盾した存在として特徴づけられ得る。神経組織損傷は、脳、脊髄又は末梢神経に見出され得る。症状は、その状態に依存して変化し、そして痛感鋭敏(苦痛域値の低下、及び有害刺激への増加反応)、異痛(非有害刺激、例えば冷感、熱感、又は接触による痛みの喚起)、痛覚過敏(刺激強度が知覚の閾値を超える場合、増強された知覚検出閾値を伴って、皮膚領域から突然喚起される爆発的疼痛反応)、痙攣(自発的に、又は無毒の触覚の刺激又はありのままの圧力による刺激に続いて生じるシューテング、電気的、ショック−様又は速い痛みにより特徴づけられるタイプの喚起された疼痛)、錯感覚(自発的であるか又は喚起され、しばしばピン及び針として記載される、異常であるば、しかし、非苦痛性感覚)、知覚異常(自発的であるか又は外部刺激により引き起こされ得る、異常で不快であるが、しかし必ずしも苦痛な感覚ではない)、 関連痛及び異常疼痛輻射(疼痛の異常な蔓延)、 及びワインド・アップ様疼痛及び残感覚(苦痛な刺激の終結の後、長い疼痛の持続)として明白である。
【0164】
神経因性疼痛を有する患者の典型的には、燃えるような、電撃的な、突き刺すような、締め付けるような、痛み、 及び/又は時々、しっかりと締めつけているような疼痛を表す。その痛みは、発作性であるが又は一定であり得る。末梢神経、脊髄及び脳への病理学的変化は、慢性神経因性疼痛の誘発及び維持に関係されて来た。神経因性疼痛を有する患者は典型的には、現在の薬物療法に対して難治性であり、そして彼らの生命の質に深く影響を及ぼす慢性的衰弱性エピソードに耐えている。三環状抗うつ剤及びガバペンチルを包含する、神経因性疼痛のための現在入手できる処理は、典型的には、大部分の患者において制限された効能を示す。
【0165】
いくつかのタイプの神経因性疼痛が存在する。苦痛を伴った神経障害を引起す損傷のタイプ又は関連する病理生理学に関連する分類は、次のものを包含する:機械的な神経損傷に関連する神経障害、例えば手根管症候群、脊椎骨の椎間板ヘルニア、絞扼性神経障害、尺骨の神経障害、及び神経原性胸郭口症候群; 神経障害に関連する代謝性疾患、例えば糖尿病性多発神経障害; 神経向性のウイルス性疾病に関連する神経障害、例えば帯状疱疹及びヒト免疫不全ウィルス(HIV) 疾患; 神経毒性作用に関連する神経障害、例えば癌腫又は結核の化学療法、放射線療法、薬物誘発性神経障害、及びアルコール神経炎; 炎症及び/又は免疫機構に関連する神経障害、例えば多発性硬化症、抗スルファチド抗体神経障害、モノクローナル高ガンマグロブリン血症に関連する神経障害、シェーグレン病、狼蒼、脈管性神経障害、ポリクローナル炎症性神経障害、ギラン・バレー症候群、慢性炎症性脱髄性神経障害、多病巣性運動神経障害、傍腫瘍自律性ニューロパシー、神経節アセチルコリン受容体抗体自律性ニューロパシー、ランバート=イートン筋無力症症候群及び重症筋無力症; 神経系巣状虚血に関連する神経障害、例えば視床症候群(有痛知覚消失); 多発神経伝達物質系機能不全に関連する神経障害、例えば複雑な局所疼痛症候群(CRPS); 慢性的な/神経因性疼痛に関連する神経障害、例えば変形性関節症、腰痛、線維筋痛症、癌腫骨の疼痛、慢性的断端痛、幻肢痛、及び腫瘍随伴性神経障害;
【0166】
毒性神経炎(例えば、 化学物質への暴露、例えばアクリルアミド、3- クロロフェン、カルバメート、二硫化炭素、 酸化エチレン、 n-ヘキサン、 メチルn-ブチルケトン、臭化メチル、有機リン酸塩、ポリ塩化ビフェニール、 ピリミニル、トリクロロエチレン、 又はジクロロアセチレンへの暴露)、焦点の外傷性の神経障害、幻肢痛及び断端痛、単-神経根疾患、及び三叉神経痛; 中枢神経障害、例えば虚血性脳血管性損傷(脳卒中)、多発性硬化症、脊髄損傷、パーキンソン病、筋萎縮性側索硬化症、脊髄空洞症、腫瘍、クモ膜炎、及び術後痛; 混合された神経障害、例えば糖尿病性神経障害(左右対称の多発神経病変、例えば知覚又は知覚運動性多発神経病変、選択的小ファイバ多発神経病変、及び自律性ニューロパシー; 及び焦点の及び多病巣性の神経障害、例えば頭側神経障害、手足モノニューロパシー、 トランクモノニューロパシー(trunk mononeuropathy)、多発性単神経炎、及び非対称の下周辺運動神経障害)及び 好意的に維持される疼痛。他の神経障害は、焦点の神経障害; 舌咽神経痛; 虚血性疼痛; 三叉神経痛; ファブリー病、小児脂肪便症、遺伝性感覚性ニューロパシー又はB12-欠陥に関連する非定型顔面痛; モノニューロパシー; 多発神経病変; 遺伝性末梢性ニューロパシー、例えばCarcot-Marie-歯疾患、レフサム病、シュトルンペル-ローライン病、及び網膜色素変性症; 急性多発神経根筋障害; 及び慢性的多発神経根筋障害を包含する。
【0167】
腫瘍随伴性神経障害は、腫瘍随伴性亜急性知覚神経障害、 腫瘍随伴性運動神経疾患、腫瘍随伴性神経筋緊張症、腫瘍随伴性脱髄性神経障害、腫瘍随伴性 血管炎性神経障害、 及び 腫瘍随伴性自律神経機能不全症を包含する。本発明の開示により供給されるGABABアゴニストのプロドラッグは、前記タイプの神経因性疼痛のいずれかを処理するために使用され得る。ある態様においては、神経因性疼痛は、帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症から選択される。ある態様においては、神経因性疼痛は、帯状疱疹後神経痛及び三叉神経痛から選択される。
【0168】
臨床研究においては、鞘内バクロフェン投与は、脊髄損傷及び多発性硬化症に関連する神経因性疼痛(Herman et al., Clin J Pain 1992, 8(4), 338-345;及びTaira et al., Stereotactic Funct Neurosurg 1995, 65, 101-105)、痛みを伴った末梢異常(Gatscher et al., Acta Neurochir Suppl 2002, 79, 75-76)、及び交感神経的に維持される痛み(Van Hilten et al., N Engl J Med 2000, 343, 625-630; Becker et al., J Clin Neurosci 2000, 7, 316-319;及びZuniga et al., Reg Anesth Pain Med 2002, 27, 90-93)の処理において効果的であることが示されている。
【0169】
バクロフェンはまた、三叉神経、舌咽、迷走舌咽神経、及び眼-後ヘルペス性神経痛(Bowsher, Br Med Bull 1991, 47, 644-66; Fromm et al., Neurology 1981, 31, 683-687;及び Ringel and Roy, Ann Neurol 1987, 21, 514-515)において、及び糖尿病性神経障害を有する患者(Anghinah et al., Muscle Nerve 1994, 958-59)において有効であることも示されている。約50mg/日〜約60mg/日のバクロフェンの用量が三又神経痛の処理において有効であることが示されている(Fromm et al., Ann Neurol 1984, 15, 240-244)。
【0170】
種々のタイプの神経因性疼痛を処理するための化合物(1)の効能は、当業界に知られている技法、例えばランダム二重盲目プラシーボ制御された方法を用いて、臨床試験において評価され得る。神経因性疼痛のための臨床試験に使用されるエンドポイントは、確認された神経因性疼痛基準、例えばthe Brief Pain Inventory、 Categorical Scale、 Gracely Pain Scale、 Likert Scale、Neuropathic Pain Scale、 Numerical Pain Scale、 Short Form McGill Pain Questionnaire、Verbal Pain Scale、Visual Analog Scale (VAS)、 VAS Pain Intensity Scale及び/又はVAS Pain Relief Scaleを用いて決定され得る。
【0171】
筋骨格痛:
圧痛及び筋肉痙攣を引き起こす筋骨格状態は、線維筋痛症、緊張型頭痛、筋筋膜疼痛症候群、咬合局面関節痛、 内部椎間板離開、体性機能不全、脊椎骨折、化膿性脊椎炎、リウマチ性多発筋痛、環軸関節の不安定性、環椎後頭関節疼痛、骨粗鬆症の脊椎骨の圧縮破壊、ショイエルマン病、 脊椎分離症、脊椎辷り症、棘突起間関節形成症、仙腸関節疼痛、仙椎の圧力骨折、尾骨痛、 衰えた逆症候群、 及び機械的腰痛又は頸痛を包含する(Meleger and Krivickas, Neurol Clin 2007, 25, 419-438)。
【0172】
それらの状態においては、筋肉痙攣は、痙攣の特徴的な高められた調子又は反射を伴わないで、影響された筋肉グループを包含する局部因子に関連する。筋肉、腱、靭帯、椎間板、関節軟骨及び骨は、筋骨格痛に関与する。首及び背部痛を生成できる障害は、肉離れ、靭帯捻挫、 筋膜痛、線維筋痛症、咬合局面関節痛、内部椎間板離開、体性機能不全、脊椎骨折、 脊椎骨髄炎、及びリウマチ性多発筋痛、環軸関節の不安定性及び環椎後頭関節疼痛を包含する。
【0173】
バクロフェンは、全身的に又は中枢的に投与される場合、筋肉−弛緩効果を誘発することが知られている(Malcangio and Bowery, Trends Pharmacol Sci 1996, 17, 457-462)。従って、上位運動ニューロン症候群に関連する痙攣の処理のためへのバクロフェンの使用は十分に確立されている。バクロフェンはまた、筋肉痛及び/又は末梢筋肉骨格状態に関連する痙攣の処理のにおいて有効である研究がまた示されている。
【0174】
例えば、バクロフィンは、片頭痛(Hering-Hanit, Cephalalgia 1999, 19, 589-591; and Hering-Hanit及びGadoth, Headache 2000, 40, 48-51);及び特に、引張型頭痛(Freitag, CNS Drugs 2003, 17(6), 373-381);並びに腰痛及び神経根症(Zuniga et al., Anesthesiology 2000, 92, 876-880; Vatine et al., Pain Clin 1989, 2, 207-217; Dapas et al., Spine 1985, 10(4), 345-349; Raphael et al., BMC Musculoskeletal Disorders 2002, June 20, 3(17);及びMagora et al., Pain Clin 1988, 2, 81-85)の処理において効果的であることが示されている。
【0175】
1又は複数のタイプの筋骨格痛の処理のために化合物(1)のプロドラッグの効能は、神経因性疼痛の動物モデル及び臨床試験において評価され得る。
【0176】
背痛
化合物(1)は、頸部、胸部及び/又は腰背骨領域における背痛を包含する背痛みの処理に使用される。背痛は急性又は慢性であり得る。急性背痛は、4週よりも少ない間、存在する低背痛みとして定義され、時々、3ヶ月よりも少ない間、存在する症状を伴って、亜急性背痛により分類される。慢性低背痛は、3ヶ月以上の間、存在する低背痛として定義される。
【0177】
腰痛:
腰痛は一般的に、腰椎L1−L5の位置における背部のコシ領域において発生する。腰痛は、捻挫、緊張、 又は筋、腱、面関節、 及び/又は背部における仙腸関節の1つに対する痙攣; 背骨捻挫又は過剰圧縮; 又は脊椎板破壊又は湾曲部により引起され得る。腰痛はまた、神経又は筋肉刺激又は骨損傷に影響を及ぼすことができる。ほとんどの腰痛は背部への損傷又は外傷を従えるが、しかし疼痛はまた、変性状態、例えば関節炎又は脊椎板疾患、骨粗鬆症、又は他の骨疾患、ウイルス感染,関節及び脊椎板に対する刺激、又は背骨における先天性異常により引起され得る。
【0178】
肥満、喫煙、妊娠の間の体重増、 ストレス、 不良な物理的状態、 実施される活動のために不適切な姿勢、及び不十分な睡眠位置がまた、腰痛にも寄与している。さらに、外傷の背部がそれ自体治癒する場合に創造される瘢痕組織は、正常組織の強さ又は柔軟性を有さない。反復された外傷からの瘢痕組織の再生は結果的には、背部を弱くし、そしてより重度の外傷を導く。時折、腰痛はより重度の医学的問題を示す。発熱又は腸管又は膀胱制御の損失に伴う疼痛、咳する場合の疼痛、及び脚における進行性衰弱は、狭まれた神経又は他の重度の状態を示すことができる。糖尿病を有する人々は、重度の腰痛、又は神経障害に関連する脚の下側まで広がる疼痛を有する。
【0179】
腰痛は、隆起した脊椎板 (例えば、突出した、ヘルニアを起こした、又は破裂した脊椎板)、坐骨神経痛、背骨の変性、頚椎以外の脊柱管狭窄症、骨粗鬆症、変形性関節症、圧縮破壊、骨格の不規則、線維筋痛症、椎弓分離症及び/又は脊椎辷り症により引起され得る。腰痛を引起すことができるまれな脊椎状態は、強直性脊椎炎、細菌感染、頭蓋骨骨髄炎、背骨の腫瘍、パジェット病、及びショイエルマン病を包含する。臨床結果は、GABABアゴニスト、例えばバクロフェンが腰痛の処理において効果的であり得ることを示唆する(Dapas et al., Spine 1985, 10(4), 345-349; and Raphael et al., BMC Musculoskeletal Disorders 2002, June 20, 3(17))。例えば、約20mg/日〜約80mg/日の用量のバクロフェンが、急性腰痛の処理において有効であることが示されている(Dapas et al., Spine 1985, 10(4), 345-9)。
【0180】
ある態様においては、本発明の開示により提供される、腰痛の処理方法は、腰痛、例えば筋肉痙攣に関連する障害、状態及び/又は症状を処理することを含んで成る。腰痛の症状は、その原因に依存する。例えば、逆捻挫又は背部緊張の症状は、背部及び臀部痙攣、痙縮、硬直及び疼痛を包含する。神経根圧の症状は、坐骨神経痛として言及される脚痛、及び神経関連の集積、例えば1本の脚又は足、下脚、又は両脚における刺痛、しびれ感又は衰弱を包含する。背骨の関節炎の症状は、背部及び股関節において悪化する疼痛及び硬直を包含する。
【0181】
苦痛を伴う急性筋骨格状態に関連する筋肉痙攣:
筋肉痙攣は、苦痛を伴う急性筋骨格状態に関連している。腰痛及び頸痛はそのような状態の共通する表れである。背部の急性筋骨格痙攣は、局在化された疼痛、硬直、低められた移動性、毎日の生活の活動消出、及び睡眠障害を引起す共通する障害である。急性腰痛又は頚痛のほとんどのエピソードは、非特異的である。ほとんどの対象は、腰痛及び頸痛、例えば有意な痙攣、癌、感染又は運動衰弱を示す基準を満たさない。非特異的な腰痛は、機械的背痛、個眼関節痛、変形性関節炎、筋捻挫及び筋痙攣として定義される。腰痛は、傍脊椎筋群における反射性痙攣により引起され得る。急性背痛は不本意であり、そしてしばしば、頸部、胸部及び/又は腰髄部を包含する背部の筋肉の苦痛を伴った収縮である。腰椎に関連する痙攣はまた、下背痙攣としても言及される。
【0182】
急性頸痛及び腰痛についての典型的な薬理学的処理剤は、NSAIDS、アセトアミノフェン及び筋肉弛緩薬である。最近のプラシーボ制御された研究は、バクロフェンが、2週間以下の傍背柱筋痙攣及び機能的不能性の現象を有する急性下背症候群の処理において、効果的で、安全で且つ十分に許容されていることを結論づけた。従って、化合物(1)は、苦痛を伴う筋骨格状態、例えば急性背部痙攣及びより特定には急性腰痙攣に関係する筋肉痙攣の処理に使用され得る。
【0183】
線維筋痛:
線維筋痛は、身体じゅう、但し特に背骨を通して、筋肉、腱及び関節における痛み及び疼痛により特徴づけられる状態である。身体はまた、圧点又はトリガー点として言及される特定領域において接触しにくい。線維筋痛の他の症状は、睡眠妨害、鬱病、昼間の疲労、頭痛、下痢便秘交代症、手足の麻痺及び刺痛、脱力感、 記憶困難、及び眩暈感を包含する。線維筋痛の病因学は知られていないが、ストレス、乱れた睡眼パターン、神経系における疼痛関連の化学物質の異常生成、及び/又は低レベルの成長ホルモンが、線維筋痛の開始に寄与していると思われる。
【0184】
線維筋痛の現在の処理は、疼痛の緩和、睡眼の回復、及び生活の一般的質の改善する目的を伴っての症状に基づかれる。いくつかの非薬理学的処理は、運動、教育、行動的及び物理的治療を包含する。薬理学的処理は、三環状化合物、セロトニン再摂取インヒビター、鎮痛薬、筋弛緩薬、及びACEインヒビターを包含する。バクロフェンが線維筋痛症状の改善において有用であることを示す証拠が存在する(Taylor-Gjevre and Gjevre, Lupis 2005, 14(6), 486-8)。
【0185】
線維筋痛の処理のための本発明の開示により供給される化合物の投与の効能は、線維筋痛の動物及びヒトモデルを用いて、及び臨床試験において評価され得る。神経因性疼痛の動物モデル、又は異なったタイプの神経因性疼痛の臨床学的に適切な研究が、線維筋痛の処理のための治療活動の評価においてい有用であることが見出された。
【0186】
神経因性疼痛、筋骨格痛、腰痛、急性の苦痛を伴った筋骨格状態に関連する筋肉痙攣、及び線維筋症の処理のためへの化合物(1)及び他のR−バクロフェンプロドラッグの使用は、Bensonなど., (US 2009/0118365号)(その全内容の引用により本明細書に組込まれる)に開示されている。
【0187】
尿失禁:
尿失禁は、尿のいずれかの不本意な漏れであり、そして切迫性尿失禁、ストレス性失禁、奇異性尿失禁、機能的な失禁、及び混合汚泥失禁を包含する状態のパターンに基づいて5種のタイプに分類され得る(Abrams et al., Neurology and Urodynamics 2002, 21, 167-178)。
【0188】
切迫性尿失禁は、制御され得ない、排尿する突然で且つ激しい行動であり、続いて制御できない排尿が存在する。切迫性尿失禁は、排尿を阻害する前頭葉における脳の部分の変化のために一部、膀胱筋肉の不良なスクイージング能力と共に、膀胱における筋肉の過剰活性の組合せにより引起され得る。膀胱筋肉の不本意な作用は、膀胱の神経、脊髄及び脳を包含する神経系、又は筋肉自体への損傷のために生じる。筋肉及び神経への損傷は、脳卒中、 手術、又は脳障害、例えば多発性硬化症、パーキンソン病、アルツハイマー病の結果として生じる。
【0189】
ストレス性失禁は、咳をし、りきみ、くしゃみをし、重いものを持ち上げ、又は腹内圧迫を突然、高め、そして一般的に、直腸床筋の不十分な強さにより引起されるいずれかの操作を実施する場合、少量の尿の制御できない排尿である。前立腺手術に続く失禁は、男性におけるストレス性失禁の最も共通する形である。女性においては、ストレス性失禁は、妊娠、出産、閉経又は骨盤の手術に関連する物理的変化に起因する。
【0190】
奇異性尿失禁は、あるタイプの閉塞、又は膀胱筋肉の弱い収縮により通常、引起される少量の尿の制御できない漏れである。奇異性尿失禁は、前立腺外科、前立腺肥大症、便秘、神経損傷、神経メッセージを妨害する脳又は脊髄に影響を及ぼす薬物、糖尿病、 多発性硬化症、腫瘍、脊髄損傷、神経系障害、 及び疾病 、例えば 膀胱からの神経シグナル又は排尿筋による尿の圧出を低めることができる多発性硬化症により引起され得る。
【0191】
機能的失禁は、制限された移動性のために物理的に又は不本意には、トレイに行けないことに起因する排尿を言及する。機能的失禁の原因は、次のものを包含する:混乱、痴呆、 不良な視力、不良な移動性、 不良な機敏性、鬱病によるトイレへの気がすすまないこと、不安感、怒り、泥酔、 又は物理的不可能性 、例えば車椅子の人。非可動性を引起す状態は、脳卒中、重度の関節炎、 及び精神機能を妨害する論争、例えばアルツハイマー病及び重度の鬱病による痴呆を包含する。
【0192】
混合汚尿失禁は、1つよりも多くのタイプの失禁を包含する。
尿失禁はまた、夜尿症又は遺尿を包含する。
【0193】
尿失禁はまた、活動亢進性膀胱を包含する。活動亢進性膀胱は、緊急の失禁を伴って又は伴わないで、通常、頻度及び夜間多尿症を伴って、緊急である(Abrams, Urology 2003, 62 (Suppl 5B), 28-37; Ouslander N Engl J Med 2004, 350, 786-99; and Wein and Rovner, Urology 2002 (Suppl 5A), 7-12)。緊急は、排泄の突然の強制的所望の不満であり;頻度は、男性/女性が日中あまりにも頻繁に排泄すると思われる患者による不満であり;そして夜間多尿症は、個人が排泄のために夜間、約1度よりも多く目をさますべきである不満である。
【0194】
活動亢進性膀胱を有する患者は典型的には、延期するのに困難である排尿の突然の及び強制的な必要性(緊急)、緊急を感じる尿の不本意な漏れ(尿失禁の促進)、頻度(24時間で8回以上の排尿)及び夜間多尿症(排尿するために夜、1度以上の目覚め)の症状を提供する。活動亢進性膀胱の症状は、排尿サイクルの充満期の間、排尿筋の付随意的収縮のためである。それらの付随意的収縮は、排尿筋過剰活性と呼ばれ、そして膀胱ムスカリン作動受容体のアセチルコリン−誘発された刺激により仲介される(Andersson, Urology 1997, 50(Suppl. 6A), 74-84)。排尿筋過剰活性は、自発的であるか又は引起され得る充満期の間、不本意な排尿筋収縮により特徴づけられる尿力学的観察である。排尿筋過剰活性は、適切な神経学的状態が存在する、定義された根本にある原因及び排尿筋過剰活性が存在しない、特発性排尿筋過剰活性として特徴づけられ得る。
【0195】
苦痛を伴った膀胱症候群とも呼ばれる間質性膀胱炎は、尿失禁に関連する障害である。間質性膀胱炎は、多くの因子、例えば自己免疫、アレルギー及び感染症病因学により引起されると思われる膀胱の慢性炎症状態である。間質性膀胱炎の症状は、患者が排尿した後でさえ、排尿する過度の緊急性、1日当たり平均16又はそれ以上の尿頻度、夜間での排尿、恥骨結合上(膀胱/骨盤/会陰)疼痛、及び/又は性交疼痛症を包含する。ある状態においては、本発明の開示により供給される投与形は、間質性膀胱炎の処理に使用され得る。
【0196】
二重盲検クロスオーバー試験においては、1日当たり4度、5mgの用量で投与されるバクロフェンは、不安定な膀胱症候群を有する患者において昼間及び夜間頻尿、及び失禁の重症度を有意に改善することが示されている(Taylor and Bates, British J Urology 1979, 51, 504-505)。従って、化合物(1)は、尿失禁、例えば活動亢進性膀胱及び/又は排尿筋過剰活性の処理において有用であることが予測される。
【0197】
投与量:
R−バクロフェンの持続した全身性濃度を提供する錠剤投与形は、現在、1日当たり3度、投与される即時放出性非プロドラッグ形、すなわち患者のために不便であり、そして患者にとって覚えるのが困難であるレジメに比較して、患者のコンプライアンスを増強するであろうと思われる。さらに、本発明の開示により供給される経口投与形錠剤の使用は、低められた副作用、例えば傾眠状態、衰弱、頭痛、発作、嘔気、嘔吐、低血圧、便秘、精神錯乱、呼吸抑制、不眠、及び頻尿又は尿閉を伴って、増強された効能を提供するであろうと思われる。
【0198】
本明細書に開示される特定の疾病の処理において効果的であろう化合物(1)の量は、疾病の性質に、少なくとも一部、依存し、そして当業界において知られている標準の臨床学的技法により決定され得る。さらに、インビトロ又はインビボアッセイは、最適な投与量範囲の同定を助けるために使用され得る。投与レジメ及び投与間隔はまた、当業者に知られている方法により決定され得る。投与される化合物(1)の量は、他の因子の中でも、処理される対象、対象の体重、疾病の重症者、投与路、及び処方医者の判断に依存することができる。
【0199】
全身性投与に関しては、治療的有効用量は、最初、インビトロアッセイから評価され得る。初期用量はまた、インビボデータ、例えば動物モデルから、当業界において知られている技法を用いて評価され得る。そのような情報は、ヒトにおける有用な用量を、より正確に決定するために使用され得る。当業者は、動物データに基づいて、ヒトへの投与を最適化することができる。
【0200】
化合物(1)の用量は、等モル量又は等価質量のR−バクロフェンを供給するよう調節され得る。用量は、本発明の開示により供給される、複数用量形を含んで成ることができる。ある態様においては、R−バクロフェンの治療的有効用量は一般的に、1日当たり約0.03mg〜約1mg/kg体重である。ある態様においては、毎日の用量は、約1mg〜約100mg;ある態様においては、約5mg〜約80mg;ある態様においては、約5mg〜約60mg;及びある態様においては、約10mg〜約40mgの範囲の等価質量のR−バクロフェンを含んで成ることができる。ある態様においては、化合物(1)の用量は、患者において、中位の鎮静及び運動活動の障害を引起す用量以下である。化合物(1)の用量及び適切な投与間隔は、患者におけるR−バクロフェンの持続した治療的有効濃度を、ある態様においては、最小悪影響濃度を越えないで、維持するよう選択され得る。
【0201】
ある態様においては、本発明の開示により供給される投与形は、1日1度、1日2度、及びある態様においては、1日1度以上の間隔で投与され得る。投与は、単独で、及び他の薬物と組合して提供され、そして疾病の効果的処理のために必要とされる限り、続けることができる。投与は、哺乳類、例えばヒトに、食物を食べた状態又は絶食された状態で投与形を投与することを包含する。
【0202】
用量は、単一投与形で、又は複数の投与形で投与され得る。複数の投与形が使用される場合、その複数投与形の個々内に含まれる化合物(1)の量は同じであっても、又は異なっていても良い。
【0203】
ある態様においては、投与される用量は、毒性用量以下である。本明細書に記載される組成物の毒性は、細胞培養物又は実験運動において標準の医薬方法により、例えばLD50(集団の50%が致死する用量)又はLD100(集団の100%が致死する用量)により決定され得る。毒性効果:治療効果の用量比は、治療指数を示す。それらの細胞培養アッセイ及び動物研究から得られるデータは、ヒトへの使用のために毒性でない投与範囲の配合に使用され得る。化合物(1)の用量は、治療的に有効であり、鎮静用量以下であり、そして/又は毒性をほとんどか又はまったく示さない、血液、血漿又は中枢神経系における一定範囲内の循環濃度であり得る。
【0204】
処理の間、用量及び投与スケジュールは、疾病を処理するために十分な又は定常状態の全身性濃度のR−バクロフェンを提供することができる。ある態様においては、徐々に増大する用量が投与され得る。
【0205】
組合せ療法:
本発明の開示により供給される投与形はさらに、化合物(1)の他に、1又は複数の医薬的化合物を含んで成ることができる。そのような化合物は、化合物(1)により処理される疾病と同じ疾病又は異なった疾病を処理するために供給され得る。
【0206】
ある態様においては、化合物(1)は、少なくとも1つの他の治療剤と組合して使用され得る。ある態様においては、化合物(1)は、移動障害、例えば痙攣、消化障害、例えば胃食道逆流病及び嘔吐、 又は中毒又は乱用障害、 例えば ニコチン中毒又は乱用、 アルコール 中毒又は乱用、麻薬中毒又は乱用、 咳、神経因性疼痛、筋骨格痛、又は尿失禁を処理するために、他の化合物と一緒に患者に投与され得る。ある態様においては、少なくとも1つの他の治療剤は、異なったR−バクロフェンプロドラッグであり得る。種々の観点においては化合物(1)及び少なくとも1つの他の治療剤は、加算的に、及びある態様においては、相乗的に作用することができる。
【0207】
少なくとも1つの追加の治療剤は、化合物(1)を含んで成る同じ投与形に含まれ得るか、又は別の投与形に存在することができる。従って、本発明の開示により提供される方法はさらに、化合物(1)を投与する他に、化合物(1)により処理される疾病と同じか又は異なった疾病を処理するために有効な1又は複数の治療剤を投与することを包含する。本発明の開示により提供される方法は、化合物(1)、及び1又は複数の他の治療剤の投与を包含し、但し組合された投与は化合物(1)の治療効能を阻害せず、そして/又は副組合せ作用を生成しない。
【0208】
ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)を含んで成る投与形と同じ投与形の一部であるか、又は異なった投与形に存在する、他の治療剤の投与と同時に投与され得る。化合物(1)は、他の治療剤の投与の前又は続いて投与され得る。組合せ療法のある態様においては、組合せ療法は、例えば特定薬物に関連する薬物副作用を最少にするために、化合物(1)と他の治療剤を含んで成る組成物との間の投与を交換することを含んで成る。化合物(1)が毒性(但し、それだけには限定されない)を包含する薬物副作用を実質的に生成する他の治療剤と同時に投与され得る場合、前記他の治療剤は好都合には、有害薬物反応が誘発される閾値以下に低下する用量で投与され得る。
【0209】
ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)の放出性、生物学的利用能、治療効能、治療能力、安定性及び同様のものを増強するか、調節するか、そして/又は制御するために1又は複数の物質と共に投与され得る。例えば、化合物(1)又はその代謝物の治療効能を増強するために、R−バクロフェン及び化合物(1)は、同時に投与され得るか、又は化合物(1)を含んで成る投与形は、胃腸管から全身循環への化合物(1)又はR−バクロフェンの吸収又は拡散を高めるために、又は患者の血液における化合物(1)又はR−バクロフェンの分解を阻止するために、1又は複数の剤を含んで成ることができる。ある態様においては、化合物(1)を含んで成る投与形は、化合物(1)の治療効能を増強する薬理学的効果を有する活性剤と共に同時投与され得る。
【0210】
さらに、本発明の開示により供給される投与形は、副作用として、痙攣、胃食道逆流病、嘔吐、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、嘔吐、咳、神経因性疼痛、及び/又は筋骨格痛を、それら自体引起すことが知られている他の薬物と組合して使用され得、これにより、その副作用の発生を妨げるか、又は低めることができる。
【0211】
ある態様においては、本発明の開示により供給される投与形は、移動障害、例えば痙攣を処理するために、治療剤、又は移動障害、例えば痙攣の処理において効果的であることが知られているか、又は思われる他の治療剤と組合して、患者に投与され得る。移動障害、例えば痙攣の処理のための、及び化合物の(1)と共に投与され得る薬物の例は、次のものを包含する:レボドパ、軽い鎮静剤、例えばベンゾジアゼピン、例えばアルプラゾラム、クロルジアゼポキシド、クロナゼパム、 クロラゼパート、ジアゼパム、ロラゼパム、及びオキザゼパム; 筋弛緩剤、例えばバクロフェン、抗コリン作動薬、例えばトリヘキシフェニジル、アトロピン、スコポラミン、及びジフェンヒドラミン; 抗精神病薬、例えばクロルプロマジン、フルフェナジン、ハロペリドール、ロキサピン、メソリダジン、モリンドン、ペルフェナジン、ピモジド、チオリダジン、チオチキセン、トリフルオロペラジン、 アリピラゾール、クロザピン、オランザピン、 クエチアピン、 リスペリドン、及びジプラシドン;及び抗うつ剤 、例えばアミトリプチン。
【0212】
ある態様においては、本発明の開示により供給される投与形は、胃腸障害、例えば胃食道逆流病を処理するために、治療剤、又は胃腸障害、例えば胃食道逆流病の処理において効果的であることが知られているか、又は思われる他の治療剤と組合して、患者に投与され得る。胃腸障害、例えば胃食道逆流病の処理のための、及び化合物の(1)と共に投与され得る薬物の例は、次のものを包含する:H2 インヒビター、例えばシメチジン、ファモチジン、 ニザチジン、及び ラニチジン; プロトンポンプインヒビター、例えばオメプラゾール、 ランソプラゾール、 パントプラゾール、 ラベプラゾール、 及びエキソメプラゾール; 及び消化管運動促進剤、例えばシサプリド、ベタンコール、及びメトクロプラミド。
【0213】
ある態様においては、本発明の開示により供給される投与形は、嘔吐を処理するために、治療剤、又は嘔吐の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。嘔吐(悪心及び吐気)を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ベンズアミン、例えばメトクロプラミド; フェノチアジン、例えばプロクロルペラジン、ペルフェナジン、クロルプロマジン、プロメタジン、及びトリエチルペラジン; ブチロフェノン、例えばドロペリドール及びハロペリドール;ドーパミン2 アンタゴニスト、例えば メトクロルパミド; 5-HT3 アンタゴニスト、例えばオンダンセトロン、グラニセトロン、 ドラセトロン、パロノセトロン; NK-1 受容体アンタゴニスト、例えばアプレピタント、コルチコステロイド、例えばデキサメタゾン; 抗ヒスタミン、例えばジフェンヒドラミン及びヒドロキシジン; カンナビノイド、例えばドロナビノール; 及びベンゾジアゼピン、例えばロラゼパム、ミダゾラム、アルプラゾラム、及びオランザピン。
【0214】
ある態様においては、本発明の開示により供給される投与形は、アルコール中毒症又は乱用を処理するために、治療剤、又はアルコール中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。アルコール中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:アンタビュース、ナルトレキソン、クロニジン、メサドン、 1-α-アセチルメタドール、ブプレノルフィン、及びブプロピオン。
【0215】
ある態様においては、本発明の開示により供給される投与形は、麻薬中毒症又は乱用を処理するために、治療剤、又は麻薬中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。麻薬中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ブプレノルフィン、トラマドール、メサドン、及びナルトレキソン。
【0216】
ある態様においては、本発明の開示により供給される投与形は、ニコチン中毒症又は乱用を処理するために、治療剤、又はニコチン中毒症又は乱用の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。ニコチン中毒症又は乱用を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:ブプロピオン、クロニジン、及びニコチン。
【0217】
ある態様においては、本発明の開示により供給される投与形は、咳を処理するために、治療剤、又は咳の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。咳を処理するための、及び化合物(1)と共に投与され得る薬物の例は、次のものを包含する:デクストロメトルファン、グアイフェネシン、ヒドロコドン、ベンゾナタート、ジフェンヒドラミン、プソイドエフェドリン、パラセタモール、及びカルビノキサミン。
【0218】
ある態様においては、本発明の開示により供給される投与形は、神経因性疼痛を処理するために、治療剤、又は神経因性疼痛の処理のために効果的であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。神経因性疼痛を処理するために有用な薬物の例は、次のものを包含する:オピオイド鎮痛薬、例えばモルフィン、コデイン、フェンタニール、メペリディン、メサドン、プロポキシフェン、 レボルフェノール、ヒドロモルフォン、オキシコドン、オキシモルフォン、トラマドール及びペンタゾシン; 非オピオイド鎮痛薬、例えばアスピリン、イブプロフェン、ケトプロフェン、ナプロキセン、及びパラセタモール; 非ステロイド性抗炎症薬、例えばアスペリン、コリンサリチル酸マグネシウム、ジフルニサール、サルサレート、 セレコキシブ、 ロフェコキシブ、バルデコキシブ、ジクロフェナック、エトドラク、フェノプロフェン、フルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、 ケトロラック、 メクロファナメート、メフェナム酸、 メロキシカム、ナブメトン、オキサプロジン、ピロキシカム、スリンダク、及びトメチン;
【0219】
抗癲癇薬、例えばガバペンチン、 プレガバリン、カルバマゼピン、フェニトイン、 ラモトリジン、 及びトピラメート; 抗抑うつ剤、例えばジュロキセチン、アミトリプチン、 ベンラファキシン、 ノルトリプチリン、イミプラミン、及びデシプラミン; 局部麻酔剤、例えばリドカイン、及びメキシレチン; NMDA 受容体アンタゴニスト、例えばデキストロメトルファン、メマンチン、及びケタミン; N-タイプカルシウムチャネル遮断薬、例えばジコノチド; バニロイド受容体-1 モジュレーター、例えばカプサイシン; カンナビノイド受容体モジュレーター、例えばサチベックス; ノイロキニン受容体アンタゴニスト 、例えばラネピタント; 他の痛み止め、例えばノイロトロピン;及び他の薬物、例えばデシプラミン、クロナゼパム、ジバルプロエクス、オキスカルバゼピン、ジバルプロエクス、ブトルファノール、バルデコキシブ、ビコプロフェン、ペンタゾシン、プロポキシヘン、フェノプロフェン、ピロキシカム、 インドメタシン、ヒドロキシジン、ブプレノルフィン、ベンゾカイン、クロニジン、フルルビプロフェン、メペリディン、ラコサミド、 デスベンラファキシン、及び ビシファジン。
【0220】
ある態様においては、神経因性疼痛の処理に有効な薬物は、プロポキシフェン、メペリディン、ヒドロモルフォン、ヒドロコドン、モルフィン、コデイン、 2-ピペリジノール-1-アルカノール、 エリプロジル、イフェンプロジル、 ロフェコキシブ、 セロコキシブ、サリチル酸、ジクロフェナック、ピロキシカムインドメタシン、イブプロフェン、ナプロキセン、 ガバペンチン、 カルベマゼピン、 プレガバリン、トピラメート、バルプロン酸、スマトリプタン、 エリトリプタン、リザトリプタン、ゾルミトリプタン、 ナラトリプタン、フレクセリル、カリソプロドール、ロバキシサル、 ノルゲシック(norgesic)、ダントリウム、ジアゼパム、クロルジアゼポキシド、アルプラゾラム、ロラゼパム、パラセタモール、亜酸化窒素、ハロセン、リドカイン、エチドカイン、ロピバカイン、塩化プロカイン、サラピン、ブピバカイン、カプシシン、デシプラミン、アミトリプチン、ドキセピン、ペルフェナジン、プロトリプチリン、トラニルシプロミン、バクロフェン、クロニジン、メキセリチン( mexelitine)、ジフェンヒドラミン、ヒドロキシジン、カフェイン、プレドニソン、 メチル-プレドニソン、デカドロン、セルトラリン、パロキセチン、フルオキセチン、トラマドール、レボドパ、デクストロメトルファン、 サブスタンス Pアンタゴニスト、及びボツリヌス毒素から選択される。
【0221】
ある態様においては、神経因性疾病の処理にお有用な薬物は、ニコチン受容体部分作動薬及び鎮痛薬から選択され得る。
神経因性疼痛の処理のための非薬理学的療法は、経皮的電気刺激、経皮の電気的神経刺激、及び刺鍼術を包含する。
【0222】
ある態様においては、本発明の開示により供給される投与形は、線維筋痛症を処理するために、治療剤、又は線維筋痛症、又はある態様においては、線維筋痛症に関連する疾病、障害又は状態の処理において効果的であることが知られているいるか、又は思われる他の治療剤と組合して患者に投与され得る。線維筋痛症についての薬物療法は、線維筋痛症の重症度及び頻度に適応され得る。時折のエピソードに関しては、急性処理が示され得る。1月あたり複数回、発生する線維筋痛症エピソードに関しては、又は攻撃が患者の毎日の生活に非常に影響を及ぼす場合、進行に基づいて慢性療法が適切である。
【0223】
エピソードの頻度を低める、繊維筋痛症のための処理は、非ステロイド性抗炎症薬(NSAIDs)、アドレナリン性β遮断剤、カルシウムチャンネル阻害剤、三環系抗うつ薬、選択的セロトニン再取込み阻害薬、抗痙攣薬、 NMDA 受容体アンタゴニスト、ドーパミンアゴニスト、選択的5-HT3受容体アゴニスト、 オピオイド、 筋弛緩薬、催眠鎮静薬、 及び他の治療剤を包含する。線維筋痛症の処理に有用なNSAIDの例は、アスピリン、イブプロフェン、フェノプロフェン、フルルビプロフェン、ケトプロフェン、メフェナム酸、及びナプロキセンを包含する。線維筋痛症の処理に有用なアドレナリン性β遮断剤の例は、アセブトロール、アテノロール、イミロール、メトプロロール、ナドロール、ピンドロール、プロプラノロール、及びチモロールを包含する。
【0224】
線維筋痛症の処理に有用なチャンネル遮断剤の例は、アムロジピン、カルシウム拮抗物質ジルチアゼム、ドタリジン、フェロジピン、フルナリジン、ニカルジピン、ニフェジピン、ニモジピン、ニソルジピン、及びベラパミルを包含する。線維筋痛症の処理のために有用な三環式抗うつ薬の例は、アミトリプチン、デシプラミン、ドキセピン、イミプラミン、ノルトリプチリン、シクロベンザプリン、及びプロトリプチリンを包含する。線維筋痛症の処理のために有用な選択的セロトニン再摂取インヒビターの例は、フルオキセチン、メチセルギド、ネファゾドン、パロキセチン、セルトラリン、及びシタロプラムを包含する。線維筋痛症の処理のために有用な他の抗うつ薬の例は、ブプロピオン、ネファゾドン、ノルエピネフリン、バラファキシン、ジュロキセチン、及びトラゾドンを包含する。
【0225】
線維筋痛症の処理のために有用な抗痙攣薬(抗癲癇薬)の例は、ジバルプロックス・ナトリウム、フェルバメート、 ガバペンチン、 ラモトリジン、 レベチラセタム、オキスカルバゼピン、チアガビン、トピラメート、バルプロエート、及びゾニサミドを包含する。線維筋痛症の処理のために有用なNMDA受容体アンタゴニストの例は、デクストロメトルファン、マグネシウム及びケタミンを包含する。線維筋痛症の処理のために有用なドーパミンアゴニストの例は、α−ジヒドロエルゴクリプチンを包含する。線維筋痛症を妨げるために有用なオピオイドの例は、トラマドール、オキシコドン及びメサドンである。
【0226】
線維筋痛症の処理のために有用な筋弛緩薬の例は、シクロベンザプリンである。線維筋痛症の処理のために有用な治療薬の例は、運動、インターフェロン、成長ホルモン、 ホルモン療法、 動物脂肪が低くそして繊維に富んでいる食物、 及び相補療法、例えばカウンセリング/精神療法、リラクゼーション・トレーニング、段階的筋弛緩法、誘導されたイメージ、横隔膜の呼吸、バイオフィードバック、刺鍼術、及び物理的及びマッサージ療法を包含する。
【0227】
筋肉/骨格痛の重症度及びいずれかの関連する症状を排除するか又は低めるための急性線維筋痛症処理は、セロトニン受容体作動薬、例えば トリプタンス (5-ヒドロキシトリプトファン(5-HT) アゴニスト)、 例えば、アルモトリプタン、 エレトリプタン、 フロバトリプタン、 ナラトリプタン、 リザトリプタン、 スマトリプタン、及びザルミトリプタン; エルゴタミン-に基づく化合物、例えばジヒドロエルゴタミン及びエルゴタミン; 鎮吐薬、例えばメトクロプラミド及びプロクロルペラジン;及び鎮痛効果を提供する化合物を包含する。
【0228】
線維筋痛症の処理において有用な薬物の他の例は、パラセタモール、アスピリン、カフェイン、シプロヘプタジン、メチセルギド、バルプロン酸、 NSAIDs、例えばジクロフェナック、フルルビプロフェン、ケタプロフェン、 ケトロラック、イブプロフェン、インドメタシン、メクロフェナメート 、及びナプロキセンナトリウム; オピオイド、例えばコデイン、メペリディン、及びオキシコドン;及びグルココルチコイ、例えばデキサメサゾン、プレドニソン、及びメチルプレドニゾロンを包含する。
【0229】
ある態様においては、本発明の開示により供給される投与形は筋骨格痛の処理のために、治療剤、又は筋骨格痛の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。筋骨格痛の処理のために有用な薬物の例は、シクロベンザプリン、ダントロレン、メトカルバモール、オーフェナドリン、 トリザニドリン、メタキサロン、カリソプロドール、クロルフェネシン、クロルゾキサゾン、アルプラゾラム、ブロマゼパム、クロルジアゼポキシド、クロラゼプ酸、ジアゼパム、フルニトリアゼパム、ロラゼパム、メダゼパム、ミダゾラム、オキザゼパム、プラゼパム、トリアゾラム、テマゼパム、及びボツリヌス毒素を包含する。ある態様においては、神経因性疼痛の処理のために有用ないずれかの薬物が、筋骨格痛の処理のために化合物(1)と共に同時投与され得る。
【0230】
ある態様においては、本発明の開示により供給される投与形は腰痛の処理のために、治療剤、又は腰痛の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。腰痛の処理のために有用な薬物の例は、NSAIDs、例えばアスピリン、ナプロキセン、及びイブプロフェン; 抗痙攣薬、抗抑うつ薬、例えばアミトリプチン及びデシプラミン; 及びオピオイド、例えばコデイン、オキシコドン、ヒドロコドン、及びモルフィンを包含する。
【0231】
ある態様においては、神経因性疼痛の処理のために有用ないずれかの薬物が、腰痛の処理のためにGABAB アゴニストのプロドラッグと共に同時投与され得る。腰痛のための治療は、冷たくて熱い湿布の使用、床上安静、 運動、背骨の操作、刺鍼術、バイオフィードバック、介入療法、牽引、経皮的電気刺激、超音波、椎体療法、椎骨形成術、椎間板切除、椎間孔拡大術、 椎間板内電気療法、経皮的高周波椎間板減圧術、ラジオ周波外傷、脊椎固定、 及び背骨ラミネクトミーを包含する。
【0232】
ある態様においては、本発明の開示により供給される投与形は、腰痛の処理のために、治療剤、又は筋肉痙攣、例えば腰痛に関連する筋肉痙攣を処理するための他の治療剤、例えば筋肉弛緩剤を組合して患者に投与され得る。筋肉痙攣を処理するための筋肉弛緩剤として有用な薬物の例は、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、ジアゼパム、メタキサロン、メトカルバモール、オーフェナドリン、及びチザニジンを包含する。
【0233】
ある態様においては、本発明の開示により供給される投与形は尿失禁の処理のために、治療剤、又は尿失禁の処理において有効であることが知られているか、又は思われる他の治療剤と組合して患者に投与され得る。尿失禁の処理のために有用な薬物の例はアミトリプチン、ベラドンナ、darifenacin、デスモプレッシン、ジュロキセチン、発情物質、 フェソテロジン、フラボキサート、ヒヨスチアミン、イミダフェナシン、イミプラミン、ニトロフラントイン、オキシブチニン、 プロピベリン、 ソラベロン、ソリフェナシン、タムスロシン塩酸塩、タムスロシン、トルテロジン、トロスピウム、 タイプ Aボツリヌス毒素、 及びバルデナフィル塩酸塩を包含する。
【0234】
尿失禁及び特に活動亢進性膀胱の処理のために可能性を示す他の薬物は、K+ チャネルに対して作用する薬物、例えば NS-8、KW-7158、 ZD-0947; 5-HT3 アンタゴニスト; 5-HT1aアンタゴニスト、例えばREC-0545; P2X アンタゴニスト; NK1 受容体アンタゴニスト、例えばSSR-240600、TA-5538、及びアプレピタント; β3-アゴニスト、例えばGW-427353及びKUC-7483、 YM-178; 及び 他のもの、例えばDDP-200 (オキシブチニン及びガバペンチン)、ニトロフルルビプロフェン、エロカルシトール、 NCX-2111、及びビピリジンを包含する(Colli et al., Expert Opin Investig Drugs 2007, 16(7), 999-1007)。
【0235】
β3−アドレノ受容体アゴニストは、活動亢進性膀胱の処理のために最近提案されて来た(Tyagi et al., Drugs of the Future 2009, 34(8), 635-640)。尿失禁の処理のために有用な他の薬物は、Robinson and Cardozo, Maturitas 2010 doi:10.1016/j.maturitas.2009.12.022に開示されている。
【実施例】
【0236】
次の例は、化合物(1)を含んで成る経口錠剤投与形を詳細に記載する。材料及び方法に関する多くの修飾は本発明の開示の範囲内で実施され得ることは当業者に明らかであろう。
【0237】
例1:化合物(1)乾燥粉末の調製及び特徴化:
化合物(1)ロット70を、次の方法を用いて結晶化した。機械撹拌機、テフロン(登録商標)被覆されたサーモカップル、還流冷却器及び窒素入口を備えた、5Lの三ツ首丸底フラスコに、化合物(1)(72g)、アセトン(186ml)及びヘキサン(672ml)を添加した。その混合物を、撹拌し(60%の速度)、そして50℃に1時間、加熱した。すべての材料は溶解した。追加のヘキサン(1081ml)を、50℃で温度を維持しながら、1時間にわたって添加した。その溶液を45℃に冷却し、そして45℃で5時間、維持し、この時点で固形物が形成した。溶液をさらに、40℃に12時間、次に35℃に12時間、冷却した。次に、その溶液を22〜25℃に2時間、冷却した。生成物を焼結ガラス漏斗を通しての濾過(早い)により集め、そしてアセトン(100ml)及びヘキサン(900ml)の溶液によりすすいだ。湿ったフィルターケークを真空オーブンに移し、そして40℃で12時間、乾燥し、白色固形物として結晶性化合物(1)(ロット70、70g、97%)を得た。
【0238】
化合物(1)ロット71を、次の方法を用いて結晶化した。機械撹拌機、テフロン(登録商標)被覆されたサーモカップル、還流冷却器及び窒素入口を備えた、5Lの三ツ首丸底フラスコに、化合物(1)(72g)、アセトン(186ml)及びヘキサン(672ml)を添加した。その混合物を、撹拌し(60%の速度)、そして50℃に1時間、加熱した。すべての材料は溶解した。追加のヘキサン(1081ml)を、2分間にわたって添加し、そして温度を39℃に下げ、固形物を形成した。その溶液を22〜25℃に1時間にわたって冷却し、そして次に、0〜5℃に1時間、冷却した。生成物を焼結ガラス漏斗を通しての濾過(遅い)により集め、そしてアセトン(100ml)及びヘキサン(900ml)の溶液によりすすいだ。湿ったフィルターケークを真空オーブンに移し、そして40℃で12時間、乾燥し、白色固形物として結晶性化合物(1)(ロット71、70g、97%)を得た。
【0239】
結晶性化合物(1)ロット4を、同じ濃度及び溶媒を用いて調製した(但し、2.5時間にわたって50℃〜0℃の線状除熱傾斜を用いた)。
遅い結晶化(ロット70)、早い結晶化(ロット71)、及び中間速度の結晶化(ロット4)により調製された結晶性化合物(1)の種々の性質が表2に示されている。
【0240】
【表2】
【0241】
化合物(1)サンプルの粒子サイズ及び形状を、走査電子顕微鏡(SEM)分析により特徴づけた。3種のロットの個々からのサンプルを両面炭素テープ上に取り付け、白金の薄層によりスパター被覆し、そして次に、Hitachi−4700SEMを用いて試験した。500倍率での3種のロットのSEMマイクログラフが図1に示される。10,000倍率での3種のロットの対応する像が図2に示される。
【0242】
ロット4(図1A及び2A)は、約25〜50ミクロンの直径の丸い凝集体を含んで成る。対照的に、ロット70(図1B及び2B)は約100ミクロンの長さの線状体を含んで成る。ロット71(図1C及び2C)は、約20〜50ミクロンの長/幅の不規則な形状の凝集体により特徴づけられるさらにもう1つの形態学を含んで成る。
【0243】
例2. 乾燥粉末の流動特性:
乾燥粉末の流れを、FLODEX(商標) Powder Flowability Index Test Instrument (Hanson Research Corporation, Chatsworth, CA)を用いて特徴づけた。その器具は、流動試験の前、試験粉末を保持する円柱状の金属貯蔵物を備えた。その円柱状貯蔵物は、5.7cmの内径及び7.4mmの長さを有する。貯蔵物の底端は、取りはずせる金属ディスクにより密封され得る。個々のディスクは、そのディスクの中心に丸型開口部を有する。開口部の直径は、1mmの増分で4mm〜10mmの範囲であり、そして2mmの増分で10mm〜34mmの範囲である。流動試験の前、その開口部は閉じられる。
【0244】
次に、粉末を、その閉じられた開口部上に配置する。開口部が閉じられていない場合、粉末は、開口部の直径が十分に大きい場合、重力下でその開口部を通して流動することができる。小さな開口部を通して流れる粉末は、錠剤のために有用な流動性質を有すると思われる。例えば、約24mm以下のFlodex測定(Flodex)が典型的には、商業的規模で高速錠剤化操作のために使用される。約20mm以下のFlodexは、高速錠剤化操作のために有用である。18mm又はそれ以下のFlodexは、特に高速錠剤化操作のために有用であると思われる。
【0245】
重度の積層を回避し、そして粉末層を振動又はタッピングしないで、底での開口部を閉鎖しないで、Flodexを、約70ccの試験粉末により保存物をまず軽く満たすことにより決定する。次に、開口部を開放する。これは、前記装置により供給されるシャッターを開口することにより達成され得る。他方では、シャッターが使用されない場合、ディスクにより固定された粉末充填保存物を、開口部を閉じるために乾燥した平らな表面上に設置する。次に、ゆっくり且つ均等に、貯蔵物を持ち上げ、粉末の流動を可能にする。
【0246】
いずれかの方法においては、粉末が開口部を通して流れる場合、透明なチャネルが粉末層内に放置される。粉末が開口部を通して流れない場合、粉末層内のアーチ形状の腔が開口部上に形成され、そしてアーチとして言及される。流動試験は、良好な流れのための最小開口部サイズが同定されるまで(Flodexとして言及される)、種々の開口部サイズで実施される。Flodexは、粉末が少なくとも3回の測定試験において開口部を通して流れないよりも多く開口部を通して流れる最小開口部直径である。
【0247】
例3.化合物(1)及び賦形剤の流動性質:
純粋化合物(1)及び種々の純粋錠剤化賦形剤の3種のロットについてのFlodexが表3に示される。純粋化合物(1)の3種のロットは化学的に同等であるが、しかし個々のロットの粒子サイズ及び形状により異なる(例1、図1及び2を参照のこと)。表3に示されるように、ロット4は最低のFlodexを示す。低いFlodexは丸型の粒子形状に寄与し、それは、もつれるロット70の特徴的な繊維状粒子塊状物、又は運動下で高い粒子間摩擦力を付与するロット71の特徴的な不規則形状の粒子よりも、より滑らかに流れる。それらの測定は、Flodexが、異なった形状学を有する粒子を含んで成る粉末の流動性質における差異を検出し、そして定量化するために十分に異なることを示す。
【0248】
【表3】
【0249】
表3に列挙されるAVICEL(商標) PH200は、190ミクロンの平均サイズを有する微晶性セルロースである(FMC Biopolymer Corporation, Philadelphia, PA)。METHOCELTM K4M SPは、ヒドロキシプロピルメチルセルロース(METHOCELTM K4M Standard Premium, Dow Chemical Company, Midland, MI)である。この置換されたセルロースポリマーは、約8重量%のヒドロキシプロピル含有物、約22重量%のメトキシル含有物、2%濃度で約4,000センチポアズの水中、呼称粘度、及び少なくとも75重量%が149ミクロ以下になるような粒子サイズを有する。METHOCELTM K4M (DC) (Dow Wolff Cellulosics, Midland, MI)は、METHOCELTM K4M SPと化学的に同一であるが、しかし約250ミクロンの大きな粒子サイズを有する。
【0250】
EUDRAGIT(商標) RLPO (Evonik Industries AG, Darmstadt, DE)は、ポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)(1:2:0.2)を含んで成るコポリマーである(USP/NFアッセイ、乾燥物質に基づいて8.85〜11.96%のアンモニオメタクリレート)。このアクリレートポリマーは、1モル当たり約150,000gの平均分子量、及び少なくとも90重量%が315ミクロン以下であるような粒子サイズを有する。EUDRAGIT(商標) RSPO (Evonik Industries AG, Darmstadt, DE)は、エチルアクリレート、メチルメタクリレート及び低含有率のメタクリル酸エステルのコポリマー(四級アンモニウム基を有する)である(USP/NFアッセイ、乾燥物質に基づいて4.48〜6.77%のアンモニオメタクリレート単位)。DI-TAB(商標)は、180ミクロンの呼称粒子サイズを有する二塩基リン酸カルシウム・二水和物(Innophos, Inc., Cranbury, NJ)である。
【0251】
表3に示されるように、錠剤配合物に使用される賦形剤のFlodexは、有意に異なった流動性質を示すことができる。低い〜高いFlodexの順位は、DI-TAB(商標)、AVICEL(商標) PH200、EUDRAGIT(商標) RLPO、METHOCELTM K4M SP及び METHOCELTM K4M DCである。
【0252】
例4.乾燥粉末ブレンドの流れ:
錠剤化賦形剤の2種の乾燥ブレンドを調製し、そして流動性質を決定した。ブレンドA08−011を、1インチ当たり20のワイヤを有する篩を通して、42.0 gの AVICEL(商標) PH200、43.4 g のMETHOCELTM K4M (SP)及び25.7 gのEUDRAGIT(商標) RLPOを容器中に連続的に通すことにより調製した。36gのDI−TAB(商標)及び1.5gのコロイド状二酸化珪素(CAB-O-SILTM M-5P, Cabot Corporation, Billerica, MA)を予備混合し、そして次に、同じスクリーンを通して容器中に通した。コロイド状二酸化珪素は、流動促進剤としてブレンドに含まれた。
【0253】
サイズ分けされた粉末を、5分間、V−ブレンダーにおいてタンブル−混合した。最終的に、40ワイア/インチを有する篩を前もって通された、1.5gのステアリン酸マグネシウム(Univar)を、粉末混合物に添加し、そして全組成を、5分間、V−ブレンドした。ステアリン酸マグネシウムは、錠剤圧縮の間、摩擦を低め、そして錠剤の滑らかな射出を促進するために錠剤化滑剤として作用した。得られるブレンド(A08−011)の流れは、20mmのFladexを示した。
【0254】
ブレンドA08−016を、ブレンドA08−011を調節するために使用される方法に類似する方法を用いて調製した。ブレンドA08−016は、ブレンドA08−011と組成において同等であるが、但しMETHOCELTM K4M SPがMETHOCELTM K4M DCにより置換された。得られるブレンドA08−016の流れを測定した。
【0255】
2種のブレンドの組成及びそれぞれのFlodexは、表4に提供される。純粋なMETHOCELTM K4M SPは28mmの高いFlodexを示し、そして純粋なMETHOCELTM K4M DCの流れは30mmのさらに高いFlodexを示すが(表3)、組合わされた賦形剤のブレンドは、それぞれ20及び15mmの値を示した。この結果は、表4に列挙される個々の成分のFlodexに基づいて予測されなかった。
【0256】
【表4】
【0257】
例5.化合物(1)を含む乾燥粉末ブレンドの流れ:
賦形剤及び単一ロットの化合物(1)により配合された2種の乾燥粉末ブレンドを調製し、そして流動性質を決定した。30.0gの化合物(1)、69.0 g の AVICEL(商標) PH200、86.7 g のMETHOCELTM K4M SP、及び51.3 g のEUDRAGIT(商標) RLPOを、20メッシュの篩を通してボウル中に連続的に通した。57.0gのDI-TAB(商標)及び3.0gの二酸化珪素を予備混合し、そして同じ20メッシュの篩を通してボウル中に通した。得られるサイズ分けされた粉末を、2クオートのV−ブレンダにおいて5分間タンブル−ブレンドした。最終的に、40メッシュの篩を通して前もってサイズ分けされた、3.0gのステアリン酸マグネシウムを、前記混合された粉末に添加し、そして5分間タンブル混合した。得られるブレンドA08−020のFlodexは22mmであった。
【0258】
第2ブレンドAO8−017を、ブレンドA08-020を調製するために使用される方法に類似する方法を用いて調製した。ブレンドA08-017の組成は、A08-020の組成と同等であったが、但しMETHOCELTM K4M DCをMETHOCELTM K4M SPにより置換した。2種のブレンドの組成及び流動性質が表5に要約されている。METHOCELTM K4M DCにより配合される化合物(1)を含むブレンドの流れは、22mmでMETHOCELTM K4M SPにより配合された同じブレンドに比較して、16mmのFlodexで有意に良好であった。この結果は、METHOCELTM K4M SP and METHOCELTM K4M DCがそれぞれ28mm及び30mmの類似する流動性質を有することを示す、表3に列挙される個々の成分の流れの結果に基づいて単に予測されなかった。
【0259】
【表5】
【0260】
例6.異なったロットからの化合物(1)を含む乾燥粉末ブレンドの流れ:
異なったロットの化合物(1)によりそれぞれ配合された、3種の乾燥粉末ブレンドを調製し、そして得られるブレンドの流動性質を比較した。ブレンドを調製するために使用されるサイズ分け及び混合方法は、例5に記載されるのと同じであった。組成及び対応するFlodex数が表6に要約されている。ブレンドB08-019及びBO8-020はそれぞれ、21mm及び13mmのFlodex数を示した。化合物(1)の個々のロットについてのFlodex数に基づいて(表3)、化合物(1)ロット70により調製されたブレンドが、化合物(1)ロット71により調製されたブレンドのFlodexに相当するFlodexを示したことが予測される。しかしながら、ロット71により配合されたブレンドのFlodexは、ロット70により配合されたブレンドのFlodexよりも有意に良好であり;それぞれ13mm対21mmであった。
【0261】
【表6】
【0262】
例7.化合物(1)及びEUDRAGIT(商標) RLPO又は EUDRAGIT(商標) RSPOを含む乾燥粉末ブレンドの流れ:
異なった品質のEUDRAGIT(商標)によりそれぞれ配合された2種の乾燥粉末ブレンドを調製した。16.5gのAVICEL(商標) PH200、32.9 g の METHOCELTM K4M DC、及び22.1 gのEUDRAGIT(商標) RLPOを、20メッシュの篩を通して通常の容器中に連続的に通した。16.5gのDI-TAB(商標)及び1.0gのコロイド状二酸化珪素を予備混合し、20メッシュの篩を通してサイズ分けし、そしてそのサイズ分けされた粉末に添加した。
【0263】
粉末をV−ブレンダーにおいて5分間タンブル混合した。次に、混合され、そしてサイズ分けされた粉末の半分を除いた。20%メッシュの篩を通して前もってサイズ分けされた10.0gの化合物(1)を、前記半分の粉末上に均一層として広げた。除かれた半分の粉末層を、化合物(1)の層上に適用し、そして得られる三層組成物を5分間タンブル混合した。最終的に、40メッシュの篩を通して前もってサイズ分けされた1.0gのステアリン酸マグネシウムを、前記混合物に添加し、そしてV−ブレンダーにおいて3.5分間タンブル混合し、ブレンドE08-007を形成した。
【0264】
第2ブレンドを、類似する混合方法を用いて調製した。ブレンドE08-013は、組成において同一であったが、但し、EUDRAGIT(商標) RSPOがEUDRAGIT(商標) RLPOにより置換されている。両ブレンドの組成が表7に提供される。
【0265】
【表7】
【0266】
例8.錠剤配合物の溶解プロフィール:
例5及び6に記載されるブレンドを、1/4−インチの丸型標準量凸錠剤型押し及びダイスを用いて、100mgの重量の錠剤に圧縮した。個々のタイプの錠剤を、37℃の温度で、50mMの一塩基性リン酸ナトリウム(pH6.8)900mlにおけるUSA櫂装置(タイプII)における薬物の溶解性について試験した。櫂撹拌速度は75回転/分であった。溶解試験の間、錠剤は、個々の容器の底で錠剤を位置決定するためにステンレス鋼製カゴ内に含まれた。
【0267】
EUDRAGIT(商標) RLPOを含んで成る錠剤(すなわち、 Blend E08-007)はまた、SR4−10錠剤配合物としても言及される。
【0268】
例6に記載されるブレンドを用いて調製された錠剤に関して、一定時間にわたって錠剤から放出される累積%化合物(1)を示す溶解プロフィールが図3に示されている。
【0269】
例7に記載されるブレンドを用いて調製された錠剤に関して、一定時間にわたって錠剤から放出される累積%化合物(1)を示す溶解プロフィールが図4に示されている。EUDRAGIT(商標) RSPOにより配合される錠剤の溶解プロフィールは、そのプロフィールにおいて三角形の記号により示され、そしてEUDRAGIT(商標) RLPOにより配合される錠剤の溶解プロフィールにおいて円形の記号により示される。
【0270】
例9.20mgの化合物(1)錠剤(SR4-20)の調製及び特徴化:
錠剤バッチを、パイロット規模で製造し、ブレンド及び錠剤化操作において許容できる錠剤重量及び薬物含有率の維持の実行可能性を評価した。粉末ブレンドを、20メッシュのスクリーンを備えたRussell Finex 篩 (Russell Finex, Pineville, NC)を通して、順に1,140 g の AVICEL(商標) PH200、1,974 g のMETHOCELTM K4M DC、1,026 g のEUDRAGIT(商標) RLPO、1,140 gのDI-TAB(商標)、及び60gのコロイド状二酸化珪素を通すことにより調製した。
【0271】
サイズ分けされた粉末を、1立方フィートのV−ブレンダーに移し、そして5分間タンブル混合した。約2,670gのブレンドを除き、そしてそばに置いた。20メッシュの篩を通して前もって手動的に通された、600gの化合物(1)(ロット4)を、ブレンダーに残る半分の粉末上に均等層を広げた。2,670gの前もって除かれた粉末をブレンダーに戻し、そしてその三層混合物を5分間ブレンドした。最終的に、40メッシュスクリーンを通され、前もってふるいにかけられた60gのステアリン酸マグネシウムをブレンドに添加し、そしてその混合物を4分間ブレンドし、6,000gのバッチの混合及びブレンド操作を完結した。
【0272】
2,400gの得られるブレンドを、5/16−インチの標準の丸型凹パンチ用具を備えたKilian T100 回転錠剤プレス(Kilian & Co., Inc., Horsham, PA)のホッパーに移した。錠剤を、1分当たり180個の錠剤の速度で圧縮し、それらは錠剤当たり200mgの呼称重量を有する。錠剤のサンプルを約5分ごとに集め、そして錠剤の重量を測定した。10個の錠剤の平均重量を個々の時点で決定した。図5に示される錠剤重量ヒストグラムは、錠剤重量が十分に維持されていることを示す。平均重量は、200mgの錠剤重量(錠剤当たり20mgの化合物(1)の±5%以内に維持された。
【0273】
錠剤における化合物(1)の含有率を決定した。9の点の個々で集められた3個の錠剤を、1.2ml/分の流速で35℃で、Alltech Platinum EPS C18 カラム (4.6 × 150 mm, 3 μm, 100Å) (Alltech Associates, Inc., Deerfield, IL)を用いて、高圧液体クロマトグラフィーにより化合物(1)について分析した。移動相は、0.02 M KH2PO4, pH 2.5/水/アセトニトリル, 461/44/495 (v/v/v)であり、そして検出波長は220nmであった。錠剤を、アセトニトリル:水(80:20)中、1%(w/v)ドデシル硫酸ナトリウム(SDS)に溶解し、そして30分間、音波処理した(標的濃度0.2mg/mlの化合物(1))。すべてのサンプルを、分析の前、0.45μmのナイロンフィルターにより濾過した。少量の界面活性剤(すなわち、ドデシル硫酸ナトリウム(SDS))添加は、薬物の不溶性賦形剤への結合を最少にすることが示されており、改良された薬物抽出効率をもたらした。
【0274】
錠剤についての化合物(1)含有率ヒストグラムが図6に示されており、そして20mgの標的化合物(1)含有量に基づかれている。錠剤サンプルを、錠剤化実施の間、一定間隔で入手し、ここでその間隔は合計錠剤化実施(100%)の%として同定される。錠剤当たり20mgの化合物(1)含有量は、グラフの縦座標に基づいて100%に等しい。錠剤当たりの平均化合物(1)含有量は、すべての時点で標的用量の±5%以内に維持された。個々の錠剤における化合物(1)含有率の範囲は、20mgの標的化合物(1)含有率の92.6%低値〜105.7%の高値の範囲であった。
【0275】
錠剤の厚さをダイヤルキャリパにより測定し、約4.02mmであることがわかった。錠剤の圧縮強さを、錠剤硬度計上で測定し、約8.1kポンドであることがわかった。錠剤を例6に記載される方法を用いて、溶解について試験した。例8に従って決定される、18時間で20mgの錠剤からの化合物(1)の平均累積放出性は、図7に示されるように約45%であった。
【0276】
例10.30mg化合物(1)錠剤(SR4-30)の調製及び特徴化:
例9に記載されるブレンドの 3,600gを、3/8−インチの標準の丸型凹パンチ用具を備えたKilian T100 回転錠剤プレスのホッパーに移した。錠剤を、1分当たり180個の錠剤の速度で圧縮し、それらは錠剤当たり300mg(錠剤当たり30mgの化合物(1))の呼称重量を有する。錠剤のサンプルを約5分ごとに集め、そして錠剤の重量を測定した。10個の錠剤の平均重量を個々の時点で決定した。図8における錠剤重量ヒストグラムは、錠剤重量が300mgの標的重量の5%以内に維持されたことを示す。
【0277】
錠剤における化合物(1)の含有量をまたモニターした。12時点の個々で集められた3個の錠剤を、例9に記載されるようにHPLCを用いて化合物(1)について分析した。化合物(1)含有率のヒストグラムが図9に示されている。錠剤当たり30mgの化合物(1)含有率は、グラフ縦軸上で100%の標的含有率に等しい。錠剤当たりの平均薬物含有率を、錠剤化実施を通して30mgの標的量の±5%以内に維持した。個々の錠剤における薬物含有率の範囲は、30mgの標的重量の95.0%の低値〜103.6%の高値の範囲であった。
【0278】
錠剤の厚さをダイヤルキャリパにより測定し、約4.18mmであることがわかった。錠剤の圧縮強さを、錠剤硬度計上で測定し、約9.5kポンドであることがわかった。錠剤を例8に記載される方法を用いて、溶解について試験した。例8に従って決定される、18時間で30mgの錠剤からの化合物(1)の平均累積放出性は、図10に示されるように約39%であった。
【0279】
例11.10mgの化合物(1)の錠剤(SR4-10)の調製及び特徴化:
10mgの化合物(1)を含んで成り、そして約175.1mgの重量の持効性放出錠剤を例9及び10に記載される方法に類似する方法を用いて調製した。SR4錠剤投与形の組成物は、表8に要約されている。
【0280】
【表8】
【0281】
例12.10mgの化合物(1)SR3錠剤の調製:
化合物(1)及びアンモニオアルキルメタクリレートポリマー、EUDRAGIT(商標) RL 30D, (SR3)を含んで成る持効性放出錠剤を、300gのバッチサイズで調製した。
【0282】
次の量の成分を用いて、300gのバッチを調製した:12.0 g の化合物(1)、128.7 g の 微晶性セルロース (AVICEL(商標) PH200, FMC Corp., Philadelphia, PA)、106.5 gの ヒドロキシプロピルメチルセルロースMETHOCELTM K4M (METHOCELTM K4M, Dow Chemical)、51.3 g のアンモニオアルキルメタクリレートコポリマー(EUDRAGIT(商標) RL 30D, Degussa)、及び1.5 g のステアリン酸マグネシウム (HYQUAL(商標)植物源, Mallinckrodt, Phillipsburg, NJ)。10mgの化合物(1)及びアンモニオアルキルメタクリレートポリマーを含んで成る持効性放出錠剤(SR3)における成分の量が表9に提供される。
【0283】
【表9】
【0284】
化合物 (1) (12 g)、微晶性セルロース(AVICEL(商標)PH200, FMC Biopolymer) (127.8 g)、 及びヒドロキシプロピルメチルセルロース(METHOCELTM K4M SP, Dow Chemical Co.) (106.5 g)を計量し、#20%メッシュのステンレス鋼製スクリーンを通してスクリーンし、そしてV−ブレンダー(2 quart, Model MB-1, Globepharma, New Brunswick, NJ)において5分間、混合した。
【0285】
ブレンドを放し、そして1 Lのボウルを有する KG-5 Mixer/Granulator (Key International, Englishtown, NJ)を用いて、高剪断で湿式粒状化した。湿式粒状化は、100mlの水、1mmの管直径、250rpmのインペラー速度、及び1500rpmのチョッパー速度を用いて実施された。
【0286】
湿った粒状物を、25 SCFMの入口、45℃の入口空気温度、30℃以下の出口空気温度、及び200〜900mmH2Oのフィルター圧力を用いて、Fluid Bed Model 0002 (Fluid Air, Aurora, IL)グラニュレーター/ドライヤーにおいて乾燥した。乾燥後の標的重量損失は、約3%以下であった。
【0287】
乾燥生成物を、0.079インチのガータ型スクリーン(識別番号7L079G03123-(2007)0503)及びステンレス鋼、150粗粒(Ra1.06)表面仕上げを用いて、2500rpmの操作速度でComil Model U5ミル (Quadro Engineering, Inc., Millburn, NJ)に通し、さらなる圧縮のための微粉砕された材料を得た。
【0288】
粒状物を、KG-5 ミキサー/グラニュレーターに戻し、そして250rpmのインペラ速度及び1500rpmのチョッパー速度を混合しながら、2.4ml/分で、171gのEUDRAGIT(商標)RL 30D(Degussaから入手できる、約125,000ドルトン〜約175,000ドルトンの分子量により特徴づけられる、タイプA、アンモニオアルキルメタクリレートコポリマー30%水性分散体)を添加することにより、アンモニオアルキルメタクリレートコポリマー及び賦形剤を含んで成るブレンドにより被覆した。次に粒状物を乾燥した。
【0289】
ステアリン酸マグネシウム(1.5g)(HYQUAL(商標)植物源)を計量し、そして#40メッシュスクリーンに通した。微粉砕された材料及びステアリン酸マグネシウムを、V−ブレンダーに添加し、そして25rpmで5分間ブレンドした。
【0290】
ブレンドされた材料を放し、そして圧縮し、250mgの合計重量及び10mg(4重量%)の化合物(1)を有する錠剤を形成した。5/16−インチ直径のIPT標準同心の上下パンチ、及び5/16−インチ(ID)×1.1875ODストレート穴鉄鋼ダイを備えた10ステーション、Mini Press-IIBD (Globepharma, New Brunswick, NJ)を用いて、錠剤を圧縮した。錠剤は、約6kp〜約9kp(59〜88ニュートン)の最終平均硬度を有した。
【0291】
例13.錠剤配合物における化合物(1)の化学的安定性:
温度及び湿度の種々の条件下での化合物(1)の開放皿化学安定性を、SR3-10及びSR4-10錠剤配合物について決定した。錠剤を、温度及び湿度に対して3ヶ月までの間、暴露し、そしてR−バクロフェンの量及びラクタム分解(R−(4−クロロ−フェニル)−ピロリジン−2−オン)を決定した。結果は表10に示される。SR4-10配合物は、3ヶ月での個々の貯蔵条件で一定した低いラクタムレベルにより示されるように、SR3-10配合物に比較して、卓越した化学安定性を示した。
【0292】
【表10】
【0293】
例14.化合物(1)を含んで成る錠剤投与形の投与に続くヒト患者におけるR−バクロフェンの定常状態薬物力学:
健康な成人ボランティアにおける10mgのSR3及び10mg、20mg及び30mgのSR4錠剤配合物の定常状態薬物力学を比較する、ランダム化された、多重投与、4−処理、4−期間クロスオーバー研究を実施した。
【0294】
研究1日目での投与の前、患者を4系列の1つにランダム化した。1日目で、すべての患者は朝食中の10分以内にSR3錠剤として20mg(2×10mg)の用量の化合物(1)を受けた。2日目で、すべての患者は、朝食の完結の後10分以内に、SR3錠剤として30mg(3×10mg)の用量の化合物(1)を受けた。3日目で、すべての患者は、朝食の完結の後10分以内にSR3錠剤として40mg(4×10mg)の用量の化合物を受けた。
【0295】
患者は、期間1(4日目〜7日目)、期間2(8日目〜11日目)、期間3(12日目〜15日目)及び期間4(16日目〜19日目)の間、ランダム化された態様で次の4種の処理の1つを受けた:
【0296】
処理A:朝食の完結後10分以内に1日1度(QD)、4日間、6×10mgの化合物(1)SR3錠剤;
処理B:朝食の完結後10分以内に1日1度(QD)、4日間、6×10mgの化合物(1)SR4-10錠剤;
処理C:朝食の完結後10分以内に1日1度(QD)、4日間、3×20mgの化合物(1)SR4-20錠剤;及び
処理D:朝食の完結後10分以内に1日1度(QD)、4日間、2×30mgの化合物(1)SR4-30錠剤。
個々の処理グループは、16人の健康な成人ボランティアを含んだ。
【0297】
血液サンプル(約4ml)を、投与の前、及びK2EDTAを含む管中への投与後、一定間隔で患者から集めた。血液サンプルアリコートを、メタノールによりすぐに急冷し、化合物(1)のさらなる加水分解を妨げた。2つのアリコート(それぞれ1ml)をすぐに、Nalgene管に移し、そして3mlのメタノールにより急冷した。血液サンプルアリコートを、−80℃で冷凍庫に貯蔵した。血液サンプルアリコートを、敏感で且つ特異的なLC-MS/MS方法を用いて、完全な血液上清液におけるR−バクロフェン及び化合物(1)について分析した。
【0298】
血液中のR−バクロフェン及び化合物(1)についての濃度データを、WINNONLIN(商標) Software バージョン4.1 (Pharsight Corporation, Mountain View, CA)を用いて、非コンパートメント方法により分析した。濃度データ及び薬物力学パラメーターを、SIGMAPLOTTM バージョン9.0 (Systat Software Inc., Point Richmond, CA)を用いてプロットした。実時間点を、薬物力学パラメーターの計算のために使用した。
【0299】
最大の観察される薬物の濃度(Cmax)及びCmaxまでの時間(Tmax)を観察により入手した。見かけ排除半減期(T1/2)を、末端過程における3又はそれ以上の対数−転換されたデータ点の直線回帰法により決定した(In(2)/Kelとして計算され、ここでKelは、対数濃度対時間曲線の末端直線部分の直線回帰により計算される末端排除速度定数である)。直線回帰モデル下の領域が、Windows(登録商標) (SAS Institute, Cary, NC)についてのSASTMバージョン9.1を用いて、AUC対用量及びCmax対用量のために適合された。両モデルにおいては、用量効果が、不等間隔値のための直交多項式係数を用いてパラメーター化された。
【0300】
血液サンプルを、7, 11, 15及び19日目に入手した。期間4に続いて、患者は、4日間にわたって養生法から除外された。研究の間、患者は、約2000kカロリーの毎日の合計カロリー量を有する標準化された臨床食事(脂質からのエネルギーの約30%)を供給された。
4種の処理の間、定常状態で決定される、血液中のR−バクロフェンについての平均薬物力学的パラメーターが表1に要約され、そして薬物力学的パラメーターが図11に示される。3種のSR4配合物についてのAUCss, 24値は、SR3配合物について約1500ng×時/mlのAUCss, 24に比較して、約1750ng×時/ml〜約1850ng×時/mlの範囲であった。
【0301】
例15. 不統一粉末組成物を用いて調製されたSR4錠剤(ベンチスケール):
500gの粉末ブレンドを調製した。最初に、28.6gの化合物(1)中、ヘラにより20−メッシュ篩に通し、そして得られるサイズ分けされた薬物をそばに置いた。次に、105.7 g のAVICEL(商標) PH200、164.5 gのMETHOCELTM K4M 直接圧縮グレード(DC)、85.4 g のEUDRAGIT(商標) RLPO、 5.2 g の二酸化珪素グレード M5P、 及び105.7 g のDI-TAB(商標)としての二塩基性リン酸カルシウム・二水和物を、ヘラを用いて、20−メッシュ篩に連続的に通した。次に、サイズ分けされた賦形剤粉末を、2クォーツのツイン−シェルブレンダーに移し、そして25回転/分の回転速度で5分間タンブル混合した。
【0302】
233gの得られるサイズ分けされ、そして混合された賦形剤粉末を除いた。次に、サイズ分けされた化合物(1)を、ミキサーにおける半分の粉末層上に均一の厚さの層として広げた。233gの除去された粉末を、ブレンダーのシェルに戻し、そして薄層組成物を25rpmで5分間ブレンドした。次に、5.2gのステアリン酸マグネシウムを、60メッシュの篩にヘラにより通し、そして粉末層に添加した。最終的に、粉末の層を、25rpmで4分間タンブル混合した。これは、不統一粉末ブレンド形成した。
【0303】
得られる不統一粉末ブレンドを、5/16インチの丸型標準凹形型押しを備えた2つのステーションを有するKorsch XL 100錠剤プレス(Korsch America Inc., South Easton, MA)のホッパーに移した。粉末を、個々の錠剤が10mgの呼称含有量の化合物(1)を含むよう、175mgの呼称重量を有する錠剤に圧縮した。開始で及び圧縮工程の間、錠剤のサンプルを集めた。得られる錠剤サンプルを、高性能液体クロマトグラフィーにより化合物(1)の含有率について分析した。さらに、錠剤サンプルを、イオンクロマトグラフィーを用いて、DI-TAB(商標)含有率について分析した。
【0304】
得られる錠剤含有率データは図12にプロットされている。図12Aは、錠剤化実施の間、錠剤における化合物(1)の含有率を示す。個々のデータ点は、錠剤化実施の間、特定点でサンプリングされ、そして完全な錠剤化実施(100%)の%として表される10.0mgの化合物(1)の標的量に対する、3個の錠剤における化合物(1)の平均含有率を表す。個々のデータ点についての誤差バーは、個々の時点での3種の測定される値の相対的偏差を表す。化合物(1)含有率データへの直線回帰適合の傾斜は、+0.121であった。その実施を通して化合物(1)の含有率の相対的標準偏差は6.34%であった。図12Bは、同じ錠剤化実施の間、個々の錠剤内のDI−TAB(商標)の含有率を示す。DI-TAB(商標)含有率データに対する直線回帰適合の傾斜は、-0.099であった。その実施を通してのDI-TAB(商標)含有率の相対的標準偏差は、5.60%であった。
【0305】
それらのヒストグラムは、錠剤における化合物(1)の含有率が錠剤化実施の開始で低いことを示す。さらに、錠剤内化合物(1)の含有率は、錠剤化実施の間、上昇する。化合物(1)の含有率はその実施の最後で高いことはまた明白である。さらに、錠剤内のDI-TAB(商標)の含有率は、反対の傾向に従う。DI-TAB(商標)の含有率はその実施の開始で高く、そして錠剤化実施を通して着実に低下する。
【0306】
この現象は、それらの錠剤を調製するために使用される粉末ブレンドの不統一性質及びブレンドを含んで成る主要成分の嵩密度により説明される。この不統一ブレンド内の主要成分の嵩密度値が表11に要約されている。
【0307】
【表11】
【0308】
DI-TAG(商標)の密度は、いずれかの他の成分の密度を越える。それは、ブレンド内のいずれか他の成分の密度の2倍以上である。従って、不統一粉末ブレンド内で、そのブレンドの単純な取り扱い及び加工が、ブレンドの底への重力下での密なDI-TAB(商標)成分の沈降を引起すのに十分な振動を誘発する。そのようにする際、低い密度の成分、例えばバクロフェンプロドラッグ化合物(1)が、ブレンドの上部に置換される。ブレンドが錠剤プレスのホッパーに供給される場合、比較的高い画分のDI-TAB(商標)及び比較的低い画分の化合物(1)を含む、不統一粉末層の最低部分がまず、錠剤に転換される。従って、実施の開始での錠剤は、低い薬分含有率及び高いDI-TAB(商標)含有率を有する。対照的に、錠剤化実施の最後での錠剤は、比較的高い薬物含有率及び比較的低いDI-TAB(商標)含有率を有する。従って、図12におけるデータの直接回帰分析は、化合物(1)の含有率を表す曲線の正の傾斜、及びDI-TAB(商標)の含有率を表す曲線について類似する大きさの対応する負の傾斜に影響を及ぼす。
【0309】
例16.整構造の粉末組成物を用いて調製されるSR4錠剤(ベンチスケール):
整構造の粉末ブレンドを調製した。最初に、33.0gのバクロフェンプロドラッグ化合物(1)、5.94gの二酸化珪素M5P、及び122.1gの DI-TAB(商標)を、ポリエチレンバッグに配置し、そして2分間タンブル混合した。次に、予備混合された粉末を、円錐状ミル(Quadro Comil Model U5, Quadro Engineering Corp., Waterloo, Ontario, Canada)に通した。その円錐上ミルは、457ミクロンの直径を有する丸型メッシュ開口部を有するスクリーンを固定され、そして3000rpmのインペラ速度で作動した。
【0310】
この微紛化工程は、バクロフェンプロドラッグ化合物(1)により被覆され、そして整構造の粉末ブレンドを調製するために使用されるDI-TAB(商標)粒子をもたらした。
【0311】
整構造の粉末ブレンドの調製を続けるために、172.8gのMETHOCELTM K4M DCを、20メッシュの篩に通し、そして2−クォーツの双シェルブレンダー中に充填した。次に、146.4gの円錐状ミル粉砕された化合物(1)/DI-TAB(商標)/二酸化珪素混合物を、ヒドロキシプロピルメチルセルロース上に均等な厚い層として双シェル混合物中に充填した。次に、89.7 g of EUDRAGIT(商標)RLPO 及び 111.0 g のAVICEL(商標)PH200を、20メッシュの篩を通してサイズ分けし、そして双シェルミキサー中に充填した。次に、組成物を25rpmで10分間タンブル混合した。次に、100gのその混合物を除いた。5.4gのステアリン酸マグネシウムを、40メッシュの篩に通し、そして100gのブレンド中にヘラにより撹拌した。最終的に、ステアリン酸マグネシウムを含むサンプル100gをブレンドに添加し、そして全バッチを25rpmで4分間タンブル混合した。これは整構造の粉末ブレンドを形成した。
【0312】
得られる整構造の粉末ブレンドを、Korsch XL100錠剤プレスのホッパーに移した。プレスは、5/16インチの丸型標準凹型パンチ及び2ステーションで設定されるダイを適合された。ブレンドを、10mgの化合物(1)のユニット用量を供給するために、175mgの呼称標的重量を用いて錠剤に圧縮した。錠剤サンプルを、例15に記載される方法に従って、集め、そして分析した。得られるデータは、図13に示される。図13は、全錠剤化実施(100%)の間、一定間隔で、化合物(1)の標的含有率(±SD)に標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰適合の傾斜は、+0.028であった。バッチを通しての化合物(1)の含有率の相対的標準偏差は1.00%であった。
【0313】
図12における不統一ブレンドの含有率結果と図13における整構造ブレンドの結果との比較は、整構造粉末ブレンドにより提供される化合物(1)含有率均質性において有意な改良性を示す。整構造ブレンドから製造された錠剤中の化合物(1)含有率についての直線回帰の傾斜は、+0.121の不統一ブレンドから製造された錠剤中の化合物(1)含有率についての傾斜に比較して、+0.028である。当業者は、傾斜がゼロに近いほど、バッチを通しての平均含有率がより均等になることを理解しているであろう。同様に、整構造粉末ブレンドから製造された錠剤についての含有率の相対的標準偏差(RSD)は、1.00%であった。これは、6.34%の相対的標準偏差を有した不統一ブレンドから製造された錠剤についてよりも、錠剤内の化合物(1)含有率における、より良好な含有率均一性を示唆する。
【0314】
例17.不統一粉末ブレンドを用いて調製されたSR4錠剤(パイロット製造スケール):
次の工程を用いて、6kgの不統一粉末ブレンドを調製した。最初に、0.340kgの化合物(1)を、1インチ当たり20のワイヤのメッシュを有する篩を通してスクリーンした。次に、1.258 kg のAVICEL(商標)PH200、0.061 kgのコロイド状二酸化珪素、 1.958 kgのMETHOCELTM K4M (DC)、1.258 kgのDI-TAB(商標)、及び 1.017 kgのEUDRAGIT(商標)RLPOを、20メッシュ篩を備えたFinex電気シフター(Russell Finex Inc.)に通した。サイズ分けされた賦形剤を、1立方フィートのV−ブレンダー中に充填し、そして25rpmの回転速度で5分間タンブル混合した。得られる賦形剤ブレンドの半分を除いた。
【0315】
次に、サイズ分けされたプロドラッグ化合物(1)を、前記半分の粉末層上に積層した。残りの半分の賦形剤層を化合物(1)上に積層した。次に、得られる3層組成物を、25rpmの回転速度で5分間V−ブレンダーにおいてタンブル混合した。ステアリン酸マグネシウム(0.061kg)を、1インチ当たり40のワイヤを有するメッシュに通し、そして次に、粉末層に添加した。得られる組成物を、25rpmで4分間タンブル混合した。これは、高い剪断混合を伴わないで調製されたSR4ブレンドを形成した。
【0316】
得られる6kgのブレンドを、5/16インチの標準凹型丸形パンチ及びダイの9ステーションを備えたKilian T100回転錠剤プレス(IMA Kilian GmbH & Co. KG, Koeln, DE)に供給した。約175mgの重量及び約7kNの硬度を有する錠剤を、25rpmの速度で圧縮した。個々の錠剤は、10mgの呼称含有量の化合物(1)を含んだ。錠剤サンプルを、約37,000個の錠剤ごとに集めた。化合物(1)の含有率を、個々のサンプリング点で3個の錠剤において高性能液体クロマトグラフィーを用いて決定した。
【0317】
得られるヒストグラムが図14に示されている。図14は、合計の錠剤化実施(100%)の間、一定間隔で化合物(1)の標的含有率(±SD)に対して標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰適合の傾斜は、+0.051であった。約340,000個の錠剤のバッチを通しての化合物(1)含有率の相対的標準偏差は、4.94%であった。
【0318】
例18.整構造粉末ブレンドを用いて調製されたSR4錠剤(パイロット製造スケール):
次の工程を用いて、6kgの整構造粉末ブレンドを調製した。A−TAB(商標)の粒子を、次の方法を用いて、化合物(1)により被覆した。最初に、0.600kgのA−TAB(商標)を、8クオートのV−ブレンダーに充填した。次に、0.350kgの化合物(1)を、A-TAB(商標)上に積層した。0.695kgの追加のA-TAB(商標)を、化合物(1)上に1つの層として適用した。次に、3層組成物を、25rpmで4分間、ブレンダーによりタンブル混合した。得られる混合物を、813ミクロンの直径を有する丸形開口部を有するスクリーン、長方形のインペラー及び0.225インチのインペラースペーサーを備えたQuadro Comil Model 197円錐形ミルに通した。インペラーは、2000rpmの速度で回転した。混合物を、1分当たり約0.3kgの速度でミルにすくい取り、A−TAB(商標)により被覆された化合物(1)を生成した。
【0319】
次に、1.000 kg のMETHOCELTM K4M DC及び 0.063 kg の二酸化珪素を、8クォーツのV−ブレンダーに添加し、そして25rpmで2分間、混合した。得られるヒドロキシプロピルメチルセルロース(HPMC)混合物を、A−TAB(商標)により被覆された化合物(1)を調製するために使用されたのと同じパラメーターを用いて、円錐形ミルに通した。
【0320】
HPMC混合物を、1 ft3 Loedige 高-剪断ブレンダー(Gebrueder Loedige Maschinenbau GmbH, Paderborn, DE)に移した。次に化合物(1)を含む混合物を、HPMC混合物上に1つの層として適用した。次に、1.309 kgのAVICEL(商標)PH200、 及び1.046 kgの EUDRAGIT(商標)RLPOを、ブレンダーに層として添加した。得られる4層混合物を、高い剪断で5分間ブレンドした。
【0321】
得られる混合物を、1ft3のV−ブレンダーに移した。40メッシュの篩を前もって通されたステアリン酸マグネシウム(0.046kg)を混合物に添加し、そして25rpmで4分間ブレンドした。その混合物をドラムに放し、整構造粉末ブレンドの形成を完成した。
【0322】
得られる整構造粉末ブレンドを、フィードフレーム中、5/16標準凹型丸形パンチ及びダイ及び2−櫂フィダーの9ステーションを備えたkilian T100を用いて、錠剤に圧縮した。プレスの砲塔は 25rpmで回転し、そして櫂供給装置は6rpmで作動した。ブレンドをプレスのホッパーにすくい取った。175mgの呼称標的重量及び10.0mgの呼称標的化合物(1)含有量を有する錠剤を圧縮した。錠剤の硬度は約7kpであり、そして錠剤の厚さは約3.6mmであった。錠剤の破砕性は0.3%であった。
【0323】
錠剤サンプルを、圧縮の実施の間、約3,375個の錠剤の間隔で集めた。3個の錠剤を、集められた間隔の個々で化合物(1)含有率について分析した。得られるデータを、図15に表されるヒストグラムにプロットした。図15は、合計の錠剤化実施(100%)の間、一定間隔での化合物(1)の標的含有率(±SD)に対して標準化された化合物(1)の錠剤含有率を示す。データに対する直線回帰の傾斜は+0.033であり、そして相対的標準偏差は1.95%であった。
【0324】
例18に記載される整構造粉末ブレンドから製造された錠剤の均一性に比較して、例17に記載されるような不統一の粉末ブレンドにより製造された錠剤における含有物均一性が表12に要約されている。ヒストグラムの傾斜がゼロに近づくほど、バッチの開始から最後までの薬物含有率はより均一である。相対的標準偏差が小さいほど、バッチ内の薬物含有率はより均一である。整構造粉末ブレンドから製造された錠剤は、両測定によれば、化合物(1)の有意に良好な均一性を示す。
【0325】
円錐形ミルへの化合物(1)及びA−TAB(商標)のブレンドの通過は、化合物(1)により被覆されたA−TAB(商標)コアー粒子を生成した。それらの被覆された粒子は、ブレンドの他の成分に付着し、A−TAB(商標)の分離を低めた三次元構造体を形成した。高い剪断ブレンドはさらに、粒子サイズを低め、そしてブレンドを均質化した。
【0326】
【表12】
【0327】
例19.整構造粉末ブレンドを用いて調製されたSR4錠剤の溶解:
錠剤を、丸形標準凹形装置を備えたKorsch XL100錠剤プレスを用いて、10kN〜20kNの圧力で調製した。10mg、20mg、30mg及び40mgの化合物(1)を含むSR4錠剤の組成が表13に要約されている。SR4錠剤についての溶解プロフィールが表14に要約されている。
【0328】
【表13】
【0329】
【表14】
【0330】
最終的に、本明細書に開示される態様を実現するための他の手段が存在することは注目されるべきである。従って、本発明の態様は、例示的であって、制限的ではない。さらに、請求項は本明細書に与えられる詳細に制限されるものではなく、そしてそれらの十分な範囲及びその同等物に権利が与えられる。
【図1A】
【図1B】
【図1C】
【図2A】
【図2B】
【図2C】
【特許請求の範囲】
【請求項1】
投与形の合計重量に基づいて;
約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約15重量%〜約40重量%の微晶性セルロース;
約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び
約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る経口錠剤投与形。
【請求項2】
投与形の合計重量に基づいて;
約5重量%〜約15重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約17重量%〜約33重量%の微晶性セルロース;
約20重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び
約5重量%〜約20重量%の放出速度−制御ポリマーを含んで成る、請求項1記載の投与形。
【請求項3】
希釈剤、充填剤、滑剤及びそれらのいずれかの組合せから選択された1又は複数の賦形剤を含んで成る、請求項1又は2記載の投与形。
【請求項4】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形で存在する、請求項1〜3のいずれか1項記載の投与形。
【請求項5】
前記遊離酸形で存在する(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が結晶性である、請求項4記載の投与形。
【請求項6】
前記放出速度−制御ポリマーが、乾燥物質基準に基づいて約8.8%〜約12.0%のアンモニオメタクリレート単位を有するポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)コポリマーである、請求項1〜5のいずれか1項記載の投与形。
【請求項7】
前記放出速度−制御ポリマーが、乾燥物質基準に基づいて約4.4%〜約6.8%のアンモニオメタクリレート単位を有するポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)コポリマーである、請求項1〜5のいずれか1項記載の投与形。
【請求項8】
約0.5重量%〜約1.5重量%のステアリン酸マグネシウム、約0.5重量%〜約1.5重量%のコロイド状二酸化珪素及び約15重量%〜約30重量%の二塩基性無水リン酸カルシウムを含んで成る、請求項1〜7のいずれか1項記載の投与形。
【請求項9】
前記投与形の合計重量が約100mg〜約600mgである、請求項1〜8のいずれか1項記載の投与形。
【請求項10】
前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, Type II )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:
約10%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
約15%〜約35%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;
約20%〜約50%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして
約30%〜約80%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される、請求項1〜9のいずれか1項記載の投与形。
【請求項11】
前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, Type II )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:
約10%〜約20%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
約20%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;
約25%〜約45%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして
約35%〜約55%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される、請求項1〜9のいずれか1項記載の投与形。
【請求項12】
約60mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量での16人の健康な成人患者への経口投与に続いて、約202±56ng/mlのCss, max;約3.9±1.0時間のTss, max;約19ng/mlのCss, 12;約10.9±3.8時間のTss, 1/2;及び1803±420ng・時/mlのAUCss, 24により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールが提供される、請求項1〜11のいずれか1項記載の経口投与形。
【請求項13】
約60mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量での16人の健康な成人患者への経口投与に続いて、約8〜約15のCss, max/Css,12により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールが提供される、請求項1〜12のいずれか1項記載の経口投与形。
【請求項14】
USP1216に従って決定される場合、約0.5重量%以下の破砕性を有する、請求項1〜13のいずれか1項記載の経口投与形。
【請求項15】
約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約15重量%〜約40重量%の微晶性セルロース;
約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び
約3重量%〜約30重量%の放出速度−制御ポリマー(ここで前記重量%は投与形の合計重量に基づかれる)、を含んで成る配合物を乾式ブレンドし、;そして
前記ブレンドされた配合物を圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法。
【請求項16】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の乾燥粉末が、約20μm〜約60μmの直径を有する丸型凝集体により特徴づけられる、請求項15記載の方法。
【請求項17】
前記ブレンドされた配合物が、約22mm以下のFlodexを示す、請求項15又は16記載の方法。
【請求項18】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形で存在する、請求項15〜17のいずれか1項記載の方法。
【請求項19】
請求項15〜18のいずれか1項記載の方法により調製される、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩を含んで成る経口錠剤投与形。
【請求項20】
リン酸二カルシウム及び(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩をブレンドし、第1ブレンドを供給し;
前記第1ブレンドを円錐型ミルに通し;
ヒドロキシプロピルメチルセルロース及びコロイド状二酸化珪素を乾燥ブレンドし、第2ブレンドを形成し;
前記第2ブレンドを円錐形ミルに通し;
前記第1ブレンド、前記第2ブレンド、微晶性セルロース、及び放出速度−制御ポリマーを、高剪断ブレンダーにおいてブレンドし、第3ブレンドを形成し;
前記第3ブレンドと共にステアリン酸マグネシウムをブレンドし;そして
前記第3ブレンドを圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法。
【請求項21】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形及び結晶形で存在する、請求項20記載の方法。
【請求項22】
請求項20又は21のいずれか1項記載の方法により調製される、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩を含んで成る経口錠剤投与形。
【請求項23】
痙攣、胃食道逆流病、嘔吐、咳嗽、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、神経因性疼痛、筋骨格痛及び尿失禁から選択された、患者における疾病の処理方法であって、少なくとも1つの請求項1〜13のいずれか1項記載の経口投与形を、そのような処理の必要な患者に経口投与することを含んで成る方法。
【請求項24】
前記疾病が痙攣である、請求項23記載の方法。
【請求項25】
前記疾病が胃食道逆流病である、請求項23記載の方法。
【請求項26】
前記疾病が神経因性疼痛であり、そして前記神経因性疼痛が帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症から選択される、請求項23記載の方法。
【請求項27】
前記疾病が尿失禁である、請求項23記載の方法。
【請求項1】
投与形の合計重量に基づいて;
約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約15重量%〜約40重量%の微晶性セルロース;
約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び
約3重量%〜約30重量%の放出速度−制御ポリマーを含んで成る経口錠剤投与形。
【請求項2】
投与形の合計重量に基づいて;
約5重量%〜約15重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約17重量%〜約33重量%の微晶性セルロース;
約20重量%〜約35重量%のヒドロキシプロピルメチルセルロース;及び
約5重量%〜約20重量%の放出速度−制御ポリマーを含んで成る、請求項1記載の投与形。
【請求項3】
希釈剤、充填剤、滑剤及びそれらのいずれかの組合せから選択された1又は複数の賦形剤を含んで成る、請求項1又は2記載の投与形。
【請求項4】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形で存在する、請求項1〜3のいずれか1項記載の投与形。
【請求項5】
前記遊離酸形で存在する(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が結晶性である、請求項4記載の投与形。
【請求項6】
前記放出速度−制御ポリマーが、乾燥物質基準に基づいて約8.8%〜約12.0%のアンモニオメタクリレート単位を有するポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)コポリマーである、請求項1〜5のいずれか1項記載の投与形。
【請求項7】
前記放出速度−制御ポリマーが、乾燥物質基準に基づいて約4.4%〜約6.8%のアンモニオメタクリレート単位を有するポリ(エチルアクリレート、メチルメタクリレート、トリメチルアンモニオエチルメタクリレートクロライド)コポリマーである、請求項1〜5のいずれか1項記載の投与形。
【請求項8】
約0.5重量%〜約1.5重量%のステアリン酸マグネシウム、約0.5重量%〜約1.5重量%のコロイド状二酸化珪素及び約15重量%〜約30重量%の二塩基性無水リン酸カルシウムを含んで成る、請求項1〜7のいずれか1項記載の投与形。
【請求項9】
前記投与形の合計重量が約100mg〜約600mgである、請求項1〜8のいずれか1項記載の投与形。
【請求項10】
前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, Type II )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:
約10%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
約15%〜約35%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;
約20%〜約50%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして
約30%〜約80%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される、請求項1〜9のいずれか1項記載の投与形。
【請求項11】
前記経口投与形からの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の放出が、75rpm(USP, Type II )で撹拌される、37℃での50mMのリン酸ナトリウム緩衝液(pH6.8)において、次の通りにインビトロ溶解プロフィールを示し:
約10%〜約20%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約4時間以内に放出され;
約20%〜約30%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約8時間以内に放出され;
約25%〜約45%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約12時間以内に放出され;そして
約35%〜約55%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩が約18時間以内に放出される、請求項1〜9のいずれか1項記載の投与形。
【請求項12】
約60mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量での16人の健康な成人患者への経口投与に続いて、約202±56ng/mlのCss, max;約3.9±1.0時間のTss, max;約19ng/mlのCss, 12;約10.9±3.8時間のTss, 1/2;及び1803±420ng・時/mlのAUCss, 24により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールが提供される、請求項1〜11のいずれか1項記載の経口投与形。
【請求項13】
約60mgの(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸の用量での16人の健康な成人患者への経口投与に続いて、約8〜約15のCss, max/Css,12により特徴づけられる、前記患者の血液における(R)−3−アミノ−3−(4−クロロフェニル)ブタン酸の平均定常状態薬物力学プロフィールが提供される、請求項1〜12のいずれか1項記載の経口投与形。
【請求項14】
USP1216に従って決定される場合、約0.5重量%以下の破砕性を有する、請求項1〜13のいずれか1項記載の経口投与形。
【請求項15】
約3重量%〜約20重量%の(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩;
約15重量%〜約40重量%の微晶性セルロース;
約15重量%〜約40重量%のヒドロキシプロピルメチルセルロース;及び
約3重量%〜約30重量%の放出速度−制御ポリマー(ここで前記重量%は投与形の合計重量に基づかれる)、を含んで成る配合物を乾式ブレンドし、;そして
前記ブレンドされた配合物を圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法。
【請求項16】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩の乾燥粉末が、約20μm〜約60μmの直径を有する丸型凝集体により特徴づけられる、請求項15記載の方法。
【請求項17】
前記ブレンドされた配合物が、約22mm以下のFlodexを示す、請求項15又は16記載の方法。
【請求項18】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形で存在する、請求項15〜17のいずれか1項記載の方法。
【請求項19】
請求項15〜18のいずれか1項記載の方法により調製される、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩を含んで成る経口錠剤投与形。
【請求項20】
リン酸二カルシウム及び(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩をブレンドし、第1ブレンドを供給し;
前記第1ブレンドを円錐型ミルに通し;
ヒドロキシプロピルメチルセルロース及びコロイド状二酸化珪素を乾燥ブレンドし、第2ブレンドを形成し;
前記第2ブレンドを円錐形ミルに通し;
前記第1ブレンド、前記第2ブレンド、微晶性セルロース、及び放出速度−制御ポリマーを、高剪断ブレンダーにおいてブレンドし、第3ブレンドを形成し;
前記第3ブレンドと共にステアリン酸マグネシウムをブレンドし;そして
前記第3ブレンドを圧縮し、経口錠剤投与形を供給することを含んで成る、経口錠剤投与形の調製方法。
【請求項21】
前記(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸が遊離酸形及び結晶形で存在する、請求項20記載の方法。
【請求項22】
請求項20又は21のいずれか1項記載の方法により調製される、(3R)−4−{[(1S)−2−メチル−1−(2−メチルプロパノイルオキシ)プロポキシ]カルボニルアミノ}−3−(4−クロロフェニル)ブタン酸又は医薬的に許容できるその塩を含んで成る経口錠剤投与形。
【請求項23】
痙攣、胃食道逆流病、嘔吐、咳嗽、麻薬中毒又は乱用、アルコール中毒症又は乱用、ニコチン中毒又は乱用、神経因性疼痛、筋骨格痛及び尿失禁から選択された、患者における疾病の処理方法であって、少なくとも1つの請求項1〜13のいずれか1項記載の経口投与形を、そのような処理の必要な患者に経口投与することを含んで成る方法。
【請求項24】
前記疾病が痙攣である、請求項23記載の方法。
【請求項25】
前記疾病が胃食道逆流病である、請求項23記載の方法。
【請求項26】
前記疾病が神経因性疼痛であり、そして前記神経因性疼痛が帯状疱疹後神経痛、末梢ニューロパシー、三叉神経痛、苦痛性糖尿病性神経障害、HIV関連の神経因性疼痛、癌関連の疼痛及び線維筋痛症から選択される、請求項23記載の方法。
【請求項27】
前記疾病が尿失禁である、請求項23記載の方法。
【図3】

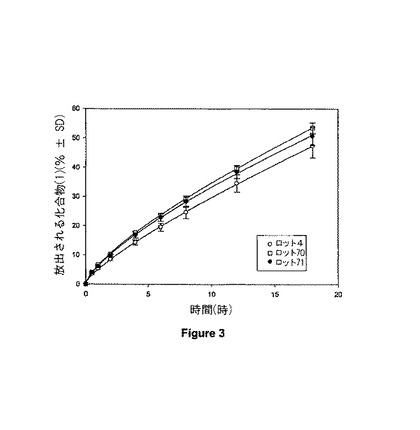
【図4】
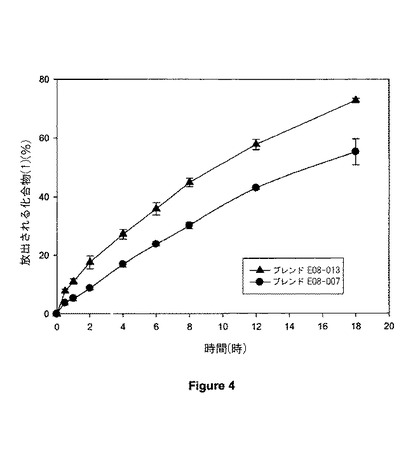
【図5】
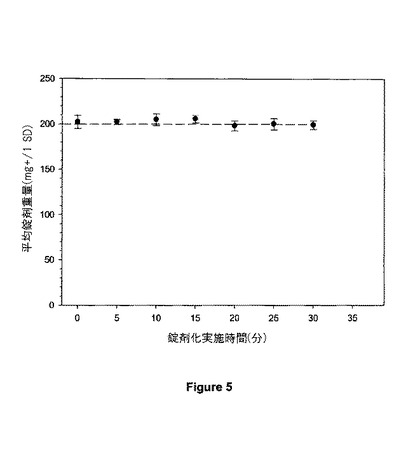
【図6】
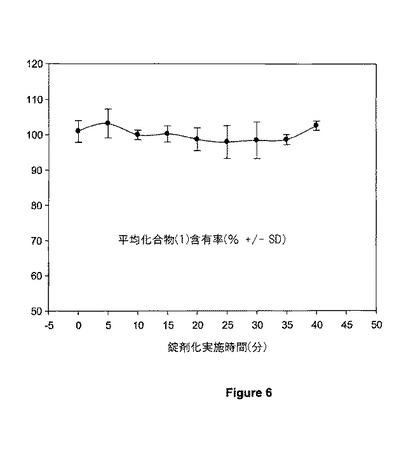
【図7】
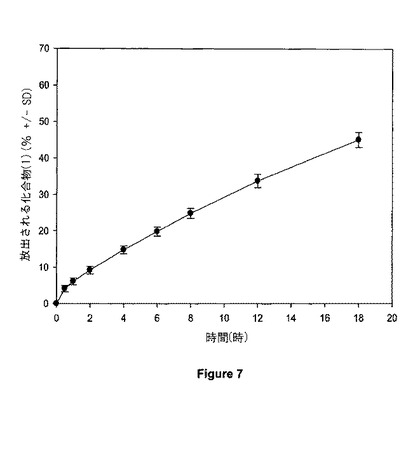
【図8】
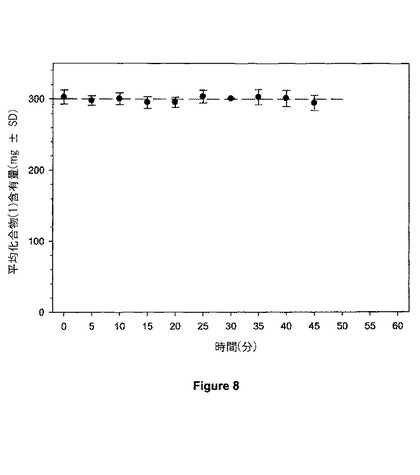
【図9】
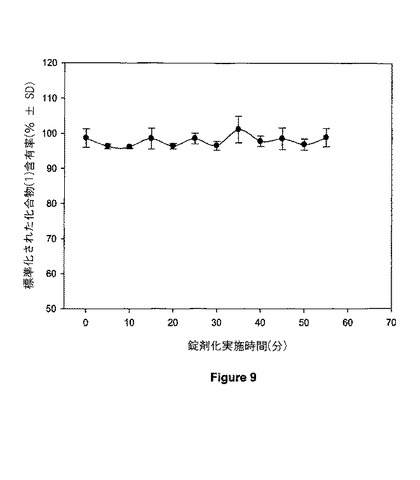
【図10】
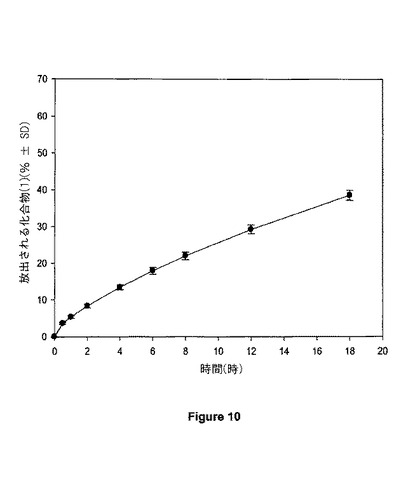
【図11】
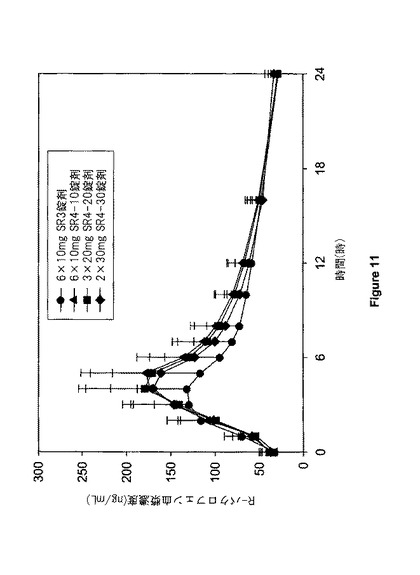
【図12】
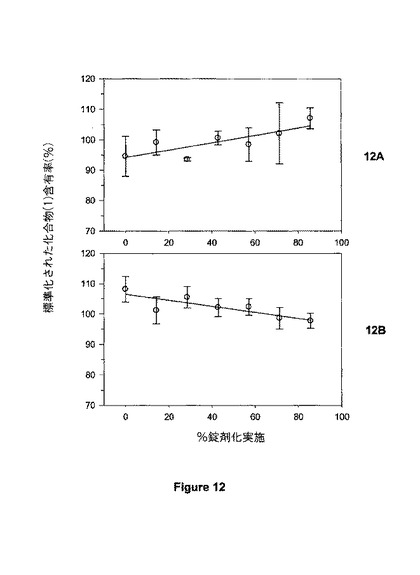
【図13】
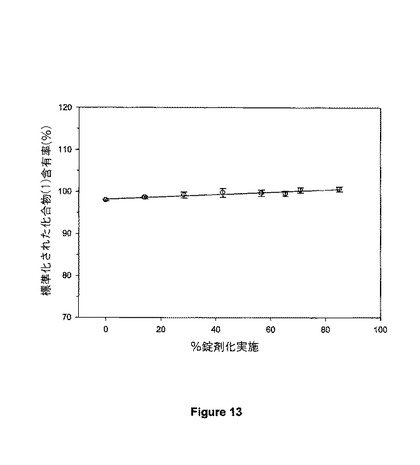
【図14】
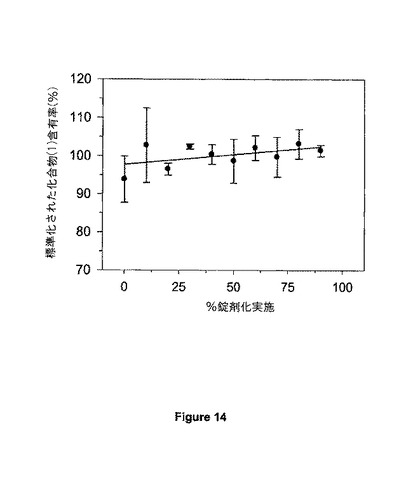
【図15】
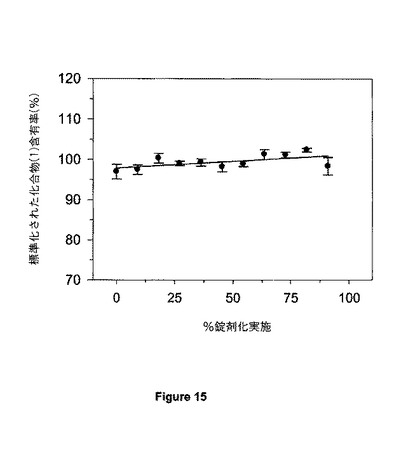
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2012−519212(P2012−519212A)
【公表日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願番号】特願2011−553098(P2011−553098)
【出願日】平成22年3月3日(2010.3.3)
【国際出願番号】PCT/US2010/026133
【国際公開番号】WO2010/102071
【国際公開日】平成22年9月10日(2010.9.10)
【出願人】(503455503)ゼノポート,インコーポレイティド (22)
【Fターム(参考)】
【公表日】平成24年8月23日(2012.8.23)
【国際特許分類】
【出願日】平成22年3月3日(2010.3.3)
【国際出願番号】PCT/US2010/026133
【国際公開番号】WO2010/102071
【国際公開日】平成22年9月10日(2010.9.10)
【出願人】(503455503)ゼノポート,インコーポレイティド (22)
【Fターム(参考)】
[ Back to top ]