
α−グルコシダーゼ阻害剤
【課題】新規でかつ活性の高いα−グルコシダーゼ阻害剤の提供。
【解決手段】コタラヒムブツ(Salacia reticulata)の根又は/及び地上部から抽出した下記の一般式に示す化合物。

【解決手段】コタラヒムブツ(Salacia reticulata)の根又は/及び地上部から抽出した下記の一般式に示す化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なα−グルコシダーゼ阻害剤に関する。
【背景技術】
【0002】
α−グルコシダーゼは、α−グルカン、短鎖オリゴ糖などの非還元末端側からグルコースを解離するエキソ型加水分解酵素である。この酵素は、植物におけるデンプン代謝や動物の消化作用並びにグリコーゲン代謝に関与し、糖質代謝系において重要な役割を果たしている。中でも、ヒト小腸における糖吸収を制御することから、その阻害剤については多くの研究がなされており、高血糖症状を原因とする糖尿病、高脂血症、肥満などの予防や治療において有効とされる。
【0003】
事実、α−グルコシダーゼ阻害剤は糖吸収阻害の他に、耐糖能異常者(IGT)への長期投与によってインスリン感受性の改善や2型糖尿病の発症が有意に抑制されるだけでなく、心血管疾患の発生も有意に抑制されることが示され、更には、高血圧発症においても有意に抑制されることがわかってくるなど、優れた有効性が明らかにされてきている(非特許文献1、2)。
【0004】
α−グルコシダーゼ阻害剤は、1966年にStreptomyces sp.の代謝産物として分離されたノジリマイシンの構造が明らかにされて以来(非特許文献3)、多数の類縁化合物が単離され、またそれらをモチーフとした化合物の合成研究がなされてきた(非特許文献4)。これらの化合物は、単糖のピラノース内の酸素原子が窒素に置換されたアザ糖に属する擬似糖質の一種である点を共通の特徴としている。これらの他に、硫黄、リン、炭素原子などで置換された化合物がチオ糖、フォスファ糖、カルバ糖として知られており、既に医薬品となっているアカルボース、ボグリボース、ミグリトールなどもこれらの化合物群に属する。
【0005】
中でも、最も研究されているのはアザ糖類であり、前記のミグリトールはノジリマイシンより生まれた薬剤である。ノジリマイシンは、優れたα−及びβ−グルコシダーゼ阻害成分ではあったが、不安定であることから、まずC−1位の水酸基を還元したデオキシノジリマイシンが合成された。その結果、更に安定性及び阻害活性を増したが、in vivo において活性が低下するという欠点があり、更なる誘導体の合成、検討が成され、ミグリトールが誕生した。最も阻害活性の強いボグリボースについても同様で、アカルボースの発見に端を発し、構造活性相関の研究が進められ、バリダミン、バリエナミン、バリオールアミンなどカルバ糖アミン類をリード化合物とした化学修飾により合成に至っている。
【0006】
一方、グルコシダーゼ酵素の活性部位については、コンズリトールBエポキシド(CBE)の特異的結合反応を利用した研究が行われており、Asp−カルボキシル基の関与が明らかとなってきている(非特許文献5)。また、デオキシノジリマイシン、その異性体であるイソファゴミンの選択的阻害活性からもイミノ基と酵素活性部位とのイオン対形成により作用していると考えられている。
【0007】
これらの擬似糖質と異なる阻害成分としては、アクリドンアルカロイド(非特許文献6)、クマリン配糖体(非特許文献7)、フラボノイド類(非特許文献8、9)、タンニン類(非特許文献10、11)など多くの植物由来成分が知られているが、これらの化合物群は阻害レベルが非常に弱いことから、実用化には至っていない。
【0008】
このような状況下、近年特に有効性の高いものとしてコタラヒムブツ(Salacia reticulata WIGHT)が注目されている。コタラヒムブツは、デチンムル科(Hippocrateaceae)サラキア属(Salacia)のツル性植物であり、スリランカでは伝承医学アーユルヴェーダにおいて、リューマチ、皮膚病に加え糖尿病に有効な薬用植物として昔から利用されてきたと言われている(非特許文献12)。利用部位は、主に根や幹部とされており、太い幹などになるとくり抜いてコップを作り、食事の際お湯やお水を入れて使うと糖尿病予防にいいと伝えられているほどである。
【0009】
その有効性については、1984年にコロンボ大学のE.H.Karunanayake等によって糖負荷試験による血糖上昇抑制作用が確認されて以来(非特許文献13)、多くの研究が成されてきており、他にも抗肥満、肝保護などといった作用について明らかにされてきている(非特許文献14、15、16、17、18)。また、変異原性や急性毒性試験など(非特許文献19、20、21)、安全性についても検討がなされており、コタラヒムブツが優れた有効性と高い安全性を併せ持つ植物であるという科学的裏付けが得られてきている。
【0010】
このコタラヒムブツのα−グルコシダーゼ阻害活性に関しては、有効成分として分子内に硫酸基を有するチオ糖類のサラシノール(特許文献1、非特許文献22)やコタラノール(非特許文献23)及びレティキュラノール(特許文献2)が既に明らかにされている。また、医薬品として上市されているアカルボースを凌ぐ非常に強い阻害活性を有していることから、この成分をリード化合物として、異性体合成などが盛んに行われている(非特許文献24、25)。
【0011】
しかしながら、一方では現在明らかとなっている前記3成分の含有量や活性比較から考えると、これら3成分からではコタラヒムブツのもつ阻害活性を充分に説明することが出来ないとの報告例もあり(非特許文献26)、更なる有効成分の存在が示唆された。
【0012】
【特許文献1】特開平11−29472号公報
【特許文献2】特開2004−323420号公報
【非特許文献1】Chiasson JL. et al., JAMA,. 290, p486-494, 2003、
【非特許文献2】Yuichi Shinoda et al., Metabolism., 55, p935−939, 2006
【非特許文献3】Inoue S et al., J. Antibiot., 19, p288−292, 1966
【非特許文献4】Naoki Asano, Glycobiology., 13(10), p 93−104, 2003
【非特許文献5】Monique M.P. Hermans et al., J. Biol. Chem., 266(21), p13507−13512, 1991
【非特許文献6】Jean Duplex Wansi et al., Chem. Pharm. Bull., 54(3), p292−296, 2006
【非特許文献7】Sung Ok Lee et al., Arch.Pharm.Res., 27(12), p1207−1210, 2004
【非特許文献8】Jun Kawabata et al., Biosci. Biotechnol. Biochem., 67(2), p445−447, 2003.
【非特許文献9】Kiran Iqbal et al., Chem. Pharm. Bull., 52(7), p785−789, 2004
【非特許文献10】Miou Toda et al., Biosci. Biotechnol. Biochem., 64(2), p294−298, 2000、
【非特許文献11】Miou Toda et al., Biosci. Biotechnol. Biochem., 65(3), p542−547, 2001
【非特許文献12】Attygalle, Sinhalese Materia Medica., p43, 1917
【非特許文献13】E.H.Karunanayake et al., J Ethonopharmacol., 11, p223−231, 1984
【非特許文献14】Osami Kjimoto et al., 日本栄養・食糧学会誌, 53(5), p199−205, 2000
【非特許文献15】Masayuki Yoshikawa et al., J Nutr., 132, p1819−1824, 2002
【非特許文献16】Masayuki Yoshikawa et al., Biol.Pharm.Bull.,25, p72−76, 2002
【非特許文献17】Masayuki Yoshikawa et al., 薬学雑誌, 123(10), p871−880, 2003、
【非特許文献18】Hidehiko Beppu et al., 日本食品新素材研究会誌, 8(2), p105−117, 2005
【非特許文献19】Hiroshi Shimoda et al., 食衛誌, 40(3), p198−205, 1999
【非特許文献20】Hiroshi Shimoda et al., 食衛誌, 42(2), p144−147, 2001
【非特許文献21】Hiroshi Shimoda et al., 医学と薬学, 46(4), p527−540, 2001
【非特許文献22】Masayuki Yoshikawa et al., Tetrahedron Letters., 38(48), p8367−8370, 1997
【非特許文献23】Masayuki Yoshikawa et al., Chem.Pharm.Bull., 46(8), p1339−1340, 1998
【非特許文献24】Ahmad Ghavami et al., J. Org. Chem., 66, p2312−2317, 2001
【非特許文献25】Monica G.Szczepina et al., J. Am. Chem. Soc., 126( 39 ), p12458−12469, 2004
【非特許文献26】Masayuki Yoshikawa et al., 薬学雑誌, 121(5), p371−378, 2001
【発明の開示】
【発明が解決しようとする課題】
【0013】
本願発明者らは、上記背景技術に基づき、さらに研究を重ねた結果、コタラヒムブツから新たなα−グルコシダーゼ阻害活性物質を見出し、本発明を完成させるに至った。
【課題を解決するための手段】
【0014】
本発明の化合物はこれまでに知られていなかった式1で示される新規の化合物であって、α−グルコシダーゼ阻害活性を有する。
【発明の効果】
【0015】
本発明によると、これまでに知られたα−グルコシダーゼ阻害剤よりも強い作用を有するα−グルコシダーゼ阻害剤が提供され、抗糖尿病薬や抗脂血症薬など抗高血糖を原因とする各症状への臨床適用やいわゆる効能表示が認められた特定保健用食品などへの応用が期待される。
【発明を実施するための最良の形態】
【0016】
本発明に係る新規化合物は次の化学式(式1)の構造を有するもので、デチンムル科サラキア属植物の根及び地上部に多く含まれている。この化合物は、非常に優れたα−グルコシダーゼ阻害活性を有する。この化合物は、主として、デチンムル科サラキア属植物の根部及び地上部の溶媒抽出物から得られ、サラキア属植物として、例えば、サラキア・プリノイデス(Salacia prinoides)、サラキア・オブロンガ(Salacia oblonga)、サラキア・キネンシス(Salacia chinensis)が例示される。もっとも、本発明の化合物はこれら以外の部位や当該植物以外の植物から抽出してもよいのは言うまでもない。
【化1】
【0017】
その精製方法は特に限定されるものではなく、例えば種々の公知の方法を組み合わせることによって得られる。例えば、サラキア属植物の根や地上部の切断物等に対して抽出溶媒を加えて抽出し、その後濾液を取り、濃縮後、溶媒やカラムを用いた分画操作により精製を加えることによって得られる。
【0018】
さらに具体的な一例を挙げると、サラキア属植物の根及び/又は地上部に対して、重量比で約2〜20倍量の抽出溶媒を加える。抽出溶媒としては、水、あるいはアルコール類、あるいは水とアルコール類又はアセトンなどの親水溶媒との混合溶媒から選択してもよいが、特に水が好ましい。また、超臨界抽出法を用いることもできる。
【0019】
抽出時間は適宜定められるが、好ましくは一晩以上還流する。その後、遠心分離やろ過処理により残渣を除去する。得られた濾液を減圧濃縮後、凍結乾燥する。以上の操作により、出発原料となるコタラヒムブツエキス末が得られる。尚、前記残渣に再度溶媒を添加し、同一の抽出操作を繰り返しても良い。
【0020】
このコタラヒムブツエキス末を用いて水溶液を調整し、当量のエタノールを添加してエタノール沈殿処理を行う。沈殿溶媒としては、他にメタノール、イソプロパノール、アセトニトリルなどを使用しても良い。その後、遠心分離やろ過処理により沈殿を除去する。そして、減圧濃縮により有機溶媒を除去したものを用いてカラムクロマトグラフィーなどの分離操作を行うことにより、本発明の新規α−グルコシダーゼ阻害物質を得ることができる。
【0021】
こうして得られたα−グルコシダーゼ阻害剤は単一成分としてそのまま用いることもできるが、各種の食品やお茶などの飲料に添加剤として用いたり、食後の血糖の上昇を抑えたり、糖の吸収を抑えたりするなどと言った効能効果が標榜できる特定保健用食品として提供される。また、適当な賦形剤や保存料、香料などと共に錠剤、カプセル剤、軟カプセル剤、散剤、顆粒剤等の内服剤や注射剤といった形状に製剤化してもよい。また、医薬品としての用途のみならず、いわゆる医薬品の形態に類似した健康食品としても利用することができる。これらに用いられる賦形剤としては、例えば、乳糖、でんぷん、リン酸カルシウム、結晶セルロース、マンニット、タルク、ステアリン酸マグネシウム、水、生理食塩水が例示される。
【0022】
これらの食品や医薬品において、α−グルコシダーゼ阻害活性が有効に発揮されるためには、全重量に対して、本発明成分を少なくとも0.0001%、より好ましくは0.005〜0.1%程度が必要とされる。もっとも、この量は投与対象となる患者や動物の症状、年齢、摂取形態等によって適宜設定されうる。
【0023】
以下、実施例に基づいて本発明についてさらに詳細に説明するが、本発明は以下の実施例に限定されないのは言うまでもない。
【実施例1】
【0024】
〔コタラヒムブツ抽出物の粗分画〕
コタラヒムブツ(Salacia reticulata)の乾燥した幹 約1.1kgに10倍量の水を加えて、一晩(14〜15時間)熱水抽出を行った。得られた抽出液中の不溶物は、遠心分離(8,000rpm/30min)及びろ過処理により除去した。これらの操作により、コタラヒムブツ熱水エキス末100gを得た。これに水を添加し、500mlの水溶液を調整した。その後、攪拌しながら当量のエタノールを添加し、生じた沈殿物は遠心分離(8,000rpm/30min)により除去を行った。得られた上清は、減圧濃縮によりエタノール除去を行った(収量69.0g)。これを用いて再度500mlの水溶液を調整した後、HP−20カラム処理(使用担体:三菱化学社製合成吸着剤HP−20を1kg充填、カラム:8×35cm)を行った。溶出は、水(5L)、20容量%、40容量%、60容量%、80容量%のメタノール水溶液(各2.5L)を用いて、ステップワイズ溶出を行い、水溶出画分(「HP−20W」画分;収量34.1g)、20容量%メタノール画分(「HP−20−20」画分;収量5.89g)、40容量%メタノール画分(「HP−20−40」画分;収量9.48g)、60容量%メタノール画分(「HP−20−60」画分;収量4.85g)、80容量%メタノール画分(「HP−20−60」画分;収量0.67g)を得た(全回収率 86.0%)。得られた各分画物について、α−グルコシダーゼ阻害活性試験を実施した。阻害活性試験は、以下の操作方法に従い実施した。その結果を表1に示す。
【0025】
〔α−グルコシダーゼ阻害活性測定(In vitro)〕
(粗酵素溶液の調製)
試薬として、試薬1:ラット小腸アセトンパウダー(SIGMA社製)、試薬2:100mMリン酸カリウム緩衝液(5mMEDTA、pH7.0)、試薬3:100mMリン酸カリウム緩衝液(0.1%Triton、pH7.0)、試薬4:10mMリン酸カリウム緩衝液(pH7.0)を用いる。
【0026】
まず、試薬1の5gに試薬2の50mlを加え、氷冷しながらホモジナイズ(1分×3回)を行った。得られた溶液を遠心分離(15,000×g/60minあるいは21,000×g/60min)し上清を除去した。その後、沈殿に試薬3の50mLを加え、攪拌可溶化させ、4℃下に60分間放置した。これを更に遠心分離(15,000×g/90minあるいは110,000×g/90min)し、上清を得た。得られた上清について、試薬4を用いて24時間透析を行った。透析後に得られた内液を粗酵素溶液として試験に用いた。
【0027】
(α−グルコシダーゼ阻害活性試験)
試験用試薬として、試薬1:粗酵素溶液、試薬2:DMSO、試薬3:100mMスクロース水溶液(25mMマルトース水溶液)、試薬4:2MTris-HCl緩衝液(pH7.0)、試薬5:グルコースCIIテストワコー発色液(和光純薬製)を準備した。試験サンプル溶液は、被験物質(10mg)をDMSO(10mL)にて溶解させたものを(サンプルの阻害活性の強さに応じて適宜希釈する)用いた。また、1検体あたりn=3で測定を行い、検体毎にそれぞれブランクをおいた。次いで、以下の操作により阻害活性を測定した。
【0028】
96穴プレートにサンプル溶液10μL及び試薬1の25μLを加えて恒温槽で5分間プレインキュベートした。この時、ブランクには試薬1を添加する前に、反応が進行しないよう試薬4の50μLを加えておく。その後、試薬3の20μLを加えて反応を開始する。
【0029】
反応開始30分後、ブランク以外の反応液に試薬4の50μLを添加し反応停止させた。それから、各反応液に試薬5の150μLを加えて10分間発色させた後、マイクロプレートリーダーを用いて492nmにおける吸光度を測定した。得られた測定結果より、下記の式1にて阻害率を算出した。
阻害率(%)={(Glc0−Glcx)/Glc0}×100・・・・式1
ただし、Glc0:コントロール(サンプルと同量のDMSO)吸光度の平均値(n=3)より相当するブランクの吸光度の平均値(n=3)を差し引いたもの
Glcx:サンプルの吸光度の平均値(n=3)より相当するブランクの吸光度の平均値(n=3)を差し引いたもの
【0030】
【表1】
【0031】
表1から理解されるように、エキス内の阻害活性成分は、殆んどが水溶出画分に回収されており、メタノール濃度を高めるに従って阻害活性は低下していった。60容量%以上の濃度では阻害活性が認められなくなった。そこで、顕著な活性が認められたHP−20W画分について更に分離を進めることとした。
【0032】
〔コタラヒムブツ粗分画物の分画・精製〕
分画物を200mLの水溶液とした後、当量のエタノールを添加攪拌し不溶部分を遠心除去した(7,500rpm/30min)。得られた上清は、減圧濃縮によりエタノールを除去した。これを40mLの水溶液とし、アミノカラム(使用担体 :富士シリシア化学社製NH-DM1020SGを950g充填したカラム:8×49cm)にアプライした。
【0033】
溶出は、80容量%(6L)、70容量%(9L)、60容量%、40容量%、20%容量アセトニトリル水溶液(各6L)、水(12L)を用いて、ステップワイズ溶出を行い、80容量%アセトニトリル溶出画分(「80%CN」画分;収量4.43g)、70%アセトニトリル溶出画分(「70%CN」画分;収量12.10g)、60%アセトニトリル溶出画分(「60%CN」画分;収量2.78g)、40%アセトニトリル溶出画分(「40%CN」画分;収量6.75g)、20%アセトニトリル溶出画分(「20%CN」画分;収量1.90g)、水溶出画分(「W」画分;4.20g)を得た(全回収率94.3%)。得られた各分画物について、上記方法にてα−グルコシダーゼ阻害活性試験を実施した。その結果を表2に示した。
【0034】
【表2】
【0035】
試験結果より、70%CN画分以外の溶出部分では殆んど阻害活性が認められず、主要な阻害活性成分は70%CN画分に存在していることが明らかとなった。尚、前処理時に生じた沈殿について、阻害活性試験を実施したところ活性は認められなかった。
【0036】
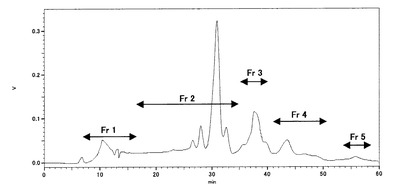
次に、この分画物を用いてHPLC分取を行った。分取条件は、使用カラム:YMC pack polyamine−II(20×250mm)、移動相:65容量%アセトニトリル、流速:4.5mL/min、カラム温度:30℃、検出器:RIで行った。分離用サンプルは、移動相溶媒に溶解したものを用いた。しかし、この時大量の白色沈殿が生じたため、これを遠心除去した後得られる上清部分を使用した(白色沈殿について、阻害活性試験を実施したところ活性は認められなかった)。このときに得られたHPLC溶出液から、それぞれFr1(7−15min/収量433.0mg)、Fr2(25−35min/収量1744.0mg)、Fr3(35−40min/収量526.7mg)、Fr4(40−50min/収量256.7mg)、Fr5(53−57min/収量33.6mg)と5つの分画物を得た。このときのHPLCチャートを図1に示す。更に、カラム吸着成分の回収のために20容量%アセトニトリル水溶液にて洗浄を行った(全回収率88.2%)。得られた分画物について、上記方法にてα−グルコシダーゼ阻害活性試験を実施した。その結果を表3に示した。
【0037】
【表3】
【0038】
表3から理解されるように、Fr1及びFr3〜5について阻害活性が認められ、Fr2及び20%アセトニトリル回収部においては、全く阻害活性が認められなかった。Fr3〜5においても比較的強い阻害活性が認められたが、特にFr1が強い阻害活性を示したことから、これを更に分離していくこととした。なお、移動相として50〜70容量%アセトニトリル水溶液を用いることによってもFr1を分離することができる。この場合保持時間が変動するが10〜25min以内のピークを回収することで他の活性成分画分(Fr3〜5)と分離できる。
【0039】
YMC pack polyamine−IIカラムでは、図1に示したスペクトルの通り、ピークがブロードになることから更に分離していくことは非常に困難であると考え、次に使用カラムをODSに変更し、さらに分画を進めた。
【0040】
分取条件は、使用カラム:DAISOPAK SP-120-5-ODS-BP(20×250mm)、移動相:水、流速:5.0mL/min、カラム温度:30℃、検出器:RIで実施した。得られたスペクトルより、ODSFr1(10−10.7min/収量230.4mg)、ODSFr2(10.7−11.2min/5.8mg)、ODSFr3(11.2−13min/70.8mg)と3つの分画物を得た。これらの分画物について阻害活性試験を実施した。その結果を表4に示す。
【0041】
【表4】
【0042】
〔分画・精製物の構造決定〕
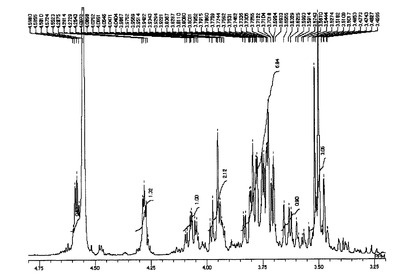
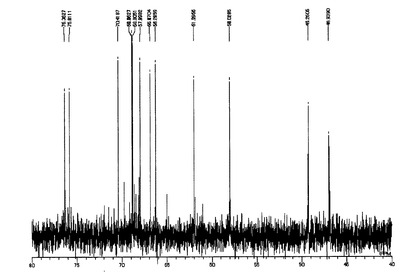
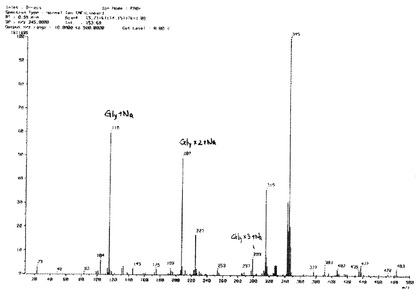
ODSFr2画分についてさらに数回同様の分取操作を繰り返してほぼ単一の物質を得た。この物質の構造を明らかとするために各種NMR分析(1H、13C、1H−1HCOSY、HMQC及びHMBC)並びにFAB−MS分析を実施し、得られた各種スペクトルデータを用いて構造解析を行った。NMR分析(1D−及び2D−)は、分析装置:JNM AL−400(日本電子製、400MHz)、溶媒:D2O(内部標準試薬としてアセトンを使用)、分析温度:25℃にて行った。FAB−MS分析は、分析装置:7000QQ型(日本電子製)、イオン化法:高速原子衝撃法(FAB)、加速電圧:6kV、マトリックス:グリセリン、測定質量範囲:〜m/z1000にて実施した。図2に1H−NMRスペクトル(inD2O/400MHz)を、図3に13C−NMRスペクトル(inD2O/400MHz)を、図4にFAB−MSスペクトルを示した。
【0043】
以上の結果を基に、解析を行った結果、ODSFr2から得られた成分は図5に示すようなスルフォキシドを有するシクリトールであることが示唆された。しかし、得られたスペクトルデータからでは、スルフォキシドの存在を確認することが出来ないことから、更にHR−MS分析により、元素組成を明らかにすることとした。分析条件は、分析装置:JMS−T100LC(日本電子製)、イオン化法:APCI、測定質量範囲:〜m/z1000にて実施した。この分析から、表5に示す結果が得られた。
【0044】
【表5】
【0045】
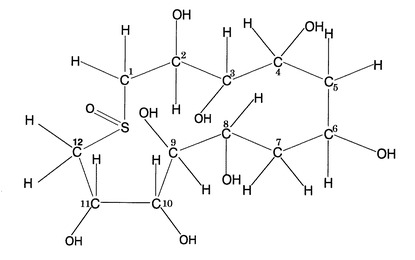
以上のことより、図5に示した構造との整合性が得られたことから、ODSFr2で得られた成分は、次の化学式(化1)に示すようにスルフォキシドを有する13員環シクリトール構造であることが明らかとなった。
【0046】
【化2】
【0047】
〔分画・精製物の活性比較〕
上記構造決定されたα−グルコシダーゼ阻害剤について、コタラヒムブツから得られた既報成分(非特許文献26から引用)及び医薬品として販売が認められているボグリボースとその活性を比較した。その結果を表6に示す。この表から分かるように、上市されているボグリボースとほぼ同程度の活性強度を示し、同じ植物から得られた既報成分と比較して、マルターゼ阻害で約71〜103倍、スクラーゼ阻害では約19〜34倍もの阻害活性強度を示した。このように、本発明によるα−グルコシダーゼ阻害剤は、既報成分をはるかに凌ぐ阻害活性を示す成分であることが明らかとなり、コタラヒムブツ抽出物における主たる活性成分であると考えられる。
【0048】
【表6】
【0049】
以上のように、化学式(化1)で示される化合物は優れたα−グルコシダーゼ阻害活性を有していることが確認された。この化合物のα−グルコシダーゼ阻害活性は、これまで報告がなされているコタラヒムブツ中に存在しているサラシノールやコタラノールに比べるとかなり高い比活性を有し、コタラヒムブツ抽出物中における本発明に係る化合物の寄与率は高いものと考えられる。
【実施例2】
【0050】
次に、この成分を使った処方例を示す。
〔製造例1:錠剤〕
本発明のα−グルコシダーゼ阻害剤 0.07g
マルトシルサイクロデキストリン 600g
ショ糖脂肪酸エステル 50g
炭酸カルシウム 15g
還元麦芽糖水あめ 75g
無水乳酸 110g
結晶セルロース 100g
ミルクカルシウム 50g
上記処方に従って、常法により錠剤5000錠を製造した。
【産業上の利用可能性】
【0051】
本発明の化合物は、新規でかつ活性の高いα−グルコシダーゼ阻害作用を有し、これにより、抗糖尿病患者や糖尿病予備群の人に対してより有効な抗糖尿病薬や特定保健用食品などが提供される。
【図面の簡単な説明】
【0052】
【図1】コタラヒムブツ水抽出物のHP−20分画物をアミノカラムで分画した70%アセトニトリル溶出物のHPLCチャートである。
【図2】コタラヒムブツ抽出物の最終分画物の1H−NMRスペクトルである。
【図3】コタラヒムブツ抽出物の最終分画物の13C−NMRスペクトルである。
【図4】コタラヒムブツ抽出物の最終分画物のFAB−MSスペクトルである。
【図5】コタラヒムブツ抽出物の最終分画物の推定平面構造図である。
【技術分野】
【0001】
本発明は、新規なα−グルコシダーゼ阻害剤に関する。
【背景技術】
【0002】
α−グルコシダーゼは、α−グルカン、短鎖オリゴ糖などの非還元末端側からグルコースを解離するエキソ型加水分解酵素である。この酵素は、植物におけるデンプン代謝や動物の消化作用並びにグリコーゲン代謝に関与し、糖質代謝系において重要な役割を果たしている。中でも、ヒト小腸における糖吸収を制御することから、その阻害剤については多くの研究がなされており、高血糖症状を原因とする糖尿病、高脂血症、肥満などの予防や治療において有効とされる。
【0003】
事実、α−グルコシダーゼ阻害剤は糖吸収阻害の他に、耐糖能異常者(IGT)への長期投与によってインスリン感受性の改善や2型糖尿病の発症が有意に抑制されるだけでなく、心血管疾患の発生も有意に抑制されることが示され、更には、高血圧発症においても有意に抑制されることがわかってくるなど、優れた有効性が明らかにされてきている(非特許文献1、2)。
【0004】
α−グルコシダーゼ阻害剤は、1966年にStreptomyces sp.の代謝産物として分離されたノジリマイシンの構造が明らかにされて以来(非特許文献3)、多数の類縁化合物が単離され、またそれらをモチーフとした化合物の合成研究がなされてきた(非特許文献4)。これらの化合物は、単糖のピラノース内の酸素原子が窒素に置換されたアザ糖に属する擬似糖質の一種である点を共通の特徴としている。これらの他に、硫黄、リン、炭素原子などで置換された化合物がチオ糖、フォスファ糖、カルバ糖として知られており、既に医薬品となっているアカルボース、ボグリボース、ミグリトールなどもこれらの化合物群に属する。
【0005】
中でも、最も研究されているのはアザ糖類であり、前記のミグリトールはノジリマイシンより生まれた薬剤である。ノジリマイシンは、優れたα−及びβ−グルコシダーゼ阻害成分ではあったが、不安定であることから、まずC−1位の水酸基を還元したデオキシノジリマイシンが合成された。その結果、更に安定性及び阻害活性を増したが、in vivo において活性が低下するという欠点があり、更なる誘導体の合成、検討が成され、ミグリトールが誕生した。最も阻害活性の強いボグリボースについても同様で、アカルボースの発見に端を発し、構造活性相関の研究が進められ、バリダミン、バリエナミン、バリオールアミンなどカルバ糖アミン類をリード化合物とした化学修飾により合成に至っている。
【0006】
一方、グルコシダーゼ酵素の活性部位については、コンズリトールBエポキシド(CBE)の特異的結合反応を利用した研究が行われており、Asp−カルボキシル基の関与が明らかとなってきている(非特許文献5)。また、デオキシノジリマイシン、その異性体であるイソファゴミンの選択的阻害活性からもイミノ基と酵素活性部位とのイオン対形成により作用していると考えられている。
【0007】
これらの擬似糖質と異なる阻害成分としては、アクリドンアルカロイド(非特許文献6)、クマリン配糖体(非特許文献7)、フラボノイド類(非特許文献8、9)、タンニン類(非特許文献10、11)など多くの植物由来成分が知られているが、これらの化合物群は阻害レベルが非常に弱いことから、実用化には至っていない。
【0008】
このような状況下、近年特に有効性の高いものとしてコタラヒムブツ(Salacia reticulata WIGHT)が注目されている。コタラヒムブツは、デチンムル科(Hippocrateaceae)サラキア属(Salacia)のツル性植物であり、スリランカでは伝承医学アーユルヴェーダにおいて、リューマチ、皮膚病に加え糖尿病に有効な薬用植物として昔から利用されてきたと言われている(非特許文献12)。利用部位は、主に根や幹部とされており、太い幹などになるとくり抜いてコップを作り、食事の際お湯やお水を入れて使うと糖尿病予防にいいと伝えられているほどである。
【0009】
その有効性については、1984年にコロンボ大学のE.H.Karunanayake等によって糖負荷試験による血糖上昇抑制作用が確認されて以来(非特許文献13)、多くの研究が成されてきており、他にも抗肥満、肝保護などといった作用について明らかにされてきている(非特許文献14、15、16、17、18)。また、変異原性や急性毒性試験など(非特許文献19、20、21)、安全性についても検討がなされており、コタラヒムブツが優れた有効性と高い安全性を併せ持つ植物であるという科学的裏付けが得られてきている。
【0010】
このコタラヒムブツのα−グルコシダーゼ阻害活性に関しては、有効成分として分子内に硫酸基を有するチオ糖類のサラシノール(特許文献1、非特許文献22)やコタラノール(非特許文献23)及びレティキュラノール(特許文献2)が既に明らかにされている。また、医薬品として上市されているアカルボースを凌ぐ非常に強い阻害活性を有していることから、この成分をリード化合物として、異性体合成などが盛んに行われている(非特許文献24、25)。
【0011】
しかしながら、一方では現在明らかとなっている前記3成分の含有量や活性比較から考えると、これら3成分からではコタラヒムブツのもつ阻害活性を充分に説明することが出来ないとの報告例もあり(非特許文献26)、更なる有効成分の存在が示唆された。
【0012】
【特許文献1】特開平11−29472号公報
【特許文献2】特開2004−323420号公報
【非特許文献1】Chiasson JL. et al., JAMA,. 290, p486-494, 2003、
【非特許文献2】Yuichi Shinoda et al., Metabolism., 55, p935−939, 2006
【非特許文献3】Inoue S et al., J. Antibiot., 19, p288−292, 1966
【非特許文献4】Naoki Asano, Glycobiology., 13(10), p 93−104, 2003
【非特許文献5】Monique M.P. Hermans et al., J. Biol. Chem., 266(21), p13507−13512, 1991
【非特許文献6】Jean Duplex Wansi et al., Chem. Pharm. Bull., 54(3), p292−296, 2006
【非特許文献7】Sung Ok Lee et al., Arch.Pharm.Res., 27(12), p1207−1210, 2004
【非特許文献8】Jun Kawabata et al., Biosci. Biotechnol. Biochem., 67(2), p445−447, 2003.
【非特許文献9】Kiran Iqbal et al., Chem. Pharm. Bull., 52(7), p785−789, 2004
【非特許文献10】Miou Toda et al., Biosci. Biotechnol. Biochem., 64(2), p294−298, 2000、
【非特許文献11】Miou Toda et al., Biosci. Biotechnol. Biochem., 65(3), p542−547, 2001
【非特許文献12】Attygalle, Sinhalese Materia Medica., p43, 1917
【非特許文献13】E.H.Karunanayake et al., J Ethonopharmacol., 11, p223−231, 1984
【非特許文献14】Osami Kjimoto et al., 日本栄養・食糧学会誌, 53(5), p199−205, 2000
【非特許文献15】Masayuki Yoshikawa et al., J Nutr., 132, p1819−1824, 2002
【非特許文献16】Masayuki Yoshikawa et al., Biol.Pharm.Bull.,25, p72−76, 2002
【非特許文献17】Masayuki Yoshikawa et al., 薬学雑誌, 123(10), p871−880, 2003、
【非特許文献18】Hidehiko Beppu et al., 日本食品新素材研究会誌, 8(2), p105−117, 2005
【非特許文献19】Hiroshi Shimoda et al., 食衛誌, 40(3), p198−205, 1999
【非特許文献20】Hiroshi Shimoda et al., 食衛誌, 42(2), p144−147, 2001
【非特許文献21】Hiroshi Shimoda et al., 医学と薬学, 46(4), p527−540, 2001
【非特許文献22】Masayuki Yoshikawa et al., Tetrahedron Letters., 38(48), p8367−8370, 1997
【非特許文献23】Masayuki Yoshikawa et al., Chem.Pharm.Bull., 46(8), p1339−1340, 1998
【非特許文献24】Ahmad Ghavami et al., J. Org. Chem., 66, p2312−2317, 2001
【非特許文献25】Monica G.Szczepina et al., J. Am. Chem. Soc., 126( 39 ), p12458−12469, 2004
【非特許文献26】Masayuki Yoshikawa et al., 薬学雑誌, 121(5), p371−378, 2001
【発明の開示】
【発明が解決しようとする課題】
【0013】
本願発明者らは、上記背景技術に基づき、さらに研究を重ねた結果、コタラヒムブツから新たなα−グルコシダーゼ阻害活性物質を見出し、本発明を完成させるに至った。
【課題を解決するための手段】
【0014】
本発明の化合物はこれまでに知られていなかった式1で示される新規の化合物であって、α−グルコシダーゼ阻害活性を有する。
【発明の効果】
【0015】
本発明によると、これまでに知られたα−グルコシダーゼ阻害剤よりも強い作用を有するα−グルコシダーゼ阻害剤が提供され、抗糖尿病薬や抗脂血症薬など抗高血糖を原因とする各症状への臨床適用やいわゆる効能表示が認められた特定保健用食品などへの応用が期待される。
【発明を実施するための最良の形態】
【0016】
本発明に係る新規化合物は次の化学式(式1)の構造を有するもので、デチンムル科サラキア属植物の根及び地上部に多く含まれている。この化合物は、非常に優れたα−グルコシダーゼ阻害活性を有する。この化合物は、主として、デチンムル科サラキア属植物の根部及び地上部の溶媒抽出物から得られ、サラキア属植物として、例えば、サラキア・プリノイデス(Salacia prinoides)、サラキア・オブロンガ(Salacia oblonga)、サラキア・キネンシス(Salacia chinensis)が例示される。もっとも、本発明の化合物はこれら以外の部位や当該植物以外の植物から抽出してもよいのは言うまでもない。
【化1】
【0017】
その精製方法は特に限定されるものではなく、例えば種々の公知の方法を組み合わせることによって得られる。例えば、サラキア属植物の根や地上部の切断物等に対して抽出溶媒を加えて抽出し、その後濾液を取り、濃縮後、溶媒やカラムを用いた分画操作により精製を加えることによって得られる。
【0018】
さらに具体的な一例を挙げると、サラキア属植物の根及び/又は地上部に対して、重量比で約2〜20倍量の抽出溶媒を加える。抽出溶媒としては、水、あるいはアルコール類、あるいは水とアルコール類又はアセトンなどの親水溶媒との混合溶媒から選択してもよいが、特に水が好ましい。また、超臨界抽出法を用いることもできる。
【0019】
抽出時間は適宜定められるが、好ましくは一晩以上還流する。その後、遠心分離やろ過処理により残渣を除去する。得られた濾液を減圧濃縮後、凍結乾燥する。以上の操作により、出発原料となるコタラヒムブツエキス末が得られる。尚、前記残渣に再度溶媒を添加し、同一の抽出操作を繰り返しても良い。
【0020】
このコタラヒムブツエキス末を用いて水溶液を調整し、当量のエタノールを添加してエタノール沈殿処理を行う。沈殿溶媒としては、他にメタノール、イソプロパノール、アセトニトリルなどを使用しても良い。その後、遠心分離やろ過処理により沈殿を除去する。そして、減圧濃縮により有機溶媒を除去したものを用いてカラムクロマトグラフィーなどの分離操作を行うことにより、本発明の新規α−グルコシダーゼ阻害物質を得ることができる。
【0021】
こうして得られたα−グルコシダーゼ阻害剤は単一成分としてそのまま用いることもできるが、各種の食品やお茶などの飲料に添加剤として用いたり、食後の血糖の上昇を抑えたり、糖の吸収を抑えたりするなどと言った効能効果が標榜できる特定保健用食品として提供される。また、適当な賦形剤や保存料、香料などと共に錠剤、カプセル剤、軟カプセル剤、散剤、顆粒剤等の内服剤や注射剤といった形状に製剤化してもよい。また、医薬品としての用途のみならず、いわゆる医薬品の形態に類似した健康食品としても利用することができる。これらに用いられる賦形剤としては、例えば、乳糖、でんぷん、リン酸カルシウム、結晶セルロース、マンニット、タルク、ステアリン酸マグネシウム、水、生理食塩水が例示される。
【0022】
これらの食品や医薬品において、α−グルコシダーゼ阻害活性が有効に発揮されるためには、全重量に対して、本発明成分を少なくとも0.0001%、より好ましくは0.005〜0.1%程度が必要とされる。もっとも、この量は投与対象となる患者や動物の症状、年齢、摂取形態等によって適宜設定されうる。
【0023】
以下、実施例に基づいて本発明についてさらに詳細に説明するが、本発明は以下の実施例に限定されないのは言うまでもない。
【実施例1】
【0024】
〔コタラヒムブツ抽出物の粗分画〕
コタラヒムブツ(Salacia reticulata)の乾燥した幹 約1.1kgに10倍量の水を加えて、一晩(14〜15時間)熱水抽出を行った。得られた抽出液中の不溶物は、遠心分離(8,000rpm/30min)及びろ過処理により除去した。これらの操作により、コタラヒムブツ熱水エキス末100gを得た。これに水を添加し、500mlの水溶液を調整した。その後、攪拌しながら当量のエタノールを添加し、生じた沈殿物は遠心分離(8,000rpm/30min)により除去を行った。得られた上清は、減圧濃縮によりエタノール除去を行った(収量69.0g)。これを用いて再度500mlの水溶液を調整した後、HP−20カラム処理(使用担体:三菱化学社製合成吸着剤HP−20を1kg充填、カラム:8×35cm)を行った。溶出は、水(5L)、20容量%、40容量%、60容量%、80容量%のメタノール水溶液(各2.5L)を用いて、ステップワイズ溶出を行い、水溶出画分(「HP−20W」画分;収量34.1g)、20容量%メタノール画分(「HP−20−20」画分;収量5.89g)、40容量%メタノール画分(「HP−20−40」画分;収量9.48g)、60容量%メタノール画分(「HP−20−60」画分;収量4.85g)、80容量%メタノール画分(「HP−20−60」画分;収量0.67g)を得た(全回収率 86.0%)。得られた各分画物について、α−グルコシダーゼ阻害活性試験を実施した。阻害活性試験は、以下の操作方法に従い実施した。その結果を表1に示す。
【0025】
〔α−グルコシダーゼ阻害活性測定(In vitro)〕
(粗酵素溶液の調製)
試薬として、試薬1:ラット小腸アセトンパウダー(SIGMA社製)、試薬2:100mMリン酸カリウム緩衝液(5mMEDTA、pH7.0)、試薬3:100mMリン酸カリウム緩衝液(0.1%Triton、pH7.0)、試薬4:10mMリン酸カリウム緩衝液(pH7.0)を用いる。
【0026】
まず、試薬1の5gに試薬2の50mlを加え、氷冷しながらホモジナイズ(1分×3回)を行った。得られた溶液を遠心分離(15,000×g/60minあるいは21,000×g/60min)し上清を除去した。その後、沈殿に試薬3の50mLを加え、攪拌可溶化させ、4℃下に60分間放置した。これを更に遠心分離(15,000×g/90minあるいは110,000×g/90min)し、上清を得た。得られた上清について、試薬4を用いて24時間透析を行った。透析後に得られた内液を粗酵素溶液として試験に用いた。
【0027】
(α−グルコシダーゼ阻害活性試験)
試験用試薬として、試薬1:粗酵素溶液、試薬2:DMSO、試薬3:100mMスクロース水溶液(25mMマルトース水溶液)、試薬4:2MTris-HCl緩衝液(pH7.0)、試薬5:グルコースCIIテストワコー発色液(和光純薬製)を準備した。試験サンプル溶液は、被験物質(10mg)をDMSO(10mL)にて溶解させたものを(サンプルの阻害活性の強さに応じて適宜希釈する)用いた。また、1検体あたりn=3で測定を行い、検体毎にそれぞれブランクをおいた。次いで、以下の操作により阻害活性を測定した。
【0028】
96穴プレートにサンプル溶液10μL及び試薬1の25μLを加えて恒温槽で5分間プレインキュベートした。この時、ブランクには試薬1を添加する前に、反応が進行しないよう試薬4の50μLを加えておく。その後、試薬3の20μLを加えて反応を開始する。
【0029】
反応開始30分後、ブランク以外の反応液に試薬4の50μLを添加し反応停止させた。それから、各反応液に試薬5の150μLを加えて10分間発色させた後、マイクロプレートリーダーを用いて492nmにおける吸光度を測定した。得られた測定結果より、下記の式1にて阻害率を算出した。
阻害率(%)={(Glc0−Glcx)/Glc0}×100・・・・式1
ただし、Glc0:コントロール(サンプルと同量のDMSO)吸光度の平均値(n=3)より相当するブランクの吸光度の平均値(n=3)を差し引いたもの
Glcx:サンプルの吸光度の平均値(n=3)より相当するブランクの吸光度の平均値(n=3)を差し引いたもの
【0030】
【表1】
【0031】
表1から理解されるように、エキス内の阻害活性成分は、殆んどが水溶出画分に回収されており、メタノール濃度を高めるに従って阻害活性は低下していった。60容量%以上の濃度では阻害活性が認められなくなった。そこで、顕著な活性が認められたHP−20W画分について更に分離を進めることとした。
【0032】
〔コタラヒムブツ粗分画物の分画・精製〕
分画物を200mLの水溶液とした後、当量のエタノールを添加攪拌し不溶部分を遠心除去した(7,500rpm/30min)。得られた上清は、減圧濃縮によりエタノールを除去した。これを40mLの水溶液とし、アミノカラム(使用担体 :富士シリシア化学社製NH-DM1020SGを950g充填したカラム:8×49cm)にアプライした。
【0033】
溶出は、80容量%(6L)、70容量%(9L)、60容量%、40容量%、20%容量アセトニトリル水溶液(各6L)、水(12L)を用いて、ステップワイズ溶出を行い、80容量%アセトニトリル溶出画分(「80%CN」画分;収量4.43g)、70%アセトニトリル溶出画分(「70%CN」画分;収量12.10g)、60%アセトニトリル溶出画分(「60%CN」画分;収量2.78g)、40%アセトニトリル溶出画分(「40%CN」画分;収量6.75g)、20%アセトニトリル溶出画分(「20%CN」画分;収量1.90g)、水溶出画分(「W」画分;4.20g)を得た(全回収率94.3%)。得られた各分画物について、上記方法にてα−グルコシダーゼ阻害活性試験を実施した。その結果を表2に示した。
【0034】
【表2】
【0035】
試験結果より、70%CN画分以外の溶出部分では殆んど阻害活性が認められず、主要な阻害活性成分は70%CN画分に存在していることが明らかとなった。尚、前処理時に生じた沈殿について、阻害活性試験を実施したところ活性は認められなかった。
【0036】
次に、この分画物を用いてHPLC分取を行った。分取条件は、使用カラム:YMC pack polyamine−II(20×250mm)、移動相:65容量%アセトニトリル、流速:4.5mL/min、カラム温度:30℃、検出器:RIで行った。分離用サンプルは、移動相溶媒に溶解したものを用いた。しかし、この時大量の白色沈殿が生じたため、これを遠心除去した後得られる上清部分を使用した(白色沈殿について、阻害活性試験を実施したところ活性は認められなかった)。このときに得られたHPLC溶出液から、それぞれFr1(7−15min/収量433.0mg)、Fr2(25−35min/収量1744.0mg)、Fr3(35−40min/収量526.7mg)、Fr4(40−50min/収量256.7mg)、Fr5(53−57min/収量33.6mg)と5つの分画物を得た。このときのHPLCチャートを図1に示す。更に、カラム吸着成分の回収のために20容量%アセトニトリル水溶液にて洗浄を行った(全回収率88.2%)。得られた分画物について、上記方法にてα−グルコシダーゼ阻害活性試験を実施した。その結果を表3に示した。
【0037】
【表3】
【0038】
表3から理解されるように、Fr1及びFr3〜5について阻害活性が認められ、Fr2及び20%アセトニトリル回収部においては、全く阻害活性が認められなかった。Fr3〜5においても比較的強い阻害活性が認められたが、特にFr1が強い阻害活性を示したことから、これを更に分離していくこととした。なお、移動相として50〜70容量%アセトニトリル水溶液を用いることによってもFr1を分離することができる。この場合保持時間が変動するが10〜25min以内のピークを回収することで他の活性成分画分(Fr3〜5)と分離できる。
【0039】
YMC pack polyamine−IIカラムでは、図1に示したスペクトルの通り、ピークがブロードになることから更に分離していくことは非常に困難であると考え、次に使用カラムをODSに変更し、さらに分画を進めた。
【0040】
分取条件は、使用カラム:DAISOPAK SP-120-5-ODS-BP(20×250mm)、移動相:水、流速:5.0mL/min、カラム温度:30℃、検出器:RIで実施した。得られたスペクトルより、ODSFr1(10−10.7min/収量230.4mg)、ODSFr2(10.7−11.2min/5.8mg)、ODSFr3(11.2−13min/70.8mg)と3つの分画物を得た。これらの分画物について阻害活性試験を実施した。その結果を表4に示す。
【0041】
【表4】
【0042】
〔分画・精製物の構造決定〕
ODSFr2画分についてさらに数回同様の分取操作を繰り返してほぼ単一の物質を得た。この物質の構造を明らかとするために各種NMR分析(1H、13C、1H−1HCOSY、HMQC及びHMBC)並びにFAB−MS分析を実施し、得られた各種スペクトルデータを用いて構造解析を行った。NMR分析(1D−及び2D−)は、分析装置:JNM AL−400(日本電子製、400MHz)、溶媒:D2O(内部標準試薬としてアセトンを使用)、分析温度:25℃にて行った。FAB−MS分析は、分析装置:7000QQ型(日本電子製)、イオン化法:高速原子衝撃法(FAB)、加速電圧:6kV、マトリックス:グリセリン、測定質量範囲:〜m/z1000にて実施した。図2に1H−NMRスペクトル(inD2O/400MHz)を、図3に13C−NMRスペクトル(inD2O/400MHz)を、図4にFAB−MSスペクトルを示した。
【0043】
以上の結果を基に、解析を行った結果、ODSFr2から得られた成分は図5に示すようなスルフォキシドを有するシクリトールであることが示唆された。しかし、得られたスペクトルデータからでは、スルフォキシドの存在を確認することが出来ないことから、更にHR−MS分析により、元素組成を明らかにすることとした。分析条件は、分析装置:JMS−T100LC(日本電子製)、イオン化法:APCI、測定質量範囲:〜m/z1000にて実施した。この分析から、表5に示す結果が得られた。
【0044】
【表5】
【0045】
以上のことより、図5に示した構造との整合性が得られたことから、ODSFr2で得られた成分は、次の化学式(化1)に示すようにスルフォキシドを有する13員環シクリトール構造であることが明らかとなった。
【0046】
【化2】
【0047】
〔分画・精製物の活性比較〕
上記構造決定されたα−グルコシダーゼ阻害剤について、コタラヒムブツから得られた既報成分(非特許文献26から引用)及び医薬品として販売が認められているボグリボースとその活性を比較した。その結果を表6に示す。この表から分かるように、上市されているボグリボースとほぼ同程度の活性強度を示し、同じ植物から得られた既報成分と比較して、マルターゼ阻害で約71〜103倍、スクラーゼ阻害では約19〜34倍もの阻害活性強度を示した。このように、本発明によるα−グルコシダーゼ阻害剤は、既報成分をはるかに凌ぐ阻害活性を示す成分であることが明らかとなり、コタラヒムブツ抽出物における主たる活性成分であると考えられる。
【0048】
【表6】
【0049】
以上のように、化学式(化1)で示される化合物は優れたα−グルコシダーゼ阻害活性を有していることが確認された。この化合物のα−グルコシダーゼ阻害活性は、これまで報告がなされているコタラヒムブツ中に存在しているサラシノールやコタラノールに比べるとかなり高い比活性を有し、コタラヒムブツ抽出物中における本発明に係る化合物の寄与率は高いものと考えられる。
【実施例2】
【0050】
次に、この成分を使った処方例を示す。
〔製造例1:錠剤〕
本発明のα−グルコシダーゼ阻害剤 0.07g
マルトシルサイクロデキストリン 600g
ショ糖脂肪酸エステル 50g
炭酸カルシウム 15g
還元麦芽糖水あめ 75g
無水乳酸 110g
結晶セルロース 100g
ミルクカルシウム 50g
上記処方に従って、常法により錠剤5000錠を製造した。
【産業上の利用可能性】
【0051】
本発明の化合物は、新規でかつ活性の高いα−グルコシダーゼ阻害作用を有し、これにより、抗糖尿病患者や糖尿病予備群の人に対してより有効な抗糖尿病薬や特定保健用食品などが提供される。
【図面の簡単な説明】
【0052】
【図1】コタラヒムブツ水抽出物のHP−20分画物をアミノカラムで分画した70%アセトニトリル溶出物のHPLCチャートである。
【図2】コタラヒムブツ抽出物の最終分画物の1H−NMRスペクトルである。
【図3】コタラヒムブツ抽出物の最終分画物の13C−NMRスペクトルである。
【図4】コタラヒムブツ抽出物の最終分画物のFAB−MSスペクトルである。
【図5】コタラヒムブツ抽出物の最終分画物の推定平面構造図である。
【特許請求の範囲】
【請求項1】
次の化学式(化1)で示される化合物。
【化1】
【請求項2】
次の化学式(化1)からなることを特徴とするα−グルコシダーゼ阻害剤。
【化1】
【請求項3】
請求項2に記載のα−グルコシダーゼ阻害剤を含有することを特徴とする抗糖尿病薬。
【請求項4】
請求項2に記載のα−グルコシダーゼ阻害剤を活性成分とする糖吸収抑制効果を標榜することが可能な食品。
【請求項1】
次の化学式(化1)で示される化合物。
【化1】
【請求項2】
次の化学式(化1)からなることを特徴とするα−グルコシダーゼ阻害剤。
【化1】
【請求項3】
請求項2に記載のα−グルコシダーゼ阻害剤を含有することを特徴とする抗糖尿病薬。
【請求項4】
請求項2に記載のα−グルコシダーゼ阻害剤を活性成分とする糖吸収抑制効果を標榜することが可能な食品。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2008−137925(P2008−137925A)
【公開日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願番号】特願2006−324346(P2006−324346)
【出願日】平成18年11月30日(2006.11.30)
【出願人】(591046892)富士産業株式会社 (12)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
【公開日】平成20年6月19日(2008.6.19)
【国際特許分類】
【出願日】平成18年11月30日(2006.11.30)
【出願人】(591046892)富士産業株式会社 (12)
【出願人】(505127721)公立大学法人大阪府立大学 (688)
【Fターム(参考)】
[ Back to top ]