
α−(フェノキシ)フェニル酢酸誘導体の分割
本発明は、式(I)(式中、R1はアルキルまたはハロアルキル、およびXはハライドである)で示されるエナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸化合物を、そのエナンチオマー混合物から生成するための方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、α-(フェノキシ)フェニル酢酸をそのエナンチオマー混合物から分離するためのエナンチオ選択的分割方法に関する。
【背景技術】
【0002】
発明の背景
ハロフェナートなどのα-(フェノキシ)フェニル酢酸のエステルおよびアミド誘導体はキラル化合物であり、例えばII型糖尿病および高脂血症のような血液脂質沈着に関連する疾患を含む様々な生理学的疾患の改善に役立つ。例えば、米国特許第3,517,050号および第6,262,118号を参照のこと。α-(フェノキシ)フェニル酢酸には、単一のキラル中心がカルボニル炭素原子に対してαの位置の非対称的に置換された炭素原子に存在し、したがって2つのエナンチオマーの形が存在する。
【0003】
チトクロームP450 2C9は、特定の薬物代謝に重要な役割を果たすことが知られている酵素である。チトクロームP450酵素の阻害によって起こる薬物代謝の変化が患者に対して重大な悪影響を引き起こす高い可能性を有していることは、当業者に公知である。例えばハロフェン酸のようなラセミ体のα-(フェノキシ)フェニル酢酸がチトクロームP450 2C9を阻害することも、また公知である。例えば、米国特許第6,262,118号 を参照のこと。したがってハロフェン酸またはその誘導体などのラセミ体のα-(フェノキシ)フェニル酢酸の投与により、この酵素によって代謝される抗凝血剤、抗炎症剤およびその他の薬剤を含む他の薬物との様々な薬物相互作用問題が引き起こされる可能性がある。ハロフェン酸の(-)-エナンチオマーは、チトクロームP450 2C9を阻害する能力が(+)-エナンチオマーに比べて約20倍弱いことが発見された(同文献)。したがって、実質的に(+)-エナンチオマーを含まないハロフェン酸の(-)-エナンチオマーまたはその誘導体を投与することが、薬物相互作用の可能性を減少させるために望ましい。
【0004】
したがって、例えば(-)-ハロフェン酸のような、α-(フェノキシ)フェニル酢酸の所望のエナンチオマーを濃縮した生成物を生産するための効率的な方法が必要である。
【発明の開示】
【0005】
発明の概要
本発明の一つの局面において、式:
(式中、
R1は、アルキルまたはハロアルキル、および
Xは、ハライドである)
で示されるエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸化合物を、第1および第2のエナンチオマーを含むα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物から生成するための方法を提供する。ある特定な実施態様においては、エナンチオマー混合物はラセミ混合物である。
【0006】
本発明の方法は、次の工程を含む:
(a) 第1エナンチオマーがエナンチオマー濃縮された固体の酸塩基塩を含む溶液を、α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物を0.5モル当量未満のエナンチオマー濃縮したキラルアミン化合物と、溶液中におけるフリーの第1エナンチオマーの量とフリーの第2エナンチオマーの量の比を約1対3とするのに十分な条件下で接触させることにより、生成する工程;および、
(b) 第1エナンチオマーの固体酸塩基塩を溶液から、α-(フェノキシ)フェニル酢酸化合物の第2エナンチオマーの酸塩基塩の濃度がその飽和点の近くまたはその飽和点より低い温度で分離する工程。
【0007】
第2エナンチオマーの少なくとも一部を、例えば、第2エナンチオマーを塩基と接触させることによりラセミ化させ、第1エナンチオマーに変換することができる。その結果生じるエナンチオマー混合物を再利用し、同様のエナンチオマー濃縮処理に供して第1エナンチオマー酸塩基塩の収率を増加させることができる。
【0008】
ある特別の実施態様においては、キラルアミン化合物は、式:
(式中、
R2およびR3は、それぞれ独立に水素またはアルキルであるか;またはR2およびR3はそれらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素またはアルキルであるか、あるいはR5またはR6のうちの1つは、アミン保護基であり;および
Arは、アリールである)
で示される化合物である。
【0009】
詳細な説明
I. 定義
「アルキル」とは、1〜10個の炭素原子の、好ましくは1〜6個の炭素原子の、より好ましくは1〜4個の炭素原子の直鎖状または分岐した脂肪族炭化水素鎖基を指す。例示的なアルキル基にはメチル、エチル、n-プロピル、2-プロピル、tert-ブチル、ペンチルその他が含まれるが、それらに限定されるわけではない。
【0010】
「アリール」とは、一価の6〜10個の炭素環原子の一環または二環から成る芳香族炭化水素部分を指す。特に別の記述または表示をしない限り、アリール基を1つまたは複数の置換基、好ましくは1個、2個または3個の置換基、より好ましくは、アルキル、ハロアルキル、ニトロおよびハロから選択される1個または2個の置換基で置換することができる。より具体的には、用語「アリール」は、フェニル、1-ナフチル、および2-ナフチルその他を含むが、それらに限定されるわけではなく、その各々は上に述べた1つまたは複数の置換基で置換されてもよい。
【0011】
「CAF D塩基」とは、クロラムフェニコールD塩基、すなわちD-threo-(-)-2-アミノ-1-(ニトロフェニル)-1,3-プロパンジオールを指す。
【0012】
「キラル」または「キラル中心」とは、4つの異なる置換基を有する炭素原子を指す。しかしながら、キラリティの最終判断基準は鏡像が重ね合わせられないことである。
【0013】
「CPTA」および「ハロフェン酸」という用語は、本明細書において交換して用いることができ、(4-クロロフェニル)(3-トリフルオロメチルフェノキシ)酢酸を指す。
【0014】
「エナンチオマー混合物」とは、ラセミ混合物を含むエナンチオマーの混合物を有するキラル化合物を意味する。好ましくは、エナンチオマー混合物とは、実質的に等しい量の各エナンチオマーを有するキラル化合物を指す。より好ましくは、エナンチオマー混合物は各エナンチオマーが当量存在するラセミ混合物を指す。
【0015】
「エナンチオマー濃縮された」とは、1つのエナンチオマーがそれを分離工程に付す前よりも多量に存在する組成物を指す。
【0016】
「エナンチオマー過剰率」または「%ee」とは、第1エナンチオマーと第2エナンチオマーの間の差異の量を指す。エナンチオマー過剰率は、式:%ee = (第1エナンチオマーの%)-(第2エナンチオマーの%)と定義される。したがって、組成物が98%の第1エナンチオマーおよび2%の第2エナンチオマーを含む場合、第1エナンチオマーのエナンチオマー過剰率は98%-2%すなわち96%である。
【0017】
用語「ハライド」および「ハロ」は本明細書中で交換して用いることができ、F、Cl、BrおよびI、ならびに-CNおよび-SCNなどの偽ハロゲン化物を含むハロゲンを指す。
【0018】
「ハロアルキル」とは、1つまたは複数の水素原子がトリフルオロメチルなどのペルハロアルキルを含むハロゲンで置換されている、本明細書で定義のアルキル基を指す。
【0019】
「ハロフェナート」とは、2-アセトアミドエチル4-クロロフェニル-(3-トリフルオロメチル-フェノキシ)アセタート(すなわち、4-クロロ-α-(3-(トリフルオロメチル)フェノキシ)ベンゼン酢酸、2-(アセチルアミノ)エチルエステル、または (4-クロロフェニル)(3-トリフルオロメチルフェノキシ)酢酸、2-(アセチルアミノ)エチルエステル)を指す。
【0020】
「ヘテロアルキル」とは、1または複数の異種原子、または1または複数の異種原子を含む置換基を含む、分岐したまたは分岐のない非環状の飽和アルキル部分を意味し、ここで異種原子とはO、NまたはSである。典型的な異種原子を含む置換基には、=O、-ORa、-C(=O)Ra、-NRaRb、-N(Ra)C(=O)Rb、-C(=O)NRaRb および-S(O)nRa(ここでnは0〜2までの整数である)が含まれる。各RaおよびRbは独立に水素、アルキル、ハロアルキル、アリールまたはアラルキルである。ヘテロアルキルの代表的な例には、例えば、N-アセチル2-アミノエチル(すなわち、-CH2CH2NHC(=O)CH3)が挙げられる。
【0021】
用語「ヘテロサイクリル」と「複素環」は交換して用いることができ、3〜8個の環原子の非芳香性環部分を指し、ここで1、2、または3個の環原子はN、OまたはS(O)n(ここでnは、0〜2の整数である)から選択される異種原子であり、残る環原子は、Cであり、1または2個のC原子はカルボニル基で置換されてもよい。もし別の記述または表示がなければヘテロサイクリル環を、ハロゲン、アルキル、アリール、ヒドロキシ、アミノ、またはアルコキシから選択される1、2または3個の置換基で任意に独立して置換されてもよい。より具体的には、用語「ヘテロサイクリル」には、1,3-ジオキサンおよびその誘導体その他が含まれるが、それに限定されるわけではない。
【0022】
「脱離基」は、従来合成有機化学においてそれに関連する意味、すなわち、求核基によって置き換えられることのできる原子または基の意味を有し、ハロ(クロロ、ブロモおよびヨードなど)、アルカンスルホニルオキシ、アレーンスルホニルオキシ、アルキルカルボニルオキシ(例えば、アセトキシ)、アリールカルボニルオキシ、メシルオキシ、トシルオキシ、トリフルオロメタンスルホニルオキシ、アリールオキシ(例えば、2,4-ジニトロフェノキシ)、メトキシ、N,O-ジメチルヒドロキシルアミノ、その他を含む。
【0023】
用語「金属」には、第I、II族金属、および遷移金属、ならびにBおよびSiなどの主族金属が、含まれる。
【0024】
「光学純度」とは、組成物中に存在する特定のエナンチオマーの量を指す。例えば、ある組成物が98%の第1エナンチオマーおよび2%の第2エナンチオマーを含む場合、第1エナンチオマーの光学純度は98%である。
【0025】
もし別の記述がなければ、用語「フェニル」とは、置換されていてもよいフェニル基を指す。適切なフェニル置換基は「アリール」の定義に記述されたものと同一である。同様に、用語「フェノキシ」とは、式-OAraの部分を指し、Araは本明細書で定義されるフェニル基である。したがって、用語「α-(フェノキシ)フェニル酢酸」とは、置換されていてもよいフェニルおよび置換されていてもよいフェノキシ部分(残基)によって2位が置換された酢酸を指す。
【0026】
「保護基」とは、分子中の反応性基に結合された時その反応性を遮蔽、減少、または阻止する部分を指す。保護基の例は、T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York, 1999, および、 Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Vols.1-8 (John Wiley and Sons, 1971-1996)に見出され、それらの全体が参照により本明細書組み入れられる。代表的なヒドロキシ保護基には、アシル基、ベンジルおよびトリチルエーテル、テトラヒドロピラニルエーテル、トリアルキルシリルエーテルおよびアリルエーテルが含まれる。代表的なアミノ保護基には、ホルミル、アセチル、トリフルオロアセチル、ベンジル、ベンジルオキシカルボニル(CBZ)、tert-ブトキシカルボニル(Boc)、トリメチルシリル(TMS)、2-トリメチルシリル-エタンスルホニル(SES)、トリチルおよび置換されたトリチル基、アリルオキシカルボニル、9-フルオレニルメチルオキシカルボニル(FMOC)、ニトロ-ベラトリルオキシカルボニル(NVOC)その他が含まれる。
【0027】
用語「速度」とは、塩の生成に言及している場合は、速度論的および/または熱力学的速度を指す。
【0028】
本明細書において用いられる用語「処理する」、「接触させる」、または「反応させる」とは、指示されたおよび/または所望の生成物を産生するために適切な条件下で、2以上の反応物を加えることまたは混合することを指す。指示されたおよび/または所望の生成物を産生する反応は、必ずしも最初に加えられた2つの反応物の組み合わせから直接得られなくてもよく、すなわち、混合物中で1または複数の中間生成物が生成され、それが最終的には指示されたおよび/または所望の生成物の生成をもたらしてもよいことを認識するべきである。
【0029】
本明細書において用いられる用語「上に定義されたもの」および「本明細書において定義されたもの」には、変数に言及する場合には、もしあれば変数の好ましい、より好ましい、および最も好ましい定義は勿論、変数の広い定義を参照により組み入れる。
【0030】
多くの有機化合物は光学活性型で存在する、すなわちそれらは平面偏光の面を回転させる能力を有している。光学活性化合物について記述する際には、接頭辞RおよびSを用いて分子のキラル中心に関する分子の絶対立体配置を表示する。接頭辞「d」および「l」あるいは(+)および(-)を、化合物による平面偏光の回転の符号を指定するために採用し、(-)または(l)は化合物が「左旋性」であることを意味し、また(+)または(d)は化合物が「右旋性」であることを意味する。エナンチオマーの絶対立体化学の命名法と回転の命名法との間には相関が無い。与えられた化学構造に対して、「立体異性体」と呼ばれるこれらの化合物は、それらが互いの鏡像である以外は同一である。特定の立体異性体は「エナンチオマー」と呼ぶこともできる。またそのような異性体の混合物をしばしば、「エナンチオマー」混合物または「ラセミ」混合物と呼ぶ。例えば、Streitwiesser, A. & Heathcock, C.H., INTRODUCTION TO ORGANIC CHEMISTRY, 2nd Edition, Chapter 7 (MacMillan Publishing Co., U.S.A. 1981) を参照のこと。
【0031】
用語「その(+)-立体異性体を実質的に含まない」と「その(+)-エナンチオマーを実質的に含まない」とは、本明細書において交換して用いることができ、組成物が(+)-異性体と比較して実質的に(-)-異性体をより大きな割合で含んでいることを意味する。好ましい実施態様において、用語「その(+)立体異性体を実質的に含まない」とは、組成物が重量で少なくとも90%の(-)-異性体および重量で10%以下の(+)-異性体であることを意味する。より好ましい実施態様において、用語「その(+)-立体異性体を実質的に含まない」とは、組成物が重量で少なくとも99%の(-)-異性体を含み、重量で1%以下の(+)-異性体を含むことを意味する。最も好ましい実施態様においては、用語「その(+)-立体異性体を実質的に含まない」とは、組成物が重量で99%を越える(-)-異性体を含むことを意味する。これらの百分率は、組成物中の異性体の総量に基づく。
【0032】
II. 序論
キラル合成は近年大きな進歩をしているが、ラセミ体の分割が、光学活性化合物すなわちキラル化合物の調製のための工業的方法において、いまだに一般的に好まれる方法である。通常は、キラル化合物をラセミ体の形で合成し、最終生産物を分割してエナンチオマー濃縮された化合物を産生する。
【0033】
最終生産物を分割するこの方法が、薬学的活性のあるキラル化合物の大規模調製に特に役立つ。キラル化合物のエナンチオマーは正確に同じ化学結合を持つが、エナンチオマー中の原子の空間的配向が異なる。したがって、キラル薬剤の1つのエナンチオマーがもう一方のエナンチオマーより望ましい活性を発揮し、副作用は著しく少ないことがしばしば起こる。光学活性薬剤のキラリティーとその副作用のそのような関係は以前から知られているが、いまだに多くのキラル薬剤はラセミ体の形で投与されている。
【0034】
ジアステレオマーの結晶化が工業的な規模において広く用いられる。ジアステレオマーの結晶化による分割の理論的な1回全工程を通しての収率は50パーセントである。しかし通常は、十分な光学純度を有する組成物を産生するためには2回以上の再結晶工程が必要である。
【0035】
本発明は、例えばハロフェン酸のようなα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物、好ましくはラセミ混合物、をエナンチオマー濃縮する方法を提供する。好ましくは、本発明の方法は、α-(フェノキシ)フェニル酢酸化合物の(-)-エナンチオマーの固体の酸塩基塩を提供する。このようにして、(-)-エナンチオマーを溶液から容易に分離することができる。
【0036】
エナンチオマー濃縮したα-(フェノキシ)フェニル酢酸のカルボン酸基を、次にカルボン酸活性化基によって活性化し、活性化されたα-(フェノキシ)フェニル酢酸を生成することができ、これを、アルコール、アミン、チオール、また他の求核性の化合物と反応させて、それぞれエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸エステル、アミド、チオエステルまたは他の誘導体を産生することができる。したがって、本発明の方法を用いて生成されたエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸化合物は、米国特許第3,517,050号に開示されたようなα-(フェノキシ)フェニル酢酸誘導体を産生するために有用である。特に、本発明の方法は、(-)-ハロフェナートを産生するために役立つ。
【0037】
III. エナンチオ選択的結晶化
上述したように、大部分のエナンチオ選択的結晶化方法においては、十分な光学純度を有する組成物を生成するためには、2回以上の再結晶化工程が必要である。しかしながら、本発明者らは、本明細書において開示する一定の条件下において、単一の結晶化工程により十分な光学純度のα-(フェノキシ)フェニル酢酸化合物を生成することができることを見出した。したがってある局面において、本発明の方法は、α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物をキラルアミン化合物を用いてエナンチオマー濃縮することができるという、本発明者らによる驚くべきかつ予期しない発見に基づいている。特に本発明の方法は、光学純度が少なくとも約90%、好ましくは少なくとも約95%、より好ましくは少なくとも約97%、そして最も好ましくは少なくとも約98%の、α-(フェノキシ)フェニル酢酸化合物の所望のエナンチオマーを提供する。
【0038】
ある実施態様において、本発明の方法は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライドである)
で示されるα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物、好ましくはラセミ混合物のエナンチオマー濃縮を提供する。工程は一般に、キラルアミン化合物を用いて、α-(フェノキシ)フェニル酢酸化合物の固体のエナンチオマー濃縮された酸塩基塩を形成する工程を含む。
【0039】
特に本発明の方法は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライドである)
で示されるα-(フェノキシ)フェニル酢酸、例えばハロフェン酸(この場合R1はCF3、およびXはCl)、の分割を目標とする。
【0040】
ある特定の実施態様では、本発明の方法は、Xがクロロである式Iまたは好ましくは式IIのα-(フェノキシ)フェニル酢酸の分割を目標とする。
【0041】
さらに別の実施態様では、本発明の方法は、R1がハロアルキル、好ましくはトリフルオロメチルである、式Iまたは好ましくは式IIのα-(フェノキシ)フェニル酢酸の分割を目標とする。
【0042】
ある特定の実施態様では、α-(フェノキシ)フェニル酢酸をキラル塩基を用いて結晶化する。下記の実施例の項で開示する塩基を含め、種々様々なキラル塩基を用いることができる。好ましくは、用いたキラル塩基により、α-(フェノキシ)フェニル酢酸の(-)-エナンチオマーの固体の酸塩基塩が得られる。このようにして、(-)-エナンチオマーが例えばろ過により容易に溶液から分離される。ある特定の実施態様では、キラル塩基は、式:
(式中、各R2およびR3は、独立して水素、アルキルまたはヒドロキシ保護基であるか;あるいはR2およびR3は、それらが結合している原子と共に複素環部分を形成し;R4は、水素またはアルキルであり;各R5およびR6は、独立して水素またはアルキルであるか、R5またはR6のうちの1つがアミン保護基であり;またArは、アリールである)
で示されるアミン化合物である。
【0043】
ある特定の実施態様では、R2とR3はそれらが結合する酸素原子と共に1,3-ジオキサン、置換された1,3-ジオキサン(例えば、5,5-ジメチル-1,3-ジオキサンなどのジアルキルで置換された1,3-ジオキサン)またはその誘導体を形成する。
【0044】
別の実施態様では、R2とR3は水素である。
【0045】
さらに別の実施態様では、R4は水素である。
【0046】
また別の実施態様では、Arは置換されたアリールである。特に好ましいAr部分は、置換されていてもよいフェニルである。特別に好ましいAr部分は、4-ニトロフェニルである。
【0047】
またさらに、上述の好ましい基の組合せが他の好ましい実施態様を形成することになる。例えば、特に好ましいキラル塩基は上記式IIIのアミン化合物であって、式中、R2、R3、R4、R5およびR6は、水素であり;またArは、4-ニトロフェニルである。また特に好ましいα-(フェノキシ)フェニル酢酸化合物は、上記の式IIの化合物であり、式中、R1はトリフルオロメチルであり、Xはクロロである。このようにして、多種多様な好ましいキラル塩基およびα-(フェノキシ)フェニル酢酸化合物が本発明において具体化される。
【0048】
本発明者らは、α-(フェノキシ)フェニル酢酸の結晶化に用いられるキラル塩基の量が、エナンチオマー濃縮の光学純度に重要な影響があることを見出した。例えば、式:
(式中、R2、R3、R4およびArは、本明細書において定義されたものである)
で示されるキラルアミン化合物をα-(フェノキシ)フェニル酢酸化合物の結晶化で用いる場合は、より高い%eeが、キラルアミン化合物を0.5モル当量未満、好ましくは約0.48モル当量以下、より好ましくは約0.47モル当量以下、そして最も好ましくは約0.45モル当量以下の量を用いることにより、得られる。高度にエナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸誘導体を生成するためには、キラルアミン化合物それ自身がエナンチオマーとして十分純粋でなければならないことを認識するべきである。
【0049】
結晶化は、通常、α-(フェノキシ)フェニル酢酸の2つのエナンチオマーとキラルアミン間で形成される塩が異なる溶解度を有する溶媒中で実施する。このようにして、ジアステレオマー塩のうちの1つが、溶液から優先的に沈殿する。適当な結晶化溶媒には、アルコールなどのプロトン性溶媒が含まれる。特に好ましい結晶化溶媒はイソプロピルアルコールである。
【0050】
エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸の収率はまた、とりわけ用いる結晶化溶媒の量に依存する。例えば、大量の結晶化溶媒を用いる場合、混合物が稀薄になりすぎ、固体の形成が減少する。用いる結晶化溶媒の量が少なすぎる場合は、溶液が望まれないジアステレオマー塩で過飽和することになり、望ましくないジアステレオマー塩の結晶化をもたらす可能性があり、そのために所望のエナンチオマーの光学純度が減少する。したがって、結晶化溶媒としてイソプロパノールを用いる場合、用いる結晶化溶媒の量は、1グラムのα-(フェノキシ)フェニル酢酸化合物当り、好ましくは約2グラム〜約6グラム、より好ましくは約3グラム〜約5グラム、さらにより好ましくは約3.5グラム〜約4.5グラム、そして最も好ましくは約4グラムである。
【0051】
ある実施態様では、結晶化工程は、両方のエナンチオマーの核形成温度より高い温度へ結晶化溶液混合物を加熱して、両方のエナンチオマーを実質的にすべて溶解する工程を含む。例えば、結晶化溶液を約60℃から溶液の沸点までの範囲の温度に、好ましくは約70℃から約80℃までの範囲の温度に加熱する。より好ましくは、結晶化溶液を約75℃に加熱する。溶液は、キラルアミン化合物を加える前および/または後に加熱してもよい。加熱は固形物が実質的に完全に溶解するまで行い、それは通常約0.5〜約16時間、好ましくは約1〜約8時間の間である。
【0052】
その後結晶化溶液を、第1ジアステレオマー塩の、例えば、α-(フェノキシ)フェニル酢酸の(-)-エナンチオマー塩の核形成温度以下になるまで、しかし好ましくは第2ジアステレオマー塩の、例えばα-(フェノキシ)フェニル酢酸の(+)-エナンチオマー塩の核形成温度より高い温度まで冷却する。これによって、第1エナンチオマーとキラルアミン化合物の固体酸塩基塩の形成が可能になる。どのような理論にも拘束されずに、キラルミン化合物を使用することにより、一方のエナンチオマーの酸塩基塩の形成が、他方のエナンチオマーの酸塩基塩の形成より著しく速い速度で起こることになると信じられている。この速度は、2つのエナンチオマー間の速度論的および/または熱力学的速度差による可能性がある。典型的な化合物の場合と同じく、本発明のα-(フェノキシ)フェニル酢酸化合物の溶解度プロフィールは、高い温度でより高い溶解度を有している。したがって、第2ジアステレオマー塩の核形成温度のちょうど上まで結晶化溶液を冷却することにより、固体の第1ジアステレオマー塩の回収率がより高くなる。
【0053】
スラリーが形成された後、さらに結晶化溶液を溶液の温度が第2ジアステレオマー塩の飽和点の近くまたは上になるまで、冷やすことができる。これが、第1エナンチオマーのジアステレオマーの固体酸塩基塩の形成を増加させると共に、第2エナンチオマーからのジアステレオマー固体酸塩基塩の形成を防ぐ。
【0054】
結晶化溶液の冷却速度が、形成される固体酸塩基塩の光学純度に影響する可能性がある。例えば、結晶化溶液を速く冷却しすぎる場合には、望ましいエナンチオマーの固体酸塩基塩の格子内に望ましくないエナンチオマーがトラップされる可能性がある。しかしながら、遅すぎる冷却速度は生産時間とコストを増加させる。したがって、光学純度の損失を最小限にするが、しかし経済的に十分な速度で結晶化溶液を冷却するべきである。通常、結晶化溶液冷却速度は約0.05℃/min〜約1℃/min、好ましくは約0.1℃/min〜約0.7℃/min、およびより好ましくは約0.25℃/min〜約0.4℃である。その後結晶化溶液は、第2のすなわち望ましくないエナンチオマーの固体酸塩基塩の飽和点より高く維持される。通常、結晶化溶液をこの温度で、約1〜約72時間、好ましくは約2〜約48時間、より好ましくは約3〜約30時間維持する。
【0055】
予想通りに、少量のキラルアミン化合物を用いることにより、第1エナンチオマーの固体酸塩基塩の選択的な形成が可能になる。しかしその結果、収率が相応して小さくなる。理論上、ラセミ混合物からの所望のエナンチオマーの収率は、50%である。したがって、0.5モル当量のキラルアミン化合物を用いる場合、理論的収率は全α-(フェノキシ)フェニル酢酸の50%(あるいは所望のエナンチオマーの100%)である。経済的に望ましいためには、本発明の方法は所望のエナンチオマーの少なくとも約50%、好ましくは少なくとも約60%、より好ましくは少なくとも約70%、そして最も好ましくは少なくとも約75%の収率を提供する。100%の選択性を仮定すると、これらの収率は約0.25、0.30、0.35および0.375モル当量のキラルアミン化合物を加えることに相当し、それらは結晶化溶液に加える必要のあるキラルアミン化合物の最少量を表わす。
【0056】
第2エナンチオマーがキラルアミン化合物と固体酸塩基塩を形成する傾向が、従来の結晶化工程の変動の主な原因の1つであると信じられている。したがって第2のすなわち望ましくないエナンチオマーの過飽和点を測定することにより、第2エナンチオマーの固体酸塩基形成の予測不可能性を最小限にするか防止することができる。当業者は、例えば溶解度実験により過飽和点を容易に決定することができる。
【0057】
本発明の方法をラセミ混合物中に存在する(-)-エナンチオマーの濃縮に関して議論しているが、本発明の方法はまた(+)-エナンチオマーの濃縮にも適用可能であることに注意するべきである。本発明の方法は、基本的に(-)-エナンチオマーが濃縮された固体の沈殿、および(+)-エナンチオマーが濃縮されたろ過液すなわち母液を提供する。沈殿した塩からの所望の(-)-エナンチオマーの遊離およびキラルアミン化合物の回収を、例えば従来からこの性質の塩を加水分解することが知られている薄い鉱酸もしくは他の任意の無機酸または有機酸により塩を酸性化することにより、容易に遂行することができる。この手法は望まない副産物としてろ過液を残しているので、ろ過液をさらに酸または好ましくは塩基で処理して、(+)-エナンチオマーが濃縮されたろ過液をラセミ混合物に変換することができる。例えば、(+)-エナンチオマーを、水酸化ナトリウム水溶液を用いてラセミ体化することができる。次にこのラセミ混合物を再使用、すなわちリサイクルすることができる。さらに、キラルアミン化合物もまた上記の変換工程から回収してリサイクルすることができる。したがって本発明の方法は容易にリサイクル型の方法となる。
【0058】
IV. ラセミ体α-(フェノキシ)フェニル酢酸の合成
式Iのα-(フェノキシ)フェニル酢酸のラセミ混合物を生成する1つの方法を以下のスキームIに示す。
【0059】
このようにして、フェニル酢酸1の、例えば酸塩化物などの活性化カルボン酸誘導体への変換と、それに続くα-臭素化により、α-ブロモフェニルアセチルクロリド(示さず)を得た。次に酸塩化物をエステル2に変換し、式中Rは、通常はアルキルである。酸塩化物をエステル2に変換するために用いられるアルコールROHは、次の反応で溶媒として用いられるアルコールと同じアルコールであることが好ましい。このようにして、異なる溶媒型の数を最小限にする。さらに、次の反応で同じROHを溶媒として用いることにより、例えばトランスエステル化などによるの副産物の形成量が最小限になる。例えばイソプロピルエステル2は、すなわちRがイソプロピルである場合は、次の反応がイソプロパノール溶媒中で便利に行なわれるため、特に有利である。水酸化物(例えば水酸化カリウム)などの塩基の存在下におけるエステル2のフェノール化合物3による置換反応によって、α-(フェノキシ)フェニル酢酸エステル4を得た。α-(フェノキシ)フェニル酢酸エステル4の加水分解が、α-(フェノキシ)フェニル酢酸Iを生じた。
【0060】
このようにして、ヘプタンからの結晶化に続いて、(4-クロロフェニル)-(3-トリフルオロメチルフェノキシ)-酢酸、すなわちCPTAを、中間に単離のない5ステップで、収率約85%で調製することができる。
【0061】
V. エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸の有用性
エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸化合物は、米国特許第3,517,050号に開示されたα-(フェノキシ)フェニル酢酸化合物を含む様々な薬学的に活性な化合物を調製する際に有用な中間物である。したがって、本発明の別の局面は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライド、およびR7はヘテロアルキル、好ましくはN-アセチル2-アミノエチル(すなわち、式:-CH2CH2NHC(=O)CH3部分)である)で示されるα-(フェノキシ)フェニル酢酸化合物を、式Iのα-(フェノキシ)フェニル酢酸化合物のラセミ混合物からエナンチオ選択的に生成する方法を提供する。本方法は、式Iのα-(フェノキシ)フェニル酢酸化合物のラセミ混合物を上述のように分割する工程、およびエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸をカルボン酸活性化試薬と反応させることによりエナンチオマー濃縮した活性化α-(フェノキシ)フェニル酢酸を生産する工程、を含む。適当なカルボン酸活性化試薬には、チオニルハライド(例えば、チオニルクロリド)、無水物、チオエステルを生成する試薬、および当業者に公知の他のカルボン酸活性化試薬が含まれる。
【0062】
次に活性化α-(フェノキシ)フェニル酢酸は式(R7-O)wMの化合物、例えばN-アセチルエタノールアミン誘導体と反応させて、エナンチオマー濃縮した式IIIのα-(フェノキシ)酢酸フェニル化合物を生成し、式中、R7は上に定義した通りで、Mは水素、または例えばNa、K、Li、Ca、Mg、Csなどの金属、および添字wはMの酸化状態である。本発明者らは、有意なラセミ化なしに、活性化された酸と式(R7-O)wMの化合物の反応を行なうことができることを発見した。
【0063】
本発明のさらなる目的、利点および新規な特徴が、本発明の次の実施例を検討することによって当業者に明白になるであろうが、実施例により制限するものではない。
【0064】
実施例
試薬と実験装置
もし他の記載がなければ、試薬および溶媒は Aldrich ChemicalまたはFisher Scientificから購入した。またN-アセチルエタノールアミンをLancaster Synthesisから得た。ラセミ体のCPTA、すなわちハロフェン酸を、米国特許第3,517,050号および第6,262,118号(そのすべてが参照により全体を本明細書に組み入れられる)に開示された製法により調製した。(1R,2R)-(-)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオール(すなわちCAF D塩基)はTCI Americasから得た。
【0065】
操作は陽圧窒素雰囲気下で行った。再循環加熱および冷却システムに付属したCamileプロセス制御コンピュータを用いて、ジャケットで覆った直壁底部排水管ガラス反応器のジャケット温度を調節した。特に表記しない限り、溶媒をBuchi回転蒸発装置を用い、15〜25torr、浴槽温度40℃以下で除去した。固体試料を真空オーブン中40℃、15〜25torrで乾燥させた。Cenco HYVAC真空ポンプを、真空蒸留用に1torr未満の真空を供給するために用いた。水位を、カールフィッシャー解析によりMetrohm 756 KF電量計およびHYDRANAL Coulomat AG試薬を用いて測定した。融点を、Mettler Toledo FP62融点測定装置を用いて測定した。pHを、較正されたOrion Model 290A pHメーターを用いて測定した。プロトンおよび13CのNMRスペクトルを、Bruker Avance 300 MHzスペクトロメーターで記録した。
【0066】
キラルHPLC解析を、λ=240nmで、可動相に溶解した10μlの試料を(R,R)WHELK-O 1.5μm 250×4.6mmカラム (Regis Technologies) に注入し、95/5/0.4 (v/v/v) のヘキサン/2-プロパノール/酢酸により、1.0 ml/分の流速で溶出して実施した。CPTA/CAF D塩基ジアステレオマー塩の固体試料については、固体を塩酸水溶液に加えて、CPTAをメチレンクロリド中へ抽出し、メチレンクロリド層から溶媒を除去した後に、残留物を解析のために可動相に溶解した。
【0067】
非キラルHPLC解析を、λ=220nmで、可動相に溶解した5μlの試料をPhenomenex LUNA 5μm C18(2) 250×4.6 mmカラムに注入して、25℃で実施した。1.5 ml/分の流量で、66体積%の水/34体積%のアセトニトリル/0.1体積%のトリフルオロ酢酸からスタートし、20分で26体積%の水/74体積%のアセトニトリル/0.1体積%のトリフルオロ酢酸に直線的に増加する勾配を用いた。
【0068】
ハロフェナートなどのエステルの酸性溶液の解析には、アセトニトリルを注入用溶媒として用いた。測定に際しては、CPTAおよびハロフェナートの生成物濃度をHPLC分析により、外部標準法および非キラル解析手法を用い、2.5 mg/ml未満の試料濃度で評価した。
【0069】
実施例1
以前のCPTAの分割は米国特許第3,517,050号に報告されたものであり、そこでキラル塩基としてシンコニジンを用い、CPTAの(+)-エナンチオマーがジアステレオマー塩として沈殿した。この手法の主な短所の1つは、所望の(-)-エナンチオマーが母液に残り、純粋な(-)-エナンチオマー画分の分離を困難にしたことであった。
【0070】
本実施例は、様々な異なるキラル塩基を用いてCPTAのラセミ混合物を分割し、エナンチオマー濃縮した固体の(-)-異性体を得た結果を示す。以前の方法と異なり、本発明の方法によって、エナンチオマー濃縮した固体の(-)-CPTAを容易に溶液から分離することができる。
【0071】
ラセミ体CPTAを、ラセミ体ハロフェナートの水酸化カリウム加水分解により調製した。キラル塩基による選別をするために、CPTAとキラル塩基の等モル混合物をガラスバイアルに入れたエタノール、メタノールおよびアセトン中で混合し、溶液を静穏に保った。終夜周囲温度に保持した後、溶液中に残存している試料を5℃で冷蔵庫中に置いた。冷蔵庫に終夜保持した後、エタノール溶液中に残存している試料に少量の水を加えた。周囲温度で4日間置いた後、エタノール水溶液を冷蔵庫に戻した。試料をすべて冷蔵庫に残し、沈殿形成を1か月間に亘って定期的に点検した。表1に、検討した塩基および溶媒条件、ならびに結晶の塩が見出された温度のリストを示す。
【0072】
(表1)CPTA分割のために検討した塩基
E - 評価した
C - 結晶化(温度)
* - 1モル/モル水酸化ナトリウム水溶液で
【0073】
4つのキラル塩基、キニーネ、L-チロシンヒドラジド、(-)-シンコニジン、および2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールの両方のエナンチオマーが、ラセミ体CPTAから結晶塩を生ずることが見出された。結晶した試料について、固体をろ過により分離し、固相と母液の両方をキラルHPLCにより解析し、双方の系統のエナンチオマー組成を測定した。表2にスクリーニングの結果を示す。表2に示す塩基のうちの3つが、固相中の(+)-エナンチオマー濃縮をもたらした。
【0074】
(表2)キラル塩基スクリーニングの結果
* -より希薄
**-より遅い冷却プロフィール
【0075】
表2には、固体と母液の系統中の異性体比率から計算した固体のパーセント収率が含まれている。用いた方程式を下に示す。100%の異性体純度を有する場合の最大の理論的収率は50%である。50%以上の収率は、他の異性体が含まれることを示す。
異性体比率から収率を計算する方程式。
設定する: a=出発物質中の成分1の面積%; b=出発物質中の成分2の面積%; x=単離された物質中の成分1の面積%; y=単離された物質中の成分2の面積%; w=母液中の成分1の面積%; z=母液中の成分2の面積%; E=単離された物質のグラム数; F=母液中の物質のグラム数
また:a + b=100%; E + F=1
そして:xE + wF=a; yE + zF =b
解くと:xE + w(1-E)= a; yE + z(1-E) =b
E = 単離された物質の収率 = (a-w)/(x-w) = (b-z)/(y-z)
【0076】
実施例2
本実施例は、エタノールおよび2-プロパノール中でCPTAをCAF D塩基を用いて分割した結果を示す。
【0077】
下の表3にエタノールおよび2-プロパノールの結果を要約する。この評価のためにスラリーを冷却プロフィール中の様々なポイントでサンプリングし、固体と溶液の両相のエナンチオマー組成を測定した。この情報から、固相の%eeおよび期待される重量パーセント収率(100%eeで最大50%収率)が異性体比率から計算され、決定された。表3には(-)-CPTAの収率が含まれ、それは重量パーセント収率および固相の(-)-CPTA含量から導出される(100%eeで最大100%の収率)。
【0078】
この特定の研究では、エタノール中における最良結果は、CPTA 1モル当たりCAF D塩基1モルを用いた。(-)-CPTA CAF D塩基塩のおよそ72%の収率を、固相中の(-)-CPTA塩の87.6%eeと共に、両相のキラル組成から計算した。2-プロパノール中で同様の濃度でCAF D塩基1モル当量を使用すると、より低い分割度となった。CPTA 1モル当たりCAF D塩基を0.55モル用いた時に、より高いエナンチオマー濃縮を達成した。これらの条件下で、(-)-CPTA CAF D塩基塩のおよそ76〜79%の収率を、固相中の(-)-CPTAの87〜90%eeと共に、相組成から計算した。物理的な損失を考慮に入れていない計算された重量パーセント収率は41〜42%だったが、実際に秤量した単離収率は37〜39%であった。
【0079】
(表3)CAF D塩基を用いたCPTAの分割
【0080】
2-プロパノールからのCPTA CAF D塩基塩の再結晶化により、光学純度が、およそ87%eeから98%eeに増加し、87%の質量回収率、または供給した(-)-CPTA含量に基づくと93%の回収率であった(表4)。
【0081】
(表4)(-)-CPTA CAF D塩基の2-プロパノールからの再結晶化
【0082】
全体として、ラセミ体CPTAから、最大50%の内のおよそ35%の収率で、およそ98%eeの光学純度を有する(-)-CPTA CAF D塩基塩を得た。
【0083】
光学的に濃縮されたエナンチオマーの結晶化によって、しばしばキラル純度が増加した。分割剤の除去に続いて、メチルシクロヘキサノンから(-)-CPTAを結晶化させることも、ある程度光学純度を増加させることになる。ある実験では、(+)-CPTAの結晶化により光学純度が99.1%eeから100%eeに増加した;母液は95%eeであった。
【0084】
実施例3
本実施例は、2-プロパノール中における、CPTAの(+)-および(-)-異性体のCAF D塩基塩の溶解度プロフィールを示す。
【0085】
CAF D塩基を用いたCPTA分割の最適化を補助するために、2-プロパノール中における両方のジアステレオマー塩の溶解度プロフィールを測定した。結果を図1に示す。(+)-CPTA CAF D塩基塩はシンコニジンで分割した(+)-CPTAを用いて調製した。図1に示すように、所望の(-)-CPTAジアステレオマーは(+)-CPTA形よりおよそ3倍溶解度が低い。図に含まれる溶解度を記述する式を、最小二乗法により計算した(R2>0.99)。(-)-CPTA塩に対する82℃のデータ点は方程式の決定に含まれていなかったが、計算された溶解度によく一致している。
【0086】
望ましくないCPTAエナンチオマーのラセミ化物を工程に戻し入れてリサイクルすることができた。このようにエナンチオマー濃縮されたCPTAの望ましくない異性体を1Nの水酸化ナトリウム水溶液中で還流させながら加熱すると、1時間未満でそれがラセミ化されることを見出した。他のどのような副産物も、単離したCPTAのHPLC解析によって検出されなかった。
【0087】
実施例4
本実施例は、(+)-CPTAを得る方法を示す。
【0088】
オーバーヘッドスターラーを備えた2L丸底フラスコに、33.0 gの粗(+)-CPTA-シンコニジン塩、610 mlのエタノールおよび125 mlのメタノールを入れた。スラリーを加熱還流して溶液とし、次に冷却した。非常に厚いスラリーが42℃で形成された。スラリーを68℃に加熱して軽いスラリーとし、次に周囲温度まで放冷した。その混合物を26℃でろ過し、150 mlのエタノールですすぎ、40℃真空下で乾燥させた後、23.48 gの(+)-CPTA-シンコニジン塩を得た。600 mlのエタノールおよび120 mlのメタノールで再結晶手順を繰り返し、18.23 gの(+)-CPTA-シンコニジン塩(2回の結晶化から55%回収率)を得た。分離度が良くないために低レベルの評価は不可能であったけれども(ハロフェナートのキラル解析条件をこの時にCPTAの解析にも用いた)、キラルクロマトグラフィーによっては、(-)-CPTAは検出されなかった。
【0089】
3.61 gの精製した塩試料を水50 mlおよびトルエン50 mlと混合し、2.9 gの硫酸を加えた。有機相を30 mlの水で洗い、次に蒸発させて残留物を得た。残留物を20 mlのシクロヘキサンから結晶化させて、1.22 gの(+)-CPTAを得た。あるいは、6.3 gの(+)-CPTA-シンコニジン塩(10.2 mmol)を、56 gのジエチルエーテルおよび29gの水と混合し、硫酸の滴加によりpH1.9に酸性化した。有機相を25 mlの水で洗い、乾燥させ(硫酸マグネシウム)、ろ過して蒸発させ、残留物とした。残留物を22 mlのメチルシクロヘキサンと周囲温度で攪拌し、スラリーを形成させた。スラリーを40℃に温め、次に氷浴で冷却し、固体をろ過によって分離して、40℃真空下で乾燥させた後に、2.62 gの(+)-CPTA(7.92 mmol、収率78%)を得た。
【0090】
実施例5
本実施例は、(+)-CPTAから(+)-ハロフェナートを合成する方法を示す。
【0091】
25 ml丸底フラスコに0.91gの(+)-CPTAおよび2.6 gのチオニルクロリドを入れ、混合物を加熱還流し、溶液とした。メタノールによって試料をクエンチし、生成物をHPLCによって解析して、酸塩化物への変換をモニターした。酸塩化物溶液に、4.8 gのジエチルエーテルを加え、そしてこの溶液を、氷浴で冷却した0.37 gのピリジンを含む12 mlのN,N-ジメチルホルムアミド(DMF)中の2.0 gのN-アセチルエタノールアミンに加えた。その結果の溶液を、25 mlの水のおよび30 mlのジエチルエーテルに加えた。有機相を分離し、25 mlの水で洗浄し、乾燥させ(MgS04)、ろ過し、溶媒を除去した後に0.92 gの油状物を得た。HPLC解析は、45面積%のハロフェナートおよび50面積%のCPTAを示した。キラルHPLC解析によって、ハロフェナートは(+)-エナンチオマーが99.78%eeであることが示された。
【0092】
実施例6
本実施例は、ラセミ体CPTAを調製する方法を示す。
【0093】
オーバーヘッドスターラーを備えた2L丸底フラスコに、102.7 gのハロフェナート、500 mlの水、および16.3 gの2-プロパノールを入れた。スラリーを撹拌し、32.3 gの45%水酸化カリウム水溶液を加えた。1時間の加熱還流後、溶液を周囲温度に冷却し、380 mlのヘキサンを加えた。24.57 gの37%塩酸で、pHを12.5から2に調節した。三相混合物を60℃に加熱して二相とした。下部の水相を取り出し、50 mlのヘキサンで抽出した。合わせた有機層を加熱して大気圧で蒸留し、100 mlの濁った蒸留物を除いた。溶液を30℃に冷却しCPTAの種を入れた。スラリーが生じた。スラリーを氷浴で冷却し、固体をろ過により単離して64.0 g(収率78.4%)のラセミ体CPTA、すなわち(4-クロロフェニル)(3-トリフルオロ-メチルフェノキシ)酢酸を得た。
【0094】
実施例7
本実施例は、様々なキラル塩基を用いて、エタノール中でキラル分割スクリーニングを行った代表的結果を示す。
【0095】
1.16 g(3.51 mmol)のCPTAの試料を6.98 gのエタノールに溶解し、溶液(0.431 mmol/g)を得た。ガラスバイアルに個別に、表5に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTAのエタノール溶液を加えた。ある場合には、CPTA溶液を加える前に少量のエタノールを加えて塩基を湿らせた。バイアルを終夜周囲温度に置いた。バイアル7Gおよび7Iが沈殿を生じた。各上清液の試料を取り出し、キラルHPLC解析により解析した。固体をろ過して分離し、解析もした。結果のいくつかを表2に示す(上記実施例1を参照)。残りのバイアルを5℃の冷蔵庫に置いた。1日後に7Eが沈殿を生じた。前述のように試料を解析した。残りのバイアルに、50μlの水を加え、周囲温度に3日間保持した後冷蔵庫に置いた。1か月後にさらに沈殿を生じていたものは無かった。
【0096】
(表5)エタノール中における塩基スクリーニング
【0097】
実施例8
本実施例は、様々なキラル塩基を用いて、アセトン中でキラル分割スクリーニングを行った代表的結果を示す。
【0098】
1.67gのCPTAの試料を7.57 gのHPLC級アセトンに溶解し、溶液とした。ガラスバイアルに個別に、表6に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTA溶液を加えた。ある場合には、少量のアセトンを加え、混合物を約40℃に温めて溶液とした。さらに、バイアル16Mに0.300 mlの1Nの水酸化ナトリウムを加えた。バイアルを終夜周囲温度に置いた。バイアル16Dが沈殿を形成し、上述のように解析した。表2中に結果のいくつかがまとめられている(実施例1を参照)。残りのバイアルを冷蔵庫に置いた。バイアル16Nが沈殿を形成し、解析した。バイアル16Gがわずかに沈殿を形成した。1週間後に、バイアル16Lが沈殿を生じたことが分かった。前に示したように試料を解析した。さらなる沈殿は認められなかった。
【0099】
(表6)アセトン中の塩基スクリーニング
【0100】
実施例9
本実施例は、様々なキラル塩基を用いて、メタノール中でキラル分割スクリーニングを行った代表的結果を示す。
【0101】
2.00 gのCPTAの試料を8.03 gのHPLC級メタノールに溶解し、溶液とした。ガラスバイアルに、個別に表7に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTA溶液を加えた。さらに、バイアル27Jに0.300 mlの1N水酸化ナトリウムを加えた。バイアルを、終夜周囲温度に置いた。バイアル27Bは固形化し、300μlのメタノールをさらに加えた後、上述のように試料を解析した。残りのバイアルを冷蔵庫に置いた。1か月後にさらなる沈殿は認められなかった。
【0102】
(表7)メタノール中の塩基スクリーニング
【0103】
実施例10
本実施例は、CPTAをキニーネによって分割した結果を示す。
【0104】
150 mlのジャケット付底部排液管フラスコに、2.70 g(8.17 mmol)のCPTA、2.65 g(8.17 mmol)のキニーネおよび50 mlの2-プロパノールを入れた。混合物を70℃に加熱して溶液とし、その後、0.2℃/分の速度で30℃に冷却し、2時間保持してスラリーを得た。試料のキラルHPLC解析により、固相中で(+)および(-)-CPTAがそれぞれ42.88および56.47面積%であり、また溶液中で(+)および(-)-CPTAがそれぞれ61.54および34.19面積%であることが示された。60℃にスラリーを加熱し、次に0.04℃/分の速度で30℃に冷却し、終夜保持してスラリーを得た。キラルHPLC解析によって、固相中で(+)および(-)-CPTAがそれぞれ29.94および44.19面積%であり、また溶液中で(+)および(-)-CPTAがそれぞれ77.54および20.88面積%であることが示された。スラリーを50 mlの2-プロパノールで希釈し、57℃に加熱して溶液とし、次に0.2℃/分の速度で30℃に冷却した。スラリーが1時間後に30℃で生じ始めた。その混合物を周囲温度で2日間攪拌し、次に固体をろ過により分離し、2-プロパノールですすぎ、真空下で乾燥させた後、2.89 g(質量収率54%)のCPTAキニーネ塩を得た。キラルHPLC解析によって、固相中で(+)および(-)-CPTAがそれぞれ42.25および57.75面積%であり、また母液中で(+)および(-)-CPTAがそれぞれ56.56および39.20面積%であることが明らかになった。表2に結果が含まれている(実施例1を参照)。
【0105】
実施例11
本実施例は、CPTAをCAF D塩基によって分割した結果を示す。
【0106】
150 mlの底部排液管フラスコに、19.54 gのCPTA、6.82 gのCAF D塩基(すなわち、D-threo-(-)-2-アミノ-1-(ニトロフェニル)-1,3-プロパンジオール)および80.2 gの2-プロパノールを入れた。混合物を70℃に加熱して溶液とし、その後、0.1℃/分の速度でジャケット温度5℃まで冷却した。混合物が62℃で混濁した。6℃に9時間保持した後に、固体をろ過により単離し、5 mlの2-プロパノールですすぎ、真空下40℃で乾燥させて、(-)-CPTA CAF D塩基塩12.03 g(収率37.4重量%)を得た。固体のキラルHPLC解析により、6.34面積%の(+)-CPTAおよび93.46面積%の(-)-CPTAが明らかになり;母液は、81.41面積%の(+)-CPTAおよび17.76面積%の(-)-CPTAを含んでいた。
【0107】
実施例12
本実施例は、(-)-CPTA CAF D塩基塩の再結晶の結果を示す。
【0108】
150 mlの底部排液管フラスコに、8.00 gの(-)-CPTA CAF D塩基塩(上記実施例11による)および54.2 gの2-プロパノールを入れた。混合物を加熱還流して溶液とし、その後、0.1℃/分の速度でジャケット温度20℃まで冷却し、内部温度を22℃で6時間保持した。固体をろ過して単離し、2-プロパノールですすぎ、真空下40℃で乾燥させて6.93 g(回収率86.6重量%)の(-)-CPTA CAF D塩基塩(融点184〜185℃)を得た。固体は0.995面積%の(+)-CPTAおよび99.01面積%の(-)-CPTAを含み;母液は44.53面積%の(+)-CPTAおよび54.47面積%の(-)-CPTAを含んでいた。反応容器をアセトンで洗い出した。アセトンを蒸発させ、0.27 g(3.4重量%)の残留物を得た。
【0109】
実施例13
本実施例は、(+)-CPTA CAF D塩基塩を調製する方法を示す。
【0110】
1 Lのフラスコに10.94 g(17.5 mmol)の(+)-CPTAシンコニジン塩、200 mlの水および100 mlのメチレンクロリドを入れた。1.8 gの硫酸を加えてpHを1.9に調節した。有機層を100 mlずつの希硫酸水溶液で3回洗浄し、乾燥(硫酸マグネシウム)させ、ろ過し、蒸発させて5.79 gの残留物を得た。残留物を22.2 gの2-プロパノールに溶解し、3.5 gのCAF D塩基を加えた。得られたスラリーを加熱還流して溶液とし、次に周囲温度に冷却し、スラリーを3時間撹拌した。氷浴で冷却した後に、固体を真空ろ過により分離し、5 mlの2-プロパノールですすぎ、真空下40℃で乾燥させて、7.39 g(収率80%)の(+)-CPTA CAF D塩基塩(融点172〜173℃)を得た。
【0111】
実施例14
本実施例は、2-プロパノール中のジアステレオマーCPTA-CAF D塩基塩の溶解度を示す。
【0112】
(-)-CPTA CAF D塩基および(+)-CPTA CAF D塩基(>98%ee)の試料を、表8中に示す量の2-プロパノールに加え、超音波浴を用いて混合した。全試料はスラリーのままであった。表に記した温度でスラリーを終夜保持し、次に上清液の試料を取り出し、定量的HPLC解析によって解析してCPTA濃度を決定した。結果を表および図1に示す。さらに、8.00 gの(-)-CPTA CAF D塩基塩を82℃で溶液(14.7重量%)とするために54.2 gの2-プロパノールが必要であった。このデータ点は図1には含めたが、溶解度方程式には含めなかった。
【0113】
(表8)2-プロパノール中の溶解度
【0114】
実施例15
本実施例は、エナンチオマー濃縮されたCPTAをラセミ化する方法を示す。
【0115】
50 ml丸底フラスコに、0.31 gの(-)-CPTA(68.7%ee)および9.4 gの1N水酸化ナトリウムを入れた。溶液を1時間加熱還流し、その後周囲温度に冷却し1 gの37%塩酸で酸性化した。CPTAをメチレンクロリドへ抽出し、溶媒を蒸発させ、0.46gの油状物を得た。HPLC解析によって、99.4面積%のCPTAが見出され、またキラルHPLC解析によって、CPTAエナンチオマーの50/50混合物が見出された。
【0116】
実施例16
本実施例は、様々な結晶化条件下において、CAF D塩基を用いてCPTAのラセミ混合物を分割する方法を示す。
【0117】
一般的な結晶化手順は、CPTA、CAF D塩基、および2-プロパノールを室温で加え、約75℃で加熱して溶液とすることであった。溶液を約60℃に冷却し、核形成が起るまで保持した。いくつかのバッチに、(-)-塩(すなわち(-)-CPTAおよびCAF D塩基の塩)を種として入れ、核形成を誘起した。スラリーを約1時間に亘って成長させた後、容器を単離温度に冷却した。図2中の最初の5つの実験では、約0.05〜0.10℃/分の遅い冷却速度を用いて単離温度に到達させた。他の実験では、0.25〜0.40℃/分のより速い冷却速度を用いた。ファイバー光学プローブを結晶化装置に直接挿入してスラリー密度を測定する。
【0118】
加えたCAF D塩基の量および溶質濃度は、最終的なバッチ組成をもたらす重要な変数である。(+)-塩(すなわち、(+)-CPTAおよびCAF D塩基の塩)が様々な時間過飽和状態であり続ける傾向が、いくつかの実験における変動の主な原因であると考えられる。図2中の実験5でこれが例証され、この場合、スラリーを8時間13℃で保持して、高純度結晶(99.7%(-)-塩)が生成された。3時間後に、ファイバー光学プローブの信号の増加が、(+)-塩の可能性が高い核形成を示した。さらに27時間の後、スラリーを単離し、結晶生成物は83.3/16.7%の(-/+)-CPTA比率であった。HPLCによる結晶生成物の解析によって、(-)-CPTAおよび(+)-CPTAの比率が与えられる。溶液中のフリーCPTAは飽和濃度以下であるので、結晶の解析はジアステレオマー塩比率を与える。母液は、溶解した塩およびフリーCPTAの両方を含む。HPLC解析は、各エナンチオマーを合計した量をCPTAとして報告する。同様に、図2の実験6が、スラリーを20時間1℃で保持して、高純度塩(>98%の(-)-CPTA)が生成されたことを示している。17℃に加熱した後に(+)-塩が核形成され、より低品質の生成物を得た[(-/+)-CPTA=81.2/18.8%]。
【0119】
他の試行では、図2の実験2、8および10におけるように、(+)-塩の核形成がより急速に生じた。結晶化は、(+)-塩の飽和温度の近くで、好ましくは飽和温度のすぐ上で単離を行うことができることが望ましい。
【0120】
CPTA 1グラム当りおよびCAF D塩基の0.45当量で、3.9 gの2-プロパノールを加えた場合、室温における単離は、(+)-塩の飽和レベル(または準安定域内)に非常に近いように見える。図2中の実験12は、0.43当量の塩基で開始され、(+)-塩の種を入れた後でさえ、21℃における結晶生成物は純粋なままであった(>99% (-)-塩)。CAF D塩基を加えて0.45当量とした後に、スラリーを14時間保持し、その後(+)-塩の種を入れた後にさらに6時間保持した。結晶生成物を解析すると、98.7%の(-)-CPTA比率であった。全塩基を0.47当量に増加すると、(+)-塩成分が ゆっくり増加して(-/+)-CPTA=92.3/7.7%の結晶生成物が得られた。
【0121】
図2の実験11(CPTA 1グラム当り3.9gの2-プロパノール、0.45当量の塩基)では、14時間後に高純度の(-)-塩(99.1%)が維持されていたが、しかし、さらに塩基を追加して0.48当量とすると、生成物比率が(-/+)-塩=89.2/10.8%となった。図2の実験9(0.45当量の塩基)では、22℃16時間後に99.5%の(-)-塩純度が維持されていた。これらの条件下の3つのバッチから計算された(-)-CPTAの収率は、70.7〜71.6%であった。計算収率は、結晶中および母液中の(-)-CPTAおよび(+)-CPTAの組成を知ることにより、ラセミ体CPTA原料を用いたことによる強制的な物質収支から導出される。
【0122】
このようにCPTA 1グラム当り約0.45当量のCAF D塩基および約4 gの2-プロパノールを加えることが、高純度(-)-塩(>98.5%)生成物をもたらし、それ以上の再結晶をしないで使用することができる。
【0123】
実施例17
本実施例は、CPTA塩の分割/結晶化について記述するためのモデルを提供する。
【0124】
フリーのCPTAの濃度は、加える塩基の量および溶媒の量に依存する。例えば、4.0グラムの2-プロパノールおよび0.50当量のCAF D塩基を加えることによるCPTAの分割においては、11%のフリーCPTAを含む2-プロパノール中において塩形成が行われる結果になる。この溶媒は、(-)-塩および(+)-塩の両方に対してより大きな溶解度を有し、溶解度を図3に示すように測定した。図3はさらに純粋な2-プロパノール中の溶解度データを含んでおり、それは2-プロパノール1グラム当りの成分グラム数で表現されている。図3が示すように、それぞれの塩の曲線は類似の形である。
【0125】
CPTA、CAF D塩基および2-プロパノールの配合の他の組合せによっても、図4に示すように、2-プロパノール中11.0%のフリーCPTAとなる系に到達できる。図4が示すように、図2の様々な実験の配合では、通常正確にこの直線に乗ることはなかった。しかし(-)-塩および(+)-塩の溶解度を以下のようにして推定することができる:「11.0%のフリーCPTA」直線より上の点となる配合は、より稀薄(すなわち2-プロパノール中で<11%のフリーCPTA)となり、「11.0%」直線より低い溶解度を示す。反対に、「11.0%」直線より下の点は、>11.0%のフリーCPTAを含む溶媒となり、塩の溶解度は図3で決定された溶解度より大きい。成分の溶解度を推定するために、定乗数kを用いた。(-)-塩および(+)-塩に対する修正溶解度方程式はしたがって、S(-) = 0.01421ke0.02613T および S(+)= 0.02868ke0.02771Tである。
【0126】
kを調節して(-)-塩および(+)-塩の溶解度の良い推定を行っても、他の未知数が分割剤塩基の添加によって形成される(-)-塩と(+)-塩の比率であるので、結晶化を記述することができない。図5に、より詳細な実験の1つを示す(図2も参照のこと)。この実験では、0.75当量の塩基を用い、21.5℃で試料を採取したときに、66.4/33.6%の(-/+)-塩比率の生成物が得られた。スラリーを加熱し、試料を採取し続けることによって、溶媒中の(-)-塩および(+)-塩の両方に対する飽和直線を追跡することができる。
【0127】
溶解度モデルを実際のデータに一致させるために、回帰技法を用い、測定値と一致した解答(すなわち結晶組成、母液組成および結晶の収率)を得るように、溶解度因子kならびに(-)-塩および(+)-塩の供給比率を操作した。k=0.68および0.75当量の塩の供給比率を58.1%(-)-塩/41.9%(+)-塩(すなわち0.436当量の(-)-塩および0.314当量の(+)-塩がCAF D塩基の添加によって形成された)に選ぶことによって、よい一致を得た。図6に比較を示す。溶解度モデルによって、単離のための完全な物質収支:結晶中の(-)-塩および(+)-塩の量、母液中の(-)-塩および(+)-塩の量、およびさらに母液中の(-)-フリーCPTAおよび(+)-フリーCPTAの量の計算が可能になる。溶解度差を用いた、抽出精密測定(extractive work-up)による母液中の(-/+)-塩および(-/+)-フリーCPTAを定量化するための一方法を下記の実施例19において提供する。
【0128】
溶解度モデルについての回帰技法を種々の量の分割剤を加えた他の実験に対して適用した。溶解度因子k、および材料としての塩の組成(すなわち、塩基の添加で形成された(-)塩および(+)塩の比率)の組合せを用いることにより、モデルは実験結果に適合する一意的な解に到達した。これらから、図7のグラフを構成した。この結果は、より多くの分割剤(外挿した最小点0.34当量より多く)を加えると、(+)-塩の形成量が増加することを示す。いくつかの実施態様では、0.34当量未満を加えると、CAF D塩基は実質的に(-)-CPTAのみと整合し、ほとんど排他的に(-)-塩を形成すると、いかなる理論にもとらわれずに信じられている。さらに、図7の曲線を用いて、(-)-CPTAおよび(+)-CPTA(遊離酸)の量を計算することができる。0.35〜0.75当量の間の塩基を添加した場合の、{(-)-CPTA/総CPTA遊離酸}の%比率は約25%(23.3〜27.1%)である。形成される(-/+)-塩の比率の「選択性」は、このようにして(溶液中に)残存するフリーの(-)-CPTAの量に依存し、それは約(-)-CPTA/(+)-CPTA=1/3である終点に至る。約0.34当量の塩基を添加することによって、一旦(-)-CPTA濃度が減少して(-/+)-CPTA比率が1/3となると、塩基の継続的な追加は(溶液中においてフリーの(-/+)-CPTAを一定の1/3の比率に保持するために)(-/+)-塩を1/3の比率で形成すると信じられている。
【0129】
実施例18
本実施例は、CPTAのラセミ混合物の分割を示す。
【0130】
200 ml容器に17.0 gのCPTA(51.4 mmol)、4.91gのCAF D塩基(23.1 mmol、0.450当量)および85 mlの2-プロパノールを入れた。その混合物を78℃で加熱して溶液とし、次に、0.5℃/分で54℃まで冷却した。約1/2時間後に、その溶液に(-)-塩の種を入れ、核形成を引き起した。54℃に約1・1/2時間保持した後、スラリーを0.25℃/分で22℃に冷却した。14時間22℃で保持した後に、小量の試料(約5 ml)を取り出して、15 mlの中型フリット化漏斗で分離した。母液の重量を計り保存し、固体を2 mlの2-プロパノールで洗浄した。洗浄液の重量を計って保存し、吸引を続けて結晶を乾燥させた。標準HPLCシステムでの解析によって、(-)-CPTAおよび(+)-CPTAの各処理系統における重量%の計算が可能になった。この試料についての物質収支(結晶、母液および洗浄液中のCPTAの総量が0.85 gとなるべき)から、総CPTAから単離した結晶生成物の収率31.9%が得られた。結晶の純度は重量で、99.1/0.9% = (-/+)-CPTA比率であった。図8の四角の囲み中に分析結果および物質収支の結果を示す。円内に、原材料/母液/結晶の組成に基づいて計算された収率(CPTAからの)を示す。図8中の略語は以下のとおりである: R.A.= 分割試薬、xまたはxtal = 結晶、ML = 母液、Yld = 収率。
【0131】
容器に(+)-塩を含む結晶の種を数回入れ、約2時間後に0.31 gのCAF D塩基(1.46 mmol、約0.03当量)を加えた。容器から試料を2回採取した後、60 mlの中型フリット化漏斗上に最終的に単離した(図8を参照)。母液は、透明の淡イエローゴールドで、59.1gであった。固体を19.2 gの2-プロパノールで洗浄し、18.8 gの洗浄溶液を回収した。洗浄した固体(10.07 g)をさらに1時間ろうと上で吸引して乾燥し、8.36 g(15.4 mmolの塩)を得た。最終単離からすべての系統を解析して、13.45g(40.67 mmol)のCPTAが計算された。単離した収率(-)-CPTA=33.8%(CPTAから)に対して、最終の結晶生成物の比率は、(-/+)-CPTA=89.2/10.8%であった。原材料、母液および結晶の組成に基づいて計算された(-)-CPTAの収率は、35.0%であった。
【0132】
実施例19
本実施例は、母液中の(-/+)-塩および(-/+)-CPTAを定量化するための抽出精密測定法を示す。
【0133】
(-/+)-塩の80/20混合物は、メチレンクロリドに約0.016%がようやく可溶であり、一方、ラセミ体CPTAは相当程度溶解し、少し3.4%に足らなかった。図2の実験4において55.3℃で分離した最終母液(図2および図5を参照)0.1286gを蒸発させて0.0242 gのガラス状残留物にすることにより解析した。残留物を5 mlのメチレンクロリドに溶解し、(-/+)-塩 = 80/20の種を入れて、終夜放置した。上清液の大部分を取り出し、3 mlのメチレンクロリドを加え、また液体の大部分を取り出して最初の抽出液と合わせた。メチレンクロリド抽出物を蒸発させ、ガラス状の固体0.0074 g得て、次にHPLCにより解析した。残りの濃いスラリーを蒸発させて0.0162 gとし、HPLCにより解析した。抽出精密測定法の結果は、図9に示すように、概して溶解度モデルによって予測される組成に類似している。
【0134】
実施例20
本実施例は、(-)-および(+)-CPTA・CAF D塩基塩の、CPTAを含むアルコール中における溶解度を示す。
【0135】
2.40 gのラセミ体CPTAを19.42 gの2-プロパノール(Fisher、HPLC級)に、または4.90 gのラセミ体CPTAを31.4 gのエタノールに溶解することにより「溶媒」を調製した。溶液中のCPTAの濃度はそれぞれ11.0%および13.5%であった。(-)-CPTA・CAF D塩基塩(すなわち(-)-塩)または(+)-CPTA・CAF D塩基塩(すなわち(+)-塩)の溶解度を重量法により決定した。与えられた温度で、上清液の一部を飽和溶液から重量が分かっているバイアルへ取り出した。溶液重量を測定し、窒素を吹き込んで揮発性溶媒を蒸発させた。固体が一定重量になるまで、約50℃/1 mm Hgの真空オーブン中でさらに乾燥させた。再度バイアルの重量を計り、揮発性溶媒の消失および残存固体の重量を測定した。これにより、「溶媒」から溶けていたCPTAの量を計算することができた。CPTAから全固形物の重量を引くことによって、溶媒中の可溶塩の重量が得られた。図10Aおよび10B中にデータを示す。
【0136】
実施例21
本実施例は、エナンチオマー濃縮された(-)-ハロフェナートを調製する方法を示す。
【0137】
CPTAを5工程で、上に述べたように、中間の単離なしで、ヘプタンからの結晶化により約85%の収率で調製した。分割によって、>98%光学純度の(-)-CPTAジアステレオマー塩が平均32%の収率(最大50%)で得られた。分割剤を除去した後に、(-)-CPTAをチオニルクロリドおよびN-アセチルエタノールアミンを用いてエステル化し、約55%の収率で(-)-ハロフェナートを得た。水酸化ナトリウム水溶液で母液残留物を加水分解して、(-)-CPTAを最終生産物母液から回収することができ、工程の再循環に戻すことができる。分割剤を、水からpH調整により約90%回収率で単離した。水酸化ナトリウム水溶液を用いた(+)-CPTAの回収とラセミ化により、約90%の回収率が得られた。全体として、4-クロロフェニル酢酸からの初回通過の収率は15〜17%だった。8工程の全工程で、3種の有機溶媒および3回の固体単離工程を用いた。
【0138】
実施例22
本実施例は、CPTAの調製方法を示す。
【0139】
CPTAの合成ルートの略図を上に示す。酸塩化物1を1,2-ジクロロエタン中で臭素化して2を得、続いて2-プロパノールを加えて、イソプロピルエステル3を得た。α,α,α-トリフルオロ-m-クレゾールによる置換反応を、2-プロパノール中で水酸化カリウムを用いて実施した。水によるクエンチと、洗浄および1,2-ジクロロエタンの除去に続いて、液体3をα,α,α-トリフルオロ-m-クレゾールおよび水酸化カリウムの2-プロパノール溶液に加え、4を得た。2-プロパノール溶媒を除去し、水酸化ナトリウム水溶液と加熱することにより加水分解が完了し、CPTAを得た。
【0140】
CPTAのナトリウム塩を、単に反応液を冷やすことにより固体として単離することができる。しかし、カルボン酸を単離することによってより良好な単離収率を得た。単離のために、塩基性のCPTA反応混合水溶液を塩酸で酸性化し、CPTAを1,2-ジクロロエタン中へ抽出した。分離した有機相の溶媒を1,2-ジクロロエタンからヘプタンへ交換することによって、CPTAが白色固体としておよそ85%の収率で4-クロロフェニル酢酸から得られた。
【0141】
実施例23
本実施例は、CPTAの1,2-ジクロロエタンとヘプタン中の溶解度を示す。
【0142】
ラセミ体CPTAの1,2-ジクロロエタンおよびヘプタン中の溶解度を、図11および12にそれぞれ示す。図には、データに最小二乗適合させた方程式が含まれる。
【0143】
図11の溶解度プロフィールに基づいて、1,2-ジクロロエタン中のおよそ25重量%CPTAの濃度を温度およそ35℃で、CPTAの抽出条件として選んだ。
【0144】
ヘプタンからのCPTA結晶化は発熱性であった。およそ170 gのCPTAの500 mlヘプタン溶液に46℃で種を入れると、結晶化が進行すると共に、54℃へ温度が上昇した。結晶化は、HPLC解析によって決定されるCPTA純度を、93〜95面積%から>99面積%まで増加させた。結晶化母液のHPLC分析(それは15面積%のCPTAを含んでいた)によって、母液への収率損失は3%より少ないことがわかった。結晶化によって純度が改善するとともに、単離収率は高く、母液への損失は小さかった。
【0145】
実施例24
本実施例は、様々な結晶化条件下のCPTA分割の収率を示す。
【0146】
様々な結晶化条件下におけるCAF D塩基を用いたCPTA分割の結果を図13に示す。0回、1回または2回の再結晶後に得られた各標品の最終のキラル純度を、太字で示す。CAF D塩基のモル比を0.5〜0.56まで変えた。結晶化および再結晶化のために表に示した2-プロパノール溶媒の量は共にラセミ体CPTAの最初の添加量に基づいている。単離した固体および母液の両方のキラルHPLCの結果を100%に標準化してある。計算収率および通算収率は、単離した固体および母液中の(+)-エナンチオマー型および(-)-エナンチオマー型の比率から計算する。最後の行の実際のパーセント収率は、計量した乾燥した物質のものであり、50%の最大収率に基づいている。
【0147】
>98%の光学純度のジアステレオマー塩の通算収率は、28〜35%の範囲であり、平均32%であった。1つの場合では、分割剤の最低の比率を用い、再結晶なしでこれが得られた(図13の実験2)。最初に単離した固体のキラル純度は73〜98%の範囲であった。1回の再結晶が一般に所望の光学純度を得るのに十分だった。高い通算収率が、母液が(-)-CPTAの(+)-CPTAに対する比率が20/80に達した時に得られた。
【0148】
図14は、(-)-CPTAの収率が減少する順に並べた分割結晶化のための冷却プロフィールを示す。図14の実験番号は図13の実験番号に対応している。(-)-CPTAの単離収率は、図13の計算収率および単離した物質中の(-)-CPTAのパーセントを用いて決定した。一般に、より低温のより長い保持時間が収率の増加をもたらした。
【0149】
0.45モル当量のCAF D塩基の使用により一貫して、再結晶の必要なしに、>98%の光学純度の物質の35〜37%の収率が得られた。
【0150】
実施例25
本実施例は、(-)-CPTAをCAF D塩基から分離する方法を示す。
【0151】
(-)-CPTAをCAF D塩基から分離するために、ジアステレオマー塩を1,2-ジクロロエタンと混合し、塩酸水溶液を加えて水相のpHを約2未満にした。CAF D塩基の塩酸塩を含む水相を分離した。有機相の水洗浄の後に、1,2-ジクロロエタンの大部分を蒸留によって除去し、残留水を取り除いた。完全な溶剤除去によって油状物が得られた。
【0152】
実施例26
本実施例は、有意なラセミ化なしに、(-)-CPTAをエステル化する方法を示す。
【0153】
(-)-CPTAを1,2-ジクロロエタン中で還流してチオニルクロリドと反応させ、対応する酸塩化物を生成した。反応の進行をHPLC解析によってモニターできる。少量の蒸留物を除去して、過剰のチオニルクロリドを除いた。混合物を冷却し、大過剰の真空蒸留したN-アセチルエタノールアミンを加えた。周囲温度で攪拌し、(-)-ハロフェナートを得た。
【0154】
反応混合物のエステル化を、反応混合物を炭酸カリウム水溶液に加えることによりクエンチした。6:1のヘプタン:2-プロパノールへ溶媒を交換し、そこから結晶化することによって(-)-ハロフェナートを単離した。結果を図15に要約する。
【0155】
第1の収穫の単離収率は、47〜59%の範囲であり、平均55%であった。この単離収率は、この工程の75〜80%の反応収率を表わす。第2の収穫はより高い通算収率をもたらした;しかしながら、第2の収穫は製品品質がより劣っていた。
【0156】
単離したハロフェナートおよび母液中のハロフェナートおよびCPTAとして見出された、加えたCPTAのモルアカウンタビリティーは90〜99%の範囲であった。
【0157】
実施例27
本実施例は、(+)-CPTAを回収して再循環させる方法を示す。
【0158】
塩基水溶液中でCPTAを加熱するとラセミ化が起った。実施例25の分割工程で残ったCPTAはおよそ47%eeの(+)-エナンチオマーであり、またそれはさらに残りのCAF D塩基を含んでいる。
【0159】
(+)-CPTAを回収しラセミ化するために、2-プロパノール溶媒を除去し、1,2-ジクロロエタンと交換した。pH約2未満の水で洗浄して、後に続く回収のためにCAF-D塩基を除去した。水酸化ナトリウム水溶液を加え、水溶液を加熱還流した。1,2-ジクロロエタンを、塩基溶液の添加に先立つ蒸留、または塩基溶液の添加に続く相分離によって除去した。1.4モル当量の水酸化ナトリウムと水溶液を4時間加熱した後に、ヘプタンから89%の収率のラセミ体CPTAを単離した。結晶化した中間物としてCPTAを単離することにより、分割工程に対してより一貫した品質の供給が可能になった。
【0160】
酸型として測定し表示した水中のラセミ体CPTAナトリウム塩の溶解度を図16に示す。単離したナトリウム塩を水に加えると、約9.5のpHとなり、上の溶解度曲線で示される溶解度プロフィールが得られた。少量の水酸化ナトリウムの追加によって約12.6のpHとすると、下部の曲線によって示されるように水への溶解度が減少した。
【0161】
実施例28
本実施例は、(+)-ハロフェナートからCPTAを生成する方法を示す。
【0162】
1〜3モル当量の水酸化ナトリウムを水中の約10重量%の87%ee(+)-ハロフェナートへ加えて50〜60℃に加熱すると、実質的に完全なCPTAの加水分解が起こった。部分的ラセミ化が生じ、およそ70%eeの(+)-CPTAを生じた(図17の、時間=0)。溶液を加熱還流し、エナンチオマー比率を時間を追ってモニターした。3モル当量の塩基により、ほとんど完全なラセミ化が(キラルHPLC分析法により<3%ee)2時間未満の還流で生じた。ラセミ化の間にpHが12.8から12.6まで低下した。2モル当量(pH12.6から11.6へ)ではわずかに長い反応時間が必要であった。1モル当量では、ラセミ化はおよそ60〜70%eeで止まり、最終pHは9.4となった。
【0163】
0.5モル当量の水酸化ナトリウムを用いると、およそ40%のハロフェナートが60℃で2時間後に加水分解されずに残り;終夜加熱還流すると、およそ1%のハロフェナートが残り、最終pHは4.8であった。これは有意にラセミ化を最小化しなかった。生成されたCPTAの量は72.6%eeの(+)-エナンチオマーであった。
【0164】
実施例29
本実施例は、(-)-ハロフェナート結晶化母液から(-)-CPTAを回収する方法を示す。
【0165】
以前に述べ、また図15に示したように、(-)-ハロフェナート結晶化母液は大量の(-)-ハロフェナートおよび(-)-CPTAを含む。(-)-ハロフェナートの加水分解により、さらに(-)-CPTAを分割工程の材料として生成することができる。
【0166】
(-)-ハロフェナート結晶化母液(88.3%eeの(-)-ハロフェナート)の50℃および最終pH12.7の加水分解により、急速に65.8%eeの(-)-CPTAが得られた。(-)-CPTAを、CAF D塩基を2-プロパノール溶液へ加えることによりCAF D塩基ジアステレオマー塩(96.4%ee)として回収した。最初に加えたジアステレオマー塩の量から55 mol%を(-)-ハロフェナートとして得、28%を(-)-CPTA/CAF D塩基塩として回収し、また14 mol%が母液中にCPTAとして残った。
【0167】
実施例30
本実施例は、CAF D塩基を回収する方法を示す。
【0168】
CAF D塩基は、ジアステレオマー塩から(-)-CPTAを分離した酸性相中に、および分割母液からのCPTA回収の酸性洗浄工程から得られた酸性相中に見出される。水酸化ナトリウム水溶液で約12を越えるpHへ塩基性化することにより、容易にろ過される形で良好に回収できる沈殿となった。図18に結果を示す。ジアステレオマー塩からの回収率は一般に90%を越えたが;分割母液からの回収率はより低かった。水溶液中の濃度は約5〜20%の範囲であった。
【0169】
CAF D塩基のエナンチオマー純度を、DSCによる融点の注意深い解析により決定することができる(D. Pitre, M. Nebuloni, and V. Ferri; Arch. Pharm. (Weinheim) 324, 525 (1991))。(+)-と(-)-形の混晶、例えばラセミ化合物は、純粋なエナンチオマーより20℃以上低い温度で溶融するので、融点がエナンチオマー純度を評価するための敏感な方法であることが分かった。しかし、誘導体のクロマトグラフ分離による2つの試料のエナンチオマー純度を測定しても、キラル純度の低下は示されなかった。回収されたCAF D塩基のエナンチオマー純度は、HPLC分析法の検出限界の近くで、原料物質と判別することが不可能であった。
【0170】
実施例31
本実施例は、ラセミ体CPTAを調製する別の方法を示す。
【0171】
加熱マントル中にありオーバーヘッドスターラーおよび冷却器を備えたが500 ml丸底フラスコに、73.28 g(0.430 mol)の4-クロロフェニル酢酸、70 mlの1,2-ジクロロエタンおよび41 ml(0.56 mol)のチオニルクロリドを入れた。混合物を50〜55℃で19時間加熱した。反応混合物をHPLC解析により解析した。酸塩化物の溶液に29 ml(0.57 mol)の臭素を加え、溶液を70〜75℃で20時間加熱した。生じたα-ブロモ生成物を氷浴で冷却し、100 ml(1.31 mol)の2-プロパノールを滴下して加えた。達した最高温度は17℃であった。4℃に冷却した後に、反応混合物を水に加えた。溶液を周囲温度に温め、水層を除去した。有機相を37 mlの水で洗浄した。分離された1,2-ジクロロエタン溶液を蒸発させ、134.1 gの油状物を得た。
【0172】
オーバーヘッドスターラーを備えた1 Lの丸底フラスコに、34.0 g(0.515 mol)の85%水酸化カリウムおよび370 mlの2-プロパノールを入れた。その混合物を41℃に水浴を用いて温め、固体の大部分を溶解させた。その混合物を氷浴で冷却し、73.8 g(0.455 mol)のα,α,α-トリフルオロ-m-クレゾールを滴下して加えた。最高温度は13℃に達した。溶液を5℃に冷却した後、上で得られた134.1 gの油状物を滴下して加えた。材料を18 gの2-プロパノールですすいだ。スラリーを蒸発させて残留物とし、次に250 mlの水および42.8 g(0.535 mol)の50%水酸化ナトリウム水溶液を加えた。混合物を1時間加熱還流した。
【0173】
周囲温度に冷却した後に、混合物を250 mlの1,2-ジクロロエタンで希釈し、71 g(0.72 mol)の37%塩酸の滴下による添加によりpHを0.3まで下げた。相分離の後、溶媒を1,2-ジクロロエタン相から除去し、202.2 gの残留物を得た。残留物を131 gのヘプタンで処理し、蒸発させて164 gの残留物を得た。97 gのヘプタンでこの手順を繰り返し、160 gの油状物を得た。残留した油状物を周囲温度で257 gのヘプタンと攪拌してスラリーを得、これを氷浴で冷却した後、ろ過して固体を単離した。ろ過ケーキを49 gのヘプタンで洗浄し、次に真空下で乾燥させ、125.58 g(0.380 mol、収率88%)のCPTAを得た。
【0174】
実施例32
本実施例は、実施例22の化合物4のラセミ混合物を調製する方法を示す。
【0175】
マグネティックスターラーと還流冷却器を備えた50 ml丸底フラスコに、2.10 g(6.35 mmol)のラセミ体CPTA、21 gの2-プロパノールおよび0.50 g(4.2 mmol)のチオニルクロリドを入れた。90分還流後のHPLC解析によって、84.2面積%の7および12.7面積%のCPTAが明らかになった。さらに1.0 g(8.4 mmol)のチオニルクロリドを加えると、CPTAが1面積%未満となった。溶液を周囲温度に冷却し、1.0 g(12 mmol)の固体の炭酸水素ナトリウムで処理した。溶媒を蒸発させ、残留物を25 mlのトルエンに溶解した。水(2×10 ml)で洗浄した後、溶媒を蒸発させ、残留物として実施例22の化合物4を2.31 g(6.2 mmol、収率98%)を得た(95.8面積%の7と2.4面積%のトルエン)。
【0176】
実施例33
本実施例は、ラセミ体CPTAの溶解度を測定する方法を示す。
【0177】
マグネティックスターラーを備えた100 mlのウォータジャケット付き樹脂ポットを再循環水浴に接続し、9.44 gのラセミ体CPTAおよび16.78 gの1,2-ジクロロエタンを入れた。浴温を35℃に温め、スラリーを1時間撹拌した。撹拌器を止め、固体を30分沈殿させた。0.1360 gの上清試料を取り出しアセトニトリルで25.00 mlに希釈し、溶液をHPLC解析によって分析した。この結果および一連の他の測定の結果を図11および19に示す。約2℃で解析するために、1.92 gの1,2-ジクロロエタン中の0.54 gのCPTA試料を終夜冷蔵庫に格納した後、上清液をHPLC解析により分析した。図12で示される図19に含まれるヘプタン中のCPTAの溶解度を同様の方法で測定した。
【0178】
実施例34
本実施例は、CPTAのラセミ混合物を分割する方法を示す。
【0179】
1 Lの底部排液管反応器に、48.2 g(146 mmol)のCPTA、16.4 g(77.3 mmol)の(1R,2R)-(-)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオール(CAF D-塩基)および193 gの2-プロパノールを入れた。スラリーを70℃に加熱して溶液とし、次に60℃に冷却し、1時間保持した。生じたスラリーを0.25℃/分でジャケット温度2℃へ冷却し、14時間保持した;内部温度は4℃であった。固体を真空ろ過によって分離し、27 gの2-プロパノールですすいだ。母液および洗浄溶液をHPLC解析のために採取し、その結果を図13に示す。50.48 gのウエットケーキを193 gの2-プロパノールと共に1 Lの反応器に再び入れ、スラリーを85℃のジャケット温度で温めて穏やかに還流して溶液とした。溶液をHPLC解析のために採取した;図13に結果を一覧にして示す。65℃に冷却することによってスラリーが形成された。68℃に30分間温めた後に、スラリーを0.25℃/分で40℃に、次に0.4℃/分で18℃に、その後1℃/分で2℃に冷却した。(他の調製では、図14に記録した直線的な冷却速度を用いた)。固体を真空ろ過によって分離し、18 gの2-プロパノールですすぎ、真空下で乾燥させて27.29 g(50.4 mmol、収率34.5%)の(-)-CPTA/CAF D塩基を得た。図13に、単離した固体および母液、および洗浄液のHPLC解析の結果が含まれる。
【0180】
実施例35
本実施例は、ハロフェナートからのラセミ体CPTAの調製および分割を示す。
【0181】
オーバーヘッドスターラー付きの1 L丸底フラスコに、129.75 g(0.312 mol)のラセミ体ハロフェナート、325 gの水および32.6 g(0.408 mol)の50%水酸化ナトリウム水溶液を入れた。スラリーを60℃に1時間加熱して溶液とし、次に冷却した。温度40℃で、328.5 gの1,2-ジクロロエタンおよび44 g(0.45 mol)の37%塩酸を加え、二相混合物を29℃に冷却した。水相のpHは0.85であった。有機相を分離し水250 mlで洗浄し、次に蒸発させ118.2 gの残留物を得た。2-プロパノール(149 g)を加えて蒸発させ、131.2 gの残留物とした。加えたハロフェナートの量に基づいて理論上103.2 gのラセミ体CPTAを含む残留物を、1 L底部排液管反応器に、33.10 g(0.1556 mol)のCAF D塩基および400 gの2-プロパノールと共に入れた。混合物を67℃に温めて、軽いスラリーを生成させ、次に0.075℃/分で1℃まで冷却した。混合物を-7℃に冷やし、固体を真空ろ過によって単離し、60 mlの2-プロパノールで洗浄した。単離した固体および492.8 gの母液および洗浄溶液のHPLC解析結果を図13(実験9)に示す。92.74 gのウエットケーキを再び1 L反応器に477 gの2-プロパノールと共に加え、混合物を75℃に加熱して溶液とした。溶液を0.5℃/分で5℃に冷却し、結晶化した固体を真空ろ過によって単離し、60 mlの2-プロパノールですすぎ、乾燥させて、51.81 g(0.0956 mol、収率31%)の(-)-CPTA CAF D塩基ジアステレオマー塩を得た。単離した固体、529.9 gの母液および洗浄溶液のHPLC解析の結果が図13に含まれる。
【0182】
実施例36
本実施例は、(+)-CPTAをラセミ化する方法およびラセミ体CPTAを回収する方法を示す。
【0183】
単離したジアステレオマー塩の収率および純度に基づいて71.6 g(0.217 mol)のCPTA(44%eeの(+)-エナンチオマー)を含む、上の実施例35に記載された103.2 gのCPTAの分割から得られた分割母液および再結晶化母液を蒸発させ、108.7 gの残留物を得た。残留物を176 gの1,2-ジクロロエタン、35.2 gの水および6.8 gの37%塩酸で処理した。有機相を取り出して蒸発させ、79.0 gの残留物とした。水(80 g)を加え、溶媒を蒸発させ78.1 gの残留物とした。残留物を141.9 gの水および24.6 g(0.308 mol)の50%水酸化ナトリウム水溶液で処理し、溶液を4時間加熱還流し、キラルHPLC解析によるラセミ化合物を得た。溶液を冷却し、140 mlの1,2-ジクロロエタンおよび32.0 g(0.325 mol)の37%塩酸で処理した。有機相を取り出して蒸発させて80.1 gの残留物とし、それを40℃水浴中、250 mlヘプタンで処理し、スラリーを得た。固体を真空ろ過により単離し、乾燥させて、63.83 g(0.193 mol、収率89%)のラセミ体CPTAを得た。試料の分割については、新鮮なCPTAの分割と一致する結果を得た(図13の実験10)。
【0184】
実施例37
本実施例は、ジアステレオマー塩から(-)-CPTAを分離する方法を示す。
【0185】
マグネティックスターラー付き500 mlフラスコに、40.0 g(73.7 mmol)の(-)-CPTA/CAF D塩基、100 gの1,2-ジクロロエタン、40 gの水および7.6 g(77 mmol)の37%塩酸を入れた。固体が完全に溶解した後、下部の有機相を取り出し、10 mlの水で洗浄した。合わせた水相のpHは0.9であった。128.2 gの有機相のHPLC解析により、1,2-ジクロロエタン溶液として24.32 gの(-)-CPTA(73.6 mmol、理論値の99.8%)が見出された。
【0186】
実施例38
本実施例は、N-アセチルエタノールアミンの真空精製を示す。
【0187】
マグネティックスターラー、加熱マントルおよび短経路蒸留ヘッドを装備した50 ml丸底フラスコに、29.09 gのN-アセチルエタノールアミンを入れ、およそ0.8 torrの真空下に置いた。凝縮物が集まることはなかったが、液体を加熱すると泡が形成された。およそ130℃のヘッド温度で蒸留物を集め、澄んだ液体として26.71 g(回収率92%)の N-アセチルエタノールアミンを得た。
【0188】
実施例39
本実施例は、(-)-ハロフェナートを生成する方法を示す。
【0189】
マグネティックスターラー付き500 ml丸底フラスコに、35.5 g(65.4 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩(99.4%ee)、89.0 gの1,2-ジクロロエタンおよび35.5 mlの水を入れた。スラリーに、6.7 g(68 mmol)の37%塩酸を加えて混合物を周囲温度で攪拌し、透明な2相を得た。下部の有機相を取り出し、7.0 gの水で洗浄した。有機相を蒸発させて26.13 gの残留物を得て、次に55.6 gの1,2-ジクロロエタンに溶解し、加熱マントル中のマグネティックスターラーおよび還流/蒸留ヘッド付き250 ml丸底フラスコに入れた。溶液のHPLC解析によって22.06 g(66.7 mmol、理論値の102%)のCPTAが存在することが分かった。溶液に7.5 ml(100 mmol)のチオニルクロリドを加え、溶液を2時間加熱還流した。加熱を続け、6.1 gの蒸留液を集めた。溶液を周囲温度に冷却し、次に氷浴で冷却して、蒸留した25.85 g(251 mmol)のN-アセチルエタノールアミン(KF解析で1176および1288 ppmの水)を加えた。添加後に温度が約26℃に上昇した。溶液を、氷浴中の36 gの水中の9.90 g(71.6 mmol)の炭酸カリウムに攪拌しながらゆっくり加えた。達した最高温度は15℃であった。反応混合液を5 mlの1,2-ジクロロエタンですすいだ。下部の有機相を取り出し、37 mlの水で洗浄した。溶液を蒸発させ、油状物(32.84 g)を得た。油状物を54 gのヘプタンで処理し、溶媒を除去して31.56 gの固形残留物を得た。固体に、76 gのヘプタンを加え、溶媒を除去して29.19 gの固形残留物を得た。固体を40℃で28 mlの2-プロパノールに溶解し、次にさらに、28 mlの20プロパノールおよび334 mlのヘプタンで希釈した。周囲温度に冷却すると薄いスラリーが得られた。氷浴で冷却すると厚いスラリーが形成された。2時間撹拌した後に、真空ろ過により固体を単離し、29 gのヘプタンですすぎ、乾燥させて、14.21 g(34.2 mmol、収率52.3%)の(-)-ハロフェナートを得た。キラルHPLC解析によって(+)-ハロフェナートは検出されなかった(>99.8%ee)。
【0190】
294.1 gの母液および洗浄液のHPLC解析によって、11.2 gのハロフェナートおよび1.26 gのCPTAが見出された。溶媒を蒸発させ、12.47 gの残留物を14 mlの2-プロパノールに溶解した。84 mlのヘプタンを加え、終夜周囲温度で撹拌した後にスラリーを得た。スラリーを氷浴で冷却し、固体を集めて、9 gのヘプタンですすぎ、乾燥させ、5.64 g(13.6 mmol、収率20.7%、HPLC解析による89.9%のハロフェナートおよび3.9%のCPTA、99.6%ee)の(-)-ハロフェナートを得た。81.74 gの母液および洗浄液のHPLC解析によって、3.66 g(8.8 mmol、13.5%)のハロフェナートおよび0.93 g(2.8 mmol、4.8%)のCPTAの存在が分かった。
【0191】
実施例40
本実施例は、ラセミ体CPTAナトリウム塩を単離する方法を示す。
【0192】
分割回収率に基づき理論上は63.9 g(0.193 mol)のCPTAを含む分割結晶化および再結晶化の母液を蒸発させ、91 gの残留物を得た。残留物を146 gの1,2-ジクロロエタンに溶解し、28.6 gの水および6.3 gの37%塩酸で40℃にて処理した。219 gの有機相を蒸発させ、71.86 gの残留物を得た。残留物に、120 gの水および21.5 g(0.269 mol)の50%水酸化ナトリウムを加えた。溶液を加熱還流し、次に周囲温度に放冷して厚いスラリーを得た。冷却によって生じた固体を真空ろ過によって単離し、25 mlの水ですすいで次に乾燥させ、31.78 g(0.0901 mol、回収率46.7%)のCPTAナトリウム塩を得た。キラルHPLC解析により、物質はラセミ体であることが分かった。188.6 gの母液および洗浄液のHPLC解析により28.3 g(0.0856 mol、44.4%)のCPTAが存在することが判明した。
【0193】
実施例41
本実施例は、ラセミ体CPTAナトリウム塩の溶解度を測定する方法を示す。
【0194】
マグネティックスターラーを備えた100 mlのウォータージャケット付き樹脂ポットを再循環水浴に接続し、3.48 gのラセミ体CPTAナトリウム塩および20.0 gの水を加えた。浴温を35℃に温め、スラリーを1時間撹拌した。撹拌器を止め、固体を30分間静置した。pHは9.4であった。0.3036 gの上清試料を取り出し、アセトニトリルで25.00 mlに希釈して、溶液をHPLC解析により分析した。解析を47℃、および19℃で繰り返した。さらに3.01 gのCPTAナトリウム塩を加えてスラリーを高温で維持し、25 gの水を加えて、より低温でより薄いスラリーを得た。50%の水酸化ナトリウム水溶液の添加により、周囲温度でpHを12.7に高め、13.5、25、34、および42℃で解析を続けた。結果を図16および20に示す。
【0195】
実施例42
本実施例は、(+)-ハロフェナートの加水分解およびラセミ化を示す。
【0196】
マグネティックスターラーと加熱マントルを備えた250 ml丸底フラスコに、7.28 g(17.5 mmol)の(+)-ハロフェナート(86.9%ee)、72.2 gの水および4.21 g(52.6 mmol)50%水酸化ナトリウム水溶液を入れた。スラリーを50〜60℃に加熱した。生じた溶液のpHは12.8であった。キラルHPLC解析により、80.4%の(+)-CPTAおよび10.5%の(-)-CPTAが示された。溶液を90分間加熱還流した。キラルHPLC解析が、49.6%の(+)-CPTAおよび47.0%の(-)-CPTAを示した。pHは12.6であった。周囲温度に冷却した後に、およそ50 mlの1,2-ジクロロエタンを加え、7.3 g(74 mmol)の37%塩酸を加えてpHを0.8に調節した。有機相を蒸発させ、6.0 gの残留物を得た。残留物を25 mlのヘプタンで処理し、温めて油状物を溶解し、次に氷浴で冷却した。固体を真空ろ過によって集め、乾燥させて5.10 g(15.4 mmol、収率88%)のラセミ体CPTAを得た。このデータおよび2つの同様の加水分解のデータを図17および21に示す。
【0197】
同様に、6.75 g(16.3 mmol)の(+)-ハロフェナートを、0.65 g(8.1 mmol)の50%水酸化ナトリウム水溶液と67.5 gの水と共に2時間60℃で熱することにより、37.5%のハロフェナートおよび54.2%のCPTAを得た。終夜加熱還流することにより92.1%のCPTAおよび1.1%のハロフェナートが得られ、最終pHは4.8であった。キラルHPLC解析によって、(+)/(-)-CPTAが80.3/12.8の比率であることが分かった。
【0198】
実施例43
本実施例は、(-)-ハロフェナート結晶化母液から(-)-CPTA/CAF D塩基ジアステレオマー塩を回収することによる(-)-ハロフェナートの調製を示す。
【0199】
マグネティックスターラー付き1 L丸底フラスコに、50.0 g(92.3 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩(97.1%ee)、124 gの1,2-ジクロロエタン、50 mlの水および9.6 g(98 mmol)の37%塩酸を入れた。有機相を分離して50 mlの水で洗浄し、次に加熱マントル中のマグネティックスターラー付き250 ml丸底フラスコに入れた。還流/蒸留ヘッドを結合し、溶液を加熱蒸留して35.4 gの蒸留液を除去した。40℃に冷却した後に、溶液を25 mlの1,2-ジクロロエタンで希釈し、11 ml(150 mmol)のチオニルクロリドを加えた。2時間加熱還流し、22.6 gの蒸留液を除去した後、溶液を氷浴で冷却して、38.6 g(374 mmol)の蒸留したN-アセチルエタノールアミンを滴下して加えた。添加中に反応温度が7℃から18℃まで上昇した。周囲温度で終夜撹拌した後、溶液を、氷浴で冷却した51 mlの水中の12.7 gの炭酸カリウムに撹拌しながら加えた。有機相を取り出し、51 gの水で洗浄した。有機相(HPLC解析により、85.2%ハロフェナートおよび6.1% CPTA)を蒸発させて44.3 gの油状物とし、133 gのヘプタンで処理し、次に蒸発させて43.3 gの固体にした。固形残留物を61.5 gの2-プロパノールに溶解し、320 gのヘプタンと共に1 Lの底部排液管反応器に入れ、50℃に温め、そして3℃/分で20℃まで、その後1℃/分で-3℃まで冷却した。溶液は27℃で濁り、15℃で厚いスラリーが形成された。固体を真空ろ過によって単離し、5 mlの2-プロパノールを含む40 mlのヘプタンで洗浄し、乾燥させて21.01 g(50.6 mmol、収率55%、HPLCにより98.93%)の(-)-ハロフェナート(99.9%ee)を得た。HPLC分析によれば14.65 g(35.3 mmol)のハロフェナート(88.3%ee)および1.78 g(5.4 mmol)のCPTAを含む395.7 gの母液および洗浄溶液を蒸発させ、21.57 gの残留物を得た。残留物を100 mlの水および5.0 g(63 mmol)の50%水酸化ナトリウム水溶液と共に50℃に加熱し、溶液とした。約10分後のHPLC解析により、83.6%のCPTAおよび0.3%のハロフェナートの存在が分かった。溶液を冷却して50 mlの1,2-ジクロロエタンで希釈し、7.3 g(74 mmol)の37%塩酸によってpHを12.7から1.6まで低下させた。30 mlの水で洗った後に、HPLC分析により11.32 g(34.2 mmol)のCPTAを含む72.9 gの有機相を蒸発させて残留物とし、36 gのヘプタンで処理し、次に蒸発させて14.9 gの残留物とした。油状の残留物を38 gのヘプタンに加熱して溶解した。冷却して油状物を得た。溶媒を除去し、残油を34.8 gのメチルシクロヘキサンに溶解した。冷却によって油状物が形成された。溶媒を除去し、45.6 gの2-プロパノールに交換した。キラルHPLC解析により、65.8%eeの(-)-CPTA(16.9/81.6の(+/-)-比率)であることが分かった。周囲温度の溶液に6.50 g(30.6 mmol)のCAF D塩基を加えた。厚いスラリーが急速に形成された。スラリーを撹拌しながら40℃に温め、その後氷浴で冷却して、固体を真空ろ過によって分離し、7 gの2-プロパノールで洗浄し、乾燥させて13.91 g(25.7 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩を得、それは最初に加えた50.0 gの塩の28%の回収率に相当する。(+)/(-)-CPTA比率は1.77/97.86であった。45.34 gの母液および洗浄溶液のHPLC解析によって、4.34 g(13.1 mmol)のCPTAを見出し、それは最初に加えた50.0 gの塩の14 mol%に相当する。
【0200】
実施例44
本実施例は、CPTA/CAF D塩基塩からCAF D塩基を回収する方法を示す。
【0201】
マグネティックスターラー付き1 L丸底フラスコに80.16 g(0.148 mol)の(-)-CPTA/CAF D塩基塩、237 gの1,2-ジクロロエタンおよび80mlの水を入れた。スラリーに15.2 g(0.154 mol)の37%塩酸を加え、透明な2相を得た。水層のpHは1.2であった。下部の有機層を取り出し、16 mlの水で洗浄した。合併した水相(140.7 g)を12.9 g(0.161 mol)の50%水酸化ナトリウム水溶液で処理し、pHを12.1とした。生じたスラリーをろ過し、固体を25mlの水ですすぎ、乾燥させて30.79 g(0.145 mol、回収率98%)のCAF D塩基(融点160.4〜161.0℃)を得た。
【0202】
実施例45
本実施例は、分割母液からCAF D塩基を回収する方法を示す。
【0203】
60.0 gのラセミ体CPTA試料を、上述のように20.88 gのCAF D塩基によって240 gの2-プロパノール中で分割し、74.7 gのウエットケーキを得た。ウエットケーキを218 gの2-プロパノール中で再結晶し、32.35 g(収率32.8%)の(-)-CPTA/CAF D塩基塩を得た。得られた塩の量から理論上40.32 gのCPTAおよび8.23 g(38.8 mmol)のCAF D塩基を含む結晶化および再結晶化からの母液および洗浄溶液を蒸発させ、72.9 gの残留物とした。残留物を、265 gの1,2-ジクロロエタン、50mlの水および4.0 g(40.6 mmol)の37%塩酸に溶解した。水層を分離し、3.88 g(48.5 mmol)の50%水酸化ナトリウム水溶液を加えてpHを0.6から12.3に高めた。生じたスラリーをろ過し、固体を集めて、水ですすぎ、7.12 g(33.6 mmol、回収率87%)のCAF D塩基(融点162.4〜163.0℃)を得た。
【0204】
本明細書に記述された実施例と実施態様はただ説明目的のみのためであり、またそれらを考慮に入れた様々な修正または変更は当業者に示唆されており、本出願の精神と範囲および添付の請求項の範囲内に含まれることとなると理解される。本明細書に引用されたすべての出版物、特許および特許出願は、参照によりその全体をあらゆる目的のために本明細書に組み入れられる。
【図面の簡単な説明】
【0205】
【図1】(-)-および(+)-CPTA/CAF D-塩基塩の2-プロパノール中における溶解度プロフィールを示すグラフである。
【図2】CAF D塩基を用いたCPTAのラセミ混合物を分割するための方法の、様々な結晶化条件における結果を示す。
【図3】(-)-および(+)-CPTA/CAF D-塩基塩の、純粋なイソプロパノール中、およびイソプロパノールとCPTA(11%)の混合物を含む溶液中における溶解度を示すグラフである。
【図4】様々な量の各成分の混合物の組成を示すグラフである。
【図5】結晶化および加熱に対する(-/+)-塩の飽和プロフィールを示すグラフである。
【図6】図2の実験4におけるモデル予測と実験結果の比較を示す表である。
【図7】加えたCAF D塩基の量の関数として生成された(+)-塩の量を示すグラフである。
【図8】図2の実験11に示す分割の実験データのグラフ表示である。
【図9】母液中のCPTAの実際量と計算量、および(+)-CPTA塩の計算百分率と実験データとのグラフによる比較を示す。
【図10A】図7(すなわち、図2の実験13)の実験データおよび溶解度モデル計算を示す表である。
【図10B】図2の実験4の28.3℃における実験データと溶解度モデル計算を示す表である。
【図11】1,2-ジクロロエタン中の、様々な温度におけるラセミ体CPTAの溶解度を示すグラフである。
【図12】ヘプタン中の、様々な温度におけるラセミ体CPTAの溶解度を示すグラフである。
【図13】様々な結晶化条件下でCAF D-塩基を用いたCPTA分割の収率を示す実施例24の結果の表である。
【図14】図2の様々な実験における分割結晶化の冷却プロフィールを示す。
【図15】実施例26における(-)-CPTA塩からの(-)-ハロフェナートの収率を示す表である。
【図16】水中での様々な温度におけるラセミ体CPTAナトリウム塩の溶解度を示すグラフである。
【図17】(-)-ハロフェナートの加水分解中の様々なpHにおけるCPTAラセミ化プロフィールを示すグラフである。
【図18】実施例30記載の様々なpHにおけるCAF D-塩基回収の結果を示す表である。
【図19】実施例33において測定された1,2-ジクロロエタンおよびヘプタン中のラセミ体CPTAの溶解度測定の実験結果である。
【図20】実施例41において測定された水中のラセミ体CPTAナトリウム塩の溶解度測定の実験結果である。
【図21】実施例42において測定された(+)-ハロフェナートの塩基性加水分解の実験結果である。
【技術分野】
【0001】
発明の分野
本発明は、α-(フェノキシ)フェニル酢酸をそのエナンチオマー混合物から分離するためのエナンチオ選択的分割方法に関する。
【背景技術】
【0002】
発明の背景
ハロフェナートなどのα-(フェノキシ)フェニル酢酸のエステルおよびアミド誘導体はキラル化合物であり、例えばII型糖尿病および高脂血症のような血液脂質沈着に関連する疾患を含む様々な生理学的疾患の改善に役立つ。例えば、米国特許第3,517,050号および第6,262,118号を参照のこと。α-(フェノキシ)フェニル酢酸には、単一のキラル中心がカルボニル炭素原子に対してαの位置の非対称的に置換された炭素原子に存在し、したがって2つのエナンチオマーの形が存在する。
【0003】
チトクロームP450 2C9は、特定の薬物代謝に重要な役割を果たすことが知られている酵素である。チトクロームP450酵素の阻害によって起こる薬物代謝の変化が患者に対して重大な悪影響を引き起こす高い可能性を有していることは、当業者に公知である。例えばハロフェン酸のようなラセミ体のα-(フェノキシ)フェニル酢酸がチトクロームP450 2C9を阻害することも、また公知である。例えば、米国特許第6,262,118号 を参照のこと。したがってハロフェン酸またはその誘導体などのラセミ体のα-(フェノキシ)フェニル酢酸の投与により、この酵素によって代謝される抗凝血剤、抗炎症剤およびその他の薬剤を含む他の薬物との様々な薬物相互作用問題が引き起こされる可能性がある。ハロフェン酸の(-)-エナンチオマーは、チトクロームP450 2C9を阻害する能力が(+)-エナンチオマーに比べて約20倍弱いことが発見された(同文献)。したがって、実質的に(+)-エナンチオマーを含まないハロフェン酸の(-)-エナンチオマーまたはその誘導体を投与することが、薬物相互作用の可能性を減少させるために望ましい。
【0004】
したがって、例えば(-)-ハロフェン酸のような、α-(フェノキシ)フェニル酢酸の所望のエナンチオマーを濃縮した生成物を生産するための効率的な方法が必要である。
【発明の開示】
【0005】
発明の概要
本発明の一つの局面において、式:
(式中、
R1は、アルキルまたはハロアルキル、および
Xは、ハライドである)
で示されるエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸化合物を、第1および第2のエナンチオマーを含むα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物から生成するための方法を提供する。ある特定な実施態様においては、エナンチオマー混合物はラセミ混合物である。
【0006】
本発明の方法は、次の工程を含む:
(a) 第1エナンチオマーがエナンチオマー濃縮された固体の酸塩基塩を含む溶液を、α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物を0.5モル当量未満のエナンチオマー濃縮したキラルアミン化合物と、溶液中におけるフリーの第1エナンチオマーの量とフリーの第2エナンチオマーの量の比を約1対3とするのに十分な条件下で接触させることにより、生成する工程;および、
(b) 第1エナンチオマーの固体酸塩基塩を溶液から、α-(フェノキシ)フェニル酢酸化合物の第2エナンチオマーの酸塩基塩の濃度がその飽和点の近くまたはその飽和点より低い温度で分離する工程。
【0007】
第2エナンチオマーの少なくとも一部を、例えば、第2エナンチオマーを塩基と接触させることによりラセミ化させ、第1エナンチオマーに変換することができる。その結果生じるエナンチオマー混合物を再利用し、同様のエナンチオマー濃縮処理に供して第1エナンチオマー酸塩基塩の収率を増加させることができる。
【0008】
ある特別の実施態様においては、キラルアミン化合物は、式:
(式中、
R2およびR3は、それぞれ独立に水素またはアルキルであるか;またはR2およびR3はそれらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素またはアルキルであるか、あるいはR5またはR6のうちの1つは、アミン保護基であり;および
Arは、アリールである)
で示される化合物である。
【0009】
詳細な説明
I. 定義
「アルキル」とは、1〜10個の炭素原子の、好ましくは1〜6個の炭素原子の、より好ましくは1〜4個の炭素原子の直鎖状または分岐した脂肪族炭化水素鎖基を指す。例示的なアルキル基にはメチル、エチル、n-プロピル、2-プロピル、tert-ブチル、ペンチルその他が含まれるが、それらに限定されるわけではない。
【0010】
「アリール」とは、一価の6〜10個の炭素環原子の一環または二環から成る芳香族炭化水素部分を指す。特に別の記述または表示をしない限り、アリール基を1つまたは複数の置換基、好ましくは1個、2個または3個の置換基、より好ましくは、アルキル、ハロアルキル、ニトロおよびハロから選択される1個または2個の置換基で置換することができる。より具体的には、用語「アリール」は、フェニル、1-ナフチル、および2-ナフチルその他を含むが、それらに限定されるわけではなく、その各々は上に述べた1つまたは複数の置換基で置換されてもよい。
【0011】
「CAF D塩基」とは、クロラムフェニコールD塩基、すなわちD-threo-(-)-2-アミノ-1-(ニトロフェニル)-1,3-プロパンジオールを指す。
【0012】
「キラル」または「キラル中心」とは、4つの異なる置換基を有する炭素原子を指す。しかしながら、キラリティの最終判断基準は鏡像が重ね合わせられないことである。
【0013】
「CPTA」および「ハロフェン酸」という用語は、本明細書において交換して用いることができ、(4-クロロフェニル)(3-トリフルオロメチルフェノキシ)酢酸を指す。
【0014】
「エナンチオマー混合物」とは、ラセミ混合物を含むエナンチオマーの混合物を有するキラル化合物を意味する。好ましくは、エナンチオマー混合物とは、実質的に等しい量の各エナンチオマーを有するキラル化合物を指す。より好ましくは、エナンチオマー混合物は各エナンチオマーが当量存在するラセミ混合物を指す。
【0015】
「エナンチオマー濃縮された」とは、1つのエナンチオマーがそれを分離工程に付す前よりも多量に存在する組成物を指す。
【0016】
「エナンチオマー過剰率」または「%ee」とは、第1エナンチオマーと第2エナンチオマーの間の差異の量を指す。エナンチオマー過剰率は、式:%ee = (第1エナンチオマーの%)-(第2エナンチオマーの%)と定義される。したがって、組成物が98%の第1エナンチオマーおよび2%の第2エナンチオマーを含む場合、第1エナンチオマーのエナンチオマー過剰率は98%-2%すなわち96%である。
【0017】
用語「ハライド」および「ハロ」は本明細書中で交換して用いることができ、F、Cl、BrおよびI、ならびに-CNおよび-SCNなどの偽ハロゲン化物を含むハロゲンを指す。
【0018】
「ハロアルキル」とは、1つまたは複数の水素原子がトリフルオロメチルなどのペルハロアルキルを含むハロゲンで置換されている、本明細書で定義のアルキル基を指す。
【0019】
「ハロフェナート」とは、2-アセトアミドエチル4-クロロフェニル-(3-トリフルオロメチル-フェノキシ)アセタート(すなわち、4-クロロ-α-(3-(トリフルオロメチル)フェノキシ)ベンゼン酢酸、2-(アセチルアミノ)エチルエステル、または (4-クロロフェニル)(3-トリフルオロメチルフェノキシ)酢酸、2-(アセチルアミノ)エチルエステル)を指す。
【0020】
「ヘテロアルキル」とは、1または複数の異種原子、または1または複数の異種原子を含む置換基を含む、分岐したまたは分岐のない非環状の飽和アルキル部分を意味し、ここで異種原子とはO、NまたはSである。典型的な異種原子を含む置換基には、=O、-ORa、-C(=O)Ra、-NRaRb、-N(Ra)C(=O)Rb、-C(=O)NRaRb および-S(O)nRa(ここでnは0〜2までの整数である)が含まれる。各RaおよびRbは独立に水素、アルキル、ハロアルキル、アリールまたはアラルキルである。ヘテロアルキルの代表的な例には、例えば、N-アセチル2-アミノエチル(すなわち、-CH2CH2NHC(=O)CH3)が挙げられる。
【0021】
用語「ヘテロサイクリル」と「複素環」は交換して用いることができ、3〜8個の環原子の非芳香性環部分を指し、ここで1、2、または3個の環原子はN、OまたはS(O)n(ここでnは、0〜2の整数である)から選択される異種原子であり、残る環原子は、Cであり、1または2個のC原子はカルボニル基で置換されてもよい。もし別の記述または表示がなければヘテロサイクリル環を、ハロゲン、アルキル、アリール、ヒドロキシ、アミノ、またはアルコキシから選択される1、2または3個の置換基で任意に独立して置換されてもよい。より具体的には、用語「ヘテロサイクリル」には、1,3-ジオキサンおよびその誘導体その他が含まれるが、それに限定されるわけではない。
【0022】
「脱離基」は、従来合成有機化学においてそれに関連する意味、すなわち、求核基によって置き換えられることのできる原子または基の意味を有し、ハロ(クロロ、ブロモおよびヨードなど)、アルカンスルホニルオキシ、アレーンスルホニルオキシ、アルキルカルボニルオキシ(例えば、アセトキシ)、アリールカルボニルオキシ、メシルオキシ、トシルオキシ、トリフルオロメタンスルホニルオキシ、アリールオキシ(例えば、2,4-ジニトロフェノキシ)、メトキシ、N,O-ジメチルヒドロキシルアミノ、その他を含む。
【0023】
用語「金属」には、第I、II族金属、および遷移金属、ならびにBおよびSiなどの主族金属が、含まれる。
【0024】
「光学純度」とは、組成物中に存在する特定のエナンチオマーの量を指す。例えば、ある組成物が98%の第1エナンチオマーおよび2%の第2エナンチオマーを含む場合、第1エナンチオマーの光学純度は98%である。
【0025】
もし別の記述がなければ、用語「フェニル」とは、置換されていてもよいフェニル基を指す。適切なフェニル置換基は「アリール」の定義に記述されたものと同一である。同様に、用語「フェノキシ」とは、式-OAraの部分を指し、Araは本明細書で定義されるフェニル基である。したがって、用語「α-(フェノキシ)フェニル酢酸」とは、置換されていてもよいフェニルおよび置換されていてもよいフェノキシ部分(残基)によって2位が置換された酢酸を指す。
【0026】
「保護基」とは、分子中の反応性基に結合された時その反応性を遮蔽、減少、または阻止する部分を指す。保護基の例は、T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edition, John Wiley & Sons, New York, 1999, および、 Harrison and Harrison et al., Compendium of Synthetic Organic Methods, Vols.1-8 (John Wiley and Sons, 1971-1996)に見出され、それらの全体が参照により本明細書組み入れられる。代表的なヒドロキシ保護基には、アシル基、ベンジルおよびトリチルエーテル、テトラヒドロピラニルエーテル、トリアルキルシリルエーテルおよびアリルエーテルが含まれる。代表的なアミノ保護基には、ホルミル、アセチル、トリフルオロアセチル、ベンジル、ベンジルオキシカルボニル(CBZ)、tert-ブトキシカルボニル(Boc)、トリメチルシリル(TMS)、2-トリメチルシリル-エタンスルホニル(SES)、トリチルおよび置換されたトリチル基、アリルオキシカルボニル、9-フルオレニルメチルオキシカルボニル(FMOC)、ニトロ-ベラトリルオキシカルボニル(NVOC)その他が含まれる。
【0027】
用語「速度」とは、塩の生成に言及している場合は、速度論的および/または熱力学的速度を指す。
【0028】
本明細書において用いられる用語「処理する」、「接触させる」、または「反応させる」とは、指示されたおよび/または所望の生成物を産生するために適切な条件下で、2以上の反応物を加えることまたは混合することを指す。指示されたおよび/または所望の生成物を産生する反応は、必ずしも最初に加えられた2つの反応物の組み合わせから直接得られなくてもよく、すなわち、混合物中で1または複数の中間生成物が生成され、それが最終的には指示されたおよび/または所望の生成物の生成をもたらしてもよいことを認識するべきである。
【0029】
本明細書において用いられる用語「上に定義されたもの」および「本明細書において定義されたもの」には、変数に言及する場合には、もしあれば変数の好ましい、より好ましい、および最も好ましい定義は勿論、変数の広い定義を参照により組み入れる。
【0030】
多くの有機化合物は光学活性型で存在する、すなわちそれらは平面偏光の面を回転させる能力を有している。光学活性化合物について記述する際には、接頭辞RおよびSを用いて分子のキラル中心に関する分子の絶対立体配置を表示する。接頭辞「d」および「l」あるいは(+)および(-)を、化合物による平面偏光の回転の符号を指定するために採用し、(-)または(l)は化合物が「左旋性」であることを意味し、また(+)または(d)は化合物が「右旋性」であることを意味する。エナンチオマーの絶対立体化学の命名法と回転の命名法との間には相関が無い。与えられた化学構造に対して、「立体異性体」と呼ばれるこれらの化合物は、それらが互いの鏡像である以外は同一である。特定の立体異性体は「エナンチオマー」と呼ぶこともできる。またそのような異性体の混合物をしばしば、「エナンチオマー」混合物または「ラセミ」混合物と呼ぶ。例えば、Streitwiesser, A. & Heathcock, C.H., INTRODUCTION TO ORGANIC CHEMISTRY, 2nd Edition, Chapter 7 (MacMillan Publishing Co., U.S.A. 1981) を参照のこと。
【0031】
用語「その(+)-立体異性体を実質的に含まない」と「その(+)-エナンチオマーを実質的に含まない」とは、本明細書において交換して用いることができ、組成物が(+)-異性体と比較して実質的に(-)-異性体をより大きな割合で含んでいることを意味する。好ましい実施態様において、用語「その(+)立体異性体を実質的に含まない」とは、組成物が重量で少なくとも90%の(-)-異性体および重量で10%以下の(+)-異性体であることを意味する。より好ましい実施態様において、用語「その(+)-立体異性体を実質的に含まない」とは、組成物が重量で少なくとも99%の(-)-異性体を含み、重量で1%以下の(+)-異性体を含むことを意味する。最も好ましい実施態様においては、用語「その(+)-立体異性体を実質的に含まない」とは、組成物が重量で99%を越える(-)-異性体を含むことを意味する。これらの百分率は、組成物中の異性体の総量に基づく。
【0032】
II. 序論
キラル合成は近年大きな進歩をしているが、ラセミ体の分割が、光学活性化合物すなわちキラル化合物の調製のための工業的方法において、いまだに一般的に好まれる方法である。通常は、キラル化合物をラセミ体の形で合成し、最終生産物を分割してエナンチオマー濃縮された化合物を産生する。
【0033】
最終生産物を分割するこの方法が、薬学的活性のあるキラル化合物の大規模調製に特に役立つ。キラル化合物のエナンチオマーは正確に同じ化学結合を持つが、エナンチオマー中の原子の空間的配向が異なる。したがって、キラル薬剤の1つのエナンチオマーがもう一方のエナンチオマーより望ましい活性を発揮し、副作用は著しく少ないことがしばしば起こる。光学活性薬剤のキラリティーとその副作用のそのような関係は以前から知られているが、いまだに多くのキラル薬剤はラセミ体の形で投与されている。
【0034】
ジアステレオマーの結晶化が工業的な規模において広く用いられる。ジアステレオマーの結晶化による分割の理論的な1回全工程を通しての収率は50パーセントである。しかし通常は、十分な光学純度を有する組成物を産生するためには2回以上の再結晶工程が必要である。
【0035】
本発明は、例えばハロフェン酸のようなα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物、好ましくはラセミ混合物、をエナンチオマー濃縮する方法を提供する。好ましくは、本発明の方法は、α-(フェノキシ)フェニル酢酸化合物の(-)-エナンチオマーの固体の酸塩基塩を提供する。このようにして、(-)-エナンチオマーを溶液から容易に分離することができる。
【0036】
エナンチオマー濃縮したα-(フェノキシ)フェニル酢酸のカルボン酸基を、次にカルボン酸活性化基によって活性化し、活性化されたα-(フェノキシ)フェニル酢酸を生成することができ、これを、アルコール、アミン、チオール、また他の求核性の化合物と反応させて、それぞれエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸エステル、アミド、チオエステルまたは他の誘導体を産生することができる。したがって、本発明の方法を用いて生成されたエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸化合物は、米国特許第3,517,050号に開示されたようなα-(フェノキシ)フェニル酢酸誘導体を産生するために有用である。特に、本発明の方法は、(-)-ハロフェナートを産生するために役立つ。
【0037】
III. エナンチオ選択的結晶化
上述したように、大部分のエナンチオ選択的結晶化方法においては、十分な光学純度を有する組成物を生成するためには、2回以上の再結晶化工程が必要である。しかしながら、本発明者らは、本明細書において開示する一定の条件下において、単一の結晶化工程により十分な光学純度のα-(フェノキシ)フェニル酢酸化合物を生成することができることを見出した。したがってある局面において、本発明の方法は、α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物をキラルアミン化合物を用いてエナンチオマー濃縮することができるという、本発明者らによる驚くべきかつ予期しない発見に基づいている。特に本発明の方法は、光学純度が少なくとも約90%、好ましくは少なくとも約95%、より好ましくは少なくとも約97%、そして最も好ましくは少なくとも約98%の、α-(フェノキシ)フェニル酢酸化合物の所望のエナンチオマーを提供する。
【0038】
ある実施態様において、本発明の方法は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライドである)
で示されるα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物、好ましくはラセミ混合物のエナンチオマー濃縮を提供する。工程は一般に、キラルアミン化合物を用いて、α-(フェノキシ)フェニル酢酸化合物の固体のエナンチオマー濃縮された酸塩基塩を形成する工程を含む。
【0039】
特に本発明の方法は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライドである)
で示されるα-(フェノキシ)フェニル酢酸、例えばハロフェン酸(この場合R1はCF3、およびXはCl)、の分割を目標とする。
【0040】
ある特定の実施態様では、本発明の方法は、Xがクロロである式Iまたは好ましくは式IIのα-(フェノキシ)フェニル酢酸の分割を目標とする。
【0041】
さらに別の実施態様では、本発明の方法は、R1がハロアルキル、好ましくはトリフルオロメチルである、式Iまたは好ましくは式IIのα-(フェノキシ)フェニル酢酸の分割を目標とする。
【0042】
ある特定の実施態様では、α-(フェノキシ)フェニル酢酸をキラル塩基を用いて結晶化する。下記の実施例の項で開示する塩基を含め、種々様々なキラル塩基を用いることができる。好ましくは、用いたキラル塩基により、α-(フェノキシ)フェニル酢酸の(-)-エナンチオマーの固体の酸塩基塩が得られる。このようにして、(-)-エナンチオマーが例えばろ過により容易に溶液から分離される。ある特定の実施態様では、キラル塩基は、式:
(式中、各R2およびR3は、独立して水素、アルキルまたはヒドロキシ保護基であるか;あるいはR2およびR3は、それらが結合している原子と共に複素環部分を形成し;R4は、水素またはアルキルであり;各R5およびR6は、独立して水素またはアルキルであるか、R5またはR6のうちの1つがアミン保護基であり;またArは、アリールである)
で示されるアミン化合物である。
【0043】
ある特定の実施態様では、R2とR3はそれらが結合する酸素原子と共に1,3-ジオキサン、置換された1,3-ジオキサン(例えば、5,5-ジメチル-1,3-ジオキサンなどのジアルキルで置換された1,3-ジオキサン)またはその誘導体を形成する。
【0044】
別の実施態様では、R2とR3は水素である。
【0045】
さらに別の実施態様では、R4は水素である。
【0046】
また別の実施態様では、Arは置換されたアリールである。特に好ましいAr部分は、置換されていてもよいフェニルである。特別に好ましいAr部分は、4-ニトロフェニルである。
【0047】
またさらに、上述の好ましい基の組合せが他の好ましい実施態様を形成することになる。例えば、特に好ましいキラル塩基は上記式IIIのアミン化合物であって、式中、R2、R3、R4、R5およびR6は、水素であり;またArは、4-ニトロフェニルである。また特に好ましいα-(フェノキシ)フェニル酢酸化合物は、上記の式IIの化合物であり、式中、R1はトリフルオロメチルであり、Xはクロロである。このようにして、多種多様な好ましいキラル塩基およびα-(フェノキシ)フェニル酢酸化合物が本発明において具体化される。
【0048】
本発明者らは、α-(フェノキシ)フェニル酢酸の結晶化に用いられるキラル塩基の量が、エナンチオマー濃縮の光学純度に重要な影響があることを見出した。例えば、式:
(式中、R2、R3、R4およびArは、本明細書において定義されたものである)
で示されるキラルアミン化合物をα-(フェノキシ)フェニル酢酸化合物の結晶化で用いる場合は、より高い%eeが、キラルアミン化合物を0.5モル当量未満、好ましくは約0.48モル当量以下、より好ましくは約0.47モル当量以下、そして最も好ましくは約0.45モル当量以下の量を用いることにより、得られる。高度にエナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸誘導体を生成するためには、キラルアミン化合物それ自身がエナンチオマーとして十分純粋でなければならないことを認識するべきである。
【0049】
結晶化は、通常、α-(フェノキシ)フェニル酢酸の2つのエナンチオマーとキラルアミン間で形成される塩が異なる溶解度を有する溶媒中で実施する。このようにして、ジアステレオマー塩のうちの1つが、溶液から優先的に沈殿する。適当な結晶化溶媒には、アルコールなどのプロトン性溶媒が含まれる。特に好ましい結晶化溶媒はイソプロピルアルコールである。
【0050】
エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸の収率はまた、とりわけ用いる結晶化溶媒の量に依存する。例えば、大量の結晶化溶媒を用いる場合、混合物が稀薄になりすぎ、固体の形成が減少する。用いる結晶化溶媒の量が少なすぎる場合は、溶液が望まれないジアステレオマー塩で過飽和することになり、望ましくないジアステレオマー塩の結晶化をもたらす可能性があり、そのために所望のエナンチオマーの光学純度が減少する。したがって、結晶化溶媒としてイソプロパノールを用いる場合、用いる結晶化溶媒の量は、1グラムのα-(フェノキシ)フェニル酢酸化合物当り、好ましくは約2グラム〜約6グラム、より好ましくは約3グラム〜約5グラム、さらにより好ましくは約3.5グラム〜約4.5グラム、そして最も好ましくは約4グラムである。
【0051】
ある実施態様では、結晶化工程は、両方のエナンチオマーの核形成温度より高い温度へ結晶化溶液混合物を加熱して、両方のエナンチオマーを実質的にすべて溶解する工程を含む。例えば、結晶化溶液を約60℃から溶液の沸点までの範囲の温度に、好ましくは約70℃から約80℃までの範囲の温度に加熱する。より好ましくは、結晶化溶液を約75℃に加熱する。溶液は、キラルアミン化合物を加える前および/または後に加熱してもよい。加熱は固形物が実質的に完全に溶解するまで行い、それは通常約0.5〜約16時間、好ましくは約1〜約8時間の間である。
【0052】
その後結晶化溶液を、第1ジアステレオマー塩の、例えば、α-(フェノキシ)フェニル酢酸の(-)-エナンチオマー塩の核形成温度以下になるまで、しかし好ましくは第2ジアステレオマー塩の、例えばα-(フェノキシ)フェニル酢酸の(+)-エナンチオマー塩の核形成温度より高い温度まで冷却する。これによって、第1エナンチオマーとキラルアミン化合物の固体酸塩基塩の形成が可能になる。どのような理論にも拘束されずに、キラルミン化合物を使用することにより、一方のエナンチオマーの酸塩基塩の形成が、他方のエナンチオマーの酸塩基塩の形成より著しく速い速度で起こることになると信じられている。この速度は、2つのエナンチオマー間の速度論的および/または熱力学的速度差による可能性がある。典型的な化合物の場合と同じく、本発明のα-(フェノキシ)フェニル酢酸化合物の溶解度プロフィールは、高い温度でより高い溶解度を有している。したがって、第2ジアステレオマー塩の核形成温度のちょうど上まで結晶化溶液を冷却することにより、固体の第1ジアステレオマー塩の回収率がより高くなる。
【0053】
スラリーが形成された後、さらに結晶化溶液を溶液の温度が第2ジアステレオマー塩の飽和点の近くまたは上になるまで、冷やすことができる。これが、第1エナンチオマーのジアステレオマーの固体酸塩基塩の形成を増加させると共に、第2エナンチオマーからのジアステレオマー固体酸塩基塩の形成を防ぐ。
【0054】
結晶化溶液の冷却速度が、形成される固体酸塩基塩の光学純度に影響する可能性がある。例えば、結晶化溶液を速く冷却しすぎる場合には、望ましいエナンチオマーの固体酸塩基塩の格子内に望ましくないエナンチオマーがトラップされる可能性がある。しかしながら、遅すぎる冷却速度は生産時間とコストを増加させる。したがって、光学純度の損失を最小限にするが、しかし経済的に十分な速度で結晶化溶液を冷却するべきである。通常、結晶化溶液冷却速度は約0.05℃/min〜約1℃/min、好ましくは約0.1℃/min〜約0.7℃/min、およびより好ましくは約0.25℃/min〜約0.4℃である。その後結晶化溶液は、第2のすなわち望ましくないエナンチオマーの固体酸塩基塩の飽和点より高く維持される。通常、結晶化溶液をこの温度で、約1〜約72時間、好ましくは約2〜約48時間、より好ましくは約3〜約30時間維持する。
【0055】
予想通りに、少量のキラルアミン化合物を用いることにより、第1エナンチオマーの固体酸塩基塩の選択的な形成が可能になる。しかしその結果、収率が相応して小さくなる。理論上、ラセミ混合物からの所望のエナンチオマーの収率は、50%である。したがって、0.5モル当量のキラルアミン化合物を用いる場合、理論的収率は全α-(フェノキシ)フェニル酢酸の50%(あるいは所望のエナンチオマーの100%)である。経済的に望ましいためには、本発明の方法は所望のエナンチオマーの少なくとも約50%、好ましくは少なくとも約60%、より好ましくは少なくとも約70%、そして最も好ましくは少なくとも約75%の収率を提供する。100%の選択性を仮定すると、これらの収率は約0.25、0.30、0.35および0.375モル当量のキラルアミン化合物を加えることに相当し、それらは結晶化溶液に加える必要のあるキラルアミン化合物の最少量を表わす。
【0056】
第2エナンチオマーがキラルアミン化合物と固体酸塩基塩を形成する傾向が、従来の結晶化工程の変動の主な原因の1つであると信じられている。したがって第2のすなわち望ましくないエナンチオマーの過飽和点を測定することにより、第2エナンチオマーの固体酸塩基形成の予測不可能性を最小限にするか防止することができる。当業者は、例えば溶解度実験により過飽和点を容易に決定することができる。
【0057】
本発明の方法をラセミ混合物中に存在する(-)-エナンチオマーの濃縮に関して議論しているが、本発明の方法はまた(+)-エナンチオマーの濃縮にも適用可能であることに注意するべきである。本発明の方法は、基本的に(-)-エナンチオマーが濃縮された固体の沈殿、および(+)-エナンチオマーが濃縮されたろ過液すなわち母液を提供する。沈殿した塩からの所望の(-)-エナンチオマーの遊離およびキラルアミン化合物の回収を、例えば従来からこの性質の塩を加水分解することが知られている薄い鉱酸もしくは他の任意の無機酸または有機酸により塩を酸性化することにより、容易に遂行することができる。この手法は望まない副産物としてろ過液を残しているので、ろ過液をさらに酸または好ましくは塩基で処理して、(+)-エナンチオマーが濃縮されたろ過液をラセミ混合物に変換することができる。例えば、(+)-エナンチオマーを、水酸化ナトリウム水溶液を用いてラセミ体化することができる。次にこのラセミ混合物を再使用、すなわちリサイクルすることができる。さらに、キラルアミン化合物もまた上記の変換工程から回収してリサイクルすることができる。したがって本発明の方法は容易にリサイクル型の方法となる。
【0058】
IV. ラセミ体α-(フェノキシ)フェニル酢酸の合成
式Iのα-(フェノキシ)フェニル酢酸のラセミ混合物を生成する1つの方法を以下のスキームIに示す。
【0059】
このようにして、フェニル酢酸1の、例えば酸塩化物などの活性化カルボン酸誘導体への変換と、それに続くα-臭素化により、α-ブロモフェニルアセチルクロリド(示さず)を得た。次に酸塩化物をエステル2に変換し、式中Rは、通常はアルキルである。酸塩化物をエステル2に変換するために用いられるアルコールROHは、次の反応で溶媒として用いられるアルコールと同じアルコールであることが好ましい。このようにして、異なる溶媒型の数を最小限にする。さらに、次の反応で同じROHを溶媒として用いることにより、例えばトランスエステル化などによるの副産物の形成量が最小限になる。例えばイソプロピルエステル2は、すなわちRがイソプロピルである場合は、次の反応がイソプロパノール溶媒中で便利に行なわれるため、特に有利である。水酸化物(例えば水酸化カリウム)などの塩基の存在下におけるエステル2のフェノール化合物3による置換反応によって、α-(フェノキシ)フェニル酢酸エステル4を得た。α-(フェノキシ)フェニル酢酸エステル4の加水分解が、α-(フェノキシ)フェニル酢酸Iを生じた。
【0060】
このようにして、ヘプタンからの結晶化に続いて、(4-クロロフェニル)-(3-トリフルオロメチルフェノキシ)-酢酸、すなわちCPTAを、中間に単離のない5ステップで、収率約85%で調製することができる。
【0061】
V. エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸の有用性
エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸化合物は、米国特許第3,517,050号に開示されたα-(フェノキシ)フェニル酢酸化合物を含む様々な薬学的に活性な化合物を調製する際に有用な中間物である。したがって、本発明の別の局面は、式:
(式中、R1はアルキルまたはハロアルキル、Xはハライド、およびR7はヘテロアルキル、好ましくはN-アセチル2-アミノエチル(すなわち、式:-CH2CH2NHC(=O)CH3部分)である)で示されるα-(フェノキシ)フェニル酢酸化合物を、式Iのα-(フェノキシ)フェニル酢酸化合物のラセミ混合物からエナンチオ選択的に生成する方法を提供する。本方法は、式Iのα-(フェノキシ)フェニル酢酸化合物のラセミ混合物を上述のように分割する工程、およびエナンチオマー濃縮したα-(フェノキシ)フェニル酢酸をカルボン酸活性化試薬と反応させることによりエナンチオマー濃縮した活性化α-(フェノキシ)フェニル酢酸を生産する工程、を含む。適当なカルボン酸活性化試薬には、チオニルハライド(例えば、チオニルクロリド)、無水物、チオエステルを生成する試薬、および当業者に公知の他のカルボン酸活性化試薬が含まれる。
【0062】
次に活性化α-(フェノキシ)フェニル酢酸は式(R7-O)wMの化合物、例えばN-アセチルエタノールアミン誘導体と反応させて、エナンチオマー濃縮した式IIIのα-(フェノキシ)酢酸フェニル化合物を生成し、式中、R7は上に定義した通りで、Mは水素、または例えばNa、K、Li、Ca、Mg、Csなどの金属、および添字wはMの酸化状態である。本発明者らは、有意なラセミ化なしに、活性化された酸と式(R7-O)wMの化合物の反応を行なうことができることを発見した。
【0063】
本発明のさらなる目的、利点および新規な特徴が、本発明の次の実施例を検討することによって当業者に明白になるであろうが、実施例により制限するものではない。
【0064】
実施例
試薬と実験装置
もし他の記載がなければ、試薬および溶媒は Aldrich ChemicalまたはFisher Scientificから購入した。またN-アセチルエタノールアミンをLancaster Synthesisから得た。ラセミ体のCPTA、すなわちハロフェン酸を、米国特許第3,517,050号および第6,262,118号(そのすべてが参照により全体を本明細書に組み入れられる)に開示された製法により調製した。(1R,2R)-(-)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオール(すなわちCAF D塩基)はTCI Americasから得た。
【0065】
操作は陽圧窒素雰囲気下で行った。再循環加熱および冷却システムに付属したCamileプロセス制御コンピュータを用いて、ジャケットで覆った直壁底部排水管ガラス反応器のジャケット温度を調節した。特に表記しない限り、溶媒をBuchi回転蒸発装置を用い、15〜25torr、浴槽温度40℃以下で除去した。固体試料を真空オーブン中40℃、15〜25torrで乾燥させた。Cenco HYVAC真空ポンプを、真空蒸留用に1torr未満の真空を供給するために用いた。水位を、カールフィッシャー解析によりMetrohm 756 KF電量計およびHYDRANAL Coulomat AG試薬を用いて測定した。融点を、Mettler Toledo FP62融点測定装置を用いて測定した。pHを、較正されたOrion Model 290A pHメーターを用いて測定した。プロトンおよび13CのNMRスペクトルを、Bruker Avance 300 MHzスペクトロメーターで記録した。
【0066】
キラルHPLC解析を、λ=240nmで、可動相に溶解した10μlの試料を(R,R)WHELK-O 1.5μm 250×4.6mmカラム (Regis Technologies) に注入し、95/5/0.4 (v/v/v) のヘキサン/2-プロパノール/酢酸により、1.0 ml/分の流速で溶出して実施した。CPTA/CAF D塩基ジアステレオマー塩の固体試料については、固体を塩酸水溶液に加えて、CPTAをメチレンクロリド中へ抽出し、メチレンクロリド層から溶媒を除去した後に、残留物を解析のために可動相に溶解した。
【0067】
非キラルHPLC解析を、λ=220nmで、可動相に溶解した5μlの試料をPhenomenex LUNA 5μm C18(2) 250×4.6 mmカラムに注入して、25℃で実施した。1.5 ml/分の流量で、66体積%の水/34体積%のアセトニトリル/0.1体積%のトリフルオロ酢酸からスタートし、20分で26体積%の水/74体積%のアセトニトリル/0.1体積%のトリフルオロ酢酸に直線的に増加する勾配を用いた。
【0068】
ハロフェナートなどのエステルの酸性溶液の解析には、アセトニトリルを注入用溶媒として用いた。測定に際しては、CPTAおよびハロフェナートの生成物濃度をHPLC分析により、外部標準法および非キラル解析手法を用い、2.5 mg/ml未満の試料濃度で評価した。
【0069】
実施例1
以前のCPTAの分割は米国特許第3,517,050号に報告されたものであり、そこでキラル塩基としてシンコニジンを用い、CPTAの(+)-エナンチオマーがジアステレオマー塩として沈殿した。この手法の主な短所の1つは、所望の(-)-エナンチオマーが母液に残り、純粋な(-)-エナンチオマー画分の分離を困難にしたことであった。
【0070】
本実施例は、様々な異なるキラル塩基を用いてCPTAのラセミ混合物を分割し、エナンチオマー濃縮した固体の(-)-異性体を得た結果を示す。以前の方法と異なり、本発明の方法によって、エナンチオマー濃縮した固体の(-)-CPTAを容易に溶液から分離することができる。
【0071】
ラセミ体CPTAを、ラセミ体ハロフェナートの水酸化カリウム加水分解により調製した。キラル塩基による選別をするために、CPTAとキラル塩基の等モル混合物をガラスバイアルに入れたエタノール、メタノールおよびアセトン中で混合し、溶液を静穏に保った。終夜周囲温度に保持した後、溶液中に残存している試料を5℃で冷蔵庫中に置いた。冷蔵庫に終夜保持した後、エタノール溶液中に残存している試料に少量の水を加えた。周囲温度で4日間置いた後、エタノール水溶液を冷蔵庫に戻した。試料をすべて冷蔵庫に残し、沈殿形成を1か月間に亘って定期的に点検した。表1に、検討した塩基および溶媒条件、ならびに結晶の塩が見出された温度のリストを示す。
【0072】
(表1)CPTA分割のために検討した塩基
E - 評価した
C - 結晶化(温度)
* - 1モル/モル水酸化ナトリウム水溶液で
【0073】
4つのキラル塩基、キニーネ、L-チロシンヒドラジド、(-)-シンコニジン、および2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールの両方のエナンチオマーが、ラセミ体CPTAから結晶塩を生ずることが見出された。結晶した試料について、固体をろ過により分離し、固相と母液の両方をキラルHPLCにより解析し、双方の系統のエナンチオマー組成を測定した。表2にスクリーニングの結果を示す。表2に示す塩基のうちの3つが、固相中の(+)-エナンチオマー濃縮をもたらした。
【0074】
(表2)キラル塩基スクリーニングの結果
* -より希薄
**-より遅い冷却プロフィール
【0075】
表2には、固体と母液の系統中の異性体比率から計算した固体のパーセント収率が含まれている。用いた方程式を下に示す。100%の異性体純度を有する場合の最大の理論的収率は50%である。50%以上の収率は、他の異性体が含まれることを示す。
異性体比率から収率を計算する方程式。
設定する: a=出発物質中の成分1の面積%; b=出発物質中の成分2の面積%; x=単離された物質中の成分1の面積%; y=単離された物質中の成分2の面積%; w=母液中の成分1の面積%; z=母液中の成分2の面積%; E=単離された物質のグラム数; F=母液中の物質のグラム数
また:a + b=100%; E + F=1
そして:xE + wF=a; yE + zF =b
解くと:xE + w(1-E)= a; yE + z(1-E) =b
E = 単離された物質の収率 = (a-w)/(x-w) = (b-z)/(y-z)
【0076】
実施例2
本実施例は、エタノールおよび2-プロパノール中でCPTAをCAF D塩基を用いて分割した結果を示す。
【0077】
下の表3にエタノールおよび2-プロパノールの結果を要約する。この評価のためにスラリーを冷却プロフィール中の様々なポイントでサンプリングし、固体と溶液の両相のエナンチオマー組成を測定した。この情報から、固相の%eeおよび期待される重量パーセント収率(100%eeで最大50%収率)が異性体比率から計算され、決定された。表3には(-)-CPTAの収率が含まれ、それは重量パーセント収率および固相の(-)-CPTA含量から導出される(100%eeで最大100%の収率)。
【0078】
この特定の研究では、エタノール中における最良結果は、CPTA 1モル当たりCAF D塩基1モルを用いた。(-)-CPTA CAF D塩基塩のおよそ72%の収率を、固相中の(-)-CPTA塩の87.6%eeと共に、両相のキラル組成から計算した。2-プロパノール中で同様の濃度でCAF D塩基1モル当量を使用すると、より低い分割度となった。CPTA 1モル当たりCAF D塩基を0.55モル用いた時に、より高いエナンチオマー濃縮を達成した。これらの条件下で、(-)-CPTA CAF D塩基塩のおよそ76〜79%の収率を、固相中の(-)-CPTAの87〜90%eeと共に、相組成から計算した。物理的な損失を考慮に入れていない計算された重量パーセント収率は41〜42%だったが、実際に秤量した単離収率は37〜39%であった。
【0079】
(表3)CAF D塩基を用いたCPTAの分割
【0080】
2-プロパノールからのCPTA CAF D塩基塩の再結晶化により、光学純度が、およそ87%eeから98%eeに増加し、87%の質量回収率、または供給した(-)-CPTA含量に基づくと93%の回収率であった(表4)。
【0081】
(表4)(-)-CPTA CAF D塩基の2-プロパノールからの再結晶化
【0082】
全体として、ラセミ体CPTAから、最大50%の内のおよそ35%の収率で、およそ98%eeの光学純度を有する(-)-CPTA CAF D塩基塩を得た。
【0083】
光学的に濃縮されたエナンチオマーの結晶化によって、しばしばキラル純度が増加した。分割剤の除去に続いて、メチルシクロヘキサノンから(-)-CPTAを結晶化させることも、ある程度光学純度を増加させることになる。ある実験では、(+)-CPTAの結晶化により光学純度が99.1%eeから100%eeに増加した;母液は95%eeであった。
【0084】
実施例3
本実施例は、2-プロパノール中における、CPTAの(+)-および(-)-異性体のCAF D塩基塩の溶解度プロフィールを示す。
【0085】
CAF D塩基を用いたCPTA分割の最適化を補助するために、2-プロパノール中における両方のジアステレオマー塩の溶解度プロフィールを測定した。結果を図1に示す。(+)-CPTA CAF D塩基塩はシンコニジンで分割した(+)-CPTAを用いて調製した。図1に示すように、所望の(-)-CPTAジアステレオマーは(+)-CPTA形よりおよそ3倍溶解度が低い。図に含まれる溶解度を記述する式を、最小二乗法により計算した(R2>0.99)。(-)-CPTA塩に対する82℃のデータ点は方程式の決定に含まれていなかったが、計算された溶解度によく一致している。
【0086】
望ましくないCPTAエナンチオマーのラセミ化物を工程に戻し入れてリサイクルすることができた。このようにエナンチオマー濃縮されたCPTAの望ましくない異性体を1Nの水酸化ナトリウム水溶液中で還流させながら加熱すると、1時間未満でそれがラセミ化されることを見出した。他のどのような副産物も、単離したCPTAのHPLC解析によって検出されなかった。
【0087】
実施例4
本実施例は、(+)-CPTAを得る方法を示す。
【0088】
オーバーヘッドスターラーを備えた2L丸底フラスコに、33.0 gの粗(+)-CPTA-シンコニジン塩、610 mlのエタノールおよび125 mlのメタノールを入れた。スラリーを加熱還流して溶液とし、次に冷却した。非常に厚いスラリーが42℃で形成された。スラリーを68℃に加熱して軽いスラリーとし、次に周囲温度まで放冷した。その混合物を26℃でろ過し、150 mlのエタノールですすぎ、40℃真空下で乾燥させた後、23.48 gの(+)-CPTA-シンコニジン塩を得た。600 mlのエタノールおよび120 mlのメタノールで再結晶手順を繰り返し、18.23 gの(+)-CPTA-シンコニジン塩(2回の結晶化から55%回収率)を得た。分離度が良くないために低レベルの評価は不可能であったけれども(ハロフェナートのキラル解析条件をこの時にCPTAの解析にも用いた)、キラルクロマトグラフィーによっては、(-)-CPTAは検出されなかった。
【0089】
3.61 gの精製した塩試料を水50 mlおよびトルエン50 mlと混合し、2.9 gの硫酸を加えた。有機相を30 mlの水で洗い、次に蒸発させて残留物を得た。残留物を20 mlのシクロヘキサンから結晶化させて、1.22 gの(+)-CPTAを得た。あるいは、6.3 gの(+)-CPTA-シンコニジン塩(10.2 mmol)を、56 gのジエチルエーテルおよび29gの水と混合し、硫酸の滴加によりpH1.9に酸性化した。有機相を25 mlの水で洗い、乾燥させ(硫酸マグネシウム)、ろ過して蒸発させ、残留物とした。残留物を22 mlのメチルシクロヘキサンと周囲温度で攪拌し、スラリーを形成させた。スラリーを40℃に温め、次に氷浴で冷却し、固体をろ過によって分離して、40℃真空下で乾燥させた後に、2.62 gの(+)-CPTA(7.92 mmol、収率78%)を得た。
【0090】
実施例5
本実施例は、(+)-CPTAから(+)-ハロフェナートを合成する方法を示す。
【0091】
25 ml丸底フラスコに0.91gの(+)-CPTAおよび2.6 gのチオニルクロリドを入れ、混合物を加熱還流し、溶液とした。メタノールによって試料をクエンチし、生成物をHPLCによって解析して、酸塩化物への変換をモニターした。酸塩化物溶液に、4.8 gのジエチルエーテルを加え、そしてこの溶液を、氷浴で冷却した0.37 gのピリジンを含む12 mlのN,N-ジメチルホルムアミド(DMF)中の2.0 gのN-アセチルエタノールアミンに加えた。その結果の溶液を、25 mlの水のおよび30 mlのジエチルエーテルに加えた。有機相を分離し、25 mlの水で洗浄し、乾燥させ(MgS04)、ろ過し、溶媒を除去した後に0.92 gの油状物を得た。HPLC解析は、45面積%のハロフェナートおよび50面積%のCPTAを示した。キラルHPLC解析によって、ハロフェナートは(+)-エナンチオマーが99.78%eeであることが示された。
【0092】
実施例6
本実施例は、ラセミ体CPTAを調製する方法を示す。
【0093】
オーバーヘッドスターラーを備えた2L丸底フラスコに、102.7 gのハロフェナート、500 mlの水、および16.3 gの2-プロパノールを入れた。スラリーを撹拌し、32.3 gの45%水酸化カリウム水溶液を加えた。1時間の加熱還流後、溶液を周囲温度に冷却し、380 mlのヘキサンを加えた。24.57 gの37%塩酸で、pHを12.5から2に調節した。三相混合物を60℃に加熱して二相とした。下部の水相を取り出し、50 mlのヘキサンで抽出した。合わせた有機層を加熱して大気圧で蒸留し、100 mlの濁った蒸留物を除いた。溶液を30℃に冷却しCPTAの種を入れた。スラリーが生じた。スラリーを氷浴で冷却し、固体をろ過により単離して64.0 g(収率78.4%)のラセミ体CPTA、すなわち(4-クロロフェニル)(3-トリフルオロ-メチルフェノキシ)酢酸を得た。
【0094】
実施例7
本実施例は、様々なキラル塩基を用いて、エタノール中でキラル分割スクリーニングを行った代表的結果を示す。
【0095】
1.16 g(3.51 mmol)のCPTAの試料を6.98 gのエタノールに溶解し、溶液(0.431 mmol/g)を得た。ガラスバイアルに個別に、表5に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTAのエタノール溶液を加えた。ある場合には、CPTA溶液を加える前に少量のエタノールを加えて塩基を湿らせた。バイアルを終夜周囲温度に置いた。バイアル7Gおよび7Iが沈殿を生じた。各上清液の試料を取り出し、キラルHPLC解析により解析した。固体をろ過して分離し、解析もした。結果のいくつかを表2に示す(上記実施例1を参照)。残りのバイアルを5℃の冷蔵庫に置いた。1日後に7Eが沈殿を生じた。前述のように試料を解析した。残りのバイアルに、50μlの水を加え、周囲温度に3日間保持した後冷蔵庫に置いた。1か月後にさらに沈殿を生じていたものは無かった。
【0096】
(表5)エタノール中における塩基スクリーニング
【0097】
実施例8
本実施例は、様々なキラル塩基を用いて、アセトン中でキラル分割スクリーニングを行った代表的結果を示す。
【0098】
1.67gのCPTAの試料を7.57 gのHPLC級アセトンに溶解し、溶液とした。ガラスバイアルに個別に、表6に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTA溶液を加えた。ある場合には、少量のアセトンを加え、混合物を約40℃に温めて溶液とした。さらに、バイアル16Mに0.300 mlの1Nの水酸化ナトリウムを加えた。バイアルを終夜周囲温度に置いた。バイアル16Dが沈殿を形成し、上述のように解析した。表2中に結果のいくつかがまとめられている(実施例1を参照)。残りのバイアルを冷蔵庫に置いた。バイアル16Nが沈殿を形成し、解析した。バイアル16Gがわずかに沈殿を形成した。1週間後に、バイアル16Lが沈殿を生じたことが分かった。前に示したように試料を解析した。さらなる沈殿は認められなかった。
【0099】
(表6)アセトン中の塩基スクリーニング
【0100】
実施例9
本実施例は、様々なキラル塩基を用いて、メタノール中でキラル分割スクリーニングを行った代表的結果を示す。
【0101】
2.00 gのCPTAの試料を8.03 gのHPLC級メタノールに溶解し、溶液とした。ガラスバイアルに、個別に表7に示した量の各塩基を入れ、酸対塩基が1対1のモル比となるように計算した量のCPTA溶液を加えた。さらに、バイアル27Jに0.300 mlの1N水酸化ナトリウムを加えた。バイアルを、終夜周囲温度に置いた。バイアル27Bは固形化し、300μlのメタノールをさらに加えた後、上述のように試料を解析した。残りのバイアルを冷蔵庫に置いた。1か月後にさらなる沈殿は認められなかった。
【0102】
(表7)メタノール中の塩基スクリーニング
【0103】
実施例10
本実施例は、CPTAをキニーネによって分割した結果を示す。
【0104】
150 mlのジャケット付底部排液管フラスコに、2.70 g(8.17 mmol)のCPTA、2.65 g(8.17 mmol)のキニーネおよび50 mlの2-プロパノールを入れた。混合物を70℃に加熱して溶液とし、その後、0.2℃/分の速度で30℃に冷却し、2時間保持してスラリーを得た。試料のキラルHPLC解析により、固相中で(+)および(-)-CPTAがそれぞれ42.88および56.47面積%であり、また溶液中で(+)および(-)-CPTAがそれぞれ61.54および34.19面積%であることが示された。60℃にスラリーを加熱し、次に0.04℃/分の速度で30℃に冷却し、終夜保持してスラリーを得た。キラルHPLC解析によって、固相中で(+)および(-)-CPTAがそれぞれ29.94および44.19面積%であり、また溶液中で(+)および(-)-CPTAがそれぞれ77.54および20.88面積%であることが示された。スラリーを50 mlの2-プロパノールで希釈し、57℃に加熱して溶液とし、次に0.2℃/分の速度で30℃に冷却した。スラリーが1時間後に30℃で生じ始めた。その混合物を周囲温度で2日間攪拌し、次に固体をろ過により分離し、2-プロパノールですすぎ、真空下で乾燥させた後、2.89 g(質量収率54%)のCPTAキニーネ塩を得た。キラルHPLC解析によって、固相中で(+)および(-)-CPTAがそれぞれ42.25および57.75面積%であり、また母液中で(+)および(-)-CPTAがそれぞれ56.56および39.20面積%であることが明らかになった。表2に結果が含まれている(実施例1を参照)。
【0105】
実施例11
本実施例は、CPTAをCAF D塩基によって分割した結果を示す。
【0106】
150 mlの底部排液管フラスコに、19.54 gのCPTA、6.82 gのCAF D塩基(すなわち、D-threo-(-)-2-アミノ-1-(ニトロフェニル)-1,3-プロパンジオール)および80.2 gの2-プロパノールを入れた。混合物を70℃に加熱して溶液とし、その後、0.1℃/分の速度でジャケット温度5℃まで冷却した。混合物が62℃で混濁した。6℃に9時間保持した後に、固体をろ過により単離し、5 mlの2-プロパノールですすぎ、真空下40℃で乾燥させて、(-)-CPTA CAF D塩基塩12.03 g(収率37.4重量%)を得た。固体のキラルHPLC解析により、6.34面積%の(+)-CPTAおよび93.46面積%の(-)-CPTAが明らかになり;母液は、81.41面積%の(+)-CPTAおよび17.76面積%の(-)-CPTAを含んでいた。
【0107】
実施例12
本実施例は、(-)-CPTA CAF D塩基塩の再結晶の結果を示す。
【0108】
150 mlの底部排液管フラスコに、8.00 gの(-)-CPTA CAF D塩基塩(上記実施例11による)および54.2 gの2-プロパノールを入れた。混合物を加熱還流して溶液とし、その後、0.1℃/分の速度でジャケット温度20℃まで冷却し、内部温度を22℃で6時間保持した。固体をろ過して単離し、2-プロパノールですすぎ、真空下40℃で乾燥させて6.93 g(回収率86.6重量%)の(-)-CPTA CAF D塩基塩(融点184〜185℃)を得た。固体は0.995面積%の(+)-CPTAおよび99.01面積%の(-)-CPTAを含み;母液は44.53面積%の(+)-CPTAおよび54.47面積%の(-)-CPTAを含んでいた。反応容器をアセトンで洗い出した。アセトンを蒸発させ、0.27 g(3.4重量%)の残留物を得た。
【0109】
実施例13
本実施例は、(+)-CPTA CAF D塩基塩を調製する方法を示す。
【0110】
1 Lのフラスコに10.94 g(17.5 mmol)の(+)-CPTAシンコニジン塩、200 mlの水および100 mlのメチレンクロリドを入れた。1.8 gの硫酸を加えてpHを1.9に調節した。有機層を100 mlずつの希硫酸水溶液で3回洗浄し、乾燥(硫酸マグネシウム)させ、ろ過し、蒸発させて5.79 gの残留物を得た。残留物を22.2 gの2-プロパノールに溶解し、3.5 gのCAF D塩基を加えた。得られたスラリーを加熱還流して溶液とし、次に周囲温度に冷却し、スラリーを3時間撹拌した。氷浴で冷却した後に、固体を真空ろ過により分離し、5 mlの2-プロパノールですすぎ、真空下40℃で乾燥させて、7.39 g(収率80%)の(+)-CPTA CAF D塩基塩(融点172〜173℃)を得た。
【0111】
実施例14
本実施例は、2-プロパノール中のジアステレオマーCPTA-CAF D塩基塩の溶解度を示す。
【0112】
(-)-CPTA CAF D塩基および(+)-CPTA CAF D塩基(>98%ee)の試料を、表8中に示す量の2-プロパノールに加え、超音波浴を用いて混合した。全試料はスラリーのままであった。表に記した温度でスラリーを終夜保持し、次に上清液の試料を取り出し、定量的HPLC解析によって解析してCPTA濃度を決定した。結果を表および図1に示す。さらに、8.00 gの(-)-CPTA CAF D塩基塩を82℃で溶液(14.7重量%)とするために54.2 gの2-プロパノールが必要であった。このデータ点は図1には含めたが、溶解度方程式には含めなかった。
【0113】
(表8)2-プロパノール中の溶解度
【0114】
実施例15
本実施例は、エナンチオマー濃縮されたCPTAをラセミ化する方法を示す。
【0115】
50 ml丸底フラスコに、0.31 gの(-)-CPTA(68.7%ee)および9.4 gの1N水酸化ナトリウムを入れた。溶液を1時間加熱還流し、その後周囲温度に冷却し1 gの37%塩酸で酸性化した。CPTAをメチレンクロリドへ抽出し、溶媒を蒸発させ、0.46gの油状物を得た。HPLC解析によって、99.4面積%のCPTAが見出され、またキラルHPLC解析によって、CPTAエナンチオマーの50/50混合物が見出された。
【0116】
実施例16
本実施例は、様々な結晶化条件下において、CAF D塩基を用いてCPTAのラセミ混合物を分割する方法を示す。
【0117】
一般的な結晶化手順は、CPTA、CAF D塩基、および2-プロパノールを室温で加え、約75℃で加熱して溶液とすることであった。溶液を約60℃に冷却し、核形成が起るまで保持した。いくつかのバッチに、(-)-塩(すなわち(-)-CPTAおよびCAF D塩基の塩)を種として入れ、核形成を誘起した。スラリーを約1時間に亘って成長させた後、容器を単離温度に冷却した。図2中の最初の5つの実験では、約0.05〜0.10℃/分の遅い冷却速度を用いて単離温度に到達させた。他の実験では、0.25〜0.40℃/分のより速い冷却速度を用いた。ファイバー光学プローブを結晶化装置に直接挿入してスラリー密度を測定する。
【0118】
加えたCAF D塩基の量および溶質濃度は、最終的なバッチ組成をもたらす重要な変数である。(+)-塩(すなわち、(+)-CPTAおよびCAF D塩基の塩)が様々な時間過飽和状態であり続ける傾向が、いくつかの実験における変動の主な原因であると考えられる。図2中の実験5でこれが例証され、この場合、スラリーを8時間13℃で保持して、高純度結晶(99.7%(-)-塩)が生成された。3時間後に、ファイバー光学プローブの信号の増加が、(+)-塩の可能性が高い核形成を示した。さらに27時間の後、スラリーを単離し、結晶生成物は83.3/16.7%の(-/+)-CPTA比率であった。HPLCによる結晶生成物の解析によって、(-)-CPTAおよび(+)-CPTAの比率が与えられる。溶液中のフリーCPTAは飽和濃度以下であるので、結晶の解析はジアステレオマー塩比率を与える。母液は、溶解した塩およびフリーCPTAの両方を含む。HPLC解析は、各エナンチオマーを合計した量をCPTAとして報告する。同様に、図2の実験6が、スラリーを20時間1℃で保持して、高純度塩(>98%の(-)-CPTA)が生成されたことを示している。17℃に加熱した後に(+)-塩が核形成され、より低品質の生成物を得た[(-/+)-CPTA=81.2/18.8%]。
【0119】
他の試行では、図2の実験2、8および10におけるように、(+)-塩の核形成がより急速に生じた。結晶化は、(+)-塩の飽和温度の近くで、好ましくは飽和温度のすぐ上で単離を行うことができることが望ましい。
【0120】
CPTA 1グラム当りおよびCAF D塩基の0.45当量で、3.9 gの2-プロパノールを加えた場合、室温における単離は、(+)-塩の飽和レベル(または準安定域内)に非常に近いように見える。図2中の実験12は、0.43当量の塩基で開始され、(+)-塩の種を入れた後でさえ、21℃における結晶生成物は純粋なままであった(>99% (-)-塩)。CAF D塩基を加えて0.45当量とした後に、スラリーを14時間保持し、その後(+)-塩の種を入れた後にさらに6時間保持した。結晶生成物を解析すると、98.7%の(-)-CPTA比率であった。全塩基を0.47当量に増加すると、(+)-塩成分が ゆっくり増加して(-/+)-CPTA=92.3/7.7%の結晶生成物が得られた。
【0121】
図2の実験11(CPTA 1グラム当り3.9gの2-プロパノール、0.45当量の塩基)では、14時間後に高純度の(-)-塩(99.1%)が維持されていたが、しかし、さらに塩基を追加して0.48当量とすると、生成物比率が(-/+)-塩=89.2/10.8%となった。図2の実験9(0.45当量の塩基)では、22℃16時間後に99.5%の(-)-塩純度が維持されていた。これらの条件下の3つのバッチから計算された(-)-CPTAの収率は、70.7〜71.6%であった。計算収率は、結晶中および母液中の(-)-CPTAおよび(+)-CPTAの組成を知ることにより、ラセミ体CPTA原料を用いたことによる強制的な物質収支から導出される。
【0122】
このようにCPTA 1グラム当り約0.45当量のCAF D塩基および約4 gの2-プロパノールを加えることが、高純度(-)-塩(>98.5%)生成物をもたらし、それ以上の再結晶をしないで使用することができる。
【0123】
実施例17
本実施例は、CPTA塩の分割/結晶化について記述するためのモデルを提供する。
【0124】
フリーのCPTAの濃度は、加える塩基の量および溶媒の量に依存する。例えば、4.0グラムの2-プロパノールおよび0.50当量のCAF D塩基を加えることによるCPTAの分割においては、11%のフリーCPTAを含む2-プロパノール中において塩形成が行われる結果になる。この溶媒は、(-)-塩および(+)-塩の両方に対してより大きな溶解度を有し、溶解度を図3に示すように測定した。図3はさらに純粋な2-プロパノール中の溶解度データを含んでおり、それは2-プロパノール1グラム当りの成分グラム数で表現されている。図3が示すように、それぞれの塩の曲線は類似の形である。
【0125】
CPTA、CAF D塩基および2-プロパノールの配合の他の組合せによっても、図4に示すように、2-プロパノール中11.0%のフリーCPTAとなる系に到達できる。図4が示すように、図2の様々な実験の配合では、通常正確にこの直線に乗ることはなかった。しかし(-)-塩および(+)-塩の溶解度を以下のようにして推定することができる:「11.0%のフリーCPTA」直線より上の点となる配合は、より稀薄(すなわち2-プロパノール中で<11%のフリーCPTA)となり、「11.0%」直線より低い溶解度を示す。反対に、「11.0%」直線より下の点は、>11.0%のフリーCPTAを含む溶媒となり、塩の溶解度は図3で決定された溶解度より大きい。成分の溶解度を推定するために、定乗数kを用いた。(-)-塩および(+)-塩に対する修正溶解度方程式はしたがって、S(-) = 0.01421ke0.02613T および S(+)= 0.02868ke0.02771Tである。
【0126】
kを調節して(-)-塩および(+)-塩の溶解度の良い推定を行っても、他の未知数が分割剤塩基の添加によって形成される(-)-塩と(+)-塩の比率であるので、結晶化を記述することができない。図5に、より詳細な実験の1つを示す(図2も参照のこと)。この実験では、0.75当量の塩基を用い、21.5℃で試料を採取したときに、66.4/33.6%の(-/+)-塩比率の生成物が得られた。スラリーを加熱し、試料を採取し続けることによって、溶媒中の(-)-塩および(+)-塩の両方に対する飽和直線を追跡することができる。
【0127】
溶解度モデルを実際のデータに一致させるために、回帰技法を用い、測定値と一致した解答(すなわち結晶組成、母液組成および結晶の収率)を得るように、溶解度因子kならびに(-)-塩および(+)-塩の供給比率を操作した。k=0.68および0.75当量の塩の供給比率を58.1%(-)-塩/41.9%(+)-塩(すなわち0.436当量の(-)-塩および0.314当量の(+)-塩がCAF D塩基の添加によって形成された)に選ぶことによって、よい一致を得た。図6に比較を示す。溶解度モデルによって、単離のための完全な物質収支:結晶中の(-)-塩および(+)-塩の量、母液中の(-)-塩および(+)-塩の量、およびさらに母液中の(-)-フリーCPTAおよび(+)-フリーCPTAの量の計算が可能になる。溶解度差を用いた、抽出精密測定(extractive work-up)による母液中の(-/+)-塩および(-/+)-フリーCPTAを定量化するための一方法を下記の実施例19において提供する。
【0128】
溶解度モデルについての回帰技法を種々の量の分割剤を加えた他の実験に対して適用した。溶解度因子k、および材料としての塩の組成(すなわち、塩基の添加で形成された(-)塩および(+)塩の比率)の組合せを用いることにより、モデルは実験結果に適合する一意的な解に到達した。これらから、図7のグラフを構成した。この結果は、より多くの分割剤(外挿した最小点0.34当量より多く)を加えると、(+)-塩の形成量が増加することを示す。いくつかの実施態様では、0.34当量未満を加えると、CAF D塩基は実質的に(-)-CPTAのみと整合し、ほとんど排他的に(-)-塩を形成すると、いかなる理論にもとらわれずに信じられている。さらに、図7の曲線を用いて、(-)-CPTAおよび(+)-CPTA(遊離酸)の量を計算することができる。0.35〜0.75当量の間の塩基を添加した場合の、{(-)-CPTA/総CPTA遊離酸}の%比率は約25%(23.3〜27.1%)である。形成される(-/+)-塩の比率の「選択性」は、このようにして(溶液中に)残存するフリーの(-)-CPTAの量に依存し、それは約(-)-CPTA/(+)-CPTA=1/3である終点に至る。約0.34当量の塩基を添加することによって、一旦(-)-CPTA濃度が減少して(-/+)-CPTA比率が1/3となると、塩基の継続的な追加は(溶液中においてフリーの(-/+)-CPTAを一定の1/3の比率に保持するために)(-/+)-塩を1/3の比率で形成すると信じられている。
【0129】
実施例18
本実施例は、CPTAのラセミ混合物の分割を示す。
【0130】
200 ml容器に17.0 gのCPTA(51.4 mmol)、4.91gのCAF D塩基(23.1 mmol、0.450当量)および85 mlの2-プロパノールを入れた。その混合物を78℃で加熱して溶液とし、次に、0.5℃/分で54℃まで冷却した。約1/2時間後に、その溶液に(-)-塩の種を入れ、核形成を引き起した。54℃に約1・1/2時間保持した後、スラリーを0.25℃/分で22℃に冷却した。14時間22℃で保持した後に、小量の試料(約5 ml)を取り出して、15 mlの中型フリット化漏斗で分離した。母液の重量を計り保存し、固体を2 mlの2-プロパノールで洗浄した。洗浄液の重量を計って保存し、吸引を続けて結晶を乾燥させた。標準HPLCシステムでの解析によって、(-)-CPTAおよび(+)-CPTAの各処理系統における重量%の計算が可能になった。この試料についての物質収支(結晶、母液および洗浄液中のCPTAの総量が0.85 gとなるべき)から、総CPTAから単離した結晶生成物の収率31.9%が得られた。結晶の純度は重量で、99.1/0.9% = (-/+)-CPTA比率であった。図8の四角の囲み中に分析結果および物質収支の結果を示す。円内に、原材料/母液/結晶の組成に基づいて計算された収率(CPTAからの)を示す。図8中の略語は以下のとおりである: R.A.= 分割試薬、xまたはxtal = 結晶、ML = 母液、Yld = 収率。
【0131】
容器に(+)-塩を含む結晶の種を数回入れ、約2時間後に0.31 gのCAF D塩基(1.46 mmol、約0.03当量)を加えた。容器から試料を2回採取した後、60 mlの中型フリット化漏斗上に最終的に単離した(図8を参照)。母液は、透明の淡イエローゴールドで、59.1gであった。固体を19.2 gの2-プロパノールで洗浄し、18.8 gの洗浄溶液を回収した。洗浄した固体(10.07 g)をさらに1時間ろうと上で吸引して乾燥し、8.36 g(15.4 mmolの塩)を得た。最終単離からすべての系統を解析して、13.45g(40.67 mmol)のCPTAが計算された。単離した収率(-)-CPTA=33.8%(CPTAから)に対して、最終の結晶生成物の比率は、(-/+)-CPTA=89.2/10.8%であった。原材料、母液および結晶の組成に基づいて計算された(-)-CPTAの収率は、35.0%であった。
【0132】
実施例19
本実施例は、母液中の(-/+)-塩および(-/+)-CPTAを定量化するための抽出精密測定法を示す。
【0133】
(-/+)-塩の80/20混合物は、メチレンクロリドに約0.016%がようやく可溶であり、一方、ラセミ体CPTAは相当程度溶解し、少し3.4%に足らなかった。図2の実験4において55.3℃で分離した最終母液(図2および図5を参照)0.1286gを蒸発させて0.0242 gのガラス状残留物にすることにより解析した。残留物を5 mlのメチレンクロリドに溶解し、(-/+)-塩 = 80/20の種を入れて、終夜放置した。上清液の大部分を取り出し、3 mlのメチレンクロリドを加え、また液体の大部分を取り出して最初の抽出液と合わせた。メチレンクロリド抽出物を蒸発させ、ガラス状の固体0.0074 g得て、次にHPLCにより解析した。残りの濃いスラリーを蒸発させて0.0162 gとし、HPLCにより解析した。抽出精密測定法の結果は、図9に示すように、概して溶解度モデルによって予測される組成に類似している。
【0134】
実施例20
本実施例は、(-)-および(+)-CPTA・CAF D塩基塩の、CPTAを含むアルコール中における溶解度を示す。
【0135】
2.40 gのラセミ体CPTAを19.42 gの2-プロパノール(Fisher、HPLC級)に、または4.90 gのラセミ体CPTAを31.4 gのエタノールに溶解することにより「溶媒」を調製した。溶液中のCPTAの濃度はそれぞれ11.0%および13.5%であった。(-)-CPTA・CAF D塩基塩(すなわち(-)-塩)または(+)-CPTA・CAF D塩基塩(すなわち(+)-塩)の溶解度を重量法により決定した。与えられた温度で、上清液の一部を飽和溶液から重量が分かっているバイアルへ取り出した。溶液重量を測定し、窒素を吹き込んで揮発性溶媒を蒸発させた。固体が一定重量になるまで、約50℃/1 mm Hgの真空オーブン中でさらに乾燥させた。再度バイアルの重量を計り、揮発性溶媒の消失および残存固体の重量を測定した。これにより、「溶媒」から溶けていたCPTAの量を計算することができた。CPTAから全固形物の重量を引くことによって、溶媒中の可溶塩の重量が得られた。図10Aおよび10B中にデータを示す。
【0136】
実施例21
本実施例は、エナンチオマー濃縮された(-)-ハロフェナートを調製する方法を示す。
【0137】
CPTAを5工程で、上に述べたように、中間の単離なしで、ヘプタンからの結晶化により約85%の収率で調製した。分割によって、>98%光学純度の(-)-CPTAジアステレオマー塩が平均32%の収率(最大50%)で得られた。分割剤を除去した後に、(-)-CPTAをチオニルクロリドおよびN-アセチルエタノールアミンを用いてエステル化し、約55%の収率で(-)-ハロフェナートを得た。水酸化ナトリウム水溶液で母液残留物を加水分解して、(-)-CPTAを最終生産物母液から回収することができ、工程の再循環に戻すことができる。分割剤を、水からpH調整により約90%回収率で単離した。水酸化ナトリウム水溶液を用いた(+)-CPTAの回収とラセミ化により、約90%の回収率が得られた。全体として、4-クロロフェニル酢酸からの初回通過の収率は15〜17%だった。8工程の全工程で、3種の有機溶媒および3回の固体単離工程を用いた。
【0138】
実施例22
本実施例は、CPTAの調製方法を示す。
【0139】
CPTAの合成ルートの略図を上に示す。酸塩化物1を1,2-ジクロロエタン中で臭素化して2を得、続いて2-プロパノールを加えて、イソプロピルエステル3を得た。α,α,α-トリフルオロ-m-クレゾールによる置換反応を、2-プロパノール中で水酸化カリウムを用いて実施した。水によるクエンチと、洗浄および1,2-ジクロロエタンの除去に続いて、液体3をα,α,α-トリフルオロ-m-クレゾールおよび水酸化カリウムの2-プロパノール溶液に加え、4を得た。2-プロパノール溶媒を除去し、水酸化ナトリウム水溶液と加熱することにより加水分解が完了し、CPTAを得た。
【0140】
CPTAのナトリウム塩を、単に反応液を冷やすことにより固体として単離することができる。しかし、カルボン酸を単離することによってより良好な単離収率を得た。単離のために、塩基性のCPTA反応混合水溶液を塩酸で酸性化し、CPTAを1,2-ジクロロエタン中へ抽出した。分離した有機相の溶媒を1,2-ジクロロエタンからヘプタンへ交換することによって、CPTAが白色固体としておよそ85%の収率で4-クロロフェニル酢酸から得られた。
【0141】
実施例23
本実施例は、CPTAの1,2-ジクロロエタンとヘプタン中の溶解度を示す。
【0142】
ラセミ体CPTAの1,2-ジクロロエタンおよびヘプタン中の溶解度を、図11および12にそれぞれ示す。図には、データに最小二乗適合させた方程式が含まれる。
【0143】
図11の溶解度プロフィールに基づいて、1,2-ジクロロエタン中のおよそ25重量%CPTAの濃度を温度およそ35℃で、CPTAの抽出条件として選んだ。
【0144】
ヘプタンからのCPTA結晶化は発熱性であった。およそ170 gのCPTAの500 mlヘプタン溶液に46℃で種を入れると、結晶化が進行すると共に、54℃へ温度が上昇した。結晶化は、HPLC解析によって決定されるCPTA純度を、93〜95面積%から>99面積%まで増加させた。結晶化母液のHPLC分析(それは15面積%のCPTAを含んでいた)によって、母液への収率損失は3%より少ないことがわかった。結晶化によって純度が改善するとともに、単離収率は高く、母液への損失は小さかった。
【0145】
実施例24
本実施例は、様々な結晶化条件下のCPTA分割の収率を示す。
【0146】
様々な結晶化条件下におけるCAF D塩基を用いたCPTA分割の結果を図13に示す。0回、1回または2回の再結晶後に得られた各標品の最終のキラル純度を、太字で示す。CAF D塩基のモル比を0.5〜0.56まで変えた。結晶化および再結晶化のために表に示した2-プロパノール溶媒の量は共にラセミ体CPTAの最初の添加量に基づいている。単離した固体および母液の両方のキラルHPLCの結果を100%に標準化してある。計算収率および通算収率は、単離した固体および母液中の(+)-エナンチオマー型および(-)-エナンチオマー型の比率から計算する。最後の行の実際のパーセント収率は、計量した乾燥した物質のものであり、50%の最大収率に基づいている。
【0147】
>98%の光学純度のジアステレオマー塩の通算収率は、28〜35%の範囲であり、平均32%であった。1つの場合では、分割剤の最低の比率を用い、再結晶なしでこれが得られた(図13の実験2)。最初に単離した固体のキラル純度は73〜98%の範囲であった。1回の再結晶が一般に所望の光学純度を得るのに十分だった。高い通算収率が、母液が(-)-CPTAの(+)-CPTAに対する比率が20/80に達した時に得られた。
【0148】
図14は、(-)-CPTAの収率が減少する順に並べた分割結晶化のための冷却プロフィールを示す。図14の実験番号は図13の実験番号に対応している。(-)-CPTAの単離収率は、図13の計算収率および単離した物質中の(-)-CPTAのパーセントを用いて決定した。一般に、より低温のより長い保持時間が収率の増加をもたらした。
【0149】
0.45モル当量のCAF D塩基の使用により一貫して、再結晶の必要なしに、>98%の光学純度の物質の35〜37%の収率が得られた。
【0150】
実施例25
本実施例は、(-)-CPTAをCAF D塩基から分離する方法を示す。
【0151】
(-)-CPTAをCAF D塩基から分離するために、ジアステレオマー塩を1,2-ジクロロエタンと混合し、塩酸水溶液を加えて水相のpHを約2未満にした。CAF D塩基の塩酸塩を含む水相を分離した。有機相の水洗浄の後に、1,2-ジクロロエタンの大部分を蒸留によって除去し、残留水を取り除いた。完全な溶剤除去によって油状物が得られた。
【0152】
実施例26
本実施例は、有意なラセミ化なしに、(-)-CPTAをエステル化する方法を示す。
【0153】
(-)-CPTAを1,2-ジクロロエタン中で還流してチオニルクロリドと反応させ、対応する酸塩化物を生成した。反応の進行をHPLC解析によってモニターできる。少量の蒸留物を除去して、過剰のチオニルクロリドを除いた。混合物を冷却し、大過剰の真空蒸留したN-アセチルエタノールアミンを加えた。周囲温度で攪拌し、(-)-ハロフェナートを得た。
【0154】
反応混合物のエステル化を、反応混合物を炭酸カリウム水溶液に加えることによりクエンチした。6:1のヘプタン:2-プロパノールへ溶媒を交換し、そこから結晶化することによって(-)-ハロフェナートを単離した。結果を図15に要約する。
【0155】
第1の収穫の単離収率は、47〜59%の範囲であり、平均55%であった。この単離収率は、この工程の75〜80%の反応収率を表わす。第2の収穫はより高い通算収率をもたらした;しかしながら、第2の収穫は製品品質がより劣っていた。
【0156】
単離したハロフェナートおよび母液中のハロフェナートおよびCPTAとして見出された、加えたCPTAのモルアカウンタビリティーは90〜99%の範囲であった。
【0157】
実施例27
本実施例は、(+)-CPTAを回収して再循環させる方法を示す。
【0158】
塩基水溶液中でCPTAを加熱するとラセミ化が起った。実施例25の分割工程で残ったCPTAはおよそ47%eeの(+)-エナンチオマーであり、またそれはさらに残りのCAF D塩基を含んでいる。
【0159】
(+)-CPTAを回収しラセミ化するために、2-プロパノール溶媒を除去し、1,2-ジクロロエタンと交換した。pH約2未満の水で洗浄して、後に続く回収のためにCAF-D塩基を除去した。水酸化ナトリウム水溶液を加え、水溶液を加熱還流した。1,2-ジクロロエタンを、塩基溶液の添加に先立つ蒸留、または塩基溶液の添加に続く相分離によって除去した。1.4モル当量の水酸化ナトリウムと水溶液を4時間加熱した後に、ヘプタンから89%の収率のラセミ体CPTAを単離した。結晶化した中間物としてCPTAを単離することにより、分割工程に対してより一貫した品質の供給が可能になった。
【0160】
酸型として測定し表示した水中のラセミ体CPTAナトリウム塩の溶解度を図16に示す。単離したナトリウム塩を水に加えると、約9.5のpHとなり、上の溶解度曲線で示される溶解度プロフィールが得られた。少量の水酸化ナトリウムの追加によって約12.6のpHとすると、下部の曲線によって示されるように水への溶解度が減少した。
【0161】
実施例28
本実施例は、(+)-ハロフェナートからCPTAを生成する方法を示す。
【0162】
1〜3モル当量の水酸化ナトリウムを水中の約10重量%の87%ee(+)-ハロフェナートへ加えて50〜60℃に加熱すると、実質的に完全なCPTAの加水分解が起こった。部分的ラセミ化が生じ、およそ70%eeの(+)-CPTAを生じた(図17の、時間=0)。溶液を加熱還流し、エナンチオマー比率を時間を追ってモニターした。3モル当量の塩基により、ほとんど完全なラセミ化が(キラルHPLC分析法により<3%ee)2時間未満の還流で生じた。ラセミ化の間にpHが12.8から12.6まで低下した。2モル当量(pH12.6から11.6へ)ではわずかに長い反応時間が必要であった。1モル当量では、ラセミ化はおよそ60〜70%eeで止まり、最終pHは9.4となった。
【0163】
0.5モル当量の水酸化ナトリウムを用いると、およそ40%のハロフェナートが60℃で2時間後に加水分解されずに残り;終夜加熱還流すると、およそ1%のハロフェナートが残り、最終pHは4.8であった。これは有意にラセミ化を最小化しなかった。生成されたCPTAの量は72.6%eeの(+)-エナンチオマーであった。
【0164】
実施例29
本実施例は、(-)-ハロフェナート結晶化母液から(-)-CPTAを回収する方法を示す。
【0165】
以前に述べ、また図15に示したように、(-)-ハロフェナート結晶化母液は大量の(-)-ハロフェナートおよび(-)-CPTAを含む。(-)-ハロフェナートの加水分解により、さらに(-)-CPTAを分割工程の材料として生成することができる。
【0166】
(-)-ハロフェナート結晶化母液(88.3%eeの(-)-ハロフェナート)の50℃および最終pH12.7の加水分解により、急速に65.8%eeの(-)-CPTAが得られた。(-)-CPTAを、CAF D塩基を2-プロパノール溶液へ加えることによりCAF D塩基ジアステレオマー塩(96.4%ee)として回収した。最初に加えたジアステレオマー塩の量から55 mol%を(-)-ハロフェナートとして得、28%を(-)-CPTA/CAF D塩基塩として回収し、また14 mol%が母液中にCPTAとして残った。
【0167】
実施例30
本実施例は、CAF D塩基を回収する方法を示す。
【0168】
CAF D塩基は、ジアステレオマー塩から(-)-CPTAを分離した酸性相中に、および分割母液からのCPTA回収の酸性洗浄工程から得られた酸性相中に見出される。水酸化ナトリウム水溶液で約12を越えるpHへ塩基性化することにより、容易にろ過される形で良好に回収できる沈殿となった。図18に結果を示す。ジアステレオマー塩からの回収率は一般に90%を越えたが;分割母液からの回収率はより低かった。水溶液中の濃度は約5〜20%の範囲であった。
【0169】
CAF D塩基のエナンチオマー純度を、DSCによる融点の注意深い解析により決定することができる(D. Pitre, M. Nebuloni, and V. Ferri; Arch. Pharm. (Weinheim) 324, 525 (1991))。(+)-と(-)-形の混晶、例えばラセミ化合物は、純粋なエナンチオマーより20℃以上低い温度で溶融するので、融点がエナンチオマー純度を評価するための敏感な方法であることが分かった。しかし、誘導体のクロマトグラフ分離による2つの試料のエナンチオマー純度を測定しても、キラル純度の低下は示されなかった。回収されたCAF D塩基のエナンチオマー純度は、HPLC分析法の検出限界の近くで、原料物質と判別することが不可能であった。
【0170】
実施例31
本実施例は、ラセミ体CPTAを調製する別の方法を示す。
【0171】
加熱マントル中にありオーバーヘッドスターラーおよび冷却器を備えたが500 ml丸底フラスコに、73.28 g(0.430 mol)の4-クロロフェニル酢酸、70 mlの1,2-ジクロロエタンおよび41 ml(0.56 mol)のチオニルクロリドを入れた。混合物を50〜55℃で19時間加熱した。反応混合物をHPLC解析により解析した。酸塩化物の溶液に29 ml(0.57 mol)の臭素を加え、溶液を70〜75℃で20時間加熱した。生じたα-ブロモ生成物を氷浴で冷却し、100 ml(1.31 mol)の2-プロパノールを滴下して加えた。達した最高温度は17℃であった。4℃に冷却した後に、反応混合物を水に加えた。溶液を周囲温度に温め、水層を除去した。有機相を37 mlの水で洗浄した。分離された1,2-ジクロロエタン溶液を蒸発させ、134.1 gの油状物を得た。
【0172】
オーバーヘッドスターラーを備えた1 Lの丸底フラスコに、34.0 g(0.515 mol)の85%水酸化カリウムおよび370 mlの2-プロパノールを入れた。その混合物を41℃に水浴を用いて温め、固体の大部分を溶解させた。その混合物を氷浴で冷却し、73.8 g(0.455 mol)のα,α,α-トリフルオロ-m-クレゾールを滴下して加えた。最高温度は13℃に達した。溶液を5℃に冷却した後、上で得られた134.1 gの油状物を滴下して加えた。材料を18 gの2-プロパノールですすいだ。スラリーを蒸発させて残留物とし、次に250 mlの水および42.8 g(0.535 mol)の50%水酸化ナトリウム水溶液を加えた。混合物を1時間加熱還流した。
【0173】
周囲温度に冷却した後に、混合物を250 mlの1,2-ジクロロエタンで希釈し、71 g(0.72 mol)の37%塩酸の滴下による添加によりpHを0.3まで下げた。相分離の後、溶媒を1,2-ジクロロエタン相から除去し、202.2 gの残留物を得た。残留物を131 gのヘプタンで処理し、蒸発させて164 gの残留物を得た。97 gのヘプタンでこの手順を繰り返し、160 gの油状物を得た。残留した油状物を周囲温度で257 gのヘプタンと攪拌してスラリーを得、これを氷浴で冷却した後、ろ過して固体を単離した。ろ過ケーキを49 gのヘプタンで洗浄し、次に真空下で乾燥させ、125.58 g(0.380 mol、収率88%)のCPTAを得た。
【0174】
実施例32
本実施例は、実施例22の化合物4のラセミ混合物を調製する方法を示す。
【0175】
マグネティックスターラーと還流冷却器を備えた50 ml丸底フラスコに、2.10 g(6.35 mmol)のラセミ体CPTA、21 gの2-プロパノールおよび0.50 g(4.2 mmol)のチオニルクロリドを入れた。90分還流後のHPLC解析によって、84.2面積%の7および12.7面積%のCPTAが明らかになった。さらに1.0 g(8.4 mmol)のチオニルクロリドを加えると、CPTAが1面積%未満となった。溶液を周囲温度に冷却し、1.0 g(12 mmol)の固体の炭酸水素ナトリウムで処理した。溶媒を蒸発させ、残留物を25 mlのトルエンに溶解した。水(2×10 ml)で洗浄した後、溶媒を蒸発させ、残留物として実施例22の化合物4を2.31 g(6.2 mmol、収率98%)を得た(95.8面積%の7と2.4面積%のトルエン)。
【0176】
実施例33
本実施例は、ラセミ体CPTAの溶解度を測定する方法を示す。
【0177】
マグネティックスターラーを備えた100 mlのウォータジャケット付き樹脂ポットを再循環水浴に接続し、9.44 gのラセミ体CPTAおよび16.78 gの1,2-ジクロロエタンを入れた。浴温を35℃に温め、スラリーを1時間撹拌した。撹拌器を止め、固体を30分沈殿させた。0.1360 gの上清試料を取り出しアセトニトリルで25.00 mlに希釈し、溶液をHPLC解析によって分析した。この結果および一連の他の測定の結果を図11および19に示す。約2℃で解析するために、1.92 gの1,2-ジクロロエタン中の0.54 gのCPTA試料を終夜冷蔵庫に格納した後、上清液をHPLC解析により分析した。図12で示される図19に含まれるヘプタン中のCPTAの溶解度を同様の方法で測定した。
【0178】
実施例34
本実施例は、CPTAのラセミ混合物を分割する方法を示す。
【0179】
1 Lの底部排液管反応器に、48.2 g(146 mmol)のCPTA、16.4 g(77.3 mmol)の(1R,2R)-(-)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオール(CAF D-塩基)および193 gの2-プロパノールを入れた。スラリーを70℃に加熱して溶液とし、次に60℃に冷却し、1時間保持した。生じたスラリーを0.25℃/分でジャケット温度2℃へ冷却し、14時間保持した;内部温度は4℃であった。固体を真空ろ過によって分離し、27 gの2-プロパノールですすいだ。母液および洗浄溶液をHPLC解析のために採取し、その結果を図13に示す。50.48 gのウエットケーキを193 gの2-プロパノールと共に1 Lの反応器に再び入れ、スラリーを85℃のジャケット温度で温めて穏やかに還流して溶液とした。溶液をHPLC解析のために採取した;図13に結果を一覧にして示す。65℃に冷却することによってスラリーが形成された。68℃に30分間温めた後に、スラリーを0.25℃/分で40℃に、次に0.4℃/分で18℃に、その後1℃/分で2℃に冷却した。(他の調製では、図14に記録した直線的な冷却速度を用いた)。固体を真空ろ過によって分離し、18 gの2-プロパノールですすぎ、真空下で乾燥させて27.29 g(50.4 mmol、収率34.5%)の(-)-CPTA/CAF D塩基を得た。図13に、単離した固体および母液、および洗浄液のHPLC解析の結果が含まれる。
【0180】
実施例35
本実施例は、ハロフェナートからのラセミ体CPTAの調製および分割を示す。
【0181】
オーバーヘッドスターラー付きの1 L丸底フラスコに、129.75 g(0.312 mol)のラセミ体ハロフェナート、325 gの水および32.6 g(0.408 mol)の50%水酸化ナトリウム水溶液を入れた。スラリーを60℃に1時間加熱して溶液とし、次に冷却した。温度40℃で、328.5 gの1,2-ジクロロエタンおよび44 g(0.45 mol)の37%塩酸を加え、二相混合物を29℃に冷却した。水相のpHは0.85であった。有機相を分離し水250 mlで洗浄し、次に蒸発させ118.2 gの残留物を得た。2-プロパノール(149 g)を加えて蒸発させ、131.2 gの残留物とした。加えたハロフェナートの量に基づいて理論上103.2 gのラセミ体CPTAを含む残留物を、1 L底部排液管反応器に、33.10 g(0.1556 mol)のCAF D塩基および400 gの2-プロパノールと共に入れた。混合物を67℃に温めて、軽いスラリーを生成させ、次に0.075℃/分で1℃まで冷却した。混合物を-7℃に冷やし、固体を真空ろ過によって単離し、60 mlの2-プロパノールで洗浄した。単離した固体および492.8 gの母液および洗浄溶液のHPLC解析結果を図13(実験9)に示す。92.74 gのウエットケーキを再び1 L反応器に477 gの2-プロパノールと共に加え、混合物を75℃に加熱して溶液とした。溶液を0.5℃/分で5℃に冷却し、結晶化した固体を真空ろ過によって単離し、60 mlの2-プロパノールですすぎ、乾燥させて、51.81 g(0.0956 mol、収率31%)の(-)-CPTA CAF D塩基ジアステレオマー塩を得た。単離した固体、529.9 gの母液および洗浄溶液のHPLC解析の結果が図13に含まれる。
【0182】
実施例36
本実施例は、(+)-CPTAをラセミ化する方法およびラセミ体CPTAを回収する方法を示す。
【0183】
単離したジアステレオマー塩の収率および純度に基づいて71.6 g(0.217 mol)のCPTA(44%eeの(+)-エナンチオマー)を含む、上の実施例35に記載された103.2 gのCPTAの分割から得られた分割母液および再結晶化母液を蒸発させ、108.7 gの残留物を得た。残留物を176 gの1,2-ジクロロエタン、35.2 gの水および6.8 gの37%塩酸で処理した。有機相を取り出して蒸発させ、79.0 gの残留物とした。水(80 g)を加え、溶媒を蒸発させ78.1 gの残留物とした。残留物を141.9 gの水および24.6 g(0.308 mol)の50%水酸化ナトリウム水溶液で処理し、溶液を4時間加熱還流し、キラルHPLC解析によるラセミ化合物を得た。溶液を冷却し、140 mlの1,2-ジクロロエタンおよび32.0 g(0.325 mol)の37%塩酸で処理した。有機相を取り出して蒸発させて80.1 gの残留物とし、それを40℃水浴中、250 mlヘプタンで処理し、スラリーを得た。固体を真空ろ過により単離し、乾燥させて、63.83 g(0.193 mol、収率89%)のラセミ体CPTAを得た。試料の分割については、新鮮なCPTAの分割と一致する結果を得た(図13の実験10)。
【0184】
実施例37
本実施例は、ジアステレオマー塩から(-)-CPTAを分離する方法を示す。
【0185】
マグネティックスターラー付き500 mlフラスコに、40.0 g(73.7 mmol)の(-)-CPTA/CAF D塩基、100 gの1,2-ジクロロエタン、40 gの水および7.6 g(77 mmol)の37%塩酸を入れた。固体が完全に溶解した後、下部の有機相を取り出し、10 mlの水で洗浄した。合わせた水相のpHは0.9であった。128.2 gの有機相のHPLC解析により、1,2-ジクロロエタン溶液として24.32 gの(-)-CPTA(73.6 mmol、理論値の99.8%)が見出された。
【0186】
実施例38
本実施例は、N-アセチルエタノールアミンの真空精製を示す。
【0187】
マグネティックスターラー、加熱マントルおよび短経路蒸留ヘッドを装備した50 ml丸底フラスコに、29.09 gのN-アセチルエタノールアミンを入れ、およそ0.8 torrの真空下に置いた。凝縮物が集まることはなかったが、液体を加熱すると泡が形成された。およそ130℃のヘッド温度で蒸留物を集め、澄んだ液体として26.71 g(回収率92%)の N-アセチルエタノールアミンを得た。
【0188】
実施例39
本実施例は、(-)-ハロフェナートを生成する方法を示す。
【0189】
マグネティックスターラー付き500 ml丸底フラスコに、35.5 g(65.4 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩(99.4%ee)、89.0 gの1,2-ジクロロエタンおよび35.5 mlの水を入れた。スラリーに、6.7 g(68 mmol)の37%塩酸を加えて混合物を周囲温度で攪拌し、透明な2相を得た。下部の有機相を取り出し、7.0 gの水で洗浄した。有機相を蒸発させて26.13 gの残留物を得て、次に55.6 gの1,2-ジクロロエタンに溶解し、加熱マントル中のマグネティックスターラーおよび還流/蒸留ヘッド付き250 ml丸底フラスコに入れた。溶液のHPLC解析によって22.06 g(66.7 mmol、理論値の102%)のCPTAが存在することが分かった。溶液に7.5 ml(100 mmol)のチオニルクロリドを加え、溶液を2時間加熱還流した。加熱を続け、6.1 gの蒸留液を集めた。溶液を周囲温度に冷却し、次に氷浴で冷却して、蒸留した25.85 g(251 mmol)のN-アセチルエタノールアミン(KF解析で1176および1288 ppmの水)を加えた。添加後に温度が約26℃に上昇した。溶液を、氷浴中の36 gの水中の9.90 g(71.6 mmol)の炭酸カリウムに攪拌しながらゆっくり加えた。達した最高温度は15℃であった。反応混合液を5 mlの1,2-ジクロロエタンですすいだ。下部の有機相を取り出し、37 mlの水で洗浄した。溶液を蒸発させ、油状物(32.84 g)を得た。油状物を54 gのヘプタンで処理し、溶媒を除去して31.56 gの固形残留物を得た。固体に、76 gのヘプタンを加え、溶媒を除去して29.19 gの固形残留物を得た。固体を40℃で28 mlの2-プロパノールに溶解し、次にさらに、28 mlの20プロパノールおよび334 mlのヘプタンで希釈した。周囲温度に冷却すると薄いスラリーが得られた。氷浴で冷却すると厚いスラリーが形成された。2時間撹拌した後に、真空ろ過により固体を単離し、29 gのヘプタンですすぎ、乾燥させて、14.21 g(34.2 mmol、収率52.3%)の(-)-ハロフェナートを得た。キラルHPLC解析によって(+)-ハロフェナートは検出されなかった(>99.8%ee)。
【0190】
294.1 gの母液および洗浄液のHPLC解析によって、11.2 gのハロフェナートおよび1.26 gのCPTAが見出された。溶媒を蒸発させ、12.47 gの残留物を14 mlの2-プロパノールに溶解した。84 mlのヘプタンを加え、終夜周囲温度で撹拌した後にスラリーを得た。スラリーを氷浴で冷却し、固体を集めて、9 gのヘプタンですすぎ、乾燥させ、5.64 g(13.6 mmol、収率20.7%、HPLC解析による89.9%のハロフェナートおよび3.9%のCPTA、99.6%ee)の(-)-ハロフェナートを得た。81.74 gの母液および洗浄液のHPLC解析によって、3.66 g(8.8 mmol、13.5%)のハロフェナートおよび0.93 g(2.8 mmol、4.8%)のCPTAの存在が分かった。
【0191】
実施例40
本実施例は、ラセミ体CPTAナトリウム塩を単離する方法を示す。
【0192】
分割回収率に基づき理論上は63.9 g(0.193 mol)のCPTAを含む分割結晶化および再結晶化の母液を蒸発させ、91 gの残留物を得た。残留物を146 gの1,2-ジクロロエタンに溶解し、28.6 gの水および6.3 gの37%塩酸で40℃にて処理した。219 gの有機相を蒸発させ、71.86 gの残留物を得た。残留物に、120 gの水および21.5 g(0.269 mol)の50%水酸化ナトリウムを加えた。溶液を加熱還流し、次に周囲温度に放冷して厚いスラリーを得た。冷却によって生じた固体を真空ろ過によって単離し、25 mlの水ですすいで次に乾燥させ、31.78 g(0.0901 mol、回収率46.7%)のCPTAナトリウム塩を得た。キラルHPLC解析により、物質はラセミ体であることが分かった。188.6 gの母液および洗浄液のHPLC解析により28.3 g(0.0856 mol、44.4%)のCPTAが存在することが判明した。
【0193】
実施例41
本実施例は、ラセミ体CPTAナトリウム塩の溶解度を測定する方法を示す。
【0194】
マグネティックスターラーを備えた100 mlのウォータージャケット付き樹脂ポットを再循環水浴に接続し、3.48 gのラセミ体CPTAナトリウム塩および20.0 gの水を加えた。浴温を35℃に温め、スラリーを1時間撹拌した。撹拌器を止め、固体を30分間静置した。pHは9.4であった。0.3036 gの上清試料を取り出し、アセトニトリルで25.00 mlに希釈して、溶液をHPLC解析により分析した。解析を47℃、および19℃で繰り返した。さらに3.01 gのCPTAナトリウム塩を加えてスラリーを高温で維持し、25 gの水を加えて、より低温でより薄いスラリーを得た。50%の水酸化ナトリウム水溶液の添加により、周囲温度でpHを12.7に高め、13.5、25、34、および42℃で解析を続けた。結果を図16および20に示す。
【0195】
実施例42
本実施例は、(+)-ハロフェナートの加水分解およびラセミ化を示す。
【0196】
マグネティックスターラーと加熱マントルを備えた250 ml丸底フラスコに、7.28 g(17.5 mmol)の(+)-ハロフェナート(86.9%ee)、72.2 gの水および4.21 g(52.6 mmol)50%水酸化ナトリウム水溶液を入れた。スラリーを50〜60℃に加熱した。生じた溶液のpHは12.8であった。キラルHPLC解析により、80.4%の(+)-CPTAおよび10.5%の(-)-CPTAが示された。溶液を90分間加熱還流した。キラルHPLC解析が、49.6%の(+)-CPTAおよび47.0%の(-)-CPTAを示した。pHは12.6であった。周囲温度に冷却した後に、およそ50 mlの1,2-ジクロロエタンを加え、7.3 g(74 mmol)の37%塩酸を加えてpHを0.8に調節した。有機相を蒸発させ、6.0 gの残留物を得た。残留物を25 mlのヘプタンで処理し、温めて油状物を溶解し、次に氷浴で冷却した。固体を真空ろ過によって集め、乾燥させて5.10 g(15.4 mmol、収率88%)のラセミ体CPTAを得た。このデータおよび2つの同様の加水分解のデータを図17および21に示す。
【0197】
同様に、6.75 g(16.3 mmol)の(+)-ハロフェナートを、0.65 g(8.1 mmol)の50%水酸化ナトリウム水溶液と67.5 gの水と共に2時間60℃で熱することにより、37.5%のハロフェナートおよび54.2%のCPTAを得た。終夜加熱還流することにより92.1%のCPTAおよび1.1%のハロフェナートが得られ、最終pHは4.8であった。キラルHPLC解析によって、(+)/(-)-CPTAが80.3/12.8の比率であることが分かった。
【0198】
実施例43
本実施例は、(-)-ハロフェナート結晶化母液から(-)-CPTA/CAF D塩基ジアステレオマー塩を回収することによる(-)-ハロフェナートの調製を示す。
【0199】
マグネティックスターラー付き1 L丸底フラスコに、50.0 g(92.3 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩(97.1%ee)、124 gの1,2-ジクロロエタン、50 mlの水および9.6 g(98 mmol)の37%塩酸を入れた。有機相を分離して50 mlの水で洗浄し、次に加熱マントル中のマグネティックスターラー付き250 ml丸底フラスコに入れた。還流/蒸留ヘッドを結合し、溶液を加熱蒸留して35.4 gの蒸留液を除去した。40℃に冷却した後に、溶液を25 mlの1,2-ジクロロエタンで希釈し、11 ml(150 mmol)のチオニルクロリドを加えた。2時間加熱還流し、22.6 gの蒸留液を除去した後、溶液を氷浴で冷却して、38.6 g(374 mmol)の蒸留したN-アセチルエタノールアミンを滴下して加えた。添加中に反応温度が7℃から18℃まで上昇した。周囲温度で終夜撹拌した後、溶液を、氷浴で冷却した51 mlの水中の12.7 gの炭酸カリウムに撹拌しながら加えた。有機相を取り出し、51 gの水で洗浄した。有機相(HPLC解析により、85.2%ハロフェナートおよび6.1% CPTA)を蒸発させて44.3 gの油状物とし、133 gのヘプタンで処理し、次に蒸発させて43.3 gの固体にした。固形残留物を61.5 gの2-プロパノールに溶解し、320 gのヘプタンと共に1 Lの底部排液管反応器に入れ、50℃に温め、そして3℃/分で20℃まで、その後1℃/分で-3℃まで冷却した。溶液は27℃で濁り、15℃で厚いスラリーが形成された。固体を真空ろ過によって単離し、5 mlの2-プロパノールを含む40 mlのヘプタンで洗浄し、乾燥させて21.01 g(50.6 mmol、収率55%、HPLCにより98.93%)の(-)-ハロフェナート(99.9%ee)を得た。HPLC分析によれば14.65 g(35.3 mmol)のハロフェナート(88.3%ee)および1.78 g(5.4 mmol)のCPTAを含む395.7 gの母液および洗浄溶液を蒸発させ、21.57 gの残留物を得た。残留物を100 mlの水および5.0 g(63 mmol)の50%水酸化ナトリウム水溶液と共に50℃に加熱し、溶液とした。約10分後のHPLC解析により、83.6%のCPTAおよび0.3%のハロフェナートの存在が分かった。溶液を冷却して50 mlの1,2-ジクロロエタンで希釈し、7.3 g(74 mmol)の37%塩酸によってpHを12.7から1.6まで低下させた。30 mlの水で洗った後に、HPLC分析により11.32 g(34.2 mmol)のCPTAを含む72.9 gの有機相を蒸発させて残留物とし、36 gのヘプタンで処理し、次に蒸発させて14.9 gの残留物とした。油状の残留物を38 gのヘプタンに加熱して溶解した。冷却して油状物を得た。溶媒を除去し、残油を34.8 gのメチルシクロヘキサンに溶解した。冷却によって油状物が形成された。溶媒を除去し、45.6 gの2-プロパノールに交換した。キラルHPLC解析により、65.8%eeの(-)-CPTA(16.9/81.6の(+/-)-比率)であることが分かった。周囲温度の溶液に6.50 g(30.6 mmol)のCAF D塩基を加えた。厚いスラリーが急速に形成された。スラリーを撹拌しながら40℃に温め、その後氷浴で冷却して、固体を真空ろ過によって分離し、7 gの2-プロパノールで洗浄し、乾燥させて13.91 g(25.7 mmol)の(-)-CPTA/CAF D塩基ジアステレオマー塩を得、それは最初に加えた50.0 gの塩の28%の回収率に相当する。(+)/(-)-CPTA比率は1.77/97.86であった。45.34 gの母液および洗浄溶液のHPLC解析によって、4.34 g(13.1 mmol)のCPTAを見出し、それは最初に加えた50.0 gの塩の14 mol%に相当する。
【0200】
実施例44
本実施例は、CPTA/CAF D塩基塩からCAF D塩基を回収する方法を示す。
【0201】
マグネティックスターラー付き1 L丸底フラスコに80.16 g(0.148 mol)の(-)-CPTA/CAF D塩基塩、237 gの1,2-ジクロロエタンおよび80mlの水を入れた。スラリーに15.2 g(0.154 mol)の37%塩酸を加え、透明な2相を得た。水層のpHは1.2であった。下部の有機層を取り出し、16 mlの水で洗浄した。合併した水相(140.7 g)を12.9 g(0.161 mol)の50%水酸化ナトリウム水溶液で処理し、pHを12.1とした。生じたスラリーをろ過し、固体を25mlの水ですすぎ、乾燥させて30.79 g(0.145 mol、回収率98%)のCAF D塩基(融点160.4〜161.0℃)を得た。
【0202】
実施例45
本実施例は、分割母液からCAF D塩基を回収する方法を示す。
【0203】
60.0 gのラセミ体CPTA試料を、上述のように20.88 gのCAF D塩基によって240 gの2-プロパノール中で分割し、74.7 gのウエットケーキを得た。ウエットケーキを218 gの2-プロパノール中で再結晶し、32.35 g(収率32.8%)の(-)-CPTA/CAF D塩基塩を得た。得られた塩の量から理論上40.32 gのCPTAおよび8.23 g(38.8 mmol)のCAF D塩基を含む結晶化および再結晶化からの母液および洗浄溶液を蒸発させ、72.9 gの残留物とした。残留物を、265 gの1,2-ジクロロエタン、50mlの水および4.0 g(40.6 mmol)の37%塩酸に溶解した。水層を分離し、3.88 g(48.5 mmol)の50%水酸化ナトリウム水溶液を加えてpHを0.6から12.3に高めた。生じたスラリーをろ過し、固体を集めて、水ですすぎ、7.12 g(33.6 mmol、回収率87%)のCAF D塩基(融点162.4〜163.0℃)を得た。
【0204】
本明細書に記述された実施例と実施態様はただ説明目的のみのためであり、またそれらを考慮に入れた様々な修正または変更は当業者に示唆されており、本出願の精神と範囲および添付の請求項の範囲内に含まれることとなると理解される。本明細書に引用されたすべての出版物、特許および特許出願は、参照によりその全体をあらゆる目的のために本明細書に組み入れられる。
【図面の簡単な説明】
【0205】
【図1】(-)-および(+)-CPTA/CAF D-塩基塩の2-プロパノール中における溶解度プロフィールを示すグラフである。
【図2】CAF D塩基を用いたCPTAのラセミ混合物を分割するための方法の、様々な結晶化条件における結果を示す。
【図3】(-)-および(+)-CPTA/CAF D-塩基塩の、純粋なイソプロパノール中、およびイソプロパノールとCPTA(11%)の混合物を含む溶液中における溶解度を示すグラフである。
【図4】様々な量の各成分の混合物の組成を示すグラフである。
【図5】結晶化および加熱に対する(-/+)-塩の飽和プロフィールを示すグラフである。
【図6】図2の実験4におけるモデル予測と実験結果の比較を示す表である。
【図7】加えたCAF D塩基の量の関数として生成された(+)-塩の量を示すグラフである。
【図8】図2の実験11に示す分割の実験データのグラフ表示である。
【図9】母液中のCPTAの実際量と計算量、および(+)-CPTA塩の計算百分率と実験データとのグラフによる比較を示す。
【図10A】図7(すなわち、図2の実験13)の実験データおよび溶解度モデル計算を示す表である。
【図10B】図2の実験4の28.3℃における実験データと溶解度モデル計算を示す表である。
【図11】1,2-ジクロロエタン中の、様々な温度におけるラセミ体CPTAの溶解度を示すグラフである。
【図12】ヘプタン中の、様々な温度におけるラセミ体CPTAの溶解度を示すグラフである。
【図13】様々な結晶化条件下でCAF D-塩基を用いたCPTA分割の収率を示す実施例24の結果の表である。
【図14】図2の様々な実験における分割結晶化の冷却プロフィールを示す。
【図15】実施例26における(-)-CPTA塩からの(-)-ハロフェナートの収率を示す表である。
【図16】水中での様々な温度におけるラセミ体CPTAナトリウム塩の溶解度を示すグラフである。
【図17】(-)-ハロフェナートの加水分解中の様々なpHにおけるCPTAラセミ化プロフィールを示すグラフである。
【図18】実施例30記載の様々なpHにおけるCAF D-塩基回収の結果を示す表である。
【図19】実施例33において測定された1,2-ジクロロエタンおよびヘプタン中のラセミ体CPTAの溶解度測定の実験結果である。
【図20】実施例41において測定された水中のラセミ体CPTAナトリウム塩の溶解度測定の実験結果である。
【図21】実施例42において測定された(+)-ハロフェナートの塩基性加水分解の実験結果である。
【特許請求の範囲】
【請求項1】
エナンチオマー濃縮された式:
(式中、R1は、アルキルまたはハロアルキルであり、
Xは、ハライドである)
のα-(フェノキシ)フェニル酢酸化合物を、第1および第2エナンチオマーを含むα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物から生成するための方法であって、以下の工程を含む方法:
(a) α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物を、0.5モル当量未満のエナンチオマー濃縮されたキラルアミン化合物と、溶液中のフリー第1エナンチオマー量対フリー第2エナンチオマー量の比率を約1対3にするのに十分な条件下で接触させることにより、第1エナンチオマーの固体のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程;および、
(b) α-(フェノキシ)フェニル酢酸化合物の第2エナンチオマーの酸塩基塩の濃度がその飽和点の近くまたはその飽和点より低い温度で、溶液から第1エナンチオマーの固体の酸塩基塩を分離する工程。
【請求項2】
第1エナンチオマーの固体のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程(a)が:
(i) 溶液を第1エナンチオマーの核形成温度より上の温度に加熱する工程;および
(ii) 溶液温度を第1エナンチオマーの核形成温度以下に低下させて、第1エナンチオマーの固体の酸塩基塩を生成する工程
を含む、請求項1記載の方法。
【請求項3】
第1エナンチオマーの固体酸塩基塩を分離する工程(b)が、第2エナンチオマーの酸塩基塩の飽和温度の近くまたは飽和温度より上の温度で実施される、請求項2記載の方法。
【請求項4】
分離された第1エナンチオマーの固体酸塩基塩からキラルアミン化合物を取り出すことにより、キラルアミン化合物を回収する工程をさらに含む、請求項1記載の方法。
【請求項5】
工程(a)の酸塩基塩を生成する際に使用されるエナンチオマー濃縮されたキラルアミン化合物が、回収されたキラルアミン化合物を含む、請求項4記載の方法。
【請求項6】
第2エナンチオマーを塩基と接触させることにより、分離された溶液中の第2エナンチオマーの少なくとも一部をラセミ化させる工程をさらに含む、請求項1記載の方法。
【請求項7】
工程(a)中で用いられるα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物が、ラセミ化されたα-(フェノキシ)フェニル酢酸化合物を含む、請求項6記載の方法。
【請求項8】
キラルアミン化合物が、式:
(式中、
R2およびR3は、それぞれ独立して水素もしくはアルキルであるか;またはR2およびR3はそれらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素もしくはアルキル、またはR5またはR6のうちの1つがアミン保護基であり;および
Arは、アリールである)
で示される化合物である、請求項1記載の方法。
【請求項9】
4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物から、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸をエナンチオマー濃縮するための方法であって、以下の工程を含む方法:
(a) 4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物を、0.5モル当量未満のエナンチオマー濃縮された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールと、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸1グラム当り約4グラムのアルコール性溶媒中で接触させることにより、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程;
(b) (+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸が濃縮された溶液からエナンチオマー濃縮された酸塩基塩を分離する工程;および
(c) (1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールを酸塩基塩から除去して、エナンチオマー濃縮された(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸を生成する工程。
【請求項10】
アルコール性溶媒がイソプロパノールである、請求項9記載の方法。
【請求項11】
酸塩基塩を生成するために、約0.47モル当量以下の(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールを用いる、請求項10記載の方法。
【請求項12】
(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程(a)が、(-)-酸塩基塩の核形成温度以上の温度へ溶液混合物を加熱する工程を含む、請求項11記載の方法。
【請求項13】
エナンチオマー濃縮された酸塩基塩を分離する工程(b)を、(+)-エナンチオマーの酸塩基塩の飽和温度の近くまたは飽和温度より上の温度で実施する、請求項12記載の方法。
【請求項14】
エナンチオマー濃縮された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールが、工程(c)の酸塩基塩から取り出された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールの少なくとも一部を含む、請求項10記載の方法。
【請求項15】
工程(b)で得られた(+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸の少なくとも一部をラセミ化する工程をさらに含む、請求項10記載の方法。
【請求項16】
4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物が、ラセミ化された(+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸の少なくとも一部を含む、請求項15記載の方法。
【請求項17】
式:
で示されるα-(フェノキシ)フェニル酢酸化合物;および、
式:
で示されるキラルアミン化合物、
(式中、
R1は、アルキルまたはハロアルキルであり;
Xは、ハライドであり;
R2およびR3は、それぞれ独立して水素もしくはアルキルであるか;またはR2およびR3は、それらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素もしくはアルキルであるか、またはR5もしくはR6のうちの1つがアミン保護基であり;および
Arは、アリールである)
から誘導される酸塩基塩。
【請求項18】
α-(フェノキシ)フェニル酢酸化合物およびキラルアミン化合物がエナンチオマー濃縮されている、請求項17記載の酸塩基塩。
【請求項19】
α-(フェノキシ)フェニル酢酸化合物が(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸である、請求項18記載の酸塩基塩。
【請求項20】
キラルアミン化合物が(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールである、請求項18記載の酸塩基塩。
【請求項21】
少なくとも約95%のエナンチオマー過剰率を有するエナンチオマー濃縮された(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸。
【請求項22】
エナンチオ選択的に、式:
で示されるα-(フェノキシ)フェニル酢酸化合物を生成するための方法であって、以下の工程を含む方法:
(a) 式:
で示されるα-(フェノキシ)フェニル酢酸のラセミ混合物を生成する工程;
(b) 0.5モル当量未満のエナンチオマー濃縮されたキラルアミン化合物を用いてα-(フェノキシ)フェニル酢酸のラセミ混合物を分割し、エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸を生成する工程;
(c) エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸をカルボン酸活性化試薬と接触させることにより、エナンチオマー濃縮された活性化α-(フェノキシ)フェニル酢酸を生成する工程;および、
(d) エナンチオマー濃縮された活性化α-(フェノキシ)フェニル酢酸を式(R5-O)wMの化合物と接触させて、α-(フェノキシ)フェニル酢酸化合物を生成する工程
(式中、
R1は、アルキルまたはハロアルキルであり;
Xは、ハライドであり;
R7は、ヘテロアルキルであり;
Mは、水素または金属であり;および
下付添字wはMの酸化状態である)。
【請求項23】
α-(フェノキシ)フェニル酢酸化合物が(-)-ハロフェナートである、請求項22記載の方法。
【請求項1】
エナンチオマー濃縮された式:
(式中、R1は、アルキルまたはハロアルキルであり、
Xは、ハライドである)
のα-(フェノキシ)フェニル酢酸化合物を、第1および第2エナンチオマーを含むα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物から生成するための方法であって、以下の工程を含む方法:
(a) α-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物を、0.5モル当量未満のエナンチオマー濃縮されたキラルアミン化合物と、溶液中のフリー第1エナンチオマー量対フリー第2エナンチオマー量の比率を約1対3にするのに十分な条件下で接触させることにより、第1エナンチオマーの固体のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程;および、
(b) α-(フェノキシ)フェニル酢酸化合物の第2エナンチオマーの酸塩基塩の濃度がその飽和点の近くまたはその飽和点より低い温度で、溶液から第1エナンチオマーの固体の酸塩基塩を分離する工程。
【請求項2】
第1エナンチオマーの固体のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程(a)が:
(i) 溶液を第1エナンチオマーの核形成温度より上の温度に加熱する工程;および
(ii) 溶液温度を第1エナンチオマーの核形成温度以下に低下させて、第1エナンチオマーの固体の酸塩基塩を生成する工程
を含む、請求項1記載の方法。
【請求項3】
第1エナンチオマーの固体酸塩基塩を分離する工程(b)が、第2エナンチオマーの酸塩基塩の飽和温度の近くまたは飽和温度より上の温度で実施される、請求項2記載の方法。
【請求項4】
分離された第1エナンチオマーの固体酸塩基塩からキラルアミン化合物を取り出すことにより、キラルアミン化合物を回収する工程をさらに含む、請求項1記載の方法。
【請求項5】
工程(a)の酸塩基塩を生成する際に使用されるエナンチオマー濃縮されたキラルアミン化合物が、回収されたキラルアミン化合物を含む、請求項4記載の方法。
【請求項6】
第2エナンチオマーを塩基と接触させることにより、分離された溶液中の第2エナンチオマーの少なくとも一部をラセミ化させる工程をさらに含む、請求項1記載の方法。
【請求項7】
工程(a)中で用いられるα-(フェノキシ)フェニル酢酸化合物のエナンチオマー混合物が、ラセミ化されたα-(フェノキシ)フェニル酢酸化合物を含む、請求項6記載の方法。
【請求項8】
キラルアミン化合物が、式:
(式中、
R2およびR3は、それぞれ独立して水素もしくはアルキルであるか;またはR2およびR3はそれらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素もしくはアルキル、またはR5またはR6のうちの1つがアミン保護基であり;および
Arは、アリールである)
で示される化合物である、請求項1記載の方法。
【請求項9】
4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物から、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸をエナンチオマー濃縮するための方法であって、以下の工程を含む方法:
(a) 4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物を、0.5モル当量未満のエナンチオマー濃縮された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールと、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸1グラム当り約4グラムのアルコール性溶媒中で接触させることにより、(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程;
(b) (+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸が濃縮された溶液からエナンチオマー濃縮された酸塩基塩を分離する工程;および
(c) (1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールを酸塩基塩から除去して、エナンチオマー濃縮された(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸を生成する工程。
【請求項10】
アルコール性溶媒がイソプロパノールである、請求項9記載の方法。
【請求項11】
酸塩基塩を生成するために、約0.47モル当量以下の(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールを用いる、請求項10記載の方法。
【請求項12】
(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー濃縮された酸塩基塩を含む溶液を生成する工程(a)が、(-)-酸塩基塩の核形成温度以上の温度へ溶液混合物を加熱する工程を含む、請求項11記載の方法。
【請求項13】
エナンチオマー濃縮された酸塩基塩を分離する工程(b)を、(+)-エナンチオマーの酸塩基塩の飽和温度の近くまたは飽和温度より上の温度で実施する、請求項12記載の方法。
【請求項14】
エナンチオマー濃縮された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールが、工程(c)の酸塩基塩から取り出された(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールの少なくとも一部を含む、請求項10記載の方法。
【請求項15】
工程(b)で得られた(+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸の少なくとも一部をラセミ化する工程をさらに含む、請求項10記載の方法。
【請求項16】
4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸のエナンチオマー混合物が、ラセミ化された(+)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸の少なくとも一部を含む、請求項15記載の方法。
【請求項17】
式:
で示されるα-(フェノキシ)フェニル酢酸化合物;および、
式:
で示されるキラルアミン化合物、
(式中、
R1は、アルキルまたはハロアルキルであり;
Xは、ハライドであり;
R2およびR3は、それぞれ独立して水素もしくはアルキルであるか;またはR2およびR3は、それらが結合している原子と共に複素環部分を形成し;
R4は、水素またはアルキルであり;
R5およびR6は、それぞれ独立して水素もしくはアルキルであるか、またはR5もしくはR6のうちの1つがアミン保護基であり;および
Arは、アリールである)
から誘導される酸塩基塩。
【請求項18】
α-(フェノキシ)フェニル酢酸化合物およびキラルアミン化合物がエナンチオマー濃縮されている、請求項17記載の酸塩基塩。
【請求項19】
α-(フェノキシ)フェニル酢酸化合物が(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸である、請求項18記載の酸塩基塩。
【請求項20】
キラルアミン化合物が(1R,2R)-2-アミノ-1-(4-ニトロフェニル)-1,3-プロパンジオールである、請求項18記載の酸塩基塩。
【請求項21】
少なくとも約95%のエナンチオマー過剰率を有するエナンチオマー濃縮された(-)-4-クロロ-α-(3-トリフルオロメチルフェノキシ)フェニル酢酸。
【請求項22】
エナンチオ選択的に、式:
で示されるα-(フェノキシ)フェニル酢酸化合物を生成するための方法であって、以下の工程を含む方法:
(a) 式:
で示されるα-(フェノキシ)フェニル酢酸のラセミ混合物を生成する工程;
(b) 0.5モル当量未満のエナンチオマー濃縮されたキラルアミン化合物を用いてα-(フェノキシ)フェニル酢酸のラセミ混合物を分割し、エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸を生成する工程;
(c) エナンチオマー濃縮されたα-(フェノキシ)フェニル酢酸をカルボン酸活性化試薬と接触させることにより、エナンチオマー濃縮された活性化α-(フェノキシ)フェニル酢酸を生成する工程;および、
(d) エナンチオマー濃縮された活性化α-(フェノキシ)フェニル酢酸を式(R5-O)wMの化合物と接触させて、α-(フェノキシ)フェニル酢酸化合物を生成する工程
(式中、
R1は、アルキルまたはハロアルキルであり;
Xは、ハライドであり;
R7は、ヘテロアルキルであり;
Mは、水素または金属であり;および
下付添字wはMの酸化状態である)。
【請求項23】
α-(フェノキシ)フェニル酢酸化合物が(-)-ハロフェナートである、請求項22記載の方法。
【図1】

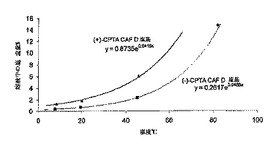
【図2】
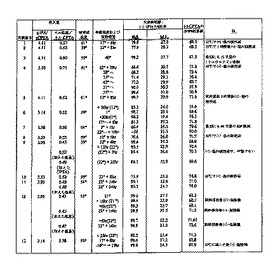
【図3】
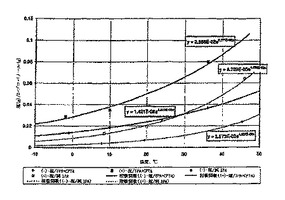
【図4】
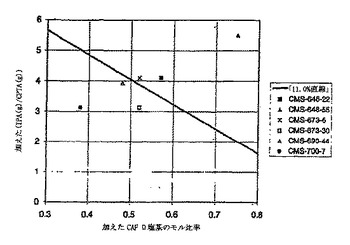
【図5】
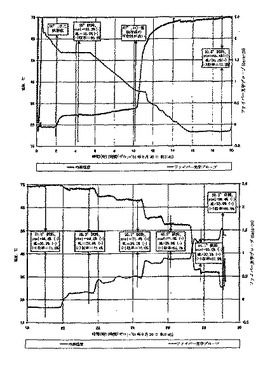
【図6】
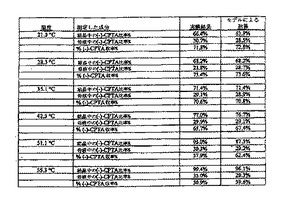
【図7】
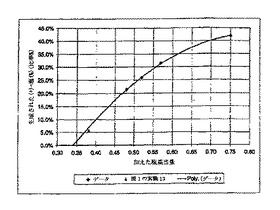
【図8】
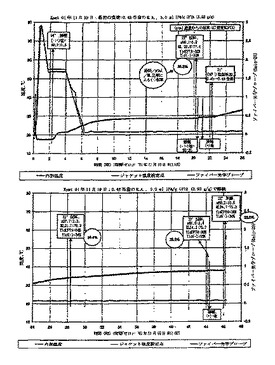
【図9】
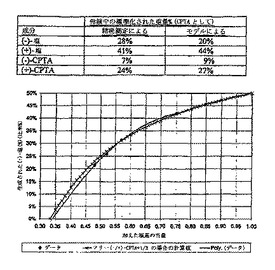
【図10A】
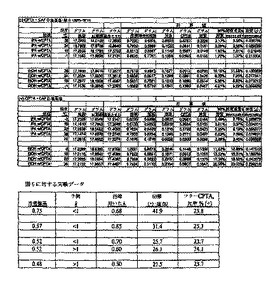
【図10B】

【図11】
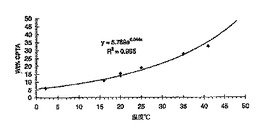
【図12】
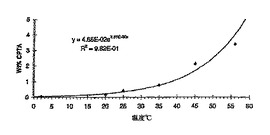
【図13】
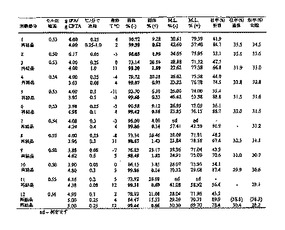
【図14】
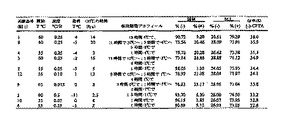
【図15】
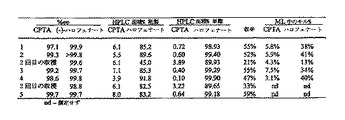
【図16】
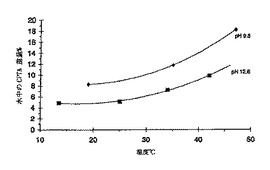
【図17】
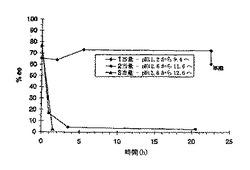
【図18】

【図19】
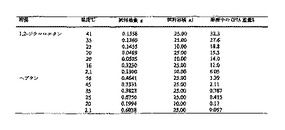
【図20】
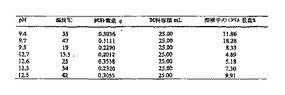
【図21】
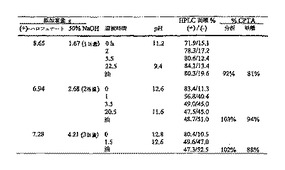
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【公表番号】特表2007−524614(P2007−524614A)
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願番号】特願2006−517439(P2006−517439)
【出願日】平成16年6月18日(2004.6.18)
【国際出願番号】PCT/US2004/019616
【国際公開番号】WO2004/112774
【国際公開日】平成16年12月29日(2004.12.29)
【出願人】(504214280)メタボレックス インコーポレーティッド (21)
【Fターム(参考)】
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願日】平成16年6月18日(2004.6.18)
【国際出願番号】PCT/US2004/019616
【国際公開番号】WO2004/112774
【国際公開日】平成16年12月29日(2004.12.29)
【出願人】(504214280)メタボレックス インコーポレーティッド (21)
【Fターム(参考)】
[ Back to top ]