
αウイルス構造タンパク質の発現のためのプロモーターレスカセット
本発明は、a)少なくとも1つの開始コドンが除去されているアルファウイルス5’複製認識配列と、b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、c)アルファウイルス3’複製認識配列とを含む単離されたRNA分子であって、当該RNA分子が、(b)のヌクレオチド配列の転写を誘導するプロモーターを含まず、(a)および(c)のアルファウイルス5’および3’複製認識配列が、アルファウイルス非構造タンパク質の存在下において、当該RNA分子の複製を誘導する、RNA分子を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
優先権の声明
この出願は、米国特許法§119(e)の下、2007年6月21日に出願された米国仮出願第60/936,637号(この全体の内容は、参考として本明細書に援用される)の利益を主張する。
【0002】
政府の支援の声明
本発明の局面は、国立衛生研究所(National Institutes of Health)からの助成金第5UO1A1057286−03の下での資金拠出によって援助された。米国政府は、本発明に特定の権利を有する。
【0003】
発明の分野
本発明は、組換えアルファウイルス粒子のための改良された構築物およびその製造方法に関する。
【背景技術】
【0004】
アルファウイルスは、現在、感染性疾患および癌のワクチンを開発するためのベクタープラットフォームとして用いられている(例えば、特許文献1;特許文献2;特許文献3;特許文献4;特許文献5;特許文献6;特許文献7;特許文献8;特許文献9;特許文献10;特許文献11;特許文献12;非特許文献1、非特許文献2;非特許文献3を参照のこと)。アルファウイルスは、Togaviridae科の属を含み、当該属のメンバーは、世界中の至る所で脊椎および無脊椎の双方の宿主において見出される。ベクタープラットフォームのためにアルファウイルスの中で最も研究されているのは、当該属のプロトタイプのメンバーであるVenezuelan Equine Encephalitis(VEE)Virus、Semliki Forest Virus(SFV)、およびSindbis Virus(SV)である。
【0005】
そのようなベクタープラットフォームの1つは、Garoffらの米国特許第6,190,666号、Johnstonらの特許文献1および特許文献2、Dubenskyらの米国特許第5,814,482,同第5,843,723号、特許文献10、特許文献11、同第6,105,686号、および同第6,376,236号、米国特許出願公開第2002−0015945号(Poloら)、米国特許出願公開第2001−0016199(Johnstonら)、Frolovら(1996)Proc.Natl.Acad.Sci.USA 93:11371−11377、並びに非特許文献1に記載されているアルファウイルスレプリコンシステムである。アルファウイルスレプリコンベクターは、対象となる1つ以上の核酸を含有しかつ発現するように操作され、この場合、対象となる核酸は、例えば、抗原、サイトカイン、リボザイム、または酵素をコードし得る。アルファウイルスレプリコンベクターは、任意のアルファウイルス由来であってもよく、中でも、例えばVenezuelan Equine Encephalitis(VEE)ウイルス、Sindbisウイルス、例えばTR339株、South African ArboウイルスNo.86、およびSemliki Forestウイルス由来であってもよい。当該ベクターは、次に培養中の細胞に導入され、それによってアルファウイルスの複製が可能になり、さらにこの細胞においてアルファウイルス構造タンパク質も発現され、その結果、当該ベクターは、アルファウイルス構造タンパク質によってアルファウイルスレプリコン粒子(ARP)中にパッケージされる。次いで、ARPが培養物から収穫され、様々な治療目的のために被験体へと提供される。
【0006】
ワクチン用途におけるARPシステムの免疫原性および有効性を高めるために、様々な構築物が開発されている。これらの構築物の多くは、さらに、ゲノム断片の組換えによって複製能力のあるアルファウイルスが形成される可能性を低減するように設計されている。Johnstonら(特許文献1および特許文献2)は、単一のヘルパーシステム(このシステムにおいて、アルファウイルスの構造タンパク質遺伝子の完全なセットが1つのRNA分子上にあり、非構造タンパク質遺伝子および対象となる非相同核酸は別のレプリコンRNA上にある)からの組換えの可能性を認識しており、したがって、構造タンパク質をコードする2つのヘルパーRNAを採用した「二重ヘルパー」システムを設計した。Dubenskyら(特許文献10)およびPoloら(米国特許第6,242,259号)は、パッケージング細胞の培養物中への複製性アルファウイルスベクターの導入に関してこれら構造タンパク質を発現するRNAの産生によってアルファウイルスベクターをパッケージするための、パッケージング細胞株に安定してトランスフォームされる2つのDNAアルファウイルス構造タンパク質発現カセットの使用について記載している。Liljestromおよび共同研究者は、「単一のヘルパーシステム」が、野生型アルファウイルス粒子を生成することを確認するデータを提示している(Berglund,et al. Biotechnology 11(8):916−920(1993))。スミスらは、構造タンパク質の発現を誘導する他の新規のRNAヘルパーについて記載している(国際公開第2004/085660号)。
【0007】
上記のように、3つのうちの2つがヘルパーシステムを含む3つの核酸間にウイルスのコード配列を分配することにより、単一ヘルパーシステムと比較して、複製能力のあるウイルス(「RCV(replication−competent virus)」を生じる組換えの理論的頻度が著しく減少する。これらのシステムは、非依存性転写単位として機能する構造物を提供するために、しばしば26Sプロモーターまたはウイルス接合領域プロモーターとも呼ばれるアルファウイルスサブゲノムプロモーターの使用、並びにアルファウイルスRNAポリメラーゼ認識シグナルの使用を含み、それによって、当該ヘルパーシステムは、ヘルパー機能の増幅および効率的な発現のためにアルファウイルス複製機構の存在を利用することができる。
【0008】
既存のシステムでは、既知のパッケージングシグナルは、通常はレプリコンRNAに含まれており、ヘルパー構築物からは除かれる。しかしながら、それでもなお、ヘルパーRNAは、低頻度ではあるがパッケージングまたは同時パッケージングされ(Lu and Silver.J.Virol Methods,91(1):59−65(2001))、末端認識シグナルを有するヘルパー構築物が、レプリコンの存在下において増幅および発現され、他のヘルパー分子もしくはレプリコンRNAの組換え現象を生じる可能性がある。
【0009】
アルファウイルスレプリコン粒子による動物研究では、105〜108の範囲の投与量が採用されており、非ヒト霊長動物においては107、5×107、および108が効果的に採用され、当該投与量は、ヒトの臨床試験において使用される。さらに、2×108、5×108、および109などの多量の投与量も、ヒトに対する適用において有用である。そのような投与量は、大規模の製造手段を必要とし、そのような規模では、統計的に、複製能力のあるアルファウイルスが既存のRNAヘルパーシステムによって生成され得る可能性がある。
【0010】
したがって、複製能力のあるアルファウイルスの形成に対する予測される頻度をさらに減少させ、並びに製造戦略およびコストを最適化するために、アルファウイルスレプリコン粒子を製造するための改善されたシステムが当技術分野において依然として必要とされている。
【0011】
本発明は、アルファウイルス構造タンパク質をコードするアルファウイルスRNAヘルパー分子であって、プロモーター配列を欠失しているためにヘルパー分子とレプリコンベクターとの間で生じ得る機能的組換え現象の理論上の回数を著しく減少させ、結果として、組換えアルファウイルス粒子の製造中に複製能力のあるアルファウイルスが形成される割合の理論的予測が減少する、アルファウイルスRNAヘルパー分子を提供する。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】米国特許第5,792,462号明細書
【特許文献2】米国特許第6,156,558号明細書
【特許文献3】米国特許第5,811,407号明細書
【特許文献4】米国特許第6,531,135号明細書
【特許文献5】米国特許第6,541,010号明細書
【特許文献6】米国特許第6,783,939号明細書
【特許文献7】米国特許第6,844,188号明細書
【特許文献8】米国特許第6,982,087号明細書
【特許文献9】米国特許第7,045,335号明細書
【特許文献10】米国特許第5,789,245号明細書
【特許文献11】米国特許第6,015,694号明細書
【特許文献12】米国特許第5,739,026号明細書
【非特許文献】
【0013】
【非特許文献1】Pushkoら、Virology(1997)239(2):389−401
【非特許文献2】Frolovら、J.Virol.(1997)71(1):248−258
【非特許文献3】SmerdouおよびLiljestrom,J.Virol.(1999)73(2):1092−1098
【発明の概要】
【課題を解決するための手段】
【0014】
本発明は、a)開始コドンが除去されているアルファウイルス5’複製認識配列と、b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、c)アルファウイルス3’複製認識配列とを含む単離されたRNA分子であって、当該RNA分子が、(b)のヌクレオチド配列の転写を誘導するプロモーターを含まず、アルファウイルス5’および3’複製認識配列がアルファウイルス非構造タンパク質の存在下において当該RNA分子全体の複製を誘導する、RNA分子を提供する。
【0015】
さらに、本明細書において、本発明のRNA分子の1つ以上を細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によって、アルファウイルスレプリコン粒子が産生される条件下において、RNA分子の組み合わせが、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生のために必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0016】
さらに、a)アルファウイルスレプリコンRNAと、b)本発明のRNA分子の1つ以上と、c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物とを細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によって、アルファウイルスレプリコン粒子が産生される条件下において、(b)のRNA分子と(c)のヘルパー構築物の組み合わせが、アルファウイルスレプリコン粒子の産生のために必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0017】
さらなる実施形態において、本発明は、アルファウイルスレプリコン粒子の集団であって、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含有しない集団を提供する。
【0018】
さらに本明細書において、培養における許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含まないアルファウイルスレプリコン粒子の集団であって、当該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異(attenuating mutation)を含む集団が提供される。
【0019】
さらに、本発明は、被験体において免疫応答を誘発する方法であって、有効量の本発明のアルファウイルスレプリコン粒子の集団を当該被験体に投与する段階を含む方法を提供する。
【図面の簡単な説明】
【0020】
【図1】完全長(FL:full length)プロモーターレスヘルパー分子の5’複製認識配列(RRS:replication recognition sequence)の構築物を示す。5’複製認識配列内のカプシドまたは糖タンパク質(GP:glycoprotein)開始コドンの上流の出発コドンの位置を、枠線と黒線で示す。枠線は、カプシドまたはGPのためのコード配列とインフレームである出発コドンを示す。黒線は、カプシドまたはGPのためのコード配列とリーディングフレーム外である出発コドンを示す。線の真下の数値は、当該分子の5’末端から付番された、5’複製認識配列における推定上の出発コドンに対する最初のヌクレオチド配列の位置を示す。
【図2】プロモーターレスヘルパー分子における5’複製認識配列欠失の構造を示す。枠線のボックスは、各構築物に残存している5’複製認識配列を示し、ボックス内の数値は、配列のヌクレオチドの長さを示す。黒い細線は、各構造物から削除されている5’複製認識配列を示す。縞の斜線のボックスは、カプシドまたはGPのいずれかのためのコード配列の位置を表す。
【図3】ノーザンブロット分析を示す。全細胞RNAを、30μgのpERK/342/MS/BoNT AレプリコンRNAおよび30μgのdHE1−6M1(プロモーターレスE1ヘルパー)もしくは26Sプロモーター(13.4.6)を含有する30μgのGPヘルパーRNAのいずれかによってエレクトポレーションしたベロ細胞から抽出した。各試料(5μg)のRNAを1%のグリオキサールゲル上で展開し、BrightStar(登録商標)膜(Ambion社;Austin、TX)に転写した。ゲノムのセンスアルファウイルスRNA3’末端に特異的なプローブを使用して、ヘルパーの複製を検出した。レーン1:RNA分子量マーカー、レーン2:dHE1−6M1ヘルパー+BoNT Aレプリコン、レーン3:プロモーター補助GPヘルパー+BoNT Aレプリコン。
【図4】ユビキチン化構築物(dHcapUおよびdHgpU)または標準構築物(dHcapおよびdHgp)の、ユビキチン単量体のC末端アミノ酸およびヌクレオチド配列、並びにアルファウイルスカプシドおよび糖タンパク質コード配列のN末端残基を示すダイヤグラムを示す。これらの配列の右端の「Met Phe」、「Pro Met Phe」、「Pro Thr Met Ser」、および「Thr Met Ser」は、カプシドおよびGPタンパク質のN末端に見られるアミノ酸を表す。ユビキチン化構築物は、13.2.2および13.4.6ヘルパーには見られないさらなるN末端残基を有する。右側のボックスは、ヌクレオチド一次配列の結果としてコードされた3’RsrII制限部位およびアミノ酸を示す。左側のボックスは、VEE構造タンパク質からのユビキチンの切断に重要な残基を表す。
【図5】パッケージングの実験(表10)においてVRPを作製するためにエレクトポレーションされた細胞から生じた細胞溶解物のウェスタンブロット分析(一方はカプシド−特異抗体を使用し、他方は糖タンパク質(GP)特異抗体を使用する)を示す。以下に示すように、レプリコンと共に2つのRNAヘルパーを細胞中にエレクトポレーションした:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6;レーン9、Hcap4および13.4.6レーン10、分子量マーカー。
【図6】以下に示すように、レプリコンと共に2つのRNAヘルパーを細胞中にエレクトポレーションしたベロ細胞で産生されたカプシドヘルパーRNAのノーザンブロット分析を示す:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6。各レーンにおける翻訳可能なカプシドRNA分子は、アスタリスクでマークしてある。
【図7】以下に示すように、レプリコンに加えて、2つのRNAヘルパーを細胞中にエレクトポレーションしたベロ細胞で産生された糖タンパク質(GP)ヘルパーRNAのノーザンブロット分析を示す:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6。各レーンにおける翻訳可能な糖タンパク質RNA分子は、アスタリスクでマークしてある。
【発明を実施するための形態】
【0021】
本明細書で使用される場合、「1つの(”a”、”an”)」および「当該(”the”)」は、それが使用される状況に応じて、1つまたは2つ以上を意味し得る。例えば、「1つの(“a”)」細胞は、1つの細胞または複数の細胞を意味し得る。
【0022】
さらに、本明細書で使用される場合、「および/または(”and/or”)」は、1つ以上の関連する列挙された項目の任意のかつ全ての可能な組み合わせ、並びに代替(「または」)において解釈される場合は組み合わせの欠如を意味しかつ包含する。
【0023】
さらに、本明細書で使用される場合、「約(”about”)」なる用語は、例えば、本発明の化合物または薬剤の量、投与量、時間、温度などの測定可能な値を意味する場合、指定された量の±20%、±10%、±5%、±1%、±0.5%、またはさらに±0.1%の変動を包含することを意味する。
【0024】
「5’アルファウイルス複製認識配列」、「3’アルファウイルス複製認識配列」、「5’複製認識配列」、および「3’複製認識配列」なる用語は、アルファウイルス中に見られるRNA配列、そこから誘導される配列、または様々なアルファウイルス間で保存される配列をベースとする合成配列を意味し、非構造アルファウイルスレプリカーゼタンパク質によって認識されてウイルスRNAの複製を引き起こす。いくつかの実施形態において、これらの配列は、VRPを作製するための本発明の構築物、プラスミド、および核酸の調製、突然変異、および/または操作を容易にするために、DNAの形態であってもよい。これらの配列はさらに、「5’および3’末端」とも呼ばれ、非構造タンパク質媒介性増幅に必要な5’および3’ウイルス配列、非構造タンパク質媒介性増幅に必要な5’および3’配列、5’または3’保存配列要素(CSE:conserved sequence element)、5’または3’非コード領域、5’または3’非コード領域配列、複製のためにシス型において必要な5’または3’ウイルス配列、アルファウイルスの転写を開始する5’または3’配列、並びに/あるいはアルファウイルス5’および3’配列、を意味し、5’および3’の指定はアルファウイルスゲノムにおけるそれらの位置を意味している。本発明の核酸分子において、これらの5’および3’末端を使用して、2つの末端の間でコードされるRNA配列の複製および/または転写が行われる。これらの配列を標準的な分子生物学的技術によって修飾することにより(例えば、開始(すなわち、出発)コドンを除去するために、または翻訳性を高めるために、どちらかの末端で切端および/または修飾することにより)、組換えの可能性のさらなる最小化および/またはクローニング部位の導入などが可能であるが、ただし、当該配列は、依然としてアルファウイルス複製機構によって認識されなければならない。
【0025】
本明細書で使用される場合、「開始コドン」または「出発コドン」は、機能タンパク質の翻訳において使用されてもされなくてもよいコドンを意味し、これはRNAではAUGであり、DNAではATGである。
【0026】
「アルファウイルス構造タンパク質/タンパク質(単数または複数)」なる用語は、アルファウイルスによってコードされる構造タンパク質の1つまたは組み合わせを意味する。これらは、ポリタンパク質として野生型ウイルスにより産生され、一般的に、文献においてC−E3−E2−6k−E1として記載される。E3および6kは、2個の糖タンパク質であるE2およびE1の膜貫通/輸送シグナルとして機能する。したがって、本明細書におけるE1なる用語の使用は、E1、E3−E1、6k−E1、またはE3−6k−E1を意味し得、並びに本明細書でのE2なる用語の使用は、E2、E3−E2、6k−E2、PE2、p62、またはE3−6k−E2を意味し得る。「糖タンパク質ヘルパー」または「GPヘルパー」なる用語は、通常、本明細書において、E2およびE1糖タンパク質の両方をコードするヘルパー分子を意味し、本発明のある特定の実施形態において、E1およびE2は、別々のヘルパー分子でコードされる。
【0027】
「ヘルパー(単数または複数)」および「ヘルパー分子」なる用語は、互換的に使用され、1つ以上のアルファウイルス構造タンパク質をコードする核酸を発現する核酸分子を意味する。
【0028】
「ヘルパー細胞」および「パッケージング細胞」なる用語は、本明細書において互換的に使用され、アルファウイルスレプリコン粒子を産生する細胞を意味する。ヘルパー細胞は、1つ以上のアルファウイルス構造タンパク質をコードする、本明細書に記載されているようなヘルパー分子および/またはヘルパー構築物のセットを含む。ヘルパーは、RNAもしくはDNAであってもよく、またはその両方であってもよい。ヘルパー細胞またはパッキング細胞は、アルファウイルス許容性の、すなわちレプリコンRNAの導入においてアルファウイルス粒子を産生することができる任意の細胞であってもよい。アルファウイルス許容性細胞としては、これに限定されるものではないが、ベロ細胞、ベビーハムスター腎臓(BHK:baby hamster kidney)細胞、293細胞、293T/17(ATCCアクセッション番号CRL−11268)細胞、ニワトリ胚繊維芽細胞(CEF:chicken embryo fibroblast)、UMNSAH/DF−1(ATCCアクセッション番号CRL−12203)細胞、およびチャイニーズハムスター卵巣(CHO:Chinese hamster ovary)細胞などが挙げられる。
【0029】
本明細書で使用される場合、「プロモーター」は、RNA分子の転写を誘導する核酸配列である。
【0030】
本明細書で使用される場合、「単離された細胞」は、細胞が天然環境から分離され、および/または細胞がその自然環境において発生した状態から変更もしくは修飾された細胞または細胞の集団である。本発明の単離された細胞は、例えば、細胞培養物中の細胞であってもよい。本発明の単離された細胞はさらに、動物の細胞および/または動物に導入された細胞であってもよく、この場合、当該細胞は、例えば、本発明のアルファウイルス粒子の細胞中への導入によって変更もしくは修飾されている。
【0031】
本明細書で使用される場合、「アルファウイルスサブゲノムプロモーター」または「26Sプロモーター」は、アルファウイルス複製プロセスの一部としてサブゲノムのメッセンジャーRNAの転写を誘導する野生型アルファウイルスゲノムにおいて本来定義されるようなプロモーターである。
【0032】
本発明のいくつかの実施形態において使用される非相同核酸(例えば、対象となる遺伝子もしくは「GOI(gene of interest)」、または対象となる核酸もしくは「NOI(nucleic acid of interest)」)は、野生型アルファウイルスのゲノム内には存在しない核酸、および/または野生型アルファウイルスのゲノム内には本発明の組換え核酸中に存在する順序と同じ順序では存在しない核酸である。例えば、ある特定の実施形態において、NOIは、それらが、感染性の複製能欠損アルファウイルス粒子(例えば、アルファウイルスレプリコン粒子)のアセンブリにおいてヘルパー核酸として使用される場合、あるいは、ある特定のアルファウイルスによって引き起こされた疾病に対するワクチンのための免疫原として使用される場合に、1つ以上アルファウイルス構造タンパク質(例えば、C、PE2/E2、E1、E3、6K)をコードし得る。
【0033】
本発明は、アルファウイルス構造タンパク質(単数または複数)並びにアルファウイルス5’および3’配列をコードするヌクレオチド配列を含むRNA分子であって、この場合、開始コドンが5’複製認識配列から除去されており、しかし、プロモーター配列(例えば、サブゲノムアルファウイルスプロモーター配列であり、場合によって26Sプロモーターもしくはウイルス接合領域プロモーターとも呼ばれる)を欠失しているRNA分子が、培養細胞株におけるアルファウイルスレプリコン粒子の産生に対して、完全長プラス鎖RNAが効率的に翻訳され、十分な量のアルファウイルス構造タンパク質がトランス型において産生されるように複製され得るという驚くべき予想されない発見に基づいている。場合によって本明細書で「Δ26Sヘルパー」とも呼ばれるこれらの「プロモーターレス」RNA分子は、ヘルパー分子とレプリコンベクターの間で生じる機能的組換え現象の発生の予測された理論上の頻度を減少させることにより、(例えば、ワクチンまたはアジュバントとしての使用のために製造された)アルファウイルスレプリコン粒子の集団における理論上の安全域を増加させる。
【0034】
いずれのスプリットヘルパーシステムも、複製能力のあるアルファウイルス(RCV)を生成するために、最小限の2つの独立した組換え現象を必要とする。相同組換えも報告されているが、アルファウイルスでは、組換えは主にRNA複製複合体によるランダムな鎖スイッチングの結果であると考えられている(Weissら、1991)。第一の組換え現象では、複製複合体は、例えば、文献(例えば、Johnstonら米国特許第5,792,462号)において開示されているスプリットヘルパー/レプリコンパッケージングシステムにおけるRNAヘルパー分子の3’末端であり得る。当該複合体が、26Sまたはウイルス接合領域を介してこのヘルパーRNAの複製を継続し、次いで、鋳型としてのレプリコンRNAの外来ヌクレオチド配列にスイッチングし、レプリコン5’末端を介して複製を完了するのであれば、結果として得られる「組換えレプリコン中間体」は、すべての非構造タンパク質をコードする配列と、領域をコードする外来の対象となる核酸(NOI)を含有する転写単位のいくつかまたはすべてと、アルファウイルス構造タンパク質の1つを発現する新しい挿入された転写単位とを含有するであろう。RCVが産生されるためには、続く第二の組換え現象が、(上に記載された)組換えレプリコン中間体の3’複製認識配列への鎖スイッチング現象によって生じなければならない。なぜなら、これが、挿入突然変異誘発を伴わないで、配列をコードする全ての非構造タンパク質および構造タンパク質のための機能的な転写単位が保持される唯一の位置であるからである。ヘルパーRNA分子が26Sプロモーターを含有しているために、理論上、そのような2つの組換え現象によってRCVが生じ得る。各組換え挿入体は、非依存性転写単位であろうから、正確な組み換え点は重要ではないだろう。
【0035】
本発明のプロモーターレスヘルパー分子を使用したRCVの生成も、最小限の2つの非依存性組換え事象を必要とするだろうが、機能的な遺伝子組換え体が得られる制約は、文献において知られているRNAヘルパー分子の場合よりもはるかに高く、したがって、RCVが生成される理論上の頻度ははるかに低い。これは、26Sプロモーターの不在下では、ほとんどの組換え現象により、結果として、アルファウイルス構造タンパク質を発現し得る機能的な転写単位が生じるためであろう。したがって、プロモーターレスヘルパー分子を使用するRCVの生成は、構造的ポリプロテインのオープンリーディングフレームの再生を必要とするであろうし(すなわち、これらのヘルパーが由来する野生型ウイルスに見られる構造と実質的に同様に)、これ自体が、2回の必要な組換え現象が特異的な順序においてかつ非常に特異的なヌクレオチド位置において生じることを必要とする。最初の組換え現象は、カプシドヘルパーコード配列を伴わなければならない。なぜなら、それ自体が分裂し、機能的なカプシドタンパク質を生成するためには、糖タンパク質に対して適切な(すなわち、5’)位置になければならないためである。カプシドヘルパーは、レプリコン26Sプロモーターからカプシドタンパク質が発現する遺伝子組換えを達成するために、ヌクレオチドの完全組換え現象またはヌクレオチドのほぼ完全組換え現象を介して、レプリコンベクターで組換えられなければならない。すなわち、1)レプリコン26Sプロモーターのすぐ下流であり、2)非相同的GOIの任意の残りとインフレームであり、並びに3)結果としてGOI/アルファウイルスカプシド融合タンパク質を生成しない(したがって、不活発なアルファウイルスカプシドを産生する)組換えのみが、機能的であるだろう。アルファウイルス糖タンパク質ヘルパーによる第二の組換え現象は、3’複製認識配列での発生に制限されることに加えて、第一の組換え事象と同じ制約下にある。したがって、プロモーターレスヘルパー分子を用いて粒子を製造する本発明の方法では、RCVが生成される頻度は、理論上、文献において知られているヘルパー分子よりはるかに低いであろうし、非常に低い頻度であるために、本発明の方法ではそのようなRCVが検出されなかった。
【0036】
本発明の驚くべき特徴は、アルファウイルス粒子をアセンブルするためのヘルパー/レプリコンシステムを製造する従来の試みが、アセンブリのための構造タンパク質を翻訳するのに十分なRNA分子を得るために、強力なプロモーター、たいていの場合はアルファウイルス26Sサブゲノムプロモーター、の使用に頼っていたという事実に存する。既存の文献との明確な対比において、本発明者は、通常、野生型アルファウイルスの増殖、および文献において知られているヘルパーRNAシステムにおいて伴われる、26Sプロモーターからのより小さなメッセンジャーRNAの転写と当該メッセンジャーの増幅とがなくても、完全長分子として直接翻訳され得る新規のRNAヘルパー分子を利用することができるということを発見した。したがって、本発明のヘルパーRNAの直接翻訳は、細胞のリボソーム装置による完全長RNAの5’末端におけるキャップの認識を介して達成される。真核細胞内では、mRNAに由来する翻訳の開始は、リボソームサブユニットのmRNAへの補充を可能にする厳しく制御された一連の現象を伴う。キャップ依存性翻訳の場合、「キャップ」として知られる、mRNAの5’末端に存在するメチル−7−G(5’)pppN構造が、eIF4E、eIF4G、およびeIF4Aから構成される開始因子eIF4Fによって認識される(Hershey&Merrick.Translational Control of Gene Expression,pp.33−88.Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press.2000において概説されている)。
【0037】
アルファウイルスは、プラス鎖RNAウイルスであり、すなわち、ウイルスRNAが細胞に入ると、このRNAから非構造アルファウイルスタンパク質(nsP1、nsP2、nsP3、およびnsP4)の翻訳が生じ、これらのタンパク質が、それぞれ、完全長マイナス鎖RNA鋳型を生成する。次いで、マイナス鎖RNAが複製され、完全長(「ゲノム」)プラス鎖RNAと、26Sプロモーターで始まるより小さい(「サブゲノム」)プラス鎖とを産生する。プラス鎖が産生される場合、アルファウイルスの非構造タンパク質も、RNAをキャップして、リボソームによる翻訳のために細胞質において利用できるようにする。「キャップ」は、RNAの5末端に付加したメチル化残基を意味する。本発明の特定の実施形態において、インビトロにおいて産生されるヘルパーRNA分子(これは、プラス鎖RNAである)は、キャップされていない。プラス鎖ヘルパーRNAが、レプリコンRNAを含有する真核細胞に導入されると、主にマイナス鎖の合成が最初に生じる。非構造タンパク質を合成するアルファウイルスレプリコンRNAの存在下において、マイナス鎖鋳型(ヘルパーとレプリコンの両方)は、次にキャップされたプラス鎖RNAへと複製される。本発明の他の実施形態において、ヘルパーRNAは、当技術分野において周知の試薬および、例えば、Promega社(Madison、WI)およびAmbion社(Austin、TX)から市販の試薬を使用して、インビトロでキャップされ得る。キャップは、例えば、Gキャップ、Cキャップ、Aキャップ、メチル化G(m7G(5’ppp(5’)pppG(5)A);非メチル化G(G(5’ppp(5’)A);ARCA(抗−リバースキャップ類似物、3−O−Me−m7G(5’)pppG(5));トリメチル化(m2、2,7G(5’ppp(5’)pppG)、2ウェイキャップ(m7G(5’ppp(5’)m7G)を含み得る。いくつかの実施形態において、ARPの最大収量のために、ARPを産生するために使用される本発明のすべてのヘルパーは、キャップされているかまたはキャップされていないかのいずれかである。最も高い収量は、キャップされたヘルパーRNAによって記録されているが、ある特定の実施形態において、キャップされていないヘルパーRNAも、著しく高い収量において生成し得るので、キャッピングのさらなるコストを避けることができる。ヘルパーRNAの一方のみをキャップすることも可能であるが、これも、場合によってARPの収量を制限する場合がある。概して、本発明者は、キャップの使用、転写混合物中のNTPに対するキャップ類似体の比率、およびARPを生成するために使用されるRNAの比率の変更などの、いくつかの変数を調べる通常の実験によってARP収量を最適化することができるということを示している。
【0038】
したがって、特定の実施形態において、本発明は、a)少なくとも1つの開始コドンが除去されているアルファウイルス5’複製認識配列と、b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、c)アルファウイルス3’複製認識配列とを含む、から実質的に成る、および/またはから成る単離されたRNA分子であって、ただし、当該RNA分子は、(b)のヌクレオチド配列の転写を誘導するプロモーターを含まず、アルファウイルス5’および3’複製認識配列が、アルファウイルス非構造タンパク質の存在下においてRNA分子全体の複製を誘導する、RNA分子を提供する。
【0039】
様々な核酸配列が、本発明の核酸構築物において5’末端および3’末端の機能を満たし得る。例えば、当該配列は、VEEアルファウイルスに対して、上で例示されているような、アルファウイルス5’複製認識配列および他の隣接する配列を含み得る。さらに、天然の5’アルファウイルスの末端において欠失を生じさせて、ある特定な二次構造要素、例えば、ステムループ構造など、を除去することもできる。ある特定の実施形態において、これらのステムループ構造の1つ以上が、本発明のヘルパー構築物から除去され得る。あるいは、例えばSindbisウイルスの場合の、tRNAアスパラギンに対するヌクレオチド10〜75(Schlesingerら、米国特許第5,091,309号)など、同様の機能的能力を維持しながら、この要素として非アルファウイルスまたは他の配列を利用することもできる。
【0040】
いくつかの実施形態において、3’アルファウイルス複製認識配列は、長さが約300ヌクレオチドであり得、実質的に天然のアルファウイルス3’複製認識配列を含有する。アルファウイルス間で保存される最小の3’複製認識配列は、19ヌクレオチド配列である(Hillら、Journal of Virology,2693−2704(1997))。加えて、Sindbisウイルスでは、3’複製認識配列のすぐ後に続くポリ(A)テールは、長さが少なくとも11〜12の残基でなければならず、並びに、3’複製認識配列の3’13ntは、効率的なマイナス鎖RNAの合成にとって重要であるということが示されている(Hardy and Rice, Journal of Virology,79:4630−4639(2005))。したがって、3’末端のための配列は、完全なアルファウイルス3’複製認識配列、または依然として認識配列としての機能を維持する3’複製認識配列の切端した領域、または長さが25〜325ヌクレオチドでありかつ3’複製認識配列のすぐ後に続く最小長さ11〜12ntのポリ(A)ランを含む3’末端を含み得る。このコンテキストにおいて使用することができる配列の他の例としては、これらに限定されるものではないが、非アルファウイルス、または同様の機能的能力を維持してマイナス鎖RNA合成の開始を可能にする他の配列(例えば、GeorgeらJ Virol.74:9776−9785(2000)に記載されている配列)が挙げられる。
【0041】
本発明のRNA分子において使用される5’および3’複製認識配列は、任意の組み合わせにおける同一のまたは異なるアルファウイルスに由来してもよく、これらは、同一のまたは異なるアルファウイルスに由来するレプリコンベクターとの任意の組み合わせにおいて使用することができる。
【0042】
本発明のある特定の実施形態において、ヘルパーRNA分子の5’および3’配列は、VRPの生成においてヘルパーの性能を最大にし、かつRCVの生成に対する理論上の可能性を最小にするように選択される。特定の実施形態は、5’および3’配列の修飾、並びに本明細書にその例が記載されている、アルファウイルス由来の元来の5’および3’配列の一部の欠失を含み得る。本発明に記載された5’および3’配列の、非常に多くの組み合わせがあり、各ヘルパー分子に対して様々な組み合わせを使用することができる。VRPの生成におけるそれらの性能を決定するために、本明細書において教示する修飾および欠失の様々な組み合わせを試験することは、当業者の範囲内である。
【0043】
本発明のRNAヘルパー分子は、アルファウイルス構造タンパク質の翻訳をそれらの天然メチオニン出発コドンで開始するために、5’キャップ構造から5’複製認識配列までスキャンするリボソームに頼っている。これらの領域にさらなる開始コドンが存在すると、構造タンパク質に対して天然の出発コドン以外の部位で翻訳を開始することを可能にし、その結果、リボソームがmRNAに沿ってアルファウイルス構造タンパク質コード領域中へと移動する場合の融合タンパク質か、または続いてリボソームが5’複製認識配列の停止コドンに達した場合の短い非機能性ペプチドか、どちらかが産生されるため、これらのヘルパーの有効性が低下する。したがって、これらのヘルパーにおいて完全な5’アルファウイルス非コード領域(すなわち、野生型アルファウイルスの5’末端から26Sサブゲノムプロモーターの最初のコドンまでの全配列)を使用することは、この領域に多く存在する出発コドンおよび停止コドンのために最適ではない。したがって、特定の実施形態において、本発明のRNA分子は、5’複製認識配列から除去される1つ以上の開始コドンを有し得る。1つ以上とは、当技術分野において標準的な方法に従って除去または不活性化される2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、10つ、11つ、12つ、またはそれ以上の開始コドン(すなわち、出発コドン)を意味する。
【0044】
したがって、本発明は、例えばAUGからGUGへの変異によって、5’複製認識配列から1つ以上開始コドンが除去されている本発明のRNA分子を提供する。ある特定の実施形態では、RNA分子は、例えばAUGからGUGへの変異によって、5’複製認識配列から全ての開始コドンが除去されて提供される。例えば、図1に示されるような、以下の位置:12、45、148、154、160、258、294、299、331、390、411、441、および499での任意の組み合わせにおける1つ以上の開始コドンが除去、例えば変異、され得る。
【0045】
開始コドンの除去とは、(例えば、本明細書に記載された方法および当技術分野において知られているような方法に従って)ヌクレオチド配列を修飾して、開始コドンを削除または変更し、その結果、その部位での開始または活性(例えば、翻訳活性)を除去または変更することを意味する。いくつかの実施形態において、開始コドンの大部分は、除去され得るが、特定のヘルパー構築物の5’領域のそのようなコドンのほんのいくつかだけを、リボソームによって通常認識されるコンテキストに存在させることが可能である。したがって、特定の5’配列では、可能な10〜12コドンの外側のそのような2〜3のコドンの除去により、結果として、すべての10〜12のコドンが除去されている構築物とあまり変わらないレベルの発現が生じ得る。本発明のヘルパー分子において使用される場合に、結果としてパッケージング細胞またはヘルパー細胞内において十分発現し、アルファウイルスレプリコン粒子の許容可能な収量を提供するであろう、野生型アルファウイルス配列に由来する多数の特定の5’配列が存在することは、本発明の範囲内である。
【0046】
本発明のRNA分子は、1)アルファウイルスカプシドタンパク質、2)任意の順序のアルファウイルスE1およびE2タンパク質、3)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE1タンパク質、4)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE2タンパク質、5)アルファウイルスE2タンパク質、並びに/あるいは6)アルファウイルスE1タンパク質をコードするヌクレオチド配列を含み得る。他の実施形態において、本発明の単一のRNA分子は、3つのアルファウイルス構造タンパク質、すなわち、順不同にカプシドタンパク質、アルファウイルスE1タンパク質、およびアルファウイルスE2タンパク質をコードし得る。いくつかの実施形態において、本発明のRNA分子は、アルファウイルス構造タンパク質をコードするヌクレオチド配列を特異的に排除することができる(例えば、当該分子は、カプシド、アルファウイルスE1タンパク質、アルファウイルスE2タンパク質、あるいはカプシド、E1タンパク質、およびE2タンパク質の任意の組み合わせをコードするヌクレオチド配列を特異的に排除することができる)。
【0047】
本発明の様々な実施形態において、当該RNA分子は、5’複製認識配列を含む、Venezuelan equine encephalitis(VEE)ウイルスの5’末端由来の配列を含み得る。Pushkoら(1997)によって記載されているように、VEEプロモーター補助ヘルパーの5’複製認識配列は、通常、VEE配列の575ヌクレオチド(nt)から成る。最初の519は接在し、44ntの非翻訳領域(UTR:untranslated region)およびnsP1の最初の475ntを表す(44+475=519)。残りの56ntは、nsP4遺伝子の最後の21nt(TAA停止コドンを含む)、最小26Sプロモーターの7nt(nsP4遺伝子と部分的に重なる配列を有する)、およびVEE構造タンパク質遺伝子開始コドンの上流の28ntリーダー配列をコードする(21+7+28=56)。
【0048】
したがって、Pushkoら(1997)によって記載されているプロモーター補助ヘルパーに対する完全な5’複製認識配列は、VEE配列の575ntから成る。本発明のプロモーターレスヘルパーは、上に記載したプロモーター補助ヘルパーにおいて見られる最初の514のヌクレオチド(nt)の全てもしくは一部をコードする。上に記載した514ntに加えて、RsrII制限酵素部位(7nt)をコードする配列も、構造タンパク質のコード配列開始部位(DNAではATGであり、RNAではAUGである)のすぐ上流に存在する。これらのntの封入体は、完全長カプシドヘルパー(dHcap(FL)のための5’複製認識配列を521nt(開始コドンのA残基を含まない)に増加させる。
【0049】
プロモーターレスカプシドヘルパーのいくつかの例およびプロモーターレス糖タンパク質ヘルパーのすべての例において、構造タンパク質コード配列開始コドンのすぐ上流の、しかしRsrII配列の下流に近コンセンサスコーザック配列(3nt(ACC))を含むためのさらなる修飾が加えてある。コーザック修飾のために、完全長糖タンパク質ヘルパー(dHgp(FL)は、524ntの5’複製認識配列を有する。これらのヌクレオチド配列は、以下の説明のために、プロモーターレスカプシドおよび糖タンパク質ヘルパーに対して「完全長」(「FL」)5’VEE配列として定義され、この配列における欠失は、結果として5’複製認識配列を包含する他の実施形態へとつながる。これらの実施形態は、少なくとも、VEEヌクレオチド配列のヌクレオチド1〜141(開始コドンのA残基を含まない)を含む。5’配列の最初の200のヌクレオチド内に、RNAにおける4つのステムループ(SL:stem−loop)構造が予測される。
【0050】
本発明のヘルパー構築物において有用な5’配列の実施形態は、この領域において、SL構造の1、2、3、または全てを含み得る。SL2領域を取り除き、かつSL1、SL3、およびSL4構造を保有する実施形態は、本発明のヘルパー構築物において有用である。SL構造1およびSL構造2は、最初の145のヌクレオチドに含まれており、すなわち、SL3およびSL4は、ヌクレオチド145と200との間に存在している。したがって、いくつかの実施形態において、5’複製認識配列は、長さが524ヌクレオチド(例えば、図2のdHgp(FL))である構築物の5’非コード領域に含まれ、他の実施形態において、5’複製認識配列は、いずれかの、長さ70ヌクレオチド(例えば、SL1、SL3、およびSL4を含む)〜524ヌクレオチドである5’非コード領域に含まれ得る。例えば、5’複製認識配列は、長さ141、144(dH#8)200、203(dH#7)、248、249(dH#6)、309、312(dH#5)、351、354(dH#4)、412、415(dH#3)450、452(dH#2)、499、または502(dH#1)ヌクレオチドであり得、本明細書において特に列挙はしていない70〜524の間の任意の数(例えば、237、379、444など)を含む。正確なヌクレオチド番号と長さは、異なったアルファウイルス間、および所定のアルファウイルスの異種系統の間でいくらか変わることに注意するべきである。本明細書に記載されたヌクレオチドの対応する位置を、任意のアルファウイルスの対応する構造および/または機能並びに/あるいは本明細書に記載された二次構造の対応する構造および/または機能に基づいて特定し、任意のアルファウイルスの一次ヌクレオチド配列から、本発明のRNAヘルパー分子並びに上に記載された修飾を作製することは、当業者の能力の範囲内である。
【0051】
本発明のRNAヘルパー分子はさらに、アルファウイルスの3’末端由来の配列を含み、当該アルファウイルスは、特定の実施形態において、アルファウイルス3’複製認識配列を含むVenezuelan equine encephalitisウイルスであり得るが、これに限定されるものではない。全てのアルファウイルスの3’末端19ヌクレオチドは高度に保存され、一方、E1糖タンパク質の最後のコドンと高度に保存された19ヌクレオチドの間の3’配列は、アルファウイルス間において長さおよび配列の両方に関してほとんど保存されない。3’非コード領域の長さ(本明細書において、配列番号52である、保存された19ヌクレオチドを含む)は、25〜325ヌクレオチドの範囲であり得る。本発明のある特定の実施形態において、3’配列は、VEE3’末端の73〜117ヌクレオチドの間である。特定の実施形態において、本発明のアルファウイルス3’複製認識配列は、配列番号55(dHcap(FL)からdHcap7まで;dHcap(FL)mmからdHcap7mmまで、dHcap(FL)mut1からdHcap7−mut1まで)、配列番号56(Hgp(FL)からdHgp7まで、dHgp(FL)mmからdHgp7−mmまで、dHgp(FL)mut1からdHgp7−mut1まで)、配列番号57(dHcap6mut1(W/停止)、配列番号58(dHcap7mut1(W/停止)+19ntおよびdHgp7mut1− S+19nt)、並びに配列番号59(dHcap6mut1(W−停止)のヌクレオチド配列を含み得、から実質的に成り得、および/またはから成り得る。
【0052】
特定の実施形態において、本発明のアルファウイルス5’複製認識配列は、
【0053】
【化1】
のヌクレオチド配列を含み得、から実質的に成り得、および/またはから成り得る。これら5’配列の例が合成されている個別のヘルパーを括弧内に記載する。当該配列は、追加的なヌクレオチドの使用のために長さがわずかに変わり得、いくつかのヘルパー構築物において構造タンパク質コード配列の翻訳を高める、最適に近いコーザックコンセンサス配列が得られる(構造タンバク質コード配列のためのコード領域のATG(RNAではAUG)は、これら5’配列には含まれない)。上記において特定されたヌクレオチド配列を含む本発明のRNA分子は、任意の組み合わせ、任意の順序、および/または任意の多様性において、アルファウイルスレプリコン粒子の製造のための本発明の方法で利用することができる。
【0054】
本発明は、さらに、本発明のRNA分子を含むベクターおよび/または核酸構築物を提供する。さらに、本発明の1つ以上のRNA分子と1つ以上アルファウイルス複製単位ベクターとを含む細胞を提供する。1つ以上とは、1つ、2つ、3つ、4つ、5つ、6つ、7つなどを意味する。本発明の細胞は、アルファウイルスタンパク質をコードする核酸構築物を発現させることができる任意の細胞である。本発明の細胞の例としては、これらに限定されるものではないが、ベロ細胞、ベビーハムスター腎臓(BHK)細胞、293細胞、293T/17(ATCCアクセッション番号CRL−11268)細胞、ニワトリ胚繊維芽細胞(CEF)、UMNSAH/DF−1(ATCCアクセッション番号CRL−12203)細胞、PERC.6細胞、およびチャイニーズハムスター卵巣(CHO)細胞などが挙げられる。
【0055】
さらに、本明細書において、本発明の1つ以上のRNA分子を細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によってRNA分子の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生に必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。本発明のいくつかの実施形態において、当該アルファウイルス粒子は、天然アルファウイルスの構造的な構成を模倣しており、ここでレプリコンRNAがカプシドタンパク質によって覆われ、次いでアルファウイルス糖タンパク質を含有する細胞膜でエンベロープされる。このような実施形態において、アルファウイルス構造タンパク質は、すべて同じアルファウイルスに由来する。代替的な実施態様において、アルファウイルスタンパク質は、異なるアルファウイルスに由来していてもよく、ただし、これらの異なるタンパク質が、粒子アセンブリ中にお互いを「認識」するか、あるいは、それらが、お互いを認識できるように修飾される(文献に記載されているように)場合に限る。
【0056】
本発明の方法のいくつかの実施形態において、本発明の2つのRNA分子が本発明の細胞に導入され、この場合、当該2つのRNA分子は、アルファウイルスレプリコン粒子を産生するために、パッケージング細胞において全ての必要な構造タンバク質が産生される組み合わせにおいて、異なるアルファウイルス構造タンパク質をコードする。したがって、本発明は、細胞に2つのRNA分子を導入し、当該2つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードするが、構造タンパク質のすべてはコードせず、当該2つのRNA分子の第二のRNA分子が、第一のRNA分子によってコードされない1つ以上のアルファウイルス構造タンパク質をコードする方法を提供する。
【0057】
さらに、本発明は、本発明の3つのRNA分子を細胞に導入し、アルファウイルスレプリコン粒子を産生するために必要な構造タンパク質のすべてが当該細胞において産生される組み合わせにおいて、当該3つのRNA分子のそれぞれが、異なるアルファウイルス構造タンパク質をコードする方法を提供する。したがって、本発明の3つのRNA分子を細胞に導入し、当該3つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードするが、構造タンパク質のすべてはコードせず、当該3つのRNA分子の第二のRNA分子が、第一のRNA分子によってコードされたアルファウイルス構造タンパク質とは異なる1つ以上のアルファウイルス構造タンパク質をコードし、当該3つのRNA分子の第三のRNA分子が、第一のRNA分子および第二のRNA分子によってコードされるアルファウイルス構造タンパク質とは異なる1つ以上のアルファウイルス構造タンパク質をコードする方法を提供する。例えば、一実施形態において、当該第一のRNA分子は、アルファウイルスカプシドタンパク質をコードすることができ、当該第二のRNA分子は、アルファウイルス糖タンパク質E1をコードすることができ、並びに当該第三のRNA分子は、アルファウイルス糖タンパク質E2をコードすることができる。
【0058】
いくつかの実施形態において、1つ以上ではあるが全てではないアルファウイルス構造タンパク質が、アルファウイルス構造タンパク質によってパッケージされるレプリコンRNAによってコードされ得る。例えば、本明細書において特許請求されたアルファウイルスレプリコン粒子の製造方法において使用される組換えRNAは、対象となる核酸として、および/または対象となる核酸に加えて、1つのアルファウイルス構造タンパク質または2つ以上のアルファウイルス構造タンパク質をコードする核酸配列を含み得る。したがって、特定の実施形態において、レプリコンRNAは、1つのアルファウイルス構造タンパク質または2つ以上のアルファウイルス構造タンパク質をコードする。このレプリコンRNAは、本発明の1つ以上のRNAヘルパー分子と一緒に細胞の集団中に導入され得る。このレプリコンRNAは、当該レプリコンRNAおよび本発明のRNAヘルパー分子(単数または複数)が全てのアルファウイルス構造タンパク質を産生し、当該レプリコンRNAが当該細胞において粒子内にパッケージされるように、本発明の1つ以上のRNAヘルパー分子と一緒に細胞の集団中に導入することができる。
【0059】
さらなる実施形態において、a)アルファウイルスレプリコンRNAと、b)本発明の1つ以上のRNA分子と、c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物とを細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入により、(b)のRNA分子と(c)のヘルパー構築物の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下においてアルファウイルスレプリコン粒子の産生に必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0060】
したがって、本発明のさらなる実施形態において、「プロモーター補助ヘルパー構築物」、すなわち、例えば26Sプロモーターなどのプロモーターの誘導の下で1つ以上のアルファウイルス構造タンパク質を発現する組換えDNAまたはRNA分子は、本発明のヘルパー分子と組み合わせて使用される。1セットのRNA分子の実施形態において、当該「プロモーター補助ヘルパー構築物」は、(i)5’アルファウイルス複製認識配列と、(ii)転写プロモーターと、(iii)1つ以上のアルファウイルス構造タンパク質をコードする核酸配列と、(iv)3’アルファウイルス複製認識配列とをコードする第一の核酸配列を含む。
【0061】
別のセットのRNA分子の実施形態において、当該「プロモーター補助ヘルパー構築物」は、5’アルファウイルス複製認識配列をコードする核酸配列と、IRES要素のすぐ上流にあるアルファウイルスサブゲノムプロモーターと、アルファウイルス構造タンパク質をコードする少なくとも1つの核酸と、3’アルファウイルス複製認識配列をコードする核酸とを含む、国際公開第2004/085660号(2004年10月7日に公開、当該特許は、参照により本明細書に組み込まれる)に記載されているような、組換えヘルパー核酸である。さらなる実施形態において、これらのプロモーター補助ヘルパー構築物は、サブゲノムプロモーターのすぐ下流かつIRES要素のすぐ上流に位置するスペーサー核酸を含み得る。当該スペーサー核酸は、構造タンパク質の翻訳の一部または全部が、次にIRESによって誘導されるように、メッセンジャーRNAの5’キャップからの少なくとも一部の翻訳、およびいくつかの実施形態では全部の翻訳を防止するのに十分な長さの任意のランダムもしくは特異的非コード核酸配列を含み得、またはから成り得る。あるいは、スペーサー核酸が、メッセンジャーRNAの5’キャップからの少なくとも一部および可能であれば全部の翻訳活性を防止するのに十分な二次構造を核酸に付与する長さおよび配列構造であり得る。本発明において使用されるプロモーター補助ヘルパー構築物はさらに、ヘルパー細胞のゲノムに安定して組み込まれ得るDNA分子であり得るか、または有意に組み込まれること無しにエピソーム(例えば、プラスミド)から一過的に発現され得るDNA分子であり得る。本発明のDNA分子は、DNAベクターであってもよく、例えば、これに限定されるものではないが、プラスミドなどの非組み込み型DNAベクター、またはウイルスベクターが挙げられる。
【0062】
本明細書に記載されるような「ヘルパー細胞」または「パッケージング細胞」を利用しかつ本発明のプロモーターレスRNA分子を含む本発明の実施形態において、当該ヘルパー細胞は、本発明のアルファウイルスレプリコン粒子を産生するのに十分なアルファウイルス構造タンパク質をコードするヌクレオチド配列の組み合わせを当該ヘルパー細胞が含むような任意の組み合わせにおいて、プロモーター補助ヘルパー構築物(RNAおよび/またはDNA)をさらに含み得る。ある特定の実施形態において、E1およびE2糖タンパク質は、第一のヘルパー構築物によってコードされ、カプシドタンパク質は、第二のヘルパー構築物によってコードされる。別の実施形態において、E1糖タンパク質、E2糖タンパク質、およびカプシドタンパク質は、それぞれ別個の(例えば、第一、第二、および第三の)ヘルパー構築物によってコードされる。さらに他の実施形態において、カプシドタンパク質および糖タンパク質E1もしくはE2のどちらかが、第一のヘルパー構築物によってコードされ、第一のヘルパー構築物に含まれない残りの糖タンパク質E1もしくはE2は、カプシドコード配列の有無にかかわらず、第二のヘルパー構築物によってコードされる。さらなる実施形態において、アルファウイルス糖タンパク質E1およびE2、並びにカプシドタンバク質は、任意の順序および/または任意の多様性において、全て1つのヘルパー構築物でコードされ得る。本発明に含まれる実施形態では、所定のアルファウイルス構造タンパク質を、2つ以上のヘルパー構築物によって発現させることも可能である。本発明のプロモーターレスRNAヘルパーは、必要に応じて本明細書に記載されているような他の既知のヘルパーとの組み合わせにおいて、任意の組み合わせ、任意の順序、および/または任意の多様性において、アルファウイルス−許容細胞中に導入することができる。
【0063】
本発明のいくつかの実施形態において(例えば、プロモーターレスRNA分子またはプロモーターによって補助されるRNAヘルパー構築物をコードするDNA構築物のための)、DNAからのRNAの転写を誘導するためのプロモーター、すなわちDNA依存性RNAポリメラーゼが、インビトロ転写反応においてRNAを合成するために利用され、この使用のために好適な特定のプロモーターとしては、SP6、T7、およびT3 RNAポリメラーゼプロモーターが挙げられるが、これらに限定されるものではない。
【0064】
本発明の全ての実施形態において、プロモーターレスヘルパー分子および/またはプロモーター補助ヘルパー構築物および/またはレプリコンベクターによってコードされる少なくとも1つのアルファウイルス構造タンパク質および/または非構造タンパク質、並びにレプリコン核酸の非翻訳領域は、本明細書に記載されているように、任意の組み合わせにおいて、1つ以上の弱毒化突然変異を含有し得ることが想到される。
【0065】
本発明はさらに、アルファウイルスレプリコン粒子108個当たりに1個未満の複製能力のあるアルファウイルス粒子を含有するアルファウイルスレプリコン粒子の集団を提供する。さらなる実施形態において、当該集団は、アルファウイルスレプリコン粒子109個、1010個、1011個、1012個、または1013個当たりに1個未満の複製能力のあるアルファウイルス粒子を含有する。本発明は、さらに、当技術分野で周知の方法に従って、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含有しないアルファウイルスレプリコン粒子の集団を提供する。
【0066】
さらに本明細書において、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるアルファウイルス粒子をアルファウイルスレプリコン粒子108個、109個、1010個、1011個、1012個、または1013個当たりに含まないかもしくは1個未満の複製能力のあるアルファウイルス粒子を含むアルファウイルスレプリコン粒子の集団であって、この場合、当該アルファウイルスレプリコン粒子は、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む集団が提供される。さらに、アルファウイルスレプリコン粒子の集団であって、当該集団が、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含まず、当該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む集団が提供される。
【0067】
同定可能な「パッケージングシグナル」の欠失にもかかわらず、本発明のヘルパーRNA、並びに文献に記載されているヘルパーRNAは、時に文献で報告される頻度よりかなり高い頻度で、培養細胞においてアルファウイルス構造タンパク質によってパッケージされる。したがって、本発明のアルファウイルスレプリコン粒子の集団は、本発明の新規のヘルパー分子がパッケージされる集団における粒子のサブセットの存在により、文献に記載されている粒子と区別される。
【0068】
「アルファウイルスレプリコン粒子」、「ARP」、「ウイルスレプリコン粒子」、または「組換えアルファウイルス粒子」なる用語は、本明細書において互換的に用いられ、1つ以上の非相同RNA配列を発現するアルファウイルスレプリコンRNAを取り込むビリオン様構造複合体を意味する。通常、ビリオン様構造複合体は、脂質エンベロープに包埋されている1つ以上のアルファウイルス構造タンパク質を含み、当該脂質エンベロープはヌクレオカプシドを封入し、当該ヌクレオカプシドはRNAを封入する。当該脂質エンベロープは、通常、当該粒子が産生される細胞の原形質膜に由来する。ある特定の実施形態において、当該アルファウイルスレプリコンRNAは、アルファウイルスッカプシドタンパク質から成るヌクレオカプシド構造によって取り囲まれており、アルファウイルス糖タンパク質は細胞由来の脂質エンベロープに包埋されている。構造タンパク質およびレプリコンRNAは、同じアルファウイルスまたは異なるアルファウイルスに由来してもよい。特定の実施形態において、レプリコンRNAは、VEE由来であり、構造タンパク質は、Sindbisウイルス由来である(例えば、Dubenskyら、米国特許第6,376,236号を参照のこと)。アルファウイルスレプリコン粒子は、感染性であるが、伝播性が欠損しており、すなわち、レプリコンRNAは、アルファウイルス構造タンパク質をコードするヘルパー核酸(単数または複数)の不在下において粒子が最初に感染した宿主細胞を越えて伝播することができない。
【0069】
「アルファウイルスRNAレプリコン」、「アルファウイルスレプリコンRNA」、「アルファウイルスRNAベクターレプリコン」、および「ベクターレプリコンRNA」なる用語は、互換的に用いられ、自身の複製(増幅)を誘導することができ、少なくとも、5’および3’アルファウイルス複製認識配列(当該配列は、上で定義されるように最小配列であり得るが、代わりに、アルファウイルス由来の領域全体であり得る)、アルファウイルス非構造タンパク質のコード配列、およびポリアデニル化トラクトを含むように非構造タンパク質遺伝子を発現するRNA分子を意味する。これはさらに、プロモーターおよび/またはIRESを含有し得る。さらにこれは、アルファウイルス構造タンパク質を発現するように操作することもできる。JohnstonらおよびPoloらは、このようなアルファウイルスRNAレプリコンのための多くの構築物について記載しており、このような構築物は、参照により本明細書に組み込まれる。アルファウイルスレプリコンRNAの一実施形態において、アルファウイルス非構造タンパク質は、米国特許出願公開第2003−0119182号に記載されているように、2つの別々の翻訳単位に分離されている。なお、当該特許は、参照により本明細書に組み込まれる。
【0070】
非相同配列を持たないアルファウイルスレプリコンRNA、すなわち、空のレプリコンは、アジュバント組成物を産生するために、アルファウイルスレプリコン粒子において使用され得る。あるいは、アルファウイルスレプリコンRNAは、アルファウイルス構造タンパク質および/または他の非相同核酸配列をコードする核酸を発現することができ、この後者は、ウイルス、原始核細胞、および/または真核細胞に由来する広範囲の配列から選択することができる。非相同配列のカテゴリの例としては、これらに限定されるものではないが、免疫原(例えば、天然、修飾または合成の抗原性タンパク質、ペプチド、免疫原性断片、またはエピトープ)、サイトカイン、トキシン、治療用タンパク質、酵素、アンチセンス配列、および免疫応答調節因子が挙げられる。特定の用途に適切かつ望ましい場合、当該転写されたmRNAは、翻訳され、すなわち、タンパク質が合成されるか、または機能性RNAが産生される。これらのmRNAは、真核細胞内に「キャップ」されており、すなわち、メチル−7−グアノシン(5’)pppN構造が、mRNAの5’末端に存在し(「キャップ」または「5’キャップ」)、このキャップが、mRNAからタンパク質を合成する翻訳開始因子によって認識される。したがって、26Sプロモーターが、転写を誘導し、「キャップ」が、翻訳のための開始シグナルを提供する。
【0071】
いくつかの実施形態において、レプリコンRNAは、任意のアルファウイルス構造タンパク質(単数または複数)をコードする核酸を欠失し得る。他の実施形態において、アルファウイルスレプリコンRNAは、1つまたは2つのアルファウイルス構造タンパク質をコードする核酸を含み得るが、当該レプリコンRNAは、アルファウイルス構造タンパク質の全てをコードする核酸を含有しない。したがって、結果として得られる本発明のアルファウイルスレプリコン粒子は、当該レプリコンRNAがそのカプシド形成および感染性ビリオンのアセンブリに必要な全ての構造タンパク質をコードしないが故に、伝播性が欠損している。
【0072】
特許請求される本発明において利用されるアルファウイルスRNAレプリコンの特定の実施形態は、本明細書に詳細に記載されているような1つ以上の弱毒化突然変異を含み得る。弱毒化ヌクレオチド置換の例としては、本明細書に記載されているVEEの5’末端におけるヌクレオチド3での突然変異、並びにアルファウイルスS.A.AR86におけるnsP1アミノ酸位置538、nsP2アミノ酸位置96、またはnsP2アミノ酸位置372での突然変異が挙げられる。
【0073】
本発明のアルファウイルスレプリコン粒子は、任意のアルファウイルスに由来するレプリコンRNAを含み得る。その上、本発明のアルファウイルスレプリコン粒子は、本発明のアルファウイルスのいずれかに由来するアルファウイルス構造タンパク質を含み得る。したがって、当該レプリコン粒子は、同じアルファウイルスあるいは異なるアルファウイルスに由来するレプリコンRNAおよび構造タンパク質から製造することができ、これらの後者は、キメラアルファウイルスレプリコン粒子(例えば、VEEウイルスベースのレプリコンRNAおよびSindbisウイルス構造タンパク質を含む粒子)であるであろう。
【0074】
本発明の特定の実施形態において、本発明のアルファウイルス構造タンパク質は、Sindbisウイルス構造タンパク質、SFV構造タンパク質、VEE構造タンパク質、ロスリバーウイルス構造タンパク質、EEE構造タンパク質、および/またはWEE構造タンパク質であってもよい。これらは、お互いの任意の組み合わせにおいて存在し得、並びに、非構造タンパク質および他のアルファウイルス配列、例えば、5’アルファウイルス複製認識配列、アルファウイルスのサブゲノムプロモーター、および3’アルファウイルス複製認識配列など、の組み合わせにおいて存在し得、これらまたは他のアルファウイルスのいずれかから、本発明のキメラ組換えアルファウイルスレプリコン粒子および/またはキメラ組換え核酸を産生する。
【0075】
本発明のいくつかの実施形態において、本発明は、アルファウイルス核酸、アルファウイルスタンパク質、アルファウイルスレプリコンRNAおよび/または1つ以上の弱毒化突然変異を含むアルファウイルスレプリコン粒子、ヌクレオチド欠失、ヌクレオチド付加、および/または1つ以上のヌクレオチドの置換として定義される弱毒化突然変異、あるいは、適切な野生型アルファウイルスと比較して、突然変異を含有する生存ウイルスにおいて毒性が喪失されるに至る再構成またはキメラ構築物を含む突然変異を含み得る。
【0076】
適切な弱毒化突然変異は、使用されるアルファウイルスに応じて変わり、そのような弱毒化突然変異は、当業者には既知であろう。代表的な弱毒化突然変異としては、これらに限定されるものではないが、Johnstonらの米国特許第5,505,947号、Johnstonらの米国特許第5,185,440号、Davisらの米国特許第5,643,576号、Johnstonらの米国特許第5,792,462号、同第6,156,558号、および同第5,639,650号に記載されているものが挙げられ、なお、これらの各特許の開示は、参照によりその全体が本明細書に組み込まれる。
【0077】
VEE E1糖タンパク質に対する特定の弱毒化突然変異は、E1アミノ酸位置81,272または253の任意の1つでの弱毒化突然変異を含み得る。VEE−3042突然変異体から製造されたアルファウイルスレプリコン粒子は、E1−81にイソロイシン置換を含有し、VEE−3040突然変異体から作製されたウイルスレプリコン粒子は、E1−253に弱毒化突然変異を含有する。VEE E2糖タンパク質に対する特定の弱毒化突然変異は、E2アミノ酸位置76、120、または209の任意の1つに弱毒化突然変異を含み得る。VEE−3014突然変異体から製造されたアルファウイルスレプリコン粒子は、E1−272およびE2−209の両方に弱毒化突然変異を含有する(米国特許第5,792,492号を参照のこと)。VEE E3糖タンパク質に対する特定の弱毒化突然変異は、E3アミノ酸56〜59の欠失からなる弱毒化突然変異を含む。VEE−3526突然変異体から製造されたウイルスレプリコン粒子は、E3(aa56〜59)にこの欠失を含有し、E1−253に第二の弱毒化突然変異を含有する。S.A.AR86 E2糖タンパク質に対する特定の弱毒化突然変異は、E2アミノ酸位置304、314、372、または376の任意の1つに弱毒化突然変異を含む。あるいは、当該弱毒化突然変異は、例えば、アミノ酸位置158、159、160、161、および162の任意の組み合わせにおける任意の1つ以上での、E2糖タンパク質におけるアミノ酸の置換、欠失、または挿入であってもよい(Poloら国際公開第00/61772号を参照のこと)。あるいは、本発明のRNA分子は、VEEのワクチン株であるTC83から誘導することもできる(国際公開第2005/113782号を参照のこと。なお、当該特許は参照により本明細書に組み込まれる)。
【0078】
本発明の別の弱毒化突然変異は、VEEゲノムRNAのヌクレオチド3、すなわち、5’メチル化キャップの後の三番目のヌクレオチドでの弱毒化突然変異であってもよい(例えば、nt3でのG→C突然変異について記載されている米国特許第5,643,576号を参照のこと)。ウイルスまたはレプリコンの非コード配列に位置するこの突然変異は、いくつかの実施形態において、G→AまたはG→Uの突然変異であり得る。アルファウイルス構造タンパク質および/または非構造タンパク質が、S.A.AR86に由来する場合、当該構造タンパク質および/または非構造タンパク質における代表的な弱毒化突然変異が、文献に記載されている(例えば、米国特許第5,639,650号および同第6,982,087を参照のこと。なお、これらの開示は、参照によりその全体が本明細書に組み込まれる)。
【0079】
本発明のアルファウイルスは、Sindbisウイルス株(例えば、TR339)、VEE(例えば、メチル化キャップまたはTC83に続くゲノムRNAのヌクレオチド3に突然変異を有する)、S.A.AR86ウイルス、Girdwood S.A.ウイルス、Ockelboウイルス、および/またはそれらのキメラウイルスであってもよい。完全ゲノム配列、ならびに様々な構造タンパク質および非構造タンパク質の配列は、多数のアルファウイルスに対して文献から利用可能であり、その例としては、Sindbisウイルスゲノム配列(GenBankアクセッション番号J02363、NCBIアクセッション番号NC_001547)、S.A.AR86ゲノム配列(GenBankアクセッション番号U38305)、VEEゲノム配列(GenBankアクセッション番号L04653、NCBIアクセッション番号NC_001449)、VEEのTC−83ワクチン株(Kinney RMら(1989)Virology 170:19−30;Kinney RMら(1993) J.Virol.67(3):1269−1277において補正)、Girdwood S.Aゲノム配列(GenBankアクセッション番号U38304)、Semliki Forestウイルスゲノム配列(GenBankアクセッション番号X04129、NCBIアクセッション番号NC_003215)、およびTR339ゲノム配列(Klimstraら(1988)、J.Virol.72:7357;McKnightら(1996)、J.Virol.70:1981)が挙げられる。
【0080】
アルファウイルスレプリコン粒子は、当業者に公知の技術の組み合わせにおいて、本明細書に開示する方法に従って調製される。当該方法は、最初に、選択されたヘルパー(単数または複数)およびアルファウイルスレプリコンRNAをアルファウイルス許容細胞の集団に導入する工程と、次いでアルファウイルスレプリコン粒子の産生を可能にする、当技術分野において周知の条件下において、細胞をインキュベートする工程とを含む。ヘルパー(単数または複数)およびアルファウイルスレプリコンRNAをヘルパー細胞の集団に導入する工程は、本明細書に開示されているような、および一般的な当業者に公知のような、いずれかの好適な手段によって実施することができる。
【0081】
アルファウイルスレプリコン粒子の集団は、例えば、米国特許第7,078,218号に記載されているような方法に従って、ヘルパー細胞またはパッケージング細胞から収集する。なお当該米国特許の内容は、参照によりその全体が本明細書に組み込まれる。あるいは、当業者に公知の他の技術を用いてパッケージング細胞から収集することもできる(例えば、米国特許第5,492,462号および同第6,156,558号)。これらの集団は、本明細書に記載されているような方法および文献において公知のような方法に従って、複製能力のあるウイルス(RCV)の存在に対して評価される。本発明の集団は、培養中のアルファウイルス許容細胞での継代により特定されるような、検出可能なRCVを含有しない。
【0082】
いくつかの実施形態において、本発明は、被験体において免疫原性ポリペプチドをコードするアルファウイルスRNAレプリコンをパッケージするため(例えば、ワクチン接種のため)、免疫療法のため(例えば、癌もしくは腫瘍を有する被験体を治療する)、または免疫調節因子のため(例えば、ARPまたは他のワクチン様相をアジュバンドするため)に用いることができる。本発明は、被験体において免疫応答を誘発するまたは高める方法であって、本発明のヘルパー構築物によって粒子内にパッケージされた有効量の核酸を当該被験体に投与する段階を含む方法を提供する。
【0083】
本明細書で使用される場合、「免疫応答を誘発する」および「被験体を免疫化する」には、被験体における、本発明のタンパク質および/またはポリペプチド(例えば、免疫原、抗原、免疫原性ペプチド、および/または1つ以上のエピトープ)に対する体液性および/または細胞性免疫反応の発生が含まれる。「体液性」免疫反応は、この用語が当分野において周知であるように、抗体を含む免疫反応を意味するが、一方で「細胞性」免疫反応は、この用語が当分野において周知であるように、T−リンパ球および他の白血球を含む免疫反応、特に、HLA拘束性細胞溶解性T細胞、すなわち「CTL」による免疫原特異的応答を意味する。
【0084】
さらに、本発明の核酸、粒子、集団、および医薬組成物は、被験体における細胞であり得る細胞へ、対象となるNOIを送達する方法において採用され得ることも想到される。したがって、本発明は、非相同核酸を細胞に送達する方法であって、本発明のヘルパー構築物でパッケージされた有効量の粒子、集団、および/または組成物を当該細胞内に導入する段階を含む方法を提供する。さらに、被験体において非相同核酸を細胞に送達する方法であって、本発明のヘルパー構築物でパッケージされた有効量の粒子、集団、および/または組成物を被験体に送達する段階を含む方法が提供される。当該細胞は、外因性の核酸を取り込みかつ発現する任意の細胞であり得る。当該細胞は、非相同核酸が発現して非相同核酸によってコードされるタンパク質、ペプチド、または他のコード配列生成物(例えば、機能性RNA配列)を産生する条件下で維持される。免疫療法および/または遺伝子療法のための周知のプロトコールに従って、本発明の細胞および/または被験体に治療効果を与えるために、このような方法を利用することができる。
【0085】
本発明の「被験体」としては、これらに限定されるものではないが、例えば、ヒト、非ヒト霊長動物、ウマ、ウシ、ネコ、イヌ、ブタ、ラット、およびマウスなどの温血動物が挙げられる。
【0086】
本発明はさらに、薬学的に許容される担体中に本発明の粒子および/または粒子の集団を含む組成物(例えば、医薬組成物)を提供する。「薬学的に許容される」とは、生物学的でない物質または他の点で不所望でない物質、すなわち、実質的に有害な生物学的効果を引き起こさずに、またはそれが含有される組成物の他の成分のいずれとも有害な方法で相互作用することなく、選択された粒子および/またはその集団と一緒に被験体に投与することができる物質を意味する。薬学的に許容される担体は、ヒトおよび本発明の他の被験体への投与または送達に好適である。当該担体は、当然のことながら、当業者に周知であるように、有効成分の任意の分解を最小にするように、並びに被験体において任意の有害な副作用を最小にするように選択されるであろう(例えば、Remington’s Pharmaceutical Science; 最新版を参照のこと)。本発明の、例えばワクチンまたは別の免疫原性組成物などの医薬製剤は、薬学的に許容される担体と組み合わせて、本発明のヘルパー構築物を使用して作製された免疫原性量の感染性の伝播欠損性アルファウイルスレプリコン粒子を含む。代表的な薬学的に許容される担体としては、これに限定されるものではないが、滅菌パイロジェン非含水および滅菌パイロジェン非含生理食塩水が挙げられる。
【0087】
「免疫原性量」は、投与または送達される被験体において、免疫応答を誘起するのに十分な、本発明の集団における感染性アルファウイルス粒子の量である。本明細書に記載されたアッセイによって特定されるような、投与量当たり約104〜約109、特に106〜108の量の感染ユニットまたは「IU(infectious unit)」が、治療される被験体の年齢および種に応じて好適であると考えられる。投与は、例えば、腹腔内、筋肉内、鼻腔内、腟内、静脈内、皮内(例えば、遺伝子銃により)、直腸内、および/または皮下などの任意の好適な方法によって行われ得る。本明細書の組成物は、皮膚乱切法、および/またはパッチもしくは液体により経皮的に投与してもよい。当該組成物は、ある期間にわたって組成物を放出する生分解性物質の形態において皮下により送達することができる。
【0088】
本明細書で使用される場合、「有効量」は、治療効果であり得る所望の効果を得るのに十分な、本発明の集団または組成物または製剤の量を意味する。当該有効量は、被験体の年齢、全身状態、治療される状態の重症度、投与される特定の薬剤、治療の期間、任意の併用治療の性質、使用される薬学的に許容される担体、当業者の知識および専門的技術の範囲内の因子などにより変わるであろう。必要に応じて、任意の個々の場合における「有効量」は、適切な教示書および文献を参照することにより、および/または通常の実験により、当業者が決定することができる(例えば、Remington,The Science And Practice of Pharmacy(20th ed.2000)を参照のこと)。
【0089】
あるいは、本発明の医薬製剤は、被験体の粘膜への投与(例えば、鼻腔内投与、口腔内投与、および/または吸入)に好適であり得る。当該製剤は、単位投与剤形において好都合に調製され得、並びに当技術分野において周知の任意の方法によって調製され得る。
【0090】
さらに、本発明の当該組成物は、当技術分野において周知の方法に従って被験体の細胞から単離または増殖された樹枝状細胞、またはバルク末梢血単核細胞(PBMC:peripheral blood mononuclear cell)、あるいは被験体に由来するそれらの様々な細胞サブフラクションに感染またはトランスフェクトするために使用され得る。
【0091】
エキソビボ法を用いる場合、細胞または組織は、当技術分野において周知の標準的なプロトコールに従って、体外に取り出され、本発明の組成物を当該細胞または組織中に導入する間、維持され得る。
【0092】
本発明の粒子の集団を含む免疫原性組成物(当該粒子は、当該組成物がヒトまたは動物に投与された場合に、対象となる核酸配列(単数または複数)の発現を誘導する)は、当技術分野において公知の任意の方法によって製剤化され得る。そのような組成物、特にワクチンは、通常、注射液として、液体溶液または懸濁液のいずれかとして調製される。さらに注射前の液体への溶解または懸濁に好適な固体形態も調製され得る。凍結乾燥された製剤も好適である。
【0093】
活性免疫原性成分(例えば、アルファウイルスレプリコン粒子)は、多くの場合、有効成分に対して薬学的に許容されるおよび/または適合する賦形剤および/または担体と混合される。好適な賦形剤としては、これらに限定されるものではないが、滅菌水、塩水、デキストロース、グリセロール、エタノールなど、およびそれらの組み合せ、並びに安定化剤、例えばHSAまたは他の適切なタンパク質および還元糖が挙げられる。
【0094】
さらに、所望の場合、当該ワクチンは、少量の、例えば湿潤剤および/または乳濁剤などの補助物質、pH緩衝剤、および/またはワクチンの有効性を高めるアジュバントを含み得る。効果的であり得るアジュバントの例としては、これらに限定されるものではないが、QS−21、フロイントアジュバント(完全および不完全)、アルミニウム塩(ミョウバン)、リン酸アルミニウム、水酸化アルミニウム;N−アセチル−ムラミル−L− トレオニル−D−イソグルタミン(thr−MDP);N−アセチル−ノルームラミル−L−アラニル−D−イソグルタミン(CGP 11637、nor−MDPとも呼ばれる);N−アセチルムラミル−L−アラニル−D−イソグルタミニル−L−アラニン−2−(1’−2’−ジパルミトイル−sn−グリセロ−3ヒドロキシホスホリルオキシ))−エチルアミン(CGP 19835A、MTP−PEとも呼ばれる);およびバクテリアから抽出された3つの成分のモノホスホリルリピドA、トレハロースジミコレート、および細胞壁骨格(MPL+TDM+CWS)を2%のスクアレン/Tween 80エマルジョン中に含有するRIBIが挙げられる。
【0095】
アジュバントのさらなる例には、これらに限定されるものではないが、水中油エマルジョン配合物、免疫促進物質、例えばバクテリア細胞壁成分もしくは合成分子など、またはオリゴヌクレオチド(例えば、CpG)、および、例えばポリビニルポリマーなどの別の骨格部分を組み込むことのできる核酸ポリマー(二本鎖および一本鎖のRNAおよびDNAの両方)が含まれ得る。
【0096】
アジュバントの有効性は、アジュバンドもしくはアジュバンドの組み合わせも含むワクチン製剤中の当該粒子含有組成物の投与によって生じる、アルファウイルスレプリコン粒子の免疫原性産物に対する抗体または細胞障害性T細胞の量を測定することによって決定され得る。当該技術分野において既知の、そのようなさらなる製剤および投与の様式も使用され得る。
【0097】
アジュバントは、本発明の組成物、または本発明の組成物と組み合わせて使用できる他のワクチン配合物のどちらかと組み合わせることができる。
【0098】
本発明の組成物は、さらに、他の薬剤、医薬品、担体、および希釈剤を含み得る。
【0099】
本発明の組成物を、最適化し、他のワクチン接種レジメンと組み合わせることにより、広範囲の(すなわち、上記に記載された機能を含む、免疫応答の全ての態様を網羅する)可能な細胞応答および体液応答を得ることができる。ある特定の実施形態において、これは、病原菌または腫瘍に由来する免疫原、組換え免疫原、裸の核酸、脂質含有成分により調製された核酸、非アルファウイルスベクター(これらに限定されるものではないが、痘疹ベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、水疱性口内炎ウイルスベクター、パラミクソウイルスベクター、パルボウイルスベクター、パポーバウイルスベクター、レトロウイルスベクター、レンチウイルスベクターを含む)、および他のアルファウイルスベクターの1つ以上を含む組成物との組み合わせにおいて本発明の組成物が使用される非相同プライムブースト法の使用を含み得る。当該ウイルスベクターは、ウイルス様粒子もしくは核酸であり得る。代表的なアルファウイルスベクターは、レプリコン含有粒子、DNAベースのレプリコン含有ベクター(場合によって「ELVIS」システムとも呼ばれ、例えば、米国特許第5,814,482号を参照のこと)、および/または裸のRNAベクターであり得る。
【0100】
本発明の免疫原性(またはそうでなければ、生理的に活性な)アルファウイルス粒子含有集団および組成物は、投与剤形と適合する方法において、並びに予防上および/または治療上有効なそのような量において投与される。投与される量は、一般的に、投与量1mL当たり約104〜109感染ユニットの範囲であり、治療される被験体、当該粒子が投与または送達される経路、発現産物の免疫原性、所望されるエフェクター免疫応答のタイプ、並びに所望される予防の程度に応じて変わる。いくつかの実施形態において、約106、107、および108I.U.(infectious unit)の投与量は、ヒト被験体において特に効果的である。投与または送達に必要とされる有効成分の有効量は、医師、獣医師、または別の保健実務者の判断に応じて変わり、所定の被験体に対して固有であり得るが、そのような決定は、そのような実務者の技術の範囲内である。
【0101】
本発明の組成物および配合物は、1回投与または複数回投与のスケジュールにおいて与えられ得る。複数回投与スケジュールは、投与の初回治療単位が、1〜10またはそれ以上の分割投与量を含み、その後に、所望する効果(例えば、免疫応答)を維持および/または増強するために必要とされるような時間間隔、例えば、毎週または第二の投与に対して1〜4ヶ月目に他の投与量が投与され、並びに必要であれば、さらに数ヶ月(例えば、4または6ヶ月)/数年後に次の投与量(単数または複数)が投与されるスケジュールである。
【0102】
本発明の治療方法の効果は、本発明の障害の治療効果を評価するための周知のプロトコールに従って決定され得る。治療効果の決定因子としては、これらに限定されるものではないが、当技術分野において周知であるように、全生存率、無病生存率、症状の改善、進行までの期間、および/または生活の質などが挙げられる。
【0103】
「治療する」または「治療すること」または「治療」は、障害、疾病、もしくは病気で苦しむ被験体に、例えば有益効果であり得る調節作用を付与する任意の種類の処置を意味し、当技術分野において周知であるような、被験体の状態(例えば、1つ以上症状)の改善、状態の進行の遅延または軽減、障害、疾病、もしくは病気の兆候の予防または遅延、並びに/あるいは障害、疾病、もしくは病気に関する臨床的指標のいずれかにおける変化などが挙げられる。
【0104】
前述の詳細な説明は、単に例示のために提供されるものであり、本発明の精神および範囲から逸脱せずに修飾および変更を加えることができることは理解される。
【実施例】
【0105】
(実施例1)
dHcapおよびdHgpヘルパーの構築
米国特許第5,792,462号、Pushkoら、1997(Virology 239:389−401)、および国際公開第02/03917号(Olmstedら)に記載されているように、VEEヘルパープラスミド(カプシドヘルパーに対しては「13.2.2」とも呼ばれ、糖タンパク質ヘルパーに対しては「13.4.6」とも呼ばれる)からカプシドおよび糖タンパク質(GP)遺伝子を増幅するようにプライマーを設計した(カプシドF(配列番号98)、GP F(配列番号60)、および13−101.pr4(配列番号61)(表1)。これらのプライマーは、RsrII制限部位を提供し、さらに、それぞれ、カプシドまたは糖タンパク質コード配列の開始位置に結合する。上記に引用された参照文献に記載されているDNAプラスミドは、例えばPCR増幅により構造タンパク質コード断片を得るための便利な供給源であり得る。あるいは、これらのコード断片は、VEEの完全長クローンまたはその弱毒化変異体から得ることができる(米国特許第5,185,440号;同第5,505,947号を参照のこと)。
【0106】
これらのプライマーによる増幅は、結果として、PCR産物の5’末端から3’末端へと列挙される以下の要素:5’〜RsrII制限部位、VEE構造タンパク質コード配列ORF、3’UTR、SphI制限部位〜3’、を有する断片を生じる。次いで、当該PCR産物は、米国特許第5,792,462号、Pushkoら、1997(Virology 239:389−401)、および国際公開第02/03917号(Olmstedら)に記載されるように、RsrIIおよびSphI制限酵素によって消化され、空のVEEレプリコンベクターにライゲーションする。このレプリコンRNAは、VEE非構造遺伝子および後に多重クローニング部位(MCS:multiple cloning site)が続く26SサブゲノムRNAプロモーターの単一のコピーを含む。ワクチン構築物では、免疫原をコードする1つ以上のコード配列がこのクローニング部位に挿入される。このベクターは、RsrIIおよびSphIによって消化され(nsPlの大部分およびnsPs2〜4のすべてを除去する)、ライゲーションにより、完全なアルファウイルス5’および3’末端、すなわち「完全長」末端を含むヘルパーが生成する。したがって、これら2つのヘルパーは、dHcap(FL)およびdHgp(FL)と称され、それぞれ、配列番号1および配列番号10の5’配列、並びにそれぞれ、配列番号55および配列番号56の3’配列を有する(図1)。
【0107】
続いて、dHcap(FL)およびdHgp(FL)ヘルパーの両方に存在する552nt5’末端に、それぞれおよそ50ntの8つの連続した欠失部位を作製した(図2)。この手順は、2段階にて実施した。第一に、8つの異なるリバースプライマー(dHelp1〜8R、配列番号63〜70)を、(上に記載された)13.2.2および13.4.6ヘルパーの位置502までに、5’末端に対して相補的に設計し、RsrII制限部位をさらに含有するように、それぞれを操作した(表1)。任意のリバースプライマーと組み合わせた場合に、以下の要素(5’から3’へと列挙):5’〜XbaI制限部位、T7プロモーター、5’の切断された末端、RsrII制限部位〜3’、を有する断片を増幅するように、フォワードプライマー(3−16.1.1(配列番号62)を設計した。第二に、増幅された5’の切断された末端断片を、XbaIおよびRsrIIによって直線化されたdHcap(FL)およびdHgp(FL)ヘルパー中にクローン化した。これにより、それぞれ配列番号2〜9および配列番号11〜18の5’配列を有する、dHcap1〜8およびdHgp1〜8と称される5’の切断された末端ヘルパー構築物の8つのセットが生成した。dHcapシリーズの各メンバーの3’配列は、本明細書において配列番号55として提供され、dHgpシリーズの各メンバーの3’配列は、本明細書において配列番号56として提供される。
【0108】
(実施例2)
プロモーターレスヘルパー発現カセットの発現解析方法
本明細書に記載されたΔ26Sヘルパー構造体がVEE構造タンパク質をどの程度発現するかを決定するために、上に記載されたようなVEEレプリコンベクターと一緒に各ヘルパーをベロ細胞中にエレクトポレーションした。本発明の新規のプロモーターレス構造タンパク質発現カセットの能力を実証するために、GFPまたはボツリヌス神経毒素コード配列を、VEEレプリコンベクターのクローニング部位に挿入することにより、VEEレプリコンを構築した。本明細書に記載されたプロモーターレス構造タンパク質発現カセットの様々な組み合わせによって作製された粒子からのこれらのコード配列の発現により、これらのカセットの有用性および新規性が実証される。
【0109】
メーカーの指示に従ってRiboMAX T7 Express(登録商標)転写キット(Promega Corporation社、Madison、WI)を使用して、ラン−オフ転写により、各ヘルパーおよびレプリコンベクターからRNAを転写した。エレクトロポレーションの前に、ヘルパーおよびレプリコンRNAを、シリカベースのクロマトグラフィによって精製した。30マイクログラム(30μg)の各ヘルパーおよびレプリコンRNAを混合し、3〜5X107のベロ細胞中にエレクトポレーションした。エレクトロポレーションした細胞を培地で希釈し、25cm2のフラスコまたは96ウェルプレートに播種した。次いで、エレクトロポレーションした細胞を37℃で16〜24時間インキュベートした。
【0110】
A.IFA分析
96ウェルプレート中に播種されたエレクトロポレーションした細胞を、リン酸緩衝食塩水(PBS:phosphate buffered saline)で1回洗浄し、次いで、室温で5時間かけてアセトン:メタノール(1:1)で固定化した。次に、当該細胞を、構造タンパク質に特異的なマウス抗体を使用して、VEEカプシドまたはGPタンパク質の発現について分析した。一次抗体をPBS:FBS(1:1)で希釈し、各ウェルに100μlを加えた。プレートを37℃で30分間インキュベートし、150μlのPBSで3回洗浄し、次いでAlexa Fluor 488ヤギ抗マウス二次抗体(Invitrogen社、Carlsbad、CA)により37℃で30分間インキュベートした。インキュベーション後、細胞を上に記載されているように再び洗浄し、各ウェルに最終容積が100μlになるようにPBSを加え、紫外蛍光顕微鏡(Nikon Eclipse TE300)によって検査した。
【0111】
B.ノーザン分析
エレクトロポレーションした細胞を、25cm2のフラスコに播種し、PBSで洗浄し、次いで、RNAwizRNA(登録商標)単離試薬(Ambion社、Austin、TX)を使用し、メーカーの提案するプロトコールに従って全細胞RNAを抽出した。RNA濃度は吸光分光分析法によりで決定した。各試料の5マイクログラム(5μg)を、1%グリオキサールアガロースゲルにより電気泳動して、RNAをBrightStar Plus(登録商標)(Ambion社)膜に受動移入させた。ノーザン分析は、BrightStar BioDetect(登録商標)キット(Ambion社)を使用し、メーカーの提案するプロトコールに従って、VEEの3’複製認識配列のブラス鎖に特異的なビオチニル化DNAオリゴで実施した。化学発光は、処理された膜をフィルムに露光して検出した。
【0112】
C.dHcap(FL)およびdHgp(FL)発現の分析
完全長Δ26Sヘルパー(dHcap(FL)およびdHgp(FL))が複製され、タンパク質を発現し得ることを実証するために、これらのヘルパーRNAを、ヘルパーRNAの複製を促進するアルファウイルス非構造タンパク質を得るために必要なレプリコンベクターと一緒に、細胞中へエレクトポレーションした。ベロ細胞を、30μgのレプリコンRNAと組み合わせた30μgまたは60μgのdHcap(FL)またはdHgp(FL)ヘルパーRNAによりエレクトポレーションした。当該エレクトポレーションした細胞を、上に記載されたようなIFA、ウエスタンブロット、およびノーザン分析ために処理した。
【0113】
(実施例3)
完全長および切端されたΔ26Sヘルパーの発現分析
dHcap(FL)およびdHgp(FL)ヘルパーは、IFAおよびウエスタンブロットによって決定されるように、タンパク質を発現し、ノーザンブロットによって図示されるように、効率的に複製された。
【0114】
カプシドおよびGPの両方に対する切端されたΔ26Sヘルパー(欠失1〜8)の完全なセットが、それぞれがどの程度良好に発現されかつ複製されるかを評価するため、IFAおよびノーザンブロットにより当該セットのタンパク質発現を分析した。それぞれのdHcapヘルパーRNAをVEEレプリコンRNAおよび13.4.6糖タンパク質ヘルパーRNAと組み合わせて、3つのRNAをベロ細胞中にエレクトロポレーションした。ノーザン分析およびIFAは、上に記載されたようにして実施した。カプシド特異的抗体を使用したIFAの結果を表2に示す。すべてのdHcapヘルパーは、IFAによりカプシド発現に対して陽性であったが、dHcap8ヘルパーのみは、弱い陽性であった。
【0115】
エレクトポレーションした細胞から抽出されたRNAのノーザン分析は、切端されたカプシドΔ26Sヘルパーが、dHcap8を除いて、全てよく複製することを示した。
【0116】
dHgpヘルパーについても同様の方法で実験したが、この実験には13.2.2カプシドヘルパーは含めなかった。各dHgpヘルパーをVEEレプリコンRNA組み合わせて、細胞中にエレクトポレーションし、IFAおよびノーザン分析のために上記と同様にして試料を作製した。抗−GP IFAの結果を表3に示す。dHcapヘルパーと同様に、dHgp8を除いて、全てのdHgpヘルパーが陽性であった。dHgp8を除いて、全てのdHgpヘルパーが、良好に複製した。
【0117】
(実施例4)
修飾されたプロモーターレスヘルパー構築物
本発明者は、ウェスタンブロット法によって明らかにされたように、dHcap(FL)およびdHgp(FL)ヘルパーが融合タンパク質を発現する点に注目した。このような融合タンパク質は、Δ26Sヘルパーの転写の際に、カプシドまたはGPのための出発コドンの上流にあるインフレームの出発コドンで翻訳が開始された結果であり得る。そのような上流のコドンの1つは、VEE nsP1に対する天然の出発コドン(VEEウイルスゲノムのヌクレオチド45に位置する)であり、これは、dHcapヘルパーおよびdHgpヘルパーの両方の5’末端に存在し、翻訳の開始に対して有利なコンテキストにある(例えば、コーザック共通配列)。有利なコーザック環境の出発コドンは、これらのヘルパーのキャップされた5’末端からのリボソームのスキャンによって使用され得、それによって、機能的ではない融合タンパク質が生成され、さらなる下流に位置する適切な出発コドンからの機能的カプシドおよび糖タンパク質ポリペプチドの産生が減少する可能性がある。
【0118】
そのような融合タンパク質の産生量を減少させ、かつ完全長カプシドおよび糖タンパク質の発現を増加させるために2つのアプローチを用いた。第一に、上に記載された有利な出発コドンを、TAG停止コドンへ変異させ、残りの出発コドンは変更せずそのまま残した。このアプローチは、5’末端配列を天然のVEEゲノムに存在する配列のできるだけ近くに維持して、これらのヘルパーにとって必要とされる複製要素を維持するために実施した。第二に、nt3の下流の全ての出発コドン(nsP1(有利な)出発コドンを含む)およびカプシドもしくはタンパク質コード配列のオープンリーディングフレーム(ORF:open reading frame)を、AUGからGUGに変更した(全部で12のそのような変更があった)。このアプローチは、有利なAGTコドン(RNAではAUG)が、完全長カプシドもしくは糖タンパク質発現の産生に対して有害効果を有していないかどうかを調べるために実施した。
【0119】
A.dHcap−mut1およびdHgp−mut1ヘルパーの構築
有利なnsP1出発コドンをTAG停止コドンへと変えるdHcap−mut1およびdHgp−mut1ヘルパーを生成するために、各dHcapヘルパーおよびdHgpヘルパーに部位特異的変異を実施し、dHcap−mut1ヘルパーおよびdHgp−mut1ヘルパーと称される、それぞれ配列番号35〜42および43〜50として本明細書において提供されるような5’配列を有する変異したヘルパーの完全なセット(FLおよび切端物1〜7)を生成させた。部位特異的変異は、Quikchange XL(登録商標)部位特異的変異キット(Stratagene社、La Jolla、CA)により、メーカーのプロトコールに従って、表4のフォワード(配列番号71)プライマーおよびリバース(配列番号72)プライマーを使用して実施した。
【0120】
B.dHcap−mmヘルパーおよびdHgp−mmヘルパーの構築
nt3およびカプシドもしくはGP ORF出発コドンの下流に出発コドンを持たない5’末端を有するdHcapヘルパーおよびdHgpヘルパーを生成させるため、部位特異的変異を実施して、介在する全てATG(RNAではAUG)をGTG(RNAではGUG)コドンに変えた。dHgp(FL)構築物を、部位特異的変異のための鋳型として使用した。Quikchange(登録商標)複数部位特異的変異キット(Stratagene社)を使用し、メーカーのプロトコールに従ってコドン変更を導入した。コドン変更を導入するために使用したプライマーを表5に示す(配列番号73〜82)。全てのコドン変更を含有するdHgp(FL)構築物を、dHgp(FL)−mm(配列番号19として本明細書において提供される5’配列を有する)と称した。全てのコドン変更が存在することを配列確認した後に、このDNAを使用して、dHcap(FL)5’複製認識配列をdHgp−mmに由来する5’複製認識配列で置換することにより、dHcap(FL)−mm構築物を生成させた。これは、両方のDNAをRsrIIおよびNotI酵素で消化することにより達成した。次いで、dHgp(FL)−mm RsrII/NotI5’複製認識配列断片を、直線化されたdHcap(FL)DNAでライゲーションさせ、dHcap(FL)−mm(配列番号27として本明細書において提供される5’配列を有する)を生成させた。さらに、dHgp(FL)−mmDNAを鋳型として使用して、dHcap1〜7およびdHgp1〜7に対して上に記載された方法およびプライマーを使用してカプシドヘルパーおよびGPヘルパーの両方に対する切端された5’末端のセットを生成させた。新規のヘルパーは、dHcap1mm 〜dHcap7mm(配列番号20〜26として本明細書において提供される5’配列を有する)およびdHgp1mm〜dHgp7mm(配列番号28〜34として本明細書において提供される5’配列を有する)と称した。
【0121】
C.mut1およびmmのプロモーターレスヘルパーの発現分析
上に記載されたΔ26Sヘルパーのmut1およびmmの様々な変形からのタンパク質産生を分析した。この実験において、dHcap6−mut1(配列番号41として本明細書において提供される5’配列を有する)、dHcap6−mm(配列番号25として本明細書において提供される5’配列を有する)、dHcap7−mm(配列番号26として本明細書において提供される5’配列を有する)、およびdHgp7−mm(配列番号27として本明細書において提供される5’配列を有する)、並びに13.2.2および13.4.6ヘルパーを、ウエスタンブロットによって分析した。mm変異を含むΔ26Sヘルパーは、わずかな検出可能な融合タンパク質を伴ってまたは伴わずに、主に完全長のカプシドタンパク質またはGPタンパク質を発現する。mut1ヘルパーは、かなりの量の完全長構造タンパク質を発現したが、依然としていくらかの融合タンパク質も発現した。
【0122】
mut1およびmmΔ26Sヘルパーの複製特性を分析するために、同じ試料についてノーザン解析を行った。結果は、dHcap6−mut1ヘルパーが、13.2.2カプシドヘルパーと同様に複製することを示している。対照的に、dHcap6−mm、dHcap7−mm、およびdHgp7−mmヘルパーの複製は、13.2.2またはmut1ヘルパーより少ない程度のようである。
【0123】
(実施例5)
Δ26SヘルパーによるVEEレプリコン粒子の生成
表6に列挙されているのは、VEEレプリコンRNAと様々なプロモーターレスカプシドヘルパーおよびGPヘルパーとを組み合わせてVEE複製粒子(VRP:VEE replicon particle)を産生させる多くの実験である。さらに、細胞中に導入された各ヘルパーRNAの量も、いくつかの実験において変えた。VRPは、5×107〜1×108個のベロ細胞を、指示された量のヘルパーRNA並びに30μgのレプリコンRNAでエレクトポレーションすることによって生成された。概して、粒子が生成される全ての実験に対し、エレクトポレーションした細胞を、血清不含培地が入った300cm2のフラスコに播種し、16〜24時間インキュベートした後、VRPを収穫した。
【0124】
VRP力価は、試料の十倍の階段希釈により、96ウェルプレートで増殖させたベロ細胞に感染させて、当該細胞を16〜18時間インキュベートし、当該細胞を固定化し、VEEのnsP2タンパク質または対象となる核酸の産物に特異的な抗体によってIFAを実施することによって決定した。VRP収量は、実験からの全収量、または20mlの調製物からの1ml当たりの量のいずれかとして示されている(表7、9〜13、15、および16)。
【0125】
これらの調製物は、さらに、細胞変性効果(CPE:cytopathic effect)アッセイにより複製能力のあるウイルス(RCV)の存在を試験した。当該CPEアッセイは、RCVの存在をスクリーンするための、細胞培養における2つの盲継代で構成された。継代1では、VRP調製物由来の試料を、ベロ細胞単層により37℃で1時間インキュベートし、次いで、試料液体を除去し、新鮮な培地と交換して、当該培養物を24時間インキュベートして、存在し得る任意のRCVの増幅を可能にした。継代2では、継代1の終了時の細胞培養の上清を新鮮なベロ細胞単層に加え、37℃で72時間インキュベートした。継代2の終了時に、倒立型光学顕微鏡を使用して、培養物のCPEを検査した。このアッセイは標準化されており、大過剰のVRPの存在下での生存ウイルスの検出における感度について評価した。このアッセイにおいてV3014またはTC−83ウイルスのいずれかを用いたスパイク試験により、1×108個のVRPをバックグランドとした3〜8PFUの検出の下限を明らかにした。このアッセイを、本発明のプロモーターレスヘルパーによって産生された1013個を超えるVRPに対して実施したところ、RCVは検出されなかった。このアッセイの検出限界にもかかわらず、RCVの産生に対する可能な組換え頻度の理論計算は、この検出限界の使用を大きく下回り、すなわち、1010、1011、1012、または1013のVRPに1個、であろう。
【0126】
(実施例6)
「スプリット糖タンパク質」プロモーターレスヘルパー
A.別々のE2および1Eのプロモーターレスヘルパーの構築
E2およびE1コード配列が別々のヘルパー上に存在する糖タンパク質のプロモーターレスヘルパーの構築は、E2および1Eの糖タンパク質カセットを、別々に、dHgp6−mut1ヘルパーのバックボーンにクローニングすることによって実施した。プライマーは、PCRによってpHCMV−Vsp由来のVEE構造上タンパク質コード領域のカプシド−E3−E2領域を増幅するように設計した(米国特許第7,045,335号を参照のこと。なお、この特許は参照により本明細書に組み入れられる)。増幅された断片は、pCR−BluntII TOPO(登録商標)ベクター(Invitrogen社)中にクローン化され、pCR−CE3E2を生成した。CE3E2カセットを配列決定して、PCR増幅中にエラーが導入されなかったことを確認した。CE3E2構造領域を含有するプロモーター補助ヘルパーを作製するため、pCR−CE3E2 DNAをSpeI制限酵素で消化し、E3E2断片を放出させた。次いで、当該E3E2(SpeI)断片を、SpeI酵素で直線化したカプシドヘルパー(13.2.2)でライゲーションした。pHCE3E2由来のE3−E2コドン領域を消化することによって、プロモーターレスE2ヘルパー(dHE2−6M1と称する)を調製した。pHCE3E2DNAプラスミドは、最初に、AscI制限酵素によって直線化し、次いで、平滑末端を作成するためにT4 DNAポリメラーゼで処理した。同様に、dHgp6−mut1 DNAプラスミドは、SphI制限酵素で直線化し、T4 DNAポリメラーゼ処理して、平滑末端を作成した。両方の直線化されたT4−ポリメラーゼ処理DNAは、SpeI制限酵素によって消化され、結果として生じた3.6kbのdHgp6−mut1ベクター断片および1.4kbのE3−E2断片を、それぞれゲル精製した。次いで、当該2つの精製された断片を、T4 DNAリガーゼを用いて一緒にライゲーションさせて、dHE2−6M1プロモーターレスヘルパーを作製した。
【0127】
プロモーターレスE1ヘルパーの生成は、数段階において達成した。プライマーは、次の2つの構造タンパクコード配列断片の1)カプシドE3(CE3)、および2)6K−E1(6KE1)を増幅するように設計した。PCR産物を、pCR−Blunt TOPO(登録商標)ベクター(Invitrogen社)中にクローン化し、pCR−CE3およびpCR−6KE1を生成させた。当該クローンを配列決定して、増幅中にエラーが導入されなかったことを確認した。E1糖タンパク質の上流にE3および6Kリーダー配列の両方を含有するカセットを作製するために、別の中間構築物を作製した。これは、BamHI酵素でpCR−6KE1 DNAを消化して、6KE1断片を精製することによって達成した。次いで、6KE1(BamHI)断片を、BamHI酵素で直線化したpCR−CE3 DNAでライゲーションし、pCR−CE36KE1を生成させた。CE36KE1カセットを含有するプロモーター補助ヘルパーを生成するために、pCR−CE36KE1 DNAを、SpeIおよびSphI酵素で消化させ、構造タンパク質コード配列カセットを放出させた。次いで、CE36KE1(SpeI/SphI)断片を、SpeIおよびSphIで直線化したカプシドヘルパー(13.2.2)でライゲーションして、pHCE36KE1を作製した。プロモーターレスE1ヘルパー(dHE1−6M1と称する)の生成は、pHCE36KE1プラスミド由来のE3−6K−E1コード領域を消化することによって達成した。pHCE36KE1およびdHgp6−mut1 DNAプラスミドを、SpeIおよびSphI制限酵素で消化させ、結果として得られた3.6 kbの dHgp6−mut1ベクター断片および1.7kbの E3−6K−E1断片をゲル精製した。次いで、2つの精製した断片を、T4 DNAリガーゼを使用して一緒にライゲーションさせて、dHE1−6M1プロモーターレスヘルパーを作製した。
【0128】
B.スプリット糖タンパク質プロモーターレスヘルパーの分析
個々の糖タンパク質ヘルパーをインビトロで転写させ、RNA転写物を精製した後、VEEレプリコンRNAと一緒にベロ細胞中にエレクトポレーションした。ヘルパー複製を、ノーザンブロットによって分析し、タンパク質発現は、E1およびE2糖タンパク質特異的抗体を用いてIFAによって分析した。ノーザン分析の結果は、dHE1−6M1ヘルパーおよびdHE2−6M1ヘルパーの両方が、効率的に複製することを示している。代表的なノーザンブロットを図3に示す。
【0129】
レプリコンRNAをパッケージしてVRPを作製するために2つの別々の糖タンパク質発現プロモーターレスヘルパーをΔ26Sカプシドヘルパーと組み合わせられるかどうかを決定するために、三つのヘルパーを、ボツリヌス神経毒素A断片を発現するVEEレプリコンRNAと組み合わせて、ベロ細胞中にエレクトロポレーションした。一実験からのVRP収量を表7に示す。
【0130】
(実施例7)
修飾した5’末端および3’末端プロモーターレスヘルパーカセット
A.修飾した5’末端ヘルパーカセットの構築
ほとんどのアルファウイルスのRNA5’末端(およそ最初の250nt)に予測される二次構造は、4つのステムループ(SL)構造(SL1、SL2、SL3、およびSL4)を含有する。Frolovら(RNA、7:1638−1651(2001))は、SL2をコードするヌクレオチド配列をSindbisウイルスヘルパーRNAから除去することによって、そのヘルパーの複製が増加することを実証した。
【0131】
(M倍のプログラムに基づく)VEE5末端のSL2領域であるnt46〜nt116を、以下のようにPCRによってdHcap6−mut1から除去した。2つの断片を、dHcap6−mut1 DNAから増幅した。およそ1キロベース(kb)の5’断片を、dHcap6−mut1の5’末端の45ヌクレオチドとバックボーンプラスミド配列をコードするヌクレオチドとを含有するプライマー13−82.1.9[配列番号83]およびdLS2(EcoRV)R[配列番号84]によって増幅させた(表8)。およそ1.5kb)の3’断片を、ヌクレオチド117で始まるVEE5’末端の一部とVEE3’末端を介してカプシド配列全体をコードするヌクレオチドとを含有するプライマーdSL2(EcoRV)F[配列番号85]および3−8.pr4[配列番号86]によって増幅させた(表8)。5’の約1kbのPCR断片を、XhoIおよびEcoRV制限酵素によって消化させた。3’の約1.5kbのPCR断片を、EcoRVおよびNotI制限酵素によって消化させた。プラスミドdHgp6−mut1を、XhoIおよびNotIによって消化することによって直線化し、結果として得られる約2.5kbのベクターバックボーンを精製した。本明細書において「dHcap6−mut1(dSL2)」と呼ばれる、SL2領域が削除された新しいヘルパーを生成させるため、5’(XhoI/EcoRV)断片、3’(EcoRV/NotI)断片、およびXhoI/NotI直線化ベクターを一緒にライゲーションさせた。配列が本明細書において配列番号51として提供される5’末端を有するdHcap6−mut1(dSL2)ヘルパーを完全に配列決定し、PCR増幅中にエラーが導入されなかったことを確認した。マッチングdHgp6−mut1(dSL2)ヘルパーを生成させるため、dHgp6−mut1 DNAをXhoIおよびRsrII制限酵素で消化させ、5.4kbの断片を精製した。dHcap6−mut1(dSL2)由来の修飾された5’末端は、このDNAをXhoIおよびRsrIIで消化し、1.1kbの断片を精製することにより収集した。これら二つの断片を一緒にライゲーションさせ、dHcap6−mut1(dSL2)として同じ5’末端[配列番号51]を有するdHgp6− mut1(dSL2)を生成させた。
【0132】
B.短縮された3’末端プロモーターレスヘルパーカセットの構築
これらの実施例において、カプシドヘルパー構築物のdHcap(FL)、dHcap1〜dHcap7、dHcap(FL)mm、dHcap1mm〜dHcap7mm、dHcap(FL)mut1、およびdHcap1mut1〜dHcap7mut1に対し、3’末端配列は、配列番号55として本明細書において提供される。本発明のVEEカプシドヘルパーは、完全な糖タンパク質コード領域を欠失しているが、E3タンパク質の小さな部分がカプシドヘルパー上に残っており、成熟カプシドタンパク質を産生するためにパッケージング細胞内においてキモトリプシン様切断が生じることを可能にしている。糖タンパク質ヘルパー構築物のdHgp(FL)、dHgp1〜dHgp7、dHgp(FL)mm、dHgp1mm〜dHgp7mm、dHgp(FL)mut1、およびdHgp1mut1〜dHgp7mut1に対して、成熟カプシドタンパク質を生成するための切断部位を含む3’末端配列は糖タンパク質ヘルパー構築物において必要でないため、当該配列は、より短い配列であった。これらの実施例においてこれらの糖タンパク質構築に使用される3’配列は、本明細書において配列番号56として提供される。
【0133】
さらに、より短い長さの3末端を有するプロモーターレスRNAヘルパーを構築した。アルファウイルス3’末端配列の量を減らすことにより、増殖性VEEウイルスの生成に必要であろう二次組換え現象の理論的可能性がさらに減少する。最初に、アルファウイルスの3’高度に保存された配列[配列番号52]を含む19ヌクレオチドのみを含有する機能的な26Sプロモーターを有する糖タンパク質ヘルパーを、以下の二つの段階において作製した。最初に、ユニーク5’制限部位および3’制限部位を有する糖タンパク質(GP)コード配列カセットを含有したプラスミドを作製した。3’末端のE1終止コドンのすぐ後のユニークSphI部位(「GP(SphI)R」、配列番号87、表8)、および5’末端の既存の内部SpeI部位(「3−16.1.3」、配列番号88、表8)でVEE GPを増幅するようにプライマーを設計した。増幅された断片は、pCR2.1/GP 19nt 5’を生成するpCR2.1 DNA(Invitrogen社、Carlsbad、CA)中にTAクローニングした。次に、VEE3’末端(3’ trunc(SphI)F、配列番号89、表8)の19ヌクレオチド保存配列のすぐ上流にSphI部位を導入するようにフォワードプライマーを設計した。3’末端にユニークAflII制限部位(3’trunc(AflII)R、配列番号90、表8)を含有するであろう断片を増幅するように、プラスミドバックボーン配列に特異的なリバースプライマーを設計した。これらのプライマーの増幅から得られた断片は、SphIおよびAflIIで消化され、SphIおよびAflII制限酵素で直線化されている(実施例1に記載された)13.4.6糖タンパク質ヘルパーとライゲーションし、これによって、結果としてpGPヘルパー−int1が構築された。pGPヘルパー−int1構築物は、(19nt保存配列を含む)当該ヘルパーのGP停止コドンと3’末端の間に72ヌクレオチド領域を有している。19ヌクレオチド3’末端のみによりGPヘルパーを生成させるために、pCR2.1/GP 19nt5’DNAをSpeIおよびSphIで消化させ、当該GPコード配列を、SpeIおよびSphI制限酵素で消化されたpGPヘルパーint1にライゲーションさせた。結果として得られた構築物は、pGPヘルパー19ntと命名した。
【0134】
次いで、pGPヘルパー19nt構築物を使用して、可変長の3’複製認識配列を有するΔ26Sヘルパーを作製した。当該pGPヘルパー19nt構築物を、NcoI制限酵素およびNotI制限酵素によって消化し、19nt3’末端領域を有する糖タンパク質コード配列を含有する2515塩基対断片をゲル精製した。次に、この2515塩基対(NcoI/NotI)断片を、NcoIおよびNotI制限酵素によって消化されたdHgp構築物でライゲーションし、様々なdHgp19nt構築物を生成させた。
【0135】
C.アルファウイルスカプシドタンパク質を発現する修飾されたプロモーターレスヘルパーカセットの構築
VEEウイルス感染細胞において、VEEEカプシドタンパク質は、26SサブゲノムmRNAから翻訳される構造ポリタンパク質から自身を切断する。本発明のVEEカプシドヘルパーは、完全な糖タンパク質コード領域を欠失しているが、E3タンパク質の小さな部分がカプシドヘルパー上に残っており、成熟カプシドタンパク質を産生するためにパッケージング細胞内においてキモトリプシン様切断が生じることを可能にしている。キモトリプシン様切断部位の代わりに、カプシドの3’末端での停止コドンの導入は、糖タンパク質ヘルパーによる機能的な遺伝子組換え体の産生の困難さを増加させるであろう。すなわち、本発明のdHgpヘルパーによって生じる機能的な組換え(すなわち、複製能力のあるウイルスを産生する組換え)に対して、当該組換え現象は、カプシドコード配列において、操作された停止コドンを置換して活性なカプシド切断部位を維持するために完全なヌクレオチドでなければならない。3’末端に組み込まれた停止コドンを有するdHcapヘルパーの2つの変形を作製した。1つの変形は、天然カプシドタンパク質のC末端トリプトファン残基を停止コドンで置換した、本明細書において配列番号57として提供されるdHcap6−mut1−dSL2(停止)であり、他方の変形は、C末端トリプトファン残基を保有し(本明細書において配列番号59として提供される3’配列を有するdHcap6−mut1(W−停止))、トリプトファン残基のすぐ下流に停止コドンを挿入した。カプシドコード配列を、5’末端のユニークRsrII部位(カプシド(RsrII−コーザック)F、配列番号:91、表8)および3’末端のユニークSphI部位カプシド(停止)SphI R、配列番号:92またはカプシド(W−停止)SphI R、配列番号:93、表8)を操作するために設計されたプライマーで複製した。さらに、フォワードプライマーを操作して、カプシド出発コドンをほぼ最適なコーザック共通配列に配置して(Kozak,Cell,44(2):283−292(1986))、カプシドmRNAの翻訳のリボソーム開始を高めた。増幅されたカプシドコード配列を、RsrII制限酵素およびSphI制限酵素で消化し、RsrIIおよびSphIで直線化されたΔ26Sヘルパープラスミドにライゲーションさせて、dHcap6−mut1−dSL2(stop)およびdHcap6−mut1(W−stop)構築物を作製した。
【0136】
D.アルファウイルス糖タンパク質を発現する修飾されたプロモーターヘルパーカセットの構築
VEEカプシドタンパク質は、カプシドC末端トリプトファン残基の後で切断するキモトリプシン様プロテアーゼである。キモトリプシンの切断特異性に基づいて、すべてのアミノ酸残基が、メチオニンおよびプロリン以外のトリプトファンのすぐ下流の位置において許容されることが予想される。トリプトファンのすぐ下流にこれらのアミノ酸のいずれかを有することは、キモトリプシン切断活性を著しく減じることが予想される。天然VEEウイルスには、VEE E3シグナル配列を含む18のアミノ酸がある。E3シグナル配列のアミノ酸の数を減じると同時に、E3配列のシグナリング機能は維持するように構築物を設計した。E3配列を含む18のアミノ酸のうちの16は、カプシドC末端トリプトファンの下流の位置において許容されることが予想されるため、E3シグナル配列のアミノ酸の数を減らすことにより、切断部位として機能するであろう部位がVEE構造ポリタンパク質コード配列を再構成するヌクレオチド完全組換え現象の発生の際にC末端トリプトファンのすぐ下流に位置する場合、当該部位の数が減少するであろう。そのようなアプローチの例として、通常、E3シグナル配列に存在するN末端セリン残基をPCRによって除去し、N末端基残留物としてのロイシン残基を残して、そのような修飾されたgpヘルパーがVRPをパッケージするために機能するかどうかを決定するためにdHgpのプロモーターレスヘルパーを構築した。
【0137】
E3のN末端セリン残基を除去し、ユニークRsrII制限部位を維持するように、フォワードPCRプライマー(Gp(RsrII−Ser)F、配列番号94)を設計した(表8)。ユニークSnaBI制限部位を含有するであろうgp断片を増幅するように、リバースPCRプライマー(3−16.2.14、配列番号95)を設計した(表8)。結果として得られたgpPCR断片をRsrIIおよびSnaBIで消化させ、RsrIIおよびSnaBI消化dHgp6−mut1DNAにライゲーションして、dHgp6−mut1(−S)を生成させた。
【0138】
E.5’および3’修飾Δ26Sヘルパーを使用したVRP生成実験
上記の修飾の組み合わせを含有するヘルパーも調製した。dHcapおよびdHgpプロモーターレスヘルパーの様々な組み合わせおよびRNA濃度を、VRP産生実験において分析し、それらが、VEEレプリコンRNA(ボツリヌス神経毒素断片AまたはインフルエンザHAのどちらかを発現するもの)のパッケージングにおいてどの程度効果的であるかを決定した。さらに、VRP収量に関するΔ26Sヘルパーのキャッピングの効果を、当該ヘルパーの組み合わせのサブセットに対して分析した。VRP収量の代表例は、Δ26Sヘルパーの様々な組み合わせと共に表9〜13に示す。VRPの感染力および収量を定量化する力価アッセイは、48ウェルプレートのベロ細胞単層培養物において、VRPを段階的に希釈し、ベロ細胞と共に5%のCO2中において37℃で一晩インキュベートすることによって実施した。一晩インキュベート(18〜20時間)した後、当該細胞を洗浄し、固定化し、当該固定化された単層を抗原特異的一次抗体で染色した後、FITC結合二次抗体で染色した。紫外蛍光顕微鏡(Nikon Eclipse TE300)により、FITC標識化抗原−抗体を含有する細胞を検出した。個々の抗原陽性細胞をカウントし、IU/mLで表される力価を、既知の希釈および接種量から計算する。
【0139】
(実施例8)
ユビチキン単量体を取り込むプロモーターレスヘルパー
A.ユビキチン単量体を含有するΔ26Sヘルパーの構築
真核細胞において、ユビチキンによって縮合またはタグ付けされたタンパク質は、細胞のユビキチンカルボキシル末端ヒドロラーゼ(UCH:ubiquitin carboxyl−terminal hydrolase)によってC末端グリシンのすぐ後ろで切断される(Pickart and Rose,J.Biol.Chem 260:7903−7910(1985))。ユビチキンコード配列の単量体を、カプシドおよび糖タンパク質コード配列のすぐ上流のインフレームに配置することにより、本発明のある特定のプロモーターレスヘルパー構築物によって産生される融合タンパク質を排除する(そのような融合体は、各構造タンパク質コード配列に対してATGの上流にある複数の翻訳開始部位によって生じる)。全てのインフレーム融合タンパク質は、ユビキチン単量体を含むことになり、そのため、それらがUCHによって切断され、その結果、上流に外因性タンパク質配列を有さない完全長VEE構造タンパク質が放出されるため、当該排除が生じる。増幅されたユビチキン単量体コード配列の5’および3’末端にRsrII部位を導入すると共に、UCHによるユビチキン単量体の切断に必要なArg−Gly−Gly配列(図4)を維持するように、プライマーであるubiquitin F(配列番号96)およびubiquitin R(配列番号97)(表14)を設計した。これらの特定の構築物により、切断の後に得られる構造タンパク質のそれぞれに、天然の構造タンパク質には存在しないさらなるN末端アミノ酸残基(単数または複数)が生じた(すなわち、カプシドヘルパーに対しては余分なプロリンであり、糖タンパク質ヘルパーに対しては、余分なプロリンおよびトレオニンである)(図4)。
【0140】
当該ユビキチンコード配列を、Pfu Taqポリメラーゼ(Stratagene社)を使用してPCR増幅し、dHcap(FL)およびdHgp(FL)のユニークRsrII部位にクローン化した。ユビキチン挿入の方向を決定するために、形質転換体をスクリーニングした。カプシドおよび糖タンパク質に対する陽性ユビキチンクローンは、それぞれdHcapUおよびdHgpUと称し(それぞれ、本明細書において配列番号53および54として提供される5’末端配列を有する)、これらを単離し、配列決定して、増幅されたユビチキンコード配列中にエラーが導入されていないことを確認した。エレクトロポレーションのためのRNAは、RiboMax Express RNA(登録商標)キットを使用して、dHcapU、dHgpU、dHcap(FL)、dHgp(FL)、Hcap4、および13.4.6プラスミドから別々の反応において転写し、塩化リチウムで沈殿させた。
【0141】
B.ユビキチン修飾されたΔ26Sヘルパーを使用したVRP生成実験
ベロ細胞を、HIVクレードC糖タンパク質(「DU151gp160」)を発現するVEEレプリコンRNAでエレクトロポレーションし、指示されたRNA量でプロモーターレスカプシドおよびGPヘルパーの組み合わせを選択した。いくつかの実験において、「Hcap4」カプシドヘルパーを使用した。これは、切端された5’末端を有するが(上述のdHcap4切端物に相当する)、26Sサブゲノムのプロモーター配列を保有するヘルパーであり、米国特許第7,045,335号に詳細に記載されている。なお、この特許は参照により本明細書に組み込まれる。約0.8mlの容積の0.4cmのキュベットにおいて、500V、25μF、4パルスでエレクトロポレーションを実施した。各エレクトロポレーションは、100mlのOptipro(登録商標)(Gibco社、Carlsbad、CA)と共に1つの850cm2ローラーボトル中に播種した。18時間後に、25mlの0.5MのNaClで洗浄しながら0.2μmフィルタによりVRPを収穫した。VRP塩水洗浄物質を、(HIV gp160タンパク質を認識する)抗−gp120ヤギ抗体により1:400で力価決定した。パッケージング実験の結果を表15に示す。
【0142】
続いてエレクトロポレーションを実施して、dHgp6−mut1と組み合わされたカプシドヘルパーのdHcapUまたはdHcap6−mut1(W−stop)でパッケージされた様々な核酸の力価量を比較した。VEE nsP2特異的ポリクロナール抗体を使用してVRPの力価を決定した(表16)。
【0143】
C.エレクトポレーションした細胞におけるウエスタン分析による構造タンパク質の発現
表16にまとめられたパッケージング実験においてVRPを生成するために使用した細胞から細胞溶解物を調製した。各試料からの細胞溶解物を、4〜12%のBis−Tris Novexゲルにおいて、1×MOPS中200V、400mAにて45分間電気泳動した後、1×移行緩衝液中400mAで40分間、PVDFに半乾燥移行させた。膜を、1×BMBブロック/TBS中で一晩ブロッキングした。一次抗体は、1×BMBブロック/TBS中1A4A抗−VEE GPの1:500希釈および抗−VEEカプシドの1:1500希釈であった。ウエスタンブロット結果を図5に示す。dHgpUから発現した糖タンパク質は、dHgp(FL)から発現した糖タンパク質よりもより完全なPE2およびE2GP形態へとプロセシングされる。これは、dHgp(FL)にユビキチンが存在しない場合に見られる融合タンパク質のパターンにおける違いによって実証される(図5、GP抗体を使用したウエスタンブロットのレーン3及び4を比較)。dHcapUヘルパーにおけるカプシドタンパク質のN末端でのユビキチンタンパク質の置換により、結果として、カプシド融合タンパク質が消失し(図5)、13.4.6糖タンパク質ヘルパーでパッケージングされた場合にgp160の力価において2桁以上増加する(表15)。
【0144】
D.エレクトロポレーションした細胞のノーザン分析による構造タンパク質RNAの発現
全細胞RNAを、表15にまとめられたパッケージング実験においてVRPを生成するために使用された細胞から抽出した。当該細胞をRNAwiz(登録商標)試薬(Ambion,Inc社、Austin、TX)で溶解させ、クロロホルムで抽出し、沈殿させて、カプシド特異的プローブおよびGP特異的プローブを使用してノーザン分析に供した(それぞれ、図6および図7)。すべてのRNA種が、様々な構築物から予想されるサイズと一致している。
【0145】
(実施例9)
キャップおよび非キャップΔ26Sヘルパー構築物を使用したVRP生成
A.様々なアルファウイルスの糖タンパク質を発現するVRP
VRPは、対象となる核酸(NOI)として、それぞれフューリン切断部位が欠失したVEE(3022)、Eastern equine encephalitisウイルス(EEE)(4200)、またはWestern equine encephalitisウイルス(WEE)(2100)のいずれかの糖タンパク質に対するコード配列を発現するVEEレプリコンを使用して作製した。VRPを生成するために使用されたヘルパーをコードするDNAプラスミドは、NotIにより直線化し、インビトロで、T7RiboMax(登録商標)キット(Promega社、Madison、WI)を使用して、メーカーの指示に従い、指示された場合には7.5mMのPAP類似物(Promega社)で補って転写した。キャップ類似物で作製したヘルパーは、「+Cap」として示し、キャップ類似物無しで作製されたヘルパーは「−Cap」として示した(表17)。レプリコンの組み合わせを用いてベロ細胞をエレクトロポレーションし、上述の実施例5に記載したようにしてGPヘルパーRNAおよびVRPを作製した。三つの別々の実験の結果を表17〜19に示す。
【0146】
B.インフルエンザ株ウィスコンシンのHAコード配列を発現するVRP
この実験において、VEEカプシドまたはVEE糖タンパク質のいずれかをコードするΔ26SヘルパーRNAを作製するために、転写反応においてCap類似物とGTPのモル比を変更した。当該転写反応は次のように構成した:Promega 5×転写緩衝液;rNTPミックス(6mMのUTP、CTP、ATP);GTP(表に示されるように、0〜6mMで);(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1300)およびRibom7GCap(登録商標)類似物(6mM)(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1712)。メーカーによって指定された、通常、キットによって供給される2×緩衝液で実施されるRiboMax Express(登録商標)RNA転写キット条件(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1320)を模倣するために、7.5mMのRibom7GCap(登録商標)類似物の有無において、Promega社の5×緩衝液および7.5mMのrNTPにより、さらなる反応を行った。この実験でパッケージングされたVEEレプリコンは、インフルエンザHA(A/WI/05)タンパク質をコードした。各エレクトロポレーションに対して、30μgのレプリコンRNA;10μgのΔ26SカプシドヘルパーRNA、および60μgグラムのΔ26S糖タンパク質ヘルパーRNAを使用した。ベロ細胞を増殖させ、次いで洗浄し、スクロース緩衝液中に再懸濁させて1.2×108細胞/mLとした。これらの細胞をRNAと混合し、次いで、500ボルト、25μFd、および4パルスに設定したBioRad Gene Pulse II(登録商標)装置でエレクトロポレーションした。細胞を100mLのOptiPro(登録商標)と共にローラーボトルに移し、37℃でインキュベートした。エレクトロポレーション後24時間で、VRPを収穫した。当該VRPを、ベロの48ウェルプレートで力価を決定した。その結果を表25に示す。
【0147】
(実施例10)
Δ26Sヘルパー構築物によって作製したVRPを使用したネズミにおけるボツリヌス神経毒素に対する防御
ボツリヌス神経毒素血清型AまたはB(それぞれ、BoNT AまたはBoNT B)のいずれかの重鎖の無毒のC末端断片を発現するVEEレプリコンベクターを、実施例5で記載されているように、次のいずれか:(i)それぞれ30μgのキャップされていない13.2.2および13.4.6ヘルパー、または(ii)20μgのキャップされたカプシドΔ26Sヘルパーおよび60gのキャップされた糖タンパク質Δ26Sヘルパー、を使用してVRP中にパッケージングした。これらのVRPを、0日〜28日に二回、スイスマウスにワクチン接種するために1×107IUの投与量で使用した。当該マウスを、2回目の免疫処置後1カ月において、BoNT AまたはBoNT B神経毒のいずれかの、動物の50パーセントが死亡するのに必要な投与量(1000LD50)の1000倍を用いてチャレンジ感染させた。当該チャレンジ感染実験の結果を表20にまとめる。
【0148】
(実施例11)
天然痘ウイルス由来の抗原を発現するVRPによる免疫原生および防御実験
A.Δ26Sヘルパー構築物を使用して生成させたVRPの、マウスおよび霊長動物における免疫原生
4つのワクシニアウイルス(VACV:vaccinia virus)遺伝子(L1R、B5R、A27L、およびA33R)を発現するために最適化されたVEEレプリコンベクターを、Kamrudらによって記載された方法(Virology 360(2):376−87(2007))を使用して構築した。当該4つのVACV遺伝子は、まとめて「4pox」と呼ばれる。1つは3014株をベースとし、もう一つはTC−83ワクチン株をベースとする二つの異なるVEEレプリコンベクターシステム中に、当該4pox遺伝子をクローン化した。各最適化されたVACVコード配列発現レプリコンベクターを、30μgのレプリコン、20μgのΔ26SカプシドヘルパーRNA、および60μgのΔ26SGPヘルパーRNAを組み合わせて、それらをベロ細胞中にエレクトロポレーションすることによってVRPを生成するために使用した。粒子は、実施例5に記載されているようにして作製し収集した。次いで、個々のVGACV VRPを組み合わせて、BALB/cマウスまたはカニクイザルのいずれかを免疫化するために使用する4poxVRP混合物を作製し、VACV抗原特異的ELISA分析により、体液性応答を測定した。ワクチン接種したネズミにおいて検出されるVACV特異的ELISA反応を表21に示し、予ワクチン接種したカニクイサルで検出されるVACV特異的ELISA反応を表22に示す。
【0149】
B.4pox VRPを使用した、ネズミおよび非ヒト霊長動物における防御
1.マウス
ワクシニアウイルス(IHD−J株)の2×106PFUを鼻腔内経路によりマウスにチャレンジ感染させ、その結果を表23に示す。
【0150】
2.非ヒト霊長動物
サル痘ウイルスの5×106PFUを静脈内経路により、非ヒト霊長動物にチャレンジ感染させた。世界保健機構の病巣数採点システムを使用して、重症度を判定し、その結果を表24に示す。
【0151】
当業者に理解されるように、特許請求する発明の各態様に対していくつかの実施形態および要素があり、異なる要素の全ての組み合わせが、本発明の実施形態として本明細書に組み込まれ、したがって、本明細書に例示する特定の組み合わせは、特許請求する発明の範囲の限定として解釈されない。特定の要素を除去するかまたはある組み合わせに利用できる要素の群に加える場合、要素の当該群は、そのような変更が組み込まれていると解釈される。
【0152】
非特許出版物、特許出願、および特許などの本明細書で引用されたすべの参照文献は、それぞれが個々におよび具体的に、その全部が参照文献に組み込まれていることが示される場合と同程度に、参照により本明細書に組み込まれ、その全てが本明細書で再現された。
【0153】
【表1】
【0154】
【表2】
【0155】
【表3】
【0156】
【表4】
【0157】
【表5】
【0158】
【表6−1】
【0159】
【表6−2】
【0160】
【表7】
【0161】
【表8】
【0162】
【表9】
【0163】
【表10】
【0164】
【表11】
【0165】
【表12】
【0166】
【表13】
【0167】
【表14】
【0168】
【表15】
【0169】
【表16】
【0170】
【表17】
【0171】
【表18】
【0172】
【表19】
【0173】
【表20】
【0174】
【表21】
【0175】
【表22】
【0176】
【表23】
【0177】
【表24】
【0178】
【表25−1】
【0179】
【表25−2】
【技術分野】
【0001】
優先権の声明
この出願は、米国特許法§119(e)の下、2007年6月21日に出願された米国仮出願第60/936,637号(この全体の内容は、参考として本明細書に援用される)の利益を主張する。
【0002】
政府の支援の声明
本発明の局面は、国立衛生研究所(National Institutes of Health)からの助成金第5UO1A1057286−03の下での資金拠出によって援助された。米国政府は、本発明に特定の権利を有する。
【0003】
発明の分野
本発明は、組換えアルファウイルス粒子のための改良された構築物およびその製造方法に関する。
【背景技術】
【0004】
アルファウイルスは、現在、感染性疾患および癌のワクチンを開発するためのベクタープラットフォームとして用いられている(例えば、特許文献1;特許文献2;特許文献3;特許文献4;特許文献5;特許文献6;特許文献7;特許文献8;特許文献9;特許文献10;特許文献11;特許文献12;非特許文献1、非特許文献2;非特許文献3を参照のこと)。アルファウイルスは、Togaviridae科の属を含み、当該属のメンバーは、世界中の至る所で脊椎および無脊椎の双方の宿主において見出される。ベクタープラットフォームのためにアルファウイルスの中で最も研究されているのは、当該属のプロトタイプのメンバーであるVenezuelan Equine Encephalitis(VEE)Virus、Semliki Forest Virus(SFV)、およびSindbis Virus(SV)である。
【0005】
そのようなベクタープラットフォームの1つは、Garoffらの米国特許第6,190,666号、Johnstonらの特許文献1および特許文献2、Dubenskyらの米国特許第5,814,482,同第5,843,723号、特許文献10、特許文献11、同第6,105,686号、および同第6,376,236号、米国特許出願公開第2002−0015945号(Poloら)、米国特許出願公開第2001−0016199(Johnstonら)、Frolovら(1996)Proc.Natl.Acad.Sci.USA 93:11371−11377、並びに非特許文献1に記載されているアルファウイルスレプリコンシステムである。アルファウイルスレプリコンベクターは、対象となる1つ以上の核酸を含有しかつ発現するように操作され、この場合、対象となる核酸は、例えば、抗原、サイトカイン、リボザイム、または酵素をコードし得る。アルファウイルスレプリコンベクターは、任意のアルファウイルス由来であってもよく、中でも、例えばVenezuelan Equine Encephalitis(VEE)ウイルス、Sindbisウイルス、例えばTR339株、South African ArboウイルスNo.86、およびSemliki Forestウイルス由来であってもよい。当該ベクターは、次に培養中の細胞に導入され、それによってアルファウイルスの複製が可能になり、さらにこの細胞においてアルファウイルス構造タンパク質も発現され、その結果、当該ベクターは、アルファウイルス構造タンパク質によってアルファウイルスレプリコン粒子(ARP)中にパッケージされる。次いで、ARPが培養物から収穫され、様々な治療目的のために被験体へと提供される。
【0006】
ワクチン用途におけるARPシステムの免疫原性および有効性を高めるために、様々な構築物が開発されている。これらの構築物の多くは、さらに、ゲノム断片の組換えによって複製能力のあるアルファウイルスが形成される可能性を低減するように設計されている。Johnstonら(特許文献1および特許文献2)は、単一のヘルパーシステム(このシステムにおいて、アルファウイルスの構造タンパク質遺伝子の完全なセットが1つのRNA分子上にあり、非構造タンパク質遺伝子および対象となる非相同核酸は別のレプリコンRNA上にある)からの組換えの可能性を認識しており、したがって、構造タンパク質をコードする2つのヘルパーRNAを採用した「二重ヘルパー」システムを設計した。Dubenskyら(特許文献10)およびPoloら(米国特許第6,242,259号)は、パッケージング細胞の培養物中への複製性アルファウイルスベクターの導入に関してこれら構造タンパク質を発現するRNAの産生によってアルファウイルスベクターをパッケージするための、パッケージング細胞株に安定してトランスフォームされる2つのDNAアルファウイルス構造タンパク質発現カセットの使用について記載している。Liljestromおよび共同研究者は、「単一のヘルパーシステム」が、野生型アルファウイルス粒子を生成することを確認するデータを提示している(Berglund,et al. Biotechnology 11(8):916−920(1993))。スミスらは、構造タンパク質の発現を誘導する他の新規のRNAヘルパーについて記載している(国際公開第2004/085660号)。
【0007】
上記のように、3つのうちの2つがヘルパーシステムを含む3つの核酸間にウイルスのコード配列を分配することにより、単一ヘルパーシステムと比較して、複製能力のあるウイルス(「RCV(replication−competent virus)」を生じる組換えの理論的頻度が著しく減少する。これらのシステムは、非依存性転写単位として機能する構造物を提供するために、しばしば26Sプロモーターまたはウイルス接合領域プロモーターとも呼ばれるアルファウイルスサブゲノムプロモーターの使用、並びにアルファウイルスRNAポリメラーゼ認識シグナルの使用を含み、それによって、当該ヘルパーシステムは、ヘルパー機能の増幅および効率的な発現のためにアルファウイルス複製機構の存在を利用することができる。
【0008】
既存のシステムでは、既知のパッケージングシグナルは、通常はレプリコンRNAに含まれており、ヘルパー構築物からは除かれる。しかしながら、それでもなお、ヘルパーRNAは、低頻度ではあるがパッケージングまたは同時パッケージングされ(Lu and Silver.J.Virol Methods,91(1):59−65(2001))、末端認識シグナルを有するヘルパー構築物が、レプリコンの存在下において増幅および発現され、他のヘルパー分子もしくはレプリコンRNAの組換え現象を生じる可能性がある。
【0009】
アルファウイルスレプリコン粒子による動物研究では、105〜108の範囲の投与量が採用されており、非ヒト霊長動物においては107、5×107、および108が効果的に採用され、当該投与量は、ヒトの臨床試験において使用される。さらに、2×108、5×108、および109などの多量の投与量も、ヒトに対する適用において有用である。そのような投与量は、大規模の製造手段を必要とし、そのような規模では、統計的に、複製能力のあるアルファウイルスが既存のRNAヘルパーシステムによって生成され得る可能性がある。
【0010】
したがって、複製能力のあるアルファウイルスの形成に対する予測される頻度をさらに減少させ、並びに製造戦略およびコストを最適化するために、アルファウイルスレプリコン粒子を製造するための改善されたシステムが当技術分野において依然として必要とされている。
【0011】
本発明は、アルファウイルス構造タンパク質をコードするアルファウイルスRNAヘルパー分子であって、プロモーター配列を欠失しているためにヘルパー分子とレプリコンベクターとの間で生じ得る機能的組換え現象の理論上の回数を著しく減少させ、結果として、組換えアルファウイルス粒子の製造中に複製能力のあるアルファウイルスが形成される割合の理論的予測が減少する、アルファウイルスRNAヘルパー分子を提供する。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】米国特許第5,792,462号明細書
【特許文献2】米国特許第6,156,558号明細書
【特許文献3】米国特許第5,811,407号明細書
【特許文献4】米国特許第6,531,135号明細書
【特許文献5】米国特許第6,541,010号明細書
【特許文献6】米国特許第6,783,939号明細書
【特許文献7】米国特許第6,844,188号明細書
【特許文献8】米国特許第6,982,087号明細書
【特許文献9】米国特許第7,045,335号明細書
【特許文献10】米国特許第5,789,245号明細書
【特許文献11】米国特許第6,015,694号明細書
【特許文献12】米国特許第5,739,026号明細書
【非特許文献】
【0013】
【非特許文献1】Pushkoら、Virology(1997)239(2):389−401
【非特許文献2】Frolovら、J.Virol.(1997)71(1):248−258
【非特許文献3】SmerdouおよびLiljestrom,J.Virol.(1999)73(2):1092−1098
【発明の概要】
【課題を解決するための手段】
【0014】
本発明は、a)開始コドンが除去されているアルファウイルス5’複製認識配列と、b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、c)アルファウイルス3’複製認識配列とを含む単離されたRNA分子であって、当該RNA分子が、(b)のヌクレオチド配列の転写を誘導するプロモーターを含まず、アルファウイルス5’および3’複製認識配列がアルファウイルス非構造タンパク質の存在下において当該RNA分子全体の複製を誘導する、RNA分子を提供する。
【0015】
さらに、本明細書において、本発明のRNA分子の1つ以上を細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によって、アルファウイルスレプリコン粒子が産生される条件下において、RNA分子の組み合わせが、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生のために必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0016】
さらに、a)アルファウイルスレプリコンRNAと、b)本発明のRNA分子の1つ以上と、c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物とを細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によって、アルファウイルスレプリコン粒子が産生される条件下において、(b)のRNA分子と(c)のヘルパー構築物の組み合わせが、アルファウイルスレプリコン粒子の産生のために必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0017】
さらなる実施形態において、本発明は、アルファウイルスレプリコン粒子の集団であって、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含有しない集団を提供する。
【0018】
さらに本明細書において、培養における許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含まないアルファウイルスレプリコン粒子の集団であって、当該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異(attenuating mutation)を含む集団が提供される。
【0019】
さらに、本発明は、被験体において免疫応答を誘発する方法であって、有効量の本発明のアルファウイルスレプリコン粒子の集団を当該被験体に投与する段階を含む方法を提供する。
【図面の簡単な説明】
【0020】
【図1】完全長(FL:full length)プロモーターレスヘルパー分子の5’複製認識配列(RRS:replication recognition sequence)の構築物を示す。5’複製認識配列内のカプシドまたは糖タンパク質(GP:glycoprotein)開始コドンの上流の出発コドンの位置を、枠線と黒線で示す。枠線は、カプシドまたはGPのためのコード配列とインフレームである出発コドンを示す。黒線は、カプシドまたはGPのためのコード配列とリーディングフレーム外である出発コドンを示す。線の真下の数値は、当該分子の5’末端から付番された、5’複製認識配列における推定上の出発コドンに対する最初のヌクレオチド配列の位置を示す。
【図2】プロモーターレスヘルパー分子における5’複製認識配列欠失の構造を示す。枠線のボックスは、各構築物に残存している5’複製認識配列を示し、ボックス内の数値は、配列のヌクレオチドの長さを示す。黒い細線は、各構造物から削除されている5’複製認識配列を示す。縞の斜線のボックスは、カプシドまたはGPのいずれかのためのコード配列の位置を表す。
【図3】ノーザンブロット分析を示す。全細胞RNAを、30μgのpERK/342/MS/BoNT AレプリコンRNAおよび30μgのdHE1−6M1(プロモーターレスE1ヘルパー)もしくは26Sプロモーター(13.4.6)を含有する30μgのGPヘルパーRNAのいずれかによってエレクトポレーションしたベロ細胞から抽出した。各試料(5μg)のRNAを1%のグリオキサールゲル上で展開し、BrightStar(登録商標)膜(Ambion社;Austin、TX)に転写した。ゲノムのセンスアルファウイルスRNA3’末端に特異的なプローブを使用して、ヘルパーの複製を検出した。レーン1:RNA分子量マーカー、レーン2:dHE1−6M1ヘルパー+BoNT Aレプリコン、レーン3:プロモーター補助GPヘルパー+BoNT Aレプリコン。
【図4】ユビキチン化構築物(dHcapUおよびdHgpU)または標準構築物(dHcapおよびdHgp)の、ユビキチン単量体のC末端アミノ酸およびヌクレオチド配列、並びにアルファウイルスカプシドおよび糖タンパク質コード配列のN末端残基を示すダイヤグラムを示す。これらの配列の右端の「Met Phe」、「Pro Met Phe」、「Pro Thr Met Ser」、および「Thr Met Ser」は、カプシドおよびGPタンパク質のN末端に見られるアミノ酸を表す。ユビキチン化構築物は、13.2.2および13.4.6ヘルパーには見られないさらなるN末端残基を有する。右側のボックスは、ヌクレオチド一次配列の結果としてコードされた3’RsrII制限部位およびアミノ酸を示す。左側のボックスは、VEE構造タンパク質からのユビキチンの切断に重要な残基を表す。
【図5】パッケージングの実験(表10)においてVRPを作製するためにエレクトポレーションされた細胞から生じた細胞溶解物のウェスタンブロット分析(一方はカプシド−特異抗体を使用し、他方は糖タンパク質(GP)特異抗体を使用する)を示す。以下に示すように、レプリコンと共に2つのRNAヘルパーを細胞中にエレクトポレーションした:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6;レーン9、Hcap4および13.4.6レーン10、分子量マーカー。
【図6】以下に示すように、レプリコンと共に2つのRNAヘルパーを細胞中にエレクトポレーションしたベロ細胞で産生されたカプシドヘルパーRNAのノーザンブロット分析を示す:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6。各レーンにおける翻訳可能なカプシドRNA分子は、アスタリスクでマークしてある。
【図7】以下に示すように、レプリコンに加えて、2つのRNAヘルパーを細胞中にエレクトポレーションしたベロ細胞で産生された糖タンパク質(GP)ヘルパーRNAのノーザンブロット分析を示す:レーン1、dHcap6−mut1および13.4.6(GP);レーン2、Hcap4およびdHgp6−mut1;レーン3、dHcapUおよびdHgpU;レーン4、dHcap(FL)およびdHgp(FL);レーン5、Hcap4およびdHgpU;レーン6、Hcap4およびdHgp(FL);レーン7、dHcapUおよび13.4.6;レーン8、dHcap(FL)および13.4.6。各レーンにおける翻訳可能な糖タンパク質RNA分子は、アスタリスクでマークしてある。
【発明を実施するための形態】
【0021】
本明細書で使用される場合、「1つの(”a”、”an”)」および「当該(”the”)」は、それが使用される状況に応じて、1つまたは2つ以上を意味し得る。例えば、「1つの(“a”)」細胞は、1つの細胞または複数の細胞を意味し得る。
【0022】
さらに、本明細書で使用される場合、「および/または(”and/or”)」は、1つ以上の関連する列挙された項目の任意のかつ全ての可能な組み合わせ、並びに代替(「または」)において解釈される場合は組み合わせの欠如を意味しかつ包含する。
【0023】
さらに、本明細書で使用される場合、「約(”about”)」なる用語は、例えば、本発明の化合物または薬剤の量、投与量、時間、温度などの測定可能な値を意味する場合、指定された量の±20%、±10%、±5%、±1%、±0.5%、またはさらに±0.1%の変動を包含することを意味する。
【0024】
「5’アルファウイルス複製認識配列」、「3’アルファウイルス複製認識配列」、「5’複製認識配列」、および「3’複製認識配列」なる用語は、アルファウイルス中に見られるRNA配列、そこから誘導される配列、または様々なアルファウイルス間で保存される配列をベースとする合成配列を意味し、非構造アルファウイルスレプリカーゼタンパク質によって認識されてウイルスRNAの複製を引き起こす。いくつかの実施形態において、これらの配列は、VRPを作製するための本発明の構築物、プラスミド、および核酸の調製、突然変異、および/または操作を容易にするために、DNAの形態であってもよい。これらの配列はさらに、「5’および3’末端」とも呼ばれ、非構造タンパク質媒介性増幅に必要な5’および3’ウイルス配列、非構造タンパク質媒介性増幅に必要な5’および3’配列、5’または3’保存配列要素(CSE:conserved sequence element)、5’または3’非コード領域、5’または3’非コード領域配列、複製のためにシス型において必要な5’または3’ウイルス配列、アルファウイルスの転写を開始する5’または3’配列、並びに/あるいはアルファウイルス5’および3’配列、を意味し、5’および3’の指定はアルファウイルスゲノムにおけるそれらの位置を意味している。本発明の核酸分子において、これらの5’および3’末端を使用して、2つの末端の間でコードされるRNA配列の複製および/または転写が行われる。これらの配列を標準的な分子生物学的技術によって修飾することにより(例えば、開始(すなわち、出発)コドンを除去するために、または翻訳性を高めるために、どちらかの末端で切端および/または修飾することにより)、組換えの可能性のさらなる最小化および/またはクローニング部位の導入などが可能であるが、ただし、当該配列は、依然としてアルファウイルス複製機構によって認識されなければならない。
【0025】
本明細書で使用される場合、「開始コドン」または「出発コドン」は、機能タンパク質の翻訳において使用されてもされなくてもよいコドンを意味し、これはRNAではAUGであり、DNAではATGである。
【0026】
「アルファウイルス構造タンパク質/タンパク質(単数または複数)」なる用語は、アルファウイルスによってコードされる構造タンパク質の1つまたは組み合わせを意味する。これらは、ポリタンパク質として野生型ウイルスにより産生され、一般的に、文献においてC−E3−E2−6k−E1として記載される。E3および6kは、2個の糖タンパク質であるE2およびE1の膜貫通/輸送シグナルとして機能する。したがって、本明細書におけるE1なる用語の使用は、E1、E3−E1、6k−E1、またはE3−6k−E1を意味し得、並びに本明細書でのE2なる用語の使用は、E2、E3−E2、6k−E2、PE2、p62、またはE3−6k−E2を意味し得る。「糖タンパク質ヘルパー」または「GPヘルパー」なる用語は、通常、本明細書において、E2およびE1糖タンパク質の両方をコードするヘルパー分子を意味し、本発明のある特定の実施形態において、E1およびE2は、別々のヘルパー分子でコードされる。
【0027】
「ヘルパー(単数または複数)」および「ヘルパー分子」なる用語は、互換的に使用され、1つ以上のアルファウイルス構造タンパク質をコードする核酸を発現する核酸分子を意味する。
【0028】
「ヘルパー細胞」および「パッケージング細胞」なる用語は、本明細書において互換的に使用され、アルファウイルスレプリコン粒子を産生する細胞を意味する。ヘルパー細胞は、1つ以上のアルファウイルス構造タンパク質をコードする、本明細書に記載されているようなヘルパー分子および/またはヘルパー構築物のセットを含む。ヘルパーは、RNAもしくはDNAであってもよく、またはその両方であってもよい。ヘルパー細胞またはパッキング細胞は、アルファウイルス許容性の、すなわちレプリコンRNAの導入においてアルファウイルス粒子を産生することができる任意の細胞であってもよい。アルファウイルス許容性細胞としては、これに限定されるものではないが、ベロ細胞、ベビーハムスター腎臓(BHK:baby hamster kidney)細胞、293細胞、293T/17(ATCCアクセッション番号CRL−11268)細胞、ニワトリ胚繊維芽細胞(CEF:chicken embryo fibroblast)、UMNSAH/DF−1(ATCCアクセッション番号CRL−12203)細胞、およびチャイニーズハムスター卵巣(CHO:Chinese hamster ovary)細胞などが挙げられる。
【0029】
本明細書で使用される場合、「プロモーター」は、RNA分子の転写を誘導する核酸配列である。
【0030】
本明細書で使用される場合、「単離された細胞」は、細胞が天然環境から分離され、および/または細胞がその自然環境において発生した状態から変更もしくは修飾された細胞または細胞の集団である。本発明の単離された細胞は、例えば、細胞培養物中の細胞であってもよい。本発明の単離された細胞はさらに、動物の細胞および/または動物に導入された細胞であってもよく、この場合、当該細胞は、例えば、本発明のアルファウイルス粒子の細胞中への導入によって変更もしくは修飾されている。
【0031】
本明細書で使用される場合、「アルファウイルスサブゲノムプロモーター」または「26Sプロモーター」は、アルファウイルス複製プロセスの一部としてサブゲノムのメッセンジャーRNAの転写を誘導する野生型アルファウイルスゲノムにおいて本来定義されるようなプロモーターである。
【0032】
本発明のいくつかの実施形態において使用される非相同核酸(例えば、対象となる遺伝子もしくは「GOI(gene of interest)」、または対象となる核酸もしくは「NOI(nucleic acid of interest)」)は、野生型アルファウイルスのゲノム内には存在しない核酸、および/または野生型アルファウイルスのゲノム内には本発明の組換え核酸中に存在する順序と同じ順序では存在しない核酸である。例えば、ある特定の実施形態において、NOIは、それらが、感染性の複製能欠損アルファウイルス粒子(例えば、アルファウイルスレプリコン粒子)のアセンブリにおいてヘルパー核酸として使用される場合、あるいは、ある特定のアルファウイルスによって引き起こされた疾病に対するワクチンのための免疫原として使用される場合に、1つ以上アルファウイルス構造タンパク質(例えば、C、PE2/E2、E1、E3、6K)をコードし得る。
【0033】
本発明は、アルファウイルス構造タンパク質(単数または複数)並びにアルファウイルス5’および3’配列をコードするヌクレオチド配列を含むRNA分子であって、この場合、開始コドンが5’複製認識配列から除去されており、しかし、プロモーター配列(例えば、サブゲノムアルファウイルスプロモーター配列であり、場合によって26Sプロモーターもしくはウイルス接合領域プロモーターとも呼ばれる)を欠失しているRNA分子が、培養細胞株におけるアルファウイルスレプリコン粒子の産生に対して、完全長プラス鎖RNAが効率的に翻訳され、十分な量のアルファウイルス構造タンパク質がトランス型において産生されるように複製され得るという驚くべき予想されない発見に基づいている。場合によって本明細書で「Δ26Sヘルパー」とも呼ばれるこれらの「プロモーターレス」RNA分子は、ヘルパー分子とレプリコンベクターの間で生じる機能的組換え現象の発生の予測された理論上の頻度を減少させることにより、(例えば、ワクチンまたはアジュバントとしての使用のために製造された)アルファウイルスレプリコン粒子の集団における理論上の安全域を増加させる。
【0034】
いずれのスプリットヘルパーシステムも、複製能力のあるアルファウイルス(RCV)を生成するために、最小限の2つの独立した組換え現象を必要とする。相同組換えも報告されているが、アルファウイルスでは、組換えは主にRNA複製複合体によるランダムな鎖スイッチングの結果であると考えられている(Weissら、1991)。第一の組換え現象では、複製複合体は、例えば、文献(例えば、Johnstonら米国特許第5,792,462号)において開示されているスプリットヘルパー/レプリコンパッケージングシステムにおけるRNAヘルパー分子の3’末端であり得る。当該複合体が、26Sまたはウイルス接合領域を介してこのヘルパーRNAの複製を継続し、次いで、鋳型としてのレプリコンRNAの外来ヌクレオチド配列にスイッチングし、レプリコン5’末端を介して複製を完了するのであれば、結果として得られる「組換えレプリコン中間体」は、すべての非構造タンパク質をコードする配列と、領域をコードする外来の対象となる核酸(NOI)を含有する転写単位のいくつかまたはすべてと、アルファウイルス構造タンパク質の1つを発現する新しい挿入された転写単位とを含有するであろう。RCVが産生されるためには、続く第二の組換え現象が、(上に記載された)組換えレプリコン中間体の3’複製認識配列への鎖スイッチング現象によって生じなければならない。なぜなら、これが、挿入突然変異誘発を伴わないで、配列をコードする全ての非構造タンパク質および構造タンパク質のための機能的な転写単位が保持される唯一の位置であるからである。ヘルパーRNA分子が26Sプロモーターを含有しているために、理論上、そのような2つの組換え現象によってRCVが生じ得る。各組換え挿入体は、非依存性転写単位であろうから、正確な組み換え点は重要ではないだろう。
【0035】
本発明のプロモーターレスヘルパー分子を使用したRCVの生成も、最小限の2つの非依存性組換え事象を必要とするだろうが、機能的な遺伝子組換え体が得られる制約は、文献において知られているRNAヘルパー分子の場合よりもはるかに高く、したがって、RCVが生成される理論上の頻度ははるかに低い。これは、26Sプロモーターの不在下では、ほとんどの組換え現象により、結果として、アルファウイルス構造タンパク質を発現し得る機能的な転写単位が生じるためであろう。したがって、プロモーターレスヘルパー分子を使用するRCVの生成は、構造的ポリプロテインのオープンリーディングフレームの再生を必要とするであろうし(すなわち、これらのヘルパーが由来する野生型ウイルスに見られる構造と実質的に同様に)、これ自体が、2回の必要な組換え現象が特異的な順序においてかつ非常に特異的なヌクレオチド位置において生じることを必要とする。最初の組換え現象は、カプシドヘルパーコード配列を伴わなければならない。なぜなら、それ自体が分裂し、機能的なカプシドタンパク質を生成するためには、糖タンパク質に対して適切な(すなわち、5’)位置になければならないためである。カプシドヘルパーは、レプリコン26Sプロモーターからカプシドタンパク質が発現する遺伝子組換えを達成するために、ヌクレオチドの完全組換え現象またはヌクレオチドのほぼ完全組換え現象を介して、レプリコンベクターで組換えられなければならない。すなわち、1)レプリコン26Sプロモーターのすぐ下流であり、2)非相同的GOIの任意の残りとインフレームであり、並びに3)結果としてGOI/アルファウイルスカプシド融合タンパク質を生成しない(したがって、不活発なアルファウイルスカプシドを産生する)組換えのみが、機能的であるだろう。アルファウイルス糖タンパク質ヘルパーによる第二の組換え現象は、3’複製認識配列での発生に制限されることに加えて、第一の組換え事象と同じ制約下にある。したがって、プロモーターレスヘルパー分子を用いて粒子を製造する本発明の方法では、RCVが生成される頻度は、理論上、文献において知られているヘルパー分子よりはるかに低いであろうし、非常に低い頻度であるために、本発明の方法ではそのようなRCVが検出されなかった。
【0036】
本発明の驚くべき特徴は、アルファウイルス粒子をアセンブルするためのヘルパー/レプリコンシステムを製造する従来の試みが、アセンブリのための構造タンパク質を翻訳するのに十分なRNA分子を得るために、強力なプロモーター、たいていの場合はアルファウイルス26Sサブゲノムプロモーター、の使用に頼っていたという事実に存する。既存の文献との明確な対比において、本発明者は、通常、野生型アルファウイルスの増殖、および文献において知られているヘルパーRNAシステムにおいて伴われる、26Sプロモーターからのより小さなメッセンジャーRNAの転写と当該メッセンジャーの増幅とがなくても、完全長分子として直接翻訳され得る新規のRNAヘルパー分子を利用することができるということを発見した。したがって、本発明のヘルパーRNAの直接翻訳は、細胞のリボソーム装置による完全長RNAの5’末端におけるキャップの認識を介して達成される。真核細胞内では、mRNAに由来する翻訳の開始は、リボソームサブユニットのmRNAへの補充を可能にする厳しく制御された一連の現象を伴う。キャップ依存性翻訳の場合、「キャップ」として知られる、mRNAの5’末端に存在するメチル−7−G(5’)pppN構造が、eIF4E、eIF4G、およびeIF4Aから構成される開始因子eIF4Fによって認識される(Hershey&Merrick.Translational Control of Gene Expression,pp.33−88.Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press.2000において概説されている)。
【0037】
アルファウイルスは、プラス鎖RNAウイルスであり、すなわち、ウイルスRNAが細胞に入ると、このRNAから非構造アルファウイルスタンパク質(nsP1、nsP2、nsP3、およびnsP4)の翻訳が生じ、これらのタンパク質が、それぞれ、完全長マイナス鎖RNA鋳型を生成する。次いで、マイナス鎖RNAが複製され、完全長(「ゲノム」)プラス鎖RNAと、26Sプロモーターで始まるより小さい(「サブゲノム」)プラス鎖とを産生する。プラス鎖が産生される場合、アルファウイルスの非構造タンパク質も、RNAをキャップして、リボソームによる翻訳のために細胞質において利用できるようにする。「キャップ」は、RNAの5末端に付加したメチル化残基を意味する。本発明の特定の実施形態において、インビトロにおいて産生されるヘルパーRNA分子(これは、プラス鎖RNAである)は、キャップされていない。プラス鎖ヘルパーRNAが、レプリコンRNAを含有する真核細胞に導入されると、主にマイナス鎖の合成が最初に生じる。非構造タンパク質を合成するアルファウイルスレプリコンRNAの存在下において、マイナス鎖鋳型(ヘルパーとレプリコンの両方)は、次にキャップされたプラス鎖RNAへと複製される。本発明の他の実施形態において、ヘルパーRNAは、当技術分野において周知の試薬および、例えば、Promega社(Madison、WI)およびAmbion社(Austin、TX)から市販の試薬を使用して、インビトロでキャップされ得る。キャップは、例えば、Gキャップ、Cキャップ、Aキャップ、メチル化G(m7G(5’ppp(5’)pppG(5)A);非メチル化G(G(5’ppp(5’)A);ARCA(抗−リバースキャップ類似物、3−O−Me−m7G(5’)pppG(5));トリメチル化(m2、2,7G(5’ppp(5’)pppG)、2ウェイキャップ(m7G(5’ppp(5’)m7G)を含み得る。いくつかの実施形態において、ARPの最大収量のために、ARPを産生するために使用される本発明のすべてのヘルパーは、キャップされているかまたはキャップされていないかのいずれかである。最も高い収量は、キャップされたヘルパーRNAによって記録されているが、ある特定の実施形態において、キャップされていないヘルパーRNAも、著しく高い収量において生成し得るので、キャッピングのさらなるコストを避けることができる。ヘルパーRNAの一方のみをキャップすることも可能であるが、これも、場合によってARPの収量を制限する場合がある。概して、本発明者は、キャップの使用、転写混合物中のNTPに対するキャップ類似体の比率、およびARPを生成するために使用されるRNAの比率の変更などの、いくつかの変数を調べる通常の実験によってARP収量を最適化することができるということを示している。
【0038】
したがって、特定の実施形態において、本発明は、a)少なくとも1つの開始コドンが除去されているアルファウイルス5’複製認識配列と、b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、c)アルファウイルス3’複製認識配列とを含む、から実質的に成る、および/またはから成る単離されたRNA分子であって、ただし、当該RNA分子は、(b)のヌクレオチド配列の転写を誘導するプロモーターを含まず、アルファウイルス5’および3’複製認識配列が、アルファウイルス非構造タンパク質の存在下においてRNA分子全体の複製を誘導する、RNA分子を提供する。
【0039】
様々な核酸配列が、本発明の核酸構築物において5’末端および3’末端の機能を満たし得る。例えば、当該配列は、VEEアルファウイルスに対して、上で例示されているような、アルファウイルス5’複製認識配列および他の隣接する配列を含み得る。さらに、天然の5’アルファウイルスの末端において欠失を生じさせて、ある特定な二次構造要素、例えば、ステムループ構造など、を除去することもできる。ある特定の実施形態において、これらのステムループ構造の1つ以上が、本発明のヘルパー構築物から除去され得る。あるいは、例えばSindbisウイルスの場合の、tRNAアスパラギンに対するヌクレオチド10〜75(Schlesingerら、米国特許第5,091,309号)など、同様の機能的能力を維持しながら、この要素として非アルファウイルスまたは他の配列を利用することもできる。
【0040】
いくつかの実施形態において、3’アルファウイルス複製認識配列は、長さが約300ヌクレオチドであり得、実質的に天然のアルファウイルス3’複製認識配列を含有する。アルファウイルス間で保存される最小の3’複製認識配列は、19ヌクレオチド配列である(Hillら、Journal of Virology,2693−2704(1997))。加えて、Sindbisウイルスでは、3’複製認識配列のすぐ後に続くポリ(A)テールは、長さが少なくとも11〜12の残基でなければならず、並びに、3’複製認識配列の3’13ntは、効率的なマイナス鎖RNAの合成にとって重要であるということが示されている(Hardy and Rice, Journal of Virology,79:4630−4639(2005))。したがって、3’末端のための配列は、完全なアルファウイルス3’複製認識配列、または依然として認識配列としての機能を維持する3’複製認識配列の切端した領域、または長さが25〜325ヌクレオチドでありかつ3’複製認識配列のすぐ後に続く最小長さ11〜12ntのポリ(A)ランを含む3’末端を含み得る。このコンテキストにおいて使用することができる配列の他の例としては、これらに限定されるものではないが、非アルファウイルス、または同様の機能的能力を維持してマイナス鎖RNA合成の開始を可能にする他の配列(例えば、GeorgeらJ Virol.74:9776−9785(2000)に記載されている配列)が挙げられる。
【0041】
本発明のRNA分子において使用される5’および3’複製認識配列は、任意の組み合わせにおける同一のまたは異なるアルファウイルスに由来してもよく、これらは、同一のまたは異なるアルファウイルスに由来するレプリコンベクターとの任意の組み合わせにおいて使用することができる。
【0042】
本発明のある特定の実施形態において、ヘルパーRNA分子の5’および3’配列は、VRPの生成においてヘルパーの性能を最大にし、かつRCVの生成に対する理論上の可能性を最小にするように選択される。特定の実施形態は、5’および3’配列の修飾、並びに本明細書にその例が記載されている、アルファウイルス由来の元来の5’および3’配列の一部の欠失を含み得る。本発明に記載された5’および3’配列の、非常に多くの組み合わせがあり、各ヘルパー分子に対して様々な組み合わせを使用することができる。VRPの生成におけるそれらの性能を決定するために、本明細書において教示する修飾および欠失の様々な組み合わせを試験することは、当業者の範囲内である。
【0043】
本発明のRNAヘルパー分子は、アルファウイルス構造タンパク質の翻訳をそれらの天然メチオニン出発コドンで開始するために、5’キャップ構造から5’複製認識配列までスキャンするリボソームに頼っている。これらの領域にさらなる開始コドンが存在すると、構造タンパク質に対して天然の出発コドン以外の部位で翻訳を開始することを可能にし、その結果、リボソームがmRNAに沿ってアルファウイルス構造タンパク質コード領域中へと移動する場合の融合タンパク質か、または続いてリボソームが5’複製認識配列の停止コドンに達した場合の短い非機能性ペプチドか、どちらかが産生されるため、これらのヘルパーの有効性が低下する。したがって、これらのヘルパーにおいて完全な5’アルファウイルス非コード領域(すなわち、野生型アルファウイルスの5’末端から26Sサブゲノムプロモーターの最初のコドンまでの全配列)を使用することは、この領域に多く存在する出発コドンおよび停止コドンのために最適ではない。したがって、特定の実施形態において、本発明のRNA分子は、5’複製認識配列から除去される1つ以上の開始コドンを有し得る。1つ以上とは、当技術分野において標準的な方法に従って除去または不活性化される2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つ、10つ、11つ、12つ、またはそれ以上の開始コドン(すなわち、出発コドン)を意味する。
【0044】
したがって、本発明は、例えばAUGからGUGへの変異によって、5’複製認識配列から1つ以上開始コドンが除去されている本発明のRNA分子を提供する。ある特定の実施形態では、RNA分子は、例えばAUGからGUGへの変異によって、5’複製認識配列から全ての開始コドンが除去されて提供される。例えば、図1に示されるような、以下の位置:12、45、148、154、160、258、294、299、331、390、411、441、および499での任意の組み合わせにおける1つ以上の開始コドンが除去、例えば変異、され得る。
【0045】
開始コドンの除去とは、(例えば、本明細書に記載された方法および当技術分野において知られているような方法に従って)ヌクレオチド配列を修飾して、開始コドンを削除または変更し、その結果、その部位での開始または活性(例えば、翻訳活性)を除去または変更することを意味する。いくつかの実施形態において、開始コドンの大部分は、除去され得るが、特定のヘルパー構築物の5’領域のそのようなコドンのほんのいくつかだけを、リボソームによって通常認識されるコンテキストに存在させることが可能である。したがって、特定の5’配列では、可能な10〜12コドンの外側のそのような2〜3のコドンの除去により、結果として、すべての10〜12のコドンが除去されている構築物とあまり変わらないレベルの発現が生じ得る。本発明のヘルパー分子において使用される場合に、結果としてパッケージング細胞またはヘルパー細胞内において十分発現し、アルファウイルスレプリコン粒子の許容可能な収量を提供するであろう、野生型アルファウイルス配列に由来する多数の特定の5’配列が存在することは、本発明の範囲内である。
【0046】
本発明のRNA分子は、1)アルファウイルスカプシドタンパク質、2)任意の順序のアルファウイルスE1およびE2タンパク質、3)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE1タンパク質、4)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE2タンパク質、5)アルファウイルスE2タンパク質、並びに/あるいは6)アルファウイルスE1タンパク質をコードするヌクレオチド配列を含み得る。他の実施形態において、本発明の単一のRNA分子は、3つのアルファウイルス構造タンパク質、すなわち、順不同にカプシドタンパク質、アルファウイルスE1タンパク質、およびアルファウイルスE2タンパク質をコードし得る。いくつかの実施形態において、本発明のRNA分子は、アルファウイルス構造タンパク質をコードするヌクレオチド配列を特異的に排除することができる(例えば、当該分子は、カプシド、アルファウイルスE1タンパク質、アルファウイルスE2タンパク質、あるいはカプシド、E1タンパク質、およびE2タンパク質の任意の組み合わせをコードするヌクレオチド配列を特異的に排除することができる)。
【0047】
本発明の様々な実施形態において、当該RNA分子は、5’複製認識配列を含む、Venezuelan equine encephalitis(VEE)ウイルスの5’末端由来の配列を含み得る。Pushkoら(1997)によって記載されているように、VEEプロモーター補助ヘルパーの5’複製認識配列は、通常、VEE配列の575ヌクレオチド(nt)から成る。最初の519は接在し、44ntの非翻訳領域(UTR:untranslated region)およびnsP1の最初の475ntを表す(44+475=519)。残りの56ntは、nsP4遺伝子の最後の21nt(TAA停止コドンを含む)、最小26Sプロモーターの7nt(nsP4遺伝子と部分的に重なる配列を有する)、およびVEE構造タンパク質遺伝子開始コドンの上流の28ntリーダー配列をコードする(21+7+28=56)。
【0048】
したがって、Pushkoら(1997)によって記載されているプロモーター補助ヘルパーに対する完全な5’複製認識配列は、VEE配列の575ntから成る。本発明のプロモーターレスヘルパーは、上に記載したプロモーター補助ヘルパーにおいて見られる最初の514のヌクレオチド(nt)の全てもしくは一部をコードする。上に記載した514ntに加えて、RsrII制限酵素部位(7nt)をコードする配列も、構造タンパク質のコード配列開始部位(DNAではATGであり、RNAではAUGである)のすぐ上流に存在する。これらのntの封入体は、完全長カプシドヘルパー(dHcap(FL)のための5’複製認識配列を521nt(開始コドンのA残基を含まない)に増加させる。
【0049】
プロモーターレスカプシドヘルパーのいくつかの例およびプロモーターレス糖タンパク質ヘルパーのすべての例において、構造タンパク質コード配列開始コドンのすぐ上流の、しかしRsrII配列の下流に近コンセンサスコーザック配列(3nt(ACC))を含むためのさらなる修飾が加えてある。コーザック修飾のために、完全長糖タンパク質ヘルパー(dHgp(FL)は、524ntの5’複製認識配列を有する。これらのヌクレオチド配列は、以下の説明のために、プロモーターレスカプシドおよび糖タンパク質ヘルパーに対して「完全長」(「FL」)5’VEE配列として定義され、この配列における欠失は、結果として5’複製認識配列を包含する他の実施形態へとつながる。これらの実施形態は、少なくとも、VEEヌクレオチド配列のヌクレオチド1〜141(開始コドンのA残基を含まない)を含む。5’配列の最初の200のヌクレオチド内に、RNAにおける4つのステムループ(SL:stem−loop)構造が予測される。
【0050】
本発明のヘルパー構築物において有用な5’配列の実施形態は、この領域において、SL構造の1、2、3、または全てを含み得る。SL2領域を取り除き、かつSL1、SL3、およびSL4構造を保有する実施形態は、本発明のヘルパー構築物において有用である。SL構造1およびSL構造2は、最初の145のヌクレオチドに含まれており、すなわち、SL3およびSL4は、ヌクレオチド145と200との間に存在している。したがって、いくつかの実施形態において、5’複製認識配列は、長さが524ヌクレオチド(例えば、図2のdHgp(FL))である構築物の5’非コード領域に含まれ、他の実施形態において、5’複製認識配列は、いずれかの、長さ70ヌクレオチド(例えば、SL1、SL3、およびSL4を含む)〜524ヌクレオチドである5’非コード領域に含まれ得る。例えば、5’複製認識配列は、長さ141、144(dH#8)200、203(dH#7)、248、249(dH#6)、309、312(dH#5)、351、354(dH#4)、412、415(dH#3)450、452(dH#2)、499、または502(dH#1)ヌクレオチドであり得、本明細書において特に列挙はしていない70〜524の間の任意の数(例えば、237、379、444など)を含む。正確なヌクレオチド番号と長さは、異なったアルファウイルス間、および所定のアルファウイルスの異種系統の間でいくらか変わることに注意するべきである。本明細書に記載されたヌクレオチドの対応する位置を、任意のアルファウイルスの対応する構造および/または機能並びに/あるいは本明細書に記載された二次構造の対応する構造および/または機能に基づいて特定し、任意のアルファウイルスの一次ヌクレオチド配列から、本発明のRNAヘルパー分子並びに上に記載された修飾を作製することは、当業者の能力の範囲内である。
【0051】
本発明のRNAヘルパー分子はさらに、アルファウイルスの3’末端由来の配列を含み、当該アルファウイルスは、特定の実施形態において、アルファウイルス3’複製認識配列を含むVenezuelan equine encephalitisウイルスであり得るが、これに限定されるものではない。全てのアルファウイルスの3’末端19ヌクレオチドは高度に保存され、一方、E1糖タンパク質の最後のコドンと高度に保存された19ヌクレオチドの間の3’配列は、アルファウイルス間において長さおよび配列の両方に関してほとんど保存されない。3’非コード領域の長さ(本明細書において、配列番号52である、保存された19ヌクレオチドを含む)は、25〜325ヌクレオチドの範囲であり得る。本発明のある特定の実施形態において、3’配列は、VEE3’末端の73〜117ヌクレオチドの間である。特定の実施形態において、本発明のアルファウイルス3’複製認識配列は、配列番号55(dHcap(FL)からdHcap7まで;dHcap(FL)mmからdHcap7mmまで、dHcap(FL)mut1からdHcap7−mut1まで)、配列番号56(Hgp(FL)からdHgp7まで、dHgp(FL)mmからdHgp7−mmまで、dHgp(FL)mut1からdHgp7−mut1まで)、配列番号57(dHcap6mut1(W/停止)、配列番号58(dHcap7mut1(W/停止)+19ntおよびdHgp7mut1− S+19nt)、並びに配列番号59(dHcap6mut1(W−停止)のヌクレオチド配列を含み得、から実質的に成り得、および/またはから成り得る。
【0052】
特定の実施形態において、本発明のアルファウイルス5’複製認識配列は、
【0053】
【化1】
のヌクレオチド配列を含み得、から実質的に成り得、および/またはから成り得る。これら5’配列の例が合成されている個別のヘルパーを括弧内に記載する。当該配列は、追加的なヌクレオチドの使用のために長さがわずかに変わり得、いくつかのヘルパー構築物において構造タンパク質コード配列の翻訳を高める、最適に近いコーザックコンセンサス配列が得られる(構造タンバク質コード配列のためのコード領域のATG(RNAではAUG)は、これら5’配列には含まれない)。上記において特定されたヌクレオチド配列を含む本発明のRNA分子は、任意の組み合わせ、任意の順序、および/または任意の多様性において、アルファウイルスレプリコン粒子の製造のための本発明の方法で利用することができる。
【0054】
本発明は、さらに、本発明のRNA分子を含むベクターおよび/または核酸構築物を提供する。さらに、本発明の1つ以上のRNA分子と1つ以上アルファウイルス複製単位ベクターとを含む細胞を提供する。1つ以上とは、1つ、2つ、3つ、4つ、5つ、6つ、7つなどを意味する。本発明の細胞は、アルファウイルスタンパク質をコードする核酸構築物を発現させることができる任意の細胞である。本発明の細胞の例としては、これらに限定されるものではないが、ベロ細胞、ベビーハムスター腎臓(BHK)細胞、293細胞、293T/17(ATCCアクセッション番号CRL−11268)細胞、ニワトリ胚繊維芽細胞(CEF)、UMNSAH/DF−1(ATCCアクセッション番号CRL−12203)細胞、PERC.6細胞、およびチャイニーズハムスター卵巣(CHO)細胞などが挙げられる。
【0055】
さらに、本明細書において、本発明の1つ以上のRNA分子を細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入によってRNA分子の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生に必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。本発明のいくつかの実施形態において、当該アルファウイルス粒子は、天然アルファウイルスの構造的な構成を模倣しており、ここでレプリコンRNAがカプシドタンパク質によって覆われ、次いでアルファウイルス糖タンパク質を含有する細胞膜でエンベロープされる。このような実施形態において、アルファウイルス構造タンパク質は、すべて同じアルファウイルスに由来する。代替的な実施態様において、アルファウイルスタンパク質は、異なるアルファウイルスに由来していてもよく、ただし、これらの異なるタンパク質が、粒子アセンブリ中にお互いを「認識」するか、あるいは、それらが、お互いを認識できるように修飾される(文献に記載されているように)場合に限る。
【0056】
本発明の方法のいくつかの実施形態において、本発明の2つのRNA分子が本発明の細胞に導入され、この場合、当該2つのRNA分子は、アルファウイルスレプリコン粒子を産生するために、パッケージング細胞において全ての必要な構造タンバク質が産生される組み合わせにおいて、異なるアルファウイルス構造タンパク質をコードする。したがって、本発明は、細胞に2つのRNA分子を導入し、当該2つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードするが、構造タンパク質のすべてはコードせず、当該2つのRNA分子の第二のRNA分子が、第一のRNA分子によってコードされない1つ以上のアルファウイルス構造タンパク質をコードする方法を提供する。
【0057】
さらに、本発明は、本発明の3つのRNA分子を細胞に導入し、アルファウイルスレプリコン粒子を産生するために必要な構造タンパク質のすべてが当該細胞において産生される組み合わせにおいて、当該3つのRNA分子のそれぞれが、異なるアルファウイルス構造タンパク質をコードする方法を提供する。したがって、本発明の3つのRNA分子を細胞に導入し、当該3つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードするが、構造タンパク質のすべてはコードせず、当該3つのRNA分子の第二のRNA分子が、第一のRNA分子によってコードされたアルファウイルス構造タンパク質とは異なる1つ以上のアルファウイルス構造タンパク質をコードし、当該3つのRNA分子の第三のRNA分子が、第一のRNA分子および第二のRNA分子によってコードされるアルファウイルス構造タンパク質とは異なる1つ以上のアルファウイルス構造タンパク質をコードする方法を提供する。例えば、一実施形態において、当該第一のRNA分子は、アルファウイルスカプシドタンパク質をコードすることができ、当該第二のRNA分子は、アルファウイルス糖タンパク質E1をコードすることができ、並びに当該第三のRNA分子は、アルファウイルス糖タンパク質E2をコードすることができる。
【0058】
いくつかの実施形態において、1つ以上ではあるが全てではないアルファウイルス構造タンパク質が、アルファウイルス構造タンパク質によってパッケージされるレプリコンRNAによってコードされ得る。例えば、本明細書において特許請求されたアルファウイルスレプリコン粒子の製造方法において使用される組換えRNAは、対象となる核酸として、および/または対象となる核酸に加えて、1つのアルファウイルス構造タンパク質または2つ以上のアルファウイルス構造タンパク質をコードする核酸配列を含み得る。したがって、特定の実施形態において、レプリコンRNAは、1つのアルファウイルス構造タンパク質または2つ以上のアルファウイルス構造タンパク質をコードする。このレプリコンRNAは、本発明の1つ以上のRNAヘルパー分子と一緒に細胞の集団中に導入され得る。このレプリコンRNAは、当該レプリコンRNAおよび本発明のRNAヘルパー分子(単数または複数)が全てのアルファウイルス構造タンパク質を産生し、当該レプリコンRNAが当該細胞において粒子内にパッケージされるように、本発明の1つ以上のRNAヘルパー分子と一緒に細胞の集団中に導入することができる。
【0059】
さらなる実施形態において、a)アルファウイルスレプリコンRNAと、b)本発明の1つ以上のRNA分子と、c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物とを細胞へ導入する工程を含む、アルファウイルスレプリコン粒子を製造する方法であって、当該導入により、(b)のRNA分子と(c)のヘルパー構築物の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下においてアルファウイルスレプリコン粒子の産生に必要なすべてのアルファウイルス構造タンパク質をコードする方法が提供される。
【0060】
したがって、本発明のさらなる実施形態において、「プロモーター補助ヘルパー構築物」、すなわち、例えば26Sプロモーターなどのプロモーターの誘導の下で1つ以上のアルファウイルス構造タンパク質を発現する組換えDNAまたはRNA分子は、本発明のヘルパー分子と組み合わせて使用される。1セットのRNA分子の実施形態において、当該「プロモーター補助ヘルパー構築物」は、(i)5’アルファウイルス複製認識配列と、(ii)転写プロモーターと、(iii)1つ以上のアルファウイルス構造タンパク質をコードする核酸配列と、(iv)3’アルファウイルス複製認識配列とをコードする第一の核酸配列を含む。
【0061】
別のセットのRNA分子の実施形態において、当該「プロモーター補助ヘルパー構築物」は、5’アルファウイルス複製認識配列をコードする核酸配列と、IRES要素のすぐ上流にあるアルファウイルスサブゲノムプロモーターと、アルファウイルス構造タンパク質をコードする少なくとも1つの核酸と、3’アルファウイルス複製認識配列をコードする核酸とを含む、国際公開第2004/085660号(2004年10月7日に公開、当該特許は、参照により本明細書に組み込まれる)に記載されているような、組換えヘルパー核酸である。さらなる実施形態において、これらのプロモーター補助ヘルパー構築物は、サブゲノムプロモーターのすぐ下流かつIRES要素のすぐ上流に位置するスペーサー核酸を含み得る。当該スペーサー核酸は、構造タンパク質の翻訳の一部または全部が、次にIRESによって誘導されるように、メッセンジャーRNAの5’キャップからの少なくとも一部の翻訳、およびいくつかの実施形態では全部の翻訳を防止するのに十分な長さの任意のランダムもしくは特異的非コード核酸配列を含み得、またはから成り得る。あるいは、スペーサー核酸が、メッセンジャーRNAの5’キャップからの少なくとも一部および可能であれば全部の翻訳活性を防止するのに十分な二次構造を核酸に付与する長さおよび配列構造であり得る。本発明において使用されるプロモーター補助ヘルパー構築物はさらに、ヘルパー細胞のゲノムに安定して組み込まれ得るDNA分子であり得るか、または有意に組み込まれること無しにエピソーム(例えば、プラスミド)から一過的に発現され得るDNA分子であり得る。本発明のDNA分子は、DNAベクターであってもよく、例えば、これに限定されるものではないが、プラスミドなどの非組み込み型DNAベクター、またはウイルスベクターが挙げられる。
【0062】
本明細書に記載されるような「ヘルパー細胞」または「パッケージング細胞」を利用しかつ本発明のプロモーターレスRNA分子を含む本発明の実施形態において、当該ヘルパー細胞は、本発明のアルファウイルスレプリコン粒子を産生するのに十分なアルファウイルス構造タンパク質をコードするヌクレオチド配列の組み合わせを当該ヘルパー細胞が含むような任意の組み合わせにおいて、プロモーター補助ヘルパー構築物(RNAおよび/またはDNA)をさらに含み得る。ある特定の実施形態において、E1およびE2糖タンパク質は、第一のヘルパー構築物によってコードされ、カプシドタンパク質は、第二のヘルパー構築物によってコードされる。別の実施形態において、E1糖タンパク質、E2糖タンパク質、およびカプシドタンパク質は、それぞれ別個の(例えば、第一、第二、および第三の)ヘルパー構築物によってコードされる。さらに他の実施形態において、カプシドタンパク質および糖タンパク質E1もしくはE2のどちらかが、第一のヘルパー構築物によってコードされ、第一のヘルパー構築物に含まれない残りの糖タンパク質E1もしくはE2は、カプシドコード配列の有無にかかわらず、第二のヘルパー構築物によってコードされる。さらなる実施形態において、アルファウイルス糖タンパク質E1およびE2、並びにカプシドタンバク質は、任意の順序および/または任意の多様性において、全て1つのヘルパー構築物でコードされ得る。本発明に含まれる実施形態では、所定のアルファウイルス構造タンパク質を、2つ以上のヘルパー構築物によって発現させることも可能である。本発明のプロモーターレスRNAヘルパーは、必要に応じて本明細書に記載されているような他の既知のヘルパーとの組み合わせにおいて、任意の組み合わせ、任意の順序、および/または任意の多様性において、アルファウイルス−許容細胞中に導入することができる。
【0063】
本発明のいくつかの実施形態において(例えば、プロモーターレスRNA分子またはプロモーターによって補助されるRNAヘルパー構築物をコードするDNA構築物のための)、DNAからのRNAの転写を誘導するためのプロモーター、すなわちDNA依存性RNAポリメラーゼが、インビトロ転写反応においてRNAを合成するために利用され、この使用のために好適な特定のプロモーターとしては、SP6、T7、およびT3 RNAポリメラーゼプロモーターが挙げられるが、これらに限定されるものではない。
【0064】
本発明の全ての実施形態において、プロモーターレスヘルパー分子および/またはプロモーター補助ヘルパー構築物および/またはレプリコンベクターによってコードされる少なくとも1つのアルファウイルス構造タンパク質および/または非構造タンパク質、並びにレプリコン核酸の非翻訳領域は、本明細書に記載されているように、任意の組み合わせにおいて、1つ以上の弱毒化突然変異を含有し得ることが想到される。
【0065】
本発明はさらに、アルファウイルスレプリコン粒子108個当たりに1個未満の複製能力のあるアルファウイルス粒子を含有するアルファウイルスレプリコン粒子の集団を提供する。さらなる実施形態において、当該集団は、アルファウイルスレプリコン粒子109個、1010個、1011個、1012個、または1013個当たりに1個未満の複製能力のあるアルファウイルス粒子を含有する。本発明は、さらに、当技術分野で周知の方法に従って、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含有しないアルファウイルスレプリコン粒子の集団を提供する。
【0066】
さらに本明細書において、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるアルファウイルス粒子をアルファウイルスレプリコン粒子108個、109個、1010個、1011個、1012個、または1013個当たりに含まないかもしくは1個未満の複製能力のあるアルファウイルス粒子を含むアルファウイルスレプリコン粒子の集団であって、この場合、当該アルファウイルスレプリコン粒子は、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む集団が提供される。さらに、アルファウイルスレプリコン粒子の集団であって、当該集団が、培養中の許容細胞での継代によって決定されるような、検出可能な複製能力のあるウイルス粒子を含まず、当該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む集団が提供される。
【0067】
同定可能な「パッケージングシグナル」の欠失にもかかわらず、本発明のヘルパーRNA、並びに文献に記載されているヘルパーRNAは、時に文献で報告される頻度よりかなり高い頻度で、培養細胞においてアルファウイルス構造タンパク質によってパッケージされる。したがって、本発明のアルファウイルスレプリコン粒子の集団は、本発明の新規のヘルパー分子がパッケージされる集団における粒子のサブセットの存在により、文献に記載されている粒子と区別される。
【0068】
「アルファウイルスレプリコン粒子」、「ARP」、「ウイルスレプリコン粒子」、または「組換えアルファウイルス粒子」なる用語は、本明細書において互換的に用いられ、1つ以上の非相同RNA配列を発現するアルファウイルスレプリコンRNAを取り込むビリオン様構造複合体を意味する。通常、ビリオン様構造複合体は、脂質エンベロープに包埋されている1つ以上のアルファウイルス構造タンパク質を含み、当該脂質エンベロープはヌクレオカプシドを封入し、当該ヌクレオカプシドはRNAを封入する。当該脂質エンベロープは、通常、当該粒子が産生される細胞の原形質膜に由来する。ある特定の実施形態において、当該アルファウイルスレプリコンRNAは、アルファウイルスッカプシドタンパク質から成るヌクレオカプシド構造によって取り囲まれており、アルファウイルス糖タンパク質は細胞由来の脂質エンベロープに包埋されている。構造タンパク質およびレプリコンRNAは、同じアルファウイルスまたは異なるアルファウイルスに由来してもよい。特定の実施形態において、レプリコンRNAは、VEE由来であり、構造タンパク質は、Sindbisウイルス由来である(例えば、Dubenskyら、米国特許第6,376,236号を参照のこと)。アルファウイルスレプリコン粒子は、感染性であるが、伝播性が欠損しており、すなわち、レプリコンRNAは、アルファウイルス構造タンパク質をコードするヘルパー核酸(単数または複数)の不在下において粒子が最初に感染した宿主細胞を越えて伝播することができない。
【0069】
「アルファウイルスRNAレプリコン」、「アルファウイルスレプリコンRNA」、「アルファウイルスRNAベクターレプリコン」、および「ベクターレプリコンRNA」なる用語は、互換的に用いられ、自身の複製(増幅)を誘導することができ、少なくとも、5’および3’アルファウイルス複製認識配列(当該配列は、上で定義されるように最小配列であり得るが、代わりに、アルファウイルス由来の領域全体であり得る)、アルファウイルス非構造タンパク質のコード配列、およびポリアデニル化トラクトを含むように非構造タンパク質遺伝子を発現するRNA分子を意味する。これはさらに、プロモーターおよび/またはIRESを含有し得る。さらにこれは、アルファウイルス構造タンパク質を発現するように操作することもできる。JohnstonらおよびPoloらは、このようなアルファウイルスRNAレプリコンのための多くの構築物について記載しており、このような構築物は、参照により本明細書に組み込まれる。アルファウイルスレプリコンRNAの一実施形態において、アルファウイルス非構造タンパク質は、米国特許出願公開第2003−0119182号に記載されているように、2つの別々の翻訳単位に分離されている。なお、当該特許は、参照により本明細書に組み込まれる。
【0070】
非相同配列を持たないアルファウイルスレプリコンRNA、すなわち、空のレプリコンは、アジュバント組成物を産生するために、アルファウイルスレプリコン粒子において使用され得る。あるいは、アルファウイルスレプリコンRNAは、アルファウイルス構造タンパク質および/または他の非相同核酸配列をコードする核酸を発現することができ、この後者は、ウイルス、原始核細胞、および/または真核細胞に由来する広範囲の配列から選択することができる。非相同配列のカテゴリの例としては、これらに限定されるものではないが、免疫原(例えば、天然、修飾または合成の抗原性タンパク質、ペプチド、免疫原性断片、またはエピトープ)、サイトカイン、トキシン、治療用タンパク質、酵素、アンチセンス配列、および免疫応答調節因子が挙げられる。特定の用途に適切かつ望ましい場合、当該転写されたmRNAは、翻訳され、すなわち、タンパク質が合成されるか、または機能性RNAが産生される。これらのmRNAは、真核細胞内に「キャップ」されており、すなわち、メチル−7−グアノシン(5’)pppN構造が、mRNAの5’末端に存在し(「キャップ」または「5’キャップ」)、このキャップが、mRNAからタンパク質を合成する翻訳開始因子によって認識される。したがって、26Sプロモーターが、転写を誘導し、「キャップ」が、翻訳のための開始シグナルを提供する。
【0071】
いくつかの実施形態において、レプリコンRNAは、任意のアルファウイルス構造タンパク質(単数または複数)をコードする核酸を欠失し得る。他の実施形態において、アルファウイルスレプリコンRNAは、1つまたは2つのアルファウイルス構造タンパク質をコードする核酸を含み得るが、当該レプリコンRNAは、アルファウイルス構造タンパク質の全てをコードする核酸を含有しない。したがって、結果として得られる本発明のアルファウイルスレプリコン粒子は、当該レプリコンRNAがそのカプシド形成および感染性ビリオンのアセンブリに必要な全ての構造タンパク質をコードしないが故に、伝播性が欠損している。
【0072】
特許請求される本発明において利用されるアルファウイルスRNAレプリコンの特定の実施形態は、本明細書に詳細に記載されているような1つ以上の弱毒化突然変異を含み得る。弱毒化ヌクレオチド置換の例としては、本明細書に記載されているVEEの5’末端におけるヌクレオチド3での突然変異、並びにアルファウイルスS.A.AR86におけるnsP1アミノ酸位置538、nsP2アミノ酸位置96、またはnsP2アミノ酸位置372での突然変異が挙げられる。
【0073】
本発明のアルファウイルスレプリコン粒子は、任意のアルファウイルスに由来するレプリコンRNAを含み得る。その上、本発明のアルファウイルスレプリコン粒子は、本発明のアルファウイルスのいずれかに由来するアルファウイルス構造タンパク質を含み得る。したがって、当該レプリコン粒子は、同じアルファウイルスあるいは異なるアルファウイルスに由来するレプリコンRNAおよび構造タンパク質から製造することができ、これらの後者は、キメラアルファウイルスレプリコン粒子(例えば、VEEウイルスベースのレプリコンRNAおよびSindbisウイルス構造タンパク質を含む粒子)であるであろう。
【0074】
本発明の特定の実施形態において、本発明のアルファウイルス構造タンパク質は、Sindbisウイルス構造タンパク質、SFV構造タンパク質、VEE構造タンパク質、ロスリバーウイルス構造タンパク質、EEE構造タンパク質、および/またはWEE構造タンパク質であってもよい。これらは、お互いの任意の組み合わせにおいて存在し得、並びに、非構造タンパク質および他のアルファウイルス配列、例えば、5’アルファウイルス複製認識配列、アルファウイルスのサブゲノムプロモーター、および3’アルファウイルス複製認識配列など、の組み合わせにおいて存在し得、これらまたは他のアルファウイルスのいずれかから、本発明のキメラ組換えアルファウイルスレプリコン粒子および/またはキメラ組換え核酸を産生する。
【0075】
本発明のいくつかの実施形態において、本発明は、アルファウイルス核酸、アルファウイルスタンパク質、アルファウイルスレプリコンRNAおよび/または1つ以上の弱毒化突然変異を含むアルファウイルスレプリコン粒子、ヌクレオチド欠失、ヌクレオチド付加、および/または1つ以上のヌクレオチドの置換として定義される弱毒化突然変異、あるいは、適切な野生型アルファウイルスと比較して、突然変異を含有する生存ウイルスにおいて毒性が喪失されるに至る再構成またはキメラ構築物を含む突然変異を含み得る。
【0076】
適切な弱毒化突然変異は、使用されるアルファウイルスに応じて変わり、そのような弱毒化突然変異は、当業者には既知であろう。代表的な弱毒化突然変異としては、これらに限定されるものではないが、Johnstonらの米国特許第5,505,947号、Johnstonらの米国特許第5,185,440号、Davisらの米国特許第5,643,576号、Johnstonらの米国特許第5,792,462号、同第6,156,558号、および同第5,639,650号に記載されているものが挙げられ、なお、これらの各特許の開示は、参照によりその全体が本明細書に組み込まれる。
【0077】
VEE E1糖タンパク質に対する特定の弱毒化突然変異は、E1アミノ酸位置81,272または253の任意の1つでの弱毒化突然変異を含み得る。VEE−3042突然変異体から製造されたアルファウイルスレプリコン粒子は、E1−81にイソロイシン置換を含有し、VEE−3040突然変異体から作製されたウイルスレプリコン粒子は、E1−253に弱毒化突然変異を含有する。VEE E2糖タンパク質に対する特定の弱毒化突然変異は、E2アミノ酸位置76、120、または209の任意の1つに弱毒化突然変異を含み得る。VEE−3014突然変異体から製造されたアルファウイルスレプリコン粒子は、E1−272およびE2−209の両方に弱毒化突然変異を含有する(米国特許第5,792,492号を参照のこと)。VEE E3糖タンパク質に対する特定の弱毒化突然変異は、E3アミノ酸56〜59の欠失からなる弱毒化突然変異を含む。VEE−3526突然変異体から製造されたウイルスレプリコン粒子は、E3(aa56〜59)にこの欠失を含有し、E1−253に第二の弱毒化突然変異を含有する。S.A.AR86 E2糖タンパク質に対する特定の弱毒化突然変異は、E2アミノ酸位置304、314、372、または376の任意の1つに弱毒化突然変異を含む。あるいは、当該弱毒化突然変異は、例えば、アミノ酸位置158、159、160、161、および162の任意の組み合わせにおける任意の1つ以上での、E2糖タンパク質におけるアミノ酸の置換、欠失、または挿入であってもよい(Poloら国際公開第00/61772号を参照のこと)。あるいは、本発明のRNA分子は、VEEのワクチン株であるTC83から誘導することもできる(国際公開第2005/113782号を参照のこと。なお、当該特許は参照により本明細書に組み込まれる)。
【0078】
本発明の別の弱毒化突然変異は、VEEゲノムRNAのヌクレオチド3、すなわち、5’メチル化キャップの後の三番目のヌクレオチドでの弱毒化突然変異であってもよい(例えば、nt3でのG→C突然変異について記載されている米国特許第5,643,576号を参照のこと)。ウイルスまたはレプリコンの非コード配列に位置するこの突然変異は、いくつかの実施形態において、G→AまたはG→Uの突然変異であり得る。アルファウイルス構造タンパク質および/または非構造タンパク質が、S.A.AR86に由来する場合、当該構造タンパク質および/または非構造タンパク質における代表的な弱毒化突然変異が、文献に記載されている(例えば、米国特許第5,639,650号および同第6,982,087を参照のこと。なお、これらの開示は、参照によりその全体が本明細書に組み込まれる)。
【0079】
本発明のアルファウイルスは、Sindbisウイルス株(例えば、TR339)、VEE(例えば、メチル化キャップまたはTC83に続くゲノムRNAのヌクレオチド3に突然変異を有する)、S.A.AR86ウイルス、Girdwood S.A.ウイルス、Ockelboウイルス、および/またはそれらのキメラウイルスであってもよい。完全ゲノム配列、ならびに様々な構造タンパク質および非構造タンパク質の配列は、多数のアルファウイルスに対して文献から利用可能であり、その例としては、Sindbisウイルスゲノム配列(GenBankアクセッション番号J02363、NCBIアクセッション番号NC_001547)、S.A.AR86ゲノム配列(GenBankアクセッション番号U38305)、VEEゲノム配列(GenBankアクセッション番号L04653、NCBIアクセッション番号NC_001449)、VEEのTC−83ワクチン株(Kinney RMら(1989)Virology 170:19−30;Kinney RMら(1993) J.Virol.67(3):1269−1277において補正)、Girdwood S.Aゲノム配列(GenBankアクセッション番号U38304)、Semliki Forestウイルスゲノム配列(GenBankアクセッション番号X04129、NCBIアクセッション番号NC_003215)、およびTR339ゲノム配列(Klimstraら(1988)、J.Virol.72:7357;McKnightら(1996)、J.Virol.70:1981)が挙げられる。
【0080】
アルファウイルスレプリコン粒子は、当業者に公知の技術の組み合わせにおいて、本明細書に開示する方法に従って調製される。当該方法は、最初に、選択されたヘルパー(単数または複数)およびアルファウイルスレプリコンRNAをアルファウイルス許容細胞の集団に導入する工程と、次いでアルファウイルスレプリコン粒子の産生を可能にする、当技術分野において周知の条件下において、細胞をインキュベートする工程とを含む。ヘルパー(単数または複数)およびアルファウイルスレプリコンRNAをヘルパー細胞の集団に導入する工程は、本明細書に開示されているような、および一般的な当業者に公知のような、いずれかの好適な手段によって実施することができる。
【0081】
アルファウイルスレプリコン粒子の集団は、例えば、米国特許第7,078,218号に記載されているような方法に従って、ヘルパー細胞またはパッケージング細胞から収集する。なお当該米国特許の内容は、参照によりその全体が本明細書に組み込まれる。あるいは、当業者に公知の他の技術を用いてパッケージング細胞から収集することもできる(例えば、米国特許第5,492,462号および同第6,156,558号)。これらの集団は、本明細書に記載されているような方法および文献において公知のような方法に従って、複製能力のあるウイルス(RCV)の存在に対して評価される。本発明の集団は、培養中のアルファウイルス許容細胞での継代により特定されるような、検出可能なRCVを含有しない。
【0082】
いくつかの実施形態において、本発明は、被験体において免疫原性ポリペプチドをコードするアルファウイルスRNAレプリコンをパッケージするため(例えば、ワクチン接種のため)、免疫療法のため(例えば、癌もしくは腫瘍を有する被験体を治療する)、または免疫調節因子のため(例えば、ARPまたは他のワクチン様相をアジュバンドするため)に用いることができる。本発明は、被験体において免疫応答を誘発するまたは高める方法であって、本発明のヘルパー構築物によって粒子内にパッケージされた有効量の核酸を当該被験体に投与する段階を含む方法を提供する。
【0083】
本明細書で使用される場合、「免疫応答を誘発する」および「被験体を免疫化する」には、被験体における、本発明のタンパク質および/またはポリペプチド(例えば、免疫原、抗原、免疫原性ペプチド、および/または1つ以上のエピトープ)に対する体液性および/または細胞性免疫反応の発生が含まれる。「体液性」免疫反応は、この用語が当分野において周知であるように、抗体を含む免疫反応を意味するが、一方で「細胞性」免疫反応は、この用語が当分野において周知であるように、T−リンパ球および他の白血球を含む免疫反応、特に、HLA拘束性細胞溶解性T細胞、すなわち「CTL」による免疫原特異的応答を意味する。
【0084】
さらに、本発明の核酸、粒子、集団、および医薬組成物は、被験体における細胞であり得る細胞へ、対象となるNOIを送達する方法において採用され得ることも想到される。したがって、本発明は、非相同核酸を細胞に送達する方法であって、本発明のヘルパー構築物でパッケージされた有効量の粒子、集団、および/または組成物を当該細胞内に導入する段階を含む方法を提供する。さらに、被験体において非相同核酸を細胞に送達する方法であって、本発明のヘルパー構築物でパッケージされた有効量の粒子、集団、および/または組成物を被験体に送達する段階を含む方法が提供される。当該細胞は、外因性の核酸を取り込みかつ発現する任意の細胞であり得る。当該細胞は、非相同核酸が発現して非相同核酸によってコードされるタンパク質、ペプチド、または他のコード配列生成物(例えば、機能性RNA配列)を産生する条件下で維持される。免疫療法および/または遺伝子療法のための周知のプロトコールに従って、本発明の細胞および/または被験体に治療効果を与えるために、このような方法を利用することができる。
【0085】
本発明の「被験体」としては、これらに限定されるものではないが、例えば、ヒト、非ヒト霊長動物、ウマ、ウシ、ネコ、イヌ、ブタ、ラット、およびマウスなどの温血動物が挙げられる。
【0086】
本発明はさらに、薬学的に許容される担体中に本発明の粒子および/または粒子の集団を含む組成物(例えば、医薬組成物)を提供する。「薬学的に許容される」とは、生物学的でない物質または他の点で不所望でない物質、すなわち、実質的に有害な生物学的効果を引き起こさずに、またはそれが含有される組成物の他の成分のいずれとも有害な方法で相互作用することなく、選択された粒子および/またはその集団と一緒に被験体に投与することができる物質を意味する。薬学的に許容される担体は、ヒトおよび本発明の他の被験体への投与または送達に好適である。当該担体は、当然のことながら、当業者に周知であるように、有効成分の任意の分解を最小にするように、並びに被験体において任意の有害な副作用を最小にするように選択されるであろう(例えば、Remington’s Pharmaceutical Science; 最新版を参照のこと)。本発明の、例えばワクチンまたは別の免疫原性組成物などの医薬製剤は、薬学的に許容される担体と組み合わせて、本発明のヘルパー構築物を使用して作製された免疫原性量の感染性の伝播欠損性アルファウイルスレプリコン粒子を含む。代表的な薬学的に許容される担体としては、これに限定されるものではないが、滅菌パイロジェン非含水および滅菌パイロジェン非含生理食塩水が挙げられる。
【0087】
「免疫原性量」は、投与または送達される被験体において、免疫応答を誘起するのに十分な、本発明の集団における感染性アルファウイルス粒子の量である。本明細書に記載されたアッセイによって特定されるような、投与量当たり約104〜約109、特に106〜108の量の感染ユニットまたは「IU(infectious unit)」が、治療される被験体の年齢および種に応じて好適であると考えられる。投与は、例えば、腹腔内、筋肉内、鼻腔内、腟内、静脈内、皮内(例えば、遺伝子銃により)、直腸内、および/または皮下などの任意の好適な方法によって行われ得る。本明細書の組成物は、皮膚乱切法、および/またはパッチもしくは液体により経皮的に投与してもよい。当該組成物は、ある期間にわたって組成物を放出する生分解性物質の形態において皮下により送達することができる。
【0088】
本明細書で使用される場合、「有効量」は、治療効果であり得る所望の効果を得るのに十分な、本発明の集団または組成物または製剤の量を意味する。当該有効量は、被験体の年齢、全身状態、治療される状態の重症度、投与される特定の薬剤、治療の期間、任意の併用治療の性質、使用される薬学的に許容される担体、当業者の知識および専門的技術の範囲内の因子などにより変わるであろう。必要に応じて、任意の個々の場合における「有効量」は、適切な教示書および文献を参照することにより、および/または通常の実験により、当業者が決定することができる(例えば、Remington,The Science And Practice of Pharmacy(20th ed.2000)を参照のこと)。
【0089】
あるいは、本発明の医薬製剤は、被験体の粘膜への投与(例えば、鼻腔内投与、口腔内投与、および/または吸入)に好適であり得る。当該製剤は、単位投与剤形において好都合に調製され得、並びに当技術分野において周知の任意の方法によって調製され得る。
【0090】
さらに、本発明の当該組成物は、当技術分野において周知の方法に従って被験体の細胞から単離または増殖された樹枝状細胞、またはバルク末梢血単核細胞(PBMC:peripheral blood mononuclear cell)、あるいは被験体に由来するそれらの様々な細胞サブフラクションに感染またはトランスフェクトするために使用され得る。
【0091】
エキソビボ法を用いる場合、細胞または組織は、当技術分野において周知の標準的なプロトコールに従って、体外に取り出され、本発明の組成物を当該細胞または組織中に導入する間、維持され得る。
【0092】
本発明の粒子の集団を含む免疫原性組成物(当該粒子は、当該組成物がヒトまたは動物に投与された場合に、対象となる核酸配列(単数または複数)の発現を誘導する)は、当技術分野において公知の任意の方法によって製剤化され得る。そのような組成物、特にワクチンは、通常、注射液として、液体溶液または懸濁液のいずれかとして調製される。さらに注射前の液体への溶解または懸濁に好適な固体形態も調製され得る。凍結乾燥された製剤も好適である。
【0093】
活性免疫原性成分(例えば、アルファウイルスレプリコン粒子)は、多くの場合、有効成分に対して薬学的に許容されるおよび/または適合する賦形剤および/または担体と混合される。好適な賦形剤としては、これらに限定されるものではないが、滅菌水、塩水、デキストロース、グリセロール、エタノールなど、およびそれらの組み合せ、並びに安定化剤、例えばHSAまたは他の適切なタンパク質および還元糖が挙げられる。
【0094】
さらに、所望の場合、当該ワクチンは、少量の、例えば湿潤剤および/または乳濁剤などの補助物質、pH緩衝剤、および/またはワクチンの有効性を高めるアジュバントを含み得る。効果的であり得るアジュバントの例としては、これらに限定されるものではないが、QS−21、フロイントアジュバント(完全および不完全)、アルミニウム塩(ミョウバン)、リン酸アルミニウム、水酸化アルミニウム;N−アセチル−ムラミル−L− トレオニル−D−イソグルタミン(thr−MDP);N−アセチル−ノルームラミル−L−アラニル−D−イソグルタミン(CGP 11637、nor−MDPとも呼ばれる);N−アセチルムラミル−L−アラニル−D−イソグルタミニル−L−アラニン−2−(1’−2’−ジパルミトイル−sn−グリセロ−3ヒドロキシホスホリルオキシ))−エチルアミン(CGP 19835A、MTP−PEとも呼ばれる);およびバクテリアから抽出された3つの成分のモノホスホリルリピドA、トレハロースジミコレート、および細胞壁骨格(MPL+TDM+CWS)を2%のスクアレン/Tween 80エマルジョン中に含有するRIBIが挙げられる。
【0095】
アジュバントのさらなる例には、これらに限定されるものではないが、水中油エマルジョン配合物、免疫促進物質、例えばバクテリア細胞壁成分もしくは合成分子など、またはオリゴヌクレオチド(例えば、CpG)、および、例えばポリビニルポリマーなどの別の骨格部分を組み込むことのできる核酸ポリマー(二本鎖および一本鎖のRNAおよびDNAの両方)が含まれ得る。
【0096】
アジュバントの有効性は、アジュバンドもしくはアジュバンドの組み合わせも含むワクチン製剤中の当該粒子含有組成物の投与によって生じる、アルファウイルスレプリコン粒子の免疫原性産物に対する抗体または細胞障害性T細胞の量を測定することによって決定され得る。当該技術分野において既知の、そのようなさらなる製剤および投与の様式も使用され得る。
【0097】
アジュバントは、本発明の組成物、または本発明の組成物と組み合わせて使用できる他のワクチン配合物のどちらかと組み合わせることができる。
【0098】
本発明の組成物は、さらに、他の薬剤、医薬品、担体、および希釈剤を含み得る。
【0099】
本発明の組成物を、最適化し、他のワクチン接種レジメンと組み合わせることにより、広範囲の(すなわち、上記に記載された機能を含む、免疫応答の全ての態様を網羅する)可能な細胞応答および体液応答を得ることができる。ある特定の実施形態において、これは、病原菌または腫瘍に由来する免疫原、組換え免疫原、裸の核酸、脂質含有成分により調製された核酸、非アルファウイルスベクター(これらに限定されるものではないが、痘疹ベクター、アデノウイルスベクター、アデノ随伴ウイルスベクター、ヘルペスウイルスベクター、水疱性口内炎ウイルスベクター、パラミクソウイルスベクター、パルボウイルスベクター、パポーバウイルスベクター、レトロウイルスベクター、レンチウイルスベクターを含む)、および他のアルファウイルスベクターの1つ以上を含む組成物との組み合わせにおいて本発明の組成物が使用される非相同プライムブースト法の使用を含み得る。当該ウイルスベクターは、ウイルス様粒子もしくは核酸であり得る。代表的なアルファウイルスベクターは、レプリコン含有粒子、DNAベースのレプリコン含有ベクター(場合によって「ELVIS」システムとも呼ばれ、例えば、米国特許第5,814,482号を参照のこと)、および/または裸のRNAベクターであり得る。
【0100】
本発明の免疫原性(またはそうでなければ、生理的に活性な)アルファウイルス粒子含有集団および組成物は、投与剤形と適合する方法において、並びに予防上および/または治療上有効なそのような量において投与される。投与される量は、一般的に、投与量1mL当たり約104〜109感染ユニットの範囲であり、治療される被験体、当該粒子が投与または送達される経路、発現産物の免疫原性、所望されるエフェクター免疫応答のタイプ、並びに所望される予防の程度に応じて変わる。いくつかの実施形態において、約106、107、および108I.U.(infectious unit)の投与量は、ヒト被験体において特に効果的である。投与または送達に必要とされる有効成分の有効量は、医師、獣医師、または別の保健実務者の判断に応じて変わり、所定の被験体に対して固有であり得るが、そのような決定は、そのような実務者の技術の範囲内である。
【0101】
本発明の組成物および配合物は、1回投与または複数回投与のスケジュールにおいて与えられ得る。複数回投与スケジュールは、投与の初回治療単位が、1〜10またはそれ以上の分割投与量を含み、その後に、所望する効果(例えば、免疫応答)を維持および/または増強するために必要とされるような時間間隔、例えば、毎週または第二の投与に対して1〜4ヶ月目に他の投与量が投与され、並びに必要であれば、さらに数ヶ月(例えば、4または6ヶ月)/数年後に次の投与量(単数または複数)が投与されるスケジュールである。
【0102】
本発明の治療方法の効果は、本発明の障害の治療効果を評価するための周知のプロトコールに従って決定され得る。治療効果の決定因子としては、これらに限定されるものではないが、当技術分野において周知であるように、全生存率、無病生存率、症状の改善、進行までの期間、および/または生活の質などが挙げられる。
【0103】
「治療する」または「治療すること」または「治療」は、障害、疾病、もしくは病気で苦しむ被験体に、例えば有益効果であり得る調節作用を付与する任意の種類の処置を意味し、当技術分野において周知であるような、被験体の状態(例えば、1つ以上症状)の改善、状態の進行の遅延または軽減、障害、疾病、もしくは病気の兆候の予防または遅延、並びに/あるいは障害、疾病、もしくは病気に関する臨床的指標のいずれかにおける変化などが挙げられる。
【0104】
前述の詳細な説明は、単に例示のために提供されるものであり、本発明の精神および範囲から逸脱せずに修飾および変更を加えることができることは理解される。
【実施例】
【0105】
(実施例1)
dHcapおよびdHgpヘルパーの構築
米国特許第5,792,462号、Pushkoら、1997(Virology 239:389−401)、および国際公開第02/03917号(Olmstedら)に記載されているように、VEEヘルパープラスミド(カプシドヘルパーに対しては「13.2.2」とも呼ばれ、糖タンパク質ヘルパーに対しては「13.4.6」とも呼ばれる)からカプシドおよび糖タンパク質(GP)遺伝子を増幅するようにプライマーを設計した(カプシドF(配列番号98)、GP F(配列番号60)、および13−101.pr4(配列番号61)(表1)。これらのプライマーは、RsrII制限部位を提供し、さらに、それぞれ、カプシドまたは糖タンパク質コード配列の開始位置に結合する。上記に引用された参照文献に記載されているDNAプラスミドは、例えばPCR増幅により構造タンパク質コード断片を得るための便利な供給源であり得る。あるいは、これらのコード断片は、VEEの完全長クローンまたはその弱毒化変異体から得ることができる(米国特許第5,185,440号;同第5,505,947号を参照のこと)。
【0106】
これらのプライマーによる増幅は、結果として、PCR産物の5’末端から3’末端へと列挙される以下の要素:5’〜RsrII制限部位、VEE構造タンパク質コード配列ORF、3’UTR、SphI制限部位〜3’、を有する断片を生じる。次いで、当該PCR産物は、米国特許第5,792,462号、Pushkoら、1997(Virology 239:389−401)、および国際公開第02/03917号(Olmstedら)に記載されるように、RsrIIおよびSphI制限酵素によって消化され、空のVEEレプリコンベクターにライゲーションする。このレプリコンRNAは、VEE非構造遺伝子および後に多重クローニング部位(MCS:multiple cloning site)が続く26SサブゲノムRNAプロモーターの単一のコピーを含む。ワクチン構築物では、免疫原をコードする1つ以上のコード配列がこのクローニング部位に挿入される。このベクターは、RsrIIおよびSphIによって消化され(nsPlの大部分およびnsPs2〜4のすべてを除去する)、ライゲーションにより、完全なアルファウイルス5’および3’末端、すなわち「完全長」末端を含むヘルパーが生成する。したがって、これら2つのヘルパーは、dHcap(FL)およびdHgp(FL)と称され、それぞれ、配列番号1および配列番号10の5’配列、並びにそれぞれ、配列番号55および配列番号56の3’配列を有する(図1)。
【0107】
続いて、dHcap(FL)およびdHgp(FL)ヘルパーの両方に存在する552nt5’末端に、それぞれおよそ50ntの8つの連続した欠失部位を作製した(図2)。この手順は、2段階にて実施した。第一に、8つの異なるリバースプライマー(dHelp1〜8R、配列番号63〜70)を、(上に記載された)13.2.2および13.4.6ヘルパーの位置502までに、5’末端に対して相補的に設計し、RsrII制限部位をさらに含有するように、それぞれを操作した(表1)。任意のリバースプライマーと組み合わせた場合に、以下の要素(5’から3’へと列挙):5’〜XbaI制限部位、T7プロモーター、5’の切断された末端、RsrII制限部位〜3’、を有する断片を増幅するように、フォワードプライマー(3−16.1.1(配列番号62)を設計した。第二に、増幅された5’の切断された末端断片を、XbaIおよびRsrIIによって直線化されたdHcap(FL)およびdHgp(FL)ヘルパー中にクローン化した。これにより、それぞれ配列番号2〜9および配列番号11〜18の5’配列を有する、dHcap1〜8およびdHgp1〜8と称される5’の切断された末端ヘルパー構築物の8つのセットが生成した。dHcapシリーズの各メンバーの3’配列は、本明細書において配列番号55として提供され、dHgpシリーズの各メンバーの3’配列は、本明細書において配列番号56として提供される。
【0108】
(実施例2)
プロモーターレスヘルパー発現カセットの発現解析方法
本明細書に記載されたΔ26Sヘルパー構造体がVEE構造タンパク質をどの程度発現するかを決定するために、上に記載されたようなVEEレプリコンベクターと一緒に各ヘルパーをベロ細胞中にエレクトポレーションした。本発明の新規のプロモーターレス構造タンパク質発現カセットの能力を実証するために、GFPまたはボツリヌス神経毒素コード配列を、VEEレプリコンベクターのクローニング部位に挿入することにより、VEEレプリコンを構築した。本明細書に記載されたプロモーターレス構造タンパク質発現カセットの様々な組み合わせによって作製された粒子からのこれらのコード配列の発現により、これらのカセットの有用性および新規性が実証される。
【0109】
メーカーの指示に従ってRiboMAX T7 Express(登録商標)転写キット(Promega Corporation社、Madison、WI)を使用して、ラン−オフ転写により、各ヘルパーおよびレプリコンベクターからRNAを転写した。エレクトロポレーションの前に、ヘルパーおよびレプリコンRNAを、シリカベースのクロマトグラフィによって精製した。30マイクログラム(30μg)の各ヘルパーおよびレプリコンRNAを混合し、3〜5X107のベロ細胞中にエレクトポレーションした。エレクトロポレーションした細胞を培地で希釈し、25cm2のフラスコまたは96ウェルプレートに播種した。次いで、エレクトロポレーションした細胞を37℃で16〜24時間インキュベートした。
【0110】
A.IFA分析
96ウェルプレート中に播種されたエレクトロポレーションした細胞を、リン酸緩衝食塩水(PBS:phosphate buffered saline)で1回洗浄し、次いで、室温で5時間かけてアセトン:メタノール(1:1)で固定化した。次に、当該細胞を、構造タンパク質に特異的なマウス抗体を使用して、VEEカプシドまたはGPタンパク質の発現について分析した。一次抗体をPBS:FBS(1:1)で希釈し、各ウェルに100μlを加えた。プレートを37℃で30分間インキュベートし、150μlのPBSで3回洗浄し、次いでAlexa Fluor 488ヤギ抗マウス二次抗体(Invitrogen社、Carlsbad、CA)により37℃で30分間インキュベートした。インキュベーション後、細胞を上に記載されているように再び洗浄し、各ウェルに最終容積が100μlになるようにPBSを加え、紫外蛍光顕微鏡(Nikon Eclipse TE300)によって検査した。
【0111】
B.ノーザン分析
エレクトロポレーションした細胞を、25cm2のフラスコに播種し、PBSで洗浄し、次いで、RNAwizRNA(登録商標)単離試薬(Ambion社、Austin、TX)を使用し、メーカーの提案するプロトコールに従って全細胞RNAを抽出した。RNA濃度は吸光分光分析法によりで決定した。各試料の5マイクログラム(5μg)を、1%グリオキサールアガロースゲルにより電気泳動して、RNAをBrightStar Plus(登録商標)(Ambion社)膜に受動移入させた。ノーザン分析は、BrightStar BioDetect(登録商標)キット(Ambion社)を使用し、メーカーの提案するプロトコールに従って、VEEの3’複製認識配列のブラス鎖に特異的なビオチニル化DNAオリゴで実施した。化学発光は、処理された膜をフィルムに露光して検出した。
【0112】
C.dHcap(FL)およびdHgp(FL)発現の分析
完全長Δ26Sヘルパー(dHcap(FL)およびdHgp(FL))が複製され、タンパク質を発現し得ることを実証するために、これらのヘルパーRNAを、ヘルパーRNAの複製を促進するアルファウイルス非構造タンパク質を得るために必要なレプリコンベクターと一緒に、細胞中へエレクトポレーションした。ベロ細胞を、30μgのレプリコンRNAと組み合わせた30μgまたは60μgのdHcap(FL)またはdHgp(FL)ヘルパーRNAによりエレクトポレーションした。当該エレクトポレーションした細胞を、上に記載されたようなIFA、ウエスタンブロット、およびノーザン分析ために処理した。
【0113】
(実施例3)
完全長および切端されたΔ26Sヘルパーの発現分析
dHcap(FL)およびdHgp(FL)ヘルパーは、IFAおよびウエスタンブロットによって決定されるように、タンパク質を発現し、ノーザンブロットによって図示されるように、効率的に複製された。
【0114】
カプシドおよびGPの両方に対する切端されたΔ26Sヘルパー(欠失1〜8)の完全なセットが、それぞれがどの程度良好に発現されかつ複製されるかを評価するため、IFAおよびノーザンブロットにより当該セットのタンパク質発現を分析した。それぞれのdHcapヘルパーRNAをVEEレプリコンRNAおよび13.4.6糖タンパク質ヘルパーRNAと組み合わせて、3つのRNAをベロ細胞中にエレクトロポレーションした。ノーザン分析およびIFAは、上に記載されたようにして実施した。カプシド特異的抗体を使用したIFAの結果を表2に示す。すべてのdHcapヘルパーは、IFAによりカプシド発現に対して陽性であったが、dHcap8ヘルパーのみは、弱い陽性であった。
【0115】
エレクトポレーションした細胞から抽出されたRNAのノーザン分析は、切端されたカプシドΔ26Sヘルパーが、dHcap8を除いて、全てよく複製することを示した。
【0116】
dHgpヘルパーについても同様の方法で実験したが、この実験には13.2.2カプシドヘルパーは含めなかった。各dHgpヘルパーをVEEレプリコンRNA組み合わせて、細胞中にエレクトポレーションし、IFAおよびノーザン分析のために上記と同様にして試料を作製した。抗−GP IFAの結果を表3に示す。dHcapヘルパーと同様に、dHgp8を除いて、全てのdHgpヘルパーが陽性であった。dHgp8を除いて、全てのdHgpヘルパーが、良好に複製した。
【0117】
(実施例4)
修飾されたプロモーターレスヘルパー構築物
本発明者は、ウェスタンブロット法によって明らかにされたように、dHcap(FL)およびdHgp(FL)ヘルパーが融合タンパク質を発現する点に注目した。このような融合タンパク質は、Δ26Sヘルパーの転写の際に、カプシドまたはGPのための出発コドンの上流にあるインフレームの出発コドンで翻訳が開始された結果であり得る。そのような上流のコドンの1つは、VEE nsP1に対する天然の出発コドン(VEEウイルスゲノムのヌクレオチド45に位置する)であり、これは、dHcapヘルパーおよびdHgpヘルパーの両方の5’末端に存在し、翻訳の開始に対して有利なコンテキストにある(例えば、コーザック共通配列)。有利なコーザック環境の出発コドンは、これらのヘルパーのキャップされた5’末端からのリボソームのスキャンによって使用され得、それによって、機能的ではない融合タンパク質が生成され、さらなる下流に位置する適切な出発コドンからの機能的カプシドおよび糖タンパク質ポリペプチドの産生が減少する可能性がある。
【0118】
そのような融合タンパク質の産生量を減少させ、かつ完全長カプシドおよび糖タンパク質の発現を増加させるために2つのアプローチを用いた。第一に、上に記載された有利な出発コドンを、TAG停止コドンへ変異させ、残りの出発コドンは変更せずそのまま残した。このアプローチは、5’末端配列を天然のVEEゲノムに存在する配列のできるだけ近くに維持して、これらのヘルパーにとって必要とされる複製要素を維持するために実施した。第二に、nt3の下流の全ての出発コドン(nsP1(有利な)出発コドンを含む)およびカプシドもしくはタンパク質コード配列のオープンリーディングフレーム(ORF:open reading frame)を、AUGからGUGに変更した(全部で12のそのような変更があった)。このアプローチは、有利なAGTコドン(RNAではAUG)が、完全長カプシドもしくは糖タンパク質発現の産生に対して有害効果を有していないかどうかを調べるために実施した。
【0119】
A.dHcap−mut1およびdHgp−mut1ヘルパーの構築
有利なnsP1出発コドンをTAG停止コドンへと変えるdHcap−mut1およびdHgp−mut1ヘルパーを生成するために、各dHcapヘルパーおよびdHgpヘルパーに部位特異的変異を実施し、dHcap−mut1ヘルパーおよびdHgp−mut1ヘルパーと称される、それぞれ配列番号35〜42および43〜50として本明細書において提供されるような5’配列を有する変異したヘルパーの完全なセット(FLおよび切端物1〜7)を生成させた。部位特異的変異は、Quikchange XL(登録商標)部位特異的変異キット(Stratagene社、La Jolla、CA)により、メーカーのプロトコールに従って、表4のフォワード(配列番号71)プライマーおよびリバース(配列番号72)プライマーを使用して実施した。
【0120】
B.dHcap−mmヘルパーおよびdHgp−mmヘルパーの構築
nt3およびカプシドもしくはGP ORF出発コドンの下流に出発コドンを持たない5’末端を有するdHcapヘルパーおよびdHgpヘルパーを生成させるため、部位特異的変異を実施して、介在する全てATG(RNAではAUG)をGTG(RNAではGUG)コドンに変えた。dHgp(FL)構築物を、部位特異的変異のための鋳型として使用した。Quikchange(登録商標)複数部位特異的変異キット(Stratagene社)を使用し、メーカーのプロトコールに従ってコドン変更を導入した。コドン変更を導入するために使用したプライマーを表5に示す(配列番号73〜82)。全てのコドン変更を含有するdHgp(FL)構築物を、dHgp(FL)−mm(配列番号19として本明細書において提供される5’配列を有する)と称した。全てのコドン変更が存在することを配列確認した後に、このDNAを使用して、dHcap(FL)5’複製認識配列をdHgp−mmに由来する5’複製認識配列で置換することにより、dHcap(FL)−mm構築物を生成させた。これは、両方のDNAをRsrIIおよびNotI酵素で消化することにより達成した。次いで、dHgp(FL)−mm RsrII/NotI5’複製認識配列断片を、直線化されたdHcap(FL)DNAでライゲーションさせ、dHcap(FL)−mm(配列番号27として本明細書において提供される5’配列を有する)を生成させた。さらに、dHgp(FL)−mmDNAを鋳型として使用して、dHcap1〜7およびdHgp1〜7に対して上に記載された方法およびプライマーを使用してカプシドヘルパーおよびGPヘルパーの両方に対する切端された5’末端のセットを生成させた。新規のヘルパーは、dHcap1mm 〜dHcap7mm(配列番号20〜26として本明細書において提供される5’配列を有する)およびdHgp1mm〜dHgp7mm(配列番号28〜34として本明細書において提供される5’配列を有する)と称した。
【0121】
C.mut1およびmmのプロモーターレスヘルパーの発現分析
上に記載されたΔ26Sヘルパーのmut1およびmmの様々な変形からのタンパク質産生を分析した。この実験において、dHcap6−mut1(配列番号41として本明細書において提供される5’配列を有する)、dHcap6−mm(配列番号25として本明細書において提供される5’配列を有する)、dHcap7−mm(配列番号26として本明細書において提供される5’配列を有する)、およびdHgp7−mm(配列番号27として本明細書において提供される5’配列を有する)、並びに13.2.2および13.4.6ヘルパーを、ウエスタンブロットによって分析した。mm変異を含むΔ26Sヘルパーは、わずかな検出可能な融合タンパク質を伴ってまたは伴わずに、主に完全長のカプシドタンパク質またはGPタンパク質を発現する。mut1ヘルパーは、かなりの量の完全長構造タンパク質を発現したが、依然としていくらかの融合タンパク質も発現した。
【0122】
mut1およびmmΔ26Sヘルパーの複製特性を分析するために、同じ試料についてノーザン解析を行った。結果は、dHcap6−mut1ヘルパーが、13.2.2カプシドヘルパーと同様に複製することを示している。対照的に、dHcap6−mm、dHcap7−mm、およびdHgp7−mmヘルパーの複製は、13.2.2またはmut1ヘルパーより少ない程度のようである。
【0123】
(実施例5)
Δ26SヘルパーによるVEEレプリコン粒子の生成
表6に列挙されているのは、VEEレプリコンRNAと様々なプロモーターレスカプシドヘルパーおよびGPヘルパーとを組み合わせてVEE複製粒子(VRP:VEE replicon particle)を産生させる多くの実験である。さらに、細胞中に導入された各ヘルパーRNAの量も、いくつかの実験において変えた。VRPは、5×107〜1×108個のベロ細胞を、指示された量のヘルパーRNA並びに30μgのレプリコンRNAでエレクトポレーションすることによって生成された。概して、粒子が生成される全ての実験に対し、エレクトポレーションした細胞を、血清不含培地が入った300cm2のフラスコに播種し、16〜24時間インキュベートした後、VRPを収穫した。
【0124】
VRP力価は、試料の十倍の階段希釈により、96ウェルプレートで増殖させたベロ細胞に感染させて、当該細胞を16〜18時間インキュベートし、当該細胞を固定化し、VEEのnsP2タンパク質または対象となる核酸の産物に特異的な抗体によってIFAを実施することによって決定した。VRP収量は、実験からの全収量、または20mlの調製物からの1ml当たりの量のいずれかとして示されている(表7、9〜13、15、および16)。
【0125】
これらの調製物は、さらに、細胞変性効果(CPE:cytopathic effect)アッセイにより複製能力のあるウイルス(RCV)の存在を試験した。当該CPEアッセイは、RCVの存在をスクリーンするための、細胞培養における2つの盲継代で構成された。継代1では、VRP調製物由来の試料を、ベロ細胞単層により37℃で1時間インキュベートし、次いで、試料液体を除去し、新鮮な培地と交換して、当該培養物を24時間インキュベートして、存在し得る任意のRCVの増幅を可能にした。継代2では、継代1の終了時の細胞培養の上清を新鮮なベロ細胞単層に加え、37℃で72時間インキュベートした。継代2の終了時に、倒立型光学顕微鏡を使用して、培養物のCPEを検査した。このアッセイは標準化されており、大過剰のVRPの存在下での生存ウイルスの検出における感度について評価した。このアッセイにおいてV3014またはTC−83ウイルスのいずれかを用いたスパイク試験により、1×108個のVRPをバックグランドとした3〜8PFUの検出の下限を明らかにした。このアッセイを、本発明のプロモーターレスヘルパーによって産生された1013個を超えるVRPに対して実施したところ、RCVは検出されなかった。このアッセイの検出限界にもかかわらず、RCVの産生に対する可能な組換え頻度の理論計算は、この検出限界の使用を大きく下回り、すなわち、1010、1011、1012、または1013のVRPに1個、であろう。
【0126】
(実施例6)
「スプリット糖タンパク質」プロモーターレスヘルパー
A.別々のE2および1Eのプロモーターレスヘルパーの構築
E2およびE1コード配列が別々のヘルパー上に存在する糖タンパク質のプロモーターレスヘルパーの構築は、E2および1Eの糖タンパク質カセットを、別々に、dHgp6−mut1ヘルパーのバックボーンにクローニングすることによって実施した。プライマーは、PCRによってpHCMV−Vsp由来のVEE構造上タンパク質コード領域のカプシド−E3−E2領域を増幅するように設計した(米国特許第7,045,335号を参照のこと。なお、この特許は参照により本明細書に組み入れられる)。増幅された断片は、pCR−BluntII TOPO(登録商標)ベクター(Invitrogen社)中にクローン化され、pCR−CE3E2を生成した。CE3E2カセットを配列決定して、PCR増幅中にエラーが導入されなかったことを確認した。CE3E2構造領域を含有するプロモーター補助ヘルパーを作製するため、pCR−CE3E2 DNAをSpeI制限酵素で消化し、E3E2断片を放出させた。次いで、当該E3E2(SpeI)断片を、SpeI酵素で直線化したカプシドヘルパー(13.2.2)でライゲーションした。pHCE3E2由来のE3−E2コドン領域を消化することによって、プロモーターレスE2ヘルパー(dHE2−6M1と称する)を調製した。pHCE3E2DNAプラスミドは、最初に、AscI制限酵素によって直線化し、次いで、平滑末端を作成するためにT4 DNAポリメラーゼで処理した。同様に、dHgp6−mut1 DNAプラスミドは、SphI制限酵素で直線化し、T4 DNAポリメラーゼ処理して、平滑末端を作成した。両方の直線化されたT4−ポリメラーゼ処理DNAは、SpeI制限酵素によって消化され、結果として生じた3.6kbのdHgp6−mut1ベクター断片および1.4kbのE3−E2断片を、それぞれゲル精製した。次いで、当該2つの精製された断片を、T4 DNAリガーゼを用いて一緒にライゲーションさせて、dHE2−6M1プロモーターレスヘルパーを作製した。
【0127】
プロモーターレスE1ヘルパーの生成は、数段階において達成した。プライマーは、次の2つの構造タンパクコード配列断片の1)カプシドE3(CE3)、および2)6K−E1(6KE1)を増幅するように設計した。PCR産物を、pCR−Blunt TOPO(登録商標)ベクター(Invitrogen社)中にクローン化し、pCR−CE3およびpCR−6KE1を生成させた。当該クローンを配列決定して、増幅中にエラーが導入されなかったことを確認した。E1糖タンパク質の上流にE3および6Kリーダー配列の両方を含有するカセットを作製するために、別の中間構築物を作製した。これは、BamHI酵素でpCR−6KE1 DNAを消化して、6KE1断片を精製することによって達成した。次いで、6KE1(BamHI)断片を、BamHI酵素で直線化したpCR−CE3 DNAでライゲーションし、pCR−CE36KE1を生成させた。CE36KE1カセットを含有するプロモーター補助ヘルパーを生成するために、pCR−CE36KE1 DNAを、SpeIおよびSphI酵素で消化させ、構造タンパク質コード配列カセットを放出させた。次いで、CE36KE1(SpeI/SphI)断片を、SpeIおよびSphIで直線化したカプシドヘルパー(13.2.2)でライゲーションして、pHCE36KE1を作製した。プロモーターレスE1ヘルパー(dHE1−6M1と称する)の生成は、pHCE36KE1プラスミド由来のE3−6K−E1コード領域を消化することによって達成した。pHCE36KE1およびdHgp6−mut1 DNAプラスミドを、SpeIおよびSphI制限酵素で消化させ、結果として得られた3.6 kbの dHgp6−mut1ベクター断片および1.7kbの E3−6K−E1断片をゲル精製した。次いで、2つの精製した断片を、T4 DNAリガーゼを使用して一緒にライゲーションさせて、dHE1−6M1プロモーターレスヘルパーを作製した。
【0128】
B.スプリット糖タンパク質プロモーターレスヘルパーの分析
個々の糖タンパク質ヘルパーをインビトロで転写させ、RNA転写物を精製した後、VEEレプリコンRNAと一緒にベロ細胞中にエレクトポレーションした。ヘルパー複製を、ノーザンブロットによって分析し、タンパク質発現は、E1およびE2糖タンパク質特異的抗体を用いてIFAによって分析した。ノーザン分析の結果は、dHE1−6M1ヘルパーおよびdHE2−6M1ヘルパーの両方が、効率的に複製することを示している。代表的なノーザンブロットを図3に示す。
【0129】
レプリコンRNAをパッケージしてVRPを作製するために2つの別々の糖タンパク質発現プロモーターレスヘルパーをΔ26Sカプシドヘルパーと組み合わせられるかどうかを決定するために、三つのヘルパーを、ボツリヌス神経毒素A断片を発現するVEEレプリコンRNAと組み合わせて、ベロ細胞中にエレクトロポレーションした。一実験からのVRP収量を表7に示す。
【0130】
(実施例7)
修飾した5’末端および3’末端プロモーターレスヘルパーカセット
A.修飾した5’末端ヘルパーカセットの構築
ほとんどのアルファウイルスのRNA5’末端(およそ最初の250nt)に予測される二次構造は、4つのステムループ(SL)構造(SL1、SL2、SL3、およびSL4)を含有する。Frolovら(RNA、7:1638−1651(2001))は、SL2をコードするヌクレオチド配列をSindbisウイルスヘルパーRNAから除去することによって、そのヘルパーの複製が増加することを実証した。
【0131】
(M倍のプログラムに基づく)VEE5末端のSL2領域であるnt46〜nt116を、以下のようにPCRによってdHcap6−mut1から除去した。2つの断片を、dHcap6−mut1 DNAから増幅した。およそ1キロベース(kb)の5’断片を、dHcap6−mut1の5’末端の45ヌクレオチドとバックボーンプラスミド配列をコードするヌクレオチドとを含有するプライマー13−82.1.9[配列番号83]およびdLS2(EcoRV)R[配列番号84]によって増幅させた(表8)。およそ1.5kb)の3’断片を、ヌクレオチド117で始まるVEE5’末端の一部とVEE3’末端を介してカプシド配列全体をコードするヌクレオチドとを含有するプライマーdSL2(EcoRV)F[配列番号85]および3−8.pr4[配列番号86]によって増幅させた(表8)。5’の約1kbのPCR断片を、XhoIおよびEcoRV制限酵素によって消化させた。3’の約1.5kbのPCR断片を、EcoRVおよびNotI制限酵素によって消化させた。プラスミドdHgp6−mut1を、XhoIおよびNotIによって消化することによって直線化し、結果として得られる約2.5kbのベクターバックボーンを精製した。本明細書において「dHcap6−mut1(dSL2)」と呼ばれる、SL2領域が削除された新しいヘルパーを生成させるため、5’(XhoI/EcoRV)断片、3’(EcoRV/NotI)断片、およびXhoI/NotI直線化ベクターを一緒にライゲーションさせた。配列が本明細書において配列番号51として提供される5’末端を有するdHcap6−mut1(dSL2)ヘルパーを完全に配列決定し、PCR増幅中にエラーが導入されなかったことを確認した。マッチングdHgp6−mut1(dSL2)ヘルパーを生成させるため、dHgp6−mut1 DNAをXhoIおよびRsrII制限酵素で消化させ、5.4kbの断片を精製した。dHcap6−mut1(dSL2)由来の修飾された5’末端は、このDNAをXhoIおよびRsrIIで消化し、1.1kbの断片を精製することにより収集した。これら二つの断片を一緒にライゲーションさせ、dHcap6−mut1(dSL2)として同じ5’末端[配列番号51]を有するdHgp6− mut1(dSL2)を生成させた。
【0132】
B.短縮された3’末端プロモーターレスヘルパーカセットの構築
これらの実施例において、カプシドヘルパー構築物のdHcap(FL)、dHcap1〜dHcap7、dHcap(FL)mm、dHcap1mm〜dHcap7mm、dHcap(FL)mut1、およびdHcap1mut1〜dHcap7mut1に対し、3’末端配列は、配列番号55として本明細書において提供される。本発明のVEEカプシドヘルパーは、完全な糖タンパク質コード領域を欠失しているが、E3タンパク質の小さな部分がカプシドヘルパー上に残っており、成熟カプシドタンパク質を産生するためにパッケージング細胞内においてキモトリプシン様切断が生じることを可能にしている。糖タンパク質ヘルパー構築物のdHgp(FL)、dHgp1〜dHgp7、dHgp(FL)mm、dHgp1mm〜dHgp7mm、dHgp(FL)mut1、およびdHgp1mut1〜dHgp7mut1に対して、成熟カプシドタンパク質を生成するための切断部位を含む3’末端配列は糖タンパク質ヘルパー構築物において必要でないため、当該配列は、より短い配列であった。これらの実施例においてこれらの糖タンパク質構築に使用される3’配列は、本明細書において配列番号56として提供される。
【0133】
さらに、より短い長さの3末端を有するプロモーターレスRNAヘルパーを構築した。アルファウイルス3’末端配列の量を減らすことにより、増殖性VEEウイルスの生成に必要であろう二次組換え現象の理論的可能性がさらに減少する。最初に、アルファウイルスの3’高度に保存された配列[配列番号52]を含む19ヌクレオチドのみを含有する機能的な26Sプロモーターを有する糖タンパク質ヘルパーを、以下の二つの段階において作製した。最初に、ユニーク5’制限部位および3’制限部位を有する糖タンパク質(GP)コード配列カセットを含有したプラスミドを作製した。3’末端のE1終止コドンのすぐ後のユニークSphI部位(「GP(SphI)R」、配列番号87、表8)、および5’末端の既存の内部SpeI部位(「3−16.1.3」、配列番号88、表8)でVEE GPを増幅するようにプライマーを設計した。増幅された断片は、pCR2.1/GP 19nt 5’を生成するpCR2.1 DNA(Invitrogen社、Carlsbad、CA)中にTAクローニングした。次に、VEE3’末端(3’ trunc(SphI)F、配列番号89、表8)の19ヌクレオチド保存配列のすぐ上流にSphI部位を導入するようにフォワードプライマーを設計した。3’末端にユニークAflII制限部位(3’trunc(AflII)R、配列番号90、表8)を含有するであろう断片を増幅するように、プラスミドバックボーン配列に特異的なリバースプライマーを設計した。これらのプライマーの増幅から得られた断片は、SphIおよびAflIIで消化され、SphIおよびAflII制限酵素で直線化されている(実施例1に記載された)13.4.6糖タンパク質ヘルパーとライゲーションし、これによって、結果としてpGPヘルパー−int1が構築された。pGPヘルパー−int1構築物は、(19nt保存配列を含む)当該ヘルパーのGP停止コドンと3’末端の間に72ヌクレオチド領域を有している。19ヌクレオチド3’末端のみによりGPヘルパーを生成させるために、pCR2.1/GP 19nt5’DNAをSpeIおよびSphIで消化させ、当該GPコード配列を、SpeIおよびSphI制限酵素で消化されたpGPヘルパーint1にライゲーションさせた。結果として得られた構築物は、pGPヘルパー19ntと命名した。
【0134】
次いで、pGPヘルパー19nt構築物を使用して、可変長の3’複製認識配列を有するΔ26Sヘルパーを作製した。当該pGPヘルパー19nt構築物を、NcoI制限酵素およびNotI制限酵素によって消化し、19nt3’末端領域を有する糖タンパク質コード配列を含有する2515塩基対断片をゲル精製した。次に、この2515塩基対(NcoI/NotI)断片を、NcoIおよびNotI制限酵素によって消化されたdHgp構築物でライゲーションし、様々なdHgp19nt構築物を生成させた。
【0135】
C.アルファウイルスカプシドタンパク質を発現する修飾されたプロモーターレスヘルパーカセットの構築
VEEウイルス感染細胞において、VEEEカプシドタンパク質は、26SサブゲノムmRNAから翻訳される構造ポリタンパク質から自身を切断する。本発明のVEEカプシドヘルパーは、完全な糖タンパク質コード領域を欠失しているが、E3タンパク質の小さな部分がカプシドヘルパー上に残っており、成熟カプシドタンパク質を産生するためにパッケージング細胞内においてキモトリプシン様切断が生じることを可能にしている。キモトリプシン様切断部位の代わりに、カプシドの3’末端での停止コドンの導入は、糖タンパク質ヘルパーによる機能的な遺伝子組換え体の産生の困難さを増加させるであろう。すなわち、本発明のdHgpヘルパーによって生じる機能的な組換え(すなわち、複製能力のあるウイルスを産生する組換え)に対して、当該組換え現象は、カプシドコード配列において、操作された停止コドンを置換して活性なカプシド切断部位を維持するために完全なヌクレオチドでなければならない。3’末端に組み込まれた停止コドンを有するdHcapヘルパーの2つの変形を作製した。1つの変形は、天然カプシドタンパク質のC末端トリプトファン残基を停止コドンで置換した、本明細書において配列番号57として提供されるdHcap6−mut1−dSL2(停止)であり、他方の変形は、C末端トリプトファン残基を保有し(本明細書において配列番号59として提供される3’配列を有するdHcap6−mut1(W−停止))、トリプトファン残基のすぐ下流に停止コドンを挿入した。カプシドコード配列を、5’末端のユニークRsrII部位(カプシド(RsrII−コーザック)F、配列番号:91、表8)および3’末端のユニークSphI部位カプシド(停止)SphI R、配列番号:92またはカプシド(W−停止)SphI R、配列番号:93、表8)を操作するために設計されたプライマーで複製した。さらに、フォワードプライマーを操作して、カプシド出発コドンをほぼ最適なコーザック共通配列に配置して(Kozak,Cell,44(2):283−292(1986))、カプシドmRNAの翻訳のリボソーム開始を高めた。増幅されたカプシドコード配列を、RsrII制限酵素およびSphI制限酵素で消化し、RsrIIおよびSphIで直線化されたΔ26Sヘルパープラスミドにライゲーションさせて、dHcap6−mut1−dSL2(stop)およびdHcap6−mut1(W−stop)構築物を作製した。
【0136】
D.アルファウイルス糖タンパク質を発現する修飾されたプロモーターヘルパーカセットの構築
VEEカプシドタンパク質は、カプシドC末端トリプトファン残基の後で切断するキモトリプシン様プロテアーゼである。キモトリプシンの切断特異性に基づいて、すべてのアミノ酸残基が、メチオニンおよびプロリン以外のトリプトファンのすぐ下流の位置において許容されることが予想される。トリプトファンのすぐ下流にこれらのアミノ酸のいずれかを有することは、キモトリプシン切断活性を著しく減じることが予想される。天然VEEウイルスには、VEE E3シグナル配列を含む18のアミノ酸がある。E3シグナル配列のアミノ酸の数を減じると同時に、E3配列のシグナリング機能は維持するように構築物を設計した。E3配列を含む18のアミノ酸のうちの16は、カプシドC末端トリプトファンの下流の位置において許容されることが予想されるため、E3シグナル配列のアミノ酸の数を減らすことにより、切断部位として機能するであろう部位がVEE構造ポリタンパク質コード配列を再構成するヌクレオチド完全組換え現象の発生の際にC末端トリプトファンのすぐ下流に位置する場合、当該部位の数が減少するであろう。そのようなアプローチの例として、通常、E3シグナル配列に存在するN末端セリン残基をPCRによって除去し、N末端基残留物としてのロイシン残基を残して、そのような修飾されたgpヘルパーがVRPをパッケージするために機能するかどうかを決定するためにdHgpのプロモーターレスヘルパーを構築した。
【0137】
E3のN末端セリン残基を除去し、ユニークRsrII制限部位を維持するように、フォワードPCRプライマー(Gp(RsrII−Ser)F、配列番号94)を設計した(表8)。ユニークSnaBI制限部位を含有するであろうgp断片を増幅するように、リバースPCRプライマー(3−16.2.14、配列番号95)を設計した(表8)。結果として得られたgpPCR断片をRsrIIおよびSnaBIで消化させ、RsrIIおよびSnaBI消化dHgp6−mut1DNAにライゲーションして、dHgp6−mut1(−S)を生成させた。
【0138】
E.5’および3’修飾Δ26Sヘルパーを使用したVRP生成実験
上記の修飾の組み合わせを含有するヘルパーも調製した。dHcapおよびdHgpプロモーターレスヘルパーの様々な組み合わせおよびRNA濃度を、VRP産生実験において分析し、それらが、VEEレプリコンRNA(ボツリヌス神経毒素断片AまたはインフルエンザHAのどちらかを発現するもの)のパッケージングにおいてどの程度効果的であるかを決定した。さらに、VRP収量に関するΔ26Sヘルパーのキャッピングの効果を、当該ヘルパーの組み合わせのサブセットに対して分析した。VRP収量の代表例は、Δ26Sヘルパーの様々な組み合わせと共に表9〜13に示す。VRPの感染力および収量を定量化する力価アッセイは、48ウェルプレートのベロ細胞単層培養物において、VRPを段階的に希釈し、ベロ細胞と共に5%のCO2中において37℃で一晩インキュベートすることによって実施した。一晩インキュベート(18〜20時間)した後、当該細胞を洗浄し、固定化し、当該固定化された単層を抗原特異的一次抗体で染色した後、FITC結合二次抗体で染色した。紫外蛍光顕微鏡(Nikon Eclipse TE300)により、FITC標識化抗原−抗体を含有する細胞を検出した。個々の抗原陽性細胞をカウントし、IU/mLで表される力価を、既知の希釈および接種量から計算する。
【0139】
(実施例8)
ユビチキン単量体を取り込むプロモーターレスヘルパー
A.ユビキチン単量体を含有するΔ26Sヘルパーの構築
真核細胞において、ユビチキンによって縮合またはタグ付けされたタンパク質は、細胞のユビキチンカルボキシル末端ヒドロラーゼ(UCH:ubiquitin carboxyl−terminal hydrolase)によってC末端グリシンのすぐ後ろで切断される(Pickart and Rose,J.Biol.Chem 260:7903−7910(1985))。ユビチキンコード配列の単量体を、カプシドおよび糖タンパク質コード配列のすぐ上流のインフレームに配置することにより、本発明のある特定のプロモーターレスヘルパー構築物によって産生される融合タンパク質を排除する(そのような融合体は、各構造タンパク質コード配列に対してATGの上流にある複数の翻訳開始部位によって生じる)。全てのインフレーム融合タンパク質は、ユビキチン単量体を含むことになり、そのため、それらがUCHによって切断され、その結果、上流に外因性タンパク質配列を有さない完全長VEE構造タンパク質が放出されるため、当該排除が生じる。増幅されたユビチキン単量体コード配列の5’および3’末端にRsrII部位を導入すると共に、UCHによるユビチキン単量体の切断に必要なArg−Gly−Gly配列(図4)を維持するように、プライマーであるubiquitin F(配列番号96)およびubiquitin R(配列番号97)(表14)を設計した。これらの特定の構築物により、切断の後に得られる構造タンパク質のそれぞれに、天然の構造タンパク質には存在しないさらなるN末端アミノ酸残基(単数または複数)が生じた(すなわち、カプシドヘルパーに対しては余分なプロリンであり、糖タンパク質ヘルパーに対しては、余分なプロリンおよびトレオニンである)(図4)。
【0140】
当該ユビキチンコード配列を、Pfu Taqポリメラーゼ(Stratagene社)を使用してPCR増幅し、dHcap(FL)およびdHgp(FL)のユニークRsrII部位にクローン化した。ユビキチン挿入の方向を決定するために、形質転換体をスクリーニングした。カプシドおよび糖タンパク質に対する陽性ユビキチンクローンは、それぞれdHcapUおよびdHgpUと称し(それぞれ、本明細書において配列番号53および54として提供される5’末端配列を有する)、これらを単離し、配列決定して、増幅されたユビチキンコード配列中にエラーが導入されていないことを確認した。エレクトロポレーションのためのRNAは、RiboMax Express RNA(登録商標)キットを使用して、dHcapU、dHgpU、dHcap(FL)、dHgp(FL)、Hcap4、および13.4.6プラスミドから別々の反応において転写し、塩化リチウムで沈殿させた。
【0141】
B.ユビキチン修飾されたΔ26Sヘルパーを使用したVRP生成実験
ベロ細胞を、HIVクレードC糖タンパク質(「DU151gp160」)を発現するVEEレプリコンRNAでエレクトロポレーションし、指示されたRNA量でプロモーターレスカプシドおよびGPヘルパーの組み合わせを選択した。いくつかの実験において、「Hcap4」カプシドヘルパーを使用した。これは、切端された5’末端を有するが(上述のdHcap4切端物に相当する)、26Sサブゲノムのプロモーター配列を保有するヘルパーであり、米国特許第7,045,335号に詳細に記載されている。なお、この特許は参照により本明細書に組み込まれる。約0.8mlの容積の0.4cmのキュベットにおいて、500V、25μF、4パルスでエレクトロポレーションを実施した。各エレクトロポレーションは、100mlのOptipro(登録商標)(Gibco社、Carlsbad、CA)と共に1つの850cm2ローラーボトル中に播種した。18時間後に、25mlの0.5MのNaClで洗浄しながら0.2μmフィルタによりVRPを収穫した。VRP塩水洗浄物質を、(HIV gp160タンパク質を認識する)抗−gp120ヤギ抗体により1:400で力価決定した。パッケージング実験の結果を表15に示す。
【0142】
続いてエレクトロポレーションを実施して、dHgp6−mut1と組み合わされたカプシドヘルパーのdHcapUまたはdHcap6−mut1(W−stop)でパッケージされた様々な核酸の力価量を比較した。VEE nsP2特異的ポリクロナール抗体を使用してVRPの力価を決定した(表16)。
【0143】
C.エレクトポレーションした細胞におけるウエスタン分析による構造タンパク質の発現
表16にまとめられたパッケージング実験においてVRPを生成するために使用した細胞から細胞溶解物を調製した。各試料からの細胞溶解物を、4〜12%のBis−Tris Novexゲルにおいて、1×MOPS中200V、400mAにて45分間電気泳動した後、1×移行緩衝液中400mAで40分間、PVDFに半乾燥移行させた。膜を、1×BMBブロック/TBS中で一晩ブロッキングした。一次抗体は、1×BMBブロック/TBS中1A4A抗−VEE GPの1:500希釈および抗−VEEカプシドの1:1500希釈であった。ウエスタンブロット結果を図5に示す。dHgpUから発現した糖タンパク質は、dHgp(FL)から発現した糖タンパク質よりもより完全なPE2およびE2GP形態へとプロセシングされる。これは、dHgp(FL)にユビキチンが存在しない場合に見られる融合タンパク質のパターンにおける違いによって実証される(図5、GP抗体を使用したウエスタンブロットのレーン3及び4を比較)。dHcapUヘルパーにおけるカプシドタンパク質のN末端でのユビキチンタンパク質の置換により、結果として、カプシド融合タンパク質が消失し(図5)、13.4.6糖タンパク質ヘルパーでパッケージングされた場合にgp160の力価において2桁以上増加する(表15)。
【0144】
D.エレクトロポレーションした細胞のノーザン分析による構造タンパク質RNAの発現
全細胞RNAを、表15にまとめられたパッケージング実験においてVRPを生成するために使用された細胞から抽出した。当該細胞をRNAwiz(登録商標)試薬(Ambion,Inc社、Austin、TX)で溶解させ、クロロホルムで抽出し、沈殿させて、カプシド特異的プローブおよびGP特異的プローブを使用してノーザン分析に供した(それぞれ、図6および図7)。すべてのRNA種が、様々な構築物から予想されるサイズと一致している。
【0145】
(実施例9)
キャップおよび非キャップΔ26Sヘルパー構築物を使用したVRP生成
A.様々なアルファウイルスの糖タンパク質を発現するVRP
VRPは、対象となる核酸(NOI)として、それぞれフューリン切断部位が欠失したVEE(3022)、Eastern equine encephalitisウイルス(EEE)(4200)、またはWestern equine encephalitisウイルス(WEE)(2100)のいずれかの糖タンパク質に対するコード配列を発現するVEEレプリコンを使用して作製した。VRPを生成するために使用されたヘルパーをコードするDNAプラスミドは、NotIにより直線化し、インビトロで、T7RiboMax(登録商標)キット(Promega社、Madison、WI)を使用して、メーカーの指示に従い、指示された場合には7.5mMのPAP類似物(Promega社)で補って転写した。キャップ類似物で作製したヘルパーは、「+Cap」として示し、キャップ類似物無しで作製されたヘルパーは「−Cap」として示した(表17)。レプリコンの組み合わせを用いてベロ細胞をエレクトロポレーションし、上述の実施例5に記載したようにしてGPヘルパーRNAおよびVRPを作製した。三つの別々の実験の結果を表17〜19に示す。
【0146】
B.インフルエンザ株ウィスコンシンのHAコード配列を発現するVRP
この実験において、VEEカプシドまたはVEE糖タンパク質のいずれかをコードするΔ26SヘルパーRNAを作製するために、転写反応においてCap類似物とGTPのモル比を変更した。当該転写反応は次のように構成した:Promega 5×転写緩衝液;rNTPミックス(6mMのUTP、CTP、ATP);GTP(表に示されるように、0〜6mMで);(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1300)およびRibom7GCap(登録商標)類似物(6mM)(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1712)。メーカーによって指定された、通常、キットによって供給される2×緩衝液で実施されるRiboMax Express(登録商標)RNA転写キット条件(Promega Corporation Woods Hollow Rd.社, Madison、WI、カタログ番号#P1320)を模倣するために、7.5mMのRibom7GCap(登録商標)類似物の有無において、Promega社の5×緩衝液および7.5mMのrNTPにより、さらなる反応を行った。この実験でパッケージングされたVEEレプリコンは、インフルエンザHA(A/WI/05)タンパク質をコードした。各エレクトロポレーションに対して、30μgのレプリコンRNA;10μgのΔ26SカプシドヘルパーRNA、および60μgグラムのΔ26S糖タンパク質ヘルパーRNAを使用した。ベロ細胞を増殖させ、次いで洗浄し、スクロース緩衝液中に再懸濁させて1.2×108細胞/mLとした。これらの細胞をRNAと混合し、次いで、500ボルト、25μFd、および4パルスに設定したBioRad Gene Pulse II(登録商標)装置でエレクトロポレーションした。細胞を100mLのOptiPro(登録商標)と共にローラーボトルに移し、37℃でインキュベートした。エレクトロポレーション後24時間で、VRPを収穫した。当該VRPを、ベロの48ウェルプレートで力価を決定した。その結果を表25に示す。
【0147】
(実施例10)
Δ26Sヘルパー構築物によって作製したVRPを使用したネズミにおけるボツリヌス神経毒素に対する防御
ボツリヌス神経毒素血清型AまたはB(それぞれ、BoNT AまたはBoNT B)のいずれかの重鎖の無毒のC末端断片を発現するVEEレプリコンベクターを、実施例5で記載されているように、次のいずれか:(i)それぞれ30μgのキャップされていない13.2.2および13.4.6ヘルパー、または(ii)20μgのキャップされたカプシドΔ26Sヘルパーおよび60gのキャップされた糖タンパク質Δ26Sヘルパー、を使用してVRP中にパッケージングした。これらのVRPを、0日〜28日に二回、スイスマウスにワクチン接種するために1×107IUの投与量で使用した。当該マウスを、2回目の免疫処置後1カ月において、BoNT AまたはBoNT B神経毒のいずれかの、動物の50パーセントが死亡するのに必要な投与量(1000LD50)の1000倍を用いてチャレンジ感染させた。当該チャレンジ感染実験の結果を表20にまとめる。
【0148】
(実施例11)
天然痘ウイルス由来の抗原を発現するVRPによる免疫原生および防御実験
A.Δ26Sヘルパー構築物を使用して生成させたVRPの、マウスおよび霊長動物における免疫原生
4つのワクシニアウイルス(VACV:vaccinia virus)遺伝子(L1R、B5R、A27L、およびA33R)を発現するために最適化されたVEEレプリコンベクターを、Kamrudらによって記載された方法(Virology 360(2):376−87(2007))を使用して構築した。当該4つのVACV遺伝子は、まとめて「4pox」と呼ばれる。1つは3014株をベースとし、もう一つはTC−83ワクチン株をベースとする二つの異なるVEEレプリコンベクターシステム中に、当該4pox遺伝子をクローン化した。各最適化されたVACVコード配列発現レプリコンベクターを、30μgのレプリコン、20μgのΔ26SカプシドヘルパーRNA、および60μgのΔ26SGPヘルパーRNAを組み合わせて、それらをベロ細胞中にエレクトロポレーションすることによってVRPを生成するために使用した。粒子は、実施例5に記載されているようにして作製し収集した。次いで、個々のVGACV VRPを組み合わせて、BALB/cマウスまたはカニクイザルのいずれかを免疫化するために使用する4poxVRP混合物を作製し、VACV抗原特異的ELISA分析により、体液性応答を測定した。ワクチン接種したネズミにおいて検出されるVACV特異的ELISA反応を表21に示し、予ワクチン接種したカニクイサルで検出されるVACV特異的ELISA反応を表22に示す。
【0149】
B.4pox VRPを使用した、ネズミおよび非ヒト霊長動物における防御
1.マウス
ワクシニアウイルス(IHD−J株)の2×106PFUを鼻腔内経路によりマウスにチャレンジ感染させ、その結果を表23に示す。
【0150】
2.非ヒト霊長動物
サル痘ウイルスの5×106PFUを静脈内経路により、非ヒト霊長動物にチャレンジ感染させた。世界保健機構の病巣数採点システムを使用して、重症度を判定し、その結果を表24に示す。
【0151】
当業者に理解されるように、特許請求する発明の各態様に対していくつかの実施形態および要素があり、異なる要素の全ての組み合わせが、本発明の実施形態として本明細書に組み込まれ、したがって、本明細書に例示する特定の組み合わせは、特許請求する発明の範囲の限定として解釈されない。特定の要素を除去するかまたはある組み合わせに利用できる要素の群に加える場合、要素の当該群は、そのような変更が組み込まれていると解釈される。
【0152】
非特許出版物、特許出願、および特許などの本明細書で引用されたすべの参照文献は、それぞれが個々におよび具体的に、その全部が参照文献に組み込まれていることが示される場合と同程度に、参照により本明細書に組み込まれ、その全てが本明細書で再現された。
【0153】
【表1】
【0154】
【表2】
【0155】
【表3】
【0156】
【表4】
【0157】
【表5】
【0158】
【表6−1】
【0159】
【表6−2】
【0160】
【表7】
【0161】
【表8】
【0162】
【表9】
【0163】
【表10】
【0164】
【表11】
【0165】
【表12】
【0166】
【表13】
【0167】
【表14】
【0168】
【表15】
【0169】
【表16】
【0170】
【表17】
【0171】
【表18】
【0172】
【表19】
【0173】
【表20】
【0174】
【表21】
【0175】
【表22】
【0176】
【表23】
【0177】
【表24】
【0178】
【表25−1】
【0179】
【表25−2】
【特許請求の範囲】
【請求項1】
a)少なくとも一つの開始コドンが除去されているアルファウイルス5’複製認識配列と、
b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、
c)アルファウイルス3’複製認識配列と
を含み、(b)の該ヌクレオチド配列の転写を誘導するプロモーターを含まないことを条件とする、単離されたRNA分子であって、
(a)および(c)の該アルファウイルス5’および3’複製認識配列が、アルファウイルス非構造タンパク質の存在下において該RNA分子全体の複製を誘導する、RNA分子。
【請求項2】
前記アルファウイルス構造タンパク質をコードする前記ヌクレオチド配列が、1)アルファウイルスカプシドタンパク質、2)任意の順序のアルファウイルスE1およびE2タンパク質、3)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE1タンパク質、5)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE2タンパク質、6)アルファウイルスE2タンパク質、並びに7)アルファウイルスE1タンパク質をコードするヌクレオチド配列からなる群より選択される、請求項1に記載のRNA分子。
【請求項3】
前記アルファウイルス5’複製認識配列が、Venezuelan equine encephalitisウイルスの5’複製認識配列である、請求項1に記載のRNA分子。
【請求項4】
前記5’複製認識配列が、70ヌクレオチドと524ヌクレオチドの間の長さである、請求項3に記載のRNA分子。
【請求項5】
前記アルファウイルス3’複製認識配列が、Venezuelan equine encephalitisウイルスの3’複製認識配列である、請求項1または3に記載のRNA分子。
【請求項6】
前記3’複製認識配列が、19ヌクレオチドから325ヌクレオチドまでの長さである、請求項1に記載のRNA分子。
【請求項7】
前記少なくとも一つの開始コドンが、非構造タンパク質1(nsp1)のための開始コドンである、請求項1に記載のRNA分子。
【請求項8】
全ての開始コドンが、前記5’複製認識配列から除去されている、請求項1に記載のRNA分子。
【請求項9】
前記RNAが、5’末端でキャップされている、請求項1に記載のRNA分子。
【請求項10】
アルファウイルスレプリコン粒子を製造する方法であって、該方法は、請求項1に記載のRNA分子の1つ以上を細胞に導入する工程を含み、これによって、RNA分子の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生に必要な全てのアルファウイルス構造タンパク質をコードする、方法。
【請求項11】
2つのRNA分子が前記細胞中に導入され、該2つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、該2つのRNA分子の第二のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なる、請求項10に記載の方法。
【請求項12】
3つのRNA分子が前記細胞中に導入され、該3つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、該3つのRNA分子の第二のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なり、第三のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子および該第二のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なる、請求項10に記載の方法。
【請求項13】
前記1つ以上のRNA分子の少なくとも1つが、5’末端でキャップされている、請求項10に記載の方法。
【請求項14】
アルファウイルスレプリコン粒子を製造する方法であって、該方法は、
a)アルファウイルスレプリコンRNAと、
b)請求項1に記載のRNA分子の1つ以上と、
c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物と
を細胞に導入する工程を含み、
これによって、(b)のRNA分子と(c)のヘルパー構築物との組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコン粒子の産生に必要な全てのアルファウイルス構造タンパク質をコードする、方法。
【請求項15】
前記1つ以上のRNA分子の少なくとも1つが、5’末端でキャップされている、請求項14に記載の方法。
【請求項16】
請求項1に記載のRNA分子を含む粒子のサブセットを含む、アルファウイルスレプリコン粒子の集団であって、該集団が、培養における許容細胞での継代によって決定されるような、アルファウイルスレプリコン粒子108個当たりに検出可能な複製能力のあるアルファウイルスウイルス粒子を含まない、集団。
【請求項17】
請求項1に記載のRNA分子を含む粒子のサブセットを含む、アルファウイルスレプリコン粒子の集団であって、該集団が、培養における許容細胞での継代によって決定されるような、アルファウイルスレプリコン粒子108個当たりに検出可能な複製能力のあるアルファウイルス粒子を含まず、該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む、集団。
【請求項18】
薬学的に許容される担体中に、請求項16または17に記載の集団を含む組成物。
【請求項19】
被験体において免疫応答を誘起する方法であって、該被験体に請求項16または17に記載の集団の有効量を投与する工程を含む、方法。
【請求項20】
請求項1に記載のRNA分子を含む細胞。
【請求項21】
請求項1に記載のRNA分子を含むベクター。
【請求項22】
請求項1に記載のRNA分子を含む核酸構築物。
【請求項23】
請求項21に記載のベクターを含む細胞。
【請求項24】
請求項22に記載の核酸構築物を含む細胞。
【請求項1】
a)少なくとも一つの開始コドンが除去されているアルファウイルス5’複製認識配列と、
b)アルファウイルス構造タンパク質をコードするヌクレオチド配列と、
c)アルファウイルス3’複製認識配列と
を含み、(b)の該ヌクレオチド配列の転写を誘導するプロモーターを含まないことを条件とする、単離されたRNA分子であって、
(a)および(c)の該アルファウイルス5’および3’複製認識配列が、アルファウイルス非構造タンパク質の存在下において該RNA分子全体の複製を誘導する、RNA分子。
【請求項2】
前記アルファウイルス構造タンパク質をコードする前記ヌクレオチド配列が、1)アルファウイルスカプシドタンパク質、2)任意の順序のアルファウイルスE1およびE2タンパク質、3)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE1タンパク質、5)任意の順序のアルファウイルスカプシドタンパク質およびアルファウイルスE2タンパク質、6)アルファウイルスE2タンパク質、並びに7)アルファウイルスE1タンパク質をコードするヌクレオチド配列からなる群より選択される、請求項1に記載のRNA分子。
【請求項3】
前記アルファウイルス5’複製認識配列が、Venezuelan equine encephalitisウイルスの5’複製認識配列である、請求項1に記載のRNA分子。
【請求項4】
前記5’複製認識配列が、70ヌクレオチドと524ヌクレオチドの間の長さである、請求項3に記載のRNA分子。
【請求項5】
前記アルファウイルス3’複製認識配列が、Venezuelan equine encephalitisウイルスの3’複製認識配列である、請求項1または3に記載のRNA分子。
【請求項6】
前記3’複製認識配列が、19ヌクレオチドから325ヌクレオチドまでの長さである、請求項1に記載のRNA分子。
【請求項7】
前記少なくとも一つの開始コドンが、非構造タンパク質1(nsp1)のための開始コドンである、請求項1に記載のRNA分子。
【請求項8】
全ての開始コドンが、前記5’複製認識配列から除去されている、請求項1に記載のRNA分子。
【請求項9】
前記RNAが、5’末端でキャップされている、請求項1に記載のRNA分子。
【請求項10】
アルファウイルスレプリコン粒子を製造する方法であって、該方法は、請求項1に記載のRNA分子の1つ以上を細胞に導入する工程を含み、これによって、RNA分子の組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコンRNAと共に、アルファウイルスレプリコン粒子の産生に必要な全てのアルファウイルス構造タンパク質をコードする、方法。
【請求項11】
2つのRNA分子が前記細胞中に導入され、該2つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、該2つのRNA分子の第二のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なる、請求項10に記載の方法。
【請求項12】
3つのRNA分子が前記細胞中に導入され、該3つのRNA分子の第一のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、該3つのRNA分子の第二のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なり、第三のRNA分子が、1つ以上のアルファウイルス構造タンパク質をコードし、そのうちの少なくとも1つが、該第一のRNA分子および該第二のRNA分子によってコードされる該アルファウイルス構造タンパク質とは異なる、請求項10に記載の方法。
【請求項13】
前記1つ以上のRNA分子の少なくとも1つが、5’末端でキャップされている、請求項10に記載の方法。
【請求項14】
アルファウイルスレプリコン粒子を製造する方法であって、該方法は、
a)アルファウイルスレプリコンRNAと、
b)請求項1に記載のRNA分子の1つ以上と、
c)1つ以上のプロモーター補助アルファウイルスヘルパー構築物と
を細胞に導入する工程を含み、
これによって、(b)のRNA分子と(c)のヘルパー構築物との組み合わせが、アルファウイルスレプリコン粒子が産生される条件下において、アルファウイルスレプリコン粒子の産生に必要な全てのアルファウイルス構造タンパク質をコードする、方法。
【請求項15】
前記1つ以上のRNA分子の少なくとも1つが、5’末端でキャップされている、請求項14に記載の方法。
【請求項16】
請求項1に記載のRNA分子を含む粒子のサブセットを含む、アルファウイルスレプリコン粒子の集団であって、該集団が、培養における許容細胞での継代によって決定されるような、アルファウイルスレプリコン粒子108個当たりに検出可能な複製能力のあるアルファウイルスウイルス粒子を含まない、集団。
【請求項17】
請求項1に記載のRNA分子を含む粒子のサブセットを含む、アルファウイルスレプリコン粒子の集団であって、該集団が、培養における許容細胞での継代によって決定されるような、アルファウイルスレプリコン粒子108個当たりに検出可能な複製能力のあるアルファウイルス粒子を含まず、該アルファウイルスレプリコン粒子が、アルファウイルス構造タンパク質またはアルファウイルス非構造タンパク質のいずれか、あるいはアルファウイルス構造タンパク質およびアルファウイルス非構造タンパク質の両方に、1つ以上の弱毒化突然変異を含む、集団。
【請求項18】
薬学的に許容される担体中に、請求項16または17に記載の集団を含む組成物。
【請求項19】
被験体において免疫応答を誘起する方法であって、該被験体に請求項16または17に記載の集団の有効量を投与する工程を含む、方法。
【請求項20】
請求項1に記載のRNA分子を含む細胞。
【請求項21】
請求項1に記載のRNA分子を含むベクター。
【請求項22】
請求項1に記載のRNA分子を含む核酸構築物。
【請求項23】
請求項21に記載のベクターを含む細胞。
【請求項24】
請求項22に記載の核酸構築物を含む細胞。
【図1】

【図2】
【図3】
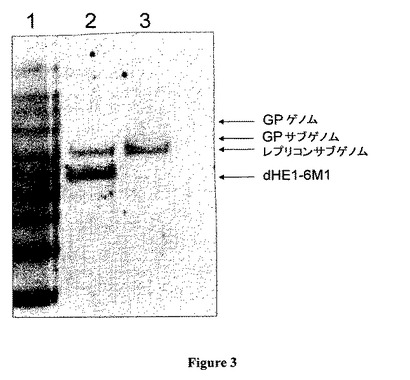
【図4】
【図5】
【図6】

【図7】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2010−530245(P2010−530245A)
【公表日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願番号】特願2010−513254(P2010−513254)
【出願日】平成20年6月20日(2008.6.20)
【国際出願番号】PCT/US2008/007701
【国際公開番号】WO2008/156829
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(509350147)アルファバックス, インコーポレイテッド (1)
【Fターム(参考)】
【公表日】平成22年9月9日(2010.9.9)
【国際特許分類】
【出願日】平成20年6月20日(2008.6.20)
【国際出願番号】PCT/US2008/007701
【国際公開番号】WO2008/156829
【国際公開日】平成20年12月24日(2008.12.24)
【出願人】(509350147)アルファバックス, インコーポレイテッド (1)
【Fターム(参考)】
[ Back to top ]