
α(1,3)−ガラクトシルトランスフェラーゼを発現する同種異系腫瘍細胞を用いる抗腫瘍ワクチン接種
本発明は腫瘍細胞の選択的ターゲティングおよび死滅を引き起こす方法と組成物に関する。ex vivo遺伝子療法プロトコルを介して、α(1,3)ガラクトシルエピトープを発現するように腫瘍細胞を遺伝子操作する。次いで細胞を照射するかさもなくば死滅させて、患者に投与する。αガラクトシルエピトープは腫瘍細胞のオプソニン化を引き起こして、オプソニン化腫瘍細胞の抗原提示細胞による取込みを増進し、その結果、腫瘍特異的抗原提示を増進する。このように動物の免疫系は刺激されて、動物中に存在する腫瘍細胞を攻撃しかつ死滅させうる腫瘍特異的細胞傷害性細胞および抗体を産生する。
【発明の詳細な説明】
【技術分野】
【0001】
関係出願の相互参照
本出願は、2002年10月9日に出願した仮出願60/417,343の、米国特許法第119(e)条(35 U.S.C. §119(e))の規定による利益を主張する。
【0002】
発明の分野
本発明は、腫瘍細胞に対する体液性および細胞性免疫応答を刺激することにより癌を治療する方法および組成物に関する。特に、本発明は、補体が媒介する腫瘍細胞の破壊を刺激しかつ腫瘍特異的抗体産生と腫瘍特異的細胞傷害性細胞を同時刺激する方法に関する。
【背景技術】
【0003】
発明の背景
異種移植に対する大きな障害は、本質的に超急性異種移植片拒絶(HAR)を引き起こす外来組織中に存在する糖鎖エピトープの即時認識にある。この反応は再潅流すると直ぐ始まり、一旦始まると、外来組織を数分ないし数時間内に破壊する。或るドナー(donor)とレシピエント(recipient)の組合わせでHARが存在する一方、他のドナーとレシピエントの組合わせでは存在しないこの現象は、2つの主な因子、すなわち、a)レシピエントの異種反応性天然抗体が移植片中の抗原もしくは内皮細胞と結合すること、およびb)移植片の補体調節タンパク質とレシピエントの補体系が不適合であるために補体の無制御な活性化が容認されることに関係があると仮定されている。ヒト血清中の補体結合性天然抗体の1%超は、単構造Galα(1-3)Galβ(1,4)GlcNAc-Rを認識する。Galα(1-3)Galβ(1,4)GlcNAc-Rの合成は、酵素α(1,3)ガラクトシルトランスフェラーゼ(αGT)が触媒する。
【0004】
この酵素は様々な非霊長類哺乳動物由来の細胞のゴルジ体において次の反応:
Galβ(1,4)GlcNAc-R+UDP-Gal→Galα(1-3)Galβ(1,4)GlcNAc-R
によるα-ガラクトシル(αGal)エピトープの合成を触媒する。
【0005】
この酵素は新世界サルで活性を有するが旧世界サルおよびヒトでは活性がないことが見出されている。αGTのcDNAは、ウシおよびマウスcDNAライブラリーからクローニングされている。Larson, R. D.ら, (1989) 「マウスUDPガラクトース;.β-D-ガラクトシル-(1,4)-Nアセチル-D-グルコサミンα-(1,3)ガラクトシルトランスフェラーゼをコードするcDNAの単離:遺伝子導入による発現クローニング(Isolation of a cDNA Encoding Murine UDP galactose; .β-D-galactosyl-(1,4)-N Acetyl-D-Glucosamine α-(1,3) Galactosyl Transferase: Expression Cloning by Gene Transfer)」, PNAS, USA 86:8227;およびJoziasse, D. H.ら, (1989) 「ウシα-(1,3)ガラクトシルトランスフェラーゼ:cDNAクローンの単離と特徴付け、ヒトゲノムDNA中の相同的配列の同定(Bovine α-(1,3) Galactosyl Transferase: Isolation and Characterization of a cDNA Clone, Identification of Homologous Sequences in Human Genomic DNA)」, J. Biol Chem 264:14290。
【0006】
この遺伝子はヒトゲノム中に存在するが、転写は検出されていない。代わりに、2つのフレームシフト突然変異(未熟な停止コドンを作製する欠失)がこの酵素をコードするヒトエキソン中に見出された。一般的には、Galili, Uri 「ヒト天然抗α-ガラクトシルIgG(抗αGal)抗体の病態生理学の進展(Evolution in Pathophysiology of the Human Natural anti- α-Galactosyl IgG (anti- αGal) Antibody)」, Springer Semin. Immunopathol. (1993) 15:155-171を参照。
【0007】
全てのヒトに存在する天然抗体である抗αGalは、糖鎖エピトープGalα(1-3)Galβ(1,4)GlcNAc-R(αGalエピトープ)と特異的に相互作用する。この抗体は、哺乳類動物細胞が産生する他の既知の糖鎖エピトープとは相互作用しない(Galili, 1993, Springer Seminar Immunopathology 15:153)。抗αGalは循環するIgGのほぼ1%を構成して(Galiliら, 1984, J. Exp. Med. 160:1519)、IgAおよびIgMの形態でも見出される(Davineら, 1987, Kidney Int. 31:1132;Sandrinら, 1993, Proc. Natl. Acad. Sci. USA 90:11391)。これは1%の循環Bリンパ球により産生される(Galiliら, 1993, Blood 82:2485)。ヒトにおけるこの天然抗αGal Abの産生は、腸および肺細菌叢中に存在するαGal糖鎖残基の存在により常に刺激されている。ヒトでは、超急性異種移植拒絶において抗αGalがこのエピトープの存在と反応し、補体は速やかにかつ確実に数分〜数時間で外来組織の破壊をもたらす。
【0008】
本発明の目的は治療用癌ワクチンを開発することであり、それは、αGTをコードする遺伝子を腫瘍細胞中に導入してこのワクチン腫瘍細胞にαGalエピトープを付加し、天然抗αGal抗体によるワクチン細胞のオプソニン作用の増強を可能にし、かつ、腫瘍抗原提示を刺激して腫瘍特異的抗原に対する体液性および細胞性免疫応答を誘導することによる。
【0009】
本発明のさらなる目的は、αGTを発現しかつプロセシングして細胞上にαGalエピトープを作製する組換え細胞を含む上記医薬組成物を提供することである。
【0010】
本発明のさらなる目的は、増殖しかつ細胞性および体液性免疫応答を逃れる腫瘍、ウイルス、新生物細胞、または他の細胞を治療するための組成物および方法を提供することである。
【0011】
本発明の他の目的は以下の本発明の説明から明らかになるであろう。
【発明の開示】
【0012】
発明の概要
本発明は、腫瘍細胞を選択的にターゲティングして死滅させる免疫応答を引き起こす方法および組成物に関する。腫瘍細胞を、ex vivo遺伝子療法プロトコルを介して、αGalエピトープを発現するように遺伝子操作する。次いで細胞を死滅させ(γ線または紫外線照射、熱、ホルムアルデヒドなどにより)そして患者に投与する。該αGalエピトープは腫瘍細胞のオプソニン作用を誘発し、腫瘍細胞全体中に存在する抗原の腫瘍特異的抗原提示を亢進する。本発明の重要な特徴は、本発明の医薬組成物に細胞全体の利用を含むことである。これによって腫瘍細胞全体内に存在する腫瘍関連抗原のプロセシングが行われ、これらのタンパク質がαGalエピトープの付加による影響を受けているか否かには関わらない。αGal改変は、細胞表面上の複数の糖タンパク質および糖脂質に影響を与えるので、動物の免疫系は、腫瘍特異的抗原を検出し、プロセシングしそして上記抗原に対する抗体および細胞性免疫応答を産生する機会を増加しうる。このようにして動物の免疫系は、刺激されて腫瘍特異的抗体および免疫細胞を産生し、これらは、動物中に存在して遺伝子操作された細胞全体ワクチンにより与えられたものと共通の腫瘍関連抗原を保持するαGalネガティブ腫瘍細胞を攻撃しかつ死滅させうる。
【0013】
本発明によれば、医薬組成物は、発現するとマウスαGTをコードするポリヌクレオチド配列をex vivoで腫瘍細胞全体に導入することにより作製する。レシピエント腫瘍細胞は同系(syngenic)、同質異系(allogenic)、または自己(autologous)であってもよい。該配列はいずれかのヌクレオチド導入ビヒクルを介して導入し、上記ビヒクルはウイルスもしくは非ウイルスベクター、プラスミド、または活性ウイルス粒子を産生するベクター産生細胞であってもよい。これらの遺伝子導入ビヒクルは腫瘍細胞を形質転換し、挿入された外来遺伝子材料を上記細胞内に発現する。発現された遺伝子産物は、上記細胞上に存在する細胞表面糖タンパク質および糖脂質上のαGalエピトープ合成を触媒する。本発明は、予め存在する抗αGal抗体によるαGalエピトープの結合を最大化するための複数の細胞表面糖タンパク質をもつ腫瘍細胞全体の利用、こうしてこの複合体の抗原提示細胞上に存在するFc受容体との結合の亢進、およびこうして上記ワクチン腫瘍細胞に存在する複数の腫瘍に関連する抗原の抗原提示のトリガリング(triggering)を意図する。
【0014】
さらに好ましい実施形態においては、同じ組織タイプ、または癌タイプ由来の複数タイプの形質転換した細胞を投与して、異なるエピトープの数をさらに増加させて個体に存在する腫瘍細胞の完全な改善の確率を増加する。
【0015】
本発明は、医薬組成物および上記医薬組成物を作る方法を含んでなり、ここで上記組成物は弱毒腫瘍細胞混合物の混合物の治療上有効な量と担体を含んでなり、上記混合物は複数の細胞表面糖タンパク質を含んでなり、そして上記糖タンパク質はαGalエピトープを含んでなる。好ましい実施形態においては、細胞は細胞全体である。上記組成物を作る方法は、生腫瘍細胞のコレクションを取得し、発現するとαGalをコードするヌクレオチド配列を用いて上記細胞を形質転換してαGalが上記細胞の細胞表面糖タンパク質上に提示されるようにすることを含んでなる。次いで細胞を死滅させて、投与のための製薬担体と組合わせる。
【0016】
定義
本発明の組成物と方法に関係する様々な用語が、本明細書でこれまでに用いられたしまた明細書および請求の範囲の全体にわたっても用いられる。
【0017】
単位、接頭辞、および記号はそのSIで許容された型で記述することができる。特に断らない限り、核酸は左から右へ5'から3'方向に記し;アミノ酸配列は左から右へアミノからカルボキシ方向にそれぞれ記す。数字の範囲は範囲を規定する数字を含み、かつそれぞれの整数を規定された範囲内に含むものである。アミノ酸は、本明細書において、IUPAC-IUB生化学命名委員会(IUPAC-IUB Biochemical nomenclature Commission)が推奨する、一般的に知られる3文字記号または1文字記号により参照することができる。ヌクレオチドも同様に一般的に受入れられる1文字コードにより参照することができる。特に断りのない限り、本明細書に用いるソフトウエア、電気工学、および電子工学用語は新しい電気工学および電子工学用語の新IEEE標準辞書(New IEEE Standard Dictionary of Electrical and Electronics Terms)第5版、1993に定義されたものである。以下に定義される用語は、本明細書全体への参照によりさらに詳しく定義される。
【0018】
用語「α-(1,3)ガラクトシルトランスフェラーゼをコードする配列」、または「αGTをコードする配列」は、次の反応:
Galβ(1,4)GlcNAc-R+UDP-Gal→Galα(1-3)Galβ(1,4)GlcNAc-R
によりα-ガラクトシル(αGal)エピトープを生成するタンパク質をコードする、ポリヌクレオチド配列を意味する。
【0019】
この配列は、変異体、改変体、末端切断体など、ならびにマウス配列、ウシ配列または当業者に公知のおよびGenbank、他の開示物またはデータベースで利用しうるいずれかの他の供給源由来であって、上記反応の機能を保持する配列を含むことができる。典型的には、かかる配列は、本明細書に記載のマウスまたはウシαGT配列と少なくとも80%以上相同的でありうる。
【0020】
「増幅した」は、少なくとも1つの核酸配列をテンプレートとして用いる、核酸配列の多コピーまたは核酸配列と相補的な多コピーの構築を意味する。増幅系としては、ポリメラーゼ連鎖反応(PCR)系、リガーゼ連鎖反応(LCR)系、核酸配列に基づく増幅(NASBA, Canteen, Mississauga, Ontario)、Q-Betaレプリカーゼ系、転写に基づく増幅系(TAS)、およびストランドディスプレースメント増幅(strand displacement amplification)(SDA)が挙げられる。例えば、「診断分子ミクロ生物学:原理と応用(Diagnostic Molecular Microbiology: Principles and Applications)」, D.H. Persingら, 編, American Society for Microbiology, Washington, D.C. (1993)を参照。増幅産物をアンプリコンと呼ぶ。
【0021】
本明細書に用いる用語「動物」は抗αGal抗体を合成する全ての動物と解釈することとし、抗αGal抗体を合成するかどうかが未知の動物も含む。例えば、鳥類の複数種の動物はαGalエピトープを合成するかどうかが未知である。抗αGal抗体またはαGalエピトープのいずれかを合成する動物の間にはユニークな相反関係があるので、αGalエピトープが不在である未試験の多くの動物は抗αGal抗体を合成する動物であることを立証しうると考えられる。本発明はこれらの動物を包含する。
【0022】
用語「抗体」は抗体の抗原結合型(例えば、Fab、F(ab)2)を意味する。用語「抗体」は、しばしば、免疫グロブリン遺伝子により実質的にコードされるポリペプチド、または分析質(抗原)と特異的に結合しかつ認識するそのフラグメントを意味する。しかし、様々な抗体フラグメントは無傷の抗体の消化方法で定義することができる一方、当業者はかかるフラグメントを化学的にまたは組換えDNA技法を用いることによりde novo合成できることを理解している。従って、本明細書に用いる用語の抗体はまた、抗体フラグメント、例えば1本鎖Fv、キメラ抗体(すなわち、色々な種由来の定常および可変領域を含む)、ヒト化抗体(すなわち、非ヒト由来の相補性決定領域(CDR)を含む)および異種複合体抗体(例えば、二特異的抗体)を含む。
【0023】
用語「抗αGal」は、αGalエピトープを認識する免疫グロブリン、例えばIgG、IgA、IgEまたはIgM抗αGal抗体のいずれのタイプまたはサブタイプも含む。
【0024】
本明細書に用いる用語「抗原」は、それ自身またはその部分に対する免疫応答を誘発することができるいずれかの生物学的分子(タンパク質、ペプチド、脂質、グリカン、糖タンパク質、糖脂質その他)を意味し、限定されるものでないが、腫瘍関連抗原およびウイルス、細菌、寄生虫および真菌の抗原を含む。
【0025】
本明細書に用いる用語「抗原提示」は、マクロファージ、樹状細胞、B細胞および他のタイプの抗原提示細胞が内部または外部抗原をこれらの分子のサブフラグメントにプロセシングして、それらを細胞表面上のクラスIもしくはクラスII主要組織適合複合体またはCD1分子と複合して提示する生物学的機構を意味する。このプロセスは、これらの複合体を特異的に認識してこれらの抗原もしくはこれらの抗原を提示する細胞に対する免疫応答を媒介することができる免疫系の他のタイプの細胞(CD4+、CD8+、BおよびNK細胞などの)の増殖を刺激する。
【0026】
用語「保存的に改変された変異体」は、ある特定の配列を参照する場合、アミノ酸と核酸配列の両方に適用しかつ両方を含むことを意図する。特定の核酸配列について保存的に改変された変異体は、同一または保存的に改変されたアミノ酸配列の変異体をコードする核酸配列を意味する。遺伝コードの縮重のために、多数の機能的に同じ核酸が所与のタンパク質をコードする。例えば、コドンGCA、GCC、GCGおよびGCUはすべて、アミノ酸アラニンをコードする。すなわち、アラニンがコドンにより規定されるすべての位置で、そのコドンを、コードされたポリペプチドを改変することなく、記載の対応するコドンのいずれかに改変することができる。そのような核酸変化は「サイレントな変化」であり、これは保存的に改変された変化の一種を表す。従って、あるポリペプチドをコードする本明細書に記載の全ての核酸配列はまた、遺伝子コードを参照して、その核酸のすべての可能なサイレントな変化を記載する。当業者は(通常、メチオニンの唯一のコドンであるAUG;および通常、トリプトファンの唯一のコドンであるUUGを除いて)核酸中のそれぞれのコドンを改変して機能的に同じ分子を得ることができるのを理解するであろう。従って、本発明のポリペプチドをコードする核酸のそれぞれのサイレントな変化は、暗黙のままそれぞれの記載されたポリペプチド配列に含まれ、本発明の範囲に含まれる。
【0027】
アミノ酸配列に関して、コードされた配列中の単一アミノ酸もしくは小パーセントのアミノ酸を改変、付加または欠失する、核酸、ペプチド、ポリペプチド、またはタンパク質配列に対する個々の置換、欠失または付加は、改変が化学的に同様なアミノ酸によるアミノ酸の置換をもたらす場合、「保存的に改変された変異体」であることを当業者は理解するであろう。以下の6グループは、それぞれ、互いにとって保存的置換であるアミノ酸を含有する:
1)アラニン(A)、セリン(S)、トレオニン(T);
2)アスパラギン酸(D)、グルタミン酸(E);
3)アスパラギン(N)、グルタミン(Q);
4)アルギニン(R)、リジン(K);
5)イソロイシン(I)、ロイシン(L)、メチオニン(M)、バリン(V);および
6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W)。
【0028】
Creighton (1984) 「タンパク質(Proteins)」 W.H. Freeman and Companyも参照すること。
【0029】
本発明者らは、2つのアミノ酸配列の「配列同一性パーセント」を、対合アラインメント後の2つのアミノ酸配列が共有する同一アミノ酸の数をその対の最短配列の全長により除して定義する。
【0030】
本発明者らは、2つのアミノ酸配列の「配列類似性パーセント」を、対合アラインメント後の2つのアミノ酸配列が共有する同一アミノ酸+保存的アミノ酸置換の数をその対の最短配列の全長により除して定義する。
【0031】
規定した核酸について「コードする」または「コードした」は、規定したタンパク質に翻訳するための情報を含むことを意味する。タンパク質をコードする核酸は、非翻訳配列(例えば、イントロン)を核酸の翻訳領域内に含んでもよく、またはかかる介在する非翻訳配列を欠いてもよい(例えば、cDNAにおけるように)。タンパク質をコードする情報はコドンを利用して規定される。典型的にはアミノ酸配列は、核酸により「ユニバーサル」遺伝子コードを用いてコードされる。
【0032】
核酸を合成により調製するかまたは改変するとき、核酸が発現される意図する宿主の既知のコドン優先性をとると有利である。
【0033】
タンパク質またはペプチドについて、用語「単離されたタンパク質(またはペプチド)」が本明細書で時々用いられる。この用語は、天然で随伴する他のタンパク質から十分分離されていて、「実質的に純粋な」形態で存在するタンパク質を意味する。あるいは、この用語は単離された核酸分子の発現により産生されるタンパク質を意味してもよい。
【0034】
核酸分子を参照して、用語「単離された核酸」が時々用いられる。DNAに応用される場合、この用語は、それが誘導された生物の天然のゲノムにおいて(5'および3'方向に)直接連続している配列から分離されたDNA分子を意味する。例えば、「単離された核酸」は、プラスミドまたはウイルスベクターなどのベクター中に挿入されたまたは原核生物または真核生物のゲノムDNA中に組込まれたDNA分子を含む。「単離された核酸分子」はまたcDNA分子も含む。RNA分子についての用語「単離された核酸」は主に上に定義した単離されたDNA分子がコードするRNA分子を意味する。あるいは、この用語は、その天然状態で(すなわち、細胞または組織中で)それと随伴していたであろうRNA分子から十分分離されていて、「実質的に純粋な」形態で存在するRNA分子を意味することができる(用語「実質的に純粋な」は以下に定義する)。
【0035】
本明細書において核酸を参照して用いる「異種(heterologous)」は、外来種に起源するか、または、同じ種由来であれば、意図的な人為的介入によってその天然の形態から組成および/またはゲノム遺伝子座が実質的に改変されている核酸である。例えば、異種構造遺伝子と機能しうる形で連結されたプロモーターは、その構造遺伝子が誘導された種と異なる種由来であるか、または、もし同じ種由来であれば、片方または両方がそれらの元来の形態から実質的に改変されている。異種タンパク質は外来種からの起源であっても、または、もし同じ種であれば、その元来の形態から意図的な人為的介入により実質的に改変されていてもよい。
【0036】
「宿主細胞」は、ベクターを含有しかつベクターの複製および/または発現を支援する細胞を意味する。宿主細胞は原核生物細胞、例えば大腸菌(E.coli)、または真核生物細胞、例えば酵母、昆虫、両生類、または哺乳類細胞であってもよい。
【0037】
用語「導入された」は、核酸を細胞中に挿入する文脈において「トランスフェクション」または「形質転換」または「形質導入」を意味し、核酸の真核生物または原核生物細胞中への組込みを意味し、この場合、核酸は細胞のゲノム中に組込まれて自律性レプリコンに変換されても(例えば、染色体、プラスミド、プラスチドまたはミトコンドリアDNA)または一過的に発現されても(例えば、トランスフェクトされたmRNA)よいことを意味する。
【0038】
本明細書で用いられる「マーカー」は、染色体上のユニークな位置を同定するのに役立つ染色体上の遺伝子座の意味を含む。「多形マーカー」は、複数の形態(対立遺伝子)で現れるマーカーの意味を含み、それらが相同的な対(homologous pair)で存在するとき、マーカーの異なる形態はその対の染色体のそれぞれの伝達を追跡することを可能にする。遺伝形質を1以上のマーカーを用いて定義してもよい。
【0039】
本明細書に用いられる「核酸」は、1本鎖または2本鎖形態のデオキシリボヌクレオチドまたはリボヌクレオチドの意味を含み、特に断らない限り、それらが天然ヌクレオチドと同様な方法で1本鎖核酸とハイブリダイズする点において天然ヌクレオチドの本質的性質を有する既知の類似体(例えば、ペプチド核酸)を包含する。
【0040】
抗原または腫瘍細胞の用語「オプソニン作用」は、抗原にまたは腫瘍細胞表面上に存在するαGalエピトープの抗αGal抗体との結合と、それによる、抗体のFc部分の抗原提示細胞の表面上に存在するFc受容体との結合を介する、マクロファージ、樹状細胞、B細胞または他のタイプの抗原提示細胞によるオプソニン化された抗原または腫瘍細胞の食作用の増強を意味する。
【0041】
本明細書に用いられる「ポリヌクレオチド」は、ストリンジェントなハイブリダイゼーション条件下で、天然のヌクレオチドと実質的に同じヌクレオチド配列とハイブリダイズする、および/または、天然のヌクレオチドと同じアミノ酸に翻訳されるという点において、天然リボヌクレオチドの本質的性質を有するデオキシリボポリヌクレオチド、リボポリヌクレオチド、またはそれらの類似体の意味を含む。ポリヌクレオチドは、未変性または異種の構造または調節遺伝子の全長またはサブ配列であってもよい。特に断らない限り、この用語は規定した配列、ならびにその相補的配列の意味を含む。従って、安定性のためまたは他の理由のために主鎖が改変されたDNAまたはRNAは、本明細書で意図される「ポリヌクレオチド」である。
【0042】
用語「ポリペプチド」、「ペプチド」および「タンパク質」は、本明細書において互換的にアミノ酸残基のポリマーを意味する。本用語は、1以上のアミノ酸残基が対応する天然アミノ酸、ならびに天然アミノ酸ポリマーの人為的な化学類似体であるアミノ酸ポリマーに適用される。かかる天然アミノ酸の類似体の本質的な性質とは、タンパク質中に組み込まれると、そのタンパク質が、同じタンパク質であるが全て天然アミノ酸から構成されるタンパク質に対して誘発される抗体に対して特異的に反応性を有することである。用語「ポリペプチド」、「ペプチド」および「タンパク質」はまた、限定されるものでないが、リン酸化、グリコシル化、脂質結合、硫酸化、グルタミン酸残基のγカルボキシル化、ヒドロキシル化およびADPリボシル化を含む改変体も包含する。
【0043】
本明細書に用いられる「組換え体」は、異種核酸の導入により改変されているかまたは細胞がそのように改変された細胞から誘導された、細胞またはベクターの意味を含む。従って、例えば、組換え細胞は、細胞の未変性(非組換え)形態内の同一形態に見出されない遺伝子を発現するかまたはそうでなければ、意図的な人為的介入の結果として異常に発現された、発現不足であるか、または全く発現されない未変性遺伝子を発現する。本明細書に用いられる用語「組換え」は、意図的な人為的介入なしに生じる改変のような、天然事象(例えば、自発性突然変異、天然の形質転換/形質導入/遺伝子転移)による細胞またはベクターの改変を包含しない。
【0044】
本明細書に用いられる「組換え発現カセット」は、宿主細胞において特定の核酸の転写を可能にする一連の規定した核酸エレメントを用いて、組換えによりまたは合成により作製される核酸構築物である。組換え発現カセットを、プラスミド、染色体、ミトコンドリアDNA、プラスチドDNA、ウイルス、または核酸断片中に組み込んでもよい。典型的には、発現ベクターの組換え発現カセット部分は、他の配列の中に、転写される核酸およびプロモーターを含む。
【0045】
用語「残基」または「アミノ酸残基」または「アミノ酸」は、本明細書中では互換的に用いられ、タンパク質、ポリペプチド、またはペプチド(まとめて「タンパク質」)中に組み込まれたアミノ酸を意味する。アミノ酸は天然のアミノ酸であってもよく、特に限定しない限り、天然アミノ酸と同様な方法で機能しうる天然アミノ酸の非天然類似体を包含してもよい。
【0046】
用語「実質的に同じ」は、タンパク質の性質(すなわち、タンパク質の構造、安定性特性、基質特異性および/または生物学的活性)に実質的に影響を与えることのない配列変化を有する核酸またはアミノ酸配列を意味する。特に核酸配列を参照する用語「実質的に同じ」は、コード領域および発現を支配する保存的配列を意味することを意図し、そして主に同じアミノ酸をコードする縮重コドン、またはコードされたポリペプチドに保存的置換アミノ酸をコードする代わりのコドンを意味する。アミノ酸配列を参照する用語「実質的に同じ」は、一般的に構造または機能の決定に関わらないポリペプチドの領域における保存的置換および/または変化を意味する。
【0047】
抗体に関連して、用語「免疫学的に特異的な」は、抗原性生物学的分子の混合集団を含有するサンプル中の目的のタンパク質の1以上のエピトープと結合するが、他の分子を実質的に認識せずかつ結合しない抗体を意味する。
【0048】
「コード配列」または「コード領域」は、配列が発現されるときに遺伝子産物を産生するために必要な配列情報を有する核酸分子を意味する。
【0049】
用語「機能しうる形で連結された」または「機能しうる形で挿入された」は、コード配列の発現に必要な調節配列を、コード配列の発現ができるようにするために、核酸分子中のコード領域と相対的に適当な位置に配置することを意味する。この同じ定義は、時々、発現ベクター中の他の転写制御エレメント(例えば、エンハンサー)の配列に適用される。
【0050】
転写および翻訳制御配列は、宿主細胞中でコード配列を発現させるDNA調節配列、例えばプロモーター、エンハンサー、ポリアデニル化シグナル、ターミネーターなどである。
【0051】
用語「プロモーター」、「プロモーター領域」または「プロモーター配列」は、一般的に、コード領域の5'または3'サイドに、またはコード領域内に、またはイントロン内に見出しうる遺伝子の転写調節領域を意味する。典型的には、プロモーターは、細胞内でRNAポリメラーゼと結合して下流(3'方向)コード配列の転写を開始することができるDNA調節領域である。典型的な5'プロモーター配列は、その3'端末で転写開始部位と境界を接し、上流(5'方向)に伸びてバックグラウンドを超える検出可能なレベルの転写を開始するために必要な最小限の数の塩基またはエレメントを含む。プロモーター配列内には、転写開始部位(便宜的にヌクレアーゼS1によるマッピングにより規定した)、ならびにRNAポリメラーゼの結合に関わるタンパク質結合ドメイン(コンセンサス配列)がある。
【0052】
用語「治療上有効な量」は、限定されるものでないが、本明細書に記載の技術により測定可能である、予め存在する腫瘍細胞の数、量または複製の測定可能な減少をもたらすために十分な治療組成物の量を意味する。
【0053】
用語「腫瘍細胞」は、動物における腫瘍の構成要素である細胞、または動物における腫瘍の構成要素になることが定まっている細胞、すなわち、前癌病変の構成要素である細胞を意味する。この定義には、固体腫瘍を形成しない造血系の悪性腫瘍細胞、例えば、白血病、リンパ腫および骨髄腫が含まれる。
【0054】
用語「腫瘍」は、進行性で正常細胞と置換わるかまたは破壊する浸潤性集団を形成しうる1以上の腫瘍細胞として定義される。
【0055】
用語「悪性腫瘍」は、その元来の発生部位を越えて播種する特性を発揮しうる腫瘍細胞により形成される腫瘍として定義される。
【0056】
「ベクター」は、それに対して他の核酸セグメントを機能しうる形で挿入して上記セグメントを複製または発現させる、レプリコン、例えばプラスミド、ファージ、コスミド、またはウイルスである。
【0057】
用語「核酸構築物」または「DNA構築物」は、時々、細胞を形質転換するために適当な調節配列と機能しうる形で連結されてベクター中に挿入されるコード配列を意味するために用いられる。この用語は、用語「形質転換用DNA」と互換的に利用しうる。かかる核酸構築物は、選択マーカー遺伝子および/またはレポーター遺伝子とともに目的の遺伝子産物に対するコード配列を含有しうる。
【0058】
用語「選択マーカー遺伝子」は、発現されると、形質転換した細胞に選択表現型、例えば抗生物質耐性を与える産物をコードする遺伝子を意味する。
【0059】
用語「レポーター遺伝子」は、標準的方法により直接または間接に検出可能である産物をコードする遺伝子を意味する。
【0060】
細胞が外因性または異種DNAにより「形質転換」または「トランスフェクト」されているというのは、上記DNAが細胞内部に導入されていることを意味する。形質転換用DNAは細胞のゲノム中に(共有結合により)組込まれていてもいなくてもよい。原核生物、酵母、および哺乳類動物細胞において、例えば、形質転換用DNAはプラスミドなどのエピソーム上に維持されてもよい。真核生物細胞については、安定して形質転換された細胞は、形質転換用DNAが染色体中に組込まれ、染色体複製を介して娘細胞に継承される細胞である。この安定性は、形質転換用DNAを含有する娘細胞の集団から構成される培養細胞株またはクローンを確立する真核生物細胞の能力により実証される。
【0061】
「クローン」は単一細胞または共通の祖先から有糸分裂により導入された細胞集団である。「培養細胞株」は、in vitroで多世代にわたって安定した増殖能力を有する一次細胞のクローンである。
【0062】
腫瘍細胞に関する用語「治療する」は、上記細胞の進行を停止するか、増殖を減速するか、上記細胞の存在に伴う症状の退行または改善を誘導することを意味する。
【0063】
用語「異種(xenogeneic)細胞」は、移植またはワクチン接種操作において、レシピエント動物宿主になる動物種と異なる動物種に由来する細胞を意味する。
【0064】
用語「同種異系(allogeneic)細胞」は、同じ動物種であるが、「レシピエント宿主」になる動物と1以上の遺伝子座において遺伝的に異なる細胞を意味する。この用語は通常、ある動物から同種の他の同一でない動物へ移植される細胞に適用される。
【0065】
用語「同系(syngeneic)細胞」は、移植またはワクチン接種操作において、同じ動物種である細胞であり、かつほとんどの遺伝型および表現型マーカーについてその培養細胞株のレシピエント宿主になる動物と同じ遺伝子組成を有する細胞を意味する。この用語は通常、同一の双生児から移植されるかまたは高度に近交系の動物間で移植される細胞に適用される。
【発明を実施するための最良の形態】
【0066】
発明の詳細な説明
腫瘍に対する免疫療法の基本原理は、腫瘍関連抗原(TAA)に対する有効な免疫応答の誘導であり、これが、次に、これらの抗原を発現する増殖腫瘍細胞の免疫が媒介する破壊をもたらすことにある。TAAを含むタンパク質に対して有効である免疫応答のために、これらの抗原は最初に抗原提示細胞(APC)、例えばマクロファージ、樹状細胞およびB細胞による飲食作用を受けなければならない。TAAは、APC内のリソソーム区画中で分解され、生じたペプチドがマクロファージ細胞膜の表面上に、ほとんどMHCクラスII分子と一緒に、しかしまたMHCクラスI分子とも一緒に発現される。この発現は、特異的CD4+ヘルパーT細胞による認識およびこれらの細胞の続いての活性化を媒介して免疫応答を果たさせる(Stevenson, 1991, FASEB J. 5:2250;Lanzavecchia, 1993, Science 260:937;Pardoll, 1993, Immunol. Today 14:310)。ヒトTAA分子の大多数は分子的用語で定義されていないので、これらを薬物療法の標的としてまたは抗腫瘍ワクチンとして利用することができない。
【0067】
予防ワクチン接種型のプロトコルにおいて腫瘍特異的応答を誘導する上でのαGalエピトープの利用は、当技術分野の他研究者から提案されており、同じ腫瘍細胞による後期のチャレンジに対して保護を生じることが示されている。しかし予防免疫応答を生じた特異的TAAが、続いて発生した腫瘍細胞に対して有効である保証はない。予防ワクチン接種を介して予防免疫応答を生じたTAAの特定のエピトープは、その腫瘍にだけ、そのタイプの癌だけに、その患者だけに、またはその組織だけに等々、特異的でありうる。全ての癌に発現される普遍的なTAAの同定は依然として捉えがたく、癌予防用の予防ワクチン接種計画の広汎な利用を妨げている。
【0068】
一般的予防手法の代わりに、再発性または診断された腫瘍をもつ個体の免疫治療はより直接的に標的を定めた方法を提供しうる、しかしワクチン技法が既に存在する腫瘍細胞に対する治療として効力がありうるという一致した予測はない。複数の報告は、予防モデルに有効であるが、その同じ処置が治療手法としては効果が低いかまたは無いことを報じている。例えば、マウスTRP-1をコードする組換えワクシニアウイルスを用いる予防ワクチン接種は、その後の生腫瘍B16細胞による皮下チャレンジに対する保護に成功した。この治療は肺黒色腫転移の予防に部分的にだけ有効であり、予め確立した皮下黒色腫の治療には無効であった(Overwijkら, Proc. Natl, Acad. Sci. USA (1999) 96: 2982-2987)
類似の研究において、非メチル化シトシン-グアニンモチーフ(CpG-ODN)およびCTLA-4遮断剤を含有する合成オリゴデオキシヌクレオチド(ODN)アジュバントを用いて、ペプチドワクチンの有効性を予防、治療および両処置の組合わせにおいて評価した。この研究の結論は、B16黒色腫に対する予防または予防方式の治療は無効というものであった。その著者は、マウスにワクチン接種し、B16をチャレンジし、続いてさらなるワクチンの治療用量を与えると、マウスの有効な治療と延命が観察されることを実証した。しかし、この研究において、ワクチン接種は、予め形成された疾患の治療に対しては、部分的にしか有効でなかった、というのは、全てのマウスは、予防および治療腫瘍の両方において、黒色腫で死亡したからである(Davila, KennedyおよびCelis, Cancer Research (2003), 63:3281-3288)。
【0069】
多数の他の研究は、皮下黒色腫の予防治療にだけ焦点を合わせているが、これらのごく少数しか腫瘍増殖速度について有意でかつ有効な治療効果を実証していない、しかし、腫瘍増殖速度の低減は動物の生存率に反映されなかった(Wangら, Cancer Research (2003) 63:2553-2560;Current Protocols of Immunology, Chapter 20, 「ヒト黒色腫のモデルとしてのB16(B16 as a Model for Human Melanoma”)」)。
【0070】
問題をさらに複雑にするのは、腫瘍特異的リンパ球の存在は腫瘍破壊を誘導するのに不十分であることが示されていることである。実際、最近、大量の腫瘍特異的T細胞は腫瘍をもつマウスを延命するのに十分でないことが実証されている(Overwijkら, Journal of Experimental Medicine, (2003) 198: 569-580)。著者は、天然自己ペプチドよりむしろ改変したペプチドリガンドによる抗原特異的ワクチン接種を介するT細胞刺激、ならびにT細胞増殖および活性化因子の同時投与が、腫瘍退行を誘導するための厳密に必要な3要素であることを実証した。
【0071】
ほとんどの腫瘍細胞はユニークなTAA(腫瘍関連抗原)の発現プロファイルを有するが、多くの場合、これらのTAAは未知であるかまたは個々の腫瘍に対して確認するまたは単離することが非常に困難である。さらに、免疫監視を逃げるほとんどの腫瘍については、これらの抗原が細胞「危険」シグナルの文脈で提示されないので、または免疫系がこれらの抗原を認容して「自己」抗原と認識するので、免疫系はこれらのTAAを外来抗原として認識しない。細胞全体ワクチンの使用はこれらの抗原を単離または特徴付けることなくTAAの全レパートリーを提供するので、この第一の困難を緩和する。同種異系(allelic)細胞全体ワクチンの使用は個体間の同種異系差を提示し得るTAAを提供するので、それ故に、これらのTAAに対する免疫系の認容を壊すことができよう。細胞全体ワクチンはまた、GM-CSFなどの免疫応答を増強する分子を発現するように遺伝的に改変されている(Dranoffら, Proc. Natl. Acad. Sci. USA (1993) 90:3539)。遺伝的に改変したまたは改変してない細胞全体同種異系癌ワクチンは抗腫瘍活性および生存についての利点を臨床試験で示しており、それにより、研究室で産生したヒト癌培養細胞株の免疫拒絶が患者の悪性腫瘍の破壊を誘導できるという仮説を実証している。これらのワクチンは、細胞傷害性Tリンパ球およびナチュラルキラー細胞を直接活性化する腫瘍ワクチンクラスI MHCの助けで、TAAの直接提示による細胞免疫エフェクターの直接刺激により機能する(van Elsasら, J. Exp. Medicine (1999) 190:355-366)。従来述べられた手法を用いる抗腫瘍応答の刺激は、しばしば、腫瘍組織と同じ細胞型の正常組織に対する自己免疫疾患の発症と関連している。さらに、これらのワクチンは、TAAを認識しかつ抗原提示および腫瘍細胞の補体が介在する破壊を増強する免疫系の体液性免疫機構を利用しない。強い体液性免疫応答がより有効な抗腫瘍応答を産生する理由はいくつかある:すなわち、I)細胞表面上の特異的抗原と結合することによりワクチン細胞をオプソニン化する抗体は、抗体Fc部の抗原提示細胞上のFc受容体との結合により、マクロファージによるそれらの食作用を促進しうる。II)Fc受容体を標的とすることは、有効なワクチン性能にとって重要ないくつかの機能、例えば、MHCクラスIおよびII両方の抗原提示のための抗原の効率的摂取の促進;APC活性化の促進および樹状細胞の成熟の促進を達成する。III)Fc受容体が介在する食作用経由の抗原提示細胞によるオプソニン化細胞の摂取は、有効な抗腫瘍CTL応答を産生するのに重要な意味を持ちうる、何故なら、これがCD8+T細胞によるMHCクラスIに制限された応答の活性化を交差提示経路を介して促進するからである。IV)体液性免疫応答を刺激することが出来ないワクチンは、HLA制限により細胞性免疫を誘導する能力が限られている。CTLはHLA制限され、自己クラスI MHC分子上の腫瘍抗原を提示するワクチン細胞を破壊するだけであろう。NK細胞は、もしMHC相互作用が低ければ、低い免疫応答を産生する腫瘍ワクチン細胞を破壊しうる。V)APCを活性化するシグナルは直接または間接に、天然に獲得した免疫応答から来る。腫瘍ワクチン細胞を摂取するAPCは、抗原を効果的に提示しうる前に、活性化されていなくてはならない。さらに、所要の活性化シグナルなしに未熟APCに抗原を提示することはその免疫応答を抑制しうる。さらに、活性化されたAPCは、CTLを活性化することができる(CTLは活性化されていないと、たとえCTLがHLA適合腫瘍細胞上にそのコグネイト抗原を認識しても死滅させることができない)。
【0072】
上記の考察から、革新的な腫瘍ワクチンが腫瘍ワクチン分野に必要とされていることは明白であり、ここで特に必要なことは、I)予防ワクチン接種治療計画を必要としない治療法の設定において、既に存在しかつ播種性の腫瘍の退行を生じうるかまたは少なくとも腫瘍増殖速度を減速させうる腫瘍ワクチンを開発すること、II)免疫系の細胞性および体液性部門の両方を刺激するワクチンを開発すること、および、III)自己免疫疾患などの望ましくない二次的影響を有しないワクチンを開発することである。
【0073】
本出願人らの発明は、これらの要件を満たす治療用腫瘍ワクチン製剤を提供する。本発明は、α(1,3)-ガラクトシルエピトープをin vitroで合成する腫瘍細胞を作製する遺伝子導入技術の利用を含むものである。細胞自身の機構の利用およびこのように遺伝子操作された複数の同種異系または同系腫瘍細胞の混合物の利用により、エピトープを多数の細胞表面糖タンパク質および糖脂質上に作製して、抗体が個々のまたは別々の細胞上のTAAに対して産生する多数の機会を提供する。これらのワクチンに用いられる細胞は、100万〜200万個のαGalエピトープを含有すると見積もられる。抗αGal Abに対するこの多数の天然に予め存在する結合部位により、高密度のオプソニン作用が得られ、次いで、体液性および細胞性部門の両方の免疫系を活性化する様々なプロセスを開始する補体破壊が起こる。同種異系腫瘍細胞表面上のかかる高密度のαGal残基の存在は、改変した腫瘍細胞の注入部位において異種移植超急性拒絶と類似した超免疫応答を誘発する。さらに、これらの癌ワクチンは多価であり、多重の腫瘍抗原標的を免疫系に提示することを意味する。このことは、いくつかのTAAが提示される点でより効率的な治療、および提示されるTAAの数の増加とともに色々な個々の腫瘍からのエピトープが恐らく重複されるのでより広く効果的な治療をもたらす。オプソニン化された細胞は容易に貪食細胞により摂取されて、腫瘍抗原のほとんどが同時に適応性免疫系に提示されうる機構を提供する。これらの細胞内で癌ワクチン細胞からのタンパク質は消化されて、癌細胞中の突然変異タンパク質エピトープをT細胞監視に曝すクラスII MHC提示がなされる。さらに、抗原提示細胞(APC)による、Fc受容体が媒介する飲食作用(endocytosis)を経由するオプソニン化細胞の取込みは、CD8+細胞による交差提示経路を介するMHCクラスI制限応答の活性化を容易にしうる。このプロセスにより運動を始める免疫系カスケードは、確立されたヒト悪性腫瘍由来のナイーブ腫瘍細胞を破壊する特異的T細胞応答を誘導する刺激を与える。さらに、一次免疫応答により誘導される炎症性環境は、サイトカイン、ヒスタミンおよび他のアップレギュレートされた分子が媒介するT細胞応答をブーストする増幅効果をもたらす。この方法で活性化されたT細胞は直接、癌細胞を死滅させることができる。重要な所見は、腫瘍ワクチン中に存在する糖タンパク質および糖脂質に対するαGalエピトープの付加は、グリコシル化された抗原に対してだけでなく、グリコシル化による影響の有無に関わらず、腫瘍細胞内に存在する全ての抗原に対する免疫応答を発生させうることである。
【0074】
この種の細胞全体ワクチンの治療用腫瘍免疫誘導における有効性をノックアウト(KO)マウスを用いる動物モデルを立証した。αGT遺伝子の機能を欠くKOマウスを作製し、腫瘍培養細胞株上のαGalエピトープに対するin vivo免疫応答を研究するための小動物モデルを得た。これらのマウスを、ウサギ赤血球(rRBC)により免疫感作してヒト血清中に見出される高力価の抗αGal抗体を刺激することができる。これらのマウスを用いて、本出願人らは、該免疫系がαGalポジティブ腫瘍細胞を拒絶しかつこの拒絶がαGalネガティブ腫瘍細胞に拡大されたT細胞免疫を発生させることを実証した。この動物モデルを用いてまた、予め形成されたヒト悪性腫瘍をシミュレートするために、ワクチンによる治療前にαGalネガティブ腫瘍細胞を動物に与える臨床応用のシミュレーションを行った。このタイプの実験により、本出願人らは、本発明により遺伝子操作した細胞を用いて免疫治療したマウスに誘導された細胞が媒介する免疫は、皮下および肺の予め形成された局所および播種性腫瘍を有効に治療することができ、腫瘍増殖速度を減速するだけでなく、さらに重要なことは、治療したマウスの長期間生存をもたらすことを示した。養子細胞移入(adoptive cell transfer)実験は、最終的に、予め形成された腫瘍の拒絶の細胞依存構成要素を示した。他の手法を用いるときに記載された細胞全体腫瘍ワクチン摂取に関連する典型的な自己免疫色素脱失は、この治療を受けたマウスにおいては決して観察されず、異なる免疫刺激法が与える免疫応答の最終成果に対する影響の重要性を強調した。
【0075】
従って本出願人の発明は、予め形成された腫瘍細胞の選択的ターゲティングおよび死滅を引き起こす方法と組成物に関する。ex vivo遺伝子療法プロトコルを介して、腫瘍細胞を遺伝子操作してαGalエピトープを発現させる。次いで細胞を照射するかまたは死滅させて患者に投与する。天然に予め存在する抗αGal抗体によるαGalエピトープの結合は、腫瘍細胞のオプソニン化を引き起こし、腫瘍特異的抗原提示を増強する。本発明の重要な特徴は、本発明の医薬組成物中の細胞全体、および形質移入された複数の細胞の混合物の利用を意図することである。αGal改変は細胞表面上の複数の糖タンパク質に影響を与えるので、動物免疫系は検出し、プロセシングし、そして腫瘍特異的抗原に対する抗体を作製する機会の増加を有しうる。
【0076】
本発明によれば、医薬組成物は、発現するとαGT酵素をコードするポリヌクレオチド配列を腫瘍細胞に導入することにより作製される。該配列はウイルスまたは非ウイルスベクターを含んでもよいいずれかのヌクレオチド導入ビヒクルを介して導入される。これらの遺伝子導入ビヒクルは腫瘍細胞を形質転換し、そこに挿入される外来遺伝子材料の発現を引き起こす。得られる遺伝子産物は、上記細胞に存在する細胞表面糖タンパク質上のαGalエピトープの合成を触媒する。本発明は、多数の細胞表面糖タンパク質をもつ腫瘍細胞全体を利用して予め存在する抗αGal抗体によるαGalエピトープとの結合を最大化し、そして、この複合体と抗原提示細胞上に存在するFc受容体との結合を増強し、その結果、上記ワクチン腫瘍細胞中に存在する多数の腫瘍関連抗原の抗原提示をトリガーすることを意図する。
【0077】
本発明はまた、医薬組成物および上記医薬組成物を作る方法も含むものであり、ここで上記医薬組成物は、αGalエピトープを含む複数の細胞表面糖タンパク質を含有する弱毒化腫瘍細胞の混合物の治療上有効な量および担体を含む。好ましい実施形態においては、細胞は細胞全体である。組成物を作る方法は、生腫瘍細胞のコレクションを取得し、発現するとαGTをコードするヌクレオチド配列を用いて上記細胞を形質転換し、そして上記細胞の細胞表面糖タンパク質上にαGalエピトープを提示させることを含んでなる。次いで細胞を、好ましくは、照射により死滅させ、投与のための製薬担体と組合わせる。
【0078】
本発明のさらに他の実施形態は、腫瘍細胞上にαGalエピトープを作製しうるポリヌクレオチドによる腫瘍細胞の形質転換を含んでなる。本発明の一実施形態は、発現すると酵素α-(1,3)-ガラクトシルトランスフェラーゼ(αGT)をコードするヌクレオチド配列による腫瘍細胞の形質転換を含んでなる。αGT cDNAはウシおよびマウスcDNAライブラリーからクローニングされている。Larson, R. D.ら, (1989) 「マウスUDPガラクトースをコードするcDNAの単離;β-D-ガラクトシル-1,4-Nアセトール-D-グルコサミンα1-3ガラクトシルトランスフェラーゼ:遺伝子導入による発現クローニング(Isolation of a cDNA Encoding Murine UDP galactose; β-D-galactosyl-1, 4-N Acetol-D-Glucosamine α1-3 Galactosyl Transferase : Expression Cloning by Gene Transfer)」, PNAS, USA 86:8227;およびJoziasse, D. H.ら, (1989) 「ウシα1-3ガラクトシルトランスフェラーゼ(Bovine α1-3 Galactosyl Transferase)」: 「cDNAクローンの単離と特徴付け、ヒトゲノムDNA中の相同的配列の同定(Isolation and Characterization of a cDNA Clone, Identification of Homologous Sequences in Human Genomic DNA)」, J. Biol Chem 264:14290。同様に、細胞表面上にαGalエピトープを発現する腫瘍細胞を生じうる、例えばこの反応を触媒する他の酵素に対する、または、恐らくはさらに、細胞表面上にさらなる糖タンパク質を存在させて免疫系に提示しうるTAAを人為的に作製する遺伝子操作に対する他のヌクレオチド配列を利用することができる。
【0079】
本発明の医薬組成物に利用する腫瘍細胞は、自己または、好ましい実施形態においては、同種異系もしくは同系であってもよい。治療する形質転換細胞および腫瘍細胞は、普通は少なくとも1以上のエピトープを有しなければならないが、好ましくは多数を有しうる。一般的なまたは重複するエピトープまたはTAAが異なる癌の間に存在する限り、本医薬組成物を非常に広く応用することができる。
【0080】
本発明はまた、癌細胞に限定されるものでなく、細胞表面糖タンパク質を含有するいずれの細胞、またはαGalエピトープに対する部位を有する細胞構成要素も含むことができる。これはある特定のウイルス、新生物細胞などを含んでもよい。
【0081】
αGalエピトープを作製するタンパク質をコードするヌクレオチド配列は、腫瘍細胞を形質導入する適当な発現ビヒクル中に含有される。かかる発現ベクターとしては、限定されるものでないが、真核生物ベクター、原核生物ベクター(例えば、細菌ベクターなど)、およびウイルスベクターが挙げられる。
【0082】
一実施形態においては、発現ベクターはウイルスベクターである。使用しうるウイルスベクターは、限定されるものでないが、レトロウイルスベクター、アデノウイルスベクター、ヘルペスウイルスベクター、およびアデノ随伴ウイルスベクター、またはDNA複合体が挙げられる。
【0083】
好ましい実施形態においては、ウイルスベクターパッケージング培養細胞株に、抗体結合および補体活性化により腫瘍細胞の破壊を誘導する作用物質(agent)をコードする核酸配列を含有するウイルスベクターを形質導入する。パッケージング培養細胞株により産生されるウイルス粒子を収穫して、抗腫瘍ワクチンとして投与する腫瘍細胞を形質導入するために用いる。
【0084】
伝統的に、ウイルスベクターはレトロウイルスまたはアデノウイルスベクターである。利用しうるレトロウイルスベクターの例としては、限定されるものでないが、モロニーマウス白血病ウイルス、脾臓壊死ウイルス、およびレトロウイルス、例えばラウス肉腫ウイルス、ハーベイ(Harvey)肉腫ウイルス、鳥類白血症ウイルス、ヒト免疫不全ウイルス、骨髄増殖性肉腫ウイルス、および哺乳類動物腫瘍ウイルス由来のベクターが挙げられる
レトロウイルスベクターは、真核生物細胞中にレトロウイルス媒介遺伝子導入を媒介する作用物質として有用である。レトロウイルスベクターは、一般的に、ウイルスの構造遺伝子をコードする配列の大多数を欠失させ、そして目的の遺伝子により置換えて構築する。最も多いのは、構造遺伝子(すなわち、gag、pol、およびenv)をレトロウイルス主鎖から当技術分野で公知の遺伝子操作技法によって取除く。
【0085】
これらの新しい遺伝子は、いくつかの一般的方法でプロウイルス主鎖中に組込んだ。最も簡単な構築物は、レトロウイルスの構造遺伝子を単一遺伝子により置換え、次いでそれを長末端反復配列(LTR)内のウイルス調節配列の制御下で転写したものである。標的細胞中に複数の遺伝子を導入することができるレトロウイルスベクターを構築しておいてもよい。通常、このようなベクターでは、1つの遺伝子はウイルスLTRの調節下にある一方、第2の遺伝子はスプライスされたメッセージとして発現されないかまたはそれ自身の内部プロモーターの調節下にある。
【0086】
主に、ベクターとパッケージング細胞内のパッケージング欠損ヘルパーウイルスの間の組換えの機会を減少させる努力として、ウイルス主鎖のウイルス構成要素を最小限にする努力がなされている。パッケージング欠損ヘルパーウイルスは、ベクター自身から欠失されたレトロウイルスの構造遺伝子を与えるために必要である。
【0087】
一実施形態においては、レトロウイルスベクターは、Benderら, J. Virol. 61:1639-1649 (1987)に記載の一連のベクターの1つであってもよく、これはN2ベクター(Armentanoら, J. Virol., 61:1647-1650)ベースであり、ベクターとパッケージング系の間の相同性を絶対的最小値に低下させる一連の欠失および置換を含有する。これらの変化はまた、ウイルスタンパク質が発現されうる可能性も低下させた。これらのベクターの最初のものであるLNL-XHCでは、位置指定突然変異誘発により天然ATG開始コドンをgagからTAGに改変し、それによりこの点からの意図しないタンパク質合成を排除した。
【0088】
モロニーマウス白血病ウイルス(MoMuLV)においては、真正gagスタートの5'に、他のグリコシル化タンパク質(pPr80.sup.gag)の発現を可能にするオープンリーディングフレームが存在する。モロニーマウス肉腫ウイルス(MoMuSV)はこの5'にフレームシフトおよびグリコシル化部位の消失を含む変化があり、pPr80.sup.gagのアミノ末端の発現の可能性を除去する。従って、LNL-XHCの改変されたATGとMoMuSVの5'部分の両方を組込んだベクターLNL6を作った。このようにLNベクターシリーズの5'構造は遺伝的に形質導入された標的細胞におけるレトロウイルスリーディングフレームの発現とともに続いてのウイルス抗原産生の可能性を排除する。パッケージング欠損ヘルパーウイルスとの重複を少なくする最終的改変において、MillerはLNベクター中の3'LTRのすぐ前の余分なenv配列を排除した(Millerら, Biotechniques, 7:980-990, 1989)。本発明によって使用しうるベクターの一例を図1に示し、その配列を図2、配列番号1に示す。
【0089】
遺伝子療法に応用するために遺伝子導入系が満たさなければならない最高の要件は安全である。安全は、ベクターゲノム構造と、感染性ベクター産生に用いるパッケージング系との組合わせから誘導される。Millerらは、レトロウイルス構造タンパク質を発現するpPAM3プラスミド(パッケージング欠損ヘルパーゲノム)とベクターパッケージング系を作るLNベクターシリーズとの組合わせ(すなわち、LNとpPAM3)を開発し、ベクターゲノムとパッケージング欠損ヘルパーゲノムの間のほとんど全ての組換えの部位を排除することを介して組換え野生型レトロウイルスの生成を最小限に低下させた。
【0090】
一実施形態においては、レトロウイルスベクターは、上述のおよびさらにBenderら(1987)およびMillerら(1989)に記載されたベクターなどのモロニーマウス白血病ウイルスのLNシリーズのベクターであってもよい。かかるベクターはマウス肉腫ウイルスから誘導されたパッケージングシグナルの一部分、および突然変異したgag開始コドンを有する。本明細書に用いる用語「突然変異した」は、gag開始コドンが欠失しているかまたは改変されてgagタンパク質またはその断片もしくは末端切断部が発現されないことを意味する。
【0091】
該ベクターは1以上のプロモーターを含む。使用しうる好適なプロモーターとしては、限定されるものでないが、Millerら, Biotechniques, Vol. 7, No. 9, 980-990 (1989)に記載のレトロウイルスLTR;SV40プロモーター;およびヒトサイトメガロウイルス(CMV)プロモーター、またはいずれか他のプロモーター(例えば、限定されるものでないが、ヒストン、pol III、およびβ-アクチンプロモーターを含む真核生物細胞性プロモーターなどの細胞性プロモーター)が挙げられる。使用しうる他のウイルスプロモーターとしては、限定されるものでないが、アデノウイルスプロモーター、TKプロモーター、およびB19パルボウイルスプロモーターが挙げられる。
【0092】
他の実施形態においては、本発明は誘導プロモーターを含む。かかるプロモーターの1つはテトラサイクリン制御トランス活性化因子(tTA)応答性プロモーター(tet系)、哺乳類動物細胞における利用に適応させている原核生物誘導プロモーター系である。tet系はレトロウイルスベクター内に組織化し、高レベルの構成的に産生されるtTA mRNAがtTAタンパク質の産生だけでなく、アンチセンス抑制による応答ユニットの低い基底発現にも機能するようにした。Paulus, W.ら, 「自己に含有される、哺乳類動物細胞への遺伝子送達用テトラサイクリン調節レトロウイルスベクター系(Self-Contained, Tetracycline-Regulated Retroviral Vector System for Gene Delivery to Mammalian Cells)」, J of Virology, January. 1996, Vol. 70, No. 1, pp. 62-67を参照すること。好適なプロモーターは、本明細書に含まれる教示から当業者には明らかであろう。
【0093】
次いでベクターを用いてパッケージング培養細胞株に形質導入して、生産培養細胞株を作製する。トランスフェクトしうるパッケージング細胞の例としては、限定されるものでないが、PE501、PA317、.psi.2、.psi.-AM、PA12、T19-14X、VT-19-17-H2、.psi.CRE、.psi.CRIP、GP+E-86、GP+envAM12、DANおよびAMIZ培養細胞株が挙げられる。作用物質をコードする核酸配列が発現すると、腫瘍細胞を破壊しかつ補体カスケードを活性化することができる作用物質をコードする核酸配列を含有するベクターは、当業者に公知のいずれかの方法を介してパッケージング細胞に形質導入することができる。かかる方法は、限定されるものでないが、エレクトロポレーション、リポソームの利用、およびCaPO4沈降を含む。
【0094】
好ましい実施形態においては、本発明は、通常ヒトおよびヒトに基づくパッケージング培養細胞株に感染するウイルスベクターを含む。例えば、ヘルペスウイルス、エプスタインバーウイルスなどのヒトに通常感染するウイルスから誘導されたベクターを利用することができる。
【0095】
本発明により治療することができる腫瘍は悪性および非悪性腫瘍を含む。治療しうる悪性(原発性および転移性を含む)腫瘍としては、限定されるものでないが、副腎;膀胱;骨;乳房;頚部;内分泌腺(甲状腺、脳下垂体、および膵臓を含む);大腸;直腸;心臓;造血性組織;腎臓;肝臓;肺;筋肉,筋;神経系;脳;眼;口腔;咽頭;喉頭;卵巣;陰茎;前立腺;皮膚(黒色腫を含む);精巣;胸腺;および子宮に発生する腫瘍が挙げられる。かかる腫瘍の例としては、APUD細胞腫(apudoma)、分離腫、鰓腫(branchioma)、悪性カルチノイド症状、カルチノイド心疾患、癌(例えば、Walker、基底細胞、基底有棘細胞、ブラウン-ピアス(Brown-Pearce)、腺管(ductal)、エールリッヒ(Ehrlich)腫瘍、in situ、クレブス(Krebs)2、メルケル(Merkel)細胞、粘液性、非小細胞肺(non-small cell lung)、燕麦細胞、乳頭、硬性、細気管支、気管支原性、扁平細胞、および移行細胞)、プラズマ細胞腫、黒色腫、軟骨芽細胞腫、軟骨腫、軟骨肉腫、線維腫、線維肉腫、巨細胞腫瘍、組織球腫、脂肪腫、脂肪肉腫、中皮腫、粘液腫、粘液肉腫、骨腫、骨肉腫、ユーイング(Ewing's) 肉腫、骨膜腫、腺線維腫、腺リンパ腫、癌肉腫、脊索腫、間葉細胞腫、中腎腫、筋肉腫、エナメル上皮腫、セメント質腫、歯牙腫、奇形腫、胸腺腫、栄養膜腫瘍、腺癌、腺腫、胆管腫、胆脂腫、円柱腫、嚢胞腺癌、嚢胞腺腫、顆粒膜細胞腫、半陰ポジティブ卵巣腫瘍(gynandroblastoma)、肝臓癌、汗腺腫、島細胞腫、Leydig細胞腫、乳頭腫、セルトーリ(Sertoli)細胞腫、卵胞膜細胞腫、平滑筋腫、平滑筋肉腫、筋芽細胞腫、筋腫、筋肉腫、横紋筋腫、横紋筋肉腫、上衣細胞腫、神経節神経腫、神経膠腫、髄芽腫、髄膜腫、神経鞘腫、神経芽細胞腫、神経上皮腫、神経線維腫、神経腫、傍神経節腫、非クロム親和性傍神経節腫、被角血管腫、好酸球増加随伴性血管類リンパ組織増殖症、硬化性血管腫(angioma sclerosing)、血管腫症、グロムス血管腫(glomangioma)、血管内皮腫、血管腫、血管外皮細胞腫、血管肉腫、リンパ管腫、リンパ管筋腫、リンパ管肉腫、松果体腫、癌肉腫、軟骨肉腫、葉状嚢肉腫、線維肉腫、血管肉腫、平滑筋肉腫、白血肉腫、脂肪肉腫、リンパ管肉腫、筋肉腫、粘液肉腫、卵巣癌、横紋筋肉腫、肉腫(例えば、ユーイング(Ewing's)実験的、カポージ(Kaposi's)、および肥満細胞)、新生物および他のかかる細胞に対する腫瘍が挙げられる。
【0096】
医薬調製物
本発明に従って、弱毒化αGalを発現する腫瘍細胞を、腫瘍を処置するための予防または治療ワクチンとして使用する。従って、本発明はまた、これらのトランスジェニック腫瘍細胞(HA1、HA2などとして表される、表1を参照)に関わるヒトおよび動物に対する医薬調製物も含む。医学分野の業者は、医薬組成物の投与量およびスケジュールがヒトおよび動物の年齢、健康、性別、サイズおよび体重に依存して変化しうることを容易に理解するであろう。これらのパラメーターは、それぞれの系に対してフェーズI、IIおよびIII臨床試験における十分確立された方法および分析によりならびに本明細書に記載の例を総括することにより決定することができる。
【0097】
投与に際して、弱毒化腫瘍細胞を、適当な液体ビヒクルまたは賦形剤および任意の補助添加剤などの製薬上許容される担体と組合わせることができる。液体ビヒクルおよび賦形剤は市販されている。それらの説明のための例は、蒸留水、生理学的食塩水、ブドウ糖水溶液などである。
【0098】
非経口、皮下、皮内、筋肉内、経口または腹腔内投与用の好適な製剤としては、水溶性または水分散性形態の活性化合物の水溶液が挙げられる。さらに、油性注射懸濁液としての活性化合物の懸濁液を投与することができる。好適な親油性溶媒またはビヒクルとしては、脂肪油、例えばゴマ油、または合成脂肪酸エステル、例えばオレイン酸エチルまたはトリグリセリドが挙げられる。水性注射懸濁液は、懸濁液の粘度を増加する物質、例えば、カルボキシメチルセルロースナトリウム、ソルビトールおよび/またはデキストランを含有してもよく、場合によっては、懸濁液は安定剤を含有してもよい。また、腫瘍ワクチン細胞を、当技術分野で公知の免疫アジュバント、例えばフロイントの完全アジュバント、無機塩、例えば塩化亜鉛、リン酸カルシウム、水酸化アルミニウム、リン酸アルミニウム、サポニン、ポリマー、脂質または脂質画分(脂質A、モノホスホリル脂質A)、改変オリゴヌクレオチドなどと混合してもよい。
【0099】
通常の担体を用いる投与だけでなく、活性成分を、当業者に公知の様々な特殊な送達薬物技術により投与することができる。以下の例は、説明の目的で記載されたものであり、決して本発明を限定することを意図するものではない。
【実施例】
【0100】
本発明は、これから、以下の実施例について記載する、しかし、本発明の範囲はこれにより限定されることを意図するものでない。特許および刊行物に対する全ての引用は、本明細書に参照により明白に組み入れられるものである。
【0101】
実施例1
αGTを発現するレトロウイルスベクター、pLNCKGの作製
マウスαGT遺伝子の1,077bp断片をαGTの翻訳を促進するKozak配列を含有するフォワードプライマー、5’-ACAAAAGCTTGACATGGATGTCAAGGGAAAAGTAAT-3’、およびリバースプライマー、5’-AATTATCGATTCAGACATTATTTCTAAC-3’によりPCR増幅し、次いでpLNCXのClaIおよびHindIII部位にクローニングしてpLNCKGレトロウイルスベクターを作製した(図1)。このベクターをパッケージング培養細胞株293.AMIZ(YoungおよびLink 「レトロウイルスパッケージング細胞の改良のためのDNAメチル化を排除するキメラレトロウイルスヘルパーウイルスとピコルナウイルスIRES配列(Chimeric retroviral helper virus and picornavirus IRES sequence to eliminate DNA methylation for improved retroviral packaging cells)」 J. Virol. (2000) 74: 5242-5249)中にトランスフェクトして、ベクター生産培養細胞株293.AMIZ/LNCKGを作製した。トランスフェクトした細胞をG418およびZeocinTMの存在のもとで2週間選択した。選択された細胞の混合集団を限定希釈によりサブクローニングした。単一細胞由来のVPCを、色々な組織から確立したヒト上皮癌培養細胞株に効果的に形質導入する能力についてスクリーニングした。その上清が一貫して最高の形質導入効率および1パネルのヒト上皮癌培養細胞株上のαGT発現を生じるクローンを同定して293Z.CKG VPCと名付けた。293Z.CKG VPCはシードバンク(seed bank)の1バイアルから起こしたので、マスター細胞バンク、ワーキング細胞バンク、および生産ロットを作製し、フラスコ内で37℃±1℃、5%±1%CO2中にて拡大した。培地は10%胎児ウシ血清(FBS)および2mm L-グルタミンを補充したRPMI-1640であった。293Z.CKG VPCが十分な密度に到達すると、培養液(上清)を収穫し、濾過し、そして無菌容器中に収穫し、濾過し、そしてプールした。プールを十分混合し、次いで無菌状態で標識した、無菌のプラスチックボトルに満たした(標識は製品名、ロット番号および充填日を含む)。充填したボトルを凍結して-60℃にて貯蔵する。アリコートを安全試験にかける。293Z.CKG VPCからのレトロウイルスを含有する上清を用いて色々なヒト癌培養細胞株を形質導入して(以下の実施例に記述する)αGal(+)細胞全体ワクチンを確立した。
【0102】
実施例2
LNCKGレトロウイルスベクターによる、B16.BL6黒色腫細胞の形質導入
αGal(+)B16細胞を作製するために、2x106細胞を、2x106tu/mLの感染性力価をもつLNCKGレトロウイルスを含有する上清2mLを用いて形質導入した。細胞をネオマイシン耐性に対してG418 1mg/mLを補充した培地中で2週間選択により選択した。この選択期間後に、細胞をαGalエピトープの発現についてニワトリ抗αGalポリクローナル抗体を用いて染色し、蛍光活性化細胞ソーターによりソートした。
【0103】
実施例3
ウサギ赤血球を用いる免疫感作による、α(1,3)-ガラクトシルトランスフェラーゼノックアウト(αGT KO)マウスにおける抗αGal抗体の誘導
雌性および雄性8〜14週齢α(1,3)ガラクトシルトランスフェラーゼ(αGT)ノックアウト(KO)マウスを本研究に用いた。マウスは最初、混合ハプロタイプ(H-2 b/d)であって、増殖と選択によりH-2 b/bハプロタイプのF4同系交配世代からなるαGT KOマウスの現コロニーを得た。これらの動物はαGalエピトープに対して低力価の天然抗体を産生する。抗αGal Abの力価を増加するために、1x108ウサギ赤血球を用いて2回、2週を隔ててマウスを腹腔内(i.p.)に免疫感作した。最後のRRBC注射の1週後に抗αGal Abの力価をチェックして、研究中の全マウスは高い抗αGal Abを有することを実証した。本研究に用いた全マウスは、ELISAにより測定すると1:500希釈より大きい高い抗αGal Ab力価を有する。代表的な実験を図3に示す。
【0104】
実施例4
非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の生存試験
本実験の目的は、癌細胞中のαGalエピトープの発現が、予め存在する抗αGal Abをもつ宿主においてin vivo超急性拒絶を起こしうるかどうかを確認することであった。従って、RRBC注射したαGT KOマウスに、αGalエピトープを発現するまたは発現しない105 非照射B16黒色腫細胞をチャレンジし、腫瘍発生を測定した(図4)。
【0105】
図5はチャレンジ後15日の腫瘍サイズを示す。予想通り、天然αGal(-) B16を受けた19マウスのうちの11マウスは、チャレンジ後2週に測定可能な腫瘍を発生した。同様に、B16.LNL αGal(-) B16細胞を受けた20マウスのうちの10マウスは腫瘍を発生した(それぞれ、57%および50%、カイ自乗p>0.05で、有意でない)。しかし、マウスがαGal(+)B16細胞を受けた時は、有意なより少数の動物が腫瘍を発生した。20動物のうち5動物(25%)だけが明白な腫瘍を発生した(カイ自乗、p=0.03、対照と有意差あり)。さらに、対照および模擬(mock)形質導入した細胞を用いてチャレンジした動物では、αGal発現細胞を受けたマウスにおいて発生した腫瘍と比較して、腫瘍が相当大きかった。平均サイズ200mm3の腫瘍が両方の対象グループに観察され、グループ間の差は統計的に有意でなかった(F試験、p=0.38)。しかし、B16.αGal(+)細胞を受けたグループの腫瘍サイズに有意差が観察された。試験グループの平均サイズ腫瘍は36mm3であって、対照動物と比較して腫瘍サイズは約80%の低下を示した(F試験、p=0.002)。
【0106】
この結果はαGalを発現するB16細胞を用いてチャレンジするとより少数の動物が小さい腫瘍を発生したことを実証するものであり、抗αGalを前感作したマウスは、αGalを発現する細胞のより効果的なin vivoクリアランスを、天然または模擬(mock)形質導入したαGalネガティブB16対照のそれと比較して媒介できることを示す。
【0107】
腫瘍をチャレンジ後30日に測定したときも、比較しうる結果を観察した。B16野生型を受けたグループでは17マウスのうちの13マウスが腫瘍を有した(76%)。模擬(mock)ベクターを用いて形質導入した細胞を受けたグループでは18マウスのうちの13マウスが腫瘍を有した(72%)。しかし、αGal発現細胞を受けたグループではマウスの50%だけが測定しうる腫瘍を有した。さらに重要なこととして、対照グループのそれぞれ2動物、計4動物を大きな腫瘍が発生したため安楽死させなければならなかった。αGalを発現する細胞を受けた動物は腫瘍移植後30日までに死亡したものはなく、αGalを発現する細胞を用いてチャレンジした動物の生存率の増加を示した。
【0108】
図6は、対照、模擬(mock)およびαGalを発現するB16細胞によるチャレンジ後の腫瘍発生の動力学を示す。両方の対照αGal(-)グループの腫瘍は、極度に速く増殖し7日間で容積がほとんど2倍になった。これに反して、αGal(+)B16細胞を用いてチャレンジしたマウス由来の腫瘍は早い時点で有意に遅い速度で増殖した(p=0.03)。これは、免疫機構が腫瘍の増殖を制限し、腫瘍を保持するマウスの生存を延ばすのを助けうることを示唆する。
【0109】
αGal(+)B16細胞による致死的チャレンジ後のマウスの長期生存
対照αGal(-)、模擬(mock)形質導入αGal(-)B16およびαGal(+)B16を受けたグループからのマウスを90日間、毎週観察した(図7)。図に示したように、天然B16を受けた10動物のうちのどれもチャレンジで生き残らず、模擬(mock)形質導入したαGal(-)B16細胞を受けた11マウスのうちの1マウスだけが皮下注射で生き残った。両方のグループは非常に類似する生存曲線であり、NeoR遺伝子産物はB16の拒絶および/または免疫原性に有意な変化を誘導しないことを示した(ログランク(Logrank)試験p=0.5、差は統計学的に有意でない)。これに反して、αGalを発現する非照射B16細胞を受ける動物の47%が、致死量チャレンジで生き残った。19マウスのうちの9マウスは皮下注射後80日間、無腫瘍のままであった。Kaplan-Meier生存分析を用いると、対照グループと比較して、αGal(+)B16細胞を皮下注射したグループの生存率に有意な増加が観察された(ログランク(Logrank)試験、p<0.02)。
【0110】
αGal(-)B16を用いて注射した両方の対照グループでは、動物の60%近くはチャレンジ後最初の30日に死亡した。これに反して、αGal(+)B16を用いて注射した実験グループは、5動物だけ(26%)を、チャレンジ後の最初の月に安楽死させねばならなかった。
【0111】
第2の独立実験において、同様な結果を得た。天然αGal(-)B16を皮下注射した19マウスのうちの2マウスだけが生き残った。比較として、21マウスのうちの4マウスだけが模擬(mock)形質導入αGal(-)B16細胞の致死的皮下チャレンジで生き残った。これに反して、天然αGal(+)B16を皮下注射した20マウスのうちの8マウスが生き残り、80日を越えて無腫瘍のままであった。
【0112】
全体としてこの結果は、αGT遺伝子の発現とB16のグリコシル化パターンの変化が生癌細胞のin vivo破壊を誘導して腫瘍増殖が低下するのを助けて腫瘍保持マウスを延命することを実証する。さらに重要なこととして、チャレンジしたマウスのほぼ40%が無腫瘍のままであるので、αGal発現細胞の注射は、致死性の高いαGal(-)黒色腫細胞の増殖を制御することができる強い免疫応答を誘導しうる。
【0113】
非照射αGal(+)B16細胞による致死量チャレンジ後の生存は、野生型αGal(-)B16に対する記憶保護免疫を誘導する。
本発明者らは、非照射αGal(+) B16細胞の皮下注射から生き残った動物は天然αGal(-) B16腫瘍に拡大した細胞性免疫を発生するという仮説を立てた。この仮説を試験するために、全ての生き残った無腫瘍マウスに野生型B16(αGalネガティブ)を再チャレンジした。年齢の適合したマウスを対照に加えて、αGal(-)B16を注射した(図8)。予想通り、αGal(-)B16をチャレンジした12対照マウスのうちの11マウスは大きな着色腫瘍を発生する皮下黒色腫によって死亡した(図9)。その一方で、非照射αGal(+)B16黒色腫を最初に受けて生き残ったマウスはいずれも腫瘍を発生しなかった。8マウスは全て、70日間無腫瘍のままであり、最初にαGal(+)B16細胞を拒絶したマウスの生存率を有意に増加した(ログランク(logrank)検定、p<0.001)。興味深いことに、B16黒色腫に対する保護は、先に他の研究者が記載した自己免疫色素脱失(白斑)と関係がなかった(Overwijkら, 「マウスにおいて、自己抗原をコードする組換えワクシニアウイルスによるワクチン接種は自己免疫白斑および腫瘍細胞破壊を誘導する:CD4+ Tリンパ球の必要性(Vaccination with a recombinant vaccinia virus encoding a self antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4+ T lymphocytes)」 Proc. Natl. Acad. Sci. USA (1999) 96:2982-2987;Overwijkら, 「自己反応性CD8+ T細胞の機能的寛容状態の逆転後における腫瘍退行と自己免疫(Tumor regression and autoimmunity after reversal of functionally tolerant state of self-reactive CD8+ T cells)」 J. Exp. Med. (2003) 198:569-580;van Elsasら, 「抗CTLA-4およびGM-CSF産生ワクチンを用いるB16黒色腫の併用免疫療法は、自己免疫色素脱失を伴う皮下および転移性腫瘍の拒絶を誘導する(Combination immunotherapy of B16 melanoma using anti-CTLA-4 and GM-CSF-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation)」 J. Exp. Med. (1999) 190:355-366)。
【0114】
この結果は、αGal(+)細胞の致死的チャレンジで生き残ったマウスが天然αGal(-)腫瘍に対して強い免疫を発生することを実証した。この細胞性および恐らくは体液性の免疫応答は生き残ったマウスを野生腫瘍による第2の致死的チャレンジから保護し、全ての保護されたマウスにおいて免疫応答が天然αGal(-)腫瘍B16に拡大されたことを示す。
【0115】
さらに、T細胞の媒介する免疫が保護されたマウスに誘導されたという仮説を実証するために、黒色腫特異的T細胞培養物を作製してαGal(-)B16黒色腫および非特異的培養細胞株EL-4およびMC38に対して試験した(図10)。
【0116】
図11に示したように、B16黒色腫を特異的に溶解することができる強いCTLが保護されたマウスに誘導されることを標的細胞中のLDH放出により測定した。
【0117】
全体として、これらの結果は、αGal(+)細胞の注射がマウスの生存率を増加することを実証する。さらに、αGal(+)B16細胞を拒絶しかつ最初の致死的注射で生き残ったマウスは天然αGal(-)B16黒色腫腫瘍に対して拡大した強い免疫を発生することができる。これは、αGal(+)B16細胞の拒絶後に記憶T細胞が媒介する免疫が誘導されて、αGal(-)B16腫瘍を認識して致死的腫瘍投与からマウスを保護することができるのを示す。
【0118】
実施例5
非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死量の静脈注射による、αGTノックアウトマウスの播種性黒色腫転移モデル
さらに、高力価の抗αGal AbをもつマウスがαGal(+) B16黒色腫細胞を拒絶できることを実証するために、肺黒色腫転移モデルを用いた。マウスに、105非照射αGal(-)またはαGal(+)B16黒色腫細胞を静脈(i.v)注射した。3週後、肺黒色腫転移を数えた(図12)。αGal(-)B16細胞を注射したマウスは多数の肺転移を有する。これに反して、αGal(+)B16細胞を注射したマウスは肺腫瘍量が低下した(図13)。さらに5マウスのうちの2マウスは無腫瘍であった。この結果は、予め存在する抗αGal AbがαGal(+)B16細胞のクリアランスに大きな役割を果たすことを示す。
【0119】
実施例6
αGal(+)細胞を用いる予防ワクチン接種による皮下腫瘍発生の予防
本実験の目的は、皮下腫瘍の予防を、先に記載のαGal(+)ワクチンを用いるワクチン接種により達成できるかどうかを決定することである(LaTempleら, 「抗αGal抗体と複合した腫瘍ワクチンの増加した免疫原性:α(1,3)ガラクトシルトランスフェラーゼのノックアウトマウスにおける研究(Increased immunogenicity of tumor vaccines complexed with anti-αGal: studies in knockout mice for α(1,3) galatosyl transferase)」, Cancer Res. (1999) 59:3417-3423)。マウスをRRBCを用いて先に記載のとおり免疫感作した。最後のRRBC注射後1週に、マウスは照射細胞ワクチンの第1用量を受けた。マウスに、照射天然αGal(-)B16、対照ベクター(pLNL、ネオマイシン耐性遺伝子をコードする)を用いて形質導入したαGal(-)B16、またはαGal(+)B16(ネオマイシン耐性遺伝子およびαGT遺伝子をコードするベクターを用いて形質導入した)のいずれかを注射した。複数のマウスは照射細胞ワクチンを受けなかった。細胞ワクチン接種を2週後に繰返した。最後のワクチン接種後2週に、マウスに105非照射αGal(-)B16細胞を皮下注射した(図14)。腫瘍を週2回、90日間測定した。図15に示すように、B16細胞ワクチンを受けなかった10マウスのうちチャレンジで生き残ったものはゼロであり、チャレンジ後50日までに死亡した。天然αGal(-)B16およびαGal(-)B16/NeoRワクチンをワクチン接種したマウスには若干の保護が観察された。B16チャレンジで14マウスのうち4マウスおよび12マウスのうち5マウスがそれぞれ生き残った。B16とB16/NeoRをワクチン接種したマウス間に統計学的な差はなく、この条件のもとでNeoR遺伝子産物はB16の免疫原性を増加しないことを示す(p>0.2、ログランク(Logrank)試験)。これに反して、マウスにαGal(+)B16細胞をワクチン接種すると、20マウスのうちの12マウスがチャレンジで生き残り、かつ90日間無腫瘍のままであって、有意なさらに大きい保護が観察された(p<0.001、ANOVA、p=0.08 ログランク(Logrank)試験)。この結果は、照射αGal(+)細胞によるワクチン接種がαGal(+)B16ワクチン接種マウスの60%において皮下黒色腫腫瘍の発生を防止しかつαGal(-)B16黒色腫をチャレンジしたマウスを有意に延命することを実証する。
【0120】
本発明者らは、生αGal(-)B16による注射から保護されたマウスでは、天然αGal(-)B16細胞を認識できるT細胞がαGal(+)B16ワクチンによるワクチン接種後に誘導されたという仮説をたてた。この仮説を試験するために、αGal(+)ワクチンを用いてワクチン接種したマウスから脾細胞を収穫し、刺激の存在または不在のもとで6時間培養した。刺激を最大限にするために、PMA/Ca++イオノフォアを用いた。細胞を特異的認識を測定するために105照射B16細胞ともにまたはCA320M、同一のH-2 b/bハプロタイプをもつ非特異的αGal(-)培養細胞株とともに培養した。インキュベーション後、細胞を収穫して細胞内TNF-αを染色した。前方散乱プロットでリンパ球のFACSゲーティングによる検出を実施した(図16)。αGal(+)B16ワクチン接種したマウスから収穫したT細胞はPMA/Ca++イオノフォアにより効率的に活性化された。このポリクローナルアクチベーター技法により活性化されたリンパ球のパーセントは本実験で検出された最大活性化と考えられる。休止(刺激されない)T細胞とCA320Mにより刺激されたT細胞はTNF-aを産生することができず、B16-αGal(+)ワクチン接種後にCA320Mの抗原を認識できるT細胞前駆体が誘導されなかったことを示した。これに反して、B16-αGal(+)を用いるワクチン接種は、in vitroでB16-αGal(-)を特異的に認識するT細胞前駆体を誘導した。この結果は、αGal(+)B16を用いるワクチン接種後に誘導されたT細胞がαGal(+)B16処置したマウスのほぼ半分の腫瘍予防の原因でありうることを示唆する。
【0121】
実施例7
αGal(+)細胞ワクチン接種による、予め形成された皮下黒色腫腫瘍の治療
天然αGal(-)B16およびαGal(-)模擬(mock)形質導入B16/NeoRを用いるワクチン接種が以上の実験において類似したデータを生じたので、統計学的解析力を増加するネガティブ対照として照射αGal(-)B16/NeoRを単独で用いるワクチン接種による次の実験を実施した。
【0122】
次の実験の目的は、予め形成された皮下黒色腫腫瘍の治療をαGal(+)B16ワクチンによるワクチン接種により達成しうるかどうかを決定することであった。有効な予防治療が予め形成された腫瘍の治療にも有効でありうるかは明らかでない。腫瘍モデルとしてB16黒色腫の特定の事例において、複数の治療計画が免疫感作の予防スケジュールに有効であったこと、そして予め形成された皮下黒色腫腫瘍の治療に与える効果は低いかまたは無かったことが立証されている。例えば、マウスmTRP-1をコードする組換えワクシニアウイルスを用いるワクチン接種は皮下黒色腫腫瘍の発生を予防するが、予め形成された皮下腫瘍の治療に効果がない(Overwijkら, 「マウスにおいて自己抗原をコードする組換えワクシニアウイルスを用いるワクチン接種は自己白斑および腫瘍細胞破壊を誘導する:CD4+Tリンパ球の必要性(Vaccination with a recombinant vaccinia virus encoding a self antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4+ T lymphocytes)」” Proc. Natl. Acad. Sci. USA (1999) 96:2982-2987)。同様に、ペプチド特異的免疫感作は強いT細胞免疫を誘導するが、予め形成された腫瘍に対する治療に有効なことは稀である(Davilaら, 「細胞傷害性Tリンパ球エピトープペプチドワクチン接種、CpGオリゴデオキシヌクレオチドアジュバントおよびCTLA-4遮断による抗腫瘍免疫の産生)(Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG oligodeoxynucleotide adjuvant and CTLA-4 blockade)」 Cancer Research (2003) 63:3281-3288)。さらに、腫瘍特異的T細胞の唯一の存在は、腫瘍根絶を効果的に誘導するための必要条件であるが十分ではない。
【0123】
本発明者らは、αGal(+)ワクチンは、予め形成されたαGal(-)腫瘍を拒絶できる強い細胞依存性腫瘍免疫を誘導することができるという仮説を立てた。この仮説を試験するために、本発明者らは予め形成された腫瘍を治療するように設計した実験を実施した(図17)。
【0124】
マウスに、105非照射B16細胞を皮下注射して無作為化した。チャレンジ後3日に、それらのマウスに、照射αGal(-)B16/NeoRまたは照射αGal(+)B16を皮下にワクチン接種した。3日後、照射細胞ワクチンを用いるワクチン接種を繰返した。基線対照マウスは非照射B16による皮下注射を受け、照射細胞ワクチン治療を受けなかった(無ワクチングループ)。図18aおよびbは、ワクチン接種しなかったマウス(n=11)、αGal(-)B16/NeoRによるワクチン接種したマウス(n=24)およびαGal(+)B16ワクチンを受けたマウス(n=23)の腫瘍増殖の動力学を示す。データは平均および誤差バー、SEMを表す。統計学的解析は、対照マウスとαGal(+)B16細胞ワクチンを受けたマウスを比較した時、勾配の有意差があるのを示す(p<0.009)。この結果は、非照射αGal(+)B16ワクチンを用いてワクチン接種したマウスが、増殖のより遅い腫瘍をより少数発生することを示す。
【0125】
実験#2においては、マウスはさらなる用量の細胞ワクチンを受けた。非照射αGal(-)B16による皮下注射だけを受けたマウスは、観察の最初の月に非常に増殖の早い大きな腫瘍を発生した(n=15)。同様に、対照αGal(-) B16ワクチンを注射したマウスは非常に増殖の早い大きな腫瘍を発生した(n=29)。これらの2グループのスロープの統計学的比較は、これらに有意差のないことを示した(p=0.17)。これは、αGal(-) B16/NeoRによるワクチン接種は皮下に予め形成された黒色腫腫瘍の治療に影響を与えないことを示す。これに反して、αGal(+)B16を受けたマウスは増殖のより遅い腫瘍をより少数発生した(n=33)。このグループの腫瘍増殖を対照グループと比較すると、統計学的に有意差があった(p<0.03)。
【0126】
これらの2つの実験は、αGal(-)B16細胞を用いるワクチン接種は予め形成された皮下黒色腫腫瘍を治療することができないことを示した。一方、予め形成された皮下黒色腫腫瘍はαGal(+)B16ワクチンを用いるワクチン接種により成功裏に治療することができる。
【0127】
αGal(+)B16ワクチンを用いて処置した、予め形成された皮下腫瘍をもつマウスの生存分析
予め形成されたαGal(-)B16皮下腫瘍をもち、αGal(+)B16刺激した細胞をワクチン接種したマウスは、非ワクチン接種対照およびαGal(-)模擬(mock)ワクチン接種したグループと比較すると、生存の延長を示した(図19)。非ワクチン接種対照およびαGal(-)模擬(mock)ワクチン接種動物ではそれぞれ9マウスのうちのゼロマウスおよび20マウスのうちの1マウスだけが皮下チャレンジで生き残ったが、αGal発現B16細胞で処置した19マウスのうち5マウスがチャレンジ後70日より長く生存した。非ワクチンおよび模擬(mock)ワクチングループのメジアン生存時間はそれぞれ27および26日であった。これに反して、αGal(+)B16ワクチンを受けたマウスのメジアン生存時間は有意に増加した(39日)。この結果は、αGal(+)B16ワクチン細胞が予め形成された皮下黒色腫腫瘍を有効に治療できることを、生存動物数の増加(26%対5%)およびメジアン生存時間の延長(39対26日)により実証する。
【0128】
第2の実験においては、マウスは3用量の細胞ワクチンを、非照射αGal(-)B16による最初の皮下注射後、4、11および18日に受けた(図20)。それぞれの時間のワクチン用量は3x105細胞であった。マウスに非ワクチン接種(n=12)、αGal(-)B16によるワクチン接種(n=23)またはαGal(+)B16(n=26)によるワクチン接種のいずれかを実施した。
【0129】
図20は観察の70日後のKaplan-Meier生存分析を示す。生存曲線のログランク(logrank)検定比較は、対照非ワクチン接種およびαGal(-)B16ワクチン接種マウスと比較して、αGal(+)B16ワクチンを用いて処置した皮下黒色腫腫瘍を保持する生存マウスの数に有意差を示した(p<0.005)。非ワクチン接種の12マウスのうちB16による皮下注射で生き残ったものはなかった。同様に、αGal(-)B16ワクチンをワクチン接種した23マウスのうちB16による皮下注射で生き残ったものはなかった。これに反して、αGal(+)B16ワクチンを受けた26マウス(42%)のうち11マウスは、αGal(-)B16黒色腫の致死的皮下注射後、70日間生き残った。
【0130】
対照と模擬(mock)ワクチン接種したマウスのメジアン生存に有意差はなかった(それぞれ38および42日、ログランク(logrank)検定でp=0.43、ns)。これに反して、αGal(+)B16で処置したマウスのメジアン生存は、60日より長かった。これはαGalワクチン接種グループのメジアン生存の有意な増加を表した(p<0.005)。
【0131】
この結果はさらに、αGal(+)B16ワクチンによる予め形成された皮下黒色腫腫瘍の治療は、αGal(-)ワクチン接種または無処置と比較して有意に有効であることを実証した。
【0132】
実施例8
αGal(+)B16細胞ワクチンによる肺黒色腫転移の治療
本発明者らはさらに、播種性疾患のモデルとして肺黒色腫転移の治療における有効性を評価した。この実験は、腫瘍を保持する患者が外科的に除去できない播種性疾患によりほとんど死亡しうるので、臨床の視点から非常に重要である。そこで、RRBC免疫感作マウスに105非照射αGal(-)B16を静脈内にチャレンジして、無作為化した。マウスに照射B16/NeoRワクチン(αGal(-)、n=6)またはαGal(+)B16ワクチン(n=7)を皮下に処置した。ワクチン(2x105照射細胞)は、B16のi.v.注射後、4、11および21日に、皮下投与した。マウスをチャレンジ後30日に犠牲にして、黒色腫転移の数を数えた(図22)。肺転移の数は、3マウスで「数え切れないほど多数」(任意の値>250腫瘍)であり、他のマウスでは30腫瘍を数える一方、2マウスだけは無腫瘍であった。さらに、このグループの動物の1つは腹腔内に3つのさらなる転移性小結節を示し、該疾患の肺以外の他の場所への播種を実証した。
【0133】
これに反して、αGal(+)B16ワクチンを用いて処置したマウスは、全て無腫瘍であり、予め形成された腫瘍がαGal(+)を発現するワクチン療法により非常に成功裏に治療されたことを実証した。
【0134】
第2の独立した実験(図23)においては非照射αGal(-)B16の5倍増加用量上昇を用いた。マウスに5x105生B16をi.v.注射して肺黒色腫転移を予め確立した。4日後に、対照およびαGal(+)B16細胞を用いるワクチン接種を先の通り実施した。マウスは非ワクチン処置(n=10)、αGal(-)B16によるワクチン接種(n=11)またはαGal(+)B16(n=11)の3用量によるワクチンのいずれかを受けた。
【0135】
B16によるi.v.チャレンジ後30日に、マウスを安楽死させて肺黒色腫転移を数えた(図23)。腫瘍増殖も肺腫瘍負荷の計数によりおよび肺組織重量測定により評価した(図24)。
【0136】
多数の肺黒色腫転移が、非ワクチン接種マウスならびに対照ワクチン接種グループの肺に見出された。統計学的比較を実施するため、肺組織の重量を用いた。対照と非ワクチン接種グループの肺負荷の間の差は統計学的に差がなかった(アンペアードt検定、p=0.66, ns)。これに反して、αGal発現B16ワクチンによりワクチン接種したマウスには肺負荷の有意な低下が観察された(ワンウエイANOVA、p<0.006)。
【0137】
第1実験の観察と同様に、対照および非ワクチン接種グループからのいくつかのマウスは「数え切れないほど多数の」黒色腫転移を有した。さらに、2動物は、肺組織の他に、肺外に散乱した黒色腫腫瘍を有し、播種性疾患であることを示した。αGal(+)B16ワクチン接種したマウスは、「数え切れないほど多数の」黒色腫腫瘍を有するものはなく、肺外の腫瘍も有しなかった。これは、αGal(+)B16ワクチンを用いるワクチン接種が播種性転移性黒色腫を効果的に治療できることを示す。さらに、αGal(+)B16ワクチンによるワクチン接種は全身疾患のさらなる拡大を予防することができる。
【0138】
実施例9
αGal(+)B16細胞によるワクチン接種後の、αGal(-)B16特異的T細胞前駆体の誘導を実証するためのT細胞研究
この技術の基本的疑問の1つは、αGal(+)細胞を用いるワクチン接種がαGal(-)腫瘍に対して反応しうるT細胞が媒介する免疫を誘導できるかということである。上記の実験において、本発明者らは、αGal(+)B16ワクチンが、予め形成されたαGal(-)腫瘍のクリアランスを媒介しうる強い免疫を誘導することを実証した。この免疫は、マウスをαGal(-)B16ワクチンを用いてワクチン接種したときには誘導されない。さらに、αGal(-)B16腫瘍に特異的なT細胞前駆体がαGal(+)B16ワクチンを用いるワクチン接種後に誘導されたことを実証するために、in vivo T細胞研究を実施した。これらの実験の目的は、T細胞によるαGal(-)B16細胞の特異的認識を実証することにあった。T細胞を対照ワクチンαGal(-)B16を用いてワクチン接種したマウスグループまたはαGal(+)B16照射細胞を用いてワクチン接種したマウスから収穫した(図25)。2つのタイプの研究を実施して、異なる方法により、同じ結論、すなわち、照射αGal(+)B16細胞を用いてワクチン接種したマウスはαGal(-)B16腫瘍細胞を特異的に認識しうるT細胞前駆体の増加した数を有することを実証した。
【0139】
実験の第1セットにおいては、細胞内サイトカインTNF-αをFACSにより検出した。細胞を照射αGal(-)B16細胞を用いてまたはαGal(+)B16細胞を用いてワクチン接種したマウスから、最後の皮下ワクチン接種後2週に収穫した。脾細胞をネガティブ対照として刺激を与えずに培養した。刺激については、脾細胞をネガティブ対照としてCA320M(αGal(-)同系H-2 b/bハプロタイプ)とともに、またはαGal(-)B16細胞とともに共培養した。6時間の刺激後に、細胞を収穫して細胞内TNFαを染色した。TNFα(+)リンパ球のパーセント増加を、特異的にαGal(-)B16細胞を認識するαGal(+)B16細胞を用いてワクチン接種したマウスの脾臓に検出した(図26)。これらのT細胞は、CA320Mととも培養すると、TNFαを産生せず、これらはB16を特異的に認識するが、無関係な同系培養細胞株を認識しないことを示した。黒色腫に特異的T細胞前駆体の数の増加に加えて、これらの細胞が産生するTNFαの量は、FACSにより検出した平均蛍光強度により測定すると、有意に増加した(図26)。TNFα(+)細胞におけるMFIの4倍増加を検出した。模擬(mock)ワクチン接種マウスからの脾細胞をαGal(-)B16とともに培養すると、TNFα(+)細胞の4%しか検出されず、MFIは17であった。これに反して、αGal(+)B16細胞ワクチンを受けたマウスからのT細胞をαGal(-)B16とともに培養すると、7%のTNFα(+)細胞が検出され、MFIは69であった。これは、脾臓中に存在するT細胞前駆体のパーセントが2倍増加したことを表す。この実験を全3回繰返して同様な結果を得た。
【0140】
この結果は、αGal(+)B16細胞を用いるワクチン接種は黒色腫標的のT細胞特異的認識によりin vitroで検出された強いT細胞免疫を誘導し、そしてαGal(-)B16を用いるワクチン接種はこれを誘導しないことを実証した。
【0141】
異なるセットの実験においては、細胞表面活性化マーカーを用いてαGal(-)B16黒色腫培養細胞株の特異的T細胞認識を測定した。T細胞受容体(TCR)が関与すると、T細胞が複数の細胞表面分子をアップレギュレートしてリンパ球の活性化状態を示すことはよく記載されている。これらの分子の1つはIL-2受容体α鎖またはCD25である。TCRが関与すると、CD25がアップレギュレートされて、活性化後1日で、FACSにより検出されうる。同様に、T細胞が活性化するとCD69(または極初期活性化抗原(VEA))がアップレギュレートされる。CD69はシグナル伝達受容体として色々な細胞において機能し、リンパ球活性化の初期事象に関わり、色々なサイトカイン、およびその受容体の合成を誘導することによるT細胞活性化に寄与する。両方の活性化マーカー(CD25およびCD69)は休止T細胞では非常に低レベルでしか発現されない。αGal(+)B16細胞を用いるワクチン接種がαGal(-)B16を特異的に認識することができるT細胞前駆体を誘導したことを実証するために、活性化マーカーのアップレギュレーションを、認識および活性化を測定するパラメーターとして用いた。細胞をαGal(-)B16を用いてワクチン接種した、またはαGal(+)B16を用いてワクチン接種したマウスから収穫した。これらの細胞を無刺激でまたはネガティブ対照培養細胞株(CA320M)を用いてまたはαGal(-)B16を用いて培養した。培養の24時間後、細胞を収穫し、染色してCD25またはCD69を検出した。取得は、生命染色(vital staining)7-AAD(live細胞)を排除する細胞をゲーティングして実施した。予想されるように、休止T細胞(無刺激)および同系非黒色腫培養細胞株CA320Mにより刺激した細胞は非常に低レベルの活性化マーカーしか発現しない(図27aおよび27b)。αGal(-)B16を受けたマウスからの脾細胞をB16とともに培養すると、活性化マーカーのいくらかのアップレギュレーションが観察された。これはマウスが天然αGal(-)B16ワクチンを受けると低い程度の免疫反応性が観察されうることを示した文献からの従来の報告を実証する。しかし、この反応性は予め形成された黒色腫腫瘍を予防および/または治療するのには十分でない。他方、T細胞をαGal(-)B16を用いて培養したとき、αGal(+)B16をワクチン接種したマウスからリンパ球活性化の増加が検出され、CD25(+)およびCD69(+)細胞の数の増加が測定された。
【0142】
この結果も再び、αGal(+)B16細胞を用いるワクチン接種は、αGal(-)B16黒色腫細胞を特異的に認識できるT細胞前駆体を誘導したことを実証した。
【0143】
実施例10
αGal(+)またはαGal(-)B16細胞ワクチンを用いてワクチン接種したマウスからの養子T細胞導入による、予め形成された転移性黒色腫腫瘍の治療
上記のin vitro実験は、αGal(-)B16ワクチン接種を受けたマウスと比較すると、αGal(+)B16細胞を用いてワクチン接種したマウスではより多くの量と質の黒色腫特異的T細胞が誘導されることを実証した。これらの黒色腫特異的T細胞は数が増加し(脾細胞中により多くのT細胞が見出された)、それらはより多くのTNFαを産生した。また、B16を用いて共培養するとより多くの脾細胞が活性化された(CD25およびCD69のアップレギュレーション)。以上に示した実験において、皮下および肺性転移の両方を保持してαGal(+)B16を受けたマウスは、より長い生存と増加した肺腫瘍のクリアランスを示した。これらの2グループのデータがあるので、本発明者らは事実としてαGal(+)B16ワクチン接種により誘導されたT細胞が予め形成された黒色腫腫瘍の治療の原因であると推論することができよう。しかし、大量の黒色腫特異的T細胞は、それらが寛容状態(tolerant state)にあるので予め形成された皮下黒色腫腫瘍を治療するのには不十分であることが示されており、この推論が正しいのかは明らかではない(Overwijkら, 「自己反応性CD8+ T細胞の機能的寛容状態の逆転後の腫瘍退行と自己免疫(Tumor regression and autoimmunity after reversal of functionally tolerant state of self-reactive CD8+ T cells)」 J. Exp. Med. (2003) 198:569-580)。本発明者らは、αGal(+)B16細胞を用いるワクチン接種は記憶を呼び戻すと急速に活性化されうる細胞の媒介する強い免疫を誘導し、これが予め形成された疾患を保持するマウスにおける腫瘍クリアランスの原因であるという仮説を立てた。この仮説を実証するために、養子細胞導入実験を実施した(図28)。ドナーマウスを、先に記載したように、照射αGal(+)B16またはαGal(-)B16ワクチンの3用量を受けるワクチン接種を実施した。レシピエントマウスに生αGal(-)B16をi.v.注射して肺黒色腫転移を確立し、そして無作為化した。非照射B16のi.v.注射後4日にマウスは、αGal(+)またはαGal(-)B16細胞をワクチン接種したドナーからのT細胞を受けるかまたは受けなかった。4週後、肺黒色腫転移負荷を、肺腫瘍を数えることにより、およびブロックで得た肺の重量を測ることにより測定した。実験を2回実施し、両者の結果を図29Aおよび29Bに示した。実験#1は、T細胞療法を施さないB16をi.v.に受けたマウス(n=16)、αGal(-)B16ワクチン接種したマウスからのT細胞を受けたマウス(n=15)、およびαGal(+)B16ワクチン接種したマウスからのT細胞を受けたマウス(n=17)の平均肺重量を示す。バーは肺重量の平均および誤差バー、SEMを表す。対照マウスにおけるおよび模擬(mock)ワクチン接種したマウスにおける大腫瘍および肺負荷の有意な増加を実証した。T細胞を受けないマウス(対照)と模擬(mock)ワクチン接種したマウスからのT細胞を受けたマウスの間の肺黒色腫転移の差は統計学的に有意でなかった(p>0.05)。これに反して、肺黒色腫負荷の有意な低下がαGal(+)B16ワクチン接種したマウスからT細胞を受けたマウスに観察された(ワンウエイANOVA、p<0.006)。同様な結果が第2の独立実験において観察された。T細胞を受けないマウスは64肺黒色腫腫瘍の平均値を有する(n=7)。αGal(+)B16ワクチン接種したマウスからの細胞を受けたマウスは多数の肺黒色腫転移を有した(平均=90、n=10、p=>0.05)。対照的に、αGal(+)B16細胞を用いてワクチン接種したマウスからの脾細胞を受けたマウスには31肺黒色腫腫瘍しか観察されなかった。これは、αGal(+)B16細胞を用いてワクチン接種したマウスからのT細胞を受けたマウスの肺黒色腫負荷における有意な低下を表す(アンペアードt検定p<0.05)。
【0144】
αGal(+)ワクチン接種したマウスからの細胞の唯一の治療による、予め形成された肺黒色腫転移低下の著しい成功は、始めて、強い細胞が媒介する免疫がαGal(+)細胞ワクチンによって誘導され、そしてαGal(-)ワクチンによって誘導されないことを実証するものである。この強い細胞依存性腫瘍免疫は、ほとんど間違いなく、播種性の予め形成された疾患の治療の原因である。
【0145】
実施例11
非照射αGal(-)CA320M細胞の致死用量の皮下注射後の、αGal(-)またはαGal(+)照射CA320M肉腫細胞を用いてワクチン接種したαGal(-)ノックアウトマウスの生存
上記B16黒色腫細胞を用いる予防ワクチン接種実験を、αGal(-)ノックアウトマウスから誘導した、従って、宿主細胞と完全に同系である色々な腫瘍培養細胞株を用いて繰返した。この腫瘍培養細胞株はCA320Mであって、9,10-ジメチル-1,2-ベンズ-アントラセン(DMBA)2mgおよび3-メチルコラントレン(3-MC)1mgを溶解した250μlのオリーブ油を、αGTノックアウトマウス中に2週間隔で腹腔内注射することにより取得した。1マウスは、最初の注射後8ヶ月に腫瘍が現れた。腫瘍はほぼ2cm幅、3cm長さそして1.5cm深さの大きな塊で腹腔内に局在した。腸間膜に位置する転移性小結節が認められた。原発塊(primary mass)と腸管を採取し、培養して組織病理学試験を実施した。腫瘍の原発部位に随伴する腸間膜から採取した転移性小結節の培養に成功し、CA320Mと名付けた。原発および移植腫瘍の凍結およびパラフィンセクションの両方の組織病理学試験は、それぞれ、分化の低い肉腫と一致する形態であることを実証した。ヘマトキシリンおよびエオシン染色ならびに免疫組織化学分析は、CA320Mが小腸の肉腫のような消化管間質腫瘍のファミリーに分類されうることを示唆した。αGT KOマウスに誘導されたCA320M細胞はαGalエピトープを特異的に検出するIB4イソレクチンと結合せず、このマウス腫瘍は、予想通り、機能性αGT酵素を欠き、それ故にαGalエピトープを欠くことを立証する。CA320M細胞はHSV-1に基づくベクターによる感染に感受性があり、HE7αGal1による形質導入後にαGalエピトープを発現した。
【0146】
GT KOマウスを、14日間隔で2回の2x107ウサギ全血球の皮下注射により初回刺激し、αGalエピトープに対する免疫を発生させた。CA320M細胞に、10 MOIのHDKgalまたはHDKgalΔsalIのいずれかを用いて8時間形質導入して、CA320MαGal(+)またはαGal(-)細胞をそれぞれを得た。簡単に説明すると、HDKgalはαGT遺伝子をKozak配列とともにpHD1アンプリコンHSV-1ベクター中に挿入することにより発生させた。従って、pHDKgalは、CMVプロモーターの制御下にある単一の真核生物遺伝子、αGTを保持する。突然変異αGT遺伝子を構築するために、αGT酵素の対応する触媒ドメインに配置したユニークなSalI制限酵素切断部位を切断し、クレノー(Klenow)およびdNTPを用いて満たした。得られるフレームシフト突然変異は酵素を非機能化するポリペプチドの未熟な末端を生じる。突然変異αGT遺伝子を、pHD1アンプリコン中にもクローニングし、両方のアンプリコンを、ヘルパーを含まないヘルペスウイルス系を用いて感染性HSV中にパッケージングした。形質導入したCA320M細胞を照射し(25Gy)、1x103細胞を15動物中に注射した。21日後、動物に1x107生CA320M細胞をチャレンジし、その後、腫瘍増殖および生存分析を行った。αGal(+) CA320Mワクチン接種した動物の93パーセントが、突然変異αGal(-)CA320M(33%)およびゼロ(38%)対照と比較して、致死量の腫瘍チャレンジで、60日まで生き残った(>400mm3腫瘍)(図30)。この結果は、αGal(+)同系細胞を用いたワクチン接種が、対応するαGal(-)細胞に対する保護免疫応答を誘導できることを立証する。
【0147】
全体的に考えて、全てのこれらの結果は、始めて、細胞全体ワクチン中のα-ガラクトシルエピトープの存在がαGalエピトープの発現を欠く予め形成された腫瘍を治療するT細胞の媒介する免疫を誘導する強い免疫アジュバントであることを実証するものである。これらの結果はまた、このワクチンの臨床での利用がヒトの医療に大きなインパクトを与えうることを示す。さらに、癌患者に対する予防ワクチンは現在可能でなく、従って、本明細書に記載の技術のような治療手法は迅速に応用する現実的価値がありかつ癌患者に利益をもたらす可能性を有する。
【0148】
実施例12
ヒト細胞全体癌ワクチン(HyperAcute(商標))の開発
HyperAcute(商標)細胞全体癌ワクチンは、マウスα(1,3)ガラクトシルトランスフェラーゼ遺伝子を発現するように遺伝子操作した同種異系(allogeneic)癌培養細胞株から成る。複数の腫瘍組織型由来の複数の独立した癌培養細胞株を遺伝子操作してそれらの表面上にαGalエピトープを発現させている。ある治療用細胞全体癌ワクチンは、同じ腫瘍組織型に属する複数の遺伝子操作した癌細胞型の、照射した個々の培養細胞株または混合物の注射剤から成る。表1は、HyperAcute(商標)細胞全体癌ワクチンを作り出すために用いた癌培養細胞株を示す。
【表1】

【0149】
HyperAcute(商標)細胞全体癌ワクチンは、表1に示した培養細胞株の、プロタミン硫酸10g/mLの存在で293Z.CKGからのレトロウイルス上清を用いた形質導入により確立した。形質導入した細胞の集団をαGal細胞表面エピトープについて0-2605抗αGal抗体(NewLink)を用いて染色した。細胞表面上のαGalエピトープ発現強度が最高である5%の細胞集団を、FACSソーターを用いて別々の亜集団にソート(sort)した。ソートされた最高のαGalエピトープ発現をもつ形質導入細胞の亜集団が、表1に記載されたものである。これらの細胞を拡大して(分割比1:5)、マスター細胞バンク(MCB)を作製した。増殖パターン、形態学的外見、およびαGalエピトープ発現の平均強度をモニターしたが、培養の全増殖期間にわたって安定であった。MCBは、細胞をフラスコ中で37℃、5%±1%CO2にて拡大することにより開発した。培地は10%胎児ウシ血清(FBS)および2mmのL-グルタミンを補充したRPMI-1640であった。各継代において細胞をトリプシン処理し、数え、そしてその生存をトリパンブルー排除により評価した。細胞を増殖して10億個の細胞とし、収穫し、プールし、100個の凍結用バイアル(cryovial)中に分配し、そしてプログラム速度凍結室(programmed rate freezing chamber)を用いて凍結した。細胞を液体窒素貯蔵タンクの蒸気相に貯蔵する。各培養細胞株に対する作業用細胞バンク(WCB)をMCBから作製した。WCBの拡大、収穫、凍結および貯蔵の条件はMCBについて記載した通りである。それぞれのHyperAcute(商標)培養癌細胞株の生産ロットはそれぞれのWCBに由来する。生産ロットからの細胞が十分な密度に到達すると、培養液(上清)を収穫し、濾過し、そして無菌容器中にプールする。プールを十分に混合し、次いで無菌状態で標識した無菌のプラスチックボトルに充填する(標識には、製品名、ロット番号および充填日が記載される)。満たしたボトルを凍結して-60℃以下で貯蔵する。安全試験用のアリコートをとる。生産ロットに対する細胞をトリプシン処理により収穫し、プールし、そして完全培地に再懸濁させる。プールした細胞を150〜200Greyで照射する。細胞を、Varian
Clinic 2100C医療用直線加速器(medical linear accelerator)を6MV光子モードで用いて照射する。機械の照射出力は、AAPM TG-51較正プロトコル(calibration protocol)を用いて水中で較正する。出力一貫性は毎日;出力較正は毎月、NIST較正トレーサブルイオンチャンバー/エレクトロメーター線量計を用いてモニターする。照射細胞を遠心分離し、そして5%グリセロールおよびヒト血清アルブミンから構成される注射用最終剤形に再懸濁する。次いで0.4mlの細胞を、用量レベルに対応して1x106/0.2ml、3.5x106/0.2ml、1x107/0.2mlおよび3.5x107細胞/0.2mlの濃度で無菌の凍結用バイアル中に分配する。HyperAcute(商標)細胞MCB、WCB、およびPL品質管理試験は細胞および上清の両方を実施する。HyperAcute(商標)癌ワクチンMCB、WCBおよびPL細胞の許容判定基準は、ヌードマウスにおける腫瘍原性に対するアッセイから成る。
【0150】
実施例13
ヒト患者に対する投与量レベルと投与スケジュール
次のデータは投与量レベルとワクチン接種スケジュールの提案を支持するものである。マウスについての前臨床毒性学的研究は、α-gal発現同種異系および同系腫瘍は、70kgヒト1人当たり3.5x109細胞と等価である1マウス当たり1x106細胞までの十分な耐性を有することを示している。細胞表面上にα-galエピトープを自然発現するマウスベクター産生細胞(MVPCもしくはマウスVPC)についてのフェーズI研究(第2.1節を参照)は、マウスVPCが患者1人当たり7x109細胞の投与量に十分な耐性を有することを示している。フェーズIIマウスVPC試験において、患者は各サイクルに7x109細胞の注射を受ける3サイクルの治療を受けた。患者はこの治療に十分に耐性があった。マウスVPC注射を6週の間隔で繰返した。提案した投与レベルはマウスのα-galワクチンの十分に耐えられる投与量の範囲内にある(患者1人当たり最大4x108細胞)。ワクチン注射の間の4週の間隔は、治療の毒性の評価および免疫応答の成熟を評価するのに十分な時間でありうる。フェーズIマウスVPC研究からのデータは、抗αGal免疫応答は、αGal発現細胞注射後14〜21日の間にその最高値に到達することを示唆する。
【0151】
実施例14
マウスαGT遺伝子を保持するレトロウイルスベクターを用いて形質導入した同系腫瘍ワクチンを用いる毒性学研究
最初の研究では、マウスをαGal(+)B16黒色腫細胞を用いて1マウス当たり1x105細胞の用量で皮下注射した。αGal(+)生存黒色腫細胞を拒絶した動物にはαGal(-)B16細胞を再チャレンジしてαGal(+)黒色腫細胞の拒絶が腫瘍免疫を誘導することを立証した。全てのマウスがB16による第2チャレンジに生き残った。これらの黒色腫保護された動物(第1実験でn=8、および第2実験でn=9)について6ヶ月間、長期毒性研究を実施した。毒性学的研究についての組織学的、血液学的、臨床的観察を保護されたマウスのいくつかについて実施した。組織病理学的試験は、主な潅流器官:腎臓、脾臓および肝臓、ならびに潜在的標的組織、皮膚および乳腺を含んだ。安全を評価する組織学的研究は、試験した全てのサンプル組織(皮膚、乳腺、腎臓、脾臓および肝臓)に注目すべき病変を示さなかった。複数の動物(対照マウスを含む)は炎症性細胞の僅かな浸潤を伴う腎脈管周囲炎を示した。試験グループのこの観察頻度は対照グループの動物の頻度と同様であった。血液学研究結果は、全ての研究値が正常値の範囲内にあることを示した。正常値は、挙動を含むナイーブ無遺伝子操作α-gal KOマウス臨床観察から考察された結果であり、可能性のある二次的副作用としての自己免疫色素脱失(白斑)または柔皮変化の発生は、黒色腫保護されたマウス(n=17)に研究期間中観察されなかった。
【0152】
第2研究においては、マウスに同種異系αGal(+)EMT-6乳房細胞を1マウス当たり1x106細胞の用量で皮下注射した。ワクチン接種後、2、4および6週に、血液および組織サンプルを取得して毒性学的研究を実施した。組織学的および血液学液研究を実施した。組織病理学的試験は、主な潅流器官:腎臓、脾臓および肝臓、ならびに潜在的標的組織、皮膚および乳腺を含んだ。安全を評価する組織学的研究は、試験した全てのサンプル組織(皮膚、乳腺、腎臓、脾臓および肝臓)に注目すべき病変を示さなかった。複数の動物(対照マウスを含む)は炎症性細胞の僅かな浸潤を伴う腎脈管周囲炎を示した。試験グループのこの観察頻度は対照グループの動物の頻度と同様であった。血液学研究結果は、全ての研究値が正常値の範囲内にあることを示した。正常値は、挙動を含むナイーブ無遺伝子操作α-gal KOマウス臨床観察から考察された結果であった。一過性好酸球増加症が同種異系ワクチン接種後、2週に観察された。毒性学研究に用いた最大用量は1x106細胞/マウスであった。この用量の用量/kgに基づいてヒトにスケールアップすると、マウスを20グラムと仮定して、用量/マウスが70kgヒトにおいて3.5x109細胞と等価になる。また、ヒトの平均寿命の70年と比較してマウスの平均寿命がほぼ2年であることも考慮すると、長期毒性学研究(6ヶ月)はマウス寿命の4分の1と等価であり、ほぼヒトの17年にあたる。これらの研究は、16週の治療期間全体にわたる1患者当たり4x108の現行投与量スケジュールを支持する。
【0153】
実施例15
HyperAcute(商標)癌ワクチンの調製および患者への投与スケジュール
この方法は、細胞全体癌ワクチン(本発明の医薬組成物)を調製する方法およびヒト患者へ投与する方法を記載する。細胞は、それらを調製した直後に、患者に注射しなければならない。投与される細胞は凍結前に致死的に照射されているので、特別な安全上の注意事項は不要である。最初に各ワクチン培養細胞株の凍結用バイアルを液体窒素容器から取り出す。次いで、全てのバイアルを、37℃の水浴に凍結内容レベルを超えるまで浸漬して同時に解凍する。バイアルが解凍されると直ぐに、それらの表面を70%アルコールを用いてリンス洗いする。ワクチンの各培養細胞株成分による等量のバイアルの内容物を注射用の1シリンジ中に一緒にして、直ぐ、患者中に注射する。ワクチン細胞を25ゲージ針を備えたツベルクリンシリンジを用いて皮内に(i.d.)注射しうる。注射は腕および脚に回転ベース上で行うべきである。HABワクチン細胞を1、29、57、および85日に投与しうる。それぞれの注射後2時間、外来患者診療所において看護スタッフにより患者をモニターしうる。患者モニタリングは:ワクチンの投与前30分間以内の温度(T)、脈拍(P)、血圧(BP)および呼吸数(R)を含み、ならびに、次いでワクチン接種後、毎15分間x4回、次いで毎30分間x2回のチェックによるべきである。温度は診療所から退所する前にチェックしうる。さらに、患者を、局所または播種性皮膚発疹および他の副作用を含む急性反応症状についてモニターしうる。グレードII以上の急性副作用を経験した患者は、さらに1〜2時間診療所において、その症状がグレードII未満に回復するまでモニターしてもよい。観察および/または治療にも関わらず、もしグレードII以上が4時間を越えて続くのであれば、観察を続けるか、治療を調査(institute)または改変するか、または患者を入院させるかどうかの決定をする。
【0154】
実施例16
最大耐量の決定
治療は適格な3患者の投与コホートで進めうる。最大耐量(MD;maximum tolerated dose)は、用量制限毒性(DLG;dose-limiting toxicity)が見られる投与コホートより低い投与コホートとして定義される。もしある投与コホートで患者の>33%(すなわち、1/3、または2/4-6)がDLTを訴えれば、MTDは決定されうるので、さらなる用量増加は許容されないであろう。もしある投与コホートにおいて3患者の1患者(1/3)でDLTが認められれば、そのコホートを拡大して全部で6患者となるまで拡大しうる。もしさらに1つのDLTが観察されれば、コホートを閉じて、MTDを定義してさらなる用量増加は許容されないであろう。もしさらなるDLTがそのコホートで観察されなければ、次のより高い投与コホートへの増加を開始しうる。さらなる患者を本研究のフェーズII部分でMTDにて治療しうる。
【0155】
ある投与コホートにおける最後の患者の開始と次のより高い投与コホートの開始の間には最小限4週の遅れがありうる。
【表2】
【0156】
本研究における応答と進行は、固体腫瘍の応答評価判定基準委員会(Response Evaluation Criteria in Solid Tumors (RECIST) Committee)の提案による新しい国際判定基準を用いて評価する。腫瘍病変の最大直径(一次元測定)だけの変化をRECIST判定基準では利用する。病変は、以下に記載する判定基準を用いて測定可能または測定不可能である。測定可能性の意味での用語「評価できる(evaluable)」は、さらなる意味または正確度を与えるものでないので、使用しないであろう。測定可能な病変は、慣用技術(CT、MRI、X線)を用いて>20mmのように、またはらせんCTスキャンを用いて>10mmのように、少なくとも1次元(記録すべき最長直径)で正確に測定できる病変として定義される。全ての主要測定はミリメートル(またはセンチメートルの10分の1分率)で記録しなければならない。小さい病変(慣用技術を用いて最長直径<20mmまたはらせんCTスキャンを用いて<10mm)を含む他の病変(または疾患部位)は測定不可能な疾患と考えられる。骨病変、軟髄膜疾患、腹水、胸膜または心嚢貯留液、リンパ管炎皮膚または肺、炎症性乳房疾患、腹腔塊(CTまたはMRIによりフォローされない)、および嚢胞性病変は全て測定不可能と考えられる。全ての関わる器官の代表として、最大1器官当たり5病変および全部で10病変までの全ての測定可能な病変は、標的病変として同定しかつ基線で記録しかつ測定すべきである。標的病変は、そのサイズ(最長直径をもつ病変)および正確な反復測定値(イメージング技術によりまたは臨床的のいずれか)の適性に基づいて選択すべきである。全ての標的病変に対する最長直径和(LD)を計算し、基線和LDとして報じる。基線和LDは、それにより目的腫瘍応答を特徴付ける意味で用いる。全ての他の病変(または疾患部位)は非標的病変と同定すべきであり、これらも基線において記録すべきである。非標的病変は、1器官当たり最大数または全ての関わる器官の全体を超える測定可能な病変、ならびに測定不可能な病変を含む。これらの病変の測定値は必要としないが、それぞれの存在または不在はフォローアップ全体を通して記入すべきである。全測定値を、ルーラーまたは好ましくはカリパーを用いて採取し、メートル表示で記録すべきである。全ての基線評価を、治療の開始まで、そして治療開始前2週間を超えないように可能な限り綿密に実施すべきである。先の照射領域に位置する腫瘍病変は測定可能と考えない。臨床病変は、それらが表面的(例えば、皮膚小節および明白なリンパ節)であるときだけ、測定可能と考えられる。皮膚病変の場合、病変のサイズを求めるルーラーを含む、デジタルカラー写真による記録を推奨する。胸部の病変の場合、X線は、明確に規定されかつ通気した肺に囲まれているときは測定可能な病変として受入れられる。しかし、CTが好ましい。慣用のコンピューター断層撮影(CT)および磁気共鳴イメージング(MRI)を、スライス厚みが10mm以下のカットで連続的に実施すべきである。らせんCTは5mm連続再構築アルゴリズムを用いて実施すべきである。これは、胸部、腹部、および骨盤の腫瘍に適用する。頭および首腫瘍ならびに四肢の腫瘍は特定のプロトコルを必要とする。研究の主な目的が対象の応答評価であるときは、超音波(US)を利用する。USは腫瘍病変を測定するために用いるべきでない。しかし、表面上の明白なリンパ節、皮下病変および甲状腺節(thyroid nodules)を臨床測定するための代替としては可能である。USはまた、通常、臨床試験により評価する表面の病変の完全な消滅を確認するためにも有用である。内視鏡検査および腹腔鏡検査は腫瘍評価がまだ十分にかつ広汎に実証されていない対象にとって有用である。この特定の筋道でのこれらの利用は完備した設備と高レベルの熟練者が必要であり、いくつかのセンターにおいてのみ利用可能である。従って、対象腫瘍応答のためのかかる技術の利用は、関係センターにおける実証目的に制限すべきである。しかし、かかる技術は、生検が得られるときは、完全な病理学的応答を確証するために有用でありうる。腫瘍マーカーだけを用いて乳房腫瘍応答を評価することはできない。稀な場合(例えば、既知の残余良性腫瘍が残留しうる、生殖細胞腫瘍などの腫瘍型における残余病変)には、細胞学と組織学を用いて部分的応答(PR)と完全応答(CR)の間の相違を識別することができる。
【0157】
測定可能な腫瘍が応答または安定な疾患に対する判定基準に会ったとき、治療中に現れるまたは悪化するいずれかの浸出液の新生物起源の細胞病理学的確証が、応答または安定な疾患(浸出液が治療に伴う影響であってもよい)および進行性疾患の間の相違を識別するために必須である。
【0158】
以上の記載から、本発明が少なくともその目的の全てを達成することを理解することができる。本明細書に引用した全ての参考文献は、本明細書にその全てが組込まれる。
【図面の簡単な説明】
【0159】
【図1】本発明によって使用しうるpLNC-KGプラスミドの描写である。
【図2A】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である。
【図2B】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である(図2Aの続き)。
【図2C】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である(図2Bの続き)。
【図3】ウサギ赤血球を用いた免疫感作による、α(1,3)-ガラクトシルトランスフェラーゼノックアウト(αGT KO)マウスにおける抗αGal抗体誘導の模式図である。抗αGal抗体(Ab)を誘導するために、マウスの腹腔内に108ウサギ赤血球(RRBC)を用いて2回、2週間隔てて注射した。最後の免疫感作の1週後に血液サンプルを得て、抗αGal抗体力価をELISAにより測定した。本研究に用いた全てのマウスは高い力価の抗αGal Abを発生した。
【図4】非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の生存試験を示す模式図である。雌性および雄性8〜14週齢H-2 b/bハプロタイプαGT KOマウスをこの研究で用いた。全マウスはRRBCを用いて図3に示したように免疫感作した。最後の免疫感作の1週後に、マウスは3種の培養細胞株:a)野生型B16.BL6黒色腫培養細胞株(αGal(-))、b)Neo-R遺伝子を発現するベクターを用いてレトロウイルスにより形質導入したB16細胞(B16.LNL、模擬(mock)対照αGal(-))、またはc)NeoR遺伝子とαGTの両方を発現するベクターを用いてレトロウイルスにより形質導入したB16細胞(B16.αGal(+)、pLNCKG)、の致死的皮下チャレンジ1x105を受けた。チャレンジ後、腫瘍を盲検方式で1週間当たり2回測定した。
【図5】αGTノックアウトマウスにおけるαGal(+)またはαGal(-)B16黒色腫細胞の致死的非照射皮下注射後の腫瘍サイズ(5a:チャレンジ後15日、5b:チャレンジ後30日)を示すグラフである。明白な皮下腫瘍をカリパーを用いて3垂直軸に沿って測定して容積を計算し、mm3で表した。図は、図4に記載したそれぞれのB16培養細胞株による皮下注射後15および30日における腫瘍サイズを示す。バーは平均および誤差バーSEMを表す。
【図6】αGT KOマウスにおいて、非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の腫瘍増殖動力学を示すグラフである。腫瘍サイズは図5のように野生型B16(αGal(-))、B16.LNL(対照αGal(-))およびαGal(+)B16によるチャレンジ後15、23および29日に測定した。
【図7】非照射αGal(-)またはαGal(+)B16黒色腫細胞の致死量を用いて注射したαGT KOマウスの生存分析を示すグラフである。図4に記載のように処置したマウスを90日間研究した。Kaplan-Meier生存分析および長ランク生存曲線比較を統計学ソフトウエアを用いて実施した。
【図8】非照射αGal(+)B16黒色腫細胞の皮下注射で生き残ったマウスに非照射αGal(-)B16黒色腫細胞の致死量を皮下注射した後の生存試験に関する実験設計を記述する模式図である。
【図9】ノックアウトマウスの生存分析を示すグラフである。図7からのαGal(+)B16細胞による第1チャレンジで生き残ったマウスを、続いて天然αGal(-)B16の第2皮下投与によりチャレンジした(図8)。Kaplan-Meier分析を、αGal(-)B16の注射後、60日間実施した。αGal(-)B16を皮下に受けたナイーブマウスを対照として用いた。
【図10】非照射αGal(+)細胞の致死量注射で生き残ったマウスにおける黒色腫特異的細胞傷害性T細胞の誘導を示す模式図である。図7からのαGal(+)B16細胞による第1チャレンジで生き残った2マウスから採集した脾細胞を、細胞傷害性Tリンパ球(CTL)をin vitroで作製するために利用した。αGal(+)B16の注射後90日に無腫瘍マウスから脾細胞を収穫し、そして脾細胞を照射αGal(-)B16細胞とともにIL-2不在のもとで5日間培養することにより、黒色腫特異的培養物を作製した。CTLを収穫し、特異的標的αGal(-)B16および非特異的同系培養細胞株大腸癌MC38およびT細胞リンパ腫EL-4に対して試験した。CTLの特異的および非特異的標的とのインキュベーションの4時間後に、培養上清におけるLDH放出を測定することにより、特異的細胞傷害性を決定した。
【図11】図10に記述した、非照射αGal(+)細胞の致死量の投与で生き残ったマウスにおけるB16黒色腫-特異的細胞傷害性T細胞の誘導結果を示すグラフである。
【図12】αGTノックアウトマウスにおいて、非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死量の静脈注射による播種性黒色腫転移モデルの実験設計を示す模式図である。雌性および雄性αGT KOマウスを、図3の通り、RRBCを用いて免疫感作して抗αGal Ab力価を増加した。最後の免疫感作後1週に、マウスの尾静脈にαGal(-)B16またはαGal(+)B16を静脈注射した。注射後3週に、肺を収穫して肺黒色腫転移を数えた。
【図13】図12の実験の統計学的結果を示すグラフである。
【図14】照射αGal(+)B16黒色腫細胞を用いたワクチン接種後のαGTノックアウトマウスにおける皮下αGal(-)黒色腫腫瘍発生の予防に対する実験設計を示す模式図である。細胞ワクチンは、先の実験に記載したB16由来の培養細胞株:すなわち、天然αGal(-)B16、対照ベクターを形質導入したαGal(-)B16.LNL、およびマウスαGT遺伝子をコードするベクターを形質導入したαGal(+)B16細胞を用いて調製した。細胞ワクチンをγ線照射(250 Gy)により調製して細胞増殖を防止し、使用するまで凍結媒質中で保存した。注射前に細胞ワクチンを解凍し、洗浄し、計数し、そしてハンクス溶液(HBSS;Hanks' Balanced Salt Solution)中に懸濁して皮下注射した。使用した全てのαGT KOマウスに、図3のようにRRBCを注射した。最後のRRBC注射後1週にマウスは第1用量の細胞ワクチンを受けた。2週後に細胞ワクチン接種を繰返した。それぞれのワクチンの用量は1マウス当たり106細胞を皮下投与した。細胞の細胞ワクチン後2週に、マウスに105非照射天然αGal(-)B16細胞を皮下注射し、そして腫瘍発生を週2回、90日間観察した。
【図15】図14に記載した実験のKaplan-Meier生存分析を示すグラフである。
【図16】TNF-αを認識するためのFACS分析結果を示す。αGal(-)B16黒色腫細胞のin vitro認識後に、照射αGal(+)B16細胞を用いてワクチン接種したマウスに誘導された、黒色腫特異的T細胞による細胞内TNF-αの検出。無腫瘍マウスからの脾細胞は、αGal(+)B16ワクチンを用いてワクチン接種したマウスから、αGal(-)B16黒色腫の注射後90日に収穫した(図15で生き残ったマウス)。αGal(-)B16のin vitro認識を測定するために、細胞内TNF-αをFACSにより検出した。T細胞を、6時間、サイトカインの分泌をブロックするブレフェルジンA(Brefeldin A)による刺激の存在または非存在のもとで培養した。活性化を最大限にするためにPMA/Ca++イオノフォアを用いた。細胞を特異的認識を測定するために105照射B16細胞とともに、または非黒色腫同系ネガティブ対照培養細胞株として105 CA320Mとともに培養した。インキュベーション後、細胞を収穫し、透過化処理し、固定し、そしてPE標識した抗TNF-αモノクローナルAbを用いて細胞内TNF-αを染色した。細胞をCoulterフローサイトメーターを用いて分析し、少なくとも10.000ゲーテッド(gated)リンパ球を取得した。
【図17】照射黒色腫細胞を用いるワクチン接種を介する予め形成された皮下腫瘍を治療処置する実験設計を示す模式図である。予め形成された皮下αGal(-)黒色腫腫瘍の、照射αGal(+)またはαGal(-)B16黒色腫細胞を用いたワクチン接種を介する治療処置。マウスに図3に説明したようにRRBCを注射した。最後のRRBC注射後1週、マウスに105非照射αGal(-)B16黒色腫細胞を皮下注射して無作為化した。実験#1においては、2用量の細胞ワクチンをαGal(-)B16の皮下注射後、3および6日に投与した。実験#2においては、マウスは生αGal(-)B16細胞の皮下注射後、4、11および18日に3用量の照射細胞ワクチンを受けた。これらの実験において、2種の細胞ワクチン、すなわち、対照ベクターを形質導入した照射αGal(-)B16、または照射αGal(+)B16細胞を用いた。
【図18】αGal(+)またはαGal(-)B16黒色腫細胞によるワクチン接種を受けた、予め形成されたαGal(-)皮下黒色腫腫瘍を有するマウスにおける腫瘍増殖動力学の2つの異なる実験の結果を示す。腫瘍サイズは、細胞ワクチンを受けたまたは受けないマウスにおいて早朝の時点に測定した。数字はそれぞれのグループにおける全マウスの腫瘍サイズの平均を表す。
【図19】照射αGal(+)またはαGal(-)B16黒色腫細胞による治療用ワクチン接種を受けた、予め形成された皮下αGal(-)B16黒色腫腫瘍を保持するマウスのKaplan-Meier生存分析を示すグラフである。αGal(-)B16を注射し、αGal(+)またはαGal(-)B16黒色腫ワクチンにより治療したまたはしなかったマウスを75日間生存について評価した(実験#1)。
【図20】照射αGal(+)またはαGal(-)B16黒色腫細胞による治療用ワクチン接種を受けた、予め形成された皮下αGal(-)B16黒色腫腫瘍を保持するマウスのKaplan-Meier生存分析を示すグラフである。αGal(-)B16を注射し、αGal(+)またはαGal(-)B16黒色腫ワクチンにより処置したまたはしなかったマウスを70日間生存について評価した(実験#2)。
【図21】予め形成された肺転移性腫瘍を治療する実験設計を示す模式図である。予め形成された播種性肺転移性αGal(-)黒色腫腫瘍の、照射αGal(+)またはαGal(-)B16黒色腫細胞を用いるワクチン接種を介する治療。マウスに、図3に説明したようにRRBCを注射した。最後のRRBCの注射後1週に、マウスに非照射αGal(-)B16黒色腫細胞を静脈内(i.v.)注射して無作為化した。これらのマウスに、続いてαGal(-)B16またはαGal(+)B16ワクチン細胞のいずれかを、播種性転移の確立後4、11および21日にワクチン接種した。第30日に動物を犠牲にして、その肺腫瘍負荷を評価した。
【図22】図21に示したプロトコルの実験1における腫瘍負荷を示すグラフである。実験#1において、マウスは105αGal(-)B16黒色腫細胞を受けた。非照射黒色腫細胞のi.v.注射後30日に、マウスを犠牲にして肺黒色腫転移を数えた。
【図23】予め形成された播種性転移性黒色腫を治療する第2の実験設計を示す模式図である。実験#2において、マウスは5x105αGal(-)B16黒色腫細胞を受けた。この後、マウスを皮下に黒色腫細胞ワクチンを用いて処理した。これらのマウスは、非照射αGal(-)B16のi.v.注射後4、11および21日に、対照ベクターを用いて形質導入した照射αGal(-)B16または照射αGal(+)B16細胞の2x105の3用量を受けた。
【図24】図23に記載した実験の結果を示すグラフであり、肺の平均重量(24A)としてまたは平均肺腫瘍負荷(24B)として表した。
【図25】αGal(+)またはαGal(-)B16細胞を用いるワクチン接種後の、αGal(-)B16黒色腫に特異的なT細胞免疫応答のin vitro誘導を実証する実験設計を示す模式図である。マウスに図3のようにRRBCを注射した。最後のRRBC注射後2週にマウスは、対照ベクターを用いて形質導入した照射αGal(-)B16細胞または照射αGal(+)B16細胞ワクチンの2x105の3回の皮下注射を受けた。最後の細胞ワクチン後2週に、脾細胞を収穫し、T細胞研究を実施した。αGal(-)B16の特異的認識を確認するために、細胞内TNF-αおよび活性化マーカーCD25およびCD69のアップレギュレーションを測定した。
【図26】B16抗原を特異的に認識するワクチン接種した動物(図25)のT細胞前駆体中のTNF-αの特異的誘導を示すグラフである。TNF-αを検出するために、T細胞を6時間、サイトカインの分泌をブロックするブレフェルジンA(Brefeldin A)による刺激の存在または非存在において培養した。最大刺激のために、PMA/Ca++イオノフォアをポジティブ対照として用いた。細胞を、特異的認識を測定するためにαGal(-)B16とともに、またはネガティブ対照としてCA320M(非黒色腫同系(H-2 b/b)小腸培養細胞株)とともに培養した。インキュベーションの後、細胞を収穫し、細胞内TNF-αを染色した。ポジティブ細胞を、少なくとも10.000ゲーテッドイベントの取得後の前方散乱プロット中のリンパ球のFACSゲーティングにより検出した。プロットはTNF-α(+)細胞の平均蛍光強度(MFI)を表す。
【図27】B16抗原を特異的に認識する、αGal(+)細胞を用いてワクチン接種した動物からのT細胞前駆体における活性化マーカーCD25およびCD69のアップレギュレーションを示すグラフである。測定は、図26に記載したのと同様の条件下で、ブレフェルジンA(Brefeldin A)の不在のもとでの培養の1日後に実施した。インキュベーション後に、細胞を収穫し、PE標識したモノクローナルAb抗CD-25または抗CD-69を用いて染色した。取得はCoulterフローサイトメーターを用いて実施した。プロットはポジティブCD25またはCD69細胞を示す。
【図28】照射αGal(+) またはαGal(-)B16細胞を用いてワクチン接種したドナーマウスからの養子T細胞導入による、特異的T細胞が媒介する免疫のαGal(-)B16黒色腫の予め形成された播種性転移に対する治療有効性をin vivo実証する実験の模式図を示す。ドナーマウスにRRBCを図3の通り注射した。最後のRRBC注射後2週に、マウスは対照ベクターを形質導入した照射αGal(-)B16または照射αGal(+)B16細胞ワクチンの2x105皮下注射を3回受けた。最後の細胞ワクチンの後に、脾細胞を収穫して性別の整合したレシピエントに導入した。細胞導入の4日前に、レシピエントに非照射αGal(-)B16をi.v.注射して肺黒色腫転移を確立し、そして無作為化した。T細胞導入後30日に、レシピエントを安楽死されて肺黒色腫転移負荷を、肺重量を測ることによりおよび黒色腫腫瘍を数えることにより決定した。
【図29】図28に記載した実験の結果を示す。バーは平均および誤差バー、SEMを表す。2つの独立実験を実施してそれらを示した。
【図30】αGal(+)またはαGal(-)照射CA320M肉腫細胞をワクチン接種したαGT KOマウスの、非照射αGal(-)CA320M肉腫細胞の致死用量を皮下注射した後の生存分析を示すグラフである。マウスに図3のようにRRBCを注射した。最後のRRBC注射後2週に、それらのマウスに、10 MOI HDKgalΔsalI(αGal(-)ベクター)またはHDKgal1(αGal(+)ベクター)を用いて形質導入したCA320Mの1x103を皮下にワクチン接種し、次いで25Gyを照射した。21日後、マウスに1x107非照射CA320M細胞を皮下注射した。対照のNullはワクチンを含まなかった。60日の観察期間の生存分析を実施した。
【技術分野】
【0001】
関係出願の相互参照
本出願は、2002年10月9日に出願した仮出願60/417,343の、米国特許法第119(e)条(35 U.S.C. §119(e))の規定による利益を主張する。
【0002】
発明の分野
本発明は、腫瘍細胞に対する体液性および細胞性免疫応答を刺激することにより癌を治療する方法および組成物に関する。特に、本発明は、補体が媒介する腫瘍細胞の破壊を刺激しかつ腫瘍特異的抗体産生と腫瘍特異的細胞傷害性細胞を同時刺激する方法に関する。
【背景技術】
【0003】
発明の背景
異種移植に対する大きな障害は、本質的に超急性異種移植片拒絶(HAR)を引き起こす外来組織中に存在する糖鎖エピトープの即時認識にある。この反応は再潅流すると直ぐ始まり、一旦始まると、外来組織を数分ないし数時間内に破壊する。或るドナー(donor)とレシピエント(recipient)の組合わせでHARが存在する一方、他のドナーとレシピエントの組合わせでは存在しないこの現象は、2つの主な因子、すなわち、a)レシピエントの異種反応性天然抗体が移植片中の抗原もしくは内皮細胞と結合すること、およびb)移植片の補体調節タンパク質とレシピエントの補体系が不適合であるために補体の無制御な活性化が容認されることに関係があると仮定されている。ヒト血清中の補体結合性天然抗体の1%超は、単構造Galα(1-3)Galβ(1,4)GlcNAc-Rを認識する。Galα(1-3)Galβ(1,4)GlcNAc-Rの合成は、酵素α(1,3)ガラクトシルトランスフェラーゼ(αGT)が触媒する。
【0004】
この酵素は様々な非霊長類哺乳動物由来の細胞のゴルジ体において次の反応:
Galβ(1,4)GlcNAc-R+UDP-Gal→Galα(1-3)Galβ(1,4)GlcNAc-R
によるα-ガラクトシル(αGal)エピトープの合成を触媒する。
【0005】
この酵素は新世界サルで活性を有するが旧世界サルおよびヒトでは活性がないことが見出されている。αGTのcDNAは、ウシおよびマウスcDNAライブラリーからクローニングされている。Larson, R. D.ら, (1989) 「マウスUDPガラクトース;.β-D-ガラクトシル-(1,4)-Nアセチル-D-グルコサミンα-(1,3)ガラクトシルトランスフェラーゼをコードするcDNAの単離:遺伝子導入による発現クローニング(Isolation of a cDNA Encoding Murine UDP galactose; .β-D-galactosyl-(1,4)-N Acetyl-D-Glucosamine α-(1,3) Galactosyl Transferase: Expression Cloning by Gene Transfer)」, PNAS, USA 86:8227;およびJoziasse, D. H.ら, (1989) 「ウシα-(1,3)ガラクトシルトランスフェラーゼ:cDNAクローンの単離と特徴付け、ヒトゲノムDNA中の相同的配列の同定(Bovine α-(1,3) Galactosyl Transferase: Isolation and Characterization of a cDNA Clone, Identification of Homologous Sequences in Human Genomic DNA)」, J. Biol Chem 264:14290。
【0006】
この遺伝子はヒトゲノム中に存在するが、転写は検出されていない。代わりに、2つのフレームシフト突然変異(未熟な停止コドンを作製する欠失)がこの酵素をコードするヒトエキソン中に見出された。一般的には、Galili, Uri 「ヒト天然抗α-ガラクトシルIgG(抗αGal)抗体の病態生理学の進展(Evolution in Pathophysiology of the Human Natural anti- α-Galactosyl IgG (anti- αGal) Antibody)」, Springer Semin. Immunopathol. (1993) 15:155-171を参照。
【0007】
全てのヒトに存在する天然抗体である抗αGalは、糖鎖エピトープGalα(1-3)Galβ(1,4)GlcNAc-R(αGalエピトープ)と特異的に相互作用する。この抗体は、哺乳類動物細胞が産生する他の既知の糖鎖エピトープとは相互作用しない(Galili, 1993, Springer Seminar Immunopathology 15:153)。抗αGalは循環するIgGのほぼ1%を構成して(Galiliら, 1984, J. Exp. Med. 160:1519)、IgAおよびIgMの形態でも見出される(Davineら, 1987, Kidney Int. 31:1132;Sandrinら, 1993, Proc. Natl. Acad. Sci. USA 90:11391)。これは1%の循環Bリンパ球により産生される(Galiliら, 1993, Blood 82:2485)。ヒトにおけるこの天然抗αGal Abの産生は、腸および肺細菌叢中に存在するαGal糖鎖残基の存在により常に刺激されている。ヒトでは、超急性異種移植拒絶において抗αGalがこのエピトープの存在と反応し、補体は速やかにかつ確実に数分〜数時間で外来組織の破壊をもたらす。
【0008】
本発明の目的は治療用癌ワクチンを開発することであり、それは、αGTをコードする遺伝子を腫瘍細胞中に導入してこのワクチン腫瘍細胞にαGalエピトープを付加し、天然抗αGal抗体によるワクチン細胞のオプソニン作用の増強を可能にし、かつ、腫瘍抗原提示を刺激して腫瘍特異的抗原に対する体液性および細胞性免疫応答を誘導することによる。
【0009】
本発明のさらなる目的は、αGTを発現しかつプロセシングして細胞上にαGalエピトープを作製する組換え細胞を含む上記医薬組成物を提供することである。
【0010】
本発明のさらなる目的は、増殖しかつ細胞性および体液性免疫応答を逃れる腫瘍、ウイルス、新生物細胞、または他の細胞を治療するための組成物および方法を提供することである。
【0011】
本発明の他の目的は以下の本発明の説明から明らかになるであろう。
【発明の開示】
【0012】
発明の概要
本発明は、腫瘍細胞を選択的にターゲティングして死滅させる免疫応答を引き起こす方法および組成物に関する。腫瘍細胞を、ex vivo遺伝子療法プロトコルを介して、αGalエピトープを発現するように遺伝子操作する。次いで細胞を死滅させ(γ線または紫外線照射、熱、ホルムアルデヒドなどにより)そして患者に投与する。該αGalエピトープは腫瘍細胞のオプソニン作用を誘発し、腫瘍細胞全体中に存在する抗原の腫瘍特異的抗原提示を亢進する。本発明の重要な特徴は、本発明の医薬組成物に細胞全体の利用を含むことである。これによって腫瘍細胞全体内に存在する腫瘍関連抗原のプロセシングが行われ、これらのタンパク質がαGalエピトープの付加による影響を受けているか否かには関わらない。αGal改変は、細胞表面上の複数の糖タンパク質および糖脂質に影響を与えるので、動物の免疫系は、腫瘍特異的抗原を検出し、プロセシングしそして上記抗原に対する抗体および細胞性免疫応答を産生する機会を増加しうる。このようにして動物の免疫系は、刺激されて腫瘍特異的抗体および免疫細胞を産生し、これらは、動物中に存在して遺伝子操作された細胞全体ワクチンにより与えられたものと共通の腫瘍関連抗原を保持するαGalネガティブ腫瘍細胞を攻撃しかつ死滅させうる。
【0013】
本発明によれば、医薬組成物は、発現するとマウスαGTをコードするポリヌクレオチド配列をex vivoで腫瘍細胞全体に導入することにより作製する。レシピエント腫瘍細胞は同系(syngenic)、同質異系(allogenic)、または自己(autologous)であってもよい。該配列はいずれかのヌクレオチド導入ビヒクルを介して導入し、上記ビヒクルはウイルスもしくは非ウイルスベクター、プラスミド、または活性ウイルス粒子を産生するベクター産生細胞であってもよい。これらの遺伝子導入ビヒクルは腫瘍細胞を形質転換し、挿入された外来遺伝子材料を上記細胞内に発現する。発現された遺伝子産物は、上記細胞上に存在する細胞表面糖タンパク質および糖脂質上のαGalエピトープ合成を触媒する。本発明は、予め存在する抗αGal抗体によるαGalエピトープの結合を最大化するための複数の細胞表面糖タンパク質をもつ腫瘍細胞全体の利用、こうしてこの複合体の抗原提示細胞上に存在するFc受容体との結合の亢進、およびこうして上記ワクチン腫瘍細胞に存在する複数の腫瘍に関連する抗原の抗原提示のトリガリング(triggering)を意図する。
【0014】
さらに好ましい実施形態においては、同じ組織タイプ、または癌タイプ由来の複数タイプの形質転換した細胞を投与して、異なるエピトープの数をさらに増加させて個体に存在する腫瘍細胞の完全な改善の確率を増加する。
【0015】
本発明は、医薬組成物および上記医薬組成物を作る方法を含んでなり、ここで上記組成物は弱毒腫瘍細胞混合物の混合物の治療上有効な量と担体を含んでなり、上記混合物は複数の細胞表面糖タンパク質を含んでなり、そして上記糖タンパク質はαGalエピトープを含んでなる。好ましい実施形態においては、細胞は細胞全体である。上記組成物を作る方法は、生腫瘍細胞のコレクションを取得し、発現するとαGalをコードするヌクレオチド配列を用いて上記細胞を形質転換してαGalが上記細胞の細胞表面糖タンパク質上に提示されるようにすることを含んでなる。次いで細胞を死滅させて、投与のための製薬担体と組合わせる。
【0016】
定義
本発明の組成物と方法に関係する様々な用語が、本明細書でこれまでに用いられたしまた明細書および請求の範囲の全体にわたっても用いられる。
【0017】
単位、接頭辞、および記号はそのSIで許容された型で記述することができる。特に断らない限り、核酸は左から右へ5'から3'方向に記し;アミノ酸配列は左から右へアミノからカルボキシ方向にそれぞれ記す。数字の範囲は範囲を規定する数字を含み、かつそれぞれの整数を規定された範囲内に含むものである。アミノ酸は、本明細書において、IUPAC-IUB生化学命名委員会(IUPAC-IUB Biochemical nomenclature Commission)が推奨する、一般的に知られる3文字記号または1文字記号により参照することができる。ヌクレオチドも同様に一般的に受入れられる1文字コードにより参照することができる。特に断りのない限り、本明細書に用いるソフトウエア、電気工学、および電子工学用語は新しい電気工学および電子工学用語の新IEEE標準辞書(New IEEE Standard Dictionary of Electrical and Electronics Terms)第5版、1993に定義されたものである。以下に定義される用語は、本明細書全体への参照によりさらに詳しく定義される。
【0018】
用語「α-(1,3)ガラクトシルトランスフェラーゼをコードする配列」、または「αGTをコードする配列」は、次の反応:
Galβ(1,4)GlcNAc-R+UDP-Gal→Galα(1-3)Galβ(1,4)GlcNAc-R
によりα-ガラクトシル(αGal)エピトープを生成するタンパク質をコードする、ポリヌクレオチド配列を意味する。
【0019】
この配列は、変異体、改変体、末端切断体など、ならびにマウス配列、ウシ配列または当業者に公知のおよびGenbank、他の開示物またはデータベースで利用しうるいずれかの他の供給源由来であって、上記反応の機能を保持する配列を含むことができる。典型的には、かかる配列は、本明細書に記載のマウスまたはウシαGT配列と少なくとも80%以上相同的でありうる。
【0020】
「増幅した」は、少なくとも1つの核酸配列をテンプレートとして用いる、核酸配列の多コピーまたは核酸配列と相補的な多コピーの構築を意味する。増幅系としては、ポリメラーゼ連鎖反応(PCR)系、リガーゼ連鎖反応(LCR)系、核酸配列に基づく増幅(NASBA, Canteen, Mississauga, Ontario)、Q-Betaレプリカーゼ系、転写に基づく増幅系(TAS)、およびストランドディスプレースメント増幅(strand displacement amplification)(SDA)が挙げられる。例えば、「診断分子ミクロ生物学:原理と応用(Diagnostic Molecular Microbiology: Principles and Applications)」, D.H. Persingら, 編, American Society for Microbiology, Washington, D.C. (1993)を参照。増幅産物をアンプリコンと呼ぶ。
【0021】
本明細書に用いる用語「動物」は抗αGal抗体を合成する全ての動物と解釈することとし、抗αGal抗体を合成するかどうかが未知の動物も含む。例えば、鳥類の複数種の動物はαGalエピトープを合成するかどうかが未知である。抗αGal抗体またはαGalエピトープのいずれかを合成する動物の間にはユニークな相反関係があるので、αGalエピトープが不在である未試験の多くの動物は抗αGal抗体を合成する動物であることを立証しうると考えられる。本発明はこれらの動物を包含する。
【0022】
用語「抗体」は抗体の抗原結合型(例えば、Fab、F(ab)2)を意味する。用語「抗体」は、しばしば、免疫グロブリン遺伝子により実質的にコードされるポリペプチド、または分析質(抗原)と特異的に結合しかつ認識するそのフラグメントを意味する。しかし、様々な抗体フラグメントは無傷の抗体の消化方法で定義することができる一方、当業者はかかるフラグメントを化学的にまたは組換えDNA技法を用いることによりde novo合成できることを理解している。従って、本明細書に用いる用語の抗体はまた、抗体フラグメント、例えば1本鎖Fv、キメラ抗体(すなわち、色々な種由来の定常および可変領域を含む)、ヒト化抗体(すなわち、非ヒト由来の相補性決定領域(CDR)を含む)および異種複合体抗体(例えば、二特異的抗体)を含む。
【0023】
用語「抗αGal」は、αGalエピトープを認識する免疫グロブリン、例えばIgG、IgA、IgEまたはIgM抗αGal抗体のいずれのタイプまたはサブタイプも含む。
【0024】
本明細書に用いる用語「抗原」は、それ自身またはその部分に対する免疫応答を誘発することができるいずれかの生物学的分子(タンパク質、ペプチド、脂質、グリカン、糖タンパク質、糖脂質その他)を意味し、限定されるものでないが、腫瘍関連抗原およびウイルス、細菌、寄生虫および真菌の抗原を含む。
【0025】
本明細書に用いる用語「抗原提示」は、マクロファージ、樹状細胞、B細胞および他のタイプの抗原提示細胞が内部または外部抗原をこれらの分子のサブフラグメントにプロセシングして、それらを細胞表面上のクラスIもしくはクラスII主要組織適合複合体またはCD1分子と複合して提示する生物学的機構を意味する。このプロセスは、これらの複合体を特異的に認識してこれらの抗原もしくはこれらの抗原を提示する細胞に対する免疫応答を媒介することができる免疫系の他のタイプの細胞(CD4+、CD8+、BおよびNK細胞などの)の増殖を刺激する。
【0026】
用語「保存的に改変された変異体」は、ある特定の配列を参照する場合、アミノ酸と核酸配列の両方に適用しかつ両方を含むことを意図する。特定の核酸配列について保存的に改変された変異体は、同一または保存的に改変されたアミノ酸配列の変異体をコードする核酸配列を意味する。遺伝コードの縮重のために、多数の機能的に同じ核酸が所与のタンパク質をコードする。例えば、コドンGCA、GCC、GCGおよびGCUはすべて、アミノ酸アラニンをコードする。すなわち、アラニンがコドンにより規定されるすべての位置で、そのコドンを、コードされたポリペプチドを改変することなく、記載の対応するコドンのいずれかに改変することができる。そのような核酸変化は「サイレントな変化」であり、これは保存的に改変された変化の一種を表す。従って、あるポリペプチドをコードする本明細書に記載の全ての核酸配列はまた、遺伝子コードを参照して、その核酸のすべての可能なサイレントな変化を記載する。当業者は(通常、メチオニンの唯一のコドンであるAUG;および通常、トリプトファンの唯一のコドンであるUUGを除いて)核酸中のそれぞれのコドンを改変して機能的に同じ分子を得ることができるのを理解するであろう。従って、本発明のポリペプチドをコードする核酸のそれぞれのサイレントな変化は、暗黙のままそれぞれの記載されたポリペプチド配列に含まれ、本発明の範囲に含まれる。
【0027】
アミノ酸配列に関して、コードされた配列中の単一アミノ酸もしくは小パーセントのアミノ酸を改変、付加または欠失する、核酸、ペプチド、ポリペプチド、またはタンパク質配列に対する個々の置換、欠失または付加は、改変が化学的に同様なアミノ酸によるアミノ酸の置換をもたらす場合、「保存的に改変された変異体」であることを当業者は理解するであろう。以下の6グループは、それぞれ、互いにとって保存的置換であるアミノ酸を含有する:
1)アラニン(A)、セリン(S)、トレオニン(T);
2)アスパラギン酸(D)、グルタミン酸(E);
3)アスパラギン(N)、グルタミン(Q);
4)アルギニン(R)、リジン(K);
5)イソロイシン(I)、ロイシン(L)、メチオニン(M)、バリン(V);および
6)フェニルアラニン(F)、チロシン(Y)、トリプトファン(W)。
【0028】
Creighton (1984) 「タンパク質(Proteins)」 W.H. Freeman and Companyも参照すること。
【0029】
本発明者らは、2つのアミノ酸配列の「配列同一性パーセント」を、対合アラインメント後の2つのアミノ酸配列が共有する同一アミノ酸の数をその対の最短配列の全長により除して定義する。
【0030】
本発明者らは、2つのアミノ酸配列の「配列類似性パーセント」を、対合アラインメント後の2つのアミノ酸配列が共有する同一アミノ酸+保存的アミノ酸置換の数をその対の最短配列の全長により除して定義する。
【0031】
規定した核酸について「コードする」または「コードした」は、規定したタンパク質に翻訳するための情報を含むことを意味する。タンパク質をコードする核酸は、非翻訳配列(例えば、イントロン)を核酸の翻訳領域内に含んでもよく、またはかかる介在する非翻訳配列を欠いてもよい(例えば、cDNAにおけるように)。タンパク質をコードする情報はコドンを利用して規定される。典型的にはアミノ酸配列は、核酸により「ユニバーサル」遺伝子コードを用いてコードされる。
【0032】
核酸を合成により調製するかまたは改変するとき、核酸が発現される意図する宿主の既知のコドン優先性をとると有利である。
【0033】
タンパク質またはペプチドについて、用語「単離されたタンパク質(またはペプチド)」が本明細書で時々用いられる。この用語は、天然で随伴する他のタンパク質から十分分離されていて、「実質的に純粋な」形態で存在するタンパク質を意味する。あるいは、この用語は単離された核酸分子の発現により産生されるタンパク質を意味してもよい。
【0034】
核酸分子を参照して、用語「単離された核酸」が時々用いられる。DNAに応用される場合、この用語は、それが誘導された生物の天然のゲノムにおいて(5'および3'方向に)直接連続している配列から分離されたDNA分子を意味する。例えば、「単離された核酸」は、プラスミドまたはウイルスベクターなどのベクター中に挿入されたまたは原核生物または真核生物のゲノムDNA中に組込まれたDNA分子を含む。「単離された核酸分子」はまたcDNA分子も含む。RNA分子についての用語「単離された核酸」は主に上に定義した単離されたDNA分子がコードするRNA分子を意味する。あるいは、この用語は、その天然状態で(すなわち、細胞または組織中で)それと随伴していたであろうRNA分子から十分分離されていて、「実質的に純粋な」形態で存在するRNA分子を意味することができる(用語「実質的に純粋な」は以下に定義する)。
【0035】
本明細書において核酸を参照して用いる「異種(heterologous)」は、外来種に起源するか、または、同じ種由来であれば、意図的な人為的介入によってその天然の形態から組成および/またはゲノム遺伝子座が実質的に改変されている核酸である。例えば、異種構造遺伝子と機能しうる形で連結されたプロモーターは、その構造遺伝子が誘導された種と異なる種由来であるか、または、もし同じ種由来であれば、片方または両方がそれらの元来の形態から実質的に改変されている。異種タンパク質は外来種からの起源であっても、または、もし同じ種であれば、その元来の形態から意図的な人為的介入により実質的に改変されていてもよい。
【0036】
「宿主細胞」は、ベクターを含有しかつベクターの複製および/または発現を支援する細胞を意味する。宿主細胞は原核生物細胞、例えば大腸菌(E.coli)、または真核生物細胞、例えば酵母、昆虫、両生類、または哺乳類細胞であってもよい。
【0037】
用語「導入された」は、核酸を細胞中に挿入する文脈において「トランスフェクション」または「形質転換」または「形質導入」を意味し、核酸の真核生物または原核生物細胞中への組込みを意味し、この場合、核酸は細胞のゲノム中に組込まれて自律性レプリコンに変換されても(例えば、染色体、プラスミド、プラスチドまたはミトコンドリアDNA)または一過的に発現されても(例えば、トランスフェクトされたmRNA)よいことを意味する。
【0038】
本明細書で用いられる「マーカー」は、染色体上のユニークな位置を同定するのに役立つ染色体上の遺伝子座の意味を含む。「多形マーカー」は、複数の形態(対立遺伝子)で現れるマーカーの意味を含み、それらが相同的な対(homologous pair)で存在するとき、マーカーの異なる形態はその対の染色体のそれぞれの伝達を追跡することを可能にする。遺伝形質を1以上のマーカーを用いて定義してもよい。
【0039】
本明細書に用いられる「核酸」は、1本鎖または2本鎖形態のデオキシリボヌクレオチドまたはリボヌクレオチドの意味を含み、特に断らない限り、それらが天然ヌクレオチドと同様な方法で1本鎖核酸とハイブリダイズする点において天然ヌクレオチドの本質的性質を有する既知の類似体(例えば、ペプチド核酸)を包含する。
【0040】
抗原または腫瘍細胞の用語「オプソニン作用」は、抗原にまたは腫瘍細胞表面上に存在するαGalエピトープの抗αGal抗体との結合と、それによる、抗体のFc部分の抗原提示細胞の表面上に存在するFc受容体との結合を介する、マクロファージ、樹状細胞、B細胞または他のタイプの抗原提示細胞によるオプソニン化された抗原または腫瘍細胞の食作用の増強を意味する。
【0041】
本明細書に用いられる「ポリヌクレオチド」は、ストリンジェントなハイブリダイゼーション条件下で、天然のヌクレオチドと実質的に同じヌクレオチド配列とハイブリダイズする、および/または、天然のヌクレオチドと同じアミノ酸に翻訳されるという点において、天然リボヌクレオチドの本質的性質を有するデオキシリボポリヌクレオチド、リボポリヌクレオチド、またはそれらの類似体の意味を含む。ポリヌクレオチドは、未変性または異種の構造または調節遺伝子の全長またはサブ配列であってもよい。特に断らない限り、この用語は規定した配列、ならびにその相補的配列の意味を含む。従って、安定性のためまたは他の理由のために主鎖が改変されたDNAまたはRNAは、本明細書で意図される「ポリヌクレオチド」である。
【0042】
用語「ポリペプチド」、「ペプチド」および「タンパク質」は、本明細書において互換的にアミノ酸残基のポリマーを意味する。本用語は、1以上のアミノ酸残基が対応する天然アミノ酸、ならびに天然アミノ酸ポリマーの人為的な化学類似体であるアミノ酸ポリマーに適用される。かかる天然アミノ酸の類似体の本質的な性質とは、タンパク質中に組み込まれると、そのタンパク質が、同じタンパク質であるが全て天然アミノ酸から構成されるタンパク質に対して誘発される抗体に対して特異的に反応性を有することである。用語「ポリペプチド」、「ペプチド」および「タンパク質」はまた、限定されるものでないが、リン酸化、グリコシル化、脂質結合、硫酸化、グルタミン酸残基のγカルボキシル化、ヒドロキシル化およびADPリボシル化を含む改変体も包含する。
【0043】
本明細書に用いられる「組換え体」は、異種核酸の導入により改変されているかまたは細胞がそのように改変された細胞から誘導された、細胞またはベクターの意味を含む。従って、例えば、組換え細胞は、細胞の未変性(非組換え)形態内の同一形態に見出されない遺伝子を発現するかまたはそうでなければ、意図的な人為的介入の結果として異常に発現された、発現不足であるか、または全く発現されない未変性遺伝子を発現する。本明細書に用いられる用語「組換え」は、意図的な人為的介入なしに生じる改変のような、天然事象(例えば、自発性突然変異、天然の形質転換/形質導入/遺伝子転移)による細胞またはベクターの改変を包含しない。
【0044】
本明細書に用いられる「組換え発現カセット」は、宿主細胞において特定の核酸の転写を可能にする一連の規定した核酸エレメントを用いて、組換えによりまたは合成により作製される核酸構築物である。組換え発現カセットを、プラスミド、染色体、ミトコンドリアDNA、プラスチドDNA、ウイルス、または核酸断片中に組み込んでもよい。典型的には、発現ベクターの組換え発現カセット部分は、他の配列の中に、転写される核酸およびプロモーターを含む。
【0045】
用語「残基」または「アミノ酸残基」または「アミノ酸」は、本明細書中では互換的に用いられ、タンパク質、ポリペプチド、またはペプチド(まとめて「タンパク質」)中に組み込まれたアミノ酸を意味する。アミノ酸は天然のアミノ酸であってもよく、特に限定しない限り、天然アミノ酸と同様な方法で機能しうる天然アミノ酸の非天然類似体を包含してもよい。
【0046】
用語「実質的に同じ」は、タンパク質の性質(すなわち、タンパク質の構造、安定性特性、基質特異性および/または生物学的活性)に実質的に影響を与えることのない配列変化を有する核酸またはアミノ酸配列を意味する。特に核酸配列を参照する用語「実質的に同じ」は、コード領域および発現を支配する保存的配列を意味することを意図し、そして主に同じアミノ酸をコードする縮重コドン、またはコードされたポリペプチドに保存的置換アミノ酸をコードする代わりのコドンを意味する。アミノ酸配列を参照する用語「実質的に同じ」は、一般的に構造または機能の決定に関わらないポリペプチドの領域における保存的置換および/または変化を意味する。
【0047】
抗体に関連して、用語「免疫学的に特異的な」は、抗原性生物学的分子の混合集団を含有するサンプル中の目的のタンパク質の1以上のエピトープと結合するが、他の分子を実質的に認識せずかつ結合しない抗体を意味する。
【0048】
「コード配列」または「コード領域」は、配列が発現されるときに遺伝子産物を産生するために必要な配列情報を有する核酸分子を意味する。
【0049】
用語「機能しうる形で連結された」または「機能しうる形で挿入された」は、コード配列の発現に必要な調節配列を、コード配列の発現ができるようにするために、核酸分子中のコード領域と相対的に適当な位置に配置することを意味する。この同じ定義は、時々、発現ベクター中の他の転写制御エレメント(例えば、エンハンサー)の配列に適用される。
【0050】
転写および翻訳制御配列は、宿主細胞中でコード配列を発現させるDNA調節配列、例えばプロモーター、エンハンサー、ポリアデニル化シグナル、ターミネーターなどである。
【0051】
用語「プロモーター」、「プロモーター領域」または「プロモーター配列」は、一般的に、コード領域の5'または3'サイドに、またはコード領域内に、またはイントロン内に見出しうる遺伝子の転写調節領域を意味する。典型的には、プロモーターは、細胞内でRNAポリメラーゼと結合して下流(3'方向)コード配列の転写を開始することができるDNA調節領域である。典型的な5'プロモーター配列は、その3'端末で転写開始部位と境界を接し、上流(5'方向)に伸びてバックグラウンドを超える検出可能なレベルの転写を開始するために必要な最小限の数の塩基またはエレメントを含む。プロモーター配列内には、転写開始部位(便宜的にヌクレアーゼS1によるマッピングにより規定した)、ならびにRNAポリメラーゼの結合に関わるタンパク質結合ドメイン(コンセンサス配列)がある。
【0052】
用語「治療上有効な量」は、限定されるものでないが、本明細書に記載の技術により測定可能である、予め存在する腫瘍細胞の数、量または複製の測定可能な減少をもたらすために十分な治療組成物の量を意味する。
【0053】
用語「腫瘍細胞」は、動物における腫瘍の構成要素である細胞、または動物における腫瘍の構成要素になることが定まっている細胞、すなわち、前癌病変の構成要素である細胞を意味する。この定義には、固体腫瘍を形成しない造血系の悪性腫瘍細胞、例えば、白血病、リンパ腫および骨髄腫が含まれる。
【0054】
用語「腫瘍」は、進行性で正常細胞と置換わるかまたは破壊する浸潤性集団を形成しうる1以上の腫瘍細胞として定義される。
【0055】
用語「悪性腫瘍」は、その元来の発生部位を越えて播種する特性を発揮しうる腫瘍細胞により形成される腫瘍として定義される。
【0056】
「ベクター」は、それに対して他の核酸セグメントを機能しうる形で挿入して上記セグメントを複製または発現させる、レプリコン、例えばプラスミド、ファージ、コスミド、またはウイルスである。
【0057】
用語「核酸構築物」または「DNA構築物」は、時々、細胞を形質転換するために適当な調節配列と機能しうる形で連結されてベクター中に挿入されるコード配列を意味するために用いられる。この用語は、用語「形質転換用DNA」と互換的に利用しうる。かかる核酸構築物は、選択マーカー遺伝子および/またはレポーター遺伝子とともに目的の遺伝子産物に対するコード配列を含有しうる。
【0058】
用語「選択マーカー遺伝子」は、発現されると、形質転換した細胞に選択表現型、例えば抗生物質耐性を与える産物をコードする遺伝子を意味する。
【0059】
用語「レポーター遺伝子」は、標準的方法により直接または間接に検出可能である産物をコードする遺伝子を意味する。
【0060】
細胞が外因性または異種DNAにより「形質転換」または「トランスフェクト」されているというのは、上記DNAが細胞内部に導入されていることを意味する。形質転換用DNAは細胞のゲノム中に(共有結合により)組込まれていてもいなくてもよい。原核生物、酵母、および哺乳類動物細胞において、例えば、形質転換用DNAはプラスミドなどのエピソーム上に維持されてもよい。真核生物細胞については、安定して形質転換された細胞は、形質転換用DNAが染色体中に組込まれ、染色体複製を介して娘細胞に継承される細胞である。この安定性は、形質転換用DNAを含有する娘細胞の集団から構成される培養細胞株またはクローンを確立する真核生物細胞の能力により実証される。
【0061】
「クローン」は単一細胞または共通の祖先から有糸分裂により導入された細胞集団である。「培養細胞株」は、in vitroで多世代にわたって安定した増殖能力を有する一次細胞のクローンである。
【0062】
腫瘍細胞に関する用語「治療する」は、上記細胞の進行を停止するか、増殖を減速するか、上記細胞の存在に伴う症状の退行または改善を誘導することを意味する。
【0063】
用語「異種(xenogeneic)細胞」は、移植またはワクチン接種操作において、レシピエント動物宿主になる動物種と異なる動物種に由来する細胞を意味する。
【0064】
用語「同種異系(allogeneic)細胞」は、同じ動物種であるが、「レシピエント宿主」になる動物と1以上の遺伝子座において遺伝的に異なる細胞を意味する。この用語は通常、ある動物から同種の他の同一でない動物へ移植される細胞に適用される。
【0065】
用語「同系(syngeneic)細胞」は、移植またはワクチン接種操作において、同じ動物種である細胞であり、かつほとんどの遺伝型および表現型マーカーについてその培養細胞株のレシピエント宿主になる動物と同じ遺伝子組成を有する細胞を意味する。この用語は通常、同一の双生児から移植されるかまたは高度に近交系の動物間で移植される細胞に適用される。
【発明を実施するための最良の形態】
【0066】
発明の詳細な説明
腫瘍に対する免疫療法の基本原理は、腫瘍関連抗原(TAA)に対する有効な免疫応答の誘導であり、これが、次に、これらの抗原を発現する増殖腫瘍細胞の免疫が媒介する破壊をもたらすことにある。TAAを含むタンパク質に対して有効である免疫応答のために、これらの抗原は最初に抗原提示細胞(APC)、例えばマクロファージ、樹状細胞およびB細胞による飲食作用を受けなければならない。TAAは、APC内のリソソーム区画中で分解され、生じたペプチドがマクロファージ細胞膜の表面上に、ほとんどMHCクラスII分子と一緒に、しかしまたMHCクラスI分子とも一緒に発現される。この発現は、特異的CD4+ヘルパーT細胞による認識およびこれらの細胞の続いての活性化を媒介して免疫応答を果たさせる(Stevenson, 1991, FASEB J. 5:2250;Lanzavecchia, 1993, Science 260:937;Pardoll, 1993, Immunol. Today 14:310)。ヒトTAA分子の大多数は分子的用語で定義されていないので、これらを薬物療法の標的としてまたは抗腫瘍ワクチンとして利用することができない。
【0067】
予防ワクチン接種型のプロトコルにおいて腫瘍特異的応答を誘導する上でのαGalエピトープの利用は、当技術分野の他研究者から提案されており、同じ腫瘍細胞による後期のチャレンジに対して保護を生じることが示されている。しかし予防免疫応答を生じた特異的TAAが、続いて発生した腫瘍細胞に対して有効である保証はない。予防ワクチン接種を介して予防免疫応答を生じたTAAの特定のエピトープは、その腫瘍にだけ、そのタイプの癌だけに、その患者だけに、またはその組織だけに等々、特異的でありうる。全ての癌に発現される普遍的なTAAの同定は依然として捉えがたく、癌予防用の予防ワクチン接種計画の広汎な利用を妨げている。
【0068】
一般的予防手法の代わりに、再発性または診断された腫瘍をもつ個体の免疫治療はより直接的に標的を定めた方法を提供しうる、しかしワクチン技法が既に存在する腫瘍細胞に対する治療として効力がありうるという一致した予測はない。複数の報告は、予防モデルに有効であるが、その同じ処置が治療手法としては効果が低いかまたは無いことを報じている。例えば、マウスTRP-1をコードする組換えワクシニアウイルスを用いる予防ワクチン接種は、その後の生腫瘍B16細胞による皮下チャレンジに対する保護に成功した。この治療は肺黒色腫転移の予防に部分的にだけ有効であり、予め確立した皮下黒色腫の治療には無効であった(Overwijkら, Proc. Natl, Acad. Sci. USA (1999) 96: 2982-2987)
類似の研究において、非メチル化シトシン-グアニンモチーフ(CpG-ODN)およびCTLA-4遮断剤を含有する合成オリゴデオキシヌクレオチド(ODN)アジュバントを用いて、ペプチドワクチンの有効性を予防、治療および両処置の組合わせにおいて評価した。この研究の結論は、B16黒色腫に対する予防または予防方式の治療は無効というものであった。その著者は、マウスにワクチン接種し、B16をチャレンジし、続いてさらなるワクチンの治療用量を与えると、マウスの有効な治療と延命が観察されることを実証した。しかし、この研究において、ワクチン接種は、予め形成された疾患の治療に対しては、部分的にしか有効でなかった、というのは、全てのマウスは、予防および治療腫瘍の両方において、黒色腫で死亡したからである(Davila, KennedyおよびCelis, Cancer Research (2003), 63:3281-3288)。
【0069】
多数の他の研究は、皮下黒色腫の予防治療にだけ焦点を合わせているが、これらのごく少数しか腫瘍増殖速度について有意でかつ有効な治療効果を実証していない、しかし、腫瘍増殖速度の低減は動物の生存率に反映されなかった(Wangら, Cancer Research (2003) 63:2553-2560;Current Protocols of Immunology, Chapter 20, 「ヒト黒色腫のモデルとしてのB16(B16 as a Model for Human Melanoma”)」)。
【0070】
問題をさらに複雑にするのは、腫瘍特異的リンパ球の存在は腫瘍破壊を誘導するのに不十分であることが示されていることである。実際、最近、大量の腫瘍特異的T細胞は腫瘍をもつマウスを延命するのに十分でないことが実証されている(Overwijkら, Journal of Experimental Medicine, (2003) 198: 569-580)。著者は、天然自己ペプチドよりむしろ改変したペプチドリガンドによる抗原特異的ワクチン接種を介するT細胞刺激、ならびにT細胞増殖および活性化因子の同時投与が、腫瘍退行を誘導するための厳密に必要な3要素であることを実証した。
【0071】
ほとんどの腫瘍細胞はユニークなTAA(腫瘍関連抗原)の発現プロファイルを有するが、多くの場合、これらのTAAは未知であるかまたは個々の腫瘍に対して確認するまたは単離することが非常に困難である。さらに、免疫監視を逃げるほとんどの腫瘍については、これらの抗原が細胞「危険」シグナルの文脈で提示されないので、または免疫系がこれらの抗原を認容して「自己」抗原と認識するので、免疫系はこれらのTAAを外来抗原として認識しない。細胞全体ワクチンの使用はこれらの抗原を単離または特徴付けることなくTAAの全レパートリーを提供するので、この第一の困難を緩和する。同種異系(allelic)細胞全体ワクチンの使用は個体間の同種異系差を提示し得るTAAを提供するので、それ故に、これらのTAAに対する免疫系の認容を壊すことができよう。細胞全体ワクチンはまた、GM-CSFなどの免疫応答を増強する分子を発現するように遺伝的に改変されている(Dranoffら, Proc. Natl. Acad. Sci. USA (1993) 90:3539)。遺伝的に改変したまたは改変してない細胞全体同種異系癌ワクチンは抗腫瘍活性および生存についての利点を臨床試験で示しており、それにより、研究室で産生したヒト癌培養細胞株の免疫拒絶が患者の悪性腫瘍の破壊を誘導できるという仮説を実証している。これらのワクチンは、細胞傷害性Tリンパ球およびナチュラルキラー細胞を直接活性化する腫瘍ワクチンクラスI MHCの助けで、TAAの直接提示による細胞免疫エフェクターの直接刺激により機能する(van Elsasら, J. Exp. Medicine (1999) 190:355-366)。従来述べられた手法を用いる抗腫瘍応答の刺激は、しばしば、腫瘍組織と同じ細胞型の正常組織に対する自己免疫疾患の発症と関連している。さらに、これらのワクチンは、TAAを認識しかつ抗原提示および腫瘍細胞の補体が介在する破壊を増強する免疫系の体液性免疫機構を利用しない。強い体液性免疫応答がより有効な抗腫瘍応答を産生する理由はいくつかある:すなわち、I)細胞表面上の特異的抗原と結合することによりワクチン細胞をオプソニン化する抗体は、抗体Fc部の抗原提示細胞上のFc受容体との結合により、マクロファージによるそれらの食作用を促進しうる。II)Fc受容体を標的とすることは、有効なワクチン性能にとって重要ないくつかの機能、例えば、MHCクラスIおよびII両方の抗原提示のための抗原の効率的摂取の促進;APC活性化の促進および樹状細胞の成熟の促進を達成する。III)Fc受容体が介在する食作用経由の抗原提示細胞によるオプソニン化細胞の摂取は、有効な抗腫瘍CTL応答を産生するのに重要な意味を持ちうる、何故なら、これがCD8+T細胞によるMHCクラスIに制限された応答の活性化を交差提示経路を介して促進するからである。IV)体液性免疫応答を刺激することが出来ないワクチンは、HLA制限により細胞性免疫を誘導する能力が限られている。CTLはHLA制限され、自己クラスI MHC分子上の腫瘍抗原を提示するワクチン細胞を破壊するだけであろう。NK細胞は、もしMHC相互作用が低ければ、低い免疫応答を産生する腫瘍ワクチン細胞を破壊しうる。V)APCを活性化するシグナルは直接または間接に、天然に獲得した免疫応答から来る。腫瘍ワクチン細胞を摂取するAPCは、抗原を効果的に提示しうる前に、活性化されていなくてはならない。さらに、所要の活性化シグナルなしに未熟APCに抗原を提示することはその免疫応答を抑制しうる。さらに、活性化されたAPCは、CTLを活性化することができる(CTLは活性化されていないと、たとえCTLがHLA適合腫瘍細胞上にそのコグネイト抗原を認識しても死滅させることができない)。
【0072】
上記の考察から、革新的な腫瘍ワクチンが腫瘍ワクチン分野に必要とされていることは明白であり、ここで特に必要なことは、I)予防ワクチン接種治療計画を必要としない治療法の設定において、既に存在しかつ播種性の腫瘍の退行を生じうるかまたは少なくとも腫瘍増殖速度を減速させうる腫瘍ワクチンを開発すること、II)免疫系の細胞性および体液性部門の両方を刺激するワクチンを開発すること、および、III)自己免疫疾患などの望ましくない二次的影響を有しないワクチンを開発することである。
【0073】
本出願人らの発明は、これらの要件を満たす治療用腫瘍ワクチン製剤を提供する。本発明は、α(1,3)-ガラクトシルエピトープをin vitroで合成する腫瘍細胞を作製する遺伝子導入技術の利用を含むものである。細胞自身の機構の利用およびこのように遺伝子操作された複数の同種異系または同系腫瘍細胞の混合物の利用により、エピトープを多数の細胞表面糖タンパク質および糖脂質上に作製して、抗体が個々のまたは別々の細胞上のTAAに対して産生する多数の機会を提供する。これらのワクチンに用いられる細胞は、100万〜200万個のαGalエピトープを含有すると見積もられる。抗αGal Abに対するこの多数の天然に予め存在する結合部位により、高密度のオプソニン作用が得られ、次いで、体液性および細胞性部門の両方の免疫系を活性化する様々なプロセスを開始する補体破壊が起こる。同種異系腫瘍細胞表面上のかかる高密度のαGal残基の存在は、改変した腫瘍細胞の注入部位において異種移植超急性拒絶と類似した超免疫応答を誘発する。さらに、これらの癌ワクチンは多価であり、多重の腫瘍抗原標的を免疫系に提示することを意味する。このことは、いくつかのTAAが提示される点でより効率的な治療、および提示されるTAAの数の増加とともに色々な個々の腫瘍からのエピトープが恐らく重複されるのでより広く効果的な治療をもたらす。オプソニン化された細胞は容易に貪食細胞により摂取されて、腫瘍抗原のほとんどが同時に適応性免疫系に提示されうる機構を提供する。これらの細胞内で癌ワクチン細胞からのタンパク質は消化されて、癌細胞中の突然変異タンパク質エピトープをT細胞監視に曝すクラスII MHC提示がなされる。さらに、抗原提示細胞(APC)による、Fc受容体が媒介する飲食作用(endocytosis)を経由するオプソニン化細胞の取込みは、CD8+細胞による交差提示経路を介するMHCクラスI制限応答の活性化を容易にしうる。このプロセスにより運動を始める免疫系カスケードは、確立されたヒト悪性腫瘍由来のナイーブ腫瘍細胞を破壊する特異的T細胞応答を誘導する刺激を与える。さらに、一次免疫応答により誘導される炎症性環境は、サイトカイン、ヒスタミンおよび他のアップレギュレートされた分子が媒介するT細胞応答をブーストする増幅効果をもたらす。この方法で活性化されたT細胞は直接、癌細胞を死滅させることができる。重要な所見は、腫瘍ワクチン中に存在する糖タンパク質および糖脂質に対するαGalエピトープの付加は、グリコシル化された抗原に対してだけでなく、グリコシル化による影響の有無に関わらず、腫瘍細胞内に存在する全ての抗原に対する免疫応答を発生させうることである。
【0074】
この種の細胞全体ワクチンの治療用腫瘍免疫誘導における有効性をノックアウト(KO)マウスを用いる動物モデルを立証した。αGT遺伝子の機能を欠くKOマウスを作製し、腫瘍培養細胞株上のαGalエピトープに対するin vivo免疫応答を研究するための小動物モデルを得た。これらのマウスを、ウサギ赤血球(rRBC)により免疫感作してヒト血清中に見出される高力価の抗αGal抗体を刺激することができる。これらのマウスを用いて、本出願人らは、該免疫系がαGalポジティブ腫瘍細胞を拒絶しかつこの拒絶がαGalネガティブ腫瘍細胞に拡大されたT細胞免疫を発生させることを実証した。この動物モデルを用いてまた、予め形成されたヒト悪性腫瘍をシミュレートするために、ワクチンによる治療前にαGalネガティブ腫瘍細胞を動物に与える臨床応用のシミュレーションを行った。このタイプの実験により、本出願人らは、本発明により遺伝子操作した細胞を用いて免疫治療したマウスに誘導された細胞が媒介する免疫は、皮下および肺の予め形成された局所および播種性腫瘍を有効に治療することができ、腫瘍増殖速度を減速するだけでなく、さらに重要なことは、治療したマウスの長期間生存をもたらすことを示した。養子細胞移入(adoptive cell transfer)実験は、最終的に、予め形成された腫瘍の拒絶の細胞依存構成要素を示した。他の手法を用いるときに記載された細胞全体腫瘍ワクチン摂取に関連する典型的な自己免疫色素脱失は、この治療を受けたマウスにおいては決して観察されず、異なる免疫刺激法が与える免疫応答の最終成果に対する影響の重要性を強調した。
【0075】
従って本出願人の発明は、予め形成された腫瘍細胞の選択的ターゲティングおよび死滅を引き起こす方法と組成物に関する。ex vivo遺伝子療法プロトコルを介して、腫瘍細胞を遺伝子操作してαGalエピトープを発現させる。次いで細胞を照射するかまたは死滅させて患者に投与する。天然に予め存在する抗αGal抗体によるαGalエピトープの結合は、腫瘍細胞のオプソニン化を引き起こし、腫瘍特異的抗原提示を増強する。本発明の重要な特徴は、本発明の医薬組成物中の細胞全体、および形質移入された複数の細胞の混合物の利用を意図することである。αGal改変は細胞表面上の複数の糖タンパク質に影響を与えるので、動物免疫系は検出し、プロセシングし、そして腫瘍特異的抗原に対する抗体を作製する機会の増加を有しうる。
【0076】
本発明によれば、医薬組成物は、発現するとαGT酵素をコードするポリヌクレオチド配列を腫瘍細胞に導入することにより作製される。該配列はウイルスまたは非ウイルスベクターを含んでもよいいずれかのヌクレオチド導入ビヒクルを介して導入される。これらの遺伝子導入ビヒクルは腫瘍細胞を形質転換し、そこに挿入される外来遺伝子材料の発現を引き起こす。得られる遺伝子産物は、上記細胞に存在する細胞表面糖タンパク質上のαGalエピトープの合成を触媒する。本発明は、多数の細胞表面糖タンパク質をもつ腫瘍細胞全体を利用して予め存在する抗αGal抗体によるαGalエピトープとの結合を最大化し、そして、この複合体と抗原提示細胞上に存在するFc受容体との結合を増強し、その結果、上記ワクチン腫瘍細胞中に存在する多数の腫瘍関連抗原の抗原提示をトリガーすることを意図する。
【0077】
本発明はまた、医薬組成物および上記医薬組成物を作る方法も含むものであり、ここで上記医薬組成物は、αGalエピトープを含む複数の細胞表面糖タンパク質を含有する弱毒化腫瘍細胞の混合物の治療上有効な量および担体を含む。好ましい実施形態においては、細胞は細胞全体である。組成物を作る方法は、生腫瘍細胞のコレクションを取得し、発現するとαGTをコードするヌクレオチド配列を用いて上記細胞を形質転換し、そして上記細胞の細胞表面糖タンパク質上にαGalエピトープを提示させることを含んでなる。次いで細胞を、好ましくは、照射により死滅させ、投与のための製薬担体と組合わせる。
【0078】
本発明のさらに他の実施形態は、腫瘍細胞上にαGalエピトープを作製しうるポリヌクレオチドによる腫瘍細胞の形質転換を含んでなる。本発明の一実施形態は、発現すると酵素α-(1,3)-ガラクトシルトランスフェラーゼ(αGT)をコードするヌクレオチド配列による腫瘍細胞の形質転換を含んでなる。αGT cDNAはウシおよびマウスcDNAライブラリーからクローニングされている。Larson, R. D.ら, (1989) 「マウスUDPガラクトースをコードするcDNAの単離;β-D-ガラクトシル-1,4-Nアセトール-D-グルコサミンα1-3ガラクトシルトランスフェラーゼ:遺伝子導入による発現クローニング(Isolation of a cDNA Encoding Murine UDP galactose; β-D-galactosyl-1, 4-N Acetol-D-Glucosamine α1-3 Galactosyl Transferase : Expression Cloning by Gene Transfer)」, PNAS, USA 86:8227;およびJoziasse, D. H.ら, (1989) 「ウシα1-3ガラクトシルトランスフェラーゼ(Bovine α1-3 Galactosyl Transferase)」: 「cDNAクローンの単離と特徴付け、ヒトゲノムDNA中の相同的配列の同定(Isolation and Characterization of a cDNA Clone, Identification of Homologous Sequences in Human Genomic DNA)」, J. Biol Chem 264:14290。同様に、細胞表面上にαGalエピトープを発現する腫瘍細胞を生じうる、例えばこの反応を触媒する他の酵素に対する、または、恐らくはさらに、細胞表面上にさらなる糖タンパク質を存在させて免疫系に提示しうるTAAを人為的に作製する遺伝子操作に対する他のヌクレオチド配列を利用することができる。
【0079】
本発明の医薬組成物に利用する腫瘍細胞は、自己または、好ましい実施形態においては、同種異系もしくは同系であってもよい。治療する形質転換細胞および腫瘍細胞は、普通は少なくとも1以上のエピトープを有しなければならないが、好ましくは多数を有しうる。一般的なまたは重複するエピトープまたはTAAが異なる癌の間に存在する限り、本医薬組成物を非常に広く応用することができる。
【0080】
本発明はまた、癌細胞に限定されるものでなく、細胞表面糖タンパク質を含有するいずれの細胞、またはαGalエピトープに対する部位を有する細胞構成要素も含むことができる。これはある特定のウイルス、新生物細胞などを含んでもよい。
【0081】
αGalエピトープを作製するタンパク質をコードするヌクレオチド配列は、腫瘍細胞を形質導入する適当な発現ビヒクル中に含有される。かかる発現ベクターとしては、限定されるものでないが、真核生物ベクター、原核生物ベクター(例えば、細菌ベクターなど)、およびウイルスベクターが挙げられる。
【0082】
一実施形態においては、発現ベクターはウイルスベクターである。使用しうるウイルスベクターは、限定されるものでないが、レトロウイルスベクター、アデノウイルスベクター、ヘルペスウイルスベクター、およびアデノ随伴ウイルスベクター、またはDNA複合体が挙げられる。
【0083】
好ましい実施形態においては、ウイルスベクターパッケージング培養細胞株に、抗体結合および補体活性化により腫瘍細胞の破壊を誘導する作用物質(agent)をコードする核酸配列を含有するウイルスベクターを形質導入する。パッケージング培養細胞株により産生されるウイルス粒子を収穫して、抗腫瘍ワクチンとして投与する腫瘍細胞を形質導入するために用いる。
【0084】
伝統的に、ウイルスベクターはレトロウイルスまたはアデノウイルスベクターである。利用しうるレトロウイルスベクターの例としては、限定されるものでないが、モロニーマウス白血病ウイルス、脾臓壊死ウイルス、およびレトロウイルス、例えばラウス肉腫ウイルス、ハーベイ(Harvey)肉腫ウイルス、鳥類白血症ウイルス、ヒト免疫不全ウイルス、骨髄増殖性肉腫ウイルス、および哺乳類動物腫瘍ウイルス由来のベクターが挙げられる
レトロウイルスベクターは、真核生物細胞中にレトロウイルス媒介遺伝子導入を媒介する作用物質として有用である。レトロウイルスベクターは、一般的に、ウイルスの構造遺伝子をコードする配列の大多数を欠失させ、そして目的の遺伝子により置換えて構築する。最も多いのは、構造遺伝子(すなわち、gag、pol、およびenv)をレトロウイルス主鎖から当技術分野で公知の遺伝子操作技法によって取除く。
【0085】
これらの新しい遺伝子は、いくつかの一般的方法でプロウイルス主鎖中に組込んだ。最も簡単な構築物は、レトロウイルスの構造遺伝子を単一遺伝子により置換え、次いでそれを長末端反復配列(LTR)内のウイルス調節配列の制御下で転写したものである。標的細胞中に複数の遺伝子を導入することができるレトロウイルスベクターを構築しておいてもよい。通常、このようなベクターでは、1つの遺伝子はウイルスLTRの調節下にある一方、第2の遺伝子はスプライスされたメッセージとして発現されないかまたはそれ自身の内部プロモーターの調節下にある。
【0086】
主に、ベクターとパッケージング細胞内のパッケージング欠損ヘルパーウイルスの間の組換えの機会を減少させる努力として、ウイルス主鎖のウイルス構成要素を最小限にする努力がなされている。パッケージング欠損ヘルパーウイルスは、ベクター自身から欠失されたレトロウイルスの構造遺伝子を与えるために必要である。
【0087】
一実施形態においては、レトロウイルスベクターは、Benderら, J. Virol. 61:1639-1649 (1987)に記載の一連のベクターの1つであってもよく、これはN2ベクター(Armentanoら, J. Virol., 61:1647-1650)ベースであり、ベクターとパッケージング系の間の相同性を絶対的最小値に低下させる一連の欠失および置換を含有する。これらの変化はまた、ウイルスタンパク質が発現されうる可能性も低下させた。これらのベクターの最初のものであるLNL-XHCでは、位置指定突然変異誘発により天然ATG開始コドンをgagからTAGに改変し、それによりこの点からの意図しないタンパク質合成を排除した。
【0088】
モロニーマウス白血病ウイルス(MoMuLV)においては、真正gagスタートの5'に、他のグリコシル化タンパク質(pPr80.sup.gag)の発現を可能にするオープンリーディングフレームが存在する。モロニーマウス肉腫ウイルス(MoMuSV)はこの5'にフレームシフトおよびグリコシル化部位の消失を含む変化があり、pPr80.sup.gagのアミノ末端の発現の可能性を除去する。従って、LNL-XHCの改変されたATGとMoMuSVの5'部分の両方を組込んだベクターLNL6を作った。このようにLNベクターシリーズの5'構造は遺伝的に形質導入された標的細胞におけるレトロウイルスリーディングフレームの発現とともに続いてのウイルス抗原産生の可能性を排除する。パッケージング欠損ヘルパーウイルスとの重複を少なくする最終的改変において、MillerはLNベクター中の3'LTRのすぐ前の余分なenv配列を排除した(Millerら, Biotechniques, 7:980-990, 1989)。本発明によって使用しうるベクターの一例を図1に示し、その配列を図2、配列番号1に示す。
【0089】
遺伝子療法に応用するために遺伝子導入系が満たさなければならない最高の要件は安全である。安全は、ベクターゲノム構造と、感染性ベクター産生に用いるパッケージング系との組合わせから誘導される。Millerらは、レトロウイルス構造タンパク質を発現するpPAM3プラスミド(パッケージング欠損ヘルパーゲノム)とベクターパッケージング系を作るLNベクターシリーズとの組合わせ(すなわち、LNとpPAM3)を開発し、ベクターゲノムとパッケージング欠損ヘルパーゲノムの間のほとんど全ての組換えの部位を排除することを介して組換え野生型レトロウイルスの生成を最小限に低下させた。
【0090】
一実施形態においては、レトロウイルスベクターは、上述のおよびさらにBenderら(1987)およびMillerら(1989)に記載されたベクターなどのモロニーマウス白血病ウイルスのLNシリーズのベクターであってもよい。かかるベクターはマウス肉腫ウイルスから誘導されたパッケージングシグナルの一部分、および突然変異したgag開始コドンを有する。本明細書に用いる用語「突然変異した」は、gag開始コドンが欠失しているかまたは改変されてgagタンパク質またはその断片もしくは末端切断部が発現されないことを意味する。
【0091】
該ベクターは1以上のプロモーターを含む。使用しうる好適なプロモーターとしては、限定されるものでないが、Millerら, Biotechniques, Vol. 7, No. 9, 980-990 (1989)に記載のレトロウイルスLTR;SV40プロモーター;およびヒトサイトメガロウイルス(CMV)プロモーター、またはいずれか他のプロモーター(例えば、限定されるものでないが、ヒストン、pol III、およびβ-アクチンプロモーターを含む真核生物細胞性プロモーターなどの細胞性プロモーター)が挙げられる。使用しうる他のウイルスプロモーターとしては、限定されるものでないが、アデノウイルスプロモーター、TKプロモーター、およびB19パルボウイルスプロモーターが挙げられる。
【0092】
他の実施形態においては、本発明は誘導プロモーターを含む。かかるプロモーターの1つはテトラサイクリン制御トランス活性化因子(tTA)応答性プロモーター(tet系)、哺乳類動物細胞における利用に適応させている原核生物誘導プロモーター系である。tet系はレトロウイルスベクター内に組織化し、高レベルの構成的に産生されるtTA mRNAがtTAタンパク質の産生だけでなく、アンチセンス抑制による応答ユニットの低い基底発現にも機能するようにした。Paulus, W.ら, 「自己に含有される、哺乳類動物細胞への遺伝子送達用テトラサイクリン調節レトロウイルスベクター系(Self-Contained, Tetracycline-Regulated Retroviral Vector System for Gene Delivery to Mammalian Cells)」, J of Virology, January. 1996, Vol. 70, No. 1, pp. 62-67を参照すること。好適なプロモーターは、本明細書に含まれる教示から当業者には明らかであろう。
【0093】
次いでベクターを用いてパッケージング培養細胞株に形質導入して、生産培養細胞株を作製する。トランスフェクトしうるパッケージング細胞の例としては、限定されるものでないが、PE501、PA317、.psi.2、.psi.-AM、PA12、T19-14X、VT-19-17-H2、.psi.CRE、.psi.CRIP、GP+E-86、GP+envAM12、DANおよびAMIZ培養細胞株が挙げられる。作用物質をコードする核酸配列が発現すると、腫瘍細胞を破壊しかつ補体カスケードを活性化することができる作用物質をコードする核酸配列を含有するベクターは、当業者に公知のいずれかの方法を介してパッケージング細胞に形質導入することができる。かかる方法は、限定されるものでないが、エレクトロポレーション、リポソームの利用、およびCaPO4沈降を含む。
【0094】
好ましい実施形態においては、本発明は、通常ヒトおよびヒトに基づくパッケージング培養細胞株に感染するウイルスベクターを含む。例えば、ヘルペスウイルス、エプスタインバーウイルスなどのヒトに通常感染するウイルスから誘導されたベクターを利用することができる。
【0095】
本発明により治療することができる腫瘍は悪性および非悪性腫瘍を含む。治療しうる悪性(原発性および転移性を含む)腫瘍としては、限定されるものでないが、副腎;膀胱;骨;乳房;頚部;内分泌腺(甲状腺、脳下垂体、および膵臓を含む);大腸;直腸;心臓;造血性組織;腎臓;肝臓;肺;筋肉,筋;神経系;脳;眼;口腔;咽頭;喉頭;卵巣;陰茎;前立腺;皮膚(黒色腫を含む);精巣;胸腺;および子宮に発生する腫瘍が挙げられる。かかる腫瘍の例としては、APUD細胞腫(apudoma)、分離腫、鰓腫(branchioma)、悪性カルチノイド症状、カルチノイド心疾患、癌(例えば、Walker、基底細胞、基底有棘細胞、ブラウン-ピアス(Brown-Pearce)、腺管(ductal)、エールリッヒ(Ehrlich)腫瘍、in situ、クレブス(Krebs)2、メルケル(Merkel)細胞、粘液性、非小細胞肺(non-small cell lung)、燕麦細胞、乳頭、硬性、細気管支、気管支原性、扁平細胞、および移行細胞)、プラズマ細胞腫、黒色腫、軟骨芽細胞腫、軟骨腫、軟骨肉腫、線維腫、線維肉腫、巨細胞腫瘍、組織球腫、脂肪腫、脂肪肉腫、中皮腫、粘液腫、粘液肉腫、骨腫、骨肉腫、ユーイング(Ewing's) 肉腫、骨膜腫、腺線維腫、腺リンパ腫、癌肉腫、脊索腫、間葉細胞腫、中腎腫、筋肉腫、エナメル上皮腫、セメント質腫、歯牙腫、奇形腫、胸腺腫、栄養膜腫瘍、腺癌、腺腫、胆管腫、胆脂腫、円柱腫、嚢胞腺癌、嚢胞腺腫、顆粒膜細胞腫、半陰ポジティブ卵巣腫瘍(gynandroblastoma)、肝臓癌、汗腺腫、島細胞腫、Leydig細胞腫、乳頭腫、セルトーリ(Sertoli)細胞腫、卵胞膜細胞腫、平滑筋腫、平滑筋肉腫、筋芽細胞腫、筋腫、筋肉腫、横紋筋腫、横紋筋肉腫、上衣細胞腫、神経節神経腫、神経膠腫、髄芽腫、髄膜腫、神経鞘腫、神経芽細胞腫、神経上皮腫、神経線維腫、神経腫、傍神経節腫、非クロム親和性傍神経節腫、被角血管腫、好酸球増加随伴性血管類リンパ組織増殖症、硬化性血管腫(angioma sclerosing)、血管腫症、グロムス血管腫(glomangioma)、血管内皮腫、血管腫、血管外皮細胞腫、血管肉腫、リンパ管腫、リンパ管筋腫、リンパ管肉腫、松果体腫、癌肉腫、軟骨肉腫、葉状嚢肉腫、線維肉腫、血管肉腫、平滑筋肉腫、白血肉腫、脂肪肉腫、リンパ管肉腫、筋肉腫、粘液肉腫、卵巣癌、横紋筋肉腫、肉腫(例えば、ユーイング(Ewing's)実験的、カポージ(Kaposi's)、および肥満細胞)、新生物および他のかかる細胞に対する腫瘍が挙げられる。
【0096】
医薬調製物
本発明に従って、弱毒化αGalを発現する腫瘍細胞を、腫瘍を処置するための予防または治療ワクチンとして使用する。従って、本発明はまた、これらのトランスジェニック腫瘍細胞(HA1、HA2などとして表される、表1を参照)に関わるヒトおよび動物に対する医薬調製物も含む。医学分野の業者は、医薬組成物の投与量およびスケジュールがヒトおよび動物の年齢、健康、性別、サイズおよび体重に依存して変化しうることを容易に理解するであろう。これらのパラメーターは、それぞれの系に対してフェーズI、IIおよびIII臨床試験における十分確立された方法および分析によりならびに本明細書に記載の例を総括することにより決定することができる。
【0097】
投与に際して、弱毒化腫瘍細胞を、適当な液体ビヒクルまたは賦形剤および任意の補助添加剤などの製薬上許容される担体と組合わせることができる。液体ビヒクルおよび賦形剤は市販されている。それらの説明のための例は、蒸留水、生理学的食塩水、ブドウ糖水溶液などである。
【0098】
非経口、皮下、皮内、筋肉内、経口または腹腔内投与用の好適な製剤としては、水溶性または水分散性形態の活性化合物の水溶液が挙げられる。さらに、油性注射懸濁液としての活性化合物の懸濁液を投与することができる。好適な親油性溶媒またはビヒクルとしては、脂肪油、例えばゴマ油、または合成脂肪酸エステル、例えばオレイン酸エチルまたはトリグリセリドが挙げられる。水性注射懸濁液は、懸濁液の粘度を増加する物質、例えば、カルボキシメチルセルロースナトリウム、ソルビトールおよび/またはデキストランを含有してもよく、場合によっては、懸濁液は安定剤を含有してもよい。また、腫瘍ワクチン細胞を、当技術分野で公知の免疫アジュバント、例えばフロイントの完全アジュバント、無機塩、例えば塩化亜鉛、リン酸カルシウム、水酸化アルミニウム、リン酸アルミニウム、サポニン、ポリマー、脂質または脂質画分(脂質A、モノホスホリル脂質A)、改変オリゴヌクレオチドなどと混合してもよい。
【0099】
通常の担体を用いる投与だけでなく、活性成分を、当業者に公知の様々な特殊な送達薬物技術により投与することができる。以下の例は、説明の目的で記載されたものであり、決して本発明を限定することを意図するものではない。
【実施例】
【0100】
本発明は、これから、以下の実施例について記載する、しかし、本発明の範囲はこれにより限定されることを意図するものでない。特許および刊行物に対する全ての引用は、本明細書に参照により明白に組み入れられるものである。
【0101】
実施例1
αGTを発現するレトロウイルスベクター、pLNCKGの作製
マウスαGT遺伝子の1,077bp断片をαGTの翻訳を促進するKozak配列を含有するフォワードプライマー、5’-ACAAAAGCTTGACATGGATGTCAAGGGAAAAGTAAT-3’、およびリバースプライマー、5’-AATTATCGATTCAGACATTATTTCTAAC-3’によりPCR増幅し、次いでpLNCXのClaIおよびHindIII部位にクローニングしてpLNCKGレトロウイルスベクターを作製した(図1)。このベクターをパッケージング培養細胞株293.AMIZ(YoungおよびLink 「レトロウイルスパッケージング細胞の改良のためのDNAメチル化を排除するキメラレトロウイルスヘルパーウイルスとピコルナウイルスIRES配列(Chimeric retroviral helper virus and picornavirus IRES sequence to eliminate DNA methylation for improved retroviral packaging cells)」 J. Virol. (2000) 74: 5242-5249)中にトランスフェクトして、ベクター生産培養細胞株293.AMIZ/LNCKGを作製した。トランスフェクトした細胞をG418およびZeocinTMの存在のもとで2週間選択した。選択された細胞の混合集団を限定希釈によりサブクローニングした。単一細胞由来のVPCを、色々な組織から確立したヒト上皮癌培養細胞株に効果的に形質導入する能力についてスクリーニングした。その上清が一貫して最高の形質導入効率および1パネルのヒト上皮癌培養細胞株上のαGT発現を生じるクローンを同定して293Z.CKG VPCと名付けた。293Z.CKG VPCはシードバンク(seed bank)の1バイアルから起こしたので、マスター細胞バンク、ワーキング細胞バンク、および生産ロットを作製し、フラスコ内で37℃±1℃、5%±1%CO2中にて拡大した。培地は10%胎児ウシ血清(FBS)および2mm L-グルタミンを補充したRPMI-1640であった。293Z.CKG VPCが十分な密度に到達すると、培養液(上清)を収穫し、濾過し、そして無菌容器中に収穫し、濾過し、そしてプールした。プールを十分混合し、次いで無菌状態で標識した、無菌のプラスチックボトルに満たした(標識は製品名、ロット番号および充填日を含む)。充填したボトルを凍結して-60℃にて貯蔵する。アリコートを安全試験にかける。293Z.CKG VPCからのレトロウイルスを含有する上清を用いて色々なヒト癌培養細胞株を形質導入して(以下の実施例に記述する)αGal(+)細胞全体ワクチンを確立した。
【0102】
実施例2
LNCKGレトロウイルスベクターによる、B16.BL6黒色腫細胞の形質導入
αGal(+)B16細胞を作製するために、2x106細胞を、2x106tu/mLの感染性力価をもつLNCKGレトロウイルスを含有する上清2mLを用いて形質導入した。細胞をネオマイシン耐性に対してG418 1mg/mLを補充した培地中で2週間選択により選択した。この選択期間後に、細胞をαGalエピトープの発現についてニワトリ抗αGalポリクローナル抗体を用いて染色し、蛍光活性化細胞ソーターによりソートした。
【0103】
実施例3
ウサギ赤血球を用いる免疫感作による、α(1,3)-ガラクトシルトランスフェラーゼノックアウト(αGT KO)マウスにおける抗αGal抗体の誘導
雌性および雄性8〜14週齢α(1,3)ガラクトシルトランスフェラーゼ(αGT)ノックアウト(KO)マウスを本研究に用いた。マウスは最初、混合ハプロタイプ(H-2 b/d)であって、増殖と選択によりH-2 b/bハプロタイプのF4同系交配世代からなるαGT KOマウスの現コロニーを得た。これらの動物はαGalエピトープに対して低力価の天然抗体を産生する。抗αGal Abの力価を増加するために、1x108ウサギ赤血球を用いて2回、2週を隔ててマウスを腹腔内(i.p.)に免疫感作した。最後のRRBC注射の1週後に抗αGal Abの力価をチェックして、研究中の全マウスは高い抗αGal Abを有することを実証した。本研究に用いた全マウスは、ELISAにより測定すると1:500希釈より大きい高い抗αGal Ab力価を有する。代表的な実験を図3に示す。
【0104】
実施例4
非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の生存試験
本実験の目的は、癌細胞中のαGalエピトープの発現が、予め存在する抗αGal Abをもつ宿主においてin vivo超急性拒絶を起こしうるかどうかを確認することであった。従って、RRBC注射したαGT KOマウスに、αGalエピトープを発現するまたは発現しない105 非照射B16黒色腫細胞をチャレンジし、腫瘍発生を測定した(図4)。
【0105】
図5はチャレンジ後15日の腫瘍サイズを示す。予想通り、天然αGal(-) B16を受けた19マウスのうちの11マウスは、チャレンジ後2週に測定可能な腫瘍を発生した。同様に、B16.LNL αGal(-) B16細胞を受けた20マウスのうちの10マウスは腫瘍を発生した(それぞれ、57%および50%、カイ自乗p>0.05で、有意でない)。しかし、マウスがαGal(+)B16細胞を受けた時は、有意なより少数の動物が腫瘍を発生した。20動物のうち5動物(25%)だけが明白な腫瘍を発生した(カイ自乗、p=0.03、対照と有意差あり)。さらに、対照および模擬(mock)形質導入した細胞を用いてチャレンジした動物では、αGal発現細胞を受けたマウスにおいて発生した腫瘍と比較して、腫瘍が相当大きかった。平均サイズ200mm3の腫瘍が両方の対象グループに観察され、グループ間の差は統計的に有意でなかった(F試験、p=0.38)。しかし、B16.αGal(+)細胞を受けたグループの腫瘍サイズに有意差が観察された。試験グループの平均サイズ腫瘍は36mm3であって、対照動物と比較して腫瘍サイズは約80%の低下を示した(F試験、p=0.002)。
【0106】
この結果はαGalを発現するB16細胞を用いてチャレンジするとより少数の動物が小さい腫瘍を発生したことを実証するものであり、抗αGalを前感作したマウスは、αGalを発現する細胞のより効果的なin vivoクリアランスを、天然または模擬(mock)形質導入したαGalネガティブB16対照のそれと比較して媒介できることを示す。
【0107】
腫瘍をチャレンジ後30日に測定したときも、比較しうる結果を観察した。B16野生型を受けたグループでは17マウスのうちの13マウスが腫瘍を有した(76%)。模擬(mock)ベクターを用いて形質導入した細胞を受けたグループでは18マウスのうちの13マウスが腫瘍を有した(72%)。しかし、αGal発現細胞を受けたグループではマウスの50%だけが測定しうる腫瘍を有した。さらに重要なこととして、対照グループのそれぞれ2動物、計4動物を大きな腫瘍が発生したため安楽死させなければならなかった。αGalを発現する細胞を受けた動物は腫瘍移植後30日までに死亡したものはなく、αGalを発現する細胞を用いてチャレンジした動物の生存率の増加を示した。
【0108】
図6は、対照、模擬(mock)およびαGalを発現するB16細胞によるチャレンジ後の腫瘍発生の動力学を示す。両方の対照αGal(-)グループの腫瘍は、極度に速く増殖し7日間で容積がほとんど2倍になった。これに反して、αGal(+)B16細胞を用いてチャレンジしたマウス由来の腫瘍は早い時点で有意に遅い速度で増殖した(p=0.03)。これは、免疫機構が腫瘍の増殖を制限し、腫瘍を保持するマウスの生存を延ばすのを助けうることを示唆する。
【0109】
αGal(+)B16細胞による致死的チャレンジ後のマウスの長期生存
対照αGal(-)、模擬(mock)形質導入αGal(-)B16およびαGal(+)B16を受けたグループからのマウスを90日間、毎週観察した(図7)。図に示したように、天然B16を受けた10動物のうちのどれもチャレンジで生き残らず、模擬(mock)形質導入したαGal(-)B16細胞を受けた11マウスのうちの1マウスだけが皮下注射で生き残った。両方のグループは非常に類似する生存曲線であり、NeoR遺伝子産物はB16の拒絶および/または免疫原性に有意な変化を誘導しないことを示した(ログランク(Logrank)試験p=0.5、差は統計学的に有意でない)。これに反して、αGalを発現する非照射B16細胞を受ける動物の47%が、致死量チャレンジで生き残った。19マウスのうちの9マウスは皮下注射後80日間、無腫瘍のままであった。Kaplan-Meier生存分析を用いると、対照グループと比較して、αGal(+)B16細胞を皮下注射したグループの生存率に有意な増加が観察された(ログランク(Logrank)試験、p<0.02)。
【0110】
αGal(-)B16を用いて注射した両方の対照グループでは、動物の60%近くはチャレンジ後最初の30日に死亡した。これに反して、αGal(+)B16を用いて注射した実験グループは、5動物だけ(26%)を、チャレンジ後の最初の月に安楽死させねばならなかった。
【0111】
第2の独立実験において、同様な結果を得た。天然αGal(-)B16を皮下注射した19マウスのうちの2マウスだけが生き残った。比較として、21マウスのうちの4マウスだけが模擬(mock)形質導入αGal(-)B16細胞の致死的皮下チャレンジで生き残った。これに反して、天然αGal(+)B16を皮下注射した20マウスのうちの8マウスが生き残り、80日を越えて無腫瘍のままであった。
【0112】
全体としてこの結果は、αGT遺伝子の発現とB16のグリコシル化パターンの変化が生癌細胞のin vivo破壊を誘導して腫瘍増殖が低下するのを助けて腫瘍保持マウスを延命することを実証する。さらに重要なこととして、チャレンジしたマウスのほぼ40%が無腫瘍のままであるので、αGal発現細胞の注射は、致死性の高いαGal(-)黒色腫細胞の増殖を制御することができる強い免疫応答を誘導しうる。
【0113】
非照射αGal(+)B16細胞による致死量チャレンジ後の生存は、野生型αGal(-)B16に対する記憶保護免疫を誘導する。
本発明者らは、非照射αGal(+) B16細胞の皮下注射から生き残った動物は天然αGal(-) B16腫瘍に拡大した細胞性免疫を発生するという仮説を立てた。この仮説を試験するために、全ての生き残った無腫瘍マウスに野生型B16(αGalネガティブ)を再チャレンジした。年齢の適合したマウスを対照に加えて、αGal(-)B16を注射した(図8)。予想通り、αGal(-)B16をチャレンジした12対照マウスのうちの11マウスは大きな着色腫瘍を発生する皮下黒色腫によって死亡した(図9)。その一方で、非照射αGal(+)B16黒色腫を最初に受けて生き残ったマウスはいずれも腫瘍を発生しなかった。8マウスは全て、70日間無腫瘍のままであり、最初にαGal(+)B16細胞を拒絶したマウスの生存率を有意に増加した(ログランク(logrank)検定、p<0.001)。興味深いことに、B16黒色腫に対する保護は、先に他の研究者が記載した自己免疫色素脱失(白斑)と関係がなかった(Overwijkら, 「マウスにおいて、自己抗原をコードする組換えワクシニアウイルスによるワクチン接種は自己免疫白斑および腫瘍細胞破壊を誘導する:CD4+ Tリンパ球の必要性(Vaccination with a recombinant vaccinia virus encoding a self antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4+ T lymphocytes)」 Proc. Natl. Acad. Sci. USA (1999) 96:2982-2987;Overwijkら, 「自己反応性CD8+ T細胞の機能的寛容状態の逆転後における腫瘍退行と自己免疫(Tumor regression and autoimmunity after reversal of functionally tolerant state of self-reactive CD8+ T cells)」 J. Exp. Med. (2003) 198:569-580;van Elsasら, 「抗CTLA-4およびGM-CSF産生ワクチンを用いるB16黒色腫の併用免疫療法は、自己免疫色素脱失を伴う皮下および転移性腫瘍の拒絶を誘導する(Combination immunotherapy of B16 melanoma using anti-CTLA-4 and GM-CSF-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation)」 J. Exp. Med. (1999) 190:355-366)。
【0114】
この結果は、αGal(+)細胞の致死的チャレンジで生き残ったマウスが天然αGal(-)腫瘍に対して強い免疫を発生することを実証した。この細胞性および恐らくは体液性の免疫応答は生き残ったマウスを野生腫瘍による第2の致死的チャレンジから保護し、全ての保護されたマウスにおいて免疫応答が天然αGal(-)腫瘍B16に拡大されたことを示す。
【0115】
さらに、T細胞の媒介する免疫が保護されたマウスに誘導されたという仮説を実証するために、黒色腫特異的T細胞培養物を作製してαGal(-)B16黒色腫および非特異的培養細胞株EL-4およびMC38に対して試験した(図10)。
【0116】
図11に示したように、B16黒色腫を特異的に溶解することができる強いCTLが保護されたマウスに誘導されることを標的細胞中のLDH放出により測定した。
【0117】
全体として、これらの結果は、αGal(+)細胞の注射がマウスの生存率を増加することを実証する。さらに、αGal(+)B16細胞を拒絶しかつ最初の致死的注射で生き残ったマウスは天然αGal(-)B16黒色腫腫瘍に対して拡大した強い免疫を発生することができる。これは、αGal(+)B16細胞の拒絶後に記憶T細胞が媒介する免疫が誘導されて、αGal(-)B16腫瘍を認識して致死的腫瘍投与からマウスを保護することができるのを示す。
【0118】
実施例5
非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死量の静脈注射による、αGTノックアウトマウスの播種性黒色腫転移モデル
さらに、高力価の抗αGal AbをもつマウスがαGal(+) B16黒色腫細胞を拒絶できることを実証するために、肺黒色腫転移モデルを用いた。マウスに、105非照射αGal(-)またはαGal(+)B16黒色腫細胞を静脈(i.v)注射した。3週後、肺黒色腫転移を数えた(図12)。αGal(-)B16細胞を注射したマウスは多数の肺転移を有する。これに反して、αGal(+)B16細胞を注射したマウスは肺腫瘍量が低下した(図13)。さらに5マウスのうちの2マウスは無腫瘍であった。この結果は、予め存在する抗αGal AbがαGal(+)B16細胞のクリアランスに大きな役割を果たすことを示す。
【0119】
実施例6
αGal(+)細胞を用いる予防ワクチン接種による皮下腫瘍発生の予防
本実験の目的は、皮下腫瘍の予防を、先に記載のαGal(+)ワクチンを用いるワクチン接種により達成できるかどうかを決定することである(LaTempleら, 「抗αGal抗体と複合した腫瘍ワクチンの増加した免疫原性:α(1,3)ガラクトシルトランスフェラーゼのノックアウトマウスにおける研究(Increased immunogenicity of tumor vaccines complexed with anti-αGal: studies in knockout mice for α(1,3) galatosyl transferase)」, Cancer Res. (1999) 59:3417-3423)。マウスをRRBCを用いて先に記載のとおり免疫感作した。最後のRRBC注射後1週に、マウスは照射細胞ワクチンの第1用量を受けた。マウスに、照射天然αGal(-)B16、対照ベクター(pLNL、ネオマイシン耐性遺伝子をコードする)を用いて形質導入したαGal(-)B16、またはαGal(+)B16(ネオマイシン耐性遺伝子およびαGT遺伝子をコードするベクターを用いて形質導入した)のいずれかを注射した。複数のマウスは照射細胞ワクチンを受けなかった。細胞ワクチン接種を2週後に繰返した。最後のワクチン接種後2週に、マウスに105非照射αGal(-)B16細胞を皮下注射した(図14)。腫瘍を週2回、90日間測定した。図15に示すように、B16細胞ワクチンを受けなかった10マウスのうちチャレンジで生き残ったものはゼロであり、チャレンジ後50日までに死亡した。天然αGal(-)B16およびαGal(-)B16/NeoRワクチンをワクチン接種したマウスには若干の保護が観察された。B16チャレンジで14マウスのうち4マウスおよび12マウスのうち5マウスがそれぞれ生き残った。B16とB16/NeoRをワクチン接種したマウス間に統計学的な差はなく、この条件のもとでNeoR遺伝子産物はB16の免疫原性を増加しないことを示す(p>0.2、ログランク(Logrank)試験)。これに反して、マウスにαGal(+)B16細胞をワクチン接種すると、20マウスのうちの12マウスがチャレンジで生き残り、かつ90日間無腫瘍のままであって、有意なさらに大きい保護が観察された(p<0.001、ANOVA、p=0.08 ログランク(Logrank)試験)。この結果は、照射αGal(+)細胞によるワクチン接種がαGal(+)B16ワクチン接種マウスの60%において皮下黒色腫腫瘍の発生を防止しかつαGal(-)B16黒色腫をチャレンジしたマウスを有意に延命することを実証する。
【0120】
本発明者らは、生αGal(-)B16による注射から保護されたマウスでは、天然αGal(-)B16細胞を認識できるT細胞がαGal(+)B16ワクチンによるワクチン接種後に誘導されたという仮説をたてた。この仮説を試験するために、αGal(+)ワクチンを用いてワクチン接種したマウスから脾細胞を収穫し、刺激の存在または不在のもとで6時間培養した。刺激を最大限にするために、PMA/Ca++イオノフォアを用いた。細胞を特異的認識を測定するために105照射B16細胞ともにまたはCA320M、同一のH-2 b/bハプロタイプをもつ非特異的αGal(-)培養細胞株とともに培養した。インキュベーション後、細胞を収穫して細胞内TNF-αを染色した。前方散乱プロットでリンパ球のFACSゲーティングによる検出を実施した(図16)。αGal(+)B16ワクチン接種したマウスから収穫したT細胞はPMA/Ca++イオノフォアにより効率的に活性化された。このポリクローナルアクチベーター技法により活性化されたリンパ球のパーセントは本実験で検出された最大活性化と考えられる。休止(刺激されない)T細胞とCA320Mにより刺激されたT細胞はTNF-aを産生することができず、B16-αGal(+)ワクチン接種後にCA320Mの抗原を認識できるT細胞前駆体が誘導されなかったことを示した。これに反して、B16-αGal(+)を用いるワクチン接種は、in vitroでB16-αGal(-)を特異的に認識するT細胞前駆体を誘導した。この結果は、αGal(+)B16を用いるワクチン接種後に誘導されたT細胞がαGal(+)B16処置したマウスのほぼ半分の腫瘍予防の原因でありうることを示唆する。
【0121】
実施例7
αGal(+)細胞ワクチン接種による、予め形成された皮下黒色腫腫瘍の治療
天然αGal(-)B16およびαGal(-)模擬(mock)形質導入B16/NeoRを用いるワクチン接種が以上の実験において類似したデータを生じたので、統計学的解析力を増加するネガティブ対照として照射αGal(-)B16/NeoRを単独で用いるワクチン接種による次の実験を実施した。
【0122】
次の実験の目的は、予め形成された皮下黒色腫腫瘍の治療をαGal(+)B16ワクチンによるワクチン接種により達成しうるかどうかを決定することであった。有効な予防治療が予め形成された腫瘍の治療にも有効でありうるかは明らかでない。腫瘍モデルとしてB16黒色腫の特定の事例において、複数の治療計画が免疫感作の予防スケジュールに有効であったこと、そして予め形成された皮下黒色腫腫瘍の治療に与える効果は低いかまたは無かったことが立証されている。例えば、マウスmTRP-1をコードする組換えワクシニアウイルスを用いるワクチン接種は皮下黒色腫腫瘍の発生を予防するが、予め形成された皮下腫瘍の治療に効果がない(Overwijkら, 「マウスにおいて自己抗原をコードする組換えワクシニアウイルスを用いるワクチン接種は自己白斑および腫瘍細胞破壊を誘導する:CD4+Tリンパ球の必要性(Vaccination with a recombinant vaccinia virus encoding a self antigen induces autoimmune vitiligo and tumor cell destruction in mice: requirement for CD4+ T lymphocytes)」” Proc. Natl. Acad. Sci. USA (1999) 96:2982-2987)。同様に、ペプチド特異的免疫感作は強いT細胞免疫を誘導するが、予め形成された腫瘍に対する治療に有効なことは稀である(Davilaら, 「細胞傷害性Tリンパ球エピトープペプチドワクチン接種、CpGオリゴデオキシヌクレオチドアジュバントおよびCTLA-4遮断による抗腫瘍免疫の産生)(Generation of antitumor immunity by cytotoxic T lymphocyte epitope peptide vaccination, CpG oligodeoxynucleotide adjuvant and CTLA-4 blockade)」 Cancer Research (2003) 63:3281-3288)。さらに、腫瘍特異的T細胞の唯一の存在は、腫瘍根絶を効果的に誘導するための必要条件であるが十分ではない。
【0123】
本発明者らは、αGal(+)ワクチンは、予め形成されたαGal(-)腫瘍を拒絶できる強い細胞依存性腫瘍免疫を誘導することができるという仮説を立てた。この仮説を試験するために、本発明者らは予め形成された腫瘍を治療するように設計した実験を実施した(図17)。
【0124】
マウスに、105非照射B16細胞を皮下注射して無作為化した。チャレンジ後3日に、それらのマウスに、照射αGal(-)B16/NeoRまたは照射αGal(+)B16を皮下にワクチン接種した。3日後、照射細胞ワクチンを用いるワクチン接種を繰返した。基線対照マウスは非照射B16による皮下注射を受け、照射細胞ワクチン治療を受けなかった(無ワクチングループ)。図18aおよびbは、ワクチン接種しなかったマウス(n=11)、αGal(-)B16/NeoRによるワクチン接種したマウス(n=24)およびαGal(+)B16ワクチンを受けたマウス(n=23)の腫瘍増殖の動力学を示す。データは平均および誤差バー、SEMを表す。統計学的解析は、対照マウスとαGal(+)B16細胞ワクチンを受けたマウスを比較した時、勾配の有意差があるのを示す(p<0.009)。この結果は、非照射αGal(+)B16ワクチンを用いてワクチン接種したマウスが、増殖のより遅い腫瘍をより少数発生することを示す。
【0125】
実験#2においては、マウスはさらなる用量の細胞ワクチンを受けた。非照射αGal(-)B16による皮下注射だけを受けたマウスは、観察の最初の月に非常に増殖の早い大きな腫瘍を発生した(n=15)。同様に、対照αGal(-) B16ワクチンを注射したマウスは非常に増殖の早い大きな腫瘍を発生した(n=29)。これらの2グループのスロープの統計学的比較は、これらに有意差のないことを示した(p=0.17)。これは、αGal(-) B16/NeoRによるワクチン接種は皮下に予め形成された黒色腫腫瘍の治療に影響を与えないことを示す。これに反して、αGal(+)B16を受けたマウスは増殖のより遅い腫瘍をより少数発生した(n=33)。このグループの腫瘍増殖を対照グループと比較すると、統計学的に有意差があった(p<0.03)。
【0126】
これらの2つの実験は、αGal(-)B16細胞を用いるワクチン接種は予め形成された皮下黒色腫腫瘍を治療することができないことを示した。一方、予め形成された皮下黒色腫腫瘍はαGal(+)B16ワクチンを用いるワクチン接種により成功裏に治療することができる。
【0127】
αGal(+)B16ワクチンを用いて処置した、予め形成された皮下腫瘍をもつマウスの生存分析
予め形成されたαGal(-)B16皮下腫瘍をもち、αGal(+)B16刺激した細胞をワクチン接種したマウスは、非ワクチン接種対照およびαGal(-)模擬(mock)ワクチン接種したグループと比較すると、生存の延長を示した(図19)。非ワクチン接種対照およびαGal(-)模擬(mock)ワクチン接種動物ではそれぞれ9マウスのうちのゼロマウスおよび20マウスのうちの1マウスだけが皮下チャレンジで生き残ったが、αGal発現B16細胞で処置した19マウスのうち5マウスがチャレンジ後70日より長く生存した。非ワクチンおよび模擬(mock)ワクチングループのメジアン生存時間はそれぞれ27および26日であった。これに反して、αGal(+)B16ワクチンを受けたマウスのメジアン生存時間は有意に増加した(39日)。この結果は、αGal(+)B16ワクチン細胞が予め形成された皮下黒色腫腫瘍を有効に治療できることを、生存動物数の増加(26%対5%)およびメジアン生存時間の延長(39対26日)により実証する。
【0128】
第2の実験においては、マウスは3用量の細胞ワクチンを、非照射αGal(-)B16による最初の皮下注射後、4、11および18日に受けた(図20)。それぞれの時間のワクチン用量は3x105細胞であった。マウスに非ワクチン接種(n=12)、αGal(-)B16によるワクチン接種(n=23)またはαGal(+)B16(n=26)によるワクチン接種のいずれかを実施した。
【0129】
図20は観察の70日後のKaplan-Meier生存分析を示す。生存曲線のログランク(logrank)検定比較は、対照非ワクチン接種およびαGal(-)B16ワクチン接種マウスと比較して、αGal(+)B16ワクチンを用いて処置した皮下黒色腫腫瘍を保持する生存マウスの数に有意差を示した(p<0.005)。非ワクチン接種の12マウスのうちB16による皮下注射で生き残ったものはなかった。同様に、αGal(-)B16ワクチンをワクチン接種した23マウスのうちB16による皮下注射で生き残ったものはなかった。これに反して、αGal(+)B16ワクチンを受けた26マウス(42%)のうち11マウスは、αGal(-)B16黒色腫の致死的皮下注射後、70日間生き残った。
【0130】
対照と模擬(mock)ワクチン接種したマウスのメジアン生存に有意差はなかった(それぞれ38および42日、ログランク(logrank)検定でp=0.43、ns)。これに反して、αGal(+)B16で処置したマウスのメジアン生存は、60日より長かった。これはαGalワクチン接種グループのメジアン生存の有意な増加を表した(p<0.005)。
【0131】
この結果はさらに、αGal(+)B16ワクチンによる予め形成された皮下黒色腫腫瘍の治療は、αGal(-)ワクチン接種または無処置と比較して有意に有効であることを実証した。
【0132】
実施例8
αGal(+)B16細胞ワクチンによる肺黒色腫転移の治療
本発明者らはさらに、播種性疾患のモデルとして肺黒色腫転移の治療における有効性を評価した。この実験は、腫瘍を保持する患者が外科的に除去できない播種性疾患によりほとんど死亡しうるので、臨床の視点から非常に重要である。そこで、RRBC免疫感作マウスに105非照射αGal(-)B16を静脈内にチャレンジして、無作為化した。マウスに照射B16/NeoRワクチン(αGal(-)、n=6)またはαGal(+)B16ワクチン(n=7)を皮下に処置した。ワクチン(2x105照射細胞)は、B16のi.v.注射後、4、11および21日に、皮下投与した。マウスをチャレンジ後30日に犠牲にして、黒色腫転移の数を数えた(図22)。肺転移の数は、3マウスで「数え切れないほど多数」(任意の値>250腫瘍)であり、他のマウスでは30腫瘍を数える一方、2マウスだけは無腫瘍であった。さらに、このグループの動物の1つは腹腔内に3つのさらなる転移性小結節を示し、該疾患の肺以外の他の場所への播種を実証した。
【0133】
これに反して、αGal(+)B16ワクチンを用いて処置したマウスは、全て無腫瘍であり、予め形成された腫瘍がαGal(+)を発現するワクチン療法により非常に成功裏に治療されたことを実証した。
【0134】
第2の独立した実験(図23)においては非照射αGal(-)B16の5倍増加用量上昇を用いた。マウスに5x105生B16をi.v.注射して肺黒色腫転移を予め確立した。4日後に、対照およびαGal(+)B16細胞を用いるワクチン接種を先の通り実施した。マウスは非ワクチン処置(n=10)、αGal(-)B16によるワクチン接種(n=11)またはαGal(+)B16(n=11)の3用量によるワクチンのいずれかを受けた。
【0135】
B16によるi.v.チャレンジ後30日に、マウスを安楽死させて肺黒色腫転移を数えた(図23)。腫瘍増殖も肺腫瘍負荷の計数によりおよび肺組織重量測定により評価した(図24)。
【0136】
多数の肺黒色腫転移が、非ワクチン接種マウスならびに対照ワクチン接種グループの肺に見出された。統計学的比較を実施するため、肺組織の重量を用いた。対照と非ワクチン接種グループの肺負荷の間の差は統計学的に差がなかった(アンペアードt検定、p=0.66, ns)。これに反して、αGal発現B16ワクチンによりワクチン接種したマウスには肺負荷の有意な低下が観察された(ワンウエイANOVA、p<0.006)。
【0137】
第1実験の観察と同様に、対照および非ワクチン接種グループからのいくつかのマウスは「数え切れないほど多数の」黒色腫転移を有した。さらに、2動物は、肺組織の他に、肺外に散乱した黒色腫腫瘍を有し、播種性疾患であることを示した。αGal(+)B16ワクチン接種したマウスは、「数え切れないほど多数の」黒色腫腫瘍を有するものはなく、肺外の腫瘍も有しなかった。これは、αGal(+)B16ワクチンを用いるワクチン接種が播種性転移性黒色腫を効果的に治療できることを示す。さらに、αGal(+)B16ワクチンによるワクチン接種は全身疾患のさらなる拡大を予防することができる。
【0138】
実施例9
αGal(+)B16細胞によるワクチン接種後の、αGal(-)B16特異的T細胞前駆体の誘導を実証するためのT細胞研究
この技術の基本的疑問の1つは、αGal(+)細胞を用いるワクチン接種がαGal(-)腫瘍に対して反応しうるT細胞が媒介する免疫を誘導できるかということである。上記の実験において、本発明者らは、αGal(+)B16ワクチンが、予め形成されたαGal(-)腫瘍のクリアランスを媒介しうる強い免疫を誘導することを実証した。この免疫は、マウスをαGal(-)B16ワクチンを用いてワクチン接種したときには誘導されない。さらに、αGal(-)B16腫瘍に特異的なT細胞前駆体がαGal(+)B16ワクチンを用いるワクチン接種後に誘導されたことを実証するために、in vivo T細胞研究を実施した。これらの実験の目的は、T細胞によるαGal(-)B16細胞の特異的認識を実証することにあった。T細胞を対照ワクチンαGal(-)B16を用いてワクチン接種したマウスグループまたはαGal(+)B16照射細胞を用いてワクチン接種したマウスから収穫した(図25)。2つのタイプの研究を実施して、異なる方法により、同じ結論、すなわち、照射αGal(+)B16細胞を用いてワクチン接種したマウスはαGal(-)B16腫瘍細胞を特異的に認識しうるT細胞前駆体の増加した数を有することを実証した。
【0139】
実験の第1セットにおいては、細胞内サイトカインTNF-αをFACSにより検出した。細胞を照射αGal(-)B16細胞を用いてまたはαGal(+)B16細胞を用いてワクチン接種したマウスから、最後の皮下ワクチン接種後2週に収穫した。脾細胞をネガティブ対照として刺激を与えずに培養した。刺激については、脾細胞をネガティブ対照としてCA320M(αGal(-)同系H-2 b/bハプロタイプ)とともに、またはαGal(-)B16細胞とともに共培養した。6時間の刺激後に、細胞を収穫して細胞内TNFαを染色した。TNFα(+)リンパ球のパーセント増加を、特異的にαGal(-)B16細胞を認識するαGal(+)B16細胞を用いてワクチン接種したマウスの脾臓に検出した(図26)。これらのT細胞は、CA320Mととも培養すると、TNFαを産生せず、これらはB16を特異的に認識するが、無関係な同系培養細胞株を認識しないことを示した。黒色腫に特異的T細胞前駆体の数の増加に加えて、これらの細胞が産生するTNFαの量は、FACSにより検出した平均蛍光強度により測定すると、有意に増加した(図26)。TNFα(+)細胞におけるMFIの4倍増加を検出した。模擬(mock)ワクチン接種マウスからの脾細胞をαGal(-)B16とともに培養すると、TNFα(+)細胞の4%しか検出されず、MFIは17であった。これに反して、αGal(+)B16細胞ワクチンを受けたマウスからのT細胞をαGal(-)B16とともに培養すると、7%のTNFα(+)細胞が検出され、MFIは69であった。これは、脾臓中に存在するT細胞前駆体のパーセントが2倍増加したことを表す。この実験を全3回繰返して同様な結果を得た。
【0140】
この結果は、αGal(+)B16細胞を用いるワクチン接種は黒色腫標的のT細胞特異的認識によりin vitroで検出された強いT細胞免疫を誘導し、そしてαGal(-)B16を用いるワクチン接種はこれを誘導しないことを実証した。
【0141】
異なるセットの実験においては、細胞表面活性化マーカーを用いてαGal(-)B16黒色腫培養細胞株の特異的T細胞認識を測定した。T細胞受容体(TCR)が関与すると、T細胞が複数の細胞表面分子をアップレギュレートしてリンパ球の活性化状態を示すことはよく記載されている。これらの分子の1つはIL-2受容体α鎖またはCD25である。TCRが関与すると、CD25がアップレギュレートされて、活性化後1日で、FACSにより検出されうる。同様に、T細胞が活性化するとCD69(または極初期活性化抗原(VEA))がアップレギュレートされる。CD69はシグナル伝達受容体として色々な細胞において機能し、リンパ球活性化の初期事象に関わり、色々なサイトカイン、およびその受容体の合成を誘導することによるT細胞活性化に寄与する。両方の活性化マーカー(CD25およびCD69)は休止T細胞では非常に低レベルでしか発現されない。αGal(+)B16細胞を用いるワクチン接種がαGal(-)B16を特異的に認識することができるT細胞前駆体を誘導したことを実証するために、活性化マーカーのアップレギュレーションを、認識および活性化を測定するパラメーターとして用いた。細胞をαGal(-)B16を用いてワクチン接種した、またはαGal(+)B16を用いてワクチン接種したマウスから収穫した。これらの細胞を無刺激でまたはネガティブ対照培養細胞株(CA320M)を用いてまたはαGal(-)B16を用いて培養した。培養の24時間後、細胞を収穫し、染色してCD25またはCD69を検出した。取得は、生命染色(vital staining)7-AAD(live細胞)を排除する細胞をゲーティングして実施した。予想されるように、休止T細胞(無刺激)および同系非黒色腫培養細胞株CA320Mにより刺激した細胞は非常に低レベルの活性化マーカーしか発現しない(図27aおよび27b)。αGal(-)B16を受けたマウスからの脾細胞をB16とともに培養すると、活性化マーカーのいくらかのアップレギュレーションが観察された。これはマウスが天然αGal(-)B16ワクチンを受けると低い程度の免疫反応性が観察されうることを示した文献からの従来の報告を実証する。しかし、この反応性は予め形成された黒色腫腫瘍を予防および/または治療するのには十分でない。他方、T細胞をαGal(-)B16を用いて培養したとき、αGal(+)B16をワクチン接種したマウスからリンパ球活性化の増加が検出され、CD25(+)およびCD69(+)細胞の数の増加が測定された。
【0142】
この結果も再び、αGal(+)B16細胞を用いるワクチン接種は、αGal(-)B16黒色腫細胞を特異的に認識できるT細胞前駆体を誘導したことを実証した。
【0143】
実施例10
αGal(+)またはαGal(-)B16細胞ワクチンを用いてワクチン接種したマウスからの養子T細胞導入による、予め形成された転移性黒色腫腫瘍の治療
上記のin vitro実験は、αGal(-)B16ワクチン接種を受けたマウスと比較すると、αGal(+)B16細胞を用いてワクチン接種したマウスではより多くの量と質の黒色腫特異的T細胞が誘導されることを実証した。これらの黒色腫特異的T細胞は数が増加し(脾細胞中により多くのT細胞が見出された)、それらはより多くのTNFαを産生した。また、B16を用いて共培養するとより多くの脾細胞が活性化された(CD25およびCD69のアップレギュレーション)。以上に示した実験において、皮下および肺性転移の両方を保持してαGal(+)B16を受けたマウスは、より長い生存と増加した肺腫瘍のクリアランスを示した。これらの2グループのデータがあるので、本発明者らは事実としてαGal(+)B16ワクチン接種により誘導されたT細胞が予め形成された黒色腫腫瘍の治療の原因であると推論することができよう。しかし、大量の黒色腫特異的T細胞は、それらが寛容状態(tolerant state)にあるので予め形成された皮下黒色腫腫瘍を治療するのには不十分であることが示されており、この推論が正しいのかは明らかではない(Overwijkら, 「自己反応性CD8+ T細胞の機能的寛容状態の逆転後の腫瘍退行と自己免疫(Tumor regression and autoimmunity after reversal of functionally tolerant state of self-reactive CD8+ T cells)」 J. Exp. Med. (2003) 198:569-580)。本発明者らは、αGal(+)B16細胞を用いるワクチン接種は記憶を呼び戻すと急速に活性化されうる細胞の媒介する強い免疫を誘導し、これが予め形成された疾患を保持するマウスにおける腫瘍クリアランスの原因であるという仮説を立てた。この仮説を実証するために、養子細胞導入実験を実施した(図28)。ドナーマウスを、先に記載したように、照射αGal(+)B16またはαGal(-)B16ワクチンの3用量を受けるワクチン接種を実施した。レシピエントマウスに生αGal(-)B16をi.v.注射して肺黒色腫転移を確立し、そして無作為化した。非照射B16のi.v.注射後4日にマウスは、αGal(+)またはαGal(-)B16細胞をワクチン接種したドナーからのT細胞を受けるかまたは受けなかった。4週後、肺黒色腫転移負荷を、肺腫瘍を数えることにより、およびブロックで得た肺の重量を測ることにより測定した。実験を2回実施し、両者の結果を図29Aおよび29Bに示した。実験#1は、T細胞療法を施さないB16をi.v.に受けたマウス(n=16)、αGal(-)B16ワクチン接種したマウスからのT細胞を受けたマウス(n=15)、およびαGal(+)B16ワクチン接種したマウスからのT細胞を受けたマウス(n=17)の平均肺重量を示す。バーは肺重量の平均および誤差バー、SEMを表す。対照マウスにおけるおよび模擬(mock)ワクチン接種したマウスにおける大腫瘍および肺負荷の有意な増加を実証した。T細胞を受けないマウス(対照)と模擬(mock)ワクチン接種したマウスからのT細胞を受けたマウスの間の肺黒色腫転移の差は統計学的に有意でなかった(p>0.05)。これに反して、肺黒色腫負荷の有意な低下がαGal(+)B16ワクチン接種したマウスからT細胞を受けたマウスに観察された(ワンウエイANOVA、p<0.006)。同様な結果が第2の独立実験において観察された。T細胞を受けないマウスは64肺黒色腫腫瘍の平均値を有する(n=7)。αGal(+)B16ワクチン接種したマウスからの細胞を受けたマウスは多数の肺黒色腫転移を有した(平均=90、n=10、p=>0.05)。対照的に、αGal(+)B16細胞を用いてワクチン接種したマウスからの脾細胞を受けたマウスには31肺黒色腫腫瘍しか観察されなかった。これは、αGal(+)B16細胞を用いてワクチン接種したマウスからのT細胞を受けたマウスの肺黒色腫負荷における有意な低下を表す(アンペアードt検定p<0.05)。
【0144】
αGal(+)ワクチン接種したマウスからの細胞の唯一の治療による、予め形成された肺黒色腫転移低下の著しい成功は、始めて、強い細胞が媒介する免疫がαGal(+)細胞ワクチンによって誘導され、そしてαGal(-)ワクチンによって誘導されないことを実証するものである。この強い細胞依存性腫瘍免疫は、ほとんど間違いなく、播種性の予め形成された疾患の治療の原因である。
【0145】
実施例11
非照射αGal(-)CA320M細胞の致死用量の皮下注射後の、αGal(-)またはαGal(+)照射CA320M肉腫細胞を用いてワクチン接種したαGal(-)ノックアウトマウスの生存
上記B16黒色腫細胞を用いる予防ワクチン接種実験を、αGal(-)ノックアウトマウスから誘導した、従って、宿主細胞と完全に同系である色々な腫瘍培養細胞株を用いて繰返した。この腫瘍培養細胞株はCA320Mであって、9,10-ジメチル-1,2-ベンズ-アントラセン(DMBA)2mgおよび3-メチルコラントレン(3-MC)1mgを溶解した250μlのオリーブ油を、αGTノックアウトマウス中に2週間隔で腹腔内注射することにより取得した。1マウスは、最初の注射後8ヶ月に腫瘍が現れた。腫瘍はほぼ2cm幅、3cm長さそして1.5cm深さの大きな塊で腹腔内に局在した。腸間膜に位置する転移性小結節が認められた。原発塊(primary mass)と腸管を採取し、培養して組織病理学試験を実施した。腫瘍の原発部位に随伴する腸間膜から採取した転移性小結節の培養に成功し、CA320Mと名付けた。原発および移植腫瘍の凍結およびパラフィンセクションの両方の組織病理学試験は、それぞれ、分化の低い肉腫と一致する形態であることを実証した。ヘマトキシリンおよびエオシン染色ならびに免疫組織化学分析は、CA320Mが小腸の肉腫のような消化管間質腫瘍のファミリーに分類されうることを示唆した。αGT KOマウスに誘導されたCA320M細胞はαGalエピトープを特異的に検出するIB4イソレクチンと結合せず、このマウス腫瘍は、予想通り、機能性αGT酵素を欠き、それ故にαGalエピトープを欠くことを立証する。CA320M細胞はHSV-1に基づくベクターによる感染に感受性があり、HE7αGal1による形質導入後にαGalエピトープを発現した。
【0146】
GT KOマウスを、14日間隔で2回の2x107ウサギ全血球の皮下注射により初回刺激し、αGalエピトープに対する免疫を発生させた。CA320M細胞に、10 MOIのHDKgalまたはHDKgalΔsalIのいずれかを用いて8時間形質導入して、CA320MαGal(+)またはαGal(-)細胞をそれぞれを得た。簡単に説明すると、HDKgalはαGT遺伝子をKozak配列とともにpHD1アンプリコンHSV-1ベクター中に挿入することにより発生させた。従って、pHDKgalは、CMVプロモーターの制御下にある単一の真核生物遺伝子、αGTを保持する。突然変異αGT遺伝子を構築するために、αGT酵素の対応する触媒ドメインに配置したユニークなSalI制限酵素切断部位を切断し、クレノー(Klenow)およびdNTPを用いて満たした。得られるフレームシフト突然変異は酵素を非機能化するポリペプチドの未熟な末端を生じる。突然変異αGT遺伝子を、pHD1アンプリコン中にもクローニングし、両方のアンプリコンを、ヘルパーを含まないヘルペスウイルス系を用いて感染性HSV中にパッケージングした。形質導入したCA320M細胞を照射し(25Gy)、1x103細胞を15動物中に注射した。21日後、動物に1x107生CA320M細胞をチャレンジし、その後、腫瘍増殖および生存分析を行った。αGal(+) CA320Mワクチン接種した動物の93パーセントが、突然変異αGal(-)CA320M(33%)およびゼロ(38%)対照と比較して、致死量の腫瘍チャレンジで、60日まで生き残った(>400mm3腫瘍)(図30)。この結果は、αGal(+)同系細胞を用いたワクチン接種が、対応するαGal(-)細胞に対する保護免疫応答を誘導できることを立証する。
【0147】
全体的に考えて、全てのこれらの結果は、始めて、細胞全体ワクチン中のα-ガラクトシルエピトープの存在がαGalエピトープの発現を欠く予め形成された腫瘍を治療するT細胞の媒介する免疫を誘導する強い免疫アジュバントであることを実証するものである。これらの結果はまた、このワクチンの臨床での利用がヒトの医療に大きなインパクトを与えうることを示す。さらに、癌患者に対する予防ワクチンは現在可能でなく、従って、本明細書に記載の技術のような治療手法は迅速に応用する現実的価値がありかつ癌患者に利益をもたらす可能性を有する。
【0148】
実施例12
ヒト細胞全体癌ワクチン(HyperAcute(商標))の開発
HyperAcute(商標)細胞全体癌ワクチンは、マウスα(1,3)ガラクトシルトランスフェラーゼ遺伝子を発現するように遺伝子操作した同種異系(allogeneic)癌培養細胞株から成る。複数の腫瘍組織型由来の複数の独立した癌培養細胞株を遺伝子操作してそれらの表面上にαGalエピトープを発現させている。ある治療用細胞全体癌ワクチンは、同じ腫瘍組織型に属する複数の遺伝子操作した癌細胞型の、照射した個々の培養細胞株または混合物の注射剤から成る。表1は、HyperAcute(商標)細胞全体癌ワクチンを作り出すために用いた癌培養細胞株を示す。
【表1】
【0149】
HyperAcute(商標)細胞全体癌ワクチンは、表1に示した培養細胞株の、プロタミン硫酸10g/mLの存在で293Z.CKGからのレトロウイルス上清を用いた形質導入により確立した。形質導入した細胞の集団をαGal細胞表面エピトープについて0-2605抗αGal抗体(NewLink)を用いて染色した。細胞表面上のαGalエピトープ発現強度が最高である5%の細胞集団を、FACSソーターを用いて別々の亜集団にソート(sort)した。ソートされた最高のαGalエピトープ発現をもつ形質導入細胞の亜集団が、表1に記載されたものである。これらの細胞を拡大して(分割比1:5)、マスター細胞バンク(MCB)を作製した。増殖パターン、形態学的外見、およびαGalエピトープ発現の平均強度をモニターしたが、培養の全増殖期間にわたって安定であった。MCBは、細胞をフラスコ中で37℃、5%±1%CO2にて拡大することにより開発した。培地は10%胎児ウシ血清(FBS)および2mmのL-グルタミンを補充したRPMI-1640であった。各継代において細胞をトリプシン処理し、数え、そしてその生存をトリパンブルー排除により評価した。細胞を増殖して10億個の細胞とし、収穫し、プールし、100個の凍結用バイアル(cryovial)中に分配し、そしてプログラム速度凍結室(programmed rate freezing chamber)を用いて凍結した。細胞を液体窒素貯蔵タンクの蒸気相に貯蔵する。各培養細胞株に対する作業用細胞バンク(WCB)をMCBから作製した。WCBの拡大、収穫、凍結および貯蔵の条件はMCBについて記載した通りである。それぞれのHyperAcute(商標)培養癌細胞株の生産ロットはそれぞれのWCBに由来する。生産ロットからの細胞が十分な密度に到達すると、培養液(上清)を収穫し、濾過し、そして無菌容器中にプールする。プールを十分に混合し、次いで無菌状態で標識した無菌のプラスチックボトルに充填する(標識には、製品名、ロット番号および充填日が記載される)。満たしたボトルを凍結して-60℃以下で貯蔵する。安全試験用のアリコートをとる。生産ロットに対する細胞をトリプシン処理により収穫し、プールし、そして完全培地に再懸濁させる。プールした細胞を150〜200Greyで照射する。細胞を、Varian
Clinic 2100C医療用直線加速器(medical linear accelerator)を6MV光子モードで用いて照射する。機械の照射出力は、AAPM TG-51較正プロトコル(calibration protocol)を用いて水中で較正する。出力一貫性は毎日;出力較正は毎月、NIST較正トレーサブルイオンチャンバー/エレクトロメーター線量計を用いてモニターする。照射細胞を遠心分離し、そして5%グリセロールおよびヒト血清アルブミンから構成される注射用最終剤形に再懸濁する。次いで0.4mlの細胞を、用量レベルに対応して1x106/0.2ml、3.5x106/0.2ml、1x107/0.2mlおよび3.5x107細胞/0.2mlの濃度で無菌の凍結用バイアル中に分配する。HyperAcute(商標)細胞MCB、WCB、およびPL品質管理試験は細胞および上清の両方を実施する。HyperAcute(商標)癌ワクチンMCB、WCBおよびPL細胞の許容判定基準は、ヌードマウスにおける腫瘍原性に対するアッセイから成る。
【0150】
実施例13
ヒト患者に対する投与量レベルと投与スケジュール
次のデータは投与量レベルとワクチン接種スケジュールの提案を支持するものである。マウスについての前臨床毒性学的研究は、α-gal発現同種異系および同系腫瘍は、70kgヒト1人当たり3.5x109細胞と等価である1マウス当たり1x106細胞までの十分な耐性を有することを示している。細胞表面上にα-galエピトープを自然発現するマウスベクター産生細胞(MVPCもしくはマウスVPC)についてのフェーズI研究(第2.1節を参照)は、マウスVPCが患者1人当たり7x109細胞の投与量に十分な耐性を有することを示している。フェーズIIマウスVPC試験において、患者は各サイクルに7x109細胞の注射を受ける3サイクルの治療を受けた。患者はこの治療に十分に耐性があった。マウスVPC注射を6週の間隔で繰返した。提案した投与レベルはマウスのα-galワクチンの十分に耐えられる投与量の範囲内にある(患者1人当たり最大4x108細胞)。ワクチン注射の間の4週の間隔は、治療の毒性の評価および免疫応答の成熟を評価するのに十分な時間でありうる。フェーズIマウスVPC研究からのデータは、抗αGal免疫応答は、αGal発現細胞注射後14〜21日の間にその最高値に到達することを示唆する。
【0151】
実施例14
マウスαGT遺伝子を保持するレトロウイルスベクターを用いて形質導入した同系腫瘍ワクチンを用いる毒性学研究
最初の研究では、マウスをαGal(+)B16黒色腫細胞を用いて1マウス当たり1x105細胞の用量で皮下注射した。αGal(+)生存黒色腫細胞を拒絶した動物にはαGal(-)B16細胞を再チャレンジしてαGal(+)黒色腫細胞の拒絶が腫瘍免疫を誘導することを立証した。全てのマウスがB16による第2チャレンジに生き残った。これらの黒色腫保護された動物(第1実験でn=8、および第2実験でn=9)について6ヶ月間、長期毒性研究を実施した。毒性学的研究についての組織学的、血液学的、臨床的観察を保護されたマウスのいくつかについて実施した。組織病理学的試験は、主な潅流器官:腎臓、脾臓および肝臓、ならびに潜在的標的組織、皮膚および乳腺を含んだ。安全を評価する組織学的研究は、試験した全てのサンプル組織(皮膚、乳腺、腎臓、脾臓および肝臓)に注目すべき病変を示さなかった。複数の動物(対照マウスを含む)は炎症性細胞の僅かな浸潤を伴う腎脈管周囲炎を示した。試験グループのこの観察頻度は対照グループの動物の頻度と同様であった。血液学研究結果は、全ての研究値が正常値の範囲内にあることを示した。正常値は、挙動を含むナイーブ無遺伝子操作α-gal KOマウス臨床観察から考察された結果であり、可能性のある二次的副作用としての自己免疫色素脱失(白斑)または柔皮変化の発生は、黒色腫保護されたマウス(n=17)に研究期間中観察されなかった。
【0152】
第2研究においては、マウスに同種異系αGal(+)EMT-6乳房細胞を1マウス当たり1x106細胞の用量で皮下注射した。ワクチン接種後、2、4および6週に、血液および組織サンプルを取得して毒性学的研究を実施した。組織学的および血液学液研究を実施した。組織病理学的試験は、主な潅流器官:腎臓、脾臓および肝臓、ならびに潜在的標的組織、皮膚および乳腺を含んだ。安全を評価する組織学的研究は、試験した全てのサンプル組織(皮膚、乳腺、腎臓、脾臓および肝臓)に注目すべき病変を示さなかった。複数の動物(対照マウスを含む)は炎症性細胞の僅かな浸潤を伴う腎脈管周囲炎を示した。試験グループのこの観察頻度は対照グループの動物の頻度と同様であった。血液学研究結果は、全ての研究値が正常値の範囲内にあることを示した。正常値は、挙動を含むナイーブ無遺伝子操作α-gal KOマウス臨床観察から考察された結果であった。一過性好酸球増加症が同種異系ワクチン接種後、2週に観察された。毒性学研究に用いた最大用量は1x106細胞/マウスであった。この用量の用量/kgに基づいてヒトにスケールアップすると、マウスを20グラムと仮定して、用量/マウスが70kgヒトにおいて3.5x109細胞と等価になる。また、ヒトの平均寿命の70年と比較してマウスの平均寿命がほぼ2年であることも考慮すると、長期毒性学研究(6ヶ月)はマウス寿命の4分の1と等価であり、ほぼヒトの17年にあたる。これらの研究は、16週の治療期間全体にわたる1患者当たり4x108の現行投与量スケジュールを支持する。
【0153】
実施例15
HyperAcute(商標)癌ワクチンの調製および患者への投与スケジュール
この方法は、細胞全体癌ワクチン(本発明の医薬組成物)を調製する方法およびヒト患者へ投与する方法を記載する。細胞は、それらを調製した直後に、患者に注射しなければならない。投与される細胞は凍結前に致死的に照射されているので、特別な安全上の注意事項は不要である。最初に各ワクチン培養細胞株の凍結用バイアルを液体窒素容器から取り出す。次いで、全てのバイアルを、37℃の水浴に凍結内容レベルを超えるまで浸漬して同時に解凍する。バイアルが解凍されると直ぐに、それらの表面を70%アルコールを用いてリンス洗いする。ワクチンの各培養細胞株成分による等量のバイアルの内容物を注射用の1シリンジ中に一緒にして、直ぐ、患者中に注射する。ワクチン細胞を25ゲージ針を備えたツベルクリンシリンジを用いて皮内に(i.d.)注射しうる。注射は腕および脚に回転ベース上で行うべきである。HABワクチン細胞を1、29、57、および85日に投与しうる。それぞれの注射後2時間、外来患者診療所において看護スタッフにより患者をモニターしうる。患者モニタリングは:ワクチンの投与前30分間以内の温度(T)、脈拍(P)、血圧(BP)および呼吸数(R)を含み、ならびに、次いでワクチン接種後、毎15分間x4回、次いで毎30分間x2回のチェックによるべきである。温度は診療所から退所する前にチェックしうる。さらに、患者を、局所または播種性皮膚発疹および他の副作用を含む急性反応症状についてモニターしうる。グレードII以上の急性副作用を経験した患者は、さらに1〜2時間診療所において、その症状がグレードII未満に回復するまでモニターしてもよい。観察および/または治療にも関わらず、もしグレードII以上が4時間を越えて続くのであれば、観察を続けるか、治療を調査(institute)または改変するか、または患者を入院させるかどうかの決定をする。
【0154】
実施例16
最大耐量の決定
治療は適格な3患者の投与コホートで進めうる。最大耐量(MD;maximum tolerated dose)は、用量制限毒性(DLG;dose-limiting toxicity)が見られる投与コホートより低い投与コホートとして定義される。もしある投与コホートで患者の>33%(すなわち、1/3、または2/4-6)がDLTを訴えれば、MTDは決定されうるので、さらなる用量増加は許容されないであろう。もしある投与コホートにおいて3患者の1患者(1/3)でDLTが認められれば、そのコホートを拡大して全部で6患者となるまで拡大しうる。もしさらに1つのDLTが観察されれば、コホートを閉じて、MTDを定義してさらなる用量増加は許容されないであろう。もしさらなるDLTがそのコホートで観察されなければ、次のより高い投与コホートへの増加を開始しうる。さらなる患者を本研究のフェーズII部分でMTDにて治療しうる。
【0155】
ある投与コホートにおける最後の患者の開始と次のより高い投与コホートの開始の間には最小限4週の遅れがありうる。
【表2】
【0156】
本研究における応答と進行は、固体腫瘍の応答評価判定基準委員会(Response Evaluation Criteria in Solid Tumors (RECIST) Committee)の提案による新しい国際判定基準を用いて評価する。腫瘍病変の最大直径(一次元測定)だけの変化をRECIST判定基準では利用する。病変は、以下に記載する判定基準を用いて測定可能または測定不可能である。測定可能性の意味での用語「評価できる(evaluable)」は、さらなる意味または正確度を与えるものでないので、使用しないであろう。測定可能な病変は、慣用技術(CT、MRI、X線)を用いて>20mmのように、またはらせんCTスキャンを用いて>10mmのように、少なくとも1次元(記録すべき最長直径)で正確に測定できる病変として定義される。全ての主要測定はミリメートル(またはセンチメートルの10分の1分率)で記録しなければならない。小さい病変(慣用技術を用いて最長直径<20mmまたはらせんCTスキャンを用いて<10mm)を含む他の病変(または疾患部位)は測定不可能な疾患と考えられる。骨病変、軟髄膜疾患、腹水、胸膜または心嚢貯留液、リンパ管炎皮膚または肺、炎症性乳房疾患、腹腔塊(CTまたはMRIによりフォローされない)、および嚢胞性病変は全て測定不可能と考えられる。全ての関わる器官の代表として、最大1器官当たり5病変および全部で10病変までの全ての測定可能な病変は、標的病変として同定しかつ基線で記録しかつ測定すべきである。標的病変は、そのサイズ(最長直径をもつ病変)および正確な反復測定値(イメージング技術によりまたは臨床的のいずれか)の適性に基づいて選択すべきである。全ての標的病変に対する最長直径和(LD)を計算し、基線和LDとして報じる。基線和LDは、それにより目的腫瘍応答を特徴付ける意味で用いる。全ての他の病変(または疾患部位)は非標的病変と同定すべきであり、これらも基線において記録すべきである。非標的病変は、1器官当たり最大数または全ての関わる器官の全体を超える測定可能な病変、ならびに測定不可能な病変を含む。これらの病変の測定値は必要としないが、それぞれの存在または不在はフォローアップ全体を通して記入すべきである。全測定値を、ルーラーまたは好ましくはカリパーを用いて採取し、メートル表示で記録すべきである。全ての基線評価を、治療の開始まで、そして治療開始前2週間を超えないように可能な限り綿密に実施すべきである。先の照射領域に位置する腫瘍病変は測定可能と考えない。臨床病変は、それらが表面的(例えば、皮膚小節および明白なリンパ節)であるときだけ、測定可能と考えられる。皮膚病変の場合、病変のサイズを求めるルーラーを含む、デジタルカラー写真による記録を推奨する。胸部の病変の場合、X線は、明確に規定されかつ通気した肺に囲まれているときは測定可能な病変として受入れられる。しかし、CTが好ましい。慣用のコンピューター断層撮影(CT)および磁気共鳴イメージング(MRI)を、スライス厚みが10mm以下のカットで連続的に実施すべきである。らせんCTは5mm連続再構築アルゴリズムを用いて実施すべきである。これは、胸部、腹部、および骨盤の腫瘍に適用する。頭および首腫瘍ならびに四肢の腫瘍は特定のプロトコルを必要とする。研究の主な目的が対象の応答評価であるときは、超音波(US)を利用する。USは腫瘍病変を測定するために用いるべきでない。しかし、表面上の明白なリンパ節、皮下病変および甲状腺節(thyroid nodules)を臨床測定するための代替としては可能である。USはまた、通常、臨床試験により評価する表面の病変の完全な消滅を確認するためにも有用である。内視鏡検査および腹腔鏡検査は腫瘍評価がまだ十分にかつ広汎に実証されていない対象にとって有用である。この特定の筋道でのこれらの利用は完備した設備と高レベルの熟練者が必要であり、いくつかのセンターにおいてのみ利用可能である。従って、対象腫瘍応答のためのかかる技術の利用は、関係センターにおける実証目的に制限すべきである。しかし、かかる技術は、生検が得られるときは、完全な病理学的応答を確証するために有用でありうる。腫瘍マーカーだけを用いて乳房腫瘍応答を評価することはできない。稀な場合(例えば、既知の残余良性腫瘍が残留しうる、生殖細胞腫瘍などの腫瘍型における残余病変)には、細胞学と組織学を用いて部分的応答(PR)と完全応答(CR)の間の相違を識別することができる。
【0157】
測定可能な腫瘍が応答または安定な疾患に対する判定基準に会ったとき、治療中に現れるまたは悪化するいずれかの浸出液の新生物起源の細胞病理学的確証が、応答または安定な疾患(浸出液が治療に伴う影響であってもよい)および進行性疾患の間の相違を識別するために必須である。
【0158】
以上の記載から、本発明が少なくともその目的の全てを達成することを理解することができる。本明細書に引用した全ての参考文献は、本明細書にその全てが組込まれる。
【図面の簡単な説明】
【0159】
【図1】本発明によって使用しうるpLNC-KGプラスミドの描写である。
【図2A】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である。
【図2B】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である(図2Aの続き)。
【図2C】配列番号1、すなわち図1に描写したpLNC-KGプラスミドの配列である(図2Bの続き)。
【図3】ウサギ赤血球を用いた免疫感作による、α(1,3)-ガラクトシルトランスフェラーゼノックアウト(αGT KO)マウスにおける抗αGal抗体誘導の模式図である。抗αGal抗体(Ab)を誘導するために、マウスの腹腔内に108ウサギ赤血球(RRBC)を用いて2回、2週間隔てて注射した。最後の免疫感作の1週後に血液サンプルを得て、抗αGal抗体力価をELISAにより測定した。本研究に用いた全てのマウスは高い力価の抗αGal Abを発生した。
【図4】非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の生存試験を示す模式図である。雌性および雄性8〜14週齢H-2 b/bハプロタイプαGT KOマウスをこの研究で用いた。全マウスはRRBCを用いて図3に示したように免疫感作した。最後の免疫感作の1週後に、マウスは3種の培養細胞株:a)野生型B16.BL6黒色腫培養細胞株(αGal(-))、b)Neo-R遺伝子を発現するベクターを用いてレトロウイルスにより形質導入したB16細胞(B16.LNL、模擬(mock)対照αGal(-))、またはc)NeoR遺伝子とαGTの両方を発現するベクターを用いてレトロウイルスにより形質導入したB16細胞(B16.αGal(+)、pLNCKG)、の致死的皮下チャレンジ1x105を受けた。チャレンジ後、腫瘍を盲検方式で1週間当たり2回測定した。
【図5】αGTノックアウトマウスにおけるαGal(+)またはαGal(-)B16黒色腫細胞の致死的非照射皮下注射後の腫瘍サイズ(5a:チャレンジ後15日、5b:チャレンジ後30日)を示すグラフである。明白な皮下腫瘍をカリパーを用いて3垂直軸に沿って測定して容積を計算し、mm3で表した。図は、図4に記載したそれぞれのB16培養細胞株による皮下注射後15および30日における腫瘍サイズを示す。バーは平均および誤差バーSEMを表す。
【図6】αGT KOマウスにおいて、非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死的皮下注射後の腫瘍増殖動力学を示すグラフである。腫瘍サイズは図5のように野生型B16(αGal(-))、B16.LNL(対照αGal(-))およびαGal(+)B16によるチャレンジ後15、23および29日に測定した。
【図7】非照射αGal(-)またはαGal(+)B16黒色腫細胞の致死量を用いて注射したαGT KOマウスの生存分析を示すグラフである。図4に記載のように処置したマウスを90日間研究した。Kaplan-Meier生存分析および長ランク生存曲線比較を統計学ソフトウエアを用いて実施した。
【図8】非照射αGal(+)B16黒色腫細胞の皮下注射で生き残ったマウスに非照射αGal(-)B16黒色腫細胞の致死量を皮下注射した後の生存試験に関する実験設計を記述する模式図である。
【図9】ノックアウトマウスの生存分析を示すグラフである。図7からのαGal(+)B16細胞による第1チャレンジで生き残ったマウスを、続いて天然αGal(-)B16の第2皮下投与によりチャレンジした(図8)。Kaplan-Meier分析を、αGal(-)B16の注射後、60日間実施した。αGal(-)B16を皮下に受けたナイーブマウスを対照として用いた。
【図10】非照射αGal(+)細胞の致死量注射で生き残ったマウスにおける黒色腫特異的細胞傷害性T細胞の誘導を示す模式図である。図7からのαGal(+)B16細胞による第1チャレンジで生き残った2マウスから採集した脾細胞を、細胞傷害性Tリンパ球(CTL)をin vitroで作製するために利用した。αGal(+)B16の注射後90日に無腫瘍マウスから脾細胞を収穫し、そして脾細胞を照射αGal(-)B16細胞とともにIL-2不在のもとで5日間培養することにより、黒色腫特異的培養物を作製した。CTLを収穫し、特異的標的αGal(-)B16および非特異的同系培養細胞株大腸癌MC38およびT細胞リンパ腫EL-4に対して試験した。CTLの特異的および非特異的標的とのインキュベーションの4時間後に、培養上清におけるLDH放出を測定することにより、特異的細胞傷害性を決定した。
【図11】図10に記述した、非照射αGal(+)細胞の致死量の投与で生き残ったマウスにおけるB16黒色腫-特異的細胞傷害性T細胞の誘導結果を示すグラフである。
【図12】αGTノックアウトマウスにおいて、非照射αGal(+)またはαGal(-)B16黒色腫細胞の致死量の静脈注射による播種性黒色腫転移モデルの実験設計を示す模式図である。雌性および雄性αGT KOマウスを、図3の通り、RRBCを用いて免疫感作して抗αGal Ab力価を増加した。最後の免疫感作後1週に、マウスの尾静脈にαGal(-)B16またはαGal(+)B16を静脈注射した。注射後3週に、肺を収穫して肺黒色腫転移を数えた。
【図13】図12の実験の統計学的結果を示すグラフである。
【図14】照射αGal(+)B16黒色腫細胞を用いたワクチン接種後のαGTノックアウトマウスにおける皮下αGal(-)黒色腫腫瘍発生の予防に対する実験設計を示す模式図である。細胞ワクチンは、先の実験に記載したB16由来の培養細胞株:すなわち、天然αGal(-)B16、対照ベクターを形質導入したαGal(-)B16.LNL、およびマウスαGT遺伝子をコードするベクターを形質導入したαGal(+)B16細胞を用いて調製した。細胞ワクチンをγ線照射(250 Gy)により調製して細胞増殖を防止し、使用するまで凍結媒質中で保存した。注射前に細胞ワクチンを解凍し、洗浄し、計数し、そしてハンクス溶液(HBSS;Hanks' Balanced Salt Solution)中に懸濁して皮下注射した。使用した全てのαGT KOマウスに、図3のようにRRBCを注射した。最後のRRBC注射後1週にマウスは第1用量の細胞ワクチンを受けた。2週後に細胞ワクチン接種を繰返した。それぞれのワクチンの用量は1マウス当たり106細胞を皮下投与した。細胞の細胞ワクチン後2週に、マウスに105非照射天然αGal(-)B16細胞を皮下注射し、そして腫瘍発生を週2回、90日間観察した。
【図15】図14に記載した実験のKaplan-Meier生存分析を示すグラフである。
【図16】TNF-αを認識するためのFACS分析結果を示す。αGal(-)B16黒色腫細胞のin vitro認識後に、照射αGal(+)B16細胞を用いてワクチン接種したマウスに誘導された、黒色腫特異的T細胞による細胞内TNF-αの検出。無腫瘍マウスからの脾細胞は、αGal(+)B16ワクチンを用いてワクチン接種したマウスから、αGal(-)B16黒色腫の注射後90日に収穫した(図15で生き残ったマウス)。αGal(-)B16のin vitro認識を測定するために、細胞内TNF-αをFACSにより検出した。T細胞を、6時間、サイトカインの分泌をブロックするブレフェルジンA(Brefeldin A)による刺激の存在または非存在のもとで培養した。活性化を最大限にするためにPMA/Ca++イオノフォアを用いた。細胞を特異的認識を測定するために105照射B16細胞とともに、または非黒色腫同系ネガティブ対照培養細胞株として105 CA320Mとともに培養した。インキュベーション後、細胞を収穫し、透過化処理し、固定し、そしてPE標識した抗TNF-αモノクローナルAbを用いて細胞内TNF-αを染色した。細胞をCoulterフローサイトメーターを用いて分析し、少なくとも10.000ゲーテッド(gated)リンパ球を取得した。
【図17】照射黒色腫細胞を用いるワクチン接種を介する予め形成された皮下腫瘍を治療処置する実験設計を示す模式図である。予め形成された皮下αGal(-)黒色腫腫瘍の、照射αGal(+)またはαGal(-)B16黒色腫細胞を用いたワクチン接種を介する治療処置。マウスに図3に説明したようにRRBCを注射した。最後のRRBC注射後1週、マウスに105非照射αGal(-)B16黒色腫細胞を皮下注射して無作為化した。実験#1においては、2用量の細胞ワクチンをαGal(-)B16の皮下注射後、3および6日に投与した。実験#2においては、マウスは生αGal(-)B16細胞の皮下注射後、4、11および18日に3用量の照射細胞ワクチンを受けた。これらの実験において、2種の細胞ワクチン、すなわち、対照ベクターを形質導入した照射αGal(-)B16、または照射αGal(+)B16細胞を用いた。
【図18】αGal(+)またはαGal(-)B16黒色腫細胞によるワクチン接種を受けた、予め形成されたαGal(-)皮下黒色腫腫瘍を有するマウスにおける腫瘍増殖動力学の2つの異なる実験の結果を示す。腫瘍サイズは、細胞ワクチンを受けたまたは受けないマウスにおいて早朝の時点に測定した。数字はそれぞれのグループにおける全マウスの腫瘍サイズの平均を表す。
【図19】照射αGal(+)またはαGal(-)B16黒色腫細胞による治療用ワクチン接種を受けた、予め形成された皮下αGal(-)B16黒色腫腫瘍を保持するマウスのKaplan-Meier生存分析を示すグラフである。αGal(-)B16を注射し、αGal(+)またはαGal(-)B16黒色腫ワクチンにより治療したまたはしなかったマウスを75日間生存について評価した(実験#1)。
【図20】照射αGal(+)またはαGal(-)B16黒色腫細胞による治療用ワクチン接種を受けた、予め形成された皮下αGal(-)B16黒色腫腫瘍を保持するマウスのKaplan-Meier生存分析を示すグラフである。αGal(-)B16を注射し、αGal(+)またはαGal(-)B16黒色腫ワクチンにより処置したまたはしなかったマウスを70日間生存について評価した(実験#2)。
【図21】予め形成された肺転移性腫瘍を治療する実験設計を示す模式図である。予め形成された播種性肺転移性αGal(-)黒色腫腫瘍の、照射αGal(+)またはαGal(-)B16黒色腫細胞を用いるワクチン接種を介する治療。マウスに、図3に説明したようにRRBCを注射した。最後のRRBCの注射後1週に、マウスに非照射αGal(-)B16黒色腫細胞を静脈内(i.v.)注射して無作為化した。これらのマウスに、続いてαGal(-)B16またはαGal(+)B16ワクチン細胞のいずれかを、播種性転移の確立後4、11および21日にワクチン接種した。第30日に動物を犠牲にして、その肺腫瘍負荷を評価した。
【図22】図21に示したプロトコルの実験1における腫瘍負荷を示すグラフである。実験#1において、マウスは105αGal(-)B16黒色腫細胞を受けた。非照射黒色腫細胞のi.v.注射後30日に、マウスを犠牲にして肺黒色腫転移を数えた。
【図23】予め形成された播種性転移性黒色腫を治療する第2の実験設計を示す模式図である。実験#2において、マウスは5x105αGal(-)B16黒色腫細胞を受けた。この後、マウスを皮下に黒色腫細胞ワクチンを用いて処理した。これらのマウスは、非照射αGal(-)B16のi.v.注射後4、11および21日に、対照ベクターを用いて形質導入した照射αGal(-)B16または照射αGal(+)B16細胞の2x105の3用量を受けた。
【図24】図23に記載した実験の結果を示すグラフであり、肺の平均重量(24A)としてまたは平均肺腫瘍負荷(24B)として表した。
【図25】αGal(+)またはαGal(-)B16細胞を用いるワクチン接種後の、αGal(-)B16黒色腫に特異的なT細胞免疫応答のin vitro誘導を実証する実験設計を示す模式図である。マウスに図3のようにRRBCを注射した。最後のRRBC注射後2週にマウスは、対照ベクターを用いて形質導入した照射αGal(-)B16細胞または照射αGal(+)B16細胞ワクチンの2x105の3回の皮下注射を受けた。最後の細胞ワクチン後2週に、脾細胞を収穫し、T細胞研究を実施した。αGal(-)B16の特異的認識を確認するために、細胞内TNF-αおよび活性化マーカーCD25およびCD69のアップレギュレーションを測定した。
【図26】B16抗原を特異的に認識するワクチン接種した動物(図25)のT細胞前駆体中のTNF-αの特異的誘導を示すグラフである。TNF-αを検出するために、T細胞を6時間、サイトカインの分泌をブロックするブレフェルジンA(Brefeldin A)による刺激の存在または非存在において培養した。最大刺激のために、PMA/Ca++イオノフォアをポジティブ対照として用いた。細胞を、特異的認識を測定するためにαGal(-)B16とともに、またはネガティブ対照としてCA320M(非黒色腫同系(H-2 b/b)小腸培養細胞株)とともに培養した。インキュベーションの後、細胞を収穫し、細胞内TNF-αを染色した。ポジティブ細胞を、少なくとも10.000ゲーテッドイベントの取得後の前方散乱プロット中のリンパ球のFACSゲーティングにより検出した。プロットはTNF-α(+)細胞の平均蛍光強度(MFI)を表す。
【図27】B16抗原を特異的に認識する、αGal(+)細胞を用いてワクチン接種した動物からのT細胞前駆体における活性化マーカーCD25およびCD69のアップレギュレーションを示すグラフである。測定は、図26に記載したのと同様の条件下で、ブレフェルジンA(Brefeldin A)の不在のもとでの培養の1日後に実施した。インキュベーション後に、細胞を収穫し、PE標識したモノクローナルAb抗CD-25または抗CD-69を用いて染色した。取得はCoulterフローサイトメーターを用いて実施した。プロットはポジティブCD25またはCD69細胞を示す。
【図28】照射αGal(+) またはαGal(-)B16細胞を用いてワクチン接種したドナーマウスからの養子T細胞導入による、特異的T細胞が媒介する免疫のαGal(-)B16黒色腫の予め形成された播種性転移に対する治療有効性をin vivo実証する実験の模式図を示す。ドナーマウスにRRBCを図3の通り注射した。最後のRRBC注射後2週に、マウスは対照ベクターを形質導入した照射αGal(-)B16または照射αGal(+)B16細胞ワクチンの2x105皮下注射を3回受けた。最後の細胞ワクチンの後に、脾細胞を収穫して性別の整合したレシピエントに導入した。細胞導入の4日前に、レシピエントに非照射αGal(-)B16をi.v.注射して肺黒色腫転移を確立し、そして無作為化した。T細胞導入後30日に、レシピエントを安楽死されて肺黒色腫転移負荷を、肺重量を測ることによりおよび黒色腫腫瘍を数えることにより決定した。
【図29】図28に記載した実験の結果を示す。バーは平均および誤差バー、SEMを表す。2つの独立実験を実施してそれらを示した。
【図30】αGal(+)またはαGal(-)照射CA320M肉腫細胞をワクチン接種したαGT KOマウスの、非照射αGal(-)CA320M肉腫細胞の致死用量を皮下注射した後の生存分析を示すグラフである。マウスに図3のようにRRBCを注射した。最後のRRBC注射後2週に、それらのマウスに、10 MOI HDKgalΔsalI(αGal(-)ベクター)またはHDKgal1(αGal(+)ベクター)を用いて形質導入したCA320Mの1x103を皮下にワクチン接種し、次いで25Gyを照射した。21日後、マウスに1x107非照射CA320M細胞を皮下注射した。対照のNullはワクチンを含まなかった。60日の観察期間の生存分析を実施した。
【特許請求の範囲】
【請求項1】
抗αGalを合成する動物に存在する腫瘍細胞を抑制するための医薬組成物であって、α(1,3)ガラクトシルトランスフェラーゼをコードする配列およびその上にαGalエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含む弱毒化したワクチン腫瘍細胞の混合物、ならびに担体を含んでなる上記医薬組成物。
【請求項2】
抗αGalを合成する動物において複数の腫瘍抗原に対する免疫応答を、それらのグリコシル化状態に関わらず、同時に刺激するための医薬組成物であって、α(1,3)ガラクトシルトランスフェラーゼタンパク質をコードする配列およびその上にαGalエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含み、腫瘍細胞全体中の抗原を免疫系に提示することができる弱毒化した全ワクチン腫瘍細胞の混合物、ならびに好適な担体を含んでなる上記医薬組成物。
【請求項3】
上記腫瘍細胞が、大細胞癌、扁平細胞癌、腹水炎腺癌(ascitis adenocarcinoma)、胸水癌、原発腺管上皮癌、転移性腺癌、原発腺癌、腎臓明細胞癌(clear cell carcinoma)、卵巣腺癌、卵巣奇形癌、悪性黒色腫、結腸直腸癌、結腸直腸腺癌、および結腸直腸腺癌グレードIIからなる群から選択される、請求項1に記載の組成物。
【請求項4】
上記腫瘍細胞がA549細胞由来である、請求項1に記載の組成物。
【請求項5】
上記腫瘍細胞がNCI-H460細胞由来である、請求項1に記載の組成物。
【請求項6】
上記ワクチン腫瘍細胞がNCI-H520細胞由来である、請求項1に記載の組成物。
【請求項7】
上記ワクチン腫瘍細胞がMCF-7細胞由来である、請求項1に記載の組成物。
【請求項8】
上記ワクチン腫瘍細胞BT-20細胞が由来である、請求項1に記載の組成物。
【請求項9】
上記ワクチン腫瘍細胞がHPAF-II由来である、請求項1に記載の組成物。
【請求項10】
上記ワクチン腫瘍細胞がPANC-1細胞由来である、請求項1に記載の組成物。
【請求項11】
上記ワクチン腫瘍細胞がASPC-1細胞由来である、請求項1に記載の組成物。
【請求項12】
上記ワクチン腫瘍細胞がBxPC3細胞由来である、請求項1に記載の組成物。
【請求項13】
上記ワクチン腫瘍細胞がIGROV細胞由来である、請求項1に記載の組成物。
【請求項14】
上記ワクチン腫瘍細胞がES-2細胞由来である、請求項1に記載の組成物。
【請求項15】
上記ワクチン腫瘍細胞がNIH:OVCAR3細胞由来である、請求項1に記載の組成物。
【請求項16】
上記ワクチン腫瘍細胞がCOLO829細胞由来である、請求項1に記載の組成物。
【請求項17】
上記ワクチン腫瘍細胞がG-361細胞由来である、請求項1に記載の組成物。
【請求項18】
上記ワクチン腫瘍細胞がHCT-116細胞由来である、請求項1に記載の組成物。
【請求項19】
上記ワクチン腫瘍細胞がCOLO205細胞由来である、請求項1に記載の組成物。
【請求項20】
上記ワクチン腫瘍細胞がLoVo細胞由来である、請求項1に記載の組成物。
【請求項21】
上記ワクチン腫瘍細胞がWiDr細胞由来である、請求項1に記載の組成物。
【請求項22】
上記ワクチン腫瘍細胞がDLD-1細胞由来である、請求項1に記載の組成物。
【請求項23】
上記ワクチン腫瘍細胞がHCT-15細胞由来である、請求項1に記載の組成物。
【請求項24】
上記ワクチン腫瘍細胞がSW620細胞由来である、請求項1に記載の組成物。
【請求項25】
上記ワクチン腫瘍細胞がSW480細胞由来である、請求項1に記載の組成物。
【請求項26】
上記ワクチン腫瘍細胞がSW1116細胞由来である、請求項1に記載の組成物。
【請求項27】
上記ワクチン腫瘍細胞がHT-29細胞由来である、請求項1に記載の組成物。
【請求項28】
上記ワクチン腫瘍細胞がFHC細胞由来である、請求項1に記載の組成物。
【請求項29】
上記ワクチン腫瘍細胞がCCD841CoN細胞由来である、請求項1に記載の組成物。
【請求項30】
上記ワクチン腫瘍細胞がPC-3細胞由来である、請求項1に記載の組成物。
【請求項31】
上記ワクチン腫瘍細胞がLNCaPFGC細胞由来である、請求項1に記載の組成物。
【請求項32】
上記ワクチン腫瘍細胞がMDAPca2b細胞由来である、請求項1に記載の組成物。
【請求項33】
上記ワクチン腫瘍細胞がDU145細胞由来である、請求項1に記載の組成物。
【請求項34】
上記組成物がHAL1である、請求項1に記載の組成物。
【請求項35】
上記組成物がHAL2である、請求項1に記載の組成物。
【請求項36】
上記組成物がHAL3である、請求項1に記載の組成物。
【請求項37】
上記組成物がHAB1である、請求項1に記載の組成物。
【請求項38】
上記組成物がHAB2である、請求項1に記載の組成物。
【請求項39】
上記組成物がHAPA1である、請求項1に記載の組成物。
【請求項40】
上記組成物がHAPA2である、請求項1に記載の組成物。
【請求項41】
上記組成物がHAPA3である、請求項1に記載の組成物。
【請求項42】
上記組成物がHAPA4である、請求項1に記載の組成物。
【請求項43】
上記組成物がHAO1である、請求項1に記載の組成物。
【請求項44】
上記組成物がHAO2である、請求項1に記載の組成物。
【請求項45】
上記組成物がHAO3である、請求項1に記載の組成物。
【請求項46】
上記組成物がHAO4である、請求項1に記載の組成物。
【請求項47】
上記組成物がHAM1である、請求項1に記載の組成物。
【請求項48】
上記組成物がHAM2である、請求項1に記載の組成物。
【請求項49】
上記組成物がHAM3である、請求項1に記載の組成物。
【請求項50】
上記組成物がHAC1である、請求項1に記載の組成物。
【請求項51】
上記組成物がHAC2である、請求項1に記載の組成物。
【請求項52】
上記組成物がHAC3である、請求項1に記載の組成物。
【請求項53】
上記組成物がHAC4である、請求項1に記載の組成物。
【請求項54】
上記組成物がHAC5である、請求項1に記載の組成物。
【請求項55】
上記組成物がHAC6である、請求項1に記載の組成物。
【請求項56】
上記組成物がHAC7である、請求項1に記載の組成物。
【請求項57】
上記組成物がHAC8である、請求項1に記載の組成物。
【請求項58】
上記組成物がHAC9である、請求項1に記載の組成物。
【請求項59】
上記組成物がHAC10である、請求項1に記載の組成物。
【請求項60】
上記組成物がHAC11である、請求項1に記載の組成物。
【請求項61】
上記組成物がHAC12である、請求項1に記載の組成物。
【請求項62】
上記組成物がHAPR1である、請求項1に記載の組成物。
【請求項63】
上記組成物がHAPR2である、請求項1に記載の組成物。
【請求項64】
上記組成物がHAPR3である、請求項1に記載の組成物。
【請求項65】
上記組成物がHAPR4である、請求項1に記載の組成物。
【請求項66】
さらにアジュバントを含む、請求項1に記載の組成物。
【請求項67】
抗α-galを合成する動物において腫瘍細胞に対する免疫応答を誘導するための医薬組成物であって、A549、NCI-H460、NCI-H520、MCF-7、BT-20、HPAF-II、PANC-1、ASPC-1、BxPC3、IGROV、ES-2、NIH:OVCAR3、PA-1、A375、COLO829、G-361、HCT-116、COLO205、LoVo、WiDr、DLD-1、HCT-15、SW620、SW480、SW116、HT-29、FHC、CCD841CoN、PC-3、LNCaPFGC、MDAPca2b、およびDU145からなる群から選択される、α(1,3)ガラクトシルトランスフェラーゼをコードする配列およびその上にαガラクトシルエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含む弱毒化ワクチン腫瘍細胞の混合物;ならびに担体を含んでなる上記医薬組成物。
【請求項68】
HAL1、HAL2またはHAL3の群から選択される1以上の培養細胞株を含んでなる、動物において肺組織腫瘍細胞を治療するための医薬組成物。
【請求項69】
HAB1またはHAB2の群から選択される1以上の培養細胞株を含んでなる、動物において乳房組織腫瘍細胞を治療するための医薬組成物。
【請求項70】
HAPA1、HAPA2、HAPA3、またはHAPA4の群から選択される1以上の培養細胞株を含んでなる、動物において膵臓組織腫瘍細胞を治療するための医薬組成物。
【請求項71】
HAO1、HAO2、HAO3、またはHAO4の群から選択される1以上の培養細胞株を含んでなる、動物において卵巣組織腫瘍細胞を治療するための医薬組成物。
【請求項72】
HAM1、HAM2、またはHAM3の群から選択される1以上の培養細胞株を含んでなる、動物において黒色腫組織腫瘍細胞を治療するための医薬組成物。
【請求項73】
HAC1、HAC2、HAC3、HAC4、HAC5、HAC6、HAC7、HAC8、HAC9、HAC10、HAC11、またはHAC12の群から選択される1以上の培養細胞株を含んでなる、動物において大腸組織腫瘍細胞を治療するための医薬組成物。
【請求項74】
HAPR1、HAPR2、HAPR3、またはHAPR4の群から選択される1以上の培養細胞株を含んでなる、動物において前立腺組織腫瘍細胞を治療するための医薬組成物。
【請求項75】
治療用医薬組成物を作る方法であって、生腫瘍細胞のコレクションを取得するステップ、発現するとα(1,3)ガラクトシルトランスフェラーゼをコードするヌクレオチド配列を用いて上記細胞を形質転換して、その結果、上記α(1,3)ガラクトシルトランスフェラーゼが上記細胞上の複数の細胞表面糖タンパク質上にαガラクトシルエピトープを合成するステップ、上記腫瘍細胞を採集してαガラクトシルエピトープを有する複数の糖タンパク質をもつ細胞の混合物を作るステップ、および上記細胞を死滅させるステップを含んでなる上記方法。
【請求項76】
上記細胞が同種異系である、請求項75に記載の方法。
【請求項77】
上記細胞が同系である、請求項75に記載の方法。
【請求項78】
上記細胞が自己である、請求項75に記載の方法。
【請求項79】
上記α(1,3)ガラクトシルトランスフェラーゼをコードする配列がマウス配列である、請求項75に記載の方法。
【請求項80】
細胞を死滅させる上記ステップがγ線または紫外線照射によるものである、請求項75に記載の方法。
【請求項81】
上記形質転換のステップは、上記α(1,3)ガラクトシルトランスフェラーゼをコードするポリヌクレオチド配列の導入がベクターにより媒介されるステップである、請求項75に記載の方法。
【請求項82】
ベクターがプラスミドベクターである、請求項81に記載の方法。
【請求項83】
ベクターがウイルスベクターである、請求項81に記載の方法。
【請求項84】
ウイルスベクターがレトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターからなる群から選択される、請求項83に記載の方法。
【請求項85】
請求項75に記載の方法により作られる治療用医薬組成物。
【請求項86】
動物において免疫の媒介する腫瘍細胞破壊を誘導する方法であって、上記細胞をもつ動物に、その上にαGalエピトープが存在する複数の細胞表面糖タンパク質を含む弱毒化腫瘍細胞の混合物を投与することを含んでなる上記方法。
【請求項87】
腫瘍細胞をもつ動物を治療する方法であって、遺伝的にα(1,3)ガラクトシルトランスフェラーゼタンパク質をコードするポリヌクレオチドを用いて改変された弱毒化腫瘍細胞の混合物を含む組成物の治療上有効な量を上記動物に投与することを含んでなり、上記細胞が自己、同系、および同種異系腫瘍細胞からなる群から選択される、上記方法。
【請求項88】
上記α(1,3)ガラクトシルトランスフェラーゼポリヌクレオチドがマウス起源である、請求項87に記載の方法。
【請求項89】
上記ポリヌクレオチドが配列番号2のアミノ酸配列に対して少なくとも70%アミノ酸配列同一性または85%アミノ酸配列類似性をもつα(1,3)ガラクトシルトランスフェラーゼをコードする、請求項87に記載の方法。
【請求項90】
α(1,3)ガラクトシルトランスフェラーゼ遺伝子をベクターにより細胞中に導入する、請求項87に記載の方法。
【請求項91】
ベクターがウイルスベクターである、請求項90に記載の方法。
【請求項92】
ウイルスベクターがマウスレトロウイルスベクター、レンチウイルスベクターs、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターベースのものである、請求項91に記載の方法。
【請求項93】
動物において予め形成された腫瘍細胞を治療する方法であって、上記動物に、α(1,3)ガラクトシルトランスフェラーゼ遺伝子を発現する致死照射した同種異系、同系または自己腫瘍細胞の治療上有効な用量を導入するステップを含んでなる上記方法。
【請求項94】
α(1,3)ガラクトシルトランスフェラーゼポリヌクレオチドがマウス起源である、請求項93に記載の方法。
【請求項95】
上記遺伝子が配列番号2のアミノ酸配列に対して少なくとも70%アミノ酸配列同一性または85%アミノ酸配列類似性をもつα(1,3)ガラクトシルトランスフェラーゼをコードする、請求項93に記載の方法。
【請求項96】
α(1,3) ガラクトシルトランスフェラーゼ遺伝子をベクターにより細胞中に導入する、請求項93に記載の方法。
【請求項97】
ベクターがウイルスベクターである、請求項96に記載の方法。
【請求項98】
ウイルスベクターがマウスレトロウイルスベクター、レンチウイルスベクターs、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターベースのものである、請求項97に記載の方法。
【請求項1】
抗αGalを合成する動物に存在する腫瘍細胞を抑制するための医薬組成物であって、α(1,3)ガラクトシルトランスフェラーゼをコードする配列およびその上にαGalエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含む弱毒化したワクチン腫瘍細胞の混合物、ならびに担体を含んでなる上記医薬組成物。
【請求項2】
抗αGalを合成する動物において複数の腫瘍抗原に対する免疫応答を、それらのグリコシル化状態に関わらず、同時に刺激するための医薬組成物であって、α(1,3)ガラクトシルトランスフェラーゼタンパク質をコードする配列およびその上にαGalエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含み、腫瘍細胞全体中の抗原を免疫系に提示することができる弱毒化した全ワクチン腫瘍細胞の混合物、ならびに好適な担体を含んでなる上記医薬組成物。
【請求項3】
上記腫瘍細胞が、大細胞癌、扁平細胞癌、腹水炎腺癌(ascitis adenocarcinoma)、胸水癌、原発腺管上皮癌、転移性腺癌、原発腺癌、腎臓明細胞癌(clear cell carcinoma)、卵巣腺癌、卵巣奇形癌、悪性黒色腫、結腸直腸癌、結腸直腸腺癌、および結腸直腸腺癌グレードIIからなる群から選択される、請求項1に記載の組成物。
【請求項4】
上記腫瘍細胞がA549細胞由来である、請求項1に記載の組成物。
【請求項5】
上記腫瘍細胞がNCI-H460細胞由来である、請求項1に記載の組成物。
【請求項6】
上記ワクチン腫瘍細胞がNCI-H520細胞由来である、請求項1に記載の組成物。
【請求項7】
上記ワクチン腫瘍細胞がMCF-7細胞由来である、請求項1に記載の組成物。
【請求項8】
上記ワクチン腫瘍細胞BT-20細胞が由来である、請求項1に記載の組成物。
【請求項9】
上記ワクチン腫瘍細胞がHPAF-II由来である、請求項1に記載の組成物。
【請求項10】
上記ワクチン腫瘍細胞がPANC-1細胞由来である、請求項1に記載の組成物。
【請求項11】
上記ワクチン腫瘍細胞がASPC-1細胞由来である、請求項1に記載の組成物。
【請求項12】
上記ワクチン腫瘍細胞がBxPC3細胞由来である、請求項1に記載の組成物。
【請求項13】
上記ワクチン腫瘍細胞がIGROV細胞由来である、請求項1に記載の組成物。
【請求項14】
上記ワクチン腫瘍細胞がES-2細胞由来である、請求項1に記載の組成物。
【請求項15】
上記ワクチン腫瘍細胞がNIH:OVCAR3細胞由来である、請求項1に記載の組成物。
【請求項16】
上記ワクチン腫瘍細胞がCOLO829細胞由来である、請求項1に記載の組成物。
【請求項17】
上記ワクチン腫瘍細胞がG-361細胞由来である、請求項1に記載の組成物。
【請求項18】
上記ワクチン腫瘍細胞がHCT-116細胞由来である、請求項1に記載の組成物。
【請求項19】
上記ワクチン腫瘍細胞がCOLO205細胞由来である、請求項1に記載の組成物。
【請求項20】
上記ワクチン腫瘍細胞がLoVo細胞由来である、請求項1に記載の組成物。
【請求項21】
上記ワクチン腫瘍細胞がWiDr細胞由来である、請求項1に記載の組成物。
【請求項22】
上記ワクチン腫瘍細胞がDLD-1細胞由来である、請求項1に記載の組成物。
【請求項23】
上記ワクチン腫瘍細胞がHCT-15細胞由来である、請求項1に記載の組成物。
【請求項24】
上記ワクチン腫瘍細胞がSW620細胞由来である、請求項1に記載の組成物。
【請求項25】
上記ワクチン腫瘍細胞がSW480細胞由来である、請求項1に記載の組成物。
【請求項26】
上記ワクチン腫瘍細胞がSW1116細胞由来である、請求項1に記載の組成物。
【請求項27】
上記ワクチン腫瘍細胞がHT-29細胞由来である、請求項1に記載の組成物。
【請求項28】
上記ワクチン腫瘍細胞がFHC細胞由来である、請求項1に記載の組成物。
【請求項29】
上記ワクチン腫瘍細胞がCCD841CoN細胞由来である、請求項1に記載の組成物。
【請求項30】
上記ワクチン腫瘍細胞がPC-3細胞由来である、請求項1に記載の組成物。
【請求項31】
上記ワクチン腫瘍細胞がLNCaPFGC細胞由来である、請求項1に記載の組成物。
【請求項32】
上記ワクチン腫瘍細胞がMDAPca2b細胞由来である、請求項1に記載の組成物。
【請求項33】
上記ワクチン腫瘍細胞がDU145細胞由来である、請求項1に記載の組成物。
【請求項34】
上記組成物がHAL1である、請求項1に記載の組成物。
【請求項35】
上記組成物がHAL2である、請求項1に記載の組成物。
【請求項36】
上記組成物がHAL3である、請求項1に記載の組成物。
【請求項37】
上記組成物がHAB1である、請求項1に記載の組成物。
【請求項38】
上記組成物がHAB2である、請求項1に記載の組成物。
【請求項39】
上記組成物がHAPA1である、請求項1に記載の組成物。
【請求項40】
上記組成物がHAPA2である、請求項1に記載の組成物。
【請求項41】
上記組成物がHAPA3である、請求項1に記載の組成物。
【請求項42】
上記組成物がHAPA4である、請求項1に記載の組成物。
【請求項43】
上記組成物がHAO1である、請求項1に記載の組成物。
【請求項44】
上記組成物がHAO2である、請求項1に記載の組成物。
【請求項45】
上記組成物がHAO3である、請求項1に記載の組成物。
【請求項46】
上記組成物がHAO4である、請求項1に記載の組成物。
【請求項47】
上記組成物がHAM1である、請求項1に記載の組成物。
【請求項48】
上記組成物がHAM2である、請求項1に記載の組成物。
【請求項49】
上記組成物がHAM3である、請求項1に記載の組成物。
【請求項50】
上記組成物がHAC1である、請求項1に記載の組成物。
【請求項51】
上記組成物がHAC2である、請求項1に記載の組成物。
【請求項52】
上記組成物がHAC3である、請求項1に記載の組成物。
【請求項53】
上記組成物がHAC4である、請求項1に記載の組成物。
【請求項54】
上記組成物がHAC5である、請求項1に記載の組成物。
【請求項55】
上記組成物がHAC6である、請求項1に記載の組成物。
【請求項56】
上記組成物がHAC7である、請求項1に記載の組成物。
【請求項57】
上記組成物がHAC8である、請求項1に記載の組成物。
【請求項58】
上記組成物がHAC9である、請求項1に記載の組成物。
【請求項59】
上記組成物がHAC10である、請求項1に記載の組成物。
【請求項60】
上記組成物がHAC11である、請求項1に記載の組成物。
【請求項61】
上記組成物がHAC12である、請求項1に記載の組成物。
【請求項62】
上記組成物がHAPR1である、請求項1に記載の組成物。
【請求項63】
上記組成物がHAPR2である、請求項1に記載の組成物。
【請求項64】
上記組成物がHAPR3である、請求項1に記載の組成物。
【請求項65】
上記組成物がHAPR4である、請求項1に記載の組成物。
【請求項66】
さらにアジュバントを含む、請求項1に記載の組成物。
【請求項67】
抗α-galを合成する動物において腫瘍細胞に対する免疫応答を誘導するための医薬組成物であって、A549、NCI-H460、NCI-H520、MCF-7、BT-20、HPAF-II、PANC-1、ASPC-1、BxPC3、IGROV、ES-2、NIH:OVCAR3、PA-1、A375、COLO829、G-361、HCT-116、COLO205、LoVo、WiDr、DLD-1、HCT-15、SW620、SW480、SW116、HT-29、FHC、CCD841CoN、PC-3、LNCaPFGC、MDAPca2b、およびDU145からなる群から選択される、α(1,3)ガラクトシルトランスフェラーゼをコードする配列およびその上にαガラクトシルエピトープが存在する複数の細胞表面糖タンパク質または糖脂質を含む弱毒化ワクチン腫瘍細胞の混合物;ならびに担体を含んでなる上記医薬組成物。
【請求項68】
HAL1、HAL2またはHAL3の群から選択される1以上の培養細胞株を含んでなる、動物において肺組織腫瘍細胞を治療するための医薬組成物。
【請求項69】
HAB1またはHAB2の群から選択される1以上の培養細胞株を含んでなる、動物において乳房組織腫瘍細胞を治療するための医薬組成物。
【請求項70】
HAPA1、HAPA2、HAPA3、またはHAPA4の群から選択される1以上の培養細胞株を含んでなる、動物において膵臓組織腫瘍細胞を治療するための医薬組成物。
【請求項71】
HAO1、HAO2、HAO3、またはHAO4の群から選択される1以上の培養細胞株を含んでなる、動物において卵巣組織腫瘍細胞を治療するための医薬組成物。
【請求項72】
HAM1、HAM2、またはHAM3の群から選択される1以上の培養細胞株を含んでなる、動物において黒色腫組織腫瘍細胞を治療するための医薬組成物。
【請求項73】
HAC1、HAC2、HAC3、HAC4、HAC5、HAC6、HAC7、HAC8、HAC9、HAC10、HAC11、またはHAC12の群から選択される1以上の培養細胞株を含んでなる、動物において大腸組織腫瘍細胞を治療するための医薬組成物。
【請求項74】
HAPR1、HAPR2、HAPR3、またはHAPR4の群から選択される1以上の培養細胞株を含んでなる、動物において前立腺組織腫瘍細胞を治療するための医薬組成物。
【請求項75】
治療用医薬組成物を作る方法であって、生腫瘍細胞のコレクションを取得するステップ、発現するとα(1,3)ガラクトシルトランスフェラーゼをコードするヌクレオチド配列を用いて上記細胞を形質転換して、その結果、上記α(1,3)ガラクトシルトランスフェラーゼが上記細胞上の複数の細胞表面糖タンパク質上にαガラクトシルエピトープを合成するステップ、上記腫瘍細胞を採集してαガラクトシルエピトープを有する複数の糖タンパク質をもつ細胞の混合物を作るステップ、および上記細胞を死滅させるステップを含んでなる上記方法。
【請求項76】
上記細胞が同種異系である、請求項75に記載の方法。
【請求項77】
上記細胞が同系である、請求項75に記載の方法。
【請求項78】
上記細胞が自己である、請求項75に記載の方法。
【請求項79】
上記α(1,3)ガラクトシルトランスフェラーゼをコードする配列がマウス配列である、請求項75に記載の方法。
【請求項80】
細胞を死滅させる上記ステップがγ線または紫外線照射によるものである、請求項75に記載の方法。
【請求項81】
上記形質転換のステップは、上記α(1,3)ガラクトシルトランスフェラーゼをコードするポリヌクレオチド配列の導入がベクターにより媒介されるステップである、請求項75に記載の方法。
【請求項82】
ベクターがプラスミドベクターである、請求項81に記載の方法。
【請求項83】
ベクターがウイルスベクターである、請求項81に記載の方法。
【請求項84】
ウイルスベクターがレトロウイルスベクター、レンチウイルスベクター、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターからなる群から選択される、請求項83に記載の方法。
【請求項85】
請求項75に記載の方法により作られる治療用医薬組成物。
【請求項86】
動物において免疫の媒介する腫瘍細胞破壊を誘導する方法であって、上記細胞をもつ動物に、その上にαGalエピトープが存在する複数の細胞表面糖タンパク質を含む弱毒化腫瘍細胞の混合物を投与することを含んでなる上記方法。
【請求項87】
腫瘍細胞をもつ動物を治療する方法であって、遺伝的にα(1,3)ガラクトシルトランスフェラーゼタンパク質をコードするポリヌクレオチドを用いて改変された弱毒化腫瘍細胞の混合物を含む組成物の治療上有効な量を上記動物に投与することを含んでなり、上記細胞が自己、同系、および同種異系腫瘍細胞からなる群から選択される、上記方法。
【請求項88】
上記α(1,3)ガラクトシルトランスフェラーゼポリヌクレオチドがマウス起源である、請求項87に記載の方法。
【請求項89】
上記ポリヌクレオチドが配列番号2のアミノ酸配列に対して少なくとも70%アミノ酸配列同一性または85%アミノ酸配列類似性をもつα(1,3)ガラクトシルトランスフェラーゼをコードする、請求項87に記載の方法。
【請求項90】
α(1,3)ガラクトシルトランスフェラーゼ遺伝子をベクターにより細胞中に導入する、請求項87に記載の方法。
【請求項91】
ベクターがウイルスベクターである、請求項90に記載の方法。
【請求項92】
ウイルスベクターがマウスレトロウイルスベクター、レンチウイルスベクターs、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターベースのものである、請求項91に記載の方法。
【請求項93】
動物において予め形成された腫瘍細胞を治療する方法であって、上記動物に、α(1,3)ガラクトシルトランスフェラーゼ遺伝子を発現する致死照射した同種異系、同系または自己腫瘍細胞の治療上有効な用量を導入するステップを含んでなる上記方法。
【請求項94】
α(1,3)ガラクトシルトランスフェラーゼポリヌクレオチドがマウス起源である、請求項93に記載の方法。
【請求項95】
上記遺伝子が配列番号2のアミノ酸配列に対して少なくとも70%アミノ酸配列同一性または85%アミノ酸配列類似性をもつα(1,3)ガラクトシルトランスフェラーゼをコードする、請求項93に記載の方法。
【請求項96】
α(1,3) ガラクトシルトランスフェラーゼ遺伝子をベクターにより細胞中に導入する、請求項93に記載の方法。
【請求項97】
ベクターがウイルスベクターである、請求項96に記載の方法。
【請求項98】
ウイルスベクターがマウスレトロウイルスベクター、レンチウイルスベクターs、アデノウイルスベクター、アデノ随伴ウイルスベクターおよびヘルペスシンプレックスウイルスベクターベースのものである、請求項97に記載の方法。
【図1】

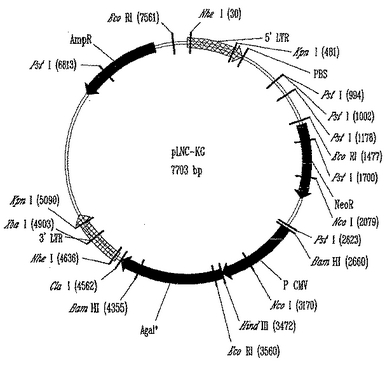
【図2A】

【図2B】

【図2C】

【図3】
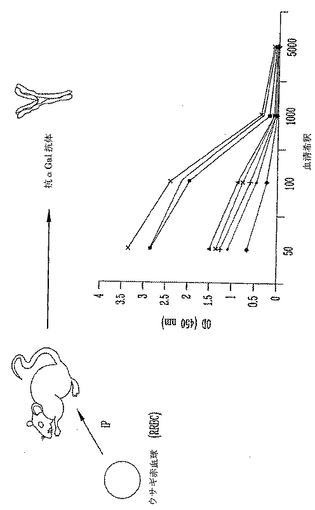
【図4】
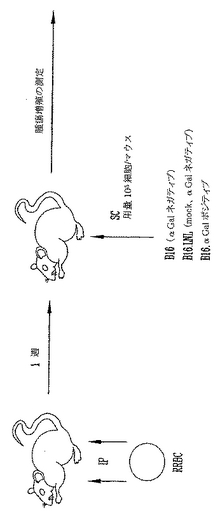
【図5】
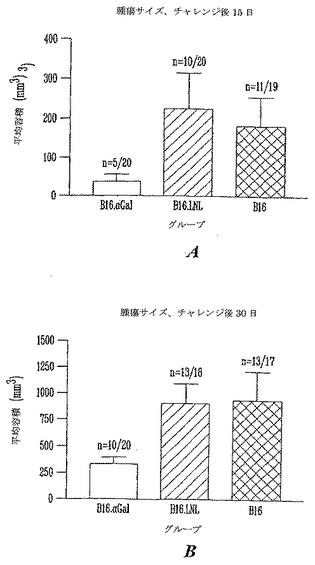
【図6】
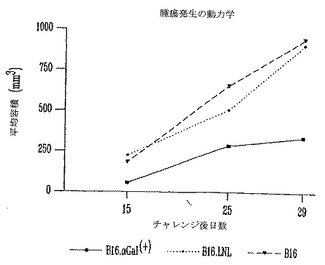
【図7】
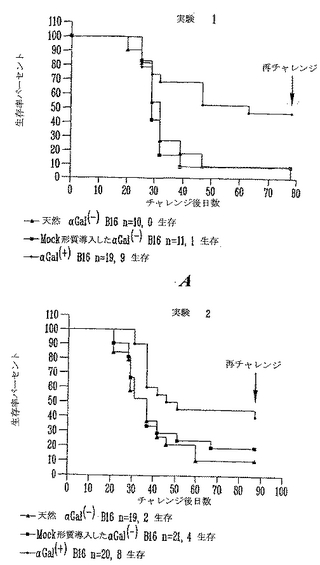
【図8】
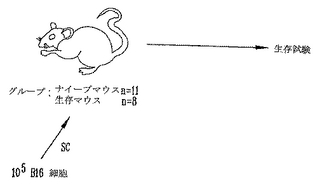
【図9】
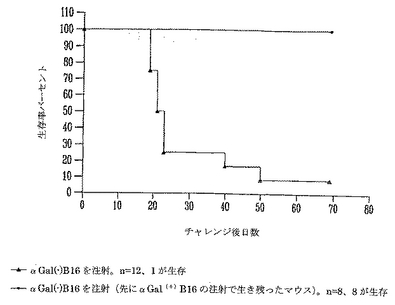
【図10】
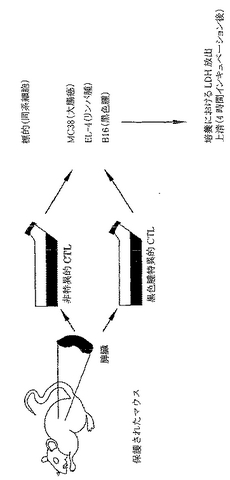
【図11】
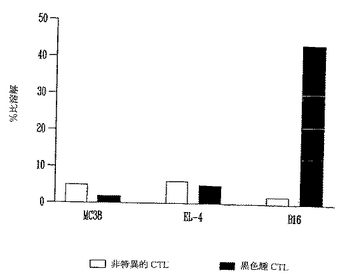
【図12】
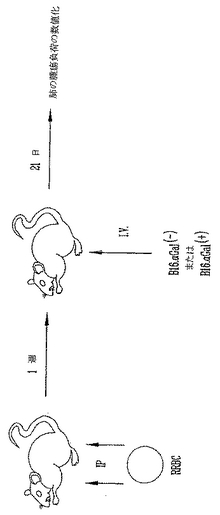
【図13】
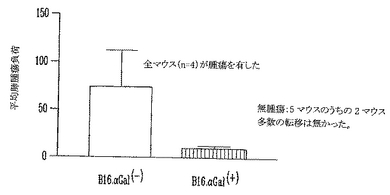
【図14】
【図15】
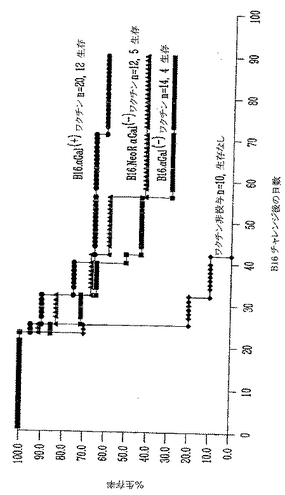
【図16】
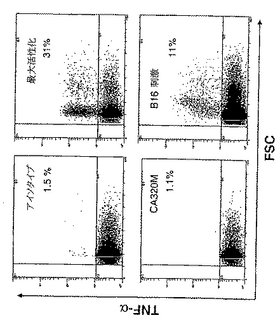
【図17】
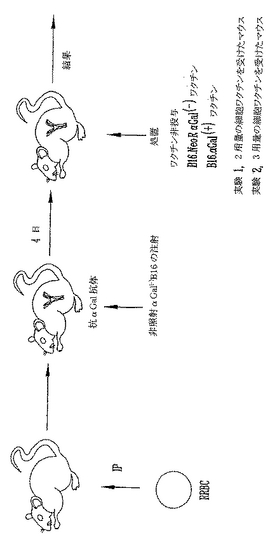
【図18】
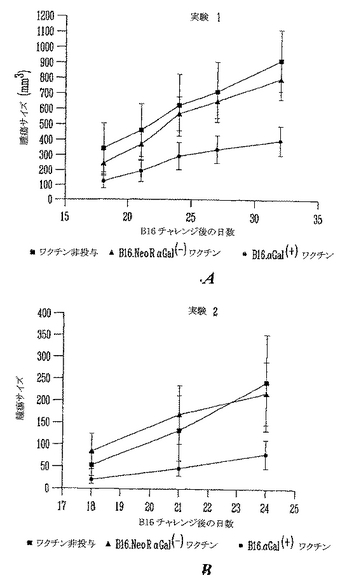
【図19】
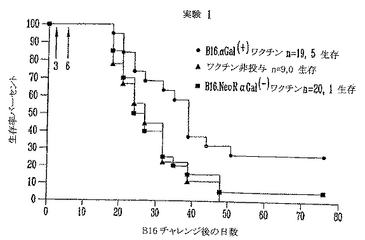
【図20】
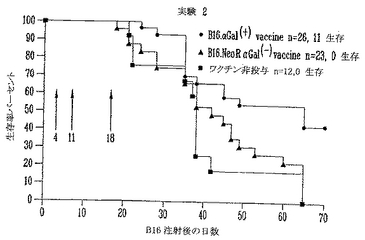
【図21】
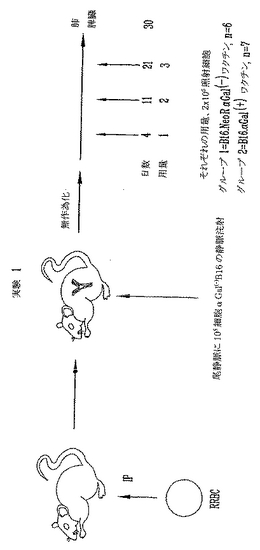
【図22】
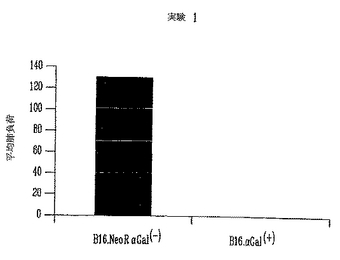
【図23】
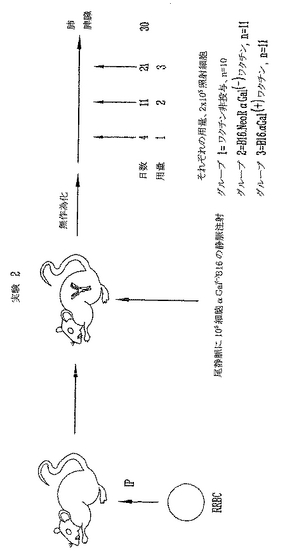
【図24】
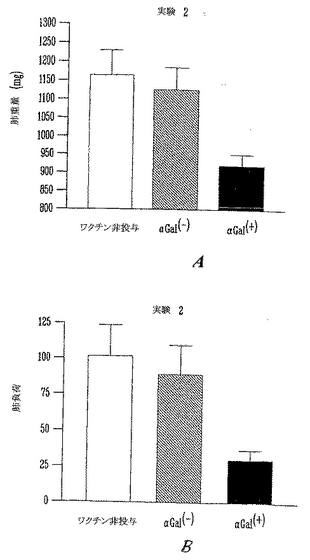
【図25】
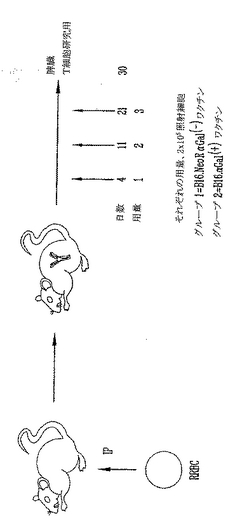
【図26】
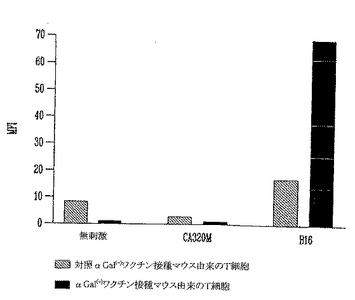
【図27】
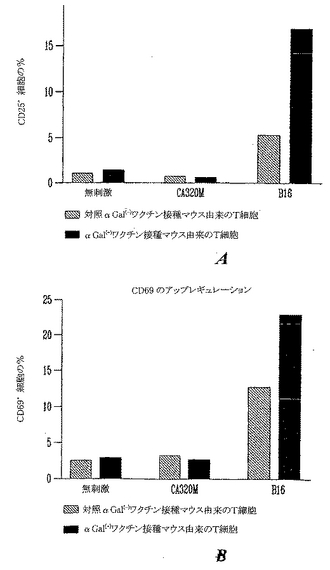
【図28】
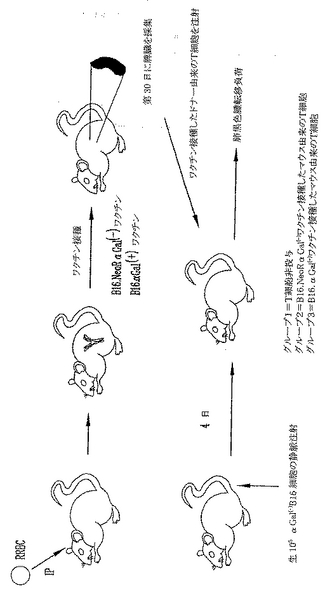
【図29】
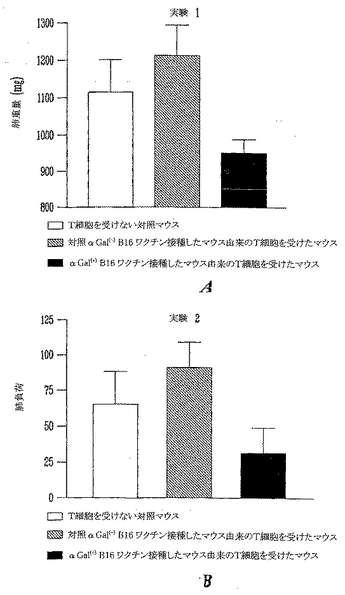
【図30】
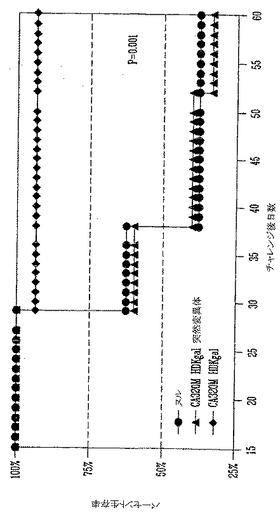
【図2A】
【図2B】
【図2C】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【図28】
【図29】
【図30】
【公表番号】特表2006−507267(P2006−507267A)
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願番号】特願2004−543606(P2004−543606)
【出願日】平成15年10月9日(2003.10.9)
【国際出願番号】PCT/US2003/032036
【国際公開番号】WO2004/032865
【国際公開日】平成16年4月22日(2004.4.22)
【出願人】(505129378)セントラル アイオワ ヘルス システム (1)
【Fターム(参考)】
【公表日】平成18年3月2日(2006.3.2)
【国際特許分類】
【出願日】平成15年10月9日(2003.10.9)
【国際出願番号】PCT/US2003/032036
【国際公開番号】WO2004/032865
【国際公開日】平成16年4月22日(2004.4.22)
【出願人】(505129378)セントラル アイオワ ヘルス システム (1)
【Fターム(参考)】
[ Back to top ]