
α,ω−3級ジアミノ化合物の製造方法
【課題】臭気が低く活性も高い高品質のウレタンフォーム製造用触媒等に用いられ、炭素数6〜12のメチレン鎖が3級ジアミン骨格を有するN,N,N’,N’−テトラメチル−1,10−デカンジアミン等のα,ω−3級ジアミンや、α,ω−3級ジアミンの中間体であるN,N,N’,N’−テトラメチル−1,10−デカナミド等のα,ω−3級ジアミドを、経済的に高収率で量産できるα,ω−3級アミノ化合物の製造方法を提供することを目的とする。
【解決手段】本発明のα,ω−3級ジアミノ化合物の製造方法は、酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得る構成を有している。
【解決手段】本発明のα,ω−3級ジアミノ化合物の製造方法は、酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得る構成を有している。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、ウレタンフォームの製造用触媒等に用いられるα,ω−3級ジアミンや、α,ω−3級ジアミンの中間体であるα,ω−3級ジアミドからのα,ω−3級ジアミノ化合物の製造方法に関するものである。
【背景技術】
【0002】
従来より、ウレタンフォームの製造用触媒として3級アミンが好適であることが知られている。このような3級アミンには、高温においても蒸気圧が低く、臭気も低く長期間安定した性質を示すことが要求される。また、ウレタンフォームの製造用触媒として優れた触媒活性を有していることも要求される。
このような3級アミンとしては、(特許文献1)に「炭素数9〜12のメチレン鎖が3級ジアミン骨格を有するポリウレタン製造用触媒」が記載されている。そして、「ポリウレタン製造用触媒は、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカンの還元メチル化、あるいは1,8−オクタンジオール、1,9−ノナンジオール、1,10−デカンジオール、1,11−ウンデカンジオール、1,12−ドデカンジオールとジメチルアミンの反応等により、通常公知の方法で合成できること」も記載されている。
(特許文献2)には、「N,N−ジメチル型の3級アミド(モノ・アミド)の接触水素還元による対応するN,N−ジメチル型の3級アミン(モノ・アミン)の製造方法」が記載されている。そして実施例には、「ソジウム・メチラート(NOCH3)等の塩基触媒の存在下、高級脂肪酸メチルエステル(ラウリン酸メチルとミリスチン酸メチルの混合物)とジメチルアミンとを80℃で反応させ、脱水下にアミド化反応を進行させて、対応するN,N−ジメチル型の3級アミド(モノ・アミド)を製造すること」も記載されている。さらに、「高級脂肪酸メチルエステルから合成したN,N,N’,N’−型の3級ジアミドが、対応する3級ジアミンの原料となり得ること」も記載されている。
【特許文献1】特開平7−90040号公報
【特許文献2】米国特許第5840985号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
しかしながら上記従来の技術においては、以下のような課題を有していた。
(1)(特許文献1)に開示されたウレタンフォーム製造用触媒の原料となる1,8−オクタンジオール、1,9−ノナンジオール等は石油化学原料由来であり、省資源性に欠けるという課題を有していた。また、これらのジオールは非常に高価なため、経済的に量産できないという課題を有していた。
(2)(特許文献2)には「高級脂肪酸メチルエステルから合成したN,N,N’,N’−型の3級ジアミドが、対応する3級ジアミンの原料となり得ること」が記載されてはいるが、その実施例は記載されていなかった。そこで、特許文献2に記載された方法に準拠して、本発明者らが高級脂肪酸メチルエステル(具体的にはセバシン酸ジメチルエステル)からN,N,N’,N’−型の3級ジアミド(具体的にはN,N,N’,N’−テトラメチル−1,10−デカナミド)の合成を試みたが、後述するようにアミド化反応(3級モノアミド及び3級ジアミドの生成)の進行は皆無に近く、目的とする3級ジアミドを全く製造できなかった。
このように従来は、ウレタンフォームの製造用触媒等に有用なα,ω−3級ジアミンや、その中間体であるα,ω−3級ジアミドを経済的に量産する技術は皆無であった。
【0004】
本発明は上記従来の課題を解決するもので、ウレタンフォームの製造用触媒であるα,ω−3級ジアミンの中間体や、電解コンデンサの電解質等として可能性のあるα,ω−3級ジアミドを経済的に高収率で量産できるα,ω−3級アミノ化合物の製造方法の提供を目的とする。
【課題を解決するための手段】
【0005】
上記従来の課題を解決するために本発明のα,ω−3級ジアミノ化合物の製造方法は、以下の構成を有している。
本発明の請求項1に記載のα,ω−3級ジアミノ化合物の製造方法は、酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得る構成を有している。
この構成により、以下のような作用が得られる。
(1)高級脂肪酸メチルエステルと比較すると反応性は若干劣るが安価なα,ω−ジカルボン酸を原料として、対応するN,N,N’,N’−型のα,ω−3級ジアミドを経済的に高収率で量産することができる。
【0006】
ここで、α,ω−ジカルボン酸は酸触媒の存在下で、式(1)及び式(2)に示した2段の逐次反応によってアミド化が進行し、α,ω−ジカルボン酸に対応するα,ω−3級ジアミドを製造することができる。
反応の進行は、反応水の留出、酸価、ガスクロマトグラフィー、赤外スペクトルによって追跡することができる。赤外スペクトルでは、アミド化反応の進行とともにカルボン酸のカルボキシル基のカルボニル基の伸縮振動の吸収スペクトルの強度が激減し、代わって3級ジアミドのカルボニル基の伸縮振動に由来する特徴的吸収が低波数側に出現する。このスペクトルの強度を追跡することができる。
【0007】
【化1】

【0008】
α,ω−ジカルボン酸としては、HOOC−(CH2)n−COOH(但し、nは自然数。好ましくは、nは4〜10。)の一般式で示される。
具体的には、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸,ウンデカン酸、ドデカニ酸等が用いられる。
【0009】
酸触媒としては、均一触媒、不均一触媒(固体触媒)のいずれも用いることができる。また、ルイス酸、ブレンステッド酸のいずれも用いることができる。固体触媒としては、アルミナ、シリカ・アルミナ、ゼオライト、ジルコニア、酸化亜鉛、シリカ・チタニア、シリカ・マグネシア、チタニア・ジルコニア等のアルミニウム,ケイ素,チタン,亜鉛,ジルコニウム等の1種以上の元素を含む金属化合物を用いることができる。ヘテロポリ酸(日本新金属製)や陽イオン交換樹脂(ナフィオン)等も用いることができる。均一触媒としては、周期律表第4a族のチタンやジルコニウム等のアルコキシドであるチタンイソプロポキシドやジルコニウムイソプロポキシド等を用いることができる。これらのアルコキシドは、触媒担体に担持させて固体触媒として用いることもできる。
【0010】
酸触媒は、出発原料のα,ω−ジカルボン酸に対し0.001〜50wt%好ましくは0.01〜20wt%が添加される。酸触媒の添加量が0.01wt%より少なくなるにつれアミド化反応が進行し難くなり、20wt%を超えるにつれ触媒のランニングコストが増加し経済的に有効でなくなる傾向がみられる。特に、0.001wt%より少なくなるか50wt%より多くなると、これらの傾向が著しくなるため、いずれも好ましくない。
【0011】
アミド化反応の反応温度としては、α,ω−ジカルボン酸の融点以上300℃以下、好ましくは150〜260℃、より好ましくは170〜230℃が好適である。反応温度が低くなるにつれアミド化反応速度が低下する傾向がみられる。反応温度が300℃を超えると、生成したα,ω−3級ジアミドが熱劣化し品質の低下をもたらすため好ましくない。
【0012】
ジメチルアミンは、微小気泡に分散できるスパージャー(多孔ノズル)を通して連続的に若しくは間歇的に常圧でバブリング導入するのが好ましい。
過剰に供給されたジメチルアミンは、反応水に溶解して反応系外に流出する。反応水に溶解したジメチルアミンは回収して再使用することができる。ジメチルアミンの供給速度は、アミド化反応の進行とジメチルアミンの回収系の負荷を考慮して、適宜設定することができる。
なお、ジメチルアミンの供給速度が低い場合、反応器に供給されたジメチルアミンが完全消費されるため、ジメチルアミンでは反応水を反応器外に搬出することができなくなる。この場合は反応器内で反応水の突沸が起こり反応速度の低下をきたすため、必要に応じて、窒素等の不活性ガスをジメチルアミンと混合して供給することもできる。
【0013】
反応時の圧力は常圧でよい。アミド化反応は常圧で効率よく進行するからである。加圧条件は、脱水反応を抑制する方向に作用するため好ましくない。
【0014】
反応は回分式でも連続式でも実施することができる。回分式の場合は、撹拌槽型反応器やインジェクター方式の反応器を使用することができる。連続式の場合、酸触媒が不均一触媒の場合は固定床反応器を、均一触媒の場合はCSTR(continuous stirred tank reactor:連続式撹拌槽型反応器)なども使用することができる。
【0015】
反応時間は触媒の種類、触媒の添加量、反応温度、ジメチルアミンの供給速度、不活性ガス等の反応水搬出促進ガスの供給速度等に大きく依存するが、回分反応においては1〜20時間で反応を完結させることができる。
【0016】
反応後、得られたα,ω−3級ジアミドは、不均一触媒(固体触媒)を使用した場合は濾過によって触媒を分離することができる。均一触媒を使用した場合は蒸留に付して、蒸留残渣に残存させて分離できることがある。
【0017】
本発明の請求項2に記載の発明は、請求項1に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを蒸留精製する構成を有している。
この構成により、請求項1で得られる作用に加え、以下のような作用が得られる。
(1)生産性に欠ける再結晶法とは異なり、蒸留精製するので生産性に優れる。これまで、α,ω−3級ジアミドは蒸留精製できないとされ、文献に記載がなく、石油エーテルからの再結晶精製法が記載されているに過ぎなかった。本発明者らは鋭意検討することで、生産性に優れる蒸留精製に成功した。
(2)アミド化反応に均一触媒を使用した場合でも、触媒を蒸留残渣に残存させて分離しα,ω−3級ジアミドを精製できるため生産性に優れる。
【0018】
ここで、α,ω−3級ジアミドは減圧蒸留するのが好ましい。α,ω−3級ジアミドが熱劣化するのを防止できるからである。
【0019】
本発明の請求項3に記載の発明は、請求項1又は2に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを接触水素還元して対応する3級ジアミンを得る構成を有している。
この構成により、請求項1又は2で得られる作用に加え、以下のような作用が得られる。
(1)比較的穏和な条件でα,ω−3級ジアミドのカルボニル基が容易に還元されて、対応するN,N,N’,N’−型のα,ω−3級ジアミンを製造することができ生産性に優れる。
【0020】
ここで、3級ジアミドは、均一触媒の存在下で生成したものである場合、蒸留精製したもの、精製処理をしていないもののいずれも用いることができる。不均一触媒の存在下で生成したものである場合には、濾過分離したものを用いる。
【0021】
接触水素化用触媒としては、高級アルコール製造時の高級脂肪酸メチルエステル還元用の銅系触媒を使用することができる。銅系触媒としては、銅クロム、銅/亜鉛/アルミナ、銅/鉄/アルミナ等が好適に使用される。なお、アルコールのアミノ化反応に使用する銅/ニッケル系触媒は活性が低く、本発明における接触水素化用触媒としては有効ではない。
水素圧としては、比較的穏和な0.1〜20MPa(0.99〜198Kg/cm2)が好適である。なお、高級脂肪酸メチルエステルからの高級アルコール製造時のような高い水素圧(15〜25MPa)は不要であり、0.1〜3MPa程度の水素圧下でも、3級ジアミドを3級ジアミンに変換させることができる。
反応温度としては、150〜300℃好ましくは180〜250℃が好適である。反応温度が180℃より低くなるにつれ反応に長時間を要し生産性が低下する傾向がみられ、250℃より高くなるにつれ水素化分解による副反応が増大する傾向がみられる。特に、150℃より低くなるか300℃より高くなると、これらの傾向が著しくなるためいずれも好ましくない。
α,ω−3級ジアミドのカルボニル基は、式(3)、式(4)で示される逐次反応に従って容易に還元されて、対応するα,ω−3級ジアミンに変換される。反応水は反応系外に除去する。反応水が反応系内に存在すると触媒毒として作用し、触媒活性が低下するからである。
【0022】
【化2】
【0023】
α,ω−3級ジアミドの接触水素還元は、式(3)、式(4)に示したように2段の逐次反応によって進行するため、3級モノアミドの接触水素還元の場合と比較して、接触水素化触媒に対する負荷が2倍以上に増加する。このため、接触水素還元に先立って、接触水素化用触媒を還元活性化する必要がある。通常、反応温度までの昇温中に接触水素化用触媒の還元活性化が進行する。接触水素化用触媒の還元活性化に伴って水が生成し、これが触媒毒として作用するので、この触媒還元水を連続的に反応系外に除去する。
【0024】
接触水素還元の際、副反応として、式(5)、式(6)で示されるジメチルアミド基の水素化分解によるアルデヒド経由のアルコールの副生がある。
【0025】
【化3】
【0026】
式(5)、式(6)で示されるアルコール体は、生成物であるα,ω−3級ジアミンに対し不純物となるため、高純度のα,ω−3級ジアミンが必要な場合には除去する必要がある。
アルコール体の除去方法としては、アルコール体のリン酸エステルを生成させ蒸留分離する方法、アルコール体とジメチルアミンとのアミノ化反応によりアルコール体を3級ジアミンにまで変換する方法を用いることができる。前者はアルコール体を回収再使用することができ、後者は3級ジアミンの歩留向上をもたらすため、いずれも有効な方法である。
【0027】
本発明の請求項4に記載の発明は、請求項3に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを接触水素還元して生成した反応混合物とジメチルアミンとを、触媒の存在下、反応させる構成を有している。
この構成により、請求項3で得られる作用に加え、以下のような作用が得られる。
(1)3級ジアミドを接触水素還元して生成した反応混合物には、目的とする3級ジアミンの他、前述の式(5)、式(6)で示される副反応によって生成されるアルコール体が含まれており、3級ジアミンの収率を低下させる原因となっている。これは、3級ジアミドは分子両末端に一個ずつ、合計2個の3級アミド基を有するため、アミド結合の解裂(水素化分解)によるアルデヒド経由のアルコール体副生の確率が、分子の一末端に一個の3級アミド基を有する3級モノアミドの場合に比べて著しく高いことによる。反応混合物に含まれるアルコール体とジメチルアミンとのアミノ化反応により、アルコール体を3級ジアミンにまで変換させることができるため、目的とする3級ジアミンの収率を高めることができる。
【0028】
ここで、触媒としては、多価アルコールのアミノ化反応に用いられる公知の触媒を用いることができる。例えば、特公昭60−11020号公報に記載された(a)銅のカルボン酸塩又は銅の分子内錯体として銅アセチルアセトン錯体と、(b)周期律表第8族元素、マンガン及び亜鉛から選ばれる金属のカルボン酸塩又は分子内錯体としてアセチルアセトン錯体の1種又は2種以上と、(c)カルボン酸又はカルボン酸のアルカリ金属塩若しくはアルカリ土類金属塩の1種又は2種以上と、の混合物を水素とアミンの混合物又は他の還元剤で還元処理した触媒、特公平3−4534号公報や特開平5−39338号公報に記載された銅−ニッケル−第8族白金元素触媒等を用いることができる。
しかし、本発明者らが新たに開発した、銅、ニッケル、カルシウム、アルカリ金属又はアルカリ土類金属(カルシウムを除く)を必須成分とする触媒を用い、その触媒の存在下でアミノ化反応を実施することにより、高い反応速度で選択性よく3級ジアミンを製造できる。
【0029】
銅、ニッケル、カルシウム及びアルカリ土類金属(カルシウムを除く)を必須成分とする触媒の触媒原料としては、(a)銅のカルボン酸塩又は銅の分子内錯体の1種又は2種以上、(b)ニッケルのカルボン酸塩又はニッケルの分子内錯体の1種又は2種以上、(c)カルシウムのカルボン酸塩又はカルシウム錯体の1種又は2種以上、(d)アルカリ金属又はアルカリ土類金属(カルシウムを除く)のカルボン酸塩の1種又は2種以上の混合物が用いられる。
アミノ化反応を実施するには、まず触媒原料を還元活性化させる必要がある。例えば、反応混合物に触媒原料を加熱溶解させ、水素又は他の還元剤を導入して還元活性化させた後(以下、還元活性化処理という。)、ジメチルアミンを導入することでアミノ化反応を進行させることができる。還元活性化処理によって得られた触媒は、見掛け上均一なコロイド状触媒(銅/ニッケル粒子径は約1nm)となる。
【0030】
触媒原料中、(a)銅のカルボン酸塩及び銅の分子内錯体は、還元活性化処理の過程で、金属銅にまで還元される。銅のカルボン酸塩を形成するカルボン酸としては、分子中にカルボキシル基を有するものであれば芳香族系であっても、分岐を有するものでも、直鎖アルキル基に複数のカルボキシル基や、他の置換基を有するものであってもよく、例えば、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、ウンデカン酸、ラウリン酸、トリデカン酸、ミリスチン酸、ペンタデカン酸、パルミチン酸、ステアリン酸、オレイン酸等を挙げることができる。好ましいのは炭素数6〜36のカルボン酸であり、特に好ましくは炭素数12〜24のカルボン酸である。
炭素数5以下のカルボン酸塩は還元中、遊離したカルボン酸の影響によって金属コロイドが凝集し易く活性が低下し易いからである。また、炭素数の大きなカルボン酸は入手が困難になるからである。銅の分子内錯体としては、例えば、アセチルアセトン錯体やジメチルグリオキシム錯体等、イオウを含有しない一般のキレート化合物を挙げることができる。
【0031】
また、触媒原料中、(b)ニッケルのカルボン酸塩及びニッケルの分子内錯体も、還元活性化処理の過程で還元される。カルボン酸塩、分子内錯体としては、前記カルボン酸、前記分子内錯体と同様の有機配位子を例示できる。なお、カルボン酸としては炭素数6〜36のものが好ましい。炭素数5以下のカルボン酸塩は還元中、遊離したカルボン酸の影響によって金属コロイドが凝集し易く活性が低下し易いからである。また、炭素数の大きなカルボン酸は入手が困難になるからである。
【0032】
また、触媒原料中、(c)カルシウムのカルボン酸塩及びカルシウム錯体は、アミノ化反応中に次第に還元され、銅及びニッケルと共に強力な触媒作用を発現する。カルボン酸としては前記カルボン酸と同様のものを例示できる。カルシウム錯体としては、例えば、アセチルアセトン錯体、ジメチルグリオキシム錯体等、無機陰イオンを持たない一般のキレート化合物が挙げられる。
なお、カルボン酸としては、前述したように炭素数6〜36のものが好ましい。銅、ニッケルの場合と同様に入手が容易であるとともに、カルボン酸を遊離させ、銅/ニッケル金属コロイドを凝集させ易く活性を低下させ易いからである。
【0033】
触媒原料中、(d)アルカリ金属又はアルカリ土類金属(カルシウムを除く)のカルボン酸塩は、還元活性化処理及びアミノ化反応中も還元されることなく、銅−ニッケル−カルシウム系コロイド触媒の安定化剤として機能する。なかでもアルカリ土類金属のカルボン酸塩、特にバリウムのカルボン酸塩が有効である。バリウムは、銅やニッケルと比較して特に還元され難く、触媒の活性を維持する安定化剤として特に有効に機能するからである。
カルボン酸塩としては、前記したものと同様のものを例示でき、例えばステアリン酸バリウム、ラウリン酸バリウム、ステアリン酸ナトリウム等を挙げることができる。なかでも炭素数8〜30好ましくは10〜24特に18〜24のステアリン酸、ベヘニン酸、リグノセリン酸等が好適に用いられる。容易に入手できるとともに、本発明者らの実験の結果、炭素数8〜30のカルボン酸塩は、カルボキシル基の鎖長効果により銅/ニッケル金属コロイドの凝集抑制効果が高く、高い触媒活性を与えることがわかったからである。
なお、銅、ニッケル、アルカリ金属、アルカリ土類金属のカルボン酸塩は、特公昭59−27617号公報等に記載された周知の方法を用いて製造できる。
【0034】
触媒原料の還元活性化処理で用いる水素以外の他の還元剤としては、Al(C2H5)3、(C2H5)2Al(OC2H5)等を用いることができる。
触媒原料の還元活性化処理では、反応混合物中に触媒原料を投入し、昇温と同時に水素等の還元剤を連続的に供給する。160℃付近から2価の銅の還元が始まり、200℃未満で触媒の活性化が完了する。還元活性化処理後、アミノ化反応の進行とともにコロイド状触媒を含有する反応混合物の色調が淡黄色(透明)から黒色へと変化し、次第に赤褐色の均一なコロイド状触媒へと変化し、高活性を発現するようになる。
【0035】
還元活性化処理後は、反応器を100〜250℃好ましくは150〜220℃より好ましくは180〜220℃に設定し、反応混合物にジメチルアミンを導入し、アミノ化反応を開始させる。反応温度が180℃より低くなるにつれ反応速度が低下する傾向がみられ、150℃より低くなるにつれこの傾向が顕著になり、100℃より低いと生産性に著しく欠けるため好ましくない。反応温度が220℃より高くなるにつれ副反応が加速される傾向がみられ、250℃より高くなると顕著になるため好ましくない。
【0036】
アミノ化反応は、−5〜10気圧(−506〜1013kPa)好ましくは常圧〜5気圧(101〜506kPa)より好ましくは常圧〜3気圧(101〜304kPa)の範囲で行うのが好適である。アミノ化反応は脱水反応であるため、加圧条件下では反応速度の低下を招くためである。特に、5気圧より高くなるとこの傾向が増大し、10気圧を超えると顕著になるため好ましくない。
なお、前述の米国特許第5840985号公報(特許文献2)では、実施例1に、副反応物としてのアルコール体の処理を目的として、アミド還元時に還元剤としての水素に加えジメチルアミンを供給し、200〜400psi(1379〜2758kPa)の加圧下で、副生アルコール体をアミド還元と同時にアミンに変換する例が記載されている。
しかしながら、アルコール体のアミノ化反応は、アルコール体の脱水素に始まり生成アルデヒドへのジメチルアミン付加、次いでジメチルアミン付加体の水素化分解反応によって進行するため、200〜400psi(1379〜2758kPa)の加圧下では、アルコールの脱水素に対し、さらに生成水の留出除去に対し不利であり、反応速度の低下を招く。
アルコール体のアミノ化反応の本質的反応機構に基づき、アミノ化反応を−5〜10気圧(−506〜1013kPa)好ましくは常圧〜5気圧(101〜506kPa)より好ましくは常圧〜3気圧(101〜304kPa)の範囲で行うことにより、副生アルコール体を対応する3級ジアミンに効率良く変換させることができる。
【0037】
アミノ化反応では、ジメチルアミンを導入すると数分間の誘導期の後、水が留出し始め反応の進行を確認できる。触媒の活性化後は、アミノ化反応は水素を導入しない条件下でも進行する。反応混合物の脱水素によって発生した活性化水素が反応に使われるからである。しかし、水素を導入して水素の存在下で反応を行うのが好ましい。反応時間を若干短縮できるとともに、導入された水素が水の系外への搬出を助けるからである。水を系外へ効率よく搬出させるため、反応器内に導入された水搬出用の水素,窒素や不活性ガス等は消費されないので、水素に代えて、又は水素に混合して、窒素や希ガス等の不活性ガスを反応器内に導入することもできる。
水の生成とともに生成物である油分も留出するので、常法によって油水分離し、必要に応じて油分を反応器に戻しアミノ化反応を進行させる。反応の進行は、アミン価、水酸基価、或いはガスクロマトグラフィー分析によって追跡することができる。
アミノ化反応が終了すると水の留出も停止する。反応温度、触媒の濃度、ジメチルアミンの供給速度にもよるが、アミノ化反応は2〜10時間で完了させることができる。
【0038】
触媒の濃度としては、金属銅を基準にして0.001〜10wt%(反応混合物中のアルコール体に対して)好ましくは0.01〜5wt%より好ましくは0.05〜2wt%が好適である。濃度が0.05wt%より低くなるにつれ反応速度が低下する傾向がみられ、0.001wt%より低くなると生産性に著しく欠けるため好ましくない。濃度が2wt%より高くなるにつれ副反応が促進される傾向がみられ、10wt%を超えると顕著になるため好ましくない。
【0039】
アミノ化反応において、反応器内に供給されるジメチルアミンの単位時間当たりの供給速度(L/時間)としては、標準状態で、反応混合物中のアルコール体の水酸基1モル当たり0.01〜100モル/時間、好ましくは0.1〜10モル/時間、特に好ましくは0.2〜5モル/時間がよい。0.01モル/時間未満では反応速度が遅く生産性に著しく欠けるため好ましくない。100モル/時間を越える場合は、ジメチルアミンによる触媒被毒が顕著になって反応速度と収率の低下をきたし、さらに不均化も大きく促進されるため好ましくない。
【0040】
本発明の請求項5に記載の発明は、請求項1乃至4の内いずれか1に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記α,ω−ジカルボン酸のメチレン鎖の炭素数が、4〜10である構成を有している。
この構成により、請求項1乃至4の内いずれか1で得られる作用に加え、以下のような作用が得られる。
(1)得られるα,ω−3級ジアミンの沸点が高いことに由来して臭気が低く、眼粘膜刺激性も低く、それゆえ、作業環境の向上をもたらすとともに、ウレタンフォーム製造用触媒としての活性にも優れるため応用性に優れる。
【0041】
ここで、α,ω−ジカルボン酸のメチレン鎖の炭素数が4未満であると、得られるα,ω−3級ジアミンが低沸点のため臭気が激しく、炭素数が10を超えると、得られるα,ω−3級ジアミンのウレタンフォーム製造用触媒としての活性低下が大きくなるため、いずれも好ましくない。
特に、α、ω―ジカルボン酸のセバシン酸(メチレン鎖の炭素数8)は、その2官能性により広範囲の用途を有し、インドを最大の生産国とするヒマシ油からのオレオ・インダストリーとして今後、飛躍的な成長と発展が期待されるため好適である。
【発明の効果】
【0042】
以上のように、本発明のα,ω−3級ジアミノ化合物の製造方法によれば、以下のような有利な効果が得られる。
請求項1に記載の発明によれば、
(1)α,ω−ジカルボン酸を原料として、対応するα,ω−3級ジアミドを経済的に高収率で量産できるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0043】
請求項2に記載の発明によれば、請求項1の効果に加え、
(1)生産性に欠ける再結晶法とは異なり、3級ジアミドを蒸留精製するので生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
(2)アミド化反応に均一触媒を使用した場合でも、触媒を蒸留残渣に残存させて分離しα,ω−3級ジアミドを精製でき生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0044】
請求項3に記載の発明によれば、請求項1又は2の効果に加え、
(1)比較的穏和な条件でα,ω−3級ジアミドのカルボニル基が容易に還元されて、対応するα,ω−3級ジアミンを製造することができ生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0045】
請求項4に記載の発明によれば、請求項3の効果に加え、
(1)反応混合物に含まれる副反応生成物のアルコール体とジメチルアミンとのアミノ化反応により、目的とする3級ジアミンを高い収率で製造できるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0046】
請求項5に記載の発明によれば、請求項1乃至4の内いずれか1の効果に加え、
(1)得られるα,ω−3級ジアミンの沸点が高いことに由来して臭気が低く、眼粘膜刺激性も低く、それゆえ、作業環境の向上をもたらすとともに、ウレタンフォーム製造用触媒としての活性にも優れるため応用性に優れたα,ω−3級ジアミンが得られるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【発明を実施するための最良の形態】
【0047】
以下、本発明を実施例により具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
本実施例では、冷却器、撹拌機、温度指示制御器、原料ガス導入管(ガラス素焼き製のスパージャー)、サンプリング器を設けた500mlのセパラブル・フラスコ(反応器)に、α,ω−ジカルボン酸としてのセバシン酸(酸価556、小倉合成工業製)と、固体酸触媒(200〜300メッシュ通過品)又は均一触媒を所定量仕込み、所定の反応温度(180〜220℃)まで昇温し、ジメチルアミンと窒素との混合ガスを常圧でそれぞれ所定の流速で連続的に供給した。数分〜30分の誘導期間が発生するが、この間はセバシン酸とジメチルアミンとの間にアンモニウム塩が形成されている。
その後、アンモニウムの脱水反応により反応水の留出を伴って2段逐次のアミド化反応が進行し、α,ω−3級ジアミドとしてのN,N,N’,N’−テトラメチル1,10−デカナミドが生成する。セバシン酸の転化率は99.8%以上を達成することができる。
なお、生成物の分析にはガスクロマトグラフィー(島津製作所製、GC−8A)を用いた。測定条件は以下のとおりである。
カラム:DB−17、カラム温度:Inj.250℃、initial:100℃、final:290℃、 昇温速度:10℃/分、試料濃度:10%、試料注入量:0.2μL。
また、酸価及び水酸基価の測定は、JIS K0070−1992に準拠して行なった。
【0048】
(実施例1)
反応器に、セバシン酸を200g、ルイス酸触媒としての均一触媒であるチタニウム・テトラ・イソプロポキシド(Ti(O-i-Pr)4)を40mg(セバシン酸に対し0.02wt%)仕込んだ。反応系は均一な無色透明溶液を形成した。撹拌下、窒素を常圧で9L/時の流速で連続供給し、設定反応温度の180℃まで昇温した。その後、窒素の供給速度を維持したまま、ジメチルアミンを常圧で15L/時の流速で連続供給した。
約30分の誘導期間の後、反応水の留出を伴ってアミド化反応が定常的に進行した。セバシン酸とジメチルアミンとのアミド化反応は2段の逐次反応で進行した。反応9時間での酸価は21.0であり、セバシン酸転化率96.2%であった。
得られた反応混合物を減圧蒸留した結果、留分の留出温度は0.4KPaで201〜215℃であった。このとき、蒸留缶の温度は215〜223℃、N,N,N’,N’−テトラメチル−1,10−デカナミドの蒸留収率は97.2%であった。
従来、N,N,N’,N’−テトラメチル−1,10−デカナミドのようなα、ω−3級ジアミドの蒸留精製の事例はないが、蒸留精製できることを今回初めて確認した。
【0049】
(実施例2)
酸触媒として、日揮化学製の脱水反応用シリカ・アルミナ固体触媒(商品名:N633HN、粒子径250メッシュ通過、表面積380m2/g、嵩比重0.4)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった。
反応15時間での反応混合物の酸価は16.2であり、セバシン酸転化率は97.1%であった。
【0050】
(実施例3)
酸触媒として、日揮化学製の脱水反応用シリカ・アルミナ固体触媒(商品名:N633L)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった(反応温度180℃)。
反応18時間で酸価は1.0であり、セバシン酸転化率は99.8%であった。得られた反応混合物を実施例1と同様に蒸留精製した結果、酸価は0.7(純度99.9%)にまで向上した。
なお、反応温度を220℃にした場合も、反応混合物の酸価は1.0であり、セバシン酸転化率99.8%に相当した。
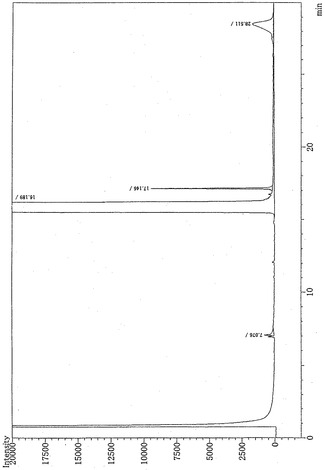
図1は、反応混合物(反応温度180℃)を蒸留精製した精製物のガスクロマトグラフィーのチャートである。ピーク面積から換算すると、N,N,N’,N’−テトラメチル−1,10−デカナミドの純度は99.9%以上であることが確認された。
【0051】
(実施例4)
酸触媒として、日揮化学製の脱水反応用アルミナ固体触媒(商品名:N613N、嵩比重0.5)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった。
反応18時間で酸価は9.0であり、セバシン酸転化率は98.5%であった。
【0052】
(実施例5)
反応温度が220℃、ジメチルアミン供給速度が35L/時である以外は、実施例4と同様にしてアミド化反応を行なった。
反応7.5時間で酸価は4.9であり、セバシン酸転化率は99.1%であった。
【0053】
(比較例1)
(特許文献2)に記載された「塩基触媒としてのナトリウム・メトキシド(NaOCH3)の存在下のラウリン酸メチルエステルとジメチルアミンとのアミド化反応(反応温度80℃)」に準じて、セバシン酸ジメチルエステルとジメチルアミンとを出発原料とするアミド化反応を行った。
反応器にセバシン酸ジメチルエステルを250g、塩基触媒としてのナトリウム・メトキシド3.0gを10.3gのメチルアルコールに分散した溶液を仕込み、撹拌下、設定反応温度である75℃に昇温した。
ここで、設定反応温度を文献に記載された80℃より低めに設定した理由は、触媒としてのナトリウム・メトキシドがメチルアルコールに分散していることから、メチルアルコールの蒸発によりナトリウム・メトキシドが凝集し触媒活性が激減する可能性があるからである。
メチルアルコールの還流下に7時間反応を行なった結果、セバシン酸とジメチルアミンとの第4級アンモニウム塩は生成するものの、4級塩からの脱水反応がほとんど進行しないことが判明し、(特許文献2)に記載された方法ではセバシン酸からの3級ジアミドを製造し得ないことが判明した。
一般にα、ω−型の2官能性化合物を合成する場合、逐次反応系の2段目の反応が進行し難いという一般的傾向がある。(特許文献2)の出発原料は一価のカルボン酸であり、本発明の出発原料であるジカルボン酸とは構造が全く異なるため、(特許文献2)に記載された方法では、4級アンモニウム塩の脱水反応を進行させることができず、3級ジアミドを製造できないことが判明した。
【0054】
(比較例2)
反応性が大きなα,ω−ジカルボン酸ジクロリド(ここではセバシン酸のジ酸クロリド)を石油エーテルに溶解させ、常温でジメチルアミンをバブリング導入してアミド化反応を2時間行なった。その結果、腐食性の酸クロリドが定量的に反応せず、未反応の酸クロリドの除去と粗3級ジアミドの精製に多大な負荷がかかることが判明した。これにより、α,ω−ジカルボン酸ジクロリドを出発原料として、工業的にα,ω−3級ジアミドを製造できないことが判明した。
【0055】
(比較例3)
触媒として、固体酸ではないシリカゲルを用いた以外は、実施例2と同様にしてα,ω−3級ジアミドの製造を試みた。しかし、安定な4級塩が主に生成するのみであった。4級塩を主成分とする反応混合物は水溶性が強く、220〜250℃でも固体であり均一溶液を形成することができず、4級塩からの脱水反応を観測することができなかった。
【0056】
以上の実施例によれば、酸触媒の存在下でセバシン酸から、対応するα,ω−3級ジアミドとしてのN,N,N’,N’−テトラメチル1,10−デカナミドを容易に高い転化率で製造できることが明らかになった。また、蒸留精製によって高純度のα,ω−3級ジアミドが得られることも初めて確認することができた。
【0057】
(実施例6)
実施例6では、N,N,N’,N’−テトラメチル−1,10−デカナミドの接触水素還元を行った。反応器としては、撹拌機、温度指示制御器、水素供給ライン及び排ガスラインを設けた0.3Lのオートクレーブを用いた。
この反応器に、実施例3で得られた3級ジアミドであるN,N,N’,N’−テトラメチル−1,10−デカナミド(酸価1.0)の蒸留生成物(酸価0.7、純度99.9%)を87g、銅/亜鉛/アルミナ触媒(日揮化学製、CuO:ZnO:Al2O3=43:44:5〜6wt%、Cu:Zn=1:1)4.35g(3級ジアミドに対し5wt%)を仕込んだ。
水素供給ラインから反応器内に水素を連続的に供給するとともに(約30L/時、水素圧0.5MPa)、排ガスラインから触媒の還元によって生成した還元水及び排ガスを連続的に排出しながら昇温し、触媒の還元活性化を行なった。本実施例で用いる銅/亜鉛/アルミナ触媒は、約135℃から水素還元が進行し、約160℃で還元が完了することを事前に確認している。反応器に水素を連続的に供給し排ガス及び還元水を連続的に排出する水素流通系を適用した理由は、還元水による触媒の被毒を最大限回避するためである。さらに水素流通下に昇温を続け、設定反応温度である230℃に達してから水素圧を2.5MPaに上げ、この水素圧を維持しながら、3級ジアミドの還元によって生成した反応水を含む排ガスを連続的に排出して3級ジアミドの水素還元を行なった。反応中、排ガスラインが反応混合物で閉塞傾向になるので、排ガス及び反応水の排出を妨げないように排ガスラインを十分に保温した。
反応4時間目の3級ジアミドの転化率は97%、反応6時間目の3級ジアミドの転化率は99.9%であった。反応6時間目の反応混合物の分析結果を(表1)の「3級ジアミド還元」と表記した欄に示した。
【0058】
【表1】
【0059】
(表1)に示すように、目的とするN,N,N’,N’−テトラメチル−1,10−デカンジアミンMe2N−(CH2)10−NMe2(3級ジアミン)は反応混合物に対して56.8%生成しており、Me2N−(CH2)10−OH等のアルコール体(水酸基含有物)が副反応生成物として約30%生成していることがわかった。
【0060】
次に、反応混合物をオートクレーブから取り出した後、生成した水を分離するための凝縮器及び分離器、反応混合物サンプリング器、排ガス出口管、原料ガス導入管(多孔質ガラス製のスパージャー)、撹拌器、温度計を設けたフラスコ(反応器)に87g仕込んだ。次に、触媒原料としてステアリン酸銅1.74g(アルコール体に対する金属銅0.1wt%)、ステアリン酸ニッケル0.35g(アルコール体に対する金属ニッケル0.02wt%)、ステアリン酸カルシウム0.35g(アルコール体に対する金属カルシウム0.02wt%)、ステアリン酸バリウム0.35g(アルコール体に対する金属バリウム0.02wt%)を加えた後、撹拌器を回転させ反応器内を窒素で置換し昇温した。触媒原料の4種類の金属石鹸は、100℃に達するまでに均一に溶解した。100℃に達したら窒素を水素に切換え、水素ガスを、流量計を通じて22L/時間の流速でフラスコ内にバブリングさせて還元活性化処理を行った。170〜190℃で2価の銅とニッケルの特徴的な緑色が次第に淡色化し、触媒原料が還元され見掛け上均一なコロイド状触媒となった。反応温度を210℃に保ち、ジメチルアミンを常圧で20〜30L/時間の流速で水素(流速22L/時間)との混合ガスとして連続供給し反応させた。
反応2時間目と反応6時間目の反応混合物の分析結果を(表1)の「アルコール アミノ化」と表記した欄に示した。
【0061】
反応2時間目では、Me2N−(CH2)10−OH等のアルコール体(水酸基含有物)はアミノ化反応によって約6%に減少し、目的とするMe2N−(CH2)10−NMe2(3級ジアミン)は反応混合物に対して73.6%生成していることがわかった。反応を続けたところ、反応6時間目で3級ジアミンの収率は76.1%に達していた。
なお、本実施例においては、反応器に水素を連続的に供給し排ガス及び還元水を連続的に排出する水素流通系を適用したため、3級ジアミドを接触水素還元したとき、及び、副生アルコール体をアミノ化したときに、目的の3級ジアミン含量の高い低沸点の油分が一部留出していた。これは、留出油分の成分分析によって確認することができた。
従って、この留出油分を連続的に反応器に戻すならば、3級ジアミンの収率は90%程度になることがわかった。
以上のように本実施例によれば、反応混合物に含まれる副反応生成物のアルコール体とジメチルアミンとのアミノ化反応により、目的とする3級ジアミンを高収率で製造できることが明らかになった。
【0062】
以上の実施例においては、α,ω−ジカルボン酸としてのセバシン酸を用いた場合について説明したが、一般式HOOC−(CH2)n−COOH(但し、nは自然数。)で示される他のα,ω−ジカルボン酸からも、同様に対応するα,ω−3級ジアミド及びα,ω−3級ジアミンを製造できることを確認した。
【産業上の利用可能性】
【0063】
本発明は、電解コンデンサの電解質等として可能性のあるα,ω−3級ジアミドや、ウレタンフォームの製造用触媒等として有用なα,ω−3級ジアミンを経済的に高収率で量産できるα,ω−3級ジアミノ化合物の製造方法を提供できる。本発明は、椰子、パームに次ぐ第3のオレオ・インダストリー構築のための技術要素を形成する必要不可欠な技術である。
【図面の簡単な説明】
【0064】
【図1】反応混合物を蒸留精製した精製物のガスクロマトグラフィーのチャート
【技術分野】
【0001】
本発明は、ウレタンフォームの製造用触媒等に用いられるα,ω−3級ジアミンや、α,ω−3級ジアミンの中間体であるα,ω−3級ジアミドからのα,ω−3級ジアミノ化合物の製造方法に関するものである。
【背景技術】
【0002】
従来より、ウレタンフォームの製造用触媒として3級アミンが好適であることが知られている。このような3級アミンには、高温においても蒸気圧が低く、臭気も低く長期間安定した性質を示すことが要求される。また、ウレタンフォームの製造用触媒として優れた触媒活性を有していることも要求される。
このような3級アミンとしては、(特許文献1)に「炭素数9〜12のメチレン鎖が3級ジアミン骨格を有するポリウレタン製造用触媒」が記載されている。そして、「ポリウレタン製造用触媒は、1,8−ジアミノオクタン、1,9−ジアミノノナン、1,10−ジアミノデカン、1,11−ジアミノウンデカン、1,12−ジアミノドデカンの還元メチル化、あるいは1,8−オクタンジオール、1,9−ノナンジオール、1,10−デカンジオール、1,11−ウンデカンジオール、1,12−ドデカンジオールとジメチルアミンの反応等により、通常公知の方法で合成できること」も記載されている。
(特許文献2)には、「N,N−ジメチル型の3級アミド(モノ・アミド)の接触水素還元による対応するN,N−ジメチル型の3級アミン(モノ・アミン)の製造方法」が記載されている。そして実施例には、「ソジウム・メチラート(NOCH3)等の塩基触媒の存在下、高級脂肪酸メチルエステル(ラウリン酸メチルとミリスチン酸メチルの混合物)とジメチルアミンとを80℃で反応させ、脱水下にアミド化反応を進行させて、対応するN,N−ジメチル型の3級アミド(モノ・アミド)を製造すること」も記載されている。さらに、「高級脂肪酸メチルエステルから合成したN,N,N’,N’−型の3級ジアミドが、対応する3級ジアミンの原料となり得ること」も記載されている。
【特許文献1】特開平7−90040号公報
【特許文献2】米国特許第5840985号公報
【発明の開示】
【発明が解決しようとする課題】
【0003】
しかしながら上記従来の技術においては、以下のような課題を有していた。
(1)(特許文献1)に開示されたウレタンフォーム製造用触媒の原料となる1,8−オクタンジオール、1,9−ノナンジオール等は石油化学原料由来であり、省資源性に欠けるという課題を有していた。また、これらのジオールは非常に高価なため、経済的に量産できないという課題を有していた。
(2)(特許文献2)には「高級脂肪酸メチルエステルから合成したN,N,N’,N’−型の3級ジアミドが、対応する3級ジアミンの原料となり得ること」が記載されてはいるが、その実施例は記載されていなかった。そこで、特許文献2に記載された方法に準拠して、本発明者らが高級脂肪酸メチルエステル(具体的にはセバシン酸ジメチルエステル)からN,N,N’,N’−型の3級ジアミド(具体的にはN,N,N’,N’−テトラメチル−1,10−デカナミド)の合成を試みたが、後述するようにアミド化反応(3級モノアミド及び3級ジアミドの生成)の進行は皆無に近く、目的とする3級ジアミドを全く製造できなかった。
このように従来は、ウレタンフォームの製造用触媒等に有用なα,ω−3級ジアミンや、その中間体であるα,ω−3級ジアミドを経済的に量産する技術は皆無であった。
【0004】
本発明は上記従来の課題を解決するもので、ウレタンフォームの製造用触媒であるα,ω−3級ジアミンの中間体や、電解コンデンサの電解質等として可能性のあるα,ω−3級ジアミドを経済的に高収率で量産できるα,ω−3級アミノ化合物の製造方法の提供を目的とする。
【課題を解決するための手段】
【0005】
上記従来の課題を解決するために本発明のα,ω−3級ジアミノ化合物の製造方法は、以下の構成を有している。
本発明の請求項1に記載のα,ω−3級ジアミノ化合物の製造方法は、酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得る構成を有している。
この構成により、以下のような作用が得られる。
(1)高級脂肪酸メチルエステルと比較すると反応性は若干劣るが安価なα,ω−ジカルボン酸を原料として、対応するN,N,N’,N’−型のα,ω−3級ジアミドを経済的に高収率で量産することができる。
【0006】
ここで、α,ω−ジカルボン酸は酸触媒の存在下で、式(1)及び式(2)に示した2段の逐次反応によってアミド化が進行し、α,ω−ジカルボン酸に対応するα,ω−3級ジアミドを製造することができる。
反応の進行は、反応水の留出、酸価、ガスクロマトグラフィー、赤外スペクトルによって追跡することができる。赤外スペクトルでは、アミド化反応の進行とともにカルボン酸のカルボキシル基のカルボニル基の伸縮振動の吸収スペクトルの強度が激減し、代わって3級ジアミドのカルボニル基の伸縮振動に由来する特徴的吸収が低波数側に出現する。このスペクトルの強度を追跡することができる。
【0007】
【化1】
【0008】
α,ω−ジカルボン酸としては、HOOC−(CH2)n−COOH(但し、nは自然数。好ましくは、nは4〜10。)の一般式で示される。
具体的には、アジピン酸、ピメリン酸、スベリン酸、アゼライン酸、セバシン酸,ウンデカン酸、ドデカニ酸等が用いられる。
【0009】
酸触媒としては、均一触媒、不均一触媒(固体触媒)のいずれも用いることができる。また、ルイス酸、ブレンステッド酸のいずれも用いることができる。固体触媒としては、アルミナ、シリカ・アルミナ、ゼオライト、ジルコニア、酸化亜鉛、シリカ・チタニア、シリカ・マグネシア、チタニア・ジルコニア等のアルミニウム,ケイ素,チタン,亜鉛,ジルコニウム等の1種以上の元素を含む金属化合物を用いることができる。ヘテロポリ酸(日本新金属製)や陽イオン交換樹脂(ナフィオン)等も用いることができる。均一触媒としては、周期律表第4a族のチタンやジルコニウム等のアルコキシドであるチタンイソプロポキシドやジルコニウムイソプロポキシド等を用いることができる。これらのアルコキシドは、触媒担体に担持させて固体触媒として用いることもできる。
【0010】
酸触媒は、出発原料のα,ω−ジカルボン酸に対し0.001〜50wt%好ましくは0.01〜20wt%が添加される。酸触媒の添加量が0.01wt%より少なくなるにつれアミド化反応が進行し難くなり、20wt%を超えるにつれ触媒のランニングコストが増加し経済的に有効でなくなる傾向がみられる。特に、0.001wt%より少なくなるか50wt%より多くなると、これらの傾向が著しくなるため、いずれも好ましくない。
【0011】
アミド化反応の反応温度としては、α,ω−ジカルボン酸の融点以上300℃以下、好ましくは150〜260℃、より好ましくは170〜230℃が好適である。反応温度が低くなるにつれアミド化反応速度が低下する傾向がみられる。反応温度が300℃を超えると、生成したα,ω−3級ジアミドが熱劣化し品質の低下をもたらすため好ましくない。
【0012】
ジメチルアミンは、微小気泡に分散できるスパージャー(多孔ノズル)を通して連続的に若しくは間歇的に常圧でバブリング導入するのが好ましい。
過剰に供給されたジメチルアミンは、反応水に溶解して反応系外に流出する。反応水に溶解したジメチルアミンは回収して再使用することができる。ジメチルアミンの供給速度は、アミド化反応の進行とジメチルアミンの回収系の負荷を考慮して、適宜設定することができる。
なお、ジメチルアミンの供給速度が低い場合、反応器に供給されたジメチルアミンが完全消費されるため、ジメチルアミンでは反応水を反応器外に搬出することができなくなる。この場合は反応器内で反応水の突沸が起こり反応速度の低下をきたすため、必要に応じて、窒素等の不活性ガスをジメチルアミンと混合して供給することもできる。
【0013】
反応時の圧力は常圧でよい。アミド化反応は常圧で効率よく進行するからである。加圧条件は、脱水反応を抑制する方向に作用するため好ましくない。
【0014】
反応は回分式でも連続式でも実施することができる。回分式の場合は、撹拌槽型反応器やインジェクター方式の反応器を使用することができる。連続式の場合、酸触媒が不均一触媒の場合は固定床反応器を、均一触媒の場合はCSTR(continuous stirred tank reactor:連続式撹拌槽型反応器)なども使用することができる。
【0015】
反応時間は触媒の種類、触媒の添加量、反応温度、ジメチルアミンの供給速度、不活性ガス等の反応水搬出促進ガスの供給速度等に大きく依存するが、回分反応においては1〜20時間で反応を完結させることができる。
【0016】
反応後、得られたα,ω−3級ジアミドは、不均一触媒(固体触媒)を使用した場合は濾過によって触媒を分離することができる。均一触媒を使用した場合は蒸留に付して、蒸留残渣に残存させて分離できることがある。
【0017】
本発明の請求項2に記載の発明は、請求項1に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを蒸留精製する構成を有している。
この構成により、請求項1で得られる作用に加え、以下のような作用が得られる。
(1)生産性に欠ける再結晶法とは異なり、蒸留精製するので生産性に優れる。これまで、α,ω−3級ジアミドは蒸留精製できないとされ、文献に記載がなく、石油エーテルからの再結晶精製法が記載されているに過ぎなかった。本発明者らは鋭意検討することで、生産性に優れる蒸留精製に成功した。
(2)アミド化反応に均一触媒を使用した場合でも、触媒を蒸留残渣に残存させて分離しα,ω−3級ジアミドを精製できるため生産性に優れる。
【0018】
ここで、α,ω−3級ジアミドは減圧蒸留するのが好ましい。α,ω−3級ジアミドが熱劣化するのを防止できるからである。
【0019】
本発明の請求項3に記載の発明は、請求項1又は2に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを接触水素還元して対応する3級ジアミンを得る構成を有している。
この構成により、請求項1又は2で得られる作用に加え、以下のような作用が得られる。
(1)比較的穏和な条件でα,ω−3級ジアミドのカルボニル基が容易に還元されて、対応するN,N,N’,N’−型のα,ω−3級ジアミンを製造することができ生産性に優れる。
【0020】
ここで、3級ジアミドは、均一触媒の存在下で生成したものである場合、蒸留精製したもの、精製処理をしていないもののいずれも用いることができる。不均一触媒の存在下で生成したものである場合には、濾過分離したものを用いる。
【0021】
接触水素化用触媒としては、高級アルコール製造時の高級脂肪酸メチルエステル還元用の銅系触媒を使用することができる。銅系触媒としては、銅クロム、銅/亜鉛/アルミナ、銅/鉄/アルミナ等が好適に使用される。なお、アルコールのアミノ化反応に使用する銅/ニッケル系触媒は活性が低く、本発明における接触水素化用触媒としては有効ではない。
水素圧としては、比較的穏和な0.1〜20MPa(0.99〜198Kg/cm2)が好適である。なお、高級脂肪酸メチルエステルからの高級アルコール製造時のような高い水素圧(15〜25MPa)は不要であり、0.1〜3MPa程度の水素圧下でも、3級ジアミドを3級ジアミンに変換させることができる。
反応温度としては、150〜300℃好ましくは180〜250℃が好適である。反応温度が180℃より低くなるにつれ反応に長時間を要し生産性が低下する傾向がみられ、250℃より高くなるにつれ水素化分解による副反応が増大する傾向がみられる。特に、150℃より低くなるか300℃より高くなると、これらの傾向が著しくなるためいずれも好ましくない。
α,ω−3級ジアミドのカルボニル基は、式(3)、式(4)で示される逐次反応に従って容易に還元されて、対応するα,ω−3級ジアミンに変換される。反応水は反応系外に除去する。反応水が反応系内に存在すると触媒毒として作用し、触媒活性が低下するからである。
【0022】
【化2】
【0023】
α,ω−3級ジアミドの接触水素還元は、式(3)、式(4)に示したように2段の逐次反応によって進行するため、3級モノアミドの接触水素還元の場合と比較して、接触水素化触媒に対する負荷が2倍以上に増加する。このため、接触水素還元に先立って、接触水素化用触媒を還元活性化する必要がある。通常、反応温度までの昇温中に接触水素化用触媒の還元活性化が進行する。接触水素化用触媒の還元活性化に伴って水が生成し、これが触媒毒として作用するので、この触媒還元水を連続的に反応系外に除去する。
【0024】
接触水素還元の際、副反応として、式(5)、式(6)で示されるジメチルアミド基の水素化分解によるアルデヒド経由のアルコールの副生がある。
【0025】
【化3】
【0026】
式(5)、式(6)で示されるアルコール体は、生成物であるα,ω−3級ジアミンに対し不純物となるため、高純度のα,ω−3級ジアミンが必要な場合には除去する必要がある。
アルコール体の除去方法としては、アルコール体のリン酸エステルを生成させ蒸留分離する方法、アルコール体とジメチルアミンとのアミノ化反応によりアルコール体を3級ジアミンにまで変換する方法を用いることができる。前者はアルコール体を回収再使用することができ、後者は3級ジアミンの歩留向上をもたらすため、いずれも有効な方法である。
【0027】
本発明の請求項4に記載の発明は、請求項3に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記3級ジアミドを接触水素還元して生成した反応混合物とジメチルアミンとを、触媒の存在下、反応させる構成を有している。
この構成により、請求項3で得られる作用に加え、以下のような作用が得られる。
(1)3級ジアミドを接触水素還元して生成した反応混合物には、目的とする3級ジアミンの他、前述の式(5)、式(6)で示される副反応によって生成されるアルコール体が含まれており、3級ジアミンの収率を低下させる原因となっている。これは、3級ジアミドは分子両末端に一個ずつ、合計2個の3級アミド基を有するため、アミド結合の解裂(水素化分解)によるアルデヒド経由のアルコール体副生の確率が、分子の一末端に一個の3級アミド基を有する3級モノアミドの場合に比べて著しく高いことによる。反応混合物に含まれるアルコール体とジメチルアミンとのアミノ化反応により、アルコール体を3級ジアミンにまで変換させることができるため、目的とする3級ジアミンの収率を高めることができる。
【0028】
ここで、触媒としては、多価アルコールのアミノ化反応に用いられる公知の触媒を用いることができる。例えば、特公昭60−11020号公報に記載された(a)銅のカルボン酸塩又は銅の分子内錯体として銅アセチルアセトン錯体と、(b)周期律表第8族元素、マンガン及び亜鉛から選ばれる金属のカルボン酸塩又は分子内錯体としてアセチルアセトン錯体の1種又は2種以上と、(c)カルボン酸又はカルボン酸のアルカリ金属塩若しくはアルカリ土類金属塩の1種又は2種以上と、の混合物を水素とアミンの混合物又は他の還元剤で還元処理した触媒、特公平3−4534号公報や特開平5−39338号公報に記載された銅−ニッケル−第8族白金元素触媒等を用いることができる。
しかし、本発明者らが新たに開発した、銅、ニッケル、カルシウム、アルカリ金属又はアルカリ土類金属(カルシウムを除く)を必須成分とする触媒を用い、その触媒の存在下でアミノ化反応を実施することにより、高い反応速度で選択性よく3級ジアミンを製造できる。
【0029】
銅、ニッケル、カルシウム及びアルカリ土類金属(カルシウムを除く)を必須成分とする触媒の触媒原料としては、(a)銅のカルボン酸塩又は銅の分子内錯体の1種又は2種以上、(b)ニッケルのカルボン酸塩又はニッケルの分子内錯体の1種又は2種以上、(c)カルシウムのカルボン酸塩又はカルシウム錯体の1種又は2種以上、(d)アルカリ金属又はアルカリ土類金属(カルシウムを除く)のカルボン酸塩の1種又は2種以上の混合物が用いられる。
アミノ化反応を実施するには、まず触媒原料を還元活性化させる必要がある。例えば、反応混合物に触媒原料を加熱溶解させ、水素又は他の還元剤を導入して還元活性化させた後(以下、還元活性化処理という。)、ジメチルアミンを導入することでアミノ化反応を進行させることができる。還元活性化処理によって得られた触媒は、見掛け上均一なコロイド状触媒(銅/ニッケル粒子径は約1nm)となる。
【0030】
触媒原料中、(a)銅のカルボン酸塩及び銅の分子内錯体は、還元活性化処理の過程で、金属銅にまで還元される。銅のカルボン酸塩を形成するカルボン酸としては、分子中にカルボキシル基を有するものであれば芳香族系であっても、分岐を有するものでも、直鎖アルキル基に複数のカルボキシル基や、他の置換基を有するものであってもよく、例えば、カプロン酸、エナント酸、カプリル酸、ペラルゴン酸、カプリン酸、ウンデカン酸、ラウリン酸、トリデカン酸、ミリスチン酸、ペンタデカン酸、パルミチン酸、ステアリン酸、オレイン酸等を挙げることができる。好ましいのは炭素数6〜36のカルボン酸であり、特に好ましくは炭素数12〜24のカルボン酸である。
炭素数5以下のカルボン酸塩は還元中、遊離したカルボン酸の影響によって金属コロイドが凝集し易く活性が低下し易いからである。また、炭素数の大きなカルボン酸は入手が困難になるからである。銅の分子内錯体としては、例えば、アセチルアセトン錯体やジメチルグリオキシム錯体等、イオウを含有しない一般のキレート化合物を挙げることができる。
【0031】
また、触媒原料中、(b)ニッケルのカルボン酸塩及びニッケルの分子内錯体も、還元活性化処理の過程で還元される。カルボン酸塩、分子内錯体としては、前記カルボン酸、前記分子内錯体と同様の有機配位子を例示できる。なお、カルボン酸としては炭素数6〜36のものが好ましい。炭素数5以下のカルボン酸塩は還元中、遊離したカルボン酸の影響によって金属コロイドが凝集し易く活性が低下し易いからである。また、炭素数の大きなカルボン酸は入手が困難になるからである。
【0032】
また、触媒原料中、(c)カルシウムのカルボン酸塩及びカルシウム錯体は、アミノ化反応中に次第に還元され、銅及びニッケルと共に強力な触媒作用を発現する。カルボン酸としては前記カルボン酸と同様のものを例示できる。カルシウム錯体としては、例えば、アセチルアセトン錯体、ジメチルグリオキシム錯体等、無機陰イオンを持たない一般のキレート化合物が挙げられる。
なお、カルボン酸としては、前述したように炭素数6〜36のものが好ましい。銅、ニッケルの場合と同様に入手が容易であるとともに、カルボン酸を遊離させ、銅/ニッケル金属コロイドを凝集させ易く活性を低下させ易いからである。
【0033】
触媒原料中、(d)アルカリ金属又はアルカリ土類金属(カルシウムを除く)のカルボン酸塩は、還元活性化処理及びアミノ化反応中も還元されることなく、銅−ニッケル−カルシウム系コロイド触媒の安定化剤として機能する。なかでもアルカリ土類金属のカルボン酸塩、特にバリウムのカルボン酸塩が有効である。バリウムは、銅やニッケルと比較して特に還元され難く、触媒の活性を維持する安定化剤として特に有効に機能するからである。
カルボン酸塩としては、前記したものと同様のものを例示でき、例えばステアリン酸バリウム、ラウリン酸バリウム、ステアリン酸ナトリウム等を挙げることができる。なかでも炭素数8〜30好ましくは10〜24特に18〜24のステアリン酸、ベヘニン酸、リグノセリン酸等が好適に用いられる。容易に入手できるとともに、本発明者らの実験の結果、炭素数8〜30のカルボン酸塩は、カルボキシル基の鎖長効果により銅/ニッケル金属コロイドの凝集抑制効果が高く、高い触媒活性を与えることがわかったからである。
なお、銅、ニッケル、アルカリ金属、アルカリ土類金属のカルボン酸塩は、特公昭59−27617号公報等に記載された周知の方法を用いて製造できる。
【0034】
触媒原料の還元活性化処理で用いる水素以外の他の還元剤としては、Al(C2H5)3、(C2H5)2Al(OC2H5)等を用いることができる。
触媒原料の還元活性化処理では、反応混合物中に触媒原料を投入し、昇温と同時に水素等の還元剤を連続的に供給する。160℃付近から2価の銅の還元が始まり、200℃未満で触媒の活性化が完了する。還元活性化処理後、アミノ化反応の進行とともにコロイド状触媒を含有する反応混合物の色調が淡黄色(透明)から黒色へと変化し、次第に赤褐色の均一なコロイド状触媒へと変化し、高活性を発現するようになる。
【0035】
還元活性化処理後は、反応器を100〜250℃好ましくは150〜220℃より好ましくは180〜220℃に設定し、反応混合物にジメチルアミンを導入し、アミノ化反応を開始させる。反応温度が180℃より低くなるにつれ反応速度が低下する傾向がみられ、150℃より低くなるにつれこの傾向が顕著になり、100℃より低いと生産性に著しく欠けるため好ましくない。反応温度が220℃より高くなるにつれ副反応が加速される傾向がみられ、250℃より高くなると顕著になるため好ましくない。
【0036】
アミノ化反応は、−5〜10気圧(−506〜1013kPa)好ましくは常圧〜5気圧(101〜506kPa)より好ましくは常圧〜3気圧(101〜304kPa)の範囲で行うのが好適である。アミノ化反応は脱水反応であるため、加圧条件下では反応速度の低下を招くためである。特に、5気圧より高くなるとこの傾向が増大し、10気圧を超えると顕著になるため好ましくない。
なお、前述の米国特許第5840985号公報(特許文献2)では、実施例1に、副反応物としてのアルコール体の処理を目的として、アミド還元時に還元剤としての水素に加えジメチルアミンを供給し、200〜400psi(1379〜2758kPa)の加圧下で、副生アルコール体をアミド還元と同時にアミンに変換する例が記載されている。
しかしながら、アルコール体のアミノ化反応は、アルコール体の脱水素に始まり生成アルデヒドへのジメチルアミン付加、次いでジメチルアミン付加体の水素化分解反応によって進行するため、200〜400psi(1379〜2758kPa)の加圧下では、アルコールの脱水素に対し、さらに生成水の留出除去に対し不利であり、反応速度の低下を招く。
アルコール体のアミノ化反応の本質的反応機構に基づき、アミノ化反応を−5〜10気圧(−506〜1013kPa)好ましくは常圧〜5気圧(101〜506kPa)より好ましくは常圧〜3気圧(101〜304kPa)の範囲で行うことにより、副生アルコール体を対応する3級ジアミンに効率良く変換させることができる。
【0037】
アミノ化反応では、ジメチルアミンを導入すると数分間の誘導期の後、水が留出し始め反応の進行を確認できる。触媒の活性化後は、アミノ化反応は水素を導入しない条件下でも進行する。反応混合物の脱水素によって発生した活性化水素が反応に使われるからである。しかし、水素を導入して水素の存在下で反応を行うのが好ましい。反応時間を若干短縮できるとともに、導入された水素が水の系外への搬出を助けるからである。水を系外へ効率よく搬出させるため、反応器内に導入された水搬出用の水素,窒素や不活性ガス等は消費されないので、水素に代えて、又は水素に混合して、窒素や希ガス等の不活性ガスを反応器内に導入することもできる。
水の生成とともに生成物である油分も留出するので、常法によって油水分離し、必要に応じて油分を反応器に戻しアミノ化反応を進行させる。反応の進行は、アミン価、水酸基価、或いはガスクロマトグラフィー分析によって追跡することができる。
アミノ化反応が終了すると水の留出も停止する。反応温度、触媒の濃度、ジメチルアミンの供給速度にもよるが、アミノ化反応は2〜10時間で完了させることができる。
【0038】
触媒の濃度としては、金属銅を基準にして0.001〜10wt%(反応混合物中のアルコール体に対して)好ましくは0.01〜5wt%より好ましくは0.05〜2wt%が好適である。濃度が0.05wt%より低くなるにつれ反応速度が低下する傾向がみられ、0.001wt%より低くなると生産性に著しく欠けるため好ましくない。濃度が2wt%より高くなるにつれ副反応が促進される傾向がみられ、10wt%を超えると顕著になるため好ましくない。
【0039】
アミノ化反応において、反応器内に供給されるジメチルアミンの単位時間当たりの供給速度(L/時間)としては、標準状態で、反応混合物中のアルコール体の水酸基1モル当たり0.01〜100モル/時間、好ましくは0.1〜10モル/時間、特に好ましくは0.2〜5モル/時間がよい。0.01モル/時間未満では反応速度が遅く生産性に著しく欠けるため好ましくない。100モル/時間を越える場合は、ジメチルアミンによる触媒被毒が顕著になって反応速度と収率の低下をきたし、さらに不均化も大きく促進されるため好ましくない。
【0040】
本発明の請求項5に記載の発明は、請求項1乃至4の内いずれか1に記載のα,ω−3級ジアミノ化合物の製造方法であって、前記α,ω−ジカルボン酸のメチレン鎖の炭素数が、4〜10である構成を有している。
この構成により、請求項1乃至4の内いずれか1で得られる作用に加え、以下のような作用が得られる。
(1)得られるα,ω−3級ジアミンの沸点が高いことに由来して臭気が低く、眼粘膜刺激性も低く、それゆえ、作業環境の向上をもたらすとともに、ウレタンフォーム製造用触媒としての活性にも優れるため応用性に優れる。
【0041】
ここで、α,ω−ジカルボン酸のメチレン鎖の炭素数が4未満であると、得られるα,ω−3級ジアミンが低沸点のため臭気が激しく、炭素数が10を超えると、得られるα,ω−3級ジアミンのウレタンフォーム製造用触媒としての活性低下が大きくなるため、いずれも好ましくない。
特に、α、ω―ジカルボン酸のセバシン酸(メチレン鎖の炭素数8)は、その2官能性により広範囲の用途を有し、インドを最大の生産国とするヒマシ油からのオレオ・インダストリーとして今後、飛躍的な成長と発展が期待されるため好適である。
【発明の効果】
【0042】
以上のように、本発明のα,ω−3級ジアミノ化合物の製造方法によれば、以下のような有利な効果が得られる。
請求項1に記載の発明によれば、
(1)α,ω−ジカルボン酸を原料として、対応するα,ω−3級ジアミドを経済的に高収率で量産できるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0043】
請求項2に記載の発明によれば、請求項1の効果に加え、
(1)生産性に欠ける再結晶法とは異なり、3級ジアミドを蒸留精製するので生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
(2)アミド化反応に均一触媒を使用した場合でも、触媒を蒸留残渣に残存させて分離しα,ω−3級ジアミドを精製でき生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0044】
請求項3に記載の発明によれば、請求項1又は2の効果に加え、
(1)比較的穏和な条件でα,ω−3級ジアミドのカルボニル基が容易に還元されて、対応するα,ω−3級ジアミンを製造することができ生産性に優れたα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0045】
請求項4に記載の発明によれば、請求項3の効果に加え、
(1)反応混合物に含まれる副反応生成物のアルコール体とジメチルアミンとのアミノ化反応により、目的とする3級ジアミンを高い収率で製造できるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【0046】
請求項5に記載の発明によれば、請求項1乃至4の内いずれか1の効果に加え、
(1)得られるα,ω−3級ジアミンの沸点が高いことに由来して臭気が低く、眼粘膜刺激性も低く、それゆえ、作業環境の向上をもたらすとともに、ウレタンフォーム製造用触媒としての活性にも優れるため応用性に優れたα,ω−3級ジアミンが得られるα,ω−3級ジアミノ化合物の製造方法を提供できる。
【発明を実施するための最良の形態】
【0047】
以下、本発明を実施例により具体的に説明する。なお、本発明はこれらの実施例に限定されるものではない。
本実施例では、冷却器、撹拌機、温度指示制御器、原料ガス導入管(ガラス素焼き製のスパージャー)、サンプリング器を設けた500mlのセパラブル・フラスコ(反応器)に、α,ω−ジカルボン酸としてのセバシン酸(酸価556、小倉合成工業製)と、固体酸触媒(200〜300メッシュ通過品)又は均一触媒を所定量仕込み、所定の反応温度(180〜220℃)まで昇温し、ジメチルアミンと窒素との混合ガスを常圧でそれぞれ所定の流速で連続的に供給した。数分〜30分の誘導期間が発生するが、この間はセバシン酸とジメチルアミンとの間にアンモニウム塩が形成されている。
その後、アンモニウムの脱水反応により反応水の留出を伴って2段逐次のアミド化反応が進行し、α,ω−3級ジアミドとしてのN,N,N’,N’−テトラメチル1,10−デカナミドが生成する。セバシン酸の転化率は99.8%以上を達成することができる。
なお、生成物の分析にはガスクロマトグラフィー(島津製作所製、GC−8A)を用いた。測定条件は以下のとおりである。
カラム:DB−17、カラム温度:Inj.250℃、initial:100℃、final:290℃、 昇温速度:10℃/分、試料濃度:10%、試料注入量:0.2μL。
また、酸価及び水酸基価の測定は、JIS K0070−1992に準拠して行なった。
【0048】
(実施例1)
反応器に、セバシン酸を200g、ルイス酸触媒としての均一触媒であるチタニウム・テトラ・イソプロポキシド(Ti(O-i-Pr)4)を40mg(セバシン酸に対し0.02wt%)仕込んだ。反応系は均一な無色透明溶液を形成した。撹拌下、窒素を常圧で9L/時の流速で連続供給し、設定反応温度の180℃まで昇温した。その後、窒素の供給速度を維持したまま、ジメチルアミンを常圧で15L/時の流速で連続供給した。
約30分の誘導期間の後、反応水の留出を伴ってアミド化反応が定常的に進行した。セバシン酸とジメチルアミンとのアミド化反応は2段の逐次反応で進行した。反応9時間での酸価は21.0であり、セバシン酸転化率96.2%であった。
得られた反応混合物を減圧蒸留した結果、留分の留出温度は0.4KPaで201〜215℃であった。このとき、蒸留缶の温度は215〜223℃、N,N,N’,N’−テトラメチル−1,10−デカナミドの蒸留収率は97.2%であった。
従来、N,N,N’,N’−テトラメチル−1,10−デカナミドのようなα、ω−3級ジアミドの蒸留精製の事例はないが、蒸留精製できることを今回初めて確認した。
【0049】
(実施例2)
酸触媒として、日揮化学製の脱水反応用シリカ・アルミナ固体触媒(商品名:N633HN、粒子径250メッシュ通過、表面積380m2/g、嵩比重0.4)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった。
反応15時間での反応混合物の酸価は16.2であり、セバシン酸転化率は97.1%であった。
【0050】
(実施例3)
酸触媒として、日揮化学製の脱水反応用シリカ・アルミナ固体触媒(商品名:N633L)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった(反応温度180℃)。
反応18時間で酸価は1.0であり、セバシン酸転化率は99.8%であった。得られた反応混合物を実施例1と同様に蒸留精製した結果、酸価は0.7(純度99.9%)にまで向上した。
なお、反応温度を220℃にした場合も、反応混合物の酸価は1.0であり、セバシン酸転化率99.8%に相当した。
図1は、反応混合物(反応温度180℃)を蒸留精製した精製物のガスクロマトグラフィーのチャートである。ピーク面積から換算すると、N,N,N’,N’−テトラメチル−1,10−デカナミドの純度は99.9%以上であることが確認された。
【0051】
(実施例4)
酸触媒として、日揮化学製の脱水反応用アルミナ固体触媒(商品名:N613N、嵩比重0.5)をセバシン酸に対し2wt%仕込んだ以外は、実施例1と同様にしてアミド化反応を行なった。
反応18時間で酸価は9.0であり、セバシン酸転化率は98.5%であった。
【0052】
(実施例5)
反応温度が220℃、ジメチルアミン供給速度が35L/時である以外は、実施例4と同様にしてアミド化反応を行なった。
反応7.5時間で酸価は4.9であり、セバシン酸転化率は99.1%であった。
【0053】
(比較例1)
(特許文献2)に記載された「塩基触媒としてのナトリウム・メトキシド(NaOCH3)の存在下のラウリン酸メチルエステルとジメチルアミンとのアミド化反応(反応温度80℃)」に準じて、セバシン酸ジメチルエステルとジメチルアミンとを出発原料とするアミド化反応を行った。
反応器にセバシン酸ジメチルエステルを250g、塩基触媒としてのナトリウム・メトキシド3.0gを10.3gのメチルアルコールに分散した溶液を仕込み、撹拌下、設定反応温度である75℃に昇温した。
ここで、設定反応温度を文献に記載された80℃より低めに設定した理由は、触媒としてのナトリウム・メトキシドがメチルアルコールに分散していることから、メチルアルコールの蒸発によりナトリウム・メトキシドが凝集し触媒活性が激減する可能性があるからである。
メチルアルコールの還流下に7時間反応を行なった結果、セバシン酸とジメチルアミンとの第4級アンモニウム塩は生成するものの、4級塩からの脱水反応がほとんど進行しないことが判明し、(特許文献2)に記載された方法ではセバシン酸からの3級ジアミドを製造し得ないことが判明した。
一般にα、ω−型の2官能性化合物を合成する場合、逐次反応系の2段目の反応が進行し難いという一般的傾向がある。(特許文献2)の出発原料は一価のカルボン酸であり、本発明の出発原料であるジカルボン酸とは構造が全く異なるため、(特許文献2)に記載された方法では、4級アンモニウム塩の脱水反応を進行させることができず、3級ジアミドを製造できないことが判明した。
【0054】
(比較例2)
反応性が大きなα,ω−ジカルボン酸ジクロリド(ここではセバシン酸のジ酸クロリド)を石油エーテルに溶解させ、常温でジメチルアミンをバブリング導入してアミド化反応を2時間行なった。その結果、腐食性の酸クロリドが定量的に反応せず、未反応の酸クロリドの除去と粗3級ジアミドの精製に多大な負荷がかかることが判明した。これにより、α,ω−ジカルボン酸ジクロリドを出発原料として、工業的にα,ω−3級ジアミドを製造できないことが判明した。
【0055】
(比較例3)
触媒として、固体酸ではないシリカゲルを用いた以外は、実施例2と同様にしてα,ω−3級ジアミドの製造を試みた。しかし、安定な4級塩が主に生成するのみであった。4級塩を主成分とする反応混合物は水溶性が強く、220〜250℃でも固体であり均一溶液を形成することができず、4級塩からの脱水反応を観測することができなかった。
【0056】
以上の実施例によれば、酸触媒の存在下でセバシン酸から、対応するα,ω−3級ジアミドとしてのN,N,N’,N’−テトラメチル1,10−デカナミドを容易に高い転化率で製造できることが明らかになった。また、蒸留精製によって高純度のα,ω−3級ジアミドが得られることも初めて確認することができた。
【0057】
(実施例6)
実施例6では、N,N,N’,N’−テトラメチル−1,10−デカナミドの接触水素還元を行った。反応器としては、撹拌機、温度指示制御器、水素供給ライン及び排ガスラインを設けた0.3Lのオートクレーブを用いた。
この反応器に、実施例3で得られた3級ジアミドであるN,N,N’,N’−テトラメチル−1,10−デカナミド(酸価1.0)の蒸留生成物(酸価0.7、純度99.9%)を87g、銅/亜鉛/アルミナ触媒(日揮化学製、CuO:ZnO:Al2O3=43:44:5〜6wt%、Cu:Zn=1:1)4.35g(3級ジアミドに対し5wt%)を仕込んだ。
水素供給ラインから反応器内に水素を連続的に供給するとともに(約30L/時、水素圧0.5MPa)、排ガスラインから触媒の還元によって生成した還元水及び排ガスを連続的に排出しながら昇温し、触媒の還元活性化を行なった。本実施例で用いる銅/亜鉛/アルミナ触媒は、約135℃から水素還元が進行し、約160℃で還元が完了することを事前に確認している。反応器に水素を連続的に供給し排ガス及び還元水を連続的に排出する水素流通系を適用した理由は、還元水による触媒の被毒を最大限回避するためである。さらに水素流通下に昇温を続け、設定反応温度である230℃に達してから水素圧を2.5MPaに上げ、この水素圧を維持しながら、3級ジアミドの還元によって生成した反応水を含む排ガスを連続的に排出して3級ジアミドの水素還元を行なった。反応中、排ガスラインが反応混合物で閉塞傾向になるので、排ガス及び反応水の排出を妨げないように排ガスラインを十分に保温した。
反応4時間目の3級ジアミドの転化率は97%、反応6時間目の3級ジアミドの転化率は99.9%であった。反応6時間目の反応混合物の分析結果を(表1)の「3級ジアミド還元」と表記した欄に示した。
【0058】
【表1】
【0059】
(表1)に示すように、目的とするN,N,N’,N’−テトラメチル−1,10−デカンジアミンMe2N−(CH2)10−NMe2(3級ジアミン)は反応混合物に対して56.8%生成しており、Me2N−(CH2)10−OH等のアルコール体(水酸基含有物)が副反応生成物として約30%生成していることがわかった。
【0060】
次に、反応混合物をオートクレーブから取り出した後、生成した水を分離するための凝縮器及び分離器、反応混合物サンプリング器、排ガス出口管、原料ガス導入管(多孔質ガラス製のスパージャー)、撹拌器、温度計を設けたフラスコ(反応器)に87g仕込んだ。次に、触媒原料としてステアリン酸銅1.74g(アルコール体に対する金属銅0.1wt%)、ステアリン酸ニッケル0.35g(アルコール体に対する金属ニッケル0.02wt%)、ステアリン酸カルシウム0.35g(アルコール体に対する金属カルシウム0.02wt%)、ステアリン酸バリウム0.35g(アルコール体に対する金属バリウム0.02wt%)を加えた後、撹拌器を回転させ反応器内を窒素で置換し昇温した。触媒原料の4種類の金属石鹸は、100℃に達するまでに均一に溶解した。100℃に達したら窒素を水素に切換え、水素ガスを、流量計を通じて22L/時間の流速でフラスコ内にバブリングさせて還元活性化処理を行った。170〜190℃で2価の銅とニッケルの特徴的な緑色が次第に淡色化し、触媒原料が還元され見掛け上均一なコロイド状触媒となった。反応温度を210℃に保ち、ジメチルアミンを常圧で20〜30L/時間の流速で水素(流速22L/時間)との混合ガスとして連続供給し反応させた。
反応2時間目と反応6時間目の反応混合物の分析結果を(表1)の「アルコール アミノ化」と表記した欄に示した。
【0061】
反応2時間目では、Me2N−(CH2)10−OH等のアルコール体(水酸基含有物)はアミノ化反応によって約6%に減少し、目的とするMe2N−(CH2)10−NMe2(3級ジアミン)は反応混合物に対して73.6%生成していることがわかった。反応を続けたところ、反応6時間目で3級ジアミンの収率は76.1%に達していた。
なお、本実施例においては、反応器に水素を連続的に供給し排ガス及び還元水を連続的に排出する水素流通系を適用したため、3級ジアミドを接触水素還元したとき、及び、副生アルコール体をアミノ化したときに、目的の3級ジアミン含量の高い低沸点の油分が一部留出していた。これは、留出油分の成分分析によって確認することができた。
従って、この留出油分を連続的に反応器に戻すならば、3級ジアミンの収率は90%程度になることがわかった。
以上のように本実施例によれば、反応混合物に含まれる副反応生成物のアルコール体とジメチルアミンとのアミノ化反応により、目的とする3級ジアミンを高収率で製造できることが明らかになった。
【0062】
以上の実施例においては、α,ω−ジカルボン酸としてのセバシン酸を用いた場合について説明したが、一般式HOOC−(CH2)n−COOH(但し、nは自然数。)で示される他のα,ω−ジカルボン酸からも、同様に対応するα,ω−3級ジアミド及びα,ω−3級ジアミンを製造できることを確認した。
【産業上の利用可能性】
【0063】
本発明は、電解コンデンサの電解質等として可能性のあるα,ω−3級ジアミドや、ウレタンフォームの製造用触媒等として有用なα,ω−3級ジアミンを経済的に高収率で量産できるα,ω−3級ジアミノ化合物の製造方法を提供できる。本発明は、椰子、パームに次ぐ第3のオレオ・インダストリー構築のための技術要素を形成する必要不可欠な技術である。
【図面の簡単な説明】
【0064】
【図1】反応混合物を蒸留精製した精製物のガスクロマトグラフィーのチャート
【特許請求の範囲】
【請求項1】
酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得ることを特徴とするα,ω−3級ジアミノ化合物の製造方法。
【請求項2】
前記3級ジアミドを蒸留精製することを特徴とする請求項1に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項3】
前記3級ジアミドを接触水素還元して対応する3級ジアミンを得ることを特徴とする請求項1又は2に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項4】
前記3級ジアミドを接触水素還元して生成した反応混合物とジメチルアミンとを、触媒の存在下、反応させることを特徴とする請求項3に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項5】
前記α,ω−ジカルボン酸のメチレン鎖の炭素数が、4〜10であることを特徴とする請求項1乃至4の内いずれか1に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項1】
酸触媒の存在下、α,ω−ジカルボン酸とジメチルアミンとを反応させて3級ジアミドを得ることを特徴とするα,ω−3級ジアミノ化合物の製造方法。
【請求項2】
前記3級ジアミドを蒸留精製することを特徴とする請求項1に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項3】
前記3級ジアミドを接触水素還元して対応する3級ジアミンを得ることを特徴とする請求項1又は2に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項4】
前記3級ジアミドを接触水素還元して生成した反応混合物とジメチルアミンとを、触媒の存在下、反応させることを特徴とする請求項3に記載のα,ω−3級ジアミノ化合物の製造方法。
【請求項5】
前記α,ω−ジカルボン酸のメチレン鎖の炭素数が、4〜10であることを特徴とする請求項1乃至4の内いずれか1に記載のα,ω−3級ジアミノ化合物の製造方法。
【図1】

【公開番号】特開2008−169153(P2008−169153A)
【公開日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願番号】特願2007−4481(P2007−4481)
【出願日】平成19年1月12日(2007.1.12)
【出願人】(592135041)小倉合成工業株式会社 (4)
【Fターム(参考)】
【公開日】平成20年7月24日(2008.7.24)
【国際特許分類】
【出願日】平成19年1月12日(2007.1.12)
【出願人】(592135041)小倉合成工業株式会社 (4)
【Fターム(参考)】
[ Back to top ]