
β−アミロイド斑に作用物質を結合させる方法
β-アミロイド斑や神経原繊維変化などの構造を、in vivo 又はin vitroで、ラベルする方法が提供され、この方法は、脳組織を一つ以上の化合物、好ましくは陽電子断層撮影法(PET)による検出のために放射性ラベルされた化合物、と接触するステップを含む。

【発明の詳細な説明】
【技術分野】
【0001】
政府補助に対する謝辞
本発明は、米国エネルギー省による補助金No.DE-FC0387-ER60615の下で、政府の補助によってなされた。政府はこの発明に一定の権利を有する。
【0002】
関連出願への相互参照
本出願は、2003年5月20日に出願された米国特許出願No. 60/471,945の優先権を主張するものであり、その出願の内容全体は(付録AとBを含めて)、全体が記載されたと同様に参照によって本明細書に組み込まれる。
【背景技術】
【0003】
発明の背景
アルツハイマー病は、米国やその他の工業国において最も急速に増加している年齢層である80歳以上の人口の約20〜40%の人たちがかかる病気である。アルツハイマー病患者の脳によく見られる特徴として、多量のニューロン内神経原繊維変化(NFT)と細胞外のアミロイドに富むβ-アミロイド斑などがある。NFTsは、主として過剰にリン酸化されたタウ蛋白質が対らせんフィラメントと呼ばれる周期的に制限されたアミロイド繊維の形に組み合わされた凝集体から構成される。アミロイド斑の主要な成分はペプチドであり、これはもっと大きなアミロイド前駆蛋白質の開裂によって生成される長さが39−43アミノ酸の小さなβ-アミロイドペプチドである。しかし、ほとんど全くB-アミロイドから成るぼやけた(diffuse)斑を除き、アミロイド斑は多数の細胞生成物を含む複雑な病変である。42アミノ酸形態のこのペプチドの産生を増やす突然変異はアルツハイマー病の常染色体優性家族形態と遺伝的にリンクしている。β-アミロイドの沈着は疾病過程の非常に早い時期に、臨床的な症状が現れるよりもずっと前に起こっている。この突然変異は病原性があり遺伝子導入マウスにアルツハイマー病を生ずるように見えるので、β-アミロイドはこの病気において原因としての役割を果たしていると広く信じられている。アミロイドの沈着が原因であるかどうかに関わりなく、それが診断の重要な部分であることは確かである。さらに、アミロイド斑は疾病の初期に見られるので、沈着を画像化できる能力は、早期の診断と疾病の予防のための好適なマーカー、並びに治療方式の有効性をモニターする方法を提供する。
【0004】
アルツハイマー病は、現在、死後の脳から切片を採取して新皮質のアミロイド沈着を定量化することによって確定的に診断される。残念ながら、アミロイド沈着及び/又はNFTsを検出する現行の方法は、死後の分析又は生検分析を必要とする。例えば、in vitroの脳切片におけるアミロイドのチオフラビン蛍光標識法は脳の評価のために現在広く用いられている方法である。別の可能なアミロイド・プローブとして、Chrysamine-G、コンゴーレッド誘導体、も開発されている。コンゴーレッドは荷電分子であり、したがって、血液−脳関門を越えて拡散するのに十分な疎水性を欠いており、in vivoラベルとして用いることができない。K;unk et al, Neurobiology of Aging, 16: 541-548 (1995), 及びPCT Publication No. WO 96/34853,を見よ。Chrysamine-Gは、コンゴーレッドよりも良く血液−脳関門を通過するが、アルツハイマー病の脳のアミロイド斑をラベルする能力は弱いように見える。例えば、H. Han, C-G Cho and P. T. Lansbury, Jr., J. Am. Chem. Soc. 118, 4506 (1996); N. A. Dezutter et al., J. Label. Compd. Radiopharm, 42, 309 (1999), を参照のこと。同様に、β-アミロイドのin vivo画像化のためのプローブとしてモノクローナル抗体を用いる初期の試みは、それが血液−脳関門を通過する能力が限られていることによって妨げられた。R. E. Majocha et al., J. Nucl. Med. 33, 2184 (1992) を見よ。最近では、血液−脳関門を通過できる1251-Aβ 1-41のモノビオチン化複合体の利用も提案されているが(Y. Saito et al., Proc. Natl. Acad. Sci. USA 22, 2288 (1991)を見よ)、それがin vivoでβ-アミロイド及び/又はNFTsをラベルする能力はまだ実証されていない。アミロイドの沈着をin vivoで定量化することは現在利用できるプローブによっては不可能である。したがって、アルツハイマー病の早期診断のための好適なマーカーに対するニーズが存在している。
【0005】
陽電子放出型断層撮影(PET)による局所脳グルコース代謝率(rCMRGI)のin vivoの、非侵襲的測定は、アルツハイマー病患者における脳機能の評価のための重要な手段になっている。2-[F-18]-2-デオキシ-D-グルコース(DFG)を用いた多くの研究は、側頭頭頂及び前頭連合野における代謝低下の特徴的な代謝パタンをはっきりと示している。これらの研究のうちのいくつかは、rCMRGIと死後の局所ニューロン病理とを比較した。それらの結果、及びアルツハイマー病の病原カスケードの不確かさは、これらの患者におけるアミロイドと神経原繊維の沈着をin vivoで非侵襲的に評価することの重要性を浮き彫りにしている。
【発明を実施するための最良の形態】
【0006】
発明の詳細な説明
ある実施形態では、本発明はβ-アミロイド斑と神経原繊維変化から成る群から選択される構造を、in vivo 又はin vitroでラベルする方法であって、脳組織と化学式(IA)又は(IB):
【化1】
(式中;
Qは、-NR2R3, -OR9, 及びSR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N及びCR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9, 及び-C(R9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及び-SR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、Nと-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化2】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル- R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、-C(O)O-アルキル、-C(O)O-アルケニル、-C(O)O-アルキニル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング、-OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル、-O-アルケニル、-O-アルキニル、-S-アルキル、-S-アルケニル、及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;
R9は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成する;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではない)
の化合物を接触させるステップを含む方法に向けられる。上記の化合物において、1又は複数の水素、ハロゲン、又は炭素原子を任意に放射性ラベルで置き換えることができる。
【0007】
本発明は、また、上記の方法で有用な、化学式(IA)又は(IB)の化合物を含む組成物:β-アミロイド斑と神経原繊維変化から成る群から選択される構造を、in vivo 又はin vitroでラベルする方法であって、脳組織と化学式(IA)又は(IB):
【化3】
(式中;
Qは、-NR2R3, -OR9, 及び-SR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N及びCR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9及び-C(CR9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及びSR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N及び-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化4】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル- R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング, -C(O)O-アルキル, -C(O)O-アルケニル, -C(O)O-アルキニル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素、アルキル、アルケニル, アルキニル, 環式リング, 複素環式リング, -OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル, -O-アルケニル, -O-アルキニル, -S-アルキル, -S-アルケニル, 及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R9は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素, アルキル, アルケニル, アルキニル, 環式リング、複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成する;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではなく;
さらに、一つ以上の水素原子が任意に放射性ラベルで置き換えられる)
の化合物を接触させるステップを含む方法に向けられる。
【0008】
諸定義:
本明細書で用いられる場合、“アルキル”という用語は、飽和炭素原子と水素原子の直鎖又は枝分かれ鎖の一価ラジカル、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、t-ブチル、ペンチル、及びヘキシル、などを指し、置換されていても置換されていなくてもよい。“低級アルキル”という用語は、1〜4個の飽和炭素原子と水素原子を有する直鎖又は枝分かれ鎖の一価ラジカル、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、及びt-ブチル、などを指し、置換されていても置換されていなくてもよい。
【0009】
本明細書で用いられる場合、“アルキレニル”という用語は、少なくとも一つの-(CH2)-を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、エチレニル及びプロピレニルなど、を指し、置換されていても置換されていなくてもよい。
【0010】
本明細書で用いられる場合、“アルケニル”という用語は、少なくとも一つの炭素−炭素二重結合を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、ブテニル及びペンテニルなど、を指し、置換されていても置換されていなくてもよい。
【0011】
本明細書で用いられる場合、“アルキニル”という用語は、少なくとも一つの炭素−炭素三重結合を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、エチニル、プロピニル、ブチニル及びペンチニルなど、を指し、置換されていても置換されていなくてもよい。
【0012】
本明細書で用いられる場合、“環式リング(cyclic ring)”という用語は、それぞれが飽和又は不飽和の3, 4, 5, 6, 7, 8, 9, 10, 11, 又は12個のリング原子を含む単環式又は多環式ラジカル、例えばシクロブチル、シクロペンチル、シクロヘキシル、シクロヘキセニル、及びフェニルなど、を指し、置換されていても置換されていなくてもよい。
【0013】
本明細書で用いられる場合、“複素環式リング(heterocyclic ring)”という用語は、窒素、酸素、及びイオウから選択される1, 2, 3, 4, 又は5個のヘテロ原子を含む、それぞれが飽和又は不飽和の3, 4, 5, 6, 7, 8, 9, 10, 11, 又は12個のリング原子を含む単環式又は多環式ラジカルを指し、置換されていても置換されていなくてもよい。非限定的な例は、アジリジン、アゼチジン、ピロリジン、ピペリジン(piperidine)、及びピペリジン(piperizine)などである。
【0014】
上で注意したように、“アルキル、”“アルケニル、”“アルキニル、”“環式リング、”“複素環式リング”という用語は、置換されたアルキル、アルケニル、アルキニル、環式リング、及び複素環式リング・グループを含み、それらのグループは適当な置換基、例えば、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、ハロゲン、-OH, -OTs, O-アルキル、O-アルケニル、O-アルキニル、O-環式リング、O-複素環式リング、アシル、チオアシル、スルフォニル、メルカプト、アルキルチオ、アミノ、アルキルアミノ、ジアルキルアミノ、及びカルボモイル基、などによって置換できる。
【0015】
化学式(I)及び化学式(II)の化合物では、好ましくはR2とR3はそれぞれ独立にアルキルであり、さらに好ましくは低級アルキルである。化学式(II)の化合物では、好ましくはR9は低級アルキルであり、さらに好ましくはメチル又はエチル、アリール、及び置換されたアリールである。本発明に関連して用いるのに特に好ましい化合物は、2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)と2-(1,1-ジシアノプロペン-2-イル)-6-(エチル)(メチル)(アミノ)ナフタレンであり、どちらも任意に放射性ラベル標識できる。別の好ましい化合物、特にin vivoで用いるのに好ましい化合物は、2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)である。
【0016】
本発明は、また、in vitro又はin vivoでβ-アミロイド斑や神経原繊維変化などの構造を検出する方法に向けられる。“構造”という用語は、疾病病理の一部として生ずるペプチドやその他の細胞物質を含む生物物質の凝集体を指す。“ペプチド”という用語は蛋白質を含む。
【0017】
上述の化合物は約470から610 nmまでの範囲で蛍光活性を有する。ある応用では、本発明は脳組織におけるβ-アミロイド斑や神経原繊維変化をラベル標識する。すなわち、アルツハイマー病のin vitro検出のために、この化合物を脳組織に接触させ、脳組織が蛍光顕微鏡で観察される。
【0018】
in vivo検出のためには、化合物を放射性ラベル標識することが好ましい。好ましい放射性ラベルは18Fである。これは陽電子断層撮影法のための半減期が約2時間である。別の放射性ラベルは放射性ヨウ素、例えば123Iであり、シングルフォトン断層撮影法(SPECT)で用いられる。あるいはまた、他の放射性ラベル、例えば11C, 13N, 及び15O, が用いられるが、これらの放射性ラベルは比較的寿命が短いのであまり望ましくない。化合物のどの原子も適当な放射性ラベルで置き換えることができる。放射性ラベル標識は、当業者に公知のどんな方法で行ってもよい。例えば、K2CO3 (0.75 mg)中のドライ[F-18]フッ化物イオン[18O (p, n) 18F]とKryptofix 2.2.2TM (19 mg)が化学式(I)又は化学式(II)の化合物の溶液(1 mL CH3CN中4 mg)に加えられる。混合物はオイル浴で85℃に約10〜40分熱せられる。冷却して水で希釈した後、放射性ラベルされた生成物は分取HPLCによって精製される。Kryptofix 2.2.2TM はAldrich Chemical Co. (Milwauky, Wisconsin)から入手できるクラウンエーテルである。
【0019】
次に、放射性ラベルされた化合物を含む溶液が患者に注射される。本明細書で用いられる場合、“患者”という用語は、ヒト、ラット、マウス、イヌ、及びネコを含む哺乳類を指す。神経解剖的領域は、MRIスキャンを用いて手作業で、例えば、Tela磁石を用いて、次に、アミロイドPET(陽電子断層撮影)とEDG-PET(フルオロデオキシグルコース-PET)でMRIスキャンとの重ね合わせによって、決定できる。PETは現在2〜3分という分解能で脳における放射性ラベルされた化合物の沈着を動的に決定することができ、異常な区域を検出することができる。
【0020】
上述の方法によって、β-アミロイド斑や神経原繊維変化の蓄積によって特徴づけられるアルツハイマー病やその他の脳の劣化に関連した疾病を検出することができる。
【0021】
本発明は、また、患者におけるアルツハイマー病を治療又は予防する治療物質の能力を決定する方法に向けられる。“アルツハイマー病を予防する”というフレーズは、アルツハイマー病のリスクを軽減すること及び/又は発病を送らせることを含む。この方法は、β-アミロイドペプチドをその治療物質及び化学式(IA)又は(IB):
【化5】
(式中:
X1は、O, S, -NR4, -CH2, -CHアルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N-(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化6】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、アリール, 複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2, 及び-アルキレニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になってアリール又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になってアリール又は複素環式リングをA1によってR1, X3又はZ4以外の炭素のところで置換されたものを形成し;
R4は、アルキル, 置換されたアルキル, アリール, 置換されたアリール, -C(O)O-アルキル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル(O-アルキル)2, -O-アルケニル, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルケニル, 及び-S-アルキレニル- R4から成る群から選択されるラジカルであり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;任意に、アルキル, アルコキシ、OH, OTs, ハロゲン、アルキレニル-R5, カルボニル、スピペロン、スピペロンケタール、及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換され;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成する;
ここで、1又は複数の水素、ハロゲン、又は炭素原子が放射性ラベルによって置き換えられる)
による放射性ラベルされた化合物と、in vivo又はin vitroで接触させるステップを含む。
【0022】
この方法によれば、放射性ラベルされた化合物の少なくとも一部はβ-アミロイドペプチドと結合する。この方法に関連して、β-アミロイドペプチドという用語は、β-アミロイド凝集体又は繊維、並びにβ-アミロイド老人斑を含む。治療物質と放射性ラベルされた化合物は、両方を同時にβ-アミロイドペプチドと接触させることも、放射性ラベルされた化合物をβ-アミロイドペプチドと接触させる前及び/又は後に治療物質をβ-アミロイドペプチドと接触させることもできる。
【0023】
次に、β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を測定して、それにより放射能をもたない作用物質がβ-アミロイドペプチドと結合したかどうか、どの程度結合したかを決定する。ある好ましい実施形態では、放射性ラベルされた化合物だけを(すなわち、放射能のない作用物質が存在しない状態で)β-アミロイドペプチドと接触させて、100 %特異的結合を決定する。
【0024】
この方法では、治療物質の濃度を変えて、どの程度治療物質がβ-アミロイドペプチドと結合できるかを決定することができる。
【0025】
本発明の別の方法では、治療物質の抗凝集効果を評価することができる。抗凝集効果とは、治療物質がβ-アミロイドペプチドを破壊及び/又はその形成を妨害する能力を指す。この方法では、患者におけるβ-アミロイドペプチドの量が、上で一般的に述べたように放射性ラベルされた化合物を用いて決定される。その後で、治療物質がある期間、例えば1週間、1ヶ月、又はそれ以上、にわたって患者に投与され、治療物質がβ-アミロイドペプチドと接触するようにする。その期間の後、放射性ラベルされた化合物を再びβ-アミロイドペプチドと接触させて患者におけるβ-アミロイドペプチドの量を決定することができる。治療物質がβ-アミロイドペプチドの形成をどの程度妨害できるかを決定するためには、アルツハイマー病にかかりそうな、しかしこの病気があまり進行していない一人以上の患者を評価することが有効である。
【0026】
本発明は、また、患者におけるアルツハイマー病を治療又は予防する方法に向けられる。この方法は、治療的に有効な量の、上述の化学式(IA)又は(IB)による作用物質を患者に投与するステップを含む。
【実施例】
【0027】
実施例
実施例1
以下の本発明による組成物が調製された。NMRスペクトルはBruker AM 360 WB又はDPX 300スペクトロメーターで得られた。1H化学シフトは内部標準としてTMSからのppmダウンフィールド(downfield)で報告される。19F化学シフトは外部のフルオロトリクロロメタンに対して報告される。別に断らない限り、ジューテリオクロロフォルムが溶媒として用いられた。融点はElectrothermal Melting Point Apparatusで測定され、補正されなかった。元素分析は、Galbraith Laboratories, Inc., Knoxville, TX,又はMs. Metka Kastelic at the Faculty of Chemistry and Chemical Technology, University of Ljublijana, によって行われた。放射状クロマトグラフィーは、Chromatron (Harrison Research, 840 Moana Court, Palo Alto, CA 94306)で行われた。ローターはHarrison Researchが勧告したようにE. Merck Silica Gel (Cat. No. 7749-3)を用いて作成された。HPLCはAlltech Econosil C-18 5μm, 4.6x250mmカラムで、溶媒として水:アセトニトリル:トリエチルアミンの40:60:2混合物を用いて行われた。254nmにおけるUV検出が用いられた。溶媒と試薬はFisher, Aldrich又はFlukaから入手し、別に断らない限り入手した状態で用いた。
【0028】
実施例1(a)−2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)の調製
【化7】
29 mLの新しく蒸留されたヘキサメチルリン酸ヨリアミド(HMPT)中の5.26 g (117 mmol)のジメチルアミンの溶液に、31 mLの無水トルエンと780 mg (117 mmol) の小さな片のLiが加えられた。混合物は室温でアルゴンの下で1.5時間攪拌された。2-アセチル-6-メトキシナフタレンがArsenijevic et al., Org. Synth. Coll. 1988, 6:34-36に記述されたように調製された。この文献の開示内容は参照によって本明細書に組み込まれる。2-アセチル-6-メトキシナフタレン(5.57 g, 27.8 mmol) が一度に加えられ、攪拌が20時間続けられた。混合物は氷水浴で冷却されて冷たい水/酢酸エチル混合物(各300 mL)に注がれた。十分な攪拌の後、層が分離し、水層は225 mLの酢酸エチルで2回抽出された。有機抽出物を一緒にし、乾燥、蒸発させて黄色の固体が得られた。エタノールからの再結晶化によって2-アセチル-6-(ジメチルアミノ)ナフタレン(ADMAN)が黄色固体として得られ、153.5-155℃で融解した:1H NMR (CDCl3, TMS) δ2.67 (s, 3H, COCH3), 3.15 (s, 6H, N(CH3)2), 6.87 (d, 1H, H-5), 7.17 (dd, 1H, H-7), 7.63 (d, 1H, H-4), 7.80 (d, 1H, H-8), 7.92 (dd, 1H, H-3), 8.32 (bs, 1H, H-1), J1,3=2.3 Hz, J3,4=8.7 Hz, J5,7=2.4 Hz, J7,8=9.3 Hz. MS (M+) 213: 実測:213. 分析. C14H15NOで計算:C, 78.84; H, 7.09; N, 6.57. 実測:C, 78.96; H, 7.10; N, 6.45.
【0029】
20 mLのピリジン中でマロニトリル(436 mg, 6.6 mmol)とADMAN (1.278 g, 6.6 mmol)の混合物が110℃で19時間熱せられた。冷却後、残った赤色固体が100 mLの塩化メチレンに溶解され、10 gのフラッシュ・シリカget (230〜400メッシュ)に吸着され、トルエンでクロマトグラフィー分析された。適当な分画を一緒にして、蒸発させて1.12 g (72%) の2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)が得られた。ベンゼン−ヘキサンからの再結晶化によって赤色針状体が得られ、154.5〜155℃で融解した:1H NMR (CDCl3, TMS) δ2.69 (s, 3H, CH3), 3.11 (s, 6H, N(CH3)2), 6.85 (d, 1H, H-5), 7.18 (dd, 1H, H-7), 7.56 (dd, 1H, H-3), 7.66 (d, 1H, H-4), 7.76 (dd, 1H, H-8), 8.02 (d, 1H, H-1). J1,3=2.04 Hz, J3,4=9.13 Hz, J5,7=2.5 Hz, J7,8=9.11 Hz. IR (CHCl3) 2250 cm-1 (CN伸張). MS (M+) 261: 実測:262. 分析値. C17H15N3で計算:C, 78.13; H, 5.79; N, 18.08. 実測:C, 78.17; H, 5.68; N, 17.91.
【0030】
実施例1(b)−2-(1-{6[エチル-(2-{8-[4-(4-フルオロフェニル)-4-オキソブチル]-4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4.5]デシ-3-イル}エチル)-アミノ]-2-ナフチル}エチリデン)マロノニトリルの調製
【化8】
還流冷却器と滴下漏斗を備えた3Lの2首丸底フラスコで、2Lの塩化水素酸(d=1.16)を攪拌し、沸騰まで加熱した。最小量のジクロロメタン中の6.06 g (30.3 mmol)の1-(6-メトキシ-2-ナフチル)-1-エタノン(Arsenijevic et al., Org. Synth. Coll. 6:34 (1988)に記述されたように調製、この開示内容は参照によって本明細書に組み込まれる)の溶液が加えられ、混合物は攪拌され還流で2時間加熱された。熱い溶液がミネラルウール栓を通して濾過されて油性の残渣が除かれた。冷却後に分離した固体がガラス・フリットで濾過され、130 mLの酢酸エチルに溶解された。溶液を塩水で洗浄し、無水硫酸マグネシウムで乾燥し、蒸発させて5 g(89%)の1-(6-ヒドロキシ-2-ナフチル)-1-エタノンが得られた。
【0031】
1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(744 mg, 3.92 mmol)、硫酸水素ナトリウム(IV)(1.66 g, 16 mmol)、2-エチルアミノエタノール(2 mL)、及び水(5 mL)の混合物がスチールボンベ中で130〜140℃で3日間加熱された。冷却後、混合物を水と酢酸エチルの間で分配し、有機層を塩水で洗浄し、乾燥し、蒸発させた。残渣はアセトンに溶解され、4 mmのドライシリカプレートに装荷され、放射状クロマトグラフィーにかけられた。プレートは石油エーテルと酢酸エチルの1:1混合物で溶出された。適当な分画を集めて、蒸発させて125 mg (12%) の1-{6-[エチル-(2-ヒドロキシエチル)-アミノ]-2-ナフチル}エタノンが得られた。
【0032】
ピリジン(3.5 mL)中の1-{6-[エチル-(2-ヒドロキシエチル)-アミノ]-2-ナフチル}エタノン(125 mg, 0.486 mmol) の溶液が-15℃に冷却され、p-トルエン無水スルフォン酸(252 mg, 0.81 mmol)がアルゴン下で攪拌しながら加えられた。反応混合物は放置されてゆっくりと室温まで暖められ、攪拌が24時間続けられた。TLC (シリカ、石油エーテル中10%酢酸エチル) によって出発物質がまだ存在することが明らかになったので、さらにp-トルエン無水スルフォン酸(252 mg, 0.81 mmol)が加えられ、攪拌がさらに24時間続けられた。次に、混合物は氷水浴中で冷却され、塩水とエーテルに分配された。有機層を乾燥、蒸発させ、オイル状の残渣が残された。生成物、6-アセチル-2-(エチル−2[(4-メチルフェニル)-スルフォニルオキシ]-エチルアミノ)-ナフタレン、は放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン)で単離され、収率は30%だった。HRMS C23H25NO4S計算値: 411.1504. 実測値:411.1514. 1H NMR δ1.25 (t, 3H, CH2CH3), 2.33 (s, 3H, PhCH3), 2.67 (s, 3H, COCH3), 3.49 (q, 2H, CH2CH3), 3.75 (t, 2H, NCH2CH2O), 4.25 (t, 2H, NCH2CH2O), 6.97 (d, 1H, 5-H), 7.01 (dd, 1H, 7-H), 7.18 and 7.20 (d, 2H, 3’-H, 5”-H), 7.56 (d, 1H, 4-H), 7.69 and 7.72 (d, 2H, 2’-H, 6’-H), 7.75 (d, 1H, 8-H). 7.93 (dd, 1H, 3-H), 8.29 (d, 1H, 1-H). J1,3=1.6 Hz, J2’,6=J3’,5=8.5 Hz, J7,5=2.5 Hz, J7,8=9.2 Hz, J3,4=8.7 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.2 Hz.
【0033】
水(2 mL)中の水酸化ナトリウム(1 g)とテトラ-n-ブチルアンモニウム硫酸水素塩(VI)(50 mg, 0.15 mmol)の溶液に、スピペロンケタール(8-3[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-1-フェニル-1,3,8-トリアザスピロ[4.5]デカン-4-オン (これは、米国特許第3,839,342号, Chem. Abstr. 82: 43416, 及びKiesewetter et al., Appl. Radiat. Isot. 37: 1181 (1986), に記述されているように調製できる。これらの文献の内容は参照によって本明細書に組み込まれる)(15 mg, 0.034 mmol)が加えられ、強く攪拌された。10分後、トルエン(3 mL)中の6-アセチル-2(エチル-2-[(4-メチルフェニル)-スルフォニルオキシ-エチルアミノ]-ナフタレン)(12 mg, 0.03 mmol)の溶液が加えられ、反応混合物は攪拌され1時間90℃で加熱された。冷却後、反応混合物は水とジクロロメタンに分配され、有機層は塩水で洗浄され、乾燥、蒸発されてオイル状の残渣が残された。放射状クロマトグラフィー(1 mmシリカ, ジクロロメタン中2%メタノール)によって5 mg (25%) の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-4-フェニル-2,4,8-トリアザスピロ[4.5]デセ-1-en-1-イル)-オキシ]-エチルアミノ)-2-ナフチル]-1-エタノン(化合物A)と11 mg (56%) の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ)-2-ナフチル]-1-エタノン(化合物B)が得られた。
【0034】
化合物A−HRMS C41H47FN4O4に対する計算値:678.3581. 実測値:678.3605. 1H NMR: δ1.45-2.24 (m, 11H, スピペロンCH2, CH2CH3), 2.35-2.84 (m, 6H, スピペロン), 2.65 (s, 3H, OCH3), 3.59 (q, 2H, NCH2CH3), 2.35-2.84 (M, 6H, スピペロン), 2.65 (s, 3H, OCH3), 3.59 (q, 2H, NCH2CH3), 3.76 in 4.05 (m, 4H, OCH2CH2O), 3.85 (t, 2H, NCH2CH2O), 4.52 (t, 2H, NCH2CH2O), 4.99 (s, 2H, NCH2N), 6.76-6.83 9m, 3H, フェニル, フルオロフェニル), 6.93 (d, 1H, 5-H), 6.95-7.04 (M, 2H, フェニル、フルオロフェニル), 7.19 (dd, 1H, 7-H), 7.21-7.26 (m, 2H, フェニル、フルオロフェニル), 7.61 (d, 1H, 4-H), 7.78 (d, 1H, 8-H,), 7.93 (dd, 1H, 3-H). 8.30 (d, 1H, 1-H). J1,3=1.5 Hz, J5,7=2.4 Hz, J3,4=9.5 Hz, J7,8=9.2 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.3 Hz.
【0035】
化合物B−HRMS C41H47FN4O4に対する計算値:678.3581. 実測値:678.3603. 1H NMR: δ1.20-1.94 (m, 17H, スピペロンCH2, CH2CH3), 2.66 (s, 3H, COCH3), 3.56 (q, 2H, NCH2CH3), 3.66 and 4.02 (m, 4H, OCH2CH2O), 3.71-3.81 (m, 4H, NCH2CH2N), 4.68 (s, 2H, NCH2N), 6.82-6.90 (m, 2H, フェニル, フルオロフェニル), 6.94 (d, 1H, 5-H), 6.98-7.04 (m, 2H, フェニル、フルオロフェニル), 7.18 (dd, 1H, H-7), 7.21-7.26 (m, 3H, フェニル、フルオロフェニル), 7.39-7.45 (m, 2H, フェニル, フルオロフェニル), 7.60 (d, 1H, 4-H), 7.78 (d, 1H, 8-H,), 7.93 (dd, 1H, 3-H). 8.29 (d, 1H, 1-H). J1,3=1.6 Hz, J3,4=9.8 Hz, J5,7=2.4 Hz, J7,8=10.4 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.3 Hz.
【0036】
ピリジン(3 mL)中の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ)-2-ナフチル]-1-エタノン(13 mg, 0.018 mmol)とマロノニトリル(6 mg, 0.09 mmol)の溶液がアルゴンの下で85℃で24時間加熱された。ピリジンが室温において真空で除去された後、残渣が塩水とジクロロメタンに分配され、有機層が乾燥、蒸発された。生成物、2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリル、が放射状クロマトグラフィー(1 mmシリカ, ジクロロメタン中2.5%メタノール;13.5 mg, 97%)によって単離された。
【0037】
HRMS C44H48FN6O3に対する計算値:727.37719. 実測値:727.3772. 1H NMR: δ1.25-1.93 (m, 17H, スピペロンCH2, CH2CH3), 2.70 (s, 3H, C=C-CH3), 3.57 (q, 2H, NCH2CH3), 3.64及び4.03 (m, 4H, OCH2CH2O), 3.71-3.78 (m, 4H, NCH2CH2N), 4.68 (s, 2H, NCH2N), 6.83-6.88 (m, 2H, フェニル, フルオロフェニル), 6.94 (d, 1H, 5-H), 6.96-7.04 (m, 2H, フェニル、フルオロフェニル), 7.56 (dd, 1H, 3-H), 7.63 (d, 1H, 4-H), 7.76 (dd, 1H, 4-H,), 7.76 (dd, 1H, 9-H). 8.00 (d, 1H, 1-H). J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.4 Hz, J7,8=9.3 Hz, J(CH2CH3)=7.1 Hz.
【0038】
一滴の濃塩酸を含むメタノール(1 mL)中で、2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリル(13.5 mg, 0.0186 mmol)を室温で3時間攪拌することによってケタール保護基が除去された。反応混合物はジクロロメタンで希釈され、炭酸水素ナトリウムの飽和溶液で洗浄された。真空中での蒸発の後、残渣は放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中2%メタノール)によって精製されて10 mg (79%)の2-[1-(6-(エチル-[2-8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルエチル]-アミノ)-2-ナフチルエチリデン)-マロノニトリルが得られた。HRMS C42H44FN6O2(M+H)に対する計算値:683.35. 実測値:683. 1H NMR: δ1.21-3.02 (m, 17H, スピペロンCH2, CH3), 2.71 (s, 3H, C=C-CH3), 3.56 (q, 2H, NCH2CH3), 3.69 (m, 4H, NCH2CH2N), 4.67 (s, 2H, NCH2N), 6.79-7.23 (m, 7H, フェニル, フルオロフェニル), 6.95 (d, 1H, 5-H),.5.19 (dd, 1H, 7-H), 7.56 (dd, 1H, 3-H), 7.65 (d, 1H, 4-H,), 7.76 (d, 1H, 8-H). 7.97-8.04 (m, 3H, フルオロフェニル、1-H). J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.5 Hz, J7,8=9.1 Hz, J(CH2CH3)=7.1 Hz.
【0039】
実施例1(c)−2-(1-6-[4-(8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)ピペリジノ]-2-ナフチルエチリデン)-マロノニトリル
【化9】
1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(653 mg, 3.5 mmol)(実施例1(b)で述べたように調製された)、硫酸水素ナトリウム(IV)(1.6 g, 15.5 mmol)、4-ピペリジルメタノール(2 g, 17.6 mmol)(Bradbury et al., J. Med. Chem. 34: 1073 (1991)に記述されているように調製された、この内容は参照によって本明細書に組み込まれる)、及び水(6 mL)の混合物がスチール・ボンベ中で135〜142℃で16日間加熱された。冷却後、反応混合物は酢酸エチルで抽出された。生成物の一部はまだ残渣に残っていたので、さらにジクロロメタン中5%メタノールで抽出された。有機抽出物を一緒にして、乾燥、蒸発させた。残渣は放射状クロマトグラフィー(2 mmシリカ、ジクロロメタン中2%メタノール)によって精製されて139 mg (14%) の1-6-[(4-ヒドロキシメチル)-ピペリジノ]-2-ナフチル-1-エタノンが得られた。酢酸エチルから再結晶化させた後、この化合物は180〜182℃で融解した。1H NMR: δ1.44 (dddd, 2H, 3’a-H, 5’a-H), 1.76 (m, 1H, 4’a-H), 1.91 (bd, 2H, 3’e-H, 5’c-H), 2.68 (s, 3H, COCH3), 2.89 (ddd, 2H, 2’a-H, 6’a-H), 3.58 (d, 2H, OCH2), 3.94 (bd, 2H, 27e-H, 67e-H), 7.10 (d, 1H, 5-H), 7.32 (dd, 1H, 7-H), 7.66 (d, 1H, 4-H), 7.80 (d, 1H, 8-H), 7.94 (dd, 1H, 3-H), 8.32 (d, 1H, 1-H), J3’a,3’c= J5’a,5’c =12.5 Hz, J2’a,3’a=J6’a,5’a=12.5Hz, J3’a,4’a=J5’a,4’a=12.5 Hz, J2’e,3’a=J6’e,5’a=4.0Hz, J2’a,2’c= J6’a,6’c =12.5 Hz, J2’a,3’e=J6’a,5e=2.6Hz, J4a,OCH2=6.4Hz, J1,3=1.9 Hz, J3,4=8.9 Hz, J5,7=2.3 Hz, J7,8=9.0 Hz.
【0040】
ピリジン(3.mL)中の1-6-[(4-ヒドロキシメチル)-ピペリジノ]-2-ナフチル-1-エタノン(59 mg, 0.2 mmol) の溶液が-15℃に冷却され、p-トルエン無水スルフォン酸(205 mg, 0.6 mmol)がアルゴン下で攪拌しながら加えられた。反応混合物は放置されてゆっくりと1時間で室温まで暖められた。それが再び冷却され、塩水とエーテルの間で分配された。有機層を塩水で洗浄し、乾燥、蒸発させると、83 mg (91%)の粗1-(6-アセチル-2-ナフチル)-4-[(4-メチルフェニル)-スルフォニルオキシ]メチルピペリジンが残された。
【0041】
水(2 mL)中の水酸化ナトリウム(1 g)とテトラ-n-ブチルアンモニウム硫酸水素塩(VI)(50 mg, 0.15 mmol)の溶液に、スピペロンケタール(100 mg, 0.2 mmol)が加えられ、激しく攪拌された。10分後、トルエン(10 mL)中の1-(6-アセチル-2-ナフチル)-4-[(4-メチルフェニル)-スルフォニルオキシ]メチルピペリジン(98 mg, 0.2 mmol)の溶液が加えられ、反応混合物は室温で11日間攪拌された。反応混合物は塩水とジクロロメタンの間で分配され、有機層は乾燥され蒸発されて190 mgのオイル状の残渣が残された。放射状クロマトグラフィー(1 mmシリカ、ジクロロメタンのあとにジクロロメタン中2%メタノール)によって、27 mg (17%) の1-[6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-4-フェニル-2,4,8-トリアザスピロ[4.5]デセ-1-エン-1-イル]-オキシル-メチルピペリジノ)-2-ナフチル)-1-エタノン(化合物3)と92 mg (58%) の1-[6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル]-メチル)ピペリジノ-2-ナフチル)-1-エタノン(化合物4)が得られた。
【0042】
化合物3:HRMS C43H49FN4O4に対する計算値:704.3738. 実測値:704.3760. 1H NMR: δ1.46-1.90 (m, 10H, 3’a-H, 5’a-H, 3’e-H, 5’e-H, スピペロン), 1.88 (m, 1H, 4’a-H), 2.15 and 2.38 (b, 4H, スピペロン), 2.67 (s, 3H, COCH3), 2.80 (m, 4H, スピペロン), 2.95 (m, 2H, 2’a-H, 6’a-H), 3.75 (m, 2H, OCH2 CH2O), 3.87 (m, 2H, 2’e-H, 6’e-H), 3.92 (m, 2H, OCH2 CH2O), 4.19 (d, 2H, OCH2), 4.97 (s, 2H, NCH2N), 6.7-6.9 (m, 3H, Ph), 7.01 (m, 2H, Ph), 7.11 (d, 1H, 5-H), 7.23 (m, 2H, Ph), 7.32 (dd, 1H, 7-H), 7.41 (m, 2H, Ph), 7.66 (d, 1H, 4-H), 7.81 (d, 1H, 8-H), 7.95 (dd, 1H, 3-H), 8.32 (d, 1H, 1-H). J2’a,2’e= J6’a,6’e =12.4 Hz, J2’a,3’e=J6’a,5’e=2.6Hz, J4’a,OCH2=6.1Hz, J1,3=.1 Hz, J3,4=8.8 Hz, J5,7=2.1 Hz, J7,8=9.1 Hz.
【0043】
化合物4:HRMS C43H49FN4O4に対する計算値:704.3738. 実測値:704.3710. 1H NMR: δ1.50 (dddd, 2H, 3’a-H, 5’a-H), 1.55-1.70 (m, 4H, スピペロン), 1.84 (bd, 2H, 3’e-H, 5’e-H), 1.92 (m, 2H, スピペロン), 1.98 (m, 1H, 4’a-H), 2.42 (m, 2H, スピペロン), 2.67 (s, 3H, COCH1), 2.69 (m, 2H, スピペロン), 2.83 (m, 4H, スピペロン), 2.88 (m, 2H, 2’a-H, 6’a-H), 3.35 (d, 2H, 4’-CH2N), 3.75 (m, 2H, OCH2 CH2O), 3.92 (bd, 2H, 2’e-H, 6’e-H), 4.02 (m, 2H, OCH2 CH2O), 4.71 (s, 2H, NCH2N), 6.88 ((m, 1H, Ph), 6.91 (m, 2H, Ph), 7.01 (m, 2H, Ph), 7.08 (bs, 1H, 5-H), 7.27 (m, 3H, 7-H, Ph), 7.43 (m, 2H, Ph), 7.65 (d, 1H, 4-H), 7.79 (d, 1H, 8-H), 7.94 (dd, 1H, 3-H), 8.32 (bs, 1H, 1-H), J3’a,3’e= J5’a,5’e =12.4 Hz, J2’a,3’a=12.5Hz, J2’a,2’e= J6’a,6’e =12.8 Hz, J2’a,3’e= J6’a,5’e =2.4Hz, J4’a,4’CH2N=7.3Hz, J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.1 Hz, J7,8=9.2 Hz.
【0044】
2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリルの合成に関して実施例1(b)で述べた手順を用いて、1-[6-4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-メチル]-ピペリジノ-2-ナフチル]-1-エタノンが、2-[1-6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)ピペリジノ]-2-ナフチル)エチリデン]-マロノニトリルに変換された。これを、CH2Cl2中2%MeOHを溶媒とする1 mmシリカプレートでの放射状クロマトグラフィーで精製した。FAB HRMSのC46H50FN6O3(M+H)に対する計算値:753.3928. 実測値:753.3940. 1H NMR: δ1.60-2.1 (m, 11H, スピペロン、3’a-H, 3’e-H, 4’a-H, 5’a-H, 5’e-H), 2.40 (m, 2H, スピペロン), 2.71 (s, 3H, C=C-CH3), 2.60-2.80 (m, 6H, スピペロン), 2.91 (m, 2H, 2’a-H, 6’a-H), 3.37 (d, 2H, 4’-CH2N), 3.75 (m, 2H, OCH2 CH2O), 3.94 (bd, 2H, 2’e-H, 6’e-H), 4.02 (m, 2H, OCH2 CH2O), 4.72 (s, 2H, NCH2N), 6.85-6.95 ((m, 3H, Ph), 7.01 (m, 2H, フルオロフェニル), 7.07 (d, 1H, 5-H), 7.31 (m, 3H, 7-H, Ph), 7.41 (m, 2H, フルオロフェニル), 7.56 (dd, 1H, 3-H), 7.69 (d, 1H, 4-H), 7.77 (d, 1H, 8-H), 8.01 (bs, 1H, 1-H). J2’a,3’a= J5’a,6’a =12.8Hz, J2’a,2’e= J6’a,6’e =12.8Hz, J4’a,4’CH2N=7.6Hz, J1,3=1.8 Hz, J3,4=8.6 Hz, J5,7=2.2 Hz, J7,8=9.4 Hz, J2’a,3’e= J5’e,6’a =1.8 Hz, JH,F=8.7及び6.2Hz.
【0045】
実施例1(b)に記述されているようにケタール保護基を除去して2-(1-(6-[4-(8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)-ピペリジノ]-2-ナフチルエチリデン)-マロノニトリルが定量的な収率で得られた。FAB HRMSのC44H46FN6O2(M+H)に対する計算値:709.3666. 実測値:709.3689. 1H NMR: δ1.60-2.1 (m, 11H, スピペロン、3’a-H, 3’e-H, 4’a-H, 5’a-H, 5’e-H), 2.5-2.71 (m, 4H, スピペロン), 2.73 (s, 3H, C=C-CH3), 2.8-3.1 (m, 6H, スピペロン、2’a-H, 6’a-H), 3.38 (d, 2H, 4’-CH2N), 3.96 (bd, 2H, 2’e-H, 6’e-H), 4.74 (s, 2H, NCH2N), 6.91 (m, 3H, フェニル), 7.1 (d, 1H, 5-H), 7.15 (m, 2H, フルオロフェニル), 7.24-7.30 (m, 2H, Ph), 7.14 (dd, 1H, 7-H), 7.58 (dd, 1H, 3-H), 7.72 (d, 1H, 4-H), 7.79 (d, 1H, 8-H), 8.01-8.08 (m, 3H, フルオロフェニル、1-H). J1,3=2.0 Hz, J3,4=8.6 Hz, J5,7=2.4 Hz, J7,8=9.2 Hz, J4’a,4’CH2N=7.4Hz, J2’a,2’e= J6’a,6’e =13.0Hz, J2’a,3’a= J5’a,6’a =12.5Hz, J2’a,3’e= J5’e,6’a =1.9 Hz, JH,F=8.7及び6.2Hz.
【0046】
実施例1(d)−tert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレートの調製
【化10】
新しく蒸留された無水トルエンとヘキサメチル・ホスフォリックアミド(HMPA)、各25 mL、の混合物に、無水ピペラジン(7 g, 81.3 mmol; 真空乾燥器でKOH-ドライエライト上で3日間乾燥)が溶解された。この溶液に、アルゴン雰囲気中で小さな片にカットされた556 mg(80.1 mmol)のリチウム棒が加えられ、混合物はアルゴンの下で24時間攪拌され、その間にすべてのリチウムが溶解した。真空乾燥された1-(6-メトキシ-2-ナフチル)-1-エタノン(Arsenijevic et al., Org. Synth. Coll. 1988, 6:34-36に記述されたように調製された。この文献の開示内容は参照によって本明細書に組み込まれる)(3.5 g, 17.5 mmol)が加えられ、攪拌がさらに65時間続けられた。300 mLの水による反応停止、ジクロロメタンによる抽出(3 x 300 mL)、無水硫化マグネシウムによる乾燥、及び蒸発の後に、白色と黄色固体の混合物が得られた。300 mLの熱いメタノールによる抽出によって、粗生成物、1-(6-ピペラジノ-2-ナフチル)-1-エタノン、が得られ、カラム・クロマトグラフィー(70-230メッシュのシリカ、25 x 120 mm, ジクロロメタン中5%メタノール)によって精製された。収量は1.54 g(35%)であった。酢酸エチルからの再結晶後、サンプルは170.5〜172℃で融解した。
【0047】
1-(6-ピペラジノ-2-ナフチル)-1-エタノンは、また、1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(実施例1(b)に記述されているように調製された)(441 mg, 2.36 mmol)を140〜150℃で6 gのピペラジン水和物(30.9 mmol)及び244 mg (2.35 mmol)のNaHSO3と共に24 時間熱することによっても調製された。追加の亜硫酸水素ナトリウム(2 g, 19.2 mmol)が加えられた。さらに24時間後、さらに亜硫酸水素ナトリウム(1 g)が加えられ、加熱が続けられた(全反応時間は72時間)。冷却後、混合物は2 x 50 mLのメタノールで抽出された。メタノールの蒸発後の残渣は50 mLの水に懸濁され、酢酸エチルで抽出された(5 x 80 mL)。抽出物全体を乾燥させ(硫酸マグネシウム)、蒸発させて430 mg の黄色固体が得られた。放射状クロマトグラフィー(4 mmシリカ、メタノール)によって83 mg (19%)の出発ナフトールと276 mg (46%; 回収されない出発物質に基づくと56%)の1-(6-ピペラジノ-2-ナフチル)-1-エタノンが得られた。この1-(6-ピペラジノ-2-ナフチル)-1-エタノンはあらゆる点で上述した別の方法によって得られた化合物と同一であった。分析、C16H18N2Oに対する計算値:C, 75.56; H, 7.13; N, 11.01. 実測値:C, 75.82; H, 7.27; N, 10.92. 1H NMR: δ2.68 (s, 3H, CH3), 3.09 and 3.35 (t, J = 4.95Hz, 8H, ピペラジン), 7.10 (d, 1H, 5-H), 7.31 (dd, 1H, 7-H), 7.69 (d, 1H, 4-H), 7.83 (d, 1H, 8-H), 7.95 (d, 1H, 3-H), 8.34 (s, 1H, 1-H); J5,7=2Hz, J7,8=8.4Hz, J1,3=2Hz, J3,4=8.4Hz.
【0048】
1-(6-ピペラジノ-2-ナフチル)-1-エタノン(254 mg, 1 mmol)が1 g のNaOH, 100 mgのテトラ-n-ブチルアンモニウム硫酸水素塩、2 mLの水、及び6 mLのトルエンの攪拌された混合物に加えられ、続いて230 mg(1.05 mmol)のジ-tert-ブチルジカルボネートの溶液が加えられた。反応の過程はTLC (シリカ、ジクロロメタン中5%のメタノール)で追跡された。10分毎に、すべての出発物質が反応してしまうまで追加の量のジカルボネートが加えられた。全部でほぼ1.5当量が用いられた。水とジクロロメタン(各60 mL)の混合物が加えられ、十分な振蕩の後、層が分離された。水性層は、さらに別の30 mLのジクロロメタンで抽出された。有機抽出物の全体は、無水硫酸マグネシウムで乾燥された。この過程で溶液の色はピンクから淡黄色に変化した。真空中での蒸発によって295 mg (83%)のtert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレートが得られ、これをジクロロメタン−石油エーテル混合物から再結晶させると、153〜154℃で融解した。分析、C21H26N2O3に対する計算値:C, 71.16; H, 7.39; N, 7.90. 実測値:C, 71.27; H, 7.60; N, 7.86. 1H NMR: δ1.50 (s, 9H, -C(CH3)3), 2.68 (s, 3H, CH3), 3.33 and 3.64 (t, J = 4.9Hz, 8H, ピペラジン), 7.10 (d, 1H, 5-H), 7.31 (dd, 1H, 7-H), 7.70 (d, 1H, 4-H), 7.85 (d, 1H, 8-H), 7.97 (d, 1H, 3-H), 8.35 (d, 1H, 1-H); J5,7=2Hz, J7,8=9Hz, J3,4=8.7Hz.
【0049】
実施例1(e)−2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロノニトリルの調製
【化11】
実施例1(d)に記述されたように調製されたtert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレート(177 mg, 0.5 mmol)が4 mLのピリジン中で40 mg (0.6 mmol)のマロノニトリルと共に105〜110℃で熱せられた。5.5時間後、さらに24 mgのマロノニトリルが加えられ、加熱は全部で12時間40分続けられた。混合物は冷却され、真空中で蒸発された。混合物の極性成分はカラム・クロマトグラフィー(70-230メッシュのシリカ、φ20 x 120 mm, クロロフォルム)によって除去され、生成物、tert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレート、が最終的に放射状クロマトグラフィー(シリカ、2 mm, クロロフォルム)によって精製されて、155 mg(77%)のtert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレートが得られ、ジクロロメタン−石油エーテル混合物からの再結晶後、169-171℃で融解した。分析、C24H26N4O2に対する計算値:C, 71.62; H, 6.51; N, 13.92. 実測値:C, 71.62; H, 6.66; N, 13.87. 1H NMR: δ1.50 (s, 9H, -C(CH3)3), 2.72 (s, 3H, CH3), 3.34及び3.64 (t, J = 5.1Hz, 8H, ピペラジン), 7.09 (d, 1H, 5-H), 7.33 (dd, 1H, 7-H), 7.58 (dd, 1H, 3-H), 7.74 (d, 1H, 4-H), 7.81 (d, 1H, 8-H), 8.02 (d, 1H, 1-H); J5,7=2Hz, J7,8=9.1Hz, J1,3=2Hz, J3,4=9.1Hz.
【0050】
tert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレートが室温でTFA(トリフルオロ酢酸)で処理されると、TLCは反応が5分で終わったことを示し、単一の生成物、2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロノニトリルが得られた。TFAは室温で真空で除去された。1H NMR: δ2.72 (s, 3H, CH3), 3.50及び3.63 (ブロード, 8H, ピペラジン), 7.18 (ブロード s, 1H, 5-H), 7.29 (d, 1H, 7-H), 7.59 (d, 1H, 3-H), 7.79 (d, 1H, 4-H), 7.87 (d, 1H, 8-H), 8.04 (s, 1H, 1-H), 9.0 (ブロード, 1.5H, NH及び酸); J7,8=8.8Hz, J3,4=8.4Hz. 19F NMR: δ -76.2 (CF3COO).
【0051】
残渣のNMRによって、tert-ブチルオキシカルボニル基が除去され、TFAが少し残されていることが明らかになった(19F NMR)。ジクロロメタン(10 mL)が加えられ、溶液は飽和NaHCO3溶液で洗浄され、乾燥され、真空中で蒸発された。淡黄色の固体が得られ、室温で放置するとそれは暗赤色に変わった。TLCは、色の変化が2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロニトリルがいくつかの生成物に分解したためであることを示し、最も強度の大きなスポットは低-Rfの赤-オレンジ色であった。中和後のいくつかのNMR信号:δ2.72 (s, 3H, CH3), 3.09及び3.35 (t, J=5Hz, 8H, ピペラジン), 7.08 (s, 1H, 5-H), 8.02 (s, 1H, 1-H).
【0052】
実施例1(f)−2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)の調製
【化12】
4.15 g (55.5 mmol)のNaHSO3、8 mLの水、0.78 g (4.19 mmol)の1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(実施例1(b)に記述されているように調製)、及び8 mLの2-メチルアミノエタノールの混合物がスチール・ボンベの中で140℃で28時間加熱された。冷却後、混合物は酢酸エチルと水(それぞれ、500 mLと200 mL)の間で分配された。有機層を乾燥、蒸発させると粗1-(6-(2-ヒドロキシエチル-メチルアミノ)-2-ナフチル)-1-エタノン(0.749 g, 73%)が残され、それがさらに放射状クロマトグラフィー(4 mm SiO2, CH2Cl2)によって精製された。
【0053】
ピリジン(6 mL)中の201 mg (0.83 mmol)の1-(6-(2-ヒドロキシエチル-メチルアミノ)-2-ナフチル)-1-エタノンの溶液にマロノニトリル(236 mg, 3.6 mmol)が加えられ、混合物は95℃で24時間加熱された。溶媒は真空で除去され、残渣が放射状クロマトグラフィー(4 mm SiO2, 1% MeOH/CH2Cl2)にかけられ、150 mg (73%)の2-(1,1-ジシアノプロペン-2-イル)-6-(2-ヒドロキシエチル)-メチルアミノ)-ナフタレンが得られた。
【0054】
ピリジン(5 mL)中の2-(1,1-ジシアノプロペン-2-イル)-6-(2-ヒドロキシエチル)-メチルアミノ)-ナフタレン(120 mg, 0.41 mmol)の溶液に、p-トルエン無水スルフォン酸が加えられた(44 mg, 1.35 mmol)。室温で1時間攪拌した後、真空下でピリジンを除去し、残渣を放射状クロマトグラフィー(2 mm SiO2, CH2Cl2)にかけて183 mg (80%)の2-(1,1-ジシアノプロペン-2-イル)-6-(2-トシルオキシエチル)-メチルアミノ)-ナフタレンが得られた。
【0055】
サイクロ(登録商標)トロンからの放射性19Fフッ化物528.5 mCiが50μLの水と300μLのアセトニトリル中の19 mgのKryptofix 2.2.2と0.75mgの炭酸カリウムの溶液に移された。115℃で窒素の流れによって水を除去し、続いてアセトニトリルと共に蒸留した(3 x 200μL)。1 mLのアセトニトリル中のトシル化物(2-(1,1-ジシアノプロペン-2-イル)-6-(2-トシルオキシエチル)-メチルアミノ)-ナフタレン、4 mg)を加え、混合物を85〜86℃で20分加熱した。冷却後、1mLの水が加えられ、混合物はC-18 Sep-Pak Cartridgeに移され、蒸留水で洗浄され(3×4 mL)、CH2Cl2で溶出された(2×2.5 mL)。溶出物は硫酸ナトリウムを詰めたカラムを通して乾燥させ、HPLCカラム(Whatman Partisil Silica 10, 500×10 mm, mL/min CH2Cl2:ヘキサン=7 : 3, UV検出器@254 nm, 放射能検出器)に装填された。溶出物を集め、適当な分画を一緒にし、真空下で蒸発させて50.7 mCi (17%, 崩壊を補正)の注射のために製剤された表題の生成物が得られた。合成は50分で完了した。
【0056】
実施例1(g)−7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オンの調製
【化13】
7.76 mLのDMFに0℃で4.66 mL (約50 mmol)のPOCl3を(一滴ずつ)加えてVielsmeier試薬が少ない分量で調製され、混合物は同じ温度で5分間、続いて室温で30分間、攪拌された。2 mLのDMF中の3.43 g (25 mmol)の3-(ジメチルアミノ)フェノールが、Vielsmeier試薬に一滴ずつ加えられた。混合物は60〜70℃で1時間加熱され、50 gの砕いた氷に混合物を注いで過剰な試薬を消滅させた。混合物は酢酸エチルで抽出され、有機部分を無水硫酸マグネシウムで乾燥して、蒸発させた。オイル状の残渣から4-ジメチルアミノ-2-ヒドロキシベンズアルデヒドがカラム・クロマトグラフィー(シリカゲル、クロロフォルム)によって単離された。収量:2.32 g (14 mmol, 56%).
【0057】
4 mmolの4-ジメチルアミノ-2-ヒドロキシベンズアルデヒドと5 mmolの適当な1,3-ジカルボニル化合物が8 mLの無水エタノールに溶解された。16滴のピペリジンが加えられ、各混合物は2〜6時間還流で加熱された。混合物はフリーザー内で冷却され、沈殿を濾過し、エタノールで洗浄して表1に示されたような生成物が得られた。
【0058】
【表1】
【0059】
NMRスペクトル:
3-アセチル-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(1)
1H NMR (360MHz, CDCl3) δ: 2.70 (3H, s, Ac); 3.14 (6H, s, NMe2); 4.39 (2H, q, EtO); 6.48 (1H, d, 芳香族H); 6.66 (1H, dd, 芳香族H); 7.43 (1H, d, 芳香族H); 8.47 (1H, s, 4-H).
エチル7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-カルボキシレート (2)
1H NMR (360MHz, CDCl3) δ: 1.40 (3H, t, EtO-); 3.13 (6H, s, NMe2); 4.39 (2H, q, EtO); 6.48 (1H, d, 芳香族H); 6.65 (1H, dd, 芳香族H); 7.40 (1H, d, 芳香族H); 8.46 (1H, s, 4-H).
3-(2,2-ジメチルプロパノイル)-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(3)
1H NMR (360MHz, CDCl3) δ: 1.33 (9H, s, tBu); 3.10 (6H, s, NMe2); 6.50 (1H, d, 芳香族H); 6.63 (1H, dd, 芳香族H); 7.33 (1H, d, 芳香族H); 7.68 (1H, s, 4-H).
メチル-3-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]-3-オキソ-プロパノエート (4)
1H NMR (360MHz, CDCl3) δ: 3.15 (6H, s, NMe2); 3.75 (3H, s, -OMe); 4.11 (2H, s, CH2); 6.47 (1H, d, 芳香族H); 6.66 (1H, dd, 芳香族H); 7.45 (1H, d, 芳香族H); 8.53 (1H, s, 4-H).
7-(ジメチルアミノ)-3-(4-ニトロベンゾイル)-2H-1-ベンゾピラン-2-オン(5)
1H NMR (360MHz, CDCl3) δ: 3.13 (6H, s, NMe2); 6.63 (1H, br, 芳香族H); 6.84 (1H, dd, 芳香族H); 7.69 (1H, d, 芳香族H); 7.97 (2H, m, 芳香族H); 8.31 (2H, m, 芳香族H); 8.47 (1H, s, 4-H).
【0060】
実施例1(h)−ビス(7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル)ケトンの調製
【化14】
360 mg(1 mmol)のメチル-3-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]-3-オキソ-プロパノエート(実施例1(g)の化合物4)が250 mgの4-ジメチルアミノ-2-ヒドロキシベンザアルデヒドと混合され、2 mLの無水エタノールに溶解された。4滴のピペリジンが加えられ、混合物は8時間還流された。混合物は室温にされ、沈殿した固体が濾過された。179 mg (44%)のオレンジ色の固体。1H NMR (360MHz, CDCl3) δ: 3.11 (6H, s, NMe2); 6.50 (1H, d, 芳香酸 H); 6.63 (1H, dd, 芳香酸 H); 7.41 (1H, d, 芳香酸 H); 8.19 (1H, s, 4-H).
【0061】
実施例1(i)−2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの調製
【化15】
N,N-ジメチルホルムアミド・ジメチル・アセタール(DMFDMA, 5 mL)中の2-{1-[6-(ジメチルアミノ)-2-ナフチル]エチリデン}マロノニトリル(1 g, 3.8 mmol)(A. Jacobson et al., J. Am. Chem. Soc. 1996, 118, 5572-5579,に記述されているように調製)の溶液が室温で一晩攪拌された。揮発成分を真空で除去し、残渣をカラムクロマトグラフィー(シリカ70-230メッシュ、20/150 mm, クロロフォルム)にかけて、2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(1.19 g, 98%)が得られた。mp. 213〜215℃(ジクロロメタン−石油エーテル混合物から);元素分析、C20H20N4に対する計算値:C, 75.92; H, 6.37; N, 17.17. 実測値;C, 76.28; H, 6.04; N, 17.57. IR (KBrペレット): 2210 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.02 (s, 6H, NMe2); 3.1 (s, 6H, NMe2); 5.86 and 6.72 (d, 2H, CH=CH), 6.91 (d, 1H, H-5), 7.20 (dd, 1H, H-7), 7.23 (dd, 1H, H-3), 7.62 (bs, 1H, H-1), 7.69 (d, 1H, H-8), 7.73 (d, 1H, H-4). J5,7=2.3Hz, J7,8=9.1Hz, J3,4=9.0Hz, JCH=CH=12.5Hz.
【0062】
実施例1(j)−2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル
【化16】
イソプロパノール(100 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(603 mg, 1.9 mmol)(上の実施例1(i)に記述されているように調製)の溶液がrtで無水HClで飽和され、一晩攪拌された。溶媒は真空中で除去され、固体残渣はジクロロメタンに溶解され、溶液は飽和NaHCO3溶液で洗浄された。有機層を乾燥させ、蒸発させて2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(402 mg, 69%)が残された。mp. 198〜200℃(MeOHから);元素分析、C18H14N3Clに対する計算値: C, 70.24; H, 4.58; N, 13.65. 実測値:C, 69.76; H, 4.31; N, 13.38. IR (KBrペレット): 2250 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.15 (s, 6H, Me2N), 6.91 (d, 1H, H-5), 7.21 (dd, 1H, H-7), 7.47 (d, 1H, H-5’), 7.57 (dd, 1H, H-3), 7.74 (d, 1H, H-8), 7.79 (d, 1H, H-4), 7.99 (d, 1H, H-1), 8.53 (d, 1H, H-6). J5,7=2.2Hz, J7,8=9.7Hz, J1,3=2.0Hz, J3,4=9.2Hz, J5’,6’=5.1Hz.
【0063】
実施例1(k)−2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル
【化17】
MeOH (20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(100 mg, 0.32 mmol)(上の実施例1(i)に記述されているように調製)の沸騰する溶液にアンモニアガスが30分間吹き込まれた。反応の間に溶液から分離し始めた2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリルの黄色結晶が濾過された(86 mg, 93%)。mp. 256〜257℃ (MeOHから);元素分析、C18H16N4Clに対する計算値: C, 74.98; H, 5.59; N, 19.43. 実測値:C, 75.29; H, 5.31; N, 19.16. IR (KBrペレット): 2210 cm-1 (CN), 3720 cm-1 (ArNH2); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 5.27 (s, 2H, NH2), 6.87 (d, 1H, H-5), 6.92 (d, 1H, H-5), 7.20 (dd, 1H, H-7), 7.57 (dd, 1H, H-3), 7.74 (d, 1H, H-8), 7.79 (d, 1H, H-4), 7.97 (bs, 1H, H-1), 8.42 (d, 1H, H-6). J5,7=2.3Hz, J7,8=9.5Hz, J1,3=2.0Hz, J3,4=8.5Hz, J5’,6’=4.9Hz.
【0064】
実施例1(l)−2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル-1-オキシドの調製
【化18】
MeOH (20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]−2−プロペニリデン}マロノニトリル(28 mg, 0.9 mmol)(上の実施例1(i)に記述されているように調製)とヒドロキシ塩化アンモニウム(94.5 mg, 1.36 mmol)の溶液が還流で70時間加熱された。溶媒を除去し、残渣を放射状クロマトグラフィー(2 mm シリカ、ジクロロメタン中5% MeOH)にかけて2-アミノ-4-[6-(ジメチルアミノ)-2−ナフチル]ニコチノニトリル-1-オキシド(53 mg, 20%)が得られた。mp. 248〜250℃ (MeOHから);元素分析、C18H16N4OCに対する計算値: C, 71.04; H, 5.30; N, 18.41. 実測値:C, 70.59; H, 5.59; N, 18.19. IR (KBrペレット): 2220 cm-1 (CN), 3310 cm-1 (ArNH2); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 6.43 (s, 2H, NH2), 6.86 (d, 1H, H-5’), 6.91 (d, 1H, H-5), 7.21 (dd, 1H, H-7), 7.55 (dd, 1H, H-3), 7.75 (d, 1H, H-8), 7.78 (d, 1H, H-4), 7.95 (bs, 1H, H-1), 8.12 (d, 1H, H-6); J5,7=2.4Hz, J7,8=8.7Hz, J1,3=2.0Hz, J3,4=8.7Hz, J5’,6’=6.7Hz.
【0065】
実施例1(m)−4-[6-(ジメチルアミノ)-ナフチル]-2-メトキシニコチノニトリルの調製
【化19】
方法A. MeOH (15 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]−2−プロペニリデン}マロノニトリル(82.5 mg, 0.26 mmol)(上の実施例1(i)に記述されているように調製)の溶液に、ナトリウムメチラートの溶液(1 mL, mLのメタノールあたり10 mgのナトリウムを反応させることによって調製)が加えられ、反応混合物が還流で20時間加熱された。溶媒を真空で除去し、残渣をジクロロメタンと塩水の間で分配し、有機層を乾燥、蒸発させた。残渣を放射状クロマトグラフィー(1 mm シリカ、ジクロロメタン-シクロヘキサン 1:1)にかけて4-[6-(ジメチルアミノ)-2−ナフチル]-2-メトキシニコチノニトリル(40 mg, 50%)が得られた。
方法B. MeOH (10 mL)中の2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(58 mg, 0.19 mmol)(上の実施例1(j)に記述されているように調製)の溶液が、ナトリウムメチラート(0.9 mLの溶液、mLのメタノールあたり10 mgのナトリウムを反応させることによって調製、0.4 mmol)と共に6.5時間還流で加熱された。4-[6-(ジメチルアミノ)-2−ナフチル]-2-メトキシニコチノニトリルが方法Aで述べたように単離されたが、MeOHからの再結晶化によって精製された;元素分析、C19H17N3Oに対する計算値: C, 75.23; H, 5.65; N, 13.85. 実測値:C, 74.99; H, 5.59; N, 13.73. IR (KBrペレット): 2220 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 4.11 (s, 3H, OMe), 6.9 (bs, 1H, H-5), 7.12 (d, 1H, H-5’), 7.20 (bd, 1H, H-7), 7.59 (d, 1H, H-3), 7.74 (d, 1H, H-4), 7.79 (d, 1H, H-8), 7.99 (bs, 1H, H-1), 8.31 (d, 1H, H-6’); J7,8=9.2Hz, J3,4=8.6Hz, J5’,6’=5.4Hz.
【0066】
実施例1(n)−2-{(2Z)-1-[6-(ジメチルアミノ)-2-ナフチル]-3-[2-(ヒドロキシエチル)-アミノ]-2-プロペニリデン}マロノニトリルの調製
【化20】
MeOH (50 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(350 mg, 1.1 mmol)(上の実施例1(i)に記述されているように調製)と2-アミノエタノール(0.4 mL, 6.6 mmol)の溶液が還流で24 時間加熱された。溶媒を除去し、残渣を放射状クロマトグラフィー(4 mm シリカ、ジクロロメタン中5% MeOH)にかけて2-{(2Z)-1-[6-(ジメチルアミノ)-2-ナフチル]-3-[2-(ヒドロキシエチル)-アミノ]-2-プロペニリデン}マロノニトリル(327 mg, 89%)が得られた。mp. 168〜170℃ (EtOHから);1H NMR (360MHz, CDCl3) δ: 3.08 (s, 6H, Me2N), 3.98及び4.17 (bs, 4H, CH2CH2), 5.43 (b, 2H, OH, NH), 6.09及び7.24 (d, 2H, CH=CH), 6.86 (bs, 1H, H-5), 7.16 (d, 1H, H-7), 7.51 (d, 1H, H-3), 7.68 (d, 1H, H-4), 7.74 (d, 1H, H-8), 7.94 (s, 1H, H-1); J3,4=8.6Hz, J7,8=9.2Hz, JCH=CH=6.9Hz.
【0067】
実施例1(o)−4-[6-(ジメチルアミノ)-2-ナフチル]-2-[(2-ヒドロキシエチル)スルファニル]ニコチノニトリル
【化21】
ナトリウム・メチラートの溶液(5 mL, mLのMeOHあたり10 mgのナトリウムを反応させて調製、2.17 mmol)中の2-メルカプトエタノール(0.81 mL, 11.6 mmol)の溶液に、2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(273 mg, 0.89 mmol)(上の実施例1(i)に記述されているように調製)の溶液が加えられ、混合物は還流で5時間加熱された。反応混合物から分離した固体を濾過し、アセトニトリルから再結晶させて4-[6-(ジメチルアミノ)-2-ナフチル]-2-[(2-ヒドロキシエチル)スルファニル]ニコチノニトリル(271 mg, 85%)が得られた。mp. 210〜212℃. 元素分析、C20H19N3OSに対する計算値: C, 68.74; H, 5.48; N, 12.02. 実測値:C, 68.51; H, 4.99; N, 12.07. IR (KBrペレット): 2220 cm-1 (CN), 3350 cm-1 (OH); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.5 (t, 2H, CH2CH2), 3.65 (b, 1H, OH), 4.0 (b, 2H, CH2CH2), 6.91 (d, 1H, H-8), 7.20 (bd, 1H, H-7), 7.25 (d, 1H, H-5’), 7.55 (d, 1H, H-3), 7.75 (d, 1H, H-4), 7.79 (d, 1H, H-8), 7.97 (bs, 1H, H-1), 8.50 (d, 1H, H-6’); J7,8=7.8Hz, J3,4=8.6Hz, J5’,6’=5.4Hz, JCH2CH2=5.4Hz.
【0068】
実施例1(p)−2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの調製
【化22】
MeOH(20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル (100 mg, 0.316 mmol)とアミノアセトアルデヒド・ジエチル・アセタール(0.1 mL, 0.96 mmol)の溶液が還流で46時間加熱された。溶媒を真空で除去すると、オイル状の残渣が残された。それを放射状クロマトグラフィー(2 mmシリカ、ジクロロメタン、続いてジクロロメタン中5% MeOH)にかけて、2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(111 mg, 87%)が得られた。1H NMR (360MHz, CDCl3) δ: 1.22 (t, 6H, t, OCH2CH3, J=7.1Hz), 3.09 (s, 6H, Me2N), 3.59 and 3.80 (m, 4H, OCH2CH3), 4.05(d, 2H, CH2CH), 4.92 (t, 1H, CH2CH), 5.95 and 7.23 (d, 2H, CH=CH), 6.90 (bs, 1H, H-5), 7.19 (dd, 1H, H-7), 7.55 (d, 1H, H-3), 7.71 (d, 1H, H-4), 7.77 (d, 1H, H-8), 7.97 (bs, 1H, H-1); JCH=CH=7.2Hz, J7,8=9.1Hz, J3,4=8.5Hz, JCH2CH=5.1Hz.
【0069】
実施例1(q)−2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル
【化23】
MeOH(5 mL)中の2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(19 mg, 0.05 mmol)(上の実施例1(p)に記述されているように調製)の溶液にHClガスが室温で5分間吹き込まれた。反応混合物は室温でさらに30分間攪拌され、濃縮されてオイル状の残渣が残された。それを酢酸エチルと氷温の塩水の間で分配した(各40 mL)。有機層を乾燥、蒸発させて2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(12 mg, 66%)が得られた。2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルは、また、上の実施例1(i)に記述されているように調製された2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルから直接に、ジエチル・アセタールの代わりにアミノアセトアルデヒドジメチルアセタールを用い、上の実施例1(p)に記述されているような2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの合成の手順に従って調製された。mp. 158〜159℃. IR (KBrペレット): 2220 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.48 (s, 6H, OCH3), CH2CH2), 4.06 (d, 2H, CH2CH), 5.96 and 7.24 (d, 2H, CH=CH), 6.89 (d, 1H, H-5), 7.18 (dd, 1H, H-7), 7.54 (d, 1H, H-3), 7.74 (d, 1H, H-4), 7.76 (d, 1H, H-8), 7.96 (s, 1H, H-1); J5,7=2.2Hz, JCH=CH=7.0Hz, J7,8=8.9Hz, J3,4=8.5Hz, JCH2CH=5.01Hz.
【0070】
実施例1(r)−7-[6-(ジメチルアミノ)-2-ナフチル]イミダゾ[1.2-α]ピリジン-8-カルボニトリル
【化24】
MeOH(15 mL)中の2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(53 mg, 0.14 mmol)(上の実施例1(q)に記述されているように調製)の溶液が還流で2時間加熱された。最初の30分間、乾燥HClガスの流れが反応混合物を通して導かれた。反応が完了した後、溶媒を蒸発させ、固体残渣がジクロロメタンと炭酸水素ナトリウムの飽和溶液の間で分配された。有機層を乾燥し、蒸発させた。残渣を放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中5% MeOH)にかけて、7-[6-(ジメチルアミノ)-2-ナフチル]イミダゾ[1.2-α]ピリジン-8-カルボニトリル(25 mg, 57%)が得られた。mp. 268〜269℃;元素分析、C20H16N4に対する計算値: C, 76.90; H, 5.16; N, 17.94. 実測値:C, 76.231; H, 5.22; N, 17.62. IR (KBrペレット): 2230 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.07 (s, 6H, Me2N), 7.02 (d, 1H, H-5’), 7.29 (d, 1H, H-6), 7.33 (dd, 1H, H-7’), 7.69 (d, 1H, H-3’), 7.74 (s, 1H, H-3), 7.84 (d, 1H, H-4’), 7.87 (d, 1H, H-8’), 8.11 (s, 1H, H-1’), 8.14 (s, 1H, H-2), 8.9 (d, 1H, H-5); J5’,7’=2.0Hz, J7’,8’=9.4Hz, J3’,4’=8.6Hz, J5,6=7.2Hz.
【0071】
実施例1(s)−4-[6-(ジメチルアミノ)-2-ナフチル]-2-(メチルアミノ)-ニコチノニトリルの調製
【化25】
上の実施例1(j)に記述されているように調製された2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(194.5 mg, 0.63 mmol)がメチルアミンのエタノール溶液(5 mLの33%溶液)に懸濁され、還流で1時間加熱された。さらに5 mLのメチルアミンのメタノール溶液が加えられ、加熱が9時間続けられた。冷却後、4-[6-(ジメチルアミノ)-2-ナフチル]-2-(メチルアミノ)-ニコチノニトリル(147 mg, 77%)が濾過によって得られた。追加量の生成物(40 mg)が母液から放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中1%のMeOH)によって単離され、全収率は98%になった。mp. 193〜194℃.元素分析、C19H18N4に対する計算値: C, 75.470; H, 6.00; N, 18.53. 実測値:C, 75.28; H, 5.99; N, 18.48. IR (KBrペレット): 2210 cm-1 (CN), 3270 cm-1 (NH); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.12 (d, 3H, MeNH, J=4.8Hz), 5.38 (bs, 1H, NH), 6.76 (dd, 1H, H-5’, J=5.3Hz), 6.92 (d, 1H, H-5, J=2.5Hz), 7.20 (dd, 1H, H-7, J=2.5 and 9.1Hz), 7.56 (dd, 1H, H-3, J=1.9及び8.6Hz), 7.73 (d, 1H, H-4, J=8.6Hz), 7.78 (d, 1H, H-8, J=9.1Hz), 7.94 (d, 1H, H-1, J=1.9Hz), 8.29 (d, 1H, H-6’, J=5.3Hz).
【0072】
実施例1(t)−7-(ジメチルアミノ)-3-(4-フルオロベンゾイル)-2H-1-ベンゾピラン-2-オンの調製
【化26】
600 mg (1.6 mmol)のKryptofix 2.2.2と87 mg (1.5 mmol)のフッ化カリウムが1 mLの水と3 mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させた。1 mLのアセトニトリルを加え、混合物を蒸発させた。このスープが二回繰り返された。2 mLの無水ジメチル・スルホキシド中の170 mg(0.5 mmol)の3-(4-ニトロベンゾイル)-7-(ジメチルアミノ)- 2H-1-ベンゾピラン-2-オン(上の実施例1(g)で記述されているように調製)が加えられ、混合物は150℃で1時間加熱された。混合物は冷却され、10 mLの水で希釈され、固体相抽出(いくつかのC18 Sep Pakカートリッジ)で抽出され、ジクロロメタンで溶出され、生成物はジクロロメタン:メタノールによるカラム・クロマトグラフィーによって単離され、17 mg (0.5 mmol, 10%)の黄色固体が得られた。1H NMR (360MHz, CDCl3) δ:3.15 (6H, s, NMe2), 6.53 (1H, d, 芳香族H), 6.67 (1H, dd, 芳香族H), 7.13 (2H, m, 芳香族H), 7.41 (1H, d, 芳香族H), 7.87 (2H, m, 芳香族H), 8.14 (1H, s, 4-H). 19F NMR (CDCl3) δ: 34.7 ppm.
【0073】
実施例1(u)−2-{1-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]エチリデン}マロノニトリル
【化27】
233 mg (1 mmol)の3-アセチル-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(上の実施例1(g)に記述されているように調製)と70 mg (1 mmol)のマロノニトリルが2 mLのピリジンに溶解され、100℃で6時間加熱された。溶媒を蒸発させ、生成物がカラム・クロマトグラフィー(シリカゲル、ジクロロメタン)によって単離された。225 mg (81%)のオレンジ色の固体。
1H NMR (360MHz, CDCl3) δ:2.67 (3H, s, Ac); 3.15 (6H, s, NMe2), 6.51 (1H, br, aromatic H); 6.67 (1H, dd, aromatic H); 7.39 (1H, d, aromatic H), 7.95 (1H, s, 4,-H).
【0074】
実施例1(v)−7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-onesの調製
【化28】
7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを調製する合成アプローチは上の実施例1(g)においてジメチルアミノ類似体に関して記述されたアプローチと大体同様である。3-[(ヒドロキシエチル)(メチル)アミノ]フェノールは商業的に入手できないので、触媒としてのホウ酸の存在下でのレソルシノールと対応するアミンから調製する単純な方法を用いることができる。この合成手順は、また、N,N-ジ置換された3-アミノフェノールを選択したアミノ基で合成することを可能にする。
【0075】
30 g (272mmol)のレソルシノール、25 mL (311 mmol)の2-(メチルアミノ)-エタノール、及び2.0 g (32 mmol)のホウ酸が、分画カラムを備えた3首フラスコの中で還流で加熱された。9時間の過程で水が蒸留除去され、内部温度は180℃から230℃までゆっくりと上昇した。約60℃まで冷却した後、35 mLのメタノールを加え、ホウ酸メチルとメタノールの混合物をカラムでゆっくりと蒸留させた。過剰な2-(メチルアミノ)エタノールは15 mmHgで蒸留され、残ったオイルは高真空(0.4 mmHg)の下で蒸留された。少量のレソルシノールが155〜165℃までの間で蒸留され、165から175℃までの間で3-[(ヒドロキシエチル)(メチル)アミノ]フェノールが蒸留された。収量:32.5 g (215 mmol, 79%).
【0076】
3-[(ヒドロキシエチル)(メチル)アミノ]フェノールは、POCl3とDMFから0℃で調製されたVielsmeier試薬(4 mol当量)に加えられ、混合物は60-70℃で1時間加熱された。反応混合物は氷に注がれ、酢酸エチルで抽出された。有機部分を蒸発させ、残渣を希アンモニアで処理してヒドロキシエチル基を脱ホルミル化(de-formylate)した。中和して酢酸エチルで抽出した後、生成物をカラム・クロマトグラフィー(シリカゲル、クロロホルム)によって単離した。
【0077】
7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを生成するため、5 mmolの4-[(ヒドロキシエチル)(メチル)アミノ]-2-ヒドロキシベンズアルデヒドと6 mmolの適当なジカルボニル化合物が10 mLの無水エタノールに溶解された。20滴のピペリジンを加え、混合物を還流で2〜6時間加熱した。混合物をフリーザーで冷却し沈殿を濾過して取り、エタノールで洗浄した。
【0078】
実施例1(w)−2-(1-{7-[(2-ヒドロキシエチル)(メチル)アミノ]-2-オキソ-2H-1-ベンゾピラン-3-イル}エチリデン)マロノニトリルの調製
【化29】
1 mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンと1 mmolのマロノニトリルを2 mLのピリジンに溶解し、100℃で6時間加熱した。溶媒を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、ジクロロメタン)によって単離した。
【0079】
実施例1(x)−2-[(3-アセチル-2-オキソ-2H-1-ベンゾピラン-2-イル)(メチル)アミノ]エチル-4-メチルベンゼンスルフォネートの調製
【化30】
2 mLの無水ピリジン中で1 mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを3 mmolのp-トルエン無水スルフォン酸と室温で混合した。2時間後、混合物を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、酢酸エチル)によって単離した。
【0080】
実施例1(y)−2-[[3-(2,2-ジシアノ-1-メチルビニル)-2-オキソ-2H-1-ベンゾピラン-7-イル](メチル)アミノ]エチル4-メチルベンゼンスルフォネートの調製
【化31】
2 mLの無水ピリジン中で1mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを3mmolのp-トルエン無水スルフォン酸と室温で混合した。2時間後、混合物を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、酢酸エチル)によって単離した。
【0081】
実施例1(z)−3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンの調製
【化32】
2mmolのKryptofix 2.2.2と2 mmolのフッ化カリウムが1mLの水と3mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させ、1mLのアセトニトリルに再溶解して蒸発させた(3回)。2mLのアセトニトリル中の1mmolの2-[(3-アセチル-2-オキソ-2H-1-ベンゾピラン-7-イル)(メチル)アミノ]エチル-4-メチルベンゼンスルフォネートを加え、混合物を90℃で20分間加熱した。混合物を蒸発させ、生成物をカラム・クロマトグラフィーによって単離した。
【0082】
実施例1(aa)−2-(1-{7-[(2-フルオロエチル)(メチル)アミノ]-2-オキソ-2H-1-ベンゾピラン-2-イル}エチリデン)マロノニトリルの調製
【化33】
2 mmolのKryptofix 2.2.2と2 mmolのフッ化カリウムが1 mLの水と3 mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させ、1 mLのアセトニトリルに再溶解して蒸発させた(3回)。2 mLのアセトニトリル中の1 mmolの2-[[3-(2,2-ジシアノ-1-メチルビニル)-2-オキソ-2H-1-ベンゾピラン-7-イル](メチル)アミノ]エチル-4-メチルベンゼンスルフォネートを加え、混合物を90℃で20分間加熱した。混合物を蒸発させ、生成物をカラム・クロマトグラフィーによって単離した。
【0083】
実施例2
脳組織及びラットの脳を用いて、in vitro 及びin vivoでのβ-アミロイド斑の検出とラベリングが以下の手順に従って行われた。
【0084】
2.1 mg/mLのDDNPストック溶液が調製され、100%エタノールで8 mMに調整された。DDNP作業溶液はこのストック溶液を蒸留水で1:100〜1000(ストック溶液:蒸留水)という比に希釈して調製された。
【0085】
β-アミロイド250μM(蒸留水中で1.25 mg/mL)を37℃で48時間凝集させた。スライドに5μLをなすりつけ、空気乾燥し、再び蒸留水で水和させた。あるいはまた、Aβ-ポジティブな脳組織切片を蒸留水で再水和させた。DDNP作業溶液が各スライドに室温で30分間塗布された。スライドは蒸留水で5分間、3回洗浄された。スライドは蛍光保護マウンディング・メディア(VectashieldTM, Vector Labs., Burlingame, Californiaから入手可能)と共にカバーガラスをかぶせて、チオフラビンS又はFITCフィルターを用いて蛍光顕微鏡の下で観察された。
【0086】
β-アミロイド250μM(蒸留水中で1.25 mg/mL)を37℃で48時間凝集させ、塗抹によって確認される繊維(fibrils)を生成した。3匹のラットが麻酔された。3μLのAβ繊維(1.25 μg/μL)が各ラットの皮質に片側性で注射された(Bregma 0, AP -4.1 mm, ML + 2.0 mm, DV -3.1 mm)。次に、ビヒクル対照として、3μLのリン酸緩衝生理食塩水(PBS)が各ラット脳の反対側に注射された。注射後、針を5分間残して還流を防ぎ、その後頭蓋の孔を骨ろうで封止した。ラット脳にβ-アミロイドを注射してから8日後、ラットに10マイクロリットルのDDNP作業溶液を注射した(320マイクロモル)。これは、DDNPストック溶液をリン酸緩衝生理食塩水、pH 7.2),で1.5% BSA (ブタ血清アルブミン)に希釈して調製されたものである、pH 7.2)。
【0087】
1時間後、ラットにPLP固定液(0.05 Mリン酸緩衝液中4%パラホルムアルデヒド、1%ライシン、pH 7.4)を心臓灌流させた。ラット脳の追加浸漬固定がラットPLP固定液を用いて4℃で一晩行われた。ラット脳はPBSで洗浄され、10及び20%スクロースで飽和され、液体窒素によって冷却されたイソペンタン(-70℃)中でスナップ凍結された。脳は針の通路のまわりで10μMで凍結切片にサレ、グリセリンと蛍光保護剤(VectashieldTM)と共に直接カバースガラスがかぶせられた。脳切片は蛍光顕微鏡で観察された。
【0088】
図1Aから1Fまでは、AD患者及びトランスジェニック・マウスの脳からの切片におけるラベルされたアミロイド斑を示しており、DDNPがアミロイド斑をラベルできることがはっきりと示されている。図2Aから2Eまでは、ラベルされたβ-アミロイド斑を示し、DDNPがラットにおける血液−脳関門を通過することをはっきりと示している。
【0089】
DDNPは、チオフラビンSと同程度の感度レベルでAD脳組織の凍結切片及びパラフィン切片におけるアミロイド沈着を容易にラベルするということが見出された。DDNPを使用することは、チオフラビンSに比べていくつかの利点がある。すなわち、DDNPの使用は予備処理を必要とせず、チオフラビンSと異なり、最小の洗浄で、ホルマリン又はパラホルムアルデヒドによる固定なしで、また組織の区別なく使用できる。ストック溶液はフリーザーに6ヶ月保存しても1/100から1/1,000の希釈で受容できる結果が得られ、チオフラビンSラベリングのようにストックを新しく作る必要がない。
【0090】
実施例3
in vivoでのヒトβ-アミロイド斑と神経原繊維変化のラベリングが以下の手順を用いて行われた。
【0091】
患者を断層撮影装置に入れられて脳の動的PET画像を撮影した。実施例1(f)で記述されているように調製された8.0 mCiの2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)(比放射能:5〜12 Ci/マイクロモル;質量:約1ナノモル)が、患者の腕に静脈注射された。動的に取得された脳画像のデータが47の脳平面で2時間、同時に記録された。
【0092】
[F-18]FDDNPは、血液−脳関門を容易に通過し、ベータアミロイド斑と神経原繊維変化の存在と矛盾しない仕方で脳構造をラベルすることが見出された。この患者は、脳萎縮をモニターするために、以前に18F-フルオロデオキシグルコース(FDG)/陽電子断層撮影(PET)スキャン,並びにMRIスキャンを受けていた。MRIスキャンにおいて最大の萎縮が認められていた部位(下方側頭部と頭頂葉)で、[F-18]FDDNPラベルの最大の蓄積が観察された。これらの部位では低グルコース代謝(EDG/PETスキャンで測定される)も認められた。
【0093】
実施例4
in vivoでのヒトβ-アミロイド斑と神経原繊維変化のラベリングが以下の手順を用いて行われた。10人のヒト被験者、すなわち、7人のアルツハイマー病患者(71〜80歳まで)と3人の対照患者(62〜82歳まで)、が調べられた。患者はEXACT HR + 962断層撮影装置(Siemens-CTI, Knoxville, Tennessee)に仰臥で、画像面がorbito(眼窩)-meatal lineと平行になるように配置された。静脈カテーテルを取り付け、ヒト血清アルブミン中(25%)の[F-18]FDDNP(5〜10 mCi)が静脈カテーテルを経由する巨丸剤(bolus)として投与された。[F-18]FDDNPの投与直後から始めて、次のスキャン・シーケンスを用いてシーケンシャル放出スキャンが得られた:すなわち、6回の30秒スキャン、4回の3分スキャン、5回の10分スキャン、及び3回の20分スキャン、である。2人の被験者で取り付けられているカテーテルによって迅速な静脈血サンプリングを行って、インプット機能の測定と血漿中の代謝産物分析を行った。
【0094】
図3Aは、アルツハイマー病患者の海馬−扁桃体−内側嗅領−側頭皮質領域を通る脳断面のPET-[F-18]FDDNP (2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン)画像である。この画像は[F-18]FDDNPの注射後、30〜60分までに得られた走査。データから再構築された。同時に記録されたこの患者のPET-FDG (FDGとは2-[F-18]フルオロ-2-デオキシ-D-グルコースである)及びMRI (プロトン緩和時間)画像も、それぞれ図3Bと3Cに示されており、それぞれこの断面でのグルコース代謝と解剖学的構造についての情報を提供する。内側側頭領域は、[F-18]FDDNPスキャンでは暗く(遅いクリアランス)、FDGスキャンでは明るく(低いグルコース代謝)見える。
【0095】
図4は、アルツハイマー病患者における[F-18]FDDNPの推定滞留時間が、対照患者における値と異なるように見られることを示している。示された滞留時間は、アルツハイマー病の病理との関連が小さいと考えられる部位である脳橋における値を基準としている。滞留時間は、影響がある関心領域(ROI)と脳橋におけるクリアランス速度(rate)から次のように計算される:
滞留時間=[1/影響があるROIのクリアランス速度]−[1/脳橋のクリアランス速度]
内側嗅領皮質、海馬、外側側頭皮質、脳橋で別々のROIsが定められた。クリアランス速度が最も遅い領域が図4における滞留時間の計算で影響があるROIとして用いられた。
【0096】
静脈注射の後、[F-18]FDDNPは血液−脳関門を血流量に比例して容易に通過することが見出された。放射能の蓄積の後、領域によって異なる[F-18]FDDNPのクリアランスが続いた。ゆっくりしたクリアランスはβ-アミロイド斑と神経原繊維変化を蓄積することが確実に知られている脳部位で認められた、具体的には、海馬−扁桃体−内側嗅領コンプレックス、並びに、疾病がさらに進んだ状態では側頭葉及び頭頂葉皮質、に認められた。これらの被験者においてPETで測定されたrCMRGIも、予期されるβ-アミロイド斑の量、及び神経原繊維変化の存在の可能性、と矛盾していなかった。これらの患者で、低グルコース代謝の脳領域は一般に[F-18]FDDNPの高い保持率の領域と一致していた。海馬−扁桃体−内側嗅領皮質は、ほとんどの場合、症状が重篤でない患者においても、放射能([F-18]FDDNP)の高い保持率を示した。正常な82歳のボランティアは、図4に示されるように、[F-18]FDDNPによるPET研究では海馬−扁桃体−内側嗅領コンプレックスにおける放射能の沈着(deposition)を示し、同じ部位でFDGによる測定では低いrCMRGIを示した。これらの結果は、痴呆の明らかな兆候がない高齢者が内側嗅領皮質のニューロンの第二層で神経原繊維変化の病理、及び海馬形成体で斑を示すことがあるという観察と矛盾しない。症状の重篤化には、常に放射能の保持率の増加と側頭、頭頂葉、又は前頭皮質からのクリアランスの遅れが伴っており、これらの部位において予期されるAβと神経原繊維変化の沈着(deposition)と一致している。
【0097】
アルツハイマー病患者の脳試料についての[F-18]FDDNPを用いたin vitroオートラジオグラフィーも、β-アミロイド斑と神経原繊維変化の存在と合致する放射能の分布を示した。Aβ及びタウ抗体の免疫染色による結果と合致する結合が海馬、側頭、頭頂葉皮質で見られた。DNNPとその誘導体は蛍光を示すので、[F-18]FDDNPがin vitroでβ-アミロイド斑及び神経原繊維変化をラベルする能力の評価も同じ脳試料について行われた。すべてのアルツハイマー病の脳試料で、神経原繊維変化、アミロイドペプチド、及び拡散したアミロイドが、DDNPと[F-18]FDDNPの両方によってはっきりと目に見えるように視覚化され、同じサンプルについてのチオフラビンS (24)による結果と一致した。
【0098】
図5で、中央の画像は、ATS(antiphosphotau)と10G4(anti-AB1-15)によってインキュベートしたあるアルツハイマー病患者の45マイクロメーターの低温側頭皮質切片を免疫染色して得られた1 : 800の図である。挿入図は、FDDNPで染色された同じアルツハイマー病の脳試料の隣接する切片である、画像は蛍光顕微鏡を用いて生成された。グリーンの矢印は、中央の免疫染色切片に対する挿入図の大体の原位置を示す。挿入図は、左上隅から時計回りに、(1) 老人斑、(2) 拡散した斑、(3) 血管性アミロイド、(4) 密な斑と神経原繊維変化、及び(5) 密な神経原繊維変化、を示す。
【0099】
実施例5
考えられる治療物質が評価され、β-アミロイド原繊維に結合する能力が決定された。
β-アミロイド(1-40)原繊維形成
β-アミロイド(1-40)(Biosource, Camarillo, CA)原繊維が調製された。0.5 mgのβ-アミロイド(1-40)が1 mL PBS, pH 7.4, に溶解され、磁気攪拌棒で3日間37℃で攪拌され、目で見て濁った溶液が得られた。原繊維は、その生成が確認された直後に用いられた。β-アミロイド原繊維の生成はJeol 100CX透過電子顕微鏡(Jeol, Peabody, MA)による画像撮影で確認された。一滴の5μLの原繊維溶液を処理された銅のグリッド上で30 秒間落ち着かせた後に一滴の2%酢酸ウラニル溶液で洗い流した。最後に、別の一滴の2%酢酸ウラニルをグリッドに加えて原繊維をネガティブに染色(negatively stain)した。原繊維は、その生成が確認された直後に用いられた。Congo red (CR, Sigma)とチオフラビンT (TT, Sigma)を用いて原繊維の形成の別のテストも行われた。以下で述べるin vitro競合分析における濾液に原繊維が存在しないこともCRを用いた同じテストによって決定された。
【0100】
[18F]FDDNPによるNSAIDs及びチャージアミロイド色素に対するin vitro 競合分析
各放射性競合分析のために、非放射性物質、すなわち、(S)-ナプロキセン(Sigma)、(R)-イブプロフェン(BIOMOL, Plymouth Meeting, PA)、(S)- イブプロフェン(Sigma)、ジクロフェナック(Sigma)、CR 及びTT、が新たに調製された。ステンレス鋼サポート・スクリーン(Millipore)とガラスサンプルチャンバで改造された1225サンプリング・マニホールド(Millipore)でAPFFグラスファイバーフィルター(0.7μm粒子保持;Millipore, Bedford, MA)が用いられた。真空濾過では、PBS, pH 7.4 (1%エタノール)、で1時間インキュベートされた合成β-アミロイド(1-40)の0.865μg/mLのin vitro原繊維と37 MBq/mLの[18F]FDDNPを、0.1 pMから83 μMまでのいろいろな濃度の非放射性物質と共に用いた。次に、各フィルターを3 mlのPBS, pH 7.4, で2回洗浄した。フィルターに残された放射能を、Packard Cobra II Auto-Gammaガンマカウンター(Packard, Meriden, CT)で測定し、共通基準時間に減衰補正(decay-corrected)した。競合相手なしの放射性ラベルされた[18F]FDDNP のβ-アミロイド原繊維への結合は100%特異的結合と決定された。放射性[18F]FDDNPに対する100%競合は40μMの非放射性FDDNPで見られると仮定された。すべての競合分析は三重重複で行われた。競合結果が分析され、Ki値がLigand Binding Module for SigmaPlot 2001 (SPSS, Chicago, IL)を用いて計算された。
【0101】
脳組織の調製
79歳の女性の死後診断で確定されたAD患者からの脳検体が処理された。簡単に言うと、ホルマリン処理された、低温保存された脳検体が、冠状で70μm厚さの切片に切断され、ゼラチン塗布されたガラス・スライドに載せられ、放置乾燥され、キシレン中で40分間脱脂された後、エタノールで組織が洗浄された。最後に、いくつかの脳検体でリポフスチン自己蛍光は、50 mM酢酸アンモニウム緩衝液、pH 5、中の10 mM CuCl2を用いて停止させてから染色した。
【0102】
ディジタル・オートラジオグラフィー
死後診断で確定したADの脳検体(厚さ70μm)が、100 nMの新しいバッチの非放射性FDDNP及び(S)-ナプロキセン又は40μMの(R)-イブプロフェン、(S)-イブプロフェン、ジクロフェナック、CR又はTTによって、PBS, pH 7.4, 中の10%エタノールの中で60分間プレ処理され、液体を流して空にしてから[18F]FDDNPによるディジタル・オートラジオグラフィーを行った。前処理された競合相手なしの凍結切片が室温で25分間、凍結切片あたり10 mLの0.9%(w/v)塩水中の1%エタノールに溶解された3.7 GBqの[18F]FDDNPと共にインキュベートされた。インキュベーションの後、切片は、水(30 sec);区別化のために(Bancroft and Stevens, 1990)Junior Orbit Shaker (Lab-Line Instrujents, Melrose Park, IL)で40 RPMsで攪拌された60% 2-メチル-2-ブタノール(3 min; Sigma);その後で水(30 sec)によって最適に洗浄された。切片は、暖かいホットプレートで、暖かい空気の定常的な流れによって乾燥され、β+-敏感なリンプレートに40分間さらし(Fuji Film Medical Systems USA, Stanford, CT)、以前に記述されたように(Agdeppa et al., 2001a)FUJI BAS 5000 Phosphrimager (Fuji)で25μmという分解能でスキャンされた。画像が撮影された検体からの組織削り落としからの放射能がその後Packard Cobra II Auto-Gamma (Packard)によって測定され、共通基準時間に減衰され、オートラジオグラムにおける[18F]FDDNPの特異的結合の量(放射能/面積、Bq/mm2, 図2Q)を定量化するための放射性基準として用いられた。オートラジオグラフィーは、各競合相手について少なくとも三重重複で行われた。オートラジオグラムの統計解析は、Prism 3.02 (GraphPad, San Diego, CA)を用いてDunnettのポストテストによる一元配置分散分析(one-way ANOVA)を行って、非放射性物質でプレ処理されたオートラジオグラムとプレ処理なしのオートラジオグラムで、灰色物質対白色物質(図2Q)[18F]FDDNP放射能の比の差を比較した。対応のないt-テストを行って、各オートラジオグラムの灰色物質と白色物質における組織の面積あたりの測定された[18F]FDDNP放射能の差を比較した。
【0103】
蛍光顕微鏡
オートラジオグラフィーに用いたと同じ脳検体を蛍光顕微鏡を用いて観察した。組織はVectashield (Vector, Burlingame, CA)によってマウントし、Nikon Labophot 蛍光顕微鏡(Nikon USA, Melville, NY)でFITCフィルターセットを用いて観察した。
【0104】
[18F]FDDNPによるオートラジオグラフィーに以前に用いた組織の蛍光顕微鏡観察は、FDDNPの蛍光性質と残っている非放射性FDDNPによるSPのラベリングによって可能になる。合成の終わりでの非キャリア付加[18F]FDDNPの比放射能(単位質量当たりの放射能)は74〜222 GBq/μmol (2000〜6000 Ci/mmol)であり、最大理論的比放射能よりも103倍も小さい(SorensonとPhelps, 1987)。したがって、18Fの減衰後、残っているAD 脳検体におけるSPsと結合した非放射性FDDNPを蛍光顕微鏡で画像として見ることができる。
【0105】
上記のテストは、ナプロキセンとイブプロフェンがβ-アミロイド原繊維上で[18F]FDDNPと同じ結合部位を共有していることを示した。いろいろな濃度の非放射性物質を[18F]FDDNPと合成β-アミロイド原繊維と共に同時インキュベートして行われたin vitro競合におけるカーブは、(S)-ナプロキセン、(R)-イブプロフェン、及び(S)-イブプロフェン、に関する一部位結合競合を明らかにした(p=0.05)。(S)-ナプロキセン、(R)-イブプロフェン、(S)-イブプロフェン、に対する[18F]FDDNPの結合の濃度依存的な減少は、Ki値として、それぞれ、Ki=5.70±1.31 nM (±SD), Ki=44.4±17.4 μM (±SD), 及びKi=11.3±5.20 μM (±SD), という値を与え、(S)-ナプロキセンがβ-アミロイドによりしっかりと結合することを示した。ジクロフェナック、CR, TT, は[18F]FDDNPの特異的結合において量依存的な減少を示さなかった。
【0106】
さらに、ナプロキセン及びイブプロフェンによるオートラジオグラフィーは、ex vivoでの老人斑における[18F]FDDNP結合部位の完全なブロックを示した。競合相手が内場合の前頭AD脳検体の放射能の大まかなパタンは、老人斑を含む領域への[18F]FDDNPの特異的結合を明らかにした。老人斑を含む灰色物質の領域への[18F]FDDNPの特異的結合は、非放射性FDDNP、(S)-ナプロキセン、(R)-イブプロフェン、及び(S)-イブプロフェン、によるプレ処理されたAD検体において、これらの競合相手がない場合のオートラジオグラフィーに比べて著しく減少した。これらの競合相手がある場合、老人斑がある灰色物質領域と老人斑を欠く白色物質の間の放射能の差(放射能/面積、Bq/mm2;図2Q)は有意でなく(p > 0.05)、灰色物質対白色物質の放射能の比が、競合相手がない場合の[18F]FDDNPオートラジオグラムにおけるこの比に比べて有意に低い(p < 0.05)として矛盾がなかった。これらの結果は、[18F]FDDNPの特異的結合が白色物質に見られる放射能のバックグラウンドレベルまでブロックされることを示している。これらの結果はすべて同じ脳検体についての蛍光顕微鏡観察によって確認された。ジクロフェナック、CR及びTTは、同じ脳検体のオートラジオグラフィーと蛍光顕微鏡観察から判断して、老人斑への[18F]FDDNPの特異的結合に対する影響は最小であった。灰色物質と白色物質の放射能の差は、ジクロフェナック、CR及びTTに関して有意に差があり(p < 0.05)、競合相手がない場合の[18F]FDDNPオートラジオグラフィーと同様であった。ジクロフェナック、CR及びTTの間での灰色物質対白色物質の放射能の比と、競合相手がない場合の[18F]FDDNPでの比との比較は、老人斑への[18F]FDDNPの特異的結合が保存されることを示す。
【0107】
本発明の方法に関する詳細は、本明細書の付録Aと付録Bに与えられており、そこに開示されたことは参照によって本明細書に組み込まれる。
【0108】
より広い様態で、本発明は、ここで示され説明された特定の細部に限定されない。本発明の原理から外れることなく、その主な利点を失うことなく、そのような細部に変更を加えることができる。
【図面の簡単な説明】
【0109】
図面の説明
本発明のこれらの及びその他の特徴と利点は、以下の詳細な説明を添付の図面と合わせて参照することによってさらに良く理解されるであろう。添付図面のうち:
【図1】図1Aは、アルツハイマー病患者の脳皮質におけるラベルされたアミロイド斑の2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)蛍光(ex 490 nm, em 520〜530 nm)を示す(X400)。図1Bは、アルツハイマー病患者の脳皮質における斑の強いDDNPラベリングともつれの弱いDDNPラベリングを示す(X640)。図1Cは、ヒトの脳におけるアミロイドコアを有する単一の大きな斑のDDNPラベリングを示す(X640)。図1Dは、Tg2576HuAPPswトランスジェニックマウスの脳における斑のDDNPラベリングを示す(X500)。図1Eは、アルツハイマー病の人の脳におけるコアを有する斑のチオフラビンSラベリングを示す(X640)。図1Fは、図1Eに示されたと同じ人の脳切片のアミロイドβ蛋白質の4G8抗体ラベリングを示す(X640)。
【図2】図2Aは、ラットの脳に注射されたアミロイドのラベリングを示しており、ある分量のβ-アミロイド1-40を8日間37℃で放置して凝集させ、ゼラチンを塗ったスライドに乾燥させ、DDNPによってラベルし、アミロイドに合致する繊維上の蛍光がはっきりと示された。図2Bは、ラットの脳に注射されたアミロイドのラベリングを示しており、ラットの皮質に3 μgの凝集したβ-アミロイド1-40を一側性定位で注射して8日後、ラットに100μLの640μM DDNPを頸動脈に注射し、麻酔し、20分後に灌流によって屠殺し、脳を凍結切片にして蛍光を調べた:図2Bはneed trackの先端にin vivoでDDNPによって蛍光ラベルされたアミロイドをはっきりと示している(X100)。図2Cは、図2Bのin vivo DDNPラベルされた材料の高倍率図を示す(X200)。図2Dは、注射部位による切片のギ酸処理が蛍光ラベリングを除去する様子を示す(X100)。図2Eは、アミロイドが存在しないアミロイド注射部位の反対側でDDNPラベリングが弱いことを示す(X200)。
【図3】図3Aは、アルツハイマー病患者の海馬−扁桃体−内側嗅領/側頭皮質領域を通る脳断面のPET-[F-18]-FDDNP(2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン)画像である。図3Bは、図3Aの脳断面のPET-FDG(FDGは2-[F-18]フルオロ-2-デオキシ-D-グルコースである)画像である。図3Cは、図3Aの脳断面のMRI画像(陽子緩和時間)である。
【図4】図4は、患者における[F-18]-FDDNPの推定滞留時間を示すグラフである。
【図5】図5は、1:800のAT8(anti-phosphotau)と10G4(anti-AB1-15)で培養されたアルツハイマー病患者の45マイクロメーター低温側頭皮質切片を免疫染色することによって得られた画像(中央画像)を示す。挿入図は、FDDNPで染色された同じアルツハイマー病の脳検体の隣接切片であり、左上隅から時計回りに、(1)老人斑(neuritic plaque)、(2)拡散した斑、(3)血管性アミロイド、(4)レンズ斑ともつれ、及び(5)密なもつれ、を示す。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3A】
【図3B】
【図3C】
【技術分野】
【0001】
政府補助に対する謝辞
本発明は、米国エネルギー省による補助金No.DE-FC0387-ER60615の下で、政府の補助によってなされた。政府はこの発明に一定の権利を有する。
【0002】
関連出願への相互参照
本出願は、2003年5月20日に出願された米国特許出願No. 60/471,945の優先権を主張するものであり、その出願の内容全体は(付録AとBを含めて)、全体が記載されたと同様に参照によって本明細書に組み込まれる。
【背景技術】
【0003】
発明の背景
アルツハイマー病は、米国やその他の工業国において最も急速に増加している年齢層である80歳以上の人口の約20〜40%の人たちがかかる病気である。アルツハイマー病患者の脳によく見られる特徴として、多量のニューロン内神経原繊維変化(NFT)と細胞外のアミロイドに富むβ-アミロイド斑などがある。NFTsは、主として過剰にリン酸化されたタウ蛋白質が対らせんフィラメントと呼ばれる周期的に制限されたアミロイド繊維の形に組み合わされた凝集体から構成される。アミロイド斑の主要な成分はペプチドであり、これはもっと大きなアミロイド前駆蛋白質の開裂によって生成される長さが39−43アミノ酸の小さなβ-アミロイドペプチドである。しかし、ほとんど全くB-アミロイドから成るぼやけた(diffuse)斑を除き、アミロイド斑は多数の細胞生成物を含む複雑な病変である。42アミノ酸形態のこのペプチドの産生を増やす突然変異はアルツハイマー病の常染色体優性家族形態と遺伝的にリンクしている。β-アミロイドの沈着は疾病過程の非常に早い時期に、臨床的な症状が現れるよりもずっと前に起こっている。この突然変異は病原性があり遺伝子導入マウスにアルツハイマー病を生ずるように見えるので、β-アミロイドはこの病気において原因としての役割を果たしていると広く信じられている。アミロイドの沈着が原因であるかどうかに関わりなく、それが診断の重要な部分であることは確かである。さらに、アミロイド斑は疾病の初期に見られるので、沈着を画像化できる能力は、早期の診断と疾病の予防のための好適なマーカー、並びに治療方式の有効性をモニターする方法を提供する。
【0004】
アルツハイマー病は、現在、死後の脳から切片を採取して新皮質のアミロイド沈着を定量化することによって確定的に診断される。残念ながら、アミロイド沈着及び/又はNFTsを検出する現行の方法は、死後の分析又は生検分析を必要とする。例えば、in vitroの脳切片におけるアミロイドのチオフラビン蛍光標識法は脳の評価のために現在広く用いられている方法である。別の可能なアミロイド・プローブとして、Chrysamine-G、コンゴーレッド誘導体、も開発されている。コンゴーレッドは荷電分子であり、したがって、血液−脳関門を越えて拡散するのに十分な疎水性を欠いており、in vivoラベルとして用いることができない。K;unk et al, Neurobiology of Aging, 16: 541-548 (1995), 及びPCT Publication No. WO 96/34853,を見よ。Chrysamine-Gは、コンゴーレッドよりも良く血液−脳関門を通過するが、アルツハイマー病の脳のアミロイド斑をラベルする能力は弱いように見える。例えば、H. Han, C-G Cho and P. T. Lansbury, Jr., J. Am. Chem. Soc. 118, 4506 (1996); N. A. Dezutter et al., J. Label. Compd. Radiopharm, 42, 309 (1999), を参照のこと。同様に、β-アミロイドのin vivo画像化のためのプローブとしてモノクローナル抗体を用いる初期の試みは、それが血液−脳関門を通過する能力が限られていることによって妨げられた。R. E. Majocha et al., J. Nucl. Med. 33, 2184 (1992) を見よ。最近では、血液−脳関門を通過できる1251-Aβ 1-41のモノビオチン化複合体の利用も提案されているが(Y. Saito et al., Proc. Natl. Acad. Sci. USA 22, 2288 (1991)を見よ)、それがin vivoでβ-アミロイド及び/又はNFTsをラベルする能力はまだ実証されていない。アミロイドの沈着をin vivoで定量化することは現在利用できるプローブによっては不可能である。したがって、アルツハイマー病の早期診断のための好適なマーカーに対するニーズが存在している。
【0005】
陽電子放出型断層撮影(PET)による局所脳グルコース代謝率(rCMRGI)のin vivoの、非侵襲的測定は、アルツハイマー病患者における脳機能の評価のための重要な手段になっている。2-[F-18]-2-デオキシ-D-グルコース(DFG)を用いた多くの研究は、側頭頭頂及び前頭連合野における代謝低下の特徴的な代謝パタンをはっきりと示している。これらの研究のうちのいくつかは、rCMRGIと死後の局所ニューロン病理とを比較した。それらの結果、及びアルツハイマー病の病原カスケードの不確かさは、これらの患者におけるアミロイドと神経原繊維の沈着をin vivoで非侵襲的に評価することの重要性を浮き彫りにしている。
【発明を実施するための最良の形態】
【0006】
発明の詳細な説明
ある実施形態では、本発明はβ-アミロイド斑と神経原繊維変化から成る群から選択される構造を、in vivo 又はin vitroでラベルする方法であって、脳組織と化学式(IA)又は(IB):
【化1】
(式中;
Qは、-NR2R3, -OR9, 及びSR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N及びCR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9, 及び-C(R9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及び-SR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、Nと-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化2】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル- R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、-C(O)O-アルキル、-C(O)O-アルケニル、-C(O)O-アルキニル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング、-OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル、-O-アルケニル、-O-アルキニル、-S-アルキル、-S-アルケニル、及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;
R9は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成する;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではない)
の化合物を接触させるステップを含む方法に向けられる。上記の化合物において、1又は複数の水素、ハロゲン、又は炭素原子を任意に放射性ラベルで置き換えることができる。
【0007】
本発明は、また、上記の方法で有用な、化学式(IA)又は(IB)の化合物を含む組成物:β-アミロイド斑と神経原繊維変化から成る群から選択される構造を、in vivo 又はin vitroでラベルする方法であって、脳組織と化学式(IA)又は(IB):
【化3】
(式中;
Qは、-NR2R3, -OR9, 及び-SR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N及びCR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9及び-C(CR9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及びSR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N及び-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化4】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル- R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング, -C(O)O-アルキル, -C(O)O-アルケニル, -C(O)O-アルキニル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素、アルキル、アルケニル, アルキニル, 環式リング, 複素環式リング, -OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル, -O-アルケニル, -O-アルキニル, -S-アルキル, -S-アルケニル, 及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R9は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素, アルキル, アルケニル, アルキニル, 環式リング、複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成する;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではなく;
さらに、一つ以上の水素原子が任意に放射性ラベルで置き換えられる)
の化合物を接触させるステップを含む方法に向けられる。
【0008】
諸定義:
本明細書で用いられる場合、“アルキル”という用語は、飽和炭素原子と水素原子の直鎖又は枝分かれ鎖の一価ラジカル、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、t-ブチル、ペンチル、及びヘキシル、などを指し、置換されていても置換されていなくてもよい。“低級アルキル”という用語は、1〜4個の飽和炭素原子と水素原子を有する直鎖又は枝分かれ鎖の一価ラジカル、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、及びt-ブチル、などを指し、置換されていても置換されていなくてもよい。
【0009】
本明細書で用いられる場合、“アルキレニル”という用語は、少なくとも一つの-(CH2)-を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、エチレニル及びプロピレニルなど、を指し、置換されていても置換されていなくてもよい。
【0010】
本明細書で用いられる場合、“アルケニル”という用語は、少なくとも一つの炭素−炭素二重結合を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、ブテニル及びペンテニルなど、を指し、置換されていても置換されていなくてもよい。
【0011】
本明細書で用いられる場合、“アルキニル”という用語は、少なくとも一つの炭素−炭素三重結合を含む炭素原子の直鎖又は枝分かれ鎖ラジカル、例えば、エチニル、プロピニル、ブチニル及びペンチニルなど、を指し、置換されていても置換されていなくてもよい。
【0012】
本明細書で用いられる場合、“環式リング(cyclic ring)”という用語は、それぞれが飽和又は不飽和の3, 4, 5, 6, 7, 8, 9, 10, 11, 又は12個のリング原子を含む単環式又は多環式ラジカル、例えばシクロブチル、シクロペンチル、シクロヘキシル、シクロヘキセニル、及びフェニルなど、を指し、置換されていても置換されていなくてもよい。
【0013】
本明細書で用いられる場合、“複素環式リング(heterocyclic ring)”という用語は、窒素、酸素、及びイオウから選択される1, 2, 3, 4, 又は5個のヘテロ原子を含む、それぞれが飽和又は不飽和の3, 4, 5, 6, 7, 8, 9, 10, 11, 又は12個のリング原子を含む単環式又は多環式ラジカルを指し、置換されていても置換されていなくてもよい。非限定的な例は、アジリジン、アゼチジン、ピロリジン、ピペリジン(piperidine)、及びピペリジン(piperizine)などである。
【0014】
上で注意したように、“アルキル、”“アルケニル、”“アルキニル、”“環式リング、”“複素環式リング”という用語は、置換されたアルキル、アルケニル、アルキニル、環式リング、及び複素環式リング・グループを含み、それらのグループは適当な置換基、例えば、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、ハロゲン、-OH, -OTs, O-アルキル、O-アルケニル、O-アルキニル、O-環式リング、O-複素環式リング、アシル、チオアシル、スルフォニル、メルカプト、アルキルチオ、アミノ、アルキルアミノ、ジアルキルアミノ、及びカルボモイル基、などによって置換できる。
【0015】
化学式(I)及び化学式(II)の化合物では、好ましくはR2とR3はそれぞれ独立にアルキルであり、さらに好ましくは低級アルキルである。化学式(II)の化合物では、好ましくはR9は低級アルキルであり、さらに好ましくはメチル又はエチル、アリール、及び置換されたアリールである。本発明に関連して用いるのに特に好ましい化合物は、2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)と2-(1,1-ジシアノプロペン-2-イル)-6-(エチル)(メチル)(アミノ)ナフタレンであり、どちらも任意に放射性ラベル標識できる。別の好ましい化合物、特にin vivoで用いるのに好ましい化合物は、2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)である。
【0016】
本発明は、また、in vitro又はin vivoでβ-アミロイド斑や神経原繊維変化などの構造を検出する方法に向けられる。“構造”という用語は、疾病病理の一部として生ずるペプチドやその他の細胞物質を含む生物物質の凝集体を指す。“ペプチド”という用語は蛋白質を含む。
【0017】
上述の化合物は約470から610 nmまでの範囲で蛍光活性を有する。ある応用では、本発明は脳組織におけるβ-アミロイド斑や神経原繊維変化をラベル標識する。すなわち、アルツハイマー病のin vitro検出のために、この化合物を脳組織に接触させ、脳組織が蛍光顕微鏡で観察される。
【0018】
in vivo検出のためには、化合物を放射性ラベル標識することが好ましい。好ましい放射性ラベルは18Fである。これは陽電子断層撮影法のための半減期が約2時間である。別の放射性ラベルは放射性ヨウ素、例えば123Iであり、シングルフォトン断層撮影法(SPECT)で用いられる。あるいはまた、他の放射性ラベル、例えば11C, 13N, 及び15O, が用いられるが、これらの放射性ラベルは比較的寿命が短いのであまり望ましくない。化合物のどの原子も適当な放射性ラベルで置き換えることができる。放射性ラベル標識は、当業者に公知のどんな方法で行ってもよい。例えば、K2CO3 (0.75 mg)中のドライ[F-18]フッ化物イオン[18O (p, n) 18F]とKryptofix 2.2.2TM (19 mg)が化学式(I)又は化学式(II)の化合物の溶液(1 mL CH3CN中4 mg)に加えられる。混合物はオイル浴で85℃に約10〜40分熱せられる。冷却して水で希釈した後、放射性ラベルされた生成物は分取HPLCによって精製される。Kryptofix 2.2.2TM はAldrich Chemical Co. (Milwauky, Wisconsin)から入手できるクラウンエーテルである。
【0019】
次に、放射性ラベルされた化合物を含む溶液が患者に注射される。本明細書で用いられる場合、“患者”という用語は、ヒト、ラット、マウス、イヌ、及びネコを含む哺乳類を指す。神経解剖的領域は、MRIスキャンを用いて手作業で、例えば、Tela磁石を用いて、次に、アミロイドPET(陽電子断層撮影)とEDG-PET(フルオロデオキシグルコース-PET)でMRIスキャンとの重ね合わせによって、決定できる。PETは現在2〜3分という分解能で脳における放射性ラベルされた化合物の沈着を動的に決定することができ、異常な区域を検出することができる。
【0020】
上述の方法によって、β-アミロイド斑や神経原繊維変化の蓄積によって特徴づけられるアルツハイマー病やその他の脳の劣化に関連した疾病を検出することができる。
【0021】
本発明は、また、患者におけるアルツハイマー病を治療又は予防する治療物質の能力を決定する方法に向けられる。“アルツハイマー病を予防する”というフレーズは、アルツハイマー病のリスクを軽減すること及び/又は発病を送らせることを含む。この方法は、β-アミロイドペプチドをその治療物質及び化学式(IA)又は(IB):
【化5】
(式中:
X1は、O, S, -NR4, -CH2, -CHアルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N-(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化6】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、アリール, 複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2, 及び-アルキレニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になってアリール又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になってアリール又は複素環式リングをA1によってR1, X3又はZ4以外の炭素のところで置換されたものを形成し;
R4は、アルキル, 置換されたアルキル, アリール, 置換されたアリール, -C(O)O-アルキル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル(O-アルキル)2, -O-アルケニル, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルケニル, 及び-S-アルキレニル- R4から成る群から選択されるラジカルであり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;任意に、アルキル, アルコキシ、OH, OTs, ハロゲン、アルキレニル-R5, カルボニル、スピペロン、スピペロンケタール、及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換され;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成する;
ここで、1又は複数の水素、ハロゲン、又は炭素原子が放射性ラベルによって置き換えられる)
による放射性ラベルされた化合物と、in vivo又はin vitroで接触させるステップを含む。
【0022】
この方法によれば、放射性ラベルされた化合物の少なくとも一部はβ-アミロイドペプチドと結合する。この方法に関連して、β-アミロイドペプチドという用語は、β-アミロイド凝集体又は繊維、並びにβ-アミロイド老人斑を含む。治療物質と放射性ラベルされた化合物は、両方を同時にβ-アミロイドペプチドと接触させることも、放射性ラベルされた化合物をβ-アミロイドペプチドと接触させる前及び/又は後に治療物質をβ-アミロイドペプチドと接触させることもできる。
【0023】
次に、β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を測定して、それにより放射能をもたない作用物質がβ-アミロイドペプチドと結合したかどうか、どの程度結合したかを決定する。ある好ましい実施形態では、放射性ラベルされた化合物だけを(すなわち、放射能のない作用物質が存在しない状態で)β-アミロイドペプチドと接触させて、100 %特異的結合を決定する。
【0024】
この方法では、治療物質の濃度を変えて、どの程度治療物質がβ-アミロイドペプチドと結合できるかを決定することができる。
【0025】
本発明の別の方法では、治療物質の抗凝集効果を評価することができる。抗凝集効果とは、治療物質がβ-アミロイドペプチドを破壊及び/又はその形成を妨害する能力を指す。この方法では、患者におけるβ-アミロイドペプチドの量が、上で一般的に述べたように放射性ラベルされた化合物を用いて決定される。その後で、治療物質がある期間、例えば1週間、1ヶ月、又はそれ以上、にわたって患者に投与され、治療物質がβ-アミロイドペプチドと接触するようにする。その期間の後、放射性ラベルされた化合物を再びβ-アミロイドペプチドと接触させて患者におけるβ-アミロイドペプチドの量を決定することができる。治療物質がβ-アミロイドペプチドの形成をどの程度妨害できるかを決定するためには、アルツハイマー病にかかりそうな、しかしこの病気があまり進行していない一人以上の患者を評価することが有効である。
【0026】
本発明は、また、患者におけるアルツハイマー病を治療又は予防する方法に向けられる。この方法は、治療的に有効な量の、上述の化学式(IA)又は(IB)による作用物質を患者に投与するステップを含む。
【実施例】
【0027】
実施例
実施例1
以下の本発明による組成物が調製された。NMRスペクトルはBruker AM 360 WB又はDPX 300スペクトロメーターで得られた。1H化学シフトは内部標準としてTMSからのppmダウンフィールド(downfield)で報告される。19F化学シフトは外部のフルオロトリクロロメタンに対して報告される。別に断らない限り、ジューテリオクロロフォルムが溶媒として用いられた。融点はElectrothermal Melting Point Apparatusで測定され、補正されなかった。元素分析は、Galbraith Laboratories, Inc., Knoxville, TX,又はMs. Metka Kastelic at the Faculty of Chemistry and Chemical Technology, University of Ljublijana, によって行われた。放射状クロマトグラフィーは、Chromatron (Harrison Research, 840 Moana Court, Palo Alto, CA 94306)で行われた。ローターはHarrison Researchが勧告したようにE. Merck Silica Gel (Cat. No. 7749-3)を用いて作成された。HPLCはAlltech Econosil C-18 5μm, 4.6x250mmカラムで、溶媒として水:アセトニトリル:トリエチルアミンの40:60:2混合物を用いて行われた。254nmにおけるUV検出が用いられた。溶媒と試薬はFisher, Aldrich又はFlukaから入手し、別に断らない限り入手した状態で用いた。
【0028】
実施例1(a)−2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)の調製
【化7】
29 mLの新しく蒸留されたヘキサメチルリン酸ヨリアミド(HMPT)中の5.26 g (117 mmol)のジメチルアミンの溶液に、31 mLの無水トルエンと780 mg (117 mmol) の小さな片のLiが加えられた。混合物は室温でアルゴンの下で1.5時間攪拌された。2-アセチル-6-メトキシナフタレンがArsenijevic et al., Org. Synth. Coll. 1988, 6:34-36に記述されたように調製された。この文献の開示内容は参照によって本明細書に組み込まれる。2-アセチル-6-メトキシナフタレン(5.57 g, 27.8 mmol) が一度に加えられ、攪拌が20時間続けられた。混合物は氷水浴で冷却されて冷たい水/酢酸エチル混合物(各300 mL)に注がれた。十分な攪拌の後、層が分離し、水層は225 mLの酢酸エチルで2回抽出された。有機抽出物を一緒にし、乾燥、蒸発させて黄色の固体が得られた。エタノールからの再結晶化によって2-アセチル-6-(ジメチルアミノ)ナフタレン(ADMAN)が黄色固体として得られ、153.5-155℃で融解した:1H NMR (CDCl3, TMS) δ2.67 (s, 3H, COCH3), 3.15 (s, 6H, N(CH3)2), 6.87 (d, 1H, H-5), 7.17 (dd, 1H, H-7), 7.63 (d, 1H, H-4), 7.80 (d, 1H, H-8), 7.92 (dd, 1H, H-3), 8.32 (bs, 1H, H-1), J1,3=2.3 Hz, J3,4=8.7 Hz, J5,7=2.4 Hz, J7,8=9.3 Hz. MS (M+) 213: 実測:213. 分析. C14H15NOで計算:C, 78.84; H, 7.09; N, 6.57. 実測:C, 78.96; H, 7.10; N, 6.45.
【0029】
20 mLのピリジン中でマロニトリル(436 mg, 6.6 mmol)とADMAN (1.278 g, 6.6 mmol)の混合物が110℃で19時間熱せられた。冷却後、残った赤色固体が100 mLの塩化メチレンに溶解され、10 gのフラッシュ・シリカget (230〜400メッシュ)に吸着され、トルエンでクロマトグラフィー分析された。適当な分画を一緒にして、蒸発させて1.12 g (72%) の2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)が得られた。ベンゼン−ヘキサンからの再結晶化によって赤色針状体が得られ、154.5〜155℃で融解した:1H NMR (CDCl3, TMS) δ2.69 (s, 3H, CH3), 3.11 (s, 6H, N(CH3)2), 6.85 (d, 1H, H-5), 7.18 (dd, 1H, H-7), 7.56 (dd, 1H, H-3), 7.66 (d, 1H, H-4), 7.76 (dd, 1H, H-8), 8.02 (d, 1H, H-1). J1,3=2.04 Hz, J3,4=9.13 Hz, J5,7=2.5 Hz, J7,8=9.11 Hz. IR (CHCl3) 2250 cm-1 (CN伸張). MS (M+) 261: 実測:262. 分析値. C17H15N3で計算:C, 78.13; H, 5.79; N, 18.08. 実測:C, 78.17; H, 5.68; N, 17.91.
【0030】
実施例1(b)−2-(1-{6[エチル-(2-{8-[4-(4-フルオロフェニル)-4-オキソブチル]-4-オキソ-1-フェニル-1,3,8-トリアザスピロ[4.5]デシ-3-イル}エチル)-アミノ]-2-ナフチル}エチリデン)マロノニトリルの調製
【化8】
還流冷却器と滴下漏斗を備えた3Lの2首丸底フラスコで、2Lの塩化水素酸(d=1.16)を攪拌し、沸騰まで加熱した。最小量のジクロロメタン中の6.06 g (30.3 mmol)の1-(6-メトキシ-2-ナフチル)-1-エタノン(Arsenijevic et al., Org. Synth. Coll. 6:34 (1988)に記述されたように調製、この開示内容は参照によって本明細書に組み込まれる)の溶液が加えられ、混合物は攪拌され還流で2時間加熱された。熱い溶液がミネラルウール栓を通して濾過されて油性の残渣が除かれた。冷却後に分離した固体がガラス・フリットで濾過され、130 mLの酢酸エチルに溶解された。溶液を塩水で洗浄し、無水硫酸マグネシウムで乾燥し、蒸発させて5 g(89%)の1-(6-ヒドロキシ-2-ナフチル)-1-エタノンが得られた。
【0031】
1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(744 mg, 3.92 mmol)、硫酸水素ナトリウム(IV)(1.66 g, 16 mmol)、2-エチルアミノエタノール(2 mL)、及び水(5 mL)の混合物がスチールボンベ中で130〜140℃で3日間加熱された。冷却後、混合物を水と酢酸エチルの間で分配し、有機層を塩水で洗浄し、乾燥し、蒸発させた。残渣はアセトンに溶解され、4 mmのドライシリカプレートに装荷され、放射状クロマトグラフィーにかけられた。プレートは石油エーテルと酢酸エチルの1:1混合物で溶出された。適当な分画を集めて、蒸発させて125 mg (12%) の1-{6-[エチル-(2-ヒドロキシエチル)-アミノ]-2-ナフチル}エタノンが得られた。
【0032】
ピリジン(3.5 mL)中の1-{6-[エチル-(2-ヒドロキシエチル)-アミノ]-2-ナフチル}エタノン(125 mg, 0.486 mmol) の溶液が-15℃に冷却され、p-トルエン無水スルフォン酸(252 mg, 0.81 mmol)がアルゴン下で攪拌しながら加えられた。反応混合物は放置されてゆっくりと室温まで暖められ、攪拌が24時間続けられた。TLC (シリカ、石油エーテル中10%酢酸エチル) によって出発物質がまだ存在することが明らかになったので、さらにp-トルエン無水スルフォン酸(252 mg, 0.81 mmol)が加えられ、攪拌がさらに24時間続けられた。次に、混合物は氷水浴中で冷却され、塩水とエーテルに分配された。有機層を乾燥、蒸発させ、オイル状の残渣が残された。生成物、6-アセチル-2-(エチル−2[(4-メチルフェニル)-スルフォニルオキシ]-エチルアミノ)-ナフタレン、は放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン)で単離され、収率は30%だった。HRMS C23H25NO4S計算値: 411.1504. 実測値:411.1514. 1H NMR δ1.25 (t, 3H, CH2CH3), 2.33 (s, 3H, PhCH3), 2.67 (s, 3H, COCH3), 3.49 (q, 2H, CH2CH3), 3.75 (t, 2H, NCH2CH2O), 4.25 (t, 2H, NCH2CH2O), 6.97 (d, 1H, 5-H), 7.01 (dd, 1H, 7-H), 7.18 and 7.20 (d, 2H, 3’-H, 5”-H), 7.56 (d, 1H, 4-H), 7.69 and 7.72 (d, 2H, 2’-H, 6’-H), 7.75 (d, 1H, 8-H). 7.93 (dd, 1H, 3-H), 8.29 (d, 1H, 1-H). J1,3=1.6 Hz, J2’,6=J3’,5=8.5 Hz, J7,5=2.5 Hz, J7,8=9.2 Hz, J3,4=8.7 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.2 Hz.
【0033】
水(2 mL)中の水酸化ナトリウム(1 g)とテトラ-n-ブチルアンモニウム硫酸水素塩(VI)(50 mg, 0.15 mmol)の溶液に、スピペロンケタール(8-3[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-1-フェニル-1,3,8-トリアザスピロ[4.5]デカン-4-オン (これは、米国特許第3,839,342号, Chem. Abstr. 82: 43416, 及びKiesewetter et al., Appl. Radiat. Isot. 37: 1181 (1986), に記述されているように調製できる。これらの文献の内容は参照によって本明細書に組み込まれる)(15 mg, 0.034 mmol)が加えられ、強く攪拌された。10分後、トルエン(3 mL)中の6-アセチル-2(エチル-2-[(4-メチルフェニル)-スルフォニルオキシ-エチルアミノ]-ナフタレン)(12 mg, 0.03 mmol)の溶液が加えられ、反応混合物は攪拌され1時間90℃で加熱された。冷却後、反応混合物は水とジクロロメタンに分配され、有機層は塩水で洗浄され、乾燥、蒸発されてオイル状の残渣が残された。放射状クロマトグラフィー(1 mmシリカ, ジクロロメタン中2%メタノール)によって5 mg (25%) の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-4-フェニル-2,4,8-トリアザスピロ[4.5]デセ-1-en-1-イル)-オキシ]-エチルアミノ)-2-ナフチル]-1-エタノン(化合物A)と11 mg (56%) の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ)-2-ナフチル]-1-エタノン(化合物B)が得られた。
【0034】
化合物A−HRMS C41H47FN4O4に対する計算値:678.3581. 実測値:678.3605. 1H NMR: δ1.45-2.24 (m, 11H, スピペロンCH2, CH2CH3), 2.35-2.84 (m, 6H, スピペロン), 2.65 (s, 3H, OCH3), 3.59 (q, 2H, NCH2CH3), 2.35-2.84 (M, 6H, スピペロン), 2.65 (s, 3H, OCH3), 3.59 (q, 2H, NCH2CH3), 3.76 in 4.05 (m, 4H, OCH2CH2O), 3.85 (t, 2H, NCH2CH2O), 4.52 (t, 2H, NCH2CH2O), 4.99 (s, 2H, NCH2N), 6.76-6.83 9m, 3H, フェニル, フルオロフェニル), 6.93 (d, 1H, 5-H), 6.95-7.04 (M, 2H, フェニル、フルオロフェニル), 7.19 (dd, 1H, 7-H), 7.21-7.26 (m, 2H, フェニル、フルオロフェニル), 7.61 (d, 1H, 4-H), 7.78 (d, 1H, 8-H,), 7.93 (dd, 1H, 3-H). 8.30 (d, 1H, 1-H). J1,3=1.5 Hz, J5,7=2.4 Hz, J3,4=9.5 Hz, J7,8=9.2 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.3 Hz.
【0035】
化合物B−HRMS C41H47FN4O4に対する計算値:678.3581. 実測値:678.3603. 1H NMR: δ1.20-1.94 (m, 17H, スピペロンCH2, CH2CH3), 2.66 (s, 3H, COCH3), 3.56 (q, 2H, NCH2CH3), 3.66 and 4.02 (m, 4H, OCH2CH2O), 3.71-3.81 (m, 4H, NCH2CH2N), 4.68 (s, 2H, NCH2N), 6.82-6.90 (m, 2H, フェニル, フルオロフェニル), 6.94 (d, 1H, 5-H), 6.98-7.04 (m, 2H, フェニル、フルオロフェニル), 7.18 (dd, 1H, H-7), 7.21-7.26 (m, 3H, フェニル、フルオロフェニル), 7.39-7.45 (m, 2H, フェニル, フルオロフェニル), 7.60 (d, 1H, 4-H), 7.78 (d, 1H, 8-H,), 7.93 (dd, 1H, 3-H). 8.29 (d, 1H, 1-H). J1,3=1.6 Hz, J3,4=9.8 Hz, J5,7=2.4 Hz, J7,8=10.4 Hz, J(CH2CH3)=7.1 Hz, J(NCH2CH2O)=6.3 Hz.
【0036】
ピリジン(3 mL)中の1-[6-(エチル-2-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ)-2-ナフチル]-1-エタノン(13 mg, 0.018 mmol)とマロノニトリル(6 mg, 0.09 mmol)の溶液がアルゴンの下で85℃で24時間加熱された。ピリジンが室温において真空で除去された後、残渣が塩水とジクロロメタンに分配され、有機層が乾燥、蒸発された。生成物、2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリル、が放射状クロマトグラフィー(1 mmシリカ, ジクロロメタン中2.5%メタノール;13.5 mg, 97%)によって単離された。
【0037】
HRMS C44H48FN6O3に対する計算値:727.37719. 実測値:727.3772. 1H NMR: δ1.25-1.93 (m, 17H, スピペロンCH2, CH2CH3), 2.70 (s, 3H, C=C-CH3), 3.57 (q, 2H, NCH2CH3), 3.64及び4.03 (m, 4H, OCH2CH2O), 3.71-3.78 (m, 4H, NCH2CH2N), 4.68 (s, 2H, NCH2N), 6.83-6.88 (m, 2H, フェニル, フルオロフェニル), 6.94 (d, 1H, 5-H), 6.96-7.04 (m, 2H, フェニル、フルオロフェニル), 7.56 (dd, 1H, 3-H), 7.63 (d, 1H, 4-H), 7.76 (dd, 1H, 4-H,), 7.76 (dd, 1H, 9-H). 8.00 (d, 1H, 1-H). J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.4 Hz, J7,8=9.3 Hz, J(CH2CH3)=7.1 Hz.
【0038】
一滴の濃塩酸を含むメタノール(1 mL)中で、2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリル(13.5 mg, 0.0186 mmol)を室温で3時間攪拌することによってケタール保護基が除去された。反応混合物はジクロロメタンで希釈され、炭酸水素ナトリウムの飽和溶液で洗浄された。真空中での蒸発の後、残渣は放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中2%メタノール)によって精製されて10 mg (79%)の2-[1-(6-(エチル-[2-8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルエチル]-アミノ)-2-ナフチルエチリデン)-マロノニトリルが得られた。HRMS C42H44FN6O2(M+H)に対する計算値:683.35. 実測値:683. 1H NMR: δ1.21-3.02 (m, 17H, スピペロンCH2, CH3), 2.71 (s, 3H, C=C-CH3), 3.56 (q, 2H, NCH2CH3), 3.69 (m, 4H, NCH2CH2N), 4.67 (s, 2H, NCH2N), 6.79-7.23 (m, 7H, フェニル, フルオロフェニル), 6.95 (d, 1H, 5-H),.5.19 (dd, 1H, 7-H), 7.56 (dd, 1H, 3-H), 7.65 (d, 1H, 4-H,), 7.76 (d, 1H, 8-H). 7.97-8.04 (m, 3H, フルオロフェニル、1-H). J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.5 Hz, J7,8=9.1 Hz, J(CH2CH3)=7.1 Hz.
【0039】
実施例1(c)−2-(1-6-[4-(8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)ピペリジノ]-2-ナフチルエチリデン)-マロノニトリル
【化9】
1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(653 mg, 3.5 mmol)(実施例1(b)で述べたように調製された)、硫酸水素ナトリウム(IV)(1.6 g, 15.5 mmol)、4-ピペリジルメタノール(2 g, 17.6 mmol)(Bradbury et al., J. Med. Chem. 34: 1073 (1991)に記述されているように調製された、この内容は参照によって本明細書に組み込まれる)、及び水(6 mL)の混合物がスチール・ボンベ中で135〜142℃で16日間加熱された。冷却後、反応混合物は酢酸エチルで抽出された。生成物の一部はまだ残渣に残っていたので、さらにジクロロメタン中5%メタノールで抽出された。有機抽出物を一緒にして、乾燥、蒸発させた。残渣は放射状クロマトグラフィー(2 mmシリカ、ジクロロメタン中2%メタノール)によって精製されて139 mg (14%) の1-6-[(4-ヒドロキシメチル)-ピペリジノ]-2-ナフチル-1-エタノンが得られた。酢酸エチルから再結晶化させた後、この化合物は180〜182℃で融解した。1H NMR: δ1.44 (dddd, 2H, 3’a-H, 5’a-H), 1.76 (m, 1H, 4’a-H), 1.91 (bd, 2H, 3’e-H, 5’c-H), 2.68 (s, 3H, COCH3), 2.89 (ddd, 2H, 2’a-H, 6’a-H), 3.58 (d, 2H, OCH2), 3.94 (bd, 2H, 27e-H, 67e-H), 7.10 (d, 1H, 5-H), 7.32 (dd, 1H, 7-H), 7.66 (d, 1H, 4-H), 7.80 (d, 1H, 8-H), 7.94 (dd, 1H, 3-H), 8.32 (d, 1H, 1-H), J3’a,3’c= J5’a,5’c =12.5 Hz, J2’a,3’a=J6’a,5’a=12.5Hz, J3’a,4’a=J5’a,4’a=12.5 Hz, J2’e,3’a=J6’e,5’a=4.0Hz, J2’a,2’c= J6’a,6’c =12.5 Hz, J2’a,3’e=J6’a,5e=2.6Hz, J4a,OCH2=6.4Hz, J1,3=1.9 Hz, J3,4=8.9 Hz, J5,7=2.3 Hz, J7,8=9.0 Hz.
【0040】
ピリジン(3.mL)中の1-6-[(4-ヒドロキシメチル)-ピペリジノ]-2-ナフチル-1-エタノン(59 mg, 0.2 mmol) の溶液が-15℃に冷却され、p-トルエン無水スルフォン酸(205 mg, 0.6 mmol)がアルゴン下で攪拌しながら加えられた。反応混合物は放置されてゆっくりと1時間で室温まで暖められた。それが再び冷却され、塩水とエーテルの間で分配された。有機層を塩水で洗浄し、乾燥、蒸発させると、83 mg (91%)の粗1-(6-アセチル-2-ナフチル)-4-[(4-メチルフェニル)-スルフォニルオキシ]メチルピペリジンが残された。
【0041】
水(2 mL)中の水酸化ナトリウム(1 g)とテトラ-n-ブチルアンモニウム硫酸水素塩(VI)(50 mg, 0.15 mmol)の溶液に、スピペロンケタール(100 mg, 0.2 mmol)が加えられ、激しく攪拌された。10分後、トルエン(10 mL)中の1-(6-アセチル-2-ナフチル)-4-[(4-メチルフェニル)-スルフォニルオキシ]メチルピペリジン(98 mg, 0.2 mmol)の溶液が加えられ、反応混合物は室温で11日間攪拌された。反応混合物は塩水とジクロロメタンの間で分配され、有機層は乾燥され蒸発されて190 mgのオイル状の残渣が残された。放射状クロマトグラフィー(1 mmシリカ、ジクロロメタンのあとにジクロロメタン中2%メタノール)によって、27 mg (17%) の1-[6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-4-フェニル-2,4,8-トリアザスピロ[4.5]デセ-1-エン-1-イル]-オキシル-メチルピペリジノ)-2-ナフチル)-1-エタノン(化合物3)と92 mg (58%) の1-[6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル]-メチル)ピペリジノ-2-ナフチル)-1-エタノン(化合物4)が得られた。
【0042】
化合物3:HRMS C43H49FN4O4に対する計算値:704.3738. 実測値:704.3760. 1H NMR: δ1.46-1.90 (m, 10H, 3’a-H, 5’a-H, 3’e-H, 5’e-H, スピペロン), 1.88 (m, 1H, 4’a-H), 2.15 and 2.38 (b, 4H, スピペロン), 2.67 (s, 3H, COCH3), 2.80 (m, 4H, スピペロン), 2.95 (m, 2H, 2’a-H, 6’a-H), 3.75 (m, 2H, OCH2 CH2O), 3.87 (m, 2H, 2’e-H, 6’e-H), 3.92 (m, 2H, OCH2 CH2O), 4.19 (d, 2H, OCH2), 4.97 (s, 2H, NCH2N), 6.7-6.9 (m, 3H, Ph), 7.01 (m, 2H, Ph), 7.11 (d, 1H, 5-H), 7.23 (m, 2H, Ph), 7.32 (dd, 1H, 7-H), 7.41 (m, 2H, Ph), 7.66 (d, 1H, 4-H), 7.81 (d, 1H, 8-H), 7.95 (dd, 1H, 3-H), 8.32 (d, 1H, 1-H). J2’a,2’e= J6’a,6’e =12.4 Hz, J2’a,3’e=J6’a,5’e=2.6Hz, J4’a,OCH2=6.1Hz, J1,3=.1 Hz, J3,4=8.8 Hz, J5,7=2.1 Hz, J7,8=9.1 Hz.
【0043】
化合物4:HRMS C43H49FN4O4に対する計算値:704.3738. 実測値:704.3710. 1H NMR: δ1.50 (dddd, 2H, 3’a-H, 5’a-H), 1.55-1.70 (m, 4H, スピペロン), 1.84 (bd, 2H, 3’e-H, 5’e-H), 1.92 (m, 2H, スピペロン), 1.98 (m, 1H, 4’a-H), 2.42 (m, 2H, スピペロン), 2.67 (s, 3H, COCH1), 2.69 (m, 2H, スピペロン), 2.83 (m, 4H, スピペロン), 2.88 (m, 2H, 2’a-H, 6’a-H), 3.35 (d, 2H, 4’-CH2N), 3.75 (m, 2H, OCH2 CH2O), 3.92 (bd, 2H, 2’e-H, 6’e-H), 4.02 (m, 2H, OCH2 CH2O), 4.71 (s, 2H, NCH2N), 6.88 ((m, 1H, Ph), 6.91 (m, 2H, Ph), 7.01 (m, 2H, Ph), 7.08 (bs, 1H, 5-H), 7.27 (m, 3H, 7-H, Ph), 7.43 (m, 2H, Ph), 7.65 (d, 1H, 4-H), 7.79 (d, 1H, 8-H), 7.94 (dd, 1H, 3-H), 8.32 (bs, 1H, 1-H), J3’a,3’e= J5’a,5’e =12.4 Hz, J2’a,3’a=12.5Hz, J2’a,2’e= J6’a,6’e =12.8 Hz, J2’a,3’e= J6’a,5’e =2.4Hz, J4’a,4’CH2N=7.3Hz, J1,3=1.9 Hz, J3,4=8.8 Hz, J5,7=2.1 Hz, J7,8=9.2 Hz.
【0044】
2-[1-(6-(エチル-[2-(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-エチル]-アミノ-2-ナフチル)-エチリデン)-マロノニトリルの合成に関して実施例1(b)で述べた手順を用いて、1-[6-4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イル)-メチル]-ピペリジノ-2-ナフチル]-1-エタノンが、2-[1-6-(4-[(8-3-[2-(4-フルオロフェニル)-1,3-ジオキソラン-2-イル]-プロピル-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)ピペリジノ]-2-ナフチル)エチリデン]-マロノニトリルに変換された。これを、CH2Cl2中2%MeOHを溶媒とする1 mmシリカプレートでの放射状クロマトグラフィーで精製した。FAB HRMSのC46H50FN6O3(M+H)に対する計算値:753.3928. 実測値:753.3940. 1H NMR: δ1.60-2.1 (m, 11H, スピペロン、3’a-H, 3’e-H, 4’a-H, 5’a-H, 5’e-H), 2.40 (m, 2H, スピペロン), 2.71 (s, 3H, C=C-CH3), 2.60-2.80 (m, 6H, スピペロン), 2.91 (m, 2H, 2’a-H, 6’a-H), 3.37 (d, 2H, 4’-CH2N), 3.75 (m, 2H, OCH2 CH2O), 3.94 (bd, 2H, 2’e-H, 6’e-H), 4.02 (m, 2H, OCH2 CH2O), 4.72 (s, 2H, NCH2N), 6.85-6.95 ((m, 3H, Ph), 7.01 (m, 2H, フルオロフェニル), 7.07 (d, 1H, 5-H), 7.31 (m, 3H, 7-H, Ph), 7.41 (m, 2H, フルオロフェニル), 7.56 (dd, 1H, 3-H), 7.69 (d, 1H, 4-H), 7.77 (d, 1H, 8-H), 8.01 (bs, 1H, 1-H). J2’a,3’a= J5’a,6’a =12.8Hz, J2’a,2’e= J6’a,6’e =12.8Hz, J4’a,4’CH2N=7.6Hz, J1,3=1.8 Hz, J3,4=8.6 Hz, J5,7=2.2 Hz, J7,8=9.4 Hz, J2’a,3’e= J5’e,6’a =1.8 Hz, JH,F=8.7及び6.2Hz.
【0045】
実施例1(b)に記述されているようにケタール保護基を除去して2-(1-(6-[4-(8-[4-(4-フルオロフェニル)-4-オキソブチル]-1-オキソ-4-フェニル-2,4,8-トリアザスピロ[4.5]デシ-2-イルメチル)-ピペリジノ]-2-ナフチルエチリデン)-マロノニトリルが定量的な収率で得られた。FAB HRMSのC44H46FN6O2(M+H)に対する計算値:709.3666. 実測値:709.3689. 1H NMR: δ1.60-2.1 (m, 11H, スピペロン、3’a-H, 3’e-H, 4’a-H, 5’a-H, 5’e-H), 2.5-2.71 (m, 4H, スピペロン), 2.73 (s, 3H, C=C-CH3), 2.8-3.1 (m, 6H, スピペロン、2’a-H, 6’a-H), 3.38 (d, 2H, 4’-CH2N), 3.96 (bd, 2H, 2’e-H, 6’e-H), 4.74 (s, 2H, NCH2N), 6.91 (m, 3H, フェニル), 7.1 (d, 1H, 5-H), 7.15 (m, 2H, フルオロフェニル), 7.24-7.30 (m, 2H, Ph), 7.14 (dd, 1H, 7-H), 7.58 (dd, 1H, 3-H), 7.72 (d, 1H, 4-H), 7.79 (d, 1H, 8-H), 8.01-8.08 (m, 3H, フルオロフェニル、1-H). J1,3=2.0 Hz, J3,4=8.6 Hz, J5,7=2.4 Hz, J7,8=9.2 Hz, J4’a,4’CH2N=7.4Hz, J2’a,2’e= J6’a,6’e =13.0Hz, J2’a,3’a= J5’a,6’a =12.5Hz, J2’a,3’e= J5’e,6’a =1.9 Hz, JH,F=8.7及び6.2Hz.
【0046】
実施例1(d)−tert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレートの調製
【化10】
新しく蒸留された無水トルエンとヘキサメチル・ホスフォリックアミド(HMPA)、各25 mL、の混合物に、無水ピペラジン(7 g, 81.3 mmol; 真空乾燥器でKOH-ドライエライト上で3日間乾燥)が溶解された。この溶液に、アルゴン雰囲気中で小さな片にカットされた556 mg(80.1 mmol)のリチウム棒が加えられ、混合物はアルゴンの下で24時間攪拌され、その間にすべてのリチウムが溶解した。真空乾燥された1-(6-メトキシ-2-ナフチル)-1-エタノン(Arsenijevic et al., Org. Synth. Coll. 1988, 6:34-36に記述されたように調製された。この文献の開示内容は参照によって本明細書に組み込まれる)(3.5 g, 17.5 mmol)が加えられ、攪拌がさらに65時間続けられた。300 mLの水による反応停止、ジクロロメタンによる抽出(3 x 300 mL)、無水硫化マグネシウムによる乾燥、及び蒸発の後に、白色と黄色固体の混合物が得られた。300 mLの熱いメタノールによる抽出によって、粗生成物、1-(6-ピペラジノ-2-ナフチル)-1-エタノン、が得られ、カラム・クロマトグラフィー(70-230メッシュのシリカ、25 x 120 mm, ジクロロメタン中5%メタノール)によって精製された。収量は1.54 g(35%)であった。酢酸エチルからの再結晶後、サンプルは170.5〜172℃で融解した。
【0047】
1-(6-ピペラジノ-2-ナフチル)-1-エタノンは、また、1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(実施例1(b)に記述されているように調製された)(441 mg, 2.36 mmol)を140〜150℃で6 gのピペラジン水和物(30.9 mmol)及び244 mg (2.35 mmol)のNaHSO3と共に24 時間熱することによっても調製された。追加の亜硫酸水素ナトリウム(2 g, 19.2 mmol)が加えられた。さらに24時間後、さらに亜硫酸水素ナトリウム(1 g)が加えられ、加熱が続けられた(全反応時間は72時間)。冷却後、混合物は2 x 50 mLのメタノールで抽出された。メタノールの蒸発後の残渣は50 mLの水に懸濁され、酢酸エチルで抽出された(5 x 80 mL)。抽出物全体を乾燥させ(硫酸マグネシウム)、蒸発させて430 mg の黄色固体が得られた。放射状クロマトグラフィー(4 mmシリカ、メタノール)によって83 mg (19%)の出発ナフトールと276 mg (46%; 回収されない出発物質に基づくと56%)の1-(6-ピペラジノ-2-ナフチル)-1-エタノンが得られた。この1-(6-ピペラジノ-2-ナフチル)-1-エタノンはあらゆる点で上述した別の方法によって得られた化合物と同一であった。分析、C16H18N2Oに対する計算値:C, 75.56; H, 7.13; N, 11.01. 実測値:C, 75.82; H, 7.27; N, 10.92. 1H NMR: δ2.68 (s, 3H, CH3), 3.09 and 3.35 (t, J = 4.95Hz, 8H, ピペラジン), 7.10 (d, 1H, 5-H), 7.31 (dd, 1H, 7-H), 7.69 (d, 1H, 4-H), 7.83 (d, 1H, 8-H), 7.95 (d, 1H, 3-H), 8.34 (s, 1H, 1-H); J5,7=2Hz, J7,8=8.4Hz, J1,3=2Hz, J3,4=8.4Hz.
【0048】
1-(6-ピペラジノ-2-ナフチル)-1-エタノン(254 mg, 1 mmol)が1 g のNaOH, 100 mgのテトラ-n-ブチルアンモニウム硫酸水素塩、2 mLの水、及び6 mLのトルエンの攪拌された混合物に加えられ、続いて230 mg(1.05 mmol)のジ-tert-ブチルジカルボネートの溶液が加えられた。反応の過程はTLC (シリカ、ジクロロメタン中5%のメタノール)で追跡された。10分毎に、すべての出発物質が反応してしまうまで追加の量のジカルボネートが加えられた。全部でほぼ1.5当量が用いられた。水とジクロロメタン(各60 mL)の混合物が加えられ、十分な振蕩の後、層が分離された。水性層は、さらに別の30 mLのジクロロメタンで抽出された。有機抽出物の全体は、無水硫酸マグネシウムで乾燥された。この過程で溶液の色はピンクから淡黄色に変化した。真空中での蒸発によって295 mg (83%)のtert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレートが得られ、これをジクロロメタン−石油エーテル混合物から再結晶させると、153〜154℃で融解した。分析、C21H26N2O3に対する計算値:C, 71.16; H, 7.39; N, 7.90. 実測値:C, 71.27; H, 7.60; N, 7.86. 1H NMR: δ1.50 (s, 9H, -C(CH3)3), 2.68 (s, 3H, CH3), 3.33 and 3.64 (t, J = 4.9Hz, 8H, ピペラジン), 7.10 (d, 1H, 5-H), 7.31 (dd, 1H, 7-H), 7.70 (d, 1H, 4-H), 7.85 (d, 1H, 8-H), 7.97 (d, 1H, 3-H), 8.35 (d, 1H, 1-H); J5,7=2Hz, J7,8=9Hz, J3,4=8.7Hz.
【0049】
実施例1(e)−2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロノニトリルの調製
【化11】
実施例1(d)に記述されたように調製されたtert-ブチル-4-(6-アセチル-2-ナフチル)-1-ピペラジンカルボキシレート(177 mg, 0.5 mmol)が4 mLのピリジン中で40 mg (0.6 mmol)のマロノニトリルと共に105〜110℃で熱せられた。5.5時間後、さらに24 mgのマロノニトリルが加えられ、加熱は全部で12時間40分続けられた。混合物は冷却され、真空中で蒸発された。混合物の極性成分はカラム・クロマトグラフィー(70-230メッシュのシリカ、φ20 x 120 mm, クロロフォルム)によって除去され、生成物、tert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレート、が最終的に放射状クロマトグラフィー(シリカ、2 mm, クロロフォルム)によって精製されて、155 mg(77%)のtert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレートが得られ、ジクロロメタン−石油エーテル混合物からの再結晶後、169-171℃で融解した。分析、C24H26N4O2に対する計算値:C, 71.62; H, 6.51; N, 13.92. 実測値:C, 71.62; H, 6.66; N, 13.87. 1H NMR: δ1.50 (s, 9H, -C(CH3)3), 2.72 (s, 3H, CH3), 3.34及び3.64 (t, J = 5.1Hz, 8H, ピペラジン), 7.09 (d, 1H, 5-H), 7.33 (dd, 1H, 7-H), 7.58 (dd, 1H, 3-H), 7.74 (d, 1H, 4-H), 7.81 (d, 1H, 8-H), 8.02 (d, 1H, 1-H); J5,7=2Hz, J7,8=9.1Hz, J1,3=2Hz, J3,4=9.1Hz.
【0050】
tert-ブチル-4-[6-(2,2-ジシアノ-1-メチルビニル)-2-ナフチル]-1-ピペラジンカルボキシレートが室温でTFA(トリフルオロ酢酸)で処理されると、TLCは反応が5分で終わったことを示し、単一の生成物、2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロノニトリルが得られた。TFAは室温で真空で除去された。1H NMR: δ2.72 (s, 3H, CH3), 3.50及び3.63 (ブロード, 8H, ピペラジン), 7.18 (ブロード s, 1H, 5-H), 7.29 (d, 1H, 7-H), 7.59 (d, 1H, 3-H), 7.79 (d, 1H, 4-H), 7.87 (d, 1H, 8-H), 8.04 (s, 1H, 1-H), 9.0 (ブロード, 1.5H, NH及び酸); J7,8=8.8Hz, J3,4=8.4Hz. 19F NMR: δ -76.2 (CF3COO).
【0051】
残渣のNMRによって、tert-ブチルオキシカルボニル基が除去され、TFAが少し残されていることが明らかになった(19F NMR)。ジクロロメタン(10 mL)が加えられ、溶液は飽和NaHCO3溶液で洗浄され、乾燥され、真空中で蒸発された。淡黄色の固体が得られ、室温で放置するとそれは暗赤色に変わった。TLCは、色の変化が2-[1-(6-ピペラジノ-2-ナフチル)エチリデン]マロニトリルがいくつかの生成物に分解したためであることを示し、最も強度の大きなスポットは低-Rfの赤-オレンジ色であった。中和後のいくつかのNMR信号:δ2.72 (s, 3H, CH3), 3.09及び3.35 (t, J=5Hz, 8H, ピペラジン), 7.08 (s, 1H, 5-H), 8.02 (s, 1H, 1-H).
【0052】
実施例1(f)−2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)の調製
【化12】
4.15 g (55.5 mmol)のNaHSO3、8 mLの水、0.78 g (4.19 mmol)の1-(6-ヒドロキシ-2-ナフチル)-1-エタノン(実施例1(b)に記述されているように調製)、及び8 mLの2-メチルアミノエタノールの混合物がスチール・ボンベの中で140℃で28時間加熱された。冷却後、混合物は酢酸エチルと水(それぞれ、500 mLと200 mL)の間で分配された。有機層を乾燥、蒸発させると粗1-(6-(2-ヒドロキシエチル-メチルアミノ)-2-ナフチル)-1-エタノン(0.749 g, 73%)が残され、それがさらに放射状クロマトグラフィー(4 mm SiO2, CH2Cl2)によって精製された。
【0053】
ピリジン(6 mL)中の201 mg (0.83 mmol)の1-(6-(2-ヒドロキシエチル-メチルアミノ)-2-ナフチル)-1-エタノンの溶液にマロノニトリル(236 mg, 3.6 mmol)が加えられ、混合物は95℃で24時間加熱された。溶媒は真空で除去され、残渣が放射状クロマトグラフィー(4 mm SiO2, 1% MeOH/CH2Cl2)にかけられ、150 mg (73%)の2-(1,1-ジシアノプロペン-2-イル)-6-(2-ヒドロキシエチル)-メチルアミノ)-ナフタレンが得られた。
【0054】
ピリジン(5 mL)中の2-(1,1-ジシアノプロペン-2-イル)-6-(2-ヒドロキシエチル)-メチルアミノ)-ナフタレン(120 mg, 0.41 mmol)の溶液に、p-トルエン無水スルフォン酸が加えられた(44 mg, 1.35 mmol)。室温で1時間攪拌した後、真空下でピリジンを除去し、残渣を放射状クロマトグラフィー(2 mm SiO2, CH2Cl2)にかけて183 mg (80%)の2-(1,1-ジシアノプロペン-2-イル)-6-(2-トシルオキシエチル)-メチルアミノ)-ナフタレンが得られた。
【0055】
サイクロ(登録商標)トロンからの放射性19Fフッ化物528.5 mCiが50μLの水と300μLのアセトニトリル中の19 mgのKryptofix 2.2.2と0.75mgの炭酸カリウムの溶液に移された。115℃で窒素の流れによって水を除去し、続いてアセトニトリルと共に蒸留した(3 x 200μL)。1 mLのアセトニトリル中のトシル化物(2-(1,1-ジシアノプロペン-2-イル)-6-(2-トシルオキシエチル)-メチルアミノ)-ナフタレン、4 mg)を加え、混合物を85〜86℃で20分加熱した。冷却後、1mLの水が加えられ、混合物はC-18 Sep-Pak Cartridgeに移され、蒸留水で洗浄され(3×4 mL)、CH2Cl2で溶出された(2×2.5 mL)。溶出物は硫酸ナトリウムを詰めたカラムを通して乾燥させ、HPLCカラム(Whatman Partisil Silica 10, 500×10 mm, mL/min CH2Cl2:ヘキサン=7 : 3, UV検出器@254 nm, 放射能検出器)に装填された。溶出物を集め、適当な分画を一緒にし、真空下で蒸発させて50.7 mCi (17%, 崩壊を補正)の注射のために製剤された表題の生成物が得られた。合成は50分で完了した。
【0056】
実施例1(g)−7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オンの調製
【化13】
7.76 mLのDMFに0℃で4.66 mL (約50 mmol)のPOCl3を(一滴ずつ)加えてVielsmeier試薬が少ない分量で調製され、混合物は同じ温度で5分間、続いて室温で30分間、攪拌された。2 mLのDMF中の3.43 g (25 mmol)の3-(ジメチルアミノ)フェノールが、Vielsmeier試薬に一滴ずつ加えられた。混合物は60〜70℃で1時間加熱され、50 gの砕いた氷に混合物を注いで過剰な試薬を消滅させた。混合物は酢酸エチルで抽出され、有機部分を無水硫酸マグネシウムで乾燥して、蒸発させた。オイル状の残渣から4-ジメチルアミノ-2-ヒドロキシベンズアルデヒドがカラム・クロマトグラフィー(シリカゲル、クロロフォルム)によって単離された。収量:2.32 g (14 mmol, 56%).
【0057】
4 mmolの4-ジメチルアミノ-2-ヒドロキシベンズアルデヒドと5 mmolの適当な1,3-ジカルボニル化合物が8 mLの無水エタノールに溶解された。16滴のピペリジンが加えられ、各混合物は2〜6時間還流で加熱された。混合物はフリーザー内で冷却され、沈殿を濾過し、エタノールで洗浄して表1に示されたような生成物が得られた。
【0058】
【表1】
【0059】
NMRスペクトル:
3-アセチル-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(1)
1H NMR (360MHz, CDCl3) δ: 2.70 (3H, s, Ac); 3.14 (6H, s, NMe2); 4.39 (2H, q, EtO); 6.48 (1H, d, 芳香族H); 6.66 (1H, dd, 芳香族H); 7.43 (1H, d, 芳香族H); 8.47 (1H, s, 4-H).
エチル7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-カルボキシレート (2)
1H NMR (360MHz, CDCl3) δ: 1.40 (3H, t, EtO-); 3.13 (6H, s, NMe2); 4.39 (2H, q, EtO); 6.48 (1H, d, 芳香族H); 6.65 (1H, dd, 芳香族H); 7.40 (1H, d, 芳香族H); 8.46 (1H, s, 4-H).
3-(2,2-ジメチルプロパノイル)-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(3)
1H NMR (360MHz, CDCl3) δ: 1.33 (9H, s, tBu); 3.10 (6H, s, NMe2); 6.50 (1H, d, 芳香族H); 6.63 (1H, dd, 芳香族H); 7.33 (1H, d, 芳香族H); 7.68 (1H, s, 4-H).
メチル-3-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]-3-オキソ-プロパノエート (4)
1H NMR (360MHz, CDCl3) δ: 3.15 (6H, s, NMe2); 3.75 (3H, s, -OMe); 4.11 (2H, s, CH2); 6.47 (1H, d, 芳香族H); 6.66 (1H, dd, 芳香族H); 7.45 (1H, d, 芳香族H); 8.53 (1H, s, 4-H).
7-(ジメチルアミノ)-3-(4-ニトロベンゾイル)-2H-1-ベンゾピラン-2-オン(5)
1H NMR (360MHz, CDCl3) δ: 3.13 (6H, s, NMe2); 6.63 (1H, br, 芳香族H); 6.84 (1H, dd, 芳香族H); 7.69 (1H, d, 芳香族H); 7.97 (2H, m, 芳香族H); 8.31 (2H, m, 芳香族H); 8.47 (1H, s, 4-H).
【0060】
実施例1(h)−ビス(7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル)ケトンの調製
【化14】
360 mg(1 mmol)のメチル-3-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]-3-オキソ-プロパノエート(実施例1(g)の化合物4)が250 mgの4-ジメチルアミノ-2-ヒドロキシベンザアルデヒドと混合され、2 mLの無水エタノールに溶解された。4滴のピペリジンが加えられ、混合物は8時間還流された。混合物は室温にされ、沈殿した固体が濾過された。179 mg (44%)のオレンジ色の固体。1H NMR (360MHz, CDCl3) δ: 3.11 (6H, s, NMe2); 6.50 (1H, d, 芳香酸 H); 6.63 (1H, dd, 芳香酸 H); 7.41 (1H, d, 芳香酸 H); 8.19 (1H, s, 4-H).
【0061】
実施例1(i)−2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの調製
【化15】
N,N-ジメチルホルムアミド・ジメチル・アセタール(DMFDMA, 5 mL)中の2-{1-[6-(ジメチルアミノ)-2-ナフチル]エチリデン}マロノニトリル(1 g, 3.8 mmol)(A. Jacobson et al., J. Am. Chem. Soc. 1996, 118, 5572-5579,に記述されているように調製)の溶液が室温で一晩攪拌された。揮発成分を真空で除去し、残渣をカラムクロマトグラフィー(シリカ70-230メッシュ、20/150 mm, クロロフォルム)にかけて、2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(1.19 g, 98%)が得られた。mp. 213〜215℃(ジクロロメタン−石油エーテル混合物から);元素分析、C20H20N4に対する計算値:C, 75.92; H, 6.37; N, 17.17. 実測値;C, 76.28; H, 6.04; N, 17.57. IR (KBrペレット): 2210 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.02 (s, 6H, NMe2); 3.1 (s, 6H, NMe2); 5.86 and 6.72 (d, 2H, CH=CH), 6.91 (d, 1H, H-5), 7.20 (dd, 1H, H-7), 7.23 (dd, 1H, H-3), 7.62 (bs, 1H, H-1), 7.69 (d, 1H, H-8), 7.73 (d, 1H, H-4). J5,7=2.3Hz, J7,8=9.1Hz, J3,4=9.0Hz, JCH=CH=12.5Hz.
【0062】
実施例1(j)−2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル
【化16】
イソプロパノール(100 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(603 mg, 1.9 mmol)(上の実施例1(i)に記述されているように調製)の溶液がrtで無水HClで飽和され、一晩攪拌された。溶媒は真空中で除去され、固体残渣はジクロロメタンに溶解され、溶液は飽和NaHCO3溶液で洗浄された。有機層を乾燥させ、蒸発させて2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(402 mg, 69%)が残された。mp. 198〜200℃(MeOHから);元素分析、C18H14N3Clに対する計算値: C, 70.24; H, 4.58; N, 13.65. 実測値:C, 69.76; H, 4.31; N, 13.38. IR (KBrペレット): 2250 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.15 (s, 6H, Me2N), 6.91 (d, 1H, H-5), 7.21 (dd, 1H, H-7), 7.47 (d, 1H, H-5’), 7.57 (dd, 1H, H-3), 7.74 (d, 1H, H-8), 7.79 (d, 1H, H-4), 7.99 (d, 1H, H-1), 8.53 (d, 1H, H-6). J5,7=2.2Hz, J7,8=9.7Hz, J1,3=2.0Hz, J3,4=9.2Hz, J5’,6’=5.1Hz.
【0063】
実施例1(k)−2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル
【化17】
MeOH (20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(100 mg, 0.32 mmol)(上の実施例1(i)に記述されているように調製)の沸騰する溶液にアンモニアガスが30分間吹き込まれた。反応の間に溶液から分離し始めた2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリルの黄色結晶が濾過された(86 mg, 93%)。mp. 256〜257℃ (MeOHから);元素分析、C18H16N4Clに対する計算値: C, 74.98; H, 5.59; N, 19.43. 実測値:C, 75.29; H, 5.31; N, 19.16. IR (KBrペレット): 2210 cm-1 (CN), 3720 cm-1 (ArNH2); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 5.27 (s, 2H, NH2), 6.87 (d, 1H, H-5), 6.92 (d, 1H, H-5), 7.20 (dd, 1H, H-7), 7.57 (dd, 1H, H-3), 7.74 (d, 1H, H-8), 7.79 (d, 1H, H-4), 7.97 (bs, 1H, H-1), 8.42 (d, 1H, H-6). J5,7=2.3Hz, J7,8=9.5Hz, J1,3=2.0Hz, J3,4=8.5Hz, J5’,6’=4.9Hz.
【0064】
実施例1(l)−2-アミノ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル-1-オキシドの調製
【化18】
MeOH (20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]−2−プロペニリデン}マロノニトリル(28 mg, 0.9 mmol)(上の実施例1(i)に記述されているように調製)とヒドロキシ塩化アンモニウム(94.5 mg, 1.36 mmol)の溶液が還流で70時間加熱された。溶媒を除去し、残渣を放射状クロマトグラフィー(2 mm シリカ、ジクロロメタン中5% MeOH)にかけて2-アミノ-4-[6-(ジメチルアミノ)-2−ナフチル]ニコチノニトリル-1-オキシド(53 mg, 20%)が得られた。mp. 248〜250℃ (MeOHから);元素分析、C18H16N4OCに対する計算値: C, 71.04; H, 5.30; N, 18.41. 実測値:C, 70.59; H, 5.59; N, 18.19. IR (KBrペレット): 2220 cm-1 (CN), 3310 cm-1 (ArNH2); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 6.43 (s, 2H, NH2), 6.86 (d, 1H, H-5’), 6.91 (d, 1H, H-5), 7.21 (dd, 1H, H-7), 7.55 (dd, 1H, H-3), 7.75 (d, 1H, H-8), 7.78 (d, 1H, H-4), 7.95 (bs, 1H, H-1), 8.12 (d, 1H, H-6); J5,7=2.4Hz, J7,8=8.7Hz, J1,3=2.0Hz, J3,4=8.7Hz, J5’,6’=6.7Hz.
【0065】
実施例1(m)−4-[6-(ジメチルアミノ)-ナフチル]-2-メトキシニコチノニトリルの調製
【化19】
方法A. MeOH (15 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]−2−プロペニリデン}マロノニトリル(82.5 mg, 0.26 mmol)(上の実施例1(i)に記述されているように調製)の溶液に、ナトリウムメチラートの溶液(1 mL, mLのメタノールあたり10 mgのナトリウムを反応させることによって調製)が加えられ、反応混合物が還流で20時間加熱された。溶媒を真空で除去し、残渣をジクロロメタンと塩水の間で分配し、有機層を乾燥、蒸発させた。残渣を放射状クロマトグラフィー(1 mm シリカ、ジクロロメタン-シクロヘキサン 1:1)にかけて4-[6-(ジメチルアミノ)-2−ナフチル]-2-メトキシニコチノニトリル(40 mg, 50%)が得られた。
方法B. MeOH (10 mL)中の2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(58 mg, 0.19 mmol)(上の実施例1(j)に記述されているように調製)の溶液が、ナトリウムメチラート(0.9 mLの溶液、mLのメタノールあたり10 mgのナトリウムを反応させることによって調製、0.4 mmol)と共に6.5時間還流で加熱された。4-[6-(ジメチルアミノ)-2−ナフチル]-2-メトキシニコチノニトリルが方法Aで述べたように単離されたが、MeOHからの再結晶化によって精製された;元素分析、C19H17N3Oに対する計算値: C, 75.23; H, 5.65; N, 13.85. 実測値:C, 74.99; H, 5.59; N, 13.73. IR (KBrペレット): 2220 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 4.11 (s, 3H, OMe), 6.9 (bs, 1H, H-5), 7.12 (d, 1H, H-5’), 7.20 (bd, 1H, H-7), 7.59 (d, 1H, H-3), 7.74 (d, 1H, H-4), 7.79 (d, 1H, H-8), 7.99 (bs, 1H, H-1), 8.31 (d, 1H, H-6’); J7,8=9.2Hz, J3,4=8.6Hz, J5’,6’=5.4Hz.
【0066】
実施例1(n)−2-{(2Z)-1-[6-(ジメチルアミノ)-2-ナフチル]-3-[2-(ヒドロキシエチル)-アミノ]-2-プロペニリデン}マロノニトリルの調製
【化20】
MeOH (50 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(350 mg, 1.1 mmol)(上の実施例1(i)に記述されているように調製)と2-アミノエタノール(0.4 mL, 6.6 mmol)の溶液が還流で24 時間加熱された。溶媒を除去し、残渣を放射状クロマトグラフィー(4 mm シリカ、ジクロロメタン中5% MeOH)にかけて2-{(2Z)-1-[6-(ジメチルアミノ)-2-ナフチル]-3-[2-(ヒドロキシエチル)-アミノ]-2-プロペニリデン}マロノニトリル(327 mg, 89%)が得られた。mp. 168〜170℃ (EtOHから);1H NMR (360MHz, CDCl3) δ: 3.08 (s, 6H, Me2N), 3.98及び4.17 (bs, 4H, CH2CH2), 5.43 (b, 2H, OH, NH), 6.09及び7.24 (d, 2H, CH=CH), 6.86 (bs, 1H, H-5), 7.16 (d, 1H, H-7), 7.51 (d, 1H, H-3), 7.68 (d, 1H, H-4), 7.74 (d, 1H, H-8), 7.94 (s, 1H, H-1); J3,4=8.6Hz, J7,8=9.2Hz, JCH=CH=6.9Hz.
【0067】
実施例1(o)−4-[6-(ジメチルアミノ)-2-ナフチル]-2-[(2-ヒドロキシエチル)スルファニル]ニコチノニトリル
【化21】
ナトリウム・メチラートの溶液(5 mL, mLのMeOHあたり10 mgのナトリウムを反応させて調製、2.17 mmol)中の2-メルカプトエタノール(0.81 mL, 11.6 mmol)の溶液に、2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(273 mg, 0.89 mmol)(上の実施例1(i)に記述されているように調製)の溶液が加えられ、混合物は還流で5時間加熱された。反応混合物から分離した固体を濾過し、アセトニトリルから再結晶させて4-[6-(ジメチルアミノ)-2-ナフチル]-2-[(2-ヒドロキシエチル)スルファニル]ニコチノニトリル(271 mg, 85%)が得られた。mp. 210〜212℃. 元素分析、C20H19N3OSに対する計算値: C, 68.74; H, 5.48; N, 12.02. 実測値:C, 68.51; H, 4.99; N, 12.07. IR (KBrペレット): 2220 cm-1 (CN), 3350 cm-1 (OH); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.5 (t, 2H, CH2CH2), 3.65 (b, 1H, OH), 4.0 (b, 2H, CH2CH2), 6.91 (d, 1H, H-8), 7.20 (bd, 1H, H-7), 7.25 (d, 1H, H-5’), 7.55 (d, 1H, H-3), 7.75 (d, 1H, H-4), 7.79 (d, 1H, H-8), 7.97 (bs, 1H, H-1), 8.50 (d, 1H, H-6’); J7,8=7.8Hz, J3,4=8.6Hz, J5’,6’=5.4Hz, JCH2CH2=5.4Hz.
【0068】
実施例1(p)−2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの調製
【化22】
MeOH(20 mL)中の2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル (100 mg, 0.316 mmol)とアミノアセトアルデヒド・ジエチル・アセタール(0.1 mL, 0.96 mmol)の溶液が還流で46時間加熱された。溶媒を真空で除去すると、オイル状の残渣が残された。それを放射状クロマトグラフィー(2 mmシリカ、ジクロロメタン、続いてジクロロメタン中5% MeOH)にかけて、2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(111 mg, 87%)が得られた。1H NMR (360MHz, CDCl3) δ: 1.22 (t, 6H, t, OCH2CH3, J=7.1Hz), 3.09 (s, 6H, Me2N), 3.59 and 3.80 (m, 4H, OCH2CH3), 4.05(d, 2H, CH2CH), 4.92 (t, 1H, CH2CH), 5.95 and 7.23 (d, 2H, CH=CH), 6.90 (bs, 1H, H-5), 7.19 (dd, 1H, H-7), 7.55 (d, 1H, H-3), 7.71 (d, 1H, H-4), 7.77 (d, 1H, H-8), 7.97 (bs, 1H, H-1); JCH=CH=7.2Hz, J7,8=9.1Hz, J3,4=8.5Hz, JCH2CH=5.1Hz.
【0069】
実施例1(q)−2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル
【化23】
MeOH(5 mL)中の2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(19 mg, 0.05 mmol)(上の実施例1(p)に記述されているように調製)の溶液にHClガスが室温で5分間吹き込まれた。反応混合物は室温でさらに30分間攪拌され、濃縮されてオイル状の残渣が残された。それを酢酸エチルと氷温の塩水の間で分配した(各40 mL)。有機層を乾燥、蒸発させて2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(12 mg, 66%)が得られた。2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルは、また、上の実施例1(i)に記述されているように調製された2-{(2E)-3-(ジメチルアミノ)-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルから直接に、ジエチル・アセタールの代わりにアミノアセトアルデヒドジメチルアセタールを用い、上の実施例1(p)に記述されているような2-{(2Z)-3-[(2,2-ジエトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリルの合成の手順に従って調製された。mp. 158〜159℃. IR (KBrペレット): 2220 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.48 (s, 6H, OCH3), CH2CH2), 4.06 (d, 2H, CH2CH), 5.96 and 7.24 (d, 2H, CH=CH), 6.89 (d, 1H, H-5), 7.18 (dd, 1H, H-7), 7.54 (d, 1H, H-3), 7.74 (d, 1H, H-4), 7.76 (d, 1H, H-8), 7.96 (s, 1H, H-1); J5,7=2.2Hz, JCH=CH=7.0Hz, J7,8=8.9Hz, J3,4=8.5Hz, JCH2CH=5.01Hz.
【0070】
実施例1(r)−7-[6-(ジメチルアミノ)-2-ナフチル]イミダゾ[1.2-α]ピリジン-8-カルボニトリル
【化24】
MeOH(15 mL)中の2-{(2Z)-3-[(2,2-ジメトキシエチル)アミノ]-1-[6-(ジメチルアミノ)-2-ナフチル]-2-プロペニリデン}マロノニトリル(53 mg, 0.14 mmol)(上の実施例1(q)に記述されているように調製)の溶液が還流で2時間加熱された。最初の30分間、乾燥HClガスの流れが反応混合物を通して導かれた。反応が完了した後、溶媒を蒸発させ、固体残渣がジクロロメタンと炭酸水素ナトリウムの飽和溶液の間で分配された。有機層を乾燥し、蒸発させた。残渣を放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中5% MeOH)にかけて、7-[6-(ジメチルアミノ)-2-ナフチル]イミダゾ[1.2-α]ピリジン-8-カルボニトリル(25 mg, 57%)が得られた。mp. 268〜269℃;元素分析、C20H16N4に対する計算値: C, 76.90; H, 5.16; N, 17.94. 実測値:C, 76.231; H, 5.22; N, 17.62. IR (KBrペレット): 2230 cm-1 (CN); 1H NMR (360MHz, CDCl3) δ: 3.07 (s, 6H, Me2N), 7.02 (d, 1H, H-5’), 7.29 (d, 1H, H-6), 7.33 (dd, 1H, H-7’), 7.69 (d, 1H, H-3’), 7.74 (s, 1H, H-3), 7.84 (d, 1H, H-4’), 7.87 (d, 1H, H-8’), 8.11 (s, 1H, H-1’), 8.14 (s, 1H, H-2), 8.9 (d, 1H, H-5); J5’,7’=2.0Hz, J7’,8’=9.4Hz, J3’,4’=8.6Hz, J5,6=7.2Hz.
【0071】
実施例1(s)−4-[6-(ジメチルアミノ)-2-ナフチル]-2-(メチルアミノ)-ニコチノニトリルの調製
【化25】
上の実施例1(j)に記述されているように調製された2-クロロ-4-[6-(ジメチルアミノ)-2-ナフチル]ニコチノニトリル(194.5 mg, 0.63 mmol)がメチルアミンのエタノール溶液(5 mLの33%溶液)に懸濁され、還流で1時間加熱された。さらに5 mLのメチルアミンのメタノール溶液が加えられ、加熱が9時間続けられた。冷却後、4-[6-(ジメチルアミノ)-2-ナフチル]-2-(メチルアミノ)-ニコチノニトリル(147 mg, 77%)が濾過によって得られた。追加量の生成物(40 mg)が母液から放射状クロマトグラフィー(1 mmシリカ、ジクロロメタン中1%のMeOH)によって単離され、全収率は98%になった。mp. 193〜194℃.元素分析、C19H18N4に対する計算値: C, 75.470; H, 6.00; N, 18.53. 実測値:C, 75.28; H, 5.99; N, 18.48. IR (KBrペレット): 2210 cm-1 (CN), 3270 cm-1 (NH); 1H NMR (360MHz, CDCl3) δ: 3.1 (s, 6H, Me2N), 3.12 (d, 3H, MeNH, J=4.8Hz), 5.38 (bs, 1H, NH), 6.76 (dd, 1H, H-5’, J=5.3Hz), 6.92 (d, 1H, H-5, J=2.5Hz), 7.20 (dd, 1H, H-7, J=2.5 and 9.1Hz), 7.56 (dd, 1H, H-3, J=1.9及び8.6Hz), 7.73 (d, 1H, H-4, J=8.6Hz), 7.78 (d, 1H, H-8, J=9.1Hz), 7.94 (d, 1H, H-1, J=1.9Hz), 8.29 (d, 1H, H-6’, J=5.3Hz).
【0072】
実施例1(t)−7-(ジメチルアミノ)-3-(4-フルオロベンゾイル)-2H-1-ベンゾピラン-2-オンの調製
【化26】
600 mg (1.6 mmol)のKryptofix 2.2.2と87 mg (1.5 mmol)のフッ化カリウムが1 mLの水と3 mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させた。1 mLのアセトニトリルを加え、混合物を蒸発させた。このスープが二回繰り返された。2 mLの無水ジメチル・スルホキシド中の170 mg(0.5 mmol)の3-(4-ニトロベンゾイル)-7-(ジメチルアミノ)- 2H-1-ベンゾピラン-2-オン(上の実施例1(g)で記述されているように調製)が加えられ、混合物は150℃で1時間加熱された。混合物は冷却され、10 mLの水で希釈され、固体相抽出(いくつかのC18 Sep Pakカートリッジ)で抽出され、ジクロロメタンで溶出され、生成物はジクロロメタン:メタノールによるカラム・クロマトグラフィーによって単離され、17 mg (0.5 mmol, 10%)の黄色固体が得られた。1H NMR (360MHz, CDCl3) δ:3.15 (6H, s, NMe2), 6.53 (1H, d, 芳香族H), 6.67 (1H, dd, 芳香族H), 7.13 (2H, m, 芳香族H), 7.41 (1H, d, 芳香族H), 7.87 (2H, m, 芳香族H), 8.14 (1H, s, 4-H). 19F NMR (CDCl3) δ: 34.7 ppm.
【0073】
実施例1(u)−2-{1-[7-(ジメチルアミノ)-2-オキソ-2H-1-ベンゾピラン-3-イル]エチリデン}マロノニトリル
【化27】
233 mg (1 mmol)の3-アセチル-7-(ジメチルアミノ)-2H-1-ベンゾピラン-2-オン(上の実施例1(g)に記述されているように調製)と70 mg (1 mmol)のマロノニトリルが2 mLのピリジンに溶解され、100℃で6時間加熱された。溶媒を蒸発させ、生成物がカラム・クロマトグラフィー(シリカゲル、ジクロロメタン)によって単離された。225 mg (81%)のオレンジ色の固体。
1H NMR (360MHz, CDCl3) δ:2.67 (3H, s, Ac); 3.15 (6H, s, NMe2), 6.51 (1H, br, aromatic H); 6.67 (1H, dd, aromatic H); 7.39 (1H, d, aromatic H), 7.95 (1H, s, 4,-H).
【0074】
実施例1(v)−7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-onesの調製
【化28】
7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを調製する合成アプローチは上の実施例1(g)においてジメチルアミノ類似体に関して記述されたアプローチと大体同様である。3-[(ヒドロキシエチル)(メチル)アミノ]フェノールは商業的に入手できないので、触媒としてのホウ酸の存在下でのレソルシノールと対応するアミンから調製する単純な方法を用いることができる。この合成手順は、また、N,N-ジ置換された3-アミノフェノールを選択したアミノ基で合成することを可能にする。
【0075】
30 g (272mmol)のレソルシノール、25 mL (311 mmol)の2-(メチルアミノ)-エタノール、及び2.0 g (32 mmol)のホウ酸が、分画カラムを備えた3首フラスコの中で還流で加熱された。9時間の過程で水が蒸留除去され、内部温度は180℃から230℃までゆっくりと上昇した。約60℃まで冷却した後、35 mLのメタノールを加え、ホウ酸メチルとメタノールの混合物をカラムでゆっくりと蒸留させた。過剰な2-(メチルアミノ)エタノールは15 mmHgで蒸留され、残ったオイルは高真空(0.4 mmHg)の下で蒸留された。少量のレソルシノールが155〜165℃までの間で蒸留され、165から175℃までの間で3-[(ヒドロキシエチル)(メチル)アミノ]フェノールが蒸留された。収量:32.5 g (215 mmol, 79%).
【0076】
3-[(ヒドロキシエチル)(メチル)アミノ]フェノールは、POCl3とDMFから0℃で調製されたVielsmeier試薬(4 mol当量)に加えられ、混合物は60-70℃で1時間加熱された。反応混合物は氷に注がれ、酢酸エチルで抽出された。有機部分を蒸発させ、残渣を希アンモニアで処理してヒドロキシエチル基を脱ホルミル化(de-formylate)した。中和して酢酸エチルで抽出した後、生成物をカラム・クロマトグラフィー(シリカゲル、クロロホルム)によって単離した。
【0077】
7-[(2-ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを生成するため、5 mmolの4-[(ヒドロキシエチル)(メチル)アミノ]-2-ヒドロキシベンズアルデヒドと6 mmolの適当なジカルボニル化合物が10 mLの無水エタノールに溶解された。20滴のピペリジンを加え、混合物を還流で2〜6時間加熱した。混合物をフリーザーで冷却し沈殿を濾過して取り、エタノールで洗浄した。
【0078】
実施例1(w)−2-(1-{7-[(2-ヒドロキシエチル)(メチル)アミノ]-2-オキソ-2H-1-ベンゾピラン-3-イル}エチリデン)マロノニトリルの調製
【化29】
1 mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンと1 mmolのマロノニトリルを2 mLのピリジンに溶解し、100℃で6時間加熱した。溶媒を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、ジクロロメタン)によって単離した。
【0079】
実施例1(x)−2-[(3-アセチル-2-オキソ-2H-1-ベンゾピラン-2-イル)(メチル)アミノ]エチル-4-メチルベンゼンスルフォネートの調製
【化30】
2 mLの無水ピリジン中で1 mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを3 mmolのp-トルエン無水スルフォン酸と室温で混合した。2時間後、混合物を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、酢酸エチル)によって単離した。
【0080】
実施例1(y)−2-[[3-(2,2-ジシアノ-1-メチルビニル)-2-オキソ-2H-1-ベンゾピラン-7-イル](メチル)アミノ]エチル4-メチルベンゼンスルフォネートの調製
【化31】
2 mLの無水ピリジン中で1mmolの3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンを3mmolのp-トルエン無水スルフォン酸と室温で混合した。2時間後、混合物を蒸発させ、生成物をカラム・クロマトグラフィー(シリカゲル、酢酸エチル)によって単離した。
【0081】
実施例1(z)−3-アセチル-7-[(ヒドロキシエチル)(メチル)アミノ]-2H-1-ベンゾピラン-2-オンの調製
【化32】
2mmolのKryptofix 2.2.2と2 mmolのフッ化カリウムが1mLの水と3mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させ、1mLのアセトニトリルに再溶解して蒸発させた(3回)。2mLのアセトニトリル中の1mmolの2-[(3-アセチル-2-オキソ-2H-1-ベンゾピラン-7-イル)(メチル)アミノ]エチル-4-メチルベンゼンスルフォネートを加え、混合物を90℃で20分間加熱した。混合物を蒸発させ、生成物をカラム・クロマトグラフィーによって単離した。
【0082】
実施例1(aa)−2-(1-{7-[(2-フルオロエチル)(メチル)アミノ]-2-オキソ-2H-1-ベンゾピラン-2-イル}エチリデン)マロノニトリルの調製
【化33】
2 mmolのKryptofix 2.2.2と2 mmolのフッ化カリウムが1 mLの水と3 mLのアセトニトリルに溶解された。混合物を100℃でアルゴンの流れの下で蒸発させ、1 mLのアセトニトリルに再溶解して蒸発させた(3回)。2 mLのアセトニトリル中の1 mmolの2-[[3-(2,2-ジシアノ-1-メチルビニル)-2-オキソ-2H-1-ベンゾピラン-7-イル](メチル)アミノ]エチル-4-メチルベンゼンスルフォネートを加え、混合物を90℃で20分間加熱した。混合物を蒸発させ、生成物をカラム・クロマトグラフィーによって単離した。
【0083】
実施例2
脳組織及びラットの脳を用いて、in vitro 及びin vivoでのβ-アミロイド斑の検出とラベリングが以下の手順に従って行われた。
【0084】
2.1 mg/mLのDDNPストック溶液が調製され、100%エタノールで8 mMに調整された。DDNP作業溶液はこのストック溶液を蒸留水で1:100〜1000(ストック溶液:蒸留水)という比に希釈して調製された。
【0085】
β-アミロイド250μM(蒸留水中で1.25 mg/mL)を37℃で48時間凝集させた。スライドに5μLをなすりつけ、空気乾燥し、再び蒸留水で水和させた。あるいはまた、Aβ-ポジティブな脳組織切片を蒸留水で再水和させた。DDNP作業溶液が各スライドに室温で30分間塗布された。スライドは蒸留水で5分間、3回洗浄された。スライドは蛍光保護マウンディング・メディア(VectashieldTM, Vector Labs., Burlingame, Californiaから入手可能)と共にカバーガラスをかぶせて、チオフラビンS又はFITCフィルターを用いて蛍光顕微鏡の下で観察された。
【0086】
β-アミロイド250μM(蒸留水中で1.25 mg/mL)を37℃で48時間凝集させ、塗抹によって確認される繊維(fibrils)を生成した。3匹のラットが麻酔された。3μLのAβ繊維(1.25 μg/μL)が各ラットの皮質に片側性で注射された(Bregma 0, AP -4.1 mm, ML + 2.0 mm, DV -3.1 mm)。次に、ビヒクル対照として、3μLのリン酸緩衝生理食塩水(PBS)が各ラット脳の反対側に注射された。注射後、針を5分間残して還流を防ぎ、その後頭蓋の孔を骨ろうで封止した。ラット脳にβ-アミロイドを注射してから8日後、ラットに10マイクロリットルのDDNP作業溶液を注射した(320マイクロモル)。これは、DDNPストック溶液をリン酸緩衝生理食塩水、pH 7.2),で1.5% BSA (ブタ血清アルブミン)に希釈して調製されたものである、pH 7.2)。
【0087】
1時間後、ラットにPLP固定液(0.05 Mリン酸緩衝液中4%パラホルムアルデヒド、1%ライシン、pH 7.4)を心臓灌流させた。ラット脳の追加浸漬固定がラットPLP固定液を用いて4℃で一晩行われた。ラット脳はPBSで洗浄され、10及び20%スクロースで飽和され、液体窒素によって冷却されたイソペンタン(-70℃)中でスナップ凍結された。脳は針の通路のまわりで10μMで凍結切片にサレ、グリセリンと蛍光保護剤(VectashieldTM)と共に直接カバースガラスがかぶせられた。脳切片は蛍光顕微鏡で観察された。
【0088】
図1Aから1Fまでは、AD患者及びトランスジェニック・マウスの脳からの切片におけるラベルされたアミロイド斑を示しており、DDNPがアミロイド斑をラベルできることがはっきりと示されている。図2Aから2Eまでは、ラベルされたβ-アミロイド斑を示し、DDNPがラットにおける血液−脳関門を通過することをはっきりと示している。
【0089】
DDNPは、チオフラビンSと同程度の感度レベルでAD脳組織の凍結切片及びパラフィン切片におけるアミロイド沈着を容易にラベルするということが見出された。DDNPを使用することは、チオフラビンSに比べていくつかの利点がある。すなわち、DDNPの使用は予備処理を必要とせず、チオフラビンSと異なり、最小の洗浄で、ホルマリン又はパラホルムアルデヒドによる固定なしで、また組織の区別なく使用できる。ストック溶液はフリーザーに6ヶ月保存しても1/100から1/1,000の希釈で受容できる結果が得られ、チオフラビンSラベリングのようにストックを新しく作る必要がない。
【0090】
実施例3
in vivoでのヒトβ-アミロイド斑と神経原繊維変化のラベリングが以下の手順を用いて行われた。
【0091】
患者を断層撮影装置に入れられて脳の動的PET画像を撮影した。実施例1(f)で記述されているように調製された8.0 mCiの2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン([F-18]FDDNP)(比放射能:5〜12 Ci/マイクロモル;質量:約1ナノモル)が、患者の腕に静脈注射された。動的に取得された脳画像のデータが47の脳平面で2時間、同時に記録された。
【0092】
[F-18]FDDNPは、血液−脳関門を容易に通過し、ベータアミロイド斑と神経原繊維変化の存在と矛盾しない仕方で脳構造をラベルすることが見出された。この患者は、脳萎縮をモニターするために、以前に18F-フルオロデオキシグルコース(FDG)/陽電子断層撮影(PET)スキャン,並びにMRIスキャンを受けていた。MRIスキャンにおいて最大の萎縮が認められていた部位(下方側頭部と頭頂葉)で、[F-18]FDDNPラベルの最大の蓄積が観察された。これらの部位では低グルコース代謝(EDG/PETスキャンで測定される)も認められた。
【0093】
実施例4
in vivoでのヒトβ-アミロイド斑と神経原繊維変化のラベリングが以下の手順を用いて行われた。10人のヒト被験者、すなわち、7人のアルツハイマー病患者(71〜80歳まで)と3人の対照患者(62〜82歳まで)、が調べられた。患者はEXACT HR + 962断層撮影装置(Siemens-CTI, Knoxville, Tennessee)に仰臥で、画像面がorbito(眼窩)-meatal lineと平行になるように配置された。静脈カテーテルを取り付け、ヒト血清アルブミン中(25%)の[F-18]FDDNP(5〜10 mCi)が静脈カテーテルを経由する巨丸剤(bolus)として投与された。[F-18]FDDNPの投与直後から始めて、次のスキャン・シーケンスを用いてシーケンシャル放出スキャンが得られた:すなわち、6回の30秒スキャン、4回の3分スキャン、5回の10分スキャン、及び3回の20分スキャン、である。2人の被験者で取り付けられているカテーテルによって迅速な静脈血サンプリングを行って、インプット機能の測定と血漿中の代謝産物分析を行った。
【0094】
図3Aは、アルツハイマー病患者の海馬−扁桃体−内側嗅領−側頭皮質領域を通る脳断面のPET-[F-18]FDDNP (2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン)画像である。この画像は[F-18]FDDNPの注射後、30〜60分までに得られた走査。データから再構築された。同時に記録されたこの患者のPET-FDG (FDGとは2-[F-18]フルオロ-2-デオキシ-D-グルコースである)及びMRI (プロトン緩和時間)画像も、それぞれ図3Bと3Cに示されており、それぞれこの断面でのグルコース代謝と解剖学的構造についての情報を提供する。内側側頭領域は、[F-18]FDDNPスキャンでは暗く(遅いクリアランス)、FDGスキャンでは明るく(低いグルコース代謝)見える。
【0095】
図4は、アルツハイマー病患者における[F-18]FDDNPの推定滞留時間が、対照患者における値と異なるように見られることを示している。示された滞留時間は、アルツハイマー病の病理との関連が小さいと考えられる部位である脳橋における値を基準としている。滞留時間は、影響がある関心領域(ROI)と脳橋におけるクリアランス速度(rate)から次のように計算される:
滞留時間=[1/影響があるROIのクリアランス速度]−[1/脳橋のクリアランス速度]
内側嗅領皮質、海馬、外側側頭皮質、脳橋で別々のROIsが定められた。クリアランス速度が最も遅い領域が図4における滞留時間の計算で影響があるROIとして用いられた。
【0096】
静脈注射の後、[F-18]FDDNPは血液−脳関門を血流量に比例して容易に通過することが見出された。放射能の蓄積の後、領域によって異なる[F-18]FDDNPのクリアランスが続いた。ゆっくりしたクリアランスはβ-アミロイド斑と神経原繊維変化を蓄積することが確実に知られている脳部位で認められた、具体的には、海馬−扁桃体−内側嗅領コンプレックス、並びに、疾病がさらに進んだ状態では側頭葉及び頭頂葉皮質、に認められた。これらの被験者においてPETで測定されたrCMRGIも、予期されるβ-アミロイド斑の量、及び神経原繊維変化の存在の可能性、と矛盾していなかった。これらの患者で、低グルコース代謝の脳領域は一般に[F-18]FDDNPの高い保持率の領域と一致していた。海馬−扁桃体−内側嗅領皮質は、ほとんどの場合、症状が重篤でない患者においても、放射能([F-18]FDDNP)の高い保持率を示した。正常な82歳のボランティアは、図4に示されるように、[F-18]FDDNPによるPET研究では海馬−扁桃体−内側嗅領コンプレックスにおける放射能の沈着(deposition)を示し、同じ部位でFDGによる測定では低いrCMRGIを示した。これらの結果は、痴呆の明らかな兆候がない高齢者が内側嗅領皮質のニューロンの第二層で神経原繊維変化の病理、及び海馬形成体で斑を示すことがあるという観察と矛盾しない。症状の重篤化には、常に放射能の保持率の増加と側頭、頭頂葉、又は前頭皮質からのクリアランスの遅れが伴っており、これらの部位において予期されるAβと神経原繊維変化の沈着(deposition)と一致している。
【0097】
アルツハイマー病患者の脳試料についての[F-18]FDDNPを用いたin vitroオートラジオグラフィーも、β-アミロイド斑と神経原繊維変化の存在と合致する放射能の分布を示した。Aβ及びタウ抗体の免疫染色による結果と合致する結合が海馬、側頭、頭頂葉皮質で見られた。DNNPとその誘導体は蛍光を示すので、[F-18]FDDNPがin vitroでβ-アミロイド斑及び神経原繊維変化をラベルする能力の評価も同じ脳試料について行われた。すべてのアルツハイマー病の脳試料で、神経原繊維変化、アミロイドペプチド、及び拡散したアミロイドが、DDNPと[F-18]FDDNPの両方によってはっきりと目に見えるように視覚化され、同じサンプルについてのチオフラビンS (24)による結果と一致した。
【0098】
図5で、中央の画像は、ATS(antiphosphotau)と10G4(anti-AB1-15)によってインキュベートしたあるアルツハイマー病患者の45マイクロメーターの低温側頭皮質切片を免疫染色して得られた1 : 800の図である。挿入図は、FDDNPで染色された同じアルツハイマー病の脳試料の隣接する切片である、画像は蛍光顕微鏡を用いて生成された。グリーンの矢印は、中央の免疫染色切片に対する挿入図の大体の原位置を示す。挿入図は、左上隅から時計回りに、(1) 老人斑、(2) 拡散した斑、(3) 血管性アミロイド、(4) 密な斑と神経原繊維変化、及び(5) 密な神経原繊維変化、を示す。
【0099】
実施例5
考えられる治療物質が評価され、β-アミロイド原繊維に結合する能力が決定された。
β-アミロイド(1-40)原繊維形成
β-アミロイド(1-40)(Biosource, Camarillo, CA)原繊維が調製された。0.5 mgのβ-アミロイド(1-40)が1 mL PBS, pH 7.4, に溶解され、磁気攪拌棒で3日間37℃で攪拌され、目で見て濁った溶液が得られた。原繊維は、その生成が確認された直後に用いられた。β-アミロイド原繊維の生成はJeol 100CX透過電子顕微鏡(Jeol, Peabody, MA)による画像撮影で確認された。一滴の5μLの原繊維溶液を処理された銅のグリッド上で30 秒間落ち着かせた後に一滴の2%酢酸ウラニル溶液で洗い流した。最後に、別の一滴の2%酢酸ウラニルをグリッドに加えて原繊維をネガティブに染色(negatively stain)した。原繊維は、その生成が確認された直後に用いられた。Congo red (CR, Sigma)とチオフラビンT (TT, Sigma)を用いて原繊維の形成の別のテストも行われた。以下で述べるin vitro競合分析における濾液に原繊維が存在しないこともCRを用いた同じテストによって決定された。
【0100】
[18F]FDDNPによるNSAIDs及びチャージアミロイド色素に対するin vitro 競合分析
各放射性競合分析のために、非放射性物質、すなわち、(S)-ナプロキセン(Sigma)、(R)-イブプロフェン(BIOMOL, Plymouth Meeting, PA)、(S)- イブプロフェン(Sigma)、ジクロフェナック(Sigma)、CR 及びTT、が新たに調製された。ステンレス鋼サポート・スクリーン(Millipore)とガラスサンプルチャンバで改造された1225サンプリング・マニホールド(Millipore)でAPFFグラスファイバーフィルター(0.7μm粒子保持;Millipore, Bedford, MA)が用いられた。真空濾過では、PBS, pH 7.4 (1%エタノール)、で1時間インキュベートされた合成β-アミロイド(1-40)の0.865μg/mLのin vitro原繊維と37 MBq/mLの[18F]FDDNPを、0.1 pMから83 μMまでのいろいろな濃度の非放射性物質と共に用いた。次に、各フィルターを3 mlのPBS, pH 7.4, で2回洗浄した。フィルターに残された放射能を、Packard Cobra II Auto-Gammaガンマカウンター(Packard, Meriden, CT)で測定し、共通基準時間に減衰補正(decay-corrected)した。競合相手なしの放射性ラベルされた[18F]FDDNP のβ-アミロイド原繊維への結合は100%特異的結合と決定された。放射性[18F]FDDNPに対する100%競合は40μMの非放射性FDDNPで見られると仮定された。すべての競合分析は三重重複で行われた。競合結果が分析され、Ki値がLigand Binding Module for SigmaPlot 2001 (SPSS, Chicago, IL)を用いて計算された。
【0101】
脳組織の調製
79歳の女性の死後診断で確定されたAD患者からの脳検体が処理された。簡単に言うと、ホルマリン処理された、低温保存された脳検体が、冠状で70μm厚さの切片に切断され、ゼラチン塗布されたガラス・スライドに載せられ、放置乾燥され、キシレン中で40分間脱脂された後、エタノールで組織が洗浄された。最後に、いくつかの脳検体でリポフスチン自己蛍光は、50 mM酢酸アンモニウム緩衝液、pH 5、中の10 mM CuCl2を用いて停止させてから染色した。
【0102】
ディジタル・オートラジオグラフィー
死後診断で確定したADの脳検体(厚さ70μm)が、100 nMの新しいバッチの非放射性FDDNP及び(S)-ナプロキセン又は40μMの(R)-イブプロフェン、(S)-イブプロフェン、ジクロフェナック、CR又はTTによって、PBS, pH 7.4, 中の10%エタノールの中で60分間プレ処理され、液体を流して空にしてから[18F]FDDNPによるディジタル・オートラジオグラフィーを行った。前処理された競合相手なしの凍結切片が室温で25分間、凍結切片あたり10 mLの0.9%(w/v)塩水中の1%エタノールに溶解された3.7 GBqの[18F]FDDNPと共にインキュベートされた。インキュベーションの後、切片は、水(30 sec);区別化のために(Bancroft and Stevens, 1990)Junior Orbit Shaker (Lab-Line Instrujents, Melrose Park, IL)で40 RPMsで攪拌された60% 2-メチル-2-ブタノール(3 min; Sigma);その後で水(30 sec)によって最適に洗浄された。切片は、暖かいホットプレートで、暖かい空気の定常的な流れによって乾燥され、β+-敏感なリンプレートに40分間さらし(Fuji Film Medical Systems USA, Stanford, CT)、以前に記述されたように(Agdeppa et al., 2001a)FUJI BAS 5000 Phosphrimager (Fuji)で25μmという分解能でスキャンされた。画像が撮影された検体からの組織削り落としからの放射能がその後Packard Cobra II Auto-Gamma (Packard)によって測定され、共通基準時間に減衰され、オートラジオグラムにおける[18F]FDDNPの特異的結合の量(放射能/面積、Bq/mm2, 図2Q)を定量化するための放射性基準として用いられた。オートラジオグラフィーは、各競合相手について少なくとも三重重複で行われた。オートラジオグラムの統計解析は、Prism 3.02 (GraphPad, San Diego, CA)を用いてDunnettのポストテストによる一元配置分散分析(one-way ANOVA)を行って、非放射性物質でプレ処理されたオートラジオグラムとプレ処理なしのオートラジオグラムで、灰色物質対白色物質(図2Q)[18F]FDDNP放射能の比の差を比較した。対応のないt-テストを行って、各オートラジオグラムの灰色物質と白色物質における組織の面積あたりの測定された[18F]FDDNP放射能の差を比較した。
【0103】
蛍光顕微鏡
オートラジオグラフィーに用いたと同じ脳検体を蛍光顕微鏡を用いて観察した。組織はVectashield (Vector, Burlingame, CA)によってマウントし、Nikon Labophot 蛍光顕微鏡(Nikon USA, Melville, NY)でFITCフィルターセットを用いて観察した。
【0104】
[18F]FDDNPによるオートラジオグラフィーに以前に用いた組織の蛍光顕微鏡観察は、FDDNPの蛍光性質と残っている非放射性FDDNPによるSPのラベリングによって可能になる。合成の終わりでの非キャリア付加[18F]FDDNPの比放射能(単位質量当たりの放射能)は74〜222 GBq/μmol (2000〜6000 Ci/mmol)であり、最大理論的比放射能よりも103倍も小さい(SorensonとPhelps, 1987)。したがって、18Fの減衰後、残っているAD 脳検体におけるSPsと結合した非放射性FDDNPを蛍光顕微鏡で画像として見ることができる。
【0105】
上記のテストは、ナプロキセンとイブプロフェンがβ-アミロイド原繊維上で[18F]FDDNPと同じ結合部位を共有していることを示した。いろいろな濃度の非放射性物質を[18F]FDDNPと合成β-アミロイド原繊維と共に同時インキュベートして行われたin vitro競合におけるカーブは、(S)-ナプロキセン、(R)-イブプロフェン、及び(S)-イブプロフェン、に関する一部位結合競合を明らかにした(p=0.05)。(S)-ナプロキセン、(R)-イブプロフェン、(S)-イブプロフェン、に対する[18F]FDDNPの結合の濃度依存的な減少は、Ki値として、それぞれ、Ki=5.70±1.31 nM (±SD), Ki=44.4±17.4 μM (±SD), 及びKi=11.3±5.20 μM (±SD), という値を与え、(S)-ナプロキセンがβ-アミロイドによりしっかりと結合することを示した。ジクロフェナック、CR, TT, は[18F]FDDNPの特異的結合において量依存的な減少を示さなかった。
【0106】
さらに、ナプロキセン及びイブプロフェンによるオートラジオグラフィーは、ex vivoでの老人斑における[18F]FDDNP結合部位の完全なブロックを示した。競合相手が内場合の前頭AD脳検体の放射能の大まかなパタンは、老人斑を含む領域への[18F]FDDNPの特異的結合を明らかにした。老人斑を含む灰色物質の領域への[18F]FDDNPの特異的結合は、非放射性FDDNP、(S)-ナプロキセン、(R)-イブプロフェン、及び(S)-イブプロフェン、によるプレ処理されたAD検体において、これらの競合相手がない場合のオートラジオグラフィーに比べて著しく減少した。これらの競合相手がある場合、老人斑がある灰色物質領域と老人斑を欠く白色物質の間の放射能の差(放射能/面積、Bq/mm2;図2Q)は有意でなく(p > 0.05)、灰色物質対白色物質の放射能の比が、競合相手がない場合の[18F]FDDNPオートラジオグラムにおけるこの比に比べて有意に低い(p < 0.05)として矛盾がなかった。これらの結果は、[18F]FDDNPの特異的結合が白色物質に見られる放射能のバックグラウンドレベルまでブロックされることを示している。これらの結果はすべて同じ脳検体についての蛍光顕微鏡観察によって確認された。ジクロフェナック、CR及びTTは、同じ脳検体のオートラジオグラフィーと蛍光顕微鏡観察から判断して、老人斑への[18F]FDDNPの特異的結合に対する影響は最小であった。灰色物質と白色物質の放射能の差は、ジクロフェナック、CR及びTTに関して有意に差があり(p < 0.05)、競合相手がない場合の[18F]FDDNPオートラジオグラフィーと同様であった。ジクロフェナック、CR及びTTの間での灰色物質対白色物質の放射能の比と、競合相手がない場合の[18F]FDDNPでの比との比較は、老人斑への[18F]FDDNPの特異的結合が保存されることを示す。
【0107】
本発明の方法に関する詳細は、本明細書の付録Aと付録Bに与えられており、そこに開示されたことは参照によって本明細書に組み込まれる。
【0108】
より広い様態で、本発明は、ここで示され説明された特定の細部に限定されない。本発明の原理から外れることなく、その主な利点を失うことなく、そのような細部に変更を加えることができる。
【図面の簡単な説明】
【0109】
図面の説明
本発明のこれらの及びその他の特徴と利点は、以下の詳細な説明を添付の図面と合わせて参照することによってさらに良く理解されるであろう。添付図面のうち:
【図1】図1Aは、アルツハイマー病患者の脳皮質におけるラベルされたアミロイド斑の2-(1,1-ジシアノプロペン-2-イル)-6-ジメチルアミノナフタレン(DDNP)蛍光(ex 490 nm, em 520〜530 nm)を示す(X400)。図1Bは、アルツハイマー病患者の脳皮質における斑の強いDDNPラベリングともつれの弱いDDNPラベリングを示す(X640)。図1Cは、ヒトの脳におけるアミロイドコアを有する単一の大きな斑のDDNPラベリングを示す(X640)。図1Dは、Tg2576HuAPPswトランスジェニックマウスの脳における斑のDDNPラベリングを示す(X500)。図1Eは、アルツハイマー病の人の脳におけるコアを有する斑のチオフラビンSラベリングを示す(X640)。図1Fは、図1Eに示されたと同じ人の脳切片のアミロイドβ蛋白質の4G8抗体ラベリングを示す(X640)。
【図2】図2Aは、ラットの脳に注射されたアミロイドのラベリングを示しており、ある分量のβ-アミロイド1-40を8日間37℃で放置して凝集させ、ゼラチンを塗ったスライドに乾燥させ、DDNPによってラベルし、アミロイドに合致する繊維上の蛍光がはっきりと示された。図2Bは、ラットの脳に注射されたアミロイドのラベリングを示しており、ラットの皮質に3 μgの凝集したβ-アミロイド1-40を一側性定位で注射して8日後、ラットに100μLの640μM DDNPを頸動脈に注射し、麻酔し、20分後に灌流によって屠殺し、脳を凍結切片にして蛍光を調べた:図2Bはneed trackの先端にin vivoでDDNPによって蛍光ラベルされたアミロイドをはっきりと示している(X100)。図2Cは、図2Bのin vivo DDNPラベルされた材料の高倍率図を示す(X200)。図2Dは、注射部位による切片のギ酸処理が蛍光ラベリングを除去する様子を示す(X100)。図2Eは、アミロイドが存在しないアミロイド注射部位の反対側でDDNPラベリングが弱いことを示す(X200)。
【図3】図3Aは、アルツハイマー病患者の海馬−扁桃体−内側嗅領/側頭皮質領域を通る脳断面のPET-[F-18]-FDDNP(2-(1,1-ジシアノプロペン-2-イル)-6-(2-[18F]-フルオロエチル)-メチルアミノ)-ナフタレン)画像である。図3Bは、図3Aの脳断面のPET-FDG(FDGは2-[F-18]フルオロ-2-デオキシ-D-グルコースである)画像である。図3Cは、図3Aの脳断面のMRI画像(陽子緩和時間)である。
【図4】図4は、患者における[F-18]-FDDNPの推定滞留時間を示すグラフである。
【図5】図5は、1:800のAT8(anti-phosphotau)と10G4(anti-AB1-15)で培養されたアルツハイマー病患者の45マイクロメーター低温側頭皮質切片を免疫染色することによって得られた画像(中央画像)を示す。挿入図は、FDDNPで染色された同じアルツハイマー病の脳検体の隣接切片であり、左上隅から時計回りに、(1)老人斑(neuritic plaque)、(2)拡散した斑、(3)血管性アミロイド、(4)レンズ斑ともつれ、及び(5)密なもつれ、を示す。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3A】
【図3B】
【図3C】
【特許請求の範囲】
【請求項1】
β-アミロイド斑及び神経原繊維変化から成る群から選択される構造を、in vivo又はin vitroでラベルする方法であって、脳組織を次の化学式(IA)又は(IB):
【化1】
(式中;
Qは、-NR2R3, -OR9及び-SR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
X2は、N及び-CR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9, 及び-C(R9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及び-SR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N及び-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化2】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル-R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1及びA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング, -C(O)O-アルキル, -C(O)O-アルケニル, -C(O)O-アルキニル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、-OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル、-O-アルケニル、-O-アルキニル、-S-アルキル、-S-アルケニル、及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;かつ
R9は、水素、アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成し;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではなく;
さらに、ここで、1又は複数の水素原子が任意に放射性ラベルで置き換えられる)
化合物と接触させるステップを含む方法。
【請求項2】
化学式(I)の化合物を18F, 75Br, 76Br, 123I,又は124Iによって放射性ラベルすることを特徴とする請求項1に記載の方法。
【請求項3】
さらに、該構造がラベルされたかどうかを蛍光顕微鏡によって該脳組織を観察することによって決定するステップを含む、請求項1に記載の方法。
【請求項4】
請求項1に記載の該化合物を放射性ラベルすることを特徴とする請求項1に記載の方法。
【請求項5】
さらに、体の内部の放射性ラベルされた化合物の分布を検出し表すことができる方法によって該脳組織を観測することによって該構造がラベルされたかどうかを決定するステップを含む、請求項4に記載の方法。
【請求項6】
該脳組織を陽電子断層撮影法を用いて観測することを特徴とする請求項5に記載の方法。
【請求項7】
該化合物が該化学式(IA)を有することを特徴とする請求項1に記載の方法。
【請求項8】
Qが-NR2R3であることを特徴とする請求項7に記載の方法。
【請求項9】
R1が-C(A2)=C(CN2)であることを特徴とする請求項7に記載の方法。
【請求項10】
A2がアルキルであることを特徴とする請求項9に記載の方法。
【請求項11】
該化合物が該化学式(IB)を有することを特徴とする請求項1に記載の方法。
【請求項12】
Qが-NR2R3であることを特徴とする請求項11に記載の方法。
【請求項13】
R3とZ1が一緒になって複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項14】
R3とZ2が一緒になって複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項15】
化学式IBの化合物が、以下の化合物:
【化3】
(式中、nは1〜3までにわたる)
から選択されることを特徴とする請求項11に記載の方法。
【請求項16】
R2とR3が一緒になって、アルキル、アルケニル、アルキニル、アルコキシ、OH、OTs、ハロゲン、カルボニル、スピペロン、スピペロンケタール、及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項17】
アルツハイマー病を治療又は予防する治療物質の能力を決定する方法であって;
(1)β-アミロイドペプチドを、in vivo又はin vitroで、該治療物質及び化学式(IA)又は(IB):
【化4】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル、-N-(アルキル)2, -O-アルキル、及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化5】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル、アルコキシ、OH, OT, ハロゲン、アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで、1又は複数の水素、ハロゲン又は炭素原子が放射性ラベルによって置き換えられ;
それにより放射性ラベルされた化合物の少なくとも一部がβ-アミロイドペプチドと結合する)
の放射性ラベルされた化合物と接触させるステップ;及び
(2)該β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を決定し、それにより該治療物質が実際に該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む方法。
【請求項18】
該治療物質を最初に該β-アミロイドペプチドと接触させ、その後放射性ラベルされた化合物を該β-アミロイドペプチドと接触させることを特徴とする請求項17に記載の方法。
【請求項19】
最初に、該放射性ラベルされた化合物を該β-アミロイドペプチドと接触させて該放射性ラベルされた化合物が該β-アミロイドペプチドと結合する程度を決定するステップ;
その後、該治療物質をある期間にわたって投与して該治療物質が該β-アミロイドペプチドと接触できるようにするステップ;及び
再び該放射性ラベルされた化合物を該β-アミロイドペプチドと接触させて該放射性ラベルされた化合物が該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む請求項17に記載の方法。
【請求項20】
ある治療物質がβ-アミロイドペプチドと結合する能力を決定する方法であって;
(1)β-アミロイドペプチドを、in vivo又はin vitroで、該治療物質及び化学式(IA)又は(IB):
【化6】
ここで:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化7】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2, 及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル、-O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン, アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで、水素、ハロゲン又は炭素原子の一つ以上が放射性ラベルによって置き換えられ;
それにより放射性ラベルされた化合物の少なくとも一部がβ-アミロイドペプチドと結合する)
の放射性ラベルされた化合物と接触させるステップ;及び
(2)該β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を決定し、それにより該治療物質が実際に該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む方法。
【請求項21】
患者におけるアルツハイマー病を治療又は予防する方法であって、治療的に有効な量の化学式(IA)又は(IB):
【化8】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N-(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化9】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、環式リング、複素環式リング、-O-アルキレニル-R4、-NH-アルキレニル-R4、-アルキレニル-R4、-アルキレニル-NHR4、-アルキレニル-NH2、及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル、-C(O)O-アルキレニル-R4、-C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン、アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成する)
の化合物;
を該患者に投与するステップを含む方法。
【請求項22】
化学式(IA)又は(IB):
【化10】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N(アルキル)2, -O-アルキル、及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化11】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4、-アルキレニル-R4、-アルキレニル-NHR4, -アルキレニル-NH2及び-アルキレニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、アリール、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択されるラジカルであり;
R7は、O, NH, 及びSから成る群から選択される基であり;そして
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン, アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで:
該化合物が化学式(IA)の化合物であり、X1がOであり、そしてY1がOである場合、A1はOでなく;
該化合物が化学式(IB)の化合物であり、X2、Z1、Z2、Z3、及びZ4がすべて水素である場合、X3はCHでなく;
該化合物が化学式(IB)を有し且つR1が-C(=C(CN)2)-A2である場合、(1)Z1、Z2、Z3、及びZ4少なくとも一つはN又は-C-アルキルである、(2)X2はNである、又は(3)Y2は、OH、SH、-NH2、-NH-アルキル、-N(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択される;
さらに、ここで、水素、ハロゲン、又は炭素原子の一つ以上は任意に放射性ラベルで置き換えられている);
の化合物を含む組成物。
【請求項23】
化学式(I)の該化合物が18F又は123Iによって放射性ラベルされていることを特徴とする請求項22に記載の組成物。
【請求項1】
β-アミロイド斑及び神経原繊維変化から成る群から選択される構造を、in vivo又はin vitroでラベルする方法であって、脳組織を次の化学式(IA)又は(IB):
【化1】
(式中;
Qは、-NR2R3, -OR9及び-SR9から成る群から選択され;
X1は、O, S, -NR4, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
X2は、N及び-CR9から成る群から選択され;
X3は、N及び-C-Y2から成る群から選択され;
Y1は、O, S, -NR9, 及び-C(R9)2から成る群から選択され;
Y2は、R9, -N(R9)2, -OR9及び-SR9から成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N及び-CR9から成る群から選択され;
R1は、-CN, -C(=A1)-A2, アルキル-A2, アルケニル-A2, アルキニル-A2,
【化2】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR9, 及び-C(COOR9)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルケニル-R4, -アルケニル-R4, -アルケニル-NHR4, -アルケニル-NH2, -アルケニル-N(R4)2, -O-アルケニル-R4, -NH-アルキニル-R4, -アルキニル-R4, -アルキニル-NHR4, -アルキニル-NH2, -アルキニル-N(R4)2, から成る群から選択され;
又は、A1及びA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になって環式リング又は複素環式リングを形成し;
R4は、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リング, -C(O)O-アルキル, -C(O)O-アルケニル, -C(O)O-アルキニル, -OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、水素、アルキル、アルケニル、アルキニル、環式リング、複素環式リング、-OH, -OTs, -SH, ハロゲン、-N(R4)2, -O-アルキル、-O-アルケニル、-O-アルキニル、-S-アルキル、-S-アルケニル、及び-S-アルキニルから成る群から選択される基であり;
R6は、水素、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)O-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;かつ
R9は、水素、アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択される基であり;かつ
R2及びR3は、それぞれ独立に、水素, アルキル, アルケニル, アルキニル, 環式リング, 複素環式リングから成る群から選択され;
又は、R2とR3は、一緒になって複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって複素環式リングを形成し;
ここで、化合物が化学式(IB)であり、かつX2, Z1, Z2, Z3, 及びZ4がすべてCHである場合、X3はCHではなく;
さらに、ここで、1又は複数の水素原子が任意に放射性ラベルで置き換えられる)
化合物と接触させるステップを含む方法。
【請求項2】
化学式(I)の化合物を18F, 75Br, 76Br, 123I,又は124Iによって放射性ラベルすることを特徴とする請求項1に記載の方法。
【請求項3】
さらに、該構造がラベルされたかどうかを蛍光顕微鏡によって該脳組織を観察することによって決定するステップを含む、請求項1に記載の方法。
【請求項4】
請求項1に記載の該化合物を放射性ラベルすることを特徴とする請求項1に記載の方法。
【請求項5】
さらに、体の内部の放射性ラベルされた化合物の分布を検出し表すことができる方法によって該脳組織を観測することによって該構造がラベルされたかどうかを決定するステップを含む、請求項4に記載の方法。
【請求項6】
該脳組織を陽電子断層撮影法を用いて観測することを特徴とする請求項5に記載の方法。
【請求項7】
該化合物が該化学式(IA)を有することを特徴とする請求項1に記載の方法。
【請求項8】
Qが-NR2R3であることを特徴とする請求項7に記載の方法。
【請求項9】
R1が-C(A2)=C(CN2)であることを特徴とする請求項7に記載の方法。
【請求項10】
A2がアルキルであることを特徴とする請求項9に記載の方法。
【請求項11】
該化合物が該化学式(IB)を有することを特徴とする請求項1に記載の方法。
【請求項12】
Qが-NR2R3であることを特徴とする請求項11に記載の方法。
【請求項13】
R3とZ1が一緒になって複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項14】
R3とZ2が一緒になって複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項15】
化学式IBの化合物が、以下の化合物:
【化3】
(式中、nは1〜3までにわたる)
から選択されることを特徴とする請求項11に記載の方法。
【請求項16】
R2とR3が一緒になって、アルキル、アルケニル、アルキニル、アルコキシ、OH、OTs、ハロゲン、カルボニル、スピペロン、スピペロンケタール、及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成することを特徴とする請求項11に記載の方法。
【請求項17】
アルツハイマー病を治療又は予防する治療物質の能力を決定する方法であって;
(1)β-アミロイドペプチドを、in vivo又はin vitroで、該治療物質及び化学式(IA)又は(IB):
【化4】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル、-N-(アルキル)2, -O-アルキル、及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化5】
から成る群から選択され;
ここで;
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、環式リング, 複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル、アルコキシ、OH, OT, ハロゲン、アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで、1又は複数の水素、ハロゲン又は炭素原子が放射性ラベルによって置き換えられ;
それにより放射性ラベルされた化合物の少なくとも一部がβ-アミロイドペプチドと結合する)
の放射性ラベルされた化合物と接触させるステップ;及び
(2)該β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を決定し、それにより該治療物質が実際に該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む方法。
【請求項18】
該治療物質を最初に該β-アミロイドペプチドと接触させ、その後放射性ラベルされた化合物を該β-アミロイドペプチドと接触させることを特徴とする請求項17に記載の方法。
【請求項19】
最初に、該放射性ラベルされた化合物を該β-アミロイドペプチドと接触させて該放射性ラベルされた化合物が該β-アミロイドペプチドと結合する程度を決定するステップ;
その後、該治療物質をある期間にわたって投与して該治療物質が該β-アミロイドペプチドと接触できるようにするステップ;及び
再び該放射性ラベルされた化合物を該β-アミロイドペプチドと接触させて該放射性ラベルされた化合物が該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む請求項17に記載の方法。
【請求項20】
ある治療物質がβ-アミロイドペプチドと結合する能力を決定する方法であって;
(1)β-アミロイドペプチドを、in vivo又はin vitroで、該治療物質及び化学式(IA)又は(IB):
【化6】
ここで:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化7】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4, -アルキレニル-R4, -アルキレニル-NHR4, -アルキレニル-NH2, 及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル、-O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル, -C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン, アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで、水素、ハロゲン又は炭素原子の一つ以上が放射性ラベルによって置き換えられ;
それにより放射性ラベルされた化合物の少なくとも一部がβ-アミロイドペプチドと結合する)
の放射性ラベルされた化合物と接触させるステップ;及び
(2)該β-アミロイドペプチドと結合しなかった放射性ラベルされた化合物の量を決定し、それにより該治療物質が実際に該β-アミロイドペプチドと結合する程度を決定するステップ;
を含む方法。
【請求項21】
患者におけるアルツハイマー病を治療又は予防する方法であって、治療的に有効な量の化学式(IA)又は(IB):
【化8】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N-(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化9】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン、アルキル、環式リング、複素環式リング、-O-アルキレニル-R4、-NH-アルキレニル-R4、-アルキレニル-R4、-アルキレニル-NHR4、-アルキレニル-NH2、及び-アルキレニル-N(R4)2, から成る群から選択される;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成する;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、環式リング、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル-(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル、-C(O)O-アルキレニル-R4、-C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル、-C(O)NH-アルキレニル-R4から成る群から選択される基であり;
R7は、O, NH, 及びSから成る群から選択される基であり;
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン、アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成する)
の化合物;
を該患者に投与するステップを含む方法。
【請求項22】
化学式(IA)又は(IB):
【化10】
(式中:
X1は、O, S, -NR4, -CH2, -CH-アルキル、及び-C(アルキル)2から成る群から選択され;
X2は、N, -CH, 及び-C-アルキルから成る群から選択され;
X3は、O, S, -NR4, 及び-C-Y2から成る群から選択され;
Y1は、O, S, -NH, -CH2, -CH-アルキル, 及び-C(アルキル)2から成る群から選択され;
Y2は、H, OH, SH, -NH2, -NH-アルキル, -N(アルキル)2, -O-アルキル、及びアルキルから成る群から選択され;
Z1, Z2, Z3, 及びZ4は、それぞれ独立に、N, -CH, 及び-C-アルキルから成る群から選択され;
R1は、-CN, -C(=A1)-A2,
【化11】
から成る群から選択され;
ここで:
A1は、O, S, -NR4, -C(CN)2, -C(CN)COOR4, 及び-C(COOR4)2 から成る群から選択され;かつ
A2は、OH, -O-アルキル, -NH2, -NH-R4, -N(R4)2, ハロゲン, アルキル, 環式リング、複素環式リング, -O-アルキレニル-R4, -NH-アルキレニル-R4、-アルキレニル-R4、-アルキレニル-NHR4, -アルキレニル-NH2及び-アルキレニル-N(R4)2, から成る群から選択され;
又は、A1とA2は、一緒になって環式リング又は複素環式リングを形成し;
又は、R1は、X3又はZ4と一緒になってR1, X3又はZ4以外の炭素においてA1置換基によって置換された環式リング又は複素環式リングを形成し;
R4は、アルキル、置換されたアルキル、アリール、置換されたアリール、-C(O)O-アルキル、-OH, -OTs, 及びハロゲンから成る群から選択される基であり;
R5は、-NH2, -OH, -OTs, -SH, ハロゲン, -NH-アルキル, -NHR4, -NH-アルキレニル- R4, アルキル(O-アルキル)2, -O-アルキル, -O-アルキレニル- R4, -S-アルキル, 及び-S-アルキレニル- R4から成る群から選択される基であり;
R6は、-CN, -COOH, -C(O)O-アルキル, -C(O)O-アルキレニル-R4, -C(O)-アルキル、-C(O)-アルキレニル-R4, -C(O)-ハロゲン, -C(O)NH2, -C(O)NH-アルキル, -C(O)NH-アルキレニル-R4から成る群から選択されるラジカルであり;
R7は、O, NH, 及びSから成る群から選択される基であり;そして
R8はNであり;そして
R2及びR3は、それぞれ独立に、アルキル及びアルキレニル-R10から成る群から選択され;ここで、R10は-OH, -OTs, ハロゲン, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択され;
又は、R2とR3は、一緒になって、任意にアルキル, アルコキシ, OH, OTs, ハロゲン, アルキレニル-R5, カルボニル, スピペロン, スピペロンケタール, 及びスピペロン-3-イルから成る群から選択される少なくとも一つの基によって置換された複素環式リングを形成し;
又は、R3は、Z1又はZ2及びR3が結合した窒素と一緒になって、複素環式リング又は置換された複素環式リングを形成し;
ここで:
該化合物が化学式(IA)の化合物であり、X1がOであり、そしてY1がOである場合、A1はOでなく;
該化合物が化学式(IB)の化合物であり、X2、Z1、Z2、Z3、及びZ4がすべて水素である場合、X3はCHでなく;
該化合物が化学式(IB)を有し且つR1が-C(=C(CN)2)-A2である場合、(1)Z1、Z2、Z3、及びZ4少なくとも一つはN又は-C-アルキルである、(2)X2はNである、又は(3)Y2は、OH、SH、-NH2、-NH-アルキル、-N(アルキル)2, -O-アルキル, 及びアルキルから成る群から選択される;
さらに、ここで、水素、ハロゲン、又は炭素原子の一つ以上は任意に放射性ラベルで置き換えられている);
の化合物を含む組成物。
【請求項23】
化学式(I)の該化合物が18F又は123Iによって放射性ラベルされていることを特徴とする請求項22に記載の組成物。
【図4】

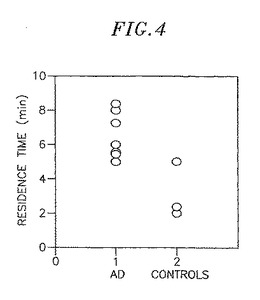
【図5】
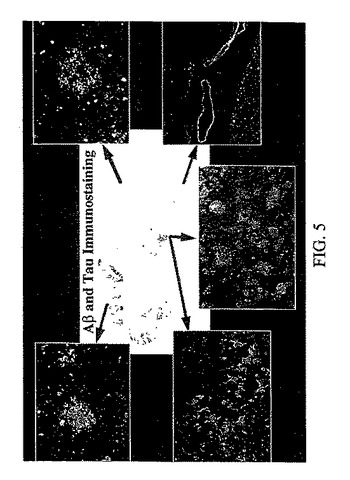
【図5】
【公表番号】特表2007−504283(P2007−504283A)
【公表日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願番号】特願2006−533296(P2006−533296)
【出願日】平成16年5月20日(2004.5.20)
【国際出願番号】PCT/US2004/016038
【国際公開番号】WO2005/040337
【国際公開日】平成17年5月6日(2005.5.6)
【出願人】(505431514)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (1)
【Fターム(参考)】
【公表日】平成19年3月1日(2007.3.1)
【国際特許分類】
【出願日】平成16年5月20日(2004.5.20)
【国際出願番号】PCT/US2004/016038
【国際公開番号】WO2005/040337
【国際公開日】平成17年5月6日(2005.5.6)
【出願人】(505431514)ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア (1)
【Fターム(参考)】
[ Back to top ]