
β3アドレナリン作動性受容体アゴニスト及び抗ムスカリン剤を用いる併用療法
本明細書に記載されるのは、過活動膀胱を治療する改善された方法であり、これにおいて該方法は、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを、それを必要とする患者に投与することを含んでなる。かかる併用療法は、効力の改善及び/又は副作用の低減を提供する。

【発明の詳細な説明】
【背景技術】
【0001】
下部尿路の機能は、尿の貯蔵及び定期的な放出である。これには、中枢及び末梢神経エフェクターメカニズムの調整と、結果として生じる自律神経系の交感神経及び副交感神経成分、並びに体性運動経路の、調和のとれた調節とをもたらす、多様な求心性及び遠心性神経経路を含む、貯蔵及び排尿反射のオーケストレーションが必要である。これらは、膀胱(排尿筋)及び尿道平滑筋、並びに尿道括約筋の収縮状態を、近位で調節する。
【0002】
過活動膀胱(OAB)は、通常は頻度及び夜間頻尿に関連する、切迫性尿失禁を伴うか伴わない、尿意切迫の症状によって特徴づけられる。米国及び欧州におけるOABの有病率は、18歳を超える女性及び男性双方の16ないし17%と推定されてきた。過活動膀胱は、最も多くの場合、特発性として分類されるが、神経学的症状、膀胱出口閉塞、及び他の原因の二次的なものでもあり得る。病理生理学的見地からは、過活動膀胱症候は、特に切迫尿失禁に付随する場合、排尿筋過活動が示唆される。切迫性は、失禁の有無にかかわらず、社会及び医療福祉の双方に負の影響を与えることが示されてきており、年間の直接及び間接的健康管理支出の面で、かなりの負担である。
【0003】
抗ムスカリン剤は、OABなどの失禁症状を治療するために使用されてきた。例えば、トルテロジン、又は(R)−N,N−ジイソプロピル−3−(2−ヒドロキシ−5−メチルフェニル)−3−フェニルプロパンアミンは、切迫尿失禁及び、他の不安定又は過活動性膀胱の症候の治療用に市販されてきた。トルテロジン及びその主要な活性代謝産物、トルテロジンの5−ヒドロキシメチル誘導体は、治療効果に貢献すると考えられている。しかしながら、抗ムスカリン剤を用いたOABのための今日の医薬療法は、しばしば多くの患者が、今日の治療に対し充分な応答を示さないこと、及び/又は今日の治療によるドライマウスなどの少なからぬ副作用に耐え得ないことから、最適とはいえない場合が多い。
【0004】
それ故、OABのより有効な治療及び/又は副作用の低減を提供する改善された療法に、継続した需要がある。
【図面の簡単な説明】
【0005】
【図1】アイソボログラム分析を示す説明図である。
【図2】CL316243の、トルテロジン(A)、オキシブチニン(B)、又はダリフェナシン(C)との併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図3】化合物12、及びトルテロジン(A)、及びダリフェナシン(B)の併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図4】メトクトラミンの前処理の有り無しでの、CL316243を示す説明図である。
【図5】メトクトラミンの前処理が有る(A)か、又は無し(B)での、CL316243及びダリフェナシンの併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図6】様々な割合での、CL316243及びオキシブチニンの併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【発明の概要】
【0006】
発明の要旨
驚くべきことに、β3アドレナリン作動性受容体アゴニスト(以降、「β3−ARアゴニスト」)、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを用いた併用療法が、過活動膀胱の治療に相乗効果をもたらすことが判明してきた。β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを含んでなる組合せ組成物もまた記載される。
【0007】
発明の詳細な記載
本明細書に記載されるのは、過活動膀胱の治療法であり、これにおいて該方法は、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを、それを必要とする患者に投与することを含んでなる。かかる併用療法は、相乗効果及びしたがって、効力の改善及び/又は副作用の低減を提供する。
【0008】
驚くべきことに、現在、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において、抗ムスカリン剤のM2拮抗作用が、OABの治療に相乗作用を与える上で重要な役割を果たし得ることが判明してきている。何らの理論にも束縛されることを望むものではないが、抗ムスカリン剤のM3拮抗作用がOAB効力にとり重要であることが、一般に信じられている(例えば、アブラムス(Abrams)及びアンデルソン(Andersson)、「BJU Int.」、2007年、第100巻、p.987〜1006参照)。今、M3拮抗作用及びβ3−ARアゴニストと一緒に作用するM2拮抗作用が、相乗作用を提供することが判明してきた。
【0009】
1つの実施態様においては、相乗効果は、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において得られており、これにおいて、抗ムスカリン剤は、約40未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は約20未満のM2/M3比を有する。
【0010】
さらに、抗ムスカリン剤が40を超えるM2/M3比を有するとき、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において、追加の選択的M2アンタゴニストを使用することにより、相乗作用が得られることがある。
【0011】
本明細書で用いるとき、「相乗作用」又は「相乗効果」は、2種以上の活性薬剤の組合せ効果が、個々の活性薬剤の合計よりも大きい場合を記載するのに使用される。言い換えれば、2種以上の活性薬剤は、一方の活性薬剤の存在が第2の効果を増強又は拡大するように相互作用し得る。対照的に、2種以上の活性薬剤の組合せ効果が、個々の活性薬剤の合計に実質的に等しい場合、組合せ効果は単純に相加的であるが、相乗的ではない。また、2種以上の活性薬剤の組合せ効果が個々の活性薬剤の合計よりも低い場合、組合せ効果は相加的以下であり、これもまた相乗的ではない。
【0012】
1つの実施態様においては、併用療法は、β3−ARアゴニスト及び抗ムスカリン剤を、それを必要とする患者に投与することを含んでなり、ここで、抗ムスカリン剤は、約40未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約30未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約20未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約15未満のM2/M3比を有する。なお別の実施態様においては、抗ムスカリン剤は、約10未満のM2/M3比を有する。さらに別の実施態様においては、抗ムスカリン剤は、約1のM2/M3比を有する。
【0013】
別の実施態様においては、併用療法における抗ムスカリン剤は、約0.1より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.5より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.8より大きいM2/M3比を有する。
【0014】
別の実施態様においては、併用療法における抗ムスカリン剤は、約0.1ないし約40のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.5ないし約30のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.8ないし約20のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約1ないし約20のM2/M3比を有する。なお別の実施態様においては、抗ムスカリン剤は、約1ないし約15のM2/M3比を有する。さらに別の実施態様においては、抗ムスカリン剤は、約1ないし約10のM2/M3比を有する。
【0015】
1つの実施態様においては、M2/M3比は、オータケ(Ohtake)ら(「Biol.Pharm.Bull.」、2007年、第30巻、p.54〜58)(これは、その記載全体が本明細書に援用される)において記載された受容体結合アッセイを使用して測定される。別の実施態様においては、M2/M3比は、ヘグデ(Hegde)ら(「Curr Opin Invest Drugs」、2004年、第5巻、p.40〜49)(これは、その記載全体が本明細書に援用される)において記載されたアッセイを使用して測定される。
【0016】
オータケ(Ohtake)ら(「Biol.Pharm.Bull.」、2007年、第30巻、p.54〜58)において報告された、いくつかの代表的な抗ムスカリン剤のM1−M4活性を、表1に示す。
【0017】
【表1】
【0018】
ヘグデ(Hegde)ら(「Curr Opin Invest Drugs」、2004年、第5巻、p.40〜49)は、いくつかの抗ムスカリン剤のM1〜M4活性を記載しており、トロスピウムのM1〜M4活性は、表2に示される。
【0019】
【表2】
【0020】
併用療法に適した抗ムスカリン剤は、これに限定されないが:トルテロジン、オキシブチニン(S−オキシブチニンを含む)、ヒオシアミン、プロパンテリン、プロピベリン、トロスピウム(塩化トロスピウムを含む)、ソリフェナシン、ダリフェナシン、ジシクロミン、イプラトロピウム、オキシトロール、イミダフェナシン、フェソテロジン、テミベリン、SVT−40776、グラクソスミスクライン(GlaxoSmithKline)による202405、TD6301、RBX9841、DDP200、及びPLD179を包含する。
【0021】
1つの実施態様においては、抗ムスカリン剤は:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301からなる群より選択される。1つの実施態様においては、適切な抗ムスカリン剤のM2/M3比は、40未満である。別の実施態様においては、M2/M3比は、30未満である。別の実施態様においては、M2/M3比は、20未満である。なお別の実施態様においては、M2/M3比は、15未満である。1つの実施態様においては、M2/M3比は、オータケ(Ohtake)ら、において記載された結合アッセイを用いて測定される。
【0022】
別の実施態様においては、抗ムスカリン剤は:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、及びトロスピウムからなる群より選択される。
【0023】
別の実施態様においては、抗ムスカリン剤は:トルテロジン及びオキシブチニンからなる群より選択される。
【0024】
なお別の実施態様においては、抗ムスカリン剤は、トルテロジンである。
【0025】
適切なβ3−ARアゴニストは、これに限定されないが、CL316243、及び表3に示した化合物を包含する。
【0026】
【表3−1】
【0027】
【表3−2】
【0028】
【表3−3】
【0029】
別の実施態様においては、β3−ARアゴニストは、表4に列記された化合物から選択される:
【0030】
【表4−1】
【0031】
【表4−2】
【0032】
別の実施態様においては、β3−ARアゴニストは、
【0033】
【化1】
【0034】
及び
【0035】
【化2】
【0036】
からなる群より選択される。
【0037】
なお別の実施態様においては、β3−ARアゴニストは、
【0038】
【化3】
【0039】
である。
【0040】
表3の化合物は、以下に記載された方法を用いて調製し得る。
【0041】
本出願全体を通して、以下の用語は、別に記されない限り、示された意味を有する。
【0042】
【化4−1】
【0043】
【化4−2】
【0044】
【化4−3】
【0045】
中間体1
ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1):
【0046】
【化5】
【0047】
工程A: エチル(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)アセタート
【0048】
【化6】
【0049】
無水DCM 250mL中のメチル(3−アミノフェニル)アセタート(25g、140mmol)の溶液に、DIEA(28.5mL、155mmol)を添加し、得られた溶液を0℃に冷却し、窒素雰囲気下に置いた。この冷却された溶液に、次いでベンジルクロロホルマート(21.1mL、148mmol)を添加し、得られた混合物を一晩攪拌して、室温に温めさせた。反応物を、1M HCl、水、及び次に食塩水で洗浄した。有機層を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。さらなる精製は不要であり、この物質(44g、99%)をそのまま次の工程反応に使用した。
LC−MS:m/z(ES)314(MH)+,336(MNa)+.
【0050】
工程B: (3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)酢酸
【0051】
【化7】
【0052】
THF、エタノール、及び水(1:1:1、1500mL)中のエチル(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)アセタート(工程Aより)44.0g(140mmol)の溶液に、固体LiOH(16.8g、700mmol)を添加し、得られた溶液を油浴により60℃で3時間加熱した。混合物を室温に一晩冷却し、次に濃HCl 40mLを、25℃未満の温度を維持しながら、溶液がおよそpH2〜3になるまで徐々に添加した。酢酸エチル(3x750mL)で抽出し、次に合わせ、有機物質を水で、次いで食塩水で洗浄した。有機物質を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。標題化合物(24.7g、87%)を、さらに精製することなく次の工程反応に使用した。
LC−MS:m/z(ES)286(MH)+,308(MNa)+.
【0053】
工程C: ベンジル(3−{2−[メトキシ(メチル)アミノ]−2−オキソエチ
ル}フェニル)カルバメート
【0054】
【化8】
【0055】
ジクロロメタン 200mL中の(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)酢酸(工程Bより)24.7g(87mmol)の懸濁液に、トリエチルアミン(30.2mL、173mmol)を添加し、これにより若干の発熱(+5℃)を生じ、懸濁液は溶液化した。10分間の冷却後、HOBt(13.2g、87mmol)、N,O−ジメチルヒドロキシルアミンHCl(8.5g、87mmol)を、続いてEDC(16.6g、87mmol)を溶液に添加し、得られた混合物を窒素雰囲気下、室温で一晩攪拌した。溶液を分液漏斗に移し、1M HClで洗浄し、これによりエマルジョンを生じた。メタノールを添加して、エマルジョンを破壊し、水性物質を分配した。有機物質を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣を70%ヘキサン(酢酸エチル中)1000mLから再結晶化して(加熱還流し、次に室温に一晩冷却)、標題化合物(21g、74%)を白色固体として得た。
LC−MS:m/z(ES)329(MH)+.
【0056】
工程D: ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1)
氷/水浴により0℃に冷却された、無水THF 1000mL中のベンジル(3−{2−[メトキシ(メチル)アミノ]−2−オキソエチル}フェニル)カルバメート(工程Cより)15g(45.7mmol)の溶液に、窒素雰囲気下で、ビニルマグネシウムブロミドの1.0M溶液(THF中100mL、100mmol)をカニューレにより滴下添加し、得られた溶液を0℃で1時間攪拌した。5℃未満の温度を保ちながら1M HCl 500mLを徐々に添加することにより、反応物をクエンチし、30分間攪拌した。次に混合物を酢酸エチルで抽出し、合わせた有機物質を水で、続いて食塩水で洗浄した。次いで有機物質を、硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣をバイオタージ(Biotage)75Mフラッシュにより、30%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(11g、78%)を明黄色の固体として得た。
LC−MS:m/z(ES)310(MH)+,332(MNa)+.1H NMR(500MHz,CDCl3)δ:7.44−7.36(m,7H),7.18(d,J=8.4Hz,2H),6.70(br s,1H),6.44(dd,J=10.5,17.6Hz,1H),6.32(dd,J=1.1,17.6Hz,1H),5.85(dd,J=1.1,10.5Hz,1H),5.22(s,2H),3.86(s,2H).
【0057】
中間体2
((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート
(i−2)
【0058】
【化9】
【0059】
工程A: 1−(3−クロロフェニル)プロパ−2−エン−1−オール
【0060】
【化10】
【0061】
無水THF 100mL中の3−クロロベンズアルデヒド(22.5g、160mmol)の冷却された溶液に、不活性窒素雰囲気下で、THF中のビニルマグネシウムクロリドの1.6M溶液(100mL、160mmol)を、シリンジにより徐々に添加し、溶液を室温に温めながら3時間攪拌した。反応物を、塩化アンモニウムの飽和溶液でクエンチし、有機層を分離し、酢酸エチル(2x200mL)で抽出し、有機層を合わせ、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。40M+シリカゲルカラムを用いたホライズン(Horizon)MPLCにより、0〜40%酢酸エチル(ヘキサン中)の勾配溶出液を用いて精製し、標題化合物(22.4g、44%)を得た。
m/z(ES)168,170(M,M+2)+,190,192(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.38(s,1H),7.35−7.22(m,3H),5.90(ddd,J=7.3,10.0,17.4Hz,1H),5.38(d,J=17.5Hz,1H),5.18(d,J=7.2Hz,1H),5.15(d,J=10.1Hz,1H),0.96(s,9H),0.18(s,3H),0.08(s,3H).
【0062】
工程B: Tert−ブチル{[1−(3−クロロフェニル)プロパ−2−エン−1−イル]オキシ}ジメチルシラン
【0063】
【化11】
【0064】
無水DMF 90mL中の1−(3−クロロフェニル)プロパ−2−エン−1−オール(工程Aより)22.4g(133mmol)の溶液に、t−ブチルジメチルシリルクロリド(20.0g、133mmol)及びイミダゾール(18.1g、266mmol)を添加し、得られた溶液を窒素下、室温で一晩攪拌した。水で洗浄し、酢酸エチルで抽出した。有機物質を分離し、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、フラッシュシリカゲルカラムにより、0〜15%酢酸エチル(ヘキサン中)の勾配溶出液で溶出して精製し、標題化合物(16.6g、46%)を得た。
m/z(ES)282,284(M,M+2)+;151,153(M−OTBS,M−OTBS+2)+.
【0065】
工程C: {[Tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)アセトアルデヒド
【0066】
【化12】
【0067】
ドライアイス/アセトン浴により−78℃に冷却された、ジクロロメタン中のtert−ブチル{[1−(3−クロロフェニル)プロパ−2−エン−1−イル]オキシ}ジメチルシラン(工程Bより)4.0g(14.2mmol)の溶液に、溶液が淡い青色を維持するまでオゾンを通気した。次に、溶液が透明になるまで窒素ガスを通気した。溶液に硫化メチルを添加し、得られた混合物を室温で一晩攪拌した。この物質を真空下で濃縮し、残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜50%酢酸エチル(ヘキサン中)の勾配溶出液で溶出して精製し、生成物(3.57g、89%)を得た。
【0068】
工程D: N−[(1E)−2−{[tert−ブチル(ジメチル)シリル]オキシ}−2−(3−クロロフェニル)エチリデン]−2−メチルプロパン−2−スルフィンアミド
【0069】
【化13】
【0070】
無水ジクロロメタン 50mL中の、{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)アセトアルデヒド(工程Cより)3.0g(10.6mmol)及び(R又はS)−2−メチル−2−プロパンスルフィンアミド 1.3g(10.6mmol)の溶液に、硫酸銅(II)(3.4g、21.2mmol)を添加し、得られた混合物を、窒素雰囲気下、室温で16時間攪拌した。反応物を水で洗浄し、ジクロロメタンで抽出した。有機物質を硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜25%酢酸エチル(ヘキサン中)の勾配溶出液系で溶出して精製し、標題化合物(3.26g、80%)を得た。
m/z(ES)387,390(M,M+2)+.
【0071】
工程E: N−{1−[{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]−プロパ−2−エン−1−イル}2−メチルプロパン−2−スルフィンアミド
【0072】
【化14】
【0073】
窒素雰囲気下で0℃に冷却された、無水THF 20mL中のN−[(1E)−2−{[tert−ブチル(ジメチル)シリル]オキシ}−2−(3−クロロフェニル)エチリデン]−2−メチルプロパン−2−スルフィンアミド(工程Dより)2.4g(6.20mmol)の溶液に、THF中のビニルマグネシウムクロリドの1.6M溶液(3.90mL、6.2mmol)をシリンジにより添加し、得られた混合物を1時間攪拌した。混合物を室温に温めさせ、さらに1時間攪拌した。反応物を塩化アンモニウム飽和溶液でクエンチし、酢酸エチルで抽出した。合わせた有機物質を、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜35%酢酸エチル(ヘキサン中)の勾配溶出液系で溶出して精製し、4つのジアステレオマーを単一の異性体として得た。
【0074】
NMRにより、得られた4つの生成物は、互いにジアステレオマーであった。異性体は、それらがシリカゲルカラムを溶出した際に標識付けした。溶出された最初の異性体を、異性体1とし、次に異性体2、3、及び最後に異性体4と命名した。
【0075】
【化15】
【0076】
異性体1:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.26(br d,J=6.2Hz,2H),7.22−7.18(m,1H),5.60(ddd,J=7.3,10.3,17.4Hz,1H),5.15(d,J=10.3Hz,1H),5.00(d,J=17.3Hz,1H),4.57(d,J=7.4Hz,1H),3.98−3.94(m,2H),1.64(br s,1H),1.23(s,9H),0.91(s,9H),0.08(s,3H),−0.18(s,3H).
異性体2:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.33−7.31(m,2H),7.26(br d,J=5.0Hz,2H),7.20−7.16(m,1H),5.44(ddd,J=7.2,10.0,17.4Hz,1H),5.26(オーバーラップd,J=7.3Hz,1H),5.25(オーバーラップd,J=17.3Hz,1H),4.84(d,J=4.4Hz,1H),4.02(dt,J=4.4,7.8Hz,1H),3.80(d,J=4.4Hz,1H),1.20(s,9H),0.94(s,9H),0.14(s,3H),−0.12(s,3H).
異性体3:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32−7.29(m,2H),7.26−7.24(m,2H),7.22−7.20(m,1H),6.04(ddd,J=7.1,10.4,17.4Hz,1H),5.40(d,J=10.2Hz,1H),5.32(d,J=17.3Hz,1H),4.80(d,J=4.0Hz,1H),3.88−3.80(m,1H),3.55(d,J=9.4Hz,1H),1.09(s,9H),0.95(s,9H),0.09(s,3H),−0.10(s,3H).
異性体4:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.27−7.25(m,2H),7.21−7.18(m,1H),5.92(ddd,J=7.1,10.3,17.4Hz,1H),5.23(d,J=10.4Hz,1H),5.18(d,J=17.4Hz,1H),4.75(d,J=4.2Hz,1H),3.88−3.82(m,1H),3.33(d,J=9.4Hz,1H),1.19(s,9H),0.94(s,9H),0.09(s,3H),−0.14(s,3H).
【0077】
工程F: ((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート(i−2)
N−{1−[{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]−プロパ−2−エン−1−イル}2−メチルプロパン−2−スルフィンアミドの異性体1(工程Eより)(510mg、2.22mmol)に、ジオキサン中の無水4M HCl 5mLを添加し、溶液を室温で15分間攪拌した。反応物を濃縮乾燥し、トルエン(2x5mL)と共沸させて、過剰のHClを除去した。次いで残渣を、窒素雰囲気下に置かれ、氷/水浴で0℃に冷却された、無水ジクロロメタン中に溶解し、次にベンジルクロロホルマート(0.32mL、2.22mmol)をシリンジにより、続いてジイソプロピルエチルアミン(1.19mL、6.66mmol)を添加し、得られた溶液を0℃で2時間攪拌した。溶液を真空下で濃縮乾燥し、残渣を分取用プレート(4x1000μM)により、20%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(703mg、71%)を得た。
m/z(ES)446,448(M,M+2)+,468,470(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.27−7.25(m,2H),7.21−7.18(m,1H),5.92(ddd,J=7.1,10.3,17.4Hz,1H),5.23(d,J=10.4Hz,1H),5.18(d,J=17.4Hz,1H),4.75(d,J=4.2Hz,1H),3.88−3.82(m,1H),3.33(d,J=9.4Hz,1H),1.19(s,9H),0.94(s,9H),0.09(s,3H),−0.14(s,3H).
【0078】
上記記載のものに関連した、立体化学を異にする中間体は、適切な出発物質から、上記記載の方法を用いて調製し得る。
【0079】
【化16】
【0080】
中間体3
Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)
【0081】
【化17】
【0082】
工程A: ベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート
【0083】
【化18】
【0084】
無水ジクロロメタン 7mL中の、ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1)(820mg、2.80mmol)及び((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート
(i−2)(500mg、1.12mmol)の溶液に、チャン(Zhan)I触媒(740mg、1.12mmol)を添加し、得られた緑色の溶液を、窒素雰囲気下、40℃で一晩加熱した。反応物を濃縮乾燥し、残渣を分取用プレート(4x1000μM)により、40%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(348mg、50%)を得た。
m/z(ES)713,715(M,M+2)+,735,737(MNa,MNa+2)+.
【0085】
工程B: 4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(フェニル)メチル]ピロリジン−2−イル}メチル)アニリン
【0086】
【化19】
【0087】
エタノール 25mL中のベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート(工程Aより)328mg(0.46mmol)の溶液に、10%パラジウム炭素を添加し、水素ガスのバルーンにより、懸濁液を水素雰囲気下に置いた。反応物を、水素下、室温で1時間攪拌した。TLCは、反応が完了したことを示した。ギルメン(Gilmen)0.45μM PTFEシリンジフィルターを用いて触媒を濾去し、エタノール(4x5mL)で洗浄した。濾液を真空下で濃縮乾燥し、残渣を分取用プレート(3x1000μM)により、5%メタノール(ジクロロメタン中)で溶出して精製し、標題化合物(121mg、66%)を得た。
m/z(ES)397(MH)+.
【0088】
工程C: Tert−ブチル(5R)−2−(4−アミノベンジル)−
5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)
無水THF 5mL中の4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(フェニル)メチル]ピロリジン−2−イル}メチル)アニリン(工程Bより)121mg(0.315mmol)の溶液に、tert−ブチルカルボナート(69mg、0.315mmol)を、続いてTEA(44μL、0.315mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。反応混合物を直接、分取用プレート(1500μM)にのせ、30%酢酸エチル(ヘキサン中)で溶出して、標題化合物(100mg、64%)を得た。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.75−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.55−3.48(m,2H),2.74(br d,J=11.8Hz,1H),2.44(br d,J=11.8Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.08(s,3H),−0.15(s,3H).
【0089】
中間体4a及び中間体4bの分離
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0090】
【化20】
【0091】
工程A: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a)及びtert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
中間体i−3(tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(シス及びトランスの4:1混合物)を、メタノール中に溶解し、バージャー(Berger)マルチグラム(Multigram)SFC(超臨界)により、30%メタノール:60%二酸化炭素の溶出液を用いて精製し、2つのジアステレオマーを分離した。カラムの最初の異性体を、副異性体1とし、2番目の異性体を主異性体2と標識した。
i−4a:m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.75−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.55−3.48(m,2H),2.74(br d,J=11.8Hz,1H),2.44(br d,J=11.8Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.92(d,J=11.8Hz,1H),0.12(br d,J=14.0Hz,3H),−0.04(s,3H).SFCで8.70minに溶出、異性体2。
i−4b:m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.76−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.60−3.46(m,2H),2.72(br d,J=12.0Hz,1H),2.44(br d,J=12.2Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.64(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.14(br d,J=13.8Hz,3H),0.09(s,3H).SFCで7.78minに溶出、異性体1。
【0092】
中間体4a及び中間体4bの合成
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0093】
【化21】
【0094】
工程A: (4S)−3−ヘキサ−5−イノイル−4−フェニル−1,3−オキサゾリジン−2−オン
【0095】
【化22】
【0096】
無水テトラヒドロフラン 1.0L中の、5−ヘキシン酸 69.0g(615mmol)及びトリエチルアミン 214mL(1540mmol)の溶液に、窒素雰囲気下、−25℃で、トリメチルアセチルクロリド 83.0mL(677mmol)を20分間にわたり添加した。添加により白色沈殿が生成され、得られた懸濁液を2時間攪拌した。次に、無水塩化リチウム 28.7g(677mmol)及び、(4S)−4−フェニル−1,3−オキサゾリジン−2−オン 100.0g(615.0mmol)を連続的に添加し、混合物を12時間で徐々に室温に温めた。全揮発性物質を真空中で除去し、残渣を水(1L)で希釈し、酢酸エチル(3x300mL)で抽出した。合わせた有機層を食塩水(250mL)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空中で濃縮した。粗残渣を、シリカゲルカラムクロマトグラフィーにより、5〜50%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色の固体として得た(135g、85.4%)。
1H NMR(500MHz,CDCl3):δ7.40−7.37(m,2H),7.36−7.32(m,1H),7.31−7.28(m,2H),5.42(dd,J=8.9,3.7Hz,1H),4.69(t,J=8.9Hz,1H),4.28(dd,J=9.2,3.7Hz,1H),3.13−3.02(m,2H),2.24−2.21(m,2H),1.94(t,J=2.6Hz,1H),1.84(quintet,J=7.1Hz,2H).
LC−MS:m/z(ES)258.2(MH)+.
【0097】
工程B: (4S)−3−{(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イノイル}−4−フェニル−1,3−オキサゾリジン−2−オン
【0098】
【化23】
【0099】
無水酢酸エチル 265mL中の上記工程Aよりの(4S)−3−ヘキサ−5−イノイル−4−フェニル−1,3−オキサゾリジン−2−オン 56.8g(221mmol)の攪拌された溶液に、窒素雰囲気下、室温で、無水塩化マグネシウム 6.31g(66.2mmol)、トリエチルアミン 61.5mL(442mmol)、ベンズアルデヒド 26.9mL(265mmol)、及びクロロトリメチルシラン 42.3mL(331mmol)を添加し、得られた混合物を72時間攪拌した。不均一な反応混合物を、シリカゲル300mLのプラグを通し、さらに酢酸エチル 1Lで溶出して濾過した。濾液を真空中で蒸発乾燥させ、残渣をメタノール 265mL及びトリフルオロ酢酸 10mL中に懸濁した。得られた混合物を、窒素下、室温で5時間攪拌し、この間に反応は均一化した。次に全揮発性物質を真空中で除去し、残渣を、シリカゲルクロマトグラフィーにより、5〜15%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を白色固体として得た(65.0g、81.2%)。
1H NMR(500MHz,CDCl3):δ7.30−7.28(m,8H),7.09−7.07(m,2H),5.42(dd,J=8.7,3.7Hz,1H),4.76−4.72(m,1H),4.72−4.67(m,1H),4.65(t,J=8.7Hz,1H),4.18(dd,J=8.7,3.7Hz,1H),3.05(d,J=7.8Hz,1H),2.24(td,J=7.1,2.5Hz,2H),2.00−1.93(m,2H),1.67−1.61(m,1H).
LC−MS:m/z(ES)346.1(MH−H2O)+,386.0(MNa)+.
【0100】
工程C: (2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イン酸
【0101】
【化24】
【0102】
無水テトラヒドロフラン対水の、20対1混合物 1050mL中の上記工程Bよりの(4S)−3−{(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イノイル}−4−フェニル−1,3−オキサゾリジン−2−オン 65.0g(179mmol)の攪拌された溶液に、窒素雰囲気下、0℃で、35%過酸化水素水溶液 77.0mL(894mmol)を、内部温度を3℃未満に維持するべく充分遅い速度で添加した。次に、1.0M 水酸化リチウム水溶液 395mL(395mmol)を、反応の内部温度を5℃未満に維持するべく充分遅い速度で添加し、得られた混合物を0℃で3時間攪拌した。混合物の内部温度を5℃未満に維持するべく充分遅い速度での1.3M 亜硫酸ナトリウム水溶液 755mL(984mmol)により、反応物をクエンチした。全揮発性物質を真空中で除去し、残留する水相を酢酸エチル(3x200mL)で抽出した。水相を0℃に冷却し、6M 塩化水素水溶液でpH3に達するまで酸性化した。次に水相を酢酸エチル(3x300mL)で抽出し、合わせた有機物質を食塩水(100ml)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。残渣を、シリカゲルクロマトグラフィーにより、5〜10%酢酸エチル及び3%酢酸(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色のガムとして得た(32.0g、82.0%)。
1H NMR(500MHz,CDCl3):δ7.39−7.28(m,5H),4.85(d,J=8.2,1H),3.03−2.97(m,1H),2.29−2.15(m,2H),1.97(t,J=2.5Hz,1H),1.93−1.82(m,1H),1.62−1.55(m,1H).
LC−MS:m/z(ES)201.0(MH−H2O)+.
【0103】
工程D: (2R)−2−[(S)−{[Tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ヘキサ−5−イン酸
【0104】
【化25】
【0105】
無水アセトニトリル 500mL中の上記工程Cよりの(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イン酸 32.0g(147mmol)の攪拌された溶液に、窒素雰囲気下、室温で、1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン 77.0mL(513mmol)、22mL、続いてtert−ブチルジメチルシリルクロリド 66.3g(440mmol)を3回に分けて10分間で添加した。反応混合物を4時間攪拌し、真空蒸発させて、全揮発性物質を除去した。残渣を、ジクロロメタン 300mL及び水 100mLで希釈した。1.0M 塩化水素水溶液を、水層においてpH3が達成されるまで混合物に添加した。相を分離し、水相をジクロロメタン(2x100mL)で抽出した。合わせた有機物質を、水(50mL)、食塩水(50mL)で洗浄し、次に硫酸マグネシウム上で乾燥した。濾過及び真空蒸発の後、残渣を、メタノール 350mL中に溶解し、0.8M炭酸カリウム水溶液 350mL(280mmol)を添加した。得られた混合物を1.5時間攪拌し、次いで真空蒸発させて、全揮発性物質を除去した。残渣を、ジクロロメタン 300mLで希釈し、水相を5.0M 塩化水素水溶液でpH3に達するまで酸性化した。相を分離し、ジクロロメタン(2x100mL)で抽出した。合わせた有機物質を、水(50mL)、食塩水(50mL)で洗浄し、次に硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。残渣を、シリカゲルクロマトグラフィーにより、3〜15%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色の固体として得た(42.3g、86.6%)。
1H NMR(500MHz,CDCl3):δ7.36−7.27(m,5H),4.78(d,J=8.7,1H),2.90−2.86(m,1H),2.19−2.11(m,1H),2.10−2.03(m,1H),1.90(t,J=2.6Hz,1H),1.75−1.67(m,1H),1.41−1.34(m,1H),0.83(s,9H),0.02(s,3H),−0.27(s,3H).
LC−MS:m/z(ES)333.2(MH)+.
【0106】
工程E: 4−メトキシベンジル {(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ペンタ−4−イン−1−イル}カルバメート
【0107】
【化26】
【0108】
無水トルエン 400mL中の、上記工程Dよりの(2R)−2−[(S)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ヘキサ−5−イン酸 40.0g(120mmol)及びトリエチルアミン 33.5mL(241mmol)の溶液に、窒素雰囲気下、室温で、ジフェニルホスホリルアジド 37.5mL(132mmol)を添加した。混合物を5時間攪拌し、次に4−メトキシベンジルアルコール 37.5mL(301mmol)を添加した。得られた混合物を105℃で16時間加熱し、室温に冷却し、次いで飽和重炭酸塩水溶液 250mLで希釈した。相を分離し、水相を酢酸エチル(2x150mL)で抽出した。合わせた有機物質を、水(100mL)、食塩水(100mL)で洗浄し、次に硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。粗残渣を、シリカゲルクロマトグラフィーにより、3〜10% 酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物を無色の油として得た(50.9g、90.5%)。
1H NMR(500MHz,CDCl3):7.28−7.21(m,7H),6.87(d,J=8.4Hz,2H),4.92(s,2H),4.77−4.59(m,2H),3.89−3.84(m,1H),3.81(s,3H),2.30−2.22(m,2H),1.95(m,1H),1.91−1.85(m,1H),1.57−1.50(m,1H),0.89(s,9H),0.06(s,3H),−0.15(s,3H).
LC−MS:m/z(ES)468.1(MH)+,490.0(MNa)+.
【0109】
工程F: 4−メトキシベンジル [(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロフェニル)ペンタ−4−イン−1−イル]カルバメート
【0110】
【化27】
【0111】
無水DMF(500ml)中の、アセチレン(工程Eより、40g、80mmol)及び4−ヨードニトロベンゼン(21.8g、88mmol)の溶液に、トリエチルアミン(111mL、797mmol)を添加した。Pd(dppf)Cl2(1.95g、2.39mmol)、及びヨウ化銅(I)(910mg、4.78mmol)を添加し、混合物を窒素で脱気し(15分間通気)、得られた溶液を室温で5時間攪拌した。混合物を水(1200ml)に注入し、EtOAc(3x300mL)で抽出した。合わせた有機物質を、次に水(2x500mL)、飽和NaCl(200mL)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空下で蒸発させた。残渣を、MPLC(ホライズン(Horizon)バイオタージ(Biotage)2xフラッシュ(Flash)65i)により、0〜30%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、41g(84%)を暗赤色の油として得た。
1H NMR(500MHz,CDCl3):8.11−8.04(m,2H),7.94−8.01(m,1H),7.38−7.21(m,8H),6.87(d,J=8.4Hz,2H),4.98(s,2H),4.77−4.59(m,2H),4.00−3.95(m,3H),3.81(s,3H),2.56(t,J=7.1Hz,H=2H),2.00−1.95(m,1H),1.66−1.61(m,1H),0.93(s,9H),0.10(s,3H),−0.10(s,3H).
LC−MS:m/z(ES)589.3(MH)+,611.2(MNa)+.
【0112】
工程G: 4−メトキシベンジル[(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロフェニル)−4−オキソペンチル]カルバメート
【0113】
【化28】
【0114】
DMF(40mL)中のニトロフェニルアセチレン(工程Fより、41g、65.5mmol)の溶液に、ピロリジン(14mL、196.5mmol)を添加し、得られた混合物を80℃で3時間加熱した。混合物を室温に冷却し、10%酢酸溶液(水中)(110ml)を添加し、得られた混合物を室温でさらに3時間攪拌した。混合物を水(300ml)に注入し、EtOAc(3x250ml)で抽出し;合わせたEtOAc層を水(2x250ml)、飽和NaCl(100ml)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、ホライズン・フラッシュ75により、100%ヘキサンから50%EtOAc(ヘキサン中)まで上昇する勾配で溶出して精製し、34g(81%)を暗橙色の油として得た。
1H NMR(500MHz,CDCl3):8.17−8.14(m,2H),7.32−7.23(m,9H),6.87(d,J=8.4Hz,2H),4.96(d,J=12.2Hz,1H),4.90(d,J=12.1Hz,1H),4.72(d,J=3Hz,1H),4.16−4.13(m,1H),3.81(s,3H),3.71−3.77(m,2H),2.65−2.52(m,2H),1.97−1.92(m,1H),1.72−1.60(m,1H),0.93(s,9H),0.05(s,3H),−0.13(s,3H).
【0115】
工程H: (2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン
【0116】
【化29】
【0117】
DCM(350ml)中のMOZ保護されたケトンアミン(工程Gより、34g、56mmol)の溶液に、TFA(256ml)を添加し、得られた混合物を室温で1.5時間攪拌した。溶液を真空下で蒸発させ、残渣をDCMと飽和NaHCO3との間で分配した。有機層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣をMeOH(750ml)中に溶解し、氷/水浴で0℃に冷却した。次にシアノ水素化ホウ素ナトリウム(21.2g、337mmol)を添加し、得られた混合物を一晩攪拌して室温に温めさせた。水の添加により混合物をクエンチし、有機物質を真空下で除去した。次いで水層をEtOAc(2x)で抽出し、合わせたEtOAc層を飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、真空下で蒸発させた。残渣をシリカ上でのカラムクロマトグラフィー(溶出液:100%ヘキサンから35%EtOAc(ヘキサン中)まで上昇する勾配)により精製し、16.4g(63.4%)の最初の異性体、(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン、及び3.1g(12%)の2番目の異性体、(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジンを得た。
異性体1:LC−MS:m/z(ES)427.3(MH)+
異性体2:LC−MS:m/z(ES)427.3(MH)+
【0118】
工程I: Tert−ブチル(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0119】
【化30】
【0120】
無水THF中のtert−ブチル(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(12g、42.5mmol)の溶液に、Boc無水物(9.3g、42.5mmol)を、続いてTEA(17.76mL、127.4mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で2時間攪拌した。混合物を水(100mL)で洗浄し、酢酸エチル(2x200mL)で抽出した。有機物質を、硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣をホライズン・バイオタージMPLC(65iシリカゲルカラム)により、20〜75%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、所望の生成物を得た。
LC−MS:m/z(ES)527.3(MH)+,549.2(MNa)+.
【0121】
工程J: Tert−ブチル(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0122】
【化31】
【0123】
シスピロリジン異性体をトランス異性体、(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(ニトロベンジル)ピロリジンで置き換えたことを除いて、工程Iと同様の方法で調製した。
LC−MS:m/z(ES)527.3(MH)+,549.2(MNa)+.
【0124】
工程K: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
【0125】
【化32】
【0126】
500mLのパー・シェイカー・フラスコに、10% Pd/c(4.75g)を装入し、これにメタノール 100mLを添加して、触媒を覆った。次いで懸濁液に、メタノール(80mL)中の工程Iからのニトロ中間体(8.5g、18.5mmol)の溶液を、続いて1.0Mの塩化水素(メタノール中)溶液 15.4mLを添加した。反応容器を50PSIの水素ガス下に置き、混合物を一晩振盪した。アリコートを採り、LC−MSにより分析し、反応完了が示された。
【0127】
セライトを用いて触媒を濾去し、メタノール(2x100mL)で洗浄した。濾液を濃縮乾燥し、生成物をホライズンMPLC(65iシリカカラム)により、0%から30%酢酸エチル(ヘキサン中)まで上昇する勾配を用いて溶出して精製し、標題化合物(6.2g、72%)を得た。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.38−7.29(m,5H),6.76−6.68(m,2H),6.55−6.50(m,2H),5.52−5.49(m,1H),5.30−5.27(m,1H),4.15−4.05(m,2H),3.86−3.81(m,1H),3.76−3.71(m,1H),3.55−3.47(m,2H),2.74(br d,J=11.7Hz,1H),2.44(br d,J=11.7Hz,1H),2.05−1.93(m,1H),1.90−1.83(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.31−1.21(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.92(d,J=11.8Hz,1H),0.13(br d,J=14.0Hz,3H),−0.05(s,3H)
【0128】
工程L: Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0129】
【化33】
【0130】
シスピロリジン異性体をトランス異性体、Tert−ブチル(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラートで置き換えたことを除いて、工程Kと同様の方法で調製した。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.41−7.30(m,5H),6.73−6.67(m,2H),6.56−6.50(m,2H),5.52−5.48(m,1H),5.33−5.28(m,1H),4.15−4.06(m,2H),3.86−3.81(m,1H),3.76−3.70(m,1H),3.59−3.46(m,2H),2.72(br d,J=12.0Hz,1H),2.44(br d,J=12.0Hz,1H),2.05−1.93(m,1H),1.90−1.82(m,1H),1.64(s,9H),1.49−1.42(m,1H),1.32−1.20(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.14(br d,J=13.7Hz,3H),0.10(s,3H).
【0131】
以下の中間体を、適切な出発物質から、中間体i−4aについて上記に記載した方法を用いて調製した。
【0132】
【化34】
【0133】
以下の中間体を、適切な出発物質から、中間体i−4bについて上記に記載した方法を用いて調製した。
【0134】
【化35】
【0135】
中間体5
Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]ピロリジン−1−カルボキシラート(i−5)
【0136】
【化36】
【0137】
工程A: 4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(3−クロロフェニル)メチル]ピロリジン−2−イル}メチル)アニリン
【0138】
【化37】
【0139】
酢酸エチル 8mL中のベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート(i−3、工程Aより)100mg(0.15mmol)の溶液に、10%パラジウム炭素を添加し、水素ガスのバルーンにより、懸濁液を水素雰囲気下に置いた。反応物を水素下、室温で8時間攪拌した。触媒を、ギルメン0.45uM PTFEシリンジフィルターを用いて濾去し、酢酸エチル(4x2mL)で洗浄した。濾液を真空下で濃縮乾燥し、残渣を分取用プレート(1000μM)により、5%メタノール(ジクロロメタン中)で溶出して精製し、標題化合物(33mg、51%)を得た。
m/z(ES)430,432(M,M+2)+.
【0140】
工程B: Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]ピロリジン−カルボキシラート(i−5)
無水THF 1mL中の4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(3−クロロフェニル)メチル]ピロリジン−2−イル}メチル)アニリン(工程Aより)33mg(0.07mmol)の溶液に、tert−ブチルカルボナート(15.3mg、0.07mmol)を、続いてTEA(13uL、0.07mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。反応混合物を直接、分取用プレート(500uM)にのせ、30%酢酸エチル(ヘキサン中)で溶出して、標題化合物(25mg、78%)を得た。
m/z(ES)530,532(M,M+2)+,430,432(M−Boc,M−Boc+2)+.
【0141】
中間体6
4−{4−[4−(トリフルオロメチル)フェニル]−1,3−チアゾール−2−イル}ベンゼンスルホニルクロリド(i−6)
【0142】
【化38】
【0143】
中間体6は、例えば、イケモト(Ikemoto)ら、「Tetrahedron」、2003年、第59巻、p.1317〜1325の公表された方法に従って調製し得る。
【0144】
中間体7
2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−7)
【0145】
【化39】
【0146】
工程A: エチル2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0147】
【化40】
【0148】
0℃に冷却されたクロロホルム(500mL)中のエチル2−オキソシクロペンタン−2−カルボキシラート(56g、359mmol)の溶液に、臭素(18.5mL、359mmol)を、約20分間で添加した。添加完了後、混合物を室温に温めさせ、一晩攪拌した。窒素ガスを、混合物を通して90分間通気して、殆どのHBrを除去した。水(500mL)、飽和NaHCO3(250mL)、飽和NaCl(200mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣をEtOH(500mL)中に溶解し、チオアセトアミド(26.9g、359mmol)を添加し、混合物を室温で1時間攪拌し、次いで一晩還流した。混合物を冷却し、蒸発させ、残渣をDCMと飽和NaHCO3との間で分配し、有機層を飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:2x FLASH65i)溶出液:100%ヘキサン(450mL)、100%ヘキサンから25%EtOAc(ヘキサン中)まで上昇する勾配(1400mL)、次いで25%EtOAc(ヘキサン中))により精製して、標題化合物(32g、42%)を暗色の油として得た。
1H NMR(500MHz,CDCl3)δ:4.22(q,J=7.0Hz,2H),3.96(m,1H),3.04(m,1H),2.88(m,1H),2.76(m,2H),2.70(s,3H),1.30(t,J=7.0Hz,3H).
【0149】
工程B: 2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−7)
THF(450mL)及びメタノール(100mL)中のエチル2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Aより)31.5g(149mmol)の溶液に、水酸化リチウム(1M溶液を149mL、149mmol)を添加し、得られた混合物を室温で3時間攪拌した。有機物質を蒸発により除去し、水性残渣をEt2O(2x250mL)で抽出し、1M 塩酸(約170mL)の添加によりpH3に酸性化し、固体NaClで飽和させた。DCM(3x250mL)で抽出し、合わせたDCM層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣をアセトニトリルで粉砕し、濾過し、乾燥させて、標題化合物(7.1g、26%)を灰白色の固体として得た。
1H NMR(500MHz,CDCl3)δ:11.75(br s,1H),4.02(m,1H),3.00(m,1H),2.90−2.66(m,6H).
【0150】
中間体8
2−[(Tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)
【0151】
【化41】
【0152】
工程A: エチル2−アミノ−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0153】
【化42】
【0154】
工程Aにおいて、チオアセトアミドをチオ尿素で置き換えることにより、中間体(i−7)と同様の方法で調製した。
1H NMR(500MHz,CDCl3)δ:5.30(br s,2H),4.21(q,J=7.0,2H),3.81(m,1H),2.91(m,1H),2.78(m,1H),2.66(m,2H),1.30(t,J=7.0,3H).
【0155】
工程B: エチル2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0156】
【化43】
【0157】
ジクロロメタン(5mL)中のエチル2−アミノ−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Aより)230mg(1.08mmol)の溶液に、ジ−tert−ブチルジカルボナート(236mg、1.08mmol)、トリエチルアミン(0.15mL、1.08mmol)、及びDMAP(13mg、0.11mmol)を添加し、得られた混合物を室温で2時間攪拌した。混合物を1N HCl(10mL)、飽和NaCl(5mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH25+S)溶出液:100%ヘキサン(100mL)、0〜15%EtOAc(ヘキサン中)の勾配(900mL)、次いで15%EtOAc(ヘキサン中)(500mL)により精製して、標題化合物(160mg、47%)を白色の泡沫として得た。
1H NMR(500MHz,CDCl3)δ:9.23(br s,1H),4.17(q,J=7.1Hz,2H),3.95(t,J=6.6Hz,1H),3.04(m,1H),2.86(m,1H),2.76(m,2H),1.55(s,9H),1.23(t,J=7.1Hz,3H).
【0158】
工程C: 2−[(Tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)
エチル2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Bより)から、中間体(i−7)工程Bに見られるものと類似の方法を用いて調製した。
1H NMR(500MHz,CDCl3)δ:3.96(m,1H),3.06(m,1H),2.88(m,2H),2.71(m,1H),1.55(s,9H).
【0159】
中間体9
2−(4−フルオロフェニル)−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−9)
【0160】
【化44】
【0161】
工程Aにおいて、チオアセトアミドを4−フルオロチオベンズアミドで置き換えることにより、中間体7(i−7)に見られるものと類似の方法を用いて調製した。
1H NMR(500MHz,DMSO−d6)δ:7.90(m,2H),7.29(t,J=8.7,2H),3.81(m,1H),2.99(m,1H),2.86(m,1H),2.70−2.58(m,2H).
【0162】
中間体10
2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−10)
【0163】
【化45】
【0164】
工程A: エチル2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート
【0165】
【化46】
【0166】
0℃に冷却された無水ジエチルエーテル(40mL)中のエチル2−オキソシクロヘキサンカルボキシラート(15g、88mmol)の溶液に、臭素(4.5mL、88mmol)を15分間にわたり滴下添加した。添加完了後、混合物を室温に90分間温めさせた。混合物をEtOAc(100mL)で希釈し、飽和NaHCO3、飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣をエタノール(100mL)中に溶解し、チオアセトアミド(6.6g、88mmol)を添加した。混合物を室温で1時間攪拌し、次いで一晩還流した。混合物を蒸発させ、残渣をNaHCO3とDCMとの間で分配した。有機層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH 65i)溶出液:100%ヘキサン(500mL)、0〜25%EtOAc(ヘキサン中)の勾配(1200mL)、次いで25%EtOAc(ヘキサン中)(1200mL)により精製して、標題化合物(6.14g、31%)を淡橙色の油として得た。
1H NMR(500MHz,CDCl3)δ:4.22(q,J=7.1,2H),3.84(t,J=5.5,1H),2.80(m,1H),2.73(m,1H),2.65(s,3H),2.18(m,1H),2.11−1.95(m,2H),1.85(m,1H),1.29(t,J=7.1,3H).
【0167】
工程B: 2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−10)
エチル2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート(工程Aより)から、中間体(i−7)工程Bに概説された方法に従って調製した。
1H NMR(500MHz,CDCl3)δ:9.26(br s,1H),3.81(q,J=7.3及び5.9,1H),2.75(m,2H),2.68(s,3H),2.24(m,1H),2.18−2.01(m,2H),1.82(m,1H).
【0168】
中間体11
2−[(Tert−ブトキシカルボニル)アミノ]−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−11)
【0169】
【化47】
【0170】
工程A: エチル2−アミノ−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート
【0171】
【化48】
【0172】
チオアセトアミドをチオ尿素で置き換えることにより、中間体10(i−10)工程Aに概説された方法に従って調製した。
1H NMR(500MHz,DMSO−d6)δ:9.28(br s,2H),4.11(q,J=7.3,2H),3.71(t,J=5.0,1H),2.57−2.39(m,2H),1.90(m,2H),1.78(m,1H),1.59(m,1H),1.17(t,J=7.3,3H).
【0173】
工程B: 2−[(Tert−ブトキシカルボニル)アミノ]−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−11)
エチル2−アミノ−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート(工程Aより)から、中間体8(i−8)工程B及びCに概説された方法に従って調製した。
1H NMR(500MHz,CDCl3)δ:3.70(t,J=5.2,1H),2.74(m,1H),2.64(m,1H),2.25(m,1H),2.10−1.94(m,2H),1.87(m,1H),1.55(s,9H).
【0174】
中間体12
インダン−1−カルボン酸(i−12)
【0175】
【化49】
【0176】
「ジャーナル・オブ・オーガニック・ケミストリー(Journal of Organic Chemistry)」、2000年、第65巻、第4号、p.1132〜1138の文献の方法に従って調製した。
【0177】
中間体13a及び中間体13b
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13b)
【0178】
【化50】
【0179】
工程A: Tert−ブチル(4R,5R)−2,2−ジメチル−4−[(1E)−3−オキソプロパ−1−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0180】
【化51】
【0181】
CH2Cl2(150mL)中のtert−ブチル(4S,5R)−4−ホルミル−2,2−ジメチル−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(20.9、89.1mmol)の溶液に、(トリフェニルホスホルアニリデン)アセトアルデヒド(27.1g、89.1mmol)を添加し、得られた混合物を室温で40時間攪拌した。溶媒の1/3を除去した後、ヘキサンをたっぷり添加し、得られた固体を濾去した。Biotage Horizon(登録商標)システム(シリカゲル、0〜20%酢酸エチル(ヘキサン中)の勾配、次に20%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物 16.3g(72%)を黄色の油として得た。
LC/MS354.3(M+23).
【0182】
工程B: Tert–ブチル(4R,5R)−2,2−ジメチル−4−(3−オキソプロピル)−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0183】
【化52】
【0184】
アセトン(150mL)中のtert−ブチル(4R,5R)−2,2−ジメチル−4−[(1E)−3−オキソプロパ−1−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(19.6g、59.1mmol)(工程Aより)の溶液に、10% Pd/C 1.9gを添加し、得られた懸濁液を水素バルーン下、室温で24時間攪拌した。固体をセライト上で濾去し、濾液を真空下で濃縮した。残渣を、Biotage Horizon(登録商標)システム(シリカゲル、0〜20%酢酸エチル(ヘキサン中)の勾配、次に20%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより精製して、標題化合物 11.5g(58%)を無色の油として得た。
LC/MS356.3(M+23).
【0185】
工程C: Tert−ブチル(4R,5R)−2,2−ジメチル−4−[(3E)−4−(4−ニトロフェニル)ブタ−3−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート及びtert−ブチル(4R,5R)−2,2−ジメチル−4−[(3Z)−4−(4−ニトロフェニル)ブタ−3−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0186】
【化53】
【0187】
CH2Cl2(200mL)中の工程Bよりのtert–ブチル(4R,5R)−2,2−ジメチル−4−(3−オキソプロピル)−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(10.0g、30.0mmol)の溶液に、(4−ニトロベンジル)トリフェニル−ホスホニウムブロミド(21.5g、45.0mmol)を、続いてEt3N(8.36mL、60.0mmol)を添加した。赤色の反応混合物を、室温で48時間攪拌した。ヘキサン(200mL)を反応混合物に注入し、固体を濾去した。Biotage Horizon(登録商標)システム(シリカゲル、0〜10%酢酸エチル(ヘキサン中)の勾配、次に10%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物(シス トランス混合物) 10.7g(79%)を淡黄色の泡沫として得た。
LC/MS475.4(M+23).
【0188】
工程D: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート及びtert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0189】
【化54】
【0190】
酢酸エチル(100mL)中の工程Cよりの上記のシス/トランス混合物(7.86g、17.4mmol)の溶液に、2N HCl溶液 50mLを添加し、得られた混合物を室温で2時間攪拌し、次に45℃で3時間加熱した。揮発性物質を、減圧下で除去した。得られた白色固体を、N,N−ジメチルホルムアミド(100mL)中に溶解し、iPr2Net 15.1mL(86.7mmol)を添加した。反応混合物を室温で7時間攪拌した。次にジ−tert−ブチルジカルボナート(4.55g、20.8mmol)を添加し、反応混合物を室温で一晩攪拌した。水(200mL)を添加し、これを酢酸エチル(200mLx3)で抽出した。合わせた有機層を、Na2SO4上で乾燥し、濾過し、減圧下で濃縮した。残渣を、Biotage Horizon(登録商標)システム(シリカゲル、0〜30%酢酸エチル(ヘキサン中)の勾配)でのフラッシュクロマトグラフィーにより精製して、標題化合物 tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(シス)1.61g(22%)及びtert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(トランス)3.9g(54%)を得た。
LC/MS435.4(M+23).
【0191】
工程E: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a)
エタノール(20mL)中の工程Dよりの、上記(シス)tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(1.51g、3.66mmol)の溶液に、10% Pd/C 0.15gを添加し、得られた懸濁液を水素バルーン下、室温で5時間攪拌した。セライトを通して濾過し、溶媒を除去して、標題化合物 1.40g(100%)を白色泡沫として得て、これをさらに精製することなく使用した。
LC/MS405.3(M+23).
【0192】
工程F: Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13b)
エタノール(40mL)中の工程Dよりの(トランス)tert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(3.90g、9.46mmol)の溶液に、10% Pd/C 0.4gを添加し、得られた懸濁液を、水素バルーン下、室温で6時間攪拌した。固体を、セライトを通して濾去した。溶媒の除去後、Biotage Horizon(登録商標)システム(シリカゲル、0〜30%酢酸エチル(ヘキサン中)の勾配、次に30%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物 2.30g(64%)を白色泡沫として得た。
LC/MS405.3(M+23).
【0193】
中間体14
(2S)−1−(1,3−ベンゾチアゾール−2−イル)ピロリジン−2−カルボン酸(i−14):
【0194】
【化55】
【0195】
N,N−ジメチルホルムアミド(3mL)中のL−プロリン 28mg(0.24mmol)の溶液に、室温で、2−ブロモベンゾチアゾール 51mg(0.24mmol)、炭酸カリウム 100mg(0.72mmol)、及びヨウ化銅 6mg(0.03mmol)を添加した。反応混合物を100℃で一晩加熱した。次に、これを濾過し、逆相HPLC(TMC Pro−Pac C18;0〜60%の、0.1%トリフルオロ酢酸(アセトニトリル中)/0.1%トリフルオロ酢酸(水中)の勾配)により精製した。純粋な分画を一晩凍結乾燥して、35mg 60%の標題化合物を明褐色の固体として得た。
1H NMR(DMSO−d6):δ7.78(d,J=8.0Hz,1H),7.45(d,J=8.0Hz,1H),7.28(t,J=7.8Hz,1H),7.08(t,J=7.8Hz,1H),4.48(d,J=7.3Hz,1H),3.52−3.61(m,2H),2.37(m,1H),2.01−2.11(m,3H).LC/MS249.3(M+1)
【0196】
中間体44
[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボン酸(i−44)の調製
【0197】
【化56】
【0198】
工程A: メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボキシラート
【0199】
【化57】
【0200】
メチル(2S)−5−メトキシ−3,4−ジヒドロ−2H−ピロール−2−カルボキシラート(4.19g、26.6mmol)及び3−アザトリシクロ[4.2.1.0.2,5]ノナ−7−エン−4−オン(2.4g、17.8mmol)を、110℃で一晩加熱した。Biotage Horizon(登録商標)システム(0〜100%酢酸エチル/ヘキサン混合物)を用いて精製し、標題化合物メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキシラート及び中間体メチル(7S)−9−オキソ−3,8−ジアザテトラシクロ[9.2.1.02,10.04,8]テトラデカ−3,12−ジエン−7−カルボキシラートを得た。中間体を150℃で45分間加熱して、さらに精製することなく標題化合物を得た。
LC/MS195.2(M+1).
【0201】
工程B: [(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボン酸
【0202】
【化58】
【0203】
テトラヒドロフラン(60mL)中の、メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキシラート(9.95g、51.2mmol)、メタノール(40mL)、及び水(40mL)中の水酸化リチウム(3.32g、77mmol)を、室温で1時間攪拌した。2N 塩酸(38.5mL)を添加して反応混合物を中和し、次にこれを直接、逆相HPLC(TMC Pro−Pac C18;0〜40% 0.1%トリフルオロ酢酸(アセトニトリル中)/0.1% トリフルオロ酢酸(水中)の勾配)により精製した。O−アルキル化生成物が、急速に溶出された。純粋な分画を収集し、一晩凍結乾燥して、標題化合物を淡黄色の固体として得た。
1H NMR(DMSO−d6):δ7.89(d,J=6.6Hz,1H),6.24(d,J=6.6Hz,1H),4.92(dd,J=10.0,3.1Hz,1H),3.12−2.99(m,2H),2.52(m,1H),2.11(m,1H).LC/MS181.2(M+1).
【0204】
中間体46
(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボン酸(i−46)
【0205】
【化59】
【0206】
工程A: (3S,9S)−5−オキソ−1,2,3,5,6,8a−ヘキサヒドロインドリジン−3−カルボン酸メチルエステル
【0207】
【化60】
【0208】
この中間体は、ハネシアン(Hanessiann,S.);サイレス(Sailes,H.);ムンロ(Munro,A.);セリアン(Therrien,E.)、「J.Org.Chem.」、2003年、第68巻、p.7219、及びバスワニ(Vaswani,R.G.);チェンバリン(Chamberlin,R.)、「J.Org.Chem.」、2008年、第73巻、p.1661に見られる方法に従って調製した。
【0209】
工程B: メチル(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキシラート
【0210】
【化61】
【0211】
ジクロロメタン 50mL中の上記工程Aよりの(3S,9S)−5−オキソ−1,2,3,5,6,8a−ヘキサヒドロインドリジン−3−カルボン酸メチルエステル 0.850g(4.06mmol)の攪拌された溶液に、酸化マンガン(IV) 6.30g(72.5mmol)を添加し、得られた混合物を還流下で12時間攪拌した。反応物を室温に冷却し、セライト(Celite)(登録商標)のパッドを通して濾過し、次に固体をジクロロメタン 100mLで洗浄した。濾液を真空中で蒸発乾燥させ、残渣をシリカゲルクロマトグラフィーにより、10〜50%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を透明なガムとして得た(0.47g、55%)。
LC−MS:m/z(ES)194(MH)+.
【0212】
工程C: (3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボン酸
【0213】
【化62】
【0214】
THF 3mL中の上記工程Bよりのメチル(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキシラート 0.200mg(1.00mmol)の攪拌された溶液に、1.0M LiOH水溶液 1.5mL(1.5mmol)を添加した。得られた混合物を室温で2時間攪拌し、次に1.0M 塩化水素水溶液 2.0mL(2.0mmol)でクエンチした。全揮発性物質を真空蒸発させ、水相を30% IPA(クロロホルム中)混合物(3x5mL)で抽出した。合わせた抽出物を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させて、標題化合物を白色固体として得た(0.17g、92%)。
1H NMR(500MHz,CD3OD):δ7.53(dd,J=8.9,7.1Hz,1H),6.38−6.35(m,2H),5.11(dd,J=9.7,2.7Hz,1H),3.23−3.12(m,2H),2.62−2.53(m,1H),2.35−2.30(m,1H).
LC−MS:m/z(ES)180(MH)+.
【0215】
中間体56
2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸(i−56)
【0216】
【化63】
【0217】
工程A: Tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアート
【0218】
【化64】
【0219】
DMF(75mL)中の3−メチル−1H−1,2,4−トリアゾール(7.3g、88mmol)の溶液に、K2CO3(60.7g、439mmol)及び2−ブロモプロピオン酸tert−ブチルエステル(14.6mL、88mmol)を添加した。反応物を室温で一晩攪拌した。混合物をEtOAc(500mL)で希釈し、水(x3)で、次に食塩水で洗浄した。MgSO4上で乾燥し、そして濃縮した。残渣をシリカゲル上でのカラムクロマトグラフィーにより、EtOAc/イソヘキサン(20〜100%)で溶出して精製し、粗生成物 13gを位置異性体の3:1混合物として得た。混合物をキラルセル(Chiralcel)ODにより、4%から30%までのIPA/ヘプタンの勾配で精製した。次に、最初の2つのピークを、Chiralcel ODカラムで、4% IPA/ヘプタンを用いてイソクラチック溶出で分離した。2番目のピークを、所望の単一立体異性体(R又はS)(2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸tert−ブチルエステル)(3.5g、19%)として収集した。
1H−NMR(500MHz,CDCl3)δ8.05(s,1H),4.90(q,J=7Hz,1H),2.35(s,3H),1.72(d,J=7Hz,3H),1.40(s,9H).ESI−MSC10H17N3O2の計算値:精密質量:211.13;実測値156.05(−tBu).
【0220】
工程B: 2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸
【0221】
【化65】
【0222】
Tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアート(1.0g、4.7mmol)を、4M HCl(ジオキサン100mL中)に溶解し、室温で一晩攪拌した。生成物を減圧下で濃縮し、高真空下で乾燥させて、(R又はS)tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアートを、HCl塩(850mg)として得た。
ESI−MSC6H9N3O2の計算値:精密質量:155.07;実測値156.05.
【0223】
化合物1
2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(5R)−[(R)−ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0224】
【化66】
【0225】
工程A: Tert−ブチル(5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}ベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート
【0226】
【化67】
【0227】
無水DMF 0.5mL中の、tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)10mg(シス/トランス 5:1混合物、0.02mmol)及び(2−アミノ−1,3−チアゾール−4−イル)酢酸(3.18mg、0.02mmol)の溶液に、0.5M HOAt溶液(DMF中)(0.04mL、0.02mmol)を、続いてEDC(5.8mg、0.03mmol)及びDIEA(3.5μL、0.02mmol)を添加した。得られた混合物を、窒素雰囲気下、室温で16時間攪拌した。混合物を水で洗浄し、ジクロロメタン(2x2mL)で抽出した。有機物質を合わせ、硫酸ナトリウム上で乾燥し、濾過し、真空中で濃縮した。残渣を、分取用TLCプレート(500uM)により、5%MeOH(ジクロロメタン中)で溶出して精製し、生成物(10.3mg、81%)を得た。
m/z(ES)637(MH)+,659(MNa)+.
【0228】
工程B: 2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(5R)−[(R)ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0229】
【化68】
【0230】
メタノール 0.20mL中のtert−ブチル(5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}ベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより)7mg(0.01mmol)の溶液に、濃HCl 0.20mLを添加し、反応混合物を室温で1時間攪拌した。トルエン(2x)で共沸して、水を除去した。残渣を、アセトニトリル/水/MeOH(9:1:1)中に溶解し、ギルソン(Gilson)HPLC上で、0〜50% アセトニトリル/水(0.05% TFAバッファを含む)の勾配で溶出して精製した。生成物を含有する分画を合わせ、凍結し、凍結乾燥して、白色の泡沫(3.3mg、71%)を得た。
m/z(ES)423(MH)+.(約5:1混合物)1H NMR(500MHz,CD3OD)δ:7.56(br d,J=8.2Hz,2H),7.44(d,7.8Hz,2H),7.39(t,J=7.6Hz,2H)7.35−7.32(m,0.8H)7.32−7.29(m,0.2H副異性体),7.26(d,J=8.0Hz,1.7H),7.14(d,J=8.1Hz,0.3H副異性体)6.67及び6.66(br s,0.2/0.8H,全1H).4.72(d,J=8.5Hz,1H),3.80−3.70(m,4H)3.14(dd,J=6.1,13.8Hz,1H),2.95(dd,J=9.1,13.8Hz,1H),2.08−2.00(m,1H),1.86−1.74(m,3H).
【0231】
本明細書に記載の生物学的アッセイを用いて、化合物1のヒトβ3機能活性は、1ないし10nMであると測定された。
【0232】
化合物2
2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(2S,5R)−[(R)−ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0233】
【化69】
【0234】
工程A: Tert−ブチル(2S,5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}べンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート
【0235】
【化70】
【0236】
標題化合物は、tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a)及び(2−アミノ−1,3−チアゾール−4−イル)酢酸から、化合物1、工程Aの方法に従って調製した。粗生成物を、分取用TLCプレートにより、5%MeOH(ジクロロメタン中)で溶出して精製し、生成物(4.1mg、21%)を得た。
m/z(ES)637(MH)+,659(MNa)+.
【0237】
工程B: 2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(2S,5R)−[(R)ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0238】
【化71】
【0239】
標題化合物は、tert−ブチル(2S,5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}べンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより)4mgから、化合物1、工程Bの方法に従って調製した。粗生成物は、Gilson HPLC上で、0〜50% アセトニトリル/水(0.05% TFAバッファを含む)の勾配で溶出して精製した。生成物を含有する分画を合わせ、凍結し、凍結乾燥して、白色の泡沫(3.3mg、71%)を得た。
m/z(ES)423(MH)+.1H NMR(500MHz,CD3OD)δ:7.55(br d,J=8.2Hz,2H),7.44(d,7.8Hz,2H),7.39(t,J=7.6Hz,2H)7.35−7.33(m,1H),7.25(d,J=8.0Hz,2H),6.65(br s,1H).4.72(d,J=8.5Hz,1H),3.80−3.72(m,4H)3.14(dd,J=6.1,13.8Hz,1H),2.96(dd,J=9.1,13.8Hz,1H),2.07−2.00(m,1H),1.85−1.73(m,3H).
【0240】
本明細書に記載の生物学的アッセイを用いて、化合物2のヒトβ3機能活性は、1ないし10nMであると測定された。
【0241】
化合物3
2−アミノ−N−[4−{((2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル)メチル}フェニル]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキサミド
【0242】
【化72】
【0243】
工程A: Tert−ブチル−(2S,5R)−2−(4[({2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]
チアゾール−4−イル}カルボニル)アミノ]ベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート
【0244】
【化73】
【0245】
無水DMF(5mL)中の、tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a)220mg(0.58mmol)及び2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)164mg(0.58mmol)の溶液に、EDC(165mg、0.86mmol)、HOBt(132mg、0.86mmol)、及びヒューニッヒ(Hunig)塩基(0.3mL、1.7mmol)を添加し、得られた混合物を室温で一晩攪拌した。水(50mL)に注入し、EtOAc(3x30mL)で抽出し、合わせたEtOAc層を水(2x50mL)、飽和NaCl(25mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH 25+M)溶出液:100%ヘキサン(100mL)、0〜35%EtOAc(ヘキサン中)の勾配(750mL)、次に35%EtOAc(ヘキサン中)(600mL)により精製した。ジアステレオ異性体を、ADカラム上でのキラルHPLC(溶出液:25%IPA(ヘプタン中))により、最初に溶出する異性体(134mg、36%)と2番目に溶出する異性体(126mg、34%)に、共に白色の泡沫として分離した。
【0246】
工程B: 2−アミノ−N−[4−{((2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル)メチル}フェニル]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキサミド
【0247】
【化74】
【0248】
DCM(3mL)中のtert−ブチル−(2S,5R)−2−(4[({2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]
チアゾール−4−イル}カルボニル)アミノ]ベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより、2番目に溶出する異性体)126mg(0.19mmol)の溶液に、トリフルオロ酢酸(3.0mL、38mmol)を添加し、得られた混合物を室温で4時間攪拌した。混合物を蒸発させ、SCXカートリッジを通し、2M NH3(メタノール中)で溶出して、塩基を除去した。生成物を、PREP−TLC 2x[20x20cmx1000ミクロン]溶出液:15% MeOH(DCM中)+1%NH4OH、により精製し、生成物を凍結乾燥して、標題化合物(65mg、75%)を白色の飛散性固体として得た。
m/z(ES)449(MH)+.1H NMR(500MHz,DMSO−d6)δ:10.00(s,1H),7.51(d,J=8.2,2H),7.30(m,4H),7.21(t,J=6.9,1H),7.12(d,J=8.2,2H),6.86(s,1H),4.23(d,J=7.3,1H),3.78(m,1H),3.21(m,1H),3.10(m,1H)2.78(m,1H),2.66(m,2H),2.57(m,2H),2.49(m,1H),1.59(m,1H),1.40(m,1H),1.39(m,2H).
【0249】
工程Aよりの生成物[最初に溶出する異性体](134mg、0.207mmol)を、同様の方法で脱保護して、標題化合物(44mg、48%)を白色の飛散性固体として得た。
m/z
(ES) 449
(MH) +.
【0250】
本明細書に記載された生物学的アッセイを用いて、化合物3のヒトβ3機能活性は、1nM未満であると測定された。
【0251】
化合物4−10
上記記載のものと同様の方法を用いて、化合物4〜10を適切な出発物質から調製した。
【0252】
本明細書に記載された生物学的アッセイを用いて、各化合物のヒトβ3機能活性が測定され、以下の表に示された。
【0253】
【化75−1】
【0254】
【化75−2】
【0255】
化合物11
N−(4−(((2S,5R)−5−((R)−ヒドロキシ(フェニル)メチル)ピロリジン−2−イル)メチル)フェニル)−2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパンアミド
【0256】
【化76】
【0257】
DMF(20mL)中の、i−13a(2.00g、5.23mmol)、2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸i−56(1.00g、5.23mmol)、HOAt(1.307mL、0.784mmol)、及びEDC(2.005g、10.46mmol)の混合物を、室温で10分間攪拌した。反応混合物を炭酸水素ナトリウム水溶液でクエンチし、EtOAcで抽出した。粗生成物をカラムクロマトグラフィー(0〜3%MeOH(10%NH4OH)(DCM中)により精製した。蒸発後、生成物をキラルHPLC(ADカラム、30%IPA/ヘプタン)によりさらに精製して、boc保護された純粋な中間体を得、これを最少体積のジオキサン中に溶解し、4M HCl(ジオキサン中)を添加した。室温で2時間後、反応混合物を減圧下で濃縮して、標題化合物のHCl塩を得た。塩基性逆相HPLC(0.1%NH4OH(H2O中)、MeCN)により、標題化合物の所望の遊離塩基を得た。
1H−NMR(500MHz,CD3OD)δ8.51(s,1H),7.49(d,J=13Hz,2H)7.35−7.29(m,4H),7.26−7.20(m,4H),5.20(q,J=7.5Hz,1H),4.20(d,J=7.5Hz,1H),3.27−3.22(m,2H),2.80−2.72(m,2H),2.34(s,3H),1.82(d,J=7.5Hz,3H),1.79−173(m,1H),1.52−1.48(m,3H).ESI−MSC24H29N5O2の計算値:精密質量:419.23,実測値420.35.
【0258】
本明細書に記載の生物学的アッセイを用いて、化合物11のヒトβ3機能活性は、1ないし10nMであると測定された。
【0259】
化合物12及び13
(3S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド(化合物12)及び(3R)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド(化合物13)
【0260】
【化77】
【0261】
工程A: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート(異性体1)及びtert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(3R)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート(異性体2)
【0262】
【化78】
【0263】
無水N,N−ジメチルホルムアミド 3.2mL中の、中間体i−13a 0.610g(1.60mmol)及び中間体i−46 0.300g(1.67mmol)の溶液に、窒素雰囲気下で、1−ヒドロキシ−7−アザベンゾトリアゾール 0.033g(0.24mmol)を、続いて1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩 0.336g(1.75mmol)を添加した。得られた懸濁液を室温で30分間攪拌し、水でクエンチし、酢酸エチル(3x10mL)で抽出した。合わせた有機層を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。粗残渣を、シリカゲルクロマトグラフィーにより、50〜100%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を、97:3の比率のジアステレオマー混合物として得た。2つのジアステレオマーを、ダイセル(Daicel)CHIRALPAK(登録商標)AD(登録商標)カラムを用いたキラルHPLC(溶出液:40%IPA(ヘプタン中))により分離した。最初に溶出するジアステレオマーは、異性体2と命名され、無色の固体である(0.020g、2.3%)。
LC−MS:m/z(ES)544.2(MH)+.
2番目に溶出するジアステレオマーは、異性体1と命名され、無色の固体である(0.650g、75%)。
LC−MS:m/z(ES)544.2(MH)+.
【0264】
工程B(化合物12): (3S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド
【0265】
【化79】
【0266】
イソプロパノール 2mL中の上記工程Aよりの異性体1 0.500g(0.920mmol)の溶液に、窒素の雰囲気下で、4.0M 無水塩化水素溶液(1,4−ジオキサン中) 4.0mLを添加した。反応混合物を1時間攪拌し、次に真空中で蒸発乾燥させた。粗反応混合物を、逆相HPLC(TMC Pro−Pac C18;0〜75% 0.01%トリフルオロ酢酸(アセトニトリル中)/0.01%トリフルオロ酢酸(水中)の勾配)により精製した。純粋な分画を一晩凍結乾燥し、次いでクロロホルム 10mLと飽和重炭酸塩水溶液 4mLとの混合物中に溶解した。二相混合物を、10分間激しく攪拌し、次に層を分離させた。水相をクロロホルム(3x10mL)で抽出し、合わせた有機層を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させて、標題化合物(化合物12)を、白色固体(0.39g、95%)として得た。
1H−NMR(500MHz,CD3OD)δ7.89(s,1H),7.54(dd,J=8.8,7.2Hz,1H),7.50(d,J=8.2Hz,2H),7.34−7.29(m,4H),7.26−7.23(m,1H),7.20(d,J=8.2Hz,2H),6.38−3.36(m,2H),5.24(dd,J=9.4,2.8Hz,1H),4.20(d,J=7.8Hz,1H),3.35−3.23(m,3H),3.19−3.12(m,1H),2.82−2.71(m,2H),2.60−2.51(m,1H),2.37−2.32(m,1H),1.79−1.72(m,1H),1.52−1.43(m,3H).
LC−MS:m/z(ES)444.0(MH)+.
【0267】
工程B(化合物13): (3R)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド
【0268】
【化80】
【0269】
上記工程Aよりの、異性体2の脱保護に用いたものと同様の方法により、標題化合物(化合物13)を単一のジアステレオマーとして得た。
LC−MS:m/z(ES)444.0(MH)+.
【0270】
本明細書に記載の生物学的アッセイを用いて、化合物12及び13のヒトβ3機能活性は、各々1ないし10nM、及び1nM未満であると測定された。
【0271】
化合物14
(6S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキサミド
【0272】
【化81】
【0273】
工程A: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート
【0274】
【化82】
【0275】
N,N−ジメチルホルムアミド(100ml)中のi−13a(21.4g、55.9mmol)の溶液に、0℃で、[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボン酸(i−44、11.1g、61.5mmol)を、続いて1−ヒドロキシベンゾトリアゾール(7.55g、55.9mmol)、N−(3−ジメチルアミノプロピル)−N’−エチルカルボジイミド塩酸塩(16.1g、84.0mmol)、及びN,N−ジイソプロピルエチルアミン(29.2ml、168mmol)を添加した。反応混合物を、0℃から室温まで2時間攪拌した。水(600ml)を添加し、これをジクロロメタン(600mlx2)で抽出した。合わせた有機層を、Na2SO4上で乾燥した。揮発性物質を除去した後、残渣を、Biotage Horizon(登録商標)システム(0〜5%、次に5%メタノール(10%アンモニア/ジクロロメタン混合物を用いて))を使用することにより精製し、副ジアステレオマーの8%を含有する標題化合物を得た。これをさらに、超臨界液体クロマトグラフィー(キラルASカラム、40%メタノール)により精製して、標題化合物を淡黄色の固体として得た(22.0g、72%)。
1H NMR(CDCl3):δ9.61(s,1H),7.93(d,J=6.6Hz,1H),7.49(d,J=8.4Hz,2H),7.35−7.28(m,5H),7.13(d,J=8.5Hz,2H),6.40(d,J=6.7Hz,1H),5.36(d,J=8.6Hz,1H),4.38(m,1H),4.12−4.04(m,2H),3.46(m,1H),3.15−3.06(m,2H),2.91(dd,J=13.1,9.0Hz,1H),2.55(m,1H),2.38(m,1H),1.71−1.49(m,13H).LC−MS567.4(M+23).
【0276】
工程B: (6S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキサミド
【0277】
【化83】
【0278】
ジクロロメタン(40ml)中の工程Aよりの中間体(2.50g、4.59mmol)の溶液に、トリフルオロ酢酸(15ml)を添加した。反応混合物を室温で1.5時間攪拌した。揮発性物質を除去した後、飽和NaHCO3を添加して、pH値を8〜9とした。次に混合物を、ジクロロメタンで抽出した。合わせた有機層を、Na2SO4上で乾燥した。濃縮後、メタノール/アセトニトリルからの結晶化により、標題化合物を白色固体として得た(1.23g、60%)。
1H NMR(DMSO−d6):δ10.40(s,1H),7.91(d,J=6.7Hz,1H),7.49(d,J=8.3Hz,2H),7.32−7.26(m,4H),7.21(m,1H),7.15(d,J=8.4Hz,2H),6.23(d,J=6.7Hz,1H),5.11(dd,J=9.6,2.9Hz,1H),5.10(br,1H),4.21(d,J=7.1Hz,1H),3.20−3.00(m,4H),2.66−2.51(m,3H),2.16(m,1H),1.57(m,1H),1.38(m,1H),1.29−1.23(m,2H).LC−MS445.3(M+1)
【0279】
本明細書に記載の生物学的アッセイを用いて、化合物14のヒトβ3機能活性は、11ないし100nMであると測定された。
【0280】
化合物15〜29は、上記記載のものと同様の方法を用いて、適切な出発物質から調製した。
【0281】
β3機能活性のための生物学的アッセイ:
以下のインビトロのアッセイは、選択的β3アゴニスト活性を有する化合物をスクリーニングするのに適している:
機能アッセイ: リガンドに応答するcAMP産生を、バートン(Barton)ら(「Agonist−induced desensitization of D2 dopamine receptors in human Y−79 retinoblastoma cells(ヒトY−79網膜芽腫細胞におけるD2ドーパミン受容体のアゴニスト誘導性脱感作),Mol.Pharmacol.」、1991年、第3229巻、p.650〜658)に従い、以下のように修正して測定する。cAMP産生は、均一時間分解蛍光共鳴エネルギー転移免疫アッセイ(ランス(LANCETM)、パーキン・エルマー(Perkin Elmer))を用いて、製造業者の指示に従って測定する。クローニングされたβ−アドレナリン作動性受容体(β1、β2、又はβ3)で安定にトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞は、継代の3日後に収集する。細胞の収穫は、無酵素解離培地(Enzyme−free Dissociation Media)(Specialty Media)を用いて行なう。次に細胞をカウントし、ホスホジエステラーゼ阻害剤(IBMX、0.6mM)を含有するアッセイバッファ(5mM HEPES、0.1%BSAを補足したハンクス平衡塩類溶液(Hank’s Balanced salt solution))中に再懸濁する。反応は、6μL中の6,000個の細胞を、アレクサフルオル(Alexa Fluor)標識cAMP抗体(LANCETMキット)6μLと混合することにより開始し、これを次に、化合物(アッセイバッファで2X最終濃度に希釈)12μLを含有するアッセイウェルに添加する。反応は、室温で30分間進行させ、検出バッファ(LANCETMキット)24μLの添加により終了させる。次にアッセイプレートを、室温で1時間インキュベートし、パーキン・エルマー・エンビジョン・リーダー(Perkin Elmer Envision reader)又は同等物により、時間分解蛍光を測定する。蛍光レベルを、cAMPの標準曲線と比較することにより、未知のcAMPレベルを測定する。
【0282】
非選択的、完全アゴニストβ−アドレナリン作動性リガンド イソプロテレノールを、全3種の受容体において使用して、最大刺激を測定する。ヒトβ3アドレナリン作動性受容体(AR)選択的リガンド(S)−N−[4−[2−[[2−ヒドロキシ−3−(4−ヒドロキシフェノキシ)プロピル]アミノ]エチル]−フェニル]−4−ヨードベンゼンスルホンアミドを、対照として全てのアッセイにおいて使用する。イソプロテレノールは、10−10Mないし10−5のアッセイ時の終濃度で滴定し、選択的リガンド(S)−N−[4−[2−[[2−ヒドロキシ−3−(4−ヒドロキシフェノキシ)プロピル]アミノ]エチル]フェニル]−4−ヨードベンゼンスルホンアミドは、10−10Mないし10−5Mの濃度のβ3受容体にて滴定する。未知のリガンドは、10−10Mないし10−5Mのアッセイ時の終濃度の全3種のβ−アドレナリン作動性受容体サブタイプについて滴定し、EC50を測定する。EC50は、それ自体の最大値の50%の活性化を与える化合物濃度として定義される。データは、マイクロソフト・エクセル(Microsoft Excel)及びグラフパッド・プリズム(Graphpad Prism)を使用して分析するか、又は自社開発したデータ分析ソフトウェアパッケージを用いて分析する。
【0283】
結合アッセイ: 化合物はまた、β1及びβ2受容体においてもアッセイし、選択性を測定する。全ての結合アッセイは、組換えによりβ1又はβ2受容体を発現するCHO細胞から調製した膜を用いて実施する。細胞は、分離後3〜4日間増殖させ;付着した細胞をPBSで洗浄し、次に氷上で、1mM トリス、pH7.2中で10分間溶解させる。フラスコをこすって細胞を取出し、次にテフロン(登録商標)(Teflon)/ガラスホモジナイザーを用いて細胞をホモジナイズする。膜は、4℃において、38,000xgで15分間の遠心分離により収集する。ペレット化された膜を、TMEバッファ(50mM トリス、pH7.4、5mM MgCl2、2mM EDTA)中に、1mgタンパク質/mLの濃度に再懸濁する。膜の大きなバッチを調製し、分注し、−70℃で1年間まで能力の損失なしに貯蔵し得る。結合アッセイは、膜(2〜5μgのタンパク質)、放射標識トレーサ125I−シアノピンドロール(125I−CYP、45pM)、200μgのWGA−PVT SPAビーズ(GEヘルスケア(Healthcare))、及び10−10Mないし10−5Mの範囲の終濃度での試験化合物と一緒に、0.1%BSAを含有する最終体積200μLのTMEバッファ中でインキュベートすることにより実施する。アッセイプレートは、室温で振盪しながら1時間インキュベートし、次にパーキン・エルマー・トリラックス(Perkin Elmer Trilux)シンチレーションカウンタに置く。プレートは、カウントする前の約10時間を、Triluxカウンター内で暗中に静置する。データは、Graphpad Prismソフトウェア又は自社開発したデータ分析パッケージを使用して、標準的な4パラメータ非線形回帰分析を用いて分析する。IC50は、放射標識されたトレーサ(125I−CYP)の結合を50%阻害し得る化合物濃度として定義される。β3受容体に対する化合物の選択性は、(IC50β1 AR、β2 AR)/(EC50β3 AR)の比を計算することにより測定してもよい。
【0284】
β3−ARアゴニスト及び抗ムスカリン剤は、500:1ないし1:50の重量比で、患者に投与し得る。1つの実施態様においては、β3−ARアゴニスト及び抗ムスカリン剤の重量比は、300:1ないし1:10である。別の実施態様においては、重量比は、300:1ないし1:1である。別の実施態様においては、重量比は、150:1ないし1:1である。別の実施態様においては、重量比は、100:1ないし1:1である。なお別の実施態様においては、重量比は、150:1である。さらに別の実施態様においては、重量比は、100:1である.
【0285】
併用療法はまた、β3−ARアゴニスト及び抗ムスカリン剤に加えて、選択的M2アンタゴニストをさらに含んでなってもよい。
【0286】
本明細書で用いるとき、用語「選択的M2アンタゴニスト」は、ムスカリンM2サブタイプを、他のムスカリンサブタイプ、例えばM3サブタイプに比較して、10倍より大きい選択性で拮抗する化合物である。デルメンド(Delmendo)、「Br J Pharmacol.」、1989年2月、第96巻、第2号、p.457〜64参照(これは、選択的アンタゴニストの議論に関し、その記載全体が本明細書に援用される)。
【0287】
1つの実施態様においては、選択的M2アンタゴニストは、メトクトラミンである。メトクトラミンは、以下の構造を有するポリメチレンテトラミン誘導体である:
【0288】
【化84】
【0289】
1つの実施態様においては、OABの治療法は、β3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストを、それを必要とする患者に投与することを含んでなる。1つの実施態様においては、抗ムスカリン剤は、約40より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約50より大きいM2/M3比を有する。1つの実施態様においては、抗ムスカリン剤は、ダリフェナシンである。1つの実施態様においては、M2/M3比は、オータケ(Ohtake)らに記載された受容体結合アッセイを用いて測定される。
【0290】
1つの実施態様においては、OABの治療法は、β3−ARアゴニスト、ダリフェナシン、及びメトクトラミンを、それを必要とする患者に投与することを含んでなる。別の実施態様においては、β3−ARアゴニストは、表3に示した化合物から選択される。別の実施態様においては、β3−ARアゴニストは、表4に示した化合物から選択される。なお別の実施態様においては、β3−ARアゴニストは:
【0291】
【化85】
【0292】
及び
【0293】
【化86】
【0294】
からなる群より選択される。
【0295】
β3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストが患者に投与される方法においては、β3−ARアゴニストは、選択的M2アンタゴニストで前処理されてもよい。1つの実施態様においては、選択的M2アンタゴニストは、メトクトラミンである。別の実施態様においては、抗ムスカリン剤は、ダリフェナシンである。別の実施態様においては、β3−ARアゴニストは、メトクトラミンで前処理される。なお別の実施態様においては、メトクトラミンで前処理されたβ3−ARアゴニストは、ダリフェナシンと同時投与される。
【0296】
1つの実施態様においては、前処理のためのメトクトラミンの濃度は、0.1ないし10μMである。別の実施態様においては、前処理のためのメトクトラミンの濃度は、1μMである。
【0297】
上記記載の併用療法においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、同時に、連続的に、又は別々に患者に投与し得る。
【0298】
1つの実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、同時に患者に投与される。別の実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、別々に患者に投与される。なお別の実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、連続的に患者に投与される。
【0299】
適した患者は、これに限定されないが、過活動膀胱又は下部尿路症状(LUTS)の患者を包含する。1つの実施態様においては、患者は、OAB症状のある女性である。別の実施態様においては、患者は、OAB症状のある閉経期の女性である。
【0300】
本発明の別の態様は、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを含んでなる、組合せ医薬組成物を提供する。適切なβ3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストは、上記記載の通りである。
【0301】
組合せ組成物中のβ3−ARアゴニストの適量は、約0.01mgないし約500mgである。1つの実施態様においては、β3−ARアゴニストの量は、約0.05mgないし約250mgである。別の実施態様においては、該量は、約0.1mgないし約150mgである。別の実施態様においては、該量は、約1ないし約100mgである。なお別の実施態様においては、該量は、約1ないし約50mgである。
【0302】
組合せ組成物中の抗ムスカリン剤の適量は、約0.01mgないし約50mgである。1つの実施態様においては、抗ムスカリン剤の量は、約0.05mgないし約12mgである。別の実施態様においては、該量は、約0.1mgないし約6mgである。別の実施態様においては、該量は、約0.2ないし約5mgである。なお別の実施態様においては、該量は、約0.2ないし約3mgである。
【0303】
組合せ組成物中の選択的M2アンタゴニストの適量は、約0.01mgないし約50mgである。1つの実施態様においては、選択的M2アンタゴニストの量は、約0.05mgないし約15mgである。
【0304】
実際の使用では、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、通常の薬学的調剤技術に従い、薬学的担体との均質な混合物中の活性成分として組合せ得る。担体は、投与、例えば、経口又は非経口(静脈内を含む)用に求められる製剤の形状に依存して、広く多様な形態をとってよい。経口剤形用の組成物の調製においては、例えば懸濁液、エリキシル、及び溶液などといった経口液体製剤の場合には、例えば水、グリコール、油、アルコール、着香剤、保存剤、着色剤などといった任意の通常の薬学的媒体を用いてもよく;又は、例えば粉末、硬及び軟カプセル、及び錠剤といった経口用固形製剤の場合には、デンプン、糖、微結晶セルロース、希釈剤、造粒剤、滑沢剤、結合剤、崩壊剤などといった担体を使用してもよく、液体製剤よりも固形経口製剤が好ましい。
【0305】
その投与の容易さから、固体の薬学的担体が使用される場合、錠剤及びカプセルが最も有利な経口用単位剤形である。所望であれば、錠剤は、標準的な水性又は非水性の技術によりコートされてもよい、かかる組合せ組成物及び製剤は、0.1〜20パーセントの各活性成分を含有し得る。これらの組合せ組成物における活性成分のパーセントは、もちろん変更されてもよく、かかる組成物中の活性成分の量は、有効な薬用量が得られるようにするものである。
【0306】
活性成分はまた、例えば液滴又はスプレーとして、鼻腔内に投与し得る。
【0307】
錠剤、丸剤、カプセルなどはまた、トラガカントガム、アラビアガム、コーンスターチ、又はゼラチンなどの結合剤;リン酸二カルシウムなどの賦形剤;コーンスターチ、ジャガイモデンプン、アルギン酸などの崩壊剤;ステアリン酸マグネシウムなどの滑沢剤;及びスクロース、ラクトース、又はサッカリンなどの甘味剤を含有してもよい。単位剤形がカプセルである場合、それは上記のタイプの物質に加えて、脂肪油などの液体担体を含有してもよい。
【0308】
様々な他の物質が、コーティングとして、又は単位剤形の物理的形態を変更するために存在してもよい。例えば、錠剤を、シェラック、糖、又はその双方でコートしてもよい。シロップ又はエリキシルは、活性成分に加えて、甘味剤としてのスクロース、保存剤としてのメチル及びプロピルパラベン、色素、及びチェリー又はオレンジフレーバーなどの着香剤を含有してもよい。
【0309】
活性成分はまた、非経口的に投与してもよい。これらの活性成分の溶液又は懸濁液は、ヒドロキシ−プロピルセルロースなどの界面活性剤と適宜混合された水中に調製し得る。分散剤はまた、グリセロール、液体ポリエチレンブリコール、及びそれらの油中混合物中に調製し得る。通常の貯蔵及び使用条件下では、これらの製剤は、微生物の増殖を防止するための保存剤を含有する。
【0310】
注射用の使用に適した組合せ組成物は、無菌の水溶液又は分散剤、及び無菌の注射用溶液又は分散剤を即時調製するための無菌の粉末を包含する。全ての場合、剤形は無菌でなければならず、注射が容易である程度の液体でなければならない。それは、製造及び貯蔵の条件下で安定でなければならず、また細菌及び真菌などの微生物の汚染作用に対抗して保存されねばならない。担体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、及び液体ポリエチレングリコール)、適切なこれらの混合物、及び植物油を含有する、溶媒又は分散媒であってもよい。
【0311】
1つの実施態様においては、組合せ組成物は、経口組成物である。別の実施態様においては、経口組成物は、カプセルゲルの状態である。なお別の実施態様においては、組合せ組成物は、経口用錠剤組成物である。さらに別の実施態様においては、組合せ組成物は、経口用ビーズ組成物である。
【0312】
1つの実施態様においては、組合せ組成物は、制御放出用組成物であって、これにおいて、抗ムスカリン剤は、組成物の投与により、24時間にわたり放出される。別の実施態様においては、抗ムスカリン剤は、10時間にわたり放出される。なお別の実施態様においては、抗ムスカリン剤は、8時間にわたり放出される。さらに別の実施態様においては、抗ムスカリン剤は、6時間にわたり放出される。
【0313】
本明細書に開示されたものはまた、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストの、過活動膀胱の治療又は予防用医薬の製造における使用も包含する。
【0314】
実施例
β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストの同時投与の効果は、以下の実施例において例示される。
【0315】
CL316243、又は二ナトリウム(R,R)−5−(2−((2−(3−クロロフェニル)−2−ヒドロキシエチル)−アミノ)プロピル)−1,3−ベンゾジオキソール−2,3−ジカルボキシラートは、β3−ARアゴニストである。CL316243は、「J.Med.Chem.」、1992年8月7日、第35巻、第16号、p.3081〜4にさらに詳細に記載されている。
【0316】
トルテロジン、又は2−[(1S)−3−(ジイソプロピルアミノ)−1−フェニルプロピル]−4−メチルフェノ−ルは、過活動膀胱の治療に使用される抗ムスカリン剤である。トルテロジンは、米国特許第5,382,600、6,630,162、6,770,295、及び6,911,217号にさらに詳細に記載されている。
【0317】
オキシブチニン、又は4−ジエチルアミノブタ−2−イニル2−シクロヘキシル−2−ヒドロキシ−2−フェニル−エタノアートは、膀胱の筋肉痙攣を低減することにより、頻尿及び排尿制御不能(切迫尿失禁)を含む、尿及び膀胱の障害を緩和するのに使用される抗ムスカリン剤である。
【0318】
ダリフェナシン、又は(S)−2−[1−[2−(2,3−ジヒドロベンゾフラン−5−イル)エチル]ピロリジン−3−イル]−2,2−ジフェニル−アセトアミドは、尿失禁を治療するために使用される抗ムスカリン剤である。ダリフェナシンは、米国特許第5,096,890号にさらに詳細に記載されている。
【0319】
実施例1〜3
材料及び方法
以下の材料及び方法を、実施例1〜3に使用した。雄成体スプラーグ・ドーリー(Sprague−Dawley)ラットを使用した。CO2ガスを用いて安楽死させた後、全膀胱を取出した。三角部外側部分の排尿筋の長軸方向の細片(約6mmx3mm)を用意した。各細片を、酸素化された(95%O2+5%CO2)クレブス(Krebs)溶液を含有する、温めた(37℃)臓器浴(25mL)中に置いた。細片は、一方の端を臓器浴につなげ、他端を等尺性の変換器(ADインスツルメンツ(Instruments))に、10mNの静止張力下で連結した。標本の応答は、多チャンネルデータ収録システム(PowerLab、AD Instruments)により、10Hzのサンプリング率で記録し、分析ソフトウェア(Chart、AD Instruments)で測定した。少なくとも60分間の平衡化時間の後、各組織細片を、60Hz;持続時間、0.3ms;3秒間;90Vでの電気的フィールド刺激(EFS)で誘発し、収縮を誘導した。EFSにより安定な収縮が得られれば、化合物溶液(25μL)を、次第に増加する様式で臓器浴中に適用した。各化合物による処理の15分後、EFSを適用した。
【0320】
アイソボログラム分析
アイソボログラム分析を使用して、併用療法の相乗効果を評価した。アイソボログラム分析は、2つの異なる薬剤の相互作用について、独立した統計解析を用いて視覚的評価を提供する。統計解析は、各化合物の単一処理及び同じ効果を得るための固定比率組合せからの、いくつかの効力指数の計算から遂行される。アイソボログラム分析は、「JPET」、2001年、第298巻、p.865〜872(これは、その記載全体が本明細書に援用される)にさらに詳細に記載されている。
【0321】
アイソボログラム分析の例示的な説明図を、図1に示す。図1の、ある特定の効果(例えば、最大の50%)についてのアイソボログラムでは、活性薬剤A単独の用量はA=20であり、活性薬剤B単独はB=100である。これらの切片を結ぶ直線(相加的直線)は、これらの効力に基づき、同じ効果をもたらすはずの、全ての用量ペアの位置である。実際の点Qなどの用量ペアは、より少ない量でこの効果を達成し、相乗的(又は超相加的)であり、一方、点Rによって示される用量ペアは、より多くの量が必要であることを示し、それ故相加的以下である。直線A−Bに近接して出現するPのような点は、単純に相加的である。相互作用の性質を実証するため、適切な統計解析がしばしば使用される。
【0322】
実施例2
β3−ARアゴニストCL316243の、トルテロジン、オキシブチニン、及びダリフェナシンから選択される抗ムスカリン剤との併用療法
個別に投与した場合、CL316243の、トルテロジン、オキシブチニン、及びダリフェナシンの各々は、EFS−誘導性の単離排尿筋収縮を阻害した。表5は、25%阻害を誘導した各化合物の濃度を示す。これらの値を、以下のアイソボログラム分析に使用した。
【0323】
【表5】
【0324】
併用療法では、CL316243は、トルテロジン、オキシブチニン、又はダリフェナシンと、固定重量比において同時投与され、アイソボログラム分析からの結果は図2に示される。図2は、CL316243の、トルテロジン(1:2、図2A)、又はオキシブチニン(1:10、図2B)との併用が、相乗効果を示したことを表している。一方、CL316243のダリフェナシンとの併用(1:2、図2C)は、単純に相加的である(すなわち、何ら相乗的ではない)ことを示す。
【0325】
何ら理論に束縛されることを望むものではないが、一般に、抗ムスカリン剤のM3拮抗作用が、OAB効力に重要であると考えられている(例えば、アブラムス(Abrams)及びアンデルソン(Andersson)「BJU Int」、2007年、第100巻、p.987〜1006参照)。驚くべきことに、今、抗ムスカリン剤のM2/M3の相対的選択性が、抗ムスカリン剤及びβ3−ARアゴニストを用いた併用療法における、OAB効力及び/又は副作用の低減に重要な役割を果たし得ることが見出された。
【0326】
上記の結果からわかるように、抗ムスカリン剤トルテロジンは、ムスカリン受容体のM2及びM3サブタイプに対し、ほぼ等しい選択性(M2/M3≒1)を有しており、トルテロジンとCL316243、β3−ARアゴニストとの、2:1の比での組合せは、相乗効果をもたらした。他の抗ムスカリン剤、約6のM2/M3比を有するオキシブチニンもまた、CL316243と10:1の比で組合せた場合、相乗効果をもたらした。
【0327】
一方、M2よりもM3にはるかに高い選択性(M2/M3≒50)をもつダリフェナシンは、CL316243と2:1の比で組合せた場合、相乗作用をもたらさなかった。
【0328】
以上をまとめれば、CL316243の、その各々が40未満のM2/M3比をもつトルテロジンは又はオキシブチニンとの併用は、排尿筋収縮の阻害に相乗作用をもたらした。一方、CL316243の、40より大きいM2/M3比をもつダリフェナシンとの併用は、相乗作用をもたらさなかった。
【0329】
実施例3
トルテロジン又はダリフェナシンとの併用における、β3−ARアゴニスト、化合物12
この実施例では、異なるβ3−ARアゴニストを使用して、β3−ARアゴニストと抗ムスカリン剤との併用療法の相乗効果を調べた。
【0330】
表3において上記記載のβ3−ARアゴニスト、化合物12は、IC25値275nMで、EFS−誘導性単離排尿筋収縮を阻害した。したがって、化合物12は、ラット膀胱細片のEFS−誘導性収縮の阻害において、CL316243(IC25 2.86nM、表5参照)よりもほぼ100倍効力が弱い。このことは、化合物12の、ラットβ3−ARにおける弱い効力と一致する。
【0331】
併用研究においては、化合物12は、トルテロジン又はダリフェナシンと、固定重量比50:1で同時投与された。アイソボログラム分析の結果を、図3に示す。図3は、化合物12の、約1のM2/M3比をもつトルテロジンとの併用が、50:1の比において(図3A)、相乗効果をもたらしたことを示す。
【0332】
一方、化合物12の、約50のM2/M3比をもつダリフェナシンとの併用が、50:1の比において(図3B)、相乗効果をもたらなかったことを示す(相加的以下)。
【0333】
上記の結果は、異なるβ3−ARアゴニスト(CL316243)が研究に使用された、実施例1で見られた結果と一致する。これらの結果は、抗ムスカリン剤が40未満のM2/M3比をもつ場合、そのβ3−ARアゴニストとの併用が、排尿筋収縮の阻害において相乗作用をもたらすことを示唆している。一方、抗ムスカリン剤が40より大きいM2/M3比をもつ場合、そのβ3−ARアゴニストとの併用は、何ら相乗作用をもたらさない。
【0334】
実施例4
併用療法の相乗効果に対する選択的M2アンタゴニストの影響
この実施例では、β3−ARアゴニストと、40より大きいM2/M3比をもつ抗ムスカリン剤との併用療法に対する、選択的M2アンタゴニストの影響を調べる。
【0335】
まず、CL316243、β3−ARアゴニストは、メトクトラミン(1μM)で未処理か又は前処理されたかのいずれかであり、結果は図4に示される。図4は、メトクトラミンによるCL316243の前処理が、EFS−誘導性膀胱収縮の阻害に関し、CL316243の効力に何ら有意な影響を及ぼさなかったことを示す。この結果は、選択的M2アンタゴニスト単独では、β3−ARアゴニスト及び抗ムスカリン剤がM3拮抗作用を用いて行うようには、予め収縮したラット膀胱細片を弛緩させないという、一般的な考えと一致する。このことは、選択的M2アンタゴニスト及びCL316243の併用では、及びM3拮抗作用なしでは、相乗作用をもたらさなかったことを示唆している。
【0336】
次に、未処理(A)及び前処理されたCL316243(B)の各々が、ダリフェナシンと各々1:2の比で組合され、その結果が図5に示される。図5は、前処理されたCL316243(1μM メトクトラミンで)と、ダリフェナシンとの1:2の比での組合せが、相乗効果をもたらしたことを示す。上記に議論されたように、ダリフェナシンは選択的M3アンタゴニストであり、約50のM2/M3比を有する。
【0337】
一方、未処理のCL316243と、ダリフェナシンとの同じ比(1:2)での組合せは、単純に相加的であった(すなわち、何ら相乗効果はない)。
【0338】
これらの結果は、β3−ARアゴニストと抗ムスカリン剤との間の組合せの相乗効果が、M2及びM3拮抗作用の双方の存在を必要とし得るという、実施例1及び2の所見と一致する。
【0339】
何ら理論に束縛されることを望むものではないが、M2受容体は、cAMP依存性のメカニズムによるアドレナリン受容体媒介性の弛緩を逆転させることにより、間接的な収縮応答の媒介において役割を果たし得ると考えられている。M2拮抗作用は、β3−ARアゴニスト誘導性のcAMP増加及びBKチャンネル開放を強化し、排尿筋のさらなる弛緩をもたらす結果となる。
【0340】
実施例5
相乗効果に対する組合せ比の影響
材料及び方法
動物:雌のSDラット(体重200〜250g)(合計7群)。
麻酔:ウレタン(1.0g/kg、ip)。
パラメータ:拡張誘導性の律動的膀胱収縮の振幅
化合物:オキシブチニン(OXY)、CL316243(CL)。
分析:ID20値(振幅を20%まで低減する用量)を用いたアイソボログラム。
【0341】
まず、以下の条件で単回投与実験を行ない、オキシブチニン(OXY)及びCL316243(CL)のID20値を計算し、得られたID20値が表6に示される。
ビヒクル(生理食塩水);
OXY、0.01、0.03、0.1mg/kg、iv;
CL、0.003、0.01、0.03mg/kg、iv。
【0342】
【表6】
【0343】
表6の結果は、オキシブチニン及びCL316243の双方が、麻酔された雌のラットにおいて、拡張誘導性の律動的膀胱収縮の振幅を低減したことを示す。
【0344】
次に、以下の表7に示した併用用法を実施して、アイソボログラムを組立てた。合計で9群あった。OXY及びCLのID20値は、各組合せ比率において計算した。
【0345】
【表7】
【0346】
図6は、CL及びOXYの、1:1及び1:10の比率の組合せについて、相乗効果が観察されたことを示す。一方、CL及びOXYの、1:3の比率の組合せは、単純な相加効果をもたらしたが、相乗効果はなかった。
【0347】
これらの結果は、β3−ARアゴニストCLと抗ムスカリン剤OXYとの間の相乗効果もまた、組合せにおける特定の組合せ比に依存することを示唆している。
【0348】
実施例6
β3−ARアゴニストと抗ムスカリン剤とを含んでなる組合せ組成物
β3−ARアゴニストと抗ムスカリン剤とを含んでなる、例示的な組合せ組成物を、表8に示す。
【0349】
【表8】
【0350】
1つの実施態様においては、β3−ARアゴニストは、表3に列記された化合物から選択される。別の実施態様においては、抗ムスカリン剤は、トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301から選択される。
【0351】
1つの実施態様においては、上記の組合せ組成物は、制御放出用(CR)製剤である。別の実施態様においては、組合せ組成物は、経口投与用のカプセルゲルの状態である。
【0352】
実施例7
CR抗ムスカリン剤及びIR β3−ARアゴニストを含んでなる組合せ組成物
制御放出(CR)部分に抗ムスカリン剤を、そして即時放出(CR)部分にβ3−ARアゴニストを含んでなる例示的な組合せ組成物を、表9に示す。
【0353】
【表9−1】
【0354】
【表9−2】
【0355】
1つの実施態様においては、β3−ARアゴニストは、表3に列記された化合物から選択される。別の実施態様においては、抗ムスカリン剤は、トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301から選択される。
【0356】
1つの実施態様においては、上記組成物は、経口投与用のカプセルゲルである。
【0357】
実施例8
アカゲザルにおける膀胱容量に対する併用療法の効果
材料及び方法
体重5.3〜6.2kg(4〜7歳齢)の成体雌のアカゲザル(Macaca mulatta)を使用した。被験者は、二匹一組又は単独のいずれかで、12時間明/12時間暗のサイクル(7:00AMに点灯)において収容した。彼女らの食餌は、2050テクラド(Teklad)(ハーラン・ラボラトリーズ(Harlan Laboratories)、インディアナ州、インディアナポリス)及び新鮮な果物又は野菜から構成された。水は自由に摂取させた。動物は全て、獣医技術者及び管理人により、健康障害の徴候について毎日観察した。被験者は、>13日の休止期間をとって反復使用された。サルは、テラゾール(Telazol)(3〜5mg/kg)又はケタミン(10〜20mg/kg)のいずれかの筋肉内注射と、その後のシリンジポンプ(552222、ハーバード・アパレイタス(Harvard Apparatus)、マサチューセッツ州、ホリストン)を用いたケタミン(0.2〜0.8mg/kg/分)の静脈内定速輸液によって麻酔した。動物を仰臥位に置き、トリプルルーメンバルーン経尿道カテーテル(7.4Fr、クック・メディカル(Cook Medical、インディアナ州、ブルーミントン)を膀胱に挿入し、バルーンを水 1mLで膨らませて、カテーテルの先端を膀胱の基底部に固定した。カテーテルは、膀胱を充填するための輸液ポンプ(ジェミニ(Gemini)PC−2TX、アラリス・メディカル・システムズ(ALARIS Medical Systems)、カリフォルニア州、サンディエゴ)、及び膀胱内圧をモニタリングするための圧力変換器に連結した。膀胱内圧は、多チャンネルデータ収録システム(Power lab、AD Instruments、バイオパック・システムズ(Biopac systems)、コロラド州、コロラドスプリングス)を用いて、20Hzのサンプリング率で連続的に記録した。超音波診断(Logiq e vet、GEメディカル・システムズ(Medical Systems)、ウィスコンシン州、ウォーキショー、図1A)により膀胱が空であることを確認した後、生理食塩水を15mL/分で膀胱内に注入した。排尿反射の圧力指標に急峻な上昇が観察されたとき、膀胱内注入を停止し、膀胱を60mlシリンジにより手作業で空にした。2回のベースラインシストメトリーの読み取りの後、薬剤を、濃度の上昇するパラダイムを用いて、各投与の10分後にシストメトリーを実施することにより、3回静脈内投与した。膀胱容量は、各シストメトリーについて測定し、ベースライン容量からの%変化を計算した。本明細書で用いるとき、「ベースライン容量」又は「ベースライン」は、2回の投与前測定値の平均膀胱容量を意味する。
【0358】
種々の用量における、トルテロジン(“TOL”)、ダリフェナシン(DAR”)、又は化合物14(Cpd 14”)の単一療法、並びに、表10に示した種々の用量及び用量比でのTOL:Cpd 14及びDAR:Cpd 14の併用療法を、アカゲザルで試験した。
【0359】
【表10】
【0360】
上記の単一療法及び併用療法を用いることによる、アカゲザルにおける膀胱容量の結果は表11に要約される。報告された結果は、4〜6匹の動物におけるベースラインからの%変化の平均値である。
【0361】
【表11】
【0362】
表11から、試験した化合物14及びトルテロジンの全ての組合せが、各個の単一療法に比較して、より大きい膀胱容量を示したことが理解される。最も低い化合物14の用量(0.003mg/kg)において、相乗効果がはるかに大きかったことが注目されるべきである。特に、0.003mg/kg:0.01mg/kgでのCpd 14:TOLの組合せは、それぞれの単一療法についての4.1%及び8.6%に比較して、28.2%の膀胱容量増加を示した。同様に、0.003mg/kg:0.03mg/kgでのCpd 14:TOLの組合せは、それぞれの単一療法についての4.1%及び16.8%に比較して、35.5%の膀胱容量増加を示した。
【0363】
化合物14とダリフェナシンとの併用療法については、より高い用量のダリフェナシン(0.03、0.1mg/kg)において、併用が優れた膀胱容量効果を示した。
【0364】
非選択的ムスカリンアンタゴニスト、トルテロジンは、調べた組合せにおいて、化合物14と、改善された効力を明示したが、選択的M3アンタゴニスト、ダリフェナシンと、化合物14との組合せでは、相加的効果は高用量にのみ限定された。これらの結果は、β3−ARアゴニストと組合された場合、抗ムスカリン剤のM2及びM3拮抗作用の双方が、改善された効力にとり重要であり得ることを示唆している。
【0365】
本発明は、そのいくつかの特定の実施態様について記載しかつ例示してきたが、当業者は、種々の変更、修飾、及び置換が、本発明の趣旨及び範囲から逸脱することなく、これにおいて行なわれ得ることを理解するであろう。例えば、上記の本明細書で示した特定の薬用量以外の有効薬用量を、上記に示した本発明において使用される活性薬剤の任意の指標について、治療される哺乳動物の応答性のバリエーションの結果として適用してもよい。同様に、観察される具体的な薬理学的応答は、選択された特定の活性化合物又は薬学的担体が存在するかどうか、並びに用いた製剤のタイプに従って、及び依存して変化してもよく、結果におけるそのような予想される変動又は差異は、本発明の目的及び実施に従って検討される。それ故、本発明は、以下の請求の範囲によって定義されること及びかかる請求の範囲が合理的である限り広く解釈されることが意図されている。
【背景技術】
【0001】
下部尿路の機能は、尿の貯蔵及び定期的な放出である。これには、中枢及び末梢神経エフェクターメカニズムの調整と、結果として生じる自律神経系の交感神経及び副交感神経成分、並びに体性運動経路の、調和のとれた調節とをもたらす、多様な求心性及び遠心性神経経路を含む、貯蔵及び排尿反射のオーケストレーションが必要である。これらは、膀胱(排尿筋)及び尿道平滑筋、並びに尿道括約筋の収縮状態を、近位で調節する。
【0002】
過活動膀胱(OAB)は、通常は頻度及び夜間頻尿に関連する、切迫性尿失禁を伴うか伴わない、尿意切迫の症状によって特徴づけられる。米国及び欧州におけるOABの有病率は、18歳を超える女性及び男性双方の16ないし17%と推定されてきた。過活動膀胱は、最も多くの場合、特発性として分類されるが、神経学的症状、膀胱出口閉塞、及び他の原因の二次的なものでもあり得る。病理生理学的見地からは、過活動膀胱症候は、特に切迫尿失禁に付随する場合、排尿筋過活動が示唆される。切迫性は、失禁の有無にかかわらず、社会及び医療福祉の双方に負の影響を与えることが示されてきており、年間の直接及び間接的健康管理支出の面で、かなりの負担である。
【0003】
抗ムスカリン剤は、OABなどの失禁症状を治療するために使用されてきた。例えば、トルテロジン、又は(R)−N,N−ジイソプロピル−3−(2−ヒドロキシ−5−メチルフェニル)−3−フェニルプロパンアミンは、切迫尿失禁及び、他の不安定又は過活動性膀胱の症候の治療用に市販されてきた。トルテロジン及びその主要な活性代謝産物、トルテロジンの5−ヒドロキシメチル誘導体は、治療効果に貢献すると考えられている。しかしながら、抗ムスカリン剤を用いたOABのための今日の医薬療法は、しばしば多くの患者が、今日の治療に対し充分な応答を示さないこと、及び/又は今日の治療によるドライマウスなどの少なからぬ副作用に耐え得ないことから、最適とはいえない場合が多い。
【0004】
それ故、OABのより有効な治療及び/又は副作用の低減を提供する改善された療法に、継続した需要がある。
【図面の簡単な説明】
【0005】
【図1】アイソボログラム分析を示す説明図である。
【図2】CL316243の、トルテロジン(A)、オキシブチニン(B)、又はダリフェナシン(C)との併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図3】化合物12、及びトルテロジン(A)、及びダリフェナシン(B)の併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図4】メトクトラミンの前処理の有り無しでの、CL316243を示す説明図である。
【図5】メトクトラミンの前処理が有る(A)か、又は無し(B)での、CL316243及びダリフェナシンの併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【図6】様々な割合での、CL316243及びオキシブチニンの併用により誘導される排尿筋収縮阻害のアイソボログラムを示す説明図である。
【発明の概要】
【0006】
発明の要旨
驚くべきことに、β3アドレナリン作動性受容体アゴニスト(以降、「β3−ARアゴニスト」)、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを用いた併用療法が、過活動膀胱の治療に相乗効果をもたらすことが判明してきた。β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを含んでなる組合せ組成物もまた記載される。
【0007】
発明の詳細な記載
本明細書に記載されるのは、過活動膀胱の治療法であり、これにおいて該方法は、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを、それを必要とする患者に投与することを含んでなる。かかる併用療法は、相乗効果及びしたがって、効力の改善及び/又は副作用の低減を提供する。
【0008】
驚くべきことに、現在、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において、抗ムスカリン剤のM2拮抗作用が、OABの治療に相乗作用を与える上で重要な役割を果たし得ることが判明してきている。何らの理論にも束縛されることを望むものではないが、抗ムスカリン剤のM3拮抗作用がOAB効力にとり重要であることが、一般に信じられている(例えば、アブラムス(Abrams)及びアンデルソン(Andersson)、「BJU Int.」、2007年、第100巻、p.987〜1006参照)。今、M3拮抗作用及びβ3−ARアゴニストと一緒に作用するM2拮抗作用が、相乗作用を提供することが判明してきた。
【0009】
1つの実施態様においては、相乗効果は、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において得られており、これにおいて、抗ムスカリン剤は、約40未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は約20未満のM2/M3比を有する。
【0010】
さらに、抗ムスカリン剤が40を超えるM2/M3比を有するとき、β3−ARアゴニスト及び抗ムスカリン剤を含んでなる併用療法において、追加の選択的M2アンタゴニストを使用することにより、相乗作用が得られることがある。
【0011】
本明細書で用いるとき、「相乗作用」又は「相乗効果」は、2種以上の活性薬剤の組合せ効果が、個々の活性薬剤の合計よりも大きい場合を記載するのに使用される。言い換えれば、2種以上の活性薬剤は、一方の活性薬剤の存在が第2の効果を増強又は拡大するように相互作用し得る。対照的に、2種以上の活性薬剤の組合せ効果が、個々の活性薬剤の合計に実質的に等しい場合、組合せ効果は単純に相加的であるが、相乗的ではない。また、2種以上の活性薬剤の組合せ効果が個々の活性薬剤の合計よりも低い場合、組合せ効果は相加的以下であり、これもまた相乗的ではない。
【0012】
1つの実施態様においては、併用療法は、β3−ARアゴニスト及び抗ムスカリン剤を、それを必要とする患者に投与することを含んでなり、ここで、抗ムスカリン剤は、約40未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約30未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約20未満のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約15未満のM2/M3比を有する。なお別の実施態様においては、抗ムスカリン剤は、約10未満のM2/M3比を有する。さらに別の実施態様においては、抗ムスカリン剤は、約1のM2/M3比を有する。
【0013】
別の実施態様においては、併用療法における抗ムスカリン剤は、約0.1より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.5より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.8より大きいM2/M3比を有する。
【0014】
別の実施態様においては、併用療法における抗ムスカリン剤は、約0.1ないし約40のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.5ないし約30のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約0.8ないし約20のM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約1ないし約20のM2/M3比を有する。なお別の実施態様においては、抗ムスカリン剤は、約1ないし約15のM2/M3比を有する。さらに別の実施態様においては、抗ムスカリン剤は、約1ないし約10のM2/M3比を有する。
【0015】
1つの実施態様においては、M2/M3比は、オータケ(Ohtake)ら(「Biol.Pharm.Bull.」、2007年、第30巻、p.54〜58)(これは、その記載全体が本明細書に援用される)において記載された受容体結合アッセイを使用して測定される。別の実施態様においては、M2/M3比は、ヘグデ(Hegde)ら(「Curr Opin Invest Drugs」、2004年、第5巻、p.40〜49)(これは、その記載全体が本明細書に援用される)において記載されたアッセイを使用して測定される。
【0016】
オータケ(Ohtake)ら(「Biol.Pharm.Bull.」、2007年、第30巻、p.54〜58)において報告された、いくつかの代表的な抗ムスカリン剤のM1−M4活性を、表1に示す。
【0017】
【表1】
【0018】
ヘグデ(Hegde)ら(「Curr Opin Invest Drugs」、2004年、第5巻、p.40〜49)は、いくつかの抗ムスカリン剤のM1〜M4活性を記載しており、トロスピウムのM1〜M4活性は、表2に示される。
【0019】
【表2】
【0020】
併用療法に適した抗ムスカリン剤は、これに限定されないが:トルテロジン、オキシブチニン(S−オキシブチニンを含む)、ヒオシアミン、プロパンテリン、プロピベリン、トロスピウム(塩化トロスピウムを含む)、ソリフェナシン、ダリフェナシン、ジシクロミン、イプラトロピウム、オキシトロール、イミダフェナシン、フェソテロジン、テミベリン、SVT−40776、グラクソスミスクライン(GlaxoSmithKline)による202405、TD6301、RBX9841、DDP200、及びPLD179を包含する。
【0021】
1つの実施態様においては、抗ムスカリン剤は:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301からなる群より選択される。1つの実施態様においては、適切な抗ムスカリン剤のM2/M3比は、40未満である。別の実施態様においては、M2/M3比は、30未満である。別の実施態様においては、M2/M3比は、20未満である。なお別の実施態様においては、M2/M3比は、15未満である。1つの実施態様においては、M2/M3比は、オータケ(Ohtake)ら、において記載された結合アッセイを用いて測定される。
【0022】
別の実施態様においては、抗ムスカリン剤は:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、及びトロスピウムからなる群より選択される。
【0023】
別の実施態様においては、抗ムスカリン剤は:トルテロジン及びオキシブチニンからなる群より選択される。
【0024】
なお別の実施態様においては、抗ムスカリン剤は、トルテロジンである。
【0025】
適切なβ3−ARアゴニストは、これに限定されないが、CL316243、及び表3に示した化合物を包含する。
【0026】
【表3−1】
【0027】
【表3−2】
【0028】
【表3−3】
【0029】
別の実施態様においては、β3−ARアゴニストは、表4に列記された化合物から選択される:
【0030】
【表4−1】
【0031】
【表4−2】
【0032】
別の実施態様においては、β3−ARアゴニストは、
【0033】
【化1】
【0034】
及び
【0035】
【化2】
【0036】
からなる群より選択される。
【0037】
なお別の実施態様においては、β3−ARアゴニストは、
【0038】
【化3】
【0039】
である。
【0040】
表3の化合物は、以下に記載された方法を用いて調製し得る。
【0041】
本出願全体を通して、以下の用語は、別に記されない限り、示された意味を有する。
【0042】
【化4−1】
【0043】
【化4−2】
【0044】
【化4−3】
【0045】
中間体1
ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1):
【0046】
【化5】
【0047】
工程A: エチル(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)アセタート
【0048】
【化6】
【0049】
無水DCM 250mL中のメチル(3−アミノフェニル)アセタート(25g、140mmol)の溶液に、DIEA(28.5mL、155mmol)を添加し、得られた溶液を0℃に冷却し、窒素雰囲気下に置いた。この冷却された溶液に、次いでベンジルクロロホルマート(21.1mL、148mmol)を添加し、得られた混合物を一晩攪拌して、室温に温めさせた。反応物を、1M HCl、水、及び次に食塩水で洗浄した。有機層を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。さらなる精製は不要であり、この物質(44g、99%)をそのまま次の工程反応に使用した。
LC−MS:m/z(ES)314(MH)+,336(MNa)+.
【0050】
工程B: (3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)酢酸
【0051】
【化7】
【0052】
THF、エタノール、及び水(1:1:1、1500mL)中のエチル(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)アセタート(工程Aより)44.0g(140mmol)の溶液に、固体LiOH(16.8g、700mmol)を添加し、得られた溶液を油浴により60℃で3時間加熱した。混合物を室温に一晩冷却し、次に濃HCl 40mLを、25℃未満の温度を維持しながら、溶液がおよそpH2〜3になるまで徐々に添加した。酢酸エチル(3x750mL)で抽出し、次に合わせ、有機物質を水で、次いで食塩水で洗浄した。有機物質を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。標題化合物(24.7g、87%)を、さらに精製することなく次の工程反応に使用した。
LC−MS:m/z(ES)286(MH)+,308(MNa)+.
【0053】
工程C: ベンジル(3−{2−[メトキシ(メチル)アミノ]−2−オキソエチ
ル}フェニル)カルバメート
【0054】
【化8】
【0055】
ジクロロメタン 200mL中の(3−{[(ベンジルオキシ)カルボニル]アミノ}フェニル)酢酸(工程Bより)24.7g(87mmol)の懸濁液に、トリエチルアミン(30.2mL、173mmol)を添加し、これにより若干の発熱(+5℃)を生じ、懸濁液は溶液化した。10分間の冷却後、HOBt(13.2g、87mmol)、N,O−ジメチルヒドロキシルアミンHCl(8.5g、87mmol)を、続いてEDC(16.6g、87mmol)を溶液に添加し、得られた混合物を窒素雰囲気下、室温で一晩攪拌した。溶液を分液漏斗に移し、1M HClで洗浄し、これによりエマルジョンを生じた。メタノールを添加して、エマルジョンを破壊し、水性物質を分配した。有機物質を硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣を70%ヘキサン(酢酸エチル中)1000mLから再結晶化して(加熱還流し、次に室温に一晩冷却)、標題化合物(21g、74%)を白色固体として得た。
LC−MS:m/z(ES)329(MH)+.
【0056】
工程D: ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1)
氷/水浴により0℃に冷却された、無水THF 1000mL中のベンジル(3−{2−[メトキシ(メチル)アミノ]−2−オキソエチル}フェニル)カルバメート(工程Cより)15g(45.7mmol)の溶液に、窒素雰囲気下で、ビニルマグネシウムブロミドの1.0M溶液(THF中100mL、100mmol)をカニューレにより滴下添加し、得られた溶液を0℃で1時間攪拌した。5℃未満の温度を保ちながら1M HCl 500mLを徐々に添加することにより、反応物をクエンチし、30分間攪拌した。次に混合物を酢酸エチルで抽出し、合わせた有機物質を水で、続いて食塩水で洗浄した。次いで有機物質を、硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣をバイオタージ(Biotage)75Mフラッシュにより、30%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(11g、78%)を明黄色の固体として得た。
LC−MS:m/z(ES)310(MH)+,332(MNa)+.1H NMR(500MHz,CDCl3)δ:7.44−7.36(m,7H),7.18(d,J=8.4Hz,2H),6.70(br s,1H),6.44(dd,J=10.5,17.6Hz,1H),6.32(dd,J=1.1,17.6Hz,1H),5.85(dd,J=1.1,10.5Hz,1H),5.22(s,2H),3.86(s,2H).
【0057】
中間体2
((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート
(i−2)
【0058】
【化9】
【0059】
工程A: 1−(3−クロロフェニル)プロパ−2−エン−1−オール
【0060】
【化10】
【0061】
無水THF 100mL中の3−クロロベンズアルデヒド(22.5g、160mmol)の冷却された溶液に、不活性窒素雰囲気下で、THF中のビニルマグネシウムクロリドの1.6M溶液(100mL、160mmol)を、シリンジにより徐々に添加し、溶液を室温に温めながら3時間攪拌した。反応物を、塩化アンモニウムの飽和溶液でクエンチし、有機層を分離し、酢酸エチル(2x200mL)で抽出し、有機層を合わせ、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。40M+シリカゲルカラムを用いたホライズン(Horizon)MPLCにより、0〜40%酢酸エチル(ヘキサン中)の勾配溶出液を用いて精製し、標題化合物(22.4g、44%)を得た。
m/z(ES)168,170(M,M+2)+,190,192(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.38(s,1H),7.35−7.22(m,3H),5.90(ddd,J=7.3,10.0,17.4Hz,1H),5.38(d,J=17.5Hz,1H),5.18(d,J=7.2Hz,1H),5.15(d,J=10.1Hz,1H),0.96(s,9H),0.18(s,3H),0.08(s,3H).
【0062】
工程B: Tert−ブチル{[1−(3−クロロフェニル)プロパ−2−エン−1−イル]オキシ}ジメチルシラン
【0063】
【化11】
【0064】
無水DMF 90mL中の1−(3−クロロフェニル)プロパ−2−エン−1−オール(工程Aより)22.4g(133mmol)の溶液に、t−ブチルジメチルシリルクロリド(20.0g、133mmol)及びイミダゾール(18.1g、266mmol)を添加し、得られた溶液を窒素下、室温で一晩攪拌した。水で洗浄し、酢酸エチルで抽出した。有機物質を分離し、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、フラッシュシリカゲルカラムにより、0〜15%酢酸エチル(ヘキサン中)の勾配溶出液で溶出して精製し、標題化合物(16.6g、46%)を得た。
m/z(ES)282,284(M,M+2)+;151,153(M−OTBS,M−OTBS+2)+.
【0065】
工程C: {[Tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)アセトアルデヒド
【0066】
【化12】
【0067】
ドライアイス/アセトン浴により−78℃に冷却された、ジクロロメタン中のtert−ブチル{[1−(3−クロロフェニル)プロパ−2−エン−1−イル]オキシ}ジメチルシラン(工程Bより)4.0g(14.2mmol)の溶液に、溶液が淡い青色を維持するまでオゾンを通気した。次に、溶液が透明になるまで窒素ガスを通気した。溶液に硫化メチルを添加し、得られた混合物を室温で一晩攪拌した。この物質を真空下で濃縮し、残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜50%酢酸エチル(ヘキサン中)の勾配溶出液で溶出して精製し、生成物(3.57g、89%)を得た。
【0068】
工程D: N−[(1E)−2−{[tert−ブチル(ジメチル)シリル]オキシ}−2−(3−クロロフェニル)エチリデン]−2−メチルプロパン−2−スルフィンアミド
【0069】
【化13】
【0070】
無水ジクロロメタン 50mL中の、{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)アセトアルデヒド(工程Cより)3.0g(10.6mmol)及び(R又はS)−2−メチル−2−プロパンスルフィンアミド 1.3g(10.6mmol)の溶液に、硫酸銅(II)(3.4g、21.2mmol)を添加し、得られた混合物を、窒素雰囲気下、室温で16時間攪拌した。反応物を水で洗浄し、ジクロロメタンで抽出した。有機物質を硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜25%酢酸エチル(ヘキサン中)の勾配溶出液系で溶出して精製し、標題化合物(3.26g、80%)を得た。
m/z(ES)387,390(M,M+2)+.
【0071】
工程E: N−{1−[{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]−プロパ−2−エン−1−イル}2−メチルプロパン−2−スルフィンアミド
【0072】
【化14】
【0073】
窒素雰囲気下で0℃に冷却された、無水THF 20mL中のN−[(1E)−2−{[tert−ブチル(ジメチル)シリル]オキシ}−2−(3−クロロフェニル)エチリデン]−2−メチルプロパン−2−スルフィンアミド(工程Dより)2.4g(6.20mmol)の溶液に、THF中のビニルマグネシウムクロリドの1.6M溶液(3.90mL、6.2mmol)をシリンジにより添加し、得られた混合物を1時間攪拌した。混合物を室温に温めさせ、さらに1時間攪拌した。反応物を塩化アンモニウム飽和溶液でクエンチし、酢酸エチルで抽出した。合わせた有機物質を、硫酸マグネシウム上で乾燥し、濾過し、真空下で濃縮した。残渣を、40M+シリカゲルカラムを用いたホライズンMPLCにより、0〜35%酢酸エチル(ヘキサン中)の勾配溶出液系で溶出して精製し、4つのジアステレオマーを単一の異性体として得た。
【0074】
NMRにより、得られた4つの生成物は、互いにジアステレオマーであった。異性体は、それらがシリカゲルカラムを溶出した際に標識付けした。溶出された最初の異性体を、異性体1とし、次に異性体2、3、及び最後に異性体4と命名した。
【0075】
【化15】
【0076】
異性体1:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.26(br d,J=6.2Hz,2H),7.22−7.18(m,1H),5.60(ddd,J=7.3,10.3,17.4Hz,1H),5.15(d,J=10.3Hz,1H),5.00(d,J=17.3Hz,1H),4.57(d,J=7.4Hz,1H),3.98−3.94(m,2H),1.64(br s,1H),1.23(s,9H),0.91(s,9H),0.08(s,3H),−0.18(s,3H).
異性体2:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.33−7.31(m,2H),7.26(br d,J=5.0Hz,2H),7.20−7.16(m,1H),5.44(ddd,J=7.2,10.0,17.4Hz,1H),5.26(オーバーラップd,J=7.3Hz,1H),5.25(オーバーラップd,J=17.3Hz,1H),4.84(d,J=4.4Hz,1H),4.02(dt,J=4.4,7.8Hz,1H),3.80(d,J=4.4Hz,1H),1.20(s,9H),0.94(s,9H),0.14(s,3H),−0.12(s,3H).
異性体3:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32−7.29(m,2H),7.26−7.24(m,2H),7.22−7.20(m,1H),6.04(ddd,J=7.1,10.4,17.4Hz,1H),5.40(d,J=10.2Hz,1H),5.32(d,J=17.3Hz,1H),4.80(d,J=4.0Hz,1H),3.88−3.80(m,1H),3.55(d,J=9.4Hz,1H),1.09(s,9H),0.95(s,9H),0.09(s,3H),−0.10(s,3H).
異性体4:m/z(ES)416,418(M,M+2)+,438,440(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.27−7.25(m,2H),7.21−7.18(m,1H),5.92(ddd,J=7.1,10.3,17.4Hz,1H),5.23(d,J=10.4Hz,1H),5.18(d,J=17.4Hz,1H),4.75(d,J=4.2Hz,1H),3.88−3.82(m,1H),3.33(d,J=9.4Hz,1H),1.19(s,9H),0.94(s,9H),0.09(s,3H),−0.14(s,3H).
【0077】
工程F: ((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート(i−2)
N−{1−[{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]−プロパ−2−エン−1−イル}2−メチルプロパン−2−スルフィンアミドの異性体1(工程Eより)(510mg、2.22mmol)に、ジオキサン中の無水4M HCl 5mLを添加し、溶液を室温で15分間攪拌した。反応物を濃縮乾燥し、トルエン(2x5mL)と共沸させて、過剰のHClを除去した。次いで残渣を、窒素雰囲気下に置かれ、氷/水浴で0℃に冷却された、無水ジクロロメタン中に溶解し、次にベンジルクロロホルマート(0.32mL、2.22mmol)をシリンジにより、続いてジイソプロピルエチルアミン(1.19mL、6.66mmol)を添加し、得られた溶液を0℃で2時間攪拌した。溶液を真空下で濃縮乾燥し、残渣を分取用プレート(4x1000μM)により、20%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(703mg、71%)を得た。
m/z(ES)446,448(M,M+2)+,468,470(MNa,MNa+2)+.1H NMR(500MHz,CDCl3)δ:7.32(s,1H),7.30(br d,J=7.5,1H),7.27−7.25(m,2H),7.21−7.18(m,1H),5.92(ddd,J=7.1,10.3,17.4Hz,1H),5.23(d,J=10.4Hz,1H),5.18(d,J=17.4Hz,1H),4.75(d,J=4.2Hz,1H),3.88−3.82(m,1H),3.33(d,J=9.4Hz,1H),1.19(s,9H),0.94(s,9H),0.09(s,3H),−0.14(s,3H).
【0078】
上記記載のものに関連した、立体化学を異にする中間体は、適切な出発物質から、上記記載の方法を用いて調製し得る。
【0079】
【化16】
【0080】
中間体3
Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)
【0081】
【化17】
【0082】
工程A: ベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート
【0083】
【化18】
【0084】
無水ジクロロメタン 7mL中の、ベンジル[3−(2−オキソブタ−3−エン−1−イル)フェニル]カルバメート(i−1)(820mg、2.80mmol)及び((1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]プロパ−2−エン−1−イル)カルバメート
(i−2)(500mg、1.12mmol)の溶液に、チャン(Zhan)I触媒(740mg、1.12mmol)を添加し、得られた緑色の溶液を、窒素雰囲気下、40℃で一晩加熱した。反応物を濃縮乾燥し、残渣を分取用プレート(4x1000μM)により、40%酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物(348mg、50%)を得た。
m/z(ES)713,715(M,M+2)+,735,737(MNa,MNa+2)+.
【0085】
工程B: 4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(フェニル)メチル]ピロリジン−2−イル}メチル)アニリン
【0086】
【化19】
【0087】
エタノール 25mL中のベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート(工程Aより)328mg(0.46mmol)の溶液に、10%パラジウム炭素を添加し、水素ガスのバルーンにより、懸濁液を水素雰囲気下に置いた。反応物を、水素下、室温で1時間攪拌した。TLCは、反応が完了したことを示した。ギルメン(Gilmen)0.45μM PTFEシリンジフィルターを用いて触媒を濾去し、エタノール(4x5mL)で洗浄した。濾液を真空下で濃縮乾燥し、残渣を分取用プレート(3x1000μM)により、5%メタノール(ジクロロメタン中)で溶出して精製し、標題化合物(121mg、66%)を得た。
m/z(ES)397(MH)+.
【0088】
工程C: Tert−ブチル(5R)−2−(4−アミノベンジル)−
5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)
無水THF 5mL中の4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(フェニル)メチル]ピロリジン−2−イル}メチル)アニリン(工程Bより)121mg(0.315mmol)の溶液に、tert−ブチルカルボナート(69mg、0.315mmol)を、続いてTEA(44μL、0.315mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。反応混合物を直接、分取用プレート(1500μM)にのせ、30%酢酸エチル(ヘキサン中)で溶出して、標題化合物(100mg、64%)を得た。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.75−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.55−3.48(m,2H),2.74(br d,J=11.8Hz,1H),2.44(br d,J=11.8Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.08(s,3H),−0.15(s,3H).
【0089】
中間体4a及び中間体4bの分離
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0090】
【化20】
【0091】
工程A: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a)及びtert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
中間体i−3(tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(シス及びトランスの4:1混合物)を、メタノール中に溶解し、バージャー(Berger)マルチグラム(Multigram)SFC(超臨界)により、30%メタノール:60%二酸化炭素の溶出液を用いて精製し、2つのジアステレオマーを分離した。カラムの最初の異性体を、副異性体1とし、2番目の異性体を主異性体2と標識した。
i−4a:m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.75−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.55−3.48(m,2H),2.74(br d,J=11.8Hz,1H),2.44(br d,J=11.8Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.92(d,J=11.8Hz,1H),0.12(br d,J=14.0Hz,3H),−0.04(s,3H).SFCで8.70minに溶出、異性体2。
i−4b:m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.40−7.30(m,5H),6.76−6.68(m,2H),6.56−6.51(m,2H),5.52−5.48(m,1H),5.32−5.28(m,1H),4.16−4.06(m,2H),3.88−3.82(m,1H),3.76−3.70(m,1H),3.60−3.46(m,2H),2.72(br d,J=12.0Hz,1H),2.44(br d,J=12.2Hz,1H),2.05−1.94(m,1H),1.90−1.82(m,1H),1.64(s,9H),1.50−1.42(m,1H),1.32−1.22(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.14(br d,J=13.8Hz,3H),0.09(s,3H).SFCで7.78minに溶出、異性体1。
【0092】
中間体4a及び中間体4bの合成
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0093】
【化21】
【0094】
工程A: (4S)−3−ヘキサ−5−イノイル−4−フェニル−1,3−オキサゾリジン−2−オン
【0095】
【化22】
【0096】
無水テトラヒドロフラン 1.0L中の、5−ヘキシン酸 69.0g(615mmol)及びトリエチルアミン 214mL(1540mmol)の溶液に、窒素雰囲気下、−25℃で、トリメチルアセチルクロリド 83.0mL(677mmol)を20分間にわたり添加した。添加により白色沈殿が生成され、得られた懸濁液を2時間攪拌した。次に、無水塩化リチウム 28.7g(677mmol)及び、(4S)−4−フェニル−1,3−オキサゾリジン−2−オン 100.0g(615.0mmol)を連続的に添加し、混合物を12時間で徐々に室温に温めた。全揮発性物質を真空中で除去し、残渣を水(1L)で希釈し、酢酸エチル(3x300mL)で抽出した。合わせた有機層を食塩水(250mL)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空中で濃縮した。粗残渣を、シリカゲルカラムクロマトグラフィーにより、5〜50%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色の固体として得た(135g、85.4%)。
1H NMR(500MHz,CDCl3):δ7.40−7.37(m,2H),7.36−7.32(m,1H),7.31−7.28(m,2H),5.42(dd,J=8.9,3.7Hz,1H),4.69(t,J=8.9Hz,1H),4.28(dd,J=9.2,3.7Hz,1H),3.13−3.02(m,2H),2.24−2.21(m,2H),1.94(t,J=2.6Hz,1H),1.84(quintet,J=7.1Hz,2H).
LC−MS:m/z(ES)258.2(MH)+.
【0097】
工程B: (4S)−3−{(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イノイル}−4−フェニル−1,3−オキサゾリジン−2−オン
【0098】
【化23】
【0099】
無水酢酸エチル 265mL中の上記工程Aよりの(4S)−3−ヘキサ−5−イノイル−4−フェニル−1,3−オキサゾリジン−2−オン 56.8g(221mmol)の攪拌された溶液に、窒素雰囲気下、室温で、無水塩化マグネシウム 6.31g(66.2mmol)、トリエチルアミン 61.5mL(442mmol)、ベンズアルデヒド 26.9mL(265mmol)、及びクロロトリメチルシラン 42.3mL(331mmol)を添加し、得られた混合物を72時間攪拌した。不均一な反応混合物を、シリカゲル300mLのプラグを通し、さらに酢酸エチル 1Lで溶出して濾過した。濾液を真空中で蒸発乾燥させ、残渣をメタノール 265mL及びトリフルオロ酢酸 10mL中に懸濁した。得られた混合物を、窒素下、室温で5時間攪拌し、この間に反応は均一化した。次に全揮発性物質を真空中で除去し、残渣を、シリカゲルクロマトグラフィーにより、5〜15%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を白色固体として得た(65.0g、81.2%)。
1H NMR(500MHz,CDCl3):δ7.30−7.28(m,8H),7.09−7.07(m,2H),5.42(dd,J=8.7,3.7Hz,1H),4.76−4.72(m,1H),4.72−4.67(m,1H),4.65(t,J=8.7Hz,1H),4.18(dd,J=8.7,3.7Hz,1H),3.05(d,J=7.8Hz,1H),2.24(td,J=7.1,2.5Hz,2H),2.00−1.93(m,2H),1.67−1.61(m,1H).
LC−MS:m/z(ES)346.1(MH−H2O)+,386.0(MNa)+.
【0100】
工程C: (2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イン酸
【0101】
【化24】
【0102】
無水テトラヒドロフラン対水の、20対1混合物 1050mL中の上記工程Bよりの(4S)−3−{(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イノイル}−4−フェニル−1,3−オキサゾリジン−2−オン 65.0g(179mmol)の攪拌された溶液に、窒素雰囲気下、0℃で、35%過酸化水素水溶液 77.0mL(894mmol)を、内部温度を3℃未満に維持するべく充分遅い速度で添加した。次に、1.0M 水酸化リチウム水溶液 395mL(395mmol)を、反応の内部温度を5℃未満に維持するべく充分遅い速度で添加し、得られた混合物を0℃で3時間攪拌した。混合物の内部温度を5℃未満に維持するべく充分遅い速度での1.3M 亜硫酸ナトリウム水溶液 755mL(984mmol)により、反応物をクエンチした。全揮発性物質を真空中で除去し、残留する水相を酢酸エチル(3x200mL)で抽出した。水相を0℃に冷却し、6M 塩化水素水溶液でpH3に達するまで酸性化した。次に水相を酢酸エチル(3x300mL)で抽出し、合わせた有機物質を食塩水(100ml)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。残渣を、シリカゲルクロマトグラフィーにより、5〜10%酢酸エチル及び3%酢酸(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色のガムとして得た(32.0g、82.0%)。
1H NMR(500MHz,CDCl3):δ7.39−7.28(m,5H),4.85(d,J=8.2,1H),3.03−2.97(m,1H),2.29−2.15(m,2H),1.97(t,J=2.5Hz,1H),1.93−1.82(m,1H),1.62−1.55(m,1H).
LC−MS:m/z(ES)201.0(MH−H2O)+.
【0103】
工程D: (2R)−2−[(S)−{[Tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ヘキサ−5−イン酸
【0104】
【化25】
【0105】
無水アセトニトリル 500mL中の上記工程Cよりの(2R)−2−[(S)−ヒドロキシ(フェニル)メチル]ヘキサ−5−イン酸 32.0g(147mmol)の攪拌された溶液に、窒素雰囲気下、室温で、1,8−ジアザビシクロ[5.4.0]ウンデカ−7−エン 77.0mL(513mmol)、22mL、続いてtert−ブチルジメチルシリルクロリド 66.3g(440mmol)を3回に分けて10分間で添加した。反応混合物を4時間攪拌し、真空蒸発させて、全揮発性物質を除去した。残渣を、ジクロロメタン 300mL及び水 100mLで希釈した。1.0M 塩化水素水溶液を、水層においてpH3が達成されるまで混合物に添加した。相を分離し、水相をジクロロメタン(2x100mL)で抽出した。合わせた有機物質を、水(50mL)、食塩水(50mL)で洗浄し、次に硫酸マグネシウム上で乾燥した。濾過及び真空蒸発の後、残渣を、メタノール 350mL中に溶解し、0.8M炭酸カリウム水溶液 350mL(280mmol)を添加した。得られた混合物を1.5時間攪拌し、次いで真空蒸発させて、全揮発性物質を除去した。残渣を、ジクロロメタン 300mLで希釈し、水相を5.0M 塩化水素水溶液でpH3に達するまで酸性化した。相を分離し、ジクロロメタン(2x100mL)で抽出した。合わせた有機物質を、水(50mL)、食塩水(50mL)で洗浄し、次に硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。残渣を、シリカゲルクロマトグラフィーにより、3〜15%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を無色の固体として得た(42.3g、86.6%)。
1H NMR(500MHz,CDCl3):δ7.36−7.27(m,5H),4.78(d,J=8.7,1H),2.90−2.86(m,1H),2.19−2.11(m,1H),2.10−2.03(m,1H),1.90(t,J=2.6Hz,1H),1.75−1.67(m,1H),1.41−1.34(m,1H),0.83(s,9H),0.02(s,3H),−0.27(s,3H).
LC−MS:m/z(ES)333.2(MH)+.
【0106】
工程E: 4−メトキシベンジル {(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ペンタ−4−イン−1−イル}カルバメート
【0107】
【化26】
【0108】
無水トルエン 400mL中の、上記工程Dよりの(2R)−2−[(S)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ヘキサ−5−イン酸 40.0g(120mmol)及びトリエチルアミン 33.5mL(241mmol)の溶液に、窒素雰囲気下、室温で、ジフェニルホスホリルアジド 37.5mL(132mmol)を添加した。混合物を5時間攪拌し、次に4−メトキシベンジルアルコール 37.5mL(301mmol)を添加した。得られた混合物を105℃で16時間加熱し、室温に冷却し、次いで飽和重炭酸塩水溶液 250mLで希釈した。相を分離し、水相を酢酸エチル(2x150mL)で抽出した。合わせた有機物質を、水(100mL)、食塩水(100mL)で洗浄し、次に硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。粗残渣を、シリカゲルクロマトグラフィーにより、3〜10% 酢酸エチル(ヘキサン中)で溶出して精製し、標題化合物を無色の油として得た(50.9g、90.5%)。
1H NMR(500MHz,CDCl3):7.28−7.21(m,7H),6.87(d,J=8.4Hz,2H),4.92(s,2H),4.77−4.59(m,2H),3.89−3.84(m,1H),3.81(s,3H),2.30−2.22(m,2H),1.95(m,1H),1.91−1.85(m,1H),1.57−1.50(m,1H),0.89(s,9H),0.06(s,3H),−0.15(s,3H).
LC−MS:m/z(ES)468.1(MH)+,490.0(MNa)+.
【0109】
工程F: 4−メトキシベンジル [(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロフェニル)ペンタ−4−イン−1−イル]カルバメート
【0110】
【化27】
【0111】
無水DMF(500ml)中の、アセチレン(工程Eより、40g、80mmol)及び4−ヨードニトロベンゼン(21.8g、88mmol)の溶液に、トリエチルアミン(111mL、797mmol)を添加した。Pd(dppf)Cl2(1.95g、2.39mmol)、及びヨウ化銅(I)(910mg、4.78mmol)を添加し、混合物を窒素で脱気し(15分間通気)、得られた溶液を室温で5時間攪拌した。混合物を水(1200ml)に注入し、EtOAc(3x300mL)で抽出した。合わせた有機物質を、次に水(2x500mL)、飽和NaCl(200mL)で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空下で蒸発させた。残渣を、MPLC(ホライズン(Horizon)バイオタージ(Biotage)2xフラッシュ(Flash)65i)により、0〜30%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、41g(84%)を暗赤色の油として得た。
1H NMR(500MHz,CDCl3):8.11−8.04(m,2H),7.94−8.01(m,1H),7.38−7.21(m,8H),6.87(d,J=8.4Hz,2H),4.98(s,2H),4.77−4.59(m,2H),4.00−3.95(m,3H),3.81(s,3H),2.56(t,J=7.1Hz,H=2H),2.00−1.95(m,1H),1.66−1.61(m,1H),0.93(s,9H),0.10(s,3H),−0.10(s,3H).
LC−MS:m/z(ES)589.3(MH)+,611.2(MNa)+.
【0112】
工程G: 4−メトキシベンジル[(1R)−1−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロフェニル)−4−オキソペンチル]カルバメート
【0113】
【化28】
【0114】
DMF(40mL)中のニトロフェニルアセチレン(工程Fより、41g、65.5mmol)の溶液に、ピロリジン(14mL、196.5mmol)を添加し、得られた混合物を80℃で3時間加熱した。混合物を室温に冷却し、10%酢酸溶液(水中)(110ml)を添加し、得られた混合物を室温でさらに3時間攪拌した。混合物を水(300ml)に注入し、EtOAc(3x250ml)で抽出し;合わせたEtOAc層を水(2x250ml)、飽和NaCl(100ml)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、ホライズン・フラッシュ75により、100%ヘキサンから50%EtOAc(ヘキサン中)まで上昇する勾配で溶出して精製し、34g(81%)を暗橙色の油として得た。
1H NMR(500MHz,CDCl3):8.17−8.14(m,2H),7.32−7.23(m,9H),6.87(d,J=8.4Hz,2H),4.96(d,J=12.2Hz,1H),4.90(d,J=12.1Hz,1H),4.72(d,J=3Hz,1H),4.16−4.13(m,1H),3.81(s,3H),3.71−3.77(m,2H),2.65−2.52(m,2H),1.97−1.92(m,1H),1.72−1.60(m,1H),0.93(s,9H),0.05(s,3H),−0.13(s,3H).
【0115】
工程H: (2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン
【0116】
【化29】
【0117】
DCM(350ml)中のMOZ保護されたケトンアミン(工程Gより、34g、56mmol)の溶液に、TFA(256ml)を添加し、得られた混合物を室温で1.5時間攪拌した。溶液を真空下で蒸発させ、残渣をDCMと飽和NaHCO3との間で分配した。有機層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣をMeOH(750ml)中に溶解し、氷/水浴で0℃に冷却した。次にシアノ水素化ホウ素ナトリウム(21.2g、337mmol)を添加し、得られた混合物を一晩攪拌して室温に温めさせた。水の添加により混合物をクエンチし、有機物質を真空下で除去した。次いで水層をEtOAc(2x)で抽出し、合わせたEtOAc層を飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、真空下で蒸発させた。残渣をシリカ上でのカラムクロマトグラフィー(溶出液:100%ヘキサンから35%EtOAc(ヘキサン中)まで上昇する勾配)により精製し、16.4g(63.4%)の最初の異性体、(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン、及び3.1g(12%)の2番目の異性体、(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジンを得た。
異性体1:LC−MS:m/z(ES)427.3(MH)+
異性体2:LC−MS:m/z(ES)427.3(MH)+
【0118】
工程I: Tert−ブチル(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0119】
【化30】
【0120】
無水THF中のtert−ブチル(2R,5S)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(12g、42.5mmol)の溶液に、Boc無水物(9.3g、42.5mmol)を、続いてTEA(17.76mL、127.4mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で2時間攪拌した。混合物を水(100mL)で洗浄し、酢酸エチル(2x200mL)で抽出した。有機物質を、硫酸ナトリウム上で乾燥し、濾過し、真空下で濃縮した。残渣をホライズン・バイオタージMPLC(65iシリカゲルカラム)により、20〜75%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、所望の生成物を得た。
LC−MS:m/z(ES)527.3(MH)+,549.2(MNa)+.
【0121】
工程J: Tert−ブチル(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0122】
【化31】
【0123】
シスピロリジン異性体をトランス異性体、(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(ニトロベンジル)ピロリジンで置き換えたことを除いて、工程Iと同様の方法で調製した。
LC−MS:m/z(ES)527.3(MH)+,549.2(MNa)+.
【0124】
工程K: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a);
【0125】
【化32】
【0126】
500mLのパー・シェイカー・フラスコに、10% Pd/c(4.75g)を装入し、これにメタノール 100mLを添加して、触媒を覆った。次いで懸濁液に、メタノール(80mL)中の工程Iからのニトロ中間体(8.5g、18.5mmol)の溶液を、続いて1.0Mの塩化水素(メタノール中)溶液 15.4mLを添加した。反応容器を50PSIの水素ガス下に置き、混合物を一晩振盪した。アリコートを採り、LC−MSにより分析し、反応完了が示された。
【0127】
セライトを用いて触媒を濾去し、メタノール(2x100mL)で洗浄した。濾液を濃縮乾燥し、生成物をホライズンMPLC(65iシリカカラム)により、0%から30%酢酸エチル(ヘキサン中)まで上昇する勾配を用いて溶出して精製し、標題化合物(6.2g、72%)を得た。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.38−7.29(m,5H),6.76−6.68(m,2H),6.55−6.50(m,2H),5.52−5.49(m,1H),5.30−5.27(m,1H),4.15−4.05(m,2H),3.86−3.81(m,1H),3.76−3.71(m,1H),3.55−3.47(m,2H),2.74(br d,J=11.7Hz,1H),2.44(br d,J=11.7Hz,1H),2.05−1.93(m,1H),1.90−1.83(m,1H),1.60(s,9H),1.50−1.42(m,1H),1.31−1.21(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.92(d,J=11.8Hz,1H),0.13(br d,J=14.0Hz,3H),−0.05(s,3H)
【0128】
工程L: Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4b)
【0129】
【化33】
【0130】
シスピロリジン異性体をトランス異性体、Tert−ブチル(2R,5R)−2−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラートで置き換えたことを除いて、工程Kと同様の方法で調製した。
m/z(ES)497(MH)+,397(M−Boc)+.1H NMR(500MHz,CDCl3)δ:7.41−7.30(m,5H),6.73−6.67(m,2H),6.56−6.50(m,2H),5.52−5.48(m,1H),5.33−5.28(m,1H),4.15−4.06(m,2H),3.86−3.81(m,1H),3.76−3.70(m,1H),3.59−3.46(m,2H),2.72(br d,J=12.0Hz,1H),2.44(br d,J=12.0Hz,1H),2.05−1.93(m,1H),1.90−1.82(m,1H),1.64(s,9H),1.49−1.42(m,1H),1.32−1.20(m,2H),1.10−1.02(m,1H),0.95(s,9H),0.14(br d,J=13.7Hz,3H),0.10(s,3H).
【0131】
以下の中間体を、適切な出発物質から、中間体i−4aについて上記に記載した方法を用いて調製した。
【0132】
【化34】
【0133】
以下の中間体を、適切な出発物質から、中間体i−4bについて上記に記載した方法を用いて調製した。
【0134】
【化35】
【0135】
中間体5
Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]ピロリジン−1−カルボキシラート(i−5)
【0136】
【化36】
【0137】
工程A: 4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(3−クロロフェニル)メチル]ピロリジン−2−イル}メチル)アニリン
【0138】
【化37】
【0139】
酢酸エチル 8mL中のベンジル{4−[(3E,5R,6R)−5−{[(ベンジルオキシ)カルボニル]アミノ−6−{[tert−ブチル(ジメチル)シリル]オキシ}−6−(3−クロロフェニル)−2−オキソヘキサ−3−エン−1−イル]フェニル}カルバメート(i−3、工程Aより)100mg(0.15mmol)の溶液に、10%パラジウム炭素を添加し、水素ガスのバルーンにより、懸濁液を水素雰囲気下に置いた。反応物を水素下、室温で8時間攪拌した。触媒を、ギルメン0.45uM PTFEシリンジフィルターを用いて濾去し、酢酸エチル(4x2mL)で洗浄した。濾液を真空下で濃縮乾燥し、残渣を分取用プレート(1000μM)により、5%メタノール(ジクロロメタン中)で溶出して精製し、標題化合物(33mg、51%)を得た。
m/z(ES)430,432(M,M+2)+.
【0140】
工程B: Tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(3−クロロフェニル)メチル]ピロリジン−カルボキシラート(i−5)
無水THF 1mL中の4−({(5R)−5−[(R)−([tert−ブチル(ジメチル)シリル]オキシ)(3−クロロフェニル)メチル]ピロリジン−2−イル}メチル)アニリン(工程Aより)33mg(0.07mmol)の溶液に、tert−ブチルカルボナート(15.3mg、0.07mmol)を、続いてTEA(13uL、0.07mmol)を添加し、得られた溶液を、窒素雰囲気下、室温で一晩攪拌した。反応混合物を直接、分取用プレート(500uM)にのせ、30%酢酸エチル(ヘキサン中)で溶出して、標題化合物(25mg、78%)を得た。
m/z(ES)530,532(M,M+2)+,430,432(M−Boc,M−Boc+2)+.
【0141】
中間体6
4−{4−[4−(トリフルオロメチル)フェニル]−1,3−チアゾール−2−イル}ベンゼンスルホニルクロリド(i−6)
【0142】
【化38】
【0143】
中間体6は、例えば、イケモト(Ikemoto)ら、「Tetrahedron」、2003年、第59巻、p.1317〜1325の公表された方法に従って調製し得る。
【0144】
中間体7
2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−7)
【0145】
【化39】
【0146】
工程A: エチル2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0147】
【化40】
【0148】
0℃に冷却されたクロロホルム(500mL)中のエチル2−オキソシクロペンタン−2−カルボキシラート(56g、359mmol)の溶液に、臭素(18.5mL、359mmol)を、約20分間で添加した。添加完了後、混合物を室温に温めさせ、一晩攪拌した。窒素ガスを、混合物を通して90分間通気して、殆どのHBrを除去した。水(500mL)、飽和NaHCO3(250mL)、飽和NaCl(200mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣をEtOH(500mL)中に溶解し、チオアセトアミド(26.9g、359mmol)を添加し、混合物を室温で1時間攪拌し、次いで一晩還流した。混合物を冷却し、蒸発させ、残渣をDCMと飽和NaHCO3との間で分配し、有機層を飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:2x FLASH65i)溶出液:100%ヘキサン(450mL)、100%ヘキサンから25%EtOAc(ヘキサン中)まで上昇する勾配(1400mL)、次いで25%EtOAc(ヘキサン中))により精製して、標題化合物(32g、42%)を暗色の油として得た。
1H NMR(500MHz,CDCl3)δ:4.22(q,J=7.0Hz,2H),3.96(m,1H),3.04(m,1H),2.88(m,1H),2.76(m,2H),2.70(s,3H),1.30(t,J=7.0Hz,3H).
【0149】
工程B: 2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−7)
THF(450mL)及びメタノール(100mL)中のエチル2−メチル−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Aより)31.5g(149mmol)の溶液に、水酸化リチウム(1M溶液を149mL、149mmol)を添加し、得られた混合物を室温で3時間攪拌した。有機物質を蒸発により除去し、水性残渣をEt2O(2x250mL)で抽出し、1M 塩酸(約170mL)の添加によりpH3に酸性化し、固体NaClで飽和させた。DCM(3x250mL)で抽出し、合わせたDCM層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣をアセトニトリルで粉砕し、濾過し、乾燥させて、標題化合物(7.1g、26%)を灰白色の固体として得た。
1H NMR(500MHz,CDCl3)δ:11.75(br s,1H),4.02(m,1H),3.00(m,1H),2.90−2.66(m,6H).
【0150】
中間体8
2−[(Tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)
【0151】
【化41】
【0152】
工程A: エチル2−アミノ−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0153】
【化42】
【0154】
工程Aにおいて、チオアセトアミドをチオ尿素で置き換えることにより、中間体(i−7)と同様の方法で調製した。
1H NMR(500MHz,CDCl3)δ:5.30(br s,2H),4.21(q,J=7.0,2H),3.81(m,1H),2.91(m,1H),2.78(m,1H),2.66(m,2H),1.30(t,J=7.0,3H).
【0155】
工程B: エチル2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート
【0156】
【化43】
【0157】
ジクロロメタン(5mL)中のエチル2−アミノ−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Aより)230mg(1.08mmol)の溶液に、ジ−tert−ブチルジカルボナート(236mg、1.08mmol)、トリエチルアミン(0.15mL、1.08mmol)、及びDMAP(13mg、0.11mmol)を添加し、得られた混合物を室温で2時間攪拌した。混合物を1N HCl(10mL)、飽和NaCl(5mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH25+S)溶出液:100%ヘキサン(100mL)、0〜15%EtOAc(ヘキサン中)の勾配(900mL)、次いで15%EtOAc(ヘキサン中)(500mL)により精製して、標題化合物(160mg、47%)を白色の泡沫として得た。
1H NMR(500MHz,CDCl3)δ:9.23(br s,1H),4.17(q,J=7.1Hz,2H),3.95(t,J=6.6Hz,1H),3.04(m,1H),2.86(m,1H),2.76(m,2H),1.55(s,9H),1.23(t,J=7.1Hz,3H).
【0158】
工程C: 2−[(Tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)
エチル2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキシラート(工程Bより)から、中間体(i−7)工程Bに見られるものと類似の方法を用いて調製した。
1H NMR(500MHz,CDCl3)δ:3.96(m,1H),3.06(m,1H),2.88(m,2H),2.71(m,1H),1.55(s,9H).
【0159】
中間体9
2−(4−フルオロフェニル)−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−9)
【0160】
【化44】
【0161】
工程Aにおいて、チオアセトアミドを4−フルオロチオベンズアミドで置き換えることにより、中間体7(i−7)に見られるものと類似の方法を用いて調製した。
1H NMR(500MHz,DMSO−d6)δ:7.90(m,2H),7.29(t,J=8.7,2H),3.81(m,1H),2.99(m,1H),2.86(m,1H),2.70−2.58(m,2H).
【0162】
中間体10
2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−10)
【0163】
【化45】
【0164】
工程A: エチル2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート
【0165】
【化46】
【0166】
0℃に冷却された無水ジエチルエーテル(40mL)中のエチル2−オキソシクロヘキサンカルボキシラート(15g、88mmol)の溶液に、臭素(4.5mL、88mmol)を15分間にわたり滴下添加した。添加完了後、混合物を室温に90分間温めさせた。混合物をEtOAc(100mL)で希釈し、飽和NaHCO3、飽和NaClで洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣をエタノール(100mL)中に溶解し、チオアセトアミド(6.6g、88mmol)を添加した。混合物を室温で1時間攪拌し、次いで一晩還流した。混合物を蒸発させ、残渣をNaHCO3とDCMとの間で分配した。有機層をMgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH 65i)溶出液:100%ヘキサン(500mL)、0〜25%EtOAc(ヘキサン中)の勾配(1200mL)、次いで25%EtOAc(ヘキサン中)(1200mL)により精製して、標題化合物(6.14g、31%)を淡橙色の油として得た。
1H NMR(500MHz,CDCl3)δ:4.22(q,J=7.1,2H),3.84(t,J=5.5,1H),2.80(m,1H),2.73(m,1H),2.65(s,3H),2.18(m,1H),2.11−1.95(m,2H),1.85(m,1H),1.29(t,J=7.1,3H).
【0167】
工程B: 2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−10)
エチル2−メチル−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート(工程Aより)から、中間体(i−7)工程Bに概説された方法に従って調製した。
1H NMR(500MHz,CDCl3)δ:9.26(br s,1H),3.81(q,J=7.3及び5.9,1H),2.75(m,2H),2.68(s,3H),2.24(m,1H),2.18−2.01(m,2H),1.82(m,1H).
【0168】
中間体11
2−[(Tert−ブトキシカルボニル)アミノ]−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−11)
【0169】
【化47】
【0170】
工程A: エチル2−アミノ−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート
【0171】
【化48】
【0172】
チオアセトアミドをチオ尿素で置き換えることにより、中間体10(i−10)工程Aに概説された方法に従って調製した。
1H NMR(500MHz,DMSO−d6)δ:9.28(br s,2H),4.11(q,J=7.3,2H),3.71(t,J=5.0,1H),2.57−2.39(m,2H),1.90(m,2H),1.78(m,1H),1.59(m,1H),1.17(t,J=7.3,3H).
【0173】
工程B: 2−[(Tert−ブトキシカルボニル)アミノ]−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボン酸(i−11)
エチル2−アミノ−4,5,6,7−テトラヒドロ−1,3−ベンゾチアゾール−4−カルボキシラート(工程Aより)から、中間体8(i−8)工程B及びCに概説された方法に従って調製した。
1H NMR(500MHz,CDCl3)δ:3.70(t,J=5.2,1H),2.74(m,1H),2.64(m,1H),2.25(m,1H),2.10−1.94(m,2H),1.87(m,1H),1.55(s,9H).
【0174】
中間体12
インダン−1−カルボン酸(i−12)
【0175】
【化49】
【0176】
「ジャーナル・オブ・オーガニック・ケミストリー(Journal of Organic Chemistry)」、2000年、第65巻、第4号、p.1132〜1138の文献の方法に従って調製した。
【0177】
中間体13a及び中間体13b
Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a);
Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13b)
【0178】
【化50】
【0179】
工程A: Tert−ブチル(4R,5R)−2,2−ジメチル−4−[(1E)−3−オキソプロパ−1−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0180】
【化51】
【0181】
CH2Cl2(150mL)中のtert−ブチル(4S,5R)−4−ホルミル−2,2−ジメチル−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(20.9、89.1mmol)の溶液に、(トリフェニルホスホルアニリデン)アセトアルデヒド(27.1g、89.1mmol)を添加し、得られた混合物を室温で40時間攪拌した。溶媒の1/3を除去した後、ヘキサンをたっぷり添加し、得られた固体を濾去した。Biotage Horizon(登録商標)システム(シリカゲル、0〜20%酢酸エチル(ヘキサン中)の勾配、次に20%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物 16.3g(72%)を黄色の油として得た。
LC/MS354.3(M+23).
【0182】
工程B: Tert–ブチル(4R,5R)−2,2−ジメチル−4−(3−オキソプロピル)−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0183】
【化52】
【0184】
アセトン(150mL)中のtert−ブチル(4R,5R)−2,2−ジメチル−4−[(1E)−3−オキソプロパ−1−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(19.6g、59.1mmol)(工程Aより)の溶液に、10% Pd/C 1.9gを添加し、得られた懸濁液を水素バルーン下、室温で24時間攪拌した。固体をセライト上で濾去し、濾液を真空下で濃縮した。残渣を、Biotage Horizon(登録商標)システム(シリカゲル、0〜20%酢酸エチル(ヘキサン中)の勾配、次に20%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより精製して、標題化合物 11.5g(58%)を無色の油として得た。
LC/MS356.3(M+23).
【0185】
工程C: Tert−ブチル(4R,5R)−2,2−ジメチル−4−[(3E)−4−(4−ニトロフェニル)ブタ−3−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート及びtert−ブチル(4R,5R)−2,2−ジメチル−4−[(3Z)−4−(4−ニトロフェニル)ブタ−3−エン−1−イル]−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート
【0186】
【化53】
【0187】
CH2Cl2(200mL)中の工程Bよりのtert–ブチル(4R,5R)−2,2−ジメチル−4−(3−オキソプロピル)−5−フェニル−1,3−オキサゾリジン−3−カルボキシラート(10.0g、30.0mmol)の溶液に、(4−ニトロベンジル)トリフェニル−ホスホニウムブロミド(21.5g、45.0mmol)を、続いてEt3N(8.36mL、60.0mmol)を添加した。赤色の反応混合物を、室温で48時間攪拌した。ヘキサン(200mL)を反応混合物に注入し、固体を濾去した。Biotage Horizon(登録商標)システム(シリカゲル、0〜10%酢酸エチル(ヘキサン中)の勾配、次に10%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物(シス トランス混合物) 10.7g(79%)を淡黄色の泡沫として得た。
LC/MS475.4(M+23).
【0188】
工程D: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート及びtert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート
【0189】
【化54】
【0190】
酢酸エチル(100mL)中の工程Cよりの上記のシス/トランス混合物(7.86g、17.4mmol)の溶液に、2N HCl溶液 50mLを添加し、得られた混合物を室温で2時間攪拌し、次に45℃で3時間加熱した。揮発性物質を、減圧下で除去した。得られた白色固体を、N,N−ジメチルホルムアミド(100mL)中に溶解し、iPr2Net 15.1mL(86.7mmol)を添加した。反応混合物を室温で7時間攪拌した。次にジ−tert−ブチルジカルボナート(4.55g、20.8mmol)を添加し、反応混合物を室温で一晩攪拌した。水(200mL)を添加し、これを酢酸エチル(200mLx3)で抽出した。合わせた有機層を、Na2SO4上で乾燥し、濾過し、減圧下で濃縮した。残渣を、Biotage Horizon(登録商標)システム(シリカゲル、0〜30%酢酸エチル(ヘキサン中)の勾配)でのフラッシュクロマトグラフィーにより精製して、標題化合物 tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(シス)1.61g(22%)及びtert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(トランス)3.9g(54%)を得た。
LC/MS435.4(M+23).
【0191】
工程E: Tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a)
エタノール(20mL)中の工程Dよりの、上記(シス)tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(1.51g、3.66mmol)の溶液に、10% Pd/C 0.15gを添加し、得られた懸濁液を水素バルーン下、室温で5時間攪拌した。セライトを通して濾過し、溶媒を除去して、標題化合物 1.40g(100%)を白色泡沫として得て、これをさらに精製することなく使用した。
LC/MS405.3(M+23).
【0192】
工程F: Tert−ブチル(2R,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13b)
エタノール(40mL)中の工程Dよりの(トランス)tert−ブチル(2R,5R)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−(4−ニトロベンジル)ピロリジン−1−カルボキシラート(3.90g、9.46mmol)の溶液に、10% Pd/C 0.4gを添加し、得られた懸濁液を、水素バルーン下、室温で6時間攪拌した。固体を、セライトを通して濾去した。溶媒の除去後、Biotage Horizon(登録商標)システム(シリカゲル、0〜30%酢酸エチル(ヘキサン中)の勾配、次に30%酢酸エチル(ヘキサン中))でのフラッシュクロマトグラフィーにより、標題化合物 2.30g(64%)を白色泡沫として得た。
LC/MS405.3(M+23).
【0193】
中間体14
(2S)−1−(1,3−ベンゾチアゾール−2−イル)ピロリジン−2−カルボン酸(i−14):
【0194】
【化55】
【0195】
N,N−ジメチルホルムアミド(3mL)中のL−プロリン 28mg(0.24mmol)の溶液に、室温で、2−ブロモベンゾチアゾール 51mg(0.24mmol)、炭酸カリウム 100mg(0.72mmol)、及びヨウ化銅 6mg(0.03mmol)を添加した。反応混合物を100℃で一晩加熱した。次に、これを濾過し、逆相HPLC(TMC Pro−Pac C18;0〜60%の、0.1%トリフルオロ酢酸(アセトニトリル中)/0.1%トリフルオロ酢酸(水中)の勾配)により精製した。純粋な分画を一晩凍結乾燥して、35mg 60%の標題化合物を明褐色の固体として得た。
1H NMR(DMSO−d6):δ7.78(d,J=8.0Hz,1H),7.45(d,J=8.0Hz,1H),7.28(t,J=7.8Hz,1H),7.08(t,J=7.8Hz,1H),4.48(d,J=7.3Hz,1H),3.52−3.61(m,2H),2.37(m,1H),2.01−2.11(m,3H).LC/MS249.3(M+1)
【0196】
中間体44
[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボン酸(i−44)の調製
【0197】
【化56】
【0198】
工程A: メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボキシラート
【0199】
【化57】
【0200】
メチル(2S)−5−メトキシ−3,4−ジヒドロ−2H−ピロール−2−カルボキシラート(4.19g、26.6mmol)及び3−アザトリシクロ[4.2.1.0.2,5]ノナ−7−エン−4−オン(2.4g、17.8mmol)を、110℃で一晩加熱した。Biotage Horizon(登録商標)システム(0〜100%酢酸エチル/ヘキサン混合物)を用いて精製し、標題化合物メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキシラート及び中間体メチル(7S)−9−オキソ−3,8−ジアザテトラシクロ[9.2.1.02,10.04,8]テトラデカ−3,12−ジエン−7−カルボキシラートを得た。中間体を150℃で45分間加熱して、さらに精製することなく標題化合物を得た。
LC/MS195.2(M+1).
【0201】
工程B: [(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−]ピリミジン−6−カルボン酸
【0202】
【化58】
【0203】
テトラヒドロフラン(60mL)中の、メチル[6(S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキシラート(9.95g、51.2mmol)、メタノール(40mL)、及び水(40mL)中の水酸化リチウム(3.32g、77mmol)を、室温で1時間攪拌した。2N 塩酸(38.5mL)を添加して反応混合物を中和し、次にこれを直接、逆相HPLC(TMC Pro−Pac C18;0〜40% 0.1%トリフルオロ酢酸(アセトニトリル中)/0.1% トリフルオロ酢酸(水中)の勾配)により精製した。O−アルキル化生成物が、急速に溶出された。純粋な分画を収集し、一晩凍結乾燥して、標題化合物を淡黄色の固体として得た。
1H NMR(DMSO−d6):δ7.89(d,J=6.6Hz,1H),6.24(d,J=6.6Hz,1H),4.92(dd,J=10.0,3.1Hz,1H),3.12−2.99(m,2H),2.52(m,1H),2.11(m,1H).LC/MS181.2(M+1).
【0204】
中間体46
(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボン酸(i−46)
【0205】
【化59】
【0206】
工程A: (3S,9S)−5−オキソ−1,2,3,5,6,8a−ヘキサヒドロインドリジン−3−カルボン酸メチルエステル
【0207】
【化60】
【0208】
この中間体は、ハネシアン(Hanessiann,S.);サイレス(Sailes,H.);ムンロ(Munro,A.);セリアン(Therrien,E.)、「J.Org.Chem.」、2003年、第68巻、p.7219、及びバスワニ(Vaswani,R.G.);チェンバリン(Chamberlin,R.)、「J.Org.Chem.」、2008年、第73巻、p.1661に見られる方法に従って調製した。
【0209】
工程B: メチル(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキシラート
【0210】
【化61】
【0211】
ジクロロメタン 50mL中の上記工程Aよりの(3S,9S)−5−オキソ−1,2,3,5,6,8a−ヘキサヒドロインドリジン−3−カルボン酸メチルエステル 0.850g(4.06mmol)の攪拌された溶液に、酸化マンガン(IV) 6.30g(72.5mmol)を添加し、得られた混合物を還流下で12時間攪拌した。反応物を室温に冷却し、セライト(Celite)(登録商標)のパッドを通して濾過し、次に固体をジクロロメタン 100mLで洗浄した。濾液を真空中で蒸発乾燥させ、残渣をシリカゲルクロマトグラフィーにより、10〜50%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を透明なガムとして得た(0.47g、55%)。
LC−MS:m/z(ES)194(MH)+.
【0212】
工程C: (3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボン酸
【0213】
【化62】
【0214】
THF 3mL中の上記工程Bよりのメチル(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキシラート 0.200mg(1.00mmol)の攪拌された溶液に、1.0M LiOH水溶液 1.5mL(1.5mmol)を添加した。得られた混合物を室温で2時間攪拌し、次に1.0M 塩化水素水溶液 2.0mL(2.0mmol)でクエンチした。全揮発性物質を真空蒸発させ、水相を30% IPA(クロロホルム中)混合物(3x5mL)で抽出した。合わせた抽出物を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させて、標題化合物を白色固体として得た(0.17g、92%)。
1H NMR(500MHz,CD3OD):δ7.53(dd,J=8.9,7.1Hz,1H),6.38−6.35(m,2H),5.11(dd,J=9.7,2.7Hz,1H),3.23−3.12(m,2H),2.62−2.53(m,1H),2.35−2.30(m,1H).
LC−MS:m/z(ES)180(MH)+.
【0215】
中間体56
2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸(i−56)
【0216】
【化63】
【0217】
工程A: Tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアート
【0218】
【化64】
【0219】
DMF(75mL)中の3−メチル−1H−1,2,4−トリアゾール(7.3g、88mmol)の溶液に、K2CO3(60.7g、439mmol)及び2−ブロモプロピオン酸tert−ブチルエステル(14.6mL、88mmol)を添加した。反応物を室温で一晩攪拌した。混合物をEtOAc(500mL)で希釈し、水(x3)で、次に食塩水で洗浄した。MgSO4上で乾燥し、そして濃縮した。残渣をシリカゲル上でのカラムクロマトグラフィーにより、EtOAc/イソヘキサン(20〜100%)で溶出して精製し、粗生成物 13gを位置異性体の3:1混合物として得た。混合物をキラルセル(Chiralcel)ODにより、4%から30%までのIPA/ヘプタンの勾配で精製した。次に、最初の2つのピークを、Chiralcel ODカラムで、4% IPA/ヘプタンを用いてイソクラチック溶出で分離した。2番目のピークを、所望の単一立体異性体(R又はS)(2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸tert−ブチルエステル)(3.5g、19%)として収集した。
1H−NMR(500MHz,CDCl3)δ8.05(s,1H),4.90(q,J=7Hz,1H),2.35(s,3H),1.72(d,J=7Hz,3H),1.40(s,9H).ESI−MSC10H17N3O2の計算値:精密質量:211.13;実測値156.05(−tBu).
【0220】
工程B: 2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸
【0221】
【化65】
【0222】
Tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアート(1.0g、4.7mmol)を、4M HCl(ジオキサン100mL中)に溶解し、室温で一晩攪拌した。生成物を減圧下で濃縮し、高真空下で乾燥させて、(R又はS)tert−ブチル2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパノアートを、HCl塩(850mg)として得た。
ESI−MSC6H9N3O2の計算値:精密質量:155.07;実測値156.05.
【0223】
化合物1
2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(5R)−[(R)−ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0224】
【化66】
【0225】
工程A: Tert−ブチル(5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}ベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート
【0226】
【化67】
【0227】
無水DMF 0.5mL中の、tert−ブチル(5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−3)10mg(シス/トランス 5:1混合物、0.02mmol)及び(2−アミノ−1,3−チアゾール−4−イル)酢酸(3.18mg、0.02mmol)の溶液に、0.5M HOAt溶液(DMF中)(0.04mL、0.02mmol)を、続いてEDC(5.8mg、0.03mmol)及びDIEA(3.5μL、0.02mmol)を添加した。得られた混合物を、窒素雰囲気下、室温で16時間攪拌した。混合物を水で洗浄し、ジクロロメタン(2x2mL)で抽出した。有機物質を合わせ、硫酸ナトリウム上で乾燥し、濾過し、真空中で濃縮した。残渣を、分取用TLCプレート(500uM)により、5%MeOH(ジクロロメタン中)で溶出して精製し、生成物(10.3mg、81%)を得た。
m/z(ES)637(MH)+,659(MNa)+.
【0228】
工程B: 2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(5R)−[(R)ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0229】
【化68】
【0230】
メタノール 0.20mL中のtert−ブチル(5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}ベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより)7mg(0.01mmol)の溶液に、濃HCl 0.20mLを添加し、反応混合物を室温で1時間攪拌した。トルエン(2x)で共沸して、水を除去した。残渣を、アセトニトリル/水/MeOH(9:1:1)中に溶解し、ギルソン(Gilson)HPLC上で、0〜50% アセトニトリル/水(0.05% TFAバッファを含む)の勾配で溶出して精製した。生成物を含有する分画を合わせ、凍結し、凍結乾燥して、白色の泡沫(3.3mg、71%)を得た。
m/z(ES)423(MH)+.(約5:1混合物)1H NMR(500MHz,CD3OD)δ:7.56(br d,J=8.2Hz,2H),7.44(d,7.8Hz,2H),7.39(t,J=7.6Hz,2H)7.35−7.32(m,0.8H)7.32−7.29(m,0.2H副異性体),7.26(d,J=8.0Hz,1.7H),7.14(d,J=8.1Hz,0.3H副異性体)6.67及び6.66(br s,0.2/0.8H,全1H).4.72(d,J=8.5Hz,1H),3.80−3.70(m,4H)3.14(dd,J=6.1,13.8Hz,1H),2.95(dd,J=9.1,13.8Hz,1H),2.08−2.00(m,1H),1.86−1.74(m,3H).
【0231】
本明細書に記載の生物学的アッセイを用いて、化合物1のヒトβ3機能活性は、1ないし10nMであると測定された。
【0232】
化合物2
2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(2S,5R)−[(R)−ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0233】
【化69】
【0234】
工程A: Tert−ブチル(2S,5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}べンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート
【0235】
【化70】
【0236】
標題化合物は、tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(i−4a)及び(2−アミノ−1,3−チアゾール−4−イル)酢酸から、化合物1、工程Aの方法に従って調製した。粗生成物を、分取用TLCプレートにより、5%MeOH(ジクロロメタン中)で溶出して精製し、生成物(4.1mg、21%)を得た。
m/z(ES)637(MH)+,659(MNa)+.
【0237】
工程B: 2−(2−アミノ−1,3−チアゾール−4−イル)−N−[4−({(2S,5R)−[(R)ヒドロキシ(フェニル)メチル]ピロリジニル}メチル)フェニル]アセトアミド
【0238】
【化71】
【0239】
標題化合物は、tert−ブチル(2S,5R)−2−(4−{[(2−アミノ−1,3−チアゾール−4−イル)アセチル]アミノ}べンジル)−5−[(R)−{[tert−ブチル(ジメチル)シリル]オキシ}(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより)4mgから、化合物1、工程Bの方法に従って調製した。粗生成物は、Gilson HPLC上で、0〜50% アセトニトリル/水(0.05% TFAバッファを含む)の勾配で溶出して精製した。生成物を含有する分画を合わせ、凍結し、凍結乾燥して、白色の泡沫(3.3mg、71%)を得た。
m/z(ES)423(MH)+.1H NMR(500MHz,CD3OD)δ:7.55(br d,J=8.2Hz,2H),7.44(d,7.8Hz,2H),7.39(t,J=7.6Hz,2H)7.35−7.33(m,1H),7.25(d,J=8.0Hz,2H),6.65(br s,1H).4.72(d,J=8.5Hz,1H),3.80−3.72(m,4H)3.14(dd,J=6.1,13.8Hz,1H),2.96(dd,J=9.1,13.8Hz,1H),2.07−2.00(m,1H),1.85−1.73(m,3H).
【0240】
本明細書に記載の生物学的アッセイを用いて、化合物2のヒトβ3機能活性は、1ないし10nMであると測定された。
【0241】
化合物3
2−アミノ−N−[4−{((2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル)メチル}フェニル]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキサミド
【0242】
【化72】
【0243】
工程A: Tert−ブチル−(2S,5R)−2−(4[({2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]
チアゾール−4−イル}カルボニル)アミノ]ベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート
【0244】
【化73】
【0245】
無水DMF(5mL)中の、tert−ブチル(2S,5R)−2−(4−アミノベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(i−13a)220mg(0.58mmol)及び2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボン酸(i−8)164mg(0.58mmol)の溶液に、EDC(165mg、0.86mmol)、HOBt(132mg、0.86mmol)、及びヒューニッヒ(Hunig)塩基(0.3mL、1.7mmol)を添加し、得られた混合物を室温で一晩攪拌した。水(50mL)に注入し、EtOAc(3x30mL)で抽出し、合わせたEtOAc層を水(2x50mL)、飽和NaCl(25mL)で洗浄し、MgSO4上で乾燥し、濾過し、蒸発させた。残渣を、MPLC(Biotage Horizon:FLASH 25+M)溶出液:100%ヘキサン(100mL)、0〜35%EtOAc(ヘキサン中)の勾配(750mL)、次に35%EtOAc(ヘキサン中)(600mL)により精製した。ジアステレオ異性体を、ADカラム上でのキラルHPLC(溶出液:25%IPA(ヘプタン中))により、最初に溶出する異性体(134mg、36%)と2番目に溶出する異性体(126mg、34%)に、共に白色の泡沫として分離した。
【0246】
工程B: 2−アミノ−N−[4−{((2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル)メチル}フェニル]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]チアゾール−4−カルボキサミド
【0247】
【化74】
【0248】
DCM(3mL)中のtert−ブチル−(2S,5R)−2−(4[({2−[(tert−ブトキシカルボニル)アミノ]−5,6−ジヒドロ−4H−シクロペンタ[α][1,3]
チアゾール−4−イル}カルボニル)アミノ]ベンジル)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−1−カルボキシラート(工程Aより、2番目に溶出する異性体)126mg(0.19mmol)の溶液に、トリフルオロ酢酸(3.0mL、38mmol)を添加し、得られた混合物を室温で4時間攪拌した。混合物を蒸発させ、SCXカートリッジを通し、2M NH3(メタノール中)で溶出して、塩基を除去した。生成物を、PREP−TLC 2x[20x20cmx1000ミクロン]溶出液:15% MeOH(DCM中)+1%NH4OH、により精製し、生成物を凍結乾燥して、標題化合物(65mg、75%)を白色の飛散性固体として得た。
m/z(ES)449(MH)+.1H NMR(500MHz,DMSO−d6)δ:10.00(s,1H),7.51(d,J=8.2,2H),7.30(m,4H),7.21(t,J=6.9,1H),7.12(d,J=8.2,2H),6.86(s,1H),4.23(d,J=7.3,1H),3.78(m,1H),3.21(m,1H),3.10(m,1H)2.78(m,1H),2.66(m,2H),2.57(m,2H),2.49(m,1H),1.59(m,1H),1.40(m,1H),1.39(m,2H).
【0249】
工程Aよりの生成物[最初に溶出する異性体](134mg、0.207mmol)を、同様の方法で脱保護して、標題化合物(44mg、48%)を白色の飛散性固体として得た。
m/z
(ES) 449
(MH) +.
【0250】
本明細書に記載された生物学的アッセイを用いて、化合物3のヒトβ3機能活性は、1nM未満であると測定された。
【0251】
化合物4−10
上記記載のものと同様の方法を用いて、化合物4〜10を適切な出発物質から調製した。
【0252】
本明細書に記載された生物学的アッセイを用いて、各化合物のヒトβ3機能活性が測定され、以下の表に示された。
【0253】
【化75−1】
【0254】
【化75−2】
【0255】
化合物11
N−(4−(((2S,5R)−5−((R)−ヒドロキシ(フェニル)メチル)ピロリジン−2−イル)メチル)フェニル)−2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパンアミド
【0256】
【化76】
【0257】
DMF(20mL)中の、i−13a(2.00g、5.23mmol)、2−(3−メチル−1H−1,2,4−トリアゾール−1−イル)プロパン酸i−56(1.00g、5.23mmol)、HOAt(1.307mL、0.784mmol)、及びEDC(2.005g、10.46mmol)の混合物を、室温で10分間攪拌した。反応混合物を炭酸水素ナトリウム水溶液でクエンチし、EtOAcで抽出した。粗生成物をカラムクロマトグラフィー(0〜3%MeOH(10%NH4OH)(DCM中)により精製した。蒸発後、生成物をキラルHPLC(ADカラム、30%IPA/ヘプタン)によりさらに精製して、boc保護された純粋な中間体を得、これを最少体積のジオキサン中に溶解し、4M HCl(ジオキサン中)を添加した。室温で2時間後、反応混合物を減圧下で濃縮して、標題化合物のHCl塩を得た。塩基性逆相HPLC(0.1%NH4OH(H2O中)、MeCN)により、標題化合物の所望の遊離塩基を得た。
1H−NMR(500MHz,CD3OD)δ8.51(s,1H),7.49(d,J=13Hz,2H)7.35−7.29(m,4H),7.26−7.20(m,4H),5.20(q,J=7.5Hz,1H),4.20(d,J=7.5Hz,1H),3.27−3.22(m,2H),2.80−2.72(m,2H),2.34(s,3H),1.82(d,J=7.5Hz,3H),1.79−173(m,1H),1.52−1.48(m,3H).ESI−MSC24H29N5O2の計算値:精密質量:419.23,実測値420.35.
【0258】
本明細書に記載の生物学的アッセイを用いて、化合物11のヒトβ3機能活性は、1ないし10nMであると測定された。
【0259】
化合物12及び13
(3S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド(化合物12)及び(3R)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド(化合物13)
【0260】
【化77】
【0261】
工程A: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(3S)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート(異性体1)及びtert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(3R)−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート(異性体2)
【0262】
【化78】
【0263】
無水N,N−ジメチルホルムアミド 3.2mL中の、中間体i−13a 0.610g(1.60mmol)及び中間体i−46 0.300g(1.67mmol)の溶液に、窒素雰囲気下で、1−ヒドロキシ−7−アザベンゾトリアゾール 0.033g(0.24mmol)を、続いて1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩 0.336g(1.75mmol)を添加した。得られた懸濁液を室温で30分間攪拌し、水でクエンチし、酢酸エチル(3x10mL)で抽出した。合わせた有機層を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させた。粗残渣を、シリカゲルクロマトグラフィーにより、50〜100%酢酸エチル(ヘキサン中)の勾配で溶出して精製し、標題化合物を、97:3の比率のジアステレオマー混合物として得た。2つのジアステレオマーを、ダイセル(Daicel)CHIRALPAK(登録商標)AD(登録商標)カラムを用いたキラルHPLC(溶出液:40%IPA(ヘプタン中))により分離した。最初に溶出するジアステレオマーは、異性体2と命名され、無色の固体である(0.020g、2.3%)。
LC−MS:m/z(ES)544.2(MH)+.
2番目に溶出するジアステレオマーは、異性体1と命名され、無色の固体である(0.650g、75%)。
LC−MS:m/z(ES)544.2(MH)+.
【0264】
工程B(化合物12): (3S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド
【0265】
【化79】
【0266】
イソプロパノール 2mL中の上記工程Aよりの異性体1 0.500g(0.920mmol)の溶液に、窒素の雰囲気下で、4.0M 無水塩化水素溶液(1,4−ジオキサン中) 4.0mLを添加した。反応混合物を1時間攪拌し、次に真空中で蒸発乾燥させた。粗反応混合物を、逆相HPLC(TMC Pro−Pac C18;0〜75% 0.01%トリフルオロ酢酸(アセトニトリル中)/0.01%トリフルオロ酢酸(水中)の勾配)により精製した。純粋な分画を一晩凍結乾燥し、次いでクロロホルム 10mLと飽和重炭酸塩水溶液 4mLとの混合物中に溶解した。二相混合物を、10分間激しく攪拌し、次に層を分離させた。水相をクロロホルム(3x10mL)で抽出し、合わせた有機層を食塩水で洗浄し、硫酸マグネシウム上で乾燥し、濾過し、真空蒸発させて、標題化合物(化合物12)を、白色固体(0.39g、95%)として得た。
1H−NMR(500MHz,CD3OD)δ7.89(s,1H),7.54(dd,J=8.8,7.2Hz,1H),7.50(d,J=8.2Hz,2H),7.34−7.29(m,4H),7.26−7.23(m,1H),7.20(d,J=8.2Hz,2H),6.38−3.36(m,2H),5.24(dd,J=9.4,2.8Hz,1H),4.20(d,J=7.8Hz,1H),3.35−3.23(m,3H),3.19−3.12(m,1H),2.82−2.71(m,2H),2.60−2.51(m,1H),2.37−2.32(m,1H),1.79−1.72(m,1H),1.52−1.43(m,3H).
LC−MS:m/z(ES)444.0(MH)+.
【0267】
工程B(化合物13): (3R)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−5−オキソ−1,2,3,5−テトラヒドロインドリジン−3−カルボキサミド
【0268】
【化80】
【0269】
上記工程Aよりの、異性体2の脱保護に用いたものと同様の方法により、標題化合物(化合物13)を単一のジアステレオマーとして得た。
LC−MS:m/z(ES)444.0(MH)+.
【0270】
本明細書に記載の生物学的アッセイを用いて、化合物12及び13のヒトβ3機能活性は、各々1ないし10nM、及び1nM未満であると測定された。
【0271】
化合物14
(6S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキサミド
【0272】
【化81】
【0273】
工程A: Tert−ブチル(2R,5S)−2−[(R)−ヒドロキシ(フェニル)メチル]−5−[4−({[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−イル]カルボニル}アミノ)ベンジル]ピロリジン−1−カルボキシラート
【0274】
【化82】
【0275】
N,N−ジメチルホルムアミド(100ml)中のi−13a(21.4g、55.9mmol)の溶液に、0℃で、[(6S)−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボン酸(i−44、11.1g、61.5mmol)を、続いて1−ヒドロキシベンゾトリアゾール(7.55g、55.9mmol)、N−(3−ジメチルアミノプロピル)−N’−エチルカルボジイミド塩酸塩(16.1g、84.0mmol)、及びN,N−ジイソプロピルエチルアミン(29.2ml、168mmol)を添加した。反応混合物を、0℃から室温まで2時間攪拌した。水(600ml)を添加し、これをジクロロメタン(600mlx2)で抽出した。合わせた有機層を、Na2SO4上で乾燥した。揮発性物質を除去した後、残渣を、Biotage Horizon(登録商標)システム(0〜5%、次に5%メタノール(10%アンモニア/ジクロロメタン混合物を用いて))を使用することにより精製し、副ジアステレオマーの8%を含有する標題化合物を得た。これをさらに、超臨界液体クロマトグラフィー(キラルASカラム、40%メタノール)により精製して、標題化合物を淡黄色の固体として得た(22.0g、72%)。
1H NMR(CDCl3):δ9.61(s,1H),7.93(d,J=6.6Hz,1H),7.49(d,J=8.4Hz,2H),7.35−7.28(m,5H),7.13(d,J=8.5Hz,2H),6.40(d,J=6.7Hz,1H),5.36(d,J=8.6Hz,1H),4.38(m,1H),4.12−4.04(m,2H),3.46(m,1H),3.15−3.06(m,2H),2.91(dd,J=13.1,9.0Hz,1H),2.55(m,1H),2.38(m,1H),1.71−1.49(m,13H).LC−MS567.4(M+23).
【0276】
工程B: (6S)−N−[4−({(2S,5R)−5−[(R)−ヒドロキシ(フェニル)メチル]ピロリジン−2−イル}メチル)フェニル]−4−オキソ−4,6,7,8−テトラヒドロピロロ[1,2−α]ピリミジン−6−カルボキサミド
【0277】
【化83】
【0278】
ジクロロメタン(40ml)中の工程Aよりの中間体(2.50g、4.59mmol)の溶液に、トリフルオロ酢酸(15ml)を添加した。反応混合物を室温で1.5時間攪拌した。揮発性物質を除去した後、飽和NaHCO3を添加して、pH値を8〜9とした。次に混合物を、ジクロロメタンで抽出した。合わせた有機層を、Na2SO4上で乾燥した。濃縮後、メタノール/アセトニトリルからの結晶化により、標題化合物を白色固体として得た(1.23g、60%)。
1H NMR(DMSO−d6):δ10.40(s,1H),7.91(d,J=6.7Hz,1H),7.49(d,J=8.3Hz,2H),7.32−7.26(m,4H),7.21(m,1H),7.15(d,J=8.4Hz,2H),6.23(d,J=6.7Hz,1H),5.11(dd,J=9.6,2.9Hz,1H),5.10(br,1H),4.21(d,J=7.1Hz,1H),3.20−3.00(m,4H),2.66−2.51(m,3H),2.16(m,1H),1.57(m,1H),1.38(m,1H),1.29−1.23(m,2H).LC−MS445.3(M+1)
【0279】
本明細書に記載の生物学的アッセイを用いて、化合物14のヒトβ3機能活性は、11ないし100nMであると測定された。
【0280】
化合物15〜29は、上記記載のものと同様の方法を用いて、適切な出発物質から調製した。
【0281】
β3機能活性のための生物学的アッセイ:
以下のインビトロのアッセイは、選択的β3アゴニスト活性を有する化合物をスクリーニングするのに適している:
機能アッセイ: リガンドに応答するcAMP産生を、バートン(Barton)ら(「Agonist−induced desensitization of D2 dopamine receptors in human Y−79 retinoblastoma cells(ヒトY−79網膜芽腫細胞におけるD2ドーパミン受容体のアゴニスト誘導性脱感作),Mol.Pharmacol.」、1991年、第3229巻、p.650〜658)に従い、以下のように修正して測定する。cAMP産生は、均一時間分解蛍光共鳴エネルギー転移免疫アッセイ(ランス(LANCETM)、パーキン・エルマー(Perkin Elmer))を用いて、製造業者の指示に従って測定する。クローニングされたβ−アドレナリン作動性受容体(β1、β2、又はβ3)で安定にトランスフェクトされたチャイニーズハムスター卵巣(CHO)細胞は、継代の3日後に収集する。細胞の収穫は、無酵素解離培地(Enzyme−free Dissociation Media)(Specialty Media)を用いて行なう。次に細胞をカウントし、ホスホジエステラーゼ阻害剤(IBMX、0.6mM)を含有するアッセイバッファ(5mM HEPES、0.1%BSAを補足したハンクス平衡塩類溶液(Hank’s Balanced salt solution))中に再懸濁する。反応は、6μL中の6,000個の細胞を、アレクサフルオル(Alexa Fluor)標識cAMP抗体(LANCETMキット)6μLと混合することにより開始し、これを次に、化合物(アッセイバッファで2X最終濃度に希釈)12μLを含有するアッセイウェルに添加する。反応は、室温で30分間進行させ、検出バッファ(LANCETMキット)24μLの添加により終了させる。次にアッセイプレートを、室温で1時間インキュベートし、パーキン・エルマー・エンビジョン・リーダー(Perkin Elmer Envision reader)又は同等物により、時間分解蛍光を測定する。蛍光レベルを、cAMPの標準曲線と比較することにより、未知のcAMPレベルを測定する。
【0282】
非選択的、完全アゴニストβ−アドレナリン作動性リガンド イソプロテレノールを、全3種の受容体において使用して、最大刺激を測定する。ヒトβ3アドレナリン作動性受容体(AR)選択的リガンド(S)−N−[4−[2−[[2−ヒドロキシ−3−(4−ヒドロキシフェノキシ)プロピル]アミノ]エチル]−フェニル]−4−ヨードベンゼンスルホンアミドを、対照として全てのアッセイにおいて使用する。イソプロテレノールは、10−10Mないし10−5のアッセイ時の終濃度で滴定し、選択的リガンド(S)−N−[4−[2−[[2−ヒドロキシ−3−(4−ヒドロキシフェノキシ)プロピル]アミノ]エチル]フェニル]−4−ヨードベンゼンスルホンアミドは、10−10Mないし10−5Mの濃度のβ3受容体にて滴定する。未知のリガンドは、10−10Mないし10−5Mのアッセイ時の終濃度の全3種のβ−アドレナリン作動性受容体サブタイプについて滴定し、EC50を測定する。EC50は、それ自体の最大値の50%の活性化を与える化合物濃度として定義される。データは、マイクロソフト・エクセル(Microsoft Excel)及びグラフパッド・プリズム(Graphpad Prism)を使用して分析するか、又は自社開発したデータ分析ソフトウェアパッケージを用いて分析する。
【0283】
結合アッセイ: 化合物はまた、β1及びβ2受容体においてもアッセイし、選択性を測定する。全ての結合アッセイは、組換えによりβ1又はβ2受容体を発現するCHO細胞から調製した膜を用いて実施する。細胞は、分離後3〜4日間増殖させ;付着した細胞をPBSで洗浄し、次に氷上で、1mM トリス、pH7.2中で10分間溶解させる。フラスコをこすって細胞を取出し、次にテフロン(登録商標)(Teflon)/ガラスホモジナイザーを用いて細胞をホモジナイズする。膜は、4℃において、38,000xgで15分間の遠心分離により収集する。ペレット化された膜を、TMEバッファ(50mM トリス、pH7.4、5mM MgCl2、2mM EDTA)中に、1mgタンパク質/mLの濃度に再懸濁する。膜の大きなバッチを調製し、分注し、−70℃で1年間まで能力の損失なしに貯蔵し得る。結合アッセイは、膜(2〜5μgのタンパク質)、放射標識トレーサ125I−シアノピンドロール(125I−CYP、45pM)、200μgのWGA−PVT SPAビーズ(GEヘルスケア(Healthcare))、及び10−10Mないし10−5Mの範囲の終濃度での試験化合物と一緒に、0.1%BSAを含有する最終体積200μLのTMEバッファ中でインキュベートすることにより実施する。アッセイプレートは、室温で振盪しながら1時間インキュベートし、次にパーキン・エルマー・トリラックス(Perkin Elmer Trilux)シンチレーションカウンタに置く。プレートは、カウントする前の約10時間を、Triluxカウンター内で暗中に静置する。データは、Graphpad Prismソフトウェア又は自社開発したデータ分析パッケージを使用して、標準的な4パラメータ非線形回帰分析を用いて分析する。IC50は、放射標識されたトレーサ(125I−CYP)の結合を50%阻害し得る化合物濃度として定義される。β3受容体に対する化合物の選択性は、(IC50β1 AR、β2 AR)/(EC50β3 AR)の比を計算することにより測定してもよい。
【0284】
β3−ARアゴニスト及び抗ムスカリン剤は、500:1ないし1:50の重量比で、患者に投与し得る。1つの実施態様においては、β3−ARアゴニスト及び抗ムスカリン剤の重量比は、300:1ないし1:10である。別の実施態様においては、重量比は、300:1ないし1:1である。別の実施態様においては、重量比は、150:1ないし1:1である。別の実施態様においては、重量比は、100:1ないし1:1である。なお別の実施態様においては、重量比は、150:1である。さらに別の実施態様においては、重量比は、100:1である.
【0285】
併用療法はまた、β3−ARアゴニスト及び抗ムスカリン剤に加えて、選択的M2アンタゴニストをさらに含んでなってもよい。
【0286】
本明細書で用いるとき、用語「選択的M2アンタゴニスト」は、ムスカリンM2サブタイプを、他のムスカリンサブタイプ、例えばM3サブタイプに比較して、10倍より大きい選択性で拮抗する化合物である。デルメンド(Delmendo)、「Br J Pharmacol.」、1989年2月、第96巻、第2号、p.457〜64参照(これは、選択的アンタゴニストの議論に関し、その記載全体が本明細書に援用される)。
【0287】
1つの実施態様においては、選択的M2アンタゴニストは、メトクトラミンである。メトクトラミンは、以下の構造を有するポリメチレンテトラミン誘導体である:
【0288】
【化84】
【0289】
1つの実施態様においては、OABの治療法は、β3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストを、それを必要とする患者に投与することを含んでなる。1つの実施態様においては、抗ムスカリン剤は、約40より大きいM2/M3比を有する。別の実施態様においては、抗ムスカリン剤は、約50より大きいM2/M3比を有する。1つの実施態様においては、抗ムスカリン剤は、ダリフェナシンである。1つの実施態様においては、M2/M3比は、オータケ(Ohtake)らに記載された受容体結合アッセイを用いて測定される。
【0290】
1つの実施態様においては、OABの治療法は、β3−ARアゴニスト、ダリフェナシン、及びメトクトラミンを、それを必要とする患者に投与することを含んでなる。別の実施態様においては、β3−ARアゴニストは、表3に示した化合物から選択される。別の実施態様においては、β3−ARアゴニストは、表4に示した化合物から選択される。なお別の実施態様においては、β3−ARアゴニストは:
【0291】
【化85】
【0292】
及び
【0293】
【化86】
【0294】
からなる群より選択される。
【0295】
β3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストが患者に投与される方法においては、β3−ARアゴニストは、選択的M2アンタゴニストで前処理されてもよい。1つの実施態様においては、選択的M2アンタゴニストは、メトクトラミンである。別の実施態様においては、抗ムスカリン剤は、ダリフェナシンである。別の実施態様においては、β3−ARアゴニストは、メトクトラミンで前処理される。なお別の実施態様においては、メトクトラミンで前処理されたβ3−ARアゴニストは、ダリフェナシンと同時投与される。
【0296】
1つの実施態様においては、前処理のためのメトクトラミンの濃度は、0.1ないし10μMである。別の実施態様においては、前処理のためのメトクトラミンの濃度は、1μMである。
【0297】
上記記載の併用療法においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、同時に、連続的に、又は別々に患者に投与し得る。
【0298】
1つの実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、同時に患者に投与される。別の実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、別々に患者に投与される。なお別の実施態様においては、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、連続的に患者に投与される。
【0299】
適した患者は、これに限定されないが、過活動膀胱又は下部尿路症状(LUTS)の患者を包含する。1つの実施態様においては、患者は、OAB症状のある女性である。別の実施態様においては、患者は、OAB症状のある閉経期の女性である。
【0300】
本発明の別の態様は、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストを含んでなる、組合せ医薬組成物を提供する。適切なβ3−ARアゴニスト、抗ムスカリン剤、及び選択的M2アンタゴニストは、上記記載の通りである。
【0301】
組合せ組成物中のβ3−ARアゴニストの適量は、約0.01mgないし約500mgである。1つの実施態様においては、β3−ARアゴニストの量は、約0.05mgないし約250mgである。別の実施態様においては、該量は、約0.1mgないし約150mgである。別の実施態様においては、該量は、約1ないし約100mgである。なお別の実施態様においては、該量は、約1ないし約50mgである。
【0302】
組合せ組成物中の抗ムスカリン剤の適量は、約0.01mgないし約50mgである。1つの実施態様においては、抗ムスカリン剤の量は、約0.05mgないし約12mgである。別の実施態様においては、該量は、約0.1mgないし約6mgである。別の実施態様においては、該量は、約0.2ないし約5mgである。なお別の実施態様においては、該量は、約0.2ないし約3mgである。
【0303】
組合せ組成物中の選択的M2アンタゴニストの適量は、約0.01mgないし約50mgである。1つの実施態様においては、選択的M2アンタゴニストの量は、約0.05mgないし約15mgである。
【0304】
実際の使用では、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストは、通常の薬学的調剤技術に従い、薬学的担体との均質な混合物中の活性成分として組合せ得る。担体は、投与、例えば、経口又は非経口(静脈内を含む)用に求められる製剤の形状に依存して、広く多様な形態をとってよい。経口剤形用の組成物の調製においては、例えば懸濁液、エリキシル、及び溶液などといった経口液体製剤の場合には、例えば水、グリコール、油、アルコール、着香剤、保存剤、着色剤などといった任意の通常の薬学的媒体を用いてもよく;又は、例えば粉末、硬及び軟カプセル、及び錠剤といった経口用固形製剤の場合には、デンプン、糖、微結晶セルロース、希釈剤、造粒剤、滑沢剤、結合剤、崩壊剤などといった担体を使用してもよく、液体製剤よりも固形経口製剤が好ましい。
【0305】
その投与の容易さから、固体の薬学的担体が使用される場合、錠剤及びカプセルが最も有利な経口用単位剤形である。所望であれば、錠剤は、標準的な水性又は非水性の技術によりコートされてもよい、かかる組合せ組成物及び製剤は、0.1〜20パーセントの各活性成分を含有し得る。これらの組合せ組成物における活性成分のパーセントは、もちろん変更されてもよく、かかる組成物中の活性成分の量は、有効な薬用量が得られるようにするものである。
【0306】
活性成分はまた、例えば液滴又はスプレーとして、鼻腔内に投与し得る。
【0307】
錠剤、丸剤、カプセルなどはまた、トラガカントガム、アラビアガム、コーンスターチ、又はゼラチンなどの結合剤;リン酸二カルシウムなどの賦形剤;コーンスターチ、ジャガイモデンプン、アルギン酸などの崩壊剤;ステアリン酸マグネシウムなどの滑沢剤;及びスクロース、ラクトース、又はサッカリンなどの甘味剤を含有してもよい。単位剤形がカプセルである場合、それは上記のタイプの物質に加えて、脂肪油などの液体担体を含有してもよい。
【0308】
様々な他の物質が、コーティングとして、又は単位剤形の物理的形態を変更するために存在してもよい。例えば、錠剤を、シェラック、糖、又はその双方でコートしてもよい。シロップ又はエリキシルは、活性成分に加えて、甘味剤としてのスクロース、保存剤としてのメチル及びプロピルパラベン、色素、及びチェリー又はオレンジフレーバーなどの着香剤を含有してもよい。
【0309】
活性成分はまた、非経口的に投与してもよい。これらの活性成分の溶液又は懸濁液は、ヒドロキシ−プロピルセルロースなどの界面活性剤と適宜混合された水中に調製し得る。分散剤はまた、グリセロール、液体ポリエチレンブリコール、及びそれらの油中混合物中に調製し得る。通常の貯蔵及び使用条件下では、これらの製剤は、微生物の増殖を防止するための保存剤を含有する。
【0310】
注射用の使用に適した組合せ組成物は、無菌の水溶液又は分散剤、及び無菌の注射用溶液又は分散剤を即時調製するための無菌の粉末を包含する。全ての場合、剤形は無菌でなければならず、注射が容易である程度の液体でなければならない。それは、製造及び貯蔵の条件下で安定でなければならず、また細菌及び真菌などの微生物の汚染作用に対抗して保存されねばならない。担体は、例えば水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコール、及び液体ポリエチレングリコール)、適切なこれらの混合物、及び植物油を含有する、溶媒又は分散媒であってもよい。
【0311】
1つの実施態様においては、組合せ組成物は、経口組成物である。別の実施態様においては、経口組成物は、カプセルゲルの状態である。なお別の実施態様においては、組合せ組成物は、経口用錠剤組成物である。さらに別の実施態様においては、組合せ組成物は、経口用ビーズ組成物である。
【0312】
1つの実施態様においては、組合せ組成物は、制御放出用組成物であって、これにおいて、抗ムスカリン剤は、組成物の投与により、24時間にわたり放出される。別の実施態様においては、抗ムスカリン剤は、10時間にわたり放出される。なお別の実施態様においては、抗ムスカリン剤は、8時間にわたり放出される。さらに別の実施態様においては、抗ムスカリン剤は、6時間にわたり放出される。
【0313】
本明細書に開示されたものはまた、β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストの、過活動膀胱の治療又は予防用医薬の製造における使用も包含する。
【0314】
実施例
β3−ARアゴニスト、抗ムスカリン剤、及び任意の選択的M2アンタゴニストの同時投与の効果は、以下の実施例において例示される。
【0315】
CL316243、又は二ナトリウム(R,R)−5−(2−((2−(3−クロロフェニル)−2−ヒドロキシエチル)−アミノ)プロピル)−1,3−ベンゾジオキソール−2,3−ジカルボキシラートは、β3−ARアゴニストである。CL316243は、「J.Med.Chem.」、1992年8月7日、第35巻、第16号、p.3081〜4にさらに詳細に記載されている。
【0316】
トルテロジン、又は2−[(1S)−3−(ジイソプロピルアミノ)−1−フェニルプロピル]−4−メチルフェノ−ルは、過活動膀胱の治療に使用される抗ムスカリン剤である。トルテロジンは、米国特許第5,382,600、6,630,162、6,770,295、及び6,911,217号にさらに詳細に記載されている。
【0317】
オキシブチニン、又は4−ジエチルアミノブタ−2−イニル2−シクロヘキシル−2−ヒドロキシ−2−フェニル−エタノアートは、膀胱の筋肉痙攣を低減することにより、頻尿及び排尿制御不能(切迫尿失禁)を含む、尿及び膀胱の障害を緩和するのに使用される抗ムスカリン剤である。
【0318】
ダリフェナシン、又は(S)−2−[1−[2−(2,3−ジヒドロベンゾフラン−5−イル)エチル]ピロリジン−3−イル]−2,2−ジフェニル−アセトアミドは、尿失禁を治療するために使用される抗ムスカリン剤である。ダリフェナシンは、米国特許第5,096,890号にさらに詳細に記載されている。
【0319】
実施例1〜3
材料及び方法
以下の材料及び方法を、実施例1〜3に使用した。雄成体スプラーグ・ドーリー(Sprague−Dawley)ラットを使用した。CO2ガスを用いて安楽死させた後、全膀胱を取出した。三角部外側部分の排尿筋の長軸方向の細片(約6mmx3mm)を用意した。各細片を、酸素化された(95%O2+5%CO2)クレブス(Krebs)溶液を含有する、温めた(37℃)臓器浴(25mL)中に置いた。細片は、一方の端を臓器浴につなげ、他端を等尺性の変換器(ADインスツルメンツ(Instruments))に、10mNの静止張力下で連結した。標本の応答は、多チャンネルデータ収録システム(PowerLab、AD Instruments)により、10Hzのサンプリング率で記録し、分析ソフトウェア(Chart、AD Instruments)で測定した。少なくとも60分間の平衡化時間の後、各組織細片を、60Hz;持続時間、0.3ms;3秒間;90Vでの電気的フィールド刺激(EFS)で誘発し、収縮を誘導した。EFSにより安定な収縮が得られれば、化合物溶液(25μL)を、次第に増加する様式で臓器浴中に適用した。各化合物による処理の15分後、EFSを適用した。
【0320】
アイソボログラム分析
アイソボログラム分析を使用して、併用療法の相乗効果を評価した。アイソボログラム分析は、2つの異なる薬剤の相互作用について、独立した統計解析を用いて視覚的評価を提供する。統計解析は、各化合物の単一処理及び同じ効果を得るための固定比率組合せからの、いくつかの効力指数の計算から遂行される。アイソボログラム分析は、「JPET」、2001年、第298巻、p.865〜872(これは、その記載全体が本明細書に援用される)にさらに詳細に記載されている。
【0321】
アイソボログラム分析の例示的な説明図を、図1に示す。図1の、ある特定の効果(例えば、最大の50%)についてのアイソボログラムでは、活性薬剤A単独の用量はA=20であり、活性薬剤B単独はB=100である。これらの切片を結ぶ直線(相加的直線)は、これらの効力に基づき、同じ効果をもたらすはずの、全ての用量ペアの位置である。実際の点Qなどの用量ペアは、より少ない量でこの効果を達成し、相乗的(又は超相加的)であり、一方、点Rによって示される用量ペアは、より多くの量が必要であることを示し、それ故相加的以下である。直線A−Bに近接して出現するPのような点は、単純に相加的である。相互作用の性質を実証するため、適切な統計解析がしばしば使用される。
【0322】
実施例2
β3−ARアゴニストCL316243の、トルテロジン、オキシブチニン、及びダリフェナシンから選択される抗ムスカリン剤との併用療法
個別に投与した場合、CL316243の、トルテロジン、オキシブチニン、及びダリフェナシンの各々は、EFS−誘導性の単離排尿筋収縮を阻害した。表5は、25%阻害を誘導した各化合物の濃度を示す。これらの値を、以下のアイソボログラム分析に使用した。
【0323】
【表5】
【0324】
併用療法では、CL316243は、トルテロジン、オキシブチニン、又はダリフェナシンと、固定重量比において同時投与され、アイソボログラム分析からの結果は図2に示される。図2は、CL316243の、トルテロジン(1:2、図2A)、又はオキシブチニン(1:10、図2B)との併用が、相乗効果を示したことを表している。一方、CL316243のダリフェナシンとの併用(1:2、図2C)は、単純に相加的である(すなわち、何ら相乗的ではない)ことを示す。
【0325】
何ら理論に束縛されることを望むものではないが、一般に、抗ムスカリン剤のM3拮抗作用が、OAB効力に重要であると考えられている(例えば、アブラムス(Abrams)及びアンデルソン(Andersson)「BJU Int」、2007年、第100巻、p.987〜1006参照)。驚くべきことに、今、抗ムスカリン剤のM2/M3の相対的選択性が、抗ムスカリン剤及びβ3−ARアゴニストを用いた併用療法における、OAB効力及び/又は副作用の低減に重要な役割を果たし得ることが見出された。
【0326】
上記の結果からわかるように、抗ムスカリン剤トルテロジンは、ムスカリン受容体のM2及びM3サブタイプに対し、ほぼ等しい選択性(M2/M3≒1)を有しており、トルテロジンとCL316243、β3−ARアゴニストとの、2:1の比での組合せは、相乗効果をもたらした。他の抗ムスカリン剤、約6のM2/M3比を有するオキシブチニンもまた、CL316243と10:1の比で組合せた場合、相乗効果をもたらした。
【0327】
一方、M2よりもM3にはるかに高い選択性(M2/M3≒50)をもつダリフェナシンは、CL316243と2:1の比で組合せた場合、相乗作用をもたらさなかった。
【0328】
以上をまとめれば、CL316243の、その各々が40未満のM2/M3比をもつトルテロジンは又はオキシブチニンとの併用は、排尿筋収縮の阻害に相乗作用をもたらした。一方、CL316243の、40より大きいM2/M3比をもつダリフェナシンとの併用は、相乗作用をもたらさなかった。
【0329】
実施例3
トルテロジン又はダリフェナシンとの併用における、β3−ARアゴニスト、化合物12
この実施例では、異なるβ3−ARアゴニストを使用して、β3−ARアゴニストと抗ムスカリン剤との併用療法の相乗効果を調べた。
【0330】
表3において上記記載のβ3−ARアゴニスト、化合物12は、IC25値275nMで、EFS−誘導性単離排尿筋収縮を阻害した。したがって、化合物12は、ラット膀胱細片のEFS−誘導性収縮の阻害において、CL316243(IC25 2.86nM、表5参照)よりもほぼ100倍効力が弱い。このことは、化合物12の、ラットβ3−ARにおける弱い効力と一致する。
【0331】
併用研究においては、化合物12は、トルテロジン又はダリフェナシンと、固定重量比50:1で同時投与された。アイソボログラム分析の結果を、図3に示す。図3は、化合物12の、約1のM2/M3比をもつトルテロジンとの併用が、50:1の比において(図3A)、相乗効果をもたらしたことを示す。
【0332】
一方、化合物12の、約50のM2/M3比をもつダリフェナシンとの併用が、50:1の比において(図3B)、相乗効果をもたらなかったことを示す(相加的以下)。
【0333】
上記の結果は、異なるβ3−ARアゴニスト(CL316243)が研究に使用された、実施例1で見られた結果と一致する。これらの結果は、抗ムスカリン剤が40未満のM2/M3比をもつ場合、そのβ3−ARアゴニストとの併用が、排尿筋収縮の阻害において相乗作用をもたらすことを示唆している。一方、抗ムスカリン剤が40より大きいM2/M3比をもつ場合、そのβ3−ARアゴニストとの併用は、何ら相乗作用をもたらさない。
【0334】
実施例4
併用療法の相乗効果に対する選択的M2アンタゴニストの影響
この実施例では、β3−ARアゴニストと、40より大きいM2/M3比をもつ抗ムスカリン剤との併用療法に対する、選択的M2アンタゴニストの影響を調べる。
【0335】
まず、CL316243、β3−ARアゴニストは、メトクトラミン(1μM)で未処理か又は前処理されたかのいずれかであり、結果は図4に示される。図4は、メトクトラミンによるCL316243の前処理が、EFS−誘導性膀胱収縮の阻害に関し、CL316243の効力に何ら有意な影響を及ぼさなかったことを示す。この結果は、選択的M2アンタゴニスト単独では、β3−ARアゴニスト及び抗ムスカリン剤がM3拮抗作用を用いて行うようには、予め収縮したラット膀胱細片を弛緩させないという、一般的な考えと一致する。このことは、選択的M2アンタゴニスト及びCL316243の併用では、及びM3拮抗作用なしでは、相乗作用をもたらさなかったことを示唆している。
【0336】
次に、未処理(A)及び前処理されたCL316243(B)の各々が、ダリフェナシンと各々1:2の比で組合され、その結果が図5に示される。図5は、前処理されたCL316243(1μM メトクトラミンで)と、ダリフェナシンとの1:2の比での組合せが、相乗効果をもたらしたことを示す。上記に議論されたように、ダリフェナシンは選択的M3アンタゴニストであり、約50のM2/M3比を有する。
【0337】
一方、未処理のCL316243と、ダリフェナシンとの同じ比(1:2)での組合せは、単純に相加的であった(すなわち、何ら相乗効果はない)。
【0338】
これらの結果は、β3−ARアゴニストと抗ムスカリン剤との間の組合せの相乗効果が、M2及びM3拮抗作用の双方の存在を必要とし得るという、実施例1及び2の所見と一致する。
【0339】
何ら理論に束縛されることを望むものではないが、M2受容体は、cAMP依存性のメカニズムによるアドレナリン受容体媒介性の弛緩を逆転させることにより、間接的な収縮応答の媒介において役割を果たし得ると考えられている。M2拮抗作用は、β3−ARアゴニスト誘導性のcAMP増加及びBKチャンネル開放を強化し、排尿筋のさらなる弛緩をもたらす結果となる。
【0340】
実施例5
相乗効果に対する組合せ比の影響
材料及び方法
動物:雌のSDラット(体重200〜250g)(合計7群)。
麻酔:ウレタン(1.0g/kg、ip)。
パラメータ:拡張誘導性の律動的膀胱収縮の振幅
化合物:オキシブチニン(OXY)、CL316243(CL)。
分析:ID20値(振幅を20%まで低減する用量)を用いたアイソボログラム。
【0341】
まず、以下の条件で単回投与実験を行ない、オキシブチニン(OXY)及びCL316243(CL)のID20値を計算し、得られたID20値が表6に示される。
ビヒクル(生理食塩水);
OXY、0.01、0.03、0.1mg/kg、iv;
CL、0.003、0.01、0.03mg/kg、iv。
【0342】
【表6】
【0343】
表6の結果は、オキシブチニン及びCL316243の双方が、麻酔された雌のラットにおいて、拡張誘導性の律動的膀胱収縮の振幅を低減したことを示す。
【0344】
次に、以下の表7に示した併用用法を実施して、アイソボログラムを組立てた。合計で9群あった。OXY及びCLのID20値は、各組合せ比率において計算した。
【0345】
【表7】
【0346】
図6は、CL及びOXYの、1:1及び1:10の比率の組合せについて、相乗効果が観察されたことを示す。一方、CL及びOXYの、1:3の比率の組合せは、単純な相加効果をもたらしたが、相乗効果はなかった。
【0347】
これらの結果は、β3−ARアゴニストCLと抗ムスカリン剤OXYとの間の相乗効果もまた、組合せにおける特定の組合せ比に依存することを示唆している。
【0348】
実施例6
β3−ARアゴニストと抗ムスカリン剤とを含んでなる組合せ組成物
β3−ARアゴニストと抗ムスカリン剤とを含んでなる、例示的な組合せ組成物を、表8に示す。
【0349】
【表8】
【0350】
1つの実施態様においては、β3−ARアゴニストは、表3に列記された化合物から選択される。別の実施態様においては、抗ムスカリン剤は、トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301から選択される。
【0351】
1つの実施態様においては、上記の組合せ組成物は、制御放出用(CR)製剤である。別の実施態様においては、組合せ組成物は、経口投与用のカプセルゲルの状態である。
【0352】
実施例7
CR抗ムスカリン剤及びIR β3−ARアゴニストを含んでなる組合せ組成物
制御放出(CR)部分に抗ムスカリン剤を、そして即時放出(CR)部分にβ3−ARアゴニストを含んでなる例示的な組合せ組成物を、表9に示す。
【0353】
【表9−1】
【0354】
【表9−2】
【0355】
1つの実施態様においては、β3−ARアゴニストは、表3に列記された化合物から選択される。別の実施態様においては、抗ムスカリン剤は、トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301から選択される。
【0356】
1つの実施態様においては、上記組成物は、経口投与用のカプセルゲルである。
【0357】
実施例8
アカゲザルにおける膀胱容量に対する併用療法の効果
材料及び方法
体重5.3〜6.2kg(4〜7歳齢)の成体雌のアカゲザル(Macaca mulatta)を使用した。被験者は、二匹一組又は単独のいずれかで、12時間明/12時間暗のサイクル(7:00AMに点灯)において収容した。彼女らの食餌は、2050テクラド(Teklad)(ハーラン・ラボラトリーズ(Harlan Laboratories)、インディアナ州、インディアナポリス)及び新鮮な果物又は野菜から構成された。水は自由に摂取させた。動物は全て、獣医技術者及び管理人により、健康障害の徴候について毎日観察した。被験者は、>13日の休止期間をとって反復使用された。サルは、テラゾール(Telazol)(3〜5mg/kg)又はケタミン(10〜20mg/kg)のいずれかの筋肉内注射と、その後のシリンジポンプ(552222、ハーバード・アパレイタス(Harvard Apparatus)、マサチューセッツ州、ホリストン)を用いたケタミン(0.2〜0.8mg/kg/分)の静脈内定速輸液によって麻酔した。動物を仰臥位に置き、トリプルルーメンバルーン経尿道カテーテル(7.4Fr、クック・メディカル(Cook Medical、インディアナ州、ブルーミントン)を膀胱に挿入し、バルーンを水 1mLで膨らませて、カテーテルの先端を膀胱の基底部に固定した。カテーテルは、膀胱を充填するための輸液ポンプ(ジェミニ(Gemini)PC−2TX、アラリス・メディカル・システムズ(ALARIS Medical Systems)、カリフォルニア州、サンディエゴ)、及び膀胱内圧をモニタリングするための圧力変換器に連結した。膀胱内圧は、多チャンネルデータ収録システム(Power lab、AD Instruments、バイオパック・システムズ(Biopac systems)、コロラド州、コロラドスプリングス)を用いて、20Hzのサンプリング率で連続的に記録した。超音波診断(Logiq e vet、GEメディカル・システムズ(Medical Systems)、ウィスコンシン州、ウォーキショー、図1A)により膀胱が空であることを確認した後、生理食塩水を15mL/分で膀胱内に注入した。排尿反射の圧力指標に急峻な上昇が観察されたとき、膀胱内注入を停止し、膀胱を60mlシリンジにより手作業で空にした。2回のベースラインシストメトリーの読み取りの後、薬剤を、濃度の上昇するパラダイムを用いて、各投与の10分後にシストメトリーを実施することにより、3回静脈内投与した。膀胱容量は、各シストメトリーについて測定し、ベースライン容量からの%変化を計算した。本明細書で用いるとき、「ベースライン容量」又は「ベースライン」は、2回の投与前測定値の平均膀胱容量を意味する。
【0358】
種々の用量における、トルテロジン(“TOL”)、ダリフェナシン(DAR”)、又は化合物14(Cpd 14”)の単一療法、並びに、表10に示した種々の用量及び用量比でのTOL:Cpd 14及びDAR:Cpd 14の併用療法を、アカゲザルで試験した。
【0359】
【表10】
【0360】
上記の単一療法及び併用療法を用いることによる、アカゲザルにおける膀胱容量の結果は表11に要約される。報告された結果は、4〜6匹の動物におけるベースラインからの%変化の平均値である。
【0361】
【表11】
【0362】
表11から、試験した化合物14及びトルテロジンの全ての組合せが、各個の単一療法に比較して、より大きい膀胱容量を示したことが理解される。最も低い化合物14の用量(0.003mg/kg)において、相乗効果がはるかに大きかったことが注目されるべきである。特に、0.003mg/kg:0.01mg/kgでのCpd 14:TOLの組合せは、それぞれの単一療法についての4.1%及び8.6%に比較して、28.2%の膀胱容量増加を示した。同様に、0.003mg/kg:0.03mg/kgでのCpd 14:TOLの組合せは、それぞれの単一療法についての4.1%及び16.8%に比較して、35.5%の膀胱容量増加を示した。
【0363】
化合物14とダリフェナシンとの併用療法については、より高い用量のダリフェナシン(0.03、0.1mg/kg)において、併用が優れた膀胱容量効果を示した。
【0364】
非選択的ムスカリンアンタゴニスト、トルテロジンは、調べた組合せにおいて、化合物14と、改善された効力を明示したが、選択的M3アンタゴニスト、ダリフェナシンと、化合物14との組合せでは、相加的効果は高用量にのみ限定された。これらの結果は、β3−ARアゴニストと組合された場合、抗ムスカリン剤のM2及びM3拮抗作用の双方が、改善された効力にとり重要であり得ることを示唆している。
【0365】
本発明は、そのいくつかの特定の実施態様について記載しかつ例示してきたが、当業者は、種々の変更、修飾、及び置換が、本発明の趣旨及び範囲から逸脱することなく、これにおいて行なわれ得ることを理解するであろう。例えば、上記の本明細書で示した特定の薬用量以外の有効薬用量を、上記に示した本発明において使用される活性薬剤の任意の指標について、治療される哺乳動物の応答性のバリエーションの結果として適用してもよい。同様に、観察される具体的な薬理学的応答は、選択された特定の活性化合物又は薬学的担体が存在するかどうか、並びに用いた製剤のタイプに従って、及び依存して変化してもよく、結果におけるそのような予想される変動又は差異は、本発明の目的及び実施に従って検討される。それ故、本発明は、以下の請求の範囲によって定義されること及びかかる請求の範囲が合理的である限り広く解釈されることが意図されている。
【特許請求の範囲】
【請求項1】
過活動膀胱を治療する方法であって、前記方法が;
β3−ARアゴニスト、
抗ムスカリン剤、及び
任意の選択的M2アンタゴニストを、
それを必要とする患者に投与することを含んでなり;
ここで、前記β3−ARアゴニストが:
【化1−1】
【化1−2】
【化1−3】
からなる群より選択される、該方法。
【請求項2】
前記抗ムスカリン剤が、40未満のM2/M3比を有する、請求項1に記載の方法。
【請求項3】
前記抗ムスカリン剤が、20未満のM2/M3比を有する、請求項2に記載の方法。
【請求項4】
前記抗ムスカリン剤が:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301からなる群より選択される、請求項2に記載の方法。
【請求項5】
前記抗ムスカリン剤が、トルテロジン又はオキシブチニンである、請求項4に記載の方法。
【請求項6】
前記β3−ARアゴニストが:
【化2】
からなる群より選択される、請求項1に記載の方法。
【請求項7】
前記β3−ARアゴニスト及び前記抗ムスカリン剤が、300:1ないし1:10の重量比で前記患者に投与される、請求項6に記載の方法。
【請求項8】
前記抗ムスカリン剤が、トルテロジンであり、かつここで、前記β3−ARアゴニスト及びトルテロジンが、300:1ないし1:1の重量比で前記患者に投与される、請求項6に記載の方法。
【請求項9】
前記方法が:
β3−ARアゴニスト、
抗ムスカリン剤、及び
選択的M2アンタゴニストを、
前記患者に投与することを含んでなる、請求項1に記載の方法。
【請求項10】
前記抗ムスカリン剤が、40より大きいM2/M3比を有する、請求項9に記載の方法。
【請求項11】
前記抗ムスカリン剤が、ダリフェナシンであり、かつ前記選択的M2アンタゴニストが、メトクトラミンである、請求項10に記載の方法。
【請求項12】
過活動膀胱を治療する方法であって、前記方法が;
CL316243及び
オキシブチニンを、
それを必要とする患者に投与することを含んでなり;
ここで、CL316243及びオキシブチニンが、1:1又は1:10の重量比で前記患者に投与される、該方法。
【請求項13】
前記β3−ARアゴニスト、前記抗ムスカリン剤、及び前記任意の選択的M2アンタゴニストが、同時に、別々に、又は連続的に投与される、請求項1に記載の方法。
【請求項14】
前記β3−ARアゴニスト、前記抗ムスカリン剤、及び前記任意の選択的M2アンタゴニストが、経口的に投与される、請求項1に記載の方法。
【請求項15】
医薬組成物であって:
β3−ARアゴニスト、
抗ムスカリン剤、及び
任意の選択的M2アンタゴニストを、
含んでなり;
ここで、前記β3−ARアゴニストが:
【化3−1】
【化3−2】
からなる群より選択される、該組成物。
【請求項16】
前記医薬組成物が:
β3−ARアゴニスト及び
抗ムスカリン剤を、
含んでなり;かつ
ここで、前記抗ムスカリン剤が、40未満のM2/M3比を有する、請求項15に記載の医薬組成物。
【請求項17】
前記抗ムスカリン剤が、トルテロジン、オキシブチニン、フェソテロジン、ソリフェナシン、プロピベリン、及びトロスピウムからなる群より選択される、請求項16に記載の医薬組成物。
【請求項18】
前記組成物が:
β3−ARアゴニスト、
抗ムスカリン剤、及び
選択的M2アンタゴニストを、
含んでなり;
ここで、前記抗ムスカリン剤が、ダリフェナシンであり、かつ
前記選択的M2アンタゴニストが、メトクトラミンである、請求項15に記載の医薬組成物。
【請求項19】
前記組成物が、経口投与用の錠剤又はカプセルである、請求項15に記載の医薬組成物。
【請求項20】
前記組成物が、抗ムスカリン剤の制御放出を提供する、請求項15に記載の医薬組成物。
【請求項1】
過活動膀胱を治療する方法であって、前記方法が;
β3−ARアゴニスト、
抗ムスカリン剤、及び
任意の選択的M2アンタゴニストを、
それを必要とする患者に投与することを含んでなり;
ここで、前記β3−ARアゴニストが:
【化1−1】
【化1−2】
【化1−3】
からなる群より選択される、該方法。
【請求項2】
前記抗ムスカリン剤が、40未満のM2/M3比を有する、請求項1に記載の方法。
【請求項3】
前記抗ムスカリン剤が、20未満のM2/M3比を有する、請求項2に記載の方法。
【請求項4】
前記抗ムスカリン剤が:トルテロジン、フェソテロジン、オキシブチニン、ソリフェナシン、プロピベリン、トロスピウム、イミダフェナシン、及びTD6301からなる群より選択される、請求項2に記載の方法。
【請求項5】
前記抗ムスカリン剤が、トルテロジン又はオキシブチニンである、請求項4に記載の方法。
【請求項6】
前記β3−ARアゴニストが:
【化2】
からなる群より選択される、請求項1に記載の方法。
【請求項7】
前記β3−ARアゴニスト及び前記抗ムスカリン剤が、300:1ないし1:10の重量比で前記患者に投与される、請求項6に記載の方法。
【請求項8】
前記抗ムスカリン剤が、トルテロジンであり、かつここで、前記β3−ARアゴニスト及びトルテロジンが、300:1ないし1:1の重量比で前記患者に投与される、請求項6に記載の方法。
【請求項9】
前記方法が:
β3−ARアゴニスト、
抗ムスカリン剤、及び
選択的M2アンタゴニストを、
前記患者に投与することを含んでなる、請求項1に記載の方法。
【請求項10】
前記抗ムスカリン剤が、40より大きいM2/M3比を有する、請求項9に記載の方法。
【請求項11】
前記抗ムスカリン剤が、ダリフェナシンであり、かつ前記選択的M2アンタゴニストが、メトクトラミンである、請求項10に記載の方法。
【請求項12】
過活動膀胱を治療する方法であって、前記方法が;
CL316243及び
オキシブチニンを、
それを必要とする患者に投与することを含んでなり;
ここで、CL316243及びオキシブチニンが、1:1又は1:10の重量比で前記患者に投与される、該方法。
【請求項13】
前記β3−ARアゴニスト、前記抗ムスカリン剤、及び前記任意の選択的M2アンタゴニストが、同時に、別々に、又は連続的に投与される、請求項1に記載の方法。
【請求項14】
前記β3−ARアゴニスト、前記抗ムスカリン剤、及び前記任意の選択的M2アンタゴニストが、経口的に投与される、請求項1に記載の方法。
【請求項15】
医薬組成物であって:
β3−ARアゴニスト、
抗ムスカリン剤、及び
任意の選択的M2アンタゴニストを、
含んでなり;
ここで、前記β3−ARアゴニストが:
【化3−1】
【化3−2】
からなる群より選択される、該組成物。
【請求項16】
前記医薬組成物が:
β3−ARアゴニスト及び
抗ムスカリン剤を、
含んでなり;かつ
ここで、前記抗ムスカリン剤が、40未満のM2/M3比を有する、請求項15に記載の医薬組成物。
【請求項17】
前記抗ムスカリン剤が、トルテロジン、オキシブチニン、フェソテロジン、ソリフェナシン、プロピベリン、及びトロスピウムからなる群より選択される、請求項16に記載の医薬組成物。
【請求項18】
前記組成物が:
β3−ARアゴニスト、
抗ムスカリン剤、及び
選択的M2アンタゴニストを、
含んでなり;
ここで、前記抗ムスカリン剤が、ダリフェナシンであり、かつ
前記選択的M2アンタゴニストが、メトクトラミンである、請求項15に記載の医薬組成物。
【請求項19】
前記組成物が、経口投与用の錠剤又はカプセルである、請求項15に記載の医薬組成物。
【請求項20】
前記組成物が、抗ムスカリン剤の制御放出を提供する、請求項15に記載の医薬組成物。
【図1】

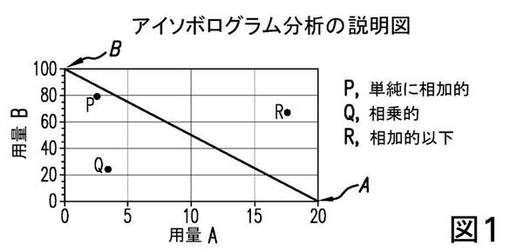
【図2A】
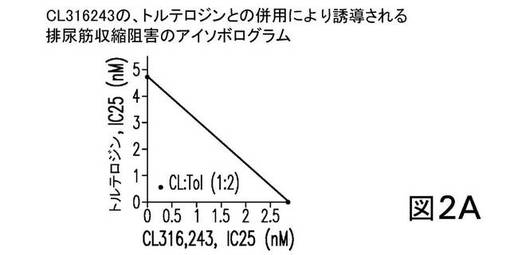
【図2B】
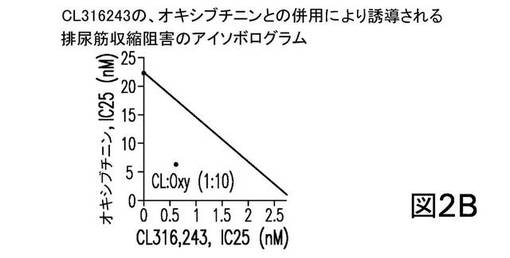
【図2C】
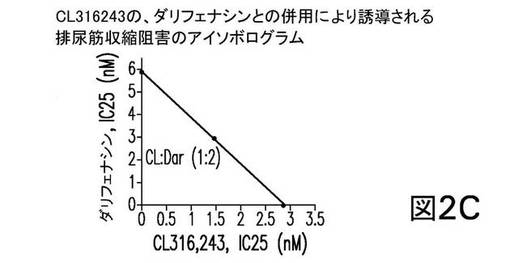
【図3A】
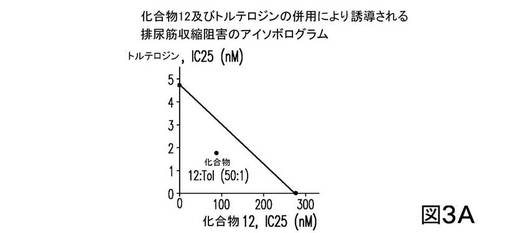
【図3B】
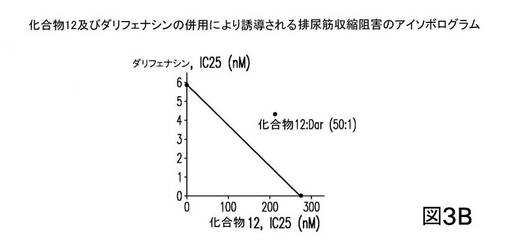
【図4】
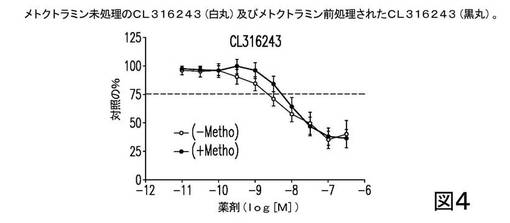
【図5A】
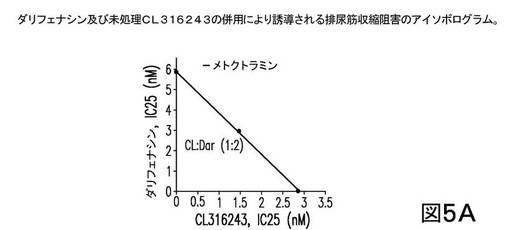
【図5B】
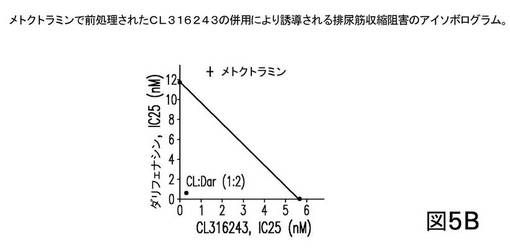
【図6】
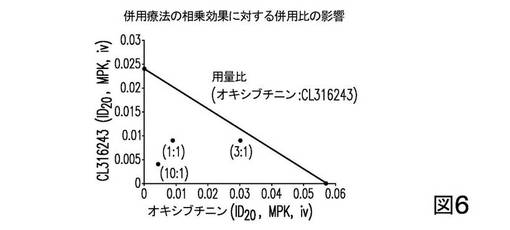
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図4】
【図5A】
【図5B】
【図6】
【公表番号】特表2013−507363(P2013−507363A)
【公表日】平成25年3月4日(2013.3.4)
【国際特許分類】
【出願番号】特願2012−533204(P2012−533204)
【出願日】平成22年9月27日(2010.9.27)
【国際出願番号】PCT/US2010/050328
【国際公開番号】WO2011/043942
【国際公開日】平成23年4月14日(2011.4.14)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
【公表日】平成25年3月4日(2013.3.4)
【国際特許分類】
【出願日】平成22年9月27日(2010.9.27)
【国際出願番号】PCT/US2010/050328
【国際公開番号】WO2011/043942
【国際公開日】平成23年4月14日(2011.4.14)
【出願人】(390023526)メルク・シャープ・エンド・ドーム・コーポレイション (924)
【Fターム(参考)】
[ Back to top ]