
γ−グルタミル化合物を含有する酵母エキスの製造方法及び当該方法に用いられる酵母
【課題】産業上有用なグルタチオン等のγ−グルタミル化合物を高濃度に蓄積する酵母、当該酵母を用いたγ−グルタミル化合物を含有する酵母エキス、および当該酵母を用いた飲食品、並びにそれらの製造方法を提供する。
【解決手段】γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを製造する。
【解決手段】γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを製造する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、γ−グルタミル化合物を含有する酵母エキスの製造方法及び当該方法に用いられる酵母に関するものである。当該酵母エキスや酵母は食品分野で有用である。
【背景技術】
【0002】
γ−グルタミル化合物の1つであるグルタチオン(γ−Glu−Cys−Gly)はすべての生物が有する細胞内に最も多量に存在する非タンパク質性チオールである。グルタチオンは生体内において抗酸化作用、免疫支援、細胞毒の除去など生体維持に必要な様々な機能を有しており、医薬品、化粧品原料として用いられる。また、グルタチオンはコク味を有し、食品添加物としても利用される。そのため、グルタチオンを安価に大量に生産することは重要である。グルタチオンは、例えば、グルタチオンを高生産する出芽酵母を分離して利用することにより、効率よく生産することが可能である(非特許文献1)。しかし、高反応性のチオール基を有するグルタチオンは、生体維持に重要である一方、過剰に蓄積されると有害であり、グルタチオンを高濃度に蓄積した細胞ではしばしば生育遅延が認められる。そのため、効率よくグルタチオンを生産するためには、グルタチオンを細胞内に高濃度に蓄積し、かつ生育が遅延しない出芽酵母の分離が必要である。
【0003】
酵母の細胞内でグルタチオンを分解する酵素としては、ECM38遺伝子にコードされるEcm38pが知られている(非特許文献2、3)。また、酵母の細胞内でグルタチオンを分解する酵素としては、近年DUG複合体が見出された(非特許文献4、5)。しかし、これらのグルタチオン分解系を弱化することにより細胞内のグルタチオン濃度が上昇したという報告は無い。その為、これらグルタチオン分解酵素の活性が低下した酵母を用いて酵母エキスを製造しようとの発想はなかった。またDUG複合体はDug1p、Dug2p、Dug3pからなるが、各DUG因子がグルタチオンの分解にどの程度寄与しているかは明らかになっていない。
【0004】
YCF1遺伝子にコードされるYcf1pは金属とキレートしたグルタチオンを液胞へ輸送するトランスポーターである(非特許文献3および6)。Ycf1pは栄養飢餓時にグルタチオンのみを液胞に輸送すると考えられているが、Ycf1pの機能が細胞内のグルタチオン濃度におよぼす影響は知られていない。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Appl Microbiol Biotechnol. DOI 10.1007/s00253-010-2968-6 (2010)
【非特許文献2】Yeast. 2003 Jul 30;20(10):857-63
【非特許文献3】FEBS Lett. 2009 May 6;583(9):1489-92
【非特許文献4】J Biol Chem. 2009 May 22;284(21):14493-502
【非特許文献5】Genetics. 2007 Mar;175(3):1137-51
【非特許文献6】FEBS Lett. 2010 Feb 19;584(4):726-32
【非特許文献7】Biometals. 2009 Apr;22(2):243-9
【非特許文献8】J Biol Chem. 2005 Feb 11; 280(6): 4851-4857
【非特許文献9】BioMetals (2006) 19:593-599
【非特許文献10】J Biol Chem. 2003 Dec 12;278(50):49920-8
【非特許文献11】Appl Microbiol Biotechnol DOI 10.1007/s00253-010-2946-z (2010)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、産業上有用なグルタチオン等のγ−グルタミル化合物を高濃度に蓄積する酵母、当該酵母を利用したγ−グルタミル化合物を含有する酵母エキス、および当該酵母エキスを含有する飲食品、並びにそれらの製造方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するために鋭意検討を行った結果、γ−グルタミルシステイン合成酵素をコードするGSH1遺伝子の高発現によってグルタチオン合成能を強化した酵母において、さらにグルタチオン分解酵素であるDUG複合体の活性を低下させることで、細胞内のグルタチオンの蓄積量が増大するにもかかわらず酵母の生育遅延を軽減できるという予想外の知見を見出した。また、本発明者は、液胞膜グルタチオントランスポーターの発現を増強し、且つ、液胞局在グルタチオン分解酵素であるEcm38pの活性を低下させることにより、グルタチオン過剰蓄積による生育遅延およびグルタチオンによるフィードバック阻害を回避し、酵母細胞内のグルタチオン蓄積量が増大することを見出した。さらに、本発明者は、γ−グルタミルシステインを蓄積する酵母においても、DUG複合体の活性低下により酵母の生育が向上すること、並びに、Ecm38pの活性低下および液胞膜グルタチオントランスポーターの発現増強により酵母細胞内のγ−グルタミルシステイン蓄積量が増大することを見出した。以上に基づき、本発明は完成された。
【0008】
すなわち、本発明は以下の通り例示できる。
[1]
γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを調製することを特徴とする、酵母エキスの製造方法。
[2]
前記グルタチオン分解酵素が下記(A)〜(D)から選ばれる1またはそれ以上のタンパク質である、前記方法。
(A)DUG1遺伝子にコードされるタンパク質
(B)DUG2遺伝子にコードされるタンパク質
(C)DUG3遺伝子にコードされるタンパク質
(D)ECM38遺伝子にコードされるタンパク質
[3]
前記酵母は、少なくとも液胞局在グルタチオン分解酵素の活性が低下し、さらに液胞グルタチオントランスポーターの発現が増大するよう改変されている、前記方法。
[4]
γ−グルタミルシステイン合成酵素の活性が増大することによりγ−グルタミル化合物合成能が増大した、前記方法。
[5]
グルタチオン合成酵素の活性が増大または低下することによりγ−グルタミル化合物合成能が増大した、前記方法。
[6]
前記γ−グルタミル化合物が、グルタチオン、γ−グルタミルシステイン、およびγ−グルタミル−α−アミノ酪酸から選択される化合物である、前記方法。
[7]
前記γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、およびペプチドから選択される化合物が添加された培地で前記酵母を培養することを特徴とする、前記方法。[8]
前記酵母がサッカロミセス属に属する酵母である、前記方法。
[9]
前記酵母がサッカロミセス・セレビシエである、前記方法。
[10]
前記方法により製造された酵母エキスを含有する飲食品。
[11]
γ−グルタミルシステイン合成酵素の活性が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母。
[12]
さらに、下記(I)および(II)から選ばれる1またはそれ以上の性質を有する前記酵母。
(I)グルタチオン合成酵素の活性の増大、または低下
(II)液胞グルタチオントランスポーターの発現の増大
【発明の効果】
【0009】
本発明により、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積し、且つ生育の良い酵母が提供される。それら酵母を原料として用いることにより、γ−グルタミル化合物を含有する酵母エキスを効率的に製造でき、また、それら酵母エキスを利用して飲食品を製造することができる。
【図面の簡単な説明】
【0010】
【図1】サッカロミセス・セレビシエY006株のウラシル非要求性株であるY003株、およびY006株に由来するGSH1発現強化株およびグルタチオン分解酵素遺伝子破壊株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006のGSH1発現強化株、d1:Y006のDUG1破壊株、d2:Y006のDUG2破壊株、d3:Y006のDUG3破壊株、e:Y006のECM38破壊株、ed:Y006のDUG2およびECM38二重破壊株。
【図2】Y003株、AG1株、およびAG1株に由来するグルタチオン分解酵素遺伝子破壊株の菌体内グルタチオン濃度を示す図。Y003:Y003株、AG1:YF006のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-ed:AG1株のDUG2およびECM38二重破壊株。
【図3】Y003株、およびY006株に由来する変異株の生育曲線を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、d2:Y006のDUG2破壊株、AG1-d:AG1株のDUG2破壊株。
【図4】DUG複合体構成因子をコードするDUG1、DUG2、およびDUG3のmRNA量を示す図。パネルa:OD600=5(中期対数増殖期)におけるDUG因子のmRNA量。パネルb:OD600=10(定常期)におけるDUG因子のmRNA量。
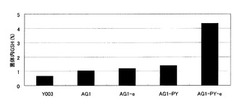
【図5】Y003株、並びに、Y006株に由来するGSH1発現強化株、グルタチオン分解酵素遺伝子破壊株、およびグルタチオン液胞トランスポーター発現強化株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、e:Y006株のECM38破壊株、d:Y006株のDUG2破壊株、PY:Y006株のYCF1発現強化株。
【図6】Y003株、AG1株、並びに、AG1株に由来するグルタチオン分解酵素遺伝子破壊株、および液胞グルタチオントランスポーター発現強化株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-PY:AG1株のYCF1発現強化株、AG1-PY-e:AG1-PY株のECM38破壊株。
【図7】Y003株、AG1株、並びに、AG1株に由来するグルタチオン分解酵素遺伝子破壊株、および液胞グルタチオントランスポーター発現強化株の生育曲線を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-PY:AG1株のYCF1発現強化株、AG1-PY-e:AG1-PY株のECM38破壊株。
【図8】γ−グルタミルシステイン蓄積酵母であるAG1-gsh2株およびAG1-d-gsh2株の生育曲線を示す図。AG1-gsh2:AG1株のGSH2破壊株、AG1-d-gsh2:AG1-d株のGSH2破壊株。
【図9】γ−グルタミルシステイン蓄積酵母であるgsh2株、AG1-gsh2株、およびAG1-d-gsh2株の細胞内γ−グルタミルシステイン濃度を示す図。gsh2:Y003株のGSH2破壊株、AG1-gsh2:AG1株のGSH2破壊株、AG1-d-gsh2:AG1-d株のGSH2破壊株。
【図10】γ−グルタミルシステイン蓄積酵母であるAG1-d-gsh2株、AG1-d-PY-gsh2株、およびAG1-d-PY-e-gsh2株の細胞内γ−グルタミルシステイン濃度を示す図。AG1-d-gsh2:AG1-d株のGSH2破壊株、AG1-d-PY-gsh2:AG1-d-PY株のGSH2破壊株、AG1-d-PY-e-gsh2:AG1-d-PY-e株のGSH2破壊株。
【図11】Abu添加培養時のAG1株、および、AG1株に由来するグルタチオン分解酵素遺伝子破壊株の生育曲線を示す図。AG1:AG1株、AG1-d:AG1株のDUG2破壊株。
【発明を実施するための形態】
【0011】
以下、本発明を詳細に説明する。
(1)本発明の酵母
(1−1)本発明の酵母
本発明の酵母は、グルタチオン等のγ−グルタミル化合物の合成能が増大し、且つ、細胞内のグルタチオン分解酵素活性が低下するように改変された、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積する酵母である。本発明の酵母は、細胞内のグルタチオン分解酵素活性が低下することにより、単にγ−グルタミル化合物の合成能が強化されただけの酵母と比較して、酵母の生育が向上するのが好ましい。更には、本発明の酵母は、細胞内のグルタチオン分解酵素活性が低下することにより、単にγ−グルタミル化合物の合成能が強化されただけの酵母と比較して、細胞内のγ−グルタミル化合物の濃度が増大するのが好ましい。本発明の酵母は、乾燥重量で、好ましくは0.5重量%以上、より好ましくは1.5重量%以上、さらに好ましくは3重量%以上、特に好ましくは5重量%以上のグルタチオン等のγ−グルタミル化合物を細胞内に蓄積できる。
【0012】
また、本発明の酵母は、液胞に局在するグルタチオン分解酵素の活性が低下し、且つ、液胞グルタチオントランスポーターの活性が増大するように改変されているのが好ましい。この場合、グルタチオン分解酵素としては、液胞局在グルタチオン分解酵素の活性のみが低下していてもよく、液胞局在グルタチオン分解酵素に加えて他のグルタチオン分解酵素の活性が低下していてもよい。高濃度のグルタチオンは生育遅延やグルタチオン合成系のフィードバック阻害を引き起こすが、液胞グルタチオントランスポーターの活性を増大させ、且つ、液胞局在グルタチオン分解酵素活性を低下させることで、グルタチオンを液胞に移送して細胞質内のグルタチオン濃度を低下させ、液胞にグルタチオンを安定に蓄積できると考えられる。これにより、グルタチオンによる生育遅延やグルタチオン合成系のフィードバック阻害を回避でき、グルタチオン蓄積量を増大させることができると考えられる。
【0013】
本発明の酵母は、酵母の適当な菌株、例えば後述する菌株を改変することで取得することができる。
【0014】
本発明の酵母は、上記のように改変されグルタチオン等のγ−グルタミル化合物を細胞内に蓄積できる限り特に制限されず、出芽酵母であってもよく、分裂酵母であってもよい。出芽酵母としては、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)等のサッカロミセス属、キャンディダ・ユティリス(Candida utilis)等のキャンディダ属、ピヒア・パストリス(Pichia pastoris)等のピヒア属、ハンゼヌラ・ポリモルファ(Hansenula polymorpha)等のハンゼヌラ属等に属する酵母を例示することができる。分裂酵母としては、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等のシゾサッカロミセス属等に属する酵母を例示することができる。中でも、酵母エキスの生産によく用いられているサッカロミセス・セレビシエやキャンディダ・ユティリスが好ましい。本発明の酵母は、1倍体でもよいし、2倍性またはそれ以上の倍数性を有するものであってもよい。
【0015】
グルタチオン生合成系はすべての生物が有しており、また、実施例で用いた液胞グルタチオントランスポーター、およびグルタチオン分解酵素のホモログは、これら種々の酵母に存在することが知られている(非特許文献5および9)。
【0016】
サッカロミセス・セレビシエとしては、具体的には、サッカロミセス・セレビシエY006株を用いることができる。サッカロミセス・セレビシエY006株は、平成22年8月18日に、産業技術総合研究所特許生物寄託センター(住所 郵便番号305−8566 茨城県つくば市東1丁目1番地1中央第6)に国際寄託され、受託番号FERM BP−11299が付与されている。
【0017】
また、サッカロミセス・セレビシエとしては、具体的には、BY4743株(ATCC201390)やS288C株(ATCC26108)を用いることができる。また、キャンディダ・ユティリスとしては、具体的には、キャンディダ・ユティリスATCC22023株を用いることができる。これらを入手するには、例えばアメリカン・タイプ・カルチャー・コレクション(住所 12301 Parklawn Drive, Rockville, Maryland 20852 P.O. Box 1549, Manassas, VA 20108, United States of America)より分譲を受けることが出来る。すなわち各菌株に対応する登録番号が付与されており、この登録番号を利用して分譲を受けることが出来る(http://www.atcc.org/参照)。各菌株に対応する登録番号は、アメリカン・タイプ・カルチャー・コレクションのカタログに記載されている。
【0018】
(1−2)グルタチオン分解酵素活性の低下
本発明の酵母は、グルタチオン分解酵素の活性が低下するように改変されている。
【0019】
「酵素の活性が低下する」とは、目的の酵素活性が野性株や親株等の非改変株と比較して減少していることを意味し、活性が完全に消失している場合を含む。
【0020】
グルタチオン分解酵素活性は、非改変株と比較して、好ましくは50%以下、より好ましくは20%以下、さらに好ましくは10%以下、特に好ましくは5%以下に低下している。また、グルタチオン分解酵素活性は、実質的に消失しているのが好ましい。
【0021】
酵素活性が低下するような改変は、例えば、突然変異処理又は遺伝子組換え技術により達成できる。
【0022】
突然変異処理としては、紫外線照射、または、N-メチル-N'-ニトロ-N-ニトロソグアニジン(MNNG)、エチルメタンスルフォネート(EMS)、メチルメタンスルフォネート(MMS)等の通常変異処理に用いられている変異剤による処理が挙げられる。
【0023】
遺伝子組換え技術としては、例えば、公知の技術(FEMS Microbiology Letters 165 (1998) 335-340、JOURNAL OF BACTERIOLOGY, Dec. 1995, p7171-7177、Curr Genet 1986; 10(8):573-578、WO 98/14600等)を利用できる。
【0024】
酵素活性が低下するような改変は、例えば、目的の酵素をコードする遺伝子の発現を低下させることにより達成できる。遺伝子の発現の低下は、例えば、遺伝子のプロモーター等の発現調節配列を改変することにより達成できる。発現調節配列を改変する場合には、発現調節配列は、好ましくは1塩基以上、より好ましくは2塩基以上、特に好ましくは3塩基以上が改変される。また、発現調節配列の一部または全部を欠失させてもよい。
【0025】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域の一部または全部を欠失させることにより達成できる。さらには、染色体上の遺伝子の前後の配列を含めて、遺伝子全体を欠失させてもよい。酵素活性の低下
が達成できる限り、欠失させる領域は、N末端領域、内部領域、C末端領域等のいずれの領域であってもよい。通常、欠失させる領域は長い方が確実に遺伝子を不活化することができる。また、欠失させる領域の前後の配列は、リーディングフレームが一致しないことが好ましい。
【0026】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域にアミノ酸置換(ミスセンス変異)を導入すること、終始コドンを導入すること(ナンセンス変異)、あるいは1〜2塩基を付加または欠失するフレームシフト変異を導入すること等によっても達成できる。
【0027】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域に他の配列を挿入することによっても達成できる。挿入部位は、遺伝子のいずれの領域であってもよいが、挿入する配列は長い方が確実に遺伝子を不活化することができる。また、挿入部位の前後の配列は、リーディングフレームが一致しないことが好ましい。他の配列としては、コードされるタンパク質の機能を低下又は消失させるものであれば特に制限されないが、例えば、マーカー遺伝子やグルタチオン等のγ−グルタミル化合物の生産に有用な遺伝子が挙げられる。
【0028】
染色体上の遺伝子を上記のように改変することは、例えば、遺伝子の部分配列を欠失し、正常に機能するタンパク質を産生しないように改変した欠失型遺伝子を作製し、該欠失型遺伝子を含む組換えDNAで酵母を形質転換して、欠失型遺伝子と染色体上の遺伝子とで相同組換えを起こさせることにより、染色体上の遺伝子を欠失型遺伝子に置換することによって達成できる。その際、組換えDNAには、宿主の栄養要求性等の形質にしたがって、マーカー遺伝子を含ませておくと操作がしやすい。また、前記組換えDNAは、制限酵素で切断する等により直鎖状にしておくと、染色体に組換えDNAが組み込まれた株を効率よく取得することができる。欠失型遺伝子によってコードされるタンパク質は、生成したとしても、野生型タンパク質とは異なる立体構造を有し、機能が低下又は消失する。
【0029】
用いる組換えDNAの構造によっては、相同組換えの結果として、野生型遺伝子と欠失型遺伝子とが組換えDNAの他の部分(例えば、ベクター部分及びマーカー遺伝子)を挟んだ状態で染色体に挿入される場合がある。この状態では野生型遺伝子が機能するため、当該2個の遺伝子間で再度相同組換えを起こさせ、1コピーの遺伝子を、ベクター部分及びマーカー遺伝子とともに染色体DNAから脱落させ、欠失型遺伝子が残ったものを選抜する必要がある。
【0030】
また、例えば、任意の配列を含む線状DNAであって、当該任意の配列の両端に染色体上の置換対象部位の上流および下流の配列を備える線状DNAで酵母を形質転換して、置換対象部位の上流および下流でそれぞれ相同組換えを起こさせることにより、1ステップで置換対象部位を当該任意の配列に置換することができる。当該任意の配列としては、例えば、マーカー遺伝子を含む配列を用いればよい。マーカー遺伝子は、その後、必要により除去してもよい。マーカー遺伝子を除去する場合には、マーカー遺伝子を効率的に除去できるよう、相同組み換え用の配列をマーカー遺伝子の両端に付加しておいてもよい。
【0031】
目的の酵素活性が低下したことの確認は、同酵素の活性を測定することによって行うことが出来る。グルタチオン分解酵素の活性は、公知の手法(Penninckx M. et al., 1980.
Eur J Biochem 104: 119-123、Ganguli D. et al., Genetics. 2007 Mar; 175(3): 1137-51等)により測定することができる。
【0032】
目的の酵素をコードする遺伝子の転写量が低下したことの確認は、同遺伝子から転写されるmRNAの量を非改変株と比較することによって行うことが出来る。mRNAの量を評価する
方法としては、ノーザンハイブリダイゼーション、RT-PCR等が挙げられる(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。mRNAの量は、非改変株と比較して、例えば、50%以下、20%以下、10%以下、5%以下、または0%に低下しているのが好ましい。
【0033】
目的の酵素の量が低下したことの確認は、抗体を用いてウェスタンブロットによって行うことが出来る(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。目的の酵素の量は、非改変株と比較して、例えば、50%以下、20%以下、10%以下、5%以下、または0%に低下しているのが好ましい。
【0034】
本発明の酵母が2倍体以上の倍数性を有する場合には、本発明の酵母は、グルタチオン等のγ−グルタミル化合物を蓄積できる限り、酵素活性が低下するように改変された遺伝子と野生型遺伝子とをヘテロで有していてもよいが、通常は、酵素活性が低下するように改変された遺伝子のホモ型であるのが好ましい。
【0035】
酵母の形質転換法としては、プロトプラスト法、KU法(H.Ito et al., J. Bateriol., 153-163 (1983))、KUR法(発酵と工業 vol.43, p.630-637 (1985))、エレクトロポレーション法(Luis et al., FEMS Micro biology Letters 165 (1998) 335-340)、キャリアDNAを用いる方法(Gietz R.D. and Schiestl R.H., Methods Mol.Cell. Biol. 5:255-269 (1995))等、通常酵母の形質転換に用いられる方法を採用することができる。また、酵母の胞子形成、1倍体酵母の分離、等の操作については、「化学と生物 実験ライン31 酵母の実験技術」、初版、廣川書店;「バイオマニュアルシリーズ10 酵母による遺伝子実験法」初版、羊土社等に記載されている。
【0036】
本発明において、グルタチオン分解酵素とはグルタチオンを分解する活性を有するタンパク質をいう。
【0037】
グルタチオン分解酵素としては、DUG1遺伝子、DUG2遺伝子、およびDUG3遺伝子によりそれぞれコードされるDug1p、Dug2p、およびDug3pが挙げられる。Dug1p、Dug2p、およびDug3pはDUG複合体として機能することが知られている。よって、「グルタチオンを分解する活性を有する」とは、遺伝子にコードされるタンパク質が単独でグルタチオンを分解する活性を示す場合だけでなく、当該タンパク質が組み込まれた酵素複合体がグルタチオンを分解する活性を示す場合を含む。DUG複合体によるグルタチオンの分解には、Dug1p、Dug2p、およびDug3pの全てが必要であることが知られており(Ganguli D. et al., Genetics.
2007 Mar; 175(3): 1137-51)、Dug1p、Dug2p、およびDug3pから選択される1またはそれ以上のタンパク質の活性を低下させることでグルタチオン分解酵素活性を低下させることができる。これらの内、少なくともDug2pの活性を低下させるのが好ましい。
【0038】
サッカロミセス・セレビシエのDUG1遺伝子、DUG2遺伝子、およびDUG3遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのDUG1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号1および2に示す。また、同様に、DUG2遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号3および4に示す。また、同様に、DUG3遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号5および6に示す。
【0039】
酵素が由来する生物によってグルタチオン分解酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、グルタチオン分解酵素をコードする遺伝子は、グルタチオンを分解する活性を有するタンパク質をコードする限り、配列番号1、3、または5に示す塩基配列のバリアントであってもよい。なお、各遺伝子のバリアントには、各遺伝子の
ホモログが含まれる。各遺伝子のホモログは、配列番号1、3、または5に示す塩基配列を問い合わせ配列として用いたBLAST検索(http://blast.genome.jp/)やFASTA検索によって公開データベースから容易に取得することができる。また、各遺伝子のホモログとしては、任意の生物、例えば酵母等の真核微生物、あるいは腸内細菌やコリネ型細菌等の細菌の染色体を鋳型にして、例えば配列番号1、3、または5の塩基配列に基づいて調製される合成オリゴヌクレオチドを用いてPCRで増幅可能な遺伝子が挙げられる。
【0040】
また、本発明に用いられる酵素をコードする遺伝子は、グルタチオンを分解する活性を有するタンパク質をコードする限り、上記のような遺伝子にコードされるタンパク質のアミノ酸配列、例えば配列番号2、4、または6のアミノ酸配列において、1若しくは数個の位置での1若しくは数個のアミノ酸の置換、欠失、挿入または付加等を含む配列を有するタンパク質をコードする遺伝子であってもよい。前記「1若しくは数個」とは、アミノ酸残基のタンパク質の立体構造における位置やアミノ酸残基の種類によっても異なるが、具体的には好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個を意味する。上記の1若しくは数個のアミノ酸の置換、欠失、挿入、または付加は、タンパク質の機能が正常に維持される保存的変異である。保存的変異の代表的なものは、保存的置換である。保存的置換とは、例えば、置換部位が芳香族アミノ酸である場合には、Phe、Trp、Tyr間で、置換部位が疎水性アミノ酸である場合には、Leu、Ile、Val間で、極性アミノ酸である場合には、Gln、Asn間で、塩基性アミノ酸である場合には、Lys、Arg、His間で、酸性アミノ酸である場合には、Asp、Glu間で、ヒドロキシル基を持つアミノ酸である場合には、Ser、Thr間でお互いに置換する変異である。保存的置換とみなされる置換としては、具体的には、AlaからSer又はThrへの置換、ArgからGln、His又はLysへの置換、AsnからGlu、Gln、Lys、His又はAspへの置換、AspからAsn、Glu又はGlnへの置換、CysからSer又はAlaへの置換、GlnからAsn、Glu、Lys、His、Asp又はArgへの置換、GluからGly、Asn、Gln、Lys又はAspへの置換、GlyからProへの置換、HisからAsn、Lys、Gln、Arg又はTyrへの置換、IleからLeu、Met、Val又はPheへの置換、LeuからIle、Met、Val又はPheへの置換、LysからAsn、Glu、Gln、His又はArgへの置換、MetからIle、Leu、Val又はPheへの置換、PheからTrp、Tyr、Met、Ile又はLeuへの置換、SerからThr又はAlaへの置換、ThrからSer又はAlaへの置換、TrpからPhe又はTyrへの置換、TyrからHis、Phe又はTrpへの置換、及び、ValからMet、Ile又はLeuへの置換が挙げられる。また、上記のようなアミノ酸の置換、欠失、挿入、付加、または逆位等には、遺伝子が由来する微生物の個体差、種の違いに基づく場合などの天然に生じる変異(mutant又はvariant)によって生じるものも含まれる。
【0041】
さらに、上記のような保存的変異を有する遺伝子は、上記のような遺伝子にコードされるタンパク質のアミノ酸配列全体、例えば配列番号2、4、または6のアミノ酸配列全体に対して、80%以上、好ましくは90%以上、より好ましくは95%以上、より好ましくは97%以上、特に好ましくは99%以上の相同性を有し、かつ、グルタチオンを分解する活性を有するタンパク質をコードする遺伝子であってもよい。尚、本明細書において、「相同性」(homology)は、「同一性」(identity)を指すことがある。
【0042】
また、本発明に用いられる酵素をコードする遺伝子は、公知の遺伝子配列から調製され得るプローブ、例えば配列番号1、3、または5に示す塩基配列の全体または一部に対する相補配列とストリンジェントな条件下でハイブリダイズし、グルタチオンを分解する活性を有するタンパク質をコードするDNAであってもよい。ここで、「ストリンジェントな条件」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。一例を示せば、相同性が高いDNA同士、例えば80%以上、好ましくは90%以上、より好ましくは95%以上、より好ましくは97%以上、特に好ましくは99%以上の相同性を有するDNA同士がハイブリダイズし、それより相同性が低いDNA同士がハイブリダイズしない条件、あるいは通常のサザンハイブリダイゼーショ
ンの洗いの条件である60℃、1×SSC、0.1% SDS、好ましくは、60℃、0.1×SSC、0.1% SDS、さらに好ましくは、68℃、0.1×SSC、0.1% SDSに相当する塩濃度および温度で、1回、より好ましくは2〜3回洗浄する条件が挙げられる。
【0043】
上述の通り、プローブは、遺伝子の相補配列の一部であってもよい。そのようなプローブは、公知の遺伝子配列に基づいて作製したオリゴヌクレオチドをプライマーとし、これらの塩基配列を含むDNA断片を鋳型とするPCRによって作製することができる。例えば、300bp程度の長さのDNA断片をプローブとして用いる場合には、ハイブリダイゼーションの洗いの条件としては、50℃、2×SSC、0.1% SDSが挙げられる。
【0044】
なお、これらのバリアントに関する記載は、本明細書中の他の遺伝子およびタンパク質についても準用されるものとする。
【0045】
また、グルタチオン分解酵素としては、液胞局在グルタチオン分解酵素が挙げられる。本発明において、液胞局在グルタチオン分解酵素とは、液胞に局在し、且つ、グルタチオンを分解する活性を有するタンパク質をいう。液胞局在グルタチオン分解酵素としては、ECM38遺伝子にコードされるEcm38pが挙げられる。Ecm38pは、具体的にはγ−グルタミルトランスペプチダーゼ(γ−GT)活性を有する。
【0046】
サッカロミセス・セレビシエのECM38遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのECM38遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号7および8に示す。
【0047】
酵素が由来する生物によって液胞局在グルタチオン分解酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、液胞局在グルタチオン分解酵素をコードする遺伝子は、液胞に局在し、グルタチオンを分解する活性を有するタンパク質をコードする限り、配列番号7に示す塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0048】
また、トリペプチドであるグルタチオンの分解には、上記のグルタチオン分解酵素の他に、LAP4遺伝子にコードされるLap4pのような各種ペプチダーゼが関与していると考えられる(非特許文献3および7)。したがって、本発明の酵母においては、そのようなペプチダーゼの活性を低下させることも有効であり得る。
【0049】
(1−3)γ−グルタミル化合物合成能の強化
本発明の酵母は、γ−グルタミル化合物合成能が増大するよう改変されている。γ−グルタミル化合物としては、γ−グルタミル基を有する化合物であれば特に制限されないが、例えば、γ‐グルタミルジペプチドやγ‐グルタミルトリペプチド等のγ−グルタミルぺプチドが挙げられる。γ‐グルタミルジペプチドとして、具体的には、例えば、γ−グルタミルシステイン(γ−Glu−Cys)やγ−グルタミル−α−アミノ酪酸(γ−Glu−Abu)が挙げられる。γ‐グルタミルトリペプチドとして、具体的には、例えば、グルタチオン(γ−Glu−Cys−Gly)やγ−グルタミル−α−アミノ酪酸−グリシン(γ−Glu−Abu−Gly)が挙げられる。なお、本発明において、γ−グルタミル化合物を構成するアミノ酸および/またはアミノ酸誘導体はいずれもL体である。
【0050】
「γ−グルタミル化合物合成能が増大する」とは、グルタチオン等のγ−グルタミル化合物の生合成が、野性株や親株等の非改変株と比較して向上していることを意味する。γ
−グルタミル化合物合成能が増大するような改変は、例えば、γ−グルタミル化合物の生合成に関わる1またはそれ以上の酵素の活性を増大させることにより達成できる。生体内では、γ−グルタミルシステイン合成酵素によりL−GluとL−Cysからγ−グルタミルシステイン(γ−Glu−Cys)が生成し、更にグルタチオン合成酵素によりL−Glyが結合してグルタチオンが生成する。よって、グルタチオン合成能が増大するような改変は、例えばγ−グルタミルシステイン合成酵素および/またはグルタチオン合成酵素の活性の増大により達成できる。また、γ−グルタミルシステイン合成能が増大するような改変は、例えばγ−グルタミルシステイン合成酵素の活性の増大により達成できる。また、γ−グルタミルシステイン合成能が増大するような改変は、グルタチオン合成酵素の活性を低下させることによっても達成できる。
【0051】
また、γ−グルタミルシステインやグルタチオン以外のγ‐グルタミルペプチドは、γ−グルタミルシステイン合成酵素および/またはグルタチオン合成酵素の副反応により生合成されうる。よって、γ‐グルタミルジペプチド合成能が増大するような改変は、例えば、γ−グルタミルシステイン合成能が増大するような改変と同様に達成できる。また、γ‐グルタミルトリペプチド合成能が増大するような改変は、例えば、グルタチオン合成能が増大するような改変と同様に達成できる。
【0052】
「酵素活性が増大する」とは、目的の酵素活性が野性株や親株等の非改変株と比較して増大していることを意味する。また、「酵素活性が増大する」とは、もともと目的の酵素活性を有する菌株において目的の酵素活性を増大させることだけでなく、もともと目的の酵素活性を有さない菌株に目的の酵素活性を付与することを含む。また、結果としてグルタチオン合成能が強化される限り、酵母が本来有するグルタチオン生合成系を弱化させた上で、好適なグルタチオン生合成系を導入してもよい。
【0053】
γ−グルタミルシステイン合成酵素活性が増大する場合には、γ−グルタミルシステイン合成酵素活性は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0054】
グルタチオン合成酵素活性が増大する場合には、グルタチオン合成酵素活性は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0055】
酵素活性が増大するような改変は、例えば、目的の酵素をコードする遺伝子の発現を増強することにより達成できる。
【0056】
遺伝子の発現の増強は、例えば、染色体上の遺伝子のプロモーターをより強力なプロモーターに置換することにより達成できる。「より強力なプロモーター」とは、遺伝子の転写が、もともと存在している野生型のプロモーターよりも向上するプロモーターを意味する。より強力なプロモーターとしては、各種レポーター遺伝子を用いることにより、在来のプロモーターの高活性型のものを取得してもよい。また、より強力なプロモーターとしては、公知の高発現プロモーター、例えば、PGK1、PDC1、TDH3、TEF1、HXT7、ADH1等の遺伝子のプロモーターを用いてもよい。なお、強力なプロモーターへの置換は、後述する遺伝子のコピー数の増加と組み合わせて利用できる。より強力なプロモーターを利用した例としては、染色体上のγ−グルタミルシステイン合成酵素遺伝子のプロモーターを強転写プロモーターで置換することによりγ−グルタミルシステイン合成酵素活性を増強する方法が開示されている(大竹康之ら、バイオサイエンスとインダストリー、第50巻第10号、第989〜994頁、1992年)。
【0057】
また、遺伝子の発現の増強は、例えば、遺伝子のコピー数を増加させることにより達成
できる。
【0058】
遺伝子のコピー数の増加は、染色体上に目的の遺伝子を導入することにより達成できる。染色体上への遺伝子の導入は、例えば相同的組み換えを利用して行うことができる。例えば、染色体中に多数のコピーが存在する配列を標的として相同的組み換えを行うことで、染色体へ遺伝子の多数のコピーを導入することができる。染色体中に多数のコピーが存在する配列としては、特有の短い繰り返し配列からなる自律複製配列(ARS)や、約150コピー存在するrDNA配列が挙げられる。ARSを含むプラスミドを用いて酵母の形質転換を行った例が、国際公開95/32289号パンフレットに記載されている。また、トランスポゾンに遺伝子を組み込み、それを染色体へ遺伝子の多数のコピーを導入するよう転移させてもよい。
【0059】
また、遺伝子のコピー数の増加は、目的遺伝子を含むベクターを宿主に導入することによっても達成できる。ベクターとしては、例えば、CEN4の複製開始点を持つプラスミドや2μm DNAの複製開始点を持つ多コピー型プラスミドを好適に用いることができる。目的遺伝子は、好適なプロモーターと組み合わせてベクターに挿入し、目的遺伝子を発現させてもよい。また、遺伝子を発現させるのに好適なプロモーターを含むベクターを用いる場合には、ベクター中のプロモーターを利用して目的遺伝子を発現させてもよい。
【0060】
γ−グルタミルシステイン合成酵素の活性の増強は、例えば米国特許第7553638号、大竹康之ら(バイオサイエンスとインダストリー、第50巻第10号、第989〜994頁、1992年)等に開示されている。その他の酵素活性も、γ−グルタミルシステイン合成酵素の活性と同様に増強させることができる。
【0061】
また、酵素活性が増大するような改変は、例えば、目的の酵素の比活性を増強することによっても達成できる。比活性が増強された酵素は、例えば、種々の生物を探索し取得することができる。また、在来の酵素に変異を導入することで高活性型のものを取得してもよい。比活性の増強は、単独で用いてもよく、上記のような遺伝子の発現を増強させる手法と任意に組み合わせて用いてもよい。
【0062】
目的の酵素活性が増大したことの確認は、同酵素の活性を測定することによって行うことが出来る。γ−グルタミルシステイン合成酵素活性は、Jacksonの方法(Jackson, R. C., Biochem.J., 111, 309 (1969))によって測定することができる。グルタチオン合成酵素活性は、Gushimaらの方法(Gushima, T. et al., J. Appl. Biochem., 5, 210 (1983))によって測定することができる。
【0063】
目的の酵素をコードする遺伝子の転写量が増大したことの確認は、同遺伝子から転写されるmRNAの量を非改変株と比較することによって行うことが出来る。mRNAの量を評価する方法としては、ノーザンハイブリダイゼーション、RT-PCR等が挙げられる(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。mRNAの量は、非改変株と比較して、例えば、1.5倍以上、2倍以上、または3倍以上に増大しているのが好ましい。
【0064】
目的の酵素の量が増大したことの確認は、抗体を用いてウェスタンブロットによって行うことが出来る(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。目的の酵素の量は、非改変株と比較して、例えば、1.5倍以上、2倍以上、または3倍以上に増大しているのが好ましい。
【0065】
本発明の酵母が2倍体以上の倍数性を有する場合であって、染色体の改変により酵素活性が増大している場合には、本発明の酵母は、グルタチオンを蓄積できる限り、酵素活性
が増大するように改変された染色体と野生型染色体とをヘテロで有していてもよく、酵素活性が増大するように改変された染色体のホモ型であってもよい。
【0066】
また、グルタチオン合成酵素活性が低下する場合には、グルタチオン合成酵素活性は、非改変株と比較して、好ましくは50%以下、より好ましくは20%以下、さらに好ましくは10%以下、特に好ましくは5%以下に低下している。また、グルタチオン合成酵素活性が低下する場合には、グルタチオン分解酵素活性は、実質的に消失しているのが好ましい。グルタチオン合成酵素活性が低下するような改変は、上述した、グルタチオン分解酵素活性を低下させるのと同様の手法により達成できる。
【0067】
本発明において、γ−グルタミルシステイン合成酵素は、L−GluおよびL−Cysを基質として認識し、γ−グルタミルシステイン(γ−Glu−Cys)を生成する反応を触媒する活性を有する限り特に限定されない。また、γ−グルタミルシステイン合成酵素は、正反応であるγ−Glu−Cysを生成する反応以外に、任意のL−アミノ酸またはアミノ酸誘導体Xを基質として認識し、L−Gluと結合させることでγ‐Glu‐Xを生成する反応を触媒する活性を有していてもよい。γ−グルタミルシステイン合成酵素としては、GSH1遺伝子によりコードされるGsh1pが挙げられる。
【0068】
本発明において、グルタチオン合成酵素は、γ−グルタミルシステインおよびGlyを基質として認識し、グルタチオン(γ−Glu−Cys−Gly)を生成する反応を触媒する活性を有する限り特に限定されない。グルタチオン合成酵素としては、GSH2遺伝子によりコードされるGsh2pが挙げられる。
【0069】
サッカロミセス・セレビシエのGSH1遺伝子およびGSH2遺伝子の塩基配列は、それぞれSaccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのGSH1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号9および10に示す。また、上記データベースより取得したサッカロミセス・セレビシエのGSH2遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号11および12に示す。
【0070】
酵素が由来する生物によってグルタチオン合成酵素またはγ−グルタミルシステイン合成酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、それらをコードする遺伝子は、グルタチオン合成酵素活性またはγ−グルタミルシステイン合成酵素活性を有するタンパク質をコードする限り、配列番号9または11に示す塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0071】
(1−4)液胞グルタチオントランスポーターの発現強化
本発明の酵母は、一態様において、液胞グルタチオントランスポーターの発現が増大するよう改変されている。
【0072】
「液胞グルタチオントランスポーターの発現が増大する」とは、液胞グルタチオントランスポーターをコードする遺伝子の発現が、野性株や親株等の非改変株と比較して増大していることを意味する。
【0073】
液胞グルタチオントランスポーターの発現を増大させる場合には、液胞グルタチオントランスポーターの発現は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0074】
また、「液胞グルタチオントランスポーターの発現が増大する」とは、もともと液胞グ
ルタチオントランスポーターが発現している菌株において発現を増大させることだけでなく、もともと液胞グルタチオントランスポーターが発現していない菌株において、液胞グルタチオントランスポーターを発現させることを含む。すなわち、例えば、液胞グルタチオントランスポーターをコードする遺伝子を保持しない菌株に液胞グルタチオントランスポーターをコードする遺伝子を導入し、液胞グルタチオントランスポーターを発現させることを含む。
【0075】
液胞グルタチオントランスポーターをコードする遺伝子の発現を増大させることは、上述した、γ−グルタミルシステイン合成酵素をコードする遺伝子やグルタチオン合成酵素をコードする遺伝子の発現を増大させるのと同様の手法により達成できる。
【0076】
本発明において、液胞グルタチオントランスポーターとは、細胞質のグルタチオンを液胞に輸送する機能を有するタンパク質を意味し、当該機能を有する限り特に限定されない。液胞グルタチオントランスポーターをコードする遺伝子としては、YCF1遺伝子およびBPT1遺伝子が挙げられる。中でも、YCF1遺伝子の発現を増大させるのが好ましい。また、液胞へのグルタチオンの輸送には、VBA1遺伝子にコードされるVba1pのような各種アミノ酸・ペプチドトランスポーターが関与していると考えられ、本発明において、液胞グルタチオントランスポーターにはそのような各種アミノ酸・ペプチドトランスポーターが含まれるものとする。
【0077】
サッカロミセス・セレビシエのYCF1遺伝子、BPT1遺伝子、およびVBA1遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。また、上記データベースより取得したサッカロミセス・セレビシエのYCF1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号13および14に示す。
【0078】
酵素が由来する生物によって液胞グルタチオントランスポーターをコードする遺伝子の塩基配列に差異が存在することがあるため、当該遺伝子は、細胞質のグルタチオンを液胞に輸送する機能を有するタンパク質をコードする限り、配列番号13に示す塩基配列のバリアント、またはSaccharomyces Genome Databaseに開示される液胞グルタチオントランスポーターをコードする遺伝子の塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0079】
(1−5)その他の改変
また、本発明の酵母は、グルタチオン等のγ−グルタミル化合物を蓄積できる限りにおいて、上記のような改変に加えて、その他の任意の改変がなされていてもよい。そのような改変としては、細胞内のγ−グルタミル化合物濃度が高まるような改変が挙げられる。例えば、酵母におけるMET25遺伝子の発現量を増大させると菌体内グルタチオン含有量が上昇することが報告されている。MET25遺伝子の発現量を増大させる方法としては、変異型MET4遺伝子(大村ら, FEBS Letters 387 (1996) 179-183、クリスet. al., Molecular Biology of the Cell., 8, 1699-1707, 1997))、変異型MET30遺伝子(DOMINIQUE et. al., MOLECULAR AND CELLUAR BIOLOGY, Dec 1995 p6526-6534、特開2004-113155)を利用する方法等が報告されている。また、アミノ基転移酵素の活性が増大していてもよい。酵母のアミノ基転移酵素として、アラニン:グリオキシル酸アミノトランフェラーゼ(Alanine:Glyoxylate aminotransferase)、分岐鎖アミノ酸トランスアミナーゼ(Branched-chain Amino acid transaminase)、アスパラギン酸アミノトランスフェラーゼ(Aspartate amino transferase)、γ−アミノ酪酸トランスアミナーゼ(Gamma-aminobutyrate transaminase)などを例示することができる。サッカロマイセス・セレビシエでは既にこれらの酵素をコードする遺伝子が特定されており、各々AGX1(システマティックネーム:YFL030W)、BAT1(システマティックネーム:YHR208W)およびBAT2(システマティックネーム:YJR148W)、AAT1(システマティックネーム:YKL106W)およびAAT2(システマティックネーム:YLR027C)、ならびにUGA1(システマティックネーム:YGR019W)にコードされている。特に、BAT1にコードされる分岐鎖アミノ酸トランスアミナーゼの活性を増大させることにより有意に細胞内のα−アミノ酪酸合成を促進することができ、細胞内でのγ-Glu-Abu(L-γ-グルタミル-L-2-アミノ酪酸)やγ-Glu-Abu-Gly(L-γ-グルタミル-L-2-アミノ酪酸-グリシン)蓄積量を増加させることができうる。アミノ基転移酵素の活性を増大させることは、上述した、γ−グルタミルシステイン合成酵素活性やグルタチオン合成酵素活性を増大させるのと同様の手法により達成できる。なお、上記各遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。
【0080】
上記のような改変、すなわちγ−グルタミル化合物合成能の増大、グルタチオン分解酵素活性の低下、液胞グルタチオントランスポーターの発現の増大、および、その他の任意の改変は、いずれの改変を先に行ってもよい。
【0081】
(2)本発明の酵母エキス
次に、酵母エキスを製造する方法を説明する。本発明の酵母エキスは、本発明の酵母を原料として用いる以外は、通常の酵母エキスと同様に製造することができる。
【0082】
まず、本発明の酵母を培地で培養する。培地は、酵母が増殖し得るものであれば特に制限されず、通常工業的に用いられる培地を利用することができる。例えば、実施例に記載したYPD培地を好適に利用することができる。
【0083】
また、グルタチオン等のγ−グルタミル化合物の原料となる化合物(以下、γ−グルタミル化合物の原料ともいう)を培地に添加してもよい。γ−グルタミル化合物の原料となる化合物とは、例えば、γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、および/またはペプチドをいう。γ−グルタミル化合物の原料を添加する場合、その添加量は、γ−グルタミル化合物の種類や、本発明の酵母が有する当該原料の生合成能等の諸条件によって適宜設定すればよいが、例えば、培養開始時の培地における終濃度として、好ましくは1ppm以上、より好ましくは10ppm以上、さらに好ましくは100ppm以上であってよい。添加量の上限は特に制限されないが、例えば費用の観点から、培養開始時の培地における終濃度として、100,000ppm以下に設定してもよい。添加量は、例えば、好ましくは10,000ppm以下、より好ましくは1,000ppm以下、さらに好ましくは500ppm以下であってよい。また、培養中に、間欠的あるいは連続的にγ−グルタミル化合物の原料を添加する場合には、添加量の累計が、上述の培養開始時の培地における終濃度と同等になるように設定すればよい。添加するγ−グルタミル化合物の原料は、1種またはそれ以上であってよい。添加するγ−グルタミル化合物の原料は、精製品(純品)であってもよく、当該原料を含む組成物であってもよい。当該原料を含む組成物は、必要量の当該原料を含む限り特に制限されない。
【0084】
培養条件は、通常の酵母エキスの製造と同様の条件を採用することができ、用いる酵母に応じて適宜変更することができる。バッチ培養、フェドバッチ培養、連続培養など任意の方法を使用することができる。サッカロミセス・セレビシエの場合は、25〜35℃で、より好ましくは、27〜33℃で、更に好ましくは28〜32℃で振とう培養等により好気的に培養することが好ましい。
【0085】
上記のようにして本発明の酵母を培養すると、酵母の細胞内にγ−グルタミル化合物が蓄積する。
【0086】
得られた酵母からの酵母エキスの調製は、通常の酵母エキスの調製と同様にして行えば
よい。酵母エキスは、酵母菌体を熱水抽出したものを処理したものでもよいし、酵母菌体を消化したものを処理したものでもよい。また、必要に応じて得られた酵母エキスを濃縮してもよいし、乾燥し粉末の形態にしてもよい。
【0087】
上記のようにして、γ−グルタミル化合物の含有量が高められた酵母エキスが得られる。好適な形態では、酵母エキスは、酵母エキス中の固形分全量に対し、γ−グルタミル化合物を乾燥重量として0.5重量%以上、より好ましくは1重量%以上、さらに好ましくは5重量%以上、特に好ましくは10重量%以上含有する。
【0088】
(3)本発明の飲食品
上記のようにして得られるγ−グルタミル化合物の含有量が高められた酵母エキスは、飲食品の製造に用いることができる。飲食品としては、例えば、アルコール飲料、パン食品、発酵食品調味料が挙げられる。
【0089】
本発明の飲食品は、本発明の酵母エキスを飲食品の原料に添加し、飲食品に加工することによって製造される。なお、酵母エキスの添加は、飲食品の製造工程のいずれの段階で行われてもよい。本発明の飲食品は、前記酵母エキスを使用すること以外は、通常の飲食品と同様の原料を用い、同様の方法によって製造することができる。このような原料としては、例えばアルコール飲料では、米、大麦、コーンスターチ等、パン食品では小麦粉、砂糖、食塩、バター、発酵用酵母菌等が、発酵食品調味料では大豆、小麦等が挙げられる。また、酵母エキスもしくはその濃縮物、またはそれらを乾燥したものは、それ自体で発酵食品調味料として用いることができる。
【実施例】
【0090】
実施例1.グルタチオン分解酵素活性低下の効果の検討
本実施例では、酵母のグルタチオン分解酵素活性を低下させ、グルタチオン蓄積能および生育速度に与える影響を検証した。
【0091】
(1)γ−グルタミルシステイン合成酵素遺伝子高発現酵母(AG1株)の作製
γ−グルタミルシステイン合成酵素をコードするGSH1遺伝子の高発現株では、細胞内グルタチオン濃度が上昇することが知られている(非特許文献10および11)。そこで、サッカロミセス・セレビシエY006株(FERM BP−11299)のGSH1遺伝子のプロモーター領域を、構成発現プロモーターであるアルコールデヒドロゲナーゼ遺伝子(以下、ADH1)のプロモーター領域に置換することにより、GSH1高発現酵母(AG1株)を作製した。手順を以下に示す。なお、Y006株はURA3遺伝子の欠損株であり、ウラシル要求性を示す。
【0092】
(1−1)ADH1プロモーター置換の為の鋳型プラスミドの作成
まず、配列番号15及び16のプライマーを用い、NBRCより分譲されたサッカロミセス・セレビシエS288C株の染色体DNAを鋳型とするPCRにより、URA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をエタノール沈殿により精製後、SphI及びEcoRIで消化し、プラスミドpUC19のSphI-EcoRI部位に挿入し、pUC19-URA3を得た。次に、配列番号17及び18のプライマーを用い、Y006株の染色体DNAを鋳型とするPCRにより、ADH1プロモーター領域(pADH1)を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をPstIで消化し、PstIで消化しCIAP処理したpUC19-URA3のPstI部位に挿入し、pUC19-pADH1-URA3を得た。同様に、配列番号19及び20のプライマーを用いて増幅したADH1プロモーター領域をAatIIで消化し、AatIIで消化しCIAP処理したpUC19-pADH1-URA3のAatII部位に挿入し、pUC19-pADH1-URA3-ADH1pを得た。
【0093】
(1−2)染色体上のGSH1遺伝子へのADH1プロモーターの導入
5'端にGSH1の上流配列をもつ配列番号21のプライマー、及びGSH1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号22のプライマーを用い、pUC19-pADH1-URA3-pADH1を鋳型にPCRを行い、ADH1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でY006株を形質転換し、ウラシルを含有しないSD平板培地に塗布した。生育した形質転換体から、GSH1プロモーターがpADH1-URA3-pADH1に置換された株を取得した。
【0094】
<SD培地組成>
グルコース 2%
Nitrogen Base 1倍濃度
(10倍濃度Nitrogen Baseは、1.7gのBacto Yeast Nitrogen Base w/o Amino Acids and
Ammonium Sulfate (Difco社)と5gの硫酸アンモニウムを混合したものを100mlの滅菌水に溶解し、pHを5.2程度に調整し、フィルター濾過滅菌したもの)
【0095】
(1−3)URA3選択マーカーの除去とGSH1遺伝子のプロモーター置換
URA3遺伝子が欠損した細胞は5−フルオロオロト酸(5-FOA)耐性を示すため、5-FOAを含有する培地を利用してURA3選択マーカーが除去された株を選抜できる。そこで、GSH1プロモーターがpADH1-URA3-pADH1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのADH1プロモーター間の相同組換えにより、URA3が除去され、GSH1プロモーターがADH1プロモーターに置換された株(AG1 ura3- 株)を取得した。
【0096】
(1−4)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、3min)、25 cycleとした。次にAG1 ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換されウラシル非要求性となった株を取得し、AG1株とした。また、Y006株を同様にこのDNA断片で形質転換し、ウラシル非要求性となった株を取得し、Y003株とした。
【0097】
(2)Y006株のグルタチオン分解酵素遺伝子破壊株の作製
グルタチオン分解活性低下の効果を検証するため、サッカロミセス・セレビシエY006株を基に、各グルタチオン分解酵素遺伝子を破壊した変異株を以下の手順で作製した。
【0098】
(2−1)Y006株のDUG1破壊株の作製
まず、DUG1の開始コドンより上流80塩基を付加した配列番号25のプライマー及びDUG1の終止コドンより下流80塩基を付加した配列番号26のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug1D株(以下、d1株)を得た。
【0099】
(2−2)Y006株のDUG2破壊株の作製
まず、DUG2の開始コドンより上流80塩基を付加した配列番号27のプライマー及びDUG2の終止コドンより下流80塩基を付加した配列番号28のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug2D株(以下、d2株)を得た。
【0100】
(2−3)Y006株のDUG3破壊株の作製
まず、DUG3の開始コドンより上流80塩基を付加した配列番号29のプライマー及びDUG3の終止コドンより下流80塩基を付加した配列番号30のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug3D株(以下、d3株)を得た。
【0101】
(2−4)Y006株のECM38破壊株の作製
まず、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のecm38D株(以下、e株)を得た。
【0102】
(2−5)dug2D株のECM38破壊株の作製
まず、BY4743を親株とするサッカロミセス・セレビシエ破壊株セット(OpenBiosystems社)のECM38破壊株を鋳型に、配列番号33のプライマー及び配列番号34のプライマーを用い、ECM38を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でdug2△株を形質転換し、終濃度50mg/LのG418を含むYPD平板培地へ塗布した。生育した形質転換体からdug2D ecm38D株(以下、ed株)を得た。
【0103】
(3)グルタチオン分解酵素破壊株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0104】
Y003、AG1、d1、d2、d3、e、およびed株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0105】
結果を図1に示す。野生型株であるY003株、GSH1の高発現株であるAG1株、並びにグルタチオン分解酵素遺伝子破壊株である、d1、d2、d3、eおよびed株の菌体内グルタチオン
濃度はそれぞれ、Y003:0.50%、AG1:0.80%、d1:0.47%、d2:0.49%、d3:0.54%、e:0.53%、ed:0.50% であった。GSH1の高発現株であるAG1株は、既に報告されているように(非特許文献10および11)、野生型株であるY003株と比較してグルタチオン濃度が向上した。一方、グルタチオン分解酵素をコードするECM38または各DUG遺伝子を破壊しても、菌体内グルタチオン濃度は影響を受けなかった。ECM38または各DUG遺伝子の単一破壊株においては、Ecm38pとDUG複合体がお互いの機能を相補するために細胞内グルタチオン濃度が影響を受けないことが予想されたが、これら2つのグルタチオン分解酵素をコードする遺伝子の二重破壊株であるed株においても細胞内グルタチオン濃度の上昇は見られなかった。
【0106】
(4)高グルタチオン蓄積形質を有する株(AG1)由来のグルタチオン分解酵素遺伝子破壊株の作製
次に、高グルタチオン蓄積形質を有する株におけるグルタチオン分解酵素活性の低下がグルタチオン蓄積能に与える影響を評価するため、AG1株を基にグルタチオン分解酵素遺伝子破壊株を作製した。手順を以下に示す。
【0107】
(4−1)AG1株のDUG2破壊株の作製
Y006株のDUG2破壊株(d2株)作製と同様に、DUG2の開始コドンより上流80塩基を付加した配列番号27のプライマー及びDUG2の終止コドンより下流80塩基を付加した配列番号28のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1 dug2D株(以下、AG1-d株)を得た。
【0108】
(4−2)AG1株のECM38破壊株の作製
Y006株のECM38破壊株(e株)作製と同様に、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1 ecm38D株(以下、AG1-e株)を得た。
【0109】
(5)AG1株由来のグルタチオン分解酵素遺伝子破壊株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0110】
Y003、AG1、AG1-d、AG1-e、および、AG1-ed株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0111】
結果を図2に示す。Y003株、AG1株、およびAG1株由来のグルタチオン分解酵素破壊株の菌体内グルタチオン濃度はそれぞれ、Y003:0.6%、AG1:1.0 %、AG1-d:2.5%、AG1-e:1.1%、AG1-ed:2.4%であった。
【0112】
単一破壊では菌体内グルタチオン濃度に影響しなかったDUG複合体の欠損は、GSH1の高発現株において菌体内グルタチオン濃度の上昇に寄与した。また、高グルタチオン蓄積形質を有するAG1はY003と比較して生育遅延が認められるが、AG-dはAG1と比較して菌体内グルタチオン濃度が高いにもかかわらず、対数増殖期における生育遅延が軽減された(図3)。
【0113】
以上の結果より、DUG複合体の欠損は、GSH1高発現株において、グルタチオン分解に寄与することが明らかになった。従って、GSH1の高発現によって、DUG複合体の活性が誘導されることが示唆された。
【0114】
(6)GSH1高発現株におけるDUG複合体構成因子の発現量の解析
GSH1の高発現によってDUG複合体の活性が誘導されるかどうかを、DUG複合体の各構成因子のmRNA量をRT-PCRで定量することにより解析した。
【0115】
Y003および、AG1株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで培養を行った。OD600=5および10の2点で1mLサンプルを分取し、滅菌水で2回リンスした後、ペレットを-80℃で冷凍した。翌日、RNA抽出キット、RNeasy(キアゲン)を用いてトータルRNAを抽出した。抽出したRNAのうち500ngを、PrimeScript RT reagent Kit(TaKaRa)を用い37℃で15分反応させ、cDNAを作製した。この際、RT-PCRのネガティブコントロールとして用いるため、酵素を入れずに反応を行ったサンプルを用意した。
【0116】
続いて、Power SYBR Green(Applied Biosystems)で、反応液を調整した。DUG1の反応には配列番号35および36に示すプライマーを用いた。DUG2の反応には配列番号37および38に示すプライマーを用いた。DUG3の反応には配列番号39および40に示すプライマーを用いた。また、配列番号41および42に示すプライマーでACT1遺伝子を増幅し、内部標準とした。cDNA液は、原液、10倍希釈液、100倍希釈液、1000倍希釈液の4つの希釈率の異なる液を用いた。RT-PCR反応には、7500 Real Time PCR System(Applied Biosystems)を用いた。
【0117】
結果を図4に示した。GSH1高発現株では、培養中期において、DUG1のmRNAがおよそ1.6倍に、DUG3のmRNAがおよそ3倍に上昇した(図4a)。その一方で、定常期では、GSH1高発現による影響は見られなかった(図4b)。
【0118】
以上の結果より、GSH1の高発現株においては、DUG構成因子の発現が誘導されることにより、グルタチオン分解酵素であるDUG複合体の活性が上昇することがわかった。また、上述の通り、Y003、AG1およびAG1-d株の生育曲線の比較より、AG1株では対数増殖期において生育遅延がみられたが、AG1-d株ではそれが軽減された(図3)。定常期に両株において差の見られないDUG遺伝子の発現量が、GSH1高発現株では対数増殖期に上昇していることから、対数増殖期のDUG遺伝子の発現量は生育に影響を及ぼすことがわかった。
【0119】
以上より、GSH1高発現株でDUG複合体の活性を低下させることにより、(1)菌体内グルタチオン濃度が上昇する、(2)菌体内グルタチオン濃度が上昇するにもかかわらず、
生育遅延が軽減するという2つの効果が得られることを明らかにした。
【0120】
実施例2.液胞グルタチオントランスポーター増強の効果の検討
本実施例では、酵母の液胞グルタチオントランスポーターの発現を増強させ、グルタチオン蓄積能および生育速度に与える影響を検証した。
【0121】
(7)グルタチオン液胞トランスポーター遺伝子高発現酵母(PY株)の作製
酵母のグルタチオン液胞トランスポーターをコードする遺伝子としてYCF1遺伝子が知られている。グルタチオン液胞トランスポーター増強の効果を検証するため、サッカロミセス・セレビシエY006株のYCF1遺伝子のプロモーター領域を、構成高発現プロモーターであるPGK1遺伝子のプロモーター領域に置換することにより、YCF1高発現酵母(PY株)を作製した。手順を以下に示す。
【0122】
(7−1)PGK1プロモーター置換の為の鋳型プラスミドの作成
まず、配列番号15及び16のプライマーを用い、サッカロミセス・セレビシエS288C株の染色体DNAを鋳型とするPCRによりURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をエタノール沈殿により精製後、SphI及びEcoRIで消化し、プラスミドpUC19のSphI-EcoRI部位に挿入し、pUC19-URA3を得た。次に、配列番号43及び44に示すプライマーを用い、Y006株の染色体DNAを鋳型とするPCRによりPGK1プロモーター領域(pPGK1)を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をPstIで消化し、PstIで消化しCIAP処理したpUC19-URA3のPstI部位に挿入し、pUC19-pPGK1-URA3を得た。同様に、配列番号45及び46に示すプライマーを用いて増幅したPGK1プロモーター領域をAatIIで消化し、AatIIで消化しCIAP処理したpUC19-pPGK1-URA3のAatII部位に挿入し、pUC19-pPGK1-URA3-pPGK1を得た。
【0123】
(7−2)染色体上のYCF1遺伝子へのPGK1プロモーターの導入
5'端にYCF1の上流配列をもつ配列番号47のプライマー、及びYCF1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号48のプライマーを用い、pUC19-pPGK1-URA3-pPGK1を鋳型にPCRを行い、PGK1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でY006株を形質転換し、ウラシルを含有しないSD平板培地に塗布し得られる形質転換体から、YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を取得した。
【0124】
(7−3)URA3選択マーカーの除去とYCF1遺伝子のプロモーター置換
YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのPGK1プロモーター間の相同組換えにより、URA3が除去され、YCF1プロモーターがPGK1プロモーターに置換した株(PY ura3- 株)を取得した。
(7−4)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。次にPY ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換された株であるPY株を取得した。
【0125】
(8)Y006株由来のグルタチオン液胞トランスポーター増強株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0126】
Y003、AG1、e、d、PY株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0127】
結果を図5に示す。Y003と、その単一変異株であるAG1、e、d、PY株の菌体内グルタチオン濃度はそれぞれ、Y003:0.75%、AG1:1.20%、e:0.70%、d2:0.75%、PY:0.65%であった。グルタチオン液胞トランスポーターであるYCF1の発現強化は細胞内グルタチオン濃度に影響を与えなかった。
【0128】
(9)高グルタチオン蓄積形質を有する株(AG1)由来のグルタチオン液胞トランスポーター増強株の作製
グルタチオン液胞トランスポーターの増強は、単独では細胞内グルタチオン濃度に影響を与えなかった。そこで次に、高グルタチオン蓄積形質を有する株におけるグルタチオン液胞トランスポーター増強の効果を評価するため、AG1株のYCF1遺伝子のプロモーター領域を、構成高発現プロモーターであるPGK1遺伝子のプロモーター領域に置換することにより、YCF1高発現酵母(AG1-PY株)を作製した。さらに、液胞局在グルタチオン分解酵素活性の低下とグルタチオン液胞トランスポーター増強との組み合わせの効果を評価するため、AG1-PY株を基に液胞局在グルタチオン分解酵素をコードするECM38が破壊された株(AG1-PY-e株)を作製した。手順を以下に示す。
【0129】
(9−1)AG1株のYCF1発現強化株の作製
【0130】
(9−1−1)AG1株の染色体上のYCF1遺伝子へのPGK1プロモーターの導入
5'端にYCF1の上流配列をもつ配列番号47のプライマー、及びYCF1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号48のプライマーを用い、pUC19-pPGK1-URA3-pPGK1を鋳型にPCRを行い、PGK1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD平板培地に塗布し得られる形質転換体から、YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を取得した。
【0131】
(9−1−2)URA3選択マーカーの除去とYCF1遺伝子のプロモーター置換
YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのPGK1プロモーター間の相同組換えにより、URA3が除去され、YCF1プロモーターがPGK1プロモーターに置換された株(AG1-PY ura3- 株)を取得した。
【0132】
(9−1−3)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。次にAG1-PY ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換された株であるAG1-PY株を取得した。
【0133】
(9−2)AG1-PY株のECM38破壊株の作製
まず、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、S288C株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1-PY ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1-PY ecm38D株(以下、AG1-PY-e株)を得た。
【0134】
(9−3)AG1-d株のYCF1発現強化株の作製
上記(9−1)でAG1株のYCF1発現強化株を作製したのと同様の手法で、AG1-d ura3- 株から、YCF1プロモーターがPGK1プロモーターに置換されたAG1-d-PY ura3- 株を取得し、さらにura3部位を野生型のURA3に置換してAG1-d-PY株を取得した。
【0135】
(9−4)AG1-d-PY株のECM38破壊株の作製
上記(9−2)でAG1-PY株のECM38破壊株を作製したのと同様の手法で、AG1-d-PY ura3- 株から、ECM38が破壊されたAG1-d-PY ecm38D株(以下、AG1-d-PY-e株)を取得した。
【0136】
(10)AG1由来各種多重変異株のグルタチオン蓄積能および生育の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株を、ジャーファーメンターで培養し、菌体内グルタチオン濃度および生育を測定した。用いた培地の組成は以下の通りである。
【0137】
<培地組成>
I)5xSD培地
D-グルコース 5g
Difco Yeast Nitrogen base w/o Amino Acids and Ammonium Sulfate 8.5 g
硫酸アンモニウム 25 g
水 1 L
【0138】
II)50%グルコース
D-グルコース 500 g
水 1 L
【0139】
Y003、AG1、AG1-d、AG1-d-e、AG1-PY、AG1-PY-e株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで36時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0140】
各株の細胞内グルタチオン濃度を図6に、生育曲線を図7に示す。それぞれの菌株の細胞内グルタチオン濃度は、Y003:0.7%、AG1:1.1%、AG1-d:2.5%、AG1-e:1.2%、AG1- PY:1.3%、AG1- PY-e:4.3%であった。細胞内の高濃度グルタチオンは生育を阻害することが知られているとおり、AG1株では細胞内グルタチオンの上昇に伴い、生育速度の低下が見られた。
【0141】
グルタチオン液胞トランスポーターをコードするYCF1の発現を強化したAG1-PY株では、細胞内グルタチオン濃度の上昇は見られなかったが、生育速度は野生型であるY003株程度まで改善した。これは、生育遅延の原因である高濃度の細胞内グルタチオンが液胞に輸送され、細胞質のグルタチオン濃度が低下したためと考えられる。
【0142】
AG1-PY株においてさらに液胞局在グルタチオン分解酵素をコードするECM38を破壊したAG1-PY-e株では、生育速度は野生型よりもはるかに速くなり、また、細胞内グルタチオン濃度も著しく上昇した。AG1-e株では細胞内グルタチオン濃度の上昇は見られなかったことから、これは、Ycf1pにより液胞に輸送されたグルタチオンが、液胞でEcm38pによって分解されずに蓄積されたほか、細胞質内のグルタチオン濃度が低下したことによってグルタチオン生合成系に対するフィードバック阻害が解消し、グルタチオン生合成能が向上したためと考えられる。
【0143】
以上の結果より、グルタチオン液胞トランスポーターをコードするYCF1を高発現することで、生育遅延やグルタチオン生合成系に対するフィードバック阻害の原因となりうる細胞質内グルタチオンを液胞へ輸送し、さらに液胞局在グルタチオン分解酵素をコードするECM38を破壊し、グルタチオンを液胞に蓄積させることで、生育遅延を回避しつつ、グルタチオン高含有株における細胞内グルタチオン濃度のさらなる上昇を達成することができることが明らかとなった。
【0144】
実施例3.酵母エキスの製造
本実施例では、上記実施例で取得した細胞内のグルタチオン濃度が上昇し、且つ、生育が改善した酵母を原料として用いて酵母エキスを製造した。
【0145】
(11)酵母エキスの製造
上記(10)で得られたAG1-d株及びAG1-PY-e株の培養ブロス約50mlを各々分取し、遠心分離により菌体と培養上清を分離した。菌体を50mlの純水に懸濁し、遠心分離により菌体を分離することにより、菌体を洗浄した。この洗浄操作を再度繰り返した後、菌体濃度が約10g / dLになるように純水に懸濁した。なお、菌体濃度は、吸光度と乾燥菌体重量の相関を調べた予備実験結果より推定した。この懸濁液を70℃10分間加熱し、その後氷上にて急速冷却した。遠心分離により抽出液と菌体残渣を分離した。このようにして、酵母エキス(抽出液)を製造した。AG1-d株からはエキス固形分あたりのGSH含量が約8.2%の酵母エキスが、AG1-PY-e株からはエキス固形分あたりのGSH含量が約13.1%の酵母エキスが得られた。
【0146】
実施例4.グルタチオン以外のγ−グルタミルペプチド生産への応用(1)
本実施例では、γ−グルタミルシステインを例として、グルタチオン以外のγ−グルタミルペプチドの蓄積においても、DUG複合体の活性低下が生育遅延の軽減に寄与し、また、グルタチオン液胞トランスポーターの増強と液胞局在グルタチオン分解酵素活性の低下との組み合わせが効果的であることを示す。γ−グルタミルシステインを蓄積する酵母は、例えば、γ−グルタミル基を有するジペプチドを生成する反応を触媒する酵素をコードするGSH1の発現を強化し、γ−グルタミルシステインとグリシンからグルタチオンを合成する反応を触媒する酵素をコードするGSH2を破壊することにより作製できる。
【0147】
(12)GSH2破壊株の作製
まず、BY4743を親株とするサッカロミセス・セレビシエ破壊株セット(OpenBiosystems社)のGSH2破壊株を鋳型に、配列番号49のプライマー及び配列番号50のプライマーを用い、GSH2を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY003株、AG1株、AG1-d株、AG1-d-PY株およびAG1-d-PY-e株を形質転換し、終濃度50mg/LのG418を含むYPD平板培地へ塗布した。生育した形質転換体からY003株のGSH2破壊株(以下gsh2株)、AG1株のGSH2破壊株(以下AG1-gsh2株)、AG1-d株のGSH2破壊株(以下AG1-d-gsh2)、AG1-d-PY株のGSH2破壊株(以下AG1-d-PY-gsh2株)、およびAG1-d-PY-e株のGSH2破壊株(以下AG1-d-PY-e-gsh2株)を得た。
【0148】
(13)各種GSH2破壊株におけるγ−グルタミルシステイン蓄積濃度と生育の評価
以上のようにして作製したサッカロミセス・セレビシエのGSH2破壊株を、ジャーファーメンターで培養し、菌体内γ−グルタミルシステイン濃度および生育を測定した。用いた培地の組成は以下の通りである。
【0149】
<培地組成>
I)5xSD培地
D-グルコース 5g
Difco Yeast Nitrogen base w/o Amino Acids and Ammonium Sulfate 8.5 g
硫酸アンモニウム 25 g
水 1 L
【0150】
II)50%グルコース
D-グルコース 500 g
水 1 L
【0151】
gsh2株、AG1-gsh2株、およびAG-d-gsh2株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでγ−グルタミルシステイン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。γ−グルタミルシステイン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してγ−グルタミルシステインのチオール基を蛍光ラベルし、HPLC分析によりγ−グルタミルシステイン量を算出した。細胞内γ−グルタミルシステイン濃度は、乾燥菌体重量あたりのγ−グルタミルシステイン量として算出した。
【0152】
生育曲線を図8に、細胞内γ−グルタミルシステイン濃度を図9に示した。GSH2の単一破壊株であるgsh2株と比較して、gsh2株のGSH1発現増強株であるAG1-gsh2株では、細胞内γ−グルタミルシステイン濃度の上昇が認められた。AG1-gsh2株においてさらにグルタチオン分解酵素をコードするDUG2を破壊したAG1-d-gsh2株では、AG1-gsh2株と比較して細胞内γ−グルタミルシステイン濃度の上昇は認められなかった。これは、DUG複合体はもともとγ−グルタミルシステイン分解に全く寄与していないか、ほとんど寄与していないためであると考えられる。しかしながら、このようにDUG2の破壊は細胞内γ−グルタミルシステイン濃度の上昇に寄与しないにもかかわらず(図9)、AG1-d-gsh2株では生育遅延が大幅に軽減された(図8)。
【0153】
以上の結果より、DUG複合体の活性低下によって、グルタチオンを高濃度に蓄積する酵母の生育遅延が軽減されるだけでなく、グルタチオン以外のγ−グルタミルペプチドであるγ−グルタミルシステインを蓄積する酵母においても生育遅延が軽減されることが明らかとなった。従ってDUG複合体の活性を低下させることにより、Gsh1pによって生成される様々なγ−グルタミルペプチドを蓄積する酵母の生育遅延を解消できると考えられる。
【0154】
(14)γ−グルタミルシステインの液胞への蓄積効果
AG1-d-gsh2株、AG-d-PY-gsh2株、およびAG-d-PY-e-gsh2株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでγ−グルタミルシステイン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。γ−グルタミルシステイン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してγ−グルタミルシステインのチオール基を蛍光ラベルし、HPLC分析によりγ−グルタミルシステイン量を算出した。細胞内γ−グルタミルシステイン濃度は、乾燥菌体重量あたりのγ−グルタミルシステイン量として算出した。
【0155】
結果を図10に示した。グルタチオン蓄積株と同様、YCF1の発現を強化し、ECM38を破壊した株であるAG1-d-PY-e-gsh2株では、AG1-d-gsh2株と比較し、細胞内γ−グルタミルシステイン濃度が大幅に上昇した。一方、野生型ECM38を有するAG1-d-PY-gsh2株では、AG1-d-gsh2株と比較し、細胞内γ−グルタミルシステイン濃度に変化が無かったことから、生成したγ−グルタミルシステインは液胞に局在するEcm38pによって分解されると考えられる。従って、AG1-d-PY-e-gsh2株において、YCF1の発現強化により液胞に移送されたγ−グルタミルシステインは、液胞に安定的に蓄積していると考えられる。
【0156】
以上の結果より、YCF1の発現強化およびECM38の活性低下により、グルタチオンだけでなく、γ−グルタミルシステインなどγ−グルタミルペプチドが液胞内で分解されずに蓄積し、結果として細胞内濃度を上昇させることができることがわかった。
【0157】
実施例5.グルタチオン以外のγ−グルタミルペプチド生産への応用(2)
γ−グルタミル基を有するジペプチドを生成する反応を触媒する酵素をコードするGSH1の発現を強化することにより、正反応であるγ−グルタミルシステインの生成に加え、各種γ−グルタミルペプチドが生成する。そこで、本実施例では、グルタチオン以外のγ−グルタミルペプチドの一例として、γ−グルタミル−α−アミノ酪酸(γ-Glu-Abu)の生産と、その蓄積が生育に与える影響を検討した。
【0158】
(15)γ-Glu-Abu蓄積濃度と生育の評価
まず、AG1株及びAG1-d株をSD培地に植菌し、30℃で振とう培養した。次に、これらのオーバーナイト培養液を100ppmのL−α−アミノ酪酸(Abu)を含むSD培地に植菌し、30℃で振とう培養した。対数増殖期に、乾燥菌体重量測定に供する培養液、および、γ−グルタミルぺプチド濃度分析に供する培養液を分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして保存した。その後、乾燥菌体重量を計測するサンプルは、適切な濃度になるように水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて乾燥菌体重量を算出した。一方、γ-Glu-Abu濃度分析に用いるサンプルは、適切な濃度になるように水に懸濁した後、70℃で10分間熱水抽出を行った。この工程にて菌体内に含まれるエキス分を抽出した。次に、遠心操作によりエキスと菌体残渣を分離した。エキスから10kDaの遠心濾過膜(MILLIPORE社:Amicon Ultra - 0.5mL 10K(カタログ番号UFC501096))を用いて細胞デブリを除去し、得られた画分を酵母抽出物として、下記に記載する方法で酵母抽出物中のAbu、γ-Glu-Abu、及びγ-Glu-Abu-Gly含量を測定した。各種化合物の細胞内濃度は、乾燥菌体重量あたりの量として算出した。
【0159】
Abu、γ-Glu-Abu、及びγ-Glu-Abu-Gly含量の測定は、これら化合物を6−アミノキノリル−N−ヒドロキシスクシンイミジルカルバメート(AQC)を用いて蛍光誘導体化し、LC-MS/MSにより検出することより行った。具体的には、適当な濃度に希釈した酵母抽出物2.5μL、又は、1μMのAbu、γ-Glu-Abu、及びγ-Glu-Abu-Glyを含む標準液2.5μLに、MillQ水2.5μL、5μM内部標準物質溶液(内部標準物質は、3-methyl-His-d2(シグマ社)、およびGly-d2(シグマ社)。いずれも安定同位体で標識されている。)5μL、硼酸緩衝液(日本ウォーターズ社製AccQ-Fluor(登録商標)試薬キット付属品)30μLを添加した。これら混合物に、AQC試薬溶液(上記AccQ-Fluor試薬キットの試薬粉末をアセトニトリル1mL中に溶解することにより調製)10μLを添加した。得られた混合物を10分間、55℃で加熱後、0.1%のギ酸水溶液100μLを加え、分析サンプルとした。
【0160】
次に前述のように調製した分析サンプルを、逆相の液体クロマトグラフィーで分離した。分離条件は下記の通りである。
【0161】
(1)HPLC:Agilent 1200シリーズ
(2)分離カラム:Unison UK-Phenyl 内径2.0mm、長さ100mm、粒子径3μm(Imtakt社製)
(3)カラム温度:40℃
(4)移動相A:25mMギ酸水溶液をアンモニア水でpH6.0に調整した水溶液
(5)移動相B:メタノール
(6)流速:0.25mL/min
(7)溶出条件:溶出は、移動相A及び移動相Bの混合液を用いて行った。混合液に対する移動相Bの比率は以下の通り。0分(5%)、0分〜17分(5%〜40%)、17分〜17.1分(40%〜80%)、17.1分〜19分(80%)、19分〜19.1分(80%〜5%)、19.1分〜27分(5%)。
【0162】
その後、前述の分離条件によって溶出されたAbu、γ-Glu-Abu、およびγ-Glu-Abu-Glyの誘導体化物を質量分析計に導入してマスクロマトグラムにより定量を行った。分析条件は下記の通りである。
【0163】
(1)質量分析装置:AB Sciex API3200 QTRAP
(2)検出モード:Selected Ion Monitoring(ポジティブイオンモード)
(3)選択イオン:表1
【0164】
【表1】

【0165】
Abu、γ-Glu-Abu、およびγ-Glu-Abu-Glyの誘導体化物の定量は、解析ソフトAnalyst v
er 1.4.2(AB Sciex)を用いて行った。定量を行うための内部標準物質として、Abuの誘導体化物の場合は3-methyl-His-d3の誘導体化物を、γ-Glu-Abu又はγ-Glu-Abu-Glyの誘導体化物の場合はGly-d2の誘導体化物を、各々用いた。なお、γ-Glu-Abuの定量の際に、極まれにサンプルによって夾雑ピークが見られた場合は、第二のマスアナライザーでの選択イオンとして、145.2、或いは、104.1を用いて定量した。その結果、表2に示すように、AG1株と比較して、AG1-d株は著量のγ-Glu-Abuを蓄積していた。
【0166】
【表2】
【0167】
次に、γ-Glu-Abuの蓄積が生育に与える影響について検討した。AG1株及びAG1-d株をSD培地に植菌し、30℃で振とう培養した。次に、これらのオーバーナイト培養液を100ppmのAbuを含むSD培地に660nmの吸光度が0.01になるように小型L型培養菅(ADVANTEC社型式TV100030)4mlに植菌し、バイオフォトレコーダー(ADVANTEC社型式TVS062CA)を用いて1時間毎に660nmの吸光度を測定し生育を確認した。培養温度は30℃、振とう数は70rpmに設定した。生育曲線を図11に示した。図に示すように、より多くのγ-Glu-Abuを蓄積したAG1-d株は、AG1株とほぼ同等の生育を示した。
【0168】
以上の結果より、DUG複合体の活性低下によって、グルタチオンを高濃度に蓄積する酵母の生育遅延が軽減されるだけでなく、グルタチオン以外のγ−グルタミルペプチドであるγ-Glu-Abuを蓄積する酵母においても生育遅延が軽減されることが明らかとなった。従ってDUG複合体の活性を低下させることにより、Gsh1pによって生成される様々なγ−グルタミルペプチドを蓄積する酵母の生育遅延を解消できると考えられる。
【0169】
本実施例1〜5のまとめとして、DUGを破壊することによる生育遅延の軽減と、YCF1の発現強化およびECM38破壊による液胞への蓄積を利用することにより、高濃度にγ−グルタミルペプチドを蓄積する酵母菌体を効率的に取得することができ、この酵母菌体を用いて高γ−グルタミルペプチド酵母エキスを製造することができることが明らかになった。
【産業上の利用可能性】
【0170】
本発明により、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積し、且つ生育の良い酵母が提供される。それら酵母を原料として用いることにより、γ−グルタミル化合物を含有する酵母エキスを効率的に製造でき、また、それら酵母エキスを利用して飲食品を製造することができる。
【0171】
配列表の説明
配列番号1:サッカロミセス・セレビシエのDUG1の塩基配列
配列番号2:サッカロミセス・セレビシエのDug1pのアミノ酸配列
配列番号3:サッカロミセス・セレビシエのDUG2の塩基配列
配列番号4:サッカロミセス・セレビシエのDug2pのアミノ酸配列
配列番号5:サッカロミセス・セレビシエのDUG3の塩基配列
配列番号6:サッカロミセス・セレビシエのDug3pのアミノ酸配列
配列番号7:サッカロミセス・セレビシエのECM38の塩基配列
配列番号8:サッカロミセス・セレビシエのEcm38pのアミノ酸配列
配列番号9:サッカロミセス・セレビシエのGSH1の塩基配列
配列番号10:サッカロミセス・セレビシエのGsh1pのアミノ酸配列
配列番号11:サッカロミセス・セレビシエのGSH2の塩基配列
配列番号12:サッカロミセス・セレビシエのGsh2pのアミノ酸配列
配列番号13:サッカロミセス・セレビシエのYCF1の塩基配列
配列番号14:サッカロミセス・セレビシエのYcf1pのアミノ酸配列
配列番号15、16:URA3増幅用プライマー
配列番号17〜20:ADH1プロモーター領域増幅用プライマー
配列番号21、22:pADH1-URA3-pADH1断片増幅用プライマー
配列番号23、24:URA3増幅用プライマー
配列番号25、26:DUG1破壊用プライマー
配列番号27、28:DUG2破壊用プライマー
配列番号29、30:DUG3破壊用プライマー
配列番号31、32:ECM38破壊用プライマー
配列番号33、34:BY4743遺伝子破壊株セットを鋳型としたECM38破壊用プライマー
配列番号35、36:RT-PCRに用いるDUG1増幅プライマー
配列番号37、38:RT-PCRに用いるDUG2増幅プライマー
配列番号39、40:RT-PCRに用いるDUG3増幅プライマー
配列番号41、42:RT-PCRに用いるACT1増幅プライマー
配列番号43〜46:PGK1プロモーター領域増幅用プライマー
配列番号47、48:pPGK1-URA3-pPGK1断片増幅用プライマー
配列番号49、50:BY4743遺伝子破壊株セットを鋳型としたGSH2破壊用プライマー
【技術分野】
【0001】
本発明は、γ−グルタミル化合物を含有する酵母エキスの製造方法及び当該方法に用いられる酵母に関するものである。当該酵母エキスや酵母は食品分野で有用である。
【背景技術】
【0002】
γ−グルタミル化合物の1つであるグルタチオン(γ−Glu−Cys−Gly)はすべての生物が有する細胞内に最も多量に存在する非タンパク質性チオールである。グルタチオンは生体内において抗酸化作用、免疫支援、細胞毒の除去など生体維持に必要な様々な機能を有しており、医薬品、化粧品原料として用いられる。また、グルタチオンはコク味を有し、食品添加物としても利用される。そのため、グルタチオンを安価に大量に生産することは重要である。グルタチオンは、例えば、グルタチオンを高生産する出芽酵母を分離して利用することにより、効率よく生産することが可能である(非特許文献1)。しかし、高反応性のチオール基を有するグルタチオンは、生体維持に重要である一方、過剰に蓄積されると有害であり、グルタチオンを高濃度に蓄積した細胞ではしばしば生育遅延が認められる。そのため、効率よくグルタチオンを生産するためには、グルタチオンを細胞内に高濃度に蓄積し、かつ生育が遅延しない出芽酵母の分離が必要である。
【0003】
酵母の細胞内でグルタチオンを分解する酵素としては、ECM38遺伝子にコードされるEcm38pが知られている(非特許文献2、3)。また、酵母の細胞内でグルタチオンを分解する酵素としては、近年DUG複合体が見出された(非特許文献4、5)。しかし、これらのグルタチオン分解系を弱化することにより細胞内のグルタチオン濃度が上昇したという報告は無い。その為、これらグルタチオン分解酵素の活性が低下した酵母を用いて酵母エキスを製造しようとの発想はなかった。またDUG複合体はDug1p、Dug2p、Dug3pからなるが、各DUG因子がグルタチオンの分解にどの程度寄与しているかは明らかになっていない。
【0004】
YCF1遺伝子にコードされるYcf1pは金属とキレートしたグルタチオンを液胞へ輸送するトランスポーターである(非特許文献3および6)。Ycf1pは栄養飢餓時にグルタチオンのみを液胞に輸送すると考えられているが、Ycf1pの機能が細胞内のグルタチオン濃度におよぼす影響は知られていない。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Appl Microbiol Biotechnol. DOI 10.1007/s00253-010-2968-6 (2010)
【非特許文献2】Yeast. 2003 Jul 30;20(10):857-63
【非特許文献3】FEBS Lett. 2009 May 6;583(9):1489-92
【非特許文献4】J Biol Chem. 2009 May 22;284(21):14493-502
【非特許文献5】Genetics. 2007 Mar;175(3):1137-51
【非特許文献6】FEBS Lett. 2010 Feb 19;584(4):726-32
【非特許文献7】Biometals. 2009 Apr;22(2):243-9
【非特許文献8】J Biol Chem. 2005 Feb 11; 280(6): 4851-4857
【非特許文献9】BioMetals (2006) 19:593-599
【非特許文献10】J Biol Chem. 2003 Dec 12;278(50):49920-8
【非特許文献11】Appl Microbiol Biotechnol DOI 10.1007/s00253-010-2946-z (2010)
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、産業上有用なグルタチオン等のγ−グルタミル化合物を高濃度に蓄積する酵母、当該酵母を利用したγ−グルタミル化合物を含有する酵母エキス、および当該酵母エキスを含有する飲食品、並びにそれらの製造方法を提供することを課題とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するために鋭意検討を行った結果、γ−グルタミルシステイン合成酵素をコードするGSH1遺伝子の高発現によってグルタチオン合成能を強化した酵母において、さらにグルタチオン分解酵素であるDUG複合体の活性を低下させることで、細胞内のグルタチオンの蓄積量が増大するにもかかわらず酵母の生育遅延を軽減できるという予想外の知見を見出した。また、本発明者は、液胞膜グルタチオントランスポーターの発現を増強し、且つ、液胞局在グルタチオン分解酵素であるEcm38pの活性を低下させることにより、グルタチオン過剰蓄積による生育遅延およびグルタチオンによるフィードバック阻害を回避し、酵母細胞内のグルタチオン蓄積量が増大することを見出した。さらに、本発明者は、γ−グルタミルシステインを蓄積する酵母においても、DUG複合体の活性低下により酵母の生育が向上すること、並びに、Ecm38pの活性低下および液胞膜グルタチオントランスポーターの発現増強により酵母細胞内のγ−グルタミルシステイン蓄積量が増大することを見出した。以上に基づき、本発明は完成された。
【0008】
すなわち、本発明は以下の通り例示できる。
[1]
γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを調製することを特徴とする、酵母エキスの製造方法。
[2]
前記グルタチオン分解酵素が下記(A)〜(D)から選ばれる1またはそれ以上のタンパク質である、前記方法。
(A)DUG1遺伝子にコードされるタンパク質
(B)DUG2遺伝子にコードされるタンパク質
(C)DUG3遺伝子にコードされるタンパク質
(D)ECM38遺伝子にコードされるタンパク質
[3]
前記酵母は、少なくとも液胞局在グルタチオン分解酵素の活性が低下し、さらに液胞グルタチオントランスポーターの発現が増大するよう改変されている、前記方法。
[4]
γ−グルタミルシステイン合成酵素の活性が増大することによりγ−グルタミル化合物合成能が増大した、前記方法。
[5]
グルタチオン合成酵素の活性が増大または低下することによりγ−グルタミル化合物合成能が増大した、前記方法。
[6]
前記γ−グルタミル化合物が、グルタチオン、γ−グルタミルシステイン、およびγ−グルタミル−α−アミノ酪酸から選択される化合物である、前記方法。
[7]
前記γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、およびペプチドから選択される化合物が添加された培地で前記酵母を培養することを特徴とする、前記方法。[8]
前記酵母がサッカロミセス属に属する酵母である、前記方法。
[9]
前記酵母がサッカロミセス・セレビシエである、前記方法。
[10]
前記方法により製造された酵母エキスを含有する飲食品。
[11]
γ−グルタミルシステイン合成酵素の活性が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母。
[12]
さらに、下記(I)および(II)から選ばれる1またはそれ以上の性質を有する前記酵母。
(I)グルタチオン合成酵素の活性の増大、または低下
(II)液胞グルタチオントランスポーターの発現の増大
【発明の効果】
【0009】
本発明により、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積し、且つ生育の良い酵母が提供される。それら酵母を原料として用いることにより、γ−グルタミル化合物を含有する酵母エキスを効率的に製造でき、また、それら酵母エキスを利用して飲食品を製造することができる。
【図面の簡単な説明】
【0010】
【図1】サッカロミセス・セレビシエY006株のウラシル非要求性株であるY003株、およびY006株に由来するGSH1発現強化株およびグルタチオン分解酵素遺伝子破壊株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006のGSH1発現強化株、d1:Y006のDUG1破壊株、d2:Y006のDUG2破壊株、d3:Y006のDUG3破壊株、e:Y006のECM38破壊株、ed:Y006のDUG2およびECM38二重破壊株。
【図2】Y003株、AG1株、およびAG1株に由来するグルタチオン分解酵素遺伝子破壊株の菌体内グルタチオン濃度を示す図。Y003:Y003株、AG1:YF006のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-ed:AG1株のDUG2およびECM38二重破壊株。
【図3】Y003株、およびY006株に由来する変異株の生育曲線を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、d2:Y006のDUG2破壊株、AG1-d:AG1株のDUG2破壊株。
【図4】DUG複合体構成因子をコードするDUG1、DUG2、およびDUG3のmRNA量を示す図。パネルa:OD600=5(中期対数増殖期)におけるDUG因子のmRNA量。パネルb:OD600=10(定常期)におけるDUG因子のmRNA量。
【図5】Y003株、並びに、Y006株に由来するGSH1発現強化株、グルタチオン分解酵素遺伝子破壊株、およびグルタチオン液胞トランスポーター発現強化株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、e:Y006株のECM38破壊株、d:Y006株のDUG2破壊株、PY:Y006株のYCF1発現強化株。
【図6】Y003株、AG1株、並びに、AG1株に由来するグルタチオン分解酵素遺伝子破壊株、および液胞グルタチオントランスポーター発現強化株の細胞内グルタチオン濃度を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-PY:AG1株のYCF1発現強化株、AG1-PY-e:AG1-PY株のECM38破壊株。
【図7】Y003株、AG1株、並びに、AG1株に由来するグルタチオン分解酵素遺伝子破壊株、および液胞グルタチオントランスポーター発現強化株の生育曲線を示す図。Y003:Y003株、AG1:Y006株のGSH1発現強化株、AG1-d:AG1株のDUG2破壊株、AG1-e:AG1株のECM38破壊株、AG1-PY:AG1株のYCF1発現強化株、AG1-PY-e:AG1-PY株のECM38破壊株。
【図8】γ−グルタミルシステイン蓄積酵母であるAG1-gsh2株およびAG1-d-gsh2株の生育曲線を示す図。AG1-gsh2:AG1株のGSH2破壊株、AG1-d-gsh2:AG1-d株のGSH2破壊株。
【図9】γ−グルタミルシステイン蓄積酵母であるgsh2株、AG1-gsh2株、およびAG1-d-gsh2株の細胞内γ−グルタミルシステイン濃度を示す図。gsh2:Y003株のGSH2破壊株、AG1-gsh2:AG1株のGSH2破壊株、AG1-d-gsh2:AG1-d株のGSH2破壊株。
【図10】γ−グルタミルシステイン蓄積酵母であるAG1-d-gsh2株、AG1-d-PY-gsh2株、およびAG1-d-PY-e-gsh2株の細胞内γ−グルタミルシステイン濃度を示す図。AG1-d-gsh2:AG1-d株のGSH2破壊株、AG1-d-PY-gsh2:AG1-d-PY株のGSH2破壊株、AG1-d-PY-e-gsh2:AG1-d-PY-e株のGSH2破壊株。
【図11】Abu添加培養時のAG1株、および、AG1株に由来するグルタチオン分解酵素遺伝子破壊株の生育曲線を示す図。AG1:AG1株、AG1-d:AG1株のDUG2破壊株。
【発明を実施するための形態】
【0011】
以下、本発明を詳細に説明する。
(1)本発明の酵母
(1−1)本発明の酵母
本発明の酵母は、グルタチオン等のγ−グルタミル化合物の合成能が増大し、且つ、細胞内のグルタチオン分解酵素活性が低下するように改変された、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積する酵母である。本発明の酵母は、細胞内のグルタチオン分解酵素活性が低下することにより、単にγ−グルタミル化合物の合成能が強化されただけの酵母と比較して、酵母の生育が向上するのが好ましい。更には、本発明の酵母は、細胞内のグルタチオン分解酵素活性が低下することにより、単にγ−グルタミル化合物の合成能が強化されただけの酵母と比較して、細胞内のγ−グルタミル化合物の濃度が増大するのが好ましい。本発明の酵母は、乾燥重量で、好ましくは0.5重量%以上、より好ましくは1.5重量%以上、さらに好ましくは3重量%以上、特に好ましくは5重量%以上のグルタチオン等のγ−グルタミル化合物を細胞内に蓄積できる。
【0012】
また、本発明の酵母は、液胞に局在するグルタチオン分解酵素の活性が低下し、且つ、液胞グルタチオントランスポーターの活性が増大するように改変されているのが好ましい。この場合、グルタチオン分解酵素としては、液胞局在グルタチオン分解酵素の活性のみが低下していてもよく、液胞局在グルタチオン分解酵素に加えて他のグルタチオン分解酵素の活性が低下していてもよい。高濃度のグルタチオンは生育遅延やグルタチオン合成系のフィードバック阻害を引き起こすが、液胞グルタチオントランスポーターの活性を増大させ、且つ、液胞局在グルタチオン分解酵素活性を低下させることで、グルタチオンを液胞に移送して細胞質内のグルタチオン濃度を低下させ、液胞にグルタチオンを安定に蓄積できると考えられる。これにより、グルタチオンによる生育遅延やグルタチオン合成系のフィードバック阻害を回避でき、グルタチオン蓄積量を増大させることができると考えられる。
【0013】
本発明の酵母は、酵母の適当な菌株、例えば後述する菌株を改変することで取得することができる。
【0014】
本発明の酵母は、上記のように改変されグルタチオン等のγ−グルタミル化合物を細胞内に蓄積できる限り特に制限されず、出芽酵母であってもよく、分裂酵母であってもよい。出芽酵母としては、サッカロミセス・セレビシエ(Saccharomyces cerevisiae)等のサッカロミセス属、キャンディダ・ユティリス(Candida utilis)等のキャンディダ属、ピヒア・パストリス(Pichia pastoris)等のピヒア属、ハンゼヌラ・ポリモルファ(Hansenula polymorpha)等のハンゼヌラ属等に属する酵母を例示することができる。分裂酵母としては、シゾサッカロミセス・ポンベ(Schizosaccharomyces pombe)等のシゾサッカロミセス属等に属する酵母を例示することができる。中でも、酵母エキスの生産によく用いられているサッカロミセス・セレビシエやキャンディダ・ユティリスが好ましい。本発明の酵母は、1倍体でもよいし、2倍性またはそれ以上の倍数性を有するものであってもよい。
【0015】
グルタチオン生合成系はすべての生物が有しており、また、実施例で用いた液胞グルタチオントランスポーター、およびグルタチオン分解酵素のホモログは、これら種々の酵母に存在することが知られている(非特許文献5および9)。
【0016】
サッカロミセス・セレビシエとしては、具体的には、サッカロミセス・セレビシエY006株を用いることができる。サッカロミセス・セレビシエY006株は、平成22年8月18日に、産業技術総合研究所特許生物寄託センター(住所 郵便番号305−8566 茨城県つくば市東1丁目1番地1中央第6)に国際寄託され、受託番号FERM BP−11299が付与されている。
【0017】
また、サッカロミセス・セレビシエとしては、具体的には、BY4743株(ATCC201390)やS288C株(ATCC26108)を用いることができる。また、キャンディダ・ユティリスとしては、具体的には、キャンディダ・ユティリスATCC22023株を用いることができる。これらを入手するには、例えばアメリカン・タイプ・カルチャー・コレクション(住所 12301 Parklawn Drive, Rockville, Maryland 20852 P.O. Box 1549, Manassas, VA 20108, United States of America)より分譲を受けることが出来る。すなわち各菌株に対応する登録番号が付与されており、この登録番号を利用して分譲を受けることが出来る(http://www.atcc.org/参照)。各菌株に対応する登録番号は、アメリカン・タイプ・カルチャー・コレクションのカタログに記載されている。
【0018】
(1−2)グルタチオン分解酵素活性の低下
本発明の酵母は、グルタチオン分解酵素の活性が低下するように改変されている。
【0019】
「酵素の活性が低下する」とは、目的の酵素活性が野性株や親株等の非改変株と比較して減少していることを意味し、活性が完全に消失している場合を含む。
【0020】
グルタチオン分解酵素活性は、非改変株と比較して、好ましくは50%以下、より好ましくは20%以下、さらに好ましくは10%以下、特に好ましくは5%以下に低下している。また、グルタチオン分解酵素活性は、実質的に消失しているのが好ましい。
【0021】
酵素活性が低下するような改変は、例えば、突然変異処理又は遺伝子組換え技術により達成できる。
【0022】
突然変異処理としては、紫外線照射、または、N-メチル-N'-ニトロ-N-ニトロソグアニジン(MNNG)、エチルメタンスルフォネート(EMS)、メチルメタンスルフォネート(MMS)等の通常変異処理に用いられている変異剤による処理が挙げられる。
【0023】
遺伝子組換え技術としては、例えば、公知の技術(FEMS Microbiology Letters 165 (1998) 335-340、JOURNAL OF BACTERIOLOGY, Dec. 1995, p7171-7177、Curr Genet 1986; 10(8):573-578、WO 98/14600等)を利用できる。
【0024】
酵素活性が低下するような改変は、例えば、目的の酵素をコードする遺伝子の発現を低下させることにより達成できる。遺伝子の発現の低下は、例えば、遺伝子のプロモーター等の発現調節配列を改変することにより達成できる。発現調節配列を改変する場合には、発現調節配列は、好ましくは1塩基以上、より好ましくは2塩基以上、特に好ましくは3塩基以上が改変される。また、発現調節配列の一部または全部を欠失させてもよい。
【0025】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域の一部または全部を欠失させることにより達成できる。さらには、染色体上の遺伝子の前後の配列を含めて、遺伝子全体を欠失させてもよい。酵素活性の低下
が達成できる限り、欠失させる領域は、N末端領域、内部領域、C末端領域等のいずれの領域であってもよい。通常、欠失させる領域は長い方が確実に遺伝子を不活化することができる。また、欠失させる領域の前後の配列は、リーディングフレームが一致しないことが好ましい。
【0026】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域にアミノ酸置換(ミスセンス変異)を導入すること、終始コドンを導入すること(ナンセンス変異)、あるいは1〜2塩基を付加または欠失するフレームシフト変異を導入すること等によっても達成できる。
【0027】
また、酵素活性が低下するような改変は、例えば、染色体上の目的の酵素をコードする遺伝子のコード領域に他の配列を挿入することによっても達成できる。挿入部位は、遺伝子のいずれの領域であってもよいが、挿入する配列は長い方が確実に遺伝子を不活化することができる。また、挿入部位の前後の配列は、リーディングフレームが一致しないことが好ましい。他の配列としては、コードされるタンパク質の機能を低下又は消失させるものであれば特に制限されないが、例えば、マーカー遺伝子やグルタチオン等のγ−グルタミル化合物の生産に有用な遺伝子が挙げられる。
【0028】
染色体上の遺伝子を上記のように改変することは、例えば、遺伝子の部分配列を欠失し、正常に機能するタンパク質を産生しないように改変した欠失型遺伝子を作製し、該欠失型遺伝子を含む組換えDNAで酵母を形質転換して、欠失型遺伝子と染色体上の遺伝子とで相同組換えを起こさせることにより、染色体上の遺伝子を欠失型遺伝子に置換することによって達成できる。その際、組換えDNAには、宿主の栄養要求性等の形質にしたがって、マーカー遺伝子を含ませておくと操作がしやすい。また、前記組換えDNAは、制限酵素で切断する等により直鎖状にしておくと、染色体に組換えDNAが組み込まれた株を効率よく取得することができる。欠失型遺伝子によってコードされるタンパク質は、生成したとしても、野生型タンパク質とは異なる立体構造を有し、機能が低下又は消失する。
【0029】
用いる組換えDNAの構造によっては、相同組換えの結果として、野生型遺伝子と欠失型遺伝子とが組換えDNAの他の部分(例えば、ベクター部分及びマーカー遺伝子)を挟んだ状態で染色体に挿入される場合がある。この状態では野生型遺伝子が機能するため、当該2個の遺伝子間で再度相同組換えを起こさせ、1コピーの遺伝子を、ベクター部分及びマーカー遺伝子とともに染色体DNAから脱落させ、欠失型遺伝子が残ったものを選抜する必要がある。
【0030】
また、例えば、任意の配列を含む線状DNAであって、当該任意の配列の両端に染色体上の置換対象部位の上流および下流の配列を備える線状DNAで酵母を形質転換して、置換対象部位の上流および下流でそれぞれ相同組換えを起こさせることにより、1ステップで置換対象部位を当該任意の配列に置換することができる。当該任意の配列としては、例えば、マーカー遺伝子を含む配列を用いればよい。マーカー遺伝子は、その後、必要により除去してもよい。マーカー遺伝子を除去する場合には、マーカー遺伝子を効率的に除去できるよう、相同組み換え用の配列をマーカー遺伝子の両端に付加しておいてもよい。
【0031】
目的の酵素活性が低下したことの確認は、同酵素の活性を測定することによって行うことが出来る。グルタチオン分解酵素の活性は、公知の手法(Penninckx M. et al., 1980.
Eur J Biochem 104: 119-123、Ganguli D. et al., Genetics. 2007 Mar; 175(3): 1137-51等)により測定することができる。
【0032】
目的の酵素をコードする遺伝子の転写量が低下したことの確認は、同遺伝子から転写されるmRNAの量を非改変株と比較することによって行うことが出来る。mRNAの量を評価する
方法としては、ノーザンハイブリダイゼーション、RT-PCR等が挙げられる(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。mRNAの量は、非改変株と比較して、例えば、50%以下、20%以下、10%以下、5%以下、または0%に低下しているのが好ましい。
【0033】
目的の酵素の量が低下したことの確認は、抗体を用いてウェスタンブロットによって行うことが出来る(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。目的の酵素の量は、非改変株と比較して、例えば、50%以下、20%以下、10%以下、5%以下、または0%に低下しているのが好ましい。
【0034】
本発明の酵母が2倍体以上の倍数性を有する場合には、本発明の酵母は、グルタチオン等のγ−グルタミル化合物を蓄積できる限り、酵素活性が低下するように改変された遺伝子と野生型遺伝子とをヘテロで有していてもよいが、通常は、酵素活性が低下するように改変された遺伝子のホモ型であるのが好ましい。
【0035】
酵母の形質転換法としては、プロトプラスト法、KU法(H.Ito et al., J. Bateriol., 153-163 (1983))、KUR法(発酵と工業 vol.43, p.630-637 (1985))、エレクトロポレーション法(Luis et al., FEMS Micro biology Letters 165 (1998) 335-340)、キャリアDNAを用いる方法(Gietz R.D. and Schiestl R.H., Methods Mol.Cell. Biol. 5:255-269 (1995))等、通常酵母の形質転換に用いられる方法を採用することができる。また、酵母の胞子形成、1倍体酵母の分離、等の操作については、「化学と生物 実験ライン31 酵母の実験技術」、初版、廣川書店;「バイオマニュアルシリーズ10 酵母による遺伝子実験法」初版、羊土社等に記載されている。
【0036】
本発明において、グルタチオン分解酵素とはグルタチオンを分解する活性を有するタンパク質をいう。
【0037】
グルタチオン分解酵素としては、DUG1遺伝子、DUG2遺伝子、およびDUG3遺伝子によりそれぞれコードされるDug1p、Dug2p、およびDug3pが挙げられる。Dug1p、Dug2p、およびDug3pはDUG複合体として機能することが知られている。よって、「グルタチオンを分解する活性を有する」とは、遺伝子にコードされるタンパク質が単独でグルタチオンを分解する活性を示す場合だけでなく、当該タンパク質が組み込まれた酵素複合体がグルタチオンを分解する活性を示す場合を含む。DUG複合体によるグルタチオンの分解には、Dug1p、Dug2p、およびDug3pの全てが必要であることが知られており(Ganguli D. et al., Genetics.
2007 Mar; 175(3): 1137-51)、Dug1p、Dug2p、およびDug3pから選択される1またはそれ以上のタンパク質の活性を低下させることでグルタチオン分解酵素活性を低下させることができる。これらの内、少なくともDug2pの活性を低下させるのが好ましい。
【0038】
サッカロミセス・セレビシエのDUG1遺伝子、DUG2遺伝子、およびDUG3遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのDUG1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号1および2に示す。また、同様に、DUG2遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号3および4に示す。また、同様に、DUG3遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号5および6に示す。
【0039】
酵素が由来する生物によってグルタチオン分解酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、グルタチオン分解酵素をコードする遺伝子は、グルタチオンを分解する活性を有するタンパク質をコードする限り、配列番号1、3、または5に示す塩基配列のバリアントであってもよい。なお、各遺伝子のバリアントには、各遺伝子の
ホモログが含まれる。各遺伝子のホモログは、配列番号1、3、または5に示す塩基配列を問い合わせ配列として用いたBLAST検索(http://blast.genome.jp/)やFASTA検索によって公開データベースから容易に取得することができる。また、各遺伝子のホモログとしては、任意の生物、例えば酵母等の真核微生物、あるいは腸内細菌やコリネ型細菌等の細菌の染色体を鋳型にして、例えば配列番号1、3、または5の塩基配列に基づいて調製される合成オリゴヌクレオチドを用いてPCRで増幅可能な遺伝子が挙げられる。
【0040】
また、本発明に用いられる酵素をコードする遺伝子は、グルタチオンを分解する活性を有するタンパク質をコードする限り、上記のような遺伝子にコードされるタンパク質のアミノ酸配列、例えば配列番号2、4、または6のアミノ酸配列において、1若しくは数個の位置での1若しくは数個のアミノ酸の置換、欠失、挿入または付加等を含む配列を有するタンパク質をコードする遺伝子であってもよい。前記「1若しくは数個」とは、アミノ酸残基のタンパク質の立体構造における位置やアミノ酸残基の種類によっても異なるが、具体的には好ましくは1〜20個、より好ましくは1〜10個、さらに好ましくは1〜5個を意味する。上記の1若しくは数個のアミノ酸の置換、欠失、挿入、または付加は、タンパク質の機能が正常に維持される保存的変異である。保存的変異の代表的なものは、保存的置換である。保存的置換とは、例えば、置換部位が芳香族アミノ酸である場合には、Phe、Trp、Tyr間で、置換部位が疎水性アミノ酸である場合には、Leu、Ile、Val間で、極性アミノ酸である場合には、Gln、Asn間で、塩基性アミノ酸である場合には、Lys、Arg、His間で、酸性アミノ酸である場合には、Asp、Glu間で、ヒドロキシル基を持つアミノ酸である場合には、Ser、Thr間でお互いに置換する変異である。保存的置換とみなされる置換としては、具体的には、AlaからSer又はThrへの置換、ArgからGln、His又はLysへの置換、AsnからGlu、Gln、Lys、His又はAspへの置換、AspからAsn、Glu又はGlnへの置換、CysからSer又はAlaへの置換、GlnからAsn、Glu、Lys、His、Asp又はArgへの置換、GluからGly、Asn、Gln、Lys又はAspへの置換、GlyからProへの置換、HisからAsn、Lys、Gln、Arg又はTyrへの置換、IleからLeu、Met、Val又はPheへの置換、LeuからIle、Met、Val又はPheへの置換、LysからAsn、Glu、Gln、His又はArgへの置換、MetからIle、Leu、Val又はPheへの置換、PheからTrp、Tyr、Met、Ile又はLeuへの置換、SerからThr又はAlaへの置換、ThrからSer又はAlaへの置換、TrpからPhe又はTyrへの置換、TyrからHis、Phe又はTrpへの置換、及び、ValからMet、Ile又はLeuへの置換が挙げられる。また、上記のようなアミノ酸の置換、欠失、挿入、付加、または逆位等には、遺伝子が由来する微生物の個体差、種の違いに基づく場合などの天然に生じる変異(mutant又はvariant)によって生じるものも含まれる。
【0041】
さらに、上記のような保存的変異を有する遺伝子は、上記のような遺伝子にコードされるタンパク質のアミノ酸配列全体、例えば配列番号2、4、または6のアミノ酸配列全体に対して、80%以上、好ましくは90%以上、より好ましくは95%以上、より好ましくは97%以上、特に好ましくは99%以上の相同性を有し、かつ、グルタチオンを分解する活性を有するタンパク質をコードする遺伝子であってもよい。尚、本明細書において、「相同性」(homology)は、「同一性」(identity)を指すことがある。
【0042】
また、本発明に用いられる酵素をコードする遺伝子は、公知の遺伝子配列から調製され得るプローブ、例えば配列番号1、3、または5に示す塩基配列の全体または一部に対する相補配列とストリンジェントな条件下でハイブリダイズし、グルタチオンを分解する活性を有するタンパク質をコードするDNAであってもよい。ここで、「ストリンジェントな条件」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。一例を示せば、相同性が高いDNA同士、例えば80%以上、好ましくは90%以上、より好ましくは95%以上、より好ましくは97%以上、特に好ましくは99%以上の相同性を有するDNA同士がハイブリダイズし、それより相同性が低いDNA同士がハイブリダイズしない条件、あるいは通常のサザンハイブリダイゼーショ
ンの洗いの条件である60℃、1×SSC、0.1% SDS、好ましくは、60℃、0.1×SSC、0.1% SDS、さらに好ましくは、68℃、0.1×SSC、0.1% SDSに相当する塩濃度および温度で、1回、より好ましくは2〜3回洗浄する条件が挙げられる。
【0043】
上述の通り、プローブは、遺伝子の相補配列の一部であってもよい。そのようなプローブは、公知の遺伝子配列に基づいて作製したオリゴヌクレオチドをプライマーとし、これらの塩基配列を含むDNA断片を鋳型とするPCRによって作製することができる。例えば、300bp程度の長さのDNA断片をプローブとして用いる場合には、ハイブリダイゼーションの洗いの条件としては、50℃、2×SSC、0.1% SDSが挙げられる。
【0044】
なお、これらのバリアントに関する記載は、本明細書中の他の遺伝子およびタンパク質についても準用されるものとする。
【0045】
また、グルタチオン分解酵素としては、液胞局在グルタチオン分解酵素が挙げられる。本発明において、液胞局在グルタチオン分解酵素とは、液胞に局在し、且つ、グルタチオンを分解する活性を有するタンパク質をいう。液胞局在グルタチオン分解酵素としては、ECM38遺伝子にコードされるEcm38pが挙げられる。Ecm38pは、具体的にはγ−グルタミルトランスペプチダーゼ(γ−GT)活性を有する。
【0046】
サッカロミセス・セレビシエのECM38遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのECM38遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号7および8に示す。
【0047】
酵素が由来する生物によって液胞局在グルタチオン分解酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、液胞局在グルタチオン分解酵素をコードする遺伝子は、液胞に局在し、グルタチオンを分解する活性を有するタンパク質をコードする限り、配列番号7に示す塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0048】
また、トリペプチドであるグルタチオンの分解には、上記のグルタチオン分解酵素の他に、LAP4遺伝子にコードされるLap4pのような各種ペプチダーゼが関与していると考えられる(非特許文献3および7)。したがって、本発明の酵母においては、そのようなペプチダーゼの活性を低下させることも有効であり得る。
【0049】
(1−3)γ−グルタミル化合物合成能の強化
本発明の酵母は、γ−グルタミル化合物合成能が増大するよう改変されている。γ−グルタミル化合物としては、γ−グルタミル基を有する化合物であれば特に制限されないが、例えば、γ‐グルタミルジペプチドやγ‐グルタミルトリペプチド等のγ−グルタミルぺプチドが挙げられる。γ‐グルタミルジペプチドとして、具体的には、例えば、γ−グルタミルシステイン(γ−Glu−Cys)やγ−グルタミル−α−アミノ酪酸(γ−Glu−Abu)が挙げられる。γ‐グルタミルトリペプチドとして、具体的には、例えば、グルタチオン(γ−Glu−Cys−Gly)やγ−グルタミル−α−アミノ酪酸−グリシン(γ−Glu−Abu−Gly)が挙げられる。なお、本発明において、γ−グルタミル化合物を構成するアミノ酸および/またはアミノ酸誘導体はいずれもL体である。
【0050】
「γ−グルタミル化合物合成能が増大する」とは、グルタチオン等のγ−グルタミル化合物の生合成が、野性株や親株等の非改変株と比較して向上していることを意味する。γ
−グルタミル化合物合成能が増大するような改変は、例えば、γ−グルタミル化合物の生合成に関わる1またはそれ以上の酵素の活性を増大させることにより達成できる。生体内では、γ−グルタミルシステイン合成酵素によりL−GluとL−Cysからγ−グルタミルシステイン(γ−Glu−Cys)が生成し、更にグルタチオン合成酵素によりL−Glyが結合してグルタチオンが生成する。よって、グルタチオン合成能が増大するような改変は、例えばγ−グルタミルシステイン合成酵素および/またはグルタチオン合成酵素の活性の増大により達成できる。また、γ−グルタミルシステイン合成能が増大するような改変は、例えばγ−グルタミルシステイン合成酵素の活性の増大により達成できる。また、γ−グルタミルシステイン合成能が増大するような改変は、グルタチオン合成酵素の活性を低下させることによっても達成できる。
【0051】
また、γ−グルタミルシステインやグルタチオン以外のγ‐グルタミルペプチドは、γ−グルタミルシステイン合成酵素および/またはグルタチオン合成酵素の副反応により生合成されうる。よって、γ‐グルタミルジペプチド合成能が増大するような改変は、例えば、γ−グルタミルシステイン合成能が増大するような改変と同様に達成できる。また、γ‐グルタミルトリペプチド合成能が増大するような改変は、例えば、グルタチオン合成能が増大するような改変と同様に達成できる。
【0052】
「酵素活性が増大する」とは、目的の酵素活性が野性株や親株等の非改変株と比較して増大していることを意味する。また、「酵素活性が増大する」とは、もともと目的の酵素活性を有する菌株において目的の酵素活性を増大させることだけでなく、もともと目的の酵素活性を有さない菌株に目的の酵素活性を付与することを含む。また、結果としてグルタチオン合成能が強化される限り、酵母が本来有するグルタチオン生合成系を弱化させた上で、好適なグルタチオン生合成系を導入してもよい。
【0053】
γ−グルタミルシステイン合成酵素活性が増大する場合には、γ−グルタミルシステイン合成酵素活性は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0054】
グルタチオン合成酵素活性が増大する場合には、グルタチオン合成酵素活性は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0055】
酵素活性が増大するような改変は、例えば、目的の酵素をコードする遺伝子の発現を増強することにより達成できる。
【0056】
遺伝子の発現の増強は、例えば、染色体上の遺伝子のプロモーターをより強力なプロモーターに置換することにより達成できる。「より強力なプロモーター」とは、遺伝子の転写が、もともと存在している野生型のプロモーターよりも向上するプロモーターを意味する。より強力なプロモーターとしては、各種レポーター遺伝子を用いることにより、在来のプロモーターの高活性型のものを取得してもよい。また、より強力なプロモーターとしては、公知の高発現プロモーター、例えば、PGK1、PDC1、TDH3、TEF1、HXT7、ADH1等の遺伝子のプロモーターを用いてもよい。なお、強力なプロモーターへの置換は、後述する遺伝子のコピー数の増加と組み合わせて利用できる。より強力なプロモーターを利用した例としては、染色体上のγ−グルタミルシステイン合成酵素遺伝子のプロモーターを強転写プロモーターで置換することによりγ−グルタミルシステイン合成酵素活性を増強する方法が開示されている(大竹康之ら、バイオサイエンスとインダストリー、第50巻第10号、第989〜994頁、1992年)。
【0057】
また、遺伝子の発現の増強は、例えば、遺伝子のコピー数を増加させることにより達成
できる。
【0058】
遺伝子のコピー数の増加は、染色体上に目的の遺伝子を導入することにより達成できる。染色体上への遺伝子の導入は、例えば相同的組み換えを利用して行うことができる。例えば、染色体中に多数のコピーが存在する配列を標的として相同的組み換えを行うことで、染色体へ遺伝子の多数のコピーを導入することができる。染色体中に多数のコピーが存在する配列としては、特有の短い繰り返し配列からなる自律複製配列(ARS)や、約150コピー存在するrDNA配列が挙げられる。ARSを含むプラスミドを用いて酵母の形質転換を行った例が、国際公開95/32289号パンフレットに記載されている。また、トランスポゾンに遺伝子を組み込み、それを染色体へ遺伝子の多数のコピーを導入するよう転移させてもよい。
【0059】
また、遺伝子のコピー数の増加は、目的遺伝子を含むベクターを宿主に導入することによっても達成できる。ベクターとしては、例えば、CEN4の複製開始点を持つプラスミドや2μm DNAの複製開始点を持つ多コピー型プラスミドを好適に用いることができる。目的遺伝子は、好適なプロモーターと組み合わせてベクターに挿入し、目的遺伝子を発現させてもよい。また、遺伝子を発現させるのに好適なプロモーターを含むベクターを用いる場合には、ベクター中のプロモーターを利用して目的遺伝子を発現させてもよい。
【0060】
γ−グルタミルシステイン合成酵素の活性の増強は、例えば米国特許第7553638号、大竹康之ら(バイオサイエンスとインダストリー、第50巻第10号、第989〜994頁、1992年)等に開示されている。その他の酵素活性も、γ−グルタミルシステイン合成酵素の活性と同様に増強させることができる。
【0061】
また、酵素活性が増大するような改変は、例えば、目的の酵素の比活性を増強することによっても達成できる。比活性が増強された酵素は、例えば、種々の生物を探索し取得することができる。また、在来の酵素に変異を導入することで高活性型のものを取得してもよい。比活性の増強は、単独で用いてもよく、上記のような遺伝子の発現を増強させる手法と任意に組み合わせて用いてもよい。
【0062】
目的の酵素活性が増大したことの確認は、同酵素の活性を測定することによって行うことが出来る。γ−グルタミルシステイン合成酵素活性は、Jacksonの方法(Jackson, R. C., Biochem.J., 111, 309 (1969))によって測定することができる。グルタチオン合成酵素活性は、Gushimaらの方法(Gushima, T. et al., J. Appl. Biochem., 5, 210 (1983))によって測定することができる。
【0063】
目的の酵素をコードする遺伝子の転写量が増大したことの確認は、同遺伝子から転写されるmRNAの量を非改変株と比較することによって行うことが出来る。mRNAの量を評価する方法としては、ノーザンハイブリダイゼーション、RT-PCR等が挙げられる(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。mRNAの量は、非改変株と比較して、例えば、1.5倍以上、2倍以上、または3倍以上に増大しているのが好ましい。
【0064】
目的の酵素の量が増大したことの確認は、抗体を用いてウェスタンブロットによって行うことが出来る(Molecular cloning(Cold spring Harbor Laboratory Press, Cold spring Harbor (USA), 2001))。目的の酵素の量は、非改変株と比較して、例えば、1.5倍以上、2倍以上、または3倍以上に増大しているのが好ましい。
【0065】
本発明の酵母が2倍体以上の倍数性を有する場合であって、染色体の改変により酵素活性が増大している場合には、本発明の酵母は、グルタチオンを蓄積できる限り、酵素活性
が増大するように改変された染色体と野生型染色体とをヘテロで有していてもよく、酵素活性が増大するように改変された染色体のホモ型であってもよい。
【0066】
また、グルタチオン合成酵素活性が低下する場合には、グルタチオン合成酵素活性は、非改変株と比較して、好ましくは50%以下、より好ましくは20%以下、さらに好ましくは10%以下、特に好ましくは5%以下に低下している。また、グルタチオン合成酵素活性が低下する場合には、グルタチオン分解酵素活性は、実質的に消失しているのが好ましい。グルタチオン合成酵素活性が低下するような改変は、上述した、グルタチオン分解酵素活性を低下させるのと同様の手法により達成できる。
【0067】
本発明において、γ−グルタミルシステイン合成酵素は、L−GluおよびL−Cysを基質として認識し、γ−グルタミルシステイン(γ−Glu−Cys)を生成する反応を触媒する活性を有する限り特に限定されない。また、γ−グルタミルシステイン合成酵素は、正反応であるγ−Glu−Cysを生成する反応以外に、任意のL−アミノ酸またはアミノ酸誘導体Xを基質として認識し、L−Gluと結合させることでγ‐Glu‐Xを生成する反応を触媒する活性を有していてもよい。γ−グルタミルシステイン合成酵素としては、GSH1遺伝子によりコードされるGsh1pが挙げられる。
【0068】
本発明において、グルタチオン合成酵素は、γ−グルタミルシステインおよびGlyを基質として認識し、グルタチオン(γ−Glu−Cys−Gly)を生成する反応を触媒する活性を有する限り特に限定されない。グルタチオン合成酵素としては、GSH2遺伝子によりコードされるGsh2pが挙げられる。
【0069】
サッカロミセス・セレビシエのGSH1遺伝子およびGSH2遺伝子の塩基配列は、それぞれSaccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。上記データベースより取得したサッカロミセス・セレビシエのGSH1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号9および10に示す。また、上記データベースより取得したサッカロミセス・セレビシエのGSH2遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号11および12に示す。
【0070】
酵素が由来する生物によってグルタチオン合成酵素またはγ−グルタミルシステイン合成酵素をコードする遺伝子の塩基配列に差異が存在することがあるため、それらをコードする遺伝子は、グルタチオン合成酵素活性またはγ−グルタミルシステイン合成酵素活性を有するタンパク質をコードする限り、配列番号9または11に示す塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0071】
(1−4)液胞グルタチオントランスポーターの発現強化
本発明の酵母は、一態様において、液胞グルタチオントランスポーターの発現が増大するよう改変されている。
【0072】
「液胞グルタチオントランスポーターの発現が増大する」とは、液胞グルタチオントランスポーターをコードする遺伝子の発現が、野性株や親株等の非改変株と比較して増大していることを意味する。
【0073】
液胞グルタチオントランスポーターの発現を増大させる場合には、液胞グルタチオントランスポーターの発現は、非改変株と比較して、好ましくは1.5倍以上、より好ましくは2倍以上、さらに好ましくは3倍以上に向上している。
【0074】
また、「液胞グルタチオントランスポーターの発現が増大する」とは、もともと液胞グ
ルタチオントランスポーターが発現している菌株において発現を増大させることだけでなく、もともと液胞グルタチオントランスポーターが発現していない菌株において、液胞グルタチオントランスポーターを発現させることを含む。すなわち、例えば、液胞グルタチオントランスポーターをコードする遺伝子を保持しない菌株に液胞グルタチオントランスポーターをコードする遺伝子を導入し、液胞グルタチオントランスポーターを発現させることを含む。
【0075】
液胞グルタチオントランスポーターをコードする遺伝子の発現を増大させることは、上述した、γ−グルタミルシステイン合成酵素をコードする遺伝子やグルタチオン合成酵素をコードする遺伝子の発現を増大させるのと同様の手法により達成できる。
【0076】
本発明において、液胞グルタチオントランスポーターとは、細胞質のグルタチオンを液胞に輸送する機能を有するタンパク質を意味し、当該機能を有する限り特に限定されない。液胞グルタチオントランスポーターをコードする遺伝子としては、YCF1遺伝子およびBPT1遺伝子が挙げられる。中でも、YCF1遺伝子の発現を増大させるのが好ましい。また、液胞へのグルタチオンの輸送には、VBA1遺伝子にコードされるVba1pのような各種アミノ酸・ペプチドトランスポーターが関与していると考えられ、本発明において、液胞グルタチオントランスポーターにはそのような各種アミノ酸・ペプチドトランスポーターが含まれるものとする。
【0077】
サッカロミセス・セレビシエのYCF1遺伝子、BPT1遺伝子、およびVBA1遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。また、上記データベースより取得したサッカロミセス・セレビシエのYCF1遺伝子の塩基配列、および同遺伝子がコードするアミノ酸配列を、それぞれ配列番号13および14に示す。
【0078】
酵素が由来する生物によって液胞グルタチオントランスポーターをコードする遺伝子の塩基配列に差異が存在することがあるため、当該遺伝子は、細胞質のグルタチオンを液胞に輸送する機能を有するタンパク質をコードする限り、配列番号13に示す塩基配列のバリアント、またはSaccharomyces Genome Databaseに開示される液胞グルタチオントランスポーターをコードする遺伝子の塩基配列のバリアントであってもよい。バリアントは、上述したDUG遺伝子群およびDUG複合体についてのバリアントの記載に準ずる。
【0079】
(1−5)その他の改変
また、本発明の酵母は、グルタチオン等のγ−グルタミル化合物を蓄積できる限りにおいて、上記のような改変に加えて、その他の任意の改変がなされていてもよい。そのような改変としては、細胞内のγ−グルタミル化合物濃度が高まるような改変が挙げられる。例えば、酵母におけるMET25遺伝子の発現量を増大させると菌体内グルタチオン含有量が上昇することが報告されている。MET25遺伝子の発現量を増大させる方法としては、変異型MET4遺伝子(大村ら, FEBS Letters 387 (1996) 179-183、クリスet. al., Molecular Biology of the Cell., 8, 1699-1707, 1997))、変異型MET30遺伝子(DOMINIQUE et. al., MOLECULAR AND CELLUAR BIOLOGY, Dec 1995 p6526-6534、特開2004-113155)を利用する方法等が報告されている。また、アミノ基転移酵素の活性が増大していてもよい。酵母のアミノ基転移酵素として、アラニン:グリオキシル酸アミノトランフェラーゼ(Alanine:Glyoxylate aminotransferase)、分岐鎖アミノ酸トランスアミナーゼ(Branched-chain Amino acid transaminase)、アスパラギン酸アミノトランスフェラーゼ(Aspartate amino transferase)、γ−アミノ酪酸トランスアミナーゼ(Gamma-aminobutyrate transaminase)などを例示することができる。サッカロマイセス・セレビシエでは既にこれらの酵素をコードする遺伝子が特定されており、各々AGX1(システマティックネーム:YFL030W)、BAT1(システマティックネーム:YHR208W)およびBAT2(システマティックネーム:YJR148W)、AAT1(システマティックネーム:YKL106W)およびAAT2(システマティックネーム:YLR027C)、ならびにUGA1(システマティックネーム:YGR019W)にコードされている。特に、BAT1にコードされる分岐鎖アミノ酸トランスアミナーゼの活性を増大させることにより有意に細胞内のα−アミノ酪酸合成を促進することができ、細胞内でのγ-Glu-Abu(L-γ-グルタミル-L-2-アミノ酪酸)やγ-Glu-Abu-Gly(L-γ-グルタミル-L-2-アミノ酪酸-グリシン)蓄積量を増加させることができうる。アミノ基転移酵素の活性を増大させることは、上述した、γ−グルタミルシステイン合成酵素活性やグルタチオン合成酵素活性を増大させるのと同様の手法により達成できる。なお、上記各遺伝子の塩基配列は、Saccharomyces Genome Database(http://www.yeastgenome.org/)に開示されている。
【0080】
上記のような改変、すなわちγ−グルタミル化合物合成能の増大、グルタチオン分解酵素活性の低下、液胞グルタチオントランスポーターの発現の増大、および、その他の任意の改変は、いずれの改変を先に行ってもよい。
【0081】
(2)本発明の酵母エキス
次に、酵母エキスを製造する方法を説明する。本発明の酵母エキスは、本発明の酵母を原料として用いる以外は、通常の酵母エキスと同様に製造することができる。
【0082】
まず、本発明の酵母を培地で培養する。培地は、酵母が増殖し得るものであれば特に制限されず、通常工業的に用いられる培地を利用することができる。例えば、実施例に記載したYPD培地を好適に利用することができる。
【0083】
また、グルタチオン等のγ−グルタミル化合物の原料となる化合物(以下、γ−グルタミル化合物の原料ともいう)を培地に添加してもよい。γ−グルタミル化合物の原料となる化合物とは、例えば、γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、および/またはペプチドをいう。γ−グルタミル化合物の原料を添加する場合、その添加量は、γ−グルタミル化合物の種類や、本発明の酵母が有する当該原料の生合成能等の諸条件によって適宜設定すればよいが、例えば、培養開始時の培地における終濃度として、好ましくは1ppm以上、より好ましくは10ppm以上、さらに好ましくは100ppm以上であってよい。添加量の上限は特に制限されないが、例えば費用の観点から、培養開始時の培地における終濃度として、100,000ppm以下に設定してもよい。添加量は、例えば、好ましくは10,000ppm以下、より好ましくは1,000ppm以下、さらに好ましくは500ppm以下であってよい。また、培養中に、間欠的あるいは連続的にγ−グルタミル化合物の原料を添加する場合には、添加量の累計が、上述の培養開始時の培地における終濃度と同等になるように設定すればよい。添加するγ−グルタミル化合物の原料は、1種またはそれ以上であってよい。添加するγ−グルタミル化合物の原料は、精製品(純品)であってもよく、当該原料を含む組成物であってもよい。当該原料を含む組成物は、必要量の当該原料を含む限り特に制限されない。
【0084】
培養条件は、通常の酵母エキスの製造と同様の条件を採用することができ、用いる酵母に応じて適宜変更することができる。バッチ培養、フェドバッチ培養、連続培養など任意の方法を使用することができる。サッカロミセス・セレビシエの場合は、25〜35℃で、より好ましくは、27〜33℃で、更に好ましくは28〜32℃で振とう培養等により好気的に培養することが好ましい。
【0085】
上記のようにして本発明の酵母を培養すると、酵母の細胞内にγ−グルタミル化合物が蓄積する。
【0086】
得られた酵母からの酵母エキスの調製は、通常の酵母エキスの調製と同様にして行えば
よい。酵母エキスは、酵母菌体を熱水抽出したものを処理したものでもよいし、酵母菌体を消化したものを処理したものでもよい。また、必要に応じて得られた酵母エキスを濃縮してもよいし、乾燥し粉末の形態にしてもよい。
【0087】
上記のようにして、γ−グルタミル化合物の含有量が高められた酵母エキスが得られる。好適な形態では、酵母エキスは、酵母エキス中の固形分全量に対し、γ−グルタミル化合物を乾燥重量として0.5重量%以上、より好ましくは1重量%以上、さらに好ましくは5重量%以上、特に好ましくは10重量%以上含有する。
【0088】
(3)本発明の飲食品
上記のようにして得られるγ−グルタミル化合物の含有量が高められた酵母エキスは、飲食品の製造に用いることができる。飲食品としては、例えば、アルコール飲料、パン食品、発酵食品調味料が挙げられる。
【0089】
本発明の飲食品は、本発明の酵母エキスを飲食品の原料に添加し、飲食品に加工することによって製造される。なお、酵母エキスの添加は、飲食品の製造工程のいずれの段階で行われてもよい。本発明の飲食品は、前記酵母エキスを使用すること以外は、通常の飲食品と同様の原料を用い、同様の方法によって製造することができる。このような原料としては、例えばアルコール飲料では、米、大麦、コーンスターチ等、パン食品では小麦粉、砂糖、食塩、バター、発酵用酵母菌等が、発酵食品調味料では大豆、小麦等が挙げられる。また、酵母エキスもしくはその濃縮物、またはそれらを乾燥したものは、それ自体で発酵食品調味料として用いることができる。
【実施例】
【0090】
実施例1.グルタチオン分解酵素活性低下の効果の検討
本実施例では、酵母のグルタチオン分解酵素活性を低下させ、グルタチオン蓄積能および生育速度に与える影響を検証した。
【0091】
(1)γ−グルタミルシステイン合成酵素遺伝子高発現酵母(AG1株)の作製
γ−グルタミルシステイン合成酵素をコードするGSH1遺伝子の高発現株では、細胞内グルタチオン濃度が上昇することが知られている(非特許文献10および11)。そこで、サッカロミセス・セレビシエY006株(FERM BP−11299)のGSH1遺伝子のプロモーター領域を、構成発現プロモーターであるアルコールデヒドロゲナーゼ遺伝子(以下、ADH1)のプロモーター領域に置換することにより、GSH1高発現酵母(AG1株)を作製した。手順を以下に示す。なお、Y006株はURA3遺伝子の欠損株であり、ウラシル要求性を示す。
【0092】
(1−1)ADH1プロモーター置換の為の鋳型プラスミドの作成
まず、配列番号15及び16のプライマーを用い、NBRCより分譲されたサッカロミセス・セレビシエS288C株の染色体DNAを鋳型とするPCRにより、URA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をエタノール沈殿により精製後、SphI及びEcoRIで消化し、プラスミドpUC19のSphI-EcoRI部位に挿入し、pUC19-URA3を得た。次に、配列番号17及び18のプライマーを用い、Y006株の染色体DNAを鋳型とするPCRにより、ADH1プロモーター領域(pADH1)を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をPstIで消化し、PstIで消化しCIAP処理したpUC19-URA3のPstI部位に挿入し、pUC19-pADH1-URA3を得た。同様に、配列番号19及び20のプライマーを用いて増幅したADH1プロモーター領域をAatIIで消化し、AatIIで消化しCIAP処理したpUC19-pADH1-URA3のAatII部位に挿入し、pUC19-pADH1-URA3-ADH1pを得た。
【0093】
(1−2)染色体上のGSH1遺伝子へのADH1プロモーターの導入
5'端にGSH1の上流配列をもつ配列番号21のプライマー、及びGSH1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号22のプライマーを用い、pUC19-pADH1-URA3-pADH1を鋳型にPCRを行い、ADH1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でY006株を形質転換し、ウラシルを含有しないSD平板培地に塗布した。生育した形質転換体から、GSH1プロモーターがpADH1-URA3-pADH1に置換された株を取得した。
【0094】
<SD培地組成>
グルコース 2%
Nitrogen Base 1倍濃度
(10倍濃度Nitrogen Baseは、1.7gのBacto Yeast Nitrogen Base w/o Amino Acids and
Ammonium Sulfate (Difco社)と5gの硫酸アンモニウムを混合したものを100mlの滅菌水に溶解し、pHを5.2程度に調整し、フィルター濾過滅菌したもの)
【0095】
(1−3)URA3選択マーカーの除去とGSH1遺伝子のプロモーター置換
URA3遺伝子が欠損した細胞は5−フルオロオロト酸(5-FOA)耐性を示すため、5-FOAを含有する培地を利用してURA3選択マーカーが除去された株を選抜できる。そこで、GSH1プロモーターがpADH1-URA3-pADH1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのADH1プロモーター間の相同組換えにより、URA3が除去され、GSH1プロモーターがADH1プロモーターに置換された株(AG1 ura3- 株)を取得した。
【0096】
(1−4)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、3min)、25 cycleとした。次にAG1 ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換されウラシル非要求性となった株を取得し、AG1株とした。また、Y006株を同様にこのDNA断片で形質転換し、ウラシル非要求性となった株を取得し、Y003株とした。
【0097】
(2)Y006株のグルタチオン分解酵素遺伝子破壊株の作製
グルタチオン分解活性低下の効果を検証するため、サッカロミセス・セレビシエY006株を基に、各グルタチオン分解酵素遺伝子を破壊した変異株を以下の手順で作製した。
【0098】
(2−1)Y006株のDUG1破壊株の作製
まず、DUG1の開始コドンより上流80塩基を付加した配列番号25のプライマー及びDUG1の終止コドンより下流80塩基を付加した配列番号26のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug1D株(以下、d1株)を得た。
【0099】
(2−2)Y006株のDUG2破壊株の作製
まず、DUG2の開始コドンより上流80塩基を付加した配列番号27のプライマー及びDUG2の終止コドンより下流80塩基を付加した配列番号28のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug2D株(以下、d2株)を得た。
【0100】
(2−3)Y006株のDUG3破壊株の作製
まず、DUG3の開始コドンより上流80塩基を付加した配列番号29のプライマー及びDUG3の終止コドンより下流80塩基を付加した配列番号30のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のdug3D株(以下、d3株)を得た。
【0101】
(2−4)Y006株のECM38破壊株の作製
まず、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY006株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からY006のecm38D株(以下、e株)を得た。
【0102】
(2−5)dug2D株のECM38破壊株の作製
まず、BY4743を親株とするサッカロミセス・セレビシエ破壊株セット(OpenBiosystems社)のECM38破壊株を鋳型に、配列番号33のプライマー及び配列番号34のプライマーを用い、ECM38を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でdug2△株を形質転換し、終濃度50mg/LのG418を含むYPD平板培地へ塗布した。生育した形質転換体からdug2D ecm38D株(以下、ed株)を得た。
【0103】
(3)グルタチオン分解酵素破壊株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0104】
Y003、AG1、d1、d2、d3、e、およびed株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0105】
結果を図1に示す。野生型株であるY003株、GSH1の高発現株であるAG1株、並びにグルタチオン分解酵素遺伝子破壊株である、d1、d2、d3、eおよびed株の菌体内グルタチオン
濃度はそれぞれ、Y003:0.50%、AG1:0.80%、d1:0.47%、d2:0.49%、d3:0.54%、e:0.53%、ed:0.50% であった。GSH1の高発現株であるAG1株は、既に報告されているように(非特許文献10および11)、野生型株であるY003株と比較してグルタチオン濃度が向上した。一方、グルタチオン分解酵素をコードするECM38または各DUG遺伝子を破壊しても、菌体内グルタチオン濃度は影響を受けなかった。ECM38または各DUG遺伝子の単一破壊株においては、Ecm38pとDUG複合体がお互いの機能を相補するために細胞内グルタチオン濃度が影響を受けないことが予想されたが、これら2つのグルタチオン分解酵素をコードする遺伝子の二重破壊株であるed株においても細胞内グルタチオン濃度の上昇は見られなかった。
【0106】
(4)高グルタチオン蓄積形質を有する株(AG1)由来のグルタチオン分解酵素遺伝子破壊株の作製
次に、高グルタチオン蓄積形質を有する株におけるグルタチオン分解酵素活性の低下がグルタチオン蓄積能に与える影響を評価するため、AG1株を基にグルタチオン分解酵素遺伝子破壊株を作製した。手順を以下に示す。
【0107】
(4−1)AG1株のDUG2破壊株の作製
Y006株のDUG2破壊株(d2株)作製と同様に、DUG2の開始コドンより上流80塩基を付加した配列番号27のプライマー及びDUG2の終止コドンより下流80塩基を付加した配列番号28のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1 dug2D株(以下、AG1-d株)を得た。
【0108】
(4−2)AG1株のECM38破壊株の作製
Y006株のECM38破壊株(e株)作製と同様に、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、Y006株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1 ecm38D株(以下、AG1-e株)を得た。
【0109】
(5)AG1株由来のグルタチオン分解酵素遺伝子破壊株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0110】
Y003、AG1、AG1-d、AG1-e、および、AG1-ed株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0111】
結果を図2に示す。Y003株、AG1株、およびAG1株由来のグルタチオン分解酵素破壊株の菌体内グルタチオン濃度はそれぞれ、Y003:0.6%、AG1:1.0 %、AG1-d:2.5%、AG1-e:1.1%、AG1-ed:2.4%であった。
【0112】
単一破壊では菌体内グルタチオン濃度に影響しなかったDUG複合体の欠損は、GSH1の高発現株において菌体内グルタチオン濃度の上昇に寄与した。また、高グルタチオン蓄積形質を有するAG1はY003と比較して生育遅延が認められるが、AG-dはAG1と比較して菌体内グルタチオン濃度が高いにもかかわらず、対数増殖期における生育遅延が軽減された(図3)。
【0113】
以上の結果より、DUG複合体の欠損は、GSH1高発現株において、グルタチオン分解に寄与することが明らかになった。従って、GSH1の高発現によって、DUG複合体の活性が誘導されることが示唆された。
【0114】
(6)GSH1高発現株におけるDUG複合体構成因子の発現量の解析
GSH1の高発現によってDUG複合体の活性が誘導されるかどうかを、DUG複合体の各構成因子のmRNA量をRT-PCRで定量することにより解析した。
【0115】
Y003および、AG1株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで培養を行った。OD600=5および10の2点で1mLサンプルを分取し、滅菌水で2回リンスした後、ペレットを-80℃で冷凍した。翌日、RNA抽出キット、RNeasy(キアゲン)を用いてトータルRNAを抽出した。抽出したRNAのうち500ngを、PrimeScript RT reagent Kit(TaKaRa)を用い37℃で15分反応させ、cDNAを作製した。この際、RT-PCRのネガティブコントロールとして用いるため、酵素を入れずに反応を行ったサンプルを用意した。
【0116】
続いて、Power SYBR Green(Applied Biosystems)で、反応液を調整した。DUG1の反応には配列番号35および36に示すプライマーを用いた。DUG2の反応には配列番号37および38に示すプライマーを用いた。DUG3の反応には配列番号39および40に示すプライマーを用いた。また、配列番号41および42に示すプライマーでACT1遺伝子を増幅し、内部標準とした。cDNA液は、原液、10倍希釈液、100倍希釈液、1000倍希釈液の4つの希釈率の異なる液を用いた。RT-PCR反応には、7500 Real Time PCR System(Applied Biosystems)を用いた。
【0117】
結果を図4に示した。GSH1高発現株では、培養中期において、DUG1のmRNAがおよそ1.6倍に、DUG3のmRNAがおよそ3倍に上昇した(図4a)。その一方で、定常期では、GSH1高発現による影響は見られなかった(図4b)。
【0118】
以上の結果より、GSH1の高発現株においては、DUG構成因子の発現が誘導されることにより、グルタチオン分解酵素であるDUG複合体の活性が上昇することがわかった。また、上述の通り、Y003、AG1およびAG1-d株の生育曲線の比較より、AG1株では対数増殖期において生育遅延がみられたが、AG1-d株ではそれが軽減された(図3)。定常期に両株において差の見られないDUG遺伝子の発現量が、GSH1高発現株では対数増殖期に上昇していることから、対数増殖期のDUG遺伝子の発現量は生育に影響を及ぼすことがわかった。
【0119】
以上より、GSH1高発現株でDUG複合体の活性を低下させることにより、(1)菌体内グルタチオン濃度が上昇する、(2)菌体内グルタチオン濃度が上昇するにもかかわらず、
生育遅延が軽減するという2つの効果が得られることを明らかにした。
【0120】
実施例2.液胞グルタチオントランスポーター増強の効果の検討
本実施例では、酵母の液胞グルタチオントランスポーターの発現を増強させ、グルタチオン蓄積能および生育速度に与える影響を検証した。
【0121】
(7)グルタチオン液胞トランスポーター遺伝子高発現酵母(PY株)の作製
酵母のグルタチオン液胞トランスポーターをコードする遺伝子としてYCF1遺伝子が知られている。グルタチオン液胞トランスポーター増強の効果を検証するため、サッカロミセス・セレビシエY006株のYCF1遺伝子のプロモーター領域を、構成高発現プロモーターであるPGK1遺伝子のプロモーター領域に置換することにより、YCF1高発現酵母(PY株)を作製した。手順を以下に示す。
【0122】
(7−1)PGK1プロモーター置換の為の鋳型プラスミドの作成
まず、配列番号15及び16のプライマーを用い、サッカロミセス・セレビシエS288C株の染色体DNAを鋳型とするPCRによりURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をエタノール沈殿により精製後、SphI及びEcoRIで消化し、プラスミドpUC19のSphI-EcoRI部位に挿入し、pUC19-URA3を得た。次に、配列番号43及び44に示すプライマーを用い、Y006株の染色体DNAを鋳型とするPCRによりPGK1プロモーター領域(pPGK1)を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、1 min)、25 cycleとした。得られたDNA断片をPstIで消化し、PstIで消化しCIAP処理したpUC19-URA3のPstI部位に挿入し、pUC19-pPGK1-URA3を得た。同様に、配列番号45及び46に示すプライマーを用いて増幅したPGK1プロモーター領域をAatIIで消化し、AatIIで消化しCIAP処理したpUC19-pPGK1-URA3のAatII部位に挿入し、pUC19-pPGK1-URA3-pPGK1を得た。
【0123】
(7−2)染色体上のYCF1遺伝子へのPGK1プロモーターの導入
5'端にYCF1の上流配列をもつ配列番号47のプライマー、及びYCF1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号48のプライマーを用い、pUC19-pPGK1-URA3-pPGK1を鋳型にPCRを行い、PGK1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でY006株を形質転換し、ウラシルを含有しないSD平板培地に塗布し得られる形質転換体から、YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を取得した。
【0124】
(7−3)URA3選択マーカーの除去とYCF1遺伝子のプロモーター置換
YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのPGK1プロモーター間の相同組換えにより、URA3が除去され、YCF1プロモーターがPGK1プロモーターに置換した株(PY ura3- 株)を取得した。
(7−4)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。次にPY ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換された株であるPY株を取得した。
【0125】
(8)Y006株由来のグルタチオン液胞トランスポーター増強株のグルタチオン蓄積能の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株をYPD培地でフラスコ培養し、菌体内グルタチオン濃度を測定した。YPD培地の組成を以下に示す。
<YPD培地>
D-グルコース 20 g
Bacto Yeast Extract 10 g
Bacto Peptone 20 g
水 1 L
【0126】
Y003、AG1、e、d、PY株のSD培地オーバーナイト培養液2.5mLを、それぞれ50mLのYPD培地に植菌し、フラスコで18時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mL、およびHPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、100mLの水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃で10分間熱水抽出を行った。熱水抽出液を4-フルオロ-7-スルファモイルベンゾフラザン(ABD-F)反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0127】
結果を図5に示す。Y003と、その単一変異株であるAG1、e、d、PY株の菌体内グルタチオン濃度はそれぞれ、Y003:0.75%、AG1:1.20%、e:0.70%、d2:0.75%、PY:0.65%であった。グルタチオン液胞トランスポーターであるYCF1の発現強化は細胞内グルタチオン濃度に影響を与えなかった。
【0128】
(9)高グルタチオン蓄積形質を有する株(AG1)由来のグルタチオン液胞トランスポーター増強株の作製
グルタチオン液胞トランスポーターの増強は、単独では細胞内グルタチオン濃度に影響を与えなかった。そこで次に、高グルタチオン蓄積形質を有する株におけるグルタチオン液胞トランスポーター増強の効果を評価するため、AG1株のYCF1遺伝子のプロモーター領域を、構成高発現プロモーターであるPGK1遺伝子のプロモーター領域に置換することにより、YCF1高発現酵母(AG1-PY株)を作製した。さらに、液胞局在グルタチオン分解酵素活性の低下とグルタチオン液胞トランスポーター増強との組み合わせの効果を評価するため、AG1-PY株を基に液胞局在グルタチオン分解酵素をコードするECM38が破壊された株(AG1-PY-e株)を作製した。手順を以下に示す。
【0129】
(9−1)AG1株のYCF1発現強化株の作製
【0130】
(9−1−1)AG1株の染色体上のYCF1遺伝子へのPGK1プロモーターの導入
5'端にYCF1の上流配列をもつ配列番号47のプライマー、及びYCF1遺伝子の開始コドンから始まる一部のORF内配列を持つ配列番号48のプライマーを用い、pUC19-pPGK1-URA3-pPGK1を鋳型にPCRを行い、PGK1プロモーターに挟まれたURA3を有するDNA断片を得た。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(60℃、10 sec)、伸張(72℃、4 min)、25 cycleとした。このDNA断片でAG1 ura3- 株を形質転換し、ウラシルを含有しないSD平板培地に塗布し得られる形質転換体から、YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を取得した。
【0131】
(9−1−2)URA3選択マーカーの除去とYCF1遺伝子のプロモーター置換
YCF1プロモーターがpPGK1-URA3-pPGK1に置換された株を、ウラシル添加SD培地で一晩培養し、適量を5-FOA平板培地に塗布した。生育したコロニーから、導入された2つのPGK1プロモーター間の相同組換えにより、URA3が除去され、YCF1プロモーターがPGK1プロモーターに置換された株(AG1-PY ura3- 株)を取得した。
【0132】
(9−1−3)ura3部位へのURA3遺伝子の挿入
サッカロミセス・セレビシエS288Cのゲノムを鋳型に、配列番号23および24に示すプライマーを用い、URA3遺伝子部位を増幅した。次にAG1-PY ura3- 株をこのDNA断片で形質転換し、ura3部位が野生型のURA3に置換された株であるAG1-PY株を取得した。
【0133】
(9−2)AG1-PY株のECM38破壊株の作製
まず、ECM38の開始コドンより上流80塩基を付加した配列番号31のプライマー及びECM38の終止コドンより下流80塩基を付加した配列番号32のプライマーを用い、S288C株のURA3遺伝子を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でAG1-PY ura3- 株を形質転換し、ウラシルを含有しないSD培地に塗布した。生育した形質転換体からAG1-PY ecm38D株(以下、AG1-PY-e株)を得た。
【0134】
(9−3)AG1-d株のYCF1発現強化株の作製
上記(9−1)でAG1株のYCF1発現強化株を作製したのと同様の手法で、AG1-d ura3- 株から、YCF1プロモーターがPGK1プロモーターに置換されたAG1-d-PY ura3- 株を取得し、さらにura3部位を野生型のURA3に置換してAG1-d-PY株を取得した。
【0135】
(9−4)AG1-d-PY株のECM38破壊株の作製
上記(9−2)でAG1-PY株のECM38破壊株を作製したのと同様の手法で、AG1-d-PY ura3- 株から、ECM38が破壊されたAG1-d-PY ecm38D株(以下、AG1-d-PY-e株)を取得した。
【0136】
(10)AG1由来各種多重変異株のグルタチオン蓄積能および生育の評価
以上のようにして作製したサッカロミセス・セレビシエ遺伝子変異株を、ジャーファーメンターで培養し、菌体内グルタチオン濃度および生育を測定した。用いた培地の組成は以下の通りである。
【0137】
<培地組成>
I)5xSD培地
D-グルコース 5g
Difco Yeast Nitrogen base w/o Amino Acids and Ammonium Sulfate 8.5 g
硫酸アンモニウム 25 g
水 1 L
【0138】
II)50%グルコース
D-グルコース 500 g
水 1 L
【0139】
Y003、AG1、AG1-d、AG1-d-e、AG1-PY、AG1-PY-e株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで36時間培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでグルタチオン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。グルタチオン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してグルタチオンのチオール基を蛍光ラベルし、HPLC分析によりグルタチオン量を算出した。細胞内グルタチオン濃度は、乾燥菌体重量あたりのグルタチオン量として算出した。
【0140】
各株の細胞内グルタチオン濃度を図6に、生育曲線を図7に示す。それぞれの菌株の細胞内グルタチオン濃度は、Y003:0.7%、AG1:1.1%、AG1-d:2.5%、AG1-e:1.2%、AG1- PY:1.3%、AG1- PY-e:4.3%であった。細胞内の高濃度グルタチオンは生育を阻害することが知られているとおり、AG1株では細胞内グルタチオンの上昇に伴い、生育速度の低下が見られた。
【0141】
グルタチオン液胞トランスポーターをコードするYCF1の発現を強化したAG1-PY株では、細胞内グルタチオン濃度の上昇は見られなかったが、生育速度は野生型であるY003株程度まで改善した。これは、生育遅延の原因である高濃度の細胞内グルタチオンが液胞に輸送され、細胞質のグルタチオン濃度が低下したためと考えられる。
【0142】
AG1-PY株においてさらに液胞局在グルタチオン分解酵素をコードするECM38を破壊したAG1-PY-e株では、生育速度は野生型よりもはるかに速くなり、また、細胞内グルタチオン濃度も著しく上昇した。AG1-e株では細胞内グルタチオン濃度の上昇は見られなかったことから、これは、Ycf1pにより液胞に輸送されたグルタチオンが、液胞でEcm38pによって分解されずに蓄積されたほか、細胞質内のグルタチオン濃度が低下したことによってグルタチオン生合成系に対するフィードバック阻害が解消し、グルタチオン生合成能が向上したためと考えられる。
【0143】
以上の結果より、グルタチオン液胞トランスポーターをコードするYCF1を高発現することで、生育遅延やグルタチオン生合成系に対するフィードバック阻害の原因となりうる細胞質内グルタチオンを液胞へ輸送し、さらに液胞局在グルタチオン分解酵素をコードするECM38を破壊し、グルタチオンを液胞に蓄積させることで、生育遅延を回避しつつ、グルタチオン高含有株における細胞内グルタチオン濃度のさらなる上昇を達成することができることが明らかとなった。
【0144】
実施例3.酵母エキスの製造
本実施例では、上記実施例で取得した細胞内のグルタチオン濃度が上昇し、且つ、生育が改善した酵母を原料として用いて酵母エキスを製造した。
【0145】
(11)酵母エキスの製造
上記(10)で得られたAG1-d株及びAG1-PY-e株の培養ブロス約50mlを各々分取し、遠心分離により菌体と培養上清を分離した。菌体を50mlの純水に懸濁し、遠心分離により菌体を分離することにより、菌体を洗浄した。この洗浄操作を再度繰り返した後、菌体濃度が約10g / dLになるように純水に懸濁した。なお、菌体濃度は、吸光度と乾燥菌体重量の相関を調べた予備実験結果より推定した。この懸濁液を70℃10分間加熱し、その後氷上にて急速冷却した。遠心分離により抽出液と菌体残渣を分離した。このようにして、酵母エキス(抽出液)を製造した。AG1-d株からはエキス固形分あたりのGSH含量が約8.2%の酵母エキスが、AG1-PY-e株からはエキス固形分あたりのGSH含量が約13.1%の酵母エキスが得られた。
【0146】
実施例4.グルタチオン以外のγ−グルタミルペプチド生産への応用(1)
本実施例では、γ−グルタミルシステインを例として、グルタチオン以外のγ−グルタミルペプチドの蓄積においても、DUG複合体の活性低下が生育遅延の軽減に寄与し、また、グルタチオン液胞トランスポーターの増強と液胞局在グルタチオン分解酵素活性の低下との組み合わせが効果的であることを示す。γ−グルタミルシステインを蓄積する酵母は、例えば、γ−グルタミル基を有するジペプチドを生成する反応を触媒する酵素をコードするGSH1の発現を強化し、γ−グルタミルシステインとグリシンからグルタチオンを合成する反応を触媒する酵素をコードするGSH2を破壊することにより作製できる。
【0147】
(12)GSH2破壊株の作製
まず、BY4743を親株とするサッカロミセス・セレビシエ破壊株セット(OpenBiosystems社)のGSH2破壊株を鋳型に、配列番号49のプライマー及び配列番号50のプライマーを用い、GSH2を増幅した。PCRの条件は、熱変性(94℃、10 sec)、アニーリング(50℃、10 sec)、伸張(72℃、2 min)、25 cycleとした。得られたDNA断片でY003株、AG1株、AG1-d株、AG1-d-PY株およびAG1-d-PY-e株を形質転換し、終濃度50mg/LのG418を含むYPD平板培地へ塗布した。生育した形質転換体からY003株のGSH2破壊株(以下gsh2株)、AG1株のGSH2破壊株(以下AG1-gsh2株)、AG1-d株のGSH2破壊株(以下AG1-d-gsh2)、AG1-d-PY株のGSH2破壊株(以下AG1-d-PY-gsh2株)、およびAG1-d-PY-e株のGSH2破壊株(以下AG1-d-PY-e-gsh2株)を得た。
【0148】
(13)各種GSH2破壊株におけるγ−グルタミルシステイン蓄積濃度と生育の評価
以上のようにして作製したサッカロミセス・セレビシエのGSH2破壊株を、ジャーファーメンターで培養し、菌体内γ−グルタミルシステイン濃度および生育を測定した。用いた培地の組成は以下の通りである。
【0149】
<培地組成>
I)5xSD培地
D-グルコース 5g
Difco Yeast Nitrogen base w/o Amino Acids and Ammonium Sulfate 8.5 g
硫酸アンモニウム 25 g
水 1 L
【0150】
II)50%グルコース
D-グルコース 500 g
水 1 L
【0151】
gsh2株、AG1-gsh2株、およびAG-d-gsh2株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでγ−グルタミルシステイン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。γ−グルタミルシステイン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してγ−グルタミルシステインのチオール基を蛍光ラベルし、HPLC分析によりγ−グルタミルシステイン量を算出した。細胞内γ−グルタミルシステイン濃度は、乾燥菌体重量あたりのγ−グルタミルシステイン量として算出した。
【0152】
生育曲線を図8に、細胞内γ−グルタミルシステイン濃度を図9に示した。GSH2の単一破壊株であるgsh2株と比較して、gsh2株のGSH1発現増強株であるAG1-gsh2株では、細胞内γ−グルタミルシステイン濃度の上昇が認められた。AG1-gsh2株においてさらにグルタチオン分解酵素をコードするDUG2を破壊したAG1-d-gsh2株では、AG1-gsh2株と比較して細胞内γ−グルタミルシステイン濃度の上昇は認められなかった。これは、DUG複合体はもともとγ−グルタミルシステイン分解に全く寄与していないか、ほとんど寄与していないためであると考えられる。しかしながら、このようにDUG2の破壊は細胞内γ−グルタミルシステイン濃度の上昇に寄与しないにもかかわらず(図9)、AG1-d-gsh2株では生育遅延が大幅に軽減された(図8)。
【0153】
以上の結果より、DUG複合体の活性低下によって、グルタチオンを高濃度に蓄積する酵母の生育遅延が軽減されるだけでなく、グルタチオン以外のγ−グルタミルペプチドであるγ−グルタミルシステインを蓄積する酵母においても生育遅延が軽減されることが明らかとなった。従ってDUG複合体の活性を低下させることにより、Gsh1pによって生成される様々なγ−グルタミルペプチドを蓄積する酵母の生育遅延を解消できると考えられる。
【0154】
(14)γ−グルタミルシステインの液胞への蓄積効果
AG1-d-gsh2株、AG-d-PY-gsh2株、およびAG-d-PY-e-gsh2株のSD培地オーバーナイト培養液30mLを、それぞれ270mLの5xSD培地に植菌し、必要に応じて50%グルコース溶液を滴下しながらジャーファーメンターで培養を行った。培養終了後、乾燥菌体重量測定に供する培養液5mLおよび、HPLCでγ−グルタミルシステイン濃度分析に供する培養液1mLをそれぞれ分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして-80℃で保存した。その後、乾燥菌体重量を計測するサンプルは、3日間、42℃の真空乾燥機で菌体を完全に乾燥させ、培養液5mL当たりの乾燥菌体重量を算出した。γ−グルタミルシステイン濃度分析に用いるサンプルは、10mLの純水に懸濁し、70℃10分間熱水抽出を行った。熱水抽出液はABD-F反応液と混合してγ−グルタミルシステインのチオール基を蛍光ラベルし、HPLC分析によりγ−グルタミルシステイン量を算出した。細胞内γ−グルタミルシステイン濃度は、乾燥菌体重量あたりのγ−グルタミルシステイン量として算出した。
【0155】
結果を図10に示した。グルタチオン蓄積株と同様、YCF1の発現を強化し、ECM38を破壊した株であるAG1-d-PY-e-gsh2株では、AG1-d-gsh2株と比較し、細胞内γ−グルタミルシステイン濃度が大幅に上昇した。一方、野生型ECM38を有するAG1-d-PY-gsh2株では、AG1-d-gsh2株と比較し、細胞内γ−グルタミルシステイン濃度に変化が無かったことから、生成したγ−グルタミルシステインは液胞に局在するEcm38pによって分解されると考えられる。従って、AG1-d-PY-e-gsh2株において、YCF1の発現強化により液胞に移送されたγ−グルタミルシステインは、液胞に安定的に蓄積していると考えられる。
【0156】
以上の結果より、YCF1の発現強化およびECM38の活性低下により、グルタチオンだけでなく、γ−グルタミルシステインなどγ−グルタミルペプチドが液胞内で分解されずに蓄積し、結果として細胞内濃度を上昇させることができることがわかった。
【0157】
実施例5.グルタチオン以外のγ−グルタミルペプチド生産への応用(2)
γ−グルタミル基を有するジペプチドを生成する反応を触媒する酵素をコードするGSH1の発現を強化することにより、正反応であるγ−グルタミルシステインの生成に加え、各種γ−グルタミルペプチドが生成する。そこで、本実施例では、グルタチオン以外のγ−グルタミルペプチドの一例として、γ−グルタミル−α−アミノ酪酸(γ-Glu-Abu)の生産と、その蓄積が生育に与える影響を検討した。
【0158】
(15)γ-Glu-Abu蓄積濃度と生育の評価
まず、AG1株及びAG1-d株をSD培地に植菌し、30℃で振とう培養した。次に、これらのオーバーナイト培養液を100ppmのL−α−アミノ酪酸(Abu)を含むSD培地に植菌し、30℃で振とう培養した。対数増殖期に、乾燥菌体重量測定に供する培養液、および、γ−グルタミルぺプチド濃度分析に供する培養液を分取した。培養液から菌体を集菌し、純水で一回リンスした後、サンプルとして保存した。その後、乾燥菌体重量を計測するサンプルは、適切な濃度になるように水に懸濁した後、予め乾燥重量を計測したろ紙に塗布し、4時間乾燥させて乾燥菌体重量を算出した。一方、γ-Glu-Abu濃度分析に用いるサンプルは、適切な濃度になるように水に懸濁した後、70℃で10分間熱水抽出を行った。この工程にて菌体内に含まれるエキス分を抽出した。次に、遠心操作によりエキスと菌体残渣を分離した。エキスから10kDaの遠心濾過膜(MILLIPORE社:Amicon Ultra - 0.5mL 10K(カタログ番号UFC501096))を用いて細胞デブリを除去し、得られた画分を酵母抽出物として、下記に記載する方法で酵母抽出物中のAbu、γ-Glu-Abu、及びγ-Glu-Abu-Gly含量を測定した。各種化合物の細胞内濃度は、乾燥菌体重量あたりの量として算出した。
【0159】
Abu、γ-Glu-Abu、及びγ-Glu-Abu-Gly含量の測定は、これら化合物を6−アミノキノリル−N−ヒドロキシスクシンイミジルカルバメート(AQC)を用いて蛍光誘導体化し、LC-MS/MSにより検出することより行った。具体的には、適当な濃度に希釈した酵母抽出物2.5μL、又は、1μMのAbu、γ-Glu-Abu、及びγ-Glu-Abu-Glyを含む標準液2.5μLに、MillQ水2.5μL、5μM内部標準物質溶液(内部標準物質は、3-methyl-His-d2(シグマ社)、およびGly-d2(シグマ社)。いずれも安定同位体で標識されている。)5μL、硼酸緩衝液(日本ウォーターズ社製AccQ-Fluor(登録商標)試薬キット付属品)30μLを添加した。これら混合物に、AQC試薬溶液(上記AccQ-Fluor試薬キットの試薬粉末をアセトニトリル1mL中に溶解することにより調製)10μLを添加した。得られた混合物を10分間、55℃で加熱後、0.1%のギ酸水溶液100μLを加え、分析サンプルとした。
【0160】
次に前述のように調製した分析サンプルを、逆相の液体クロマトグラフィーで分離した。分離条件は下記の通りである。
【0161】
(1)HPLC:Agilent 1200シリーズ
(2)分離カラム:Unison UK-Phenyl 内径2.0mm、長さ100mm、粒子径3μm(Imtakt社製)
(3)カラム温度:40℃
(4)移動相A:25mMギ酸水溶液をアンモニア水でpH6.0に調整した水溶液
(5)移動相B:メタノール
(6)流速:0.25mL/min
(7)溶出条件:溶出は、移動相A及び移動相Bの混合液を用いて行った。混合液に対する移動相Bの比率は以下の通り。0分(5%)、0分〜17分(5%〜40%)、17分〜17.1分(40%〜80%)、17.1分〜19分(80%)、19分〜19.1分(80%〜5%)、19.1分〜27分(5%)。
【0162】
その後、前述の分離条件によって溶出されたAbu、γ-Glu-Abu、およびγ-Glu-Abu-Glyの誘導体化物を質量分析計に導入してマスクロマトグラムにより定量を行った。分析条件は下記の通りである。
【0163】
(1)質量分析装置:AB Sciex API3200 QTRAP
(2)検出モード:Selected Ion Monitoring(ポジティブイオンモード)
(3)選択イオン:表1
【0164】
【表1】
【0165】
Abu、γ-Glu-Abu、およびγ-Glu-Abu-Glyの誘導体化物の定量は、解析ソフトAnalyst v
er 1.4.2(AB Sciex)を用いて行った。定量を行うための内部標準物質として、Abuの誘導体化物の場合は3-methyl-His-d3の誘導体化物を、γ-Glu-Abu又はγ-Glu-Abu-Glyの誘導体化物の場合はGly-d2の誘導体化物を、各々用いた。なお、γ-Glu-Abuの定量の際に、極まれにサンプルによって夾雑ピークが見られた場合は、第二のマスアナライザーでの選択イオンとして、145.2、或いは、104.1を用いて定量した。その結果、表2に示すように、AG1株と比較して、AG1-d株は著量のγ-Glu-Abuを蓄積していた。
【0166】
【表2】
【0167】
次に、γ-Glu-Abuの蓄積が生育に与える影響について検討した。AG1株及びAG1-d株をSD培地に植菌し、30℃で振とう培養した。次に、これらのオーバーナイト培養液を100ppmのAbuを含むSD培地に660nmの吸光度が0.01になるように小型L型培養菅(ADVANTEC社型式TV100030)4mlに植菌し、バイオフォトレコーダー(ADVANTEC社型式TVS062CA)を用いて1時間毎に660nmの吸光度を測定し生育を確認した。培養温度は30℃、振とう数は70rpmに設定した。生育曲線を図11に示した。図に示すように、より多くのγ-Glu-Abuを蓄積したAG1-d株は、AG1株とほぼ同等の生育を示した。
【0168】
以上の結果より、DUG複合体の活性低下によって、グルタチオンを高濃度に蓄積する酵母の生育遅延が軽減されるだけでなく、グルタチオン以外のγ−グルタミルペプチドであるγ-Glu-Abuを蓄積する酵母においても生育遅延が軽減されることが明らかとなった。従ってDUG複合体の活性を低下させることにより、Gsh1pによって生成される様々なγ−グルタミルペプチドを蓄積する酵母の生育遅延を解消できると考えられる。
【0169】
本実施例1〜5のまとめとして、DUGを破壊することによる生育遅延の軽減と、YCF1の発現強化およびECM38破壊による液胞への蓄積を利用することにより、高濃度にγ−グルタミルペプチドを蓄積する酵母菌体を効率的に取得することができ、この酵母菌体を用いて高γ−グルタミルペプチド酵母エキスを製造することができることが明らかになった。
【産業上の利用可能性】
【0170】
本発明により、細胞内にグルタチオン等のγ−グルタミル化合物を蓄積し、且つ生育の良い酵母が提供される。それら酵母を原料として用いることにより、γ−グルタミル化合物を含有する酵母エキスを効率的に製造でき、また、それら酵母エキスを利用して飲食品を製造することができる。
【0171】
配列表の説明
配列番号1:サッカロミセス・セレビシエのDUG1の塩基配列
配列番号2:サッカロミセス・セレビシエのDug1pのアミノ酸配列
配列番号3:サッカロミセス・セレビシエのDUG2の塩基配列
配列番号4:サッカロミセス・セレビシエのDug2pのアミノ酸配列
配列番号5:サッカロミセス・セレビシエのDUG3の塩基配列
配列番号6:サッカロミセス・セレビシエのDug3pのアミノ酸配列
配列番号7:サッカロミセス・セレビシエのECM38の塩基配列
配列番号8:サッカロミセス・セレビシエのEcm38pのアミノ酸配列
配列番号9:サッカロミセス・セレビシエのGSH1の塩基配列
配列番号10:サッカロミセス・セレビシエのGsh1pのアミノ酸配列
配列番号11:サッカロミセス・セレビシエのGSH2の塩基配列
配列番号12:サッカロミセス・セレビシエのGsh2pのアミノ酸配列
配列番号13:サッカロミセス・セレビシエのYCF1の塩基配列
配列番号14:サッカロミセス・セレビシエのYcf1pのアミノ酸配列
配列番号15、16:URA3増幅用プライマー
配列番号17〜20:ADH1プロモーター領域増幅用プライマー
配列番号21、22:pADH1-URA3-pADH1断片増幅用プライマー
配列番号23、24:URA3増幅用プライマー
配列番号25、26:DUG1破壊用プライマー
配列番号27、28:DUG2破壊用プライマー
配列番号29、30:DUG3破壊用プライマー
配列番号31、32:ECM38破壊用プライマー
配列番号33、34:BY4743遺伝子破壊株セットを鋳型としたECM38破壊用プライマー
配列番号35、36:RT-PCRに用いるDUG1増幅プライマー
配列番号37、38:RT-PCRに用いるDUG2増幅プライマー
配列番号39、40:RT-PCRに用いるDUG3増幅プライマー
配列番号41、42:RT-PCRに用いるACT1増幅プライマー
配列番号43〜46:PGK1プロモーター領域増幅用プライマー
配列番号47、48:pPGK1-URA3-pPGK1断片増幅用プライマー
配列番号49、50:BY4743遺伝子破壊株セットを鋳型としたGSH2破壊用プライマー
【特許請求の範囲】
【請求項1】
γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを調製することを特徴とする、酵母エキスの製造方法。
【請求項2】
前記グルタチオン分解酵素が下記(A)〜(D)から選ばれる1またはそれ以上のタンパク質である、請求項1記載の方法。
(A)DUG1遺伝子にコードされるタンパク質
(B)DUG2遺伝子にコードされるタンパク質
(C)DUG3遺伝子にコードされるタンパク質
(D)ECM38遺伝子にコードされるタンパク質
【請求項3】
前記酵母は、少なくとも液胞局在グルタチオン分解酵素の活性が低下し、さらに液胞グルタチオントランスポーターの発現が増大するよう改変されている、請求項1または2記載の方法。
【請求項4】
γ−グルタミルシステイン合成酵素の活性が増大することによりγ−グルタミル化合物合成能が増大した、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
グルタチオン合成酵素の活性が増大または低下することによりγ−グルタミル化合物合成能が増大した、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記γ−グルタミル化合物が、グルタチオン、γ−グルタミルシステイン、およびγ−グルタミル−α−アミノ酪酸から選択される化合物である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、およびペプチドから選択される化合物が添加された培地で前記酵母を培養することを特徴とする、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記酵母がサッカロミセス属に属する酵母である、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記酵母がサッカロミセス・セレビシエである、請求項8に記載の方法。
【請求項10】
請求項1〜9のいずれか1項に記載の方法により製造された酵母エキスを含有する飲食品。
【請求項11】
γ−グルタミルシステイン合成酵素の活性が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母。
【請求項12】
さらに、下記(I)および(II)から選ばれる1またはそれ以上の性質を有する請求項11に記載の酵母。
(I)グルタチオン合成酵素の活性の増大、または低下
(II)液胞グルタチオントランスポーターの発現の増大
【請求項1】
γ−グルタミル化合物合成能が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母を原料として用いて酵母エキスを調製することを特徴とする、酵母エキスの製造方法。
【請求項2】
前記グルタチオン分解酵素が下記(A)〜(D)から選ばれる1またはそれ以上のタンパク質である、請求項1記載の方法。
(A)DUG1遺伝子にコードされるタンパク質
(B)DUG2遺伝子にコードされるタンパク質
(C)DUG3遺伝子にコードされるタンパク質
(D)ECM38遺伝子にコードされるタンパク質
【請求項3】
前記酵母は、少なくとも液胞局在グルタチオン分解酵素の活性が低下し、さらに液胞グルタチオントランスポーターの発現が増大するよう改変されている、請求項1または2記載の方法。
【請求項4】
γ−グルタミルシステイン合成酵素の活性が増大することによりγ−グルタミル化合物合成能が増大した、請求項1〜3のいずれか1項に記載の方法。
【請求項5】
グルタチオン合成酵素の活性が増大または低下することによりγ−グルタミル化合物合成能が増大した、請求項1〜4のいずれか1項に記載の方法。
【請求項6】
前記γ−グルタミル化合物が、グルタチオン、γ−グルタミルシステイン、およびγ−グルタミル−α−アミノ酪酸から選択される化合物である、請求項1〜5のいずれか1項に記載の方法。
【請求項7】
前記γ−グルタミル化合物を構成するアミノ酸、アミノ酸誘導体、およびペプチドから選択される化合物が添加された培地で前記酵母を培養することを特徴とする、請求項1〜6のいずれか1項に記載の方法。
【請求項8】
前記酵母がサッカロミセス属に属する酵母である、請求項1〜7のいずれか1項に記載の方法。
【請求項9】
前記酵母がサッカロミセス・セレビシエである、請求項8に記載の方法。
【請求項10】
請求項1〜9のいずれか1項に記載の方法により製造された酵母エキスを含有する飲食品。
【請求項11】
γ−グルタミルシステイン合成酵素の活性が増大し、且つ、グルタチオン分解酵素の活性が低下するよう改変された酵母。
【請求項12】
さらに、下記(I)および(II)から選ばれる1またはそれ以上の性質を有する請求項11に記載の酵母。
(I)グルタチオン合成酵素の活性の増大、または低下
(II)液胞グルタチオントランスポーターの発現の増大
【図1】

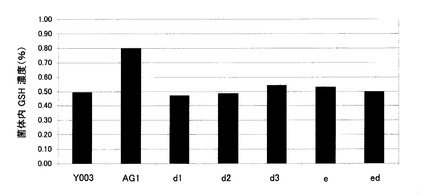
【図2】
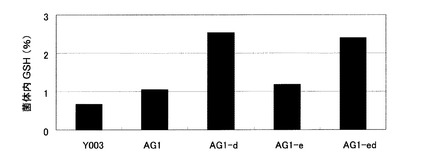
【図3】
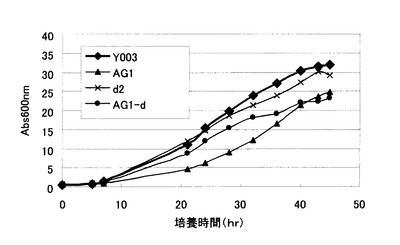
【図4】
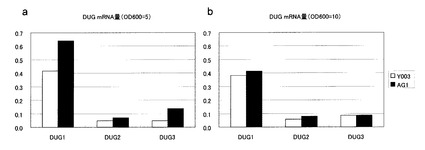
【図5】
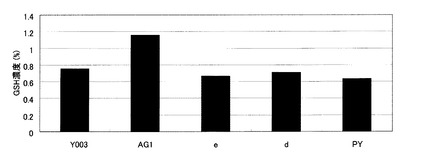
【図6】
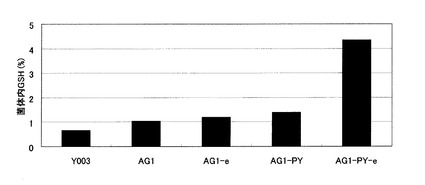
【図7】
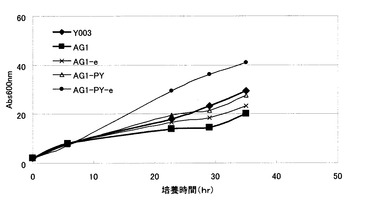
【図8】
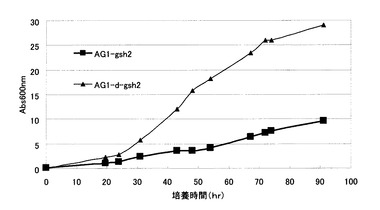
【図9】
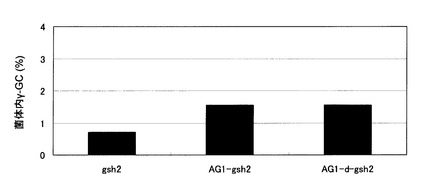
【図10】
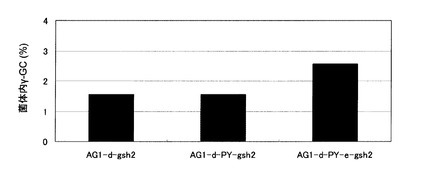
【図11】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2012−213376(P2012−213376A)
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願番号】特願2011−220222(P2011−220222)
【出願日】平成23年10月4日(2011.10.4)
【出願人】(000000066)味の素株式会社 (887)
【Fターム(参考)】
【公開日】平成24年11月8日(2012.11.8)
【国際特許分類】
【出願日】平成23年10月4日(2011.10.4)
【出願人】(000000066)味の素株式会社 (887)
【Fターム(参考)】
[ Back to top ]