
「擬似」天然型化学的ライゲーション
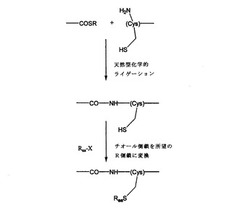
【課題】アミド結合を介し、より広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にするために天然型化学的ライゲーションの技法を拡大するための方法および組成物の提供。
【解決手段】側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質。
【解決手段】側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アミド結合を介し、より広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にするために天然型化学的ライゲーションの技法を拡大するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途を提供する。
【0002】
(関連出願の相互参照)
本出願は、米国特許出願第60/231330号(2000年9月8日出願)および第60/236377号(2000年9月29日出願)の一部継続出願であり、これら双方の出願全体を参照により本明細書に組み込むものとする。
【背景技術】
【0003】
過去30年の間に、疾患を治療するための治療用薬剤として天然に産生されたタンパク質を用いることの可能性に医学上の注目がますます向けられるようになった。
【0004】
組換えDNA技法は、細菌および他の宿主細胞において多量に産生することができるので、多くのポリペプチドおよびタンパク質の商業的産生の主要な方法となっている。組換えタンパク質の産生には、所望の外生タンパク質をコード化するDNAを用いて宿主細胞をトランスフェクトまたは形質転換すること、およびその組換えタンパク質の発現に有利な条件下で細胞を増殖することが含まれる。組換えタンパク質を高力価で産生させることができることから、大腸菌および酵母が宿主として好都合である(米国特許第5756672号(Builder他)を参照のこと)。
【0005】
組換えDNAコード化タンパク質の一般的な細菌の発現に関して、多数の米国特許が発行されている(たとえば、米国特許第4565785号、第4673641号、第4738921号、第4795706号、第4710473号を参照のこと)。残念ながら、組換えDNA技法は例外なく成功したわけではない。いくつかの条件下で、細菌宿主から多量に発現したある種の異種タンパク質は、細胞内で濃縮な凝集物として沈降し、位相差顕微鏡において細胞内部に可視の輝点として認識されることになる。これらの沈降タンパク質の凝集物は「屈折体」と呼ばれ、細胞タンパク質全体のかなり大きな部分を構成する可能性がある(Brems等、Biochemistry(1985)24:7662)。
【0006】
これらの物体からのタンパク質の回収は多数の問題を提起しており、たとえば細胞内に包含されたタンパク質を、それを収容している細胞材料およびタンパク質からどのように分離するのか、その封入体タンパク質を生体活性形態でどのように回収するのかなどの問題がある。屈折体に関する全般的な概説は、Marston、上記;MitrakiおよびKing、Bio/Technology、7:690(1989);MarstonおよびHartley、Methods in Enzymol.、182:264〜276(1990);Wetzel、「Protein Aggregation In Vivo:Bacterial Inclusion Bodies and Mammalian Amyloid」、Stability of Potein Pharmaceuticals:In Vivo Pathways of Degradation and Strategies for Protein Stabilization、AhernおよびManning(編)(Plenum Press、1991);Wetzel、「Enhanced Folding and Stabilization of Proteins by Suppression of Aggregation In Vitro and In Vivo」、Protein Engineering−A Practical Approach、Rees、A.R.等(編)(IRL Press、Oxford University Press、Oxford、1991)を参照されたい。したがって、生物活性タンパク質を産生する別の方法が求められている。
【0007】
タンパク質を組換え産生する別の方法の1つには、タンパク質を合成するために有機化学の原理を用いることが含まれる。現在、タンパク質を化学合成するための方法には、段階固相合成、および溶液中での、または固相でのフラグメント縮合が含まれる。メリフィールド(Merrifield)の古典的な段階固相合成には、所望のペプチド鎖のカルボキシ末端アミノ酸に対応するアミノ酸を固体支持体に共有結合すること、および活性化カルボキシル基を有する活性化アミノ酸誘導体の段階カップリングによってアミノ末端に向かってポリペプチド鎖を延長することが含まれる。完全に保護された固相結合ペプチド鎖のアセンブリが完了した後、ペプチド−固相共有結合を適切な化学方法によって開裂し、保護基を除去して生成物のポリペプチドを得る。
【0008】
残念ながら、段階固相合成法には多くの欠点があり、それには各サイクルのカップリングおよび脱保護での不完全な反応に起因する生成物によって結合した固相の形成が含まれる。ペプチド鎖が長くなるにつれて、高純度の意図した生成物を得ることがより難しくなる。この方法によるタンパク質および大型のポリペプチドの合成は、時間のかかる困難な作業である。
【0009】
固相フラグメント縮合アプローチ(セグメント縮合としても知られる)は、固相段階合成法で長いポリペプチドを得るときの困難を克服するように考案されたものである。このセグメント縮合法には、固相段階法によって数個のペプチドセグメントを調製すること、続いて、最大限に保護されたセグメントを固相から開裂し、精製することが含まれる。保護セグメントは、第1のセグメントに1つずつ縮合され、それが固相に結合する。しかしながら多くの場合、固相セグメント縮合の多くのステップで技術的問題が生じている。E.Atherton等、「Solid Phase Fragment Condensation−The Problems」、Innovation and Perspectives in Solid Phase Synthesis 11〜25(R.Epton等、1990)を参照されたい。たとえば、望ましくないライゲーション反応をブロックするためのセグメント上での保護基の使用によって、しばしば保護セグメントの可溶性はわずかとなり、カルボキシル基の有効な活性化が妨げられる可能性がある。保護セグメントの限定された溶解性はさらに、保護セグメントの精製を妨げる可能性がある。K.Akaji等、Chem.Pharm.Bull.(Tokyo)33:184〜102(1985)を参照されたい。保護セグメントは、純度、共有構造に関して特性付けが困難であり、高分解能分析ESMS(エレクトロスプレー質量分析法)(電荷に基づく)を施すことができない。ライゲーションがグリシン残基で行われる場合を除いて、各活性化ペプチドセグメントのC末端残基のラセミ化も問題である。さらに、完全にアセンブルされた固相結合ポリペプチドの固相からの開裂および保護基の除去は、しばしば、完全にアセンブルされたポリペプチドの分解をもたらす厳しい化学手法および長い反応時間を必要とすることになる。
【0010】
セグメント縮合は、固相上ではなく、溶液中で行うことができる。H.Muramatsu等、Biochem.and Biophys.Res.Commn.203(2):1131〜1139(1994)を参照されたい。しかしながら、溶液中のセグメント縮合は、ライゲーションに先立ってセグメントを精製する必要があり、且つ複数の望ましくない副反応を防ぐために一連の異なる側鎖官能基上で保護基を用いる必要がある。さらに、溶液中のライゲーションでは、固相ライゲーションで提供される簡便な精製および洗浄ステップが行えない。さらに、保護ペプチドセグメントおよび保護ペプチド中間反応生成物の溶解性に関する限定が悪化してくる。
【0011】
最大限に保護されたペプチドでしばしば生じる溶解性の問題を克服するために、ペプチドセグメントの化学的ライゲーションが探求されてきた。化学的ライゲーションには、第1の化学成分と第2の化学成分との間の選択的共有結合の形成が含まれる。第1および第2の成分に存在する一意性の相互反応性官能基を用いて、ライゲーション反応化学選択性を付与することができる。たとえば、ペプチドおよびポリペプチドの化学的ライゲーションには、適合性の、一意性の相互反応性C末端およびN末端アミノ酸残基を有するペプチドまたはポリペプチドセグメントの化学選択的反応が含まれる。この目的のために、いくつかの異なる化学方法が用いられており、その例としては、天然型化学的ライゲーション(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434)、オキシム形成化学的ライゲーション(Rose等、J.Amer.Chem.Soc.(1994)116:30〜34)、チオエステル形成ライゲーション(Schnolzer等、Science(1992)256:221〜225)、チオエーテル形成ライゲーション(Englebretsen等、Tet.Letts.(1995)36(48):8871〜8874)、ヒドラゾン形成ライゲーション(Gaertner等、Bioconj.Chem.(1994)5(4):333〜338)、ならびにチアゾリジン形成ライゲーションおよびオキサゾリジン形成ライゲーション(Zhang等、Proc.Natl.Acad.Sci.(1998)95(16):9184〜9189;Tam他、WO95/00846;米国特許第5589356号)、または他の方法によるもの(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」J.Am.Chem.Soc.2001、123、526〜533;Gieselnan等、Org.Lett.2001、3(9):1331〜1334;Saxon、E.等、「Traceless」Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds.Org.Lett.2000、2、2141〜2143)が挙げられる。
【0012】
これらの方法のなかで、天然型化学的ライゲーション法のみが、ライゲーション部位に天然アミド(すなわちペプチド)結合を有するライゲーション生成物を生じさせる。初期の天然型化学的ライゲーション方法論(Dawson等、上記、およびWO96/34878)は、ライゲーション部位で天然アミド結合を生じるための強固な方法を証明した。天然型化学的ライゲーションには、C末端α−カルボキシチオエステル部分を有する第1のペプチドまたはポリペプチドセグメントと、N末端システイン残基を有する第2のペプチドまたはポリペプチドとの間の化学選択的反応が含まれる。チオール交換反応は、最初のチオエステル結合中間体を生じさせ、これは自発的に転位して、ライゲーション部位に天然アミド結合をもたらし、同時にシステイン側鎖チオールを再生する。初期の天然型化学的ライゲーション法の主な欠点は、N末端システインを必要とすることであり、すなわちこの方法ではN末端システインを有するペプチドおよびポリペプチドセグメントの結合のみが可能となる。
【0013】
このような欠点があるにもかかわらず、システイン以外のN末端アミノ酸を有するペプチドの天然型化学的ライゲーションが提案されている(WO98/28434)。この提案においては、ライゲーションは、C末端α−カルボキシチオエステルを有する第1のペプチドまたはポリペプチドセグメント、および式HS−CH2−CH2−O−NH−[ペプチド]によって表されるN末端のN−{チオール置換補助}基を有する第2のペプチドまたはポリペプチドセグメントを用いて行われる。ライゲーションに続いて、N−{チオール置換補助}基が、HS−CH2−CH2−O−補助基の開裂によって除去され、ライゲーション部位に天然アミド結合が生じる。この方法の1つの限界は、メルカプトエトキシ補助基を使用すると、グリシン残基においてのみ、アミド結合が形成され得ることである。これは、開裂によって第2のペプチドまたはポリペプチドセグメントのN置換アミノ酸の位置にグリシン残基を生じるライゲーション生成物を産生することになる。そのため、この方法の実施は、反応後のライゲーション生成物がそのような位置にグリシン残基を含有することを所望する場合にのみ適しており、いずれにしても、ライゲーション収率、前駆体の安定性、およびO結合補助基を除去できる可能性が問題となり得る。反応をグリシン残基でのライゲーションに限ることなく、他の補助基、たとえばHSCH2CH2NH−[ペプチド]を用いることができるが、そのような補助基は、結合された生成物から除去することができない。
【発明の開示】
【発明が解決しようとする課題】
【0014】
したがって、有効で容易に除去可能なチオール含有補助基を用いることによって、天然型化学的ライゲーションを多種多様な異なるアミノ酸残基、ペプチド、ポリペプチド、ポリマーおよび他の分子に拡大し、且つライゲーション部位においてそのような分子を天然アミド結合で結合する、広い適用範囲を持った、強固な化学的ライゲーション法が求められている。本発明は、このような要望および他の要望に応えるものである。
【課題を解決するための手段】
【0015】
本発明は、アミド結合を介するより広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にするために天然型化学的ライゲーションの技法を拡張するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途を提供する。本発明は、特に、随意ポリマー修飾された合成生物活性タンパク質、およびそのようなタンパク質を含有する薬剤組成物の合成における使用に好適なものである。
【0016】
詳細には、本発明は、側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質を提供する。
【0017】
本発明は、実施態様として、特に、そのタンパク質がリボソーム特定生物活性タンパク質の持つ生物活性を有する前述の合成タンパク質を提供する。
【0018】
本発明は、約25kDaを超えるモノマー分子量を有する前述のの合成タンパク質に関する。
【0019】
本発明は、さらに、タンパク質の複数のアミノ酸残基の少なくとも1つが隣接アミノ酸残基に非アミド結合(たとえば、チオエステル結合、チオエーテル結合およびオキシム結合)によって結合している前述の合成タンパク質に関する。
【0020】
本発明は、さらに、リボソーム特定生物活性タンパク質が哺乳動物のタンパク質(たとえば、ヒト、サル、ウシ、マウス、ブタ、ヒツジ、またはウマのタンパク質)である前述の合成タンパク質に関する。
【0021】
本発明は、さらに、タンパク質が、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカイン(特に、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択されるサイトカイン)の選択された生物活性を有する前述の合成タンパク質に関する。
【0022】
本発明は、さらに、合成生物活性タンパク質がSEP−0、SEP−1およびSEP−3から選択される合成タンパク質に関する。
【0023】
本発明は、さらに、1つまたは複数のそれらのアミノ酸残基が1つまたは複数のポリマー付加物によって修飾されているもの、特に合成タンパク質が、リボソームコード化生物活性タンパク質の少なくとも1つのグリコシル化部位に対応するアミノ酸残基において、1つまたは複数のポリマー付加物によって修飾されている1つまたは複数のアミノ酸残基を含むすべての前述の合成タンパク質に関する。
【0024】
本発明は、さらに、タンパク質がリボソーム特定生物活性哺乳動物タンパク質に関連する生物活性を模倣する生物活性を有し、約25kDaを超えるモノマー分子量を有し、且つ側鎖が式−S−Raaを有し、該式中、Raaはリボソーム特定アミノ酸側鎖の随意置換された末端部分、またはその類似体からなる群から選択される擬似アミノ酸残基を含有する、合成タンパク質を含む分子的に均一な薬剤組成物を提供する。
【0025】
本発明は、さらに、有効量の薬剤組成物を治療を必要としている個体に投与することを含む、ヒトの疾患または症状を治療する方法を提供するものであって、該薬剤組成物は、それぞれが合成タンパク質を含む1つまたは複数の分子的に均一な薬剤組成物を含み、該合成タンパク質は、約25kDaを超えるモノマー分子量を有し、且つ側鎖が式−S−Raaを有し、該式中、Raaは、リボソーム特定アミノ酸側鎖の随意置換された末端部分、またはその類似体からなる群から選択されるものである擬似アミノ酸残基を含有し、該合成タンパク質は、リボソーム特定生物活性ヒトタンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインの生物活性を模倣する生物活性を有するものである。
【0026】
本発明は、さらに、所望のポリペプチドを合成する方法およびそのような方法によって製造された所望のポリペプチドを提供し、該所望のポリペプチドは、以下の式を有し、
aaNH2−Q−aax−aay−W−aaCOOH
該式中、QおよびWは、それぞれ任意で存在する1つまたは複数の追加のアミノ酸残基を示し、aaNH2は、ポリペプチドのN末端アミノ酸残基を示し、aaxおよびaayは、それぞれ側鎖xおよびyを有する内部隣接アミノ酸残基を示し、aaCOOHは、ポリペプチドのC末端アミノ酸残基を示し、該方法は、
(A)式aaNH2−Q−aax−COSRを有し、該式中、Rは、アリール、ベンジルおよびアルキル基を非限定的に含むチオエステル基に適合する任意の基である第1のペプチドを、式Cys−W−aaCOOHを有する第2のペプチドに結合し、それによりポリペプチドaaNH2−Q−aax−Cys−W−aaCOOHを形成すること、および
(B)該ポリペプチドを試薬Raa−Xの存在下でインキュベートすることを含むものであって、該式中、Raaは、アミノ酸残基aayの側鎖の末端部分を模倣する構造を有する基であり、Xは、良好な離脱基(特に、F、IまたはBrなどのハロゲン)であり、該インキュベーションは、所望のポリペプチドを形成するのに充分な条件下で行われる。
【0027】
本発明は、さらに、アミノ酸残基aayが、擬似アルギニン、擬似アスパラギン、擬似アスパラギン酸塩、擬似ドパミン、擬似グルタミン酸塩、擬似グルタミン、擬似ヒスチジン、擬似イソロイシン、擬似ロイシン、擬似リジン、擬似メチオニン、擬似フェニルアラニン、擬似セリン、擬似トレオニン、擬似トリプトファン、擬似チロシンおよび擬似バリンからなる群から選択される前述の方法およびポリペプチドの実施態様を提供する。
【0028】
本発明は、アミド結合を介するより広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にし、それによってそのような分子の化学合成を促進するために天然型化学的ライゲーションの技法を拡張するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途をも提供する。本発明は、特に、好ましくは約25kDaを超えるモノマー分子量を有する随意ポリマー修飾された合成生物活性タンパク質およびそのようなタンパク質を含有する薬剤組成物の合成における使用にも好適である。
【発明を実施するための最良の形態】
【0029】
I.生物活性ペプチドおよびタンパク質の化学合成
一般的に、生物活性ペプチドおよびタンパク質の合成は、標準的な自動ペプチド合成装置を使用する1960年代初期に開発された「メリフィールド」化学反応の段階固相ペプチド合成プロトコルを用いる。そのような合成に固相もしくは液相ライゲーション方法を採用してもよい。そのような化学反応は、多くのポリペプチドを生成するために容易に用いることができるが、それに伴う収量損失、副生成物の生成、および不完全な反応のために、タンパク質または大きいポリペプチドの生成には適さない。
【0030】
これらの制限に対処するために、化学的ライゲーションの技法は、より大きなポリペプチドおよびタンパク質の合成を達成するために、あらかじめ形成されたペプチドフラグメントを合わせて結合できるように開発されてきた。
【0031】
化学的ライゲーションには、第1の化学成分と第2の化学成分との間の選択的共有結合の形成が含まれる。第1および第2の成分に存在する一意性の相互反応性官能基を用いて、ライゲーション反応化学選択性を付与することができる。たとえば、ペプチドおよびポリペプチドの化学的ライゲーションには、適合性の、一意性の相互反応性C末端およびN末端アミノ酸残基を有するペプチドまたはポリペプチドセグメントの化学選択的反応が含まれる。この目的のために、いくつかの異なる化学反応が用いられており、その例としては、天然型化学的ライゲーション(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434)、オキシム形成化学的ライゲーション(Rose等、J.Amer.Chem.Soc.(1994)116:30〜34)、チオエステル形成ライゲーション(Schnolzer等、Science(1992)256:221〜225)、チオエーテル形成ライゲーション(Englebretsen等、Tet.Letts.(1995)36(48):8871〜8874)、ヒドラゾン形成ライゲーション(Gaertner等、Bioconj.Chem.(1994)5(4):333〜338)、ならびにチアゾリジン形成ライゲーションおよびオキサゾリジン形成ライゲーション(Zhang等、Proc.Natl.Acad.Sci.(1998)95(16):9184〜9189;Tam他、WO95/00846;米国特許第5589356号)、または他の方法によるもの(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」J.Am.Chem.Soc.2001、123、526〜533;Gieselnan等、Org.Lett.2001、3(9):1331〜1334;Saxon、E.等、「Traceless」Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds.Org.Lett.2000、2、2141〜2143)が挙げられる。
【0032】
ライゲーションが、N末端システイン残基を有するポリペプチドの結合反応を含む場合、好ましくは天然型化学的ライゲーションの手順が用いられる(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434;米国特許出願第09/097094、これらすべてを参照により本明細書に組み入れるものとする)。天然型化学的ライゲーションには、C末端α−カルボキシチオエステル部分を有する第1のペプチドまたはポリペプチドセグメントと、N末端システイン残基を有する第2のペプチドまたはポリペプチドとの間の化学選択的反応が含まれる。チオール交換反応は、最初のチオエステル結合中間体を生じ、これは自発的に転位して、ライゲーション部位で天然アミド結合をもたらし、同時にシステイン側鎖チオールを再生する。多くの場合、この天然タンパク質の配列は、N末端システイン残基を有するポリペプチドフラグメントが天然型化学的ライゲーション反応で合成され、用いられることができるように、適切に配置されたシステイン残基を含むことになる。
【0033】
この方法は、大きいポリペプチドおよびタンパク質の合成に実用的で強固で有用であることが証明されたが、ライゲーション部位におけるシステイン残基に関する要件がその適応性を制限する。システインは、もっとも一般的でないアミノ酸の1つである。典型的なタンパク質ドメインは、150〜200のアミノ酸を含み、多くのタンパク質は、システイン残基を含有しない長さ60超のアミノ酸領域を含有する。この問題の1つの解決策には、タンパク質の1つまたは複数の天然アミノ酸残基がシステイン残基で置換された非天然タンパク質を設計し、それによって天然型化学的ライゲーション法の使用を可能にすることが含まれる。この解決策は多くの場合、システイン残基の導入がタンパク質の構造または機能に及ぼす可能性のある予測できない影響によって複雑なものとなる。そのようなシステイン残基は、合成されたタンパク質を共有的に固定化するのに用いることができる。
【0034】
本発明は、個別に、または組み合わせて用いることのできる、この問題に対する2つの別の解決策を提供する。第1の解決策(本明細書では「擬似天然型化学的ライゲーション」と呼ぶ)には、タンパク質合成に用いられるペプチドのあらかじめ選択された位置で、非天然擬似アミノ酸残基を用いることが含まれる。そのような擬似アミノ酸の構造は、システインの構造、および合成されるタンパク質のあらかじめ選択された位置で天然に見出されるアミノ酸の構造の両方を模倣したものである。第2の解決策(本明細書では「拡張天然型化学的ライゲーション」と呼ぶ)は、参照により本明細書に組み込むものとする米国特許出願第60/231339号(Kent他、2000年9月8日出願)に記載されており、カルボキシルチオエステル、より好ましくはα−カルボキシルチオエステルを含む第1の成分を、酸安定性N置換、好ましくはNα−置換、2または3炭素鎖アミノアルキルまたはアリールチオールを含む第2の成分と結合することを含む。これらの解決策は、それぞれ以下に詳しく説明する。
【0035】
A.擬似天然型化学的ライゲーション
擬似天然型化学的ライゲーションは、天然型化学的ライゲーションによってライゲーション部位に生じるシステイン側鎖のチオアルキル化を対象とする。好ましい態様は、少なくとも1つのペプチドが、適切な保護基で保護されたチオール側鎖を有する天然システインを含有する、システインライゲーション部位のチオアルキル化である。
【0036】
本発明の好ましい実施形態において、システイン基のチオール部分は、所望の側鎖、たとえばリボソーム特定アミノ酸の側鎖、そのようなアミノ酸の類似体、非リボソーム特定アミノ酸に修飾されている。本明細書では、リボソーム特定アミノ酸とは、タンパク質翻訳のプロセスにおいてリボソームによって認識され、リボソーム生成タンパク質に組み込むことのできるアミノ酸である。相当数の公表された文献が存在し、システイン側鎖チオール部分の化学修飾を記載している(たとえば、「Current Protocols in Protein Science」John E.Coligan等編、John Wiley&Sons、NY(2000)を参照のこと)。Kaiser、E.T.は、天然アミノ酸側鎖の化学特性を模倣するシステイン残基側鎖の変換を記載している(たとえば、Kaiser、E.T.等、「Chemical Mutation Of Enzyme Active Sites」、Science、1984 Nov 2;226(4674):505〜11を参照のこと)。さらに、標識をペプチドまたはタンパク質に導入するためのシステイン側鎖の使用が記載されている。システイン側鎖修飾は、Chemistry of Protein Conjugation and Crosslinking、S.S.Wong(1991、CRC Press);Chemical Modification of Proteins、Gary E.Means等(1971、Holden−Day)、Chemical Modification of Proteins:Selected methods and analytical procedures、Glazer、A.N.等(1975、Elsevier);Chemical Reagents for Protein Modification、RL Lundblad(1991、CRC Press)に概説されている。Tam等(Biopolymers(1998)46:319〜327)は、非システイン天然型化学的ライゲーションでのホモシステイン(−CH2−CH2−SH)の使用、それに続くメチルp−ニトロベンゼンスルホネート(メチル化試薬)を用いるチオアルキル化によるホモシステイン側鎖の天然メチオニン側鎖(−CH2−CH2−S−CH3)への変換を開示している。本発明はまた、ホモシステインを擬似アミノ酸、すなわちメチオニン以外のアミノ酸に変換するために用いることもできる。しかしながら、本明細書に記載のシステインの変換と同様に、本発明によれば、変換を望まない少なくとも1つの天然システインを含有するペプチドの場合、ジスルフィドペアリングに関わる天然システインの分解を回避するために、保護基を用いる必要がある。適切な保護基は以下に記載する。
【0037】
擬似天然型化学的ライゲーション法は、ある種のリボソーム特定アミノ酸の側鎖(たとえば、グリシン、アラニン、バリンおよびプロリンの側鎖)の模倣を促進しないが(しかしながら、アラニンの側鎖は脱硫反応によって形成され得る(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」、J.Am.Chem.Soc.2001、123、526〜533))、多くのリボソーム特定または非コード化アミノ酸を模倣する側鎖を形成するために用いることができる。本発明の擬似天然型化学的ライゲーション法によって生成されたアミノ酸は、チオエーテル結合を含有し、β分岐を持たない(それらのアミノ酸はすべてベータ位でメチル基を含む、すなわちaa−βCH2−S−であるため)。したがって、β分岐アミノ酸、イソロイシンおよびトレオニンの擬似アミノ酸種は、β配置および付随の制約を有することなく、ペンダント側鎖構造を持たせることができる。
【0038】
注目に値すべきなのは、本発明の方法は、リボソーム特定アミノ酸と同じ長さ、あるいはそれよりも長い、または短いアミノ酸側鎖を形成するために用いることができることである。側鎖長のそのような変更は、三次元配座(コンフォメーション)を安定にして(または、不安定にして)、タンパク質安定性を増大する(または、タンパク質がその配座を変え、それによって天然タンパク質に受容されるものと異なる範囲の基質、阻害因子、受容体、リガンドなどを受容する能力を高める)ために用いることができる。たとえば、Cys−CH2−SH + Br−CH2−COOHは、Cys−CH2−S−CH2−COOHを生じる(そのような「擬似グルタミン酸塩」は、1つの追加の側鎖原子、すなわち−S−基を有し、あるいはアスパラギン酸塩の代わりに用いられた場合、2つの追加の側鎖原子、すなわち−CH2−S−基を有する)。他の側鎖は、側鎖に同じ数の原子を有するが、チオエーテル結合(−S−)を包含する点で異なる。たとえば、Cys−CH2−SH + Br−CH2−CH2−NH−PG、それに続くPGの除去によって、Cys−CH2−S−CH2−CH2−NH2が生じる。結果として生じた構造は、側鎖に追加の原子を持たないが、1つの−CH2−基が、−S−で置換されている。メチオニンは別の例であり、Cys−CH2−SH + I−CH2−CH3によって、Cys−CH2−S−CH2−CH3が生じ(天然Met構造、Met−CH2−CH2−S−CH3に対して)、したがってチオエーテルが置き換えられている。アルギニンも同様に、Cys−CH2−SH + Br−CH2−NH−CH((−NH2)(=NH2+))によって、Cys−CH2−S−CH2−NH−CH((−NH2)(=NH2+))が生じる。好ましくは、所望でない副反応を回避するために、特に擬似リジンを構成する場合は、反応性アミノ基の保護を用いることができる。ひとたびチオアルキル化反応が遂行されたら、その保護基を除去できる。
【0039】
一般に、できる限り厳密に天然タンパク質を模倣することを望む場合、タンパク質のそのような位置に通常存在するリボソーム特定アミノ酸と同じ長さの側鎖長を有する擬似アミノ酸分子を用いるのがもっとも好ましく、リボソーム特定アミノ酸より1原子長い側鎖長を有する擬似アミノ酸分子の使用は好ましさに劣り、リボソーム特定アミノ酸より2原子長い側鎖長を有する擬似アミノ酸分子の使用はさらに好ましさに劣る。さらに、遺伝子変化が機能を崩壊する見込みがないか、あるいは関連タンパク質のその部位でアミノ酸が保存されているような位置のシステインライゲーション部位を選択することが好ましい。そのような部位は、アラニンスキャニング、ホモロジーモデリングおよび他の方法によって同定できる。
【0040】
擬似天然型化学的ライゲーションでは、天然型化学的ライゲーションと同様に、アミノ末端システイン残基を含有するペプチドを、カルボキシ末端チオエステルを有するペプチドに結合する。その後、システインのチオール側鎖を、式Raa−Xであって、該式中、Xは良好な離脱基であり、Raaは、その構造がリボソーム特定アミノ酸または合成アミノ酸の側鎖の末端部分を模倣する基である化合物と反応させる。
【0041】
注目に値すべきなのは、擬似天然型化学的ライゲーション反応は、システイン側鎖の天然L立体配置、またはD立体配置を用いて作用することである。D立体配置の使用によって、そのライゲーション部位にプロテアーゼ耐性を付与することができ、したがってタンパク分解に対する高い安定性が所望であるとき、その使用が望ましい可能性がある。しかしながら、D−システインの使用においては、その側の主鎖構造が変更されることになる。そのような変更は、プロテアーゼ耐性に加えて、生物活性を変えるのに望ましい可能性がある。しかしながら、生物活性への影響力を最小にするために、その分子の無秩序末端の無秩序領域など可撓性の高い部位、たとえば結果として生じる折りたたまれた分子の表面に位置することになる無秩序ループなどにD−システインを配置することが好ましい。望ましくは、擬似天然型化学的ライゲーション反応は、合成分子の表面上に大きな荷電側鎖(たとえば、Lys、Arg、Asp、またはGluの側鎖)を配置するために用いることができる。
【0042】
適切な良好な離脱基、X、の例には、ハロゲン、特にヨウ素および臭素が含まれる。Raa基の例には、PO4、COOH、COO、CONH2、グアニジニウム、アミン、アルキル、置換アルキル、アリール、置換アリール、イミダゾール、アルキル化イミダゾール、インドール、またはアルキル化インドール基が含まれる。
【0043】
用いるRaaの選択は、特定の位置に存在することを所望するアミノ酸側鎖に応じて決まることになる。したがって、たとえば、
aaNH2−Q−aax−aay−W−aaCOOH
であって、該式中、QおよびWは、それぞれ任意で存在するか、または存在しない追加のアミノ酸残基を示し、aaxおよびaayは、内部隣接残基を示し(それぞれ側鎖xおよびyを有する)、aaNH2およびaaCOOHは、それぞれポリペプチドまたはタンパク質のアミノ(N−)末端残基およびカルボキシ(C−)末端残基を示すアミノ酸配列を有する所望のポリペプチドまたはタンパク質は、それぞれ
aaNH2−Q−aax−COSR および Cys−W−aaCOOH
であって、該式中、Cysは、システインによるaayの置換を示し、Rは、これに限定されるものではないが、アリール、ベンジルおよびアルキル基を含む、チオエステル基と適合する任意の基である上記の2つのペプチドフラグメントを調製することによって合成できる。Rの例には、3−カルボキシ−4−ニトロフェニルチオエステル、ベンジルエステルおよびメルカプトプロピオン酸ロイシンエステルが含まれる(たとえば、Dawson等、Science(1994)266:776〜779;Canne等、Tetrahedron Lett.(1995)36:1217〜1220;Kent他、WO96/34878、Kent他、WO98/28434;Ingenito等、JACS(1999)121(49):11369〜11374;Hackeng等、Proc.Natl.Acad.Sci.U.S.A.(1999)96:10068〜10073を参照のこと)。他の例には、同様によく知られているインテイン媒介の生物学的技法によって生成することのできるジチオトレイトール、あるいはアルキルまたはアリールチオエステルが含まれ(たとえば、Chong等、Gene(1997)192:277〜281;Chong等、Nucl.Acids.Res.(1998)26:5109〜5115;Evans等、Protein Science(1998)7:2256〜2264;Cotton等、Chemistry&Biology(1999)6(9):247〜256を参照のこと)、次いでそれらのフラグメントを合わせて結合することによって以下が形成される。
aaNH2−Q−aax−Cys−W−aaCOOH
【0044】
次いで、結合されたフラグメントを、式中、Ryは側鎖yの構造を模倣する側基であるRy−Xと反応させる。この反応は、システインのチオール基を「擬似y」側鎖に変換するのに充分な条件下で行われる。たとえば、RaaがCH2−COOHまたは(CH2)2−COOHとなるように選択される場合、この反応は、アスパラギン酸塩(「擬似Asp」)またはグルタミン酸塩(「擬似Glu」)の構造および機能を模倣するアミノ酸残基を形成することになる。理解されるように、上記の説明を考慮すれば、3つ以上のペプチドフラグメントを用いてより複雑な合成を行うことができる。
【0045】
本発明による「擬似アミノ酸」を含む天然型化学的ライゲーションは、タンパク質を化学合成するための天然型化学的ライゲーション方法の適応性を著しく拡大する。「擬似アミノ酸」を含む天然型化学的ライゲーションは、ライゲーションステップにおいて同じ化学反応を用いて、天然チオール含有側鎖官能基とCys残基でペプチド結合を形成し、一意性の第2のステップにおいて、ライゲーション部位のCysの天然チオール含有側鎖が化学選択的反応によって変換されて、遺伝コード化アミノ酸と同じ官能基を含有する非天然側鎖を生じる。
【0046】
たとえば、ライゲーション部位のシステインは、水中、pH中性でブロモ酢酸(または他のハロ酢酸)と反応させることができ、ライゲーション部位のCysの非保護チオールのみがこのような条件下で反応して、ライゲーション部位にS−カルボキシメチル化残基を含有するポリペプチド、すなわち、
【化1】

を生じる。
【0047】
この非天然アミノ酸は、側鎖にカルボキシル成分、すなわちグルタミン酸残基およびアスパラギン酸残基と同様な官能基、を含有する。このS−カルボキシメチル化Cysを、本明細書では、(1つの過剰な側鎖原子が存在し、チオエーテル結合を形成する−S−基である場合)「擬似グルタミン酸」(「擬似グルタメート(glutamate)」「擬似Glu」)と呼ぶ(2つの過剰な側鎖原子が存在し、その1つがチオエーテル結合を形成する−S−基である場合、「擬似アスパラギン酸」と呼ぶことができる)。多くのタンパク質の場合、この「擬似アミノ酸」であっても荷電カルボキシル化側鎖の単純保存は、タンパク質に所望の特性を保持または付与するのに充分であろう。重要な相違は、それが、適切なシステインのない位置で天然型化学的ライゲーション部位として機能することである。
【0048】
ライゲーション部位でCys残基の側鎖チオールを修飾するために他の試薬を用いることによって、他の「擬似アミノ酸」残基が生じ得る。たとえば、チオールのハロアセトアミド、たとえばブロモアセトアミドとの反応によって、ライゲーション部位に「擬似グルタミン」、すなわち、
【化2】
が生じる。
同様に、N−保護ハロアルキルアミン、たとえば、
【化3】
との反応、それに続くアミノ保護基の除去によって、「擬似リジン」残基、すなわち、
【化4】
が生じる。
ライゲーション部位のCys残基の適切なハロアルカン、たとえば、
【化5】
との反応によって、「擬似ロイシン」アミノ酸、すなわち、
【化6】
が生じる。
Rは、H、OH、アリールなどであり得る、他の適切に選択されたアリール含有ハロゲン化物、たとえば、
【化7】
を、Phe、Tyr、またはTrp残基に対応する擬似アミノ酸、すなわち、
【化8】
を生成するために同様に用いることができる。
【0049】
このアプローチの著しい特徴は、ライゲーション部位に存在するもの以外のCys残基の化学修飾を防ぐために、反応セグメントの修飾を望まないシステイン残基が、たとえばCys(Acm)として側鎖保護されること、または反応セグメントの他のCys残基が、ライゲーション後の化学修飾反応による同一「擬似アミノ酸」の同時形成に向けられることである。同じ原理がライゲーション部位でのホモシステインの使用に適用されることが理解されるであろう。さらに、セレノシステインがシステイン媒介天然型化学的ライゲーションによってライゲーション部位で用いられ、類似のアルキル化反応によって変換されて、セレノエーテル基がチオエーテル基に置き換わる本発明の擬似アミノ酸が生成されることも理解されるであろう。
【0050】
本明細書では、記号φは、ベンジル基を示し、IMは、イミダゾール基を示し、INは、インドール基を示し、PGは、保護基を示す。以下は、本発明によって擬似アミノ酸残基を含有するペプチドの合成に用いることのできるRaa側基の概要である(Xがハロゲン(I、Br、Cl、F)である場合であり、IおよびBrは、ほとんどの場合に好ましく、Fは、φ結合に好ましい)。
【0051】
塩基性アミノ酸:
Lys(余分な原子無し)
−CH2−SH + X−CH2−CH2−NH−PG、続いて脱保護することにより、−CH2−S−CH2−CH2−NH2が得られる。
Arg(余分な原子無し)
−CH2−SH + X−CH2−NH−C((NH2)(=NH2+))により
−CH2−S−CH2−NH−C((NH2)(=NH2+))が得られる。
His(余分な原子2個)
−CH2−SH + X−CH2−IM−PG、続いて脱保護することにより
−CH2−S−CH2−IMが得られる。
【0052】
酸性アミノ酸:
Asp(余分な原子2個)
−CH2−SH + X−CH2−COOHにより−CH2−S−CH2−COOHが得られる。
Glu(余分な原子1個)
−CH2−SH + X−CH2−COOHにより−CH2−S−CH2−COOHが得られる。
【0053】
非荷電極性アミノ酸:
Tyr(余分な原子無しまたは1個)
−CH2−SH + F−φ−pOHにより−CH2−S−φ−pOHが得られる(余分な原子無し、同じ幾何配置)。
−CH2−SH + Br/I−CH2−φ−pOHにより−CH2−S−CH2−φ−pOHが得られる(余分な原子1個)。
Gln(余分な原子1個)
−CH2−SH + X−CH2−C(O)(NH2)により−CH2−S−CH2−C(O)(NH2)が得られる。
Asn(余分な原子2個)
−CH2−SH + X−CH2−C(O)(NH2)により−CH2−S−CH2−C(O)(NH2)が得られる。
Ser(余分な原子2ないし3個)
−CH2−SH + X−CH2OHにより−CH2−S−CH2OHが得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH2OHにより−CH2−S−CH2−CH2OHが得られる(余分な原子3個)。
Thr(余分な原子2ないし3個、β分岐が無い)
−CH2−SH + X−CH((CH3)(O−PG))に続いてPGを除去することにより−CH2−S−CH((CH3)(OH))が得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH((CH3)(O−PG))に続いてPGを除去することにより−CH2−S−CH2−CH((CH3)(OH))が得られる(余分な原子3個)。
【0054】
非極性アミノ酸:
Leu(余分な原子1ないし2個)
−CH2−SH + X−CH((CH3)(CH3))により−CH2−S−CH((CH3)(CH3))が得られる(余分な原子1個)。
−CH2−SH + X−CH2−((CH3)(CH3))により−CH2−S−CH2−CH((CH3)(CH3))が得られる(余分な原子2個)。
Ile(余分な原子2ないし3個、β分岐無し)
−CH2−SH + X−CH(CH3)−CH2−CH3により−CH2−S−CH(CH3)−CH2−CH3が得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH(CH3)−CH2−CH3により−CH2−S−CH2−CH(CH3)−CH2−CH3が得られる(余分な原子3個)
Phe(余分な原子1ないし2個)。
−CH2−SH + F−φにより−CH2−S−φが得られる(余分な原子1個)。
−CH2−SH + Br/I−CH2−φにより−CH2−S−CH2−φが得られる(余分な原子2個)。
Met(余分な原子無し)
−CH2−SH + I−CH2−CH3により−CH2−SH−CH2−CH3が得られる。
Trp(余分な原子1個)
−CH2−SH + F−INにより−CH2−S−INが得られる。
【0055】
一般的な擬似化学的ライゲーション反応を図1に例示する。好ましい擬似アミノ酸を形成するために用いることのできる好ましいRaaおよびX基を表Iに示す。残基は非荷電として示すが、NH2またはCOOHなどのイオン性基は、それらの荷電形態であるか、または塩として提供されてもよいことが理解されるであろう。擬似リジンを形成するために用いられる反応を除いて、すべての反応は水性であり、pH8〜9で行われ、反応の化学量および最適化は、標準的なプロトコルに従って調整することができる。擬似リジンは、pH8〜9で行われる水性反応、それに続くピペリジン/H2O(トリフルオロ酢酸(TFA))中の反応で形成される。
【表1】
【表2】
【表3】
【表4】
【0056】
本発明のさらなる実施形態において、システイン残基のチオール基は、酸化するか(−SH→−SO3−)、または酸素と置換してセリンを形成することができる(−SH→−OH)。そのような置換は、システインをトシル−Cl(Tos−Cl)と反応させ、それによってチオール基−SHを、−S−Tos基に変換することによって達成できる。強塩基の存在下、OHはTos置換基に代わり、それによって所望の−OH基が形成される。
【0057】
さらに、システインのチオール基は、以下に示すように、マイケル付加反応によって修飾することができ、
【化9】
該式中、Rは、H、アルキル、アリール、アルキルアリール、アミノであるか、またはポリマー分子であって、分岐または非分岐、ランダムまたはブロックであってよい。
【0058】
本発明のさらなる態様は、アミノ末端システイン残基がアルキルアミノ(好ましくはメチルアミノ)部分を含むように修飾されたペプチドフラグメントの使用を対象とする。
【化10】
【0059】
アルキルアミノ部分のアミノ基を保護することによって、ペプチドAを一般式ペプチドB−αCOSRを有する第2のペプチドと結合することができる。保護基の除去によって、一般式ペプチドC−αCOSRを有するさらなるペプチドを、前に結合したペプチドに結合し、分岐ペプチド複合体を形成することができる。
【化11】
【0060】
この分岐タンパク質は依然としてシステインのチオール基を含有するので、上に記載した様式でRaa−Xと反応し、それによって側鎖のさらなる修飾が可能となる。特に、R置換基としてアルキルアミドを用いることによって、別のアミノ基を分子に導入することができ、それによってさらなる分岐、またはさらなるライゲーション反応が可能となる。
【化12】
【0061】
本発明のさらなる実施形態において、システイン側鎖の修飾を用いて、任意の種々の標識、または結合分子を導入することができる。たとえば、電子スピン共鳴、蛍光(たとえば、アンモニウム4−クロロ−7−スルホベンゾ−フラザン(SBF)Pierce Chemical Co.)、または磁気画像法によって検出可能な標識を用いることができる。そのような標識付けは、疾患および症状の診断において、ならびに分子相互作用の分析(たとえば、内在性膜タンパク質における距離の測定)において有用である。本発明の方法は、標識および結合分子の使用に関して、いくつかの顕著な利点を有する。そのような置換基は、部位特異的、非ランダム様式で組み込むことができる。さらに、この標識部分は、続いて起こる化学的ライゲーションステップ(存在する場合)に対して安定であることだけが必要とされる。そのような反応は一般に、非常に穏やかな条件下で行われる(たとえば、天然型化学的ライゲーション、他のシステイン残基のチオール基を保護するために随意用いられる可能性のあるAcmまたは他の保護基の除去(Hg(CH2COOH)2など)、および逆相HPLC精製に用いられるものなど)。そのような追加のチオール基の保護は、Cys含有ペプチドの酸化性重合を妨げ、したがってそれらの精製および取扱いを容易にする。
【0062】
B.拡張天然型化学的ライゲーション
上述したように、天然タンパク質の配列が適切なシステイン残基を含有せず、ポリペプチドのN末端にシステイン残基を導入するように天然タンパク質配列を修飾することが不都合であるか、または望ましくない場合、そのN末端がN置換、好ましくはNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含有し、それによってポリペプチド欠失システイン残基に天然型化学的ライゲーションの原理を用いることができるように修飾されたポリペプチドを結合するために、「拡張天然型化学的ライゲーション」の方法を用いることができる(参照により本明細書に組み込むものとする米国特許出願第60/231339号を参照のこと)。
【0063】
「拡張天然型化学的ライゲーション」の方法には、カルボキシルチオエステル、より好ましくはα−カルボキシルチオエステルを含む第1の成分を、酸安定性N置換、好ましくはNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含む第2の成分と結合することが含まれる。第1成分のカルボキシチオエステルと、第2成分のN置換2または3炭素鎖アルキルまたはアリールチオールのチオールとの間の化学選択的反応は、チオエステル結合中間体を経て進み、最初のライゲーション生成物に分解する。より具体的には、COSRチオエステル成分とアミノアルキルチオール成分との間で起こるチオール交換は、チオエステル結合中間体ライゲーション生成物を生じ、これは自発転位の後、アミノアルキルチオール成分がそれぞれ式IまたはIIを有するかどうかに応じて、5員または6員環中間体を経て最初のアミド結合ライゲーション生成物を生じ、
該式中、J1は、随意保護された1つまたは複数のアミノ酸側鎖を有するペプチドまたはポリペプチド、あるいはそのようなペプチドまたはポリペプチドの部分、ポリマー、色素、適切に機能化された表面、リンカーまたは検出可能マーカー、あるいは化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の他の任意の化学部分であり、R1、R2およびR3は、独立して、H、またはC1と共役した電子供与基であるが、ただしR1、R2およびR3の少なくとも1つは、C1と共役した電子供与基を含み、J2は、随意保護された1つまたは複数のアミノ酸側鎖を有するペプチドまたはポリペプチド、あるいはそのようなペプチドまたはポリペプチドの部分、ポリマー、色素、適切に機能化された表面、リンカーまたは検出可能マーカー、あるいは化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の他の任意の化学部分である。
【0064】
ライゲーション部位のN置換2または3炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]または[HS−(C3(R3)−C2(R2)−C1(R1)−]は、生成物への損傷なしに、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式IIIの最終ライゲーション生成物を生じる。
該式中、J1、J2、R1、R2およびR3は、上に定義したとおりである。
【0065】
R1、R2およびR3基は、ペプチド適合性開裂条件下でN−C1結合の開裂を促進するように選択される。たとえば、電子供与基は、特にC1に共役している場合、C1で開裂を促進する共鳴安定化カチオンを形成するために用いることができる。この化学的ライゲーション反応は、好ましくは、賦形剤としてチオール触媒を含み、中性に近いpH条件、水性または混合有機−水性条件で行われる。第1および第2成分の化学的ライゲーションは、5または6員環を経て進行することができ、5または6員環は自発転位を受けて、N置換アミド結合ライゲーション生成物を生じる。第1および第2の成分がペプチドまたはポリペプチドである場合、N置換アミド結合ライゲーション生成物は式IVまたはVを有し、
該式中、J1、J2およびR1、R2、R3は、上に定義したとおりであり、Z2は、アミノ酸側鎖、またはその側鎖の誘導体である。
【0066】
N置換アミド結合ライゲーション生成物の共役電子供与基R1、R2、またはR3は、N−C1結合の開裂、およびN置換アミド結合ライゲーション生成物からの2または3炭素鎖アルキルまたはアリールチオールの除去を促進する。ペプチド適合性開裂条件下でのNのアルキルまたはアリールチオール鎖の除去によって、ライゲーション部位に天然アミド結合を有するライゲーション生成物が生じる。第1および第2成分がペプチドまたはポリペプチドである場合、ライゲーション生成物は以下の式を有することになる。
【0067】
式IのR1置換基の例を表IIに示す。
【表5】
【0068】
式IIのR1、R2およびR3置換基の例を表IIIに示す。
【表6】
【0069】
N置換2炭素鎖化合物と同様に、オルトまたはパラ位におけるベンジルおよびピコリル電子供与置換基R1’、R3’およびR5’の位置決定は、ライゲーションに続くNα−C1結合の強固な開裂のためにC1炭素に対する電子共役を維持する必要がある。しかしながら、R2およびR3がC2およびC3と共にベンジル基を形成するとき、R1’およびR3’の少なくとも1つは、強い電子供与基を含み、ここでR1’またはR3’は、メトキシ(−OCH3)、チオール(−SH)、ヒドロキシル(−OH)およびチオメチル(−SCH3)から選択される。R2およびR3が水素であるN置換3炭素鎖チオールの場合、R1は、R1’、R3’およびR5’が強い電子供与基または中強度の電子供与基、あるいはそれらの組み合わせを含むベンジルまたはピコリル基を含む。N置換2炭素鎖アルキルまたはアリールチオールと同様に、強い電子供与基は、ライゲーションに続く開裂に対する3炭素鎖アルキルまたはアリールチオールの感受性を高める。したがって、特定の電子供与基またはその組み合わせはそれに応じて選択できる。
【0070】
N置換2炭素鎖化合物と同様に、本発明のN置換3炭素鎖化合物は、対象となる構成において置換に利用できるとき、R1’およびR5’位のR1の置換基としてチオールを含むことができる。ここでも、電子供与チオール基はC1と共役し、これらの位置におけるその導入は、化合物が6員環のライゲーション事象形成のための2つの経路を有することを可能にする。これはさらに、α−カルボキシチオエステルとの反応に利用できるチオールの局所濃度を高め、構造的制約の点から別の配位を提供し、ライゲーションを向上することができる。
【0071】
本発明のN末端N置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸の合成は、たとえばスキームIおよびスキームIIによって本明細書に記載のとおり、且つ当分野で知られている標準的な有機化学技法に従って実行することができる。たとえば、「Advanced Organic Chemistry,Reactions,Mechanisms,and Structure」、第4版、J.March編、John Wiley&Sons、New York、NY、1992;「Comprehensive Organic Transformations,A Guide to Functional Group Preparations」、R.Larock編、VCH Publishers、New York、NY、1989を参照されたい。それらのアミノ酸は、溶液中で、ポリマー担持型合成によって、またはその組み合わせによって合成することができる。好ましいアプローチは、Nα−保護Nアルキル化S保護アミノアルキルまたはアリールチオールアミノ酸前駆体を用いる。合成に用いられる試薬は、多くの製造業者から入手できる。さらに、出発成分および種々の中間体、たとえば個々のアミノ酸誘導体などは、キットなどで提供され、後の使用のために貯蔵できることが充分に理解されるであろう。
【0072】
本発明のN末端Nα−置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸の調製において、保護基手順が用いられる。種々の合成手順一般において用いられる好ましい保護基(PG)は、固相ペプチド合成(「SPPS」)に適合性である。場合によってはさらに、異なる条件下で除去可能な直交性の保護基を用いる必要がある。多くのそのような保護基が知られており、この目的に適している(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。例には、ベンジルオキシカルボニル(Z)、Boc、Bpoc、Trt、Nps、FmocCl−Z、Br−Z、NSC、MSC、Ddeなどが含まれる。硫黄部分の場合、適切な保護基の例には、これに限定されるものではないが、ベンジル、4−メチルベンジル、4−メトキシベンジル、トリチル、ACM、TACAM、キサンチル、ジスルフィド誘導体、ピコリルおよびフェナシルが含まれる。
【0073】
より具体的には、このNα−置換2または3炭素鎖アルキルまたはアリールチオールは、スキームI(Nα−置換前駆体の固相調製)、スキームII(Nα−置換前駆体の液相調製)に従って調製することができる。スキームIにおいて、Nα置換−2または3炭素鎖アルキルまたはアリールチオールは、ポリマー担持型有機合成の標準方法を用いて固相上で直接組み上げされるが、一方スキームIIのNα−保護、Nアルキル化、S保護、アミノアルキルまたはアリールチオールアミノ酸前駆体は、標準的なカップリングプロトコルを用いて樹脂に結合される。ラセミまたはジアステレオマー生成物が生成される場合、拡張天然型化学的ライゲーションに用いる前に、標準的な方法によるこれらの分離が必要となる可能性がある。
スキームI
ECL補助誘導化分子の固相合成
【化13】
スキームII
【化14】
【0074】
α−カルボキシチオエステルは、実施例を含む本明細書で記載の技法など、当分野で知られている標準的な技法に従う化学または生物学法によって生成することができる。化学合成の場合、α−カルボキシチオエステルペプチドは、溶液中、またはチオエステル生成樹脂から合成することができ、その技法はよく知られている(たとえば、Dawson等、上記;Canne等、上記;Hackeng等、上記、Aimoto等を参照のこと)。たとえば、化学合成チオエステルペプチドは対応するペプチドα−チオ酸から製造することができ、ペプチドα−チオ酸は、樹脂法が好ましいが、チオエステル樹脂上、または溶液中で合成することができる。このペプチドα−チオ酸は、対応する3−カルボキシ−4−ニトロフェニルチオエステル、対応するベンジルエステル、または任意の多様なアルキルチオエステルに変換することができる。これらのチオエステルはすべて、ライゲーション反応のための充分良好な離脱基を提供し、3−カルボキシ−4−ニトロフェニルチオエステルは、アルキルチオエステルに比べてより反応性である可能性のある、対応するベンジルチオエステルよりもいくらか速い反応速度を示す。別の例として、トリチル関連メルカプトプロピオン酸ロイシンチオエステル生成樹脂は、C末端チオエステルを構成するために用いることができる(Hackeng等、上記)。C末端チオエステル合成も、ジアゾメタンまたはヨードアセトニトリルによる活性化、それに続く適切なチオールによる置換によって、3−カルボキシプロパンスルホンアミド安全捕捉(セイフティーキャッチ)リンカーを用いて達成することができる(Ingenito他、上記;Bertozzi等)。
【0075】
本発明のN置換2または3炭素鎖アルキルまたはアリールチオール成分の、第1カルボキシチオエステル成分とのライゲーションによって、ライゲーション部位にN置換アミド結合を有するライゲーション生成物が生じる。この反応のライゲーション条件は、チオエステルのN置換2または3炭素鎖アルキルまたはアリールチオール部分との選択的反応性を維持するように選択される。好ましい実施形態において、ライゲーション反応は、pH6〜8を有する緩衝液中で実行され、好ましいpHの範囲は6.5〜7.5である。この緩衝液は、水性、有機性、またはその混合物であることができる。ライゲーション反応はさらに、1種または複数の触媒および/または1種または複数の還元剤、脂質、洗浄剤、他の変性剤または可溶化試薬などを含むことができる。好ましい触媒の例は、チオールおよびホスフィン含有部分、たとえばチオフェノール、ベンジルメルカプタン、TCEPおよびアルキルホスフィンなどである。変性剤および/または可溶化剤の例には、グアニジニウム、尿素の水またはTFE、HFIP、DMF、NMPなど有機溶媒溶液、水、またはグアニジニウムおよび尿素水溶液と混合したアセトニトリルが含まれる。温度もライゲーション反応の速度を調節するために利用することができ、通常は5℃から55℃の間であり、好ましい温度は15℃から40℃の間である。例として、ライゲーション反応は、pH6.8から7.8の間で、6Mグアニジニウム中の2%チオフェノールを用いる反応系において良好に進行する。
【0076】
N置換2炭素鎖アルキルまたはアリールチオールの場合、ライゲーション事象は、COSRチオエステル成分とアミノアルキルチオール成分との間に起こるチオール交換に起因する。この交換によってチオエステル結合中間体ライゲーション生成物が生成され、これは自発転位の後、5員環中間体を経て、ライゲーション部位に除去可能なN置換2炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]を有し、置換基は上に定義したとおりである式J1−HN−CH(Z1)−C(O)−Nα(C1(R1)−C2−SH)−CH(Z2)−J2の最初のライゲーション生成物を生じる。このライゲーション部位のN置換2炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]は、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式J1−HN−CH(Z1)−CO−NH−CH(Z2)−CO−J2の最終ライゲーション生成物を生じる。
【0077】
N置換3炭素鎖アリールまたはアルキルチオールの場合、COSRチオエステル成分とアミノアルキルチオール成分との間のチオール交換によってチオエステル結合中間体ライゲーション生成物が生成され、これは自発転位の後、6員環中間体を経て、ライゲーション部位に除去可能なN置換3炭素鎖アルキルまたはアリールチオール[HS−C3(R3)−C2(R2)−C1(R1)−]を有する式J1−HN−CH(Z1)−C(O)−Nα(C1−C2(R2)−C3(R3)−SH)−CH(Z2)−J2の最初のライゲーション生成物を生じる。このライゲーション部位のN置換3炭素鎖アリールチオール[HS−C3(R3)−C2(R2)−C1(R1)−]は、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式J1−HN−CH(Z1)−CO−NH−CH(Z2)−CO−J2の最終ライゲーション生成物を生じる。
【0078】
N置換アルキルまたはアリールチオール基の除去は、N−C1結合の開裂を促進し、ライゲーション部位に安定化非置換アミド結合を得るために、好ましくは酸性条件で行われる。「ペプチド適合性開裂条件」とは、ペプチドに適合性であり、ライゲーション生成物からのアルキルまたはアリールチオール部分の開裂に適した物理的化学的条件を意味する。一般にペプチド適合性開裂条件は、用いられるα−置換化合物に応じて選択され、これは通常のよく知られたアプローチによって容易に推測できる(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。
【0079】
たとえば、R1、R2、またはR3置換基が、メトキシ、ヒドロキシ、チオールまたはチオメチル、メチルなどを含む場合、除去のためのより普遍的な方法は、ペプチド合成化学に典型的な酸性開裂条件を含む。これには、還元試薬および/またはスカベンジャ系(たとえば、無水フッ化水素(HF)、トリフルオロ酢酸(TFA)、またはトリメチルスルホニルフルオロ酢酸(TMSFA)などの酸)を含むか、または含まない、強酸性条件または水−酸性条件下でのN−C1結合の開裂が含まれる。より具体的な酸性開裂系は、所与の構成体に関してアリールまたはアルキルチオール部分を除去するためにNα−C1結合の開裂を最適化するように選択することができる。そのような条件はよく知られており、ペプチドの完全性の維持に適合する。特にペプチドまたはポリペプチド配列にトリプトファンが存在する場合、トリプトファン側鎖の遊離アリールまたはアルキルチオール部分との反応を回避するために、チオールスカベンジャを含むことができる。チオールスカベンジャの例には、エタンジオール、システイン、β−メルカプトエタノールおよびチオクレゾールが含まれる。
【0080】
他の特殊な開裂条件には、ピコリル基が置換基であるときの光または還元型開裂条件が含まれる。例として、R1、またはR2およびR3置換基がピコリル部分を含むとき、光分解(たとえば、紫外光)、亜鉛/酢酸、または電気分解還元を、標準的なプロトコルに従って開裂のために用いることができる。N置換2炭素鎖チオールのR1がR1にチオメタンを含む場合、水銀またはHF開裂を用いることができる。この開裂系はさらに、固体支持体からの同時開裂のために、および/または第1または第2ライゲーション成分が他の保護基を含むときの脱保護試薬として用いることができる。
【0081】
本発明の一実施形態において、Nα−置換2または3炭素鎖アルキルまたはアリールチオールは、合成生物活性タンパク質を生じる拡張化学的ライゲーションの基質として用いることのできる適切にN末端誘導化されたポリペプチドを得るためのペプチド合成ステップ(特に自動ペプチド合成、ならびに直交型および収斂型ライゲーションストラテジー)において用いられる。そのような化合物は、式(PG1)S−C2−C1(R1)−Nα(PG2)−CH(Z2)−C(O)−J2、または(PG1)S−C3(R3)−C2(R2)−C1(R1)−Nα(PG2)−CH(Z2)−C(O)−J2の完全に保護された、または部分的に保護された、または完全に保護されていない酸安定性のNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含み、それらを以下の表IVおよび表Vに示す。特に、R1、R2およびR3の1つまたは複数はC1に共役した電子供与基を含み、これはNα−置換アミノアルキルまたはアリールチオールのNα置換−アミドアルキルまたはアリールチオールへの変換に続いて、ペプチド適合性開裂条件下でNα−C1結合の開裂を促進する共鳴安定化カチオンをC1において形成することができる。Z2が化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の任意の化学部分であり、J2が化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の任意の化学部分である場合、PG1およびPG2は個別にまたは組み合わせで存在するか、あるいは存在しない保護基であり、同一であるか、または異なることができる。PG1(またはX1)は、アミンを保護するための基である。PG2(またはX2)は、チオールを保護する基である。多くのそのような保護基が知られており、この目的に適している。(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。
【表7】
【0082】
PG1およびX1の好ましい保護基の例には、これに限定されるものではないが、Boc(t−ブチルカルバメート)、Troc(2,2,2−トリクロロエチルカルバメート)、Fmoc(9−フルオレニルメチルカルバメート)、Br−ZまたはCl−Z(Br−またはCl−ベンジルカルバメート)、Dde(4,4−ジメチル−2,6−ジオキソシクロヘキシル−イリデン)、MsZ(4−メチルスルフィニルベンジルカルバメート)、Msc(2−メチルスルホエチルカルバメート)、Nsc(4−ニトロフェニルエチルスルホニル−エトキシ−カルボニル)が含まれる。好ましいPG1およびX1保護基は、「Protective Groups in Organic Synthesis」、GreenおよびWuts、第3版、Wiley−Interscience(1999)から選択され、もっとも好ましいのはFmocおよびNscである。PG2の好ましい保護基の例には、これに限定されるものではないが、[Acm(アセトアミドメチル)、MeoBzlまたはMob(p−メトキシベンジル)、Meb(p−メチルベンジル)、Trt(トリチル)、Xan(キサンテニル)、tButhio(s−t−ブチル)、Mmt(p−メトキシトリチル)、2または4ピコリル(2または4−ピリジル)、Fm(9−フルオレニル−メチル)、tbut(t−ブチル)、Tacam(トリメチルアセトアミドメチル)]が含まれる。好ましい保護基PG2およびX2は、「Protective Groups in Organic Synthesis」、GreenおよびWuts、第3版、Wiley−Interscience(1999)から選択され、もっとも好ましいのはAcm、Mob、MeB、ピコリルである。
【表8】
【0083】
本発明のNα−置換2または3鎖アルキルまたはアリールチオールの保護形態は上述のスキームIおよびIIのように調製できる。
【0084】
本発明の化合物は、任意の多様な手段によって生成することができ、ハロゲン媒介アミノアルキル化、還元アミノ化、および固相アミノ酸合成法に適合するNα−保護Nアルキル化S保護アミノアルキルまたはアリールチオールアミノ酸前駆体の調製を含む。所望であるとき、許容されるキラル純度の化合物を得るために、生成されたラセミ体またはジアステレオマーの分割を標準的な方法によって行うことができる。
【0085】
II.本発明の生物活性ペプチドおよびタンパク質
A.合成生物活性タンパク質
上述の方法は、合成生物活性ペプチドおよびタンパク質を合成するために用いることができる。本明細書では、そのアミノ酸残基のいくつか、もっとも好ましくはそのすべてを重合するために非組換え技術が用いられた場合、生物活性タンパク質は「合成」であると言う。「非組換え技術」という用語は、有機化学および他の合成重合アプローチを含む技術を、in vivoまたはin vitroでのRNAのタンパク質への翻訳を含む技術と区別することを意図する。もっとも好ましくは、本発明の合成生物活性タンパク質は、有機化学の原理、特に本明細書に記載の「天然型化学的ライゲーション」および「拡張天然型化学的ライゲーション」の方法を用いてin vitroで生成される。そのような化学合成は、好ましくは、ポリペプチドフラグメントが合成され、次いで互いに結合されて合成生物活性タンパク質が形成される収斂型合成アプローチを用いて行われる。別法として、本発明の合成生物活性タンパク質は、単一の非収斂型合成で合成することができる。
【0086】
上述したように、本発明の合成生物活性タンパク質は、好ましくは約25kDaを超える相当の分子量を有する。本発明の目的のために、そのような分子量の決定は、変性SDSポリアクリルアミド電気泳動によってなされる。「モノマー分子量」という用語は、タンパク質またはポリペプチドの複数のコピーを有する可能性のある合成タンパク質と区別して、モノマー合成タンパク質の分子量を指すことを意図する。
【0087】
本明細書では、合成タンパク質の構造またはアミノ酸配列に依存し、そのような構造または配列の変化がタンパク質の生物活性を増強、修飾、または低減するような認識できる生物活性を有する場合、合成タンパク質は「生物活性」を有すると言う。限定するものではないが、そのような「生物活性」には、触媒、シグナル、または誘発反応を媒介するタンパク質の能力が含まれる。本明細書では、それ自体が消費されたり、永久的に修飾されたりすることなく、基質を生成物に変換することによって、タンパク質は「触媒反応を媒介する」と言う。タンパク質の存在が原因となって、生体、あるいはその組織または細胞が、遺伝子発現、細胞分化、または細胞増殖を開始、持続、増強、修飾、または低減する場合、タンパク質は「シグナルまたは誘発反応を媒介する」と言う。そのようなシグナルまたは誘発反応の例には、サイトカイン発現またはサイトカイン発現に対する応答の誘発、赤血球生成の誘発、炎症および/または炎症性過程の誘発または緩和、新脈管形成または血管新生の開始、アポトーシスの誘発、細胞周期への影響などが含まれる。
【0088】
本発明の合成生物活性タンパク質の生物活性はさらに、タンパク質が特異的に受容体、リガンド、または抗体に結合する能力を含む。本明細書では、「特異的」結合という用語は、電荷、溶解度、疎水性などに基づく「非特異的」結合と区別して、ホルモンとその受容体との間、または抗体と抗原エピトープとの間に存在するものなど、構造に基づく結合相互作用を指すことを意図する。
【0089】
本発明の合成生物活性タンパク質は、好ましくはアミノ酸残基の縮合によって合成される。そのようなアミノ酸残基は、核酸コード化リボソーム導入アミノ酸、例えば、アラニン、アルギニン、アスパラギン、アスパラギン酸塩、システイン、グルタミン酸塩、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リジン、メチオニン、フェニルアラニン、プロリン、セリン、トレオニン、トリプトファン、チロシンおよびバリンであることができる。一実施形態において、特定の所望の合成生物活性タンパク質のために選択されたアミノ酸配列は、そのタンパク質の天然のまたは自然の配列となる。別の実施形態において、選択されたアミノ酸配列は、そのタンパク質の天然のまたは自然の配列に存在するアミノ酸残基の代わりにN末端システイン残基を含有することのできるポリペプチドフラグメントからなり、すべて構成されることになる。そのような残基を含むことによって、そのポリペプチドが本明細書に記載の天然型化学的ライゲーションの原理を用いて(カルボキシチオエステル基を含有するように修飾された)別のポリペプチドと結合されることを可能にし、それによって合成生物活性タンパク質を生成する収斂型合成の使用が容易になる。
【0090】
さらなる実施形態において、本発明の合成生物活性タンパク質は、「不規則」アミノ酸残基を含有することができる。本明細書では、「不規則」アミノ酸残基という用語は、RNAによってコード化されておらず、リボソーム導入されていないアミノ酸を指すものである。この点に関して、本発明は、合成生物活性タンパク質の設計および/または構成において広い選択性および柔軟性が可能となる。本発明によって用いることのできる非リボソーム導入アミノ酸の例には、D−アミノ酸、β−アミノ酸、擬似グルタミン酸塩、γ−アミノ酪酸、オルニチン、ホモシステイン、N置換アミノ酸(R.Simon等、Proc.Natl.Acad,Sci.U.S.A.(1992)89:9367〜71;WO91/19735(Bartlett他)、米国特許第5646285号(Baindur)、α−アミノメチレンオキシ酢酸(アミノ酸−Glyジペプチド同配体)およびα―アミノオキシ酸などが含まれる。チオアミド、ビニル性アミド、ヒドラジノ、メチレンオキシ、チオメチレン、ホスホンアミド、オキシアミド、ヒドロキシエチレン、還元アミドおよび置換還元アミド同配体、ならびにβ−スルホンアミドを含むペプチド類似体を用いることができる。
【0091】
特に、R鎖の修飾が合成タンパク質へのポリマー付加物の結合を可能にするので、擬似グルタミン酸塩の使用が有利である。同様に、N末端Nα−置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸を用いることができる。そのような残基(末端またはポリペプチドに存在する)は、本明細書に記載の拡張天然型化学的ライゲーションの方法に従って、そのポリペプチドを、カルボキシチオエステル部分を有するポリペプチドに結合するために有利に用いることができる。
【0092】
一実施形態において、合成生物活性タンパク質のアミノ酸残基すべては、ペプチド結合(すなわち、アミド結合)によって互いに結合することができる。別法として、2つのアミノ酸残基(または2つのポリペプチドのC末端およびN末端残基)を、非アミド結合(チオエステル結合、オキシム結合、チオエーテル結合、有向ジスルフィド結合、チオゾリジン結合など)によって互いに結合することができる(Schnolzer、M.およびKent、S.B.H.、Science(1992)256:221〜225;Rose、K.、J.Amer.Chem.Soc.(1994)116:30〜33;Englebretsen、D.R.等、Tetrahedron Lett.36:8871〜8874;Baca、M.等、J.Amer.Chem.Soc.(1995)117:1881〜1887;Liu、C.F.等、J.Amer.Chem.Soc.(1994)116:4149〜4153;Liu、C.F.等、J.Amer.Chem.Soc.(1996)118:307〜312;Dawson、P.E.等、(1994)Science 266:776〜779)。したがって本発明は、生物活性タンパク質の調製における多様なペプチド結合の修飾、代用、および等配電子性置換の利用を可能にする。
【0093】
本発明の合成生物活性タンパク質は、哺乳動物(ヒト、サル、ウシ、マウス、ブタ、ヒツジ、ウマなどを含む)、鳥類、または魚類タンパク質に関連する生物活性を模倣する生物活性を有するように設計できる。本明細書では、第1のタンパク質が、第2のタンパク質の生物活性に関して修飾、増強、または低減された生物活性を有する場合、第1のタンパク質が第2のタンパク質を模倣していると言う。生物活性は、その存在が直接または間接的にタンパク質のアミノ酸配列に依存している場合、そのタンパク質に「関連している」と言う。
【0094】
別の実施形態において、本発明の生物活性タンパク質は、対応する天然生物活性相当物を参照せずに、全体的または部分的に操作することができる。本発明の好ましい合成生物活性タンパク質には、受容体、タンパク質受容体リガンドが含まれる。そのようなタンパク質の例には、副腎皮質刺激ホルモン受容体およびその生物活性フラグメント、アンギオテンシン受容体、心房性ナトリウム利尿受容体、ブラジキニニン受容体、成長ホルモン受容体、化学走性受容体、ジノルフィン受容体、エンドルフィン受容体、β−リポトロピンの受容体およびその生物活性フラグメント、エンケファリン受容体、酵素阻害因子受容体、フィブロネクチン受容体およびその生物活性フラグメント、消化管および成長ホルモン放出ペプチド受容体、黄体化ホルモン放出ペプチドの受容体、メラニン細胞刺激ホルモンの受容体、ニューロテンシン受容体、オピオイド受容体、オキシトシン受容体、バソプレッシン受容体、バソトシン受容体、副甲状腺ホルモンの受容体およびフラグメント、プロテインキナーゼ受容体、ソマトスタチン受容体、サブスタンスP受容体が含まれる。
【0095】
本発明の合成生物活性タンパク質にはさらに、酵素、および構造分子、たとえばキチン、またはコラーゲンなどが含まれる。
【0096】
さらなる実施形態において、本発明の合成生物活性タンパク質には、サイトカインが含まれる。サイトカインは、細胞によって放出され、細胞−細胞相互作用、連絡、および他の細胞の挙動への特異的影響を媒介する小タンパク質または生物因子(5〜20kDaの範囲)である。本明細書では、「サイトカイン」という用語は、インターロイキン(IL)(IL−1、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、IL−12、IL−13、IL−14など)、リンホカインおよび信号分子、たとえば赤血球産生刺激タンパク質(たとえば、エリスロポエチン(EPO))、腫瘍壊死因子(TNF)、インターフェロンなど、増殖因子、たとえばトランスホーミング増殖因子、神経発育因子、脳由来増殖因子、ニューロトロフィン−3、ニューロトロフィン−4、肝細胞増殖因子、T細胞増殖因子(TGF、TGF−β1、TGF−β2、TGF−β3など)、コロニー刺激因子(G−CSF、GM−CSF、M−CSFなど)、上皮増殖因子(EGF、LIF、KGF、OSM、PDGF、IGF−Iなど)、線維芽細胞増殖因子(αFGF、βFGFなど)、およびホルモン、特に単一ペプチドホルモン、たとえば成長ホルモンなどを含むことを意図する。
【0097】
サイトカイン、特に化学走性サイトカインはさらに、本発明の範囲内の合成生物活性タンパク質の設計を助けるためにそのアミノ酸配列を用いることのできる特に好ましいタンパク質の例を含む。サイトカインは、白血球補充を直接または間接的に媒介することによって、あるいは微生物侵入の特異部位に蓄積された白血球の活性化において重要なシグナルとして働くことによって、先天または後天免疫応答に関与する。炎症性サイトカイン、インターロイキン−1β(IL−1β)およびインターロイキン−6(IL−6)、媒介物質は、好中球補充および活性化をもたらし、可溶性細胞間接着分子、インターロイキン−8を含む(Kotecha S.、European Journal of Pediatrics(1996)155、補遺2:S14〜17)。インターロイキン−6は、主として急性期応答、B細胞分化、および抗体産生に関わる多官能サイトカインである(Akira S.等、Interleukin−6 in biology and medicine、Adv Immunol(1993)54:1〜78)。このサイトカインは、単球、マクロファージ、TおよびB細胞、内皮細胞、および線維芽細胞を含む多くの異なる細胞によって合成され、分泌される。IL−6は、しばしば多くの警告状態において炎症性サイトカインTNFα、およびIL−1によって誘発され、循環IL−6は、急性期反応の誘発において重要な役割を果たす(Xing Z.等、J.Clin.Invest.(1998)101(2):311〜20)。白血球補充および活性化の両方に、直接または間接的に影響を及ぼすサイトカインには、TNF−α、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、およびケモカインファミリーのメンバーが含まれる。主として白血球活性化の状態を調節するサイトカインには、インターフェロンγ(IFN−γ)、インターロイキン−10(IL−10)、およびインターロイキン−12(IL−12)が含まれる。サイトカインはまた、他のエフェクタ分子の発現を誘発または調節することによって微生物クリアランスに影響を及ぼすことができ、これはオートクリンまたはパラクリンいずれかの様式で起こり得る。
【0098】
コロニー刺激因子は、造血幹細胞の増殖および成熟に必要とされる多様な骨髄性細胞および間質性細胞によって産生されるサイトカインである。さらに、研究は、これらの因子が成熟白血球集団のエフェクタ細胞活性を高めることを示している。具体的には、G−CSFは、in vitroでの好中球の生存を延長し、好中球食細胞活性を高め、好中球呼吸バーストを増大する(Standiford TJ等、J.Invest.Med.(19097)45:335〜345)。対照的に、GM−CSFは、好中球およびマクロファージ両方の増殖および成熟を促進し、このサイトカインの活性化特性は、主として成熟マクロファージ集団に向けられる(Chen GH等、J Immunol(1994)152:724〜734)。
【0099】
ケモカインと呼ばれる化学走性サイトカインの密接に関連する6種のサブファミリーが今では特徴づけられた。これらのなかで少なくとも2つのサブファミリーのメンバーが、肺の抗菌宿主防御に寄与する(Mandujano J等、Amer.J.Respir.Crit.Care Med.(1995)151:1233〜1238;Baggiolini M等、Ann.Rev.Immunol.(1997)15:675〜705)。IL−8、MIP(マクロファージ炎症タンパク)−2、KC、ENA−78およびNAP−2を含むC−X−Cケモカインファミリーのメンバーは、優勢な好中球刺激活性および化学走性活性を有し、それに対してMCP(単球化学誘導タンパク)−1、MCP−2、MCP−3、RANTES、MIP−1αおよびMIP−1βを含むC−Cファミリーは、マクロファージ、リンパ球および好酸球に優勢な化学走性および/または活性化作用を及ぼす(Huffnagle GB等、The Role Of Chemokines In Pneumonia、Koch A、Strieter R編、Chemokines in Disease、Texas、R.G.Landis Co、1996:151〜168)。
【0100】
インターフェロンγは、肺病原体の広域スペクトルに対する細胞媒介性免疫に寄与する、T細胞(α、β、およびγ、δ T細胞の両方)およびNK細胞によって産生されるサイトカインである。このサイトカインは、呼吸バーストの刺激、抗原を提供する能力、マクロファージ由来TNF放出のプライミング、および細菌、真菌、マイコバクテリア生体に対するin vitroマクロファージ抗菌活性の強化を含むいくつかのマクロファージエフェクタ細胞機能を活性化する。
【0101】
インターロイキン−12は、Th1型免疫応答を促進し、Th2型免疫応答を阻害するサイトカインである。具体的には、IL−12は、Th1T細胞およびNK細胞の発生を刺激し、CD8+細胞およびNK細胞の細胞溶解活性を増大する。もっとも重要なことに、IL−12は、T細胞およびNK細胞からのIFN−γの主要誘発因子として働く。
【0102】
IL−12とは対照的に、IL−10は、Th2型免疫応答の発達を促進し、細胞媒介性(Th1型)免疫を阻害する(Howard M等、J.Clin.Immunol.(1992)12:239〜247)。このサイトカインは、一部には好中球およびマクロファージを直接不活性化することによって、且つTNF、IFN−γ、ならびにC−X−CおよびC−C両方のケモカインファミリーのメンバーの発現をダウンレギュレートすることによって、効力のある抗炎症特性を与えることが示された。IL−10は、全身免疫細胞活性化の状態において炎症性サイトカインの望ましくない生成を減ずるのに寄与する。
【0103】
EPOは、赤血球産生刺激活性を有する合成生物活性タンパク質の設計を助けるために、そのアミノ酸配列を用いることのできるタンパク質の特に好ましい例である。EPOは、定常状態における赤血球産生の調節、および出血後の赤血球塊回復の促進に関与する主要な因子である(Jelkmann、W.(1992)Physiol.Reviews 72:449;Krantz、S.B.(1991)Blood 77:419;Porter、D.L.およびM.A.Goldberg(1993)Exp.Hematol.21:399;Nissenson、A.R.(1994)Blood Purif.12:6)。ヒトEPOの環状形態は、およそ30000の分子量を有する165アミノ酸(aa)糖タンパク質である(Sawyer、S.T.等(1994)Hematol.Oncol.Clinics NA 8:895;Jacobs、K.J.等、Nature(1985)313:806;Lin、F−K.等、Proc.Natl.Acad.Sci.USA(1985)82:7580)。EPOのcDNAは、166アミノ酸残基を有する分子を予測するが、カルボキシ末端アルギニンは、翻訳後修飾において除去される(12)。N結合グリコシル化のための3つの潜在的な部位があり、それらはすべて占められている。1つのO結合炭水化物部分も存在する(13)。グリコシル化の作用は複雑である。非グリコシル化大腸菌由来EPOはin vitroで完全な生物活性を示すが、in vivoでの完全な活性のためにはグリコシル化が必要なようである。したがって、大腸菌産生、脱グリコシル化、天然EPOは、動物研究で非常に低い活性を示す(Krantz、S.B.(1991)Blood 77:419;Sasaki、H.等(1987)J.Biol.Chem.262:12059;Lowy、P.H.等(1960)Nature 185:102;Wojchowski、D.M.等、Biochem.Biophys.Acta 910:224;Dordal、M.S.等(1985)Endocrinology 116:2293)。
【0104】
上記に矛盾することなく、可変のグリコシル化様式は、可変の効果を示す。たとえば、脱シアル化EPOは、in vitroでの増強された活性、およびin vivoでの低減された活性の両方を示し、肝細胞によって認識、結合、除去されるガラクトース残基の暴露に帰する作用である(Spivak、J.L.およびB.B.Hogans(1989)Blood 73v:90;Goldwasser、E.等(1974)J.Biol.Chem.249:4202)。完全にシアル化されたEPOの分岐パターンも、生物活性に違いをもたらす。主として4枝に分岐したEPOは、「標準の」EPOと同等の活性を示し、一方、主として2枝に分岐したEPOは、in vitroで標準の3倍の活性を示すが、in vivoではわずか15%の活性を示す(Takeuchi、M.等(1989)Proc.Natl.Acad.Sci.USA 86:7819)。研究は、O結合糖ではなく、N結合糖のみがEPOの機能に重要であることを示している(Higuchi、M.等(1992)J.Biol.Chem.267:770319)。
【0105】
コロニー刺激因子、およびRANTESも、本発明の範囲内の合成生物活性タンパク質の設計を助けるためにそのアミノ酸配列を用いることのできる特に好ましいタンパク質の例を含む。
【0106】
B.ポリマー修飾タンパク質およびポリペプチド
本発明の第2の特質は、あらかじめ選択された位置に構造の定義された1つまたは複数のポリマー付加物を含むように、タンパク質またはポリペプチドを修飾する能力に関する。ポリマー修飾タンパク質は以前に記載されているが、考えられる本発明のポリマー修飾の性質および方式は、そのような従来の研究とは著しく異なる。
【0107】
本発明の合成生物活性タンパク質は、全体または部分的に化学合成されるので、ポリペプチド、タンパク質主鎖の、使用者が選択し、使用者が定義した1つまたは複数の特定の部位にポリマー(たとえばポリマーまたは糖、ポリエチレングリコール、グリコール、ヒアルロン酸など)を共有結合できるような方式でこのようなタンパク質を合成することが可能である。さらに、本発明のタンパク質の合成的生産は、調製品中の各分子の使用者選択、使用者定義部位のそれぞれに確実にそのような修飾を存在させることができる。合成のそのような一様性および制御は本発明を、従来技術の方法の使用によって可能となる無作為な修飾と明らかに区別する。注目に値すべきなのは、本発明は、任意の重要でない残基を誘導化してポリマー付加物を含有することのできる合成タンパク質の設計を可能にすることである。さらに、そのような使用者選択、使用者定義の各部位に関して、それを介して付加物がポリペプチドまたはタンパク質主鎖に結合される正確な結合(アミド、チオエステル、チオエーテル、オキシムなど)を使用者が定義することができる。さらに、存在するすべてのタンパク質またはポリペプチドが正確に同じ誘導部位に正確に同じ誘導付加物を含有する最終調製物が得られるように、特定の部位に存在を所望する特定のポリマー付加物を変更および制御することができる。したがって、本発明は、ポリマー修飾ポリペプチドおよびタンパク質の均一な調製物の形成を可能にする。
【0108】
ポリマーが結合することのできる結合部位の位置および数を変更する手段を提供することに加えて、本発明は、結合ポリマーの性質を変えることを可能にする。使用者選択、使用者定義された任意の特定部位に組み込むことのできるポリマー付加物は、任意の定義された長さであることができる。同様に、本発明の方法に矛盾することなく、異なる部位において異なる長さのポリマーを用いることが可能である。したがって、一実施形態において、本発明の合成生物活性タンパク質は、ポリマー付加物による「単一修飾」または「多修飾」であることができる。複数のポリマー付加物を特定のポリペプチドまたはタンパク質に導入した場合、その使用ポリマーは、「単一種分化」「多種分化」「一様に種分化」または「多様に種分化」されていることができる。本明細書では、「単一種分化」という用語は、単一のポリマー種によって修飾されたポリペプチドまたはタンパク質を指すことを意図する。対照的に、「多種分化」という用語は、複数のポリマー種によって修飾されたポリペプチドまたはタンパク質を指すものである。そのような多種分化ポリペプチドまたはタンパク質は、そのポリペプチドまたはタンパク質の各修飾部位に同じ単一種のポリマーが存在する場合、「一様に種分化」されていると言う。対照的に、ポリペプチドまたはタンパク質の修飾部位が異なるポリマー種によって修飾されている場合、種分化ポリペプチドまたはタンパク質は「多様に種分化」されていると言う。
【0109】
さらに、使用者定義、使用者選択されたそれぞれの部位において、ポリマー付加物の直線性または分岐性の程度を変えることが可能である。したがって、ポリマー付加物は、直線状、分岐状、または一様に分岐状であることができる。「一様に分岐状」という用語は、特定部位においてポリマーのすべての分岐が、同一の構造および長さを有することを意味するものである。本発明者等の理解のとおり、本発明は、個々の分岐の長さ、ならびにそのような分岐点に存在するポリマーの構造の両方を、独立して変更することを可能にする。
【0110】
要約すれば、本発明は、ポリペプチドまたはタンパク質主鎖におけるポリマー修飾、使用者選択、使用者定義部位の(1)位置、および(2)頻度を定義すること、ならびにそのような各部位に存在する分岐の(3)長さ、(4)種、および(5)程度を制御することを可能にする。さらに、複数のポリマー修飾を有するポリペプチドおよびタンパク質に関して、本発明は、各部位について上に特定した5つの変数のそれぞれを独立して定義することを可能にする。さらに、分岐ポリマー修飾を有するポリペプチドおよびタンパク質に関して、本発明は、各分岐点について上に特定した5つの変数のそれぞれを独立して定義することを可能にする。したがって、本発明は、ポリマー修飾の実質的な適応性を提供する。
【0111】
C.タンパク質およびペプチドのデリバリー
本発明の随意ポリマー修飾された合成生物活性ポリペプチドおよびタンパク質は、疾患および症状の治療を行うために薬剤として用いることができる。もっとも好ましくは、治療を必要としている患者または個体に投与されるとき、そのような合成生物活性ポリペプチドおよび/またはタンパク質は、薬剤デリバリーシステムを用いて投与される。そのようなシステムの使用は、患者が薬剤を受け入れることができ、且つ所望の生物活性の有効性を高めるような様式で患者に薬剤を提供することを可能にする。本発明の目的の好ましい薬剤デリバリーシステムには、経口、経鼻、または吸入経路によって、あるいは筋肉内、皮下、経皮、静脈内、耳内、または眼内に、ポリペプチドまたはタンパク質を投与できるシステムが含まれる。
【0112】
そのような薬剤デリバリーシステムは、部位特異放出を提供する製剤、または腸粘膜の保護を増強する製剤を含むことができる。適切な製剤には、乾燥粉末製剤、ウイルス粒子カプセルによるデリバリー、リポソームカプセル、経皮パッチ、電気援用輸送(電気穿孔療法)、およびポリマー/ナイアゾン(niazone)複合体が含まれる。
【0113】
好ましい実施形態において、そのような薬剤デリバリー装置は生物環境の変化に応答し、それらの変化に基づいて薬剤をデリバリーし、またはデリバリーを中止する。薬剤および他の活性剤の放出を制御するために一連の材料が用いられてきており、ポリ(ウレタン)、ポリ(シロキサン)、ポリ(メチルメタクリレート)、吸水性および強度のためのポリ(ビニルアルコール)、ポリ(エチレン)、ポリ(ビニルピロリドン)、ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(n−ビニルピロリドン)、ポリ(メチルメタクリレート)、ポリ(ビニルアルコール)、ポリ(アクリル酸)、ポリアクリルアミド、ポリ(エチレン−コ−酢酸ビニル)、ポリ(エチレングリコール)、ポリ(メタクリル酸)などである。さらなる好ましい実施形態において、薬剤デリバリーを促進するために、生分解性ポリマーが用いられる。そのようなポリマーには、ポリラクチド(PLA)、ポリグリコリド(PGA)、ポリ(ラクチド−コ−グリコリド)(PLGA)、ポリ無水物、およびポリオルトエステルが含まれる。薬剤デリバリー装置、およびその使用のための方法は、米国特許第6072041号、第6041253号、第6018678号、第6017318号、第6002961号、第5879712号、第5849331号、第5792451号、第5783212号、第5766633号、第5759566号、第5690954号、第5681811号、第5654000号、第5641511号、第5438040号、第4810499号および第4659558号に記載されている。
【0114】
III.合成生物活性タンパク質の生成
本発明の合成生物活性タンパク質の生成は、以下のステップまたは構成要素、すなわち設計、合成、ペプチドライゲーション、タンパク質フォールディング(折りたたみ)、生物活性の評価、ポリペプチド主鎖のポリマー修飾を有するものとして考えることができる(図2)。
【0115】
A.好ましい合成生物活性タンパク質の設計
特に好ましい実施形態において、本発明の合成生物活性タンパク質は、赤血球産生刺激タンパク質である。本明細書では、赤血球産生刺激タンパク質は、赤血球の生成を媒介するタンパク質である。タンパク質の赤血球産生刺激生物活性は、たとえばin vivo投与後の赤血球生成を追跡する、EPO依存細胞系の増殖を媒介するタンパク質の能力を検定するなど、多様な任意の手段によって判別することができる。
【0116】
好ましい赤血球産生刺激タンパク質は、哺乳動物、もっとも好ましくはヒトのEPOおよびその類似体である。エリスロポエチンは主として、腎臓の尿細管および尿細管近接毛細管内皮細胞および間質細胞によって合成および分泌される。慢性腎臓疾患は、腎臓のEPO産生細胞の破壊をもたらす。結果として生じるEPOの欠乏は、しばしば貧血を誘発する。したがって、EPOの主要な臨床上の使用は、通常は輸血も受ける重篤な腎不全(ヘマトクリット0.3以下)を有する患者の治療、ならびに化学療法後の貧血患者の治療である。典型的に、EPOは臨床使用のためにハムスター卵巣細胞において組換えによって合成される。EPOは、その成熟形態で165アミノ酸残基を有する比較的熱安定性、且つpH安定性の酸性(pI=4.5)タンパク質である。EPOは、EPO受容体を介してその活性を伝える。EPOの分子量のおよそ40パーセントは、グリコシル化によるものである。グリコシル化は、in vivoでのEPOの薬物動態挙動を決定する重要な要因である。非グリコシル化EPOは、極端に短い生物学的半減期を有する。これはin vitroにおいて依然として受容体に結合し、より高い特異的活性を有する可能性もある。しかしながら、最近の実験によって、EPOのPEG化はその寿命を著しく増大するが、細胞に基づくアッセイシステムにおいてEPO活性を著しく低減することが示された。
【0117】
本発明の合成赤血球産生刺激タンパク質(「SEP」)は、好ましくは、ヒト尿EPOの化学的に修飾された合成類似体である。好ましい実施形態において、SEPは、チオエステル結合、オキシム結合、アミド結合、または他の結合を介して、1つまたは複数のペプチド残基に結合している1つまたは複数ポリマー部分(もっとも好ましくはpPEG部分)を含有する。非常に好ましい実施形態において、そのような結合は、ヒト尿EPOにおいて天然にグリコシル化されている1つまたは複数の位置に存在する(すなわち、24、38、83および126位)。もっとも好ましくは、SEP分子は、2つ以上のそのような位置にpPEG部分を有する。別の実施形態において、他のタンパク質残基は、ポリマー部分によって修飾されることができる。本発明の合成生物活性タンパク質のための修飾位置には、タンパク質の無秩序ループ、領域、またはドメイン、あるいは潜在性プロテアーゼ開裂部位またはその近傍部位に位置する残基が含まれる。たとえば、ポリマー修飾は、EPOの9、69、および/または125位の1つまたは複数の位置に導入することができる。本発明のSEP分子の全分子量は、約25から約150kDa、より好ましくは約30から約80kDaであることができる。分子量は、所与の類似体の修飾に用いられるポリマー(pPEGなど)の数および構造を増大または低減することによって制御できる。より大きな構成体のpPEG媒介流体力学MWは約100kDaを超える。in vitroにおけるヒトEPO受容体発現細胞でのアッセイは、本発明のSEPが、組換えによって産生されたグリコシル化ヒトEPOと同等のED50を有することを示す。
【0118】
オキシム結合pPEG類似体は、好ましくは、側鎖アミノオキシ、ケトン、またはアルデヒド官能基を有する非天然コード化アミノ酸において、SEPタンパク質に、アミノオキシ、ケトン、またはアルデヒド官能化pPEGを結合することによって構成される。たとえば、SEP−0およびSEP−1の89および117位は、擬似グルタミン酸塩((グルタミン酸塩側鎖−CHα−CH2−CH2−COOHに対して)式−CHα−CH2−S−COOHの側鎖を有する非天然コード化アミノ酸)を含有する。チオエーテル結合を用いるSEP類似体は、好ましくは、チオールを有する側鎖を備えたシステインまたは非天然アミノ酸によって提供されるチオール官能基を含有するように構成される。図3は、好ましい合成赤血球産生刺激タンパク質の1つの型の基本的な構造を示す。
【0119】
別の実施形態において、本発明のSEP分子は、EPOの天然アミノおよびカルボキシ末端が再配置された「環状順列化」EPO類似体を含むことができる。より好ましくは、そのような再配置は、アミノおよびカルボキシ末端を、無秩序ループなど、構造的制約の低い位置に移動する。125および126位(天然EPO残基ナンバリングシステムを基準として)近くの無秩序ループは、そのような再配置部位の例である。もっとも好ましくは、そのようなSEPはジスルフィドを含まず、あらかじめ選択された残基にポリマー部分を含有するように化学的に修飾される。
【0120】
あるいは、SEP分子は、天然グリコシル化部位、またはグリコシル化を施すことのできる他の部位、たとえば126および125位などに再配置されたアミノおよびカルボキシ末端を有することができる。SEP分子は、電荷を除去または変更するためのアミノおよびカルボキシ末端の修飾(カルボキシアミド化などによる)を含むこともできる。
【0121】
そのような環状順列化分子の好ましい例において、新しいNおよびC末端は、それぞれ126および125位によって提供される。7、29、32および161位の天然ジスルフィド形成システインは、好ましくは、ジスルフィド架橋を形成することのできない非天然コード化アミノ酸、L−α−N−酪酸(Aba)に置換されている。残基R166、E37およびV82は、好ましくは、生成を向上させるためにアラニンに置換されている。さらに、好ましくは追加のシステインが、天然EPOナンバリングスキームを基準として1位と166位の間に挿入され、以下「0」と番号を付す。結果として生じた分子は、4つのシステインを含有し(126、0、38および83位(N末端からC末端方向に読み取り))、それらは(1)ライゲーション部位、および(2)チオエーテル形成pPEG結合部位として用いられる。随意、システインは、追加のpPEG結合部位を提供するためにA125を置換することができる。本発明のそのようなSEP分子の全分子量は、約25から約150kDa、より好ましくは約30から約80kDaであることができる。分子量は、所与の類似体の修飾に用いられるポリマー(pPEGなど)の数および構造を増大または低減することによって制御できる。より大きな構成体のpPEG媒介流体力学MWは約100kDaを超える。任意のpPEG結合部位は、125、9および24位(N末端からC末端方向に読み取り)に位置する。さらなるSEP類似体設計は、新しいNおよび/またはC末端からの無秩序ループ、領域、および/または切断残基に代わりのNおよびC末端を有する。好ましい循環順列化分子の基本構造を図4に示す。
【0122】
別の実施形態において、本発明の合成生物活性タンパク質は、哺乳動物、もっとも好ましくはヒトのC−GSFである。G−CSFは、好中球性顆粒球の急速な増殖および血流への放出を誘発し、それによって感染との戦いに治療的効果をもたらす。ヒトGCSFタンパク質は、アミノ酸長174であり(特許EP0459630(Camble、R他)、その配列の変種がすでに単離されている。このタンパク質は4つのシステイン残基を有し、トレオニン133位にO−グリコシル化部位を有する。
【0123】
そのような合成GCSF分子の一実施形態において、分子のアミノ酸配列は、1、2、または3位の1つまたは複数の位置に天然、またはより好ましくは非天然コード化疎水残基を含有するように、天然GCSFの配列に対して変更される。随意、そのような分子は、GCSFの5位、および/または173位および/または174位に追加の天然、より好ましくは非天然コード化疎水残基を含有するようにさらに修飾される。
【0124】
さらなる好ましい実施形態において、合成GCSF分子は、63、64、および/または133位の1つまたは複数の位置、および/または1、および174位の1つまたは複数の位置でポリマー修飾(好ましくはpPEG)される。このpPEGポリマーは、線状または分岐状、荷電、非荷電、または混合であることができ、約5から約80Da、より好ましくは約40から約70kDaの分子量の範囲であることができ、pPEG全MW寄与は修飾に用いられるpPEGの数および構造に依存する。流体力学半径は、in vivoでより大きなMW効果を示す(たとえば、10kDa分岐pPEGは約55kDaの有効分子量を示す)。推定pPEG媒介流体力学MWは、100kDaを超える。
【0125】
オキシム結合を用いる類似体の場合、pPEGは、好ましくは、側鎖アミノオキシ、ケトン、またはアルデヒド官能基を有する非天然コード化残基に結合されている。チオエーテル結合を用いる類似体の場合、pPEGは、好ましくは、チオールを有する側鎖を備えたシステインまたは非天然アミノ酸に結合されている。アミド結合を用いる類似体の場合、pPEGは、反応性側鎖アミンを有する天然または非天然アミノ酸に結合されている。
【0126】
合成生物活性GCSFタンパク質の基本構造を図5に示す。この図において、「J」は、疎水性側鎖を有する非天然コード化残基を示す。
【0127】
そのような合成生物活性GCSFタンパク質の生物活性は、たとえば因子依存性ヒトまたはマウス細胞系(たとえばNFS60細胞系)の増殖を媒介するそれらの能力を評価することによって、あるいは20日超の半減期/放出をターゲットとするデリバリーシステムを用いて、または用いずに、in vivoでの好中球刺激、半減期、および免疫原性を測定することによってなど、通常の手段で評価することができる。
【0128】
別の実施形態において、本発明の合成生物活性タンパク質は、哺乳動物、もっとも好ましくはヒトのケモカイン、RANTESである。RANTESは、主要受容体CCR5を経るマクロファージ親和性HIV株の侵入を遮断し、さらに喘息、移植拒絶、および創傷治癒に関わる炎症経路をダウンモジュレートする(Kalinkovich A.等、Immunol Lett.(1999)68:281〜287)。本発明の合成RANTES分子は、好ましくは、(1)RANTES1〜68の1位に認められるN末端セリンが、n−ノナノイル(「NNY」)基で置換されており、(2)3位のチロシンが、疎水性側鎖を有する非天然コード化アミノ酸で置換されているように化学修飾されている点でRANTESケモカインと異なる。そのような化合物は、組換え「Met−RANTES1〜68」のミリモル範囲に比べて、ピコモル範囲のED50を有し、極めて有効であることが見出されている。
【0129】
疎水性側鎖を有する非天然コード化アミノ酸で2位のプロリンを置換する、分子のC末端に脂肪酸を付加するなど、上記のRANTESに対して1つまたは複数の追加の変更を有する有効なRANTES類似体が作られた。RANTES残基12〜20に対応するNループ領域への修飾によって、効力と合わせて受容体特異性が向上した。さらなる好ましい実施形態において、本発明の合成RANTES分子は、特にin vivo半減期を改善するためにNまたはC末端にポリマー部分(pPEGなど、あるいはノナノイル(「NNY」)またはY3Xを組み込む。このpPEG部分は、好ましくはオキシム結合を介して非天然コード化アミノ酸に結合され、これは66、67、68位、または68位のリンカーに導入され、67位が好ましい。脂肪酸は、好ましくはオキシム結合(好ましくはリンカーを経て)を介して、アミノ−オキシ修飾アミノ酸ジ−またはトリ−ペプチドリンカーなど残基68に結合される。他の結合化学を別法として用いることができる。そのような合成RANTES類似体のより大きな構成体の分子量は、pPEG付加物の性質および構造に応じて約25kDaから約45kDaの範囲であり、より大きい構成体では、60kDaを超えるpPEG媒介流体力学MWを有する。好ましい合成RANTES類似体の構造を図6に示す。
【0130】
B.合成生物活性タンパク質およびペプチドの定義された特定のポリマー修飾
本発明のさらなる態様は、非修飾タンパク質に比べて改善された特性を有するタンパク質薬剤を生じるようにタンパク質を修飾するための化学部分およびポリマー、特に水溶性ポリマー、ならびにそれらの使用に関する。そのような修飾の予期される有益な効果には、これに限定されるものではないが、非修飾分子と比べて(a)効力の向上、(b)タンパク分解安定性の増大、およびクリアランス速度の低減による循環におけるタンパク質薬剤存続期間の延長、(c)免疫原性の低減、および(d)識別的ターゲッティングの可能性が含まれる。
【0131】
上に論じたように、タンパク質を修飾するためにポリエチレングリコールが用いられてきたが(たとえば、反応性アミン基によるポリエチレングリコール分子の結合を可能にするために、G−CSFのリジン残基を修飾することに関する米国特許第6027720号、Kuga等を参照のこと)、そのような修飾は分子異質性、および分子多様性を伴う。
【0132】
本明細書では、「分子異質性」という用語は、タンパク質製剤の各タンパク質に結合されたポリマー分子の数のばらつきを指すものである。「分子多様性」という用語は、(a)そのポリマーによって修飾されるタンパク質の部位のばらつき、(b)タンパク質製剤の同一タンパク質の異なる部位、または異なるタンパク質の同一部位のポリマー付加物の長さのばらつき、または(c)タンパク質製剤の同一タンパク質の異なる部位、または異なるタンパク質の同一部位のポリマー付加物の、任意の分岐の程度および/または性質のばらつきを指すものである。本明細書では、「分子的に均一」という用語は、すべてのタンパク質分子が同じ位置にアミノ酸配列および同一のポリマー修飾を含有するタンパク質の製剤を指すものである。2つ以上の「分子的に均一」なタンパク質製剤の混合物は、本明細書では、「分子的に定義された」製剤と呼ぶ。
【0133】
本発明のこの態様に従って、上に特定したポリマー異質性、多様性、および不適合性の問題の解決策には、ポリペプチド(厳密な長さ、好都合な合成)および「pPEG」(「精密PEG」)(適応性、両親媒性、非免疫原性、プロテアーゼ感受性でないポリマー)両方の利点を兼ね備える新しい類の生体適合性ポリマーを製造することが含まれる(Rose、K.等、参照により本明細書に組み込むものとする米国特許出願第09/379297号)。この新しい類の生体適合性ポリマーは以下の式を有し、
−[CO−X−CO−NH−Y−NH]n−
nは、整数であって、好ましくは1〜100、より好ましくは2〜100であり、該式中、XおよびYは、アミド結合によって結合した正確な構造の生体適合性反復要素である。好ましくは、XおよびYは、反応性官能基を欠く2価有機基であるか、または不在であり、同一であるか、または異なり、且つ各反復単位によって独立して異なることができる。好ましくは、n=2であるとき、XまたはYの少なくとも1つは、置換、非置換、分岐状、および線状脂肪族および芳香族基からなる群から選択される。より好ましくは、2価有機基XまたはYの少なくとも1つは、フェニル、C1〜C10アルキレン部分、C1〜C10アルキル基、ヘテロ原子含有フェニル、ヘテロ原子含有C1〜C10アルキレン部分、ヘテロ原子含有C1〜C10アルキル基、およびそれらの組み合わせからなる群から選択される。
【0134】
特に好ましいpPEG部分は以下の式を有し、
−{CO−(CH2)2−CO−NH−(CH2)3−(OCH2CH2)3−CH2−NH}n−
該式中、nは、好ましくは1〜100、より好ましくは2〜100であり、または、
−{CO−(CH2)2−CO−NH−(CH2)6−NH−CO−(CH2)2−CO−NH−(CH2)3−(OCH2CH2)3−CH2−NH}n−
を有し、該式中、nは、好ましくは1〜50、より好ましくは2〜50である。
【0135】
そのようなpPEG部分は、多様な方法のいずれかで合成することができる。しかしながら、そのような部分は、好ましくは、重合法よりも、単位の固相段階的鎖アセンブリ法を用いて生成される。そのようなアセンブリ法の使用によって、製剤のその部分が、それらの長さ、XおよびY置換基の性質、(もしあれば)分岐点の位置、ならびに分岐の長さ、XおよびY置換基、および位置に関して定義された均一な構造を有することが可能になる。
【0136】
好ましくは、そのような部分は、
(a)式HOOC−X−COOHを有する2酸の誘導体のモル過剰量を用いて、該式中、Zは、H2N−またはHO−であり、Qは、リンカーまたは標的分子であり、支持体は、固相、マトリクス、または表面である式Z−Q−支持体の化合物のアミノまたはヒドロキシル基をアシル化するステップ、
(b)ステップ(a)の生成物の遊離カルボキシル基を活性化するステップ、
(c)式NH2−Y−NH2を有するモル過剰量のジアミンを用いて、ステップ(b)の生成物をアミノ分解するステップ、および
(d)HOOC−X−COOHおよびNH2−Y−NH2を用いてステップ(a)〜(c)を随意繰り返すステップであって、前記XおよびYは、2価有機基であるか、または不在であり、同一であるか、または異なり、且つ前記の任意選択の各反復単位によって独立して異なることができ、且つ先のアシル化およびアミノ分解ステップのいずれかで用いたXおよびY置換基と同じか、または異なっているステップなどのステップによって合成される。
【0137】
好ましい実施形態において、上述の反復単位の6マー、12マー、18マー、および32マーが用いられる。所望である場合、この反復単位は、たとえば分岐pPEG構造を形成するためにリジンのアミノ基と共に用いることができる。pPEGは、チオエーテル、オキシム、およびアミド結合形成を含む多様な化学によって、本発明の合成タンパク質に結合することができる。
【0138】
一実施形態において、単位の固相段階的鎖アセンブリは、以下のステップ1〜5を含む。
ステップ1: 保護または非保護2酸を樹脂上でアミノに結合して、結合部位にアミド結合を生じる(PGは、用いられる2酸に応じて存在するか、または存在しない保護基である):
PG−OOC−X−COOH + NH2−Y−NH−樹脂
ステップ2: 存在する場合、樹脂上で保護基(PG)を除去する:
HOOC−X−CO−NH−Y−NH−樹脂
ステップ3: 保護または非保護ジアミノを樹脂上でカルボキシに結合して、結合部位にアミド結合を生じる(PGは用いられるジアミノに応じて存在するか、または存在しない):
PG−NH−Y−NH + HOOC−X−CO−NH−Y−NH−樹脂
ステップ4: 存在する場合、樹脂上で保護基(PG)を除去する:
−NH−Y−NH−OC−X−CO−NH−Y−NH−樹脂
ステップ5: 「n」単位を付加するためにステップ1〜4を「n」回繰り返し、次いで樹脂から−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−を開裂する。
【0139】
上述したように、線状または分岐状pPEG構成体は、本発明の合成生物活性分子に結合するのに好ましい水溶性ポリマーである。用いられるpPEGは、生理的条件下で荷電または中性であり、用いるpPEG構造に応じて結合化学、長さ、分岐、および溶解性を変えることのできるペンダント基を有する。上記のとおり、本発明の好ましいpPEGは、XおよびYの1つまたは両方が水溶性反復単位、もっとも好ましくは「PEG」ベースの反復単位を含む、反復単位−CO−X−CO−NH−Y−NH−を有する水溶性ポリアミドを含む。アミノ樹脂、NH2−樹脂、および対称ジアミンNH2−Y−NH2の場合に関して下に示したように、原理的には簡単な2ステップ固相手順を用いてオリゴ尿素を利用でき、カルボニルジイミダゾールによる活性化→im−CO−NH−樹脂
ジアミンNH2−Y−NH2によるアミノ分解→NH2−Y−NH−CO−NH−樹脂
これら2つのステップを何度か繰り返して、反復単位−NH−Y−NH−CO−を有するオリゴ尿素を得ることができるが、このアプローチは好ましさに劣る。これは、上記ステップの収率が、室温において非常に過剰の試薬と長い反応時間を用いても、量的でない可能性があるためである。したがって、アミノ樹脂、NH2樹脂の場合を以下に示した、ポリアミドを形成する3ステップの固相手順を用いることが好ましい。試薬HO2C−X−CO2H、およびNH2−Y−NH2は、異性体生成物を回避するために、対称であるべきである。
二酸HO2C−X−CO2Hを用いたアシル化→HO−CO−X−CO−NH−樹脂
カルボニルジイミダゾールを用いた活性化→im−CO−OCO−X−CO−NH−樹脂
ジアミンNH2−Y−NH2を用いたアミノ分解→NH2−Y−NH−CO−X−CO−NH−樹脂
【0140】
これらの3ステップは、反復単位−NH−Y−NH−CO−X−CO−を有するポリアミドを得るために、何度か連続して繰り返すことができる。このポリマーは、正確な数のモノマー単位を含有し、XおよびYは、各ステップにおいて独立して異なることができ、末端基は、随意に選択できる。たとえば、アシル化ステップに無水コハク酸(「Succ」)を用い、アミノ分解ステップに4,7,10−トリオキサ−1,13−トリデカンジアミン(「EDA」または「TTD」とも呼ばれる)を用いることによって、Xが−CH2CH2−であり、Yが−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH−であり、反復単位が−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH−COCH2CH2CO−である「PEG」ベースのポリアミドが形成される。この手順には保護基を持たない2価の試薬が含まれるにもかかわらず、以下に言及する分岐構成体を製造するための分岐Lysコアの場合を除いて、標準的な市販のペプチド合成樹脂が用いられるとき、架橋結合は問題とならない(Rose他、米国特許出願第09/379297号、およびRose等、J.Am.Chem.Soc.(1999)121:7034)。
【0141】
たとえば、Rose他(米国特許出願第09/379297号)、およびRose、K.およびVizzavona、J.(J.Am.Chem.Soc.(1999)121:7034)に記載されている「化学抗体(chemobody)」構成体と類似の方法で、分岐pPEG構成体を製造することができる。したがって、分岐pPEG構成体はリジン分岐コアなどの分岐コアを有するように製造することができ、分岐コアはオキシムリンカーを介して−(COCH2CH2CO−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH)n−などの好ましい水溶性ポリアミドに結合している。たとえば、オキシム結合は、ポリアミドの他端にあるLys側鎖のアミノオキシアセチル基と、リジンコア、たとえばテトラマーコア(O=CHCO)4Lys2Lys−のグリオキシリル基との間に容易に形成できる。したがって、各リジン分岐点のオキシムリンカーは、構造−Lys(COCH2ON=CHCO−)アミド−として調製することができる。別の構成は、以下に示す4価分岐構成体を生じるために、好ましくはモノ保護ジアミン、およびポリアミドカップリング後の脱保護を用いて、オキシム結合をポリアミドとポリアミドの遊離ペンダント基との間に配置することができる。
[O=CHCO−(NH−CH2CH2CH2−[OCH2CH2]3−CH2−NH−COCH2CH2CO−)n]4Lys2Lys−
【0142】
オキシム化学は、ダイマーおよびテトラマー分岐構成体の製造に用いるだけでなく、オクタマー分岐構成体のアセンブリにも適している(Rose、K.、J.Am.Chem.Soc.(1994)116:30;Rose、K.等、Bioconj.Chem.(1996)7:552)。当然ながら、そのようなpPEGポリアミドを用いるとき、その目的のために他の化学を用いることができる(たとえば図7を参照のこと)。さらに、ポリアミド形成は、BocまたはFmoc化学を含むペプチド合成のための合成スキームに組み入れることができるが、そのようなスキームを練り上げるとき、アミノ分解ステップはFmoc基を除去し、Boc化学が用いられる場合にはインドールのホルミル保護を除去することを留意しなければならない。
【0143】
したがって、そのようなpPEGは、好ましい結合(たとえばアミド、チオエーテル、オキシムなど)を介して線状水溶性ポリアミド(たとえば−(Succ−TTD)n−等)に結合した種々の分岐コアを有するように製造することができ、ここで各線状水溶性ポリアミドの自由端はそれぞれ所望のペンダント基(たとえばカルボキシレート、アミノ、アミド等)を用いてキャップして所望の電荷を提供することができる。さらに、本発明の線状および分岐状pPEGは、ユニークで相互反応性の1つまたは複数の官能基を有する合成タンパク質に結合するためにユニークな官能基を含むように製造することができ、pPEGのペンダント基は、好ましくは非抗原性である(たとえば図7を参照のこと)。
【0144】
本明細書で言及したすべての刊行物および特許出願は、それぞれ個々の刊行物または特許出願が参照により組み込まれると具体的に且つ個別に指示されるのと同程度に、参照により本明細書に組み込むものとする。本発明の内容を説明するために、本明細書において本発明の背景の考察が含まれる。これは、請求の範囲のいずれかの優先権主張日の時点で、参照された資料のいずれかが世界のいずれかの場所で公表されているか、知られている、あるいは従来技術または周知の事柄の一部であることを認めるものであるとみなされるものではない。ここに本発明を一般的に説明したが、例示的なものとして提供され、明記されない限り本発明を限定するものではない以下の実施例を参照することによって、本発明はより容易に理解されるであろう。
【0145】
(実施例)
実施例1
合成赤血球産生刺激タンパク質SEP−0の合成
合成赤血球産生刺激タンパク質(SEP)を合成した。完全長合成タンパク質(「SEP−0(1〜166)」とする)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAASAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:1)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示す。このSEP−0タンパク質は、以下の4つのポリペプチドセグメントから溶液中で合成された。
セグメントSEP−0:1(GRFN1711、配列番号:1の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA
EH−チオエステル
セグメントSEP−0:2(GRFN1712、配列番号:1の残基33〜88からなる):
CSLNEKITVP DTKVNFYAWK RMEVGQQAVE
VWQGLALLSE AVLRGQALLV KSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−0:3(GRFN1713、配列番号:1の残基89〜116からなる):
CPLQLHVDKA VSGLRSLTTL LRALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−0:4(GRFN1714、配列番号:1の残基117〜166からなる):
CAISPPDAAS AAPLRTITAD TFRKLFRVYS
NFLRGKLKLY TGEACRTGDR−カルボキシレート(C末端システイン(Cys161)はピコリル(pico)保護基を有する)
【0146】
ペプチドSEP−0:1およびSEP−0:2およびSEP−0:3を、ABI433A自動ペプチド合成装置で、または手作業の鎖アセンブリで、Boc(tert−ブトキシカルボニル)化学のためのin situ中和プロトコル、および確立されたSPPS、側鎖保護、およびチオエステル樹脂法を用いる段階的固相ペプチド合成(SPPS)(Hackeng等、PNAS(1999)96:10068〜10073、およびSchnolzer等、Int.J.Pept.Prot.Res.(1992)40:180〜193)によってチオエステル生成樹脂上で合成するか、あるいは供給業者に注文し、入手した。たとえば、BocSPPS保護基の標準セットを用い、すなわちArg(Tos)、Asp(cHex)、Cys(4MeBzl)およびCys(Acm)、Glu(cHex)、His(DNP)、Lys(CIZ)、Ser(Bzl)、Thr(Bzl)、Trp(ホルミル)、Tyr(BrZ)であり、Met、Asn、Glnは非保護側鎖であった。セグメントSEP−0:4は、−OCH2−Pam−樹脂上で同様に合成した。これらのペプチドを、標準的なBoc化学手順に従い、HF/p−クレゾールを用いて脱保護し、同時に樹脂支持体から開裂したが、しかしながらHF/p−クレゾール中で除去されない保護基を含有するペプチドの場合、保護基は維持された。これらのペプチドを、分取C4逆相高圧液体クロマトグラフィ(HPLC)で精製した。純粋ペプチドを含有する分画を、ES−MS(エレクトロスプレーイオン化質量分析法)を用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0147】
ステップ1:ライゲーション#1 セグメントSEP−0:4およびセグメントSEP−0:3を、濃度15mMでTFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する飽和リン酸緩衝液(pH7.9)を添加し、ペプチドセグメントの透明溶液を得た。ライゲーション後、ライゲーション混合物を、TFE(トリフルオロエタノール)2ml、6M塩化グアニジニウム6ml、25%β−メルカプトエタノールを含有する100mMリン酸塩の溶液に添加し、20分間インキュベートした。その溶液を、15mg/mlのTCEP(トリス(2−カルボキシエチル)ホスフィンHCl)氷酢酸溶液で酸性にし、分取C4逆相HPLCカラム(直径1インチ)に充填した。次いで、そのペプチドを分取勾配逆相HPLCによって精製した。所望の結合生成物SEP−0:Acm+SEP−0:3+SEP−0:4を含有する分画を、ES−MSによって同定し、プールした。
【0148】
ステップ2:Acm除去#1 Acm除去のために、プールしたSEP−0:Acm+SEP−0:3+SEP−0:4分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−0:3+SEP−0:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0149】
ステップ3:ライゲーション#2 等量のSEP−0:3+SEP−0:4およびSEP−0:2を合わせて、15mM濃度で純TFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6M塩化グアニジニウム20ml、pH4の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0150】
ステップ4:カルボキシメチル化 SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4を、15mM濃度でTFEに溶解した。6M塩化グアニジニウムを含有する2倍過剰(v/v)の200mMリン酸緩衝液(pH7.9)を添加し、ペプチドセグメントの透明溶液を得た。最小量のメタノールに溶解した25倍過剰のブロモ酢酸を添加し、その溶液を2時間反応させた。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、段階勾配で精製した。所望のカルボキシメチル化生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Etを含有する分画をES−MSによって同定し、プールした。
【0151】
ステップ5:ピコリル除去 亜鉛末を2MのHCl中で30分間活性化した。ペプチドSEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Etを、約10mg/mlの濃度で純TFEに溶解した。その溶液を(新たに添加した)35mg/mlのL−メチオニンおよび35mg/mlのドデシルサルコシン(すなわちN−ドデカノイルサルコシンナトリウム)を含有する4x(v/v、TFEに対して)の6M塩化グアニジニウム、100mM酢酸塩、pH4で希釈した。その溶液を、活性化Zn粉末に添加した。反応は〜1時間間隔でモニターし、5時間後に完了する。完了後、上澄みを除去し、残存するZn粉末を、20%TFEを含有する35mg/mlL−メチオニンおよび35mg/mlドデシルサルコシン含有6M塩化グアニジニウム、pH4、100mM酢酸塩で5分間2回洗浄し、20%β−メルカプトエタノールを含有する同じ溶液で1回洗浄した。混合生成物を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の修飾生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoを含有する分画をES−MSによって同定し、プールした。
【0152】
ステップ6:Acm除去#2 プールしたSEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoの溶液を、HPLCグレードの水で3x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoを含有する分画をES−MSによって同定し、2x(w/w、ペプチド質量に対して)のDPC(ドデシルホスホコリン)を含有する水で2x(v/v)希釈し、一晩凍結乾燥した。
【0153】
ステップ7:ライゲーション#3 等量のSEP−0:2+SEP−0:3+SEP−0:4+Et−PicoおよびSEP−0:1を合わせて、15mM濃度で純TFEに溶解し、6M塩化グアニジニウムを含有する250mMのリン酸緩衝液(pH7.5)を添加した。その溶液に、1%チオフェノールを添加した。1日のライゲーション後、ライゲーション混合物を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6M塩化グアニジニウム20ml、pH4の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−0(1〜166)(配列番号:1)を含有する分画をエレクトロスプレー質量分析法によって同定し、2x(w/wペプチド質量に対して)のドデシルサルコシンを含有する水で2x(v/v)希釈し、凍結乾燥した。
【0154】
ステップ8:フォールディング(折りたたみ) 完全長結合ペプチドSEP−0(1〜166)を、6M塩化グアニジニウム、20%TFE、10倍モル過剰(タンパク質のCys残基に対して)のシステインを含有する200mMトリス緩衝液(pH8.7)に溶解した。この溶液を、3M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温で一晩透析した。次いでその溶液を、1M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温4℃で4時間透析し、最後に10mMリン酸緩衝液(pH7.0)に対して、4℃で4時間透析して、最終フォールド生成物を得た。フォールディングは、エレクトロスプレーES−MSおよびCD(円偏光二色性)分光法によって確認した。
【0155】
ステップ9:精製 折りたたまれたポリペプチドを、セントリコン濃縮バイアルで5x濃縮し、10mMリン酸塩、pH7.0で平衡させたResource Sカチオン交換カラムに充填した。折りたたまれたタンパク質を、500mMのNaClへの10分の直線塩勾配で溶出した。所望の折りたたまれた生成物SEP−0(1〜166)を含有する分画を、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)によって同定、凍結し、−80℃で貯蔵した。折りたたまれたタンパク質生成物の分析逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、折りたたまれたタンパク質の存在を実証した。
【0156】
実施例2
合成赤血球産生刺激タンパク質SEP−1−L30の合成
2番目の合成赤血球産生刺激タンパク質(SEP−1−L30とする)を、SEP−0の24および126位にオキシム形成基を含有するように合成した。次いでこれらの基を用いて、線状(EDA−Succ)18カルボキシレート(EDA=(4,7,10)−トリオキサトリデカン−1,13ジアミンであり、TTDとも呼ばれる。Succ=−CO−CH2CH2CO−)ポリマーがそのタンパク質主鎖に結合しているSEP−1−L30を形成した。完全長SEP−1(1〜166)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0157】
A.GRFNP32によるGRFN1776およびGRFN1711のオキシム化
オキシム形成を行って、アミノオキシアセチル基を有する水溶性ポリマーを、ケトンカルボニル基を有するペプチドに結合した。これを達成するために、以下のペプチドセグメントを合成した。
セグメントSEP−1:4(GRFN1776、配列番号:2の残基117〜166からなる):
CAISPPDAAK AAPLRTITAD TFRKLFRVYS NFLRGKLKLY TGEACRTGDR−カルボキシレート(Lys126はε−アミノ基においてレブリン酸残基で修飾され、Cys117はAcm保護されている)
セグメントSEP−1:1(GRFN1711、配列番号:2の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA EH−チオエステル(Lys24はレブリン酸残基で修飾されている)
【0158】
実施例1のとおり、セグメントSEP−1:1(GRFN1711)をチオエステル生成樹脂上で、セグメントSEP−1:4(GRFN1776)を−OCH2−Pam−樹脂上で合成した。これら2つのペプチドセグメントのリジン24および126を、初めにε−アミノ基においてFmoc基で保護した。鎖アセンブリの完了後、Fmocを有するアミノ基を標準的なFmoc脱保護手順に従って脱保護し、各ペプチド樹脂にそれぞれレブリン酸を結合することによって修飾した。次いでこれらのペプチドを、実施例1に記載のとおり、脱保護し、同時に樹脂支持体から開裂した。ペプチドを、分取C4逆相HPLCによって精製した。純粋ペプチドを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。GRFNP32[−(EDA−Succ)18カルボキシレート]を、標準的なプロトコルに従って、Sasrinカルボキシル生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。5倍過剰の活性化Boc−アミノオキシ酢酸をカップリングすることによって、アミノオキシアセチル(AoA)部分をポリマーのN末端アミノ基に結合した。そのポリマー鎖を、典型的なFmoc化学手順を用いて樹脂支持体から開裂した。ポリマー鎖を、分取C4逆相HPLCによって精製した。純粋なポリマーを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0159】
セグメントSEP−1:4およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP32を含有する分画をES−MSによって同定し、プール、凍結乾燥した。
【0160】
セグメントSEP−1:1およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP32を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0161】
B.合成赤血球産生刺激タンパク質SEP−1−L30の合成
SEP−1−L30を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP32(GRFN1711+GRFNP32、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP32(GRFN1776+GRFNP32、配列番号:2の残基117〜166に対応):
CAIS PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システインはピコリル(pico)保護基を有する)
【0162】
追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を、実施例1に記載のとおり行い、完全長フォールドSEP−1−L30を得た。フォールドタンパク質生成物の分析逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、フォールドタンパク質の存在を実証した。
【0163】
実施例3
合成赤血球産生刺激タンパク質SEP−1−L26の合成
3番目の合成赤血球産生刺激タンパク質(SEP−1−L26とする)を、SEP−0の24および126位にオキシム形成基を含有するように合成した。次いで、これらの基を用いて、線状ポリマー(EDA−Succ)18カルボキシレートおよび(EDA−Succ)6−アミドが、それぞれ24位および126位においてオキシム結合を介してタンパク質主鎖に結合しているSEP−1−L26を形成した。完全長SEP−1(1−166)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0164】
SEP−1−L30と対照的に、SEP−1−L26構成体は、126位に結合されたより小さな非荷電水溶性ポリマーを有するように設計された。24位に結合されたポリマーは、SEP−1−L30と同じである。完全長生成物のアセンブリは、実施例2に記載したとおりである。
【0165】
A.GRFNP6によるGRFN1776のオキシム化およびGRFNP32によるGRFN1711のオキシム化
オキシム形成を行い、アミノオキシアセチル基を有する水溶性ポリマーを、ケトンカルボニル基を有するペプチドに結合した。これを達成するために、以下のペプチドセグメントを合成した。
セグメントSEP−1:4(GRFN1776、配列番号:2の残基117〜166からなる):
CAISPPDAAK AAPLRTITAD TFRKLFRVYS
NFLRGKLKLY TGEACRTGDR−カルボキシレート(Lys126はε−アミノ基においてレブリン酸残基で修飾され、Cys117はAcm保護されている)
セグメントSEP−1:1(GRFN1711、配列番号:2の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA
EH−チオエステル(Lys24はレブリン酸残基で修飾されている)
【0166】
実施例1のとおり、セグメントSEP−1:1(GRFN1711)をチオエステル生成樹脂上で、セグメントSEP−1:4(GRFN1776)を−OCH2−Pam−樹脂上で合成した。これら2つのペプチドセグメントのリジン24および126を、初めにε−アミノ基においてFmoc基で保護した。鎖アセンブリの完了後、Fmocを有するアミノ基を標準的なFmoc脱保護手順に従って脱保護し、標準的にカップリングプロトコルに従って各ペプチド樹脂にそれぞれレブリン酸を結合することによって修飾した。次いで、実施例1に記載のBoc化学手順によって、これらのペプチドを脱保護し、同時に樹脂支持体から開裂した。ペプチドを、分取C4逆相HPLCによって個別に精製した。各ペプチドに関して、純粋ペプチドを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0167】
水溶性ポリマー(EDA−Succ)18カルボキシレート(GRFNP32)を、標準的なプロトコルに従って、Sasrinカルボキシル生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。水溶性ポリマー(EDA−Succ)6−アミド(GRFNP6)を、標準的なプロトコルに従って、Sieberアミド生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。5倍過剰の活性化Boc−アミノオキシ酢酸をカップリングすることによって、アミノオキシアセチル(AoA)部分を樹脂結合ポリマーそれぞれのN末端アミノ基に結合した。その2つのポリマー鎖を個々に、典型的なFmoc化学手順を用いてそれぞれの樹脂支持体から開裂した。各ポリマー鎖を、分取逆相HPLCによって精製した。各ポリマーに関して、純粋ポリマーを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0168】
セグメントSEP−1:4およびGRFNP6を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP6を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0169】
セグメントSEP−1:1およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP32を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0170】
B.合成赤血球産生刺激タンパク質SEP−1−L26の合成
SEP−1−L26を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP32(GRFN1711+GRFNP32、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP6(GRFN1776+GRFNP6、配列番号:2の残基117〜166に対応)
CAIS PPDAAKOXAAPL RTITADTFRK
LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP6にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システイン(すなわち、Cys161)はピコリル(pico)保護基を有する)
【0171】
追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を、実施例1および2に記載のとおり行い、完全長フォールドSEP−1−L26を得た。フォールドタンパク質生成物の分析C4逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、フォールドタンパク質の存在を実証した。
【0172】
実施例4
合成赤血球産生刺激タンパク質SEP−1−B50の合成
4番目の合成赤血球産生刺激タンパク質(SEP−1−B50とする)を合成した。完全長SEP−1−B50のアミノ酸配列は、SEP−1−L30の配列と同じであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0173】
しかしながら、このタンパク質は、SEP−1−L30の線状(Succ−EDA)18ポリマーではなく、4つの線状(Succ−EDA)12部分を有する分岐ポリマー構成体で誘導化した。誘導化は、オキシム結合によって達成した。完全長生成物のアセンブリは、実施例2に記載のとおりであった。
【0174】
A.複数のチオール基を有するテンプレートGRFNP17の合成
テンプレートGRFNP17を、0.4mmolスケールのアミド生成(4−メチル)ベンズヒドリルアミン(MBHA)樹脂上で手作業によって合成した。Fmoc−Lys(Boc)−OHを、標準的なカップリングプロトコルを用いてカップリングした(Schnolzer、M、Int.J.Pept.Protein Res.(1992)40:180〜93)。2.1mmolアミノ酸、0.5MのHBTU3.8ml中の10%DIEA、すなわち5倍過剰のアミノ酸を用いた。Fmoc保護基を除去した後、Fmoc−Lys(Fmoc)−0Hを、標準的なアミノ酸カップリングプロトコルを用いてカップリングした(2.1mmolアミノ酸、0.5MのHBTU3.8ml中の10%DIEA、すなわち5倍過剰のアミノ酸)。第2のFmoc除去ステップの後、Fmoc−Lys(Fmoc)−0Hを、標準的なアミノ酸カップリングプロトコルを用いてカップリングした(4.2mmolアミノ酸、0.5MのHBTU7.6ml中の10%DIEA、すなわち遊離アミンに対して5倍過剰のアミノ酸)。最後のFmoc脱保護ステップの後、5倍過剰(遊離アミンに対して)のDMF中S−アセチルチオグリコール酸ペンタフルオロフェニルエステル(SAMA−oPfp)を30分間カップリングした。C末端リシル残基の側鎖のBoc保護基を、純TFAで1分のバッチ洗浄を2回行うことによって除去し、次いでDMF中10%DIEAで洗浄することによって樹脂を中和した。2mmolのBoc−アミノオキシ酢酸、および2mmolのN−ヒドロキシスクシンイミド(NHS)を、DMF3mlに溶解した。2mmolのDIC(ジイソプロピルカルボジイミド)を添加した後、この酸を30〜60分間活性化した。その溶液を中和樹脂に添加し、1時間カップリングした。最後に、S結合アセチル基を、30分間DMF中の20%ピペリジンによって除去した。遊離アルデヒドのスカベンジャとしてシステインの存在下、標準的なBoc化学手順に従って、HF/p−クレゾールを用い、そのテンプレートを脱保護し、同時に樹脂支持体から開裂した(Schnolzer、M、Int.J.Pept.Protein Res.(1992)40:180〜93)。50%B(すなわち、0.1%TFAを含有する50%アセトニトリル水溶液)中の回収ポリアミド(アルデヒドを含まない)を、凍結し、凍結乾燥した。精製のために、そのテンプレート粗生成物を2mlの50%Bに溶解し、100mlの100%A(すなわち、0.1%TFA水溶液)を添加して試料を希釈した(アルデヒドの付加が確実であるため、塩化グアニジニウムまたはアセテートの添加は回避)。そのテンプレートを、3%B、T=40℃で平衡したC4分取逆相HPLCカラムに充填した。塩を定組成溶離し、所望のテンプレートGRFNP17を直線勾配で精製した。所望の生成物を含有する分画を、ES−MSによって同定し、プールし、凍結乾燥した。
【0175】
B.分岐水溶性ポリマーGRNP29の合成
分子量15kDaの分岐(EDA−Succ)12ポリマーGRFNP29を、精製チオール含有テンプレートGRFNP17、および線状ポリマーGRFNP31、Br−アセチル−(EDA−Succ)12カルボキシレートのチオエーテル生成ライゲーションによって合成したが、ここでGRFNP31は標準的なプロトコルに従ってSasrinカルボキシ生成樹脂上で合成した(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。
【0176】
1.3xモル過剰(全チオールに関して)の精製GRFNP31、Br−アセチル化(EDA−Succ)12、および精製チオール含有テンプレートGRFNP17を合わせて、0.1Mトリス−HCl/6M塩化グアニジニウムpH8.7に濃度〜10mMで溶解した。溶解後、その溶液を0.1Mトリス−HCl、pH8.7緩衝液で3倍(v/v)に希釈した。ライゲーション混合物を室温で攪拌し、反応を逆相HPLCおよびES/MSでモニターした。追加のGRFNP31反応物を、所望の反応生成物が主生成物となるまで必要に応じて添加した。仕上げとして、3x(v/v、ライゲーション混合物に対して)の0.1M酢酸塩/6M塩化グアニジニウム、pH4を添加し、その溶液を分取C4逆相HPLCカラムに充填して、直線勾配で精製した。純粋GRFNP29構成体を含有する分画をES−MSを用いて同定し、プール、凍結乾燥した。
【0177】
C.GRFNP29によるGRFN1776およびGRFN1711のオキシム化
セグメントSEP−1:4およびセグメントSEP−1:1を、実施例2に記載のとおり合成した。セグメントSEP−1:4およびGRFNP29を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。その溶液を凍結乾燥した。その乾燥粉末を、分取逆相HPLCカラム(直径1インチ)に充填した。ポリマー修飾ペプチドを、分取勾配C4逆相HPLCによって非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP29を含有する分画を、ES−MSによって同定し、プールした。
【0178】
セグメントSEP−1:1およびGRFNP29を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。その溶液を凍結および凍結乾燥した。その乾燥粉末を、0.1%TFAを含有する50%アセトニトリル水溶液に溶解し、分取GPC(ゲル透過クロマトグラフィ)カラム(直径1インチ)に充填した。ポリマー修飾ペプチドを、定組成溶離によって非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP29を含有する分画を、ES−MSによって同定し、プールした。
【0179】
D.合成赤血球産生刺激タンパク質SEP−1−B50の合成
SEP−1−B50(配列番号:2)を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP29(GRFN1711+GRFNP29、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP29にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP29(GRFN1776+GRFNP29、配列番号:2の残基117〜166に対応):
CAIS PPDAAKOXAAPL RTITADTFRK
LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP29にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システインはピコリル(pico)保護基を有する)
【0180】
精製をResourceQカラムで行ったことを除いて、追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を実施例1および2に記載のとおり行い、完全長折りたたみSEP−1−B50(配列番号:2)を得た。折りたたみタンパク質生成物SEP−1−B50の分析C4逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、折りたたみタンパク質の存在を実証した。
【0181】
実施例5
合成赤血球産生刺激タンパク質SEP−3−L42の合成
5番目の合成赤血球産生刺激タンパク質(SEP−3−L42とする)を合成した。完全長SEP−3タンパク質のアミノ酸配列は以下のとおりである。
APPRLICDSR VLERYLLEAK EAECITTGCA
EHCSLNECIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LACSSQPWEP
LQLHVDKAVS GLRSLTTLLR ALGAQKEAIS
PPDAACAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:3)
【0182】
24、38、83、および126位のシステイン残基を、マレイミド官能化(EDA−Succ)18(GRFNP32)ポリマー単位で修飾して(マイケル付加反応によって)、SEP−3−L42を形成した。
【0183】
詳細には、SEP−3を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−3:1(GRFN1747、配列番号:3の残基1〜37に対応):
APPRLICDSR VLERYLLEAK EAECITTGCA
EHCSLNE−チオエステル(Cys7、Cys29、Cys33はAcm保護されている)
セグメントSEP−3:2(GRFN1774、配列番号:3の残基38〜82に対応):
CIT VPDTKVNFYA WKRMEVGQQA VEVWQGLALLSEAVLRGQAL LA−チオエステル(Cys38はAcm基で側鎖保護されている)
セグメントSEP−3:3(GRFN1749、配列番号:3の残基83〜125に対応):
CSSQPWEP LQLHVDKAVS GLRSLTTLLR
ALGAQKEAIS PPDAA−チオエステル(Cys83はAcm基で側鎖保護されている)
セグメントSEP−3:4(GRFN1750、配列番号:3の残基126〜166に対応):
CAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR−カルボキシレート(Cys161は、Pbo(すなわち、4−(CH3S(O)−)ベンジル−)保護されている)
【0184】
ペプチド合成: ペプチドSEP−3:1およびSEP−3:2およびSEP−3:3を、上述の特定のペプチドに示したように保護基方法を変更して、実施例1に記載の確立された側鎖保護法を用い、Boc化学SPPSのためのin situ中和プロトコルによってチオエステル生成樹脂上で合成した。セグメントSEP−3:4を、−OCH2−Pam−樹脂上で同様に合成した。これらのペプチドを、実施例1に記載のとおり脱保護し、同時に樹脂支持体から開裂した。上に記載の結果として生じたペプチドを、分取逆相HPLCによって精製した。純粋ペプチドを含有する分画を、ES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0185】
ステップ1:ライゲーション#1 セグメントSEP−3:4およびセグメントSEP−3:3を、濃度15mMでTFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する飽和リン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。ライゲーション後、ライゲーション混合物(1体積とする)を、2体積の{TFE2ml、6M塩化グアニジニウム6ml、25%β−メルカプトエタノール含有100mMリン酸塩}溶液に添加し、20分間インキュベートした。その溶液を、15mg/mlのTCEP氷酢酸溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:Acm+SEP−3:3+SEP−3:4を含有する分画を、ES−MSによって同定し、プールした。
【0186】
ステップ2:Acm除去#1 Acm除去のために、プールしたSEP−3:Acm+SEP−3:3+SEP−3:4分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取C4逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0187】
ステップ3:ライゲーション#2 等量のSEP−3:3+SEP−3:4およびSEP−3:2を合わせて、15mM濃度で純TFEに溶解した。6Mグアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物(1体積とする)を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6Mグアニジニウム20ml、pH4の2体積の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:Acm+SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0188】
ステップ4:Acm除去 Acm除去のために、プールしたSEP−3:Acm+SEP−3:2+SEP−3:3+SEP−3:4の分画を含有するアセトニトリル水溶液を、HPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、C4半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の結合生成物SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0189】
ステップ5:ライゲーション#3 等量のSEP−3:2+SEP−3:3+SEP−3:4およびSEP−3:1を合わせて、15mM濃度で純TFEに溶解した。6Mグアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物(1体積とする)を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6Mグアニジニウム20ml、pH4の2体積の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0190】
ステップ6:ポリマーGRFNP32の結合 GRFNP32−マレイミドと称するマレイミド官能化線状(EDA−Succ)18ポリマーを、マレイミド官能化線状(EDA−Succ)18ポリマー(すなわち、マレイミド−(EDA−Succ)18)を形成するための製造業者プロトコルに従って、GRFNP32をBMPS(3−マレイミドプロピオン酸NHSエステル、Pierce、USA)で機能化することによって調製した。SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4を、必要最小量のTFEに溶解した。3倍過剰のGRFNP32−マレイミドを、6M塩化グアニジニウム、100mMリン酸塩、pH7.5に溶解し、TFE溶液に添加した。マイケル付加反応の進行を、分析逆相HPLCによって追跡した。反応が完了した後、その溶液を分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望のポリマー修飾生成物SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+pPEG(すなわち、Cys24、Cys38、Cys83、およびCys126の側鎖チオールに結合したGRFNP32の4つの複製を有する結合完全長166残基ポリペプチド鎖であり、したがってSEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32とも呼ばれる)を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0191】
ステップ7:Pbo除去 Pbo除去のために、SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32の凍結乾燥粉末を、5%エタンジチオールを含有する純TFAに溶解した。次いで、Pbo基を、10%チオアニソールおよび15%ブロモトリメチルシランを添加することによって30分間開裂した。その溶液を回転式エバポレータで乾燥し、0.1%TFAを含有するアセトニトリル水溶液に取った。結果として生じた溶液を、半分取逆相HPLCカラムに充填し、段階勾配によって精製した。所望のCys161脱保護生成物SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32−Pboを含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0192】
ステップ8:Acm除去 Cys7、Cys29、およびCys33の側鎖からの最終Acm除去のために、プールしたSEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32−Pboの分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取C4逆相HPLCカラムに充填し、段階勾配で精製した。所望の結合ポリマー修飾生成物SEP−3(1〜166)を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0193】
ステップ9:フォールディング 完全長結合ポリマー修飾ペプチドSEP−3(1〜166)を、6M塩化グアニジニウム、20%TFE、10倍モル過剰(SEP−3のCys残基に対して)のシステインを含有する200mMトリス緩衝液(pH8.7)に溶解した。その溶液を、3M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温で一晩透析した。次いでその溶液を、1M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、4℃で4時間透析し、最後に10mMリン酸緩衝液(pH7.0)に対して、4℃で4時間透析して、最終フォールド生成物を得た。フォールディングは、エレクトロスプレーES−MSおよびCD分光法によって確認した。
【0194】
ステップ10:精製 折りたたまれたポリペプチドを、セントリコン濃縮バイアルで5x濃縮し、10mMリン酸塩、pH7.0で平衡させたResource Sカチオン交換カラムに充填した。折りたたまれたタンパク質を、500mMのNaClへの10分の直線塩勾配で溶出した。所望の折りたたまれた生成物SEP−3−L42を含有する分画をSDS−PAGEによって同定、凍結し、−80℃で貯蔵した。
【0195】
実施例6
合成赤血球産生刺激タンパク質の生物活性アッセイ
フォールド合成赤血球産生刺激タンパク質、SEP−0、SEP−1−L26、SEP−1−L30、SEP−1−B50、およびSEP−3−L42の生物活性を、コントロール標準として市販の組換えエリスロポエチンを用いる因子依存細胞系増殖アッセイにおいて、UT/7および32D103197細胞系を用い判定した。UT−7は、増殖および生存に関してインターロイキン−3、顆粒球マクロファージコロニー刺激因子(GM−CSF)、またはエリスロポエチン(EPO)の1つに絶対依存性を有するヒト巨核芽球白血病細胞系である(Miura Y.等、Acta Haematol(1998)99:180〜184;Komatsu N.等、Cancer Res.(1991)51:341〜8)。32D103197は、マウス造血細胞系である(Metcalf、D.Int J Cell Cloning(1992)10:116〜25)。
【0196】
イスコフ改変ダルベッコ培地(IMDM)、10%FBS(ウシ胎児血清)、グルタミン、およびPenstrep中でSEP構成体の保存溶液を作製し、その保存溶液の連続2x希釈液をマルチウェルプレートに添加し、5000細胞/50μlの濃度でヒトUT/7EPO細胞を加えた。プレートを5%CO2の存在下、37℃でインキュベートし、増殖を毎日モニターした。4日後、PBS(リン酸緩衝化食塩水)中の2.5mg/mlMTT(メチルチアゾールテトラゾリウム)20μlを添加し、プレートを4時間インキュベートした。IPA150μlを加え、各ウェルの吸光度を562nmで読み取った。SEP化合物のED50(最大効果の50%に達する有効量)値を求め、CHO(Chineseハムスター卵巣)細胞産生rhEPO(組換えヒトエリスロポエチン)の値と比較した。これらの実験の結果は、すべての合成赤血球産生刺激タンパク質が生物活性を示すことを実証した。SEP−0、SEP−1−L26、SEP−1−L30、およびSEP−1−B50のED50の結果を表VIに示す。
【表9】
【0197】
本発明をその特定の実施形態に関して述べたが、さらなる変更が可能であり、本出願が、一般に本発明の原理に従い、本発明の属する技術分野において知られ、慣例的に行われている範囲内であって、且つ本明細書に記載の本質的な特徴に適用できるような本発明の開示からの逸脱を含む本発明の変形、使用、または適応を含むものであることは理解されるであろう。
【図面の簡単な説明】
【0198】
【図1】擬似天然型化学的ライゲーションの全体的な反応を例示する図である。
【図2】本発明の合成生物活性タンパク質を調製する全体的なプロセスを例示する図である。
【図3】合成赤血球産生刺激タンパク質の好ましい型の基本構造を示す図である。
【図4】遊離アミノ末端またはカルボキシ末端を持たない環状順列化SEP類似体の一般的な構造を示す図である。pPEG(本明細書に記載の「精密PEG」)結合部位は、SEP類似体と位置的に同一である。SEP類似体は、P126CおよびA125Xなどの、新しいN/C末端位置間のオキシム結合によって環化される。SEP類似体の全分子量は、所与の類似体の修飾に用いられるpPEGに応じて約50〜80kDaの範囲となり、pPEGを介した流体力学分子量は約100kDaと推定される。
【図5】合成生物活性GCSFタンパク質の基本構造を示す図である。この図において、「J」は、疎水性側鎖を有する非天然コード化残基を示す。
【図6】好ましい合成RANTES類似体の構造を示す図である。この図において、NNY=ノナノイル、X=疎水性側鎖pPEGを有する非天然コード化アミノ酸、FA=脂肪酸である。
【図7】分岐鎖pPEGポリマーの形成を図式的に示す図である。
【技術分野】
【0001】
本発明は、アミド結合を介し、より広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にするために天然型化学的ライゲーションの技法を拡大するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途を提供する。
【0002】
(関連出願の相互参照)
本出願は、米国特許出願第60/231330号(2000年9月8日出願)および第60/236377号(2000年9月29日出願)の一部継続出願であり、これら双方の出願全体を参照により本明細書に組み込むものとする。
【背景技術】
【0003】
過去30年の間に、疾患を治療するための治療用薬剤として天然に産生されたタンパク質を用いることの可能性に医学上の注目がますます向けられるようになった。
【0004】
組換えDNA技法は、細菌および他の宿主細胞において多量に産生することができるので、多くのポリペプチドおよびタンパク質の商業的産生の主要な方法となっている。組換えタンパク質の産生には、所望の外生タンパク質をコード化するDNAを用いて宿主細胞をトランスフェクトまたは形質転換すること、およびその組換えタンパク質の発現に有利な条件下で細胞を増殖することが含まれる。組換えタンパク質を高力価で産生させることができることから、大腸菌および酵母が宿主として好都合である(米国特許第5756672号(Builder他)を参照のこと)。
【0005】
組換えDNAコード化タンパク質の一般的な細菌の発現に関して、多数の米国特許が発行されている(たとえば、米国特許第4565785号、第4673641号、第4738921号、第4795706号、第4710473号を参照のこと)。残念ながら、組換えDNA技法は例外なく成功したわけではない。いくつかの条件下で、細菌宿主から多量に発現したある種の異種タンパク質は、細胞内で濃縮な凝集物として沈降し、位相差顕微鏡において細胞内部に可視の輝点として認識されることになる。これらの沈降タンパク質の凝集物は「屈折体」と呼ばれ、細胞タンパク質全体のかなり大きな部分を構成する可能性がある(Brems等、Biochemistry(1985)24:7662)。
【0006】
これらの物体からのタンパク質の回収は多数の問題を提起しており、たとえば細胞内に包含されたタンパク質を、それを収容している細胞材料およびタンパク質からどのように分離するのか、その封入体タンパク質を生体活性形態でどのように回収するのかなどの問題がある。屈折体に関する全般的な概説は、Marston、上記;MitrakiおよびKing、Bio/Technology、7:690(1989);MarstonおよびHartley、Methods in Enzymol.、182:264〜276(1990);Wetzel、「Protein Aggregation In Vivo:Bacterial Inclusion Bodies and Mammalian Amyloid」、Stability of Potein Pharmaceuticals:In Vivo Pathways of Degradation and Strategies for Protein Stabilization、AhernおよびManning(編)(Plenum Press、1991);Wetzel、「Enhanced Folding and Stabilization of Proteins by Suppression of Aggregation In Vitro and In Vivo」、Protein Engineering−A Practical Approach、Rees、A.R.等(編)(IRL Press、Oxford University Press、Oxford、1991)を参照されたい。したがって、生物活性タンパク質を産生する別の方法が求められている。
【0007】
タンパク質を組換え産生する別の方法の1つには、タンパク質を合成するために有機化学の原理を用いることが含まれる。現在、タンパク質を化学合成するための方法には、段階固相合成、および溶液中での、または固相でのフラグメント縮合が含まれる。メリフィールド(Merrifield)の古典的な段階固相合成には、所望のペプチド鎖のカルボキシ末端アミノ酸に対応するアミノ酸を固体支持体に共有結合すること、および活性化カルボキシル基を有する活性化アミノ酸誘導体の段階カップリングによってアミノ末端に向かってポリペプチド鎖を延長することが含まれる。完全に保護された固相結合ペプチド鎖のアセンブリが完了した後、ペプチド−固相共有結合を適切な化学方法によって開裂し、保護基を除去して生成物のポリペプチドを得る。
【0008】
残念ながら、段階固相合成法には多くの欠点があり、それには各サイクルのカップリングおよび脱保護での不完全な反応に起因する生成物によって結合した固相の形成が含まれる。ペプチド鎖が長くなるにつれて、高純度の意図した生成物を得ることがより難しくなる。この方法によるタンパク質および大型のポリペプチドの合成は、時間のかかる困難な作業である。
【0009】
固相フラグメント縮合アプローチ(セグメント縮合としても知られる)は、固相段階合成法で長いポリペプチドを得るときの困難を克服するように考案されたものである。このセグメント縮合法には、固相段階法によって数個のペプチドセグメントを調製すること、続いて、最大限に保護されたセグメントを固相から開裂し、精製することが含まれる。保護セグメントは、第1のセグメントに1つずつ縮合され、それが固相に結合する。しかしながら多くの場合、固相セグメント縮合の多くのステップで技術的問題が生じている。E.Atherton等、「Solid Phase Fragment Condensation−The Problems」、Innovation and Perspectives in Solid Phase Synthesis 11〜25(R.Epton等、1990)を参照されたい。たとえば、望ましくないライゲーション反応をブロックするためのセグメント上での保護基の使用によって、しばしば保護セグメントの可溶性はわずかとなり、カルボキシル基の有効な活性化が妨げられる可能性がある。保護セグメントの限定された溶解性はさらに、保護セグメントの精製を妨げる可能性がある。K.Akaji等、Chem.Pharm.Bull.(Tokyo)33:184〜102(1985)を参照されたい。保護セグメントは、純度、共有構造に関して特性付けが困難であり、高分解能分析ESMS(エレクトロスプレー質量分析法)(電荷に基づく)を施すことができない。ライゲーションがグリシン残基で行われる場合を除いて、各活性化ペプチドセグメントのC末端残基のラセミ化も問題である。さらに、完全にアセンブルされた固相結合ポリペプチドの固相からの開裂および保護基の除去は、しばしば、完全にアセンブルされたポリペプチドの分解をもたらす厳しい化学手法および長い反応時間を必要とすることになる。
【0010】
セグメント縮合は、固相上ではなく、溶液中で行うことができる。H.Muramatsu等、Biochem.and Biophys.Res.Commn.203(2):1131〜1139(1994)を参照されたい。しかしながら、溶液中のセグメント縮合は、ライゲーションに先立ってセグメントを精製する必要があり、且つ複数の望ましくない副反応を防ぐために一連の異なる側鎖官能基上で保護基を用いる必要がある。さらに、溶液中のライゲーションでは、固相ライゲーションで提供される簡便な精製および洗浄ステップが行えない。さらに、保護ペプチドセグメントおよび保護ペプチド中間反応生成物の溶解性に関する限定が悪化してくる。
【0011】
最大限に保護されたペプチドでしばしば生じる溶解性の問題を克服するために、ペプチドセグメントの化学的ライゲーションが探求されてきた。化学的ライゲーションには、第1の化学成分と第2の化学成分との間の選択的共有結合の形成が含まれる。第1および第2の成分に存在する一意性の相互反応性官能基を用いて、ライゲーション反応化学選択性を付与することができる。たとえば、ペプチドおよびポリペプチドの化学的ライゲーションには、適合性の、一意性の相互反応性C末端およびN末端アミノ酸残基を有するペプチドまたはポリペプチドセグメントの化学選択的反応が含まれる。この目的のために、いくつかの異なる化学方法が用いられており、その例としては、天然型化学的ライゲーション(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434)、オキシム形成化学的ライゲーション(Rose等、J.Amer.Chem.Soc.(1994)116:30〜34)、チオエステル形成ライゲーション(Schnolzer等、Science(1992)256:221〜225)、チオエーテル形成ライゲーション(Englebretsen等、Tet.Letts.(1995)36(48):8871〜8874)、ヒドラゾン形成ライゲーション(Gaertner等、Bioconj.Chem.(1994)5(4):333〜338)、ならびにチアゾリジン形成ライゲーションおよびオキサゾリジン形成ライゲーション(Zhang等、Proc.Natl.Acad.Sci.(1998)95(16):9184〜9189;Tam他、WO95/00846;米国特許第5589356号)、または他の方法によるもの(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」J.Am.Chem.Soc.2001、123、526〜533;Gieselnan等、Org.Lett.2001、3(9):1331〜1334;Saxon、E.等、「Traceless」Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds.Org.Lett.2000、2、2141〜2143)が挙げられる。
【0012】
これらの方法のなかで、天然型化学的ライゲーション法のみが、ライゲーション部位に天然アミド(すなわちペプチド)結合を有するライゲーション生成物を生じさせる。初期の天然型化学的ライゲーション方法論(Dawson等、上記、およびWO96/34878)は、ライゲーション部位で天然アミド結合を生じるための強固な方法を証明した。天然型化学的ライゲーションには、C末端α−カルボキシチオエステル部分を有する第1のペプチドまたはポリペプチドセグメントと、N末端システイン残基を有する第2のペプチドまたはポリペプチドとの間の化学選択的反応が含まれる。チオール交換反応は、最初のチオエステル結合中間体を生じさせ、これは自発的に転位して、ライゲーション部位に天然アミド結合をもたらし、同時にシステイン側鎖チオールを再生する。初期の天然型化学的ライゲーション法の主な欠点は、N末端システインを必要とすることであり、すなわちこの方法ではN末端システインを有するペプチドおよびポリペプチドセグメントの結合のみが可能となる。
【0013】
このような欠点があるにもかかわらず、システイン以外のN末端アミノ酸を有するペプチドの天然型化学的ライゲーションが提案されている(WO98/28434)。この提案においては、ライゲーションは、C末端α−カルボキシチオエステルを有する第1のペプチドまたはポリペプチドセグメント、および式HS−CH2−CH2−O−NH−[ペプチド]によって表されるN末端のN−{チオール置換補助}基を有する第2のペプチドまたはポリペプチドセグメントを用いて行われる。ライゲーションに続いて、N−{チオール置換補助}基が、HS−CH2−CH2−O−補助基の開裂によって除去され、ライゲーション部位に天然アミド結合が生じる。この方法の1つの限界は、メルカプトエトキシ補助基を使用すると、グリシン残基においてのみ、アミド結合が形成され得ることである。これは、開裂によって第2のペプチドまたはポリペプチドセグメントのN置換アミノ酸の位置にグリシン残基を生じるライゲーション生成物を産生することになる。そのため、この方法の実施は、反応後のライゲーション生成物がそのような位置にグリシン残基を含有することを所望する場合にのみ適しており、いずれにしても、ライゲーション収率、前駆体の安定性、およびO結合補助基を除去できる可能性が問題となり得る。反応をグリシン残基でのライゲーションに限ることなく、他の補助基、たとえばHSCH2CH2NH−[ペプチド]を用いることができるが、そのような補助基は、結合された生成物から除去することができない。
【発明の開示】
【発明が解決しようとする課題】
【0014】
したがって、有効で容易に除去可能なチオール含有補助基を用いることによって、天然型化学的ライゲーションを多種多様な異なるアミノ酸残基、ペプチド、ポリペプチド、ポリマーおよび他の分子に拡大し、且つライゲーション部位においてそのような分子を天然アミド結合で結合する、広い適用範囲を持った、強固な化学的ライゲーション法が求められている。本発明は、このような要望および他の要望に応えるものである。
【課題を解決するための手段】
【0015】
本発明は、アミド結合を介するより広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にするために天然型化学的ライゲーションの技法を拡張するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途を提供する。本発明は、特に、随意ポリマー修飾された合成生物活性タンパク質、およびそのようなタンパク質を含有する薬剤組成物の合成における使用に好適なものである。
【0016】
詳細には、本発明は、側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質を提供する。
【0017】
本発明は、実施態様として、特に、そのタンパク質がリボソーム特定生物活性タンパク質の持つ生物活性を有する前述の合成タンパク質を提供する。
【0018】
本発明は、約25kDaを超えるモノマー分子量を有する前述のの合成タンパク質に関する。
【0019】
本発明は、さらに、タンパク質の複数のアミノ酸残基の少なくとも1つが隣接アミノ酸残基に非アミド結合(たとえば、チオエステル結合、チオエーテル結合およびオキシム結合)によって結合している前述の合成タンパク質に関する。
【0020】
本発明は、さらに、リボソーム特定生物活性タンパク質が哺乳動物のタンパク質(たとえば、ヒト、サル、ウシ、マウス、ブタ、ヒツジ、またはウマのタンパク質)である前述の合成タンパク質に関する。
【0021】
本発明は、さらに、タンパク質が、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカイン(特に、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択されるサイトカイン)の選択された生物活性を有する前述の合成タンパク質に関する。
【0022】
本発明は、さらに、合成生物活性タンパク質がSEP−0、SEP−1およびSEP−3から選択される合成タンパク質に関する。
【0023】
本発明は、さらに、1つまたは複数のそれらのアミノ酸残基が1つまたは複数のポリマー付加物によって修飾されているもの、特に合成タンパク質が、リボソームコード化生物活性タンパク質の少なくとも1つのグリコシル化部位に対応するアミノ酸残基において、1つまたは複数のポリマー付加物によって修飾されている1つまたは複数のアミノ酸残基を含むすべての前述の合成タンパク質に関する。
【0024】
本発明は、さらに、タンパク質がリボソーム特定生物活性哺乳動物タンパク質に関連する生物活性を模倣する生物活性を有し、約25kDaを超えるモノマー分子量を有し、且つ側鎖が式−S−Raaを有し、該式中、Raaはリボソーム特定アミノ酸側鎖の随意置換された末端部分、またはその類似体からなる群から選択される擬似アミノ酸残基を含有する、合成タンパク質を含む分子的に均一な薬剤組成物を提供する。
【0025】
本発明は、さらに、有効量の薬剤組成物を治療を必要としている個体に投与することを含む、ヒトの疾患または症状を治療する方法を提供するものであって、該薬剤組成物は、それぞれが合成タンパク質を含む1つまたは複数の分子的に均一な薬剤組成物を含み、該合成タンパク質は、約25kDaを超えるモノマー分子量を有し、且つ側鎖が式−S−Raaを有し、該式中、Raaは、リボソーム特定アミノ酸側鎖の随意置換された末端部分、またはその類似体からなる群から選択されるものである擬似アミノ酸残基を含有し、該合成タンパク質は、リボソーム特定生物活性ヒトタンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインの生物活性を模倣する生物活性を有するものである。
【0026】
本発明は、さらに、所望のポリペプチドを合成する方法およびそのような方法によって製造された所望のポリペプチドを提供し、該所望のポリペプチドは、以下の式を有し、
aaNH2−Q−aax−aay−W−aaCOOH
該式中、QおよびWは、それぞれ任意で存在する1つまたは複数の追加のアミノ酸残基を示し、aaNH2は、ポリペプチドのN末端アミノ酸残基を示し、aaxおよびaayは、それぞれ側鎖xおよびyを有する内部隣接アミノ酸残基を示し、aaCOOHは、ポリペプチドのC末端アミノ酸残基を示し、該方法は、
(A)式aaNH2−Q−aax−COSRを有し、該式中、Rは、アリール、ベンジルおよびアルキル基を非限定的に含むチオエステル基に適合する任意の基である第1のペプチドを、式Cys−W−aaCOOHを有する第2のペプチドに結合し、それによりポリペプチドaaNH2−Q−aax−Cys−W−aaCOOHを形成すること、および
(B)該ポリペプチドを試薬Raa−Xの存在下でインキュベートすることを含むものであって、該式中、Raaは、アミノ酸残基aayの側鎖の末端部分を模倣する構造を有する基であり、Xは、良好な離脱基(特に、F、IまたはBrなどのハロゲン)であり、該インキュベーションは、所望のポリペプチドを形成するのに充分な条件下で行われる。
【0027】
本発明は、さらに、アミノ酸残基aayが、擬似アルギニン、擬似アスパラギン、擬似アスパラギン酸塩、擬似ドパミン、擬似グルタミン酸塩、擬似グルタミン、擬似ヒスチジン、擬似イソロイシン、擬似ロイシン、擬似リジン、擬似メチオニン、擬似フェニルアラニン、擬似セリン、擬似トレオニン、擬似トリプトファン、擬似チロシンおよび擬似バリンからなる群から選択される前述の方法およびポリペプチドの実施態様を提供する。
【0028】
本発明は、アミド結合を介するより広範なペプチド、ポリペプチド、他のポリマーおよび他の分子のライゲーションを可能にし、それによってそのような分子の化学合成を促進するために天然型化学的ライゲーションの技法を拡張するための方法および組成物に関する。本発明は、さらに、そのようなタンパク質および誘導化タンパク質のための方法および用途をも提供する。本発明は、特に、好ましくは約25kDaを超えるモノマー分子量を有する随意ポリマー修飾された合成生物活性タンパク質およびそのようなタンパク質を含有する薬剤組成物の合成における使用にも好適である。
【発明を実施するための最良の形態】
【0029】
I.生物活性ペプチドおよびタンパク質の化学合成
一般的に、生物活性ペプチドおよびタンパク質の合成は、標準的な自動ペプチド合成装置を使用する1960年代初期に開発された「メリフィールド」化学反応の段階固相ペプチド合成プロトコルを用いる。そのような合成に固相もしくは液相ライゲーション方法を採用してもよい。そのような化学反応は、多くのポリペプチドを生成するために容易に用いることができるが、それに伴う収量損失、副生成物の生成、および不完全な反応のために、タンパク質または大きいポリペプチドの生成には適さない。
【0030】
これらの制限に対処するために、化学的ライゲーションの技法は、より大きなポリペプチドおよびタンパク質の合成を達成するために、あらかじめ形成されたペプチドフラグメントを合わせて結合できるように開発されてきた。
【0031】
化学的ライゲーションには、第1の化学成分と第2の化学成分との間の選択的共有結合の形成が含まれる。第1および第2の成分に存在する一意性の相互反応性官能基を用いて、ライゲーション反応化学選択性を付与することができる。たとえば、ペプチドおよびポリペプチドの化学的ライゲーションには、適合性の、一意性の相互反応性C末端およびN末端アミノ酸残基を有するペプチドまたはポリペプチドセグメントの化学選択的反応が含まれる。この目的のために、いくつかの異なる化学反応が用いられており、その例としては、天然型化学的ライゲーション(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434)、オキシム形成化学的ライゲーション(Rose等、J.Amer.Chem.Soc.(1994)116:30〜34)、チオエステル形成ライゲーション(Schnolzer等、Science(1992)256:221〜225)、チオエーテル形成ライゲーション(Englebretsen等、Tet.Letts.(1995)36(48):8871〜8874)、ヒドラゾン形成ライゲーション(Gaertner等、Bioconj.Chem.(1994)5(4):333〜338)、ならびにチアゾリジン形成ライゲーションおよびオキサゾリジン形成ライゲーション(Zhang等、Proc.Natl.Acad.Sci.(1998)95(16):9184〜9189;Tam他、WO95/00846;米国特許第5589356号)、または他の方法によるもの(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」J.Am.Chem.Soc.2001、123、526〜533;Gieselnan等、Org.Lett.2001、3(9):1331〜1334;Saxon、E.等、「Traceless」Staudinger Ligation for the Chemoselective Synthesis of Amide Bonds.Org.Lett.2000、2、2141〜2143)が挙げられる。
【0032】
ライゲーションが、N末端システイン残基を有するポリペプチドの結合反応を含む場合、好ましくは天然型化学的ライゲーションの手順が用いられる(Dawson等、Science(1994)266:776〜779;Kent他、WO96/34878;Kent他、WO98/28434;米国特許出願第09/097094、これらすべてを参照により本明細書に組み入れるものとする)。天然型化学的ライゲーションには、C末端α−カルボキシチオエステル部分を有する第1のペプチドまたはポリペプチドセグメントと、N末端システイン残基を有する第2のペプチドまたはポリペプチドとの間の化学選択的反応が含まれる。チオール交換反応は、最初のチオエステル結合中間体を生じ、これは自発的に転位して、ライゲーション部位で天然アミド結合をもたらし、同時にシステイン側鎖チオールを再生する。多くの場合、この天然タンパク質の配列は、N末端システイン残基を有するポリペプチドフラグメントが天然型化学的ライゲーション反応で合成され、用いられることができるように、適切に配置されたシステイン残基を含むことになる。
【0033】
この方法は、大きいポリペプチドおよびタンパク質の合成に実用的で強固で有用であることが証明されたが、ライゲーション部位におけるシステイン残基に関する要件がその適応性を制限する。システインは、もっとも一般的でないアミノ酸の1つである。典型的なタンパク質ドメインは、150〜200のアミノ酸を含み、多くのタンパク質は、システイン残基を含有しない長さ60超のアミノ酸領域を含有する。この問題の1つの解決策には、タンパク質の1つまたは複数の天然アミノ酸残基がシステイン残基で置換された非天然タンパク質を設計し、それによって天然型化学的ライゲーション法の使用を可能にすることが含まれる。この解決策は多くの場合、システイン残基の導入がタンパク質の構造または機能に及ぼす可能性のある予測できない影響によって複雑なものとなる。そのようなシステイン残基は、合成されたタンパク質を共有的に固定化するのに用いることができる。
【0034】
本発明は、個別に、または組み合わせて用いることのできる、この問題に対する2つの別の解決策を提供する。第1の解決策(本明細書では「擬似天然型化学的ライゲーション」と呼ぶ)には、タンパク質合成に用いられるペプチドのあらかじめ選択された位置で、非天然擬似アミノ酸残基を用いることが含まれる。そのような擬似アミノ酸の構造は、システインの構造、および合成されるタンパク質のあらかじめ選択された位置で天然に見出されるアミノ酸の構造の両方を模倣したものである。第2の解決策(本明細書では「拡張天然型化学的ライゲーション」と呼ぶ)は、参照により本明細書に組み込むものとする米国特許出願第60/231339号(Kent他、2000年9月8日出願)に記載されており、カルボキシルチオエステル、より好ましくはα−カルボキシルチオエステルを含む第1の成分を、酸安定性N置換、好ましくはNα−置換、2または3炭素鎖アミノアルキルまたはアリールチオールを含む第2の成分と結合することを含む。これらの解決策は、それぞれ以下に詳しく説明する。
【0035】
A.擬似天然型化学的ライゲーション
擬似天然型化学的ライゲーションは、天然型化学的ライゲーションによってライゲーション部位に生じるシステイン側鎖のチオアルキル化を対象とする。好ましい態様は、少なくとも1つのペプチドが、適切な保護基で保護されたチオール側鎖を有する天然システインを含有する、システインライゲーション部位のチオアルキル化である。
【0036】
本発明の好ましい実施形態において、システイン基のチオール部分は、所望の側鎖、たとえばリボソーム特定アミノ酸の側鎖、そのようなアミノ酸の類似体、非リボソーム特定アミノ酸に修飾されている。本明細書では、リボソーム特定アミノ酸とは、タンパク質翻訳のプロセスにおいてリボソームによって認識され、リボソーム生成タンパク質に組み込むことのできるアミノ酸である。相当数の公表された文献が存在し、システイン側鎖チオール部分の化学修飾を記載している(たとえば、「Current Protocols in Protein Science」John E.Coligan等編、John Wiley&Sons、NY(2000)を参照のこと)。Kaiser、E.T.は、天然アミノ酸側鎖の化学特性を模倣するシステイン残基側鎖の変換を記載している(たとえば、Kaiser、E.T.等、「Chemical Mutation Of Enzyme Active Sites」、Science、1984 Nov 2;226(4674):505〜11を参照のこと)。さらに、標識をペプチドまたはタンパク質に導入するためのシステイン側鎖の使用が記載されている。システイン側鎖修飾は、Chemistry of Protein Conjugation and Crosslinking、S.S.Wong(1991、CRC Press);Chemical Modification of Proteins、Gary E.Means等(1971、Holden−Day)、Chemical Modification of Proteins:Selected methods and analytical procedures、Glazer、A.N.等(1975、Elsevier);Chemical Reagents for Protein Modification、RL Lundblad(1991、CRC Press)に概説されている。Tam等(Biopolymers(1998)46:319〜327)は、非システイン天然型化学的ライゲーションでのホモシステイン(−CH2−CH2−SH)の使用、それに続くメチルp−ニトロベンゼンスルホネート(メチル化試薬)を用いるチオアルキル化によるホモシステイン側鎖の天然メチオニン側鎖(−CH2−CH2−S−CH3)への変換を開示している。本発明はまた、ホモシステインを擬似アミノ酸、すなわちメチオニン以外のアミノ酸に変換するために用いることもできる。しかしながら、本明細書に記載のシステインの変換と同様に、本発明によれば、変換を望まない少なくとも1つの天然システインを含有するペプチドの場合、ジスルフィドペアリングに関わる天然システインの分解を回避するために、保護基を用いる必要がある。適切な保護基は以下に記載する。
【0037】
擬似天然型化学的ライゲーション法は、ある種のリボソーム特定アミノ酸の側鎖(たとえば、グリシン、アラニン、バリンおよびプロリンの側鎖)の模倣を促進しないが(しかしながら、アラニンの側鎖は脱硫反応によって形成され得る(参照により本明細書に組み込むものとするYan、L.Z.およびDawson、P.E.の「Synthesis of Peptides and Proteins without Cysteine Residues by Native Chemical Ligation Combined with Desulfurization」、J.Am.Chem.Soc.2001、123、526〜533))、多くのリボソーム特定または非コード化アミノ酸を模倣する側鎖を形成するために用いることができる。本発明の擬似天然型化学的ライゲーション法によって生成されたアミノ酸は、チオエーテル結合を含有し、β分岐を持たない(それらのアミノ酸はすべてベータ位でメチル基を含む、すなわちaa−βCH2−S−であるため)。したがって、β分岐アミノ酸、イソロイシンおよびトレオニンの擬似アミノ酸種は、β配置および付随の制約を有することなく、ペンダント側鎖構造を持たせることができる。
【0038】
注目に値すべきなのは、本発明の方法は、リボソーム特定アミノ酸と同じ長さ、あるいはそれよりも長い、または短いアミノ酸側鎖を形成するために用いることができることである。側鎖長のそのような変更は、三次元配座(コンフォメーション)を安定にして(または、不安定にして)、タンパク質安定性を増大する(または、タンパク質がその配座を変え、それによって天然タンパク質に受容されるものと異なる範囲の基質、阻害因子、受容体、リガンドなどを受容する能力を高める)ために用いることができる。たとえば、Cys−CH2−SH + Br−CH2−COOHは、Cys−CH2−S−CH2−COOHを生じる(そのような「擬似グルタミン酸塩」は、1つの追加の側鎖原子、すなわち−S−基を有し、あるいはアスパラギン酸塩の代わりに用いられた場合、2つの追加の側鎖原子、すなわち−CH2−S−基を有する)。他の側鎖は、側鎖に同じ数の原子を有するが、チオエーテル結合(−S−)を包含する点で異なる。たとえば、Cys−CH2−SH + Br−CH2−CH2−NH−PG、それに続くPGの除去によって、Cys−CH2−S−CH2−CH2−NH2が生じる。結果として生じた構造は、側鎖に追加の原子を持たないが、1つの−CH2−基が、−S−で置換されている。メチオニンは別の例であり、Cys−CH2−SH + I−CH2−CH3によって、Cys−CH2−S−CH2−CH3が生じ(天然Met構造、Met−CH2−CH2−S−CH3に対して)、したがってチオエーテルが置き換えられている。アルギニンも同様に、Cys−CH2−SH + Br−CH2−NH−CH((−NH2)(=NH2+))によって、Cys−CH2−S−CH2−NH−CH((−NH2)(=NH2+))が生じる。好ましくは、所望でない副反応を回避するために、特に擬似リジンを構成する場合は、反応性アミノ基の保護を用いることができる。ひとたびチオアルキル化反応が遂行されたら、その保護基を除去できる。
【0039】
一般に、できる限り厳密に天然タンパク質を模倣することを望む場合、タンパク質のそのような位置に通常存在するリボソーム特定アミノ酸と同じ長さの側鎖長を有する擬似アミノ酸分子を用いるのがもっとも好ましく、リボソーム特定アミノ酸より1原子長い側鎖長を有する擬似アミノ酸分子の使用は好ましさに劣り、リボソーム特定アミノ酸より2原子長い側鎖長を有する擬似アミノ酸分子の使用はさらに好ましさに劣る。さらに、遺伝子変化が機能を崩壊する見込みがないか、あるいは関連タンパク質のその部位でアミノ酸が保存されているような位置のシステインライゲーション部位を選択することが好ましい。そのような部位は、アラニンスキャニング、ホモロジーモデリングおよび他の方法によって同定できる。
【0040】
擬似天然型化学的ライゲーションでは、天然型化学的ライゲーションと同様に、アミノ末端システイン残基を含有するペプチドを、カルボキシ末端チオエステルを有するペプチドに結合する。その後、システインのチオール側鎖を、式Raa−Xであって、該式中、Xは良好な離脱基であり、Raaは、その構造がリボソーム特定アミノ酸または合成アミノ酸の側鎖の末端部分を模倣する基である化合物と反応させる。
【0041】
注目に値すべきなのは、擬似天然型化学的ライゲーション反応は、システイン側鎖の天然L立体配置、またはD立体配置を用いて作用することである。D立体配置の使用によって、そのライゲーション部位にプロテアーゼ耐性を付与することができ、したがってタンパク分解に対する高い安定性が所望であるとき、その使用が望ましい可能性がある。しかしながら、D−システインの使用においては、その側の主鎖構造が変更されることになる。そのような変更は、プロテアーゼ耐性に加えて、生物活性を変えるのに望ましい可能性がある。しかしながら、生物活性への影響力を最小にするために、その分子の無秩序末端の無秩序領域など可撓性の高い部位、たとえば結果として生じる折りたたまれた分子の表面に位置することになる無秩序ループなどにD−システインを配置することが好ましい。望ましくは、擬似天然型化学的ライゲーション反応は、合成分子の表面上に大きな荷電側鎖(たとえば、Lys、Arg、Asp、またはGluの側鎖)を配置するために用いることができる。
【0042】
適切な良好な離脱基、X、の例には、ハロゲン、特にヨウ素および臭素が含まれる。Raa基の例には、PO4、COOH、COO、CONH2、グアニジニウム、アミン、アルキル、置換アルキル、アリール、置換アリール、イミダゾール、アルキル化イミダゾール、インドール、またはアルキル化インドール基が含まれる。
【0043】
用いるRaaの選択は、特定の位置に存在することを所望するアミノ酸側鎖に応じて決まることになる。したがって、たとえば、
aaNH2−Q−aax−aay−W−aaCOOH
であって、該式中、QおよびWは、それぞれ任意で存在するか、または存在しない追加のアミノ酸残基を示し、aaxおよびaayは、内部隣接残基を示し(それぞれ側鎖xおよびyを有する)、aaNH2およびaaCOOHは、それぞれポリペプチドまたはタンパク質のアミノ(N−)末端残基およびカルボキシ(C−)末端残基を示すアミノ酸配列を有する所望のポリペプチドまたはタンパク質は、それぞれ
aaNH2−Q−aax−COSR および Cys−W−aaCOOH
であって、該式中、Cysは、システインによるaayの置換を示し、Rは、これに限定されるものではないが、アリール、ベンジルおよびアルキル基を含む、チオエステル基と適合する任意の基である上記の2つのペプチドフラグメントを調製することによって合成できる。Rの例には、3−カルボキシ−4−ニトロフェニルチオエステル、ベンジルエステルおよびメルカプトプロピオン酸ロイシンエステルが含まれる(たとえば、Dawson等、Science(1994)266:776〜779;Canne等、Tetrahedron Lett.(1995)36:1217〜1220;Kent他、WO96/34878、Kent他、WO98/28434;Ingenito等、JACS(1999)121(49):11369〜11374;Hackeng等、Proc.Natl.Acad.Sci.U.S.A.(1999)96:10068〜10073を参照のこと)。他の例には、同様によく知られているインテイン媒介の生物学的技法によって生成することのできるジチオトレイトール、あるいはアルキルまたはアリールチオエステルが含まれ(たとえば、Chong等、Gene(1997)192:277〜281;Chong等、Nucl.Acids.Res.(1998)26:5109〜5115;Evans等、Protein Science(1998)7:2256〜2264;Cotton等、Chemistry&Biology(1999)6(9):247〜256を参照のこと)、次いでそれらのフラグメントを合わせて結合することによって以下が形成される。
aaNH2−Q−aax−Cys−W−aaCOOH
【0044】
次いで、結合されたフラグメントを、式中、Ryは側鎖yの構造を模倣する側基であるRy−Xと反応させる。この反応は、システインのチオール基を「擬似y」側鎖に変換するのに充分な条件下で行われる。たとえば、RaaがCH2−COOHまたは(CH2)2−COOHとなるように選択される場合、この反応は、アスパラギン酸塩(「擬似Asp」)またはグルタミン酸塩(「擬似Glu」)の構造および機能を模倣するアミノ酸残基を形成することになる。理解されるように、上記の説明を考慮すれば、3つ以上のペプチドフラグメントを用いてより複雑な合成を行うことができる。
【0045】
本発明による「擬似アミノ酸」を含む天然型化学的ライゲーションは、タンパク質を化学合成するための天然型化学的ライゲーション方法の適応性を著しく拡大する。「擬似アミノ酸」を含む天然型化学的ライゲーションは、ライゲーションステップにおいて同じ化学反応を用いて、天然チオール含有側鎖官能基とCys残基でペプチド結合を形成し、一意性の第2のステップにおいて、ライゲーション部位のCysの天然チオール含有側鎖が化学選択的反応によって変換されて、遺伝コード化アミノ酸と同じ官能基を含有する非天然側鎖を生じる。
【0046】
たとえば、ライゲーション部位のシステインは、水中、pH中性でブロモ酢酸(または他のハロ酢酸)と反応させることができ、ライゲーション部位のCysの非保護チオールのみがこのような条件下で反応して、ライゲーション部位にS−カルボキシメチル化残基を含有するポリペプチド、すなわち、
【化1】
を生じる。
【0047】
この非天然アミノ酸は、側鎖にカルボキシル成分、すなわちグルタミン酸残基およびアスパラギン酸残基と同様な官能基、を含有する。このS−カルボキシメチル化Cysを、本明細書では、(1つの過剰な側鎖原子が存在し、チオエーテル結合を形成する−S−基である場合)「擬似グルタミン酸」(「擬似グルタメート(glutamate)」「擬似Glu」)と呼ぶ(2つの過剰な側鎖原子が存在し、その1つがチオエーテル結合を形成する−S−基である場合、「擬似アスパラギン酸」と呼ぶことができる)。多くのタンパク質の場合、この「擬似アミノ酸」であっても荷電カルボキシル化側鎖の単純保存は、タンパク質に所望の特性を保持または付与するのに充分であろう。重要な相違は、それが、適切なシステインのない位置で天然型化学的ライゲーション部位として機能することである。
【0048】
ライゲーション部位でCys残基の側鎖チオールを修飾するために他の試薬を用いることによって、他の「擬似アミノ酸」残基が生じ得る。たとえば、チオールのハロアセトアミド、たとえばブロモアセトアミドとの反応によって、ライゲーション部位に「擬似グルタミン」、すなわち、
【化2】
が生じる。
同様に、N−保護ハロアルキルアミン、たとえば、
【化3】
との反応、それに続くアミノ保護基の除去によって、「擬似リジン」残基、すなわち、
【化4】
が生じる。
ライゲーション部位のCys残基の適切なハロアルカン、たとえば、
【化5】
との反応によって、「擬似ロイシン」アミノ酸、すなわち、
【化6】
が生じる。
Rは、H、OH、アリールなどであり得る、他の適切に選択されたアリール含有ハロゲン化物、たとえば、
【化7】
を、Phe、Tyr、またはTrp残基に対応する擬似アミノ酸、すなわち、
【化8】
を生成するために同様に用いることができる。
【0049】
このアプローチの著しい特徴は、ライゲーション部位に存在するもの以外のCys残基の化学修飾を防ぐために、反応セグメントの修飾を望まないシステイン残基が、たとえばCys(Acm)として側鎖保護されること、または反応セグメントの他のCys残基が、ライゲーション後の化学修飾反応による同一「擬似アミノ酸」の同時形成に向けられることである。同じ原理がライゲーション部位でのホモシステインの使用に適用されることが理解されるであろう。さらに、セレノシステインがシステイン媒介天然型化学的ライゲーションによってライゲーション部位で用いられ、類似のアルキル化反応によって変換されて、セレノエーテル基がチオエーテル基に置き換わる本発明の擬似アミノ酸が生成されることも理解されるであろう。
【0050】
本明細書では、記号φは、ベンジル基を示し、IMは、イミダゾール基を示し、INは、インドール基を示し、PGは、保護基を示す。以下は、本発明によって擬似アミノ酸残基を含有するペプチドの合成に用いることのできるRaa側基の概要である(Xがハロゲン(I、Br、Cl、F)である場合であり、IおよびBrは、ほとんどの場合に好ましく、Fは、φ結合に好ましい)。
【0051】
塩基性アミノ酸:
Lys(余分な原子無し)
−CH2−SH + X−CH2−CH2−NH−PG、続いて脱保護することにより、−CH2−S−CH2−CH2−NH2が得られる。
Arg(余分な原子無し)
−CH2−SH + X−CH2−NH−C((NH2)(=NH2+))により
−CH2−S−CH2−NH−C((NH2)(=NH2+))が得られる。
His(余分な原子2個)
−CH2−SH + X−CH2−IM−PG、続いて脱保護することにより
−CH2−S−CH2−IMが得られる。
【0052】
酸性アミノ酸:
Asp(余分な原子2個)
−CH2−SH + X−CH2−COOHにより−CH2−S−CH2−COOHが得られる。
Glu(余分な原子1個)
−CH2−SH + X−CH2−COOHにより−CH2−S−CH2−COOHが得られる。
【0053】
非荷電極性アミノ酸:
Tyr(余分な原子無しまたは1個)
−CH2−SH + F−φ−pOHにより−CH2−S−φ−pOHが得られる(余分な原子無し、同じ幾何配置)。
−CH2−SH + Br/I−CH2−φ−pOHにより−CH2−S−CH2−φ−pOHが得られる(余分な原子1個)。
Gln(余分な原子1個)
−CH2−SH + X−CH2−C(O)(NH2)により−CH2−S−CH2−C(O)(NH2)が得られる。
Asn(余分な原子2個)
−CH2−SH + X−CH2−C(O)(NH2)により−CH2−S−CH2−C(O)(NH2)が得られる。
Ser(余分な原子2ないし3個)
−CH2−SH + X−CH2OHにより−CH2−S−CH2OHが得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH2OHにより−CH2−S−CH2−CH2OHが得られる(余分な原子3個)。
Thr(余分な原子2ないし3個、β分岐が無い)
−CH2−SH + X−CH((CH3)(O−PG))に続いてPGを除去することにより−CH2−S−CH((CH3)(OH))が得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH((CH3)(O−PG))に続いてPGを除去することにより−CH2−S−CH2−CH((CH3)(OH))が得られる(余分な原子3個)。
【0054】
非極性アミノ酸:
Leu(余分な原子1ないし2個)
−CH2−SH + X−CH((CH3)(CH3))により−CH2−S−CH((CH3)(CH3))が得られる(余分な原子1個)。
−CH2−SH + X−CH2−((CH3)(CH3))により−CH2−S−CH2−CH((CH3)(CH3))が得られる(余分な原子2個)。
Ile(余分な原子2ないし3個、β分岐無し)
−CH2−SH + X−CH(CH3)−CH2−CH3により−CH2−S−CH(CH3)−CH2−CH3が得られる(余分な原子2個)。
−CH2−SH + X−CH2−CH(CH3)−CH2−CH3により−CH2−S−CH2−CH(CH3)−CH2−CH3が得られる(余分な原子3個)
Phe(余分な原子1ないし2個)。
−CH2−SH + F−φにより−CH2−S−φが得られる(余分な原子1個)。
−CH2−SH + Br/I−CH2−φにより−CH2−S−CH2−φが得られる(余分な原子2個)。
Met(余分な原子無し)
−CH2−SH + I−CH2−CH3により−CH2−SH−CH2−CH3が得られる。
Trp(余分な原子1個)
−CH2−SH + F−INにより−CH2−S−INが得られる。
【0055】
一般的な擬似化学的ライゲーション反応を図1に例示する。好ましい擬似アミノ酸を形成するために用いることのできる好ましいRaaおよびX基を表Iに示す。残基は非荷電として示すが、NH2またはCOOHなどのイオン性基は、それらの荷電形態であるか、または塩として提供されてもよいことが理解されるであろう。擬似リジンを形成するために用いられる反応を除いて、すべての反応は水性であり、pH8〜9で行われ、反応の化学量および最適化は、標準的なプロトコルに従って調整することができる。擬似リジンは、pH8〜9で行われる水性反応、それに続くピペリジン/H2O(トリフルオロ酢酸(TFA))中の反応で形成される。
【表1】
【表2】
【表3】
【表4】
【0056】
本発明のさらなる実施形態において、システイン残基のチオール基は、酸化するか(−SH→−SO3−)、または酸素と置換してセリンを形成することができる(−SH→−OH)。そのような置換は、システインをトシル−Cl(Tos−Cl)と反応させ、それによってチオール基−SHを、−S−Tos基に変換することによって達成できる。強塩基の存在下、OHはTos置換基に代わり、それによって所望の−OH基が形成される。
【0057】
さらに、システインのチオール基は、以下に示すように、マイケル付加反応によって修飾することができ、
【化9】
該式中、Rは、H、アルキル、アリール、アルキルアリール、アミノであるか、またはポリマー分子であって、分岐または非分岐、ランダムまたはブロックであってよい。
【0058】
本発明のさらなる態様は、アミノ末端システイン残基がアルキルアミノ(好ましくはメチルアミノ)部分を含むように修飾されたペプチドフラグメントの使用を対象とする。
【化10】
【0059】
アルキルアミノ部分のアミノ基を保護することによって、ペプチドAを一般式ペプチドB−αCOSRを有する第2のペプチドと結合することができる。保護基の除去によって、一般式ペプチドC−αCOSRを有するさらなるペプチドを、前に結合したペプチドに結合し、分岐ペプチド複合体を形成することができる。
【化11】
【0060】
この分岐タンパク質は依然としてシステインのチオール基を含有するので、上に記載した様式でRaa−Xと反応し、それによって側鎖のさらなる修飾が可能となる。特に、R置換基としてアルキルアミドを用いることによって、別のアミノ基を分子に導入することができ、それによってさらなる分岐、またはさらなるライゲーション反応が可能となる。
【化12】
【0061】
本発明のさらなる実施形態において、システイン側鎖の修飾を用いて、任意の種々の標識、または結合分子を導入することができる。たとえば、電子スピン共鳴、蛍光(たとえば、アンモニウム4−クロロ−7−スルホベンゾ−フラザン(SBF)Pierce Chemical Co.)、または磁気画像法によって検出可能な標識を用いることができる。そのような標識付けは、疾患および症状の診断において、ならびに分子相互作用の分析(たとえば、内在性膜タンパク質における距離の測定)において有用である。本発明の方法は、標識および結合分子の使用に関して、いくつかの顕著な利点を有する。そのような置換基は、部位特異的、非ランダム様式で組み込むことができる。さらに、この標識部分は、続いて起こる化学的ライゲーションステップ(存在する場合)に対して安定であることだけが必要とされる。そのような反応は一般に、非常に穏やかな条件下で行われる(たとえば、天然型化学的ライゲーション、他のシステイン残基のチオール基を保護するために随意用いられる可能性のあるAcmまたは他の保護基の除去(Hg(CH2COOH)2など)、および逆相HPLC精製に用いられるものなど)。そのような追加のチオール基の保護は、Cys含有ペプチドの酸化性重合を妨げ、したがってそれらの精製および取扱いを容易にする。
【0062】
B.拡張天然型化学的ライゲーション
上述したように、天然タンパク質の配列が適切なシステイン残基を含有せず、ポリペプチドのN末端にシステイン残基を導入するように天然タンパク質配列を修飾することが不都合であるか、または望ましくない場合、そのN末端がN置換、好ましくはNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含有し、それによってポリペプチド欠失システイン残基に天然型化学的ライゲーションの原理を用いることができるように修飾されたポリペプチドを結合するために、「拡張天然型化学的ライゲーション」の方法を用いることができる(参照により本明細書に組み込むものとする米国特許出願第60/231339号を参照のこと)。
【0063】
「拡張天然型化学的ライゲーション」の方法には、カルボキシルチオエステル、より好ましくはα−カルボキシルチオエステルを含む第1の成分を、酸安定性N置換、好ましくはNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含む第2の成分と結合することが含まれる。第1成分のカルボキシチオエステルと、第2成分のN置換2または3炭素鎖アルキルまたはアリールチオールのチオールとの間の化学選択的反応は、チオエステル結合中間体を経て進み、最初のライゲーション生成物に分解する。より具体的には、COSRチオエステル成分とアミノアルキルチオール成分との間で起こるチオール交換は、チオエステル結合中間体ライゲーション生成物を生じ、これは自発転位の後、アミノアルキルチオール成分がそれぞれ式IまたはIIを有するかどうかに応じて、5員または6員環中間体を経て最初のアミド結合ライゲーション生成物を生じ、
該式中、J1は、随意保護された1つまたは複数のアミノ酸側鎖を有するペプチドまたはポリペプチド、あるいはそのようなペプチドまたはポリペプチドの部分、ポリマー、色素、適切に機能化された表面、リンカーまたは検出可能マーカー、あるいは化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の他の任意の化学部分であり、R1、R2およびR3は、独立して、H、またはC1と共役した電子供与基であるが、ただしR1、R2およびR3の少なくとも1つは、C1と共役した電子供与基を含み、J2は、随意保護された1つまたは複数のアミノ酸側鎖を有するペプチドまたはポリペプチド、あるいはそのようなペプチドまたはポリペプチドの部分、ポリマー、色素、適切に機能化された表面、リンカーまたは検出可能マーカー、あるいは化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の他の任意の化学部分である。
【0064】
ライゲーション部位のN置換2または3炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]または[HS−(C3(R3)−C2(R2)−C1(R1)−]は、生成物への損傷なしに、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式IIIの最終ライゲーション生成物を生じる。
該式中、J1、J2、R1、R2およびR3は、上に定義したとおりである。
【0065】
R1、R2およびR3基は、ペプチド適合性開裂条件下でN−C1結合の開裂を促進するように選択される。たとえば、電子供与基は、特にC1に共役している場合、C1で開裂を促進する共鳴安定化カチオンを形成するために用いることができる。この化学的ライゲーション反応は、好ましくは、賦形剤としてチオール触媒を含み、中性に近いpH条件、水性または混合有機−水性条件で行われる。第1および第2成分の化学的ライゲーションは、5または6員環を経て進行することができ、5または6員環は自発転位を受けて、N置換アミド結合ライゲーション生成物を生じる。第1および第2の成分がペプチドまたはポリペプチドである場合、N置換アミド結合ライゲーション生成物は式IVまたはVを有し、
該式中、J1、J2およびR1、R2、R3は、上に定義したとおりであり、Z2は、アミノ酸側鎖、またはその側鎖の誘導体である。
【0066】
N置換アミド結合ライゲーション生成物の共役電子供与基R1、R2、またはR3は、N−C1結合の開裂、およびN置換アミド結合ライゲーション生成物からの2または3炭素鎖アルキルまたはアリールチオールの除去を促進する。ペプチド適合性開裂条件下でのNのアルキルまたはアリールチオール鎖の除去によって、ライゲーション部位に天然アミド結合を有するライゲーション生成物が生じる。第1および第2成分がペプチドまたはポリペプチドである場合、ライゲーション生成物は以下の式を有することになる。
【0067】
式IのR1置換基の例を表IIに示す。
【表5】
【0068】
式IIのR1、R2およびR3置換基の例を表IIIに示す。
【表6】
【0069】
N置換2炭素鎖化合物と同様に、オルトまたはパラ位におけるベンジルおよびピコリル電子供与置換基R1’、R3’およびR5’の位置決定は、ライゲーションに続くNα−C1結合の強固な開裂のためにC1炭素に対する電子共役を維持する必要がある。しかしながら、R2およびR3がC2およびC3と共にベンジル基を形成するとき、R1’およびR3’の少なくとも1つは、強い電子供与基を含み、ここでR1’またはR3’は、メトキシ(−OCH3)、チオール(−SH)、ヒドロキシル(−OH)およびチオメチル(−SCH3)から選択される。R2およびR3が水素であるN置換3炭素鎖チオールの場合、R1は、R1’、R3’およびR5’が強い電子供与基または中強度の電子供与基、あるいはそれらの組み合わせを含むベンジルまたはピコリル基を含む。N置換2炭素鎖アルキルまたはアリールチオールと同様に、強い電子供与基は、ライゲーションに続く開裂に対する3炭素鎖アルキルまたはアリールチオールの感受性を高める。したがって、特定の電子供与基またはその組み合わせはそれに応じて選択できる。
【0070】
N置換2炭素鎖化合物と同様に、本発明のN置換3炭素鎖化合物は、対象となる構成において置換に利用できるとき、R1’およびR5’位のR1の置換基としてチオールを含むことができる。ここでも、電子供与チオール基はC1と共役し、これらの位置におけるその導入は、化合物が6員環のライゲーション事象形成のための2つの経路を有することを可能にする。これはさらに、α−カルボキシチオエステルとの反応に利用できるチオールの局所濃度を高め、構造的制約の点から別の配位を提供し、ライゲーションを向上することができる。
【0071】
本発明のN末端N置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸の合成は、たとえばスキームIおよびスキームIIによって本明細書に記載のとおり、且つ当分野で知られている標準的な有機化学技法に従って実行することができる。たとえば、「Advanced Organic Chemistry,Reactions,Mechanisms,and Structure」、第4版、J.March編、John Wiley&Sons、New York、NY、1992;「Comprehensive Organic Transformations,A Guide to Functional Group Preparations」、R.Larock編、VCH Publishers、New York、NY、1989を参照されたい。それらのアミノ酸は、溶液中で、ポリマー担持型合成によって、またはその組み合わせによって合成することができる。好ましいアプローチは、Nα−保護Nアルキル化S保護アミノアルキルまたはアリールチオールアミノ酸前駆体を用いる。合成に用いられる試薬は、多くの製造業者から入手できる。さらに、出発成分および種々の中間体、たとえば個々のアミノ酸誘導体などは、キットなどで提供され、後の使用のために貯蔵できることが充分に理解されるであろう。
【0072】
本発明のN末端Nα−置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸の調製において、保護基手順が用いられる。種々の合成手順一般において用いられる好ましい保護基(PG)は、固相ペプチド合成(「SPPS」)に適合性である。場合によってはさらに、異なる条件下で除去可能な直交性の保護基を用いる必要がある。多くのそのような保護基が知られており、この目的に適している(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。例には、ベンジルオキシカルボニル(Z)、Boc、Bpoc、Trt、Nps、FmocCl−Z、Br−Z、NSC、MSC、Ddeなどが含まれる。硫黄部分の場合、適切な保護基の例には、これに限定されるものではないが、ベンジル、4−メチルベンジル、4−メトキシベンジル、トリチル、ACM、TACAM、キサンチル、ジスルフィド誘導体、ピコリルおよびフェナシルが含まれる。
【0073】
より具体的には、このNα−置換2または3炭素鎖アルキルまたはアリールチオールは、スキームI(Nα−置換前駆体の固相調製)、スキームII(Nα−置換前駆体の液相調製)に従って調製することができる。スキームIにおいて、Nα置換−2または3炭素鎖アルキルまたはアリールチオールは、ポリマー担持型有機合成の標準方法を用いて固相上で直接組み上げされるが、一方スキームIIのNα−保護、Nアルキル化、S保護、アミノアルキルまたはアリールチオールアミノ酸前駆体は、標準的なカップリングプロトコルを用いて樹脂に結合される。ラセミまたはジアステレオマー生成物が生成される場合、拡張天然型化学的ライゲーションに用いる前に、標準的な方法によるこれらの分離が必要となる可能性がある。
スキームI
ECL補助誘導化分子の固相合成
【化13】
スキームII
【化14】
【0074】
α−カルボキシチオエステルは、実施例を含む本明細書で記載の技法など、当分野で知られている標準的な技法に従う化学または生物学法によって生成することができる。化学合成の場合、α−カルボキシチオエステルペプチドは、溶液中、またはチオエステル生成樹脂から合成することができ、その技法はよく知られている(たとえば、Dawson等、上記;Canne等、上記;Hackeng等、上記、Aimoto等を参照のこと)。たとえば、化学合成チオエステルペプチドは対応するペプチドα−チオ酸から製造することができ、ペプチドα−チオ酸は、樹脂法が好ましいが、チオエステル樹脂上、または溶液中で合成することができる。このペプチドα−チオ酸は、対応する3−カルボキシ−4−ニトロフェニルチオエステル、対応するベンジルエステル、または任意の多様なアルキルチオエステルに変換することができる。これらのチオエステルはすべて、ライゲーション反応のための充分良好な離脱基を提供し、3−カルボキシ−4−ニトロフェニルチオエステルは、アルキルチオエステルに比べてより反応性である可能性のある、対応するベンジルチオエステルよりもいくらか速い反応速度を示す。別の例として、トリチル関連メルカプトプロピオン酸ロイシンチオエステル生成樹脂は、C末端チオエステルを構成するために用いることができる(Hackeng等、上記)。C末端チオエステル合成も、ジアゾメタンまたはヨードアセトニトリルによる活性化、それに続く適切なチオールによる置換によって、3−カルボキシプロパンスルホンアミド安全捕捉(セイフティーキャッチ)リンカーを用いて達成することができる(Ingenito他、上記;Bertozzi等)。
【0075】
本発明のN置換2または3炭素鎖アルキルまたはアリールチオール成分の、第1カルボキシチオエステル成分とのライゲーションによって、ライゲーション部位にN置換アミド結合を有するライゲーション生成物が生じる。この反応のライゲーション条件は、チオエステルのN置換2または3炭素鎖アルキルまたはアリールチオール部分との選択的反応性を維持するように選択される。好ましい実施形態において、ライゲーション反応は、pH6〜8を有する緩衝液中で実行され、好ましいpHの範囲は6.5〜7.5である。この緩衝液は、水性、有機性、またはその混合物であることができる。ライゲーション反応はさらに、1種または複数の触媒および/または1種または複数の還元剤、脂質、洗浄剤、他の変性剤または可溶化試薬などを含むことができる。好ましい触媒の例は、チオールおよびホスフィン含有部分、たとえばチオフェノール、ベンジルメルカプタン、TCEPおよびアルキルホスフィンなどである。変性剤および/または可溶化剤の例には、グアニジニウム、尿素の水またはTFE、HFIP、DMF、NMPなど有機溶媒溶液、水、またはグアニジニウムおよび尿素水溶液と混合したアセトニトリルが含まれる。温度もライゲーション反応の速度を調節するために利用することができ、通常は5℃から55℃の間であり、好ましい温度は15℃から40℃の間である。例として、ライゲーション反応は、pH6.8から7.8の間で、6Mグアニジニウム中の2%チオフェノールを用いる反応系において良好に進行する。
【0076】
N置換2炭素鎖アルキルまたはアリールチオールの場合、ライゲーション事象は、COSRチオエステル成分とアミノアルキルチオール成分との間に起こるチオール交換に起因する。この交換によってチオエステル結合中間体ライゲーション生成物が生成され、これは自発転位の後、5員環中間体を経て、ライゲーション部位に除去可能なN置換2炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]を有し、置換基は上に定義したとおりである式J1−HN−CH(Z1)−C(O)−Nα(C1(R1)−C2−SH)−CH(Z2)−J2の最初のライゲーション生成物を生じる。このライゲーション部位のN置換2炭素鎖アルキルまたはアリールチオール[HS−C2−C1(R1)−]は、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式J1−HN−CH(Z1)−CO−NH−CH(Z2)−CO−J2の最終ライゲーション生成物を生じる。
【0077】
N置換3炭素鎖アリールまたはアルキルチオールの場合、COSRチオエステル成分とアミノアルキルチオール成分との間のチオール交換によってチオエステル結合中間体ライゲーション生成物が生成され、これは自発転位の後、6員環中間体を経て、ライゲーション部位に除去可能なN置換3炭素鎖アルキルまたはアリールチオール[HS−C3(R3)−C2(R2)−C1(R1)−]を有する式J1−HN−CH(Z1)−C(O)−Nα(C1−C2(R2)−C3(R3)−SH)−CH(Z2)−J2の最初のライゲーション生成物を生じる。このライゲーション部位のN置換3炭素鎖アリールチオール[HS−C3(R3)−C2(R2)−C1(R1)−]は、ペプチド適合性条件下で除去することができ、ライゲーション部位に天然アミド結合を有する式J1−HN−CH(Z1)−CO−NH−CH(Z2)−CO−J2の最終ライゲーション生成物を生じる。
【0078】
N置換アルキルまたはアリールチオール基の除去は、N−C1結合の開裂を促進し、ライゲーション部位に安定化非置換アミド結合を得るために、好ましくは酸性条件で行われる。「ペプチド適合性開裂条件」とは、ペプチドに適合性であり、ライゲーション生成物からのアルキルまたはアリールチオール部分の開裂に適した物理的化学的条件を意味する。一般にペプチド適合性開裂条件は、用いられるα−置換化合物に応じて選択され、これは通常のよく知られたアプローチによって容易に推測できる(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。
【0079】
たとえば、R1、R2、またはR3置換基が、メトキシ、ヒドロキシ、チオールまたはチオメチル、メチルなどを含む場合、除去のためのより普遍的な方法は、ペプチド合成化学に典型的な酸性開裂条件を含む。これには、還元試薬および/またはスカベンジャ系(たとえば、無水フッ化水素(HF)、トリフルオロ酢酸(TFA)、またはトリメチルスルホニルフルオロ酢酸(TMSFA)などの酸)を含むか、または含まない、強酸性条件または水−酸性条件下でのN−C1結合の開裂が含まれる。より具体的な酸性開裂系は、所与の構成体に関してアリールまたはアルキルチオール部分を除去するためにNα−C1結合の開裂を最適化するように選択することができる。そのような条件はよく知られており、ペプチドの完全性の維持に適合する。特にペプチドまたはポリペプチド配列にトリプトファンが存在する場合、トリプトファン側鎖の遊離アリールまたはアルキルチオール部分との反応を回避するために、チオールスカベンジャを含むことができる。チオールスカベンジャの例には、エタンジオール、システイン、β−メルカプトエタノールおよびチオクレゾールが含まれる。
【0080】
他の特殊な開裂条件には、ピコリル基が置換基であるときの光または還元型開裂条件が含まれる。例として、R1、またはR2およびR3置換基がピコリル部分を含むとき、光分解(たとえば、紫外光)、亜鉛/酢酸、または電気分解還元を、標準的なプロトコルに従って開裂のために用いることができる。N置換2炭素鎖チオールのR1がR1にチオメタンを含む場合、水銀またはHF開裂を用いることができる。この開裂系はさらに、固体支持体からの同時開裂のために、および/または第1または第2ライゲーション成分が他の保護基を含むときの脱保護試薬として用いることができる。
【0081】
本発明の一実施形態において、Nα−置換2または3炭素鎖アルキルまたはアリールチオールは、合成生物活性タンパク質を生じる拡張化学的ライゲーションの基質として用いることのできる適切にN末端誘導化されたポリペプチドを得るためのペプチド合成ステップ(特に自動ペプチド合成、ならびに直交型および収斂型ライゲーションストラテジー)において用いられる。そのような化合物は、式(PG1)S−C2−C1(R1)−Nα(PG2)−CH(Z2)−C(O)−J2、または(PG1)S−C3(R3)−C2(R2)−C1(R1)−Nα(PG2)−CH(Z2)−C(O)−J2の完全に保護された、または部分的に保護された、または完全に保護されていない酸安定性のNα−置換2または3炭素鎖アミノアルキルまたはアリールチオールを含み、それらを以下の表IVおよび表Vに示す。特に、R1、R2およびR3の1つまたは複数はC1に共役した電子供与基を含み、これはNα−置換アミノアルキルまたはアリールチオールのNα置換−アミドアルキルまたはアリールチオールへの変換に続いて、ペプチド適合性開裂条件下でNα−C1結合の開裂を促進する共鳴安定化カチオンをC1において形成することができる。Z2が化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の任意の化学部分であり、J2が化学ペプチド合成または拡張天然型化学的ライゲーションに適合性の任意の化学部分である場合、PG1およびPG2は個別にまたは組み合わせで存在するか、あるいは存在しない保護基であり、同一であるか、または異なることができる。PG1(またはX1)は、アミンを保護するための基である。PG2(またはX2)は、チオールを保護する基である。多くのそのような保護基が知られており、この目的に適している。(たとえば、「Protecting Groups in Organic Synthesis」、第3版、T.W.Greene、P.G.M.Wuts編、John Wiley&Sons,Inc.、1999;NovaBiochem Catalog 2000;「Synthetic Peptides,A User’s Guide」、G.A.Grant編、W.H.Freeman&Company、New York、NY、1992;「Advanced Chemtech Handbook of Combinatorial&Solid Phase Organic Chemistry」、W.D.Bennet、J.W.Christensen、L.K.Hamaker、M.L.Peterson、M.R.Rhodes、H.H.Saneii編、Advanced Chemtech、1998;「Priciples of Peptide Synthesis、第2版」、M.Bodanszky編、Springer−Verlag、1993;「The Practice of Peptide Synthesis、第2版」M.Bodanszky、A.Bodanszky編、Springer−Verlag、1994;および「Protecting Groups」、P.J.Kocienski編、Georg Thieme Verlag、Stuttgart、Germany、1994を参照のこと)。
【表7】
【0082】
PG1およびX1の好ましい保護基の例には、これに限定されるものではないが、Boc(t−ブチルカルバメート)、Troc(2,2,2−トリクロロエチルカルバメート)、Fmoc(9−フルオレニルメチルカルバメート)、Br−ZまたはCl−Z(Br−またはCl−ベンジルカルバメート)、Dde(4,4−ジメチル−2,6−ジオキソシクロヘキシル−イリデン)、MsZ(4−メチルスルフィニルベンジルカルバメート)、Msc(2−メチルスルホエチルカルバメート)、Nsc(4−ニトロフェニルエチルスルホニル−エトキシ−カルボニル)が含まれる。好ましいPG1およびX1保護基は、「Protective Groups in Organic Synthesis」、GreenおよびWuts、第3版、Wiley−Interscience(1999)から選択され、もっとも好ましいのはFmocおよびNscである。PG2の好ましい保護基の例には、これに限定されるものではないが、[Acm(アセトアミドメチル)、MeoBzlまたはMob(p−メトキシベンジル)、Meb(p−メチルベンジル)、Trt(トリチル)、Xan(キサンテニル)、tButhio(s−t−ブチル)、Mmt(p−メトキシトリチル)、2または4ピコリル(2または4−ピリジル)、Fm(9−フルオレニル−メチル)、tbut(t−ブチル)、Tacam(トリメチルアセトアミドメチル)]が含まれる。好ましい保護基PG2およびX2は、「Protective Groups in Organic Synthesis」、GreenおよびWuts、第3版、Wiley−Interscience(1999)から選択され、もっとも好ましいのはAcm、Mob、MeB、ピコリルである。
【表8】
【0083】
本発明のNα−置換2または3鎖アルキルまたはアリールチオールの保護形態は上述のスキームIおよびIIのように調製できる。
【0084】
本発明の化合物は、任意の多様な手段によって生成することができ、ハロゲン媒介アミノアルキル化、還元アミノ化、および固相アミノ酸合成法に適合するNα−保護Nアルキル化S保護アミノアルキルまたはアリールチオールアミノ酸前駆体の調製を含む。所望であるとき、許容されるキラル純度の化合物を得るために、生成されたラセミ体またはジアステレオマーの分割を標準的な方法によって行うことができる。
【0085】
II.本発明の生物活性ペプチドおよびタンパク質
A.合成生物活性タンパク質
上述の方法は、合成生物活性ペプチドおよびタンパク質を合成するために用いることができる。本明細書では、そのアミノ酸残基のいくつか、もっとも好ましくはそのすべてを重合するために非組換え技術が用いられた場合、生物活性タンパク質は「合成」であると言う。「非組換え技術」という用語は、有機化学および他の合成重合アプローチを含む技術を、in vivoまたはin vitroでのRNAのタンパク質への翻訳を含む技術と区別することを意図する。もっとも好ましくは、本発明の合成生物活性タンパク質は、有機化学の原理、特に本明細書に記載の「天然型化学的ライゲーション」および「拡張天然型化学的ライゲーション」の方法を用いてin vitroで生成される。そのような化学合成は、好ましくは、ポリペプチドフラグメントが合成され、次いで互いに結合されて合成生物活性タンパク質が形成される収斂型合成アプローチを用いて行われる。別法として、本発明の合成生物活性タンパク質は、単一の非収斂型合成で合成することができる。
【0086】
上述したように、本発明の合成生物活性タンパク質は、好ましくは約25kDaを超える相当の分子量を有する。本発明の目的のために、そのような分子量の決定は、変性SDSポリアクリルアミド電気泳動によってなされる。「モノマー分子量」という用語は、タンパク質またはポリペプチドの複数のコピーを有する可能性のある合成タンパク質と区別して、モノマー合成タンパク質の分子量を指すことを意図する。
【0087】
本明細書では、合成タンパク質の構造またはアミノ酸配列に依存し、そのような構造または配列の変化がタンパク質の生物活性を増強、修飾、または低減するような認識できる生物活性を有する場合、合成タンパク質は「生物活性」を有すると言う。限定するものではないが、そのような「生物活性」には、触媒、シグナル、または誘発反応を媒介するタンパク質の能力が含まれる。本明細書では、それ自体が消費されたり、永久的に修飾されたりすることなく、基質を生成物に変換することによって、タンパク質は「触媒反応を媒介する」と言う。タンパク質の存在が原因となって、生体、あるいはその組織または細胞が、遺伝子発現、細胞分化、または細胞増殖を開始、持続、増強、修飾、または低減する場合、タンパク質は「シグナルまたは誘発反応を媒介する」と言う。そのようなシグナルまたは誘発反応の例には、サイトカイン発現またはサイトカイン発現に対する応答の誘発、赤血球生成の誘発、炎症および/または炎症性過程の誘発または緩和、新脈管形成または血管新生の開始、アポトーシスの誘発、細胞周期への影響などが含まれる。
【0088】
本発明の合成生物活性タンパク質の生物活性はさらに、タンパク質が特異的に受容体、リガンド、または抗体に結合する能力を含む。本明細書では、「特異的」結合という用語は、電荷、溶解度、疎水性などに基づく「非特異的」結合と区別して、ホルモンとその受容体との間、または抗体と抗原エピトープとの間に存在するものなど、構造に基づく結合相互作用を指すことを意図する。
【0089】
本発明の合成生物活性タンパク質は、好ましくはアミノ酸残基の縮合によって合成される。そのようなアミノ酸残基は、核酸コード化リボソーム導入アミノ酸、例えば、アラニン、アルギニン、アスパラギン、アスパラギン酸塩、システイン、グルタミン酸塩、グルタミン、グリシン、ヒスチジン、イソロイシン、ロイシン、リジン、メチオニン、フェニルアラニン、プロリン、セリン、トレオニン、トリプトファン、チロシンおよびバリンであることができる。一実施形態において、特定の所望の合成生物活性タンパク質のために選択されたアミノ酸配列は、そのタンパク質の天然のまたは自然の配列となる。別の実施形態において、選択されたアミノ酸配列は、そのタンパク質の天然のまたは自然の配列に存在するアミノ酸残基の代わりにN末端システイン残基を含有することのできるポリペプチドフラグメントからなり、すべて構成されることになる。そのような残基を含むことによって、そのポリペプチドが本明細書に記載の天然型化学的ライゲーションの原理を用いて(カルボキシチオエステル基を含有するように修飾された)別のポリペプチドと結合されることを可能にし、それによって合成生物活性タンパク質を生成する収斂型合成の使用が容易になる。
【0090】
さらなる実施形態において、本発明の合成生物活性タンパク質は、「不規則」アミノ酸残基を含有することができる。本明細書では、「不規則」アミノ酸残基という用語は、RNAによってコード化されておらず、リボソーム導入されていないアミノ酸を指すものである。この点に関して、本発明は、合成生物活性タンパク質の設計および/または構成において広い選択性および柔軟性が可能となる。本発明によって用いることのできる非リボソーム導入アミノ酸の例には、D−アミノ酸、β−アミノ酸、擬似グルタミン酸塩、γ−アミノ酪酸、オルニチン、ホモシステイン、N置換アミノ酸(R.Simon等、Proc.Natl.Acad,Sci.U.S.A.(1992)89:9367〜71;WO91/19735(Bartlett他)、米国特許第5646285号(Baindur)、α−アミノメチレンオキシ酢酸(アミノ酸−Glyジペプチド同配体)およびα―アミノオキシ酸などが含まれる。チオアミド、ビニル性アミド、ヒドラジノ、メチレンオキシ、チオメチレン、ホスホンアミド、オキシアミド、ヒドロキシエチレン、還元アミドおよび置換還元アミド同配体、ならびにβ−スルホンアミドを含むペプチド類似体を用いることができる。
【0091】
特に、R鎖の修飾が合成タンパク質へのポリマー付加物の結合を可能にするので、擬似グルタミン酸塩の使用が有利である。同様に、N末端Nα−置換2または3炭素鎖アルキルまたはアリールチオールアミノ酸を用いることができる。そのような残基(末端またはポリペプチドに存在する)は、本明細書に記載の拡張天然型化学的ライゲーションの方法に従って、そのポリペプチドを、カルボキシチオエステル部分を有するポリペプチドに結合するために有利に用いることができる。
【0092】
一実施形態において、合成生物活性タンパク質のアミノ酸残基すべては、ペプチド結合(すなわち、アミド結合)によって互いに結合することができる。別法として、2つのアミノ酸残基(または2つのポリペプチドのC末端およびN末端残基)を、非アミド結合(チオエステル結合、オキシム結合、チオエーテル結合、有向ジスルフィド結合、チオゾリジン結合など)によって互いに結合することができる(Schnolzer、M.およびKent、S.B.H.、Science(1992)256:221〜225;Rose、K.、J.Amer.Chem.Soc.(1994)116:30〜33;Englebretsen、D.R.等、Tetrahedron Lett.36:8871〜8874;Baca、M.等、J.Amer.Chem.Soc.(1995)117:1881〜1887;Liu、C.F.等、J.Amer.Chem.Soc.(1994)116:4149〜4153;Liu、C.F.等、J.Amer.Chem.Soc.(1996)118:307〜312;Dawson、P.E.等、(1994)Science 266:776〜779)。したがって本発明は、生物活性タンパク質の調製における多様なペプチド結合の修飾、代用、および等配電子性置換の利用を可能にする。
【0093】
本発明の合成生物活性タンパク質は、哺乳動物(ヒト、サル、ウシ、マウス、ブタ、ヒツジ、ウマなどを含む)、鳥類、または魚類タンパク質に関連する生物活性を模倣する生物活性を有するように設計できる。本明細書では、第1のタンパク質が、第2のタンパク質の生物活性に関して修飾、増強、または低減された生物活性を有する場合、第1のタンパク質が第2のタンパク質を模倣していると言う。生物活性は、その存在が直接または間接的にタンパク質のアミノ酸配列に依存している場合、そのタンパク質に「関連している」と言う。
【0094】
別の実施形態において、本発明の生物活性タンパク質は、対応する天然生物活性相当物を参照せずに、全体的または部分的に操作することができる。本発明の好ましい合成生物活性タンパク質には、受容体、タンパク質受容体リガンドが含まれる。そのようなタンパク質の例には、副腎皮質刺激ホルモン受容体およびその生物活性フラグメント、アンギオテンシン受容体、心房性ナトリウム利尿受容体、ブラジキニニン受容体、成長ホルモン受容体、化学走性受容体、ジノルフィン受容体、エンドルフィン受容体、β−リポトロピンの受容体およびその生物活性フラグメント、エンケファリン受容体、酵素阻害因子受容体、フィブロネクチン受容体およびその生物活性フラグメント、消化管および成長ホルモン放出ペプチド受容体、黄体化ホルモン放出ペプチドの受容体、メラニン細胞刺激ホルモンの受容体、ニューロテンシン受容体、オピオイド受容体、オキシトシン受容体、バソプレッシン受容体、バソトシン受容体、副甲状腺ホルモンの受容体およびフラグメント、プロテインキナーゼ受容体、ソマトスタチン受容体、サブスタンスP受容体が含まれる。
【0095】
本発明の合成生物活性タンパク質にはさらに、酵素、および構造分子、たとえばキチン、またはコラーゲンなどが含まれる。
【0096】
さらなる実施形態において、本発明の合成生物活性タンパク質には、サイトカインが含まれる。サイトカインは、細胞によって放出され、細胞−細胞相互作用、連絡、および他の細胞の挙動への特異的影響を媒介する小タンパク質または生物因子(5〜20kDaの範囲)である。本明細書では、「サイトカイン」という用語は、インターロイキン(IL)(IL−1、IL−2、IL−3、IL−4、IL−5、IL−6、IL−7、IL−8、IL−9、IL−10、IL−11、IL−12、IL−13、IL−14など)、リンホカインおよび信号分子、たとえば赤血球産生刺激タンパク質(たとえば、エリスロポエチン(EPO))、腫瘍壊死因子(TNF)、インターフェロンなど、増殖因子、たとえばトランスホーミング増殖因子、神経発育因子、脳由来増殖因子、ニューロトロフィン−3、ニューロトロフィン−4、肝細胞増殖因子、T細胞増殖因子(TGF、TGF−β1、TGF−β2、TGF−β3など)、コロニー刺激因子(G−CSF、GM−CSF、M−CSFなど)、上皮増殖因子(EGF、LIF、KGF、OSM、PDGF、IGF−Iなど)、線維芽細胞増殖因子(αFGF、βFGFなど)、およびホルモン、特に単一ペプチドホルモン、たとえば成長ホルモンなどを含むことを意図する。
【0097】
サイトカイン、特に化学走性サイトカインはさらに、本発明の範囲内の合成生物活性タンパク質の設計を助けるためにそのアミノ酸配列を用いることのできる特に好ましいタンパク質の例を含む。サイトカインは、白血球補充を直接または間接的に媒介することによって、あるいは微生物侵入の特異部位に蓄積された白血球の活性化において重要なシグナルとして働くことによって、先天または後天免疫応答に関与する。炎症性サイトカイン、インターロイキン−1β(IL−1β)およびインターロイキン−6(IL−6)、媒介物質は、好中球補充および活性化をもたらし、可溶性細胞間接着分子、インターロイキン−8を含む(Kotecha S.、European Journal of Pediatrics(1996)155、補遺2:S14〜17)。インターロイキン−6は、主として急性期応答、B細胞分化、および抗体産生に関わる多官能サイトカインである(Akira S.等、Interleukin−6 in biology and medicine、Adv Immunol(1993)54:1〜78)。このサイトカインは、単球、マクロファージ、TおよびB細胞、内皮細胞、および線維芽細胞を含む多くの異なる細胞によって合成され、分泌される。IL−6は、しばしば多くの警告状態において炎症性サイトカインTNFα、およびIL−1によって誘発され、循環IL−6は、急性期反応の誘発において重要な役割を果たす(Xing Z.等、J.Clin.Invest.(1998)101(2):311〜20)。白血球補充および活性化の両方に、直接または間接的に影響を及ぼすサイトカインには、TNF−α、顆粒球コロニー刺激因子(G−CSF)、顆粒球マクロファージコロニー刺激因子(GM−CSF)、およびケモカインファミリーのメンバーが含まれる。主として白血球活性化の状態を調節するサイトカインには、インターフェロンγ(IFN−γ)、インターロイキン−10(IL−10)、およびインターロイキン−12(IL−12)が含まれる。サイトカインはまた、他のエフェクタ分子の発現を誘発または調節することによって微生物クリアランスに影響を及ぼすことができ、これはオートクリンまたはパラクリンいずれかの様式で起こり得る。
【0098】
コロニー刺激因子は、造血幹細胞の増殖および成熟に必要とされる多様な骨髄性細胞および間質性細胞によって産生されるサイトカインである。さらに、研究は、これらの因子が成熟白血球集団のエフェクタ細胞活性を高めることを示している。具体的には、G−CSFは、in vitroでの好中球の生存を延長し、好中球食細胞活性を高め、好中球呼吸バーストを増大する(Standiford TJ等、J.Invest.Med.(19097)45:335〜345)。対照的に、GM−CSFは、好中球およびマクロファージ両方の増殖および成熟を促進し、このサイトカインの活性化特性は、主として成熟マクロファージ集団に向けられる(Chen GH等、J Immunol(1994)152:724〜734)。
【0099】
ケモカインと呼ばれる化学走性サイトカインの密接に関連する6種のサブファミリーが今では特徴づけられた。これらのなかで少なくとも2つのサブファミリーのメンバーが、肺の抗菌宿主防御に寄与する(Mandujano J等、Amer.J.Respir.Crit.Care Med.(1995)151:1233〜1238;Baggiolini M等、Ann.Rev.Immunol.(1997)15:675〜705)。IL−8、MIP(マクロファージ炎症タンパク)−2、KC、ENA−78およびNAP−2を含むC−X−Cケモカインファミリーのメンバーは、優勢な好中球刺激活性および化学走性活性を有し、それに対してMCP(単球化学誘導タンパク)−1、MCP−2、MCP−3、RANTES、MIP−1αおよびMIP−1βを含むC−Cファミリーは、マクロファージ、リンパ球および好酸球に優勢な化学走性および/または活性化作用を及ぼす(Huffnagle GB等、The Role Of Chemokines In Pneumonia、Koch A、Strieter R編、Chemokines in Disease、Texas、R.G.Landis Co、1996:151〜168)。
【0100】
インターフェロンγは、肺病原体の広域スペクトルに対する細胞媒介性免疫に寄与する、T細胞(α、β、およびγ、δ T細胞の両方)およびNK細胞によって産生されるサイトカインである。このサイトカインは、呼吸バーストの刺激、抗原を提供する能力、マクロファージ由来TNF放出のプライミング、および細菌、真菌、マイコバクテリア生体に対するin vitroマクロファージ抗菌活性の強化を含むいくつかのマクロファージエフェクタ細胞機能を活性化する。
【0101】
インターロイキン−12は、Th1型免疫応答を促進し、Th2型免疫応答を阻害するサイトカインである。具体的には、IL−12は、Th1T細胞およびNK細胞の発生を刺激し、CD8+細胞およびNK細胞の細胞溶解活性を増大する。もっとも重要なことに、IL−12は、T細胞およびNK細胞からのIFN−γの主要誘発因子として働く。
【0102】
IL−12とは対照的に、IL−10は、Th2型免疫応答の発達を促進し、細胞媒介性(Th1型)免疫を阻害する(Howard M等、J.Clin.Immunol.(1992)12:239〜247)。このサイトカインは、一部には好中球およびマクロファージを直接不活性化することによって、且つTNF、IFN−γ、ならびにC−X−CおよびC−C両方のケモカインファミリーのメンバーの発現をダウンレギュレートすることによって、効力のある抗炎症特性を与えることが示された。IL−10は、全身免疫細胞活性化の状態において炎症性サイトカインの望ましくない生成を減ずるのに寄与する。
【0103】
EPOは、赤血球産生刺激活性を有する合成生物活性タンパク質の設計を助けるために、そのアミノ酸配列を用いることのできるタンパク質の特に好ましい例である。EPOは、定常状態における赤血球産生の調節、および出血後の赤血球塊回復の促進に関与する主要な因子である(Jelkmann、W.(1992)Physiol.Reviews 72:449;Krantz、S.B.(1991)Blood 77:419;Porter、D.L.およびM.A.Goldberg(1993)Exp.Hematol.21:399;Nissenson、A.R.(1994)Blood Purif.12:6)。ヒトEPOの環状形態は、およそ30000の分子量を有する165アミノ酸(aa)糖タンパク質である(Sawyer、S.T.等(1994)Hematol.Oncol.Clinics NA 8:895;Jacobs、K.J.等、Nature(1985)313:806;Lin、F−K.等、Proc.Natl.Acad.Sci.USA(1985)82:7580)。EPOのcDNAは、166アミノ酸残基を有する分子を予測するが、カルボキシ末端アルギニンは、翻訳後修飾において除去される(12)。N結合グリコシル化のための3つの潜在的な部位があり、それらはすべて占められている。1つのO結合炭水化物部分も存在する(13)。グリコシル化の作用は複雑である。非グリコシル化大腸菌由来EPOはin vitroで完全な生物活性を示すが、in vivoでの完全な活性のためにはグリコシル化が必要なようである。したがって、大腸菌産生、脱グリコシル化、天然EPOは、動物研究で非常に低い活性を示す(Krantz、S.B.(1991)Blood 77:419;Sasaki、H.等(1987)J.Biol.Chem.262:12059;Lowy、P.H.等(1960)Nature 185:102;Wojchowski、D.M.等、Biochem.Biophys.Acta 910:224;Dordal、M.S.等(1985)Endocrinology 116:2293)。
【0104】
上記に矛盾することなく、可変のグリコシル化様式は、可変の効果を示す。たとえば、脱シアル化EPOは、in vitroでの増強された活性、およびin vivoでの低減された活性の両方を示し、肝細胞によって認識、結合、除去されるガラクトース残基の暴露に帰する作用である(Spivak、J.L.およびB.B.Hogans(1989)Blood 73v:90;Goldwasser、E.等(1974)J.Biol.Chem.249:4202)。完全にシアル化されたEPOの分岐パターンも、生物活性に違いをもたらす。主として4枝に分岐したEPOは、「標準の」EPOと同等の活性を示し、一方、主として2枝に分岐したEPOは、in vitroで標準の3倍の活性を示すが、in vivoではわずか15%の活性を示す(Takeuchi、M.等(1989)Proc.Natl.Acad.Sci.USA 86:7819)。研究は、O結合糖ではなく、N結合糖のみがEPOの機能に重要であることを示している(Higuchi、M.等(1992)J.Biol.Chem.267:770319)。
【0105】
コロニー刺激因子、およびRANTESも、本発明の範囲内の合成生物活性タンパク質の設計を助けるためにそのアミノ酸配列を用いることのできる特に好ましいタンパク質の例を含む。
【0106】
B.ポリマー修飾タンパク質およびポリペプチド
本発明の第2の特質は、あらかじめ選択された位置に構造の定義された1つまたは複数のポリマー付加物を含むように、タンパク質またはポリペプチドを修飾する能力に関する。ポリマー修飾タンパク質は以前に記載されているが、考えられる本発明のポリマー修飾の性質および方式は、そのような従来の研究とは著しく異なる。
【0107】
本発明の合成生物活性タンパク質は、全体または部分的に化学合成されるので、ポリペプチド、タンパク質主鎖の、使用者が選択し、使用者が定義した1つまたは複数の特定の部位にポリマー(たとえばポリマーまたは糖、ポリエチレングリコール、グリコール、ヒアルロン酸など)を共有結合できるような方式でこのようなタンパク質を合成することが可能である。さらに、本発明のタンパク質の合成的生産は、調製品中の各分子の使用者選択、使用者定義部位のそれぞれに確実にそのような修飾を存在させることができる。合成のそのような一様性および制御は本発明を、従来技術の方法の使用によって可能となる無作為な修飾と明らかに区別する。注目に値すべきなのは、本発明は、任意の重要でない残基を誘導化してポリマー付加物を含有することのできる合成タンパク質の設計を可能にすることである。さらに、そのような使用者選択、使用者定義の各部位に関して、それを介して付加物がポリペプチドまたはタンパク質主鎖に結合される正確な結合(アミド、チオエステル、チオエーテル、オキシムなど)を使用者が定義することができる。さらに、存在するすべてのタンパク質またはポリペプチドが正確に同じ誘導部位に正確に同じ誘導付加物を含有する最終調製物が得られるように、特定の部位に存在を所望する特定のポリマー付加物を変更および制御することができる。したがって、本発明は、ポリマー修飾ポリペプチドおよびタンパク質の均一な調製物の形成を可能にする。
【0108】
ポリマーが結合することのできる結合部位の位置および数を変更する手段を提供することに加えて、本発明は、結合ポリマーの性質を変えることを可能にする。使用者選択、使用者定義された任意の特定部位に組み込むことのできるポリマー付加物は、任意の定義された長さであることができる。同様に、本発明の方法に矛盾することなく、異なる部位において異なる長さのポリマーを用いることが可能である。したがって、一実施形態において、本発明の合成生物活性タンパク質は、ポリマー付加物による「単一修飾」または「多修飾」であることができる。複数のポリマー付加物を特定のポリペプチドまたはタンパク質に導入した場合、その使用ポリマーは、「単一種分化」「多種分化」「一様に種分化」または「多様に種分化」されていることができる。本明細書では、「単一種分化」という用語は、単一のポリマー種によって修飾されたポリペプチドまたはタンパク質を指すことを意図する。対照的に、「多種分化」という用語は、複数のポリマー種によって修飾されたポリペプチドまたはタンパク質を指すものである。そのような多種分化ポリペプチドまたはタンパク質は、そのポリペプチドまたはタンパク質の各修飾部位に同じ単一種のポリマーが存在する場合、「一様に種分化」されていると言う。対照的に、ポリペプチドまたはタンパク質の修飾部位が異なるポリマー種によって修飾されている場合、種分化ポリペプチドまたはタンパク質は「多様に種分化」されていると言う。
【0109】
さらに、使用者定義、使用者選択されたそれぞれの部位において、ポリマー付加物の直線性または分岐性の程度を変えることが可能である。したがって、ポリマー付加物は、直線状、分岐状、または一様に分岐状であることができる。「一様に分岐状」という用語は、特定部位においてポリマーのすべての分岐が、同一の構造および長さを有することを意味するものである。本発明者等の理解のとおり、本発明は、個々の分岐の長さ、ならびにそのような分岐点に存在するポリマーの構造の両方を、独立して変更することを可能にする。
【0110】
要約すれば、本発明は、ポリペプチドまたはタンパク質主鎖におけるポリマー修飾、使用者選択、使用者定義部位の(1)位置、および(2)頻度を定義すること、ならびにそのような各部位に存在する分岐の(3)長さ、(4)種、および(5)程度を制御することを可能にする。さらに、複数のポリマー修飾を有するポリペプチドおよびタンパク質に関して、本発明は、各部位について上に特定した5つの変数のそれぞれを独立して定義することを可能にする。さらに、分岐ポリマー修飾を有するポリペプチドおよびタンパク質に関して、本発明は、各分岐点について上に特定した5つの変数のそれぞれを独立して定義することを可能にする。したがって、本発明は、ポリマー修飾の実質的な適応性を提供する。
【0111】
C.タンパク質およびペプチドのデリバリー
本発明の随意ポリマー修飾された合成生物活性ポリペプチドおよびタンパク質は、疾患および症状の治療を行うために薬剤として用いることができる。もっとも好ましくは、治療を必要としている患者または個体に投与されるとき、そのような合成生物活性ポリペプチドおよび/またはタンパク質は、薬剤デリバリーシステムを用いて投与される。そのようなシステムの使用は、患者が薬剤を受け入れることができ、且つ所望の生物活性の有効性を高めるような様式で患者に薬剤を提供することを可能にする。本発明の目的の好ましい薬剤デリバリーシステムには、経口、経鼻、または吸入経路によって、あるいは筋肉内、皮下、経皮、静脈内、耳内、または眼内に、ポリペプチドまたはタンパク質を投与できるシステムが含まれる。
【0112】
そのような薬剤デリバリーシステムは、部位特異放出を提供する製剤、または腸粘膜の保護を増強する製剤を含むことができる。適切な製剤には、乾燥粉末製剤、ウイルス粒子カプセルによるデリバリー、リポソームカプセル、経皮パッチ、電気援用輸送(電気穿孔療法)、およびポリマー/ナイアゾン(niazone)複合体が含まれる。
【0113】
好ましい実施形態において、そのような薬剤デリバリー装置は生物環境の変化に応答し、それらの変化に基づいて薬剤をデリバリーし、またはデリバリーを中止する。薬剤および他の活性剤の放出を制御するために一連の材料が用いられてきており、ポリ(ウレタン)、ポリ(シロキサン)、ポリ(メチルメタクリレート)、吸水性および強度のためのポリ(ビニルアルコール)、ポリ(エチレン)、ポリ(ビニルピロリドン)、ポリ(2−ヒドロキシエチルメタクリレート)、ポリ(n−ビニルピロリドン)、ポリ(メチルメタクリレート)、ポリ(ビニルアルコール)、ポリ(アクリル酸)、ポリアクリルアミド、ポリ(エチレン−コ−酢酸ビニル)、ポリ(エチレングリコール)、ポリ(メタクリル酸)などである。さらなる好ましい実施形態において、薬剤デリバリーを促進するために、生分解性ポリマーが用いられる。そのようなポリマーには、ポリラクチド(PLA)、ポリグリコリド(PGA)、ポリ(ラクチド−コ−グリコリド)(PLGA)、ポリ無水物、およびポリオルトエステルが含まれる。薬剤デリバリー装置、およびその使用のための方法は、米国特許第6072041号、第6041253号、第6018678号、第6017318号、第6002961号、第5879712号、第5849331号、第5792451号、第5783212号、第5766633号、第5759566号、第5690954号、第5681811号、第5654000号、第5641511号、第5438040号、第4810499号および第4659558号に記載されている。
【0114】
III.合成生物活性タンパク質の生成
本発明の合成生物活性タンパク質の生成は、以下のステップまたは構成要素、すなわち設計、合成、ペプチドライゲーション、タンパク質フォールディング(折りたたみ)、生物活性の評価、ポリペプチド主鎖のポリマー修飾を有するものとして考えることができる(図2)。
【0115】
A.好ましい合成生物活性タンパク質の設計
特に好ましい実施形態において、本発明の合成生物活性タンパク質は、赤血球産生刺激タンパク質である。本明細書では、赤血球産生刺激タンパク質は、赤血球の生成を媒介するタンパク質である。タンパク質の赤血球産生刺激生物活性は、たとえばin vivo投与後の赤血球生成を追跡する、EPO依存細胞系の増殖を媒介するタンパク質の能力を検定するなど、多様な任意の手段によって判別することができる。
【0116】
好ましい赤血球産生刺激タンパク質は、哺乳動物、もっとも好ましくはヒトのEPOおよびその類似体である。エリスロポエチンは主として、腎臓の尿細管および尿細管近接毛細管内皮細胞および間質細胞によって合成および分泌される。慢性腎臓疾患は、腎臓のEPO産生細胞の破壊をもたらす。結果として生じるEPOの欠乏は、しばしば貧血を誘発する。したがって、EPOの主要な臨床上の使用は、通常は輸血も受ける重篤な腎不全(ヘマトクリット0.3以下)を有する患者の治療、ならびに化学療法後の貧血患者の治療である。典型的に、EPOは臨床使用のためにハムスター卵巣細胞において組換えによって合成される。EPOは、その成熟形態で165アミノ酸残基を有する比較的熱安定性、且つpH安定性の酸性(pI=4.5)タンパク質である。EPOは、EPO受容体を介してその活性を伝える。EPOの分子量のおよそ40パーセントは、グリコシル化によるものである。グリコシル化は、in vivoでのEPOの薬物動態挙動を決定する重要な要因である。非グリコシル化EPOは、極端に短い生物学的半減期を有する。これはin vitroにおいて依然として受容体に結合し、より高い特異的活性を有する可能性もある。しかしながら、最近の実験によって、EPOのPEG化はその寿命を著しく増大するが、細胞に基づくアッセイシステムにおいてEPO活性を著しく低減することが示された。
【0117】
本発明の合成赤血球産生刺激タンパク質(「SEP」)は、好ましくは、ヒト尿EPOの化学的に修飾された合成類似体である。好ましい実施形態において、SEPは、チオエステル結合、オキシム結合、アミド結合、または他の結合を介して、1つまたは複数のペプチド残基に結合している1つまたは複数ポリマー部分(もっとも好ましくはpPEG部分)を含有する。非常に好ましい実施形態において、そのような結合は、ヒト尿EPOにおいて天然にグリコシル化されている1つまたは複数の位置に存在する(すなわち、24、38、83および126位)。もっとも好ましくは、SEP分子は、2つ以上のそのような位置にpPEG部分を有する。別の実施形態において、他のタンパク質残基は、ポリマー部分によって修飾されることができる。本発明の合成生物活性タンパク質のための修飾位置には、タンパク質の無秩序ループ、領域、またはドメイン、あるいは潜在性プロテアーゼ開裂部位またはその近傍部位に位置する残基が含まれる。たとえば、ポリマー修飾は、EPOの9、69、および/または125位の1つまたは複数の位置に導入することができる。本発明のSEP分子の全分子量は、約25から約150kDa、より好ましくは約30から約80kDaであることができる。分子量は、所与の類似体の修飾に用いられるポリマー(pPEGなど)の数および構造を増大または低減することによって制御できる。より大きな構成体のpPEG媒介流体力学MWは約100kDaを超える。in vitroにおけるヒトEPO受容体発現細胞でのアッセイは、本発明のSEPが、組換えによって産生されたグリコシル化ヒトEPOと同等のED50を有することを示す。
【0118】
オキシム結合pPEG類似体は、好ましくは、側鎖アミノオキシ、ケトン、またはアルデヒド官能基を有する非天然コード化アミノ酸において、SEPタンパク質に、アミノオキシ、ケトン、またはアルデヒド官能化pPEGを結合することによって構成される。たとえば、SEP−0およびSEP−1の89および117位は、擬似グルタミン酸塩((グルタミン酸塩側鎖−CHα−CH2−CH2−COOHに対して)式−CHα−CH2−S−COOHの側鎖を有する非天然コード化アミノ酸)を含有する。チオエーテル結合を用いるSEP類似体は、好ましくは、チオールを有する側鎖を備えたシステインまたは非天然アミノ酸によって提供されるチオール官能基を含有するように構成される。図3は、好ましい合成赤血球産生刺激タンパク質の1つの型の基本的な構造を示す。
【0119】
別の実施形態において、本発明のSEP分子は、EPOの天然アミノおよびカルボキシ末端が再配置された「環状順列化」EPO類似体を含むことができる。より好ましくは、そのような再配置は、アミノおよびカルボキシ末端を、無秩序ループなど、構造的制約の低い位置に移動する。125および126位(天然EPO残基ナンバリングシステムを基準として)近くの無秩序ループは、そのような再配置部位の例である。もっとも好ましくは、そのようなSEPはジスルフィドを含まず、あらかじめ選択された残基にポリマー部分を含有するように化学的に修飾される。
【0120】
あるいは、SEP分子は、天然グリコシル化部位、またはグリコシル化を施すことのできる他の部位、たとえば126および125位などに再配置されたアミノおよびカルボキシ末端を有することができる。SEP分子は、電荷を除去または変更するためのアミノおよびカルボキシ末端の修飾(カルボキシアミド化などによる)を含むこともできる。
【0121】
そのような環状順列化分子の好ましい例において、新しいNおよびC末端は、それぞれ126および125位によって提供される。7、29、32および161位の天然ジスルフィド形成システインは、好ましくは、ジスルフィド架橋を形成することのできない非天然コード化アミノ酸、L−α−N−酪酸(Aba)に置換されている。残基R166、E37およびV82は、好ましくは、生成を向上させるためにアラニンに置換されている。さらに、好ましくは追加のシステインが、天然EPOナンバリングスキームを基準として1位と166位の間に挿入され、以下「0」と番号を付す。結果として生じた分子は、4つのシステインを含有し(126、0、38および83位(N末端からC末端方向に読み取り))、それらは(1)ライゲーション部位、および(2)チオエーテル形成pPEG結合部位として用いられる。随意、システインは、追加のpPEG結合部位を提供するためにA125を置換することができる。本発明のそのようなSEP分子の全分子量は、約25から約150kDa、より好ましくは約30から約80kDaであることができる。分子量は、所与の類似体の修飾に用いられるポリマー(pPEGなど)の数および構造を増大または低減することによって制御できる。より大きな構成体のpPEG媒介流体力学MWは約100kDaを超える。任意のpPEG結合部位は、125、9および24位(N末端からC末端方向に読み取り)に位置する。さらなるSEP類似体設計は、新しいNおよび/またはC末端からの無秩序ループ、領域、および/または切断残基に代わりのNおよびC末端を有する。好ましい循環順列化分子の基本構造を図4に示す。
【0122】
別の実施形態において、本発明の合成生物活性タンパク質は、哺乳動物、もっとも好ましくはヒトのC−GSFである。G−CSFは、好中球性顆粒球の急速な増殖および血流への放出を誘発し、それによって感染との戦いに治療的効果をもたらす。ヒトGCSFタンパク質は、アミノ酸長174であり(特許EP0459630(Camble、R他)、その配列の変種がすでに単離されている。このタンパク質は4つのシステイン残基を有し、トレオニン133位にO−グリコシル化部位を有する。
【0123】
そのような合成GCSF分子の一実施形態において、分子のアミノ酸配列は、1、2、または3位の1つまたは複数の位置に天然、またはより好ましくは非天然コード化疎水残基を含有するように、天然GCSFの配列に対して変更される。随意、そのような分子は、GCSFの5位、および/または173位および/または174位に追加の天然、より好ましくは非天然コード化疎水残基を含有するようにさらに修飾される。
【0124】
さらなる好ましい実施形態において、合成GCSF分子は、63、64、および/または133位の1つまたは複数の位置、および/または1、および174位の1つまたは複数の位置でポリマー修飾(好ましくはpPEG)される。このpPEGポリマーは、線状または分岐状、荷電、非荷電、または混合であることができ、約5から約80Da、より好ましくは約40から約70kDaの分子量の範囲であることができ、pPEG全MW寄与は修飾に用いられるpPEGの数および構造に依存する。流体力学半径は、in vivoでより大きなMW効果を示す(たとえば、10kDa分岐pPEGは約55kDaの有効分子量を示す)。推定pPEG媒介流体力学MWは、100kDaを超える。
【0125】
オキシム結合を用いる類似体の場合、pPEGは、好ましくは、側鎖アミノオキシ、ケトン、またはアルデヒド官能基を有する非天然コード化残基に結合されている。チオエーテル結合を用いる類似体の場合、pPEGは、好ましくは、チオールを有する側鎖を備えたシステインまたは非天然アミノ酸に結合されている。アミド結合を用いる類似体の場合、pPEGは、反応性側鎖アミンを有する天然または非天然アミノ酸に結合されている。
【0126】
合成生物活性GCSFタンパク質の基本構造を図5に示す。この図において、「J」は、疎水性側鎖を有する非天然コード化残基を示す。
【0127】
そのような合成生物活性GCSFタンパク質の生物活性は、たとえば因子依存性ヒトまたはマウス細胞系(たとえばNFS60細胞系)の増殖を媒介するそれらの能力を評価することによって、あるいは20日超の半減期/放出をターゲットとするデリバリーシステムを用いて、または用いずに、in vivoでの好中球刺激、半減期、および免疫原性を測定することによってなど、通常の手段で評価することができる。
【0128】
別の実施形態において、本発明の合成生物活性タンパク質は、哺乳動物、もっとも好ましくはヒトのケモカイン、RANTESである。RANTESは、主要受容体CCR5を経るマクロファージ親和性HIV株の侵入を遮断し、さらに喘息、移植拒絶、および創傷治癒に関わる炎症経路をダウンモジュレートする(Kalinkovich A.等、Immunol Lett.(1999)68:281〜287)。本発明の合成RANTES分子は、好ましくは、(1)RANTES1〜68の1位に認められるN末端セリンが、n−ノナノイル(「NNY」)基で置換されており、(2)3位のチロシンが、疎水性側鎖を有する非天然コード化アミノ酸で置換されているように化学修飾されている点でRANTESケモカインと異なる。そのような化合物は、組換え「Met−RANTES1〜68」のミリモル範囲に比べて、ピコモル範囲のED50を有し、極めて有効であることが見出されている。
【0129】
疎水性側鎖を有する非天然コード化アミノ酸で2位のプロリンを置換する、分子のC末端に脂肪酸を付加するなど、上記のRANTESに対して1つまたは複数の追加の変更を有する有効なRANTES類似体が作られた。RANTES残基12〜20に対応するNループ領域への修飾によって、効力と合わせて受容体特異性が向上した。さらなる好ましい実施形態において、本発明の合成RANTES分子は、特にin vivo半減期を改善するためにNまたはC末端にポリマー部分(pPEGなど、あるいはノナノイル(「NNY」)またはY3Xを組み込む。このpPEG部分は、好ましくはオキシム結合を介して非天然コード化アミノ酸に結合され、これは66、67、68位、または68位のリンカーに導入され、67位が好ましい。脂肪酸は、好ましくはオキシム結合(好ましくはリンカーを経て)を介して、アミノ−オキシ修飾アミノ酸ジ−またはトリ−ペプチドリンカーなど残基68に結合される。他の結合化学を別法として用いることができる。そのような合成RANTES類似体のより大きな構成体の分子量は、pPEG付加物の性質および構造に応じて約25kDaから約45kDaの範囲であり、より大きい構成体では、60kDaを超えるpPEG媒介流体力学MWを有する。好ましい合成RANTES類似体の構造を図6に示す。
【0130】
B.合成生物活性タンパク質およびペプチドの定義された特定のポリマー修飾
本発明のさらなる態様は、非修飾タンパク質に比べて改善された特性を有するタンパク質薬剤を生じるようにタンパク質を修飾するための化学部分およびポリマー、特に水溶性ポリマー、ならびにそれらの使用に関する。そのような修飾の予期される有益な効果には、これに限定されるものではないが、非修飾分子と比べて(a)効力の向上、(b)タンパク分解安定性の増大、およびクリアランス速度の低減による循環におけるタンパク質薬剤存続期間の延長、(c)免疫原性の低減、および(d)識別的ターゲッティングの可能性が含まれる。
【0131】
上に論じたように、タンパク質を修飾するためにポリエチレングリコールが用いられてきたが(たとえば、反応性アミン基によるポリエチレングリコール分子の結合を可能にするために、G−CSFのリジン残基を修飾することに関する米国特許第6027720号、Kuga等を参照のこと)、そのような修飾は分子異質性、および分子多様性を伴う。
【0132】
本明細書では、「分子異質性」という用語は、タンパク質製剤の各タンパク質に結合されたポリマー分子の数のばらつきを指すものである。「分子多様性」という用語は、(a)そのポリマーによって修飾されるタンパク質の部位のばらつき、(b)タンパク質製剤の同一タンパク質の異なる部位、または異なるタンパク質の同一部位のポリマー付加物の長さのばらつき、または(c)タンパク質製剤の同一タンパク質の異なる部位、または異なるタンパク質の同一部位のポリマー付加物の、任意の分岐の程度および/または性質のばらつきを指すものである。本明細書では、「分子的に均一」という用語は、すべてのタンパク質分子が同じ位置にアミノ酸配列および同一のポリマー修飾を含有するタンパク質の製剤を指すものである。2つ以上の「分子的に均一」なタンパク質製剤の混合物は、本明細書では、「分子的に定義された」製剤と呼ぶ。
【0133】
本発明のこの態様に従って、上に特定したポリマー異質性、多様性、および不適合性の問題の解決策には、ポリペプチド(厳密な長さ、好都合な合成)および「pPEG」(「精密PEG」)(適応性、両親媒性、非免疫原性、プロテアーゼ感受性でないポリマー)両方の利点を兼ね備える新しい類の生体適合性ポリマーを製造することが含まれる(Rose、K.等、参照により本明細書に組み込むものとする米国特許出願第09/379297号)。この新しい類の生体適合性ポリマーは以下の式を有し、
−[CO−X−CO−NH−Y−NH]n−
nは、整数であって、好ましくは1〜100、より好ましくは2〜100であり、該式中、XおよびYは、アミド結合によって結合した正確な構造の生体適合性反復要素である。好ましくは、XおよびYは、反応性官能基を欠く2価有機基であるか、または不在であり、同一であるか、または異なり、且つ各反復単位によって独立して異なることができる。好ましくは、n=2であるとき、XまたはYの少なくとも1つは、置換、非置換、分岐状、および線状脂肪族および芳香族基からなる群から選択される。より好ましくは、2価有機基XまたはYの少なくとも1つは、フェニル、C1〜C10アルキレン部分、C1〜C10アルキル基、ヘテロ原子含有フェニル、ヘテロ原子含有C1〜C10アルキレン部分、ヘテロ原子含有C1〜C10アルキル基、およびそれらの組み合わせからなる群から選択される。
【0134】
特に好ましいpPEG部分は以下の式を有し、
−{CO−(CH2)2−CO−NH−(CH2)3−(OCH2CH2)3−CH2−NH}n−
該式中、nは、好ましくは1〜100、より好ましくは2〜100であり、または、
−{CO−(CH2)2−CO−NH−(CH2)6−NH−CO−(CH2)2−CO−NH−(CH2)3−(OCH2CH2)3−CH2−NH}n−
を有し、該式中、nは、好ましくは1〜50、より好ましくは2〜50である。
【0135】
そのようなpPEG部分は、多様な方法のいずれかで合成することができる。しかしながら、そのような部分は、好ましくは、重合法よりも、単位の固相段階的鎖アセンブリ法を用いて生成される。そのようなアセンブリ法の使用によって、製剤のその部分が、それらの長さ、XおよびY置換基の性質、(もしあれば)分岐点の位置、ならびに分岐の長さ、XおよびY置換基、および位置に関して定義された均一な構造を有することが可能になる。
【0136】
好ましくは、そのような部分は、
(a)式HOOC−X−COOHを有する2酸の誘導体のモル過剰量を用いて、該式中、Zは、H2N−またはHO−であり、Qは、リンカーまたは標的分子であり、支持体は、固相、マトリクス、または表面である式Z−Q−支持体の化合物のアミノまたはヒドロキシル基をアシル化するステップ、
(b)ステップ(a)の生成物の遊離カルボキシル基を活性化するステップ、
(c)式NH2−Y−NH2を有するモル過剰量のジアミンを用いて、ステップ(b)の生成物をアミノ分解するステップ、および
(d)HOOC−X−COOHおよびNH2−Y−NH2を用いてステップ(a)〜(c)を随意繰り返すステップであって、前記XおよびYは、2価有機基であるか、または不在であり、同一であるか、または異なり、且つ前記の任意選択の各反復単位によって独立して異なることができ、且つ先のアシル化およびアミノ分解ステップのいずれかで用いたXおよびY置換基と同じか、または異なっているステップなどのステップによって合成される。
【0137】
好ましい実施形態において、上述の反復単位の6マー、12マー、18マー、および32マーが用いられる。所望である場合、この反復単位は、たとえば分岐pPEG構造を形成するためにリジンのアミノ基と共に用いることができる。pPEGは、チオエーテル、オキシム、およびアミド結合形成を含む多様な化学によって、本発明の合成タンパク質に結合することができる。
【0138】
一実施形態において、単位の固相段階的鎖アセンブリは、以下のステップ1〜5を含む。
ステップ1: 保護または非保護2酸を樹脂上でアミノに結合して、結合部位にアミド結合を生じる(PGは、用いられる2酸に応じて存在するか、または存在しない保護基である):
PG−OOC−X−COOH + NH2−Y−NH−樹脂
ステップ2: 存在する場合、樹脂上で保護基(PG)を除去する:
HOOC−X−CO−NH−Y−NH−樹脂
ステップ3: 保護または非保護ジアミノを樹脂上でカルボキシに結合して、結合部位にアミド結合を生じる(PGは用いられるジアミノに応じて存在するか、または存在しない):
PG−NH−Y−NH + HOOC−X−CO−NH−Y−NH−樹脂
ステップ4: 存在する場合、樹脂上で保護基(PG)を除去する:
−NH−Y−NH−OC−X−CO−NH−Y−NH−樹脂
ステップ5: 「n」単位を付加するためにステップ1〜4を「n」回繰り返し、次いで樹脂から−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−[CO−X−CO−NH−Y−NH]−を開裂する。
【0139】
上述したように、線状または分岐状pPEG構成体は、本発明の合成生物活性分子に結合するのに好ましい水溶性ポリマーである。用いられるpPEGは、生理的条件下で荷電または中性であり、用いるpPEG構造に応じて結合化学、長さ、分岐、および溶解性を変えることのできるペンダント基を有する。上記のとおり、本発明の好ましいpPEGは、XおよびYの1つまたは両方が水溶性反復単位、もっとも好ましくは「PEG」ベースの反復単位を含む、反復単位−CO−X−CO−NH−Y−NH−を有する水溶性ポリアミドを含む。アミノ樹脂、NH2−樹脂、および対称ジアミンNH2−Y−NH2の場合に関して下に示したように、原理的には簡単な2ステップ固相手順を用いてオリゴ尿素を利用でき、カルボニルジイミダゾールによる活性化→im−CO−NH−樹脂
ジアミンNH2−Y−NH2によるアミノ分解→NH2−Y−NH−CO−NH−樹脂
これら2つのステップを何度か繰り返して、反復単位−NH−Y−NH−CO−を有するオリゴ尿素を得ることができるが、このアプローチは好ましさに劣る。これは、上記ステップの収率が、室温において非常に過剰の試薬と長い反応時間を用いても、量的でない可能性があるためである。したがって、アミノ樹脂、NH2樹脂の場合を以下に示した、ポリアミドを形成する3ステップの固相手順を用いることが好ましい。試薬HO2C−X−CO2H、およびNH2−Y−NH2は、異性体生成物を回避するために、対称であるべきである。
二酸HO2C−X−CO2Hを用いたアシル化→HO−CO−X−CO−NH−樹脂
カルボニルジイミダゾールを用いた活性化→im−CO−OCO−X−CO−NH−樹脂
ジアミンNH2−Y−NH2を用いたアミノ分解→NH2−Y−NH−CO−X−CO−NH−樹脂
【0140】
これらの3ステップは、反復単位−NH−Y−NH−CO−X−CO−を有するポリアミドを得るために、何度か連続して繰り返すことができる。このポリマーは、正確な数のモノマー単位を含有し、XおよびYは、各ステップにおいて独立して異なることができ、末端基は、随意に選択できる。たとえば、アシル化ステップに無水コハク酸(「Succ」)を用い、アミノ分解ステップに4,7,10−トリオキサ−1,13−トリデカンジアミン(「EDA」または「TTD」とも呼ばれる)を用いることによって、Xが−CH2CH2−であり、Yが−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH−であり、反復単位が−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH−COCH2CH2CO−である「PEG」ベースのポリアミドが形成される。この手順には保護基を持たない2価の試薬が含まれるにもかかわらず、以下に言及する分岐構成体を製造するための分岐Lysコアの場合を除いて、標準的な市販のペプチド合成樹脂が用いられるとき、架橋結合は問題とならない(Rose他、米国特許出願第09/379297号、およびRose等、J.Am.Chem.Soc.(1999)121:7034)。
【0141】
たとえば、Rose他(米国特許出願第09/379297号)、およびRose、K.およびVizzavona、J.(J.Am.Chem.Soc.(1999)121:7034)に記載されている「化学抗体(chemobody)」構成体と類似の方法で、分岐pPEG構成体を製造することができる。したがって、分岐pPEG構成体はリジン分岐コアなどの分岐コアを有するように製造することができ、分岐コアはオキシムリンカーを介して−(COCH2CH2CO−NH−CH2CH2CH2−(OCH2CH2)3−CH2−NH)n−などの好ましい水溶性ポリアミドに結合している。たとえば、オキシム結合は、ポリアミドの他端にあるLys側鎖のアミノオキシアセチル基と、リジンコア、たとえばテトラマーコア(O=CHCO)4Lys2Lys−のグリオキシリル基との間に容易に形成できる。したがって、各リジン分岐点のオキシムリンカーは、構造−Lys(COCH2ON=CHCO−)アミド−として調製することができる。別の構成は、以下に示す4価分岐構成体を生じるために、好ましくはモノ保護ジアミン、およびポリアミドカップリング後の脱保護を用いて、オキシム結合をポリアミドとポリアミドの遊離ペンダント基との間に配置することができる。
[O=CHCO−(NH−CH2CH2CH2−[OCH2CH2]3−CH2−NH−COCH2CH2CO−)n]4Lys2Lys−
【0142】
オキシム化学は、ダイマーおよびテトラマー分岐構成体の製造に用いるだけでなく、オクタマー分岐構成体のアセンブリにも適している(Rose、K.、J.Am.Chem.Soc.(1994)116:30;Rose、K.等、Bioconj.Chem.(1996)7:552)。当然ながら、そのようなpPEGポリアミドを用いるとき、その目的のために他の化学を用いることができる(たとえば図7を参照のこと)。さらに、ポリアミド形成は、BocまたはFmoc化学を含むペプチド合成のための合成スキームに組み入れることができるが、そのようなスキームを練り上げるとき、アミノ分解ステップはFmoc基を除去し、Boc化学が用いられる場合にはインドールのホルミル保護を除去することを留意しなければならない。
【0143】
したがって、そのようなpPEGは、好ましい結合(たとえばアミド、チオエーテル、オキシムなど)を介して線状水溶性ポリアミド(たとえば−(Succ−TTD)n−等)に結合した種々の分岐コアを有するように製造することができ、ここで各線状水溶性ポリアミドの自由端はそれぞれ所望のペンダント基(たとえばカルボキシレート、アミノ、アミド等)を用いてキャップして所望の電荷を提供することができる。さらに、本発明の線状および分岐状pPEGは、ユニークで相互反応性の1つまたは複数の官能基を有する合成タンパク質に結合するためにユニークな官能基を含むように製造することができ、pPEGのペンダント基は、好ましくは非抗原性である(たとえば図7を参照のこと)。
【0144】
本明細書で言及したすべての刊行物および特許出願は、それぞれ個々の刊行物または特許出願が参照により組み込まれると具体的に且つ個別に指示されるのと同程度に、参照により本明細書に組み込むものとする。本発明の内容を説明するために、本明細書において本発明の背景の考察が含まれる。これは、請求の範囲のいずれかの優先権主張日の時点で、参照された資料のいずれかが世界のいずれかの場所で公表されているか、知られている、あるいは従来技術または周知の事柄の一部であることを認めるものであるとみなされるものではない。ここに本発明を一般的に説明したが、例示的なものとして提供され、明記されない限り本発明を限定するものではない以下の実施例を参照することによって、本発明はより容易に理解されるであろう。
【0145】
(実施例)
実施例1
合成赤血球産生刺激タンパク質SEP−0の合成
合成赤血球産生刺激タンパク質(SEP)を合成した。完全長合成タンパク質(「SEP−0(1〜166)」とする)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAASAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:1)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示す。このSEP−0タンパク質は、以下の4つのポリペプチドセグメントから溶液中で合成された。
セグメントSEP−0:1(GRFN1711、配列番号:1の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA
EH−チオエステル
セグメントSEP−0:2(GRFN1712、配列番号:1の残基33〜88からなる):
CSLNEKITVP DTKVNFYAWK RMEVGQQAVE
VWQGLALLSE AVLRGQALLV KSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−0:3(GRFN1713、配列番号:1の残基89〜116からなる):
CPLQLHVDKA VSGLRSLTTL LRALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−0:4(GRFN1714、配列番号:1の残基117〜166からなる):
CAISPPDAAS AAPLRTITAD TFRKLFRVYS
NFLRGKLKLY TGEACRTGDR−カルボキシレート(C末端システイン(Cys161)はピコリル(pico)保護基を有する)
【0146】
ペプチドSEP−0:1およびSEP−0:2およびSEP−0:3を、ABI433A自動ペプチド合成装置で、または手作業の鎖アセンブリで、Boc(tert−ブトキシカルボニル)化学のためのin situ中和プロトコル、および確立されたSPPS、側鎖保護、およびチオエステル樹脂法を用いる段階的固相ペプチド合成(SPPS)(Hackeng等、PNAS(1999)96:10068〜10073、およびSchnolzer等、Int.J.Pept.Prot.Res.(1992)40:180〜193)によってチオエステル生成樹脂上で合成するか、あるいは供給業者に注文し、入手した。たとえば、BocSPPS保護基の標準セットを用い、すなわちArg(Tos)、Asp(cHex)、Cys(4MeBzl)およびCys(Acm)、Glu(cHex)、His(DNP)、Lys(CIZ)、Ser(Bzl)、Thr(Bzl)、Trp(ホルミル)、Tyr(BrZ)であり、Met、Asn、Glnは非保護側鎖であった。セグメントSEP−0:4は、−OCH2−Pam−樹脂上で同様に合成した。これらのペプチドを、標準的なBoc化学手順に従い、HF/p−クレゾールを用いて脱保護し、同時に樹脂支持体から開裂したが、しかしながらHF/p−クレゾール中で除去されない保護基を含有するペプチドの場合、保護基は維持された。これらのペプチドを、分取C4逆相高圧液体クロマトグラフィ(HPLC)で精製した。純粋ペプチドを含有する分画を、ES−MS(エレクトロスプレーイオン化質量分析法)を用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0147】
ステップ1:ライゲーション#1 セグメントSEP−0:4およびセグメントSEP−0:3を、濃度15mMでTFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する飽和リン酸緩衝液(pH7.9)を添加し、ペプチドセグメントの透明溶液を得た。ライゲーション後、ライゲーション混合物を、TFE(トリフルオロエタノール)2ml、6M塩化グアニジニウム6ml、25%β−メルカプトエタノールを含有する100mMリン酸塩の溶液に添加し、20分間インキュベートした。その溶液を、15mg/mlのTCEP(トリス(2−カルボキシエチル)ホスフィンHCl)氷酢酸溶液で酸性にし、分取C4逆相HPLCカラム(直径1インチ)に充填した。次いで、そのペプチドを分取勾配逆相HPLCによって精製した。所望の結合生成物SEP−0:Acm+SEP−0:3+SEP−0:4を含有する分画を、ES−MSによって同定し、プールした。
【0148】
ステップ2:Acm除去#1 Acm除去のために、プールしたSEP−0:Acm+SEP−0:3+SEP−0:4分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−0:3+SEP−0:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0149】
ステップ3:ライゲーション#2 等量のSEP−0:3+SEP−0:4およびSEP−0:2を合わせて、15mM濃度で純TFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6M塩化グアニジニウム20ml、pH4の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0150】
ステップ4:カルボキシメチル化 SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4を、15mM濃度でTFEに溶解した。6M塩化グアニジニウムを含有する2倍過剰(v/v)の200mMリン酸緩衝液(pH7.9)を添加し、ペプチドセグメントの透明溶液を得た。最小量のメタノールに溶解した25倍過剰のブロモ酢酸を添加し、その溶液を2時間反応させた。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、段階勾配で精製した。所望のカルボキシメチル化生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Etを含有する分画をES−MSによって同定し、プールした。
【0151】
ステップ5:ピコリル除去 亜鉛末を2MのHCl中で30分間活性化した。ペプチドSEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Etを、約10mg/mlの濃度で純TFEに溶解した。その溶液を(新たに添加した)35mg/mlのL−メチオニンおよび35mg/mlのドデシルサルコシン(すなわちN−ドデカノイルサルコシンナトリウム)を含有する4x(v/v、TFEに対して)の6M塩化グアニジニウム、100mM酢酸塩、pH4で希釈した。その溶液を、活性化Zn粉末に添加した。反応は〜1時間間隔でモニターし、5時間後に完了する。完了後、上澄みを除去し、残存するZn粉末を、20%TFEを含有する35mg/mlL−メチオニンおよび35mg/mlドデシルサルコシン含有6M塩化グアニジニウム、pH4、100mM酢酸塩で5分間2回洗浄し、20%β−メルカプトエタノールを含有する同じ溶液で1回洗浄した。混合生成物を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の修飾生成物SEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoを含有する分画をES−MSによって同定し、プールした。
【0152】
ステップ6:Acm除去#2 プールしたSEP−0:Acm+SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoの溶液を、HPLCグレードの水で3x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−0:2+SEP−0:3+SEP−0:4+Et−Picoを含有する分画をES−MSによって同定し、2x(w/w、ペプチド質量に対して)のDPC(ドデシルホスホコリン)を含有する水で2x(v/v)希釈し、一晩凍結乾燥した。
【0153】
ステップ7:ライゲーション#3 等量のSEP−0:2+SEP−0:3+SEP−0:4+Et−PicoおよびSEP−0:1を合わせて、15mM濃度で純TFEに溶解し、6M塩化グアニジニウムを含有する250mMのリン酸緩衝液(pH7.5)を添加した。その溶液に、1%チオフェノールを添加した。1日のライゲーション後、ライゲーション混合物を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6M塩化グアニジニウム20ml、pH4の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−0(1〜166)(配列番号:1)を含有する分画をエレクトロスプレー質量分析法によって同定し、2x(w/wペプチド質量に対して)のドデシルサルコシンを含有する水で2x(v/v)希釈し、凍結乾燥した。
【0154】
ステップ8:フォールディング(折りたたみ) 完全長結合ペプチドSEP−0(1〜166)を、6M塩化グアニジニウム、20%TFE、10倍モル過剰(タンパク質のCys残基に対して)のシステインを含有する200mMトリス緩衝液(pH8.7)に溶解した。この溶液を、3M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温で一晩透析した。次いでその溶液を、1M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温4℃で4時間透析し、最後に10mMリン酸緩衝液(pH7.0)に対して、4℃で4時間透析して、最終フォールド生成物を得た。フォールディングは、エレクトロスプレーES−MSおよびCD(円偏光二色性)分光法によって確認した。
【0155】
ステップ9:精製 折りたたまれたポリペプチドを、セントリコン濃縮バイアルで5x濃縮し、10mMリン酸塩、pH7.0で平衡させたResource Sカチオン交換カラムに充填した。折りたたまれたタンパク質を、500mMのNaClへの10分の直線塩勾配で溶出した。所望の折りたたまれた生成物SEP−0(1〜166)を含有する分画を、SDSポリアクリルアミドゲル電気泳動(SDS−PAGE)によって同定、凍結し、−80℃で貯蔵した。折りたたまれたタンパク質生成物の分析逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、折りたたまれたタンパク質の存在を実証した。
【0156】
実施例2
合成赤血球産生刺激タンパク質SEP−1−L30の合成
2番目の合成赤血球産生刺激タンパク質(SEP−1−L30とする)を、SEP−0の24および126位にオキシム形成基を含有するように合成した。次いでこれらの基を用いて、線状(EDA−Succ)18カルボキシレート(EDA=(4,7,10)−トリオキサトリデカン−1,13ジアミンであり、TTDとも呼ばれる。Succ=−CO−CH2CH2CO−)ポリマーがそのタンパク質主鎖に結合しているSEP−1−L30を形成した。完全長SEP−1(1〜166)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0157】
A.GRFNP32によるGRFN1776およびGRFN1711のオキシム化
オキシム形成を行って、アミノオキシアセチル基を有する水溶性ポリマーを、ケトンカルボニル基を有するペプチドに結合した。これを達成するために、以下のペプチドセグメントを合成した。
セグメントSEP−1:4(GRFN1776、配列番号:2の残基117〜166からなる):
CAISPPDAAK AAPLRTITAD TFRKLFRVYS NFLRGKLKLY TGEACRTGDR−カルボキシレート(Lys126はε−アミノ基においてレブリン酸残基で修飾され、Cys117はAcm保護されている)
セグメントSEP−1:1(GRFN1711、配列番号:2の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA EH−チオエステル(Lys24はレブリン酸残基で修飾されている)
【0158】
実施例1のとおり、セグメントSEP−1:1(GRFN1711)をチオエステル生成樹脂上で、セグメントSEP−1:4(GRFN1776)を−OCH2−Pam−樹脂上で合成した。これら2つのペプチドセグメントのリジン24および126を、初めにε−アミノ基においてFmoc基で保護した。鎖アセンブリの完了後、Fmocを有するアミノ基を標準的なFmoc脱保護手順に従って脱保護し、各ペプチド樹脂にそれぞれレブリン酸を結合することによって修飾した。次いでこれらのペプチドを、実施例1に記載のとおり、脱保護し、同時に樹脂支持体から開裂した。ペプチドを、分取C4逆相HPLCによって精製した。純粋ペプチドを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。GRFNP32[−(EDA−Succ)18カルボキシレート]を、標準的なプロトコルに従って、Sasrinカルボキシル生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。5倍過剰の活性化Boc−アミノオキシ酢酸をカップリングすることによって、アミノオキシアセチル(AoA)部分をポリマーのN末端アミノ基に結合した。そのポリマー鎖を、典型的なFmoc化学手順を用いて樹脂支持体から開裂した。ポリマー鎖を、分取C4逆相HPLCによって精製した。純粋なポリマーを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0159】
セグメントSEP−1:4およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP32を含有する分画をES−MSによって同定し、プール、凍結乾燥した。
【0160】
セグメントSEP−1:1およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP32を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0161】
B.合成赤血球産生刺激タンパク質SEP−1−L30の合成
SEP−1−L30を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP32(GRFN1711+GRFNP32、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP32(GRFN1776+GRFNP32、配列番号:2の残基117〜166に対応):
CAIS PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システインはピコリル(pico)保護基を有する)
【0162】
追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を、実施例1に記載のとおり行い、完全長フォールドSEP−1−L30を得た。フォールドタンパク質生成物の分析逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、フォールドタンパク質の存在を実証した。
【0163】
実施例3
合成赤血球産生刺激タンパク質SEP−1−L26の合成
3番目の合成赤血球産生刺激タンパク質(SEP−1−L26とする)を、SEP−0の24および126位にオキシム形成基を含有するように合成した。次いで、これらの基を用いて、線状ポリマー(EDA−Succ)18カルボキシレートおよび(EDA−Succ)6−アミドが、それぞれ24位および126位においてオキシム結合を介してタンパク質主鎖に結合しているSEP−1−L26を形成した。完全長SEP−1(1−166)の配列は以下のとおりであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0164】
SEP−1−L30と対照的に、SEP−1−L26構成体は、126位に結合されたより小さな非荷電水溶性ポリマーを有するように設計された。24位に結合されたポリマーは、SEP−1−L30と同じである。完全長生成物のアセンブリは、実施例2に記載したとおりである。
【0165】
A.GRFNP6によるGRFN1776のオキシム化およびGRFNP32によるGRFN1711のオキシム化
オキシム形成を行い、アミノオキシアセチル基を有する水溶性ポリマーを、ケトンカルボニル基を有するペプチドに結合した。これを達成するために、以下のペプチドセグメントを合成した。
セグメントSEP−1:4(GRFN1776、配列番号:2の残基117〜166からなる):
CAISPPDAAK AAPLRTITAD TFRKLFRVYS
NFLRGKLKLY TGEACRTGDR−カルボキシレート(Lys126はε−アミノ基においてレブリン酸残基で修飾され、Cys117はAcm保護されている)
セグメントSEP−1:1(GRFN1711、配列番号:2の残基1〜32からなる):
APPRLICDSR VLERYLLEAK EAEKITTGCA
EH−チオエステル(Lys24はレブリン酸残基で修飾されている)
【0166】
実施例1のとおり、セグメントSEP−1:1(GRFN1711)をチオエステル生成樹脂上で、セグメントSEP−1:4(GRFN1776)を−OCH2−Pam−樹脂上で合成した。これら2つのペプチドセグメントのリジン24および126を、初めにε−アミノ基においてFmoc基で保護した。鎖アセンブリの完了後、Fmocを有するアミノ基を標準的なFmoc脱保護手順に従って脱保護し、標準的にカップリングプロトコルに従って各ペプチド樹脂にそれぞれレブリン酸を結合することによって修飾した。次いで、実施例1に記載のBoc化学手順によって、これらのペプチドを脱保護し、同時に樹脂支持体から開裂した。ペプチドを、分取C4逆相HPLCによって個別に精製した。各ペプチドに関して、純粋ペプチドを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0167】
水溶性ポリマー(EDA−Succ)18カルボキシレート(GRFNP32)を、標準的なプロトコルに従って、Sasrinカルボキシル生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。水溶性ポリマー(EDA−Succ)6−アミド(GRFNP6)を、標準的なプロトコルに従って、Sieberアミド生成樹脂上でアセンブルした(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。5倍過剰の活性化Boc−アミノオキシ酢酸をカップリングすることによって、アミノオキシアセチル(AoA)部分を樹脂結合ポリマーそれぞれのN末端アミノ基に結合した。その2つのポリマー鎖を個々に、典型的なFmoc化学手順を用いてそれぞれの樹脂支持体から開裂した。各ポリマー鎖を、分取逆相HPLCによって精製した。各ポリマーに関して、純粋ポリマーを含有する分画をES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0168】
セグメントSEP−1:4およびGRFNP6を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP6を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0169】
セグメントSEP−1:1およびGRFNP32を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。次いで、その溶液を凍結乾燥した。その乾燥粉末を溶解し、分取勾配C4逆相HPLCによってポリマー修飾ペプチドを未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP32を含有する分画を、ES−MSによって同定し、プール、凍結乾燥した。
【0170】
B.合成赤血球産生刺激タンパク質SEP−1−L26の合成
SEP−1−L26を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP32(GRFN1711+GRFNP32、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP32にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP6(GRFN1776+GRFNP6、配列番号:2の残基117〜166に対応)
CAIS PPDAAKOXAAPL RTITADTFRK
LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP6にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システイン(すなわち、Cys161)はピコリル(pico)保護基を有する)
【0171】
追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を、実施例1および2に記載のとおり行い、完全長フォールドSEP−1−L26を得た。フォールドタンパク質生成物の分析C4逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、フォールドタンパク質の存在を実証した。
【0172】
実施例4
合成赤血球産生刺激タンパク質SEP−1−B50の合成
4番目の合成赤血球産生刺激タンパク質(SEP−1−B50とする)を合成した。完全長SEP−1−B50のアミノ酸配列は、SEP−1−L30の配列と同じであり、
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EHCSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPWΨP
LQLHVDKAVS GLRSLTTLLR ALGAQKΨAIS
PPDAAKOXAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:2)
配列中、Ψは、スルフヒドリル基においてカルボキシメチル化されているシステインからなる非天然アミノ酸残基を示し、KOXは、オキシム結合を介して指定の水溶性ポリマーにカップリングしたオキシムリンカー基でε−アミノ基において化学修飾されている非天然リジンを示す。
【0173】
しかしながら、このタンパク質は、SEP−1−L30の線状(Succ−EDA)18ポリマーではなく、4つの線状(Succ−EDA)12部分を有する分岐ポリマー構成体で誘導化した。誘導化は、オキシム結合によって達成した。完全長生成物のアセンブリは、実施例2に記載のとおりであった。
【0174】
A.複数のチオール基を有するテンプレートGRFNP17の合成
テンプレートGRFNP17を、0.4mmolスケールのアミド生成(4−メチル)ベンズヒドリルアミン(MBHA)樹脂上で手作業によって合成した。Fmoc−Lys(Boc)−OHを、標準的なカップリングプロトコルを用いてカップリングした(Schnolzer、M、Int.J.Pept.Protein Res.(1992)40:180〜93)。2.1mmolアミノ酸、0.5MのHBTU3.8ml中の10%DIEA、すなわち5倍過剰のアミノ酸を用いた。Fmoc保護基を除去した後、Fmoc−Lys(Fmoc)−0Hを、標準的なアミノ酸カップリングプロトコルを用いてカップリングした(2.1mmolアミノ酸、0.5MのHBTU3.8ml中の10%DIEA、すなわち5倍過剰のアミノ酸)。第2のFmoc除去ステップの後、Fmoc−Lys(Fmoc)−0Hを、標準的なアミノ酸カップリングプロトコルを用いてカップリングした(4.2mmolアミノ酸、0.5MのHBTU7.6ml中の10%DIEA、すなわち遊離アミンに対して5倍過剰のアミノ酸)。最後のFmoc脱保護ステップの後、5倍過剰(遊離アミンに対して)のDMF中S−アセチルチオグリコール酸ペンタフルオロフェニルエステル(SAMA−oPfp)を30分間カップリングした。C末端リシル残基の側鎖のBoc保護基を、純TFAで1分のバッチ洗浄を2回行うことによって除去し、次いでDMF中10%DIEAで洗浄することによって樹脂を中和した。2mmolのBoc−アミノオキシ酢酸、および2mmolのN−ヒドロキシスクシンイミド(NHS)を、DMF3mlに溶解した。2mmolのDIC(ジイソプロピルカルボジイミド)を添加した後、この酸を30〜60分間活性化した。その溶液を中和樹脂に添加し、1時間カップリングした。最後に、S結合アセチル基を、30分間DMF中の20%ピペリジンによって除去した。遊離アルデヒドのスカベンジャとしてシステインの存在下、標準的なBoc化学手順に従って、HF/p−クレゾールを用い、そのテンプレートを脱保護し、同時に樹脂支持体から開裂した(Schnolzer、M、Int.J.Pept.Protein Res.(1992)40:180〜93)。50%B(すなわち、0.1%TFAを含有する50%アセトニトリル水溶液)中の回収ポリアミド(アルデヒドを含まない)を、凍結し、凍結乾燥した。精製のために、そのテンプレート粗生成物を2mlの50%Bに溶解し、100mlの100%A(すなわち、0.1%TFA水溶液)を添加して試料を希釈した(アルデヒドの付加が確実であるため、塩化グアニジニウムまたはアセテートの添加は回避)。そのテンプレートを、3%B、T=40℃で平衡したC4分取逆相HPLCカラムに充填した。塩を定組成溶離し、所望のテンプレートGRFNP17を直線勾配で精製した。所望の生成物を含有する分画を、ES−MSによって同定し、プールし、凍結乾燥した。
【0175】
B.分岐水溶性ポリマーGRNP29の合成
分子量15kDaの分岐(EDA−Succ)12ポリマーGRFNP29を、精製チオール含有テンプレートGRFNP17、および線状ポリマーGRFNP31、Br−アセチル−(EDA−Succ)12カルボキシレートのチオエーテル生成ライゲーションによって合成したが、ここでGRFNP31は標準的なプロトコルに従ってSasrinカルボキシ生成樹脂上で合成した(Rose、K.他、米国特許出願第09/379297号;Rose等、J.Am.Chem.Soc.(1999)121:7034)。
【0176】
1.3xモル過剰(全チオールに関して)の精製GRFNP31、Br−アセチル化(EDA−Succ)12、および精製チオール含有テンプレートGRFNP17を合わせて、0.1Mトリス−HCl/6M塩化グアニジニウムpH8.7に濃度〜10mMで溶解した。溶解後、その溶液を0.1Mトリス−HCl、pH8.7緩衝液で3倍(v/v)に希釈した。ライゲーション混合物を室温で攪拌し、反応を逆相HPLCおよびES/MSでモニターした。追加のGRFNP31反応物を、所望の反応生成物が主生成物となるまで必要に応じて添加した。仕上げとして、3x(v/v、ライゲーション混合物に対して)の0.1M酢酸塩/6M塩化グアニジニウム、pH4を添加し、その溶液を分取C4逆相HPLCカラムに充填して、直線勾配で精製した。純粋GRFNP29構成体を含有する分画をES−MSを用いて同定し、プール、凍結乾燥した。
【0177】
C.GRFNP29によるGRFN1776およびGRFN1711のオキシム化
セグメントSEP−1:4およびセグメントSEP−1:1を、実施例2に記載のとおり合成した。セグメントSEP−1:4およびGRFNP29を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。その溶液を凍結乾燥した。その乾燥粉末を、分取逆相HPLCカラム(直径1インチ)に充填した。ポリマー修飾ペプチドを、分取勾配C4逆相HPLCによって非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:4+GP29を含有する分画を、ES−MSによって同定し、プールした。
【0178】
セグメントSEP−1:1およびGRFNP29を等モル比で、0.1%TFAを含有する50%アセトニトリル水溶液に、合わせて溶解した。その溶液を凍結および凍結乾燥した。その乾燥粉末を、0.1%TFAを含有する50%アセトニトリル水溶液に溶解し、分取GPC(ゲル透過クロマトグラフィ)カラム(直径1インチ)に充填した。ポリマー修飾ペプチドを、定組成溶離によって非修飾ペプチドおよび未反応ポリマーから分離した。所望のオキシム化生成物SEP−1:1+GP29を含有する分画を、ES−MSによって同定し、プールした。
【0179】
D.合成赤血球産生刺激タンパク質SEP−1−B50の合成
SEP−1−B50(配列番号:2)を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−1:1+GP29(GRFN1711+GRFNP29、配列番号:2の残基1〜32に対応):
APPRLICDSR VLERYLLEAK EAEKOXITTGCA
EH−チオエステル(Lys24はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP29にカップリングしているレブリン酸オキシムリンカー基で修飾されている)
セグメントSEP−1:2(GRFN1712、配列番号:2の残基33〜88に対応):
CSLNEKIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LVKSSQPW−チオエステル(Cys33はAcm保護されている)
セグメントSEP−1:3(GRFN1713、配列番号:2の残基89〜116に対応):
CP LQLHVDKAVS GLRSLTTLLR ALGAQK−チオエステル(Cys89はAcm保護されている)
セグメントSEP−1:4+GP29(GRFN1776+GRFNP29、配列番号:2の残基117〜166に対応):
CAIS PPDAAKOXAAPL RTITADTFRK
LFRVYSNFLR GKLKLYTGEA CRTGDR−カルボキシレート(Lys126はε−アミノ基において、KOXと示されるレブリン酸−アミノオキシアセチル(Lev−AoA)オキシム結合を介してGRFNP29にカップリングしているレブリン酸オキシムリンカー基で修飾されており、C末端システインはピコリル(pico)保護基を有する)
【0180】
精製をResourceQカラムで行ったことを除いて、追加のペプチドの合成、ライゲーション反応、カルボキシメチル化、保護基除去反応、フォールディング、および精製を実施例1および2に記載のとおり行い、完全長折りたたみSEP−1−B50(配列番号:2)を得た。折りたたみタンパク質生成物SEP−1−B50の分析C4逆相HPLCクロマトグラムおよびES−MSスペクトル、ならびにCDスペクトルは、折りたたみタンパク質の存在を実証した。
【0181】
実施例5
合成赤血球産生刺激タンパク質SEP−3−L42の合成
5番目の合成赤血球産生刺激タンパク質(SEP−3−L42とする)を合成した。完全長SEP−3タンパク質のアミノ酸配列は以下のとおりである。
APPRLICDSR VLERYLLEAK EAECITTGCA
EHCSLNECIT VPDTKVNFYA WKRMEVGQQA
VEVWQGLALL SEAVLRGQAL LACSSQPWEP
LQLHVDKAVS GLRSLTTLLR ALGAQKEAIS
PPDAACAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR (配列番号:3)
【0182】
24、38、83、および126位のシステイン残基を、マレイミド官能化(EDA−Succ)18(GRFNP32)ポリマー単位で修飾して(マイケル付加反応によって)、SEP−3−L42を形成した。
【0183】
詳細には、SEP−3を以下の4つのポリペプチドセグメントから溶液中で合成した。
セグメントSEP−3:1(GRFN1747、配列番号:3の残基1〜37に対応):
APPRLICDSR VLERYLLEAK EAECITTGCA
EHCSLNE−チオエステル(Cys7、Cys29、Cys33はAcm保護されている)
セグメントSEP−3:2(GRFN1774、配列番号:3の残基38〜82に対応):
CIT VPDTKVNFYA WKRMEVGQQA VEVWQGLALLSEAVLRGQAL LA−チオエステル(Cys38はAcm基で側鎖保護されている)
セグメントSEP−3:3(GRFN1749、配列番号:3の残基83〜125に対応):
CSSQPWEP LQLHVDKAVS GLRSLTTLLR
ALGAQKEAIS PPDAA−チオエステル(Cys83はAcm基で側鎖保護されている)
セグメントSEP−3:4(GRFN1750、配列番号:3の残基126〜166に対応):
CAAPL RTITADTFRK LFRVYSNFLR
GKLKLYTGEA CRTGDR−カルボキシレート(Cys161は、Pbo(すなわち、4−(CH3S(O)−)ベンジル−)保護されている)
【0184】
ペプチド合成: ペプチドSEP−3:1およびSEP−3:2およびSEP−3:3を、上述の特定のペプチドに示したように保護基方法を変更して、実施例1に記載の確立された側鎖保護法を用い、Boc化学SPPSのためのin situ中和プロトコルによってチオエステル生成樹脂上で合成した。セグメントSEP−3:4を、−OCH2−Pam−樹脂上で同様に合成した。これらのペプチドを、実施例1に記載のとおり脱保護し、同時に樹脂支持体から開裂した。上に記載の結果として生じたペプチドを、分取逆相HPLCによって精製した。純粋ペプチドを含有する分画を、ES−MSを用いて同定し、続くライゲーションのためにプールし、凍結乾燥した。
【0185】
ステップ1:ライゲーション#1 セグメントSEP−3:4およびセグメントSEP−3:3を、濃度15mMでTFEに溶解した。6M塩化グアニジニウムおよび1%チオフェノールを含有する飽和リン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。ライゲーション後、ライゲーション混合物(1体積とする)を、2体積の{TFE2ml、6M塩化グアニジニウム6ml、25%β−メルカプトエタノール含有100mMリン酸塩}溶液に添加し、20分間インキュベートした。その溶液を、15mg/mlのTCEP氷酢酸溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:Acm+SEP−3:3+SEP−3:4を含有する分画を、ES−MSによって同定し、プールした。
【0186】
ステップ2:Acm除去#1 Acm除去のために、プールしたSEP−3:Acm+SEP−3:3+SEP−3:4分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取C4逆相HPLCカラムに充填し、段階勾配で精製した。所望の生成物SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0187】
ステップ3:ライゲーション#2 等量のSEP−3:3+SEP−3:4およびSEP−3:2を合わせて、15mM濃度で純TFEに溶解した。6Mグアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物(1体積とする)を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6Mグアニジニウム20ml、pH4の2体積の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:Acm+SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0188】
ステップ4:Acm除去 Acm除去のために、プールしたSEP−3:Acm+SEP−3:2+SEP−3:3+SEP−3:4の分画を含有するアセトニトリル水溶液を、HPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、C4半分取逆相HPLCカラムに充填し、段階勾配で精製した。所望の結合生成物SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0189】
ステップ5:ライゲーション#3 等量のSEP−3:2+SEP−3:3+SEP−3:4およびSEP−3:1を合わせて、15mM濃度で純TFEに溶解した。6Mグアニジニウムおよび1%チオフェノールを含有する250mMのリン酸緩衝液(pH7.5)を添加し、ペプチドセグメントの透明溶液を得た。1日のライゲーション後、ライゲーション混合物(1体積とする)を、TFE10ml、β−メルカプトエタノール10ml、ピペリジン10ml、および6Mグアニジニウム20ml、pH4の2体積の溶液に添加し、20分間インキュベートして、残存する保護基を除去した。その溶液を、20%酢酸水溶液中の15mg/mlTCEP溶液で酸性にし、分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望の結合生成物SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0190】
ステップ6:ポリマーGRFNP32の結合 GRFNP32−マレイミドと称するマレイミド官能化線状(EDA−Succ)18ポリマーを、マレイミド官能化線状(EDA−Succ)18ポリマー(すなわち、マレイミド−(EDA−Succ)18)を形成するための製造業者プロトコルに従って、GRFNP32をBMPS(3−マレイミドプロピオン酸NHSエステル、Pierce、USA)で機能化することによって調製した。SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4を、必要最小量のTFEに溶解した。3倍過剰のGRFNP32−マレイミドを、6M塩化グアニジニウム、100mMリン酸塩、pH7.5に溶解し、TFE溶液に添加した。マイケル付加反応の進行を、分析逆相HPLCによって追跡した。反応が完了した後、その溶液を分取C4逆相HPLCカラムに充填し、直線勾配で精製した。所望のポリマー修飾生成物SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+pPEG(すなわち、Cys24、Cys38、Cys83、およびCys126の側鎖チオールに結合したGRFNP32の4つの複製を有する結合完全長166残基ポリペプチド鎖であり、したがってSEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32とも呼ばれる)を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0191】
ステップ7:Pbo除去 Pbo除去のために、SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32の凍結乾燥粉末を、5%エタンジチオールを含有する純TFAに溶解した。次いで、Pbo基を、10%チオアニソールおよび15%ブロモトリメチルシランを添加することによって30分間開裂した。その溶液を回転式エバポレータで乾燥し、0.1%TFAを含有するアセトニトリル水溶液に取った。結果として生じた溶液を、半分取逆相HPLCカラムに充填し、段階勾配によって精製した。所望のCys161脱保護生成物SEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32−Pboを含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0192】
ステップ8:Acm除去 Cys7、Cys29、およびCys33の側鎖からの最終Acm除去のために、プールしたSEP3:Acm+SEP−3:1+SEP−3:2+SEP−3:3+SEP−3:4+GP32−Pboの分画を含有するアセトニトリル水溶液をHPLCグレードの水で1x希釈し、最終濃度2モルで固形尿素を添加した。3倍モル過剰(予期される全システイン濃度に対して)の3%酢酸水溶液中30mg/ml(酢酸)2Hg溶液を添加し、その溶液を1時間攪拌した。次いで、その溶液をβ−メルカプトエタノール中で20%とし、半分取C4逆相HPLCカラムに充填し、段階勾配で精製した。所望の結合ポリマー修飾生成物SEP−3(1〜166)を含有する分画をES−MSによって同定し、一晩凍結乾燥した。
【0193】
ステップ9:フォールディング 完全長結合ポリマー修飾ペプチドSEP−3(1〜166)を、6M塩化グアニジニウム、20%TFE、10倍モル過剰(SEP−3のCys残基に対して)のシステインを含有する200mMトリス緩衝液(pH8.7)に溶解した。その溶液を、3M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、室温で一晩透析した。次いでその溶液を、1M塩化グアニジニウムを含有する200mMトリス緩衝液(pH8.7)溶液に対して、4℃で4時間透析し、最後に10mMリン酸緩衝液(pH7.0)に対して、4℃で4時間透析して、最終フォールド生成物を得た。フォールディングは、エレクトロスプレーES−MSおよびCD分光法によって確認した。
【0194】
ステップ10:精製 折りたたまれたポリペプチドを、セントリコン濃縮バイアルで5x濃縮し、10mMリン酸塩、pH7.0で平衡させたResource Sカチオン交換カラムに充填した。折りたたまれたタンパク質を、500mMのNaClへの10分の直線塩勾配で溶出した。所望の折りたたまれた生成物SEP−3−L42を含有する分画をSDS−PAGEによって同定、凍結し、−80℃で貯蔵した。
【0195】
実施例6
合成赤血球産生刺激タンパク質の生物活性アッセイ
フォールド合成赤血球産生刺激タンパク質、SEP−0、SEP−1−L26、SEP−1−L30、SEP−1−B50、およびSEP−3−L42の生物活性を、コントロール標準として市販の組換えエリスロポエチンを用いる因子依存細胞系増殖アッセイにおいて、UT/7および32D103197細胞系を用い判定した。UT−7は、増殖および生存に関してインターロイキン−3、顆粒球マクロファージコロニー刺激因子(GM−CSF)、またはエリスロポエチン(EPO)の1つに絶対依存性を有するヒト巨核芽球白血病細胞系である(Miura Y.等、Acta Haematol(1998)99:180〜184;Komatsu N.等、Cancer Res.(1991)51:341〜8)。32D103197は、マウス造血細胞系である(Metcalf、D.Int J Cell Cloning(1992)10:116〜25)。
【0196】
イスコフ改変ダルベッコ培地(IMDM)、10%FBS(ウシ胎児血清)、グルタミン、およびPenstrep中でSEP構成体の保存溶液を作製し、その保存溶液の連続2x希釈液をマルチウェルプレートに添加し、5000細胞/50μlの濃度でヒトUT/7EPO細胞を加えた。プレートを5%CO2の存在下、37℃でインキュベートし、増殖を毎日モニターした。4日後、PBS(リン酸緩衝化食塩水)中の2.5mg/mlMTT(メチルチアゾールテトラゾリウム)20μlを添加し、プレートを4時間インキュベートした。IPA150μlを加え、各ウェルの吸光度を562nmで読み取った。SEP化合物のED50(最大効果の50%に達する有効量)値を求め、CHO(Chineseハムスター卵巣)細胞産生rhEPO(組換えヒトエリスロポエチン)の値と比較した。これらの実験の結果は、すべての合成赤血球産生刺激タンパク質が生物活性を示すことを実証した。SEP−0、SEP−1−L26、SEP−1−L30、およびSEP−1−B50のED50の結果を表VIに示す。
【表9】
【0197】
本発明をその特定の実施形態に関して述べたが、さらなる変更が可能であり、本出願が、一般に本発明の原理に従い、本発明の属する技術分野において知られ、慣例的に行われている範囲内であって、且つ本明細書に記載の本質的な特徴に適用できるような本発明の開示からの逸脱を含む本発明の変形、使用、または適応を含むものであることは理解されるであろう。
【図面の簡単な説明】
【0198】
【図1】擬似天然型化学的ライゲーションの全体的な反応を例示する図である。
【図2】本発明の合成生物活性タンパク質を調製する全体的なプロセスを例示する図である。
【図3】合成赤血球産生刺激タンパク質の好ましい型の基本構造を示す図である。
【図4】遊離アミノ末端またはカルボキシ末端を持たない環状順列化SEP類似体の一般的な構造を示す図である。pPEG(本明細書に記載の「精密PEG」)結合部位は、SEP類似体と位置的に同一である。SEP類似体は、P126CおよびA125Xなどの、新しいN/C末端位置間のオキシム結合によって環化される。SEP類似体の全分子量は、所与の類似体の修飾に用いられるpPEGに応じて約50〜80kDaの範囲となり、pPEGを介した流体力学分子量は約100kDaと推定される。
【図5】合成生物活性GCSFタンパク質の基本構造を示す図である。この図において、「J」は、疎水性側鎖を有する非天然コード化残基を示す。
【図6】好ましい合成RANTES類似体の構造を示す図である。この図において、NNY=ノナノイル、X=疎水性側鎖pPEGを有する非天然コード化アミノ酸、FA=脂肪酸である。
【図7】分岐鎖pPEGポリマーの形成を図式的に示す図である。
【特許請求の範囲】
【請求項1】
側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質。
【請求項2】
当該タンパク質は、リボソーム特定生物活性タンパク質の持つ生物活性を有する、請求項1に記載の合成タンパク質。
【請求項3】
当該合成タンパク質は、約25kDaを超えるモノマー分子量を有する、請求項2に記載の合成タンパク質。
【請求項4】
当該タンパク質の複数のアミノ酸残基の少なくとも1つは、隣接アミノ酸残基に非アミド結合によって結合している、請求項1に記載の合成タンパク質。
【請求項5】
当該非アミド結合は、チオエステル結合、チオエーテル結合およびオキシム結合からなる群から選択される、請求項4に記載の合成タンパク質。
【請求項6】
リボソーム産生生物活性タンパク質は、哺乳動物のタンパク質である、請求項2に記載の合成タンパク質。
【請求項7】
当該哺乳動物のタンパク質は、ヒト、サル、ウシ、マウス、ブタ、ヒツジおよびウマの各タンパク質からなる群から選択される、請求項6に記載の合成タンパク質。
【請求項8】
当該哺乳動物のタンパク質は、ヒトのタンパク質である、請求項7に記載の合成タンパク質。
【請求項9】
当該タンパク質は、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインからなる群から選択される、請求項1から8のいずれか1つに記載の合成タンパク質。
【請求項10】
当該合成生物活性タンパク質は、サイトカインの生物活性を有する、請求項1に記載の合成タンパク質。
【請求項11】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項10に記載の合成タンパク質。
【請求項12】
当該サイトカインは、赤血球産生刺激タンパク質である、請求項11に記載の合成タンパク質。
【請求項13】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項12に記載の合成タンパク質。
【請求項14】
当該合成生物活性タンパク質は、SEP−0、SEP−1およびSEP−3からなる群から選択される、請求項12に記載の合成タンパク質。
【請求項15】
当該サイトカインは、RANTESタンパク質である、請求項10に記載の合成タンパク質。
【請求項16】
当該サイトカインは、増殖因子である、請求項10に記載の合成タンパク質。
【請求項17】
当該増殖因子は、G−CSFである、請求項16に記載の合成タンパク質。
【請求項18】
当該リボソーム特定生物活性タンパク質は、1つまたは複数のグリコシル化部位でグリコシル化されている、請求項2に記載の合成タンパク質。
【請求項19】
薬剤組成物を必要としている人の治療の為の薬剤組成物の製造における、請求項1に記載の化合物の使用方法。
【請求項20】
当該合成タンパク質は、約25kDaを超えるモノマー分子量を有する、請求項19に記載の使用方法。
【請求項21】
当該リボソーム特定生物活性タンパク質は、ヒトのタンパク質である、請求項19に記載の使用方法。
【請求項22】
当該タンパク質は、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインの生物活性を有する、請求項19に記載の使用方法。
【請求項23】
当該合成タンパク質は、サイトカインの生物活性を有する、請求項22に記載の使用方法。
【請求項24】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項23に記載の使用方法。
【請求項25】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項24に記載の使用方法。
【請求項26】
当該合成生物活性タンパク質は、SEP−0、SEP−1およびSEP−3からなる群から選択される、請求項25に記載の使用方法。
【請求項27】
当該サイトカインは、RANTESタンパク質である、請求項24に記載の使用方法。
【請求項28】
当該サイトカインは、増殖因子である、請求項24に記載の使用方法。
【請求項29】
当該増殖因子は、G−CSFである、請求項28に記載の使用方法。
【請求項30】
以下の式の所望のポリペプチドを合成する方法であって、
aaNH2−Q−aax−aay−W−aaCOOH
該式中、QおよびWは、それぞれ任意で存在する1つまたは複数の追加のアミノ酸残基を示し、aaNH2は、ポリペプチドのN末端アミノ酸残基を示し、aaxおよびaayは、それぞれ側鎖xおよびyを有する内部隣接アミノ酸残基を示し、aaCOOHは、ポリペプチドのC末端アミノ酸残基を示し、
該方法は、
(A)式aaNH2−Q−aax−COSRを有し、該式中、Rは、アリール、ベンジルおよびアルキル基を非限定的に含むチオエステル基に適合する任意の基である第1のペプチドを、式Cys−W−aaCOOHを有する第2のペプチドに結合し、それによりポリペプチドaaNH2−Q−aax−Cys−W−aaCOOHを形成すること、および
(B)該ポリペプチドを試薬Raa−Xの存在下でインキュベートすることを含むものであって、該式中、Raaは、CH2−COOH、(CH2)−C(O)NH2、CH2OH、CHOHCH3、CH2CHOHCH3、CH2CH2OH、(CH2)2NH−PG、CH2NH−C(NH2)2、(CH2)2NH−C(NH2)2、CH2CH3、φ、CH2φ、φ−OH、CH2φ−OH、CH2φ−OPO3、CH2φ−(OH)2、CH(COOH)2、CH2−IM−PG、CH2−IN、CH−(CH3)2、CH2CH−(CH3)2、CH(CH3)−CH2−CH3、CH2−CH(CH3)−CH2−CH3、(CH2)2NH−C(O)CF3、アンモニウム4−クロロ−7−スルホベンゾ−フラザン、CH2−アンモニウム4−クロロ−7−スルホベンゾ−フラザン、からなる郡から選択される基であり、
Xは、良好な離脱基であり、
該インキュベーションは、所望のポリペプチドを形成するのに充分な条件下で行われる方法。
【請求項31】
Xは、ハロゲンである、請求項30に記載の方法。
【請求項32】
当該ハロゲンは、F、IまたはBrである、請求項31に記載の方法。
【請求項33】
当該アミノ酸aayは、擬似アミノ酸である、請求項30に記載の方法。
【請求項34】
当該ポリペプチドは、リボソーム産生生物活性タンパク質により所有されている生物活性を有する、請求項30に記載の方法。
【請求項35】
当該ポリペプチドは、約25kDaを超えるモノマー分子量を有する、請求項30に記載の方法。
【請求項36】
当該ポリペプチドは、複数のアミノ酸残基から成り、該アミノ酸残基の少なくとも1つが、チオエステル結合、チオエーテル結合およびオキシム結合からなる群から選択される結合により、隣接アミノ酸残基に結合される、請求項30に記載の方法。
【請求項37】
当該リボソーム産生生物活性タンパク質は、哺乳動物のタンパク質である、請求項34に記載の方法。
【請求項38】
当該哺乳動物のタンパク質は、ヒト、サル、ウシ、マウス、ブタ、ヒツジおよびウマの各タンパク質からなる群から選択される、請求項37に記載の方法。
【請求項39】
当該ポリペプチドは、サイトカインである、請求項30から38のいずれか1つに記載の方法。
【請求項40】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項39に記載の方法。
【請求項41】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項40に記載の方法。
【請求項42】
当該ポリペプチドは、1つまたは複数のグリコシル化部位でグリコシル化されている、請求項30に記載の方法。
【請求項1】
側鎖が式−S−Raaを有し、該式中、Raaは、表Iの擬似Asp、擬似Gln、擬似Asn、擬似Ser、擬似Thr、擬似Lys、擬似Arg、擬似Met、擬似Phe、擬似Tyr、擬似ホスホtyr、擬似Dopa、擬似Gla、擬似His、擬似Trp、擬似Leu、擬似Val、擬似Ile、擬似N−(エチル)トリフルオロアセトアミド、擬似SBF、及び擬似メチルSBF、から成る群より選択される擬似アミノ酸残基を含有する合成タンパク質。
【請求項2】
当該タンパク質は、リボソーム特定生物活性タンパク質の持つ生物活性を有する、請求項1に記載の合成タンパク質。
【請求項3】
当該合成タンパク質は、約25kDaを超えるモノマー分子量を有する、請求項2に記載の合成タンパク質。
【請求項4】
当該タンパク質の複数のアミノ酸残基の少なくとも1つは、隣接アミノ酸残基に非アミド結合によって結合している、請求項1に記載の合成タンパク質。
【請求項5】
当該非アミド結合は、チオエステル結合、チオエーテル結合およびオキシム結合からなる群から選択される、請求項4に記載の合成タンパク質。
【請求項6】
リボソーム産生生物活性タンパク質は、哺乳動物のタンパク質である、請求項2に記載の合成タンパク質。
【請求項7】
当該哺乳動物のタンパク質は、ヒト、サル、ウシ、マウス、ブタ、ヒツジおよびウマの各タンパク質からなる群から選択される、請求項6に記載の合成タンパク質。
【請求項8】
当該哺乳動物のタンパク質は、ヒトのタンパク質である、請求項7に記載の合成タンパク質。
【請求項9】
当該タンパク質は、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインからなる群から選択される、請求項1から8のいずれか1つに記載の合成タンパク質。
【請求項10】
当該合成生物活性タンパク質は、サイトカインの生物活性を有する、請求項1に記載の合成タンパク質。
【請求項11】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項10に記載の合成タンパク質。
【請求項12】
当該サイトカインは、赤血球産生刺激タンパク質である、請求項11に記載の合成タンパク質。
【請求項13】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項12に記載の合成タンパク質。
【請求項14】
当該合成生物活性タンパク質は、SEP−0、SEP−1およびSEP−3からなる群から選択される、請求項12に記載の合成タンパク質。
【請求項15】
当該サイトカインは、RANTESタンパク質である、請求項10に記載の合成タンパク質。
【請求項16】
当該サイトカインは、増殖因子である、請求項10に記載の合成タンパク質。
【請求項17】
当該増殖因子は、G−CSFである、請求項16に記載の合成タンパク質。
【請求項18】
当該リボソーム特定生物活性タンパク質は、1つまたは複数のグリコシル化部位でグリコシル化されている、請求項2に記載の合成タンパク質。
【請求項19】
薬剤組成物を必要としている人の治療の為の薬剤組成物の製造における、請求項1に記載の化合物の使用方法。
【請求項20】
当該合成タンパク質は、約25kDaを超えるモノマー分子量を有する、請求項19に記載の使用方法。
【請求項21】
当該リボソーム特定生物活性タンパク質は、ヒトのタンパク質である、請求項19に記載の使用方法。
【請求項22】
当該タンパク質は、タンパク質受容体もしくはそのフラグメント、タンパク質受容体リガンドもしくはそのフラグメント、またはサイトカインの生物活性を有する、請求項19に記載の使用方法。
【請求項23】
当該合成タンパク質は、サイトカインの生物活性を有する、請求項22に記載の使用方法。
【請求項24】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項23に記載の使用方法。
【請求項25】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項24に記載の使用方法。
【請求項26】
当該合成生物活性タンパク質は、SEP−0、SEP−1およびSEP−3からなる群から選択される、請求項25に記載の使用方法。
【請求項27】
当該サイトカインは、RANTESタンパク質である、請求項24に記載の使用方法。
【請求項28】
当該サイトカインは、増殖因子である、請求項24に記載の使用方法。
【請求項29】
当該増殖因子は、G−CSFである、請求項28に記載の使用方法。
【請求項30】
以下の式の所望のポリペプチドを合成する方法であって、
aaNH2−Q−aax−aay−W−aaCOOH
該式中、QおよびWは、それぞれ任意で存在する1つまたは複数の追加のアミノ酸残基を示し、aaNH2は、ポリペプチドのN末端アミノ酸残基を示し、aaxおよびaayは、それぞれ側鎖xおよびyを有する内部隣接アミノ酸残基を示し、aaCOOHは、ポリペプチドのC末端アミノ酸残基を示し、
該方法は、
(A)式aaNH2−Q−aax−COSRを有し、該式中、Rは、アリール、ベンジルおよびアルキル基を非限定的に含むチオエステル基に適合する任意の基である第1のペプチドを、式Cys−W−aaCOOHを有する第2のペプチドに結合し、それによりポリペプチドaaNH2−Q−aax−Cys−W−aaCOOHを形成すること、および
(B)該ポリペプチドを試薬Raa−Xの存在下でインキュベートすることを含むものであって、該式中、Raaは、CH2−COOH、(CH2)−C(O)NH2、CH2OH、CHOHCH3、CH2CHOHCH3、CH2CH2OH、(CH2)2NH−PG、CH2NH−C(NH2)2、(CH2)2NH−C(NH2)2、CH2CH3、φ、CH2φ、φ−OH、CH2φ−OH、CH2φ−OPO3、CH2φ−(OH)2、CH(COOH)2、CH2−IM−PG、CH2−IN、CH−(CH3)2、CH2CH−(CH3)2、CH(CH3)−CH2−CH3、CH2−CH(CH3)−CH2−CH3、(CH2)2NH−C(O)CF3、アンモニウム4−クロロ−7−スルホベンゾ−フラザン、CH2−アンモニウム4−クロロ−7−スルホベンゾ−フラザン、からなる郡から選択される基であり、
Xは、良好な離脱基であり、
該インキュベーションは、所望のポリペプチドを形成するのに充分な条件下で行われる方法。
【請求項31】
Xは、ハロゲンである、請求項30に記載の方法。
【請求項32】
当該ハロゲンは、F、IまたはBrである、請求項31に記載の方法。
【請求項33】
当該アミノ酸aayは、擬似アミノ酸である、請求項30に記載の方法。
【請求項34】
当該ポリペプチドは、リボソーム産生生物活性タンパク質により所有されている生物活性を有する、請求項30に記載の方法。
【請求項35】
当該ポリペプチドは、約25kDaを超えるモノマー分子量を有する、請求項30に記載の方法。
【請求項36】
当該ポリペプチドは、複数のアミノ酸残基から成り、該アミノ酸残基の少なくとも1つが、チオエステル結合、チオエーテル結合およびオキシム結合からなる群から選択される結合により、隣接アミノ酸残基に結合される、請求項30に記載の方法。
【請求項37】
当該リボソーム産生生物活性タンパク質は、哺乳動物のタンパク質である、請求項34に記載の方法。
【請求項38】
当該哺乳動物のタンパク質は、ヒト、サル、ウシ、マウス、ブタ、ヒツジおよびウマの各タンパク質からなる群から選択される、請求項37に記載の方法。
【請求項39】
当該ポリペプチドは、サイトカインである、請求項30から38のいずれか1つに記載の方法。
【請求項40】
当該サイトカインは、インターロイキン、リンホカイン、RANTESタンパク質、赤血球産生刺激タンパク質、腫瘍壊死因子(TNF)、インターフェロン、増殖因子および単一ペプチドホルモンからなる群から選択される、請求項39に記載の方法。
【請求項41】
当該赤血球産生刺激タンパク質は、エリスロポエチンである、請求項40に記載の方法。
【請求項42】
当該ポリペプチドは、1つまたは複数のグリコシル化部位でグリコシル化されている、請求項30に記載の方法。
【図1】

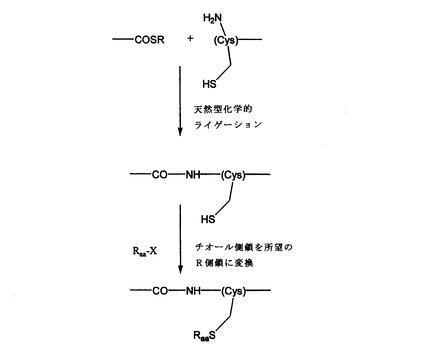
【図2】
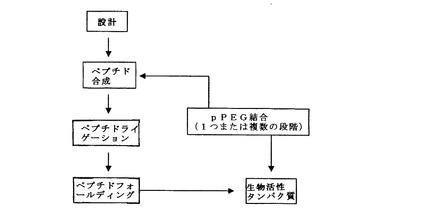
【図3】
【図4】
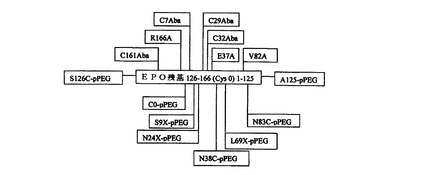
【図5】
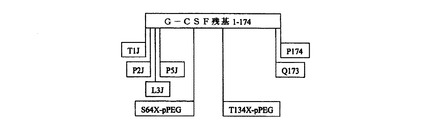
【図6】
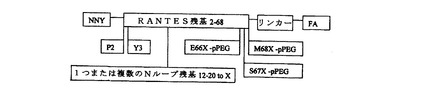
【図7】
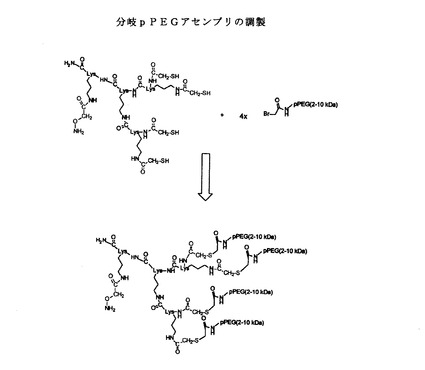
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公開番号】特開2007−269799(P2007−269799A)
【公開日】平成19年10月18日(2007.10.18)
【国際特許分類】
【出願番号】特願2007−125104(P2007−125104)
【出願日】平成19年5月9日(2007.5.9)
【分割の表示】特願2002−524517(P2002−524517)の分割
【原出願日】平成13年7月12日(2001.7.12)
【出願人】(501084271)グリフォン セラピューティクス,インコーポレーテッド (4)
【Fターム(参考)】
【公開日】平成19年10月18日(2007.10.18)
【国際特許分類】
【出願日】平成19年5月9日(2007.5.9)
【分割の表示】特願2002−524517(P2002−524517)の分割
【原出願日】平成13年7月12日(2001.7.12)
【出願人】(501084271)グリフォン セラピューティクス,インコーポレーテッド (4)
【Fターム(参考)】
[ Back to top ]