
び慢性大脳白質形成不全症患者又は保因者の検出方法
【課題】び慢性大脳白質形成不全症(HCAHC)の新規原因遺伝子を同定し、より多くの症例を適切に診断できる手段を提供すること。
【解決手段】血縁関係のない3家系に由来するHCAHC患者3名を対象に、次世代シークエンサーを用いて全エキソーム配列解析を行ない、膨大なリード配列を鋭意解析した結果、低分子非コードRNAの転写に関与するRNAポリメラーゼIIIのサブユニットをコードするPOLR3B遺伝子及びPOLR3A遺伝子の変異がHCAHCの病因変異であることが判明した。HCAHCは常染色体劣性疾患であり、患者は変異をホモ又は複合ヘテロで有する。
【解決手段】血縁関係のない3家系に由来するHCAHC患者3名を対象に、次世代シークエンサーを用いて全エキソーム配列解析を行ない、膨大なリード配列を鋭意解析した結果、低分子非コードRNAの転写に関与するRNAポリメラーゼIIIのサブユニットをコードするPOLR3B遺伝子及びPOLR3A遺伝子の変異がHCAHCの病因変異であることが判明した。HCAHCは常染色体劣性疾患であり、患者は変異をホモ又は複合ヘテロで有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、び慢性大脳白質形成不全症患者又は保因者の検出方法に関する。
【背景技術】
【0002】
先天性ミエリン形成不全症は、白質ジストロフィーに分類される多様な疾患群であり、ミエリン形成異常を特徴とする。これらの病態は脳のMRIにより認識することができるが、正しく診断されない症例が多い(非特許文献1)。これまでに、歯数不足症及び低ゴナドトロピン性性腺機能低下症を伴うミエリン形成不全(4H)症候群(MIM 612440)、大脳基底核及び小脳の萎縮を伴うミエリン形成不全(H-ABC)(MIM 612438)等の、ミエリン形成に影響する新たな症候群が数種報告されている(非特許文献2〜5)。
【0003】
小脳委縮と脳梁低形成を伴うび慢性の大脳白質形成不全症(cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum; HCAHC)は、近年報告された新たなミエリン形成不全症候群であり、び慢性のミエリン形成不全と小脳及び脳梁の低形成を特徴とする(非特許文献6)。HCAHC、4H症候群、及びH-ABCの3症候群には、び慢性のミエリン形成不全、小脳及び脳梁の委縮ないしは低形成という共通した特徴が認められるが、HCAHCでは4H症候群及びH-ABCにおいて見られる歯数不足や大脳基底核の萎縮が認められない。
【0004】
び慢性大脳白質形成不全症の原因遺伝子としては、4つの遺伝子(PLP1, MBP, GJC2, FAM126)の関与が知られていたが、これら遺伝子の変異で説明ができない症例が数多く残されていた。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Schiffmann, R., and van der Knaap, M.S. (2009). Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750-759.
【非特許文献2】Timmons, M., Tsokos, M., Asab, M.A., Seminara, S.B., Zirzow, G.C., Kaneski, C.R., Heiss, J.D., van der Knaap, M.S., Vanier, M.T., Schiffmann, R., et al. (2006). Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 67, 2066-2069.
【非特許文献3】Wolf, N.I., Harting, I., Boltshauser, E., Wiegand, G., Koch, M.J., Schmitt-Mechelke, T., Martin, E., Zschocke, J., Uhlenberg, B., Hoffmann, G.F., et al. (2005). Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 64, 1461-1464.
【非特許文献4】Wolf, N.I., Harting, I., Innes, A.M., Patzer, S., Zeitler, P., Schneider, A., Wolff, A., Baier, K., Zschocke, J., Ebinger, F., et al. (2007). Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics 38, 64-70.
【非特許文献5】van der Knaap, M.S., Naidu, S., Pouwels, P.J., Bonavita, S., van Coster, R., Lagae, L., Sperner, J., Surtees, R., Schiffmann, R., and Valk, J. (2002). New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 23, 1466-1474.
【非特許文献6】Sasaki, M., Takanashi, J., Tada, H., Sakuma, H., Furushima, W., and Sato, N. (2009). Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 31, 582-587.
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、び慢性大脳白質形成不全症の新規原因遺伝子を同定し、より多くの症例を適切に診断できる手段を提供することにある。
【課題を解決するための手段】
【0007】
本願発明者らは、血縁関係のない3家系に由来するHCAHC患者3名を対象に、次世代シークエンサーを用いて全エキソーム配列解析を行ない、膨大なリード配列を鋭意解析した結果、低分子非コードRNAの転写に関与するRNAポリメラーゼIIIのサブユニットをコードするPOLR3B遺伝子及びPOLR3A遺伝子において、数百例の健常コントロールサンプルには認められず、SNPデータベース等にも登録がないまれな新規変異が複合ヘテロ接合で存在することを見出し、本願発明を完成した。
【0008】
すなわち、本発明は、被検者から分離した試料を用いて、該被検者がPOLR3B遺伝子及びPOLR3A遺伝子のうちの少なくともいずれかに変異を有するか否かを調べることを含む、び慢性大脳白質形成不全症患者又は保因者の検出方法であって、ホモ接合又は複合ヘテロ接合の前記変異が検出された場合に患者が検出され、ヘテロ接合の前記変異が検出された場合に保因者が検出される、方法を提供する。また、本発明は、低分子非コードRNAの転写の促進を指標として化合物を選択することを含む、び慢性大脳白質形成不全症の治療薬のスクリーニング方法を提供する。
【発明の効果】
【0009】
本発明により、HCAHCの新規な原因遺伝子が同定された。これにより、該疾患への関与が既に知られている4遺伝子に変異が認められない症例の多くを診断可能になる。HCAHCの原因遺伝子として同定されたPOLR3B遺伝子及びPOLR3A遺伝子は、低分子非コードRNAの転写を行なっている遺伝子であり、変異により転写が低下することでHCAHCが発症すると考えられる。低分子非コードRNAの転写を促進する薬剤をスクリーニングすることでHCAHCの治療薬の創薬が期待される。
【図面の簡単な説明】
【0010】
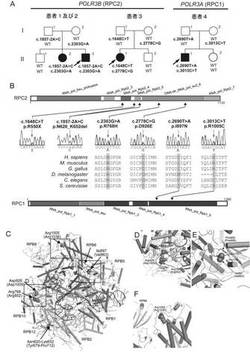
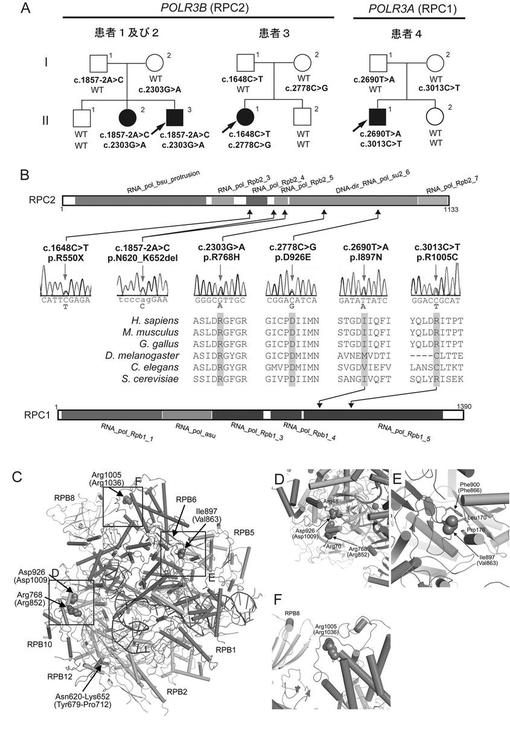
【図1】実施例で同定されたPOLR3B遺伝子及びPOLR3A遺伝子中の変異を説明する図である。(A) HCAHC患者を生じた3家系の家系図である。血縁関係のない2家系由来の患者3名において、RPC2をコードするPOLR3B遺伝子中に4種の変異を同定した。また、1家系において、RPC1をコードするPOLR3A遺伝子中に2種の変異を同定した。各変異の分離を併せて図中に示す。(B) Pfamドメイン(データベースEnsembl [http://uswest.ensembl.org/index.html]より)を有するRPC2タンパク質(上段)及びRPC1タンパク質(下段)の略図である。アミノ酸を改変する各変異の位置を波形データと共に示す。ミスセンス変異は全て進化的に保存されたアミノ酸で生じていた。CLUSTALWを用いて相同配列を整列化した。(C-F) RPC1及びRPC2の変異箇所の3次元表示である。RPC1及びRPC2中の変異アミノ酸を、RNAポリメラーゼIIの相同なサブユニットRPB1及びRPB2において相当する位置を付記して示した(括弧書きのアミノ酸と数値)。変異の構造と位置は結晶構造(PDB ID 3GTP)を用いてPyMOLにより描写した。RNAポリメラーゼIIに特異的なサブユニットであるRPB3, RPB9及びRPB11は図中では省略した。変異アミノ酸と相互作用するアミノ酸も示した。
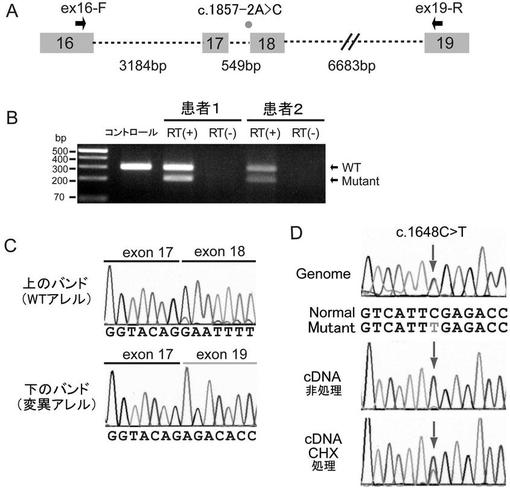
【図2】POLR3B遺伝子中のスプライスサイト変異及びナンセンス変異の影響を調べた結果を示す図である。(A) POLR3B遺伝子のエクソン16〜19のゲノム構造を示す略図である。エクソン、イントロン及びプライマーをそれぞれボックス、破線及び矢印で示す。イントロン17中のc.1857-2A>C変異を点で記す。(B) c.1857-2A>C変異を有する患者1及び患者2並びにコントロール健常者のRT-PCR解析の結果である。患者のcDNAでは2種のPCR産物が検出された。上のバンドが野生型(WT)転写物であり、下のバンドが変異転写物である。コントロールではWTの単一バンドのみが検出された。(C) WT転写物及び変異転写物の配列解析の結果である。変異アレルではエクソン18の読み飛ばしが明らかに確認された。(D) c.1648C>T変異の解析結果である。上段;ゲノムDNAから増幅したPCR産物の配列、中段;CHX非処理細胞のcDNAから増幅したPCR産物の配列、下段;CHX処理細胞のcDNAから増幅したPCR産物の配列。非処理細胞ではc.1648C>T変異アレルの発現が極めて低いのに対し、CHX処理でナンセンス変異依存mRNA分解(NMD)を阻害した処理細胞では変異アレルの発現が有意に増大していた。
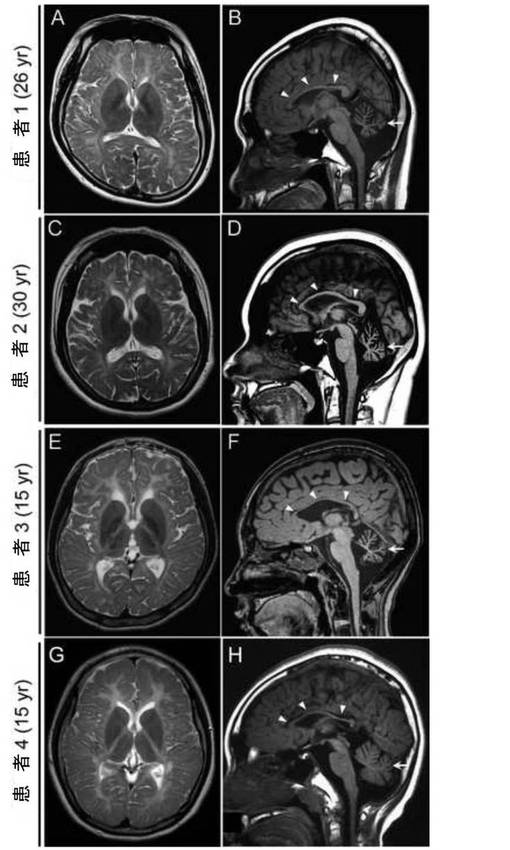
【図3】POLR3B変異又はPOLR3A変異を有する患者の脳MRI像である。(A, C, E, F, G) 大脳基底核を通るT2強調横断像である。全ての患者で白質中に高強度領域を認めた。 (B, D, F, H) T1強調正中矢状断像である。全ての患者で脳梁低形成(矢頭)と小脳の委縮(矢印)を認めた。
【発明を実施するための形態】
【0011】
本発明が対象とするび慢性大脳白質形成不全症とは、非特許文献6で報告されている、小脳委縮と脳梁低形成を伴うび慢性の大脳白質形成不全を特徴とする新規な先天性ミエリン形成不全症である。本明細書では該疾患を「HCAHC」と呼ぶことがある。
【0012】
POLR3B(GenBankアクセッション番号NM_018082)及びPOLR3A(GenBankアクセッション番号NM_007055)は、RNAポリメラーゼIIIのサブユニットRPC2及びRPC1をそれぞれコードする遺伝子である。RNAポリメラーゼIIIは、17のサブユニットから成り、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)等の低分子非コードRNAの転写に関与している。配列表の配列番号1及び2に示す配列は、POLR3B遺伝子のコード領域のcDNA配列及びアミノ酸配列であり、配列番号3及び4は、POLR3A遺伝子のコード領域のcDNA配列及びアミノ酸配列である。なお、配列番号48及び49は、GenBankに上記の番号で登録されているPOLR3B及びPOLR3AのmRNA配列である。配列番号5〜47には、POLR3B及びPOLR3Aの各エクソン及びその近傍のイントロンの塩基配列を下記表1、2の通りに示した。
【0013】
【表1】

【0014】
【表2】
【0015】
本願発明者らにより、POLR3B遺伝子又はPOLR3A遺伝子がHCAHCの原因遺伝子であることが判明した。HCAHC患者は、POLR3B遺伝子又はPOLR3A遺伝子中に変異をホモ又は複合ヘテロで有し、HCAHC保因者は該変異をヘテロで有する。複合ヘテロとは、POLR3B遺伝子又はPOLR3A遺伝子の両アリルにそれぞれ異なる変異を有することを意味する。
【0016】
ここでいう変異には、POLR3B遺伝子がコードするRPC2又はPOLR3AがコードするRPC1のごく少数のアミノ酸の変化のほか、これらサブユニットの少なくとも一部の領域を欠失するような変化をもたらす塩基配列の変異が包含される。そのような塩基配列の変異は、例えば、エクソン又はイントロン領域内での塩基の置換、欠失、挿入、重複等によるミスセンス変異、ナンセンス変異、フレームシフト変異、アミノ酸欠失変異、スプライシング異常変異等であり得る。
【0017】
配列表の配列番号2、4に示された野生型のアミノ酸配列からなるタンパク質は、正常なサブユニットタンパク質(RPC2、RPC1)の典型例である。本発明では、変異の有無は、配列番号1〜4に示されたPOLR3B遺伝子及びPOLR3A遺伝子の配列を基準にして判断され得る。アミノ酸配列の変化を生じるPOLR3B又はPOLR3A遺伝子の変異は、HCAHCの病因変異であると考えることができるが、中でも、進化的に保存性の高いアミノ酸を変化させる遺伝子変異は、正常な機能が損なわれたサブユニットタンパク質を生じる蓋然性が高く、HCAHC病因変異の典型例である。あるいはまた、検出された塩基の変異が、NCBIのdbSNP等の塩基配列の多様性に関する周知のデータベースには登録されていないまれな塩基変異の場合も、HCAHC病因変異と考えてよい。病因変異は、POLR3B又はPOLR3Aにコードされるサブユニットタンパク質それ自体の立体構造や、サブユニット間の相互作用に影響すると考えられる。
【0018】
下記表3の変異は、実施例において血縁関係のない3家系から同定されたHCAHC病因変異である。これらの変異は、dbSNP等の塩基配列の多様性に関するデータベースに登録されておらず、また健常者集団においてもほとんど認められないまれな変異であった。POLR3B遺伝子の変異4種のうちのいずれか2つを有する場合、HCAHC患者と判断することができる。もっとも、当該3家系以外の家系には当然ながら異なる変異が存在し得るので、本発明の範囲はこれらの具体例に限定されるものではない。
【0019】
【表3】
【0020】
POLR3B又はPOLR3A遺伝子の変異は、ゲノムDNAやRNA等の核酸試料を用いて塩基配列を解析することで検出可能である。とりわけ、ゲノムDNA試料を用いてゲノム配列の解析を行なうことが最も確実で望ましい。ゲノムDNA等の核酸試料は、末梢血や口腔粘膜スワブ等から常法により容易に調製することができる。
【0021】
タンパク質のアミノ酸配列は、エクソン領域だけではなくイントロン領域における変異によっても影響され得るが、遺伝子検査では通常、エクソン及びその近傍数十〜数百塩基程度、例えば30〜50塩基程度のイントロン領域を含めて検査するのが一般的である。本発明でもエクソン及びその近傍のイントロンを対象に配列解析を行えばよい。ゲノム配列の解析により変異を検出する場合には、本願配列表の配列番号5〜47や公知のデータベースから入手可能なPOLR3B及びPOLR3A遺伝子のゲノム配列を参照して適宜プライマーを設計し、ゲノムDNA試料を用いて常法によりシークエンシングを行えばよい。対象生体ゲノムDNA上のPOLR3B及びPOLR3A遺伝子の塩基配列を決定し、これを野生型配列と比較することにより、変異を詳細に同定できる。決定した塩基配列は、例えばSeqScape (登録商標) 等の公知のソフトウェアを用いて解析することにより、変異の検出やプロファイリングを容易に行うことができる。
【0022】
変異がホモかヘテロかは、シークエンスの波形データから確認できる。ヘテロ変異がある場合、同一部位に2種類のシグナルが重なることになる。ヘテロ変異が2箇所以上ある場合、変異が複合ヘテロであるかどうか(すなわち、変異が異なる染色体上に存在するかどうか)は、患者の両親の検査を行い、2つの変異がそれぞれ父親と母親由来であることを調べることで確認できる。
【0023】
ヘテロ二本鎖の検出により、POLR3B遺伝子及びPOLR3A遺伝子の変異のスクリーニングを行なうことも有効である。ヘテロや複合ヘテロの変異が存在する場合、ゲノムDNA試料を熱変性後に再会合させることにより、正常型DNAと変異DNAとがハイブリダイズしたヘテロ二本鎖が生じる。ヘテロ二本鎖は、(1)非変性ポリアクリルアミドゲル中で異なる移動度を示す、(2)ミスマッチ部分の塩基は化学物質や酵素による切断を受けやすい、(3)変性の際に異なる変性温度を示す、といった特性を有する。これらの特性を利用してヘテロ二本鎖を検出する方法がこの分野において公知であり、変異の検査方法として実用化もされている。具体的には、例えば、変性高速液体クロマトグラフィー(dHPLC)を用いてヘテロ二本鎖を検出する方法や、High Resolution Melt法が知られている。
【0024】
High Resolution Melt法とは、二本鎖DNAに高密度で結合する蛍光色素(SYTO(登録商標)9, LC Green(登録商標), EvaGreen(商標)等)を用いて、二本鎖DNAの融解(熱変性)の過程を蛍光強度の変化としてとらえ、ヘテロ二本鎖を検出する方法である。すなわち、二本鎖DNAに高密度で結合する蛍光色素を用いて二本鎖DNAを染色すると、該二本鎖DNAを融解(熱変性)させたとき、二本鎖が解離した部位から蛍光色素が脱落するため、二本鎖DNAからの蛍光シグナルの量が減少する。従って、そのような蛍光色素を用いることで、二本鎖DNAの熱変性の過程を蛍光強度の変化として視覚的にとらえることができる。温度−蛍光のデータを高密度で取得し解析することで、ヘテロ二本鎖の検出を迅速に高感度で行うことができる。市販の機器類及びキット等を用いて容易に実施可能である。変異がホモである場合、High resolution melt法の検出感度は低くなると想定されるが(検出できないわけではない)、び慢性大脳白質形成不全症では、患者両親の血族婚がない場合ではまず複合ヘテロ変異が想定されるため、High Resolution Melt法等のヘテロ二本鎖検出は本発明でも有効な検査方法となり得る。
【0025】
本発明では、POLR3B及びPOLR3Aのエクソン+近傍イントロン領域を全て対象として塩基配列を決定し、変異の有無を調べてもよい。また、例えば、ヘテロ二本鎖の検出により塩基配列を決定すべき領域を絞り込み、その後に対象領域の塩基配列を決定することで、検査をより効率的に実施することができる。POLR3BとPOLR3Aのいずれかのみを先に調べて変異が発見された場合、他方は必ずしも調べることを要しないが、POLR3B及びPOLR3Aの両者を調べることが好ましい。
【0026】
被検試料の塩基配列と野生型配列とを対比し、アミノ酸配列の変化を生じるような変異が見つかった場合、HCAHCの病因変異ありと判断することができる。適宜、NCBIのdbSNPや1000 Genomes Project等の塩基配列多様性に関するデータベースを参照し、登録のないまれな変異であることを確認してもよい。また、進化的に保存性の高いアミノ酸を変化させる変異であれば、HCAHCの病因変異と考えることができる。種々の動物のPOLR3Bタンパク質(RPC2)及びPOLR3Aタンパク質(RPC1)の配列が公知であり、GenBank等の各種データベースに登録されているので、当業者であれば容易に配列情報を入手して常法により各アミノ酸の進化的保存性を調べることができる。
【0027】
タンパク質立体構造予測からサブユニットタンパク質の機能が損なわれる変異かどうかを推定することも可能である。タンパク質の構造予測は公知のデータベース及びソフトウェア(例えば、データベースEnsembl、RCSB Protein Data Bank、ソフトウェアPyMOL等、下記実施例参照)を用いて容易に実施できる。サブユニット自身の構造に影響するアミノ酸の変異や、サブユニット間の相互作用に影響するアミノ酸の変異は、HCAHCの病因変異と考えられる。
【0028】
HCAHCは乳児期早期は正常に発育するため、乳児期早期に本発明の方法を実施することで、該被検者がHCAHCを将来発症するかどうかを調べることができる(HCAHC患者の検出)。HCAHCが疑われる被検者について本発明の方法を実施した場合、該被検者がHCAHC患者であるか否かの確定診断が可能である(HCAHC患者の検出)。また、健常者について本発明の方法を実施することで、病因変異を有する保因者であるかどうかを調べることができる(保因者の検出)。患者両親の変異確認で本発明の検出方法を実施する場合は保因者の検出に該当する。
【0029】
本発明により、POLR3B及びPOLR3Aがび慢性大脳白質形成不全症の原因遺伝子であることが明らかとなったが、これらはそれぞれRNAポリメラーゼIIIのサブユニットRPC2及びRPC1をそれぞれコードする遺伝子である。RNAポリメラーゼIIIは、上述した通り、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)等の低分子非コードRNAの転写に関与している。そのため、POLR3B遺伝子又はPOLR3A遺伝子の変異によりこれら低分子非コードRNAの転写が低下することでHCAHCが発症すると考えられる。従って、こうした低分子非コードRNAの転写の促進を指標として化合物を選択することにより、HCAHCの治療薬をスクリーニングすることができ、治療薬の開発が可能になる。
【実施例】
【0030】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0031】
1.原因遺伝子及び病因変異の同定
血縁関係のない3家系に由来するHCAHC患者4名を対象に原因遺伝子の解析を行なった(図1A; 表4)。
【0032】
【表4】
【0033】
臨床情報及び末梢血又は唾液サンプルは、書面によるインフォームドコンセントを得た後に各家族員から取得した。実験プロトコルは横浜市立大学の施設内審査委員会に承認された。病原性変異の同定のため、血縁関係のない3家系由来の発端者3名について全エキソーム配列解析を行なった(患者1、3及び4)。SureSelect Human All エクソン50Mb Kit (アジレントテクノロジーズ社、米国カリフォルニア州サンタクララ) を用いてDNAを捕捉し、Illumina GAIIx(イルミナ社、米国カリフォルニア州サンディエゴ)を用いて1サンプルあたり1レーンを使用してリード長108 bpでpaired endで配列決定した。画像解析とベースコーリングは、Sequence Control Software real-time analysis及びCASAVA software v1.7 (イルミナ社) を用いて行なった。合計で90,014,368 (患者1)、86,942,264 (患者3)、及び92,168,758 (患者4) のpaired-endリードが得られた。得られたリード配列をMAQ (Li, H., Ruan, J., and Durbin, R. (2008). Genome Res 18, 1851-1858.) 及びNextGENe software v2.00(ソフトジェネティクス社、米国ペンシルベニア州ステートカレッジ)を用いてヒト参照ゲノム配列(GRCh37/hg19)に対してアラインメントした。この手法により、88%以上のターゲットエキソームが10以上のリードでカバーされた(表5参照)。一塩基変異(single nucleotide variant; SNV)はMAQ及びNextGENeを用いてコールした。短い挿入・欠失はNextGENeを用いて検出した。コールされたSNVはSeattleSeq Annotationを用いてアノテートした。
【0034】
【表5】
【0035】
近年の研究で用いた手法と同様の優先順位付けスキームを採用し(表6)(Doi, H. et al., (2011) Am J Hum Genet 89, 320-327.; Pierce, S.B. et al., (2010) Am J Hum Genet 87, 282-288.; Gilissen, C. et al., (2010) Am J Hum Genet 87, 418-423.)、各患者の病原性変異を同定した。まずはじめに、検出された全ての塩基変異から、dbSNPや1000 Genome projectに登録されている変異を除外した。次いで、MAQ解析とNextGENe解析で共通して検出されたSNVを、信頼性の高い変異として選択した。各患者につき、非同義の(NS)又はカノニカルなスプライスサイト(SP)の変化のうちの364〜370個のSNVが、113〜124個の短い挿入・欠失(indel)と共に同定された。また、我々の55のin-houseエキソームで発見された、健常者12名及び無関係な疾患を有する患者43名に由来する変異も除外し、候補の変異の数を1患者当たり250以下に絞り込んだ。1家系における2名の罹患者(患者1及び2)に基づいてHCAHCが常染色体劣性の遺伝疾患であるものと仮定し、dbSNPにも我々のin-houseの55エキソームにも登録されていないまれなヘテロ接合の変異に着目した。
【0036】
各患者の全遺伝子を、2個以上のNS、SP又はindel変異について調べたところ、患者当たり7〜10の候補遺伝子が見出された(表6)。これらのうち、RNAポリメラーゼIII(Pol III)の2番目に大きいサブユニットRPC2をコードするPOLR3B遺伝子のみが2名の患者で共通していた(患者1及び3)。
【0037】
【表6】
【0038】
POLR3B遺伝子(transcript variant 1, GenBank accession number NM_018082.5)中の変異の遺伝をサンガ―法で調べた。患者1において、エクソン18の2 bp上流のカノニカルスプライスサイト変異(c.1857-2A>C)が父親から遺伝していること、エクソン21のミスセンス変異(c.2303G>A, p.R768H)が母親から遺伝していることが確認された(図1A)。この2つの変異は罹患者である姉(患者2)にも存在していたが、健常な兄には存在しなかった。患者3では、エクソン16のナンセンス変異(c.1648C>T, p.R550X)が父親から遺伝していること、エクソン24のミスセンス変異(c.2778C>G, p.D926E)が母親から遺伝していることが確認された(図1A)。この2つの変異は健常な弟には存在しなかった。
【0039】
c.1857-2A>C及びc.1648C>Tの変異の影響を調べるため、患者由来のリンパ芽球様細胞から抽出した全RNAを用いて既報(Saitsu, H. et al., (2010) Epilepsia 51, 2397-2405.)に従い逆転写PCR(c.1857-2A>C変異については配列番号50、51に示す塩基配列からなるプライマー、c.1648C>T変異については配列番号52、53に示す塩基配列からなるプライマーを使用)と配列決定を行なった。その結果、c.1867-2A>C変異がPOLR3BのmRNAからエクソン18を欠失させ (図2A−2C)、RPC2でインフレームの33アミノ酸の欠失(p.N620_K652del)をもたらすことが確認された(図1B)。さらに、ナンセンス変異(c.1648C>T)を含む変異転写物の発現が野生型転写物の発現よりもはるかに低レベルであることが見出された(図2D)。ナンセンス変異依存mRNA分解(NMD)の阻害剤であるシクロヘキシミド(CHX、30μMで使用)で処理すると変異転写物の発現レベルが増大したことから、変異転写物がNMDを受けることが示された(図2D)。3名の患者で発見された2つのミスセンス変異(p.R768H及びp.D926E)は、進化的に保存されたアミノ酸で生じていた(図1B)。
【0040】
患者4では、POLR3Bの変異が全く発見されなかったが、常染色体劣性モデルの候補遺伝子が9つ存在した。これらのうち、2つのミスセンス変異を含むPOLR3A (NM_007055.3)は、Pol IIIの最も大きいサブユニット(RPC1)をコードしていることから、該遺伝子が第1の候補と考えられた(図1A及び表6)。サンガ―法により、エクソン20中のミスセンス変異 (c.2690T>A, p.I897H)が父親から遺伝したこと、エクソン23中のもう一つのミスセンス変異 (c.3013C>T, p.R1005C) が母親から遺伝したことが確認された (図1A)。これら2つの変異は健常者である妹には存在しなかった。これら2つのミスセンス変異 (p.I897H及びp.R1005C) は比較的保存されたアミノ酸で生じていた (図1B)。
【0041】
以上の通り、POLR3B遺伝子中に4種の変異、POLR3A遺伝子中に2種の変異が発見された。540例の日本人コントロール染色体サンプルのうち1つのアレルでc.2303G>A変異が発見され、残りの5種の変異は日本人コントロール染色体サンプル540例で検出されなかった。このことは、これらの変異が日本人集団の中で非常にまれであることを示している。
【0042】
2.変異サブユニットの構造予測
Pol III(Jasiak, A.J. et al., (2006) Mol Cell 23, 71-81.; Fernandez-Tornero, C. et al., (2007). Mol Cell 25, 813-823.)及びPol II(Cramer, P. et al., (2001). Science 292, 1863-1876.; Gnatt, A.L. et al., (2001). Science 292, 1876-1882.)の構造は相同性が高く、最も大きなサブユニットにおいて特に相同性が高い。そこで、酵母Pol II (Protein data bank ID, 3GTP)(Gnatt, A.L. et al., (2001). Science 292, 1876-1882.)の構造上でRPC1又はRPC2の変異の影響を推定した (図1C)。
【0043】
酵母Pol IIのRPB1サブユニット及びRPB2サブユニットは、それぞれPol IIIのRPC1及びRPC2と相同である。RPC2におけるAsn620_Lys652はRPB2におけるTyr679_Lys712に相当する。Asn620_Lys652 (Tyr679_Lys712) の欠失はRPB2の構造的中心部を破壊し、RPB2の機能喪失をもたらすものと推察された。また、Arg768 (RPB2におけるArg852) は、Pol IIのRPB12サブユニットのArg70の主鎖カルボニル基と相互作用し、Asp926 (RPB2におけるAsp1009) は、Pol IIのRPB10サブユニットのArg48の側鎖と相互作用するが (図1D)、アミノ酸置換Arg768His (Arg852His)及びAsp926Glu (Asp1009Glu) は、これらのサブユニット相互作用を妨げ、ポリメラーゼの機能障害をもたらすものと考えられた。以上の構造予測は、POLR3B (RPC2) 中の変異がPol IIIの機能に影響を与え得ることを示唆している。
【0044】
一方、RPC1におけるIle897及びArg1005は、それぞれRPB1におけるVal863及びArg1036に相当する。Ile897 (Val863) は、Pol IIのRPB5サブユニットのLeu170及びPro176、並びにPol IIのRPB1サブユニットのPhe900 (Phe866) と疎水的相互作用を示すが (図1E)、アミノ酸置換Ile897Asn (Val863Asn) はおそらくこの相互作用を妨げるものと考えられた。また、Arg1005 (Arg1036) はRPB1サブユニットとRPB8サブユニットとの間の相互作用を安定化するが (図1F)、アミノ酸置換Arg1005Cys (Arg1036Cys) は、この相互作用を不安定にするものと考えられた。以上の通り、POLR3A遺伝子中に見出された変異もまた、Pol IIIの機能に影響を与えるものと予測される。
【0045】
3.HCAHC患者の臨床所見
POLR3A遺伝子又はPOLR3B遺伝子の変異を有する患者の臨床像を表4(上掲)に示す。MRIの結果、患者4名の全てで、T2強調画像で白質中に強度の高い領域、小脳委縮と脳梁低形成が確認された (図3)。
【0046】
患者1及び2は極めて類似した臨床経過を示した。患者1、2は、乳児期早期には正常に発育した;それぞれ、独歩15ヶ月及び14ヶ月、数語の発語12ヶ月及び13ヶ月。3歳齢以降、患者1は歩行不安定、頻繁なつまずきと転倒を呈し、患者2は運動が不得手になった。両者いずれも強度の近視であった(矯正視力でそれぞれ最大0.7及び0.5)。両患者は小学校、中学校及び高校を低成績で卒業し、患者2の知能指数(IQ)は52であった(WAIS-III)。患者1では、歩行不安定が18歳頃から顕著になり、運動失調のために自転車に乗ることができなかったが、自動車の運転は可能であった。患者2では無月経が認められ、ホルモン療法が奏効した。患者1は、腋毛及び髭の欠如、薄い陰毛(Tanner II)、並びに正常な27歳齢の値よりも低いテストステロン、濾胞刺激ホルモン及び黄体形成ホルモンの血清レベルといった、性腺機能低下のいくつかの兆候を示した。両患者の神経学的検査により、軽度の水平眼振、滑動性追従眼球運動の緩慢化、特に垂直方向における注視の制限、筋緊張低下、軽度に亢進した深部腱反射(膝蓋腱反射及びアキレス腱反射)、バビンスキー反射は陰性、並びに失調性言語、開脚失調性歩行、拮抗運動反復不全及び測定障害等の小脳性の兆候及び症状が確認された。
【0047】
患者3の臨床情報は既報の通りであり(非特許文献6)、さらなる所見として、滑動性追従眼球運動の緩慢化、垂直方向における注視の制限、正常な聴性脳幹反応(ABR)、軽度の痙縮を伴う脳症状、知的障害(IQ43, WISC-III)、近視はないが遠視性乱視が確認された。軽度の嚥下障害の他に増悪は認められず、養護学校に自力歩行で通学した。
【0048】
患者4は、乳児期早期は正常に発育した;頚定3ヶ月、数語の発語12ヶ月、独歩14ヶ月。4歳頃に両親が軽度の振戦に気付いた。身長、体重、頭囲は正常であった。強度の近視であったが、眼球運動は円滑で制限や眼振は認めなかった。左側に感覚神経性難聴があった。筋緊張は正常で痙縮や硬直は認めなかった。腱反射はわずかに上昇、バビンスキー反射は陰性。表出性失調性爆発性言語、企図振戦、指鼻試験拙劣、拮抗運動反復不全、測定障害及び開脚失調性歩行といった小脳性の兆候が見られた。知能指数は57(WISC-III)。末梢神経伝導速度は正常範囲であり、ABRは右側で正常であった。その後、14歳頃に運動機能の低下が起こり、車椅子生活となった。
【技術分野】
【0001】
本発明は、び慢性大脳白質形成不全症患者又は保因者の検出方法に関する。
【背景技術】
【0002】
先天性ミエリン形成不全症は、白質ジストロフィーに分類される多様な疾患群であり、ミエリン形成異常を特徴とする。これらの病態は脳のMRIにより認識することができるが、正しく診断されない症例が多い(非特許文献1)。これまでに、歯数不足症及び低ゴナドトロピン性性腺機能低下症を伴うミエリン形成不全(4H)症候群(MIM 612440)、大脳基底核及び小脳の萎縮を伴うミエリン形成不全(H-ABC)(MIM 612438)等の、ミエリン形成に影響する新たな症候群が数種報告されている(非特許文献2〜5)。
【0003】
小脳委縮と脳梁低形成を伴うび慢性の大脳白質形成不全症(cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum; HCAHC)は、近年報告された新たなミエリン形成不全症候群であり、び慢性のミエリン形成不全と小脳及び脳梁の低形成を特徴とする(非特許文献6)。HCAHC、4H症候群、及びH-ABCの3症候群には、び慢性のミエリン形成不全、小脳及び脳梁の委縮ないしは低形成という共通した特徴が認められるが、HCAHCでは4H症候群及びH-ABCにおいて見られる歯数不足や大脳基底核の萎縮が認められない。
【0004】
び慢性大脳白質形成不全症の原因遺伝子としては、4つの遺伝子(PLP1, MBP, GJC2, FAM126)の関与が知られていたが、これら遺伝子の変異で説明ができない症例が数多く残されていた。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Schiffmann, R., and van der Knaap, M.S. (2009). Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 72, 750-759.
【非特許文献2】Timmons, M., Tsokos, M., Asab, M.A., Seminara, S.B., Zirzow, G.C., Kaneski, C.R., Heiss, J.D., van der Knaap, M.S., Vanier, M.T., Schiffmann, R., et al. (2006). Peripheral and central hypomyelination with hypogonadotropic hypogonadism and hypodontia. Neurology 67, 2066-2069.
【非特許文献3】Wolf, N.I., Harting, I., Boltshauser, E., Wiegand, G., Koch, M.J., Schmitt-Mechelke, T., Martin, E., Zschocke, J., Uhlenberg, B., Hoffmann, G.F., et al. (2005). Leukoencephalopathy with ataxia, hypodontia, and hypomyelination. Neurology 64, 1461-1464.
【非特許文献4】Wolf, N.I., Harting, I., Innes, A.M., Patzer, S., Zeitler, P., Schneider, A., Wolff, A., Baier, K., Zschocke, J., Ebinger, F., et al. (2007). Ataxia, delayed dentition and hypomyelination: a novel leukoencephalopathy. Neuropediatrics 38, 64-70.
【非特許文献5】van der Knaap, M.S., Naidu, S., Pouwels, P.J., Bonavita, S., van Coster, R., Lagae, L., Sperner, J., Surtees, R., Schiffmann, R., and Valk, J. (2002). New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 23, 1466-1474.
【非特許文献6】Sasaki, M., Takanashi, J., Tada, H., Sakuma, H., Furushima, W., and Sato, N. (2009). Diffuse cerebral hypomyelination with cerebellar atrophy and hypoplasia of the corpus callosum. Brain Dev 31, 582-587.
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明の目的は、び慢性大脳白質形成不全症の新規原因遺伝子を同定し、より多くの症例を適切に診断できる手段を提供することにある。
【課題を解決するための手段】
【0007】
本願発明者らは、血縁関係のない3家系に由来するHCAHC患者3名を対象に、次世代シークエンサーを用いて全エキソーム配列解析を行ない、膨大なリード配列を鋭意解析した結果、低分子非コードRNAの転写に関与するRNAポリメラーゼIIIのサブユニットをコードするPOLR3B遺伝子及びPOLR3A遺伝子において、数百例の健常コントロールサンプルには認められず、SNPデータベース等にも登録がないまれな新規変異が複合ヘテロ接合で存在することを見出し、本願発明を完成した。
【0008】
すなわち、本発明は、被検者から分離した試料を用いて、該被検者がPOLR3B遺伝子及びPOLR3A遺伝子のうちの少なくともいずれかに変異を有するか否かを調べることを含む、び慢性大脳白質形成不全症患者又は保因者の検出方法であって、ホモ接合又は複合ヘテロ接合の前記変異が検出された場合に患者が検出され、ヘテロ接合の前記変異が検出された場合に保因者が検出される、方法を提供する。また、本発明は、低分子非コードRNAの転写の促進を指標として化合物を選択することを含む、び慢性大脳白質形成不全症の治療薬のスクリーニング方法を提供する。
【発明の効果】
【0009】
本発明により、HCAHCの新規な原因遺伝子が同定された。これにより、該疾患への関与が既に知られている4遺伝子に変異が認められない症例の多くを診断可能になる。HCAHCの原因遺伝子として同定されたPOLR3B遺伝子及びPOLR3A遺伝子は、低分子非コードRNAの転写を行なっている遺伝子であり、変異により転写が低下することでHCAHCが発症すると考えられる。低分子非コードRNAの転写を促進する薬剤をスクリーニングすることでHCAHCの治療薬の創薬が期待される。
【図面の簡単な説明】
【0010】
【図1】実施例で同定されたPOLR3B遺伝子及びPOLR3A遺伝子中の変異を説明する図である。(A) HCAHC患者を生じた3家系の家系図である。血縁関係のない2家系由来の患者3名において、RPC2をコードするPOLR3B遺伝子中に4種の変異を同定した。また、1家系において、RPC1をコードするPOLR3A遺伝子中に2種の変異を同定した。各変異の分離を併せて図中に示す。(B) Pfamドメイン(データベースEnsembl [http://uswest.ensembl.org/index.html]より)を有するRPC2タンパク質(上段)及びRPC1タンパク質(下段)の略図である。アミノ酸を改変する各変異の位置を波形データと共に示す。ミスセンス変異は全て進化的に保存されたアミノ酸で生じていた。CLUSTALWを用いて相同配列を整列化した。(C-F) RPC1及びRPC2の変異箇所の3次元表示である。RPC1及びRPC2中の変異アミノ酸を、RNAポリメラーゼIIの相同なサブユニットRPB1及びRPB2において相当する位置を付記して示した(括弧書きのアミノ酸と数値)。変異の構造と位置は結晶構造(PDB ID 3GTP)を用いてPyMOLにより描写した。RNAポリメラーゼIIに特異的なサブユニットであるRPB3, RPB9及びRPB11は図中では省略した。変異アミノ酸と相互作用するアミノ酸も示した。
【図2】POLR3B遺伝子中のスプライスサイト変異及びナンセンス変異の影響を調べた結果を示す図である。(A) POLR3B遺伝子のエクソン16〜19のゲノム構造を示す略図である。エクソン、イントロン及びプライマーをそれぞれボックス、破線及び矢印で示す。イントロン17中のc.1857-2A>C変異を点で記す。(B) c.1857-2A>C変異を有する患者1及び患者2並びにコントロール健常者のRT-PCR解析の結果である。患者のcDNAでは2種のPCR産物が検出された。上のバンドが野生型(WT)転写物であり、下のバンドが変異転写物である。コントロールではWTの単一バンドのみが検出された。(C) WT転写物及び変異転写物の配列解析の結果である。変異アレルではエクソン18の読み飛ばしが明らかに確認された。(D) c.1648C>T変異の解析結果である。上段;ゲノムDNAから増幅したPCR産物の配列、中段;CHX非処理細胞のcDNAから増幅したPCR産物の配列、下段;CHX処理細胞のcDNAから増幅したPCR産物の配列。非処理細胞ではc.1648C>T変異アレルの発現が極めて低いのに対し、CHX処理でナンセンス変異依存mRNA分解(NMD)を阻害した処理細胞では変異アレルの発現が有意に増大していた。
【図3】POLR3B変異又はPOLR3A変異を有する患者の脳MRI像である。(A, C, E, F, G) 大脳基底核を通るT2強調横断像である。全ての患者で白質中に高強度領域を認めた。 (B, D, F, H) T1強調正中矢状断像である。全ての患者で脳梁低形成(矢頭)と小脳の委縮(矢印)を認めた。
【発明を実施するための形態】
【0011】
本発明が対象とするび慢性大脳白質形成不全症とは、非特許文献6で報告されている、小脳委縮と脳梁低形成を伴うび慢性の大脳白質形成不全を特徴とする新規な先天性ミエリン形成不全症である。本明細書では該疾患を「HCAHC」と呼ぶことがある。
【0012】
POLR3B(GenBankアクセッション番号NM_018082)及びPOLR3A(GenBankアクセッション番号NM_007055)は、RNAポリメラーゼIIIのサブユニットRPC2及びRPC1をそれぞれコードする遺伝子である。RNAポリメラーゼIIIは、17のサブユニットから成り、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)等の低分子非コードRNAの転写に関与している。配列表の配列番号1及び2に示す配列は、POLR3B遺伝子のコード領域のcDNA配列及びアミノ酸配列であり、配列番号3及び4は、POLR3A遺伝子のコード領域のcDNA配列及びアミノ酸配列である。なお、配列番号48及び49は、GenBankに上記の番号で登録されているPOLR3B及びPOLR3AのmRNA配列である。配列番号5〜47には、POLR3B及びPOLR3Aの各エクソン及びその近傍のイントロンの塩基配列を下記表1、2の通りに示した。
【0013】
【表1】
【0014】
【表2】
【0015】
本願発明者らにより、POLR3B遺伝子又はPOLR3A遺伝子がHCAHCの原因遺伝子であることが判明した。HCAHC患者は、POLR3B遺伝子又はPOLR3A遺伝子中に変異をホモ又は複合ヘテロで有し、HCAHC保因者は該変異をヘテロで有する。複合ヘテロとは、POLR3B遺伝子又はPOLR3A遺伝子の両アリルにそれぞれ異なる変異を有することを意味する。
【0016】
ここでいう変異には、POLR3B遺伝子がコードするRPC2又はPOLR3AがコードするRPC1のごく少数のアミノ酸の変化のほか、これらサブユニットの少なくとも一部の領域を欠失するような変化をもたらす塩基配列の変異が包含される。そのような塩基配列の変異は、例えば、エクソン又はイントロン領域内での塩基の置換、欠失、挿入、重複等によるミスセンス変異、ナンセンス変異、フレームシフト変異、アミノ酸欠失変異、スプライシング異常変異等であり得る。
【0017】
配列表の配列番号2、4に示された野生型のアミノ酸配列からなるタンパク質は、正常なサブユニットタンパク質(RPC2、RPC1)の典型例である。本発明では、変異の有無は、配列番号1〜4に示されたPOLR3B遺伝子及びPOLR3A遺伝子の配列を基準にして判断され得る。アミノ酸配列の変化を生じるPOLR3B又はPOLR3A遺伝子の変異は、HCAHCの病因変異であると考えることができるが、中でも、進化的に保存性の高いアミノ酸を変化させる遺伝子変異は、正常な機能が損なわれたサブユニットタンパク質を生じる蓋然性が高く、HCAHC病因変異の典型例である。あるいはまた、検出された塩基の変異が、NCBIのdbSNP等の塩基配列の多様性に関する周知のデータベースには登録されていないまれな塩基変異の場合も、HCAHC病因変異と考えてよい。病因変異は、POLR3B又はPOLR3Aにコードされるサブユニットタンパク質それ自体の立体構造や、サブユニット間の相互作用に影響すると考えられる。
【0018】
下記表3の変異は、実施例において血縁関係のない3家系から同定されたHCAHC病因変異である。これらの変異は、dbSNP等の塩基配列の多様性に関するデータベースに登録されておらず、また健常者集団においてもほとんど認められないまれな変異であった。POLR3B遺伝子の変異4種のうちのいずれか2つを有する場合、HCAHC患者と判断することができる。もっとも、当該3家系以外の家系には当然ながら異なる変異が存在し得るので、本発明の範囲はこれらの具体例に限定されるものではない。
【0019】
【表3】
【0020】
POLR3B又はPOLR3A遺伝子の変異は、ゲノムDNAやRNA等の核酸試料を用いて塩基配列を解析することで検出可能である。とりわけ、ゲノムDNA試料を用いてゲノム配列の解析を行なうことが最も確実で望ましい。ゲノムDNA等の核酸試料は、末梢血や口腔粘膜スワブ等から常法により容易に調製することができる。
【0021】
タンパク質のアミノ酸配列は、エクソン領域だけではなくイントロン領域における変異によっても影響され得るが、遺伝子検査では通常、エクソン及びその近傍数十〜数百塩基程度、例えば30〜50塩基程度のイントロン領域を含めて検査するのが一般的である。本発明でもエクソン及びその近傍のイントロンを対象に配列解析を行えばよい。ゲノム配列の解析により変異を検出する場合には、本願配列表の配列番号5〜47や公知のデータベースから入手可能なPOLR3B及びPOLR3A遺伝子のゲノム配列を参照して適宜プライマーを設計し、ゲノムDNA試料を用いて常法によりシークエンシングを行えばよい。対象生体ゲノムDNA上のPOLR3B及びPOLR3A遺伝子の塩基配列を決定し、これを野生型配列と比較することにより、変異を詳細に同定できる。決定した塩基配列は、例えばSeqScape (登録商標) 等の公知のソフトウェアを用いて解析することにより、変異の検出やプロファイリングを容易に行うことができる。
【0022】
変異がホモかヘテロかは、シークエンスの波形データから確認できる。ヘテロ変異がある場合、同一部位に2種類のシグナルが重なることになる。ヘテロ変異が2箇所以上ある場合、変異が複合ヘテロであるかどうか(すなわち、変異が異なる染色体上に存在するかどうか)は、患者の両親の検査を行い、2つの変異がそれぞれ父親と母親由来であることを調べることで確認できる。
【0023】
ヘテロ二本鎖の検出により、POLR3B遺伝子及びPOLR3A遺伝子の変異のスクリーニングを行なうことも有効である。ヘテロや複合ヘテロの変異が存在する場合、ゲノムDNA試料を熱変性後に再会合させることにより、正常型DNAと変異DNAとがハイブリダイズしたヘテロ二本鎖が生じる。ヘテロ二本鎖は、(1)非変性ポリアクリルアミドゲル中で異なる移動度を示す、(2)ミスマッチ部分の塩基は化学物質や酵素による切断を受けやすい、(3)変性の際に異なる変性温度を示す、といった特性を有する。これらの特性を利用してヘテロ二本鎖を検出する方法がこの分野において公知であり、変異の検査方法として実用化もされている。具体的には、例えば、変性高速液体クロマトグラフィー(dHPLC)を用いてヘテロ二本鎖を検出する方法や、High Resolution Melt法が知られている。
【0024】
High Resolution Melt法とは、二本鎖DNAに高密度で結合する蛍光色素(SYTO(登録商標)9, LC Green(登録商標), EvaGreen(商標)等)を用いて、二本鎖DNAの融解(熱変性)の過程を蛍光強度の変化としてとらえ、ヘテロ二本鎖を検出する方法である。すなわち、二本鎖DNAに高密度で結合する蛍光色素を用いて二本鎖DNAを染色すると、該二本鎖DNAを融解(熱変性)させたとき、二本鎖が解離した部位から蛍光色素が脱落するため、二本鎖DNAからの蛍光シグナルの量が減少する。従って、そのような蛍光色素を用いることで、二本鎖DNAの熱変性の過程を蛍光強度の変化として視覚的にとらえることができる。温度−蛍光のデータを高密度で取得し解析することで、ヘテロ二本鎖の検出を迅速に高感度で行うことができる。市販の機器類及びキット等を用いて容易に実施可能である。変異がホモである場合、High resolution melt法の検出感度は低くなると想定されるが(検出できないわけではない)、び慢性大脳白質形成不全症では、患者両親の血族婚がない場合ではまず複合ヘテロ変異が想定されるため、High Resolution Melt法等のヘテロ二本鎖検出は本発明でも有効な検査方法となり得る。
【0025】
本発明では、POLR3B及びPOLR3Aのエクソン+近傍イントロン領域を全て対象として塩基配列を決定し、変異の有無を調べてもよい。また、例えば、ヘテロ二本鎖の検出により塩基配列を決定すべき領域を絞り込み、その後に対象領域の塩基配列を決定することで、検査をより効率的に実施することができる。POLR3BとPOLR3Aのいずれかのみを先に調べて変異が発見された場合、他方は必ずしも調べることを要しないが、POLR3B及びPOLR3Aの両者を調べることが好ましい。
【0026】
被検試料の塩基配列と野生型配列とを対比し、アミノ酸配列の変化を生じるような変異が見つかった場合、HCAHCの病因変異ありと判断することができる。適宜、NCBIのdbSNPや1000 Genomes Project等の塩基配列多様性に関するデータベースを参照し、登録のないまれな変異であることを確認してもよい。また、進化的に保存性の高いアミノ酸を変化させる変異であれば、HCAHCの病因変異と考えることができる。種々の動物のPOLR3Bタンパク質(RPC2)及びPOLR3Aタンパク質(RPC1)の配列が公知であり、GenBank等の各種データベースに登録されているので、当業者であれば容易に配列情報を入手して常法により各アミノ酸の進化的保存性を調べることができる。
【0027】
タンパク質立体構造予測からサブユニットタンパク質の機能が損なわれる変異かどうかを推定することも可能である。タンパク質の構造予測は公知のデータベース及びソフトウェア(例えば、データベースEnsembl、RCSB Protein Data Bank、ソフトウェアPyMOL等、下記実施例参照)を用いて容易に実施できる。サブユニット自身の構造に影響するアミノ酸の変異や、サブユニット間の相互作用に影響するアミノ酸の変異は、HCAHCの病因変異と考えられる。
【0028】
HCAHCは乳児期早期は正常に発育するため、乳児期早期に本発明の方法を実施することで、該被検者がHCAHCを将来発症するかどうかを調べることができる(HCAHC患者の検出)。HCAHCが疑われる被検者について本発明の方法を実施した場合、該被検者がHCAHC患者であるか否かの確定診断が可能である(HCAHC患者の検出)。また、健常者について本発明の方法を実施することで、病因変異を有する保因者であるかどうかを調べることができる(保因者の検出)。患者両親の変異確認で本発明の検出方法を実施する場合は保因者の検出に該当する。
【0029】
本発明により、POLR3B及びPOLR3Aがび慢性大脳白質形成不全症の原因遺伝子であることが明らかとなったが、これらはそれぞれRNAポリメラーゼIIIのサブユニットRPC2及びRPC1をそれぞれコードする遺伝子である。RNAポリメラーゼIIIは、上述した通り、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)等の低分子非コードRNAの転写に関与している。そのため、POLR3B遺伝子又はPOLR3A遺伝子の変異によりこれら低分子非コードRNAの転写が低下することでHCAHCが発症すると考えられる。従って、こうした低分子非コードRNAの転写の促進を指標として化合物を選択することにより、HCAHCの治療薬をスクリーニングすることができ、治療薬の開発が可能になる。
【実施例】
【0030】
以下、本発明を実施例に基づきより具体的に説明する。もっとも、本発明は下記実施例に限定されるものではない。
【0031】
1.原因遺伝子及び病因変異の同定
血縁関係のない3家系に由来するHCAHC患者4名を対象に原因遺伝子の解析を行なった(図1A; 表4)。
【0032】
【表4】
【0033】
臨床情報及び末梢血又は唾液サンプルは、書面によるインフォームドコンセントを得た後に各家族員から取得した。実験プロトコルは横浜市立大学の施設内審査委員会に承認された。病原性変異の同定のため、血縁関係のない3家系由来の発端者3名について全エキソーム配列解析を行なった(患者1、3及び4)。SureSelect Human All エクソン50Mb Kit (アジレントテクノロジーズ社、米国カリフォルニア州サンタクララ) を用いてDNAを捕捉し、Illumina GAIIx(イルミナ社、米国カリフォルニア州サンディエゴ)を用いて1サンプルあたり1レーンを使用してリード長108 bpでpaired endで配列決定した。画像解析とベースコーリングは、Sequence Control Software real-time analysis及びCASAVA software v1.7 (イルミナ社) を用いて行なった。合計で90,014,368 (患者1)、86,942,264 (患者3)、及び92,168,758 (患者4) のpaired-endリードが得られた。得られたリード配列をMAQ (Li, H., Ruan, J., and Durbin, R. (2008). Genome Res 18, 1851-1858.) 及びNextGENe software v2.00(ソフトジェネティクス社、米国ペンシルベニア州ステートカレッジ)を用いてヒト参照ゲノム配列(GRCh37/hg19)に対してアラインメントした。この手法により、88%以上のターゲットエキソームが10以上のリードでカバーされた(表5参照)。一塩基変異(single nucleotide variant; SNV)はMAQ及びNextGENeを用いてコールした。短い挿入・欠失はNextGENeを用いて検出した。コールされたSNVはSeattleSeq Annotationを用いてアノテートした。
【0034】
【表5】
【0035】
近年の研究で用いた手法と同様の優先順位付けスキームを採用し(表6)(Doi, H. et al., (2011) Am J Hum Genet 89, 320-327.; Pierce, S.B. et al., (2010) Am J Hum Genet 87, 282-288.; Gilissen, C. et al., (2010) Am J Hum Genet 87, 418-423.)、各患者の病原性変異を同定した。まずはじめに、検出された全ての塩基変異から、dbSNPや1000 Genome projectに登録されている変異を除外した。次いで、MAQ解析とNextGENe解析で共通して検出されたSNVを、信頼性の高い変異として選択した。各患者につき、非同義の(NS)又はカノニカルなスプライスサイト(SP)の変化のうちの364〜370個のSNVが、113〜124個の短い挿入・欠失(indel)と共に同定された。また、我々の55のin-houseエキソームで発見された、健常者12名及び無関係な疾患を有する患者43名に由来する変異も除外し、候補の変異の数を1患者当たり250以下に絞り込んだ。1家系における2名の罹患者(患者1及び2)に基づいてHCAHCが常染色体劣性の遺伝疾患であるものと仮定し、dbSNPにも我々のin-houseの55エキソームにも登録されていないまれなヘテロ接合の変異に着目した。
【0036】
各患者の全遺伝子を、2個以上のNS、SP又はindel変異について調べたところ、患者当たり7〜10の候補遺伝子が見出された(表6)。これらのうち、RNAポリメラーゼIII(Pol III)の2番目に大きいサブユニットRPC2をコードするPOLR3B遺伝子のみが2名の患者で共通していた(患者1及び3)。
【0037】
【表6】
【0038】
POLR3B遺伝子(transcript variant 1, GenBank accession number NM_018082.5)中の変異の遺伝をサンガ―法で調べた。患者1において、エクソン18の2 bp上流のカノニカルスプライスサイト変異(c.1857-2A>C)が父親から遺伝していること、エクソン21のミスセンス変異(c.2303G>A, p.R768H)が母親から遺伝していることが確認された(図1A)。この2つの変異は罹患者である姉(患者2)にも存在していたが、健常な兄には存在しなかった。患者3では、エクソン16のナンセンス変異(c.1648C>T, p.R550X)が父親から遺伝していること、エクソン24のミスセンス変異(c.2778C>G, p.D926E)が母親から遺伝していることが確認された(図1A)。この2つの変異は健常な弟には存在しなかった。
【0039】
c.1857-2A>C及びc.1648C>Tの変異の影響を調べるため、患者由来のリンパ芽球様細胞から抽出した全RNAを用いて既報(Saitsu, H. et al., (2010) Epilepsia 51, 2397-2405.)に従い逆転写PCR(c.1857-2A>C変異については配列番号50、51に示す塩基配列からなるプライマー、c.1648C>T変異については配列番号52、53に示す塩基配列からなるプライマーを使用)と配列決定を行なった。その結果、c.1867-2A>C変異がPOLR3BのmRNAからエクソン18を欠失させ (図2A−2C)、RPC2でインフレームの33アミノ酸の欠失(p.N620_K652del)をもたらすことが確認された(図1B)。さらに、ナンセンス変異(c.1648C>T)を含む変異転写物の発現が野生型転写物の発現よりもはるかに低レベルであることが見出された(図2D)。ナンセンス変異依存mRNA分解(NMD)の阻害剤であるシクロヘキシミド(CHX、30μMで使用)で処理すると変異転写物の発現レベルが増大したことから、変異転写物がNMDを受けることが示された(図2D)。3名の患者で発見された2つのミスセンス変異(p.R768H及びp.D926E)は、進化的に保存されたアミノ酸で生じていた(図1B)。
【0040】
患者4では、POLR3Bの変異が全く発見されなかったが、常染色体劣性モデルの候補遺伝子が9つ存在した。これらのうち、2つのミスセンス変異を含むPOLR3A (NM_007055.3)は、Pol IIIの最も大きいサブユニット(RPC1)をコードしていることから、該遺伝子が第1の候補と考えられた(図1A及び表6)。サンガ―法により、エクソン20中のミスセンス変異 (c.2690T>A, p.I897H)が父親から遺伝したこと、エクソン23中のもう一つのミスセンス変異 (c.3013C>T, p.R1005C) が母親から遺伝したことが確認された (図1A)。これら2つの変異は健常者である妹には存在しなかった。これら2つのミスセンス変異 (p.I897H及びp.R1005C) は比較的保存されたアミノ酸で生じていた (図1B)。
【0041】
以上の通り、POLR3B遺伝子中に4種の変異、POLR3A遺伝子中に2種の変異が発見された。540例の日本人コントロール染色体サンプルのうち1つのアレルでc.2303G>A変異が発見され、残りの5種の変異は日本人コントロール染色体サンプル540例で検出されなかった。このことは、これらの変異が日本人集団の中で非常にまれであることを示している。
【0042】
2.変異サブユニットの構造予測
Pol III(Jasiak, A.J. et al., (2006) Mol Cell 23, 71-81.; Fernandez-Tornero, C. et al., (2007). Mol Cell 25, 813-823.)及びPol II(Cramer, P. et al., (2001). Science 292, 1863-1876.; Gnatt, A.L. et al., (2001). Science 292, 1876-1882.)の構造は相同性が高く、最も大きなサブユニットにおいて特に相同性が高い。そこで、酵母Pol II (Protein data bank ID, 3GTP)(Gnatt, A.L. et al., (2001). Science 292, 1876-1882.)の構造上でRPC1又はRPC2の変異の影響を推定した (図1C)。
【0043】
酵母Pol IIのRPB1サブユニット及びRPB2サブユニットは、それぞれPol IIIのRPC1及びRPC2と相同である。RPC2におけるAsn620_Lys652はRPB2におけるTyr679_Lys712に相当する。Asn620_Lys652 (Tyr679_Lys712) の欠失はRPB2の構造的中心部を破壊し、RPB2の機能喪失をもたらすものと推察された。また、Arg768 (RPB2におけるArg852) は、Pol IIのRPB12サブユニットのArg70の主鎖カルボニル基と相互作用し、Asp926 (RPB2におけるAsp1009) は、Pol IIのRPB10サブユニットのArg48の側鎖と相互作用するが (図1D)、アミノ酸置換Arg768His (Arg852His)及びAsp926Glu (Asp1009Glu) は、これらのサブユニット相互作用を妨げ、ポリメラーゼの機能障害をもたらすものと考えられた。以上の構造予測は、POLR3B (RPC2) 中の変異がPol IIIの機能に影響を与え得ることを示唆している。
【0044】
一方、RPC1におけるIle897及びArg1005は、それぞれRPB1におけるVal863及びArg1036に相当する。Ile897 (Val863) は、Pol IIのRPB5サブユニットのLeu170及びPro176、並びにPol IIのRPB1サブユニットのPhe900 (Phe866) と疎水的相互作用を示すが (図1E)、アミノ酸置換Ile897Asn (Val863Asn) はおそらくこの相互作用を妨げるものと考えられた。また、Arg1005 (Arg1036) はRPB1サブユニットとRPB8サブユニットとの間の相互作用を安定化するが (図1F)、アミノ酸置換Arg1005Cys (Arg1036Cys) は、この相互作用を不安定にするものと考えられた。以上の通り、POLR3A遺伝子中に見出された変異もまた、Pol IIIの機能に影響を与えるものと予測される。
【0045】
3.HCAHC患者の臨床所見
POLR3A遺伝子又はPOLR3B遺伝子の変異を有する患者の臨床像を表4(上掲)に示す。MRIの結果、患者4名の全てで、T2強調画像で白質中に強度の高い領域、小脳委縮と脳梁低形成が確認された (図3)。
【0046】
患者1及び2は極めて類似した臨床経過を示した。患者1、2は、乳児期早期には正常に発育した;それぞれ、独歩15ヶ月及び14ヶ月、数語の発語12ヶ月及び13ヶ月。3歳齢以降、患者1は歩行不安定、頻繁なつまずきと転倒を呈し、患者2は運動が不得手になった。両者いずれも強度の近視であった(矯正視力でそれぞれ最大0.7及び0.5)。両患者は小学校、中学校及び高校を低成績で卒業し、患者2の知能指数(IQ)は52であった(WAIS-III)。患者1では、歩行不安定が18歳頃から顕著になり、運動失調のために自転車に乗ることができなかったが、自動車の運転は可能であった。患者2では無月経が認められ、ホルモン療法が奏効した。患者1は、腋毛及び髭の欠如、薄い陰毛(Tanner II)、並びに正常な27歳齢の値よりも低いテストステロン、濾胞刺激ホルモン及び黄体形成ホルモンの血清レベルといった、性腺機能低下のいくつかの兆候を示した。両患者の神経学的検査により、軽度の水平眼振、滑動性追従眼球運動の緩慢化、特に垂直方向における注視の制限、筋緊張低下、軽度に亢進した深部腱反射(膝蓋腱反射及びアキレス腱反射)、バビンスキー反射は陰性、並びに失調性言語、開脚失調性歩行、拮抗運動反復不全及び測定障害等の小脳性の兆候及び症状が確認された。
【0047】
患者3の臨床情報は既報の通りであり(非特許文献6)、さらなる所見として、滑動性追従眼球運動の緩慢化、垂直方向における注視の制限、正常な聴性脳幹反応(ABR)、軽度の痙縮を伴う脳症状、知的障害(IQ43, WISC-III)、近視はないが遠視性乱視が確認された。軽度の嚥下障害の他に増悪は認められず、養護学校に自力歩行で通学した。
【0048】
患者4は、乳児期早期は正常に発育した;頚定3ヶ月、数語の発語12ヶ月、独歩14ヶ月。4歳頃に両親が軽度の振戦に気付いた。身長、体重、頭囲は正常であった。強度の近視であったが、眼球運動は円滑で制限や眼振は認めなかった。左側に感覚神経性難聴があった。筋緊張は正常で痙縮や硬直は認めなかった。腱反射はわずかに上昇、バビンスキー反射は陰性。表出性失調性爆発性言語、企図振戦、指鼻試験拙劣、拮抗運動反復不全、測定障害及び開脚失調性歩行といった小脳性の兆候が見られた。知能指数は57(WISC-III)。末梢神経伝導速度は正常範囲であり、ABRは右側で正常であった。その後、14歳頃に運動機能の低下が起こり、車椅子生活となった。
【特許請求の範囲】
【請求項1】
被検者から分離した試料を用いて、該被検者がPOLR3B遺伝子及びPOLR3A遺伝子のうちの少なくともいずれかに変異を有するか否かを調べることを含む、び慢性大脳白質形成不全症患者又は保因者の検出方法であって、ホモ接合又は複合ヘテロ接合の前記変異が検出された場合に患者が検出され、ヘテロ接合の前記変異が検出された場合に保因者が検出される、方法。
【請求項2】
ゲノムDNA試料を用いてゲノム配列を調べることにより行なわれる請求項1記載の方法。
【請求項3】
前記変異はミスセンス変異、ナンセンス変異、フレームシフト変異又はスプライシング異常を生じる変異である請求項1又は2記載の方法。
【請求項4】
POLR3B遺伝子変異の少なくとも一つが以下のいずれかから選択される請求項3記載の方法。
(1) コード領域の第1648位のC(配列番号18中の第321位)がTになる変異
(2) エクソン18の2塩基上流のA(配列番号19中の第923位)がCになる変異
(3) コード領域の第2303位のG(配列番号22中の第310位)がAになる変異
(4) コード領域の第2778位のC(配列番号25中の第365位)がGになる変異
【請求項5】
POLR3A遺伝子変異の少なくとも一つが以下のいずれかから選択される請求項3記載の方法。
(5) コード領域の第2690位のT(配列番号41中の第374位)がAになる変異
(6) コード領域の第3013位のC(配列番号43中の第499位)がTになる変異
【請求項6】
低分子非コードRNAの転写の促進を指標として化合物を選択することを含む、び慢性大脳白質形成不全症の治療薬のスクリーニング方法。
【請求項7】
前記低分子非コードRNAは、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)からなる群より選択される少なくとも1種である請求項6記載のスクリーニング方法。
【請求項1】
被検者から分離した試料を用いて、該被検者がPOLR3B遺伝子及びPOLR3A遺伝子のうちの少なくともいずれかに変異を有するか否かを調べることを含む、び慢性大脳白質形成不全症患者又は保因者の検出方法であって、ホモ接合又は複合ヘテロ接合の前記変異が検出された場合に患者が検出され、ヘテロ接合の前記変異が検出された場合に保因者が検出される、方法。
【請求項2】
ゲノムDNA試料を用いてゲノム配列を調べることにより行なわれる請求項1記載の方法。
【請求項3】
前記変異はミスセンス変異、ナンセンス変異、フレームシフト変異又はスプライシング異常を生じる変異である請求項1又は2記載の方法。
【請求項4】
POLR3B遺伝子変異の少なくとも一つが以下のいずれかから選択される請求項3記載の方法。
(1) コード領域の第1648位のC(配列番号18中の第321位)がTになる変異
(2) エクソン18の2塩基上流のA(配列番号19中の第923位)がCになる変異
(3) コード領域の第2303位のG(配列番号22中の第310位)がAになる変異
(4) コード領域の第2778位のC(配列番号25中の第365位)がGになる変異
【請求項5】
POLR3A遺伝子変異の少なくとも一つが以下のいずれかから選択される請求項3記載の方法。
(5) コード領域の第2690位のT(配列番号41中の第374位)がAになる変異
(6) コード領域の第3013位のC(配列番号43中の第499位)がTになる変異
【請求項6】
低分子非コードRNAの転写の促進を指標として化合物を選択することを含む、び慢性大脳白質形成不全症の治療薬のスクリーニング方法。
【請求項7】
前記低分子非コードRNAは、5SリボソームRNA (rRNA)、U6核内低分子RNA (snRNA)、7SL RNA、RNase P、RNase MRP、短鎖散在性反復配列 (SINE)及び転移RNA (tRNA)からなる群より選択される少なくとも1種である請求項6記載のスクリーニング方法。
【図1】

【図2】
【図3】
【図2】
【図3】
【公開番号】特開2013−85489(P2013−85489A)
【公開日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願番号】特願2011−226488(P2011−226488)
【出願日】平成23年10月14日(2011.10.14)
【出願人】(505155528)公立大学法人横浜市立大学 (101)
【Fターム(参考)】
【公開日】平成25年5月13日(2013.5.13)
【国際特許分類】
【出願日】平成23年10月14日(2011.10.14)
【出願人】(505155528)公立大学法人横浜市立大学 (101)
【Fターム(参考)】
[ Back to top ]