
アクチノニン類縁体
【課題】本発明は、アクチノニンをリード化合物とし、既存のアクチノニンに比べて簡便に合成可能であり、既存のアクチノニン化合物と同等又はそれ以上の生理活性を有する新規化合物を提供することを課題とする。
【解決手段】アクチノニン中の不斉コハク酸構造をアミノ酸などにより代替することによる、新規アクチノニン類縁体による。本発明の化合物は、プロテアーゼ阻害活性、具体的にはペプチド脱ホルミル化酵素(PDF)阻害やアミノペプチダーゼ(APN)阻害活性を有する。
【解決手段】アクチノニン中の不斉コハク酸構造をアミノ酸などにより代替することによる、新規アクチノニン類縁体による。本発明の化合物は、プロテアーゼ阻害活性、具体的にはペプチド脱ホルミル化酵素(PDF)阻害やアミノペプチダーゼ(APN)阻害活性を有する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規アクチノニン類縁体に関する。さらにはその合成法に関する。
【背景技術】
【0002】
アクチノニンは、1962年に放線菌の培養液より単離・精製され多くの細菌に対して高い抗菌活性を発現することが見出された抗生物質である。アクチノニンの示す抗菌活性については、詳細に検討されており、ペプチド脱ホルミル化酵素(PDF)を阻害して発現していることが明らかとなっている(非特許文献1)。
【0003】
2001年には、マラリアに関する遺伝子解析より、熱帯熱マラリア原虫がPDFを有していることが発見され、(非特許文献2)アクチノニンが、熱帯熱マラリア原虫に対して、IC50値が2.5 μMの活性があることが判明している(非特許文献3)。
【0004】
さらに、アクチノニンはアミノペプチダーゼ N(APN)を阻害することが発見されている(非特許文献4)。APNは、癌細胞の浸潤や運動 (転移)、また潰瘍性大腸炎などの炎症性消化器疾患に関与する酵素であることから、アクチノニンによる抗がん作用、さらには抗炎症作用が報告されている(非特許文献5、6)。
【0005】
アクチノニンの分子構造は式IVに示す構造式のごときである。
【化4】

【0006】
アクチノニンは、ヒドロキシプロリン、バリンを構成要素として有するが、加えて、ペンチル基を側鎖に有する不斉コハク酸構造を含有する。プロリノール、バリンなどは、アミノ酸であることから安価に入手可能であるが、不斉コハク酸化合物は、数工程の不斉合成を必要とする。そのため、構造変換は容易ではなく、アクチノニンを上回る化合物への変換、またアクチノニンの有する生理活性を分離した新規化合物の創出は困難であった。
【0007】
アクチノニン類縁体は、いくつかのグループにより報告されており、ヒドロキシプロリン部位について、ベンズイミダゾールへ変換し、抗菌活性を見出したことに関する報告がある(非特許文献6)。しかしながら、アクチノニン分子中、バリンのアミノ基と、ヒドロキシルアミンとの間に相当する部分、すなわち不斉コハク酸構造部位をアミノ酸へ置き換えた報告はない。
【非特許文献1】Biochemistry, 39 (6), 1256 -1262, 2000
【非特許文献2】Archives of Biochemistry and Biophysics, 396 (12), 162-170, 2001
【非特許文献3】Antimicrob Agents Chemother. 47(8), 2545-2550, 2003
【非特許文献4】J. Clin. Invest. 114(8), 1107-1116, 2004
【非特許文献5】Journal of Investigative Dermatology 127, 1042-1051, 2007
【非特許文献6】International Immunopharmacology, 6, 1935-1942, 2006
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、抗がん作用、抗炎症作用、抗菌作用、抗マラリア作用を有する化合物として公知のアクチノニンをリード化合物とし、既存のアクチノニンに比べて簡便に合成可能であり、既存のアクチノニン化合物と同等又はそれ以上の生理活性を有する新規化合物を提供することを課題とする。
【課題を解決するための手段】
【0009】
発明者らは、鋭意研究を重ねた結果、アクチノニン中の不斉コハク酸構造をアミノ酸などにより代替し、さらには、バリンを種々のアミノ酸へと置き換えることで、アクチノニンと同様な生理活性を有する新規アクチノニン誘導体が合成され、本発明を完成した。
【0010】
即ち本発明は、以下よりなる。
1.下記の一般式Iで表される化合物。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
2.下記の一般式IIで表される化合物。
下記の一般式IIで表される化合物。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
3.下記の一般式IIIで表される化合物。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
4.不斉合成工程含まない工程により合成される前項1〜3のいずれか1に記載の化合物の合成方法。
5.以下の式IVで表される化合物のうち、不斉コハク酸構造部位をアミノ酸により代替する工程を含む、前項4に記載の合成方法。
式IV:
【化4】
6.前項1〜3のいずれか1に記載の化合物を有効成分として含有する薬剤。
7.有効成分が、プロテアーゼ阻害活性を有する化合物である前項6に記載の薬剤。
8.前項6又は7に記載の薬剤、並びに薬理学的及び製剤学的に許容される担体を含む医薬組成物。
【発明の効果】
【0011】
本発明のアクチノニン類縁体化合物は、アクチノニンそのものと異なり、不斉炭素を有するコハク酸を別途合成する必要がないため、合成工程数の大幅な削減が見込め、さらには天然のアミノ酸を主原料に合成可能である。またあるものはPDF阻害活性を有し、あるものはAPN阻害活性を有する。本発明の化合物のうち、PDF阻害活性を有する化合物は、新たな抗菌剤、抗マラリア剤としての応用が期待できる。また、APN阻害活性を有する化合物は、抗がん剤、抗炎症剤としての応用が期待出来る。
【発明を実施するための最良の形態】
【0012】
本発明の化合物は、以下の一般式Iで表される。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
【0013】
また、本発明の化合物は、下記の一般式IIで表すこともできる。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
【0014】
さらに、本発明の化合物は、下記の一般式IIIで表すこともできる。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
【0015】
具体的には、本発明の化合物は、前項で示す構造式で表され、具体的には表1に示す構造式で表される。
【表1】
【0016】
より具体的には、以下の実施例で示す化合物のうち、化合物9a,9bに示す化合物が挙げられる。本発明の化合物のうち、PDF阻害薬として、化合物9a,9bに示す化合物が挙げられる。
【0017】
本発明の化合物は、以下の実施例に示す方法により合成することができる。本発明の化合物であるアクチノニン類縁体の合成方法は、従来のアクチノニン合成方法と異なり、アクチノニン分子中、バリンのアミノ基とヒドロキシルアミンとの間に相当する部分、すなわち不斉炭素を有するコハク酸を別途合成する必要がない点が特徴である。すなわち、本発明の化合物は、不斉合成工程を含まないで合成することができる。具体的には、従来のアクチノニン合成方法において、不斉合成工程を含まず、不斉コハク酸構造部位をアミノ酸へ置き換えることで、合成することができる。
【0018】
本発明において、一般式I〜IIIのいずれかで表される化合物は、さらに、薬学的に許容される塩であってもよい。また、一般式I〜IIIのいずれかの化合物又はその塩において、異性体(例えば光学異性体、幾何異性体及び互換異性体)などが存在する場合は、本発明はそれらの異性体を包含し、また溶媒和物、水和物及び種々の形状の結晶を包含するものである。
【0019】
本発明において、薬学的に許容される塩とは、薬理学的及び製剤学的に許容される一般的な塩が挙げられる。そのような塩として、具体的には以下が例示される。
塩基性付加塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩;例えばカルシウム塩、マグネシウム塩等のアルカリ土類金属塩;例えばアンモニウム塩;例えばトリメチルアミン塩、トリエチルアミン塩;ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩;たとえばN,N−ジベンジルエチレンジアミン等のアラルキルアミン塩;例えばピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等の複素環芳香族アミン塩;例えばテトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩;リジン塩等の塩基性アミノ酸塩等が挙げられる。
【0020】
酸付加塩としては、例えば塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;例えば酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;例えばメタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;例えばアスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等を挙げることができる。
【0021】
本発明において、一般式I〜IIIのいずれかで表される化合物は、PDFやAPNに対し拮抗作用を有する。本発明の化合物がこれらの酵素阻害作用のいずれを有するかは、本明細書の実験例に具体的に示した方法に従って容易に検定可能である。具体的には、例えば以下の方法で、検定することができる。
【0022】
酵素阻害活性については、例えばAPN阻害活性は、動物細胞、例えばHL−60細胞を用いて、本発明の化合物による蛍光性基質(Ala-MCA)の酵素的加水分解を指標として行うことができる。具体的には、5mLプラスチックチューブに50mM Tris−HClpH7.4を790μL分注し、合成化合物のDMSO溶液を10μLを加えた後、50mM Tris−HClpH7.4に懸濁した2×106cells/mL に調整したHL−60 PBS(−)細胞懸濁液を100μL加えた後、37℃に調整した恒温水槽にて10分間インキュベーションし、1mMに調整した蛍光性基質Ala-MCAの50mM Tris−HClpH7.4を100μLを加えた後、37℃に調整した恒温水槽にて30分間インキュベーションした後、1M AcONa-AcOH pH 4.0を3mL加え、酵素反応を停止し、励起波長380nm,蛍光波長420nmでの蛍光強度を測定することができる。
【0023】
本発明において、一般式I〜IIIのいずれかで表される化合物は、プロテアーゼ阻害活性を有する。プロテアーゼ阻害活性とは、具体的にはペプチド脱ホルミル化酵素(PDF)阻害活性やアミノペプチダーゼ N(APN)阻害活性をいう。
【0024】
本発明は、本発明の化合物を有効成分とする試薬又は医薬等の薬剤も、本発明の範囲に含まれる。医薬品として用いる場合には、プロテアーゼ阻害活性、例えば、PDF阻害活性やAPN阻害活性により治療効果を発揮しうる疾患に対して使用することができる。このような疾患としては、がん、炎症などが挙げられる。
【0025】
本発明の化合物を有効成分とする医薬として用いる場合には、投与量は特に限定されない。例えばPDF阻害活性剤やAPN阻害活性剤として本発明の薬剤を投与する場合は、あらゆる投与方法において適宜の投与量が容易に選択できる。例えば、経口投与の場合には有効成分を成人一日あたり0.01〜1000mg程度の範囲で用いることができる。抗がん剤、抗炎症剤等を有効成分として含む医薬と本発明の薬剤とを併用する場合には、これらの薬剤の投与期間中、及び/又はその前若しくは後の期間のいずれにおいても本発明の薬剤を投与することが可能である。
【0026】
本発明の薬剤として、上記一般式I〜IIIのいずれかで表される化合物から選ばれる1種又は2種以上の物質をそのまま投与してもよいが、好ましくは、上記の物質の1種又は2種以上を含む、経口用あるいは非経口用の医薬組成物として投与することが好ましい。経口用あるいは非経口用の医薬組成物は、当業者に利用可能な製剤用添加物、即ち薬理学的及び製剤学的に許容しうる担体を用いて製造することができる。
【0027】
経口投与に適する医薬用組成物としては、例えば、錠剤、カプセル剤、散剤、細粒剤、顆粒剤、液剤、及びシロップ剤等を挙げることができ、非経口投与に適する医薬組成物としては、例えば、注射剤、点滴剤、坐剤、吸入剤、点眼剤、点鼻剤、軟膏剤、クリーム剤、及び貼付剤等を挙げることができる。上記の医薬組成物の製造に用いられる薬理学的及び製剤学的に許容しうる担体としては、例えば、賦形剤、崩壊剤ないし崩壊補助剤、結合剤、滑沢剤、コーティング剤、色素、希釈剤、基剤、溶解剤ないし溶解補助剤、等張化剤、pH調節剤、安定化剤、噴射剤、及び粘着剤等を挙げることができる。
【0028】
本明細書の実施例に、本発明の式Iに示される好ましい化合物の製造方法を具体的に説明する。これらの製造方法において用いられた出発原料及び試薬、並びに反応条件などを適宜修飾ないし改変することにより、本発明の範囲に包含される化合物はいずれも製造可能である。本発明の化合物の製造方法は、実施例に具体的に説明されたものに限定されるものではない。
【実施例】
【0029】
以下、本発明を実施例によりさらに具体的に説明するが、本発明は下記の実施例の範囲に限定されることはない。
【0030】
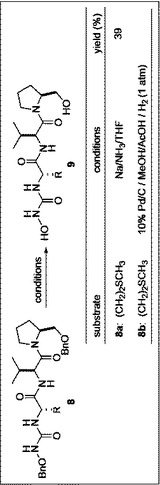
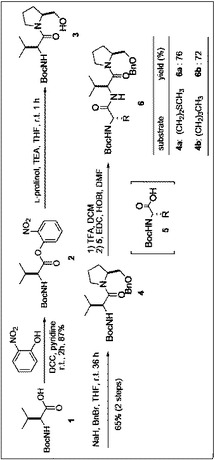
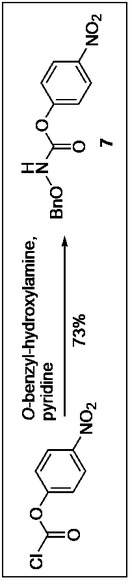
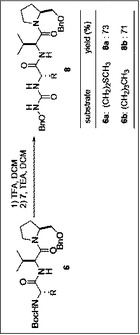
[実施例]目的化合物の合成
本実施例における最終生成物9a、9bを得るまでの製造方法のスキームを図1から図4に示した。
【0031】
1)中間体 N−ボック−L−バリン O−ニトロフェノールエステル(2)の合成
【化5】
【0032】
N−ボック−L−バリン (1)(18.8g,87.5mmol)、O−ニトロフェノール(21.5g,157.5mmol)をピリジン(87mL)に溶解させ、DCC(19.5mg,87.5mmol)を加え室温で2時間撹拌し、TLCプレート (酢酸エチル:ヘキサン=1:10 ニンヒドリン試薬で呈色)で反応終了を確認した。析出したDCUを吸引ろ過により除去した。ろ液に1N HCl溶液(170mL)で酸性とし、ジエチルエーテル(200mL)で抽出した。有機層を飽和炭酸水素水(100mL×2)、水(100mL×2)、飽和食塩水(100mL)で洗浄、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去、黄色オイル状残渣を得た。この黄色オイル状残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:7)で単離し、ヘキサンで結晶化を行い、表題化合物(淡黄色結晶)を得た。収率87%。
【0033】
融点53.6−55.0℃。IR(KBr)cm−1:3360(NH),1755(C=O),1690(C=O)。[α]D23−39.4 (c0.15,CHCl3)。
1H NMR(CDCl3,300MHz)δ: 8.07 (1H,dd,Jo=8.0Hz,Jm=1.5Hz,ArH ortho to NO2),7.66(1H,td,Jo=8.0Hz,Jm=1.5Hz,ArH para to NO2),7.41(1H,td,Jo=8.0Hz,Jm=1.0Hz,ArH para to O),7.28 (1H,d, Jo=8.0 Hz,ArH ortho to O), 5.04 (1H,d,J=8.5Hz,NH),4.37(1H,q,J=9.0Hz,J=4.5Hz,NHCHCO),2.45(1H,m,CHMe2),1.47(9H,s,CMe3),1.11(3H,d, J=7.0Hz,CHMeMe),1.04 (3H,d,J=7.0 Hz,CHMeMe)。
【0034】
2)中間体 N−ボック−L−バリルプロリノール (3)の合成
【化6】
【0035】
(S)−(+)−2−ピロリジンメタノール(505mg,5.0mmol)とトリエチルアミン(3.0mL)を無水テトラヒドロフラン(70mL)に溶解させたものに、N−ボック−L−バリン O−ニトロフェノールエステル (2)(1776mg,5.25mmol)を無水テトラヒドロフラン(15mL)に溶解させたものを滴下、室温で1時間攪拌した。反応終了後、減圧下溶媒を留去、酢酸エチル(500mL)にあけた。有機層を2N HCl 溶液(200mL)、水(200mL)、飽和炭酸水素水(200mL)、飽和食塩水(200mL)で洗浄後、無水硫酸マグネシウムにより乾燥し,溶媒を減圧下留去し、黄色オイル状残渣を得た。このオイル状残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=2:1)で単離し,表題化合物(黄色オイル)を得た。収率98%。
【0036】
[α]D24 −32.2(c 0.42,CHCl3)。IR(neat)cm−1: 3400br(OH),1700(C=O),1630(C=O)。
1H−NMR(300MHz,CDCl3):δ5.23(1H,d,J=9.5Hz,NH),4.62(1H,d,J=5.5Hz,OH),4.28〜4.23(2H,m,NHCHCO,NCHCH2OH),3.84 3.79(1H,M,NHH'),3.66〜3.63(1H,m,NHH'),3.56〜3.53(1H,m,CHH'),3.48〜3.43(1H,m,CHH'),2.06〜1.85(4H,m,NCH2CH2CH2),1.62〜1.52(1H,m,CHMe2),1.41 (9H,s,CMe3),0.97(3H,d,CHMeMe),0.91(3H,d,CHMeMe)
【0037】
3)中間体 O−ベンジル−N−ボック−L−バリルプロリノール (4)の合成
【化7】
【0038】
合成法(I)
N−ボック−L−バリルプロリノール (3)(1408mg,4.69mmol)と水素化ナトリウム(50% in oil,270mg,5.62mmol)、臭化ベンジル(0.55mL)を無水テトラヒドロフラン(34mL)に溶解させ、室温で36時間攪拌した。反応終了後、溶媒を減圧下留去し、酢酸エチル (400mL)にあける。有機層を2N HCl溶液(160mL)、水(160mL)、飽和炭酸水素水(160mL)、飽和食塩水(160mL)で洗浄後、無水硫酸マグネシウムにより乾燥し,溶媒を減圧下留去した。淡黄色オイル状残渣を得た。その残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で単離し,表題化合物(黄色オイル)を得た。収率84%。
【0039】
合成法(II)
(S)−(+)−2−ピロリジンメタノール(549.2mg,5.4mmol)とトリエチルアミン(3.0mL)を無水テトラヒドロフラン(70mL)に溶解させたものに、N−ボック−L−バリンO−ニトロフェノールエステル (3)(1839.8mg,5.4mmol)を無水テトラヒドロフラン(15mL)に溶解させたものを滴下し、室温で2時間撹拌した。
【0040】
反応終了後、氷冷下水素化ナトリウム(50% in oil 628mg,13mmol)、臭化ベンジル(1.6mL,13mmol)を加え、室温で36時間撹拌した。反応終了後、減圧下溶媒を留去し、酢酸エチル(50mL)にあけた。有機層を2N HCl(10mL)で酸性とし、飽和炭酸水素水(30mL×2)、水(30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。黄色オイル状残渣(2810mg)を得た。黄色オイル状残渣 (2810mg)をフラッシュカラムクロマトグラフィーを行った。酢酸エチル:ヘキサン=1:1より表題化合物(黄色オイル)を得た。収率65%。
【0041】
[α]D26 −60.0(c 1.0,CHCl3) 。IR(neat)cm−1:3300(NH),1750(C=O),1640 (C=O)。
1H−NMR(300MHz,CDCl3):δ7.33〜7.27(5H,m,ArH),5.28(1H,d,J=9.3Hz,NH),4.49(2H,d,J=2.4Hz,CH2Ph),4.35〜4.24(2H,m,NHCHCO,NCHCH2O),3.68〜3.46(4H,m,NCH2,NCHCH2O),2.04〜1.88(5H,m,NCH2CH2CH,CHMe2),1.42(9H,s,CMe3),0.95(3H,d,J=7.0Hz,CHMeMe),0.89(3H,d,J=7.0Hz,CHMeMe)。
【0042】
4)中間体 O−ベンジル−N−ボック−L−メチオニルバリルプロリノール (6a)の合成
【化8】
【0043】
O−ベンジル−N−ボック−L−バリルプロリノール (4)(1543mg,3.95mmol)を塩化メチレン(7.11mL)に溶解させ,トリフルオロ酢酸(4.3mL)を滴下し,室温で1時間攪拌する。反応終了後,酢酸エチル(400mL)にあけ,飽和炭酸水素水,水(各160mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣のO−ベンジル−L−バリルプロリノール (1166mg)を得た。
【0044】
O−ベンジル−N−ボック−L−バリルプロリノール(1166mg)、N−ボック−L−メチオニン(1083mg,4.34mmol)、HOBt(587mg,4.34mmol)をジメチルホルムアイド(27mL)に溶解後,EDC(830mg,4.34mmol)を加え,2時間攪拌する。反応終了後,酢酸エチル(400mL)にあけ,飽和炭酸水素水,水,飽和食塩水(各160mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィーを行い、酢酸エチル:ヘキサン=2:3溶出部より表題化合物を得た。収率76%。
【0045】
[α]D26 56.1 (c 1.0,CHCl3) 。IR(neat)cm−1 3300(NH),1640(C=O)。
1H−NMR(300MHz,CDCl3):店 δ732〜7.29(5H,m,ArH),6.81 (1H,d,J=10.8Hz, NH),5.16 (1H,d,J=8.4Hz,NH),4.59−4.50 (1H,m,NHCHCO),4.48(2H,d,J=2.1Hz,CH2Ph),4.30〜4.26(2H,m,NHCHCO,NCHCH2O),3.69〜3.46(4H,m,NCH2,NCHCH2O),2.55(2H,t,J=6.9Hz,SCH2CH2),2.10(1H,s,CH3S),2.08〜1.89(7H,m,NCH2CH2CH,CHMe2,SCH2CH2),1.44(9H,s,CMe3),0.94 (3H,d,J=7.0Hz,CHMeMe),0.89(3H,d,J=7.0Hz,CHMeMe)。
【0046】
13C−NMR(75MHz,CDCl3):171.4,170.1,155.5,138.4,128.4,127.6,127.5,80.0,73.2,70.1,56.8,56.8,55.9,53.6,47.8,31.9,31.6,30.2,28.4,27.4,24.6,19.5。
【0047】
5)中間体 O−ベンジル−O−ベンジルオキシカルボニル−L−メチオニルバリルプロリノール (8a)の合成
【化9】
【0048】
O−ベンジル−N−ボック−L−メチオニルバリルプロリノール (6a)(624mg,1.2mmol) を塩化メチレン(2mL)に溶解させ,トリフルオロ酢酸(1.3mL)を滴下し,室温で1時間攪拌する。反応終了後,酢酸エチル(150mL)にあけ,飽和炭酸水素水(60mL),飽和食塩水(30mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣のO−ベンジル−L−メチオニルバリルプロリノールを得た。
【0049】
O−ベンジル−L−メチオニルバリルプロリノール(459mg), 4−ニトロフェニル−N−(O−ベンジルヒドロキシ)カルバメート(345mg,1.2mmol),トリエチルアミン(0.167mL)をCH2Cl2(14)に溶解し,1.5時間攪拌した。反応終了後,1M 水酸化ナトリウム溶液(30mL×3),1M HCl(30mL), 水(30mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィーを酢酸エチル:ヘキサン=1:1で表題化合物を得た。収率73%。
【0050】
IR(CHCl3)cm−1:3400(NH),1640(C=O),1680(C=O)。
1H−NMR(300 MHz,CDCl3):7.38〜7.29(10H,m,ArH)。
7.04(1H,d,J=9.0Hz,NH),6.33(1H,d,J=9.0Hz,NH),4.80(2H,d,J=5.1Hz,CH2Ph),4.56〜4.51(2H,m,NHCHCO,NHCHCO),4.47(2H,s,CH2Ph),4.30(1H,s,NCHCH2O),3.70〜3.33(4H,m,NCH2,NCHCH2O),2.44(2H,t,J=7.2Hz,SCH2CH2),2.07(3H,s,CH3S),2.04〜1.86(7H,m,NCH2CH2CH2,CHMe2,SCH2CH2),0.93(3H,d,J=6.6Hz,CHMeMe),0.87(3H,d,J=6.6Hz,CHMeMe)。
【0051】
13C−NMR (75MHz,CDCl3):171.3,170.3,159.6,138.5,135.6,129.4,128.8,128.4,127.8,127.5,78.7,73.3,70.1,56.8,56.1,52.4,47.9,32.5,31.5,30.1,27.4,24.6,19.4,17.9,15.4。
【0052】
6)O−ベンジル−N−(N−(O−ベンジルヒドロキシカルボニル)−L−メチオニル−バリル)(9a)L−プロリノールの合成
【化10】
【0053】
O−ベンジル−O−ベンジルオキシカルボニル−L−メチオニルバリルプロリノール(8a)(228mg,0.4mmol)を無水テトラヒドロフラン(8mL)に溶解させ,−78℃冷却下,液体アンモニア(約45mL)を滴下する。反応液の色が,青くなるまでナトリウムを少しずつ加える。反応液の青色が消失するまで塩化アンモニウムを加える。液体アンモニウムを留去後,室温まで昇温する。残渣を酢酸エチルにあけ,ろ過する。無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去する。無色オイル状残渣を得た。塩化メチレンに溶解させ,フラッシュカラムクロマトグラフィーを行い,酢酸エチル:2−プロパノール = 20:1で表題化合物を得た。収率39%。
【0054】
IR(CHCl3)cm−1:3400(OH),1720(C=O),1670(C=O),1620(C=O)。1H−NMR(300MHz,CDCl3):8.48(1H,s,OH),7.89(1H,s,OH),7.74(1H,d,J=8.7Hz,NH),6.76(1H,d,J=8.4Hz,NH),4.70〜4.52(2H,m,NHCHCO,NHCHCO),4.42(1H,br s,NH),4.24(1Hbr s,NCHCH2OH),3.90〜3.86,3.64〜3.49(4H,m,NCH2,NCHCH2OH),2.55(2H,m,SCH2CH2),2.08(3H,s,CH3S),2.14〜1.73(7H,m,NCH2CH2CH2,CHMe2,SCH2CH2),0.96〜0.92(6H,m,CHMeMe)。
【0055】
13C−NMR(75 MHz,CDCl3): 172.5,172.2,161.8,65.1,60.8,56.5,53.2,48.4,32.2,31.4,30.3,27.7,24.3,19.4,18.3。
【0056】
7)中間体 O−ベンジル−N−(N−ボック−L−バリル−L−ノルロイシル)−L−プロリノール (6b)の合成
【化11】
【0057】
O−ベンジル−N−(N−ボック−L−バリル)−L−プロリノール (4)(301.1mg,0.77mmol)を塩化メチレン(1.4mL)に溶解させ、トリフルオロ酢酸(0.85mL)を滴下し、室温で1時間撹拌した。反応終了後、酢酸エチル(30mL)にあけ、飽和炭酸水素水、水(各30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒留去した。無色オイル状残渣を得た。
O−ベンジル−L−バリル−L−プロリノール(200mg,0.68mmol)、ノルロイシン(175mg,0.76mmol)、HOBt(101.1mg,0.74mmol)をジメチルホルムアミド(5mL)に溶解後、EDC(142.9mg,0.75mmol)を加え、1時間半攪拌した。反応終了後、酢酸エチル(20mL)にあけ、飽和炭酸水素水、水(各20mL×2)、飽和食塩水(20mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒留去した。無色オイル状残渣を得た。無色オイル状残渣をフラッシュカラムクロマトグラフィーを行った。酢酸エチル:ヘキサン=1:1溶出部より表題化合物を得た。収率68%。
【0058】
1H−NMR(300MHz,CDCl3)δ:7.29〜7.23(5H,m,ArH'),6.80(1H,d,J=8.7Hz,NH),5.06(1H,d,J=8.4 Hz,NH),4.45(1H,d,J=1.8Hz,CHAHBPh),4.45(1H,d,J=1.8Hz,CHAHBPh),4.31〜4.27(1H,m,NHCHCO),4.10〜4.05(1H,m,NCH2CH2CH2CH),3.67〜3.44(4H,m,CH2,OCH2Ph,NCH2CH2CH2CH),2.01〜1.55,1.27〜1.22(11H,m,CH(Me)2,CHCH2CH2CH2CH3,NCH2CH2CH2CH),1.42(9H,s,CMe3),0.92〜0.85(9H,m,CHMe2,CHCH2CH2CH2CH3)。IR(KBr)cm−1:3300(NH),1710(C=O),1630(C=O)。
【0059】
8)中間体 O−ベンジル−O−ベンジルオキシカルバモイル−L−ノルロイシルバリルプロリノール(8b)の合成
【化12】
【0060】
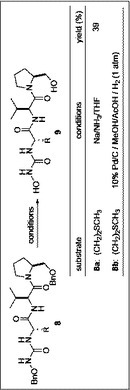
O−ベンジル−N−(N−ボック−L−バリル−L−ノルロイシル)−L−プロリノール(6b)(262.3mg,0.52mmol)を塩化メチレン(1mL)に溶解させ、トリフルオロ酢酸(0.6mL)を滴下し、室温で1時間撹拌した。反応終了後、酢酸エチル(30mL)にあけ、飽和炭酸水素水、水(各30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。O−ベンジル−N−(L−バリル−L−ノルロイシル)−L−プロリノール(228.4mg,0.57mmol)、4−ニトロフェニル−N−(O−ベンジルオキシ)カルバメ−ト(63.5mg,0.57mmol)、トリエチルアミン(0.08mL,0.57mmol)を塩化メチレン(7mL)に溶解し、2時間攪拌した。反応終了後、1M 水酸化ナトリウム(20×3mL)、1M HCl水溶液(20mL)、水(20mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。無色オイル状残渣をフラッシュカラムクロマトグラフィーで酢酸エチル:ヘキサン=1:1溶出部より表題化合物を得た。収率71%。
【0061】
1H−NMR(300MHz,CDCl3)δ:8.21(1H,d,J=9.0Hz,NH),7.89(1H,d,J=7.5Hz,NH),7.40〜7.26(10H,m,ArH'),6.12(1H,d,J=8.7Hz,NH),4.75(4H,d,J=1.8Hz,CH2Ph,CH2Ph),4.28〜4.29(1H,m,NHCHCO),3.57〜3.55(4H,m,CH2,OCH2Ph,NCH2CH2CH2CH),2.44〜2.43(1H,m,NHCHCO),2.07〜1.88(4H,m,NCH2CH2CH2CH),1.83〜1.14(6H,m,CHCH2CH2CH2CH3),0.87〜0.83(9H,m,CHMe2,CHCH2CH2CH2CH3)。
【0062】
13C−NMR(75MHz,CDCl3)δ:172.3,170.5,159.8,138.4,135.7,129.3,128.3,128.3,127.5,127.4,129.3,128.6,128.4,128.3,127.5,127.4,78.5,73.1,70.0,56.7,56.0,53.0,47.8,33.0,31.3,28.3,27.5,27.3,24.4,22.4,19.3,18.1,14.0。
【0063】
9)O−ベンジル−N−(N−(O−ベンジルハイドロキシカルボニル)−L−ノルロイシル―バリル)−L−プロリノール (9b)の合成
【化13】
【0064】
O−ベンジル−O−ベンジルオキシカルバモイル−L−ノルロイシルバリルプロリノール(8b)をメタノール(5mL)に溶解させ、酢酸(0.6mL)を加えた後、10%パラジウム/カーボン(27mg)を加え,水素(1atm)雰囲気下、室温で6時間撹拌した。反応終了後、10%パラジウム/カーボンをろ過し、水(20mL×2)で抽出し、炭酸水素水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィー を行い。酢酸エチル:イソプロピルアルコール=1:1溶出部より表題化合物を得た。
【0065】
[実験例]酵素阻害活性の測定
酵素阻害活性については、例えばAPN阻害活性は、HL−60細胞を用いて、これらによる蛍光性基質(Ala-MCA)の酵素的加水分解を指標として行った。具体的には、5mLプラスチックチューブに50mM Tris−HClpH7.4を790μLを分注し、合成化合物のDMSO溶液を10μL加えた後、50mM Tris−HClpH7.4に懸濁した2×106cells/mL に調整したHL−60 PBS(−)細胞懸濁液を100μL加えた後、37℃に調整した恒温水槽にて10分間インキュベーションし、1mMに調整した蛍光性基質Ala-MCAの50mM Tris−HClpH7.4を100μL加えた後、37℃に調整した恒温水槽にて30分間インキュベーションした後、1M AcONa-AcOH pH 4.0を3mL加え、酵素反応を停止し、励起波長380nm,蛍光波長420nmでの蛍光強度を測定することができる。
【産業上の利用可能性】
【0066】
以上詳述したように、本発明の化合物(アクチノニン類縁体)は、プロテアーゼ阻害活性、具体的にはPDF阻害活性やAPN阻害活性を有するので、PDF阻害活性やAPN阻害活性により治療効果を発揮しうる疾患に対して使用することができる。このような疾患としては、例えば、がん、炎症などが挙げられ、このような疾患に対する医薬として利用することができる。また、生化学試験用試薬としても利用することができる。
【図面の簡単な説明】
【0067】
【図1】中間体6の化合物の合成スキームを示す図である。
【図2】中間体7の化合物の合成スキームを示す図である。
【図3】中間体8の化合物の合成スキームを示す図である。
【図4】目的化合物9の合成スキームを示す図である。
【技術分野】
【0001】
本発明は、新規アクチノニン類縁体に関する。さらにはその合成法に関する。
【背景技術】
【0002】
アクチノニンは、1962年に放線菌の培養液より単離・精製され多くの細菌に対して高い抗菌活性を発現することが見出された抗生物質である。アクチノニンの示す抗菌活性については、詳細に検討されており、ペプチド脱ホルミル化酵素(PDF)を阻害して発現していることが明らかとなっている(非特許文献1)。
【0003】
2001年には、マラリアに関する遺伝子解析より、熱帯熱マラリア原虫がPDFを有していることが発見され、(非特許文献2)アクチノニンが、熱帯熱マラリア原虫に対して、IC50値が2.5 μMの活性があることが判明している(非特許文献3)。
【0004】
さらに、アクチノニンはアミノペプチダーゼ N(APN)を阻害することが発見されている(非特許文献4)。APNは、癌細胞の浸潤や運動 (転移)、また潰瘍性大腸炎などの炎症性消化器疾患に関与する酵素であることから、アクチノニンによる抗がん作用、さらには抗炎症作用が報告されている(非特許文献5、6)。
【0005】
アクチノニンの分子構造は式IVに示す構造式のごときである。
【化4】
【0006】
アクチノニンは、ヒドロキシプロリン、バリンを構成要素として有するが、加えて、ペンチル基を側鎖に有する不斉コハク酸構造を含有する。プロリノール、バリンなどは、アミノ酸であることから安価に入手可能であるが、不斉コハク酸化合物は、数工程の不斉合成を必要とする。そのため、構造変換は容易ではなく、アクチノニンを上回る化合物への変換、またアクチノニンの有する生理活性を分離した新規化合物の創出は困難であった。
【0007】
アクチノニン類縁体は、いくつかのグループにより報告されており、ヒドロキシプロリン部位について、ベンズイミダゾールへ変換し、抗菌活性を見出したことに関する報告がある(非特許文献6)。しかしながら、アクチノニン分子中、バリンのアミノ基と、ヒドロキシルアミンとの間に相当する部分、すなわち不斉コハク酸構造部位をアミノ酸へ置き換えた報告はない。
【非特許文献1】Biochemistry, 39 (6), 1256 -1262, 2000
【非特許文献2】Archives of Biochemistry and Biophysics, 396 (12), 162-170, 2001
【非特許文献3】Antimicrob Agents Chemother. 47(8), 2545-2550, 2003
【非特許文献4】J. Clin. Invest. 114(8), 1107-1116, 2004
【非特許文献5】Journal of Investigative Dermatology 127, 1042-1051, 2007
【非特許文献6】International Immunopharmacology, 6, 1935-1942, 2006
【発明の開示】
【発明が解決しようとする課題】
【0008】
本発明は、抗がん作用、抗炎症作用、抗菌作用、抗マラリア作用を有する化合物として公知のアクチノニンをリード化合物とし、既存のアクチノニンに比べて簡便に合成可能であり、既存のアクチノニン化合物と同等又はそれ以上の生理活性を有する新規化合物を提供することを課題とする。
【課題を解決するための手段】
【0009】
発明者らは、鋭意研究を重ねた結果、アクチノニン中の不斉コハク酸構造をアミノ酸などにより代替し、さらには、バリンを種々のアミノ酸へと置き換えることで、アクチノニンと同様な生理活性を有する新規アクチノニン誘導体が合成され、本発明を完成した。
【0010】
即ち本発明は、以下よりなる。
1.下記の一般式Iで表される化合物。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
2.下記の一般式IIで表される化合物。
下記の一般式IIで表される化合物。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
3.下記の一般式IIIで表される化合物。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
4.不斉合成工程含まない工程により合成される前項1〜3のいずれか1に記載の化合物の合成方法。
5.以下の式IVで表される化合物のうち、不斉コハク酸構造部位をアミノ酸により代替する工程を含む、前項4に記載の合成方法。
式IV:
【化4】
6.前項1〜3のいずれか1に記載の化合物を有効成分として含有する薬剤。
7.有効成分が、プロテアーゼ阻害活性を有する化合物である前項6に記載の薬剤。
8.前項6又は7に記載の薬剤、並びに薬理学的及び製剤学的に許容される担体を含む医薬組成物。
【発明の効果】
【0011】
本発明のアクチノニン類縁体化合物は、アクチノニンそのものと異なり、不斉炭素を有するコハク酸を別途合成する必要がないため、合成工程数の大幅な削減が見込め、さらには天然のアミノ酸を主原料に合成可能である。またあるものはPDF阻害活性を有し、あるものはAPN阻害活性を有する。本発明の化合物のうち、PDF阻害活性を有する化合物は、新たな抗菌剤、抗マラリア剤としての応用が期待できる。また、APN阻害活性を有する化合物は、抗がん剤、抗炎症剤としての応用が期待出来る。
【発明を実施するための最良の形態】
【0012】
本発明の化合物は、以下の一般式Iで表される。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
【0013】
また、本発明の化合物は、下記の一般式IIで表すこともできる。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
【0014】
さらに、本発明の化合物は、下記の一般式IIIで表すこともできる。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
【0015】
具体的には、本発明の化合物は、前項で示す構造式で表され、具体的には表1に示す構造式で表される。
【表1】
【0016】
より具体的には、以下の実施例で示す化合物のうち、化合物9a,9bに示す化合物が挙げられる。本発明の化合物のうち、PDF阻害薬として、化合物9a,9bに示す化合物が挙げられる。
【0017】
本発明の化合物は、以下の実施例に示す方法により合成することができる。本発明の化合物であるアクチノニン類縁体の合成方法は、従来のアクチノニン合成方法と異なり、アクチノニン分子中、バリンのアミノ基とヒドロキシルアミンとの間に相当する部分、すなわち不斉炭素を有するコハク酸を別途合成する必要がない点が特徴である。すなわち、本発明の化合物は、不斉合成工程を含まないで合成することができる。具体的には、従来のアクチノニン合成方法において、不斉合成工程を含まず、不斉コハク酸構造部位をアミノ酸へ置き換えることで、合成することができる。
【0018】
本発明において、一般式I〜IIIのいずれかで表される化合物は、さらに、薬学的に許容される塩であってもよい。また、一般式I〜IIIのいずれかの化合物又はその塩において、異性体(例えば光学異性体、幾何異性体及び互換異性体)などが存在する場合は、本発明はそれらの異性体を包含し、また溶媒和物、水和物及び種々の形状の結晶を包含するものである。
【0019】
本発明において、薬学的に許容される塩とは、薬理学的及び製剤学的に許容される一般的な塩が挙げられる。そのような塩として、具体的には以下が例示される。
塩基性付加塩としては、例えばナトリウム塩、カリウム塩等のアルカリ金属塩;例えばカルシウム塩、マグネシウム塩等のアルカリ土類金属塩;例えばアンモニウム塩;例えばトリメチルアミン塩、トリエチルアミン塩;ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、ブロカイン塩等の脂肪族アミン塩;たとえばN,N−ジベンジルエチレンジアミン等のアラルキルアミン塩;例えばピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等の複素環芳香族アミン塩;例えばテトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩;リジン塩等の塩基性アミノ酸塩等が挙げられる。
【0020】
酸付加塩としては、例えば塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;例えば酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;例えばメタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;例えばアスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等を挙げることができる。
【0021】
本発明において、一般式I〜IIIのいずれかで表される化合物は、PDFやAPNに対し拮抗作用を有する。本発明の化合物がこれらの酵素阻害作用のいずれを有するかは、本明細書の実験例に具体的に示した方法に従って容易に検定可能である。具体的には、例えば以下の方法で、検定することができる。
【0022】
酵素阻害活性については、例えばAPN阻害活性は、動物細胞、例えばHL−60細胞を用いて、本発明の化合物による蛍光性基質(Ala-MCA)の酵素的加水分解を指標として行うことができる。具体的には、5mLプラスチックチューブに50mM Tris−HClpH7.4を790μL分注し、合成化合物のDMSO溶液を10μLを加えた後、50mM Tris−HClpH7.4に懸濁した2×106cells/mL に調整したHL−60 PBS(−)細胞懸濁液を100μL加えた後、37℃に調整した恒温水槽にて10分間インキュベーションし、1mMに調整した蛍光性基質Ala-MCAの50mM Tris−HClpH7.4を100μLを加えた後、37℃に調整した恒温水槽にて30分間インキュベーションした後、1M AcONa-AcOH pH 4.0を3mL加え、酵素反応を停止し、励起波長380nm,蛍光波長420nmでの蛍光強度を測定することができる。
【0023】
本発明において、一般式I〜IIIのいずれかで表される化合物は、プロテアーゼ阻害活性を有する。プロテアーゼ阻害活性とは、具体的にはペプチド脱ホルミル化酵素(PDF)阻害活性やアミノペプチダーゼ N(APN)阻害活性をいう。
【0024】
本発明は、本発明の化合物を有効成分とする試薬又は医薬等の薬剤も、本発明の範囲に含まれる。医薬品として用いる場合には、プロテアーゼ阻害活性、例えば、PDF阻害活性やAPN阻害活性により治療効果を発揮しうる疾患に対して使用することができる。このような疾患としては、がん、炎症などが挙げられる。
【0025】
本発明の化合物を有効成分とする医薬として用いる場合には、投与量は特に限定されない。例えばPDF阻害活性剤やAPN阻害活性剤として本発明の薬剤を投与する場合は、あらゆる投与方法において適宜の投与量が容易に選択できる。例えば、経口投与の場合には有効成分を成人一日あたり0.01〜1000mg程度の範囲で用いることができる。抗がん剤、抗炎症剤等を有効成分として含む医薬と本発明の薬剤とを併用する場合には、これらの薬剤の投与期間中、及び/又はその前若しくは後の期間のいずれにおいても本発明の薬剤を投与することが可能である。
【0026】
本発明の薬剤として、上記一般式I〜IIIのいずれかで表される化合物から選ばれる1種又は2種以上の物質をそのまま投与してもよいが、好ましくは、上記の物質の1種又は2種以上を含む、経口用あるいは非経口用の医薬組成物として投与することが好ましい。経口用あるいは非経口用の医薬組成物は、当業者に利用可能な製剤用添加物、即ち薬理学的及び製剤学的に許容しうる担体を用いて製造することができる。
【0027】
経口投与に適する医薬用組成物としては、例えば、錠剤、カプセル剤、散剤、細粒剤、顆粒剤、液剤、及びシロップ剤等を挙げることができ、非経口投与に適する医薬組成物としては、例えば、注射剤、点滴剤、坐剤、吸入剤、点眼剤、点鼻剤、軟膏剤、クリーム剤、及び貼付剤等を挙げることができる。上記の医薬組成物の製造に用いられる薬理学的及び製剤学的に許容しうる担体としては、例えば、賦形剤、崩壊剤ないし崩壊補助剤、結合剤、滑沢剤、コーティング剤、色素、希釈剤、基剤、溶解剤ないし溶解補助剤、等張化剤、pH調節剤、安定化剤、噴射剤、及び粘着剤等を挙げることができる。
【0028】
本明細書の実施例に、本発明の式Iに示される好ましい化合物の製造方法を具体的に説明する。これらの製造方法において用いられた出発原料及び試薬、並びに反応条件などを適宜修飾ないし改変することにより、本発明の範囲に包含される化合物はいずれも製造可能である。本発明の化合物の製造方法は、実施例に具体的に説明されたものに限定されるものではない。
【実施例】
【0029】
以下、本発明を実施例によりさらに具体的に説明するが、本発明は下記の実施例の範囲に限定されることはない。
【0030】
[実施例]目的化合物の合成
本実施例における最終生成物9a、9bを得るまでの製造方法のスキームを図1から図4に示した。
【0031】
1)中間体 N−ボック−L−バリン O−ニトロフェノールエステル(2)の合成
【化5】
【0032】
N−ボック−L−バリン (1)(18.8g,87.5mmol)、O−ニトロフェノール(21.5g,157.5mmol)をピリジン(87mL)に溶解させ、DCC(19.5mg,87.5mmol)を加え室温で2時間撹拌し、TLCプレート (酢酸エチル:ヘキサン=1:10 ニンヒドリン試薬で呈色)で反応終了を確認した。析出したDCUを吸引ろ過により除去した。ろ液に1N HCl溶液(170mL)で酸性とし、ジエチルエーテル(200mL)で抽出した。有機層を飽和炭酸水素水(100mL×2)、水(100mL×2)、飽和食塩水(100mL)で洗浄、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去、黄色オイル状残渣を得た。この黄色オイル状残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:7)で単離し、ヘキサンで結晶化を行い、表題化合物(淡黄色結晶)を得た。収率87%。
【0033】
融点53.6−55.0℃。IR(KBr)cm−1:3360(NH),1755(C=O),1690(C=O)。[α]D23−39.4 (c0.15,CHCl3)。
1H NMR(CDCl3,300MHz)δ: 8.07 (1H,dd,Jo=8.0Hz,Jm=1.5Hz,ArH ortho to NO2),7.66(1H,td,Jo=8.0Hz,Jm=1.5Hz,ArH para to NO2),7.41(1H,td,Jo=8.0Hz,Jm=1.0Hz,ArH para to O),7.28 (1H,d, Jo=8.0 Hz,ArH ortho to O), 5.04 (1H,d,J=8.5Hz,NH),4.37(1H,q,J=9.0Hz,J=4.5Hz,NHCHCO),2.45(1H,m,CHMe2),1.47(9H,s,CMe3),1.11(3H,d, J=7.0Hz,CHMeMe),1.04 (3H,d,J=7.0 Hz,CHMeMe)。
【0034】
2)中間体 N−ボック−L−バリルプロリノール (3)の合成
【化6】
【0035】
(S)−(+)−2−ピロリジンメタノール(505mg,5.0mmol)とトリエチルアミン(3.0mL)を無水テトラヒドロフラン(70mL)に溶解させたものに、N−ボック−L−バリン O−ニトロフェノールエステル (2)(1776mg,5.25mmol)を無水テトラヒドロフラン(15mL)に溶解させたものを滴下、室温で1時間攪拌した。反応終了後、減圧下溶媒を留去、酢酸エチル(500mL)にあけた。有機層を2N HCl 溶液(200mL)、水(200mL)、飽和炭酸水素水(200mL)、飽和食塩水(200mL)で洗浄後、無水硫酸マグネシウムにより乾燥し,溶媒を減圧下留去し、黄色オイル状残渣を得た。このオイル状残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=2:1)で単離し,表題化合物(黄色オイル)を得た。収率98%。
【0036】
[α]D24 −32.2(c 0.42,CHCl3)。IR(neat)cm−1: 3400br(OH),1700(C=O),1630(C=O)。
1H−NMR(300MHz,CDCl3):δ5.23(1H,d,J=9.5Hz,NH),4.62(1H,d,J=5.5Hz,OH),4.28〜4.23(2H,m,NHCHCO,NCHCH2OH),3.84 3.79(1H,M,NHH'),3.66〜3.63(1H,m,NHH'),3.56〜3.53(1H,m,CHH'),3.48〜3.43(1H,m,CHH'),2.06〜1.85(4H,m,NCH2CH2CH2),1.62〜1.52(1H,m,CHMe2),1.41 (9H,s,CMe3),0.97(3H,d,CHMeMe),0.91(3H,d,CHMeMe)
【0037】
3)中間体 O−ベンジル−N−ボック−L−バリルプロリノール (4)の合成
【化7】
【0038】
合成法(I)
N−ボック−L−バリルプロリノール (3)(1408mg,4.69mmol)と水素化ナトリウム(50% in oil,270mg,5.62mmol)、臭化ベンジル(0.55mL)を無水テトラヒドロフラン(34mL)に溶解させ、室温で36時間攪拌した。反応終了後、溶媒を減圧下留去し、酢酸エチル (400mL)にあける。有機層を2N HCl溶液(160mL)、水(160mL)、飽和炭酸水素水(160mL)、飽和食塩水(160mL)で洗浄後、無水硫酸マグネシウムにより乾燥し,溶媒を減圧下留去した。淡黄色オイル状残渣を得た。その残渣をフラッシュカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で単離し,表題化合物(黄色オイル)を得た。収率84%。
【0039】
合成法(II)
(S)−(+)−2−ピロリジンメタノール(549.2mg,5.4mmol)とトリエチルアミン(3.0mL)を無水テトラヒドロフラン(70mL)に溶解させたものに、N−ボック−L−バリンO−ニトロフェノールエステル (3)(1839.8mg,5.4mmol)を無水テトラヒドロフラン(15mL)に溶解させたものを滴下し、室温で2時間撹拌した。
【0040】
反応終了後、氷冷下水素化ナトリウム(50% in oil 628mg,13mmol)、臭化ベンジル(1.6mL,13mmol)を加え、室温で36時間撹拌した。反応終了後、減圧下溶媒を留去し、酢酸エチル(50mL)にあけた。有機層を2N HCl(10mL)で酸性とし、飽和炭酸水素水(30mL×2)、水(30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。黄色オイル状残渣(2810mg)を得た。黄色オイル状残渣 (2810mg)をフラッシュカラムクロマトグラフィーを行った。酢酸エチル:ヘキサン=1:1より表題化合物(黄色オイル)を得た。収率65%。
【0041】
[α]D26 −60.0(c 1.0,CHCl3) 。IR(neat)cm−1:3300(NH),1750(C=O),1640 (C=O)。
1H−NMR(300MHz,CDCl3):δ7.33〜7.27(5H,m,ArH),5.28(1H,d,J=9.3Hz,NH),4.49(2H,d,J=2.4Hz,CH2Ph),4.35〜4.24(2H,m,NHCHCO,NCHCH2O),3.68〜3.46(4H,m,NCH2,NCHCH2O),2.04〜1.88(5H,m,NCH2CH2CH,CHMe2),1.42(9H,s,CMe3),0.95(3H,d,J=7.0Hz,CHMeMe),0.89(3H,d,J=7.0Hz,CHMeMe)。
【0042】
4)中間体 O−ベンジル−N−ボック−L−メチオニルバリルプロリノール (6a)の合成
【化8】
【0043】
O−ベンジル−N−ボック−L−バリルプロリノール (4)(1543mg,3.95mmol)を塩化メチレン(7.11mL)に溶解させ,トリフルオロ酢酸(4.3mL)を滴下し,室温で1時間攪拌する。反応終了後,酢酸エチル(400mL)にあけ,飽和炭酸水素水,水(各160mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣のO−ベンジル−L−バリルプロリノール (1166mg)を得た。
【0044】
O−ベンジル−N−ボック−L−バリルプロリノール(1166mg)、N−ボック−L−メチオニン(1083mg,4.34mmol)、HOBt(587mg,4.34mmol)をジメチルホルムアイド(27mL)に溶解後,EDC(830mg,4.34mmol)を加え,2時間攪拌する。反応終了後,酢酸エチル(400mL)にあけ,飽和炭酸水素水,水,飽和食塩水(各160mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィーを行い、酢酸エチル:ヘキサン=2:3溶出部より表題化合物を得た。収率76%。
【0045】
[α]D26 56.1 (c 1.0,CHCl3) 。IR(neat)cm−1 3300(NH),1640(C=O)。
1H−NMR(300MHz,CDCl3):店 δ732〜7.29(5H,m,ArH),6.81 (1H,d,J=10.8Hz, NH),5.16 (1H,d,J=8.4Hz,NH),4.59−4.50 (1H,m,NHCHCO),4.48(2H,d,J=2.1Hz,CH2Ph),4.30〜4.26(2H,m,NHCHCO,NCHCH2O),3.69〜3.46(4H,m,NCH2,NCHCH2O),2.55(2H,t,J=6.9Hz,SCH2CH2),2.10(1H,s,CH3S),2.08〜1.89(7H,m,NCH2CH2CH,CHMe2,SCH2CH2),1.44(9H,s,CMe3),0.94 (3H,d,J=7.0Hz,CHMeMe),0.89(3H,d,J=7.0Hz,CHMeMe)。
【0046】
13C−NMR(75MHz,CDCl3):171.4,170.1,155.5,138.4,128.4,127.6,127.5,80.0,73.2,70.1,56.8,56.8,55.9,53.6,47.8,31.9,31.6,30.2,28.4,27.4,24.6,19.5。
【0047】
5)中間体 O−ベンジル−O−ベンジルオキシカルボニル−L−メチオニルバリルプロリノール (8a)の合成
【化9】
【0048】
O−ベンジル−N−ボック−L−メチオニルバリルプロリノール (6a)(624mg,1.2mmol) を塩化メチレン(2mL)に溶解させ,トリフルオロ酢酸(1.3mL)を滴下し,室温で1時間攪拌する。反応終了後,酢酸エチル(150mL)にあけ,飽和炭酸水素水(60mL),飽和食塩水(30mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣のO−ベンジル−L−メチオニルバリルプロリノールを得た。
【0049】
O−ベンジル−L−メチオニルバリルプロリノール(459mg), 4−ニトロフェニル−N−(O−ベンジルヒドロキシ)カルバメート(345mg,1.2mmol),トリエチルアミン(0.167mL)をCH2Cl2(14)に溶解し,1.5時間攪拌した。反応終了後,1M 水酸化ナトリウム溶液(30mL×3),1M HCl(30mL), 水(30mL)で洗浄後,無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィーを酢酸エチル:ヘキサン=1:1で表題化合物を得た。収率73%。
【0050】
IR(CHCl3)cm−1:3400(NH),1640(C=O),1680(C=O)。
1H−NMR(300 MHz,CDCl3):7.38〜7.29(10H,m,ArH)。
7.04(1H,d,J=9.0Hz,NH),6.33(1H,d,J=9.0Hz,NH),4.80(2H,d,J=5.1Hz,CH2Ph),4.56〜4.51(2H,m,NHCHCO,NHCHCO),4.47(2H,s,CH2Ph),4.30(1H,s,NCHCH2O),3.70〜3.33(4H,m,NCH2,NCHCH2O),2.44(2H,t,J=7.2Hz,SCH2CH2),2.07(3H,s,CH3S),2.04〜1.86(7H,m,NCH2CH2CH2,CHMe2,SCH2CH2),0.93(3H,d,J=6.6Hz,CHMeMe),0.87(3H,d,J=6.6Hz,CHMeMe)。
【0051】
13C−NMR (75MHz,CDCl3):171.3,170.3,159.6,138.5,135.6,129.4,128.8,128.4,127.8,127.5,78.7,73.3,70.1,56.8,56.1,52.4,47.9,32.5,31.5,30.1,27.4,24.6,19.4,17.9,15.4。
【0052】
6)O−ベンジル−N−(N−(O−ベンジルヒドロキシカルボニル)−L−メチオニル−バリル)(9a)L−プロリノールの合成
【化10】
【0053】
O−ベンジル−O−ベンジルオキシカルボニル−L−メチオニルバリルプロリノール(8a)(228mg,0.4mmol)を無水テトラヒドロフラン(8mL)に溶解させ,−78℃冷却下,液体アンモニア(約45mL)を滴下する。反応液の色が,青くなるまでナトリウムを少しずつ加える。反応液の青色が消失するまで塩化アンモニウムを加える。液体アンモニウムを留去後,室温まで昇温する。残渣を酢酸エチルにあけ,ろ過する。無水硫酸マグネシウムにより乾燥し,減圧下溶媒を留去する。無色オイル状残渣を得た。塩化メチレンに溶解させ,フラッシュカラムクロマトグラフィーを行い,酢酸エチル:2−プロパノール = 20:1で表題化合物を得た。収率39%。
【0054】
IR(CHCl3)cm−1:3400(OH),1720(C=O),1670(C=O),1620(C=O)。1H−NMR(300MHz,CDCl3):8.48(1H,s,OH),7.89(1H,s,OH),7.74(1H,d,J=8.7Hz,NH),6.76(1H,d,J=8.4Hz,NH),4.70〜4.52(2H,m,NHCHCO,NHCHCO),4.42(1H,br s,NH),4.24(1Hbr s,NCHCH2OH),3.90〜3.86,3.64〜3.49(4H,m,NCH2,NCHCH2OH),2.55(2H,m,SCH2CH2),2.08(3H,s,CH3S),2.14〜1.73(7H,m,NCH2CH2CH2,CHMe2,SCH2CH2),0.96〜0.92(6H,m,CHMeMe)。
【0055】
13C−NMR(75 MHz,CDCl3): 172.5,172.2,161.8,65.1,60.8,56.5,53.2,48.4,32.2,31.4,30.3,27.7,24.3,19.4,18.3。
【0056】
7)中間体 O−ベンジル−N−(N−ボック−L−バリル−L−ノルロイシル)−L−プロリノール (6b)の合成
【化11】
【0057】
O−ベンジル−N−(N−ボック−L−バリル)−L−プロリノール (4)(301.1mg,0.77mmol)を塩化メチレン(1.4mL)に溶解させ、トリフルオロ酢酸(0.85mL)を滴下し、室温で1時間撹拌した。反応終了後、酢酸エチル(30mL)にあけ、飽和炭酸水素水、水(各30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒留去した。無色オイル状残渣を得た。
O−ベンジル−L−バリル−L−プロリノール(200mg,0.68mmol)、ノルロイシン(175mg,0.76mmol)、HOBt(101.1mg,0.74mmol)をジメチルホルムアミド(5mL)に溶解後、EDC(142.9mg,0.75mmol)を加え、1時間半攪拌した。反応終了後、酢酸エチル(20mL)にあけ、飽和炭酸水素水、水(各20mL×2)、飽和食塩水(20mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒留去した。無色オイル状残渣を得た。無色オイル状残渣をフラッシュカラムクロマトグラフィーを行った。酢酸エチル:ヘキサン=1:1溶出部より表題化合物を得た。収率68%。
【0058】
1H−NMR(300MHz,CDCl3)δ:7.29〜7.23(5H,m,ArH'),6.80(1H,d,J=8.7Hz,NH),5.06(1H,d,J=8.4 Hz,NH),4.45(1H,d,J=1.8Hz,CHAHBPh),4.45(1H,d,J=1.8Hz,CHAHBPh),4.31〜4.27(1H,m,NHCHCO),4.10〜4.05(1H,m,NCH2CH2CH2CH),3.67〜3.44(4H,m,CH2,OCH2Ph,NCH2CH2CH2CH),2.01〜1.55,1.27〜1.22(11H,m,CH(Me)2,CHCH2CH2CH2CH3,NCH2CH2CH2CH),1.42(9H,s,CMe3),0.92〜0.85(9H,m,CHMe2,CHCH2CH2CH2CH3)。IR(KBr)cm−1:3300(NH),1710(C=O),1630(C=O)。
【0059】
8)中間体 O−ベンジル−O−ベンジルオキシカルバモイル−L−ノルロイシルバリルプロリノール(8b)の合成
【化12】
【0060】
O−ベンジル−N−(N−ボック−L−バリル−L−ノルロイシル)−L−プロリノール(6b)(262.3mg,0.52mmol)を塩化メチレン(1mL)に溶解させ、トリフルオロ酢酸(0.6mL)を滴下し、室温で1時間撹拌した。反応終了後、酢酸エチル(30mL)にあけ、飽和炭酸水素水、水(各30mL×2)、飽和食塩水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。O−ベンジル−N−(L−バリル−L−ノルロイシル)−L−プロリノール(228.4mg,0.57mmol)、4−ニトロフェニル−N−(O−ベンジルオキシ)カルバメ−ト(63.5mg,0.57mmol)、トリエチルアミン(0.08mL,0.57mmol)を塩化メチレン(7mL)に溶解し、2時間攪拌した。反応終了後、1M 水酸化ナトリウム(20×3mL)、1M HCl水溶液(20mL)、水(20mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。無色オイル状残渣をフラッシュカラムクロマトグラフィーで酢酸エチル:ヘキサン=1:1溶出部より表題化合物を得た。収率71%。
【0061】
1H−NMR(300MHz,CDCl3)δ:8.21(1H,d,J=9.0Hz,NH),7.89(1H,d,J=7.5Hz,NH),7.40〜7.26(10H,m,ArH'),6.12(1H,d,J=8.7Hz,NH),4.75(4H,d,J=1.8Hz,CH2Ph,CH2Ph),4.28〜4.29(1H,m,NHCHCO),3.57〜3.55(4H,m,CH2,OCH2Ph,NCH2CH2CH2CH),2.44〜2.43(1H,m,NHCHCO),2.07〜1.88(4H,m,NCH2CH2CH2CH),1.83〜1.14(6H,m,CHCH2CH2CH2CH3),0.87〜0.83(9H,m,CHMe2,CHCH2CH2CH2CH3)。
【0062】
13C−NMR(75MHz,CDCl3)δ:172.3,170.5,159.8,138.4,135.7,129.3,128.3,128.3,127.5,127.4,129.3,128.6,128.4,128.3,127.5,127.4,78.5,73.1,70.0,56.7,56.0,53.0,47.8,33.0,31.3,28.3,27.5,27.3,24.4,22.4,19.3,18.1,14.0。
【0063】
9)O−ベンジル−N−(N−(O−ベンジルハイドロキシカルボニル)−L−ノルロイシル―バリル)−L−プロリノール (9b)の合成
【化13】
【0064】
O−ベンジル−O−ベンジルオキシカルバモイル−L−ノルロイシルバリルプロリノール(8b)をメタノール(5mL)に溶解させ、酢酸(0.6mL)を加えた後、10%パラジウム/カーボン(27mg)を加え,水素(1atm)雰囲気下、室温で6時間撹拌した。反応終了後、10%パラジウム/カーボンをろ過し、水(20mL×2)で抽出し、炭酸水素水(30mL)で洗浄後、無水硫酸マグネシウムにより乾燥し、減圧下溶媒を留去した。無色オイル状残渣を得た。フラッシュカラムクロマトグラフィー を行い。酢酸エチル:イソプロピルアルコール=1:1溶出部より表題化合物を得た。
【0065】
[実験例]酵素阻害活性の測定
酵素阻害活性については、例えばAPN阻害活性は、HL−60細胞を用いて、これらによる蛍光性基質(Ala-MCA)の酵素的加水分解を指標として行った。具体的には、5mLプラスチックチューブに50mM Tris−HClpH7.4を790μLを分注し、合成化合物のDMSO溶液を10μL加えた後、50mM Tris−HClpH7.4に懸濁した2×106cells/mL に調整したHL−60 PBS(−)細胞懸濁液を100μL加えた後、37℃に調整した恒温水槽にて10分間インキュベーションし、1mMに調整した蛍光性基質Ala-MCAの50mM Tris−HClpH7.4を100μL加えた後、37℃に調整した恒温水槽にて30分間インキュベーションした後、1M AcONa-AcOH pH 4.0を3mL加え、酵素反応を停止し、励起波長380nm,蛍光波長420nmでの蛍光強度を測定することができる。
【産業上の利用可能性】
【0066】
以上詳述したように、本発明の化合物(アクチノニン類縁体)は、プロテアーゼ阻害活性、具体的にはPDF阻害活性やAPN阻害活性を有するので、PDF阻害活性やAPN阻害活性により治療効果を発揮しうる疾患に対して使用することができる。このような疾患としては、例えば、がん、炎症などが挙げられ、このような疾患に対する医薬として利用することができる。また、生化学試験用試薬としても利用することができる。
【図面の簡単な説明】
【0067】
【図1】中間体6の化合物の合成スキームを示す図である。
【図2】中間体7の化合物の合成スキームを示す図である。
【図3】中間体8の化合物の合成スキームを示す図である。
【図4】目的化合物9の合成スキームを示す図である。
【特許請求の範囲】
【請求項1】
下記の一般式Iで表される化合物。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
【請求項2】
下記の一般式IIで表される化合物。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
【請求項3】
下記の一般式IIIで表される化合物。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
【請求項4】
不斉合成工程含まない工程により合成される請求項1〜3のいずれか1に記載の化合物の合成方法。
【請求項5】
以下の式IVで表される化合物のうち、不斉コハク酸構造部位をアミノ酸により代替する工程を含む、請求項4に記載の合成方法。
式IV:
【化4】
【請求項6】
請求項1〜3のいずれか1に記載の化合物を有効成分として含有する薬剤。
【請求項7】
有効成分が、プロテアーゼ阻害活性を有する化合物である請求項6に記載の薬剤。
【請求項8】
請求項6又は7に記載の薬剤、並びに薬理学的及び製剤学的に許容される担体を含む医薬組成物。
【請求項1】
下記の一般式Iで表される化合物。
一般式I:
【化1】
(式中、Aは、C1-3のアルキル、アミノ基等から選択され、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。但し、AがCH2、R1がC5H11、R2がi-Pr及びWがCONHOHの場合を除く。)
【請求項2】
下記の一般式IIで表される化合物。
一般式II:
【化2】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択され、Wは、アルキルチオール、カルボン酸エステル、並びにカルボキシル基、ヒドロキサム酸及びそれらの塩から選択される化合物。)
【請求項3】
下記の一般式IIIで表される化合物。
一般式III:
【化3】
(式中、R1, R2は、各々直鎖アルキル、分枝アルキル、及び芳香環を有するアルキル、ヒドロキシル基、アミノ基、カルボキシル基、アミド基、チオール基、グアニジノ基などを有するアルキルから選択される化合物。)
【請求項4】
不斉合成工程含まない工程により合成される請求項1〜3のいずれか1に記載の化合物の合成方法。
【請求項5】
以下の式IVで表される化合物のうち、不斉コハク酸構造部位をアミノ酸により代替する工程を含む、請求項4に記載の合成方法。
式IV:
【化4】
【請求項6】
請求項1〜3のいずれか1に記載の化合物を有効成分として含有する薬剤。
【請求項7】
有効成分が、プロテアーゼ阻害活性を有する化合物である請求項6に記載の薬剤。
【請求項8】
請求項6又は7に記載の薬剤、並びに薬理学的及び製剤学的に許容される担体を含む医薬組成物。
【図1】

【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2010−111628(P2010−111628A)
【公開日】平成22年5月20日(2010.5.20)
【国際特許分類】
【出願番号】特願2008−286015(P2008−286015)
【出願日】平成20年11月7日(2008.11.7)
【出願人】(504147243)国立大学法人 岡山大学 (444)
【Fターム(参考)】
【公開日】平成22年5月20日(2010.5.20)
【国際特許分類】
【出願日】平成20年11月7日(2008.11.7)
【出願人】(504147243)国立大学法人 岡山大学 (444)
【Fターム(参考)】
[ Back to top ]