
アクチン結合タンパク質の細胞運動関連疾患への利用
【課題】アクチン結合タンパク質の利用に関し、特に、セリン/スレオニンキナーゼAktの新規な基質でありアクチン結合タンパク質であるGirdin(Akt Phosphorylation Enhancer)の当該タンパク質が関連する疾患等への利用に関する。Aktと細胞運動との分子機構に関わるタンパク質を同定するとともにその機能を解明し、その利用を図る。
【解決手段】特定のアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いて細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩をスクリーニングする。
【解決手段】特定のアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いて細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩をスクリーニングする。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アクチン結合タンパク質の利用に関し、特に、セリン/スレオニンキナーゼAktの新規な基質でありアクチン結合タンパク質であるGirdin(Akt Phosphorylation Enhancer)の当該タンパク質が関連する疾患等への利用に関する。
【背景技術】
【0002】
細胞運動の制御機構は、発生における形態形成、創傷治癒や血管新生のほか、癌細胞の浸潤、動脈硬化、免疫疾患などの病態に密接に関わっている。細胞運動の制御分子としては、低分子量のGタンパク質を始め、数多く同定されている(Ridley A, al., Science(2003)302、p1702-1709)。セリン/スレオニンキナーゼであるAkt(プロテインキナーゼB:PKBともいう。)は、細胞の生存や増殖を制御する重要な因子として知られているが、細胞運動に必須であることも哺乳類や細胞性粘菌などの多くの種で示されている(Higuchi M, al., Curr. Biol(2001)11, p1958-1962、Merlot S, al., J. Cell Sci(2003)116, p3471-3478)。Aktの発現が顕著である悪性腫瘍は浸潤性が高い傾向にある。
【発明の概要】
【発明が解決しようとする課題】
【0003】
しかしながら、Aktが細胞運動を促進する分子機構については全く知られていない。本発明は、Aktと細胞運動との分子機構に関わるタンパク質を同定するとともにその機能を解明し、その利用を図ることを一つの目的とする。
【課題を解決するための手段】
【0004】
本発明者らは、酵母two−hybrid法を用いてAktの新規基質を見出すとともに、この基質がアクチン結合タンパク質であって、Aktによってリン酸化されると移動する細胞の先端部(リーディングエッジ)に局在が変化される一方、このアクチン結合タンパク質の変異体を細胞に発現させると細胞の形態に変化が生じて増殖因子の刺激に依存した細胞運動が障害されることを見出し、このアクチン結合タンパク質がアクチン細胞骨格の統合及び細胞運動によって必須であることを見出し、本発明を完成した。すなわち、本発明者らの知見によれば、以下の手段が提供される。
【0005】
(1)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
(2)前記タンパク質は、配列番号2で表されるアミノ酸配列からなるタンパク質である、(1)に記載のスクリーニング方法。
(3)前記部分ペプチドは、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域(CT1領域及び/又はCT2領域)である、(1)に記載のスクリーニング方法。
(4)前記タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている、(1)又は(2)に記載のスクリーニング方法。
(5)セリン/スレオニンキナーゼAktと配列番号1で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含むタンパク質又はその一部との結合活性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(6)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(7)アクチンフィラメント結合能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(8)アクチンストレスファイバー形成能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(9)ラポリメディアにおけるアクチンメッシュワークの形成能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(10)細胞膜結合性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(11)フォスフォイノシタイド結合性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(12)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質をコードするポリヌクレオチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
(13)細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬のスクリーニング方法である、(1)〜(12)のいずれかに記載のスクリーニング方法。
(14)前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、(1)〜(13)のいずれかに記載のスクリーニング方法。
(15)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかあるいは2種以上を発現する化合物又はその塩である、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合の阻害活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の阻害活性
c)アクチンフィラメントへの結合の阻害活性
d)アクチンストレスファイバー形成の阻害活性
e)ラポリメディアにおけるアクチンメッシュワークの形成の阻害活性
f)細胞膜結合の阻害活性
g)フォスフォイノシタイドへの結合の阻害活性
(16)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質の発現量を抑制する化合物である、細胞運動、細胞移動又は血管形成のいずれかが関連する疾患の予防・治療薬。
(17)前記化合物は、配列番号2で表されるアミノ酸配列をコードするポリヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するポリヌクレオチドである、(16)に記載の予防・治療薬。
(18)RNA干渉を発現するRNAである、(17)に記載の予防・治療薬。
(19)前記(18)に記載のRNAをコードするDNAである、(16)に記載の予防・治療薬。
(20)前記(19)に記載のDNAを含有する組換えベクターである、(16)に記載の予防・治療薬。
(21)前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、(16)〜(20)のいずれかに記載の予防・治療薬。
(22)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された細胞。
(23)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された動物。
(24)前記遺伝子の発現量の抑制はノックダウンによる(23)に記載の動物。
(25)細胞運動、細胞移動及び血管形成のいずれかに関連する疾患の研究用、予防・治
療薬のスクリーニング用である、(23)又は(24)に記載の動物。
(26)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定性的分析方法。
(27)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定量方法。
(28)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに特異的な抗体。
(29)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに特異的な抗体。
(30)前記(28)又は(29)に記載の抗体を含む、細胞運動、細胞移動及び血管形成の活性化又は阻害の研究用試薬。
(31)前記(28)又は(29)に記載の抗体を含む、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断薬。
(32)疾患部位の細胞における配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかを利用する、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断方法。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化活性
c)アクチンフィラメントへの結合活性
d)アクチンストレスファイバー形成活性
e)ラポリメディアにおけるアクチンメッシュワークの形成活性
f)細胞膜結合活性
g)フォスフォイノシタイドへの結合活性
【図面の簡単な説明】
【0006】
【図1a】図1aは、 酵母two-hybridアッセイに用いた各種ベイト(おとり)のコンストラクト(いろいろな遺伝子を組み込んだ発現ベクター)を示す。ヒトAkt1の各種cDNAフラグメントをGAL4転写因子のDNA結合ドメインに融合させるようにpAS2ベクター内に構築した。PH, プレクストリンホモロジードメイン; KD, キナーゼ活性ドメイン; RD, 調節ドメイン。
【図1b】図1bは、 酵母two-hybridアッセイにおけるヒトAkt1とクローンF-10およびF-12の結合の様式を示す。pAS, ネガティブコントロール; pVA3-pTD1, ポジティブコントロール, His, ヒスチジン。
【図1c】図1cは、 Girdinの一次構造とCOILSアルゴリズムによって予測されるGirdinの構造を示す。
【図1d】図1dは、ヒト各種組織におけるGirdinのメッセンジャーRNA(mRNA)の普遍的な発現を示す。クロンテック社の多組織ノーザンブロットメンブレンをGirdin cDNAの3’領域内のプローブによってハイブリダイズした。
【図1e】図1eは、ヒト脳および精巣における内因性Girdinの発現を抗Girdinポリクローナル抗体を用いて還元条件のウェスタンブロット法で検出した結果を示す。(対照の)ウサギIgGでは検出されなかったことを示す。
【図1f】図1fは、内因性Girdinによって形成される大きなタンパク質複合体の検出を示す。核成分を除いたCOS7細胞を100μM BS3で架橋した条件、および架橋しない条件下で抗Girdin抗体を用いてウェスタンブロット法を行った。架橋後(矢印)では架橋しない場合(矢頭)に比べてかなり大きいサイズのバンドが検出された。
【図1g】図1gは、GirdinのN末端ドメイン(NT)は多量体を形成することを示す。V5タグ(NT-V5)およびmycタグを付加したNT(NT-myc)をCOS7細胞にトランスフェクトし、抗myc抗体で免疫沈降したところ、NT-V5がNT-mycと共沈した。下のパネルにみられる複数のバンドはNT-mycの分解産物を示しているのかもしれない。
【図1h】Girdinの推測されるアミノ酸配列を示す。α−ヘリカルコイルドコイル構造を形成すると考えられる、135ヘプタドリピート(abcdefg)135におけるポジションa及びdにおけるアミノ酸残基は、それぞれ赤と緑で示している。フォスフォイノシタイドへの推定結合サイトを下線を施して示す。ボックスは、推定されるAktリン酸化サイトを示す。
【図1i】Girdinは細胞内でオリゴマーを形成し、アクチンフィラメントと大きなタンパク質複合体を形成することを示す図であり、Aは、COS7細胞溶解物Superose6PC3.2/20(Amersham)でゲルろ過し、フラクション後、抗Girdin抗体(上段)及び抗アクチン抗体(下段)でウエスタンブロット分析により各フラクションを確認し、それらの溶出特性を確認した図である。アクチンは二つのピークとして溶出し、最初の一つは、ボイドボリュームに溶出し、アクチン(F-アクチン)の重合体に対応していた。アクチンの主要な溶出ピークは67kDa未満の低分子量に対応する位置にあり、これは、部分的に解離しているかあるいは単量体(G-アクチン)を示唆した。これらのデータは、Girdinが各種の長さのF-アクチンと大きな複合体を形成していることを示している。Bは、COS7細胞を、V5エピトープタグ付きGFP−Girdin N末端フラグメント(NT)(GFP−NT−V5)(上段)又はmycエピトープタグ付きNT(NT−myc)(下段)のいずれかでトランスフェクトし、細胞溶解物をゲルろ過のためにSuperose 6PC処理した。フラクション後、各フラクションを抗V5抗体及び抗myc抗体を用いてウエスタンブロット分析した。GFT−NT−V5又はNT−mycモノマーの算出した分子量は、それぞれ58kDa又は28kDaであり、これらのデータはNTドメインは二量体を形成していることを示唆した。方法:ディッシュ中のCOS7細胞は冷PBSで洗浄し、0.5mlの25mMTris-HCl,pH8.0、250mM、NaCl5mM、1mMEDTA、1mMDTT.懸濁液は、超音波処理し4℃、60分間100000gで遠心分離した。上清をSuperose6PC3.2/20(Amersham)で処理した。溶出は、40μl/minの流速で行った。50μlのフラクションを収集した。使用したタンパク質マーカーは、タイログロブリン(669K)、フェリチン(440K)及びウシ血清アルブミン(67K)であった。
【図2a】図2a中、aは、GirdinのC末端ドメイン(CT)の1416番目セリンがAktのリン酸化サイトであることを示す。bは、Girdin CT-V5の野生型(WT)およびSA変異体(1416番目のセリンをアラニンに置換した変異体)をCOS7細胞にトランスフェクトし、抗V5抗体で免疫沈降し、放射性標識されたATPの存在下において、活性型および非活性型の組換えAktとインキュベーションした。リン酸化されたGirdin CTはオートラジオグラフィーで(上段)、免疫沈降産物はウェスタンブロット法(下段)によって確認したことを示す。
【図2b】図2b中、aは、COS7細胞にトランスフェクトしたV5タグが付加された様々なGirdinのフラグメントを示す。bは、免疫沈降後、放射性標識されたATPの存在下において、活性型Aktとインキュベーションした結果を示す。免疫沈降産物はウェスタンブロット法(左)によって、リン酸化されたGirdin CTはオートラジオグラフィーで(右)によって確認したことを示す。
【図2c】図2cは、 Girdin CT WTおよびSAを各種Akt変異体と共にCOS7細胞へトランスフェクトし、免疫沈降後、抗リン酸化Girdin抗体(anti-P-Girdin)を用いてウェスタンブロット法を示す(上段)。Girdin CTと各種Aktの発現はウェスタンブロット法によるモニター結果を示す(中および下段)。
【図2d】図2dは、 DMSO、PD98059, LY294002およびWortmanninの存在下においてCOS7細胞を上皮増殖因子(EGF)で刺激した。抗Girdin抗体で免疫沈降後、抗リン酸化Girdin抗体および抗Girdin抗体を用いてウェスタンブロット法を行った。Aktの活性化は抗リン酸化Akt抗体を用いて検出した。
【図2e】図2eは、 Akt WT及びAkt CT及びAkt DNのCOS7形質転換体におけるGirdinのリン酸化の誘導を示す。COS7細胞にAktの各種変異体をトランスフェクトした。内因性Girdinを抗Girdin抗体を用いて免疫沈降後、抗リン酸化Girdin抗体および抗Girdin抗体を用いてウェスタンブロット法を行った。
【図2f】図2f中、aは、内因性Girdinのリン酸化におけるAktのノックダウンの影響を示す。COS7細胞にコントロールあるいはAkt siRNAをトランスフェクトし、48時間培養した。その後細胞をEGFで30分刺激し、(E)で記述した方法を用いてGirdinのリン酸化を検出した。bは、COS7細胞におけるsiRNAによるAktの発現抑制を示す。
【図3a】図3a中、a及びbは、刺激前(a)あるいはEGFで刺激後(b)のVero細胞をAlexa488標識のファロイジン(アクチン線維に結合する試薬)および抗Cortactin抗体、抗Girdin抗体で二重染色を行った結果を示す。矢頭はリーディングエッジ(移動する細胞の先端部)におけるラメリポディア(葉状仮足:細胞運動の先端部に形成されるアクチン線維の豊富な構造体)を示す。
【図3b】]図3bは、Girdinの各種フラグメントの細胞内局在。GFP(緑色の蛍光色素遺伝子)と融合させたGirdinの各種フラグメントをVero細胞に発現させ、固定後、それぞれのプローブによって染色した結果を示す。矢印は発現したタンパク質の凝集体を示している。
【図3c】図3cは、Girdin CT2フラグメントはin vitro(試験管内)において直接アクチン線維と結合する。精製されたGST、GST-CT2およびalpha-アクチニンをin vitroで調整されたアクチン線維とインキュベートした。引き続きF-アクチン線維を超遠心によってペレット化し、F-アクチンと共に沈降した各種タンパク質をゲルのCBB染色法(クーマシー染色法)によって検出した結果を示す。
【図3d】図3dは、 GST-CT2とアクチン繊維の結合様式を示す。一定量のGST-CT2を様々な量のF-アクチンと混合し、引き続き超遠心を行った。非結合型、および結合型のF-アクチンを定量し、結合型のGST-CT2をF-アクチンの濃度に対してプロットした。Scatchard解析によってKd値を計算した。
【図3e】図3e中、aは、 Girdinの各種ドメインの局在と機能のまとめを示す。bは、提案されるGirdinの構造を示す。
【図4a】図4aは、Vero細胞におけるsiRNAによるGirdinの発現抑制Aktを示す。コントロールsiRNAおよびGirdin siRNAをトランスフェクトされたVero細胞の細胞抽出液を抗Girdin抗体、抗Akt抗体、および抗アクチン抗体を用いて検出した。
【図4b】図4bは、Vero細胞にコントロールsiRNAおよびGirdin siRNAをトランスフェクトし、72時間後に固定し、Alexa488標識のファロイジンおよび抗Girdin抗体で二重染色を行った。矢頭は突起の先端におけるラメリポディアを示す。
【図4c】図4cは、 各種siRNAのトランスフェクション後、複数の突起を形成している細胞数を抗Girdin抗体およびAlexa488標識のファロイジンによる染色によって定量した。それぞれの群で100個以上の細胞を計測した。結果は平均±標準誤差で示す。
【図4d】図4dは、Vero細胞にGFP-アクチンと各種siRNAをトランスフェクトし48時間培養した。細胞をフィブロネクチンでコートしたガラスのカバースリップにまき、細胞運動を誘発するために細胞層に先の尖ったチップで傷を加えた。細胞イメージの取得は細胞層に傷を加えてから2時間後に開始し、5-6時間、90秒ごとに行った。矢印は傷の方向に向かって細胞が運動する方向を示す。
【図4e】図4eは、Girdinのノックダウンがストレスファイバーの形成及びRETチロシンキナーゼSK−N−MC細胞のラメリポディアへ与える影響を示す。Aは、SK−N−MC(RET)細胞を、siRNAのトランスフェクション後、72時間後に収集した。細胞溶解物は、SDS−PAGEで分析し、抗Girdin抗体及び抗アクチン抗体を用いてウエスタンブロット分析した結果を示す。Bは、siRNAでトランスフェクトしたSK−N−MC(RET)細胞を、50ng/mlのGDNFで60分間刺激し、固定化し、その後Alexa−488ファロイディンで染色した。Girdinがノックダウンされた細胞は、コントロール細胞と比較して多数の突起を伴う顕著な形態変化を呈した。矢印は、リーディングエッジにおけるラメリポディアを示している。Girdinノックダウン細胞は、ラメリポディアを形成するものの、コントロール細胞に比較して極性が弱く伸長性も低かったことを示す。
【図5a】図5aは、抗Girdin抗体を用いた免疫電子顕微鏡による解析結果を示す。
【図5b】図5bは、円で示したGirdin分子の免疫金コロイドシグナルを拡大して示す。矢印は免疫金コロイドシグナルを示す。
【図5c】図5cは、コントロールあるいはGirdin siRNAをトランスフェクトしたVero細胞のアクチン線維の微細構造を示す。
【図6a】図6aは、刺激前、EGFで刺激後、あるいはGirdin siRNAをトランスフェクトしたVero細胞を固定し、Alexa488標識のファロイジンおよび抗リン酸化Girdin抗体で二重染色を行った。矢印はリン酸化Girdinが集積しているラメリポディアを示す。
【図6b】図6bは、 移動中のVero細胞を抗Cortactin抗体と抗リン酸化Girdin抗体で二重染色を行った結果を示す。
【図6c】図6c中、aは GirdinのAktリン酸化サイトの上流に位置している想定されるイノシトールリン脂質結合サイトを示す。bは、GFP-CT1あるいはGFP-CT1 ΔPB(想定されるイノシトールリン脂質結合サイトを除いてある)をVero細胞にトランスフェクトした。矢印は細胞膜におけるGFPシグナルを示す。
【図6d】図6dは、GST-CTの精製とタンパク質-脂質結合アッセイを示す。aは、精製されたGST-CTあるいはGST-CT ΔPBを抗Girdin抗体を用いたウェスタンブロット法で解析した結果を示す。bは、in vitroにおける組換えAktによるGST-CTのリン酸化を示す。In vitroキナーゼアッセイにおけるGST-CTのリン酸化は抗リン酸化Girdin抗体で検出した。
【図6e】図6eは、GST-CT、GST-CT ΔPBおよびリン酸化GST-CTを用いたタンパク質-脂質結合アッセイを示す。GST-CTおよびGST-CT ΔPBの脂質に対する結合は抗Girdin抗体で検出し、またリン酸化GST-CTの脂質に対する結合は抗リン酸化Girdin抗体で検出した。メンブレンにスポットされた各種脂質を(Ed)に示す。
【図6f】図6fは、リン酸化GST-CTはF-アクチンに結合することを示す。GST-CTをin vitroにおいてAktによりリン酸化させ、続いてアクチン沈降アッセイに用いた。一定量のGST-CTをリン酸化させ、様々な量のF-アクチンと混合し、超遠心を行った。沈殿画分に含まれるF-アクチンに結合したリン酸化GST-CTは抗リン酸化Girdin抗体を用いたウェスタンブロット法で検出した。
【図6g】図6gは、活性Aktとリン酸化GirdinとがEGF刺激ベロ細胞のラメリポディアに共局在することを示す。24時間血清飢餓状態においたベロ細胞を未処理A及び50ng/mlのEGF処理Bのいずれかで30分間処理し、固定化し、抗リン酸化Girdinモノクローナル抗体(緑、セルシグナリングテクノロジー)及び抗リン酸化Girdin抗体(赤)で染色した結果を示す。イメージは、共焦点顕微鏡で視覚化した。矢印は、リン酸化Aktとリン酸化Girdinのリーディングエッジのラメリポディアでの共局在を示す。白いボックス内の領域を拡大して示す。
【図6h】図6hは、CT1ドメインのアミノ酸置換のGirdinの機能における影響を示す。Aは、推定されるフォスフォイノシタイド結合(PB)サイトにおける正の電荷が細胞膜におけるCT1ドメインの局在に必要であることを示す。各種のCT1変異体、すなわち、リジン及びアルギニンの正電荷アミノ酸残基のいずれかをアラニンで置換した変異体(PBala、PBalaF、PBalaR)Ser−1416をアラニン又はアスパラギン酸によって置換した変異体(SA、SD)をaに示すようにして作製し、そしてこれらのベロ細胞内における局在性を調べた(b)。矢じりは、細胞膜におけるGFP−CT1の局在を示しており、矢印は、変異体が細胞膜へ局在していないことを示す。Bは、タンパク質−脂質オーバーレイアッセイの結果を示す。GST−CT野生型(ST)及びPBサイトの正電荷アミノ酸残基をアラニンで置換したGSTCT−PBalaをBL−21CodonPlus細胞(Stratagene)で発現させ、精製し(a)、そして、タンパク質−脂質オーバーレイアッセイに供した結果を示す(b,c)。Cは、GFP(0.5μg)、siRNA(20pmol)及び示されたsiRNA抵抗性コンストラクト(siRNAr)(1μg)で同時トランスフェクトし、48時間インキュベートし、ボイデンチャンバー法によりアッセイした。アステリスクは、WTと比較した統計上の有意差(スッチューデントt−検定*P<0.05)を示す。
【図6i】図6iは、Latrunculin処理細胞におけるEGFによる全Girdinの膜相互作用の調節を示す。Aは、COS7細胞をGFP−全長Girdin野生型(WT)又はSer−1416がアラニンに置換されたSA変異体でトランスフェクトし、ガラス製ディッシュ上で24時間インキュベーションした。EGF(50ng/ml)の存在下又は不存在下で60分間インキュベーションした細胞をLatrunculin(1μM、Calbiochem)で10分間処理し、固定化した。COS7細胞で過剰発現されたGirdinWT又はGirdinSAは、核を除いた細胞全体にわたって分散して存在した(上段)。アクチンを破壊するLatrunculinで処理したときには(中段)、GirdinWTとSAとは、細胞質と同様に細胞膜にて検出された(黄色い矢じり)。EGFで刺激した細胞をLatrunculinで処理したときには(下段)、GirdinWTの多くは、細胞膜から解離して点状に細胞質に蓄積した(矢印)。反対に、GirdinSAは、EGFで刺激後も以前として細胞膜と相互作用を維持した(白抜き矢じり)。Bは、COS7細胞を、GFP−全長GirdinPBala変異体でトランスフェクトし、Latrunculinでその後処理した結果を示す。変異体の細胞膜との相互作用は、EGFの不存在下でもLatrunculinによる処理後はほとんど観察されなかった。
【図7a】図7aは、Girdinは細胞運動中に先端部(リーディングエッジ)でリン酸化される。aは、Vero細胞のモノレーヤー(単層にまかれた細胞)をスクラッチした後、障害された面に向いている細胞を固定し、抗リン酸化Girdin抗体で染色した。矢頭はGirdinのリン酸化シグナルを示す。bは、リーディングエッジにおけるGirdinのリン酸化は時間依存性に増加することを示す。
【図7b】図7bは、GFP遺伝子と各種siRNAおよびGirdin CTをトランスフェクトしたVero細胞を48時間培養し、ボイデンチャンバーアッセイに使用した結果を示す。下段チャンバーにはEGFを加える条件と加えない条件で行った。アスタリスクはコントロールと比較して有意差があることを示す。囲みはVero細胞におけるsiRNAによるAktの発現抑制を示す。結果は平均±標準誤差で示した。
【図7c】図7cは、GirdinのsiRNA抵抗性変異体の作製及びそれを利用した評価結果を示す。aは、 Girdin siRNAの標的配列とsiRNA抵抗性変異体の作製のための塩基置換を示す。bは、示された各種のsiRNAとコンストラクトをトランスフェクションしたCOS7細胞の細胞溶解液を抗V5抗体を用いたウェスタンブロット法で解析した(Girdinの各種コンストラクトのC末端にはV5タグが付加してあるため、この方法で検出が可能)結果を示す。
【図7d】図7dは、GFP遺伝子と各種siRNAおよびコンストラクトをVero細胞にトランスフェクトし、48時間培養後にボイデンチャンバーアッセイを行った結果を示す。Girdinの各種変異体の発現は細胞移動アッセイの前にウェスタンブロット法で解析し、発現レベルが同様であることを確認した(結果は示さず)。結果は平均±標準誤差で示した。
【図7e】図7eは、Girdinのリン酸化によって細胞の運動性が制御される提唱モデルを示す。静止した細胞ではGirdinはアクチン線維を架橋し、また細胞辺縁部のアクチン(cortical actin)を細胞膜にアンカーする。一方、細胞運動中にはAktによるリン酸化によりGirdinはリーディングエッジに局在を変化させ、ラメリポディアにおける短く枝分かれしたアクチン線維を架橋する。
【図7f】図7fは、ヒト線維肉腫細胞株HT-1080細胞における指向性移動に必須であることを示す。Aは、HT1080細胞でのGirdinの枯渇を示す。SiRNAを含まないコントロール細胞及びsiRNAがトランスフェクトされたHT-1080細胞からの細胞全溶解物は、抗Girdin抗体、抗Akt抗体、及び抗アクチン抗体を用いたウエスタンブロット分析に供した。Bは、Girdinの枯渇は、HT-1080細胞の指向性のある細胞移動を損なうことを示す。siRNAがトランスフェクトされたHT-1080細胞をボイデンチャンバー法に供した。下段のチャンバーには、血清(5%)が添加された。対コントロールで*Pは<0.05であった。Cは、Girdinの枯渇により細胞移動が障害された細胞は、siRNA抵抗性(siRNAr)型のGirdinを再添加することによって回復することを示す。GFP、示されたsiRNA(20pmol)及び各種コンストラクト(2.5μg)をHT-1080細胞に同時トランスフェクトし、トランスフェクト後、48時間ボイデンチャンバー法に供した。SiRNAr-Girdin野生型(WT)は完全に細胞移動性を回復させた一方、SA及び△PBは、回復させなかった。示した結果は、2つの独立した実験の代表例である。
【図7g】図7gは、Girdin変異対の発現のベロ細胞の指向性のある移動性に対する影響を示す。Aは、損傷治癒アッセイによるGirdin変異体を発現するベロ細胞の移動の観察結果を示す。フィブロネクチン(10μg/ml)でコートしたグラス製ディッシュ上のコンフルエント状態のベロ細胞に、Girdin siRNAとGirdin siRNA抵抗性(siRNAr)野生型GFP-Girdin(WT)と示された変異体(SA、△PB及びPbala)を同時トランスフェクトし(a)、トランスフェクションから48時間経過後、細胞の単層をスクラッチして損傷部への細胞移動を開始させ(b)、GFP-Girdinを発現し、損傷部と向き合う細胞を低速度イメージングで分析した(c)。Bは、野生型Girdin(WT)及び各種変異体(SA、△PB及びPbala)を発現するベロ細胞の移動の低速度イメージングを示す。イメージは、スクラッチから2時間経過後から7時間の間90秒毎に取得した。白い破線は、損傷ラインを示し、矢じりは移動する細胞の核を示す。これらの結果は、二つの独立した実験の代表的なイメージである。
【図8a】図8aは、Girdin siRNAでHUVEC細胞を処理してボイデンチャンバー法による遊走能の定量結果を示す。
【図8b】図8bは、遊走した細胞を染色した画像を示す。
【図9a】図9aは、管腔形成能の観察結果を示す。
【図9b】図9bは、抗Girdin抗体によるウエスタンブロット分析結果を示す。
【図10a】図10aは、管腔形成の回復を示す。
【図10b】図10bは、抗Girdin抗体によるウエスタンブロット分析結果を示す。
【図11】図11は、マトリゲルプラグアッセイの結果を示す図であり、上段がコントロールであり下段がノックダウンベクターを含むマトリゲルを注入した結果である。
【図12】図12は、乳癌及び子宮頸癌におけるGirdinの組織免疫染色結果を示す図である。発明を実施するための最良の形態
【0007】
本発明は、セリン/スレオニンキナーゼAkt(以下、単にAktという。)によってリン酸化されるタンパク質(Girdin:本発明者らによりGirdinと称される。)でありアクチン結合タンパク質であるGirdinに関し、特に、配列番号2で表されるアミノ酸配列と同一のアミノ酸配列(Girdinのアミノ酸配列である。)を有するタンパク質若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチド(以下、前記タンパク質及び前記部分ペプチドを総称して本タンパク質という。)に関する。Girdinは、通常、ヒト受精卵から身体各部が形成される過程において細胞の運動、細胞の移動や血管形成に関与している。具体的には、腫瘍細胞の運動や移動は癌の進行、転移、浸潤に関連し、神経細胞の移動等は神経形成に関連し、血管内皮細胞などの血管構成細胞の移動は、血管形成に関連する。したがって、本タンパク質を用いることにより、細胞運動、細胞移動及び血管形成のいずれか関連する疾患に有効な予防・治療薬を探索し、提供することができる。また、本タンパク質を用いることにより、これらの疾患に関する診断も可能となる。
【0008】
本発明は、また、本タンパク質とAktとの関係において、細胞運動等に現れる各種活性を促進又は阻害する化合物又はその塩に関している。こうした化合物又はその塩は、細胞運動、細胞移動及び血管形成のいずれかを促進又は阻害する化合物又はその塩として利用できる。こうした本発明の化合物によれば、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害することができるため、例えば、これらの細胞動態が関連する疾患の治療、予防に有用である。また、本タンパク質に試験化合物を供給して、本タンパク質に対する作用を検出することにより、細胞運動、細胞移動及び血管形成等に関連する疾患の予防・治療剤として有用な化合物をスクリーニングすることも可能となる。さらに、本タンパク質に特異的な抗体等は、細胞における本タンパク質の量的な測定又は局在を検出することができるため、細胞運動、細胞移動及び血管形成等が関連する疾患の予防や治療のための診断も可能となる。以下、本発明の実施の形態について詳細に説明する。
【0009】
(本タンパク質) 本タンパク質は、配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有することができる。これらのアミノ酸配列からなるものであってもよい。配列番号2で表されるアミノ酸配列と実質的に同一であるアミノ酸配列としては、配列番号2で表されると約50%以上、好ましくは約60%以上、さらに好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、最も好ましくは約95%以上の相同性を有するアミノ酸配列などが挙げられる。相同性(同一性)(%)は、当該分野で慣用のホモロジー検索プログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。なお、相同性(%)は、当該分野で公知の任意のアルゴリズム、例えば、Needlemanら(1970)(J.Mol.Biol.48:444−453)、Myers及びMiller(CABIOS,1988,4:11−17)のアルゴリズム等を使用して決定してもよい。
【0010】
本タンパク質は、配列番号2で表されるアミノ酸配列中の1個又は2個以上(好ましくは、1〜30個程度、好ましくは1〜10個程度、さらに好ましくは数(1〜5)個)のアミノ酸が欠失したアミノ酸配列、又は同数のアミノ酸が付加したアミノ酸配列、又は同数のアミノ酸が挿入されたアミノ酸配列、又は同数のアミノ酸が置換されたアミノ酸配列、また、同数のアミノ酸が欠失、付加、挿入及び置換から選択される2種類以上を組み合わせた態様で改変されたアミノ酸配列を有し、あるいはこうしたアミノ酸配列からなっていてもよい。こうしたアミノ酸の改変部位は、本タンパク質の活性が喪失しない範囲であれば特に限定されない。
【0011】
また、配列番号2で表されるアミノ酸配列と実質的に同一のタンパク質としては、配列番号2で表されるアミノ酸配列からなるタンパク質と同質の活性を有していることが好ましい。同質の活性とは、活性の程度は問わないことを意味している。したがって、活性の程度は、配列番号2で表されるアミノ酸配列からなるタンパク質と同程度未満であることもあり、同程度以上であることもある。こうした活性としては、例えば、本タンパク質のAktの基質としての活性であり、特に、配列番号2のアミノ酸配列における第1416位のセリンあるいはそれに相当する位置のセリンがAktによってリン酸化される活性を有していることが好ましい。また、同質の活性としては上記リン酸化活性は好ましいものとして挙げられるがは上記活性に限定するものではなく、配列番号2で表されるアミノ酸配列からなるタンパク質の他のいずれかの活性であってもよく、2種類以上を組み合わせたものであってもよい。これらの活性については後段で詳述する。
【0012】
また、本タンパク質は、配列番号2で表されるタンパク質と同質の活性を有している限り、その部分ペプチドを包含している。部分ペプチドの領域は特に限定しないが、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域CT領域である、配列番号2のアミノ酸配列の第1376位〜第1870位のアミノ酸(CT1領域及び/又はCT2領域)を含んでいることが好ましい。CT1領域は、配列番号2のアミノ酸配列の第前半部分の領域であり、少なくとも第1389位〜第1407位あるいは当該アミノ酸配列に対応する配列及び第1411位〜1416位の配列を含んでいることが好ましく、例えば、第1375位〜第1622位のアミノ酸配列を有することができる。また、CT2領域は、CT領域の後半部分であり、例えば、第1623位〜第1870位のアミノ酸配列を有することができる。部分ペプチドは、これらの双方の領域を有していてもよいし、いずれか一方のみであってもよい。好ましいCT1領域は、Aktリン酸化サイトを有するとともに細胞膜と相互作用し、好ましいCT2領域はアクチンフィラメントと結合することができる。
【0013】
こうしたタンパク質としては、ヒトにおけるGirdinのオルソログ又はホモログやGirdin(NCBIホームページ(http://www.ncbi.nlm.nih.gov/、アクセッション番号BAE44387にてアミノ酸配列を取得可能である。)等の配列に基づいて人工的に改変したタンパク質が挙げられる。また、Girdinのトランスクリプショナルバリアントとして、既に、同様にNCBIホームページにおいてアクセッション番号NP_060554に記載のアミノ酸配列(1843残基)からなるタンパク質が挙げられる。
【0014】
本タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている態様のタンパク質又は部分ペプチドも包含している。このリン酸化は、Aktによるものであって、Girdinは、このリン酸化により、細胞のリーディングエッジに局在が濃縮され、細胞運動に関与するものと推論される。なお、特に、リン酸化されていない態様とリン酸化された態様とを区別するときには、それぞれを、リン酸化されていない本タンパク質及びリン酸化された本タンパク質と表現するものとする。
【0015】
本タンパク質としては、配列番号2で表されるアミノ酸配列からなる天然由来のタンパク質である上記Girdinが挙げられるが、本タンパク質に包含されるタンパク質又はペプチドには、天然のアミノ酸のみからなっていなくてもよく、本タンパク質の活性が喪失されない範囲で各種の修飾が施されていてもよい。タンパク質及びアミノ酸に対する各種の修飾はよく知られているが、例えば、カルボキシル基に対しては、カルボキシレート(−COO−)、アミド(−CONH2)またはエステル(−COOR)の何れとすることができる。ここでエステルにおけるRとしては、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチルなどのC1−6アルキル基、例えば、シクロペンチル、シクロヘキシルなどのC3−8シクロアルキル基、例えば、フェニル、α−ナフチルなどのC6−12アリール基、例えば、ベンジル、フェネチルなどのフェニル−C1−2アルキル基もしくはα−ナフチルメチルなどのα−ナフチル−C1−2アルキル基などのC7−14アラルキル基、ピバロイルオキシメチル基などが用いられる。本発明で用いられるタンパク質がC末端以外にカルボキシル基(またはカルボキシレート)を有している場合、カルボキシル基がアミド化またはエステル化されているものも本発明で用いられるタンパク質に含まれる。この場合のエステルとしては、例えば上記したC末端のエステルなどが用いられる。さらに、N末端のアミノ酸残基(例、メチオニン残基)などのアミノ基が保護基(例えば、ホルミル基、アセチル基などのC1−6アルカノイルなどのC1−6アシル基など)で保護されていてもよいし、生体内で切断されて生成するN末端のグルタミン残基がピログルタミン酸化したもの、分子内のアミノ酸の側鎖上の置換基(例えば−OH、−SH、アミノ基、イミダゾール基、インドール基、グアニジノ基など)が適当な保護基(例えば、ホルミル基、アセチル基などのC1−6アルカノイル基などのC1−6アシル基など)で保護されていてもよいし、糖鎖が結合したいわゆる糖タンパク質などであってもよい。
【0016】
なお、リン酸化されていない本タンパク質においては、配列番号:1のアミノ酸配列の第1416位のセリンあるいはそれに対応するセリンにおいて、Aktによるリン酸化が可能な範囲に修飾されている。
【0017】
本タンパク質は、塩の形態を採っていてもよい。例えば、生理学的に許容される酸(例、無機酸、有機酸)や塩基(例、アルカリ金属塩)などとの塩が用いられ、とりわけ生理学的に許容される酸付加塩が好ましい。この様な塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩などが用いられる。
【0018】
本タンパク質は、従来公知の方法で取得することができる。例えば、本タンパク質は、哺乳類細胞においてユビキタスに存在するタンパク質であるので、これら細胞あるいはこれらの細胞を含む組織から公知のタンパク質の精製方法により取得することもできる。さらに、本タンパク質をコードするDNAを取得し、該DNAを含有する形質転換体を作製し、培養することによっても得ることができる。タンパク質は、硫安沈殿、エタノール沈殿、酸抽出、イオン交換クロマトグラフィー、疎水クロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、逆相クロマトグラフィー、ゲル濾過クロマトグラフィーなどのクロマトグラフィーを組み合わせることにより精製単離することができる。また、本発明のタンパク質は、公知のペプチドの固相合成法(矢島治明および榊原俊平、生化学実験講座1、タンパク質 の化学IV、205、(1977年)等)によって合成可能な場合もある。ペプチド合成に使用する官能基の保護ならびに保護基及びその保護基の脱離、反応に関与する官能基の活性化などは公知の基または公知の手段から適宜選択して実施すればよい。また、本タンパク質は、後述するように本タンパク質を発現する外来DNAを保持する細胞を培養したり、当該外来DNAを保持するトランスジェニック動物の細胞や組織から取得することもできる。
【0019】
次に、本タンパク質の活性、すなわち、配列番号2で表されるアミノ酸配列からなるタンパク質であるGirdinの活性について説明する。Girdinは、以下の活性を有しており、本タンパク質は、これらのいずれかの活性を有している。すなわち、(1)Aktの基質としての活性(本タンパク質のAktにより配列番号2における第1416位あるいはそれに対応する部位のセリンがリン酸化される。)、(2)アクチン結合タンパク質活性(アクチンフィラメントに結合しこれらを架橋する。)、及び(3)細胞膜結合活性、を有している。(1)のAkt基質活性は、Aktとの結合(複合体の形成)、Aktによるリン酸化として把握され、(2)のアクチン結合タンパク質活性は、本タンパク質によるアクチンストレスファイバー形成やラポリメディアにおけるアクチンメッシュワークの形成として把握され、(3)の細胞膜結合活性は、細胞膜中のフォスフォイノシタイドへの結合(相互作用)として把握される。
【0020】
こうした(1)〜(3)の各種活性は、本発明者らによれば、Aktによる細胞運動制御に関連し、ひいては細胞移動及び血管形成に関連し、これらの細胞動態の活性化・促進に寄与することが確認されている。なお、上記(1)及び(3)の活性は、リン酸化されていないGirdinの活性であり、(2)の活性は、リン酸化の有無に関わらないでGirdinが有する活性である。こうした各種の活性は、後述する実施例において具体的に説明するように、いずれも、インビトロ又はインビボにおける実験系において検出することができる。
【0021】
(本タンパク質をコードするヌクレオチド)
本発明において、配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を有するタンパク質又はその部分ペプチドをコードするヌクレオチドは、各タンパク質等をコードする塩基配列を有していれば、特に限定されない。ヌクレオチドは、こうした塩基配列を有するDNA、RNA、DNA/RNAキメラとすることができ、1本鎖であっても2本鎖であってもよい。2本鎖の場合には、DNA2本鎖、RNA2本鎖のほか、DNA/RNAハイブリッドであってもよい。1本鎖の場合には、センス鎖及びアンチセンス鎖のいずれであってもよい。具体的には、ゲノムDNA、cDNA、mRNA、あるいはPCR産物,化学合成DNA等といった形態でこうしたヌクレオチドを取得することができる。
【0022】
配列番号2で表されるアミノ酸配列をコードする塩基配列として配列番号1で表される塩基配列が挙げられる。この塩基配列はGirdinをコードする塩基配列である。上記したように、本タンパク質をコードするヌクレオチドは、配列番号1で表される塩基配列と同一の塩基配列に限定されないで、この塩基配列とは異なるコドン用法によって同一のアミノ酸配列をコードする塩基配列であってもよい。また、本ヌクレオチドの塩基配列は、配列番号1で表される塩基配列をストリンジェントな条件でハイブリダイズするDNA等のヌクレオチドであって、本タンパク質と同質な活性を有するタンパク質をコードするヌクレオチドであってもよい。
【0023】
なお、ストリンジェントな条件下でハイブリダイズできるDNAとしては、例えば、各配列番号で表される塩基配列と約50%以上、好ましくは約60%以上、さらに好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、最も好ましくは約95%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。ハイブリダイゼーションは、公知の方法あるいはそれに準じる方法、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al., Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行うことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行うことができる。より好ましくは、ハイストリンジェントな条件に従って行うことができる。ハイストリンジェントな条件とは、例えば、ナトリウム濃度が約19〜40mM、好ましくは約19〜20mMで、温度が約50〜70℃、好ましくは約60〜65℃の条件を示す。特に、ナトリウム濃度が約19mMで温度が約65℃の場合が最も好ましい。
【0024】
Girdinをコードするヌクレオチドとしては、例えば、配列番号1で表される塩基配列を有するヌクレオチド又は配列番号1で表される塩基配列とハイストリンジェントな条件でハイブリダイズする塩基配列を有し、配列番号2で表されるタンパク質と同質の活性を有するタンパク質をコードするものであればよい。
【0025】
本タンパク質をコードするヌクレオチドは、形質転換用核酸構築物としての形態を採ることができる。すなわち、哺乳類動物細胞などの宿主細胞等において本タンパク質を発現させるための核酸構築物としての各種形態(Naked DNA、各種発現ベクター、人工染色体等)を採ることができる。こうした核酸構築物は、本タンパク質を発現可能にプロモーター、ターミネーター他、各種のエレメントを適宜備えることができる。
【0026】
(本タンパク質に特異的な抗体) 本タンパク質に対して特異的な抗体は、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに対して特異的な抗体及びリン酸化されたGirdinなどの配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに対して特異的な抗体を包含している。
【0027】
本発明の抗体は、ポリクローナル抗体であってもモノクローナル抗体であってもよい。抗体は、適当な抗原物質を温血動物に投与して抗体を産生させた上、当該温血動物から取得することができ、モノクローナル抗体は、最終的に作製したモノクローナル産生ハイブリドーマから取得できる。
【0028】
ポリクローナル抗体は、例えば、免疫原性のある抗原(タンパク質抗原)とキャリアー蛋白質との複合体をつくり、適宜温血動物に免疫を行い、該免疫動物からポリクローナル抗体含有物を採取して、抗体の分離精製を行なうことにより製造することができる。キャリアタンパクとしては、従来公知のものを用いることができるが、ウシ血清アルブミン、ウシサイログロブリン、ヘモシアニン等を用いることができ、免疫原と適当な比率で複合体を形成すればよい。なお、ハプテンとキャリアーとのカップリングには、一般的には、グルタルアルデヒドやカルボジイミド、マレイミド活性エステル、チオール基、ジチオビリジル基を含有する活性エステル試薬を用いることができる。こうした複合体の投与にあたっては、適宜、完全フロイントアジュバントや不完全フロイントアジュバントを同時に投与することができる。投与は、通常約2〜6週毎に1回ずつ、計約3〜10回程度行なうことができる。ポリクローナル抗体は、上記の方法で免疫された温血動物の血液、腹水など、好ましくは血液から採取することができる。抗血清中のポリクローナル抗体価の測定は、ELISAなどのEIA等を適宜用いて測定すればよく、抗体の分離精製は、公知の抗体の分離精製法に従って行えばよい。
【0029】
モノクローナル抗体は、例えば、ポリクローナル抗体の作製時と同様に温血動物を免疫して、抗体価の認められた個体を選択し最終免疫の適数日後に脾臓またはリンパ節を採取し、それらに含まれる抗体産生細胞をミエローマと融合させることにより、モノクローナル抗体産生ハイブリドーマを調製し、スクリーニングされたモノクローナル抗体産生ハイブリドーマを培養し、培養物からモノクローナル抗体を分離精製することで得ることができる。なお、細胞融合操作、モノクローナル抗体の選別は、従来公知の手法を用いることができる。
【0030】
本発明の抗体は、担体に固定化した形態を採ることができる。担体の形態は特に限定しないで、ビーズ状、平板状、マイクロタイタープレート等とすることができる。ビーズ状の担体に固定化された抗体は、免疫沈降、アフィニティクロマトグラフィ等に利用でき、また、平板状の担体やマイクロタイタープレート等に固定化された抗体は、抗体アレイあるいはELISA等のEIAやRIAアッセイプレートとして用いることができる。
【0031】
(本タンパク質をコードするヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するヌクレオチド) この態様の1つのヌクレオチドとしては、Girdinなど本タンパク質をコードするDNAに対するアンチジーンヌクレオチド(好ましくはDNA)又は本タンパク質をコードするmRNAに対するアンチセンスヌクレオチド(好ましくはRNA)が挙げられる。こうしたヌクレオチドとしては、本タンパク質をコードするヌクレオチドと同様の各種の態様を採ることができ、本タンパク質の発現を抑制できるものであれば、その長さや塩基や糖部分などの修飾形態は特に限定されないで各種の修飾形態を採用できる。ここで、「相補的な塩基配列」とは、例えば、本タンパク質又は部分ペプチドをコードするDNAまたはmRNAの全塩基配列または部分塩基配列と約40%以上、好ましくは約60%以上、より好ましくは約80%以上、さらに好ましくは約90%以上の相同性を有する塩基配列などが挙げられる。特に、本発明のDNAまたはmRNAの全塩基配列うち、本発明のタンパク質のN末端部位をコードする部分の塩基配列(例えば、開始コドン付近の塩基配列など)と約40%以上、好ましくは約60%以上、より好ましくは約80%以上、さらに好ましくは約90%以上の相同性を有するアンチセンスDNAが好適である。これらのアンチセンスDNAは、公知のDNA合成装置などを用いて製造することができる。
【0032】
また、この態様の他の1つのヌクレオチドとしては、Girdinなど本タンパク質をコードする遺伝子転写産物に対してRNA干渉を発現するヌクレオチドが挙げられる。こうしたヌクレオチドは、本タンパク質、特には、GirdinをコードするDNAのmRNA等の転写産物の少なくとも一部を標的としてこれらGirdinの発現を抑制可能に構築されている。こうしたヌクレオチドの一つの態様は、互いにハイブリダイズするオリゴリボヌクレオチドの二本鎖構造を有するヌクレオチドである。具体的には、突出した3’末端をそれぞれ有するあるいは有しない比較的短い二本鎖オリゴリボヌクレオチド(small interfering RNA :siRNA)及びヘアピン構造を形成する(又は有する)単一のオリゴリボヌクレオチド(short hairpin RNA : shRNA)が挙げられる。なお、ヘアピン構造を形成しない一本鎖オリゴヌリボヌクレオチドもRNA干渉を発現することができる。
【0033】
本態様のヌクレオチドにおけるセンス配列及びアンチセンス配列が対合する二重鎖の長さは、RNA干渉効果が得られる限り特に限定しないが、好ましくは50塩基対以下であり、典型的には13〜28塩基対であることが好ましく、より好ましくは13〜27塩基対の範囲であり、さらに好ましくは19〜21塩基対である。最も好ましくは、19又は20塩基対である。また、二重鎖を形成しない3’側構造を含めたセンス配列及びアンチセンス配列は、典型的には15〜30ntであることが好ましく、より好ましくは15〜29ntの範囲であり、さらに好ましくは21〜23ntである。最も好ましくは、21又は22ntである。また、siRNAにおいて突出する3'末端は、2〜4ntであることが好ましく、より好ましくは2ntである。さらに、shRNAにおけるループ部位は、こうした態様の二重鎖(shRNAにおいてはステム)及び3’末端構造を形成し維持するのを妨げない程度のループ部位の長さを備えていればよい。
【0034】
siRNAやshRNAは、公開されているルールなど適宜適用してターゲット配列を決定し、それに基づいて設計すればよい。さらに、ターゲット配列の決定方法を含むsiRNAの設計は、http://design.rnai.jp/sidirect/index.php、http://www.rockefeller.edu/labheads/tuschl/sirna.htmlやRational siRNA design for RNA interference (Nature Biotechnology , vol. 22, 326 - 330 (2004), Angela Reynolds, Devin Leake, Queta Boese,Stephen Scaringe, William S Marshall & Anastasia Khvorova)、Improved and automated prediction of effective siRNA (Biochem Biophys Res Commun. 2004 Jun 18;319(1):264-74、Chalk AM, Wahlestedt C, Sonnhammer EL.)等公開された各種のルールを適宜適用して行うことができる。
【0035】
RNA干渉を発現するヌクレオチドは、塩基や糖部分などの修飾形態は特に限定されないで各種の修飾形態を採用できる。
【0036】
(RNA干渉を発現するヌクレオチドをコードするヌクレオチド)
本発明によれば、Girdinなど本タンパク質をコードする遺伝子転写産物に対してRNA干渉を発現ヌクレオチドをコードするヌクレオチド(好ましくはDNA)及びこのヌクレオチドを含有するベクターを含むことができる。すなわち、本タンパク質をコードする遺伝子転写産物に対するsiRNAやshRNAを発現可能にコードするベクターの態様である。こうした態様のヌクレオチドは、継続性のあるRNA干渉を実現でき、ノックダウン動物を作製できる点において好ましい。この態様において、shRNA発現ベクターは、アンチセンス配列及びセンス配列の他ループ配列を、細胞内転写によりshRNAを構築可能な連続した一本鎖RNAが転写されるように構成することができる。また、siRNA発現ベクターは、所定のセンス配列及びアンチセンス配列を有するRNAが転写されるように構成すればよい。siRNA発現ベクターにおいては、センス配列及びアンチセンス配列は単一のベクターによって発現されるようにしてもよいし、また、それぞれ異なるベクターから発現されるようにしてもよい。
【0037】
発現ベクターは、プラスミドベクターやウイルスベクターの形態を採ることができる。ベクターの種類は特に問わないで、導入したい細胞などに対応して選択することができる。例えば、哺乳動物細胞では、例えば、レトロウイルスベクター、アデノウイルスベクター、アデノ関連ウイルスベクター、ワクシニアウイルスベクター、レンチウイルスベクター、ヘルペスウイルスベクター、アルファウイルスベクター、EBウイルスベクター、パピローマウイルスベクター、フォーミーウイルスベクターなどのウイルスベクターが挙げられる。また、こうした発現ベクターに用いるプロモーターとしては、上記各DNAより対応するRNAを産生し得るものであれば、polII系、polIII系のいずれであってもよい。
【0038】
(細胞) 本発明は、1つの態様として、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を有するタンパク質をコードする遺伝子の発現量が抑制された細胞を含んでいる。この細胞は、本タンパク質をコードする塩基配列に基づいて作成されたノックアウトベクターや上記したRNA干渉を発現するヌクレオチドや該ヌクレオチドをコードするヌクレオチド(ベクター)を細胞に導入することによりあるいは後述する非ヒト哺乳類ノックアウト動物や非ヒト哺乳類ノックダウン動物から取得できる。
【0039】
また、本発明は、他の1つの態様として、Girdinなどの配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする外来DNAを保持した細胞を含んでいる。こうした細胞は、本タンパク質を発現するための核酸構築物を細胞に導入することによりあるいは後述する非ヒト哺乳類トランスジェニック動物を取得源として取得できる。
【0040】
さらに、本発明は、他の1つの態様として、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質が変異された本変異型タンパク質をコードする外来DNAを保持した細胞を含んでいる。こうした細胞は、本変異型タンパク質を発現するための核酸構築物を細胞に導入することによりあるいは後述する非ヒト哺乳類トランスジェニック動物を取得源として取得できる。
【0041】
これらの各種細胞は、例えば、ヒト(トランスジェニック動物、ノックアウト動物及びノックダウン動物を取得源とする場合を含まない。)及び非ヒト哺乳類細胞とすることができる。また、マウス、ラットなどゲッ歯類を始めとする非ヒト哺乳類胚性幹細胞としてもよいし、体性幹細胞や体細胞であてもよい。さらに、神経細胞、血管内皮細胞、癌細胞など、本タンパク質が関連する疾患の標的細胞であってもよい。こうした本発明の各種細胞は、本発明の予防・治療剤のスクリーニング、評価、細胞運動等の評価に有用である。
【0042】
以下、本タンパク質及び本タンパク質に関連して得られるヌクレオチド、抗体、細胞等の用途について説明する。
【0043】
(本タンパク質が関連する疾患の予防・治療剤) 本タンパク質は、上記(1)〜(3)のいずれかの活性を有し、これらの活性は、細胞運動、細胞移動及び血管形成を活性化するから、本タンパク質についての上記(1)〜(3)のいずれか又は2種以上の活性を阻害する活性を有する化合物又はその塩は、Girdinなど本タンパク質が関連する疾患、具体的には細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤として利用できる。また、本タンパク質の発現量を抑制する化合物又はその塩も、Girdinなど本タンパク質が関連する疾患、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤として利用できる。
【0044】
ここで、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患としては、こうした現象が関連する疾患の全てを包含しているが、例えば、以下の疾患を例示することができる。
【0045】
(1)癌
例えば、原発性、転移性または再発性の、乳癌、前立腺癌、膵癌、胃癌、肺癌、大腸癌(結腸癌、直腸癌、肛門癌)、食道癌、十二指腸癌、頭頚部癌(舌癌、咽頭癌、喉頭癌)、脳腫瘍、神経鞘腫、非小細胞肺癌、肺小細胞癌、肝臓癌、腎臓癌、胆管癌、子宮癌(子宮体癌、子宮頸癌)、卵巣癌、膀胱癌、皮膚癌、血管腫、悪性リンパ腫、悪性黒色腫、甲状腺癌、骨腫瘍、血管腫、血管線維腫、網膜肉腫、陰茎癌、小児固形癌、カポジ肉腫、AIDSに起因するカポジ肉腫、上顎洞腫瘍、線維性組織球腫、平滑筋肉腫、横紋筋肉腫、脂肪肉腫、子宮筋腫、骨芽細胞腫、骨肉腫、軟骨肉腫、癌性の中皮腫瘍、白血病などの腫瘍、ホジキン病など
【0046】
(2)動脈硬化性疾患
例えば、急性冠動脈症候群(例、急性心筋梗塞、不安定狭心症など)、末梢動脈閉塞症、冠動脈インターベンション(経皮的冠動脈形成術(PTCA)、アテレクトミー(DCA)、ステント留置等)後の再狭搾、冠動脈バイパス手術後の再狭窄、その他の末梢動脈におけるインターベンション(血管形成術、アテレクトミー、ステント留置等)及びバイパス手術後の再狭窄、虚血性心疾患(例、心筋梗塞、狭心症など)、心筋炎、間歇性跛行、ラクネ梗塞、動脈硬化症(例、アテローム性動脈硬化症など)、心不全(急性心不全、うっ血性を含む慢性心不全)、不整脈、動脈硬化巣の進展、血栓症、高血圧症、高血圧性耳鳴り、低血圧症など
【0047】
(3)中枢および末梢神経障害
例えば、頭部外傷、脊髄損傷、脳浮腫、知覚機能障害、知覚機能異常、自律神経機能障害、自律神経機能異常、むち打ち症など
【0048】
本発明の予防・治療剤として用いることができる化合物又はその塩を得るための指標としては、具体的には、以下の活性を阻害する活性が挙げられる。なお、下記a)〜b)は、上記(1)のAkt基質活性の阻害に関連し、下記c)〜e)は、上記(2)のアクチン結合タンパク質活性の阻害に関連し、下記f)〜g)は、上記(3)の細胞膜結合活性の阻害に関連する。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化
c)アクチンフィラメントへの結合
d)アクチンストレスファイバー形成
e)ラポリメディアにおけるアクチンメッシュワークの形成
f)細胞膜への結合
g)フォスフォイノシタイドへの結合
【0049】
こうした阻害活性を有する化合物は、後述するように、上記a)〜g)を阻害する化合物を取得するスクリーニング方法に基づいて得ることができる。こうした化合物としては、例えば、本タンパク質に特異的な抗体等が挙げられる。また、Aktによるリン酸化サイトに変異を有する本タンパク質(例えばGirdinなど)の変異体及びフォスフォイノシタイド結合サイトに変異を有する本タンパク質(例えばGirdinなど)の変異体であってもよい。
【0050】
こうした化合物の塩としては、生理学的に許容される酸(例、無機酸、有機酸)や塩基(例えばアルカリ金属)等との塩が用いられ、とりわけ生理学的に許容される酸付加塩が好ましい。この様な塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩等が用いられる。
【0051】
スクリーニング方法から得られた化合物又はその塩を、本発明の予防・治療剤として用いる場合、その製剤形態は特に限定されない。経口剤、注射剤、注入剤等の非経口剤、局所適用に適した外用剤等、いずれの形態も採用できる。例えば、必要に応じて糖衣を施した錠剤、カプセル剤、エリキシル剤、マイクロカプセル剤などとして経口的に、あるいは水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、または懸濁液剤などの注射剤の形で非経口的に使用できる。こうした各種の製剤も本発明の予防・治療剤の一態様である。例えば、生理学的に認められる担体、香味剤、賦形剤、ビヒクル、防腐剤、安定剤、結合剤などとともに一般に認められた製剤実施に要求される単位用量形態で混和することによって製造することができる。これら製剤における有効成分量は指示された範囲の適当な用量が得られるようにするものである。なお、上記製剤を得るための各種添加剤は、当該分野において公知のものを適宜選択して用いればよい。こうして得られる製剤は、ヒトのほか、非ヒト動物である、例えば、ラット、マウス、モルモット、ウサギ、トリ、ヒツジ、ブタ、ウシ、ウマ、ネコ、イヌ、サルなどに対して投与することができる。上記化合物の投与量は、症状及患者の個体差などを勘案して設定することができる。
【0052】
本発明の予防・治療剤としては、スクリーニングされた化合物のほか、本タンパク質の発現量を抑制する化合物又はその塩であってもよい。本タンパク質の細胞内で発現量を抑制する化合物又はその塩としては、「本タンパク質をコードするヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するヌクレオチド」に含まれる各種態様のヌクレオチドが挙げられる。これらのヌクレオチドは、細胞内においてアンチジーン、アンチセンス、RAN干渉により、本タンパク質の発現を抑制するため、結果として、上記a)〜g)を阻害することができ、上記a)〜g)による細胞運動、細胞移動及び血管形成を活性化を抑制し、ひいては、これらのいずれかが関連する疾患を予防・治療剤として作用することができる。
【0053】
また、本タンパク質の発現量を抑制する化合物又はその塩としては、本タンパク質をコードする遺伝子を破壊可能なDNA構築物が挙げられる。こうしたDNA構築物は、本タンパク質をコードするヌクレオチドの塩基配列に基づいて作製することができる。
【0054】
こうしたヌクレオチド構築物を含有する予防・治療剤は、適切な担体に含有させて用いることにより、より一層、細胞への導入が容易になる。このため、本発明の予防・治療剤としては、こうした担体を含有するものも包含する。担体としては、細胞への導入に適したベクター、リポソーム、金属粒子、正電荷ポリマー、リン酸カルシウム、DEAEデキストランなどが挙げられる。また、担体は、リポソーム、金属粒子、正電荷ポリマー、リン酸カルシウム又はDEAEデキストランのいずれかに、本発明のヌクレオチド等を保持するベクターを担持させたものでもよい。細胞への導入のためのベクターは、非ウイルス性ベクターやウイルス性ベクターを用いることができる。さらに、本予防・治療剤は、通常の薬理学的に許容されうる担体、賦形剤、結合剤、安定剤、緩衝剤、溶解補助剤、等張剤などを含有してもよい。
【0055】
投与は、特に標的細胞における導入を達成して、本タンパク質の発現量の抑制を実現できるものであれば特に限定されないが、例えば、以下の手法が挙げられる。すなわち、線維芽細胞等の各種細胞に対して体外で本発明の遺伝子を導入し、遺伝子導入が成功した細胞(本発明の細胞でもある。)を選択したうえで、標的部位に移植するexvivoの方法や、ウイルスベクターやリポソームを用いて直接標的部位に局所注入し、in vivoで本発明の遺伝子の導入を行なうアプローチが挙げられる。さらに、徐放性の製剤を調製し、患部近くに埋め込む手法挙げられる。なお、こうしたヌクレオチド構築物を含有する予防・治療剤を細胞や組織等に導入する方法としては、リン酸−カルシウム共沈法;微小ガラス管を用いた核酸の直接注入法;内包型リポソームによる遺伝子導入法;静電気型リポソームによる遺伝子導入法;HVJ−リポソーム法、改良型HVJ−リポソーム法(HVJ−AVEリポソーム法);受容体介在性遺伝子導入法;パーティクル銃で担体(金属粒子)とともに核酸分子を細胞に移入する方法;naked−DNAの直接導入法;正電荷ポリマーによる導入法等が挙げられる。
【0056】
こうしたヌクレオチド構築物を含有する予防・治療剤は、投与目的、個体の年齢、体重、状態等に応じて、本タンパク質の発現量を低下させる範囲で適宜設定され得る。
【0057】
(本タンパク質が関連する疾患の予防・治療方法)
本発明の予防・治療剤は、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療方法としても実施できる。すなわち、本発明の予防・治療剤をヒト又は非ヒト温血動物を予防又は治療のために有効量投与することは、上記疾患の予防・治療方法である。
【0058】
(スクリーニング方法) 本発明によれば、本タンパク質及び本タンパク質をコードするヌクレオチドを用いる、本タンパク質の各種活性を促進又は阻害し、あるいは本タンパク質の発現量を増加又は低下させる化合物又はその塩のスクリーニング方法が提供される。このスクリーニング方法によって得られる化合物又はその塩は、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物として使用できる。また、本タンパク質の各種活性を阻害し又は発現量を低下させる化合物又はその塩は、本タンパク質が関連する疾患の予防・治療剤として使用でき、特には、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤としても使用できる。
【0059】
本スクリーニング方法は、試験化合物を本タンパク質、本タンパク質を発現する細胞に供給して、試験化合物を供給しないときに比較して、本タンパク質の上記した活性(1)〜(3)、具体的には上記(a)〜(g)を促進するか又は阻害するかを指標とすることにより、本タンパク質の活性を促進又は阻害する化合物又はその塩を取得できる。また、試験化合物を本タンパク質を発現する細胞に供給して、試験化合物を供給しないときに比較して本タンパク質の発現量を増加させるか低下させるかを検出することによれば、本タンパク質の発現量を増加又は低下させる化合物又はその塩を得ることができる。なお、前記作用の検出にあたって、スクリーニング系に供給される試験化合物としては、本タンパク質に対する作用するもののほか、本タンパク質をコードするヌクレオチドあるいは当該ヌクレオチドに作用するものであってもよい。本タンパク質の上記活性を阻害したり、本タンパク質の発現量を低下させる化合物又はその塩は、細胞運動、細胞移動及び血管形成を抑制するため、これらの細胞動態のいずれかが関連する疾患の予防・治療剤として用いることができる。
【0060】
こうした試験化合物としては、例えば、ペプチド、タンパク、非ペプチド性化合物、合成化合物、発酵生産物、細胞抽出液、植物抽出液、動物組織抽出液、遺伝子(ゲノムDNA、cDNA)などが挙げられ、これら化合物は新規な化合物であってもよいし、公知の化合物であってもよい。
【0061】
本スクリーニング方法において、本タンパク質の発現量や上記a)〜g)の各種指標を検出するには、例えば、以下の手法を採用できる。 例えば、Aktとリン酸化されていない本タンパク質との結合を指標とするには、Aktによるリン酸化されていない本タンパク質のリン酸化を指標とすることが好ましく、例えば、Girdin(タグ付きGirdinを発現するCOS7細胞からの細胞溶解物をタグで精製したものであってもよい。)と構成的活性型であるなどの活性型AktとP32等で標識したATPとを含むインビトロキナーゼアッセイ系に試験化合物を供給してインキュベーションし、得られるリン酸化物をオートラジオグラフィーで検出するなどすることができる。あるいは、Girdinを発現する細胞にEGFを加えてAktを活性化するとともに、試験化合物を供給してインキュベーションし、その後、細胞溶解物を抗Girdin抗体で免疫沈降後、リン酸化Girdin抗体で免疫沈降し、抗リン酸化Girdin抗体などでウエスタンブロット分析すればよい。また、本タンパク質のアクチンフィラメントへの結合を指標とする場合には、GSTなどでタグを付加したGirdinのCT2ドメイン又はGirdinを含むアクチン共沈アッセイ系に試験化合物を供給し、インキュベーション後、共沈させたタンパク質を回収後、SDS−PAGEで分離してクマシーブリリアントブルー(CBB)、抗GST抗体(Cell Signaling Technology)又は抗リン酸化Girdin抗体を用いたウエスタンブロット分析を行えばよい。さらに、アクチンストレスファイバー形成を指標とする場合には、例えば、Girdinを発現する細胞にEGFなどAktを活性化する刺激と試験化合物とを加えた上で、Girdin発現細胞をリン酸化Girdinを特異的に認識する抗リン酸化Girdin抗体などを用いて免疫染色することで、リン酸化Girdinによるアクチンメッシュワークの形成を観察してもよいし、さらに、Girdinを発現する細胞に試験化合物をコンフルエント状態の単層の細胞培養物にスクラッチにより損傷を付与したとき(損傷治癒アッセイ)の、リーディングエッジにおけるリン酸化Girdinを免疫染色により検出してもよい。リーディングエッジにおけるリン酸化Girdinはアクチンメッシュワークに存在する。さらにまた、Girdinを発現する細胞についてボイデンチャンバー法における細胞移動として検出してもよい。また、細胞膜やフォスフォイノシタイドへの結合性を指標とする場合には、タグなどを付加したGirdinを含むタンパク質−脂質オーバーレイ法のアッセイ系に試験化合物を供給して、リン脂質に結合するタンパク質を検出するか、あるいは、Girdinを発現する細胞に試験化合物を供給し、抗Girdin抗体を用いてGirdinを観察することによってもよい。
【0062】
本スクリーニング方法によって得られる化合物又はその塩としては、本タンパク質に特異的に結合する抗体が挙げられる。また、本タンパク質のリン酸化部位の立体構造を変化させて、Aktとの結合活性、Akt-本タンパク質との複合体の安定性などを低下させてAktのリン酸化を抑制する化合物、Aktの基質結合部位に可逆的に結合するAkt阻害剤などが挙げられる。さらに、アクチンフィラメントに対する本タンパク質の架橋を阻害したり架橋状態の解除を促進する化合物又はその塩、アクチンストレスファイバーの形成を阻害したりアクチンストレスファイバー解除を促進する化合物又はその塩、ラポリメディアにおけるアクチンメッシュワークの形成を阻害したり、その解除を促進する化合物又はその塩、細胞膜結合を阻害するか結合解除を促進する化合物又はその塩、ホスホイノシダイドへの結合を阻害するか又は結合解除を促進する化合物又はその塩が挙げられる。さらに、既に説明したように、本タンパク質の発現をノックアウトやノックダウンにより抑制する各種ヌクレオチド構築物が挙げられる。
【0063】
(トランスジェニック動物)
本発明によれば、本タンパク質をコードする外来性DNAを有する非ヒト哺乳動物が提供される。このトランスジェニック動物によれば、外来性DNAによって本タンパク質を発現しているため、本タンパク質の活性の研究や、本タンパク質が関連する疾患の予防・治療剤のスクリーニングや評価等に適した動物となっている。ここで非ヒト哺乳動物としては、例えば、ウシ、ブタ、ヒツジ、ヤギ、ウサギ、イヌ、ネコ、モルモット、ハムスター、マウス、ラットなどが用いられる。なかでも、マウスやラットなどのゲッ歯動物が好ましい。
【0064】
トランスジェニック動物は、未受精卵、受精卵、精子およびその始原細胞を含む胚芽細胞などに対して、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法などにより本外来性DNA等を導入するなどし、該細胞を用いて胚を作製することにより得ることができる。
【0065】
本トランスジェニック動物の作製に用いる外来性DNAは、本タンパク質をコードする領域を、各種哺乳動物(例えば、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)由来のDNAを発現させうる各種プロモーターの制御下に有し、さらに、ターミネーター等を備えるDNA構築物とすることができる。こうしたDNA構築物は、DNAの導入方法の種類に応じて適宜選択することができ、Naked DNAとすることができるほか、従来公知の発現ベクターの形態を採ることができる。
【0066】
本トランスジェニック動物は、本外来DNAをヘテロで有することもでき、ホモで有することもできる。また、本トランスジェニック動物は、こうした外来DNAを宿主染色体においてランダムに導入されたものであってもよいし、所望の部位にノックイン形態で導入されたものであってもよい。本トランスジェニック動物は、通常よりも本タンパク質を過剰に発現する傾向があるため、本トランスジェニック動物を用いることで、例えば、本タンパク質の機能亢進症を発症した場合には、当該病態モデル動物として使用できることもある。
【0067】
また、本発明によれば、本タンパク質の変異型、例えば、1416位のセリンがアラニンに置換されてAktの基質となりえず本タンパク質と同質の活性を有しない変異型タンパク質をコードする外来DNAを保持するトランスジェニック動物も提供される。この変異型タンパク質を発現するトランスジェニック動物は、本タンパク質の外来DNAが導入されたトランスジェニック動物と同様、ヘテロであってもホモであってもよいが、ランダムに変異型外来DNAが導入された場合には、多くの場合本タンパク質以外にその変異型タンパク質を発現するものとなっており、本タンパク質をコードする宿主染色体を破壊するように変異型外来DNAが導入された場合には、変異型タンパク質のみを発現するものとなる。こうした変異型タンパク質発現トランスジェニック動物は、本タンパク質の機能不活性型不応症の病態機序の解明及びこうした疾患を治療方法の研究用途に用いることができる。また、本タンパク質の機能不活性型不応症に対する治療薬スクリーニングや評価にも利用できる。
【0068】
なお、こうしたトランスジェニック動物は、組織培養のための細胞源、組織観察用の組織源、薬剤スクリーニング用細胞源、本タンパク質又はその変異型タンパク質の取得源として利用することもできる。
【0069】
(ノックアウト動物)
本発明によれば、本タンパク質をコードするDNAの発現が抑制された非ヒト動物が提供される。こうした動物によれば、本タンパク質の発現が抑制されているため、本タンパク質が関わる各種の活性を欠失する可能性があり、本タンパク質の生物活性の不活性化を原因とする疾病のモデルとすることができる。
【0070】
なお、非ヒト哺乳動物としては、前記トランスジェニック動物と同様のものを用いることができる。また、本タンパク質をコードするDNAがノックアウトにより破壊されたノックアウト動物を得るには、本タンパク質をコードするゲノムやコード領域あるいはその近傍の塩基配列を利用してターゲティングベクターを構築して、これを胚作製に用いればよい。胚作製にあたっては、ES細胞あるいは非ヒトクローン動物の作製が可能な体細胞を用いることが好ましい。また、上記したRNA干渉を発現可能な核酸構築物を導入した胚を作製することにより、本タンパク質をコードするDNAの発現がRNA干渉により抑制されたノックダウン動物を得ることができる。
【0071】
(分析方法等、診断剤、診断方法、診断キット)
本発明の抗体は、本タンパク質又はリン酸化された本タンパク質を特異的に認識することができる。本発明の抗体を用いることにより、被検試料中の本タンパク質又はリン酸化された本タンパク質を、特にELISAなどのEIAなどの抗体を利用した酵素抗体法等などの測定方法により定量することができる。すなわち、本発明の抗体は本タンパク質又はリン酸化された本タンパク質の定量用試薬として用いることができる。もちろん、本タンパク質の定性用試薬としても用いることができる。さらに、細胞や組織などの被験試料中の本タンパク質又はリン酸化された本タンパク質の量は、本タンパク質が関連する疾患、例えば、細胞運動、細胞移動及び血管形成に関連する疾患の診断に関連するため、本発明の抗体は、こうした疾患の診断剤として使用できる。
【0072】
また、Aktが本タンパク質をリン酸化することを利用し、このリン酸化の活性化程度又は阻害程度から本タンパク質の各種活性を活性化し阻害する化合物の濃度を定性的に検出できる。すなわち、Aktと本タンパク質とを含有しAktによる本タンパク質のリン酸化が可能なアッセイ系を準備しておき、このアッセイ系に試験化合物を供給して、基質である本タンパク質又は反応生成物であるリン酸化された本タンパク質を検出することにより、この試験化合物がAktによる本タンパク質のリン酸化を活性化する化合物であるか阻害する化合物であるかを定性的に検出することができる。なお、本タンパク質又はリン酸化された本タンパク質の検出は、本タンパク質に特異的な抗体又はリン酸化された本タンパク質に特異的な抗体を用いた酵素抗体法などによることができる。また、同様の系を利用し、標準曲線等を作製することにより、試験化合物がAktによる本タンパク質のリン酸化を活性化するあるいは阻害する化合物であるとき、試験化合物を定量することができる。こうしたアッセイ系による試験化合物の定性的又は定量的分析は、本発明のスクリーニング方法や本タンパク質の関連する疾患の診断方法に利用できる。
【0073】
なお、本発明の抗体を用いる本発明のタンパク質及びリン酸化された抗体の定量法は、特に制限されるべきものではなく、被測定液中の抗原量(例えば、タンパク質 量)に対応した抗体、抗原もしくは抗体−抗原複合体の量を化学的または物理的手段により検出し、これを既知量の抗原を含む標準液を用いて作製した標準曲線より算出する測定法であれば、いずれの測定法を用いてもよい。例えば、ネフロメトリー、競合法、イムノメトリック法およびサンドイッチ法が好適に用いられるが、感度、特異性の点で、サンドイッチ法を用いるのが特に好ましい。標識物質を用いる測定法に用いられる標識剤としては、例えば、放射性同位元素、酵素、蛍光物質、発光物質などが用いられる。放射性同位元素としては、例えば、〔125I〕、〔131I〕などが、上記酵素としては、安定で比活性の大きなものが好ましく、例えば、β−ガラクトシダーゼ、β−グルコシダーゼ、アルカリフォスファターゼ、パーオキシダーゼ、リンゴ酸脱水素酵素などが、蛍光物質としては、例えば、フルオレスカミン、フルオレッセンイソチオシアネートなどが、発光物質としては、例えば、ルミノール、ルミノール誘導体、ルシフェリン、ルシゲニンなどがそれぞれ用いられる。さらに、抗体あるいは抗原と標識剤との結合にビオチン−アビジン系を用いることもできる。担体としては、例えば、アガロース、デキストラン、セルロースなどの不溶性多糖類、ポリスチレン、ポリアクリルアミド、シリコン等の合成樹脂、あるいはガラスなどが用いられる。また、こうした検出においては、抗体分子そのものを用いてもよく、その一部を用いてもよい。
【0074】
本発明の診断キットは、本発明の抗体及びAkt及びGirdinなどの本タンパク質を含むAktリン酸化アッセイ系エレメントから選択される1種又は2種以上を含むことができる。以上説明した、本発明の抗体、Aktリン酸化アッセイ系は、診断用途のみならず、細胞運動、細胞移動及び血管形成の研究用途に用いることができる。
[実施例]
【0075】
以下、本発明を挙げて説明するが、本発明は以下の実施例に限定されるものではない。なお、以下の実施例において用いた実験方法等について以下に説明し、次いで個別実施例について説明する。
【0076】
1.酵母2-ハイブリッド試験
Aktと相互作用するプロテインを特定するため、Akt1のfull-length cDNAをpAS2ベクター(Clontech)に挿入した。それによって得られたコンストラクトを先述のヒト胎児の脳MARCHMAKER cDNAライブラリー(Murakamiほか、2002)をスクリーニングするための材料(ベイト)とした。挿入されたcDNAを含む2つの陽性プラスミドを選び、配列を解析した。それら2つのプラスミドは、Girdinの C末端領域 (残基1217-1870)をコードするcDNA 断片を含んでいた。Girdinをコードするfull-length cDNAについて、その末端を5’ 迅速増幅(5’-RACE システム, Invitrogen)することによりヒト胎児の脳ポリA+ RNA から分離した。さらに、ヒトAkt1とGirdin cDNAの断片を含む精製したpASとpACでもって酵母2-ハイブリッド結合試験を行った。
【0077】
2.プラスミド
構成的に活性で優性阻害の特性を持つ野生のヒトAkt1はY. Gotoh氏(東京大学)の好意により入手した。そしてpcDNA3.1-, pGEX-, pEGFP- Girdin断片のコンストラクトを作製した(Murakamiほか、2002)。EGFPをタンパク質のアミノ末端に、V5とmyc tagとをタンパク質のカルボキシル末端に融合した。Girdin変異体はQuick Change site-directed mutagenesis kit(Stratagene)を使って、製造者プロトコールに従って作製した。Girdinのヌクレオチド780-800(5’-AAGAAGGCTACGGCAGGAATT-3’)(下線部は変異遺伝子)に2つのサイレント変異を導入することで抗siRNA Girdinを作製した。
【0078】
3.抗体
Girdinのカルボキシル末端の19アミノ酸に対してウサギ抗Girdinポリクローナル抗体を生成させ、免疫ペプチドでアフィニティー精製した。抗リン酸化Girdinポリクローナル抗体は、Kumamoto Immunochemical Laboratory, Transgenic, Inc.(熊本県)から提供を受けたもので、Girdinのアミノ酸配列1408-1420(CDINRERQKpSLTLT)に対応する、キーホールリンペットヘモシアニンに結合したリン酸化ペプチドでウサギを免疫することにより取得した。抗血清はリン酸化ペプチドに結合したカラムに結合したフラクションとして精製した。実験で使用した他の抗体としては、抗Aktポリクローナル抗体(Cell Signaling Technology)、抗リン酸化Aktポリクローナル抗体(Cell Signaling Technology)、抗コータクチンモノクローナル抗体(Upstate)などがある。
【0079】
4.キナーゼアッセイ
Girdin CT-V5 WT、SAまたは生成したGST-STを発現しているCOS7細胞から採取した抗V5抗体(Invitrogen)による免疫沈降物を、活性もしくは不活性組み換えAkt(500ng)(Upstate)と共に、キナーゼ緩衝液(20 mM MOPS、25 mM β-glycerophosphate、5mM EGTA、15mM MgCl2)に10μCi[γ-32P]ATP(3000 Ci/mmol、Amersham)を添加した中で培養した。混合物を30℃で30分間培養し、Laemmliドデシル硫酸ナトリウム(SDS)サンプル希釈緩衝液(20 mM Tris-HCl [pH 6.8]、2 mM EDTA、2% SDS、10% sucrose、20μg/ml bromophenol blue、80mM dithiothreitol)を加えることによって、反応を終わらせた。
【0080】
5.免疫蛍光染色法
ベロ細胞をフィブロネクチン(10μg/ml、Sigma)とコラーゲンI(10μg/ml、Upstate)でコートしたカバースリップもしくはガラス製ディッシュにのせて固定し、上述した抗体で染色した。蛍光染色は共焦点レーザ顕微鏡(fluoview FV500、Olympus)で確認した。
【0081】
6.アクチン共沈アッセイ
F-アクチン共沈降試験を製造者プロトコール(Cytoskeleton)に従って行った。精製したGST融合プロテイン、GST、α-アクチン(Cytoskeleton)を40μgの精製アクチンフィラメントと共に室温で30分間培養した。F-アクチンの最終濃度は18μMであった。フィラメントは、その後、遠心分離(100000×g)(ベックマン)でペレット化した。共沈させたタンパク質は、SDS−PAGEで分離してクマシーブリリアントブルー(CBB)又は抗GST抗体(Cell Signaling Technology)又は抗リン酸化Girdin抗体を用いたウエスタンブロット分析で確認した。 定量的アッセイには、所定濃度のGST-Girdin CT2(1μM)を、重合性緩衝液中で各種濃度のF-アクチン(0〜2.5μM)を混合し、室温で30分間インキュベートした。タンパク質を上記と同様にして遠心分離し、全てのペレットと上清をそれぞれSDS-PAGEを施した。タンパク質のバンドをCBB染色で検出してスキャンし、WinROOF(Mitani Corp., Fukui,Japan)で解析した。
【0082】
7.RNA干渉
GirdinとAktのsiRNAによるノックダウンは既に記載された方法を用いて実施した(Watanabe et al., 2004)。Girdinの発現のサイレンシングを媒介したターゲット配列は、以下のとおり、5-AACCAGGTCATGCTCCAAATT-3(145-165ヌクレオチド、GirdinsiRNA A)及び5-AAGAAGGCTTAGGCAGGAATT-3(780-800ヌクレオチド,GirdinsiRNA B)であった。これらの21ヌクレオチドの合成二重鎖はQiagenにより作製された。また、ヒトAkt1に特異的なsiRNAはQiagenより入手した。ベロ細胞を、リポフェクタミン2000(Invitrogen)を用いてこれらのsiRNA又はコントロールとしての無関係な21RNA(Qiagen)により製造者プロトコールに従ってトランスフェクトした。
【0083】
8.細胞膜表面(細胞質側)の凍結レプリカ電子顕微鏡
細胞質膜の細胞質側表面の電子顕微鏡観察は、以前に報告された方法に従った(Heuser, 1989, 2000; Usukura, 1993)。ガラスカバースリップ(直径3mm, スタンダード#1, Matunami、Osaka, Japan)上で培養したベロ細胞を、siRNAでトランスフェクトした。上層側の細胞膜から割断(アンルーフ)した後、直ちに細胞を2.5%グルタルアルデヒドの緩衝液A溶液(緩衝液A;70mM KCl, 5mM MgCl2、3mMEDTA、30mM HEPES緩衝液,pH7.4(KOHにて))中で固定化した。緩衝液と蒸留水との混液で洗浄した後、直ちに試料を液体ヘリウムで急速冷凍装置(Eiko, Tokyo, Japan)で凍結した。試料は、その後、フリーズエッチ装置(FR2000、HITACHI、Ibaraki、Japan)を用いてプラチナ−カーボンでロータリーシャドウィングした。Girdin分子の免疫ラベルのために、アンルーフした細胞を4%パラホルムアルデヒド/0.5%グルタルアルデヒドの緩衝液A溶液中で固定化した。緩衝液B(100mM NaCl、30mM HEPES、2mMcaCl2)で3回洗浄後、試料をクエンチしブロックして、1%BSAを含む緩衝液B中で一次又は二次金結合抗体(Amersham)で37℃で1時間ラベルした。最終的に、上記と同様の方法で速やかに凍結し、フリーズエッチした。
【0084】
9.スクラッチ誘導細胞移動及び低速度イメージング
ベロ細胞における方向性のある細胞移動をインビトロスクラッチ損傷アッセイ(Watanabe et al., 2004)を用いて単層状態で刺激した。ベロ細胞を、フィブロネクチンがコートされたカバースリップか又は35mmのガラス製ディッシュに播種し、siRNAをトランスフェクトした。トランスフェクション48時間経過後、コンフルーエント状態のベロ細胞を、200μlのディスポーザブルピペットチップの先端で引っ掻いて(スクラッチ)、損傷方向に移動可能な状態とした。細胞は、示された時間ごとに免疫染色のために固定化した。低速度観察のためには、ベロ細胞にはsiRNAとGFP-アクチンとの双方をトランスフェクトして、スクラッチ損傷アッセイに供した。損傷側エッジの細胞は、共焦点レーザ走査電子顕微鏡(Fluoview, FV500, Olympus)にて観察した。
【0085】
10.三次元細胞移動アッセイ
各種のコンストラクトやsiRNAでトランスフェクトした細胞の移動性を評価するために、ボイデンチャンバー細胞移動アッセイを改変して、HTS FluoroBlok Insert(8.0μM pores 24-ウエル、BD)を用いる蛍光色素ブロッキングマイクロポアメンブレンを通過するGFPでラベルした細胞の移動を計測することができるようにした。すなわち、メンブレンの両サイドを10μg/mlのフィブロネクチンを用いて12時間37℃でコートし、PBSで洗浄した。その後、チャンバーを、0.1%のBSAを含有しヒト組換えEGF(20ng/ml)を含む又は含まないDMEMが満たされた24ウェルディッシュ内に配置した。HT-1080を用いた移動アッセイには、10%FBSが下側のチャンバーに添加された。24ウェルのプレート内において、細胞(1×105)にGFP(0.5μg(対個々の細胞)、示されたコンストラクト(2.5μg)及びsiRNA(20pmol)をトランスフェクトし、チャンバー上部に置いて、4時間メンブレンの孔部を通過して移動させるようにした。細胞の移動性は、膜を通過して移動したGFP陽性細胞を蛍光顕微鏡でカウントすることで定量した。
【0086】
11.タンパク質−脂質オーバーレイアッセイ
GST融合タンパク質をDH5α又はBL21−CodonPlus細胞(Stratagene)中に発現させ、常法にて精製した。リン脂質への精製GST-CTの結合は、製造者プロトコールに従って、PPIP-Strip(Echolen Biosciences)を用いて4℃で確認した。結合したGST-CTは、抗Girdin抗体又は抗リン酸化Girdin抗体のいずれかを用いて検出した。
【0087】
(実施例1: Girdinの同定、一次構造及びその発現様式)
Aktの新規基質を同定するために、ヒトAkt1の全長をベイトコンストラクトとして酵母two-hybrid法を行って、ヒト胎児脳cDNAライブラリを用いて相互作用するタンパク質を探索した(図1a参照)。その結果、二つのクローン(F-10、F-12)を検出した。図1bに、ヒトAkt1とクローンF-10及びF-12との結合様式を示す。次いで、これらのcDNAを酵母で発現させると、Akt1のC末端調節領域とのみ相互作用することがわかった。さらに、cDNAの5'-末端につき、5'-RACEを行うことで、5'末端に連続する3.6kbのcDNAを含む全コード領域を備えるクローンを取得した。得られた全長cDNAは5610bpのオープンリーディングフレームを含んでおり、1870アミノ酸残基を有するタンパク質をコードしていた。また、データベース探索の結果、マウス、ラットとショウジョウバエにおいてホモログが存在することがわかった。
【0088】
図1cには、Girdinの一次構造とCOILSアルゴリズムによって予測されるGirdinの構造を示す。図1cに示すように、Girdinは、N末端領域(NT)とα−ヘリカルコイルド−コイル領域(Ala253〜Lys-1375)とにより二量体を形成することが予測された。予測されるコイルドーコイル領域は、当該領域に典型的な7アミノ酸リピートを135個有していた(図1h参照)。
【0089】
また、GirdinのmRNAの発現をノザンブロッティングにより分析した。結果を図1dに示す。図1dに示すように、Girdinは多種のヒト組織において普遍的に発現していることがわかった。さらに、GirdinのC末端の19アミノ酸に対するポリクローナル抗体を用いてウエスタンブロッティングを行ったところ、250kDaのタンパク質として脳及び精巣溶解物中に確認することができた(図1e参照)。
【0090】
また、Girdinのオリゴマー形態を確認するため、COS7細胞溶解物の100μMスルホスクシニルイミドによる架橋前後につき、抗Girdin抗体を用いてウエスタンブロッティングにより分析した。結果は、図1fに示すように、架橋剤処理後では、Girdinは無傷の細胞内において高分子の複合体として存在することが強く示唆された。さらに、COS7細胞溶解物をゲルろ過に供した結果によれば、Girdinは、大きなタンパク質複合体を形成していることがわかった。さらに、NT領域とCT領域とがオリゴマー形成に関与するかどうかを調べるために、V5エピトープタグ付きNT(NT-V5)とmycエピトープタグ付きNT領域(NT-myc)をCOS7細胞で発現させると、図1gに示すように、これらの二つのNT領域の複合体が細胞内にて観察された。このことはNT領域がGirdinのオリゴマー形成に寄与していることを示唆していた。一方、CT領域はオリゴマー形成に寄与していないことがわかった(データ示さず)。NT領域を発現するCOS7細胞溶解物のゲルろ過結果によれば、NT領域は二量体を形成していると予測された(図1i参照)。
【0091】
(実施例2:Aktによる試験管内(in vitro)および生体内(in vivo)におけるGirdinのリン酸化)
AktとGirdinとが物理的に相互作用するかどうかをインビボ及びインビトロによるアッセイで確認した。しかしながら、インビトロにおいても免疫沈降によっても両タンパク質の安定的な複合体を検出できなかった(データ示さず)。このことは、両者には、例えば、プロテインキナーゼとその基質の相互作用に観察されるような極めて一過性の様式によって通常の相互作用が生じていることを示唆した。Aktのリン酸化サイト基質はR-x-R-x-x-S/Tサイトを含むものとされており、GirdinのCT領域の1416位のセリン(下線)に近接するアミノ酸配列(RERQKS)がこのアミノ酸配列に相当していた。また、このサイトが唯一のコンセンサス領域であった。推定されるリン酸化領域であるCT領域は、他の種類のGirdinホモログにおいても保存されていたため、AktがGirdinをリン酸化するかどうかを検討することとした。
【0092】
図2aに示すインビトロキナーゼアッセイにより、GirdinCT野生型(WT)がAktによりリン酸化されるが、Ser-1416がAlaに置換したSA変異体がリン酸化されないことは、Aktは直接1416-Serをリン酸化していることを示しているといえた。一方、NT領域及びコイル領域はいずれもインビトロではGirdinによりリン酸化されなかった(図2b参照)。
【0093】
次に、Ser-1416がAktによって触媒されるリン酸化部位であることをインビボで確認するためにリン酸化されたSer-1416を含むペプチドに特異的に結合する抗体を作製し、この抗体を用いて、図2cに示すウエスタンブロッティング分析を行った。図2cに示すように、抗リン酸化Ser-1416抗体は、野生型Akt(Akt WT)又は構成的活性型Akt(Akt CA)の共発現下GirdinCT(WT)を認識した。しかしながら、ドミナントネガティブ(優勢阻害)Akt(Akt DN)では認識しなかった。また、抗リン酸化Ser-1416抗体は、Akt CAの共発現下であっても、Girdin CT(SA)変異体を認識しなかった。これらの結果から、抗リン酸化Ser-1416抗体は、Ser-1416がリン酸化されたGirdin CTを特異的に認識することがわかった。
【0094】
次いで、抗リン酸化Ser-1416抗体が、生理学的に内在性Aktを活性化する外部刺激に応答してリン酸化された内在性Girdinを認識できるかどうかを検討した。図2dに示すように、EGFを供給すると、免疫沈降によってGirdinと推定される250kdaのタンパク質のリン酸化が誘導された。この誘導のタイムコースは、Aktの活性化のタイムコースに類似していた。さらに、Girdinのリン酸化は、細胞がPI3KインヒビターであるLY294002及びWortmanninにより処理されとき阻害されたが、ミトゲン活性化プロテインキナーゼキナーゼ1(MEK1)インヒビターであるPD98059により処理されたときには阻害されなかった。これらの結果から、Girdinのリン酸化は、PI3K依存的に誘導されると結論した。また、図2eに示すように、Akt WT及びAkt CAの発現は、Girdinの有意なリン酸化を誘導するが、Akt DNはリン酸化を誘導しなかったことは、活性Aktのみが細胞においてリン酸化を誘導できること示している。さらに、Girdinのリン酸化は、Akt RNA干渉短鎖RNA(Akt siRNA)が導入された細胞においては認められなかった。これらの知見は、Aktの活性化がGirdinのリン酸化に必要であることを支持していた。
【0095】
(実施例3:GirdinのC末端ドメインを介したアクチン線維への結合) Girdinの細胞内局在を確認するために、抗Girdin抗体により、ベロ線維芽細胞を免疫蛍光染色した。図3aのaに示すように、Girdinは、静止細胞においてはアクチンストレスファイバーとともに局在していた。EGF(50ng/ml)により細胞が刺激されると、これらは極在化し始め、その後ラメリポディア(葉状仮足)の指向性のある伸長が生じた。EGFにより刺激された細胞では、Girdinはアクチンストレスファイバーのみならずリーディングエッジにおけるラメリポディアにも局在した。リーディングエッジは、図3aのbにおいて、Arp2/3結合タンパク質コータクチンによる染色域として示されている。
【0096】
Girdinの局在は、GirdinがF−アクチン結合タンパク質であることを示唆していることから、Girdinのアクチン結合ドメインを確認するため、断片をコードする各種の融合GFPタンパク質、NT(GFP-NT)、コイルド-コイル領域についてのN末端ハーフ(GFP-M1)及びC末端ハーフ(GFP-M2)、CT領域についてのN末端ハーフ(GFP-CT1)及びC末端ハーフ(GFP-CT2)を作製し、これらの細胞内局在性を確認した(図3bのa参照)。図3bのb〜dに示すように、ベロ細胞にて発現されたとき、GFP-NT、GFP-M1及びGFP-M2は、細胞質に局在した。図3bのeに示すように、GFP-CT1は、核と、カルボシアニンメンブレンプローブであるCM-Dilの共局在によって示されるように細胞膜との双方に局在した。この一方、図3bのfに示すように、GFP-CT2は、明らかにストレスファイバー上に局在した。これらの結果は、GirdinはそのCT2領域を介してアクチンフィラメントに局在するが、CT1領域を介して細胞膜を相互作用することを示唆していた。なお、内在性Girdinの核への蓄積が観察されないことから(図3a参照)、GFP-CT1の核局在は人工的なものであると考えられた。
【0097】
次に、アクチン共沈アッセイによりCT2ドメインのアクチン結合能力について検討した。図3c及び図3dに示すように、精製したグルタチオンSトランスフェラーゼ(GST)融合CT2(GST-CT2)はα-アクチニンと同様、F-アクチンと共沈したが、GST単独では共沈しなかった。このことは、GST-CT2は、F-アクチンに直接結合していることを示していた。F-アクチンの量が増えると、図3dのaに示すように、F-アクチンと結合するGST-CT2の量は飽和した。図3dのbに示すように、F-アクチンの解離係数(Kd)は、1.03μMであった。このことは、GirdinがF-アクチンに対して比較的低い親和性しか有していないことを示している。Girdinの各種のドメイン、予測される機能及びGirdinの推測される構造を図3eに示す。
【0098】
(実施例4:Girdinのストレスファイバーの形成と細胞運動における重要性) Girdinの機能を評価するために、ベロ細胞でのGirdin発現を抑制(ノックダウン)するためにRNA干渉を採用した。いくつかのGirdin siRNA(21ヌクレオチド)とコントロールsiRNAとをベロ細胞に導入した。図4aに示すように、ウエスタンブロット分析によれば、Girdin siRNAを導入した細胞では、Akt及びアクチンの活性に影響を及ぼすことなくGirdinの発現レベルを90%以上抑制した。ノックダウン効果の特異性を確認するために、2種類のGirdin siRNA(A及びB)を用いたウエスタンブロット分析を行い、他のファンクションアッセイを行った。
【0099】
Girdinがアクチンフィラメントと架橋するのに機能するかどうかを確認するため、アクチン細胞骨格の組織化におけるGirdinのノックダウン効果を調べた。図4bのaに示すように、抗Girdin抗体による免疫蛍光染色によれば、Girdin siRNAが導入された細胞では、Girdinは非常に少量であった。また、図4bのbに示すように、ファロイジンによるF-アクチンリッチ構造の染色によれば、Girdin siRNAが導入された細胞ではアクチンストレスファイバーが破壊されていた。このことはGirdinが、アクチンストレスファイバーの形成に本質的であることを示している。
【0100】
高倍率でGirdin siRNAが導入された細胞を観察すると、この細胞は、薄く短いストレスファイバーが顕著な低下が観察された。さらに、これらは本来の形態を失い、多数の突起(リーディングエッジ)の形成ででこぼこした境界を形成していた(図4bのbの下側及び図4c参照)。
【0101】
さらに、アクチン動態におけるGirdinの役割を明らかにするために、試験管内創傷治癒アッセイにおける細胞移動におけるGirdinが抑制された細胞の振る舞いを調べた。図4dに示すように、創傷に接触したGirdin siRNAが導入された細胞は、そのリーディングエッジにおいて伸長したラメリポディアを形成することができず、突起が繰り返し伸びたり縮んだりするような細胞運動異常を示した。これらの結果は、Girdinは細胞移動の間のアクチンフィラメントの組織化に不可欠であることがわかった。
【0102】
Girdinのノックダウン効果の特異性を確認するため、他のセルラインであり、RETレセプターチロシンキナーゼを安定的に発現しているSK-N-MC神経芽腫細胞にGirdin siRNAを導入した。Girdin siRNAが導入されたSK-N-MC(RET)細胞では、ストレスファイバーの破壊とグリア細胞系列由来神経成長因子(GDNF)であるRETリガンドに応答した制限的なラメリポディアの形成を示した。
【0103】
(実施例5:電子顕微鏡によるGirdinの局在とその発現抑制がアクチン機構に与える影響の解析)
さらに、インビボでのGirdinとアクチンフィラメントとの相互作用とアクチン組織化のける役割を確認するために、「アンルーフ」ベロ細胞についてのディープエッチ電子顕微鏡による超構造分析を行った。図5aに示すように、免疫金コロイドラベル分析によって、Girdin分子は、アクチンフィラメントとともに局在することわかった。図5bに示すように、高倍率画像から、Girdinがアクチンフィラメントと間の連結部分とともに局在することがわかった。この免疫蛍光分析結果と整合して、Girdinが枯渇したベロ細胞の細胞膜に接した細胞質界面における電子顕微鏡像において視認される表層のアクチンフィラメント密度は、コントロール細胞よりも低かった(図5c参照)。また、コントロール細胞では、より強固に組織化されたアクチンファイバーが優勢であった。コントロールsiRNAを導入した細胞では、フィラメントは相互に隣接して分離することなく同方向を指向して、たくさんの分子によって強固に架橋された太いケーブルを形成していた。いくつかのフィラメントが他のアクチンケーブルに対して鉛直に指向して、結果として、織物様で高密度なアクチンネットワークを形成していた。
【0104】
(実施例6:Girdinのリン酸化が局在およびイノシトールリン脂質(ホスホイノシチド)との相互作用に与える影響) AktによるGirdinのリン酸の役割についての知見を得るために、抗リン酸化Girdin抗体による染色を用いてベロ細胞中のリン酸化Girdinの局在を調べた。図6aの最上図に示すように、血清飢餓状態の静止細胞では、Girdinのリン酸化は細胞全体を通じてほとんど観察されなかった。この結果は、図2dに示すウエスタンブロット分析結果に整合している。一方、細胞がEGF(50ng/ml)で刺激されると、免疫染色によれば、移動する細胞のリーディングエッジにおけるラメリポディアにリン酸化されたGirdinが現れることを示している(図6a中段)。また、リン酸化されたGirdinは、リーディングエッジに存在するコータクチンと共局在する(図6b参照)。さらに、EGFで刺激されたベロ細胞は、リーディングエッジのラメリポディアにおいて、リン酸化されたGirdinとともに活性化されたAktの蓄積と局在とが増強されていることを観察した(図6g)。また、Girdin siRNAが導入された細胞では、EGFで刺激されると、多数のリーディングエッジが誘導されるが、これらのリーディングエッジでは、リン酸化されたGirdinのシグナルは、予想されたように存在しなかった(図6a下段)。この結果は免疫染色の特異性を支持している。
【0105】
次に、リン酸化されたGirdinとリン酸化されていないGirdinの局在性の違いを決定するメカニズムを解明するために、GirdinのCT1ドメインは細胞膜に局在しリン酸化サイトを有していることから、Girdinのリン酸化によって制御されているのは、Girdinの細胞膜との相互作用であると仮定して実験を行った。本発明者らは、図6cのaに示すように、リン酸化サイトの上流側にプラスに荷電した19アミノ酸残基(Arg-1389からLys-1407)からなり、フォスファチジルイノシトール4,5−ビスリン酸が結合するためのコンセンサス配列に類似した配列を見出した。このフォスフォイノシタイド結合サイトを欠損させたGFP-CT1融合タンパク質をベロ細胞に導入すると、この誘導タンパク質の細胞膜への局在は観察されなかった(図6cのb参照)。この結果は、CT1ドメインは、フォスフォイノシタイドへの結合を介して細胞膜に固定されることを示唆している。
【0106】
Girdinのフォスフォイノシタイド結合能力を確認するために、Girdinのフォスフォイノシタイド結合サイトを含む精製GST融合タンパク質を用いたタンパク質−脂質結合アッセイを実施した。なお、この実験においては、GST-CT1融合タンパク質は、発現及び精製操作において容易に分解してしまうため、CT1ドメインに替えてCTドメインとGSTとを融合させて用いた。図6eのaに示すように、GST-CTは、選択的にPI(4)Pに結合し、PI(3)Pには弱く結合したが、他のフォスフォイノシタイドやリン脂質には結合しなかった。また、図6eのbに示すように、GST-CT融合タンパク質のPI(4)P及びPI(3)Pへの結合能力は、フォスフォイノシタイド結合サイトが除去されたとき喪失した。さらに、CT1ドメインの変異体(プラス電荷を有する塩基性アミノ酸残基がアラニンで置換されたもの)は、フォスフォイノシタイドに結合できないこと及び細胞膜局在性を有しないことを見出した。これらの結果は、フォスフォイノシタイド結合モチーフにおいて塩基性アミノ酸残基によって生じるプラス電荷がGirdinの細胞膜への相互作用に必要であることを示唆した(図6h参照)。
【0107】
次に、フォスフォイノシタイド結合サイトは、Aktリン酸化サイトの近傍に存在する(図6cのa参照)が、Aktはそのフォスフォイノシタイド結合能力を調節することでGirdinの局在性をコントロールしているのかどうかを確認するため、リン酸化されたGST-CT融合タンパク質のフォスフォイノシタイド結合能力を調べた。精製されたGST-CT融合タンパク質は、当然にAktによりインビトロでリン酸化された(図6dのb参照)。次いでこのリン酸化されたGST-CTを、タンパク質-脂質結合アッセイに供した。なお、このタンパク質−脂質結合は、抗リン酸化Girdin抗体により検出した。図6eのcに示すように、リン酸化したGST-CTは、PI4PにもPI3Pにも結合しないことがわかった。アクチン共沈アッセイによれば、GST-CTのリン酸化は、F-アクチンへの親和性を減衰させるものではなく、その結合動態は、GST-CT2に類似したものであることがわかった(図6f、図3d参照)。これらの知見は、フォスフォイノシタイドに対するGirdinの結合はリン酸化により減衰されるが、F-アクチンに対する結合は減衰されないことを示唆した。
【0108】
さらに、Girdinの細胞膜相互作用が、EGF処理によって調節されているかどうかを、GFPと全長Girdin(WT)又はSA変異体との融合タンパク質のいずれかを発現するCOS7細胞を用いて調べた。図6iに示すように、AktによるSer-1416のリン酸化が、細胞膜からのGirdinの脱局在化に必要であることがわかった。
【0109】
(実施例7:Girdinのリン酸化による細胞移動の調節) リーディングエッジにおけるリン酸化Girdinの局在化がGirdinのリン酸化は細胞運動により引き起こされるかどうかを確認するために、以下の実験を行った。すなわち、ベロ細胞のコンフルーエントの単層に損傷を与えた後、抗リン酸化Girdin抗体で免疫染色すると、損傷を受けた端部においては細胞内のGirdinリン酸化レベルが損傷後速やかに上昇し、8時間後に最大に達し、その後少なくとも12時間継続した(図7a参照)。このことは、Girdinのリン酸化が細胞運動に重要な役割を果たしていることを示唆した。したがって、細胞運動におけるGirdinの役割をボイデンチャンバー法により調べた。図7bに示すように、Girdinのノックダウンは、有意にベロ細胞が供給されたEGF(50ng/ml)に応答して下流のチャンバーに移動するのを抑制した。また、図7bに示すように、ベロ細胞におけるCTドメインの発現が細胞移動により有意に減衰したことを見出した。このことは、内在性Girdinとアクチンフィラメントとの相互作用が統合的な細胞移動に重要であることを支持している。先行する研究と整合するように、siRNAによるAktの枯渇も、細胞移動を減衰させた(図7b)。このことは、本来的なAktの活性が、ベロ細胞がこのアッセイ方法の孔部を介して移動するのに必要であることを示唆した。
【0110】
次に、外来性Girdinをノックダウン細胞に供給して当該細胞における細胞移動傷害を復帰させるかどうかを調べた。このため、サイレントミューテーションを有してsiRNA抵抗性Girdin(siRNAr Girdin)コンストラクトを構築した(図7c参照)。図7dに示すように、siRNAr Girdin(WT)をノックダウン細胞において発現させることにより、EGF応答性の細胞移動性を完全に回復させた。これに反して、Ser-1416がAlaに置換されたsiRNAr Girdin変異体であるsiRNAr Girdin(SA)、フォスフォイノシタイド結合サイトを欠損させたsiRNAr Girdi変異体であるsiRNAr Girdin(ΔPB)、フォスフォイノシタイド結合サイトがアラニンで置換されたsiRNAr Girdin変異体であるsiRNAr Girdin(PB ala)は、細胞移動傷害を補うことはできなかった。(図7d及び図6hのc参照)。これらの結果は、ヒト線維肉腫細胞株HT1080を用いた細胞移動アッセイにより確認した(図7f)。
【0111】
最終的に、Girdin変異体を発現するベロ細胞の移動を損傷治癒アッセイを用いて直接確認した(図7g参照)。GirdinWTを発現する細胞は、損傷部位に容易にか速やかに移動したが、GirdinSAを発現する細胞は、延長された形態を示すに留まった。さらに、後者の細胞における核は固定され移動性がないようであった。このことは、これらの細胞がマトリックスから分離できないことを示唆している。GirdinΔPBを発現する細胞及びGirdin PBalaを発現する細胞は、Girdin WTを発現する細胞よりも移動能力は低かった。これらの結果は、Girdin変異体の発現は、細胞移動の適切な方向性を損なうことを示している。図6a〜f及び図6iにおける結果を合わせて考慮すると、これらのデータは、AktによるGirdinと細胞膜との相互作用の調節が細胞移動に必須であることを示している。
【0112】
(実施例8:Girdin siRNAによるヒト臍帯血管内皮細胞HUVECの遊走能の阻害) HUVEC細胞をコントロールsiRNAおよびGirdin siRNAで48時間処理した後、ボイデンチャンバー法(Boyden Chamber法)にてVEGF(血管内皮成長因子)による遊走能を定量した。図8aに示すように、VEGF存在下においてGirdin siRNAを処理したHUVEC細胞の遊走能が著明に低下することが明らかになった。また、図8bには、遊走した細胞が紫色に染色された図を示す。図8bに示すように、Girdin siRNAで処理したHUVEC細胞の遊走細胞数が低下していることが明らかであった。
【0113】
(実施例9:Girdin siRNAによるヒト臍帯血管内皮細胞HUVECの管腔形成能の阻害) 実施例8と同様にしてHUVEC細胞をGirdin siRNAで処理した後、管腔形成能を観察した。観察結果を図9に示す。図9aに示すように、未処理のHUVEC細胞及びコントロールsiRNAで処理したHUVEC細胞は通常の培養条件で管腔形成を示すが、Girdin siRNAで処理したHUVEC細胞では管腔形成は著明に阻害された。図9bには、図9aの各細胞におけるGirdinの発現を抗Girdin抗体で検出したウェスタンブロティング結果を示しているが、図9bに示すように、Girdin siRNAを導入したHUVEC細胞では、Girdinの発現が顕著に抑制されていた。
【0114】
(実施例10:siRNA抵抗性のGirdin遺伝子の発現による管腔形成能の回復) アデノウィルスベクターにsiRNA抵抗性のGirdin遺伝子cDNAを導入した(Ad-rGirdin WT-V5)。またGirdinのAktリン酸化部位であるセリン1416をアラニンに置換したGirdin cDNAを導入したベクターも作製した(Ad-rGirdin SA-V5)。図10aに示すように、siRNAとAd-rGirdin WT-V5をHUVEC細胞に同時に導入すると、管腔形成が回復した(図10aの中央)。Aktによるリン酸化部位を変異させると、管腔形成は回復せず(図10a右)、AktによるGirdinのリン酸化がその機能に重要であることが明らかになった。図10bは、図10aの各細胞におけるGirdinの発現を抗Girdin抗体と抗V5抗体で検出したウェスタンブロティング結果を示している。なお、コントロールとしてVEGFR2(血管内皮増殖因子レセプター2)の発現を示す。
【0115】
以上の結果よりGirdinは血管形成にも重要な役割をはたしていることが判明し、腫瘍内の血管新生の阻害を目指したAkt-Girdin系の阻害化合物の開発による、抗腫瘍薬の開発の可能性がある。
【0116】
(実施例11:マトリゲルプラグアッセイ)
アデノウイルスベクターを用いてショートヘアピン型Girdin siRNAのためのノックダウンベクターを構築した。Girdin遺伝子におけるターゲット配列は、gaaggagaggcaactggat(配列番号3)とした。このアデノウイルス(1×1010個/ml)100μlをVEGF100ng/mlを含有するマトリゲル(BDマトリゲル(製品名))400μlに混合し、その全量をマウス腹部の皮下に注入した。1週間経過後、マトリゲル注入部位を取り出してヘマトキシリン・エオジン(HE)染色した。また、コントロールとして、ノックダウンカセットに替えてEGFPを保持したアデノウイルスを用いる以外は実施例と同様に操作した。結果を図11に示す。
【0117】
図11には、上段にコントロールの結果を示し、下段に本実施例の結果を示す。コントロールでは血管新生が観察されたのに対し、実施例では、ほとんど血管新生が観察されなかった。Girdinの発現が抑制された結果、血管新生が阻害されたことがわかった。なお、本実施例で用いたノックダウンベクターはGirdinの発現を抑制することが確認されている。
【0118】
(実施例12:腫瘍血管におけるGirdinの発現)ヒトの乳癌と子宮頸癌の組織を採取し、Girdinを定法により免疫染色した結果を図12に示す。なお、Girdin組織免疫染色にあたっては、キシレンを用いてパラフィン包理された組織切片の脱パラフィン処理を行った後、脱水処理し、0.3%過酸化水素水による内因性peroxidase活性の不活化処理を行い、ブロッキングをした。その後、抗Girdin抗体(BSAを含むPBSで50倍希釈) を加えて、保湿箱内でインキュベート(4度、一晩)し、PBSで洗浄後、二次抗体を加えて室温でインキュベート(15分)した。PBSで洗浄後、酵素標識ストレプトアビジンを加えて、室温で15分インキュベートし、洗浄し、DAB基質を添加した。PBSに浸して反応を停止させた後、ヘマトキシレン染色で核染色し、洗浄及び脱水した後キシレン処理した。図12において血管内皮細胞にGirdinが強く染色されていることがわかる(矢印部分がGirdinに基づく強い染色部分)。これらの結果から、Girdinは腫瘍血管の内皮細胞で強く発現していることがわかった。
産業上の利用可能性
【0119】
本発明は、各種疾患の予防・治療のための薬剤、そのスクリーニング及び試薬として利用することができる。
【配列表フリーテキスト】
【0120】
配列番号3:Girdin siRNAのターゲット配列
【技術分野】
【0001】
本発明は、アクチン結合タンパク質の利用に関し、特に、セリン/スレオニンキナーゼAktの新規な基質でありアクチン結合タンパク質であるGirdin(Akt Phosphorylation Enhancer)の当該タンパク質が関連する疾患等への利用に関する。
【背景技術】
【0002】
細胞運動の制御機構は、発生における形態形成、創傷治癒や血管新生のほか、癌細胞の浸潤、動脈硬化、免疫疾患などの病態に密接に関わっている。細胞運動の制御分子としては、低分子量のGタンパク質を始め、数多く同定されている(Ridley A, al., Science(2003)302、p1702-1709)。セリン/スレオニンキナーゼであるAkt(プロテインキナーゼB:PKBともいう。)は、細胞の生存や増殖を制御する重要な因子として知られているが、細胞運動に必須であることも哺乳類や細胞性粘菌などの多くの種で示されている(Higuchi M, al., Curr. Biol(2001)11, p1958-1962、Merlot S, al., J. Cell Sci(2003)116, p3471-3478)。Aktの発現が顕著である悪性腫瘍は浸潤性が高い傾向にある。
【発明の概要】
【発明が解決しようとする課題】
【0003】
しかしながら、Aktが細胞運動を促進する分子機構については全く知られていない。本発明は、Aktと細胞運動との分子機構に関わるタンパク質を同定するとともにその機能を解明し、その利用を図ることを一つの目的とする。
【課題を解決するための手段】
【0004】
本発明者らは、酵母two−hybrid法を用いてAktの新規基質を見出すとともに、この基質がアクチン結合タンパク質であって、Aktによってリン酸化されると移動する細胞の先端部(リーディングエッジ)に局在が変化される一方、このアクチン結合タンパク質の変異体を細胞に発現させると細胞の形態に変化が生じて増殖因子の刺激に依存した細胞運動が障害されることを見出し、このアクチン結合タンパク質がアクチン細胞骨格の統合及び細胞運動によって必須であることを見出し、本発明を完成した。すなわち、本発明者らの知見によれば、以下の手段が提供される。
【0005】
(1)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
(2)前記タンパク質は、配列番号2で表されるアミノ酸配列からなるタンパク質である、(1)に記載のスクリーニング方法。
(3)前記部分ペプチドは、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域(CT1領域及び/又はCT2領域)である、(1)に記載のスクリーニング方法。
(4)前記タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている、(1)又は(2)に記載のスクリーニング方法。
(5)セリン/スレオニンキナーゼAktと配列番号1で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含むタンパク質又はその一部との結合活性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(6)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(7)アクチンフィラメント結合能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(8)アクチンストレスファイバー形成能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(9)ラポリメディアにおけるアクチンメッシュワークの形成能の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(10)細胞膜結合性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(11)フォスフォイノシタイド結合性の促進又は阻害を指標とする、(1)〜(4)のいずれかに記載のスクリーニング方法。
(12)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質をコードするポリヌクレオチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
(13)細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬のスクリーニング方法である、(1)〜(12)のいずれかに記載のスクリーニング方法。
(14)前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、(1)〜(13)のいずれかに記載のスクリーニング方法。
(15)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかあるいは2種以上を発現する化合物又はその塩である、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合の阻害活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の阻害活性
c)アクチンフィラメントへの結合の阻害活性
d)アクチンストレスファイバー形成の阻害活性
e)ラポリメディアにおけるアクチンメッシュワークの形成の阻害活性
f)細胞膜結合の阻害活性
g)フォスフォイノシタイドへの結合の阻害活性
(16)配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質の発現量を抑制する化合物である、細胞運動、細胞移動又は血管形成のいずれかが関連する疾患の予防・治療薬。
(17)前記化合物は、配列番号2で表されるアミノ酸配列をコードするポリヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するポリヌクレオチドである、(16)に記載の予防・治療薬。
(18)RNA干渉を発現するRNAである、(17)に記載の予防・治療薬。
(19)前記(18)に記載のRNAをコードするDNAである、(16)に記載の予防・治療薬。
(20)前記(19)に記載のDNAを含有する組換えベクターである、(16)に記載の予防・治療薬。
(21)前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、(16)〜(20)のいずれかに記載の予防・治療薬。
(22)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された細胞。
(23)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された動物。
(24)前記遺伝子の発現量の抑制はノックダウンによる(23)に記載の動物。
(25)細胞運動、細胞移動及び血管形成のいずれかに関連する疾患の研究用、予防・治
療薬のスクリーニング用である、(23)又は(24)に記載の動物。
(26)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定性的分析方法。
(27)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定量方法。
(28)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに特異的な抗体。
(29)配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに特異的な抗体。
(30)前記(28)又は(29)に記載の抗体を含む、細胞運動、細胞移動及び血管形成の活性化又は阻害の研究用試薬。
(31)前記(28)又は(29)に記載の抗体を含む、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断薬。
(32)疾患部位の細胞における配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかを利用する、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断方法。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化活性
c)アクチンフィラメントへの結合活性
d)アクチンストレスファイバー形成活性
e)ラポリメディアにおけるアクチンメッシュワークの形成活性
f)細胞膜結合活性
g)フォスフォイノシタイドへの結合活性
【図面の簡単な説明】
【0006】
【図1a】図1aは、 酵母two-hybridアッセイに用いた各種ベイト(おとり)のコンストラクト(いろいろな遺伝子を組み込んだ発現ベクター)を示す。ヒトAkt1の各種cDNAフラグメントをGAL4転写因子のDNA結合ドメインに融合させるようにpAS2ベクター内に構築した。PH, プレクストリンホモロジードメイン; KD, キナーゼ活性ドメイン; RD, 調節ドメイン。
【図1b】図1bは、 酵母two-hybridアッセイにおけるヒトAkt1とクローンF-10およびF-12の結合の様式を示す。pAS, ネガティブコントロール; pVA3-pTD1, ポジティブコントロール, His, ヒスチジン。
【図1c】図1cは、 Girdinの一次構造とCOILSアルゴリズムによって予測されるGirdinの構造を示す。
【図1d】図1dは、ヒト各種組織におけるGirdinのメッセンジャーRNA(mRNA)の普遍的な発現を示す。クロンテック社の多組織ノーザンブロットメンブレンをGirdin cDNAの3’領域内のプローブによってハイブリダイズした。
【図1e】図1eは、ヒト脳および精巣における内因性Girdinの発現を抗Girdinポリクローナル抗体を用いて還元条件のウェスタンブロット法で検出した結果を示す。(対照の)ウサギIgGでは検出されなかったことを示す。
【図1f】図1fは、内因性Girdinによって形成される大きなタンパク質複合体の検出を示す。核成分を除いたCOS7細胞を100μM BS3で架橋した条件、および架橋しない条件下で抗Girdin抗体を用いてウェスタンブロット法を行った。架橋後(矢印)では架橋しない場合(矢頭)に比べてかなり大きいサイズのバンドが検出された。
【図1g】図1gは、GirdinのN末端ドメイン(NT)は多量体を形成することを示す。V5タグ(NT-V5)およびmycタグを付加したNT(NT-myc)をCOS7細胞にトランスフェクトし、抗myc抗体で免疫沈降したところ、NT-V5がNT-mycと共沈した。下のパネルにみられる複数のバンドはNT-mycの分解産物を示しているのかもしれない。
【図1h】Girdinの推測されるアミノ酸配列を示す。α−ヘリカルコイルドコイル構造を形成すると考えられる、135ヘプタドリピート(abcdefg)135におけるポジションa及びdにおけるアミノ酸残基は、それぞれ赤と緑で示している。フォスフォイノシタイドへの推定結合サイトを下線を施して示す。ボックスは、推定されるAktリン酸化サイトを示す。
【図1i】Girdinは細胞内でオリゴマーを形成し、アクチンフィラメントと大きなタンパク質複合体を形成することを示す図であり、Aは、COS7細胞溶解物Superose6PC3.2/20(Amersham)でゲルろ過し、フラクション後、抗Girdin抗体(上段)及び抗アクチン抗体(下段)でウエスタンブロット分析により各フラクションを確認し、それらの溶出特性を確認した図である。アクチンは二つのピークとして溶出し、最初の一つは、ボイドボリュームに溶出し、アクチン(F-アクチン)の重合体に対応していた。アクチンの主要な溶出ピークは67kDa未満の低分子量に対応する位置にあり、これは、部分的に解離しているかあるいは単量体(G-アクチン)を示唆した。これらのデータは、Girdinが各種の長さのF-アクチンと大きな複合体を形成していることを示している。Bは、COS7細胞を、V5エピトープタグ付きGFP−Girdin N末端フラグメント(NT)(GFP−NT−V5)(上段)又はmycエピトープタグ付きNT(NT−myc)(下段)のいずれかでトランスフェクトし、細胞溶解物をゲルろ過のためにSuperose 6PC処理した。フラクション後、各フラクションを抗V5抗体及び抗myc抗体を用いてウエスタンブロット分析した。GFT−NT−V5又はNT−mycモノマーの算出した分子量は、それぞれ58kDa又は28kDaであり、これらのデータはNTドメインは二量体を形成していることを示唆した。方法:ディッシュ中のCOS7細胞は冷PBSで洗浄し、0.5mlの25mMTris-HCl,pH8.0、250mM、NaCl5mM、1mMEDTA、1mMDTT.懸濁液は、超音波処理し4℃、60分間100000gで遠心分離した。上清をSuperose6PC3.2/20(Amersham)で処理した。溶出は、40μl/minの流速で行った。50μlのフラクションを収集した。使用したタンパク質マーカーは、タイログロブリン(669K)、フェリチン(440K)及びウシ血清アルブミン(67K)であった。
【図2a】図2a中、aは、GirdinのC末端ドメイン(CT)の1416番目セリンがAktのリン酸化サイトであることを示す。bは、Girdin CT-V5の野生型(WT)およびSA変異体(1416番目のセリンをアラニンに置換した変異体)をCOS7細胞にトランスフェクトし、抗V5抗体で免疫沈降し、放射性標識されたATPの存在下において、活性型および非活性型の組換えAktとインキュベーションした。リン酸化されたGirdin CTはオートラジオグラフィーで(上段)、免疫沈降産物はウェスタンブロット法(下段)によって確認したことを示す。
【図2b】図2b中、aは、COS7細胞にトランスフェクトしたV5タグが付加された様々なGirdinのフラグメントを示す。bは、免疫沈降後、放射性標識されたATPの存在下において、活性型Aktとインキュベーションした結果を示す。免疫沈降産物はウェスタンブロット法(左)によって、リン酸化されたGirdin CTはオートラジオグラフィーで(右)によって確認したことを示す。
【図2c】図2cは、 Girdin CT WTおよびSAを各種Akt変異体と共にCOS7細胞へトランスフェクトし、免疫沈降後、抗リン酸化Girdin抗体(anti-P-Girdin)を用いてウェスタンブロット法を示す(上段)。Girdin CTと各種Aktの発現はウェスタンブロット法によるモニター結果を示す(中および下段)。
【図2d】図2dは、 DMSO、PD98059, LY294002およびWortmanninの存在下においてCOS7細胞を上皮増殖因子(EGF)で刺激した。抗Girdin抗体で免疫沈降後、抗リン酸化Girdin抗体および抗Girdin抗体を用いてウェスタンブロット法を行った。Aktの活性化は抗リン酸化Akt抗体を用いて検出した。
【図2e】図2eは、 Akt WT及びAkt CT及びAkt DNのCOS7形質転換体におけるGirdinのリン酸化の誘導を示す。COS7細胞にAktの各種変異体をトランスフェクトした。内因性Girdinを抗Girdin抗体を用いて免疫沈降後、抗リン酸化Girdin抗体および抗Girdin抗体を用いてウェスタンブロット法を行った。
【図2f】図2f中、aは、内因性Girdinのリン酸化におけるAktのノックダウンの影響を示す。COS7細胞にコントロールあるいはAkt siRNAをトランスフェクトし、48時間培養した。その後細胞をEGFで30分刺激し、(E)で記述した方法を用いてGirdinのリン酸化を検出した。bは、COS7細胞におけるsiRNAによるAktの発現抑制を示す。
【図3a】図3a中、a及びbは、刺激前(a)あるいはEGFで刺激後(b)のVero細胞をAlexa488標識のファロイジン(アクチン線維に結合する試薬)および抗Cortactin抗体、抗Girdin抗体で二重染色を行った結果を示す。矢頭はリーディングエッジ(移動する細胞の先端部)におけるラメリポディア(葉状仮足:細胞運動の先端部に形成されるアクチン線維の豊富な構造体)を示す。
【図3b】]図3bは、Girdinの各種フラグメントの細胞内局在。GFP(緑色の蛍光色素遺伝子)と融合させたGirdinの各種フラグメントをVero細胞に発現させ、固定後、それぞれのプローブによって染色した結果を示す。矢印は発現したタンパク質の凝集体を示している。
【図3c】図3cは、Girdin CT2フラグメントはin vitro(試験管内)において直接アクチン線維と結合する。精製されたGST、GST-CT2およびalpha-アクチニンをin vitroで調整されたアクチン線維とインキュベートした。引き続きF-アクチン線維を超遠心によってペレット化し、F-アクチンと共に沈降した各種タンパク質をゲルのCBB染色法(クーマシー染色法)によって検出した結果を示す。
【図3d】図3dは、 GST-CT2とアクチン繊維の結合様式を示す。一定量のGST-CT2を様々な量のF-アクチンと混合し、引き続き超遠心を行った。非結合型、および結合型のF-アクチンを定量し、結合型のGST-CT2をF-アクチンの濃度に対してプロットした。Scatchard解析によってKd値を計算した。
【図3e】図3e中、aは、 Girdinの各種ドメインの局在と機能のまとめを示す。bは、提案されるGirdinの構造を示す。
【図4a】図4aは、Vero細胞におけるsiRNAによるGirdinの発現抑制Aktを示す。コントロールsiRNAおよびGirdin siRNAをトランスフェクトされたVero細胞の細胞抽出液を抗Girdin抗体、抗Akt抗体、および抗アクチン抗体を用いて検出した。
【図4b】図4bは、Vero細胞にコントロールsiRNAおよびGirdin siRNAをトランスフェクトし、72時間後に固定し、Alexa488標識のファロイジンおよび抗Girdin抗体で二重染色を行った。矢頭は突起の先端におけるラメリポディアを示す。
【図4c】図4cは、 各種siRNAのトランスフェクション後、複数の突起を形成している細胞数を抗Girdin抗体およびAlexa488標識のファロイジンによる染色によって定量した。それぞれの群で100個以上の細胞を計測した。結果は平均±標準誤差で示す。
【図4d】図4dは、Vero細胞にGFP-アクチンと各種siRNAをトランスフェクトし48時間培養した。細胞をフィブロネクチンでコートしたガラスのカバースリップにまき、細胞運動を誘発するために細胞層に先の尖ったチップで傷を加えた。細胞イメージの取得は細胞層に傷を加えてから2時間後に開始し、5-6時間、90秒ごとに行った。矢印は傷の方向に向かって細胞が運動する方向を示す。
【図4e】図4eは、Girdinのノックダウンがストレスファイバーの形成及びRETチロシンキナーゼSK−N−MC細胞のラメリポディアへ与える影響を示す。Aは、SK−N−MC(RET)細胞を、siRNAのトランスフェクション後、72時間後に収集した。細胞溶解物は、SDS−PAGEで分析し、抗Girdin抗体及び抗アクチン抗体を用いてウエスタンブロット分析した結果を示す。Bは、siRNAでトランスフェクトしたSK−N−MC(RET)細胞を、50ng/mlのGDNFで60分間刺激し、固定化し、その後Alexa−488ファロイディンで染色した。Girdinがノックダウンされた細胞は、コントロール細胞と比較して多数の突起を伴う顕著な形態変化を呈した。矢印は、リーディングエッジにおけるラメリポディアを示している。Girdinノックダウン細胞は、ラメリポディアを形成するものの、コントロール細胞に比較して極性が弱く伸長性も低かったことを示す。
【図5a】図5aは、抗Girdin抗体を用いた免疫電子顕微鏡による解析結果を示す。
【図5b】図5bは、円で示したGirdin分子の免疫金コロイドシグナルを拡大して示す。矢印は免疫金コロイドシグナルを示す。
【図5c】図5cは、コントロールあるいはGirdin siRNAをトランスフェクトしたVero細胞のアクチン線維の微細構造を示す。
【図6a】図6aは、刺激前、EGFで刺激後、あるいはGirdin siRNAをトランスフェクトしたVero細胞を固定し、Alexa488標識のファロイジンおよび抗リン酸化Girdin抗体で二重染色を行った。矢印はリン酸化Girdinが集積しているラメリポディアを示す。
【図6b】図6bは、 移動中のVero細胞を抗Cortactin抗体と抗リン酸化Girdin抗体で二重染色を行った結果を示す。
【図6c】図6c中、aは GirdinのAktリン酸化サイトの上流に位置している想定されるイノシトールリン脂質結合サイトを示す。bは、GFP-CT1あるいはGFP-CT1 ΔPB(想定されるイノシトールリン脂質結合サイトを除いてある)をVero細胞にトランスフェクトした。矢印は細胞膜におけるGFPシグナルを示す。
【図6d】図6dは、GST-CTの精製とタンパク質-脂質結合アッセイを示す。aは、精製されたGST-CTあるいはGST-CT ΔPBを抗Girdin抗体を用いたウェスタンブロット法で解析した結果を示す。bは、in vitroにおける組換えAktによるGST-CTのリン酸化を示す。In vitroキナーゼアッセイにおけるGST-CTのリン酸化は抗リン酸化Girdin抗体で検出した。
【図6e】図6eは、GST-CT、GST-CT ΔPBおよびリン酸化GST-CTを用いたタンパク質-脂質結合アッセイを示す。GST-CTおよびGST-CT ΔPBの脂質に対する結合は抗Girdin抗体で検出し、またリン酸化GST-CTの脂質に対する結合は抗リン酸化Girdin抗体で検出した。メンブレンにスポットされた各種脂質を(Ed)に示す。
【図6f】図6fは、リン酸化GST-CTはF-アクチンに結合することを示す。GST-CTをin vitroにおいてAktによりリン酸化させ、続いてアクチン沈降アッセイに用いた。一定量のGST-CTをリン酸化させ、様々な量のF-アクチンと混合し、超遠心を行った。沈殿画分に含まれるF-アクチンに結合したリン酸化GST-CTは抗リン酸化Girdin抗体を用いたウェスタンブロット法で検出した。
【図6g】図6gは、活性Aktとリン酸化GirdinとがEGF刺激ベロ細胞のラメリポディアに共局在することを示す。24時間血清飢餓状態においたベロ細胞を未処理A及び50ng/mlのEGF処理Bのいずれかで30分間処理し、固定化し、抗リン酸化Girdinモノクローナル抗体(緑、セルシグナリングテクノロジー)及び抗リン酸化Girdin抗体(赤)で染色した結果を示す。イメージは、共焦点顕微鏡で視覚化した。矢印は、リン酸化Aktとリン酸化Girdinのリーディングエッジのラメリポディアでの共局在を示す。白いボックス内の領域を拡大して示す。
【図6h】図6hは、CT1ドメインのアミノ酸置換のGirdinの機能における影響を示す。Aは、推定されるフォスフォイノシタイド結合(PB)サイトにおける正の電荷が細胞膜におけるCT1ドメインの局在に必要であることを示す。各種のCT1変異体、すなわち、リジン及びアルギニンの正電荷アミノ酸残基のいずれかをアラニンで置換した変異体(PBala、PBalaF、PBalaR)Ser−1416をアラニン又はアスパラギン酸によって置換した変異体(SA、SD)をaに示すようにして作製し、そしてこれらのベロ細胞内における局在性を調べた(b)。矢じりは、細胞膜におけるGFP−CT1の局在を示しており、矢印は、変異体が細胞膜へ局在していないことを示す。Bは、タンパク質−脂質オーバーレイアッセイの結果を示す。GST−CT野生型(ST)及びPBサイトの正電荷アミノ酸残基をアラニンで置換したGSTCT−PBalaをBL−21CodonPlus細胞(Stratagene)で発現させ、精製し(a)、そして、タンパク質−脂質オーバーレイアッセイに供した結果を示す(b,c)。Cは、GFP(0.5μg)、siRNA(20pmol)及び示されたsiRNA抵抗性コンストラクト(siRNAr)(1μg)で同時トランスフェクトし、48時間インキュベートし、ボイデンチャンバー法によりアッセイした。アステリスクは、WTと比較した統計上の有意差(スッチューデントt−検定*P<0.05)を示す。
【図6i】図6iは、Latrunculin処理細胞におけるEGFによる全Girdinの膜相互作用の調節を示す。Aは、COS7細胞をGFP−全長Girdin野生型(WT)又はSer−1416がアラニンに置換されたSA変異体でトランスフェクトし、ガラス製ディッシュ上で24時間インキュベーションした。EGF(50ng/ml)の存在下又は不存在下で60分間インキュベーションした細胞をLatrunculin(1μM、Calbiochem)で10分間処理し、固定化した。COS7細胞で過剰発現されたGirdinWT又はGirdinSAは、核を除いた細胞全体にわたって分散して存在した(上段)。アクチンを破壊するLatrunculinで処理したときには(中段)、GirdinWTとSAとは、細胞質と同様に細胞膜にて検出された(黄色い矢じり)。EGFで刺激した細胞をLatrunculinで処理したときには(下段)、GirdinWTの多くは、細胞膜から解離して点状に細胞質に蓄積した(矢印)。反対に、GirdinSAは、EGFで刺激後も以前として細胞膜と相互作用を維持した(白抜き矢じり)。Bは、COS7細胞を、GFP−全長GirdinPBala変異体でトランスフェクトし、Latrunculinでその後処理した結果を示す。変異体の細胞膜との相互作用は、EGFの不存在下でもLatrunculinによる処理後はほとんど観察されなかった。
【図7a】図7aは、Girdinは細胞運動中に先端部(リーディングエッジ)でリン酸化される。aは、Vero細胞のモノレーヤー(単層にまかれた細胞)をスクラッチした後、障害された面に向いている細胞を固定し、抗リン酸化Girdin抗体で染色した。矢頭はGirdinのリン酸化シグナルを示す。bは、リーディングエッジにおけるGirdinのリン酸化は時間依存性に増加することを示す。
【図7b】図7bは、GFP遺伝子と各種siRNAおよびGirdin CTをトランスフェクトしたVero細胞を48時間培養し、ボイデンチャンバーアッセイに使用した結果を示す。下段チャンバーにはEGFを加える条件と加えない条件で行った。アスタリスクはコントロールと比較して有意差があることを示す。囲みはVero細胞におけるsiRNAによるAktの発現抑制を示す。結果は平均±標準誤差で示した。
【図7c】図7cは、GirdinのsiRNA抵抗性変異体の作製及びそれを利用した評価結果を示す。aは、 Girdin siRNAの標的配列とsiRNA抵抗性変異体の作製のための塩基置換を示す。bは、示された各種のsiRNAとコンストラクトをトランスフェクションしたCOS7細胞の細胞溶解液を抗V5抗体を用いたウェスタンブロット法で解析した(Girdinの各種コンストラクトのC末端にはV5タグが付加してあるため、この方法で検出が可能)結果を示す。
【図7d】図7dは、GFP遺伝子と各種siRNAおよびコンストラクトをVero細胞にトランスフェクトし、48時間培養後にボイデンチャンバーアッセイを行った結果を示す。Girdinの各種変異体の発現は細胞移動アッセイの前にウェスタンブロット法で解析し、発現レベルが同様であることを確認した(結果は示さず)。結果は平均±標準誤差で示した。
【図7e】図7eは、Girdinのリン酸化によって細胞の運動性が制御される提唱モデルを示す。静止した細胞ではGirdinはアクチン線維を架橋し、また細胞辺縁部のアクチン(cortical actin)を細胞膜にアンカーする。一方、細胞運動中にはAktによるリン酸化によりGirdinはリーディングエッジに局在を変化させ、ラメリポディアにおける短く枝分かれしたアクチン線維を架橋する。
【図7f】図7fは、ヒト線維肉腫細胞株HT-1080細胞における指向性移動に必須であることを示す。Aは、HT1080細胞でのGirdinの枯渇を示す。SiRNAを含まないコントロール細胞及びsiRNAがトランスフェクトされたHT-1080細胞からの細胞全溶解物は、抗Girdin抗体、抗Akt抗体、及び抗アクチン抗体を用いたウエスタンブロット分析に供した。Bは、Girdinの枯渇は、HT-1080細胞の指向性のある細胞移動を損なうことを示す。siRNAがトランスフェクトされたHT-1080細胞をボイデンチャンバー法に供した。下段のチャンバーには、血清(5%)が添加された。対コントロールで*Pは<0.05であった。Cは、Girdinの枯渇により細胞移動が障害された細胞は、siRNA抵抗性(siRNAr)型のGirdinを再添加することによって回復することを示す。GFP、示されたsiRNA(20pmol)及び各種コンストラクト(2.5μg)をHT-1080細胞に同時トランスフェクトし、トランスフェクト後、48時間ボイデンチャンバー法に供した。SiRNAr-Girdin野生型(WT)は完全に細胞移動性を回復させた一方、SA及び△PBは、回復させなかった。示した結果は、2つの独立した実験の代表例である。
【図7g】図7gは、Girdin変異対の発現のベロ細胞の指向性のある移動性に対する影響を示す。Aは、損傷治癒アッセイによるGirdin変異体を発現するベロ細胞の移動の観察結果を示す。フィブロネクチン(10μg/ml)でコートしたグラス製ディッシュ上のコンフルエント状態のベロ細胞に、Girdin siRNAとGirdin siRNA抵抗性(siRNAr)野生型GFP-Girdin(WT)と示された変異体(SA、△PB及びPbala)を同時トランスフェクトし(a)、トランスフェクションから48時間経過後、細胞の単層をスクラッチして損傷部への細胞移動を開始させ(b)、GFP-Girdinを発現し、損傷部と向き合う細胞を低速度イメージングで分析した(c)。Bは、野生型Girdin(WT)及び各種変異体(SA、△PB及びPbala)を発現するベロ細胞の移動の低速度イメージングを示す。イメージは、スクラッチから2時間経過後から7時間の間90秒毎に取得した。白い破線は、損傷ラインを示し、矢じりは移動する細胞の核を示す。これらの結果は、二つの独立した実験の代表的なイメージである。
【図8a】図8aは、Girdin siRNAでHUVEC細胞を処理してボイデンチャンバー法による遊走能の定量結果を示す。
【図8b】図8bは、遊走した細胞を染色した画像を示す。
【図9a】図9aは、管腔形成能の観察結果を示す。
【図9b】図9bは、抗Girdin抗体によるウエスタンブロット分析結果を示す。
【図10a】図10aは、管腔形成の回復を示す。
【図10b】図10bは、抗Girdin抗体によるウエスタンブロット分析結果を示す。
【図11】図11は、マトリゲルプラグアッセイの結果を示す図であり、上段がコントロールであり下段がノックダウンベクターを含むマトリゲルを注入した結果である。
【図12】図12は、乳癌及び子宮頸癌におけるGirdinの組織免疫染色結果を示す図である。発明を実施するための最良の形態
【0007】
本発明は、セリン/スレオニンキナーゼAkt(以下、単にAktという。)によってリン酸化されるタンパク質(Girdin:本発明者らによりGirdinと称される。)でありアクチン結合タンパク質であるGirdinに関し、特に、配列番号2で表されるアミノ酸配列と同一のアミノ酸配列(Girdinのアミノ酸配列である。)を有するタンパク質若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチド(以下、前記タンパク質及び前記部分ペプチドを総称して本タンパク質という。)に関する。Girdinは、通常、ヒト受精卵から身体各部が形成される過程において細胞の運動、細胞の移動や血管形成に関与している。具体的には、腫瘍細胞の運動や移動は癌の進行、転移、浸潤に関連し、神経細胞の移動等は神経形成に関連し、血管内皮細胞などの血管構成細胞の移動は、血管形成に関連する。したがって、本タンパク質を用いることにより、細胞運動、細胞移動及び血管形成のいずれか関連する疾患に有効な予防・治療薬を探索し、提供することができる。また、本タンパク質を用いることにより、これらの疾患に関する診断も可能となる。
【0008】
本発明は、また、本タンパク質とAktとの関係において、細胞運動等に現れる各種活性を促進又は阻害する化合物又はその塩に関している。こうした化合物又はその塩は、細胞運動、細胞移動及び血管形成のいずれかを促進又は阻害する化合物又はその塩として利用できる。こうした本発明の化合物によれば、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害することができるため、例えば、これらの細胞動態が関連する疾患の治療、予防に有用である。また、本タンパク質に試験化合物を供給して、本タンパク質に対する作用を検出することにより、細胞運動、細胞移動及び血管形成等に関連する疾患の予防・治療剤として有用な化合物をスクリーニングすることも可能となる。さらに、本タンパク質に特異的な抗体等は、細胞における本タンパク質の量的な測定又は局在を検出することができるため、細胞運動、細胞移動及び血管形成等が関連する疾患の予防や治療のための診断も可能となる。以下、本発明の実施の形態について詳細に説明する。
【0009】
(本タンパク質) 本タンパク質は、配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有することができる。これらのアミノ酸配列からなるものであってもよい。配列番号2で表されるアミノ酸配列と実質的に同一であるアミノ酸配列としては、配列番号2で表されると約50%以上、好ましくは約60%以上、さらに好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、最も好ましくは約95%以上の相同性を有するアミノ酸配列などが挙げられる。相同性(同一性)(%)は、当該分野で慣用のホモロジー検索プログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。なお、相同性(%)は、当該分野で公知の任意のアルゴリズム、例えば、Needlemanら(1970)(J.Mol.Biol.48:444−453)、Myers及びMiller(CABIOS,1988,4:11−17)のアルゴリズム等を使用して決定してもよい。
【0010】
本タンパク質は、配列番号2で表されるアミノ酸配列中の1個又は2個以上(好ましくは、1〜30個程度、好ましくは1〜10個程度、さらに好ましくは数(1〜5)個)のアミノ酸が欠失したアミノ酸配列、又は同数のアミノ酸が付加したアミノ酸配列、又は同数のアミノ酸が挿入されたアミノ酸配列、又は同数のアミノ酸が置換されたアミノ酸配列、また、同数のアミノ酸が欠失、付加、挿入及び置換から選択される2種類以上を組み合わせた態様で改変されたアミノ酸配列を有し、あるいはこうしたアミノ酸配列からなっていてもよい。こうしたアミノ酸の改変部位は、本タンパク質の活性が喪失しない範囲であれば特に限定されない。
【0011】
また、配列番号2で表されるアミノ酸配列と実質的に同一のタンパク質としては、配列番号2で表されるアミノ酸配列からなるタンパク質と同質の活性を有していることが好ましい。同質の活性とは、活性の程度は問わないことを意味している。したがって、活性の程度は、配列番号2で表されるアミノ酸配列からなるタンパク質と同程度未満であることもあり、同程度以上であることもある。こうした活性としては、例えば、本タンパク質のAktの基質としての活性であり、特に、配列番号2のアミノ酸配列における第1416位のセリンあるいはそれに相当する位置のセリンがAktによってリン酸化される活性を有していることが好ましい。また、同質の活性としては上記リン酸化活性は好ましいものとして挙げられるがは上記活性に限定するものではなく、配列番号2で表されるアミノ酸配列からなるタンパク質の他のいずれかの活性であってもよく、2種類以上を組み合わせたものであってもよい。これらの活性については後段で詳述する。
【0012】
また、本タンパク質は、配列番号2で表されるタンパク質と同質の活性を有している限り、その部分ペプチドを包含している。部分ペプチドの領域は特に限定しないが、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域CT領域である、配列番号2のアミノ酸配列の第1376位〜第1870位のアミノ酸(CT1領域及び/又はCT2領域)を含んでいることが好ましい。CT1領域は、配列番号2のアミノ酸配列の第前半部分の領域であり、少なくとも第1389位〜第1407位あるいは当該アミノ酸配列に対応する配列及び第1411位〜1416位の配列を含んでいることが好ましく、例えば、第1375位〜第1622位のアミノ酸配列を有することができる。また、CT2領域は、CT領域の後半部分であり、例えば、第1623位〜第1870位のアミノ酸配列を有することができる。部分ペプチドは、これらの双方の領域を有していてもよいし、いずれか一方のみであってもよい。好ましいCT1領域は、Aktリン酸化サイトを有するとともに細胞膜と相互作用し、好ましいCT2領域はアクチンフィラメントと結合することができる。
【0013】
こうしたタンパク質としては、ヒトにおけるGirdinのオルソログ又はホモログやGirdin(NCBIホームページ(http://www.ncbi.nlm.nih.gov/、アクセッション番号BAE44387にてアミノ酸配列を取得可能である。)等の配列に基づいて人工的に改変したタンパク質が挙げられる。また、Girdinのトランスクリプショナルバリアントとして、既に、同様にNCBIホームページにおいてアクセッション番号NP_060554に記載のアミノ酸配列(1843残基)からなるタンパク質が挙げられる。
【0014】
本タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている態様のタンパク質又は部分ペプチドも包含している。このリン酸化は、Aktによるものであって、Girdinは、このリン酸化により、細胞のリーディングエッジに局在が濃縮され、細胞運動に関与するものと推論される。なお、特に、リン酸化されていない態様とリン酸化された態様とを区別するときには、それぞれを、リン酸化されていない本タンパク質及びリン酸化された本タンパク質と表現するものとする。
【0015】
本タンパク質としては、配列番号2で表されるアミノ酸配列からなる天然由来のタンパク質である上記Girdinが挙げられるが、本タンパク質に包含されるタンパク質又はペプチドには、天然のアミノ酸のみからなっていなくてもよく、本タンパク質の活性が喪失されない範囲で各種の修飾が施されていてもよい。タンパク質及びアミノ酸に対する各種の修飾はよく知られているが、例えば、カルボキシル基に対しては、カルボキシレート(−COO−)、アミド(−CONH2)またはエステル(−COOR)の何れとすることができる。ここでエステルにおけるRとしては、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチルなどのC1−6アルキル基、例えば、シクロペンチル、シクロヘキシルなどのC3−8シクロアルキル基、例えば、フェニル、α−ナフチルなどのC6−12アリール基、例えば、ベンジル、フェネチルなどのフェニル−C1−2アルキル基もしくはα−ナフチルメチルなどのα−ナフチル−C1−2アルキル基などのC7−14アラルキル基、ピバロイルオキシメチル基などが用いられる。本発明で用いられるタンパク質がC末端以外にカルボキシル基(またはカルボキシレート)を有している場合、カルボキシル基がアミド化またはエステル化されているものも本発明で用いられるタンパク質に含まれる。この場合のエステルとしては、例えば上記したC末端のエステルなどが用いられる。さらに、N末端のアミノ酸残基(例、メチオニン残基)などのアミノ基が保護基(例えば、ホルミル基、アセチル基などのC1−6アルカノイルなどのC1−6アシル基など)で保護されていてもよいし、生体内で切断されて生成するN末端のグルタミン残基がピログルタミン酸化したもの、分子内のアミノ酸の側鎖上の置換基(例えば−OH、−SH、アミノ基、イミダゾール基、インドール基、グアニジノ基など)が適当な保護基(例えば、ホルミル基、アセチル基などのC1−6アルカノイル基などのC1−6アシル基など)で保護されていてもよいし、糖鎖が結合したいわゆる糖タンパク質などであってもよい。
【0016】
なお、リン酸化されていない本タンパク質においては、配列番号:1のアミノ酸配列の第1416位のセリンあるいはそれに対応するセリンにおいて、Aktによるリン酸化が可能な範囲に修飾されている。
【0017】
本タンパク質は、塩の形態を採っていてもよい。例えば、生理学的に許容される酸(例、無機酸、有機酸)や塩基(例、アルカリ金属塩)などとの塩が用いられ、とりわけ生理学的に許容される酸付加塩が好ましい。この様な塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩などが用いられる。
【0018】
本タンパク質は、従来公知の方法で取得することができる。例えば、本タンパク質は、哺乳類細胞においてユビキタスに存在するタンパク質であるので、これら細胞あるいはこれらの細胞を含む組織から公知のタンパク質の精製方法により取得することもできる。さらに、本タンパク質をコードするDNAを取得し、該DNAを含有する形質転換体を作製し、培養することによっても得ることができる。タンパク質は、硫安沈殿、エタノール沈殿、酸抽出、イオン交換クロマトグラフィー、疎水クロマトグラフィー、ヒドロキシアパタイトクロマトグラフィー、逆相クロマトグラフィー、ゲル濾過クロマトグラフィーなどのクロマトグラフィーを組み合わせることにより精製単離することができる。また、本発明のタンパク質は、公知のペプチドの固相合成法(矢島治明および榊原俊平、生化学実験講座1、タンパク質 の化学IV、205、(1977年)等)によって合成可能な場合もある。ペプチド合成に使用する官能基の保護ならびに保護基及びその保護基の脱離、反応に関与する官能基の活性化などは公知の基または公知の手段から適宜選択して実施すればよい。また、本タンパク質は、後述するように本タンパク質を発現する外来DNAを保持する細胞を培養したり、当該外来DNAを保持するトランスジェニック動物の細胞や組織から取得することもできる。
【0019】
次に、本タンパク質の活性、すなわち、配列番号2で表されるアミノ酸配列からなるタンパク質であるGirdinの活性について説明する。Girdinは、以下の活性を有しており、本タンパク質は、これらのいずれかの活性を有している。すなわち、(1)Aktの基質としての活性(本タンパク質のAktにより配列番号2における第1416位あるいはそれに対応する部位のセリンがリン酸化される。)、(2)アクチン結合タンパク質活性(アクチンフィラメントに結合しこれらを架橋する。)、及び(3)細胞膜結合活性、を有している。(1)のAkt基質活性は、Aktとの結合(複合体の形成)、Aktによるリン酸化として把握され、(2)のアクチン結合タンパク質活性は、本タンパク質によるアクチンストレスファイバー形成やラポリメディアにおけるアクチンメッシュワークの形成として把握され、(3)の細胞膜結合活性は、細胞膜中のフォスフォイノシタイドへの結合(相互作用)として把握される。
【0020】
こうした(1)〜(3)の各種活性は、本発明者らによれば、Aktによる細胞運動制御に関連し、ひいては細胞移動及び血管形成に関連し、これらの細胞動態の活性化・促進に寄与することが確認されている。なお、上記(1)及び(3)の活性は、リン酸化されていないGirdinの活性であり、(2)の活性は、リン酸化の有無に関わらないでGirdinが有する活性である。こうした各種の活性は、後述する実施例において具体的に説明するように、いずれも、インビトロ又はインビボにおける実験系において検出することができる。
【0021】
(本タンパク質をコードするヌクレオチド)
本発明において、配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を有するタンパク質又はその部分ペプチドをコードするヌクレオチドは、各タンパク質等をコードする塩基配列を有していれば、特に限定されない。ヌクレオチドは、こうした塩基配列を有するDNA、RNA、DNA/RNAキメラとすることができ、1本鎖であっても2本鎖であってもよい。2本鎖の場合には、DNA2本鎖、RNA2本鎖のほか、DNA/RNAハイブリッドであってもよい。1本鎖の場合には、センス鎖及びアンチセンス鎖のいずれであってもよい。具体的には、ゲノムDNA、cDNA、mRNA、あるいはPCR産物,化学合成DNA等といった形態でこうしたヌクレオチドを取得することができる。
【0022】
配列番号2で表されるアミノ酸配列をコードする塩基配列として配列番号1で表される塩基配列が挙げられる。この塩基配列はGirdinをコードする塩基配列である。上記したように、本タンパク質をコードするヌクレオチドは、配列番号1で表される塩基配列と同一の塩基配列に限定されないで、この塩基配列とは異なるコドン用法によって同一のアミノ酸配列をコードする塩基配列であってもよい。また、本ヌクレオチドの塩基配列は、配列番号1で表される塩基配列をストリンジェントな条件でハイブリダイズするDNA等のヌクレオチドであって、本タンパク質と同質な活性を有するタンパク質をコードするヌクレオチドであってもよい。
【0023】
なお、ストリンジェントな条件下でハイブリダイズできるDNAとしては、例えば、各配列番号で表される塩基配列と約50%以上、好ましくは約60%以上、さらに好ましくは約70%以上、より好ましくは約80%以上、特に好ましくは約90%以上、最も好ましくは約95%以上の相同性を有する塩基配列を含有するDNAなどが用いられる。ハイブリダイゼーションは、公知の方法あるいはそれに準じる方法、例えば、モレキュラー・クローニング(Molecular Cloning)2nd(J. Sambrook et al., Cold Spring Harbor Lab. Press, 1989)に記載の方法などに従って行うことができる。また、市販のライブラリーを使用する場合、添付の使用説明書に記載の方法に従って行うことができる。より好ましくは、ハイストリンジェントな条件に従って行うことができる。ハイストリンジェントな条件とは、例えば、ナトリウム濃度が約19〜40mM、好ましくは約19〜20mMで、温度が約50〜70℃、好ましくは約60〜65℃の条件を示す。特に、ナトリウム濃度が約19mMで温度が約65℃の場合が最も好ましい。
【0024】
Girdinをコードするヌクレオチドとしては、例えば、配列番号1で表される塩基配列を有するヌクレオチド又は配列番号1で表される塩基配列とハイストリンジェントな条件でハイブリダイズする塩基配列を有し、配列番号2で表されるタンパク質と同質の活性を有するタンパク質をコードするものであればよい。
【0025】
本タンパク質をコードするヌクレオチドは、形質転換用核酸構築物としての形態を採ることができる。すなわち、哺乳類動物細胞などの宿主細胞等において本タンパク質を発現させるための核酸構築物としての各種形態(Naked DNA、各種発現ベクター、人工染色体等)を採ることができる。こうした核酸構築物は、本タンパク質を発現可能にプロモーター、ターミネーター他、各種のエレメントを適宜備えることができる。
【0026】
(本タンパク質に特異的な抗体) 本タンパク質に対して特異的な抗体は、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに対して特異的な抗体及びリン酸化されたGirdinなどの配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに対して特異的な抗体を包含している。
【0027】
本発明の抗体は、ポリクローナル抗体であってもモノクローナル抗体であってもよい。抗体は、適当な抗原物質を温血動物に投与して抗体を産生させた上、当該温血動物から取得することができ、モノクローナル抗体は、最終的に作製したモノクローナル産生ハイブリドーマから取得できる。
【0028】
ポリクローナル抗体は、例えば、免疫原性のある抗原(タンパク質抗原)とキャリアー蛋白質との複合体をつくり、適宜温血動物に免疫を行い、該免疫動物からポリクローナル抗体含有物を採取して、抗体の分離精製を行なうことにより製造することができる。キャリアタンパクとしては、従来公知のものを用いることができるが、ウシ血清アルブミン、ウシサイログロブリン、ヘモシアニン等を用いることができ、免疫原と適当な比率で複合体を形成すればよい。なお、ハプテンとキャリアーとのカップリングには、一般的には、グルタルアルデヒドやカルボジイミド、マレイミド活性エステル、チオール基、ジチオビリジル基を含有する活性エステル試薬を用いることができる。こうした複合体の投与にあたっては、適宜、完全フロイントアジュバントや不完全フロイントアジュバントを同時に投与することができる。投与は、通常約2〜6週毎に1回ずつ、計約3〜10回程度行なうことができる。ポリクローナル抗体は、上記の方法で免疫された温血動物の血液、腹水など、好ましくは血液から採取することができる。抗血清中のポリクローナル抗体価の測定は、ELISAなどのEIA等を適宜用いて測定すればよく、抗体の分離精製は、公知の抗体の分離精製法に従って行えばよい。
【0029】
モノクローナル抗体は、例えば、ポリクローナル抗体の作製時と同様に温血動物を免疫して、抗体価の認められた個体を選択し最終免疫の適数日後に脾臓またはリンパ節を採取し、それらに含まれる抗体産生細胞をミエローマと融合させることにより、モノクローナル抗体産生ハイブリドーマを調製し、スクリーニングされたモノクローナル抗体産生ハイブリドーマを培養し、培養物からモノクローナル抗体を分離精製することで得ることができる。なお、細胞融合操作、モノクローナル抗体の選別は、従来公知の手法を用いることができる。
【0030】
本発明の抗体は、担体に固定化した形態を採ることができる。担体の形態は特に限定しないで、ビーズ状、平板状、マイクロタイタープレート等とすることができる。ビーズ状の担体に固定化された抗体は、免疫沈降、アフィニティクロマトグラフィ等に利用でき、また、平板状の担体やマイクロタイタープレート等に固定化された抗体は、抗体アレイあるいはELISA等のEIAやRIAアッセイプレートとして用いることができる。
【0031】
(本タンパク質をコードするヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するヌクレオチド) この態様の1つのヌクレオチドとしては、Girdinなど本タンパク質をコードするDNAに対するアンチジーンヌクレオチド(好ましくはDNA)又は本タンパク質をコードするmRNAに対するアンチセンスヌクレオチド(好ましくはRNA)が挙げられる。こうしたヌクレオチドとしては、本タンパク質をコードするヌクレオチドと同様の各種の態様を採ることができ、本タンパク質の発現を抑制できるものであれば、その長さや塩基や糖部分などの修飾形態は特に限定されないで各種の修飾形態を採用できる。ここで、「相補的な塩基配列」とは、例えば、本タンパク質又は部分ペプチドをコードするDNAまたはmRNAの全塩基配列または部分塩基配列と約40%以上、好ましくは約60%以上、より好ましくは約80%以上、さらに好ましくは約90%以上の相同性を有する塩基配列などが挙げられる。特に、本発明のDNAまたはmRNAの全塩基配列うち、本発明のタンパク質のN末端部位をコードする部分の塩基配列(例えば、開始コドン付近の塩基配列など)と約40%以上、好ましくは約60%以上、より好ましくは約80%以上、さらに好ましくは約90%以上の相同性を有するアンチセンスDNAが好適である。これらのアンチセンスDNAは、公知のDNA合成装置などを用いて製造することができる。
【0032】
また、この態様の他の1つのヌクレオチドとしては、Girdinなど本タンパク質をコードする遺伝子転写産物に対してRNA干渉を発現するヌクレオチドが挙げられる。こうしたヌクレオチドは、本タンパク質、特には、GirdinをコードするDNAのmRNA等の転写産物の少なくとも一部を標的としてこれらGirdinの発現を抑制可能に構築されている。こうしたヌクレオチドの一つの態様は、互いにハイブリダイズするオリゴリボヌクレオチドの二本鎖構造を有するヌクレオチドである。具体的には、突出した3’末端をそれぞれ有するあるいは有しない比較的短い二本鎖オリゴリボヌクレオチド(small interfering RNA :siRNA)及びヘアピン構造を形成する(又は有する)単一のオリゴリボヌクレオチド(short hairpin RNA : shRNA)が挙げられる。なお、ヘアピン構造を形成しない一本鎖オリゴヌリボヌクレオチドもRNA干渉を発現することができる。
【0033】
本態様のヌクレオチドにおけるセンス配列及びアンチセンス配列が対合する二重鎖の長さは、RNA干渉効果が得られる限り特に限定しないが、好ましくは50塩基対以下であり、典型的には13〜28塩基対であることが好ましく、より好ましくは13〜27塩基対の範囲であり、さらに好ましくは19〜21塩基対である。最も好ましくは、19又は20塩基対である。また、二重鎖を形成しない3’側構造を含めたセンス配列及びアンチセンス配列は、典型的には15〜30ntであることが好ましく、より好ましくは15〜29ntの範囲であり、さらに好ましくは21〜23ntである。最も好ましくは、21又は22ntである。また、siRNAにおいて突出する3'末端は、2〜4ntであることが好ましく、より好ましくは2ntである。さらに、shRNAにおけるループ部位は、こうした態様の二重鎖(shRNAにおいてはステム)及び3’末端構造を形成し維持するのを妨げない程度のループ部位の長さを備えていればよい。
【0034】
siRNAやshRNAは、公開されているルールなど適宜適用してターゲット配列を決定し、それに基づいて設計すればよい。さらに、ターゲット配列の決定方法を含むsiRNAの設計は、http://design.rnai.jp/sidirect/index.php、http://www.rockefeller.edu/labheads/tuschl/sirna.htmlやRational siRNA design for RNA interference (Nature Biotechnology , vol. 22, 326 - 330 (2004), Angela Reynolds, Devin Leake, Queta Boese,Stephen Scaringe, William S Marshall & Anastasia Khvorova)、Improved and automated prediction of effective siRNA (Biochem Biophys Res Commun. 2004 Jun 18;319(1):264-74、Chalk AM, Wahlestedt C, Sonnhammer EL.)等公開された各種のルールを適宜適用して行うことができる。
【0035】
RNA干渉を発現するヌクレオチドは、塩基や糖部分などの修飾形態は特に限定されないで各種の修飾形態を採用できる。
【0036】
(RNA干渉を発現するヌクレオチドをコードするヌクレオチド)
本発明によれば、Girdinなど本タンパク質をコードする遺伝子転写産物に対してRNA干渉を発現ヌクレオチドをコードするヌクレオチド(好ましくはDNA)及びこのヌクレオチドを含有するベクターを含むことができる。すなわち、本タンパク質をコードする遺伝子転写産物に対するsiRNAやshRNAを発現可能にコードするベクターの態様である。こうした態様のヌクレオチドは、継続性のあるRNA干渉を実現でき、ノックダウン動物を作製できる点において好ましい。この態様において、shRNA発現ベクターは、アンチセンス配列及びセンス配列の他ループ配列を、細胞内転写によりshRNAを構築可能な連続した一本鎖RNAが転写されるように構成することができる。また、siRNA発現ベクターは、所定のセンス配列及びアンチセンス配列を有するRNAが転写されるように構成すればよい。siRNA発現ベクターにおいては、センス配列及びアンチセンス配列は単一のベクターによって発現されるようにしてもよいし、また、それぞれ異なるベクターから発現されるようにしてもよい。
【0037】
発現ベクターは、プラスミドベクターやウイルスベクターの形態を採ることができる。ベクターの種類は特に問わないで、導入したい細胞などに対応して選択することができる。例えば、哺乳動物細胞では、例えば、レトロウイルスベクター、アデノウイルスベクター、アデノ関連ウイルスベクター、ワクシニアウイルスベクター、レンチウイルスベクター、ヘルペスウイルスベクター、アルファウイルスベクター、EBウイルスベクター、パピローマウイルスベクター、フォーミーウイルスベクターなどのウイルスベクターが挙げられる。また、こうした発現ベクターに用いるプロモーターとしては、上記各DNAより対応するRNAを産生し得るものであれば、polII系、polIII系のいずれであってもよい。
【0038】
(細胞) 本発明は、1つの態様として、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を有するタンパク質をコードする遺伝子の発現量が抑制された細胞を含んでいる。この細胞は、本タンパク質をコードする塩基配列に基づいて作成されたノックアウトベクターや上記したRNA干渉を発現するヌクレオチドや該ヌクレオチドをコードするヌクレオチド(ベクター)を細胞に導入することによりあるいは後述する非ヒト哺乳類ノックアウト動物や非ヒト哺乳類ノックダウン動物から取得できる。
【0039】
また、本発明は、他の1つの態様として、Girdinなどの配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする外来DNAを保持した細胞を含んでいる。こうした細胞は、本タンパク質を発現するための核酸構築物を細胞に導入することによりあるいは後述する非ヒト哺乳類トランスジェニック動物を取得源として取得できる。
【0040】
さらに、本発明は、他の1つの態様として、Girdinなど配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質が変異された本変異型タンパク質をコードする外来DNAを保持した細胞を含んでいる。こうした細胞は、本変異型タンパク質を発現するための核酸構築物を細胞に導入することによりあるいは後述する非ヒト哺乳類トランスジェニック動物を取得源として取得できる。
【0041】
これらの各種細胞は、例えば、ヒト(トランスジェニック動物、ノックアウト動物及びノックダウン動物を取得源とする場合を含まない。)及び非ヒト哺乳類細胞とすることができる。また、マウス、ラットなどゲッ歯類を始めとする非ヒト哺乳類胚性幹細胞としてもよいし、体性幹細胞や体細胞であてもよい。さらに、神経細胞、血管内皮細胞、癌細胞など、本タンパク質が関連する疾患の標的細胞であってもよい。こうした本発明の各種細胞は、本発明の予防・治療剤のスクリーニング、評価、細胞運動等の評価に有用である。
【0042】
以下、本タンパク質及び本タンパク質に関連して得られるヌクレオチド、抗体、細胞等の用途について説明する。
【0043】
(本タンパク質が関連する疾患の予防・治療剤) 本タンパク質は、上記(1)〜(3)のいずれかの活性を有し、これらの活性は、細胞運動、細胞移動及び血管形成を活性化するから、本タンパク質についての上記(1)〜(3)のいずれか又は2種以上の活性を阻害する活性を有する化合物又はその塩は、Girdinなど本タンパク質が関連する疾患、具体的には細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤として利用できる。また、本タンパク質の発現量を抑制する化合物又はその塩も、Girdinなど本タンパク質が関連する疾患、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤として利用できる。
【0044】
ここで、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患としては、こうした現象が関連する疾患の全てを包含しているが、例えば、以下の疾患を例示することができる。
【0045】
(1)癌
例えば、原発性、転移性または再発性の、乳癌、前立腺癌、膵癌、胃癌、肺癌、大腸癌(結腸癌、直腸癌、肛門癌)、食道癌、十二指腸癌、頭頚部癌(舌癌、咽頭癌、喉頭癌)、脳腫瘍、神経鞘腫、非小細胞肺癌、肺小細胞癌、肝臓癌、腎臓癌、胆管癌、子宮癌(子宮体癌、子宮頸癌)、卵巣癌、膀胱癌、皮膚癌、血管腫、悪性リンパ腫、悪性黒色腫、甲状腺癌、骨腫瘍、血管腫、血管線維腫、網膜肉腫、陰茎癌、小児固形癌、カポジ肉腫、AIDSに起因するカポジ肉腫、上顎洞腫瘍、線維性組織球腫、平滑筋肉腫、横紋筋肉腫、脂肪肉腫、子宮筋腫、骨芽細胞腫、骨肉腫、軟骨肉腫、癌性の中皮腫瘍、白血病などの腫瘍、ホジキン病など
【0046】
(2)動脈硬化性疾患
例えば、急性冠動脈症候群(例、急性心筋梗塞、不安定狭心症など)、末梢動脈閉塞症、冠動脈インターベンション(経皮的冠動脈形成術(PTCA)、アテレクトミー(DCA)、ステント留置等)後の再狭搾、冠動脈バイパス手術後の再狭窄、その他の末梢動脈におけるインターベンション(血管形成術、アテレクトミー、ステント留置等)及びバイパス手術後の再狭窄、虚血性心疾患(例、心筋梗塞、狭心症など)、心筋炎、間歇性跛行、ラクネ梗塞、動脈硬化症(例、アテローム性動脈硬化症など)、心不全(急性心不全、うっ血性を含む慢性心不全)、不整脈、動脈硬化巣の進展、血栓症、高血圧症、高血圧性耳鳴り、低血圧症など
【0047】
(3)中枢および末梢神経障害
例えば、頭部外傷、脊髄損傷、脳浮腫、知覚機能障害、知覚機能異常、自律神経機能障害、自律神経機能異常、むち打ち症など
【0048】
本発明の予防・治療剤として用いることができる化合物又はその塩を得るための指標としては、具体的には、以下の活性を阻害する活性が挙げられる。なお、下記a)〜b)は、上記(1)のAkt基質活性の阻害に関連し、下記c)〜e)は、上記(2)のアクチン結合タンパク質活性の阻害に関連し、下記f)〜g)は、上記(3)の細胞膜結合活性の阻害に関連する。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化
c)アクチンフィラメントへの結合
d)アクチンストレスファイバー形成
e)ラポリメディアにおけるアクチンメッシュワークの形成
f)細胞膜への結合
g)フォスフォイノシタイドへの結合
【0049】
こうした阻害活性を有する化合物は、後述するように、上記a)〜g)を阻害する化合物を取得するスクリーニング方法に基づいて得ることができる。こうした化合物としては、例えば、本タンパク質に特異的な抗体等が挙げられる。また、Aktによるリン酸化サイトに変異を有する本タンパク質(例えばGirdinなど)の変異体及びフォスフォイノシタイド結合サイトに変異を有する本タンパク質(例えばGirdinなど)の変異体であってもよい。
【0050】
こうした化合物の塩としては、生理学的に許容される酸(例、無機酸、有機酸)や塩基(例えばアルカリ金属)等との塩が用いられ、とりわけ生理学的に許容される酸付加塩が好ましい。この様な塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩等が用いられる。
【0051】
スクリーニング方法から得られた化合物又はその塩を、本発明の予防・治療剤として用いる場合、その製剤形態は特に限定されない。経口剤、注射剤、注入剤等の非経口剤、局所適用に適した外用剤等、いずれの形態も採用できる。例えば、必要に応じて糖衣を施した錠剤、カプセル剤、エリキシル剤、マイクロカプセル剤などとして経口的に、あるいは水もしくはそれ以外の薬学的に許容し得る液との無菌性溶液、または懸濁液剤などの注射剤の形で非経口的に使用できる。こうした各種の製剤も本発明の予防・治療剤の一態様である。例えば、生理学的に認められる担体、香味剤、賦形剤、ビヒクル、防腐剤、安定剤、結合剤などとともに一般に認められた製剤実施に要求される単位用量形態で混和することによって製造することができる。これら製剤における有効成分量は指示された範囲の適当な用量が得られるようにするものである。なお、上記製剤を得るための各種添加剤は、当該分野において公知のものを適宜選択して用いればよい。こうして得られる製剤は、ヒトのほか、非ヒト動物である、例えば、ラット、マウス、モルモット、ウサギ、トリ、ヒツジ、ブタ、ウシ、ウマ、ネコ、イヌ、サルなどに対して投与することができる。上記化合物の投与量は、症状及患者の個体差などを勘案して設定することができる。
【0052】
本発明の予防・治療剤としては、スクリーニングされた化合物のほか、本タンパク質の発現量を抑制する化合物又はその塩であってもよい。本タンパク質の細胞内で発現量を抑制する化合物又はその塩としては、「本タンパク質をコードするヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するヌクレオチド」に含まれる各種態様のヌクレオチドが挙げられる。これらのヌクレオチドは、細胞内においてアンチジーン、アンチセンス、RAN干渉により、本タンパク質の発現を抑制するため、結果として、上記a)〜g)を阻害することができ、上記a)〜g)による細胞運動、細胞移動及び血管形成を活性化を抑制し、ひいては、これらのいずれかが関連する疾患を予防・治療剤として作用することができる。
【0053】
また、本タンパク質の発現量を抑制する化合物又はその塩としては、本タンパク質をコードする遺伝子を破壊可能なDNA構築物が挙げられる。こうしたDNA構築物は、本タンパク質をコードするヌクレオチドの塩基配列に基づいて作製することができる。
【0054】
こうしたヌクレオチド構築物を含有する予防・治療剤は、適切な担体に含有させて用いることにより、より一層、細胞への導入が容易になる。このため、本発明の予防・治療剤としては、こうした担体を含有するものも包含する。担体としては、細胞への導入に適したベクター、リポソーム、金属粒子、正電荷ポリマー、リン酸カルシウム、DEAEデキストランなどが挙げられる。また、担体は、リポソーム、金属粒子、正電荷ポリマー、リン酸カルシウム又はDEAEデキストランのいずれかに、本発明のヌクレオチド等を保持するベクターを担持させたものでもよい。細胞への導入のためのベクターは、非ウイルス性ベクターやウイルス性ベクターを用いることができる。さらに、本予防・治療剤は、通常の薬理学的に許容されうる担体、賦形剤、結合剤、安定剤、緩衝剤、溶解補助剤、等張剤などを含有してもよい。
【0055】
投与は、特に標的細胞における導入を達成して、本タンパク質の発現量の抑制を実現できるものであれば特に限定されないが、例えば、以下の手法が挙げられる。すなわち、線維芽細胞等の各種細胞に対して体外で本発明の遺伝子を導入し、遺伝子導入が成功した細胞(本発明の細胞でもある。)を選択したうえで、標的部位に移植するexvivoの方法や、ウイルスベクターやリポソームを用いて直接標的部位に局所注入し、in vivoで本発明の遺伝子の導入を行なうアプローチが挙げられる。さらに、徐放性の製剤を調製し、患部近くに埋め込む手法挙げられる。なお、こうしたヌクレオチド構築物を含有する予防・治療剤を細胞や組織等に導入する方法としては、リン酸−カルシウム共沈法;微小ガラス管を用いた核酸の直接注入法;内包型リポソームによる遺伝子導入法;静電気型リポソームによる遺伝子導入法;HVJ−リポソーム法、改良型HVJ−リポソーム法(HVJ−AVEリポソーム法);受容体介在性遺伝子導入法;パーティクル銃で担体(金属粒子)とともに核酸分子を細胞に移入する方法;naked−DNAの直接導入法;正電荷ポリマーによる導入法等が挙げられる。
【0056】
こうしたヌクレオチド構築物を含有する予防・治療剤は、投与目的、個体の年齢、体重、状態等に応じて、本タンパク質の発現量を低下させる範囲で適宜設定され得る。
【0057】
(本タンパク質が関連する疾患の予防・治療方法)
本発明の予防・治療剤は、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療方法としても実施できる。すなわち、本発明の予防・治療剤をヒト又は非ヒト温血動物を予防又は治療のために有効量投与することは、上記疾患の予防・治療方法である。
【0058】
(スクリーニング方法) 本発明によれば、本タンパク質及び本タンパク質をコードするヌクレオチドを用いる、本タンパク質の各種活性を促進又は阻害し、あるいは本タンパク質の発現量を増加又は低下させる化合物又はその塩のスクリーニング方法が提供される。このスクリーニング方法によって得られる化合物又はその塩は、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物として使用できる。また、本タンパク質の各種活性を阻害し又は発現量を低下させる化合物又はその塩は、本タンパク質が関連する疾患の予防・治療剤として使用でき、特には、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療剤としても使用できる。
【0059】
本スクリーニング方法は、試験化合物を本タンパク質、本タンパク質を発現する細胞に供給して、試験化合物を供給しないときに比較して、本タンパク質の上記した活性(1)〜(3)、具体的には上記(a)〜(g)を促進するか又は阻害するかを指標とすることにより、本タンパク質の活性を促進又は阻害する化合物又はその塩を取得できる。また、試験化合物を本タンパク質を発現する細胞に供給して、試験化合物を供給しないときに比較して本タンパク質の発現量を増加させるか低下させるかを検出することによれば、本タンパク質の発現量を増加又は低下させる化合物又はその塩を得ることができる。なお、前記作用の検出にあたって、スクリーニング系に供給される試験化合物としては、本タンパク質に対する作用するもののほか、本タンパク質をコードするヌクレオチドあるいは当該ヌクレオチドに作用するものであってもよい。本タンパク質の上記活性を阻害したり、本タンパク質の発現量を低下させる化合物又はその塩は、細胞運動、細胞移動及び血管形成を抑制するため、これらの細胞動態のいずれかが関連する疾患の予防・治療剤として用いることができる。
【0060】
こうした試験化合物としては、例えば、ペプチド、タンパク、非ペプチド性化合物、合成化合物、発酵生産物、細胞抽出液、植物抽出液、動物組織抽出液、遺伝子(ゲノムDNA、cDNA)などが挙げられ、これら化合物は新規な化合物であってもよいし、公知の化合物であってもよい。
【0061】
本スクリーニング方法において、本タンパク質の発現量や上記a)〜g)の各種指標を検出するには、例えば、以下の手法を採用できる。 例えば、Aktとリン酸化されていない本タンパク質との結合を指標とするには、Aktによるリン酸化されていない本タンパク質のリン酸化を指標とすることが好ましく、例えば、Girdin(タグ付きGirdinを発現するCOS7細胞からの細胞溶解物をタグで精製したものであってもよい。)と構成的活性型であるなどの活性型AktとP32等で標識したATPとを含むインビトロキナーゼアッセイ系に試験化合物を供給してインキュベーションし、得られるリン酸化物をオートラジオグラフィーで検出するなどすることができる。あるいは、Girdinを発現する細胞にEGFを加えてAktを活性化するとともに、試験化合物を供給してインキュベーションし、その後、細胞溶解物を抗Girdin抗体で免疫沈降後、リン酸化Girdin抗体で免疫沈降し、抗リン酸化Girdin抗体などでウエスタンブロット分析すればよい。また、本タンパク質のアクチンフィラメントへの結合を指標とする場合には、GSTなどでタグを付加したGirdinのCT2ドメイン又はGirdinを含むアクチン共沈アッセイ系に試験化合物を供給し、インキュベーション後、共沈させたタンパク質を回収後、SDS−PAGEで分離してクマシーブリリアントブルー(CBB)、抗GST抗体(Cell Signaling Technology)又は抗リン酸化Girdin抗体を用いたウエスタンブロット分析を行えばよい。さらに、アクチンストレスファイバー形成を指標とする場合には、例えば、Girdinを発現する細胞にEGFなどAktを活性化する刺激と試験化合物とを加えた上で、Girdin発現細胞をリン酸化Girdinを特異的に認識する抗リン酸化Girdin抗体などを用いて免疫染色することで、リン酸化Girdinによるアクチンメッシュワークの形成を観察してもよいし、さらに、Girdinを発現する細胞に試験化合物をコンフルエント状態の単層の細胞培養物にスクラッチにより損傷を付与したとき(損傷治癒アッセイ)の、リーディングエッジにおけるリン酸化Girdinを免疫染色により検出してもよい。リーディングエッジにおけるリン酸化Girdinはアクチンメッシュワークに存在する。さらにまた、Girdinを発現する細胞についてボイデンチャンバー法における細胞移動として検出してもよい。また、細胞膜やフォスフォイノシタイドへの結合性を指標とする場合には、タグなどを付加したGirdinを含むタンパク質−脂質オーバーレイ法のアッセイ系に試験化合物を供給して、リン脂質に結合するタンパク質を検出するか、あるいは、Girdinを発現する細胞に試験化合物を供給し、抗Girdin抗体を用いてGirdinを観察することによってもよい。
【0062】
本スクリーニング方法によって得られる化合物又はその塩としては、本タンパク質に特異的に結合する抗体が挙げられる。また、本タンパク質のリン酸化部位の立体構造を変化させて、Aktとの結合活性、Akt-本タンパク質との複合体の安定性などを低下させてAktのリン酸化を抑制する化合物、Aktの基質結合部位に可逆的に結合するAkt阻害剤などが挙げられる。さらに、アクチンフィラメントに対する本タンパク質の架橋を阻害したり架橋状態の解除を促進する化合物又はその塩、アクチンストレスファイバーの形成を阻害したりアクチンストレスファイバー解除を促進する化合物又はその塩、ラポリメディアにおけるアクチンメッシュワークの形成を阻害したり、その解除を促進する化合物又はその塩、細胞膜結合を阻害するか結合解除を促進する化合物又はその塩、ホスホイノシダイドへの結合を阻害するか又は結合解除を促進する化合物又はその塩が挙げられる。さらに、既に説明したように、本タンパク質の発現をノックアウトやノックダウンにより抑制する各種ヌクレオチド構築物が挙げられる。
【0063】
(トランスジェニック動物)
本発明によれば、本タンパク質をコードする外来性DNAを有する非ヒト哺乳動物が提供される。このトランスジェニック動物によれば、外来性DNAによって本タンパク質を発現しているため、本タンパク質の活性の研究や、本タンパク質が関連する疾患の予防・治療剤のスクリーニングや評価等に適した動物となっている。ここで非ヒト哺乳動物としては、例えば、ウシ、ブタ、ヒツジ、ヤギ、ウサギ、イヌ、ネコ、モルモット、ハムスター、マウス、ラットなどが用いられる。なかでも、マウスやラットなどのゲッ歯動物が好ましい。
【0064】
トランスジェニック動物は、未受精卵、受精卵、精子およびその始原細胞を含む胚芽細胞などに対して、リン酸カルシウム法、電気パルス法、リポフェクション法、凝集法、マイクロインジェクション法、パーティクルガン法、DEAE−デキストラン法などにより本外来性DNA等を導入するなどし、該細胞を用いて胚を作製することにより得ることができる。
【0065】
本トランスジェニック動物の作製に用いる外来性DNAは、本タンパク質をコードする領域を、各種哺乳動物(例えば、ウサギ、イヌ、ネコ、モルモット、ハムスター、ラット、マウスなど)由来のDNAを発現させうる各種プロモーターの制御下に有し、さらに、ターミネーター等を備えるDNA構築物とすることができる。こうしたDNA構築物は、DNAの導入方法の種類に応じて適宜選択することができ、Naked DNAとすることができるほか、従来公知の発現ベクターの形態を採ることができる。
【0066】
本トランスジェニック動物は、本外来DNAをヘテロで有することもでき、ホモで有することもできる。また、本トランスジェニック動物は、こうした外来DNAを宿主染色体においてランダムに導入されたものであってもよいし、所望の部位にノックイン形態で導入されたものであってもよい。本トランスジェニック動物は、通常よりも本タンパク質を過剰に発現する傾向があるため、本トランスジェニック動物を用いることで、例えば、本タンパク質の機能亢進症を発症した場合には、当該病態モデル動物として使用できることもある。
【0067】
また、本発明によれば、本タンパク質の変異型、例えば、1416位のセリンがアラニンに置換されてAktの基質となりえず本タンパク質と同質の活性を有しない変異型タンパク質をコードする外来DNAを保持するトランスジェニック動物も提供される。この変異型タンパク質を発現するトランスジェニック動物は、本タンパク質の外来DNAが導入されたトランスジェニック動物と同様、ヘテロであってもホモであってもよいが、ランダムに変異型外来DNAが導入された場合には、多くの場合本タンパク質以外にその変異型タンパク質を発現するものとなっており、本タンパク質をコードする宿主染色体を破壊するように変異型外来DNAが導入された場合には、変異型タンパク質のみを発現するものとなる。こうした変異型タンパク質発現トランスジェニック動物は、本タンパク質の機能不活性型不応症の病態機序の解明及びこうした疾患を治療方法の研究用途に用いることができる。また、本タンパク質の機能不活性型不応症に対する治療薬スクリーニングや評価にも利用できる。
【0068】
なお、こうしたトランスジェニック動物は、組織培養のための細胞源、組織観察用の組織源、薬剤スクリーニング用細胞源、本タンパク質又はその変異型タンパク質の取得源として利用することもできる。
【0069】
(ノックアウト動物)
本発明によれば、本タンパク質をコードするDNAの発現が抑制された非ヒト動物が提供される。こうした動物によれば、本タンパク質の発現が抑制されているため、本タンパク質が関わる各種の活性を欠失する可能性があり、本タンパク質の生物活性の不活性化を原因とする疾病のモデルとすることができる。
【0070】
なお、非ヒト哺乳動物としては、前記トランスジェニック動物と同様のものを用いることができる。また、本タンパク質をコードするDNAがノックアウトにより破壊されたノックアウト動物を得るには、本タンパク質をコードするゲノムやコード領域あるいはその近傍の塩基配列を利用してターゲティングベクターを構築して、これを胚作製に用いればよい。胚作製にあたっては、ES細胞あるいは非ヒトクローン動物の作製が可能な体細胞を用いることが好ましい。また、上記したRNA干渉を発現可能な核酸構築物を導入した胚を作製することにより、本タンパク質をコードするDNAの発現がRNA干渉により抑制されたノックダウン動物を得ることができる。
【0071】
(分析方法等、診断剤、診断方法、診断キット)
本発明の抗体は、本タンパク質又はリン酸化された本タンパク質を特異的に認識することができる。本発明の抗体を用いることにより、被検試料中の本タンパク質又はリン酸化された本タンパク質を、特にELISAなどのEIAなどの抗体を利用した酵素抗体法等などの測定方法により定量することができる。すなわち、本発明の抗体は本タンパク質又はリン酸化された本タンパク質の定量用試薬として用いることができる。もちろん、本タンパク質の定性用試薬としても用いることができる。さらに、細胞や組織などの被験試料中の本タンパク質又はリン酸化された本タンパク質の量は、本タンパク質が関連する疾患、例えば、細胞運動、細胞移動及び血管形成に関連する疾患の診断に関連するため、本発明の抗体は、こうした疾患の診断剤として使用できる。
【0072】
また、Aktが本タンパク質をリン酸化することを利用し、このリン酸化の活性化程度又は阻害程度から本タンパク質の各種活性を活性化し阻害する化合物の濃度を定性的に検出できる。すなわち、Aktと本タンパク質とを含有しAktによる本タンパク質のリン酸化が可能なアッセイ系を準備しておき、このアッセイ系に試験化合物を供給して、基質である本タンパク質又は反応生成物であるリン酸化された本タンパク質を検出することにより、この試験化合物がAktによる本タンパク質のリン酸化を活性化する化合物であるか阻害する化合物であるかを定性的に検出することができる。なお、本タンパク質又はリン酸化された本タンパク質の検出は、本タンパク質に特異的な抗体又はリン酸化された本タンパク質に特異的な抗体を用いた酵素抗体法などによることができる。また、同様の系を利用し、標準曲線等を作製することにより、試験化合物がAktによる本タンパク質のリン酸化を活性化するあるいは阻害する化合物であるとき、試験化合物を定量することができる。こうしたアッセイ系による試験化合物の定性的又は定量的分析は、本発明のスクリーニング方法や本タンパク質の関連する疾患の診断方法に利用できる。
【0073】
なお、本発明の抗体を用いる本発明のタンパク質及びリン酸化された抗体の定量法は、特に制限されるべきものではなく、被測定液中の抗原量(例えば、タンパク質 量)に対応した抗体、抗原もしくは抗体−抗原複合体の量を化学的または物理的手段により検出し、これを既知量の抗原を含む標準液を用いて作製した標準曲線より算出する測定法であれば、いずれの測定法を用いてもよい。例えば、ネフロメトリー、競合法、イムノメトリック法およびサンドイッチ法が好適に用いられるが、感度、特異性の点で、サンドイッチ法を用いるのが特に好ましい。標識物質を用いる測定法に用いられる標識剤としては、例えば、放射性同位元素、酵素、蛍光物質、発光物質などが用いられる。放射性同位元素としては、例えば、〔125I〕、〔131I〕などが、上記酵素としては、安定で比活性の大きなものが好ましく、例えば、β−ガラクトシダーゼ、β−グルコシダーゼ、アルカリフォスファターゼ、パーオキシダーゼ、リンゴ酸脱水素酵素などが、蛍光物質としては、例えば、フルオレスカミン、フルオレッセンイソチオシアネートなどが、発光物質としては、例えば、ルミノール、ルミノール誘導体、ルシフェリン、ルシゲニンなどがそれぞれ用いられる。さらに、抗体あるいは抗原と標識剤との結合にビオチン−アビジン系を用いることもできる。担体としては、例えば、アガロース、デキストラン、セルロースなどの不溶性多糖類、ポリスチレン、ポリアクリルアミド、シリコン等の合成樹脂、あるいはガラスなどが用いられる。また、こうした検出においては、抗体分子そのものを用いてもよく、その一部を用いてもよい。
【0074】
本発明の診断キットは、本発明の抗体及びAkt及びGirdinなどの本タンパク質を含むAktリン酸化アッセイ系エレメントから選択される1種又は2種以上を含むことができる。以上説明した、本発明の抗体、Aktリン酸化アッセイ系は、診断用途のみならず、細胞運動、細胞移動及び血管形成の研究用途に用いることができる。
[実施例]
【0075】
以下、本発明を挙げて説明するが、本発明は以下の実施例に限定されるものではない。なお、以下の実施例において用いた実験方法等について以下に説明し、次いで個別実施例について説明する。
【0076】
1.酵母2-ハイブリッド試験
Aktと相互作用するプロテインを特定するため、Akt1のfull-length cDNAをpAS2ベクター(Clontech)に挿入した。それによって得られたコンストラクトを先述のヒト胎児の脳MARCHMAKER cDNAライブラリー(Murakamiほか、2002)をスクリーニングするための材料(ベイト)とした。挿入されたcDNAを含む2つの陽性プラスミドを選び、配列を解析した。それら2つのプラスミドは、Girdinの C末端領域 (残基1217-1870)をコードするcDNA 断片を含んでいた。Girdinをコードするfull-length cDNAについて、その末端を5’ 迅速増幅(5’-RACE システム, Invitrogen)することによりヒト胎児の脳ポリA+ RNA から分離した。さらに、ヒトAkt1とGirdin cDNAの断片を含む精製したpASとpACでもって酵母2-ハイブリッド結合試験を行った。
【0077】
2.プラスミド
構成的に活性で優性阻害の特性を持つ野生のヒトAkt1はY. Gotoh氏(東京大学)の好意により入手した。そしてpcDNA3.1-, pGEX-, pEGFP- Girdin断片のコンストラクトを作製した(Murakamiほか、2002)。EGFPをタンパク質のアミノ末端に、V5とmyc tagとをタンパク質のカルボキシル末端に融合した。Girdin変異体はQuick Change site-directed mutagenesis kit(Stratagene)を使って、製造者プロトコールに従って作製した。Girdinのヌクレオチド780-800(5’-AAGAAGGCTACGGCAGGAATT-3’)(下線部は変異遺伝子)に2つのサイレント変異を導入することで抗siRNA Girdinを作製した。
【0078】
3.抗体
Girdinのカルボキシル末端の19アミノ酸に対してウサギ抗Girdinポリクローナル抗体を生成させ、免疫ペプチドでアフィニティー精製した。抗リン酸化Girdinポリクローナル抗体は、Kumamoto Immunochemical Laboratory, Transgenic, Inc.(熊本県)から提供を受けたもので、Girdinのアミノ酸配列1408-1420(CDINRERQKpSLTLT)に対応する、キーホールリンペットヘモシアニンに結合したリン酸化ペプチドでウサギを免疫することにより取得した。抗血清はリン酸化ペプチドに結合したカラムに結合したフラクションとして精製した。実験で使用した他の抗体としては、抗Aktポリクローナル抗体(Cell Signaling Technology)、抗リン酸化Aktポリクローナル抗体(Cell Signaling Technology)、抗コータクチンモノクローナル抗体(Upstate)などがある。
【0079】
4.キナーゼアッセイ
Girdin CT-V5 WT、SAまたは生成したGST-STを発現しているCOS7細胞から採取した抗V5抗体(Invitrogen)による免疫沈降物を、活性もしくは不活性組み換えAkt(500ng)(Upstate)と共に、キナーゼ緩衝液(20 mM MOPS、25 mM β-glycerophosphate、5mM EGTA、15mM MgCl2)に10μCi[γ-32P]ATP(3000 Ci/mmol、Amersham)を添加した中で培養した。混合物を30℃で30分間培養し、Laemmliドデシル硫酸ナトリウム(SDS)サンプル希釈緩衝液(20 mM Tris-HCl [pH 6.8]、2 mM EDTA、2% SDS、10% sucrose、20μg/ml bromophenol blue、80mM dithiothreitol)を加えることによって、反応を終わらせた。
【0080】
5.免疫蛍光染色法
ベロ細胞をフィブロネクチン(10μg/ml、Sigma)とコラーゲンI(10μg/ml、Upstate)でコートしたカバースリップもしくはガラス製ディッシュにのせて固定し、上述した抗体で染色した。蛍光染色は共焦点レーザ顕微鏡(fluoview FV500、Olympus)で確認した。
【0081】
6.アクチン共沈アッセイ
F-アクチン共沈降試験を製造者プロトコール(Cytoskeleton)に従って行った。精製したGST融合プロテイン、GST、α-アクチン(Cytoskeleton)を40μgの精製アクチンフィラメントと共に室温で30分間培養した。F-アクチンの最終濃度は18μMであった。フィラメントは、その後、遠心分離(100000×g)(ベックマン)でペレット化した。共沈させたタンパク質は、SDS−PAGEで分離してクマシーブリリアントブルー(CBB)又は抗GST抗体(Cell Signaling Technology)又は抗リン酸化Girdin抗体を用いたウエスタンブロット分析で確認した。 定量的アッセイには、所定濃度のGST-Girdin CT2(1μM)を、重合性緩衝液中で各種濃度のF-アクチン(0〜2.5μM)を混合し、室温で30分間インキュベートした。タンパク質を上記と同様にして遠心分離し、全てのペレットと上清をそれぞれSDS-PAGEを施した。タンパク質のバンドをCBB染色で検出してスキャンし、WinROOF(Mitani Corp., Fukui,Japan)で解析した。
【0082】
7.RNA干渉
GirdinとAktのsiRNAによるノックダウンは既に記載された方法を用いて実施した(Watanabe et al., 2004)。Girdinの発現のサイレンシングを媒介したターゲット配列は、以下のとおり、5-AACCAGGTCATGCTCCAAATT-3(145-165ヌクレオチド、GirdinsiRNA A)及び5-AAGAAGGCTTAGGCAGGAATT-3(780-800ヌクレオチド,GirdinsiRNA B)であった。これらの21ヌクレオチドの合成二重鎖はQiagenにより作製された。また、ヒトAkt1に特異的なsiRNAはQiagenより入手した。ベロ細胞を、リポフェクタミン2000(Invitrogen)を用いてこれらのsiRNA又はコントロールとしての無関係な21RNA(Qiagen)により製造者プロトコールに従ってトランスフェクトした。
【0083】
8.細胞膜表面(細胞質側)の凍結レプリカ電子顕微鏡
細胞質膜の細胞質側表面の電子顕微鏡観察は、以前に報告された方法に従った(Heuser, 1989, 2000; Usukura, 1993)。ガラスカバースリップ(直径3mm, スタンダード#1, Matunami、Osaka, Japan)上で培養したベロ細胞を、siRNAでトランスフェクトした。上層側の細胞膜から割断(アンルーフ)した後、直ちに細胞を2.5%グルタルアルデヒドの緩衝液A溶液(緩衝液A;70mM KCl, 5mM MgCl2、3mMEDTA、30mM HEPES緩衝液,pH7.4(KOHにて))中で固定化した。緩衝液と蒸留水との混液で洗浄した後、直ちに試料を液体ヘリウムで急速冷凍装置(Eiko, Tokyo, Japan)で凍結した。試料は、その後、フリーズエッチ装置(FR2000、HITACHI、Ibaraki、Japan)を用いてプラチナ−カーボンでロータリーシャドウィングした。Girdin分子の免疫ラベルのために、アンルーフした細胞を4%パラホルムアルデヒド/0.5%グルタルアルデヒドの緩衝液A溶液中で固定化した。緩衝液B(100mM NaCl、30mM HEPES、2mMcaCl2)で3回洗浄後、試料をクエンチしブロックして、1%BSAを含む緩衝液B中で一次又は二次金結合抗体(Amersham)で37℃で1時間ラベルした。最終的に、上記と同様の方法で速やかに凍結し、フリーズエッチした。
【0084】
9.スクラッチ誘導細胞移動及び低速度イメージング
ベロ細胞における方向性のある細胞移動をインビトロスクラッチ損傷アッセイ(Watanabe et al., 2004)を用いて単層状態で刺激した。ベロ細胞を、フィブロネクチンがコートされたカバースリップか又は35mmのガラス製ディッシュに播種し、siRNAをトランスフェクトした。トランスフェクション48時間経過後、コンフルーエント状態のベロ細胞を、200μlのディスポーザブルピペットチップの先端で引っ掻いて(スクラッチ)、損傷方向に移動可能な状態とした。細胞は、示された時間ごとに免疫染色のために固定化した。低速度観察のためには、ベロ細胞にはsiRNAとGFP-アクチンとの双方をトランスフェクトして、スクラッチ損傷アッセイに供した。損傷側エッジの細胞は、共焦点レーザ走査電子顕微鏡(Fluoview, FV500, Olympus)にて観察した。
【0085】
10.三次元細胞移動アッセイ
各種のコンストラクトやsiRNAでトランスフェクトした細胞の移動性を評価するために、ボイデンチャンバー細胞移動アッセイを改変して、HTS FluoroBlok Insert(8.0μM pores 24-ウエル、BD)を用いる蛍光色素ブロッキングマイクロポアメンブレンを通過するGFPでラベルした細胞の移動を計測することができるようにした。すなわち、メンブレンの両サイドを10μg/mlのフィブロネクチンを用いて12時間37℃でコートし、PBSで洗浄した。その後、チャンバーを、0.1%のBSAを含有しヒト組換えEGF(20ng/ml)を含む又は含まないDMEMが満たされた24ウェルディッシュ内に配置した。HT-1080を用いた移動アッセイには、10%FBSが下側のチャンバーに添加された。24ウェルのプレート内において、細胞(1×105)にGFP(0.5μg(対個々の細胞)、示されたコンストラクト(2.5μg)及びsiRNA(20pmol)をトランスフェクトし、チャンバー上部に置いて、4時間メンブレンの孔部を通過して移動させるようにした。細胞の移動性は、膜を通過して移動したGFP陽性細胞を蛍光顕微鏡でカウントすることで定量した。
【0086】
11.タンパク質−脂質オーバーレイアッセイ
GST融合タンパク質をDH5α又はBL21−CodonPlus細胞(Stratagene)中に発現させ、常法にて精製した。リン脂質への精製GST-CTの結合は、製造者プロトコールに従って、PPIP-Strip(Echolen Biosciences)を用いて4℃で確認した。結合したGST-CTは、抗Girdin抗体又は抗リン酸化Girdin抗体のいずれかを用いて検出した。
【0087】
(実施例1: Girdinの同定、一次構造及びその発現様式)
Aktの新規基質を同定するために、ヒトAkt1の全長をベイトコンストラクトとして酵母two-hybrid法を行って、ヒト胎児脳cDNAライブラリを用いて相互作用するタンパク質を探索した(図1a参照)。その結果、二つのクローン(F-10、F-12)を検出した。図1bに、ヒトAkt1とクローンF-10及びF-12との結合様式を示す。次いで、これらのcDNAを酵母で発現させると、Akt1のC末端調節領域とのみ相互作用することがわかった。さらに、cDNAの5'-末端につき、5'-RACEを行うことで、5'末端に連続する3.6kbのcDNAを含む全コード領域を備えるクローンを取得した。得られた全長cDNAは5610bpのオープンリーディングフレームを含んでおり、1870アミノ酸残基を有するタンパク質をコードしていた。また、データベース探索の結果、マウス、ラットとショウジョウバエにおいてホモログが存在することがわかった。
【0088】
図1cには、Girdinの一次構造とCOILSアルゴリズムによって予測されるGirdinの構造を示す。図1cに示すように、Girdinは、N末端領域(NT)とα−ヘリカルコイルド−コイル領域(Ala253〜Lys-1375)とにより二量体を形成することが予測された。予測されるコイルドーコイル領域は、当該領域に典型的な7アミノ酸リピートを135個有していた(図1h参照)。
【0089】
また、GirdinのmRNAの発現をノザンブロッティングにより分析した。結果を図1dに示す。図1dに示すように、Girdinは多種のヒト組織において普遍的に発現していることがわかった。さらに、GirdinのC末端の19アミノ酸に対するポリクローナル抗体を用いてウエスタンブロッティングを行ったところ、250kDaのタンパク質として脳及び精巣溶解物中に確認することができた(図1e参照)。
【0090】
また、Girdinのオリゴマー形態を確認するため、COS7細胞溶解物の100μMスルホスクシニルイミドによる架橋前後につき、抗Girdin抗体を用いてウエスタンブロッティングにより分析した。結果は、図1fに示すように、架橋剤処理後では、Girdinは無傷の細胞内において高分子の複合体として存在することが強く示唆された。さらに、COS7細胞溶解物をゲルろ過に供した結果によれば、Girdinは、大きなタンパク質複合体を形成していることがわかった。さらに、NT領域とCT領域とがオリゴマー形成に関与するかどうかを調べるために、V5エピトープタグ付きNT(NT-V5)とmycエピトープタグ付きNT領域(NT-myc)をCOS7細胞で発現させると、図1gに示すように、これらの二つのNT領域の複合体が細胞内にて観察された。このことはNT領域がGirdinのオリゴマー形成に寄与していることを示唆していた。一方、CT領域はオリゴマー形成に寄与していないことがわかった(データ示さず)。NT領域を発現するCOS7細胞溶解物のゲルろ過結果によれば、NT領域は二量体を形成していると予測された(図1i参照)。
【0091】
(実施例2:Aktによる試験管内(in vitro)および生体内(in vivo)におけるGirdinのリン酸化)
AktとGirdinとが物理的に相互作用するかどうかをインビボ及びインビトロによるアッセイで確認した。しかしながら、インビトロにおいても免疫沈降によっても両タンパク質の安定的な複合体を検出できなかった(データ示さず)。このことは、両者には、例えば、プロテインキナーゼとその基質の相互作用に観察されるような極めて一過性の様式によって通常の相互作用が生じていることを示唆した。Aktのリン酸化サイト基質はR-x-R-x-x-S/Tサイトを含むものとされており、GirdinのCT領域の1416位のセリン(下線)に近接するアミノ酸配列(RERQKS)がこのアミノ酸配列に相当していた。また、このサイトが唯一のコンセンサス領域であった。推定されるリン酸化領域であるCT領域は、他の種類のGirdinホモログにおいても保存されていたため、AktがGirdinをリン酸化するかどうかを検討することとした。
【0092】
図2aに示すインビトロキナーゼアッセイにより、GirdinCT野生型(WT)がAktによりリン酸化されるが、Ser-1416がAlaに置換したSA変異体がリン酸化されないことは、Aktは直接1416-Serをリン酸化していることを示しているといえた。一方、NT領域及びコイル領域はいずれもインビトロではGirdinによりリン酸化されなかった(図2b参照)。
【0093】
次に、Ser-1416がAktによって触媒されるリン酸化部位であることをインビボで確認するためにリン酸化されたSer-1416を含むペプチドに特異的に結合する抗体を作製し、この抗体を用いて、図2cに示すウエスタンブロッティング分析を行った。図2cに示すように、抗リン酸化Ser-1416抗体は、野生型Akt(Akt WT)又は構成的活性型Akt(Akt CA)の共発現下GirdinCT(WT)を認識した。しかしながら、ドミナントネガティブ(優勢阻害)Akt(Akt DN)では認識しなかった。また、抗リン酸化Ser-1416抗体は、Akt CAの共発現下であっても、Girdin CT(SA)変異体を認識しなかった。これらの結果から、抗リン酸化Ser-1416抗体は、Ser-1416がリン酸化されたGirdin CTを特異的に認識することがわかった。
【0094】
次いで、抗リン酸化Ser-1416抗体が、生理学的に内在性Aktを活性化する外部刺激に応答してリン酸化された内在性Girdinを認識できるかどうかを検討した。図2dに示すように、EGFを供給すると、免疫沈降によってGirdinと推定される250kdaのタンパク質のリン酸化が誘導された。この誘導のタイムコースは、Aktの活性化のタイムコースに類似していた。さらに、Girdinのリン酸化は、細胞がPI3KインヒビターであるLY294002及びWortmanninにより処理されとき阻害されたが、ミトゲン活性化プロテインキナーゼキナーゼ1(MEK1)インヒビターであるPD98059により処理されたときには阻害されなかった。これらの結果から、Girdinのリン酸化は、PI3K依存的に誘導されると結論した。また、図2eに示すように、Akt WT及びAkt CAの発現は、Girdinの有意なリン酸化を誘導するが、Akt DNはリン酸化を誘導しなかったことは、活性Aktのみが細胞においてリン酸化を誘導できること示している。さらに、Girdinのリン酸化は、Akt RNA干渉短鎖RNA(Akt siRNA)が導入された細胞においては認められなかった。これらの知見は、Aktの活性化がGirdinのリン酸化に必要であることを支持していた。
【0095】
(実施例3:GirdinのC末端ドメインを介したアクチン線維への結合) Girdinの細胞内局在を確認するために、抗Girdin抗体により、ベロ線維芽細胞を免疫蛍光染色した。図3aのaに示すように、Girdinは、静止細胞においてはアクチンストレスファイバーとともに局在していた。EGF(50ng/ml)により細胞が刺激されると、これらは極在化し始め、その後ラメリポディア(葉状仮足)の指向性のある伸長が生じた。EGFにより刺激された細胞では、Girdinはアクチンストレスファイバーのみならずリーディングエッジにおけるラメリポディアにも局在した。リーディングエッジは、図3aのbにおいて、Arp2/3結合タンパク質コータクチンによる染色域として示されている。
【0096】
Girdinの局在は、GirdinがF−アクチン結合タンパク質であることを示唆していることから、Girdinのアクチン結合ドメインを確認するため、断片をコードする各種の融合GFPタンパク質、NT(GFP-NT)、コイルド-コイル領域についてのN末端ハーフ(GFP-M1)及びC末端ハーフ(GFP-M2)、CT領域についてのN末端ハーフ(GFP-CT1)及びC末端ハーフ(GFP-CT2)を作製し、これらの細胞内局在性を確認した(図3bのa参照)。図3bのb〜dに示すように、ベロ細胞にて発現されたとき、GFP-NT、GFP-M1及びGFP-M2は、細胞質に局在した。図3bのeに示すように、GFP-CT1は、核と、カルボシアニンメンブレンプローブであるCM-Dilの共局在によって示されるように細胞膜との双方に局在した。この一方、図3bのfに示すように、GFP-CT2は、明らかにストレスファイバー上に局在した。これらの結果は、GirdinはそのCT2領域を介してアクチンフィラメントに局在するが、CT1領域を介して細胞膜を相互作用することを示唆していた。なお、内在性Girdinの核への蓄積が観察されないことから(図3a参照)、GFP-CT1の核局在は人工的なものであると考えられた。
【0097】
次に、アクチン共沈アッセイによりCT2ドメインのアクチン結合能力について検討した。図3c及び図3dに示すように、精製したグルタチオンSトランスフェラーゼ(GST)融合CT2(GST-CT2)はα-アクチニンと同様、F-アクチンと共沈したが、GST単独では共沈しなかった。このことは、GST-CT2は、F-アクチンに直接結合していることを示していた。F-アクチンの量が増えると、図3dのaに示すように、F-アクチンと結合するGST-CT2の量は飽和した。図3dのbに示すように、F-アクチンの解離係数(Kd)は、1.03μMであった。このことは、GirdinがF-アクチンに対して比較的低い親和性しか有していないことを示している。Girdinの各種のドメイン、予測される機能及びGirdinの推測される構造を図3eに示す。
【0098】
(実施例4:Girdinのストレスファイバーの形成と細胞運動における重要性) Girdinの機能を評価するために、ベロ細胞でのGirdin発現を抑制(ノックダウン)するためにRNA干渉を採用した。いくつかのGirdin siRNA(21ヌクレオチド)とコントロールsiRNAとをベロ細胞に導入した。図4aに示すように、ウエスタンブロット分析によれば、Girdin siRNAを導入した細胞では、Akt及びアクチンの活性に影響を及ぼすことなくGirdinの発現レベルを90%以上抑制した。ノックダウン効果の特異性を確認するために、2種類のGirdin siRNA(A及びB)を用いたウエスタンブロット分析を行い、他のファンクションアッセイを行った。
【0099】
Girdinがアクチンフィラメントと架橋するのに機能するかどうかを確認するため、アクチン細胞骨格の組織化におけるGirdinのノックダウン効果を調べた。図4bのaに示すように、抗Girdin抗体による免疫蛍光染色によれば、Girdin siRNAが導入された細胞では、Girdinは非常に少量であった。また、図4bのbに示すように、ファロイジンによるF-アクチンリッチ構造の染色によれば、Girdin siRNAが導入された細胞ではアクチンストレスファイバーが破壊されていた。このことはGirdinが、アクチンストレスファイバーの形成に本質的であることを示している。
【0100】
高倍率でGirdin siRNAが導入された細胞を観察すると、この細胞は、薄く短いストレスファイバーが顕著な低下が観察された。さらに、これらは本来の形態を失い、多数の突起(リーディングエッジ)の形成ででこぼこした境界を形成していた(図4bのbの下側及び図4c参照)。
【0101】
さらに、アクチン動態におけるGirdinの役割を明らかにするために、試験管内創傷治癒アッセイにおける細胞移動におけるGirdinが抑制された細胞の振る舞いを調べた。図4dに示すように、創傷に接触したGirdin siRNAが導入された細胞は、そのリーディングエッジにおいて伸長したラメリポディアを形成することができず、突起が繰り返し伸びたり縮んだりするような細胞運動異常を示した。これらの結果は、Girdinは細胞移動の間のアクチンフィラメントの組織化に不可欠であることがわかった。
【0102】
Girdinのノックダウン効果の特異性を確認するため、他のセルラインであり、RETレセプターチロシンキナーゼを安定的に発現しているSK-N-MC神経芽腫細胞にGirdin siRNAを導入した。Girdin siRNAが導入されたSK-N-MC(RET)細胞では、ストレスファイバーの破壊とグリア細胞系列由来神経成長因子(GDNF)であるRETリガンドに応答した制限的なラメリポディアの形成を示した。
【0103】
(実施例5:電子顕微鏡によるGirdinの局在とその発現抑制がアクチン機構に与える影響の解析)
さらに、インビボでのGirdinとアクチンフィラメントとの相互作用とアクチン組織化のける役割を確認するために、「アンルーフ」ベロ細胞についてのディープエッチ電子顕微鏡による超構造分析を行った。図5aに示すように、免疫金コロイドラベル分析によって、Girdin分子は、アクチンフィラメントとともに局在することわかった。図5bに示すように、高倍率画像から、Girdinがアクチンフィラメントと間の連結部分とともに局在することがわかった。この免疫蛍光分析結果と整合して、Girdinが枯渇したベロ細胞の細胞膜に接した細胞質界面における電子顕微鏡像において視認される表層のアクチンフィラメント密度は、コントロール細胞よりも低かった(図5c参照)。また、コントロール細胞では、より強固に組織化されたアクチンファイバーが優勢であった。コントロールsiRNAを導入した細胞では、フィラメントは相互に隣接して分離することなく同方向を指向して、たくさんの分子によって強固に架橋された太いケーブルを形成していた。いくつかのフィラメントが他のアクチンケーブルに対して鉛直に指向して、結果として、織物様で高密度なアクチンネットワークを形成していた。
【0104】
(実施例6:Girdinのリン酸化が局在およびイノシトールリン脂質(ホスホイノシチド)との相互作用に与える影響) AktによるGirdinのリン酸の役割についての知見を得るために、抗リン酸化Girdin抗体による染色を用いてベロ細胞中のリン酸化Girdinの局在を調べた。図6aの最上図に示すように、血清飢餓状態の静止細胞では、Girdinのリン酸化は細胞全体を通じてほとんど観察されなかった。この結果は、図2dに示すウエスタンブロット分析結果に整合している。一方、細胞がEGF(50ng/ml)で刺激されると、免疫染色によれば、移動する細胞のリーディングエッジにおけるラメリポディアにリン酸化されたGirdinが現れることを示している(図6a中段)。また、リン酸化されたGirdinは、リーディングエッジに存在するコータクチンと共局在する(図6b参照)。さらに、EGFで刺激されたベロ細胞は、リーディングエッジのラメリポディアにおいて、リン酸化されたGirdinとともに活性化されたAktの蓄積と局在とが増強されていることを観察した(図6g)。また、Girdin siRNAが導入された細胞では、EGFで刺激されると、多数のリーディングエッジが誘導されるが、これらのリーディングエッジでは、リン酸化されたGirdinのシグナルは、予想されたように存在しなかった(図6a下段)。この結果は免疫染色の特異性を支持している。
【0105】
次に、リン酸化されたGirdinとリン酸化されていないGirdinの局在性の違いを決定するメカニズムを解明するために、GirdinのCT1ドメインは細胞膜に局在しリン酸化サイトを有していることから、Girdinのリン酸化によって制御されているのは、Girdinの細胞膜との相互作用であると仮定して実験を行った。本発明者らは、図6cのaに示すように、リン酸化サイトの上流側にプラスに荷電した19アミノ酸残基(Arg-1389からLys-1407)からなり、フォスファチジルイノシトール4,5−ビスリン酸が結合するためのコンセンサス配列に類似した配列を見出した。このフォスフォイノシタイド結合サイトを欠損させたGFP-CT1融合タンパク質をベロ細胞に導入すると、この誘導タンパク質の細胞膜への局在は観察されなかった(図6cのb参照)。この結果は、CT1ドメインは、フォスフォイノシタイドへの結合を介して細胞膜に固定されることを示唆している。
【0106】
Girdinのフォスフォイノシタイド結合能力を確認するために、Girdinのフォスフォイノシタイド結合サイトを含む精製GST融合タンパク質を用いたタンパク質−脂質結合アッセイを実施した。なお、この実験においては、GST-CT1融合タンパク質は、発現及び精製操作において容易に分解してしまうため、CT1ドメインに替えてCTドメインとGSTとを融合させて用いた。図6eのaに示すように、GST-CTは、選択的にPI(4)Pに結合し、PI(3)Pには弱く結合したが、他のフォスフォイノシタイドやリン脂質には結合しなかった。また、図6eのbに示すように、GST-CT融合タンパク質のPI(4)P及びPI(3)Pへの結合能力は、フォスフォイノシタイド結合サイトが除去されたとき喪失した。さらに、CT1ドメインの変異体(プラス電荷を有する塩基性アミノ酸残基がアラニンで置換されたもの)は、フォスフォイノシタイドに結合できないこと及び細胞膜局在性を有しないことを見出した。これらの結果は、フォスフォイノシタイド結合モチーフにおいて塩基性アミノ酸残基によって生じるプラス電荷がGirdinの細胞膜への相互作用に必要であることを示唆した(図6h参照)。
【0107】
次に、フォスフォイノシタイド結合サイトは、Aktリン酸化サイトの近傍に存在する(図6cのa参照)が、Aktはそのフォスフォイノシタイド結合能力を調節することでGirdinの局在性をコントロールしているのかどうかを確認するため、リン酸化されたGST-CT融合タンパク質のフォスフォイノシタイド結合能力を調べた。精製されたGST-CT融合タンパク質は、当然にAktによりインビトロでリン酸化された(図6dのb参照)。次いでこのリン酸化されたGST-CTを、タンパク質-脂質結合アッセイに供した。なお、このタンパク質−脂質結合は、抗リン酸化Girdin抗体により検出した。図6eのcに示すように、リン酸化したGST-CTは、PI4PにもPI3Pにも結合しないことがわかった。アクチン共沈アッセイによれば、GST-CTのリン酸化は、F-アクチンへの親和性を減衰させるものではなく、その結合動態は、GST-CT2に類似したものであることがわかった(図6f、図3d参照)。これらの知見は、フォスフォイノシタイドに対するGirdinの結合はリン酸化により減衰されるが、F-アクチンに対する結合は減衰されないことを示唆した。
【0108】
さらに、Girdinの細胞膜相互作用が、EGF処理によって調節されているかどうかを、GFPと全長Girdin(WT)又はSA変異体との融合タンパク質のいずれかを発現するCOS7細胞を用いて調べた。図6iに示すように、AktによるSer-1416のリン酸化が、細胞膜からのGirdinの脱局在化に必要であることがわかった。
【0109】
(実施例7:Girdinのリン酸化による細胞移動の調節) リーディングエッジにおけるリン酸化Girdinの局在化がGirdinのリン酸化は細胞運動により引き起こされるかどうかを確認するために、以下の実験を行った。すなわち、ベロ細胞のコンフルーエントの単層に損傷を与えた後、抗リン酸化Girdin抗体で免疫染色すると、損傷を受けた端部においては細胞内のGirdinリン酸化レベルが損傷後速やかに上昇し、8時間後に最大に達し、その後少なくとも12時間継続した(図7a参照)。このことは、Girdinのリン酸化が細胞運動に重要な役割を果たしていることを示唆した。したがって、細胞運動におけるGirdinの役割をボイデンチャンバー法により調べた。図7bに示すように、Girdinのノックダウンは、有意にベロ細胞が供給されたEGF(50ng/ml)に応答して下流のチャンバーに移動するのを抑制した。また、図7bに示すように、ベロ細胞におけるCTドメインの発現が細胞移動により有意に減衰したことを見出した。このことは、内在性Girdinとアクチンフィラメントとの相互作用が統合的な細胞移動に重要であることを支持している。先行する研究と整合するように、siRNAによるAktの枯渇も、細胞移動を減衰させた(図7b)。このことは、本来的なAktの活性が、ベロ細胞がこのアッセイ方法の孔部を介して移動するのに必要であることを示唆した。
【0110】
次に、外来性Girdinをノックダウン細胞に供給して当該細胞における細胞移動傷害を復帰させるかどうかを調べた。このため、サイレントミューテーションを有してsiRNA抵抗性Girdin(siRNAr Girdin)コンストラクトを構築した(図7c参照)。図7dに示すように、siRNAr Girdin(WT)をノックダウン細胞において発現させることにより、EGF応答性の細胞移動性を完全に回復させた。これに反して、Ser-1416がAlaに置換されたsiRNAr Girdin変異体であるsiRNAr Girdin(SA)、フォスフォイノシタイド結合サイトを欠損させたsiRNAr Girdi変異体であるsiRNAr Girdin(ΔPB)、フォスフォイノシタイド結合サイトがアラニンで置換されたsiRNAr Girdin変異体であるsiRNAr Girdin(PB ala)は、細胞移動傷害を補うことはできなかった。(図7d及び図6hのc参照)。これらの結果は、ヒト線維肉腫細胞株HT1080を用いた細胞移動アッセイにより確認した(図7f)。
【0111】
最終的に、Girdin変異体を発現するベロ細胞の移動を損傷治癒アッセイを用いて直接確認した(図7g参照)。GirdinWTを発現する細胞は、損傷部位に容易にか速やかに移動したが、GirdinSAを発現する細胞は、延長された形態を示すに留まった。さらに、後者の細胞における核は固定され移動性がないようであった。このことは、これらの細胞がマトリックスから分離できないことを示唆している。GirdinΔPBを発現する細胞及びGirdin PBalaを発現する細胞は、Girdin WTを発現する細胞よりも移動能力は低かった。これらの結果は、Girdin変異体の発現は、細胞移動の適切な方向性を損なうことを示している。図6a〜f及び図6iにおける結果を合わせて考慮すると、これらのデータは、AktによるGirdinと細胞膜との相互作用の調節が細胞移動に必須であることを示している。
【0112】
(実施例8:Girdin siRNAによるヒト臍帯血管内皮細胞HUVECの遊走能の阻害) HUVEC細胞をコントロールsiRNAおよびGirdin siRNAで48時間処理した後、ボイデンチャンバー法(Boyden Chamber法)にてVEGF(血管内皮成長因子)による遊走能を定量した。図8aに示すように、VEGF存在下においてGirdin siRNAを処理したHUVEC細胞の遊走能が著明に低下することが明らかになった。また、図8bには、遊走した細胞が紫色に染色された図を示す。図8bに示すように、Girdin siRNAで処理したHUVEC細胞の遊走細胞数が低下していることが明らかであった。
【0113】
(実施例9:Girdin siRNAによるヒト臍帯血管内皮細胞HUVECの管腔形成能の阻害) 実施例8と同様にしてHUVEC細胞をGirdin siRNAで処理した後、管腔形成能を観察した。観察結果を図9に示す。図9aに示すように、未処理のHUVEC細胞及びコントロールsiRNAで処理したHUVEC細胞は通常の培養条件で管腔形成を示すが、Girdin siRNAで処理したHUVEC細胞では管腔形成は著明に阻害された。図9bには、図9aの各細胞におけるGirdinの発現を抗Girdin抗体で検出したウェスタンブロティング結果を示しているが、図9bに示すように、Girdin siRNAを導入したHUVEC細胞では、Girdinの発現が顕著に抑制されていた。
【0114】
(実施例10:siRNA抵抗性のGirdin遺伝子の発現による管腔形成能の回復) アデノウィルスベクターにsiRNA抵抗性のGirdin遺伝子cDNAを導入した(Ad-rGirdin WT-V5)。またGirdinのAktリン酸化部位であるセリン1416をアラニンに置換したGirdin cDNAを導入したベクターも作製した(Ad-rGirdin SA-V5)。図10aに示すように、siRNAとAd-rGirdin WT-V5をHUVEC細胞に同時に導入すると、管腔形成が回復した(図10aの中央)。Aktによるリン酸化部位を変異させると、管腔形成は回復せず(図10a右)、AktによるGirdinのリン酸化がその機能に重要であることが明らかになった。図10bは、図10aの各細胞におけるGirdinの発現を抗Girdin抗体と抗V5抗体で検出したウェスタンブロティング結果を示している。なお、コントロールとしてVEGFR2(血管内皮増殖因子レセプター2)の発現を示す。
【0115】
以上の結果よりGirdinは血管形成にも重要な役割をはたしていることが判明し、腫瘍内の血管新生の阻害を目指したAkt-Girdin系の阻害化合物の開発による、抗腫瘍薬の開発の可能性がある。
【0116】
(実施例11:マトリゲルプラグアッセイ)
アデノウイルスベクターを用いてショートヘアピン型Girdin siRNAのためのノックダウンベクターを構築した。Girdin遺伝子におけるターゲット配列は、gaaggagaggcaactggat(配列番号3)とした。このアデノウイルス(1×1010個/ml)100μlをVEGF100ng/mlを含有するマトリゲル(BDマトリゲル(製品名))400μlに混合し、その全量をマウス腹部の皮下に注入した。1週間経過後、マトリゲル注入部位を取り出してヘマトキシリン・エオジン(HE)染色した。また、コントロールとして、ノックダウンカセットに替えてEGFPを保持したアデノウイルスを用いる以外は実施例と同様に操作した。結果を図11に示す。
【0117】
図11には、上段にコントロールの結果を示し、下段に本実施例の結果を示す。コントロールでは血管新生が観察されたのに対し、実施例では、ほとんど血管新生が観察されなかった。Girdinの発現が抑制された結果、血管新生が阻害されたことがわかった。なお、本実施例で用いたノックダウンベクターはGirdinの発現を抑制することが確認されている。
【0118】
(実施例12:腫瘍血管におけるGirdinの発現)ヒトの乳癌と子宮頸癌の組織を採取し、Girdinを定法により免疫染色した結果を図12に示す。なお、Girdin組織免疫染色にあたっては、キシレンを用いてパラフィン包理された組織切片の脱パラフィン処理を行った後、脱水処理し、0.3%過酸化水素水による内因性peroxidase活性の不活化処理を行い、ブロッキングをした。その後、抗Girdin抗体(BSAを含むPBSで50倍希釈) を加えて、保湿箱内でインキュベート(4度、一晩)し、PBSで洗浄後、二次抗体を加えて室温でインキュベート(15分)した。PBSで洗浄後、酵素標識ストレプトアビジンを加えて、室温で15分インキュベートし、洗浄し、DAB基質を添加した。PBSに浸して反応を停止させた後、ヘマトキシレン染色で核染色し、洗浄及び脱水した後キシレン処理した。図12において血管内皮細胞にGirdinが強く染色されていることがわかる(矢印部分がGirdinに基づく強い染色部分)。これらの結果から、Girdinは腫瘍血管の内皮細胞で強く発現していることがわかった。
産業上の利用可能性
【0119】
本発明は、各種疾患の予防・治療のための薬剤、そのスクリーニング及び試薬として利用することができる。
【配列表フリーテキスト】
【0120】
配列番号3:Girdin siRNAのターゲット配列
【特許請求の範囲】
【請求項1】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
【請求項2】
前記タンパク質は、配列番号2で表されるアミノ酸配列からなるタンパク質である、請求項1に記載のスクリーニング方法。
【請求項3】
前記部分ペプチドは、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域(CT1領域及び/又はCT2領域)である、請求項1に記載のスクリーニング方法。
【請求項4】
前記タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている、請求項1又は2に記載のスクリーニング方法。
【請求項5】
セリン/スレオニンキナーゼAktと配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含むタンパク質又はその一部との結合活性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項6】
セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項7】
アクチンフィラメント結合能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項8】
アクチンストレスファイバー形成能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項9】
ラポリメディアにおけるアクチンメッシュワークの形成能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項10】
細胞膜結合性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項11】
フォスフォイノシタイド結合性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項12】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質をコードするポリヌクレオチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
【請求項13】
細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬のスクリーニング方法である、請求項1〜12のいずれかに記載のスクリーニング方法。
【請求項14】
前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、請求項1〜13のいずれかに記載のスクリーニング方法。
【請求項15】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかあるいは2種以上を発現する化合物又はその塩である、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合の阻害活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の阻害活性
c)アクチンフィラメントへの結合の阻害活性
d)アクチンストレスファイバー形成の阻害活性
e)ラポリメディアにおけるアクチンメッシュワークの形成の阻害活性
f)細胞膜結合の阻害活性
g)フォスフォイノシタイドへの結合の阻害活性
【請求項16】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質の発現量を抑制する化合物である、細胞運動、細胞移動又は血管形成のいずれかが関連する疾患の予防・治療薬。
【請求項17】
前記化合物は、配列番号2で表されるアミノ酸配列をコードするポリヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するポリヌクレオチドである、請求項16に記載の予防・治療薬。
【請求項18】
RNA干渉を発現するRNAである、請求項17に記載の予防・治療薬。
【請求項19】
請求項18に記載のRNAをコードするDNAである、請求項16に記載の予防・治療薬。
【請求項20】
請求項19に記載のDNAを含有する組換えベクターである、請求項16に記載の予防・治療薬。
【請求項21】
前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、請求項16〜20のいずれかに記載の予防・治療薬。
【請求項22】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された細胞。
【請求項23】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された動物。
【請求項24】
前記遺伝子の発現量の抑制はノックダウンによる請求項23に記載の動物。
【請求項25】
細胞運動、細胞移動及び血管形成のいずれかに関連する疾患の研究用、予防・治療薬のスクリーニング用である、請求項23又は24に記載の動物。
【請求項26】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定性的分析方法。
【請求項27】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定量方法。
【請求項28】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに特異的な抗体。
【請求項29】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに特異的な抗体。
【請求項30】
請求項28又は29に記載の抗体を含む、細胞運動、細胞移動及び血管形成の活性化又は阻害の研究用試薬。
【請求項31】
請求項28又は29に記載の抗体を含む、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断薬。
【請求項32】
疾患部位の細胞における配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかを利用する、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断方法。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化活性
c)アクチンフィラメントへの結合活性
d)アクチンストレスファイバー形成活性
e)ラポリメディアにおけるアクチンメッシュワークの形成活性
f)細胞膜結合活性
g)フォスフォイノシタイドへの結合活性
【請求項1】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
【請求項2】
前記タンパク質は、配列番号2で表されるアミノ酸配列からなるタンパク質である、請求項1に記載のスクリーニング方法。
【請求項3】
前記部分ペプチドは、配列番号2で表されるアミノ酸配列からなるタンパク質のC末端側領域(CT1領域及び/又はCT2領域)である、請求項1に記載のスクリーニング方法。
【請求項4】
前記タンパク質は、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に相当するセリン残基がリン酸化されている、請求項1又は2に記載のスクリーニング方法。
【請求項5】
セリン/スレオニンキナーゼAktと配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含むタンパク質又はその一部との結合活性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項6】
セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項7】
アクチンフィラメント結合能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項8】
アクチンストレスファイバー形成能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項9】
ラポリメディアにおけるアクチンメッシュワークの形成能の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項10】
細胞膜結合性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項11】
フォスフォイノシタイド結合性の促進又は阻害を指標とする、請求項1〜4のいずれかに記載のスクリーニング方法。
【請求項12】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質をコードするポリヌクレオチドを用いることを特徴とする、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物又はその塩のスクリーニング方法。
【請求項13】
細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬のスクリーニング方法である、請求項1〜12のいずれかに記載のスクリーニング方法。
【請求項14】
前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、請求項1〜13のいずれかに記載のスクリーニング方法。
【請求項15】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかあるいは2種以上を発現する化合物又はその塩である、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の予防・治療薬。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合の阻害活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化の阻害活性
c)アクチンフィラメントへの結合の阻害活性
d)アクチンストレスファイバー形成の阻害活性
e)ラポリメディアにおけるアクチンメッシュワークの形成の阻害活性
f)細胞膜結合の阻害活性
g)フォスフォイノシタイドへの結合の阻害活性
【請求項16】
配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質の発現量を抑制する化合物である、細胞運動、細胞移動又は血管形成のいずれかが関連する疾患の予防・治療薬。
【請求項17】
前記化合物は、配列番号2で表されるアミノ酸配列をコードするポリヌクレオチドの塩基配列に相補的若しくは実質的に相補的な塩基配列又はその一部を有するポリヌクレオチドである、請求項16に記載の予防・治療薬。
【請求項18】
RNA干渉を発現するRNAである、請求項17に記載の予防・治療薬。
【請求項19】
請求項18に記載のRNAをコードするDNAである、請求項16に記載の予防・治療薬。
【請求項20】
請求項19に記載のDNAを含有する組換えベクターである、請求項16に記載の予防・治療薬。
【請求項21】
前記疾患は、癌、動脈硬化性疾患並びに中枢及び末梢神経障害から選択されるいずれかである、請求項16〜20のいずれかに記載の予防・治療薬。
【請求項22】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された細胞。
【請求項23】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質をコードする遺伝子の発現量が抑制された動物。
【請求項24】
前記遺伝子の発現量の抑制はノックダウンによる請求項23に記載の動物。
【請求項25】
細胞運動、細胞移動及び血管形成のいずれかに関連する疾患の研究用、予防・治療薬のスクリーニング用である、請求項23又は24に記載の動物。
【請求項26】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定性的分析方法。
【請求項27】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドとセリン/スレオニンキナーゼAktとの反応を利用する、細胞運動、細胞移動及び血管形成のいずれかを活性化又は阻害する化合物の定量方法。
【請求項28】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質又はその部分ペプチドに特異的な抗体。
【請求項29】
配列番号2で表されるアミノ酸配列と同一又は実質的に同一のアミノ酸配列を含むタンパク質であって、配列番号2で表されるアミノ酸配列の第1416位のセリン残基又は該セリン残基に対応するセリン残基がリン酸化されているタンパク質又は当該リン酸化部分を含む部分ペプチドに特異的な抗体。
【請求項30】
請求項28又は29に記載の抗体を含む、細胞運動、細胞移動及び血管形成の活性化又は阻害の研究用試薬。
【請求項31】
請求項28又は29に記載の抗体を含む、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断薬。
【請求項32】
疾患部位の細胞における配列番号2で表されるアミノ酸配列と同一若しくは実質的に同一のアミノ酸配列を含有するタンパク質又はその部分ペプチドについて以下の反応性のいずれかを利用する、細胞運動、細胞移動及び血管形成のいずれかが関連する疾患の診断方法。
a)セリン/スレオニンキナーゼAktと前記タンパク質と結合活性
b)セリン/スレオニンキナーゼAktによる前記タンパク質のリン酸化活性
c)アクチンフィラメントへの結合活性
d)アクチンストレスファイバー形成活性
e)ラポリメディアにおけるアクチンメッシュワークの形成活性
f)細胞膜結合活性
g)フォスフォイノシタイドへの結合活性
【図1a】



【図1b】
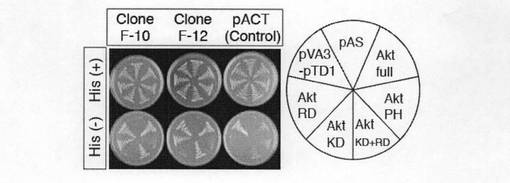
【図1c】
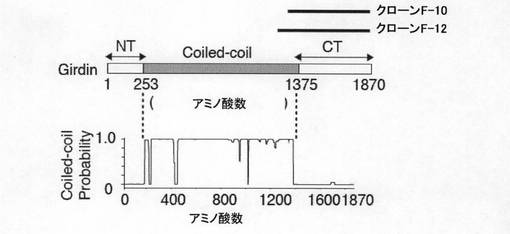
【図1d】
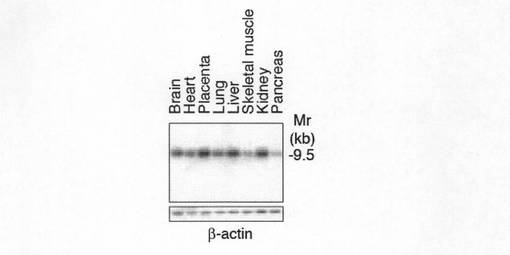
【図1e】
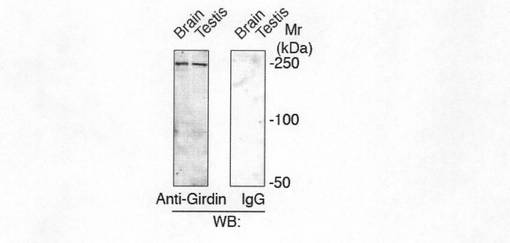
【図1f】
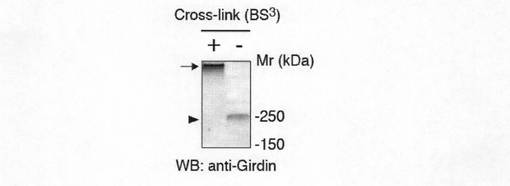
【図1g】
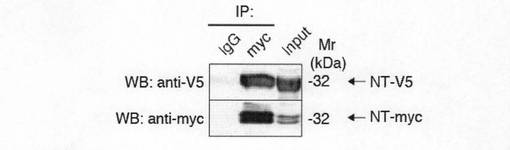
【図1h】

【図1i】
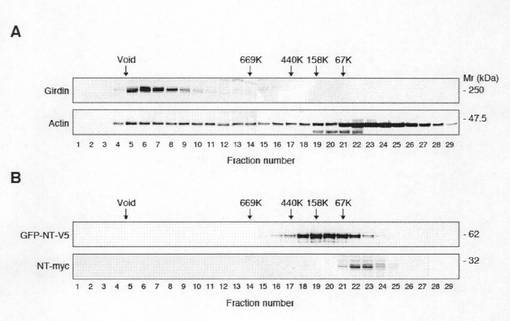
【図2a】
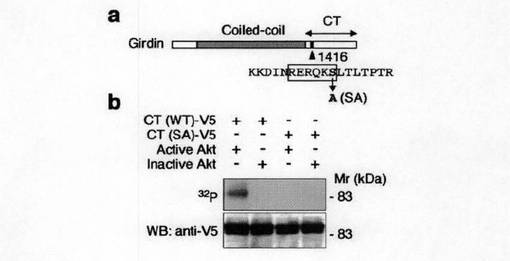
【図2b】
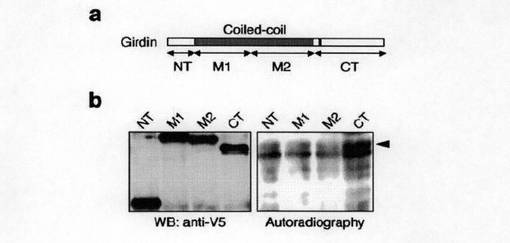
【図2c】
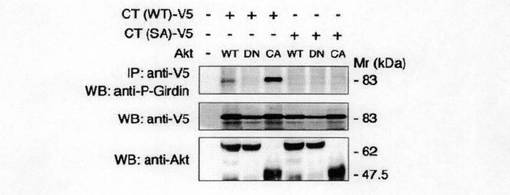
【図2d】
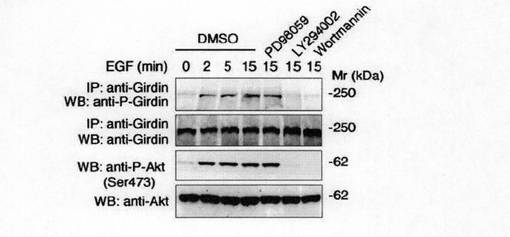
【図2e】
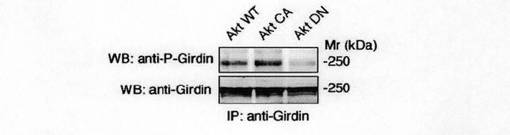
【図2f】
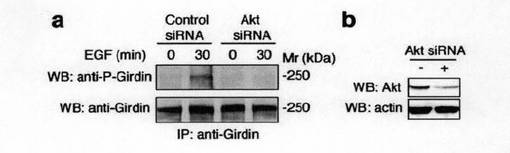
【図3a】
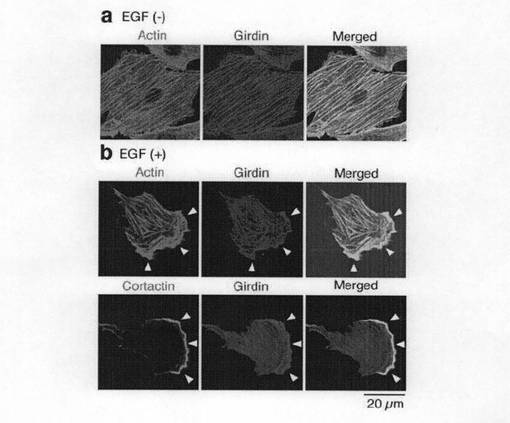
【図3b】
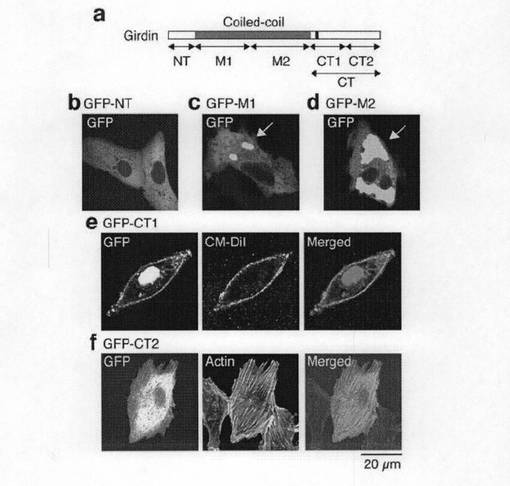
【図3c】
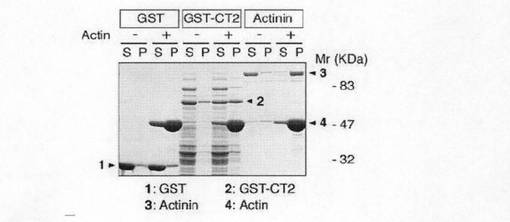
【図3d】
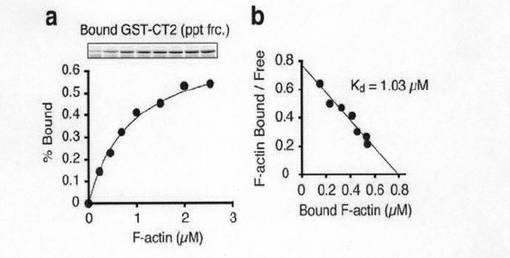
【図3e】
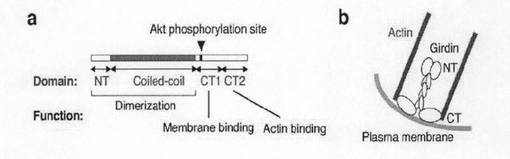
【図4a】
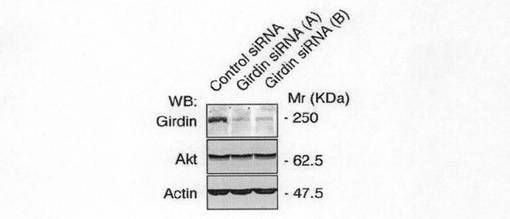
【図4b】
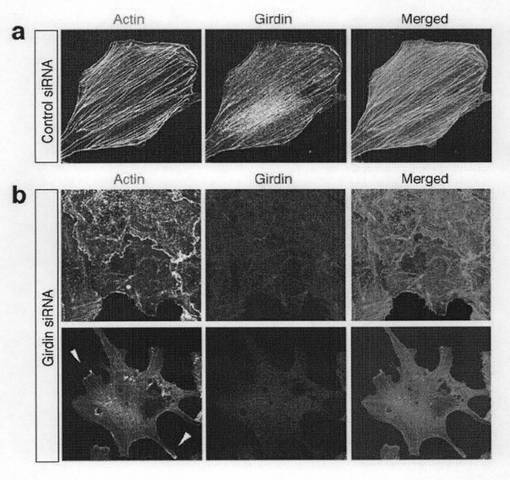
【図4c】
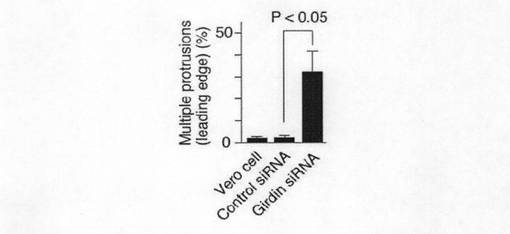
【図4d】
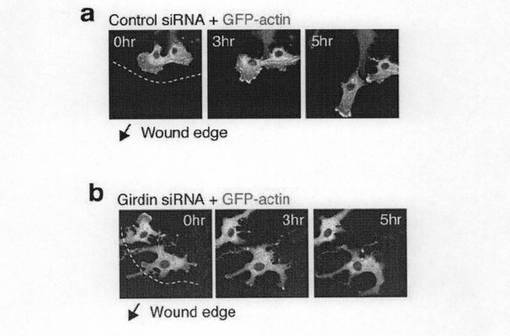
【図4e】
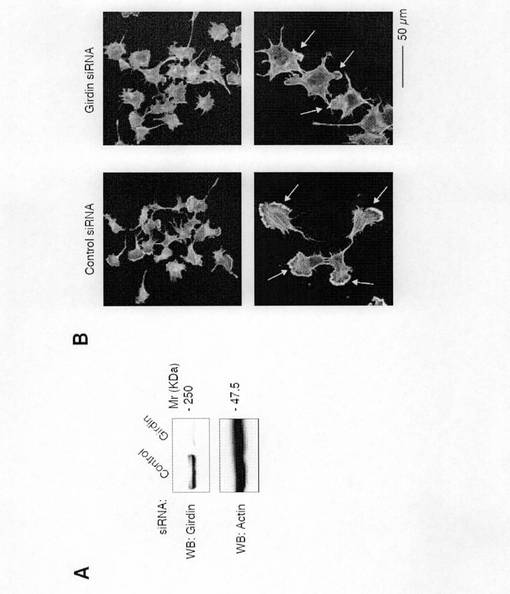
【図5a】

【図5b】
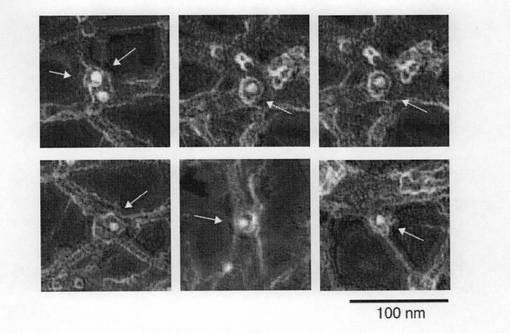
【図5c】
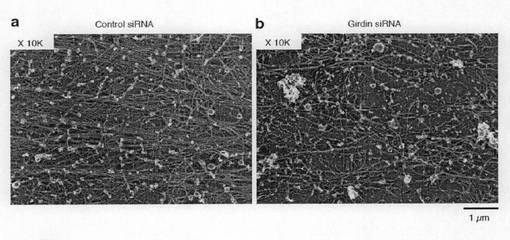
【図6a】

【図6b】
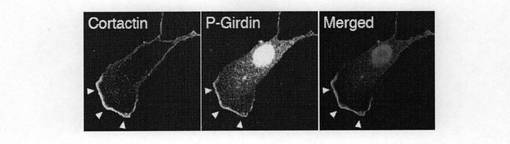
【図6c】
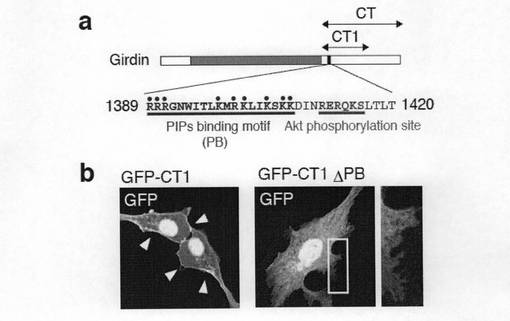
【図6d】
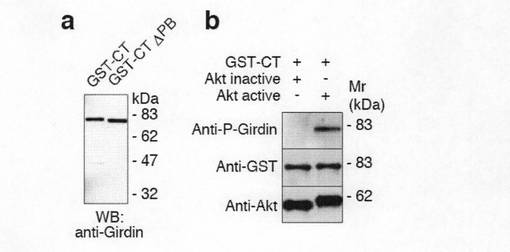
【図6e】
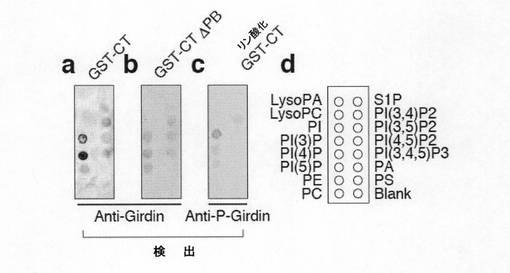
【図6f】
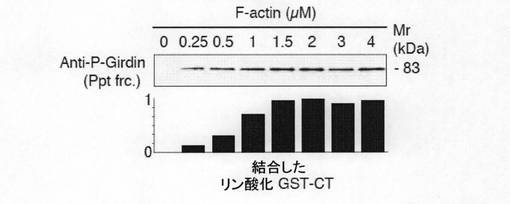
【図6g】
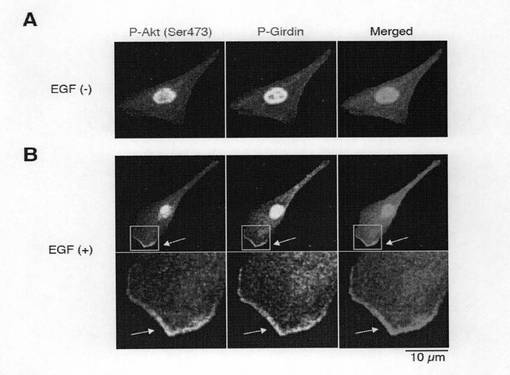
【図6h】
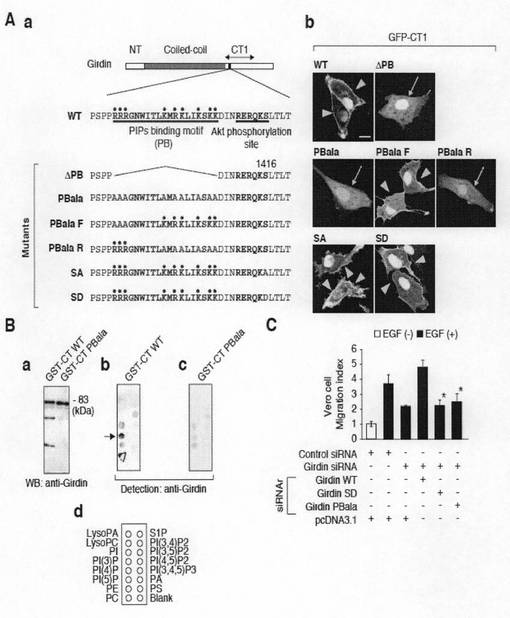
【図6i】
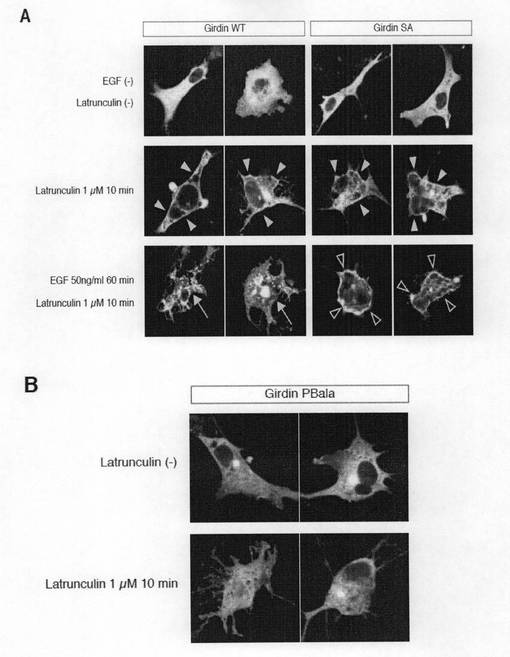
【図7a】
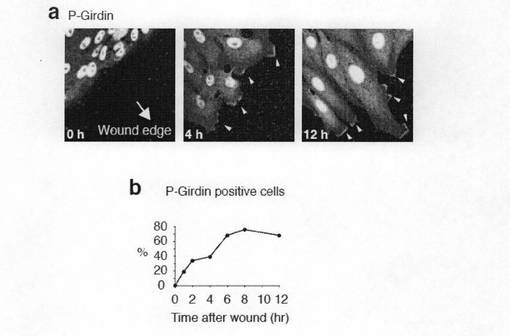
【図7b】
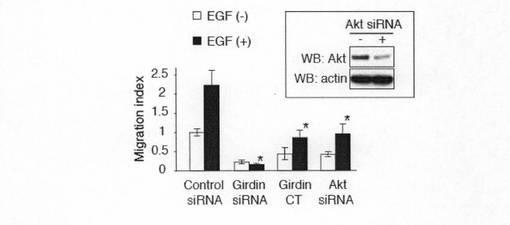
【図7c】
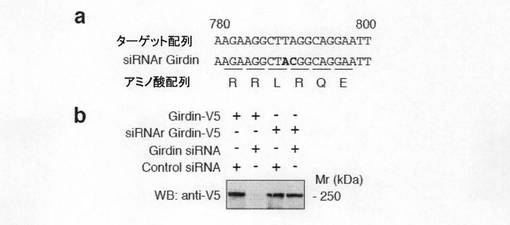
【図7d】
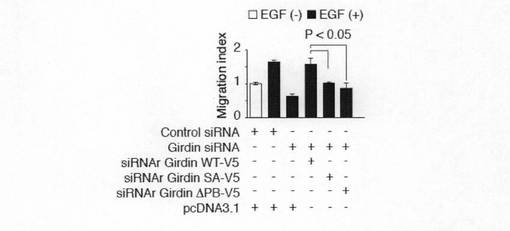
【図7e】
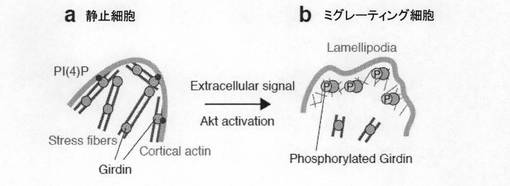
【図7f】
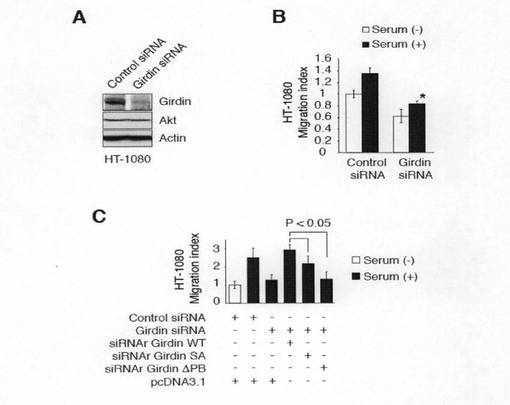
【図7g】

【図8a】
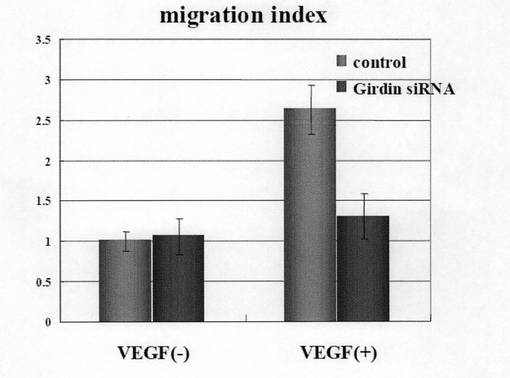
【図8b】
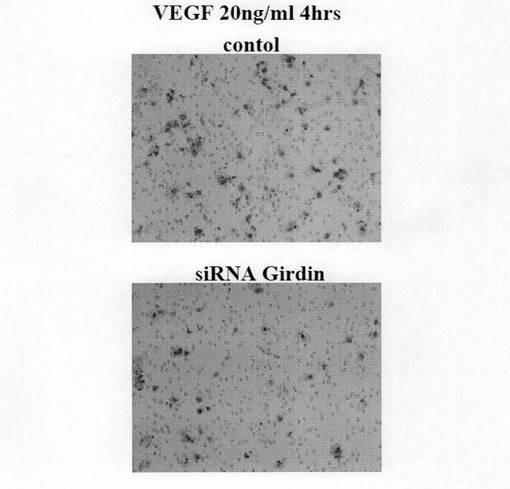
【図9a】

【図9b】
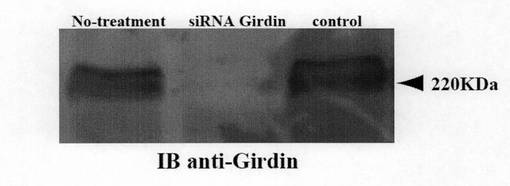
【図10a】
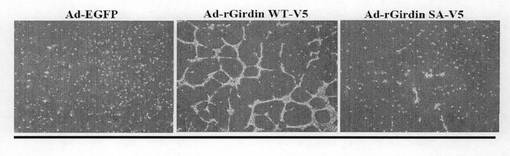
【図10b】
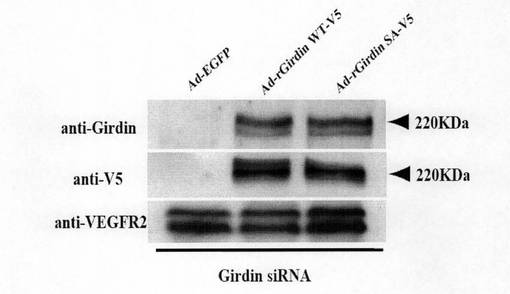
【図11】
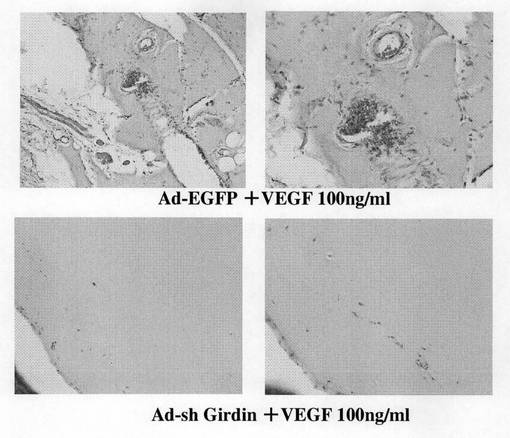
【図12】
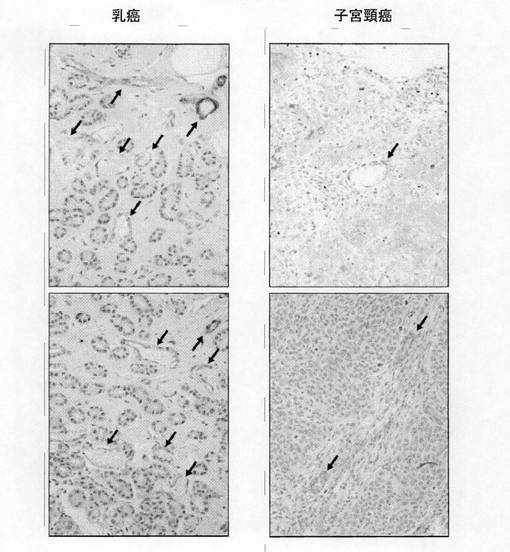
【図1b】
【図1c】
【図1d】
【図1e】
【図1f】
【図1g】
【図1h】
【図1i】
【図2a】
【図2b】
【図2c】
【図2d】
【図2e】
【図2f】
【図3a】
【図3b】
【図3c】
【図3d】
【図3e】
【図4a】
【図4b】
【図4c】
【図4d】
【図4e】
【図5a】
【図5b】
【図5c】
【図6a】
【図6b】
【図6c】
【図6d】
【図6e】
【図6f】
【図6g】
【図6h】
【図6i】
【図7a】
【図7b】
【図7c】
【図7d】
【図7e】
【図7f】
【図7g】
【図8a】
【図8b】
【図9a】
【図9b】
【図10a】
【図10b】
【図11】
【図12】
【公開番号】特開2013−31443(P2013−31443A)
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願番号】特願2012−176352(P2012−176352)
【出願日】平成24年8月8日(2012.8.8)
【分割の表示】特願2007−546426(P2007−546426)の分割
【原出願日】平成18年11月10日(2006.11.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 特許法第30条第1項適用、Developmental Cell、第9巻第3号(平成17年9月6日)Elsevier発行第389−402頁に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 特許法第30条第1項適用、細胞工学、第24巻第11号(平成17年10月22日)秀潤社発行第1192、1193頁に発表
【出願人】(504139662)国立大学法人名古屋大学 (996)
【Fターム(参考)】
【公開日】平成25年2月14日(2013.2.14)
【国際特許分類】
【出願日】平成24年8月8日(2012.8.8)
【分割の表示】特願2007−546426(P2007−546426)の分割
【原出願日】平成18年11月10日(2006.11.10)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 特許法第30条第1項適用、Developmental Cell、第9巻第3号(平成17年9月6日)Elsevier発行第389−402頁に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 特許法第30条第1項適用、細胞工学、第24巻第11号(平成17年10月22日)秀潤社発行第1192、1193頁に発表
【出願人】(504139662)国立大学法人名古屋大学 (996)
【Fターム(参考)】
[ Back to top ]