
アシクロビル改変組成物及びその製造方法

【課題】アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供する。
【解決手段】ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することにより、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスを製造する。前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることが好ましく、前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることが好ましい。この様な共結晶、アモルファスとしては、アシクロビルと酒石酸の共結晶と、アシクロビルとクエン酸からなる、アモルファスとが好ましく例示できる。
【解決手段】ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することにより、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスを製造する。前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることが好ましく、前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることが好ましい。この様な共結晶、アモルファスとしては、アシクロビルと酒石酸の共結晶と、アシクロビルとクエン酸からなる、アモルファスとが好ましく例示できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アシクロビルの結晶を改変した組成物及び当該組成物の製造方法に関する。
【背景技術】
【0002】
医薬品において、その有効成分、APIの性状をコントロールすることは重要な課題となる。例えば、結晶系をコントロールし、医薬品として好ましい性状を得る技術は既に医薬品特許の分野では、「結晶多形特許」、「水和物特許」というパターンを形成している。また、研究の分野でも、結晶系、溶媒和などをコントロールし、薬動力学的利用性を高める試みが盛んに為されている。(例えば、特許文献1、特許文献2、特許文献3、特許文献4、非特許文献1、非特許文献2、非特許文献3を参照)その反面、生体利用性が低く、しかも、アモルファス化しにくく、更には、好ましい他の結晶系の結晶も存しない薬剤は存在し、その生体利用性を高めることは一つの課題となっている。この様な物質としては、アシクロビル、ガンシクロビル、ビダラビン、5−FUのような、ヌクレオシド系の医薬、特に、抗ウイルス剤が典型的な物質として、好適に例示できる。この様な物質に対して、物理的性状を変えて、その生体利用性を高める手段としては、例えば、クレアチニンやグリシンとともに凍結乾燥し、アモルファス化する方法が考えられるが(例えば、特許文献5を参照)、通常のAPIではコストがかかりすぎ、コストメリットバランス上、実施に至る物はほとんどないのが現状である。
【0003】
一方、他の結晶性成分とともに、共結晶形成せしめ、物理的性状を変える試みも為されている(例えば、特許文献6、特許文献7を参照)が、アシクロビルとポリヒドロキシカルボン酸との共結晶も、アモルファス物質も知られていない。また、この様な共結晶やアモルファスを形成することにより、溶解度や生体利用性が高まることも全く知られていない。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開平5−43590号公報
【特許文献2】特開平7−316141号公報
【特許文献3】特開平01−38096号公報
【特許文献4】特開平01−250384号公報
【特許文献5】WO/2008/152764
【特許文献6】特表2011−516567号公報
【特許文献7】特表2007−524596号公報
【非特許文献】
【0005】
【非特許文献1】Berge,S.M.,Bighley,L.D.,Monkhouse,D.C.,1977.Pharmaceutical salts.J.Pharm.Sci.66,1−19
【非特許文献2】Kato,Y.,Okamoto,Y.,Nagasawa,S.,Ueki,T.,1981.Solubility of a new polymorph of phenobarbital obtained by crystallization in the presence of phenytoin.Chem.Pharm.Bull.29,3410−3413
【非特許文献3】Serajuddin,A.T.M.,1999,Solid dispersion of poorly water−soluble drugs:early promises,subsequent problems,and recent breakthroughs.J.Pharm.Sci.88,1058−1066.
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、この様な状況下為されたものであり、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供すること課題とする。
【課題を解決するための手段】
【0007】
この様な状況に鑑みて、本発明者らは、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を求めて、鋭意研究努力を重ねた結果、極性溶媒を用いて、ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを加熱溶解、冷却析出させることにより、当該医薬原体とポリヒドロキシカルボン酸の共結晶乃至はアモルファスを得ることが出来、かかる共結晶、アモルファスが非常に好ましい物性を有していることを見出し、発明を完成させるに至った。即ち、本発明は、以下に示すとおりである。
<1>ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することを特徴とする、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<2>前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることを特徴とする、<1>に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸との共結晶又はアモルファスの製造法。
<3>前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることを特徴とする、<1>又は<2>に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<4>前記極性溶媒は、酢酸及び/又はジアルキルホルムアミドを含むものであることを特徴とする、<1>〜<3>何れか1項に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<5>アシクロビルと酒石酸の共結晶。
<6>次の物性を有することを特徴とする、<5>に記載の共結晶。
分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストローム
<7><1>〜<4>何れか1項に記載の製造方法で製造された物であることを特徴とする、<5>または<6>に記載の共結晶。
<8>アシクロビルとクエン酸からなる、アモルファス。
<9>示差熱分析において、60〜80℃にガラス転移点を有することを特徴とする、<8>に記載のアモルファス。
<10><1>〜<4>何れか1項に記載の製造方法で製造された物であることを特徴とする、<8>または<9>に記載のアモルファス。
<11><5>〜<7>何れか1項に記載の共結晶及び/又は<8>〜<10>何れか1項に記載のアモルファスを有効成分として含有することを特徴とする、医薬組成物。
<12>皮膚外用剤であることを特徴とする、<11>に記載の医薬組成物。
【発明の効果】
【0008】
本発明によれば、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供できる。
【図面の簡単な説明】
【0009】
【図1】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、アシクロビル2/3水和物(c)、アシクロビル2水和物(d)、酒石酸(e)、アシクロビル・酒石酸共結晶(f)、アシクロビル・クエン酸アモルファス(g)の粉末X線解析を示す図である。
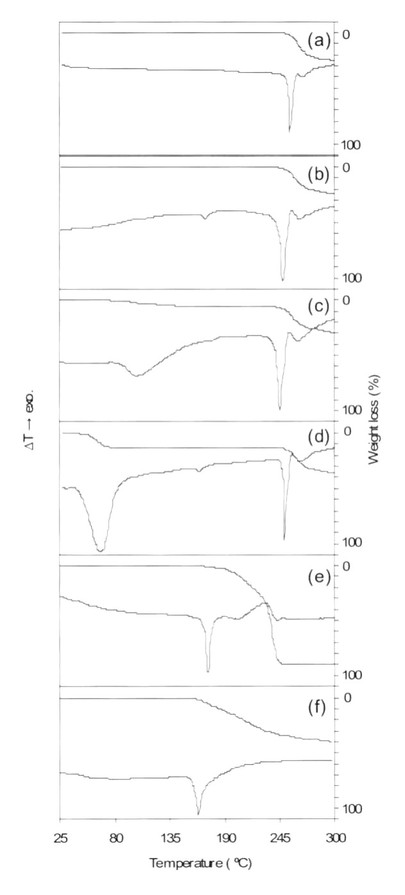
【図2】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、アシクロビル2/3水和物(c)、アシクロビル2水和物(d)、酒石酸(e)、アシクロビル・酒石酸共結晶(f)のTG/DTAを示す図である。
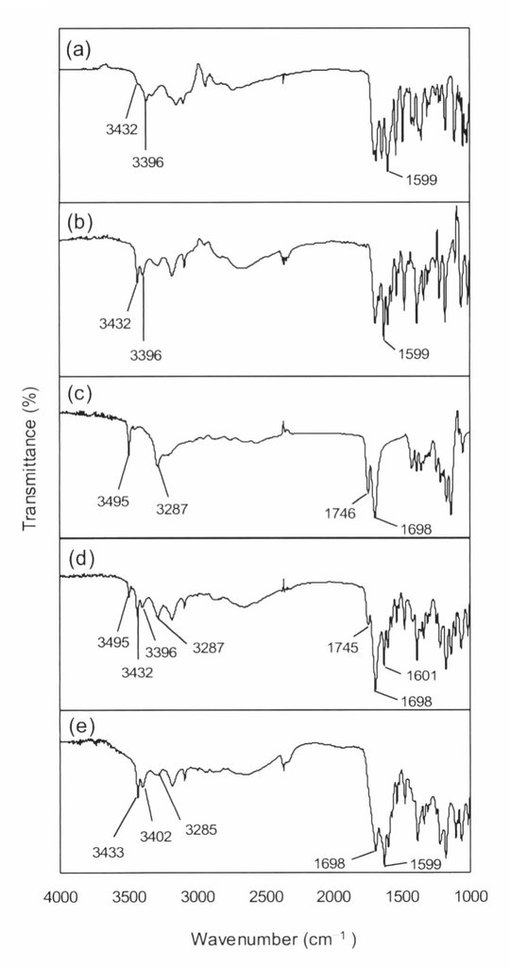
【図3】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、酒石酸(c)、アシクロビル・酒石酸共結晶(d)のIRスペクトルを示す図である。
【図4】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、クエン酸(c)、アシクロビル・クエン酸物理的混合物(d)、アシクロビル・クエン酸アモルファス(e)のIRスペクトルを示す図である。
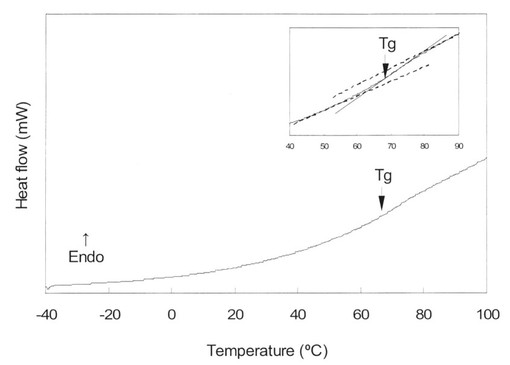
【図5】アシクロビル・クエン酸アモルファスのDSCの測定結果を示す図である。
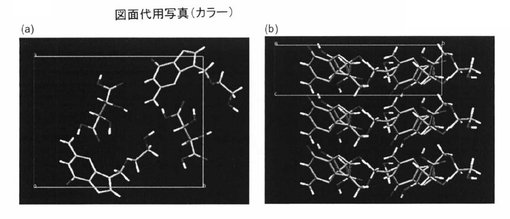
【図6】アシクロビル・酒石酸共結晶のab平面と、bc平面を示す図である。
【図7】アシクロビル無水晶系2、アシクロビル・酒石酸物理的混合物、アシクロビル・酒石酸共結晶の蒸留水、リン酸バッファー、エタノールに対する飽和溶解度を示す図である。
【図8】アシクロビル無水晶系1、アシクロビル無水晶系2、アシクロビル2/3水和物、アシクロビル2水和物、アシクロビル・酒石酸物理的混合物、アシクロビル・酒石酸共結晶のリン酸バッファーにおける溶解速度比較を示す図である。
【図9】アシクロビル結晶、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスのPEG−400に対する飽和溶解度を示す図である。
【図10】PEG軟膏の顕微鏡観察の結果を示す図である。(図面代用写真)
【図11】PEG軟膏からのアシクロビルの放出特性を示す図である。
【図12】PEG軟膏における、アシクロビルのヘアレスマウスの皮膚からの透過特性を示す図である。
【発明を実施するための形態】
【0010】
<1>本発明の製造法の対象とする医薬原体
本発明は、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供することを特徴とし、その対象となる医薬原体はヌクレオシド系医薬原体である。ヌクレオシド系医薬原体とは、プリン塩基またはピリミジン塩基乃至はそれらの誘導体とそれと結合した直鎖乃至は環状の糖からなる構造を有するものであり、糖の水酸基はリン酸化されていない物を意味する。この様な医薬原体としては、例えば、アシクロビル、ガンシクロビル、ビダラビン、AZT、テガフールなどが好適に例示できる。この中では、アシクロビルが特に好ましい。これは、その溶解性の悪さ故に効果発現が抑制されていると考えられるためである。
【0011】
<2>本発明において医薬原体と対になるポリヒドロキシカルボン酸
本発明においては、前記ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを共結晶またはアモルファスに加工することを主旨とする。ここで、ポリヒドロキシカルボン酸としては、2個以上の水酸基を有し、且つ、カルボキシル基も有するものが好適に例示でき、具体的には、酒石酸、リンゴ酸、グルコン酸、クエン酸などが好適に例示でき、特に好ましくは、酒石酸またはクエン酸である。かかる成分は、アモルファスの場合、前記ヌクレオシド系医薬原体とほぼ2:1で複合体を形成するため、ヌクレオシド系医薬原体の少なくとも2質量倍含有されることが好ましい。また、共結晶の場合、前記ヌクレオシド系医薬原体とほぼ1:1で複合体を形成するため、ヌクレオシド系医薬原体の少なくとも等質量含有されることが好ましい。
【0012】
<3>本発明において使用される極性溶剤
本発明において、前記ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを共結晶乃至はアモルファスにするために使用される極性溶剤としては、水と混和しうる溶剤であって、親油性の成分も溶かしうる物が好ましく、具体的には、ギ酸、酢酸、プロピオン酸などの低鎖長カルボン酸、ジメチルホルムアミド、ジエチルホルムアミド、ジメチルスルホキシド、エチルセルソルブ、メチルセルソルブなどが好適に例示でき、酢酸などの低鎖長カルボン酸及び/又はジメチルホルムアミドなどのジアルキルホルムアミドが特に好適に例示できる。かかる極性溶媒を、ヌクレオシド系医薬原体の5〜20質量倍用い、加温して溶解後、氷等で冷却して、析出させる方法で製造する。この様な操作をすることにより、ヌクレオシド1分子にポリヒドロキシ酸2分子が付加した形の共結晶乃至はアモルファスを得ることが出来る。加温は水浴上沸点付近まで行い、冷却は氷浴中で室温以下程度まで行うことが好ましい。斯くして得られた共結晶またはアモルファスは、優れた水溶性を示すとともに、皮膚から体内への移行性に非常に優れる。この様な効果により、医薬原体の生体利用性を高めることが出来る。
【0013】
斯くして得られた、共結晶、アモルファスは本発明の共結晶、アモルファスである。この様な共結晶、アモルファスとしては、例えば、アシクロビル・酒石酸共結晶、アシクロビル・クエン酸アモルファスが好適に例示できる。これらを特徴づける、物性値としては、例えば、アシクロビル・酒石酸共結晶であれば、分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストロームと言った共結晶を特徴付ける物性値を上げることが出来、アシクロビル・クエン酸アモルファスであれば、示差熱分析において、60〜80℃にガラス転移点を有することが挙げられる。尚、本発明に言うアモルファスとは、固形であって、粉末X線解析において、ピークを認めないものと定義できる。
【0014】
斯くして得られた、共結晶、アモルファスは、これを医薬原薬、或いは、中間処理品として、医薬組成物に配合することにより、溶解性に優れ、生体内移行性に優れる医薬組成物に加工することが出来る。この様な加工において、上記成分以外に、医薬組成物で使用される、可溶化剤、分散剤、賦形剤、結合剤、崩壊剤、被覆剤、矯味矯臭剤など添加することも出来る。また、前記加工は常法に従えばよい。
【0015】
以下に、実施例を示しながら、本発明について更に詳細に説明を加える。
【実施例1】
【0016】
<アシクロビル結晶多形の調整>
アシクロビル無水晶系1は、2/3水和物を溶解させたN,N−ジメチルホルムアミドからアセトニトリルを貧溶媒として再結晶により調製した。アシクロビル無水晶系2は、2/3水和物を180℃で1時間乾燥することにより調製した。アシクロビル2水和物は、無水晶系2を溶解させた水からの再結晶により調製した。
【0017】
<共結晶、アモルファスの調製>
アシクロビル無水晶系2を薬物として用いた。いくつかの共結晶formerが適切なモル比でジカルボン酸、高級脂肪酸、アミノ酸、尿素、ニコチンアミド、サッカリンの中から選択された。溶液法(溶媒の冷却もしくはSlow evaporation)とLiquid−assisted(Solvent−drop)共粉砕法が共結晶とアモルファスのスクリーニングに用いられた。溶媒として、水、メタノール、エタノール、アセトニトリル、クロロホルム、n−ヘキサン、シクロヘキサン、N,N−ジメチルホルムアミド、酢酸を用いた。いくつかの可能性のある共結晶が得られたが、再現良く得られる共結晶lはアシクロビル・酒石酸共結晶のみであった。N,N−ジメチルホルムアミドを用いた溶媒法により、アシクロビルとクエン酸の間でアモルファス性の複合体が得られた。
【0018】
<アシクロビル・酒石酸共結晶の調製>
アシクロビル無水晶系2(225.21mg,1mmol)とL−酒石酸(150.09mg,1mmol)の1:1混合物を30mLの酢酸に60℃で一定の攪拌で溶解させた。得られた溶液をろ過し、室温まで冷却した。得られた結晶をろ過し、室温においてシリカゲルを充填したデシケータ中で乾燥させた。アシクロビル・酒石酸共結晶はまたN,N−ジメチルホルムアミドを用いたSolvent−drop共粉砕法でも調製できた。
【0019】
<アシクロビル・クエン酸からなるアモルファスの調製>
アシクロビル無水晶系2(225.21mg,1mmol)と過剰量の無水クエン酸(1921.24mg,10mmol)を30mLのN,N−ジメチルホルムアミドに60℃で一定の攪拌で懸濁させた。得られたホットサスペンジョンを素早くろ過し、それから溶媒をゆっくり蒸発させた。得られた共沈物を室温においてシリカゲルを充填したデシケータ中で乾燥させて、表記のアシクロビル・クエン酸からなるアモルファスを得た。
【0020】
<物理的混合物の調製>
アシクロビル無水晶系2とL−酒石酸を1:1でボルテックスミキサーを用いて一定振幅で10分間混合し、アシクロビル・酒石酸物理的混合物(ACV−TA−PM)とした。
アシクロビル無水晶系2と無水クエン酸を1:2でボルテックスミキサーを用いて一定振幅で10分間混合し、アシクロビル・クエン酸物理的混合物(ACV−CA−PM)とした。
【0021】
<検体の固体分析>
上記の手技で得られた各種結晶、共結晶、アモルファスを以下のように固体分析を行った。
【0022】
<粉末X線回折測定>
全ての試料のPXRDパターンは、グラファイトモノクロメーターと0.3mmシングルピンホールコリメーターで線源にCuKαを用いたBruker D8 Discover with a GADDS CS(Bruker AXS GMBH、カールスルーエ、ドイツ)を用いて反射法により収集された。管電圧と管電流はそれぞれ40kVと40mAにセットした。この回折計はXYZサンプルステージと試料から25cm(2θ=5〜25°)の距離に設置されたHi−STAR二次元検出器を装備している。捕捉時間は180秒/フレームとした。
【0023】
<TG/DTA(示差熱天秤)による分析>
TG/DTAはThermo Plus TG−8120(リガク、東京、日本)を用いて行われた。全ての測定はオープンAlパンを用いて、昇温速度10℃/minで窒素パージの下、25〜300℃の範囲で行われた。全ての試料は正確に秤量された(1〜7mg)。
【0024】
<赤外吸収スペクトル>
試料のIRスペクトルを収集するためにFT/IR−4100(日本分光、東京、日本)がATR法により用いられた。各試料のスペクトルは1000〜4000cm−1の範囲でスキャン回数32回、分解能4cm−1で収集された。
【0025】
<示差熱量計(DSC)による分析>
Perkin Elmer DSC−7(パーキンエルマー(株)、マサチューセッツ、米国)がアモルファス性複合体のTgを決定するために用いられた。試料(3mg)が非密閉Alパンにクランプされ、昇温速度10℃/minで窒素パージの下、−40〜100℃までスキャンされた。
【0026】
<薬剤/ポリヒドロキシカルボン酸の質量比>
薬物/添加物の配合比を決定するために、共結晶またはアモルファス中の薬物含量がHPLCより分析された。添加物含量は総重量から薬物含量を差し引くことで算出された。
【0027】
<放射光粉末X線回折測定>
アシクロビル・酒石酸共結晶の放射光粉末X線回折データがPhoton Factory(筑波、日本)のビームラインBL4B2で多連装検出器を用いて波長1.19601オングストローム、温度300Kの条件で収集された。結晶構造はシミュレーテッド・アニーリング法によりDASHを用いて解明された。この計算において、測定されたパウダーパターンは関連する積分強度を抽出するために、空間群P21でPawley法により精密化された。データに最もフィットした構造が、シミュレーテッド・アニーリング法で得られた分率座標をリートベルト法により精密化することでバリデートされた。リートベルト法による精密化はGSASを用いて行った。
【0028】
<結果>
スクリーニングの結果を表1にまとめた。アシクロビルと酒石酸の組み合わせによるスクリーニングから共結晶が形成されることを確認した。その新しい共結晶はもたらした溶媒はN,N−ジメチルホルムアミドであった。また、アシクロビルとクエン酸の組み合わせでアモルファス性の複合体が得られた。アシクロビルとクエン酸はN,N−ジメチルホルムアミドの効果によって、アモルファス化した。アシクロビル(無水晶系1及び無水晶系2、2/3水和物、2水和物)、酒石酸、アシクロビル・酒石酸共結晶(ACV−TAcocrystal)及びアシクロビル・クエン酸アモルファス(ACV−CA amorphous)のPXRDパターンを図1に示した。アシクロビル・酒石酸共結晶(ACV−TA cocrystal)特有のPXRDパターンはアシクロビルや酒石酸と区別できるものであった。アシクロビル・クエン酸アモルファスにおいては2θが5°から25°の範囲でハローパターンを除くピークが観察されなかった。これより、共結晶乃至はアモルファスはポリヒドロキシカルボン酸を対成分とした場合にのみ現れること、及び、溶剤としては、水と混和しうる有機溶剤であるジメチルホルムアミドのような溶剤の時のみに現れることが判る。
【0029】
【表1】

【0030】
アシクロビル・酒石酸共結晶の物性がTG/DTAとIRによって、より詳細に調べられた。図2に示したように、アシクロビル・酒石酸共結晶のTG/DTA曲線もまたアシクロビルや酒石酸と区別できるものであった。さらに、TG/DTA曲線は溶媒和結晶でないことを示した。アシクロビル(無水晶系1及び無水晶系2)、酒石酸、アシクロビル・酒石酸共結晶のIRスペクトルを図3に示した。アシクロビル無水物のIRスペクトルにおいて、3300〜3500cm−1の領域に第1級アミンと第2級アミンに相当する2つのピークがあった。さらに、1600cm−1の領域にアミドによるピークが示された(図3a,b)。酒石酸のスペクトルにおいては、OH基に由来する2つのピークが3405cm−1と3336cm−1に存在する。さらに、1737cm−1はC=Oの伸縮振動を示している(図3c)。アシクロビルの第1級アミンに由来するN−H伸縮振動と酒石酸のカルボキシル基に由来するO−H伸縮振動がアシクロビル・酒石酸共結晶においてそれぞれ3476cm−1と3328cm−1に観察された(図3d)。このことはアシクロビルと酒石酸の分子が新しい相に存在していることを示している。アシクロビル無水物に由来するN−H伸縮振動が3432cm−1から3476cm−1に増加していることは、第1級アミンが弱い水素結合に関与していることを暗に示している。さらに、アシクロビル無水物に由来するN−Hの変角振動(アミド)が1599cm−1から1592cm−1に減少していることは、アミドのN−H基が強い水素結合に関与していることを暗に示している。次に、酒石酸に由来するO−H伸縮振動が3336cm−1から3328cm−1に減少していることは、カルボキシル基が強い水素結合に関与していることを示している。酒石酸におけるC=Oの伸縮振動が1737cm−1から1710cm−1に深色シフトしているがアシクロビル・酒石酸共結晶の形成をさらに説明している。
アシクロビル(無水晶系1及び無水晶系2)、クエン酸、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスのIRスペクトルを図4に示した。クエン酸のIRスペクトルにおいて、3495cm−1と3287cm−1に無水クエン酸に特徴的なOH基に由来するピークが確認された(図4c)。中間の振動領域においては、1698cm−1で無水クエン酸のC=O伸縮振動を検出した。1690〜1720cm−1のC=O伸縮振動の領域では通常二量体化が起こっている。1746cm−1のピークは水素結合により低波数シフトしたO−H伸縮振動に関連している。アシクロビル・クエン酸物理的混合物のIRスペクトルにおいてはピークシフトは確認されなかった(図4d)。このことは、物理的混合物のスペクトルがアシクロビルとクエン酸の単なる合算であり、アシクロビルのクエン酸の間で相互作用が起こっていないことを示している。一方、アシクロビル・クエン酸アモルファスにおいては、クエン酸のOH基に由来する3495cm−1のピークが消失し、アシクロビルのN−H伸縮振動に由来する3396cm−1のピークが3402cm−1へシフトした(図5e)。さらに、クエン酸のO−H伸縮振動に由来する1746cm−1のピークが消失もしくはC=O伸縮振動のピークと重複した。これらの結果は、アシクロビル・クエン酸アモルファスにおいて、アシクロビルとクエン酸の間で水素結合を介した相互作用が起こっている可能性を示している。
【0031】
図5はアシクロビル・クエン酸アモルファスのDSC曲線を示している。DSC曲線はこの複合体について1つのガラス転移点を示したが、これはアシクロビルとクエン酸がアモルファス状態で混和していることを示唆している。アシクロビル・クエン酸アモルファスのガラス転移温度(Tg)は68℃であった。このことは、この2成分系アモルファスが室温より高いことを示している。つまり、この2成分系アモルファスは室温で安定であると考えられた。
【0032】
アシクロビル・酒石酸共結晶とアシクロビル・クエン酸アモルファスにおけるアシクロビル含量がHPLCにより決定された。共結晶中の薬物含量は59.44±0.81(%)であった。その結果、添加物含量は40.56±0.81(%)と算出された。2つの成分の理論含量が60.01%と39.99%であることから、アシクロビルと酒石酸のモル比は1:1であると決定された。一方、アモルファス中のアシクロビルとクエン酸の含量はそれぞれ37.26±0.73(%)と62.74±0.73(%)であった。2つの成分の理論含量が36.95%と63.05%であることから、アシクロビルとクエン酸のモル比は1:2であると決定された。
【実施例2】
【0033】
実施例1で得た、アシクロビル・酒石酸共結晶について、その溶解特性を調べた。即ち、蒸留水、pH6.8リン酸バッファー、エタノールに対するアシクロビルとアシクロビル・酒石酸共結晶の飽和溶解度を決定するために、約500mgの各試料が10mLの各溶媒に加えられた。その後、得られた懸濁液を、マグネティックスターラーを用いて500rpmで25℃において24時間攪拌した。懸濁液の一部を0.45μmのフィルターでろ過した。ろ過した試料1mLを蒸留水で希釈して100mLとした。それから、アシクロビル濃度を決定するために、その希釈液をHPLCで分析した。
【0034】
共結晶の初期溶出速度を決定するために、溶出試験機(TDS−30、富山産業(株)、大阪、日本)を用いた溶出試験が行われた。この溶出試験において、約100mgの各試料(アシクロビル多形、アシクロビル・酒石酸共結晶)を直径0.45cmの穴を持つ金型を用いて、1ton/gで1分間圧力をかけることによって、0.2cm2のディスクに圧縮成型した。このディスクは金型の片面で平滑な表面を曝露している。もう一方の面は試験液に触れないように密閉した。各実験において、3連の1000mL溶出試験ベッセルにpH6.8のリン酸バッファーを900mL入れ、37.0℃、パドル回転数50rpmで平衡化した。全ての試料について、0〜5分のデータを用いて初期溶出速度を算出した。薬物の濃度はHPLCにより分析した。
【0035】
本発明で用いられたHPLC分析の条件は、以下の通り。高速液体クロマトグラフィーシステム(HPLC;Agilent 1100,Agilent Technologies,Inc.、カリフォルニア、米国)が用いられた。このHPLCシステムではC18カラム(Inertsil ODS−3 5μm 4.6×250mm、ジーエルサイエンス(株)、東京、日本)を用いた。水/アセトニトリル/酢酸(959:40:1)から成る移動相がカラムに注入された。UV検出は254nmで行われた。
【0036】
蒸留水、pH6.8リン酸バッファー、エタノールに対するアシクロビル・酒石酸共結晶の溶解性をアシクロビル無水晶系2及びアシクロビル・酒石酸物理的混合物のそれと比較するために、HPLCにより、アシクロビル・酒石酸共結晶、アシクロビル・酒石酸物理的混合物、アシクロビル無水晶系2の各溶媒に対する飽和溶解度が決定された。図7に示したように、アシクロビル・酒石酸共結晶の各溶媒に対する飽和溶解度は、それぞれ6.83±0.36(mg/mL)、5.96±0.01(mg/mL)、0.93±0.01(mg/mL)であった。一方で、アシクロビル無水晶系2の各溶媒に対する飽和溶解度は1.09±0.01(mg/mL)、1.08±0.07(mg/mL)、0.13±0.00(mg/mL)であった。アシクロビル・酒石酸物理的混合物の各溶媒に対する飽和溶解度は6.03±0.61(mg/mL)、5.46±0.69(mg/mL)、0.89±0.07(mg/mL)であった。溶液中でも相互作用が起こっていると考えられるため、平衡状態における溶解度では共結晶と物理的混合物でほぼ同等であった。共結晶化することにより、優れた溶解度を示すことが判る。
【0037】
アシクロビル・酒石酸共結晶とアシクロビル多形及びアシクロビル・酒石酸物理的混合物の初期溶出速度を比較するために、溶出試験が行われた。図8に各試料の初期溶出プロファイルを示した。溶出プロファイルで示したように、アシクロビル・酒石酸共結晶はアシクロビル多形及び物理的混合物と比較すると即座に溶解した。その初期溶出速度はアシクロビル多形及び物理的混合物の3〜6倍高かった。
【0038】
これらの結果はアシクロビルの溶解性が共結晶化によって改善されたことを示している。
【実施例3】
【0039】
PEG400に対するアシクロビル結晶、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスの飽和溶解度を決定するために、約50mgの各試料が5mLのPEG400に加えられた。その後、得られた懸濁液を、マグネティックスターラーを用いて500rpmで25℃において24時間攪拌した。懸濁液の一部を0.45μmのフィルターでろ過した。ろ過した試料1mLを蒸留水で希釈して10mLとした。それから、アシクロビル濃度を決定するために、その希釈液をHPLCで分析した。
【0040】
マクロゴール軟膏基剤に対するアシクロビル・クエン酸アモルファスの溶解性を結晶性アシクロビルのそれと比較するために、HPLCにより、アシクロビル・クエン酸アモルファスと結晶性アシクロビルと結晶性アシクロビルとクエン酸の物理的混合物のPEG400に対する飽和溶解度が決定された。図9に示したように、アシクロビル・クエン酸アモルファスのPEG400に対する25℃での飽和溶解度は結晶性アシクロビルによりかなり高い結果となった。アシクロビル・クエン酸アモルファス、結晶性アシクロビル、物理的混合物の飽和溶解度はそれぞれ0.64±0.01(mg/mL)、0.12±0.01(mg/mL)、0.09±0.00(mg/mL)であった。物理的混合物の飽和溶解度は結晶性アシクロビルとほぼ同程度であった。
【実施例4】
【0041】
以下の処方に従って、軟膏を作成した。即ち、80℃で処方成分を溶解し、攪拌可溶化した後、攪拌冷却して軟膏(マクロゴール軟膏)を得た。また、このもののアシクロビル・クエン酸アモルファスを結晶性アシクロビルに置換した比較例及びアシクロビル・クエン酸物理的混合物に置換した比較例の軟膏も同様に作成した。
【0042】
【表2】
【0043】
5%アシクロビルまたは5%アシクロビル・クエン酸物理的混合物または5%アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏の光学顕微鏡写真を図10に示した。5%アシクロビル含有マクロゴール軟膏と5%アシクロビル・クエン酸物理的混合物含有マクロゴール軟膏において、薬物は軟膏基剤に対する溶解性が低いために、ほとんどが結晶化していた。一方、5%アシクロビル・クエン酸アモルファス含有マクロゴール軟膏において、薬物は一部結晶化していたが、軟膏基剤中にほとんど溶解していた。斯くの如くに、アモルファスとすることにより、優れた溶解特性を示すことが判る。
【0044】
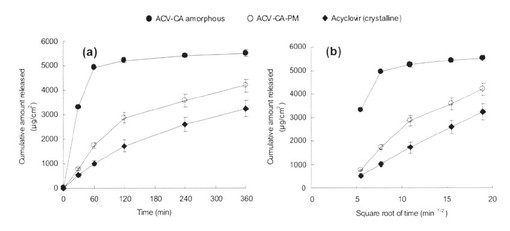
この様に作成した、アシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro放出性を比較するため、フランツ型拡散セル(HansonResearch,Inc.、カリフォルニア、米国)を用いて3回繰り返し実験を行った。約300mgの各製剤(5%アシクロビル軟膏、5%アシクロビル・クエン酸物理的混合物軟膏5%、アシクロビル・クエン酸アモルファス軟膏)を用いた。ナイロン膜(HNWP02500、厚さ170μm)孔径0.45μm(日本ミリポア(株)、東京、日本)を用いた。拡散セルの温度は32℃に保たれたウォータージャケットによって維持され、それから、32℃に保持された7mLの放出液(pH6.8リン酸バッファー)をレセプターチャンバーに満たした。実験の間、放出液はマグネティックスターラーを用いて500rpmで連続的に攪拌した。既定の時間で1mLの放出液をレセプターチャンバーから回収し、32℃に保持された同じ容量の放出液をチャンバー内に戻した。この放出実験は360分まで継続した。アシクロビルの累積放出量を算出するために、回収された放出液中のアシクロビル濃度をHPLCにより測定した。マクロゴール軟膏中からのアシクロビル放出速度を得るために、アシクロビル累積放出量が時間の平方根に対してプロットされた。アモルファス化による促進効果を評価するために、アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏からのアシクロビル放出速度を結晶性のアシクロビルを含有するマクロゴール軟膏からの放出速度と比較した。
【0045】
アシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro放出プロファイルを図11aに示す。この放出プロファイルからアシクロビル・クエン酸アモルファスの薬物放出速度が結晶性アシクロビルと比較して顕著に高く、放出率は60分でほぼ100%となることがわかった。全てのデータにHiguchi式を適用し、図11bにおいて累積放出量を時間の平方根に対してプロットした。アシクロビル・クエン酸アモルファスにおいて、leveling−offがプロファイルの後半のポイントで観察された。この現象は軟膏基剤中からの固体薬物の消失によるものと考えられた。このことはアモルファスの溶解度が結晶より高かったことにより、後半のポイントでアモルファス固体が既に排出しきっていることを示唆している。斯くの如くに、アシクロビル・クエン酸アモルファスを含有する軟膏は、優れた薬剤放出特性を有していることが判る。
【実施例5】
【0046】
薬物のアモルファス化による促進効果を評価するために、in vitroにおいて、アモルファスのアシクロビル(アシクロビル・クエン酸アモルファス)を含有するマクロゴール軟膏の皮膚透過性が、結晶性のアシクロビルを含有するマクロゴール軟膏と比較された。ラボスキン(星野試験動物飼育所(株)、茨城、日本)が皮膚試料として用いられた。ラボスキンは、7週齢の雄のヘアレスマウス(Hos:HR−1)の背部から剥離した皮膚試料であり、−20℃で冷凍保存されている。実験の直前に皮膚を室温でゆっくり解凍し、フランツ型拡散セルにマウントするために切り分けた。拡散セルは放出試験で用いたものと同じものを用いた。レセプター液もまた放出試験で用いたものと同じものを用いた(つまり、pH6.8リン酸バッファー)。拡散セルの温度は37℃に保たれたウォータージャケットによって維持された。透過は約200mgのマクロゴール軟膏を皮膚の上に適用することで開始された。それから、規定の時間で1mLのレセプター液をレセプターチャンバーから回収し、37℃に保持された同じ容量の放出液をチャンバー内に戻した。この透過試験は26時間まで継続した。実験の間、レセプター液はマグネティックスターラーを用いて500rpmで連続的に攪拌した。HPLCにより測定された回収したレセプター液中のアシクロビル濃度から、皮膚単位面積当たりのアシクロビル累積透過量が算出された。各マクロゴール軟膏について、3回透過試験が行われた。アシクロビル累積透過量は透過プロファイルを得るためにサンプリング時間に対してプロットされ、その透過プロファイルの直線部分から皮膚透過速度を算出した。アモルファス化による促進効果を評価するために、アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏の透過速度と結晶性のアシクロビルを含有するマクロゴール軟膏の透過速度を比較した。
【0047】
結果は、図12にアシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro皮膚透過プロファイルとして示す。図12の挿入図は結晶性アシクロビルと物理的混合物のプロファイルの拡大図である。アシクロビル・クエン酸アモルファスのプロファイルを結晶性アシクロビルと物理的混合物のそれと比較した。透過における定常状態はアモルファスにおいて2〜26時間の間で観察された。アモルファスの定常状態における透過速度は2.06μg/cm2/hであった。一方、結晶性アシクロビルまたはアシクロビル・クエン酸物理的混合物のマクロゴール軟膏中からの送達は非常に遅く、透過における定常状態は22〜26時間の間で確認された。結晶性アシクロビルの透過速度はそれぞれ0.02μg/cm2/h、0.09μg/cm2/hであった。透過速度は薬物のアモルファス化により顕著に増加した。これはin vitro薬物放出性の結果とも相関していた。
即ち、本発明のアシクロビル・クエン酸アモルファス体は皮膚外用剤に好ましい、優れた皮膚透過性を有していることが判る。
【産業上の利用可能性】
【0048】
本発明は、皮膚外用医薬などに応用できる。
【技術分野】
【0001】
本発明は、アシクロビルの結晶を改変した組成物及び当該組成物の製造方法に関する。
【背景技術】
【0002】
医薬品において、その有効成分、APIの性状をコントロールすることは重要な課題となる。例えば、結晶系をコントロールし、医薬品として好ましい性状を得る技術は既に医薬品特許の分野では、「結晶多形特許」、「水和物特許」というパターンを形成している。また、研究の分野でも、結晶系、溶媒和などをコントロールし、薬動力学的利用性を高める試みが盛んに為されている。(例えば、特許文献1、特許文献2、特許文献3、特許文献4、非特許文献1、非特許文献2、非特許文献3を参照)その反面、生体利用性が低く、しかも、アモルファス化しにくく、更には、好ましい他の結晶系の結晶も存しない薬剤は存在し、その生体利用性を高めることは一つの課題となっている。この様な物質としては、アシクロビル、ガンシクロビル、ビダラビン、5−FUのような、ヌクレオシド系の医薬、特に、抗ウイルス剤が典型的な物質として、好適に例示できる。この様な物質に対して、物理的性状を変えて、その生体利用性を高める手段としては、例えば、クレアチニンやグリシンとともに凍結乾燥し、アモルファス化する方法が考えられるが(例えば、特許文献5を参照)、通常のAPIではコストがかかりすぎ、コストメリットバランス上、実施に至る物はほとんどないのが現状である。
【0003】
一方、他の結晶性成分とともに、共結晶形成せしめ、物理的性状を変える試みも為されている(例えば、特許文献6、特許文献7を参照)が、アシクロビルとポリヒドロキシカルボン酸との共結晶も、アモルファス物質も知られていない。また、この様な共結晶やアモルファスを形成することにより、溶解度や生体利用性が高まることも全く知られていない。
【先行技術文献】
【特許文献】
【0004】
【特許文献1】特開平5−43590号公報
【特許文献2】特開平7−316141号公報
【特許文献3】特開平01−38096号公報
【特許文献4】特開平01−250384号公報
【特許文献5】WO/2008/152764
【特許文献6】特表2011−516567号公報
【特許文献7】特表2007−524596号公報
【非特許文献】
【0005】
【非特許文献1】Berge,S.M.,Bighley,L.D.,Monkhouse,D.C.,1977.Pharmaceutical salts.J.Pharm.Sci.66,1−19
【非特許文献2】Kato,Y.,Okamoto,Y.,Nagasawa,S.,Ueki,T.,1981.Solubility of a new polymorph of phenobarbital obtained by crystallization in the presence of phenytoin.Chem.Pharm.Bull.29,3410−3413
【非特許文献3】Serajuddin,A.T.M.,1999,Solid dispersion of poorly water−soluble drugs:early promises,subsequent problems,and recent breakthroughs.J.Pharm.Sci.88,1058−1066.
【発明の概要】
【発明が解決しようとする課題】
【0006】
本発明は、この様な状況下為されたものであり、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供すること課題とする。
【課題を解決するための手段】
【0007】
この様な状況に鑑みて、本発明者らは、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を求めて、鋭意研究努力を重ねた結果、極性溶媒を用いて、ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを加熱溶解、冷却析出させることにより、当該医薬原体とポリヒドロキシカルボン酸の共結晶乃至はアモルファスを得ることが出来、かかる共結晶、アモルファスが非常に好ましい物性を有していることを見出し、発明を完成させるに至った。即ち、本発明は、以下に示すとおりである。
<1>ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することを特徴とする、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<2>前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることを特徴とする、<1>に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸との共結晶又はアモルファスの製造法。
<3>前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることを特徴とする、<1>又は<2>に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<4>前記極性溶媒は、酢酸及び/又はジアルキルホルムアミドを含むものであることを特徴とする、<1>〜<3>何れか1項に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
<5>アシクロビルと酒石酸の共結晶。
<6>次の物性を有することを特徴とする、<5>に記載の共結晶。
分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストローム
<7><1>〜<4>何れか1項に記載の製造方法で製造された物であることを特徴とする、<5>または<6>に記載の共結晶。
<8>アシクロビルとクエン酸からなる、アモルファス。
<9>示差熱分析において、60〜80℃にガラス転移点を有することを特徴とする、<8>に記載のアモルファス。
<10><1>〜<4>何れか1項に記載の製造方法で製造された物であることを特徴とする、<8>または<9>に記載のアモルファス。
<11><5>〜<7>何れか1項に記載の共結晶及び/又は<8>〜<10>何れか1項に記載のアモルファスを有効成分として含有することを特徴とする、医薬組成物。
<12>皮膚外用剤であることを特徴とする、<11>に記載の医薬組成物。
【発明の効果】
【0008】
本発明によれば、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供できる。
【図面の簡単な説明】
【0009】
【図1】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、アシクロビル2/3水和物(c)、アシクロビル2水和物(d)、酒石酸(e)、アシクロビル・酒石酸共結晶(f)、アシクロビル・クエン酸アモルファス(g)の粉末X線解析を示す図である。
【図2】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、アシクロビル2/3水和物(c)、アシクロビル2水和物(d)、酒石酸(e)、アシクロビル・酒石酸共結晶(f)のTG/DTAを示す図である。
【図3】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、酒石酸(c)、アシクロビル・酒石酸共結晶(d)のIRスペクトルを示す図である。
【図4】アシクロビル無水晶系1(a)、アシクロビル無水晶系2(b)、クエン酸(c)、アシクロビル・クエン酸物理的混合物(d)、アシクロビル・クエン酸アモルファス(e)のIRスペクトルを示す図である。
【図5】アシクロビル・クエン酸アモルファスのDSCの測定結果を示す図である。
【図6】アシクロビル・酒石酸共結晶のab平面と、bc平面を示す図である。
【図7】アシクロビル無水晶系2、アシクロビル・酒石酸物理的混合物、アシクロビル・酒石酸共結晶の蒸留水、リン酸バッファー、エタノールに対する飽和溶解度を示す図である。
【図8】アシクロビル無水晶系1、アシクロビル無水晶系2、アシクロビル2/3水和物、アシクロビル2水和物、アシクロビル・酒石酸物理的混合物、アシクロビル・酒石酸共結晶のリン酸バッファーにおける溶解速度比較を示す図である。
【図9】アシクロビル結晶、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスのPEG−400に対する飽和溶解度を示す図である。
【図10】PEG軟膏の顕微鏡観察の結果を示す図である。(図面代用写真)
【図11】PEG軟膏からのアシクロビルの放出特性を示す図である。
【図12】PEG軟膏における、アシクロビルのヘアレスマウスの皮膚からの透過特性を示す図である。
【発明を実施するための形態】
【0010】
<1>本発明の製造法の対象とする医薬原体
本発明は、アシクロビル等のヌクレオシド系の医薬の物理的性状を改変し、生体利用性を高める手段を提供することを特徴とし、その対象となる医薬原体はヌクレオシド系医薬原体である。ヌクレオシド系医薬原体とは、プリン塩基またはピリミジン塩基乃至はそれらの誘導体とそれと結合した直鎖乃至は環状の糖からなる構造を有するものであり、糖の水酸基はリン酸化されていない物を意味する。この様な医薬原体としては、例えば、アシクロビル、ガンシクロビル、ビダラビン、AZT、テガフールなどが好適に例示できる。この中では、アシクロビルが特に好ましい。これは、その溶解性の悪さ故に効果発現が抑制されていると考えられるためである。
【0011】
<2>本発明において医薬原体と対になるポリヒドロキシカルボン酸
本発明においては、前記ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを共結晶またはアモルファスに加工することを主旨とする。ここで、ポリヒドロキシカルボン酸としては、2個以上の水酸基を有し、且つ、カルボキシル基も有するものが好適に例示でき、具体的には、酒石酸、リンゴ酸、グルコン酸、クエン酸などが好適に例示でき、特に好ましくは、酒石酸またはクエン酸である。かかる成分は、アモルファスの場合、前記ヌクレオシド系医薬原体とほぼ2:1で複合体を形成するため、ヌクレオシド系医薬原体の少なくとも2質量倍含有されることが好ましい。また、共結晶の場合、前記ヌクレオシド系医薬原体とほぼ1:1で複合体を形成するため、ヌクレオシド系医薬原体の少なくとも等質量含有されることが好ましい。
【0012】
<3>本発明において使用される極性溶剤
本発明において、前記ヌクレオシド系医薬原体と、ポリヒドロキシカルボン酸とを共結晶乃至はアモルファスにするために使用される極性溶剤としては、水と混和しうる溶剤であって、親油性の成分も溶かしうる物が好ましく、具体的には、ギ酸、酢酸、プロピオン酸などの低鎖長カルボン酸、ジメチルホルムアミド、ジエチルホルムアミド、ジメチルスルホキシド、エチルセルソルブ、メチルセルソルブなどが好適に例示でき、酢酸などの低鎖長カルボン酸及び/又はジメチルホルムアミドなどのジアルキルホルムアミドが特に好適に例示できる。かかる極性溶媒を、ヌクレオシド系医薬原体の5〜20質量倍用い、加温して溶解後、氷等で冷却して、析出させる方法で製造する。この様な操作をすることにより、ヌクレオシド1分子にポリヒドロキシ酸2分子が付加した形の共結晶乃至はアモルファスを得ることが出来る。加温は水浴上沸点付近まで行い、冷却は氷浴中で室温以下程度まで行うことが好ましい。斯くして得られた共結晶またはアモルファスは、優れた水溶性を示すとともに、皮膚から体内への移行性に非常に優れる。この様な効果により、医薬原体の生体利用性を高めることが出来る。
【0013】
斯くして得られた、共結晶、アモルファスは本発明の共結晶、アモルファスである。この様な共結晶、アモルファスとしては、例えば、アシクロビル・酒石酸共結晶、アシクロビル・クエン酸アモルファスが好適に例示できる。これらを特徴づける、物性値としては、例えば、アシクロビル・酒石酸共結晶であれば、分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストロームと言った共結晶を特徴付ける物性値を上げることが出来、アシクロビル・クエン酸アモルファスであれば、示差熱分析において、60〜80℃にガラス転移点を有することが挙げられる。尚、本発明に言うアモルファスとは、固形であって、粉末X線解析において、ピークを認めないものと定義できる。
【0014】
斯くして得られた、共結晶、アモルファスは、これを医薬原薬、或いは、中間処理品として、医薬組成物に配合することにより、溶解性に優れ、生体内移行性に優れる医薬組成物に加工することが出来る。この様な加工において、上記成分以外に、医薬組成物で使用される、可溶化剤、分散剤、賦形剤、結合剤、崩壊剤、被覆剤、矯味矯臭剤など添加することも出来る。また、前記加工は常法に従えばよい。
【0015】
以下に、実施例を示しながら、本発明について更に詳細に説明を加える。
【実施例1】
【0016】
<アシクロビル結晶多形の調整>
アシクロビル無水晶系1は、2/3水和物を溶解させたN,N−ジメチルホルムアミドからアセトニトリルを貧溶媒として再結晶により調製した。アシクロビル無水晶系2は、2/3水和物を180℃で1時間乾燥することにより調製した。アシクロビル2水和物は、無水晶系2を溶解させた水からの再結晶により調製した。
【0017】
<共結晶、アモルファスの調製>
アシクロビル無水晶系2を薬物として用いた。いくつかの共結晶formerが適切なモル比でジカルボン酸、高級脂肪酸、アミノ酸、尿素、ニコチンアミド、サッカリンの中から選択された。溶液法(溶媒の冷却もしくはSlow evaporation)とLiquid−assisted(Solvent−drop)共粉砕法が共結晶とアモルファスのスクリーニングに用いられた。溶媒として、水、メタノール、エタノール、アセトニトリル、クロロホルム、n−ヘキサン、シクロヘキサン、N,N−ジメチルホルムアミド、酢酸を用いた。いくつかの可能性のある共結晶が得られたが、再現良く得られる共結晶lはアシクロビル・酒石酸共結晶のみであった。N,N−ジメチルホルムアミドを用いた溶媒法により、アシクロビルとクエン酸の間でアモルファス性の複合体が得られた。
【0018】
<アシクロビル・酒石酸共結晶の調製>
アシクロビル無水晶系2(225.21mg,1mmol)とL−酒石酸(150.09mg,1mmol)の1:1混合物を30mLの酢酸に60℃で一定の攪拌で溶解させた。得られた溶液をろ過し、室温まで冷却した。得られた結晶をろ過し、室温においてシリカゲルを充填したデシケータ中で乾燥させた。アシクロビル・酒石酸共結晶はまたN,N−ジメチルホルムアミドを用いたSolvent−drop共粉砕法でも調製できた。
【0019】
<アシクロビル・クエン酸からなるアモルファスの調製>
アシクロビル無水晶系2(225.21mg,1mmol)と過剰量の無水クエン酸(1921.24mg,10mmol)を30mLのN,N−ジメチルホルムアミドに60℃で一定の攪拌で懸濁させた。得られたホットサスペンジョンを素早くろ過し、それから溶媒をゆっくり蒸発させた。得られた共沈物を室温においてシリカゲルを充填したデシケータ中で乾燥させて、表記のアシクロビル・クエン酸からなるアモルファスを得た。
【0020】
<物理的混合物の調製>
アシクロビル無水晶系2とL−酒石酸を1:1でボルテックスミキサーを用いて一定振幅で10分間混合し、アシクロビル・酒石酸物理的混合物(ACV−TA−PM)とした。
アシクロビル無水晶系2と無水クエン酸を1:2でボルテックスミキサーを用いて一定振幅で10分間混合し、アシクロビル・クエン酸物理的混合物(ACV−CA−PM)とした。
【0021】
<検体の固体分析>
上記の手技で得られた各種結晶、共結晶、アモルファスを以下のように固体分析を行った。
【0022】
<粉末X線回折測定>
全ての試料のPXRDパターンは、グラファイトモノクロメーターと0.3mmシングルピンホールコリメーターで線源にCuKαを用いたBruker D8 Discover with a GADDS CS(Bruker AXS GMBH、カールスルーエ、ドイツ)を用いて反射法により収集された。管電圧と管電流はそれぞれ40kVと40mAにセットした。この回折計はXYZサンプルステージと試料から25cm(2θ=5〜25°)の距離に設置されたHi−STAR二次元検出器を装備している。捕捉時間は180秒/フレームとした。
【0023】
<TG/DTA(示差熱天秤)による分析>
TG/DTAはThermo Plus TG−8120(リガク、東京、日本)を用いて行われた。全ての測定はオープンAlパンを用いて、昇温速度10℃/minで窒素パージの下、25〜300℃の範囲で行われた。全ての試料は正確に秤量された(1〜7mg)。
【0024】
<赤外吸収スペクトル>
試料のIRスペクトルを収集するためにFT/IR−4100(日本分光、東京、日本)がATR法により用いられた。各試料のスペクトルは1000〜4000cm−1の範囲でスキャン回数32回、分解能4cm−1で収集された。
【0025】
<示差熱量計(DSC)による分析>
Perkin Elmer DSC−7(パーキンエルマー(株)、マサチューセッツ、米国)がアモルファス性複合体のTgを決定するために用いられた。試料(3mg)が非密閉Alパンにクランプされ、昇温速度10℃/minで窒素パージの下、−40〜100℃までスキャンされた。
【0026】
<薬剤/ポリヒドロキシカルボン酸の質量比>
薬物/添加物の配合比を決定するために、共結晶またはアモルファス中の薬物含量がHPLCより分析された。添加物含量は総重量から薬物含量を差し引くことで算出された。
【0027】
<放射光粉末X線回折測定>
アシクロビル・酒石酸共結晶の放射光粉末X線回折データがPhoton Factory(筑波、日本)のビームラインBL4B2で多連装検出器を用いて波長1.19601オングストローム、温度300Kの条件で収集された。結晶構造はシミュレーテッド・アニーリング法によりDASHを用いて解明された。この計算において、測定されたパウダーパターンは関連する積分強度を抽出するために、空間群P21でPawley法により精密化された。データに最もフィットした構造が、シミュレーテッド・アニーリング法で得られた分率座標をリートベルト法により精密化することでバリデートされた。リートベルト法による精密化はGSASを用いて行った。
【0028】
<結果>
スクリーニングの結果を表1にまとめた。アシクロビルと酒石酸の組み合わせによるスクリーニングから共結晶が形成されることを確認した。その新しい共結晶はもたらした溶媒はN,N−ジメチルホルムアミドであった。また、アシクロビルとクエン酸の組み合わせでアモルファス性の複合体が得られた。アシクロビルとクエン酸はN,N−ジメチルホルムアミドの効果によって、アモルファス化した。アシクロビル(無水晶系1及び無水晶系2、2/3水和物、2水和物)、酒石酸、アシクロビル・酒石酸共結晶(ACV−TAcocrystal)及びアシクロビル・クエン酸アモルファス(ACV−CA amorphous)のPXRDパターンを図1に示した。アシクロビル・酒石酸共結晶(ACV−TA cocrystal)特有のPXRDパターンはアシクロビルや酒石酸と区別できるものであった。アシクロビル・クエン酸アモルファスにおいては2θが5°から25°の範囲でハローパターンを除くピークが観察されなかった。これより、共結晶乃至はアモルファスはポリヒドロキシカルボン酸を対成分とした場合にのみ現れること、及び、溶剤としては、水と混和しうる有機溶剤であるジメチルホルムアミドのような溶剤の時のみに現れることが判る。
【0029】
【表1】
【0030】
アシクロビル・酒石酸共結晶の物性がTG/DTAとIRによって、より詳細に調べられた。図2に示したように、アシクロビル・酒石酸共結晶のTG/DTA曲線もまたアシクロビルや酒石酸と区別できるものであった。さらに、TG/DTA曲線は溶媒和結晶でないことを示した。アシクロビル(無水晶系1及び無水晶系2)、酒石酸、アシクロビル・酒石酸共結晶のIRスペクトルを図3に示した。アシクロビル無水物のIRスペクトルにおいて、3300〜3500cm−1の領域に第1級アミンと第2級アミンに相当する2つのピークがあった。さらに、1600cm−1の領域にアミドによるピークが示された(図3a,b)。酒石酸のスペクトルにおいては、OH基に由来する2つのピークが3405cm−1と3336cm−1に存在する。さらに、1737cm−1はC=Oの伸縮振動を示している(図3c)。アシクロビルの第1級アミンに由来するN−H伸縮振動と酒石酸のカルボキシル基に由来するO−H伸縮振動がアシクロビル・酒石酸共結晶においてそれぞれ3476cm−1と3328cm−1に観察された(図3d)。このことはアシクロビルと酒石酸の分子が新しい相に存在していることを示している。アシクロビル無水物に由来するN−H伸縮振動が3432cm−1から3476cm−1に増加していることは、第1級アミンが弱い水素結合に関与していることを暗に示している。さらに、アシクロビル無水物に由来するN−Hの変角振動(アミド)が1599cm−1から1592cm−1に減少していることは、アミドのN−H基が強い水素結合に関与していることを暗に示している。次に、酒石酸に由来するO−H伸縮振動が3336cm−1から3328cm−1に減少していることは、カルボキシル基が強い水素結合に関与していることを示している。酒石酸におけるC=Oの伸縮振動が1737cm−1から1710cm−1に深色シフトしているがアシクロビル・酒石酸共結晶の形成をさらに説明している。
アシクロビル(無水晶系1及び無水晶系2)、クエン酸、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスのIRスペクトルを図4に示した。クエン酸のIRスペクトルにおいて、3495cm−1と3287cm−1に無水クエン酸に特徴的なOH基に由来するピークが確認された(図4c)。中間の振動領域においては、1698cm−1で無水クエン酸のC=O伸縮振動を検出した。1690〜1720cm−1のC=O伸縮振動の領域では通常二量体化が起こっている。1746cm−1のピークは水素結合により低波数シフトしたO−H伸縮振動に関連している。アシクロビル・クエン酸物理的混合物のIRスペクトルにおいてはピークシフトは確認されなかった(図4d)。このことは、物理的混合物のスペクトルがアシクロビルとクエン酸の単なる合算であり、アシクロビルのクエン酸の間で相互作用が起こっていないことを示している。一方、アシクロビル・クエン酸アモルファスにおいては、クエン酸のOH基に由来する3495cm−1のピークが消失し、アシクロビルのN−H伸縮振動に由来する3396cm−1のピークが3402cm−1へシフトした(図5e)。さらに、クエン酸のO−H伸縮振動に由来する1746cm−1のピークが消失もしくはC=O伸縮振動のピークと重複した。これらの結果は、アシクロビル・クエン酸アモルファスにおいて、アシクロビルとクエン酸の間で水素結合を介した相互作用が起こっている可能性を示している。
【0031】
図5はアシクロビル・クエン酸アモルファスのDSC曲線を示している。DSC曲線はこの複合体について1つのガラス転移点を示したが、これはアシクロビルとクエン酸がアモルファス状態で混和していることを示唆している。アシクロビル・クエン酸アモルファスのガラス転移温度(Tg)は68℃であった。このことは、この2成分系アモルファスが室温より高いことを示している。つまり、この2成分系アモルファスは室温で安定であると考えられた。
【0032】
アシクロビル・酒石酸共結晶とアシクロビル・クエン酸アモルファスにおけるアシクロビル含量がHPLCにより決定された。共結晶中の薬物含量は59.44±0.81(%)であった。その結果、添加物含量は40.56±0.81(%)と算出された。2つの成分の理論含量が60.01%と39.99%であることから、アシクロビルと酒石酸のモル比は1:1であると決定された。一方、アモルファス中のアシクロビルとクエン酸の含量はそれぞれ37.26±0.73(%)と62.74±0.73(%)であった。2つの成分の理論含量が36.95%と63.05%であることから、アシクロビルとクエン酸のモル比は1:2であると決定された。
【実施例2】
【0033】
実施例1で得た、アシクロビル・酒石酸共結晶について、その溶解特性を調べた。即ち、蒸留水、pH6.8リン酸バッファー、エタノールに対するアシクロビルとアシクロビル・酒石酸共結晶の飽和溶解度を決定するために、約500mgの各試料が10mLの各溶媒に加えられた。その後、得られた懸濁液を、マグネティックスターラーを用いて500rpmで25℃において24時間攪拌した。懸濁液の一部を0.45μmのフィルターでろ過した。ろ過した試料1mLを蒸留水で希釈して100mLとした。それから、アシクロビル濃度を決定するために、その希釈液をHPLCで分析した。
【0034】
共結晶の初期溶出速度を決定するために、溶出試験機(TDS−30、富山産業(株)、大阪、日本)を用いた溶出試験が行われた。この溶出試験において、約100mgの各試料(アシクロビル多形、アシクロビル・酒石酸共結晶)を直径0.45cmの穴を持つ金型を用いて、1ton/gで1分間圧力をかけることによって、0.2cm2のディスクに圧縮成型した。このディスクは金型の片面で平滑な表面を曝露している。もう一方の面は試験液に触れないように密閉した。各実験において、3連の1000mL溶出試験ベッセルにpH6.8のリン酸バッファーを900mL入れ、37.0℃、パドル回転数50rpmで平衡化した。全ての試料について、0〜5分のデータを用いて初期溶出速度を算出した。薬物の濃度はHPLCにより分析した。
【0035】
本発明で用いられたHPLC分析の条件は、以下の通り。高速液体クロマトグラフィーシステム(HPLC;Agilent 1100,Agilent Technologies,Inc.、カリフォルニア、米国)が用いられた。このHPLCシステムではC18カラム(Inertsil ODS−3 5μm 4.6×250mm、ジーエルサイエンス(株)、東京、日本)を用いた。水/アセトニトリル/酢酸(959:40:1)から成る移動相がカラムに注入された。UV検出は254nmで行われた。
【0036】
蒸留水、pH6.8リン酸バッファー、エタノールに対するアシクロビル・酒石酸共結晶の溶解性をアシクロビル無水晶系2及びアシクロビル・酒石酸物理的混合物のそれと比較するために、HPLCにより、アシクロビル・酒石酸共結晶、アシクロビル・酒石酸物理的混合物、アシクロビル無水晶系2の各溶媒に対する飽和溶解度が決定された。図7に示したように、アシクロビル・酒石酸共結晶の各溶媒に対する飽和溶解度は、それぞれ6.83±0.36(mg/mL)、5.96±0.01(mg/mL)、0.93±0.01(mg/mL)であった。一方で、アシクロビル無水晶系2の各溶媒に対する飽和溶解度は1.09±0.01(mg/mL)、1.08±0.07(mg/mL)、0.13±0.00(mg/mL)であった。アシクロビル・酒石酸物理的混合物の各溶媒に対する飽和溶解度は6.03±0.61(mg/mL)、5.46±0.69(mg/mL)、0.89±0.07(mg/mL)であった。溶液中でも相互作用が起こっていると考えられるため、平衡状態における溶解度では共結晶と物理的混合物でほぼ同等であった。共結晶化することにより、優れた溶解度を示すことが判る。
【0037】
アシクロビル・酒石酸共結晶とアシクロビル多形及びアシクロビル・酒石酸物理的混合物の初期溶出速度を比較するために、溶出試験が行われた。図8に各試料の初期溶出プロファイルを示した。溶出プロファイルで示したように、アシクロビル・酒石酸共結晶はアシクロビル多形及び物理的混合物と比較すると即座に溶解した。その初期溶出速度はアシクロビル多形及び物理的混合物の3〜6倍高かった。
【0038】
これらの結果はアシクロビルの溶解性が共結晶化によって改善されたことを示している。
【実施例3】
【0039】
PEG400に対するアシクロビル結晶、アシクロビル・クエン酸物理的混合物、アシクロビル・クエン酸アモルファスの飽和溶解度を決定するために、約50mgの各試料が5mLのPEG400に加えられた。その後、得られた懸濁液を、マグネティックスターラーを用いて500rpmで25℃において24時間攪拌した。懸濁液の一部を0.45μmのフィルターでろ過した。ろ過した試料1mLを蒸留水で希釈して10mLとした。それから、アシクロビル濃度を決定するために、その希釈液をHPLCで分析した。
【0040】
マクロゴール軟膏基剤に対するアシクロビル・クエン酸アモルファスの溶解性を結晶性アシクロビルのそれと比較するために、HPLCにより、アシクロビル・クエン酸アモルファスと結晶性アシクロビルと結晶性アシクロビルとクエン酸の物理的混合物のPEG400に対する飽和溶解度が決定された。図9に示したように、アシクロビル・クエン酸アモルファスのPEG400に対する25℃での飽和溶解度は結晶性アシクロビルによりかなり高い結果となった。アシクロビル・クエン酸アモルファス、結晶性アシクロビル、物理的混合物の飽和溶解度はそれぞれ0.64±0.01(mg/mL)、0.12±0.01(mg/mL)、0.09±0.00(mg/mL)であった。物理的混合物の飽和溶解度は結晶性アシクロビルとほぼ同程度であった。
【実施例4】
【0041】
以下の処方に従って、軟膏を作成した。即ち、80℃で処方成分を溶解し、攪拌可溶化した後、攪拌冷却して軟膏(マクロゴール軟膏)を得た。また、このもののアシクロビル・クエン酸アモルファスを結晶性アシクロビルに置換した比較例及びアシクロビル・クエン酸物理的混合物に置換した比較例の軟膏も同様に作成した。
【0042】
【表2】
【0043】
5%アシクロビルまたは5%アシクロビル・クエン酸物理的混合物または5%アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏の光学顕微鏡写真を図10に示した。5%アシクロビル含有マクロゴール軟膏と5%アシクロビル・クエン酸物理的混合物含有マクロゴール軟膏において、薬物は軟膏基剤に対する溶解性が低いために、ほとんどが結晶化していた。一方、5%アシクロビル・クエン酸アモルファス含有マクロゴール軟膏において、薬物は一部結晶化していたが、軟膏基剤中にほとんど溶解していた。斯くの如くに、アモルファスとすることにより、優れた溶解特性を示すことが判る。
【0044】
この様に作成した、アシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro放出性を比較するため、フランツ型拡散セル(HansonResearch,Inc.、カリフォルニア、米国)を用いて3回繰り返し実験を行った。約300mgの各製剤(5%アシクロビル軟膏、5%アシクロビル・クエン酸物理的混合物軟膏5%、アシクロビル・クエン酸アモルファス軟膏)を用いた。ナイロン膜(HNWP02500、厚さ170μm)孔径0.45μm(日本ミリポア(株)、東京、日本)を用いた。拡散セルの温度は32℃に保たれたウォータージャケットによって維持され、それから、32℃に保持された7mLの放出液(pH6.8リン酸バッファー)をレセプターチャンバーに満たした。実験の間、放出液はマグネティックスターラーを用いて500rpmで連続的に攪拌した。既定の時間で1mLの放出液をレセプターチャンバーから回収し、32℃に保持された同じ容量の放出液をチャンバー内に戻した。この放出実験は360分まで継続した。アシクロビルの累積放出量を算出するために、回収された放出液中のアシクロビル濃度をHPLCにより測定した。マクロゴール軟膏中からのアシクロビル放出速度を得るために、アシクロビル累積放出量が時間の平方根に対してプロットされた。アモルファス化による促進効果を評価するために、アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏からのアシクロビル放出速度を結晶性のアシクロビルを含有するマクロゴール軟膏からの放出速度と比較した。
【0045】
アシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro放出プロファイルを図11aに示す。この放出プロファイルからアシクロビル・クエン酸アモルファスの薬物放出速度が結晶性アシクロビルと比較して顕著に高く、放出率は60分でほぼ100%となることがわかった。全てのデータにHiguchi式を適用し、図11bにおいて累積放出量を時間の平方根に対してプロットした。アシクロビル・クエン酸アモルファスにおいて、leveling−offがプロファイルの後半のポイントで観察された。この現象は軟膏基剤中からの固体薬物の消失によるものと考えられた。このことはアモルファスの溶解度が結晶より高かったことにより、後半のポイントでアモルファス固体が既に排出しきっていることを示唆している。斯くの如くに、アシクロビル・クエン酸アモルファスを含有する軟膏は、優れた薬剤放出特性を有していることが判る。
【実施例5】
【0046】
薬物のアモルファス化による促進効果を評価するために、in vitroにおいて、アモルファスのアシクロビル(アシクロビル・クエン酸アモルファス)を含有するマクロゴール軟膏の皮膚透過性が、結晶性のアシクロビルを含有するマクロゴール軟膏と比較された。ラボスキン(星野試験動物飼育所(株)、茨城、日本)が皮膚試料として用いられた。ラボスキンは、7週齢の雄のヘアレスマウス(Hos:HR−1)の背部から剥離した皮膚試料であり、−20℃で冷凍保存されている。実験の直前に皮膚を室温でゆっくり解凍し、フランツ型拡散セルにマウントするために切り分けた。拡散セルは放出試験で用いたものと同じものを用いた。レセプター液もまた放出試験で用いたものと同じものを用いた(つまり、pH6.8リン酸バッファー)。拡散セルの温度は37℃に保たれたウォータージャケットによって維持された。透過は約200mgのマクロゴール軟膏を皮膚の上に適用することで開始された。それから、規定の時間で1mLのレセプター液をレセプターチャンバーから回収し、37℃に保持された同じ容量の放出液をチャンバー内に戻した。この透過試験は26時間まで継続した。実験の間、レセプター液はマグネティックスターラーを用いて500rpmで連続的に攪拌した。HPLCにより測定された回収したレセプター液中のアシクロビル濃度から、皮膚単位面積当たりのアシクロビル累積透過量が算出された。各マクロゴール軟膏について、3回透過試験が行われた。アシクロビル累積透過量は透過プロファイルを得るためにサンプリング時間に対してプロットされ、その透過プロファイルの直線部分から皮膚透過速度を算出した。アモルファス化による促進効果を評価するために、アシクロビル・クエン酸アモルファスを含有するマクロゴール軟膏の透過速度と結晶性のアシクロビルを含有するマクロゴール軟膏の透過速度を比較した。
【0047】
結果は、図12にアシクロビルの2つの形態(結晶とアモルファス)のマクロゴール軟膏からのin vitro皮膚透過プロファイルとして示す。図12の挿入図は結晶性アシクロビルと物理的混合物のプロファイルの拡大図である。アシクロビル・クエン酸アモルファスのプロファイルを結晶性アシクロビルと物理的混合物のそれと比較した。透過における定常状態はアモルファスにおいて2〜26時間の間で観察された。アモルファスの定常状態における透過速度は2.06μg/cm2/hであった。一方、結晶性アシクロビルまたはアシクロビル・クエン酸物理的混合物のマクロゴール軟膏中からの送達は非常に遅く、透過における定常状態は22〜26時間の間で確認された。結晶性アシクロビルの透過速度はそれぞれ0.02μg/cm2/h、0.09μg/cm2/hであった。透過速度は薬物のアモルファス化により顕著に増加した。これはin vitro薬物放出性の結果とも相関していた。
即ち、本発明のアシクロビル・クエン酸アモルファス体は皮膚外用剤に好ましい、優れた皮膚透過性を有していることが判る。
【産業上の利用可能性】
【0048】
本発明は、皮膚外用医薬などに応用できる。
【特許請求の範囲】
【請求項1】
ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することを特徴とする、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項2】
前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることを特徴とする、請求項1に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸との共結晶又はアモルファスの製造法。
【請求項3】
前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることを特徴とする、請求項1又は2に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項4】
前記極性溶媒は、酢酸及び/又はジアルキルホルムアミドを含むものであることを特徴とする、請求項1〜3何れか1項に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項5】
アシクロビルと酒石酸の共結晶。
【請求項6】
次の物性を有することを特徴とする、請求項5に記載の共結晶。分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストローム
【請求項7】
請求項1〜4何れか1項に記載の製造方法で製造された物であることを特徴とする、請求項5または6に記載の共結晶。
【請求項8】
アシクロビルとクエン酸からなる、アモルファス。
【請求項9】
示差熱分析において、60〜80℃にガラス転移点を有することを特徴とする、請求項8に記載のアモルファス。
【請求項10】
請求項1〜4何れか1項に記載の製造方法で製造された物であることを特徴とする、請求項8または9に記載のアモルファス。
【請求項11】
請求項5〜7何れか1項に記載の共結晶及び/又は請求項8〜10何れか1項に記載のアモルファスを有効成分として含有することを特徴とする、医薬組成物。
【請求項12】
皮膚外用剤であることを特徴とする、請求項11に記載の医薬組成物。
【請求項1】
ヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸とを極性溶媒中で加熱溶解し、しかる後に冷却することを特徴とする、ヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項2】
前記ヌクレオシド系抗ウイルス剤は、アシクロビルであることを特徴とする、請求項1に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシカルボン酸との共結晶又はアモルファスの製造法。
【請求項3】
前記ポリヒドロキシカルボン酸は、酒石酸又はクエン酸であることを特徴とする、請求項1又は2に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項4】
前記極性溶媒は、酢酸及び/又はジアルキルホルムアミドを含むものであることを特徴とする、請求項1〜3何れか1項に記載のヌクレオシド系抗ウイルス剤とポリヒドロキシ有機酸との共結晶又はアモルファスの製造法。
【請求項5】
アシクロビルと酒石酸の共結晶。
【請求項6】
次の物性を有することを特徴とする、請求項5に記載の共結晶。分子量:370〜380、結晶系:単斜晶、空間群:P21、a:10〜12オングストローム、b:14〜16オングストローム、c:4〜6オングストローム、β:90〜110度、共結晶の体積:780〜800立方オングストローム
【請求項7】
請求項1〜4何れか1項に記載の製造方法で製造された物であることを特徴とする、請求項5または6に記載の共結晶。
【請求項8】
アシクロビルとクエン酸からなる、アモルファス。
【請求項9】
示差熱分析において、60〜80℃にガラス転移点を有することを特徴とする、請求項8に記載のアモルファス。
【請求項10】
請求項1〜4何れか1項に記載の製造方法で製造された物であることを特徴とする、請求項8または9に記載のアモルファス。
【請求項11】
請求項5〜7何れか1項に記載の共結晶及び/又は請求項8〜10何れか1項に記載のアモルファスを有効成分として含有することを特徴とする、医薬組成物。
【請求項12】
皮膚外用剤であることを特徴とする、請求項11に記載の医薬組成物。
【図1】


【図2】
【図3】

【図4】
【図5】
【図6】
【図7】

【図8】

【図9】

【図10】

【図11】
【図12】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公開番号】特開2013−43886(P2013−43886A)
【公開日】平成25年3月4日(2013.3.4)
【国際特許分類】
【出願番号】特願2011−193322(P2011−193322)
【出願日】平成23年8月19日(2011.8.19)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年2月21日掲載の「http://pfwww.kek.jp/pf−sympo/28/posterprogram.html」及び「http://pfwww.kek.jp/pf−sympo/28/abst/P−UG06−04.pdf」
【出願人】(511216983)
【Fターム(参考)】
【公開日】平成25年3月4日(2013.3.4)
【国際特許分類】
【出願日】平成23年8月19日(2011.8.19)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年2月21日掲載の「http://pfwww.kek.jp/pf−sympo/28/posterprogram.html」及び「http://pfwww.kek.jp/pf−sympo/28/abst/P−UG06−04.pdf」
【出願人】(511216983)
【Fターム(参考)】
[ Back to top ]