
アシルアミノフェニル基を有する抗がん剤
【課題】本発明は、大腸がんや乳がん等のがんの治療薬として非常に有望な候補化合物を提供することを課題とする。
【解決手段】
以下の構造を有する化合物:

であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物。
【解決手段】
以下の構造を有する化合物:
であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アシルアミノフェニル基を有する化合物に関する。より詳細には、本発明は、医薬品:抗がん剤、抗炎症剤、胃壁保護剤、心筋梗塞予防剤、脳梗塞予防剤、慢性動脈閉塞症(PAOD)、NOS2発現阻害脳卒中予防剤、アルツハイマー進行抑制・予防剤、血液抗凝固剤に関する。
【背景技術】
【0002】
転移性・悪性乳がん細胞や大腸がん細胞上に過剰発現しているErbB/Her受容体ファミリーの一つ、Her2タンパク質、を認識し、その2量体化を阻害して乳がん細胞の増殖を抑制する制癌剤として抗体医薬品Herceptin等が知られている。しかし、タンパク質製剤であるため、代謝速度は速い、投与に長時間を要する点滴製剤である、高価である、等の様々なデメリットをもっている。一方、特に大腸がん細胞では、Wntシグナル上のタンパク質β−カテニンが異常に蓄積し、高レベルのβ−カテニン/TCF複合体がサイクリンD1[非特許文献1,非特許文献2]やc−myc[非特許文献3]の遺伝子発現を促進し、大腸がんの細胞増殖を活発化させることが知られている。最近、このErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害する、悪性乳がんや大腸がんの治療薬の開発研究が活発化している。
【0003】
アスピリンは、心筋梗塞および脳卒中の予防、がん疼痛の緩解に使用されている。また、最近では、アルツハイマーや結腸がんのリスクを減らす効果があることも知られてきた。しかし、消化管出血や潰瘍穿孔などの消化管毒性があることが障害となっている。最近、4−(ニトロオキシメチル)フェニル基を有するNO供与型アスピリン(NO−ASA):NCX4040やNCX4016[非特許文献4、非特許文献5および非特許文献6]などが見出された。これらは、アスピリンの有益な効果を維持しつつ、消化管毒性の少ないことが知られている。特に、前者NCX4040は、大腸がんのがん化初期に関わる、Wntシグナル上のタンパク質複合体β−カテニン/TCFの結合を阻害する効果を示す他、NF−κBのDNA結合阻害、NOS2発現阻害、COX2発現誘導、プロスタグランジンE2レベルの上昇、等の効果を示すことから、複数経路で大腸がんなどの予防・治療効果を発揮すると推定されている[非特許文献4]。本薬剤はNO供与型化合物としてデザインされた化合物であるが、最近の研究で、実は、主としてキノンメサイド(以下の化1のスキーム1参照)を経由するアルキル化剤として働き、グルタチオンなどのSH含有生体化合物に結合することにより、酸化ストレスを亢進し、がん細胞の増殖を抑制する効果を発揮しているという説が有力となった[非特許文献7,非特許文献8]。NO−ASA系化合物は、大腸がんへの治療薬として、単独、或いは、既存の抗がん剤との併用で、それぞれ前臨床試験、第1相臨床試験が実施されている。
【0004】
(スキーム 1)
【0005】
【化1】
【非特許文献1】Shtutman M., Zhurinsky J., Simcha I., Albanese C., D’Amico M., Pestell R, Ben−Ze’evA., Proc. Natl. Acad. Sci. USA, 96, 5522−5527 (1999).
【非特許文献2】Tetsu O. & McCormick F., Nature, 398, 422−426 (1999).
【非特許文献3】He T.C., Sparks A.B., Rago C., Hermeking H., Zawel L., da Costa L.T., MorinP.J., Vogelstein B., Kinzlcr K.W., Science, 281, 1509−1512 (1998).
【非特許文献4】Williams J.L., Nath N., Chen J., Hundley T.R., Gao J., Kopelovich L., KashfiK., Rigas B., Cancer Res., 63, 7613−7618 (2003).
【非特許文献5】Nath N., Kashfi K.,Chen J., Rigas B., Proc. Natl. Acad. Sci. USA, 100, 12584−12589 (2003).
【非特許文献6】Hulsman N., MedemaJ.P., Bos C., Jongejan A., Leurs R., Smit M.J., de Esch I.J., Richel D.,Wijtmans M., J. Med. Chem., 50, 2424−2431 (2007).
【非特許文献7】Hulsman N., MedemaJ.P., Bos C., Jongejan A., Leurs R., Smit M.J., de Esch I.J., Richel D.,Wijtmans M., J. Med. Chem., 50, 2424−2431 (2007).
【非特許文献8】Dunlap T.,Chandrasena R.E.P., Wang Z., Sinha V., Wang Z., Thatcher G.R.J., Chem. Res.Toxxicol., 20, 1903−1912 (2007).
【非特許文献9】Okamoto H., YonemoriF., Wakitani K.,分間owa T., Maeda K.,Shinkai H., Nature, 406, 203−207 (2000).
【非特許文献10】Shinkai H., Maeda K.,Yamasaki T., Okamoto H., Uchida I., J. Med. Chem., 43, 3566−3572 (2000).
【非特許文献11】Yamakawa S., DemizuA., Kawaratani Y., Nagaoka Y., Terada Y., Maruyama S., Uesato S., Biol. Pharm. Bull., 31 (5), 910−920 (2008).
【発明の開示】
【発明が解決しようとする課題】
【0006】
一般に、タンパク質同士の結合を低分子で阻害することは、困難であると考えられていた。発明者は、Her2/Herceptin間、β−カテニン/TCF間などの結合部位に存在するCys残基に着目し、Cysと‐S‐S‐結合をする低分子化合物bis[2−(アシルアミノ)フェニル]ジスルフィド及びS−[2−(アシルアミノ)フェニル]アルカンチオレート[非特許文献9,非特許文献10]を評価した。その結果、Her2高発現型乳がん細胞株SKBR−3や大腸がん細胞株HCT116等に高い細胞増殖抑制活性を示す低分子化合物K−153およびK−154を見出した。
(K−153)
【0007】
【化2】
【0008】
(K−154)
【0009】
【化3】
【0010】
特に前者は、β−カテニン/TCF の結合を阻害し、また、ストレス応答の主要MAPKsを活性化し、がん細胞の増殖を抑制していることが推定された[非特許文献11]。
【0011】
そこで、本発明は、大腸がんや乳がん等のがんの治療薬として非常に有望な候補化合物を提供することを課題とする。
特に、NCX4040
【0012】
【化4】
【0013】
は、β−カテニン/TCF結合の阻害効果の他、NF−κBのDNA結合阻害、NOS2発現阻害、COX2発現誘導、プロスタグランジンE2レベルの上昇、等の効果を示し、複数経路で抗大腸がん活性を発揮する。本剤は広範ながん細胞に対して細胞増殖抑制効果を示すが、現在、大腸がん予防・治療薬としての適用で、単独、或いは、既存の抗がん剤との併用で、前臨床試験、第1相臨床試験を実施中である。しかし、NCX4040は、ある種の大腸がん細胞株(HCT116株、SW480株など)や乳がん細胞株(SKBR−3株など)に対しは、静細胞的に作用し、その殺細胞或いは細胞増殖抑制効果(実施例の表1,2を参照)は低いという問題点があった。
【課題を解決するための手段】
【0014】
上記課題に対して、本発明者らは、前述のS−[2−(アシルアミノ)フェニル]アルカンチオレートや[2−(アシルアミノ)フェニル]アルカノエートにニトロオキシメチルフェニル基を賦与した構造の化合物を合成した。特に、K−210、K−220、K−230
(K−210)
【0015】
【化5】
【0016】
(K−220)
【0017】
【化6】
【0018】
(K−230)
【0019】
【化7】
【0020】
はアシルアミノフェニル骨格に2個のニトロオキシメチルフェニル基を有し、かつ、そのパラ位、メタ位にカルボニル基を有することから、高いNO産生能が期待された。ヒトがん細胞増殖抑制活性試験(実施例の表1,2)の結果、K210、K220などは、NCX4040では増殖抑制活性がそれほど高くない大腸がん細胞株HCT116(IC50,68.0μM)および乳がん細胞株SKBR−3(IC50,17.0μM)に対し、高い増殖抑制活性を示した[HCT116 IC50,0.67μM(K210);0.72μM(K220):SKBR−3 IC50,0.66μM(K210);0.69μM(K220)]。
【0021】
特記すべきことに、K210、K220は、NCX4040と同様、ヒト正常繊維芽細胞に対して、200μMレベルという高濃度でも事実上毒性を示さなかった。したがって、K210、K220は大腸がんおよび乳がんの治療薬として非常に有望な候補化合物であると期待される。
【0022】
免疫沈降試験(図1)で、K−210は、NCX4040とは異なり、β−カテニン/TCFの結合を阻害しないことが分かった。一方、K210は、細胞周期制御試験 (図2)で、NCX4040と比べて低濃度(5μM)で大腸がん細胞HCT116株の増殖をG2/M期で停止させることが分かった。ウェスタンブロッティングで、K210はp53変異型乳がん細胞株SKBR3において、細胞周期制御因子p21/WAF1タンパク質レベルを上昇させることから、G2/M期停止は本タンパク質による細胞周期停止効果により起こると推定された(図4)。また、TUNEL法によるアポトーシス検出試験(図3)により、5μMで顕著にアポトーシスを誘起することも分かった。K−210はウェスタンブロッティングで、ストレス応答型主要MAPKsであるp38およびJNKのリン酸化を促進したことから、この経路の活性化によってアポトーシスが誘導された可能性が強い(図4)と推定した。
【0023】
本発明は、発明を実現する技術的手段としてヒトがん細胞移植ヌードマウス実験、前臨床試験、治験、GLP、GMP、GCP、GMP対応での原末、薬剤の製造などを考慮して実施することができる。また、HCT116株などヒトがん細胞移植ヌードマウス実験により、in vivoでの有効性を確認することができる。前臨床試験、臨床試験で安全性、有効性を確認することができる。GMP対応での薬剤の製造することができる。
【0024】
以上から、本発明は以下を提供する。
【0025】
1つの局面において、本発明は、以下の構造を有する化合物:
【0026】
【化101】
【0027】
であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物またはその溶媒和物(置換基によっては酸またはアルカリになり得、これらの場合は塩を形成しうる)などのプロドラッグに関する。
【0028】
本発明は、好ましくは、以下の構造を有する化合物またはその溶媒和物:
【0029】
【化102】
【0030】
であって、
R2は、ハロゲンまたはONO2で置換されたアルキル基であり、
R3は、ハロゲンまたはONO2で置換されたアルキル基であり、
Aは、−O−、−S−または−NH−である、
化合物に関する。
【0031】
1つの好ましい実施形態において、R2およびR3は、−CH2−Clまたは−CH2−ONO2である。
【0032】
1つの好ましい実施形態において、R2およびR3は、−CH2−Clまたは−CH2−ONO2であり、パラ位に存在する。
【0033】
1つの好ましい実施形態において、R2およびR3は、−CH2−ONO2である。
【0034】
1つの好ましい実施形態において、R2およびR3は、−CH2−ONO2であり、パラ位に存在する。
【0035】
1つの好ましい実施形態において、以下からなる群より選択される、請求項1に記載の化合物。
【0036】
【化103】
【0037】
【0038】
【0039】
【0040】
【0041】
【0042】
【0043】
【0044】
またはその溶媒和物が提供される。
【0045】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、医薬組成物を提供する。
【0046】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、抗がん剤を提供する。
【0047】
好ましい局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、乳がん、結腸癌および肺がんのいずれかまたは複数に対する抗がん剤を提供する。
【0048】
別の実施形態において、本発明は、
【0049】
【化104】
【0050】
および
【0051】
【化105】
【0052】
からなる群より選択される少なくとも1つ化合物またはその溶媒和物を含む、抗乳がん剤を提供する。
【0053】
別の実施形態において、本発明は、
【0054】
【化106】
【0055】
【化107】
【0056】
および
【0057】
【化108】
【0058】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗肺がん剤を提供する。
【0059】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物の、医薬組成物の製造のための使用に関する。
【0060】
他の局面において、本発明は、本発明の上記化合物またはその溶媒和物の、抗がん剤の製造のための使用に関する。
【0061】
他の局面において、本発明は、本発明の化合物またはその溶媒和物を投与する工程を包含する治療方法に関する。
【0062】
他の局面において、本発明は、本発明の上記化合物またはその溶媒和物を投与する工程を包含するがんの治療方法に関する。
【0063】
従って、本発明のこれらおよび他の利点は、以下の詳細な説明を読めば、明白である。
【発明の効果】
【0064】
新規作用機序に基づく大腸がん、乳がんなどのがん治療薬が開発することができ、医療に対する貢献度は大きい。
【発明を実施するための最良の形態】
【0065】
以下、本発明を最良の形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の修飾語等(例えば、英語の場合は「a」、「an」、「the」等の冠詞など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
【0066】
以下に本明細書において用いられる各用語の意味を説明する。各用語は本明細書中、統一した意味で使用し、単独で用いられる場合も、または他の用語と組み合わされて用いられる場合も、同一の意味で用いられる。
【0067】
「ハロゲン」とは、フッ素、塩素、臭素およびヨウ素が挙げられる。
【0068】
「アルキル」とは、炭素数1〜10個の直鎖状又は分枝状のアルキル基を包含し、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ぺンチル、イソぺンチル、ネオペンチル、n−ヘキシル、イソヘキシル、n−ヘプチル、n−オクチル、n−ノニル、n−デシル等が挙げられる。好ましくは、炭素数1〜6または1〜4個のアルキルであり、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ぺンチル、イソペンチル、ネオペンチル、n−ヘキシル、イソヘキシルが挙げられる。
【0069】
「アルケニル」とは、上記「アルキル」に1個又はそれ以上の二重結合を有する炭素数2〜8個の直鎖状又は分枝状のアルケニルを包含し、例えば、ビニル、1−プロペニル、2−プロペニル、1−ブテニル、2−ブテニル、3−ブテニル、1,3−ブタジエニル、3−メチル−2−ブテニル等が挙げられる。
【0070】
「アルキニル」とは、上記「アルキル」に1個又はそれ以上の三重結合を有する炭素数2〜8個の直鎖状又は分枝状のアルキニルを包含し、例えば、エチニル、プロピニル、ブチニル等が挙げられる。さらに1個又はそれ以上の二重結合を有していてもよい。
【0071】
「シクロアルキル」とは、炭素数3〜15の環状飽和炭化水素基を包含し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、橋かけ環式炭化水素基、スピロ炭化水素基などが挙げられる。好ましくは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、橋かけ環式炭化水素基が挙げられる。
【0072】
「シクロアルケニル」は、炭素数3〜7個の環状の不飽和脂肪族炭化水素基を包含し、例えば、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニルが挙げられ、好ましくはシクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニルである。シクロアルケニルには、環中に不飽和結合を有する橋かけ環式炭化水素基およびスピロ炭化水素基も含む。
【0073】
「アリール」とは、単環芳香族炭化水素基(例:フェニル)及び多環芳香族炭化水素基(例:1−ナフチル、2−ナフチル、1−アントリル、2−アントリル、9−アントリル、1−フェナントリル、2−フェナントリル、3−フェナントリル、4−フェナントリル、9−フェナントリル等)を包含する。好ましくは、フェニル又はナフチル(1−ナフチル、2−ナフチル)が挙げられる。
【0074】
「ヘテロアリール」とは、単環芳香族複素環式基及び縮合芳香族複素環式基を包含する。単環芳香族複素環式基は、酸素原子、硫黄原子、および/又は窒素 原子を環内に1〜4個含んでいてもよい5〜8員の芳香環から誘導される、置換可能な任意の位置に結合手を有していてもよい基を包含する。縮合芳香族複素環式基は、酸素原子、硫黄原子、および/又は窒素原子を環内に1〜4個含んでいてもよい5〜8員の芳香環が、1〜4個の5〜8員の芳香族炭素環もしくは他の5〜8員の芳香族ヘテロ環と縮合している、置換可能な任意の位置に結合手を有していてもよい基を包含する。
【0075】
「ヘテロサイクル」とは、酸素原子、硫黄原子、及び/又は窒素原子を環内に1〜4個含んでいてもよく、置換可能な任意の位置に結合手を有していてもよい非芳香族複素環式基を包含する。また、そのような非芳香族複素環式基がさらに炭素数1〜4のアルキル鎖で架橋されていてもよく、シクロアルカン(5〜6員環が好ましい)やベンゼン環が縮合していてもよい。非芳香族であれば、飽和でも不飽和でもよい。好ましくは5〜8員環である。例えば、1−ピロリニル、2−ピロリニル、3−ピロリニル、1−ピロリジニル、2−ピロリジニル、3−ピロリジニル、ピロリジノン、1−イミダゾリニル、2−イミダゾリニル、4−イミダゾリニル、1−イミダゾリジニル、2−イミダゾリジニル、4−イミダゾリジニル、イミダゾリジノン、1−ピラゾリニル、3−ピラゾリニル、4−ピラゾリニル、1−ピラゾリジニル、3−ピラゾリジニル、4−ピラゾリジニル、ピペリジノン、ピペリジノ、2−ピペリジニル、3−ピペリジニル、4−ピペリジニル、1−ピペラジニル、2−ピペラジニル、ピペラジノン、2−モルホリニル、3−モルホリニル、モルホリノ、テトラヒドロピラニル、テトラヒドロフラニル等が挙げられる。
【0076】
「置換もしくは非置換のアルキル」、「置換もしくは非置換のアルケニル」、「置換もしくは非置換のアルキニル」、「置換もしくは非置換のアリール」、「置換もしくは非置換のシクロアルキル」、「置換もしくは非置換のシクロアルケニル」、「置換もしくは非置換のヘテロアリール」、「置換もしくは非置換のヘテロサイクル」、「置換もしくは非置換のアルコキシ」、における置換基としては、例えば、ヒドロキシ、カルボキシ、ハロゲン、ハロゲン化アルキル(例:CF3、CH2CF3、CH2CCl3)、ニトロ、ニトロソ、シアノ、アルキル(例:メチル、エチル、イソプロピル、tert−ブチル)、アルケニル(例:ビニル)、アルキニル(例:エチニル)、シクロアルキル(例:シクロプロピル、アダマンチル)、シクロアルキルアルキル(例:シクロヘキシルメチル、アダマンチルメチル)、シクロアルケニル(例:シクロプロペニル)、アリール(例:フェニル、ナフチル)、アリールアルキル(例:ベンジル、フェネチル)、ヘテロアリール(例:ピリジル、フリル)、ヘテロアリールアルキル(例:ピリジルメチル)、ヘテロサイクル(例:ピペリジル)、ヘテロサイクルアルキル(例:モルホリルメチル)、アルコキシ(例:メトキシ、エトキシ、プロポキシ、ブトキシ)、ハロゲン化アルコキシ(例:OCF3)、アルケニルオキシ(例:ビニルオキシ、アリルオキシ)、アリールオキシ(例:フェニルオキシ)、アルキルオキシカルボニル(例:メトキシカルボニル、エトキシカルボニル、tert−ブトキシカルボニル)、アリールアルキルオキシ(例:ベンジルオキシ)、アミノ(例:アルキルアミノ(例:メチルアミノ、エチルアミノ、ジメチルアミノ)、アシルアミノ(例:アセチルアミノ、ベンゾイルアミノ)、アリールアルキルアミノ(例:ベンジルアミノ、トリチルアミノ)、ヒドロキシアミノ、アルキルアミノアルキル(例:ジエチルアミノメチル)、スルファモイル、オキソ等からなる群から選択される。1〜4個の当該置換基で置換されていてもよい。
【0077】
本発明化合物の製薬上許容される塩としては、以下の塩が挙げられる。
【0078】
塩基性塩として、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、プロカイン塩、メグルミン塩、ジエタノールアミン塩またはエチレンジアミン塩等の脂肪族アミン塩;N,N−ジベンジルエチレンジアミン、ベネタミン塩等のアラルキルアミン塩;ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等のヘテロ環芳香族アミン塩;テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩、リジン塩等の塩基性アミノ酸塩等が挙げられる。
【0079】
酸性塩としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。
【0080】
「溶媒和物」とは、本発明化合物またはその製薬上許容される塩の溶媒和物を意味し、例えば、アルコール(例:エタノール)和物や水和物等が挙げられる。水和物としては、1水和物、2水和物等を挙げることができる。
【0081】
別の実施形態において、本発明は、上記のいずれかに記載の化合物、その製薬上許容される塩またはそれらの溶媒和物、あるいはそのプロドラッグ(たとえば、エステル類、アミド類)を含有する医薬組成物を提供する。
【0082】
本明細書において「プロドラッグ」、「プロドラッグ化合物」とは、化学的又は代謝的に分解し得る基を有し、加水分解や加溶媒分解によって又は生理条件下で分解することによって薬学的に活性を示す本発明化合物の誘導体である。いろいろな形のプロドラッグが、当該技術分野において知られている。このようなプロドラッグ誘導体の例については、次を参照することができる。式(I)の化合物のプロドラッグは、式(I)の化合物中に存在する官能基を、生体内で開裂すると親化合物が放出されるような修飾方法で修飾することによって製造される。たとえば、プロドラッグは、式(I)の化合物中のヒドロキシ、スルフヒドリル又はアミノ基が、生体内で開裂されるとそれぞれ遊離ヒドロキシ、アミノ、又はスルフヒドリル基を再生する基と結合している式(I)の化合物を含む。プロドラッグの例は、これらに限定されないが、式(I)の化合物中のヒドロキシ 官能基のエステル(例えば、アセタート、ホルマート、およびベンゾアート誘導体)、カルバマート(例えば、N,N−ジメチルアミノカルボニル)などを含む。
(a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods inEnzymology, Vol.42.p.309−396, edited byK.Widder,et al.(Academic Press, 1985);
(b)A Textbook of Drug Designand Development, edited by Krogsgaard−Larsen;
(c)H.Bundgaard, Chapter 5“Designand Application of Prodrugs”,by H.Bundgaard p.113−191(1991);
(d)H.Bundgaard, Advanced Drug Delivery Reviews, 8,1−38(1992) ;
(e)H.Bundgaard, et al.,Journal of Pharmaceutical Sciences, 77,285(1988);および
(f)N.Kakeya, et al., Chem Pharm Bull, 32,692(1984)。
【0083】
1つのプロドラッグ基の例は、ヒトまたは動物体内で開裂して親酸を生じる薬学的に許容しうるエステルの in vivo 開裂可能エステル基である。たとえば、プロドラッグ基は、それが結合しているカルボキシ基と一緒になって、C1−6アルキルエステルまたはC1−6シクロアルキルエステル、例えば、メチル、エチル、プロピル、イソプロピル、n−ブチルまたはシクロペンチルのエステル;C1−6アルコキシメチルエステル、例えば、メトキシメチルエステル;C1−6アルカノイルオキシメチルエステル、例えば、ピバロイルオキシメチルエステル;フタリジルエステル;C3−8シクロアルコキシカルボニルオキシC1−6アルキルエステル、例えば、1−シクロヘキシルカルボニルオキシエチルエステル;1,3−ジオキソラン−2−イルメチルエステル、例えば、5−メチル−1,3−ジオキソラン−2−イルメチルエステル;C1−6アルコキシカルボニルオキシエチルエステル、例えば、1−メトキシカルボニルオキシエチルエステル;アミノカルボニルメチルエステルおよびそのモノ−またはジ−N−(C1−6アルキル)変型、例えば、N,N−ジメチルアミノカルボニルメチルエステルおよびN−エチルアミノカルボニルメチルエステルのような薬学的に許容しうるエステル、および置換または非置換の複素環式基の薬学的に許容しうるエステルを形成する。1つの実施形態では、プロドラッグは、イソプロピルまたはシクロペンチルのようなC1−4アルキル基、またはN−メチルテトラヒドロピリジルのような置換されていてよい複素環式基より選択されるものとのエステルが挙げられる。
【0084】
記に列挙した具体的な化合物の任意の化合物を含む本発明の医薬組成物は、TTK阻害剤であることをも特徴とする。したがって、TTKの阻害を必要とする患者に投与することによって薬効を発揮する任意の医薬組成物が提供される。
【0085】
別の実施形態では、本発明は、がんまたは免疫疾患の処置または予防のための医薬であって、上記に列挙した具体的な化合物の任意の化合物を含む医薬を提供する。
【0086】
(製造方法)
本発明化合物の一般的製造法を以下に例示する。また、抽出、精製などは、通常の有機化学の実験で行う処理を行えばよい。
【0087】
以下に、本発明の化合物の製造方法を記載する。
【0088】
本発明の化合物の合成は、当該分野において公知の手法を参酌しながら実施することができる。
【0089】
原料化合物は、市販の化合物であるか、特許文献3、特許文献4、特許文献5、特許文献6、特許文献7に記載されたもの、このほか本明細書において記載されたものならびに本明細書において他に引用された文献に記載されるものならびに他に公知の化合物を利用することができる。
【0090】
本発明の化合物の中には、互変異性体が存在し得るものがあるが、本発明は、これらを含め、全ての可能な異性体およびそれらの混合物を包含する。
【0091】
本発明の化合物の塩を取得したいとき、本発明の化合物が塩の形で得られる場合には、そのまま精製すればよく、また、遊離の形で得られる場合には、適当な有機溶媒に溶解もしくは懸濁させ、酸または塩基を加えて通常の方法により塩を形成させればよい。
【0092】
また、本発明の化合物およびその製薬上許容される塩は、水あるいは各種溶媒との付加物(水和物ないし溶媒和物)の形で存在することもあるが、これら付加物も本発明に包含される。
【0093】
これらの誘導体は、体内にて変換されて活性化されるものであり、本明細書において「プロドラッグ」とも称する。プロドラッグの例としては、たとえば、上記塩、溶媒和物のほか、エステル(たとえば、アルキルエステルなど)、アミドなども含まれることが理解される。
【0094】
本発明の化合物の例は、実施例において種々列挙されており、当業者はこれらを参考にして、本発明の例示されていない化合物をも製造、使用することができる。
【0095】
本発明はまた、本発明の化合物を製造するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0096】
【化8】
【0097】
(式中、R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、)
そして、化8の化合物は、対応するアニリン誘導体と、安息香酸誘導体とからアミド結合を形成させることによって、製造することができる。
【0098】
【化9】
【0099】
(式中、各記号は前記と同義であり、Xは、ハロゲン(Clなど)、OHなどの脱離可能な基である。式(A1)シリーズ(A11、A12,A13,A14)及び式(A2)で示される化合物は公知の化合物を用いてもよく、市販の化合物を用いてもよい。)。
【0100】
すなわち、本製造工程は、式(A1)シリーズ(A11、A12,A13,A14)で示される化合物と式(A2)で示される化合物を塩基および縮合剤存在下で反応させ、式(I)で示される化合物を製造する工程である。ここで、直接合成されるものは、R1bはHであるが、前記と同様の置換基を置換することもでき、場合によっては、置換基がHの代わりに結合したものを用いて合成することもできる。
【0101】
反応溶媒としては、DMF、NMP、DMA、ジメチルスルホキシド、芳香族炭化水素類(例、トルエン、ベンゼン、キシレンなど)、飽和炭化水素類(例、シクロヘキサン、ヘキサンなど)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、1,2−ジクロロエタンなど)、エーテル類(例、テトラヒドロフラン、ジエチルエーテル、ジオキサン、1,2−ジメトキシエタンなど)、エステル類(例、酢酸メチル、酢酸エチルなど)、ケトン類(例、アセトン、メチルエチルケトンなど)、ニトリル類(例、アセトニトリルなど)、アルコール類(例、メタノール、エタノール、t−ブタノールなど)、水およびそれらの混合溶媒等が挙げられる。
【0102】
塩基としては、例えば金属水素化物(例、水素化ナトリウムなど)、金属水酸化物(例、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化バリウムなど)、金属炭酸塩(例、炭酸ナトリウム、炭酸カルシウム、炭酸セシウムなど)、金属アルコキシド(例、ナトリウムメトキシド、ナトリウムエトキシド、カリウムt−ブトキシドなど)、炭酸水素ナトリウム、金属ナトリウム、有機アミン(例、トリエチルアミン、ジイソプロピルエチルアミン、DBU、2,6−ルチジンなど)、ピリジン、アルキルリチウム(n−BuLi、sec−BuLi、tert−BuLi)等が挙げられる。塩基は必ずしも使用する必要はないが、必要に応じて用いることができる。
【0103】
上記XがOH等の場合に用いられる縮合剤としては、DCC、BOP,PyBOP、PyBrop、HATU、DPPA、WSC、DMT−MMなどを用いることができる。また、これらの試薬は、例えばHOSu、HOBt、HOAtなどと組み合わせて使用することができる。
【0104】
好ましくは、反応溶媒としてエーテル類(例、テトラヒドロフラン、ジエチルエーテル、ジオキサンなど)、N,N−ジメチルホルムアミド、N−メチルピロリドン、N,N−ジメチルアセトアミド、塩基として有機アミン(例、トリエチルアミン、ジイソプロピルエチルアミン、DBU、2,6−ルチジンなど)、縮合剤としてHATUまたはPyBOPを用いて行えばよい。反応温度、反応時間は特に限定されないが、通常は室温にて反応を実施し、反応の進行が遅い場合には加温することによって反応が促進される場合もある。
【0105】
本発明の製造は、上記好ましい実施形態を適宜改変し、あるいは組み合わせ、あるいは公知技術を付加することによって実施することができる。
【0106】
本発明の化合物は、保護基を用いて保護することができる。たとえば、代表的には、ハロゲン(I,Br,Cl、Fなど)、低級(ここでは、代表的にC1−C6を示すがこれに限定されない。)アルコキシ、低級アルキルチオ、低級アルキルスルホニルオキシ、アリールスルホニルオキシ等を表す。)において、適宜の置換基を当該分野で公知の手法により保護することによって製造することができる。このような保護基としては、例えばエトキシカルボニル、t−ブトキシカルボニル、アセチル、ベンジル等の、Protective Groups in Organic Synthesis、T.W.Green著、John Wiley & Sons Inc.(1981年)等に記載されている保護基をあげることができる。保護基の導入および脱離方法は、有機合成化学で常用される方法[例えば、Protective Groups in Organic Synthesis、T. W. Greene著、John Wiley & Sons Inc.(1981年)参照]等に記載の方法あるいはそれらに準じて得ることができる。また、各置換基に含まれる官能基の変換は、上記製造法以外にも公知の方法[例えば、Comprehensive Organic Transformations、R.C.Larock著(1989年)等]によっても行うことができ、本発明の化合物の中には、これを合成中間体としてさらに新規な誘導体へ導くことができるものもある。上記各製造法における中間体および目的化合物は、有機合成化学で常用される精製法、例えば中和、濾過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等に付して単離精製することができる。また、中間体においては、特に精製することなく次の反応に供することも可能である。
【0107】
より特定された実施形態では、式(II)の化合物:
【0108】
【化10】
【0109】
(式中、R2は、ハロゲンまたはONO2で置換されたアルキル基であり、R3は、ハロゲンまたはONO2で置換されたアルキル基であり、Aは、−O−、−S−または−NH−である)を合成する方法は、上記(A)の合成に準じて実施することができる。
【0110】
この場合、Aが−NH−である場合は、上記と同様の反応により実施可能である。
【0111】
また、Aが−O−または−S−のときは、以下の点に留意して、上記(A)の合成と同様に実施することができる。
【0112】
あるいは、Aが−O−であり、R2およびR3が同じでかつ同じ配置(たとえば、パラ位)の場合、2−アミノフェノール(o−アミノフェノール)にR2−ベンゾイルハライド(たとえば、p−クロロメチルベンゾイルクロリドなど)を用いることによって、製造することができる。
【0113】
Aが−S−であり、R2およびR3が同じでかつ同じ配置(たとえば、パラ位)の場合、2−アミノチオフェノール(o−アミノチオフェノール)にR2−ベンゾイルハライド(たとえば、p−クロロメチルベンゾイルクロリドなど)を用いることによって、製造することができる。
【0114】
R2およびR3は、たとえば、アミノフェノールまたはアミノチオフェノールとの結合後にハロゲンを別の置換基にすることもできる。たとえば、ニトロオキシを導入する場合は、アセトニトリルと結合させることも可能である。
【0115】
本発明は、スクリーニング方法により得られた化合物又はその塩として、関連して発症する疾患に対する治療又は予防作用を発揮し得る。例えば、本発明のスクリーニング方法によれば、癌、免疫疾患等に有効な治療剤又は予防剤の候補化合物をスクリーニングすることができる。
【0116】
(医薬)
本発明の化合物またはその製薬上許容される塩は、そのまま単独で投与することも可能であるが、通常各種の医薬製剤として提供するのが好ましい。また、それら医薬製剤は、動物および人に使用される。
【0117】
本発明が主に対象とする疾患である「がん」としては、固形がん、血管腫、血管内皮腫、肉腫、カポシ肉腫及び造血器腫瘍等の種々の悪性新生物が例示され、大腸がん及び肝がん等が包含され、さらにこれらがんの転移をも包含する。本発明は、上記病気を治療する医薬品を製剤化するのに特に有用になるような特定の特性を有する新規の一連の化合物の発見に成功した。特に、本化合物は、充実性腫瘍か、血液の腫瘍のような増殖性の病気の治療において、特に、結腸直腸のがん、乳がん、肺がん、前立腺がん、膵臓がんまたは膀胱がんおよび腎臓がん、同様に、白血病およびリンパ腫のような病気において有用である。
【0118】
上記医薬組成物は、本発明の化合物又はその塩を有効成分として含有することに1つの特徴がある。したがって、該医薬組成物は、発症する疾患に対して、ErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害することを介して作用し得るという優れた効果を発揮する。例えば、本発明の医薬組成物は特に、癌等に対して、ErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害することを介して作用し得るという優れた効果を発揮する。該医薬組成物を癌の治療又は予防に用いる場合は、通常の癌療法、例えば、放射線療法、化学療法、たとえば腫瘍細胞を事前感応化するためのDNA劣化剤を施すのと同時に、又はその前でも使用できる。
【0119】
上記医薬組成物中における前記化合物又はその塩の含有量は、治療目的の疾患、患者の年齢、体重等により適宜調節することができ、治療上有効量であればよく、低分子化合物又は高分子化合物の場合、例えば、0.0001〜1000mg、好ましくは、0.001〜100mg、ポリペプチド又はその誘導体の場合、例えば、0.0001〜1000mg、好ましくは、0.001〜100mg、核酸又はその誘導体の場合、例えば、0.00001〜100mg、好ましくは、0.0001〜10mgであることが望ましい。
【0120】
上記医薬組成物は、前記化合物又はその塩を安定に保持し得る種々の助剤をさらに含有してもよい。具体的には、有効成分の送達対象となる部位に到達するまでの間に、有効成分が分解することを抑制する性質を呈する薬学的に許容されうる助剤、賦形剤、結合剤、安定剤、緩衝剤、溶解補助剤、等張剤等が挙げられる。
【0121】
上記医薬組成物の投与形態は、有効成分の種類;投与対象となる個体、器官、局所部位、組織;投与対象となる個体の年齢、体重等に応じて、適宜選択される。前記投与形態としては、皮下注射、筋肉内注射、静脈内注射、局所投与等が挙げられる。
【0122】
また、上記医薬組成物の投与量も、有効成分の種類;投与対象となる個体、器官、局所部位、組織;投与対象となる個体の年齢、体重等に応じて、適宜選択される。投与としては、特に限定されないが、有効成分が、低分子化合物又は高分子化合物である場合、前記有効成分の量として、例えば、0.0001〜1000mg/kg体重、好ましくは、0.001〜100mg/kg体重、ポリペプチド又はその誘導体の場合、例えば、0.0001〜1000mg/kg体重、好ましくは、0.001〜100mg/kg体重、核酸又はその誘導体の場合、例えば、0.00001〜100mg/kg体重、好ましくは、0.0001〜10mg/kg体重の1回投与量となるように、1日につき、複数回、例えば、1〜3回投与すること等が挙げられる。
【0123】
投与経路は、治療に際し最も効果的なものを使用するのが好ましく、経口または例えば、直腸内、口腔内、皮下、筋肉内、静脈内等の非経口をあげることができる。
投与形態としては、カプセル剤、錠剤、顆粒剤、散剤、シロップ剤、乳剤、座剤、注射剤等がある。経口投与に適当な、例えば乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビット、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ゴマ油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を使用して製造できる。また、カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニット等の賦形剤、澱粉、アルギン酸ソーダ等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を用いて製造できる。非経口投与に適当な製剤は、好ましくは受容者の血液と等張である活性化合物を含む滅菌水性製剤からなる。例えば、注射剤の場合、塩溶液、ブドウ糖溶液または塩水とブドウ糖溶液の混合物からなる担体等を用いて注射用の溶液を調製する。
【0124】
局所製剤は、活性化合物を1種もしくはそれ以上の媒質、例えば鉱油、石油、多価アルコール等または局所医薬製剤に使用される他の基剤中に溶解または懸濁させて調製する。
腸内投与のための製剤は、通常の担体、例えばカカオ脂、水素化脂肪、水素化脂肪カルボン酸等を用いて調製し、座剤として提供される。本発明では、非経口剤においても、経口剤で例示したグリコール類、油類、フレーバー類、防腐剤(抗酸化剤を含む)、賦形剤、崩壊剤、滑沢剤、結合剤、界面活性剤、可塑剤等から選択される1種もしくはそれ以上の補助成分を添加することもできる。
【0125】
本発明の化合物もしくはその製薬上許容される塩の有効用量および投与回数は、投与形態、患者の年令、体重、治療すべき症状の性質もしくは重篤度等により異なるが、通常、投与量は、1日当たり0.01〜1000mg/人、好ましくは5〜500mg/人であり、投与回数は、1日1回または分割して投与するのが好ましい。
【0126】
本発明の化合物は、好ましくは、IC50が被検物質なしの場合の蛍光値を基準に、被検物質の抑制活性が1以下、好ましくは、0.1μM以下、より好ましくは、0.01μM以下の値を有するもの、あるいは、10nM〜10μMの範囲内、好ましくは、10μM未満、より好ましくは、1μM未満のIC50値をもつような化合物である。
【0127】
製剤化に関するさらなる情報については、Comprehensive Medicinal Chemistry(Corwin Hansch;Chairman of Editorial Board),Pergamon Press 1990年の第5巻の第25.2章を参照することができる。
【0128】
本発明はまた、本発明の医薬組成物を製造するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0129】
本発明はまた、本発明の化合物またはその溶媒和物を使用するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0130】
本発明化合物は、医薬としての有用性を備えた化合物である。ここで、医薬としての有用性としては、代謝安定性がよい点、薬物代謝酵素の誘導も少ない点、他の薬剤を代謝する薬物代謝酵素の阻害も小さい点、経口吸収性の高い化合物である点、クリアランスが小さい点、または、半減期が薬効を発現するために十分長い点などが含まれる。
【0131】
本明細書において引用された、科学文献、特許、特許出願などの参考文献は、その全体が、各々具体的に記載されたのと同じ程度に本明細書において参考として援用される。
【実施例】
【0132】
以下、実施例によって本発明を更に詳細に説明するが、この実施例等により本発明の技術的範囲が限定されるものではない。
【0133】
使用機器、及び測定条件などは下記に記載したものを採用した。
【0134】
(材料および方法)
細胞株:HCT116、SW620、MDA−MB−231,MCF7、SKBR3、SW480 、A549、ヒト正常繊維芽細胞 CCD−1059SKは、大日本住友製薬(Dainippon Sumitomo Pharma,Osaka, Japan)を代理店とし、アメリカンタイプカルチャーコレクション(American Type Culture Collection(ATCC); Manassas, VA)より入手した。
【0135】
McCoy’s 5A培地、Earle’s培地およびRPMI−1640 培地は、Invitrogen Corp, Carlsbad, CA, USAから入手した。
【0136】
ペニシリン−ストレプトマイシン、ウシ胎仔血清(Fetal Bovine Serum)、L−グルタミン、およびMEM 非必須アミノ酸溶液(Non−Essential Amino Acids Solution)は、それぞれ、Invitrogen Corp, Carlsbad, CA, USA、Equitech−Bio Inc.,Kerrville, TX, USA、Invitrogen Corp,Carlsbad, CA, USAおよびInvitrogen Corp,Carlsbad, CA, USAから入手した。
【0137】
CO2インキュベーターは、三洋電機(MCO−18AIC)から入手した。
【0138】
WST−1を調製(試薬Aと試薬Bを混合)した。同仁化学研究所(Dojindo Laboratories, Kumamoto, Japan)から入手した。試薬A:WST−1 (16.3 mg; 終濃度5mmol/l), HEPES (23.8 mg; 終濃度20 mmol/l; pH7.4) 試薬B:1−メトキシ PMS (0.2 mmol/l), 5 ml。
【0139】
オートプレートリーダーは、三光純薬社(Sjeia AUTOREADER III #D−ER3)から入手した。
【0140】
(評価方法)
ヒトがん細胞増殖抑制活性試験(WST−1 assay)
HCT116、SW620及びA549の細胞懸濁液[McCoy’s 5A (1% 抗生物質(ペニシリン−ストレプトマイシン及び10% ウシ胎仔血清を含む), 1.0×104 cells/ml)]、並びに、SW480、SK−BR−3、MCF7、MDA−MB−231の細胞懸濁液[RPMI−1640培地 (1% 抗生物質(ペニシリン−ストレプトマイシン及び10% ウシ胎仔血清を含む ), 5.0 × 103cells/ml)]を作製し、96−well plateに1wellあたり135μlずつ分注した後、CO2インキュベーターで24時間培養した。各濃度の薬剤培養溶液15μlを1wellあたり分注した後、CO2インキュベーターで72時間培養した。その後、WST−1を調製 (試薬Aと試薬Bを混合)し、96−well Plateに1wellあたり16μlずつ分注した後、CO2インキュベーターで4時間培養した。オートプレートリーダー(測定波長450nm、 参照波長630 nm)により吸光度を測定し、各薬剤サンプルのIC50値を算出した。
【0141】
ヒト正常繊維芽細胞CCD−1059SKに対する増殖抑制(毒性)試験
L−グルタミン200mM (100×)液(liqid) (#25030−149)を10 ml/LでEagle’s培地に添加した。MEM 非必須アミノ酸溶液 10 mM (100×)(#11140−50) を培地に対し1%添加した。56℃で30分間おきに、非働化したウシ胎仔血清を10%添加して使用した。
細胞操作は、ヒトがん細胞増殖抑制活性試験と同様の操作で行い、各薬剤サンプルのCCD−1059SK株に対するIC50を求めた。
【0142】
(製造例1:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ビス(2−メチルプロパンアミド) K−152)
(1)2−アミノフェニルジスルフィド(2.02 mM)[東京化成工業株式会社(TCI社)]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[和光純薬工業株式会社(Wako社)]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製し、白色結晶を得た。
m.p. 144.5−146.4℃
IR (KBr) cm−1: 3244.0,3186.2, 2970.2, 2869.9, 1660.6, 1577.7, 1525.6, 1460.0, 1436.9, 1380.9, 1276.8,1238.4, 1201.6, 1157.2, 1099.3, 1035.7, 948.9, 937.3, 887.2, 744.5, 686.6,596.0, 470.6
1H NMR (CDCl3) δ:1.47 (12H, d, J = 6.8Hz), 3.43 (2H, septet, J = 7.2 Hz), 7.36 (2H, dt, J = 7.2 Hz), 7.43 (2H, d, J =8.0 Hz), 7.85 (2H, d, J = 8.0 Hz), 7.98 (2H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z: 389.14(M + H)+。
【0143】
(製造例2:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ビス(2,2−ジメチルプロパンアミド) K−153)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)トリメチルアセチルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 86.0−87.0℃
IR (KBr) cm−1: 3386.8, 3247.9,2869.9, 2391.6, 1678.0, 1643.2, 1575.7, 1467.7, 1433.0, 1361.7, 1305.7, 1226.6, 1161.1, 1058.8, 1035.7, 931.6, 916.1, 771.5, 750.3, 626.8, 455.2.
1H NMR (CDCl3) δ: 1.25 (18H, s), 6.94(2H, dt, J = 1.5 and 7.8 Hz), 7.21 (2H, dd, J = 1.5 and 7.8 Hz), 7.40 (2H, dd,J = 1.5 and 7.8 Hz), 8.46 (2H, dd, J = 1.5 and 8.4 Hz), 8.52 (2H, br s).
ESI−MS (positive mode) m/z: 417.17(M + H)+。
【0144】
(製造例3:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジオクタンアミド K154)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加える。
(3)n−オクタノイルクロリド(4.04 mM)[関東化学株式会社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 79.0−81.0℃
IR (KBr) cm−1: 3267.2, 3192.0,2923.9, 2848.7, 2405.1, 1662.5, 1577.7, 1523.7, 1465.8, 1438.8, 1409.9, 1284.5,1234.4, 1033.8, 962.4, 732.9, 686.6, 464.8, 405.0.
1H NMR (CDCl3) δ: 0.89 (6H, m), 1.30(16H, m), 1.65 (4H, m), 2.16 (4H, t, J = 7.6 Hz), 6.99 (2H, dt, J = 7.6 Hz),7.35 − 7.43 (4H, m), 7.97 (2H, brs), 8.40 (2H, brd, J = 7.6 Hz).
ESI−MS (positive mode) m/z: 501.26(M + H)+。
【0145】
(製造例4:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジシクロペンタンカルボキサミド K−201)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 157−158℃
1H NMR (CDCl3) δ: 1.55−1.86 (16H, m), 2.47 (4H, quintet, J = 7.6 Hz), 6.98 (2H,brt, J -= 7.6 Hz), 7.35 (2H, brd, J = 8.0 Hz), 7.40 (2H, brt, J = 8.0 Hz), 8.06(2H, brs), 8.42 (2H, brd, J = 8.4 Hz)。
【0146】
(製造例5:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジベンズアミド K−202)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)塩化ベンゾイル(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 144−145℃
1H NMR (CDCl3) δ: 6.95 (2H, dt, J = 1.5and 7.8 Hz), 7.31 (2H, dt, J = 1.5 and 8.4 Hz), 7.40 − 7.60 (8H, m), 7.69 (4H, dd, J = 1.5 and 8.4 Hz), 8.50(2H, dd, J = 1.5 and 8.4 Hz), 8.93 (2H, br s)。
【0147】
(製造例6:S−2−イソブチルアミドフェニル 2−メチルプロパンチオレート K−178)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[Wako社]をCH2Cl2(5 ml)で希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 92−94℃
IR (KBr) cm−1: 3286.5, 2968.2,2871.8, 1753.2, 1658.7, 1606.6, 1250.4, 1348.1, 1298.0, 1255.6, 1180.4, 1099.3,920.0, 842.8, 758.0, 694.3.
1H NMR (CDCl3) δ: 1.24 (6H, d, J= 6.9 Hz), 1.33 (6H, d, J = 6.9 Hz), 2.51 (1H, septet, J = 7.2 Hz), 2.94 (1H,septet, J = 7.2 Hz), 7.14 (1H, t, J = 7.6 Hz), 7.38 (1H, d, J = 6.4 Hz), 7.43(1H, t, J = 8.4 Hz), 7.76 (1H, br s), 8.35 (1H, d, J = 8.0 Hz)。
【0148】
(製造例7:S−2−ピバルアミドフェニル 2−メチルプロパンチオレート K−179)
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3) トリメチルアセチルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮し、得られた残渣をMeOH(5ml)に溶解させた。
(8)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(9)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(10)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(11)有機層を乾燥後し、5ml程度に濃縮した。
(12)この溶液にピリジン(400μl)を加えた。
(13)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に希釈し、上記の溶液に滴下した。
(14)常温、常圧で1時間撹拌した。
(15)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(16)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(17)有機層を乾燥し、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
オイル状の化合物を得た。
m.p. 88.5−90.6℃
1H NMR (CDCl3) δ: 1.25 (9H, s), 1.31 (15H,m), 2.93 (1H, septet, J = 7.0 Hz), 7.11 (1H, dt, J = 1.0 and 8.0 Hz), 7.38 (1H,dd, J = 8.0 and 1.0 Hz), 7.44 (1H, dt, J = 1.0 and 8.5 Hz), 8.05 (1H, brs),8.35 (1H, brd, J = 9.0 Hz).
ESI−MS (positive mode) m/z: 294.1(M + H)+。
【0149】
(製造例8:S−2−(シクロペンタンカルボキサミド)フェニル 2−メチルプロパンチオレート K−180)
本製造例は、製造例7に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)上記の中間体をMeOH(5ml)に溶解させた。
(9)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(10)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(11)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(12)有機層を乾燥し、5ml程度に濃縮した。
(13)この溶液にピリジン(400μl)を加えた。
(14)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に希釈、上記の溶液に滴下した。
(15)常温、常圧で1時間撹拌した。
(16)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(17)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(18)有機層を減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。白色結晶の化合物を得た。
m.p. 86−87℃
1H NMR (CDCl3) δ: 1.29 (6H, d, J = 6.8Hz), 1.58 − 1.93 (8H, m), 2.69 (1H,quintet, J = 8.4 Hz), 2.94 (1H, septet, J = 7.2 Hz), 7.12 (1H, t, J = 7.6 Hz), 7.26 − 7.47 (2H, m), 7.74 (1H, brs), 8.34 (1H, d, J = 8.0 Hz)。
【0150】
(製造例9:S−2−ベンズアミドフェニル 2−メチルプロパンチオレート K−181)
本製造例は、製造例7に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した。(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)上記の中間体をメタノール(MeOH)(5ml)に溶解させた。
(9)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(10)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(11)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(12)有機層を乾燥後、5ml程度に濃縮した。
(13)この溶液にピリジン(400μl)を加えた。
(14)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に加え、上記の溶液に滴下した。
(15)常温、常圧で1時間撹拌した。
(16)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(17)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(18)有機層を乾燥後、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。白色結晶の化合物を得た。
m.p. 65−66℃
1H NMR (CDCl3) δ: 1.27 (6H, d, J = 6.8Hz), 2.95 (1H, septet, J = 6.8 Hz), 7.42 − 7.57 (7H, m), 7.85(1H, d, J = 8.0 Hz), 8.48 (1H, d, J = 8.8 Hz), 8.51 (1H、 brs)。
【0151】
(製造例10:2−イソブチルアミドフェニル イソブチレートK−205)
(1)2−アミノフェノール(1.87mmol)[TCI社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[Wako社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥後、減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 88.5−90.6℃
1H NMR (CDCl3) δ: 1.25 (3H, d, J = 7.0Hz), 1.37 (3H, J = 6.0 Hz), 2.51 (1H, J = 7.0 Hz), 2.88 (1H, J = 7.0 Hz), 7.11(1H, d, J = 3.7 Hz), 7.22 (1H, m), 7.26 (1H, brs), 8.19 (1H, d, J = 7.8 Hz).
ESI−MS (positive mode) m/z:250 (M +H)+。
【0152】
(製造例11:N−(2−ヒドロキシフェニル)イソブチルアミド K−204)
(1)上記の反応によって得られたK−205をMeOH(5 ml)に溶解させた。
(2)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(3)その反応液をHClで酸性(pH 5〜6)にし、CHCl3で抽出した。
(4)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(5)有機層を乾燥後、減圧濃縮。白色結晶を得た。
m.p. 107.4−109.2℃
1H NMR (CDCl3) δ: 1.30 (6H, d, J = 6.9Hz), 2.65 (1H, septet, J = 7.0 Hz), 6.90 (1H, brt, J = 7.0 Hz), 7.01 (1H, brd,J = 7.0 Hz), 7.11 (1H, brt, J = 7.0 Hz), 7.54 (1H, br s), 8.89 (1H, br s)。
【0153】
(製造例12:3−イソブチリルベンゾ[d]チアゾール−2(3H)−オン K−209)
(1)2−ヒドロキシベンゾチアゾール(2.02 mM)[Wako社]を秤量しCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)イソプロピルクロリド(3.03mM)[Wako社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
油状化合物を得た。
油状物
1H NMR (CDCl3) δ: 1.28 (6H, d, J = 6.8Hz), 3.89 (6H, d, J = 6.8 Hz), 7.15 - 7.40 (3H, m), 8.10 (1H, d, J = 8.0 Hz)。
【0154】
(実施例13:2−(4−(クロロメチル)ベンズアミド)フェニル 4−(クロロメチル)ベンゾエートK−211)
2−アミノフェノール 0.51mlをCHCl3 10 mlに溶かし、ピリジン1.86 mlを加えた。これに、p−クロロメチルベンゾイルクロリド 2.29 gをCHCl310 mlに溶かした溶液を滴下し、氷浴中2時間攪拌した。反応液をCHCl3 30 mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、メタノール(MeOH)から再結晶し、目的化合物0.78gを得た。
m.p. 138.5℃
IR (KBr) cm−1:3347.2, 1782.1, 1715.6, 1659.6, 1531.4, 1445.5, 1416.6, 1171.7, 1077.2, 1014.5,750.3, 704.0, 634.5, 441.7.
1H NMR (CDCl3) δ: 4.57 (2H,s), 4.66 (2H, brs), 7.43 (2H, d, J = 8.0 Hz), 7.55 − 7.76 (4H, m), 7.75 (2H, d,J = 8.0 Hz), 8.14 (2H, d, J = 8.4 Hz), 8.22 (2H, d, J = 8.4 Hz).
ESI−MS(positive mode) m/z: 414.3 (M + H)+
FAB−MS m/z: 414(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18Cl2NO3,414.0664; found, 414.0659。
【0155】
(実施例14:2−(4−(ニトロオキシメチル)ベンズアミド)フェニル 4−(ニトロオキシメチル)ベンゾエートK−210)
アセトニトリル20mlにK−211を2.49 g 溶かし、これにAgNO3のアセトニトリル10 ml懸濁液を加えた。遮光下アルゴン気流中65 ℃(外浴)で8時間攪拌した。沈殿物を減圧下濾取し、少量のCHCl3で洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査をCHCl3:酢酸エチル:n−ヘキサン(1:3:1)を展開溶媒として、シリカゲルカラムクロマトグラフィーに付し、白色結晶のK−210を2.18 g得た。
m.p. 136.2℃
IR (KBr) cm−1:3348.2, 3041.5, 2895.9, 2544.9, 1920.0, 1715.6, 1597.9, 1504.4, 1454.2, 1258.5,1179.4, 1116.7, 1020.3, 857.3, 749.3, 631.6, 593.1, 450.3.
1H NMR (CDCl3) δ: 5.45 (2H,s), 5.53 (2H, s), 7.43 (2H, d, J = 8.0 Hz), 7.57 (2H, d, J = 8.4 Hz), 7.77 (2H,d, J = 8.0 Hz), 7.97 (1H, brs), 8.26 (3H, m).
ESI−MS(positive mode) m/z: 468.1 (M + H)+
FAB−MS m/z: 468(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18N3O9,468.1044; found, 468.1050。
【0156】
(実施例15:S− 2−(4−(クロロメチル)ベンズアミド)フェニル 4−(クロロメチル)ベンゾチオエートK−221)
アルゴン気流下、o−アミノチオフェノール0.64 mlをCH2Cl2 15 mlに溶解させ、この溶液にピリジン1.5 mlを加えた。これに、p−クロロメチルベンゾイルクロリド2.84 gをCH2Cl2 5 ml に溶かした溶液を加え、氷温で1時間撹拌した。反応液をCHCl3 80mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、シリカゲルカラムクロマトグラフィー(酢酸エチル:トルエン=1:8) により精製し、減圧濃縮し、白色結晶を得た。
m.p. 158.8−159.4℃
IR (KBr) cm−1: 3384.8,2974.0, 1674.1, 1502.4, 1409.9, 1101.3, 1018.3, 896.8, 804.3, 677.0, 541.7,495.7, 470.6, 435.9, 412.7.
1H NMR (CDCl3)δ: 4.59 (2H, s),4.65 (2H, s), 7.44 (2H, d, J = 8.0 Hz), 7.53 − 7.60 (4H, m), 7.79 (2H, d,J = 8.4 Hz), 8.07 (2H, d, J = 8.4 Hz), 8.51(1H, d, J = 8.4 Hz), 8.57 (1H, brs).
ESI−MS(negative mode) m/z: 430.2 (M − H)−
FAB−MS m/z: 430(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18Cl2NO2S,430.0436; found, 430.0439。
【0157】
(実施例16:S− 2−(4−(ニトロオキシメチル)ベンズアミド)フェニル 4−(ニトロオキシメチル)ベンゾチオエートK−220)
アセトニトリル30mlにK−221を100 mg 溶かし、これにAgNO3 800 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。沈殿物を減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、PLCにより精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK−220を7.5mg得た。
m.p. 171.8−172.9℃
IR (KBr) cm−1: 3859.3,3843.9, 3826.5, 3679.9, 3639.4, 2349.1, 1610.5, 1535.2, 1438.8, 752.2, 690.5,538.1, 453.2, 422.4.
1H NMR (CDCl3) δ: 5.45 (2H, s), 5.52(2H, s), 7.45 (2H, d, J = 8.0 Hz), 7.49−7.69 (4H, m), 7.83 (2H,d, J = 8.4 Hz), 8.10 (2H, d, J = 8.4 Hz), 8.49 (1H, d, J = 8.0 Hz),8.56 (1H, brs).
ESI−MS (negative mode) m/z:482.7 (M - H)−
FAB−MS m/z: 483 (M+H)+。
【0158】
(実施例17:2−(3−(クロロメチル)ベンズアミド)フェニル 3−(クロロメチル)ベンゾエート K−231)
アルゴン気流下、o−アミノチオフェノール109 mgをCHCl3 5 mlに溶解させ、この溶液にピリジン500μlを加えた。これに、m−(クロロメチル)ベンゾイルクロリド341μlを加え、室温で1時間撹拌した。反応液をCHCl3 5mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、シリカゲルカラムクロマトグラフィー(酢酸エチル:トルエン=1:8) により精製し、減圧濃縮し、目的の白色結晶を235.0 mg得た。
m.p. 155.5−157.4℃
IR (CHCl3) cm−1: 3018.4,2399.3, 1521.7, 1475.4, 1423.4, 1215.1, 1045.3, 927.7, 754.1, 669.3, 626.8.
1H NMR (CDCl3) δ: 4.66 (2H, s), 4.71(2H, s), 7.24−7.37 (4H, m), 7.41 (1H, t, J =7.6 Hz), 7.52 (1H, brd, J = 7.6 Hz), 7.56 (1H, t, J = 8.0 Hz), 7.72 (1H, brd, J= 8.0 Hz), 7.77 (1H, brs), 8.04 (1H, brs), 8.20 (1H, dd, J = 1.2 and 7.6 Hz),8.26 (1H, s), 8.32 (1H, d, J = 7.6 Hz).
ESI−MS (negative mode) m/z:430.5 (M − H)+ 。
【0159】
(実施例18:2−(3−(ニトロオキシメチル)ベンズアミド)フェニル 3−(ニトロオキシメチル)ベンゾエート K−230)
アセトニトリル15mlにK−231 を870 mg 溶かし、これにAgNO3 3915 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。沈殿物を、セライトを用いて減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、分取液体クロマトグラフィー(PLC)により精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK230を422.4mg得た。
m.p. 132.7−135.8℃
IR (CHCl3) cm−1: 3681.9,3620.1, 3018.4, 2399.3, 1521.7, 1423.4, 1215.1, 1031.8, 929.6, 754.1, 669.3,626.8.
1H NMR (CDCl3) δ: 5.38 (2H, s), 5.50(2H, s), 7.26−7.40 (3H, m), 7.47 (1H, t, J =7.6 Hz), 7.54 (1H, brd, J = 7.2 Hz), 7.61 (1H, t, J = 7.6 Hz), 7.72 (1H, J =7.6 Hz), 7.77 (2H, s), 7.99 (1H, brs), 8.27 (3H, s).
ESI−MS m/z: 468.4 (M+H)+。
【0160】
(実施例19:4−(クロロメチル)−N−フェニルベンズアミド K−241)
アニリン0.27 mlをCHCl3 10 mlに溶かし、ピリジン0.29 mlを加えた。これに、p−クロロメチルベンゾイルクロリド 680 mgをCHCl310 mlに溶かした溶液を滴下し、氷浴中2時間攪拌した。反応液をクロロホルム(CHCl3)30 mlで希釈後、順次1% 塩酸(HCl)、飽和炭酸水素ナトリウム(NaHCO3)、飽和食塩水で洗浄した。硫酸ナトリウム(Na2SO4)で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、クロロホルム、ヘキサン混液から再結晶し、目的化合物670mgを得た。
m.p. 164.5−166.3℃
IR (KBr) cm−1: 3346.3,1654.8, 1598.9,1529.4, 1438.8,1325.0, 1303.8, 1267.1, 769.5, 754.1, 713.6,690.5, 675.0, 651.9.
1H NMR (CDCl3) δ:.4.64 (2H, s),7.17 (1H, t, J = 8.0 Hz), 7.39 (2H, t, J = 8.0 Hz), 7.52 (2H, d, J = 8.0 Hz),7.64 (2H, d, J = 8.0 Hz), 7.78 (1H, brs), 7.87 (1H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z:246.6(M+ H)+。
【0161】
(実施例20:4−(フェニルカルバモイル)ベンジル 硝酸塩 K−240)
アセトニトリル20 mlにK−241 を245.7mg 溶かし、これに硝酸銀(AgNO3)849.35 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。反応液を、セライトを用いて減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、PLCにより精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK−240を14.5mg得た。
m.p. 168.0−170.9℃
IR (CHCl3) cm−1: 3681.9,3620.1, 3018.4, 2399.3, 1523.7, 1477.4, 1423.4, 1215.1, 1031.8, 929.6, 786.9,754.1, 669.3, 626.8.
1H NMR (CDCl3) δ:.5.50 (2H, s),7.18 (1H, t, J = 6.8 Hz), 7.39 (2H, t, J = 8.0 Hz), 7.53 (2H, d, J = 7.6 Hz),7.63 (2H, d, J = 8.0 Hz), 7.80 (1H, brs), 7.91 (1H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z: 273.5(M + H)+。
【0162】
(実施例21:生物学的試験)
各製造例および実施例に記載の化合物について、上記したヒトがん細胞増殖抑制活性試験およびヒト正常繊維芽細胞CCD−1059SKに対する増殖抑制(毒性)試験を行った。
【0163】
その結果を、表1及び表2に示す。
表1 bis[2−(アシルアミノ)フェニル]ジスルフィド及びS−[2−(アシルアミノ)フェニル]アルカンチオレート、並びにその類縁体のヒト乳がん細胞並びにヒト正常繊維芽細胞に対する細胞増殖抑制活性(IC50)
【0164】
【表1】
【0165】
【0166】
【0167】
(表2 ヒト大腸がん細胞並びにヒト正常繊維芽細胞に対する増殖抑制活性(IC50))
【0168】
【表2】
【0169】
(実施例21:免疫沈降試験)
本発明の化合物の薬理活性について、そのメカニズムを探るために免疫沈降実験を行った。その手順は以下のとおりである。
【0170】
1.0×106cell/mlのHCT116細胞懸濁液或いは2.0×106cell/mlのSW620細胞懸濁液を60mm ディッシュに播種し、24時間培養後無血清培地 1mlで2回ディッシュ内を洗浄した。ついで、5 mlの無血清培地で再度洗浄し、24時間更に培養した。被検物質を培地に加え、18時間培養した。細胞を採取し、2度PBSで洗浄し、溶解用緩衝液(lysis buffer)(1M Tris−HCl (pH 8.0) 1ml, 5M NaCl, 1% nonidet P−40, 1 mg/ml アプロチニン(aprotinin),1 mg/ml ロイペプチン(leupeptin), 1 mg/ml ペプスタチン(pepstatin), 200 mM Na3VO4,200 mM NaF, 200 mM Na4P2O7, グリセロール(glycerol), および200mM PMSF) 400μlで再懸濁させた。得られた溶解物(lysates)は15000 × g, 4°Cで15分遠心処理した。得られた上清のタンパク質溶液のタンパク質濃度はBio−Radタンパク質アッセイキット(protein assay kit)で測定し、 その一部200μg (220μl) に対し、Anti−TCF4 モノクローナル抗体(2.5μl)及び600μl Protein G−Sepharose (2 mg/ml) を加え、4度で1時間インキュベーションした。ビーズを溶解用緩衝液で3度洗浄し、タンパク質をロード用緩衝液(loadingbuffer)に溶かし、常法にしたがいSDS/PAGEにより、ウェスタンブロッティングを行った。最終の検出は 「ECL Plus reagents」により行った。その結果を以下の表および図1に示す。
【0171】
【表3A】
【0172】
【表3B】
【0173】
図1の結果からも明らかなように、NCX4040は10μMで、β−カテニン/TCF4複合体レベルを約30%まで、20μMで10%まで減少させた。一方、K153のβ−カテニン/TCF4複合体形成阻害効果のIC50は約150μMであった。
【0174】
以上の結果から、本発明の化合物は、β−カテニン/TCF4複合体形成阻害に関連する任意の疾患における用途にも使用されることが理解される。
【0175】
(実施例22:細胞周期制御試験)
本発明の化合物の薬理活性について、そのメカニズムを探るために細胞周期制御試験を行った。その手順は以下のとおりである。
【0176】
1.5×106cell/mlのHCT116細胞懸濁液を作製し、24枚のディッシュ(コントロール 2枚、K−210添加濃度:1μM、5μM、10μM、20μM、40μM、60μM; NCX−4040添加濃度:10μM、20μM、30μM、50μMのディッシュを、それぞれ24hと48h用2枚ずつ)に予め培地を入れ、細胞懸濁液を1ml加えた。抗生物質(ペニシリン−ストレプトマイシン)を培地の量に対して1/100になるように加え、細胞の様子を顕微鏡で確認してからCO2インキュベーターで24時間培養した。
【0177】
ディッシュの培地をピペットマンで取り、無血清培地1mlで2回ディッシュ内を洗浄した。ディッシュに培地を5ml加え、CO2インキュベーターで24時間培養した。
【0178】
がん細胞に添加する薬剤サンプルK−210とNCX−4040を秤量し、DMSO(100μl)に溶解させ、培地(900μl)加えた後、培地で希釈し前培養で用意したディッシュの通りのサンプル溶液を製作した。ディッシュの培地を捨て、4mlの無血清培地を加えた。0時間以外の各ディッシュにサンプル溶液1mlを加え、24時間、また48時間培養した。培養した細胞をトリプシン3mlで剥がし、遠沈管に入れた培地に懸濁し、1000rpmで、5分間、遠心分離した。上清を捨て、常温に戻した「Buffer Solution」 (BD社 CycleTEST Plus)を1mlずつ加え、ボルテックスで穏やかに懸濁させた後、300×g5分間で遠心分離した。これを2回行った。上清を捨て、常温に戻したSolution A (BD社 CycleTEST Plus)を250μl加え、穏やかに攪拌、懸濁させ、室温で10分間静置した。室温に戻したSolutionB (BD社 CycleTEST Plus)を200μl加え、穏やかに攪拌、懸濁させ、室温で10分間静置した。氷温に解凍した「Solution C 」(BD社CycleTEST Plus)を200μl加え、完全に遮光し、氷上で10分間静置後、フローサイトメーター(Beckton Dickinson社FACScan)で測定した。その測定結果を図2(図2Aおよび図2B)に示す。
【0179】
図2の結果からも明らかなように、K210はヒト大腸がん細胞HCT116株の細胞周期をG2/M期で停止させていることが分かった。
【0180】
以上の結果から、本発明の化合物は、細胞周期をG2/M期で停止させることによって処置しうる任意の疾患における用途にも使用されることが理解される。
【0181】
(実施例23:TUNEL法によるアポトーシスの検出試験)
本発明の化合物の薬理活性について、そのメカニズムを探るためにTUNEL法によるアポトーシスの検出試験を行った。TUNEL法によるアポトーシスの検出試験は、Promega社 DeadEndTMFluorometric TUNEL System (#G3250)を用いて行った。その手順は以下のとおりである。
【0182】
薬剤サンプルを調製し、コントロールを除く、各60 mm ディッシュ(70 - 80% コンフルエンス(confluence)) に0.5 mlずつ加えCO2インキュベーターで24時間培養した。各ディッシュの培地をすべて、個々の遠沈管に回収した。
【0183】
残りの接着している細胞を、氷冷1×PBS (NaCl, 8.006 g; KCN, 0.1997 g; NaHPO4・12H20,2.897 g; KH2PO4, 0.200 g) 1 mlで2回洗浄した。37 ℃で温めたトリプシン1mlを加え、細胞をピペッティングにより剥がし、上記で取った培地に加え、300×gにて10分間遠心分離した。上清を捨てた。氷冷1×PBS3 mlを加え、ボルテックスで振とうした後、300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。
【0184】
氷冷1×PBS0.5 ml加え、4% パラホルムアルデヒドを加え、十分にピペッティングした。氷中に20分間置き、細胞を固定した。300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた。氷冷1×PBS3 mlを加え、ボルテックスで振とうした後、300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。
【0185】
氷冷1×PBS0.5 ml加え、70% エタノールを5ml加え、ピペッティングをした。
−20 ℃にて4時間放置。300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた。氷冷1×PBS3mlを加え、ボルテックス振とうした後300×gにて10分間遠心分離した。
【0186】
細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。BSA (5 mg/ml) およびTriton X−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。基準とするサンプル(24時間コントロール)に含まれる細胞数を、血球計算版を用いて計測した。全サンプルのOD600値を、吸光度計を用いて測定した。
【0187】
最も細胞数の少ないサンプルに合わせて、全サンプルを一定量にし、1.5 mlのエッペンドルフチューブに移した。420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
【0188】
80μlの平衡化用緩衝液(Equilibration buffer)を加えて懸濁し、室温で5分間静置した。
【0189】
TdTインキュベーションバッファーを以下の要領で作成した。
【0190】
Equilibration buffer 45μl×サンプル数
Nucleotide Mix 5μl×サンプル数
rTdT Enzyme 1μl×サンプル数。
【0191】
全量を混ぜて、遮光して氷中に置いておく。
【0192】
420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
各サンプルに、TdTインキュベーションバッファーを50μlずつ加え、懸濁した。
完全に遮光し、37℃のヒートブロック上で90分間インキュベートした(その間、エッペンドルフチューブを15分おきにボルテックス振とうし、遠心分離器によりチューブ壁に付着した沈殿物と溶液を底に集めた)。20mM EDTA溶液を1 ml加え、穏やかにボルテックスすることで、反応を停止させた。
【0193】
420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
BSA (5 mg/ml) およびTritonX−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。
【0194】
細胞が沈殿していることを確認した後、上清を捨てた。BSA (5 mg/ml) およびTriton X−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。細胞が沈殿していることを確認した後、上清を捨てた。5μg/mlにMilli−Qで200倍希釈したPIを0.5 ml加え、ピペッティングにより懸濁後、各サンプルに25μlのRNaseを加え攪拌した。5μg/ml にMilli−Qで200倍希釈したPIを0.5ml加え、ピペッティングにより懸濁後、各サンプルに25μlのRNaseを加え攪拌した。調製した溶液の全量を穏やかにボルテックスで撹拌し、セルストレーナーを通して細胞塊を取り除いた。セルストレーナーをFACScanにセットし、測定した。測定結果を図3に示す。
【0195】
図3に示すように、K210はヒト大腸がん細胞株HCT116株に対し、アポトーシスを誘導することが分かった。
【0196】
以上の結果から、本発明の化合物は、アポトーシスの誘導によって処置しうる任意の疾患における用途にも使用されることが理解される。
【0197】
(実施例24:ウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出)
本発明の化合物の薬理活性について、そのメカニズムを探るためにウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出を行った。その手順は以下のとおりである。
【0198】
HCT116細胞を60mmディッシュに添加(2.0 × 106 cells/ディッシュ)し、CO2インキュベータで24時間培養した。細胞を無血清培地(McCoy’s5A) 1 mlで2回洗浄後、直ちに無血清培地 5 mlを加え、更に24時間培養した。細胞を、各ディッシュ毎に無血清培地4 mlで洗浄し、K−210 (20μM,40μM, 60μM)を、0分間, 15分間, 30分間, 1時間, 2時間, 4時間, 6時間, 8時間の各時間培養した。同様に、SKBR3細胞を60 mmディッシュに添加(1.0 × 106 cells/ディッシュ)し、CO2インキュベータで24時間培養した。細胞を無血清培地(RPMI−1640)1 mlで2回洗浄後、直ちに無血清培地 5 mlを加え、更に24時間培養した。細胞を、各ディッシュ毎に無血清培地4 mlで洗浄し、K−210 (1.0,5.0, 10, 20, 40 and 60μM)を、24時間培養した。各接着細胞をPBSで二回洗浄し、Covance Laboratories lysisbuffer (Immunochemistry Protocols for use with Innovative Antibody Products byCovance Research Products. http://store.crpinc.com/prot_western.aspx r) (ただし、pH7.4の代わりにpH 8.0 、1% Triton−X 100の代わりに1% Nonidet P−40 を採用した) を200μMを加え、懸濁した。細胞溶解液は4度で15分間遠心処理(14000× g )し、得られたタンパク溶液の濃度をBio−Rad protein assay kitにより測定した。均一濃度のたんぱく質溶液をSDS−PAGE に展開し、PVDFメンブレンにトランスファーした。一次抗体として、抗アクチン(actin)モノクローナル抗体、ERKモノクローナル抗体, p−ERKモノクローナル抗体, JNKモノクローナル抗体,p−JNKモノクローナル抗体, p38モノクローナル抗体, p−p38モノクローナル抗体, p21/WAF1モノクローナル抗体(Sigma, St.Louis, MOから入手可能)を使用し、2,500倍に希釈した。また、セイヨウワサビ ペルオキシダーゼ標識二次抗体を2,500倍に希釈し、各バンドの蛍光強度をECL化学発光法(chemiluminescence)で測定した。結果を図4に示す。図4の結果から、ストレス応答型主要MAPKsであるp38およびJNKのリン酸化体のタンパクレベルが上昇したことにより、K−210等はこの経路によってアポトーシスを誘導した可能性が高いと推定された。
【0199】
以上の結果から、本発明の化合物は、主要MAPKs、ERK、JNK、p38、およびそのリン酸化の誘導によって処置しうる任意の疾患における用途にも使用されることが理解される。
【0200】
以上のように、本発明の好ましい実施形態を用いて本発明を例示してきたが、本発明は、この実施形態に限定して解釈されるべきものではない。本発明は、特許請求の範囲によってのみその範囲が解釈されるべきであることが理解される。当業者は、本発明の具体的な好ましい実施形態の記載から、本発明の記載および技術常識に基づいて等価な範囲を実施することができることが理解される。本明細書において引用した特許、特許出願および文献は、その内容自体が具体的に本明細書に記載されているのと同様にその内容が本明細書に対する参考として援用されるべきであることが理解される。
【産業上の利用可能性】
【0201】
本発明は、β−カテニン/TCF複合体の形成に関与する疾患、たとえば、がんを処置するための医薬、それに使用される化合物またはその溶媒和物を提供する。本発明化合物は実施例の記載の通り、優れた抗がん活性を示す。
【図面の簡単な説明】
【0202】
【図1】図1は、免疫沈降試験(実施例21)の結果を示す。左側は大腸がん細胞株HCT116を示し、右側は大腸がん細胞SW620を示す。二段目には各化合物の記号を示す。上のウェスタンブロッティングの結果はβ−カテニンの結果、下はTCF4での結果を示す。下のグラフには、それぞれ左側は大腸がん細胞株HCT116を示し、右側は大腸がん細胞SW620のβ−カテニン−TCF複合体量の濃度依存性を示したグラフを示す。
【図2A】図2Aは、細胞周期制御試験実験(実施例22)の結果(フローサイトメトリ)を示す。左上はコントロール、右上は、K210 1μM,左下にはK210 5μM、右下にはK210 10μMのものを示す。
【図2B】図2Bは、細胞周期制御試験実験(実施例22)の結果(フローサイトメトリ)を示す。参考物質のNCX−4040について、左上はコントロール、右上は、10μM,左下には20μM、中下には30μM、右下には50μMのものを示す。
【図3】図3は、TUNEL法によるアポトーシスの検出試験(実施例23)の結果を示す。左上はコントロール、右上は、K210 1μM,左下にはK210 5μM、右下にはK210 10μMのものを示す。横軸は、FL2−H (PI)を示し、縦軸 FL1−H (フルオレセイン−12−dUTP)を示す。
【図4】図4は、ウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出(実施例24)の結果を示す。各々についての説明は、以下のとおりである。左パネルに20μM、中パネルに40μM、右パネルに60μMを示す。マーカーは、上から、p0ERK1、pERK2、pJNK1、pJNK2、p−p38、p38、アクチン、アクチン、p21/WAF1を示す。ストレス応答型主要MAPKsであるp38およびJNKのリン酸化体のタンパクレベルが上昇したことにより、K−210等はこの経路によってアポトーシスを誘導した可能性が高いと推定された。
【技術分野】
【0001】
本発明は、アシルアミノフェニル基を有する化合物に関する。より詳細には、本発明は、医薬品:抗がん剤、抗炎症剤、胃壁保護剤、心筋梗塞予防剤、脳梗塞予防剤、慢性動脈閉塞症(PAOD)、NOS2発現阻害脳卒中予防剤、アルツハイマー進行抑制・予防剤、血液抗凝固剤に関する。
【背景技術】
【0002】
転移性・悪性乳がん細胞や大腸がん細胞上に過剰発現しているErbB/Her受容体ファミリーの一つ、Her2タンパク質、を認識し、その2量体化を阻害して乳がん細胞の増殖を抑制する制癌剤として抗体医薬品Herceptin等が知られている。しかし、タンパク質製剤であるため、代謝速度は速い、投与に長時間を要する点滴製剤である、高価である、等の様々なデメリットをもっている。一方、特に大腸がん細胞では、Wntシグナル上のタンパク質β−カテニンが異常に蓄積し、高レベルのβ−カテニン/TCF複合体がサイクリンD1[非特許文献1,非特許文献2]やc−myc[非特許文献3]の遺伝子発現を促進し、大腸がんの細胞増殖を活発化させることが知られている。最近、このErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害する、悪性乳がんや大腸がんの治療薬の開発研究が活発化している。
【0003】
アスピリンは、心筋梗塞および脳卒中の予防、がん疼痛の緩解に使用されている。また、最近では、アルツハイマーや結腸がんのリスクを減らす効果があることも知られてきた。しかし、消化管出血や潰瘍穿孔などの消化管毒性があることが障害となっている。最近、4−(ニトロオキシメチル)フェニル基を有するNO供与型アスピリン(NO−ASA):NCX4040やNCX4016[非特許文献4、非特許文献5および非特許文献6]などが見出された。これらは、アスピリンの有益な効果を維持しつつ、消化管毒性の少ないことが知られている。特に、前者NCX4040は、大腸がんのがん化初期に関わる、Wntシグナル上のタンパク質複合体β−カテニン/TCFの結合を阻害する効果を示す他、NF−κBのDNA結合阻害、NOS2発現阻害、COX2発現誘導、プロスタグランジンE2レベルの上昇、等の効果を示すことから、複数経路で大腸がんなどの予防・治療効果を発揮すると推定されている[非特許文献4]。本薬剤はNO供与型化合物としてデザインされた化合物であるが、最近の研究で、実は、主としてキノンメサイド(以下の化1のスキーム1参照)を経由するアルキル化剤として働き、グルタチオンなどのSH含有生体化合物に結合することにより、酸化ストレスを亢進し、がん細胞の増殖を抑制する効果を発揮しているという説が有力となった[非特許文献7,非特許文献8]。NO−ASA系化合物は、大腸がんへの治療薬として、単独、或いは、既存の抗がん剤との併用で、それぞれ前臨床試験、第1相臨床試験が実施されている。
【0004】
(スキーム 1)
【0005】
【化1】
【非特許文献1】Shtutman M., Zhurinsky J., Simcha I., Albanese C., D’Amico M., Pestell R, Ben−Ze’evA., Proc. Natl. Acad. Sci. USA, 96, 5522−5527 (1999).
【非特許文献2】Tetsu O. & McCormick F., Nature, 398, 422−426 (1999).
【非特許文献3】He T.C., Sparks A.B., Rago C., Hermeking H., Zawel L., da Costa L.T., MorinP.J., Vogelstein B., Kinzlcr K.W., Science, 281, 1509−1512 (1998).
【非特許文献4】Williams J.L., Nath N., Chen J., Hundley T.R., Gao J., Kopelovich L., KashfiK., Rigas B., Cancer Res., 63, 7613−7618 (2003).
【非特許文献5】Nath N., Kashfi K.,Chen J., Rigas B., Proc. Natl. Acad. Sci. USA, 100, 12584−12589 (2003).
【非特許文献6】Hulsman N., MedemaJ.P., Bos C., Jongejan A., Leurs R., Smit M.J., de Esch I.J., Richel D.,Wijtmans M., J. Med. Chem., 50, 2424−2431 (2007).
【非特許文献7】Hulsman N., MedemaJ.P., Bos C., Jongejan A., Leurs R., Smit M.J., de Esch I.J., Richel D.,Wijtmans M., J. Med. Chem., 50, 2424−2431 (2007).
【非特許文献8】Dunlap T.,Chandrasena R.E.P., Wang Z., Sinha V., Wang Z., Thatcher G.R.J., Chem. Res.Toxxicol., 20, 1903−1912 (2007).
【非特許文献9】Okamoto H., YonemoriF., Wakitani K.,分間owa T., Maeda K.,Shinkai H., Nature, 406, 203−207 (2000).
【非特許文献10】Shinkai H., Maeda K.,Yamasaki T., Okamoto H., Uchida I., J. Med. Chem., 43, 3566−3572 (2000).
【非特許文献11】Yamakawa S., DemizuA., Kawaratani Y., Nagaoka Y., Terada Y., Maruyama S., Uesato S., Biol. Pharm. Bull., 31 (5), 910−920 (2008).
【発明の開示】
【発明が解決しようとする課題】
【0006】
一般に、タンパク質同士の結合を低分子で阻害することは、困難であると考えられていた。発明者は、Her2/Herceptin間、β−カテニン/TCF間などの結合部位に存在するCys残基に着目し、Cysと‐S‐S‐結合をする低分子化合物bis[2−(アシルアミノ)フェニル]ジスルフィド及びS−[2−(アシルアミノ)フェニル]アルカンチオレート[非特許文献9,非特許文献10]を評価した。その結果、Her2高発現型乳がん細胞株SKBR−3や大腸がん細胞株HCT116等に高い細胞増殖抑制活性を示す低分子化合物K−153およびK−154を見出した。
(K−153)
【0007】
【化2】
【0008】
(K−154)
【0009】
【化3】
【0010】
特に前者は、β−カテニン/TCF の結合を阻害し、また、ストレス応答の主要MAPKsを活性化し、がん細胞の増殖を抑制していることが推定された[非特許文献11]。
【0011】
そこで、本発明は、大腸がんや乳がん等のがんの治療薬として非常に有望な候補化合物を提供することを課題とする。
特に、NCX4040
【0012】
【化4】
【0013】
は、β−カテニン/TCF結合の阻害効果の他、NF−κBのDNA結合阻害、NOS2発現阻害、COX2発現誘導、プロスタグランジンE2レベルの上昇、等の効果を示し、複数経路で抗大腸がん活性を発揮する。本剤は広範ながん細胞に対して細胞増殖抑制効果を示すが、現在、大腸がん予防・治療薬としての適用で、単独、或いは、既存の抗がん剤との併用で、前臨床試験、第1相臨床試験を実施中である。しかし、NCX4040は、ある種の大腸がん細胞株(HCT116株、SW480株など)や乳がん細胞株(SKBR−3株など)に対しは、静細胞的に作用し、その殺細胞或いは細胞増殖抑制効果(実施例の表1,2を参照)は低いという問題点があった。
【課題を解決するための手段】
【0014】
上記課題に対して、本発明者らは、前述のS−[2−(アシルアミノ)フェニル]アルカンチオレートや[2−(アシルアミノ)フェニル]アルカノエートにニトロオキシメチルフェニル基を賦与した構造の化合物を合成した。特に、K−210、K−220、K−230
(K−210)
【0015】
【化5】
【0016】
(K−220)
【0017】
【化6】
【0018】
(K−230)
【0019】
【化7】
【0020】
はアシルアミノフェニル骨格に2個のニトロオキシメチルフェニル基を有し、かつ、そのパラ位、メタ位にカルボニル基を有することから、高いNO産生能が期待された。ヒトがん細胞増殖抑制活性試験(実施例の表1,2)の結果、K210、K220などは、NCX4040では増殖抑制活性がそれほど高くない大腸がん細胞株HCT116(IC50,68.0μM)および乳がん細胞株SKBR−3(IC50,17.0μM)に対し、高い増殖抑制活性を示した[HCT116 IC50,0.67μM(K210);0.72μM(K220):SKBR−3 IC50,0.66μM(K210);0.69μM(K220)]。
【0021】
特記すべきことに、K210、K220は、NCX4040と同様、ヒト正常繊維芽細胞に対して、200μMレベルという高濃度でも事実上毒性を示さなかった。したがって、K210、K220は大腸がんおよび乳がんの治療薬として非常に有望な候補化合物であると期待される。
【0022】
免疫沈降試験(図1)で、K−210は、NCX4040とは異なり、β−カテニン/TCFの結合を阻害しないことが分かった。一方、K210は、細胞周期制御試験 (図2)で、NCX4040と比べて低濃度(5μM)で大腸がん細胞HCT116株の増殖をG2/M期で停止させることが分かった。ウェスタンブロッティングで、K210はp53変異型乳がん細胞株SKBR3において、細胞周期制御因子p21/WAF1タンパク質レベルを上昇させることから、G2/M期停止は本タンパク質による細胞周期停止効果により起こると推定された(図4)。また、TUNEL法によるアポトーシス検出試験(図3)により、5μMで顕著にアポトーシスを誘起することも分かった。K−210はウェスタンブロッティングで、ストレス応答型主要MAPKsであるp38およびJNKのリン酸化を促進したことから、この経路の活性化によってアポトーシスが誘導された可能性が強い(図4)と推定した。
【0023】
本発明は、発明を実現する技術的手段としてヒトがん細胞移植ヌードマウス実験、前臨床試験、治験、GLP、GMP、GCP、GMP対応での原末、薬剤の製造などを考慮して実施することができる。また、HCT116株などヒトがん細胞移植ヌードマウス実験により、in vivoでの有効性を確認することができる。前臨床試験、臨床試験で安全性、有効性を確認することができる。GMP対応での薬剤の製造することができる。
【0024】
以上から、本発明は以下を提供する。
【0025】
1つの局面において、本発明は、以下の構造を有する化合物:
【0026】
【化101】
【0027】
であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物またはその溶媒和物(置換基によっては酸またはアルカリになり得、これらの場合は塩を形成しうる)などのプロドラッグに関する。
【0028】
本発明は、好ましくは、以下の構造を有する化合物またはその溶媒和物:
【0029】
【化102】
【0030】
であって、
R2は、ハロゲンまたはONO2で置換されたアルキル基であり、
R3は、ハロゲンまたはONO2で置換されたアルキル基であり、
Aは、−O−、−S−または−NH−である、
化合物に関する。
【0031】
1つの好ましい実施形態において、R2およびR3は、−CH2−Clまたは−CH2−ONO2である。
【0032】
1つの好ましい実施形態において、R2およびR3は、−CH2−Clまたは−CH2−ONO2であり、パラ位に存在する。
【0033】
1つの好ましい実施形態において、R2およびR3は、−CH2−ONO2である。
【0034】
1つの好ましい実施形態において、R2およびR3は、−CH2−ONO2であり、パラ位に存在する。
【0035】
1つの好ましい実施形態において、以下からなる群より選択される、請求項1に記載の化合物。
【0036】
【化103】
【0037】
【0038】
【0039】
【0040】
【0041】
【0042】
【0043】
【0044】
またはその溶媒和物が提供される。
【0045】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、医薬組成物を提供する。
【0046】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、抗がん剤を提供する。
【0047】
好ましい局面において、本発明は、本発明の上記化合物またはその溶媒和物を含む、乳がん、結腸癌および肺がんのいずれかまたは複数に対する抗がん剤を提供する。
【0048】
別の実施形態において、本発明は、
【0049】
【化104】
【0050】
および
【0051】
【化105】
【0052】
からなる群より選択される少なくとも1つ化合物またはその溶媒和物を含む、抗乳がん剤を提供する。
【0053】
別の実施形態において、本発明は、
【0054】
【化106】
【0055】
【化107】
【0056】
および
【0057】
【化108】
【0058】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗肺がん剤を提供する。
【0059】
別の局面において、本発明は、本発明の上記化合物またはその溶媒和物の、医薬組成物の製造のための使用に関する。
【0060】
他の局面において、本発明は、本発明の上記化合物またはその溶媒和物の、抗がん剤の製造のための使用に関する。
【0061】
他の局面において、本発明は、本発明の化合物またはその溶媒和物を投与する工程を包含する治療方法に関する。
【0062】
他の局面において、本発明は、本発明の上記化合物またはその溶媒和物を投与する工程を包含するがんの治療方法に関する。
【0063】
従って、本発明のこれらおよび他の利点は、以下の詳細な説明を読めば、明白である。
【発明の効果】
【0064】
新規作用機序に基づく大腸がん、乳がんなどのがん治療薬が開発することができ、医療に対する貢献度は大きい。
【発明を実施するための最良の形態】
【0065】
以下、本発明を最良の形態を示しながら説明する。本明細書の全体にわたり、単数形の表現は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。従って、単数形の修飾語等(例えば、英語の場合は「a」、「an」、「the」等の冠詞など)は、特に言及しない限り、その複数形の概念をも含むことが理解されるべきである。また、本明細書において使用される用語は、特に言及しない限り、当上記分野で通常用いられる意味で用いられることが理解されるべきである。したがって、他に定義されない限り、本明細書中で使用される全ての専門用語および科学技術用語は、本発明の属する分野の当業者によって一般的に理解されるのと同じ意味を有する。矛盾する場合、本明細書(定義を含めて)が優先する。
【0066】
以下に本明細書において用いられる各用語の意味を説明する。各用語は本明細書中、統一した意味で使用し、単独で用いられる場合も、または他の用語と組み合わされて用いられる場合も、同一の意味で用いられる。
【0067】
「ハロゲン」とは、フッ素、塩素、臭素およびヨウ素が挙げられる。
【0068】
「アルキル」とは、炭素数1〜10個の直鎖状又は分枝状のアルキル基を包含し、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ぺンチル、イソぺンチル、ネオペンチル、n−ヘキシル、イソヘキシル、n−ヘプチル、n−オクチル、n−ノニル、n−デシル等が挙げられる。好ましくは、炭素数1〜6または1〜4個のアルキルであり、例えば、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、n−ぺンチル、イソペンチル、ネオペンチル、n−ヘキシル、イソヘキシルが挙げられる。
【0069】
「アルケニル」とは、上記「アルキル」に1個又はそれ以上の二重結合を有する炭素数2〜8個の直鎖状又は分枝状のアルケニルを包含し、例えば、ビニル、1−プロペニル、2−プロペニル、1−ブテニル、2−ブテニル、3−ブテニル、1,3−ブタジエニル、3−メチル−2−ブテニル等が挙げられる。
【0070】
「アルキニル」とは、上記「アルキル」に1個又はそれ以上の三重結合を有する炭素数2〜8個の直鎖状又は分枝状のアルキニルを包含し、例えば、エチニル、プロピニル、ブチニル等が挙げられる。さらに1個又はそれ以上の二重結合を有していてもよい。
【0071】
「シクロアルキル」とは、炭素数3〜15の環状飽和炭化水素基を包含し、例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル、橋かけ環式炭化水素基、スピロ炭化水素基などが挙げられる。好ましくは、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、橋かけ環式炭化水素基が挙げられる。
【0072】
「シクロアルケニル」は、炭素数3〜7個の環状の不飽和脂肪族炭化水素基を包含し、例えば、シクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニル、シクロヘプテニルが挙げられ、好ましくはシクロプロペニル、シクロブテニル、シクロペンテニル、シクロヘキセニルである。シクロアルケニルには、環中に不飽和結合を有する橋かけ環式炭化水素基およびスピロ炭化水素基も含む。
【0073】
「アリール」とは、単環芳香族炭化水素基(例:フェニル)及び多環芳香族炭化水素基(例:1−ナフチル、2−ナフチル、1−アントリル、2−アントリル、9−アントリル、1−フェナントリル、2−フェナントリル、3−フェナントリル、4−フェナントリル、9−フェナントリル等)を包含する。好ましくは、フェニル又はナフチル(1−ナフチル、2−ナフチル)が挙げられる。
【0074】
「ヘテロアリール」とは、単環芳香族複素環式基及び縮合芳香族複素環式基を包含する。単環芳香族複素環式基は、酸素原子、硫黄原子、および/又は窒素 原子を環内に1〜4個含んでいてもよい5〜8員の芳香環から誘導される、置換可能な任意の位置に結合手を有していてもよい基を包含する。縮合芳香族複素環式基は、酸素原子、硫黄原子、および/又は窒素原子を環内に1〜4個含んでいてもよい5〜8員の芳香環が、1〜4個の5〜8員の芳香族炭素環もしくは他の5〜8員の芳香族ヘテロ環と縮合している、置換可能な任意の位置に結合手を有していてもよい基を包含する。
【0075】
「ヘテロサイクル」とは、酸素原子、硫黄原子、及び/又は窒素原子を環内に1〜4個含んでいてもよく、置換可能な任意の位置に結合手を有していてもよい非芳香族複素環式基を包含する。また、そのような非芳香族複素環式基がさらに炭素数1〜4のアルキル鎖で架橋されていてもよく、シクロアルカン(5〜6員環が好ましい)やベンゼン環が縮合していてもよい。非芳香族であれば、飽和でも不飽和でもよい。好ましくは5〜8員環である。例えば、1−ピロリニル、2−ピロリニル、3−ピロリニル、1−ピロリジニル、2−ピロリジニル、3−ピロリジニル、ピロリジノン、1−イミダゾリニル、2−イミダゾリニル、4−イミダゾリニル、1−イミダゾリジニル、2−イミダゾリジニル、4−イミダゾリジニル、イミダゾリジノン、1−ピラゾリニル、3−ピラゾリニル、4−ピラゾリニル、1−ピラゾリジニル、3−ピラゾリジニル、4−ピラゾリジニル、ピペリジノン、ピペリジノ、2−ピペリジニル、3−ピペリジニル、4−ピペリジニル、1−ピペラジニル、2−ピペラジニル、ピペラジノン、2−モルホリニル、3−モルホリニル、モルホリノ、テトラヒドロピラニル、テトラヒドロフラニル等が挙げられる。
【0076】
「置換もしくは非置換のアルキル」、「置換もしくは非置換のアルケニル」、「置換もしくは非置換のアルキニル」、「置換もしくは非置換のアリール」、「置換もしくは非置換のシクロアルキル」、「置換もしくは非置換のシクロアルケニル」、「置換もしくは非置換のヘテロアリール」、「置換もしくは非置換のヘテロサイクル」、「置換もしくは非置換のアルコキシ」、における置換基としては、例えば、ヒドロキシ、カルボキシ、ハロゲン、ハロゲン化アルキル(例:CF3、CH2CF3、CH2CCl3)、ニトロ、ニトロソ、シアノ、アルキル(例:メチル、エチル、イソプロピル、tert−ブチル)、アルケニル(例:ビニル)、アルキニル(例:エチニル)、シクロアルキル(例:シクロプロピル、アダマンチル)、シクロアルキルアルキル(例:シクロヘキシルメチル、アダマンチルメチル)、シクロアルケニル(例:シクロプロペニル)、アリール(例:フェニル、ナフチル)、アリールアルキル(例:ベンジル、フェネチル)、ヘテロアリール(例:ピリジル、フリル)、ヘテロアリールアルキル(例:ピリジルメチル)、ヘテロサイクル(例:ピペリジル)、ヘテロサイクルアルキル(例:モルホリルメチル)、アルコキシ(例:メトキシ、エトキシ、プロポキシ、ブトキシ)、ハロゲン化アルコキシ(例:OCF3)、アルケニルオキシ(例:ビニルオキシ、アリルオキシ)、アリールオキシ(例:フェニルオキシ)、アルキルオキシカルボニル(例:メトキシカルボニル、エトキシカルボニル、tert−ブトキシカルボニル)、アリールアルキルオキシ(例:ベンジルオキシ)、アミノ(例:アルキルアミノ(例:メチルアミノ、エチルアミノ、ジメチルアミノ)、アシルアミノ(例:アセチルアミノ、ベンゾイルアミノ)、アリールアルキルアミノ(例:ベンジルアミノ、トリチルアミノ)、ヒドロキシアミノ、アルキルアミノアルキル(例:ジエチルアミノメチル)、スルファモイル、オキソ等からなる群から選択される。1〜4個の当該置換基で置換されていてもよい。
【0077】
本発明化合物の製薬上許容される塩としては、以下の塩が挙げられる。
【0078】
塩基性塩として、例えば、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アンモニウム塩;トリメチルアミン塩、トリエチルアミン塩、ジシクロヘキシルアミン塩、エタノールアミン塩、ジエタノールアミン塩、トリエタノールアミン塩、プロカイン塩、メグルミン塩、ジエタノールアミン塩またはエチレンジアミン塩等の脂肪族アミン塩;N,N−ジベンジルエチレンジアミン、ベネタミン塩等のアラルキルアミン塩;ピリジン塩、ピコリン塩、キノリン塩、イソキノリン塩等のヘテロ環芳香族アミン塩;テトラメチルアンモニウム塩、テトラエチルアモニウム塩、ベンジルトリメチルアンモニウム塩、ベンジルトリエチルアンモニウム塩、ベンジルトリブチルアンモニウム塩、メチルトリオクチルアンモニウム塩、テトラブチルアンモニウム塩等の第4級アンモニウム塩;アルギニン塩、リジン塩等の塩基性アミノ酸塩等が挙げられる。
【0079】
酸性塩としては、例えば、塩酸塩、硫酸塩、硝酸塩、リン酸塩、炭酸塩、炭酸水素塩、過塩素酸塩等の無機酸塩;酢酸塩、プロピオン酸塩、乳酸塩、マレイン酸塩、フマール酸塩、酒石酸塩、リンゴ酸塩、クエン酸塩、アスコルビン酸塩等の有機酸塩;メタンスルホン酸塩、イセチオン酸塩、ベンゼンスルホン酸塩、p−トルエンスルホン酸塩等のスルホン酸塩;アスパラギン酸塩、グルタミン酸塩等の酸性アミノ酸等が挙げられる。
【0080】
「溶媒和物」とは、本発明化合物またはその製薬上許容される塩の溶媒和物を意味し、例えば、アルコール(例:エタノール)和物や水和物等が挙げられる。水和物としては、1水和物、2水和物等を挙げることができる。
【0081】
別の実施形態において、本発明は、上記のいずれかに記載の化合物、その製薬上許容される塩またはそれらの溶媒和物、あるいはそのプロドラッグ(たとえば、エステル類、アミド類)を含有する医薬組成物を提供する。
【0082】
本明細書において「プロドラッグ」、「プロドラッグ化合物」とは、化学的又は代謝的に分解し得る基を有し、加水分解や加溶媒分解によって又は生理条件下で分解することによって薬学的に活性を示す本発明化合物の誘導体である。いろいろな形のプロドラッグが、当該技術分野において知られている。このようなプロドラッグ誘導体の例については、次を参照することができる。式(I)の化合物のプロドラッグは、式(I)の化合物中に存在する官能基を、生体内で開裂すると親化合物が放出されるような修飾方法で修飾することによって製造される。たとえば、プロドラッグは、式(I)の化合物中のヒドロキシ、スルフヒドリル又はアミノ基が、生体内で開裂されるとそれぞれ遊離ヒドロキシ、アミノ、又はスルフヒドリル基を再生する基と結合している式(I)の化合物を含む。プロドラッグの例は、これらに限定されないが、式(I)の化合物中のヒドロキシ 官能基のエステル(例えば、アセタート、ホルマート、およびベンゾアート誘導体)、カルバマート(例えば、N,N−ジメチルアミノカルボニル)などを含む。
(a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods inEnzymology, Vol.42.p.309−396, edited byK.Widder,et al.(Academic Press, 1985);
(b)A Textbook of Drug Designand Development, edited by Krogsgaard−Larsen;
(c)H.Bundgaard, Chapter 5“Designand Application of Prodrugs”,by H.Bundgaard p.113−191(1991);
(d)H.Bundgaard, Advanced Drug Delivery Reviews, 8,1−38(1992) ;
(e)H.Bundgaard, et al.,Journal of Pharmaceutical Sciences, 77,285(1988);および
(f)N.Kakeya, et al., Chem Pharm Bull, 32,692(1984)。
【0083】
1つのプロドラッグ基の例は、ヒトまたは動物体内で開裂して親酸を生じる薬学的に許容しうるエステルの in vivo 開裂可能エステル基である。たとえば、プロドラッグ基は、それが結合しているカルボキシ基と一緒になって、C1−6アルキルエステルまたはC1−6シクロアルキルエステル、例えば、メチル、エチル、プロピル、イソプロピル、n−ブチルまたはシクロペンチルのエステル;C1−6アルコキシメチルエステル、例えば、メトキシメチルエステル;C1−6アルカノイルオキシメチルエステル、例えば、ピバロイルオキシメチルエステル;フタリジルエステル;C3−8シクロアルコキシカルボニルオキシC1−6アルキルエステル、例えば、1−シクロヘキシルカルボニルオキシエチルエステル;1,3−ジオキソラン−2−イルメチルエステル、例えば、5−メチル−1,3−ジオキソラン−2−イルメチルエステル;C1−6アルコキシカルボニルオキシエチルエステル、例えば、1−メトキシカルボニルオキシエチルエステル;アミノカルボニルメチルエステルおよびそのモノ−またはジ−N−(C1−6アルキル)変型、例えば、N,N−ジメチルアミノカルボニルメチルエステルおよびN−エチルアミノカルボニルメチルエステルのような薬学的に許容しうるエステル、および置換または非置換の複素環式基の薬学的に許容しうるエステルを形成する。1つの実施形態では、プロドラッグは、イソプロピルまたはシクロペンチルのようなC1−4アルキル基、またはN−メチルテトラヒドロピリジルのような置換されていてよい複素環式基より選択されるものとのエステルが挙げられる。
【0084】
記に列挙した具体的な化合物の任意の化合物を含む本発明の医薬組成物は、TTK阻害剤であることをも特徴とする。したがって、TTKの阻害を必要とする患者に投与することによって薬効を発揮する任意の医薬組成物が提供される。
【0085】
別の実施形態では、本発明は、がんまたは免疫疾患の処置または予防のための医薬であって、上記に列挙した具体的な化合物の任意の化合物を含む医薬を提供する。
【0086】
(製造方法)
本発明化合物の一般的製造法を以下に例示する。また、抽出、精製などは、通常の有機化学の実験で行う処理を行えばよい。
【0087】
以下に、本発明の化合物の製造方法を記載する。
【0088】
本発明の化合物の合成は、当該分野において公知の手法を参酌しながら実施することができる。
【0089】
原料化合物は、市販の化合物であるか、特許文献3、特許文献4、特許文献5、特許文献6、特許文献7に記載されたもの、このほか本明細書において記載されたものならびに本明細書において他に引用された文献に記載されるものならびに他に公知の化合物を利用することができる。
【0090】
本発明の化合物の中には、互変異性体が存在し得るものがあるが、本発明は、これらを含め、全ての可能な異性体およびそれらの混合物を包含する。
【0091】
本発明の化合物の塩を取得したいとき、本発明の化合物が塩の形で得られる場合には、そのまま精製すればよく、また、遊離の形で得られる場合には、適当な有機溶媒に溶解もしくは懸濁させ、酸または塩基を加えて通常の方法により塩を形成させればよい。
【0092】
また、本発明の化合物およびその製薬上許容される塩は、水あるいは各種溶媒との付加物(水和物ないし溶媒和物)の形で存在することもあるが、これら付加物も本発明に包含される。
【0093】
これらの誘導体は、体内にて変換されて活性化されるものであり、本明細書において「プロドラッグ」とも称する。プロドラッグの例としては、たとえば、上記塩、溶媒和物のほか、エステル(たとえば、アルキルエステルなど)、アミドなども含まれることが理解される。
【0094】
本発明の化合物の例は、実施例において種々列挙されており、当業者はこれらを参考にして、本発明の例示されていない化合物をも製造、使用することができる。
【0095】
本発明はまた、本発明の化合物を製造するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0096】
【化8】
【0097】
(式中、R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、)
そして、化8の化合物は、対応するアニリン誘導体と、安息香酸誘導体とからアミド結合を形成させることによって、製造することができる。
【0098】
【化9】
【0099】
(式中、各記号は前記と同義であり、Xは、ハロゲン(Clなど)、OHなどの脱離可能な基である。式(A1)シリーズ(A11、A12,A13,A14)及び式(A2)で示される化合物は公知の化合物を用いてもよく、市販の化合物を用いてもよい。)。
【0100】
すなわち、本製造工程は、式(A1)シリーズ(A11、A12,A13,A14)で示される化合物と式(A2)で示される化合物を塩基および縮合剤存在下で反応させ、式(I)で示される化合物を製造する工程である。ここで、直接合成されるものは、R1bはHであるが、前記と同様の置換基を置換することもでき、場合によっては、置換基がHの代わりに結合したものを用いて合成することもできる。
【0101】
反応溶媒としては、DMF、NMP、DMA、ジメチルスルホキシド、芳香族炭化水素類(例、トルエン、ベンゼン、キシレンなど)、飽和炭化水素類(例、シクロヘキサン、ヘキサンなど)、ハロゲン化炭化水素類(例、ジクロロメタン、クロロホルム、1,2−ジクロロエタンなど)、エーテル類(例、テトラヒドロフラン、ジエチルエーテル、ジオキサン、1,2−ジメトキシエタンなど)、エステル類(例、酢酸メチル、酢酸エチルなど)、ケトン類(例、アセトン、メチルエチルケトンなど)、ニトリル類(例、アセトニトリルなど)、アルコール類(例、メタノール、エタノール、t−ブタノールなど)、水およびそれらの混合溶媒等が挙げられる。
【0102】
塩基としては、例えば金属水素化物(例、水素化ナトリウムなど)、金属水酸化物(例、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、水酸化バリウムなど)、金属炭酸塩(例、炭酸ナトリウム、炭酸カルシウム、炭酸セシウムなど)、金属アルコキシド(例、ナトリウムメトキシド、ナトリウムエトキシド、カリウムt−ブトキシドなど)、炭酸水素ナトリウム、金属ナトリウム、有機アミン(例、トリエチルアミン、ジイソプロピルエチルアミン、DBU、2,6−ルチジンなど)、ピリジン、アルキルリチウム(n−BuLi、sec−BuLi、tert−BuLi)等が挙げられる。塩基は必ずしも使用する必要はないが、必要に応じて用いることができる。
【0103】
上記XがOH等の場合に用いられる縮合剤としては、DCC、BOP,PyBOP、PyBrop、HATU、DPPA、WSC、DMT−MMなどを用いることができる。また、これらの試薬は、例えばHOSu、HOBt、HOAtなどと組み合わせて使用することができる。
【0104】
好ましくは、反応溶媒としてエーテル類(例、テトラヒドロフラン、ジエチルエーテル、ジオキサンなど)、N,N−ジメチルホルムアミド、N−メチルピロリドン、N,N−ジメチルアセトアミド、塩基として有機アミン(例、トリエチルアミン、ジイソプロピルエチルアミン、DBU、2,6−ルチジンなど)、縮合剤としてHATUまたはPyBOPを用いて行えばよい。反応温度、反応時間は特に限定されないが、通常は室温にて反応を実施し、反応の進行が遅い場合には加温することによって反応が促進される場合もある。
【0105】
本発明の製造は、上記好ましい実施形態を適宜改変し、あるいは組み合わせ、あるいは公知技術を付加することによって実施することができる。
【0106】
本発明の化合物は、保護基を用いて保護することができる。たとえば、代表的には、ハロゲン(I,Br,Cl、Fなど)、低級(ここでは、代表的にC1−C6を示すがこれに限定されない。)アルコキシ、低級アルキルチオ、低級アルキルスルホニルオキシ、アリールスルホニルオキシ等を表す。)において、適宜の置換基を当該分野で公知の手法により保護することによって製造することができる。このような保護基としては、例えばエトキシカルボニル、t−ブトキシカルボニル、アセチル、ベンジル等の、Protective Groups in Organic Synthesis、T.W.Green著、John Wiley & Sons Inc.(1981年)等に記載されている保護基をあげることができる。保護基の導入および脱離方法は、有機合成化学で常用される方法[例えば、Protective Groups in Organic Synthesis、T. W. Greene著、John Wiley & Sons Inc.(1981年)参照]等に記載の方法あるいはそれらに準じて得ることができる。また、各置換基に含まれる官能基の変換は、上記製造法以外にも公知の方法[例えば、Comprehensive Organic Transformations、R.C.Larock著(1989年)等]によっても行うことができ、本発明の化合物の中には、これを合成中間体としてさらに新規な誘導体へ導くことができるものもある。上記各製造法における中間体および目的化合物は、有機合成化学で常用される精製法、例えば中和、濾過、抽出、洗浄、乾燥、濃縮、再結晶、各種クロマトグラフィー等に付して単離精製することができる。また、中間体においては、特に精製することなく次の反応に供することも可能である。
【0107】
より特定された実施形態では、式(II)の化合物:
【0108】
【化10】
【0109】
(式中、R2は、ハロゲンまたはONO2で置換されたアルキル基であり、R3は、ハロゲンまたはONO2で置換されたアルキル基であり、Aは、−O−、−S−または−NH−である)を合成する方法は、上記(A)の合成に準じて実施することができる。
【0110】
この場合、Aが−NH−である場合は、上記と同様の反応により実施可能である。
【0111】
また、Aが−O−または−S−のときは、以下の点に留意して、上記(A)の合成と同様に実施することができる。
【0112】
あるいは、Aが−O−であり、R2およびR3が同じでかつ同じ配置(たとえば、パラ位)の場合、2−アミノフェノール(o−アミノフェノール)にR2−ベンゾイルハライド(たとえば、p−クロロメチルベンゾイルクロリドなど)を用いることによって、製造することができる。
【0113】
Aが−S−であり、R2およびR3が同じでかつ同じ配置(たとえば、パラ位)の場合、2−アミノチオフェノール(o−アミノチオフェノール)にR2−ベンゾイルハライド(たとえば、p−クロロメチルベンゾイルクロリドなど)を用いることによって、製造することができる。
【0114】
R2およびR3は、たとえば、アミノフェノールまたはアミノチオフェノールとの結合後にハロゲンを別の置換基にすることもできる。たとえば、ニトロオキシを導入する場合は、アセトニトリルと結合させることも可能である。
【0115】
本発明は、スクリーニング方法により得られた化合物又はその塩として、関連して発症する疾患に対する治療又は予防作用を発揮し得る。例えば、本発明のスクリーニング方法によれば、癌、免疫疾患等に有効な治療剤又は予防剤の候補化合物をスクリーニングすることができる。
【0116】
(医薬)
本発明の化合物またはその製薬上許容される塩は、そのまま単独で投与することも可能であるが、通常各種の医薬製剤として提供するのが好ましい。また、それら医薬製剤は、動物および人に使用される。
【0117】
本発明が主に対象とする疾患である「がん」としては、固形がん、血管腫、血管内皮腫、肉腫、カポシ肉腫及び造血器腫瘍等の種々の悪性新生物が例示され、大腸がん及び肝がん等が包含され、さらにこれらがんの転移をも包含する。本発明は、上記病気を治療する医薬品を製剤化するのに特に有用になるような特定の特性を有する新規の一連の化合物の発見に成功した。特に、本化合物は、充実性腫瘍か、血液の腫瘍のような増殖性の病気の治療において、特に、結腸直腸のがん、乳がん、肺がん、前立腺がん、膵臓がんまたは膀胱がんおよび腎臓がん、同様に、白血病およびリンパ腫のような病気において有用である。
【0118】
上記医薬組成物は、本発明の化合物又はその塩を有効成分として含有することに1つの特徴がある。したがって、該医薬組成物は、発症する疾患に対して、ErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害することを介して作用し得るという優れた効果を発揮する。例えば、本発明の医薬組成物は特に、癌等に対して、ErbB/Her受容体ファミリーの2量体化やβ−カテニンとTCFの結合を阻害することを介して作用し得るという優れた効果を発揮する。該医薬組成物を癌の治療又は予防に用いる場合は、通常の癌療法、例えば、放射線療法、化学療法、たとえば腫瘍細胞を事前感応化するためのDNA劣化剤を施すのと同時に、又はその前でも使用できる。
【0119】
上記医薬組成物中における前記化合物又はその塩の含有量は、治療目的の疾患、患者の年齢、体重等により適宜調節することができ、治療上有効量であればよく、低分子化合物又は高分子化合物の場合、例えば、0.0001〜1000mg、好ましくは、0.001〜100mg、ポリペプチド又はその誘導体の場合、例えば、0.0001〜1000mg、好ましくは、0.001〜100mg、核酸又はその誘導体の場合、例えば、0.00001〜100mg、好ましくは、0.0001〜10mgであることが望ましい。
【0120】
上記医薬組成物は、前記化合物又はその塩を安定に保持し得る種々の助剤をさらに含有してもよい。具体的には、有効成分の送達対象となる部位に到達するまでの間に、有効成分が分解することを抑制する性質を呈する薬学的に許容されうる助剤、賦形剤、結合剤、安定剤、緩衝剤、溶解補助剤、等張剤等が挙げられる。
【0121】
上記医薬組成物の投与形態は、有効成分の種類;投与対象となる個体、器官、局所部位、組織;投与対象となる個体の年齢、体重等に応じて、適宜選択される。前記投与形態としては、皮下注射、筋肉内注射、静脈内注射、局所投与等が挙げられる。
【0122】
また、上記医薬組成物の投与量も、有効成分の種類;投与対象となる個体、器官、局所部位、組織;投与対象となる個体の年齢、体重等に応じて、適宜選択される。投与としては、特に限定されないが、有効成分が、低分子化合物又は高分子化合物である場合、前記有効成分の量として、例えば、0.0001〜1000mg/kg体重、好ましくは、0.001〜100mg/kg体重、ポリペプチド又はその誘導体の場合、例えば、0.0001〜1000mg/kg体重、好ましくは、0.001〜100mg/kg体重、核酸又はその誘導体の場合、例えば、0.00001〜100mg/kg体重、好ましくは、0.0001〜10mg/kg体重の1回投与量となるように、1日につき、複数回、例えば、1〜3回投与すること等が挙げられる。
【0123】
投与経路は、治療に際し最も効果的なものを使用するのが好ましく、経口または例えば、直腸内、口腔内、皮下、筋肉内、静脈内等の非経口をあげることができる。
投与形態としては、カプセル剤、錠剤、顆粒剤、散剤、シロップ剤、乳剤、座剤、注射剤等がある。経口投与に適当な、例えば乳剤およびシロップ剤のような液体調製物は、水、ショ糖、ソルビット、果糖等の糖類、ポリエチレングリコール、プロピレングリコール等のグリコール類、ゴマ油、オリーブ油、大豆油等の油類、p−ヒドロキシ安息香酸エステル類等の防腐剤、ストロベリーフレーバー、ペパーミント等のフレーバー類等を使用して製造できる。また、カプセル剤、錠剤、散剤、顆粒剤等は、乳糖、ブドウ糖、ショ糖、マンニット等の賦形剤、澱粉、アルギン酸ソーダ等の崩壊剤、ステアリン酸マグネシウム、タルク等の滑沢剤、ポリビニルアルコール、ヒドロキシプロピルセルロース、ゼラチン等の結合剤、脂肪酸エステル等の界面活性剤、グリセリン等の可塑剤等を用いて製造できる。非経口投与に適当な製剤は、好ましくは受容者の血液と等張である活性化合物を含む滅菌水性製剤からなる。例えば、注射剤の場合、塩溶液、ブドウ糖溶液または塩水とブドウ糖溶液の混合物からなる担体等を用いて注射用の溶液を調製する。
【0124】
局所製剤は、活性化合物を1種もしくはそれ以上の媒質、例えば鉱油、石油、多価アルコール等または局所医薬製剤に使用される他の基剤中に溶解または懸濁させて調製する。
腸内投与のための製剤は、通常の担体、例えばカカオ脂、水素化脂肪、水素化脂肪カルボン酸等を用いて調製し、座剤として提供される。本発明では、非経口剤においても、経口剤で例示したグリコール類、油類、フレーバー類、防腐剤(抗酸化剤を含む)、賦形剤、崩壊剤、滑沢剤、結合剤、界面活性剤、可塑剤等から選択される1種もしくはそれ以上の補助成分を添加することもできる。
【0125】
本発明の化合物もしくはその製薬上許容される塩の有効用量および投与回数は、投与形態、患者の年令、体重、治療すべき症状の性質もしくは重篤度等により異なるが、通常、投与量は、1日当たり0.01〜1000mg/人、好ましくは5〜500mg/人であり、投与回数は、1日1回または分割して投与するのが好ましい。
【0126】
本発明の化合物は、好ましくは、IC50が被検物質なしの場合の蛍光値を基準に、被検物質の抑制活性が1以下、好ましくは、0.1μM以下、より好ましくは、0.01μM以下の値を有するもの、あるいは、10nM〜10μMの範囲内、好ましくは、10μM未満、より好ましくは、1μM未満のIC50値をもつような化合物である。
【0127】
製剤化に関するさらなる情報については、Comprehensive Medicinal Chemistry(Corwin Hansch;Chairman of Editorial Board),Pergamon Press 1990年の第5巻の第25.2章を参照することができる。
【0128】
本発明はまた、本発明の医薬組成物を製造するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0129】
本発明はまた、本発明の化合物またはその溶媒和物を使用するシステム、装置、キットにも関する。そのようなシステム、装置、キットの構成要件は、当該分野において公知のものを利用することができ、当業者は適宜設計することができることが理解される。
【0130】
本発明化合物は、医薬としての有用性を備えた化合物である。ここで、医薬としての有用性としては、代謝安定性がよい点、薬物代謝酵素の誘導も少ない点、他の薬剤を代謝する薬物代謝酵素の阻害も小さい点、経口吸収性の高い化合物である点、クリアランスが小さい点、または、半減期が薬効を発現するために十分長い点などが含まれる。
【0131】
本明細書において引用された、科学文献、特許、特許出願などの参考文献は、その全体が、各々具体的に記載されたのと同じ程度に本明細書において参考として援用される。
【実施例】
【0132】
以下、実施例によって本発明を更に詳細に説明するが、この実施例等により本発明の技術的範囲が限定されるものではない。
【0133】
使用機器、及び測定条件などは下記に記載したものを採用した。
【0134】
(材料および方法)
細胞株:HCT116、SW620、MDA−MB−231,MCF7、SKBR3、SW480 、A549、ヒト正常繊維芽細胞 CCD−1059SKは、大日本住友製薬(Dainippon Sumitomo Pharma,Osaka, Japan)を代理店とし、アメリカンタイプカルチャーコレクション(American Type Culture Collection(ATCC); Manassas, VA)より入手した。
【0135】
McCoy’s 5A培地、Earle’s培地およびRPMI−1640 培地は、Invitrogen Corp, Carlsbad, CA, USAから入手した。
【0136】
ペニシリン−ストレプトマイシン、ウシ胎仔血清(Fetal Bovine Serum)、L−グルタミン、およびMEM 非必須アミノ酸溶液(Non−Essential Amino Acids Solution)は、それぞれ、Invitrogen Corp, Carlsbad, CA, USA、Equitech−Bio Inc.,Kerrville, TX, USA、Invitrogen Corp,Carlsbad, CA, USAおよびInvitrogen Corp,Carlsbad, CA, USAから入手した。
【0137】
CO2インキュベーターは、三洋電機(MCO−18AIC)から入手した。
【0138】
WST−1を調製(試薬Aと試薬Bを混合)した。同仁化学研究所(Dojindo Laboratories, Kumamoto, Japan)から入手した。試薬A:WST−1 (16.3 mg; 終濃度5mmol/l), HEPES (23.8 mg; 終濃度20 mmol/l; pH7.4) 試薬B:1−メトキシ PMS (0.2 mmol/l), 5 ml。
【0139】
オートプレートリーダーは、三光純薬社(Sjeia AUTOREADER III #D−ER3)から入手した。
【0140】
(評価方法)
ヒトがん細胞増殖抑制活性試験(WST−1 assay)
HCT116、SW620及びA549の細胞懸濁液[McCoy’s 5A (1% 抗生物質(ペニシリン−ストレプトマイシン及び10% ウシ胎仔血清を含む), 1.0×104 cells/ml)]、並びに、SW480、SK−BR−3、MCF7、MDA−MB−231の細胞懸濁液[RPMI−1640培地 (1% 抗生物質(ペニシリン−ストレプトマイシン及び10% ウシ胎仔血清を含む ), 5.0 × 103cells/ml)]を作製し、96−well plateに1wellあたり135μlずつ分注した後、CO2インキュベーターで24時間培養した。各濃度の薬剤培養溶液15μlを1wellあたり分注した後、CO2インキュベーターで72時間培養した。その後、WST−1を調製 (試薬Aと試薬Bを混合)し、96−well Plateに1wellあたり16μlずつ分注した後、CO2インキュベーターで4時間培養した。オートプレートリーダー(測定波長450nm、 参照波長630 nm)により吸光度を測定し、各薬剤サンプルのIC50値を算出した。
【0141】
ヒト正常繊維芽細胞CCD−1059SKに対する増殖抑制(毒性)試験
L−グルタミン200mM (100×)液(liqid) (#25030−149)を10 ml/LでEagle’s培地に添加した。MEM 非必須アミノ酸溶液 10 mM (100×)(#11140−50) を培地に対し1%添加した。56℃で30分間おきに、非働化したウシ胎仔血清を10%添加して使用した。
細胞操作は、ヒトがん細胞増殖抑制活性試験と同様の操作で行い、各薬剤サンプルのCCD−1059SK株に対するIC50を求めた。
【0142】
(製造例1:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ビス(2−メチルプロパンアミド) K−152)
(1)2−アミノフェニルジスルフィド(2.02 mM)[東京化成工業株式会社(TCI社)]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[和光純薬工業株式会社(Wako社)]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製し、白色結晶を得た。
m.p. 144.5−146.4℃
IR (KBr) cm−1: 3244.0,3186.2, 2970.2, 2869.9, 1660.6, 1577.7, 1525.6, 1460.0, 1436.9, 1380.9, 1276.8,1238.4, 1201.6, 1157.2, 1099.3, 1035.7, 948.9, 937.3, 887.2, 744.5, 686.6,596.0, 470.6
1H NMR (CDCl3) δ:1.47 (12H, d, J = 6.8Hz), 3.43 (2H, septet, J = 7.2 Hz), 7.36 (2H, dt, J = 7.2 Hz), 7.43 (2H, d, J =8.0 Hz), 7.85 (2H, d, J = 8.0 Hz), 7.98 (2H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z: 389.14(M + H)+。
【0143】
(製造例2:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ビス(2,2−ジメチルプロパンアミド) K−153)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)トリメチルアセチルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 86.0−87.0℃
IR (KBr) cm−1: 3386.8, 3247.9,2869.9, 2391.6, 1678.0, 1643.2, 1575.7, 1467.7, 1433.0, 1361.7, 1305.7, 1226.6, 1161.1, 1058.8, 1035.7, 931.6, 916.1, 771.5, 750.3, 626.8, 455.2.
1H NMR (CDCl3) δ: 1.25 (18H, s), 6.94(2H, dt, J = 1.5 and 7.8 Hz), 7.21 (2H, dd, J = 1.5 and 7.8 Hz), 7.40 (2H, dd,J = 1.5 and 7.8 Hz), 8.46 (2H, dd, J = 1.5 and 8.4 Hz), 8.52 (2H, br s).
ESI−MS (positive mode) m/z: 417.17(M + H)+。
【0144】
(製造例3:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジオクタンアミド K154)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加える。
(3)n−オクタノイルクロリド(4.04 mM)[関東化学株式会社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 79.0−81.0℃
IR (KBr) cm−1: 3267.2, 3192.0,2923.9, 2848.7, 2405.1, 1662.5, 1577.7, 1523.7, 1465.8, 1438.8, 1409.9, 1284.5,1234.4, 1033.8, 962.4, 732.9, 686.6, 464.8, 405.0.
1H NMR (CDCl3) δ: 0.89 (6H, m), 1.30(16H, m), 1.65 (4H, m), 2.16 (4H, t, J = 7.6 Hz), 6.99 (2H, dt, J = 7.6 Hz),7.35 − 7.43 (4H, m), 7.97 (2H, brs), 8.40 (2H, brd, J = 7.6 Hz).
ESI−MS (positive mode) m/z: 501.26(M + H)+。
【0145】
(製造例4:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジシクロペンタンカルボキサミド K−201)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。得られた結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 157−158℃
1H NMR (CDCl3) δ: 1.55−1.86 (16H, m), 2.47 (4H, quintet, J = 7.6 Hz), 6.98 (2H,brt, J -= 7.6 Hz), 7.35 (2H, brd, J = 8.0 Hz), 7.40 (2H, brt, J = 8.0 Hz), 8.06(2H, brs), 8.42 (2H, brd, J = 8.4 Hz)。
【0146】
(製造例5:N,N’−(2,2’−ジスルファンジイルビス(2,1−フェニレン)ジベンズアミド K−202)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)2−アミノフェニルジスルフィド(2.02 mM)[TCI社]をCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)塩化ベンゾイル(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に溶かし、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 144−145℃
1H NMR (CDCl3) δ: 6.95 (2H, dt, J = 1.5and 7.8 Hz), 7.31 (2H, dt, J = 1.5 and 8.4 Hz), 7.40 − 7.60 (8H, m), 7.69 (4H, dd, J = 1.5 and 8.4 Hz), 8.50(2H, dd, J = 1.5 and 8.4 Hz), 8.93 (2H, br s)。
【0147】
(製造例6:S−2−イソブチルアミドフェニル 2−メチルプロパンチオレート K−178)
本製造例は、製造例1に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[Wako社]をCH2Cl2(5 ml)で希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 92−94℃
IR (KBr) cm−1: 3286.5, 2968.2,2871.8, 1753.2, 1658.7, 1606.6, 1250.4, 1348.1, 1298.0, 1255.6, 1180.4, 1099.3,920.0, 842.8, 758.0, 694.3.
1H NMR (CDCl3) δ: 1.24 (6H, d, J= 6.9 Hz), 1.33 (6H, d, J = 6.9 Hz), 2.51 (1H, septet, J = 7.2 Hz), 2.94 (1H,septet, J = 7.2 Hz), 7.14 (1H, t, J = 7.6 Hz), 7.38 (1H, d, J = 6.4 Hz), 7.43(1H, t, J = 8.4 Hz), 7.76 (1H, br s), 8.35 (1H, d, J = 8.0 Hz)。
【0148】
(製造例7:S−2−ピバルアミドフェニル 2−メチルプロパンチオレート K−179)
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3) トリメチルアセチルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を減圧濃縮し、得られた残渣をMeOH(5ml)に溶解させた。
(8)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(9)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(10)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(11)有機層を乾燥後し、5ml程度に濃縮した。
(12)この溶液にピリジン(400μl)を加えた。
(13)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に希釈し、上記の溶液に滴下した。
(14)常温、常圧で1時間撹拌した。
(15)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(16)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(17)有機層を乾燥し、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
オイル状の化合物を得た。
m.p. 88.5−90.6℃
1H NMR (CDCl3) δ: 1.25 (9H, s), 1.31 (15H,m), 2.93 (1H, septet, J = 7.0 Hz), 7.11 (1H, dt, J = 1.0 and 8.0 Hz), 7.38 (1H,dd, J = 8.0 and 1.0 Hz), 7.44 (1H, dt, J = 1.0 and 8.5 Hz), 8.05 (1H, brs),8.35 (1H, brd, J = 9.0 Hz).
ESI−MS (positive mode) m/z: 294.1(M + H)+。
【0149】
(製造例8:S−2−(シクロペンタンカルボキサミド)フェニル 2−メチルプロパンチオレート K−180)
本製造例は、製造例7に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)上記の中間体をMeOH(5ml)に溶解させた。
(9)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(10)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(11)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(12)有機層を乾燥し、5ml程度に濃縮した。
(13)この溶液にピリジン(400μl)を加えた。
(14)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に希釈、上記の溶液に滴下した。
(15)常温、常圧で1時間撹拌した。
(16)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(17)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(18)有機層を減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。白色結晶の化合物を得た。
m.p. 86−87℃
1H NMR (CDCl3) δ: 1.29 (6H, d, J = 6.8Hz), 1.58 − 1.93 (8H, m), 2.69 (1H,quintet, J = 8.4 Hz), 2.94 (1H, septet, J = 7.2 Hz), 7.12 (1H, t, J = 7.6 Hz), 7.26 − 7.47 (2H, m), 7.74 (1H, brs), 8.34 (1H, d, J = 8.0 Hz)。
【0150】
(製造例9:S−2−ベンズアミドフェニル 2−メチルプロパンチオレート K−181)
本製造例は、製造例7に準じて実施した。具体的には以下のとおりである。
(1)o−アミノチオフェノール(1.87mmol)[Wako社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)シクロペンタンカルボニルクロリド(4.04 mM)[Aldrich社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した。(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)上記の中間体をメタノール(MeOH)(5ml)に溶解させた。
(9)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(10)その反応液をHClで酸性(ph5〜6)にし、CHCl3で抽出した。
(11)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(12)有機層を乾燥後、5ml程度に濃縮した。
(13)この溶液にピリジン(400μl)を加えた。
(14)イソプロピルクロリド(2.02mM)[Wako社]をCH2Cl2(5ml)に加え、上記の溶液に滴下した。
(15)常温、常圧で1時間撹拌した。
(16)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(17)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(18)有機層を乾燥後、減圧濃縮。析出した結晶をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。白色結晶の化合物を得た。
m.p. 65−66℃
1H NMR (CDCl3) δ: 1.27 (6H, d, J = 6.8Hz), 2.95 (1H, septet, J = 6.8 Hz), 7.42 − 7.57 (7H, m), 7.85(1H, d, J = 8.0 Hz), 8.48 (1H, d, J = 8.8 Hz), 8.51 (1H、 brs)。
【0151】
(製造例10:2−イソブチルアミドフェニル イソブチレートK−205)
(1)2−アミノフェノール(1.87mmol)[TCI社]をCHCl3(5ml)に溶解させた。
(2)この溶液にピリジン(350μl)を加えた。
(3)イソプロピルクロリド(4.04 mM)[Wako社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1〜2時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を、順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥後、減圧濃縮し、得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
m.p. 88.5−90.6℃
1H NMR (CDCl3) δ: 1.25 (3H, d, J = 7.0Hz), 1.37 (3H, J = 6.0 Hz), 2.51 (1H, J = 7.0 Hz), 2.88 (1H, J = 7.0 Hz), 7.11(1H, d, J = 3.7 Hz), 7.22 (1H, m), 7.26 (1H, brs), 8.19 (1H, d, J = 7.8 Hz).
ESI−MS (positive mode) m/z:250 (M +H)+。
【0152】
(製造例11:N−(2−ヒドロキシフェニル)イソブチルアミド K−204)
(1)上記の反応によって得られたK−205をMeOH(5 ml)に溶解させた。
(2)5N KOH(1 ml)を加え、常温常圧で2時間撹拌を行った。
(3)その反応液をHClで酸性(pH 5〜6)にし、CHCl3で抽出した。
(4)順次、飽和NaHCO3、飽和食塩水で洗浄した。
(5)有機層を乾燥後、減圧濃縮。白色結晶を得た。
m.p. 107.4−109.2℃
1H NMR (CDCl3) δ: 1.30 (6H, d, J = 6.9Hz), 2.65 (1H, septet, J = 7.0 Hz), 6.90 (1H, brt, J = 7.0 Hz), 7.01 (1H, brd,J = 7.0 Hz), 7.11 (1H, brt, J = 7.0 Hz), 7.54 (1H, br s), 8.89 (1H, br s)。
【0153】
(製造例12:3−イソブチリルベンゾ[d]チアゾール−2(3H)−オン K−209)
(1)2−ヒドロキシベンゾチアゾール(2.02 mM)[Wako社]を秤量しCH2Cl2(5 ml)に溶解した。
(2)この溶液にピリジン(400μl)を加えた。
(3)イソプロピルクロリド(3.03mM)[Wako社]をCH2Cl2(5 ml)に希釈し、氷冷下、上記の溶液に滴下した。
(4)常温、常圧で1時間撹拌した。
(5)反応液をCHCl3で希釈した(このときの容量は約100mlになるようにした)。
(6)反応液を順次、1%HCl、水、飽和NaHCO3、飽和食塩水で洗浄した。
(7)有機層を乾燥し、減圧濃縮。
(8)残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:5)により精製した。
油状化合物を得た。
油状物
1H NMR (CDCl3) δ: 1.28 (6H, d, J = 6.8Hz), 3.89 (6H, d, J = 6.8 Hz), 7.15 - 7.40 (3H, m), 8.10 (1H, d, J = 8.0 Hz)。
【0154】
(実施例13:2−(4−(クロロメチル)ベンズアミド)フェニル 4−(クロロメチル)ベンゾエートK−211)
2−アミノフェノール 0.51mlをCHCl3 10 mlに溶かし、ピリジン1.86 mlを加えた。これに、p−クロロメチルベンゾイルクロリド 2.29 gをCHCl310 mlに溶かした溶液を滴下し、氷浴中2時間攪拌した。反応液をCHCl3 30 mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、メタノール(MeOH)から再結晶し、目的化合物0.78gを得た。
m.p. 138.5℃
IR (KBr) cm−1:3347.2, 1782.1, 1715.6, 1659.6, 1531.4, 1445.5, 1416.6, 1171.7, 1077.2, 1014.5,750.3, 704.0, 634.5, 441.7.
1H NMR (CDCl3) δ: 4.57 (2H,s), 4.66 (2H, brs), 7.43 (2H, d, J = 8.0 Hz), 7.55 − 7.76 (4H, m), 7.75 (2H, d,J = 8.0 Hz), 8.14 (2H, d, J = 8.4 Hz), 8.22 (2H, d, J = 8.4 Hz).
ESI−MS(positive mode) m/z: 414.3 (M + H)+
FAB−MS m/z: 414(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18Cl2NO3,414.0664; found, 414.0659。
【0155】
(実施例14:2−(4−(ニトロオキシメチル)ベンズアミド)フェニル 4−(ニトロオキシメチル)ベンゾエートK−210)
アセトニトリル20mlにK−211を2.49 g 溶かし、これにAgNO3のアセトニトリル10 ml懸濁液を加えた。遮光下アルゴン気流中65 ℃(外浴)で8時間攪拌した。沈殿物を減圧下濾取し、少量のCHCl3で洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査をCHCl3:酢酸エチル:n−ヘキサン(1:3:1)を展開溶媒として、シリカゲルカラムクロマトグラフィーに付し、白色結晶のK−210を2.18 g得た。
m.p. 136.2℃
IR (KBr) cm−1:3348.2, 3041.5, 2895.9, 2544.9, 1920.0, 1715.6, 1597.9, 1504.4, 1454.2, 1258.5,1179.4, 1116.7, 1020.3, 857.3, 749.3, 631.6, 593.1, 450.3.
1H NMR (CDCl3) δ: 5.45 (2H,s), 5.53 (2H, s), 7.43 (2H, d, J = 8.0 Hz), 7.57 (2H, d, J = 8.4 Hz), 7.77 (2H,d, J = 8.0 Hz), 7.97 (1H, brs), 8.26 (3H, m).
ESI−MS(positive mode) m/z: 468.1 (M + H)+
FAB−MS m/z: 468(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18N3O9,468.1044; found, 468.1050。
【0156】
(実施例15:S− 2−(4−(クロロメチル)ベンズアミド)フェニル 4−(クロロメチル)ベンゾチオエートK−221)
アルゴン気流下、o−アミノチオフェノール0.64 mlをCH2Cl2 15 mlに溶解させ、この溶液にピリジン1.5 mlを加えた。これに、p−クロロメチルベンゾイルクロリド2.84 gをCH2Cl2 5 ml に溶かした溶液を加え、氷温で1時間撹拌した。反応液をCHCl3 80mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、シリカゲルカラムクロマトグラフィー(酢酸エチル:トルエン=1:8) により精製し、減圧濃縮し、白色結晶を得た。
m.p. 158.8−159.4℃
IR (KBr) cm−1: 3384.8,2974.0, 1674.1, 1502.4, 1409.9, 1101.3, 1018.3, 896.8, 804.3, 677.0, 541.7,495.7, 470.6, 435.9, 412.7.
1H NMR (CDCl3)δ: 4.59 (2H, s),4.65 (2H, s), 7.44 (2H, d, J = 8.0 Hz), 7.53 − 7.60 (4H, m), 7.79 (2H, d,J = 8.4 Hz), 8.07 (2H, d, J = 8.4 Hz), 8.51(1H, d, J = 8.4 Hz), 8.57 (1H, brs).
ESI−MS(negative mode) m/z: 430.2 (M − H)−
FAB−MS m/z: 430(M+H)+; HR−FAB−MS m/z: (M+H)+ calcd for C22H18Cl2NO2S,430.0436; found, 430.0439。
【0157】
(実施例16:S− 2−(4−(ニトロオキシメチル)ベンズアミド)フェニル 4−(ニトロオキシメチル)ベンゾチオエートK−220)
アセトニトリル30mlにK−221を100 mg 溶かし、これにAgNO3 800 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。沈殿物を減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、PLCにより精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK−220を7.5mg得た。
m.p. 171.8−172.9℃
IR (KBr) cm−1: 3859.3,3843.9, 3826.5, 3679.9, 3639.4, 2349.1, 1610.5, 1535.2, 1438.8, 752.2, 690.5,538.1, 453.2, 422.4.
1H NMR (CDCl3) δ: 5.45 (2H, s), 5.52(2H, s), 7.45 (2H, d, J = 8.0 Hz), 7.49−7.69 (4H, m), 7.83 (2H,d, J = 8.4 Hz), 8.10 (2H, d, J = 8.4 Hz), 8.49 (1H, d, J = 8.0 Hz),8.56 (1H, brs).
ESI−MS (negative mode) m/z:482.7 (M - H)−
FAB−MS m/z: 483 (M+H)+。
【0158】
(実施例17:2−(3−(クロロメチル)ベンズアミド)フェニル 3−(クロロメチル)ベンゾエート K−231)
アルゴン気流下、o−アミノチオフェノール109 mgをCHCl3 5 mlに溶解させ、この溶液にピリジン500μlを加えた。これに、m−(クロロメチル)ベンゾイルクロリド341μlを加え、室温で1時間撹拌した。反応液をCHCl3 5mlで希釈後、順次1% HCl、飽和NaHCO3、飽和食塩水で洗浄した。Na2SO4で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、シリカゲルカラムクロマトグラフィー(酢酸エチル:トルエン=1:8) により精製し、減圧濃縮し、目的の白色結晶を235.0 mg得た。
m.p. 155.5−157.4℃
IR (CHCl3) cm−1: 3018.4,2399.3, 1521.7, 1475.4, 1423.4, 1215.1, 1045.3, 927.7, 754.1, 669.3, 626.8.
1H NMR (CDCl3) δ: 4.66 (2H, s), 4.71(2H, s), 7.24−7.37 (4H, m), 7.41 (1H, t, J =7.6 Hz), 7.52 (1H, brd, J = 7.6 Hz), 7.56 (1H, t, J = 8.0 Hz), 7.72 (1H, brd, J= 8.0 Hz), 7.77 (1H, brs), 8.04 (1H, brs), 8.20 (1H, dd, J = 1.2 and 7.6 Hz),8.26 (1H, s), 8.32 (1H, d, J = 7.6 Hz).
ESI−MS (negative mode) m/z:430.5 (M − H)+ 。
【0159】
(実施例18:2−(3−(ニトロオキシメチル)ベンズアミド)フェニル 3−(ニトロオキシメチル)ベンゾエート K−230)
アセトニトリル15mlにK−231 を870 mg 溶かし、これにAgNO3 3915 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。沈殿物を、セライトを用いて減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、分取液体クロマトグラフィー(PLC)により精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK230を422.4mg得た。
m.p. 132.7−135.8℃
IR (CHCl3) cm−1: 3681.9,3620.1, 3018.4, 2399.3, 1521.7, 1423.4, 1215.1, 1031.8, 929.6, 754.1, 669.3,626.8.
1H NMR (CDCl3) δ: 5.38 (2H, s), 5.50(2H, s), 7.26−7.40 (3H, m), 7.47 (1H, t, J =7.6 Hz), 7.54 (1H, brd, J = 7.2 Hz), 7.61 (1H, t, J = 7.6 Hz), 7.72 (1H, J =7.6 Hz), 7.77 (2H, s), 7.99 (1H, brs), 8.27 (3H, s).
ESI−MS m/z: 468.4 (M+H)+。
【0160】
(実施例19:4−(クロロメチル)−N−フェニルベンズアミド K−241)
アニリン0.27 mlをCHCl3 10 mlに溶かし、ピリジン0.29 mlを加えた。これに、p−クロロメチルベンゾイルクロリド 680 mgをCHCl310 mlに溶かした溶液を滴下し、氷浴中2時間攪拌した。反応液をクロロホルム(CHCl3)30 mlで希釈後、順次1% 塩酸(HCl)、飽和炭酸水素ナトリウム(NaHCO3)、飽和食塩水で洗浄した。硫酸ナトリウム(Na2SO4)で乾燥後減圧濃縮すると、固形物が残査として得られた。これを、クロロホルム、ヘキサン混液から再結晶し、目的化合物670mgを得た。
m.p. 164.5−166.3℃
IR (KBr) cm−1: 3346.3,1654.8, 1598.9,1529.4, 1438.8,1325.0, 1303.8, 1267.1, 769.5, 754.1, 713.6,690.5, 675.0, 651.9.
1H NMR (CDCl3) δ:.4.64 (2H, s),7.17 (1H, t, J = 8.0 Hz), 7.39 (2H, t, J = 8.0 Hz), 7.52 (2H, d, J = 8.0 Hz),7.64 (2H, d, J = 8.0 Hz), 7.78 (1H, brs), 7.87 (1H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z:246.6(M+ H)+。
【0161】
(実施例20:4−(フェニルカルバモイル)ベンジル 硝酸塩 K−240)
アセトニトリル20 mlにK−241 を245.7mg 溶かし、これに硝酸銀(AgNO3)849.35 mgを加えた。遮光下アルゴン気流中60 ℃(外浴)で4時間攪拌した。反応液を、セライトを用いて減圧下濾取し、少量の酢酸エチルで洗浄した。洗液と濾液とを合し、減圧濃縮した。得られた残査を酢酸エチル:トルエン:ヘキサン=1:3:3を展開溶媒として、PLCにより精製した。得られた白色結晶を酢酸エチル、ヘキサン混液から再結晶し、目的のK−240を14.5mg得た。
m.p. 168.0−170.9℃
IR (CHCl3) cm−1: 3681.9,3620.1, 3018.4, 2399.3, 1523.7, 1477.4, 1423.4, 1215.1, 1031.8, 929.6, 786.9,754.1, 669.3, 626.8.
1H NMR (CDCl3) δ:.5.50 (2H, s),7.18 (1H, t, J = 6.8 Hz), 7.39 (2H, t, J = 8.0 Hz), 7.53 (2H, d, J = 7.6 Hz),7.63 (2H, d, J = 8.0 Hz), 7.80 (1H, brs), 7.91 (1H, d, J = 8.0 Hz).
ESI−MS (positive mode) m/z: 273.5(M + H)+。
【0162】
(実施例21:生物学的試験)
各製造例および実施例に記載の化合物について、上記したヒトがん細胞増殖抑制活性試験およびヒト正常繊維芽細胞CCD−1059SKに対する増殖抑制(毒性)試験を行った。
【0163】
その結果を、表1及び表2に示す。
表1 bis[2−(アシルアミノ)フェニル]ジスルフィド及びS−[2−(アシルアミノ)フェニル]アルカンチオレート、並びにその類縁体のヒト乳がん細胞並びにヒト正常繊維芽細胞に対する細胞増殖抑制活性(IC50)
【0164】
【表1】
【0165】
【0166】
【0167】
(表2 ヒト大腸がん細胞並びにヒト正常繊維芽細胞に対する増殖抑制活性(IC50))
【0168】
【表2】
【0169】
(実施例21:免疫沈降試験)
本発明の化合物の薬理活性について、そのメカニズムを探るために免疫沈降実験を行った。その手順は以下のとおりである。
【0170】
1.0×106cell/mlのHCT116細胞懸濁液或いは2.0×106cell/mlのSW620細胞懸濁液を60mm ディッシュに播種し、24時間培養後無血清培地 1mlで2回ディッシュ内を洗浄した。ついで、5 mlの無血清培地で再度洗浄し、24時間更に培養した。被検物質を培地に加え、18時間培養した。細胞を採取し、2度PBSで洗浄し、溶解用緩衝液(lysis buffer)(1M Tris−HCl (pH 8.0) 1ml, 5M NaCl, 1% nonidet P−40, 1 mg/ml アプロチニン(aprotinin),1 mg/ml ロイペプチン(leupeptin), 1 mg/ml ペプスタチン(pepstatin), 200 mM Na3VO4,200 mM NaF, 200 mM Na4P2O7, グリセロール(glycerol), および200mM PMSF) 400μlで再懸濁させた。得られた溶解物(lysates)は15000 × g, 4°Cで15分遠心処理した。得られた上清のタンパク質溶液のタンパク質濃度はBio−Radタンパク質アッセイキット(protein assay kit)で測定し、 その一部200μg (220μl) に対し、Anti−TCF4 モノクローナル抗体(2.5μl)及び600μl Protein G−Sepharose (2 mg/ml) を加え、4度で1時間インキュベーションした。ビーズを溶解用緩衝液で3度洗浄し、タンパク質をロード用緩衝液(loadingbuffer)に溶かし、常法にしたがいSDS/PAGEにより、ウェスタンブロッティングを行った。最終の検出は 「ECL Plus reagents」により行った。その結果を以下の表および図1に示す。
【0171】
【表3A】
【0172】
【表3B】
【0173】
図1の結果からも明らかなように、NCX4040は10μMで、β−カテニン/TCF4複合体レベルを約30%まで、20μMで10%まで減少させた。一方、K153のβ−カテニン/TCF4複合体形成阻害効果のIC50は約150μMであった。
【0174】
以上の結果から、本発明の化合物は、β−カテニン/TCF4複合体形成阻害に関連する任意の疾患における用途にも使用されることが理解される。
【0175】
(実施例22:細胞周期制御試験)
本発明の化合物の薬理活性について、そのメカニズムを探るために細胞周期制御試験を行った。その手順は以下のとおりである。
【0176】
1.5×106cell/mlのHCT116細胞懸濁液を作製し、24枚のディッシュ(コントロール 2枚、K−210添加濃度:1μM、5μM、10μM、20μM、40μM、60μM; NCX−4040添加濃度:10μM、20μM、30μM、50μMのディッシュを、それぞれ24hと48h用2枚ずつ)に予め培地を入れ、細胞懸濁液を1ml加えた。抗生物質(ペニシリン−ストレプトマイシン)を培地の量に対して1/100になるように加え、細胞の様子を顕微鏡で確認してからCO2インキュベーターで24時間培養した。
【0177】
ディッシュの培地をピペットマンで取り、無血清培地1mlで2回ディッシュ内を洗浄した。ディッシュに培地を5ml加え、CO2インキュベーターで24時間培養した。
【0178】
がん細胞に添加する薬剤サンプルK−210とNCX−4040を秤量し、DMSO(100μl)に溶解させ、培地(900μl)加えた後、培地で希釈し前培養で用意したディッシュの通りのサンプル溶液を製作した。ディッシュの培地を捨て、4mlの無血清培地を加えた。0時間以外の各ディッシュにサンプル溶液1mlを加え、24時間、また48時間培養した。培養した細胞をトリプシン3mlで剥がし、遠沈管に入れた培地に懸濁し、1000rpmで、5分間、遠心分離した。上清を捨て、常温に戻した「Buffer Solution」 (BD社 CycleTEST Plus)を1mlずつ加え、ボルテックスで穏やかに懸濁させた後、300×g5分間で遠心分離した。これを2回行った。上清を捨て、常温に戻したSolution A (BD社 CycleTEST Plus)を250μl加え、穏やかに攪拌、懸濁させ、室温で10分間静置した。室温に戻したSolutionB (BD社 CycleTEST Plus)を200μl加え、穏やかに攪拌、懸濁させ、室温で10分間静置した。氷温に解凍した「Solution C 」(BD社CycleTEST Plus)を200μl加え、完全に遮光し、氷上で10分間静置後、フローサイトメーター(Beckton Dickinson社FACScan)で測定した。その測定結果を図2(図2Aおよび図2B)に示す。
【0179】
図2の結果からも明らかなように、K210はヒト大腸がん細胞HCT116株の細胞周期をG2/M期で停止させていることが分かった。
【0180】
以上の結果から、本発明の化合物は、細胞周期をG2/M期で停止させることによって処置しうる任意の疾患における用途にも使用されることが理解される。
【0181】
(実施例23:TUNEL法によるアポトーシスの検出試験)
本発明の化合物の薬理活性について、そのメカニズムを探るためにTUNEL法によるアポトーシスの検出試験を行った。TUNEL法によるアポトーシスの検出試験は、Promega社 DeadEndTMFluorometric TUNEL System (#G3250)を用いて行った。その手順は以下のとおりである。
【0182】
薬剤サンプルを調製し、コントロールを除く、各60 mm ディッシュ(70 - 80% コンフルエンス(confluence)) に0.5 mlずつ加えCO2インキュベーターで24時間培養した。各ディッシュの培地をすべて、個々の遠沈管に回収した。
【0183】
残りの接着している細胞を、氷冷1×PBS (NaCl, 8.006 g; KCN, 0.1997 g; NaHPO4・12H20,2.897 g; KH2PO4, 0.200 g) 1 mlで2回洗浄した。37 ℃で温めたトリプシン1mlを加え、細胞をピペッティングにより剥がし、上記で取った培地に加え、300×gにて10分間遠心分離した。上清を捨てた。氷冷1×PBS3 mlを加え、ボルテックスで振とうした後、300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。
【0184】
氷冷1×PBS0.5 ml加え、4% パラホルムアルデヒドを加え、十分にピペッティングした。氷中に20分間置き、細胞を固定した。300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた。氷冷1×PBS3 mlを加え、ボルテックスで振とうした後、300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。
【0185】
氷冷1×PBS0.5 ml加え、70% エタノールを5ml加え、ピペッティングをした。
−20 ℃にて4時間放置。300×gにて10分間遠心分離した。細胞が沈殿していることを確認した後、上清を捨てた。氷冷1×PBS3mlを加え、ボルテックス振とうした後300×gにて10分間遠心分離した。
【0186】
細胞が沈殿していることを確認した後、上清を捨てた(2回行った)。BSA (5 mg/ml) およびTriton X−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。基準とするサンプル(24時間コントロール)に含まれる細胞数を、血球計算版を用いて計測した。全サンプルのOD600値を、吸光度計を用いて測定した。
【0187】
最も細胞数の少ないサンプルに合わせて、全サンプルを一定量にし、1.5 mlのエッペンドルフチューブに移した。420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
【0188】
80μlの平衡化用緩衝液(Equilibration buffer)を加えて懸濁し、室温で5分間静置した。
【0189】
TdTインキュベーションバッファーを以下の要領で作成した。
【0190】
Equilibration buffer 45μl×サンプル数
Nucleotide Mix 5μl×サンプル数
rTdT Enzyme 1μl×サンプル数。
【0191】
全量を混ぜて、遮光して氷中に置いておく。
【0192】
420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
各サンプルに、TdTインキュベーションバッファーを50μlずつ加え、懸濁した。
完全に遮光し、37℃のヒートブロック上で90分間インキュベートした(その間、エッペンドルフチューブを15分おきにボルテックス振とうし、遠心分離器によりチューブ壁に付着した沈殿物と溶液を底に集めた)。20mM EDTA溶液を1 ml加え、穏やかにボルテックスすることで、反応を停止させた。
【0193】
420×gにて10分間遠心分離し、細胞が沈殿していることを確認した後、上清を捨てた。
BSA (5 mg/ml) およびTritonX−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。
【0194】
細胞が沈殿していることを確認した後、上清を捨てた。BSA (5 mg/ml) およびTriton X−100 (0.1 %) を含む1×PBS 1mlを加え、ピペッティングをした。細胞が沈殿していることを確認した後、上清を捨てた。5μg/mlにMilli−Qで200倍希釈したPIを0.5 ml加え、ピペッティングにより懸濁後、各サンプルに25μlのRNaseを加え攪拌した。5μg/ml にMilli−Qで200倍希釈したPIを0.5ml加え、ピペッティングにより懸濁後、各サンプルに25μlのRNaseを加え攪拌した。調製した溶液の全量を穏やかにボルテックスで撹拌し、セルストレーナーを通して細胞塊を取り除いた。セルストレーナーをFACScanにセットし、測定した。測定結果を図3に示す。
【0195】
図3に示すように、K210はヒト大腸がん細胞株HCT116株に対し、アポトーシスを誘導することが分かった。
【0196】
以上の結果から、本発明の化合物は、アポトーシスの誘導によって処置しうる任意の疾患における用途にも使用されることが理解される。
【0197】
(実施例24:ウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出)
本発明の化合物の薬理活性について、そのメカニズムを探るためにウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出を行った。その手順は以下のとおりである。
【0198】
HCT116細胞を60mmディッシュに添加(2.0 × 106 cells/ディッシュ)し、CO2インキュベータで24時間培養した。細胞を無血清培地(McCoy’s5A) 1 mlで2回洗浄後、直ちに無血清培地 5 mlを加え、更に24時間培養した。細胞を、各ディッシュ毎に無血清培地4 mlで洗浄し、K−210 (20μM,40μM, 60μM)を、0分間, 15分間, 30分間, 1時間, 2時間, 4時間, 6時間, 8時間の各時間培養した。同様に、SKBR3細胞を60 mmディッシュに添加(1.0 × 106 cells/ディッシュ)し、CO2インキュベータで24時間培養した。細胞を無血清培地(RPMI−1640)1 mlで2回洗浄後、直ちに無血清培地 5 mlを加え、更に24時間培養した。細胞を、各ディッシュ毎に無血清培地4 mlで洗浄し、K−210 (1.0,5.0, 10, 20, 40 and 60μM)を、24時間培養した。各接着細胞をPBSで二回洗浄し、Covance Laboratories lysisbuffer (Immunochemistry Protocols for use with Innovative Antibody Products byCovance Research Products. http://store.crpinc.com/prot_western.aspx r) (ただし、pH7.4の代わりにpH 8.0 、1% Triton−X 100の代わりに1% Nonidet P−40 を採用した) を200μMを加え、懸濁した。細胞溶解液は4度で15分間遠心処理(14000× g )し、得られたタンパク溶液の濃度をBio−Rad protein assay kitにより測定した。均一濃度のたんぱく質溶液をSDS−PAGE に展開し、PVDFメンブレンにトランスファーした。一次抗体として、抗アクチン(actin)モノクローナル抗体、ERKモノクローナル抗体, p−ERKモノクローナル抗体, JNKモノクローナル抗体,p−JNKモノクローナル抗体, p38モノクローナル抗体, p−p38モノクローナル抗体, p21/WAF1モノクローナル抗体(Sigma, St.Louis, MOから入手可能)を使用し、2,500倍に希釈した。また、セイヨウワサビ ペルオキシダーゼ標識二次抗体を2,500倍に希釈し、各バンドの蛍光強度をECL化学発光法(chemiluminescence)で測定した。結果を図4に示す。図4の結果から、ストレス応答型主要MAPKsであるp38およびJNKのリン酸化体のタンパクレベルが上昇したことにより、K−210等はこの経路によってアポトーシスを誘導した可能性が高いと推定された。
【0199】
以上の結果から、本発明の化合物は、主要MAPKs、ERK、JNK、p38、およびそのリン酸化の誘導によって処置しうる任意の疾患における用途にも使用されることが理解される。
【0200】
以上のように、本発明の好ましい実施形態を用いて本発明を例示してきたが、本発明は、この実施形態に限定して解釈されるべきものではない。本発明は、特許請求の範囲によってのみその範囲が解釈されるべきであることが理解される。当業者は、本発明の具体的な好ましい実施形態の記載から、本発明の記載および技術常識に基づいて等価な範囲を実施することができることが理解される。本明細書において引用した特許、特許出願および文献は、その内容自体が具体的に本明細書に記載されているのと同様にその内容が本明細書に対する参考として援用されるべきであることが理解される。
【産業上の利用可能性】
【0201】
本発明は、β−カテニン/TCF複合体の形成に関与する疾患、たとえば、がんを処置するための医薬、それに使用される化合物またはその溶媒和物を提供する。本発明化合物は実施例の記載の通り、優れた抗がん活性を示す。
【図面の簡単な説明】
【0202】
【図1】図1は、免疫沈降試験(実施例21)の結果を示す。左側は大腸がん細胞株HCT116を示し、右側は大腸がん細胞SW620を示す。二段目には各化合物の記号を示す。上のウェスタンブロッティングの結果はβ−カテニンの結果、下はTCF4での結果を示す。下のグラフには、それぞれ左側は大腸がん細胞株HCT116を示し、右側は大腸がん細胞SW620のβ−カテニン−TCF複合体量の濃度依存性を示したグラフを示す。
【図2A】図2Aは、細胞周期制御試験実験(実施例22)の結果(フローサイトメトリ)を示す。左上はコントロール、右上は、K210 1μM,左下にはK210 5μM、右下にはK210 10μMのものを示す。
【図2B】図2Bは、細胞周期制御試験実験(実施例22)の結果(フローサイトメトリ)を示す。参考物質のNCX−4040について、左上はコントロール、右上は、10μM,左下には20μM、中下には30μM、右下には50μMのものを示す。
【図3】図3は、TUNEL法によるアポトーシスの検出試験(実施例23)の結果を示す。左上はコントロール、右上は、K210 1μM,左下にはK210 5μM、右下にはK210 10μMのものを示す。横軸は、FL2−H (PI)を示し、縦軸 FL1−H (フルオレセイン−12−dUTP)を示す。
【図4】図4は、ウェスタンブロッティングによる主要MAPKs、ERK、JNK、p38、およびそのリン酸化体の検出(実施例24)の結果を示す。各々についての説明は、以下のとおりである。左パネルに20μM、中パネルに40μM、右パネルに60μMを示す。マーカーは、上から、p0ERK1、pERK2、pJNK1、pJNK2、p−p38、p38、アクチン、アクチン、p21/WAF1を示す。ストレス応答型主要MAPKsであるp38およびJNKのリン酸化体のタンパクレベルが上昇したことにより、K−210等はこの経路によってアポトーシスを誘導した可能性が高いと推定された。
【特許請求の範囲】
【請求項1】
以下の構造を有する化合物:
【化1】
であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物またはその溶媒和物。
【請求項2】
以下の構造を有する化合物:
【化2】
であって、
R2は、ハロゲンまたはONO2で置換されたアルキル基であり、
R3は、ハロゲンまたはONO2で置換されたアルキル基であり、
Aは、−O−、−S−または−NH−である、
請求項1に記載の化合物またはその溶媒和物。
【請求項3】
R2およびR3は、−CH2−Clまたは−CH2−ONO2である、請求項2に記載の化合物またはその溶媒和物。
【請求項4】
R2およびR3は、−CH2−Clまたは−CH2−ONO2であり、パラ位に存在する、請求項2に記載の化合物またはその溶媒和物。
【請求項5】
R2およびR3は、−CH2−ONO2である、請求項2に記載の化合物またはその溶媒和物。
【請求項6】
R2およびR3は、−CH2−ONO2であり、パラ位に存在する、請求項2に記載の化合物またはその溶媒和物。
【請求項7】
以下からなる群より選択される、請求項1に記載の化合物またはその溶媒和物。
【化3】
。
【請求項8】
請求項1〜6のいずれか1項に記載の化合物またはその溶媒和物を含む、医薬組成物。
【請求項9】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を含む、抗がん剤。
【請求項10】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を含む、乳がん、結腸癌および肺がんのいずれかまたは複数に対する抗がん剤。
【請求項11】
【化4】
および
【化5】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗乳がん剤。
【請求項12】
【化6】
【化7】
および
【化8】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗肺がん剤。
【請求項13】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物の、医薬組成物の製造のための使用。
【請求項14】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物の、抗がん剤の製造のための使用。
【請求項15】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を投与する工程を包含する治療方法。
【請求項16】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を投与する工程を包含するがんの治療方法。
【請求項1】
以下の構造を有する化合物:
【化1】
であって、
R1は、Hまたは−R1aであり、
R1aは、−A−R1bであり、
Aは、−O−、−S−または−NH−であり、
R1bは、−C(=O)R1cであり、
R1cは、水素、置換または非置換のアルキル基、置換または非置換のアルケニル基、置換または非置換のアルキニル基、置換または非置換のシクロアルキル基、置換または非置換のシクロアルケニル基、および置換または非置換のアリール基、ヘテロアリール基またはヘテロサイクル基からなる群より選択され、
R2は、ハロゲンまたはONO2で置換されたアルキル基である、
化合物またはその溶媒和物。
【請求項2】
以下の構造を有する化合物:
【化2】
であって、
R2は、ハロゲンまたはONO2で置換されたアルキル基であり、
R3は、ハロゲンまたはONO2で置換されたアルキル基であり、
Aは、−O−、−S−または−NH−である、
請求項1に記載の化合物またはその溶媒和物。
【請求項3】
R2およびR3は、−CH2−Clまたは−CH2−ONO2である、請求項2に記載の化合物またはその溶媒和物。
【請求項4】
R2およびR3は、−CH2−Clまたは−CH2−ONO2であり、パラ位に存在する、請求項2に記載の化合物またはその溶媒和物。
【請求項5】
R2およびR3は、−CH2−ONO2である、請求項2に記載の化合物またはその溶媒和物。
【請求項6】
R2およびR3は、−CH2−ONO2であり、パラ位に存在する、請求項2に記載の化合物またはその溶媒和物。
【請求項7】
以下からなる群より選択される、請求項1に記載の化合物またはその溶媒和物。
【化3】
。
【請求項8】
請求項1〜6のいずれか1項に記載の化合物またはその溶媒和物を含む、医薬組成物。
【請求項9】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を含む、抗がん剤。
【請求項10】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を含む、乳がん、結腸癌および肺がんのいずれかまたは複数に対する抗がん剤。
【請求項11】
【化4】
および
【化5】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗乳がん剤。
【請求項12】
【化6】
【化7】
および
【化8】
からなる群より選択される少なくとも1つの化合物またはその溶媒和物を含む、抗肺がん剤。
【請求項13】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物の、医薬組成物の製造のための使用。
【請求項14】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物の、抗がん剤の製造のための使用。
【請求項15】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を投与する工程を包含する治療方法。
【請求項16】
請求項1〜7のいずれか1項に記載の化合物またはその溶媒和物を投与する工程を包含するがんの治療方法。
【図1】

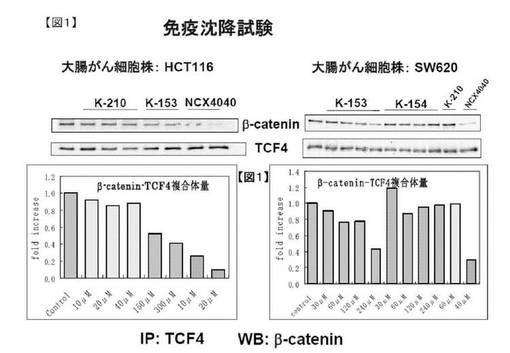
【図2A】
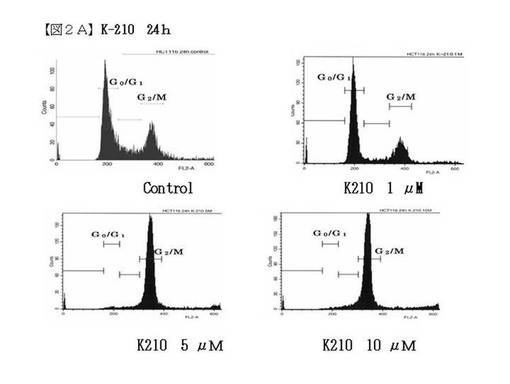
【図2B】
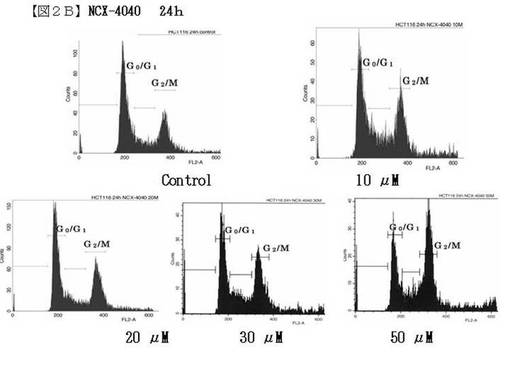
【図3】
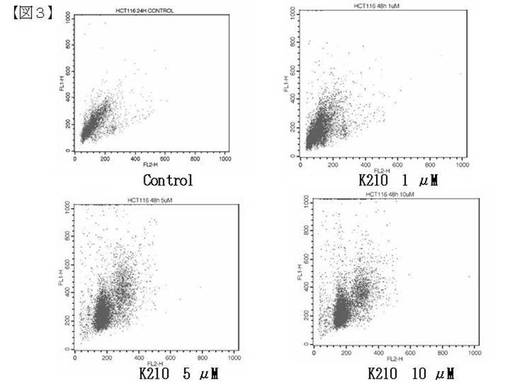
【図4】
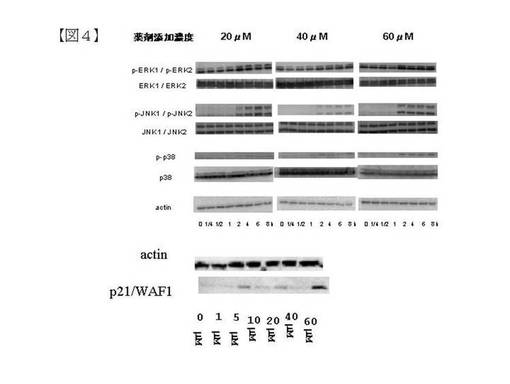
【図2A】
【図2B】
【図3】
【図4】
【公開番号】特開2010−53060(P2010−53060A)
【公開日】平成22年3月11日(2010.3.11)
【国際特許分類】
【出願番号】特願2008−218662(P2008−218662)
【出願日】平成20年8月27日(2008.8.27)
【出願人】(399030060)学校法人 関西大学 (208)
【Fターム(参考)】
【公開日】平成22年3月11日(2010.3.11)
【国際特許分類】
【出願日】平成20年8月27日(2008.8.27)
【出願人】(399030060)学校法人 関西大学 (208)
【Fターム(参考)】
[ Back to top ]