
アシルオキサゾロン誘導体及びβ−ケトアミノ酸誘導体の製造方法
【課題】β−ケトアミノ酸誘導体に変換可能なアシルオキサゾロン化合物を短工程、かつ強塩基や高価な試薬、極低温を必要としない安価な条件で効率的・高収率、かつ後処理・精製が容易な方法で製造することができる、アシルオキサゾロン化合物の工業的製造方法を提供すること。
【解決手段】オキサゾール化合物にルイス酸の存在下、酸ハライド、酸無水物等のアシル化剤を作用させることにより、アシルオキサゾロン化合物を得ることを特徴とするアシルオキサゾロン化合物の製造方法、及び上記アシルオキサゾロン化合物からβ−ケトアミノ酸誘導体を製造する方法。
【解決手段】オキサゾール化合物にルイス酸の存在下、酸ハライド、酸無水物等のアシル化剤を作用させることにより、アシルオキサゾロン化合物を得ることを特徴とするアシルオキサゾロン化合物の製造方法、及び上記アシルオキサゾロン化合物からβ−ケトアミノ酸誘導体を製造する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アシルオキサゾロン化合物及びβ−ケトアミノ酸誘導体の製造方法に関する。本発明の方法で得られるβ−ケトアミノ酸誘導体は、変換して光学活性β−ヒドロキシアミノ酸を経由して、抗HIVウィルス薬として有用なことが知られている薬剤の中間体であるD−エリスロ−3−シクロヘキシルセリンに誘導可能であることから、医農薬中間体として有用な化合物である。
【背景技術】
【0002】
アシルオキサゾロン類の合成方法としては、馬尿酸又は2−フェニルオキサゾール−5−オンをトリフルオロ酢酸存在下、γ−ピコリン中、酸クロリドと反応させる方法(非特許文献1)、及び馬尿酸ナトリウム塩と酸無水物をβ−ピコリン中反応させる方法(非特許文献2及び3)が知られている。しかし、いずれの反応も酸クロリド又は酸無水物を過剰に用いる必要があり、高価な酸クロリド又は酸無水物を使用する必要がある場合、製造コストが高額になるという問題点があった。
【0003】
また、2−フェニルオキサゾール−5−オンとアシル化剤等量を用いる方法(非特許文献4)も報告されている。この文献では高価なγ−ピコリンを溶媒として用いているが、γ−ピコリンは高沸点かつ水溶性も高いため回収が困難であるので製造コストが高くなり、また、大量の溶媒を用い、反応時間に1日を要する等実用的ではない。一方、γ−ピコリンを用いない方法(特許文献1及び2)も報告されているが、いずれも用いる酸クロリドはコハク酸モノエステルクロライド誘導体に限定され、一般性が高いとは言えず、反応時間も1日〜3日と長時間を要する。
【0004】
また、これまでに報告されているアシルオキサゾロン類の合成例は単純な化合物又は類似した化合物が多く、特に立体障害の大きいα位に置換基を持つ酸クロリド又は酸無水物を用いた例はわずかしかない上(特許文献1、非特許文献3)、そのいずれも収率30%程度と低収率であり、工業的に安価かつ汎用性の高いアシルオキサゾロン類の製造には課題が残されていた。
【0005】
【非特許文献1】Bull.Acad.Sci.USSR Div.Chem.Sci.1982,31,2223.
【非特許文献2】J.Chem.Soc.1948,3,310−318
【非特許文献3】J.Chem.Soc.1960,968−973
【非特許文献4】Bull.Acad.Sci.USSR Div.Chem.Sci.1984,33,384.
【特許文献1】特開平1−305071号公報
【特許文献2】国際公開第98/54162号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、β−ケトアミノ酸誘導体に変換可能なアシルオキサゾロン化合物を短工程、かつ強塩基や高価な試薬、極低温を必要としない安価な条件で効率的・高収率、かつ後処理・精製が容易な方法で製造することができる、アシルオキサゾロン化合物の工業的製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するために鋭意検討した結果、オキサゾール化合物をアシル化することでアシルオキサゾロン化合物を合成でき、このアシルオキサゾロン化合物をアルコール類と反応させることでβ−ケトアミノ酸化合物を一貫して温和な条件で効率的に合成できることを見出し、本発明を完成するに至った。
【0008】
即ち、本発明によれば、以下の発明が提供される。
(1) 下記一般式(1)
【化1】

(式中、R1及びR2はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。)で表されるオキサゾール化合物にルイス酸の存在下、アシル化剤を作用させることにより、下記一般式(2)
【化2】
(式中、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるアシルオキサゾロン化合物を製造する工程を含む、アシルオキサゾロン化合物の製造方法。
【0009】
(2) アシル化剤が、酸ハライド又は酸無水物である、(1)に記載の方法。
【0010】
(3) (1)の方法により製造した一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【化3】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表される化合物を製造する工程を含む、β−ケトアミノ酸誘導体の製造方法。
【0011】
(4) 下記一般式(4)
【化4】
(式中、R1は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるシクロヘキシルメチリデニルオキサゾロン化合物。
【0012】
(5) (i)(3)に記載の方法により一般式(3)で表されるβ−ケトアミノ酸誘導体を製造する工程、及び
(ii)上記工程(i)で得られた一般式(3)で表されるβ−ケトアミノ酸誘導体に微生物の菌体及び/又は該菌体処理物を作用させて下記一般式(5)
【化5】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるα−N−アシルアミノ−β−ヒドロキシエステルを得る工程を含む、α−N−アシルアミノ−β−ヒドロキシエステルを製造する方法。
【0013】
(6) (i)(5)に記載の方法により一般式(5)で表されるN−アシルアミノ−β−ヒドロキシエステルを製造する工程、
(ii)上記工程(i)で得られたα−N−アシルアミノ−β−ヒドロキシエステルに酸又は塩基を作用させて加水分解を行い、下記一般式(6)
【化6】
(式中、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるβ−ヒドロキシアミノ酸を得る工程;及び
(iii)上記工程(ii)で得られたβ−ヒドロキシアミノ酸にt−ブトキシカルボニル化剤を作用させて下記一般式(7)
【化7】
(式中、R3及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物を製造する工程、を含むN−t−ブトキシカルボニル−β−ヒドロキシアミノ酸の製造方法。
【発明の効果】
【0014】
本発明によれば、安価かつ温和な工業的製造法で、医農薬中間体として有用な化合物であるアシルオキサゾロン化合物及びβ−ケトアミノ酸化合物を効率よく製造することができる。
【発明を実施するための最良の形態】
【0015】
以下、本発明を詳細に説明する。
<オキサゾ−ル化合物、アシルオキサゾロン化合物>
本発明の製造方法は、上記一般式(1)で表されるオキサゾール化合物にルイス酸の存在下、酸ハライド又は酸無水物などのアシル化剤を作用させることにより、上記一般式(2)で表されるアシルオキサゾロン化合物を得ることを特徴とするものである。
【0016】
上記一般式(1)において、R1は、置換していてもよいアルキル基又は置換してもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくは炭素数1〜6の直鎖状若しくは分岐状のアルキル基であり、より好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、例えば、メチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、シクロヘキシル基であり、最も好ましくはメチル基である。置換してもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記のアルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、アルキル基、アリール基、アルコキシ基、フッ素原子、塩素原子、臭素原子等のハロゲン原子、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられる。好ましくはハロゲン原子、アルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0017】
上記一般式(1)において、R2は置換していてもよいアルキル基又は置換していてもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくは炭素数1〜6の直鎖状若しくは分岐状のアルキル基であり、より好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、例えば、メチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、又はシクロヘキシル基であり、最も好ましくはメチル基である。置換してもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記アルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、アルキル基、アリール基、アルコキシ基、フッ素原子、塩素原子、臭素原子等のハロゲン原子、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられる。好ましくはハロゲン原子、アルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0018】
一般式(1)は、好ましくはR1がフェニル基で、R2がメチル基又はフェニル基であることが好ましい。
【0019】
上記一般式(2)において、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくはメチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基等の炭素数1〜7の直鎖状、分岐状若しくは環状のアルキル基、特に好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基又は炭素数5〜7の環状のアルキル基であり、最も好ましくは炭素数5〜7の環状のアルキル基である。置換していてもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記アルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、フッ素原子、塩素原子、臭素原子等のハロゲン原子、アルキル基、アリール基、アルコキシ基、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられ、好ましくはアルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0020】
上記一般式(2)において、Xは水素原子又は金属原子を示す。金属原子は特に限定はないが、Li、Na、K、Rb等のアルカリ金属原子、Be、Mg、Ca、Sr、Ba等のアルカリ土類金属、Ti、Mn、Fe、Ni、Zn、Zr、Hg、Pd等の遷移金属原子、Sn、Pb、Al等の典型金属が挙げられる。金属原子として好ましいのは、アルカリ金属であるLi、Na、K、Rb、又はアルカリ土類金属であるBe、Mg、Ca、Sr、Baである。Xとして、好ましいのは水素原子である。
【0021】
上記一般式(2)において、Xが水素原子の場合、金属化合物と錯体を形成していてもよい。具体的には、一般式(2)のヒドロキシル基とカルボニル基が金属化合物と配位結合をして錯体を形成する場合や、一般式(2)のヒドロキシル基と窒素原子が金属化合物と配位結合をして錯体を形成する場合が挙げられる。また、金属に対して、一般式(2)は1:1配位であってもよく、1:2配位であってもかまわない。
【0022】
金属化合物は特に限定はないが、金属原子は、Ti、Mn、Fe、Ni、Zn、 Zr、Hg、Pd等の遷移金属原子、Sn、Pb、Al等の典型金属が挙げられる。また、金属原子として好ましいのは、安価かつ本反応で種々の基質に対して汎用性のある触媒となりうるSn、Al、Fe、Ni、Zn等が好ましい。
【0023】
錯体として、好ましいのは反応に用いるルイス酸又はその派生物がヒドロキシル基とカルボニル基と配位結合をしている化合物である。具体的には、塩化スズ(IV)錯体、塩化アルミニウム錯体、塩化鉄(III)錯体、塩化ニッケル錯体等が挙げられる。好ましくは塩化スズ(IV)錯体、塩化アルミニウム錯体である。
【0024】
一般式(2)において好ましくは、R1がフェニル基であり、R3がシクロヘキシル基であり、Xが水素、Na、K、又はCaであるか、又はXが水素でSn、Al、Fe、Ni、Zn錯体である。より好ましくは、R1がフェニル基であり、R3がシクロヘキシル基であり、Xが水素であるか、又はXが水素でSn、Al、Fe、Ni、Zn錯体である。
【0025】
また、一般式(2)及び(4)で表される化合物のうち、Xが水素原子の場合は、ケト型の互変異性体であってもよい。
【0026】
本発明においては、一般式(1)のオキサゾール化合物をルイス酸の存在下、酸ハライド(例えば、酸クロライド)又は酸無水物などのアシル化剤を作用させて一般式(2)のアシルオキサゾロン化合物を得るものである。かくして製造される一般式(2)のアシルオキサゾロン化合物のうち、下記一般式(4)
【0027】
【化8】
【0028】
(式中、R1及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物は新規化合物である。
【0029】
一般式(4)の具体例としては、次のような化合物が挙げられる。
【0030】
【化9】
【0031】
式(4a)〜(4e)中、R1はメチル基、フェニル基を、M1はNa、K、Caを、M2はSnCl4、AlCl3、FeCl3、NiCl2、ZnCl2を表す。
【0032】
<アシルオキサゾロン誘導体の製造方法>
本発明は、一般式(1)のオキサゾール化合物をルイス酸の存在下、酸ハライド或いは酸無水物などのアシル化剤を作用させて一般式(2)のアシルオキサゾロン化合物を得るものである。
【0033】
ルイス酸としては、特に限定はないが、具体的には塩化アルミニウム(III)、四塩化スズ(IV)、塩化鉄(III)、塩化亜鉛(II)、塩化ニッケル(II)、三フッ化ホウ素エーテル錯体等が挙げられ、好ましくは工業的に入手が容易である四塩化スズ(IV)、塩化鉄(III)、塩化ニッケル(II)、塩化アルミニウム(III)が好ましい。
【0034】
ルイス酸の使用量としては特に限定されないが、通常は原料である一般式(1)に対して0.02モルから10モル倍で反応は進行するが、生成物とルイス酸が錯体を形成しやすい性質があるため1モル倍以上の使用が好ましく、コストの観点から1〜3モル倍の使用が好ましい。
【0035】
本発明で使用するアシル化剤は、一般式(1)のオキサゾール化合物に作用させることによって一般式(2)のアシルオキサゾロン化合物を製造することができるものであれば特に限定されないが、好ましくは酸ハライド又は酸無水物である。酸ハライド又は酸無水物としては、特に限定されないが、具体的には、アセチルクロリド、イソ酪酸クロリド、ピバロイルクロリド、シクロヘキサノイルクロリド、ベンゾイルクロリド、4−メトキシベンゾイルクロリド等の酸クロリド;アセチルブロミド、イソプロピオン酸ブロミド、ピバロイルブロミド、シクロヘキシルブロミド、ベンゾイルブロミド、4−メトキシベンゾイルブロミド等の酸ブロミド;アセチルヨージド、イソ酪酸ヨージド、ピバロイルヨージド、シクロヘキサノイルヨージド、ベンゾイルヨージド、4−メトキシベンゾイルヨージド等の酸ヨージドが挙げられ、好ましくはコスト的に安価である酸クロリド、酸ブロミドが好ましく、更に好ましくはシクロヘキサノイルクロリドやイソ酪酸クロリド、ベンゾイルクロリド等である。
【0036】
酸無水物としては、無水酢酸、プロピオン酸無水物、ピバリン酸無水物、シクロヘキサン酸カルボン酸無水物、安息香酸無水物等があり、好ましくはシクロヘキサン酸カルボン酸無水物、プロピオン酸無水物、ピバリン酸無水物である。
【0037】
酸ハライドの使用量としては特に限定されないが、通常は原料である化合物(1)に対して0.8〜5倍モル、コストや化合物(2)の精製のしやすさの観点から一般式(1)と同モルに近い0.9〜2倍モルの使用が好ましい。
【0038】
反応を行う条件のうち、用いる溶媒としては反応に悪影響を与えないものであれば特に限定はないが、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒;エチルエーテル、プロピルエーテル、ブチルエーテル、テトラヒドロフラン等のエーテル系溶媒;ジクロロメタン、クロロホルム、ジクロロエタン、クロロベンゼン等のハロゲン系溶媒;酢酸エチル、酢酸ブチル等のエステル系溶媒、アセトニトリル等のニトリル系溶媒、ニトロベンゼン、ニトロメタン等のニトロ系溶媒;等が挙げられる。これらから選ばれる複数の溶媒を任意の割合に混合して用いてもよい。好ましい溶媒は、一般的にアシル化反応に影響を及ぼさないジクロロメタン、クロロベンゼン等のハロゲン系溶媒、アセトニトリル等のニトリル系溶媒、ニトロベンゼン、ニトロメタン等のニトロ系溶媒、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒である。更に好ましくは、アセトニトリル、ジクロロメタン、クロロホルム、クロロベンゼン、ニトロベンゼン、ニトロメタン及びトルエンである。
【0039】
また、溶媒の使用量としては、任意の量の溶媒を用いることができるが、通常は原料基質に対して2〜50倍体積量、好ましくは3〜10倍体積量である。
【0040】
この反応を実施する温度としては、特に限定されないが−50℃から100℃程度であり、好ましくは−10℃から40℃程度が好ましい。実施する圧力に関しては特に加圧下で行う必要はないが、反応時間を短縮する等の観点から常圧から10気圧程度の圧力をかけてもよい。反応時間は、10分〜数日の範囲で行うことができるが、製造コストを抑える観点から24時間以内に終了させることが好ましく、更に好ましくは10分から8時間である。
【0041】
一般式(1)のオキサゾール化合物は公知の方法で製造すれば特に限定はないが、下記一般式(8)
【0042】
【化10】
【0043】
で表されるオキサゾロン化合物とベンゾイルクロリドやアセチルクロリド等のアシル化剤を作用させる、あるいは下記一般式(9)
【0044】
【化11】
【0045】
で表される馬尿酸とベンゾイルクロライドやアセチルクロライド等のアシル化剤を作用させることで合成可能である。ここで、一般式(8)及び(9)におけるR1は、一般式(1)におけるR1と同義である。
【0046】
<β−ケトアミノ酸誘導体の製造方法>
上記一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【0047】
【化12】
【0048】
(式中、R1及びR3は前記と同義である。R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表されるβ−ケトアミノ酸誘導体を製造することができる。
【0049】
アルキル基としては、好ましくは炭素数1〜6の直鎖状、分岐状又は環状のアルキル基であり、更に好ましくはメチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基等の炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、より好ましくはメチル基、エチル基、イソプロピル基、t−ブチル基である。
【0050】
アラルキル基としては、ベンジル基が好ましい。
【0051】
上記アルキル基及びアラルキル基は置換していてもよく、置換基としては特に限定はないが、フッ素原子、塩素原子、臭素原子等のハロゲン原子、アルキル基、アルコキシ基、ニトロ基、アリールオキシ基、アミノ基、アリール基等が挙げられる。好ましくは塩素原子、アルコキシ基である。置換してもよいアラルキル基としては、ベンジル基、4−メトキシベンジル基、4−フルオロベンジル基、4−クロロベンジル基、3,4−ジメチルベンジル基が好ましく、更に好ましくはベンジル基、4−クロロベンジル基である。
【0052】
一般式(3)は、好ましくはR1がフェニル基又はメチル基であり、R3がシクロヘキシル基であり、R4がメチル基、エチル基、イソプロピル基、t−ブチル基、又はベンジル基であることが好ましい。
【0053】
上記一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより、上記一般式(3)で表されるβ−ケトアミノ酸誘導体を製造することができる。
【0054】
一般式(2)で表されるオキサゾロン化合物は、アルコールと反応させるにあたり、各種有機溶媒にて抽出及び/又は晶析等の方法で精製・単離することが好ましい。好ましくは、ろ過操作にて原料を取得する或いは塩酸等の酸の水溶液にて反応を終結後、有機溶媒にて抽出・洗浄操作を行い、反応に用いたルイス酸を除去したのち、晶析操作を行うことが望ましい。
【0055】
反応に用いるアルコールとしては、特に限定されるものではないが、具体的にはメタノール、エタノール、ノルマルプロピルアルコール、イソプロピルアルコール、ノルマルブチルアルコール、イソブチルアルコール、t−ブチルアルコール、ベンジルアルコール等であり、好ましくはエタノール、イソプロピルアルコール、t−ブチルアルコール、ベンジルアルコールである。
【0056】
アルコールの使用量は、一般式(2)のオキサゾロン化合物に対して1倍量から溶媒量まで使用することができるが、好ましくは1〜5倍量で使用することがコスト的に好ましい。
【0057】
反応の温度としては、特に限定はされないが、通常室温から各種溶媒の沸点付近まで、好ましくは20℃から100℃がよい。この反応は常圧、大気中で行うことができ、特に窒素雰囲気下で行う必要はないが、必要に応じて窒素、ヘリウム等の不活性気体中で加圧下にて実施することもできる。
【0058】
反応時間は、10分〜数日の範囲で行うことができるが、製造コストを抑える観点から24時間以内に終了させることが好ましく、更に好ましくは10分から8時間である。
【0059】
上記にて製造した一般式(3)のβ−ケトアミノ酸化合物は、反応液を濃縮するだけで特に精製は必要ないが、用いるアルコールの沸点が高い場合、用いた一般式(2)のオキサゾロン化合物の純度が低かった場合等は必要に応じて適宜、有機溶媒と水による分液操作での単離又は晶析による精製を行うことができる。
【0060】
<β−ケトアミノ酸化合物の用途>
上記一般式(3)で表されるβ−ケトアミノ酸化合物は、それ自体或いは各種の手法にて誘導化し、種々の医薬や農薬の中間体として有用である。特に、一般式(7)で表される化合物において、R3がシクロヘキシル基である化合物に相当する下記式(10)
【0061】
【化13】
で表される化合物は抗HIV活性をもつ薬剤(WO01/40227) の中間体として工業的に有用である。上記式(10)やその類縁体への変換の手法としては、上記一般式(3)で表される化合物を下記一般式(5)
【0062】
【化14】
【0063】
(式中、R1、R3、R4は前記と同義。)で表される化合物に立体選択的に変換する。
【0064】
この変換の手法としては、微生物の菌体及び/又は該菌体処理物を作用させる方法、或いは特許文献(WO04/005371A1)や非特許文献(Tetrahedron Asymmetry 2001,12,1757-1762)に記載のルテニウム等の金属触媒を用いた不斉水素化反応が利用可能である。好ましくは、重金属混入の恐れがない微生物の菌体及び/又は該菌体処理物を作用させる方法である。
【0065】
微生物の菌体及び/又は該菌体処理物を作用させる方法としては、特に限定はないが、アグロバクテリウム属又はエクシグオバクテリウム(Exiguobacterium)属に属する微生物を使用するか、不斉還元反応を触媒するタンパク質をコードする遺伝子を導入した形質転換細胞を作製し、該細胞、該細胞の調製物、又は該細胞を培養して得られた培養液を原料と作用させてもかまわない。
【0066】
上記一般式(5)で表される化合物を下記一般式(6)
【0067】
【化15】
表される化合物或いはその塩に変換する。
【0068】
この変換の手法としては、酸又は塩基の存在下での加水分解、又は加水分解酵素にて加水分解を行う手法が挙げられる。
【0069】
上記一般式(6)で表される化合物を塩基条件下、t−ブトキシカルボニル化を行うことにより上記一般式(7)で表される化合物(例えば、式(10)で表される化合物)へと変換可能である。
【0070】
以下、本発明を実施例により更に詳細に説明するが、本発明はこれらによって限定されるものではない。
【実施例】
【0071】
[実施例1a]4−(1−シクロヘキシル−1−ヒドロキシメチリデン)−2−フェニルオキサゾール−5−オンの合成
100mLのフラスコにジクロロメタン43mL、5−アセトキシ−2−フェニルオキサゾール4.28g(21.1mmol)を仕込み、シクロヘキサンカルボン酸クロリド3.10mL(23.2mmol)を添加し、しばらく撹拌した後に四塩化スズ3.87mL(73.2mmol)を10分間かけて滴下した。24時間後、TLCにて原料の消失を確認し、4N塩酸水溶液10mL及び酢酸エチル150mLを添加し、有機層を分離した後、更に水50mLで2回、飽和食塩水50mLにて洗浄し、硫酸マグネシウムにて乾燥後、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル150g、 ヘキサン:酢酸エチル=100:2〜100:4)にて精製し、黄色固体として目的物3.20g(純度換算後の収率52%)を得た。
1H−NMR(400MHz,DMSO−d):δ1.14−1.51(5H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),7.53−7.57(3H,m),7.96−8.01(2H,m)
【0072】
[実施例1b]4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの合成
1Lのフラスコに5−ベンゾイルオキシ−2−フェニルオキサゾール47.0g(177mmol)、ジクロロメタン380mLを仕込み、氷冷下にてシクロヘキサンカルボン酸クロリド33.0mL(248mmol)を添加し、しばらく撹拌した後に四塩化スズ29.0mL(248mmol)を2分間かけて滴下した。室温に昇温した後、24時間後にTLCにて原料の消失を確認し、反応液をろ過して得られた4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの塩化スズ錯体をLCにて定量分析したところ、4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンとしての収率は89%であった。この錯体に2N塩酸160mL及び酢酸エチル500mLを添加し、有機層を分離した後に更に2N塩酸160mLで2回洗浄し、硫酸マグネシウムにて乾燥後、溶媒を留去した。得られた残渣をジクロロメタン15mL、ヘキサン15mLにて懸洗し、ろ過して得られた黄色固体として目的物4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オン15.5g(純度98%、収率32%)を得た。
1H−NMR(400MHz,DMSO−d):δ1.14−1.51(5H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),7.53−7.57(3H,m),7.96−8.01(2H,m).
【0073】
[実施例1c]4−(1−シクロヘキシル-1-ヒドロキシメチリデン)−2−フェニルオキサゾール-5-オンの合成
200mlのフラスコにジクロロメタン94ml、5−ベンゾイルオキシ-2−フェニルオキサゾール 13.4g(50.5mmol)を仕込み、氷冷下にてシクロヘキサンカルボン酸クロリド13.5ml(101mmol)を添加し、しばらく攪拌した後に四塩化すず16.5ml(2.8mmol)を10分間かけて滴下した。室温に昇温させて16時間攪拌後、TLCにて原料の消失を確認し、反応液をろ過し、固体部分を塩化メチレン:n-ヘキサンの1:1の混合溶液20mlにて洗浄後、淡黄色固体として目的物のすず塩34.6g(純度換算後の収率97%)を得た。
1H-NMR(400MHz, DMSO-d):δ1.14-1.51(5H,m), 1.68-1.79(5H, m), 3.31-3.37(1H, m), 7.53-7.57(3H,m), 7.96-8.01(2H, m)
【0074】
[実施例2a]イソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸の合成
実施例1bで合成した4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オン47.5g(175mmol)、及びイソプロピルアルコール238mLを仕込み、加熱還流下2時間撹拌した後、減圧濃縮し、白色固体としてイソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸を66.1g(純分として53.9g、収率93%)得た。
1H−NMR(400MHz,CDCl3):δ1.20−1.38(9H,m),1.45−1.56(2H,m),1.69−1.85(4H,m),2.08−2.10(1H,br−s),2.86−2.93(1H,m),5.10−5.17(1H,m),5.52(1H,d,J=6.6Hz),7.29(1H,d,J=6.1Hz),7.43−7.55(3H,m),7.83−7.85(m,2H).
【0075】
[実施例2b]tert-ブチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 150mg(0.553mmol)、およびtert-ブチルアルコール3.0mlを仕込み、油浴にて90度に加熱し、2時間攪拌した後、減圧濃縮し、白色固体としてtert-ブチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を248mg(純分を換算後、収率99%)得た。
1H-NMR(400MHz,CDCl3):δ
1.22-1.85(19H,m), 2.10-2.12(1H, m), 2.90-2.93(1H, m), 5.47 (1H,d, J=6.6Hz), 7.26(1H, br-s) , 7.42-7.54(3H, m), 7.83-7.85(m, 2H)
【0076】
[実施例2c]ベンジル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 150mg(0.553mmol)、およびベンジルアルコール3.0mlを仕込み、油浴にて100度に加熱し、2時間攪拌した後、減圧濃縮し、白色固体としてベンジル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を253mg(純分を換算後、収率:定量的)得た。
1H-NMR(400MHz,CDCl3):δ
0.95-1.97(10H, m), 2.65-2.74(1H, m), 5.19(2H, AB-q, J=12Hz), 5.52(1H, d, J=6.6Hz), 7.19-7.30 (6H,m) , 7.36-7.48(3H, m), 7.76-7.78(m, 2H)
【0077】
[実施例2d]エチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 100mg(0.369mmol)、およびエチルアルコール2.0mlを仕込み、油浴にて加熱還流させ、2時間攪拌した後減圧濃縮し、白色固体としてエチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を131mg(純分を換算後、収率99%)得た。
1H-NMR(400MHz,CDCl3):δ
1.15-1.78(13H,m), 2.00-2.03(1H, m), 2.79-2.86(1H, m), 4.14-4.29(m、2H), 5.49 (1H, d, J=6.6Hz), 7.24(1H, m) , 7.37-7.48(3H, m), 7.76-7.79(m, 2H)
【0078】
[実施例2e]イソプロピル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1cで合成した4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オンのずず塩34.5g(純分として49.2mmol)、およびイソプロピルアルコール53.4ml、ピリジン9.3mlを仕込み、加熱還流下4時間30分攪拌した後、反応液を室温にもどし、酢酸エチル20mlにて3回希釈し、ろ過して得られた溶液を減圧濃縮し、白色固体としてイソプロピル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を19.4g(純分として13.6 g、収率84%)を得た。
1H-NMR(400MHz,CDCl3):δ
1.20-1.38(9H,m), 1.45-1.56(2H, m), 1.69-1.85((4H, m), 2.08-2.10(1H, br-s), 2.86-2.93(1H, m), 5.10-5.17 (1H,m), 5.52(1H, d, J=6.6Hz), 7.29(1H,d,J=6.1Hz) , 7.43-7.55(3H, m), 7.83-7.85(m, 2H)
【0079】
[実施例3]イソプロピル(2R,3R)−2−ベンゾイルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸エステルの合成
[1]エクシグオバクテリウム(Exiguobacterium)sp.MCI4322由来のカルボニルレダクターゼ遺伝子で形質転換された形質転換細胞の作製
エクシグオバクテリウム(Exiguobacterium)sp.MCI4322由来のカルボニルレダクターゼ(Accession No.3016330A、配列番号2)をコードするDNA配列(genbank Accession No.gp:AB154409、配列番号1)をもとに配列番号3及び4に記載のプライマーを合成した。
【0080】
配列番号1:
atgaaatata cggtgattac tggagcaagt tcaggaattg gatatgagac ggcaaaacta 60
ctcgcaggaa aaggaaaatc actcgttctc gtcgcgcgac ggacgtctga gctcgaaaaa 120
cttcgggatg aagtcaaaca aatctcacca gatagtgatg tcatcctcaa gtcggtcgat 180
ctcgcagata accaaaatgt ccatgattta tatgagggat taaaggaact cgacatcgaa 240
acgtggatca acaatgctgg attcggcgat tttgatctcg tccaggacat tgagctcggg 300
aaaatcgaga aaatgcttcg cttgaacatc gaggcgctaa cgattctatc gagtctgttc 360
gtccgcgatc atcatgacat cgaaggaacg acgctcgtca atatctcgtc agcaggtggc 420
taccggatcg tgccgaacgc ggtcacgtat tgcgcgacga agttctatgt cagtgcttat 480
acagaagggc tcgcgcaaga actgcaaaaa ggtggggcaa aacttcgggc gaaagtactg 540
gcaccagctg cgactgagac agagtttgcg gatcgttcgc gcggcgaagc aggtttcgac 600
tacagcaaga acgtcaaaaa gtaccatacg gcggctgaaa tggcaggctt cctgcatcag 660
ttgattgaaa gcgacgcgat cgtcggcatc gtcgacggtg agacgtatga gttcgaattg 720
cgtggtccat tgttcaacta cgcaggataa 750
【0081】
配列番号2:
Met Lys Tyr Thr Val Ile Thr Gly Ala Ser Ser Gly Ile Gly Tyr Glu
Thr Ala Lys Leu Leu Ala Gly Lys Gly Lys Ser Leu Val Leu Val Ala
Arg Arg Thr Ser Glu Leu Glu Lys Leu Arg Asp Glu Val Lys Gln Ile
Ser Pro Asp Ser Asp Val Ile Leu Lys Ser Val Asp Leu Ala Asp Asn
Gln Asn Val His Asp Leu Tyr Glu Gly Leu Lys Glu Leu Asp Ile Glu
Thr Trp Ile Asn Asn Ala Gly Phe Gly Asp Phe Asp Leu Val Gln Asp
Ile Glu Leu Gly Lys Ile Glu Lys Met Leu Arg Leu Asn Ile Glu Ala
Leu Thr Ile Leu Ser Ser Leu Phe Val Arg Asp His His Asp Ile Glu
Gly Thr Thr Leu Val Asn Ile Ser Ser Ala Gly Gly Tyr Arg Ile Val
Pro Asn Ala Val Thr Tyr Cys Ala Thr Lys Phe Tyr Val Ser Ala Tyr
Thr Glu Gly Leu Ala Gln Glu Leu Gln Lys Gly Gly Ala Lys Leu Arg
Ala Lys Val Leu Ala Pro Ala Ala Thr Glu Thr Glu Phe Ala Asp Arg
Ser Arg Gly Glu Ala Gly Phe Asp Tyr Ser Lys Asn Val Lys Lys Tyr
His Thr Ala Ala Glu Met Ala Gly Phe Leu His Gln Leu Ile Glu Ser
Asp Ala Ile Val Gly Ile Val Asp Gly Glu Thr Tyr Glu Phe Glu Leu
Arg Gly Pro Leu Phe Asn Tyr Ala Gly
【0082】
配列番号3:ggaggcgaat tcatgaaata tacggtgatt
配列番号4:taatcactgc agttatcctg cgtagttgaa
【0083】
これらのプライマーを各15pmol、dNTP各10nmol、エクシグオバクテリウム(Exiguobacterium)sp.MCI4322のゲノムDNA 25ng、KOD−plus−用10×緩衝液(東洋紡績社製)5μL、KOD−plus−1ユニット(東洋紡績社製)を含む50μLの反応液を用い、変性(94℃、15秒)、アニール(57℃、30秒)、伸長(68℃、2分)を30サイクルで、PTC−200(MJ Research社製)を用いてPCR反応を行った。なお、エクシグオバクテリウム sp(Exiguobacterium sp)MCI4322株は、FERM-P-19239として独立行政法人産業総合研究所特許生物寄託センターから入手可能である。PCR反応液の一部をアガロースゲル電気泳動により解析した結果、特異的と思われるバンドが検出できた。
【0084】
上記反応液をMinElute PCR Purification kit(Qiagen社製)にて精製した。精製したDNA断片を制限酵素EcoRIとPstIで消化し、アガロースゲル電気泳動を行い、目的とするバンドの部分を切り出し、Qiagen Gel Extraction kit(Qiagen社製)により精製後回収した。得られたDNA断片を、EcoRI、及びPstIで消化したpKK223−3とTakara Ligation Kitを用いてライゲーションし、大腸菌JM109株を形質転換した。
【0085】
形質転換細胞をアンピシリン(50μg/mL)を含むLB寒天培地上で生育させ、コロニーダイレクトPCRを行い、挿入断片のサイズを確認した。目的とするDNA断片が挿入されていると考えられる形質転換細胞を50μg/mLのアンピシリンを含むLB培地で培養し、QIAPrepSpin Mini Prep kit(Qiagen社製)を用いてプラスミドを精製し、pExSDR1とした。プラスミドに挿入したDNAの塩基配列をダイターミネーター法により解析したところ、挿入されたDNA断片は、配列番号1の塩基配列と一致した。
【0086】
[2]カルボニルレダクターゼをコードするDNAで形質転換した大腸菌を用いた イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルの合成
上記[1]で得られた形質転換体をアンピシリン(50μg/mL)を含むCircleGrow培地(国産化学社製)で30℃で1.7時間生育させ、0.1mMになるようにイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、更に30℃で20時間培養した。得られた菌体ブロス1mLを遠心分離により集菌し、100μLの緩衝液(10%硫酸ナトリウム、10%グルコースを含む100mMトリス(最終pH9.9))を加えよく混合した。これに30μLの10g/L NADP+(オリエンタル酵母社製)、30μLのグルコースデヒドロゲナーゼ(天野製薬社製、1unit/mL)、実施例2aで得られた20μLの10g/Lイソプロピル(2R,3R)−2−ベンゾイルアミノ−3−シクロヘキシル−3−オキソプロピオン酸エステル(DMSO溶液)を添加後40℃で18時間振とう反応させた。反応終了後の反応液を酢酸エチル抽出、濃縮し、200μLのイソプロピルアルコールで溶かし、イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルを定量した。定量は高速液体クロマトグラフィー(HPLC)を用いて測定した。HPLCの条件は以下のとおりである。
【0087】
カラム:ODSカラム Acentis Amide(Spelco社製、4.6mmx10cm)、40℃
溶離液:10mM酢酸アンモニウム/アセトニトリル=55/45
流速:1mL/min
検出:UV254nm
【0088】
この結果、イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルの収量は14.1μgであった。ジアステレオ選択性は90%d.e.であった。また、該遺伝子を含まないプラスミドpKK223−3をもつ大腸菌を上記と同様の方法で培養し、反応させてみたが上記生成物は認められなかった。次に光学純度の測定をHPLCを用いて行なった。HPLCの条件は以下のとおりである。
【0089】
カラム:OD−RH(ダイセル社製、4.6mmx15cm)、30℃
溶離液:40%アセトニトリル
流速:0.5mL/min
検出:UV254nm
光学純度は100%e.e.であった。
【0090】
[実施例4](2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸の合成法
500mLのナスフラスコに、実施例3と同様に、ただし反応スケールを1000倍にあげて実施して得られたイソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸エステル12.4g(37.0mmol)を仕込み、6N塩酸水溶液を添加して110℃に加温しながら6.5時間反応させた。室温に冷却後、トルエン50mLにて2回洗浄し、水層部を更に110℃に昇温し、5時間反応させた。室温に戻し、トルエン50mLにて2回洗浄し、得られた水層に25wt%の水酸化ナトリウム水溶液をpH5.68になるまで添加した。析出した固体をろ過し、減圧乾燥後に純度90%程度の白色固体として(2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸を8.40g取得した。この化合物を塩酸塩に変換後、下記スペクトルを取得した。
1H−NMR(400MHz,CD3OD):δ0.80−1.27(5H,m),1.58−1.79(5H,m),1.96−1.99(1H,m),3.42(1H,m),4.00(1H,d,J=2.3Hz).
【0091】
[実施例5](2R,3R)−2−t−ブトキシカルボニルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸の合成
実施例4で取得した(2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸8.40gを1N水酸化ナトリウム水溶液35mL及びTHF35mLに溶解させ、ジ−t−ブチルジカルボナート8.89g(40.7mmol)を添加した。室温下で6時間反応させ、反応液を減圧下でTHFを留去した。トルエン20mLを添加し、有機層と水層を分離し、更に有機層を1N水酸化ナトリウム水溶液5mLで3回抽出した。水層を併せ、1N塩酸60mLにて酸性に調整し、酢酸エチル70mL、30mLでそれぞれ抽出した。得られた有機層を硫酸マグネシウムで乾燥・ろ過し、得られた液を濃縮し、(2R,3R)−2−t−ブトキシカルボニルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸7.09g(純度96%、イソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸エステルからの換算収率67%)を得た。
1H−NMR(400MHz,CD3OD):δ0.88−1.24(5H,m),1.62−1.68(5H,m),1.79−1.83(1H,m),3.37(1H,t,J=6.2Hz),4.07−4.18(1H,m).
【0092】
[比較例1]4−(1−ヒドロキシメチリデン)−2−フェニルオキサゾール−5−オンの合成(非特許文献2及び非特許文献3)
馬尿酸1.00g(5.58mmol)をエタノール5mLに懸濁させ、20%ナトリウムエトキシドエタノール溶液を4.76g(14mmol)添加した。2時間撹拌後、反応液を減圧乾燥して馬尿酸のナトリウム塩1.12とした。この馬尿酸ナトリウム塩にβ−ピコリン1.70mL、無水酢酸(1.67mL)を添加し、室温にて2時間反応後、更に40℃にて5時間反応させた。反応液を室温に冷却し、LCにて定量分析したところ収率は15%、変換率は50%であった。
1H−NMR(400MHz,CDCl3):δ2.50(3H,s),7.52−7.57(3H,m),7.88−7.94(2H,m).
【0093】
[比較例2]4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの合成(非特許文献2及び非特許文献3)
2−フェニルオキサゾール−5−オン200mg(1.24mmol)をβ−ピコリン0.38mLに懸濁させ、氷冷下、シクロヘキサンカルボン酸クロライド0.16mLを滴下して室温に戻して20時間反応させた。反応液に2N塩酸水20mLを加え、ジクロロメタン30mLにて2回抽出し、得られた有機層をLCにて定量分析したところ収率は18%、変換率は100%であった。反応副生物である4−(2−N−ベンゾイルアミノ−1−ヒドロキシルメチリデン)−イル−2−フェニルオキサゾール−5−オンが50%生成していた。
1H−NMR(400MHz,CDCl3):δ4.46(3H,m),7.43−7.66(7H,m),7.83(2H),7.90(1H,s),12.55(1H,s).
【図面の簡単な説明】
【0094】
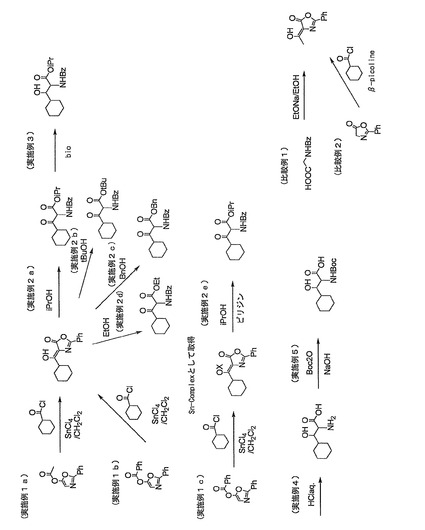
【図1】図1は、実施例及び比較例における反応の概要を示す。
【配列表フリーテキスト】
【0095】
[配列表]
SEQUENCE LISTING
<110> API Corporation
<120> A method for producing an acyloxazolone derivative and beta-ketoamino acid derivative
<130> A61441A
<160> 4
<210> 1
<211> 750
<212> DNA
<213> Exiguobacterium sp.
<400> 1
atgaaatata cggtgattac tggagcaagt tcaggaattg gatatgagac ggcaaaacta 60
ctcgcaggaa aaggaaaatc actcgttctc gtcgcgcgac ggacgtctga gctcgaaaaa 120
cttcgggatg aagtcaaaca aatctcacca gatagtgatg tcatcctcaa gtcggtcgat 180
ctcgcagata accaaaatgt ccatgattta tatgagggat taaaggaact cgacatcgaa 240
acgtggatca acaatgctgg attcggcgat tttgatctcg tccaggacat tgagctcggg 300
aaaatcgaga aaatgcttcg cttgaacatc gaggcgctaa cgattctatc gagtctgttc 360
gtccgcgatc atcatgacat cgaaggaacg acgctcgtca atatctcgtc agcaggtggc 420
taccggatcg tgccgaacgc ggtcacgtat tgcgcgacga agttctatgt cagtgcttat 480
acagaagggc tcgcgcaaga actgcaaaaa ggtggggcaa aacttcgggc gaaagtactg 540
gcaccagctg cgactgagac agagtttgcg gatcgttcgc gcggcgaagc aggtttcgac 600
tacagcaaga acgtcaaaaa gtaccatacg gcggctgaaa tggcaggctt cctgcatcag 660
ttgattgaaa gcgacgcgat cgtcggcatc gtcgacggtg agacgtatga gttcgaattg 720
cgtggtccat tgttcaacta cgcaggataa 750
<210> 2
<211> 249
<212> PRT
<213> Exiguobacterium sp.
<400> 2
Met Lys Tyr Thr Val Ile Thr Gly Ala Ser Ser Gly Ile Gly Tyr Glu
1 5 10 15
Thr Ala Lys Leu Leu Ala Gly Lys Gly Lys Ser Leu Val Leu Val Ala
20 25 30
Arg Arg Thr Ser Glu Leu Glu Lys Leu Arg Asp Glu Val Lys Gln Ile
35 40 45
Ser Pro Asp Ser Asp Val Ile Leu Lys Ser Val Asp Leu Ala Asp Asn
50 55 60
Gln Asn Val His Asp Leu Tyr Glu Gly Leu Lys Glu Leu Asp Ile Glu
65 70 75 80
Thr Trp Ile Asn Asn Ala Gly Phe Gly Asp Phe Asp Leu Val Gln Asp
85 90 95
Ile Glu Leu Gly Lys Ile Glu Lys Met Leu Arg Leu Asn Ile Glu Ala
100 105 110
Leu Thr Ile Leu Ser Ser Leu Phe Val Arg Asp His His Asp Ile Glu
115 120 125
Gly Thr Thr Leu Val Asn Ile Ser Ser Ala Gly Gly Tyr Arg Ile Val
130 135 140
Pro Asn Ala Val Thr Tyr Cys Ala Thr Lys Phe Tyr Val Ser Ala Tyr
145 150 155 160
Thr Glu Gly Leu Ala Gln Glu Leu Gln Lys Gly Gly Ala Lys Leu Arg
165 170 175
Ala Lys Val Leu Ala Pro Ala Ala Thr Glu Thr Glu Phe Ala Asp Arg
180 185 190
Ser Arg Gly Glu Ala Gly Phe Asp Tyr Ser Lys Asn Val Lys Lys Tyr
195 200 205
His Thr Ala Ala Glu Met Ala Gly Phe Leu His Gln Leu Ile Glu Ser
210 215 220
Asp Ala Ile Val Gly Ile Val Asp Gly Glu Thr Tyr Glu Phe Glu Leu
225 230 235 240
Arg Gly Pro Leu Phe Asn Tyr Ala Gly
245
<210> 3
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> PCR primer
<400> 3
ggaggcgaat tcatgaaata tacggtgatt 30
<210> 4
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> PCR primer
<400> 4
taatcactgc agttatcctg cgtagttgaa 30
【技術分野】
【0001】
本発明は、アシルオキサゾロン化合物及びβ−ケトアミノ酸誘導体の製造方法に関する。本発明の方法で得られるβ−ケトアミノ酸誘導体は、変換して光学活性β−ヒドロキシアミノ酸を経由して、抗HIVウィルス薬として有用なことが知られている薬剤の中間体であるD−エリスロ−3−シクロヘキシルセリンに誘導可能であることから、医農薬中間体として有用な化合物である。
【背景技術】
【0002】
アシルオキサゾロン類の合成方法としては、馬尿酸又は2−フェニルオキサゾール−5−オンをトリフルオロ酢酸存在下、γ−ピコリン中、酸クロリドと反応させる方法(非特許文献1)、及び馬尿酸ナトリウム塩と酸無水物をβ−ピコリン中反応させる方法(非特許文献2及び3)が知られている。しかし、いずれの反応も酸クロリド又は酸無水物を過剰に用いる必要があり、高価な酸クロリド又は酸無水物を使用する必要がある場合、製造コストが高額になるという問題点があった。
【0003】
また、2−フェニルオキサゾール−5−オンとアシル化剤等量を用いる方法(非特許文献4)も報告されている。この文献では高価なγ−ピコリンを溶媒として用いているが、γ−ピコリンは高沸点かつ水溶性も高いため回収が困難であるので製造コストが高くなり、また、大量の溶媒を用い、反応時間に1日を要する等実用的ではない。一方、γ−ピコリンを用いない方法(特許文献1及び2)も報告されているが、いずれも用いる酸クロリドはコハク酸モノエステルクロライド誘導体に限定され、一般性が高いとは言えず、反応時間も1日〜3日と長時間を要する。
【0004】
また、これまでに報告されているアシルオキサゾロン類の合成例は単純な化合物又は類似した化合物が多く、特に立体障害の大きいα位に置換基を持つ酸クロリド又は酸無水物を用いた例はわずかしかない上(特許文献1、非特許文献3)、そのいずれも収率30%程度と低収率であり、工業的に安価かつ汎用性の高いアシルオキサゾロン類の製造には課題が残されていた。
【0005】
【非特許文献1】Bull.Acad.Sci.USSR Div.Chem.Sci.1982,31,2223.
【非特許文献2】J.Chem.Soc.1948,3,310−318
【非特許文献3】J.Chem.Soc.1960,968−973
【非特許文献4】Bull.Acad.Sci.USSR Div.Chem.Sci.1984,33,384.
【特許文献1】特開平1−305071号公報
【特許文献2】国際公開第98/54162号パンフレット
【発明の開示】
【発明が解決しようとする課題】
【0006】
本発明は、β−ケトアミノ酸誘導体に変換可能なアシルオキサゾロン化合物を短工程、かつ強塩基や高価な試薬、極低温を必要としない安価な条件で効率的・高収率、かつ後処理・精製が容易な方法で製造することができる、アシルオキサゾロン化合物の工業的製造方法を提供することを目的とする。
【課題を解決するための手段】
【0007】
本発明者は、上記課題を解決するために鋭意検討した結果、オキサゾール化合物をアシル化することでアシルオキサゾロン化合物を合成でき、このアシルオキサゾロン化合物をアルコール類と反応させることでβ−ケトアミノ酸化合物を一貫して温和な条件で効率的に合成できることを見出し、本発明を完成するに至った。
【0008】
即ち、本発明によれば、以下の発明が提供される。
(1) 下記一般式(1)
【化1】
(式中、R1及びR2はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。)で表されるオキサゾール化合物にルイス酸の存在下、アシル化剤を作用させることにより、下記一般式(2)
【化2】
(式中、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるアシルオキサゾロン化合物を製造する工程を含む、アシルオキサゾロン化合物の製造方法。
【0009】
(2) アシル化剤が、酸ハライド又は酸無水物である、(1)に記載の方法。
【0010】
(3) (1)の方法により製造した一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【化3】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表される化合物を製造する工程を含む、β−ケトアミノ酸誘導体の製造方法。
【0011】
(4) 下記一般式(4)
【化4】
(式中、R1は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるシクロヘキシルメチリデニルオキサゾロン化合物。
【0012】
(5) (i)(3)に記載の方法により一般式(3)で表されるβ−ケトアミノ酸誘導体を製造する工程、及び
(ii)上記工程(i)で得られた一般式(3)で表されるβ−ケトアミノ酸誘導体に微生物の菌体及び/又は該菌体処理物を作用させて下記一般式(5)
【化5】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるα−N−アシルアミノ−β−ヒドロキシエステルを得る工程を含む、α−N−アシルアミノ−β−ヒドロキシエステルを製造する方法。
【0013】
(6) (i)(5)に記載の方法により一般式(5)で表されるN−アシルアミノ−β−ヒドロキシエステルを製造する工程、
(ii)上記工程(i)で得られたα−N−アシルアミノ−β−ヒドロキシエステルに酸又は塩基を作用させて加水分解を行い、下記一般式(6)
【化6】
(式中、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるβ−ヒドロキシアミノ酸を得る工程;及び
(iii)上記工程(ii)で得られたβ−ヒドロキシアミノ酸にt−ブトキシカルボニル化剤を作用させて下記一般式(7)
【化7】
(式中、R3及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物を製造する工程、を含むN−t−ブトキシカルボニル−β−ヒドロキシアミノ酸の製造方法。
【発明の効果】
【0014】
本発明によれば、安価かつ温和な工業的製造法で、医農薬中間体として有用な化合物であるアシルオキサゾロン化合物及びβ−ケトアミノ酸化合物を効率よく製造することができる。
【発明を実施するための最良の形態】
【0015】
以下、本発明を詳細に説明する。
<オキサゾ−ル化合物、アシルオキサゾロン化合物>
本発明の製造方法は、上記一般式(1)で表されるオキサゾール化合物にルイス酸の存在下、酸ハライド又は酸無水物などのアシル化剤を作用させることにより、上記一般式(2)で表されるアシルオキサゾロン化合物を得ることを特徴とするものである。
【0016】
上記一般式(1)において、R1は、置換していてもよいアルキル基又は置換してもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくは炭素数1〜6の直鎖状若しくは分岐状のアルキル基であり、より好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、例えば、メチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、シクロヘキシル基であり、最も好ましくはメチル基である。置換してもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記のアルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、アルキル基、アリール基、アルコキシ基、フッ素原子、塩素原子、臭素原子等のハロゲン原子、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられる。好ましくはハロゲン原子、アルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0017】
上記一般式(1)において、R2は置換していてもよいアルキル基又は置換していてもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくは炭素数1〜6の直鎖状若しくは分岐状のアルキル基であり、より好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、例えば、メチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、又はシクロヘキシル基であり、最も好ましくはメチル基である。置換してもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記アルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、アルキル基、アリール基、アルコキシ基、フッ素原子、塩素原子、臭素原子等のハロゲン原子、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられる。好ましくはハロゲン原子、アルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0018】
一般式(1)は、好ましくはR1がフェニル基で、R2がメチル基又はフェニル基であることが好ましい。
【0019】
上記一般式(2)において、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。アルキル基としては、好ましくは炭素数1〜10の直鎖状、分岐状若しくは環状のアルキル基であり、更に好ましくはメチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基、シクロプロピル基、シクロブチル基、シクロペンチル基、シクロヘキシル基、シクロヘプチル基等の炭素数1〜7の直鎖状、分岐状若しくは環状のアルキル基、特に好ましくは炭素数1〜4の直鎖状若しくは分岐状のアルキル基又は炭素数5〜7の環状のアルキル基であり、最も好ましくは炭素数5〜7の環状のアルキル基である。置換していてもよいアリール基のアリール基としては、ナフチル基、フェニル基等であり、最も好ましくはフェニル基である。上記アルキル基及びアリール基は置換していてもよく、置換基としては特に限定はないが、フッ素原子、塩素原子、臭素原子等のハロゲン原子、アルキル基、アリール基、アルコキシ基、ニトロ基、水酸基、カルボキシル基、アミノ基等が挙げられ、好ましくはアルコキシ基である。置換してもよいアリール基としては、フェニル基や4−メトキシフェニル基が好ましく、更に好ましくはフェニル基である。
【0020】
上記一般式(2)において、Xは水素原子又は金属原子を示す。金属原子は特に限定はないが、Li、Na、K、Rb等のアルカリ金属原子、Be、Mg、Ca、Sr、Ba等のアルカリ土類金属、Ti、Mn、Fe、Ni、Zn、Zr、Hg、Pd等の遷移金属原子、Sn、Pb、Al等の典型金属が挙げられる。金属原子として好ましいのは、アルカリ金属であるLi、Na、K、Rb、又はアルカリ土類金属であるBe、Mg、Ca、Sr、Baである。Xとして、好ましいのは水素原子である。
【0021】
上記一般式(2)において、Xが水素原子の場合、金属化合物と錯体を形成していてもよい。具体的には、一般式(2)のヒドロキシル基とカルボニル基が金属化合物と配位結合をして錯体を形成する場合や、一般式(2)のヒドロキシル基と窒素原子が金属化合物と配位結合をして錯体を形成する場合が挙げられる。また、金属に対して、一般式(2)は1:1配位であってもよく、1:2配位であってもかまわない。
【0022】
金属化合物は特に限定はないが、金属原子は、Ti、Mn、Fe、Ni、Zn、 Zr、Hg、Pd等の遷移金属原子、Sn、Pb、Al等の典型金属が挙げられる。また、金属原子として好ましいのは、安価かつ本反応で種々の基質に対して汎用性のある触媒となりうるSn、Al、Fe、Ni、Zn等が好ましい。
【0023】
錯体として、好ましいのは反応に用いるルイス酸又はその派生物がヒドロキシル基とカルボニル基と配位結合をしている化合物である。具体的には、塩化スズ(IV)錯体、塩化アルミニウム錯体、塩化鉄(III)錯体、塩化ニッケル錯体等が挙げられる。好ましくは塩化スズ(IV)錯体、塩化アルミニウム錯体である。
【0024】
一般式(2)において好ましくは、R1がフェニル基であり、R3がシクロヘキシル基であり、Xが水素、Na、K、又はCaであるか、又はXが水素でSn、Al、Fe、Ni、Zn錯体である。より好ましくは、R1がフェニル基であり、R3がシクロヘキシル基であり、Xが水素であるか、又はXが水素でSn、Al、Fe、Ni、Zn錯体である。
【0025】
また、一般式(2)及び(4)で表される化合物のうち、Xが水素原子の場合は、ケト型の互変異性体であってもよい。
【0026】
本発明においては、一般式(1)のオキサゾール化合物をルイス酸の存在下、酸ハライド(例えば、酸クロライド)又は酸無水物などのアシル化剤を作用させて一般式(2)のアシルオキサゾロン化合物を得るものである。かくして製造される一般式(2)のアシルオキサゾロン化合物のうち、下記一般式(4)
【0027】
【化8】
【0028】
(式中、R1及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物は新規化合物である。
【0029】
一般式(4)の具体例としては、次のような化合物が挙げられる。
【0030】
【化9】
【0031】
式(4a)〜(4e)中、R1はメチル基、フェニル基を、M1はNa、K、Caを、M2はSnCl4、AlCl3、FeCl3、NiCl2、ZnCl2を表す。
【0032】
<アシルオキサゾロン誘導体の製造方法>
本発明は、一般式(1)のオキサゾール化合物をルイス酸の存在下、酸ハライド或いは酸無水物などのアシル化剤を作用させて一般式(2)のアシルオキサゾロン化合物を得るものである。
【0033】
ルイス酸としては、特に限定はないが、具体的には塩化アルミニウム(III)、四塩化スズ(IV)、塩化鉄(III)、塩化亜鉛(II)、塩化ニッケル(II)、三フッ化ホウ素エーテル錯体等が挙げられ、好ましくは工業的に入手が容易である四塩化スズ(IV)、塩化鉄(III)、塩化ニッケル(II)、塩化アルミニウム(III)が好ましい。
【0034】
ルイス酸の使用量としては特に限定されないが、通常は原料である一般式(1)に対して0.02モルから10モル倍で反応は進行するが、生成物とルイス酸が錯体を形成しやすい性質があるため1モル倍以上の使用が好ましく、コストの観点から1〜3モル倍の使用が好ましい。
【0035】
本発明で使用するアシル化剤は、一般式(1)のオキサゾール化合物に作用させることによって一般式(2)のアシルオキサゾロン化合物を製造することができるものであれば特に限定されないが、好ましくは酸ハライド又は酸無水物である。酸ハライド又は酸無水物としては、特に限定されないが、具体的には、アセチルクロリド、イソ酪酸クロリド、ピバロイルクロリド、シクロヘキサノイルクロリド、ベンゾイルクロリド、4−メトキシベンゾイルクロリド等の酸クロリド;アセチルブロミド、イソプロピオン酸ブロミド、ピバロイルブロミド、シクロヘキシルブロミド、ベンゾイルブロミド、4−メトキシベンゾイルブロミド等の酸ブロミド;アセチルヨージド、イソ酪酸ヨージド、ピバロイルヨージド、シクロヘキサノイルヨージド、ベンゾイルヨージド、4−メトキシベンゾイルヨージド等の酸ヨージドが挙げられ、好ましくはコスト的に安価である酸クロリド、酸ブロミドが好ましく、更に好ましくはシクロヘキサノイルクロリドやイソ酪酸クロリド、ベンゾイルクロリド等である。
【0036】
酸無水物としては、無水酢酸、プロピオン酸無水物、ピバリン酸無水物、シクロヘキサン酸カルボン酸無水物、安息香酸無水物等があり、好ましくはシクロヘキサン酸カルボン酸無水物、プロピオン酸無水物、ピバリン酸無水物である。
【0037】
酸ハライドの使用量としては特に限定されないが、通常は原料である化合物(1)に対して0.8〜5倍モル、コストや化合物(2)の精製のしやすさの観点から一般式(1)と同モルに近い0.9〜2倍モルの使用が好ましい。
【0038】
反応を行う条件のうち、用いる溶媒としては反応に悪影響を与えないものであれば特に限定はないが、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒;エチルエーテル、プロピルエーテル、ブチルエーテル、テトラヒドロフラン等のエーテル系溶媒;ジクロロメタン、クロロホルム、ジクロロエタン、クロロベンゼン等のハロゲン系溶媒;酢酸エチル、酢酸ブチル等のエステル系溶媒、アセトニトリル等のニトリル系溶媒、ニトロベンゼン、ニトロメタン等のニトロ系溶媒;等が挙げられる。これらから選ばれる複数の溶媒を任意の割合に混合して用いてもよい。好ましい溶媒は、一般的にアシル化反応に影響を及ぼさないジクロロメタン、クロロベンゼン等のハロゲン系溶媒、アセトニトリル等のニトリル系溶媒、ニトロベンゼン、ニトロメタン等のニトロ系溶媒、ヘキサン、ベンゼン、トルエン等の炭化水素系溶媒である。更に好ましくは、アセトニトリル、ジクロロメタン、クロロホルム、クロロベンゼン、ニトロベンゼン、ニトロメタン及びトルエンである。
【0039】
また、溶媒の使用量としては、任意の量の溶媒を用いることができるが、通常は原料基質に対して2〜50倍体積量、好ましくは3〜10倍体積量である。
【0040】
この反応を実施する温度としては、特に限定されないが−50℃から100℃程度であり、好ましくは−10℃から40℃程度が好ましい。実施する圧力に関しては特に加圧下で行う必要はないが、反応時間を短縮する等の観点から常圧から10気圧程度の圧力をかけてもよい。反応時間は、10分〜数日の範囲で行うことができるが、製造コストを抑える観点から24時間以内に終了させることが好ましく、更に好ましくは10分から8時間である。
【0041】
一般式(1)のオキサゾール化合物は公知の方法で製造すれば特に限定はないが、下記一般式(8)
【0042】
【化10】
【0043】
で表されるオキサゾロン化合物とベンゾイルクロリドやアセチルクロリド等のアシル化剤を作用させる、あるいは下記一般式(9)
【0044】
【化11】
【0045】
で表される馬尿酸とベンゾイルクロライドやアセチルクロライド等のアシル化剤を作用させることで合成可能である。ここで、一般式(8)及び(9)におけるR1は、一般式(1)におけるR1と同義である。
【0046】
<β−ケトアミノ酸誘導体の製造方法>
上記一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【0047】
【化12】
【0048】
(式中、R1及びR3は前記と同義である。R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表されるβ−ケトアミノ酸誘導体を製造することができる。
【0049】
アルキル基としては、好ましくは炭素数1〜6の直鎖状、分岐状又は環状のアルキル基であり、更に好ましくはメチル基、エチル基、イソプロピル基、ノルマルプロピル基、ノルマルブチル基、t−ブチル基等の炭素数1〜4の直鎖状若しくは分岐状のアルキル基であり、より好ましくはメチル基、エチル基、イソプロピル基、t−ブチル基である。
【0050】
アラルキル基としては、ベンジル基が好ましい。
【0051】
上記アルキル基及びアラルキル基は置換していてもよく、置換基としては特に限定はないが、フッ素原子、塩素原子、臭素原子等のハロゲン原子、アルキル基、アルコキシ基、ニトロ基、アリールオキシ基、アミノ基、アリール基等が挙げられる。好ましくは塩素原子、アルコキシ基である。置換してもよいアラルキル基としては、ベンジル基、4−メトキシベンジル基、4−フルオロベンジル基、4−クロロベンジル基、3,4−ジメチルベンジル基が好ましく、更に好ましくはベンジル基、4−クロロベンジル基である。
【0052】
一般式(3)は、好ましくはR1がフェニル基又はメチル基であり、R3がシクロヘキシル基であり、R4がメチル基、エチル基、イソプロピル基、t−ブチル基、又はベンジル基であることが好ましい。
【0053】
上記一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより、上記一般式(3)で表されるβ−ケトアミノ酸誘導体を製造することができる。
【0054】
一般式(2)で表されるオキサゾロン化合物は、アルコールと反応させるにあたり、各種有機溶媒にて抽出及び/又は晶析等の方法で精製・単離することが好ましい。好ましくは、ろ過操作にて原料を取得する或いは塩酸等の酸の水溶液にて反応を終結後、有機溶媒にて抽出・洗浄操作を行い、反応に用いたルイス酸を除去したのち、晶析操作を行うことが望ましい。
【0055】
反応に用いるアルコールとしては、特に限定されるものではないが、具体的にはメタノール、エタノール、ノルマルプロピルアルコール、イソプロピルアルコール、ノルマルブチルアルコール、イソブチルアルコール、t−ブチルアルコール、ベンジルアルコール等であり、好ましくはエタノール、イソプロピルアルコール、t−ブチルアルコール、ベンジルアルコールである。
【0056】
アルコールの使用量は、一般式(2)のオキサゾロン化合物に対して1倍量から溶媒量まで使用することができるが、好ましくは1〜5倍量で使用することがコスト的に好ましい。
【0057】
反応の温度としては、特に限定はされないが、通常室温から各種溶媒の沸点付近まで、好ましくは20℃から100℃がよい。この反応は常圧、大気中で行うことができ、特に窒素雰囲気下で行う必要はないが、必要に応じて窒素、ヘリウム等の不活性気体中で加圧下にて実施することもできる。
【0058】
反応時間は、10分〜数日の範囲で行うことができるが、製造コストを抑える観点から24時間以内に終了させることが好ましく、更に好ましくは10分から8時間である。
【0059】
上記にて製造した一般式(3)のβ−ケトアミノ酸化合物は、反応液を濃縮するだけで特に精製は必要ないが、用いるアルコールの沸点が高い場合、用いた一般式(2)のオキサゾロン化合物の純度が低かった場合等は必要に応じて適宜、有機溶媒と水による分液操作での単離又は晶析による精製を行うことができる。
【0060】
<β−ケトアミノ酸化合物の用途>
上記一般式(3)で表されるβ−ケトアミノ酸化合物は、それ自体或いは各種の手法にて誘導化し、種々の医薬や農薬の中間体として有用である。特に、一般式(7)で表される化合物において、R3がシクロヘキシル基である化合物に相当する下記式(10)
【0061】
【化13】
で表される化合物は抗HIV活性をもつ薬剤(WO01/40227) の中間体として工業的に有用である。上記式(10)やその類縁体への変換の手法としては、上記一般式(3)で表される化合物を下記一般式(5)
【0062】
【化14】
【0063】
(式中、R1、R3、R4は前記と同義。)で表される化合物に立体選択的に変換する。
【0064】
この変換の手法としては、微生物の菌体及び/又は該菌体処理物を作用させる方法、或いは特許文献(WO04/005371A1)や非特許文献(Tetrahedron Asymmetry 2001,12,1757-1762)に記載のルテニウム等の金属触媒を用いた不斉水素化反応が利用可能である。好ましくは、重金属混入の恐れがない微生物の菌体及び/又は該菌体処理物を作用させる方法である。
【0065】
微生物の菌体及び/又は該菌体処理物を作用させる方法としては、特に限定はないが、アグロバクテリウム属又はエクシグオバクテリウム(Exiguobacterium)属に属する微生物を使用するか、不斉還元反応を触媒するタンパク質をコードする遺伝子を導入した形質転換細胞を作製し、該細胞、該細胞の調製物、又は該細胞を培養して得られた培養液を原料と作用させてもかまわない。
【0066】
上記一般式(5)で表される化合物を下記一般式(6)
【0067】
【化15】
表される化合物或いはその塩に変換する。
【0068】
この変換の手法としては、酸又は塩基の存在下での加水分解、又は加水分解酵素にて加水分解を行う手法が挙げられる。
【0069】
上記一般式(6)で表される化合物を塩基条件下、t−ブトキシカルボニル化を行うことにより上記一般式(7)で表される化合物(例えば、式(10)で表される化合物)へと変換可能である。
【0070】
以下、本発明を実施例により更に詳細に説明するが、本発明はこれらによって限定されるものではない。
【実施例】
【0071】
[実施例1a]4−(1−シクロヘキシル−1−ヒドロキシメチリデン)−2−フェニルオキサゾール−5−オンの合成
100mLのフラスコにジクロロメタン43mL、5−アセトキシ−2−フェニルオキサゾール4.28g(21.1mmol)を仕込み、シクロヘキサンカルボン酸クロリド3.10mL(23.2mmol)を添加し、しばらく撹拌した後に四塩化スズ3.87mL(73.2mmol)を10分間かけて滴下した。24時間後、TLCにて原料の消失を確認し、4N塩酸水溶液10mL及び酢酸エチル150mLを添加し、有機層を分離した後、更に水50mLで2回、飽和食塩水50mLにて洗浄し、硫酸マグネシウムにて乾燥後、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(シリカゲル150g、 ヘキサン:酢酸エチル=100:2〜100:4)にて精製し、黄色固体として目的物3.20g(純度換算後の収率52%)を得た。
1H−NMR(400MHz,DMSO−d):δ1.14−1.51(5H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),7.53−7.57(3H,m),7.96−8.01(2H,m)
【0072】
[実施例1b]4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの合成
1Lのフラスコに5−ベンゾイルオキシ−2−フェニルオキサゾール47.0g(177mmol)、ジクロロメタン380mLを仕込み、氷冷下にてシクロヘキサンカルボン酸クロリド33.0mL(248mmol)を添加し、しばらく撹拌した後に四塩化スズ29.0mL(248mmol)を2分間かけて滴下した。室温に昇温した後、24時間後にTLCにて原料の消失を確認し、反応液をろ過して得られた4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの塩化スズ錯体をLCにて定量分析したところ、4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンとしての収率は89%であった。この錯体に2N塩酸160mL及び酢酸エチル500mLを添加し、有機層を分離した後に更に2N塩酸160mLで2回洗浄し、硫酸マグネシウムにて乾燥後、溶媒を留去した。得られた残渣をジクロロメタン15mL、ヘキサン15mLにて懸洗し、ろ過して得られた黄色固体として目的物4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オン15.5g(純度98%、収率32%)を得た。
1H−NMR(400MHz,DMSO−d):δ1.14−1.51(5H,m),1.68−1.79(5H,m),3.31−3.37(1H,m),7.53−7.57(3H,m),7.96−8.01(2H,m).
【0073】
[実施例1c]4−(1−シクロヘキシル-1-ヒドロキシメチリデン)−2−フェニルオキサゾール-5-オンの合成
200mlのフラスコにジクロロメタン94ml、5−ベンゾイルオキシ-2−フェニルオキサゾール 13.4g(50.5mmol)を仕込み、氷冷下にてシクロヘキサンカルボン酸クロリド13.5ml(101mmol)を添加し、しばらく攪拌した後に四塩化すず16.5ml(2.8mmol)を10分間かけて滴下した。室温に昇温させて16時間攪拌後、TLCにて原料の消失を確認し、反応液をろ過し、固体部分を塩化メチレン:n-ヘキサンの1:1の混合溶液20mlにて洗浄後、淡黄色固体として目的物のすず塩34.6g(純度換算後の収率97%)を得た。
1H-NMR(400MHz, DMSO-d):δ1.14-1.51(5H,m), 1.68-1.79(5H, m), 3.31-3.37(1H, m), 7.53-7.57(3H,m), 7.96-8.01(2H, m)
【0074】
[実施例2a]イソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸の合成
実施例1bで合成した4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オン47.5g(175mmol)、及びイソプロピルアルコール238mLを仕込み、加熱還流下2時間撹拌した後、減圧濃縮し、白色固体としてイソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸を66.1g(純分として53.9g、収率93%)得た。
1H−NMR(400MHz,CDCl3):δ1.20−1.38(9H,m),1.45−1.56(2H,m),1.69−1.85(4H,m),2.08−2.10(1H,br−s),2.86−2.93(1H,m),5.10−5.17(1H,m),5.52(1H,d,J=6.6Hz),7.29(1H,d,J=6.1Hz),7.43−7.55(3H,m),7.83−7.85(m,2H).
【0075】
[実施例2b]tert-ブチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 150mg(0.553mmol)、およびtert-ブチルアルコール3.0mlを仕込み、油浴にて90度に加熱し、2時間攪拌した後、減圧濃縮し、白色固体としてtert-ブチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を248mg(純分を換算後、収率99%)得た。
1H-NMR(400MHz,CDCl3):δ
1.22-1.85(19H,m), 2.10-2.12(1H, m), 2.90-2.93(1H, m), 5.47 (1H,d, J=6.6Hz), 7.26(1H, br-s) , 7.42-7.54(3H, m), 7.83-7.85(m, 2H)
【0076】
[実施例2c]ベンジル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 150mg(0.553mmol)、およびベンジルアルコール3.0mlを仕込み、油浴にて100度に加熱し、2時間攪拌した後、減圧濃縮し、白色固体としてベンジル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を253mg(純分を換算後、収率:定量的)得た。
1H-NMR(400MHz,CDCl3):δ
0.95-1.97(10H, m), 2.65-2.74(1H, m), 5.19(2H, AB-q, J=12Hz), 5.52(1H, d, J=6.6Hz), 7.19-7.30 (6H,m) , 7.36-7.48(3H, m), 7.76-7.78(m, 2H)
【0077】
[実施例2d]エチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1bで合成した( 4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オン 100mg(0.369mmol)、およびエチルアルコール2.0mlを仕込み、油浴にて加熱還流させ、2時間攪拌した後減圧濃縮し、白色固体としてエチル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を131mg(純分を換算後、収率99%)得た。
1H-NMR(400MHz,CDCl3):δ
1.15-1.78(13H,m), 2.00-2.03(1H, m), 2.79-2.86(1H, m), 4.14-4.29(m、2H), 5.49 (1H, d, J=6.6Hz), 7.24(1H, m) , 7.37-7.48(3H, m), 7.76-7.79(m, 2H)
【0078】
[実施例2e]イソプロピル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸の合成
実施例1cで合成した4−〔1−シクロヘキシル-1-ヒドロキシメチリデン〕−2−フェニルオキサゾール-5-オンのずず塩34.5g(純分として49.2mmol)、およびイソプロピルアルコール53.4ml、ピリジン9.3mlを仕込み、加熱還流下4時間30分攪拌した後、反応液を室温にもどし、酢酸エチル20mlにて3回希釈し、ろ過して得られた溶液を減圧濃縮し、白色固体としてイソプロピル-2-ベンゾイルアミノ -3-シクロヘキサノイル -3-オキソプロピオン酸を19.4g(純分として13.6 g、収率84%)を得た。
1H-NMR(400MHz,CDCl3):δ
1.20-1.38(9H,m), 1.45-1.56(2H, m), 1.69-1.85((4H, m), 2.08-2.10(1H, br-s), 2.86-2.93(1H, m), 5.10-5.17 (1H,m), 5.52(1H, d, J=6.6Hz), 7.29(1H,d,J=6.1Hz) , 7.43-7.55(3H, m), 7.83-7.85(m, 2H)
【0079】
[実施例3]イソプロピル(2R,3R)−2−ベンゾイルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸エステルの合成
[1]エクシグオバクテリウム(Exiguobacterium)sp.MCI4322由来のカルボニルレダクターゼ遺伝子で形質転換された形質転換細胞の作製
エクシグオバクテリウム(Exiguobacterium)sp.MCI4322由来のカルボニルレダクターゼ(Accession No.3016330A、配列番号2)をコードするDNA配列(genbank Accession No.gp:AB154409、配列番号1)をもとに配列番号3及び4に記載のプライマーを合成した。
【0080】
配列番号1:
atgaaatata cggtgattac tggagcaagt tcaggaattg gatatgagac ggcaaaacta 60
ctcgcaggaa aaggaaaatc actcgttctc gtcgcgcgac ggacgtctga gctcgaaaaa 120
cttcgggatg aagtcaaaca aatctcacca gatagtgatg tcatcctcaa gtcggtcgat 180
ctcgcagata accaaaatgt ccatgattta tatgagggat taaaggaact cgacatcgaa 240
acgtggatca acaatgctgg attcggcgat tttgatctcg tccaggacat tgagctcggg 300
aaaatcgaga aaatgcttcg cttgaacatc gaggcgctaa cgattctatc gagtctgttc 360
gtccgcgatc atcatgacat cgaaggaacg acgctcgtca atatctcgtc agcaggtggc 420
taccggatcg tgccgaacgc ggtcacgtat tgcgcgacga agttctatgt cagtgcttat 480
acagaagggc tcgcgcaaga actgcaaaaa ggtggggcaa aacttcgggc gaaagtactg 540
gcaccagctg cgactgagac agagtttgcg gatcgttcgc gcggcgaagc aggtttcgac 600
tacagcaaga acgtcaaaaa gtaccatacg gcggctgaaa tggcaggctt cctgcatcag 660
ttgattgaaa gcgacgcgat cgtcggcatc gtcgacggtg agacgtatga gttcgaattg 720
cgtggtccat tgttcaacta cgcaggataa 750
【0081】
配列番号2:
Met Lys Tyr Thr Val Ile Thr Gly Ala Ser Ser Gly Ile Gly Tyr Glu
Thr Ala Lys Leu Leu Ala Gly Lys Gly Lys Ser Leu Val Leu Val Ala
Arg Arg Thr Ser Glu Leu Glu Lys Leu Arg Asp Glu Val Lys Gln Ile
Ser Pro Asp Ser Asp Val Ile Leu Lys Ser Val Asp Leu Ala Asp Asn
Gln Asn Val His Asp Leu Tyr Glu Gly Leu Lys Glu Leu Asp Ile Glu
Thr Trp Ile Asn Asn Ala Gly Phe Gly Asp Phe Asp Leu Val Gln Asp
Ile Glu Leu Gly Lys Ile Glu Lys Met Leu Arg Leu Asn Ile Glu Ala
Leu Thr Ile Leu Ser Ser Leu Phe Val Arg Asp His His Asp Ile Glu
Gly Thr Thr Leu Val Asn Ile Ser Ser Ala Gly Gly Tyr Arg Ile Val
Pro Asn Ala Val Thr Tyr Cys Ala Thr Lys Phe Tyr Val Ser Ala Tyr
Thr Glu Gly Leu Ala Gln Glu Leu Gln Lys Gly Gly Ala Lys Leu Arg
Ala Lys Val Leu Ala Pro Ala Ala Thr Glu Thr Glu Phe Ala Asp Arg
Ser Arg Gly Glu Ala Gly Phe Asp Tyr Ser Lys Asn Val Lys Lys Tyr
His Thr Ala Ala Glu Met Ala Gly Phe Leu His Gln Leu Ile Glu Ser
Asp Ala Ile Val Gly Ile Val Asp Gly Glu Thr Tyr Glu Phe Glu Leu
Arg Gly Pro Leu Phe Asn Tyr Ala Gly
【0082】
配列番号3:ggaggcgaat tcatgaaata tacggtgatt
配列番号4:taatcactgc agttatcctg cgtagttgaa
【0083】
これらのプライマーを各15pmol、dNTP各10nmol、エクシグオバクテリウム(Exiguobacterium)sp.MCI4322のゲノムDNA 25ng、KOD−plus−用10×緩衝液(東洋紡績社製)5μL、KOD−plus−1ユニット(東洋紡績社製)を含む50μLの反応液を用い、変性(94℃、15秒)、アニール(57℃、30秒)、伸長(68℃、2分)を30サイクルで、PTC−200(MJ Research社製)を用いてPCR反応を行った。なお、エクシグオバクテリウム sp(Exiguobacterium sp)MCI4322株は、FERM-P-19239として独立行政法人産業総合研究所特許生物寄託センターから入手可能である。PCR反応液の一部をアガロースゲル電気泳動により解析した結果、特異的と思われるバンドが検出できた。
【0084】
上記反応液をMinElute PCR Purification kit(Qiagen社製)にて精製した。精製したDNA断片を制限酵素EcoRIとPstIで消化し、アガロースゲル電気泳動を行い、目的とするバンドの部分を切り出し、Qiagen Gel Extraction kit(Qiagen社製)により精製後回収した。得られたDNA断片を、EcoRI、及びPstIで消化したpKK223−3とTakara Ligation Kitを用いてライゲーションし、大腸菌JM109株を形質転換した。
【0085】
形質転換細胞をアンピシリン(50μg/mL)を含むLB寒天培地上で生育させ、コロニーダイレクトPCRを行い、挿入断片のサイズを確認した。目的とするDNA断片が挿入されていると考えられる形質転換細胞を50μg/mLのアンピシリンを含むLB培地で培養し、QIAPrepSpin Mini Prep kit(Qiagen社製)を用いてプラスミドを精製し、pExSDR1とした。プラスミドに挿入したDNAの塩基配列をダイターミネーター法により解析したところ、挿入されたDNA断片は、配列番号1の塩基配列と一致した。
【0086】
[2]カルボニルレダクターゼをコードするDNAで形質転換した大腸菌を用いた イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルの合成
上記[1]で得られた形質転換体をアンピシリン(50μg/mL)を含むCircleGrow培地(国産化学社製)で30℃で1.7時間生育させ、0.1mMになるようにイソプロピル−β−D−チオガラクトピラノシド(IPTG)を添加し、更に30℃で20時間培養した。得られた菌体ブロス1mLを遠心分離により集菌し、100μLの緩衝液(10%硫酸ナトリウム、10%グルコースを含む100mMトリス(最終pH9.9))を加えよく混合した。これに30μLの10g/L NADP+(オリエンタル酵母社製)、30μLのグルコースデヒドロゲナーゼ(天野製薬社製、1unit/mL)、実施例2aで得られた20μLの10g/Lイソプロピル(2R,3R)−2−ベンゾイルアミノ−3−シクロヘキシル−3−オキソプロピオン酸エステル(DMSO溶液)を添加後40℃で18時間振とう反応させた。反応終了後の反応液を酢酸エチル抽出、濃縮し、200μLのイソプロピルアルコールで溶かし、イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルを定量した。定量は高速液体クロマトグラフィー(HPLC)を用いて測定した。HPLCの条件は以下のとおりである。
【0087】
カラム:ODSカラム Acentis Amide(Spelco社製、4.6mmx10cm)、40℃
溶離液:10mM酢酸アンモニウム/アセトニトリル=55/45
流速:1mL/min
検出:UV254nm
【0088】
この結果、イソプロピル(2R,3R)−3−シクロヘキシル−3−ヒドロキシ−2−アミノベンゾイルプロピオン酸エステルの収量は14.1μgであった。ジアステレオ選択性は90%d.e.であった。また、該遺伝子を含まないプラスミドpKK223−3をもつ大腸菌を上記と同様の方法で培養し、反応させてみたが上記生成物は認められなかった。次に光学純度の測定をHPLCを用いて行なった。HPLCの条件は以下のとおりである。
【0089】
カラム:OD−RH(ダイセル社製、4.6mmx15cm)、30℃
溶離液:40%アセトニトリル
流速:0.5mL/min
検出:UV254nm
光学純度は100%e.e.であった。
【0090】
[実施例4](2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸の合成法
500mLのナスフラスコに、実施例3と同様に、ただし反応スケールを1000倍にあげて実施して得られたイソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸エステル12.4g(37.0mmol)を仕込み、6N塩酸水溶液を添加して110℃に加温しながら6.5時間反応させた。室温に冷却後、トルエン50mLにて2回洗浄し、水層部を更に110℃に昇温し、5時間反応させた。室温に戻し、トルエン50mLにて2回洗浄し、得られた水層に25wt%の水酸化ナトリウム水溶液をpH5.68になるまで添加した。析出した固体をろ過し、減圧乾燥後に純度90%程度の白色固体として(2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸を8.40g取得した。この化合物を塩酸塩に変換後、下記スペクトルを取得した。
1H−NMR(400MHz,CD3OD):δ0.80−1.27(5H,m),1.58−1.79(5H,m),1.96−1.99(1H,m),3.42(1H,m),4.00(1H,d,J=2.3Hz).
【0091】
[実施例5](2R,3R)−2−t−ブトキシカルボニルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸の合成
実施例4で取得した(2R,3R)−2−アミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸8.40gを1N水酸化ナトリウム水溶液35mL及びTHF35mLに溶解させ、ジ−t−ブチルジカルボナート8.89g(40.7mmol)を添加した。室温下で6時間反応させ、反応液を減圧下でTHFを留去した。トルエン20mLを添加し、有機層と水層を分離し、更に有機層を1N水酸化ナトリウム水溶液5mLで3回抽出した。水層を併せ、1N塩酸60mLにて酸性に調整し、酢酸エチル70mL、30mLでそれぞれ抽出した。得られた有機層を硫酸マグネシウムで乾燥・ろ過し、得られた液を濃縮し、(2R,3R)−2−t−ブトキシカルボニルアミノ−3−シクロヘキシル−3−ヒドロキシプロピオン酸7.09g(純度96%、イソプロピル−2−ベンゾイルアミノ−3−シクロヘキサノイル−3−オキソプロピオン酸エステルからの換算収率67%)を得た。
1H−NMR(400MHz,CD3OD):δ0.88−1.24(5H,m),1.62−1.68(5H,m),1.79−1.83(1H,m),3.37(1H,t,J=6.2Hz),4.07−4.18(1H,m).
【0092】
[比較例1]4−(1−ヒドロキシメチリデン)−2−フェニルオキサゾール−5−オンの合成(非特許文献2及び非特許文献3)
馬尿酸1.00g(5.58mmol)をエタノール5mLに懸濁させ、20%ナトリウムエトキシドエタノール溶液を4.76g(14mmol)添加した。2時間撹拌後、反応液を減圧乾燥して馬尿酸のナトリウム塩1.12とした。この馬尿酸ナトリウム塩にβ−ピコリン1.70mL、無水酢酸(1.67mL)を添加し、室温にて2時間反応後、更に40℃にて5時間反応させた。反応液を室温に冷却し、LCにて定量分析したところ収率は15%、変換率は50%であった。
1H−NMR(400MHz,CDCl3):δ2.50(3H,s),7.52−7.57(3H,m),7.88−7.94(2H,m).
【0093】
[比較例2]4−〔1−シクロヘキシル−1−ヒドロキシメチリデン〕−2−フェニルオキサゾール−5−オンの合成(非特許文献2及び非特許文献3)
2−フェニルオキサゾール−5−オン200mg(1.24mmol)をβ−ピコリン0.38mLに懸濁させ、氷冷下、シクロヘキサンカルボン酸クロライド0.16mLを滴下して室温に戻して20時間反応させた。反応液に2N塩酸水20mLを加え、ジクロロメタン30mLにて2回抽出し、得られた有機層をLCにて定量分析したところ収率は18%、変換率は100%であった。反応副生物である4−(2−N−ベンゾイルアミノ−1−ヒドロキシルメチリデン)−イル−2−フェニルオキサゾール−5−オンが50%生成していた。
1H−NMR(400MHz,CDCl3):δ4.46(3H,m),7.43−7.66(7H,m),7.83(2H),7.90(1H,s),12.55(1H,s).
【図面の簡単な説明】
【0094】
【図1】図1は、実施例及び比較例における反応の概要を示す。
【配列表フリーテキスト】
【0095】
[配列表]
SEQUENCE LISTING
<110> API Corporation
<120> A method for producing an acyloxazolone derivative and beta-ketoamino acid derivative
<130> A61441A
<160> 4
<210> 1
<211> 750
<212> DNA
<213> Exiguobacterium sp.
<400> 1
atgaaatata cggtgattac tggagcaagt tcaggaattg gatatgagac ggcaaaacta 60
ctcgcaggaa aaggaaaatc actcgttctc gtcgcgcgac ggacgtctga gctcgaaaaa 120
cttcgggatg aagtcaaaca aatctcacca gatagtgatg tcatcctcaa gtcggtcgat 180
ctcgcagata accaaaatgt ccatgattta tatgagggat taaaggaact cgacatcgaa 240
acgtggatca acaatgctgg attcggcgat tttgatctcg tccaggacat tgagctcggg 300
aaaatcgaga aaatgcttcg cttgaacatc gaggcgctaa cgattctatc gagtctgttc 360
gtccgcgatc atcatgacat cgaaggaacg acgctcgtca atatctcgtc agcaggtggc 420
taccggatcg tgccgaacgc ggtcacgtat tgcgcgacga agttctatgt cagtgcttat 480
acagaagggc tcgcgcaaga actgcaaaaa ggtggggcaa aacttcgggc gaaagtactg 540
gcaccagctg cgactgagac agagtttgcg gatcgttcgc gcggcgaagc aggtttcgac 600
tacagcaaga acgtcaaaaa gtaccatacg gcggctgaaa tggcaggctt cctgcatcag 660
ttgattgaaa gcgacgcgat cgtcggcatc gtcgacggtg agacgtatga gttcgaattg 720
cgtggtccat tgttcaacta cgcaggataa 750
<210> 2
<211> 249
<212> PRT
<213> Exiguobacterium sp.
<400> 2
Met Lys Tyr Thr Val Ile Thr Gly Ala Ser Ser Gly Ile Gly Tyr Glu
1 5 10 15
Thr Ala Lys Leu Leu Ala Gly Lys Gly Lys Ser Leu Val Leu Val Ala
20 25 30
Arg Arg Thr Ser Glu Leu Glu Lys Leu Arg Asp Glu Val Lys Gln Ile
35 40 45
Ser Pro Asp Ser Asp Val Ile Leu Lys Ser Val Asp Leu Ala Asp Asn
50 55 60
Gln Asn Val His Asp Leu Tyr Glu Gly Leu Lys Glu Leu Asp Ile Glu
65 70 75 80
Thr Trp Ile Asn Asn Ala Gly Phe Gly Asp Phe Asp Leu Val Gln Asp
85 90 95
Ile Glu Leu Gly Lys Ile Glu Lys Met Leu Arg Leu Asn Ile Glu Ala
100 105 110
Leu Thr Ile Leu Ser Ser Leu Phe Val Arg Asp His His Asp Ile Glu
115 120 125
Gly Thr Thr Leu Val Asn Ile Ser Ser Ala Gly Gly Tyr Arg Ile Val
130 135 140
Pro Asn Ala Val Thr Tyr Cys Ala Thr Lys Phe Tyr Val Ser Ala Tyr
145 150 155 160
Thr Glu Gly Leu Ala Gln Glu Leu Gln Lys Gly Gly Ala Lys Leu Arg
165 170 175
Ala Lys Val Leu Ala Pro Ala Ala Thr Glu Thr Glu Phe Ala Asp Arg
180 185 190
Ser Arg Gly Glu Ala Gly Phe Asp Tyr Ser Lys Asn Val Lys Lys Tyr
195 200 205
His Thr Ala Ala Glu Met Ala Gly Phe Leu His Gln Leu Ile Glu Ser
210 215 220
Asp Ala Ile Val Gly Ile Val Asp Gly Glu Thr Tyr Glu Phe Glu Leu
225 230 235 240
Arg Gly Pro Leu Phe Asn Tyr Ala Gly
245
<210> 3
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> PCR primer
<400> 3
ggaggcgaat tcatgaaata tacggtgatt 30
<210> 4
<211> 30
<212> DNA
<213> Artificial Sequence
<220>
<223> PCR primer
<400> 4
taatcactgc agttatcctg cgtagttgaa 30
【特許請求の範囲】
【請求項1】
下記一般式(1)
【化1】
(式中、R1及びR2はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。)で表されるオキサゾール化合物にルイス酸の存在下、アシル化剤を作用させることにより、下記一般式(2)
【化2】
(式中、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるアシルオキサゾロン化合物を製造する工程を含む、アシルオキサゾロン化合物の製造方法。
【請求項2】
アシル化剤が、酸ハライド又は酸無水物である、請求項1に記載の方法。
【請求項3】
請求項1の方法により製造した一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【化3】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表される化合物を製造する工程を含む、β−ケトアミノ酸誘導体の製造方法。
【請求項4】
下記一般式(4)
【化4】
(式中、R1は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるシクロヘキシルメチリデニルオキサゾロン化合物。
【請求項5】
(i)請求項3に記載の方法により一般式(3)で表されるβ−ケトアミノ酸誘導体を製造する工程、及び
(ii)上記工程(i)で得られた一般式(3)で表されるβ−ケトアミノ酸誘導体に微生物の菌体及び/又は該菌体処理物を作用させて下記一般式(5)
【化5】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるα−N−アシルアミノ−β−ヒドロキシエステルを得る工程を含む、α−N−アシルアミノ−β−ヒドロキシエステルを製造する方法。
【請求項6】
(i)請求項5に記載の方法により一般式(5)で表されるN−アシルアミノ−β−ヒドロキシエステルを製造する工程、
(ii)上記工程(i)で得られたα−N−アシルアミノ−β−ヒドロキシエステルに酸又は塩基を作用させて加水分解を行い、下記一般式(6)
【化6】
(式中、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるβ−ヒドロキシアミノ酸を得る工程;及び
(iii)上記工程(ii)で得られたβ−ヒドロキシアミノ酸にt−ブトキシカルボニル化剤を作用させて下記一般式(7)
【化7】
(式中、R3及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物を製造する工程、を含むN−t−ブトキシカルボニル−β−ヒドロキシアミノ酸の製造方法。
【請求項1】
下記一般式(1)
【化1】
(式中、R1及びR2はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示す。)で表されるオキサゾール化合物にルイス酸の存在下、アシル化剤を作用させることにより、下記一般式(2)
【化2】
(式中、R1は前記と同義であり、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるアシルオキサゾロン化合物を製造する工程を含む、アシルオキサゾロン化合物の製造方法。
【請求項2】
アシル化剤が、酸ハライド又は酸無水物である、請求項1に記載の方法。
【請求項3】
請求項1の方法により製造した一般式(2)で表されるオキサゾロン化合物にアルコール類を作用させることにより下記一般式(3)
【化3】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示す。)で表される化合物を製造する工程を含む、β−ケトアミノ酸誘導体の製造方法。
【請求項4】
下記一般式(4)
【化4】
(式中、R1は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるシクロヘキシルメチリデニルオキサゾロン化合物。
【請求項5】
(i)請求項3に記載の方法により一般式(3)で表されるβ−ケトアミノ酸誘導体を製造する工程、及び
(ii)上記工程(i)で得られた一般式(3)で表されるβ−ケトアミノ酸誘導体に微生物の菌体及び/又は該菌体処理物を作用させて下記一般式(5)
【化5】
(式中、R1及びR3はそれぞれ独立して、置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、R4は置換してもよいアルキル基、又は置換していてもよいアラルキル基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるα−N−アシルアミノ−β−ヒドロキシエステルを得る工程を含む、α−N−アシルアミノ−β−ヒドロキシエステルを製造する方法。
【請求項6】
(i)請求項5に記載の方法により一般式(5)で表されるN−アシルアミノ−β−ヒドロキシエステルを製造する工程、
(ii)上記工程(i)で得られたα−N−アシルアミノ−β−ヒドロキシエステルに酸又は塩基を作用させて加水分解を行い、下記一般式(6)
【化6】
(式中、R3は置換していてもよいアルキル基、又は置換していてもよいアリール基を示し、Xは水素原子又は金属原子を示す。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表されるβ−ヒドロキシアミノ酸を得る工程;及び
(iii)上記工程(ii)で得られたβ−ヒドロキシアミノ酸にt−ブトキシカルボニル化剤を作用させて下記一般式(7)
【化7】
(式中、R3及びXは前記と同義。Xが水素原子の場合、金属化合物と錯体を形成していてもよい。)で表される化合物を製造する工程、を含むN−t−ブトキシカルボニル−β−ヒドロキシアミノ酸の製造方法。
【図1】

【公開番号】特開2007−31428(P2007−31428A)
【公開日】平成19年2月8日(2007.2.8)
【国際特許分類】
【出願番号】特願2006−172191(P2006−172191)
【出願日】平成18年6月22日(2006.6.22)
【出願人】(396020464)株式会社エーピーアイ コーポレーション (39)
【Fターム(参考)】
【公開日】平成19年2月8日(2007.2.8)
【国際特許分類】
【出願日】平成18年6月22日(2006.6.22)
【出願人】(396020464)株式会社エーピーアイ コーポレーション (39)
【Fターム(参考)】
[ Back to top ]