
アシルオキシ化反応用触媒およびその製造方法
【課題】アシルオキシ化反応を効率よく行い、アシルオキシ化生成物化合物を経済的に製造するための触媒およびその製造方法を提供する。
【解決手段】(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とを(d)担体に担持させてアシルオキシ化反応用触媒を得る。
【解決手段】(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とを(d)担体に担持させてアシルオキシ化反応用触媒を得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アシルオキシ化反応用触媒の製造方法、それにより得られるアシルオキシ化反応用触媒および該触媒を用いたアシルオキシ化反応方法に関する。
アシルオキシ化反応は、香料や医農薬原料として、有機合成中間体として、さらに重合性材料として用いられる種々の有用な化合物の合成反応である。
【背景技術】
【0002】
トルエン、キシレン等のベンジル位に水素原子を有する化合物またはプロピレン、シクロヘキセン等のアリル位に水素原子を有する化合物と、酢酸等のカルボン酸および酸素とによるアシルオキシ化反応は、従来より公知である。
かかるアシルオキシ化反応用の触媒として、酢酸パラジウム等の均一系触媒およびパラジウムをシリカ等の担体に担持した不均一系触媒が開発されている。
【0003】
特開2001−269577号公報には、活性炭などの担体に、パラジウムなどの周期律表第8〜11族の金属、ナトリウムやカリウムなどのアルカリ金属および/またはアンチモン、ビスマス、テルルなどの周期律表第12〜16族の金属を担持した触媒を用いることによりアシルオキシ化合物を経済的に製造する方法が開示されている。
【0004】
特表2001−521817号公報には、パラジウムおよび金に加えて、マグネシウム、カルシウム、バリウム、ジルコニウム、セリウムなどの第三金属をその酸化物またはその酸化物とその金属との混合物として担体中に含む触媒が開示され、またこれらのパラジウム、金および第三金属の水溶性塩の溶液で担体を含浸し、その後アルカリ性化合物との反応により非水溶性化合物として固定し、そして固定されたパラジウムおよび金をそれらの金属状態に、また第三金属をその酸化物またはその酸化物と金属の混合物に還元する工程が開示されている。
【0005】
また、特開2005−296858号公報には、パラジウム、金、亜鉛などの両性金属およびアルカリ金属を含有する触媒が開示され、亜鉛を添加することにより液相還元法によった場合にも気相還元法による場合に匹敵するほどの貴金属表面積が得られるということが報告されている。
【0006】
【特許文献1】特開2001−269577号公報
【特許文献2】特表2001−521817号公報
【特許文献3】特開2005−296858号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、上記した従来のアシルオキシ化反応用触媒は、反応初期の活性、選択性および活性の持続性の性能バランスが不十分である等の問題を有していた。
【0008】
本発明は、上記のごとき状況に鑑みてなされたものであり、上記の問題を解決し、アシルオキシ化合物を経済的に製造するための触媒およびその製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0009】
本願発明者らは、上記課題を解決するため鋭意検討を重ねた結果、少なくとも周期律表の第8、9、10または11族の元素を含む第一成分、前記第一成分の元素とは異なる元素を含む第二成分および前記第一成分および第二成分の元素とは異なる元素を含む第三成分を担体に担持させることにより、アシルオキシ化反応を効率よく行うことができ、アシルオキシ化生成物を経済的に製造することができる触媒が得られることを見出し、この知見に基づき本発明を完成するに至ったものである。
【0010】
よって、本発明は、例えば、以下の(1)〜(14)からなる。
(1)(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とをいっしょに(d)担体に担持させる工程を含むことを特徴とするアシルオキシ化反応用触媒の製造方法。
【0011】
(2)前記(d)担体に担持させる工程に次いで、さらに非水溶化処理を行うことを特徴とする前記(1)に記載のアシルオキシ化反応用触媒の製造方法。
(3)前記(d)担体に担持させる工程に次いで、さらに還元処理を行うことを特徴とする前記(1)または(2)に記載のアシルオキシ化反応用触媒の製造方法。
【0012】
(4)前記還元処理に次いで、酸および/またはキレート剤を接触させることを特徴とする前記(3)に記載のアシルオキシ化反応用触媒の製造方法。
(5)さらに(e)周期律表の第1および2族の元素(水素を除く)の少なくとも1種を含む第四成分を添加することを特徴とする前記(1)〜(4)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0013】
(6)前記(a)第一成分がパラジウムを含むことを特徴とする前記(1)〜(5)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(7)前記(b)第二成分が周期律表の第11族元素を含むことを特徴とする前記(1)〜(6)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0014】
(8)前記(b)第二成分が金および銅から選ばれる少なくとも1種を含むことを特徴とする前記(7)に記載のアシルオキシ化反応用触媒の製造方法。
(9)前記(c)第三成分が周期律表の第3〜13族元素の1種を含むことを特徴とする前記(1)〜(8)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0015】
(10)前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比が、0.4〜1.5であることを特徴とする前記(1)〜(9)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(11)前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比が、0.1〜0.5であることを特徴とする前記(1)〜(10)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(12)前記(1)〜(11)のいずれかに記載した方法により得られるアシルオキシ化反応用触媒。
【0016】
(13)前記(1)〜(11)のいずれかに記載した方法により得られる触媒の存在下に、一般式(1):CHR1R2−X(式中、R1およびR2は、それぞれ独立して、水素原子または有機残基を表し、Xは置換基を有していてもよい芳香族炭化水素残基または置換基を有していてもよいオレフィン残基を表す)で表される化合物またはエチレンと、一般式(2):R3−COOH(式中、R3は水素原子または有機残基を表す)で表されるカルボン酸類とおよび酸素とを反応させて、一般式(3):R3−COO−CR1R2−X(式中、R1、R2、R3およびXは前記規定に同一のものを表す)で表される化合物または酢酸ビニルを生成させることを特徴とするアシルオキシ化合物の製造方法。
【0017】
(14)前記反応が、さらに塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の存在下に行われることを特徴とする前記(13)に記載のアシルオキシ化合物の製造方法。
【発明の効果】
【0018】
本発明によれば、反応初期の活性、選択性および活性の持続性の性能バランスに優れたアシルオキシ化反応用触媒を得ることができ、それによって効率的かつ経済的にアシルオキシ化合物を製造することができる。
【発明を実施するための最良の形態】
【0019】
以下に本発明の好ましい実施の形態について説明するが、本発明はこれらの形態のみに限定されるものではなく、その精神と実施の範囲内において様々な変形が可能であることを理解されたい。
本発明のアシルオキシ化反応用触媒は、担体に担持物質が担持されてなるアシルオキシ化反応用触媒である。
【0020】
本発明において、(a)第一成分および(b)第二成分の元素とは、IUPAC無機化学命名法改訂版(1989)による周期律表の第8〜11族の元素を指し、例えば、鉄、ルテニウム、オスミウム、コバルト、ロジウム、イリジウム、ニッケル、パラジウム、白金、銅、銀および金が挙げられる。これらのうちでは、(a)第一成分の元素としてはルテニウム、ロジウム、パラジウムまたは銀が好ましく、パラジウムがさらに好ましく、(b)第二成分の元素としてはオスミウム、イリジウム、白金、銅または金が好ましく、金がさらに好ましい。(a)第一成分は反応に欠かせない主触媒に関わる元素を含み、また(b)第二成分は反応効率を高める助触媒に関わる元素を含む。(a)第一成分および(b)第二成分の元素はそれぞれ1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0021】
(a)第一成分および(b)第二成分としては、パラジウム元素を例に挙げれば、パラジウム金属、ヘキサクロロパラジウム酸アンモニウム、ヘキサクロロパラジウム酸カリウム、ヘキサクロロパラジウム酸ナトリウム、テトラクロロパラジウム酸アンモニウム、テトラクロロパラジウム酸カリウム、テトラクロロパラジウム酸ナトリウム、テトラブロモパラジウム酸カリウム、酸化パラジウム、塩化パラジウム、臭化パラジウム、ヨウ化パラジウム、硝酸パラジウム、硫酸パラジウム、酢酸パラジウム、ジニトロサルファイトパラジウム酸カリウム、クロロカルボニルパラジウム、ジニトロジアンミンパラジウム、テトラアンミンパラジウム塩化物、テトラアンミンパラジウム硝酸塩、テトラアンミンパラジウム水酸化物、cis−ジクロロジアンミンパラジウム、trans−ジクロロジアンミンパラジウム、ジクロロ(エチレンジアミン)パラジウム、テトラシアノパラジウム酸カリウム等を挙げることができる。
【0022】
(a)第一成分および(b)第二成分の元素の担持量は、完成された触媒に対して、それぞれ、好ましくは0.01〜20質量%、より好ましくは0.1〜10質量%である。この担持量は、完成された触媒を酸に溶解して、測定対象の元素の含有量をICP分析により求めることができる。
【0023】
(a)第一成分、(b)第二成分および(c)第三成分は、それぞれ、例えば、水溶液の状態で(d)担体に含浸させて担持させることができる。この場合、(a)第一成分、(b)第二成分および(c)第三成分をいっしょに1つの水溶液中に含有させ、この水溶液を(d)担体に含浸させて担持させることが好ましい。
【0024】
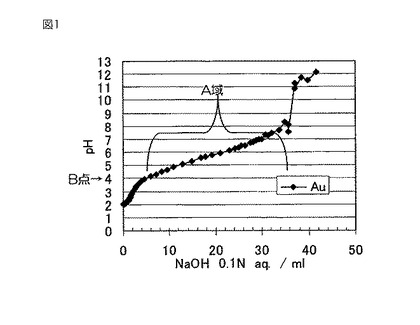
本発明における沈澱開始pHは、次の方法で決定することができる。すなわち、各種金属または金属化合物を酸性水溶液に溶解した後、アルカリ性水溶液を滴下することでpHが上昇していき、ある領域でpH上昇が鈍化するが、その鈍化の開始点のpHを沈澱開始pHと定義する。例えば、塩化金酸水溶液に水酸化ナトリウム水溶液を滴下しながら水溶液のpHを測定し、その滴下量に対するpHをプロットすることにより、図1に示すような滴定曲線が描かれる。ある量を滴下すると図中のA域のように水酸化ナトリウム水溶液を滴下してもpHがあまり上昇しないような比較的安定した状態が確認される。この安定状態の開始点(B点)のpHが沈澱開始pHである。この測定における注意点は、沈殿開始前に当該金属および金属化合物が溶解して均一な水溶液を形成している状態で滴定を開始する必要がある点にある。
【0025】
なお、この滴定に用いられるアルカリ性水溶液は、各種酸性水溶液のpH変化が確認できるアルカリ性水溶液であれば、その化学種、濃度や滴下量などは特に制限されない。ただし、一連の比較測定においては、同一のアルカリ水溶液を用いて各酸性水溶液の滴定を行う必要がある。この滴定には、例えば、水酸化ナトリウム水溶液が好適に用いられる。
【0026】
こうして測定された沈澱開始pHをもとに、(a)第一成分および(b)第二成分の沈澱開始pH以下の沈殿開始pHを与える成分であって、第一成分および第二成分の元素とは異なる元素を含む成分を(c)第三成分として選択することができる。例えば、パラジウム元素を含む(a)第一成分よりも沈澱開始pHが低い第三成分としては、スカンジウム、チタン、モリブデン、タングステンまたは鉄を含む化合物またはそれらの単体を挙げることができる。化合物としては、酸化物、水酸化物、ハロゲン化物、オキシハロゲン化物、アルコキシド、酢酸塩等の脂肪族カルボン酸塩、硝酸塩、炭酸塩、リン酸塩、ホウ酸塩等とそれらの水和物が挙げられる。鉄を例に挙げれば、例えば、鉄金属、水酸化鉄、塩化鉄、臭化鉄、ヨウ化鉄、過塩素酸鉄、鉄メトキシド、鉄エトキシド、酢酸鉄、プロピオン酸鉄、アクリル酸鉄、硝酸鉄、炭酸鉄、硫酸鉄、リン酸鉄、酢酸鉄、くえん酸鉄、グルコン酸鉄、フマル酸鉄、シュウ酸鉄、ホウ酸鉄等とそれらの水和物である。また、(c)第三成分としては、これらのうちでスカンジウム、チタンおよび鉄から選ばれる少なくとも1種を含むものが好ましい。(c)第三成分は1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0027】
上記の方法で選定されるc)第三成分を添加することにより、(a)第一成分と(b)第二成分との共存化もしくは均一混合化が促され、より高い活性、選択性および活性低下抑制効果が得られるのであると考えられる。
【0028】
本発明の触媒において、(c)第三成分の元素の担持量は、完成された触媒に対して、好ましくは0.001〜20質量%、より好ましくは0.01〜10質量%である。この担持量は、前述の(a)第一成分や(b)第二成分の元素の場合と同様に、完成された触媒を酸に溶解して、測定対象の元素の含有量をICP分析により求めることができる。
【0029】
前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比は、0.4〜1.5であるのが好ましく、0.8〜1.2であるのがさらに好ましい。また、前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比は、0.1〜0.5であるのが好ましく、0.2〜0.4であるのがさらに好ましい。
なお、本発明の触媒の製造方法においては酸洗浄の工程を行ってもよく、それによって担持量が低下することがあるが、上記担持量は酸洗浄前の値である。
【0030】
本発明の触媒の(d)担体としては、例えば、シリカ、アルミナ、ジルコニア、チタニア等の金属酸化物、炭素、木炭、活性炭、アスベスト、シリカ−アルミナ、ゼオライト、オルガノゾルゲル、イオン交換樹脂、クレー、炭酸塩、炭酸塩等が挙げられる。これらのうちでは、シリカ、アルミナ、ジルコニア、チタニア等の金属酸化物、活性炭およびゼオライトが好ましい。
担体の比表面積は特に制限されるものではないが、触媒成分の分散化と担体の機械的強度とのバランスの観点からは、B.E.T.法で測定した値が10〜1500m2/gであることが好ましく、100〜500m2/gであることがさらに好ましい。
【0031】
前記活性炭の原料は、木質、ヤシ殻、もみ殻、有機高分子等が挙げられる。これらのうちでも、木質およびヤシ殻が好ましい。担体として用いられる活性炭の性質としては、比表面積が800〜2500m2/gの範囲、特に1000〜1800m2/gの範囲であるのが、触媒成分の分散化と担体の機械的強度とのバランスの観点から好ましい。
担体の形態としては、例えば、固定床方式、流動床方式、懸濁触媒方式等の実施する反応の形態等により、粉末状、破砕状、粒子状、柱状等を適宜選ぶことができる。
【0032】
(a)第一成分、(b)第二成分および(c)第三成分を(d)担体に沈積させるために非水溶化処理を行うことが好ましい。
非水溶化処理とは、(a)第一成分、(b)第二成分および(c)第三成分を溶解した水溶液を(d)担体に含浸させた触媒前駆体を、酸性あるいはアルカリ性水溶液に接触させ、(a)第一成分、(b)第二成分および(c)第三成分を沈澱させる工程をいう。ここで、触媒前駆体とは、担体に担持物質を接触あるいは担持させた時点から各種工程を経て最終の触媒として完成するまでの途中の中間状態の触媒を指す。(a)第一成分、(b)第二成分および(c)第三成分を溶解した水溶液が酸性であればアルカリ性水溶液と接触させ、アルカリ性であれば酸性水溶液と接触させる。本発明においては、主にアルカリ性水溶液と接触させる。
【0033】
酸性水溶液に使用される酸としては、特に限定されるものではないが、例えば、塩酸、硫酸、硝酸、ヘテロポリ酸などの無機酸や、酢酸、リン酸、シュウ酸、クエン酸、グルコン酸などの有機酸が挙げられる。
【0034】
アルカリ性水溶液としては、例えば、アルカリ金属やアルカリ土類金属の水酸化物、アルカリ金属やアルカリ土類金属の重炭酸塩、アルカリ金属やアルカリ土類金属の炭酸塩、アルカリ金属やアルカリ土類金属のケイ酸塩等のアルカリ性化合物の水溶液が挙げられる。好ましいアルカリ金属としては、リチウム、ナトリウム、カリウムなどがある。好ましいアルカリ土類金属としては、バリウム、マグネシウムなどがある。特に好適には、メタケイ酸ナトリウム、メタケイ酸カリウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウムなどが用いられる。
【0035】
非水溶化処理の方法としては、例えば、前記触媒前駆体を酸性水溶液またはアルカリ水溶液に浸漬するか、または前記触媒前駆体に酸性水溶液またはアルカリ水溶液を滴下することにより、両者を接触させる方法を挙げることができる。
非水溶化処理で使用される酸性水溶液またはアルカリ水溶液の使用量および水溶液の濃度は特に限定されない。ただし、処理後の液相のpHが7〜11の範囲となるように、調整することが好ましい。
【0036】
非水溶化処理のための酸性水溶液またはアルカリ水溶液との接触時間は、0.5〜100時間、特に3〜50時間が好ましい。0.5時間未満では非水溶化が十分に行えない場合がある。また、100時間を超えると、洗浄液の種類や担体の種類によっては(d)担体がダメージを受けたり、(a)第一成分、(b)第二成分の再溶解が進むため好ましくない。
接触温度は、10〜80℃、特に20〜60℃が好ましい。10℃より低いと反応が遅く、処理時間の延長につながるため好ましくない。80℃を超えると(a)第一成分、(b)第二成分の凝集が進む恐れがあるため好ましくない。
【0037】
本発明において、(a)第一成分、(b)第二成分および(c)第三成分の担持工程の後、もしくは前記非水溶化処理の工程の後に、特に(a)第一成分、(b)第二成分の溶出を防ぐために還元処理を行うことが好ましい。
例えば、液相還元では、アルコールや炭化水素類を用いた非水系、水系溶液が用いられる。還元剤として、カルボン酸およびその塩、アルデヒド、過酸化水素、糖類、ジボラン、アミン、ヒドラジンなどが用いられる。具体的には、シュウ酸、シュウ酸カリウム、ギ酸、ギ酸カリウム、クエン酸アンモニウム、グルコース、多価フェノール、ヒドラジン、ホルムアルデヒド、アセトアルデヒド、ハイドロキノン、水素化ホウ素ナトリウム、クエン酸カリウムなどが挙げられる。特に好ましくはヒドラジンが用いられる。
【0038】
気相還元では、水素、一酸化炭素、アルコール、アルデヒド、エチレン、プロペン、およびイソブテンなどのオレフィンが還元ガスとして用いられる。好ましくは水素が用いられる。気相還元では、希釈剤として不活性ガスを用いてもよい。不活性ガスとしては、例えば、ヘリウム、アルゴン、窒素などがある。
【0039】
前記(c)第三成分を除去したほうが触媒の性能が向上する場合がある。除去する場合は、例えば、還元処理後に洗浄を行うことで除去することが好ましい。(c)第三成分の洗浄除去は、例えば、酸やキレート剤などと接触させて溶解除去することにより、行うことができる。
前記酸としては、例えば、塩酸、硫酸、硝酸、ヘテロポリ酸などの無機酸や、酢酸、リン酸、シュウ酸、クエン酸、グルコン酸などの有機酸が挙げられる。
【0040】
(c)第三成分の洗浄除去には、上記の酸以外に、以下のようなキレート剤を使用しても良い。また、酸処理を含めた複数の処理を併用しても良い。
ここで、キレート剤とは、金属イオンに配位結合することができる電子供与体(配位子)を有する化合物をいい、金属イオンと反応することにより金属錯塩あるいは金属キレート化合物を形成する物質をいう。
【0041】
有用なキレート剤としては、ニトリロ三酢酸、ジエチレントリアミン五酢酸、ヒドロキシエチルエチレンジアミン三酢酸、トリエチレンテトラアミン六酢酸、1,3−プロパンジアミン四酢酸、1,3−ジアミノ−2−ヒドロキシプロパン四酢酸、ヒドロキシルイミノ二酢酸、ジヒドロキシルグリシン、グリコールエーテルジアミン四酢酸、L−グルタミン酸二酢酸などが挙げられる。
また、洗浄に使用するため、キレート剤を水に溶かして水溶液として用いても良い。多くの場合、水酸化ナトリウム水溶液やアンモニア水等のアルカリ性水溶液に容易に溶けるが、アルコールなどの有機溶剤に溶かしてもよい。
【0042】
(c)第三成分の洗浄除去方法に制限はないが、一般には、触媒前駆体を洗浄液に浸漬させて行うのがよい。浸漬時間は、0.5〜100時間、特に3〜50時間が好ましい。0.5時間未満では洗浄が十分に行えないことがある。また、100時間を超えると、洗浄液の種類や担体の種類によっては(d)担体がダメージを受けたり、(a)第一成分、(b)第二成分の再溶解が進むことがあり、好ましくない。
接触温度は、特に制限はないが、10〜80℃、特に20〜60℃が好ましい。10℃より低いと反応が遅く、処理時間の延長につながることがあるため好ましくない。80℃を超えると(a)第一成分、(b)第二成分)の凝集が進む恐れがあり、好ましくない。
【0043】
前記触媒には、(e)周期律表第1および/または2族の元素(水素を除く)を含む第四成分をさらに追加添加することが好ましい。この添加は、触媒製造時に担持することにより行ってもよいし、触媒製造後反応への使用前に反応系内に添加してもよいし、反応の間に追加添加してもよい。
(e)第四成分を、本発明のアシルオキシ化反応用触媒に担持し、あるいは反応前または反応の間に反応系内に添加することにより、アシルオキシ化反応における転化率、選択率が向上し、アシルオキシ化生成物を経済的に製造することができる場合がある。
【0044】
(e)第四成分の周期律表第1および/または2族の元素としては、例えば、リチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウム、ベリリウム、マグネシウム、カルシウム、ストロンチウムおよびバリウムが挙げられる。これらの内では、リチウム、ナトリウム、カリウム、セシウムが好ましい。前記金属原子は1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0045】
(e)第四成分を与える原料としては、具体的には、周期律表第1および2族の元素の金属、酸化物、水酸化物、ハロゲン化物、オキシハロゲン化物、アルコキシド、酢酸塩等の脂肪族カルボン酸塩、硝酸塩、炭酸塩、リン酸塩、ホウ酸塩等が挙げられる。カリウムを例に挙げれば、カリウム金属、水酸化カリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、硫化カリウム、酢酸カリウム、硝酸カリウム、安息香酸カリウム、炭酸カリウム、リン酸カリウム、ホウ酸カリウム等を挙げることができる。
【0046】
(e)第四成分に含まれる上記元素の担持量は、完成された触媒に対して、好ましくは0.001〜40質量%、より好ましくは0.01〜10質量%である。この担持量は、前述の(a)第一成分や(b)第二成分の元素の場合と同様に、完成した触媒を酸に溶かし、測定対象の元素の含有量をICP分析により求めることができる。この担持量の範囲が転化率および選択率の点および経済性の点で好ましい。
(e)第四成分の担持は、例えば、含浸法、イオン交換法、共沈法、沈着法、混練法で行うことができるが、(e)第四成分を含む水溶液を担体に含浸させて担持するのが好ましい。
【0047】
本発明の触媒は、特に、トルエン、キシレン等のベンジル位に水素原子を有する化合物またはエチレン、プロピレン、シクロヘキセン等のビニル位もしくはアリル位に水素原子を有する化合物と、酢酸等のカルボン酸および酸素とによるアシルオキシ化反応に好適である。すなわち、本発明におけるアシルオキシ化反応は、前記一般式(1)で表される化合物またはエチレンと、前記一般式(2)で表されるカルボン酸類および酸素を反応させることによる、前記一般式(3)で表される化合物または酢酸ビニルを製造する反応である。
【0048】
反応原料として用いられる一般式(1)で表される化合物において、式中のR1、R2で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、またはアリール基が挙げられる。これらの内では、炭素数1〜10の飽和および/または不飽和アルキル基が好適に用いられる。
【0049】
Xで表される置換基を有していてもよい芳香族炭化水素残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状のアルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシ基、置換基を有していてもよいフェノキシ基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、水酸基、またはフッ素、塩素、臭素、ヨウ素等のハロゲン原子等の置換基を有していてもよいフェニル基である。
【0050】
また、Xで示される置換基を有していてもよいオレフィン残基としては、一般式(4):−CR4=CHR5(式中、R4、R5は、水素原子または有機残基を表す)で表されるものが挙げられる。ここで、R4、R5で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、アルデヒド基、脂肪族および/または芳香族ケトン基、カルボン酸基、脂肪族および/または芳香族カルボン酸エステル基がある。これらの内では、炭素数1〜10の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、カルボン酸基、脂肪族および/または芳香族カルボン酸エステル基が好適に用いられる。また、R1またはR2とR4またはR5とにより環を形成していても良い。
【0051】
一般式(1)で表される化合物の代表例としては、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸ブチル、メタクリル酸2−ヒドロキシエチルおよびメタクリル酸2−エチルヘキシル、エチレン、プロピレン、ブテン、ペンテン、ヘキセン、ヘプテン、ノネン、デセン、ブタジエン、シクロペンテン、シクロペンタジエン、シクロヘキセン、シクロヘキサジエン、シクロヘプテン、シクロオクテン、シクロノネン、シクロデセン、トルエン、エチルベンゼン、プロピルベンゼン、ブチルベンゼン、スチレン、キシレン、トリメチルベンゼン、テトラメチルベンゼン、ペンタメチルベンゼン、ヘキサメチルベンゼン、メチルビフェニル、ジメチルビフェイル、ジフェニルメタン、トリフェニルメタン、メチルフェノール、メトキシトルエン、エトキシトルエン、フェノキチトルエン等が挙げられる。これらの内で、異性体を含むものは、各異性体単独および/または各異性体混合物でもよい。これらの内では、メタクリル酸メチル、メタクリル酸ブチル、エチレン、プロピレン、ブテン、ペンテン、ヘキセン、ブタジエン、シクロペンテン、シクロペンタジエン、シクロヘキセン、シクロヘキサジエン、トルエン、キシレン、トリメチルベンゼン、メトキシトルエンおよびフェノキシトルエンが好適に用いられる。
【0052】
本発明のアシルオキシ化合物の製造方法において、原料として用いられる一般式(2)で表されるカルボン酸類は、式中のR3が水素原子または有機残基である化合物であれば、特に限定されるものではない。
R3で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数2〜20のアセトキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、置換されていてもよい芳香族基等が挙げられる。これらの内では、炭素数1〜5の飽和および/または不飽和アルキル基が好適に用いられる。
【0053】
一般式(2)で表されるカルボン酸類の代表例としては、ギ酸、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、カプリル酸、ラウリン酸、ミリスチル酸、パルミチン酸、ステアリン酸、アセト酢酸、ヒドロキシプロピオン酸、イソブタン酸、ヒドロキシイソブタン酸、t−ブチル酢酸、安息香酸、アクリル酸、メタクリル酸等が挙げられる。これらの内では、酢酸、プロピオン酸、酪酸、安息香酸、アクリル酸およびメタクリル酸が好ましく、酢酸、プロピオン酸、アクリル酸およびメタクリル酸が特に好ましい。
【0054】
一般式(3)で表される化合物の例としては、例えば、酢酸ビニル、アクリル酸ビニル、メタクリル酸ビニル、プロピオン酸ビニル、酢酸アリル、アクリル酸アリル、メタクリル酸アリル、プロピオン酸アリル、酢酸ベンジル、アクリル酸ベンジル、メタクリル酸ベンジル、プロピオン酸ベンジル、酢酸4−メチルベンジル、アクリル酸4−メチルベンジル、メタクリル酸4−メチルベンジル、プロピオン酸4−メチルベンジル、酢酸シクロヘキセン、アクリル酸シクロヘキセン、メタクリル酸シクロヘキセン、プロピオン酸シクロヘキセン、α−アセトキシメチルアクリル酸メチル、1,4−キシレンモノアセテート、1,4−キシレンジアセテートなどが挙げられる。
【0055】
前記一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類との反応初期におけるモル比は、例えば、10/1〜1/10の範囲内であってよい。上記範囲内でも6/1〜1/6の範囲内がより好ましい。一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類のどちらかを上記範囲内よりも過剰に添加しても、収率の向上および反応時間の短縮等の効果は望めず、過剰な原料の回収工程が長くなり、経済的に不利となる。
【0056】
反応に用いられる酸素は、原子状および/または分子状酸素であり、好ましくは分子状酸素である。また、分子状酸素は、窒素、アルゴン、ヘリウム、二酸化炭素等の不活性な気体との混合気体として用いられるのが好ましい。この場合、酸素の濃度は、反応系内で気体が爆発組成とならない範囲に調整して使用するのがより好ましい。
分子状酸素および分子状酸素を含む混合気体の反応系への供給は、反応系内の液相部または気相部の一方または両方に供給して行うことができる。反応系内へ分子状酸素および分子状酸素を含む混合気体を供給する場合には、酸素分圧が0.01〜20MPaの範囲内となるように供給するのがよい。
【0057】
本発明のアシルオキシ化合物の製造方法は、前記したアシルオキシ化反応を、前記した本発明のアシルオキシ化触媒の存在下で行うものである。
前記触媒の使用量は、用いる前記一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類の種類や組み合わせにもよるが、前記(a)第一成分が一般式(1)で表される化合物1モルに対して0.01ミリモル〜100モルの範囲内になる量であってよい。この範囲で前記触媒を使用するのが、収率の点および経済性の点で好ましい。
【0058】
また、本発明のアシルオキシ化合物の製造方法において、反応系中に塩基性化合物、窒素含有化合物およびリン含有化合物から選ばれる少なくとも1種の化合物を存在させることが転化率および選択率の点で好ましい。かかる塩基性化合物としては、例えば、ギ酸リチウム、ギ酸ナトリウム、ギ酸カリウム、ギ酸マグネシウム、ギ酸カルシウム、ギ酸バリウム、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸マグネシウム、酢酸カルシウム、酢酸バリウム、プロピオン酸リチウム、プロピオン酸ナトリウム、プロピオン酸カリウム、プロピオン酸マグネシウム、プロピオン酸カルシウム、プロピオン酸バリウム、アクリル酸リチウム、アクリル酸ナトリウム、アクリル酸カリウム、アクリル酸マグネシウム、アクリル酸カルシウム、アクリル酸バリウム、メタクリル酸リチウム、メタクリル酸ナトリウム、メタクリル酸カリウム、メタクリル酸マグネシウム、メタクリル酸カルシウム、メタクリル酸バリウム、安息香酸リチウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸マグネシウム、安息香酸カルシウム、安息香酸バリウム等のアルカリ金属および/またはアルカリ土類金属カルボン酸塩;水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化マグネシウム、水酸化カルシウム、水酸化バリウム等のアルカリ金属および/またはアルカリ土類金属水酸化物;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸マグネシウム、炭酸カルシウム、炭酸バリウム等のアルカリ金属および/またはアルカリ土類金属炭酸塩;リン酸リチウム、リン酸ナトリウム、リン酸カリウム、リン酸マグネシウム、リン酸カルシウム、リン酸バリウム等のアルカリ金属および/またはアルカリ土類金属炭酸塩;ほう酸リチウム、ほう酸ナトリウム、ほう酸カリウム、ほう酸マグネシウム、ほう酸カルシウム、ほう酸バリウム等のアルカリ金属および/またはアルカリ土類金属ほう酸塩等が挙げられる。
【0059】
前記窒素含有化合物の具体例としては、気体または水溶液等のアンモニア、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、エチレンジアミン、N,N−ジメチルエチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン等の脂肪族アミン、ピリジン、メチルピリジン、ビピリジン、ヒドロピリジン、フェナントロリン等の複素環アミン、アニリン、ジフェニルアミン、トリフェニルアミン等の芳香族アミン等が挙げられる。
【0060】
また、前記リン含有化合物の具体例としては、トリメチルホスフィン、トリエチルホスフィン等のトリアルキルホスフィン、トリフェニルホスフィン、トリス(2−メトキシフェニル)ホスフィン等のトリアリールホスフィン、ジフェニルメチルホスフィン、ジフェニルエチルホスフィン等のジアリールアルキルホスフィン等の一座ホスフィン、1,2−ジフェニルホスフィノエタン、1,4−ビスジフェニルホスフィノブタン等の二座ホスフィン、トリメチルホスファイトトリエチルホスファイト、トリブチルホスファイト等のトリアルキルホスファイト、トリフェニルホスファイト等のトリアリールホスファイト等が挙げられる。
これらは単独でも、あるいは2種以上を適宜組み合わせて使用することもできる。
【0061】
これらの中でも、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、プロピオン酸リチウム、プロピオン酸ナトリウム、プロピオン酸カリウム、アクリル酸リチウム、アクリル酸ナトリウム、アクリル酸カリウム、メタクリル酸リチウム、メタクリル酸ナトリウム、メタクリル酸カリウム、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、ピリジン、フェナントロリン、トリメチルホスフィン、トリエチルホスフィン、トリフェニルホスフィンおよびトリス(2−メトキシフェニル)ホスフィンが好適に用いられる。特に、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、トリメチルホスフィンおよびトリフェニルホスフィンがより好適に用いられる。
【0062】
上記塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の添加総量は、用いる原料の種類にもよるが、前記一般式(1)で表される化合物1モルに対して、好ましくは0.001〜3モル、より好ましくは0.01〜2モル、さらに好ましくは0.05〜1.8モル、特に好ましくは0.1〜1.5モルの範囲内であるのがよい。この範囲の量で添加することが、収率の点および経済性の点で好ましい。
また、上記塩基性化合物、窒素含有化合物およびリン含有化合物から選ばれる化合物は、触媒製造時に触媒に担持されてもよいし、触媒製造後反応の前に反応系内に添加されてもよいし、反応の間に追加添加されてもよい。
【0063】
本発明のアシルオキシ化合物の製造方法では、特に溶剤を使用する必要は無いが、有機溶剤を使用することもできる。有機溶剤としては、例えば、ベンゼン等の芳香族炭化水素類;ペンタン、ヘキサン、シクロヘキサン、ヘプタン等の脂肪族炭化水素類;ジエチルエーテル、ジイソプロピルエーテル等のエーテル類;クロロホルム、塩化メチレン、ジクロロエタン、クロロベンゼン等のハロゲン化炭化水素類;酢酸メチル、酢酸エチル、プロピオン酸メチル、プロピオン酸エチル、(メタ)アクリル酸メチルおよび(メタ)アクリル酸エチル等のカルボン酸エステル類等が挙げられる。
上記有機溶剤の使用量は、用いる原料にもよるが、原料の総量の0〜200質量%、好ましくは0〜100質量%、さらに好ましくは0〜80質量%、特に好ましくは0〜70質量%の範囲内であってよい。この範囲の量で使用することが、収率の点および経済性の点で好ましい。
【0064】
また、本発明のアシルオキシ化合物の製造方法において、原料である前記一般式(1)で表される化合物および得られるアシルオキシ化生成物が重合性化合物である場合、重合禁止剤の存在下で反応させることが、これらの化合物の重合を抑制し、収率を向上させる点で好ましい。かかる重合禁止剤としては、ヒドロキノン、メトキシヒドロキノン、ベンゾキノン、p−tert−ブチルカテコール等のキノン系重合禁止剤;2,6−ジ−tert−ブチルフェノール、2,4−ジ−tert−ブチルフェノール、2−tert−ブチル−4,6−ジメチルフェノール、2,6―ジ―tert−ブチル−4−メチルフェノール、2,4,6−トリ−tert−ブチルフェノール等のアルキルフェノール系重合禁止剤;アルキル化ジフェニルアミン、N,N’−ジフェニル−p−フェニレンジアミン、フェノチアジン等のアミン系重合禁止剤;ジメチルジチオカルバミン酸銅、ジエチルジチオカルバミン酸銅、ジブチルジチオカルバミン酸銅等のジチオカルバミン酸銅系重合禁止剤等が挙げられる。これらは単独でも、あるいは2種以上を適宜組み合わせて使用することができる。これらのなかでも、キノン系重合禁止剤、特にヒドロキノン、メトキシヒドロキノン、ベンゾキノン、p−tert−ブチルカテコールおよびフェノチアジンが好適に用いられる。
【0065】
上記重合禁止剤の添加量は、用いる前記一般式(1)で表される化合物の種類にもよるが、好ましくは該重合性化合物の0.001〜5質量%、より好ましくは0.005〜1質量%、特に好ましくは0.01〜0.1質量%の範囲内となる量であってよい。この範囲の量で添加することが、重合の抑制および経済性の点で好ましい。
反応温度は、0〜500℃の範囲内が好ましく、30〜300℃の範囲内が特に好ましい。反応時間は、上記反応が完結するように原料、触媒および有機溶剤の種類や組み合わせ、使用量等に応じて適宜設定すればよい。反応圧力は、原料と反応温度の組み合わせにもよるが、常圧(大気圧)、加圧の何れであってもよい。また、反応方式としては、例えば、回分式、半回分式、連続式等がある。
【0066】
本発明の方法にしたがって製造されたアシルオキシ化合物化生成物は、使用した前記触媒を分離した後、反応溶液を精製することによって得ることができる。精製手段は特に限定されるものではないが、蒸留法、抽出法、カラムクロマト法等によって分離、精製を行うことができる。これらの方法を組み合わせて実施してもよい。これらの内では、蒸留法おとび抽出法が特に好ましい。
上記精製工程で分離された原料および有機溶剤を、再び反応に用いることができる。また、分離した触媒も、再び反応に用いることができる。
【実施例】
【0067】
以下、実施例により、本発明をさらに具体的に説明するが、本発明はこれらにより何ら限定されるものではない。
アシルオキシ化反応用触媒の製造
【0068】
実施例1 触媒A−1の製造
シリカ球体担体(球体直径5mm、比表面積160m2/g、吸水率0.75g/g、上海海源化工科技有限公司のHSV−I)を用いて、触媒A−1を次の手順で製造した。
【0069】
工程1.担体23g(吸水量19.7g)に、56質量%Na2PdCl4水溶液1.5gおよび17質量%HAuCl4水溶液1.5g、20質量%FeCl3・6H2O水溶液0.7gを含む担体吸水量相当の水溶液を含浸し、触媒前駆体を得た。
工程2.工程1で得られた担体触媒前駆体をNa2SiO3・9H2O3gを含む水溶液に浸漬し、室温で20時間静置した。
【0070】
工程3.工程2の水溶液に53質量%ヒドラジン水和物水溶液4mlを加え、静かに混合し、室温で4時間静置させた。この還元処理後の触媒前駆体を塩化物イオンが無くなるまで流水で洗浄した。洗浄した触媒前駆体を、約110℃で4時間乾燥した。
工程4.工程3で得られた触媒前駆体を1質量%硫酸水溶液2Lに浸漬し、一晩静置した。この後、一晩流水で水洗し、110℃で4時間乾燥した。
【0071】
工程5.工程4で得られた酸処理後の触媒前駆体を、2gの酢酸カリウムを溶かした担体吸水量相当水溶液に含浸し、110℃で4時間乾燥した。
Na2PdCl4水溶液、HAuCl4水溶液、および、FeCl3・6H2O水溶液の沈澱開始pHを測定した結果、それぞれ、4.5、4.0、2.5であった。
【0072】
実施例2 触媒A−2の製造
工程4を行わなかった以外は実施例1と同じ操作を行い、触媒A−2を製造した。
実施例3 触媒B−1の製造
工程1で用いたFeCl3・6H2O水溶液をTiCl4水溶液とした以外は実施例1と同じ操作を行い、触媒B−1を製造した。TiCl4水溶液の沈澱開始pHを測定した結果、1.5であった。
【0073】
実施例4 触媒B−2の製造
工程4を行わなかった以外は実施例3と同じ操作を行い、触媒B−2を製造した。
実施例5 触媒C−1の製造
工程1で用いたFeCl3・6H2O水溶液をScCl3・6H2O水溶液とし、工程4の1質量%硫酸水溶液2Lを1質量%リン酸水溶液200mlとした以外は実施例1と同じ操作を行い、触媒C−1を製造した。ScCl3・6H2O水溶液の沈澱開始pHを測定した結果、4.0であった。
【0074】
実施例6 触媒G−1の製造
工程1のHAuCl4水溶液の添加量を3.0gとし、FeCl3・6H2O水溶液の添加量を1.4gとした以外は実施例2と同じ操作を行い、触媒G−1を製造した。
【0075】
比較例1 触媒D−1の製造
工程1で用いたFeCl3・6H2O水溶液を添加しなかったこと以外は実施例2と同じ操作を行い、触媒D−1を製造した。
比較例2 触媒E−1の製造
工程1で用いたFeCl3・6H2O水溶液をZnCl2水溶液とした以外は実施例1と同じ操作を行い、触媒E−1を製造した。ZnCl2水溶液の沈澱開始pHを測定した結果、7.0であった。
【0076】
比較例3 触媒E−2の製造
工程4を行わなかった以外は比較例2と同じ操作を行い、触媒E−2を製造した。
比較例4 触媒F−1の製造
工程1で用いられたFeCl3・6H2O水溶液をBaCl2・2H2O水溶液とした以外は実施例2と同じ操作を行い、触媒F−1を製造した。BaCl2・2H2O水溶液の沈澱開始pHを測定した結果、11.0であった。
【0077】
触媒の評価
以下の方法により、上記で得られた各触媒の反応評価を行った。
【0078】
触媒活性評価
触媒3ccをガラスビーズ75ccで希釈して反応管(SUS316L製、内径22mm、長さ480mm)に充填した。反応温度150℃、反応圧力0.6MPaG、ガス組成C2H4/O2/H2O/HOAc/N2=47.3/6.1/5.6/26.3/14.7(mol%)のガスを流量20nL/hで流通させ、反応を行った。
反応開始後2hから4h反応後までの間生成ガスおよび生成液を採取し、4h反応サンプルとした。また、反応開始後96hから98h反応後までの間生成ガスおよび生成液を採取し、98h反応サンプルとした。次いで、以下の分析を行い、生成酢酸ビニルの活性および選択率を算出した。
【0079】
反応器出口ガスの分析を、以下の方法を用いて行った。
1.酸素
絶対検量線法を用い、流出ガスを50ml採取し、ガスクロマトグラフィーに付属する1mlのガスサンプラーに全量流し、以下の条件で分析を行った。
ガスクロマトグラフィー:島津ガスクロマトグラフ用ガスサンプラ−(MGS−4:計量管1ml)付ガスクロマトグラフィー(島津製作所製GC−14(B)
カラム:MS−5A IS 60/80mesh(3mmΦ×3m)
キャリアーガス:ヘリウム(流量20ml/min.)
温度条件:検出器温度、気化室温度が110℃、カラム温度は70℃一定
検出器:TCD(He圧70kPaG、Current100m(A)
【0080】
2.酢酸
内部標準法を用い、反応液10mlに対し、内部標準として1,4−ジオキサンを1ml添加したものを分析液として、そのうちの0.2μlを注入して以下の条件で分析を行った。
ガスクロマトグラフィー:島津製作所製GC−14B
カラム:パックドカラムThermon 3000(長さ3m、内径0.3mm)
キャリアーガス:窒素(流量20ml/min.)
温度条件:検出器温度、気化室温度が180℃、カラム温度は分析開始から6分間は50℃保持、その後10℃/minの昇温速度で150℃まで昇温し、150℃で10分間保持
検出器:FID(H2 圧40kPaG、空気圧100kPaG)
【0081】
3.酢酸ビニル
内部標準法を用い、反応液6gに対し、内部標準として酢酸n−プロピルを1g添加したものを分析液として、そのうちの0.3μlを注入して以下の条件で分析した。
ガスクロマトグラフィー:島津製作所製GC−9A
カラム:キャピラリーカラムTC−WAX(長さ30m、内径0.25mm、膜厚0.5μm)
キャリアーガス:窒素(流量30ml/min.)
温度条件:検出器温度、気化室温度が200℃、カラム温度は分析開始から2分間は45℃保持、その後4℃/minの昇温速度で130℃まで昇温し、130℃で15分間保持し、その後25℃/minの昇温速度で200℃まで昇温し、200℃で10分間保持
検出器:FID(H2 圧60kPaG、空気圧100kPaG)
【0082】
各触媒(実施例1〜6および比較例1〜4)の反応評価結果を表1に示す。
表中の4h活性は、各種触媒を用いて、先述の条件で4h反応させた結果生成した酢酸ビニル量より算出した活性を、比較例D−1の活性で割った値を示している。
選択率は4h反応後の分析で算出された酢酸ビニルの選択率を示している。活性低下は98h反応後の活性を4h反応後の活性で割った値を示しており、これを活性低下の指標とした。
【0083】
尚、選択率および活性の定義は以下の通りである。
選択率(%)=(反応後の全物質量中の酢酸ビニルの量(mol))/(反応後の全物質量(mol))
活性(g/L/h)=(単位時間当たりの酢酸ビニルの生成量(g/h))/(触媒の体積(L))
【0084】
また、各触媒の製造(実施例1〜6および比較例1〜4)における各元素の担持量を表2に示す。表2において、実施例1、3、5および比較例2に対する値は酸洗浄前のものである。
【0085】
【表1】

【0086】
【表2】
【産業上の利用可能性】
【0087】
本発明は、反応初期の活性、選択性および活性の持続性の性能バランスに優れたアシルオキシ化反応用触媒を提供することができるので、産業上有用である。
【図面の簡単な説明】
【0088】
【図1】図1は、本発明で規定する沈殿開始pHの測定方法を説明するためのグラフである。
【技術分野】
【0001】
本発明は、アシルオキシ化反応用触媒の製造方法、それにより得られるアシルオキシ化反応用触媒および該触媒を用いたアシルオキシ化反応方法に関する。
アシルオキシ化反応は、香料や医農薬原料として、有機合成中間体として、さらに重合性材料として用いられる種々の有用な化合物の合成反応である。
【背景技術】
【0002】
トルエン、キシレン等のベンジル位に水素原子を有する化合物またはプロピレン、シクロヘキセン等のアリル位に水素原子を有する化合物と、酢酸等のカルボン酸および酸素とによるアシルオキシ化反応は、従来より公知である。
かかるアシルオキシ化反応用の触媒として、酢酸パラジウム等の均一系触媒およびパラジウムをシリカ等の担体に担持した不均一系触媒が開発されている。
【0003】
特開2001−269577号公報には、活性炭などの担体に、パラジウムなどの周期律表第8〜11族の金属、ナトリウムやカリウムなどのアルカリ金属および/またはアンチモン、ビスマス、テルルなどの周期律表第12〜16族の金属を担持した触媒を用いることによりアシルオキシ化合物を経済的に製造する方法が開示されている。
【0004】
特表2001−521817号公報には、パラジウムおよび金に加えて、マグネシウム、カルシウム、バリウム、ジルコニウム、セリウムなどの第三金属をその酸化物またはその酸化物とその金属との混合物として担体中に含む触媒が開示され、またこれらのパラジウム、金および第三金属の水溶性塩の溶液で担体を含浸し、その後アルカリ性化合物との反応により非水溶性化合物として固定し、そして固定されたパラジウムおよび金をそれらの金属状態に、また第三金属をその酸化物またはその酸化物と金属の混合物に還元する工程が開示されている。
【0005】
また、特開2005−296858号公報には、パラジウム、金、亜鉛などの両性金属およびアルカリ金属を含有する触媒が開示され、亜鉛を添加することにより液相還元法によった場合にも気相還元法による場合に匹敵するほどの貴金属表面積が得られるということが報告されている。
【0006】
【特許文献1】特開2001−269577号公報
【特許文献2】特表2001−521817号公報
【特許文献3】特開2005−296858号公報
【発明の開示】
【発明が解決しようとする課題】
【0007】
しかしながら、上記した従来のアシルオキシ化反応用触媒は、反応初期の活性、選択性および活性の持続性の性能バランスが不十分である等の問題を有していた。
【0008】
本発明は、上記のごとき状況に鑑みてなされたものであり、上記の問題を解決し、アシルオキシ化合物を経済的に製造するための触媒およびその製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0009】
本願発明者らは、上記課題を解決するため鋭意検討を重ねた結果、少なくとも周期律表の第8、9、10または11族の元素を含む第一成分、前記第一成分の元素とは異なる元素を含む第二成分および前記第一成分および第二成分の元素とは異なる元素を含む第三成分を担体に担持させることにより、アシルオキシ化反応を効率よく行うことができ、アシルオキシ化生成物を経済的に製造することができる触媒が得られることを見出し、この知見に基づき本発明を完成するに至ったものである。
【0010】
よって、本発明は、例えば、以下の(1)〜(14)からなる。
(1)(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とをいっしょに(d)担体に担持させる工程を含むことを特徴とするアシルオキシ化反応用触媒の製造方法。
【0011】
(2)前記(d)担体に担持させる工程に次いで、さらに非水溶化処理を行うことを特徴とする前記(1)に記載のアシルオキシ化反応用触媒の製造方法。
(3)前記(d)担体に担持させる工程に次いで、さらに還元処理を行うことを特徴とする前記(1)または(2)に記載のアシルオキシ化反応用触媒の製造方法。
【0012】
(4)前記還元処理に次いで、酸および/またはキレート剤を接触させることを特徴とする前記(3)に記載のアシルオキシ化反応用触媒の製造方法。
(5)さらに(e)周期律表の第1および2族の元素(水素を除く)の少なくとも1種を含む第四成分を添加することを特徴とする前記(1)〜(4)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0013】
(6)前記(a)第一成分がパラジウムを含むことを特徴とする前記(1)〜(5)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(7)前記(b)第二成分が周期律表の第11族元素を含むことを特徴とする前記(1)〜(6)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0014】
(8)前記(b)第二成分が金および銅から選ばれる少なくとも1種を含むことを特徴とする前記(7)に記載のアシルオキシ化反応用触媒の製造方法。
(9)前記(c)第三成分が周期律表の第3〜13族元素の1種を含むことを特徴とする前記(1)〜(8)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【0015】
(10)前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比が、0.4〜1.5であることを特徴とする前記(1)〜(9)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(11)前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比が、0.1〜0.5であることを特徴とする前記(1)〜(10)のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
(12)前記(1)〜(11)のいずれかに記載した方法により得られるアシルオキシ化反応用触媒。
【0016】
(13)前記(1)〜(11)のいずれかに記載した方法により得られる触媒の存在下に、一般式(1):CHR1R2−X(式中、R1およびR2は、それぞれ独立して、水素原子または有機残基を表し、Xは置換基を有していてもよい芳香族炭化水素残基または置換基を有していてもよいオレフィン残基を表す)で表される化合物またはエチレンと、一般式(2):R3−COOH(式中、R3は水素原子または有機残基を表す)で表されるカルボン酸類とおよび酸素とを反応させて、一般式(3):R3−COO−CR1R2−X(式中、R1、R2、R3およびXは前記規定に同一のものを表す)で表される化合物または酢酸ビニルを生成させることを特徴とするアシルオキシ化合物の製造方法。
【0017】
(14)前記反応が、さらに塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の存在下に行われることを特徴とする前記(13)に記載のアシルオキシ化合物の製造方法。
【発明の効果】
【0018】
本発明によれば、反応初期の活性、選択性および活性の持続性の性能バランスに優れたアシルオキシ化反応用触媒を得ることができ、それによって効率的かつ経済的にアシルオキシ化合物を製造することができる。
【発明を実施するための最良の形態】
【0019】
以下に本発明の好ましい実施の形態について説明するが、本発明はこれらの形態のみに限定されるものではなく、その精神と実施の範囲内において様々な変形が可能であることを理解されたい。
本発明のアシルオキシ化反応用触媒は、担体に担持物質が担持されてなるアシルオキシ化反応用触媒である。
【0020】
本発明において、(a)第一成分および(b)第二成分の元素とは、IUPAC無機化学命名法改訂版(1989)による周期律表の第8〜11族の元素を指し、例えば、鉄、ルテニウム、オスミウム、コバルト、ロジウム、イリジウム、ニッケル、パラジウム、白金、銅、銀および金が挙げられる。これらのうちでは、(a)第一成分の元素としてはルテニウム、ロジウム、パラジウムまたは銀が好ましく、パラジウムがさらに好ましく、(b)第二成分の元素としてはオスミウム、イリジウム、白金、銅または金が好ましく、金がさらに好ましい。(a)第一成分は反応に欠かせない主触媒に関わる元素を含み、また(b)第二成分は反応効率を高める助触媒に関わる元素を含む。(a)第一成分および(b)第二成分の元素はそれぞれ1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0021】
(a)第一成分および(b)第二成分としては、パラジウム元素を例に挙げれば、パラジウム金属、ヘキサクロロパラジウム酸アンモニウム、ヘキサクロロパラジウム酸カリウム、ヘキサクロロパラジウム酸ナトリウム、テトラクロロパラジウム酸アンモニウム、テトラクロロパラジウム酸カリウム、テトラクロロパラジウム酸ナトリウム、テトラブロモパラジウム酸カリウム、酸化パラジウム、塩化パラジウム、臭化パラジウム、ヨウ化パラジウム、硝酸パラジウム、硫酸パラジウム、酢酸パラジウム、ジニトロサルファイトパラジウム酸カリウム、クロロカルボニルパラジウム、ジニトロジアンミンパラジウム、テトラアンミンパラジウム塩化物、テトラアンミンパラジウム硝酸塩、テトラアンミンパラジウム水酸化物、cis−ジクロロジアンミンパラジウム、trans−ジクロロジアンミンパラジウム、ジクロロ(エチレンジアミン)パラジウム、テトラシアノパラジウム酸カリウム等を挙げることができる。
【0022】
(a)第一成分および(b)第二成分の元素の担持量は、完成された触媒に対して、それぞれ、好ましくは0.01〜20質量%、より好ましくは0.1〜10質量%である。この担持量は、完成された触媒を酸に溶解して、測定対象の元素の含有量をICP分析により求めることができる。
【0023】
(a)第一成分、(b)第二成分および(c)第三成分は、それぞれ、例えば、水溶液の状態で(d)担体に含浸させて担持させることができる。この場合、(a)第一成分、(b)第二成分および(c)第三成分をいっしょに1つの水溶液中に含有させ、この水溶液を(d)担体に含浸させて担持させることが好ましい。
【0024】
本発明における沈澱開始pHは、次の方法で決定することができる。すなわち、各種金属または金属化合物を酸性水溶液に溶解した後、アルカリ性水溶液を滴下することでpHが上昇していき、ある領域でpH上昇が鈍化するが、その鈍化の開始点のpHを沈澱開始pHと定義する。例えば、塩化金酸水溶液に水酸化ナトリウム水溶液を滴下しながら水溶液のpHを測定し、その滴下量に対するpHをプロットすることにより、図1に示すような滴定曲線が描かれる。ある量を滴下すると図中のA域のように水酸化ナトリウム水溶液を滴下してもpHがあまり上昇しないような比較的安定した状態が確認される。この安定状態の開始点(B点)のpHが沈澱開始pHである。この測定における注意点は、沈殿開始前に当該金属および金属化合物が溶解して均一な水溶液を形成している状態で滴定を開始する必要がある点にある。
【0025】
なお、この滴定に用いられるアルカリ性水溶液は、各種酸性水溶液のpH変化が確認できるアルカリ性水溶液であれば、その化学種、濃度や滴下量などは特に制限されない。ただし、一連の比較測定においては、同一のアルカリ水溶液を用いて各酸性水溶液の滴定を行う必要がある。この滴定には、例えば、水酸化ナトリウム水溶液が好適に用いられる。
【0026】
こうして測定された沈澱開始pHをもとに、(a)第一成分および(b)第二成分の沈澱開始pH以下の沈殿開始pHを与える成分であって、第一成分および第二成分の元素とは異なる元素を含む成分を(c)第三成分として選択することができる。例えば、パラジウム元素を含む(a)第一成分よりも沈澱開始pHが低い第三成分としては、スカンジウム、チタン、モリブデン、タングステンまたは鉄を含む化合物またはそれらの単体を挙げることができる。化合物としては、酸化物、水酸化物、ハロゲン化物、オキシハロゲン化物、アルコキシド、酢酸塩等の脂肪族カルボン酸塩、硝酸塩、炭酸塩、リン酸塩、ホウ酸塩等とそれらの水和物が挙げられる。鉄を例に挙げれば、例えば、鉄金属、水酸化鉄、塩化鉄、臭化鉄、ヨウ化鉄、過塩素酸鉄、鉄メトキシド、鉄エトキシド、酢酸鉄、プロピオン酸鉄、アクリル酸鉄、硝酸鉄、炭酸鉄、硫酸鉄、リン酸鉄、酢酸鉄、くえん酸鉄、グルコン酸鉄、フマル酸鉄、シュウ酸鉄、ホウ酸鉄等とそれらの水和物である。また、(c)第三成分としては、これらのうちでスカンジウム、チタンおよび鉄から選ばれる少なくとも1種を含むものが好ましい。(c)第三成分は1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0027】
上記の方法で選定されるc)第三成分を添加することにより、(a)第一成分と(b)第二成分との共存化もしくは均一混合化が促され、より高い活性、選択性および活性低下抑制効果が得られるのであると考えられる。
【0028】
本発明の触媒において、(c)第三成分の元素の担持量は、完成された触媒に対して、好ましくは0.001〜20質量%、より好ましくは0.01〜10質量%である。この担持量は、前述の(a)第一成分や(b)第二成分の元素の場合と同様に、完成された触媒を酸に溶解して、測定対象の元素の含有量をICP分析により求めることができる。
【0029】
前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比は、0.4〜1.5であるのが好ましく、0.8〜1.2であるのがさらに好ましい。また、前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比は、0.1〜0.5であるのが好ましく、0.2〜0.4であるのがさらに好ましい。
なお、本発明の触媒の製造方法においては酸洗浄の工程を行ってもよく、それによって担持量が低下することがあるが、上記担持量は酸洗浄前の値である。
【0030】
本発明の触媒の(d)担体としては、例えば、シリカ、アルミナ、ジルコニア、チタニア等の金属酸化物、炭素、木炭、活性炭、アスベスト、シリカ−アルミナ、ゼオライト、オルガノゾルゲル、イオン交換樹脂、クレー、炭酸塩、炭酸塩等が挙げられる。これらのうちでは、シリカ、アルミナ、ジルコニア、チタニア等の金属酸化物、活性炭およびゼオライトが好ましい。
担体の比表面積は特に制限されるものではないが、触媒成分の分散化と担体の機械的強度とのバランスの観点からは、B.E.T.法で測定した値が10〜1500m2/gであることが好ましく、100〜500m2/gであることがさらに好ましい。
【0031】
前記活性炭の原料は、木質、ヤシ殻、もみ殻、有機高分子等が挙げられる。これらのうちでも、木質およびヤシ殻が好ましい。担体として用いられる活性炭の性質としては、比表面積が800〜2500m2/gの範囲、特に1000〜1800m2/gの範囲であるのが、触媒成分の分散化と担体の機械的強度とのバランスの観点から好ましい。
担体の形態としては、例えば、固定床方式、流動床方式、懸濁触媒方式等の実施する反応の形態等により、粉末状、破砕状、粒子状、柱状等を適宜選ぶことができる。
【0032】
(a)第一成分、(b)第二成分および(c)第三成分を(d)担体に沈積させるために非水溶化処理を行うことが好ましい。
非水溶化処理とは、(a)第一成分、(b)第二成分および(c)第三成分を溶解した水溶液を(d)担体に含浸させた触媒前駆体を、酸性あるいはアルカリ性水溶液に接触させ、(a)第一成分、(b)第二成分および(c)第三成分を沈澱させる工程をいう。ここで、触媒前駆体とは、担体に担持物質を接触あるいは担持させた時点から各種工程を経て最終の触媒として完成するまでの途中の中間状態の触媒を指す。(a)第一成分、(b)第二成分および(c)第三成分を溶解した水溶液が酸性であればアルカリ性水溶液と接触させ、アルカリ性であれば酸性水溶液と接触させる。本発明においては、主にアルカリ性水溶液と接触させる。
【0033】
酸性水溶液に使用される酸としては、特に限定されるものではないが、例えば、塩酸、硫酸、硝酸、ヘテロポリ酸などの無機酸や、酢酸、リン酸、シュウ酸、クエン酸、グルコン酸などの有機酸が挙げられる。
【0034】
アルカリ性水溶液としては、例えば、アルカリ金属やアルカリ土類金属の水酸化物、アルカリ金属やアルカリ土類金属の重炭酸塩、アルカリ金属やアルカリ土類金属の炭酸塩、アルカリ金属やアルカリ土類金属のケイ酸塩等のアルカリ性化合物の水溶液が挙げられる。好ましいアルカリ金属としては、リチウム、ナトリウム、カリウムなどがある。好ましいアルカリ土類金属としては、バリウム、マグネシウムなどがある。特に好適には、メタケイ酸ナトリウム、メタケイ酸カリウム、水酸化ナトリウム、水酸化カリウム、水酸化バリウムなどが用いられる。
【0035】
非水溶化処理の方法としては、例えば、前記触媒前駆体を酸性水溶液またはアルカリ水溶液に浸漬するか、または前記触媒前駆体に酸性水溶液またはアルカリ水溶液を滴下することにより、両者を接触させる方法を挙げることができる。
非水溶化処理で使用される酸性水溶液またはアルカリ水溶液の使用量および水溶液の濃度は特に限定されない。ただし、処理後の液相のpHが7〜11の範囲となるように、調整することが好ましい。
【0036】
非水溶化処理のための酸性水溶液またはアルカリ水溶液との接触時間は、0.5〜100時間、特に3〜50時間が好ましい。0.5時間未満では非水溶化が十分に行えない場合がある。また、100時間を超えると、洗浄液の種類や担体の種類によっては(d)担体がダメージを受けたり、(a)第一成分、(b)第二成分の再溶解が進むため好ましくない。
接触温度は、10〜80℃、特に20〜60℃が好ましい。10℃より低いと反応が遅く、処理時間の延長につながるため好ましくない。80℃を超えると(a)第一成分、(b)第二成分の凝集が進む恐れがあるため好ましくない。
【0037】
本発明において、(a)第一成分、(b)第二成分および(c)第三成分の担持工程の後、もしくは前記非水溶化処理の工程の後に、特に(a)第一成分、(b)第二成分の溶出を防ぐために還元処理を行うことが好ましい。
例えば、液相還元では、アルコールや炭化水素類を用いた非水系、水系溶液が用いられる。還元剤として、カルボン酸およびその塩、アルデヒド、過酸化水素、糖類、ジボラン、アミン、ヒドラジンなどが用いられる。具体的には、シュウ酸、シュウ酸カリウム、ギ酸、ギ酸カリウム、クエン酸アンモニウム、グルコース、多価フェノール、ヒドラジン、ホルムアルデヒド、アセトアルデヒド、ハイドロキノン、水素化ホウ素ナトリウム、クエン酸カリウムなどが挙げられる。特に好ましくはヒドラジンが用いられる。
【0038】
気相還元では、水素、一酸化炭素、アルコール、アルデヒド、エチレン、プロペン、およびイソブテンなどのオレフィンが還元ガスとして用いられる。好ましくは水素が用いられる。気相還元では、希釈剤として不活性ガスを用いてもよい。不活性ガスとしては、例えば、ヘリウム、アルゴン、窒素などがある。
【0039】
前記(c)第三成分を除去したほうが触媒の性能が向上する場合がある。除去する場合は、例えば、還元処理後に洗浄を行うことで除去することが好ましい。(c)第三成分の洗浄除去は、例えば、酸やキレート剤などと接触させて溶解除去することにより、行うことができる。
前記酸としては、例えば、塩酸、硫酸、硝酸、ヘテロポリ酸などの無機酸や、酢酸、リン酸、シュウ酸、クエン酸、グルコン酸などの有機酸が挙げられる。
【0040】
(c)第三成分の洗浄除去には、上記の酸以外に、以下のようなキレート剤を使用しても良い。また、酸処理を含めた複数の処理を併用しても良い。
ここで、キレート剤とは、金属イオンに配位結合することができる電子供与体(配位子)を有する化合物をいい、金属イオンと反応することにより金属錯塩あるいは金属キレート化合物を形成する物質をいう。
【0041】
有用なキレート剤としては、ニトリロ三酢酸、ジエチレントリアミン五酢酸、ヒドロキシエチルエチレンジアミン三酢酸、トリエチレンテトラアミン六酢酸、1,3−プロパンジアミン四酢酸、1,3−ジアミノ−2−ヒドロキシプロパン四酢酸、ヒドロキシルイミノ二酢酸、ジヒドロキシルグリシン、グリコールエーテルジアミン四酢酸、L−グルタミン酸二酢酸などが挙げられる。
また、洗浄に使用するため、キレート剤を水に溶かして水溶液として用いても良い。多くの場合、水酸化ナトリウム水溶液やアンモニア水等のアルカリ性水溶液に容易に溶けるが、アルコールなどの有機溶剤に溶かしてもよい。
【0042】
(c)第三成分の洗浄除去方法に制限はないが、一般には、触媒前駆体を洗浄液に浸漬させて行うのがよい。浸漬時間は、0.5〜100時間、特に3〜50時間が好ましい。0.5時間未満では洗浄が十分に行えないことがある。また、100時間を超えると、洗浄液の種類や担体の種類によっては(d)担体がダメージを受けたり、(a)第一成分、(b)第二成分の再溶解が進むことがあり、好ましくない。
接触温度は、特に制限はないが、10〜80℃、特に20〜60℃が好ましい。10℃より低いと反応が遅く、処理時間の延長につながることがあるため好ましくない。80℃を超えると(a)第一成分、(b)第二成分)の凝集が進む恐れがあり、好ましくない。
【0043】
前記触媒には、(e)周期律表第1および/または2族の元素(水素を除く)を含む第四成分をさらに追加添加することが好ましい。この添加は、触媒製造時に担持することにより行ってもよいし、触媒製造後反応への使用前に反応系内に添加してもよいし、反応の間に追加添加してもよい。
(e)第四成分を、本発明のアシルオキシ化反応用触媒に担持し、あるいは反応前または反応の間に反応系内に添加することにより、アシルオキシ化反応における転化率、選択率が向上し、アシルオキシ化生成物を経済的に製造することができる場合がある。
【0044】
(e)第四成分の周期律表第1および/または2族の元素としては、例えば、リチウム、ナトリウム、カリウム、ルビジウム、セシウム、フランシウム、ベリリウム、マグネシウム、カルシウム、ストロンチウムおよびバリウムが挙げられる。これらの内では、リチウム、ナトリウム、カリウム、セシウムが好ましい。前記金属原子は1種類でも良いし、2種類以上を適宜組み合わせて使用しても良い。
【0045】
(e)第四成分を与える原料としては、具体的には、周期律表第1および2族の元素の金属、酸化物、水酸化物、ハロゲン化物、オキシハロゲン化物、アルコキシド、酢酸塩等の脂肪族カルボン酸塩、硝酸塩、炭酸塩、リン酸塩、ホウ酸塩等が挙げられる。カリウムを例に挙げれば、カリウム金属、水酸化カリウム、塩化カリウム、臭化カリウム、ヨウ化カリウム、硫化カリウム、酢酸カリウム、硝酸カリウム、安息香酸カリウム、炭酸カリウム、リン酸カリウム、ホウ酸カリウム等を挙げることができる。
【0046】
(e)第四成分に含まれる上記元素の担持量は、完成された触媒に対して、好ましくは0.001〜40質量%、より好ましくは0.01〜10質量%である。この担持量は、前述の(a)第一成分や(b)第二成分の元素の場合と同様に、完成した触媒を酸に溶かし、測定対象の元素の含有量をICP分析により求めることができる。この担持量の範囲が転化率および選択率の点および経済性の点で好ましい。
(e)第四成分の担持は、例えば、含浸法、イオン交換法、共沈法、沈着法、混練法で行うことができるが、(e)第四成分を含む水溶液を担体に含浸させて担持するのが好ましい。
【0047】
本発明の触媒は、特に、トルエン、キシレン等のベンジル位に水素原子を有する化合物またはエチレン、プロピレン、シクロヘキセン等のビニル位もしくはアリル位に水素原子を有する化合物と、酢酸等のカルボン酸および酸素とによるアシルオキシ化反応に好適である。すなわち、本発明におけるアシルオキシ化反応は、前記一般式(1)で表される化合物またはエチレンと、前記一般式(2)で表されるカルボン酸類および酸素を反応させることによる、前記一般式(3)で表される化合物または酢酸ビニルを製造する反応である。
【0048】
反応原料として用いられる一般式(1)で表される化合物において、式中のR1、R2で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、またはアリール基が挙げられる。これらの内では、炭素数1〜10の飽和および/または不飽和アルキル基が好適に用いられる。
【0049】
Xで表される置換基を有していてもよい芳香族炭化水素残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状のアルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシ基、置換基を有していてもよいフェノキシ基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、水酸基、またはフッ素、塩素、臭素、ヨウ素等のハロゲン原子等の置換基を有していてもよいフェニル基である。
【0050】
また、Xで示される置換基を有していてもよいオレフィン残基としては、一般式(4):−CR4=CHR5(式中、R4、R5は、水素原子または有機残基を表す)で表されるものが挙げられる。ここで、R4、R5で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、アルデヒド基、脂肪族および/または芳香族ケトン基、カルボン酸基、脂肪族および/または芳香族カルボン酸エステル基がある。これらの内では、炭素数1〜10の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、カルボン酸基、脂肪族および/または芳香族カルボン酸エステル基が好適に用いられる。また、R1またはR2とR4またはR5とにより環を形成していても良い。
【0051】
一般式(1)で表される化合物の代表例としては、メタクリル酸、メタクリル酸メチル、メタクリル酸エチル、メタクリル酸プロピル、メタクリル酸ブチル、メタクリル酸2−ヒドロキシエチルおよびメタクリル酸2−エチルヘキシル、エチレン、プロピレン、ブテン、ペンテン、ヘキセン、ヘプテン、ノネン、デセン、ブタジエン、シクロペンテン、シクロペンタジエン、シクロヘキセン、シクロヘキサジエン、シクロヘプテン、シクロオクテン、シクロノネン、シクロデセン、トルエン、エチルベンゼン、プロピルベンゼン、ブチルベンゼン、スチレン、キシレン、トリメチルベンゼン、テトラメチルベンゼン、ペンタメチルベンゼン、ヘキサメチルベンゼン、メチルビフェニル、ジメチルビフェイル、ジフェニルメタン、トリフェニルメタン、メチルフェノール、メトキシトルエン、エトキシトルエン、フェノキチトルエン等が挙げられる。これらの内で、異性体を含むものは、各異性体単独および/または各異性体混合物でもよい。これらの内では、メタクリル酸メチル、メタクリル酸ブチル、エチレン、プロピレン、ブテン、ペンテン、ヘキセン、ブタジエン、シクロペンテン、シクロペンタジエン、シクロヘキセン、シクロヘキサジエン、トルエン、キシレン、トリメチルベンゼン、メトキシトルエンおよびフェノキシトルエンが好適に用いられる。
【0052】
本発明のアシルオキシ化合物の製造方法において、原料として用いられる一般式(2)で表されるカルボン酸類は、式中のR3が水素原子または有機残基である化合物であれば、特に限定されるものではない。
R3で表される有機残基としては、例えば、炭素数1〜18の直鎖状、分枝状または環状の飽和および/または不飽和アルキル基、炭素数1〜8のヒドロキシアルキル基、炭素数2〜20のアルコキシアルキル基、炭素数2〜20のアセトキシアルキル基、炭素数1〜8のハロゲン化(例えば、塩素化、臭素化またはフッ素化)アルキル基、置換されていてもよい芳香族基等が挙げられる。これらの内では、炭素数1〜5の飽和および/または不飽和アルキル基が好適に用いられる。
【0053】
一般式(2)で表されるカルボン酸類の代表例としては、ギ酸、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、カプリル酸、ラウリン酸、ミリスチル酸、パルミチン酸、ステアリン酸、アセト酢酸、ヒドロキシプロピオン酸、イソブタン酸、ヒドロキシイソブタン酸、t−ブチル酢酸、安息香酸、アクリル酸、メタクリル酸等が挙げられる。これらの内では、酢酸、プロピオン酸、酪酸、安息香酸、アクリル酸およびメタクリル酸が好ましく、酢酸、プロピオン酸、アクリル酸およびメタクリル酸が特に好ましい。
【0054】
一般式(3)で表される化合物の例としては、例えば、酢酸ビニル、アクリル酸ビニル、メタクリル酸ビニル、プロピオン酸ビニル、酢酸アリル、アクリル酸アリル、メタクリル酸アリル、プロピオン酸アリル、酢酸ベンジル、アクリル酸ベンジル、メタクリル酸ベンジル、プロピオン酸ベンジル、酢酸4−メチルベンジル、アクリル酸4−メチルベンジル、メタクリル酸4−メチルベンジル、プロピオン酸4−メチルベンジル、酢酸シクロヘキセン、アクリル酸シクロヘキセン、メタクリル酸シクロヘキセン、プロピオン酸シクロヘキセン、α−アセトキシメチルアクリル酸メチル、1,4−キシレンモノアセテート、1,4−キシレンジアセテートなどが挙げられる。
【0055】
前記一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類との反応初期におけるモル比は、例えば、10/1〜1/10の範囲内であってよい。上記範囲内でも6/1〜1/6の範囲内がより好ましい。一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類のどちらかを上記範囲内よりも過剰に添加しても、収率の向上および反応時間の短縮等の効果は望めず、過剰な原料の回収工程が長くなり、経済的に不利となる。
【0056】
反応に用いられる酸素は、原子状および/または分子状酸素であり、好ましくは分子状酸素である。また、分子状酸素は、窒素、アルゴン、ヘリウム、二酸化炭素等の不活性な気体との混合気体として用いられるのが好ましい。この場合、酸素の濃度は、反応系内で気体が爆発組成とならない範囲に調整して使用するのがより好ましい。
分子状酸素および分子状酸素を含む混合気体の反応系への供給は、反応系内の液相部または気相部の一方または両方に供給して行うことができる。反応系内へ分子状酸素および分子状酸素を含む混合気体を供給する場合には、酸素分圧が0.01〜20MPaの範囲内となるように供給するのがよい。
【0057】
本発明のアシルオキシ化合物の製造方法は、前記したアシルオキシ化反応を、前記した本発明のアシルオキシ化触媒の存在下で行うものである。
前記触媒の使用量は、用いる前記一般式(1)で表される化合物と一般式(2)で表わされるカルボン酸類の種類や組み合わせにもよるが、前記(a)第一成分が一般式(1)で表される化合物1モルに対して0.01ミリモル〜100モルの範囲内になる量であってよい。この範囲で前記触媒を使用するのが、収率の点および経済性の点で好ましい。
【0058】
また、本発明のアシルオキシ化合物の製造方法において、反応系中に塩基性化合物、窒素含有化合物およびリン含有化合物から選ばれる少なくとも1種の化合物を存在させることが転化率および選択率の点で好ましい。かかる塩基性化合物としては、例えば、ギ酸リチウム、ギ酸ナトリウム、ギ酸カリウム、ギ酸マグネシウム、ギ酸カルシウム、ギ酸バリウム、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、酢酸マグネシウム、酢酸カルシウム、酢酸バリウム、プロピオン酸リチウム、プロピオン酸ナトリウム、プロピオン酸カリウム、プロピオン酸マグネシウム、プロピオン酸カルシウム、プロピオン酸バリウム、アクリル酸リチウム、アクリル酸ナトリウム、アクリル酸カリウム、アクリル酸マグネシウム、アクリル酸カルシウム、アクリル酸バリウム、メタクリル酸リチウム、メタクリル酸ナトリウム、メタクリル酸カリウム、メタクリル酸マグネシウム、メタクリル酸カルシウム、メタクリル酸バリウム、安息香酸リチウム、安息香酸ナトリウム、安息香酸カリウム、安息香酸マグネシウム、安息香酸カルシウム、安息香酸バリウム等のアルカリ金属および/またはアルカリ土類金属カルボン酸塩;水酸化リチウム、水酸化ナトリウム、水酸化カリウム、水酸化マグネシウム、水酸化カルシウム、水酸化バリウム等のアルカリ金属および/またはアルカリ土類金属水酸化物;炭酸リチウム、炭酸ナトリウム、炭酸カリウム、炭酸マグネシウム、炭酸カルシウム、炭酸バリウム等のアルカリ金属および/またはアルカリ土類金属炭酸塩;リン酸リチウム、リン酸ナトリウム、リン酸カリウム、リン酸マグネシウム、リン酸カルシウム、リン酸バリウム等のアルカリ金属および/またはアルカリ土類金属炭酸塩;ほう酸リチウム、ほう酸ナトリウム、ほう酸カリウム、ほう酸マグネシウム、ほう酸カルシウム、ほう酸バリウム等のアルカリ金属および/またはアルカリ土類金属ほう酸塩等が挙げられる。
【0059】
前記窒素含有化合物の具体例としては、気体または水溶液等のアンモニア、メチルアミン、ジメチルアミン、トリメチルアミン、エチルアミン、ジエチルアミン、トリエチルアミン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、エチレンジアミン、N,N−ジメチルエチレンジアミン、N,N,N’,N’−テトラメチルエチレンジアミン等の脂肪族アミン、ピリジン、メチルピリジン、ビピリジン、ヒドロピリジン、フェナントロリン等の複素環アミン、アニリン、ジフェニルアミン、トリフェニルアミン等の芳香族アミン等が挙げられる。
【0060】
また、前記リン含有化合物の具体例としては、トリメチルホスフィン、トリエチルホスフィン等のトリアルキルホスフィン、トリフェニルホスフィン、トリス(2−メトキシフェニル)ホスフィン等のトリアリールホスフィン、ジフェニルメチルホスフィン、ジフェニルエチルホスフィン等のジアリールアルキルホスフィン等の一座ホスフィン、1,2−ジフェニルホスフィノエタン、1,4−ビスジフェニルホスフィノブタン等の二座ホスフィン、トリメチルホスファイトトリエチルホスファイト、トリブチルホスファイト等のトリアルキルホスファイト、トリフェニルホスファイト等のトリアリールホスファイト等が挙げられる。
これらは単独でも、あるいは2種以上を適宜組み合わせて使用することもできる。
【0061】
これらの中でも、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、プロピオン酸リチウム、プロピオン酸ナトリウム、プロピオン酸カリウム、アクリル酸リチウム、アクリル酸ナトリウム、アクリル酸カリウム、メタクリル酸リチウム、メタクリル酸ナトリウム、メタクリル酸カリウム、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、ピリジン、フェナントロリン、トリメチルホスフィン、トリエチルホスフィン、トリフェニルホスフィンおよびトリス(2−メトキシフェニル)ホスフィンが好適に用いられる。特に、酢酸リチウム、酢酸ナトリウム、酢酸カリウム、炭酸リチウム、炭酸ナトリウム、炭酸カリウム、トリメチルホスフィンおよびトリフェニルホスフィンがより好適に用いられる。
【0062】
上記塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の添加総量は、用いる原料の種類にもよるが、前記一般式(1)で表される化合物1モルに対して、好ましくは0.001〜3モル、より好ましくは0.01〜2モル、さらに好ましくは0.05〜1.8モル、特に好ましくは0.1〜1.5モルの範囲内であるのがよい。この範囲の量で添加することが、収率の点および経済性の点で好ましい。
また、上記塩基性化合物、窒素含有化合物およびリン含有化合物から選ばれる化合物は、触媒製造時に触媒に担持されてもよいし、触媒製造後反応の前に反応系内に添加されてもよいし、反応の間に追加添加されてもよい。
【0063】
本発明のアシルオキシ化合物の製造方法では、特に溶剤を使用する必要は無いが、有機溶剤を使用することもできる。有機溶剤としては、例えば、ベンゼン等の芳香族炭化水素類;ペンタン、ヘキサン、シクロヘキサン、ヘプタン等の脂肪族炭化水素類;ジエチルエーテル、ジイソプロピルエーテル等のエーテル類;クロロホルム、塩化メチレン、ジクロロエタン、クロロベンゼン等のハロゲン化炭化水素類;酢酸メチル、酢酸エチル、プロピオン酸メチル、プロピオン酸エチル、(メタ)アクリル酸メチルおよび(メタ)アクリル酸エチル等のカルボン酸エステル類等が挙げられる。
上記有機溶剤の使用量は、用いる原料にもよるが、原料の総量の0〜200質量%、好ましくは0〜100質量%、さらに好ましくは0〜80質量%、特に好ましくは0〜70質量%の範囲内であってよい。この範囲の量で使用することが、収率の点および経済性の点で好ましい。
【0064】
また、本発明のアシルオキシ化合物の製造方法において、原料である前記一般式(1)で表される化合物および得られるアシルオキシ化生成物が重合性化合物である場合、重合禁止剤の存在下で反応させることが、これらの化合物の重合を抑制し、収率を向上させる点で好ましい。かかる重合禁止剤としては、ヒドロキノン、メトキシヒドロキノン、ベンゾキノン、p−tert−ブチルカテコール等のキノン系重合禁止剤;2,6−ジ−tert−ブチルフェノール、2,4−ジ−tert−ブチルフェノール、2−tert−ブチル−4,6−ジメチルフェノール、2,6―ジ―tert−ブチル−4−メチルフェノール、2,4,6−トリ−tert−ブチルフェノール等のアルキルフェノール系重合禁止剤;アルキル化ジフェニルアミン、N,N’−ジフェニル−p−フェニレンジアミン、フェノチアジン等のアミン系重合禁止剤;ジメチルジチオカルバミン酸銅、ジエチルジチオカルバミン酸銅、ジブチルジチオカルバミン酸銅等のジチオカルバミン酸銅系重合禁止剤等が挙げられる。これらは単独でも、あるいは2種以上を適宜組み合わせて使用することができる。これらのなかでも、キノン系重合禁止剤、特にヒドロキノン、メトキシヒドロキノン、ベンゾキノン、p−tert−ブチルカテコールおよびフェノチアジンが好適に用いられる。
【0065】
上記重合禁止剤の添加量は、用いる前記一般式(1)で表される化合物の種類にもよるが、好ましくは該重合性化合物の0.001〜5質量%、より好ましくは0.005〜1質量%、特に好ましくは0.01〜0.1質量%の範囲内となる量であってよい。この範囲の量で添加することが、重合の抑制および経済性の点で好ましい。
反応温度は、0〜500℃の範囲内が好ましく、30〜300℃の範囲内が特に好ましい。反応時間は、上記反応が完結するように原料、触媒および有機溶剤の種類や組み合わせ、使用量等に応じて適宜設定すればよい。反応圧力は、原料と反応温度の組み合わせにもよるが、常圧(大気圧)、加圧の何れであってもよい。また、反応方式としては、例えば、回分式、半回分式、連続式等がある。
【0066】
本発明の方法にしたがって製造されたアシルオキシ化合物化生成物は、使用した前記触媒を分離した後、反応溶液を精製することによって得ることができる。精製手段は特に限定されるものではないが、蒸留法、抽出法、カラムクロマト法等によって分離、精製を行うことができる。これらの方法を組み合わせて実施してもよい。これらの内では、蒸留法おとび抽出法が特に好ましい。
上記精製工程で分離された原料および有機溶剤を、再び反応に用いることができる。また、分離した触媒も、再び反応に用いることができる。
【実施例】
【0067】
以下、実施例により、本発明をさらに具体的に説明するが、本発明はこれらにより何ら限定されるものではない。
アシルオキシ化反応用触媒の製造
【0068】
実施例1 触媒A−1の製造
シリカ球体担体(球体直径5mm、比表面積160m2/g、吸水率0.75g/g、上海海源化工科技有限公司のHSV−I)を用いて、触媒A−1を次の手順で製造した。
【0069】
工程1.担体23g(吸水量19.7g)に、56質量%Na2PdCl4水溶液1.5gおよび17質量%HAuCl4水溶液1.5g、20質量%FeCl3・6H2O水溶液0.7gを含む担体吸水量相当の水溶液を含浸し、触媒前駆体を得た。
工程2.工程1で得られた担体触媒前駆体をNa2SiO3・9H2O3gを含む水溶液に浸漬し、室温で20時間静置した。
【0070】
工程3.工程2の水溶液に53質量%ヒドラジン水和物水溶液4mlを加え、静かに混合し、室温で4時間静置させた。この還元処理後の触媒前駆体を塩化物イオンが無くなるまで流水で洗浄した。洗浄した触媒前駆体を、約110℃で4時間乾燥した。
工程4.工程3で得られた触媒前駆体を1質量%硫酸水溶液2Lに浸漬し、一晩静置した。この後、一晩流水で水洗し、110℃で4時間乾燥した。
【0071】
工程5.工程4で得られた酸処理後の触媒前駆体を、2gの酢酸カリウムを溶かした担体吸水量相当水溶液に含浸し、110℃で4時間乾燥した。
Na2PdCl4水溶液、HAuCl4水溶液、および、FeCl3・6H2O水溶液の沈澱開始pHを測定した結果、それぞれ、4.5、4.0、2.5であった。
【0072】
実施例2 触媒A−2の製造
工程4を行わなかった以外は実施例1と同じ操作を行い、触媒A−2を製造した。
実施例3 触媒B−1の製造
工程1で用いたFeCl3・6H2O水溶液をTiCl4水溶液とした以外は実施例1と同じ操作を行い、触媒B−1を製造した。TiCl4水溶液の沈澱開始pHを測定した結果、1.5であった。
【0073】
実施例4 触媒B−2の製造
工程4を行わなかった以外は実施例3と同じ操作を行い、触媒B−2を製造した。
実施例5 触媒C−1の製造
工程1で用いたFeCl3・6H2O水溶液をScCl3・6H2O水溶液とし、工程4の1質量%硫酸水溶液2Lを1質量%リン酸水溶液200mlとした以外は実施例1と同じ操作を行い、触媒C−1を製造した。ScCl3・6H2O水溶液の沈澱開始pHを測定した結果、4.0であった。
【0074】
実施例6 触媒G−1の製造
工程1のHAuCl4水溶液の添加量を3.0gとし、FeCl3・6H2O水溶液の添加量を1.4gとした以外は実施例2と同じ操作を行い、触媒G−1を製造した。
【0075】
比較例1 触媒D−1の製造
工程1で用いたFeCl3・6H2O水溶液を添加しなかったこと以外は実施例2と同じ操作を行い、触媒D−1を製造した。
比較例2 触媒E−1の製造
工程1で用いたFeCl3・6H2O水溶液をZnCl2水溶液とした以外は実施例1と同じ操作を行い、触媒E−1を製造した。ZnCl2水溶液の沈澱開始pHを測定した結果、7.0であった。
【0076】
比較例3 触媒E−2の製造
工程4を行わなかった以外は比較例2と同じ操作を行い、触媒E−2を製造した。
比較例4 触媒F−1の製造
工程1で用いられたFeCl3・6H2O水溶液をBaCl2・2H2O水溶液とした以外は実施例2と同じ操作を行い、触媒F−1を製造した。BaCl2・2H2O水溶液の沈澱開始pHを測定した結果、11.0であった。
【0077】
触媒の評価
以下の方法により、上記で得られた各触媒の反応評価を行った。
【0078】
触媒活性評価
触媒3ccをガラスビーズ75ccで希釈して反応管(SUS316L製、内径22mm、長さ480mm)に充填した。反応温度150℃、反応圧力0.6MPaG、ガス組成C2H4/O2/H2O/HOAc/N2=47.3/6.1/5.6/26.3/14.7(mol%)のガスを流量20nL/hで流通させ、反応を行った。
反応開始後2hから4h反応後までの間生成ガスおよび生成液を採取し、4h反応サンプルとした。また、反応開始後96hから98h反応後までの間生成ガスおよび生成液を採取し、98h反応サンプルとした。次いで、以下の分析を行い、生成酢酸ビニルの活性および選択率を算出した。
【0079】
反応器出口ガスの分析を、以下の方法を用いて行った。
1.酸素
絶対検量線法を用い、流出ガスを50ml採取し、ガスクロマトグラフィーに付属する1mlのガスサンプラーに全量流し、以下の条件で分析を行った。
ガスクロマトグラフィー:島津ガスクロマトグラフ用ガスサンプラ−(MGS−4:計量管1ml)付ガスクロマトグラフィー(島津製作所製GC−14(B)
カラム:MS−5A IS 60/80mesh(3mmΦ×3m)
キャリアーガス:ヘリウム(流量20ml/min.)
温度条件:検出器温度、気化室温度が110℃、カラム温度は70℃一定
検出器:TCD(He圧70kPaG、Current100m(A)
【0080】
2.酢酸
内部標準法を用い、反応液10mlに対し、内部標準として1,4−ジオキサンを1ml添加したものを分析液として、そのうちの0.2μlを注入して以下の条件で分析を行った。
ガスクロマトグラフィー:島津製作所製GC−14B
カラム:パックドカラムThermon 3000(長さ3m、内径0.3mm)
キャリアーガス:窒素(流量20ml/min.)
温度条件:検出器温度、気化室温度が180℃、カラム温度は分析開始から6分間は50℃保持、その後10℃/minの昇温速度で150℃まで昇温し、150℃で10分間保持
検出器:FID(H2 圧40kPaG、空気圧100kPaG)
【0081】
3.酢酸ビニル
内部標準法を用い、反応液6gに対し、内部標準として酢酸n−プロピルを1g添加したものを分析液として、そのうちの0.3μlを注入して以下の条件で分析した。
ガスクロマトグラフィー:島津製作所製GC−9A
カラム:キャピラリーカラムTC−WAX(長さ30m、内径0.25mm、膜厚0.5μm)
キャリアーガス:窒素(流量30ml/min.)
温度条件:検出器温度、気化室温度が200℃、カラム温度は分析開始から2分間は45℃保持、その後4℃/minの昇温速度で130℃まで昇温し、130℃で15分間保持し、その後25℃/minの昇温速度で200℃まで昇温し、200℃で10分間保持
検出器:FID(H2 圧60kPaG、空気圧100kPaG)
【0082】
各触媒(実施例1〜6および比較例1〜4)の反応評価結果を表1に示す。
表中の4h活性は、各種触媒を用いて、先述の条件で4h反応させた結果生成した酢酸ビニル量より算出した活性を、比較例D−1の活性で割った値を示している。
選択率は4h反応後の分析で算出された酢酸ビニルの選択率を示している。活性低下は98h反応後の活性を4h反応後の活性で割った値を示しており、これを活性低下の指標とした。
【0083】
尚、選択率および活性の定義は以下の通りである。
選択率(%)=(反応後の全物質量中の酢酸ビニルの量(mol))/(反応後の全物質量(mol))
活性(g/L/h)=(単位時間当たりの酢酸ビニルの生成量(g/h))/(触媒の体積(L))
【0084】
また、各触媒の製造(実施例1〜6および比較例1〜4)における各元素の担持量を表2に示す。表2において、実施例1、3、5および比較例2に対する値は酸洗浄前のものである。
【0085】
【表1】
【0086】
【表2】
【産業上の利用可能性】
【0087】
本発明は、反応初期の活性、選択性および活性の持続性の性能バランスに優れたアシルオキシ化反応用触媒を提供することができるので、産業上有用である。
【図面の簡単な説明】
【0088】
【図1】図1は、本発明で規定する沈殿開始pHの測定方法を説明するためのグラフである。
【特許請求の範囲】
【請求項1】
(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とをいっしょに(d)担体に担持させる工程を含むことを特徴とするアシルオキシ化反応用触媒の製造方法。
【請求項2】
前記(d)担体に担持させる工程に次いで、さらに非水溶化処理を行うことを特徴とする請求項1に記載のアシルオキシ化反応用触媒の製造方法。
【請求項3】
前記(d)担体に担持させる工程に次いで、さらに還元処理を行うことを特徴とする請求項1または2に記載のアシルオキシ化反応用触媒の製造方法。
【請求項4】
前記還元処理に次いで、酸および/またはキレート剤を接触させることを特徴とする請求項3に記載のアシルオキシ化反応用触媒の製造方法。
【請求項5】
さらに(e)周期律表の第1および2族の元素(水素を除く)の少なくとも1種を含む第四成分を添加することを特徴とする請求項1〜4のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項6】
前記(a)第一成分がパラジウムを含むことを特徴とする請求項1〜5のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項7】
前記(b)第二成分が周期律表の第11族元素を含むことを特徴とする請求項1〜6のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項8】
前記(b)第二成分が金および銅から選ばれる少なくとも1種を含むことを特徴とする請求項7に記載のアシルオキシ化反応用触媒の製造方法。
【請求項9】
前記(c)第三成分が周期律表の第3〜13族元素の1種を含むことを特徴とする請求項1〜8のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項10】
前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比が、0.4〜1.5であることを特徴とする請求項1〜9のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項11】
前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比が、0.1〜0.5であることを特徴とする請求項1〜10のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項12】
請求項1〜11のいずれかに記載した方法により得られるアシルオキシ化反応用触媒。
【請求項13】
請求項1〜11のいずれかに記載した方法により得られる触媒の存在下に、一般式(1):CHR1R2−X(式中、R1およびR2は、それぞれ独立して、水素原子または有機残基を表し、Xは置換基を有していてもよい芳香族炭化水素残基または置換基を有していてもよいオレフィン残基を表す)で表される化合物またはエチレンと、一般式(2):R3−COOH(式中、R3は水素原子または有機残基を表す)で表されるカルボン酸類とおよび酸素とを反応させて、一般式(3):R3−COO−CR1R2−X(式中、R1、R2、R3およびXは前記規定に同一のものを表す)で表される化合物または酢酸ビニルを生成させることを特徴とするアシルオキシ化合物の製造方法。
【請求項14】
前記反応が、さらに塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の存在下に行われることを特徴とする請求項13に記載のアシルオキシ化合物の製造方法。
【請求項1】
(a)周期律表の第8、9、10および11族の元素の少なくとも1種を含む第一成分と、(b)周期律表の第8、9、10および11族の元素の少なくとも1種であって、前記第一成分の元素とは異なる元素を含む第二成分とおよび(c)前記第一成分および第二成分の沈澱開始pH以下の沈澱開始pHを与える成分であって、前記第一成分および第二成分の元素とは異なる元素を含む第三成分とをいっしょに(d)担体に担持させる工程を含むことを特徴とするアシルオキシ化反応用触媒の製造方法。
【請求項2】
前記(d)担体に担持させる工程に次いで、さらに非水溶化処理を行うことを特徴とする請求項1に記載のアシルオキシ化反応用触媒の製造方法。
【請求項3】
前記(d)担体に担持させる工程に次いで、さらに還元処理を行うことを特徴とする請求項1または2に記載のアシルオキシ化反応用触媒の製造方法。
【請求項4】
前記還元処理に次いで、酸および/またはキレート剤を接触させることを特徴とする請求項3に記載のアシルオキシ化反応用触媒の製造方法。
【請求項5】
さらに(e)周期律表の第1および2族の元素(水素を除く)の少なくとも1種を含む第四成分を添加することを特徴とする請求項1〜4のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項6】
前記(a)第一成分がパラジウムを含むことを特徴とする請求項1〜5のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項7】
前記(b)第二成分が周期律表の第11族元素を含むことを特徴とする請求項1〜6のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項8】
前記(b)第二成分が金および銅から選ばれる少なくとも1種を含むことを特徴とする請求項7に記載のアシルオキシ化反応用触媒の製造方法。
【請求項9】
前記(c)第三成分が周期律表の第3〜13族元素の1種を含むことを特徴とする請求項1〜8のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項10】
前記(a)第一成分の元素の担持量に対する前記(b)第二成分の元素の担持量の比が、0.4〜1.5であることを特徴とする請求項1〜9のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項11】
前記(b)第二成分の元素の担持量に対する前記(c)第三成分の元素の担持量の比が、0.1〜0.5であることを特徴とする請求項1〜10のいずれかに記載のアシルオキシ化反応用触媒の製造方法。
【請求項12】
請求項1〜11のいずれかに記載した方法により得られるアシルオキシ化反応用触媒。
【請求項13】
請求項1〜11のいずれかに記載した方法により得られる触媒の存在下に、一般式(1):CHR1R2−X(式中、R1およびR2は、それぞれ独立して、水素原子または有機残基を表し、Xは置換基を有していてもよい芳香族炭化水素残基または置換基を有していてもよいオレフィン残基を表す)で表される化合物またはエチレンと、一般式(2):R3−COOH(式中、R3は水素原子または有機残基を表す)で表されるカルボン酸類とおよび酸素とを反応させて、一般式(3):R3−COO−CR1R2−X(式中、R1、R2、R3およびXは前記規定に同一のものを表す)で表される化合物または酢酸ビニルを生成させることを特徴とするアシルオキシ化合物の製造方法。
【請求項14】
前記反応が、さらに塩基性化合物、窒素含有化合物およびリン含有化合物からなる群より選ばれる少なくとも1種の化合物の存在下に行われることを特徴とする請求項13に記載のアシルオキシ化合物の製造方法。
【図1】

【公開番号】特開2008−173629(P2008−173629A)
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願番号】特願2007−306111(P2007−306111)
【出願日】平成19年11月27日(2007.11.27)
【出願人】(000002004)昭和電工株式会社 (3,251)
【Fターム(参考)】
【公開日】平成20年7月31日(2008.7.31)
【国際特許分類】
【出願日】平成19年11月27日(2007.11.27)
【出願人】(000002004)昭和電工株式会社 (3,251)
【Fターム(参考)】
[ Back to top ]