
アシル化アミノプロパンジオール及び類似体並びにその治療上の使用
本発明は、一般式(I)(式中、R、R1、R2、R3、G2、及びG3は請求項に記載されたとおりである)を有する、アシル化アミノプロパンジオールならびにその窒素及び硫黄類似体に関する。本発明はまたこの化合物を含む医薬組成物及びその治療用途、例えば脳虚血の処置などにおける用途にも関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規アシル化アミノプロパンジオール、その窒素及び硫黄類似体、それを含む医薬組成物、その治療上の使用、特に脳虚血の処置のための使用に関する。本発明は、また、該誘導体の製造方法を提供する。
【0002】
本発明の化合物は、有利な抗酸化剤及び抗炎症医薬特性を有する。本発明は、また、該化合物及びそれを含む医薬組成物を使用する治療的処置の方法を記載する。特に、本発明の化合物は、脳卒中の予防又は治療に有用である。
【0003】
フランスでは、脳血管性疾患(年間150,000件の新規症例)は、死因の第3位であり、成人における身体障害の主要原因である。虚血性及び出血性脳卒中は、全ての脳血管障害において、それぞれ80%及び20%を占める。虚血性脳卒中は、脳血管性疾患の死亡率及び罹患率を減少させるために取り組まれなければならない、重要な治療上の問題である。虚血の急性期の処置ばかりでなく、虚血の予防においてもまた進歩があった。したがって、危険因子を同定し管理することは、この病変の治療において必須であることを留意することが重要である。
【0004】
薬剤に基づく脳虚血の処置は、異なる戦略に基づく。第1の戦略は、危険因子(高血圧、高コレステロール血症、糖尿病、心房細動など)の予防、又は血栓症の予防、特に抗血小板薬又は抗凝血薬の助けを借りることにより、脳虚血障害の発症を予防することを含む(Adams 2002) 及び (Gorelick 2002)。
【0005】
第2の戦略は、虚血の急性期を処置して、その長期の影響を減衰させることを含む(Lutsep 及び Clark 2001)。
【0006】
脳虚血の病態生理学は、下記のように記載されることができる:ニューロンが壊死している虚血性病巣と無傷の神経組織との間の中間領域である虚血性境界域は、病態生理学的カスケードの部位であり、これは、再灌流が起きないか又は神経保護が不十分である場合、数日間かけて神経細胞死を起こす。最初の事象は、最初の数時間以内に起こるが、多量のグルタミン酸を放出し、これが神経細胞脱分極及び細胞性浮腫を引き起こす。細胞内へのカルシウムの流入は、ミトコンドリアの損傷を誘導し、フリーラジカルの遊離及び神経細胞膜の分解を促進する酵素誘導を導く。カルシウム流入及びフリーラジカル産生は、次に、NF−κBなどの特定の転写因子を活性化する。該活性化は、内皮接着タンパク質の誘導、虚血病巣の多核好中球浸潤、ミクログリア活性化、酸化窒素(NO)II型シンターゼ又はII型シクロオキシゲナーゼのような酵素の誘導などの炎症過程を誘導する。これらの炎症過程は、細胞に毒性のあるNO又はプロスタノイドの放出を起こす。ともにこれらの過程は、結果として非可逆的病変を誘導するアポトーシスの現象となる(Dirnagl, Iadecola et al. 1999)。
【0007】
予防的神経保護の概念は、虚血耐性を示す動物モデルにおける実験データに基づく。事実、脳虚血を実験的に誘導する前に異なる手順を適用すると、後者の重篤度が減衰される。種々の刺激が脳虚血耐性を誘導することができる:すなわち、プレコンディショニング(遷延虚血に先立つ短期虚血)、熱ストレス、低用量の細菌性リポ多糖類の投与である(Bordet, Deplanque et al. 2000)。
【0008】
該刺激は、耐性機構を誘導し、それは保護機構を誘発する信号を活性化する。異なる誘発機構が同定されている:すなわち、サイトカイン、炎症性経路、フリーラジカル、NO、ATP−依存性カリウムチャンネル、アデノシンである。初期事象の発生と虚血耐性との間で観察される遅滞時間は、タンパク質合成の必要性に由来する。多様な種類のタンパク質が虚血耐性を誘導することが示されている:すなわち、熱ショックタンパク質、抗酸化酵素及び抗アポトーシス性タンパク質である(Nandagopal, Dawson et al. 2001)。
【0009】
したがって、アテローム動脈硬化、糖尿病、肥満などの脳血管障害の危険因子の進展を防止することができ、予防的神経保護ばかりでなく、脳虚血の急性期における積極的な神経保護もまた提供できる化合物の真の必要性がある。
【0010】
PPAR(α、β、γ)は、ホルモン活性化核内受容体ファミリーに属する。これらのリガンドとの結合により活性化される場合、これらはレチノイド−X−受容体(RXR)とヘテロ二量体化され、標的遺伝子のプロモーター配列に位置する「ペルオキシソーム増殖因子応答エレメント」(PPRE)と結合する。したがってPPARのPPREへの結合は、標的遺伝子の発現を誘導する(Fruchart, Staels et al. 2001)。
【0011】
PPARは、多種多様な器官に分布しているが、これらはPPARβを除いて全てある程度の組織特異性を示し、その発現は遍在していると思われる。PPARαの発現は、肝臓及び腸管腔において特に多く、PPARγは、主に脂肪組織及び脾臓に発現する。3つのサブタイプ(α、β、γ)は、中枢神経系に発現する。より詳細には、オリゴデンドロサイト及びアストロサイトなどの細胞が、PPARαサブタイプを発現する(Kainu, Wikstrom et al. 1994)。
【0012】
PPARの標的遺伝子は、脂質及び糖の代謝を制御する。しかし、最近の発見は、PPARが他の生物学的過程に関与していることを示唆している。PPARのそれらのリガンドによる活性化は、遺伝子の転写活性における変化を誘導し、そのことは、炎症性過程、抗酸化酵素、血管形成、細胞の増殖及び分化、アポトーシス、iNOS、MMPase及びTIMPの活性を調節する(Smith, Dipreta et al. 2001) 及び (Clark 2002)。
【0013】
フリーラジカルは、アレルギー、腫瘍のイニシエーション及びプロモーション、循環器疾患(アテローム性動脈硬化、虚血)、遺伝的及び代謝的障害(糖尿病)、感染性及び変性疾患(プリオンなど)並びに眼の疾患を含む極めて広範囲の病変において役割を果たす。
【0014】
活性酸素種(ROS)は、正常な細胞機能中に産生される。ROSは、ヒドロキシルラジカル(OH)、スーパーオキシドアニオン(O2-)、過酸化水素(H2O2)及び酸化窒素(NO)を包含する。該種は非常に不安定であり、それらの高い化学反応性のため、細胞の生物学的機能にとって脅威となる。それらは、脂質の過酸化、特定の酵素の酸化、及びタンパク質の分解をもたらす極めて広範囲に及ぶタンパク質の酸化を誘導する。脂質の過酸化に対する保護は、過酸化生成物がDNAの損傷を引き起こしうるため、好気性生物において必須のプロセスである。したがって、天然の抗酸化剤防御によるラジカル種の産生、処理及び除去の間の平衡の調節解除又は修飾によって、細胞又は生物に有害であるプロセスが確立される。
【0015】
ROSは、酵素成分と非酵素成分とを含む抗酸化剤系を介して処理される。酵素系は、下記の性質を有する幾つかの酵素から構成される:
− スーパーオキシドジスムターゼ(SOD)は、スーパーオキシドラジカルを過酸化物に変換することにより破壊する。次に過酸化物を別の酵素系に作用させる。低濃度のSODは好気的呼吸により継続的に産生される。ヒトにおいて3種のSODが同定されており、それぞれ補助因子としてCu、Zn、Fe、Mn又はNiを含有する。3つの形態のヒトSODが下記のように分布する:細胞基質Cu−Zn SOD、ミトコンドリアMn−SO及び細胞外SOD。
− カタラーゼは、過酸化水素(H2O2)を水と酸素に変換するのに非常に効率的である。過酸化水素は、好気性生物において酵素的に異化される。カタラーゼは、また、種々のヒドロペルオキシド(ROOH)の還元に触媒作用を及ぼす。
− グルタチオンペルオキシダーゼは、セレンを補助因子として使用し、グルタチオンを利用してヒドロペルオキシド(ROOH及びH2O2)の還元に触媒作用を及ぼし、それにより酸化損傷から細胞を保護する。
【0016】
細胞の非酵素的抗酸化剤防御は、合成されるか又は食事中に供給される分子を含む。
【0017】
抗酸化剤分子は、異なる細胞区画に存在する。例えば、解毒酵素はフリーラジカルを除去し、細胞の寿命にとって必須である。3つの最も重要な種類の抗酸化剤化合物は、カロチノイド、ビタミンC及びビタミンEである(Gilgun-Sherki, Melamed et al. 2001)。
【0018】
脳虚血及びその結果として起こる影響により誘導されるアポトーシスの現象を回避するため、本発明者たちは、該危険因子の進展を防止することができ、予防的神経保護活性を行うばかりでなく、脳虚血の急性期における積極的な神経保護もまた提供することができる新規化合物を開発した。
【0019】
本発明者たちは、また、PPAR活性化剤、抗酸化剤及び抗炎症特性を同時に示す本発明の化合物を示し、したがって該化合物は、脳虚血における重要な治療的又は予防的潜在能力を有する。
【0020】
したがって、本発明は、脳虚血の予防的又は治療的処置に有用な、有利な薬理学的性質を示す新規な化合物のファミリーを提供する。本発明は、また、該誘導体の製造方法も提供する。
【0021】
本発明の化合物は、一般式(I):
【0022】
【化2】
【0023】
〔式中、
G2及びG3は、独立して酸素原子、硫黄原子又はN−R4基(G2及びG3は、同時にN−R4基を表すことはない)を表し、
R及びR4は、独立して水素原子、又は飽和若しくはそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖若しくは分岐のアルキル基を表し、
R1、R2及びR3は、同一であるか又は異なって、水素原子、CO−R5基、又は式:CO−(CH2)2n+1−X−R6に対応する基(R1、R2及びR3基の少なくとも1個は、式:CO−(CH2)2n+1−X−R6に対応する基である)を表し、
R5は、飽和又はそうではない、場合により置換されている、可能であれば環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、1〜25個の炭素原子を含む)であり、
Xは、硫黄原子、セレン原子、SO基又はSO2基であり、
nは、0〜11の間に含まれる整数であり、
R6は、飽和又はそうではない、場合により置換されている、可能であれば環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは10〜23個の炭素原子を含み、場合により酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される1個以上のヘテロ基を含む)である〕により表される
(但し、G2R2及びG3R3が同時にヒドロキシル基を表す式(I)を有する化合物は除く)。
【0024】
本発明の一般式(I)により表される化合物において、R5基は、同一であるか又は異なって、好ましくは、飽和又は不飽和の、置換されているか又はされていない、直鎖又は分岐のアルキル基(その主鎖は、1〜20個の炭素原子、さらにより好ましくは7〜17個の炭素原子、さらに好ましくは14〜17個を含む)を表す。本発明の一般式(I)により表される化合物において、R5基は、同一であるか又は異なって、1〜6個の炭素原子を含む低級アルキル、例えば、特にメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ペンチル又はヘキシル基などを表すこともできる。
【0025】
本発明の一般式(I)により表される化合物において、R6基は、同一であるか又は異なって、好ましくは、飽和又は不飽和の、置換されているか又はされていない、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、さらにより好ましくは14〜17個の炭素原子、さらになお好ましくは14個の炭素原子を含む)を表す。
【0026】
R5又はR6の飽和長鎖アルキル基の具体的な例は、特にC7H15、C10H21、C11H23、C13H27、C14H29、C15H31、C16H33、C17H35基である。R5又はR6の不飽和長鎖アルキル基の具体的な例は、特にC14H27、C14H25、C15H29、C17H29、C17H31、C17H33、C19H29、C19H31、C21H31、C21H35、C21H37、C21H39、C23H45基、又はエイコサペンタン酸(EPA)C20:5(5,8,11,14,17)及びドコサヘキサン酸(DHA)C22:6(4,7,10,13,16,19)のアルキル鎖である。
【0027】
分岐長鎖アルキル基の例は、特に(CH2)n′−CH(CH3)C2H5、(CH=C(CH3)−(CH2)2)n″−CH=C(CH3)2又は(CH2)2x+1−C(CH3)2−(CH2)n″′−CH3基〔xは、1〜11と等しいか又はその間に含まれる整数であり、n′は、1〜22と等しいか又はその間に含まれる整数であり、n″は、1〜5と等しいか又はその間に含まれる整数であり、n″′は、0〜22と等しいか又はその間に含まれる整数であり、そして(2x+n″′)は、22以下、好ましくは20以下である〕である。
【0028】
前記で示したように、アルキル基R5又はR6は、場合により環式基を含むことができる。環式基の例は、特にシクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシルである。
【0029】
前記で示されるように、アルキル基R5又はR6は、場合により1個以上の置換基(同一であるか又は異なる)で置換されていることができる。置換基は、好ましくは、ハロゲン原子(ヨウ素、塩素、フッ素、臭素)及び−OH、=O、−NO2、−NH2、−CN、−O−CH3、−CH2−OH、−CH2OCH3、−CF3及び−COOZ基(Zは、水素原子又は好ましくは1〜5個の炭素原子を含むアルキル基である)からなる群より選択される。
【0030】
本発明は、また、該化合物の光学及び幾何異性体、そのラセミ体、塩、水和物、並びにこれらの混合物に関する。
【0031】
式(Ia)により表される化合物は、R1、R2又はR3基のうちのただ1個が水素原子を表す本発明の式(I)に対応する化合物である。
【0032】
式(Ib)により表される化合物は、R1、R2又はR3基のうちの2個が水素原子を表す本発明の式(I)に対応する化合物である。
【0033】
本発明は、また、式(I)により表される化合物のプロドラッグを包含し、それは、被検者に投与された後、式(I)により表される化合物に変換される、及び/又は特に脳虚血の治療において式(I)により表される化合物と同様の治療上の活性を示す式(I)により表される化合物の代謝産物に変換されるが、これらは式(I)により表される化合物に類似する。
【0034】
更に、CO−(CH2)2n+1−X−R6基において、Xは、最も好ましくは硫黄又はセレン原子、そして有利には硫黄原子を表す。
【0035】
更に、CO−(CH2)2n+1−X−R6基において、nは、好ましくは0〜3の間に含まれ、とりわけ0〜2の間に含まれ、特に0と等しい。
【0036】
本発明の一般式(I)により表される化合物において、R6は、酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される、1個以上、好ましくは0、1又は2個、より好ましくは0又は1個のヘテロ基を含むことができる。
【0037】
本発明のCO−(CH2)2n+1−X−R6基の特定の例は、CO−CH2−S−C14H29基である。
【0038】
したがって本発明の精神において好ましい化合物は、R1、R2及びR3基のうちの少なくとも1個が、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、好ましくは14〜17個、より好ましくは14〜16個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖のアルキル基である)を表す、上記記載の一般式(I)により表される化合物である。
【0039】
本発明の他の特定の化合物は、R1、R2及びR3基のうちの少なくとも2個が、同一であるか又は異なって、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表す)であるものである。
【0040】
本発明の特定の化合物は、G2が酸素原子又は硫黄原子、好ましくは酸素原子を表すものである。該化合物において、R2は、有利には上記で定義された式:CO−(CH2)2n+1−X−R6に対応する基を表す。
【0041】
特に好ましい化合物は、上記の一般式(I)
〔式中、
G3は、N−R4基(ここで、R4は、水素原子又はメチル基である)であり、そしてG2は酸素原子である;及び/又は
R2は、上記で定義されたCO−(CH2)2n+1−X−R6基を表す〕
により表される化合物である。
【0042】
他の好ましい化合物は、R1、R2及びR3が、同一であるか又は異なって、好ましくは同一で、上記で定義されたCO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、13〜17個の炭素原子、好ましくは14〜17個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖のアルキル基であり、ここでnは、好ましくは0〜3に間に含まれ、特に0と等しい)を表す、上記の一般式(I)により表される化合物である。より詳細には、好ましい化合物は、R1、R2及びR3がCO−CH2−S−C14H29基である、一般式(I)により表される化合物である。
【0043】
本発明の好ましい化合物の例は、図1に与えられている。
したがって本発明は、より詳細にはその目的として、
− 1−テトラデシルチオアセチルアミノ−2,3−(ジパルミトイルオキシ)プロパン;
− 3−テトラデシルチオアセチルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 3−パルミトイルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オール;
− 1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジオレイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパン;及び
− 1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン
から選択される一般式(I)により表される化合物を有する。
【0044】
本発明は、また、その目的として、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する上記記載の一般式(I)により表される化合物の少なくとも1個を、可能であれば別の治療剤と共に薬学的に許容されうる支持体中に含む、医薬組成物を有する。該組成物は、特に脳虚血又は出血性脳卒中などの脳血管性疾患を治療することが意図される。
【0045】
したがって本発明の別の目的は、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する、上記記載の一般式(I)により表される化合物の少なくとも1個を、薬学的に許容されうる支持体中に含む、あらゆる医薬組成物に関する。
【0046】
有利には、脳血管性の病変、より詳細には脳虚血又は脳血管性障害の治療又は予防のための医薬組成物である。事実、驚くべきことに、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する、式(I)により表される化合物は、同時に、PPAR活性化剤、抗酸化剤及び抗炎症性の特性を示し、かつ脳虚血の予防的及び治療的神経保護活性を示すことが見出された。
【0047】
本発明は、また、ヒト又は動物の治療又は予防方法の実施が意図される医薬組成物の調製における、上記で定義された化合物の使用に関する。
【0048】
本発明は、更に、脳血管性病変、より詳細には脳虚血を処置する方法であって、被検体、特にヒトに、G2R2及びR3R3基が同時にヒドロキシル基を表す一般式(I)を有する化合物を包含する、上記で定義された式(I)により表される化合物又は医薬組成物の有効投与量を投与することを含む方法に関する。
【0049】
有利には、使用される式(I)により表される化合物は、上記で定義されたものであり、また、3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオールも包含する。
【0050】
本発明の医薬組成物は、有利には、1つ以上の薬学的に許容されうる賦形剤又はビヒクルを含む。例としては、当業者に既知である、薬学的に適合されうる生理的な等張性の緩衝されている食塩水などが挙げられる。本組成物は、分散剤、可溶化剤、安定剤、界面活性剤、防腐剤などから選択される、1つ以上の作用物質又はビヒクルを含んでいてもよい。製剤(液体及び/又は注射用及び/又は固体)に使用してよい作用物質又はビヒクルは、特に、メチルセルロース、ヒドロキシメチルセルロース、カルボキシメチルセルロース、ポリソルベート80、マンニトール、ゼラチン、乳糖、植物油、アカシアなどが包含される。組成物は、注射用懸濁剤、ゲル剤、油剤、錠剤、坐剤、散剤、ゼラチンカプセル剤、カプセル剤などとして、可能であれば持続性及び/又は遅延性放出を可能にする医薬剤形又は装置に製剤化されてよい。この種類の処方には、有利にはセルロース、カーボネート又はデンプンのような作用物質が使用される。
【0051】
本発明の化合物又は組成物を、様々な方法及び様々な剤形で投与してよい。例えば、これらは、経口経路により、非経口的に、吸入により、又は例えば、静脈内、筋肉内、皮下、経皮、動脈内経路などのような注射により、全身的に投与してよい。注射には、本化合物は一般に液体懸濁剤の剤形に調製され、これはシリンジにより、又は例えば点滴により注入してよい。この点に関して、本化合物は一般に、当業者に既知の薬学的に適合されうる生理的な等張性の緩衝された食塩水などに溶解される。例えば、本組成物は、分散剤、可溶化剤、乳化剤、安定剤、界面活性剤、防腐剤、緩衝剤などから選択される、1つ以上の作用物質又はビヒクルを含んでもよい。液体及び/又は注射用製剤に使用してよい作用物質又はビヒクルは、特に、メチルセルロース、ヒドロキシメチルセルロース、カルボキシメチルセルロース、ポリソルベート80、マンニトール、ゼラチン、乳糖、植物油、アカシア、リポソームなどを包含する。
【0052】
したがって、本組成物は、ゲル剤、油剤、錠剤、坐剤、散剤、ゼラチンカプセル剤、カプセル剤、エアゾールなどの剤形で、可能であれば徐放及び/又は遅延放出を可能にする医薬剤形又は装置により投与してよい。この種類の処方には、有利にはセルロース、カーボネート又はデンプンのような作用物質が使用される。
【0053】
本化合物は、使用される作用物質又はビヒクルが、好ましくは水、ゼラチン、ゴム、乳糖、デンプン、ステアリン酸マグネシウム、タルク、油、ポリアルキレングリコールなどからなる群より選択される場合には、経口投与してもよい。
【0054】
非経口投与には、本化合物は、特に水、油又はポリアルキレングリコールを用いる、好ましくは液剤、懸濁剤又は乳剤の剤形で投与されるが、ここに保存料、安定剤、乳化剤などの他に、浸透圧を調整するための塩、緩衝剤などを加えることが可能である。
【0055】
注入速度及び/又は注入用量は、当業者により患者、病気、投与の様式などに適合されてよいことが理解される。典型的には、本化合物は、1用量当たり1μg〜2g、好ましくは1用量当たり0.1mg〜1gの範囲の用量で投与される。用量は、場合に応じて、1日1回又は1日数回投与することができる。更には、本発明の組成物は、また、他の活性物質又は活性剤を含んでもよい。
【0056】
本発明は、また、上記化合物の製造方法にも関する。本発明の化合物は、当業者に既知の化学反応の組合せを使用して、市販の製品から調製することができる。
【0057】
本発明の1つの方法によると、式(I)〔式中、(i)G2及びG3は、酸素若しくは硫黄原子、又はN−R4基であり、(ii)R及び場合によってはR4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、同一の直鎖又は分岐のアルキル基を表し、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(I)〔式中、(i)G2又はG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物と、式:A1−LG〔式中、A1は、基R又は場合によってはR4を表し、そしてLGは、例えば、Cl、Br、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得る。
【0058】
第1の実施態様において、式(I)〔式中、(i)G2及びG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であって、CO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(I)〔式中、(i)G2又はG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子である〕により表される化合物と、式:A°−CO−A〔式中、Aは、例えば、OH、Cl、O−CO−A°及びO−R7(R7はアルキル基である)からなる群より選択される反応性基であり、そしてA°は、(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得られる。
【0059】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、様々な方法により得ることができるが、その方法は、同じヘテロ原子(窒素又は酸素)に担持される基が同じ意味を有する化合物の合成を可能にする。
【0060】
第1の実施態様によると、1−アミノグリセロール、1,3−ジアミノグリセロール又は1,2−ジアミノグリセロールの分子((Morris, Atassi et al. 1997)により記載されたプロトコールを適用することにより得る)を、式:A°−CO−A1〔式中、A1は、例えば、OH、Cl及びOR7(R7はアルキル基である)からなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる。該反応は、それぞれ式(I)により表される化合物の特定の形態(化合物(IIa−c)と称する)を生じるが、(Urakami and Kakeda 1953)、(Shealy, Frye et al. 1984)、(Marx, Piantadosi et al. 1988) 及び (Rahman, Ziering et al. 1988) 又は (Nazih, Cordier et al. 1999)により記載されたプロトコールを適用して実施することができる。化合物(IIb−c)において、それぞれ同じヘテロ原子に担持されている基(R1及びR3)及び(R1及びR2)は、同じ意味を有する。
【0061】
【化3】
【0062】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(IIa−c)を有する化合物と、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得ることができる。該反応は、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R1及びR2)、(R1及びR3)又は(R2及びR3)が同じ意味を有する化合物の合成を可能にする。有利には、該反応は、例えば、(Urakami and Kakeda 1953) 及び (Nazih, Cordier et al. 1999)により記載されたプロトコールに従って実施される。
【0063】
本発明の別の特定の方法(ダイアグラム1)によると、式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R5基を表す〕により表される化合物は、下記の工程により得ることができる:
【0064】
a)1−アミノグリセロール、1,3−ジアミノグリセロール又は1,2−ジアミノグリセロールを化合物(PG)2O(ここで、PGは、保護基である)と反応させて一般式(IIIa−c)を有する化合物を得る工程。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる〔ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す〕。
【0065】
b)式(IIIa−c)を有する化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(IVa−c)〔式中、R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得る工程。
【0066】
c)化合物(IVa−c)を当業者に既知の従来の条件に従って脱保護して、一般式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3はCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得る工程。
【0067】
d)一般式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる工程。
【0068】
【化4】
【0069】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、様々な方法で得ることができる。
【0070】
第1の方法によると、本発明の一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR2は、水素原子であり、そして(iii)R1、R3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる。
【0071】
この調製方法によると、一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR2は、水素原子であり、そして(iii)R1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、上記で定義された式(IIa)により表される化合物と、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得ることができる。
【0072】
本発明の別の特定の方法によると、式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−C−R6基を表す〕により表される化合物を、本発明の式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R、R2及びR3は、水素原子を表し、そして(iii)R1は、CO−R5又はCO−(CH2)2n+1−X−R6基である〕により表される化合物(式(IIa)の化合物)から、下記の工程に従って得ることができる(ダイアグラム2):
【0073】
a)式(IIa)により表される化合物を、化合物PG−E(ここで、PGは、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させることにより、一般式(V)〔式中、R1は、CO−R5又はCO−(CH2)2n+1−X−R6基である〕により表される化合物を得る工程。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここで、PG−Eはトリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0074】
b)式(V)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(VI)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得る工程。
【0075】
c)化合物(VI)を当業者に既知の条件で脱保護することにより、一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR3は、水素原子であり、そして(iii)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得る工程。
【0076】
d)一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR3は、水素原子であり、そして(iii)R1及びR2は、同一であるか又は異なって、CO−R5又は(CO−CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる工程。
【0077】
【化5】
【0078】
有利な方法において、上記の工程は、(Marx, Piantadosi et al. 1988)により記載されたプロトコールに従って実施される。
【0079】
本発明の別の方法によると、式(I)〔式中、(i)G2又はG3は、酸素原子又はN−R4基を表し、(ii)G2又はG3の少なくとも1つは、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iv)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(I)〔式中、(i)それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iii)R1は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより得られる。
【0080】
本発明の式(I)〔式中、(i)それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iii)R1は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、本発明の式(I)〔式中、それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、上記で定義された基を表し、そして(iii)R1は、水素原子である〕により表される化合物と式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で得られる。
【0081】
第1の実施態様において、本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iv)R1及びR2は、水素原子であり、そして(v)R3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、下記の方法により得られる(ダイアグラム3):
【0082】
a)1−アミノグリセロールを、式:R−CHO〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてCHOは、アルデヒド官能基である〕に対応する化合物と、当業者に既知の還元剤の存在下で反応させて、式(VII)〔式中、Rは、上記で定義された基である〕により表される化合物を得ること。有利には、該反応は、(Antoniadou-Vyzas, Foscolos et al. 1986)により記載されたプロトコールを適用して実施することができる。
【0083】
b)式(VII)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(VIII)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる(ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す)。
【0084】
c)〔Kitchin, Bethell et al. 1994〕に記載された方法を適用して、式(VIII)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XI)により表される化合物を得ること。
【0085】
d)式(IX)により表される化合物を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕に対応する化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(X)〔式中、R及びR4は、場合により異なって、上記と同義である〕に対応する化合物を得ること。
【0086】
e)式(X)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(XI)〔式中、R及びR4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、R3は、R5基又は(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得ること。
【0087】
f)化合物(XI)を当業者に既知の条件で脱保護すること。
【0088】
【化6】
【0089】
第2の実施態様によると、本発明の式(I)〔式中、(i)G3は、酸素原子であり、(ii)G2は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iv)R1及びR3は、水素原子であり、そして(v)R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、下記の方法により得られる(ダイアグラム4):
【0090】
a)式(VIII)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH又はハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XII)〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてPGは、上記で定義された別の保護基である〕により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載された方法を適用して実行することができる(ここで、PG′−Eはトリフェニルメチルクロリド又は9−フェニルキサンテン−9−オールあるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0091】
b)(Kitchin, Bethell et al. 1994)により記載された方法を適用して、上記で定義された式(XII)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メチル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XIII)〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてPG及びPG′は、保護基である〕により表される化合物を得ること。
【0092】
c)上記で定義された式(XIII)により表される化合物を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕に対応する化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(XIV)〔式中、R及びR4は、独立して、上記と同義である〕により表される化合物を得ること。
【0093】
d)式(XIV)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(XV)〔式中、R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表し、そしてPG及びPG′は、保護基である〕により表される化合物を得ること。
【0094】
e)式(XV)により表される化合物を当業者に既知の従来の条件で脱保護して、本発明の一般式(I)〔式中、(i)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(ii)R1及びR3は、水素原子であり、そして(iii)R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0095】
【化7】
【0096】
本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、様々な方法で得ることができる。
【0097】
第1の実施態様によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す(ここで、R1、R2及び/又はR3は、同じヘテロ原子(硫黄又は窒素)により担持されている場合、同じ意味を有する)〕により表される化合物は、下記の方法により得ることができる(ダイアグラム5A):
【0098】
a)式(IIa−c)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XVIa−c)により表される化合物を得ること。
【0099】
b)式(XVIa−c)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XVIIa−c)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0100】
c)式(XVIIa−c)により表される化合物を、当業者に既知の従来の条件で、例えば、塩基性媒体中で脱保護して、一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0101】
d)一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0102】
【化8】
【0103】
同様の合成方法によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す(ここで、R1、R2及び/又はR3は、同じヘテロ原子(硫黄又は窒素)により担持されている場合、同じ意味を有する)〕を有する化合物は、下記の方法により調製することができる(ダイアグラム5B):
【0104】
a)式(IIa−c)により表される化合物を、式(LG)2〔式中、LGは、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物と、可能であれば、当業者に既知の活性化剤の存在下で反応させて、一般式(XVId−f)により表される化合物を得ること。
【0105】
b)式(XVId−f)により表される化合物を、式:HS-B+〔式中、Bは、例えば、ナトリウム又はカリウム、好ましくはナトリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0106】
c)一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0107】
【化9】
【0108】
該反応は、一般式(I)〔式中、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R2及びR3)、(R1及びR3)、並びに(R1及びR2)は同じ意味を有する〕により表される化合物の合成を可能にする。
【0109】
上記工程は、(Adams, Doyle et al. 1960) 及び (Gronowitz, Herslof et al. 1978)で記載されたプロトコールに従って有利な方法で実施することができる。
【0110】
本発明の別の方法(ダイアグラム6)によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa−c)により表される化合物から下記の方法で調製することができる:
【0111】
a)式(IIIa−c)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XVIIIa−c)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0112】
b)式(XVIIIa−c)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XIXa−c)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0113】
c)化合物(XIXa−c)の硫黄原子を当業者に既知の条件で脱保護して、一般式(XXa−c)により表される化合物を得ること。
【0114】
d)一般式(XXa−c)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXIa−c)〔式中、R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0115】
e)式(XXIa−c)により表される化合物を当業者に既知の従来の条件に従って脱保護して、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、水素原子、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0116】
f)本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、水素原子、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0117】
該反応は、一般式(I)〔式中、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R2及びR3)、(R1及びR3)及び(R1及びR2)は同じ意味を有する〕により表される化合物の合成を可能にする。
【0118】
有利な方法において、上記の工程は、(Adams, Doyle et al. 1960)、(Gronowitz, Hersloef et al. 1978)、(Bhatia and Hajdu 1987) 及び (Murata, Ikoma et al. 1991)により記載されたプロトコールに従って実施される。
【0119】
【化10】
【0120】
一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、一般式(I)〔式中、(i)G2又はG3は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、上記で定義された基を表し、(iii)R1は、水素原子であり、そして(iv)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基を表す〕に対応する化合物と、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより得る。
【0121】
一般式(I)〔式中、(i)G2及びG3基は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、上記で定義された基を表し、(iii)R1は、水素原子であり、そして、(iv)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕による表される化合物を、下記の方法により得ることができる:
【0122】
第1の実施態様において、本発明の式(I)〔式中、(i)G2基は、硫黄原子であり、(ii)G3は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表し、(iv)R1は、水素原子であり、そして(v)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、下記の方法により得る(ダイアグラム7):
【0123】
a)式(XI)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0124】
b)式(XXII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XXIII)により表される化合物を得ること。有利には、該反応を、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施する。
【0125】
c)式(XXIII)により表される化合物の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXIV)により表される化合物を得ること。
【0126】
d)一般式(XXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXV)〔式中、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0127】
e)式(XXV)の化合物を当業者に既知の条件で脱保護すること。
【0128】
【化11】
【0129】
別の方法(ダイアグラム8)に従い、本発明の式(I)〔式中、(i)G2は、N−R4基を表し、(ii)G3は、硫黄原子であり、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表し、(iv)R1は、水素原子であり、そして(v)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、下記の方法により得る:
【0130】
a)式(IX)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXVI)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0131】
b)式(XXVI)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXVII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0132】
c)化合物(XXVII)を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕により表される化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(XXVIII)〔式中、R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表す〕により表される化合物を得ること。
【0133】
d)一般式(XXVIII)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXIX)により表される化合物を得ること。
【0134】
d)式(XXIX)により表される化合物の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXX)により表される化合物を得ること。
【0135】
f)一般式(XXX)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXIV)〔式中、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0136】
g)式(XXXI)により表される化合物を当業者に既知の従来の条件で脱保護すること。
【0137】
【化12】
【0138】
本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(V)を有する化合物から下記の方法により調製することができる(ダイアグラム9A):
【0139】
a)化合物(V)を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0140】
b)式(XXXII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXIII)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0141】
c)化合物(XXIII)の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXXIV)により表される化合物を得ること。
【0142】
d)一般式(XXXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXV)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0143】
e)化合物(XXXV)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR3は、水素原子であり、そしてR1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0144】
f)一般式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)R及びR3は、水素原子であり、そして(iv)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0145】
【化13】
【0146】
同様の合成方法によると、本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(V)の化合物から下記の方法により調製することができる(ダイアグラム9B):
【0147】
a)化合物(V)を、式(LG)2〔式中、LGは、例えば、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXIIa)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0148】
b)式(XXXIIa)により表される化合物を、式:HS-B+〔式中、Bは、例えば、ナトリウム及びカリウム、好ましくはナトリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXIV)により表される化合物を得ること。
【0149】
c)一般式(XXXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXV)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0150】
d)化合物(XXXV)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR3は、水素原子であり、そしてR1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0151】
e)一般式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)R及びR3は、水素原子であり、そして(iv)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0152】
【化14】
【0153】
本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム10):
【0154】
a)式(IIIa)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XXXVI)〔式中、PGは、前に定義された別の保護基である〕により表される化合物を得ること。有利な方法において、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここでPG−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0155】
b)化合物(XXXVI)を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXVII)〔式中、PG及びPG′は、上記で定義されたような、賢明に選択された保護基を表す〕により表される化合物を得ること。
【0156】
c)式(XXXVII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXVIII)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0157】
d)化合物(XXXVIII)の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXXIX)により表される化合物を得ること。
【0158】
e)一般式(XXXIX)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XL)〔式中、R2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0159】
f)化合物(XL)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R、R1及びR3は、水素原子であり、そしてR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物(化合物XLI)を得ること。
【0160】
g)式(XLI)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(XLII)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる〔ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す〕。
【0161】
h)一般式(XLII)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、式(XLIII)により表される化合物を得ること。
【0162】
i)化合物(XLIII)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0163】
j)一般式(I)〔式中、(i)G2は、硫黄原子であり、G3は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0164】
【化15】
【0165】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R2は、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIa)を有する化合物から下記の方法により調製することができる(ダイアグラム11):
【0166】
a)上記で定義された式(IIa)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIV)により表される化合物を得ること。
【0167】
b)式(XLIV)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XLV)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0168】
c)式(XLV)により表される化合物を、化合物PG−E(ここで、PGは、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XLVI)により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここでPG−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0169】
d)化合物(XLVI)の硫黄原子を当業者に既知の条件で脱保護して、一般式(XLVII)により表される化合物を得ること。
【0170】
e)一般式(XLVII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XLVIII)〔式中、R1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0171】
f)式(XLVIII)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR3は、水素原子であり、そしてR1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0172】
g)一般式(I)〔式中、(i)G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0173】
【化16】
【0174】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、同一であるか又は異なって、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム12):
【0175】
a)上記で定義された式(IIIa)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIX)により表される化合物を得ること。
【0176】
b)式(XLIX)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(L)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0177】
c)式(L)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(LI)により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載された方法を適用して実施することができる(ここでPG′−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0178】
d)化合物(LI)の硫黄原子を当業者に既知の条件で脱保護して、一般式(LII)により表される化合物を得ること。
【0179】
e)一般式(LII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(LIII)〔式中、R3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0180】
f)式(LIII)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物(化合物LIV)を得ること。
【0181】
g)式(LIV)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(LV)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる(ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す)。
【0182】
h)一般式(LV)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、式(LVI)により表される化合物を得ること。
【0183】
i)化合物(LVI)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G3は、硫黄原子であり、G2は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0184】
j)一般式(I)〔式中、G3は、硫黄原子であり、G2は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0185】
【化17】
【0186】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R2及びR3は、同一であるか又は異なって、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R1は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム13):
【0187】
a)上記で定義された式(IIIa)により表される化合物を、式(LG)2〔式中、LGは、例えば、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIXa)により表される化合物を得ること。
【0188】
b)式(XLIXa)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(L)により表される化合物を得ること。
【0189】
c)化合物(L)の硫黄原子を当業者に既知の条件で脱保護して、一般式(LVII)により表される化合物を得ること。
【0190】
d)一般式(LVII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(LVI)〔式中、R2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0191】
e)式(LVI)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0192】
f)一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0193】
【化18】
【0194】
本発明の実行可能性、実現性及び他の利点は、以下の実施例において更に詳述されるが、これらの実施例は、説明の目的で与えられるものであり、限定を目的とするものではない。
【0195】
実施例:
文書のより容易な理解のために、活性の測定及び評価に関わる実施例で使用される本発明の化合物は、次のように省略される:例えば「Ex2」は、その調製が実施例2で記載されている本発明の化合物を示す。
【0196】
0.2mm厚のMERCKシリカゲル60F254で被覆されたプレートで薄層クロマトグラフィー(TLC)を実施した。保持係数はRfと省略される。
粒径40〜63μm(Merck参照番号9385−5000)を有するシリカゲル60でカラムクロマトグラフィーを実施した。
キャピラリー法によりBuchiB540装置で融点(MP)を測定した。
赤外線(IR)スペクトルをBruker Fourier変換スペクトロメーター(Vector 22)で記録した。
Bruker AC300スペクトロメーター(300MHz)で核磁気共鳴(NMR)スペクトルを記録した。各信号を、その化学シフト、強度、多重度(注:一重項はs、広帯一重項はsl、二重項はd、分離二重項はdd、三重項はt、分離三重項はtd、五重項はquint及び多重項はm)、及びその結合定数(J)により同定した。
Perkin Elmer Sciex API 1(ESI−MS:エレクトロスプレーイオン化質量分析(ElectroSpray lonization Mass Spectrometry))又はApplied Biosystems Voyager DE-STR製のMALDI-TOF型(マトリックス補助レーザー脱着/イオン化−飛行時間(Matrix-Assisted Laser Desorption/lonization - Time Of Flight))により質量スペクトル(MS)を測定した。
【0197】
実施例1:テトラデシルチオ酢酸の調製
水酸化カリウム(34.30g、0.611mol)、メルカプト酢酸(20.9ml、0.294mol)及び1−ブロモテトラデカン(50ml、0.184mol)をこの順序でメタノール(400ml)に加えた。この混合物を室温で一晩撹拌した。次に水(800ml)に溶解した濃塩酸溶液(60ml)を加えた。テトラデシルチオ酢酸が沈殿した。この混合物を室温で一晩撹拌した。次に沈殿物を濾過し、水で5回洗浄し、デシケーター中で乾燥した。この生成物をメタノール中で再結晶した。
【0198】
【表1】
【0199】
実施例2:3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオールの調製
テトラデシルチオ酢酸(実施例1)(14.393g、50mmol)及び3−アミノ−プロパン−1,2−ジオール(5g、55mmol)をフラスコに入れて、190℃で1時間加熱した。反応混合物を室温に冷却し、クロロホルムにとって、水で1回洗浄した。有機相を硫酸マグネシウムで乾燥し、濾過し、乾燥した。残渣をエーテル中で撹拌し、生成物を濾過により単離した。
【0200】
【表2】
【0201】
実施例3:3−(パルミトイルアミノ)プロパン−1,2−ジオール
この化合物は、3−アミノ−プロパン−1,2−ジオール及びパルミチン酸から上述の方法(実施例2)に従って合成した。
【0202】
【表3】
【0203】
実施例4:1,2−(ジパルミトイルオキシ)−3−テトラデシルチオアセチルアミノプロパンの調製
3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオール(実施例2)(1g、2.77mmol)をジクロロメタン(200ml)に溶解した。次にジシクロヘキシルカルボジイミド(1.426g、6.91mmol)、ジメチルアミノピリジン(0.845g、6.91mmol)及びパルミチン酸(1.773g、6.91mmol)を加え、混合物を室温で48時間撹拌した。沈殿したジシクロヘキシル尿素を濾過し、ジクロロメタンで洗浄した。濾液を真空留去した。残渣を、シリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 6:4)により精製した(収率:28%)。
【0204】
【表4】
【0205】
実施例5:1,2−(ジテトラデシルチオアセチルオキシ)−3−テトラデシルチオアセチルアミノプロパンの調製
この化合物は、3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオール(実施例2)及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例4)に従って合成した。
【0206】
【表5】
【0207】
実施例6:1,2−(ジテトラデシルチオアセチルオキシ)−3−パルミトイルアミノプロパンの調製
この化合物は、3−(パルミトイルアミノ)プロパン−1,2−ジオール(実施例3)及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例4)に従って合成した。
【0208】
【表6】
【0209】
実施例7:1,3−ジ(オレイルアミノ)プロパン−2−オールの調製
オレイン酸(5.698g、0.020mol)及び1,3−ジアミノプロパン−2−オール(1g、0.011mol)をフラスコに入れて、190℃で2時間加熱した。反応混合物を室温に冷却し、次にクロロホルムにとって、水で洗浄した。水相をクロロホルムで抽出し、有機相を合わせ、硫酸マグネシウムで乾燥し、濾過し、蒸発乾固して、油状の黒色残渣(6.64g)を得て、それをシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 99:1)により精製した。次に得られた生成物をエーテルで洗浄し、濾過した。
【0210】
【表7】
【0211】
実施例8:1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オールの調製
この化合物は、1,3−ジアミノプロパン−2−オール及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例7)に従って合成した。
【0212】
【表8】
【0213】
実施例9:1,3−ジ(ステアロイルアミノ)プロパン−2−オールの調製
この化合物は、1,3−ジアミノプロパン−2−オール及びステアリン酸から上記の方法(実施例7)に従って合成した。
【0214】
【表9】
【0215】
実施例10:1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン二塩酸塩の調製
1,3−ジ(tert−ブチルオキシカルボニルアミノ)プロパン−2−オールの調製(実施例10a)
1,3−ジアミノプロパン−2−オール(3g、0.033mol)をメタノール(300ml)に溶解し、続いてトリエチルアミン(33ml滴下)及びジ−tert−ブチルジカルボネート〔(BOC)2O〕(21.793g、0.100mol)を加えた。反応媒体を40〜50℃で20分間加熱し、次に室温で1時間撹拌した。溶媒を留去した後、無色の油状物残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 95:5)により精製した。反応より無色の油状物が生じ、それはゆっくりと結晶化した。
【0216】
【表10】
【0217】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(テトラデシルチオアセチルオキシ)プロパンの調製(実施例10b)
1,3−(ジ−tert−ブトキシカルボニルアミノ)−プロパン−2−オール(実施例10a)(1g、3.45mmol)、テトラデシルチオ酢酸(実施例1)(0.991g、3.45mmol)及びジメチルアミノピリジン(0.042g、0.34mmol)をジクロロメタン(40ml)に0℃で溶解した。次にジクロロメタンで希釈されたジシクロヘキシルカルボジイミド(0.709g、3.45mmol)を滴加し、混合物を0℃で30分間撹拌し、次に室温にした。20時間の反応の後、ジシクロヘキシル尿素沈殿物を濾過し、濾液を乾燥した。油状残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 5:5、続いてジクロロメタン/酢酸エチル 98:2)により精製した。
【0218】
【表11】
【0219】
1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン二塩酸塩の調製(実施例10)
1,3−(ジtert−ブトキシカルボニルアミノ)−2−テトラデシルチオアセチルオキシプロパン(実施例10b)(0.800g、1.43mmol)を、ガス状塩酸で飽和されたジエチルエーテル(50ml)に溶解した。反応媒体を室温で20時間撹拌した。次に形成された沈殿物を濾過し、エーテル洗浄した。
【0220】
【表12】
【0221】
実施例11:1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパンの調製
1,3−ジアミノ−2−テトラデシルチオアセチルオキシプロパン二塩酸塩(実施例10)(0.400g、0.92mmol)及びテトラデシルチオ酢酸(実施例1)(0.532g、1.84mmol)をジクロロメタン(50ml)に0℃で溶解し、続いてトリエチルアミン(0.3ml、2.1mmol)、ジシクロヘキシルカルボジイミド(0.571g、2.77mmol)及びヒドロキシベンゾトリアゾール(HOBt)(0.249g、1.84mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄した。濾液を真空留去した。得られた残渣(1.40g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン、続いてジクロロメタン/酢酸エチル 9:1)により精製した。
【0222】
【表13】
【0223】
実施例12:1,3−ジオレイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパンの調製
この化合物は、1,3−ジアミノ−2−テトラデシルチオアセチルオキシプロパン二塩酸塩(実施例10)及びオレイン酸から、実施例11に記載の方法に従って合成した。
【0224】
【表14】
【0225】
実施例13:2,3−ジテトラデシルチオアセチルアミノプロパン−1−オールの調製
2,3−ジアミノプロパン酸メチル二塩酸塩の調製(実施例13a)
2,3−ジアミノプロピオン酸塩酸塩(1g、7mmol)をメタノール(40ml)に溶解した。媒体を氷浴で冷却し、続いて塩化チオニル(2.08ml、28mmol)を加えた。媒体を室温にし、次に20時間還流した。溶媒を留去し、残渣をヘプタンで粉砕した。得られた沈殿物を濾過し、洗浄し、乾燥して、黄色を帯びた白色固体を得た。
【0226】
【表15】
【0227】
2,3−ジテトラデシルチオアセチルアミノプロパン酸メチルの調製(実施例13b)
2,3−ジアミノプロパン酸メチル二塩酸塩(実施例13a)(0.500g、2.62mmol)及びテトラデシルチオ酢酸(実施例1)(1.51g、5.23mmol)をジクロロメタン(80ml)に0℃で溶解し、続いてトリエチルアミン(0.79ml)、ジシクロヘキシルカルボジイミド(1.62g、7.85mmol)及びヒドロキシベンゾトリアゾール(0.707g、5.23mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(3.68g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を白色粉末の形態で得た。
【0228】
【表16】
【0229】
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オールの調製(実施例13)
水素化ホウ素ナトリウム(316mg、8.4mmol)をテトラヒドロフラン(40ml)に溶解した。反応混合物を氷浴で冷却し、続いて2,3−ジテトラデシルチオアセチルアミノプロパン酸メチル(実施例13b)(500mg、0.76mmol)を少量ずつ加えた。混合物を室温にし、撹拌した。4日間の反応の後、水20mlを加えた。沈殿した生成物を濾過し、水で洗浄し、次にデシケーター中で乾燥して、白色の粉末を得た。
【0230】
【表17】
【0231】
実施例14:2,3−ジテトラデシルチオアセチルアミノ−1−テトラデシルチオアセチルオキシプロパンの調製
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オール(実施例13)(0.200g、0.32mmol)をテトラヒドロフラン(40ml)に溶解し、続いてジシクロヘキシルカルボジイミド(65mg、0.32mmol)、ジメチルアミノピリジン(39mg、0.32mmol)及びテトラデシルチオ酢酸(実施例1)(91mg、0.32mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、テトラヒドロフランで洗浄し、濾液を留去した。得られた残渣(1g)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で生成した。
【0232】
【表18】
【0233】
実施例15:1,3−ジアミノ−2−(テトラデシルチオアセチルチオ)プロパン二塩酸塩の調製
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(p−トルエンスルホニルオキシ)プロパンの調製(実施例15a)
1,3−ジ(tert−ブチルオキシカルボニルアミノ)プロパン−2−オール(実施例10a)(2.89g、10mmol)及びトリエチルアミン(2.22ml、16mmol)を無水ジクロロメタン(100ml)に溶解した。反応混合物を氷浴で冷却し、続いてジクロロメタン(30ml)に溶解した塩化トシル(2.272g、12mmol)を滴加した。次に反応混合物を室温で72時間撹拌した。48時間後に1当量の塩化物及びトリエチルアミン1.6 を加えた。水を加えて反応を止め、媒体を沈降させた。有機相を水で数回洗浄した。水相を合わせ、ジクロロメタンで再び抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(6.44g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン、続いてジクロロメタン/メタノール 99:1)により精製して、目的化合物を白色の固体として得た。
【0234】
【表19】
【0235】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−アセチルチオプロパンの調製(実施例15b)
1,3−(ジtert−ブトキシカルボニルアミノ)−2−(p−トルエンスルホニルオキシ)プロパン(実施例15a)(0.500g、1.12mmol)及びチオ酢酸カリウム(0.161g、1.41mmol)をアセトンに溶解し、媒体を48時間還流した。24時間の還流の後で、1当量のチオ酢酸カリウムを加えた。反応を室温にし、溶媒を留去した。残渣をジエチルエーテルにとって、セライト(登録商標)で濾過した。濾液を留去した。得られた生成物(0.48g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を黄土色の固体として得た。
【0236】
【表20】
【0237】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−メルカプトプロパンの調製(実施例15c)
メタノール(10ml)で希釈された1,3−ジ(tert−ブトキシカルボニルアミノ)−2−(アセチルチオ)プロパン(実施例15b)(0.380g、1.2mmol)を、窒素流下で脱酸素化しながらメタノール中の20%炭酸カリウム溶液(2.14ml、12.4mmol)に加えた。反応混合物を窒素下、室温で20時間撹拌し、次に酢酸でpH6に酸性化した。溶媒を真空留去した。残渣を水にとって、クロロホルムで抽出した。有機相を合わせ、硫酸マグネシウムで乾燥し、次に濾過し、乾燥して、目的生成物を白色固体の形態で得て、直ちにそれを次の反応に使用した。
【0238】
【表21】
【0239】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(テトラデシルチオアセチルチオ)プロパンの調製(実施例15d)
1,3−〔ジ(tert−ブトキシカルボニルアミノ)〕−2−メルカプトプロパン(実施例15c)(0.295g、0.963mmol)をジクロロメタン(40ml)に溶解した。次にジシクロヘキシルカルボジイミド(0.199g、0.963mmol)、ジメチルアミノピリジン(0.118g、0.963mmol)及びテトラデシルチオ酢酸(実施例1)(0.278g、0.963mmol)を加えた。反応混合物を室温で撹拌し、反応の進行を、TLCで監視した。20時間の反応の後、ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(0.73g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で得た。
【0240】
【表22】
【0241】
1,3−ジアミノ−2−(テトラデシルチオアセチルチオ)プロパン二塩酸塩の調製(実施例15)
1,3−〔ジ(tert−ブトキシカルボニルアミノ)〕−2−テトラデシルチオアセチルチオプロパン(実施例15d)(0.300g、0.52mmol)を、ガス状塩酸で飽和されたエーテル(55ml)に溶解した。この混合物を室温で撹拌した。96時間の反応の後、形成された沈殿物を濾過し、ジエチルエーテルで数回洗浄し、乾燥して、目的化合物を白色粉末の形態で得た。
【0242】
【表23】
【0243】
実施例16:1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパンの調製
1,3−ジアミノ−2−テトラデシルチオアセチルチオプロパン二塩酸塩(実施例15)(100mg、0.225mmol)及びテトラデシルチオ酢酸(実施例1)(130mg、0.450)をジクロロメタン(30ml)に0℃で溶解し、続いてトリエチルアミン(68μl)、ジシクロヘキシルカルボジイミド(139mg、0.675mmol)及びヒドロキシベンゾトリアゾール(61mg、0.450mmol)を加えた。反応混合物を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(430mg)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を白色粉末の形態で得た。
【0244】
【表24】
【0245】
実施例17:1−アミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン塩酸塩の調製
1−(tert−ブチルオキシカルボニルアミノ)プロパン−2,3−ジオールの調製(実施例17a)
1−アミノプロパン−2,3−ジオール(5g、55mol)をメタノール(200ml)に溶解し、続いてトリエチルアミン(アミン1mmol当たり0.5ml)及びジ−tert−ブチルジカルボネート〔(BOC)2O〕(ここでBOCは、tertブチルオキシカルボニルである)(17.97g、82mmol)を滴加した。反応媒体を40〜50℃で20分間加熱し、次に室温で1時間撹拌した。溶媒を留去した後、無色の油状物残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 95:5)により精製して、目的化合物を無色油状物の形態で得て、それはゆっくりと結晶化した。
【0246】
【表25】
【0247】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(p−トルエンスルホニルオキシ)プロパンの調製(実施例17b)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−プロパン−2,3−ジオール(実施例17a)及びp−トルエンスルホニルクロリドから上記(実施例15a)で記載された方法に従って合成した。反応は白色の粉末を生成した。
【0248】
【表26】
【0249】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(アセチルチオ)プロパンの調製(実施例17c)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(p−トルエンスルホニルオキシ)−プロパン(実施例17b)及びチオ酢酸カリウムから上記(実施例15d)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0250】
【表27】
【0251】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジメルカプトプロパンの調製(実施例17d)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(アセチルチオ)−プロパン(実施例17c)を鹸化し、上記(実施例15c)で記載された方法に従って合成した。反応は白色の固体を生成し、直ちにそれを次の反応に使用した。
【0252】
【表28】
【0253】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(テトラデシルチオアセチルチオ)プロパンの調製(実施例17e)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジメルカプトプロパン(実施例17d)及びテトラデシルチオ酢酸(実施例1)から上記(実施例15d)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0254】
【表29】
【0255】
1−アミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン塩酸塩の調製(実施例17)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジテトラデシルチオアセチル−チオプロパン(実施例17e)から上記(実施例15)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0256】
【表30】
【0257】
実施例18:1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパンの調製
1−アミノ−2,3−ジテトラデシルチオアセチルチオプロパン塩酸塩(実施例17)(100mg、0.140mmol)及びテトラデシルチオ酢酸(実施例1)(62mg、0.210)をジクロロメタン(40ml)に0℃で溶解し、続いてトリエチルアミン(43ml)、ジシクロヘキシルカルボジイミド(59mg、0.28mmol)及びヒドロキシベンゾトリアゾール(29mg、0.210mmol)を加えた。反応混合物を0℃で1時間撹拌し、次に24時間で室温にした。次に穏やかに還流しながら48時間加熱し、次に乾燥した。得られた残渣(310mg)を、シリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 8:2)により精製して、目的化合物を白色の粉末として生成した。
【0258】
【表31】
【0259】
実施例19:1−テトラデシルチオアセチルチオ−2,3−ジ(テトラデシルチオアセチルアミノ)プロパンの調製
2,3−ジ(テトラデシルチオアセチルアミノ)−1−ヨードプロパンの調製(実施例19a)
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オール(実施例13)(0.200g、0.317mmol)をトルエン(30ml)に溶解した。次にイミダゾール(0.054g、0.792mmol)、トリフェニルホスフィン(0.208g、0.792mmol)及びヨウ素(0.161g、0.634mmol)をこの順序で加え、反応を撹拌しながら75〜80℃で加熱した。6時間の反応の後、溶媒を留去し、残留生成物を更に精製することなく使用した。
Rf(ジクロロメタン/メタノール 98:2):0.55
【0260】
2,3−ジ(テトラデシルチオアセチルアミノ)−1−メルカプトプロパンの調製(実施例19b)
硫化水素ナトリウム(0.089g、1.59mmol)を、アセトン(80ml)に溶解した2,3−ジテトラデシルチオアセチルアミノ−1−ヨードプロパン(実施例19a)(0.235g、0.32mmol)に加えた。反応媒体を70℃で16時間加熱した。溶媒を留去し、残渣を水にとって、クロロホルムで抽出した。水相を酢酸でpH6に酸性化し、次にクロロホルムで再び抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣を、更に精製することなく使用した。
【0261】
1−テトラデシルチオアセチルチオ−2,3−ジ(テトラデシルチオアセチルアミノ)プロパンの調製(実施例19)
2,3−ジテトラデシルチオアセチルアミノ−1−メルカプトプロパン(実施例19b)(0.205g、0.32mmol)をテトラヒドロフラン(50ml)に溶解した。次にジシクロヘキシルカルボジイミド(98mg、0.47mmol)、ジメチルアミノピリジン(58mg、0.47mmol)及びテトラデシルチオ酢酸(実施例1)(137mg、0.47mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、テトラヒドロフランで洗浄し、濾液を留去した。得られた残渣(1.14g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄土色粉末の形態で得た。
【0262】
【表32】
【0263】
実施例20:3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オールの調製
3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン−2−オールの調製(実施例20a)
クロロトリフェニルメタン(2.833g、10.16mmol)を、ピリジン(2.5ml)中の3−テトラデシルチオアセチルアミノプロパン−1,2−ジオール(実施例2)(3g、8.30mmol)の溶液に加えた。反応混合物を50℃で24時間撹拌し、次に溶媒を真空留去した。残渣を水にとって、ジクロロメタンで抽出した。有機相を、1N塩酸水溶液、次に塩化ナトリウムで飽和した水溶液で洗浄した。硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(6.36g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を白色粉末の形態で得た。
【0264】
【表33】
【0265】
2−ヨード−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパンの調製(実施例20b)
3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン−2−オール(実施例20a)(2g、3.31mmol)をトルエン(100ml)に溶解した。次にイミダゾール(0.564g、8.28mmol)、トリフェニルホスフィン(2.171g、8.28mmol)及びヨウ素(1.681g、6.62mmol)をこの順序で加えた。反応媒体を室温で20時間撹拌した。飽和重亜硫酸ナトリウム溶液を、反応媒体が完全に白くなるまで加えた。相を分離し、水相をトルエンで抽出した。有機相を合わせ、飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、濾過した。溶媒を留去した後で得られた残渣(4.65g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄色油状物の形態で得た。
【0266】
【表34】
【0267】
2−メルカプト−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパンの調製(実施例20c)
硫化水素ナトリウム水和物(38mg、0.68mmol)をエタノール(20ml)中の懸濁液として調製し、続いて2−ヨード−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン(実施例20b)(200mg、0.28mmol)を加えた。反応媒体を70℃で加熱した。硫化水素ナトリウム水和物238mgを数日間かけて加えた。6.5日後、溶媒を留去し、残渣をジクロロメタンにとって、水で洗浄した。水相を再抽出し、合わせた有機相を、0.5N塩酸、次に飽和塩化ナトリウム水溶液で洗浄し、次に硫酸マグネシウムで乾燥した。塩を濾過し、溶媒を留去した。得られた残渣を、更に精製することなく使用した。
【0268】
【表35】
【0269】
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオ−1−トリフェニルメチルオキシ−プロパンの調製(実施例20d)
2−メルカプト−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシトプロパン(実施例20c)(174mg、0.28mmol)をテトラヒドロフラン(20ml)に溶解した。次にジシクロヘキシルカルボジイミド(88mg、0.42mmol)、ジメチルアミノピリジン(51mg、0.42mmol)及びテトラデシルチオ酢酸(121mg、0.42mmol)を加え、反応媒体を室温で撹拌した。20時間の反応の後、溶媒を留去し、得られた残渣(45mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 3:7〜5:5)により精製して、目的化合物を白色粉末の形態で得た。
【0270】
【表36】
【0271】
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オールの調製(実施例20)
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオ−1−トリフェニルメチルオキシ−プロパン(実施例20d)(187mg、0.21mmol)を、ガス状塩酸で飽和されたエーテル(12ml)に溶解した。反応媒体を室温で20時間撹拌した。形成された沈殿物を濾過し、ジエチルエーテルで洗浄して、目的化合物を白色粉末の形態で得た。
【0272】
【表37】
【0273】
実施例21:3−テトラデシルチオアセチルアミノ−1−テトラデシルチオアセチルオキシ−2−テトラデシルチオアセチルチオプロパンの調製
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オール(実施例20a)(64mg、0.10mmol)をテトラヒドロフラン(7ml)に溶解した。次にジシクロヘキシルカルボジイミド(31mg、0.15mmol)、ジメチルアミノピリジン(18mg、0.15mmol)及びテトラデシルチオ酢酸(実施例1)(43mg、0.15mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、濾液を留去した。得られた残渣(140mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で得た。
【0274】
【表38】
【0275】
実施例22:1−アミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン塩酸塩の調製
1−tert−ブチルオキシカルボニルアミノ−3−ヨードプロパン−2−オールの調製(実施例22a)
1−〔(tert−ブチルオキシカルボニル)アミノ〕プロパン−2,3−ジオール(実施例17a)(3.88g、20mmol)をトルエン(250ml)に溶解した。次にイミダゾール(1.73g、25mmol)、トリフェニルホスフィン(6.65g、25mmol)及びヨウ素(5.15g、20mmol)をこの順序で加えた。反応媒体を室温で17時間撹拌し、0.5当量のイミダゾール、トリフェニルホスフィン及びヨウ素を加えた。21時間の反応の後、飽和亜硫酸ナトリウム溶液を、反応媒体が完全に白くなるまで加えた。相を沈降させ、水相をトルエンで2回抽出した。合わせた有機相を合わせ、飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(11.02g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を黄色のペーストとして得て、直ちにそれを次の反応に使用した。
【0276】
【表39】
【0277】
3−アセチルチオ−1−tert−ブチルオキシカルボニルアミノプロパン−2−オールの調製(実施例22b)
1−(tert−ブチルオキシカルボニルアミノ)−3−ヨードプロパン−2−オール(実施例22a)(2g、6.64mmol)及びチオ酢酸カリウム(0.948g、8.30mmol)をアセトン(30ml)に溶解し、媒体を16時間還流した。溶媒を真空留去し、残渣をジエチルエーテルにとって、次にセライト(登録商標)で濾過した。濾液を留去した。得られた残渣(1.69g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製し、次にフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄色油状物の形態で得た。
【0278】
【表40】
【0279】
1−tert−ブチルオキシカルボニルアミノ−3−メルカプトプロパン−2−オールの調製(実施例22c)
最小限のメタノール(7ml)で希釈された3−アセチルチオ−1−tert−ブチルオキシカルボニルアミノプロパン−2−オール(実施例22b)(0.307g、1.23mmol)を、窒素流下で脱酸素化しながらメタノール中の20%炭酸カリウム溶液(3.49ml、12.31mmol)に加えた。媒体を窒素流下、室温で20時間撹拌し、次に酢酸でpH6に酸性化し、濃縮乾固した。得られた残渣を水にとって、ジクロロメタンで抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、濃縮した。得られた油状残渣を、更に精製することなく直ちに次に反応に使用した。
【0280】
【表41】
【0281】
1−tert−ブチルオキシカルボニルアミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパンの調製(実施例22d)
1−(tert−ブチルオキシカルボニルアミノ)−3−メルカプトプロパン−2−オール(実施例22c)(0.200g、96mmol)をジクロロメタン(50ml)に溶解した。次にジシクロヘキシルカルボジイミド(0.398g、1.93mmol)、ジメチルアミノピリジン(0.236g、1.93mmol)及びテトラデシルチオ酢酸(実施例1)(0.557g、1.93mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(1.2g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色ペーストの形態で得た。
【0282】
【表42】
【0283】
1−アミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン塩酸塩の調製(実施例22)
1−(tert−ブトキシカルボニルアミノ)−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン(実施例22d)(300mg、0.40mmol)を、ガス状塩酸で飽和されたジエチルエーテル(70ml)に溶解し、反応媒体を室温で72時間撹拌した。形成された沈殿物を濾過し、ジエチルエーテルで洗浄し、乾燥して、目的化合物を白色粉末の形態で得た。
【0284】
【表43】
【0285】
実施例23:1−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパンの調製
3−アミノ−2−テトラデシルチオアセチルオキシ−1−テトラデシルチオアセチル−チオプロパン塩酸塩(実施例22)(100mg、0.15mmol)及びテトラデシルチオ酢酸(実施例1)(63mg、0.22)をジクロロメタン(30ml)に0℃で溶解し、続いてトリエチルアミン(0.044ml)、ジシクロヘキシルカルボジイミド(60mg、0.29mmol)及びヒドロキシベンゾトリアゾール(30mg、0.22mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(263mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を白色粉末の形態で得た。
【0286】
【表44】
【0287】
実施例24:本発明の式(I)により表される化合物の調製方法
下記実施例で記載されるインビトロ実験を実施するために、本発明の化合物を下記で記載されるように乳剤の形態で調製した。
Spoonerらにより記載されたように(Spooner, Clarkら, 1988)、本発明の化合物及びホスファチジルコリン(PC)を含む乳剤を調製した。本発明の化合物をPCと、クロロホルム中、4:1(m/m)比で混合し、この混合物を窒素下で乾燥し、次に一晩真空留去し、得られた粉末を、0.01M EDTAを含む0.16M KClにとって、次にこの脂質粒子を超音波により37℃で30分間分散させた。このようにして形成されたリポソームを次に超遠心分離(XL80超遠心分離機、Beckman Coulter, Villepinte, France)により25,000rpmで45分間分離することにより、100nmを超えてキロミクロンに近い大きさを有するリポソームを回収した。陰性対照として使用するために、PCだけからなるリポソームを同時に調製した。
本発明の化合物におけるリポソームの組成は、酵素比色トリグリセリドアッセイキットを用いて推定した。このアッセイは、脂質較正物質のCFAS、参照番号759350(Boehringer Mannheim GmbH, ドイツ)で作製した標準曲線に対して実行した。この標準曲線は、16〜500μg/mlの範囲の濃度を包含した。滴定プレート(96ウェル)上に1ウェルにつき各試料希釈液又は較正標準液100μlを滴下した。次にトリグリセリド試薬、参照番号701912(Boehringer Mannheim GmbH, ドイツ)200μlを各ウェルに加え、プレート全体を37℃で30分間インキュベートした。光学密度(OD)は、分光光度計により492nmで読みとった。各試料におけるトリグリセリド濃度は、一次関数:y=ax+b〔式中、yは、ODを表し、そしてxは、トリグリセリド濃度を表す〕としてプロットした標準曲線から計算した。
【0288】
このようにして調製された、本発明の化合物を含有するリポソームを、実施例26、27及び28に記載されているインビトロ実験で使用した。
【0289】
実施例25:本発明の化合物の抗酸化特性の評価
A−銅により誘導されるLDL酸化に対する保護:
LDLの酸化は、アテローム動脈硬化の発症及び進展において主要な役割を演じる、重要な変性である(Jurgens, Hoffら, 1987)。以下のプロトコールにより、化合物の抗酸化特性を証明することができる。他に記載がなければ、全ての試薬はシグマ(Sigma)(St Quentin, France)製である。
LDLは、Lebeauらで記載された(Lebeau, Furmanら, 2000)ように調製した。
試験化合物の溶液は、エタノール中10-2Mで調製し、最終濃度が1%(V/V)の総エタノール濃度で0.1〜100μMの範囲になるようにPBSで希釈した。
【0290】
酸化の前に、透析によりLDL調製物からEDTAを除去した。次に、16.6μM CuSO4100μlを、LDL800μl(125μgタンパク質/ml)及び試験化合物溶液100μlに加えることにより、酸化反応を30℃で行った。追跡すべき種であるジエンの形成は、銅の存在下又は不在下、本化合物により処理した試料中の234nmの光学密度により測定した。234nmの光学密度は、10分毎に8時間、サーモスタット付き分光光度計(Kontron Uvikon 930)で測定した。分析は三回繰り返した。対照試料に対して遅滞期潜伏時間をシフトするならば、化合物は抗酸化活性を有すると考えられた。発明者たちは、本発明の化合物が、LDL酸化(銅により誘導)を遅延させたことを証明したが、このことは、本発明の化合物が、固有の抗酸化活性を有することを示している。図2は、本発明の化合物により得られた結果の一例を表す。
【0291】
図2は、固有の抗酸化特性を示す、本発明の化合物Ex2、4、5、6及び11を示す。
図2aは、本発明の化合物が、遅滞期潜伏時間を化合物Ex2では13%を超えて、化合物Ex4では34.3%までシフトすることを示す。本発明の化合物は、酸化速度(図2b)又はジエン形成量(図2c)を変えるようには見えなかった。
【0292】
B−脂質過酸化に対して本発明の化合物により与えられる保護の評価:
試験した本発明の化合物は、その調製が実施例2〜23に記載されている化合物である。
LDL酸化は、TBARS法(チオバルビツール酸反応性基質)により測定した。
上述のものと同じ原理により、LDLをCuSO4の存在下で酸化させて、脂質の過酸化を以下のように評価した:
TBARSは、分光光度法により測定し、脂質ヒドロペルオキシド化は、ヨウ化物のヨウ素への脂質過酸化物依存性酸化を利用して測定した。結果は、マロンジアルデヒド(MDA)のnmolとして、又はnmolヒドロペルオキシド/mgタンパク質として表される。
共役ジエン形成の抑制を測定することにより得られた上記の結果を、LDL脂質過酸化を測定する実験により確認した。したがって、また本発明の化合物によって、銅(酸化剤)により誘導された脂質の過酸化に対するLDLの効率的な保護ができた。
【0293】
実施例26:細胞培養物に及ぼす本発明の化合物の抗酸化特性の測定
A−培養プロトコール:
ニューロン、ニューロブラストーマ(ヒト)及びPC12細胞(ラット)がこの種類の試験に使用した細胞株であった。PC12細胞は、ラットのクロム親和性細胞種から調製し、Greene 及び Tischler により特徴づけられた(Greene 及び Tischler, 1976)。これらの細胞は、通常、神経細胞の分化、信号伝達及び神経細胞死の試験に使用される。PC12細胞を前述された(Farinelli, Parkら, 1996)ように、10%ウマ血清及び5%ウシ胎仔血清を補充した完全なRPMI培地(Invitrogen)中で増殖させた。
内皮及び平滑筋細胞の初代培養物も使用した。細胞はプロモセル(Promocell)(Promocell GmBH, ハイデルベルグ)から得て、供給業者の指示書に従って培養した。
細胞を5〜100μMの範囲の異なる用量の化合物で24時間処理した。次に細胞を回収し、標的遺伝子の発現の増加を定量PCRで評価した。
【0294】
B−mRNA測定:
mRNAを、本発明の化合物で処理したか又はしなかった細胞培養物から抽出した。抽出は、Absolutely RNA RT-PCR ミニプレップキット(Stratagene, フランス)の試薬を用いて供給業者により指示されたとおりに実行した。次にmRNAを、分光法によりアッセイして、Light Cycler System (Roche, フランス) のLight Cycler Fast Start DNA Master Sybr Green Iキット(Roche)を用いる、定量RT−PCRにより定量した。抗酸化酵素スーパーオキシドジスムターゼ(SOD)、カタラーゼ及びグルタチオンペルオキシダーゼ(GPx)をコードする遺伝子に特異的なプライマー対をプローブとして使用した。アクチン及びシクロフィリン遺伝子に特異的なプライマー対を対照プローブとして使用した。
【0295】
定量RT−PCRで測定した抗酸化酵素遺伝子のmRNA発現の増加は、細胞が本発明の化合物で処理されたとき、使用した異なる細胞の種類で実証された。
【0296】
C−酸化ストレスの制御:
培養細胞における酸化種の測定:
化合物の抗酸化特性も、蛍光標識を用いて評価し、その酸化は、蛍光信号の出現により追跡した。発光した蛍光信号の強度の減少は、化合物で処理された細胞において下記の方法で測定した:上記のように培養されたPC12細胞(ブラック96ウェルプレート、透明底、Falcon)を、血清無含有培地で、H2O2の用量を増加させる(0.25mM〜1mM)と共に2及び24時間インキュベートした。インキュベーションの後、培地を除去し、細胞をPBS中の10μMジクロロジヒドロフルオレセインジアセタート溶液(DCFDA、Molecular Probes, Eugene, USA)と共に37℃、5%CO2雰囲気下で30分間インキュベートした。次に細胞をPBSですすいだ。酸化標識により発光している蛍光を、蛍光計(Tecan Ultra 384)により、励起波長495nm及び発光波長535nmで測定した。結果は、酸化対照に対する保護の百分率として表す。
蛍光強度は、未処理細胞よりも本発明の化合物とインキュベートした細胞において低かった。これらの知見は、本発明の化合物が、酸化ストレスに曝された細胞において酸化種の産生の抑制を促進することを示す。前述した抗酸化特性も、培養された細胞における抗ラジカル保護の誘導に有効である。
【0297】
D−脂質の過酸化の測定:
異なる細胞株(上記の細胞モデル)及び初代細胞培養物を前記のように処理した。処理の後、細胞上清を回収し、細胞を溶解し、タンパク質濃度の測定のために回収した。脂質の過酸化を下記のように検出した:脂質の過酸化を、マロンジアルデヒド(MDA)などのアルデヒドの脂質過酸化と反応する、チオバルビツール酸(TBA)を用いて測定した。処理のあと、細胞上清を収集し(900μl)し、ブチル化ヒドロキシトルエン90μlを加えた(Morliere, Moysanら, 1991)。15%トリクロロ酢酸を含有する0.25M塩酸中のTBAの0.375%溶液1ミリリットルもこの反応媒体に加えた。この混合物を80℃で15分間加熱し、氷上で冷却して、有機相をブタノールで抽出した。有機相を、シマヅ1501分光蛍光計(島津製作所、京都、日本)で、分光蛍光法(λexc=515nm及びλem=550nm)により分析した。TBARSを、テトラ−エトキシプロパンを標準として使用してMDA当量として表した。結果を、タンパク質濃度に対して正規化した。
本発明の化合物で処理された細胞において観察された脂質の過酸化の減少は、前記の結果を確認する。
本発明の化合物は、有利には、酸化ストレスの影響を遅延させる及び/又は抑制する、固有の抗酸化特性を示す。発明者たちは、また、本発明の化合物が、抗酸化酵素をコードする遺伝子の発現を誘導することができることも示す。これらの本発明の化合物の特定の特徴は、細胞が酸化ストレスと有効に戦うことを可能にし、したがってフリーラジカル誘発損傷から保護され得ることを可能にする。
【0298】
実施例27:本発明の化合物によるインビトロでのPPAR活性の評価
異常脂肪血症及び糖尿病の処置に病院で広く使用されている2つの主な医薬分類であるフィブラート系及びグリタゾン系(glitazones)により活性化される、PPARサブファミリーの核内受容体は、脂質及びブドウ糖のホメオスタシスにおいて重要な役割を演じる。下記の実験データは、本発明の化合物がPPARαをインビトロで活性化することを示す。
【0299】
PPAR活性化を、酵母菌gal4転写因子のDNA結合ドメインと、異なるPPARのリガンド結合ドメインとから構成されるキメラの転写活性を測定することにより、RK13線維芽細胞の細胞株又は血液細胞HepG2においてインビトロで試験した。下記の例は、HepG2細胞で示す。
【0300】
A−培養プロトコール:
HepG2細胞は、ECACC(Porton Down, UK)から得て、10%(V/V)ウシ胎仔血清、100U/mlペニシリン(Gibco, Paisley, UK)及び2mM L−グルタミン(Gibco, Paisley, UK)を補充したDMEM培地で増殖させた。培地は2日毎に取り換えた。細胞は、空気95%/CO2 5%の加湿した雰囲気中、37℃で保持した。
【0301】
B−トランスフェクションに使用したプラスミドの記載:
プラスミドpG5TkpGL3、pRL−CMV、pGal4−hPPAR、pGal4−hPPAR及びpGal4−fは、Raspeらにより記載された(Raspe、Madsenら, 1999)。pGal4−mPPARα及びpGal4−hPPARβ作成体を、マウスPPARα及びヒトPPARβ核内受容体のDEFドメインに対応するPCR増幅DNA断片を、それぞれpGal4−fベクター中にクローン化することにより得た。
【0302】
C−トランスフェクション:
HepG2細胞を24ウエル培養皿に5×104細胞/ウエルで接種し、前述のプロトコール(Raspe, Madsen ら, 1999)に従って、レポータープラスミドpG5TkpGL3(50ng/ウエル)、発現ベクターpGal4−f、pGal4−mPPARα、pGal4−hPPARα、pGal4−hPPARγ、pGal4−hPPARβ(100ng/ウエル)及びトランスフェクション効率制御ベクターpRL−CMV(1ng/ウエル)を2時間トランスフェクトし、次に試験化合物と共に36時間インキュベートした。実験の終了時に、細胞を溶解し(Gibco, Paisley, UK)、ルシフェラーゼ活性を、Dual-Luciferase(商標)Reporter Assay Systemキット(Promega, Madison, WI, USA)により、供給者の使用説明書に従って測定した。次に、Bio-Rad Protein Assayキット(Bio-Rad, Munich, Germany)で細胞抽出物中のタンパク含有量を供給業者による指示により測定した。
【0303】
発明者は、本発明の化合物で試験され、pGal4−hPPARαプラスミドをトランスフェクトされた細胞のルシフェラーゼ活性が増加したことを示す。前記のルシフェラーゼ活性の誘導は、本発明の化合物がPPARαの活性化剤であることを示す。図3は、本発明の化合物により得られた結果の例を示す。
図3:Gal4/PPARαプラスミドをトランスフェクトしたHepG2細胞を、異なる濃度(5、15、50及び100μM)の本発明の化合物(Ex2、Ex4、Ex5、Ex6、Ex11)と共に24時間、並びに本発明の化合物の濃度5、15、50及び100μM用の対照として1、2、3、4と示されている異なる濃度のビヒクル(PC)と共にインキュベートした[実施例24(本発明の式(I)により表される化合物の調製方法)で記載された4:1(m/m)比に従う]。結果は、異なる処理の後で、誘導係数(処理細胞の発光信号を未処理細胞の発光信号で除す)として表される。誘導係数が高いほど、PPARαアゴニスト活性が強力である。結果は、本発明の化合物Ex2が50μMで最大19.8倍、100μMでは19.2倍、15μMでは7.7倍、5μMでは1.5倍の発光信号の誘導を生じることを示す。本発明の化合物Ex5も、100μMで10.5、50μMで7、15μMで2.5、5μMで1.2である誘導係数の用量依存的増加を示した。本発明の化合物Ex6も、発光信号の増加を誘導し、PPARα核内受容体に対する活性を明らかにした。本発明の化合物Ex6の誘導係数は、100μMで14.5、50μMで9.6、15μMで2.2、5μMで1.1であった。一方、細胞をビヒクル(PCリポソーム)と共にインキュベートした場合、有意な誘導は観察されなかった。
これらの結果は、試験された本発明の化合物が有意なPPARαリガンド活性を示し、したがってそれらの転写活性を可能にすることを示す。
【0304】
実施例28:本発明の化合物の抗炎症特性の評価
炎症反応は、脳虚血などの多くの神経疾患において観察される。炎症は、また、神経変性の重要な因子である。脳卒中では、グリア細胞の最初の反応の1つは、サイトカイン及びフリーラジカルを放出することである。このサイトカイン及びフリーラジカルの放出により、脳における炎症性反応を生じ、これが神経細胞死を引き起こす可能性がある(Rothwell 1997)。
細胞株及び初代細胞を、上記のように培養した。
リポ多糖類(LPS)細菌性内毒素(Escherichia coli 0111 :B4)(Sigma, フランス)を蒸留水で再構成し、4℃で保存した。細胞をLPS 1μg/mlで24時間処理した。他の因子からの干渉を避けるため、培地を完全に交換した。
TNF−αは、ストレス(例えば、酸化ストレス)に対する炎症反応における重要な因子である。LPSの用量の増加による刺激に応答するTNF−α分泌を評価するため、刺激を受けた細胞の培地を除去し、TNF−αをELISA−TNF−αキット(Immunotech, フランス)によりアッセイした。試料を50倍に希釈して、標準曲線(Chang, Hudsonら, 2000)の範囲内にした。
【0305】
化合物の抗炎症特性を下記のように特徴付けした:細胞培地を完全に交換し、細胞を試験化合物と共に2時間インキュベートし、その後、LPSを最終濃度1μg/mlで培地に加えた。24時間のインキュベーションの後、細胞上清を回収し、直ちに処理しない場合は−80℃で保存した。細胞を溶解し、タンパク質をBio-Rad Protein Assayキット(Bio-Rad, Munich, Germany)で供給業者の指示書により定量した。
【0306】
試験化合物による処理で誘導されたTNF−α分泌の減少の測定値は、pg/ml/μgタンパク質として、かつ対照に対する百分率として表される。これらの結果は、本発明の化合物が、抗炎症特性を有することを示す。
【0307】
実施例29:脳虚血−再灌流モデルにおける本発明の化合物の神経保護作用の評価
A−予防モデル:
1−動物の処置
1.1動物及び化合物の投与
体重200〜350gのウィスターラットをこの実験に使用した。
動物を、12時間の明/暗サイクル、20℃±3℃の温度で維持した。水及び食餌は自由に摂取可能であった。食物摂取量及び体重増加を記録した。
中大脳動脈の閉塞により誘導される虚血の前に、動物を、ビヒクル(0.5%カルボキシセルロース(CMC)及び0.1%ツイーン)に懸濁した本発明の化合物を用いる(600mg/kg/日)か又はビヒクルのみを用いる、胃管栄養法により14日間処置した。
使用したカルボキシメチルセルロースは、中間粘度のカルボキシメチルセルロースのナトリウム塩である(Ref. C4888, Sigma-Aldrich, フランス)。使用されたツイーンは、ポリオキシエチレンソルビタンモノオレエートである(Tween 80, Ref. P8074, Sigma-Aldrich, フランス)。
【0308】
1.2中大脳動脈の腔内閉塞による虚血誘導−再灌流:
動物を抱水クローラル300mg/kgの腹腔内注入により麻酔した。直腸プローブを挿入し、体温を37℃±0.5℃に維持した。試験を通して、血圧を監視した。
外科用顕微鏡下で、頸の中央部の切開により右頸動脈を露出した。翼突口蓋動脈をその起点で結紮し、動脈切開を外頸動脈で行って、ナイロン製のモノフィラメントを挿入できるようにし、それを、総頸動脈まで穏やか進め、次に内頸動脈内に進めて、中大脳動脈の起点を閉塞するようにした。1時間後にフィラメントを抜いて再灌流を可能にした。
【0309】
2−脳梗塞量の測定:
再灌流の24時間後、本発明の化合物で予め処置したか又は処置していない動物に、ペントバルビタールの過剰投与により麻酔をかけた。
脳を急速に凍結し薄切りにした。切片をクレシルバイオレットで染色した。脳切片の未染色の帯域は、梗塞により損傷を受けたと考えられた。領域(梗塞及び2つの半球)を測定し、梗塞及び2つの半球の量を計算し、修正梗塞量を次の式:[修正梗塞量=梗塞量−(右半球の量−左半球の量)]により決定して、脳浮腫を補償した。
本発明の化合物で処置した動物の脳切片の分析によって、未処置の動物と比較して、梗塞量の著しい減少が明らかとなった。虚血の前に、本発明の化合物を動物に投与すると(予防効果)、これらは神経保護を誘導することができた。
【0310】
3−抗酸化酵素活性の測定:
ラットの脳を冷凍し、粉砕し、粉末まで小さくし、次に食塩水に再懸濁した。次に、下記の著者により記載されているように、異なる酵素活性を測定した:スーパーオキシドジスムターゼ(Flohe 及び Otting 1984);グルタチオンペルオキシダーゼ(Paglia 及び Valentine 1967);グルタチオンレダクターゼ(Spooner, Delidesら, 1981);グルタチオン−S−トランスフェラーゼ(Habig 及び Jakoby 1981);カタラーゼ(Aebi 1984)。
これら異なる酵素活性は、本発明の化合物で処置された動物の脳調製物において増加した。
【0311】
B−治療的又は急性期処置モデル:
1−中大脳動脈の腔内閉塞による虚血誘導/再灌流:
前記のような動物をこの実験に使用した。動物を抱水クローラル300mg/kgの腹腔内注入により麻酔した。直腸プローブを挿入し、体温を37℃±0.5℃に維持した。試験を通して、血圧を監視した。
外科用顕微鏡下で、頸の中央部の切開により右頸動脈を露出した。翼突口蓋動脈をその起点で結紮し、動脈切開を外頸動脈で行って、ナイロン製のモノフィラメントを挿入できるようにし、それを、総頸動脈まで穏やか進め、次に内頸動脈内に進めて、中大脳動脈の起点を閉塞するようにした。1時間後にフィラメントを抜いて再灌流を可能にした。
【0312】
2−動物の処置:
最初に虚血−再灌流に付した動物を、再灌流の後、本発明の化合物を用いて経口(前記のようなCMC+ツイーンビビクル)により1回以上処置した(600mg/kg/日又は300mg/kg/日を1日二回)。
【0313】
3−脳梗塞量の測定:
再灌流の24、48又は72時間後、化合物で予め処置したか又は処置していない動物に、ペントバルビタールの過剰投与により麻酔をかけた。
脳を急速に凍結し薄切りにした。切片をクレシルバイオレットで染色した。脳切片の未染色の帯域は、梗塞により損傷を受けたと考えられた。領域(梗塞及び2つの半球)を測定し、梗塞及び2つの半球の量を計算し、修正梗塞量を次の式:[修正梗塞量=梗塞量−(右半球の量−左半球の量)]により決定して、脳浮腫を補償した。
治療的処置の場合(急性期の処置)、本発明の化合物で処置された動物は、未処置の動物よりも脳損傷が少なかった。事実、虚血−再灌流の24時間、48時間又は72時間後、本発明の化合物を投与した場合、梗塞量は少なかった。
【0314】
したがって本発明の化合物は、急性虚血後の処置において神経保護活性を示す。
【0315】
様々な実験モデルにおける本発明の化合物の使用は、該新規化合物が固有の抗酸化活性を示し、酸化ストレスの影響を遅延させかつ減少させることができ、更に、抗酸化酵素をコードする遺伝子の発現も誘導し、その抗酸化特性と一緒になって、フリーラジカルからの保護を強化することを示す。加えて、本発明の化合物は、抗炎症活性を示し、PPARα核内受容体を活性化することができる。
最後に、本発明の化合物の動物虚血−再灌流モデルにおける使用は、保護的及び治療的でもある処置の両方の有益な神経保護作用を明らかにした。
【0316】
【表45】
【図面の簡単な説明】
【0317】
【図1】本発明の特定の化合物の構造である。その調製は、実施例2、4、5、6、8、10〜14、16、18、19、21及び23に記載されており、図においてそれぞれ1A.2、1A.4、1A.5、1A.6、1A.8、1A.10、1A.11、1A.12、1A.13、1A.14、1A.16、1A.18、1A.19、1A.21及び1A.23と示されている。
【図2】銅(Cu)によるLDL酸化に対する本発明の化合物の抗酸化特性の評価である。(2a)経時的な又は遅滞期における共役ジエン形成である。(2b)ジエン形成の速度である。(2c)共役ジエン形成最大量である。
【図3】Gal4/PPARトランス活性化系における本発明の化合物のPPARアゴニスト特性の評価である。
【技術分野】
【0001】
本発明は、新規アシル化アミノプロパンジオール、その窒素及び硫黄類似体、それを含む医薬組成物、その治療上の使用、特に脳虚血の処置のための使用に関する。本発明は、また、該誘導体の製造方法を提供する。
【0002】
本発明の化合物は、有利な抗酸化剤及び抗炎症医薬特性を有する。本発明は、また、該化合物及びそれを含む医薬組成物を使用する治療的処置の方法を記載する。特に、本発明の化合物は、脳卒中の予防又は治療に有用である。
【0003】
フランスでは、脳血管性疾患(年間150,000件の新規症例)は、死因の第3位であり、成人における身体障害の主要原因である。虚血性及び出血性脳卒中は、全ての脳血管障害において、それぞれ80%及び20%を占める。虚血性脳卒中は、脳血管性疾患の死亡率及び罹患率を減少させるために取り組まれなければならない、重要な治療上の問題である。虚血の急性期の処置ばかりでなく、虚血の予防においてもまた進歩があった。したがって、危険因子を同定し管理することは、この病変の治療において必須であることを留意することが重要である。
【0004】
薬剤に基づく脳虚血の処置は、異なる戦略に基づく。第1の戦略は、危険因子(高血圧、高コレステロール血症、糖尿病、心房細動など)の予防、又は血栓症の予防、特に抗血小板薬又は抗凝血薬の助けを借りることにより、脳虚血障害の発症を予防することを含む(Adams 2002) 及び (Gorelick 2002)。
【0005】
第2の戦略は、虚血の急性期を処置して、その長期の影響を減衰させることを含む(Lutsep 及び Clark 2001)。
【0006】
脳虚血の病態生理学は、下記のように記載されることができる:ニューロンが壊死している虚血性病巣と無傷の神経組織との間の中間領域である虚血性境界域は、病態生理学的カスケードの部位であり、これは、再灌流が起きないか又は神経保護が不十分である場合、数日間かけて神経細胞死を起こす。最初の事象は、最初の数時間以内に起こるが、多量のグルタミン酸を放出し、これが神経細胞脱分極及び細胞性浮腫を引き起こす。細胞内へのカルシウムの流入は、ミトコンドリアの損傷を誘導し、フリーラジカルの遊離及び神経細胞膜の分解を促進する酵素誘導を導く。カルシウム流入及びフリーラジカル産生は、次に、NF−κBなどの特定の転写因子を活性化する。該活性化は、内皮接着タンパク質の誘導、虚血病巣の多核好中球浸潤、ミクログリア活性化、酸化窒素(NO)II型シンターゼ又はII型シクロオキシゲナーゼのような酵素の誘導などの炎症過程を誘導する。これらの炎症過程は、細胞に毒性のあるNO又はプロスタノイドの放出を起こす。ともにこれらの過程は、結果として非可逆的病変を誘導するアポトーシスの現象となる(Dirnagl, Iadecola et al. 1999)。
【0007】
予防的神経保護の概念は、虚血耐性を示す動物モデルにおける実験データに基づく。事実、脳虚血を実験的に誘導する前に異なる手順を適用すると、後者の重篤度が減衰される。種々の刺激が脳虚血耐性を誘導することができる:すなわち、プレコンディショニング(遷延虚血に先立つ短期虚血)、熱ストレス、低用量の細菌性リポ多糖類の投与である(Bordet, Deplanque et al. 2000)。
【0008】
該刺激は、耐性機構を誘導し、それは保護機構を誘発する信号を活性化する。異なる誘発機構が同定されている:すなわち、サイトカイン、炎症性経路、フリーラジカル、NO、ATP−依存性カリウムチャンネル、アデノシンである。初期事象の発生と虚血耐性との間で観察される遅滞時間は、タンパク質合成の必要性に由来する。多様な種類のタンパク質が虚血耐性を誘導することが示されている:すなわち、熱ショックタンパク質、抗酸化酵素及び抗アポトーシス性タンパク質である(Nandagopal, Dawson et al. 2001)。
【0009】
したがって、アテローム動脈硬化、糖尿病、肥満などの脳血管障害の危険因子の進展を防止することができ、予防的神経保護ばかりでなく、脳虚血の急性期における積極的な神経保護もまた提供できる化合物の真の必要性がある。
【0010】
PPAR(α、β、γ)は、ホルモン活性化核内受容体ファミリーに属する。これらのリガンドとの結合により活性化される場合、これらはレチノイド−X−受容体(RXR)とヘテロ二量体化され、標的遺伝子のプロモーター配列に位置する「ペルオキシソーム増殖因子応答エレメント」(PPRE)と結合する。したがってPPARのPPREへの結合は、標的遺伝子の発現を誘導する(Fruchart, Staels et al. 2001)。
【0011】
PPARは、多種多様な器官に分布しているが、これらはPPARβを除いて全てある程度の組織特異性を示し、その発現は遍在していると思われる。PPARαの発現は、肝臓及び腸管腔において特に多く、PPARγは、主に脂肪組織及び脾臓に発現する。3つのサブタイプ(α、β、γ)は、中枢神経系に発現する。より詳細には、オリゴデンドロサイト及びアストロサイトなどの細胞が、PPARαサブタイプを発現する(Kainu, Wikstrom et al. 1994)。
【0012】
PPARの標的遺伝子は、脂質及び糖の代謝を制御する。しかし、最近の発見は、PPARが他の生物学的過程に関与していることを示唆している。PPARのそれらのリガンドによる活性化は、遺伝子の転写活性における変化を誘導し、そのことは、炎症性過程、抗酸化酵素、血管形成、細胞の増殖及び分化、アポトーシス、iNOS、MMPase及びTIMPの活性を調節する(Smith, Dipreta et al. 2001) 及び (Clark 2002)。
【0013】
フリーラジカルは、アレルギー、腫瘍のイニシエーション及びプロモーション、循環器疾患(アテローム性動脈硬化、虚血)、遺伝的及び代謝的障害(糖尿病)、感染性及び変性疾患(プリオンなど)並びに眼の疾患を含む極めて広範囲の病変において役割を果たす。
【0014】
活性酸素種(ROS)は、正常な細胞機能中に産生される。ROSは、ヒドロキシルラジカル(OH)、スーパーオキシドアニオン(O2-)、過酸化水素(H2O2)及び酸化窒素(NO)を包含する。該種は非常に不安定であり、それらの高い化学反応性のため、細胞の生物学的機能にとって脅威となる。それらは、脂質の過酸化、特定の酵素の酸化、及びタンパク質の分解をもたらす極めて広範囲に及ぶタンパク質の酸化を誘導する。脂質の過酸化に対する保護は、過酸化生成物がDNAの損傷を引き起こしうるため、好気性生物において必須のプロセスである。したがって、天然の抗酸化剤防御によるラジカル種の産生、処理及び除去の間の平衡の調節解除又は修飾によって、細胞又は生物に有害であるプロセスが確立される。
【0015】
ROSは、酵素成分と非酵素成分とを含む抗酸化剤系を介して処理される。酵素系は、下記の性質を有する幾つかの酵素から構成される:
− スーパーオキシドジスムターゼ(SOD)は、スーパーオキシドラジカルを過酸化物に変換することにより破壊する。次に過酸化物を別の酵素系に作用させる。低濃度のSODは好気的呼吸により継続的に産生される。ヒトにおいて3種のSODが同定されており、それぞれ補助因子としてCu、Zn、Fe、Mn又はNiを含有する。3つの形態のヒトSODが下記のように分布する:細胞基質Cu−Zn SOD、ミトコンドリアMn−SO及び細胞外SOD。
− カタラーゼは、過酸化水素(H2O2)を水と酸素に変換するのに非常に効率的である。過酸化水素は、好気性生物において酵素的に異化される。カタラーゼは、また、種々のヒドロペルオキシド(ROOH)の還元に触媒作用を及ぼす。
− グルタチオンペルオキシダーゼは、セレンを補助因子として使用し、グルタチオンを利用してヒドロペルオキシド(ROOH及びH2O2)の還元に触媒作用を及ぼし、それにより酸化損傷から細胞を保護する。
【0016】
細胞の非酵素的抗酸化剤防御は、合成されるか又は食事中に供給される分子を含む。
【0017】
抗酸化剤分子は、異なる細胞区画に存在する。例えば、解毒酵素はフリーラジカルを除去し、細胞の寿命にとって必須である。3つの最も重要な種類の抗酸化剤化合物は、カロチノイド、ビタミンC及びビタミンEである(Gilgun-Sherki, Melamed et al. 2001)。
【0018】
脳虚血及びその結果として起こる影響により誘導されるアポトーシスの現象を回避するため、本発明者たちは、該危険因子の進展を防止することができ、予防的神経保護活性を行うばかりでなく、脳虚血の急性期における積極的な神経保護もまた提供することができる新規化合物を開発した。
【0019】
本発明者たちは、また、PPAR活性化剤、抗酸化剤及び抗炎症特性を同時に示す本発明の化合物を示し、したがって該化合物は、脳虚血における重要な治療的又は予防的潜在能力を有する。
【0020】
したがって、本発明は、脳虚血の予防的又は治療的処置に有用な、有利な薬理学的性質を示す新規な化合物のファミリーを提供する。本発明は、また、該誘導体の製造方法も提供する。
【0021】
本発明の化合物は、一般式(I):
【0022】
【化2】
【0023】
〔式中、
G2及びG3は、独立して酸素原子、硫黄原子又はN−R4基(G2及びG3は、同時にN−R4基を表すことはない)を表し、
R及びR4は、独立して水素原子、又は飽和若しくはそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖若しくは分岐のアルキル基を表し、
R1、R2及びR3は、同一であるか又は異なって、水素原子、CO−R5基、又は式:CO−(CH2)2n+1−X−R6に対応する基(R1、R2及びR3基の少なくとも1個は、式:CO−(CH2)2n+1−X−R6に対応する基である)を表し、
R5は、飽和又はそうではない、場合により置換されている、可能であれば環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、1〜25個の炭素原子を含む)であり、
Xは、硫黄原子、セレン原子、SO基又はSO2基であり、
nは、0〜11の間に含まれる整数であり、
R6は、飽和又はそうではない、場合により置換されている、可能であれば環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは10〜23個の炭素原子を含み、場合により酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される1個以上のヘテロ基を含む)である〕により表される
(但し、G2R2及びG3R3が同時にヒドロキシル基を表す式(I)を有する化合物は除く)。
【0024】
本発明の一般式(I)により表される化合物において、R5基は、同一であるか又は異なって、好ましくは、飽和又は不飽和の、置換されているか又はされていない、直鎖又は分岐のアルキル基(その主鎖は、1〜20個の炭素原子、さらにより好ましくは7〜17個の炭素原子、さらに好ましくは14〜17個を含む)を表す。本発明の一般式(I)により表される化合物において、R5基は、同一であるか又は異なって、1〜6個の炭素原子を含む低級アルキル、例えば、特にメチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、ペンチル又はヘキシル基などを表すこともできる。
【0025】
本発明の一般式(I)により表される化合物において、R6基は、同一であるか又は異なって、好ましくは、飽和又は不飽和の、置換されているか又はされていない、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、さらにより好ましくは14〜17個の炭素原子、さらになお好ましくは14個の炭素原子を含む)を表す。
【0026】
R5又はR6の飽和長鎖アルキル基の具体的な例は、特にC7H15、C10H21、C11H23、C13H27、C14H29、C15H31、C16H33、C17H35基である。R5又はR6の不飽和長鎖アルキル基の具体的な例は、特にC14H27、C14H25、C15H29、C17H29、C17H31、C17H33、C19H29、C19H31、C21H31、C21H35、C21H37、C21H39、C23H45基、又はエイコサペンタン酸(EPA)C20:5(5,8,11,14,17)及びドコサヘキサン酸(DHA)C22:6(4,7,10,13,16,19)のアルキル鎖である。
【0027】
分岐長鎖アルキル基の例は、特に(CH2)n′−CH(CH3)C2H5、(CH=C(CH3)−(CH2)2)n″−CH=C(CH3)2又は(CH2)2x+1−C(CH3)2−(CH2)n″′−CH3基〔xは、1〜11と等しいか又はその間に含まれる整数であり、n′は、1〜22と等しいか又はその間に含まれる整数であり、n″は、1〜5と等しいか又はその間に含まれる整数であり、n″′は、0〜22と等しいか又はその間に含まれる整数であり、そして(2x+n″′)は、22以下、好ましくは20以下である〕である。
【0028】
前記で示したように、アルキル基R5又はR6は、場合により環式基を含むことができる。環式基の例は、特にシクロプロピル、シクロブチル、シクロペンチル及びシクロヘキシルである。
【0029】
前記で示されるように、アルキル基R5又はR6は、場合により1個以上の置換基(同一であるか又は異なる)で置換されていることができる。置換基は、好ましくは、ハロゲン原子(ヨウ素、塩素、フッ素、臭素)及び−OH、=O、−NO2、−NH2、−CN、−O−CH3、−CH2−OH、−CH2OCH3、−CF3及び−COOZ基(Zは、水素原子又は好ましくは1〜5個の炭素原子を含むアルキル基である)からなる群より選択される。
【0030】
本発明は、また、該化合物の光学及び幾何異性体、そのラセミ体、塩、水和物、並びにこれらの混合物に関する。
【0031】
式(Ia)により表される化合物は、R1、R2又はR3基のうちのただ1個が水素原子を表す本発明の式(I)に対応する化合物である。
【0032】
式(Ib)により表される化合物は、R1、R2又はR3基のうちの2個が水素原子を表す本発明の式(I)に対応する化合物である。
【0033】
本発明は、また、式(I)により表される化合物のプロドラッグを包含し、それは、被検者に投与された後、式(I)により表される化合物に変換される、及び/又は特に脳虚血の治療において式(I)により表される化合物と同様の治療上の活性を示す式(I)により表される化合物の代謝産物に変換されるが、これらは式(I)により表される化合物に類似する。
【0034】
更に、CO−(CH2)2n+1−X−R6基において、Xは、最も好ましくは硫黄又はセレン原子、そして有利には硫黄原子を表す。
【0035】
更に、CO−(CH2)2n+1−X−R6基において、nは、好ましくは0〜3の間に含まれ、とりわけ0〜2の間に含まれ、特に0と等しい。
【0036】
本発明の一般式(I)により表される化合物において、R6は、酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される、1個以上、好ましくは0、1又は2個、より好ましくは0又は1個のヘテロ基を含むことができる。
【0037】
本発明のCO−(CH2)2n+1−X−R6基の特定の例は、CO−CH2−S−C14H29基である。
【0038】
したがって本発明の精神において好ましい化合物は、R1、R2及びR3基のうちの少なくとも1個が、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、好ましくは14〜17個、より好ましくは14〜16個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖のアルキル基である)を表す、上記記載の一般式(I)により表される化合物である。
【0039】
本発明の他の特定の化合物は、R1、R2及びR3基のうちの少なくとも2個が、同一であるか又は異なって、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表す)であるものである。
【0040】
本発明の特定の化合物は、G2が酸素原子又は硫黄原子、好ましくは酸素原子を表すものである。該化合物において、R2は、有利には上記で定義された式:CO−(CH2)2n+1−X−R6に対応する基を表す。
【0041】
特に好ましい化合物は、上記の一般式(I)
〔式中、
G3は、N−R4基(ここで、R4は、水素原子又はメチル基である)であり、そしてG2は酸素原子である;及び/又は
R2は、上記で定義されたCO−(CH2)2n+1−X−R6基を表す〕
により表される化合物である。
【0042】
他の好ましい化合物は、R1、R2及びR3が、同一であるか又は異なって、好ましくは同一で、上記で定義されたCO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、13〜17個の炭素原子、好ましくは14〜17個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖のアルキル基であり、ここでnは、好ましくは0〜3に間に含まれ、特に0と等しい)を表す、上記の一般式(I)により表される化合物である。より詳細には、好ましい化合物は、R1、R2及びR3がCO−CH2−S−C14H29基である、一般式(I)により表される化合物である。
【0043】
本発明の好ましい化合物の例は、図1に与えられている。
したがって本発明は、より詳細にはその目的として、
− 1−テトラデシルチオアセチルアミノ−2,3−(ジパルミトイルオキシ)プロパン;
− 3−テトラデシルチオアセチルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 3−パルミトイルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オール;
− 1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジオレイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパン;及び
− 1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン
から選択される一般式(I)により表される化合物を有する。
【0044】
本発明は、また、その目的として、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する上記記載の一般式(I)により表される化合物の少なくとも1個を、可能であれば別の治療剤と共に薬学的に許容されうる支持体中に含む、医薬組成物を有する。該組成物は、特に脳虚血又は出血性脳卒中などの脳血管性疾患を治療することが意図される。
【0045】
したがって本発明の別の目的は、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する、上記記載の一般式(I)により表される化合物の少なくとも1個を、薬学的に許容されうる支持体中に含む、あらゆる医薬組成物に関する。
【0046】
有利には、脳血管性の病変、より詳細には脳虚血又は脳血管性障害の治療又は予防のための医薬組成物である。事実、驚くべきことに、G2R2及びR3R3基が同時にヒドロキシル基を表す式(I)を有する化合物を包含する、式(I)により表される化合物は、同時に、PPAR活性化剤、抗酸化剤及び抗炎症性の特性を示し、かつ脳虚血の予防的及び治療的神経保護活性を示すことが見出された。
【0047】
本発明は、また、ヒト又は動物の治療又は予防方法の実施が意図される医薬組成物の調製における、上記で定義された化合物の使用に関する。
【0048】
本発明は、更に、脳血管性病変、より詳細には脳虚血を処置する方法であって、被検体、特にヒトに、G2R2及びR3R3基が同時にヒドロキシル基を表す一般式(I)を有する化合物を包含する、上記で定義された式(I)により表される化合物又は医薬組成物の有効投与量を投与することを含む方法に関する。
【0049】
有利には、使用される式(I)により表される化合物は、上記で定義されたものであり、また、3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオールも包含する。
【0050】
本発明の医薬組成物は、有利には、1つ以上の薬学的に許容されうる賦形剤又はビヒクルを含む。例としては、当業者に既知である、薬学的に適合されうる生理的な等張性の緩衝されている食塩水などが挙げられる。本組成物は、分散剤、可溶化剤、安定剤、界面活性剤、防腐剤などから選択される、1つ以上の作用物質又はビヒクルを含んでいてもよい。製剤(液体及び/又は注射用及び/又は固体)に使用してよい作用物質又はビヒクルは、特に、メチルセルロース、ヒドロキシメチルセルロース、カルボキシメチルセルロース、ポリソルベート80、マンニトール、ゼラチン、乳糖、植物油、アカシアなどが包含される。組成物は、注射用懸濁剤、ゲル剤、油剤、錠剤、坐剤、散剤、ゼラチンカプセル剤、カプセル剤などとして、可能であれば持続性及び/又は遅延性放出を可能にする医薬剤形又は装置に製剤化されてよい。この種類の処方には、有利にはセルロース、カーボネート又はデンプンのような作用物質が使用される。
【0051】
本発明の化合物又は組成物を、様々な方法及び様々な剤形で投与してよい。例えば、これらは、経口経路により、非経口的に、吸入により、又は例えば、静脈内、筋肉内、皮下、経皮、動脈内経路などのような注射により、全身的に投与してよい。注射には、本化合物は一般に液体懸濁剤の剤形に調製され、これはシリンジにより、又は例えば点滴により注入してよい。この点に関して、本化合物は一般に、当業者に既知の薬学的に適合されうる生理的な等張性の緩衝された食塩水などに溶解される。例えば、本組成物は、分散剤、可溶化剤、乳化剤、安定剤、界面活性剤、防腐剤、緩衝剤などから選択される、1つ以上の作用物質又はビヒクルを含んでもよい。液体及び/又は注射用製剤に使用してよい作用物質又はビヒクルは、特に、メチルセルロース、ヒドロキシメチルセルロース、カルボキシメチルセルロース、ポリソルベート80、マンニトール、ゼラチン、乳糖、植物油、アカシア、リポソームなどを包含する。
【0052】
したがって、本組成物は、ゲル剤、油剤、錠剤、坐剤、散剤、ゼラチンカプセル剤、カプセル剤、エアゾールなどの剤形で、可能であれば徐放及び/又は遅延放出を可能にする医薬剤形又は装置により投与してよい。この種類の処方には、有利にはセルロース、カーボネート又はデンプンのような作用物質が使用される。
【0053】
本化合物は、使用される作用物質又はビヒクルが、好ましくは水、ゼラチン、ゴム、乳糖、デンプン、ステアリン酸マグネシウム、タルク、油、ポリアルキレングリコールなどからなる群より選択される場合には、経口投与してもよい。
【0054】
非経口投与には、本化合物は、特に水、油又はポリアルキレングリコールを用いる、好ましくは液剤、懸濁剤又は乳剤の剤形で投与されるが、ここに保存料、安定剤、乳化剤などの他に、浸透圧を調整するための塩、緩衝剤などを加えることが可能である。
【0055】
注入速度及び/又は注入用量は、当業者により患者、病気、投与の様式などに適合されてよいことが理解される。典型的には、本化合物は、1用量当たり1μg〜2g、好ましくは1用量当たり0.1mg〜1gの範囲の用量で投与される。用量は、場合に応じて、1日1回又は1日数回投与することができる。更には、本発明の組成物は、また、他の活性物質又は活性剤を含んでもよい。
【0056】
本発明は、また、上記化合物の製造方法にも関する。本発明の化合物は、当業者に既知の化学反応の組合せを使用して、市販の製品から調製することができる。
【0057】
本発明の1つの方法によると、式(I)〔式中、(i)G2及びG3は、酸素若しくは硫黄原子、又はN−R4基であり、(ii)R及び場合によってはR4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、同一の直鎖又は分岐のアルキル基を表し、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(I)〔式中、(i)G2又はG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物と、式:A1−LG〔式中、A1は、基R又は場合によってはR4を表し、そしてLGは、例えば、Cl、Br、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得る。
【0058】
第1の実施態様において、式(I)〔式中、(i)G2及びG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であって、CO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(I)〔式中、(i)G2又はG3は、酸素若しくは硫黄原子、又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子である〕により表される化合物と、式:A°−CO−A〔式中、Aは、例えば、OH、Cl、O−CO−A°及びO−R7(R7はアルキル基である)からなる群より選択される反応性基であり、そしてA°は、(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得られる。
【0059】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、様々な方法により得ることができるが、その方法は、同じヘテロ原子(窒素又は酸素)に担持される基が同じ意味を有する化合物の合成を可能にする。
【0060】
第1の実施態様によると、1−アミノグリセロール、1,3−ジアミノグリセロール又は1,2−ジアミノグリセロールの分子((Morris, Atassi et al. 1997)により記載されたプロトコールを適用することにより得る)を、式:A°−CO−A1〔式中、A1は、例えば、OH、Cl及びOR7(R7はアルキル基である)からなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる。該反応は、それぞれ式(I)により表される化合物の特定の形態(化合物(IIa−c)と称する)を生じるが、(Urakami and Kakeda 1953)、(Shealy, Frye et al. 1984)、(Marx, Piantadosi et al. 1988) 及び (Rahman, Ziering et al. 1988) 又は (Nazih, Cordier et al. 1999)により記載されたプロトコールを適用して実施することができる。化合物(IIb−c)において、それぞれ同じヘテロ原子に担持されている基(R1及びR3)及び(R1及びR2)は、同じ意味を有する。
【0061】
【化3】
【0062】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式(IIa−c)を有する化合物と、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得ることができる。該反応は、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R1及びR2)、(R1及びR3)又は(R2及びR3)が同じ意味を有する化合物の合成を可能にする。有利には、該反応は、例えば、(Urakami and Kakeda 1953) 及び (Nazih, Cordier et al. 1999)により記載されたプロトコールに従って実施される。
【0063】
本発明の別の特定の方法(ダイアグラム1)によると、式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R5基を表す〕により表される化合物は、下記の工程により得ることができる:
【0064】
a)1−アミノグリセロール、1,3−ジアミノグリセロール又は1,2−ジアミノグリセロールを化合物(PG)2O(ここで、PGは、保護基である)と反応させて一般式(IIIa−c)を有する化合物を得る工程。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる〔ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す〕。
【0065】
b)式(IIIa−c)を有する化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(IVa−c)〔式中、R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得る工程。
【0066】
c)化合物(IVa−c)を当業者に既知の従来の条件に従って脱保護して、一般式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3はCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得る工程。
【0067】
d)一般式(I)〔式中、(i)G2及びG3は、酸素原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる工程。
【0068】
【化4】
【0069】
本発明の式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、様々な方法で得ることができる。
【0070】
第1の方法によると、本発明の一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR2は、水素原子であり、そして(iii)R1、R3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる。
【0071】
この調製方法によると、一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR2は、水素原子であり、そして(iii)R1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、上記で定義された式(IIa)により表される化合物と、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で得ることができる。
【0072】
本発明の別の特定の方法によると、式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−C−R6基を表す〕により表される化合物を、本発明の式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R、R2及びR3は、水素原子を表し、そして(iii)R1は、CO−R5又はCO−(CH2)2n+1−X−R6基である〕により表される化合物(式(IIa)の化合物)から、下記の工程に従って得ることができる(ダイアグラム2):
【0073】
a)式(IIa)により表される化合物を、化合物PG−E(ここで、PGは、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させることにより、一般式(V)〔式中、R1は、CO−R5又はCO−(CH2)2n+1−X−R6基である〕により表される化合物を得る工程。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここで、PG−Eはトリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0074】
b)式(V)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(VI)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得る工程。
【0075】
c)化合物(VI)を当業者に既知の条件で脱保護することにより、一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR3は、水素原子であり、そして(iii)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得る工程。
【0076】
d)一般式(I)〔式中、(i)G2及びG3は、酸素原子であり、(ii)R及びR3は、水素原子であり、そして(iii)R1及びR2は、同一であるか又は異なって、CO−R5又は(CO−CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させる工程。
【0077】
【化5】
【0078】
有利な方法において、上記の工程は、(Marx, Piantadosi et al. 1988)により記載されたプロトコールに従って実施される。
【0079】
本発明の別の方法によると、式(I)〔式中、(i)G2又はG3は、酸素原子又はN−R4基を表し、(ii)G2又はG3の少なくとも1つは、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iv)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(I)〔式中、(i)それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iii)R1は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより得られる。
【0080】
本発明の式(I)〔式中、(i)それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そして(iii)R1は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、本発明の式(I)〔式中、それぞれ、G2R2又はG3R3基のうちの一方は、ヒドロキシル基を表し、G2R2又はG3R3基のうちの他方は、NR4R2又はNR4R3基を表し(ここで、R2又はR3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す)、(ii)R及びR4は、独立して、上記で定義された基を表し、そして(iii)R1は、水素原子である〕により表される化合物と式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物とから、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で得られる。
【0081】
第1の実施態様において、本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iv)R1及びR2は、水素原子であり、そして(v)R3は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、下記の方法により得られる(ダイアグラム3):
【0082】
a)1−アミノグリセロールを、式:R−CHO〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてCHOは、アルデヒド官能基である〕に対応する化合物と、当業者に既知の還元剤の存在下で反応させて、式(VII)〔式中、Rは、上記で定義された基である〕により表される化合物を得ること。有利には、該反応は、(Antoniadou-Vyzas, Foscolos et al. 1986)により記載されたプロトコールを適用して実施することができる。
【0083】
b)式(VII)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(VIII)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる(ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す)。
【0084】
c)〔Kitchin, Bethell et al. 1994〕に記載された方法を適用して、式(VIII)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XI)により表される化合物を得ること。
【0085】
d)式(IX)により表される化合物を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕に対応する化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(X)〔式中、R及びR4は、場合により異なって、上記と同義である〕に対応する化合物を得ること。
【0086】
e)式(X)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(XI)〔式中、R及びR4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、R3は、R5基又は(CH2)2n+1−X−R6基を表し、そしてPGは、保護基である〕により表される化合物を得ること。
【0087】
f)化合物(XI)を当業者に既知の条件で脱保護すること。
【0088】
【化6】
【0089】
第2の実施態様によると、本発明の式(I)〔式中、(i)G3は、酸素原子であり、(ii)G2は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iv)R1及びR3は、水素原子であり、そして(v)R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、下記の方法により得られる(ダイアグラム4):
【0090】
a)式(VIII)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH又はハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XII)〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてPGは、上記で定義された別の保護基である〕により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載された方法を適用して実行することができる(ここで、PG′−Eはトリフェニルメチルクロリド又は9−フェニルキサンテン−9−オールあるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0091】
b)(Kitchin, Bethell et al. 1994)により記載された方法を適用して、上記で定義された式(XII)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メチル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XIII)〔式中、Rは、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてPG及びPG′は、保護基である〕により表される化合物を得ること。
【0092】
c)上記で定義された式(XIII)により表される化合物を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕に対応する化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(XIV)〔式中、R及びR4は、独立して、上記と同義である〕により表される化合物を得ること。
【0093】
d)式(XIV)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより、一般式(XV)〔式中、R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表し、そしてPG及びPG′は、保護基である〕により表される化合物を得ること。
【0094】
e)式(XV)により表される化合物を当業者に既知の従来の条件で脱保護して、本発明の一般式(I)〔式中、(i)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(ii)R1及びR3は、水素原子であり、そして(iii)R2は、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0095】
【化7】
【0096】
本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、様々な方法で得ることができる。
【0097】
第1の実施態様によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す(ここで、R1、R2及び/又はR3は、同じヘテロ原子(硫黄又は窒素)により担持されている場合、同じ意味を有する)〕により表される化合物は、下記の方法により得ることができる(ダイアグラム5A):
【0098】
a)式(IIa−c)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XVIa−c)により表される化合物を得ること。
【0099】
b)式(XVIa−c)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XVIIa−c)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0100】
c)式(XVIIa−c)により表される化合物を、当業者に既知の従来の条件で、例えば、塩基性媒体中で脱保護して、一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0101】
d)一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0102】
【化8】
【0103】
同様の合成方法によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す(ここで、R1、R2及び/又はR3は、同じヘテロ原子(硫黄又は窒素)により担持されている場合、同じ意味を有する)〕を有する化合物は、下記の方法により調製することができる(ダイアグラム5B):
【0104】
a)式(IIa−c)により表される化合物を、式(LG)2〔式中、LGは、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物と、可能であれば、当業者に既知の活性化剤の存在下で反応させて、一般式(XVId−f)により表される化合物を得ること。
【0105】
b)式(XVId−f)により表される化合物を、式:HS-B+〔式中、Bは、例えば、ナトリウム又はカリウム、好ましくはナトリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0106】
c)一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、そして(ii)R1、R2及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0107】
【化9】
【0108】
該反応は、一般式(I)〔式中、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R2及びR3)、(R1及びR3)、並びに(R1及びR2)は同じ意味を有する〕により表される化合物の合成を可能にする。
【0109】
上記工程は、(Adams, Doyle et al. 1960) 及び (Gronowitz, Herslof et al. 1978)で記載されたプロトコールに従って有利な方法で実施することができる。
【0110】
本発明の別の方法(ダイアグラム6)によると、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)Rは、水素原子であり、そして(iii)R1、R2及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa−c)により表される化合物から下記の方法で調製することができる:
【0111】
a)式(IIIa−c)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XVIIIa−c)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0112】
b)式(XVIIIa−c)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XIXa−c)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0113】
c)化合物(XIXa−c)の硫黄原子を当業者に既知の条件で脱保護して、一般式(XXa−c)により表される化合物を得ること。
【0114】
d)一般式(XXa−c)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXIa−c)〔式中、R2及びR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0115】
e)式(XXIa−c)により表される化合物を当業者に既知の従来の条件に従って脱保護して、本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基であり、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、水素原子、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0116】
f)本発明の式(I)〔式中、(i)G2及びG3は、硫黄原子又はNH基を表し、(ii)R及びR1は、水素原子であり、そして(iii)R2及びR3は、水素原子、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0117】
該反応は、一般式(I)〔式中、それぞれ同じヘテロ原子(窒素又は酸素)に担持されている基(R2及びR3)、(R1及びR3)及び(R1及びR2)は同じ意味を有する〕により表される化合物の合成を可能にする。
【0118】
有利な方法において、上記の工程は、(Adams, Doyle et al. 1960)、(Gronowitz, Hersloef et al. 1978)、(Bhatia and Hajdu 1987) 及び (Murata, Ikoma et al. 1991)により記載されたプロトコールに従って実施される。
【0119】
【化10】
【0120】
一般式(I)〔式中、(i)G2及びG3は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、(iii)R1、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、一般式(I)〔式中、(i)G2又はG3は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、上記で定義された基を表し、(iii)R1は、水素原子であり、そして(iv)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基を表す〕に対応する化合物と、可能であれば当業者に既知のカップリング剤又は活性化剤の存在下で反応させることにより得る。
【0121】
一般式(I)〔式中、(i)G2及びG3基は、硫黄原子又はN−R4基を表し、(ii)R及びR4は、独立して、上記で定義された基を表し、(iii)R1は、水素原子であり、そして、(iv)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕による表される化合物を、下記の方法により得ることができる:
【0122】
第1の実施態様において、本発明の式(I)〔式中、(i)G2基は、硫黄原子であり、(ii)G3は、N−R4基を表し、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表し、(iv)R1は、水素原子であり、そして(v)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、下記の方法により得る(ダイアグラム7):
【0123】
a)式(XI)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0124】
b)式(XXII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム又はカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XXIII)により表される化合物を得ること。有利には、該反応を、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施する。
【0125】
c)式(XXIII)により表される化合物の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXIV)により表される化合物を得ること。
【0126】
d)一般式(XXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXV)〔式中、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0127】
e)式(XXV)の化合物を当業者に既知の条件で脱保護すること。
【0128】
【化11】
【0129】
別の方法(ダイアグラム8)に従い、本発明の式(I)〔式中、(i)G2は、N−R4基を表し、(ii)G3は、硫黄原子であり、(iii)R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表し、(iv)R1は、水素原子であり、そして(v)R2及びR3は、同一であるか又は異なって、CO−R5基又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、下記の方法により得る:
【0130】
a)式(IX)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXVI)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0131】
b)式(XXVI)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXVII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0132】
c)化合物(XXVII)を、式:R4−NH2〔式中、R4は、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖又は分岐のアルキル基を表し、そしてNH2は、アミン官能基を表す〕により表される化合物と、(Ramalingan, Raju et al. 1995)により記載された方法に従って反応させて、式(XXVIII)〔式中、R及びR4は、独立して、飽和又はそうでない、場合により置換されている、1〜5個の炭素原子を含む、異なる直鎖又は分岐のアルキル基を表す〕により表される化合物を得ること。
【0133】
d)一般式(XXVIII)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXIX)により表される化合物を得ること。
【0134】
d)式(XXIX)により表される化合物の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXX)により表される化合物を得ること。
【0135】
f)一般式(XXX)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXIV)〔式中、R2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0136】
g)式(XXXI)により表される化合物を当業者に既知の従来の条件で脱保護すること。
【0137】
【化12】
【0138】
本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(V)を有する化合物から下記の方法により調製することができる(ダイアグラム9A):
【0139】
a)化合物(V)を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXII)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0140】
b)式(XXXII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXIII)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0141】
c)化合物(XXIII)の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXXIV)により表される化合物を得ること。
【0142】
d)一般式(XXXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXV)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0143】
e)化合物(XXXV)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR3は、水素原子であり、そしてR1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0144】
f)一般式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)R及びR3は、水素原子であり、そして(iv)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0145】
【化13】
【0146】
同様の合成方法によると、本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(V)の化合物から下記の方法により調製することができる(ダイアグラム9B):
【0147】
a)化合物(V)を、式(LG)2〔式中、LGは、例えば、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXIIa)〔式中、PGは、保護基を表す〕により表される化合物を得ること。
【0148】
b)式(XXXIIa)により表される化合物を、式:HS-B+〔式中、Bは、例えば、ナトリウム及びカリウム、好ましくはナトリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXIV)により表される化合物を得ること。
【0149】
c)一般式(XXXIV)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XXXV)〔式中、R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0150】
d)化合物(XXXV)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR3は、水素原子であり、そしてR1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0151】
e)一般式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)R及びR3は、水素原子であり、そして(iv)R1及びR2は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0152】
【化14】
【0153】
本発明の式(I)〔式中、(i)G2は、硫黄原子であり、(ii)G3は、酸素原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、同一であるか又は異なって、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム10):
【0154】
a)式(IIIa)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XXXVI)〔式中、PGは、前に定義された別の保護基である〕により表される化合物を得ること。有利な方法において、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここでPG−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0155】
b)化合物(XXXVI)を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物と反応させて、一般式(XXXVII)〔式中、PG及びPG′は、上記で定義されたような、賢明に選択された保護基を表す〕により表される化合物を得ること。
【0156】
c)式(XXXVII)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(XXXVIII)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0157】
d)化合物(XXXVIII)の硫黄原子を当業者に既知の従来の条件で脱保護して、一般式(XXXIX)により表される化合物を得ること。
【0158】
e)一般式(XXXIX)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XL)〔式中、R2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0159】
f)化合物(XL)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R、R1及びR3は、水素原子であり、そしてR2は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物(化合物XLI)を得ること。
【0160】
g)式(XLI)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(XLII)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる〔ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す〕。
【0161】
h)一般式(XLII)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、式(XLIII)により表される化合物を得ること。
【0162】
i)化合物(XLIII)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、硫黄原子であり、G3は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0163】
j)一般式(I)〔式中、(i)G2は、硫黄原子であり、G3は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0164】
【化15】
【0165】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R2は、水素原子又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIa)を有する化合物から下記の方法により調製することができる(ダイアグラム11):
【0166】
a)上記で定義された式(IIa)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIV)により表される化合物を得ること。
【0167】
b)式(XLIV)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(XLV)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0168】
c)式(XLV)により表される化合物を、化合物PG−E(ここで、PGは、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(XLVI)により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載されたプロトコールを適用して実行することができる(ここでPG−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0169】
d)化合物(XLVI)の硫黄原子を当業者に既知の条件で脱保護して、一般式(XLVII)により表される化合物を得ること。
【0170】
e)一般式(XLVII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(XLVIII)〔式中、R1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0171】
f)式(XLVIII)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR3は、水素原子であり、そしてR1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0172】
g)一般式(I)〔式中、(i)G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR1及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0173】
【化16】
【0174】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R1及びR3は、同一であるか又は異なって、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム12):
【0175】
a)上記で定義された式(IIIa)により表される化合物を、式:LG−E〔式中、Eは、ハロゲンを表し、そしてLGは、例えば、メシル、トシルなどからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIX)により表される化合物を得ること。
【0176】
b)式(XLIX)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、式(L)により表される化合物を得ること。有利には、該反応は、(Gronowitz, Hersloef et al. 1978)により記載されたプロトコールを適用して実施することができる。
【0177】
c)式(L)により表される化合物を、化合物PG′−E(ここで、PG′は、保護基であり、そしてEは、例えば、OH及びハロゲンからなる群より選択される反応性基である)と反応させて、一般式(LI)により表される化合物を得ること。有利には、本反応は、(Marx, Piantadosi et al. 1988) 及び (Gaffney and Reese 1997) に記載された方法を適用して実施することができる(ここでPG′−Eは、トリフェニルメチルクロリド又は9−フェニルキサンテン−9−オール、あるいはまた9−クロロ−9−フェニルキサンテンを表すことができる)。
【0178】
d)化合物(LI)の硫黄原子を当業者に既知の条件で脱保護して、一般式(LII)により表される化合物を得ること。
【0179】
e)一般式(LII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(LIII)〔式中、R3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0180】
f)式(LIII)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR3は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物(化合物LIV)を得ること。
【0181】
g)式(LIV)により表される化合物を、化合物(PG)2O(ここで、PGは、保護基である)と反応させて、一般式(LV)により表される化合物を得ること。有利には、反応は、(Nazih, Cordier et al. 2000) 及び (Kotsovolou, Chiou et al. 2001)により記載されたプロトコールを適用して実施することができる(ここで、(PG)2Oは、ジ−tert−ブチルジカルボネートを表す)。
【0182】
h)一般式(LV)により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、式(LVI)により表される化合物を得ること。
【0183】
i)化合物(LVI)を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G3は、硫黄原子であり、G2は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0184】
j)一般式(I)〔式中、G3は、硫黄原子であり、G2は、酸素原子であり、R及びR1は、水素原子であり、そしてR2及びR3は、同一であるか又は異なって、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0185】
【化17】
【0186】
本発明の式(I)〔式中、(i)G2は、酸素原子であり、(ii)G3は、硫黄原子であり、(iii)Rは、水素原子であり、(iv)R2及びR3は、同一であるか又は異なって、水素原子であるか、又はCO−R5若しくはCO−(CH2)2n+1−X−R6基を表し、そして(v)R1は、CO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物は、式(IIIa)を有する化合物から下記の方法により調製することができる(ダイアグラム13):
【0187】
a)上記で定義された式(IIIa)により表される化合物を、式(LG)2〔式中、LGは、例えば、ヨウ素、臭素などからなる群より選択される反応性基である〕に対応する化合物(理論量)と反応させて、一般式(XLIXa)により表される化合物を得ること。
【0188】
b)式(XLIXa)により表される化合物を、式:Ac−S-B+〔式中、Acは、短アシル基、好ましくはアセチル基を表し、そしてBは、例えば、ナトリウム及びカリウム、好ましくはカリウムからなる群より選択される対イオンである〕に対応する化合物と反応させて、一般式(L)により表される化合物を得ること。
【0189】
c)化合物(L)の硫黄原子を当業者に既知の条件で脱保護して、一般式(LVII)により表される化合物を得ること。
【0190】
d)一般式(LVII)により表される化合物を、式A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させて、一般式(LVI)〔式中、R2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0191】
e)式(LVI)により表される化合物を当業者に既知の従来の条件で脱保護することにより、一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を得ること。
【0192】
f)一般式(I)〔式中、G2は、酸素原子であり、G3は、硫黄原子であり、R及びR2は、水素原子であり、そしてR2及びR3は、同一でCO−R5又はCO−(CH2)2n+1−X−R6基を表す〕により表される化合物を、式:A°−CO−A2〔式中、A2は、例えば、OH及びClからなる群より選択される反応性基であり、そしてA°は、R5基又は(CH2)2n+1−X−R6基である〕に対応する化合物と、可能であれば、当業者に既知のカップリング剤又は活性化剤の存在下で反応させること。
【0193】
【化18】
【0194】
本発明の実行可能性、実現性及び他の利点は、以下の実施例において更に詳述されるが、これらの実施例は、説明の目的で与えられるものであり、限定を目的とするものではない。
【0195】
実施例:
文書のより容易な理解のために、活性の測定及び評価に関わる実施例で使用される本発明の化合物は、次のように省略される:例えば「Ex2」は、その調製が実施例2で記載されている本発明の化合物を示す。
【0196】
0.2mm厚のMERCKシリカゲル60F254で被覆されたプレートで薄層クロマトグラフィー(TLC)を実施した。保持係数はRfと省略される。
粒径40〜63μm(Merck参照番号9385−5000)を有するシリカゲル60でカラムクロマトグラフィーを実施した。
キャピラリー法によりBuchiB540装置で融点(MP)を測定した。
赤外線(IR)スペクトルをBruker Fourier変換スペクトロメーター(Vector 22)で記録した。
Bruker AC300スペクトロメーター(300MHz)で核磁気共鳴(NMR)スペクトルを記録した。各信号を、その化学シフト、強度、多重度(注:一重項はs、広帯一重項はsl、二重項はd、分離二重項はdd、三重項はt、分離三重項はtd、五重項はquint及び多重項はm)、及びその結合定数(J)により同定した。
Perkin Elmer Sciex API 1(ESI−MS:エレクトロスプレーイオン化質量分析(ElectroSpray lonization Mass Spectrometry))又はApplied Biosystems Voyager DE-STR製のMALDI-TOF型(マトリックス補助レーザー脱着/イオン化−飛行時間(Matrix-Assisted Laser Desorption/lonization - Time Of Flight))により質量スペクトル(MS)を測定した。
【0197】
実施例1:テトラデシルチオ酢酸の調製
水酸化カリウム(34.30g、0.611mol)、メルカプト酢酸(20.9ml、0.294mol)及び1−ブロモテトラデカン(50ml、0.184mol)をこの順序でメタノール(400ml)に加えた。この混合物を室温で一晩撹拌した。次に水(800ml)に溶解した濃塩酸溶液(60ml)を加えた。テトラデシルチオ酢酸が沈殿した。この混合物を室温で一晩撹拌した。次に沈殿物を濾過し、水で5回洗浄し、デシケーター中で乾燥した。この生成物をメタノール中で再結晶した。
【0198】
【表1】
【0199】
実施例2:3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオールの調製
テトラデシルチオ酢酸(実施例1)(14.393g、50mmol)及び3−アミノ−プロパン−1,2−ジオール(5g、55mmol)をフラスコに入れて、190℃で1時間加熱した。反応混合物を室温に冷却し、クロロホルムにとって、水で1回洗浄した。有機相を硫酸マグネシウムで乾燥し、濾過し、乾燥した。残渣をエーテル中で撹拌し、生成物を濾過により単離した。
【0200】
【表2】
【0201】
実施例3:3−(パルミトイルアミノ)プロパン−1,2−ジオール
この化合物は、3−アミノ−プロパン−1,2−ジオール及びパルミチン酸から上述の方法(実施例2)に従って合成した。
【0202】
【表3】
【0203】
実施例4:1,2−(ジパルミトイルオキシ)−3−テトラデシルチオアセチルアミノプロパンの調製
3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオール(実施例2)(1g、2.77mmol)をジクロロメタン(200ml)に溶解した。次にジシクロヘキシルカルボジイミド(1.426g、6.91mmol)、ジメチルアミノピリジン(0.845g、6.91mmol)及びパルミチン酸(1.773g、6.91mmol)を加え、混合物を室温で48時間撹拌した。沈殿したジシクロヘキシル尿素を濾過し、ジクロロメタンで洗浄した。濾液を真空留去した。残渣を、シリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 6:4)により精製した(収率:28%)。
【0204】
【表4】
【0205】
実施例5:1,2−(ジテトラデシルチオアセチルオキシ)−3−テトラデシルチオアセチルアミノプロパンの調製
この化合物は、3−(テトラデシルチオアセチルアミノ)プロパン−1,2−ジオール(実施例2)及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例4)に従って合成した。
【0206】
【表5】
【0207】
実施例6:1,2−(ジテトラデシルチオアセチルオキシ)−3−パルミトイルアミノプロパンの調製
この化合物は、3−(パルミトイルアミノ)プロパン−1,2−ジオール(実施例3)及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例4)に従って合成した。
【0208】
【表6】
【0209】
実施例7:1,3−ジ(オレイルアミノ)プロパン−2−オールの調製
オレイン酸(5.698g、0.020mol)及び1,3−ジアミノプロパン−2−オール(1g、0.011mol)をフラスコに入れて、190℃で2時間加熱した。反応混合物を室温に冷却し、次にクロロホルムにとって、水で洗浄した。水相をクロロホルムで抽出し、有機相を合わせ、硫酸マグネシウムで乾燥し、濾過し、蒸発乾固して、油状の黒色残渣(6.64g)を得て、それをシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 99:1)により精製した。次に得られた生成物をエーテルで洗浄し、濾過した。
【0210】
【表7】
【0211】
実施例8:1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オールの調製
この化合物は、1,3−ジアミノプロパン−2−オール及びテトラデシルチオ酢酸(実施例1)から上記の方法(実施例7)に従って合成した。
【0212】
【表8】
【0213】
実施例9:1,3−ジ(ステアロイルアミノ)プロパン−2−オールの調製
この化合物は、1,3−ジアミノプロパン−2−オール及びステアリン酸から上記の方法(実施例7)に従って合成した。
【0214】
【表9】
【0215】
実施例10:1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン二塩酸塩の調製
1,3−ジ(tert−ブチルオキシカルボニルアミノ)プロパン−2−オールの調製(実施例10a)
1,3−ジアミノプロパン−2−オール(3g、0.033mol)をメタノール(300ml)に溶解し、続いてトリエチルアミン(33ml滴下)及びジ−tert−ブチルジカルボネート〔(BOC)2O〕(21.793g、0.100mol)を加えた。反応媒体を40〜50℃で20分間加熱し、次に室温で1時間撹拌した。溶媒を留去した後、無色の油状物残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 95:5)により精製した。反応より無色の油状物が生じ、それはゆっくりと結晶化した。
【0216】
【表10】
【0217】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(テトラデシルチオアセチルオキシ)プロパンの調製(実施例10b)
1,3−(ジ−tert−ブトキシカルボニルアミノ)−プロパン−2−オール(実施例10a)(1g、3.45mmol)、テトラデシルチオ酢酸(実施例1)(0.991g、3.45mmol)及びジメチルアミノピリジン(0.042g、0.34mmol)をジクロロメタン(40ml)に0℃で溶解した。次にジクロロメタンで希釈されたジシクロヘキシルカルボジイミド(0.709g、3.45mmol)を滴加し、混合物を0℃で30分間撹拌し、次に室温にした。20時間の反応の後、ジシクロヘキシル尿素沈殿物を濾過し、濾液を乾燥した。油状残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 5:5、続いてジクロロメタン/酢酸エチル 98:2)により精製した。
【0218】
【表11】
【0219】
1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン二塩酸塩の調製(実施例10)
1,3−(ジtert−ブトキシカルボニルアミノ)−2−テトラデシルチオアセチルオキシプロパン(実施例10b)(0.800g、1.43mmol)を、ガス状塩酸で飽和されたジエチルエーテル(50ml)に溶解した。反応媒体を室温で20時間撹拌した。次に形成された沈殿物を濾過し、エーテル洗浄した。
【0220】
【表12】
【0221】
実施例11:1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパンの調製
1,3−ジアミノ−2−テトラデシルチオアセチルオキシプロパン二塩酸塩(実施例10)(0.400g、0.92mmol)及びテトラデシルチオ酢酸(実施例1)(0.532g、1.84mmol)をジクロロメタン(50ml)に0℃で溶解し、続いてトリエチルアミン(0.3ml、2.1mmol)、ジシクロヘキシルカルボジイミド(0.571g、2.77mmol)及びヒドロキシベンゾトリアゾール(HOBt)(0.249g、1.84mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄した。濾液を真空留去した。得られた残渣(1.40g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン、続いてジクロロメタン/酢酸エチル 9:1)により精製した。
【0222】
【表13】
【0223】
実施例12:1,3−ジオレイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパンの調製
この化合物は、1,3−ジアミノ−2−テトラデシルチオアセチルオキシプロパン二塩酸塩(実施例10)及びオレイン酸から、実施例11に記載の方法に従って合成した。
【0224】
【表14】
【0225】
実施例13:2,3−ジテトラデシルチオアセチルアミノプロパン−1−オールの調製
2,3−ジアミノプロパン酸メチル二塩酸塩の調製(実施例13a)
2,3−ジアミノプロピオン酸塩酸塩(1g、7mmol)をメタノール(40ml)に溶解した。媒体を氷浴で冷却し、続いて塩化チオニル(2.08ml、28mmol)を加えた。媒体を室温にし、次に20時間還流した。溶媒を留去し、残渣をヘプタンで粉砕した。得られた沈殿物を濾過し、洗浄し、乾燥して、黄色を帯びた白色固体を得た。
【0226】
【表15】
【0227】
2,3−ジテトラデシルチオアセチルアミノプロパン酸メチルの調製(実施例13b)
2,3−ジアミノプロパン酸メチル二塩酸塩(実施例13a)(0.500g、2.62mmol)及びテトラデシルチオ酢酸(実施例1)(1.51g、5.23mmol)をジクロロメタン(80ml)に0℃で溶解し、続いてトリエチルアミン(0.79ml)、ジシクロヘキシルカルボジイミド(1.62g、7.85mmol)及びヒドロキシベンゾトリアゾール(0.707g、5.23mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(3.68g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を白色粉末の形態で得た。
【0228】
【表16】
【0229】
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オールの調製(実施例13)
水素化ホウ素ナトリウム(316mg、8.4mmol)をテトラヒドロフラン(40ml)に溶解した。反応混合物を氷浴で冷却し、続いて2,3−ジテトラデシルチオアセチルアミノプロパン酸メチル(実施例13b)(500mg、0.76mmol)を少量ずつ加えた。混合物を室温にし、撹拌した。4日間の反応の後、水20mlを加えた。沈殿した生成物を濾過し、水で洗浄し、次にデシケーター中で乾燥して、白色の粉末を得た。
【0230】
【表17】
【0231】
実施例14:2,3−ジテトラデシルチオアセチルアミノ−1−テトラデシルチオアセチルオキシプロパンの調製
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オール(実施例13)(0.200g、0.32mmol)をテトラヒドロフラン(40ml)に溶解し、続いてジシクロヘキシルカルボジイミド(65mg、0.32mmol)、ジメチルアミノピリジン(39mg、0.32mmol)及びテトラデシルチオ酢酸(実施例1)(91mg、0.32mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、テトラヒドロフランで洗浄し、濾液を留去した。得られた残渣(1g)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で生成した。
【0232】
【表18】
【0233】
実施例15:1,3−ジアミノ−2−(テトラデシルチオアセチルチオ)プロパン二塩酸塩の調製
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(p−トルエンスルホニルオキシ)プロパンの調製(実施例15a)
1,3−ジ(tert−ブチルオキシカルボニルアミノ)プロパン−2−オール(実施例10a)(2.89g、10mmol)及びトリエチルアミン(2.22ml、16mmol)を無水ジクロロメタン(100ml)に溶解した。反応混合物を氷浴で冷却し、続いてジクロロメタン(30ml)に溶解した塩化トシル(2.272g、12mmol)を滴加した。次に反応混合物を室温で72時間撹拌した。48時間後に1当量の塩化物及びトリエチルアミン1.6 を加えた。水を加えて反応を止め、媒体を沈降させた。有機相を水で数回洗浄した。水相を合わせ、ジクロロメタンで再び抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(6.44g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン、続いてジクロロメタン/メタノール 99:1)により精製して、目的化合物を白色の固体として得た。
【0234】
【表19】
【0235】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−アセチルチオプロパンの調製(実施例15b)
1,3−(ジtert−ブトキシカルボニルアミノ)−2−(p−トルエンスルホニルオキシ)プロパン(実施例15a)(0.500g、1.12mmol)及びチオ酢酸カリウム(0.161g、1.41mmol)をアセトンに溶解し、媒体を48時間還流した。24時間の還流の後で、1当量のチオ酢酸カリウムを加えた。反応を室温にし、溶媒を留去した。残渣をジエチルエーテルにとって、セライト(登録商標)で濾過した。濾液を留去した。得られた生成物(0.48g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を黄土色の固体として得た。
【0236】
【表20】
【0237】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−メルカプトプロパンの調製(実施例15c)
メタノール(10ml)で希釈された1,3−ジ(tert−ブトキシカルボニルアミノ)−2−(アセチルチオ)プロパン(実施例15b)(0.380g、1.2mmol)を、窒素流下で脱酸素化しながらメタノール中の20%炭酸カリウム溶液(2.14ml、12.4mmol)に加えた。反応混合物を窒素下、室温で20時間撹拌し、次に酢酸でpH6に酸性化した。溶媒を真空留去した。残渣を水にとって、クロロホルムで抽出した。有機相を合わせ、硫酸マグネシウムで乾燥し、次に濾過し、乾燥して、目的生成物を白色固体の形態で得て、直ちにそれを次の反応に使用した。
【0238】
【表21】
【0239】
1,3−ジ(tert−ブチルオキシカルボニルアミノ)−2−(テトラデシルチオアセチルチオ)プロパンの調製(実施例15d)
1,3−〔ジ(tert−ブトキシカルボニルアミノ)〕−2−メルカプトプロパン(実施例15c)(0.295g、0.963mmol)をジクロロメタン(40ml)に溶解した。次にジシクロヘキシルカルボジイミド(0.199g、0.963mmol)、ジメチルアミノピリジン(0.118g、0.963mmol)及びテトラデシルチオ酢酸(実施例1)(0.278g、0.963mmol)を加えた。反応混合物を室温で撹拌し、反応の進行を、TLCで監視した。20時間の反応の後、ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(0.73g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で得た。
【0240】
【表22】
【0241】
1,3−ジアミノ−2−(テトラデシルチオアセチルチオ)プロパン二塩酸塩の調製(実施例15)
1,3−〔ジ(tert−ブトキシカルボニルアミノ)〕−2−テトラデシルチオアセチルチオプロパン(実施例15d)(0.300g、0.52mmol)を、ガス状塩酸で飽和されたエーテル(55ml)に溶解した。この混合物を室温で撹拌した。96時間の反応の後、形成された沈殿物を濾過し、ジエチルエーテルで数回洗浄し、乾燥して、目的化合物を白色粉末の形態で得た。
【0242】
【表23】
【0243】
実施例16:1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパンの調製
1,3−ジアミノ−2−テトラデシルチオアセチルチオプロパン二塩酸塩(実施例15)(100mg、0.225mmol)及びテトラデシルチオ酢酸(実施例1)(130mg、0.450)をジクロロメタン(30ml)に0℃で溶解し、続いてトリエチルアミン(68μl)、ジシクロヘキシルカルボジイミド(139mg、0.675mmol)及びヒドロキシベンゾトリアゾール(61mg、0.450mmol)を加えた。反応混合物を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(430mg)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を白色粉末の形態で得た。
【0244】
【表24】
【0245】
実施例17:1−アミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン塩酸塩の調製
1−(tert−ブチルオキシカルボニルアミノ)プロパン−2,3−ジオールの調製(実施例17a)
1−アミノプロパン−2,3−ジオール(5g、55mol)をメタノール(200ml)に溶解し、続いてトリエチルアミン(アミン1mmol当たり0.5ml)及びジ−tert−ブチルジカルボネート〔(BOC)2O〕(ここでBOCは、tertブチルオキシカルボニルである)(17.97g、82mmol)を滴加した。反応媒体を40〜50℃で20分間加熱し、次に室温で1時間撹拌した。溶媒を留去した後、無色の油状物残渣をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/メタノール 95:5)により精製して、目的化合物を無色油状物の形態で得て、それはゆっくりと結晶化した。
【0246】
【表25】
【0247】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(p−トルエンスルホニルオキシ)プロパンの調製(実施例17b)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−プロパン−2,3−ジオール(実施例17a)及びp−トルエンスルホニルクロリドから上記(実施例15a)で記載された方法に従って合成した。反応は白色の粉末を生成した。
【0248】
【表26】
【0249】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(アセチルチオ)プロパンの調製(実施例17c)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(p−トルエンスルホニルオキシ)−プロパン(実施例17b)及びチオ酢酸カリウムから上記(実施例15d)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0250】
【表27】
【0251】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジメルカプトプロパンの調製(実施例17d)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(アセチルチオ)−プロパン(実施例17c)を鹸化し、上記(実施例15c)で記載された方法に従って合成した。反応は白色の固体を生成し、直ちにそれを次の反応に使用した。
【0252】
【表28】
【0253】
1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジ(テトラデシルチオアセチルチオ)プロパンの調製(実施例17e)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジメルカプトプロパン(実施例17d)及びテトラデシルチオ酢酸(実施例1)から上記(実施例15d)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0254】
【表29】
【0255】
1−アミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン塩酸塩の調製(実施例17)
この化合物は、1−(tert−ブチルオキシカルボニルアミノ)−2,3−ジテトラデシルチオアセチル−チオプロパン(実施例17e)から上記(実施例15)で記載された方法に従って合成した。反応は白色の固体を生成した。
【0256】
【表30】
【0257】
実施例18:1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパンの調製
1−アミノ−2,3−ジテトラデシルチオアセチルチオプロパン塩酸塩(実施例17)(100mg、0.140mmol)及びテトラデシルチオ酢酸(実施例1)(62mg、0.210)をジクロロメタン(40ml)に0℃で溶解し、続いてトリエチルアミン(43ml)、ジシクロヘキシルカルボジイミド(59mg、0.28mmol)及びヒドロキシベンゾトリアゾール(29mg、0.210mmol)を加えた。反応混合物を0℃で1時間撹拌し、次に24時間で室温にした。次に穏やかに還流しながら48時間加熱し、次に乾燥した。得られた残渣(310mg)を、シリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 8:2)により精製して、目的化合物を白色の粉末として生成した。
【0258】
【表31】
【0259】
実施例19:1−テトラデシルチオアセチルチオ−2,3−ジ(テトラデシルチオアセチルアミノ)プロパンの調製
2,3−ジ(テトラデシルチオアセチルアミノ)−1−ヨードプロパンの調製(実施例19a)
2,3−ジテトラデシルチオアセチルアミノプロパン−1−オール(実施例13)(0.200g、0.317mmol)をトルエン(30ml)に溶解した。次にイミダゾール(0.054g、0.792mmol)、トリフェニルホスフィン(0.208g、0.792mmol)及びヨウ素(0.161g、0.634mmol)をこの順序で加え、反応を撹拌しながら75〜80℃で加熱した。6時間の反応の後、溶媒を留去し、残留生成物を更に精製することなく使用した。
Rf(ジクロロメタン/メタノール 98:2):0.55
【0260】
2,3−ジ(テトラデシルチオアセチルアミノ)−1−メルカプトプロパンの調製(実施例19b)
硫化水素ナトリウム(0.089g、1.59mmol)を、アセトン(80ml)に溶解した2,3−ジテトラデシルチオアセチルアミノ−1−ヨードプロパン(実施例19a)(0.235g、0.32mmol)に加えた。反応媒体を70℃で16時間加熱した。溶媒を留去し、残渣を水にとって、クロロホルムで抽出した。水相を酢酸でpH6に酸性化し、次にクロロホルムで再び抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣を、更に精製することなく使用した。
【0261】
1−テトラデシルチオアセチルチオ−2,3−ジ(テトラデシルチオアセチルアミノ)プロパンの調製(実施例19)
2,3−ジテトラデシルチオアセチルアミノ−1−メルカプトプロパン(実施例19b)(0.205g、0.32mmol)をテトラヒドロフラン(50ml)に溶解した。次にジシクロヘキシルカルボジイミド(98mg、0.47mmol)、ジメチルアミノピリジン(58mg、0.47mmol)及びテトラデシルチオ酢酸(実施例1)(137mg、0.47mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、テトラヒドロフランで洗浄し、濾液を留去した。得られた残渣(1.14g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄土色粉末の形態で得た。
【0262】
【表32】
【0263】
実施例20:3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オールの調製
3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン−2−オールの調製(実施例20a)
クロロトリフェニルメタン(2.833g、10.16mmol)を、ピリジン(2.5ml)中の3−テトラデシルチオアセチルアミノプロパン−1,2−ジオール(実施例2)(3g、8.30mmol)の溶液に加えた。反応混合物を50℃で24時間撹拌し、次に溶媒を真空留去した。残渣を水にとって、ジクロロメタンで抽出した。有機相を、1N塩酸水溶液、次に塩化ナトリウムで飽和した水溶液で洗浄した。硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(6.36g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を白色粉末の形態で得た。
【0264】
【表33】
【0265】
2−ヨード−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパンの調製(実施例20b)
3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン−2−オール(実施例20a)(2g、3.31mmol)をトルエン(100ml)に溶解した。次にイミダゾール(0.564g、8.28mmol)、トリフェニルホスフィン(2.171g、8.28mmol)及びヨウ素(1.681g、6.62mmol)をこの順序で加えた。反応媒体を室温で20時間撹拌した。飽和重亜硫酸ナトリウム溶液を、反応媒体が完全に白くなるまで加えた。相を分離し、水相をトルエンで抽出した。有機相を合わせ、飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、濾過した。溶媒を留去した後で得られた残渣(4.65g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄色油状物の形態で得た。
【0266】
【表34】
【0267】
2−メルカプト−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパンの調製(実施例20c)
硫化水素ナトリウム水和物(38mg、0.68mmol)をエタノール(20ml)中の懸濁液として調製し、続いて2−ヨード−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシプロパン(実施例20b)(200mg、0.28mmol)を加えた。反応媒体を70℃で加熱した。硫化水素ナトリウム水和物238mgを数日間かけて加えた。6.5日後、溶媒を留去し、残渣をジクロロメタンにとって、水で洗浄した。水相を再抽出し、合わせた有機相を、0.5N塩酸、次に飽和塩化ナトリウム水溶液で洗浄し、次に硫酸マグネシウムで乾燥した。塩を濾過し、溶媒を留去した。得られた残渣を、更に精製することなく使用した。
【0268】
【表35】
【0269】
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオ−1−トリフェニルメチルオキシ−プロパンの調製(実施例20d)
2−メルカプト−3−テトラデシルチオアセチルアミノ−1−トリフェニルメチルオキシトプロパン(実施例20c)(174mg、0.28mmol)をテトラヒドロフラン(20ml)に溶解した。次にジシクロヘキシルカルボジイミド(88mg、0.42mmol)、ジメチルアミノピリジン(51mg、0.42mmol)及びテトラデシルチオ酢酸(121mg、0.42mmol)を加え、反応媒体を室温で撹拌した。20時間の反応の後、溶媒を留去し、得られた残渣(45mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン/シクロヘキサン 3:7〜5:5)により精製して、目的化合物を白色粉末の形態で得た。
【0270】
【表36】
【0271】
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オールの調製(実施例20)
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオ−1−トリフェニルメチルオキシ−プロパン(実施例20d)(187mg、0.21mmol)を、ガス状塩酸で飽和されたエーテル(12ml)に溶解した。反応媒体を室温で20時間撹拌した。形成された沈殿物を濾過し、ジエチルエーテルで洗浄して、目的化合物を白色粉末の形態で得た。
【0272】
【表37】
【0273】
実施例21:3−テトラデシルチオアセチルアミノ−1−テトラデシルチオアセチルオキシ−2−テトラデシルチオアセチルチオプロパンの調製
3−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルチオプロパン−1−オール(実施例20a)(64mg、0.10mmol)をテトラヒドロフラン(7ml)に溶解した。次にジシクロヘキシルカルボジイミド(31mg、0.15mmol)、ジメチルアミノピリジン(18mg、0.15mmol)及びテトラデシルチオ酢酸(実施例1)(43mg、0.15mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、濾液を留去した。得られた残渣(140mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色粉末の形態で得た。
【0274】
【表38】
【0275】
実施例22:1−アミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン塩酸塩の調製
1−tert−ブチルオキシカルボニルアミノ−3−ヨードプロパン−2−オールの調製(実施例22a)
1−〔(tert−ブチルオキシカルボニル)アミノ〕プロパン−2,3−ジオール(実施例17a)(3.88g、20mmol)をトルエン(250ml)に溶解した。次にイミダゾール(1.73g、25mmol)、トリフェニルホスフィン(6.65g、25mmol)及びヨウ素(5.15g、20mmol)をこの順序で加えた。反応媒体を室温で17時間撹拌し、0.5当量のイミダゾール、トリフェニルホスフィン及びヨウ素を加えた。21時間の反応の後、飽和亜硫酸ナトリウム溶液を、反応媒体が完全に白くなるまで加えた。相を沈降させ、水相をトルエンで2回抽出した。合わせた有機相を合わせ、飽和塩化ナトリウム溶液で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を留去した。得られた残渣(11.02g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 95:5)により精製して、目的化合物を黄色のペーストとして得て、直ちにそれを次の反応に使用した。
【0276】
【表39】
【0277】
3−アセチルチオ−1−tert−ブチルオキシカルボニルアミノプロパン−2−オールの調製(実施例22b)
1−(tert−ブチルオキシカルボニルアミノ)−3−ヨードプロパン−2−オール(実施例22a)(2g、6.64mmol)及びチオ酢酸カリウム(0.948g、8.30mmol)をアセトン(30ml)に溶解し、媒体を16時間還流した。溶媒を真空留去し、残渣をジエチルエーテルにとって、次にセライト(登録商標)で濾過した。濾液を留去した。得られた残渣(1.69g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製し、次にフラッシュクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を黄色油状物の形態で得た。
【0278】
【表40】
【0279】
1−tert−ブチルオキシカルボニルアミノ−3−メルカプトプロパン−2−オールの調製(実施例22c)
最小限のメタノール(7ml)で希釈された3−アセチルチオ−1−tert−ブチルオキシカルボニルアミノプロパン−2−オール(実施例22b)(0.307g、1.23mmol)を、窒素流下で脱酸素化しながらメタノール中の20%炭酸カリウム溶液(3.49ml、12.31mmol)に加えた。媒体を窒素流下、室温で20時間撹拌し、次に酢酸でpH6に酸性化し、濃縮乾固した。得られた残渣を水にとって、ジクロロメタンで抽出した。有機相を硫酸マグネシウムで乾燥し、濾過し、濃縮した。得られた油状残渣を、更に精製することなく直ちに次に反応に使用した。
【0280】
【表41】
【0281】
1−tert−ブチルオキシカルボニルアミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパンの調製(実施例22d)
1−(tert−ブチルオキシカルボニルアミノ)−3−メルカプトプロパン−2−オール(実施例22c)(0.200g、96mmol)をジクロロメタン(50ml)に溶解した。次にジシクロヘキシルカルボジイミド(0.398g、1.93mmol)、ジメチルアミノピリジン(0.236g、1.93mmol)及びテトラデシルチオ酢酸(実施例1)(0.557g、1.93mmol)を加えた。混合物を室温で20時間撹拌した。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(1.2g)をシリカゲルのクロマトグラフィー(溶離液:ジクロロメタン)により精製して、目的化合物を白色ペーストの形態で得た。
【0282】
【表42】
【0283】
1−アミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン塩酸塩の調製(実施例22)
1−(tert−ブトキシカルボニルアミノ)−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパン(実施例22d)(300mg、0.40mmol)を、ガス状塩酸で飽和されたジエチルエーテル(70ml)に溶解し、反応媒体を室温で72時間撹拌した。形成された沈殿物を濾過し、ジエチルエーテルで洗浄し、乾燥して、目的化合物を白色粉末の形態で得た。
【0284】
【表43】
【0285】
実施例23:1−テトラデシルチオアセチルアミノ−2−テトラデシルチオアセチルオキシ−3−テトラデシルチオアセチルチオプロパンの調製
3−アミノ−2−テトラデシルチオアセチルオキシ−1−テトラデシルチオアセチル−チオプロパン塩酸塩(実施例22)(100mg、0.15mmol)及びテトラデシルチオ酢酸(実施例1)(63mg、0.22)をジクロロメタン(30ml)に0℃で溶解し、続いてトリエチルアミン(0.044ml)、ジシクロヘキシルカルボジイミド(60mg、0.29mmol)及びヒドロキシベンゾトリアゾール(30mg、0.22mmol)を加えた。反応媒体を0℃で1時間撹拌し、次に48時間で室温にした。ジシクロヘキシル尿素沈殿物を濾過し、ジクロロメタンで洗浄し、濾液を留去した。得られた残渣(263mg)をフラッシュクロマトグラフィー(溶離液:ジクロロメタン/酢酸エチル 98:2)により精製して、目的化合物を白色粉末の形態で得た。
【0286】
【表44】
【0287】
実施例24:本発明の式(I)により表される化合物の調製方法
下記実施例で記載されるインビトロ実験を実施するために、本発明の化合物を下記で記載されるように乳剤の形態で調製した。
Spoonerらにより記載されたように(Spooner, Clarkら, 1988)、本発明の化合物及びホスファチジルコリン(PC)を含む乳剤を調製した。本発明の化合物をPCと、クロロホルム中、4:1(m/m)比で混合し、この混合物を窒素下で乾燥し、次に一晩真空留去し、得られた粉末を、0.01M EDTAを含む0.16M KClにとって、次にこの脂質粒子を超音波により37℃で30分間分散させた。このようにして形成されたリポソームを次に超遠心分離(XL80超遠心分離機、Beckman Coulter, Villepinte, France)により25,000rpmで45分間分離することにより、100nmを超えてキロミクロンに近い大きさを有するリポソームを回収した。陰性対照として使用するために、PCだけからなるリポソームを同時に調製した。
本発明の化合物におけるリポソームの組成は、酵素比色トリグリセリドアッセイキットを用いて推定した。このアッセイは、脂質較正物質のCFAS、参照番号759350(Boehringer Mannheim GmbH, ドイツ)で作製した標準曲線に対して実行した。この標準曲線は、16〜500μg/mlの範囲の濃度を包含した。滴定プレート(96ウェル)上に1ウェルにつき各試料希釈液又は較正標準液100μlを滴下した。次にトリグリセリド試薬、参照番号701912(Boehringer Mannheim GmbH, ドイツ)200μlを各ウェルに加え、プレート全体を37℃で30分間インキュベートした。光学密度(OD)は、分光光度計により492nmで読みとった。各試料におけるトリグリセリド濃度は、一次関数:y=ax+b〔式中、yは、ODを表し、そしてxは、トリグリセリド濃度を表す〕としてプロットした標準曲線から計算した。
【0288】
このようにして調製された、本発明の化合物を含有するリポソームを、実施例26、27及び28に記載されているインビトロ実験で使用した。
【0289】
実施例25:本発明の化合物の抗酸化特性の評価
A−銅により誘導されるLDL酸化に対する保護:
LDLの酸化は、アテローム動脈硬化の発症及び進展において主要な役割を演じる、重要な変性である(Jurgens, Hoffら, 1987)。以下のプロトコールにより、化合物の抗酸化特性を証明することができる。他に記載がなければ、全ての試薬はシグマ(Sigma)(St Quentin, France)製である。
LDLは、Lebeauらで記載された(Lebeau, Furmanら, 2000)ように調製した。
試験化合物の溶液は、エタノール中10-2Mで調製し、最終濃度が1%(V/V)の総エタノール濃度で0.1〜100μMの範囲になるようにPBSで希釈した。
【0290】
酸化の前に、透析によりLDL調製物からEDTAを除去した。次に、16.6μM CuSO4100μlを、LDL800μl(125μgタンパク質/ml)及び試験化合物溶液100μlに加えることにより、酸化反応を30℃で行った。追跡すべき種であるジエンの形成は、銅の存在下又は不在下、本化合物により処理した試料中の234nmの光学密度により測定した。234nmの光学密度は、10分毎に8時間、サーモスタット付き分光光度計(Kontron Uvikon 930)で測定した。分析は三回繰り返した。対照試料に対して遅滞期潜伏時間をシフトするならば、化合物は抗酸化活性を有すると考えられた。発明者たちは、本発明の化合物が、LDL酸化(銅により誘導)を遅延させたことを証明したが、このことは、本発明の化合物が、固有の抗酸化活性を有することを示している。図2は、本発明の化合物により得られた結果の一例を表す。
【0291】
図2は、固有の抗酸化特性を示す、本発明の化合物Ex2、4、5、6及び11を示す。
図2aは、本発明の化合物が、遅滞期潜伏時間を化合物Ex2では13%を超えて、化合物Ex4では34.3%までシフトすることを示す。本発明の化合物は、酸化速度(図2b)又はジエン形成量(図2c)を変えるようには見えなかった。
【0292】
B−脂質過酸化に対して本発明の化合物により与えられる保護の評価:
試験した本発明の化合物は、その調製が実施例2〜23に記載されている化合物である。
LDL酸化は、TBARS法(チオバルビツール酸反応性基質)により測定した。
上述のものと同じ原理により、LDLをCuSO4の存在下で酸化させて、脂質の過酸化を以下のように評価した:
TBARSは、分光光度法により測定し、脂質ヒドロペルオキシド化は、ヨウ化物のヨウ素への脂質過酸化物依存性酸化を利用して測定した。結果は、マロンジアルデヒド(MDA)のnmolとして、又はnmolヒドロペルオキシド/mgタンパク質として表される。
共役ジエン形成の抑制を測定することにより得られた上記の結果を、LDL脂質過酸化を測定する実験により確認した。したがって、また本発明の化合物によって、銅(酸化剤)により誘導された脂質の過酸化に対するLDLの効率的な保護ができた。
【0293】
実施例26:細胞培養物に及ぼす本発明の化合物の抗酸化特性の測定
A−培養プロトコール:
ニューロン、ニューロブラストーマ(ヒト)及びPC12細胞(ラット)がこの種類の試験に使用した細胞株であった。PC12細胞は、ラットのクロム親和性細胞種から調製し、Greene 及び Tischler により特徴づけられた(Greene 及び Tischler, 1976)。これらの細胞は、通常、神経細胞の分化、信号伝達及び神経細胞死の試験に使用される。PC12細胞を前述された(Farinelli, Parkら, 1996)ように、10%ウマ血清及び5%ウシ胎仔血清を補充した完全なRPMI培地(Invitrogen)中で増殖させた。
内皮及び平滑筋細胞の初代培養物も使用した。細胞はプロモセル(Promocell)(Promocell GmBH, ハイデルベルグ)から得て、供給業者の指示書に従って培養した。
細胞を5〜100μMの範囲の異なる用量の化合物で24時間処理した。次に細胞を回収し、標的遺伝子の発現の増加を定量PCRで評価した。
【0294】
B−mRNA測定:
mRNAを、本発明の化合物で処理したか又はしなかった細胞培養物から抽出した。抽出は、Absolutely RNA RT-PCR ミニプレップキット(Stratagene, フランス)の試薬を用いて供給業者により指示されたとおりに実行した。次にmRNAを、分光法によりアッセイして、Light Cycler System (Roche, フランス) のLight Cycler Fast Start DNA Master Sybr Green Iキット(Roche)を用いる、定量RT−PCRにより定量した。抗酸化酵素スーパーオキシドジスムターゼ(SOD)、カタラーゼ及びグルタチオンペルオキシダーゼ(GPx)をコードする遺伝子に特異的なプライマー対をプローブとして使用した。アクチン及びシクロフィリン遺伝子に特異的なプライマー対を対照プローブとして使用した。
【0295】
定量RT−PCRで測定した抗酸化酵素遺伝子のmRNA発現の増加は、細胞が本発明の化合物で処理されたとき、使用した異なる細胞の種類で実証された。
【0296】
C−酸化ストレスの制御:
培養細胞における酸化種の測定:
化合物の抗酸化特性も、蛍光標識を用いて評価し、その酸化は、蛍光信号の出現により追跡した。発光した蛍光信号の強度の減少は、化合物で処理された細胞において下記の方法で測定した:上記のように培養されたPC12細胞(ブラック96ウェルプレート、透明底、Falcon)を、血清無含有培地で、H2O2の用量を増加させる(0.25mM〜1mM)と共に2及び24時間インキュベートした。インキュベーションの後、培地を除去し、細胞をPBS中の10μMジクロロジヒドロフルオレセインジアセタート溶液(DCFDA、Molecular Probes, Eugene, USA)と共に37℃、5%CO2雰囲気下で30分間インキュベートした。次に細胞をPBSですすいだ。酸化標識により発光している蛍光を、蛍光計(Tecan Ultra 384)により、励起波長495nm及び発光波長535nmで測定した。結果は、酸化対照に対する保護の百分率として表す。
蛍光強度は、未処理細胞よりも本発明の化合物とインキュベートした細胞において低かった。これらの知見は、本発明の化合物が、酸化ストレスに曝された細胞において酸化種の産生の抑制を促進することを示す。前述した抗酸化特性も、培養された細胞における抗ラジカル保護の誘導に有効である。
【0297】
D−脂質の過酸化の測定:
異なる細胞株(上記の細胞モデル)及び初代細胞培養物を前記のように処理した。処理の後、細胞上清を回収し、細胞を溶解し、タンパク質濃度の測定のために回収した。脂質の過酸化を下記のように検出した:脂質の過酸化を、マロンジアルデヒド(MDA)などのアルデヒドの脂質過酸化と反応する、チオバルビツール酸(TBA)を用いて測定した。処理のあと、細胞上清を収集し(900μl)し、ブチル化ヒドロキシトルエン90μlを加えた(Morliere, Moysanら, 1991)。15%トリクロロ酢酸を含有する0.25M塩酸中のTBAの0.375%溶液1ミリリットルもこの反応媒体に加えた。この混合物を80℃で15分間加熱し、氷上で冷却して、有機相をブタノールで抽出した。有機相を、シマヅ1501分光蛍光計(島津製作所、京都、日本)で、分光蛍光法(λexc=515nm及びλem=550nm)により分析した。TBARSを、テトラ−エトキシプロパンを標準として使用してMDA当量として表した。結果を、タンパク質濃度に対して正規化した。
本発明の化合物で処理された細胞において観察された脂質の過酸化の減少は、前記の結果を確認する。
本発明の化合物は、有利には、酸化ストレスの影響を遅延させる及び/又は抑制する、固有の抗酸化特性を示す。発明者たちは、また、本発明の化合物が、抗酸化酵素をコードする遺伝子の発現を誘導することができることも示す。これらの本発明の化合物の特定の特徴は、細胞が酸化ストレスと有効に戦うことを可能にし、したがってフリーラジカル誘発損傷から保護され得ることを可能にする。
【0298】
実施例27:本発明の化合物によるインビトロでのPPAR活性の評価
異常脂肪血症及び糖尿病の処置に病院で広く使用されている2つの主な医薬分類であるフィブラート系及びグリタゾン系(glitazones)により活性化される、PPARサブファミリーの核内受容体は、脂質及びブドウ糖のホメオスタシスにおいて重要な役割を演じる。下記の実験データは、本発明の化合物がPPARαをインビトロで活性化することを示す。
【0299】
PPAR活性化を、酵母菌gal4転写因子のDNA結合ドメインと、異なるPPARのリガンド結合ドメインとから構成されるキメラの転写活性を測定することにより、RK13線維芽細胞の細胞株又は血液細胞HepG2においてインビトロで試験した。下記の例は、HepG2細胞で示す。
【0300】
A−培養プロトコール:
HepG2細胞は、ECACC(Porton Down, UK)から得て、10%(V/V)ウシ胎仔血清、100U/mlペニシリン(Gibco, Paisley, UK)及び2mM L−グルタミン(Gibco, Paisley, UK)を補充したDMEM培地で増殖させた。培地は2日毎に取り換えた。細胞は、空気95%/CO2 5%の加湿した雰囲気中、37℃で保持した。
【0301】
B−トランスフェクションに使用したプラスミドの記載:
プラスミドpG5TkpGL3、pRL−CMV、pGal4−hPPAR、pGal4−hPPAR及びpGal4−fは、Raspeらにより記載された(Raspe、Madsenら, 1999)。pGal4−mPPARα及びpGal4−hPPARβ作成体を、マウスPPARα及びヒトPPARβ核内受容体のDEFドメインに対応するPCR増幅DNA断片を、それぞれpGal4−fベクター中にクローン化することにより得た。
【0302】
C−トランスフェクション:
HepG2細胞を24ウエル培養皿に5×104細胞/ウエルで接種し、前述のプロトコール(Raspe, Madsen ら, 1999)に従って、レポータープラスミドpG5TkpGL3(50ng/ウエル)、発現ベクターpGal4−f、pGal4−mPPARα、pGal4−hPPARα、pGal4−hPPARγ、pGal4−hPPARβ(100ng/ウエル)及びトランスフェクション効率制御ベクターpRL−CMV(1ng/ウエル)を2時間トランスフェクトし、次に試験化合物と共に36時間インキュベートした。実験の終了時に、細胞を溶解し(Gibco, Paisley, UK)、ルシフェラーゼ活性を、Dual-Luciferase(商標)Reporter Assay Systemキット(Promega, Madison, WI, USA)により、供給者の使用説明書に従って測定した。次に、Bio-Rad Protein Assayキット(Bio-Rad, Munich, Germany)で細胞抽出物中のタンパク含有量を供給業者による指示により測定した。
【0303】
発明者は、本発明の化合物で試験され、pGal4−hPPARαプラスミドをトランスフェクトされた細胞のルシフェラーゼ活性が増加したことを示す。前記のルシフェラーゼ活性の誘導は、本発明の化合物がPPARαの活性化剤であることを示す。図3は、本発明の化合物により得られた結果の例を示す。
図3:Gal4/PPARαプラスミドをトランスフェクトしたHepG2細胞を、異なる濃度(5、15、50及び100μM)の本発明の化合物(Ex2、Ex4、Ex5、Ex6、Ex11)と共に24時間、並びに本発明の化合物の濃度5、15、50及び100μM用の対照として1、2、3、4と示されている異なる濃度のビヒクル(PC)と共にインキュベートした[実施例24(本発明の式(I)により表される化合物の調製方法)で記載された4:1(m/m)比に従う]。結果は、異なる処理の後で、誘導係数(処理細胞の発光信号を未処理細胞の発光信号で除す)として表される。誘導係数が高いほど、PPARαアゴニスト活性が強力である。結果は、本発明の化合物Ex2が50μMで最大19.8倍、100μMでは19.2倍、15μMでは7.7倍、5μMでは1.5倍の発光信号の誘導を生じることを示す。本発明の化合物Ex5も、100μMで10.5、50μMで7、15μMで2.5、5μMで1.2である誘導係数の用量依存的増加を示した。本発明の化合物Ex6も、発光信号の増加を誘導し、PPARα核内受容体に対する活性を明らかにした。本発明の化合物Ex6の誘導係数は、100μMで14.5、50μMで9.6、15μMで2.2、5μMで1.1であった。一方、細胞をビヒクル(PCリポソーム)と共にインキュベートした場合、有意な誘導は観察されなかった。
これらの結果は、試験された本発明の化合物が有意なPPARαリガンド活性を示し、したがってそれらの転写活性を可能にすることを示す。
【0304】
実施例28:本発明の化合物の抗炎症特性の評価
炎症反応は、脳虚血などの多くの神経疾患において観察される。炎症は、また、神経変性の重要な因子である。脳卒中では、グリア細胞の最初の反応の1つは、サイトカイン及びフリーラジカルを放出することである。このサイトカイン及びフリーラジカルの放出により、脳における炎症性反応を生じ、これが神経細胞死を引き起こす可能性がある(Rothwell 1997)。
細胞株及び初代細胞を、上記のように培養した。
リポ多糖類(LPS)細菌性内毒素(Escherichia coli 0111 :B4)(Sigma, フランス)を蒸留水で再構成し、4℃で保存した。細胞をLPS 1μg/mlで24時間処理した。他の因子からの干渉を避けるため、培地を完全に交換した。
TNF−αは、ストレス(例えば、酸化ストレス)に対する炎症反応における重要な因子である。LPSの用量の増加による刺激に応答するTNF−α分泌を評価するため、刺激を受けた細胞の培地を除去し、TNF−αをELISA−TNF−αキット(Immunotech, フランス)によりアッセイした。試料を50倍に希釈して、標準曲線(Chang, Hudsonら, 2000)の範囲内にした。
【0305】
化合物の抗炎症特性を下記のように特徴付けした:細胞培地を完全に交換し、細胞を試験化合物と共に2時間インキュベートし、その後、LPSを最終濃度1μg/mlで培地に加えた。24時間のインキュベーションの後、細胞上清を回収し、直ちに処理しない場合は−80℃で保存した。細胞を溶解し、タンパク質をBio-Rad Protein Assayキット(Bio-Rad, Munich, Germany)で供給業者の指示書により定量した。
【0306】
試験化合物による処理で誘導されたTNF−α分泌の減少の測定値は、pg/ml/μgタンパク質として、かつ対照に対する百分率として表される。これらの結果は、本発明の化合物が、抗炎症特性を有することを示す。
【0307】
実施例29:脳虚血−再灌流モデルにおける本発明の化合物の神経保護作用の評価
A−予防モデル:
1−動物の処置
1.1動物及び化合物の投与
体重200〜350gのウィスターラットをこの実験に使用した。
動物を、12時間の明/暗サイクル、20℃±3℃の温度で維持した。水及び食餌は自由に摂取可能であった。食物摂取量及び体重増加を記録した。
中大脳動脈の閉塞により誘導される虚血の前に、動物を、ビヒクル(0.5%カルボキシセルロース(CMC)及び0.1%ツイーン)に懸濁した本発明の化合物を用いる(600mg/kg/日)か又はビヒクルのみを用いる、胃管栄養法により14日間処置した。
使用したカルボキシメチルセルロースは、中間粘度のカルボキシメチルセルロースのナトリウム塩である(Ref. C4888, Sigma-Aldrich, フランス)。使用されたツイーンは、ポリオキシエチレンソルビタンモノオレエートである(Tween 80, Ref. P8074, Sigma-Aldrich, フランス)。
【0308】
1.2中大脳動脈の腔内閉塞による虚血誘導−再灌流:
動物を抱水クローラル300mg/kgの腹腔内注入により麻酔した。直腸プローブを挿入し、体温を37℃±0.5℃に維持した。試験を通して、血圧を監視した。
外科用顕微鏡下で、頸の中央部の切開により右頸動脈を露出した。翼突口蓋動脈をその起点で結紮し、動脈切開を外頸動脈で行って、ナイロン製のモノフィラメントを挿入できるようにし、それを、総頸動脈まで穏やか進め、次に内頸動脈内に進めて、中大脳動脈の起点を閉塞するようにした。1時間後にフィラメントを抜いて再灌流を可能にした。
【0309】
2−脳梗塞量の測定:
再灌流の24時間後、本発明の化合物で予め処置したか又は処置していない動物に、ペントバルビタールの過剰投与により麻酔をかけた。
脳を急速に凍結し薄切りにした。切片をクレシルバイオレットで染色した。脳切片の未染色の帯域は、梗塞により損傷を受けたと考えられた。領域(梗塞及び2つの半球)を測定し、梗塞及び2つの半球の量を計算し、修正梗塞量を次の式:[修正梗塞量=梗塞量−(右半球の量−左半球の量)]により決定して、脳浮腫を補償した。
本発明の化合物で処置した動物の脳切片の分析によって、未処置の動物と比較して、梗塞量の著しい減少が明らかとなった。虚血の前に、本発明の化合物を動物に投与すると(予防効果)、これらは神経保護を誘導することができた。
【0310】
3−抗酸化酵素活性の測定:
ラットの脳を冷凍し、粉砕し、粉末まで小さくし、次に食塩水に再懸濁した。次に、下記の著者により記載されているように、異なる酵素活性を測定した:スーパーオキシドジスムターゼ(Flohe 及び Otting 1984);グルタチオンペルオキシダーゼ(Paglia 及び Valentine 1967);グルタチオンレダクターゼ(Spooner, Delidesら, 1981);グルタチオン−S−トランスフェラーゼ(Habig 及び Jakoby 1981);カタラーゼ(Aebi 1984)。
これら異なる酵素活性は、本発明の化合物で処置された動物の脳調製物において増加した。
【0311】
B−治療的又は急性期処置モデル:
1−中大脳動脈の腔内閉塞による虚血誘導/再灌流:
前記のような動物をこの実験に使用した。動物を抱水クローラル300mg/kgの腹腔内注入により麻酔した。直腸プローブを挿入し、体温を37℃±0.5℃に維持した。試験を通して、血圧を監視した。
外科用顕微鏡下で、頸の中央部の切開により右頸動脈を露出した。翼突口蓋動脈をその起点で結紮し、動脈切開を外頸動脈で行って、ナイロン製のモノフィラメントを挿入できるようにし、それを、総頸動脈まで穏やか進め、次に内頸動脈内に進めて、中大脳動脈の起点を閉塞するようにした。1時間後にフィラメントを抜いて再灌流を可能にした。
【0312】
2−動物の処置:
最初に虚血−再灌流に付した動物を、再灌流の後、本発明の化合物を用いて経口(前記のようなCMC+ツイーンビビクル)により1回以上処置した(600mg/kg/日又は300mg/kg/日を1日二回)。
【0313】
3−脳梗塞量の測定:
再灌流の24、48又は72時間後、化合物で予め処置したか又は処置していない動物に、ペントバルビタールの過剰投与により麻酔をかけた。
脳を急速に凍結し薄切りにした。切片をクレシルバイオレットで染色した。脳切片の未染色の帯域は、梗塞により損傷を受けたと考えられた。領域(梗塞及び2つの半球)を測定し、梗塞及び2つの半球の量を計算し、修正梗塞量を次の式:[修正梗塞量=梗塞量−(右半球の量−左半球の量)]により決定して、脳浮腫を補償した。
治療的処置の場合(急性期の処置)、本発明の化合物で処置された動物は、未処置の動物よりも脳損傷が少なかった。事実、虚血−再灌流の24時間、48時間又は72時間後、本発明の化合物を投与した場合、梗塞量は少なかった。
【0314】
したがって本発明の化合物は、急性虚血後の処置において神経保護活性を示す。
【0315】
様々な実験モデルにおける本発明の化合物の使用は、該新規化合物が固有の抗酸化活性を示し、酸化ストレスの影響を遅延させかつ減少させることができ、更に、抗酸化酵素をコードする遺伝子の発現も誘導し、その抗酸化特性と一緒になって、フリーラジカルからの保護を強化することを示す。加えて、本発明の化合物は、抗炎症活性を示し、PPARα核内受容体を活性化することができる。
最後に、本発明の化合物の動物虚血−再灌流モデルにおける使用は、保護的及び治療的でもある処置の両方の有益な神経保護作用を明らかにした。
【0316】
【表45】
【図面の簡単な説明】
【0317】
【図1】本発明の特定の化合物の構造である。その調製は、実施例2、4、5、6、8、10〜14、16、18、19、21及び23に記載されており、図においてそれぞれ1A.2、1A.4、1A.5、1A.6、1A.8、1A.10、1A.11、1A.12、1A.13、1A.14、1A.16、1A.18、1A.19、1A.21及び1A.23と示されている。
【図2】銅(Cu)によるLDL酸化に対する本発明の化合物の抗酸化特性の評価である。(2a)経時的な又は遅滞期における共役ジエン形成である。(2b)ジエン形成の速度である。(2c)共役ジエン形成最大量である。
【図3】Gal4/PPARトランス活性化系における本発明の化合物のPPARアゴニスト特性の評価である。
【特許請求の範囲】
【請求項1】
一般式(I):
【化1】
〔式中、
G2及びG3は、独立して酸素原子、硫黄原子又はN−R4基(ここで、G2及びG3は、同時にN−R4基を表すことはない)を表し、
R及びR4は、独立して、水素原子、又は飽和若しくはそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖若しくは分岐のアルキル基を表し、
R1、R2及びR3は、同一であるか又は異なって、水素原子、CO−R5基、又は式:CO−(CH2)2n+1−X−R6に対応する基(ここで、R1、R2又はR3基の少なくとも1個は、式:CO−(CH2)2n+1−X−R6に対応する基である)を表し、
R5は、飽和又はそうではない、場合により置換されている、場合により環式基を含む、直鎖又は分岐のアルキル基(その主鎖は1〜25個の炭素原子を含む)であり、
Xは、硫黄原子、セレン原子、SO基又はSO2基であり、
nは、0〜11の間に含まれる整数であり、
R6は、飽和又はそうではない、場合により置換されている、場合により環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは10〜23個の炭素原子を含み、可能であれば酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される1個以上のヘテロ基を含む)である〕
により表される化合物
(但し、G2R2及びG3R3が同時にヒドロキシル基を表す式(I)により表される化合物は除く)、その光学及び幾何異性体、ラセミ体、塩、水和物、並びに混合物。
【請求項2】
R1、R2又はR3基のうちのただ1個が水素原子を表すことを特徴とする、請求項1記載の化合物。
【請求項3】
CO−(CH2)2n+1−X−R6基において、Xが、硫黄又はセレン原子、有利には硫黄原子を表すことを特徴とする、請求項1又は2のいずれかに記載の化合物。
【請求項4】
CO−(CH2)2n+1−X−R6基において、nが、0〜3の間に含まれる、とりわけ0〜2の間に含まれる、特に0と等しいことを特徴とする、請求項1、2又は3のいずれか1項記載の化合物。
【請求項5】
R6が、酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される、1個以上、好ましくは0、1又は2個、より好ましくは0又は1個のヘテロ基を含むことを特徴とする、請求項1〜4のいずれか1項記載の化合物。
【請求項6】
CO−(CH2)2n+1−X−R6がCO−CH2−S−C14H29基であることを特徴とする、請求項1〜5のいずれか1項記載の化合物。
【請求項7】
R1、R2及びR3基のうちの少なくとも1個が、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、好ましくは14〜17個、より好ましくは14〜16個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖アルキル基である)を表すことを特徴とする、請求項1〜6のいずれか1項記載の化合物。
【請求項8】
R1、R2及びR3基のうちの少なくとも2個が、同一であるか又は異なって、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表す)である、請求項1〜7のいずれか1項記載の化合物。
【請求項9】
G2が、酸素又は硫黄原子、好ましくは酸素原子を表すことを特徴とする、請求項1〜8のいずれか1項記載の化合物。
【請求項10】
R2が、式:CO−(CH2)2n+1−X−R6に対応する基を表すことを特徴とする、請求項9記載の化合物。
【請求項11】
G3が、N−R4基(ここで、R4は水素原子又はメチル基である)であり、そしてG2が酸素原子である;及び/又は
R2が、CO−(CH2)2n+1−X−R6基を表す
ことを特徴とする、請求項1〜8のいずれか1項記載の化合物。
【請求項12】
R1、R2及びR3が、同一であるか又は異なって、好ましくは同一で、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、13〜17個の炭素原子、好ましくは14〜17個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖アルキル基であり、ここでnは、好ましくは0〜3に間に含まれ、特に0と等しい)を表し、とりわけR1、R2及びR3は、CO−CH2−S−C14H29基を表すことを特徴とする、請求項1〜11のいずれか1項記載の化合物。
【請求項13】
− 1−テトラデシルチオアセチルアミノ−2,3−(ジパルミトイルオキシ)プロパン;
− 3−テトラデシルチオアセチルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 3−パルミトイルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オール;
− 1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジオレオイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパン;及び
− 1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン
からなる群より選択される、請求項1記載の式(I)により表される化合物。
【請求項14】
G2R2及びR3R3基が同時にヒドロキシル基を表す式Iにより表される化合物を包含する請求項1〜13のいずれかに記載されている式(I)で表される化合物の少なくとも1つを、薬学的に許容されうる支持体中に含む、医薬組成物。
【請求項15】
脳血管性病変、より詳細には脳虚血又は脳卒中の治療又は予防が意図される、請求項14記載の医薬組成物。
【請求項16】
ヒト又は動物における予防的又は治療的処置が意図される医薬組成物を製造するための、G2R2及びG3R3基が同時にヒドロキシル基を表す式(I)により表される化合物を包含する請求項1〜13のいずれか1項記載の式(I)により表される化合物の使用。
【請求項17】
医薬組成物が、脳血管性病変、より詳細には脳虚血又は脳卒中の治療及び/又は予防のために意図されることを特徴とする、請求項16記載の使用。
【請求項1】
一般式(I):
【化1】
〔式中、
G2及びG3は、独立して酸素原子、硫黄原子又はN−R4基(ここで、G2及びG3は、同時にN−R4基を表すことはない)を表し、
R及びR4は、独立して、水素原子、又は飽和若しくはそうでない、場合により置換されている、1〜5個の炭素原子を含む、直鎖若しくは分岐のアルキル基を表し、
R1、R2及びR3は、同一であるか又は異なって、水素原子、CO−R5基、又は式:CO−(CH2)2n+1−X−R6に対応する基(ここで、R1、R2又はR3基の少なくとも1個は、式:CO−(CH2)2n+1−X−R6に対応する基である)を表し、
R5は、飽和又はそうではない、場合により置換されている、場合により環式基を含む、直鎖又は分岐のアルキル基(その主鎖は1〜25個の炭素原子を含む)であり、
Xは、硫黄原子、セレン原子、SO基又はSO2基であり、
nは、0〜11の間に含まれる整数であり、
R6は、飽和又はそうではない、場合により置換されている、場合により環式基を含む、直鎖又は分岐のアルキル基(その主鎖は、3〜23個の炭素原子、好ましくは10〜23個の炭素原子を含み、可能であれば酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される1個以上のヘテロ基を含む)である〕
により表される化合物
(但し、G2R2及びG3R3が同時にヒドロキシル基を表す式(I)により表される化合物は除く)、その光学及び幾何異性体、ラセミ体、塩、水和物、並びに混合物。
【請求項2】
R1、R2又はR3基のうちのただ1個が水素原子を表すことを特徴とする、請求項1記載の化合物。
【請求項3】
CO−(CH2)2n+1−X−R6基において、Xが、硫黄又はセレン原子、有利には硫黄原子を表すことを特徴とする、請求項1又は2のいずれかに記載の化合物。
【請求項4】
CO−(CH2)2n+1−X−R6基において、nが、0〜3の間に含まれる、とりわけ0〜2の間に含まれる、特に0と等しいことを特徴とする、請求項1、2又は3のいずれか1項記載の化合物。
【請求項5】
R6が、酸素原子、硫黄原子、セレン原子、SO基及びSO2基からなる群より選択される、1個以上、好ましくは0、1又は2個、より好ましくは0又は1個のヘテロ基を含むことを特徴とする、請求項1〜4のいずれか1項記載の化合物。
【請求項6】
CO−(CH2)2n+1−X−R6がCO−CH2−S−C14H29基であることを特徴とする、請求項1〜5のいずれか1項記載の化合物。
【請求項7】
R1、R2及びR3基のうちの少なくとも1個が、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、3〜23個の炭素原子、好ましくは13〜20個の炭素原子、好ましくは14〜17個、より好ましくは14〜16個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖アルキル基である)を表すことを特徴とする、請求項1〜6のいずれか1項記載の化合物。
【請求項8】
R1、R2及びR3基のうちの少なくとも2個が、同一であるか又は異なって、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表す)である、請求項1〜7のいずれか1項記載の化合物。
【請求項9】
G2が、酸素又は硫黄原子、好ましくは酸素原子を表すことを特徴とする、請求項1〜8のいずれか1項記載の化合物。
【請求項10】
R2が、式:CO−(CH2)2n+1−X−R6に対応する基を表すことを特徴とする、請求項9記載の化合物。
【請求項11】
G3が、N−R4基(ここで、R4は水素原子又はメチル基である)であり、そしてG2が酸素原子である;及び/又は
R2が、CO−(CH2)2n+1−X−R6基を表す
ことを特徴とする、請求項1〜8のいずれか1項記載の化合物。
【請求項12】
R1、R2及びR3が、同一であるか又は異なって、好ましくは同一で、CO−(CH2)2n+1−X−R6基(ここで、Xは、硫黄又はセレン原子、好ましくは硫黄原子を表し、そして/又はR6は、13〜17個の炭素原子、好ましくは14〜17個、さらにより好ましくは14個の炭素原子を含む、飽和した直鎖アルキル基であり、ここでnは、好ましくは0〜3に間に含まれ、特に0と等しい)を表し、とりわけR1、R2及びR3は、CO−CH2−S−C14H29基を表すことを特徴とする、請求項1〜11のいずれか1項記載の化合物。
【請求項13】
− 1−テトラデシルチオアセチルアミノ−2,3−(ジパルミトイルオキシ)プロパン;
− 3−テトラデシルチオアセチルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 3−パルミトイルアミノ−1,2−(ジテトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジ(テトラデシルチオアセチルアミノ)プロパン−2−オール;
− 1,3−ジアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジオレオイルアミノ−2−(テトラデシルチオアセチルオキシ)プロパン;
− 1,3−ジテトラデシルチオアセチルアミノ−2−(テトラデシルチオアセチルチオ)プロパン;及び
− 1−テトラデシルチオアセチルアミノ−2,3−ジ(テトラデシルチオアセチルチオ)プロパン
からなる群より選択される、請求項1記載の式(I)により表される化合物。
【請求項14】
G2R2及びR3R3基が同時にヒドロキシル基を表す式Iにより表される化合物を包含する請求項1〜13のいずれかに記載されている式(I)で表される化合物の少なくとも1つを、薬学的に許容されうる支持体中に含む、医薬組成物。
【請求項15】
脳血管性病変、より詳細には脳虚血又は脳卒中の治療又は予防が意図される、請求項14記載の医薬組成物。
【請求項16】
ヒト又は動物における予防的又は治療的処置が意図される医薬組成物を製造するための、G2R2及びG3R3基が同時にヒドロキシル基を表す式(I)により表される化合物を包含する請求項1〜13のいずれか1項記載の式(I)により表される化合物の使用。
【請求項17】
医薬組成物が、脳血管性病変、より詳細には脳虚血又は脳卒中の治療及び/又は予防のために意図されることを特徴とする、請求項16記載の使用。
【図1】

【図2】
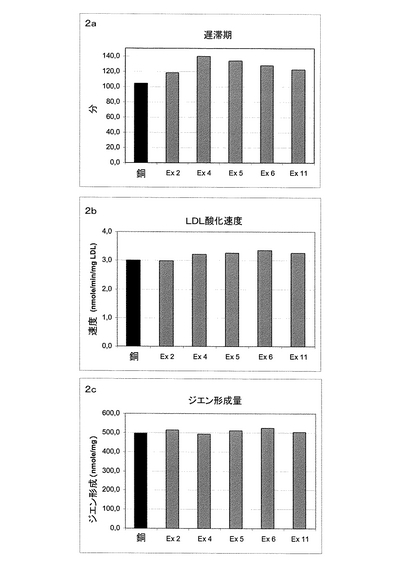
【図3】
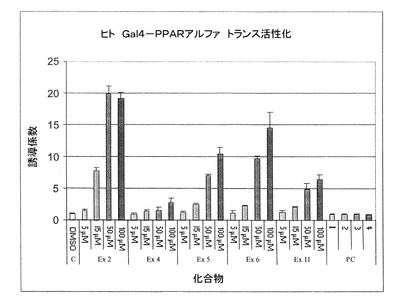
【図2】
【図3】
【公表番号】特表2006−517570(P2006−517570A)
【公表日】平成18年7月27日(2006.7.27)
【国際特許分類】
【出願番号】特願2006−502143(P2006−502143)
【出願日】平成16年2月12日(2004.2.12)
【国際出願番号】PCT/FR2004/000319
【国際公開番号】WO2004/074239
【国際公開日】平成16年9月2日(2004.9.2)
【出願人】(503067111)
【氏名又は名称原語表記】GENFIT
【Fターム(参考)】
【公表日】平成18年7月27日(2006.7.27)
【国際特許分類】
【出願日】平成16年2月12日(2004.2.12)
【国際出願番号】PCT/FR2004/000319
【国際公開番号】WO2004/074239
【国際公開日】平成16年9月2日(2004.9.2)
【出願人】(503067111)
【氏名又は名称原語表記】GENFIT
【Fターム(参考)】
[ Back to top ]