
アシル化誘導体の製造方法
【課題】医薬品、農薬、生理活性物質等の種々の分野に用いられる高い光学純度を有するアシル化誘導体を、工業的に優れた生産性で、かつ環境に低負荷に得ることが可能な製造方法を提供する。
【解決手段】アルコールとアシル化剤からアシル化誘導体を製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むアシル化誘導体の製造方法。
【解決手段】アルコールとアシル化剤からアシル化誘導体を製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むアシル化誘導体の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アシル化誘導体の製造方法に関する。より詳しくは、光学活性化合物の大規模な製造に有用で、環境に有利である速度論的分割反応によるアシル化誘導体の製造方法に関する。
【背景技術】
【0002】
光学活性化合物は、医薬品、農薬、生理活性物質等の種々の分野に用いられる化合物や中間体として有用なものである。これらの分野においてラセミ体や光学純度の低い化合物が用いられると、目的とする生物学的活性を著しく低下させたり、毒性を有するものとなったりする場合があることから、光学純度の高いものが望まれており、その製造方法においても、光学純度の高い化合物を工業的に製造できる方法が検討されている。
【0003】
従来の光学活性エステル化合物の製造方法に関し、酵素によるエステル合成反応を、二酸化炭素の臨界温度以上及び臨界圧力以上の雰囲気下で行うことについて開示されている(例えば、特許文献1参照。)。また実施例には、基質としてオレイン酸と(±)−シトロネロールを用い、超臨界二酸化炭素雰囲気下でのリパーゼによるエステル化反応を連続的に行うことが記載されている。また、超臨界流体の存在下、固体エステラーゼ酵素の存在下で有機ジオールを有機ジエステル又は有機ジカルボン酸のどちらかと反応させるポリエステルの製造方法が開示されている(例えば、特許文献2参照。)。
しかしながら、これらの製造方法においては、医薬品、農薬、生理活性物質等の種々の分野に有用であり、光学純度が高められた光学活性化合物を工業的に生産性を向上させて製造できる方法とするための工夫の余地があった。
【0004】
また光学活性化合物の製造方法に関し、(R、S)−3−ハイドロキシテトラハイドロフランのエステルと、該化合物を光学選択的に加水分解し得る酵素とを反応させる工程及び生成した光学活性3−ハイドロキシテトラハイドロフランと光学活性3−ハイドロキシテトラハイドロフランのエステルとを分離する工程を包含する製造方法(例えば、特許文献3参照。)、光学活性グリセロールα−モノカルボン酸エステルの合成活性を有する酵素の存在下、グリセロールをアシル供与体でエステル化する光学活性グリセロールα−モノカルボン酸エステルの製造方法(例えば、特許文献4参照。)、R体及びS体を含む2−ヒドロキシ脂肪酸エステルのいずれか一方の光学異性体を立体選択的にエステル化し得る酵素の存在下に、有機溶媒中でエステル化剤と作用させる光学活性エステルの製造方法(例えば、特許文献5参照。)が開示されている。
しかしながら、これらの製造方法においては、収率や収量において充分ではないことから、この点を向上させて実用的な製造方法とするとともに、光学純度が高められた光学活性化合物を工業的に製造できる方法とするための工夫の余地があった。
【特許文献1】特開平9−283号公報(第2−3頁)
【特許文献2】特開平8−256783号公報(第2頁)
【特許文献3】特開平10−337197号公報(第2頁)
【特許文献4】特開平11−113590号公報(第2頁)
【特許文献5】特開2001−128694号公報(第2頁)
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記現状に鑑みてなされたものであり、高い光学純度を有するアシル化誘導体を、工業的に優れた生産性で、かつ環境に低負荷に得ることが可能な製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0006】
本発明者等は、光学活性を有する化合物の製造方法について種々検討したところ、超臨界二酸化炭素中で酵素触媒の存在下、連続流通式の反応器を用い、(R)体と(S)体との混合物であるがアルコールのいずれか一方の光学異性体のみをアシル化剤によりアシル化(立体選択的エステル化)して、速度論的分割反応を行うことにより、高い光学純度の(R)体又は(S)体のエステル化合物を、高い空時収量で得ることができることを見いだし、上記課題をみごとに解決することができることに想到した。また、このような形態とすることにより、廃棄しなければいけない有機原料等を大幅に低減できたり、非溶媒反応とできることから、生産性にも優れ、環境面においても有利であることも見いだし、更に、アシル化されないで残存するアルコールを、ルテニウム原子を含有する触媒等によってラセミ化し、ラセミアルコールとしたうえで、再度、酵素触媒下で超臨界連続反応の原料に使用し、これを繰り返すことにより原料のアルコールをほぼ全てを(R)体又は(S)体のエステル化合物に変換できることから、高い収率で光学純度の高い化合物を得ることができることも見いだし、本発明に到達したものである。
【0007】
このように超臨界二酸化炭素を溶媒として、例えばリパーゼ(Novozyme435)等の酵素を固定床で用い立体選択的エステル化反応(アシル化反応)を連続的に行うと、バッチ法に比べて400倍程度の生産性を達成することができ、立体選択性もE>1000とすることが可能となる。このような形態においては、(S)体のアルコールと(R)体のアシル化誘導体を高い光学純度で分割取得できる。このように本発明で得られるのは、光学活性なアシル化誘導体(アシル体)と光学活性な未反応アルコールとがある。したがって、本発明は、光学活性なアシル化誘導体だけでなく、光学活性アルコールを光学分割して製造する方法でもある。また、例えば、アシル化剤であるビニルアセテート(酢酸ビニル)を少し過剰量用いることにより、アセチル化転化率を高くすることも可能であり、それによって光学純度を更に高くすることができる。
ところで、超臨界二酸化炭素は、高い反応性を有し、環境に優しい溶媒として注目されている。また、酵素は、自然界のものであり、優れた化学的選択性、位置選択性及びエナンチオ選択性を有し、再利用できる不均一系触媒であることから、このような超臨界二酸化炭素と酵素触媒とが組み合わされた反応工程は、光学活性化合物を製造するための有望な合成手段であり、グリーンケミストリー(環境に優しい化学)においても有用なものである。
【0008】
すなわち本発明は、アルコールとアシル化剤からアシル化誘導体を製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むアシル化誘導体の製造方法である。
以下に本発明を詳述する。
【0009】
本発明においては、超臨界二酸化炭素の存在下、酵素を用いて、立体選択的エステル化反応を連続的に行うことによりアルコールとアシル化剤からアシル化誘導体を製造することとなる。このような超臨界連続反応工程においては、ラセミアルコールが速度論的分割反応を経て、対応する光学的に純粋なアシル化誘導体に効率的に転化されることになることから、バッチ法に比較して生産性を向上することが可能であり、光学活性化合物の合成のための実用的手段としても有用なものである。このように、速度論的分割のために連続流通反応システムを使用することにより、長期の反応時間に対する生産性において顕著な改善をもたらすだけでなく、実質的な非溶媒反応とできることになる。
【0010】
上記立体選択的エステル化反応とは、(S)体アルコールと(R)体アルコールとの混合物である基質のアルコールを、立体選択的にアシル化する反応であり、アシル化剤を用いることにより(S)体及び(R)体のいずれか一方が選択的にアシル化されてアシル化誘導体が生成する形態が好適である。またこの場合、アシル化されない(S)体及び(R)体のいずれか一方がそのままアルコールとして残存することが好ましい。すなわち、超臨界連続反応工程の生成物は、未反応の(S)体アルコール及び生成した(R)体アシル化誘導体を含有する混合物、又は、未反応の(R)体アルコール及び生成した(S)体アシル化誘導体を含有する混合物であることが好ましい。
このような生成物においては、アシル化誘導体と未反応のアルコールのそれぞれの化学的特性や物理的特性の違いを利用して通常の方法により分割することによって、アシル化誘導体を高い光学純度で得ることができることになる。
またこのような化学的特性や物理的特性の違いを利用した分割により、光学活性なアシル化誘導体だけでなく、光学活性な未反応のアルコールを得ることもできることから、本発明は高い光学純度のアルコールを製造する場合にも好ましい方法である。すなわち、アルコールとアシル化剤とから光学活性なアシル化誘導体及び/又は光学活性なアルコールを製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含む光学活性なアシル化誘導体及び/又は光学活性なアルコールの製造方法もまた、本発明の好ましい実施形態の一つである。
上記アシル化反応とは、例えば、下記反応式(1)で表される反応をいう。なお、式中、RI〜RIIIは、アルキル基、アルケニル基、アルキニル基、アリール基等であり、Xは、ハロゲン原子である。
【0011】
【化1】

【0012】
上記アシル化誘導体の製造方法は、未反応のアルコールをラセミ体とし、超臨界連続反応工程の原料に使用することが好ましい。このように未反応のアルコールを原料として使用し、これを繰り返すことにより、初期に原料として用いたアルコールをほぼすべてアシル化誘導体に変換することができることになる。
上記未反応のアルコールをラセミ体とする方法としては、アルコールをラセミ化することができる触媒を用いることが好適であり、例えば、ルテニウム原子を含有する触媒が好ましい。
上記ルテニウム原子を含有する触媒としては、例えば、η5−C5(CH3)5RuCl−1,5−シクロオクタジエン等が好適である。
上記ラセミ化反応は、上述の立体選択的エステル化反応と交互又は同時に行うことにより、原料のアルコールを高い転化率でアシル化誘導体に変換することができることになる。
上記ラセミ化反応においては、例えば、トルエン等の溶媒中において、アミノ化合物の存在下に反応させることが好適である。反応温度としては、例えば、10〜50℃が好ましく、20〜30℃がより好ましい。
上記アミノ化合物としては、上述のルテニウム原子を含有する触媒の配位子となり得る化合物であればよく、例えば、(CH3)2N(CH2)2NH2、(C6H5)2P(CH2)2NH2、(C6H5)2P(CH2)2NHCH3、(C6H5)2P(CH2)2N(CH3)2、C6H5S(CH2)2NH2、(C6H5)2P(CH2)3NH2等が好適である。
【0013】
上記エステル化反応を連続的に行うとは、連続的に反応生成物を取り出すことが可能なように、連続的に反応基質を投入してエステル化反応を行うことであり、反応の形態としては、固定床を利用した流通式の形態等が好適である。
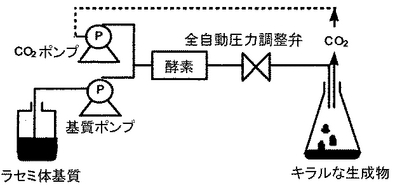
このような超臨界連続反応工程においては、例えば、図1に示すような形態で連続的に反応させることが好ましい。図1においては、ラセミ体基質であるアルコール及びアシル化剤を送液する基質ポンプと、二酸化炭素を送るCO2ポンプにより基質と二酸化炭素が酵素を有する反応器に送られることになる。二酸化炭素は、圧力が圧力調整弁により制御され、温度が加熱オーブン(図示せず)等により制御されることにより、超臨界状態となる。反応器において生成したキラルな生成物は、未反応のアルコールとともに反応器出口から流出液として回収されることになる。このような反応工程は、連続的に行われることから生産性を向上できる。
【0014】
上記超臨界二酸化炭素とは、超臨界条件にある二酸化炭素であり、超臨界条件とは、物質固有の臨界温度及び臨界圧力を超えた領域をいい、二酸化炭素においては、温度が31℃以上であり、圧力が7.3MPa以上の条件である。また、亜臨界条件にある二酸化炭素であってもよい。亜臨界条件とは、臨界点近傍の臨界圧力より低い条件領域(亜臨界条件)をいい、具体的には、温度が30℃以上で圧力が5MPa以上の領域である。
上記温度としては、30℃以上であることが好ましい。より好ましくは、35℃以上であり、更に好ましくは、40℃以上である。また、70℃以下であることが好ましい。より好ましくは、65℃以下であり、更に好ましくは、60℃以下である。また圧力としては、7MPa以上であることが好ましい。より好ましくは、10MPa以上である。また、25MPa以下であることが好ましい。より好ましくは、20MPa以下である。
【0015】
上記二酸化炭素の流量としては、例えば、1ml/hr/ml以上であることが好ましい。より好ましくは、5ml/hr/ml以上であり、更に好ましくは、10ml/hr/ml以上である。また、500ml/hr/ml以下であることが好ましい。より好ましくは、300ml/hr/ml以下であり、更に好ましくは、200ml/hr/ml以下である。
上記基質の流量としては、0.1ml/hr/ml以上であることが好ましい。より好ましくは、0.2ml/hr/ml以上であり、更に好ましくは、0.5ml/hr/ml以上である。また、50ml/hr/ml以下であることが好ましい。より好ましくは、30ml/hr/ml以下であり、更に好ましくは、20ml/hr/ml以下である。なお、二酸化酸素や基質の流量は、酵素触媒容積当りの速度(空間速度;space velocity)で表す。
上記基質であるアルコールとアシル化剤のモル比としては、例えば、アルコール/アシル化剤が1/2.0以上であることが好ましい。より好ましくは、1/1.5以上である。また、1/0.4以下であることが好ましい。より好ましくは、1/0.5以下である。
上記酵素の使用量としては、例えば、1.0g/l以上であることが好ましい。より好ましくは、10g/l以上であり、更に好ましくは、50g/l以上である。また、250g/l以下であることが好ましい。より好ましくは、200g/l以下であり、更に好ましくは、150g/l以下である。
【0016】
上記酵素としては、アルコールを立体選択的にアシル化することができる酵素であればよく、(R)体アルコールを選択的にアシル化する酵素であることが好適であり、例えば、リパーゼ又はエステラーゼであることが好ましい。このようなリパーゼやエステラーゼとしては、例えば、下記に属する微生物や、豚膵臓、豚肝臓等の哺乳類由来のものが好ましい。
クレブシエラ(Klebsiella)属、セラチア(Serratia)属、キャンディダ(Candida)属、アクロモバクター(Achromobacter)属、アルカリゲネス(Alcaligenes)属、アスペルギルス(Aspergillus)属、シュードモナス(Pseudomonas)属、バチルス(Bacillus)属、リゾプス(Rhizopus)属、ペニシリウム(Penicillium)属、ゲオトリカム(Geotrichum)属、ムコール(Mucor)属、リゾムコール(Rhizomucor)属、フミコラ(Humicola)属、バークホーデリア(Burkholderia)属、ブレビバクテリウム(Brevibacterium)属、コリネバクテリウム(Corynebacterium)属、ノカルディア(Nocardia)属、セデセア(Cedecea)属、プロテウス(Proteus)属、メチロバクテリウム(Methylobacterium)属等。
【0017】
上記微生物としては、例えば、クレブシエラ・オキシトカ(Klebsiella oxytoca)、セラチア・マルセッセンス(Serratia marcescens)、キャンディダ・シリンドラッセ(Candida cylindracea)、キャンディダ・ルゴーサ(Candida rugosa)、アスペルギルス・オリゼ(Aspergillus oryzae)、アスペルギルス・メレウス(Aspergillus melleus)、バチルス・アミロリクエファシエンス(Bacillus amyloliquefaciens)、セデセア・ダビセ(Cedecea davisae)JCM1685、ブレビバクテリウム・プロトホミエ(Brevibacterium protophomiae)IFO12128、コリネバクテリウム・アクアティカム(Corynebacterium aquaticum)IFO12154、ノカルディア・コラリナ(Nocardia corallina)IFO3338、シュードモナス・マルトフィラ(Pseudomonas maltophila)IFO12690、シュードモナス・クロロラフィス(Pseudomonas chlororaphis)IFO3523、バチルス・ステアロサーモフィラス(Bacillus stearothermophillus)IAM1035、バチルス・サチルス(Bacillus subtilis)JCM1465T、メチロバクテリウム・ラジオトレランス(Methylobacterium radiotolerans)IAM12099、プロテウス・ブルガリス(Proteus vulgaris)IAM12003等が好適である。
【0018】
上記酵素としては、例えば、下記のような市販の酵素が好適であり、その使用形態については粉末状で用いてもよく、担体に担持して用いてもよいが、固定化して用いることが好ましい。担体としては、活性炭、セライト、ゼオライト、アルミナ、シリカゲル等の金属酸化物等の無機材料;ポリスチレン、デンプン等の有機材料等が好適である。
【0019】
ノボザイム435(キャンディダ・アンタクチカ由来、ノボザイム社製)、SNSM−87(クレブシエラ・オキシトカ由来、ナガセ生化学工業社製)、SM酵素(セラチア・マルセッセンス由来、ナガセ生化学工業社製)、リパーゼOF(キャンディダ・シリンドラッセ由来、名糖産業社製)、リパーゼAL(アクロモバクター由来、名糖産業社製)、リパーゼPL−679(アルカリゲネス由来、名糖産業社製)、リパーゼAY「アマノ」(キャンディダ・ルゴーサ由来、天野製薬社製)、D−150(アスペルギルス・オリゼ由来、ナガセ生化学工業社製)、リパーゼタイプII(ブタ膵臓由来、シグマ社製)、リパーゼP原末(シュードモナス・sp由来、ナガセ生化学工業社製)、XP−488(アスペルギルス・メレウス由来、ナガセ生化学工業社製)、結晶細菌アルカリプロテアーゼ(バチルス・アミロリクエファシエンス由来、ナガセ生化学工業社製)、スミチームMP(アスペルギルス・オリゼ由来、新日本化学社製)、リパーゼQL(アルカリゲネス・sp由来、名糖産業社製)、ビオプラーゼAL−45(バチルス・ズブチリス由来、ナガセ生化学工業社製)、リパーゼAK(シュードモナス・フルオレセンス由来)、リパーゼD (リゾプス・デレマ由来)、リパーゼF−AP(リゾプス属由来)、リパーゼG(ペニシリウム・カメンバーティ由来)、リパーゼGC−20(ゲオトリカム・キャンジジューム由来)、リパーゼPZ−6(アスペルギルス属由来)、リパーゼM−AP(ムコール属由来、天野製薬社製)、キラザイムL−1(バークホーデリア属由来)、キラザイムL−4(シュードモナス属由来)、キラザイムL−6(シュードモナス属由来)、キラザイムL−8(フミコラ属由来、べーリンガー・マンハイム社製)、リポザイム(リゾムコール・ミハイ由来、Fulka社製)、リパーゼA(アスペルギルス・ニガー由来、天野製薬社製)、リパーゼP(シュードモナス・セパシア由来、天野製薬社製)、リパーゼPS(シュードモナス・セパシア由来、天野製薬社製)、リパーゼAH(シュードモナス・セパシア由来、天野製薬社製)、リパーゼAK(シュードモナス・フルオレッセンス由来、天野製薬社製)、リパーゼMY(キャンディダ・ルゴーサ由来、名糖産業社製)、CRL(キャンディダ・ルゴーサ由来、シグマ社製)、リパーゼL−9(ムコール・ミーハイ由来、ロシュ・ダイアグノスティックス社製)、MML(ムコール・ミーハイ由来、ノボノルディスク社製)等。
【0020】
上記反応においては、上述の微生物の培養物、菌体又は菌体処理物を使用することにより、酵素を触媒とした立体選択的エステル化反応する形態としてもよい。
上記微生物の培養物としては、微生物を培地中で培養して得られる培養物のそのままを用いる形態が挙げられる。菌体としては、上記培養物から遠心分離等の集菌操作によって得られる菌体が挙げられる。菌体処理物としては、凍結乾燥菌体、菌体破砕物、無細胞抽出物、無細胞抽出物からゲル濾過、イオン交換クロマトグラフィー等の分離操作により得られる粗酵素又は精製酵素等が挙げられる。これらは、常法により固定化して使用することも可能である。
【0021】
上記アルコールとしては、水酸基が結合する不斉炭素原子を有するアルコールであればよく、例えば、下記一般式(1);
【0022】
【化2】
【0023】
(式中、R1〜R3は、それぞれ異なって、水素原子、炭素数1〜30のアルキル基、アルケニル基、アルキニル基、炭素数6〜30のアリール基を表す。R1〜R3は、それぞれ置換基を有していてもよく、結合していてもよい。)で表される化合物であることが好適である。アルキル基、アルケニル基、アルキニル基は、特に炭素数3〜20が好ましく、より好ましくは炭素数5〜15である。アリール基は、特に炭素数6〜18が好ましく、より好ましくは炭素数6〜12である。
上記アルコールとしては、例えば、1−フェニルエタノール、1−フェニルプロパノール、1−フェニル−2−プロパノール、1−フェニルブタノール、1−フェニル−2−ブタノール、1−フェニルペンタノール、2−ブタノール、2−ペンタノール、2−ヘキサノール、2−ヘプタノール、2−オクタノール、2−ノナノール、2−デカノール、2−ウンデカノール、2−ドデカノール、2−トリデカノール、1,2,3,4−テトラヒドロ−1−ナフトール等が好適である。これらの中でも、1−フェニルエタノール、2−ウンデカノール、1,2,3,4−テトラヒドロ−1−ナフトールが好ましい。
【0024】
上記アシル化剤としては、有機酸エステル、有機酸無水物等を用いることができ、例えば、酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、ピバル酸ビニル、カプロン酸ビニル、ラウリン酸ビニル、ミリスチン酸ビニル、パルミチン酸ビニル、ステアリン酸ビニル、モノクロロ酢酸ビニル、アクリル酸ビニル、メタクリル酸ビニル、クロトン酸ビニル、安息香酸ビニル、2−クロロ安息香酸ビニル、4−ニトロ安息香酸ビニル、2,4−ジニトロ安息香酸ビニル、3,5−ジニトロ安息香酸ビニル、桂皮酸ビニル、4−t−ブチル安息香酸ビニル、2−フランカルボン酸ビニル、3−フランカルボン酸ビニル等のビニルエステル;酢酸プロペニル、酢酸イソプロペニル、酪酸プロペニル、安息香酸プロペニル等のプロペニルエステル;無水酢酸、無水プロピオン酸、無水酪酸、無水吉草酸、無水マイレン酸、無水フタル酸、無水シュウ酸、無水マロン酸、無水コハク酸、無水フタル酸、無水安息香酸等の無水物;酢酸1−エトキシビニル、安息香酸1−エトキシビニル、2−フランカルボン酸1−エトキシビニル、3−フランカルボン酸1−エトキシビニル、ピコリン酸1−エトキシビニル、ニコチン酸1−エトキシビニル、イソニコチン酸1−エトキシビニル、2−チオフェンカルボン酸1−エトキシビニル、3−チオフェンカルボン酸1−エトキシビニル等の1−エトキシビニルエステル;安息香酸メチル;酢酸、酪酸、アクリル酸等の2,2,2−トリフルオロエチルエステル、2,2,2−トリクロロエチルエステル、フェニルエステル等が好適である。これらの中でも、有機酸ビニルエステル、特に、酢酸ビニルが好ましい。
【0025】
本発明の立体選択的エステル化反応においては、例えば、ラセミ−1−フェニルエタノールと、アシル化剤である酢酸ビニル(ビニルアセテート)とを原料とし、酵素としてノボザイム435(ノボザイム社製)を用いて、超臨界二酸化炭素中でアシル化誘導体を生成する場合、下記反応式(2)に示されるように、(R)体アルコールがアシル化されてアシル化誘導体となり、(S)体アルコールは未反応のまま残存することになる。更に、(S)体アルコールをルテニウム原子を含有する触媒によりラセミ化する場合においては、該ラセミ化により得られるラセミ体アルコールを立体選択的エステル化反応の原料として再度用いることになる。また、アルコールとしてラセミ−2−ウンデカノールや、ラセミ−テトラヒドロナフトールを用いた場合においては、下記反応式(3)及び(4)に示されるように反応することになる。
【0026】
【化3】
【0027】
本発明において得られるアシル化誘導体の光学純度としては、98.5%ee以上であることが好ましい。より好ましくは、99.0%ee以上であり、更に好ましくは、99.5%ee以上である。またE値としては、500以上であることが好ましい。より好ましくは、1000以上であり、更に好ましくは、1500以上である。
また残存して得られるアルコールの光学純度としては、85%ee以上であることが好ましい。より好ましくは、90%ee以上であり、更に好ましくは、95%ee以上である。
本発明のアシル化誘導体の製造方法は、医薬品、農薬、生理活性物質等の種々の分野に用いられる化合物や中間体の製造方法として好適に適用することができる方法である。
【発明の効果】
【0028】
本発明のアシル化誘導体の製造方法は、上述の構成よりなり、高い光学純度を有するアシル化誘導体を、工業的に優れた生産性で、かつ環境に低負荷に得ることができるものである。
【発明を実施するための最良の形態】
【0029】
以下に実施例を掲げて本発明を更に詳細に説明するが、本発明はこれらの実施例のみに限定されるものではない。なお、特に断りのない限り、「部」は「重量部」を、「%」は「質量%」を意味するものとする。
【0030】
<分析及び試薬>
GC分析は、Shimazu GC−14B(C−R7A plus)に装備された光学活性カラム(Chrompack,Chirasil−DEX CB:25m;He 2ml/min)を用いて行った。
LC分析は、光学活性カラムOJ−H(ヘキセン/2−プロパノール=95/5,0.4ml/min,254nm,35℃)を用いて行った。
1H−NMR分析は、Brucker DPX400により400MHzで行った。
ビニルアセテートは、モリキュラーシーブズで乾燥し、蒸留してから用いた。その他の試薬は、ナカライ社、和光純薬工業社、東京化成社、アルドリッチ社、若しくは、ACROS社で市販されているものをそのまま用いた。リパーゼ(Novozym435)は、Novozymes社から購入した。また、Novozym435中の酵素タンパク含有量は、1〜10%である。
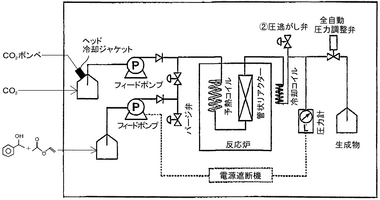
<フロー系の反応装置>
図2に示すような装置により、酵素連続流通反応を行った。
二酸化炭素の送液は、HPLCポンプ(商品名:インテリジェントHPLCポンプ PU−1580、日本分光社製)により行い、基質の送液は、HPLCポンプ(商品名:インテリジェントHPLCポンプ PU−2080、日本分光社製)により行った。
二酸化炭素の圧力は、反応器の上流と下流で測定して、全自動圧力調整弁(SCF−Bpg、日本分光社製)により制御した。
温度は、反応炉として加熱オーブン(GCオーブン、GC14B、島津製作所社製)又はサンドバス(マツキ科学社製)を用い、反応器の上下部及び外側で測定して制御した。なお、その他の装置としては、以下のものを用いた。
ヘッド冷却ジャケット:日本分光社製
圧逃し弁:NUPRO社製の型式R3A
パージ弁:日本分光社製のSCF−GetストップバルブSV−500
管状リアクター:Swagelock社製の1/2インチφステンレス製反応管(VCR継手付)
圧力計:GLサイエンス社製のデジタル圧力計(上下限リミッタ接点信号端子台付)
電源遮断機:GLサイエンス社製
【0031】
<実施例1>フェニルエタノールの反応
リパーゼ(Novozyme435)5ml(1.89g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。1−フェニルエタノール(48.8g)及びビニルアセテート(17.2g)(1−フェニルエタノール:ビニルアセテート=1:0.5(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、18ml/hr/ml(1.5ml/min)に設定した。基質溶液の流量を、0.84ml/hr/ml(0.07ml/min)に設定し、反応を開始した。約1.2時間後、装置が安定してから、反応器出口から流出液を採取し、GC分析、LC分析及びNMR分析を行った。立体配置の決定は、標準物質とGCの保持時間の比較により行った。結果を表1の1〜3に示す。
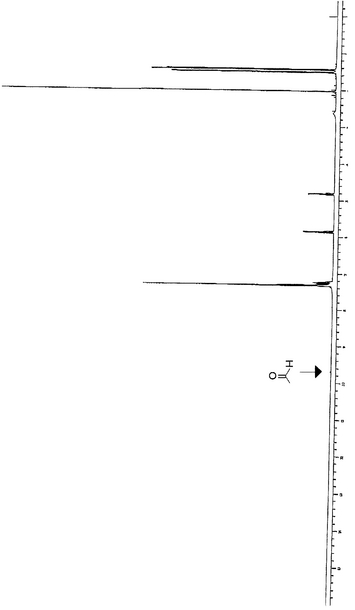
また、反応開始から3.6時間後に二酸化炭素の流量を6ml/hr/ml(0.5ml/min)に、基質溶液の流量を0.28ml/hr/ml(0.023ml/min)に変更した。5.2時間後から、反応器出口から流出液の採取を再開し、GC分析、LC分析及びNMR分析を行った。結果を表1の4〜7に示す。また、表1の3における採取物(精製は行っていない)のNMRスペクトルを図3に示す。更に、同様の実験により、圧力やビニルアセテートのモル比の検討を行った。結果を表2に示す。
【0032】
表2より、99.7%eeである所望の(R)体酢酸エステルを収率47%で得ることができた。E値は、1800を超えるものであった。また、ビニルアセテートを少し過剰量用いることにより、化学的収率が47%から50%に増加した。なお、収率は、原料である1−フェニルエタノールの初期量を基準とした(R)体酢酸エステルのモル収率である。
更に、ビニルアセテートが大量に用いられた場合であっても、(S)体アルコールの過剰反応は、同じ条件下でほとんど進行しておらず、99%ee以上の(R)体酢酸エステルと未反応の(S)体アルコールの定量的な混合物が供給され、8.9から20MPaの範囲で二酸化炭素圧力を変化させても、反応結果の顕著な変化はないことがわかった。
図3の反応混合物(表1の3における採取物)のNMRスペクトルより、副反応が生じていないことがわかった。
【0033】
上記%eeは、光学純度を示す鏡像体過剰率(%enantiomeric excess)であり、下記式(1)より求めることができる。なお、R及びSは生成物中に占めるR体及びS体の割合である。
【0034】
【数1】
【0035】
上記E値は、立体選択性を示す値であり、下記式(2)より求めることができる。なお、Cは収率であり、eeは上記鏡像体過剰率である。
【0036】
【数2】
【0037】
<実施例2>フェニルエタノールの3日間の連続運転反応
リパーゼ(Novozyme435)5ml(1.73g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。1−フェニルエタノール(293g)及びビニルアセテート(124g)(1−フェニルエタノール:ビニルアセテート=1:0.6(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、18ml/hr/ml(1.5ml/min)に設定した。基質溶液の流量を、0.84ml/hr/ml(0.07ml/min)に設定し、反応を開始した。約1時間後に装置が安定してから、反応器出口から流出液の採取を始め、GC分析、LC分析及びNMR分析を行った。
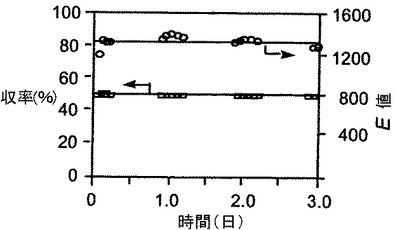
二酸化炭素及び基質溶液の流量は、3日間、一定に保ち、合計221gのラセミ体の1−フェニルエタノールを光学的に純粋な(S)体アルコールと(R)体酢酸エステルに変換した。結果を図4に示す。なお、収率は、原料である1−フェニルエタノールの初期量を基準とした(R)体酢酸エステルのモル収率である。ラセミ体アルコールからの光学活性なアシル化誘導体のモル収率の最大値は50%である。
図4に示すように、このような生触媒は、超臨界条件(12.9〜13MPa、42℃)での3日間の反応において、反応性と選択性の両方で触媒としての作用を維持し、5.0mlの反応器を用いて、(R/S)−1−フェニルエタノール(221g)が、99%eeの(S)体アルコールと99%eeの(R)体酢酸エステルに定量的に変換した。
【0038】
<実施例3>2−ウンデカノールの反応
リパーゼ(Novozyme435)1ml(0.347g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。2−ウンデカノール(43.1g)及びビニルアセテート(12.91g)(2−ウンデカノール:ビニルアセテート=1:0.6(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、180ml/hr/ml(3ml/min)に設定した。基質溶液の流量を、7.56ml/hr/ml(0.126ml/min)に設定し、反応を開始した。約0.5時間後に装置が安定してから、反応器出口から流出液を採取を始め、GC分析、LC分析及びNMR分析を行った。立体配置の決定は、標準物質とGCの保持時間の比較により行った。収率は48〜50%であり、E値は112〜137であった。なお、収率は、原料である2−ウンデカノールの初期量を基準とした(R)体酢酸エステルのモル収率である。
【0039】
<実施例4>テトラヒドロナフトールの反応
リパーゼ(ノボザイム435)5ml(1.98g)、1,2,3,4−テトラヒドロ−1−ナフトール:ビニルアセテート=1:0.6(mol/mol)、基質溶液の流量0.84ml/hr/ml(0.07ml/min)、超臨界二酸化炭素の流量18ml/hr/ml(1.5ml/min)又は36ml/hr/ml(3ml/min)、圧力13.0MPaとした以外は、実施例3と同様にして反応を行った。
E値は1500以上であった。
【0040】
<実施例5>1−フェニルエタノールの動的速度論的分割反応
フロー系のリパーゼ反応により得られた生成物(実施例1における表1の3のサンプル、図3のNMRチャート)のラセミ化反応を行った。Ru触媒を用いるラセミ化反応の結果、6割程度ラセミ化が進行した。
【0041】
<比較例1>バッチ反応
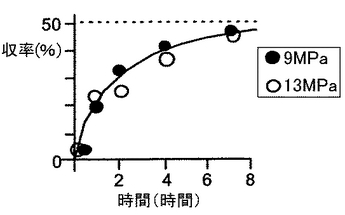
ラセミ−1−フェニルエタノール(0.83mmol)とビニルアセテート(5.4mmol)を、40℃、9MPaの二酸化炭素の存在下、酵素(Novozyme435)の存在下で反応させた。48%転化率において、99.8%eeの光学活性(R)体酢酸エステルと、90.6%eeの未反応の(S)体アルコールとの1:1の混合物が得られた。同様にして、二酸化炭素圧力を9〜13MPaの範囲を変化させて反応を行った。結果を図5に示す。
基質である(R)体アルコールが(S)体アルコールよりも早く反応し、約7時間後に反応が停止した。このようにエナンチオマーの反応性がかなり異なることから、(R)体アルコールが消費されて反応が停止したと考えられる。二酸化炭素圧力が9〜13MPaの間における反応において、収率48%、E値100以上であった。
【0042】
【表1】
【0043】
【表2】
【0044】
【表3】
【0045】
表1において、反応時間は、安定な反応時間に達した後の時間である。なお、表3は、実施例における酵素の使用量、二酸化酸素及び基質溶液(アルコール+ビニルアセテート)の流量及び空間速度(space velocity)を示す。
10mlの反応器を用いるバッチ反応においては、7時間の反応の間に、光学活性アセテート3を0.83mmol製造するが、連続流通反応器(5ml)を用いることにより、25mmol/h(3ml/h)の割合で生成物を与えることができた。
更に、ビニルアセテートを少し過剰量用いることにより、円滑に反応が進行し、反応の間の廃棄物を最小限におさえて、所望の生成物を優れた純度で供給することができた。
この合成プロセスは、光学活性アルコールの大規模の製造に特に有用である。
【図面の簡単な説明】
【0046】
【図1】図1は、本発明の超臨界連続反応工程を示す概念図である。
【図2】図2は、実施例において用いた反応装置の概念図である。
【図3】図3は、実施例における反応生成物のNMRスペクトルである。
【図4】図4は、実施例におけるフェニルエタノールの3日間の連続運転反応の結果を示すグラフである。
【図5】図5は、バッチ反応を行った場合の結果を示すグラフである。
【技術分野】
【0001】
本発明は、アシル化誘導体の製造方法に関する。より詳しくは、光学活性化合物の大規模な製造に有用で、環境に有利である速度論的分割反応によるアシル化誘導体の製造方法に関する。
【背景技術】
【0002】
光学活性化合物は、医薬品、農薬、生理活性物質等の種々の分野に用いられる化合物や中間体として有用なものである。これらの分野においてラセミ体や光学純度の低い化合物が用いられると、目的とする生物学的活性を著しく低下させたり、毒性を有するものとなったりする場合があることから、光学純度の高いものが望まれており、その製造方法においても、光学純度の高い化合物を工業的に製造できる方法が検討されている。
【0003】
従来の光学活性エステル化合物の製造方法に関し、酵素によるエステル合成反応を、二酸化炭素の臨界温度以上及び臨界圧力以上の雰囲気下で行うことについて開示されている(例えば、特許文献1参照。)。また実施例には、基質としてオレイン酸と(±)−シトロネロールを用い、超臨界二酸化炭素雰囲気下でのリパーゼによるエステル化反応を連続的に行うことが記載されている。また、超臨界流体の存在下、固体エステラーゼ酵素の存在下で有機ジオールを有機ジエステル又は有機ジカルボン酸のどちらかと反応させるポリエステルの製造方法が開示されている(例えば、特許文献2参照。)。
しかしながら、これらの製造方法においては、医薬品、農薬、生理活性物質等の種々の分野に有用であり、光学純度が高められた光学活性化合物を工業的に生産性を向上させて製造できる方法とするための工夫の余地があった。
【0004】
また光学活性化合物の製造方法に関し、(R、S)−3−ハイドロキシテトラハイドロフランのエステルと、該化合物を光学選択的に加水分解し得る酵素とを反応させる工程及び生成した光学活性3−ハイドロキシテトラハイドロフランと光学活性3−ハイドロキシテトラハイドロフランのエステルとを分離する工程を包含する製造方法(例えば、特許文献3参照。)、光学活性グリセロールα−モノカルボン酸エステルの合成活性を有する酵素の存在下、グリセロールをアシル供与体でエステル化する光学活性グリセロールα−モノカルボン酸エステルの製造方法(例えば、特許文献4参照。)、R体及びS体を含む2−ヒドロキシ脂肪酸エステルのいずれか一方の光学異性体を立体選択的にエステル化し得る酵素の存在下に、有機溶媒中でエステル化剤と作用させる光学活性エステルの製造方法(例えば、特許文献5参照。)が開示されている。
しかしながら、これらの製造方法においては、収率や収量において充分ではないことから、この点を向上させて実用的な製造方法とするとともに、光学純度が高められた光学活性化合物を工業的に製造できる方法とするための工夫の余地があった。
【特許文献1】特開平9−283号公報(第2−3頁)
【特許文献2】特開平8−256783号公報(第2頁)
【特許文献3】特開平10−337197号公報(第2頁)
【特許文献4】特開平11−113590号公報(第2頁)
【特許文献5】特開2001−128694号公報(第2頁)
【発明の開示】
【発明が解決しようとする課題】
【0005】
本発明は、上記現状に鑑みてなされたものであり、高い光学純度を有するアシル化誘導体を、工業的に優れた生産性で、かつ環境に低負荷に得ることが可能な製造方法を提供することを目的とするものである。
【課題を解決するための手段】
【0006】
本発明者等は、光学活性を有する化合物の製造方法について種々検討したところ、超臨界二酸化炭素中で酵素触媒の存在下、連続流通式の反応器を用い、(R)体と(S)体との混合物であるがアルコールのいずれか一方の光学異性体のみをアシル化剤によりアシル化(立体選択的エステル化)して、速度論的分割反応を行うことにより、高い光学純度の(R)体又は(S)体のエステル化合物を、高い空時収量で得ることができることを見いだし、上記課題をみごとに解決することができることに想到した。また、このような形態とすることにより、廃棄しなければいけない有機原料等を大幅に低減できたり、非溶媒反応とできることから、生産性にも優れ、環境面においても有利であることも見いだし、更に、アシル化されないで残存するアルコールを、ルテニウム原子を含有する触媒等によってラセミ化し、ラセミアルコールとしたうえで、再度、酵素触媒下で超臨界連続反応の原料に使用し、これを繰り返すことにより原料のアルコールをほぼ全てを(R)体又は(S)体のエステル化合物に変換できることから、高い収率で光学純度の高い化合物を得ることができることも見いだし、本発明に到達したものである。
【0007】
このように超臨界二酸化炭素を溶媒として、例えばリパーゼ(Novozyme435)等の酵素を固定床で用い立体選択的エステル化反応(アシル化反応)を連続的に行うと、バッチ法に比べて400倍程度の生産性を達成することができ、立体選択性もE>1000とすることが可能となる。このような形態においては、(S)体のアルコールと(R)体のアシル化誘導体を高い光学純度で分割取得できる。このように本発明で得られるのは、光学活性なアシル化誘導体(アシル体)と光学活性な未反応アルコールとがある。したがって、本発明は、光学活性なアシル化誘導体だけでなく、光学活性アルコールを光学分割して製造する方法でもある。また、例えば、アシル化剤であるビニルアセテート(酢酸ビニル)を少し過剰量用いることにより、アセチル化転化率を高くすることも可能であり、それによって光学純度を更に高くすることができる。
ところで、超臨界二酸化炭素は、高い反応性を有し、環境に優しい溶媒として注目されている。また、酵素は、自然界のものであり、優れた化学的選択性、位置選択性及びエナンチオ選択性を有し、再利用できる不均一系触媒であることから、このような超臨界二酸化炭素と酵素触媒とが組み合わされた反応工程は、光学活性化合物を製造するための有望な合成手段であり、グリーンケミストリー(環境に優しい化学)においても有用なものである。
【0008】
すなわち本発明は、アルコールとアシル化剤からアシル化誘導体を製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むアシル化誘導体の製造方法である。
以下に本発明を詳述する。
【0009】
本発明においては、超臨界二酸化炭素の存在下、酵素を用いて、立体選択的エステル化反応を連続的に行うことによりアルコールとアシル化剤からアシル化誘導体を製造することとなる。このような超臨界連続反応工程においては、ラセミアルコールが速度論的分割反応を経て、対応する光学的に純粋なアシル化誘導体に効率的に転化されることになることから、バッチ法に比較して生産性を向上することが可能であり、光学活性化合物の合成のための実用的手段としても有用なものである。このように、速度論的分割のために連続流通反応システムを使用することにより、長期の反応時間に対する生産性において顕著な改善をもたらすだけでなく、実質的な非溶媒反応とできることになる。
【0010】
上記立体選択的エステル化反応とは、(S)体アルコールと(R)体アルコールとの混合物である基質のアルコールを、立体選択的にアシル化する反応であり、アシル化剤を用いることにより(S)体及び(R)体のいずれか一方が選択的にアシル化されてアシル化誘導体が生成する形態が好適である。またこの場合、アシル化されない(S)体及び(R)体のいずれか一方がそのままアルコールとして残存することが好ましい。すなわち、超臨界連続反応工程の生成物は、未反応の(S)体アルコール及び生成した(R)体アシル化誘導体を含有する混合物、又は、未反応の(R)体アルコール及び生成した(S)体アシル化誘導体を含有する混合物であることが好ましい。
このような生成物においては、アシル化誘導体と未反応のアルコールのそれぞれの化学的特性や物理的特性の違いを利用して通常の方法により分割することによって、アシル化誘導体を高い光学純度で得ることができることになる。
またこのような化学的特性や物理的特性の違いを利用した分割により、光学活性なアシル化誘導体だけでなく、光学活性な未反応のアルコールを得ることもできることから、本発明は高い光学純度のアルコールを製造する場合にも好ましい方法である。すなわち、アルコールとアシル化剤とから光学活性なアシル化誘導体及び/又は光学活性なアルコールを製造する方法であって、超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含む光学活性なアシル化誘導体及び/又は光学活性なアルコールの製造方法もまた、本発明の好ましい実施形態の一つである。
上記アシル化反応とは、例えば、下記反応式(1)で表される反応をいう。なお、式中、RI〜RIIIは、アルキル基、アルケニル基、アルキニル基、アリール基等であり、Xは、ハロゲン原子である。
【0011】
【化1】
【0012】
上記アシル化誘導体の製造方法は、未反応のアルコールをラセミ体とし、超臨界連続反応工程の原料に使用することが好ましい。このように未反応のアルコールを原料として使用し、これを繰り返すことにより、初期に原料として用いたアルコールをほぼすべてアシル化誘導体に変換することができることになる。
上記未反応のアルコールをラセミ体とする方法としては、アルコールをラセミ化することができる触媒を用いることが好適であり、例えば、ルテニウム原子を含有する触媒が好ましい。
上記ルテニウム原子を含有する触媒としては、例えば、η5−C5(CH3)5RuCl−1,5−シクロオクタジエン等が好適である。
上記ラセミ化反応は、上述の立体選択的エステル化反応と交互又は同時に行うことにより、原料のアルコールを高い転化率でアシル化誘導体に変換することができることになる。
上記ラセミ化反応においては、例えば、トルエン等の溶媒中において、アミノ化合物の存在下に反応させることが好適である。反応温度としては、例えば、10〜50℃が好ましく、20〜30℃がより好ましい。
上記アミノ化合物としては、上述のルテニウム原子を含有する触媒の配位子となり得る化合物であればよく、例えば、(CH3)2N(CH2)2NH2、(C6H5)2P(CH2)2NH2、(C6H5)2P(CH2)2NHCH3、(C6H5)2P(CH2)2N(CH3)2、C6H5S(CH2)2NH2、(C6H5)2P(CH2)3NH2等が好適である。
【0013】
上記エステル化反応を連続的に行うとは、連続的に反応生成物を取り出すことが可能なように、連続的に反応基質を投入してエステル化反応を行うことであり、反応の形態としては、固定床を利用した流通式の形態等が好適である。
このような超臨界連続反応工程においては、例えば、図1に示すような形態で連続的に反応させることが好ましい。図1においては、ラセミ体基質であるアルコール及びアシル化剤を送液する基質ポンプと、二酸化炭素を送るCO2ポンプにより基質と二酸化炭素が酵素を有する反応器に送られることになる。二酸化炭素は、圧力が圧力調整弁により制御され、温度が加熱オーブン(図示せず)等により制御されることにより、超臨界状態となる。反応器において生成したキラルな生成物は、未反応のアルコールとともに反応器出口から流出液として回収されることになる。このような反応工程は、連続的に行われることから生産性を向上できる。
【0014】
上記超臨界二酸化炭素とは、超臨界条件にある二酸化炭素であり、超臨界条件とは、物質固有の臨界温度及び臨界圧力を超えた領域をいい、二酸化炭素においては、温度が31℃以上であり、圧力が7.3MPa以上の条件である。また、亜臨界条件にある二酸化炭素であってもよい。亜臨界条件とは、臨界点近傍の臨界圧力より低い条件領域(亜臨界条件)をいい、具体的には、温度が30℃以上で圧力が5MPa以上の領域である。
上記温度としては、30℃以上であることが好ましい。より好ましくは、35℃以上であり、更に好ましくは、40℃以上である。また、70℃以下であることが好ましい。より好ましくは、65℃以下であり、更に好ましくは、60℃以下である。また圧力としては、7MPa以上であることが好ましい。より好ましくは、10MPa以上である。また、25MPa以下であることが好ましい。より好ましくは、20MPa以下である。
【0015】
上記二酸化炭素の流量としては、例えば、1ml/hr/ml以上であることが好ましい。より好ましくは、5ml/hr/ml以上であり、更に好ましくは、10ml/hr/ml以上である。また、500ml/hr/ml以下であることが好ましい。より好ましくは、300ml/hr/ml以下であり、更に好ましくは、200ml/hr/ml以下である。
上記基質の流量としては、0.1ml/hr/ml以上であることが好ましい。より好ましくは、0.2ml/hr/ml以上であり、更に好ましくは、0.5ml/hr/ml以上である。また、50ml/hr/ml以下であることが好ましい。より好ましくは、30ml/hr/ml以下であり、更に好ましくは、20ml/hr/ml以下である。なお、二酸化酸素や基質の流量は、酵素触媒容積当りの速度(空間速度;space velocity)で表す。
上記基質であるアルコールとアシル化剤のモル比としては、例えば、アルコール/アシル化剤が1/2.0以上であることが好ましい。より好ましくは、1/1.5以上である。また、1/0.4以下であることが好ましい。より好ましくは、1/0.5以下である。
上記酵素の使用量としては、例えば、1.0g/l以上であることが好ましい。より好ましくは、10g/l以上であり、更に好ましくは、50g/l以上である。また、250g/l以下であることが好ましい。より好ましくは、200g/l以下であり、更に好ましくは、150g/l以下である。
【0016】
上記酵素としては、アルコールを立体選択的にアシル化することができる酵素であればよく、(R)体アルコールを選択的にアシル化する酵素であることが好適であり、例えば、リパーゼ又はエステラーゼであることが好ましい。このようなリパーゼやエステラーゼとしては、例えば、下記に属する微生物や、豚膵臓、豚肝臓等の哺乳類由来のものが好ましい。
クレブシエラ(Klebsiella)属、セラチア(Serratia)属、キャンディダ(Candida)属、アクロモバクター(Achromobacter)属、アルカリゲネス(Alcaligenes)属、アスペルギルス(Aspergillus)属、シュードモナス(Pseudomonas)属、バチルス(Bacillus)属、リゾプス(Rhizopus)属、ペニシリウム(Penicillium)属、ゲオトリカム(Geotrichum)属、ムコール(Mucor)属、リゾムコール(Rhizomucor)属、フミコラ(Humicola)属、バークホーデリア(Burkholderia)属、ブレビバクテリウム(Brevibacterium)属、コリネバクテリウム(Corynebacterium)属、ノカルディア(Nocardia)属、セデセア(Cedecea)属、プロテウス(Proteus)属、メチロバクテリウム(Methylobacterium)属等。
【0017】
上記微生物としては、例えば、クレブシエラ・オキシトカ(Klebsiella oxytoca)、セラチア・マルセッセンス(Serratia marcescens)、キャンディダ・シリンドラッセ(Candida cylindracea)、キャンディダ・ルゴーサ(Candida rugosa)、アスペルギルス・オリゼ(Aspergillus oryzae)、アスペルギルス・メレウス(Aspergillus melleus)、バチルス・アミロリクエファシエンス(Bacillus amyloliquefaciens)、セデセア・ダビセ(Cedecea davisae)JCM1685、ブレビバクテリウム・プロトホミエ(Brevibacterium protophomiae)IFO12128、コリネバクテリウム・アクアティカム(Corynebacterium aquaticum)IFO12154、ノカルディア・コラリナ(Nocardia corallina)IFO3338、シュードモナス・マルトフィラ(Pseudomonas maltophila)IFO12690、シュードモナス・クロロラフィス(Pseudomonas chlororaphis)IFO3523、バチルス・ステアロサーモフィラス(Bacillus stearothermophillus)IAM1035、バチルス・サチルス(Bacillus subtilis)JCM1465T、メチロバクテリウム・ラジオトレランス(Methylobacterium radiotolerans)IAM12099、プロテウス・ブルガリス(Proteus vulgaris)IAM12003等が好適である。
【0018】
上記酵素としては、例えば、下記のような市販の酵素が好適であり、その使用形態については粉末状で用いてもよく、担体に担持して用いてもよいが、固定化して用いることが好ましい。担体としては、活性炭、セライト、ゼオライト、アルミナ、シリカゲル等の金属酸化物等の無機材料;ポリスチレン、デンプン等の有機材料等が好適である。
【0019】
ノボザイム435(キャンディダ・アンタクチカ由来、ノボザイム社製)、SNSM−87(クレブシエラ・オキシトカ由来、ナガセ生化学工業社製)、SM酵素(セラチア・マルセッセンス由来、ナガセ生化学工業社製)、リパーゼOF(キャンディダ・シリンドラッセ由来、名糖産業社製)、リパーゼAL(アクロモバクター由来、名糖産業社製)、リパーゼPL−679(アルカリゲネス由来、名糖産業社製)、リパーゼAY「アマノ」(キャンディダ・ルゴーサ由来、天野製薬社製)、D−150(アスペルギルス・オリゼ由来、ナガセ生化学工業社製)、リパーゼタイプII(ブタ膵臓由来、シグマ社製)、リパーゼP原末(シュードモナス・sp由来、ナガセ生化学工業社製)、XP−488(アスペルギルス・メレウス由来、ナガセ生化学工業社製)、結晶細菌アルカリプロテアーゼ(バチルス・アミロリクエファシエンス由来、ナガセ生化学工業社製)、スミチームMP(アスペルギルス・オリゼ由来、新日本化学社製)、リパーゼQL(アルカリゲネス・sp由来、名糖産業社製)、ビオプラーゼAL−45(バチルス・ズブチリス由来、ナガセ生化学工業社製)、リパーゼAK(シュードモナス・フルオレセンス由来)、リパーゼD (リゾプス・デレマ由来)、リパーゼF−AP(リゾプス属由来)、リパーゼG(ペニシリウム・カメンバーティ由来)、リパーゼGC−20(ゲオトリカム・キャンジジューム由来)、リパーゼPZ−6(アスペルギルス属由来)、リパーゼM−AP(ムコール属由来、天野製薬社製)、キラザイムL−1(バークホーデリア属由来)、キラザイムL−4(シュードモナス属由来)、キラザイムL−6(シュードモナス属由来)、キラザイムL−8(フミコラ属由来、べーリンガー・マンハイム社製)、リポザイム(リゾムコール・ミハイ由来、Fulka社製)、リパーゼA(アスペルギルス・ニガー由来、天野製薬社製)、リパーゼP(シュードモナス・セパシア由来、天野製薬社製)、リパーゼPS(シュードモナス・セパシア由来、天野製薬社製)、リパーゼAH(シュードモナス・セパシア由来、天野製薬社製)、リパーゼAK(シュードモナス・フルオレッセンス由来、天野製薬社製)、リパーゼMY(キャンディダ・ルゴーサ由来、名糖産業社製)、CRL(キャンディダ・ルゴーサ由来、シグマ社製)、リパーゼL−9(ムコール・ミーハイ由来、ロシュ・ダイアグノスティックス社製)、MML(ムコール・ミーハイ由来、ノボノルディスク社製)等。
【0020】
上記反応においては、上述の微生物の培養物、菌体又は菌体処理物を使用することにより、酵素を触媒とした立体選択的エステル化反応する形態としてもよい。
上記微生物の培養物としては、微生物を培地中で培養して得られる培養物のそのままを用いる形態が挙げられる。菌体としては、上記培養物から遠心分離等の集菌操作によって得られる菌体が挙げられる。菌体処理物としては、凍結乾燥菌体、菌体破砕物、無細胞抽出物、無細胞抽出物からゲル濾過、イオン交換クロマトグラフィー等の分離操作により得られる粗酵素又は精製酵素等が挙げられる。これらは、常法により固定化して使用することも可能である。
【0021】
上記アルコールとしては、水酸基が結合する不斉炭素原子を有するアルコールであればよく、例えば、下記一般式(1);
【0022】
【化2】
【0023】
(式中、R1〜R3は、それぞれ異なって、水素原子、炭素数1〜30のアルキル基、アルケニル基、アルキニル基、炭素数6〜30のアリール基を表す。R1〜R3は、それぞれ置換基を有していてもよく、結合していてもよい。)で表される化合物であることが好適である。アルキル基、アルケニル基、アルキニル基は、特に炭素数3〜20が好ましく、より好ましくは炭素数5〜15である。アリール基は、特に炭素数6〜18が好ましく、より好ましくは炭素数6〜12である。
上記アルコールとしては、例えば、1−フェニルエタノール、1−フェニルプロパノール、1−フェニル−2−プロパノール、1−フェニルブタノール、1−フェニル−2−ブタノール、1−フェニルペンタノール、2−ブタノール、2−ペンタノール、2−ヘキサノール、2−ヘプタノール、2−オクタノール、2−ノナノール、2−デカノール、2−ウンデカノール、2−ドデカノール、2−トリデカノール、1,2,3,4−テトラヒドロ−1−ナフトール等が好適である。これらの中でも、1−フェニルエタノール、2−ウンデカノール、1,2,3,4−テトラヒドロ−1−ナフトールが好ましい。
【0024】
上記アシル化剤としては、有機酸エステル、有機酸無水物等を用いることができ、例えば、酢酸ビニル、プロピオン酸ビニル、酪酸ビニル、ピバル酸ビニル、カプロン酸ビニル、ラウリン酸ビニル、ミリスチン酸ビニル、パルミチン酸ビニル、ステアリン酸ビニル、モノクロロ酢酸ビニル、アクリル酸ビニル、メタクリル酸ビニル、クロトン酸ビニル、安息香酸ビニル、2−クロロ安息香酸ビニル、4−ニトロ安息香酸ビニル、2,4−ジニトロ安息香酸ビニル、3,5−ジニトロ安息香酸ビニル、桂皮酸ビニル、4−t−ブチル安息香酸ビニル、2−フランカルボン酸ビニル、3−フランカルボン酸ビニル等のビニルエステル;酢酸プロペニル、酢酸イソプロペニル、酪酸プロペニル、安息香酸プロペニル等のプロペニルエステル;無水酢酸、無水プロピオン酸、無水酪酸、無水吉草酸、無水マイレン酸、無水フタル酸、無水シュウ酸、無水マロン酸、無水コハク酸、無水フタル酸、無水安息香酸等の無水物;酢酸1−エトキシビニル、安息香酸1−エトキシビニル、2−フランカルボン酸1−エトキシビニル、3−フランカルボン酸1−エトキシビニル、ピコリン酸1−エトキシビニル、ニコチン酸1−エトキシビニル、イソニコチン酸1−エトキシビニル、2−チオフェンカルボン酸1−エトキシビニル、3−チオフェンカルボン酸1−エトキシビニル等の1−エトキシビニルエステル;安息香酸メチル;酢酸、酪酸、アクリル酸等の2,2,2−トリフルオロエチルエステル、2,2,2−トリクロロエチルエステル、フェニルエステル等が好適である。これらの中でも、有機酸ビニルエステル、特に、酢酸ビニルが好ましい。
【0025】
本発明の立体選択的エステル化反応においては、例えば、ラセミ−1−フェニルエタノールと、アシル化剤である酢酸ビニル(ビニルアセテート)とを原料とし、酵素としてノボザイム435(ノボザイム社製)を用いて、超臨界二酸化炭素中でアシル化誘導体を生成する場合、下記反応式(2)に示されるように、(R)体アルコールがアシル化されてアシル化誘導体となり、(S)体アルコールは未反応のまま残存することになる。更に、(S)体アルコールをルテニウム原子を含有する触媒によりラセミ化する場合においては、該ラセミ化により得られるラセミ体アルコールを立体選択的エステル化反応の原料として再度用いることになる。また、アルコールとしてラセミ−2−ウンデカノールや、ラセミ−テトラヒドロナフトールを用いた場合においては、下記反応式(3)及び(4)に示されるように反応することになる。
【0026】
【化3】
【0027】
本発明において得られるアシル化誘導体の光学純度としては、98.5%ee以上であることが好ましい。より好ましくは、99.0%ee以上であり、更に好ましくは、99.5%ee以上である。またE値としては、500以上であることが好ましい。より好ましくは、1000以上であり、更に好ましくは、1500以上である。
また残存して得られるアルコールの光学純度としては、85%ee以上であることが好ましい。より好ましくは、90%ee以上であり、更に好ましくは、95%ee以上である。
本発明のアシル化誘導体の製造方法は、医薬品、農薬、生理活性物質等の種々の分野に用いられる化合物や中間体の製造方法として好適に適用することができる方法である。
【発明の効果】
【0028】
本発明のアシル化誘導体の製造方法は、上述の構成よりなり、高い光学純度を有するアシル化誘導体を、工業的に優れた生産性で、かつ環境に低負荷に得ることができるものである。
【発明を実施するための最良の形態】
【0029】
以下に実施例を掲げて本発明を更に詳細に説明するが、本発明はこれらの実施例のみに限定されるものではない。なお、特に断りのない限り、「部」は「重量部」を、「%」は「質量%」を意味するものとする。
【0030】
<分析及び試薬>
GC分析は、Shimazu GC−14B(C−R7A plus)に装備された光学活性カラム(Chrompack,Chirasil−DEX CB:25m;He 2ml/min)を用いて行った。
LC分析は、光学活性カラムOJ−H(ヘキセン/2−プロパノール=95/5,0.4ml/min,254nm,35℃)を用いて行った。
1H−NMR分析は、Brucker DPX400により400MHzで行った。
ビニルアセテートは、モリキュラーシーブズで乾燥し、蒸留してから用いた。その他の試薬は、ナカライ社、和光純薬工業社、東京化成社、アルドリッチ社、若しくは、ACROS社で市販されているものをそのまま用いた。リパーゼ(Novozym435)は、Novozymes社から購入した。また、Novozym435中の酵素タンパク含有量は、1〜10%である。
<フロー系の反応装置>
図2に示すような装置により、酵素連続流通反応を行った。
二酸化炭素の送液は、HPLCポンプ(商品名:インテリジェントHPLCポンプ PU−1580、日本分光社製)により行い、基質の送液は、HPLCポンプ(商品名:インテリジェントHPLCポンプ PU−2080、日本分光社製)により行った。
二酸化炭素の圧力は、反応器の上流と下流で測定して、全自動圧力調整弁(SCF−Bpg、日本分光社製)により制御した。
温度は、反応炉として加熱オーブン(GCオーブン、GC14B、島津製作所社製)又はサンドバス(マツキ科学社製)を用い、反応器の上下部及び外側で測定して制御した。なお、その他の装置としては、以下のものを用いた。
ヘッド冷却ジャケット:日本分光社製
圧逃し弁:NUPRO社製の型式R3A
パージ弁:日本分光社製のSCF−GetストップバルブSV−500
管状リアクター:Swagelock社製の1/2インチφステンレス製反応管(VCR継手付)
圧力計:GLサイエンス社製のデジタル圧力計(上下限リミッタ接点信号端子台付)
電源遮断機:GLサイエンス社製
【0031】
<実施例1>フェニルエタノールの反応
リパーゼ(Novozyme435)5ml(1.89g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。1−フェニルエタノール(48.8g)及びビニルアセテート(17.2g)(1−フェニルエタノール:ビニルアセテート=1:0.5(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、18ml/hr/ml(1.5ml/min)に設定した。基質溶液の流量を、0.84ml/hr/ml(0.07ml/min)に設定し、反応を開始した。約1.2時間後、装置が安定してから、反応器出口から流出液を採取し、GC分析、LC分析及びNMR分析を行った。立体配置の決定は、標準物質とGCの保持時間の比較により行った。結果を表1の1〜3に示す。
また、反応開始から3.6時間後に二酸化炭素の流量を6ml/hr/ml(0.5ml/min)に、基質溶液の流量を0.28ml/hr/ml(0.023ml/min)に変更した。5.2時間後から、反応器出口から流出液の採取を再開し、GC分析、LC分析及びNMR分析を行った。結果を表1の4〜7に示す。また、表1の3における採取物(精製は行っていない)のNMRスペクトルを図3に示す。更に、同様の実験により、圧力やビニルアセテートのモル比の検討を行った。結果を表2に示す。
【0032】
表2より、99.7%eeである所望の(R)体酢酸エステルを収率47%で得ることができた。E値は、1800を超えるものであった。また、ビニルアセテートを少し過剰量用いることにより、化学的収率が47%から50%に増加した。なお、収率は、原料である1−フェニルエタノールの初期量を基準とした(R)体酢酸エステルのモル収率である。
更に、ビニルアセテートが大量に用いられた場合であっても、(S)体アルコールの過剰反応は、同じ条件下でほとんど進行しておらず、99%ee以上の(R)体酢酸エステルと未反応の(S)体アルコールの定量的な混合物が供給され、8.9から20MPaの範囲で二酸化炭素圧力を変化させても、反応結果の顕著な変化はないことがわかった。
図3の反応混合物(表1の3における採取物)のNMRスペクトルより、副反応が生じていないことがわかった。
【0033】
上記%eeは、光学純度を示す鏡像体過剰率(%enantiomeric excess)であり、下記式(1)より求めることができる。なお、R及びSは生成物中に占めるR体及びS体の割合である。
【0034】
【数1】
【0035】
上記E値は、立体選択性を示す値であり、下記式(2)より求めることができる。なお、Cは収率であり、eeは上記鏡像体過剰率である。
【0036】
【数2】
【0037】
<実施例2>フェニルエタノールの3日間の連続運転反応
リパーゼ(Novozyme435)5ml(1.73g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。1−フェニルエタノール(293g)及びビニルアセテート(124g)(1−フェニルエタノール:ビニルアセテート=1:0.6(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、18ml/hr/ml(1.5ml/min)に設定した。基質溶液の流量を、0.84ml/hr/ml(0.07ml/min)に設定し、反応を開始した。約1時間後に装置が安定してから、反応器出口から流出液の採取を始め、GC分析、LC分析及びNMR分析を行った。
二酸化炭素及び基質溶液の流量は、3日間、一定に保ち、合計221gのラセミ体の1−フェニルエタノールを光学的に純粋な(S)体アルコールと(R)体酢酸エステルに変換した。結果を図4に示す。なお、収率は、原料である1−フェニルエタノールの初期量を基準とした(R)体酢酸エステルのモル収率である。ラセミ体アルコールからの光学活性なアシル化誘導体のモル収率の最大値は50%である。
図4に示すように、このような生触媒は、超臨界条件(12.9〜13MPa、42℃)での3日間の反応において、反応性と選択性の両方で触媒としての作用を維持し、5.0mlの反応器を用いて、(R/S)−1−フェニルエタノール(221g)が、99%eeの(S)体アルコールと99%eeの(R)体酢酸エステルに定量的に変換した。
【0038】
<実施例3>2−ウンデカノールの反応
リパーゼ(Novozyme435)1ml(0.347g)を1/2インチφ反応管(内径10mm)に充填し、反応装置に組み込んだ。2−ウンデカノール(43.1g)及びビニルアセテート(12.91g)(2−ウンデカノール:ビニルアセテート=1:0.6(mol/mol))にモリキュラーシーブズを加え、基質溶液とし、ポンプ内を置換した。オーブンの温度を42℃まで昇温した。二酸化炭素の圧力を13.0MPaまで昇圧し、流量は、180ml/hr/ml(3ml/min)に設定した。基質溶液の流量を、7.56ml/hr/ml(0.126ml/min)に設定し、反応を開始した。約0.5時間後に装置が安定してから、反応器出口から流出液を採取を始め、GC分析、LC分析及びNMR分析を行った。立体配置の決定は、標準物質とGCの保持時間の比較により行った。収率は48〜50%であり、E値は112〜137であった。なお、収率は、原料である2−ウンデカノールの初期量を基準とした(R)体酢酸エステルのモル収率である。
【0039】
<実施例4>テトラヒドロナフトールの反応
リパーゼ(ノボザイム435)5ml(1.98g)、1,2,3,4−テトラヒドロ−1−ナフトール:ビニルアセテート=1:0.6(mol/mol)、基質溶液の流量0.84ml/hr/ml(0.07ml/min)、超臨界二酸化炭素の流量18ml/hr/ml(1.5ml/min)又は36ml/hr/ml(3ml/min)、圧力13.0MPaとした以外は、実施例3と同様にして反応を行った。
E値は1500以上であった。
【0040】
<実施例5>1−フェニルエタノールの動的速度論的分割反応
フロー系のリパーゼ反応により得られた生成物(実施例1における表1の3のサンプル、図3のNMRチャート)のラセミ化反応を行った。Ru触媒を用いるラセミ化反応の結果、6割程度ラセミ化が進行した。
【0041】
<比較例1>バッチ反応
ラセミ−1−フェニルエタノール(0.83mmol)とビニルアセテート(5.4mmol)を、40℃、9MPaの二酸化炭素の存在下、酵素(Novozyme435)の存在下で反応させた。48%転化率において、99.8%eeの光学活性(R)体酢酸エステルと、90.6%eeの未反応の(S)体アルコールとの1:1の混合物が得られた。同様にして、二酸化炭素圧力を9〜13MPaの範囲を変化させて反応を行った。結果を図5に示す。
基質である(R)体アルコールが(S)体アルコールよりも早く反応し、約7時間後に反応が停止した。このようにエナンチオマーの反応性がかなり異なることから、(R)体アルコールが消費されて反応が停止したと考えられる。二酸化炭素圧力が9〜13MPaの間における反応において、収率48%、E値100以上であった。
【0042】
【表1】
【0043】
【表2】
【0044】
【表3】
【0045】
表1において、反応時間は、安定な反応時間に達した後の時間である。なお、表3は、実施例における酵素の使用量、二酸化酸素及び基質溶液(アルコール+ビニルアセテート)の流量及び空間速度(space velocity)を示す。
10mlの反応器を用いるバッチ反応においては、7時間の反応の間に、光学活性アセテート3を0.83mmol製造するが、連続流通反応器(5ml)を用いることにより、25mmol/h(3ml/h)の割合で生成物を与えることができた。
更に、ビニルアセテートを少し過剰量用いることにより、円滑に反応が進行し、反応の間の廃棄物を最小限におさえて、所望の生成物を優れた純度で供給することができた。
この合成プロセスは、光学活性アルコールの大規模の製造に特に有用である。
【図面の簡単な説明】
【0046】
【図1】図1は、本発明の超臨界連続反応工程を示す概念図である。
【図2】図2は、実施例において用いた反応装置の概念図である。
【図3】図3は、実施例における反応生成物のNMRスペクトルである。
【図4】図4は、実施例におけるフェニルエタノールの3日間の連続運転反応の結果を示すグラフである。
【図5】図5は、バッチ反応を行った場合の結果を示すグラフである。
【特許請求の範囲】
【請求項1】
アルコールとアシル化剤からアシル化誘導体を製造する方法であって、
超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むことを特徴とするアシル化誘導体の製造方法。
【請求項2】
前記超臨界連続反応工程の生成物は、未反応の(S)体アルコール及び生成した(R)体アシル化誘導体を含有する混合物、又は、未反応の(R)体アルコール及び生成した(S)体アシル化誘導体を含有する混合物であることを特徴とする請求項1記載のアシル化誘導体の製造方法。
【請求項3】
前記アシル化誘導体の製造方法は、未反応のアルコールをラセミ体とし、超臨界連続反応工程の原料に使用することを特徴とする請求項2記載のアシル化誘導体の製造方法。
【請求項1】
アルコールとアシル化剤からアシル化誘導体を製造する方法であって、
超臨界二酸化炭素の存在下、酵素を用いて立体選択的エステル化反応を連続的に行う超臨界連続反応工程を含むことを特徴とするアシル化誘導体の製造方法。
【請求項2】
前記超臨界連続反応工程の生成物は、未反応の(S)体アルコール及び生成した(R)体アシル化誘導体を含有する混合物、又は、未反応の(R)体アルコール及び生成した(S)体アシル化誘導体を含有する混合物であることを特徴とする請求項1記載のアシル化誘導体の製造方法。
【請求項3】
前記アシル化誘導体の製造方法は、未反応のアルコールをラセミ体とし、超臨界連続反応工程の原料に使用することを特徴とする請求項2記載のアシル化誘導体の製造方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2006−14636(P2006−14636A)
【公開日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願番号】特願2004−194531(P2004−194531)
【出願日】平成16年6月30日(2004.6.30)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
【公開日】平成18年1月19日(2006.1.19)
【国際特許分類】
【出願日】平成16年6月30日(2004.6.30)
【出願人】(000004628)株式会社日本触媒 (2,292)
【Fターム(参考)】
[ Back to top ]