
アスパラギン誘導体およびその用途
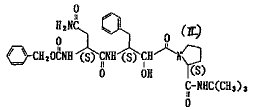
【目的】レトロウイルス性疾患の治療剤として有用なレトロウイルスプロテアーゼ阻害物質の提供。
【構成】式(I)で表わされるアスパラギン誘導体、ならびに該誘導体を含有するレトロウイルス性疾患治療剤。

[環Aはピロリジン環等の5〜7員環を、R1,R2はともに水素原子を示すか、あるいは結合して縮合環を形成してもよい。R3はエステル化もしくはアミド化されていてもよいカルボキシル基、R4は水素原子またはアシル基を示し、Xは−CH(OH)−、または−C(=O)−を示す。]式(II)の化合物は上記アスパラギン誘導体の代表的な例である。
【構成】式(I)で表わされるアスパラギン誘導体、ならびに該誘導体を含有するレトロウイルス性疾患治療剤。
[環Aはピロリジン環等の5〜7員環を、R1,R2はともに水素原子を示すか、あるいは結合して縮合環を形成してもよい。R3はエステル化もしくはアミド化されていてもよいカルボキシル基、R4は水素原子またはアシル基を示し、Xは−CH(OH)−、または−C(=O)−を示す。]式(II)の化合物は上記アスパラギン誘導体の代表的な例である。
【発明の詳細な説明】
【0001】
【産業上の利用分野】本発明は、レトロウイルスプロテアーゼ阻害活性を有する新規なアスパラギン誘導体およびその用途に関する。
【0002】
【従来の技術】レトロウイルスは、レトロウイルス科に属するウイルスであって、遺伝子としてRNAを持ち、自身の有する逆転写酵素(RNA依存性DNAポリメラーゼ)の働きによりRNAを鋳型としてDNAを合成する段階を自己複製の第1段階とするウイルスの総称である。レトロウイルス科は3つの亜科(オンコウイルス、レンチウイルス、スプマウイルス)からなる。レトロウイルスは、鳥類や哺乳類の種々の動物を宿主として感染、増殖し、肉腫、白血病、癌などを引き起こすもののあることが知られている。ヒトを宿主とするレトロウイルスとしては、成人T細胞白血病ウイルス(adult T−cell leukemia virus, ATLV)、ヒトT細胞白血病ウイルスI型(human T−cell leukemia virus typeI、HTLV−I)およびヒト免疫不全ウイルス(human immunodeficiency virus, HIV)などが報告されているが、ATLVは、HTLV−Iと同一のウイルスであることが証明された。HTLVにはこの他に、T細胞系のヘアリーセル白血病(hairycell leukemia)を誘導するII型が存在する。HIVはHIV−1およびHIV−2に分類され、HIV−1は重篤な免疫不全症である後天性免疫不全症候群(aquired immunodeficiency syndrome, AIDS)の原因ウイルスであることが分かっている。また、HIV−2もAIDSの原因ウイルスとされているが、HIV−1ほどはっきりとは解明されていない。
【0003】ATLVによって引き起こされる成人T細胞白血病は、日本の南西地域特に九州地方に多く見られる白血病であり、1977年にはじめて報告された。成人T細胞白血病患者では、T4陽性T細胞が癌化して、皮膚病変、リンパ節腫大、肝脾腫等を招来する。成人T細胞白血病に対しては有効な治療法はなく、発病するとほとんどが1年以内に死亡する。
【0004】HIV−1によって引き起こされるAIDSは、予後不良の免疫不全症として最近注目を集めている疾病であって、カリニ肺炎やカポシ肉腫などの臨床像を特徴とする。HIV−1は、T4陽性細胞に感染して細胞内に侵入、増殖し、これを破壊することにより免疫不全を招来する。現在AIDSの治療薬としてアジドチミジン(AZT)が使用されており、症状の改善および延命効果が認められている。
【0005】
【発明が解決しようとする課題】上記のように抗レトロウイルス剤としてAZTが使用されているものの、この物質は副作用として骨髄抑制を起こすなどの欠点を有しており、さらに効果が高く副作用の少ない治療薬の出現が期待されている。
【0006】
【課題を解決するための手段】レトロウイルスのゲノムにコードされているプロテアーゼは感染性成熟ウイルス粒子の形成に際して重要な役割を果たしており、このプロテアーゼ機能を欠損すると感染性粒子の形成の起こらないことが知られている[I. Katoh ら,ウィロロジー(Virology)第145巻,280頁(1985年)]。
【0007】したがって、該プロテアーゼの阻害剤はレトロウイルス性疾患の予防および治療薬に成り得るものと考えられ、最近になってペプスタチンA[フェブス レターズ(FEBS Lett.),247,349(1989)]、種々の合成ペプチド性誘導体[ネイチャー(Nature),343,90(1990)、ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.),265,14675(1990)、ジャーナル・オブ・メディシナル・ケミストリー(J. Med. Chem.), 33,1285(1990)、サイエンス(Science),247,454(1990)および248,358(1990)]や非ペプチド性化合物[サイエンス(Science),249,527(1990)、ジャーナル・オブ・メディシナル・ケミストリー(J. Med. Chem.)、33,2687(1990)および34,1 225(1991)]等のプロテアーゼ阻害剤が該治療剤として提案されている。本発明者らの一人、加藤らは広く自然界に該プロテアーゼ阻害物質を検索し、その結果、土壌中から分離した1菌株が該プロテアーゼ阻害物質を産生すること、該菌株が放線菌の中のキタサトスポリア属に属すること、該微生物の培養液から精製取得したペプチド性の新規な化合物たるRPI−856、RPI−857、RPI−858、RPI−859が試験管内で該プロテアーゼを強く阻害することを見出し、すでに特許出願した(特願平2−282025,特願平3−1450および特願平3−17604号明細書参照)。本発明者らは、これらのペプチド性化合物の化学構造に基づいてドラッグデザインを行い、関連化合物の合成研究を鋭意行った結果、以下に示されるアスパラギン誘導体が高い該プロテアーゼ阻害活性を示すことを見出し、さらに研究を進め本発明を完成した。
【0008】すなわち、本発明は、一般式(I)
【化3】
[式中、環Aは5〜7員環を示し、R1,R2はともに水素原子を示すか、または結合して縮合環を形成していてもよく、R3はエステル化もしくはアミド化されていてもよいカルボキシル基を示し、R4は水素原子またはアシル基を示し、Xは
【化4】
で表わされるアスパラギン誘導体およびその塩、および化合物(I)を含有してなるレトロウイルス性疾患治療剤を提供するものである。
【0009】化合物(I)に関し、R1,R2が結合して縮合環を形成する場合、5〜6員環を形成することが好ましく、なかでもR1,R2が結合してベンゾ縮合、パーヒドロベンゾ縮合もしくはパーヒドロシクロペンタ縮合を示すのが好ましい。この場合、具体的には式
【化5】
などを挙げることができる。
【0010】化合物(I)に関し、R1,R2で表わされる基の好ましい例は、ともに水素原子であるものおよび結合してパーヒドロベンゾ縮合であるものがあげられる。化合物(I)に関し、R3で表わされるエステル化もしくはアミド化されていてもよいカルボキシル基としては、例えばカルボキシル,カルバモイル,低級アルコキシカルボニル(例、メトキシカルボニル,エトキシカルボニル,プロポキシカルボニル,イソプロポキシカルボニル,ブトキシカルボニル,イソブトキシカルボニル,tert−ブトキシカルボニルなどのC1-4アルコキシ−CO−など),アラルキルオキシカルボニル(例、ベンジルオキシカルボニル,p−ニトロベンジルオキシカルボニル,p−メトキシベンジルオキシカルボニル,フェネチルオキシカルボニルなどの置換されていてもよいフェニル−C1-4アルコキシ−CO−など),低級アルキルアミノカルボニル(例、メチルアミノカルボニル,エチルアミノカルボニル,プロピルアミノカルボニル,イソプロピルアミノカルボニル,ブチルアミノカルボニル,イソブチルアミノカルボニル,tert−ブチルアミノカルボニルなどのN−C1-4アルキルアミノなど),アラルキルアミノカルボニル(例、ベンジルアミノカルボニル,フェネチルアミノカルボニルなどのN−フェニル−C1-4アルキルアミノ)などがあげられ、なかでも低級アルコキシカルボニルおよび低級アルキルアミノカルボニルが好ましく、とりわけ、tert−ブトキシカルボニルおよびtert−ブチルアミノカルボニルが好ましい。
【0011】化合物(I)に関し、R4で表わされるアシル基としては、例えば低級アルカノイル(例、ホルミル,アセチル,プロピオニル,ブチリル,イソブチリルなどのC1-5アルカノイルなど),低級アルコキシカルボニル(例、メトキシカルボニル,エトキシカルボニル,プロポキシカルボニル,イソプロポキシカルボニル,ブトキシカルボニル,イソブトキシカルボニル,tert−ブトキシカルボニルなどのC1-4アルコキシ−CO−など),アラルキルオキシカルボニル(例、ベンジルオキシカルボニル,p−ニトロベンジルオキシカルボニル,p−メトキシベンジルオキシカルボニル,ナフチルメチルオキシカルボニルなどの置換されていてもよいフェニル−C1-4アルコキシ−CO−など),アリールカルボニル(例、ベンゾイル,ナフトイル,トルオイルなど),ヘテロアリールカルボニル(例、フロイル,テノイル,ニコチノイル,イソニコチノイル,キノリノイル,イソキノリノイル,キナゾリノイルなど),アリールスルホニル(例、p−トルエンスルホニル,ベンゼンスルホニル,ナフタレンスルホニルなど)などがあげられ、なかでも低級アルコキシカルボニル,アラルキルオキシカルボニル,ヘテロアリールカルボニルが好ましく、とりわけ、tert−ブトキシカルボニル,ベンジルオキシカルボニル,2−キノリノイルが好ましい。
【0012】化合物(I)に関し、環Aとしては5〜6員環,とりわけピロリジン環およびピペリジン環が好ましい。本発明化合物に属する好ましい化合物の具体例としては、例えば〔表1〕に示す化合物があげられる。
【0013】
【表1】
【0014】
【化6】
[式中、各記号は前記と同意義。]で表わされる化合物を酸化することによって製造することができる。
【0015】反応は通常、ジメチルスルホキシド中、ピリジンおよびトリフルオロ酢酸の存在下に行うことが好ましく、反応温度は0〜60℃程度で行われ、反応時間は1〜24時間である。
【0016】本発明化合物(I)は、たとえば式(II)
【化7】
[式中、各記号は前記と同意義。]で表わされる化合物と、式(III)
【化8】
[式中、R4′はアシル基であり、他の記号は前記と同意義。]で表わされる化合物を縮合させることによって得ることができる。
【0017】該縮合反応条件としては、たとえば縮合試薬(例、ジシクロヘキシルカルボジイミド,カルボニルジイミダゾール,シアノリン酸ジエチル,ジフェニルリン酸アジド等)などを用いるか、または化合物(III)にたとえば2,4,5−トリクロロフェノール,ペンタクロロフェノール,ペンタフルオロフェノール,2−ニトロフェノールおよび4−ニトロフェノール等のフェノール類、または、たとえばN−ヒドロキシスクシンイミド,N−ヒドロキシ−5−ノルボルネン−2,3−ジカルボン酸イミド,1−ヒドロキシベンズトリアゾール,N−ヒドロキシピペリジン等のN−ヒドロキシ化合物をジシクロヘキシルカルボジイミド等の触媒の存在下に縮合させ、化合物(III)を活性なエステル体に変換した後、化合物(II)と反応させることが好ましい。また化合物(III)にたとえば、クロロ炭酸メチル,クロロ炭酸エチル,クロロ炭酸イソブチルなどを、好ましくは有機塩基(例、トリエチルアミン,N−メチルモルホリン)の存在下に反応させ、混合酸無水物に変換した後、化合物(II)と反応させることもできる。これらの反応において、反応温度は通常約−30℃〜+50℃であり、用いる溶媒としては、たとえばジオキサン,テトラヒドロフラン,アセトニトリル,ジエチルエーテル,N,N−ジメチルホルムアミド,N,N−ジメチルアセトアミド,ジメチルスルホキシド,N−メチルピロリドン,クロロホルム,塩化メチレンなどが挙げられ、これら溶媒は単独もしくは混合溶媒として用いてもよい。
【0018】本発明化合物(I)は、たとえば式(IV)
【化9】
[式中、R4′はアシル基であり、他の記号は前記と同意義。]で表わされる化合物と式(V)
【化10】
[式中、各記号は前記と同意義。]で表わされる化合物を縮合させることによって製造することができる。該縮合反応条件としては、上述した(II)と(III)の縮合反応において適用される条件と同様な反応条件が好ましい。
【0019】本発明化合物(I)においてR4が水素である化合物は、式(I″)で表わされる化合物を脱保護反応に付すことによって製造することができる。
【化11】
[式中、R4″はアミノ保護基を示し、他の記号は前記と同意義。]
また、本発明化合物(I)においてR3がカルボキシル基である化合物は、式(I″′)で表わされる化合物を脱保護反応に付すことによって製造することができる。
【化12】
[式中、R3′は保護されたカルボキシル基を示し、他の記号は前記と同意義。]。
【0020】該脱保護反応としては、その保護基の種類あるいは、化合物(I″),(I″′)の脱保護条件下での安定性に応じて、酸あるいは塩基による方法、または接触還元による方法を適宜選択して行うことができる。酸による方法の場合には、保護基の種類その他の条件によって異なるが、酸として例えば塩酸、臭化水素、フッ化水素、沃化水素、メタンスルホン酸、p−トルエンスルホン酸、トリフルオロメタンスルホン酸、硫酸、リン酸等の無機酸、ギ酸、酢酸、トリフルオロ酢酸、プロピオン酸等の有機酸の他、酸性イオン交換樹脂等が使用される。塩基による方法の場合には、保護基の種類その他の条件によって異なるが、塩基として例えばナトリウム,カリウム等のアルカリ金属もしくはカルシウム,マグネシウム等のアルカリ土類金属の水酸化物、炭酸塩等の無機塩基、金属アルコキサイド類、有機アミン類、第四アンモニウム塩等の有機塩基の他、塩基性イオン交換樹脂等が使用される。上記酸または塩基による方法の場合において溶媒を使用する場合には、たとえば、水,メタノール,エタノール,酢酸エチル,クロロホルム,テトラヒドロフラン,ジオキサン,ピリジン,酢酸,アセトン,塩化メチレンなどを単一、もしくは混合して用いることができる。また金属と酸による方法においては水,アセトン等が繁用されるが酸が液体のときは酸自身を溶媒として使用することもできる。酸および塩基による方法において反応温度は、通常冷却下ないし加温程度で行なわれ、反応時間は約30分ないし20時間である。
【0021】一方接触還元による方法の場合には、水またはたとえばメタノール,エタノール,ジオキサン,酢酸エチルなどの有機溶媒あるいはそれらの混合溶媒中、たとえばパラジウム−炭素などの適当な触媒の存在下に行われる。本反応は常圧または150kg/cm2程度までの圧力下、常温ないし+100℃の温度で行われるが、一般に常温,常圧で充分反応は進行する。
【0022】本発明化合物(I)は分子内に不斉原子を3個以上有しているので8個以上の立体異性体が存在し得るが、その立体異性体ならびにそれらの混合物のいずれも本発明に包含されるものである。とりわけアスパラギン残基中の不斉炭素,ベンジル基とXが結合した不斉炭素およびR3が結合した不斉炭素のいずれもがS配置である光学活性体が好ましい。
【0023】化合物(I)は塩としても得られ、該塩としては、たとえば塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、燐酸塩などの無機酸塩、たとえば酢酸塩、酒石酸塩、クエン酸塩、フマール酸塩、マレイン酸塩、トリエンスルホン酸塩、メタンスルホン酸塩などの有機酸塩、たとえばナトリウム塩、カリウム塩、カルシウム塩、アルミニウム塩などの金属塩、たとえば、トリエチルアミン塩、グアニジン塩、アンモニウム塩、ヒドラジン塩、キニーネ塩、シンコニン塩などの塩基との塩が挙げられる。
【0024】かくして得られる化合物(I)は反応混合物から通常の分離精製手段、たとえば抽出、濃縮、中和、ろ過、再結晶、カラムクロマトグラフィー、薄層クロマトグラフィーなどの手段を用いることによって単離することができる。化合物(I)は少なくとも8個の立体異性体が存在し得る。これら個々の異性体およびこれらの混合物のいずれも本発明に包含されるが、所望によりこれらの異性体を個別に製造することもできる。たとえば原料化合物(I′),(II),(III),(IV)および(V)のそれぞれの単一の異性体を用いて上記の反応を行うことにより、化合物(I)の単一の異性体を得ることができるし、また生成物が二種類以上の異性体混合物の場合には、これを通常の分離方法、たとえば光学活性酸(例、カンファースルホン酸、酒石酸など)との塩を生成させる方法や、各種のクロマトグラフィー、分別再結晶などの分離手段によってそれぞれの異性体に分離することもできる。
【0025】化合物(I)の塩は必要に応じ前記した無機酸あるいは有機酸を加えることによって生理学的に許容される塩として得ることができる。本発明における合成中間体(II)において、例えばR1,R2がともに水素原子であり、R3がtert−ブチルアミノカルボニルであり、環Aがピロリジン環であり、Xが
【化13】
である化合物(XI)は次の反応式で示す方法で合成することができる。
【0026】
【化14】
ここで用いた(IX)はジャーナル・オブ・メディシカル・ケミストリー(J. Med. Chem.), 20,510(1977)に記載された方法で合成することができる。
【0027】以下参考例および試験例においてHIV−1プロテアーゼの調製法および該プロテアーゼに対する本発明化合物(I)の阻害作用についてそれぞれ述べる。
参考例1 HIV−1プロテアーゼ遺伝子の発現プラスミドの構築HIV−1プロテアーゼはpol遺伝子にコードされた蛋白質の1つであり、感染細胞内においてはgag−polのポリプロテインとして合成されたものが、プロテアーゼ自身の作用によりプロセシングされることにより生成されると考えられている[J. M. Mc Cune ら、セル(Cell)第53巻,55頁(1988年)]。一方、プロテアーゼ領域を含むpol遺伝子の一部を大腸菌で発現させることにより、分子量11キロダルトンの活性型HIV−1プロテアーゼが自身のオートプロセシング作用により生成されることが既に知られている[M. C. Graves ら、プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc. Natl. Acad. Sci. USA)第85巻,2449頁(1988年)]。
【0028】そこで、まずHIV−1の全遺伝子を含有するプラスミドpNL4−3[A. Adachi ら、ジャーナル・オブ・ウイロロジー(J. Virol.)第59巻,284頁(1986年)]からプロテアーゼ領域を含有する1.5kb のBglII−BalI断片を取得した。次に、この断片にリンカー 5’−AATTCTATGCCA−3’ (配列番号:1)
3’−GATACGGTCTAG−5’ (配列番号:2)
を接続した後、EcoRIで消化し、さらにリンカー[5’−CTAGCTAGCTAGAATTCTAGCTAGCTAG−3’](配列番号:3)を接続した後、EcoRIで消化した。次に、この断片の切断点をクレノーフラグメントで埋めて平滑末端にした。一方、T7プロモーターを含む大腸菌用発現ベクターpET−3c[A. H. Rosenberg ら、ジーン(Gene)第56巻,125頁(1987年)]をNdeIで消化し、その切断点をT4DNAポリメラーゼで埋めて平滑末端にした。次に、これら両DNA断片をT4DNAリガーゼを用いて接続し、T7プロモーターとプロテアーゼ遺伝子の向きが同方向となったものをpHIV7004とした。
【0029】参考例2 組換え型HIV−1プロテアーゼの調製Escherichia coli MM294株にT7ファージのRNAポリメラーゼ遺伝子を組み込んだλファージDE3[F. W. Studier ら、ジャーナル・オブ・モレキュラー・バイオロジー(J. Mol. Biol.)第189巻,113頁(1986年)]を溶原化させてEscherichia coli MM294(DE3)を作製した。参考例1で得られたpHIV7004を、Escherichia coli MM294(DE3)に導入して、形質転換体Escherichia coli MM294(DE3)/pHIV7004を得た。本形質転換体をアンピシリン50μg/mlを含むLB培地に接種し、37℃で一晩振盪培養した。この培養液0.2mlを20mlの同じ培地に接種し、37℃で対数増殖期(180−200 Klett unit)まで培養した。次に、最終濃度が0.4mMになるようにイソプロピル−1−チオ−β−D−ガラクトシド(IPTG)を加え、さらに37℃で2時間培養した。培養液200mlから、菌体を5000rpm で5分間遠心することにより集め、これを〔表2〕に記載した組成を持つ緩衝液A20mlに懸濁した。懸濁液にニワトリ卵白リゾチーム2mgを加え、氷冷下で10分間放置後、ノニデットP40(NP40)を最終濃度1%になるように加え、超音波破砕装置を用いて菌体を破砕した。上記処理を行った溶菌液を、日立工機社製のRR−24Aローターを用いて16000rpm で10分間遠心分離し、HIV−1プロテアーゼを含む粗抽出液20mlを得た。
【0030】
【表2】 緩衝液Aの組成50mM トリス塩酸緩衝液pH8.05mM エチレンジアミン4酢酸(EDTA)
0.1% ノニデットP40(NP40)
0.02% 2−メルカプトエタノール(2−ME)
10% グリセリン1mM フェニルメチルスルフォニルフルオリド(PMSF)
0.1mM パラアミジノフェニルメタンスルフォニルフルオリド(APMSF)
【0031】試験例1 HIV−1プロテアーゼ阻害作用HIV−1 gagP17とP24の解裂部位を含む合成ペプチドgag−11(SQVSQNYPIVQNL)(配列番号:4)を基質とし、反応産物gag−12(SQVSQNY)(配列番号:5)の生成を指標としてHIV−1プロテアーゼ阻害活性を測定した。即ち、基質gag−11 3μg、 種々の濃度の試料、HIV−1プロテアーゼを含む5%グリセロール−1MNaCl−0.25Mリン酸ナトリウム緩衝液(pH6.2)(30μl)中で3〜5時間反応させた。0.13Mトリクロロ酢酸−0.26M酢酸ナトリウム−0.4M酢酸150μlを加えることにより反応を停止した。室温で10分間放置した後遠心上清を得た。上記の上清のうち48μlを0.1%トリフロロ酢酸で平衡化したYMC−パックFL−ODSカラム(0.46×3cm、ワイエムシィ社)に負荷し、アセトニトリルの濃度勾配で溶出した。本条件下において、gag−12の生成量を50%阻害するために必要な反応液中の試料濃度をIC50値として表わした。化合物(I)のHIV−1プロテアーゼ阻害活性を〔表3〕に示した。
【0032】
【表3】
試 料 IC50(μM)
(化合物番号) ────────────────────────── 1 0.020 2 0.034 3 0.021 4 0.031〔表3〕の結果から、本発明化合物(I)は非常に強いHIV−1プロテアーゼ阻害物質であることが明らかである。
【0033】以上のように、化合物(I)はレトロウイルスのプロテアーゼ阻害活性を有しており、また毒性も低いので、レトロウイルス性疾患治療剤として有用な物質である。 本発明化合物(I)を抗レトロウイルス剤として用いる場合、希釈剤、賦形剤、担体などと混合して、薬学的に許容される製剤とし、経口的または非経口的に投与する。例えば、注射剤として用いる場合は、化合物(I)を水溶液剤として、成人1日当り0.1〜10mg/kgを静脈内投与する。
【0034】
【実施例】以下に実施例を挙げて、本発明をさらに具体的に説明するが、これによって本発明が限定されるものではない。
実施例11−ベンジルオキシカルボニル−L−プロリン(化合物IV:23.0g,92.2mmol)をテトラヒドロフラン(150ml)に溶解し、氷冷下にトリエチルアミン(14.1ml,102mmol)を加えた。次に、クロロギ酸イソブチル(13.2ml,102mmol)のテトラヒドロフラン(43ml)溶液を滴下し、氷冷下で1時間撹拌した後、tert−ブチルアミン(19.4ml,184mmol)を加えた。氷冷下で1時間、さらに室温で一夜撹拌した後、溶媒を留去した。残渣に水,酢酸エチル(各200ml)を加えて分液し、酢酸エチル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)後、減圧留去した。得られた白色粉末を酢酸エチル−ヘキサン(1:1,40ml)から再結晶し、1−ベンジルオキシカルボニル−N−tert−ブチル−L−プロリンアミド(化合物VII:15.5g,55%)を白色結晶として得た。さらにろ液を減圧留去した後、シリカゲルカラムクロマトグラフィー(Kieselgel 60,100g:酢酸エチル−ヘキサン=1:1で溶出)で精製すると追加のプロリンアミド体(化合物VII:5.9g,21.0%)が得られた。
mp 90−91℃元素分析値: C17H24N2O3としてCalcd:C,67.08; H,7.95; N,9.20Found:C,66.85; H,7.96; N,9.45SIMS(m/z):305(MH+)
1H−NMR(d6-DMSO)δ:1.18(6H,s),1.24(3H,s),1.7-1.95(3H,m),1.95-2.2(1H,m),3.3-3.5(2H,m),4.1-4.2(1H,m),4.9-5.10(2H,m),7.2-7.5(6H,m)
【0035】実施例21−ベンジルオキシカルボニル−N−tert−ブチル−L−プロリンアミド(化合物VII:8.0g,26.3mmol)をメタノール−水(6:1,140ml)に溶解し、10%パラジウム−炭素(0.5g)を加え、水素気流中、1時間撹拌した。触媒をろ別し、触媒部分を洗浄(10%メタノール水溶液)し、ろ液と洗液を合わせて減圧留去した。得られた油状物を減圧乾燥すると結晶化した。これにジエチルエーテルと石油エーテルの混液を加えてろ取すると、N−tert−ブチル−L−プロリンアミド(化合物VIII:3.4g,75.7%)が無色粉末晶として得られた。
mp 79−80℃元素分析値: C9H18N2OとしてCalcd:C,63.49; H,10.66; N,16.45Found:C,63.45; H,10.89; N,16.151H−NMR(d6-DMSO)δ:1.26(9H,s),1.5-1.7(3H,m),1.8-2.0(1H,m),2.78(2H,ddd,J=6.1,6.5,9.7Hz),3.2-3.4(1H,m),7.57(1H,br s)
【0036】実施例3(2RS,3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブタン酸(化合物IX:2.0g,6.07mmol)をジメチルホルムアミド(20ml)に溶解した。氷冷下にN−tert−ブチル−L−プロリンアミド(化合物VIII:1.24g,7.28mmol),シアノリン酸ジエチル(DEPC)(2.09g,12.2mmol)のジエチルホルムアミド(5ml)溶液およびトリエチルアミン(1.76ml,12.2mmol)を順次加え、氷冷下に1時間、さらに室温に戻して2.5時間撹拌した。反応液に水(約5ml)を加えて1時間撹拌した後、水(80ml)を加え、酢酸エチル(150ml)で抽出した。酢酸エチル層を10%リン酸ナトリウム水溶液、5%重曹水、飽和食塩水の順で洗浄し、乾燥(Mg SO4)した後、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー (Kieselgel 60,140g:酢酸エチル−ヘキサン=2:1で溶出)で精製すると1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物X) のジアステレオマーA(極性大:0.695g)及びジアステレオマーB(極性 小:0.415g)が無色粉末晶として得られた。
ジアステレオマーA白色粉末晶mp 63−66℃元素分析値: C27H35N3O5・1/2H2OとしてCalcd:C,66.10; H,7.40; N,8.57Found:C,66.40; H,7.43; N,8.54SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.75-2.4(4H,m),2.6-2.8(2H,m),3.6-4.25(5H,m),4.3-4.65(2H,m),4.99(2H,s),5.43(1H,d,J=8.8Hz),6.49(1H,s),7.1-7.4(10H,m)ジアステレオマーB白色粉末晶mp 114−115℃元素分析値: C27H35N3O5としてCalcd:C,67.34; H,7.33; N,8.73Found:C,67.51; H,7.29; N,8.77SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.7-2.3(4H,m),2.95(2H,d,J=7.8Hz),3.0-3.4(2H,m),3.87(1H,d,J=5.6Hz),3.95(1H,d,J=3.6Hz),4.10(1H,d,J=4.6Hz),4.22(1H,m),5.03(2H,s),5.09(1H,br s),6.43(1H,br s),7.2-7.5(10H,m)
【0037】実施例41−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物X,ジアステレオマーA:300mg,0.62mmol)をメタノール−水(4:1,8ml)に溶解し、10%パラジウム−炭素(40mg)を加え、水素気流下、4時間撹拌した。触媒をろ別し、触媒部分を洗浄(50%メタノール水溶液)し、ろ液と洗液を合わせて減圧留去した。残渣にジエチルエーテルを加えて、析出した結晶をろ取すると1−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:210mg)が白色粉末晶として得られた。
mp 154.5−156.0℃元素分析値: C19H29N3O3としてCalcd:C,65.68; H,8.41; N,12.09Found:C,65.35; H,8.55; N,12.131H−NMR(CDCl3)δ:1.29(9H,s),1.4-2.2(2H,br m),1.7-2.4(4H,m),2.53(1H,dd,J=10.0,13.3Hz),2.95(1H,dd,J=3.2,13.4Hz),3.0-3.2(1H,m),3.5-3.7(2H,m),4.27(1H,d,J=5.4Hz),4.45-4.55(1H,m),6.57(1H,s),7.1-7.4(5H,m)
【0038】実施例51−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:240mg,0.69mmol)をジメチルホルムアミド(8ml)に溶解し、氷冷下にNアルフア−ベンジルオキシカルボニル−L−アスパラギンのp−ニトロフェニルエステル(267mg,0. 69mmol)を加え、氷冷下に1時間、さらに室温で4時間撹拌した。溶媒を減圧留去し た後、残渣に水,酢酸エチル(各30ml)を加えて分液した。酢酸エチ ル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順に洗浄し、乾燥(MgSO4)した後、減圧留去した。得られた黄色油状物をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル−メタノール=9:1で溶出)で精製すると1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物1:200mg,48. 7%)が白色粉 末晶として得られた。
mp 102−103℃(dec.)
元素分析値: C31H41N5O7としてCalcd:C,62.51; H,6.94; N,11.76Found:C,62.20; H,7.06; N,11.66SIMS(m/z):596(MH+)
1H−NMR(d6-DMSO)δ:1.25(9H,s),1.7−2.1(4H,m),2.2−2.45(2H,m),2.55−2.8(2H,m),3.5−3.7(2H,m),4.0−4.2(1H,m),4.2−4.4(3H,m),4.89(1H,d,J=7.2Hz),5.01(2H,s),6.87(1H,s),7.05−7.4(5H,m),7.22(1H,s),7.35(5H,s),7.52(1H,s),7.86(1H,d,J=8.2Hz)
【0039】実施例6ベンゼン(1ml)とジメチルスルホキシド(0.5ml)の混液にジシクロヘキシルカルボジイミド(104mg,0.504mmol)とピリジン(14μl,0.168mmol)を加え、次いで氷冷下に1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物1:100mg,0. 168mmol)を加えた。10分後にトリフルオロ酢酸(6.6μl,0.0 84mmol)を加え、室温で一夜撹拌した。反応液に酢酸エチル(2ml)を加え、不溶物 をろ別した。ろ液に酢酸エチル(25ml)を加え、これを水(30ml× 3)で洗浄し、乾燥(MgSO4)した後、減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,15g:酢酸エチル−塩化メチレン−メタノール=5:5:1で溶出)で精製すると1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−オキソ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物2:20mg,20.1 %)が白色粉末晶として得られた。
mp 89−93℃SIMS(m/z):594(MH+)
1H−NMR(d6-DMSO)δ:1.21(3H,s),1.24(6H,s),1.65-2.0(4H,m),2.0-2.4(2H,m),2.75-3.1(1H,m),3.1-3.3(1H,m),3.4-3.6(2H,m),4.2-4.5(2H,m),4.5-4.9(1H,m),4.9-5.1(2H,m),6.88(1H,br s),7.1-7.3(5H,m),7.33(5H,s),7.5-7.6(1H,m),8.15-8.6(1H,m)
【0040】実施例7(2RS,3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブタン酸(化合物IX:2.16g,6.55mmol)をジメチルホルムアミド(5ml)に溶解し、氷冷下にL−プロリン−tert−ブチルエステル(1.23g,7.21mmol)のジメチルホルムアミド(2.5ml)溶液,シアノリン酸ジエチル(1.23g,7.21mmol)のジメチルホルムアミド(2.5ml)溶液 およびトリエチルアミン(1.0ml,7.21mmol)を順次加えた。氷冷下に1時間、さらに室温で一夜撹拌した後、溶媒を減圧留去した。残渣を酢酸エチル(80ml)に溶解し、得られた溶液を冷5%塩酸,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)した後、減圧留去した。残留物を酢酸エチル−ヘキサン(1:1,12ml)から結晶化し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン−tert−ブチルエステルのジアステレオマーB(極性小)を白色結晶として800mg(25.3%)得た。さらにろ液を減圧留去した後、残留物をシリカゲルカラムクロ マトグラフィー(Kieselgel 60,30g:酢酸エチル−ヘキサン=1:2で溶出)で精製すると、追加のジアステレオマーB140mg(4.4%)とジアステ レオマーA(極性大,非結晶性粉末)1.03g(32.6%)が得られた。
ジアステレオマーAmp 58−60℃SIMS(m/z):483(MH+)
1H−NMR(CDCl3)δ:1.43(3H,s),1.45(6H,s),1.6-2.3(4H,m),2.5-3.0(2H,m),3.6-3.9(2H,m),4.15-4.3(1H,m),4.45-4.55(1H,m),4.60(1H,br s),5.00(2H,s),5.0-5.1(1H,m),5.22(1H,d,J=8.4Hz),7.15-7.4(5H,m),7.27(5H,s)ジアステレオマーBmp 151.0−153.5℃SIMS(m/z):483(MH+)
元素分析値: C27H34N2O6としてCalcd:C,67.20; H,7.10; N,5.80Found:C,67.11; H,7.25; N,5.531H−NMR(CDCl3)δ:1.43(9H,s),1.8-2.1(4H,m),2.94(1H,d,J=2.8Hz),2.98(1H,s),3.05-3.2(1H,m),3.15-3.4(1H,m),3.97(1H,br s),4.1-4.3(3H,m),5.03(2H,d,J=4.0Hz),5.14(1H,d,J=9.8Hz),7.2-7.4(10H,m)
【0041】実施例81−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン−tert−ブチルエステルのジアステレオマーB(840mg,1.74mmol)をジオキサン(5ml)に溶解し、4N塩酸−ジオキサン溶液(2ml)を加え、室温で24時間撹拌した。反応液にジオキサン(5ml)を加えて減圧濃縮を3回くり返した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,30g:酢酸エチル−ヘキサン=2:1で溶出)で精製し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリンの非結晶性粉末410mg(55.3%)を得た。
mp 155.5−156.0℃SIMS(m/z):427(MH+)
1H−NMR(CDCl3)δ:1.55-1.75(1H,m),1.85-2.1(2H,m),2.25-2.45(1H,m),2.85-3.15(2H,m),3.25-3.5(1H,m),3.55-3.75(1H,m),4.12(1H,dd,J=7.2,8.4Hz),4.5-4.6(1H,m),4.64(1H,d,J=5.2Hz),4.65-4.75(1H,m),5.03(2H,q,J=10.8,32.8Hz),7.27(5H,s),7.15-7.4(6H,m)
【0042】実施例91−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン(270mg,0.63mmol)をジメチルホルムアミド(4ml)に溶解し、氷冷下にn−ブチルアミン(186μl,1.90mmol),シアノリン酸ジエチル(240mg,1.39mmol)のジメチルホルムアミド(0.5ml)溶液およびトリエチルアミン(194μl,1.39mmol)を順次加え、氷冷下に1時間、さらに室温で一夜撹拌した。反応液に水(5ml)を加え、1時間撹拌した後、さらに水(30ml)を加え、酢酸エチル(40ml)で抽出した。酢酸エチル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)した後、減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル−ヘキサン=2:1で溶出)で精製した後、ヘキサンから結晶化し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−ブチル−L−プロリンアミドの白色粉末晶150mg(49.2%)を得た。
mp 66.5−68.5℃SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:0.90(3H,t,J=6.8Hz),1.2-1.6(4H,m),1.75-2.1(3H,m),2.15-2.3(1H,m),2.8-3.4(6H,m),3.87(1H,br s), 4.0-4.3(3H,m),5.03(2H,s),5.10(1H,d,J=9.4Hz),6.57(1H,br s),7.2-7.4(5H,m),7.33(5H,s)
【0043】実施例102−キノリンカルボン酸 N−ヒドロキシスクシンイミドエステル(1.08g,4.00mmol)をジメチルホルムアミド(4ml)に溶解し、これにL−アスパラギン・1水和物(600mg,4.00mmol)を加え、室温で100時間攪拌した。溶媒を減圧留去した後、残渣にジクロロメタン(18ml)を加え、室温で30分間攪拌した。生じた白色沈殿物を濾取し、ジクロロメタン(40ml)で洗浄するとNアルフア−(2−キノリルカルボニル)−L−アスパラギン(1.09g,94.9%)が白色粉末晶として得られた。
mp 189 −193 ℃(分解)
元素分析値:C14H13N3O4・0.2H2Oとして計算値 C,57.81;H,4.64;N,14.44実測値 C,57.86;H,4.66;N,14.55SIMS(m/z):288(MH+)
1H−NMR(d6-DMSO)δ:2.67-2.93(2H,m),4.78-4.87(1H,m),7.01(1H,s),7.52(1H,s),7.74(1H,t,J=7.2Hz),7.89(1H,t,J=7.2Hz), 8.09-8.21(3H,m),8.59(1H,d,J=8Hz),9.17(1H,d,J=8Hz)
【0044】実施例11Nアルフア−(2−キノリルカルボニル)−L−アスパラギン(146mg,0.509mmol)と1−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:175mg,0.509mmol)をジメチルホルムアミド(3ml)に溶解し、−5℃以下で、1−ヒドロキシベンゾトリアゾール(73mg,0.540mmol)、次いでジシクロヘキシルカルボジイミド(111mg,0.540mmol)を加え、−5℃以下で2時間、さらに室温で16時間攪拌した。生じた不溶物を濾別し、溶媒を減圧留去した。残渣に10%リン酸水溶液、酢酸エチル(各30ml)を加えて分液した。酢酸エチル層を5%重曹水,飽和食塩水の順に洗浄し、乾燥(MgSO4)した後、減圧留去した。得られた粉末をシリカゲルカラムクロマトグラフィー(Kieselgel 60, 18g:酢酸エチル:ジクロロメタン:メタノール=8:8:1で溶出)で2回精製すると1−[(3S)−2−ヒドロキシ−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物3:150mg,47.8%)が非結晶性白色粉末として得られた。
SIMS(m/z):617(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.7-2.4(4H,m),2.64-3.10(4H,m),3.69(2H,t,J=6.6Hz),4.09-4.16(1H,m),4.40-4.66(3H,m), 4.91-5.05(1H,m),5.58(1H,brs),6.10(1H,brs),6.57(1H,s),6.95-7.18(5H,m),7.64(1H,t,J=7Hz),7.75-7.91(3H,m),8.11-8.34(3H,m),9.20(1H,d,J=7.8Hz)元素分析値:C33H40N6O6・H2Oとして計算値 C,62.45;H,6.67;N,13.24実測値 C,62.20;H,6.80;N,13.19
【0045】実施例121−[(3S)−2−ヒドロキシ−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物3:50mg,0.081mmol)のベンゼン溶液(2.5ml)に、氷冷下塩酸1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(31mg,0.016mmol)、ピリジントリフルオロ酢酸塩(5.9mg,0.03mmol)、ジメチルスルホキシド(0.2ml)を順次加えた。室温で24時間攪拌した後、再度氷冷下に塩酸1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(31mg,0.016mmol)、ピリジントリフルオロ酢酸塩(5.9mg,0.03mmol)、ジメチルスルホキシド(0.2ml)を順次加えた。室温で72時間攪拌した後、反応液に酢酸エチル(20ml)を加え、これを水(10ml×2)で洗浄した。酢酸エチル層を乾燥(MgSO4)した後、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル:ジクロロメタン:メタノール=5:5:1で溶出)で2回精製すると1−[(3S)−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−2−オキソ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物4:20mg,40.2%)が白色粉末晶として得られた。
SIMS(m/z):615(MH+)
1H−NMR(CDCl3)δ:1.16(3H,s),1.20-1.37(6H,m),1.70-2.35(4H,m),2.56-3.78(6H,m),4.38-4.60(1H,m),4.92-5.1(1H,m), 5.1-5.35(1H,m),5.35-5.55(1H,m),5.80-6.12(1H,m),6.4-6.57(1H,m),6.95-7.29(5H,m),7.46-7.93(4H,m),8.09-8.35(3H,m),9.25-9.48(1H,m)mp 115−118℃
【0046】
【配列表】配列番号:1配列の長さ:12配列の型:核酸鎖の数:2本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列AATTCTATGC CA 12
【0047】配列番号:2配列の長さ:12配列の型:核酸鎖の数:2本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列GATACGGTCT AG 12
【0048】配列番号:3配列の長さ:28配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CTAGCTAGCT AGAATTCTAG CTAGCTAG 28
【0049】配列番号:4配列の長さ:13配列の型:アミノ酸トポロジー:直鎖状配列の種類:ペプチド配列Ser Gln Val Ser Gln Asn Tyr Pro Ile Val Gln Asn Leu 1 5 10
【0050】配列番号:5配列の長さ:7配列の型:アミノ酸トポロジー:直鎖状配列の種類:ペプチド
【0001】
【産業上の利用分野】本発明は、レトロウイルスプロテアーゼ阻害活性を有する新規なアスパラギン誘導体およびその用途に関する。
【0002】
【従来の技術】レトロウイルスは、レトロウイルス科に属するウイルスであって、遺伝子としてRNAを持ち、自身の有する逆転写酵素(RNA依存性DNAポリメラーゼ)の働きによりRNAを鋳型としてDNAを合成する段階を自己複製の第1段階とするウイルスの総称である。レトロウイルス科は3つの亜科(オンコウイルス、レンチウイルス、スプマウイルス)からなる。レトロウイルスは、鳥類や哺乳類の種々の動物を宿主として感染、増殖し、肉腫、白血病、癌などを引き起こすもののあることが知られている。ヒトを宿主とするレトロウイルスとしては、成人T細胞白血病ウイルス(adult T−cell leukemia virus, ATLV)、ヒトT細胞白血病ウイルスI型(human T−cell leukemia virus typeI、HTLV−I)およびヒト免疫不全ウイルス(human immunodeficiency virus, HIV)などが報告されているが、ATLVは、HTLV−Iと同一のウイルスであることが証明された。HTLVにはこの他に、T細胞系のヘアリーセル白血病(hairycell leukemia)を誘導するII型が存在する。HIVはHIV−1およびHIV−2に分類され、HIV−1は重篤な免疫不全症である後天性免疫不全症候群(aquired immunodeficiency syndrome, AIDS)の原因ウイルスであることが分かっている。また、HIV−2もAIDSの原因ウイルスとされているが、HIV−1ほどはっきりとは解明されていない。
【0003】ATLVによって引き起こされる成人T細胞白血病は、日本の南西地域特に九州地方に多く見られる白血病であり、1977年にはじめて報告された。成人T細胞白血病患者では、T4陽性T細胞が癌化して、皮膚病変、リンパ節腫大、肝脾腫等を招来する。成人T細胞白血病に対しては有効な治療法はなく、発病するとほとんどが1年以内に死亡する。
【0004】HIV−1によって引き起こされるAIDSは、予後不良の免疫不全症として最近注目を集めている疾病であって、カリニ肺炎やカポシ肉腫などの臨床像を特徴とする。HIV−1は、T4陽性細胞に感染して細胞内に侵入、増殖し、これを破壊することにより免疫不全を招来する。現在AIDSの治療薬としてアジドチミジン(AZT)が使用されており、症状の改善および延命効果が認められている。
【0005】
【発明が解決しようとする課題】上記のように抗レトロウイルス剤としてAZTが使用されているものの、この物質は副作用として骨髄抑制を起こすなどの欠点を有しており、さらに効果が高く副作用の少ない治療薬の出現が期待されている。
【0006】
【課題を解決するための手段】レトロウイルスのゲノムにコードされているプロテアーゼは感染性成熟ウイルス粒子の形成に際して重要な役割を果たしており、このプロテアーゼ機能を欠損すると感染性粒子の形成の起こらないことが知られている[I. Katoh ら,ウィロロジー(Virology)第145巻,280頁(1985年)]。
【0007】したがって、該プロテアーゼの阻害剤はレトロウイルス性疾患の予防および治療薬に成り得るものと考えられ、最近になってペプスタチンA[フェブス レターズ(FEBS Lett.),247,349(1989)]、種々の合成ペプチド性誘導体[ネイチャー(Nature),343,90(1990)、ジャーナル・オブ・バイオロジカル・ケミストリー(J. Biol. Chem.),265,14675(1990)、ジャーナル・オブ・メディシナル・ケミストリー(J. Med. Chem.), 33,1285(1990)、サイエンス(Science),247,454(1990)および248,358(1990)]や非ペプチド性化合物[サイエンス(Science),249,527(1990)、ジャーナル・オブ・メディシナル・ケミストリー(J. Med. Chem.)、33,2687(1990)および34,1 225(1991)]等のプロテアーゼ阻害剤が該治療剤として提案されている。本発明者らの一人、加藤らは広く自然界に該プロテアーゼ阻害物質を検索し、その結果、土壌中から分離した1菌株が該プロテアーゼ阻害物質を産生すること、該菌株が放線菌の中のキタサトスポリア属に属すること、該微生物の培養液から精製取得したペプチド性の新規な化合物たるRPI−856、RPI−857、RPI−858、RPI−859が試験管内で該プロテアーゼを強く阻害することを見出し、すでに特許出願した(特願平2−282025,特願平3−1450および特願平3−17604号明細書参照)。本発明者らは、これらのペプチド性化合物の化学構造に基づいてドラッグデザインを行い、関連化合物の合成研究を鋭意行った結果、以下に示されるアスパラギン誘導体が高い該プロテアーゼ阻害活性を示すことを見出し、さらに研究を進め本発明を完成した。
【0008】すなわち、本発明は、一般式(I)
【化3】
[式中、環Aは5〜7員環を示し、R1,R2はともに水素原子を示すか、または結合して縮合環を形成していてもよく、R3はエステル化もしくはアミド化されていてもよいカルボキシル基を示し、R4は水素原子またはアシル基を示し、Xは
【化4】
で表わされるアスパラギン誘導体およびその塩、および化合物(I)を含有してなるレトロウイルス性疾患治療剤を提供するものである。
【0009】化合物(I)に関し、R1,R2が結合して縮合環を形成する場合、5〜6員環を形成することが好ましく、なかでもR1,R2が結合してベンゾ縮合、パーヒドロベンゾ縮合もしくはパーヒドロシクロペンタ縮合を示すのが好ましい。この場合、具体的には式
【化5】
などを挙げることができる。
【0010】化合物(I)に関し、R1,R2で表わされる基の好ましい例は、ともに水素原子であるものおよび結合してパーヒドロベンゾ縮合であるものがあげられる。化合物(I)に関し、R3で表わされるエステル化もしくはアミド化されていてもよいカルボキシル基としては、例えばカルボキシル,カルバモイル,低級アルコキシカルボニル(例、メトキシカルボニル,エトキシカルボニル,プロポキシカルボニル,イソプロポキシカルボニル,ブトキシカルボニル,イソブトキシカルボニル,tert−ブトキシカルボニルなどのC1-4アルコキシ−CO−など),アラルキルオキシカルボニル(例、ベンジルオキシカルボニル,p−ニトロベンジルオキシカルボニル,p−メトキシベンジルオキシカルボニル,フェネチルオキシカルボニルなどの置換されていてもよいフェニル−C1-4アルコキシ−CO−など),低級アルキルアミノカルボニル(例、メチルアミノカルボニル,エチルアミノカルボニル,プロピルアミノカルボニル,イソプロピルアミノカルボニル,ブチルアミノカルボニル,イソブチルアミノカルボニル,tert−ブチルアミノカルボニルなどのN−C1-4アルキルアミノなど),アラルキルアミノカルボニル(例、ベンジルアミノカルボニル,フェネチルアミノカルボニルなどのN−フェニル−C1-4アルキルアミノ)などがあげられ、なかでも低級アルコキシカルボニルおよび低級アルキルアミノカルボニルが好ましく、とりわけ、tert−ブトキシカルボニルおよびtert−ブチルアミノカルボニルが好ましい。
【0011】化合物(I)に関し、R4で表わされるアシル基としては、例えば低級アルカノイル(例、ホルミル,アセチル,プロピオニル,ブチリル,イソブチリルなどのC1-5アルカノイルなど),低級アルコキシカルボニル(例、メトキシカルボニル,エトキシカルボニル,プロポキシカルボニル,イソプロポキシカルボニル,ブトキシカルボニル,イソブトキシカルボニル,tert−ブトキシカルボニルなどのC1-4アルコキシ−CO−など),アラルキルオキシカルボニル(例、ベンジルオキシカルボニル,p−ニトロベンジルオキシカルボニル,p−メトキシベンジルオキシカルボニル,ナフチルメチルオキシカルボニルなどの置換されていてもよいフェニル−C1-4アルコキシ−CO−など),アリールカルボニル(例、ベンゾイル,ナフトイル,トルオイルなど),ヘテロアリールカルボニル(例、フロイル,テノイル,ニコチノイル,イソニコチノイル,キノリノイル,イソキノリノイル,キナゾリノイルなど),アリールスルホニル(例、p−トルエンスルホニル,ベンゼンスルホニル,ナフタレンスルホニルなど)などがあげられ、なかでも低級アルコキシカルボニル,アラルキルオキシカルボニル,ヘテロアリールカルボニルが好ましく、とりわけ、tert−ブトキシカルボニル,ベンジルオキシカルボニル,2−キノリノイルが好ましい。
【0012】化合物(I)に関し、環Aとしては5〜6員環,とりわけピロリジン環およびピペリジン環が好ましい。本発明化合物に属する好ましい化合物の具体例としては、例えば〔表1〕に示す化合物があげられる。
【0013】
【表1】
【0014】
【化6】
[式中、各記号は前記と同意義。]で表わされる化合物を酸化することによって製造することができる。
【0015】反応は通常、ジメチルスルホキシド中、ピリジンおよびトリフルオロ酢酸の存在下に行うことが好ましく、反応温度は0〜60℃程度で行われ、反応時間は1〜24時間である。
【0016】本発明化合物(I)は、たとえば式(II)
【化7】
[式中、各記号は前記と同意義。]で表わされる化合物と、式(III)
【化8】
[式中、R4′はアシル基であり、他の記号は前記と同意義。]で表わされる化合物を縮合させることによって得ることができる。
【0017】該縮合反応条件としては、たとえば縮合試薬(例、ジシクロヘキシルカルボジイミド,カルボニルジイミダゾール,シアノリン酸ジエチル,ジフェニルリン酸アジド等)などを用いるか、または化合物(III)にたとえば2,4,5−トリクロロフェノール,ペンタクロロフェノール,ペンタフルオロフェノール,2−ニトロフェノールおよび4−ニトロフェノール等のフェノール類、または、たとえばN−ヒドロキシスクシンイミド,N−ヒドロキシ−5−ノルボルネン−2,3−ジカルボン酸イミド,1−ヒドロキシベンズトリアゾール,N−ヒドロキシピペリジン等のN−ヒドロキシ化合物をジシクロヘキシルカルボジイミド等の触媒の存在下に縮合させ、化合物(III)を活性なエステル体に変換した後、化合物(II)と反応させることが好ましい。また化合物(III)にたとえば、クロロ炭酸メチル,クロロ炭酸エチル,クロロ炭酸イソブチルなどを、好ましくは有機塩基(例、トリエチルアミン,N−メチルモルホリン)の存在下に反応させ、混合酸無水物に変換した後、化合物(II)と反応させることもできる。これらの反応において、反応温度は通常約−30℃〜+50℃であり、用いる溶媒としては、たとえばジオキサン,テトラヒドロフラン,アセトニトリル,ジエチルエーテル,N,N−ジメチルホルムアミド,N,N−ジメチルアセトアミド,ジメチルスルホキシド,N−メチルピロリドン,クロロホルム,塩化メチレンなどが挙げられ、これら溶媒は単独もしくは混合溶媒として用いてもよい。
【0018】本発明化合物(I)は、たとえば式(IV)
【化9】
[式中、R4′はアシル基であり、他の記号は前記と同意義。]で表わされる化合物と式(V)
【化10】
[式中、各記号は前記と同意義。]で表わされる化合物を縮合させることによって製造することができる。該縮合反応条件としては、上述した(II)と(III)の縮合反応において適用される条件と同様な反応条件が好ましい。
【0019】本発明化合物(I)においてR4が水素である化合物は、式(I″)で表わされる化合物を脱保護反応に付すことによって製造することができる。
【化11】
[式中、R4″はアミノ保護基を示し、他の記号は前記と同意義。]
また、本発明化合物(I)においてR3がカルボキシル基である化合物は、式(I″′)で表わされる化合物を脱保護反応に付すことによって製造することができる。
【化12】
[式中、R3′は保護されたカルボキシル基を示し、他の記号は前記と同意義。]。
【0020】該脱保護反応としては、その保護基の種類あるいは、化合物(I″),(I″′)の脱保護条件下での安定性に応じて、酸あるいは塩基による方法、または接触還元による方法を適宜選択して行うことができる。酸による方法の場合には、保護基の種類その他の条件によって異なるが、酸として例えば塩酸、臭化水素、フッ化水素、沃化水素、メタンスルホン酸、p−トルエンスルホン酸、トリフルオロメタンスルホン酸、硫酸、リン酸等の無機酸、ギ酸、酢酸、トリフルオロ酢酸、プロピオン酸等の有機酸の他、酸性イオン交換樹脂等が使用される。塩基による方法の場合には、保護基の種類その他の条件によって異なるが、塩基として例えばナトリウム,カリウム等のアルカリ金属もしくはカルシウム,マグネシウム等のアルカリ土類金属の水酸化物、炭酸塩等の無機塩基、金属アルコキサイド類、有機アミン類、第四アンモニウム塩等の有機塩基の他、塩基性イオン交換樹脂等が使用される。上記酸または塩基による方法の場合において溶媒を使用する場合には、たとえば、水,メタノール,エタノール,酢酸エチル,クロロホルム,テトラヒドロフラン,ジオキサン,ピリジン,酢酸,アセトン,塩化メチレンなどを単一、もしくは混合して用いることができる。また金属と酸による方法においては水,アセトン等が繁用されるが酸が液体のときは酸自身を溶媒として使用することもできる。酸および塩基による方法において反応温度は、通常冷却下ないし加温程度で行なわれ、反応時間は約30分ないし20時間である。
【0021】一方接触還元による方法の場合には、水またはたとえばメタノール,エタノール,ジオキサン,酢酸エチルなどの有機溶媒あるいはそれらの混合溶媒中、たとえばパラジウム−炭素などの適当な触媒の存在下に行われる。本反応は常圧または150kg/cm2程度までの圧力下、常温ないし+100℃の温度で行われるが、一般に常温,常圧で充分反応は進行する。
【0022】本発明化合物(I)は分子内に不斉原子を3個以上有しているので8個以上の立体異性体が存在し得るが、その立体異性体ならびにそれらの混合物のいずれも本発明に包含されるものである。とりわけアスパラギン残基中の不斉炭素,ベンジル基とXが結合した不斉炭素およびR3が結合した不斉炭素のいずれもがS配置である光学活性体が好ましい。
【0023】化合物(I)は塩としても得られ、該塩としては、たとえば塩酸塩、臭化水素酸塩、硫酸塩、硝酸塩、燐酸塩などの無機酸塩、たとえば酢酸塩、酒石酸塩、クエン酸塩、フマール酸塩、マレイン酸塩、トリエンスルホン酸塩、メタンスルホン酸塩などの有機酸塩、たとえばナトリウム塩、カリウム塩、カルシウム塩、アルミニウム塩などの金属塩、たとえば、トリエチルアミン塩、グアニジン塩、アンモニウム塩、ヒドラジン塩、キニーネ塩、シンコニン塩などの塩基との塩が挙げられる。
【0024】かくして得られる化合物(I)は反応混合物から通常の分離精製手段、たとえば抽出、濃縮、中和、ろ過、再結晶、カラムクロマトグラフィー、薄層クロマトグラフィーなどの手段を用いることによって単離することができる。化合物(I)は少なくとも8個の立体異性体が存在し得る。これら個々の異性体およびこれらの混合物のいずれも本発明に包含されるが、所望によりこれらの異性体を個別に製造することもできる。たとえば原料化合物(I′),(II),(III),(IV)および(V)のそれぞれの単一の異性体を用いて上記の反応を行うことにより、化合物(I)の単一の異性体を得ることができるし、また生成物が二種類以上の異性体混合物の場合には、これを通常の分離方法、たとえば光学活性酸(例、カンファースルホン酸、酒石酸など)との塩を生成させる方法や、各種のクロマトグラフィー、分別再結晶などの分離手段によってそれぞれの異性体に分離することもできる。
【0025】化合物(I)の塩は必要に応じ前記した無機酸あるいは有機酸を加えることによって生理学的に許容される塩として得ることができる。本発明における合成中間体(II)において、例えばR1,R2がともに水素原子であり、R3がtert−ブチルアミノカルボニルであり、環Aがピロリジン環であり、Xが
【化13】
である化合物(XI)は次の反応式で示す方法で合成することができる。
【0026】
【化14】
ここで用いた(IX)はジャーナル・オブ・メディシカル・ケミストリー(J. Med. Chem.), 20,510(1977)に記載された方法で合成することができる。
【0027】以下参考例および試験例においてHIV−1プロテアーゼの調製法および該プロテアーゼに対する本発明化合物(I)の阻害作用についてそれぞれ述べる。
参考例1 HIV−1プロテアーゼ遺伝子の発現プラスミドの構築HIV−1プロテアーゼはpol遺伝子にコードされた蛋白質の1つであり、感染細胞内においてはgag−polのポリプロテインとして合成されたものが、プロテアーゼ自身の作用によりプロセシングされることにより生成されると考えられている[J. M. Mc Cune ら、セル(Cell)第53巻,55頁(1988年)]。一方、プロテアーゼ領域を含むpol遺伝子の一部を大腸菌で発現させることにより、分子量11キロダルトンの活性型HIV−1プロテアーゼが自身のオートプロセシング作用により生成されることが既に知られている[M. C. Graves ら、プロシーディングス・オブ・ザ・ナショナル・アカデミー・オブ・サイエンス(Proc. Natl. Acad. Sci. USA)第85巻,2449頁(1988年)]。
【0028】そこで、まずHIV−1の全遺伝子を含有するプラスミドpNL4−3[A. Adachi ら、ジャーナル・オブ・ウイロロジー(J. Virol.)第59巻,284頁(1986年)]からプロテアーゼ領域を含有する1.5kb のBglII−BalI断片を取得した。次に、この断片にリンカー 5’−AATTCTATGCCA−3’ (配列番号:1)
3’−GATACGGTCTAG−5’ (配列番号:2)
を接続した後、EcoRIで消化し、さらにリンカー[5’−CTAGCTAGCTAGAATTCTAGCTAGCTAG−3’](配列番号:3)を接続した後、EcoRIで消化した。次に、この断片の切断点をクレノーフラグメントで埋めて平滑末端にした。一方、T7プロモーターを含む大腸菌用発現ベクターpET−3c[A. H. Rosenberg ら、ジーン(Gene)第56巻,125頁(1987年)]をNdeIで消化し、その切断点をT4DNAポリメラーゼで埋めて平滑末端にした。次に、これら両DNA断片をT4DNAリガーゼを用いて接続し、T7プロモーターとプロテアーゼ遺伝子の向きが同方向となったものをpHIV7004とした。
【0029】参考例2 組換え型HIV−1プロテアーゼの調製Escherichia coli MM294株にT7ファージのRNAポリメラーゼ遺伝子を組み込んだλファージDE3[F. W. Studier ら、ジャーナル・オブ・モレキュラー・バイオロジー(J. Mol. Biol.)第189巻,113頁(1986年)]を溶原化させてEscherichia coli MM294(DE3)を作製した。参考例1で得られたpHIV7004を、Escherichia coli MM294(DE3)に導入して、形質転換体Escherichia coli MM294(DE3)/pHIV7004を得た。本形質転換体をアンピシリン50μg/mlを含むLB培地に接種し、37℃で一晩振盪培養した。この培養液0.2mlを20mlの同じ培地に接種し、37℃で対数増殖期(180−200 Klett unit)まで培養した。次に、最終濃度が0.4mMになるようにイソプロピル−1−チオ−β−D−ガラクトシド(IPTG)を加え、さらに37℃で2時間培養した。培養液200mlから、菌体を5000rpm で5分間遠心することにより集め、これを〔表2〕に記載した組成を持つ緩衝液A20mlに懸濁した。懸濁液にニワトリ卵白リゾチーム2mgを加え、氷冷下で10分間放置後、ノニデットP40(NP40)を最終濃度1%になるように加え、超音波破砕装置を用いて菌体を破砕した。上記処理を行った溶菌液を、日立工機社製のRR−24Aローターを用いて16000rpm で10分間遠心分離し、HIV−1プロテアーゼを含む粗抽出液20mlを得た。
【0030】
【表2】 緩衝液Aの組成50mM トリス塩酸緩衝液pH8.05mM エチレンジアミン4酢酸(EDTA)
0.1% ノニデットP40(NP40)
0.02% 2−メルカプトエタノール(2−ME)
10% グリセリン1mM フェニルメチルスルフォニルフルオリド(PMSF)
0.1mM パラアミジノフェニルメタンスルフォニルフルオリド(APMSF)
【0031】試験例1 HIV−1プロテアーゼ阻害作用HIV−1 gagP17とP24の解裂部位を含む合成ペプチドgag−11(SQVSQNYPIVQNL)(配列番号:4)を基質とし、反応産物gag−12(SQVSQNY)(配列番号:5)の生成を指標としてHIV−1プロテアーゼ阻害活性を測定した。即ち、基質gag−11 3μg、 種々の濃度の試料、HIV−1プロテアーゼを含む5%グリセロール−1MNaCl−0.25Mリン酸ナトリウム緩衝液(pH6.2)(30μl)中で3〜5時間反応させた。0.13Mトリクロロ酢酸−0.26M酢酸ナトリウム−0.4M酢酸150μlを加えることにより反応を停止した。室温で10分間放置した後遠心上清を得た。上記の上清のうち48μlを0.1%トリフロロ酢酸で平衡化したYMC−パックFL−ODSカラム(0.46×3cm、ワイエムシィ社)に負荷し、アセトニトリルの濃度勾配で溶出した。本条件下において、gag−12の生成量を50%阻害するために必要な反応液中の試料濃度をIC50値として表わした。化合物(I)のHIV−1プロテアーゼ阻害活性を〔表3〕に示した。
【0032】
【表3】
試 料 IC50(μM)
(化合物番号) ────────────────────────── 1 0.020 2 0.034 3 0.021 4 0.031〔表3〕の結果から、本発明化合物(I)は非常に強いHIV−1プロテアーゼ阻害物質であることが明らかである。
【0033】以上のように、化合物(I)はレトロウイルスのプロテアーゼ阻害活性を有しており、また毒性も低いので、レトロウイルス性疾患治療剤として有用な物質である。 本発明化合物(I)を抗レトロウイルス剤として用いる場合、希釈剤、賦形剤、担体などと混合して、薬学的に許容される製剤とし、経口的または非経口的に投与する。例えば、注射剤として用いる場合は、化合物(I)を水溶液剤として、成人1日当り0.1〜10mg/kgを静脈内投与する。
【0034】
【実施例】以下に実施例を挙げて、本発明をさらに具体的に説明するが、これによって本発明が限定されるものではない。
実施例11−ベンジルオキシカルボニル−L−プロリン(化合物IV:23.0g,92.2mmol)をテトラヒドロフラン(150ml)に溶解し、氷冷下にトリエチルアミン(14.1ml,102mmol)を加えた。次に、クロロギ酸イソブチル(13.2ml,102mmol)のテトラヒドロフラン(43ml)溶液を滴下し、氷冷下で1時間撹拌した後、tert−ブチルアミン(19.4ml,184mmol)を加えた。氷冷下で1時間、さらに室温で一夜撹拌した後、溶媒を留去した。残渣に水,酢酸エチル(各200ml)を加えて分液し、酢酸エチル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)後、減圧留去した。得られた白色粉末を酢酸エチル−ヘキサン(1:1,40ml)から再結晶し、1−ベンジルオキシカルボニル−N−tert−ブチル−L−プロリンアミド(化合物VII:15.5g,55%)を白色結晶として得た。さらにろ液を減圧留去した後、シリカゲルカラムクロマトグラフィー(Kieselgel 60,100g:酢酸エチル−ヘキサン=1:1で溶出)で精製すると追加のプロリンアミド体(化合物VII:5.9g,21.0%)が得られた。
mp 90−91℃元素分析値: C17H24N2O3としてCalcd:C,67.08; H,7.95; N,9.20Found:C,66.85; H,7.96; N,9.45SIMS(m/z):305(MH+)
1H−NMR(d6-DMSO)δ:1.18(6H,s),1.24(3H,s),1.7-1.95(3H,m),1.95-2.2(1H,m),3.3-3.5(2H,m),4.1-4.2(1H,m),4.9-5.10(2H,m),7.2-7.5(6H,m)
【0035】実施例21−ベンジルオキシカルボニル−N−tert−ブチル−L−プロリンアミド(化合物VII:8.0g,26.3mmol)をメタノール−水(6:1,140ml)に溶解し、10%パラジウム−炭素(0.5g)を加え、水素気流中、1時間撹拌した。触媒をろ別し、触媒部分を洗浄(10%メタノール水溶液)し、ろ液と洗液を合わせて減圧留去した。得られた油状物を減圧乾燥すると結晶化した。これにジエチルエーテルと石油エーテルの混液を加えてろ取すると、N−tert−ブチル−L−プロリンアミド(化合物VIII:3.4g,75.7%)が無色粉末晶として得られた。
mp 79−80℃元素分析値: C9H18N2OとしてCalcd:C,63.49; H,10.66; N,16.45Found:C,63.45; H,10.89; N,16.151H−NMR(d6-DMSO)δ:1.26(9H,s),1.5-1.7(3H,m),1.8-2.0(1H,m),2.78(2H,ddd,J=6.1,6.5,9.7Hz),3.2-3.4(1H,m),7.57(1H,br s)
【0036】実施例3(2RS,3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブタン酸(化合物IX:2.0g,6.07mmol)をジメチルホルムアミド(20ml)に溶解した。氷冷下にN−tert−ブチル−L−プロリンアミド(化合物VIII:1.24g,7.28mmol),シアノリン酸ジエチル(DEPC)(2.09g,12.2mmol)のジエチルホルムアミド(5ml)溶液およびトリエチルアミン(1.76ml,12.2mmol)を順次加え、氷冷下に1時間、さらに室温に戻して2.5時間撹拌した。反応液に水(約5ml)を加えて1時間撹拌した後、水(80ml)を加え、酢酸エチル(150ml)で抽出した。酢酸エチル層を10%リン酸ナトリウム水溶液、5%重曹水、飽和食塩水の順で洗浄し、乾燥(Mg SO4)した後、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー (Kieselgel 60,140g:酢酸エチル−ヘキサン=2:1で溶出)で精製すると1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物X) のジアステレオマーA(極性大:0.695g)及びジアステレオマーB(極性 小:0.415g)が無色粉末晶として得られた。
ジアステレオマーA白色粉末晶mp 63−66℃元素分析値: C27H35N3O5・1/2H2OとしてCalcd:C,66.10; H,7.40; N,8.57Found:C,66.40; H,7.43; N,8.54SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.75-2.4(4H,m),2.6-2.8(2H,m),3.6-4.25(5H,m),4.3-4.65(2H,m),4.99(2H,s),5.43(1H,d,J=8.8Hz),6.49(1H,s),7.1-7.4(10H,m)ジアステレオマーB白色粉末晶mp 114−115℃元素分析値: C27H35N3O5としてCalcd:C,67.34; H,7.33; N,8.73Found:C,67.51; H,7.29; N,8.77SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.7-2.3(4H,m),2.95(2H,d,J=7.8Hz),3.0-3.4(2H,m),3.87(1H,d,J=5.6Hz),3.95(1H,d,J=3.6Hz),4.10(1H,d,J=4.6Hz),4.22(1H,m),5.03(2H,s),5.09(1H,br s),6.43(1H,br s),7.2-7.5(10H,m)
【0037】実施例41−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物X,ジアステレオマーA:300mg,0.62mmol)をメタノール−水(4:1,8ml)に溶解し、10%パラジウム−炭素(40mg)を加え、水素気流下、4時間撹拌した。触媒をろ別し、触媒部分を洗浄(50%メタノール水溶液)し、ろ液と洗液を合わせて減圧留去した。残渣にジエチルエーテルを加えて、析出した結晶をろ取すると1−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:210mg)が白色粉末晶として得られた。
mp 154.5−156.0℃元素分析値: C19H29N3O3としてCalcd:C,65.68; H,8.41; N,12.09Found:C,65.35; H,8.55; N,12.131H−NMR(CDCl3)δ:1.29(9H,s),1.4-2.2(2H,br m),1.7-2.4(4H,m),2.53(1H,dd,J=10.0,13.3Hz),2.95(1H,dd,J=3.2,13.4Hz),3.0-3.2(1H,m),3.5-3.7(2H,m),4.27(1H,d,J=5.4Hz),4.45-4.55(1H,m),6.57(1H,s),7.1-7.4(5H,m)
【0038】実施例51−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:240mg,0.69mmol)をジメチルホルムアミド(8ml)に溶解し、氷冷下にNアルフア−ベンジルオキシカルボニル−L−アスパラギンのp−ニトロフェニルエステル(267mg,0. 69mmol)を加え、氷冷下に1時間、さらに室温で4時間撹拌した。溶媒を減圧留去し た後、残渣に水,酢酸エチル(各30ml)を加えて分液した。酢酸エチ ル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順に洗浄し、乾燥(MgSO4)した後、減圧留去した。得られた黄色油状物をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル−メタノール=9:1で溶出)で精製すると1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物1:200mg,48. 7%)が白色粉 末晶として得られた。
mp 102−103℃(dec.)
元素分析値: C31H41N5O7としてCalcd:C,62.51; H,6.94; N,11.76Found:C,62.20; H,7.06; N,11.66SIMS(m/z):596(MH+)
1H−NMR(d6-DMSO)δ:1.25(9H,s),1.7−2.1(4H,m),2.2−2.45(2H,m),2.55−2.8(2H,m),3.5−3.7(2H,m),4.0−4.2(1H,m),4.2−4.4(3H,m),4.89(1H,d,J=7.2Hz),5.01(2H,s),6.87(1H,s),7.05−7.4(5H,m),7.22(1H,s),7.35(5H,s),7.52(1H,s),7.86(1H,d,J=8.2Hz)
【0039】実施例6ベンゼン(1ml)とジメチルスルホキシド(0.5ml)の混液にジシクロヘキシルカルボジイミド(104mg,0.504mmol)とピリジン(14μl,0.168mmol)を加え、次いで氷冷下に1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物1:100mg,0. 168mmol)を加えた。10分後にトリフルオロ酢酸(6.6μl,0.0 84mmol)を加え、室温で一夜撹拌した。反応液に酢酸エチル(2ml)を加え、不溶物 をろ別した。ろ液に酢酸エチル(25ml)を加え、これを水(30ml× 3)で洗浄し、乾燥(MgSO4)した後、減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,15g:酢酸エチル−塩化メチレン−メタノール=5:5:1で溶出)で精製すると1−[(3S)−3−(Nアルフア−ベンジルオキシカルボニル−L−アスパラギニル)アミノ−2−オキソ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物2:20mg,20.1 %)が白色粉末晶として得られた。
mp 89−93℃SIMS(m/z):594(MH+)
1H−NMR(d6-DMSO)δ:1.21(3H,s),1.24(6H,s),1.65-2.0(4H,m),2.0-2.4(2H,m),2.75-3.1(1H,m),3.1-3.3(1H,m),3.4-3.6(2H,m),4.2-4.5(2H,m),4.5-4.9(1H,m),4.9-5.1(2H,m),6.88(1H,br s),7.1-7.3(5H,m),7.33(5H,s),7.5-7.6(1H,m),8.15-8.6(1H,m)
【0040】実施例7(2RS,3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブタン酸(化合物IX:2.16g,6.55mmol)をジメチルホルムアミド(5ml)に溶解し、氷冷下にL−プロリン−tert−ブチルエステル(1.23g,7.21mmol)のジメチルホルムアミド(2.5ml)溶液,シアノリン酸ジエチル(1.23g,7.21mmol)のジメチルホルムアミド(2.5ml)溶液 およびトリエチルアミン(1.0ml,7.21mmol)を順次加えた。氷冷下に1時間、さらに室温で一夜撹拌した後、溶媒を減圧留去した。残渣を酢酸エチル(80ml)に溶解し、得られた溶液を冷5%塩酸,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)した後、減圧留去した。残留物を酢酸エチル−ヘキサン(1:1,12ml)から結晶化し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン−tert−ブチルエステルのジアステレオマーB(極性小)を白色結晶として800mg(25.3%)得た。さらにろ液を減圧留去した後、残留物をシリカゲルカラムクロ マトグラフィー(Kieselgel 60,30g:酢酸エチル−ヘキサン=1:2で溶出)で精製すると、追加のジアステレオマーB140mg(4.4%)とジアステ レオマーA(極性大,非結晶性粉末)1.03g(32.6%)が得られた。
ジアステレオマーAmp 58−60℃SIMS(m/z):483(MH+)
1H−NMR(CDCl3)δ:1.43(3H,s),1.45(6H,s),1.6-2.3(4H,m),2.5-3.0(2H,m),3.6-3.9(2H,m),4.15-4.3(1H,m),4.45-4.55(1H,m),4.60(1H,br s),5.00(2H,s),5.0-5.1(1H,m),5.22(1H,d,J=8.4Hz),7.15-7.4(5H,m),7.27(5H,s)ジアステレオマーBmp 151.0−153.5℃SIMS(m/z):483(MH+)
元素分析値: C27H34N2O6としてCalcd:C,67.20; H,7.10; N,5.80Found:C,67.11; H,7.25; N,5.531H−NMR(CDCl3)δ:1.43(9H,s),1.8-2.1(4H,m),2.94(1H,d,J=2.8Hz),2.98(1H,s),3.05-3.2(1H,m),3.15-3.4(1H,m),3.97(1H,br s),4.1-4.3(3H,m),5.03(2H,d,J=4.0Hz),5.14(1H,d,J=9.8Hz),7.2-7.4(10H,m)
【0041】実施例81−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン−tert−ブチルエステルのジアステレオマーB(840mg,1.74mmol)をジオキサン(5ml)に溶解し、4N塩酸−ジオキサン溶液(2ml)を加え、室温で24時間撹拌した。反応液にジオキサン(5ml)を加えて減圧濃縮を3回くり返した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,30g:酢酸エチル−ヘキサン=2:1で溶出)で精製し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリンの非結晶性粉末410mg(55.3%)を得た。
mp 155.5−156.0℃SIMS(m/z):427(MH+)
1H−NMR(CDCl3)δ:1.55-1.75(1H,m),1.85-2.1(2H,m),2.25-2.45(1H,m),2.85-3.15(2H,m),3.25-3.5(1H,m),3.55-3.75(1H,m),4.12(1H,dd,J=7.2,8.4Hz),4.5-4.6(1H,m),4.64(1H,d,J=5.2Hz),4.65-4.75(1H,m),5.03(2H,q,J=10.8,32.8Hz),7.27(5H,s),7.15-7.4(6H,m)
【0042】実施例91−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−L−プロリン(270mg,0.63mmol)をジメチルホルムアミド(4ml)に溶解し、氷冷下にn−ブチルアミン(186μl,1.90mmol),シアノリン酸ジエチル(240mg,1.39mmol)のジメチルホルムアミド(0.5ml)溶液およびトリエチルアミン(194μl,1.39mmol)を順次加え、氷冷下に1時間、さらに室温で一夜撹拌した。反応液に水(5ml)を加え、1時間撹拌した後、さらに水(30ml)を加え、酢酸エチル(40ml)で抽出した。酢酸エチル層を10%リン酸ナトリウム水溶液,5%重曹水,飽和食塩水の順で洗浄し、乾燥(MgSO4)した後、減圧留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル−ヘキサン=2:1で溶出)で精製した後、ヘキサンから結晶化し、1−[(3S)−3−ベンジルオキシカルボニルアミノ−2−ヒドロキシ−4−フェニルブチリル]−N−ブチル−L−プロリンアミドの白色粉末晶150mg(49.2%)を得た。
mp 66.5−68.5℃SIMS(m/z):482(MH+)
1H−NMR(CDCl3)δ:0.90(3H,t,J=6.8Hz),1.2-1.6(4H,m),1.75-2.1(3H,m),2.15-2.3(1H,m),2.8-3.4(6H,m),3.87(1H,br s), 4.0-4.3(3H,m),5.03(2H,s),5.10(1H,d,J=9.4Hz),6.57(1H,br s),7.2-7.4(5H,m),7.33(5H,s)
【0043】実施例102−キノリンカルボン酸 N−ヒドロキシスクシンイミドエステル(1.08g,4.00mmol)をジメチルホルムアミド(4ml)に溶解し、これにL−アスパラギン・1水和物(600mg,4.00mmol)を加え、室温で100時間攪拌した。溶媒を減圧留去した後、残渣にジクロロメタン(18ml)を加え、室温で30分間攪拌した。生じた白色沈殿物を濾取し、ジクロロメタン(40ml)で洗浄するとNアルフア−(2−キノリルカルボニル)−L−アスパラギン(1.09g,94.9%)が白色粉末晶として得られた。
mp 189 −193 ℃(分解)
元素分析値:C14H13N3O4・0.2H2Oとして計算値 C,57.81;H,4.64;N,14.44実測値 C,57.86;H,4.66;N,14.55SIMS(m/z):288(MH+)
1H−NMR(d6-DMSO)δ:2.67-2.93(2H,m),4.78-4.87(1H,m),7.01(1H,s),7.52(1H,s),7.74(1H,t,J=7.2Hz),7.89(1H,t,J=7.2Hz), 8.09-8.21(3H,m),8.59(1H,d,J=8Hz),9.17(1H,d,J=8Hz)
【0044】実施例11Nアルフア−(2−キノリルカルボニル)−L−アスパラギン(146mg,0.509mmol)と1−[(3S)−3−アミノ−2−ヒドロキシ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物XI:175mg,0.509mmol)をジメチルホルムアミド(3ml)に溶解し、−5℃以下で、1−ヒドロキシベンゾトリアゾール(73mg,0.540mmol)、次いでジシクロヘキシルカルボジイミド(111mg,0.540mmol)を加え、−5℃以下で2時間、さらに室温で16時間攪拌した。生じた不溶物を濾別し、溶媒を減圧留去した。残渣に10%リン酸水溶液、酢酸エチル(各30ml)を加えて分液した。酢酸エチル層を5%重曹水,飽和食塩水の順に洗浄し、乾燥(MgSO4)した後、減圧留去した。得られた粉末をシリカゲルカラムクロマトグラフィー(Kieselgel 60, 18g:酢酸エチル:ジクロロメタン:メタノール=8:8:1で溶出)で2回精製すると1−[(3S)−2−ヒドロキシ−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物3:150mg,47.8%)が非結晶性白色粉末として得られた。
SIMS(m/z):617(MH+)
1H−NMR(CDCl3)δ:1.29(9H,s),1.7-2.4(4H,m),2.64-3.10(4H,m),3.69(2H,t,J=6.6Hz),4.09-4.16(1H,m),4.40-4.66(3H,m), 4.91-5.05(1H,m),5.58(1H,brs),6.10(1H,brs),6.57(1H,s),6.95-7.18(5H,m),7.64(1H,t,J=7Hz),7.75-7.91(3H,m),8.11-8.34(3H,m),9.20(1H,d,J=7.8Hz)元素分析値:C33H40N6O6・H2Oとして計算値 C,62.45;H,6.67;N,13.24実測値 C,62.20;H,6.80;N,13.19
【0045】実施例121−[(3S)−2−ヒドロキシ−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物3:50mg,0.081mmol)のベンゼン溶液(2.5ml)に、氷冷下塩酸1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(31mg,0.016mmol)、ピリジントリフルオロ酢酸塩(5.9mg,0.03mmol)、ジメチルスルホキシド(0.2ml)を順次加えた。室温で24時間攪拌した後、再度氷冷下に塩酸1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(31mg,0.016mmol)、ピリジントリフルオロ酢酸塩(5.9mg,0.03mmol)、ジメチルスルホキシド(0.2ml)を順次加えた。室温で72時間攪拌した後、反応液に酢酸エチル(20ml)を加え、これを水(10ml×2)で洗浄した。酢酸エチル層を乾燥(MgSO4)した後、溶媒を留去した。残渣をシリカゲルカラムクロマトグラフィー(Kieselgel 60,20g:酢酸エチル:ジクロロメタン:メタノール=5:5:1で溶出)で2回精製すると1−[(3S)−3−[Nアルフア−(2−キノリルカルボニル)−L−アスパラギニル]アミノ−2−オキソ−4−フェニルブチリル]−N−tert−ブチル−L−プロリンアミド(化合物4:20mg,40.2%)が白色粉末晶として得られた。
SIMS(m/z):615(MH+)
1H−NMR(CDCl3)δ:1.16(3H,s),1.20-1.37(6H,m),1.70-2.35(4H,m),2.56-3.78(6H,m),4.38-4.60(1H,m),4.92-5.1(1H,m), 5.1-5.35(1H,m),5.35-5.55(1H,m),5.80-6.12(1H,m),6.4-6.57(1H,m),6.95-7.29(5H,m),7.46-7.93(4H,m),8.09-8.35(3H,m),9.25-9.48(1H,m)mp 115−118℃
【0046】
【配列表】配列番号:1配列の長さ:12配列の型:核酸鎖の数:2本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列AATTCTATGC CA 12
【0047】配列番号:2配列の長さ:12配列の型:核酸鎖の数:2本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列GATACGGTCT AG 12
【0048】配列番号:3配列の長さ:28配列の型:核酸鎖の数:一本鎖トポロジー:直鎖状配列の種類:他の核酸 合成DNA配列CTAGCTAGCT AGAATTCTAG CTAGCTAG 28
【0049】配列番号:4配列の長さ:13配列の型:アミノ酸トポロジー:直鎖状配列の種類:ペプチド配列Ser Gln Val Ser Gln Asn Tyr Pro Ile Val Gln Asn Leu 1 5 10
【0050】配列番号:5配列の長さ:7配列の型:アミノ酸トポロジー:直鎖状配列の種類:ペプチド
【特許請求の範囲】
【請求項1】式
【化1】
[式中、環Aは5〜7員環を示し、R1,R2はともに水素原子を示すか、または結合して縮合環を形成していてもよく、R3はエステル化もしくはアミド化されていてもよいカルボキシル基を示し、R4は水素原子またはアシル基を示し、Xは
【化2】
で表わされるアスパラギン誘導体およびその塩。
【請求項2】請求項1記載のアスパラギン誘導体を含有してなるレトロウイルス性疾患治療剤。
【請求項1】式
【化1】
[式中、環Aは5〜7員環を示し、R1,R2はともに水素原子を示すか、または結合して縮合環を形成していてもよく、R3はエステル化もしくはアミド化されていてもよいカルボキシル基を示し、R4は水素原子またはアシル基を示し、Xは
【化2】
で表わされるアスパラギン誘導体およびその塩。
【請求項2】請求項1記載のアスパラギン誘導体を含有してなるレトロウイルス性疾患治療剤。
【公開番号】特開平5−178824
【公開日】平成5年(1993)7月20日
【国際特許分類】
【出願番号】特願平4−159678
【出願日】平成4年(1992)6月18日
【出願人】(000002934)武田薬品工業株式会社 (396)
【公開日】平成5年(1993)7月20日
【国際特許分類】
【出願日】平成4年(1992)6月18日
【出願人】(000002934)武田薬品工業株式会社 (396)
[ Back to top ]