
アスパラチン様ジヒドロカルコン、未発酵ルイボスからの抽出物およびその調製方法
本発明は、式Iで示される新規な化合物ならびにその薬学的に許容される塩、誘導体およびエステルに関する。さらに本発明は、生の原料ルイボスから式Iの化合物を単離する方法に関する。さらにまた本発明は、式Iの化合物を少なくとも0.4重量%含有するルイボス抽出物に関する。さらにまた本発明は、式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステル、ならびに本発明のルイボス抽出物の薬剤としての使用に関し、特に中枢神経系の神経学的疾患および精神医学的疾患を治療するための使用に関する。「薬学的に活性な」という表現には、精神状態の主観的改善をもたらす効果も含まれるが、その場合には、薬事法における承認を必ずしも必要とするわけではない。
式Iの化合物を以下に示す。

式Iの化合物を以下に示す。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、式Iで示される新規な化合物ならびにその薬学的に許容される塩、誘導体およびエステルに関する。さらに本発明は、生の原料ルイボスから式Iの化合物を単離する方法に関する。さらにまた本発明は、式Iの化合物を少なくとも0.4重量%、好ましくは少なくとも1.5重量%含有するルイボス抽出物に関する。さらにまた本発明は、式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステル、ならびに本発明のルイボス抽出物の薬剤としての使用に関し、特に中枢神経系の神経学的疾患および精神医学的疾患、特に認知症を予防および治療するための使用に関する。「薬理学的活性のある」という表現には、精神状態の主観的改善をもたらす効果も含まれるが、その場合には、薬事法における承認を必ずしも必要とするわけではない。
【背景技術】
【0002】
ルイボス(学名:Aspalathus linearis)は南アフリカにのみ生育する植物であり、極めて強力な抗酸化物質であるアスパラチン(フラボノイドの1種)を含むものとしては、現在知られている限り、世界で唯一の植物である。ルイボスはまた、C−グリコシルフラボン(オリエンチンおよびイソオリエンチンを含む)、フラボノール−3−O−グリコシド(クエルセチン、クエルシトリン、イソクエルシトリンおよびルチンを含む)ならびにジヒドロカルコンであるノトファギンおよびアスパラチンを含有する。
【0003】
「緑色」の未発酵ルイボスは、その発酵製品と比べて、ポリフェノール類、特にアスパラチンの含有量が高いこと、および抗酸化活性が高いことで際立っている。発酵過程において抗酸化活性が低下するという結果は、紅茶と緑茶とを比較した例(Bramatiら, J. Agric. Food Chem. 2003, 51: 7472−7474)においても同様に認められる。科学的な研究により、ルイボス茶の抗酸化活性が主としてアスパラチン成分に由来するものであることが示されている。ルイボス茶の発酵過程の研究において、アスパラチンとノトファギンの含有量が発酵過程の間に減少することが明らかになった(Schulzら, Eur. Food Res. Technol. 2003, 216: 539−543)。「赤い」発酵ルイボス茶の抗酸化活性が「緑色」の未発酵ルイボス茶と比べて低いことは、これによって説明することができる。
【0004】
上述した健康増進作用のあるフラボノイドを含み、また風味が良いことから、ルイボス茶は広く消費されている。ルイボス茶の成分としてはさらに、フェノール酸、精油、ビタミンCおよび様々なミネラル、特に鉄やフッ化物が含まれている。
【0005】
できる限り高い抗酸化活性を得るためには、高いアスパラチン含有量が必要である。これに関連して、独国特許第102005004438号には、通常1〜3重量%であるアスパラチン含有量が5重量%を超え、クロロフィル含有量が0.4重量%未満と低くなっているルイボス抽出物が開示されている。独国特許第102005004438号によれば、該ルイボス抽出物は、発酵していない生の原料ルイボスをエタノールと水との比率が80対20の混合物で抽出することにより得られるものである。アスパラチン含有量の高いルイボス抽出物は、その強力な抗酸化・抗刺激・抗菌作用により、特に化粧用途、たとえばヘアケア、スキンケア、または口腔衛生のための薬剤としての使用が報告されている。
【0006】
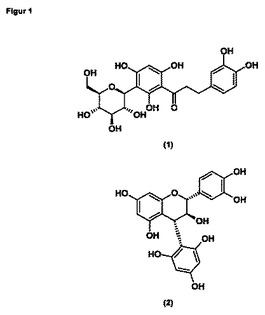
ルイボス茶には、さらなるフラボノイド、クエルセチンが含まれる。これは生の原料ルイボス中に約11mg/100g存在し、たとえばヒトの体内におけるヒスタミン放出に影響を及ぼし、アレルギー症状を緩和する。クエルセチンはさらに、モノアミン酸化酵素の産生を抑制することができ、それによって、軽度のうつ病や睡眠障害に対し、有利な影響を与えることができる(Plant extract, the nature network, issue 3 of 09.11.2005; Plantextrakt GmbH)。
【0007】
本発明の出発点は、中枢神経系の疾患、たとえば認知症、パーキンソン病、うつ病、疼痛等を治療するための活性物質を探索することであった。これらの疾患は治療が困難であり、また使用される薬剤、たとえばタクリン、ガランタミン、ネホパム等には、多岐にわたる副作用がある。
【発明の概要】
【発明が解決しようとする課題】
【0008】
したがって、本発明の目的は、これらの疾患を治療するための活性物質、組成物およびルイボス抽出物を提供することである。特に、これらの活性物質や組成物は副作用の少ないものであることが必要である。
【課題を解決するための手段】
【0009】
この目的は、本発明の請求項の主題によって達成される。
【0010】
本発明は、式Iで示される化合物:
【化1】
ならびにその薬学的に許容される塩、誘導体およびエステルに関する。ここでの好ましい誘導体とは、式Iの化合物とフェルラ酸、キナ酸、カフェ酸、グルコン酸またはクロロゲン酸とのカップリング生成物である。式Iの化合物は、その天然形態で、または塩として使用されることが好ましいが、式Iの天然形態で使用されることがより好ましい。
【0011】
好ましいエステルとして、ギ酸、酢酸、プロピオン酸、グルタル酸、酒石酸、コハク酸等のエステルが挙げられる。好ましい塩としては、カチオンの対イオン、有機または無機の対イオンとの塩、特にアルカリ金属塩、アルカリ土類金属塩、アンモニウム塩、または薬学的に許容される酸との塩、たとえばコハク酸塩、クエン酸塩、酒石酸塩等が挙げられる。
【0012】
さらに本発明は、ルイボス抽出物、好ましくは未発酵ルイボスからの抽出物に関し、該抽出物における式Iの化合物の含有量は少なくとも0.4重量%、好ましくは少なくとも1.5重量%、より好ましくは少なくとも2.5重量%、さらに好ましくは少なくとも5重量%、さらにより好ましくは少なくとも10重量%、最も好ましくは少なくとも20重量%である。
【0013】
さらに本発明は、式Iの化合物の調製方法に関し、該方法では、
−発酵していない生の原料ルイボスを乾燥および粉砕した後、メタノールおよび/または水からなる抽出剤を用いて90℃以下、好ましくは60℃以下の温度で所定の時間抽出し、
−得られた抽出物をろ過した後、減圧下で蒸発乾固させ、次いで
−複数のクロマトグラフ分離ステップ、好ましくは、2本のセファデックスLH20カラムおよび次いで1本の疎水性c18−HPLCカラムを使用した3ステップのクロマトグラフ分離により精製する。
【0014】
本発明はさらに、薬剤または補助食品としてのルイボス抽出物の使用に関する。本発明はさらにまた、薬剤または補助食品としての、式Iの化合物、ならびにその薬学的に許容される塩、誘導体およびそのエステルの使用に関する。前記補助食品または薬剤中、前記ルイボス抽出物の含有量は、たとえば補助食品1g当たりまたは薬剤の用量単位当たり少なくとも10mg、少なくとも20mgまたは少なくとも50mgであってよい。好ましくは、前記補助食品または薬剤は、補助食品1g当たりまたは薬剤の用量単位当たり少なくとも100mg、より好ましくは少なくとも200mg、さらに好ましくは少なくとも300mgのルイボス抽出物を含む。さらに好ましい一実施形態では、前記補助食品または薬剤は、補助食品1g当たりまたは薬剤の用量単位当たり少なくとも1mg、より好ましくは少なくとも2mg、さらに好ましくは少なくとも3mg、さらに好ましくは少なくとも5mg、最も好ましくは少なくとも10mgの式Iの化合物を含む。
【発明を実施するための形態】
【0015】
本発明の好ましい一実施形態では、前記ルイボス抽出物ならびに、式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステルは、中枢神経系の神経学的疾患および精神医学的疾患を予防または治療するための薬剤として、好ましくは認知症、パーキンソン病、うつ病、疼痛等、特にアルツハイマー病を予防または治療するための薬剤として、式Iの化合物の割合をより多くして使用される。
【0016】
さらにより好ましい一実施形態では、本発明は、ルイボス抽出物ならびに、式Iの化合物ならびにその薬学的に許容される塩およびエステルの、中枢神経系の神経学的疾患および精神医学的疾患を予防および/または治療するための薬剤を調製するための使用に関し、ここで中枢神経系の神経学的疾患および精神医学的疾患は認知症、パーキンソン病、うつ病、疼痛等であり、より好ましくはアルツハイマー病である。
【0017】
本発明の範囲において、薬理学的活性のある新規の天然物質をルイボス抽出物から単離し、次いで特性決定を行うことができた。意外なことに、前記の新規化合物の単離は、「緑色」の未発酵ルイボス抽出物からのみ可能であった。該化合物の特性を完全に解析した結果、発酵したルイボス抽出物に含まれる微量の該化合物についても、検出が可能となった。
【0018】
該新規な化合物を単離する方法は、実施例1において詳細に記載するが、その構造式は式Iで示される。式Iの化合物は、アスパラチンの構造ともカテキン(4α→2)フロログルシノールの構造とも類似性を有する(図1)。正確な特性決定および構造解析については、実施例2で詳細に記載する。これまでに知られているフラボノイドと比較して、式Iで示される新規化合物の分子量は大きく、740.66g/molである。このような分子量の大きい天然物質は、活性物質のスクリーニングには通常使用されない。その分子量ゆえに、血液脳関門を容易には通過できないからである。したがって、このような物質は、脳で作用するには、また中枢神経系の疾患を治療するには、不適当であると見なされる傾向がある。
【0019】
しかし、意外なことに、先入観を持たずにラットの遠隔ステレオEEG(脳波)検査を行った結果、式Iの化合物が中枢神経において顕著な薬理学的活性を示すことがわかった。この薬理学的検査の結果、驚くべきことに、ラットの遠隔ステレオEEGモデルにおける該活性は、EEG周波数の用量依存的な変化をもたらし、これは認知症、パーキンソン病および疼痛を治療するための古典的な治療薬(認知症にはたとえばガランタミンまたはタクリン、パーキンソン病にはL−DOPA、また疼痛用にはたとえばネホパム)を投与した後に見られる既知の作用と同様であった。
【0020】
本発明の範囲において、式Iで示す進歩的化合物の薬理学的活性を、ルイボス抽出物の既知の成分、たとえばアスパラチン、カテキン、または(−)−エピカテキンと比較した。実験的研究の詳細は実施例3に記載するが、意外なことに、式Iの化合物がアスパラチン、カテキン、(−)−エピカテキン等と構造的類似性を有するにもかかわらず、その作用は、ほぼ等モル量のこれら天然物質では達成し得ないものであることが示された。式Iで示す新規かつ進歩的な化合物は、これら既知のルイボス抽出物成分よりも高い薬理学的活性を有し、したがって、薬剤としての使用に特に適している。
【0021】
有利な薬理学的活性を有するこの新規な化合物の単離により、特に式Iで示す化合物に基づく薬剤または補助食品の調製(単体調製物としても、また他の活性物質と組み合わせた調製物としても)が可能となる。このようなさらなる活性物質は、同方向に作用するものでもよいが、また別の方法で臨床像に有利な影響を及ぼすようなまったく異なる特性を有するものでもよい。この化合物をルイボス中に存在する他のフラボノイドおよび他の成分とあらゆる組み合わせで組み合わせることも、調製の目的に適している。特に、ここでは、抗酸化作用を有する成分が適している。
【0022】
上述したように、式Iの化合物のin vivoデータの識別分析により、認知症、パーキンソン病、うつ病および疼痛を治療するための薬剤との関連が示された。これらの薬剤には広範な副作用があるが、天然の物質による副作用の頻度は通常低いものと考えられることから、式Iの化合物を薬剤として使用することは有利である。さらに、意外なことに、この新規な物質またはその代謝物質は、血液脳関門を通過することが明らかになっている。これは、一般に、フラボノイドとしてはまれなことである。
【0023】
さらにまた、ルイボス抽出物および新たに同定された薬理学的活性のある化合物の調製方法が提供される。本発明の方法により、上記の疾患の治療に特に適した植物抽出物の提供が可能になる。
【0024】
本発明によって調製されたルイボス抽出物は、式Iの化合物を少なくとも1重量%、より好ましくは少なくとも1.5重量%、さらにより好ましくは少なくとも2重量%、より好ましくは少なくとも2.5重量%、さらにより好ましくは少なくとも3重量%、最も好ましくは少なくとも5重量%含有している。
【0025】
非常に好ましい一実施形態では、高濃縮されたルイボス抽出物が使用される。高濃縮されたルイボス抽出物は、式Iの化合物を少なくとも10重量%、好ましくは少なくとも20重量%含む。特定の実施形態では、該高濃縮されたルイボス抽出物は、式Iの化合物を少なくとも50重量%含む。
【0026】
式Iの化合物を調製するための本発明による方法は、以下のステップを含む(実施例1も参照のこと)。
−乾燥および粉砕した、発酵していない生の原料ルイボスの提供
−提供された生の原料の、アルコール、好ましくはメタノールおよび/または水の混合物からなる抽出剤を用いた、90℃以下、好ましくは60℃以下の温度における所定の時間の抽出
−得られた抽出物のろ過
−ろ過した抽出物の減圧下における蒸発
−抽出物の3段階の精製:
−セファデックスLH20カラムクロマトグラフィーによる粗精製
−さらなるセファデックスLH20カラムクロマトグラフィーによる高精製
−疎水性c18−HPLCカラムによる分離
【0027】
特に好ましい一実施形態では、本発明の方法による抽出ステップにおいて、0.001〜1重量%、好ましくは0.01〜0.2重量%のアスコルビン酸を加える。この重量パーセントは抽出対象の薬種、すなわち乾燥した葉や茎のような植物部位の重量に基づくものである。
【0028】
式Iの化合物を多く含有する調製物を得ようとする場合、1本のクロマトグラフィーカラムのみを使用して抽出物を分離することもできる。
【0029】
提供される生のルイボス原料の水分は4%以下であることが好ましい。この条件であれば、出発原料の自己発酵が防止できるからである。
【0030】
式Iの化合物を高い収率で得るために、本発明の方法の好ましい構成における抽出剤として、アルコール/水の50:50〜80:20(比率はアルコールの種類に依存し、アルコールとしてはメタノール、エタノール、プロパノール、プロパン−2−オールが適している)混合物を使用する。好ましくは、メタノール/水の50:50混合物を使用する。本発明の方法によるこの好ましい実施形態では、原料対抽出剤の比率は好ましくは約1:6であり、また抽出ステップは、好ましくは高温(40℃以上)において1時間で達成されるが、室温で、たとえば2〜5時間かけて行うことも可能である。
【0031】
ろ過した抽出物の蒸発は、好ましくは300mbar未満の圧力下で実施される。蒸発ステップの温度は、40℃以下であることが好ましい。
【0032】
本発明によるルイボス抽出物は、上述の方法によって調製され、蒸発乾固の後に(その後のクロマトグラフィーによる精製を行うことなく)得られる。
【0033】
該ルイボス抽出物中の式Iで示す化合物の含有量を測定する方法は、実施例4で詳細に記載する。
【0034】
式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステル、ならびに本発明によるルイボス抽出物は、特に、中枢神経系の疾患、好ましくは認知症、パーキンソン病、うつ病、疼痛を治療するために、また細胞を保護する抗酸化剤または「遊離基捕捉剤」として適切である。老年性認知症等の認知症、またはアルツハイマー病の治療には、特に好適である。
【0035】
本発明によれば、該新規な化合物は、式Iの形態で、または薬学的に許容される塩もしくは複合体およびエステルもしくは誘導体として、単一の活性物質として、またはさらなる活性物質と組み合わせて使用することができる。好適な誘導体、塩、複合体およびエステル、ならびにそれらの調製については、当業者には公知である。薬学的に許容される塩(塩酸塩、コハク酸塩、クエン酸塩、酒石酸塩等)の調製も、同様に当業者には公知である。塩を形成する好適な物質は、慣習的に薬学的に許容されるすべての酸またはアニオンである。さらに、たとえばフェルラ酸、キナ酸、カフェ酸、グルコン酸、クロロゲン酸およびその関連化合物とのカップリングも可能である。特に、グルコン酸とのカップリングが好ましい。式Iの化合物と上記の酸とのカップリング生成物は、薬学的に許容される誘導体として認定されている。しかしながら、該化合物は、式Iの分子として使用されることが好ましい。
【0036】
また別の、同様に好ましい一実施形態では、該抽出物は、やや親水性の高い混合溶媒を使用して調製される。この溶媒混合液は、好ましくはメタノール、エタノール、n−プロパノール、2−プロパノールから選択される少なくとも1種のアルコールと水とを含む。そのアルコール含有量は、10〜50重量%である。この溶媒混合液を選択すると、これよりもアルコールの割合が高い溶媒混合液を使用する場合と比べて、総フラボノイドの割合が高くなる。この実施形態では、アルコールの含有量は好ましくは10〜60%(vol/vol)、好ましくは10〜50%(vol/vol)である。さらにより好ましくは、アルコールの含有量は15〜40%であり、特に好ましくは20〜30%(vol/vol)である。
【0037】
本発明の方法におけるこの実施形態では、使用する出発物質としての薬種は、残留する水分量ができる限り低い「緑色」のルイボスであり、この水分量は好ましくは5%未満(総重量当たりの水分)である。出発原料、すなわちAspalathus linearis(未発酵)の葉および若枝は、温和な条件下で著しく急速に乾燥され、次いでアルコールと水との混合物(水の割合が60%(vol/vol)より高い)で抽出される。特に好ましい実施形態では、アルコール含有量は、メタノールの場合15〜25%、非常に好ましくは20%、またエタノールの場合には25〜35%、特に好ましくは30%である。
【0038】
非常に好ましい一実施形態では、まず、乾燥した出発物質としての薬種に純粋なアルコールを加え、通常30〜60分間かかる軟化段階の後、相当する分量の水を加える。抽出後、抽出物をろ過し、ろ液を減圧下で蒸発乾固させる。この後、好ましくはクロマトグラフィーによる精製ステップが続くが、この精製は好ましくはセファデックスカラムクロマトグラフィーを使用して実施される。
【0039】
この実施形態では、式Iの化合物は実質的に完全に抽出され、同時に、総フラボノイドが高い割合で抽出される。この(クロマトグラフィー法によってさらに精製される前の)抽出物中の式Iの化合物の含有量は、乾燥抽出物の重量に対して2.5〜5%である。式Iの化合物を除く、アスパラチン類、ルトシド類およびビテキシン類の総和としてのフラボノイド総量は、乾燥抽出物に対して15〜30重量%である。ビテキシン類(=C−グリコシド)のルトシド類に対する比率は1.6である。アスパラチン類の含有量は、乾燥抽出物の重量に対して14〜25%である。
【0040】
この方法で抽出を行うと、抽出物中の総フラボノイド含有量が多くなる。通常、このような(カラムクロマトグラフィーによるさらなる精製を行わない)抽出物は、総フラボノイドを少なくとも約20重量%含有する。以下の主要成分は、抽出物中で同定され得るものである。
a)式Iの化合物、
b)カルコン(アスパラチンおよびノトファギン)からなる物質群、
c)ルトシド群(成分としてクエルセチン(すなわちルチンのアグリコン)を含有するフラボノイド、C−O−C結合した糖を含むフラボノイドを含む)、ならびに
d)ビテキシン群(C−C結合した糖を含むフラボノイド。たとえば、アグリコンがアピゲニンであるビテキシン(アピゲニン−8−C−グルコシド)、イソビテキシン(アピゲニン−6−C−グルコシド)、オリエンチン(ルテオリン−8−C−グルコシド)、イソオリエンチン(ルテオリン−6−C−グルコシド)等が挙げられる。)
【0041】
このルイボス抽出物の特質は、特に、それぞれの群と他群との重量比にある。重量割合に基づくルトシド群とビテキシン群との比率は1:2〜1:50、好ましくは1:3〜1:10である。
【0042】
本発明による抽出物は、茶飲料のような純粋な水による抽出で得られる内用の抽出物とは、言及した化合物の含有量がより高いという点で異なっている。本発明による抽出物は、たとえば化粧品のような外用を意図した抽出物とは、フラボノイド群の互いに対する比率がそれぞれ異なるという点で異なっている。外用を意図した抽出物は、エタノールの含有量を高く(エタノールが少なくとも約80%)して抽出を行うことによって得られる。80%のエタノールで得られた抽出物は、アスパラチン含有量をできる限り高くするために最適化したものである。3つのフラボノイド群(アスパラチン様、ルトシド様、およびビテキシン様すなわちC−グリコシド)の比率は、比較的疎水性の高い抽出を行ったことにより、該薬種の出発状態における比率から変化している。これは、ビテキシン様フラボノイドのルトシド様フラボノイドに対する比率を見れば特に明白である。
【0043】
本発明によるルイボス抽出物ならびに式Iの進歩的な化合物またはその塩、誘導体およびエステル、好ましくは式Iの天然に存在する化合物は、健康増進作用を有する薬剤および/または補助食品を製造するための自体公知の方法で加工することができる。本発明によるルイボス抽出物ならびに式Iの進歩的な化合物ならびにその塩、誘導体およびエステルは、たとえばタブレット、カプセル、錠剤、コーティング錠、顆粒、粉末、ロゼンジの形態、および飲料のような液体の投与形態で処方することができる。補助食品、特に熱い飲料および冷たい飲料、または可溶茶のための使用も好ましい。
【0044】
該補助食品および薬剤は、経口、局所的、非経口、静脈内、筋肉内、皮下、経鼻、吸入、直腸、または経皮により好適に投与され得る。
【0045】
本発明のルイボス抽出物および本発明の薬剤または補助食品はまた、ルイボス抽出物または式Iの進歩的な化合物もしくはその塩の効果を高めたり、前記疾患においてその機構または状態に補助的な良い影響を与えたりするさらなる活性物質(たとえば、さらなる遊離基捕捉剤、様々な酵素阻害物質、ビタミン類、レシチン、オメガ−3−脂肪酸および脳の機能に良い影響を与える物質)を含んでいてもよい。
【0046】
さらに、薬学的に許容される賦形剤および担体を使用することができる。適切な賦形剤は当業者に公知であり、たとえば、充填剤、崩壊剤、滑沢剤、結合剤、湿潤剤等が含まれる。
【0047】
適切な滑沢剤は、たとえばケイ酸塩、滑石、ステアリン酸、ステアリン酸マグネシウムもしくはステアリン酸カルシウムおよび/またはポリエチレングリコールである。
【0048】
使用してもよい結合剤は、たとえばデンプン、アラビアゴム、ゼラチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルピロリドンである。
【0049】
崩壊剤は、たとえばデンプン、アルギン酸、アルギン酸塩、デンプングリコール酸ナトリウムまたは発泡混合物である。
【0050】
使用してもよい湿潤剤は、たとえばレシチン、ポリソルベートまたはラウリル硫酸塩である。
【0051】
さらに、着色剤および甘味料もまた、製剤中に存在してもよい。
【0052】
これらの医薬製剤は、公知の方法、たとえば混合、造粒、タブレット化または糖衣もしくはオーバーコートによって調製することができる。
【0053】
経口投与用の分散液および/または溶液は、たとえば飲料、ドロップ剤、シロップ剤、乳剤および懸濁剤であってよい。
【0054】
シロップ剤は、たとえばショ糖を含んでいてもよく、またショ糖とともに担体としてのグリセリン、マンニトールおよび/もしくはソルビトールを含んでいてもよい。
【0055】
懸濁剤および乳剤は、担体としてたとえば、天然樹脂、寒天、アルギン酸ナトリウム、ペクチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルアルコールを含んでいてもよい。
【0056】
筋肉注射用の懸濁剤または液剤は、該活性物質とともに、薬学的に許容される担体、たとえば滅菌水、オリーブオイル、オレイン酸エチル、たとえばプロピレングリコール等のグリコール類、および所望により適当量の塩酸リドカインを含んでいてもよい。
【0057】
静脈注射または点滴のための液剤は、たとえば担体として滅菌水を含んでいてもよく、また好ましくは滅菌水性等張塩溶液の形態であってもよい。
【0058】
坐剤は、該活性物質とともに、薬学的に許容される担体、たとえばカカオバター、ポリエチレングリコール、ポリオキシエチレンソルビトール脂肪酸エステルまたはレシチンを含んでいてもよい。
【0059】
局所適用用組成物、たとえばクリーム、ローションまたはペーストは、活性物質と従来の含油担体または乳化担体とを混合することによって調製することができる。
【0060】
本発明によるルイボス抽出物ならびに式Iの進歩的な化合物またはその塩、誘導体およびエステルは、補助食品または薬剤に通常含まれる量で使用することができる。溶液中、式Iの進歩的な化合物またはその塩は、好ましくは0.001〜10重量%、より好ましくは0.1〜7重量%、特に好ましくは1〜5重量%で使用される。特定の一実施形態では、式Iの進歩的な化合物またはその塩は、溶液中0.02〜1重量%で使用される。
【0061】
本発明によるルイボス抽出物は、式Iの化合物の1〜1000mg、より好ましくは10〜600mg、さらに好ましくは50〜400mg、最も好ましくは50〜250mgに相当する量で使用される。
【0062】
式Iの化合物を非常に高い割合で含むルイボス抽出物を使用する場合、このような抽出物は、薬剤および補助食品において、1日当たり3〜600mg、好ましくは5〜100mg、特に好ましくは10〜50mgの用量で使用することができる。
【0063】
上述の補助食品または薬剤は、従来の方法によって調製し、薬学的に好ましい形態で投与することができる。
【0064】
本発明による好ましい固形の補助食品または薬剤は、1〜95重量%、好ましくは1〜50重量%、より好ましくは1〜20重量%、特に好ましくは1〜10重量%の充填剤をさらに含んでいてもよい。
【0065】
使用してもよい充填剤は、タブレットまたはカプセルが必要かつ所望の重量となるように原料の一部をなす1以上の化合物である。中でも、様々な粒径の微結晶性セルロース、特に平均粒子径が20μmから200μmの範囲、特に50μmから150μmの範囲にある、たとえばAvicel PH−101およびPH−102等の公知のAvicel製品のように約100μmであるものを使用することができる。さらなる好適な充填剤は、たとえばトウモロコシデンプン、ジャガイモデンプン、ラクトース、セラクトース(セルロースとラクトースとの混合物)、リン酸カルシウム、ブドウ糖、マンニトール、マルトデキストリン、イソマルト、二酸化ケイ素(Aerosil)、さらに場合によってはソルビトールおよびショ糖である。直接圧縮が意図される場合、充填剤としては、タブレットの直接圧縮に適した品質のものを選択する必要がある。市販品の場合、このような品質は製品ごとにメーカーにより明示されるものであるが、簡単な実験によって確認することもできる。最も好ましい充填剤は、微結晶性セルロース(市販品としては、たとえばAvicel、VivapurおよびEmcocel等)である。
【0066】
適切な崩壊剤は従来技術において公知である。本発明による好ましい崩壊剤は、たとえば、クロスポビドン(Kollidon CL)およびデンプンまたはあらかじめゼラチン化したデンプン、特に市販品の「Starch1500」である。さらなる好適なデンプンも市販品として入手可能であり、たとえばLycatab PGS、PrejelおよびSepistab ST 200の名称で販売されている。
【0067】
さらに、公知のいわゆる「超崩壊剤」も使用することができ、たとえばクロスカルメロースナトリウム(たとえばAc−Di−Sol等)およびカルボキシメチルデンプンナトリウム(たとえばExplotab、Primojel等)が挙げられる。Starch1500等のデンプンが特に好ましい。
【0068】
崩壊剤の含有量は、通常1〜25重量%、好ましくは1〜20重量%、特に2〜15重量%である。崩壊剤の含有量として適切な範囲はまた、たとえば2〜5重量%または15〜20重量%であり、使用する崩壊剤、充填剤および他の添加剤に依存する。
【0069】
本発明によれば、該組成物は、滑沢剤として、タブレットの調製および加工を補助する1以上の化合物を含んでいてもよい。使用してもよい滑沢剤としては、特にステアリン酸およびその誘導体、たとえばステアリン酸カルシウム、特にフマル酸ステアリルナトリウム(たとえば商品名Pruv等の市販品が入手可能)およびステアリン酸マグネシウム、モノ−、ジ−、特にトリ−ステアリン酸グリセリル、硬化植物油(たとえばLubritab、Dynasan、Sterotext)またはポリエチレングリコール(たとえばLutrol、Carbowax)が挙げられる。
【0070】
滑沢剤の含有量は、通常0.1〜4重量%、好ましくは0.2〜4重量である。
【0071】
本発明による医薬組成物は、場合により、1以上の流動調節剤を含んでいてもよい。適切な流動調節剤は、三ケイ酸マグネシウム、滑石および特に二酸化ケイ素(たとえばAerosil)である。該組成物が流動調節剤を含む場合、この流動調節剤は通常0.5〜5重量%、好ましくは1〜4重量%、特に好ましくは2〜3重量%含まれる。
【0072】
本発明による医薬組成物はさらに、活性物質のための安定化剤を含んでいてもよく、該安定化剤としてはアスコルビン酸、クエン酸、酒石酸、乳酸等が挙げられ、アスコルビン酸またはクエン酸が好ましい。安定化剤(使用するとすれば)の含有量は、通常0.1〜10重量%、0.5〜10重量%、好ましくは1〜3重量%である。
【0073】
本発明による医薬組成物はさらに、薬学的に許容される通常の添加剤および賦形剤を含んでいてもよいが、上述のもの(充填剤、崩壊剤、滑沢剤、ならびに場合により流動調節剤および安定化剤)以外の賦形剤を含まないことが好ましい。
【0074】
充填剤には、微結晶性セルロースのように、結合剤としても機能するものがある。したがって、本発明においては、結合剤としての機能を有する充填剤は、充填剤と見なすこととする。
【0075】
本発明による医薬組成物がタブレットである場合、1以上のコーティング剤でフィルムコートされていてもよい。使用してもよいコーティング剤は、セラックおよびセラックの混合物、ヒプロメロース(ヒドロキシプロピルメチルセルロース)、ポリビニルアルコール、カルボキシメチルセルロースナトリウムならびに種々のメタクリル酸重合体(オイドラジット)であり、ヒプロメロース、オイドラジット、セラックおよびセラックの混合物が好ましい。タブレットのコーティングは通常の方法で達成される。コーティング剤に加えて、タブレットのコーティングに通常使用される成分、たとえば可塑剤、着色料、孔形成剤または懸濁安定化剤がコーティング中に存在してもよく、たとえばポリエチレングリコール(PEG)、滑石、二酸化チタン、また場合によってはラクトース等が使用される。
【0076】
タブレットの重量は特に制限されないが、純粋な活性物質を使用する場合は100mg〜500mg、抽出物、植物粉末を使用する場合は250mg〜1500mg、500mg〜1500mgのタブレットが一般的である。カプセルの場合、100mg〜1000mgのものが使用される。
【0077】
薬剤または補助食品の用量単位としては、たとえば、以下のようにしてもよい。
・経口剤形の場合には、
1日用量当たり、好ましくは1〜1000mg、より好ましくは40〜800mg、特に好ましくは150〜500mg、さらに好ましくは300〜600mgのルイボス抽出物。式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステルの場合、上記の量の1/100〜1/20を使用する。1日量は毎日投与することができ、たとえば1〜3回、好ましくは2回に分けて投与される。また、式Iの化合物を含むルイボス抽出物を1日に1〜10回投与することも想定可能である。
・非経口(たとえば静脈内、皮下、筋肉内)剤形の場合には、
1日用量当たり、好ましくは3〜60mg、特に好ましくは10〜30mgの活性物質。1日量は毎日投与することができ、たとえば1〜3回に分けて、好ましくは単回で投与される。
・直腸に適用する剤形の場合には、
1日用量当たりの該抽出物中、式Iの活性物質は、好ましくは40〜80mg、特に好ましくは60mg。1日量は毎日投与することができ、たとえば1〜3回に分けて、好ましくは単回で投与される。
・皮膚または粘膜に適用する剤形(たとえば液剤、ローション、乳剤、軟膏等)の場合には、
1回当たり、好ましくは40〜80mg、特に好ましくは60mgの活性物質。調製した液剤、ローション、乳剤または軟膏に対する式Iの化合物の含有量は、このような軟膏状薬剤、クリーム状調製物に対する重量で0.05〜20重量%、好ましくは0.2〜1重量%である。1日量は毎日投与することができ、たとえば1〜6回、好ましくは1〜3回に分けて投与される。
薬学的に許容される塩の使用において、その使用量は、当業者に公知の方法で適切な量とする必要がある。
【図面の簡単な説明】
【0078】
【図1】アスパラチン(1)およびカテキン(4α→2)フロログルシノール(2)の構造式を示す。
【図2】式Iの化合物中の原子に付された番号を示す。
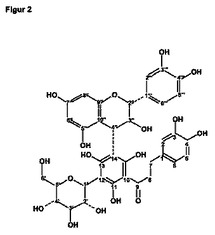
【図3】図2による番号を付した各原子に対するNMR(核磁気共鳴)シグナルの帰属を示す。 符号の説明: *)/**)/+)/++)/§)/§§)/#)##)=値は相互に入れ替え可能 A),B)=1度だけ示した基の積分値または化学シフトの和。
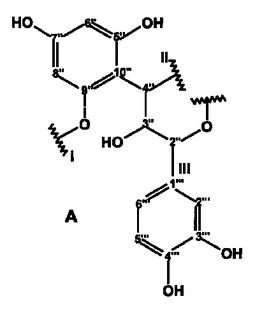
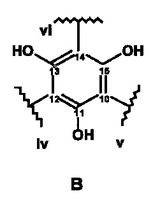
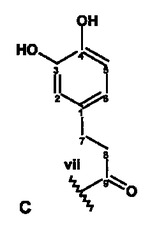
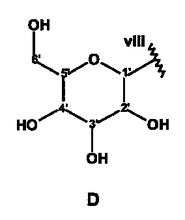
【図4】NMR実験によって特徴付けられた、式Iの化合物の構造フラグメントA〜Dを示す。
【図5】式Iの化合物の13C−NMRデータをアスパラチンおよびカテキン(4α→2)フロログルシノールのデータと比較した表2を示す。アスパラチンについては13C−NMRの文献データがないため、HWI ANALYTIK GMBHによって単離されたアスパラチンに対するシグナルの帰属は、 Ho et al., Phytochemistry 1980, 19, 476−477のデータに基づくものである。カテキン(4α→2)フロログルシノールについては、Matthews et al., J. Agric. Food. Chem. 1997, 45, 1195−1201のデータを使用した。 符号の説明: *)/**)/+)/++)/§)/§§)/#)##)=値は相互に入れ替え可能
【図6】式Iの化合物を投与した後のFischer−344ラットに食塩水を投与した際の実験条件が一定であった証拠を示す。投与後の時間(時間)をy軸に示す。x軸はデータを高速フーリエ変換した後の周波数範囲:δ、θ、α1、α2、β1およびβ2を示す。
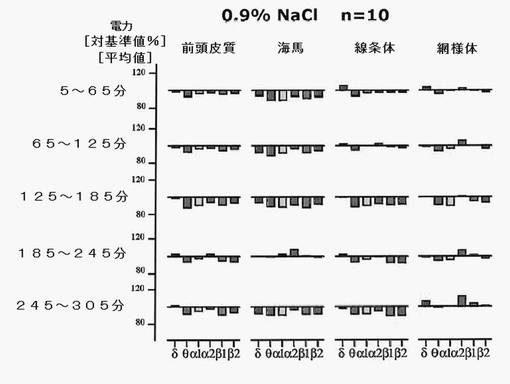
【図7】式Iの化合物3mg/kgを投与したことによるEEG周波数に対する効果を、投与後5時間まで示す。値は平均値である(n=6)。アステリスクは、AhrensおよびLauter(1974)の方法で計算された統計的有意性を表す。 *=p<0.01;**=p<0.05;***=p<0.025
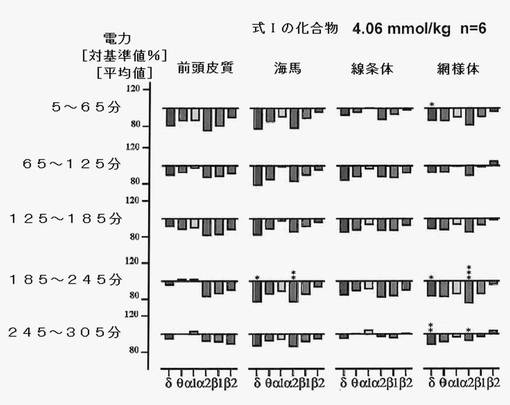
【図8】式Iの化合物6mg/kgを投与したことによるEEG周波数に対する効果を、投与後5時間まで示す。値は平均値である(n=6)。アステリスクは、AhrensおよびLauter(1974)の方法で計算された統計的有意性を表す。 *=p<0.01;**=p<0.05;***=p<0.025
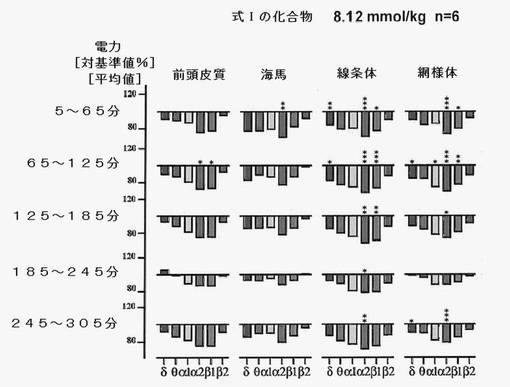
【図9】式Iの化合物の影響を、その成分分子であるアスパラチン、カテキンまたはエピカテキンと比較して示す。ほぼ等モルを投与した場合、特異構造による効果は式Iの化合物によってのみ達成され、この分子の各部分からは得られない。
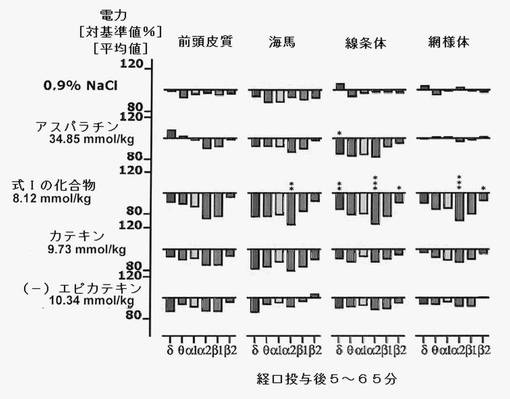
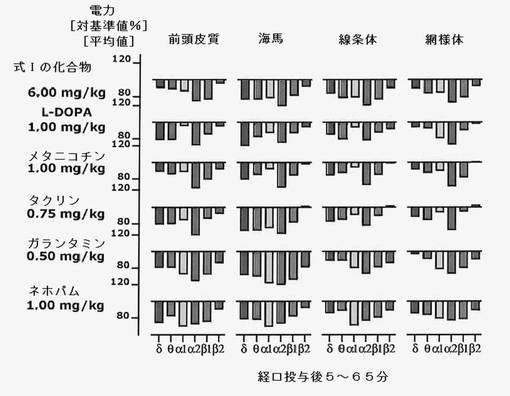
【図10】式Iの化合物6mg/kgを投与したことによる、投与後1時間のEEG周波数に対する効果を示す。値は平均値である(n=6)。認知症、パーキンソン病、うつ病および疼痛を治療するために使用される薬剤:L−DOPA(パーキンソン病)、タクリンおよびガランタミン(アルツハイマー病)、ネホパム(疼痛治療)との類似性に注目すべきである。
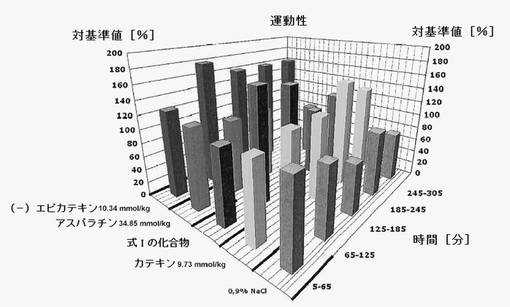
【図11】電場電位と同時に実施した運動性測定の結果を示す。データは、各物質投与前の開始値に対する%で示したものである。運動性を実質的に増加させるのは、(−)−エピカテキンのみである。
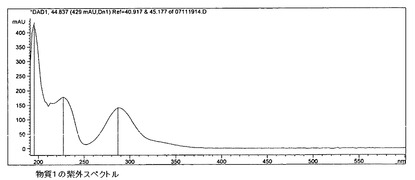
【図12】式Iの化合物(G110907SA)の紫外スペクトルを示す。
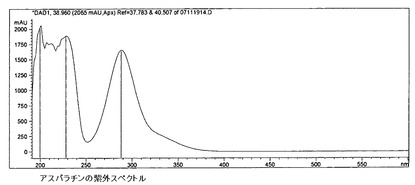
【図13】アスパラチンの紫外スペクトルを示す。
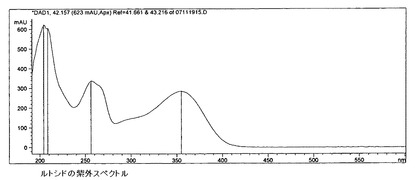
【図14】ルトシドの紫外スペクトルを示す。
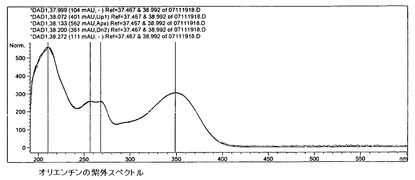
【図15】オリエンチンの紫外スペクトルを示す。
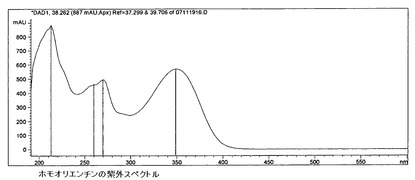
【図16】ホモオリエンチンの紫外スペクトルを示す。
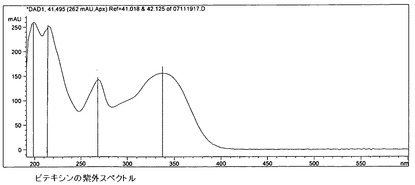
【図17】ビテキシンの紫外スペクトルを示す。
【実施例】
【0079】
本発明は、以下の実施例によって、より詳細に説明される。
【0080】
<実施例1> 式Iの化合物の単離
薬種からの抽出物を調製するために、製造業者であるルイボス社(南アフリカ、Clanwilliam)から入手した未発酵の緑のルイボスを使用する。
【0081】
発酵しないまま粉砕したAspalathus linearisの葉および/または若枝を温和な条件下で水分量が10%未満(好ましくは4%未満)になるまで乾燥し、出発原料として使用する。
【0082】
この原料の抽出を、メタノールと水との50:50(容量部)混合液を用い、回転させつつ60℃にて1時間かけて行うが、このとき原料と溶媒の比が1:7となるようにする。その後、ろ過により液体を植物部位から分離し、この植物部位を同じ方法で再度抽出し、ろ過する。
【0083】
2つのろ液を合わせ、減圧下(220mbar)55℃でメタノールを除去する。残った水溶液を、使用した乾燥植物部位の重量の5倍まで水で希釈し、液液分配に供する。
【0084】
この水溶液3Lに対し、1.5Lの水飽和したn−ブタノールを加えて振とうする操作を4回行い、ブタノール相を合わせて減圧下で乾燥させる。その収率は、使用した乾燥植物部位の量の約10%となる。
【0085】
このブタノール抽出物に対し、まず粗分離、次いで高分離を行う。
【0086】
粗分離:
約50gのブタノール抽出物を、50容量%のメタノールを用いて、セファデックスLH20カラム充填クロマトグラフィー(内径6cm、高さ80cm、2260mlのセファデックスLH20を充填)によって分離する。この目的のために、使用する50gのブタノール抽出物は、400mlの移動相に溶解した後、分離カラムに加える。カラムは、3Lの溶出液が滴下するまで、1.8mL/分の流速で洗浄する。残っている移動相が完全に滴下した後、カラムの内容物を取り出し、これを3Lの100%メタノール中で10分間十分に攪拌(抽出)する。固定相はろ過により除去し、溶出液を乾燥させる。残留物は、使用した植物部位の量の約0.5〜1%となる(=メタノール抽出物)。
【0087】
高分離1回目:
約4gのメタノール抽出物を、セファデックスLH20カラム(内径3.5cm、高さ50cm、480mlのセファデックスLH20を充填)を用いて、80容量%のメタノールでクロマトグラフィーによって分離する。この目的のために、使用する4gのブタノール抽出物は、40mlの移動相に溶解した後、分離カラムに加える。2.4mL/minの流速でカラムを操作し、10分毎の分画(=24mlの溶出液)を回収する。目的の物質は、分画48〜65に含まれる。メタノール抽出物が4gの場合、収量は約0.5〜1gとなる。
【0088】
高分離2回目:
分離カラム:250×30mm
固定相:Reprosil C18 Aqua 10μm
移動相:35%(v/v)メタノール
流速:1.5mL/分
分画サイズ:10分=15mL
物質Iは、分画51〜52に含まれる。
分画41〜52を合わせ、凍結乾燥する。
4gのメタノール抽出物から約125mgの物質Iが得られる。
クロマトグラフィーによる純度:約97%(HPLC)。
【0089】
<実施例2> 式Iの化合物の構造解析
カラムクロマトグラフィーによる分離で得た化合物について、以下に挙げる装置を使用して特性決定を行ったところ、以下に記載する測定値が得られた。
【0090】
図2に、式Iの進歩的な化合物中の原子に付された番号を示す。図3の表1に、NMRデータの帰属を示す。
【0091】
NMRデータは、以下に記載する通りに測定し、解釈した。
【0092】
1H−NMR(d6−DMSO(ジメチルスルホキシド))
DMSO中の該物質の1H−NMRスペクトルでは、重水を加えた結果、9つのフェノールプロトンおよび5つの脂肪族HOプロトンの交換がシグナル減衰(H/D交換)によって識別できた。さらに、5.5〜8.0ppmというオレフィンの範囲で8つのプロトン、また−CHxOH/−CHxOR基(x=1または2)に典型的なシフト範囲内で9つのプロトンが現れている。2つの脂肪族プロトンが2.7ppm前後において識別可能であり、またさらなる脂肪族のシグナル(3つのプロトン)が−CHxOH/−CHxORのシグナルとオーバーラップしている。シグナルには、二重線になったり、ブロードになったりしているものもある。そのため、個々の積分値は名目上異性体の1つにのみ相当するが、2つの異性体が統合されているものもある。
【0093】
13C−NMR
該物質の13C−NMRスペクトルでは、ほとんどの炭素原子のシグナルが二重線になっている。2つの異性体の比率は使用する溶媒によってわずかに変化する。25〜90ppmの範囲に脂肪族炭素原子の11のシグナルが、またオレフィン炭素原子に典型的な90〜165ppmの範囲には24のシグナル群が現れる。205ppmに見られるさらなるシグナルは溶媒とオーバーラップするが、d6−DMSO中でははっきりと識別できる。このオーバーラップのために、すべての実験をさらに二次元で、d6−DMSO中で実施した。
【0094】
DEPT(Distortionless Enhancement by Polarization Transfer)スペクトルの測定は、d6−アセトン中で実施した。DEPTスペクトルの65〜85ppmの範囲には、3つのメチレン基のみならず、脂肪族メタン基と7つの−CHOH/−CHORシグナルが存在する。95〜125ppmにはさらに8つのシグナルがあり、オレフィンのCH基に帰属される。13C−NMRシグナルとの照合により、17個の第4級炭素原子の存在が推定される。
【0095】
H/H相関(COSY(相関分光法))
相関スペクトルで評価できるシグナルはごく少数であるが、これは、相互作用の大部分が3.1〜3.5ppmの範囲で強くオーバーラップすることによる。4.0〜4.4ppmの範囲において、アルキル鎖(H7(2.7ppm)〜H8(3.3ppm))の相関と同様に、シグナル群H2”〜H4”が明確に識別できる。4.7/4.8ppmのH1’からは、3.1〜3.5ppmの範囲において1つの相関のみ識別できる。
【0096】
C/H相関スペクトル(HMQC(Heteronuclear Multiple Quantum Correlation)、HMBC(Heteronuclear Multiple Bond Correlation))
第4級炭素原子に隣接するプロトンおよび個々の構造エレメントの結合は、CH長距離相関スペクトル(HMBC)から推定できる。直接相関の帰属により、構造体の帰属の妥当性を確認することもできる。
【0097】
得られたデータの評価および文献値との比較
6.8〜6.4ppmの範囲におけるプロトンのカップリングパターンから、1,3,4位に置換基を有する非常によく似た2つの芳香族系の存在が明確に示唆される。HMBCスペクトルのカップリング、および関与する炭素原子の化学シフト(145/143/131/119/116/115ppm)から、3,4−ジヒドロキシベンジル構造が推定できる。C7に相当する2.7ppm(30ppm)のメチレン基、およびC2”に帰属される4.4ppm(82.5ppm)の−CHOR基が、結合点として識別可能である。C7を有するフラグメントについては、第2のメチレン基(C8:3.26/46.0ppm)を経てケトンC9(205ppm)まで延長できることは明らかである。しかしながら、C9以降、それ以上の相関は検出できない。C2”を含むフラグメントは、さらに別のCHOR基(4.1ppm/71ppm)に結合している。
【0098】
4.3ppmまたは37.5ppmのメタン基は、H/H相関からC4”のシグナルに帰属できる。さらに、103〜105ppmの範囲において2つのシグナルへの結合点が検出できるが、これらCHOR基と同様に、それぞれ、C14およびC10”に帰属、できる。
【0099】
5.7ppm(2H、94または96ppm)のシグナル群は、小さなメタカップリング(<3Hz)を示すのみであり、163/161/158/103ppmにおけるさらなるシグナルから、3つの酸素官能基を有する1,3,5,6−四置換芳香族に帰属できる。このように化学シフトが異なることにより、対称的置換は除外することができる。
【0100】
ここでまだ帰属されていない6つの−CHxOH/−CHOR基については、明確な帰属はできないが、これは、すべてのプロトンシグナル、いくつかの直接結合、および実質的にすべての長距離結合が3.3ppm前後のシグナル群内で重なり合うことによる。しかし、炭素原子の化学シフトから、炭水化物側鎖の存在はかなりの確実性で排除できる。
【0101】
芳香族領域の残り6つのシグナルの結合性については、これらが6つの四重シグナルであることにより、明確な説明はできない。これらは、HMBCスペクトルによっても分解できない。
【0102】
以下に示す図4の構造エレメントA〜Dは、このようにして得られるものである。
【0103】
さらに綿密な考察を行うと、これらの構造エレメントからは、2つの天然物質を構築することができる。iとiiiとを結合させることによってC4”位に置換基を有するカテキン/エピカテキンが得られ、ivとvii、さらにvとviiiとを結合させることによってC14位に置換基を有するアスパラチン誘導体が形成される。いずれの物質も、ルイボス茶に含まれることが知られている。残りの開結合C14およびC4”については、エレメントの帰属の過程で、結合していることが既に識別されている。
【0104】
アスパラチンおよびC4”位に置換基を有するカテキンの13C−NMRデータを式I(図5)による化合物のデータと比較したところ、対応する値はよく一致している。配座異性体(アトロプ異性体、回転異性体)の存在は、C14/C4”結合の自由回転の障害から、容易に説明することができる。
【0105】
回転異性体の存在によってシグナルの重なりや二重線が生じ、式Iの化合物のスペクトルが非常に複雑なものとなるため、1H−NMRデータの比較は行わない。
【0106】
この物質については現在まで文献に記載がないため、シミュレーションプログラム(ADC/C+H NMR Predictor V.10.02)によって帰属の妥当性を確認した。13C−NMRのデータの比較では、非常によい一致が見られた。
【0107】
3つの未知の立体中心、および炭水化物の明確な帰属は困難である。構造類似性から、全トランス構造またはβグルコ構造のカチノイドとして帰属される。
【0108】
質量スペクトル
装置:Micromass Quattro micro API(Waters製);LC−MS
方法:エレクトロスプレーイオン化(ESI)/二次イオン化EI
【0109】
結果
ESIスペクトルでは、m/z=741[M+H]+の分子ラジカルカチオンおよびm/z=763[M+Na]+のクラスターイオンが現れる。さらに、m/z=453[M−カテキン]+および289[M−カテキン−グリコシド]にフラグメントが現れる。質量740は、該物質に帰属する。
【0110】
紫外スペクトル
該物質の紫外スペクトルを、HPLCによるクロマトグラフィー精度の試験およびダイオードアレイ検出(DAD)(分離システム1)において記録した。
【0111】
結果
紫外スペクトルでは、285nmに吸収極大、228nmに肩が見られる。
【0112】
<実施例3> 遠隔ステレオEEG検査
生理食塩水(対照)および式Iの化合物(3mg/kg体重または6mg/kg体重)を経口投与した後、EEG(脳波記録法)周波数の変化を測定した。
【0113】
この検査は、W.Dimpfelが記載した方法(Dimpfel, W., Preclinical database of pharmaco−specific rat EEG fingerprints(Tele−Stereo−EEG).Eur. J. Med. Res.(2003)8:199−207)と同様に、以下のように実施した。
【0114】
4本の同心双極電極と1本のマイクロプラグとを共通のベースプレート上に配置したものを、6匹の6か月齢Fischer344雄性成熟ラット(昼夜逆転)に埋め込んだ。プラグは、前頭皮質、海馬、線条体および網様体からの電場電位を遠隔伝送する4チャンネルトランスミッタの記録用である。コンピュータシステム(ドイツ Linden MediSyst社製「EEG解析」ソフトウェア,OS サイエンスオペレーティングシステム、実験室用コンピュータ「LabTeam」)上でシグナルのリアルタイム高速フーリエ変換を行い、電力密度スペクトルをそれぞれ60分間測定した。スペクトルを6つの周波数領域に分割することにより、それぞれの物質の投与前にそれらの周波数帯で測定しておいた初期値に関する薬剤特異的変化の測定が可能となった。
物質の投与プロトコル:各物質は、測定開始(初期値)から45分後に経口投与した。5分後に測定を再開し、少なくともその後5時間にわたって連続分析を行い、60分毎にデータを収集した。試験物質は、用量3mg/kgおよび6mg/kg(式Iの化合物)で投与した。実験系列は、生理食塩水(対照)の投与により開始されたが、これが異常な変化をもたらすことはなかった(図6)。
【0115】
実験の結果と生理食塩水投与後の測定結果との統計的比較は、脳のあらゆる領域における個々の周波数帯内の変化に基づきこれを変数としたAhrensおよびLauter(cf. H. Ahrens, J. Lauter, “Mehrdimensionale Varianzanalyse”[“Multidimensional variance analysis”](1974), Akademie Verlag, Berlin参照)による多変量解析を用いて達成された。
【0116】
生理食塩水の投与では、初期段階の値と比較して、電気的活動(μV2/O)の変化はほとんど見られなかった(図6)。
【0117】
式Iの化合物を投与すると、脳のすべての領域で電力密度の安定的変化が見られ、特に投与後2〜3時間においては(図7,8参照)、動物数は少なかったものの、生理食塩水投与後とは顕著に異なっていた。アスパラチン、カテキンまたはエピカテキンをほぼ等モル投与してみた結果、式Iの化合物によってのみその特異構造に基づく効果が達成され、この分子のパーツによって効果を得ることはできないことがわかった(図9)。これは、この新たに発見された分子の特殊な結合構造による。運動性の増加は(−)−エピカテキン存在下においてのみ観察され(図11)、式Iの化合物の存在下では見られなかった。これにより、式Iの化合物独自の作用であることが強調される。
【0118】
式Iの化合物を試験動物に単回経口投与すると脳の電気的活動に変化をもたらすが、これはメタニコチン、ガランタミン、タクリン、L−DOPAまたはネホパムを投与した後の変化と一致する。このパターンは、認知症、パーキンソン病および疼痛の治療に既に普通に使用されている薬剤によるパターンと著しい相関があるが、他の疾患に適応される薬剤によるパターンとの相関はない(図11)。
【0119】
観測した周波数の変化を、識別分析法によって、脳の精神医学的疾患を治療するための薬剤および他の疾患に適応される薬剤の結果と比較した。活性物質の例としては、ガランタミン、ネホパム、LSD、メタニコチン、カフェイン、タクリン、アセチルサリチル酸、メサドン、メタミゾール、フェンタニール、フルボキサミン、クロルプロマジン、ハロペリドール、メトヘキシタール、メプロバメート、ミダゾラム、バルプロ酸、カルバマゼピン等が挙げられる。
【0120】
脳の4つの領域および6つの周波数帯(24の変数)に基づいた識別分析の結果、式Iの化合物の効果はガランタミン、タクリンおよびネホパムのそれに近いことがわかった。
【0121】
<実施例4> 式Iの化合物の含有量を測定する方法
ルイボスおよびルイボスからの調製物(茶、抽出物、タブレット)中の式1の化合物の含有量を、外部標準法によるHPLC/DADを用いて測定する。分析では、式Iの化合物の物質を外部標準として使用する。評価は、検出波長280nmで実施する。分析溶液中での酸化を防止するために、試料にはアスコルビン酸を添加する。
【0122】
好ましく用いられるHPLC装置はAcquity UPLC/Alliance2695;検出器:DAD(200〜400nm);カラム:Reprosil−Pur ODS−3、125×3mm、3μm(Dr. Maisch製);カラム温度:60℃である。
溶離液:溶離液A:水/ギ酸=100/0.2(v/v)
溶離液B:アセトニトリル/メタノール/水/ギ酸=50/25/25/0.2(v/v/v/v)
注入量:20μL
分析時間:95分
式Iの化合物の保持時間:39.5分
【0123】
【表1】
【0124】
以下のものを標準液として使用する。
正確に1mgを量り取った式Iの化合物と約20mgのアスコルビン酸とを2mLのメタノールに溶かし、水を加えて20.00mLとした。
必要濃度:式Iの化合物0.05mg/mL、アスコルビン酸1mg/mL
【0125】
以下のものを分析溶液として使用する。
医薬製剤:
分析試料、たとえばタブレットは、パウダーミルで粉砕し、メッシュサイズ250μmのふるいにかける。粉砕した試料約0.5gを、アスコルビン酸約50mgとともに50mLのメスフラスコに正確に量り入れ、40℃のメタノール10mLを加え、40℃の超音波浴中で10分間の抽出を行う。その後、この混合物に標線まで水を加えて激しく振り混ぜ、再度、40℃の超音波浴中で10分間の抽出を行う。冷却後、必要であれば混合物に標線まで水を加え、この溶液を9,300gで5分間の遠心分離にかける。上澄みを0.45μmメンブレンフィルタでろ過し、HPLCユニットオートサンプラー用の琥珀色をした小型ガラスびんに直接注ぐ。
【0126】
乾燥抽出物(抽出剤が水である場合):
ルイボス抽出物約125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約22mLの水を加え、この混合物を激しく振り混ぜる。必要であれば超音波浴中で処理を行い、標線まで水を加える。
【0127】
乾燥抽出物(抽出剤が水以外の場合):
ルイボス抽出物約125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約2.5mLのメタノールを加え、超音波浴中で10分間の処理を行う。次いで、この混合物に標線まで水を加えて激しく振り混ぜ、再度、超音波浴中で10分間の処理を行う。
【0128】
評価:
標準液および分析溶液については、同一条件下、直接クロマトグラフによる連続分析を行う。基準物質の紫外スペクトルを、分析クロマトグラムにおいて同じ保持時間で検知された物質の紫外スペクトルと比較し、一致が見られた場合、ピークを式Iの化合物と見なし、以下の計算式に従って計算を行う。
含有量[%]=(分析物のピーク面積´分析物希釈液(mL)´標準物質の重量(mg)´100)/(標準物質のピーク面積´標準物質希釈液(mL)´分析物(mg)の重量)
【0129】
<実施例5A> 式Iの化合物の単独製剤
各実施例において、「化合物I」は、式Iで示されるあらゆる化合物、ならびにその薬学的に許容される塩、誘導体およびエステルを示す。
フィルムコーティング錠:
式Iの化合物 50mg
アスコルビン酸 60mg
ソルビトール粉末(Karion instant) 120mg
Aerosil(ヒュームドシリカ) 3mg
タブレット賦形剤K 2.8mg
235.8mg
フィルム:
セラック 3.5mg
エタノール(セラックの溶媒) 97% 約70mg
硬カプセル:
カプセル内容物:
式Iの化合物 20mg
アスコルビン酸 60mg
マルトデキストリン 120mg
Aerosil(ヒュームドシリカ) 2mg
ステアリン酸マグネシウム 1.5mg
【0130】
<実施例5B> 式Iの化合物を含む抽出物の単独製剤
フィルムコーティング錠:
抽出物(式Iの化合物を含む)(1%) 150mg
アスコルビン酸 60mg
ソルビトール粉末(Karion instant) 80mg
Aerosil(ヒュームドシリカ) 3mg
ステアリン酸マグネシウム 2.5mg
295.5mg
フィルム:
セラック 4.2mg
エタノール(セラックの溶媒) 97% 約84mg
硬カプセル:
カプセル内容物:
抽出物(式Iの化合物を含む)(1%) 150mg
アスコルビン酸 40mg
マルトデキストリン 20mg
Aerosil(ヒュームドシリカ) 1mg
ステアリン酸マグネシウム 1.5mg
212.5mg
【0131】
<実施例6> 式Iの物質の含有量および総フラボノイド含有量の多い抽出物
a)抽出
出発物質として、温和な条件下で著しく急速に乾燥したAspalathus linearis(未発酵)の葉および若枝、すなわち残留する水分量をできる限り低くした(<5%)「緑色」のルイボスを使用した。
この抽出物は、20%、30%、または50%のエタノール水溶液といった様々な抽出剤を用いることによって得られた。あるいは、まず純粋なアルコールに薬種を加え、少なくとも30分間の軟化段階の後、相当する分量の水を加えることもできる。
これらの抽出は、式Iの物質と総フラボノイドとをできる限り完全に同時に抽出するため、最適化される。
この抽出物は、総フラボノイド含有量が高い点において、茶飲料(純粋な水抽出)に内用として使用するための現在市販で入手可能な抽出物とは異なっている。また、各フラボノイド群の比率においても、化粧品(エタノール80%)に使用するための外用抽出物とも異なっている。エタノール80%の抽出液は、アスパラチン含有量をできる限り高くするために最適化されたものである。3つのフラボノイド群(アスパラチン様、ルトシド様、およびビテキシン様すなわちC−グリコシド)の比率は、比較的疎水性の高い抽出を行ったことにより、該薬種の出発状態における比率から変化している。これは、ビテキシン様フラボノイドのルトシド様フラボノイドに対する比率を見れば特に明白である。
【0132】
b)粗製抽出物の成分を分析的に決定するための測定方法
使用したアスコルビン酸および溶媒は、Roth社(ドイツ、Karlsruhe)から、分析用品質のものを入手した。薬種は、ルイボス社(南アフリカ、Clanwilliam)から入手した。
標準のオリエンチン、ホモオリエンチン、ビテキシンは、たとえばExtrasynthese社(フランス、Genay)やRoth社(ドイツ、Karlsruhe)から市販品を入手できる。
使用したシリンジフィルタは、Roth社(ドイツ、Karlsruhe)の13mm Rotilaboシリンジフィルタ(0.45μm、PVDF)であった。
HPLC測定用には、LC 3D Rev.A.10.02(Agilent Technologies)用ChemStationソフトウェアおよびReprosil−Pur ODS−3カラム、125×3mm、3μm(Dr.Maisch、ドイツ、Ammerbuch)を備え、ダイオードアレイ検出器E−3014を有するHP190シリーズII液体クロマトグラフ装置(Hewlett Packard)を、カラム温度60℃で使用した。
移動相A:水/ギ酸=100/0.2(v/v)および
移動相B:アセトニトリル/メタノール/水/ギ酸=50/25/25/0.2(v/v/v/v)を使用し、以下の条件を選択した。
【0133】
【表2】
正確に計量した1.0mgの標準物質と約20mgのアスコルビン酸とを2.0mlのメタノールに溶解した後、水を加えて20.0mLにした。標準物質としては、ホモオリエンチン、オリエンチン、ビテキシン、ルトシド、イソクエルシトリン、フェルラ酸を使用した。
【0134】
式Iの化合物の標準液として、抽出物G110907SAのメタノール溶液を常用標準として使用した。この目的のために、抽出物G110907SAの125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約22mLの水を加えた。激しく振り混ぜ、超音波浴中で処理を行った後、この混合物に標線まで水を加えた。この抽出物における式Iの化合物の含有量は0.95%であり、これを他の抽出物の分析に使用した。
【0135】
c)測定
6.c)1)抽出:20%メタノール抽出物
抽出物0.5gを、アスコルビン酸約50mgとともに50.0mLのメスフラスコに量り入れ、40℃のメタノール10mLを加え、40℃の超音波浴中で10分間の抽出を行う。次いで、この混合物に標線まで水を加えて激しく振り混ぜ、再度、40℃の超音波浴中で10分間の抽出を行う。冷却後、必要であれば溶液に標線まで水を加え、この溶液を9,300gで5分間の遠心分離にかける。
上澄みを1mL取り、シリンジフィルタでろ過して琥珀色のガラスバイアルに入れ、HPLCで測定を行った。
【0136】
6.c)2)式Iの化合物
抽出物1(G110907SA)のクロマトグラフにおける物質1の識別は、入手可能な比較スペクトル、紫外比較スペクトル(図12)、およびオンラインの紫外スペクトルにより達成された。
様々な抽出物中の物質1の含有量は、抽出物1(G110907SA)中の既知の含有量(0.95%)を用いて、下記式によって計算した(A−面積,V−容量,m−質量,g−含有量,Ana−分析物,St−標準物質)。
相対面積relAは、以下のように計算する。
【0137】
6.c)3)物質A群のフラボノイド
物質A群のフラボノイドは、287nmおよび228nmに最大紫外吸収を有することを特徴とする。このスペクトル(図13)と実質的に一致するピークはすべてこの群に含むものとし、その含有量を下記式によって計算した。
標準物質としてルトシドを使用し、補正係数kfは0.4とする。
【0138】
6.c)4)ルトシド群フラボノイド
ルトシド比較スペクトル(図14)に基づいて、この群へのフラボノイドの帰属が行われる。含有量は以下の式により計算される。
【0139】
6.c)5)ビテキシン群フラボノイド
ビテキシン比較スペクトル(図15〜17)に基づき、この群へのフラボノイドの帰属が行われる。オリエンチンとホモオリエンチンも、この群に属している。ビテキシン(さらにオリエンチンも)が標準液の調製において溶解度に関する問題を呈したため、ビテキシンの代わりにホモオリエンチンを標準液として使用する。含有量は以下の式により計算される。
【0140】
6d)結果
分析により得られた結果を、以下の表3にまとめて示す。
本発明による抽出物の場合には、3つのフラボノイド群を出発物質としての薬種中に存在するのと可能な限り同じ比率で(薬種内の自然変動およびバッチ間変動を考慮しつつ)抽出物中に得るために努力がなされた。したがって、この比率は、高品質(低発酵)の緑色のルイボスから得られる通常の茶飲料に匹敵するものである。
【0141】
【表3】
(A)最初に純粋なアルコールで10分間抽出を行い、次いで記載のアルコール濃度になるまで水を加えた。
(B)抽出の最初から最後まで、記載のアルコール濃度を有するアルコール/水混合物を用いた。
各抽出物中に存在する各フラボノイドが、該薬種と比較して何%、また何倍に相当するかを、括弧内に示した。
【0142】
<実施例7> さらなる精製
式Iの物質および総フラボノイドの含有量がさらに高い抽出物を調製した。
該抽出物は、まず実施例6aの記載通りに調製し、次いで実施例1における純物質を得るための第1のセファデックスステップと同様に、セファデックスカラムを用いてさらに精製した。
式Iの化合物の含有量:>5%
総フラボノイド含有量(アスパラチン類+ルトシド類+ビテキシン類の総量、式Iの化合物は含まない):>35%
ビテキシン類(=C−グリコシド)のルトシド類に対する比:</=1.6
総アスパラチン:>25%
上述の条件下でのおよその保持時間(HPLC、実施例6の含有量の測定参照)は、以下の通りであった:
式Iの化合物:45分
アスパラチン:39分
ルトシド:42分
オリエンチン/ホモオリエンチン:38分
ビテキシン:41.5分
【技術分野】
【0001】
本発明は、式Iで示される新規な化合物ならびにその薬学的に許容される塩、誘導体およびエステルに関する。さらに本発明は、生の原料ルイボスから式Iの化合物を単離する方法に関する。さらにまた本発明は、式Iの化合物を少なくとも0.4重量%、好ましくは少なくとも1.5重量%含有するルイボス抽出物に関する。さらにまた本発明は、式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステル、ならびに本発明のルイボス抽出物の薬剤としての使用に関し、特に中枢神経系の神経学的疾患および精神医学的疾患、特に認知症を予防および治療するための使用に関する。「薬理学的活性のある」という表現には、精神状態の主観的改善をもたらす効果も含まれるが、その場合には、薬事法における承認を必ずしも必要とするわけではない。
【背景技術】
【0002】
ルイボス(学名:Aspalathus linearis)は南アフリカにのみ生育する植物であり、極めて強力な抗酸化物質であるアスパラチン(フラボノイドの1種)を含むものとしては、現在知られている限り、世界で唯一の植物である。ルイボスはまた、C−グリコシルフラボン(オリエンチンおよびイソオリエンチンを含む)、フラボノール−3−O−グリコシド(クエルセチン、クエルシトリン、イソクエルシトリンおよびルチンを含む)ならびにジヒドロカルコンであるノトファギンおよびアスパラチンを含有する。
【0003】
「緑色」の未発酵ルイボスは、その発酵製品と比べて、ポリフェノール類、特にアスパラチンの含有量が高いこと、および抗酸化活性が高いことで際立っている。発酵過程において抗酸化活性が低下するという結果は、紅茶と緑茶とを比較した例(Bramatiら, J. Agric. Food Chem. 2003, 51: 7472−7474)においても同様に認められる。科学的な研究により、ルイボス茶の抗酸化活性が主としてアスパラチン成分に由来するものであることが示されている。ルイボス茶の発酵過程の研究において、アスパラチンとノトファギンの含有量が発酵過程の間に減少することが明らかになった(Schulzら, Eur. Food Res. Technol. 2003, 216: 539−543)。「赤い」発酵ルイボス茶の抗酸化活性が「緑色」の未発酵ルイボス茶と比べて低いことは、これによって説明することができる。
【0004】
上述した健康増進作用のあるフラボノイドを含み、また風味が良いことから、ルイボス茶は広く消費されている。ルイボス茶の成分としてはさらに、フェノール酸、精油、ビタミンCおよび様々なミネラル、特に鉄やフッ化物が含まれている。
【0005】
できる限り高い抗酸化活性を得るためには、高いアスパラチン含有量が必要である。これに関連して、独国特許第102005004438号には、通常1〜3重量%であるアスパラチン含有量が5重量%を超え、クロロフィル含有量が0.4重量%未満と低くなっているルイボス抽出物が開示されている。独国特許第102005004438号によれば、該ルイボス抽出物は、発酵していない生の原料ルイボスをエタノールと水との比率が80対20の混合物で抽出することにより得られるものである。アスパラチン含有量の高いルイボス抽出物は、その強力な抗酸化・抗刺激・抗菌作用により、特に化粧用途、たとえばヘアケア、スキンケア、または口腔衛生のための薬剤としての使用が報告されている。
【0006】
ルイボス茶には、さらなるフラボノイド、クエルセチンが含まれる。これは生の原料ルイボス中に約11mg/100g存在し、たとえばヒトの体内におけるヒスタミン放出に影響を及ぼし、アレルギー症状を緩和する。クエルセチンはさらに、モノアミン酸化酵素の産生を抑制することができ、それによって、軽度のうつ病や睡眠障害に対し、有利な影響を与えることができる(Plant extract, the nature network, issue 3 of 09.11.2005; Plantextrakt GmbH)。
【0007】
本発明の出発点は、中枢神経系の疾患、たとえば認知症、パーキンソン病、うつ病、疼痛等を治療するための活性物質を探索することであった。これらの疾患は治療が困難であり、また使用される薬剤、たとえばタクリン、ガランタミン、ネホパム等には、多岐にわたる副作用がある。
【発明の概要】
【発明が解決しようとする課題】
【0008】
したがって、本発明の目的は、これらの疾患を治療するための活性物質、組成物およびルイボス抽出物を提供することである。特に、これらの活性物質や組成物は副作用の少ないものであることが必要である。
【課題を解決するための手段】
【0009】
この目的は、本発明の請求項の主題によって達成される。
【0010】
本発明は、式Iで示される化合物:
【化1】
ならびにその薬学的に許容される塩、誘導体およびエステルに関する。ここでの好ましい誘導体とは、式Iの化合物とフェルラ酸、キナ酸、カフェ酸、グルコン酸またはクロロゲン酸とのカップリング生成物である。式Iの化合物は、その天然形態で、または塩として使用されることが好ましいが、式Iの天然形態で使用されることがより好ましい。
【0011】
好ましいエステルとして、ギ酸、酢酸、プロピオン酸、グルタル酸、酒石酸、コハク酸等のエステルが挙げられる。好ましい塩としては、カチオンの対イオン、有機または無機の対イオンとの塩、特にアルカリ金属塩、アルカリ土類金属塩、アンモニウム塩、または薬学的に許容される酸との塩、たとえばコハク酸塩、クエン酸塩、酒石酸塩等が挙げられる。
【0012】
さらに本発明は、ルイボス抽出物、好ましくは未発酵ルイボスからの抽出物に関し、該抽出物における式Iの化合物の含有量は少なくとも0.4重量%、好ましくは少なくとも1.5重量%、より好ましくは少なくとも2.5重量%、さらに好ましくは少なくとも5重量%、さらにより好ましくは少なくとも10重量%、最も好ましくは少なくとも20重量%である。
【0013】
さらに本発明は、式Iの化合物の調製方法に関し、該方法では、
−発酵していない生の原料ルイボスを乾燥および粉砕した後、メタノールおよび/または水からなる抽出剤を用いて90℃以下、好ましくは60℃以下の温度で所定の時間抽出し、
−得られた抽出物をろ過した後、減圧下で蒸発乾固させ、次いで
−複数のクロマトグラフ分離ステップ、好ましくは、2本のセファデックスLH20カラムおよび次いで1本の疎水性c18−HPLCカラムを使用した3ステップのクロマトグラフ分離により精製する。
【0014】
本発明はさらに、薬剤または補助食品としてのルイボス抽出物の使用に関する。本発明はさらにまた、薬剤または補助食品としての、式Iの化合物、ならびにその薬学的に許容される塩、誘導体およびそのエステルの使用に関する。前記補助食品または薬剤中、前記ルイボス抽出物の含有量は、たとえば補助食品1g当たりまたは薬剤の用量単位当たり少なくとも10mg、少なくとも20mgまたは少なくとも50mgであってよい。好ましくは、前記補助食品または薬剤は、補助食品1g当たりまたは薬剤の用量単位当たり少なくとも100mg、より好ましくは少なくとも200mg、さらに好ましくは少なくとも300mgのルイボス抽出物を含む。さらに好ましい一実施形態では、前記補助食品または薬剤は、補助食品1g当たりまたは薬剤の用量単位当たり少なくとも1mg、より好ましくは少なくとも2mg、さらに好ましくは少なくとも3mg、さらに好ましくは少なくとも5mg、最も好ましくは少なくとも10mgの式Iの化合物を含む。
【発明を実施するための形態】
【0015】
本発明の好ましい一実施形態では、前記ルイボス抽出物ならびに、式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステルは、中枢神経系の神経学的疾患および精神医学的疾患を予防または治療するための薬剤として、好ましくは認知症、パーキンソン病、うつ病、疼痛等、特にアルツハイマー病を予防または治療するための薬剤として、式Iの化合物の割合をより多くして使用される。
【0016】
さらにより好ましい一実施形態では、本発明は、ルイボス抽出物ならびに、式Iの化合物ならびにその薬学的に許容される塩およびエステルの、中枢神経系の神経学的疾患および精神医学的疾患を予防および/または治療するための薬剤を調製するための使用に関し、ここで中枢神経系の神経学的疾患および精神医学的疾患は認知症、パーキンソン病、うつ病、疼痛等であり、より好ましくはアルツハイマー病である。
【0017】
本発明の範囲において、薬理学的活性のある新規の天然物質をルイボス抽出物から単離し、次いで特性決定を行うことができた。意外なことに、前記の新規化合物の単離は、「緑色」の未発酵ルイボス抽出物からのみ可能であった。該化合物の特性を完全に解析した結果、発酵したルイボス抽出物に含まれる微量の該化合物についても、検出が可能となった。
【0018】
該新規な化合物を単離する方法は、実施例1において詳細に記載するが、その構造式は式Iで示される。式Iの化合物は、アスパラチンの構造ともカテキン(4α→2)フロログルシノールの構造とも類似性を有する(図1)。正確な特性決定および構造解析については、実施例2で詳細に記載する。これまでに知られているフラボノイドと比較して、式Iで示される新規化合物の分子量は大きく、740.66g/molである。このような分子量の大きい天然物質は、活性物質のスクリーニングには通常使用されない。その分子量ゆえに、血液脳関門を容易には通過できないからである。したがって、このような物質は、脳で作用するには、また中枢神経系の疾患を治療するには、不適当であると見なされる傾向がある。
【0019】
しかし、意外なことに、先入観を持たずにラットの遠隔ステレオEEG(脳波)検査を行った結果、式Iの化合物が中枢神経において顕著な薬理学的活性を示すことがわかった。この薬理学的検査の結果、驚くべきことに、ラットの遠隔ステレオEEGモデルにおける該活性は、EEG周波数の用量依存的な変化をもたらし、これは認知症、パーキンソン病および疼痛を治療するための古典的な治療薬(認知症にはたとえばガランタミンまたはタクリン、パーキンソン病にはL−DOPA、また疼痛用にはたとえばネホパム)を投与した後に見られる既知の作用と同様であった。
【0020】
本発明の範囲において、式Iで示す進歩的化合物の薬理学的活性を、ルイボス抽出物の既知の成分、たとえばアスパラチン、カテキン、または(−)−エピカテキンと比較した。実験的研究の詳細は実施例3に記載するが、意外なことに、式Iの化合物がアスパラチン、カテキン、(−)−エピカテキン等と構造的類似性を有するにもかかわらず、その作用は、ほぼ等モル量のこれら天然物質では達成し得ないものであることが示された。式Iで示す新規かつ進歩的な化合物は、これら既知のルイボス抽出物成分よりも高い薬理学的活性を有し、したがって、薬剤としての使用に特に適している。
【0021】
有利な薬理学的活性を有するこの新規な化合物の単離により、特に式Iで示す化合物に基づく薬剤または補助食品の調製(単体調製物としても、また他の活性物質と組み合わせた調製物としても)が可能となる。このようなさらなる活性物質は、同方向に作用するものでもよいが、また別の方法で臨床像に有利な影響を及ぼすようなまったく異なる特性を有するものでもよい。この化合物をルイボス中に存在する他のフラボノイドおよび他の成分とあらゆる組み合わせで組み合わせることも、調製の目的に適している。特に、ここでは、抗酸化作用を有する成分が適している。
【0022】
上述したように、式Iの化合物のin vivoデータの識別分析により、認知症、パーキンソン病、うつ病および疼痛を治療するための薬剤との関連が示された。これらの薬剤には広範な副作用があるが、天然の物質による副作用の頻度は通常低いものと考えられることから、式Iの化合物を薬剤として使用することは有利である。さらに、意外なことに、この新規な物質またはその代謝物質は、血液脳関門を通過することが明らかになっている。これは、一般に、フラボノイドとしてはまれなことである。
【0023】
さらにまた、ルイボス抽出物および新たに同定された薬理学的活性のある化合物の調製方法が提供される。本発明の方法により、上記の疾患の治療に特に適した植物抽出物の提供が可能になる。
【0024】
本発明によって調製されたルイボス抽出物は、式Iの化合物を少なくとも1重量%、より好ましくは少なくとも1.5重量%、さらにより好ましくは少なくとも2重量%、より好ましくは少なくとも2.5重量%、さらにより好ましくは少なくとも3重量%、最も好ましくは少なくとも5重量%含有している。
【0025】
非常に好ましい一実施形態では、高濃縮されたルイボス抽出物が使用される。高濃縮されたルイボス抽出物は、式Iの化合物を少なくとも10重量%、好ましくは少なくとも20重量%含む。特定の実施形態では、該高濃縮されたルイボス抽出物は、式Iの化合物を少なくとも50重量%含む。
【0026】
式Iの化合物を調製するための本発明による方法は、以下のステップを含む(実施例1も参照のこと)。
−乾燥および粉砕した、発酵していない生の原料ルイボスの提供
−提供された生の原料の、アルコール、好ましくはメタノールおよび/または水の混合物からなる抽出剤を用いた、90℃以下、好ましくは60℃以下の温度における所定の時間の抽出
−得られた抽出物のろ過
−ろ過した抽出物の減圧下における蒸発
−抽出物の3段階の精製:
−セファデックスLH20カラムクロマトグラフィーによる粗精製
−さらなるセファデックスLH20カラムクロマトグラフィーによる高精製
−疎水性c18−HPLCカラムによる分離
【0027】
特に好ましい一実施形態では、本発明の方法による抽出ステップにおいて、0.001〜1重量%、好ましくは0.01〜0.2重量%のアスコルビン酸を加える。この重量パーセントは抽出対象の薬種、すなわち乾燥した葉や茎のような植物部位の重量に基づくものである。
【0028】
式Iの化合物を多く含有する調製物を得ようとする場合、1本のクロマトグラフィーカラムのみを使用して抽出物を分離することもできる。
【0029】
提供される生のルイボス原料の水分は4%以下であることが好ましい。この条件であれば、出発原料の自己発酵が防止できるからである。
【0030】
式Iの化合物を高い収率で得るために、本発明の方法の好ましい構成における抽出剤として、アルコール/水の50:50〜80:20(比率はアルコールの種類に依存し、アルコールとしてはメタノール、エタノール、プロパノール、プロパン−2−オールが適している)混合物を使用する。好ましくは、メタノール/水の50:50混合物を使用する。本発明の方法によるこの好ましい実施形態では、原料対抽出剤の比率は好ましくは約1:6であり、また抽出ステップは、好ましくは高温(40℃以上)において1時間で達成されるが、室温で、たとえば2〜5時間かけて行うことも可能である。
【0031】
ろ過した抽出物の蒸発は、好ましくは300mbar未満の圧力下で実施される。蒸発ステップの温度は、40℃以下であることが好ましい。
【0032】
本発明によるルイボス抽出物は、上述の方法によって調製され、蒸発乾固の後に(その後のクロマトグラフィーによる精製を行うことなく)得られる。
【0033】
該ルイボス抽出物中の式Iで示す化合物の含有量を測定する方法は、実施例4で詳細に記載する。
【0034】
式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステル、ならびに本発明によるルイボス抽出物は、特に、中枢神経系の疾患、好ましくは認知症、パーキンソン病、うつ病、疼痛を治療するために、また細胞を保護する抗酸化剤または「遊離基捕捉剤」として適切である。老年性認知症等の認知症、またはアルツハイマー病の治療には、特に好適である。
【0035】
本発明によれば、該新規な化合物は、式Iの形態で、または薬学的に許容される塩もしくは複合体およびエステルもしくは誘導体として、単一の活性物質として、またはさらなる活性物質と組み合わせて使用することができる。好適な誘導体、塩、複合体およびエステル、ならびにそれらの調製については、当業者には公知である。薬学的に許容される塩(塩酸塩、コハク酸塩、クエン酸塩、酒石酸塩等)の調製も、同様に当業者には公知である。塩を形成する好適な物質は、慣習的に薬学的に許容されるすべての酸またはアニオンである。さらに、たとえばフェルラ酸、キナ酸、カフェ酸、グルコン酸、クロロゲン酸およびその関連化合物とのカップリングも可能である。特に、グルコン酸とのカップリングが好ましい。式Iの化合物と上記の酸とのカップリング生成物は、薬学的に許容される誘導体として認定されている。しかしながら、該化合物は、式Iの分子として使用されることが好ましい。
【0036】
また別の、同様に好ましい一実施形態では、該抽出物は、やや親水性の高い混合溶媒を使用して調製される。この溶媒混合液は、好ましくはメタノール、エタノール、n−プロパノール、2−プロパノールから選択される少なくとも1種のアルコールと水とを含む。そのアルコール含有量は、10〜50重量%である。この溶媒混合液を選択すると、これよりもアルコールの割合が高い溶媒混合液を使用する場合と比べて、総フラボノイドの割合が高くなる。この実施形態では、アルコールの含有量は好ましくは10〜60%(vol/vol)、好ましくは10〜50%(vol/vol)である。さらにより好ましくは、アルコールの含有量は15〜40%であり、特に好ましくは20〜30%(vol/vol)である。
【0037】
本発明の方法におけるこの実施形態では、使用する出発物質としての薬種は、残留する水分量ができる限り低い「緑色」のルイボスであり、この水分量は好ましくは5%未満(総重量当たりの水分)である。出発原料、すなわちAspalathus linearis(未発酵)の葉および若枝は、温和な条件下で著しく急速に乾燥され、次いでアルコールと水との混合物(水の割合が60%(vol/vol)より高い)で抽出される。特に好ましい実施形態では、アルコール含有量は、メタノールの場合15〜25%、非常に好ましくは20%、またエタノールの場合には25〜35%、特に好ましくは30%である。
【0038】
非常に好ましい一実施形態では、まず、乾燥した出発物質としての薬種に純粋なアルコールを加え、通常30〜60分間かかる軟化段階の後、相当する分量の水を加える。抽出後、抽出物をろ過し、ろ液を減圧下で蒸発乾固させる。この後、好ましくはクロマトグラフィーによる精製ステップが続くが、この精製は好ましくはセファデックスカラムクロマトグラフィーを使用して実施される。
【0039】
この実施形態では、式Iの化合物は実質的に完全に抽出され、同時に、総フラボノイドが高い割合で抽出される。この(クロマトグラフィー法によってさらに精製される前の)抽出物中の式Iの化合物の含有量は、乾燥抽出物の重量に対して2.5〜5%である。式Iの化合物を除く、アスパラチン類、ルトシド類およびビテキシン類の総和としてのフラボノイド総量は、乾燥抽出物に対して15〜30重量%である。ビテキシン類(=C−グリコシド)のルトシド類に対する比率は1.6である。アスパラチン類の含有量は、乾燥抽出物の重量に対して14〜25%である。
【0040】
この方法で抽出を行うと、抽出物中の総フラボノイド含有量が多くなる。通常、このような(カラムクロマトグラフィーによるさらなる精製を行わない)抽出物は、総フラボノイドを少なくとも約20重量%含有する。以下の主要成分は、抽出物中で同定され得るものである。
a)式Iの化合物、
b)カルコン(アスパラチンおよびノトファギン)からなる物質群、
c)ルトシド群(成分としてクエルセチン(すなわちルチンのアグリコン)を含有するフラボノイド、C−O−C結合した糖を含むフラボノイドを含む)、ならびに
d)ビテキシン群(C−C結合した糖を含むフラボノイド。たとえば、アグリコンがアピゲニンであるビテキシン(アピゲニン−8−C−グルコシド)、イソビテキシン(アピゲニン−6−C−グルコシド)、オリエンチン(ルテオリン−8−C−グルコシド)、イソオリエンチン(ルテオリン−6−C−グルコシド)等が挙げられる。)
【0041】
このルイボス抽出物の特質は、特に、それぞれの群と他群との重量比にある。重量割合に基づくルトシド群とビテキシン群との比率は1:2〜1:50、好ましくは1:3〜1:10である。
【0042】
本発明による抽出物は、茶飲料のような純粋な水による抽出で得られる内用の抽出物とは、言及した化合物の含有量がより高いという点で異なっている。本発明による抽出物は、たとえば化粧品のような外用を意図した抽出物とは、フラボノイド群の互いに対する比率がそれぞれ異なるという点で異なっている。外用を意図した抽出物は、エタノールの含有量を高く(エタノールが少なくとも約80%)して抽出を行うことによって得られる。80%のエタノールで得られた抽出物は、アスパラチン含有量をできる限り高くするために最適化したものである。3つのフラボノイド群(アスパラチン様、ルトシド様、およびビテキシン様すなわちC−グリコシド)の比率は、比較的疎水性の高い抽出を行ったことにより、該薬種の出発状態における比率から変化している。これは、ビテキシン様フラボノイドのルトシド様フラボノイドに対する比率を見れば特に明白である。
【0043】
本発明によるルイボス抽出物ならびに式Iの進歩的な化合物またはその塩、誘導体およびエステル、好ましくは式Iの天然に存在する化合物は、健康増進作用を有する薬剤および/または補助食品を製造するための自体公知の方法で加工することができる。本発明によるルイボス抽出物ならびに式Iの進歩的な化合物ならびにその塩、誘導体およびエステルは、たとえばタブレット、カプセル、錠剤、コーティング錠、顆粒、粉末、ロゼンジの形態、および飲料のような液体の投与形態で処方することができる。補助食品、特に熱い飲料および冷たい飲料、または可溶茶のための使用も好ましい。
【0044】
該補助食品および薬剤は、経口、局所的、非経口、静脈内、筋肉内、皮下、経鼻、吸入、直腸、または経皮により好適に投与され得る。
【0045】
本発明のルイボス抽出物および本発明の薬剤または補助食品はまた、ルイボス抽出物または式Iの進歩的な化合物もしくはその塩の効果を高めたり、前記疾患においてその機構または状態に補助的な良い影響を与えたりするさらなる活性物質(たとえば、さらなる遊離基捕捉剤、様々な酵素阻害物質、ビタミン類、レシチン、オメガ−3−脂肪酸および脳の機能に良い影響を与える物質)を含んでいてもよい。
【0046】
さらに、薬学的に許容される賦形剤および担体を使用することができる。適切な賦形剤は当業者に公知であり、たとえば、充填剤、崩壊剤、滑沢剤、結合剤、湿潤剤等が含まれる。
【0047】
適切な滑沢剤は、たとえばケイ酸塩、滑石、ステアリン酸、ステアリン酸マグネシウムもしくはステアリン酸カルシウムおよび/またはポリエチレングリコールである。
【0048】
使用してもよい結合剤は、たとえばデンプン、アラビアゴム、ゼラチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルピロリドンである。
【0049】
崩壊剤は、たとえばデンプン、アルギン酸、アルギン酸塩、デンプングリコール酸ナトリウムまたは発泡混合物である。
【0050】
使用してもよい湿潤剤は、たとえばレシチン、ポリソルベートまたはラウリル硫酸塩である。
【0051】
さらに、着色剤および甘味料もまた、製剤中に存在してもよい。
【0052】
これらの医薬製剤は、公知の方法、たとえば混合、造粒、タブレット化または糖衣もしくはオーバーコートによって調製することができる。
【0053】
経口投与用の分散液および/または溶液は、たとえば飲料、ドロップ剤、シロップ剤、乳剤および懸濁剤であってよい。
【0054】
シロップ剤は、たとえばショ糖を含んでいてもよく、またショ糖とともに担体としてのグリセリン、マンニトールおよび/もしくはソルビトールを含んでいてもよい。
【0055】
懸濁剤および乳剤は、担体としてたとえば、天然樹脂、寒天、アルギン酸ナトリウム、ペクチン、メチルセルロース、カルボキシメチルセルロースまたはポリビニルアルコールを含んでいてもよい。
【0056】
筋肉注射用の懸濁剤または液剤は、該活性物質とともに、薬学的に許容される担体、たとえば滅菌水、オリーブオイル、オレイン酸エチル、たとえばプロピレングリコール等のグリコール類、および所望により適当量の塩酸リドカインを含んでいてもよい。
【0057】
静脈注射または点滴のための液剤は、たとえば担体として滅菌水を含んでいてもよく、また好ましくは滅菌水性等張塩溶液の形態であってもよい。
【0058】
坐剤は、該活性物質とともに、薬学的に許容される担体、たとえばカカオバター、ポリエチレングリコール、ポリオキシエチレンソルビトール脂肪酸エステルまたはレシチンを含んでいてもよい。
【0059】
局所適用用組成物、たとえばクリーム、ローションまたはペーストは、活性物質と従来の含油担体または乳化担体とを混合することによって調製することができる。
【0060】
本発明によるルイボス抽出物ならびに式Iの進歩的な化合物またはその塩、誘導体およびエステルは、補助食品または薬剤に通常含まれる量で使用することができる。溶液中、式Iの進歩的な化合物またはその塩は、好ましくは0.001〜10重量%、より好ましくは0.1〜7重量%、特に好ましくは1〜5重量%で使用される。特定の一実施形態では、式Iの進歩的な化合物またはその塩は、溶液中0.02〜1重量%で使用される。
【0061】
本発明によるルイボス抽出物は、式Iの化合物の1〜1000mg、より好ましくは10〜600mg、さらに好ましくは50〜400mg、最も好ましくは50〜250mgに相当する量で使用される。
【0062】
式Iの化合物を非常に高い割合で含むルイボス抽出物を使用する場合、このような抽出物は、薬剤および補助食品において、1日当たり3〜600mg、好ましくは5〜100mg、特に好ましくは10〜50mgの用量で使用することができる。
【0063】
上述の補助食品または薬剤は、従来の方法によって調製し、薬学的に好ましい形態で投与することができる。
【0064】
本発明による好ましい固形の補助食品または薬剤は、1〜95重量%、好ましくは1〜50重量%、より好ましくは1〜20重量%、特に好ましくは1〜10重量%の充填剤をさらに含んでいてもよい。
【0065】
使用してもよい充填剤は、タブレットまたはカプセルが必要かつ所望の重量となるように原料の一部をなす1以上の化合物である。中でも、様々な粒径の微結晶性セルロース、特に平均粒子径が20μmから200μmの範囲、特に50μmから150μmの範囲にある、たとえばAvicel PH−101およびPH−102等の公知のAvicel製品のように約100μmであるものを使用することができる。さらなる好適な充填剤は、たとえばトウモロコシデンプン、ジャガイモデンプン、ラクトース、セラクトース(セルロースとラクトースとの混合物)、リン酸カルシウム、ブドウ糖、マンニトール、マルトデキストリン、イソマルト、二酸化ケイ素(Aerosil)、さらに場合によってはソルビトールおよびショ糖である。直接圧縮が意図される場合、充填剤としては、タブレットの直接圧縮に適した品質のものを選択する必要がある。市販品の場合、このような品質は製品ごとにメーカーにより明示されるものであるが、簡単な実験によって確認することもできる。最も好ましい充填剤は、微結晶性セルロース(市販品としては、たとえばAvicel、VivapurおよびEmcocel等)である。
【0066】
適切な崩壊剤は従来技術において公知である。本発明による好ましい崩壊剤は、たとえば、クロスポビドン(Kollidon CL)およびデンプンまたはあらかじめゼラチン化したデンプン、特に市販品の「Starch1500」である。さらなる好適なデンプンも市販品として入手可能であり、たとえばLycatab PGS、PrejelおよびSepistab ST 200の名称で販売されている。
【0067】
さらに、公知のいわゆる「超崩壊剤」も使用することができ、たとえばクロスカルメロースナトリウム(たとえばAc−Di−Sol等)およびカルボキシメチルデンプンナトリウム(たとえばExplotab、Primojel等)が挙げられる。Starch1500等のデンプンが特に好ましい。
【0068】
崩壊剤の含有量は、通常1〜25重量%、好ましくは1〜20重量%、特に2〜15重量%である。崩壊剤の含有量として適切な範囲はまた、たとえば2〜5重量%または15〜20重量%であり、使用する崩壊剤、充填剤および他の添加剤に依存する。
【0069】
本発明によれば、該組成物は、滑沢剤として、タブレットの調製および加工を補助する1以上の化合物を含んでいてもよい。使用してもよい滑沢剤としては、特にステアリン酸およびその誘導体、たとえばステアリン酸カルシウム、特にフマル酸ステアリルナトリウム(たとえば商品名Pruv等の市販品が入手可能)およびステアリン酸マグネシウム、モノ−、ジ−、特にトリ−ステアリン酸グリセリル、硬化植物油(たとえばLubritab、Dynasan、Sterotext)またはポリエチレングリコール(たとえばLutrol、Carbowax)が挙げられる。
【0070】
滑沢剤の含有量は、通常0.1〜4重量%、好ましくは0.2〜4重量である。
【0071】
本発明による医薬組成物は、場合により、1以上の流動調節剤を含んでいてもよい。適切な流動調節剤は、三ケイ酸マグネシウム、滑石および特に二酸化ケイ素(たとえばAerosil)である。該組成物が流動調節剤を含む場合、この流動調節剤は通常0.5〜5重量%、好ましくは1〜4重量%、特に好ましくは2〜3重量%含まれる。
【0072】
本発明による医薬組成物はさらに、活性物質のための安定化剤を含んでいてもよく、該安定化剤としてはアスコルビン酸、クエン酸、酒石酸、乳酸等が挙げられ、アスコルビン酸またはクエン酸が好ましい。安定化剤(使用するとすれば)の含有量は、通常0.1〜10重量%、0.5〜10重量%、好ましくは1〜3重量%である。
【0073】
本発明による医薬組成物はさらに、薬学的に許容される通常の添加剤および賦形剤を含んでいてもよいが、上述のもの(充填剤、崩壊剤、滑沢剤、ならびに場合により流動調節剤および安定化剤)以外の賦形剤を含まないことが好ましい。
【0074】
充填剤には、微結晶性セルロースのように、結合剤としても機能するものがある。したがって、本発明においては、結合剤としての機能を有する充填剤は、充填剤と見なすこととする。
【0075】
本発明による医薬組成物がタブレットである場合、1以上のコーティング剤でフィルムコートされていてもよい。使用してもよいコーティング剤は、セラックおよびセラックの混合物、ヒプロメロース(ヒドロキシプロピルメチルセルロース)、ポリビニルアルコール、カルボキシメチルセルロースナトリウムならびに種々のメタクリル酸重合体(オイドラジット)であり、ヒプロメロース、オイドラジット、セラックおよびセラックの混合物が好ましい。タブレットのコーティングは通常の方法で達成される。コーティング剤に加えて、タブレットのコーティングに通常使用される成分、たとえば可塑剤、着色料、孔形成剤または懸濁安定化剤がコーティング中に存在してもよく、たとえばポリエチレングリコール(PEG)、滑石、二酸化チタン、また場合によってはラクトース等が使用される。
【0076】
タブレットの重量は特に制限されないが、純粋な活性物質を使用する場合は100mg〜500mg、抽出物、植物粉末を使用する場合は250mg〜1500mg、500mg〜1500mgのタブレットが一般的である。カプセルの場合、100mg〜1000mgのものが使用される。
【0077】
薬剤または補助食品の用量単位としては、たとえば、以下のようにしてもよい。
・経口剤形の場合には、
1日用量当たり、好ましくは1〜1000mg、より好ましくは40〜800mg、特に好ましくは150〜500mg、さらに好ましくは300〜600mgのルイボス抽出物。式Iの化合物ならびにその薬学的に許容される塩、誘導体およびエステルの場合、上記の量の1/100〜1/20を使用する。1日量は毎日投与することができ、たとえば1〜3回、好ましくは2回に分けて投与される。また、式Iの化合物を含むルイボス抽出物を1日に1〜10回投与することも想定可能である。
・非経口(たとえば静脈内、皮下、筋肉内)剤形の場合には、
1日用量当たり、好ましくは3〜60mg、特に好ましくは10〜30mgの活性物質。1日量は毎日投与することができ、たとえば1〜3回に分けて、好ましくは単回で投与される。
・直腸に適用する剤形の場合には、
1日用量当たりの該抽出物中、式Iの活性物質は、好ましくは40〜80mg、特に好ましくは60mg。1日量は毎日投与することができ、たとえば1〜3回に分けて、好ましくは単回で投与される。
・皮膚または粘膜に適用する剤形(たとえば液剤、ローション、乳剤、軟膏等)の場合には、
1回当たり、好ましくは40〜80mg、特に好ましくは60mgの活性物質。調製した液剤、ローション、乳剤または軟膏に対する式Iの化合物の含有量は、このような軟膏状薬剤、クリーム状調製物に対する重量で0.05〜20重量%、好ましくは0.2〜1重量%である。1日量は毎日投与することができ、たとえば1〜6回、好ましくは1〜3回に分けて投与される。
薬学的に許容される塩の使用において、その使用量は、当業者に公知の方法で適切な量とする必要がある。
【図面の簡単な説明】
【0078】
【図1】アスパラチン(1)およびカテキン(4α→2)フロログルシノール(2)の構造式を示す。
【図2】式Iの化合物中の原子に付された番号を示す。
【図3】図2による番号を付した各原子に対するNMR(核磁気共鳴)シグナルの帰属を示す。 符号の説明: *)/**)/+)/++)/§)/§§)/#)##)=値は相互に入れ替え可能 A),B)=1度だけ示した基の積分値または化学シフトの和。
【図4】NMR実験によって特徴付けられた、式Iの化合物の構造フラグメントA〜Dを示す。
【図5】式Iの化合物の13C−NMRデータをアスパラチンおよびカテキン(4α→2)フロログルシノールのデータと比較した表2を示す。アスパラチンについては13C−NMRの文献データがないため、HWI ANALYTIK GMBHによって単離されたアスパラチンに対するシグナルの帰属は、 Ho et al., Phytochemistry 1980, 19, 476−477のデータに基づくものである。カテキン(4α→2)フロログルシノールについては、Matthews et al., J. Agric. Food. Chem. 1997, 45, 1195−1201のデータを使用した。 符号の説明: *)/**)/+)/++)/§)/§§)/#)##)=値は相互に入れ替え可能
【図6】式Iの化合物を投与した後のFischer−344ラットに食塩水を投与した際の実験条件が一定であった証拠を示す。投与後の時間(時間)をy軸に示す。x軸はデータを高速フーリエ変換した後の周波数範囲:δ、θ、α1、α2、β1およびβ2を示す。
【図7】式Iの化合物3mg/kgを投与したことによるEEG周波数に対する効果を、投与後5時間まで示す。値は平均値である(n=6)。アステリスクは、AhrensおよびLauter(1974)の方法で計算された統計的有意性を表す。 *=p<0.01;**=p<0.05;***=p<0.025
【図8】式Iの化合物6mg/kgを投与したことによるEEG周波数に対する効果を、投与後5時間まで示す。値は平均値である(n=6)。アステリスクは、AhrensおよびLauter(1974)の方法で計算された統計的有意性を表す。 *=p<0.01;**=p<0.05;***=p<0.025
【図9】式Iの化合物の影響を、その成分分子であるアスパラチン、カテキンまたはエピカテキンと比較して示す。ほぼ等モルを投与した場合、特異構造による効果は式Iの化合物によってのみ達成され、この分子の各部分からは得られない。
【図10】式Iの化合物6mg/kgを投与したことによる、投与後1時間のEEG周波数に対する効果を示す。値は平均値である(n=6)。認知症、パーキンソン病、うつ病および疼痛を治療するために使用される薬剤:L−DOPA(パーキンソン病)、タクリンおよびガランタミン(アルツハイマー病)、ネホパム(疼痛治療)との類似性に注目すべきである。
【図11】電場電位と同時に実施した運動性測定の結果を示す。データは、各物質投与前の開始値に対する%で示したものである。運動性を実質的に増加させるのは、(−)−エピカテキンのみである。
【図12】式Iの化合物(G110907SA)の紫外スペクトルを示す。
【図13】アスパラチンの紫外スペクトルを示す。
【図14】ルトシドの紫外スペクトルを示す。
【図15】オリエンチンの紫外スペクトルを示す。
【図16】ホモオリエンチンの紫外スペクトルを示す。
【図17】ビテキシンの紫外スペクトルを示す。
【実施例】
【0079】
本発明は、以下の実施例によって、より詳細に説明される。
【0080】
<実施例1> 式Iの化合物の単離
薬種からの抽出物を調製するために、製造業者であるルイボス社(南アフリカ、Clanwilliam)から入手した未発酵の緑のルイボスを使用する。
【0081】
発酵しないまま粉砕したAspalathus linearisの葉および/または若枝を温和な条件下で水分量が10%未満(好ましくは4%未満)になるまで乾燥し、出発原料として使用する。
【0082】
この原料の抽出を、メタノールと水との50:50(容量部)混合液を用い、回転させつつ60℃にて1時間かけて行うが、このとき原料と溶媒の比が1:7となるようにする。その後、ろ過により液体を植物部位から分離し、この植物部位を同じ方法で再度抽出し、ろ過する。
【0083】
2つのろ液を合わせ、減圧下(220mbar)55℃でメタノールを除去する。残った水溶液を、使用した乾燥植物部位の重量の5倍まで水で希釈し、液液分配に供する。
【0084】
この水溶液3Lに対し、1.5Lの水飽和したn−ブタノールを加えて振とうする操作を4回行い、ブタノール相を合わせて減圧下で乾燥させる。その収率は、使用した乾燥植物部位の量の約10%となる。
【0085】
このブタノール抽出物に対し、まず粗分離、次いで高分離を行う。
【0086】
粗分離:
約50gのブタノール抽出物を、50容量%のメタノールを用いて、セファデックスLH20カラム充填クロマトグラフィー(内径6cm、高さ80cm、2260mlのセファデックスLH20を充填)によって分離する。この目的のために、使用する50gのブタノール抽出物は、400mlの移動相に溶解した後、分離カラムに加える。カラムは、3Lの溶出液が滴下するまで、1.8mL/分の流速で洗浄する。残っている移動相が完全に滴下した後、カラムの内容物を取り出し、これを3Lの100%メタノール中で10分間十分に攪拌(抽出)する。固定相はろ過により除去し、溶出液を乾燥させる。残留物は、使用した植物部位の量の約0.5〜1%となる(=メタノール抽出物)。
【0087】
高分離1回目:
約4gのメタノール抽出物を、セファデックスLH20カラム(内径3.5cm、高さ50cm、480mlのセファデックスLH20を充填)を用いて、80容量%のメタノールでクロマトグラフィーによって分離する。この目的のために、使用する4gのブタノール抽出物は、40mlの移動相に溶解した後、分離カラムに加える。2.4mL/minの流速でカラムを操作し、10分毎の分画(=24mlの溶出液)を回収する。目的の物質は、分画48〜65に含まれる。メタノール抽出物が4gの場合、収量は約0.5〜1gとなる。
【0088】
高分離2回目:
分離カラム:250×30mm
固定相:Reprosil C18 Aqua 10μm
移動相:35%(v/v)メタノール
流速:1.5mL/分
分画サイズ:10分=15mL
物質Iは、分画51〜52に含まれる。
分画41〜52を合わせ、凍結乾燥する。
4gのメタノール抽出物から約125mgの物質Iが得られる。
クロマトグラフィーによる純度:約97%(HPLC)。
【0089】
<実施例2> 式Iの化合物の構造解析
カラムクロマトグラフィーによる分離で得た化合物について、以下に挙げる装置を使用して特性決定を行ったところ、以下に記載する測定値が得られた。
【0090】
図2に、式Iの進歩的な化合物中の原子に付された番号を示す。図3の表1に、NMRデータの帰属を示す。
【0091】
NMRデータは、以下に記載する通りに測定し、解釈した。
【0092】
1H−NMR(d6−DMSO(ジメチルスルホキシド))
DMSO中の該物質の1H−NMRスペクトルでは、重水を加えた結果、9つのフェノールプロトンおよび5つの脂肪族HOプロトンの交換がシグナル減衰(H/D交換)によって識別できた。さらに、5.5〜8.0ppmというオレフィンの範囲で8つのプロトン、また−CHxOH/−CHxOR基(x=1または2)に典型的なシフト範囲内で9つのプロトンが現れている。2つの脂肪族プロトンが2.7ppm前後において識別可能であり、またさらなる脂肪族のシグナル(3つのプロトン)が−CHxOH/−CHxORのシグナルとオーバーラップしている。シグナルには、二重線になったり、ブロードになったりしているものもある。そのため、個々の積分値は名目上異性体の1つにのみ相当するが、2つの異性体が統合されているものもある。
【0093】
13C−NMR
該物質の13C−NMRスペクトルでは、ほとんどの炭素原子のシグナルが二重線になっている。2つの異性体の比率は使用する溶媒によってわずかに変化する。25〜90ppmの範囲に脂肪族炭素原子の11のシグナルが、またオレフィン炭素原子に典型的な90〜165ppmの範囲には24のシグナル群が現れる。205ppmに見られるさらなるシグナルは溶媒とオーバーラップするが、d6−DMSO中でははっきりと識別できる。このオーバーラップのために、すべての実験をさらに二次元で、d6−DMSO中で実施した。
【0094】
DEPT(Distortionless Enhancement by Polarization Transfer)スペクトルの測定は、d6−アセトン中で実施した。DEPTスペクトルの65〜85ppmの範囲には、3つのメチレン基のみならず、脂肪族メタン基と7つの−CHOH/−CHORシグナルが存在する。95〜125ppmにはさらに8つのシグナルがあり、オレフィンのCH基に帰属される。13C−NMRシグナルとの照合により、17個の第4級炭素原子の存在が推定される。
【0095】
H/H相関(COSY(相関分光法))
相関スペクトルで評価できるシグナルはごく少数であるが、これは、相互作用の大部分が3.1〜3.5ppmの範囲で強くオーバーラップすることによる。4.0〜4.4ppmの範囲において、アルキル鎖(H7(2.7ppm)〜H8(3.3ppm))の相関と同様に、シグナル群H2”〜H4”が明確に識別できる。4.7/4.8ppmのH1’からは、3.1〜3.5ppmの範囲において1つの相関のみ識別できる。
【0096】
C/H相関スペクトル(HMQC(Heteronuclear Multiple Quantum Correlation)、HMBC(Heteronuclear Multiple Bond Correlation))
第4級炭素原子に隣接するプロトンおよび個々の構造エレメントの結合は、CH長距離相関スペクトル(HMBC)から推定できる。直接相関の帰属により、構造体の帰属の妥当性を確認することもできる。
【0097】
得られたデータの評価および文献値との比較
6.8〜6.4ppmの範囲におけるプロトンのカップリングパターンから、1,3,4位に置換基を有する非常によく似た2つの芳香族系の存在が明確に示唆される。HMBCスペクトルのカップリング、および関与する炭素原子の化学シフト(145/143/131/119/116/115ppm)から、3,4−ジヒドロキシベンジル構造が推定できる。C7に相当する2.7ppm(30ppm)のメチレン基、およびC2”に帰属される4.4ppm(82.5ppm)の−CHOR基が、結合点として識別可能である。C7を有するフラグメントについては、第2のメチレン基(C8:3.26/46.0ppm)を経てケトンC9(205ppm)まで延長できることは明らかである。しかしながら、C9以降、それ以上の相関は検出できない。C2”を含むフラグメントは、さらに別のCHOR基(4.1ppm/71ppm)に結合している。
【0098】
4.3ppmまたは37.5ppmのメタン基は、H/H相関からC4”のシグナルに帰属できる。さらに、103〜105ppmの範囲において2つのシグナルへの結合点が検出できるが、これらCHOR基と同様に、それぞれ、C14およびC10”に帰属、できる。
【0099】
5.7ppm(2H、94または96ppm)のシグナル群は、小さなメタカップリング(<3Hz)を示すのみであり、163/161/158/103ppmにおけるさらなるシグナルから、3つの酸素官能基を有する1,3,5,6−四置換芳香族に帰属できる。このように化学シフトが異なることにより、対称的置換は除外することができる。
【0100】
ここでまだ帰属されていない6つの−CHxOH/−CHOR基については、明確な帰属はできないが、これは、すべてのプロトンシグナル、いくつかの直接結合、および実質的にすべての長距離結合が3.3ppm前後のシグナル群内で重なり合うことによる。しかし、炭素原子の化学シフトから、炭水化物側鎖の存在はかなりの確実性で排除できる。
【0101】
芳香族領域の残り6つのシグナルの結合性については、これらが6つの四重シグナルであることにより、明確な説明はできない。これらは、HMBCスペクトルによっても分解できない。
【0102】
以下に示す図4の構造エレメントA〜Dは、このようにして得られるものである。
【0103】
さらに綿密な考察を行うと、これらの構造エレメントからは、2つの天然物質を構築することができる。iとiiiとを結合させることによってC4”位に置換基を有するカテキン/エピカテキンが得られ、ivとvii、さらにvとviiiとを結合させることによってC14位に置換基を有するアスパラチン誘導体が形成される。いずれの物質も、ルイボス茶に含まれることが知られている。残りの開結合C14およびC4”については、エレメントの帰属の過程で、結合していることが既に識別されている。
【0104】
アスパラチンおよびC4”位に置換基を有するカテキンの13C−NMRデータを式I(図5)による化合物のデータと比較したところ、対応する値はよく一致している。配座異性体(アトロプ異性体、回転異性体)の存在は、C14/C4”結合の自由回転の障害から、容易に説明することができる。
【0105】
回転異性体の存在によってシグナルの重なりや二重線が生じ、式Iの化合物のスペクトルが非常に複雑なものとなるため、1H−NMRデータの比較は行わない。
【0106】
この物質については現在まで文献に記載がないため、シミュレーションプログラム(ADC/C+H NMR Predictor V.10.02)によって帰属の妥当性を確認した。13C−NMRのデータの比較では、非常によい一致が見られた。
【0107】
3つの未知の立体中心、および炭水化物の明確な帰属は困難である。構造類似性から、全トランス構造またはβグルコ構造のカチノイドとして帰属される。
【0108】
質量スペクトル
装置:Micromass Quattro micro API(Waters製);LC−MS
方法:エレクトロスプレーイオン化(ESI)/二次イオン化EI
【0109】
結果
ESIスペクトルでは、m/z=741[M+H]+の分子ラジカルカチオンおよびm/z=763[M+Na]+のクラスターイオンが現れる。さらに、m/z=453[M−カテキン]+および289[M−カテキン−グリコシド]にフラグメントが現れる。質量740は、該物質に帰属する。
【0110】
紫外スペクトル
該物質の紫外スペクトルを、HPLCによるクロマトグラフィー精度の試験およびダイオードアレイ検出(DAD)(分離システム1)において記録した。
【0111】
結果
紫外スペクトルでは、285nmに吸収極大、228nmに肩が見られる。
【0112】
<実施例3> 遠隔ステレオEEG検査
生理食塩水(対照)および式Iの化合物(3mg/kg体重または6mg/kg体重)を経口投与した後、EEG(脳波記録法)周波数の変化を測定した。
【0113】
この検査は、W.Dimpfelが記載した方法(Dimpfel, W., Preclinical database of pharmaco−specific rat EEG fingerprints(Tele−Stereo−EEG).Eur. J. Med. Res.(2003)8:199−207)と同様に、以下のように実施した。
【0114】
4本の同心双極電極と1本のマイクロプラグとを共通のベースプレート上に配置したものを、6匹の6か月齢Fischer344雄性成熟ラット(昼夜逆転)に埋め込んだ。プラグは、前頭皮質、海馬、線条体および網様体からの電場電位を遠隔伝送する4チャンネルトランスミッタの記録用である。コンピュータシステム(ドイツ Linden MediSyst社製「EEG解析」ソフトウェア,OS サイエンスオペレーティングシステム、実験室用コンピュータ「LabTeam」)上でシグナルのリアルタイム高速フーリエ変換を行い、電力密度スペクトルをそれぞれ60分間測定した。スペクトルを6つの周波数領域に分割することにより、それぞれの物質の投与前にそれらの周波数帯で測定しておいた初期値に関する薬剤特異的変化の測定が可能となった。
物質の投与プロトコル:各物質は、測定開始(初期値)から45分後に経口投与した。5分後に測定を再開し、少なくともその後5時間にわたって連続分析を行い、60分毎にデータを収集した。試験物質は、用量3mg/kgおよび6mg/kg(式Iの化合物)で投与した。実験系列は、生理食塩水(対照)の投与により開始されたが、これが異常な変化をもたらすことはなかった(図6)。
【0115】
実験の結果と生理食塩水投与後の測定結果との統計的比較は、脳のあらゆる領域における個々の周波数帯内の変化に基づきこれを変数としたAhrensおよびLauter(cf. H. Ahrens, J. Lauter, “Mehrdimensionale Varianzanalyse”[“Multidimensional variance analysis”](1974), Akademie Verlag, Berlin参照)による多変量解析を用いて達成された。
【0116】
生理食塩水の投与では、初期段階の値と比較して、電気的活動(μV2/O)の変化はほとんど見られなかった(図6)。
【0117】
式Iの化合物を投与すると、脳のすべての領域で電力密度の安定的変化が見られ、特に投与後2〜3時間においては(図7,8参照)、動物数は少なかったものの、生理食塩水投与後とは顕著に異なっていた。アスパラチン、カテキンまたはエピカテキンをほぼ等モル投与してみた結果、式Iの化合物によってのみその特異構造に基づく効果が達成され、この分子のパーツによって効果を得ることはできないことがわかった(図9)。これは、この新たに発見された分子の特殊な結合構造による。運動性の増加は(−)−エピカテキン存在下においてのみ観察され(図11)、式Iの化合物の存在下では見られなかった。これにより、式Iの化合物独自の作用であることが強調される。
【0118】
式Iの化合物を試験動物に単回経口投与すると脳の電気的活動に変化をもたらすが、これはメタニコチン、ガランタミン、タクリン、L−DOPAまたはネホパムを投与した後の変化と一致する。このパターンは、認知症、パーキンソン病および疼痛の治療に既に普通に使用されている薬剤によるパターンと著しい相関があるが、他の疾患に適応される薬剤によるパターンとの相関はない(図11)。
【0119】
観測した周波数の変化を、識別分析法によって、脳の精神医学的疾患を治療するための薬剤および他の疾患に適応される薬剤の結果と比較した。活性物質の例としては、ガランタミン、ネホパム、LSD、メタニコチン、カフェイン、タクリン、アセチルサリチル酸、メサドン、メタミゾール、フェンタニール、フルボキサミン、クロルプロマジン、ハロペリドール、メトヘキシタール、メプロバメート、ミダゾラム、バルプロ酸、カルバマゼピン等が挙げられる。
【0120】
脳の4つの領域および6つの周波数帯(24の変数)に基づいた識別分析の結果、式Iの化合物の効果はガランタミン、タクリンおよびネホパムのそれに近いことがわかった。
【0121】
<実施例4> 式Iの化合物の含有量を測定する方法
ルイボスおよびルイボスからの調製物(茶、抽出物、タブレット)中の式1の化合物の含有量を、外部標準法によるHPLC/DADを用いて測定する。分析では、式Iの化合物の物質を外部標準として使用する。評価は、検出波長280nmで実施する。分析溶液中での酸化を防止するために、試料にはアスコルビン酸を添加する。
【0122】
好ましく用いられるHPLC装置はAcquity UPLC/Alliance2695;検出器:DAD(200〜400nm);カラム:Reprosil−Pur ODS−3、125×3mm、3μm(Dr. Maisch製);カラム温度:60℃である。
溶離液:溶離液A:水/ギ酸=100/0.2(v/v)
溶離液B:アセトニトリル/メタノール/水/ギ酸=50/25/25/0.2(v/v/v/v)
注入量:20μL
分析時間:95分
式Iの化合物の保持時間:39.5分
【0123】
【表1】
【0124】
以下のものを標準液として使用する。
正確に1mgを量り取った式Iの化合物と約20mgのアスコルビン酸とを2mLのメタノールに溶かし、水を加えて20.00mLとした。
必要濃度:式Iの化合物0.05mg/mL、アスコルビン酸1mg/mL
【0125】
以下のものを分析溶液として使用する。
医薬製剤:
分析試料、たとえばタブレットは、パウダーミルで粉砕し、メッシュサイズ250μmのふるいにかける。粉砕した試料約0.5gを、アスコルビン酸約50mgとともに50mLのメスフラスコに正確に量り入れ、40℃のメタノール10mLを加え、40℃の超音波浴中で10分間の抽出を行う。その後、この混合物に標線まで水を加えて激しく振り混ぜ、再度、40℃の超音波浴中で10分間の抽出を行う。冷却後、必要であれば混合物に標線まで水を加え、この溶液を9,300gで5分間の遠心分離にかける。上澄みを0.45μmメンブレンフィルタでろ過し、HPLCユニットオートサンプラー用の琥珀色をした小型ガラスびんに直接注ぐ。
【0126】
乾燥抽出物(抽出剤が水である場合):
ルイボス抽出物約125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約22mLの水を加え、この混合物を激しく振り混ぜる。必要であれば超音波浴中で処理を行い、標線まで水を加える。
【0127】
乾燥抽出物(抽出剤が水以外の場合):
ルイボス抽出物約125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約2.5mLのメタノールを加え、超音波浴中で10分間の処理を行う。次いで、この混合物に標線まで水を加えて激しく振り混ぜ、再度、超音波浴中で10分間の処理を行う。
【0128】
評価:
標準液および分析溶液については、同一条件下、直接クロマトグラフによる連続分析を行う。基準物質の紫外スペクトルを、分析クロマトグラムにおいて同じ保持時間で検知された物質の紫外スペクトルと比較し、一致が見られた場合、ピークを式Iの化合物と見なし、以下の計算式に従って計算を行う。
含有量[%]=(分析物のピーク面積´分析物希釈液(mL)´標準物質の重量(mg)´100)/(標準物質のピーク面積´標準物質希釈液(mL)´分析物(mg)の重量)
【0129】
<実施例5A> 式Iの化合物の単独製剤
各実施例において、「化合物I」は、式Iで示されるあらゆる化合物、ならびにその薬学的に許容される塩、誘導体およびエステルを示す。
フィルムコーティング錠:
式Iの化合物 50mg
アスコルビン酸 60mg
ソルビトール粉末(Karion instant) 120mg
Aerosil(ヒュームドシリカ) 3mg
タブレット賦形剤K 2.8mg
235.8mg
フィルム:
セラック 3.5mg
エタノール(セラックの溶媒) 97% 約70mg
硬カプセル:
カプセル内容物:
式Iの化合物 20mg
アスコルビン酸 60mg
マルトデキストリン 120mg
Aerosil(ヒュームドシリカ) 2mg
ステアリン酸マグネシウム 1.5mg
【0130】
<実施例5B> 式Iの化合物を含む抽出物の単独製剤
フィルムコーティング錠:
抽出物(式Iの化合物を含む)(1%) 150mg
アスコルビン酸 60mg
ソルビトール粉末(Karion instant) 80mg
Aerosil(ヒュームドシリカ) 3mg
ステアリン酸マグネシウム 2.5mg
295.5mg
フィルム:
セラック 4.2mg
エタノール(セラックの溶媒) 97% 約84mg
硬カプセル:
カプセル内容物:
抽出物(式Iの化合物を含む)(1%) 150mg
アスコルビン酸 40mg
マルトデキストリン 20mg
Aerosil(ヒュームドシリカ) 1mg
ステアリン酸マグネシウム 1.5mg
212.5mg
【0131】
<実施例6> 式Iの物質の含有量および総フラボノイド含有量の多い抽出物
a)抽出
出発物質として、温和な条件下で著しく急速に乾燥したAspalathus linearis(未発酵)の葉および若枝、すなわち残留する水分量をできる限り低くした(<5%)「緑色」のルイボスを使用した。
この抽出物は、20%、30%、または50%のエタノール水溶液といった様々な抽出剤を用いることによって得られた。あるいは、まず純粋なアルコールに薬種を加え、少なくとも30分間の軟化段階の後、相当する分量の水を加えることもできる。
これらの抽出は、式Iの物質と総フラボノイドとをできる限り完全に同時に抽出するため、最適化される。
この抽出物は、総フラボノイド含有量が高い点において、茶飲料(純粋な水抽出)に内用として使用するための現在市販で入手可能な抽出物とは異なっている。また、各フラボノイド群の比率においても、化粧品(エタノール80%)に使用するための外用抽出物とも異なっている。エタノール80%の抽出液は、アスパラチン含有量をできる限り高くするために最適化されたものである。3つのフラボノイド群(アスパラチン様、ルトシド様、およびビテキシン様すなわちC−グリコシド)の比率は、比較的疎水性の高い抽出を行ったことにより、該薬種の出発状態における比率から変化している。これは、ビテキシン様フラボノイドのルトシド様フラボノイドに対する比率を見れば特に明白である。
【0132】
b)粗製抽出物の成分を分析的に決定するための測定方法
使用したアスコルビン酸および溶媒は、Roth社(ドイツ、Karlsruhe)から、分析用品質のものを入手した。薬種は、ルイボス社(南アフリカ、Clanwilliam)から入手した。
標準のオリエンチン、ホモオリエンチン、ビテキシンは、たとえばExtrasynthese社(フランス、Genay)やRoth社(ドイツ、Karlsruhe)から市販品を入手できる。
使用したシリンジフィルタは、Roth社(ドイツ、Karlsruhe)の13mm Rotilaboシリンジフィルタ(0.45μm、PVDF)であった。
HPLC測定用には、LC 3D Rev.A.10.02(Agilent Technologies)用ChemStationソフトウェアおよびReprosil−Pur ODS−3カラム、125×3mm、3μm(Dr.Maisch、ドイツ、Ammerbuch)を備え、ダイオードアレイ検出器E−3014を有するHP190シリーズII液体クロマトグラフ装置(Hewlett Packard)を、カラム温度60℃で使用した。
移動相A:水/ギ酸=100/0.2(v/v)および
移動相B:アセトニトリル/メタノール/水/ギ酸=50/25/25/0.2(v/v/v/v)を使用し、以下の条件を選択した。
【0133】
【表2】
正確に計量した1.0mgの標準物質と約20mgのアスコルビン酸とを2.0mlのメタノールに溶解した後、水を加えて20.0mLにした。標準物質としては、ホモオリエンチン、オリエンチン、ビテキシン、ルトシド、イソクエルシトリン、フェルラ酸を使用した。
【0134】
式Iの化合物の標準液として、抽出物G110907SAのメタノール溶液を常用標準として使用した。この目的のために、抽出物G110907SAの125mgを、アスコルビン酸約25mgとともに25mLのメスフラスコに量り入れ、約22mLの水を加えた。激しく振り混ぜ、超音波浴中で処理を行った後、この混合物に標線まで水を加えた。この抽出物における式Iの化合物の含有量は0.95%であり、これを他の抽出物の分析に使用した。
【0135】
c)測定
6.c)1)抽出:20%メタノール抽出物
抽出物0.5gを、アスコルビン酸約50mgとともに50.0mLのメスフラスコに量り入れ、40℃のメタノール10mLを加え、40℃の超音波浴中で10分間の抽出を行う。次いで、この混合物に標線まで水を加えて激しく振り混ぜ、再度、40℃の超音波浴中で10分間の抽出を行う。冷却後、必要であれば溶液に標線まで水を加え、この溶液を9,300gで5分間の遠心分離にかける。
上澄みを1mL取り、シリンジフィルタでろ過して琥珀色のガラスバイアルに入れ、HPLCで測定を行った。
【0136】
6.c)2)式Iの化合物
抽出物1(G110907SA)のクロマトグラフにおける物質1の識別は、入手可能な比較スペクトル、紫外比較スペクトル(図12)、およびオンラインの紫外スペクトルにより達成された。
様々な抽出物中の物質1の含有量は、抽出物1(G110907SA)中の既知の含有量(0.95%)を用いて、下記式によって計算した(A−面積,V−容量,m−質量,g−含有量,Ana−分析物,St−標準物質)。
相対面積relAは、以下のように計算する。
【0137】
6.c)3)物質A群のフラボノイド
物質A群のフラボノイドは、287nmおよび228nmに最大紫外吸収を有することを特徴とする。このスペクトル(図13)と実質的に一致するピークはすべてこの群に含むものとし、その含有量を下記式によって計算した。
標準物質としてルトシドを使用し、補正係数kfは0.4とする。
【0138】
6.c)4)ルトシド群フラボノイド
ルトシド比較スペクトル(図14)に基づいて、この群へのフラボノイドの帰属が行われる。含有量は以下の式により計算される。
【0139】
6.c)5)ビテキシン群フラボノイド
ビテキシン比較スペクトル(図15〜17)に基づき、この群へのフラボノイドの帰属が行われる。オリエンチンとホモオリエンチンも、この群に属している。ビテキシン(さらにオリエンチンも)が標準液の調製において溶解度に関する問題を呈したため、ビテキシンの代わりにホモオリエンチンを標準液として使用する。含有量は以下の式により計算される。
【0140】
6d)結果
分析により得られた結果を、以下の表3にまとめて示す。
本発明による抽出物の場合には、3つのフラボノイド群を出発物質としての薬種中に存在するのと可能な限り同じ比率で(薬種内の自然変動およびバッチ間変動を考慮しつつ)抽出物中に得るために努力がなされた。したがって、この比率は、高品質(低発酵)の緑色のルイボスから得られる通常の茶飲料に匹敵するものである。
【0141】
【表3】
(A)最初に純粋なアルコールで10分間抽出を行い、次いで記載のアルコール濃度になるまで水を加えた。
(B)抽出の最初から最後まで、記載のアルコール濃度を有するアルコール/水混合物を用いた。
各抽出物中に存在する各フラボノイドが、該薬種と比較して何%、また何倍に相当するかを、括弧内に示した。
【0142】
<実施例7> さらなる精製
式Iの物質および総フラボノイドの含有量がさらに高い抽出物を調製した。
該抽出物は、まず実施例6aの記載通りに調製し、次いで実施例1における純物質を得るための第1のセファデックスステップと同様に、セファデックスカラムを用いてさらに精製した。
式Iの化合物の含有量:>5%
総フラボノイド含有量(アスパラチン類+ルトシド類+ビテキシン類の総量、式Iの化合物は含まない):>35%
ビテキシン類(=C−グリコシド)のルトシド類に対する比:</=1.6
総アスパラチン:>25%
上述の条件下でのおよその保持時間(HPLC、実施例6の含有量の測定参照)は、以下の通りであった:
式Iの化合物:45分
アスパラチン:39分
ルトシド:42分
オリエンチン/ホモオリエンチン:38分
ビテキシン:41.5分
【特許請求の範囲】
【請求項1】
式Iで示される化合物:
【化1】
ならびにその薬学的に許容される塩、誘導体およびエステル。
【請求項2】
未発酵のルイボスから得られることを特徴とする、請求項1に記載の式Iの化合物を含むルイボス抽出物。
【請求項3】
式Iの化合物を少なくとも1.0重量%含有することを特徴とする、請求項2に記載のルイボス抽出物。
【請求項4】
式Iの化合物を少なくとも2重量%含有することを特徴とする、請求項3に記載のルイボス抽出物。
【請求項5】
総フラボノイド含有量が少なくとも20重量%であることを特徴とする、請求項2〜4のいずれかに記載のルイボス抽出物。
【請求項6】
請求項1に記載の式Iの化合物および請求項2〜5のいずれかに記載のルイボス抽出物を調製する方法であって、
−発酵していない生の原料ルイボスを乾燥および粉砕した後、アルコールおよび/または水からなる抽出剤を用いて90℃以下、特に60℃以下の温度で所定の時間抽出すること、ならびに
−得られた抽出物をろ過した後、減圧下で蒸発乾固させること
を特徴とし、さらに
前記抽出剤の10〜60%(vol/vol)がアルコールであり、残りが水であることを特徴とする方法。
【請求項7】
少なくとも1ステップのクロマトグラフ分離によってさらに実質的な精製が達成されることを特徴とする、請求項8に記載の方法。
【請求項8】
前記クロマトグラフ分離ステップがサイズ排除クロマトグラフィーであることを特徴とする、請求項7に記載の方法。
【請求項9】
請求項1に記載の式Iの化合物を、1g当たり少なくとも1mg含有することを特徴とする補助食品。
【請求項10】
請求項2〜5に記載のルイボス抽出物を、少なくとも10mg/g含有することを特徴とする補助食品。
【請求項11】
請求項1に記載の式Iの化合物を、1日用量当たり少なくとも1mg、特に少なくとも5mg含有するよう調製されていることを特徴とする、請求項9または10に記載の補助食品。
【請求項12】
請求項1に記載の式Iの化合物または請求項2〜5のいずれかに記載のルイボス抽出物を含有することを特徴とする薬剤。
【請求項13】
請求項1に記載の化合物または請求項2〜5のいずれかに記載のルイボス抽出物の、中枢神経系の神経学的疾患および精神医学的疾患を予防および/または治療するための使用。
【請求項14】
前記中枢神経系の神経学的疾患および精神医学的疾患が認知症、パーキンソン病、うつ病および疼痛であることを特徴とする、請求項13に記載の使用。
【請求項15】
前記中枢神経系の疾患がアルツハイマー病または老年性認知症であることを特徴とする、請求項14に記載の使用。
【請求項1】
式Iで示される化合物:
【化1】
ならびにその薬学的に許容される塩、誘導体およびエステル。
【請求項2】
未発酵のルイボスから得られることを特徴とする、請求項1に記載の式Iの化合物を含むルイボス抽出物。
【請求項3】
式Iの化合物を少なくとも1.0重量%含有することを特徴とする、請求項2に記載のルイボス抽出物。
【請求項4】
式Iの化合物を少なくとも2重量%含有することを特徴とする、請求項3に記載のルイボス抽出物。
【請求項5】
総フラボノイド含有量が少なくとも20重量%であることを特徴とする、請求項2〜4のいずれかに記載のルイボス抽出物。
【請求項6】
請求項1に記載の式Iの化合物および請求項2〜5のいずれかに記載のルイボス抽出物を調製する方法であって、
−発酵していない生の原料ルイボスを乾燥および粉砕した後、アルコールおよび/または水からなる抽出剤を用いて90℃以下、特に60℃以下の温度で所定の時間抽出すること、ならびに
−得られた抽出物をろ過した後、減圧下で蒸発乾固させること
を特徴とし、さらに
前記抽出剤の10〜60%(vol/vol)がアルコールであり、残りが水であることを特徴とする方法。
【請求項7】
少なくとも1ステップのクロマトグラフ分離によってさらに実質的な精製が達成されることを特徴とする、請求項8に記載の方法。
【請求項8】
前記クロマトグラフ分離ステップがサイズ排除クロマトグラフィーであることを特徴とする、請求項7に記載の方法。
【請求項9】
請求項1に記載の式Iの化合物を、1g当たり少なくとも1mg含有することを特徴とする補助食品。
【請求項10】
請求項2〜5に記載のルイボス抽出物を、少なくとも10mg/g含有することを特徴とする補助食品。
【請求項11】
請求項1に記載の式Iの化合物を、1日用量当たり少なくとも1mg、特に少なくとも5mg含有するよう調製されていることを特徴とする、請求項9または10に記載の補助食品。
【請求項12】
請求項1に記載の式Iの化合物または請求項2〜5のいずれかに記載のルイボス抽出物を含有することを特徴とする薬剤。
【請求項13】
請求項1に記載の化合物または請求項2〜5のいずれかに記載のルイボス抽出物の、中枢神経系の神経学的疾患および精神医学的疾患を予防および/または治療するための使用。
【請求項14】
前記中枢神経系の神経学的疾患および精神医学的疾患が認知症、パーキンソン病、うつ病および疼痛であることを特徴とする、請求項13に記載の使用。
【請求項15】
前記中枢神経系の疾患がアルツハイマー病または老年性認知症であることを特徴とする、請求項14に記載の使用。
【図1】

【図2】
【図4A】
【図4B】
【図4C】
【図4D】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図3】

【図5】

【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図2】
【図4A】
【図4B】
【図4C】
【図4D】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図3】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【公表番号】特表2011−501755(P2011−501755A)
【公表日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願番号】特願2010−530290(P2010−530290)
【出願日】平成20年9月5日(2008.9.5)
【国際出願番号】PCT/EP2008/007279
【国際公開番号】WO2009/052895
【国際公開日】平成21年4月30日(2009.4.30)
【出願人】(506178508)クナイプ‐ヴェルケ クナイプ‐ミッテル‐ツェントラーレ ゲゼルシャフト ミト ベシュレンクテル ハフツング ウント コムパニー カーゲー (2)
【Fターム(参考)】
【公表日】平成23年1月13日(2011.1.13)
【国際特許分類】
【出願日】平成20年9月5日(2008.9.5)
【国際出願番号】PCT/EP2008/007279
【国際公開番号】WO2009/052895
【国際公開日】平成21年4月30日(2009.4.30)
【出願人】(506178508)クナイプ‐ヴェルケ クナイプ‐ミッテル‐ツェントラーレ ゲゼルシャフト ミト ベシュレンクテル ハフツング ウント コムパニー カーゲー (2)
【Fターム(参考)】
[ Back to top ]