
アスピリンの正荷電水溶性プロドラッグ
アセチルサリチル酸及びその類似体の、式(1)で表される新規な正荷電プロドラッグを合成した。これらのプロドラッグの正に荷電されたアミノ基は、薬物の溶解性を大いに増大させるだけでなく、膜のホスファート頭基上の負電荷と結合し、プロドラッグを細胞質ゾルの中に押し込む。プロドラッグ、酢酸ジエチルアミノエチルアセチルサリチラートは、アセチルサリチル酸自体よりも〜400倍速く、エチルアセチルサリチラートよりも〜100倍速く人の皮膚を通して拡散する。プロドラッグは人又は動物において、アスピリン治療可能な状態の治療に医薬的に使用され、経口投与だけでなく経皮投与されることも可能であり、たいていのアスピリンの副作用、最も著しくは胃腸障害を回避し得る。プロドラッグの制御経皮投与システムは、アスピリンが絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、アスピリンの副作用を減少させることを可能にする。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、アスピリン又はその類似体の正に荷電された水溶性プロドラッグの調製剤、及び人又は動物において、アスピリン治療可能な状態を治療する際のその医薬用途に関する。
【背景技術】
【0002】
アセチルサリチル酸(アスピリン)は1853年に合成され、1899年に最初に医薬的に使用された。それ以来、サリチル酸の多数の誘導体が合成され、薬理学的に評価されてきたが、比較的ほとんどの誘導体は治療上有用性を達成していない。アスピリンは解熱、鎮痛及び抗炎症作用を有する。サリチラートは尿酸の排出を促進するので、痛風関節炎の治療に有益である。アスピリンはまた、潜在的に心臓発作及び脳卒中に寄与する血小板凝集を抑制し[C.H.Hennekens,et al.,N.Engl.J.Med.,321,129(1989);T.A.Gossel,U.S.Pharmacist,February,1988,p.34.]、同様に大腸癌に対して防御的となり得る[M.J.Thun,et al.,N.Engl.J.Med.,325,1393(1991)]。従って、アスピリンの治療上有用性は重要であり、増大し続けている。「米国医薬品集」(PDR Generics,1996,second edition,MedicalEconomics,Montvale,New Jersey,pg243)にはアスピリンの多くの医薬用途が記載されている。アスピリンはまた、軽度から中程度の疼痛並びに関節炎及び他の炎症性状態の長期緩和待期療法に使用されている。アスピリンは単独で又は補助薬として、川崎病、術後血栓塞栓症及び不安定狭心症の治療に使用される。アスピリンはまた急性小児胃腸炎において、便の量を減少させ、かつ体重を増加させるために、大動脈冠状動脈バイパス移植閉塞を予防する際の補助薬として使用されて、また慢性心房細動における血栓塞栓性合併症を予防するために使用される。アスピリンは、補助薬として、頚動脈内膜切除術中の血小板凝集を減らすために使用され、白内障の進行を遅らせるために処方され、冠状動脈形成術後狭窄の再発を予防するために使用され、多発脳梗塞性痴呆患者における認知機能及び大脳の血流を改善するために使用される。アスピリンは補助薬として、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含め糖尿病誘発性合併症において血漿グルコースを下げるために使用され、そして総死亡率及び心血管死亡率を減少させるために使用される。アスピリンは血液透析シャント血栓症(hemodialysis shunt thrombosis)の発生を減らすため、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生を減少させるため、及び末梢閉塞性動脈疾患の進行を遅らせるために処方される。アスピリンは大腸癌、直腸癌及び人工心臓弁患者における動脈塞栓性合併症の予防に使用される。アスピリンはまたハイリスク女性における妊娠誘発性高血圧及び妊娠中毒症の発生を減らすために処方される。
【0003】
残念ながら、多くの副作用がサリチラートの使用と関連付けられ、最も著しくは胃腸障害であり、例えば、消化不良、胃十二指腸出血、胃潰瘍及び胃炎である。「よりよい」アスピリンを見つけるための調査に多大な努力が費やされたにもかかわらず、優れた産物はまだ発見されていない。アスピリンは水中で非常に低い溶解性を有し、胃腸管に長時間滞留し得るため、胃粘膜細胞に損傷を引き起こす。O−アセチルサリチル酸と塩基性アミノ酸の安定塩は薬剤としての使用のために研究されたが(Franckowiak,et al.,U.S.Pat.No.6,773,724)、胃液のpHは1〜3であるため、おそらく塩がアセチルサリチル酸へと酸性化されるだろう。アルキル−又はアラルキル−置換アセチルサリチル酸、例えば、メチル−、エチル−、アリル−又はベンジルアセチル−サリチラートが合成され、研究された(Boghosian,et al.,U.S.Pat.No.4,244,948)。サリチル酸の高級脂肪酸誘導体及びその塩が、アスピリン治療可能な状態を治療する中で使用するために合成された(Guttag,U.S.Pat.No.5,760,261)。グアヤコールとのサルサラートのエステルが合成され、薬理学的に評価された(Nicolini,U.S.Pat.No.4,743,704)。
【0004】
アスピリンの経皮投与は多くの利点を有している(Kissel,U.S.Pat.No.5,861,170)。1.アスピリンが薬理学的活性体で循環系に直接的に導入されるため、胃腸管内での代謝を回避できること。2.胃腸の副作用の減少。3.低減量のアスピリンでの一定の治療効果。4.減少された過量投与のリスク。5.観察を必要としない外来患者の治療。6.改善された患者のコンプライアンス率。アスピリン又はその類似体を皮膚に適用する試みがすでになされている。Burton(Burton,U.S.Pat.No.4,012,508)は皮膚科学的適応の場合の中で局所応用のためにコルチコステロイドとの併用でアスピリンを用いた。Sibalis(Sibalis,U.S.Pat.No.4,640,689)は電流を用いることによってアスピリンの透過率の増大があることを記述した。しかしながら、これらの方法によって患者(host)に治療有効血漿レベルのアスピリンを送達することは難しい。Susan Milosovichらは塩酸テストステロニル−4−ジメチルアミノブチラート(testosteronyl-4-dimethylaminobutyrate.HCl)(TSBH)を設計し、調製した。これは親油性部分及び生理学的pHではプロトン化された形態で存在する第3級アミン基を有する。彼らは、プロドラッグ(TSBH)は、薬物(TS)それ自体よりも〜60倍速く人の皮膚を通して拡散することを見出した[Susan Milosovich,et al.,J.Pharm.Sci.,82,227(1993)]。
【発明の概要】
【発明が解決しようとする課題】
【0005】
アスピリンは非常に古いサリチラート薬(100年を超えている)であり、抗炎症、鎮痛、解熱及び抗リウマチ活性を示してきた。アスピリンはまた血小板凝集を抑制するために使用され、従って、心死亡率を減少させ、アスピリンの治療上有用性を増大させ続けている。「米国医薬品集」(PDR Generics,1996,second edition,Medical Economics,Montvale,New Jersey,pg243)にはアスピリンの多くの医薬用途が記載されている。残念ながら、多くの副作用がサリチラートの使用と関連付けられ、最も著しくは胃腸障害であり、例えば、消化不良、胸焼け、嘔吐、胃十二指腸出血、胃潰瘍及び胃炎である。サリチラートにより誘発される胃十二指腸出血は一般的に無痛性であるが、便による血液の喪失を招く可能性があり、持続性鉄欠乏性貧血を引き起こし得る。交差研究では、コーティングされていないアスピリン325mgの錠剤を1日に3回与えると、1日当たり平均4.33mlの便による血液の喪失を引き起こした(PDR Generics,1996,second edition,Medical Economics,Montvale,New Jersey,pg242)。コーティングされたアスピリンを同用量与えると、1日当たり平均1.5mlの便による血液の喪失を引き起こした。アスピリン及びそのエステルは水中で非常に低い溶解性を有し、胃腸管に長時間滞留し得て、胃粘膜細胞に損傷を引き起こす可能性がある。アスピリン及びそのエステルはまた非常に疎水性であり、細胞膜(疎水性層)に入ると、それらの類似性のため細胞膜の一部としてそこに留まるだろう。これらの理由から、アスピリンの吸収速度は非常に遅い。コーティングされていないアスピリンの錠剤が、サリチラートレベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。
【課題を解決するための手段】
【0006】
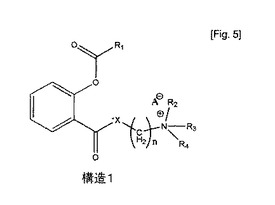
本発明は、アセチルサリチル酸又はその類似体の新規な正荷電プロドラッグの調製及びそれらの医薬用途に関する。これらのプロドラッグは式(1)の構造を有する。
【化1】
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【0007】
薬物吸収は、胃腸管からであろうと他の部位からであろうと、薬物が分子形で障壁膜を横断して通過することを必要とする。薬物は初めに溶解しなければならず、もし薬物が望ましい生物薬剤的性質を有しているならば、高濃度の領域から低濃度の領域へと膜を横断して通過し、血液循環又は体循環に入るだろう。全ての生物学的膜は主要な構成成分として脂質を含有している。膜の形態について主要な役割を果たしている分子は、全てホスファート含有高極性頭基(head group)を有し、たいていの場合、2つの高疎水性炭化水素尾部(tail)を有している。膜は2分子膜であり、その2つの親水性頭基は両側にある水性領域へと外側を向いている。非常に親水性な薬物は膜の疎水性層を通過することができず、非常に疎水性な薬物はそれらの類似性のため膜の一部として疎水性層に留まり、内側にある細胞質ゾルに能率的に入ることができないだろう。
【0008】
本発明の目的は、胃液中でのアセチルサリチル酸の溶解性を増大させること、並びにアスピリンを経皮投与可能(局所応用)にさせるであろう、膜及び皮膚障壁を通してのアスピリンの透過率を増大させることにより、アスピリンの副作用を回避することである。これらの新規なアセチルサリチル酸のプロドラッグは共通して2つの構造的特徴を有している。これらは、親油性部分及び生理学的pHではプロトン化された形態(親水性部分)で存在する第1級、第2級又は第3級アミン基を有している。膜障壁を通しての能率的な通過のためには、このような親水性親油性バランスが必要とされる[Susan Milosovich,et al.,J.Pharm.Sci.,82,227(1993)]。正に荷電されたアミノ基は薬物の溶解性を大きく増大させる。pH7ホスファート緩衝液中での酢酸ジエチルアミノエチルアセチルサリチラート(diethylaminoethyl acetylsalicylate.AcOH)及びアセチルサリチル酸の溶解性は、>300mg及び0.01mg/mlであった。多くの例では、一連の中での最も遅い又は律速段階は薬物の溶解である。アスピリンは胃液中で非常に低い溶解性を有する。アスピリンは胃腸管に長時間滞留し、胃粘膜細胞に損傷を引き起こし得る。これらの新規なプロドラッグは、錠剤、カプセル剤、液剤又は懸濁剤のような投与形態で経口投与されると、直ちに胃液中で溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、プロドラッグを半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中に押し込む。胃腸管での短い滞留のため、プロドラッグは胃粘膜細胞に損傷を引き起こさないだろう。
【0009】
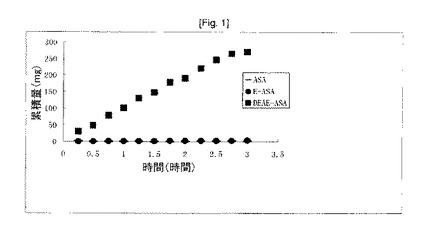
酢酸ジエチルアミノエチルアセチルサリチラート、エチルアセチルサリチラート及びアセチルサリチル酸の人の皮膚を通しての透過率は、修正フランツ(Franz)細胞を使用することによって試験管内で測定された。修正フランツ細胞は前大腿部及び後大腿部のヒト皮膚組織(厚さ360〜400μm)から単離された。受容液(receiving fluid)は生理食塩水中2%ウシ血清アルブミン10mlからなり、600rpmで撹拌した。時間に対する、皮膚を透過するアセチルサリチル酸、エチルアセチルサリチラート及び酢酸ジエチルアミノエチルアセチルサリチラートの累積量を特異的な高速液体クロマトグラフィー法により測定した。pH7.4ホスファート緩衝液(0.2M)2mL中、アセチルサリチル酸20%懸濁液、エチルアセチルサリチラート20%懸濁液、又は酢酸ジエチルアミノエチルアセチルサリチラート20%溶液のいずれかからなるドナーを使用して得た結果を図1に示す。アセチルサリチル酸、エチルアセチルサリチラート(アセチルサリチル酸の正に荷電されていない通常のエステル)及び酢酸ジエチルアミノエチルアセチルサリチラートに対して、0.25mg、1mg及び100mg/cm2/hという明白な流速値を計算した。本結果は、プロドラッグ、酢酸ジエチルアミノエチルアセチルサリチラートは、アセチルサリチル酸それ自体よりも〜400倍速く、エチルアセチルサリチラートよりも〜100倍速く人の皮膚を通して拡散することを示す。通常のエステル、エチルアセチルサリチラート、及びアセチルサリチル酸それ自体は非常に類似した透過率を有している(4倍の差にすぎない)。本結果は、ジアルキルアミノエチル基上の正電荷は、薬物が膜及び皮膚障壁を横断して通過することにおいて非常に重要な役割を有していることを示す。式(1)で表される他のプロドラッグは非常に高い透過率を有し、酢酸ジエチルアミノエチルアセチルサリチラートの透過率と非常に近似している。
【0010】
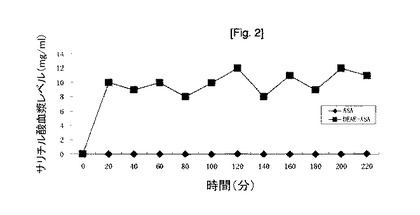
生体内での、アセチルサリチル酸及び酢酸ジエチルアミノエチルアセチルサリチラートの無処理ヘアレスマウスの皮膚を通しての透過率を比較した。イソプロパノール1mL中、アセチルサリチル酸30%懸濁液又は酢酸ジエチルアミノエチルアセチルサリチラート30%溶液のどちらかからなるドナーをヘアレスマウスの背中1cm2に塗布した。サリチル酸の血漿レベル(アセチルサリチル酸は非常に短い半減期、血漿中で〜15分、であるため、その量を測定するのは困難である)を、特異的な高速液体クロマトグラフィー法により測定した。本結果(図2)は、ドナー系の塗布後、〜20分でサリチル酸のピークレベルに達したことを示す。アスピリンを経口投与するとき、コーティングされていないアスピリンの錠剤がサリチラートレベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。サリチル酸のピークは〜0.01mg/mlであり、酢酸ジエチルアミノエチルアセチルサリチラートのピークは〜10mg/mlである(ほぼ100倍の差)。血漿中〜10mg/mlのサリチル酸は、効果的な鎮痛効果を生じるためのサリチラート血漿レベル(0.15〜0.3mg/ml)よりも33〜67倍を超えて高く、効果的な抗炎症活性を生じるためのサリチラート血漿レベル(0.2〜0.3mg/ml)よりも33〜50倍高い。これは非常に興奮させる結果である。これらのプロドラッグによって患者に治療有効血漿レベルのアスピリンを送達することは、非常に容易で迅速であろう。これらの結果は、様々な薬物治療のために、プロドラッグを経口投与だけでなく、経皮投与もし得ることを示す。生体内での、式(1)で表される他のプロドラッグの透過率は、酢酸ジエチルアミノエチルアセチルサリチラートの透過率と近似している。
【0011】
薬物によって引き起こされる胃十二指腸出血を調べるため、ラット(2つのグループがあり、それぞれのグループには10匹のラットがいる)に、1日当たりアスピリン又は酢酸ジエチルアミノエチルアセチルサリチラート100mg/kgを21日間経口投与した。アスピリンを投与したグループでは便のグラムあたり平均5mgの便血液があり、酢酸ジエチルアミノエチルアセチルサリチラートを投与したグループでは便血液がないことを見出した。
プロドラッグの急性毒性を調査した。ラットにおける経口での酢酸ジエチルアミノエチルアセチルサリチラート及び酢酸ジメチルアミノエチルアセチルサリチラートのLD50は2.3g/kg及び2.2gである。本結果は、プロドラッグはアスピリン(LD50=1.5g/kg)よりも低毒性であることを示す。
【0012】
アセチルサリチル酸は抗炎症、鎮痛、解熱及び抗リウマチ活性を示し、血小板凝集を抑制し、心死亡率を減少させるが、サリチル酸は鎮痛及び解熱活性のみ有する。
良いプロドラッグは血漿中で薬物それ自体に戻るべきである。酢酸ジエチルアミノエチルアセチルサリチラートは2つのエステル官能基を有する。一つ(アセチル)はアセチルサリチル酸それ自体からなり、一方はジエチルアミノエチルエステル(付加物)である。両方のエステル基は試験管内でヒト血漿中酵素によって速やかに切り離され得る。もしアセタート基がジエチルアミノエチルよりも早く失われるならば、プロドラッグはジエチルアミノエチルサリチラートに変化され、次いでアセチルサリチル酸の代わりにサリチル酸に変化されるだろう。酢酸ジエチルアミノエチルアセチルサリチラート及びアセチルサリチル酸は、ヒト全血中で非常に短い半減期(〜4分及び15分)を有する。どのくらいアセチルサリチル酸のプロドラッグが薬物それ自体又はジエチルアミノエチルサリチラートに変化して戻るかを測定するため、私たちはヒト全血をpH7.4ホスファート緩衝液(0.2M)で20倍に希釈した。ジエチルアミノエチルサリチラート、アセチルサリチル酸及びサリチル酸の累積量を特異的な高速液体クロマトグラフィー法によって測定した。ジエチルアミノエチルサリチラート対アセチルサリチル酸の割合は1対4であるという結果を示した。これは、プロドラッグの80%が薬物それ自体に変化して戻ったということを意味する。はるかに高い吸収率を有するプロドラッグのため、プロドラッグは同用量でアセチルサリチル酸それ自体よりも強い作用を有するだろう。
酢酸ジエチルアミノエチルアセチルサリチラートの鎮痛、解熱及び抗炎症活性を、対照としてアスピリンを使用して試験した。式(1)で表される他の化合物を同じ方法により試験したところ、酢酸ジエチルアミノエチルアセチルサリチラートの結果と非常に類似した結果を有する。
【0013】
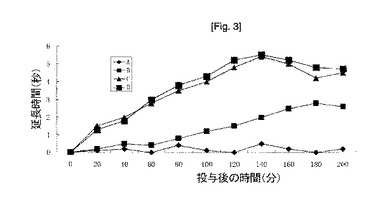
鎮痛活性:マウス尻尾の痛覚閾値延長時間をD'Amour-Smith法(J.Pharmacol.Exp.Ther.,72,74(1941))に従って測定した。アスピリン200mg/kgを経口投与、並びに酢酸ジエチルアミノエチルサリチラート200mg/kgを経口及び経皮投与した後、マウスの尻尾を熱にさらし、痛覚閾値延長時間を測定した。得られた結果を図3に示す。図3では、酢酸ジエチルアミノエチルサリチラート200mg/kgを経口投与したグループ(C)及び経皮投与したグループ(D)は、アスピリン200mg/kgを投与したグループよりも強い鎮痛活性を表すことが示された。
酢酸溶液をマウスの腹腔内に投与したときに起こるライジング数(writhing)を数え、対照群に基づく抑制率を計算した。42匹のマウスを7つのグループ(それぞれ6匹)に分けた。アスピリン(ASA,50mg及び100mg/kg)をB1及びB2グループに投与し、酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,50mg及び100mg/kg)をC1及びC2グループに経口投与した。酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,50mg及び100mg/kg)をD1及びD2グループに経皮投与した。Aグループは対照群である。酢酸溶液を投与する30分前に、マウスに試験化合物を投与した。結果を以下の表1に示す。
表1.アスピリン及びそのプロドラッグによるライジング抑制率
本結果は、酢酸ジエチルアミノエチルアセチルサリチラートはアスピリンよりも良い鎮痛活性を表すということを示す。式(1)で表される他の化合物は類似した鎮痛活性を示す。
【0014】
解熱活性:ラットは発熱物質として滅菌された大腸菌懸濁液を受容した。56匹のラットを7つのグループに分けた。対照群はグループAである。2時間後、アスピリン(ASA,B1に100mg/kg、B2に150mg/kg)及び酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,C1に100mg/kg、C2に150mg/kg)を経口投与し、酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,D1に100mg/kg、D2に150mg/kg)を経皮投与した。ラットの体温を試験化合物の投与前後90分間隔で測った。結果を以下の表2に示す。
表2.アスピリン及びそのプロドラッグの解熱活性
本結果から、酢酸ジエチルアミノエチルアセチルサリチラートは100mg/kg用量において解熱活性を示し、でアスピリンよりも良い解熱活性を示したということが分かる。本結果は、酢酸ジエチルアミノエチルアセチルサリチラートの経皮投与は経口投与よりも良いということを示す。式(1)で表される他の化合物は類似した解熱活性を示す。
【0015】
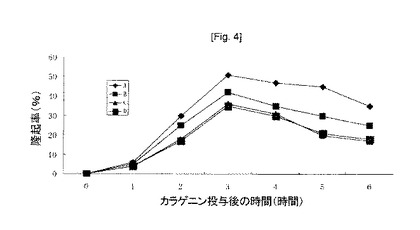
抗炎症活性:酢酸ジエチルアミノエチルアセチルサリチラート50mg/kgをラットに経口又は経皮投与し、アスピリン50mg/kgを経口投与した。60分後、カラゲニン溶液をラットの足裏に皮下投与した。後ろ足の体積をカラゲニンの投与後毎時間測定し、足の体積の増加率を計算し、隆起率(rate of swelling)(%)と称した。得られた結果を図4に示す。
本結果は、酢酸ジエチルアミノエチルアセチルサリチラートは、50mg/kgでの経口投与及び経皮投与に対して、アスピリンの抗炎症活性よりも良い抗炎症活性を表したということを示す。式(1)で表される他の化合物は類似した抗炎症活性を示す。
【0016】
高用量の経口アセチルサリチル酸はまた、シクロオキシゲナーゼ活性の抑制による抗反応−抗喘息活性を示すということも知られている(Bianco,Sebastiano,U.S.Pat.No.5,570,559)。それらの非常に高い膜透過率のため、これらのプロドラッグは患者の口又は鼻の中にスプレーすることにより喘息の治療に使用され得る。これらは抗炎症性のため、アクネを治療するために使用され得る。これらは内皮機能障害の治療及び予防に対しても同様に使用され得る。
【0017】
上記の式(1)で表される化合物はASA若しくはその類似体、又はASAの官能誘導体若しくはその類似体から調製され得る。例えば、式(2)で表される酸ハロゲン化物又は混合酸無水物である。
【化2】
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、Yはハロゲン、アルコキシカルボニル又は置換アリールオキシカルボニルオキシを表し、これは式(3)で表される化合物と反応する。
【化3】
式中、R1はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、n=0、1、2、3、4、5……である。
【0018】
上記の式(1)で表される化合物はASA又はその類似体から、カップリング試薬、例えば、N,N’−ジシクロヘキシルカルボジイミド、N,N’−ジイソプロピルカルボジイミド、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボラート、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスファート、ベンゾトリアゾール−1−イル−オキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスファート等を使用することにより、式(3)で表される化合物との反応によって調製され得る。
XがOを表すとき、上記の式(1)で表される化合物は、アセチルサリチル酸若しくはその類似体の金属塩又は有機塩基塩から、式(4)で表される化合物との反応によって調製され得る。
【化4】
式中、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、Zはハロゲン又はp−トルエンスルホニルを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【0019】
XがOを表すとき、上記の式(1)で表される化合物は、式(5)で表される、アセチルサリチル酸又はその類似体の固定化された塩基塩から調製され得る。
【化5】
式中、Rは交差結合樹脂を表し、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、Bは塩基、例えば、ピリジン、ピペリジン、トリエチルアミン又は他の塩基を表す。これは式(4)で表される化合物と反応する。
【0020】
本発明は、通例、補助剤及び賦形剤に加えて、例えば、経口投与のための錠剤、カプセル剤又は液剤の形態で、及び経皮投与のための液剤、ローション剤、軟膏剤、乳剤又はゲル剤の形態で、式(1)で表されるアセチルサリチル酸及びその類似体のプロドラッグを含む薬剤調製に関する。式(1)で表される新規な活性化合物は、人又は動物において、アスピリン治療可能な状態を治療するために、ビタミン、例えばA、B、C、E若しくはベータ−カロテン、又は他の薬物、例えば、葉酸等と併用され得る。
式(1)で表される化合物又は少なくとも式(1)で表される化合物を活性成分として含む組成物の経皮治療応用システムは、人又は動物において、アスピリン治療可能な状態を治療するために、特に血栓症及び他の心臓血管疾患の予防、並びに癌予防に対して発展され得る。これらのシステムは、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得る。最も好ましいシステムは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有する。放出の割合を制御することによって、このシステムは、アスピリンが絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、アスピリンの副作用を減少させることを可能にする。これらのシステムは手首、足首、腕、足又は体のどこでも付けることが可能である。
【発明の効果】
【0021】
これらのアスピリンのプロドラッグは、親油性部分及び親水性部分(生理学的pHではプロトン化された形態で存在するアミン基)を有する。これらのプロドラッグの正に荷電されたアミノ基は2つの優れた利点を有する。第一に、薬物の溶解性を大きく増大させる。これらの新規なプロドラッグが、錠剤、カプセル剤、液剤又は懸濁剤のような投与形態で経口投与されるとき、これらは直ちに胃液中で溶解するだろう。第二に、これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、プロドラッグを半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中に押し込む。胃腸管での短い滞留のため、プロドラッグは胃粘膜細胞に損傷を引き起こさないだろう。実験結果は、プロドラッグの80%は薬物それ自体に変化して戻ったということを示す。プロドラッグははるかに高い吸収率を有するため、従って、プロドラッグは同用量でアセチルサリチル酸それ自体よりも強い作用を有するだろう。
実験結果は、プロドラッグ、酢酸ジエチルアミノエチルアセチルサリチラートはアセチルサリチル酸それ自体よりも〜400倍速く、エチルアセチルサリチラートよりも〜100倍速く人の皮膚を通して拡散することを示す。生体内での、酢酸ジエチルアミノエチルアセチルサリチラートの無処理ヘアレスマウスの皮膚を通しての透過率は非常に高かった。アスピリンを経口投与するとき、コーティングされていないアスピリンの錠剤がサリチラート血漿レベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。しかし、酢酸ジエチルアミノエチルアセチルサリチラートは、サリチラート血漿レベルのピークに達するのにたったの20分しかかからなかった。最も興奮させる結果は、プロドラッグを経口投与だけでなく、様々な種類の薬物治療のために経皮投与することも可能であり、たいていのアスピリンの副作用、最も著しくは胃腸障害であり、例えば、消化不良、胃十二指腸出血、胃潰瘍及び胃炎、を回避するだろうということである。
【図面の簡単な説明】
【0022】
【図1】フランツ細胞(n=5)での、単離されたヒト皮膚組織を透過するアセチルサリチル酸(ASA)、エチルアセチルサリチラート(E−ASA)及び酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA)の累積量。ASA及びE−ASAを20%懸濁液として用いた。DEAE−ASAを20%溶液として用いた。それぞれ、媒体はpH7.4ホスファート緩衝液(0.2M)であった。
【図2】アセチルサリチル酸(ASA)又は酢酸ジエチルアミノエチルアセチルサリチラート300mgをヘアレスマウス(n=5)の背中に局所塗布後の、サリチル酸(SA)総血漿レベル。
【図3】アスピリン200mg/kg経口投与後(B)、酢酸ジエチルアミノエチルサリチラート200mg/kg経口投与後(C)及び経皮投与後(D)の、マウス尻尾の痛覚閾値延長時間。Aは対照線である。
【図4】カラゲニン投与後の隆起率(%)。カラゲニン投与1時間前に、アスピリン100mg/kg経口投与(B)、酢酸ジエチルアミノエチルサリチラート100mg/kg経口投与(C)及び経皮投与(D)した。Aは対照線である。 構造1.式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ又はアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ又はアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=1、2、3、4、5……である。
【発明を実施するための形態】
【0023】
酢酸ジエチルアミノエチルアセチルサリチラートの調製
o−アセチルサリチル酸18g(0.1mol)をクロロホルム180mlに溶解した。重炭酸ナトリウム12.5g(0.15mol)を溶液に加えた。水(20ml)を撹拌しながら加えた。混合物を30分間撹拌した後、無水硫酸ナトリウムを加えた。臭化水素酸ジエチルアミノエチルブロミド(diethylaminoethyl bromide.HBr)39g(0.15mol)を混合物に加え、混合物を室温で5時間撹拌した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物31g(91%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:339.38、計算%C:60.07、H:7.44、N:4.15、O:28.22、実測%C:60.16、H:7.42、N:4.13、O:28.29、1H-NMR(400MHz,CDCL3):デルタ:1.55(t,6H)、2.08(s,3H)、2.20(s,3H)、3.28(m,4H)、3.70(m,2H)、4.68(m,2H)、6.5(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【0024】
酢酸ジメチルアミノエチルアセチルサリチラートの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエタノール8.9gを反応混合物に加えた。混合物を室温で3時間撹拌する。酢酸6gを撹拌しながら反応混合物に加える。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物29g(93%)を得た。吸湿性生成物、水中での溶解性:350mg/ml、元素分析:C15H21NO6、分子量:311.33、計算%C:57.87、H:6.80、N:4.50、O:30.83、実測%C:57.82、H:6.85、N:4.48、O:30.85、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.71(m,2H)、4.69(m,2H)、6.9(b,1H)、7.18(m,1H)、7.20(m,1H)、7.47(m,1H)、7.93(m,1H)。
【0025】
酢酸S−ジメチルアミノエチルアセチルチオサリチラートの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエチルメルカプタン9.3gを反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物28g(87%)を得た。吸湿性生成物、水中での溶解性:320mg/ml、元素分析:C15H21NO5S、分子量:327.4、計算%C:55.03、H:6.47、N:4.28、O:24.43、S:9.79、実測%C:55.02、H:6.45、N:4.35、O:24.49、S:9.69、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.31(t,2H)、3.91(m,2H)、6.9(b,1H)、7.26(m,1H)、7.28(m,1H)、7.55(m,1H)、7.94(m,1H)。
【0026】
酢酸N−ジメチルアミノエチルアセチルサリチルアミドの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエチルアミン8.9gを反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物28g(90.2%)を得た。吸湿性生成物、水中での溶解性:350mg/ml、元素分析:C15H22N2O5、分子量:310.35、計算%C:58.05、H:7.15、N:9.03、O:25.78、実測%C:58.02、H:7.18、N:8.98、O:25.83、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.54(m,2H)、3.64(t,2H)、6.9(b,1H)、7.8(b,1H)、7.25(m,1H)、7.26(m,1H)、 7.48(m,1H)、7.92(m,1H)。
【0027】
酢酸S−ジエチルアミノエチルプロピオニルチオサリチラートの調製
o−アセチルサリチル酸18g(0.1mol)をジクロロメタン(DCM)100mlに溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド20.6gを反応混合物に加えた。混合物を0℃で30分間撹拌した。ジエチルアミノプロピルメルカプタン14.8g(0.1mol)を反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物32g(86.6%)を得た。吸湿性生成物、水中での溶解性:280mg/ml、元素分析:C18H27NO5S、分子量:369.48、計算%C:58.51、H:7.37、N:3.79、O:21.65、S:8.68、実測%C:58.53、H:7.39、N:3.75、O:21.68、S:8.65、1H-NMR(400MHz,CDCL3):デルタ:1.09(t,3H)、1.56(t,6H)、2.21(s,3H)、2.27(m,2H)、3.28(m,4H)、3.31(m,2H)、3.91(m,2H)、6.8(b,1H)、7.25(m,1H)、7.26(m,1H)、7.48(m,1H)、7.92(m,1H)。
【0028】
酢酸N−ジエチルアミノプロピルアセチルサリチルアミドの調製
o−アセチルサリチル酸18g(0.1mol)をアセトニトリル100mlに溶解した。O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボラート32.1g及びトリエチルアミン30mlを反応混合物に加えた。ジメチルアミノプロピルアミン13.1gを反応混合物に加えた。混合物を室温で3時間撹拌した。溶媒を蒸発させた。酢酸エチル250mlを反応混合物に加え、混合物を水(3×100ml)で洗浄した。有機溶液を無水硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去した。酢酸6gを撹拌しながら反応混合物に加えた。ヘキサン(200ml)を加えた。固形生成物を濾過により集めた。乾燥後、目的生成物32g(90.8%)を得た。吸湿性生成物、水中での溶解性:280mg/ml、元素分析:C18H28N2O5、分子量:352.43、計算%C:61.34、H:8.01、N:7.95、O:22.70、実測%C:61.25、H:8.05、N:7.96、O:22.74、1H-NMR(400MHz,CDCL3):デルタ: 1.56(t,6H)、2.03(m,2H)、2.09(s,3H)、2.21(s,3H)、3.24(m,2H)、3.20(m,2H)、3.24(m,2H)、6.9(b,1H)、7.8(b,1H)、7.25(m,1H)、7.26(m,1H)、7.48(m,1H)、7.92(m,1H)。
【0029】
酢酸ジプロピルアミノエチルアセチルサリチラートの調製
o−アセチルサリチル酸ナトリウム20.3g(0.1mol)をクロロホルム180mlに懸濁した。臭化水素酸ジプロピルアミノエチルブロミド28.8g(0.1mol)を混合物に加え、混合物を室温で5時間撹拌した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物30g(81.6%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:367.44、計算%C:62.11、H:7.96、N:3.81、O:26.13、実測%C:62.07、H:7.99、N:3.78、O:26.17、1H-NMR(400MHz,CDCL3):デルタ:0.97(t,6H)、1.77(m,4H)、2.20(s,3H)、3.25(m,4H)、3.70(m,2H)、4.69(m,2H)、6.8(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【0030】
酢酸ジプロピルアミノエチルアセチルサリチラートの調製
高分子結合トリエチルアミン(3mmol/g、100〜200mesh)60gをクロロホルム180mlに懸濁した。o−アセチルサリチル酸18g(0.1mol)を撹拌しながら混合物に加えた。臭化水素酸ジプロピルアミノエチルブロミド43g(0.15mol)を混合物に加え、混合物を室温で5時間撹拌した。高分子を濾過により除去し、テトラヒドロフラン(3×50ml)で洗浄した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物31g(91%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:339.38、計算%C:60.07、H:7.44、N:4.15、O:28.22、実測%C:60.16、H:7.42、N:4.13、O:28.29、1H-NMR(400MHz,CDCL3):デルタ:1.55(t,6H)、2.08(s,3H)、2.20(s,3H)、3.28(m,4H)、3.70(m,2H)、4.68(m,2H)、6.5(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【産業上の利用可能性】
【0031】
式(1)で表されるプロドラッグはアスピリンよりも優れている。これらは人又は動物において、アスピリン治療可能な状態の治療に医薬的に使用され得る。これらは軽度から中程度の疼痛並びに関節炎及び他の炎症性状態の長期緩和待期療法に使用され得る。これらは単独で又は補助薬として、川崎病、術後血栓塞栓症及び不安定狭心症の治療に使用され得る。これらは急性小児胃腸炎において、便の量を減少させ、かつ体重を増加させるために、大動脈冠状動脈バイパス移植閉塞を予防するために補助薬として使用され得て、また慢性心房細動における血栓塞栓性合併症を予防するために使用され得る。これらは補助薬として、頚動脈内膜切除術中の血小板凝集及びトロンボキサン抑制を減らすために使用され得、白内障の進行を遅らせるため、冠状動脈形成術後狭窄の再発を予防するため、及び多発脳梗塞性痴呆患者における認知機能及び大脳の血流を改善するために処方され得る。これらは補助薬として、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含め糖尿病誘発性合併症において血漿グルコースを下げるために使用され、そして総死亡率及び心血管死亡率を減少させるために使用され得る。これらは血液透析シャント血栓症の発生を減らすため、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生を減少させるため、及び末梢閉塞性動脈疾患の進行を遅らせるために処方され得る。これらは大腸癌及び直腸癌、並びに人工心臓弁患者における動脈塞栓性合併症の予防に使用され得る。これらはハイリスク女性における妊娠誘発性高血圧及び妊娠中毒症の発生を減らすために処方され得る。高用量の経口アセチルサリチル酸はまた、シクロオキシゲナーゼ活性の抑制による抗反応活性を示すということも知られている(Bianco,Sebastiano,U.S.Pat.No.5,570,559)。それらの非常に高い膜透過率のため、これらのプロドラッグは患者に吸入することによって喘息の治療に使用され得る。これらは抗炎症性のため、アクネを治療するために使用され得る。これらは内皮機能障害の治療及び予防に対して使用され得る。
【技術分野】
【0001】
本発明は、アスピリン又はその類似体の正に荷電された水溶性プロドラッグの調製剤、及び人又は動物において、アスピリン治療可能な状態を治療する際のその医薬用途に関する。
【背景技術】
【0002】
アセチルサリチル酸(アスピリン)は1853年に合成され、1899年に最初に医薬的に使用された。それ以来、サリチル酸の多数の誘導体が合成され、薬理学的に評価されてきたが、比較的ほとんどの誘導体は治療上有用性を達成していない。アスピリンは解熱、鎮痛及び抗炎症作用を有する。サリチラートは尿酸の排出を促進するので、痛風関節炎の治療に有益である。アスピリンはまた、潜在的に心臓発作及び脳卒中に寄与する血小板凝集を抑制し[C.H.Hennekens,et al.,N.Engl.J.Med.,321,129(1989);T.A.Gossel,U.S.Pharmacist,February,1988,p.34.]、同様に大腸癌に対して防御的となり得る[M.J.Thun,et al.,N.Engl.J.Med.,325,1393(1991)]。従って、アスピリンの治療上有用性は重要であり、増大し続けている。「米国医薬品集」(PDR Generics,1996,second edition,MedicalEconomics,Montvale,New Jersey,pg243)にはアスピリンの多くの医薬用途が記載されている。アスピリンはまた、軽度から中程度の疼痛並びに関節炎及び他の炎症性状態の長期緩和待期療法に使用されている。アスピリンは単独で又は補助薬として、川崎病、術後血栓塞栓症及び不安定狭心症の治療に使用される。アスピリンはまた急性小児胃腸炎において、便の量を減少させ、かつ体重を増加させるために、大動脈冠状動脈バイパス移植閉塞を予防する際の補助薬として使用されて、また慢性心房細動における血栓塞栓性合併症を予防するために使用される。アスピリンは、補助薬として、頚動脈内膜切除術中の血小板凝集を減らすために使用され、白内障の進行を遅らせるために処方され、冠状動脈形成術後狭窄の再発を予防するために使用され、多発脳梗塞性痴呆患者における認知機能及び大脳の血流を改善するために使用される。アスピリンは補助薬として、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含め糖尿病誘発性合併症において血漿グルコースを下げるために使用され、そして総死亡率及び心血管死亡率を減少させるために使用される。アスピリンは血液透析シャント血栓症(hemodialysis shunt thrombosis)の発生を減らすため、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生を減少させるため、及び末梢閉塞性動脈疾患の進行を遅らせるために処方される。アスピリンは大腸癌、直腸癌及び人工心臓弁患者における動脈塞栓性合併症の予防に使用される。アスピリンはまたハイリスク女性における妊娠誘発性高血圧及び妊娠中毒症の発生を減らすために処方される。
【0003】
残念ながら、多くの副作用がサリチラートの使用と関連付けられ、最も著しくは胃腸障害であり、例えば、消化不良、胃十二指腸出血、胃潰瘍及び胃炎である。「よりよい」アスピリンを見つけるための調査に多大な努力が費やされたにもかかわらず、優れた産物はまだ発見されていない。アスピリンは水中で非常に低い溶解性を有し、胃腸管に長時間滞留し得るため、胃粘膜細胞に損傷を引き起こす。O−アセチルサリチル酸と塩基性アミノ酸の安定塩は薬剤としての使用のために研究されたが(Franckowiak,et al.,U.S.Pat.No.6,773,724)、胃液のpHは1〜3であるため、おそらく塩がアセチルサリチル酸へと酸性化されるだろう。アルキル−又はアラルキル−置換アセチルサリチル酸、例えば、メチル−、エチル−、アリル−又はベンジルアセチル−サリチラートが合成され、研究された(Boghosian,et al.,U.S.Pat.No.4,244,948)。サリチル酸の高級脂肪酸誘導体及びその塩が、アスピリン治療可能な状態を治療する中で使用するために合成された(Guttag,U.S.Pat.No.5,760,261)。グアヤコールとのサルサラートのエステルが合成され、薬理学的に評価された(Nicolini,U.S.Pat.No.4,743,704)。
【0004】
アスピリンの経皮投与は多くの利点を有している(Kissel,U.S.Pat.No.5,861,170)。1.アスピリンが薬理学的活性体で循環系に直接的に導入されるため、胃腸管内での代謝を回避できること。2.胃腸の副作用の減少。3.低減量のアスピリンでの一定の治療効果。4.減少された過量投与のリスク。5.観察を必要としない外来患者の治療。6.改善された患者のコンプライアンス率。アスピリン又はその類似体を皮膚に適用する試みがすでになされている。Burton(Burton,U.S.Pat.No.4,012,508)は皮膚科学的適応の場合の中で局所応用のためにコルチコステロイドとの併用でアスピリンを用いた。Sibalis(Sibalis,U.S.Pat.No.4,640,689)は電流を用いることによってアスピリンの透過率の増大があることを記述した。しかしながら、これらの方法によって患者(host)に治療有効血漿レベルのアスピリンを送達することは難しい。Susan Milosovichらは塩酸テストステロニル−4−ジメチルアミノブチラート(testosteronyl-4-dimethylaminobutyrate.HCl)(TSBH)を設計し、調製した。これは親油性部分及び生理学的pHではプロトン化された形態で存在する第3級アミン基を有する。彼らは、プロドラッグ(TSBH)は、薬物(TS)それ自体よりも〜60倍速く人の皮膚を通して拡散することを見出した[Susan Milosovich,et al.,J.Pharm.Sci.,82,227(1993)]。
【発明の概要】
【発明が解決しようとする課題】
【0005】
アスピリンは非常に古いサリチラート薬(100年を超えている)であり、抗炎症、鎮痛、解熱及び抗リウマチ活性を示してきた。アスピリンはまた血小板凝集を抑制するために使用され、従って、心死亡率を減少させ、アスピリンの治療上有用性を増大させ続けている。「米国医薬品集」(PDR Generics,1996,second edition,Medical Economics,Montvale,New Jersey,pg243)にはアスピリンの多くの医薬用途が記載されている。残念ながら、多くの副作用がサリチラートの使用と関連付けられ、最も著しくは胃腸障害であり、例えば、消化不良、胸焼け、嘔吐、胃十二指腸出血、胃潰瘍及び胃炎である。サリチラートにより誘発される胃十二指腸出血は一般的に無痛性であるが、便による血液の喪失を招く可能性があり、持続性鉄欠乏性貧血を引き起こし得る。交差研究では、コーティングされていないアスピリン325mgの錠剤を1日に3回与えると、1日当たり平均4.33mlの便による血液の喪失を引き起こした(PDR Generics,1996,second edition,Medical Economics,Montvale,New Jersey,pg242)。コーティングされたアスピリンを同用量与えると、1日当たり平均1.5mlの便による血液の喪失を引き起こした。アスピリン及びそのエステルは水中で非常に低い溶解性を有し、胃腸管に長時間滞留し得て、胃粘膜細胞に損傷を引き起こす可能性がある。アスピリン及びそのエステルはまた非常に疎水性であり、細胞膜(疎水性層)に入ると、それらの類似性のため細胞膜の一部としてそこに留まるだろう。これらの理由から、アスピリンの吸収速度は非常に遅い。コーティングされていないアスピリンの錠剤が、サリチラートレベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。
【課題を解決するための手段】
【0006】
本発明は、アセチルサリチル酸又はその類似体の新規な正荷電プロドラッグの調製及びそれらの医薬用途に関する。これらのプロドラッグは式(1)の構造を有する。
【化1】
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【0007】
薬物吸収は、胃腸管からであろうと他の部位からであろうと、薬物が分子形で障壁膜を横断して通過することを必要とする。薬物は初めに溶解しなければならず、もし薬物が望ましい生物薬剤的性質を有しているならば、高濃度の領域から低濃度の領域へと膜を横断して通過し、血液循環又は体循環に入るだろう。全ての生物学的膜は主要な構成成分として脂質を含有している。膜の形態について主要な役割を果たしている分子は、全てホスファート含有高極性頭基(head group)を有し、たいていの場合、2つの高疎水性炭化水素尾部(tail)を有している。膜は2分子膜であり、その2つの親水性頭基は両側にある水性領域へと外側を向いている。非常に親水性な薬物は膜の疎水性層を通過することができず、非常に疎水性な薬物はそれらの類似性のため膜の一部として疎水性層に留まり、内側にある細胞質ゾルに能率的に入ることができないだろう。
【0008】
本発明の目的は、胃液中でのアセチルサリチル酸の溶解性を増大させること、並びにアスピリンを経皮投与可能(局所応用)にさせるであろう、膜及び皮膚障壁を通してのアスピリンの透過率を増大させることにより、アスピリンの副作用を回避することである。これらの新規なアセチルサリチル酸のプロドラッグは共通して2つの構造的特徴を有している。これらは、親油性部分及び生理学的pHではプロトン化された形態(親水性部分)で存在する第1級、第2級又は第3級アミン基を有している。膜障壁を通しての能率的な通過のためには、このような親水性親油性バランスが必要とされる[Susan Milosovich,et al.,J.Pharm.Sci.,82,227(1993)]。正に荷電されたアミノ基は薬物の溶解性を大きく増大させる。pH7ホスファート緩衝液中での酢酸ジエチルアミノエチルアセチルサリチラート(diethylaminoethyl acetylsalicylate.AcOH)及びアセチルサリチル酸の溶解性は、>300mg及び0.01mg/mlであった。多くの例では、一連の中での最も遅い又は律速段階は薬物の溶解である。アスピリンは胃液中で非常に低い溶解性を有する。アスピリンは胃腸管に長時間滞留し、胃粘膜細胞に損傷を引き起こし得る。これらの新規なプロドラッグは、錠剤、カプセル剤、液剤又は懸濁剤のような投与形態で経口投与されると、直ちに胃液中で溶解するだろう。これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、プロドラッグを半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中に押し込む。胃腸管での短い滞留のため、プロドラッグは胃粘膜細胞に損傷を引き起こさないだろう。
【0009】
酢酸ジエチルアミノエチルアセチルサリチラート、エチルアセチルサリチラート及びアセチルサリチル酸の人の皮膚を通しての透過率は、修正フランツ(Franz)細胞を使用することによって試験管内で測定された。修正フランツ細胞は前大腿部及び後大腿部のヒト皮膚組織(厚さ360〜400μm)から単離された。受容液(receiving fluid)は生理食塩水中2%ウシ血清アルブミン10mlからなり、600rpmで撹拌した。時間に対する、皮膚を透過するアセチルサリチル酸、エチルアセチルサリチラート及び酢酸ジエチルアミノエチルアセチルサリチラートの累積量を特異的な高速液体クロマトグラフィー法により測定した。pH7.4ホスファート緩衝液(0.2M)2mL中、アセチルサリチル酸20%懸濁液、エチルアセチルサリチラート20%懸濁液、又は酢酸ジエチルアミノエチルアセチルサリチラート20%溶液のいずれかからなるドナーを使用して得た結果を図1に示す。アセチルサリチル酸、エチルアセチルサリチラート(アセチルサリチル酸の正に荷電されていない通常のエステル)及び酢酸ジエチルアミノエチルアセチルサリチラートに対して、0.25mg、1mg及び100mg/cm2/hという明白な流速値を計算した。本結果は、プロドラッグ、酢酸ジエチルアミノエチルアセチルサリチラートは、アセチルサリチル酸それ自体よりも〜400倍速く、エチルアセチルサリチラートよりも〜100倍速く人の皮膚を通して拡散することを示す。通常のエステル、エチルアセチルサリチラート、及びアセチルサリチル酸それ自体は非常に類似した透過率を有している(4倍の差にすぎない)。本結果は、ジアルキルアミノエチル基上の正電荷は、薬物が膜及び皮膚障壁を横断して通過することにおいて非常に重要な役割を有していることを示す。式(1)で表される他のプロドラッグは非常に高い透過率を有し、酢酸ジエチルアミノエチルアセチルサリチラートの透過率と非常に近似している。
【0010】
生体内での、アセチルサリチル酸及び酢酸ジエチルアミノエチルアセチルサリチラートの無処理ヘアレスマウスの皮膚を通しての透過率を比較した。イソプロパノール1mL中、アセチルサリチル酸30%懸濁液又は酢酸ジエチルアミノエチルアセチルサリチラート30%溶液のどちらかからなるドナーをヘアレスマウスの背中1cm2に塗布した。サリチル酸の血漿レベル(アセチルサリチル酸は非常に短い半減期、血漿中で〜15分、であるため、その量を測定するのは困難である)を、特異的な高速液体クロマトグラフィー法により測定した。本結果(図2)は、ドナー系の塗布後、〜20分でサリチル酸のピークレベルに達したことを示す。アスピリンを経口投与するとき、コーティングされていないアスピリンの錠剤がサリチラートレベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。サリチル酸のピークは〜0.01mg/mlであり、酢酸ジエチルアミノエチルアセチルサリチラートのピークは〜10mg/mlである(ほぼ100倍の差)。血漿中〜10mg/mlのサリチル酸は、効果的な鎮痛効果を生じるためのサリチラート血漿レベル(0.15〜0.3mg/ml)よりも33〜67倍を超えて高く、効果的な抗炎症活性を生じるためのサリチラート血漿レベル(0.2〜0.3mg/ml)よりも33〜50倍高い。これは非常に興奮させる結果である。これらのプロドラッグによって患者に治療有効血漿レベルのアスピリンを送達することは、非常に容易で迅速であろう。これらの結果は、様々な薬物治療のために、プロドラッグを経口投与だけでなく、経皮投与もし得ることを示す。生体内での、式(1)で表される他のプロドラッグの透過率は、酢酸ジエチルアミノエチルアセチルサリチラートの透過率と近似している。
【0011】
薬物によって引き起こされる胃十二指腸出血を調べるため、ラット(2つのグループがあり、それぞれのグループには10匹のラットがいる)に、1日当たりアスピリン又は酢酸ジエチルアミノエチルアセチルサリチラート100mg/kgを21日間経口投与した。アスピリンを投与したグループでは便のグラムあたり平均5mgの便血液があり、酢酸ジエチルアミノエチルアセチルサリチラートを投与したグループでは便血液がないことを見出した。
プロドラッグの急性毒性を調査した。ラットにおける経口での酢酸ジエチルアミノエチルアセチルサリチラート及び酢酸ジメチルアミノエチルアセチルサリチラートのLD50は2.3g/kg及び2.2gである。本結果は、プロドラッグはアスピリン(LD50=1.5g/kg)よりも低毒性であることを示す。
【0012】
アセチルサリチル酸は抗炎症、鎮痛、解熱及び抗リウマチ活性を示し、血小板凝集を抑制し、心死亡率を減少させるが、サリチル酸は鎮痛及び解熱活性のみ有する。
良いプロドラッグは血漿中で薬物それ自体に戻るべきである。酢酸ジエチルアミノエチルアセチルサリチラートは2つのエステル官能基を有する。一つ(アセチル)はアセチルサリチル酸それ自体からなり、一方はジエチルアミノエチルエステル(付加物)である。両方のエステル基は試験管内でヒト血漿中酵素によって速やかに切り離され得る。もしアセタート基がジエチルアミノエチルよりも早く失われるならば、プロドラッグはジエチルアミノエチルサリチラートに変化され、次いでアセチルサリチル酸の代わりにサリチル酸に変化されるだろう。酢酸ジエチルアミノエチルアセチルサリチラート及びアセチルサリチル酸は、ヒト全血中で非常に短い半減期(〜4分及び15分)を有する。どのくらいアセチルサリチル酸のプロドラッグが薬物それ自体又はジエチルアミノエチルサリチラートに変化して戻るかを測定するため、私たちはヒト全血をpH7.4ホスファート緩衝液(0.2M)で20倍に希釈した。ジエチルアミノエチルサリチラート、アセチルサリチル酸及びサリチル酸の累積量を特異的な高速液体クロマトグラフィー法によって測定した。ジエチルアミノエチルサリチラート対アセチルサリチル酸の割合は1対4であるという結果を示した。これは、プロドラッグの80%が薬物それ自体に変化して戻ったということを意味する。はるかに高い吸収率を有するプロドラッグのため、プロドラッグは同用量でアセチルサリチル酸それ自体よりも強い作用を有するだろう。
酢酸ジエチルアミノエチルアセチルサリチラートの鎮痛、解熱及び抗炎症活性を、対照としてアスピリンを使用して試験した。式(1)で表される他の化合物を同じ方法により試験したところ、酢酸ジエチルアミノエチルアセチルサリチラートの結果と非常に類似した結果を有する。
【0013】
鎮痛活性:マウス尻尾の痛覚閾値延長時間をD'Amour-Smith法(J.Pharmacol.Exp.Ther.,72,74(1941))に従って測定した。アスピリン200mg/kgを経口投与、並びに酢酸ジエチルアミノエチルサリチラート200mg/kgを経口及び経皮投与した後、マウスの尻尾を熱にさらし、痛覚閾値延長時間を測定した。得られた結果を図3に示す。図3では、酢酸ジエチルアミノエチルサリチラート200mg/kgを経口投与したグループ(C)及び経皮投与したグループ(D)は、アスピリン200mg/kgを投与したグループよりも強い鎮痛活性を表すことが示された。
酢酸溶液をマウスの腹腔内に投与したときに起こるライジング数(writhing)を数え、対照群に基づく抑制率を計算した。42匹のマウスを7つのグループ(それぞれ6匹)に分けた。アスピリン(ASA,50mg及び100mg/kg)をB1及びB2グループに投与し、酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,50mg及び100mg/kg)をC1及びC2グループに経口投与した。酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,50mg及び100mg/kg)をD1及びD2グループに経皮投与した。Aグループは対照群である。酢酸溶液を投与する30分前に、マウスに試験化合物を投与した。結果を以下の表1に示す。
表1.アスピリン及びそのプロドラッグによるライジング抑制率
本結果は、酢酸ジエチルアミノエチルアセチルサリチラートはアスピリンよりも良い鎮痛活性を表すということを示す。式(1)で表される他の化合物は類似した鎮痛活性を示す。
【0014】
解熱活性:ラットは発熱物質として滅菌された大腸菌懸濁液を受容した。56匹のラットを7つのグループに分けた。対照群はグループAである。2時間後、アスピリン(ASA,B1に100mg/kg、B2に150mg/kg)及び酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,C1に100mg/kg、C2に150mg/kg)を経口投与し、酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA,D1に100mg/kg、D2に150mg/kg)を経皮投与した。ラットの体温を試験化合物の投与前後90分間隔で測った。結果を以下の表2に示す。
表2.アスピリン及びそのプロドラッグの解熱活性
本結果から、酢酸ジエチルアミノエチルアセチルサリチラートは100mg/kg用量において解熱活性を示し、でアスピリンよりも良い解熱活性を示したということが分かる。本結果は、酢酸ジエチルアミノエチルアセチルサリチラートの経皮投与は経口投与よりも良いということを示す。式(1)で表される他の化合物は類似した解熱活性を示す。
【0015】
抗炎症活性:酢酸ジエチルアミノエチルアセチルサリチラート50mg/kgをラットに経口又は経皮投与し、アスピリン50mg/kgを経口投与した。60分後、カラゲニン溶液をラットの足裏に皮下投与した。後ろ足の体積をカラゲニンの投与後毎時間測定し、足の体積の増加率を計算し、隆起率(rate of swelling)(%)と称した。得られた結果を図4に示す。
本結果は、酢酸ジエチルアミノエチルアセチルサリチラートは、50mg/kgでの経口投与及び経皮投与に対して、アスピリンの抗炎症活性よりも良い抗炎症活性を表したということを示す。式(1)で表される他の化合物は類似した抗炎症活性を示す。
【0016】
高用量の経口アセチルサリチル酸はまた、シクロオキシゲナーゼ活性の抑制による抗反応−抗喘息活性を示すということも知られている(Bianco,Sebastiano,U.S.Pat.No.5,570,559)。それらの非常に高い膜透過率のため、これらのプロドラッグは患者の口又は鼻の中にスプレーすることにより喘息の治療に使用され得る。これらは抗炎症性のため、アクネを治療するために使用され得る。これらは内皮機能障害の治療及び予防に対しても同様に使用され得る。
【0017】
上記の式(1)で表される化合物はASA若しくはその類似体、又はASAの官能誘導体若しくはその類似体から調製され得る。例えば、式(2)で表される酸ハロゲン化物又は混合酸無水物である。
【化2】
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、Yはハロゲン、アルコキシカルボニル又は置換アリールオキシカルボニルオキシを表し、これは式(3)で表される化合物と反応する。
【化3】
式中、R1はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、n=0、1、2、3、4、5……である。
【0018】
上記の式(1)で表される化合物はASA又はその類似体から、カップリング試薬、例えば、N,N’−ジシクロヘキシルカルボジイミド、N,N’−ジイソプロピルカルボジイミド、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボラート、O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムヘキサフルオロホスファート、ベンゾトリアゾール−1−イル−オキシ−トリス(ジメチルアミノ)ホスホニウムヘキサフルオロホスファート等を使用することにより、式(3)で表される化合物との反応によって調製され得る。
XがOを表すとき、上記の式(1)で表される化合物は、アセチルサリチル酸若しくはその類似体の金属塩又は有機塩基塩から、式(4)で表される化合物との反応によって調製され得る。
【化4】
式中、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、Zはハロゲン又はp−トルエンスルホニルを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【0019】
XがOを表すとき、上記の式(1)で表される化合物は、式(5)で表される、アセチルサリチル酸又はその類似体の固定化された塩基塩から調製され得る。
【化5】
式中、Rは交差結合樹脂を表し、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、Bは塩基、例えば、ピリジン、ピペリジン、トリエチルアミン又は他の塩基を表す。これは式(4)で表される化合物と反応する。
【0020】
本発明は、通例、補助剤及び賦形剤に加えて、例えば、経口投与のための錠剤、カプセル剤又は液剤の形態で、及び経皮投与のための液剤、ローション剤、軟膏剤、乳剤又はゲル剤の形態で、式(1)で表されるアセチルサリチル酸及びその類似体のプロドラッグを含む薬剤調製に関する。式(1)で表される新規な活性化合物は、人又は動物において、アスピリン治療可能な状態を治療するために、ビタミン、例えばA、B、C、E若しくはベータ−カロテン、又は他の薬物、例えば、葉酸等と併用され得る。
式(1)で表される化合物又は少なくとも式(1)で表される化合物を活性成分として含む組成物の経皮治療応用システムは、人又は動物において、アスピリン治療可能な状態を治療するために、特に血栓症及び他の心臓血管疾患の予防、並びに癌予防に対して発展され得る。これらのシステムは、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得る。最も好ましいシステムは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有する。放出の割合を制御することによって、このシステムは、アスピリンが絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、アスピリンの副作用を減少させることを可能にする。これらのシステムは手首、足首、腕、足又は体のどこでも付けることが可能である。
【発明の効果】
【0021】
これらのアスピリンのプロドラッグは、親油性部分及び親水性部分(生理学的pHではプロトン化された形態で存在するアミン基)を有する。これらのプロドラッグの正に荷電されたアミノ基は2つの優れた利点を有する。第一に、薬物の溶解性を大きく増大させる。これらの新規なプロドラッグが、錠剤、カプセル剤、液剤又は懸濁剤のような投与形態で経口投与されるとき、これらは直ちに胃液中で溶解するだろう。第二に、これらのプロドラッグのアミノ基上にある正電荷は、膜のホスファート頭基上にある負電荷と結合するだろう。従って、膜の外側の局所的な濃度が非常に高くなり、高濃度の領域から低濃度の領域へのこれらのプロドラッグの通過を促進するだろう。これらのプロドラッグが膜に入るとき、親水性部分は、プロドラッグを半液体状濃縮水溶液又は懸濁液である細胞質ゾルの中に押し込む。胃腸管での短い滞留のため、プロドラッグは胃粘膜細胞に損傷を引き起こさないだろう。実験結果は、プロドラッグの80%は薬物それ自体に変化して戻ったということを示す。プロドラッグははるかに高い吸収率を有するため、従って、プロドラッグは同用量でアセチルサリチル酸それ自体よりも強い作用を有するだろう。
実験結果は、プロドラッグ、酢酸ジエチルアミノエチルアセチルサリチラートはアセチルサリチル酸それ自体よりも〜400倍速く、エチルアセチルサリチラートよりも〜100倍速く人の皮膚を通して拡散することを示す。生体内での、酢酸ジエチルアミノエチルアセチルサリチラートの無処理ヘアレスマウスの皮膚を通しての透過率は非常に高かった。アスピリンを経口投与するとき、コーティングされていないアスピリンの錠剤がサリチラート血漿レベルのピークに達するのに2時間かかり、コーティングされているアスピリンの錠剤はもっと長い。しかし、酢酸ジエチルアミノエチルアセチルサリチラートは、サリチラート血漿レベルのピークに達するのにたったの20分しかかからなかった。最も興奮させる結果は、プロドラッグを経口投与だけでなく、様々な種類の薬物治療のために経皮投与することも可能であり、たいていのアスピリンの副作用、最も著しくは胃腸障害であり、例えば、消化不良、胃十二指腸出血、胃潰瘍及び胃炎、を回避するだろうということである。
【図面の簡単な説明】
【0022】
【図1】フランツ細胞(n=5)での、単離されたヒト皮膚組織を透過するアセチルサリチル酸(ASA)、エチルアセチルサリチラート(E−ASA)及び酢酸ジエチルアミノエチルアセチルサリチラート(DEAE−ASA)の累積量。ASA及びE−ASAを20%懸濁液として用いた。DEAE−ASAを20%溶液として用いた。それぞれ、媒体はpH7.4ホスファート緩衝液(0.2M)であった。
【図2】アセチルサリチル酸(ASA)又は酢酸ジエチルアミノエチルアセチルサリチラート300mgをヘアレスマウス(n=5)の背中に局所塗布後の、サリチル酸(SA)総血漿レベル。
【図3】アスピリン200mg/kg経口投与後(B)、酢酸ジエチルアミノエチルサリチラート200mg/kg経口投与後(C)及び経皮投与後(D)の、マウス尻尾の痛覚閾値延長時間。Aは対照線である。
【図4】カラゲニン投与後の隆起率(%)。カラゲニン投与1時間前に、アスピリン100mg/kg経口投与(B)、酢酸ジエチルアミノエチルサリチラート100mg/kg経口投与(C)及び経皮投与(D)した。Aは対照線である。 構造1.式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ又はアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ又はアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、シトラート、サリチラート又は陰イオンを表し、n=1、2、3、4、5……である。
【発明を実施するための形態】
【0023】
酢酸ジエチルアミノエチルアセチルサリチラートの調製
o−アセチルサリチル酸18g(0.1mol)をクロロホルム180mlに溶解した。重炭酸ナトリウム12.5g(0.15mol)を溶液に加えた。水(20ml)を撹拌しながら加えた。混合物を30分間撹拌した後、無水硫酸ナトリウムを加えた。臭化水素酸ジエチルアミノエチルブロミド(diethylaminoethyl bromide.HBr)39g(0.15mol)を混合物に加え、混合物を室温で5時間撹拌した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物31g(91%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:339.38、計算%C:60.07、H:7.44、N:4.15、O:28.22、実測%C:60.16、H:7.42、N:4.13、O:28.29、1H-NMR(400MHz,CDCL3):デルタ:1.55(t,6H)、2.08(s,3H)、2.20(s,3H)、3.28(m,4H)、3.70(m,2H)、4.68(m,2H)、6.5(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【0024】
酢酸ジメチルアミノエチルアセチルサリチラートの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエタノール8.9gを反応混合物に加えた。混合物を室温で3時間撹拌する。酢酸6gを撹拌しながら反応混合物に加える。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物29g(93%)を得た。吸湿性生成物、水中での溶解性:350mg/ml、元素分析:C15H21NO6、分子量:311.33、計算%C:57.87、H:6.80、N:4.50、O:30.83、実測%C:57.82、H:6.85、N:4.48、O:30.85、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.71(m,2H)、4.69(m,2H)、6.9(b,1H)、7.18(m,1H)、7.20(m,1H)、7.47(m,1H)、7.93(m,1H)。
【0025】
酢酸S−ジメチルアミノエチルアセチルチオサリチラートの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエチルメルカプタン9.3gを反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物28g(87%)を得た。吸湿性生成物、水中での溶解性:320mg/ml、元素分析:C15H21NO5S、分子量:327.4、計算%C:55.03、H:6.47、N:4.28、O:24.43、S:9.79、実測%C:55.02、H:6.45、N:4.35、O:24.49、S:9.69、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.31(t,2H)、3.91(m,2H)、6.9(b,1H)、7.26(m,1H)、7.28(m,1H)、7.55(m,1H)、7.94(m,1H)。
【0026】
酢酸N−ジメチルアミノエチルアセチルサリチルアミドの調製
o−アセトキシベンゾイルクロリド19.9g(0.1mol)をクロロホルム100mlに溶解した。混合物を0℃に冷却した。トリエチルアミン15ml及びジメチルアミノエチルアミン8.9gを反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×30ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物28g(90.2%)を得た。吸湿性生成物、水中での溶解性:350mg/ml、元素分析:C15H22N2O5、分子量:310.35、計算%C:58.05、H:7.15、N:9.03、O:25.78、実測%C:58.02、H:7.18、N:8.98、O:25.83、1H-NMR(400MHz,CDCL3):デルタ:2.09(s,3H)、2.21(s,3H)、2.90(s,6H)、3.54(m,2H)、3.64(t,2H)、6.9(b,1H)、7.8(b,1H)、7.25(m,1H)、7.26(m,1H)、 7.48(m,1H)、7.92(m,1H)。
【0027】
酢酸S−ジエチルアミノエチルプロピオニルチオサリチラートの調製
o−アセチルサリチル酸18g(0.1mol)をジクロロメタン(DCM)100mlに溶解した。混合物を0℃に冷却した。1,3−ジシクロヘキシルカルボジイミド20.6gを反応混合物に加えた。混合物を0℃で30分間撹拌した。ジエチルアミノプロピルメルカプタン14.8g(0.1mol)を反応混合物に加えた。混合物を室温で3時間撹拌した。酢酸6gを撹拌しながら反応混合物に加えた。固形副生成物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。有機溶液を蒸発させた。乾燥後、目的生成物32g(86.6%)を得た。吸湿性生成物、水中での溶解性:280mg/ml、元素分析:C18H27NO5S、分子量:369.48、計算%C:58.51、H:7.37、N:3.79、O:21.65、S:8.68、実測%C:58.53、H:7.39、N:3.75、O:21.68、S:8.65、1H-NMR(400MHz,CDCL3):デルタ:1.09(t,3H)、1.56(t,6H)、2.21(s,3H)、2.27(m,2H)、3.28(m,4H)、3.31(m,2H)、3.91(m,2H)、6.8(b,1H)、7.25(m,1H)、7.26(m,1H)、7.48(m,1H)、7.92(m,1H)。
【0028】
酢酸N−ジエチルアミノプロピルアセチルサリチルアミドの調製
o−アセチルサリチル酸18g(0.1mol)をアセトニトリル100mlに溶解した。O−(ベンゾトリアゾール−1−イル)−N,N,N’,N’−テトラメチルウロニウムテトラフルオロボラート32.1g及びトリエチルアミン30mlを反応混合物に加えた。ジメチルアミノプロピルアミン13.1gを反応混合物に加えた。混合物を室温で3時間撹拌した。溶媒を蒸発させた。酢酸エチル250mlを反応混合物に加え、混合物を水(3×100ml)で洗浄した。有機溶液を無水硫酸ナトリウム上で乾燥させた。硫酸ナトリウムを濾過により除去した。酢酸6gを撹拌しながら反応混合物に加えた。ヘキサン(200ml)を加えた。固形生成物を濾過により集めた。乾燥後、目的生成物32g(90.8%)を得た。吸湿性生成物、水中での溶解性:280mg/ml、元素分析:C18H28N2O5、分子量:352.43、計算%C:61.34、H:8.01、N:7.95、O:22.70、実測%C:61.25、H:8.05、N:7.96、O:22.74、1H-NMR(400MHz,CDCL3):デルタ: 1.56(t,6H)、2.03(m,2H)、2.09(s,3H)、2.21(s,3H)、3.24(m,2H)、3.20(m,2H)、3.24(m,2H)、6.9(b,1H)、7.8(b,1H)、7.25(m,1H)、7.26(m,1H)、7.48(m,1H)、7.92(m,1H)。
【0029】
酢酸ジプロピルアミノエチルアセチルサリチラートの調製
o−アセチルサリチル酸ナトリウム20.3g(0.1mol)をクロロホルム180mlに懸濁した。臭化水素酸ジプロピルアミノエチルブロミド28.8g(0.1mol)を混合物に加え、混合物を室温で5時間撹拌した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物30g(81.6%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:367.44、計算%C:62.11、H:7.96、N:3.81、O:26.13、実測%C:62.07、H:7.99、N:3.78、O:26.17、1H-NMR(400MHz,CDCL3):デルタ:0.97(t,6H)、1.77(m,4H)、2.20(s,3H)、3.25(m,4H)、3.70(m,2H)、4.69(m,2H)、6.8(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【0030】
酢酸ジプロピルアミノエチルアセチルサリチラートの調製
高分子結合トリエチルアミン(3mmol/g、100〜200mesh)60gをクロロホルム180mlに懸濁した。o−アセチルサリチル酸18g(0.1mol)を撹拌しながら混合物に加えた。臭化水素酸ジプロピルアミノエチルブロミド43g(0.15mol)を混合物に加え、混合物を室温で5時間撹拌した。高分子を濾過により除去し、テトラヒドロフラン(3×50ml)で洗浄した。酢酸ナトリウム8.2g(0.1mol)を撹拌しながら反応混合物に加えた。混合物を2時間撹拌する。固形物を濾過により除去し、クロロホルム(3×50ml)で洗浄した。溶液を真空中で100mlに濃縮する。次いでヘキサン300mlを溶液に加えた。固形生成物を濾過により集め、ヘキサン(3×100ml)で洗浄した。乾燥後、目的生成物31g(91%)を得た。吸湿性生成物、水中での溶解性:300mg/ml、元素分析:C17H25NO6、分子量:339.38、計算%C:60.07、H:7.44、N:4.15、O:28.22、実測%C:60.16、H:7.42、N:4.13、O:28.29、1H-NMR(400MHz,CDCL3):デルタ:1.55(t,6H)、2.08(s,3H)、2.20(s,3H)、3.28(m,4H)、3.70(m,2H)、4.68(m,2H)、6.5(b,1H)、7.17(m,1H)、7.19(m,1H)、7.45(m,1H)、7.94(m,1H)。
【産業上の利用可能性】
【0031】
式(1)で表されるプロドラッグはアスピリンよりも優れている。これらは人又は動物において、アスピリン治療可能な状態の治療に医薬的に使用され得る。これらは軽度から中程度の疼痛並びに関節炎及び他の炎症性状態の長期緩和待期療法に使用され得る。これらは単独で又は補助薬として、川崎病、術後血栓塞栓症及び不安定狭心症の治療に使用され得る。これらは急性小児胃腸炎において、便の量を減少させ、かつ体重を増加させるために、大動脈冠状動脈バイパス移植閉塞を予防するために補助薬として使用され得て、また慢性心房細動における血栓塞栓性合併症を予防するために使用され得る。これらは補助薬として、頚動脈内膜切除術中の血小板凝集及びトロンボキサン抑制を減らすために使用され得、白内障の進行を遅らせるため、冠状動脈形成術後狭窄の再発を予防するため、及び多発脳梗塞性痴呆患者における認知機能及び大脳の血流を改善するために処方され得る。これらは補助薬として、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含め糖尿病誘発性合併症において血漿グルコースを下げるために使用され、そして総死亡率及び心血管死亡率を減少させるために使用され得る。これらは血液透析シャント血栓症の発生を減らすため、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生を減少させるため、及び末梢閉塞性動脈疾患の進行を遅らせるために処方され得る。これらは大腸癌及び直腸癌、並びに人工心臓弁患者における動脈塞栓性合併症の予防に使用され得る。これらはハイリスク女性における妊娠誘発性高血圧及び妊娠中毒症の発生を減らすために処方され得る。高用量の経口アセチルサリチル酸はまた、シクロオキシゲナーゼ活性の抑制による抗反応活性を示すということも知られている(Bianco,Sebastiano,U.S.Pat.No.5,570,559)。それらの非常に高い膜透過率のため、これらのプロドラッグは患者に吸入することによって喘息の治療に使用され得る。これらは抗炎症性のため、アクネを治療するために使用され得る。これらは内皮機能障害の治療及び予防に対して使用され得る。
【特許請求の範囲】
【請求項1】
下式(1)で表される化合物。
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、オキサラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【請求項2】
請求項1に記載の式(1)で表される化合物を調製する方法。
【請求項3】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、人又は動物において、アスピリン治療可能な状態を治療するために、経口投与又は経皮投与可能である化合物又は組成物であって、前記アスピリン治療可能な状態が、歯痛、頭痛、関節炎及び他の炎症性疼痛、熱、子癇前症、心臓発作、川崎病、術後血栓塞栓症及び不安定狭心症、急性小児胃腸炎、大動脈冠状動脈バイパス移植閉塞、慢性心房細動における血栓塞栓性合併症、白内障、血小板凝集及びトロンボキサン抑制、冠状動脈形成術後狭窄の再発、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含む糖尿病誘発性合併症、血液透析シャント血栓症の発生、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生、末梢閉塞性動脈疾患の進行、大腸癌及び直腸癌の予防、及び人工心臓弁患者における動脈塞栓性合併症を含むが、これに限定されるものではない化合物又は組成物。
【請求項4】
身体の一部に(溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で)経皮投与して、式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物の治療有効血漿レベルを送達することにより、人又は動物において、アスピリン治療可能な状態を治療する方法。
【請求項5】
人又は動物において、疼痛、例えば、頭痛、歯痛、筋肉痛、並びに関節炎及び他の炎症性疼痛を局所的に治療する方法であって、治療有効量の式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物を炎症部位に投与することを特徴とする方法。
【請求項6】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、アクネ又は他の皮膚病を治療するために、溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で経皮投与され得る化合物又は組成物。
【請求項7】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、喘息を治療するために、口若しくは鼻又は体の他の部分を通してスプレーすることにより投与される化合物又は組成物。
【請求項8】
人又は動物において、アスピリン治療可能な状態を治療するための、特に血栓症及び他の心臓血管疾患を予防するための、並びに癌予防のための、式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物の経皮治療応用システムであって、かつ、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得、かつ、最も好ましくは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有し、放出の割合を制御することによって、アスピリンが絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、アスピリンの副作用を減少させることを可能にするシステム。
【請求項1】
下式(1)で表される化合物。
式中、R1はCH3、C2H5、C3H7又は他の低級アルキル基を表し、R2はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R3はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、R4はH、1〜6炭素原子を有するアルキル、アルキルオキシ若しくはアルケニル残基のいずれか一つ、又はアリール残基を表し、XはO、S又はNHを表し、A-はCl-、Br-、F-、I-、AcO-、アセチルサリチラート、オキサラート、シトラート、サリチラート又は陰イオンを表し、n=0、1、2、3、4、5……である。
【請求項2】
請求項1に記載の式(1)で表される化合物を調製する方法。
【請求項3】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、人又は動物において、アスピリン治療可能な状態を治療するために、経口投与又は経皮投与可能である化合物又は組成物であって、前記アスピリン治療可能な状態が、歯痛、頭痛、関節炎及び他の炎症性疼痛、熱、子癇前症、心臓発作、川崎病、術後血栓塞栓症及び不安定狭心症、急性小児胃腸炎、大動脈冠状動脈バイパス移植閉塞、慢性心房細動における血栓塞栓性合併症、白内障、血小板凝集及びトロンボキサン抑制、冠状動脈形成術後狭窄の再発、糖尿病並びに糖尿病性網膜症、壊死性潰瘍及び糖尿病性タンパク尿を含む糖尿病誘発性合併症、血液透析シャント血栓症の発生、1型膜性増殖性糸球体腎炎患者における腎機能低下及び末期腎不全の発生、末梢閉塞性動脈疾患の進行、大腸癌及び直腸癌の予防、及び人工心臓弁患者における動脈塞栓性合併症を含むが、これに限定されるものではない化合物又は組成物。
【請求項4】
身体の一部に(溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で)経皮投与して、式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物の治療有効血漿レベルを送達することにより、人又は動物において、アスピリン治療可能な状態を治療する方法。
【請求項5】
人又は動物において、疼痛、例えば、頭痛、歯痛、筋肉痛、並びに関節炎及び他の炎症性疼痛を局所的に治療する方法であって、治療有効量の式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物を炎症部位に投与することを特徴とする方法。
【請求項6】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、アクネ又は他の皮膚病を治療するために、溶液、スプレー、ローション、軟膏、エマルジョン又はゲルの形態で経皮投与され得る化合物又は組成物。
【請求項7】
式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物であって、喘息を治療するために、口若しくは鼻又は体の他の部分を通してスプレーすることにより投与される化合物又は組成物。
【請求項8】
人又は動物において、アスピリン治療可能な状態を治療するための、特に血栓症及び他の心臓血管疾患を予防するための、並びに癌予防のための、式(1)で表される化合物又は少なくとも請求項1に記載の式(1)で表される化合物を活性成分として含む組成物の経皮治療応用システムであって、かつ、活性物質含有マトリックス層及び非透過性支持層からなる包帯又はパッチであり得、かつ、最も好ましくは、活性物質貯蔵庫であり、これが皮膚に面する透過性底面を有し、放出の割合を制御することによって、アスピリンが絶えず最適治療血中レベルに達することを可能にし、効能を増大させ、アスピリンの副作用を減少させることを可能にするシステム。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2009−542797(P2009−542797A)
【公表日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願番号】特願2009−518987(P2009−518987)
【出願日】平成18年7月9日(2006.7.9)
【国際出願番号】PCT/IB2006/052318
【国際公開番号】WO2008/007171
【国際公開日】平成20年1月17日(2008.1.17)
【出願人】(509011581)テックフィールズ バイオケム カンパニー リミテッド (10)
【出願人】(509023539)
【Fターム(参考)】
【公表日】平成21年12月3日(2009.12.3)
【国際特許分類】
【出願日】平成18年7月9日(2006.7.9)
【国際出願番号】PCT/IB2006/052318
【国際公開番号】WO2008/007171
【国際公開日】平成20年1月17日(2008.1.17)
【出願人】(509011581)テックフィールズ バイオケム カンパニー リミテッド (10)
【出願人】(509023539)
【Fターム(参考)】
[ Back to top ]